Настоящее изобретение относится к пиримидинилиндольным соединениям, композициям и их применению в терапевтических целях, в частности при лечении рака и воспалительных заболеваний.
IKKβ является ключевой киназой, регулирующей сигнальные пути, связанные с воспалением и стрессом, в связи с чем полагают, что она связана с развитием различных заболеваний человека от рака до воспалительных заболеваний.
Пиримидинилиндольные соединения, которые применяют в качестве ингибиторов киназ, уже известны в данной области. См. WO 04089913 (ингибиторы IKKP), WO 06038001, и WO 06075152. Кроме того, в данной области также известны соединения пиримидинилбензотиофена, которые применяют в качестве ингибиторов IKKβ. См. WO 07092095.
Существует необходимость в эффективных ингибиторах IKKβ, которые можно применять для лечения рака или воспалительных заболеваний. Существует также потребность в таких соединениях, которые проявляют синергетический эффект в сочетании с TNFα или винкристином (VCR).
Настоящее изобретение относится к новым соединениям пиримидинилиндола для клинического применения при лечении рака и воспалительных заболеваний посредством ингибирования IKKβ. В частности, настоящее изобретение относится к новым пиримидинилиндольным соединениям формулы:
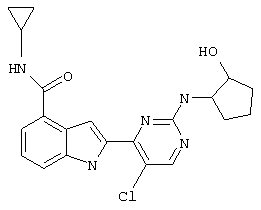
или фармацевтически приемлемым солям этого соединения.
Настоящее изобретение также относится к способу лечения ракового заболевания, выбранного из группы, состоящей из множественной миеломы, рака толстой кишки, крупноклеточного рака легкого, глиобластомы, рака поджелудочной железы и рака яичников у млекопитающего, способ включающий введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения согласно настоящему изобретению или его соли.
Настоящее изобретение также относится к способу лечения воспалительных заболеваний выбранных из группы, состоящей из ревматоидного артрита, хронической обструктивной болезни легких, бронхиальной астмы, рассеянного склероза и воспалительных заболеваний кишечника у млекопитающего, способ включающий введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения согласно настоящему изобретению или его соли.
Настоящее изобретение также относится к фармацевтическим композициям, содержащим соединение согласно настоящему изобретению или его соль в комбинации с одним или несколькими фармацевтически приемлемыми носителями, разбавителями или наполнителями. В частном варианте реализации настоящего изобретения композиция дополнительно содержит один или более других терапевтических агентов. В другом варианте реализации дополнительным терапевтическим агентом является TNFα. Согласно другому варианту реализации дополнительным терапевтическим агентом является винкристин.
Настоящее изобретение также описывает соединение или его соль для применения в терапии. Настоящее изобретение также описывает соединение или его соль для применения в лечении ракового заболевания. Дополнительно, настоящее изобретение относится к применению соединения согласно настоящему изобретению или его соли в производстве лекарственного средства для лечения ракового заболевания. В частности, раковое заболевание выбрано из группы, состоящей из множественной миеломы, рака толстой кишки, крупноклеточного рака легкого, крупноклеточный рак легкого, глиобластома, рак поджелудочной железы и рака яичников. Одним из вариантов реализации является множественная миелома. Другим вариантом реализации является рак толстой кишки. Другим вариантом реализации является крупноклеточный рак легкого. Другим вариантом реализации является глиобластома. Другим вариантом реализации является рак поджелудочной железы. Другим вариантом реализации является рак яичников. Настоящее изобретение также описывает соединения настоящего изобретения или их соли для применения при лечении воспалительных заболеваний. Кроме того, настоящее изобретение относится к применению соединения согласно настоящему изобретению или его соли в производстве лекарственного средства для лечения воспалительных заболеваний. В частности, такое воспалительное заболевание выбрано из группы, состоящей из ревматоидного артрита, хронической обструктивной болезни легких, бронхиальной астмы, рассеянного склероза и воспалительных заболеваний кишечника. Одним из вариантов реализации является ревматоидный артрит. Другим вариантом реализации является хроническая обструктивная болезнь легких. Другим вариантом реализации является астма. Другим вариантом реализации является рассеянный склероз. Другим вариантом реализации является воспалительные заболевания кишечника. Кроме того, настоящее изобретение относится к фармацевтической композиции для лечения ракового заболевания, выбранного из группы, состоящей из множественной миеломы, рака толстой кишки, крупноклеточного рака легкого, глиобластомы, рака поджелудочной железы и рака яичников, при этом активным компонентом такой композиции является соединение настоящего настоящему изобретению или его соль. Дополнительно, настоящее изобретение относится к фармацевтической композиции для лечения воспалительных заболеваний выбранных из группы, состоящей из ревматоидного артрита, хронической обструктивной болезни легких, бронхиальной астмы, рассеянного склероза и воспалительного заболевания кишечника, при этом активным компонентом такой композиции является соединение согласно настоящему изобретению или его соль.
Соединения и соли согласно настоящему изобретению получают по существу так, как проиллюстрировано как в схемах, так и в примерах. Кроме того, все соединения настоящего изобретения и их соли существуют в виде диастереомеров или энантиомеров благодаря замещениям в циклопентиловом кольце. Использование конкретных диастереомеров в качестве реагентов позволяет получить оптически чистые соединения. Оптически чистые соединения также можно получить с помощью хроматографии/хиральной хроматографии на основе смеси диастереоизомеров или рацематов, соответствующих соединениям настоящего изобретения или его солям.
Схема I
Синтез соединений согласно настоящему изобретению
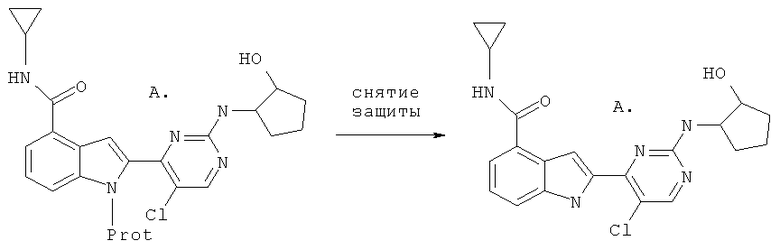
Prot=этоксиметил или трет-бутоксикарбонил
Соединения настоящего изобретения получают посредством снятия защиты с защищенных предшественников (A) с помощью обработки HCl, трифторуксусной кислотой (TFA) или п-толуолсульфокислотой (TsOH) в метаноле или этаноле.
Схема II
Синтез предшественников (А)
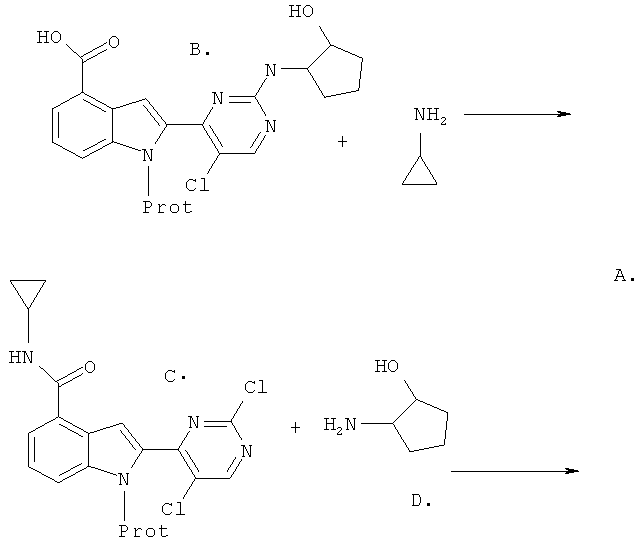
Предшественники A получают двумя путями, как показано выше. В верхней реакции, индол-4-карбоновая кислота (B) связывается с циклопропиламином в присутствии дегидратирующего агента такого, как бензотриазол-1-илокситрис(диметиламино)-фосфония гексафторфосфат (ВОР) или дициклогексилкарбодиимид. Специалистам в области органического синтеза известно, что такие реакции амидной конденсации можно проводить на любой стадии органического синтеза, ведущего к соединениям формулы (I).
В нижней реакции 2-аминоциклопентанол (D) замещает хлор в хлорпиримидиновом промежуточном продукте (C) в присутствии основания такого, как гидрид натрия, диизопропилэтиламин (DIPEA) или карбонат калия при повышенной температуре (70-130°C) в таких растворителях, как диметилсульфоксид (ДМСО) или диметилформамид (DMF). Специалистам в области органического синтеза известно, что такие реакции замещения хлора можно проводить на любой стадии органического синтеза, ведущего к соединениям настоящего изобретения (I).
Схема III
Синтез карбоновой кислоты (B)
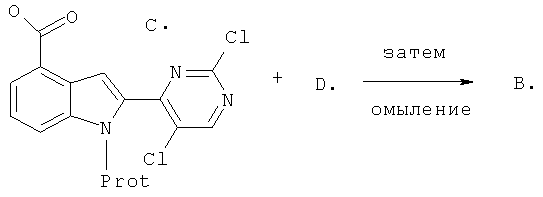
Индол-4-карбоновые кислоты (B) получают путем замещения хлора в пиримидиниловом эфире (E) на 2-аминоциклопентанол (D) аналогично нижней реакции из Схемы II с последующим омылением промежуточного эфира карбоновой кислоты.
Схема IV
Синтез пиримидинилхлоридов (C) и (E)
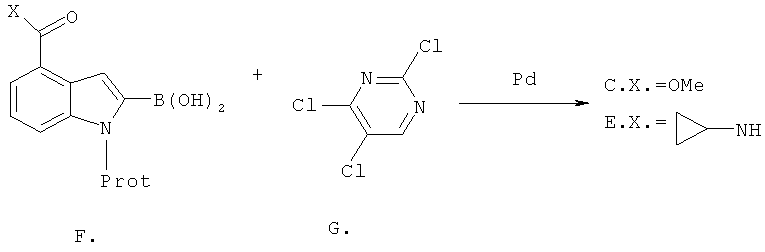
Пиримидинилхлориды (C) и (E) получают путем катализируемых палладием реакций связывания между индол-2-борной кислотой (F или их С1-C3 алкильные эфиры борной кислоты) и коммерчески доступными трихлорпиримидинами (G). Катализатором является либо Pd(OAc)2, Pd(PPh3)4, либо PdCl2(dppf), при этом реакции связывания проходят при повышенной температуре (50-110°C) в полярных апротонных растворителях, например, тетрагидрофуране (ТГФ).
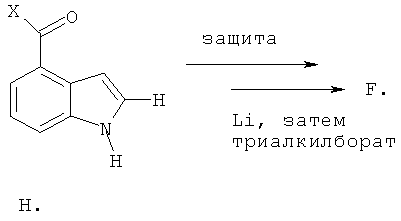
Индольные предшественники (H), которые являются коммерчески доступными или которые получают с помощью методов, описанных в литературе, сначала защищают по N1-положению в присутствии основания с помощью хлорметилэтилового эфира, C1-C3 триалкилсилилхлоридов, или ди-трет-бутилдикарбоната с последующим замещением водорода в положении 2 индола на литий и обработкой С1-C3 триалкилэфирами борной кислоты с получением борной кислоты или боратов (F).
Настоящее изобретение включает различные стереоизомеры и их смеси. Стереоизомеры представляют собой энантиомеры и диастереомеры, а также смеси энантиомеров или диастереомеров. Индивидуальные стереоизомеры соединений согласно настоящему изобретению могут быть получены синтетическим путем из коммерчески доступных исходных материалов, которые содержат асимметричные или хиральные центры, или посредством получения рацемических смесей с последующим разделением с помощью методов, хорошо известных специалистам в данной области. Примером такого разделения могут служить: (1) присоединение смеси энантиомеров к хиральному вспомогательному реагенту, разделение полученной смеси диастереомеров с помощью перекристаллизации или хроматографии, и, если это необходимо, выделение оптически чистого продукта, свободного от вспомогательного реагента, согласно методу, описанному в Furniss, Hannaford, Smith и Tatchell, "Учебник практической органической химии Вогеля", 5-е издание (1989), Longman Scientific & Technical, Essex CM20 2JE, Англия, или (2) прямое разделение смеси оптических энантиомеров на хиральных хроматографических колонках или (3) дробные методы перекристаллизации.
Для того чтобы дать названия следующим соединениям, используют ChemDraw Ultra 10.0.
Соединение 1
N-циклопропил-2-(2,5-дихлорпиримидин-4-ил)-1-(этоксиметил)-1H-индол-4-карбоксамид
(A) Получение метил-1-(этоксиметил)-1H-индол-4-карбоксилата. В атмосфере азота к раствору метил-1-(этоксиметил)-1H-индол-4-карбоксилата (80 г, 0.46 моль) в ТГФ (700 мл) добавляли гексаметилдисилазид калия (1M в ТГФ, 550 мл, 0.55 моль) по каплям при 0°C. Смесь перемешивали при 0°C в течение 30 минут (мин), после чего добавляли хлорметилэтиловый эфир (51 мл, 0.55 моль) при 0-5°С. После этого реакционную смесь перемешивали при комнатной температуре в течение 3 часов (ч), осторожно охлаждали с помощью 300 мл воды и экстрагировали этилацетатом (ЕА, 3×300 мл). Объединенные экстракты промывали насыщенным водным раствором хлорида натрия (2×400 мл), затем сушили над Na2SO4, фильтровали и концентрировали. Остаток 5 очищали с помощью хроматографии на силикагеле и получали соединение, указанное в названии (80 г, 75%). MS (m/z): 234 (М+Н)+.
(B) Получение 1-(этоксиметил)-1H-индол-4-карбоновой кислоты.
К раствору метилового эфира 1-(этоксиметил)-1H-индол-4-карбоновой кислоты (135 г, 0.58 моль) в метаноле (2 л) добавляли водный раствор гидроксида натрия (68 г, 1.74 моль в 340 мл H2O). Реакционную смесь перемешивали при 50°C в течение 3 часов. Летучие вещества удаляли в вакууме. Остаток подкисляли HCl (2 М) до pH=3-4, затем экстрагировали ЕА (2×700 мл). Объединенные экстракты промывали насыщенным водным раствором хлорида натрия (2×250 мл), сушили над безводным Na2SO4, концентрировали и получали соединение, указанное в названии (123 г, 96%). MS (m/z): 15 220(М+Н)+.
(C) Получение N-циклопропил-1-(этоксиметил)-1H-индол-4-карбоксамида.
К раствору 1-(этоксиметил)-1H-индол-4-карбоновой кислоты (123 г, 0.56 моль) в ТГФ (1,5 л) добавляли циклопропиламин (58 мл, 0.84 моль) и триэтиламин (ТЭА, 167 мл, 20 1.12 моль), а затем O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (240 г, 0.62 моль) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Летучие вещества удаляли в вакууме. Остаток перемешивали в ЕА (1.5 л) и HCl (0.5%, 1 л) в течение 10 минут. Органический слой отделяли, промывали насыщенным водным раствором хлорида натрия (3×100 мл),
25 сушили над безводным Na2SO4, фильтровали и концентрировали. Остаток очищали с помощью хроматографии на силикагеле и получали соединение, указанное в названии (110 г, 77%). MS (m/z): 259 (М+Н)+.
(D) Получение N-циклопропил-2-(2,5-дихлорпиримидин-4-ил)-1 -(этоксиметил)-1H-30 индол-4-карбоксамида.
К раствору диизопропиламина (DIPA, 147 мл, 1.04 моль) в безводном ТГФ (600 мл) добавляли н-BuLi (2.5 М в гексане, 420 мл, 1.04 моль) при температуре -50°C. После добавления смесь перемешивали при -20°C в течение 30 минут, после чего охлаждали до -70°C. Затем добавляли раствор N-циклопропил-1-(этоксиметил)-1H-индол-4-карбоксамида (60 г, 0.23 моль) и три(изопропил)бората (56,4 мл, 0.25 моль) в безводном ТГФ (300 мл). Реакционную смесь перемешивали при -70°C в течение 30 мин. Реакционную смесь медленно нагревали до комнатной температуры, перемешивали в течение 1 ч, а затем охлаждали водным раствором K3PO4·3H2O (195 г, 0.74 моль, 700 мл воды). Сырую реакционную смесь дегазировали и продували азотом три раза. Добавляли 2,4,5-трихлорпипримидин (51 г, 0.27 моль) и дифенилфосфиноферроцендихлорид палладия (PdCl2(dppf)·CH2Cl2, 19.2 г, 0.023 моль), перемешивали в атмосфере азота при температуре кипячения в течение 1.5 часов. Летучие вещества удаляли в вакууме. Остаток экстрагировали с помощью дихлорметана (DCM, 3×500 мл). Объединенные экстракты промывали насыщенным водным раствором хлорида натрия (3×100 мл), сушили над безводным Na2SO4, фильтровали и концентрировали. Остаток очищали с помощью хроматографии на силикагеле и получали соединение, указанное в названии (30 г, 32%). MS (m/z): 405 [(М+1)+, 35Cl, 35Cl], 407 [(М+1)+, 35Cl, 37Cl] и 409 [(М+1)+, 37Cl, 37Cl].
Пример 1
2-{5-Хлоро-2-[(1R,2S)-2-гидроксициклопентиламино]пиримидин-4-ил}-N-циклопропил-1H-индол-4-карбоксамид гидрохлорид
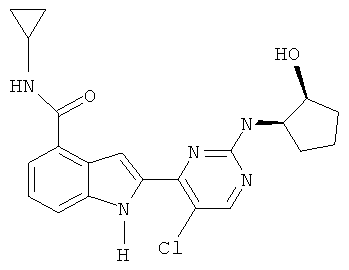
Смесь N-циклопропил-2-(2,5-дихлорпиримидин-4-ил)-1-(этоксиметил)-1H-индол-4-карбоксамид (10 г, 25 ммоль), (1S,2R)-2-аминоциклопентанол гидрохлорид (4.1 г, 30 ммоль) и DIPEA (5 мл, 30 ммоль) в ДМСО (70 мл) перемешивали при 100°C в течение 3 ч, затем выливали в воду и экстрагировали ЕА. Объединенные экстракты промывали насыщенным водным раствором хлорида натрия, сушили над Na2S04 и концентрировали в вакууме. Остаток очищали с помощью хроматографии на силикагеле и получали 2-{5-Хлоро-2-[(1R,2S)-2-гиброксициклопентилшино]пиримидин-4-ил}-N-циклопропил-1-(этоксиметил)-1H-индол-4-карбоксамид (7 г, 60.3%). MS (m/z): 470 (35Cl) и 472 (37Cl) (М+Н)+.
Полученный продукт (7 г, 14.9 ммоль) перемешивали с хлористым водородом (6 М в метаноле, 210 мл, 1.26 моль) при 45°C в течение 12 часов. Осадок отфильтровывали, промывали метанолом, сушили в вакууме и получали соединение, указанное в названии (4.7 г, 70%). MS (m/z): 412 (35Cl) и 414 (37Cl) (М+Н)+.
Пример 2
2-{5-Хлоро-2-[(1R,2R)-2-гидроксициклопентиламино]пиримидин-4-ил}-N-циклопропил-1H-индол-4-карбоксамид гидрохлорид
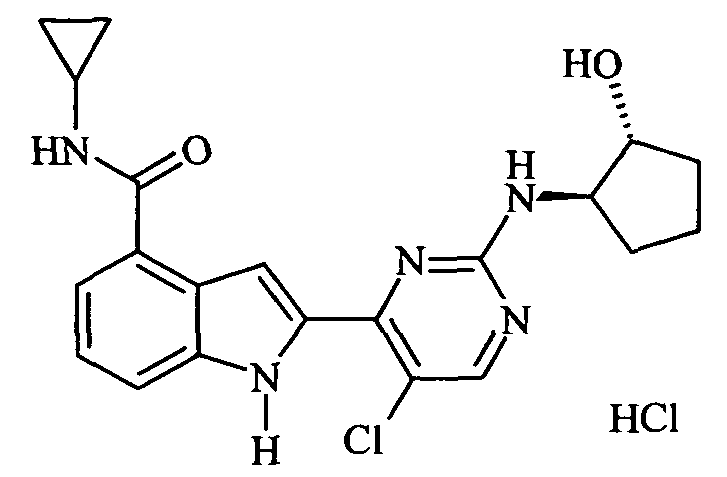
Смесь N-циклопропил-2(2,5-дихлорпиримидин-4-ил)-1-(этоксиметил)-1H-индол-4-карбоксамид (30 г, 75 ммоль), (1R,2R)-2-аминоциклопентанол гидрохлорид (12.3 г, 90 ммоль) и DIPEA (37.5 мл, 225 ммоль) в ДМСО (150 мл) перемешивали при 80°C в течение 16 ч, затем выливали в воду (1 л) и экстрагировали ЕА (2×500 мл). Объединенные экстракты промывали насыщенным водным раствором хлорида натрия (500 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали с помощью хроматографии на силикагеле и получали соединение, указанное в названии 2-{5-Хлоро-2-[(1R,2R)-2-гидроксициклопентиламино]пиримидин-4-ил}-N-циклопропил-1H-индол-4-карбоксамид гидрохлорид (33 г, 93%). МС (м/г): 470 (35Cl) и 472 (35Cl) (M+H)+.
Указанный выше продукт (33 г, 70 ммоль) перемешивали с хлористым водородом (6 М в метаноле, 1 л, 6 моль) при 45°C в течение 16 часов. Осадок отфильтровывали, промывали метанолом (2×300 мл), сушили в вакууме и получали соединение, указанное в названии (26.8 г, 85%). MS (m/z): 412 (35Cl) и 414 (37Cl) (M+H)+.
Внутрисуставное введение доминантно-негативного IKKβ значительно уменьшало тяжесть адъювант-индуцированного артрита у крыс (Tak PP et al., Arthritis Rheum. (2001) 44(8)1897-1907). IKKβ-нокаутные клетки имеют значительные дефекты в экспрессии TNFα-индуцированных цитокинов, хемокинов, или молекул адгезии. С помощью обусловленного или тканеспецифического нокаута IKKβ было обнаружено, что эта киназа необходима для выживания и пролиферации периферических В-клеток и для предотвращения апоптоза, опосредованного TNFα (Li Z-W, Omori AS, Labuda T, Karin М, Rickert RC, ″IKKβ is required for peripheral В cell survival и proliferation″ The J. Immunol, (2003), 170:4630-4637; Maeda S, Chang L, et al. ″IKKβ is required for prevention of apoptosis mediated by cell-bound but not by circulating TNFα″ Immunity, (2003), 19:725-737). Более того, удаление IKKβ из миелоидных клеток также сократило рост колит-ассоциированной опухоли (Greten FR и др., Cell, (2004), 118:285-296). Кроме того, несколько групп исследователей показало, что ингибиторы киназы IKKβ могут вызывать торможение роста клеток и/или усиление TNFα- или TRAIL-индуцированной гибели клеток у различных линий раковых клеток (Takaomi et al. Clinical Cancer Res., (2005), Vol 11:1974-82; Hideshima et al., JBC, (2002) 277:16639-47; Lam et al. Clinical Cancer Res., (2005) Vol 11:28-40).
Кроме того, WO07092095 описывает ингибиторы IKKβ, используемые для лечения множественной миеломы, рака толстой кишки, крупноклеточного рака легкого, глиобластомы, и рака яичников.
Оценка биологических свойств
Биологические свойства соединения согласно настоящему изобретению определяли с помощью следующих исследований. Ингибиторную активность соединения настоящего изобретения в отношении IKKβ оценивали с помощью ферментативного IKKβ-киназного анализа, который измеряет фосфорилирование IκВα субстрата соответствующими киназами, и с помощью анализа жизнеспособности, который измеряет способность соединений ингибировать рост клеток в различных опухолевых клеточных линиях, включая ВхРС-3 и SKOV3-luc. Противоопухолевые эффекты соединения согласно настоящему изобретению определяли как с помощью IVTI (in vivo ингибирование мишени) U87MG модели, которая позволяет оценить эффект изучаемого соединения на ингибирование экспрессии гена TNFα в U87MG ксенотрансплантатах, так и с помощью нескольких моделей эффективности ксенотрансплантата, включая влияние индивидуального соединения на рост опухоли рака яичников человека SKOV-3x-FF-Luci у голых мышей, и комбинированное изучение действия соединения с винкристином (VCR) на ксенотрансплантат рака яичника человека SKOV-3x-FF-Luci или с КПП-11 на ксенотрансплантат рака толстой кишки человека НТ-29. Противовоспалительную активность соединения настоящего изобретения определяли с помощью липополисахаридной (ЛПС) IVTI (in vivo ингибирование мишени) модели, которая позволяет оценить способность соединения ингибировать ЛПС-индуцированную продукцию цитокинов в плазме у мышей.
Противовоспалительную активность соединения настоящего изобретения исследовали как в IVTI (in vivo ингибирование мишени), так и IVEF (in vivo эффективность) моделях. Липополисахаридные (ЛПС) IVTI модели как у мышей, так и у крыс, использовали для оценки способности соединения ингибировать ЛПС-индуцированную продукцию цитокинов в плазме. Модель коллаген-индуцированного артрита (CIA) у мышей и крыс использовали для оценки противовоспалительного и анти-цитокинового эффектов. Модели овальбумин (OVA)-индуцированного воспаления легких у мышей и крыс использовали для оценки влияния соединения на OVA-индуцированное воспаление дыхательных путей. Модель экспериментального аутоиммунного энцефаломиелита (ЕАЕ) у мышей, модель рассеянного склероза у животных, и модель динитробензолсульфокислота (DNBS)-индуцированного колита у крыс также использовали для оценки противовоспалительного и анти-цитокинового эффектов.
Эти исследования показали, что Примеры 1 и 2 являются эффективными ингибиторами IKKβ, и, по меньшей мере, одно из соединений имеет противовоспалительную или противораковую активность.
IKKβ-киназный анализ
IKKβ-киназный анализ использовали для оценки влияния соединения согласно настоящему изобретению на ферментативную активность IKKβ киназы. IKKβ-киназный анализ проводили in vitro с использованием набора IKKβ-ингибиторов для скрининга (Calbiochem., Кат. №СВА044). Все реакции (50 мкл) выполняли, добавляя 10 мкл буфера для киназы (компонент набора, №JA9130), 10 мкл GST - IκВα субстрата (субстрат IKKβ, компонент набора, №JA9127), 10 мкл IKKβ (2,5 нг/лунка, компонент набора, №481404), 10 мкл испытуемого соединения (ДМСО раствор) или H2O, и 10 мкл ATP/MgCl2 (компонент набора, №JA7914), а затем инкубировали при 30°C в течение 30 мин. Содержимое лунок затем отбрасывали. Каждую лунку хорошо промывали 3 раза 200 мкл 1х буфера для промывки (компонент набора, №JA1617, разведение 1:20). 100 мкл конъюгата антител против фосфорилированного IκВα (Ser32/Ser36) (компонент набора, №JA9126) добавляли в каждую лунку и инкубировали при комнатной температуре в течение 1 часа. Затем лунки промывали 3 раза 1х буфером для промывки (компонент набора, №JA1617, разведение 1:20), 200 мл/лунка. После чего в каждую лунку добавляли по 100 мкл HRP-конъюгата (компонент набора, №JA7643) и инкубировали при комнатной температуре в течение 1 часа. Затем лунки промывали 3 раза по 200 мкл 1х буфера для промывки (компонент набора, №JA1617, разведение 1:20) и добавляли по 100 мкл конъюгата субстрата ТМБ в каждую лунку (компонент набора, №JA1608). Планшет инкубировали при комнатной температуре до изменения цвета раствора. Затем 100 мкл Stop-раствора для ИФА (компонент набора, №JA1616) добавляли в каждую лунку. Данные получали при 450 нм с опорной длиной волны 570 нм с помощью MultiScan (Thermo Labsystems). Для каждого соединения испытывали по 8 концентраций (от 10 мкМ до 0.003 мкМ), используя схему последовательных разведений 1:3. У всех примеров соединений согласно настоящему изобретению IC50<0.1 мкМ. Например, Пример 1 имеет IC50=0.015 мкМ, которая указывает на то, что соединение является эффективным ингибитором IKKβ.
В качестве альтернативы, 1ККβ-киназный анализ проводили in vitro с использованием набора для анализа Z'-Lyte™ Kit-Ser/Thr 5 Peptide (Invitrogen, Кат. №PV3178). Все реакции (20 мкл) проводили при смешивании 0,8 мкл испытуемого соединения в растворе ДМСО, 10 мкл киназно-пептидной смеси или фосфо-пептидного раствора (Invitrogen, Кат. №PV3219, разбавленного 1.33х буфером для киназы), 5 мкл 1,33×буфера для киназы (Invitrogen, Кат. №PV3189, 5х раствор, разбавленный дистиллированной водой) или раствор АТФ (5 мкМ), и 4.2 мкл дистиллированной воды. Компоненты смешивали в 384-луночном планшете для анализа (Corning, Кат. №3575) и инкубировали при комнатной температуре в течение 1 часа. Затем в каждую лунку добавляли по 10 мкл реакционного раствора [реакционный реагент В (Invitrogen, Кат. №PV3296) / реакционный буфер (Invitrogen, Кат. №PV3127)=1:128], перемешивали и инкубировали при комнатной температуре еще 1 час. После чего киназную реакцию останавливали путем добавления 10 мкл стоп-реагента (Invitrogen, Кат.№PV3094). Планшет анализировали с помощью Wallac 1420 VICTOR3 Multilabel счетчика (PERKIN ELMERTM) при длинах волн флуоресценции 445 нм и 520 нм. Анализ характеризуется MSR=2.14. Изначально протестировано 8 концентраций соединения (от 10 мкМ до 0.003 мкМ) с использованием 1:3 схемы последовательных разведений. Пример 2 имеет IC50=0.058 мкМ. Этот результат показывает, что Пример 2 также является эффективным ингибитором IKKβ.
Тест жизнеспособности клеток
Для оценки биологической активности соединения в лабораторных условиях проводится тест жизнеспособности клеток, при котором IKKβ рецептор играет важную роль в выживании и пролиферации клеток. После того, как IKKβ путь блокируется ингибиторами, клетка может пойти путем апоптоза или смерти. Тест жизнеспособности клеток предоставляет информацию о выживаемости клеток после обработки ингибиторами IKKβ.
ВхРС-3 клетки (АТСС CRL-1687; клеточная линия рака поджелудочной железы человека) выращивали в среде Roswell Park Memorial Institute (RPMI) 1640 (Gibco#A10491-01) с добавлением 10% эмбриональной бычьей сыворотки (FBS) (Gibco#10099-141). SKOV3-luc клетки (АТСС, клеточная линия карциномы яичников человека) выращивали в среде 5а Маккоя (Gibco#16600) с добавлением 10% FBS (Gibco#10099-141). Для тестирования соединения ВхРС-3 и SKOV3-luc клетки высевали в количестве 2000 и 5000 клеток/лунку соответственно в 100 мкл соответствующей среды, указанной выше для каждой клеточной линии, в 96-луночные планшеты за 20 ч до начала обработки. Клетки обрабатывали тестируемым соединением в восьми различных концентрациях в присутствии 0,5% ДМСО в течение 72 часов. Гибель клеток в каждой лунке определяли с помощью добавления 20 мкл реагента «Реагент Одного Раствора» (CELLTITER 96® «Анализ Клеточной Пролиферации Одним Водным Раствором», Promega#G3580). Через 2-4 ч инкубации при температуре 37°C оптическую плотность измеряли при 492 нм с помощью микропланшетного ридера. Ингибирование жизнеспособности клеток определяли при сравнении опытных клеток с контрольными, для обработки которых не использовали тестируемое соединение.
В процессе комбинированного исследования при использовании соединения совместно с другими противоопухолевыми агентами BxPC-3 и SKOV3-luc клетки высевали в количестве 2000 и 5000 клеток/лунку соответственно в 100 мкл соответствующей среды, указанной выше для каждой клеточной линии, в 96-луночные планшеты за 20 ч до обработки. Клетки обрабатывали испытуемым соединением в нескольких концентрациях в течение 30 минут, а затем подвергали действию 5 нг/мл TNFα или 0.15-0.6 нМ VCR (винкристин сульфат) в течение дополнительных 72 часов. Гибель клеток в каждой лунке определяли с помощью добавления 20 мкл реагента «Реагент Одного Раствора» (CELLTITER 96® Анализ Клеточной Пролиферации Одним Водным Раствором, Promega#G3580). Через 2-4 ч инкубации при температуре 37°C, оптическую плотность измеряли при 492 нм с помощью микропланшетного ридера.
Ингибирование жизнеспособности клеток определяли при сравнении опытных клеток с контрольными, для обработки которых не использовали тестируемое соединение. Например, результаты комбинированного исследования с использованием Примера 1 подробно представлены в Таблице 1. Пример 1 демонстрирует синергетические эффекты в отношении подавления роста опухолевых клеток ВхРС-3 и SKOV3 в комбинации с TNFα или с VCR (эффекты Примера 1 увеличиваются или усиливаются, см. таблицу 2 - у BxPC-3 клеточной линии: только Пример 1 (2.5 нМ) - 16,48% ингибирования; только TNFα (5 нг/мл) - 5.73% ингибирования; TNFα (5 нг/мл)+Пример 1 (2.5 мкМ) - 87.75% ингибирования; также у ВхРС-3 клеточной линии: только пример 1 (25 мкМ) - 11,5% ингибирования; только Винкристин (0.15 мкМ) - 5.73% ингибирования; Винкристин (0,15 мкМ)+Пример 1 (25 мкМ) - 80.25% ингибирования). Эти данные свидетельствуют о терапевтической пользе Примера 1 в сочетании с TNFα или VCR для лечения рака яичников и рака поджелудочной железы.
Ингибирование экспрессии гена TNFα в ксенотрансплантате U87MG
TNFα стимулирует сигнальный путь IKKβ и запускает экспрессию гена TNFα. Чтобы убедиться, что мишенью соединения in vivo является IKKβ, соединение проверяли на его способность ингибировать TNFα индуцированную экспрессию гена TNFα в ксенотрансплантате U87MG (клеточная линия глиобластомы человека). Опухолевые клетки U87MG в количестве 3×106 имплантировали подкожно в правый бок самкам бестимусных голых мышей BALB/C (6-8 недель). Через 10-12 дней, когда объем опухоли достиг 200-300 мм, животных случайным образом разделили на следующие группы: контроль (без стимуляции TNFα), модель (стимуляция TNFα), и группа, которой вводили тестируемое соединение: 10, 30, 60 и 100 мг/кг (наряду со стимуляцией TNFα). Тестируемое соединение вводили голым мышам перорально за 2 ч до извлечения опухоли. TNFα (R&D, Кат. №210-ТА) в концентрации 8 мкг/кг вводили внутривенно за 1 ч до извлечения опухоли.
Тотальную РНК экстрагировали, используя RNEASY® mini Kit (QIAGEN®, Кат. №74126). Синтез кДНК осуществляли, используя высокоэффективный набор для обратной транскрипции кДНК (ABI, Кат. №4368813). Количественный ПЦР реального времени выполняли в системе 7500 для ПЦР реального времени (Applied Biosystems), используя соответствующие праймеры/пробы для гена GAPDH человека (ABI Hs99999905_ml) и гена TNFα человека (ABI Hs00174128_ml), и главную смесь АВ gene Absolute QPCR ROX 2X master mix(AB-1139/B).
Экспрессию гена TNFα нормализовали по экспрессии гена β-актина. Уровень экспрессии генов анализировали с помощью ПЦР машины. Степень ингибирования (%)=(Уровень экспрессии гена TNFα в группе «Модель» - Уровень экспрессии гена TNFα в группе «Терапия») / (Уровень экспрессии гена TNFα в группе «Модель» - Уровень экспрессии гена TNFα в группе «Контроль»)×100%. Результаты для Примера 1 подробно изложены в Таблице 2. Пример 1 ингибирует TNFα индуцированную экспрессию гена TNFα в U87MG ксенотрансплантате дозозависимым образом.
Результаты для Примера 2 подробно изложены в Таблице 3. Пример 2 ингибирует 5 TNFα-индуцированную экспрессию гена TNFα в U87MG ксенотрансплантате дозозависимым способом.
Результаты показали, что Примеры 1 и 2 ингибируют TNFα-индуцированную экспрессию гена TNFα в U87MG ксенотрансплантате дозозависимым образом.
Противоопухолевое действие на ксенотрансплантат рака яичников человека SKOV-3x-FF-Luci
SKOV-3x-FF-Luci клеточную линию рака яичников человека (АТСС) культивировали в среде 5 а Маккоя, содержащей 10% эмбриональной телячьей сыворотки (FCS). Самкам nu/nu мышей BALB/C (6-8 недель) вводили перитонеально по 0,2 мл клеточной суспензии, содержащей 2×106 клеток. Мышей разделили на пять групп через шесть дней после имплантации клеток. Тестируемое соединение в количестве 60, 90 и 150 мг/кг перорально вводили животным последовательно в течение 21 дня, используя режимы - дважды в день (bid) или трижды в день (tid). Мышам в контрольной группе вводили среду-носитель (10% Acacia при pH 2.1, дважды в день). В конце лечения, всех мышей усыпляли с помощью CO2, а опухоли в брюшной полости, диафрагмальной мышце, брыжейке, печени, селезенке, яичниках извлекали, собирали и объединяли вместе для измерения их общего веса. Оценивали вес опухоли и степень ингибирования (IR). Степень ингибирования рассчитывали по формуле: IR=% (вес опухоли в контроле - вес опухоли после лечения) / вес опухоли в контроле ×100%. Результаты применения примера 1 подробно изложены в таблице 4. Пример 1 значительно подавляет рост опухоли яичников человека с IR=76,31% в дозах 60 мг/кг (трижды в день) у голых мышей (P<0.01, Т-критерий Стьюдента). Эти данные свидетельствуют о терапевтической пользе Примера 1 для лечения рака яичников.
νs Контроль
Кроме того, оценивали противоопухолевый эффект тестируемого соединения на рост опухоли рака яичников человека SKOV-3x-FF-Luci у голых мышей. SKOV-3x-FF-Luci клеточную линию (АТСС) культивировали в среде 5а Маккоя, содержащей 10% эмбриональной телячьей сыворотки. Самкам мышей nu/nu BALB/C (6-7 недель) перитонеально вводили по 0,2 мл клеточной суспензии, содержащей 2×106 клеток. Мышей случайным образом разделили на четыре группы через шесть дней после имплантации клеток. Тестируемое соединение в дозах 30 и 100 мг/кг перорально вводили животным два раза в день последовательно в течение 23 дней. Мышам из контрольной группы вводили среду-носитель (10% Acacia) два раза в день. Мышам из положительной контрольной группы вводили Цисплатин (4 мг/кг) через хвостовую вену один раз в неделю. В конце терапии всех мышей усыпляли с помощью CO2, а опухолевые узелки в брюшной полости, диафрагмальной мышце, брыжейке, печени, селезенке, яичниках извлекали, собирали и объединяли для измерения общего веса. Степень ингибирования: IR%=(вес опухоли в контроле - вес опухоли после терапии лекарством) / вес опухоли в контроле×100%. Пример 2 имеет степень ингибирования опухоли (IR) 59,3% и 93,9% в количестве 30 и 100 мг/кг соответственно. При проведении монотерапии Пример 2 также ингибирует рост опухоли SKOV-3x-FF-Luci. Эти данные свидетельствуют о терапевтической пользе Примера 2 для лечения рака яичников.
Противоопухолевое действие на ксенотрансплантат рака толстой кишки человека НТ-29 Показано, что ингибитор IKKβ в комбинации с СРТ-11 подавляет рост опухоли НТ-29 (Lagadec Р, E Griessinger, Nawrot MP et al. Br J. Cancer (2008) 98, 335-344). Противоопухолевый эффект соединения в комбинации с СРТ-11 исследовали в отношении ксенотрансплантата рака толстой кишки человека НТ-29. Противоопухолевые эффекты соединения изучали в отношении ксенотрансплантата рака толстой кишки человека НТ-29 при одновременном введении СРТ-11, так и без него, согласно методу, описанному в литературе (Lagadec Р, Griessinger Е, Nawrot MP, et al. Br J. Cancer (2008) 98, 335-344).
Клеточную линию аденокарциномы толстой кишки человека НТ-29 получали из АТСС и культивировали в среде 5а Маккоя, содержащей 10% эмбриональной телячьей сыворотки. Самцам мышей nu/nu BALB/C (6-7 недель) вводили подкожно в правый бок 0,1 мл клеточной суспензии, содержащей 3.0×106 клеток. Мышей разделили на шесть групп через семь дней после клеточной имплантации. Тестируемое соединение вводили перорально два раза в день в количестве 60 и 20 мг/кг, а СРТ-11 в дозе 20 мг/кг вводили перитонеально два раза в неделю. В комбинированной терапии СРТ-11 давали через 1 час после введения тестируемого соединения. Мышам из контрольной группы перорально вводили среду-носитель (10% Acacia, pH 2.1) два раза в день, и перитонеально вводили физиологический раствор два раза в неделю. Два ортогональных диаметра опухоли измеряли с помощью цифрового штангенциркуля с верньером три раза в неделю. Объем опухоли (TV) измеряли и записывали в течение периода лечения по формуле: TV=длина×ширина2/2. Ингибирование роста опухоли (TGI), исходя из абсолютного значения объема опухоли, рассчитывали по следующей формуле, где V0 - объем опухоли в день 0 (день распределения на группы), a Vt - объем опухоли в день измерения t: TGI=[1-(V-VO) группа, получавшая лекарство/(V-V0) группа, получавшая среду-носитель]×100%. Мышей усыпляли при наличии следующих условий: 1) Конец исследования (День 66); 2) Индивидуальные TV>4000 мм3, 3) изъязвление индивидуальной опухоли. Пример 1 при проведении монотерапии не препятствовал росту опухоли НТ-29 при пероральном введении в количестве 60 или 20 мг/кг, но в сочетании с СРТ-11 (20 мг/кг, внутрибрюшинно) он усиливал противоопухолевый эффект СРТ-11 в этих дозах (P<0,05, T-критерий Стьюдента).
В сочетании с СРТ-11 Пример 1 проявляет противоопухолевые эффекты в отношении модели ксенотрансплантата рака толстой кишки человека НТ-29, что указывает на участие Примера 1 в подавлении опухолевого роста. Эти данные свидетельствуют о терапевтической пользе Примера 1 в сочетании с СРТ-11 для лечения рака толстой кишки.
Противоопухолевое действие Винкристина на ксенотрансплантат SKOV-3x-FF-Luci (VCR)
В целях расширения спектра противоопухолевых эффектов проводили дополнительные исследования по комбинированной терапии. На основании обнаруженного in vitro синергетического эффекта Примера 1 и VCR исследовали противоопухолевые эффекты соединения на ксенотрансплантат SKOV -3x-FF-Luci при наличии/в отсутствие VCR. Клеточную линию рака яичников человека SKOV-3x-FF-Luci (АТСС) культивировали в среде 5а Маккоя, содержащей 10% FCS. Самкам мышей nu/nu BALB/C (6-7 недель) перитонеально вводили по 0,2 мл клеточной суспензии, содержащей 2×106 клеток. Мышей разделили на девять групп через шесть дней после имплантации клеток. Тестируемое соединение вводили перорально два раза в день по 60 мг/кг последовательно в течение 20 дней или в дозе 90 мг/кг в режиме дозирования: 2 дня - прием, 5 дней - перерыв. VCR в количестве 1 или 0.3 мг/кг вводили с помощью инъекции в хвостовую вену один раз в неделю. В группе комбинированной терапии VCR давали через 1 час после введения тестируемого соединения. Мышам из контрольной группы вводили перорально среду-носитель (10% Acacia, pH 2.1) и внутрибрюшинно вводили физиологический раствор. В конце лечения, всех мышей усыпляли с помощью CO2, а опухоли в брюшной полости, диафрагмальной мышце, брыжейке, печени, селезенке, яичниках извлекали с помощью хирургических ножниц и взвешивали. В дозах, близких к оптимальным, VCR подавлял рост опухоли на 38%; Пример 1 ингибировал рост опухоли на 42% и 36% в количестве 60 мг/кг и 90 мг/кг соответственно. В группе комбинированной терапии рост опухоли замедлялся на 76% и 74%. По сравнению с монотерапией показаны статистически значимые различия между комбинированной терапией и монотерапией. Таким образом, Пример 1 проявляет синергетический эффект в сочетании с VCR. Это позволяет предположить, что Пример 1 обладает относительно широким противоопухолевым спектром действия в сочетании с различными химиопрепаратами. В частности, эти данные демонстрируют терапевтическую пользу Примера 1 в сочетании с VCR для лечения рака яичников.
Модель продукции плазматических цитокинов мыши, индуцированной липополисахаридами (ЛПС) Чтобы оценить ингибирующий эффект соединения на воспаление in vivo, изучали способность соединения ингибировать ЛПС-индуцированную продукцию цитокинов в плазме.
Мышей BALB/C (самки, масса тела от 18 до 20 г) использовали в этих экспериментах. Зависимость эффекта от дозы тестируемого соединения и от времени эксперимента, а также фармакокинетические/фармакодинамические показатели тестируемого соединения изучали у мышей, получавших ЛПС (0,4 мг/кг). Группе из восьми мышей вводили от 1 до 100 мг/кг тестируемого соединения (суспензия в 10% Acacia) или среды-носителя через желудочный зонд за 1 ч до внутрибрюшинного введения 0,4 мг/кг ЛПС. Через девяносто минут после введения ЛПС, образцы крови собирали в пробирки, содержащие гепарин для предотвращения коагуляции. Образцы плазмы разводили 3-кратным буфером для разведения (R&D, Кат. №Часть 895206, Калибровочный буфер для разведения RD5Z).
Концентрацию TNFα измеряли с помощью ИФА (R&D, Кат. №MTA00), используя протокол производителя. Данные получали с помощью SpectraMax® Plus М2е планшетного ридера и анализировали с помощью стандартной кривой аппроксимации. Пример 1 в количестве 10 мг/кг значительно подавлял продукцию TNFα, значение которого исходно было 2346,2 пг/мл и упало до 1474,9 пг/мл (p<0,01), при этом степень ингибирования составила 37,1%.
Кроме того, соединение проверяли на его способность ингибировать ЛПС-индуцированную продукцию цитокинов в плазме крови крыс по методу, описанному в литературе (Ziegelbauer К, Gantner F, Lukacs NW, et al. Br J. Pharmacol. 2005,145 (2): 178-92). Крыс Льюиса (самцы, масса тела 140 г-180 г) использовали в экспериментах. Животным (8 особей/группа) давали от 1 до 100 мг/кг тестируемого соединения (суспензия в растворе, содержащем 35% Solutol, 60% PEG400, 5% PG) или среду-носитель через желудочный зонд за 1 ч до внутрибрюшинного введения 0,4 мг/кг ЛПС. Через девяносто минут после внутрибрюшинного введения ЛПС кровь собирали в гепарин-содержащие антикоагулянтные пробирки для измерения TNFα с помощью набора для ИФА R&D System ELISA Kit, используя обычный для этого набора протокол проведения анализа. Образец плазмы разводили в 100 раз буфером для разведения, и 50 мкл разведенного образца использовали в каждом измерении. Планшет анализировали с помощью SpectraMax® Plus M2e планшетного ридера и TNFα уровень измеряли, руководствуясь стандартной кривой. Содержание препарата в плазме крови измеряли с помощью LC-MS-MS. Пример 2 ингибировал продукцию плазменного TNFα дозозависимым образом с ED50, равным 19,8±10,9 мг/кг, и ЕС50, равным 4,042±1,28 мкг/мл.
В качестве альтернативы, соединение проверяли на его способность ингибировать ЛПС-индуцированную продукцию цитокинов в плазме мышей. В экспериментах использовали мышей BALB/C (самки, вес тела 18-20 г). Мышам (8 особей/группа) вводили от 1 до 100 мг/кг тестируемого соединения (суспензия в 10% Acacia) или среду-носитель через желудочный зонд за 1 ч до внутрибрюшинного введения ЛПС (0,4 мг/кг). Через девяносто минут после введения ЛПС кровь собирали в антикоагулянтные пробирки с гепарином для измерения уровня TNFα (R&D, Кат.№MTA00), IL-6 (R&D, Кат. №M6000B) и IL-1β (R&D, Кат. №MLB00B), используя обычные для этих наборов протоколы. Разведенные или неразведенные образцы плазмы объемом 50 мкл использовали для каждого измерения. Для измерения TNFa образец плазмы разбавляли 3-кратным буфером для разведения (R&D, Кат. №Часть 895206, Калибровочный буфер для разведения RD5Z); для измерения IL-6 образец плазмы разбавляли в 25 раз буфером для разведения (R&D, Кат. №Часть 895175, Калибровочный буфер для разведения RD5T), а для измерения IL-1β использовали неразбавленный образец плазмы. Планшет анализировали с помощью SPECTRMAX® Plus М2е планшетного ридера, данные анализировали с помощью калибровочной кривой. Пример 2 ингибировал продукцию TNFα и IL-6 в плазме дозозависимым образом с ED50, равными 4,84 мг/кг (EC50 в диапазоне до 2420 нг/мл) и 15,1 мг/кг соответственно, в то время как на продукцию IL-1β Пример 2 не оказывал никакого эффекта даже в дозе 100 мг/кг.
Показано, что оба Примера 1 и 2 являются эффективными ингибиторами IKKβ in vitro и проявляют значительную активность в мышиной модели ЛПС-индуцированной продукции цитокинов in vivo.
ЛПС-индуцированное острое воспаление суставов у крыс
Внутрисуставное введение доминантно-негативного IKKβ значительно уменьшало тяжесть адъювант-индуцированного артрита у крыс (Так РР et al., Arthritis Rheum. (2001)44(8)1897-1907). Для дальнейшего изучения влияния IKKβ ингибиторов на механизм, лежащий в основе воспаления суставов, соединение проверяли на его способность ингибировать ЛПС-индуцированное острое воспаление суставов у крыс согласно методу, описанному в литературе (Matsukawa, Yoshimura Т, Миямото K, S Ohkawara, Yoshinaga М. Lab Invest. 1997, 76 (5):629-38). А именно, в экспериментах использовали крыс Вистар (самки, масса тела 150 г-170 г). Животным (8 особей/группа) давали от 1 до 100 мг/кг тестируемого соединения [суспензия в растворе, содержащем 35% Solutol (Макрогол 15 Гидроксистеарат), 60% PEG400, 5% PG (пропиленгликоль)] или среду-носитель через желудочный зонд за 1 ч до внутрисуставного введения 10 мкг ЛПС в 10 мкл физиологического раствора в левое заднее колено каждой крысы, используя иглу, толщиной 26. В правое заднее колено каждой крысы вводили по 10 мкл физиологического раствора в качестве контроля. Через два часа после введения ЛПС каждую лодыжку крысы промывали 100 мкл физиологического раствора. Синовиальную жидкость собирали и хранили при -80 C. Плазму разбавляли в 2 раза буфером для разбавления, и 50 мкл разведенного образца использовали для измерения концентрации TNFα с помощью набора для ИФА R&D System ELISA Kit, используя обычный для этого набора протокол. Планшет анализировали с помощью SpectraMax® Plus М2е планшетного ридера, а содержание TNFα находили с помощью калибровочной кривой. Также измеряли уровень препарата с помощью жидкостной хроматографии, сопряженной с тандемной масс-спектрометрией (LC-MS-MS). Пример 2 ингибирует продукцию суставного TNFα дозозависимым образом с ED50, равным 23,4 мг/кг. Полученные данные также показали, что ингибирование продукции TNFα коррелирует с концентрацией лекарства в плазме и суставах. Таким образом, эти данные свидетельствуют о том, что соединение Примера 2 ингибирует воспаление суставов.
Протокол создания модели коллаген-индуцированного артрита (CIA) у мышей
Предыдущие исследования показали, что низкомолекулярные ингибиторы IKKβ эффективны в модели CIA у мышей (Podolin, PL et al., J. Pharm. Exp. Ther., 2005, 312:373-381). Кроме того, мыши с T-клетками, экспрессирующими доминантно-негативную форму IkBα, защищены от развития CIA (Seetharaman R. et al., J. Immunol 1999:163, 1577-1583). Эти наблюдения позволяют предположить, что ингибиторы IKKβ, вероятно, будут эффективны в лечении ревматоидного артрита. Поэтому соединение согласно настоящему изобретению тестировали на модели коллаген-индуцированного артрита (CIA) у мышей согласно методу, описанному в литературе (Rosloniec EF, Cremer М, Kang A, Myers LK. Current Protocols in Immunology. 1996,15.5.1-15.5.24).
Мышей DBA/1 иммунизировали внутридермально коллагеном II цыпленка (200 мкг/мышь), эмульгированным полным адъювантом Фрейнда (CFA, Sigma, США), в день 0 и в день 21, чтобы вызвать артрит. Тяжесть артрита оценивали с помощью системы визуальной оценки, в которой каждая лапа оценивается от 0 до 4 (0=нормальное состояние, 4=сильный отек всей лапы). Толщину задних суставов измеряли с помощью микрометра. Тестируемое соединение вводили перорально в профилактических или лечебных целях. Для исследования профилактического эффекта тестируемое соединение вводили перорально два раза в день с 1 дня по 42 день. Для исследования терапевтического эффекта медикаментозное лечение начинали после возникновения артрита, который обычно развивается в течение одной недели после второй иммунизации. Препарат вводили мышам в течение 21 дня.
Лефуномид (ЛЕФ), иммунодепрессивное средство, использовали в количестве 10 мг/кг/сут один раз в день в качестве положительного контроля. Нормальных животных, не получавших никакого лечения, использовали в качестве отрицательного контроля. Препараты вводили перорально в количестве 3, 10 и 30 мг/кг дважды в день в течение 42 дней. Статистически значимое ингибирование опухания суставов и снижение тяжести артрита наблюдали при введении 10 и 30 мг/кг дважды в день (20 и 60 мг/кг/сут) для Примера 2 с ED50, равным 4,3 мг/кг.
Эти данные свидетельствуют о терапевтической пользе Примера 2 для лечения ревматоидного артрита.
Модель коллаген П-индуцированного артрита (CIA) у крыс
Для подтверждения результатов, полученных на модели CIA у мышей, соединение также тестировали на модели CIA у крыс согласно методу, описанному в литературе (Rosloniec EF, Cremer М, Kang A, Myers LK. Current Protocols in Immunology. 1996, 15.5.1-15.5.24). А именно, крыс Вистар иммунизировали внутридермально 200 мкг бычьего коллагена II, эмульгированного в неполном адъюванте Фрейнда (IFA, Sigma, США), в день 0, и 100 мкг коллагена II, эмульгированного в IFA (неполном адъюванте Фрейнда) в день 7. Объем задней лапы измеряли до и после иммунизации. Развитие болезни на каждой из четырех лап оценивали количественно, присваивая баллы, соответствующие тяжести артрита. Крысам в нормальной и модельной группах вводили среду-носитель или лекарство (группа терапии). Крысам в группе терапии перорально вводили тестируемое соединение в дозах 3, 10, 30 мг/кг (дважды в день), с 1 дня по 21-й день после первой иммунизации. В ЛЕФ группе, используемой в качестве положительного контроля, крысам вводили перорально ЛЕФ в дозе 10 мг/кг один раз в день. Пример 2 в дозе 30 мг/кг значительно ослаблял коллаген-индуцированный артрит, об этом свидетельствовало снижение баллов тяжести артрита и объема задних лап. Эти данные свидетельствуют о терапевтической пользе Примера 2 для лечения ревматоидного артрита.
Овальбумин (ОУА)-индуцированное воспаление легких у мышей
Предыдущие исследования показали, что низкомолекулярные ингибиторы IKKβ могут подавлять аллерген-индуцированное воспаление дыхательных путей и гиперреактивность у мышей (Birrell MA et al., Am J Respir Crit Care Med, 2005, 172: 962-971). Целью данного исследования являлось изучение влияния ингибиторов IKKβ на модель антиген-зависимого воспаления дыхательных путей in vivo.
Соединение настоящего изобретения тестировали на OVA-индуцированной модели воспаления легких у мышей согласно методу, описанному в литературе (Muriel Pichavant, Sho Goya, Eckard Hamelmann, Erwin W.Gelfand, and Dale T.Umetsu. Animal Models of Airway Sensitization. Current Protocols in Immunology. (1999) 15.18.1-15.18.13). А именно, самкам мышей BALB/C весом от 18 г до 20 г делали внутрибрюшинные инъекции физиологического раствора объемом 100 мкл, содержащие 20 мкг овальбумина и 2 мг гидроксида алюминия, на 1 и 14 день. В дни 28, 29 и 30 каждую мышь подвергали воздействию 1% аэрозоля яичного альбумина (OVA) в фосфатном солевом буфере (PBS) в течение 20 минут с помощью системы дозирования массы Вuхсо. Дексаметазон в дозе 1 мг/кг, служащий положительным контролем, и тестируемое соединение в дозах 1,3, 10 и 30 мг/кг, вводили мышам перорально один раз в день с 1 дня по 31-й день. На 32-й день животных из каждой группы усыпляли с помощью 1% пентобарбитала натрия. Грудную и брюшную полости вскрывали, и собирали плазму, полученную после центрифугирования образцов. Затем трахею вскрывали и перерезали. С помощью введения в трахею катетера толщиной 18 легкие промывали стерильным раствором PBS общим объемом 1,5 мл, а затем помещали в контейнер для образцов. Путем центрифугирования BAL жидкости получали бесклеточные BAL супернатанты. Все образцы сыворотки и BAL супернатантов хранили при температуре -80°C до использования. Дифференциальный подсчет клеток осуществляли, используя полученные с помощью цитоспина препараты клеток BAL для подсчета клеток и определения цитокинов. После сбора BAL супернатантов легкие мышей-реципиентов извлекали целиком и фиксировали посредством интратрахеальной инстилляции 10% буферном раствором формалина (в PBS). От трех до шести парафиновых срезов получали из области корней легких реципиента, окрашивали гематоксилином и эозином и изучали под световым микроскопом для проведения патологического исследования.
По сравнению с нормальным контролем OVA значительно увеличивал количество эозинофилов (p<0,01) и концентрацию IL-13 (p<0,05) в BAL. Дексаметазон в дозе 1 мг/кг (КТ), служивший положительным контролем, полностью подавлял увеличение количества клеток эозинофилов и снижал уровень IL-13 на 77%. Пример 2 в дозах 1,3, 10 и 30 мг/кг при введении дважды в день уменьшал количество клеток эозинофилов на 58,6%, 49,3%, 94,1% (p<0,05) и 90,2% (p<0,05) соответственно. Пример 2 в дозах 10 и 30 мг/кг также значительно снижал продукцию IL-13 на 73,0% (p<0,05) и 85,1% (p<0,01). Кроме того, с помощью окраски гематоксилином и эозином было показано улучшение состояния легких при воспалении у мышей, получавших Пример 2 в дозах от 1 до 30 мг/кг и дексаметазон в дозе 1 мг/кг.
Это исследование еще раз демонстрирует терапевтическую пользу Примера 2 для лечения астмы.
OVA-индуцированное воспаление легких у коричневых норвежских (BN) крыс Чтобы оценить влияние ингибиторов IKKβ на вызванное антигеном воспаление дыхательных путей in vivo, соединение также тестировали в OVA-индуцированной модели воспаления легких у крыс согласно методу, описанному в литературе (Yamamoto N, Takeshita K, Shichijo М, Kokubo Т, Sato М, Nakashima K, Ishimori М, Nagai Н, Li YF, Yura T, Bacon KB. J Pharm. Exp Ther. 2003; 306(3): 1174-81). А именно, самцам крыс BN весом от 240 до 260 г внутрибрюшинно вводили 1 мл физиологического раствора, содержащего 1 мг OVA и 13 мг гидроксида алюминия, на 1, 2 и 3 день. Крысы, выступающие в качестве нормального контроля, получали по 1 мл физиологического раствора без OVA. Тестируемое соединение в дозах 0.3, 1, 3 и 30 мг/кг вводили крысам перорально дважды в день на 20 и 21 день. На 21-й день крыс подвергали воздействию 1% OVA в течение 15 минут с помощью системы дозирования массы Вuхсо. На 22 день крыс усыпляли с помощью 1% пентобарбитала натрия. Грудную и брюшную полости вскрывали. Собирали плазму, полученную после центрифугирования образцов. Трахею вскрывали и перерезали. С помощью введения в трахею катетера толщиной 18 легкие промывали стерильным раствором PBS общим объемом 15 мл, а затем помещали в контейнер для образцов. Путем центрифугирования BAL жидкости получали бесклеточные BAL супернатанты. Все образцы сыворотки и BAL супернатантов хранили при температуре -80°C до использования. Дифференциальный подсчет клеток осуществляли, используя полученные с помощью цитоспина препараты клеток BAL для подсчета клеток и определения цитокинов. OVA значительно увеличивал общее количество клеток и число эозинофилов в BAL жидкости по сравнению с нормальным контролем (p<0,01). Пример 2 в концентрации от 0,3 до 30 мг/кг значительно ингибировал рост всех клеток и эозинофилов в зависимости от дозы (p<0,05) с ED50, равным 0,49 мг/кг. Это исследование еще раз демонстрирует терапевтическую пользу Примера 2 для лечения астмы.
Модель экспериментального аутоиммунного энцефаломиелита (ЕАЕ) у мышей
CNS-ограниченное удаление NEMO или IKK2 (IKKP), но не IKK1, улучшало патологию модели рассеянного склероза у мышей (Nature Immunology, 2006; 7 (9), 954-61). Эти исследования позволяют предположить, что ингибиторы IKKβ могут быть эффективными в модели ЕАЕ у мышей.
Соединение настоящего изобретения тестировали в PLP-139-151-индуцированной ЕАЕ модели на мышах согласно методу, описанному в литературе (Miller SD, Karpus WJ. Experimental autoimmune encephalomyelitis in the mouse. Current Protocols in Immunology. 1996, 15.1.1-15.1.13). А именно, самок мышей SJL/J иммунизировали внутридермально с помощью PLP-139-151 (200 мкг/мышь), эмульгированного с H37Ra (штамм микобактерий туберкулеза) в CFA (полный адъювант Фрейнда) (300 мкг H37Ra). Тестируемое соединение в дозах 3, 10, 30 мг/кг или среду-носитель вводили перорально два раза в день с 1-го дня после иммунизации для профилактического исследования и со дня рецидива ЕАЕ в течение всей болезни для терапевтических исследований. Дексаметазон (1 мг/кг, перорально, дважды в день) использовали в качестве положительного контроля. Измерение веса тела и клиническую оценку ЕАЕ производили ежедневно. Болезнь оценивали по следующим критериям: 0, без явных признаков болезни; 1, вялый хвост или слабость задних конечностей (одно из двух, но не оба), 2, вялый хвост и слабость задних конечностей, 3, частичный паралич задних конечностей; 4, полный паралич задних конечностей, 5, состояние, близкое к смерти, или смерть. У мышей, иммунизированных PLP 139-151, наблюдались ЕАЕ-ассоциированные клинические симптомы и снижение массы тела, начиная с 11-ого дня. ЕАЕ баллы быстро возрастали и достигли максимального уровня на 15 день. Уменьшение ЕАЕ баллов происходило примерно на 20 день. После чего происходил спонтанный рецидив заболевания (модельная группа). В группах, получавших лекарство в виде Примера 2, повышенные ЕАЕ-ассоциированные клинические баллы уменьшались дозозависимым образом. Потеря массы тела замедлялась под действием Примера 2 в дозах 10 и 30 мг/кг. Пример 2 в дозах 3, 10 и 30 мг/кг снижал ЕАЕ клинические баллы с ED50, равным 3,7 мг/кг. Это исследование демонстрирует терапевтическую пользу Примера 2 для лечения рассеянного склероза.
Динитробензолсульфокислота (БКВ8)-индуцированный колит у крыс
IKKβ киназа участвует в регуляции экспрессии различных провоспалительных белков, имеющих решающее значение для патогенеза воспалительных заболеваний кишечника (IBD). Поэтому IKKβ киназа представляет собой перспективную мишень для разработки новых препаратов для лечения IBD.
Соединение изучали на модели DNBS-индуцированного колита у крыс согласно методу, описанному в литературе (Gut. 2002; 50 (3):440-1). А именно, в экспериментах использовали крыс Вистар. После индукции дистального колита посредством DNBS инсталляций внутрь толстой кишки тестируемое соединение вводили крысам перорально в дозах 3, 10, 30, 60 мг/кг дважды в день в течение шести дней. Отрицательной контрольной группе давали только среду-носитель без DNBS. В контрольной группе с носителем у крыс индуцировали колит с помощью DNBS наряду с введением среды-носителя. В группе положительного контроля крысам вводили сульфасалазин перорально в дозе 300 мг/кг в день в течение шести дней подряд. Животных усыпляли через 24 ч после последнего введения препарата. Для каждого животного рассчитывали отношение веса толстой кишки к весу тела и оценивали повреждение толстой кишки в виде баллов.
Терапия крыс с помощью носителя приводила к прогрессирующему ухудшению клинических симптомов, достигая максимальной оценки повреждений толстой кишки, равной 6.1±0.6, и показателя отношения веса толстой кишки к массе тела по отношению к длине толстой кишки, равного 0.96±0.10, на 7 день после инсталляций. В данной модели DNBS-индуцированного воспалительного заболевания кишечника у крыс, получавших Пример 2 в дозах 3, 10, 30 и 60 мг/кг, выявили существенное снижение баллов, характеризующих повреждение толстой кишки, а также снижение отношения вес толстой кишки-масса тела-длина толстой кишки более чем на 30%. Эффекты Примера 2 в количестве от 3 до 60 мг/кг/сут оказались схожи с действием сульфасалазина в дозе 300 мг/кг/сут.
Это исследование демонстрирует терапевтическую пользу Примера 2 для лечения воспалительных заболеваний кишечника.
Соединения настоящего изобретения готовили предпочтительно в виде фармацевтических композиций, используя различные способы введения. Наиболее предпочтительный способ введения таких композиций - пероральный или внутривенный. Такие фармацевтические композиции и способы их получения хорошо известны в данной области. См., например, REMINGTON: THE SCIENCE AND PRACTICE OF PHARMACY (D. Troy, et al., eds., 21st ed., Lippincott Williams & Wilkins, 2005).
Соединения настоящего изобретения, как правило, эффективны в широком диапазоне концентраций. Например, дневная дозировка обычно попадает в диапазон концентраций примерно от 0,05 до 500 мг/кг массы тела. В некоторых случаях оказывается более чем достаточно использовать дозу ниже нижнего предела вышеуказанного диапазона, в то время как в других случаях еще большие дозы могут быть использованы, не вызывая никаких вредных побочных эффектов. Следовательно, указанный выше диапазон концентраций никоим образом не ограничивает возможности изобретения. Следует иметь в виду, что количество соединения, которое будет использовано для терапии, выбирает врач, принимая во внимание текущие обстоятельства, включая подлежащее лечению состояние, выбранный путь введения, фактическое соединение или соединения для терапии, возраст, вес, индивидуальные реакции пациента, и тяжесть симптомов у пациента.
| название | год | авторы | номер документа |
|---|---|---|---|
| ВЕЗИКУЛЫ, ПОЛУЧЕННЫЕ ИЗ LACTOBACILLUS PARACASEI, И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2785709C2 |
| ПРИМЕНЕНИЕ СОЕДИНЕНИЙ МЫШЬЯКА ДЛЯ ЛЕЧЕНИЯ ОТТОРЖЕНИЯ ТКАНИ ИЛИ ОРГАНА | 2013 |
|
RU2665362C2 |
| ПРИМЕНЕНИЕ СОЕДИНЕНИЙ МЫШЬЯКА ДЛЯ ЛЕЧЕНИЯ БОЛИ И ВОСПАЛЕНИЯ | 2008 |
|
RU2630574C2 |
| ПРИМЕНЕНИЕ СОЕДИНЕНИЙ МЫШЬЯКА ДЛЯ ЛЕЧЕНИЯ БОЛИ И ВОСПАЛЕНИЯ | 2008 |
|
RU2468806C2 |
| 2,2-ДИФТОРОПРОПИОНАМИДНЫЕ ПРОИЗВОДНЫЕ БАРДОКСОЛОН-МЕТИЛА, ПОЛИМОРФНЫЕ ФОРМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2014 |
|
RU2693833C2 |
| ИНГИБИРУЮЩИЕ ASK1 ПРОИЗВОДНЫЕ ПИРРОЛОПИРИМИДИНА И ПИРРОЛОПИРИДИНА | 2018 |
|
RU2772422C2 |
| ИНГИБИТОР КИНАЗЫ | 2007 |
|
RU2440352C2 |
| ИНГИБИТОРЫ ФУКОЗИДАЗЫ | 2016 |
|
RU2765202C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ОСНОВЕ ПРОИЗВОДНОГО ТРИИНДОЛИЛМЕТАНА В КАЧЕСТВЕ ПРОТИВООПУХОЛЕВОГО СРЕДСТВА | 2012 |
|
RU2549430C2 |
| ТЕРАПЕВТИЧЕСКИЕ КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОР ПОЛИ(АДФ-РИБОЗА)ПОЛИМЕРАЗЫ | 2005 |
|
RU2361592C2 |
Изобретение относится к новому соединению формулы (I) или его фармацевтически приемлемой соли, обладающим свойствами ингибитора ΙΚΚβ и TNFα. Соединение может найти применение при лечении воспалительных заболеваний, таких как ревматоидный артрит, хроническая обструктивная болезнь легких, астма, рассеянный склероз и воспалительные заболевания кишечника, или раковых заболеваний, таких как множественная миелома, рак толстой кишки, рак поджелудочной железы и рак яичников, посредством ингибирования ΙΚΚβ. Соединение может быть использовано с дополнительным терапевтическим агентом, выбранным из винкристина, камптотецина гидрохлорида (СРТ-11), лефуномида, дексаметазона и TNFα. Предпочтительны соединения формулы (I), соответствующие 2-{5-хлор-2-[(1R,2R)-2-гидроксициклопентиламино]пиримидин-4-ил}-N-циклопропил-1H-индол-4-карбоксамиду и 2-{5-хлор-2-[(1R,2S)-2-гидроксициклопентиламино]пиримидин-4-ил}-N-циклопропил-1H-индол-4-карбоксамиду.
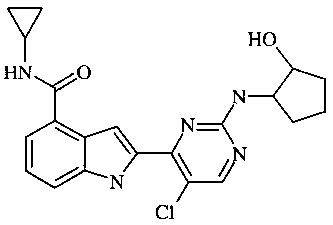 (I)
(I)
8 н. и 22 з.п. ф-лы, 4 табл., 2 пр.
1. Соединения формулы
или фармацевтически приемлемая соль этого соединения.
2. Соединение или фармацевтически приемлемая соль по п. 1, которое представляет собой 2-{5-хлор-2-[(1R, 2R)-2-гидроксициклопентиламино]пиримидин-4-ил}-N-циклопропил-1H-индол-4-карбоксамид.
3. Соединение или фармацевтически приемлемая соль по п. 1, которое представляет собой 2-{5-хлор-2-[(1R, 2S)-2-гидроксициклопентиламино]пиримидин-4-ил}-N-циклопропил-1H-индол-4-карбоксамид.
4. Фармацевтическая композиция, обладающая свойствами ингибитора ΙΚΚβ и TNFα, содержащая эффективное количество соединения или фармацевтически приемлемой соли по любому из пп. 1-3, в сочетании с одним или более фармацевтически приемлемыми носителями, разбавителями или наполнителями.
5. Фармацевтическая композиция, обладающая свойствами ингибитора ΙΚΚβ и TNFα, содержащая эффективное количество соединения или фармацевтически приемлемой соли по любому из пп. 1-3 и эффективное количество дополнительного терапевтического агента, выбранного из группы, состоящей из винкристина, камптотецина гидрохлорида (СРТ-11), лефуномида, дексаметазона и TNFα.
6. Фармацевтическая композиция по п. 5, отличающаяся тем, что терапевтическим агентом является TNFα.
7. Фармацевтическая композиция по п. 5, отличающаяся тем, что терапевтическим агентом является винкристин.
8. Способ лечения воспалительных заболеваний у млекопитающего посредством ингибирования ΙΚΚβ, включающий введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения или фармацевтически приемлемой соли по любому из пп. 1-3, причем указанное воспалительное заболевание выбрано из группы, состоящей из ревматоидного артрита, хронической обструктивной болезни легких, астмы, рассеянного склероза и воспалительных заболеваний кишечника.
9. Способ по п. 8, отличающийся тем, что воспалительным заболеванием является ревматоидный артрит.
10. Способ по п. 8, отличающийся тем, что воспалительным заболеванием является хроническое обструктивное заболевание легких.
11. Способ по п. 8, отличающийся тем, что воспалительным заболеванием является астма.
12. Способ по п. 8, отличающийся тем, что воспалительным заболеванием является рассеянный склероз.
13. Способ по п. 8, отличающийся тем, что воспалительным заболеванием является воспалительное заболевание кишечника.
14. Способ лечения ракового заболевания у млекопитающего посредством ингибирования ΙΚΚβ, включающий введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения или фармацевтически приемлемой соли по любому из пп. 1-3, причем раковое заболевание выбрано из группы, состоящей из множественной миеломы, рака толстой кишки, рака поджелудочной железы и рака яичников.
15. Способ по п. 14, отличающийся тем, что раковым заболеванием является множественная миелома.
16. Способ по п. 14, отличающийся тем, что раковым заболеванием является рак толстой кишки.
17. Способ по п. 14, отличающийся тем, что раковым заболеванием является рак поджелудочной железы.
18. Способ по п. 14, отличающийся тем, что раковым заболеванием является рак яичников.
19. Соединение или фармацевтически приемлемая соль по любому из пп. 1-3 для получения фармацевтической композиции, обладающей свойствами ингибитора ΙΚΚβ и TNFα, пригодной для лечения воспалительных и раковых заболеваний.
20. Соединение или фармацевтически приемлемая соль по любому из пп. 1-3 для применения для лечения воспалительного заболевания посредством ингибирования ΙΚΚβ, причем воспалительное заболевание выбрано из группы, состоящей из ревматоидного артрита, хронической обструктивной болезни легких, астмы, рассеянного склероза и воспалительного заболевания кишечника.
21. Соединение или фармацевтически приемлемая соль для применения в соответствии с п. 20, отличающиеся тем, что воспалительным заболеванием является ревматоидный артрит.
22. Соединение или фармацевтически приемлемая соль для применения в соответствии с п. 20, отличающиеся тем, что воспалительным заболеванием является хроническая обструктивная болезнь легких.
23. Соединение или фармацевтически приемлемая соль для применения в соответствии с п. 20, отличающиеся тем, что воспалительным заболеванием является астма.
24. Соединение или фармацевтически приемлемая соль для применения в соответствии с п. 20, отличающиеся тем, что воспалительным заболеванием является рассеянный склероз.
25. Соединение или фармацевтически приемлемая соль для применения в соответствии с п. 20, отличающиеся тем, что воспалительным заболеванием является воспалительное заболевание кишечника.
26. Соединение или фармацевтически приемлемая соль по любому из пп. 1-3 для лечения ракового заболевания посредством ингибирования ΙΚΚβ, причем раковое заболевание выбрано из группы, состоящей из множественной миеломы, рака толстой кишки, рака поджелудочной железы и рака яичников.
27. Соединение или фармацевтически приемлемая соль для применения по п. 26, отличающиеся тем, что раковым заболеванием является множественная миелома.
28. Соединение или фармацевтически приемлемая соль для применения по п. 26, отличающиеся тем, что раковым заболеванием является рак толстой кишки.
29. Соединение или фармацевтически приемлемая соль для применения по п. 26, отличающиеся тем, что раковым заболеванием является рак поджелудочной железы.
30. Соединение или фармацевтически приемлемая соль для применения по п. 26, отличающиеся тем, что раковым заболеванием является рак яичников.
| WO 2005113544 A1,01.12.2005 | |||
| WO 2004022553А1,18.03.2004 | |||
| ДИАМИНОВЫЕ ПРОИЗВОДНЫЕ | 2003 |
|
RU2333203C2 |

