Настоящее изобретение относится к пролекарству, содержащему конъюгат лекарственное средство-линкер D-L, или его фармацевтически приемлемой соли. Изобретение также относится к фармацевтическим композициям, содержащим указанные пролекарства, и их использованию в качестве лекарственных препаратов.
Для улучшения физико-химических или фармакокинетических свойств лекарственного средства in vivo оно может быть конъюгировано с носителем.
Как правило, носители в системах доставки лекарственных средств используют либо в нековалентной форме, с лекарственным средством, составленным физико-химическим образом в виде смеси носителя с растворителем, либо посредством ковалентного присоединения реагента-носителя к одной из функциональных групп лекарственного средства.
Однако нековалентный метод требует инкапсулирования высоко эффективного лекарственного средства для предотвращения его неконтролируемого, внезапного высвобождения. При ограничении диффузии несвязанной водорастворимой лекарственной молекулы необходимы сильные ван-дер-ваальсовы контакты, часто опосредованные гидрофобными частями молекулы. Многие конформационно чувствительные лекарственные средства, такие как белки или пептиды, теряют свою функциональность в процессе их инкапсулирования и/или при последующем хранении инкапсулированного лекарственного средства. Кроме того, такие содержащие аминогруппы лекарственные средства легко вступают в побочные реакции с продуктами деградации носителя (см., например, работу D.H. Lee и др., J. Contr. Rel, 2003, 92, 291-299). Более того, зависимость механизма высвобождения лекарственного средства в результате биодеградации может привести к межиндивидуальной вариабельности.
В альтернативном варианте лекарственные вещества могут быть конъюгированы с носителем посредством ковалентных связей. Этот метод применим к различным классам молекул от так называемых малых молекул, включая природные продукты вплоть до более крупных белковых молекул. Ковалентно связанные конъюгаты лекарственного средства с носителем можно подразделить на две группы. Во-первых, это конъюгаты, где ковалентная связь между носителем и лекарством обычно существует во время действия лекарственного средства ("перманентная ковалентная связь"), то есть производное лекарственного средства проявляет свои фармакологические эффекты, которые известны для данного лекарства как такового. Во-вторых, ковалентная связь по большей части предварительно разорвана с высвобождением лекарства как такового, которое может проявлять свои известные фармакологические эффекты. В последнем случае ковалентный конъюгат лекарственного средства с носителем называется пролекарством, связанным с носителем, или пролекарством с носителем.
Для обеспечения разрыва ковалентной связи между носителем и лекарственным средством необходимо легкое удаление указанной связи in vivo для высвобождения лекарственного вещества (активация пролекарства).
Активация пролекарства может являться результатом ферментативного или неферментативного расщепления указанной связи между носителем и молекулой лекарственного средства, или последовательной комбинацией обоих, т.е. через стадию ферментативной реакции, сопровождаемую неферментативной перегруппировкой.
Ферментативно индуцированная активация пролекарства характеризуется тем, что в безферментной in vitro среде, такой как водный буферный раствор, может происходить расщепление, например, сложного эфира или амида, но соответствующая скорость гидролиза может быть чрезвычайно мала и терапевтически непригодной. В in vivo среде обычно присутствуют эстеразы или амидазы, и указанные эстеразы и амидазы могут приводить к существенному каталитическому ускорению кинетики гидролиза в интервале от двукратного увеличения вплоть до величины в несколько порядков. Таким образом, разрыв ковалентной связи контролируется, главным образом, ферментативной реакцией.
Главным недостатком в основном ферментативного расщепления являются межиндивидуальная вариабельность. Уровни ферментов могут существенно различаться между отдельными лицами, что приводит к биологической изменчивости активации пролекарства под действием ферментативного гидролиза. Уровни ферментов также могут меняться в зависимости от места введения. Например, известно, что при подкожной инъекции в некоторых областях тела возникают более предсказуемые терапевтические эффекты, чем в других. Для уменьшения этого непредсказуемого эффекта неферментативное расщепление или внутримолекулярный катализ представляет особый интерес.
Таким образом, ферментнезависимое автокаталитическое расщепление связи между носителем и биологически активным фрагментом является предпочтительным. В большинстве случаев это достигается посредством соответствующим образом сконструированного линкерного фрагмента между носителем и биологически активным фрагментом, который непосредственно присоединен через ковалентную связь к функциональной группе биологически активного фрагмента.
Специфические типы линкеров известны в данной области техники. Y. Sohma и др., описывают в журнале J. Med. Chem. 46 (2003), 4124-4135 пролекарства на основе сложных эфиров, в которых носитель является водорастворимым, а биологически активный фрагмент получен из ингибитора KNI-727 протеазы ВИЧ-1. Используемый линкерный фрагмент присоединен к биологически активному фрагменту через сложноэфирную группу. Механизмом этой системы пролекарства является активация реакции циклизации, приводящая к образованию циклического имида для разрыва сложноэфирных связей.
Однако имеет недостаток из-за нестабильности сложноэфирной функциональной группы. Более того, сложноэфирные функциональные группы могут представлять собой менее хемоселективно доступные группы для конъюгирования носителя или линкера с лекарством.
A.J. Garman др., (А J. Garman, S.B. Kalindjan, FEBS Lett. 1987, 223 (2), 361-365 1987) используют ПЭГ5000-малеиновый ангидрид для обратимой модификации аминогрупп в активаторе плазминогена тканевого типа и урокиназы. Восстановление функционального фермента из конъюгата ПЭГ и uРА при инкубации в буферном растворе с pH 7,4 посредством расщепления связи малеаминовой кислоты протекает по кинетике реакции первого порядка при периоде полужизни 6,1 часа. Недостатком связи малеаминовой кислоты является отсутствие стабильности конъюгата при более низких значениях pH. Это накладывает ограничение на применимость связи малеаминовой кислоты с биологически активными веществами, которые стабильны при щелочных (высоких) значениях pH, поскольку для предотвращения преждевременного расщепления пролекарства очистку полимерного конъюгата с биологически активным агентом необходимо осуществлять в щелочных условиях (высоких значениях pH).
В WO 2004/108070 описывается система пролекарства на основе линкера амидного производного N,N-бис-(2-гидроксиэтил)глицина (бицина). В этой системе две молекулы ПЭГ-носителя соединены с бициновой молекулой, соединенной с аминогруппой молекулы лекарственного средства. Первые две стадии активации пролекарства заключаются в ферментативном расщеплении первых связей, соединяющих обе молекулы ПЭГ-носителя с гидроксильными группами бициновой активирующей группы. В этом документе описаны различные связи между ПЭГ и бицином, приводящие к различной кинетике активации пролекарства. На второй стадии активации пролекарства происходит расщепление второй связи, соединяющей бицин-активирующую группу с аминогруппой молекулы лекарственного средства. Основным недостатком этой системы является присоединение полимера к бициновому линкеру, приводящее к медленной скорости гидролиза этой второй амидной связи бицина (t1/2>3 ч в фосфатном буфере). Вследствие этого высвобождение бицин-модифицированного интермедиата пролекарства может демонстрировать различные фармакокинетические, иммуногенные, токсикологические и фармакодинамические свойства по сравнению с молекулой исходного нативного лекарственного средства.
Другая система на основе бицина описана в WO 2006/136586.
Таким образом, существует потребность в создании альтернативных связанных с носителем пролекарств, в которых линкер обеспечивал бы автокаталитическое расщепление для высвобождения лекарства в немодифицированной форме, в которой не оставалось бы остатков, происходящих из линкера.
Таким образом объектом настоящего изобретения является обеспечение таких конъюгатов лекарственное средство-линкер, где линкер ковалентно присоединен через расщепляемую связь к биологически активному фрагменту (представляющему собой лекарственное средство после его высвобождения), и где линкер также ковалентно присоединен через постоянную связь непосредственно к носителю или через спейсер с образованием пролекарства, связанного с носителем.
Эта задача решается посредством пролекарства или его фармацевтически приемлемой соли, содержащего конъюгат лекарственное средство-линкер D-L, где
-D представляет собой азотсодержащий биологически активный фрагмент; а
-L представляет собой биологически неактивный линкерный фрагмент L1, представленный формулой (I)

где пунктирная линия показывает присоединение к азоту биологически активного фрагмента посредством образования амидной связи;
X представляет собой C(R4R4a), N(R4), О, C(R4R4a)-С(R5R5a), C(R5R5a-C(R4Ra4), C(R4R4a)-N(R6), N(R6)-C(R4R4a), C(R4R4a)-O или O-C(R4R4a);
X1 представляет собой С или S(O);
X2 представляет собой C(R7, R7a) или C(R7,R7a)-C(R8,R8a);
X3 представляет собой О, S или N-CN;
R1, R1a, R2, R2a, R3, R3a, R4, R4a, R5, R5a, R6, R7, R7a, R8, R8a независимо выбирают из группы, включающей Н и C1-4алкила;
при необходимости одна или более пар R1a/R4a, R1a/R5a, R4a/R5a, R7a/R8a образуют химическую связь;
при необходимости одна или более пар R1/R1a, R2/R2a, R4/R4a, R5/R5a, R7/R7a, R8/R8a вместе с атомом, к которому они присоединены, соединяются с образованием С3-7циклоалкила или 4-7-членного гетероциклила;
при необходимости R4/R6 вместе с атомами, к которым они присоединены, соединяются с образованием 4-7-членного гетероциклила;
при необходимости одна или более пар R1/R4, R1/R5, R1/R6, R4/R5, R4/R6, R7/R8, R2/R3 вместе с атомами, к которым они присоединены, соединяются с образованием цикла А;
при необходимости R3/R3a вместе с атомом азота, к которому они присоединены, соединяются с образованием 4-7-членного гетероцикла;
А выбирают из группы, состоящей из фенила, нафтила, инденила, инданила, тетралинила, С3-10циклоалкила, 4-7-членного гетероциклила и 9-11-членного гетеробициклила; и
где L замещен 1-4 группами L2-Z и при необходимости дополнительно замещен, при условии, что водород, отмеченный символом * в формуле (I), не замещен заместителем, где
L2 представляет собой одинарную химическую связь или спейсер; и
Z представляет собой группу-носитель.
Неожиданно было обнаружено, что область применения активации циклизации посредством образования циклического имида может быть расширена в диапазоне от производных сложных эфиров вплоть до пролекарств, связанных посредством амидной связи с носителем, несмотря на значительно большую стабильность амидной связи в водных условиях. Было отмечено, что N,N'-бискарбоксамиды, соединенные посредством одной амидной связи с нуклеофильным несущим фрагментом и молекулой лекарственного средства посредством второй амидной связи, демонстрируют способность к автогидролизу в диапазоне, который можно использовать в области применений пролекарств. Кроме того, было также обнаружено, что линкеры могут быть сконструированы так, что они включают носитель, перманентно присоединенный к N,N'-бискарбоксамидному мотиву таким образом, что образование циклического имида можно использовать в качестве основы самоактивации в конструировании пролекарств, связанных с носителем через амидную связь.
Примерами таких предпочтительных циклических продуктов с расщепляемой связью являются структуры с замещенным сукцинимидным или глутаримидным кольцом. Предварительным условием такой активации реакции циклизации является присутствие аминосодержащего нуклеофила в линкерной структуре и другой амидной связи, которая не является амидной связью в пролекарстве, а представляет собой амидную связь, замещенную атомом водорода.
В случае расщепления сукцинимид- или глутаримид-активированного пролекарства аминосодержащий нуклеофил выступает в качестве соседней группы для усиления нуклеофильности азота, содержащегося в постоянной амидной связи, которая в свою очередь атакует амидную карбонильную группу пролекарства и, таким образом, индуцирует внутримолекулярное ацилирование постоянной амидной связи, приводящее к образованию циклического имидного кольца.
Следовательно, предпочтительные структуры линкера включают в себя постоянную связь с носителем, аминосодержащий нуклеофил и постоянную амидную связь водородом, присоединенным к азоту амидной связи. Связанные с соответствующим носителем пролекарства содержат линкер, содержащий постоянную связей с носителем, аминосодержащий нуклеофил и указанную постоянную амидную связь, и азотсодержащий биологически активный фрагмент молекулы, образованный из лекарственного средства, конъюгированного с линкером посредством расщепляемой амидной связи.
На Фиг. 1 показан пример расщепления, приводящего к образованию циклического имида. Азот биологически активного фрагмента представлен как водород-содержащий амин, который приводит к лекарственному средству, имеющему первичную аминную функциональную группу. При этом, также, например, вторичный амин может быть частью лекарственного средства. По соображениям упрощения не показаны от одного до четырех обязательных заместителей структуры L2-Z, в том числе носитель.
Предпочтительные свойства пролекарства настоящего изобретения приведены по периоду его полужизни при гидролизе в водном буферном растворе при pH 7,4 и 37°C в интервале от 1 часа до 3 месяцев; аналогичные скорости гидролиза в физиологических условиях в буфере и плазме крови.
Пролекарство в соответствии с настоящим изобретением может демонстрировать превосходную in vivo и in vitro корреляцию данных расщепления линкера, высокую степень независимости от фермента и его можно хранить при пониженных значениях pH (pH зависимое расщепление).
Используемые термины в объеме существа настоящего изобретения имеют следующие значения.
"Биологически активный фрагмент D" означает часть конъюгата лекарственное средство-линкер, которое после расщепления приводит к лекарственному соединению D-H известной биологической активности.
"Неактивный линкер" означает линкер, который не демонстрирует фармакологического действия лекарственного средства, образованного из биологически активного агента.
"Алкил" означает прямую или разветвленную углеродную цепь. Каждый атом водорода углерода в алкильной группе может быть замещен заместителем.
"C1-4алкил" означает алкильную цепь, содержащую от 1 до 4 атомов углерода, например, если присутствует на конце молекулы: метил, этил, н-пропил, изопропил, н-бутил, изо-бутил, втор-бутил, трет-бутил, или, например, -CH2-, -СН2-СН2-, -СН(СН3)-, - СН2-СН2-СН2-, -СН(С2Н5)-, -С(СН3)2-, если два фрагмента молекулы связаны алкильной группой. Каждый атом водорода у атома углерода в C1-4алкиле может быть замещен заместителем.
"C1-6алкил" означает алкильную цепь, содержащую от 1 до 6 атомов углерода, например, если присутствует на конце молекулы: C1-4алкил, метил, этил, н-пропил, изопропил, н-бутил, изо-бутил, втор-бутил, трет-бутил, н-пентил, н-гексил или, например, группу -СН2-, -СН2-СН2-, -СН(СН3)-, -СН2-СН2-СН2-, -CH(C2H5)-, -С(СН3)2-, если два фрагмента молекулы связаны алкильной группой. Каждый атом водорода у атома углерода в C1-6алкиле может быть замещен заместителем.
Соответственно, "C1-18алкил" означает алкильную цепь, содержащую от 1 до 18 атомов углерода, a "C8-18алкил" означает алкильную цепь, содержащую от 8 до 18 атомов углерода. Соответственно, "С1-50алкил" означает алкильную цепь, содержащую от 1 до 50 атомов углерода.
"С2-50алкенил" означает разветвленную или неразветвленную алкенильную цепь, содержащую от 2 до 50 атомов углерода, например, если присутствует на конце молекулы: -СН=СН2, -СН=СН-СН3, -СН2-СН=СН2, -СН=СН-СН2-СН3, -СН=СН-СН=СН2, или, например, -СН=СН-, если два фрагмента молекулы связаны алкениль-ной группой. Каждый атом водорода у атома углерода в C2-50алкениле может быть замещен заместителем, как дополнительно указано. Таким образом, термин "алкенил" относится к углеродной цепи с, по меньшей мере, одной двойной углерод-углеродной связью. При необходимости может встречаться одна или более тройных связей.
"С2-50алкинил" представляет собой разветвленную или неразветвленную алкиниль-ную цепь, содержащую от 2 до 50 атомов углерода, например, если присутствует на конце молекулы: -С≡СН, -СН2-С=СН, СН2-СН2-С=СН, СН2-С=С-СН3, или, например, -С≡С-, в случае, когда два фрагмента молекулы связаны алкинильной группой. Каждый атом водорода у атома углерода в C2-50алкиниле может быть замещен заместителем, как дополнительно указано. Таким образом, термин "алкинил" относится к углеродной цепи с, по меньшей мере, одной тройной углерод-углеродной связью. При необходимости может встречаться одна или несколько двойных углерод-углеродных связей.
"С3-7циклоалкил" или "С3-7циклоалкильное кольцо" означает циклическую алкильную цепь, содержащую от 3 до 7 атомов углерода, которая может содержать по меньшей мере частично насыщенные двойные углерод-углеродные связи, например, циклопропил, циклобутил, циклопентил, циклогексил циклогексенил, пиклогептил. Каждый атом водорода у атома углерода циклоалкила может быть замещен заместителем. Термин "С3-7циклоалкил" или "С3-7пиклоалкильное кольцо" также включает в себя мостиковые бициклы, аналогичные норборнану или норборнену. Соответственно, "С3-5циклоалкил" означает циклоалкил, содержащий от 3 до 5 атомов углерода.
Соответственно, "С3-10циклоалкил" означает циклический алкил, имеющий от 3 до 10 атомов углерода, например С3-7циклоалкил; циклопропил, циклобутил, циклопентил, циклогексил, циклогексенил, циклогептил, циклооктил, циклононил, циклодецил. Термин "С3-10циклоалкил" также включает в себя, по меньшей мере, частично насыщенные карбомоноциклы и бициклы.
"Галоген" означает фтор, хлор, бром или йод. Особенно предпочтительно галоген представляет собой фтор или хлор.
"4-7-членный гетероциклил" или "4-7-членный гетероцикл" представляет собой кольцо, содержащее 4, 5, 6 или 7 кольцевых атомов, которое может содержать максимально возможное число двойных связей (ароматическое или неароматическое кольцо, которое полностью насыщенное, частично насыщенное или ненасыщенное), в котором, по меньшей мере, от одного до 4 кольцевых атомов заменены гетероатомом, выбранным из группы, состоящей из серы (включая -S(O)-,-S(O)2-), кислорода и азота (включая =N(О)-), и в котором кольцо связано с остальной частью молекулы через атом углерода или азота. Примеры 4-7-членных гетероциклов включают в себя азетидин, оксетан, тиетан, фуран, тиофен, пиррол, пирролин, имидазол, имидазолин, пиразол, пиразолин, оксазол, оксазолин, изоксазол, изоксазолин, тиазол, тиазолин, изотиазол, изотиазолин, тиадиазол, тиадиазолин, тетрагидрофуран, тетрагидротиофен, пирролидин, имидазолидин, пиразолидин, оксазолидин, изоксазолидин, тиазолидин, изотиазолидин, тиадиазолидин, сульфолан, пиран, дигидропиран, тетрагидропиран, имидазолидин, пиридин, пиридазин, пиразин, пиримидин, пиперазин, пиперидин, морфолин, тетразол, триазол, триазолидин, тетразолидин, диазепан, азепин или гомопиперазин.
"9-11-членный гетеробициклил" или "9-11-членный гетеробицикл" представляет собой гетероциклическую систему, состоящую из двух колец с числом кольцевых атомов от 9 до 11, в которой, по меньшей мере, один атом кольца разделен обоими кольцами, и которая может содержать вплоть до максимально возможного числа двойные связи (полностью насыщенное, частично насыщенное или ненасыщенное ароматическое или неароматическое кольцо), где, по меньшей мере, от одного до 6 кольцевых атомов заменены гетероатомом, выбранным из группы, состоящей из серы (в том числе -S(O)-, -S(O)2-), кислорода и азота (в том числе =N(O)-), и где указанное кольцо связано с остальной частью молекулы через атом углерода или азота. В качестве примера 9-11-членного гетеробицикла могут служить индол, индолин, бензофуран, бензотиофен, бензоксазол, бензизооксазол, бензотиазол, бензизотиазол, бензимидазол, бензимидазолин, хинолин, хиназолин, дигидрохиназолин, хинолин, дигидрохинолин, тетрагидрохинолин, декагидрохинолин, изохинолин, декагидроизохинолин, тетрагидроизохинолин, дигидроизохинолин, бензазепин, пурин или птеридин. Термин 9-11-членный гетеробицикл также включает в себя спиро-структуры из двух колец, например, как 1,4-диокса-8-азаспиро[4,5]декан или гетероциклы мостиковой структуры, например, как 8-аза-бицикло[3.2.1]октан.
В случае, когда соединения формулы (I) содержат одну или несколько кислотных групп или основных групп, настоящее изобретение также содержит их соответствующие фармацевтически или токсикологически приемлемые соли, в частности их фармацевтически пригодные соли. Таким образом, в соответствии с настоящим изобретением соединения формулы (I), которые содержат кислотные группы, можно использовать в соответствии с изобретением, например, в виде солей щелочных металлов, солей щелочноземельных металлов или солей аммония. В качестве конкретных примеров таких солей могут служить соли натрия, соли калия, соли кальция, соли магния или соли, полученные из аммиака или органических аминов, такие как, например, этиламин, этаноламин, триэтаноламин или аминокислоты. Соединения формулы (I), которые содержат одну или несколько основных групп, т.е. групп, которые могут быть протонированными, могут присутствовать и их можно использовать в соответствии с изобретением в форме их аддитивных солей с неорганическими или органическими кислотами. Примеры подходящих кислот включают хлористоводородную кислоту, бромистоводородную кислоту, фосфорную кислоту, серную кислоту, азотную кислоту, метансульфоновую кислоту, паратолуолсульфоновую кислоту, нафталиндисульфоновую кислоту, щавелевую кислоту, уксусную кислоту, винную кислоту, молочную кислоту, салициловую кислоту, бензойную кислоту, муравьиную кислоту, пропионовую кислоту, пивалиновую кислоту, диэтилуксусную кислоту, малоновую кислоту, янтарную кислоту, пимелиновую кислоту, фумаровую кислоту, малеиновую кислоту, яблочную кислоту, сульфаминовую кислоту, фенилпропионовую кислоту, глюконовую кислоту, аскорбиновую кислоту, изоникотиновую кислоту, лимонную кислоту, адипиновую кислоту и другие кислоты, известные специалистам в данной области техники. Если соединения формулы (I) одновременно содержат кислотные и основные группы в молекуле, данное изобретение также включает, кроме вышеуказанных солевых форм, внутренние соли или бетаины (цвиттер-ионы). Соответствующие соли соединений в соответствии с формулой (I) можно получать традиционными способами, которые известны специалистам в данной области техники, такими как, например, путем взаимодействия их с органической или неорганической кислотой или основанием в растворителе или диспергаторе, или посредством анионного обмена или катионного обмена с другими солями. Настоящее изобретение также включает все соли соединений формулы (I), которые в силу низкой физиологической совместимости не пригодны непосредственно для использования в фармацевтических препаратах, но которые могут быть использованы, например, в качестве промежуточных химических соединений или для получения фармацевтически приемлемых солей.
Под термином "фармацевтически приемлемый" подразумевается одобренное регулирующим органом, таким как Европейское агентство по оценке медицинской продукции (ЕМЕА) и/или Управление по контролю за качеством пищевых продуктов и лекарственных препаратов США (FDA) и/или любым другим Национальным регулирующим органом использование для животных, предпочтительно для человека.
Под "фармацевтической композицией" подразумевается один или более активных ингредиентов в комбинации с одним или более инертными ингредиентами, а также любой продукт, который образуется прямо или косвенно в результате объединения, комплексообразования или агрегации любых двух или более указанных ингредиентов, или в результате диссоциации одного или более указанных ингредиентов, или других типов реакций или взаимодействий одного или нескольких ингредиентов. Таким образом, фармацевтические композиции настоящего изобретения охватывают любую композицию, полученную путем смешивания соединения настоящего изобретения с фармацевтически приемлемым наполнителем (фармацевтически приемлемым носителем).
Термин "наполнитель" относится к разбавителю, адъюванту или носителю с помощью которых вводят терапевтический препарат. В качестве примера такого фармацевтически пригодного наполнителя могут служить стерильные жидкие среды, такие как вода и масла, в том числе любое масло нефтяного, животного, растительного или синтетического происхождения, включая, но не ограничивая их объема арахисовым маслом, соевым маслом, минеральным маслом, кунжутным маслом и тому подобные. Вода является предпочтительным наполнителем, в случае, когда фармацевтическая композиция вводится перорально. Физиологические растворы и водные растворы декстрозы являются предпочтительными наполнителями, в случае, когда фармацевтическая композиция предназначена для внутривенного введения. Физиологические растворы и водные растворы декстрозы и глицерина предпочтительно используют в качестве жидких наполнителей для приготовления инъекционных растворов. Подходящие фармацевтические наполнители включают крахмал, глюкозу, лактозу, сахарозу, желатин, солод, рисовую муку, мел, силикагель, стеарат натрия, моностеарат глицерина, тальк, натрия хлорид, сухое обезжиренное молоко, глицерин, пропиленгликоль, воду, этанол и тому подобные. Композиция настоящего изобретения, по желанию, может также содержать незначительные количества смачивающих агентов или эмульгаторов, или буферных веществ для стабилизации pH. Эти композиции могут быть в форме растворов, суспензий, эмульсий, таблеток, пилюль, капсул, порошков, составов с замедленным высвобождением и тому подобное. Композиция настоящего изобретения может быть получена в форме суппозитория на основе традиционных связующих и наполнителей, таких как триглицериды. Пероральный состав может включать стандартные наполнители, такие как фармацевтические сорта маннита, лактозы, крахмала, стеарата магния, натриевой соли сахарина, целлюлозы, карбоната магния и тому подобные. Примеры подходящих фармацевтических наполнителей описаны E.W. Martin в "Remington's Pharmaceutical Sciences". Такие композиции обычно содержат терапевтически эффективное количество терапевтических средств предпочтительно в очищенном виде вместе с подходящим количеством наполнителя так, чтобы обеспечить форму для подходящего введения пациенту. Данный состав должен удовлетворять режиму его приема.
Предпочтительно X3 представляет собой О.
Предпочтительно Х представляет собой N(R4), X1 представляет С, и Х3 представляет собой О.
Предпочтительно X2 представляет собой C(R7R7a).
Предпочтительно L1 выбирают из группы, состоящей из



где R представляет собой Н или C1-4алкил; Y представляет собой NH, О или S; и R1, R1a, R2, R2a, R3, R3a, R4, X, X1, X2 имеют вышеуказанные значения;
Еще в более предпочтительном варианте L1 выбирают из группы, состоящей из





где R имеет вышеуказанное значение. По меньшей мере один (вплоть до четырех) атомов водорода заменены группой L2-Z. В случае, когда присутствует более одной группы L2-Z, каждый L и каждый Z может быть выбран независимо друг от друга. Предпочтительно, чтобы присутствовала только одна группа L2-Z, что приводит к образованию структуры формулы D-L1-L2-Z.
Обычно L2 может быть соединен с L1 в любом положении за исключением положения замены водорода, отмеченного символом * структуры формулы (I). Предпочтительно, чтобы от одного до четырех атомов водорода, заданных непосредственно радикалами R, R1-R8, или например, водород С1-4алкила или дополнительных групп и колец, задаваемых радикалами R и R1-R8, были заменены группой L2-Z.
Кроме того, L1 может при необходимости дополнительно замещаться. Обычно можно использовать любой заместитель, поскольку он не оказывает влияния на основополагающий принцип расщепления связи.
В предпочтительном варианте один или несколько дополнительных необязательных заместителей независимо выбирают из группы, состоящей из галогена, CN, COOR9, OR9, C(O)R9, C(O)N(R9R9a), S(O)2N(R9R9a), S(O)N(R9R9a), S(O)2R9, S(O)R9, N(R9)S(O)N(R9aR9b), SR9, N(R9R9a), NO2, OC(O)R9, N(R9)S(O)2R9a, N(R9)S(O)2R9a, N(R9)S(O)R9a, N(R9)C(O)OR9a, N(R9)C(O)N(R9aR9b), ОС(О)N(R9R9a), Т, C1-50алкила, C2-50алкенила или C2-50алкинила, где Т, С1-50алкил, C2-50алкенил и C2-50алкинил при необходимости замещены одним или более R10, которые одинаковые или различные, и где C1-50алкил; C2-50алкенил; и C2-50алкинил при необходимости прерваны одной или более группами, выбранными из группы, состоящей из Т, -С(O)O-, -O-, -С(O)-, - C(O)N(R11)-, -S(O)2N(R11)-, -S(O)N(R11)-, -S(O)2-, -S(O)-, -N(R11S(O)2N(R11a)-, -S-, - N(R11)-, -OC(O)R11, -N(R11)C(O)-, -N(R11)S(O)2-, -N(R11)S(O)-, -N(R11)C(O)O-, -N(R11)C(O)N(R11a)- и -OC(O)N(R11R11a);
R9, R9a, R9b независимо выбирают из группы, состоящей из Н, Т, Z и С1-50алкила, C2-50алкенила или C2-50алкинила, где Т, C1-50алкил, C2-50алкенил и C2-50алкинил при необходимости замещены одним или несколькими заместителями из R10, которые одинаковые или различные, и где С1-50алкил, C2-50алкенил и C2-50алкинил при необходимости прерываются одной или несколькими группами, выбранными из группы, состоящей из Т, -С(O)O-, -O-, -С(O)-, - C(O)N(R11)-, -S(O)2N(R11)-, -S(O)N(R11)-, -S(O)2-, -S(O)-, -N(R11)S(O)2N(R11a)-, -S-, -N(R11)-, -OC(O)R11, -N(R11)C(O)-, -N(R11)S(O)2-, -N(R11)S(O)-, -N(R11)C(O)O-, -N(Rll)C(O)N(Rlla)- и -ОС(О)R11R11a);
Т выбирают из группы, состоящей из фенила; нафтила; инденила; инданила; тетралинила; С3-10циклоалкила; 4-7-членного гетероциклила; или 9-11-членного гетеробициклила, где Т необязательно замещен одним или несколькими заместителями из R10, которые одинаковые или отличаются друг от друга;
R10 представляет собой Z, галоген, CN, оксо (=O), COOR12, OR12, C(O)R12, C(O)N(R12R12a), S(O)2N(R12R12a), S(O)N(R12R12a), S(O)2R12, S(O)R12, N(R12)S(O)2N(R12aR12b), SR12, N(R12R12a), NO2, OC(O)R12, N(R12)C(O)R12a, N(R12)S(O)2R12a, N(R12)C(O)OR12a, N(R12)C(O)OR12a, N(R12)C(O)N(R12aR12b), OC(O)N(R12R12a); или C1-6алкил, где C1-6алкил при необходимости замещен одним или несколькими атомами галогена, которые одинаковые или отличаются друг от друга;
R11, R11a, R12, R12a, R12b независимо выбирают из группы, состоящей из Н или C1-6алкила, где C1-6алкил при необходимости замещен одним или несколькими атомами галогена, которые одинаковые или различные;
Термин "прерванный" означает, что между двумя атомами углерода или на конце углеродной цепи между углеродом и водородом вставлена группа.
L2 представляет собой одинарную химическую связь или спейсер. В случае, когда L2 является спейсером, предпочтительно, чтобы он представлял собой один или несколько необязательных заместителей, определенных выше, при условии, что L2 замещен группой Z.
Соответственно, в случае когда L2 не является одинарной химической связью, L2-Z представляет собой COOR9, OR9, C(O)R9, C(O)N(R9R9a), S(O)2N(R9R9a), S(O)2N(R9R9a), S(O)2R9, S(O)R9, N(R9)S(O)2N(R9aR9b), SR9, N(R9R9a), OC(O)R9, N(R9)C(O)R9a, N(R9)S(O)2R9a, N(R9)S(O)R9a, N(R9)C(O)OR9a, N(R9)C(O)N(R9aR9b), OC(O)N(R9R9a), T, C1-50алкил, С2-50алкенил или C2-50алкинил, где Т, C1-50алкил, C2-50алкенил и C2-50алкинил являются при необходимости замещенными одним или несколькими радикалами R10, которые одинаковые или различные, и где C1-50алкил, C2-50алкенил и С2-50алкинил при необходимости прерываются одной или несколькими группами, выбранными из группы, состоящей из -Т-, -С(O)0-, -O-, -С(O)-, -C(O)N(R11)-, -S(O)2N(R11)-, -S(O)N(R11)-, -S(O)2-, -S(O)-, -N(Rll)S(O)2N(Rlla)-, -S-, -N(R11)-, -OC(O)R11, -N(R11)C(O)-, -N(R11)S(O)2-, -N(R11)S(O)-, -N(R11)C(O)O-, - N(R11)C(O)N(R11a)- и -OC(O)N(R11R11a);
R9, R98, R913 независимо выбирают из группы, состоящей из Н, Т и C1-50алкила, C2-50алкенила или C2-50алкинила, где Т, C1-50алкил, C2-50алкенил и C2-50алкинил при необходимости замещены одним или несколькими R10, которые одинаковые или различные, и где C1-50алкил, C2-50алкенил и C2-50алкинил при необходимости прерываются одной или более группами, выбранными из группы, состоящей из Т, -С(O)O-, -O-, -С(O)-, -C(O)N(R11)-, -S(O)2N(R11)-, -S(O)N(R11)-, -S(O)2-, -S(O)-, -N(R11)S(O)2N(R11a)-, -S-, -N(R11)-, -OC(O)R11, -N(R11)C(O)-, -N(R11)S(O)2-, -N(R11)S(O)-; -N(R11)C(O)O-; -N(R11)C(O)R11a)- и -OC(O)N(R11R11a);
Т выбирают из группы, состоящей из фенила, нафтила, инденила, инданила, тетралинила, С3-10циклоалкила, 4-7-членного гетероциклила или 9-11-членного гетеробициклила, где Т при необходимости замещен одним или более R10, которые одинаковые или различные;
R10 представляет собой Z, галоген, CN, оксо(=O), COOR12; OR12; C(O)R12; C(O)N(R12R12a), S(O)2N(R12R12a), S(O)N(R12R12a), S(O)2R12, S(O)R12, N(R12)S(O)2N(R12aR12b), SR12, N(R12R12a), NO2, OC(O)R12, N(R12)C(O)R12a, N(R12)S(O)2R12a, N(R12)S(O)R12a, N(R12)C(O)OR12a, N(R12)C(O)N(R12aR12b); OC(O)N(R12R12a) или C1-6алкил, где C1-6алкил при необходимости замещен одним или более атомами галогена, которые одинаковые или различные;
R11, R11a, R12, R12a, R12b независимо выбирают из группы, состоящей из Н, Z или C1-6алкила, где C1-6алкил при необходимости замещен одним или несколькими атомами галогена, которые одинаковые или различные;
при условии, что один из R9, R9a, R9b, R11, R11a, R12, R12a, R12b представляют собой группу Z.
Более предпочтительно L2 представляет собой алкильную цепь, содержащую от 1 до 20 углеродных атомов, которая при необходимости прерывается одной или более группами, независимо выбранными из -О- и C(O)N(R3aa); при необходимости замещенную одной или более группами, независимо выбранными из ОН и C(O)N(R3aaR3aaa), и где R3aa, R3aaa независимо выбирают из группы, состоящей из Н и C1-4алкила.
Предпочтительно L2 имеет молекулярную массу в интервале от 14 г/моль до 750 г/моль.
Предпочтительно L2 присоединен к Z через концевую группу, выбранную из
В случае, если L2 имеет такую концевую группу, также предпочтительно, чтобы L2 имел молекулярную массу в интервале от 14 г/моль до 500 г/моль, рассчитанную без учета такой концевой группы.
L предпочтительно представлен формулой (Ia)

где R4, L2 и Z имеют значение, указанное выше, и где R3aa, R3aaa независимо выбирают из группы, состоящей из Н и C1-4алкила? или R3aaa и R3aaa вместе с атомом азота, к которому они присоединены, соединяются с образованием 4-7-членного гетероцикла. R4 предпочтительно представляет собой Н или метил.
L предпочтительно представлен формулой (Ib)

где R1, R1a, R4, L2 и Z имеют значение, указанное выше, и где R383 представляет собой Н или C1-4алкил. R4 предпочтительно представляет собой Н или метил.
R1 в формуле (I) предпочтительно представляет собой L2-Z.
R3 в формуле (I) предпочтительно представляет собой L2-Z.
R3, R3a в формуле (I) предпочтительно вместе с атомом азота, к которому они присоединены, соединяются с образованием 4-7-членного гетероцикла, при этом указанный гетероцикл замещен L2-Z.
D-H предпочтительно представляет собой низкомолекулярный биологически активный агент или биополимер.
D-H предпочтительно представляет собой биополимер, выбранный из группы биополимеров, состоящей из белков, полипептидов, олигонуклеотидов и пептидонуклеиновых кислот.
"Олигонуклеотиды" означают ДНК, РНК, как одноцепочечные, так и двухцепочечные, siPHK (малую интерферирующую РНК), miPHK (микроРНК), аптамеры, и любые их химические модификации, которые предпочтительно имеют длину от 2 до 1000 нуклеотидов. Указанные модификации включают, но не ограничиваются теми, которые обеспечивают другие химические группы, которые придают дополнительный заряд, поляризуемость, водородное связывание, электростатическое взаимодействие и плавкость (fluxionality) основаниям нуклеиновой кислоты-лиганда или нуклеиновой кислоте-лиганду в целом. Такие модификации включают, не ограничиваются модификациями во 2'-положении сахара рибозы, модификациями в 5-положении пиримидина, модификации в 8-положении пурина, модификациями по экзоциклическим аминам, замещением 4-тиоуридина, замещением 5-бромо или 5-иодоурацила, модификациями остова, метилированиями, необычными комбинациями спаривания оснований, такими как изооснования изоцитидин и изогуанидин, и тому подобные. Модификации также могут включать в себя 3' и 5' модификации, такие как кэппирование и изменение стереохимии.
Предпочтительно D-H представляет собой полипептид, выбранный из группы полипептидов, состоящей из адренокортикотропного гормона АСТН, аденозиндезаминазы, агальзидазы, альфа-1-антитрипсина (ААТ), ингибитора альфа-1-протеиназы (API), альтеплазы, амилинов (амилина, симлина), анистреплазы, анкродсеринпротеазы, антител (моноклональных или поликлональных, и их фрагментов или продуктов слияния), антитромбина III, антитрипсинов, апротинина, аспарагиназы, атосибана, бифалина, бивалирудина, костных морфогенных белков, бычьего панкреатического трипсинового ингибитора (BPTI), фрагментов кадгерина, кальцитонина (лосося), коллагеназы, ингибитора системы комплемента с эстеразной активностью С1, конотоксинов, фрагментов цитокиновых рецепторов, ДНКазы, динорфина А, эндорфинов, энфувиртида, энкефалинов, эритропоэтинов, эксендинов, фактора VII, фактора VIIa, фактора VIII, фактора VIIIa, фактора IX, фибринолизина, фактора роста фибробластов (FGF), высвобождающего гормон роста пептида-2, (GHRP2), слитых белков, фолликулостимулирующего гормона, грамицидина, грелина, дезацилгрелина, гранулоцитарного колониестимулирующего фактора (G-CSF), галактозидазы, глюкагона, глюкагон-подобных пептидов, глюкоцереброзидазы, гранулоцитарно-макрофагального колониестамулирующего фактора (GM-CSF), белков теплового шока человека (HSP), белка, активирующего фосфолипазу, (PLAP), хорионического гонадотропина человека (hCG), гемоглобинов, вакцин против гепатита В, гирудина, ингибитора серинпротеазы человека, гиалуронидаз, идуронидазы, иммунноглобулинов, вакцин против гриппа, интерлейкинов (1-альфа, 1-бета, 2, 3, 4, 6, 10, 11, 12, 13,21), антагониста рецептора IL-1 (rhIL-Ira), инсулинов, инсулиноподобных факторов роста, белка, связывающего инсулшюподобный фактор роста (rhi GFBP), интерферонов (альфа 2а, альфа 2b, альфа 2с, бета-1a, бета 1b, гамма-1a, гамма 1b), молекулы внутриклеточной адгезии, фактора роста кератиноцитов (KGF), гликопротеинового лиганда Р-селектина (PSGL), трансформирующих факторов роста, лактазы, лептина, лейпролида, левотироксина, лютеинизирующего гормона, вакцины против болезни Лайма, натрийуретических пептидов (ANP, BNP, CNP и их фрагментов), нейропептида Y, панкрелипазы, панкреатического полипептида, папаина, паратиреоидного гормона, тромбоцитарного фактора роста (PDGF), пепсина, пептида YY, ацетилгидролазы тромбоцит-активирующего фактора (PAF-AH), пролактина, С-белка, тимальфазина, октреотида, секретина, серморелина, растворимого рецептора фактора некроза опухоли (TNFR), супероксиддисмутазы (SOD), соматропинов (гормонов роста), соматоприма, соматостатина, стрептокиназы, сахаразы, терлипрессина, фрагмента столбнячного токсина, тилактазы, тромбинов, тимозина, гормона, регулирующего деятельность щитовидной железы, тиреотропина, фактора некроза опухоли (TNF), рецептора TNF, соединенного с Fc-участком IgG, тканевого активатора плазминогена (tPA), тиреотропного гормона (TSH), уродилатина, уратоксидазы, урокиназы, вакцин, фактора роста сосудистого эндотелия (VEGF), вазоактивного интестинального пептида, вазопрессина, циконотида, лектина и рицина.
Предпочтительно D-H представляет собой белок, полученный технологиями рекомбинантных ДНК.
Предпочтительно D-H представляет собой белок, выбранный из группы белков, состоящей из фрагментов антител, одноцепочечных антиген-связывающих белков, каталитических антител и слитых белков.
Предпочтительно D-H представляет собой низкомолекулярный биологически активный агент, выбранный из группы, состоящей из агентов, воздействующих на центральную нервную систему, противоинфекционных, противоаллергических, иммуномодулирующих агентов, агентов от ожирения, антикоагулянтов, антидиабетических агентов, антиопухолевых, антибактериальных, противогрибковых, болеутоляющих, противозачаточных, противовоспалительных, стероидных, сосудорасширяющих, сосудосуживающих и сердечно-сосудистых агентов, имеющих, по меньшей мере, одну первичную или вторичную аминогруппу.
Предпочтительно D-H представляет собой низкомолекулярный биологически активный агент, выбранный из группы агентов, состоящей из акарбозы, алалроклата, алендроната, амантадина, амикацина, аминептина, аминоглютетимида, амисульприда, амлодипина, амотосалена, амоксапина, амоксициллина, амфетамина, амфотерицина В, ампициллина, ампренавира, амринона, анилеридина, апраклонидина, апрамицина, артикаина, атенолола, атомоксетина, авизафона, баклофена, беназеприла, бенсеразида, бензокаина, бетаксолола, блеомипина, бромфенака, брофаромина, карведилол, катина, катинона, карбутамида, цефалексина, клинафлоксацина, ципрофлоксацина, дефероксамина, делавирдина, дезипрамина, даунорубицина, дексметилфенидата, диафенилсульфона, дизоцилпина, допамина, добутамина, дорзоламида, доксорубицина, дулоксетина, эфлорнитина, эналаприла, адреналина, эпирубицина, эрголина, эртапенема, эсмолола, эноксацина, этамбутола, фенфлюрамина, фенолдопама, фенотерола, финголимода, флекаинида, флувоксамина, фосампренавира, фроватриптана, фуросемида, флуоксетина, габапентина, гатифлоксацина, гемифлокацина, гентамицина, грепафлоксацина, гексилкаина, гидралазина, гидрохлоротиазида, икофунгипена, идарубицина, имиквимода, инверсина, изопротеренола, исрадипина, канамицина А, кетамина, лабеталола, ламивудина, левобунолола, леводопы, левотироксина, лизиноприла, ломефлоксацина, лоракарбефа, мапротилина, мефлохина, мелфалана, мемантина, меропенема, месалазина, мескалина, метилдопы, метилендиоксиметамфетамина, метопролола, милнаципрана, митоксантрона, моксифлоксацина, норадреналина, норфлоксацина, нортриптилина, неомицина В, нистатина, осельтамивира, памидроновой кислоты, пароксетина, пазуфлоксацина, пеметрекседа, периндоприла, фенметразина, фенелзина, прегабалина, прокаина, псевдоэфедрина, протриптилина, ребоксетина, ритодрина, сабарубицина, сальбутамола, серотонина, сертралина, ситаглиптина, соталола, спектиномицина, сульфадиазина, сульфамеразина, сертралина, спректиномицина, сульфалена, сульфаметоксазола, такрина, тамсулозина, тербуталина, тимолола, тирофибана, тобрамипина, токаинида, тосульфлоксацина, трандолаприла, транексамовой кислоты, транилципромина, тримерексата, тровафлоксапина, валацикловира, валганцикловира, ванкомицина, виомицина, вилоксазина и зальцитабина.
Предпочтительно Z представляет собой полимер, имеющий молекулярную массу, по меньшей мере, 500 Да или C8-18алкильную группу.
В предпочтительном варианте Z выбирают из группы при необходимости сшитых полимеров, состоящей из поли(пропиленгликоля), поли(этиленгликоля), декстрана, хитозана, гиалуроновой кислоты, альгината, ксилана, маннана, каррагинана, агарозы, целлюлозы, крахмала, гидроксиалюилированного крахмала (HAS), поли(виниловых спиртов), поли(оксазолинов), поли(ангидридов), сложных полиортоэфиров, поликарбонатов), поли(уретанов), поли(акриловых кислот), поли(акриламидов), поли(акрилатов), поли(метакрилатов), поли(органофосфазенов), полиоксазолина, поли(силоксанов), поли(амидов), поли(винилпирролидона), поли(цианакрилатов), сложных полиэфиров, поли(иминокарбонатов), поли(аминокислот), коллагена, желатина, гидрогеля или белка плазмы крови, и их сополимеров.
В предпочтительном варианте Z представляет собой белок.
В предпочтительном варианте Z представляет собой белок, выбранный из группы, состоящей из альбумина, трансферрина, иммуноглобулина.
В предпочтительном варианте Z представляет собой линейный или разветвленный поли(этиленгликоль) с молекулярной массой от 2000 до 150000 Да.
В еще более предпочтительном варианте предлагается пролекарство настоящего изобретения, в котором D-H представляет собой агонист рецептора глюкагон-подобного пептида GLP-1, L представляет собой L1, представленный формулой (I), как указано выше, а Z представляет собой гидрогель. Еще в более предпочтительном варианте в формуле (I) Х представляет собой N(R4), X1 представляет собой С, а X3 представляет собой О. В особенно предпочтительном варианте L представлен формулой (Ia), указанной выше.
GLP-1 (глюкагон-подобный пептид-1) является одним из интестинальных пептидных гормонов, который высвобождается в кровеносную систему после приема пищи. Он усиливает постпрандиальное высвобождение инсулина в случае, когда происходит усвоение пищи (особенно углеводов), и их уровень в крови постпрандиально повышен. GLP-1 связан с сайтами рецептора GLP-1, расположенными на β-клетках поджелудочной железы, и повышает уровни эндогенного цАМФ в дозозависимой манере. В изолированных островках крыс в присутствии повьдпенных концентраций глюкозы в нормогликемическом диапазоне GLP-1 стимулирует секрецию инсулина. Ранее высказывалась возможность применения GLP-1 в терапии больных сахарным диабетом 2 типа благодаря мощному стимулирующему действию этого инсулинотропного пептида для секреции инсулина, когда повышается уровень глюкозы в крови, и прекращению его действия при возвращении в состояние нормогликемии. Антидиабетогенный эффект амида глюкагонподобного пептида-1 (7-36) у субъектов в норме и пациентов с сахарным диабетом описан, например, в журнале N. Engl. J. Med. 326 (20): стр. 1316-1322. В исследованиях in vitro и экспериментах на животных показано, что GLP-1 улучшает чувствительность к инсулину и оказывает анабо-лический эффект на панкреатические β-клетки. Также сообщалось, что у человека GLP-1 угнетает секрецию глюкагона, замедляет желудочное опустошение, и вызывает субъективное ощущение сытости, что приводит к снижению веса, если его принимают на протяжении нескольких недель и месяцев.
Сообщают, что эксендин-4 связан с рецепторами GLP-1, расположенными на панкреатических β-клетках, и имеет аффинность в 2,5 раза выше, чем GLP-1. В изолированных островках крыс и β-клетках в присутствии глюкозы эксендин усиливает секрецию инсулина в дозозависимой манере. Эксендин-4 является высоко активным агонистом, а усеченный эксендин 9-39-амид - антагонистом к рецептору на секретирующих β-клетках глюкагон-подобного пептид 1-(7-36) амида (см. J. Biol. Chem 268 (26):19650-19655). В исследованиях на грызунах с диабетом 2 типа было показано, что эксендин-4 в 5530 раз более мощный агент, чем GLP-1 по снижению уровня глюкозы в крови. Кроме того, продолжительность глюкозоснижающего действия после однократного введения эксендина-4 значительно больше, по сравнению с GLP-1 (см., например. Diabetes 48(5): 1026-1034). Сообщалось, что период полужизни в плазме эксендина-4 у людей составлял только 26 минут. Эксендин-4 снижает уровни глюкозы в крови натощак и постпрандиальной глюкозы и снижает потребление энергии у здоровых людей-добровольцев (см., например. Am. J. Physiol. Endocrinol. Metab. 281(1): Е155-61).
Используемые в соответствии с данным изобретением гидрогели известны специалистам в данной области. В качестве подходящих гидрогелей могут служить, такие которые используют в WO-A 2006/003014. В соответствии с этим, гидрогель может быть определен как трехмерные гидрофильные или амфифильных полимерные сетчатые структуры, способные поглощать большие количества воды. Такие сетчатые структуры состоят из гомополимеров или сополимеров, и они нерастворимы из-за наличия ковалентных химических или физических узлов-сшивок (в результате ионных, гидрофобных взаимодействий, переплетений). Узлы-сшивки обеспечивают сетчатую структуру и физическую целостность. Гидрогели демонстрируют термодинамическую совместимость с водой, которая обеспечивает их набухание в водной среде. Цепи указанной сетки связаны так, что имеются поры, и что основная фракция этих пор имеет размеры от 1 нм и 1000 нм.
Другим объектом настоящего изобретения является фармацевтическая композиция, содержащая в соответствии с настоящим изобретением пролекарство или его фармацевтическую соль в комбинации с фармацевтически приемлемым наполнителем.
Еще одним объектом настоящего изобретения является в соответствии с настоящим изобретением пролекарство или фармацевтическая композиция для применения в качестве лекарственного препарата.
Еще одним объектом в соответствии с настоящим изобретением является способ лечения, регулирования, замедления или предупреждения развития у пациента-млекопитающего при необходимости лечения одного или нескольких состояний, предусматривающий введение указанному пациенту терапевтически эффективного количества пролекарства в соответствии с настоящим изобретением или его фармацевтической композиции или фармацевтически приемлемой соли.
Еще другим объектом настоящего изобретения является химический предшественник пролекарства формулы Act-L, где L имеет значение, указанное выше, a Act представляет собой уходящую группу.
Act предпочтительно представляет собой хлорид, бромид, фторид, нитрофенокси, имидазолил, N-гидроксисукцинимидил, N-гидроксибензотриазолил, N-гидроксиазобензотриазолил, пентафторфенокси, 2-тиооксотиазолидинил, или N-гидроксисульфосукцинимидил.
Примеры
Материалы и методы
Материалы: защищенный в боковой цепи эксендин-4, (J. Eng et al, J. Biol.Cheni. 1992, 267, 11, 7402-7405) на амидной смоле Ринка, защищенный в боковой цепи BNP-32a, (человеческий, в котором Cys 10 и Cys 26 заменены на Ala) на хлортритильной смоле, защищенный в боковой цепи BNP-32b (человеческий, в котором Cys 10 и Cys 26 заменены на Ala), с защитной группой ivDde в боковой цепи на Lys в положении 14 на хлортритильной смоле, и амид защищенного в боковой цепи фрагмента высвобождающего фактора человеческого гормона роста 1-29 (GRF(1-29) на амидной смоле Ринка, которые синтезированы по Fmoc стратегии синтеза, получали у фирмы Peptide Specialty Laboratories GmbH, Heidelberg, Germany. В боковой цепи использовали стандартные защитные группы, за исключением того, что для Lys 27 эксендина-4 и Lys 21 соединения GRF(1-29) использовали Mmt-защитные группы боковой цепи.
40 кДа метокси(полиэтиленгликоль)малеимид-пропионамид (производное ПЭГ с молекулярной массой 40 кДа, содержащее малеимидную группу) получали у фирмы Chirotech Technology Ltd, Cambridge, UK.
2-Хлортритилхлоридную смолу, амидную смолу Зибера, и аминокислоты получали у фирмы Merck Biosciences GmbH, Schwalbach/Ts, Germany. Fmoc-D-гомоцистеин (Trt)-OH и S-тритил-3-меркаптопропионовую кислоту (TRT-MPA) получали у фирмы Bachem AG, Bubendorf, Switzerland. O-(N-Fmoc-2-аминоэтил)-O'-(2-карбоксиэтил)-ундека-этиленгликоль (Fmoc-Pop-OH) получали у фирмы Polypure AS, Oslo, Norway.
Fmoc-4-(2-аминоэтил)-1-карбоксиметилпиперазин (Fmoc-Acp-ОН) покупали у фирмы NeoMPC SA, Strasbourg, France. Цис-циклогексан-1,2-дикарбоновой кислоты ангидрид получали у фирмы Alfa Aesar GmbH & Со KG, Karlsruhe, Germany.
Все другие химические реактивы получали у фирмы Sigma-ALDRJCH Chemie GmbH, Taufldrchen, Germany.
Твердофазный синтез пептидных соединений осуществляли на 2-хлортритилхлоридной смоле с загрузкой 1,3 ммоль/г смолы или амидной смоле Зибера с загрузкой 0,55 ммоль/г смолы. В качестве реакционных сосудов использовали шприцы, оснащенные полипропиленовыми фриттами.
Загрузку первой аминокислоты в смолы осуществляли в соответствии с инструкциями производителя.
Снятие Fmoc-защиты:
Для удаления Fmoc-защищающей группы, используемую смолу перемешивали с 2/2/96 (об./об./об.) пиперидина/DBU/DMF (два раза по 10 мин каждый) и промывают DMF (десять раз).
Снятие ivDde-защиты:
Для удаления ivDde-защитной группы используемую смолу перемешивали с 98/2 (об./об.) смеси DMF/гидрат гидразина (трижды по 10 мин каждый) и промывают DMF (десять раз).
Вос-защита:
N-конец пептида защищали Вос-группой перемешиванием смолы с 30 экв. (Вос) 20 и 60 экв. пиридина в DCM. Через 1 час смолу промывают DCM (10 раз).
Стандартные условия связывания кислот:
Связывание кислот (алифатических кислот, Fmoc-аминокислот) со свободными аминогруппам на смоле проводили путем перемешивания смолы с 3 экв. кислоты, 3 экв. РуВОР и 6 экв. DEEA в отношении свободных аминогрупп на смоле (рассчитываемых на основе теоретической загрузки смолы) в ДМФА при комнатной температуре. Через 1 час смолу промывают DMF (10 раз).
Связывание 3-малеимидопропионовой кислоты:
Связывание 3-малеимидопропионовой кислоты со свободными аминогруппами на смоле проводили путем смешивания смолы с 2 экв. кислоты, 2 экв. ОПК и 2 экв. HOBt в отношении свободных аминогрупп в ДМФА при комнатной температуре. Через 30 мин смолу промывают DMF (10 раз).
Протокол стандартного синтеза производных мочевины на смоле:
Синтез производных мочевины на смоле проводили путем перемешивания смолы с 2,5 экв. бис(пентафторфенил)карбоната, 5 экв. диизопропилэтиламина (DIEA) и 0,25 экв. диметиламинопиридина (DMAP) в отношении свободных аминогрупп в смеси дихлорметана и ацетонитрила (DCM/ACN) 1/1 при комнатной температуре. Через 15 мин смолу промывают N,N-диметилформамид (DMF) (10 раз). 5 экв. амина растворяют в ДМФА. Смесь вводили в смолу и перемешивали в течение 60 мин при комнатной температуре. Смолу промывают с использованием DMF (10 раз).
Протокол расщепления для амидной смолы Зибера:
По завершении синтеза смолу промывали дихлорметаном (DCM) (10 раз), сушили под вакуумом и обрабатывали несколько раз (пять раз по 15 минут) смесью DCM/TES/TFA 97/2/1 (об./об./об.). Элюаты объединяли, летучие компоненты удаляли в потоке азота и полученный продукт очищали обращенно-фазовой ВЭЖХ. Фракции ВЭЖХ, содержащие продукт, объединяли и лиофилизировали.
Протокол расщепления для 2-хлортритилхлоридной смолы:
По завершении синтеза, смолу промывают DCM, сушили под вакуумом и обрабатывали 2 раза в течение 30 минут смесью DCM/HFIP 6/4 (об./об.). Элюаты объединяли, летучие компоненты удаляли в потоке азота и полученный продукт очищали обращенно-фазовой ВЭЖХ. Фракции ВЭЖХ, содержащие продукт, объединяли и лиофилизировали.
Протокол расщепления для амидной смолы Ринка:
По завершении синтеза смолу промывали DCM, сушили под вакуумом и обрабатывали 2 мл смеси TFA для снятия продукта (TFA/TES/HbO/DTT 95/2/2/1) на 100 мг смолы в течение 60 минут. Летучие компоненты удаляли в потоке азота. Неполярные продукты боковой цепи и защитные группы удаляли осаждением пептида из диэтилового эфира. Осажденный продукт сушили под вакуумом и растворяли в смеси ACN с водой 1/1, а затем очищали обращенно-фазовой ВЭЖХ.
Аминосодержащие продукты, полученные в виде солей трифторуксусной кислоты, превращали в соответствующие соли HCl на ионообменной смоле (Discovery DSC-SAX, Supeico, USA). Эту стадию проводили в случае, когда ожидалось, что остаточная TFA будет мешать, например, последующим реакциям связывания.
Очистка с использованием обращенно-фазовой ВЭЖХ:
Обращенно-фазовую ВЭЖХ проводили на колонке с силикагелем С 18 ReproSil-Pur 300 фаза ODS-3 5 мкм размером 100×20 или 100×40 мм (Dr. Maisch, Ammerbuch, Germany), подсоединенной к системе ВЭЖХ Waters 600 и детектору поглощения Waters 2487. Использовали линейные градиенты раствора А (0,1% TFA в Н2О) и раствора В (0,1% TFA в ацетонитриле). Фракции ВЭЖХ, содержащие продукт, лиофилизировали.
Анализ образцов: масс-спектрометрию с ионизацией электрораспылением (ESI-MS) осуществляли на измерительной системе Waters ZQ 4000 ESI и, при необходимости, масс-спектры расшифровывали с использованием программного обеспечения Waters MaxEnt.
Эксклюзионную хроматографию (SEC) проводили с использованием системы Amersham Bioscience AEKTAbasic, оснащенную колонкой Superdex200 10/300 (Amersham Bioscience/GE Healthcare), если не указано иное. В качестве подвижной фазы использовали 10 мМ фосфата натрия, 140 мМ NaCl, pH 7,4, 3 мМ EDTA.
Катионнобменную хроматографию проводили с использованием системы Amersham Bioscience AEKTAbasic, оснащенную колонкой Source 15S, заполненную HR16/10 (Amersham Bioscience/GE Healthcare).
Обессоливание проводили с использованием системы Amersham Bioscience AEKTAbasic, оснащенной колонкой для обессоливания HiPrep 26/10, и 0,1% уксусной кислоты в воде в качестве подвижной фазы.
Гидролиз линкера in vitro и высвобождение лекарственного средства: соединения растворяли в буфере А (10 мМ натрия фосфата, 140 мМ NaCl, pH 7,4, 3 мМ EDTA) или буфере В (0,1 мМ ацетата и 3 мМ EDTA, pH 4,0), и затем раствор фильтровали через фильтр 0,2 мкм и инкубировали при 37°C. Отбирали пробы с задержкой по времени и анализировали обращенно-фазовой ВЭЖХ при 215 нм и ESI-MS. УФ сигналы, коррелирующие с освобожденной молекулой лекарственного средства, объединяли и изображали на графике в зависимости от времени инкубации. В случае одинакового времени удержания пролекарства и лекарственного средства соотношение сигналов масс использовали для определения кинетики высвобождения.
В случае гидрогелевых конъюгатов соединения суспендировали в буфере А и инкубировали при температуре 37°C. Аналиты отбирали после центрифугирования суспензии и анализировали методом обращенно-фазовой ВЭЖХ при 215 нм. УФ сигналы, коррелирующие с освобожденной молекулой лекарственного средства, объединяли и изображали на графике в зависимости от времени инкубации.
Для оценки соответствующего периода полужизни при высвобождении использовали программное обеспечение для обработки кривых.
Пример 1
Синтез носителя на основе жирных кислот (1)

1 синтезируют на амидной смоле Зибера (477 мг, 0,262 ммоль), путем связывания Fmoc-Lys(ivDde)-OH, снятия Fmoc-защиты, связывания додекановой кислоты, снятия ivDde-защиты, связывания Fmoc-Pop-OH, снятия Fmoc-защиты, связывания 3-малеимидопропионовой кислоты, отщепления от смолы и последующую очистку, как показано выше и описано в разделе "Материалы и методы".
Выход: 128 мг (0,119 ммоль).
МС: м/з 1101,0=[M+Na]+ (Расчетная Мм=1078,4 г/моль).
Пример 2
Синтез линкерного реагента (2)
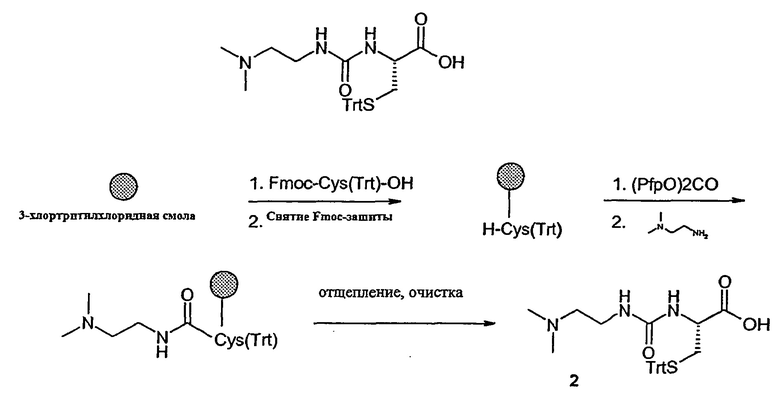
Линкерный реагент 2 синтезируют на 3-хлортритилхлоридной смоле (300 мг, 0,39 ммоль) путем нагружения смолы Fmoc-Cys (Trt)-OH, снятия Fmoc-защиты и образования на смоле производного мочевины, используя N,N-диметилэтилендиамин в качестве амина, отщепления продукта от смолы, как показано выше и описано в разделе "Материалы и методы". Для разделения методом обращенно-фазовой ВЭЖХ использовали 0,01% HCl в воде в качестве раствора А и 0,01% HCl в ацетонитриле в качестве раствора В.
Выход: 82 мг продукта в виде HCl-соли (0,16 ммоль).
МС: м/з 478,2=[М+Н]+ (Расчетная Мм=477,6 г/моль).
Пример 3
Синтез интермедиата линкера, связанного с эксендином-4 (3)

2 (14 мг, 0,027 ммоль), РуВОР (14 мг, 0,027 ммоль) и США (17 мкл, 0,10 ммоль) растворяют в 0,2 мл безводного DMF. Полученную смесь добавляют к 22 мг защищенному в боковой цепи эксендину-4 на смоле (0,1 ммоль/г, 2,2 мкмоль) и интенсивно перемешивают в течение 30 мин при комнатной температуре. Смолу промывают DMF (10 раз) и DCM (10 раз). 3 отщепляют от смолы и очищают методом обращенно-фазовой ВЭЖХ по методике, описанной в разделе "Материалы и Методы". Выход: 1,7 мг 3 в виде трифторацетатной соли (0,38 мкмоль).
МС: м/з 1468,7=[М+3Н]3+(Расчетная Мм=4403 г/моль).
Пример 4 Синтез конъюгата жирная кислота-ПЭГ-линкер-эксендин-4 (Соединение 4)

3 (1,7 мг, 0,38 ммоль) и 1 (0,6 мг, 0,58 ммоль) растворяют в 500 мкл смеси ацетонитрил/вода 7/3 (об./об.). Прибавляют 40 мкл 0,5 М фосфатного буфера (pH 7,4), и смесь инкубируют при комнатной температуре в течение 10 мин. Конъюгат 4 очищают методом обращенно-фазовой ВЭЖХ.
МС: м/з 1828,7=[М+3H]3+(Расчетная Мм=5480 г/моль).
Пример 5
Синтез линкерного интермедиата (5а)

Fmoc-Acp-OH⋅2HCl (100 мг, 0,21 ммоль) суспендируют в 400 мкл DMF/DMSO 1/1 (об./об.). Добавляют S-тритилцистеамина гидрохлорид (75 мг, 0,21 ммоль), РуВОР (109 мг, 0,21 ммоль) и DIEA (146 мкл, 0,86 ммоль) и смесь перемешивают в течение 60 мин при комнатной температуре. Fmoc-группу снимают путем добавления 75 мкл пиперидина и 25 мкл DBU. Через 15 минут смесь подвергают гидролизу и подкисляют уксусной кислотой (АсОН) и полученное соединение очищают методом обращенно-фазовой ВЭЖХ. После лиофилизапии получают 98 мг продукта (0,14 ммоль, в виде двойной трифторацетатной соли).
МС: м/з 511,6=[М+Na]+(Расчетная Мм=488,7 г/моль).
Синтез цис-циклогексан-дикарбоновой кислоты амоксапина моноамида (5b)

Амоксапин (200 мг, 0,64 ммоль) и пис-циклогексан-1,2-дикарбоновый ангидрид (108 мг, 0,70 ммоль) растворяют в 700 мкл сухого DMF. Добавляют пиридин (130 мкл, 1,6 ммоль) и смесь перемешивают в течение 60 мин при комнатной температуре. Смесь гасят 2 мл смеси ацетонитрил/уксусная кислота/вода (1/1/1) и очищают обращенно-фазовой ВЭЖХ. После лиофилизапии получают 344 мг 5b (0,49 ммоль, в виде двойной трифторацетатной соли).
МС: м/з 468,5=[М+Н]+ (Расчетная Мм=468,0 г/моль).
Синтез конъюгата линкер-амоксапин (5с)

5b (7 мг, 0,010 ммоль) предварительно активируют инкубацией с использованием РуВОР (12,5 мг, 0,024 ммоль) и DIEA (5 мкл, 0,03 ммоль) в 200 мкд безводного DMF в течение 45 мин при комнатной температуре. Добавляют 5а (20 мг, 0,028 ммоль) и DIEA (15 мкл, 0,09 ммоль) и смесь инкубируют еще 60 мин. Смесь гасят 0,5 мл смеси ацетонитрил/уксусная кислота/вода (1/1/1) и очищают методом обращенно-фазовой ВЭЖХ. После лиофилизапии получают 3 мг 5 с (0,0026 ммоль, в виде двойной трифторацетатной соли).
МС: м/з 939,3=[М+НГ (Расчетная Мм=938,6 г/моль).
Для снятия тритильной защиты лиофилизат инкубируют в 1 мл HFIP и 3 мкл TES в течение 30 мин. Смесь упаривают и полученный тиол очищают обращенно-фазовой ВЭЖХ. После лиофилизапии получают 2 мг (2,2 мкмоль, в виде двойной трифторацетатной соли) конъюгата амоксапин-линкер 5с.
МС: м/з 697,1=[М+НГ (=[М+Н:Г (Расчетная Mw=696,3 г/моль).
Синтез конъюгата жирная кислота-ПЭГ-амоксапин (5)

Конъюгат амоксапин-линкер 5с (2 мг, 2,2 мкмоль) и 1 (3,5 мг, 3,2 мкмоль) растворяют в 900 мкл смеси ацетонитрил/вода 7/3 (об./об.). Прибавляют 60 мкл 0,5 М фосфатного буфера (pH 7,4), и смесь инкубируют при комнатной температуре в течение 10 мин. 5 очищают методом обращенно-фазовой ВЭЖХ.
МС: м/з 1774,9=[М+Н]+(Расчетная Мм=1774,7 г/моль).
Пример 6
Синтез линкерного реагента (Соединение 6)

Fmoc-Asp (t-Bu)-OH (411 мг, 1 ммоль), HOBt (153 мг, 1 ммоль) и DIC (160 мкл, 1 ммоль) растворяют в 2 мл DMF и инкубируют в течение 10 мин при комнатной температуре. Прибавляют HN-диметилэтилендйамин (160 мкл, 1,5 ммоль) и перемешивают при комнатной температуре в течение 30 мин. Прибавляют уксусную кислоту (300 мкл) и Fmoc-Asp(t-Bu)-NH-(CH2)2-N(CH3)2 очищают методом обращенно-фазовой ВЭЖХ.
Выход: 220 мг (0,46 ммоль)
МС Fmoc-Asp(t-Bu)-NH-(CH2)2-N(CH3)2: м/з 504,6=[М+Na]+ (Расчетная Мм=481,6 г/моль).
Fmoc-Asp (t-Bu)-NH-(CH2)2-N(CH3)2 (220 мг, 0,46 ммоль) растворяют в 3 мл смеси TFA/TES 98/2 (об./об.). Через 30 мин растворитель удаляют в потоке азота и 6 очищают методом обращенно-фазовой ВЭЖХ с использованием 0,01% HCl в воде в качестве растворителя А и 0,01% HCl в ацетонитриле в качестве растворителя В.
Выход: 146 мг (0,32 ммоль, в виде HCl-соли).
МС: м/з 426,5=[М+Н]+ (Расчетная Мм=425,5 г/моль).
Пример 7
Синтез линкерных реагентов 7а и 7b

7a: R=Н
7b: СН3
Синтез 7а:
Fmoc-Asp (t-Bu)-OH (300 мг, 0,73 ммоль), HOBt (1112 мг, 0,73 ммоль) и DIC (117 мкл, 0,73 ммоль) растворяют в 2 мл DMF и инкубируют в течение 10 мин при комнатной температуре. Прибавляют Вос-этилендиамин (230 мг, 1,44 ммоль) и полученную смесь перемешивают при комнатной температуре в течение 30 мин. Прибавляют уксусную кислоту (300 мкл) и Fmoc-Asp(t-Bu)-NH-(CH2)2-NH-Boc очищают методом обращенно-фазовой ВЭЖХ.
Выход: 205 мг (0,37 ммоль)
МС промежуточного соединения: м/з 576,6=[М+Na]+ (Расчетная Мм=553,7 г/моль).
Fmoc-Asp (t-Bu)-NH-(CH2)2-NH-Boc (205 мг, 0,37 ммоль) растворяют в 3 мл 98/2 (об./об.) TFA/TES. Через 30 мин растворитель удаляют в потоке азота и полученный Fmoc-Asp(H)-NH-(CH2)2-NH2 очищают методом обращенно-фазовой ВЭЖХ.
Выход: 140 мг (0,27 ммоль, в виде трифторацетатной соли)
МС промежуточного соединения: м/з 398,8=[М+Н]+ (Расчетная Мм=397,4 г/моль).
Fmoc-Asp(H)-NH-(CH2)2-NH2 (140 мг, 0,27 ммоль, в виде трифторацетатной соли) растворяют в 1 мл DMF и DIEA (140 мкл, 0,81 ммоль) и прибавляют boc2O (100 мг, 0,46 ммоль). Раствор перемешивают при комнатной температуре в течение 15 минут, а затем подкисляют уксусной кислотой (300 мкл). 7а очищают методом обращенно-фазовой ВЭЖХ.
Выход 7а: 120 мг (0,24 ммоль)
МС 7а: м/з 520,5=[М+Na]+ (Расчетная Мм=497,6 г/моль).
7b получают по методике, описанной выше за исключением того, что на первой стадии синтеза используют H2N-(СН2)2-N(СН3)-boc вместо Вос-этилендиамина в качестве амина.
Выход 7b: 115 мг
МС 7b: м/з 534,5=[M+Na]+ (Расчетная Мм=511,6 г/моль).
Пример 8
Синтез конъюгатов эксендин-линкер (8а, 8b и 8с)

8a: Rl=H, R2=H
8b: Rl=H, R2=CH3
8с: R1=СН3, R2=СН3
Синтез соединения 8а:
7а (30 мг, 60 мкмоль), HOBt (9 мг, 60 мкмоль), DIEA (12 мкл, 70 мкмоль) и DIC (10 мкл, 63 мкмоль) растворяют в 200 мкл DMF, и сразу же вводят в смолу, несущую защищенный в боковой цепи эксендин-4, (40 мг, 4 мкмоль), и инкубируют в течение 1 часа при комнатной температуре. Смолу промывают десять раз DMF, а затем инкубируют в течение 5 мин в 500 мкл 1/1/2 уксусного ангидрида/пиридина/DMF. Смолу промывают 10 раз DMF и fmoc-группу удаляют. Присоединяют trt-меркаптопропионовую кислоту и 8а отщепляют от смолы и очищают методом обращенно-фазовой ВЭЖХ.
Выход: 3,6 мг
МС 8а: м/з 1108,5=[М+4Н]4+, 1477,8=[М+3Н]3+ (Расчетная Мм=4432 г/моль).
8b получают по методике синтеза 8а за исключением того, что вместо 7а используют 7b.
Выход: 3,5 мг
МС 8b: м/з 1112,5=[М+4H]4+, 1482,5=[М+3Н]3+; (Расчетная Мм=4446 г/моль).
8с получают по методике синтеза 8а за исключением того, что вместо 7а используют 6.
Выход: 3,2 мг
МС 8 с: м/з 1116,2=[М+4Н]4+, 1487,8=[М+3Н]3+ (Расчетная Мм=4460 г/моль).
Пример 9
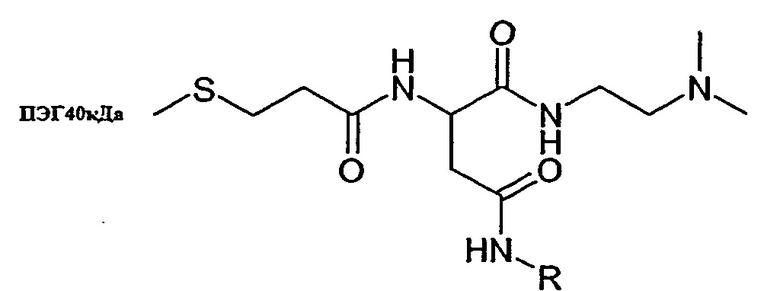
Синтез конъюгатов ПЭГ40 кДа-линкер-эксендин (9а, 9b и 9с)

9a: Rl=H, R2=H
9b: Rl=H, R2=CH3
9c: Rl=CH3, R2=CH3
Синтез 9а:
8а (3,6 мг) растворяют в 300 мкл смеси вода/ацетонитрил 2/1 и прибавляют 50 мг ПЭГ40кДа-малеимида. Прибавляют 100 мкл 0,25 М натрий-фосфатного буфера, pH 7 и через 5 минут раствор подкисляют 50 мкл уксусной кислоты.
9а очищают ионообменной хроматографией с использованием 10 мМ натрий-цитратного буфера pH3 в качестве растворителя А и 10 мМ натрий-нитратного буфера, pH 3 и 1М NaCl в качестве растворителя В и ступенчатого градиента (от 0 до 40% растворителя В). Фракции, содержащие 9а, высаливают и лиофилизируют.
Выход: 14 мг
9b синтезируют как описано выше за исключением того, что используют 8b.
Выход: 15 мг
9с синтезируют как описано выше за исключением того, что используют 8с.
Выход: 13 мг
Пример 10
Синтез конъюгата жирная кислота-линкер-эксендин (10)

8с (1 мг) растворяют в 100 мкл смеси ацетонитрил/вода, 1/1 и добавляют 1 (1 мг) в 100 мкл смеси 3/1 ацетонитрил/вода. Добавляют 100 мкл 0,25 М натрий-фосфатного буфера, реакционную смесь перемешивают в течение 5 мин, и полученное 10 очищают методом обращенно-фазовой ВЭЖХ.
Выход: 1,3 мг
МС 10: м/з 1385,9=[M+4H}4+, 1846,3=[М+3Н]3+ (Расчетная Мм=5528,3 г/моль).
Пример 11
Синтез NHS-активированного лннкерного реагента (11)

7b (20 мг, 40 мкмоль), N,N'-дициклогексилкарбодиимид (10 мг, 48 мкмоль) и N-гидроксисукцинимид (NHS) (8 мг, 70 мкмоль) растворяют в 300 мкл безводного DCM и перемешивают при комнатной температуре в течение 1 часа. Растворитель удаляют в потоке азота и 11 очищают методом обращенно-фазовой ВЭЖХ и лиофилизируют.
Выход: 22 мг (36 мкмоль)
МС: м/з 631,5=[M+Na]+ (Расчетная Мм=608,7 г/моль).
Пример 12
Синтез конъюгата линкер-эксендин (флуоресцеин) (12а) и конъюгата линкер-GRF (1-29) (флуоресцеин) (12b)

12а: R= эксендин (εК27-флуоресцеин)
12b: R=GRF (1-29) (εК21-флуоресцеин)
6 (60 мг, 130 мкмоль HCl-соль), HOBt (20 мг, 130 мкмоль), DIEA (40 мкл, 230 мкмоль) и DIC (20 мкл, 126 мкмоль) растворяют в 700 мкл DMF и сразу же вводят в защищенный в боковой цепи эксендин-4 на полимерной смоле (120 мг, 12 мкмоль), и полученную реакционную смесь инкубируют в течение 1 часа при комнатной температуре. Смолу промывают десять раз DMF, а затем инкубируют в течение 5 мин в 1 мл смеси уксусный ангидрид/пиридин/DMF (1/1/2, об./об./об.). Смолу промывают десять раз DMF и защитную группу Fmoc удаляют.Trt-меркаптопропионовую кислоту связывают по стандартной методике реакции сочетания и полученную смолу промывают пять раз DMF и затем еще десять раз DCM. Mmt-защитную группу лизина в положении 27 (Lys 27) удаляют при инкубации смолы пять раз в 2 мл смеси DCM/HFIP 9/1 (об./об.) в течение 5 мин. Смолу промывают пять раз DCM и затем еще десять раз DMF, после чего в смолу вводят 5,6-карбокси-флуоресцеин-МН8-сложный эфир (20 мг, 42 мкмоль) и DIEA (20 мкл, 115 мкл) в 300 мкл DMF и инкубируют в течение 30 мин. Полученное 12а отделяют от смолы и очищают методом обращенно-фазовой ВЭЖХ.
Выход: 12 мг
МС 12а: м/з 1205,9=[М+4Н]4+, 1607,0=[М+3Н]3+ (Расчетная Мм=4818,3 г/моль).
12b синтезируют по методике синтеза соединения 12а за исключением того, что используют GRF (1-29) на полимерной смоле (120 мг, 12 мкмоль).
Выход: 11 мг
МС 12b: м/з=998,6=[M+4H]4+, 1330,5=[М+3Н]3+ (Расчетная Мм=3989,6 г/моль).
Синтез меркаптопропионил-эксендина(флуоресцеин) (12с) и меркаптопропионил-GRF (1-29) (флуоресцеин) (12d)
12с: R= Эксендин (εК27-флуоресцеин)
12d: R=GRF (1-29) (εК27-флуоресцеин)
Trt-меркаптопропионовую кислоту связывают по стандартной методике реакции сочетания с защищенным в боковой цепи фрагментом эксендина-4 на полимерной смоле (120 мг, 12 мкмоль). Удаление Mmt-защитной группы Lys27 удаляют и реакцию сочетания 5,6-карбокси-флуоресцеин-МН8-сложного эфира проводят по методике синтеза 12а. 12 с отщепляют от смолы и очищают методом обращенно-фазовой ВЭЖХ.
Выход: 13 мг
МС 12с: м/з 1545,6=[М+3Н]3+ (Расчетная Мм=4633 г/моль).
12d получают по методике синтеза 12с, за исключением того, что используют GRF (1-29) на смоле (120 мг, 12 мкмоль).
Выход: 11 мг
МС 12d: м/з 1269,1=[М+3Н]3+ (Расчетная Мм=3804,3 г/моль).
Пример 13
Синтез обратимого ПЭГ40кДа-линкер-эксендин (флуоресцеин) конъюгата (13а) и обратимый PEG40kDa-линкер-GRF (1-29) (флуоресцеин) конъюгат (13b)
13a: R= эксендин (εК27-флуоресцеин)
13b: R=GRF(1-29) (εК21-флуоресцеин)
12а (12 мг) растворяют в 500 мкл смеси ацетонитрил/вода, 1/1 и добавляют 120 мг ПЭГ40кДа-малеимида в 1 мл смеси 1/1 ацетонитрил/вода. Затем прибавляют 300 мкл 0,25 М натрий-фосфатного буфера pH 7,0 и через 10 мин реакционный раствор подкисляют 300 мкл уксусной кислоты. 13a очищают методом катионообменной хрома-тографии, высаливают из раствора и лиофилизируют.
Выход: 51 мг
13b получают по методике синтеза 13a за исключением того, что вместо 12а используют 12b.
Выход: 46 мг
Синтез постоянного ПЭГ40кДа-эксендин (флуоресцеин) конъюгата (13с) и постоянного ПЭГ40кДа-GRF(1-29) (флуоресцеин) конъюгата (13d)

13с: R= эксендин (εК27-флуоресцеина)
13d: R=GRF (1-29) (εК21-флуоресцеина)
13с получают по методике синтеза 13a за исключением того, что вместо 12а используют 12с.
Выход: 55 мг
Соединение 13d получают по методике синтеза 13a за исключением того, что используют 12d вместо 12а.
Выход: 45 мг
Пример 14
Синтез конъюгата линкер-СКР-(1-29) (14)

14 получают по методике синтеза 8 с за исключением того, что используют защищенный в боковой GRF (1-29) на смоле.
Выход: 10 мг
МС 14: м/з 908,2=[М+4H]4+, 1211,2=[М+3Н]3+ (Расчетная Мм=3631,3 г/моль).
Пример 15
Синтез конъюгата ПЭГ40кДа-линкер-GRF (1-29) (15)

15 получают по методике синтеза 9с за исключением того, что используют 14, а для осуществления катионообменной хроматографии используют 10 мМ цитрат натрия, pH 4 в качестве растворителя А и 10 мМ цитрат натрия pH 4 и 1 М хлорид натрия в качестве растворителя В.
Выход: 11 мг
Пример 16 Синтез линкерного интермедиата (16)
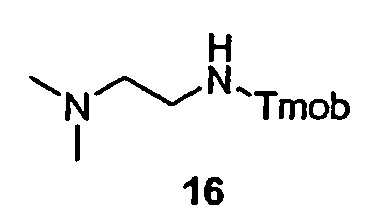
N,N-диметилэтилендиамин (198 мкл, 1,8 ммоль) и NaCNBH3 (58 мг, 0,9 ммоль) растворяют в метаноле (5 мл) и раствор доводят до pH 5,5 прибавлением уксусной кислоты (250 мкл). Прибавляют суспензию 2,4,6-триметоксибензальдегида (294 мг, 1,5 ммоль) в этаноле (5 мл) и реакционную смесь перемешивают при комнатной температуре в течение 1 часа. Добавляют 5 н HCl (0,5 мл) и смесь перемешивают в течение еще 12 часов. Растворитель упаривают при пониженном давлении, остаток растворяют в насыщенном растворе NaHCO3 и трижды экстрагируют в DCM. Объединенные органические фазы сушат над NaSO4 и растворитель упаривают при пониженном давлении.
Выход: 303 мг (1,13 ммоль)
МС: м/з 269,3=[M+H]+ (Расчетная Мм=268,4 г/моль)
Пример 17
Синтез линкера 17а и 17b

Синтез 17а:
Fmoc-Asp(OtBu)-OH (322 мг, 0,78 ммоль), Тmob-защищенный диамин (соединение 16) (150 мг, 0,56 ммоль), HATU (0-7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат) (255 мг, 0,67 ммоль) и DIEA (290 мкл, 1,68 ммоль) растворяют в DMF (1,5 мл). Смесь перемешивают в течение 30 мин, подкисляют уксусной кислотой и очищают методом обращенно-фазовой ВЭЖХ.
Выход: 463 мг (5,97 ммоль, в виде трифторацетатной соли, чистота около 90%.)
MC Fmoc-Asp(OtBu)-N(TMOB)CH2CH2N(CH3)2: м/з 662,5=[М+Н]+ (Расчетная Мм=661,8 г/моль)
Fmoc-Asp(OtBu)-N(TMOB)CH2CH2N(CH3)2 (225 мг, 0,29 ммоль) растворяют в растворе пиперидина (50 мкл) и DBU (15 шел) в DMF (1,5 мл). Реакционную смесь перемешивают при комнатной температуре в течение 1,5 часов. Прибавляют АсОН и полученное соединение H-Asp(OtBu)-N(TMOB)CH2CH2N(CH3)2 очищают методом обращенно-фазовой ВЭЖХ.
Выход: 114 мг (0,21 ммоль, в виде трифторацетатной соли)
MC H-Asp(OtBu)-N(TMOB)CH2CH2N(CH3)2: м/з 462,4=[М+Н]+ (Расчетная Mw=439,6 г/моль)
Трифторацетатную соль H-Asp(OtBu)-N(Tmob)CH2CH2N(CH3)2 (114 мг, 0,21 ммоль) растворяют в насыщенном растворе NaHCO3 (10 мл) и экстрагируют DCM (3×10 мл). Объединенные органические слои сушат над NaSO4 и растворитель упаривают при пониженном давлении. Остаток растворяют в DMF (1,0 мл), и к полученному раствору прибавляют 6-тритилметилмеркаптогексановую кислоту (121 мг, 0,31 ммоль), HATU (118 мг, 0,31 ммоль) и DIEA (108 мкл, 0,62 ммоль). Смесь перемешивают в течение 30 мин. Прибавляют АсОН (200 мкл) и TrtS(CH2)5CONH-Asp(OtBu)-N(TmOb)CH2CH2N(CH3)2 очищают методом обращенно-фазовой ВЭЖХ.
Выход: 95 мг (0,10 ммоль, в виде трифторацетатной соли)
MC TrtS(CH2)5CONH-Asp(OtBu)-N(Tmob)CH2CH2N(CH3)2: м/з 812,64=[М+Н]+ (Расчетная Мм=812,1 г/моль)
TrtS(CH2)5CONH-Asp(OtBu)-N(Tmob)CH2CH2N(CH3)2 (95 мг, 0,10 ммоль) растворяют в смеси МеОН/Н2О 3:1 (1,0 мл), прибавляют LiOH (7,4 мг, 0,31 ммоль) и реакционную смесь встряхивают в течение 5 часов при 60°C. Прибавляют АсОН (100 мкл) и 17а очищают методом обращенно-фазовой ВЭЖХ.
Выход: 64 мг (0,07 ммоль, в виде трифторацетатной соли)
MC 17а: м/з 756,5=[М+Н]+ (Расчетная Мм=756,0 г/моль)
17b синтезируют по методике, описанной выше за исключением того, что на первой стадии вместо Fmoc-Asp(OtBu)-OH используют Fmoc-NMe-Asp(OtBu)-OH.
Выход 17b: 16 мг (18 мкмоль, в виде трифторацетатной соли)
MC 17b: м/з 770,5=[М+Н]+ (Расчетная Мм=770,0 г/моль)
Пример 18
Синтез конъюгатов линкер-BNP (18а и 18b)

Синтез 18а:
17а (8,0 мг, 0,01 ммоль), РуВОР (5,2 мг, 10 мкмоль) и DIEA (7 мкл, 40 мкмоль) растворяют в DMF (400 мкл) и полученный раствор сразу же добавляют к смоле, связанной с защищенным в боковой цепи BNP-32a (50 мг, 5 мкмоль). После инкубации в течение 2 часов при комнатной температуре, смолу промывают десять раз DMF, затем еще 10 раз DCM и сушат под вакуумом. Полученный продукт отщепляют от смолы и очищают методом обращенно-фазовой ВЭЖХ.
Выход: 10,6 мг
MC 18а: м/з 930,4=[M+4H]4+, 1240,1=[М+3Н]3+ (Расчетная Мм=3717,2 г/моль)
18b синтезируют по методике, описанной выше за исключением того, что 17b используют вместо 17а.
Выход: 4,7 мг
MC 18b: м/з 933,9=[М+4Н]4+, 1244,7=[М+3Н]3+ (Расчетная Мм=3731,0 г/моль)
Пример 19
Синтез конъюгатов ПЭГ40кДа-линкер-BNP (19а и 19b)
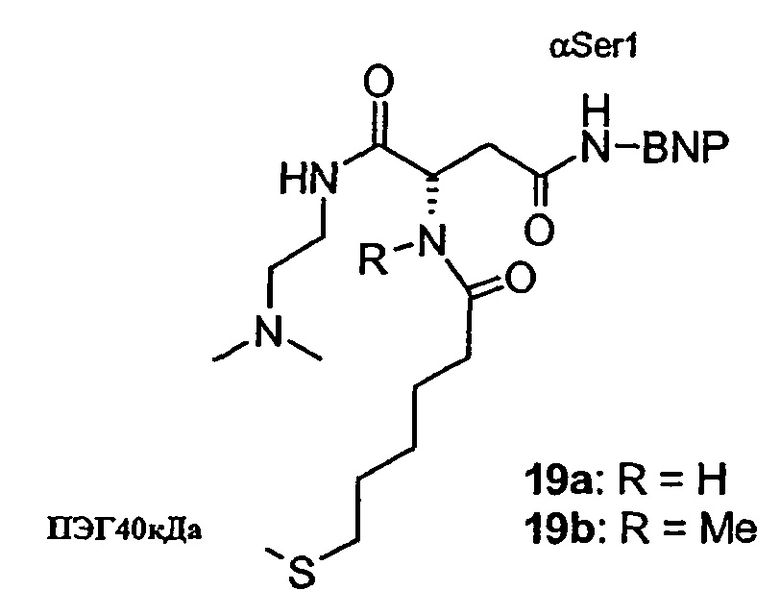
18а (5,2 мг) растворяют в смеси H2O/ацетонитрил 1:1, содержащей 0,1% TFA (200 мкл). Прибавляют раствор, содержащий ПЭГ40кДа-малеимид, (70 мг) в смеси 1:1 H2O/ацетонитрил (1,5 мл) и фосфатный буфер (30 мкл, pH 7,4, 0,5 М). Раствор инкубируют при комнатной температуре, и через 5 мин прибавляют АсОН (30 мкл). 19а очищают методом катионообменной хроматографии, высаливают из раствора и лио-филизируют.
Выход: 19,2 мг
19b получают по методике синтеза 19а за исключением того, что вместо 18а используют 18b.
Пример 20
Синтез линкера 20

Fmoc-Asp(OH)OtBu (100 мг, 0,24 ммоль), H2N-(CH2)2-N(CH3)-boc (36 мкл, 0,20 ммоль), HATU (92 мг, 0,24 ммоль) и DEEA (105 мкл, 0,60 ммоль) растворяют в 1 мл DMF. Смесь перемешивают в течение 1 часа при комнатной температуре, подкисляют уксусной кислотой (100 мкл) и очищают методом ВЭЖХ.
Выход: 91 мг (0,13 ммоль)
MC Fmoc-Asp(NH(CH2)2N(CH3)-boc)OtBu: 590,3=[M+Na]+ (Расчетная Мм=567,7 г/моль)
Fmoc-Asp(NH(CH2)2N(CH3)-boc)OtBu (91 мг, 0,13 ммоль) растворяют в DMF (1,0 мл), прибавляют пиперидин (50 мкл) и DBU (15 мкл) и полученную смесь перемешивают в течение 45 мин при комнатной температуре. Добавляют уксусную кислоту (100 мкл) и полученный NH2-Asp(NH(CH2)2N(CH3)-boc)OtBu очищают методом обращенно-фазовой ВЭЖХ.
Выход: 39 мг (0,09 ммоль, в виде трифторацетатной соли)
MC NH2-Asp(NH(CH2)2N(CH3)-boc)OtBu: м/з 368,1=[M+Na]+ (Расчетная Мм=345,4 г/моль)
NH2-Asp(NH(CH2)2N(CH3)-boc)OtBu (36 мг, 0,09 ммоль) растворяют в DMF (0,5 мл), к полученному раствору прибавляют 6-тритилметилмеркаптогексановую кислоту (55 мг, 0,14 ммоль), HATU (53 мг, 0,14 ммоль) и DIEA (49 мкл, 0,28 ммоль). Смесь перемешивают в течение 45 мин. Прибавляют АсОН (100 мкл) и TrtS(CH2)5CONH-Asp(NH(CH2)2N(CH3)-boc)OtBu очищают методом обращенно-фазовой ВЭЖХ.
Выход: 41 мг (0,06 ммоль)
MC TrtS(CH2)5CONH-Asp(NH(CH2)2N(CH3)-boc)OtBu: м/з 740,6=[M+Na]+ (Расчетная Мм=718,0 г/моль)
TrtS(CH2)5CONH-Asp(NH(CH2)2N(CH3)-boc)OtBu (41 мг, 0,06 ммоль) растворяют в смеси 1:1 диоксан/Н2О (1,0 мл), прибавляют LiOH (4,1 мг, 0,17 ммоль) и смесь перемешивают при 60°C в течение 1 часа. Прибавляют АсОН (50 мкл) и 20 очищают методом обращенно-фазовой ВЭЖХ.
Выход: 31 мг (0,05 ммоль)
MC 20: м/з 684,5=[M+Na]4+ (Расчетная Мм=661,9 г/моль)
Пример 21
Синтез конъюгата линкер-эксендин (21)

Связанный с полимерной смолой защищенный в боковой цепи эксендин (50 мг, 5 мкмоль) с Mmt-защитной группой по Lys27 сначала защищают на N-конце Вос-группой (см. Материалы и методы), а затем инкубируют пять раз (по 5 мин) 2 мл смеси DCM/HFIP 9/1 (об./об.) для снятия Mmt защитной группы с Lys27. Соединение 20 (6,6 мг, 10 мкмоль), РуВОР (5,2 мг, 10 мкмоль) и DIEA (7 мкл, 40 мкмоль) растворяют в DMF (400 мкл) и сразу же добавляют к смоле. После инкубации в течение 3 часов при комнатной температуре смолу промывают десять раз DMF и десять раз DCM и сушат под вакуумом. Полученный продукт отщепляют от смолы и очищают методом обращенно-фазовой ВЭЖХ.
Выход: 2,4 мг
МС 21: м/з 1497,2=[М+3Н]3+ (Расчетная Мм=4488,0 г/моль)
Пример 22
Синтез конъюгата жирная юнслота-линкер-эксендин (22)

21 (2,6 мг) растворяют в 200 мкл смеси ацетонитрил/вода, 1/1 и прибавляют соединение 1 (0,8 мг) в 400 мкл смеси ацетонитрил/вода, 7/3. Прибавляют 100 мкл 0,25 М натрий-фосфатного буфера, реакционную смесь перемешивают в течение 5 мин, после чего 22 очищают методом обращенно-фазовой ВЭЖХ.
Выход: 2,4 мг
МС 22: м/з 1388,3=[М+4Н]4+; 1857,1=[М+3Н]3+ (Расчетная Мм=5566,4 г/моль).
Пример 23
Синтез предшественника 23
6-Тритилмеркаптогексановую кислоту (200 мг, 0,51 ммоль), (PfpO)2CO (202 мг, 0,51 ммоль) и коллидин (340 мкл, 2,65 ммоль) растворяют в DMSO (1 мл) и перемешивают в течение 30 мин при комнатной температуре. Смесь прибавляют к раствору Fmoc-Lys-OH (170 мг, 0,46 ммоль) в смеси Н2О/пиридине/tBuOH (3:03:01, 6 мл). Реакционную смесь затем нагревают при 60°C в течение 2 часов, затем разбавляют EtOAc, экстрагируют дважды 0,1 М H2SO4, обрабатывают дважды солевым раствором и сушат на Na2SO4. Растворитель отгоняют при пониженном давлении и остаток очищают методом обращенно-фазовой ВЭЖХ.
Выход: 109 мг
МС 23: м/г 741,3=[М+Н]+ (Расчетная Мм=741,0 г/моль)
Пример 24
Синтез линкера (24а-24с)

24а: R1=Н, R2=boc
24b: R1=Me, R2=boc
24c: Rl=R2=Me
23 (186 мг, 0,25 ммоль) и DEEA (160 мкл, 0,92 ммоль) растворяют в DCM (2 мл), вводят в 2-хлортритилхлоридую смолу (312 мг, 1,3 ммоль/г) и перемешивают в течение 45 мин при комнатной температуре. Добавляют метанол (0,6 мл) и смолу инкубируют еще 15 мин. Смолу промывают DCM (10 раз) и ДМФ (10 раз). Снятие Fmoc-защиты и образование мочевины осуществляют в соответствии с общими методами синтеза (см. Материалы и методы) путем проведения взаимодействия с N-boc-этилендиамином (57 мкл, 0,34 ммоль). Полученный продукт отщепляют от смолы и очищают методом обращенно-фазовой ВЭЖХ.
Выход: 14 мг
МС 24а: м/з 705,4=[М+Н]+, 727,3=[M+Na]+ (Расчетная Мм=704,9 г/моль)
Соединение 24b получают по методике синтеза 24а за исключением того, что используют N-boc-N-метилэтилендиамин вместо N-boc-этилендиамина.
Выход: 21 мг
МС 24b: м/з 719,3=[М+Н]+, 741,4=[M+Na]+ (Расчетная Мм=719,0 г/моль)
24c получают по методике синтеза 24а за исключением того, что используют N,N-диметилэтилендиамин вместо N-boc-этилендиамина.
Выход: 10 мг
МС 24c: м/з 633,2=[М+Н]+ (Расчетная Мм=632,9 г/моль)
Пример 25
Синтез конъюгатов эксендин-линкер (25а-25с)

24а (7,0 мг, 0,01 ммоль), РуВОР (5,2 мг, 10 мкмоль) и DIEA (7 мкл, 40 мкмоль) растворяют в DMF (250 мкл), полученный раствор сразу же добавляют к смоле, связанной с защищенным в боковой цепи эксендином (50 мг, 5 мкмоль), и инкубируют в течение 2 часов при комнатной температуре. Смолу промывают DMF (10 раз), DCM (10 раз) и сушат под вакуумом. Полученный продукт отщепляют от смолы и очищают методом обращенно-фазовой ВЭЖХ.
Выход: 1,6 мг
МС 25а: м/з 1511,8=[М+3Н]3+ (Расчетная Мм=4530,8 г/моль)
25b получают по методике синтеза 25а за исключением того, что вместо 24а используют 24b.
Выход: 4,3 мг
МС 25b: м/з 1516,3=[М+3Н]3+ (Расчетная Мм=4544,8 г/моль)
25с получают по методике синтеза 25а за исключением того, что используют 24с вместо 24а.
Выход: 1,3 мг
МС 25с: м/з 1520,4=[М+3Н]3+ (Расчетная Мм=4558,8 г/моль)
Пример 26
Синтез конъюгатов жирная кислота-линкер (26а-26с)

25а (1,6 мг) растворяют в 200 мкл смеси ацетонитрил/вода, 1/1 и добавляют соединение 1 (0,11 мг) в 200 мкл ацетонитрил/вода, 7/3. В реакционную смесь добавляют 30 мкл 0,25 М натрий-фосфатного буфера, реакционную смесь перемешивают в течение 5 мин, после чего 26а очищают методом обращенно-фазовой ВЭЖХ.
МС соединения 26а: м/з 1870,0=[М+3Н]3+ (Расчетная Мм=5609,2 г/моль).
26b получают по методике синтеза 26а за исключением того, что используют 25b вместо 25а.
МС 26b: м/з 1875,9=[М+3Н]3+, 1406,7=[M+4H]4+, (Расчетная Мм=5623,2 г/моль)
26с получают по методике синтеза 26а за исключением того, что вместо 25а используют 25с.
МС 26с: м/з 1879,4=[М+3Н]3+, 1410,5=[М+4Н]4+, (Расчетная Мм=5637,2 г/моль)
Пример 27
Синтез конъюгатов ПЭГ20кДа-линкер-эксендин (Соединение 27а-27с)

25а (2,0 мг) растворяют в смеси H2O /MeCN, 1:1, содержащей 0,1% TFA (200 мкл). Добавляют раствор ПЭГ40кДа-малеимида (18 мг) в смеси 1:1 H2O/MeCN (1 мл) и фосфатный буфер (15 мкл, pH 7,4, 0,5 М). Раствор инкубируют при комнатной температуре, через 5 мин прибавляют АсОН (20 мкл) и 27а очищают методом катионо-обменной хроматографии, высаливают и лиофилизируют.
27b получают по методике синтеза 27а за исключением того, что используют 25b вместо 25а.
27с получают по методике синтеза 27а за исключением того, что используют 25с вместо 25а.
Пример 28
Синтез линкера 28
6-Бромгексаноилхлорид (46 мкл, 0,31 ммоль) растворяют в 0,2 мл CH2Cl2 и вводят в раствор, содержащий H2N-CH2-CH2-STrt (100 мг, 0,28 ммоль), DEEA (97 мкл, 0,56 ммоль) в CH2Cl2 (0,8 мл). Смесь перемешивают в течение 2 часа при комнатной температуре. Реакционную смесь подкисляют АсОН (50 мкл) и растворитель отгоняют под вакуумом. После очистки остатка на силикагеле (гептан/EtOAc=1:1) получают соединение Br-(CH2)5-CONH-(CH2)2-STrt.
Выход: 137 мг (0,276 ммоль, 98%)
МС Br-(CH2)5-CONH-(CH2)2-STrt: 518,9=[M+Na]+, (Расчетная Мм=496,5 г/моль)
N-boc-этилендиамин (81 мкл, 0,51 ммоль) добавляют к раствору, содержащему Br-(CH2)5-CONH-(CH2)2-STrt (230 мг, 0,46 ммоль) и Na2CO3 (196 мг, 1,85 ммоль) в ДМФ (0,8 мл). Реакционную смесь перемешивают в течение 10 часов при 70°C. После охлаждения до комнатной температуры смесь разбавляют 4 мл смеси MeCN/H2O, 25:75 в присутствии 1% TFA и после очистки методом обращенно-фазовой ВЭЖХ получают Boc-NH-(CH2)2-NH-(CH2)5-CONH-(CH2)2-STrt.
Выход: 189 мг (0,27 ммоль, 59%, в виде трифторацетатной соли)
МС соединения Boc-NH-(CH2)2-NH-(CH2)5-CONH-(CH2)2-STrt: 576,5=[M+H]+, (Расчетная Мм=575,5 г/моль)
Boc-NH-(CH2)2-NH-(CH2)5-CONH-(CH2)2-STrt (189 мг, 0,27 ммоль) и НСНО (35% водный раствор, 113 мкл) растворяют в MeCN (1,5 мл) и к полученной смеси добавляют СНВН3 (34 мг, 0,54 ммоль). Реакционную смесь перемешивают в течение 5 часов при комнатной температуре. После завершения реакции (при мониторинге масс-спектрометрией) раствор разбавляют H2O (5 мл) и экстрагируют CH2Cl2 (трижды по 5 мл). Объединенные органические слои сушат над MgSO4, фильтруют и растворитель отгоняют под вакуумом. После очистки остатка методом обращенно-фазовой ВЭЖХ получают Boc-NH-(CH2)2-N(CH3)-(CH2)5-CONH-(CH2)2-STrt.
Выход: 62,8 мг (0,11 ммоль, 39%)
МС Boc-NH-(CH2)2-N(CH3)-(CH2)5-CONH-(CH2)2-STrt: 590,6=[М+Н]+, (Расчетная Мм=5 89,0 г/моль)
Boc-NH-(CH2)2-N(CH3)-(CH2)5-CONH-(CH2)2-STrt (62,8 мг, 0,11 ммоль) растворяют в THF (6 мл) и прибавляют раствор HCl в диоксане (130 мкл, 4 М раствор). Реакционную смесь перемешивают в течение 12 часов при комнатной температуре. Прибавляют 200 мкл HCl в диоксане и растворитель отгоняют под вакуумом. После очистки остатка методом обращенно-фазовой ВЭЖХ получают смесь H2N-(CH2)2-N(CH3-(CH2)5-CONH-(CH2)2-STrt и не прореагировавшее исходное соединение Boc-NH-(CH2)2-N(CH3)-(CH2)5-CONH-(CH2)2-STrt.
Выход: 32,8 мг (0,062 ммоль, 44%, в виде гидрохлоридной соли) соединения 28 и 14,7 мг (0,025 ммоль, 23%, в виде трифторацетатной соли) исходного соединения МС 28: 490,5=[М+Н]+, (Расчетная Мм=489,0 г/моль)
Пример 29
Общая методика синтеза замещенных карбоновыми кислотами предшественников BNP (29а и 29b)

Цис-пиклогексан-1,2-дикарбоновой кислоты ангидрид (231 мг, 1,5 ммоль) и пиридин (271 мкл, 2 ммоль) растворяют в DCM (2 мл) и полученный раствор сразу же добавляют к смоле, связанной с защищенным в боковой цепи BNP-32a (300 мг). После инкубации в течение 1 часа при комнатной температуре полученный продукт промывают 10 раз DCM и высушивают под вакуумом.
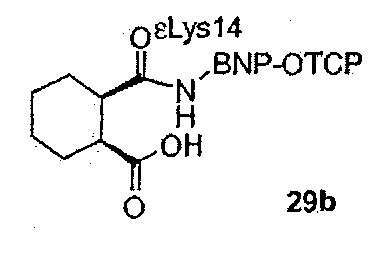
Защищенный в боковой цепи фрагмент BNP-32, связанный со смолой, несущий ivDde-защитную группу у Lys 14, вначале защищают Вос-группой на N-конце, снимают защиту в положении Lys14 (см. Материалы и методы), а затем подвергают взаимодействию с пис-пиклогексан-1,2-дикарбоновой кислоты ангидридом, как описано выше для 29а.
Пример 30
Синтез BNP-лннкер-тиолов (30а и 30b).

Соединение 28 H2N-(CH2)2-N(CH3)-(CH2)5-CONH-(CH2)2-STrt (5,2 мг, 0,01 ммоль), РуВОР (5,2 мг, 0,01 ммоль) и DIEA (7,0 мкл, 0,04 ммоль) растворяют в DMF (300 мкл) и полученный раствор добавляют к защищенному в боковой цепи BNP, связанному со смолой, 29а (50 мг, 0,005 ммоль). После инкубации в течение 2 часов при комнатной температуре смолу промывают DMF (10 раз), DCM (10 раз) и высушивают под вакуумом. Полученный продукт отщепляют от смолы и очищают методом обращенно-фазовой ВЭЖХ.
Выход: 9,8 мг
МС 30а: м/з 947,6=[M+4H]4+, 1263,1=[М+3Н]3+ (Расчетная Мм=3786,3 г/моль)

30Ь получают по методике описанной выше за исключением того, что вместо 29а используют связанное со смолой производное BNP 29b.
Выход: 7,4 мг
МС соединения 30b: м/з 947,5=[M+4H]4+, 1263,0=[М+3Н]3+ (Расчетная Мм=3786,3 г/моль)
Пример 31
Синтез конъюгатов ПЭГ40кДа-линкер-BNR (31а и 31b)

30а (4 мг) растворяют в смеси H2O/MeCN, 1:1, содержащей 0,1% TFA (200 мкл). Добавляют раствор ПЭГ40кДа-малеимида (42,2 мг) в смеси H2O/MeCN, 1:1 (1 мл) и фосфатный буфер (15 мкл, pH 7,4, 0,5 М). Полученный раствор инкубируют при комнатной температуре, через 5 мин прибавляют АсОН (20 мкл) и 31а очищают методом катионообменной хроматографии, высаливают и лиофилизируют.
Выход: 2,0 мг
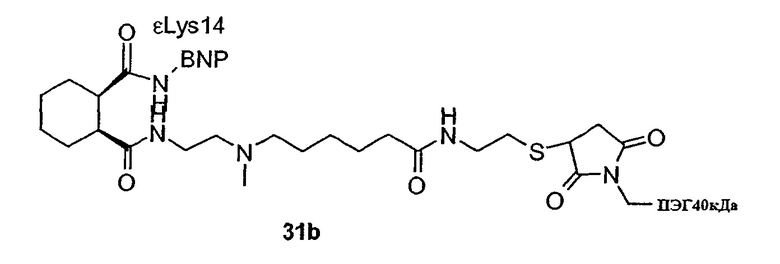
31b получают по методике синтеза 31а за исключением того, что вместо 30а используют 30b.
Выход: 16,8 мг
Пример 32
Синтез линкера 32
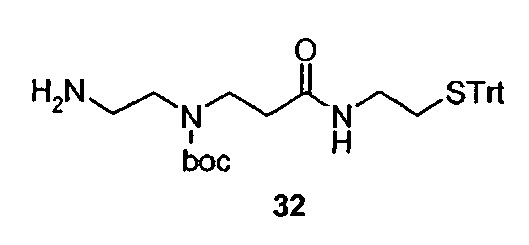
3-Бромпропионилхлорид (62,5 мкл, 0,62 ммоль) растворяют в 0,5 мл CH2Cl2 и добавляют в раствор H2N-CH2-CH2-STrt (200 мг, 0,56 ммоль), DDBA (196 мкл, 1,1 ммоль) в CH2Cl2 (1 мл). Смесь перемешивают в течение 1 часа при комнатной температуре. Реакционную смесь подкисляют АсОН (100 мкл) и растворитель удаляют под вакуумом. После очистки остатка на силикагеле (гептан/EtOAc=1:1) получают Br-(CH2)2CONH-(CH2)2-STrt.
Выход: 223 мг (0,49 ммоль, 87%)
МС Br-(CH2)2CONH-(CH2)2-STrt: 478,7=[M+Na]+, (Расчетная Мм=454,7 г/моль)
N-Alloc-этилендиамина гидрохлоридную соль (43,5 мг, 0,24 ммоль) и DIEA (38 мкл, 0,22 ммоль) прибавляют к раствору, содержащему Br-(CH2)2CONH-(CH2)2-STrt (100 мг, 0,22 ммоль) и Na2CO3 (93 мг, 0,87 ммоль) в DMF (1 мл). Реакционную смесь перемешивают в течение 10 часов при температуре 70°C. После охлаждения до комнатной температуры реакционную смесь разбавляют 4 мл (MeCNZHhO=25:75 в присутствии 0,1% TFA) и очищают ВЭЖХ с получением Alloc-NH-(CH2)2-NH-(CH2)2-CONH-(CH2)2-STrt.
Выход: 61 мг (0,096 ммоль, 44%, в виде трифторацетатной соли)
МС Alloc-NH-(CH2)2-NH-(CH2)2-CONH-(CH2)2-STrt: 540,8=[M+Na]+, (Расчетная Мм=517,8 г/моль)
Alloc-NH-(CH2)2-NH-(CH2)2-CONH-(CH2)2-STrt (60,9 мг, 0,096 ммоль) растворяют в CH2Cl2 и Boc2O (42 мг, 0,19 ммоль). Раствор перемешивают в течение 20 часов при комнатной температуре. По завершения взаимодействия реакционную смесь гасят добавлением 70 мкл АСОН и растворитель упаривают под вакуумом. Остаток разбавляют 4 мл MeCNZH2O (25:75, в присутствии 0,1% TFA) и после очистки методом обращенно-фазовой ВЭЖХ получают Alloc-NH-(CH2)2-N(Boc)-(CH2)2-CONH-(CH2)2-STrt.
Выход: 53,3 мг (0,086 ммоль, 89%)
MC Alloc-NH-(CH2)2-N(Boc)-(CH2)2-CONH-(CH2)2-STrt: 640,6=[M+Na]+, (Расчетная Мм=617,9, г/моль)
Alloc-NH-(CH2)2-N(Boc)-(CH2)2-CONH-(CH2)2-STrt (48,3 мг, 0,078 ммоль) растворяют в THF, прибавляют формиат триэтиламмония (62 мкл) и Pd (PPh3)4 (16 мг). Раствор перемешивают в течение 12 часов при комнатной температуре с осуществлением масс-спектрометрического мониторинга протекания реакции. По завершении реакции растворитель упаривают под вакуумом. Остаток растворяют в MeCN/Н2О (50:50, с 0,1% TFA) и после очистки методом обращенно-фазовой ВЭЖХ получают H2N-(CH2)2-N(Boc)-(CH2)2-CONH-(CH2)2-STrt (31).
Выход: 20,1 мг (0,031 ммоль, 40%, в виде трифторацетатной соли)
MC 31: 534,6=[М+Н]4+, 556,6=[M+Na]+, (Расчетная Мм=533,5 г/моль)
Пример 33
Синтез BNP-линкер-тиолов (Соединение 33а и 33b)
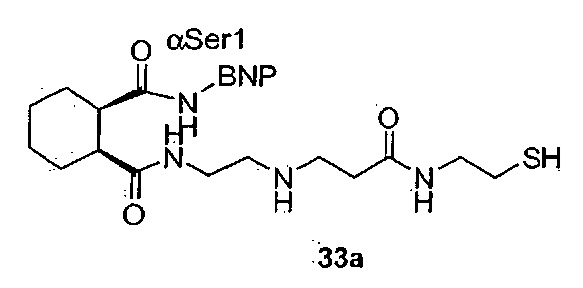
Соединение 33а получают по методике синтеза соединения 30а за исключением того, что вместо соединения 28 используют 32.
Выход: 8,0 мг
MC 33а: м/з 933,5=[М+4Н]4+, 1244,3=[М+3Н]3+ (Расчетная Мм=3729,9 г/моль)

33b получают по методике синтеза 30Ь за исключением того, что вместо 28 используют 32.
Выход: 5,0 мг
MC 33b: м/з 933,5=[М+4Н]4+, 1244,3=[М+3Н]3+ (Расчетная Мм=3715,9 г/моль)
Пример 34
Синтез конъюгатов ПЭГ40кДа-линкер-BNP (34а и 34b)

33а (4,3 мг) растворяют в смеси H2O/MeCN, 1:1, содержащей 0,1% TFA (200 мкл). Добавляют раствор ПЭГ40кДа-малеимида (42,2 мг) в смеси Н2О/МеСМ, 1:1 (1 мл) и фосфатный буфер (20 мкл, pH 7,4, 0,5 М). Полученный раствор инкубируют при комнатной температуре, через 5 мин прибавляют АсОН (20 мкл) и 34а очищают методом катионообменной хроматографии, высаливают и лиофилизируют.
Выход: 9,7 мг

34b получают по методике синтеза 34а за исключением того, что вместо 33а используют 33b.
Выход: 11,5 мг
Пример 35
Синтез линкера 35
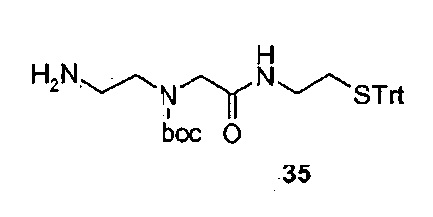
Бромацетилбромид (54 мкл, 0,62 ммоль) растворяют в 0,5 мл CH2Cl2 и добавляют к раствору, содержащему H2N-CH2-CH2-STrt (200 мг, 0,56 ммоль) и DEEA (196 мкл, 1,1 ммоль) в CH2Cl2 (1 мл). Смесь перемешивают в течение 1 часа при комнатной температуре. Реакционную смесь подкисляют уксусной кислотой (100 мкл) и растворитель упаривают под вакуумом. После очистки остатка очищают на силикагеле (гептан/EtOAc=1:), получая Br-CH2-CONH-(CH2)2-STrt.
Выход: 245 мг (0,55 ммоль, 99%)
МС Br-CH2-CONH-(CH2)2-STrt: 462,4=[M+Na]+, (Расчетная Мм=440,4 г/моль)
N-Alloc-этилендиамина гидрохлоридную соль (45 мг, 0,25 ммоль) и DIEA (79 мкл, 0,45 ммоль) прибавляют к раствору, содержащему Br-CH2-CONH-(CH2)2-STrt (100 мг, 0,23 ммоль) в DMF (1 мл). Реакционную смесь перемешивают в течение 10 часов при температуре 70°C. После охлаждения до комнатной температуры реакционную смесь разбавляют H2O/Et2O (1:1, 40 мл) и образованные слои разделяют. Водный слой экстрагируют несколько раз Et2O. Объединенные органические слои сушат над MgSO4, фильтруют и растворитель упаривают под вакуумом. Остаток очищают на силикагеле (DCM/MeOH=95:5), давая Alloc-NH-(CH2)2-NH-CH2-CONH-(CH2)2-STrt. Выход: 94 мг (0,186 ммоль, 82%, содержит остаточный DMF)
МС Alloc-NH-(CH2)2-NH-CH2-CONH-(CH2)2-STrt: 526,8=[M+Na:T, (Расчетная Мм=503,8 г/моль)
Alloc-NH-(CH2)2-NH-CH2-CONH-(CH2)2-STrt (94 мг, 0,186 ммоль, с остатком DMF) растворяют в CH2Cl2 и к полученному раствору прибавляют Boc2O (81 мг, 0,37 ммоль). Раствор перемешивают в течение 20 часов при комнатной температуре. По завершения взаимодействия реакционную смесь гасят добавлением 100 мкл АсОН и растворитель упаривают под вакуумом. Остаток разбавляют 4 мл MeCN/H2O (25:75, в присутствии 0,1% TFA) и после очистки методом обращенно-фазовой ВЭЖХ получают Alloc-NH-(CH2)2-N(Boc)-CH2-CONH-(CH2)2-STrt.
Выход: 34,7 мг (0,057 ммоль, 26%)
МС Alloc-NH-(CH2)2-N(Boc)-CH2-CONH-(CH2)2-STrt: 603,9=[M+Na:T (Расчетная Мм=603,9 г/моль)
Alloc-NH-(CH2)2-N(Boc)-CH2-CONH-(CH2)2-STrt (34,7 мг, 0,048 ммоль) растворяют в THF, прибавляют формиат триэтиламмония (38 мкл) и Pd (PPh3)4 (5 мг). Раствор перемешивают в течение 12 часов при комнатной температуре с осуществлением масс-спектрометрического мониторинга протекания реакции. По завершении реакции растворитель упаривают под вакуумом. Остаток растворяют в MeCN/H2O (50:50, с 0,1% TFA) и после очистки методом обращенно-фазовой ВЭЖХ получают H2N-(CH2)2-N(Boc)-CH2-CONH-(CH2)2-STrt соединения 35.
Выход: 12,6 мг (0,019 ммоль, 42%, в виде трифторацетатной соли)
МС 35: 520,1=[М+Н]+, 542,2=[М+Na]+, (Расчетная Мм=519,2 г/моль)
Пример 36
Синтез BNP-лннкер-тиолов (36а и 36b)

36а получают по методике синтеза 30а за исключением того, что вместо 28 используют 35.
Выход: 9,1 мг
МС 36а: м/з 930,0=[M+4H]4+, 1239,6=[М+3Н]3+ (Расчетная Мм=3715,9 г/моль)

36b получают по методике синтеза 30b за исключением того, что вместо соединения 28 используют соединение 35.
Выход: 8,0 мг
МС 36b: м/з 929,5=[М+4Н]4+, 1239,5=[М+3Н]3+ (Расчетная Мм=3715,9 г/моль)
Пример 37
Синтез конъюгатов ПЭГ40кДа-линкер-BNP (31а и 31b)

Соединение 36а (4,2 мг) растворяют в смеси H2O/MeCN, 1:1, содержащей 0,1% TFA (200 мкл). Добавляют раствор ПЭГ40кДа-малеимида (68 мг) в смеси H2O/MeCN, 1:1 (1 мл) и фосфатный буфер (20 мкл, pH 7,4, 0,5 М). Полученный раствор инкубируют при комнатной температуре, через 5 мин прибавляют АсОН (20 мкл) и 37а очищают методом ионнообменной хроматографии, высаливают и лиофилизируют.
Выход: 16 мг

37b синтезируют по методике получения 37а, за исключением того, что вместо 36а используют 36b.
Выход: 18,5 мг
Пример 38
Синтез конъюгатов линкер-эксендин
Конъюгаты линкер-эксендин получают в соответствии с общим методами синтеза С, D, Е или F.

Метод А
Синтез: Ангидрид дикислоты (0,2 ммоль) и пиридин (0,2 ммоль) растворяют в 0,3 мл сухого DMF. Полученную смесь добавляют к защищенному в боковой цепи эксен-дину-4 на смоле (2 мкмоль) и перемешивают в течение 30 мин при комнатной температуре. Смолу промывают DMF (10 раз). РуВОР (0,1 ммоль) и диамин (0,1 ммоль) растворяют в 0,3 мл сухого DMF. Полученную смесь вводят в смолу и перемешивают в течение 30 мин при комнатной температуре. Смолу промывают DMF (10 раз). Полученные конъюгаты эксендина с линкером отщепляют от смолы и очищают методом обращенно-фазовой хроматографии по методике, описанной в разделе "Материалы и методы".
Метод В
Синтез проводят по методике Метода А исключением того, что вместо ангидрида дикислоты и пиридина используют дикислоту (0,2 ммоль), HOBt (0,2 ммоль), DIC (0,2 ммоль), и коллидин (0,4 ммоль).
Метод С
Синтез: диамин (0,6 ммоль) растворяют в 1 мл сухого DCM и к полученному раствору прибавляют ангидрид дикислоты (0,4 ммоль). Смесь перемешивают в течение 60 мин при комнатной температуре. DCM упаривают, остаток растворяют в смеси ACN/вода/АсОН, полученную аминокислоту очищают методом обращенно-фазовой хроматографии и лиофилизируют.
Полученную аминокислоту (0,1 ммоль), HOBt (0,1 ммоль), DIC (0,1 ммоль) и коллидин (0,2 ммоль) растворяют в 0,3 мл сухого DMF. Полученную смесь прибавляют к эксендину-4 на смоле (2 мкмоль) и перемешивают в течение 30 мин при комнатной температуре. Смолу промывают DMF (10 раз). Полученные конъюгаты эксендина с линкером отщепляют от смолы и очищают методом обращенно-фазовой хроматографии по методике, описанной в разделе "Материалы и методы".
Метод D
Синтез: Ангидрид дикислоты (0,2 ммоль) и пиридин (0,2 ммоль) растворяют в 0,3 мл сухого DMF. Полученную смесь добавляют к эксендину-4 на смоле (2 мкмоль) и перемешивают в течение 30 мин при комнатной температуре. Смолу промывают DMF (10 раз). РуВОР (0,1 ммоль), HOBt (0,1 ммоль) и коллидин (0,4 ммоль) растворяют в 0,3 мл сухого DMF. Полученную смесь вводят в смолу и интенсивно перемешивают в течение 30 мин при комнатной температуре. Смолу промывают DMF (10 раз). Диамин (0,1 ммоль) и DIEA (0,3 ммоль) растворяют в смеси 0,4 мл DMF и 0,4 мл этанола. Полученную смесь добавляют к смоле и перемешивают в течение 30 мин при комнатной температуре. Смолу промывают DMF (10 раз). Полученные конъюгаты эксендина с линкером отщепляют от смолы и очищают методом обращенно-фазовой хроматографии по методике, описанной в разделе "Материалы и методы".
Метод Е
Синтез: Fmoc-аминокислоту (0,1 ммоль), РуВОР (0,1 ммоль) и DIEA (0,2 ммоль) растворяют в 0,3 мл сухого DMF. Полученную смесь добавляют к эксендину-4 на смоле (2 мкмоль) и перемешивают в течение 30 мин при комнатной температуре. Fmoc защитную группу удаляют путем инкубации смолы в смеси DMF/пиперидин, 4/1 (об./об.) 2 раза по 10 мин. Смолу промывают DMF (10 раз) и DCM (10 раз). Пара-нитрофенилхлорформиат (0,1 ммоль) растворяют в 0,3 мл сухого THF и DIEA (0,2 ммоль). Полученную смесь добавляют к смоле и перемешивают в течение 30 мин при комнатной температуре. Смолу промывают DCM (10 раз). Диамин (0,1 ммоль) растворяют в 0,3 мл DMF. Смесь добавляют к смоле и перемешивают в течение 30 мин при комнатной температуре. Смолу промывают DMF (10 раз). Полученные конъюгаты эксендина с линкером отщепляют от смолы и очищают методом обра-щенно-фазовой хроматографии по методике, описанной в разделе "Материалы и методы".
Метод F
Синтез проводят по методике Метода А с последующим снятием Fmoc-защиты и ацетилированием: Fmoc защитную группу удаляют посредством инкубации смолы в смеси DMF/пиперидин, 4/1 (об./об.) 2 раза по 10 мин. Смолу промывают DMF (10 раз). Ацетилирование осуществляют инкубацией смолы в смеси ангидрид уксусной кислоты/пиридин/DMF (1/1/2, (об./об./об.) в течение 30 мин. Полученную смолу промывают DMF (10 раз).
Полученные конъюгаты эксендина с линкером отщепляют от смолы и очищают методом обращенно-фазовой хроматографии по методике, описанной в разделе "Материалы и методы".
Более подробная информация, касающаяся синтезированных соединений, исходных материалов, методе синтеза, молекулярной массе (Мм) и данные масс-спектрометрии приведены на Фиг. 2.
Пример 39
Синтез конъюгатов гидрогель-линкер-эксендин (39)

Функционализированные малеиимидом гидрогелевые микрочастицы синтезируют методом, описанным в ЕР 1625856 А1.
30 мг микрочастиц малеимид-модифипированного гидрогеля (загрузка 40 мкмоль/г, 1,2 мкмоль) подвергают взаимодействию с 6 мг соединения 25а (1,32 мкмоль, 1,1 экв) в 600 мкл смеси ацетонитрил/50 Мм фосфатный буфер (pH 7,4), 20/80 (об./об.) в течение 10 минут с образованием микрочастиц гидрогеля 39, нагруженного конъю-гатом эксендина с линкером. Нагруженный гидрогель 39 промывают 5 раз смесью ацетонитрил/вода 50/50 (об./об.) и три раза водой.
Пример 40
Синтез линкера 40

Fmoc-Ala-OH (250 мг, 0,8 ммоль) и DEEA (170 мкл, 1,0 ммоль) растворяют в DCM (2 мл), полученную смесь добавляют 2-хлортритилхлоридной смоле (312 мг, 1,3 ммоль/г) и перемешивают в течение 45 мин при комнатной температуре. Прибавляют метанол (0,6 мл) и смолу инкубируют еще 15 мин. Смолу промывают DCM (10 раз) и DMF (10 раз). Снятие Fmoc-защиты и образование мочевины обеспечивают в соответствии с общей методикой проведения синтеза (см. раздел "Материалы и методы") путем проведения взаимодействия с линкерным промежуточным соединением 5а, полученный продукт отщепляют от смолы и очищают методом обращенно-фазовой ВЭЖХ.
Выход: 53 мг
МС соединения 40: м/з 604,4 [М+Н]+ (Расчетная Мм=603,8 г/моль)
Пример 41
Синтез конъюгата эксендин-линкер (41)
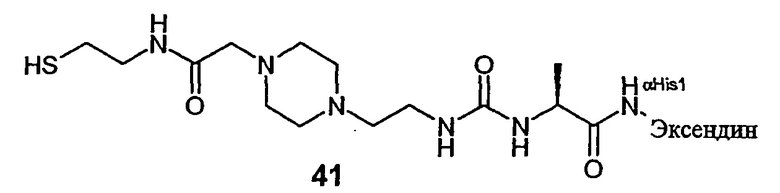
Соединение 40 (в виде гидрохлоридной соли, 14,0 мг, 0,02 ммоль), РуВОР (10,2 мг, 0,02 ммоль) и DIEA (17 мкл, 0,1 ммоль) растворяют в DMF (300 мкл), полученный раствор сразу же добавляют к защищенному в боковой цепи эксендину-4, связанному со смолой, (100 мг, 10 мкмоль) и инкубируют в течение 4 часов при комнатной температуре. Смолу промывают DMF (10x), DCM (10х) и высушивают под вакуумом. Полученный продукт отщепляют от смолы и очищают методом обращенно-фазовой ВЭЖХ. Выход: 5,4 мг МС 40: м/з 1510,9=[М+3Н]3+ (Расчетная Мм=4530,1 г/моль)
Пример 42
Синтез конъюгата жирная кислота-линкер (42)

41 (1,6 мг) растворяют в 200 мкл смеси ацетонитрил/вода, 3/1 и к полученному раствору прибавляют соединение 1 (0,11 мг) в 200 мкл смеси ацетонитрил/вода, 3/1. Затем прибавляют 30 мкл 0,25 М натрий-фосфатного буфера, реакционную смесь перемешивают в течение 5 минут, после чего 42 очищают методом обращенно-фазовой ВЭЖХ.
МС 42: м/з 1870,2=[М+3Н]3+ (Расчетная Мм=5608,4 г/моль).
Пример 43
Синтез конъюгата гадрогель-линкер-эксендин (43)

43 получают по методике синтеза 39 за исключением того, вместо 25 используют 41.
Пример 44
Синтез линкера (44)
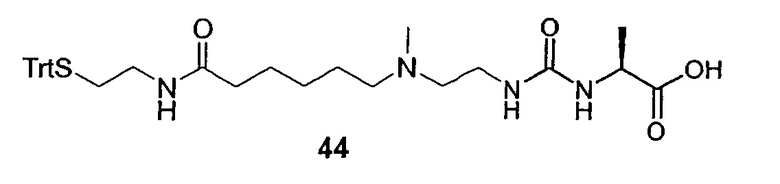
44 получают по методике синтеза 40 за исключением того, вместо 5а используют 28.
Выход: 74 мг
МС 44: м/з 605,4=[М+3Н]3+ (Расчетная Мм=604,8 г/моль)
Пример 45
Синтез конъюгата эксендин-линкер (45)
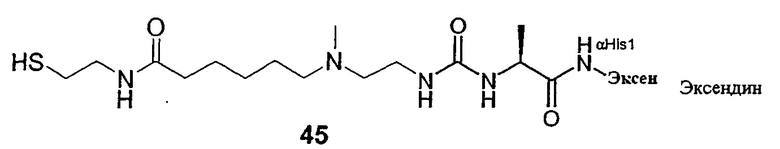
45 получают по методике синтеза 41 за исключением того, вместо 40 используют 44. Выход: 6,0 мг
МС 45: м/з 1511,3=[М+3Н]3+ (Расчетная Мм=4531,1 г/моль)
Пример 46
Синтез конъюгата жирная кислота-линкер (46)

46 получают по методике синтеза 42 за исключением того, вместо 41 используют 45. МС соединения 46: м/з 1870,5 3=(Расчетная Мм=5609,5 г/моль).
Пример 47
Синтез промежуточного линкерного соединения 47
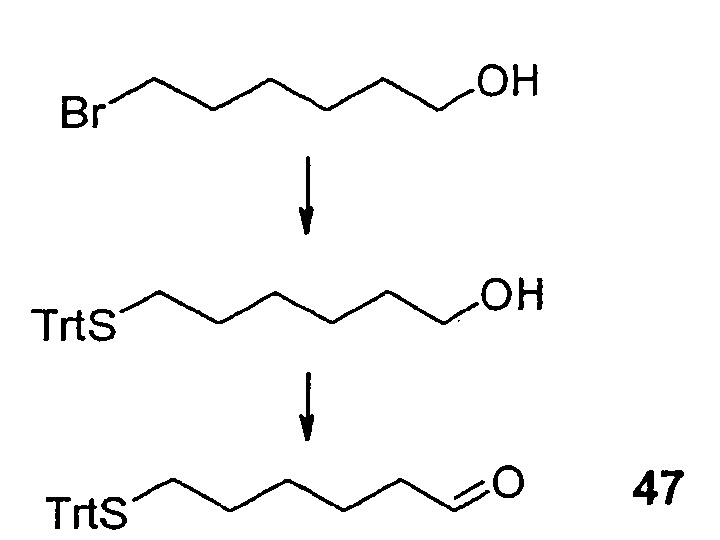
Тритилсульфид (247 мг, 0,89 ммоль) суспендируют в 1 мл DMSO. Добавляют DBU (152 мкл, 1,02 ммоль) и 6-бромегексан-1-ол (173 мг, 0,96) и полученную смесь перемешивают в течение 5 мин при комнатной температуре. Реакционную смесь растворяют в 20 мл этилацетата и промывают 1 н H2SO4 (2х) и солевым раствором (3х). Органический слой сушат (Na2SO4) и летучие компоненты упаривают в вакууме. Полученный продукт очищают флэш-хроматографией на силикагеле (гептан/AcOEt, 1/1). Выход: 283 мг (8-тритил)-6-меркаптогексан-1-ола.
(S-тритил)-6-меркаптогексан-1-ол (466 мг, 1,24 ммоль) растворяют в смеси растворителей, содержащей 3,5 мл DCM, 0,5 мл ДМСО и 0,6 мл NEt3, и затем реакционную смесь охлаждают на ледяной бане. SO3-пиридин (408 мг, 2,57 ммоль) суспендируют в 0,5 мл DMSO и добавляют к реакционной смеси. Убирают ледяную баню и реакционную смесь перемешивают в течение 60 мин при комнатной температуре. Затем реакционную смесь растворяют в 20 мл Et2O и экстрагируют с использованием 1 н H2SO4 (2х) и солевого раствора (3х). Органический слой сушат (Na2SO4) и летучие компоненты упаривают в вакууме. Полученный продукт очищают флэш-хроматографией на силикагеле (гептан /AcOEt 1/1).
Выход: 390 мг соединения 47 в форме (8-тритил)-6-меркаптогексан-1-аля.
МС 47: м/з 243,1=[Trt]+ 413,1=[М+К]+ (Расчетная Мм=374,4 г/моль)
Пример 48
Синтез линкера (48)

Fmoc-Ala-OH (250 мг, 0,8 ммоль) и DIEA (170 мкл, 1,0 ммоль) растворяют в DCM (2 мл), полученную смесь добавляют к 2-хлортритилхлоридной смоле (312 мг, 1,3 ммоль/г) и перемешивают в течение 45 мин при комнатной температуре. Прибавляют метанол (0,6 мл) и смолу инкубируют еще 15 мин. Смолу промывают DCM (10х) и DMF (10х). Снятие Fmoc-защиты и образование мочевины обеспечивают в соответствии с общей методикой проведения синтеза (см. раздел "Материалы и методы") посредством проведения взаимодействия смолы с этилендиамином. Для восстановительного алкилирования соединение 47 (299 мг, 0,8 ммоль) и Na(OAC)3BH (340 мг, 1,6 ммоль) растворяют в 0,5 мл DMF, 0,5 мл метанола и 10 мкл АсОН, добавляют к смоле и перемешивают в течение 2 часов при комнатной температуре. Смолу промывают DMF (10х) и DCM (10х). Защиту Вос-ангидридом осуществляют интенсивным перемешиванием смолы в растворе, содержащем Вос-ангидрид (218 мг, 1,0 ммоль) и DIEA (170 мкл, 1,0 ммоль) в DCM. Смолу промывают DCM (10х) и образованный продукт отщепляют от смолы и очищают методом обращенно-фазовой ВЭЖХ.
Выход: 34 мг
МС 48: м/з 634,2 [М+Н]+ (Расчетная Мм=633,9 г/моль)
Пример 49
Синтез конъюгата эксендин-лиякер (49)
49 получают по методике синтеза 41 за исключением того, вместо 40 используют 48. Выход: 4,8 мг
МС 49: м/з 1487,3=[М+3Н]3+ (Расчетная Мм=4460,0 г/моль)
Пример 50
Синтез конъюгата гидрогель-линкер-эксендин (50)

50 получают по методике синтеза 39 за исключением того, вместо 25а используют 49.
Пример 51
Кинетика высвобождения in vitro
Высвобождение молекулы лекарственного средства из 38a-38z, 38aa-38ab, 4, 5, 9а, 9b, 9с, 10, 13а, 15, 19а, 19b, 22, 26а-26с, 31а, 31b, 34а, 34b, 37а, 37b, 42, 43, 46 и 50 осуществляется в результате гидролиза в буферном растворе при pH 7,4 и температуре 37°C или pH 4 и температуре 37°C, как описано в разделе "Материалы и методы".
Пример 52
Корреляционный анализ кинетики высвобождения in vivo - in vitro/in vivo
Кинетику высвобождения in vivo определяли путем сравнения фармакокинетических параметров соединения 13а с фармакокинетическими параметрами соединения 13с и соединения 13b с соединением 13d, соответственно, после внутривенного их введения в крысу. Исследования на животных проводили в Heidelberg Pharma AG, Heidelberg, Germany.
Соединение 13а (27 мг) растворяли в 3,5 мл PBS и 500 мкл полученного раствора вводили внутривенно каждой из шести испытуемых крыс. Использовали мужские особи крыс линии SD с массой примерно 270 г. Забор образцов крови производили при Т=0,2 ч, 24 ч, 32 ч, 48 ч, 72 ч, 96 ч, 120 ч и 168 ч, после чего получали образцы плазмы крови, которые анализировали по флуоресценции флуоресцеина на спектрометре Perkin - Elmer LS 30В.
Фармакокинетику соединения 13 с определяли по методике, описанной для соединения 13а. Фармакокинегику соединений 13b и 13d определяли, как описано для соединения 13а, за исключением того, что использовали 20 мг соединения 13b и 13d, каждое в 2,5 мл PBS, и четыре крысы.
Период полужизни линкера при гидролизе рассчитывали на основе данных отношения флуоресценции соединения 13а по сравнению с флуоресценцией 13с и 13b по сравнению с 13d, соответственно, в соответствующие моменты времени.
Было определено, что период полужизни линкера при гидролизе in vivo составлял 115 и 160 часов для соединений 13а и 13b, соответственно, что является превосходной корреляцией с результатами, полученными в отношении периода полужизни линкера при гидролизе in vitro, составляющем 120 и 160 часов для соединений 13а и 13b, соответственно.
Фиг. 3 представляет данные расщепления связи линкера с соединением 13b in vivo и in vitro, где в полулогарифмическом виде показана in vivo (треугольниками) и in vitro (ромбами) кинетика расщепления.
Сокращения:
Аср-4-(2-аминоэтил)-1-карбоксиметил-пиперазин;
АсОН - уксусная кислота;
Вое - трет-бутоксикарбонил;
Dab - 2,4-диаминомасляная кислота;
DBU - 1,3-диазабицикло[5.4.0]ундецен;
DCM - дихлорметан;
Dda - додекановая кислота;
DIC - диизопропилкарбодиимид;
DIEA - диизопропилэтиламин;
DMAP - диметиламинопиридин;
DMF - N, N-диметилформамид;
DMSO - диметилсульфоксид;
EDTA - этилендиаминтетрауксусная кислота;
Eq - стехиометрический эквивалент;
Fmoc - 9-флуоренилметоксикарбонил;
HATU O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат;
HFIP - гексафторизопропанол;
HEPES - N-(2-гидроксиэтил)пиперазин-N'-2-этансульфоновая кислота;
HOBt - N-гидроксибензотриазол;
ivDde - 1-(4,4-диметил-2,6-диоксоциклогексилиден)-3-метилбутил;
LCMS - жидкостная хроматография в сочетании с масс-спектрометрией;
Ма1 3-малеимидопропионил;
Mmt - 4-метокситритил;
МС - масс-спектр;
MW - молекулярная масса;
n.d. - не установлено;
PfpOH - пентафторфенол;
РуВОР - бензотриазол-1-ил-окси-Трис-пирролидино-фосфония гексафторфосфат;
RP-HPLC - обращенно-фазовая высокоэффективная жидкостная хроматография (ОФ-ВЭЖХ);
RT - комнатная температура;
SEC - эксклюзионная хроматография молекул по размеру;
Sue - сукцинимидопропионил;
TCP - 2-хлортитилхлоридная смола;
TES - триэтилсилан;
ТМОВ - 2,4,6-триметоксибензил;
TFA - трифторуксусная кислота;
THF - тетрагидрофуран;
UV - ультрафиолетовый;
VIS - визуальный
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОЛЕКАРСТВО, СОДЕРЖАЩЕЕ САМОРАСЩЕПЛЯЕМЫЙ ЛИНКЕР | 2018 |
|
RU2798085C2 |
| ПРОЛЕКАРСТВА, СОДЕРЖАЩИЕ КОНЪЮГАТ ЭКСЕНДИН-ЛИНКЕР | 2011 |
|
RU2593774C2 |
| ПРОЛЕКАРСТВА, СОДЕРЖАЩИЕ КОНЬЮГАТ ГИАЛУРОНОВОЙ КИСЛОТЫ, ЛИНКЕРА И ДВОЙНОГО АГОНИСТА GLP-1/ГЛЮКАГОНА | 2016 |
|
RU2719482C2 |
| VEGF-НЕЙТРАЛИЗУЮЩИЕ ПРОЛЕКАРСТВА ДЛЯ ЛЕЧЕНИЯ ОФТАЛЬМОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2013 |
|
RU2673881C2 |
| ПРОЛЕКАРСТВА, СОДЕРЖАЩИЕ КОНЪЮГАТ ИНСУЛИНА И ЛИНКЕРА | 2010 |
|
RU2574667C2 |
| КОМПОЗИЦИЯ ИНСУЛИНА ДЛИТЕЛЬНОГО ДЕЙСТВИЯ | 2010 |
|
RU2556340C2 |
| НОВЫЕ ПСА-СВЯЗЫВАЮЩИЕ АГЕНТЫ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2787105C2 |
| БИОРАЗЛАГАЕМЫЕ НЕРАСТВОРИМЫЕ В ВОДЕ ГИДРОГЕЛИ НА ОСНОВЕ ПОЛИЭТИЛЕНГЛИКОЛЯ | 2010 |
|
RU2554854C9 |
| СОЕДИНЕНИЕ ПЕПТИДНОЙ ПРИРОДЫ, ОБЛАДАЮЩЕЕ СПОСОБНОСТЬЮ СВЯЗЫВАТЬСЯ С ПСМА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2021 |
|
RU2823164C2 |
| НИЗКОМОЛЕКУЛЯРНЫЕ КОНЪЮГАТЫ ДЛЯ ВНУТРИКЛЕТОЧНОЙ ДОСТАВКИ НУКЛЕИНОВЫХ КИСЛОТ | 2011 |
|
RU2582235C2 |








Настоящее изобретение относится к пролекарству, содержащему конъюгат лекарственное средство-линкер D-L, где D представляет собой азотсодержащее моноклональное или поликлональное антитело, или его фрагмент, или продукт его слияния; a L представляет собой биологически неактивный линкерный фрагмент L1, представленный структурной формулой (I)

где пунктирная линия показывает присоединение к азоту моноклонального или поликлонального антитела, или его фрагмента, или продукта его слияния посредством образования амидной связи; X представляет собой C(R4R4a), C(R4R4a)-C(R5R5a) или C(R5R5a)-C(R4R4a); X1 представляет собой С; X2 представляет собой C(R7, R7a) или C(R7, R7a)-C(R8, R8a); X3 представляет собой О; R1, R1a, R2, R2a, R3, R3a, R4, R4a, R5, R5a, R7, R7a, R8, R8a независимо выбирают из группы, состоящей из Н и С1-4 алкила; при необходимости одна или более пар R1/R4 или R2/R3 вместе с атомами, к которым они присоединены, соединяются с образованием кольца А; при необходимости R3/R3a вместе с атомом азота, к которому они присоединены, соединяются с образованием 5- или 6-членного гетероцикла; А представляет собой С6 циклоалкил и где L1 замещен одной группой L2-Z, при условии, что водород, отмеченный символом * в формуле (I), не может быть замещен заместителем, где L2 представляет собой одинарную химическую связь или алкильную цепь, имеющую от 1 до 20 углеродных атомов, которая при необходимости прерывается одной или более группами, независимо выбранными из -О- и C(O)N(R3aa); при необходимости замещенную одной или нескольких группами, независимо выбранными из ОН и C(O)N(R3aaR3aaa), и в которой R3aa, R3aaa независимо выбирают из группы, состоящей из Н и С1-4 алкила; и где L2 присоединен к Z через концевую группу, выбранную из

и Z представляет собой гидрогель. 8 з.п. ф-лы, 3 ил., 52 пр.
1. Пролекарство, содержащее конъюгат лекарственное средство-линкер D-L, где
D представляет собой азотсодержащее моноклональное или поликлональное антитело, или его фрагмент, или продукт его слияния; и
L представляет собой фрагмент биологически неактивного линкера - L1, представленного формулой (I)

где пунктирная линия показывает присоединение к азоту моноклонального или поликлонального антитела, или его фрагмента, или продукта его слияния посредством образования амидной связи;
X представляет собой C(R4R4a), C(R4R4a)-C(R5R5a) или C(R5R5a)-C(R4R4a);
X1 представляет собой С;
X2 представляет собой C(R7, R7a) или C(R7, R7a)-C(R8, R8a);
X3 представляет собой О;
R1, R1a, R2, R2a, R3, R3a, R4, R4a, R5, R5a, R7, R7a, R8, R8a независимо выбирают из группы, состоящей из Н и С1-4 алкила;
при необходимости одна или более пар R1/R4 или R2/R3 вместе с атомами, к которым они присоединены, соединяются с образованием кольца А;
при необходимости R3/R3a вместе с атомом азота, к которому они присоединены, соединяются с образованием 5- или 6-членного гетероцикла;
А представляет собой С6 циклоалкил и
где L1 замещен одной группой L2-Z при условии, что водород, отмеченный символом * в формуле (I), не может быть замещен заместителем, где
L2 представляет собой одинарную химическую связь или алкильную цепь, имеющую от 1 до 20 углеродных атомов, которая при необходимости прерывается одной или более группами, независимо выбранными из -О- и C(O)N(R3aa); при необходимости замещенную одной или нескольких группами, независимо выбранными из ОН и C(O)N(R3aaR3aaa), и в которой R3aa, R3aaa независимо выбирают из группы, состоящей из Н и С1-4 алкила; и где L2 присоединен к Z через концевую группу,
выбранную из 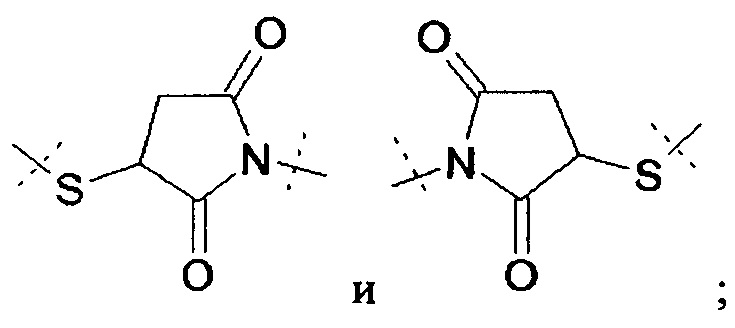
и Z представляет собой гидрогель.
2. Пролекарство по п. 1, где X2 представляет собой C(R7, R7a).
3. Пролекарство по п. 1, где L1 выбирают из группы, состоящей из


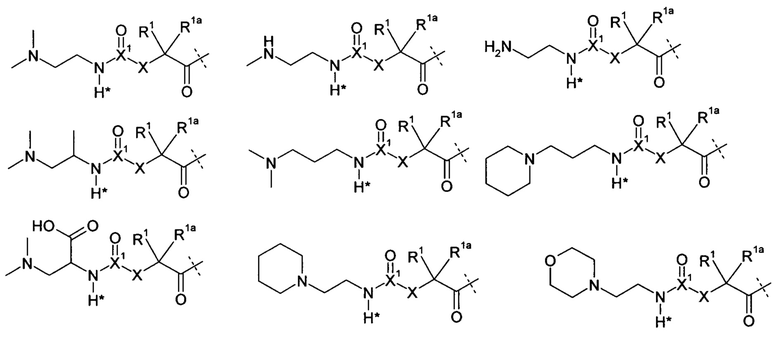

где R1, R1a, R2, R2a, R3, R3a, R4, X, X1, X2 имеют значение, указанное в п. 1.
4. Пролекарство по п. 1, где L1 выбирают из группы, состоящей из



5. Пролекарство по п. 1, где L2 имеет молекулярную массу в интервале от 14 до 750 г/моль.
6. Пролекарство по п. 1, где R1 в формуле (I) замещен L2-Z.
7. Пролекарство по п. 1, где R3 в формуле (I) замещен L2-Z.
8. Пролекарство по п. 1, где R3, R3a в формуле (I) вместе с атомом азота, к которому они присоединены, соединяются с образованием 5- или 6-членного гетероцикла, и где указанный гетероцикл замещен L2-Z.
9. Пролекарство по п. 1 для применения в качестве лекарственного препарата.
| WO 2006136586 A2, 28.12.2006 | |||
| RU 2005141550 A, 27.06.2006 | |||
| КОНЪЮГАТЫ ИНТЕРФЕРОНА | 1997 |
|
RU2180595C2 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| US 4179337 A, 18.12.1979 | |||
| Youhei Sohma et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| Способ изготовления звездочек для французской бороны-катка | 1922 |
|
SU46A1 |
| КОНДРАТЬЕВА Т.С | |||
| Технология лекарственных форм | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| БЕЛИКОВ В.Г | |||
| Фармацевтическая химия | |||
| М., Высшая школа, 1993, с | |||
| Зубчатое колесо со сменным зубчатым ободом | 1922 |
|
SU43A1 |
| Д.А | |||
| Харкевич | |||
| "Фармакология" | |||
| М., Медицина, 1987, стр | |||
| Способ очищения сернокислого глинозема от железа | 1920 |
|
SU47A1 |
| А.Н | |||
| Кудрин | |||
| "Проблемы взаимодействия лекарственных веществ", Фармация, 1983, N 2, стр | |||
| Контрольный стрелочный замок | 1920 |
|
SU71A1 |
| Bundgaard H, et al | |||
| Hydrolysis and rearrangement of phthalamic acid derivatives and assessment of their potential as prodrug forms for amines // Acta Pharm Nord | |||
| Способ приготовления консистентных мазей | 1919 |
|
SU1990A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |

