Изобретение относится к газохроматографическим методам анализа и может быть использовано для идентификации летучих компонентов различных лекарственных растений и фитопрепаратов в медицине, фармакологии, здравоохранении, пищевой, парфюмерной и других отраслях промышленности.
Известны способы и устройства для определения соответствия хроматографических пиков одному и тому же веществу на колонках с последовательно изменяющейся полярностью и многоступенчатый метод с переключением колонок (см. Вигдергауз М.С., Семенченко Л.В., Езрец В.А, Богословский Ю.Н. Качественный газохроматографический анализ. М.: Наука, 1978. С. 162-185).
Недостатками известных способов и устройств для определения соответствия хроматографических пиков одному и тому же веществу на основе независимых хроматограмм смеси, отвечающих нескольким колонкам, являются большая трудоемкость и сложность проведения эксперимента, а также отсутствие серийной хроматографической аппаратуры для получения многоэлементных спектров сорбатов.
Известны также способы определения соответствия пиков на хроматограммах с использованием дополнительной информации из одного цикла анализа от двух хроматографических детекторов: пламенно-ионизационного (ПИД) и детектора по теплопроводности (ДТП) (см. Арутюнов Ю.И., Кудряшов С.Ю., Онучак Л.А., Платонов И.А. Газохроматографический анализ смесей, содержащих неизвестные компоненты. Самара: Вестник СамГУ. Естественнонаучная серия. Изд-во «Самарский университет», 2005. №5 (39). С. 137-162).
Способ заключается в получении результатов анализа исследуемой пробы на каждой колонке с двумя детекторами на выходе в виде:
1. Расчетных концентраций
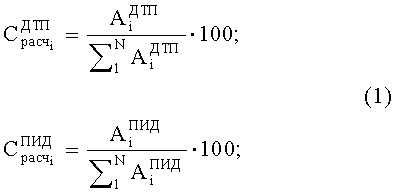
2. Относительного коэффициента чувствительности двух детекторов
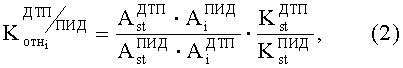
где 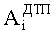
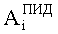
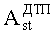
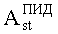
N - число пиков на хроматограммах ДТП и ПИД.
В случае когда в качестве стандарта используется бензол, множитель 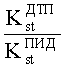
Равенство 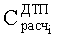
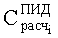
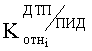
Однако в известном способе используется детектор ДТП, который из-за большой инерционности не может работать с высокоэффективными капиллярными колонками, широко применяемыми для анализа многокомпонентных сложных смесей.
Наиболее близким к заявленному изобретению по совокупности существенных признаков является хромато-распределительный метод с использованием коэффициентов распределения компонентов пробы в системе ограниченно смешиваемых растворителей «гексан-ацетонитрил», определяемых на капиллярной колонке с пламенно-ионизационным детектором при линейном программировании температуры колонки (см. Арутюнов Ю.И., Онучак Л.А., Платонов, Никитченко Н.В. Применение хромато-распределительного метода для определения молекулярной массы и температуры кипения неизвестных компонентов смеси. // Сорбционные и хроматографические процессы, 2011. Т. 11, №4. С. 502-510).
Коэффициенты распределения KCi летучих компонентов пробы определяются из хроматограмм гексанового и ацетонитрильного экстрактов на каждой колонке по уравнению
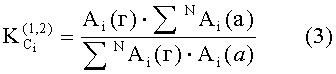
где 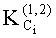
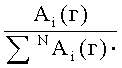
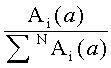
N - число пиков на хроматограммах.
Равенство KCi наряду с равенством 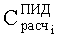
Недостатками известного способа являются уменьшение общего количества определяемых компонентов пробы за счет наложения больших пиков гексана и ацетонитрила на хроматографические пики исследуемых летучих компонентов и повышенная погрешность при определении KCi по уравнению (3) для малых количеств i-го компонента в пробе.
Наиболее близким к предлагаемому изобретению по совокупности существенных признаков является аппаратно-программный комплекс на базе хроматографа «Хроматэк-Кристалл 5000», содержащий источник газа-носителя, дозатор равновесного пара, устройство для твердофазной экстракции и термодесорбции, два аналитических блока с неполярной и полярной капиллярными колонками с делителями расхода на входе и пламенно-ионизационными детекторами на выходе, генератор водорода и компрессор (см. Сертификат об утверждении средств измерений RU. С. 39. 004A №6481).
Недостатками известного устройства на базе хроматографа «Хроматэк-Кристалл 5000» являются:
1. Невозможность проведения анализа равновесной паровой фазы исходной пробы одновременно на двух параллельно включенных капиллярных колонках с неполярной и полярной неподвижными фазами.
2. Отсутствие возможности анализа паровой фазы исходной пробы с измененными концентрациями составляющих летучих компонентов.
Задачей изобретения является увеличение количества определяемых летучих компонентов пробы и повышение сходимости определения концентраций летучих компонентов пробы.
Эта задача решается за счет того, что в способе определения соответствия хроматографических пиков одному и тому же летучему компоненту пробы, при котором исследуемую пробу анализируют не менее трех раз одновременно на хроматографических колонках с полярной и неполярной неподвижными фазами: первый анализ - исходная проба, второй и третий анализы - исходная проба с измененными концентрациями, изменение концентрации составляющих летучих компонентов исходной пробы проводят с использованием твердофазной экстракции путем анализа как газовой фазы при низкой температуре, так и конденсированной фазы при более высокой температуре.
Эта задача решается также за счет того, что в устройстве для определения соответствия хроматографических пиков одному и тому же летучему компоненту пробы, содержащем источник газа-носителя, дозатор равновесного пара, устройство для твердофазной экстракции и термодесорбции, два аналитических блока с неполярной и полярной капиллярными колонками с пламенно-ионизационными детекторами на выходе, делитель потока, генератор водорода и компрессор, устройство для твердофазной экстракции и термодесорбции выполняют в виде сменного термостатируемого патрона с зернистым сорбентом, который включают через дополнительные переключатели потока вместо дозирующей петли дозатора равновесного пара.
Соответствие пиков на хроматограммах неполярной и полярной колонок одному и тому же летучему компоненту пробы определяется в предлагаемом способе по равенству концентрации 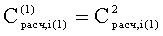
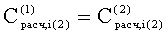
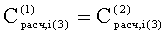
Вероятность определения соответствия пиков на двух хроматограммах одному летучему компоненту оценивается по равенству Срасч,i и времени удерживания этого компонента tRi во всех трех анализах. Вероятность уменьшается, если имеет место равенство Срасч,i и tRi только для двух или одного анализов.
При решении поставленной задачи создается технический результат, заключающийся в том, что на хроматограммах полярной и неполярной колонок отсутствуют большие пики растворителей. Это связано с тем, что вместо двух ограниченно смешивающихся жидких растворителей гексан и ацетонитрил в предлагаемом способе изменение концентраций летучих компонентов исходной пробы проводят с использованием твердофазной экстракции путем анализа как газовой фазы при низкой температуре, так и конденсированной фазы при более высокой температуре. В результате чего значительно увеличивается количество определяемых летучих компонентов пробы и сходимость определения концентрации летучих компонентов пробы.
Это позволяет сделать вывод, что заявляемые изобретения связаны между собой единым изобретательским смыслом.
Изобретение поясняется чертежом. На фигуре 1 схематически изображено устройство для определения соответствия хроматографических пиков одному и тому же летучему компоненту пробы, которое содержит источник газа-носителя 1, дозатор равновесного пара 2 с контейнером исходной пробы 3, шестипортовым газовым краном 4, дозирующей петлей 5 с переключателем потока 6 и устройством для твердофазной экстракции и термодесорбции 7 с переключателем 8, делитель потока 9, специальный микротройник 10, капиллярную колонку 11 с неполярной неподвижной фазой, капиллярную колонку 12 с полярной неподвижной фазой, пламенно-ионизационные детекторы 13.
Устройство работает следующим образом: исходную пробу помещают в контейнер 3, выдерживают при определенной температуре, равновесную паровую фазу дозируют с помощью дозирующей петли 5 переключателя 6 шестипортовым краном 4 одновременно в обе хроматографические колонки для проведения первого анализа летучих компонентов исходной пробы.
Для проведения второго анализа изменение концентрации летучих компонентов пробы проводят с помощью твердофазной экстракции и анализа газовой фазы при температуре 40°C в устройстве 7 для твердофазной экстракции и термодесорбции с переключателем 8 и шестипортовым краном 4.
Для проведения третьего независимого анализа проводят твердофазную экстракцию в устройстве для твердофазной экстракции и термодесорбции 7 путем пропускания определенного объема летучих компонентов с помощью переключателя 8 и шестипортового крана 4 определенного объема, после чего кран 8 закрывают и нагревают устройство 7 до T=100°C, после чего включают кран 8 и шестипортовый кран-дозатор дозирует десорбированную конденсированную фазу летучих компонентов в хроматографические колонки для третьего анализа.
Сравнение известного и предлагаемого способов определения соответствия хроматографических пиков одному и тому же компоненту проводили на двух примерах.
Пример 1. В качестве объекта исследования использовали модельную смесь из семи летучих компонентов, принадлежащих к различным классам органических соединений. Для этого в контейнер исходной пробы дозатора равновесного пара заливали по 0,5 см3 ацетона, изопропанола, этилацетата, изобутанола, бензола, циклогексана и гептана. Контейнер с пробой выдерживали 30 минут при температуре 40°C.
Порядок проведения эксперимента
1. Предлагаемый способ
Эксперимент проводили с помощью устройства для определения соответствия хроматографических пиков одному и тому же летучему компоненту пробы, представленного на фигуре 1. Устройство содержит дозатор равновесного пара 2 с контейнером исходной пробы 3 объемом 15 см3, дозирующую петлю 5 объемом 0,5 см3, устройство для твердофазной экстракции и термодесорбции 7, выполненное в виде трубки (7,2 см × 3,0 мм), заполненной зернистым сорбентом (трис-бета-цианоэтокси пропан 15% масс. на динохроме H, фракция 0,315-0,5 мм), делитель потока 9, специальный микротройник 10, капиллярную колонку 11 типа VF-1 фирмы Varian, США (30 м × 0,32 мм × 0,5 мкм) с полидиметилсилоксановой неподвижной фазой, капиллярную колонку 12 типа INNOWAX фирмы Agilent Technologies, США (30 м × 0,32 мм × 0,5 мкм) с полиэтиленгликолевой неподвижной фазой и два пламенно-ионизационных детектора 13.
Режим работы газохроматографического устройства:
газ-носитель - электролитический водород;
расход газа-носителя на выходе колонок 11 и 12 - 1,0 см3/мин;
температура начала анализа - 40°C;
линейное программирование со скоростью 4°C/мин;
конечная температура - 180°C;
время анализа - 40 минут;
температура детекторов - 200°C.
Для проведения первого анализа равновесную паровую фазу (РПФ) из контейнера 3 пропускали с помощью шестипортового крана 4 через дозирующую петлю 5 и открытый переключатель 6. После чего шестипортовый кран переключали во второе положение «анализ», при котором газ-носитель вытеснял пробу из петли 5 в хроматографические колонки для разделения. В делителе потока 9, поток со скоростью 2 см3/мин равномерно распределялся в специальном микротройнике 10 на две колонки 11 и 12, а поток со скоростью 80 см3/мин сбрасывался в линию сброса.
Для проведения второго анализа РПФ из контейнера 3 пропускали с помощью шестипортового крана 4 через устройство для твердофазной экстракции и термодесорбции 7 и открытый переключатель 8 при низкой температуре 40°C. При этом переключатель 6 закрыт. Затем шестипортовый кран переключали в положение «анализ», и газовая фаза после твердофазной экстракции при низкой температуре вытеснялась газом-носителем одновременно на две капиллярные колонки для анализа.
Для проведения третьего анализа устройство для твердофазной экстракции и термодесорбции 7 выдерживали 15 минут при температуре 100°C при закрытом переключателе 8 и закрытом переключателе 6. Затем открывали переключатель 8 и устанавливали шестипортовый кран 4 в положение «анализ», и газ-носитель вытеснял компоненты пробы после термодесорбции в обе капиллярные колонки для анализа.
По результатам газохроматографического анализа определяли
- Индексы удерживания Ван-Ден-Доола и Кратса 
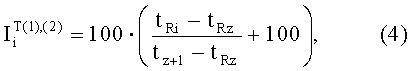
где tRi, tRz, tz+1 - время удерживания i-го компонента равновесной паровой фазы и соседних гомологов н-алканов с числом углеродных атомов в молекулах z и z+1 соответственно. Верхний индекс (1) - колонка с неполярной неподвижной фазой; верхний индекс (2) - колонка с полярной неподвижной фазой.
Для определения tz и tz+1 дополнительно хроматографировали на обеих колонках равновесную паровую фазу смеси н-алканов от пентана до тетрадекана включительно. Для этого в отдельный контейнер объемом 15 см3 заливали но 0,2 см3 пентана и гексана, по 0,3 см3 гептана и октана, по 0,5 см3 нонана и декана, по 1,0 см3 ундекана и додекана, 1,5 см3 тридекана и 2,0 см3 тетрадекана. Контейнер выдерживали 30 минут при температуре 70°C. Затем устанавливали его вместо контейнера 3 в дозатор равновесного пара 2 и проводили анализ равновесной паровой фазы н-алканов по процедуре первого анализа.
- Расчетные концентрации летучих компонентов равновесной паровой фазы исследуемой пробы 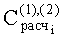
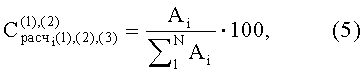
где Ai - площадь пика i-го компонента;
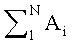
N - число пиков на хроматограмме.
Нижние индексы (1), (2), (3) отвечают результатам первого, второго и третьего анализов.
- Соответствие хроматографических пиков, полученных на колонках с полярной и неполярной неподвижными фазами, одному и тому же компоненту пробы определяли по равенству расчетных концентраций
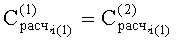
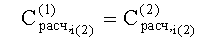
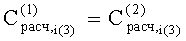
2. Известный способ
Эксперимент проводили на газовом хроматографе «Хроматэк-Кристалл 5000», содержащем дозатор равновесного пара, два аналитических блока с неполярной и полярной капиллярными колонками и пламенно-ионизационными детекторами на выходе. На входе каждой колонки имеются испарители с делителями потока. Капиллярные колонки в известном способе аналогичны описанным в предлагаемом способе.
Режим работы хроматографа:
газ-носитель - электролитический водород;
расход газа-носителя на выходе колонок 11 и 12 - 1,0 см3/мин;
температура начала анализа - 40°C;
линейное программирование со скоростью 4°C/мин;
конечная температура - 180°C;
время анализа - 40 минут;
температура детекторов - 200°C.
Первый анализ РПФ исследуемой пробы проводили аналогично описанному ранее в предлагаемом способе. При этом дозирующая петля 5 объемом 0,5 см3 соединялась шестипортовом кране без переключателя 6.
Для проведения второго и третьего анализов РПФ из контейнера в количестве 20 см3 барботировали через систему из двух ограниченно смешиваемых растворителей: 0,5 см3 гексана и 0,5 см3 ацетонитрила в отдельной емкости. Полученную смесь встряхивали в течение нескольких минут при комнатной температуре. После расслоения из каждого слоя микрошприцом отбирали пробы для газохроматографического анализа. Объем вводимой пробы в испаритель каждой колонки не более 0,5 мкл. Температура испарителя 200°C.
По результатам анализа на двух колонках определяли:
- Индексы удерживания при линейном программировании температуры колонки 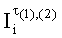
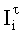
- Расчетные концентрации летучих компонентов РПФ исследуемой пробы 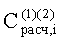
- Константы распределения компонентов РПФ в системе ограниченно смешиваемых растворителей «гексан-ацетонитрил» на колонке с неполярной фазой - 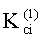
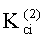
Соответствие хроматографических пиков, полученных на колонках с неполярной и полярной неподвижными фазами, одному и тому же компоненту пробы определяли по равенству констант распределения 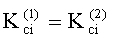
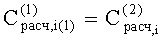
Пример 2. В качестве объекта исследования использовали воздушно-сухое сырье календулы лекарственной (Calendula officinalis) сорта Кальта, выращенное в Ботаническом саду Самарского госуниверситета (2013 г.). Измельченное сухое сырье календулы помещали в контейнер исходной пробы дозатора равновесного пара в количестве не более 12 см3. Контейнер с пробой выдерживали 40 минут при температуре 100°C.
Порядок проведения эксперимента предлагаемым и известным способом описан в примере 1.
Оценку сходимости определения расчетных концентраций проводили на примере анализа равновесной паровой фазы модельной смеси (см. Пример 1) из выборки n=5 измерений в виде относительного среднего квадратического отклонения Sz результата измерения 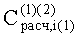
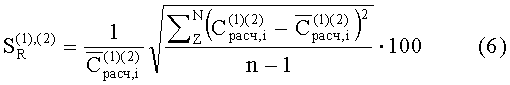
Результаты экспериментов сведены в таблицу «Сравнительные данные экспериментальной проверки известного и предлагаемого способов».
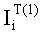
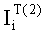
Модельная смесь, содержащая ацетон, изопропанол, этилацетат, изобутанол, бензол, циклогексан и гептан
Лекарственное растение календула (calendula officinalis), общее количество пиков при анализе равновесной паровой фазы:
неполярная фаза - 17
полярная фаза - 10
Из приведенных в таблице данных видно, что:
1. В примере 1 при анализе модельной смеси соответствие хроматографических пиков, полученных на колонках с неполярной и полярной неподвижными фазами, для всех семи летучих компонентов определено предлагаемым способом с наибольшей вероятностью по результатам всех трех анализов.
2. Сравнение полученных разностей индексов удерживания на двух колонках 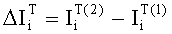
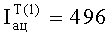
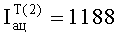
3. В примере 2 при анализе летучих компонентов равновесной паровой фазы календулы лекарственной определено соответствие хроматографических пиков одному и тому же компоненту пробы для 10 летучих компонентов предлагаемым способом и только для шести компонентов известным способом, из-за маскирования малых количеств исследуемых компонентов большим количеством гексана и ацетонитрила.
4. Восемь летучих компонентов календулы лекарственной определены с наибольшей вероятностью из трех анализов.
5. Два компонента с разностью индексов удерживания 305 и 272 определены с вероятностью из двух анализов. Это связано, по-видимому, с тем, что летучие компоненты календулы лекарственной представляют собой сложную многокомпонентную смесь из различных веществ, при хроматографическом анализе которых в одном хроматографическом пике могут присутствовать несколько неподеленных компонентов.
5. Предложенный способ обеспечивает значительно большую сходимость измерения концентрации анализируемых компонентов. Погрешность измерения SR уменьшилась практически в три раза. Связано это с тем, что в известном способе пробу на анализ отбирают из гетерогенной системы ограниченно смешиваемых жидкостей (гексан-ацетонитрил) и на результаты измерения влияет значительно большее количество внешних факторов, чем в предлагаемом способе.
Использование предлагаемого способа определения соответствия хроматографических пиков одному и тому же компоненту и устройство для его осуществления позволяет:
1. Проводить с использованием справочных данных об индексах удерживания групповую и индивидуальную идентификацию некоторых летучих компонентов лекарственных растений, фитопрепаратов и биологически активных добавок, изготовленных на их основе, для стандартизации и оценки подлинности на предприятиях при их изготовлении и реализации;
2. Проводить экспресс-анализ качества лекарственного растительного сырья и фитопрепаратов на имеющемся в лабораториях доступном оборудовании вместо дорогостоящего и сложного в эксплуатации хромато-масс-спектрометра.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОПРЕДЕЛЕНИЯ СООТВЕТСТВИЯ ХРОМАТОГРАФИЧЕСКИХ ПИКОВ, ПОЛУЧЕННЫХ НА КОЛОНКАХ С ПОЛЯРНОЙ И НЕПОЛЯРНОЙ ФАЗАМИ, ОДНОМУ И ТОМУ ЖЕ КОМПОНЕНТУ ПРОБЫ | 2014 |
|
RU2570233C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ПОДЛИННОСТИ ЛЕКАРСТВЕННОГО РАСТИТЕЛЬНОГО СЫРЬЯ | 2014 |
|
RU2582847C1 |
| СПОСОБ ОЦЕНКИ ПОДЛИННОСТИ ЛЕКАРСТВЕННОГО РАСТИТЕЛЬНОГО СЫРЬЯ | 2014 |
|
RU2582621C1 |
| Устройство подготовки пробы для анализа примесей малолетучих полярных веществ в жидких средах | 2018 |
|
RU2697575C1 |
| Способ твердофазного концентрирования комбинации водорастворимых летучих и нелетучих пластовых индикаторов | 2019 |
|
RU2720656C1 |
| Способ количественного определения содержания трихлорэтилена и тетрахлорэтилена в атмосферном воздухе методом газовой хроматографии с электронно-захватным детектированием | 2021 |
|
RU2757237C1 |
| Способ определения полярных летучих компонентов в водных средах | 2023 |
|
RU2836681C1 |
| СПОСОБ ВЫЯВЛЕНИЯ ЗОНЫ ТЕХНОГЕННОГО ХИМИЧЕСКОГО ЗАГРЯЗНЕНИЯ (ВАРИАНТЫ) | 2001 |
|
RU2208781C1 |
| Способ количественного определения N-нитрозоаминов: N-диметилнитрозоамин, N-метилэтилнитрозоамин, N-диэтилнитрозоамин, N-дибутилнитрозоамин, N-дипропилнитрозоамин, N-пиперидиннитрозоамин, N-пирролидиннитрозоамин, N-морфолиннитрозоамин, N-дифенилнитрозоамин, в пробах копченых мясопродуктов методом хромато-масс-спектрометрии | 2017 |
|
RU2657822C1 |
| Способ количественного определения N-дифенилнитрозамина в мясных пробах пищевой продукции методом хромато-масс-спектрометрии | 2017 |
|
RU2626601C1 |
Изобретение относится к газохроматографическим методам анализа и может быть использовано для идентификации летучих компонентов различных лекарственных растений и фитопрепаратов в медицине, фармакологии, здравоохранении, пищевой, парфюмерной и др. отраслях промышленности. Способ заключается в том, что исследуемую пробу одновременно трижды анализируют на хроматографических колонках с неполярной и полярной неподвижными фазами. Первый анализ - исходная проба, второй и третий анализы - исходная проба с измененными концентрациями составляющих летучих компонентов, причем изменение концентрации компонентов исходной пробы проводят с использованием твердофазной экстракции путем анализа как газовой фазы при низкой температуре, так и конденсированной фазы при более высокой температуре. Устройство для определения соответствия хроматографических пиков, содержащее устройство для твердофазной экстракции и термодесорбции, выполненное в виде сменного термостатируемого патрона с зернистым сорбентом, который включают через дополнительные переключатели потока вместо дозирующей петли дозатора равновесного пара. Техническим результатом является увеличение количества определяемых летучих компонентов пробы, а также повышение сходимости определения концентраций летучих компонентов пробы. 2 н.п. ф-лы, 1 ил., 1 табл.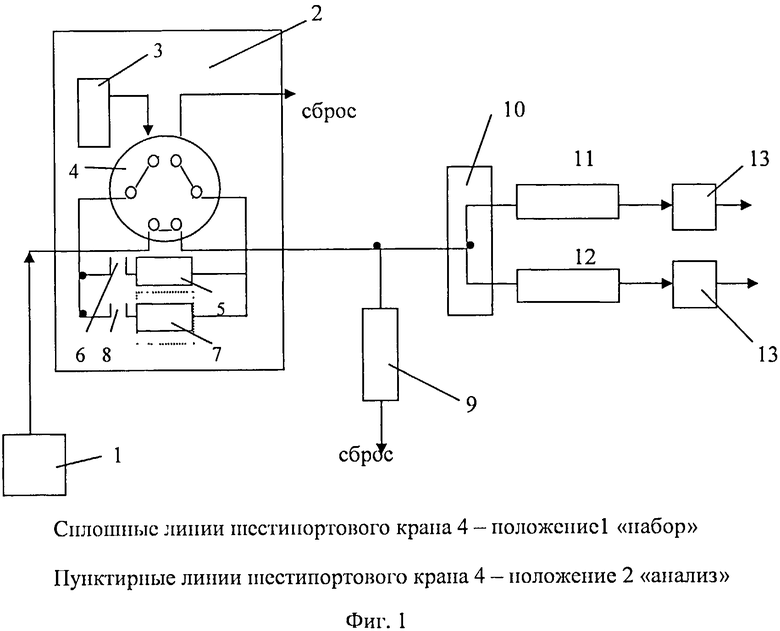
1. Способ определения соответствия хроматографических пиков одному и тому же летучему компоненту пробы, при котором проводят не менее трех анализов исследуемой пробы одновременно на хроматографических колонках с неполярной и полярной неподвижными фазами: первый анализ - исходная проба, второй и третий анализы - исходная проба с измененными концентрациями составляющих летучих компонентов, а по соотношению концентраций и времени удерживания компонентов, полученных в результате этих анализов, определяют с некоторой вероятностью соответствие хроматографических пиков на неполярной и полярной колонках одному и тому же летучему компоненту пробы, отличающийся тем, что изменение концентрации составляющих летучих компонентов исходной пробы проводят с использованием твердофазной экстракции путем анализа как газовой фазы при низкой температуре, так и конденсированной фазы при более высокой температуре.
2. Устройство для осуществления способа по п. 1, содержащее источник газа-носителя, дозатор равновесного пара, устройство для твердофазной экстракции и термодесорбции, два аналитических блока с неполярной и полярной капиллярными колонками с пламенно-ионизационными детекторами на выходе, делитель потока, генератор водорода и компрессор, отличающееся тем, что содержит устройство для твердофазной экстракции и термодесорбции, выполненное в виде сменного термостатируемого патрона с зернистым сорбентом, который включают через дополнительные переключатели потока вместо дозирующей петли дозатора равновесного пара.
| Арутюнов Ю.И | |||
| et al, Применение хромато-распределительного метода для определения молекулярной массы и температуры кипения неизвестных компонентов смеси, Сорбционные и хроматографические процессы, Т.П | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| С | |||
| Мерная кружка для жидких тел | 1914 |
|
SU502A1 |
| WO 9701755 A2, 16.01.1997 | |||
| KR 20010062385 A, 07.07.2001 | |||
| US 2013140238 A1, 06.06.2013 | |||

