ПЕРЕКРЕСТНЫЕ ССЫЛКИ НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка заявляет приоритет по предварительной заявке на патент США № 61/229695, поданной 29 июля 2009 г., и предварительной заявке на патент США № 61/255816, поданной 28 октября 2009 г., и предварительной заявке на патент США № 61/313635, поданной 12 марта 2010 г. Каждая из данных заявок включается в настоящее изобретение путем ссылки во всей своей полноте.
ССЫЛКА НА СПИСОК ПОСЛЕДОВАТЕЛЬНОСТЕЙ, ТАБЛИЦУ ИЛИ КОМПЬЮТЕРНУЮ ПРОГРАММУ
Список последовательностей подается в электронном виде через EFS в виде текстового файла, созданного 29 июля 2010 г. и названного "632008017WO00seqlist.txt" (85400 байт), содержание которого включается в настоящее изобретение путем ссылки во всей своей полноте.
ОБЛАСТЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
Настоящий объект изобретения относится к соединениям с уменьшающей уровни паратиреоидного гормона (PTH) активностью, к фармацевтическим композициям, содержащим данные соединения, и применению данных соединений и композиций в способах лечения, включая, но не ограничиваясь, лечение гиперкальциемии или гиперпаратиреоза или регулирования in vivo PTH уровней.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Гомеостаз кальция является механизмом, с помощью которого тело поддерживает приемлемые уровни кальция. Данный процесс является в высокой степени регулируемым и включает сложную взаимозависимость между поглощением, транспортом, хранением в костях, отложением в других тканях и выведением кальция. PTH представляет собой регулятор циркулирующей в крови концентрации кальция и функционирует, увеличивая концентрацию кальция в крови усилением высвобождения кальция из кости с помощью процесса резорбции кости; увеличивая реабсорбцию кальция из почечных канальцев; и усиливая поглощение кальция в кишечнике увеличением синтеза 1,25-(OH)2 витамин D, активной формы витамина D. PTH также стимулирует выведение фосфора из почек и увеличивает высвобождение из кости.
PTH секреция регулируется кальцийчувствительным рецептором (CaSR), сопряженным с G-белком рецептором, экспрессируемым несколькими типами клеток на поверхности паратиреоидных клеток, который обнаруживает небольшие отклонения концентрации внеклеточных ионов кальция (Ca2+) и реагирует изменением секреции PTH. Активация CaSR Ca2+ ингибирует PTH-секрецию в течение секунд или минут за счет ингибирования везикулярного транспорта, и данный процесс может регулироваться фосфорилированием рецептора протеинкиназой C (PKC). CaSR также экспрессируется на остеобластах и в почках, где он регулирует почечное Ca2+ выведение.
Кроме того, PTH регулирует гомеостаз фосфора, PTH стимулирует рецептор 1 паратиреоидного гормона (PTHR1) и на апикальной (мембрана щеточной каймы) и на базолатеральной мембранах клеток на GI участке. PTHR1 стимулирование приводит к увеличению мочевыделения фосфата (Pi) как следствие снижения интернализации почечного Na+/фосфат (NaPi-IIa) контранспортера на мембране щеточной каймы.
PTH также вовлечен в регуляцию остеобластов и остеокластов в кости. PTH увеличивает циркулирующий Ca2+ увеличением резорбции кости и почечной реабсорбции кальция. PTH стимулирует остеобласты к синтезу RANK лиганда (RANKL), который связывается с RANK рецептором и активирует остеокласты, приводя к увеличению резорбции кости и увеличению Ca2+ в сыворотке. Остеопротегерин (OPG) представляет собой рецептор-ловушку для RANKL, который блокирует резорбцию кости. Остеопороз вызывается нарушением баланса между процессами резорбции кости остеокластами и образования кости остеобластами.
Человеческое тело содержит приблизительно 1 кг кальция, 99% которого находится в костях. В нормальных условиях уровень циркулирующих ионов кальция (Ca2+) строго поддерживается равным приблизительно 9-10 мг/дл (т.е. 2,25-2,5 ммоль/л; ~600 мг). Приблизительно 1 г элементарного кальция (Ca2+) усваивается каждый день. Из данного количества поглощается приблизительно 200 мг/день и 800 мг/день выводится. Кроме того, приблизительно 500 мг/день высвобождается резорбцией кости или откладывается в кости. Приблизительно 10 г Ca2+ фильтруется через почки каждый день, причем приблизительно 200 мг находится в моче, а оставшаяся часть реабсорбируется.
Гиперкальциемия представляет собой повышенный уровень кальция в крови. Острая гиперкальциемия может приводить в результате к желудочно-кишечным (анорексия, тошнота, рвота); почечным (полиурия, полидипсия), нейромышечным (ослабление, помрачнение сознания, оглушение, кома) и сердечным (брадикардия, атриовентрикулярный первой степени) симптомам. Хроническая гиперкальциемия также связана с желудочно-кишечными (диспепсия, констипация, панкреатит); почечными (нефролитиаз, нефрокальциноз), нейромышечными (слабость) и сердечными (блокада сердца в результате гипертензии, дигиталисная чувствительность) симптомами. Нерегулярные сердечные сокращения могут быть результатом этого, EKG данные короткого QT интервала и уширенная T волна предполагают гиперкальциемию. Гиперкальциемия может быть бессимптомной, причем симптомы более обычно проявляются при высоких уровнях кальция (12,0 мг/дл или 3 ммоль/л). Тяжелая гиперкальциемия (выше 15-16 мг/дл или 3,75-4 ммоль/л) считается состоянием, представляющим опасность для жизни и требующим срочной медицинской помощи: при данных уровнях результатом может быть кома и остановка сердца.
Гиперкальциемия часто вызывается гиперпаратиреозом, приводя к избыточной резорбции кости и повышенным уровням кальция в сыворотке крови. При первичном спорадическом гиперпаратиреозе, PTH синтезируется в повышенном количестве единичной паратиреоидной аденомой; менее часто причиной может быть несколько аденом или диффузная гиперплазия паращитовидной железы. Повышенная PTH секреция приводит к суммарному увеличению резорбции кости, с высвобождением Ca2+ и фосфата (Pi). PTH также усиливает почечную реабсорбцию Ca2+ и ингибирует реабсорбцию фосфата (Pi), приводя в результате к суммарному увеличению содержания кальция в сыворотке крови и снижению фосфата.
Вторичный гиперпаратиреоз возникает, когда снижение циркулирующей концентрации Ca2+ стимулирует PTH секрецию. Одной причиной вторичного гиперпаратиреоза является хроническая почечная недостаточность (также называемая хроническая болезнь почек или CKD), такая как хроническая почечная недостаточность при поликистозной болезни почек или хроническом пиелонефрите, или хроническая почечная недостаточность, такая как хроническая почечная недостаточность у пациентов, находящихся на гемодиализе (также называемая болезнь почек последней стадии или ESRD). Избыток PTH может синтезироваться в ответ на гипокальциемию, являющуюся результатом слабого всасывания кальция, GI заболеваний, почечной недостаточности, дефицита витамина D и почечной гиперкальциемии. Третичный гиперпаратиреоз может возникать после долгого периода вторичного гиперпаратиреоза и гиперкальциемии.
Злокачественное образование является типичной причиной гиперкальциемии, не опосредуемой PTH. Гиперкальциемия злокачественного образования является нетипичным, но тяжелым осложнением рака, оказывает действие на 10%-20% пациентов с раком и может возникать и с солидными опухолями и лейкемией. Заболевание имеет бурное начало и очень плохой прогноз, с медианой выживаемости, составляющей только шесть недель. Факторы роста (GF) регулируют синтез белка, связанного с паратиреоидным гормоном (PTHrP) в опухолевых клетках. Опухолевые клетки можно стимулировать аутокринным GF для усиления синтеза PTHrP, приводя в результате к усилению резорбции кости. Опухолевые клетки, метастатические к кости, могут также секретировать PTHrP, который может ресорбироваться костью и высвобождать дополнительное количество GF, который, в свою очередь, действует паракринным образом, дополнительно усиливая синтез PTHrP.
Соответственно, требуются соединения с активностью, например, регулирующей PTH уровни и/или уровни кальция in vivo.
КРАТКОЕ ОПИСАНИЕ
В одном аспекте настоящее изобретение относится к соединению, имеющему формулу
X1-X2-X3-X4-X5-X6-X7,
в котором X1 представляет собой субъединицу, содержащую тиолсодержащую группу; X5 представляет собой катионную субъединицу; X6 представляет собой некатионную субъединицу; X7 представляет собой катионную субъединицу; и, по меньшей мере, одна, предпочтительно две, из X2, X3 и X4 представляет (представляют) собой независимо катионную субъединицу; и в котором соединение обладает увеличивающей концентрацию паратиреоидного гормона активностью. В одном варианте осуществления снижение концентрации паратиреоидного гормона представляет собой снижение концентрации паратиреоидного гормона в крови или в плазме у субъекта, обработанного соединением, относительно концентрации паратиреоидного гормона в крови или плазме у субъекта перед обработкой. В другом варианте осуществления снижение концентрации паратиреоидного гормона достигается в отсутствие гистаминовой реакции.
В другом варианте осуществления X3 и X4 являются некатионными, тогда как X1, X5, X6 и X7 являются катионными.
В одном варианте осуществления X1 субъединица представляет собой тиолсодержащий аминокислотный остаток. В другом варианте осуществления тиольная группа X1 субъединицы представляет собой органический тиолсодержащий фрагмент.
В другом варианте осуществления, когда X1 субъединица представляет собой тиолсодержащий аминокислотный остаток, она выбрана из группы, состоящей из L-цистеина, D-цистеина, глутатиона, N-ацетилированного цистеина, гомоцистеина и пегилированного цистеина.
В еще одном варианте осуществления органический тиолсодержащий фрагмент выбран из тиолалкильного или тиоацильного фрагмента, такого как 3-меркаптопропил или 3-меркаптопропионил, меркаптопропионовая кислота, меркаптоуксусная кислота, тиобензил или тиопропил. В еще другом варианте осуществления органический тиолсодержащий фрагмент представляет собой меркаптопропионовую кислоту.
В еще другом варианте осуществления X1 субъединица модифицирована химически так, что она содержит ацетильную группу, бензоильную группу, бутильную группу или другую аминокислоту, такую как ацетилированный бета-аланин.
В еще другом варианте осуществления, когда X1 субъединица содержит тиольный фрагмент, X1 субъединица соединена ковалентной связью со вторым тиольным фрагментом.
В другом варианте осуществления формула X1-X2-X3-X4-X5-X6-X7 состоит из непрерывной последовательности аминокислотных остатков (обозначенных в настоящем изобретении как (Xaa1)-(Xaa2)-(Xaa3)-(Xaa4)-(Xaa5)-(Xaa6)-(Xaa7) SEQ ID NO:1) или последовательности субъединиц органических соединений (неаминокислотные остатки).
В другом варианте осуществления непрерывная последовательность аминокислотных остатков представляет собой непрерывную последовательность L-аминокислотных остатков, непрерывную последовательность D-аминокислотных остатков, непрерывную последовательность смеси L-аминокислотных остатков и D-аминокислотных остатков, или смеси аминокислотных остатков и неприродных аминокислотных остатков.
В другом варианте осуществления непрерывная последовательность аминокислотных остатков соединена с соединением для облегчения транспорта через клеточную мембрану. В другом варианте осуществления непрерывная последовательность аминокислотных остатков соединена с соединением, которое улучшает доставку последовательности внутрь или через один или более слоев ткани.
В другом варианте осуществления непрерывная последовательность аминокислотных остатков содержится в последовательности аминокислотных остатков из 8-50 аминокислотных остатков, 8-40 аминокислотных остатков, 8-30 аминокислотных остатков или 8-20 аминокислотных остатков в длину. В еще другом варианте осуществления непрерывная последовательность аминокислотных остатков содержится в последовательности аминокислотных остатков из 8-19 аминокислотных остатков, 8-18 аминокислотных остатков, 8-17 аминокислотных остатков, 8-16 аминокислотных остатков, 8-15 аминокислотных остатков, 8-14 аминокислотных остатков, 8-13 аминокислотных остатков, 8-12 аминокислотных остатков, 8-11 аминокислотных остатков, 8-10 аминокислотных остатков или 8-9 аминокислотных остатков в длину.
В другом варианте осуществления X3 субъединица представляет собой катионный аминокислотный остаток.
В другом варианте осуществления X2 субъединица представляет собой некатионный аминокислотный остаток, и в другом варианте осуществления X4 субъединица представляет собой некатионный аминокислотный остаток. В одном варианте осуществления некатионный аминокислотный остаток представляет собой D-аминокислоту.
В другом варианте осуществления X3 и X4 представляют собой катионные D-аминокислотные остатки.
В другом варианте осуществления X5 субъединица представляет собой D-аминокислотный остаток.
В другом аспекте непрерывная последовательность в любом из описанных соединений ковалентно соединена через тиолсодержащую группу в X1 субъединице со второй непрерывной последовательностью. Например, вторая непрерывная последовательность может быть идентичной непрерывной последовательности (для образования димера) или может быть неидентичной, как может быть в случае при присоединении к фрагменту, который облегчает транспорт непрерывной последовательности через клеточную мембрану.
В другом аспекте настоящее изобретение относится к конъюгату, состоящему из пептида carrrar (SEQ ID NO:2), где пептид конъюгируют по его N-концевому остатку с Cys остатком.
В одном варианте осуществления пептид химически модифицируют по N-концу, C-концу или обоим.
В другом варианте осуществления N-конец пептида химически модифицируют ацетилированием и C-конец химически модифицируют амидированием.
В другом варианте осуществления конъюгат представляет собой Ac-c(C)arrrar-NH2 (SEQ ID NO:3).
В другом аспекте предусматривается способ лечения вторичного гиперпаратиреоза (SHPT) у субъекта, в котором соединение, как описано в настоящем изобретении, вводится субъекту. В различных вариантах осуществления субъект может страдать от хронической болезни почек или другого заболевания.
В другом аспекте предусматривается способ снижения концентрации паратиреоидного гормона у субъекта, в котором соединение, как описано в настоящем изобретении, вводится субъекту.
В другом аспекте настоящее изобретение относится к режиму лечения, причем режим включает введение любого соединения из соединений, описанных в настоящем изобретении, в комбинации со вторым агентом.
В одном варианте осуществления второй терапевтический агент представляет собой витамин D, аналог витамина D или гидрохлорид цинакальцета.
В любом из аспектов или вариантов осуществления, описанных в настоящем изобретении, предусматривается, что любая одна или более последовательностей отдельно исключаются или удаляются из объема формулы изобретения. В определенных вариантах осуществления пептиды, указанные любой одной или более из SEQ ID NO:162-182, отдельно или в любой комбинации, исключаются из заявленных соединений, композиций и способов.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фигура 1 представляет собой график уровня паратиреоидного гормона в виде процентов от исходного значения до введения дозы, как функцию времени, в часах, у крыс с острой почечной недостаточностью (1K1C модель), где крысам вводили дозу Ac-crrrr-NH2 (SEQ ID NO:4, ромбы), Ac-crrrrr-NH2 (SEQ ID NO:5, закрашенные квадраты), Ac-crrrrrr-NH2 (SEQ ID NO:6, треугольники), Ac-crrrrrrr-NH2 (SEQ ID NO:7, пустые квадраты), или солевой раствор в качестве контроля (x символы).
Фигура 2A представляет собой график IP1-концентрации, в нМ, как функции концентрации Ac-carrrar-NH2 (SEQ ID NO:26, квадраты) и Ac-arrrar-NH2 (SEQ ID NO:29, треугольники), в виде величины способности соединения активировать человеческий CaSR в in vitro клеточном анализе, когда человеческий CaSR экспрессируется в стабильно трансфицированной HEK-293 клеточной линии.
Фигура 2B показывает снижение PTH концентрации при in vivo введении пептидов, указанных как SEQ ID NO:26 (Ac-carrrar-NH2) (квадраты) и как SEQ ID NO:29 (Ac-arrrar-NH2) (ромбы), где пептиды вводились в виде IV болюса нормальным крысам Спрага-Доули в дозах 9 мг/кг для SEQ ID NO:29 и при 0,5 мг/кг для SEQ ID NO:26. Внутривенный (IV) болюс солевого раствора применяли в качестве контроля (пунктирная линия). Уровни PTH в плазме оценивали перед введением дозы и через 1, 2, 3 и 4 часа после введения дозы. Результаты представлены как среднее для группы ± стандартное отклонение (SD), и PTH показано в виде процентов от исходного значения до введения дозы.
Фигура 3 представляет собой гистограмму, на которой сравнивается высвобождение гистамина после IV болюсного введения различных соединений нормальным крысам Спрага-Доули, где соединения Ac-crrrr-NH2 (SEQ ID NO:4), Ac-crrrrr-NH2 (SEQ ID NO:5), Ac-crrrrrr-NH2 (SEQ ID NO:6) и Ac-crrrrrrrr-NH2 (SEQ ID NO:41) вводили в эквимолярной IV болюсной дозе 2,1 мкмоль/кг, и гистамин в плазме измеряли перед введением дозы (pre), через 5, 15 и 30 минут после введения дозы.
Фигура 4 представляет собой гистограмму, на которой сравнивается высвобождение гистамина после IV болюсного введения двух соединений нормальным крысам Спрага-Доули, где соединения Ac-c(C)arrrar-NH2 (SEQ ID NO:3, заштрихованные накрест столбцы) и Ac-crrrrrr-NH2 (SEQ ID NO:6, пустые столбцы) вводили с дозой 3 мг/кг, и гистамин в плазме измеряли до введения дозы (нулевой момент времени) и через 5, 15 и 30 минут после введения дозы.
Фигура 5 представляет собой график уровня паратиреоидного гормона в виде процентов от исходного значения до введения дозы, как функции времени, в часах, у нормальных крыс, которым вводили 0,5 мг/кг IV болюсом Ac-crrrrrr-NH2 (SEQ ID NO:6, ромбы), Ac-carrrrr-NH2 (SEQ ID NO:8, квадраты), Ac-crarrrr-NH2 (SEQ ID NO:9, треугольники), Ac-crrarrr-NH2 (SEQ ID NO:10, x символы), Ac-crrrarr-NH2 (SEQ ID NO:11, * символы), Ac-crrrrar-NH2 (SEQ ID NO:12, окружности) или Ac-crrrrra-NH2 (SEQ ID NO:13, + символы).
Фигуры 6A-6B представляют собой графики уровней паратиреоидного гормона в виде процентов от исходного значения до введения дозы, как функцию времени, в часах, у здоровых мышей, которым вводили 0,5 мг/кг IV болюсом Ac-carrrar-NH2 (SEQ ID NO:26, пустые ромбы), Ac-crrarar-NH2 (SEQ ID NO:25, пустые квадраты), Ac-caarrrr-NH2 (SEQ ID NO:22, треугольники), Ac-crraarr-NH2 (SEQ SD NO:17, закрашенные квадраты), Ac-c(C)arrrar-NH2 (SEQ ID NO:3, ромбы, фигура 6B), Ac-craarrr-NH2 (SEQ ID NO:24, x символы на фигуре 6A); Ac-c(C)rrarar-NH2 (SEQ ID NO:28, x символы, фигура 6B).
Фигура 7 показывает снижение уровней паратиреоидного гормона в крови как функции времени, для соединения Ac-c(C)arrrar-NH2 (SEQ ID NO:3), введенного в виде IV болюса нормальным крысам Спрага-Доули с дозами 1 мг/кг (ромбы), 0,5 мг/кг (квадраты), 0,3 мг/кг (треугольники) и 0,1 мг/кг (x символы). Внутривенный (IV) болюс солевого раствора (окружности) применяли в качестве контроля. Уровни PTH в плазме оценивали перед введением дозы и через 1, 2, 3 и 4 часа после введения дозы.
Фигура 8 представляет собой график уровня паратиреоидного гормона в виде процентов от исходного значения до введения дозы, как функции времени, в часах, у крыс с острой почечной недостаточностью (1K1C модель), где крысам вводили дозы с помощью IV болюса соединения Ac-c(C)arrrar-NH2 (SEQ ID NO:3) с дозой 3 мг/кг (ромбы), 1 мг/кг (треугольники), 0,5 мг/кг (квадраты) и 0,3 мг/кг (x символы) или солевой раствор (квадраты); пунктирная линия на фигуре 8 показывает исходный уровень PTH перед введением дозы.
Фигура 9 представляет собой график концентрации паратиреоидного гормона в виде процентов от исходного значения до введения дозы, как функцию времени, в часах, у крыс, которым введен внутривенно солевой раствор (x символы) или соединения Ac-crrrrrr-NH2 (SEQ ID NO:6, пустые ромбы) и Ac-carrrar-NH2 (SEQ ID NO:26, пустые квадраты) при 1 мг/кг с помощью 30-минутного IV вливания, где уровни PTH в плазме оценивали перед введением дозы, через 16 часов и 24 часа после введения дозы.
Фигура 10 представляет собой график уровня паратиреоидного гормона в виде процентов от исходного значения до введения дозы, как функцию времени, в часах, у крыс с острой почечной недостаточностью (1K1C модель), где крысам вводили с помощью IV болюса соединения Ac-c(C)arrrar-NH2 (SEQ ID NO:3, квадраты, * символы) и Ac-c(Ac-C)arrrar-NH2 (SEQ ID NO:146, треугольники, ромбы) с дозами 0,3 мг/кг (квадраты, треугольники) и 0,5 мг/кг (*, ромбы).
Фигура 11 представляет собой график уровня паратиреоидного гормона в виде процентов от исходного значения до введения дозы, как функцию времени, в часах, у крыс, обработанных с помощью облегченной микропорацией трандермальной доставки Ac-crrrrrr-NH2 (SEQ ID NO:6, двое животных, квадраты и треугольники) или солевым раствором с помощью трансдермальной доставки (ромбы).
Фигура 12 представляет собой график уровня паратиреоидного гормона в виде процентов от исходного значения до введения дозы, как функцию времени, в часах, у крыс, обработанных с помощью облегченной микропорацией трандермальной доставки Ac-c(C)arrrar-NH2 (SEQ ID NO:3).
Фигура 13 представляет собой график среднего PTH (в виде процента от исходного значения) в течение и после 6-часового IV вливания Ac-c(C)arrrar-NH2 (SEQ ID NO:3) нормальным крысам Спрага-Доули, где соединение вливали при скорости 1 мкг/кг/час (квадраты), 3 мкг/кг/час (окружности) и 10 мкг/кг/час (треугольники).
Фигура 14A показывает PTH (в виде процента от исходного значения) в течение и после 6-часового IV вливания Ac-c(C)arrrar-NH2 (SEQ ID NO:3) в 1K1C крысиной модели острой почечной недостаточности, где крысам внутривенно вливали при скоростях дозирования 30 мкг/кг/час (ромбы) и 100 мкг/кг/час (квадраты).
Фигура 14B представляет собой гистограмму, показывающую концентрацию кальция в сыворотке крови, в мг/дл, для 1K1C модельных крыс, обработанных, как на фигуре 14A.
Объект настоящего изобретения можно понять более легко со ссылкой на следующее подробное описание предпочтительных вариантов осуществления и примеры, включенные в настоящее описание.
ПОДРОБНОЕ ОПИСАНИЕ
I. Определения
В настоящей заявке, если не указано особо, определения терминов и иллюстрацию способов настоящей заявки можно найти в любом из нескольких хорошо известных источников, таких как: Sambrook, J., et al., Molecular Cloning; A Laboratory Manual, Cold Spring Harbor Laboratory Press (1989); Goeddel, D., ed., Gene Expression Technology, Methods in Enzymology, 185, Academic Press, San Diego, CA (1991); "Guide to Protein Purification" in Deutshcer, M. P., ed., Methods in Enzymology, Academic Press, San Diego, CA (1989); Innis, et al., PCR Protocols: A Guide to Methods and Applications, Academic Press, San Diego, CA (1990); Freshney, R.I., Culture of Animal Cells: A Manual of Basic Technique, 2nd Ed., Alan Liss, Inc. New York, NY (1987); Murray, EJ., ed., Gene Transfer and Expression Protocols, pp. 109-128, The Humana Press Inc., Clifton, N. J. and Lewin, B., Genes VI, Oxford University Press, New York (1997).
Как применяют в настоящем изобретении, формы единственного числа включают множественные формы, если не указано иначе. Например, пептид, являющийся модулятором, включает один или более пептидов, являющихся модуляторами.
Как применяют в настоящем изобретении, соединение обладает "снижающей уровень паратиреоидного гормона активностью" или "PTH-снижающей активностью", когда соединение, при введении субъекту, снижает уровень паратиреоидного гормона (PTH) в плазме по сравнению с PTH-концентрацией в плазме до введения соединения. В одном варианте осуществления снижение уровня PTH составляет величину по меньшей мере на 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90% или 95% меньшую, через один час после введения соединения, уровня PTH до введения соединения.
Как применяют в настоящем изобретении, "отсутствие гистаминовой реакции" или "недостаточная гистаминовая реакция" предполагает дозу соединения, которая производит меньше чем 15-кратное, 14-кратное, 13-кратное, 12-кратное, 11-кратное, 10-кратное, 9-кратное. 8-кратное, 7-кратное, 6-кратное, 5-кратное, 4-кратное или 3-кратное увеличение концентрации гистамина, измеренное в in vitro анализе, как описано в настоящем изобретении, где кратное изменение определяют на основе уровней гистамина до инкубирования с соединением и после 15 минут инкубирования с соединением.
Как применяют в настоящем изобретении, "аминокислота" относится к природным и неприродным аминокислотам. Двадцать встречающихся в природе аминокислот (L-изомеры) обозначаются трехбуквенным кодом с приставкой "L-" (за исключением глицина, который является ахиральным) или однобуквенным кодом в верхнем регистре: аланин ("L-Ala" или "A"), аргинин ("L-Arg" или "R"), аспарагин ("L-Asn" или "N"), аспарагиновая кислота (L-Asp" или "D"), цистеин ("L-Cys" или "C"), глутамин ("L-Gln" или "Q"), глутаминовая кислота ("L-Glu" или "E"), глицин ("Gly" или "G"), гистидин ("L-His" или Η"), изолейцин ("L-Ile" или "I"), лейцин ("L-Leu" или "L"), лизин ("L-Lys" или "K"), метионин ("L-Met" или "M"), фенилаланин ("L-Phe" или "F"), пролин ("L-Pro" или "P"), серин ("L-Ser" или "S"), треонин ("L-Thr или "T"), триптофан ("L-Trp" или "W"), тирозин ("L-Tyr" или "Y") и валин ("L-Val" или "V"). L-норлейцин и L-норвалин могут быть представлены как (NLeu) и (NVal), соответственно. Девятнадцать встречающихся в природе аминокислот, которые являются хиральными, имеют соответствующий D-изомер, который обозначается трехбуквенным кодом с приставкой "D-" или однобуквенным кодом в нижнем регистре: аланин ("D-Ala" или "a"), аргинин ("D-Arg" или "r"), аспарагин ("D-Asn" или "a"), аспарагиновая кислота ("D-Asp" или "d"), цистеин ("D-Cys" или "c"), глутамин ("D-Gln" или "q"), глутаминовая кислота ("D-Glu" или "e"), гистидин ("D-His" или "h"), изолейцин ("D-Ile" или "i"), лейцин ("D-Leu" или "l"), лизин ("D-Lys" или "k"), метионин ("D-Met" или "m"), фенилаланин ("D-Phe" или "f"), пролин ("D-Pro" или "p"), серин ("D-Ser" или "s"), треонин ("D-Thr" или "t"), триптофан ("D-Trp" или "w"), тирозин ("D-Tyr" или "y") и валин ("D-Val" или "v"). D-норлейцин и D-норвалин могут быть представлены в виде (dNLeu) и (dNVal), соответственно. Хотя "аминокислотный остаток" часто применяют со ссылкой на мономерную субъединицу пептида, полипептида или белка и "аминокислоту" часто применяют со ссылкой на свободную молекулу, применение данных терминов в данной области техники перекрывается и заменяется. Термин "аминокислота" и "аминокислотный остаток" применяются взаимозаменяемо, и они могут относиться к свободной молекуле или мономерной субъединице пептида, полипептида или белка, в зависимости от контекста.
Для определения процентной "гомологии" или процентной "идентичности" двух аминокислотных последовательностей последовательности выравнивают для целей оптимального сравнения (например, гэпы можно вводить в последовательность одного полипептида для оптимального сравнения с другим полипептидом). Затем сравнивают аминокислотные остатки в соответствующих положениях аминокислот. Когда положение в одной последовательности занято аналогичным аминокислотным остатком, как и в соответствующем положении в другой последовательности, молекулы являются одинаковыми по данному положению. Как применяют в настоящем изобретении, "гомология" аминокислот или нуклеиновых кислот является эквивалентной "идентичности" аминокислот или нуклеиновых кислот. Соответственно, процентная идентичность последовательности между двумя последовательностями является функцией числа идентичных положений, одинаковых для последовательностей (т.е. процентная идентичность последовательности=число идентичных положений/суммарное число положений×100). Процентная идентичность последовательности между двумя полипептидными последовательностями можно определить, применяя пакет программ Vector NTI (Invitrogen Corporation, 5791 Van Aleen Way, Carlsbad, CA 92008). Штраф за открытие гэпа 10 и штраф за продолжение гэпа 0,1 применяют для определения процентной идентичности двух полипептидов. Все другие параметры настраиваются по умолчанию.
"Катионная аминокислота" подразумевает аминокислотный остаток, который обладает общим положительным зарядом при физиологическом pH (7,4), как в случае, например, аминокислотных остатков, когда боковая цепь или "R группа" содержит аминофункциональную или другую функциональную группу, которая может принимать протон, становясь положительно заряженной при физиологическом pH, такая как гуанидин или имидазольный фрагмент. Катионные аминокислотные остатки включают аргинин, лизин, гистидин, 2,3-диаминопропионовую кислоту (Dap), 2,4-диаминомасляную кислоту (Dab), орнитин и гомоаргинин.
"Катионная субъединица" подразумевает субъединицу, которая имеет общий положительный заряд при физиологическом pH (7,4).
Как применяют в настоящем изобретении, "консервативные аминокислотные замены" представляют собой замены, которые не приводят в результате к значительному изменению активности или третичной структуры выбранного полипептида или белка. Данные замены обычно включают замену выбранного аминокислотного остатка отличным аминокислотным остатком, обладающим аналогичными физико-химическими свойствами. Распределение по группам аминокислот и аминокислотных остатков по физико-химическим свойствам является известным специалистам в данной области техники. Например, среди встречающихся в природе аминокислот, можно определить в данной области техники семейства аминокислотных остатков, имеющих аналогичные боковые цепи, и они включают основные боковые цепи (например, лизин, аргинин, гистидин), кислые боковые цепи (например, аспарагиновая кислота, глутаминовая кислота), незаряженные полярные боковые цепи (например, глицин, аспарагин, глутамин, серин, треонин, тирозин, цистеин), неполярные боковые цепи (например, аланин, валин, лейцин, изолейцин, пролин, фенилаланин, метионин, триптофан), бета-разветвленные боковые цепи (например, треонин, валин, изолейцин) и ароматические боковые цепи (например, тирозин, фенилаланин, триптофан, гистидин).
Как применяют в настоящем изобретении, "химически сшитые" относится к ковалентному присоединению двух или более молекул.
Пептид или пептидный фрагмент "получен из" исходного пептида или полипептида, если он содержит аминокислотную последовательность, которая является идентичной или гомологичной, по меньшей мере, непрерывной последовательности из пяти аминокислотных остатков, более предпочтительно восьми аминокислотных остатков, исходного пептида или полипептида.
Как применяют в настоящем изобретении, термин "гиперпаратиреоз" относится к первичному, вторичному и третичному гиперпаратиреозу, если не указано особо.
Термин "внутрикожный" подразумевает, что в способах лечения, описанных в настоящем изобретении, терапевтически эффективное количество кальциймиметического соединения наносят на кожу для доставки соединения к слоям кожи глубже рогового слоя эпидермиса и, таким образом, достигают требуемого терапевтического эффекта.
Как применяют в настоящем изобретении, "выделенный" или "очищенный" полипептид или его биологически активная часть не содержит никаких клеточных веществ при получении способами рекомбинантной ДНК, или исходные химические вещества или другие химические вещества при химическом синтезе. Формулировка "практически не содержит клеточных веществ" включает препараты полипептидов, в которых полипептид отделяют от некоторых из клеточных компонентов клеток, в которых их получают естественным образом или рекомбинантно. Когда полипептид или его биологически активную часть получают рекомбинантно, он также предпочтительно практически не содержит культуральной среды, т.е. культуральная среда присутствует в количестве менее чем приблизительно 20%, более предпочтительно менее чем приблизительно 10%, и самое предпочтительное менее чем приблизительно 5% объема полипептидного препарата. Формулировка "практически не содержит исходных химических веществ или других химических веществ" включает препараты полипептидов, в которых полипептид отделяют от исходных химических веществ или других химических веществ, которые используются или образуются при синтезе полипептида. В одном варианте осуществления формулировка "практически не содержит исходных химических веществ или других химических веществ" включает препараты полипептида, содержащие менее чем приблизительно 30% (сухой массы) исходных химических веществ или других химических веществ, предпочтительно менее чем приблизительно 20% исходных химических веществ или других химических веществ, более предпочтительно менее чем приблизительно 15% исходных химических веществ или других химических веществ, еще более предпочтительно менее чем приблизительно 10% исходных химических веществ или других химических веществ, и самое предпочтительное менее чем приблизительно 5% исходных химических веществ или других химических веществ. В предпочтительных вариантах осуществления выделенные полипептиды или их биологически активные части не содержат загрязняющих полипептидов из того же организма, из которого получают целевой полипептид.
Как применяют в настоящем изобретении, "макромолекула" относится к молекуле, такой как пептид, полипептид, белок или нуклеиновая кислота, которая обычно имеет молекулярную массу большую чем 900 дальтон.
"Некатионная аминокислота" подразумевает аминокислотный остаток, который не имеет заряда или имеет суммарный отрицательный заряд при физиологическом pH (7,4), как в случае, например, в аминокислотных остатках, когда боковая цепь или "R группа" является нейтральной (нейтральной полярной и нейтральной неполярной) и кислой. Некатионные аминокислоты включают те остатки, содержащие R группу, которая представляет собой углеводородный алкильный или ароматический фрагмент (например, валин, аланин, лейцин, изолейцин, фенилаланин); нейтральную, полярную R группу (аспарагин, цистеин, глутамин, серин, треонин, триптофан, тирозин); или нейтральную, неполярную R группу (глицин, метионин, пролин, валин, изолейцин). Некатионные аминокислоты с кислотной R группой включают аспарагиновую кислоту и глутаминовую кислоту.
"Полимер" относится к линейной цепи двух или более идентичных или неидентичных субъединиц, соединенных ковалентной связью.
Как применяют в настоящем изобретении, "пептид" и "полипептид" относится к любому полимеру, состоящему из цепи аминокислотных остатков, соединенных пептидными связями, независимо от его размера. Хотя "белок" часто применяют со ссылкой на относительно длинные полипептиды, а "пептид" часто применяют со ссылкой на небольшие полипептиды, применение данных терминов в данной области техники перекрывается и заменяется. Таким образом, для простоты, в настоящем изобретении будет применяться термин "пептид", хотя в некоторых случаях в данной области техники он может относиться к тому же полимеру как "полипептид". Если не указано особо, последовательность пептида дается в порядке от аминоконца к карбоксильному концу.
"Тиолсодержащая группа" или "тиолсодержащий фрагмент", как применяют в настоящем изобретении, подразумевает функциональную группу, содержащую связь сера-водород (-SH), и которая способна реагировать с другим тиолом при физиологических условиях, давая дисульфидную связь. Тиол, который способен образовывать дисульфидную связь с другим тиолом, называют "реакционноспособным тиолом". В предпочтительном варианте осуществления тиолсодержащая группа находится на расстоянии менее чем 6 атомов от остова соединения. В более предпочтительном варианте осуществления тиолсодержащая группа имеет структуру (-SH-CH2-CH2-C(O)-O-)-.
Как применяют в настоящем изобретении, "небольшая молекула" относится к молекуле, отличной от макромолекулы, такой как органическая молекула, и обычно имеет молекулярную массу менее чем 1000 дальтон.
Как применяют в настоящем изобретении, "субъект" относится к человеческому субъекту или животному субъекту.
"Субъединица" подразумевает мономерный блок, который соединен более чем с одним другим мономерным блоком, образуя полимерное соединение, где субъединица представляет собой самый короткий повторяющийся фрагмент элементов в полимерном соединении. Примерами субъединиц являются аминокислоты, которые при соединении образуют полимерное соединение, такое как соединение, называемое в данной области техники пептид, полипептид или белок.
Как применяют в настоящем изобретении, "терапевтически эффективное количество" представляет собой количество, требуемое для того, чтобы вызвать требуемый терапевтический эффект. Например, в способах снижения концентрации кальция в сыворотке крови у субъектов с гиперкальциемией, терапевтически эффективное количество представляет собой количество, требуемое для снижения концентрации кальция в сыворотке, по меньшей мере, на 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 11%, 12%, 13%, 14%, 15%, 16%, 17%, 18%, 19%, 20% или 25%. Кальций можно измерить как суммарный кальций или как ионизированный кальций. В качестве другого примера, в способах снижения in vivo PTH, терапевтически эффективное количество представляет собой количество, требуемое для снижения концентрации PTH, по меньшей мере, на 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 11%, 12%, 13%, 14%, 15%, 16%, 17%, 18%, 19%, 20% или 25%.
Как применяют в настоящем изобретении, термин "трансдермальный" обозначает то, что в способах лечения, описанных в настоящем изобретении, терапевтически эффективное количество кальциймиметического агента наносят на кожу для доставки соединения в системный кровоток и, таким образом, достигается требуемый терапевтический эффект.
Если не указано особо, все документы, на которые ссылаются в настоящем изобретении, включены в настоящее изобретение полностью путем ссылки.
II. Соединения
В одном аспекте настоящее изобретение относится к соединению, содержащему последовательность субъединиц X1-X2-X3-X4-X5-X6-X7, в котором X1 представляет собой субъединицу, содержащую тиольную группу; X5 представляет собой катионную субъединицу; X6 представляет собой некатионную субъединицу; X7 представляет собой катионную субъединицу; и, по меньшей мере, две из X2, X3 и X4 независимо представляют собой катионную субъединицу. Соединения обладают снижающей уровни паратиреоидного гормона и/или снижающей уровни кальция активностью в крови субъекта. Снижение уровней паратиреоидного гормона, как будет проиллюстрировано ниже, подразумевает снижение PTH-концентрации в плазме или крови у субъекта по сравнению с PTH-концентрацией в плазме или крови до обработки соединением. В одном варианте осуществления соединение достигает снижения PTH-концентрации в плазме, по меньшей мере, равного 50%, в течение одного часа после введения дозы по сравнению с PTH в плазме до введения дозы. Соединения иллюстрируются пептидами, хотя специалистам в данной области техники ясно, что непептидные соединения, которые обладают требуемой активностью, можно конструировать на основе исследований связи структура-активность, описанных в настоящем изобретении.
Как применяют в настоящем изобретении, паратиреоидный гормон или PTH представляет собой 84-аминокислотный пептид, полученный паратиреоидной железой, и его продукты разложения. Помимо PTH полной длины (который состоит из остатков 1-84 и иногда его называют "интактный" "биоактивный" PTH) различные PTH фрагменты, полученные в результате протеолиза и других путей метаболизма, присутствуют в крови. Аминоконцевая 1-34 область интактной PTH молекулы является биологически активной. Данная область молекулы содержит аминокислотную последовательность, которая позволяет PTH связываться с рецептором паратиреоидного гормона в тканях-мишенях. Считают, что средняя и карбоксиконцевая 35-84 область интактной PTH молекулы является биологически инертной, но обладает иммунологической реакционной способностью. Считают, что PTH 7-84 проявляет эффекты, которые противоположны эффектам 1-84 PTH. Разработаны различные способы анализа для измерения уровней PTH, включая различные продукты разложения, и они рассматриваются Souberbielle et al., Kidney International, 77:93-100 (2010), которая вводится в настоящее изобретение с помощью ссылки. В одном варианте осуществления соединение, обладающее снижающей PTH-концентрацию активностью, как определено в настоящем изобретении, определяют, применяя утвержденный способ количественного измерения PTH, который обнаруживает интактную биоактивную форму PTH (1-84), и имеющиеся в продаже наборы, известные в данной области техники (например, см. пример 3 в настоящем описании).
В первом исследовании получали соединения, содержащие 4-7 катионных (например, аргинин) субъединиц, и испытывали их на способность снижать PTH по сравнению с исходными PTH величинами и животными, обработанными солевым раствором. Конкретно, применяли 1K1C модель острой почечной недостаточности для характеризации PTH-снижающей активности в условиях почечной дисфункции. 1K1C модель описывают в примере 1A, и соединения, полученные для испытания, включали (i) Ac-crrrr-NH2 (SEQ ID NO:4), (ii) Ac-crrrrr-NH2 (SEQ ID NO:5), (iii) Ac-crrrrrr-NH2 (SEQ ID NO:6), (iv) Ac-crrrrrrr-NH2 (SEQ ID NO:7) и (v) контроль солевого раствора.
Как описано в примере 1B, каждое из соединений, обозначенных как SEQ ID NO:4, SEQ ID NO:5, SEQ ID NO:6 и SEQ ID NO:7, вводили 30-минутным IV вливанием животным 1K1C модели. Фигура 1 показывает снижение уровней PTH в плазме как процент концентрации до введения дозы (исходная). Все четыре соединения с дозой 3 мг/кг приводили к значительному падению PTH в плазме, но различия в эффективности и продолжительность PTH снижения предполагают связь между суммарным положительным зарядом и PTH-снижающей активностью. Например, соединение Ac-crrrrrr-NH2 (SEQ ID NO:6; треугольники) с шестью катионными (аргинин) субъединицами обладает повышенной эффективностью, а также продолжительностью действия по сравнению с соединениями Ac-crrrr-NH2 (SEQ ID NO:4; ромбы) и Ac-crrrrr-NH2 (SEQ ID NO:5; квадраты), содержащими четыре и пять катионных (аргинин) субъединиц, соответственно. Неожиданно, соединение Ac-crrrrrr-NH2 (SEQ ID NO:6; треугольники) с шестью катионными (аргинин) субъединицами обладало повышенной продолжительностью действия по сравнению с соединением Ac-crrrrrrr-NH2 (SEQ ID NO:7, пустые квадраты) с семью катионными (аргинин) остатками, предполагая, что активность или эффективность соединений не находится в простом соответствии с увеличением катионного заряда соединения. Т.е., соединение Ac-crrrrrrr-NH2 (SEQ ID NO:7) с семью катионными субъединицами (аргининовые остатки) приводит к первоначальному снижению PTH как соединения с меньшим количеством катионных остатков, но через более 24 часа после введения оно было менее эффективным, чем Ac-crrrrrr-NH2 (SEQ ID NO:6) и Ac-crrrrr-NH2 (SEQ ID NO:5). Последние два соединения приводят к среднему PTH снижению ~40% и 60% через 24 часа, соответственно. Обе величины PTH снижения и продолжительности PTH являются важными критериями для получения оптимальной терапевтической пользы для нуждающихся в лечении пациентов. Необходимо отметить, что соединения в данном исследовании вводились при той же мг/кг дозе но, из-за различия в молекулярной массе, в действительности вводили различное количество молей каждого соединения. Следовательно, Ac-crrrrrr-NH2 (SEQ ID NO:6) был значительно более эффективным, чем Ac-crrrr-NH2 (SEQ ID NO:4) и Ac-crrrrr-NH2 (SEQ ID NO:5) на основе одного моля.
Проводили дополнительное исследование для исследования связи структура-активность соединений. Соединение Ac-crrrrrr-NH2 (SEQ ID NO:6) модифицировали последовательной заменой аргининовых остатков аланиновыми остатками в каждом положении субъединицы X2-X7. Соединения характеризовали в in vitro анализе с человеческим кальцийчувствительным рецептором (CaSR), описанном в примере 2, в котором применяли HEK 293 клетки, которые экспрессируют человеческий кальцийчувствительный рецептор, для измерения активности соединений примеров. Не желая быть связанными теорией, считают, что механизм, с помощью которого описанные соединения снижают PTH in vivo, осуществляется через активацию CaSR, который экспрессируется в паратиреоидной железе и контролирует PTH секрецию. Активация CaSR приводит к увеличению внутриклеточного кальция и инозитол-3-фосфата (IP3) и последующему накоплению инозитол-фосфата-1 (IP1). Соответственно, в данном in vitro анализе определяли полумаксимальную эффективную концентрацию соединения для снижения IP1 генерации на 50% (EC50). Те же соединения также испытывали in vivo для определения их PTH-снижающей активности, как описано в примере 3. Результаты показаны в таблице 1. Число в колонке, озаглавленной "% PTH AUC (1-4 часа) солевого контроля", таблицы 1 определяет активность как снижение площади под кривой (AUC) PTH через 4 часа как процент PTH AUC, полученной для контрольных крыс, обработанных солевым раствором. Например, AUC (вводимое соединение)/AUC (солевой контроль)*100, которое равно 0, будет указывать на высокоактивное PTH-снижающее соединение, которое полностью подавляет PTH (до необнаруживаемых концентраций) через 4 часа после единичного IV введения анестезированным изофлураном (IF)-нормальным крысам. Напротив, величина AUC (введенное соединение)/AUC (солевой контроль)*100, которая равна или больше чем 100, будет указывать на неактивное соединение.
In vitro
и in vivo активность примерных соединений
** PTH снижение после 0,5 мг/кг IV введения анестезированным изофлураном нормальным крысам - PTH измеряли через 1, 2, 3 и 4 часа после введения и рассчитывали суммарную AUC. PTH данные рассчитывали согласно следующей формуле: AUCвводимое соединение/AUCсолевой контроль*100.
В таблице 1 соединения Ac-crrrrrr-NH2 (SEQ ID NO:6), Ac-carrrrr-NH2 (SEQ ID NO:8) и Ac-crrarrr-NH2 (SEQ ID NO:10) были одинаково эффективными, о чем свидетельствует снижение процентного PTH ниже обнаруживаемого предела или по существу до нуля, как измерено in vivo после однократного IV введения нормальным крысам. Замена катионного (аргинин) остатка в положениях 2, 3, 4 или 7 Ac-crrrrrr-NH2 (SEQ ID NO:6) приводило в результате к приблизительно двукратной потере in vitro эффективности. Замена в положении 5 для получения соединения Ac-crrrarr-NH2 (SEQ ID NO:11) давала 5-10 кратное снижение in vitro эффективности, хотя in vivo процентное PTH AUC снижение 45% могло быть достаточно активным для клинической терапии. Неожиданно, замена катионного аргининового остатка в положении 6 незаряженным (аланин) остатком в действительности увеличивала активность. Данные показывают, что катионные и незаряженные остатки в различных положениях не являются все эквивалентными и существуют изменения активности как результат изменения в структуре соединения.
Для дополнительной оценки влияния изменения активности как функции изменения структуры соединения, получали другой набор аналогов Ac-crrrrrr-NH2 (SEQ ID NO:6), содержащих двойные аминокислотные замены, где два катионных (аргинин) остатка заменяли незаряженными (аланин) остатками, и испытывали на активность. Данные показаны в таблице 2. Следует отметить, что данный набор соединений имел аналогичный суммарный катионный заряд как SEQ ID NO:4 (четыре катионных остатка) и, несмотря на это, неожиданно, некоторые соединения являются очень активными (SEQ ID NO:26) с очень низкой %PTH AUC солевого контроля, в то время как другие являются неактивными (например, SEQ ID NO:14). Неожиданно, это предполагает, что положение зарядов, а также суммарный катионный заряд могут влиять на снижающую PTH активность соединений. Данные, показанные в таблице 2, согласуются с данными, показанными в таблице 1, предполагая, что катионные остатки SEQ ID NO:6 являются существенными в положениях 5 и 7, но не требуется в положении 6, для PTH-снижающей активности.
In vivo
активность примерных соединений
** PTH снижение после 0,5 мг/кг IV введения анестезированным изофлураном нормальным крысам - PTH измеряли через 1, 2, 3 и 4 часа после введения и рассчитывали суммарную AUC. PTH данные рассчитывали согласно следующей формуле: AUCвводимое соединение/AUCсолевой контроль*100.
Данные в таблице 2 показывают структурные изменения, которые влияют на активность. В одном варианте осуществления соединение представляет собой Ac-caarrrr-NH2 (SEQ ID NO:22), и в другом варианте осуществления соединение представляет собой Ac-craarrr-NH2 (SEQ ID NO:24).
Дополнительные исследования связи структура-активность проводили, применяя in vitro клеточный анализ в HEK 293 клетках, которые экспрессируют человеческий кальцийчувствительный рецептор, как описано в примере 4. Способность пептидов Ac-carrrar-NH2 (SEQ ID NO:26) и Ac-arrrar-NH2 (SEQ ID NO:29) активировать человеческий CaSR устанавливали измерением накопления инозитолмонофосфата (IP1), который отражает синтез IP3. IP3 синтез является важным клеточным сигнальным вторичным мессенджером, и его синтез является непосредственным последующим в цепи вследствие CaSR активации. Накопления IP1 после IP3 синтеза можно добиться обработкой клеток, применяемых в анализе, хлоридом лития (LiCl2), который ингибирует фермент, который превращает IP1 в инозитол. В исследованиях, описанных в примере 4, накопление IP1 измеряли в присутствии примерных соединений Ac-carrrar-NH2 (SEQ ID NO:26) и Ac-arrrar-NH2 (SEQ ID NO:29). Результаты показаны на фигуре 2A.
Концентрация IP1 приводится в нМ вдоль Y-оси, и концентрация соединения SEQ ID NO:26 или SEQ ID NO:29 приводится в M вдоль X-оси. Отсутствие N-концевого D-цистеинового остатка из SEQ ID NO:29 резко снижает способность соединения активировать CaSR по сравнению с SEQ ID NO:26. Т.е., удаление N-концевого цистеинового остатка заметно снижает активность соединения, поскольку пептиды Ac-carrrar-NH (SEQ ID NO:26) и Ac-arrrar-NH2 (SEQ ID NO:29) отличаются только присутствием или отсутствием N-концевого D-цистеина.
Вклад тиолсодержащей группы в X1 субъединице соединения (например, в определенных вариантах осуществления, когда соединение представляет собой пептид на N-концевом остатке), также исследовался в in vivo исследовании. PTH-снижающая активность пептидов, обозначенных SEQ ID NO:26 (Ac-carrrar-NH2) и SEQ ID NO:29 (Ac-arrrar-NH2), оценивалась in vivo согласно способам в примере 4. Уровни PTH в плазме оценивали до введения дозы и через 1, 2, 3 и 4 часа после введения дозы. Результаты показаны на фигуре 2B. Как видно, 0,5 мг/кг доза пептида Ac-carrrar-NHz (SEQ ID NO:26) (квадраты) снижает PTH концентрацию в крови до необнаруживаемого уровня в течение вплоть до 4 часов после введения дозы. Напротив, пептид, не содержащий N-концевой остаток с тиолсодержащей группой, Ac-arrrar-NH2 (SEQ ID NO:29), ромбы, не снижал PTH концентрацию, даже при значительно большей дозе (т.е. 9 мг/кг).
Связь структура-активность тиолсодержащей группы в X1 субъединице соединения дополнительно анализировалась получением соединений с различными X1 субъединицами. Соединения, показанные в таблице 3, испытывали in vivo на нормальных крысах на снижающую PTH активность.
In vivo
активность примерных соединений
** PTH снижение после 0,5 мг/кг IV введения анестезированным изофлураном нормальным крысам - PTH измеряли через 1, 2, 3 и 4 часа после введения и рассчитывали суммарную AUC. PTH данные рассчитывали согласно следующей формуле: AUCвводимое соединение/AUCсолевой контроль*100.
Данные в таблице 3 показывают, что тиолсодержащая X1 субъединица может изменяться. Испытывали соединения со следующими остатками на N-конце - D-цистеин (cys), D-фенилаланин (dPen), D-гомоцистеин (dHcy) и меркаптопропионовая кислота (Mpa). Кроме того, природную или неприродную аминокислоту, такую как бета-аланин, можно присоединять к N-концевому тиолсодержащему остатку. Данные показывают, что катионные соединения, такие как Ac-crrrrrr-NH2 (SEQ ID NO:6), содержащие различные тиолсодержащие группы в X1 субъединице, эффективно снижают концентрацию PTH in vivo. Замена N-концевого цистеинового остатка метионином, который не содержит тиольной группы, приводила в результате к соединению с очень низкой in vivo PTH-снижающей активностью (данные не показаны).
На основе вышеуказанных исследований соединения непрерывной последовательности субъединиц X1-X2-X3-X4-X5-X6-X7, в которой X1 представляет собой субъединицу, содержащую тиолсодержащую группу, обладают снижающей уровни паратиреоидного гормона активностью. В одном варианте осуществления тиолсодержащая группа в X1 субъединице выбрана из группы, состоящей из тиолсодержащих аминокислотных остатков и органических тиолсодержащих фрагментов. В другом варианте осуществления тиолсодержащая группа способна реагировать с другой тиольной группой при физиологических pH и температуре. В определенных вариантах осуществления, когда тиолсодержащий остаток представляет собой аминокислотный остаток, X1 субъединица может быть любой одной из цистеина, глутатиона, меркаптопропионовой кислоты, N-ацилированного цистеина и пегилированного цистеина. В вариантах осуществления, в которых тиолсодержащая группа находится в неаминокислотной субъединице, такой как небольшая органическая молекула с тиолсодержащей группой, X1 субъединица может представлять собой тиолалкильный или тиоацильный фрагмент, такой как 3-меркаптопропильный или 3-меркаптопропионильный остаток. В одном варианте осуществления тиол не является гомоцистеином.
Соответственно, и в другом варианте осуществления, соединения, описанные в настоящем изобретении, обладают "клинической, снижающей концентрацию паратиреоидного гормона активностью", что подразумевает, что соединение, при введении субъекту, снижает концентрацию паратиреоидного гормона в плазме, как измерено суммарной PTH площадью под кривой (PTH AUC) через 4 часа после введения, по сравнению с PTH AUC соответствующего контрольного субъекта, обработанного носителем. Концентрацию PTH в плазме измеряют, применяя, например, имеющийся в продаже ELISA набор, который обнаруживает биоактивный интактный PTH 1-84 (см. пример 3 для конкретного набора). Соединение с клинической активностью, снижающей уровень паратиреоидного гормона, снижает PTH AUC, по меньшей мере, на 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, по сравнению с PTH AUC соответствующего контрольного субъекта, обработанного носителем.
Вышеуказанные исследования и другие, описанные ниже, иллюстрируют дополнительные варианты осуществления соединений, описанных в настоящем изобретении, в которых X1 субъединицу в некоторых вариантах осуществления можно модифицировать химически, например, химической модификацией для введения ацетильной группы, бензоильной группы, бензильной группы, бутильной группы, природной или неприродной аминокислоты, такой как ацетилированный бета-аланин, или соединять ее ковалентной связью к другому тиольному фрагменту.
Пептидные терапевтические средства могут быть чувствительными к атаке пептидазами. Экзопептидазы обычно являются неспецифическими ферментами, которые расщепляют аминокислотные остатки с амино или карбокси конца пептида или белка. Эндопептидазы, которые расщепляют внутри аминокислотной последовательности, могут также быть неспецифичными; однако эндопептидазы часто узнают конкретные аминокислотные последовательности (участки узнавания) и расщепляют пептид в или вблизи данного участка. Соответственно, предусмотрены модификации соединения для защиты его от протеолитического разрушения.
Один способ защиты пептида от протеолитического разрушения включает химическую модификацию или "кэпирование" амино и/или карбокси конца пептидов. Как применяют в настоящем изобретении, термины "химически модифицированная" или "кэпированная" применяют взаимозаменяемо для ссылки на введение блокирующей группы в конец или в оба конца соединения с помощью ковалентной модификации. Подходящие блокирующие группы служат для блокирования концов пептидов, не снижая биологическую активность пептидов. Любой остаток, расположенный на амино или карбокси конце, или обоих, описанных соединений, включая тиолсодержащие субъединицы, можно модифицировать химически.
В предпочтительном варианте осуществления амино конец соединения химически модифицируют ацетилированием для получения N-ацетилированного пептида (который может быть представлен как "Ac-" в структуре или формуле в настоящем изобретении). В предпочтительном варианте осуществления карбокси конец описанных пептидов химически модифицируют амидированием для получения первичного карбоксамида на C-конце (который может быть представлен как "-NH2" в пептидной последовательности, структуре или формуле настоящего изобретения). В предпочтительном варианте осуществления и амино конец и карбокси конец химически модифицируют ацетилированием и амидированием, соответственно. Однако возможны другие кэпирующие группы. Например, амино конец можно кэпировать ацилированием группами, такими как ацетильная группа, бензоильная группа, или природными или неприродными аминокислотами, такими как бета-аланин, кэпированный ацетильной группой; или алкилированием группами, такими как бензильная группа или бутильная группа, или сульфонилированием для получения сульфонамидов. Аналогично, карбокси конец можно этерифицировать или превращать во вторичный амид и ацилсульфонамид или подобные. В некоторых вариантах осуществления амино конец или карбокси конец может содержать сайт присоединения полиэтиленгликольного (PEG) фрагмента, т.е. амино или карбокси конец можно химически модифицировать реакцией с подходящим образом функционализированным ПЭГ (PEG).
Защита пептидов от эндопептидазы обычно включает обнаружение и устранение из пептида участка узнавания эндопептидазы. Участки протеазного узнавания являются хорошо известными специалистам в данной области техники. Таким образом, можно обнаружить потенциальный участок эндопротеазного узнавания и затем удалить данный участок изменением аминокислотной последовательности в участке узнавания. Остатки в узнаваемой последовательности можно перемещать или удалять для разрушения участка узнавания. Предпочтительно делать консервативную замену одной или более аминокислотами в установленном участке протеазного узнавания.
A. Дополнительные исследования структура-активность
Проводили дополнительные исследования связи структура-активность для дополнительной оценки влияния свойств каждой субъединицы в соединении на его терапевтическую активность. Данные исследования здесь будут описываться со ссылкой на пример 5.
Ряд соединений, в которых D-аминокислотный остаток заменяли на L-аминокислотный остаток, получали, исходя из PTH-снижающей способности Ac-c(C)arrrar-NH2 (SEQ ID NO:3). Соединения вводились субъектам, и уровни PTH в плазме оценивали до введения дозы и через 1, 2, 3 и 4 часа после введения дозы, как описано в примере 5, и AUC рассчитывали как сумму величин PTH концентраций в моменты времени 1, 2, 3 и 4 часа, нормализованную AUC для солевого контроля для тех же моментов времени, умноженную на 100. Результаты показаны в таблице 4.
Влияние L-аминокислотной замены на активность
Примерные соединения, показанные в таблице 4, химически модифицировали и по N-концу, и по C-концу, как показано Ac и NH2 обозначениями. Последовательность семи субъединиц carrrar (SEQ ID NO:3), в которых все субъединицы были D-аминокислотными остатками, модифицировали заменой одной субъединицы один раз L-аминокислотой. X1 субъединица была D-Cys остатком (или L-Cys остатком в SEQ ID NO:34), присоединенным через дисульфидную связь к L-Cys остатку, как показано обозначением в скобках (C). Данные об PTH-снижающей in vivo активности в таблице 4 показывают, что хиральность Arg и Ala влияет на активность соединений. В одном варианте осуществления предполагается соединение с последовательностью X1-X2-X3-X4-X5-X6-X7, в которой, по меньшей мере, субъединицы, обозначенные как X4 и X7, являются D-аминокислотными субъединицами. В другом варианте осуществления субъединицы, обозначенные как X4, X5, X6 и X7, являются D-аминокислотными субъединицами. В предпочтительном варианте осуществления субъединицы, обозначенные как X3, X4, X5, X6 и X7, являются D-аминокислотными субъединицами. В самых предпочтительных вариантах осуществления субъединицы, обозначенные как X2, X3, X4, X5, X6 и X7, являются D-аминокислотными субъединицами, и все из субъединиц X1, X2, X3, X4, X5, X6 и X7 являются D-аминокислотными субъединицами.
В других исследованиях также было обнаружено, что замена в пептиде всех L-аминокислот D-аминокислотами, не снижает in vitro активность испытуемых пептидов; действительно, оказалось, что пептиды, состоящие только из D-аминокислот, увеличивают активность активации CaSR. Также было показано, что некоторые из катионных (аргинин) остатков, в специфических положениях относительно цистеинового остатка, можно заменять незаряженными (аланин) остатками с минимальным влиянием на активность относительно CaSR.
Для дальнейшей характеризации связи между структурой и активностью относительно CaSR, ряд катионных пептидов с различным числом (4-8) аргининовых остатков (все из которых содержали N-концевой цистеин) испытывали, применяя HEK-293 in vitro клеточный анализ. Была обнаружена прямая зависимость между числом катионных субъединиц и активностью соединения, где об активности свидетельствовала способность активировать CaSR. Уменьшение количества катионных (например, аргинин) субъединиц с 5 до 4 приводило в результате к самому большому изменению активности (>10-кратное), предполагая, что может существовать точка перегиба активности между соединениями, содержащими данные общие заряды, и что катионная субъединица в положении субъединицы X5 предпочтительна для активности. Соответственно, предусмотрены соединения структуры X1-X2-X3-X4-X5-X6-X7, в которых X5 представляет собой катионную субъединицу. В определенных вариантах осуществления X1 представляет собой субъединицу, содержащую тиольную группу, которая способна реагировать с другой тиольной группой при физиологических условиях ("реакционноспособный тиол" предполагает тиол, который реагирует с другим тиолом (например, цистеин с цистеином) при физиологических условиях pH 7,4 и температуре тела).
Неожиданно, Ac-crrrrrr-NH2 (SEQ ID NO:6) с шестью катионными остатками, при оценке in vivo, показала большую и более продолжительную активность, чем Ac-crrrrrrrr-NH2 (SEQ ID NO:41), которая содержит восемь катионных остатков. Это противоположно наблюдению, что SEQ ID NO:41 была более активной при активации CaSR в данном in vitro клеточном анализе.
Не желая быть связанными теорией, считают, что превосходные свойства Ac-crrrrrr-NH2 (SEQ ID NO:6) in vivo могут вытекать из более хороших фармакокинетических свойств Ac-crrrrrr-NH2 (SEQ ID NO:6), потому что ожидают, что Ac-crrrrrrrr-NH2 (SEQ ID NO:41) будет поглощаться клетками посредством ее проникающей в клетки способности, и, таким образом, она будет удаляться от близости к активной части CaSR.
Для дальнейшего исследования связи структура-активность Ac-crrrrrr-NH2 (SEQ ID NO:6), некоторые из катионных (аргинин) остатков заменяли незаряженными (аланин) остатками. Было обнаружено, что замена катионных (аргинин) остатков в положениях субъединиц X2 и X4 приводила в результате к соединению (SEQ ID NO:15), которое заметно снижало активность in vitro по активации CaSR. Напротив, замена катионных (аргинин) остатков в положениях субъединиц X2 и X6 приводила в результате к соединению (SEQ ID NO:26), которое сохраняло большую часть активности, наблюдаемой для Ac-crrrrrr-NH2 (SEQ ID NO:6). Данные результаты предполагают, что положение заряженных остатков в соединении вносит свой вклад в активность и, в некоторых вариантах осуществления, может перевешивать вклад суммарного положительного заряда пептида. Также оказалось, что катионные (аргинин) остатки в определенных положениях, таких как положение субъединицы X5, делают непропорциональный вклад в активность.
Было обнаружено, что наличие N-концевого цистеина заметно усиливает активность пептидов относительно активации CaSR. CaSR представляет собой 7-трансмембранный сопряженный с G-белком рецептор с большим внеклеточным доменом, который функционирует как гомодимерный рецептор. Имеется 18 цистеиновых остатков во внеклеточном домене, некоторые из которых, как было показано полиморфизмом или анализом с введением мутаций, являются важными для рецепторной активности. Особенно следует отметить цистеины 129 и 131 области петли 2 внеклеточного домена. Считают, что цистеины 129 и 131 образуют внутримолекулярный дисульфидный мостик между двумя мономерами рецепторного комплекса, которые находятся в близкой или ингибированной конфигурации. Мутация цистеина 129 активирует CaSR, как делает ряд других мутаций, включая полную делецию области петли 2. Повышенная активность, придаваемая N-концевым цистеиновым остатком описанным соединениям, может быть результатом специфического взаимодействия с одним или более из цистеиновых остатков во внеклеточном домене CaSR.
Для дальнейшей оценки влияния хиральности аминокислотной замены на in vitro CaSR активность, был получен ряд аналогов Ac-crrrrrr-NH2 (SEQ ID NO:6), содержащих L-аминокислотную замену или замену ахиральной аминокислотой (глицин) в различных положениях, и их испытывали на активность относительно CaSR. Испытуемые аналоги включали (i) Ac-cGrrrGr-NH2 (SEQ ID NO:42), (ii) Ac-cArrrAr-NH2 (SEQ ID NO:43) и (iii) Ac-CaRrRaR-NH2 (SEQ ID NO:44). Все из вышеперечисленных аналогов обладали заметно более низкой активностью, чем Ac-crrrrrr-NH2 (SEQ ID NO:6), в диапазоне от 10-кратного отличия для SEQ ID NO:44 (самый активный из трех аналогов) и больше чем 2000-кратное отличие для SEQ ID NO:43 (наименее активный из трех аналогов). Для Ac-carrrar-NH2 (SEQ ID NO:26), где катионные D-аминокислотные остатки (D-аргининовые остатки) в положениях 2 и 6 SEQ ID NO:6 заменяли незаряженными D-аминокислотными остатками (D-аргининовыми остатками), изменение активности было гораздо меньшим (~3-кратное отличие). Таким образом, неожиданно было обнаружено, что прерывание всех D-аминокислотных остатков Ac-crrrrrr-NH2 (SEQ ID NO:6) двумя или более L-аминокислотными остатками приводило в результате к значительному снижению активности. Также неожиданным было то, что активность снижалась более чем 80-кратно, когда прерывающий остаток представлял собой незаряженный ахиральный аминокислотный остаток (глициновый остаток) по сравнению со случаем, когда он представлял собой незаряженный L-аминокислотный остаток (L-аланиновый остаток).
Также неожиданным было то, что замена двух незаряженных D-аминокислотных остатков (D-аланиновые остатки) Ac-carrrar-NH2 (SEQ ID NO:26) их L-аналогами (SEQ ID NO:43), приводила в результате к более чем 600-кратному снижению активности, тогда как замена их незаряженным ахиральным аминокислотным остатком (глициновый остаток) (SEQ ID NO:42) приводила в результате к менее чем 8-кратному снижению активности; и что замена трех катионных D-аминокислотных остатков (D-аргининовые остатки) Ac-carrrar-NH2 (SEQ ID NO:26) их L-аналогами (SEQ ID NO:44), приводила в результате к менее чем 4-кратному отличию активности.
Активность ряда пептидов и конъюгатов испытывали относительно человеческого CaSR. Данные исследования проводили измерением IP1 синтеза в HEK293-клетках, которые экспрессируют человеческий CaSR. EC50 величины показаны в таблице 5. Каждый пептид испытывали при восьми отличных концентрациях, в двух экземплярах, для получения кривой доза-реакция. Нанесение кривой осуществляли, применяя GraphPad Prism. В таблице 5 и во всем описании, остатки, показанные прописными буквами, являются L-аминокислотами, тогда как строчные буквы показывают D-аминокислоты. "Ac" показывает ацетильную кэпирующую группу, "NH2" показывает амидную кэпирующую группу, "Ac-bAla" представляет собой ацетилированный бета-аланин, "GSH" показывает восстановленный глутатион, "GS" показывает окисленный глутатион, "PEG" относится к полиэтиленгликолю, "PEG2" и "PEG5" относится к полиэтиленгликольным фрагментам 2 кДа и 5 кДа, соответственно, и "Mpa" относится к меркаптопропионовой кислоте. Группа, заключенная в круглые скобки, показывает то, что группа или фрагмент присоединен к боковой цепи предшествующей субъединицы или аминокислотного остатка.
EC
50
величины для катионных пептидов в CaSR in vitro анализе
(SEQ ID NO:47)
(SEQ ID NO:47)


В другом исследовании связи структуры с активностью, оценивали вклад некатионных аминокислот в активность пептидов получением ряда пептидов с различными D-аминокислотными остатками или глицином (таблица 6) или со стерически затрудненными неприродными аминокислотами (таблица 7), замещенными в различных положениях в пептиде Ac-carrrar-NH2 (SEQ ID NO:26) и в пептиде Ac-crrarar-NH2 (SEQ ID NO:153). Пептиды вводили в виде IV болюса нормальным крысам Спрага-Доули при дозе 0,5 мг/кг. Внутривенный (IV) болюс солевого раствора применяли в качестве контроля. Уровни PTH в плазме оценивали до введения дозы и через 1, 2, 3 и 4 часа после введения дозы. Результаты показаны в таблицах ниже, и они показывают, что: 1) небольшая аминокислота, такая как аланин, глицин или серин, является предпочтительной в положении 6 в Ac-carrrar-NH2 пептиде (SEQ ID NO:26), и 2) аланин в положении 2 в Ac-carrrar-NH2 (SEQ ID NO:26) является гораздо более позволительным для замены, и его можно заменить гидрофобными (например, D-Val, D-Leu), ароматическими (например, D-Phe) или полярными (например, D-Ser, D-Gln) природными аминокислотами, а также неприродными объемными гидрофобными аминокислотами (например, dNle, dNva), но не кислыми аминокислотами, и что 3) аланиновый остаток в положении 4 Ac-crrarar-NH2 (SEQ ID NO:25) пептида также является более пригодным для замены и может быть замещен большинством из типов природных аминокислот (а также неприродными объемными гидрофобными аминокислотами (например, dNle, dNva), но его нельзя замещать аминокислотами, которые влияют на вторичную структуру, а именно глицином или пролином или аминокислотами с кислыми боковыми цепями.
Активность примерных пептидных соединений
** PTH снижение после 0,5 мг/кг IV введения анестезированным изофлураном нормальным крысам - PTH измеряли через 1, 2, 3 и 4 часа после введения и рассчитывали суммарную AUC. PTH данные рассчитывали согласно следующей формуле: AUCвводимое соединение/AUCсолевой контроль*100.
Активность примерных пептидных соединений
** PTH снижение после 0,5 мг/кг IV введения анестезированным изофлураном нормальным крысам - PTH измеряли через 1, 2, 3 и 4 часа после введения и рассчитывали суммарную AUC. PTH данные рассчитывали согласно следующей формуле: AUCвводимое соединение/AUCсолевой контроль*100.
Активность примерных пептидных соединений

SEQ ID NO:26
SEQ ID NO:25
SEQ ID NO:26
** PTH снижение после 0,5 мг/кг IV введения анестезированным изофлураном нормальным крысам - PTH измеряли через 1, 2, 3 и 4 часа после введения и рассчитывали суммарную AUC. PTH данные рассчитывали согласно следующей формуле: AUCвводимое соединение/AUCсолевой контроль*100.
*** Соединение вводили с дозой 10 мг/кг (~молярный эквивалент 0,5 мг/кг непегилированного пептида)
**** Соединение вводили с дозой 20 мг/кг (~молярный эквивалент 0,5 мг/кг непегилированного пептида)
B. Исследование гистаминовой реакции и связи структура-активность
В литературе сообщается, что поликатионные соединения вызывают высвобождение активного биогенного амина гистамина. См. Church et al., J. Immunol., 128(5):2116-2121 (1982); Lagunoff et al., Ann. Rev. Pharmacol. Toxicol., 23:331-51 (1983). Считают, что высвобождение гистамина является результатом активации тучных клеток и базофилов, происходящей Gai зависящим способом. См. Aridor et al., J. Cell Biol., 111(3):909-17 (1990). Снижение или устранение данной физиологической реакции является желательным, в частности, для увеличения терапевтического интервала катионных пептидных кальциймиметиков для лечения SHPT.
Исследования проводили для оценки высвобождения гистамина, вызванного in vivo введением соединений, описанных в настоящем изобретении. В первом исследовании, описанном в примере 6, введение дозы IV болюсом или вливанием нормальным крысам Спрага-Доули применяли для оценки высвобождения гистамина, связанного с различными соединениями. Для оценки влияния суммарного положительного заряда на высвобождение гистамина, связанное с соединением, получали пептиды, содержащие 4-7 катионных (аргинин) остатков, и испытывали их на способность вызывать высвобождение гистамина in vivo, согласно способу, описанному в примере 6. Испытуемые пептиды включали (i) Ac-crrrr-NH2 (SEQ ID NO:4), (ii) Ac-crrrrr-NH2 (SEQ ID NO:5), (iii) Ac-crrrrrr-NH2 (SEQ ID NO:6) и (iv) Ac-crrrrrrrr-NH2 (SEQ ID NO:41).
Как показано на фигуре 3, когда эквивалентное число молей каждого пептида вводилось IV болюсом нормальным крысам, SEQ ID NO:41 (8 аргининовых остатков) показал наибольшую индукцию гистамина. Другие соединения с меньшим числом Arg остатков, включая SEQ ID NO:6 (6 аргининовых остатков), SEQ ID NO:5 (5 аргининовых остатков) и SEQ ID NO:4 (4 аргининовых остатка), также вызывали резкий рост уровня гистамина, но в меньшей степени, по сравнению с SEQ ID NO:41. SEQ ID NO:6, SEQ ID NO:5 и SEQ ID NO:4 вызывали более умеренную ответную реакцию с высвобождением гистамина (~2-3 кратную по сравнению с исходной). SEQ ID NO:5 и SEQ ID NO:4 были, однако, менее активными, чем SEQ ID NO:6 в отношении снижения PTH в плазме.
Поскольку PTH-снижающая активность Ac-crrrrrr-NH2 (SEQ ID NO:6) сопровождалась ослаблением гистаминовой реакции, дополнительные исследования проводили на основе Ac-crrrrrr-NH2 (SEQ ID NO:6) для того чтобы оценить, возможно ли еще дополнительно ослабить гистаминовую реакцию без потери в связи с этим PTH-снижающей активности. Как будет показано данными ниже, замену катионных (аргинин) остатков в Ac-crrrrrr-NH2 (SEQ ID NO:6) некатионными (аланин) остатками осуществляли для получения ряда аналогов с суммарным сниженным зарядом и пониженной плотностью заряда. Из данных аналогов, и Ac-cararrr-NH2 (SEQ ID NO:15) и Ac-carrrar-NH2 (SEQ ID NO:26) были связаны с отсутствием гистаминовой реакции при введении крысам IV болюсом. Важно, что данные два пептида сохраняли их активные кальциймиметические свойства и были способны снижать PTH секрецию и у нормальных крыс и у крыс с почечной дисфункцией.
Соединение Ac-crrrrrr-NH2, обозначенное как SEQ ID NO:6 (2,1 мкмоль/кг=2,3 мг/кг), вызывало наблюдаемую гистаминовую реакцию приблизительно в 2-3 раза большую исходной, по сравнению с 6-9-кратной для SEQ ID NO:41 при введении дозы IV болюсом (вводимый менее чем за 1 минуту) нормальным крысам. Высвобождение гистамина, вызванное Ac-crrrrrr-NH2 (SEQ ID NO:6), достигало пикового значения через 5 минут после введения дозы и возвращалось до исходной величины через 15 минут (фигура 3). Дальнейшее снижение числа заряженных субъединиц до 5 и 4 аргининовых остатков на пептид (SEQ ID NO:5 и SEQ ID NO:4, соответственно) дополнительно ослабляло гистаминовую реакцию по сравнению с более длинными олигоаргининовыми пептидами; однако 2-3-кратное увеличение концентрации гистамина по сравнению с исходным значением еще наблюдалось через 5 минут после IV болюсного введения дозы (фигура 3). Данные результаты предполагают связь между суммарным зарядом пептида и связанным с ним высвобождением гистамина. Отмечалось также, что богатые аргинином пептиды с числом остатков аргинина меньше 7 обладают достаточно ограниченной способностью проникать в клетки, предполагая, что проникновение в клетку не требуется, для того чтобы вызвать высвобождение гистамина.
Высвобождение гистамина, связанное с PTH-снижающими соединениями Ac-crrrrrr-NH2 (SEQ ID NO:6) и Ac-c(C)arrrar-NH2 (SEQ ID NO:3), оценивали in vivo. Соединение Ac-c(C)arrrar-NH2 (SEQ ID NO:3) имело следующую структуру:
 .
.
Данная конъюгатная структура обозначена в настоящем изобретении как Ac-c(C)arrrar-NH2 (SEQ ID NO:273, в которой L-Cys остаток, соединенный с тиолсодержащим остатком в X1 субъединице соединения (здесь, D-Cys остатком) через Cys-Cys дисульфидную связь, помещают в скобки в формуле. Данное обозначение применяют везде для обозначения того, что фрагмент в скобках соединен со второй тиолсодержащей группой. По сравнению с Ac-crrrrrr-NH2 (SEQ ID NO:6) соединение Ac-c(C)arrrar-NH2 (SEQ ID NO:3) содержит два катионных (аргинин) остатка, замещенных незаряженными (аланин) остатками в положениях субъединиц X2 и X6. Кроме того, D-Cys остатки в положении X1 конъюгируют с L-Cys остатком.
Данных два соединения вводились анестезированным изофлураном крысам (Спрага-Доули) при 3 мг/кг внутривенным (IV) болюсом (вводимым в течение менее чем 1 минуты). Кровь отбирали до введения дозы и через 5, 15 и 30 минут после введения дозы. Измеряли концентрацию гистамина, и кратное изменение концентрации гистамина в крови относительно концентрации гистамина в крови до введения дозы показано на фигуре 4. Соединение Ac-crrrrrr-NH2 (SEQ ID NO:6, пустые столбцы) вызывало гистаминовую реакцию, наблюдаемую в момент времени через 5 минут после введения дозы, где наблюдалось 7-кратное увеличение концентрации гистамина. Соединение Ac-c(C)arrrar-NH2 (SEQ ID NO:3, заштрихованные накрест столбцы) не вызвало явной гистаминовой реакции, как видно в моменты времени через 5, 10 и 15 минут после введения дозы, где уровень гистамина не увеличивался относительно уровня гистамина до введения дозы (нулевой момент времени).
Для дополнительной оценки связи между структурой соединения и высвобождением гистамина, получали ряд соединений и оценивали их способность вызывать индукцию гистамина в in vitro анализе, применяя перитонеальные тучные клетки крыс. В данном анализе соединения выдерживали при 10 мкМ в течение 15 минут при 37°C с клетками, выделенными из перитонеального лаважа SD крыс. После инкубирования собирали клеточную среду и определяли концентрацию гистамина. Данные показаны в таблице 9.
In vitro
индукция гистамина в перитонеальных тучных клетках крыс примерными пептидными соединениями
при 10 мкМ*
Сокращения: см. пример 7
Для дополнительной оценки связи между структурой соединения и высвобождением гистамина, получали ряд соединений и оценивали их способность вызывать индукцию гистамина в in vivo анализах. Данные показаны в таблице 10.
In vivo
индукция гистамина примерными пептидными соединениями
Соответственно, и как можно оценить ввиду PTH данных и данных о гистамине, описанных в настоящем изобретении выше, в одном варианте осуществления предусмотрено соединение, которое обладает снижающей концентрацию PTH активностью у субъекта в отсутствие гистаминовой реакции. В определенных вариантах осуществления отсутствие гистаминовой реакции подразумевает дозу соединения, которая оказывает менее чем 10-кратное, более предпочтительно 8-кратное, даже более предпочтительно 5-кратное, и даже еще более предпочтительно 3-кратное, увеличение концентрации гистамина, измеренной в in vitro анализе, как описано в настоящем изобретении, где кратное увеличение определяют на основе уровней гистамина до инкубирования с соединением и через 15 минут инкубирования с соединением. В конкретном варианте осуществления гистаминовую реакцию определяют в in vitro анализе, применяя перитонеальные тучные клетки крыс, выделенные из перитонеального лаважа нормальных крыс Спрага-Доули, и где определяют кратное изменение, исходя из концентрации гистамина перед инкубированием с соединением и после 15-минутного инкубирования с соединением. В исследованиях, проведенных в настоящем изобретении, in vitro оценку высвобождения гистамина проводили, применяя выделенные перитонеальные тучные клетки крыс, выделенные из перитонеального лаважа, используя холодный HBSS+25 мМ HEPES pH 7,4, содержащий гепарин (5 мкл/мл). Клетки промывали дважды буфером для стимуляции (HBSS+25 мМ HEPES pH 7,4) и инкубировали с 10 мкМ соединения в буфере для стимуляции (HBSS+25 мМ HEPES pH 7,4) в течение 15 минут в 96-луночном планшете (106/лунка) при 37°C. Клеточный супернатант анализировали на гистамин, применяя гистамин EIA набор (Cayman # 589651).
В другом варианте осуществления предполагается соединение, которое обладает снижающей PTH концентрацию активностью у субъекта в отсутствие клинической гистаминовой реакции. Как применяют в настоящем изобретении, отсутствие "клинической гистаминовой реакции" подразумевает, что терапевтически эффективное количество соединения, как описано в настоящем изобретении, вводят субъекту без продуцирования клинически неблагоприятного увеличения концентрации гистамина в плазме или крови, как измерено через 5-10 минут после завершения введения дозы или в процессе обработки. Например, когда соединение, требуемое для оказания требуемого терапевтического эффекта, вводят субъекту болюсом (как применяют в настоящем изобретении, "болюс" обозначает введение в течение минуты или менее), оно вызывает увеличение концентрации гистамина в плазме или крови через 5-10 минут после завершения введения дозы, которое является меньшим, чем 15-кратное, 10-кратное, 9-кратное, 8-кратное, 7-кратное, 6-кратное, 5-кратное, 4-кратное, 3-кратное, 2-кратное по сравнению с дозой до введения.
Как можно оценить из исследований, описанных выше, в одном варианте осуществления соединение содержит последовательность 3-35 аминокислотных остатков, в которой ряд субъединиц, являющихся положительно заряженными аминокислотными остатками, присутствует в последовательности. В некоторых вариантах осуществления описанные соединения содержат 5-25 субъединиц, и в предпочтительном варианте осуществления каждая субъединица представляет собой аминокислотный остаток. В других вариантах осуществления описанные соединения содержат 6-12 субъединиц. В еще других вариантах осуществления описанные соединения содержат 3-9 аминокислотных субъединиц. В альтернативных вариантах осуществления описанные соединения содержат 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34 или 35 субъединиц.
Субъединицы описанных соединений, в одном варианте осуществления, независимо выбирают из природных или неприродных аминокислот или их аналогов, и они могут иметь или L-, или D- конфигурацию (за исключением глицина, который является ахиральным). Глицин, алифатические остатки: аланин, валин, лейцин или изолейцин, пролин, содержащие гидроксильную группу остатки: серин и треонин, кислые остатки: аспарагиновая кислота и глутаминовая кислота, амидные остатки: аспарагин и глутамин, основные остатки: лизин и аргинин, гистидин, ароматические остатки: фенилаланин, тирозин и триптофан, и содержащие серу остатки: метионин и цистеин, все предусмотрены для применения в описанных соединениях. Число положительно заряженных субъединиц и их плотность могут влиять на снижающую PTH активность соединения. В некоторых вариантах осуществления положительно заряженные субъединицы разделены одной или более другими субъединицами ("разделяющие субъединицы"). В одном варианте осуществления разделяющие субъединицы представляют собой остатки аланина. В некоторых вариантах осуществления хиральность разделяющей субъединицы влияет на активность соединения.
Положительно заряженные аминокислотные остатки описанных соединений могут быть конкретными природными или неприродными остатками или их аналогом, имеющими или L- или D-конфигурацию (например, L-аргинин), которые повторяются в последовательности, или могут представлять собой ряд природных или неприродных остатков или их аналогов, имеющих или L-, или D-конфигурацию. В некоторых вариантах осуществления соединение представляет собой пептид, состоящий из 3-20 положительно заряженных аминокислотных остатков, 6-12 положительно заряженных аминокислотных остатков, 3-9 положительно заряженных аминокислотных остатков. В некоторых вариантах осуществления пептиды содержат 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 или 20 положительно заряженных аминокислотных остатков.
В некоторых вариантах осуществления положительно заряженные аминокислотные остатки независимо выбраны из природных аминокислот. В некоторых вариантах осуществления положительно заряженные аминокислотные остатки независимо выбраны из природных и/или неприродных аминокислот. В некоторых вариантах осуществления положительно заряженные аминокислотные остатки независимо выбраны из группы, состоящей из аргинина, лизина, гистидина, 2,3-диаминопропионовой кислоты (Dap), 2,4-диаминомасляной кислоты (Dab), орнитина и гомоаргинина. В предпочтительном варианте осуществления положительно заряженные аминокислотные остатки являются аргининовыми остатками.
В некоторых вариантах осуществления соединение представляет собой пептид и представляет собой одну непрерывную пептидную цепь. В других вариантах осуществления соединение представляет собой пептид, который является разветвленным. В еще других вариантах осуществления пептид конъюгирован с одним или более тиолсодержащими фрагментами (каждый, "тиолсодержащая конъюгирующая группа" или "конъюгирующая группа"). В предпочтительном варианте осуществления и в качестве просто иллюстрации, пептидное соединение конъюгировано с Cys конъюгирующей группой через (-S-S-) дисульфидную связь (например, -Cys-Cys-). Как применяют в настоящем изобретении, предполагается, что термин "соединение" включает и данные пептиды и данные конъюгаты.
Соединения обычно содержат одну или более тиольных фрагментов, предпочтительно один или более реакционноспособных тиольных фрагментов. Субъединицы, которые содержат тиольную группу, включают неаминокислотные соединения, содержащие тиольную группу, и аминокислоты с тиольной группой. Тиольная группа тиолсодержащей субъединицы может быть в конъгированной форме (например, через дисульфидную связь с конъюгирующей группой) или в неконъюгированной форме (т.е. в виде восстановленного тиола). В предпочтительном варианте осуществления, когда тиольная группа находится или в неконъюгированной или в конъюгированной форме, она способна образовывать дисульфидную связь с тиолсодержащей группой. Тиолсодержащий остаток может располагаться в любом положении вдоль цепи пептида, включая амино конец, карбокси конец, или какое-нибудь другое положение. В предпочтительном варианте осуществления тиолсодержащий остаток или субъединица может располагаться на амино конце. В других вариантах осуществления тиолсодержащий остаток или субъединица может располагаться на карбокси конце или внутри пептидной последовательности.
Некоторые типичные примеры тиолсодержащих остатков включают, без ограничения, цистеин, меркаптопропионовую кислоту, гомоцистеин и пеницилламин. Когда тиолсодержащий остаток содержит хиральный центр, он может иметь L- или D-конфигурацию. В предпочтительном варианте осуществления тиолсодержащий остаток представляет собой цистеин.
В некоторых вариантах осуществления поперечную связь между тиолсодержащей субъединицей в X1 положении в соединении и тиолсодержащей конъюгирующей группой можно расщеплять и/или обменивать на другие тиолсодержащие конъюгирующие группы, такие как цистеин (например, восстановлением дисульфидной связи) in vivo для получения биологически активной формы соединения. Таким образом, конъюгат может функционировать как пролекарство соединения. Конъюгирующую группу также можно применять для изменения физико-химических, фармакокинетических и/или фармакодинамических свойств описанных соединений (например, конъюгирование через дисульфидную связь с большим пегилированным фрагментом для улучшения фармакокинетических свойств).
В некоторых вариантах осуществления соединение представляет собой пептид, состоящий из аминокислотной последовательности (Xaa1)-(Xaa2)-(Xaa3)-(Xaa4)-(Xaa5)-(Xaa6)-(Xaa7) (SEQ ID NO:155), в которой (Xaa1) представляет собой тиолсодержащий аминокислотный остаток, (Xaa2) представляет собой некатионный аминокислотный остаток, (Xaa3) представляет собой любой аминокислотный остаток, (Xaa4) представляет собой любой аминокислотный остаток, (Xaa5) представляет собой катионный аминокислотный остаток, (Xaa6) представляет собой некатионный остаток, и (Xaa7) представляет собой любой аминокислотный остаток. Пептид можно модифицировать на N-конце, C-конце или обоих. В предпочтительном варианте осуществления пептид модифицируют и по N-концу, и по C-концу ацилированием и амидированием, соответственно.
В некоторых вариантах осуществления пептид содержит аминокислотную последовательность (D-Cys)-(Xaa2)-(Xaa3)-(Xaa4)-(Xaa5)-(Xaa6)-(Xaa7) (SEQ ID NO:156), в которой (Xaa2) представляет собой некатионный аминокислотный остаток, (Xaa3) представляет собой любой аминокислотный остаток, (Xaa4) представляет собой любой аминокислотный остаток, (Xaa5) выбран из группы, состоящей из D-Arg, L-Arg, D-Lys и L-Lys, (Xaa6) представляет собой некатионный остаток, и (Xaa7) представляет собой любой аминокислотный остаток. Пептид может содержать N-концевую кэпирующую группу, C-концевую кэпирующую группу или обе. В предпочтительном варианте осуществления пептид содержит и N-концевую кэпирующую группу, и C-концевую кэпирующую группу.
В некоторых вариантах осуществления пептид содержит аминокислотную последовательность (D-Cys)-(Xaa2)-(Xaa3)-(Xaa4)-(Xaa5)-(Xaa6)-(Xaa7) (SEQ ID NO:157), в которой (Xaa2), (Xaa3) и (Xaa4) независимо представляют собой любой аминокислотный остаток (но в предпочтительном варианте осуществления независимо выбраны из группы, состоящей из D-Ala, D-Val, D-Leu, D-NorVal и D-NorLeu), (Xaa5) и (Xaa7) независимо представляют собой любой катионный аминокислотный остаток (но в предпочтительном варианте осуществления независимо выбраны из группы, состоящей из D-Arg, L-Arg, D-Lys и L-Lys), (Xaa6) представляет собой некатионный аминокислотный остаток (в предпочтительном варианте осуществления выбран из группы, состоящей из D-Ala, D-Val, D-Leu, D-NorVal и D-NorLeu). Пептид может содержать N-концевую кэпирующую группу, C-концевую кэпирующую группу или обе. В предпочтительном варианте осуществления пептид содержит и N-концевую кэпирующую группу, и C-концевую кэпирующую группу.
В некоторых вариантах осуществления пептид содержит аминокислотную последовательность (D-Cys)-(Xaa2)-(Xaa3)-(Xaa4)-(Xaa5)-(Xaa6)-(Xaa7) (SEQ ID NO:158), в которой (Xaa2) представляет собой некатионный аминокислотный остаток, (Xaa3) представляет собой любой аминокислотный остаток, (Xaa4) представляет собой любой аминокислотный остаток, (Xaa5) выбрана из группы, состоящей из D-Arg, L-Arg, D-Lys и L-Lys, (Xaa6) представляет собой некатионный остаток, и (Xaa7) представляет собой любой аминокислотный остаток. Пептид может содержать N-концевую кэпирующую группу, C-концевую кэпирующую группу или обе. В предпочтительном варианте осуществления пептид содержит и N-концевую кэпирующую группу, и C-концевую кэпирующую группу.
В некоторых вариантах осуществления пептид содержит аминокислотную последовательность (D-Cys)-(D-Ala)-(Xaa3)-(Xaa4)- (D-Arg)-(D-Ala)-(Xaa7) (SEQ ID NO:159), в которой (Xaa3) представляет собой любой катионный аминокислотный остаток, (Xaa4) представляет собой любой катионный аминокислотный остаток, и (Xaa7) представляет собой любой катионный аминокислотный остаток. Пептид может содержать N-концевую кэпирующую группу, C-концевую кэпирующую группу или обе. В предпочтительном варианте осуществления пептид содержит и N-концевую кэпирующую группу, и C-концевую кэпирующую группу.
В некоторых вариантах осуществления пептид содержит аминокислотную последовательность (D-Cys)-(Xaa2)-(Xaa3)-(D-Ala)-(D-Arg)-(D-Ala)-(Xaa7) (SEQ ID NO:160), в которой (Xaa2), (Xaa3) и (Xaa7) независимо представляют собой любой катионный аминокислотный остаток. Пептид может содержать N-концевую кэпирующую группу, C-концевую кэпирующую группу или обе. В предпочтительном варианте осуществления пептид содержит и N-концевую кэпирующую группу, и C-концевую кэпирующую группу.
Другой вариант осуществления представляет собой кальциймиметический пептид, содержащий последовательность аминокислот, соединенных пептидными связями, в котором последовательность содержит 5-10 аминокислотных остатков, и в котором последовательность содержит амино конец, карбокси конец, по меньшей мере, один тиолсодержащий остаток, и от 3 до 9 положительно заряженных остатков. В одном варианте осуществления по меньшей мере один тиолсодержащий остаток представляет собой цистеиновый остаток. В другом аспекте цистеиновый остаток расположен на амино конце пептида. В определенном варианте осуществления цистеиновый остаток представляет собой L-Cys остаток, D-Cys остаток или L- или D-гомоCys остаток. В других вариантах осуществления аминокислотные остатки пептида представляют собой D-аминокислоты или L-аминокислоты.
Также включенными в объем заявленных соединений являются пептидомиметичекие молекулы, которые содержат приблизительно семь субъединиц, в которых, по меньшей мере, одна субъединица содержит тиольный фрагмент, предпочтительно реакционноспособный тиольный фрагмент, и другие субъединицы представляют собой ряд некатионных субъединиц, и от 1 до 4 положительно заряженных субъединиц. Данные пептидомиметические молекулы могут содержать непептидные связи между двумя или более субъединиц. Различные признаки соединений, обсуждаемые выше, в общем применимы к пептидомиметическим молекулам. Например, как обсуждалось выше, субъединицы, применяемые для получения молекул, могут представлять собой встречающиеся в природе аминокислоты или остатки с неприродными боковыми цепями. В концы конструкций можно вводить кэпирующие группы или оставлять некэпированными способом, обсуждаемым выше. Аналогично, аминокислотные остатки молекулы могут представлять собой L- или D-аминокислотные остатки. Так же, как обсуждается выше, тиолсодержащие остатки могут быть в восстановленной или окисленной форме с любым из тиолсодержащих фрагментов, обсуждаемых выше.
Многие пептидомиметические остовы и способы их получения разработаны (Babine, R. E.; Bender, S. L., Chem. Rev., 97:1359, 1997; Hanessian, S.; et al., Tetrahedron, 53:12789, 1997; Fletcher, M. D.; Cambeli, M. C., Chem. Rev., 98:763, 1998); Peptidomimetics Protocols; Kazmierski W. M., Ed.; Methods in Molecular Medicine Series, Vol. 23; Humana Press, Inc.; Totowa, NJ. (1999).
Конъюгаты
В некоторых вариантах осуществления соединение химически соединяют поперечной сшивкой с тиолсодержащей конъюгирующей группой через дисульфидную связь между тиолом соединения и тиолом конъюгирующей группы. Тиолсодержащая конъюгирующая группа может представлять собой небольшую молекулу, такую как цистеин, или макромолекулу, такую как полипептид, содержащий цистеиновый остаток. Примеры подходящих тиолсодержащих конъюгирующих групп включают цистеин, глутатион, тиоалкил, молекулы, такие как тиобензил, меркаптопропионовая кислота, N-ацетилированный цистеин, цистамид, N-ацетилцистамид, гомоцистеин, пеницилламин и модифицированные поли(этиленгликолем) (PEG) (называемые "пегилированными") тиолы, такие как пегилированный цистеин или копия соединения (т.е. для образования гомодимера, соединенного дисульфидной связью). В предпочтительном варианте осуществления тиолсодержащая конъюгирующая группа представляет собой цистеин. Другие цистеиновые гомологи также предусматриваются для применения в качестве тиолсодержащих конъюгирующих групп, или отдельно или содержащиеся в большей конъюгирующей группе. Аналогично, стереоизомеры цистеина, гомоцистеина и цистамида являются пригодными для применения в качестве тиолсодержащих фрагментов. Конъюгирующие группы можно применять для увеличения химической стабильности и, следовательно, длительности хранения фармацевтического продукта. В определенных вариантах осуществления тиолсодержащая конъюгирующая группа и пептид являются одинаковыми (т.е. конъюгат представляет собой димер), который неожиданно показал очень высокую химическую стабильность по сравнению с гетерологичной конъюгирующей группой, такой как цистеин. Не будучи связанными теорией, предполагают, что когда тиолсодержащая конъюгирующая группа и пептид являются одинаковыми, любая мутация (например, скремблирование конъюгирующей группы) будет преобразовывать исходное димерное соединение. Напротив, изменение соединения с применением гетерологичной конъюгирующей группы, такой как цистеин, может приводить к образованию гомодимеров пептида плюс цистеин (цистеин-цистеиновый гомодимер) плюс остаток исходного соединения. Гомодимерный пептид (т.е. конъюгирующая группа и пептид являются одинаковыми) будет превращаться в цистеин-конъюгированную форму пептида in vivo из-за высокой концентрации восстановленного цистеина в системном кровотоке.
В некоторых вариантах осуществления идеи включают дисульфидный конъюгат тиолсодержащей конъюгирующей группы и пептида, содержащего аминокислотную последовательность (Xaa1)-(Xaa2)-(Xaa3)-(Xaa4)-(Xaa5)-(Xaa6)-(Xaa7) (SEQ ID NO:155), в которой (Xaa1) представляет собой аминокислотный остаток c тиолсодержащим фрагментом, (Xaa2) представляет собой некатионный аминокислотный остаток, (Xaa3) представляет собой любой аминокислотный остаток, (Xaa4) представляет собой любой аминокислотный остаток, (Xaa5) представляет собой катионный аминокислотный остаток, (Xaa6) представляет собой некатионный остаток, и (Xaa7) представляет собой любой аминокислотный остаток. Пептид может содержать N-концевую кэпирующую группу, C-концевую кэпирующую группу или обе. В предпочтительном варианте осуществления пептид содержит и N-концевую кэпирующую группу, и C-концевую кэпирующую группу. В предпочтительном варианте осуществления тиолсодержащая конъюгирующая группа выбрана из группы, состоящей из D-Cys, L-Cys, пептида, содержащего D-Cys, и пептида, содержащего L-Cys. Когда тиолсодержащая конъюгирующая группа представляет собой аминокислоту или пептид, она может содержать N-концевую кэпирующую группу, C-концевую кэпирующую группу или обе. В предпочтительном варианте осуществления тиолсодержащая конъюгирующая группа содержит и N-концевую кэпирующую группу, и C-концевую кэпирующую группу. В некоторых вариантах осуществления тиолсодержащая конъюгирующая группа сама представляет собой пептид, содержащий аминокислотную последовательность SEQ ID NO:155. В некоторых вариантах осуществления тиолсодержащая конъюгирующая группа и пептид являются одинаковыми (т.е. конъюгат представляет собой димер).
В некоторых вариантах осуществления идеи включают конъюгат тиолсодержащей конъюгирующей группы и пептида, содержащего аминокислотную последовательность (D-Cys)-(Xaa2)-(Xaa3)-(Xaa4)-(Xaa5)-(Xaa6)-(Xaa7) (SEQ ID NO:156), в которой (Xaa2) представляет собой некатионный аминокислотный остаток, (Xaa3) представляет собой любой аминокислотный остаток, (Xaa4) представляет собой любой аминокислотный остаток, (Xaa5) выбрана из группы, состоящей из D-Arg, L-Arg, D-Lys и L-Lys, (Xaa6) представляет собой некатионный остаток, и (Xaa7) представляет собой любой аминокислотный остаток. Пептид может содержать N-концевую кэпирующую группу, C-концевую кэпирующую группу или обе. В предпочтительном варианте осуществления пептид содержит и N-концевую кэпирующую группу, и C-концевую кэпирующую группу. В предпочтительном варианте осуществления тиолсодержащая конъюгирующая группа выбрана из группы, состоящей из D-Cys, L-Cys, пептида, содержащего D-Cys, и пептида, содержащего L-Cys. Когда тиолсодержащая конъюгирующая группа представляет собой аминокислоту или пептид, она может содержать N-концевую кэпирующую группу, C-концевую кэпирующую группу или обе. В предпочтительном варианте осуществления, тиолсодержащая конъюгирующая группа содержит и N-концевую кэпирующую группу, и C-концевую кэпирующую группу. В некоторых вариантах осуществления тиолсодержащая конъюгирующая группа сама представляет собой пептид, содержащий аминокислотную последовательность SEQ ID NO:156. В некоторых вариантах осуществления тиолсодержащая конъюгирующая группа и пептид являются одинаковыми (т.е. конъюгат представляет собой димер).
В некоторых вариантах осуществления идеи включают конъюгат тиолсодержащей конъюгирующей группы и пептида, содержащего аминокислотную последовательность (L-Cys)-(Xaa2)-(Xaa3)-(Xaa4)-(Xaa5)-(Xaa6)-(Xaa7) (SEQ ID NO:183), в которой (Xaa2) представляет собой некатионный аминокислотный остаток, (Xaa3) представляет собой любой аминокислотный остаток, (Xaa4) представляет собой любой аминокислотный остаток, (Xaa5) выбрана из группы, состоящей из D-Arg, L-Arg, D-Lys и L-Lys, (Xaa6) представляет собой некатионный остаток, и (Xaa7) представляет собой любой аминокислотный остаток. Пептид может содержать N-концевую кэпирующую группу, C-концевую кэпирующую группу или обе. В предпочтительном варианте осуществления пептид содержит и N-концевую кэпирующую группу, и C-концевую кэпирующую группу. В предпочтительном варианте осуществления тиолсодержащая конъюгирующая группа выбрана из группы, состоящей из D-Cys, L-Cys, пептида, содержащего D-Cys, и пептида, содержащего L-Cys. Когда тиолсодержащая конъюгирующая группа представляет собой аминокислоту или пептид, она может содержать N-концевую кэпирующую группу, C-концевую кэпирующую группу или обе. В предпочтительном варианте осуществления тиолсодержащая конъюгирующая группа содержит и N-концевую кэпирующую группу, и C-концевую кэпирующую группу. В некоторых вариантах осуществления тиолсодержащая конъюгирующая группа сама представляет собой пептид, содержащий аминокислотную последовательность SEQ ID NO:183. В некоторых вариантах осуществления тиолсодержащая конъюгирующая группа и пептид являются одинаковыми (т.е. конъюгат представляет собой димер).
В некоторых вариантах осуществления идеи включают конъюгат тиолсодержащей конъюгирующей группы и пептида, содержащего аминокислотную последовательность (D-Cys)-(D-Ala)-(Xaa3)-(Xaa4)-(D-Arg)-(D-Ala)-(Xaa7) (SEQ ID NO:161), в которой (Xaa3) представляет собой любой аминокислотный остаток, (Xaa4) представляет собой любой аминокислотный остаток, и (Xaa7) представляет собой любой аминокислотный остаток. Пептид может содержать N-концевую кэпирующую группу, C-концевую кэпирующую группу или обе. В предпочтительном варианте осуществления пептид содержит и N-концевую кэпирующую группу, и C-концевую кэпирующую группу. В предпочтительном варианте осуществления тиолсодержащая конъюгирующая группа выбрана из группы, состоящей из D-Cys, L-Cys, пептида, содержащего D-Cys, и пептида, содержащего L-Cys. Когда тиолсодержащая конъюгирующая группа представляет собой аминокислоту или пептид, она может содержать N-концевую кэпирующую группу, C-концевую кэпирующую группу или обе. В предпочтительном варианте осуществления тиолсодержащий конъюгирующая группа содержит и N-концевую кэпирующую группу, и C-концевую кэпирующую группу. В некоторых вариантах осуществления тиолсодержащая конъюгирующая группа сама представляет собой пептид, содержащий аминокислотную последовательность SEQ ID NO:161. В некоторых вариантах осуществления тиолсодержащая конъюгирующая группа и пептид являются одинаковыми (т.е. конъюгат представляет собой димер).
III. Способы применения
В одном аспекте, предусмотрены способы предотвращения, лечения или облегчения гиперпаратиреоза, заболевания костей и/или других гиперкальциемических заболеваний введением соединений, описанных в настоящем изобретении. Как показано выше, соединения обладают снижающей уровни PTH и/или кальция активностью в ткани-мишени или тканях, или у субъекта. В определенных вариантах осуществления описанные соединения способны снижать уровни PTH и/или кальция, когда терапевтически эффективное количество соединения вводят нуждающемуся в данном лечении субъекту. Способы применения будут описаны здесь со ссылкой на примеры 3 и 8-11.
Снова ссылаясь на пример 3, и как обсуждается выше в отношении таблицы 1, ряд соединений, в которых катионный (аргинин) остаток последовательно заменяли некатионным остатком (аланин), вводили крысам. Фигура 5 показывает временную зависимость способности каждого соединения снижать концентрацию PTH в плазме и продолжительность действия различных соединений. На фигуре 5 соединения Ac-crrrrrr-NH2 (SEQ ID NO:6, ромбы), Ac-carrrrr-NHz (SEQ ID NO:8, квадраты) и Ac-crrarrr-NH2 (SEQ ID NO:10, x символы) и Ac-crrrrar-NH2 (SEQ ID NO:12, окружности) были активными in vivo, что подтверждено снижением процентного PTH от исходного значения до введения дозы практически до нуля, и они обеспечивали продолжительной активностью, где концентрация PTH в крови сохранялась пониженной, по меньшей мере, в течение четырех часов. Соединения Ac-crarrrr-NH2 (SEQ ID NO:9, треугольники), Ac-crrrarr-NH2 (SEQ ID NO:11, * символы) и Ac-crrrrra-NH2 (SEQ ID NO:13, + символы) снижали процентное PTH относительно исходного в течение приблизительно 2-3 часов, и после этого концентрация в крови PTH начинала расти. Замена катионного (аргинин) остатка в положениях субъединиц 5 или 7 Ac-crrrrrr-NH2 (SEQ ID NO:6) оказывала влияние на продолжительность PTH-снижающей активности.
Также оценивали графики PTH снижения для ряда соединений, содержащих две аминокислотных замены. Отобранные соединения, приведенные в таблице 2 выше, вводили нормальным крысам IV болюсом в дозе 0,5 мг/кг и оценивали снижение PTH относительно концентрации PTH в крови до введения дозы. Данные показаны на фигурах 6A-6B, где соединение обозначены следующим образом: Ac-carrrar-NH2 (SEQ ID NO:26, пустые ромбы), Ac-crrarar-NH2 (SEQ ID NO:25, пустые квадраты), Ac-caarrrr-NH2 (SEQ ID NO:22, треугольники), Ac-crraarr-NH2 (SEQ ID NO:17, окружности), Ac-c(C)arrrar-NH2 (SEQ ID NO:3, ромбы, фигура 6B), Ac-c(C)rrarar-NH2 (SEQ ID NO:28, x символы, фигура 6B).
Проводили другое исследование для оценки активности соединения Ac-c(C)arrrar-NH2 (SEQ ID NO:3). Соединение вводили внутривенно нормальным крысам, как подробно описано в примере 2, с дозами 1 мг/кг, 0,5 мг/кг, 0,3 мг/кг и 0,1 мг/кг. Уровни PTH в плазме оценивали до введения дозы и через 4 часа после введения. Фигура 7 показывает результаты, в которых PTH концентрация в крови показана как процент от исходной величины до введения дозы. Зависящее от дозы PTH снижение, наблюдаемое после единичного IV болюсного введения с самой большой дозой 1 мг/кг (ромбы), было самым большим снижением PTH, затем 0,5 мг/кг (квадраты), 0,3 мг/кг (треугольники) и 0,1 мг/кг (x символы). Солевой контроль показан окружностями. Как видно, пептид, при введении с терапевтически эффективной дозой, достигает снижения PTH большее, чем 50% относительно концентрации PTH до введения дозы ("исходная величина"). Конкретно, пептид, при введении с дозами большими, чем 0,1 мг/кг, снижает PTH концентрацию до менее чем 90% от исходной PTH концентрации через 1 час после IV введения. Данные дозы пептида, обозначенного как SEQ ID NO:3, также достигали площади под кривой (AUC) менее чем 50%, причем AUC рассчитана как сумма величин PTH концентраций в моменты времени 1, 2, 3 и 4 часа, нормализованная AUC для солевого контроля для тех же моментов времени, умноженная на 100.
То же соединение также испытывали на субъектах (крысах) с почечной недостаточностью. В данном исследовании 1K1C модель острой почечной недостаточности применяли для определения PTH-снижающей активности соединения Ac-c(C)arrrar-NH2 (SEQ ID NO:3) в условиях почечной недостаточности. Модель описана в примере 1A. Соединение вводили внутривенно в виде болюса животным (крысам) с почечными отклонениями с дозами 3 мг/кг (n=2), 1 мг/кг (n=5), 0,5 мг/кг (n=6) и 0,3 мг/кг (n=5). Контрольной группе животных вводили солевой раствор. Уровни PTH в плазме оценивали до введения дозы и в течение нескольких часов после введения. Фигура 8 показывает результаты, где обработанные солевым раствором животные (квадраты) обладали повышенной PTH концентрацией по сравнению с исходной PTH концентрацией. При различных дозах SEQ ID NO:3 наблюдали зависимое от дозы влияние на продолжительность и степень PTH снижения. Животные, обработанные самой небольшой дозой 0,3 мг/кг (x символы), показали снижение PTH в самый ранний момент времени и увеличение PTH между 1-24 часами после введения дозы. Дозы 3 мг/кг (ромбы), 1 мг/кг (треугольники) и 0,5 мг/кг (квадраты) обеспечивали пониженной концентрацией PTH в крови в течение более чем 15 часов, и для максимальной дозы - в течение более чем 24 часа.
В другом исследовании для оценки влияния замены катионных субъединиц на незаряженные субъединицы, например, аланиновыми аминокислотными остатками, в случае субъекта с почечной недостаточностью получали аналог Ac-crrrrrr-NHz (SEQ ID NO:6) и исследовали его на способность снижать PTH в 1K1C животной модели после 1 мг/кг единичного внутривенного введение. В испытуемом аналоге Ac-carrrar-NH2 (SEQ ID NO:26) катионные субъединицы в положениях X2 и X6 Ac-crrrrrr-NH2 (SEQ ID NO:6) заменяли незаряженными аминокислотами.
Как показано на фигуре 9, Ac-carrrar-NH2 (SEQ ID NO:26, пустые квадраты) обладала активностью, которая является эквивалентной Ac-crrrrrr-NH2 (SEQ ID NO:6, пустые ромбы) при испытуемой дозе (1 мг/кг) с аналогичной повышенной продолжительностью действия в течение 24 часов. Было найдено, что аналог Ac-crrrrrr-NH2 (SEQ ID NO:6) c заменой незаряженной субъединицей сохраняет активность и может, действительно, обладать in vivo активностью и продолжительностью действия, превышающими эти показатели для соединения, обозначенного как SEQ ID NO:6. В соединении Ac-carrrar-NH2 (SEQ ID NO:26) D-Arg остатки в положениях X2 и X6 заменяли D-Ala остатками по сравнению с соединением, обозначенным как SEQ ID NO:6.
Что особенно важно, как обсуждалось выше, введение соединения Ac-carrrar-NH2 (SEQ ID NO:26) не сопровождалось высвобождением гистамина, нежелательным побочным эффектом, который наблюдался для Ac-crrrrrr-NH2 (SEQ ID NO:6) и других аналогичных соединений при введении с большей дозой (>1 мг/кг) IV болюсом. Отмеченное ослабление высвобождения гистамина соединением, обозначенным SEQ ID NO:26, увеличивало терапевтический интервал между требуемой PTH-снижающей активностью и нежелательной гистамин-индуцирующей активностью после введения IV болюсом. Соответственно, в предпочтительном варианте осуществления обеспечивали соединения, обладающие снижающей in vivo концентрацию PTH активностью в отсутствие гистаминовой реакции. Соответственно, в одном варианте осуществления обеспечивали соединение, которое обладает снижающей PTH активностью, где соединение при введении субъекту, человеку или животному снижало PTH-концентрацию ниже 50% от концентрации до введения дозы в пределах одного часа после введения дозы. В конкретном варианте осуществления соединение, которое обладает заметной снижающей PTH активностью, подразумевает соединение, которое при введении нормальной крысе снижает PTH концентрацию ниже 50% от концентрации до введения в пределах одного часа после введения дозы IV болюсом.
В другом исследовании, подробно описанном в примере 8, соединения представляют собой конъюгат, в котором тиолсодержащая субъединица в положении X1 соединена через дисульфидную связь с L-Cys остатком. Данные соединения имеют следующие структуры:

В обозначениях, применяемых в настоящем изобретении, соединение, которое соединено с тиолсодержащим фрагментом в X1 субъединице, показано в скобках, где в данных типичных конъюгатах соединение L-Cys, показанное (C), соединено с тиолсодержащим фрагментом в X1 субъединице: Ac-c(C)arrrar-NH2 (SEQ ID NO:3) и Ac-c(Ac-C)arrrar-NH2 (SEQ ID NO:141). Данные соединения вводились IV болюсом животным с острой почечной недостаточностью (1K1C модель) с дозами 0,3 и 0,5 мг/кг, и результаты показаны на фигуре 10. Соединение Ac-c(C)arrrar-NH2 (SEQ ID NO:3) представлено квадратами (0,3 мг/кг, n=5) и * символами (0,5 мг/кг, n=6), и соединение Ac-c(Ac-C)arrrar-NH2 (SEQ ID NO:141) представлено треугольниками (0,3 мг/кг, n=8) и ромбами (0,5 мг/кг, n=7). Данный in vivo эффект дозы SEQ ID NO:3 показывает зависимое от дозы снижение PTH, очень схожее со снижением для Ac-crrrrrr-NH2 (SEQ ID NO:6).
В некоторых из in vivo исследований, описанных в настоящем изобретении, соединения, включая соединения в конъюгированной форме, где тиол в X1 субъединице соединен поперечной сшивкой через дисульфидную связь с другой субъединицей, вводили в виде 30-минутного IV вливания. Однако следует отметить, что менее продолжительные вливания (например, <5 минут) или доставка IV болюсом обычно оказывали сравнимое фармакодинамическое снижение PTH как и более продолжительное 30-минутное вливание. Также оказалось, что подкожное болюсное введение является эффективным путем доставки, которое обеспечивает меньшим первоначальным снижением PTH, но обладает длительным снижением PTH, аналогично профилю, наблюдаемому для IV пути введения. Как показано на фигуре 11, соединение Ac-crrrrrr-NH2 (SEQ ID NO:6) также вводили облегченной микропорацией (например, микропорация рогового слоя) трансдермальной доставкой, и оно показало отслеживаемое в течение нескольких часов снижение PTH в плазме. Соединение Ac-crrrrrr-NH2 (SEQ ID NO:6) также вводили трансдермальным путем после микропорации, что приводило в результате к снижению PTH в плазме в течение нескольких часов. Трансдермальная доставка Ac-crrrrrr-NH2 (SEQ ID NO:6) обеспечивала дополнительным вариантом клинической доставки описанных соединений.
Для оценки влияния пути введения на активность Ac-crrrrrr-NH2 (SEQ ID NO:6) в случае субъекта с почечной недостаточностью, крысам в 1K1C модели давали 1 мг/кг пептида в виде или подкожного (SC) болюса, или 30-минутного IV вливания. Оба пути введения эффективно снижали концентрацию PTH в плазме в течение более 24 часов. Когда Ac-crrrrrr-NH2 (SEQ ID NO:6) доставляли IV вливанием, PTH концентрация быстро падала на 80-90% от исходной концентрации. Через 16 часов после введения дозы PTH концентрация начинала повышаться, хотя она все еще была снижена до ~80% от исходной концентрации. Когда Ac-crrrrrr-NH2 (SEQ ID NO:6) доставляли SC болюсом, PTH концентрация показывала более умеренное первоначальное падение до ~40% от исходной концентрации, но показывала аналогичную продолжительность снижения как при доставке пептида IV путем. Через двадцать четыре часа после введения дозы, PTH концентрация у животных, которым была введена доза любым из двух путей, частично восстанавливалась, хотя оба варианта еще показывали пониженную PTH концентрацию, которая составляла ~40-60% от исходной концентрации. Результаты показывали, что данный путь введения обеспечивает аналогичным профилем относительно эффективности и продолжительности PTH снижения как IV введение, таким образом, обеспечивая альтернативным путем для клинического введения дозы (данные не показаны).
Соответственно, в предпочтительном варианте осуществления, субъекта, обладающего вторичным гиперпаратиреозом (SHPT), обрабатывали, применяя описанные соединения для снижения уровней PTH и/или кальция в плазме. Необработанные SHPT пациенты с умеренно тяжелым гиперпаратиреозом часто обладают исходной интактной циркулирующей концентрацией PTH >300 пг/мл, и концентрацией, которая может превышать 600 пг/мл. В предпочтительном варианте осуществления снижение PTH концентрации измеряют как снижение интактной концентрации PTH ниже исходной концентрации до обработки. В другом варианте осуществления требуемое снижение PTH является таким, чтобы довести PTH концентрацию в плазме до общепризнанной нормы, установленной Национальным Почечным Фондом или другими экспертами по лечению заболеваний почек и почечной недостаточности.
В другом аспекте обеспечивают способами лечения гиперпаратиреоза, гиперкальциемии и/или болезни кости, включающими введение терапевтически эффективного количества описанного соединения. В другом варианте осуществления субъекта можно обрабатывать описанным соединением в комбинации с одним или более другими терапевтически эффективными агентами.
В другом аспекте описанное соединение вводят в количестве, эффективном для снижения концентрации PTH или влияния на концентрацию PTH. В некоторых вариантах осуществления снижение PTH в плазме составляет, по меньшей мере, на 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 11%, 12%, 13%, 14%, 15%, 16%, 17%, 18%, 19%, 20%, 25% или 30% ниже исходной концентрации до обработки в течение, по меньшей мере, 10 часов после введения описанного соединения. В конкретных вариантах осуществления снижение PTH в плазме составляет, по меньшей мере, 20% через 10 часов после введения. В предпочтительных вариантах осуществления снижение PTH в плазме составляет от 15 до 40%, предпочтительно 20-50%, более предпочтительно 30-70% ниже исходной концентрации до обработки в течение, по меньшей мере, 48 часов после введения описанного соединения.
В другом аспекте описанное соединение вводят в количестве, эффективном для снижения концентрации кальция в сыворотке или влияния на концентрацию кальция. В некоторых вариантах осуществления снижение концентрации кальция в сыворотке составляет, по меньшей мере, на 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 11%, 12%, 13%, 14%, 15%, 16%, 17%, 18%, 19%, 20% или 25% ниже концентрации до обработки в течение, по меньшей мере, 10 часов после введения поликатионного пептида. В некоторых предпочтительных вариантах осуществления снижение концентрации кальция в сыворотке составляет, по меньшей мере, 5% через 10 часов после введения. В некоторых предпочтительных вариантах осуществления снижение концентрации кальция в сыворотке составляет 5-10%, предпочтительно 5-20% ниже концентрации до обработки в течение, по меньшей мере, 48 часов после введения описанного соединения.
В другом аспекте обеспечивают способом лечения гиперпаратиреоза и/или гиперкальциемии у нуждающегося в данном лечении субъекта, включающим введение терапевтически эффективного количества описанного соединения, посредством чего снижают концентрации PTH и/или кальция.
На основе связи между концентрацией кальция в сыворотке, обмена веществ в костной ткани и PTH, считают, что описанные соединения являются полезными для лечения различных форм болезни кости и/или гиперкальциемии в добавление к гиперпаратиреозу. Описанные соединения могут быть предпочтительными по сравнению с современными терапевтическими агентами, поскольку их можно вводить парентерально, и они могут быть не связаны с желудочно-кишечными побочными эффектами, не подвергаться метаболизму цитохромом P450 и могут приводить в результате к более эффективному снижению концентрации PTH и кальция в плазме.
Как обсуждается выше, описанные способы можно применять отдельно или в комбинации с одним или более другими терапевтически эффективными агентами. Данные другие терапевтически эффективные агенты включают, но не ограничиваются ими, обработку антирезорбтивными бисфосфонатными средствами, такими как алендронат и ризедронат; блокаторами интегрина, такими как αvβ3 антагонисты; конъюгированными эстрогенами, применяемыми в гормонозаместительной терапии, такими как PREMPRO™, PREMARIN™ и ENDOMETRION™; селективными модуляторами рецепторов эстрогена (SERM), такими как ралоксифен, дролоксифен, CP-336156 (Pfizer) и лазофоксифен; ингибиторами катепсина K; терапией с витамином D; аналогами витамина D, такими как ZEMPLAR™ (парикальцитол); CALCIJEX® (кальцитриол), HECTOROL® (доксеркальциферол), ONE-ALPHA® (альфакальцидол) и аналогами, находящимися на стадии разработки из цитохрома, известными как CTA-018, CTAP201 и CTAP101; другими кальциймиметиками, такими как Sensipar® (цинакальцет); ингибиторами семейства натрий-зависимых переносчиков фосфата II типа, SLC34 (включая две почечные изоформы NaPi-IIa и NaPi-IIc и интестинальный NaPi-IIb переносчик); фосфатонинами (включая FGF-23, sFRP4, MEPE или FGF-7); обработку низкими дозами PTH (с или без эстрогена); кальцитонином; ингибиторами RANK лиганда; антителами к RANK лиганду, остеопротегерином; антагонистами аденозина; и ингибиторами ATP протонного насоса.
В одном варианте осуществления описанное соединение вводят с дозой, достаточной для снижения и концентрации PTH, и концентрации кальция в сыворотке. В другом варианте осуществления описанное соединение вводят с дозой, достаточной для снижения концентрации PTH без заметного влияния на концентрацию кальция в сыворотке. В следующем варианте осуществления описанное соединение вводят с дозой, достаточной для снижения концентрации PTH без заметного влияния на концентрацию кальция в сыворотке.
Составы
Обеспечивают фармацевтической композицией, содержащей описанное соединение и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество или носитель. Способы получения данных фармацевтических композиций обычно включают стадию получения смеси описанного соединения с носителем и, необязательно, одним или более вспомогательными ингредиентами. Описанные соединения и/или содержащие их фармацевтические композиции можно формулировать в фармацевтически приемлемых лекарственных формах общепринятыми способами, известными специалистам в данной области техники. Обычно составы получают равномерным тщательным смешиванием описанного соединения с жидкими носителями или мелкоизмельченными твердыми носителями, или обоими, и затем, при необходимости, приданием формы для получения продукта.
Фармацевтические композиции по настоящему изобретению, подходящие для парентерального введения, содержат одно или более описанных соединений в комбинации с одним или более фармацевтически приемлемыми стерильными изотоническими водными или неводными растворами, дисперсиями, суспензиями или эмульсиями или стерильными порошками, которые можно растворять для получения стерильных инъецируемых растворов или дисперсий непосредственно перед применением, которые могут содержать сахара, спирты, аминокислоты, антиоксиданты, буферы, бактериостаты, растворы, которые делают состав изотоничным крови предполагаемого реципиента, или суспендирующие агенты или загустители.
Примеры подходящих водных и неводных носителей, которые можно применять в фармацевтических композициях по настоящему изобретению, включают воду, этанол, полиолы (такие как глицерин, пропиленгликоль, полиэтиленгликоль и подобные) и их подходящие смеси, растительные масла, такие как оливковое масло, и инъецируемые органические эфиры, такие как этилолеат. Подходящую текучесть можно поддерживать, например, применением материалов для нанесения покрытия, таких как лецитин, поддержанием требуемого размера частиц в случае дисперсий, и применением поверхностно-активных веществ.
Данные фармацевтические композиции могут также содержать вспомогательные лекарственные вещества, такие как консерванты, увлажняющие агенты, эмульгаторы и диспергирующие вещества. Предотвращение действия микроорганизмов на описанные соединения можно обеспечить включением различных антибактериальных или противогрибковых агентов, например, парабена, хлорбутанола, фенола, сорбиновой кислоты и подобных. Также может быть желательно включать агенты для контролирования тоничности, такие как сахара, хлорид натрия, и подобные в композиции. Кроме того, продолжительной абсорбции инъецируемой фармацевтической формы можно достичь включением агентов, который замедляют абсорбцию, таких как моностеарат алюминия и желатин.
В некоторых случаях, для того чтобы продлить эффект лекарственного средства, желательно замедлить поглощение лекарственного средства после подкожной или внутримышечной инъекции. Этого можно достичь применением жидкой суспензии кристаллического или аморфного вещества, имеющей плохую растворимость в воде. Затем скорость абсорбции лекарственного средства зависит от скорости его растворения, которая, в свою очередь, может зависеть от размера кристаллов и кристаллической формы. Альтернативно, замедленная абсорбция введенного парентерально лекарственного средства достигается растворением или суспендированием лекарственного средства в масляном носителе.
Например, описанное соединение можно доставлять человеку в форме раствора, который получен растворением твердой формы лекарственного средства в жидкости. Данный раствор можно дополнительно разбавлять жидкостью для вливания, такой как вода для инъекции, 0,9% раствор хлорида натрия, 5% раствор декстрозы и лактат Рингера. Предпочтительно применять разбавленные растворы с растворенным в них составом в пределах 4-6 часов для обеспечения максимальной активности. Альтернативно, описанное соединение можно доставлять человеку в форме таблетки или капсулы.
Инъецируемые депо-формы получают созданием микроинкапсулированной матрицы описанных соединений в биоразлагаемых полимерах, таких как полиактид-полигликолид. В зависимости от отношения лекарственного средства к полимеру и свойств применяемого конкретного полимера, можно контролировать скорость высвобождения лекарственного средства. Примеры других биоразлагаемых полимеров включают поли(ортоэфиры) и поли(ангидриды). Инъецируемые депо-составы также получают включением лекарственного средства в липосомы или микроэмульсии, которые являются совместимыми с тканью тела.
Когда описанные соединения вводят в виде лекарственного препарата, людям или животным, их можно предоставлять отдельно или в виде фармацевтической композиции, содержащей, например, 0,1-99% (более предпочтительно, 10-30%) активного ингредиента в комбинации с фармацевтически приемлемым носителем. В других вариантах осуществления фармацевтическая композиция может содержать 0,2-25%, предпочтительно 0,5-5% или 0,5-2%, активного ингредиента. Данные соединения можно вводить людям и другим животным для терапии подходящим путем введения, включая, например, подкожную инъекцию, подкожную инъекцию с замедленным всасыванием, внутривенную инъекцию, внутривенное или подкожное вливание. Данные соединения можно вводить быстро (в пределах <1 минуты) в виде болюса или более медленно в течение более продолжительного периода времени (в течение нескольких минут, часов или дней). Данные соединения можно доставлять ежедневно или в течение большего числа дней, непрерывно или периодически. В одном варианте осуществления соединения можно вводить трансдермально (например, применяя пластырь, микроиглы, микропоры, мазь, микрострую или нанострую).
Независимо от выбранного пути введения, описанные соединения, которые можно применять в подходящей гидратированной форме и/или фармацевтических композициях, формулируют в фармацевтически приемлемую лекарственную форму общепринятыми способами, известными специалистам в данной области техники.
Фактическую дозу активных ингредиентов в фармацевтических композициях можно изменять так, чтобы получить количество активного ингредиента, которое является эффективным для достижения требуемой терапевтической реакции для конкретного пациента, композиции и пути введения, без токсичности для пациента.
Выбранная доза будет зависеть от ряда факторов, включая активность конкретного активного применяемого соединения или его эфира, соли или амида, путь введения, продолжительность введения, скорость выведения или метаболизма конкретного соединения, которое будут применять, скорость и продолжительность абсорбции, продолжительность лечения, другие лекарственные средства, соединения и/или вещества, применяемые в комбинации с конкретным применяемым соединением, возраст, пол, массу, заболевание, общее состояние здоровья и предшествующую историю болезни пациента, которого будут лечить, и подобных факторов, хорошо известных в медицинской области техники.
Врач или ветеринар, являющийся специалистом в данной области техники, может легко определить и назначить эффективное количество требуемой фармацевтической композиции. Например, врач или ветеринар могут начать с доз описанных соединений, применяемых в фармацевтической композиции, меньших, чем требуется для достижения требуемого терапевтического эффекта, и постепенно увеличивать дозу до достижения требуемого эффекта.
В общем, подходящая дневная доза описанного соединения будет представлять собой такую дозу соединения, которая является наименьшей дозой, эффективной для оказания терапевтического эффекта. Данная эффективная доза будет, в общем, зависеть от факторов, описанных выше. В основном, внутривенная, внутримышечная, трансдермальная, интрацеребровентрикулярная и подкожная дозы описанных соединений для пациента, при применении для указанных эффектов, будут находиться в диапазоне от приблизительно 1 мкг до приблизительно 5 мг на килограмм массы тела в час. В других вариантах осуществления доза будет находиться в диапазоне от приблизительно 5 мкг до приблизительно 2,5 мг на килограмм массы тела в час. В следующих вариантах осуществления доза будет находиться в диапазоне от приблизительно 5 мкг до приблизительно 1 мг на килограмм массы тела в час.
При желании, эффективную дневную дозу описанного соединения можно вводить в виде двух, трех, четырех, пяти, шести или более поддоз, вводимых отдельно через подходящие промежутки времени в течение дня, необязательно, в единичных лекарственных формах. В одном варианте осуществления описанное соединение вводят в виде одной дозы в день. В следующих вариантах осуществления соединение вводят непрерывно, внутривенно или другим путем. В других вариантах осуществления соединение вводят менее часто, чем ежедневно, например, каждые 2-3 дня, в сочетании с лечением диализом, еженедельно или менее часто.
Субъект, осуществляющий данное лечение, представляет собой любое нуждающееся животное, включая приматов, в частности людей, и других млекопитающих, таких как лошади, коровы, свиньи и овцы; а также домашнюю птицу и домашних животных.
Описанные соединения можно вводить отдельно или в смеси с фармацевтически приемлемыми носителями, и их можно также вводить в комбинации с противомикробными агентами, такими как пенициллины, цефалоспорины, аминогликозиды и гликопептиды. Таким образом, комбинационная терапия включают последовательное, одновременное и раздельное введение активного соединения таким образом, чтобы терапевтические эффекты первого вводимого лекарственного средства не исчезали полностью при введении последующего.
Пути введения описанных соединений
Данные соединения можно вводить людям и животным для терапии подходящим путем введения. Как применяют в настоящем изобретении, предполагается, что термин "путь" введения включает, но не ограничивается ими, подкожную инъекцию, подкожную инъекцию с замедленным всасыванием, внутривенную инъекцию, внутривенное или подкожное вливание, внутриглазную инъекцию, внутрикожную инъекцию, внутримышечную инъекцию, внутрибрюшинную инъекцию, внутритрахеальное введение, внутриадипозное введение, внутрисуставное введение, интратекальное введение, эпидуральное введение, ингаляцию, интраназальное введение, сублингвальное введение, буккальное введение, ректальное введение, вагинальное введение, интрацистернальное введение и местное введение, трансдермальное введение или введение посредством местной доставки (например, катетер или стент).
Трасдермальная доставка лекарственного средства в тело представляет собой желательный и удобный способ для системной доставки биологически активных веществ субъекту, и в частности для доставки веществ, которые обладают плохой пероральной биодоступностью, таких как белки и пептиды. Трансдермальный путь доставки особенно эффективен с небольшими (например, меньшими чем приблизительно 1000 дальтон) липофильными соединениями, такими как скополамин и никотин, которые могут проникать через наружный роговой слой кожи, который служит в качестве эффективного барьера для проникновения веществ в тело. Под роговой оболочкой находятся живые клетки эпидермиса, которые не содержат кровеносные сосуды, но имеют некоторое количество нервных клеток. Еще ниже находится дерма, которая содержит кровеносные сосуды, лимфатические сосуды и нервы. Лекарственные средства, которые пересекают барьер из рогового слоя кожи, могут обычно диффундировать в капилляры в дерме для поглощения и распределения в организме.
Технологический прогресс способов трансдермальной доставки сфокусирован на решении необходимости в данной области техники доставлять гидрофильные, высокомолекулярные соединения, такие как белки и пептиды, через кожу. Один подход включает разрушение рогового слоя, применяя химические или физические способы для уменьшения барьера, создаваемого роговым слоем. Технология микропорации кожи, которая включает создание транспортных путей в микронных размерах (микропоры) в коже (в частности, микропоры в роговом слое), применяя минимально инвазивный способ, представляет собой более современный подход. Способы создания микропор в коже (роговом слое) включают температурную микропорацию или абляцию, матрицы из микроигл, фонофорез, лазерную абляцию и радиочастотную абляцию (Prausnitz and Langer (2008) Nat. Biotechnology 11:1261-68; Arora et al., Int. J. Pharmaceutics, 364:227 (2008); Nanda et al., Current Drug Delivery, 3:233 (2006); Meidan et al. American J. Therapeutics, 11:312 (2004)).
Как отмечалось выше, PTH секреция регулируется CaSR, который экспрессируется на клеточной поверхности паратиреоидных клеток. Таким образом, для того чтобы активировать CaSR, агент или соединение должны быть доставлены к паратиреоидной клетке. Трансдермальную доставку кальциймиметических агентов можно достигать доставкой через роговой слой и обеспечением системного воздействия для достижения паратиреоидной клетки. В настоящее время, в данной области техники не показано, можно ли доставлять кальцимиметическое соединение трансдермально в количестве, достаточном для терапевтической пользы, и, в особенности, в количестве, достаточном для снижения PTH и/или лечения, ослабления, понижения и/или облегчения гиперкальциемии.
В добавление к кальциймиметикам, аналоги 1,25-(OH)2 витамина D3 представляют собой наиболее широко применяемые соединения для лечения пациентов с гиперпаратиреозом, связанным с хроническим заболеванием почек и заболеванием почек на последней стадии. Аналоги витамина D действуют, облегчая всасывание в кишечнике кальция из пищи, и снижают уровень PTH ингибированием PTH синтеза и секреции. Тогда как внутривенную и пероральную доставку витамина D применяют терапевтически, в настоящее время, в данной области техники не показано, можно ли доставлять аналоги витамина D, такие как ZEMPLAR™ (парикальцитол), CALCIJEX* (кальцитриол), ONE-ALPHA® (альфакальцидол) и HECTOROL® (доксеркальциферол), трансдермально в количестве, достаточном для терапевтической пользы, и, в особенности, в количестве, достаточном для снижения концентрации паратиреоидного гормона (PTH). Кроме того, в данной области техники не продемонстрировано, обеспечивает ли совместное введение трансдермальной доставкой кальциймиметического агента в комбинации с аналогом витамина D (или в виде отдельных составов или в одном составе) в количестве, достаточном для терапевтической пользы, и, в особенности, в количестве, достаточном для снижения концентрации PTH, эффективное лечение пациента, страдающего от гиперпаратиреоза.
Кальциймиметические агенты можно вводить через роговой слой и/или другие слои эпидермиса, для местной или системной доставки, для снижения концентрации паратиреоидного гормона (PTH) и/или лечения гиперкальциемии. В одном варианте осуществления кальциймиметический агент доставляют с помощью микропорации. Предполагается любой один из ряда способов для микропорации, и несколько из них описываются вкратце.
Микропорации можно достигнуть механическими способами и/или внешними движущими силами для нарушения рогового слоя для доставки кальциймиметических агентов, описанных в настоящем изобретении, через поверхность кожи и в лежащие глубже слои кожи и/или кровоток.
В первом варианте осуществления способ микропорации представляет собой абляцию рогового слоя в специальной области кожи, применяя импульсное лазерное излучение, где длительность импульса, энергия импульсов, число импульсов и скорость повторения импульсов являются достаточными для разрушения рогового слоя без заметного повреждения лежащего глубже эпидермиса. Затем кальциймиметический агент наносят на область абляции. Другой способ абляционной микропорации, называемый лазерно-индуцированные волны напряжения (LISW), включает широкодиапазонные, униполярные и сжимаемые волны, генерируемые лазерами с импульсами высокой мощности. LISW взаимодействуют с тканями, разрушая липиды в роговом слое, временно создавая в роговом слое внутриклеточные каналы. Данные каналы или микропоры в роговом слое позволяют проникать кальциймиметическому агенту.
Сонофорез или фонофорез представляет собой другой способ микропорации, в котором применяют ультразвуковую энергию. Ультразвук представляет собой звуковую волну, обладающую частотой выше 20 КГц. Ультразвук можно применять или непрерывно или импульсно, и применять с различными диапазонами частот и интенсивностью (Nanda et al., Current Drug Delivery, 3:233 (2006)).
Другой способ микропорации включает применение матрицы из микроигл. Матрица из микроигл при применении на области кожи субъекта протыкает роговой слой, но не достигает глубины, которая заметно стимулирует нервные окончания или прокалывает капилляры. Таким образом, пациент не чувствует или чувствует минимальный дискомфорт или болевое ощущение при применении матрицы из микроигл для создания микропор, через которые происходит доставка кальциймиметического агента.
Предполагаются матрицы из микроигл, состоящие из полых или сплошных микроигл, где кальциймиметический агент можно наносить на внешнюю поверхность игл или распылять из внутренней части полых игл. Примеры матриц микроигл описывают, например, в Nanda et al., Current Drug Delivery, 3:233 (2006) и Meidan et al. American J. Therapeutics, 11:312 (2004). Матрицы из микроигл первого поколения состояли из сплошных кремниевых игл, которые были покрыты снаружи терапевтическим агентом. Когда микроматрицу игл прижимали к коже и удаляли после приблизительно 10 секунд, быстро достигали проникновения агента на иглах в тело. Матрицы из микроигл второго поколения состояли из сплошных или полых микроигл из оксида кремния, поликарбоната, титана или другого подходящего полимера, покрытых или заполненных раствором терапевтического соединения. Более новые поколения матриц из микроигл получают из биоразрушаемых полимеров, где кончики игл, покрытые терапевтическим агентом, остаются в роговом слое и медленно растворяются.
Микроиглы можно получить из ряда веществ, включая металлы, керамику, полупроводники, органические вещества, полимеры и сплавы. Типичные материалы для получения микроигл включают нержавеющую сталь фармацевтической категории, золото, титан, никель, железо, олово, хром, медь, палладий, платину, сплавы данных или других металлов, оксид кремния, диоксид кремния и полимеры. Типичные биоразлагаемые полимеры включают полимеры гидроксикислот, такие как полилактид молочной и гликолевой кислоты, полигликолид, полилактид-со-гликолид, и сополимеры с поли(этиленгликолем), полиангидриды, поли(орто)эфиры, полиуретаны, поли(масляную кислоту), поли(валериановую кислоту) и поли(лактид-со-капролактон). Типичные небиоразлагаемые полимеры включают поликарбонаты, полиэфиры и полиакриламиды.
Микроиглы могут иметь цилиндрическую или коническую форму. В одном варианте осуществления диаметр микроигл является самым большим у основания микроигл, и он уменьшается к точке, находящейся на противоположной стороне от основания. Можно также получить микроиглы так, чтобы они имели форму, которая включает и прямую (неконическую) часть, и коническую часть. Иглы также могут не иметь конического наконечника вообще, т.е. они могут быть просто цилиндрическими с тупыми или плоскими наконечниками. Полую микроиглу, которая имеет практически одинаковый диаметр, но которая не сужается на конце, называют "микротрубкой". Как применяют в настоящем изобретении, термин "микроигла" включает и микротрубки и конические иглы, если не указано особо.
Электропорация представляет собой другой способ для создания микропор в коже. В данном подходе применяют прикладывание микросекундных или миллисекундных электрических импульсов высокого напряжения для создания временных, проницаемых пор в роговом слое.
Другие способы микропорации включают применение радиоволн для создания микроканалов в коже. Температурная абляция представляет собой еще один другой подход для осуществления трансдермальной доставки соединений с большей молекулярной массой.
Заявители обнаружили, что низкие дозы кальциймиметических агентов можно терапевтически вводить в течение увеличенного периода времени для лечения SHPT. Это заметно отличается от современных требований к дозе других кальциймиметиков (например, гидрохлорида цинакальцета).
Трансдермальная доставка соединений, описанных в настоящем изобретении, показана в исследованиях, описанных в примерах 9-10. В первом исследовании соединение Ac-crrrrrr-NH2 (SEQ ID NO:6) вводили трансдермально крысам, в котором небольшую площадь кожи микропорировали 5 проходами 1,0-мм дерма-роллера при умеренном давлении. Раствор Ac-crrrrrr-NH2 (SEQ ID NO:6) или солевой раствор помещали на микропорированную площадь кожи. Образцы крови отбирали в течение 4 часов, и плазму анализировали на PTH концентрацию ELISA. Результаты показаны на фигуре 11, на которой концентрация PTH в плазме показана в виде процентов от исходного значения до введения дозы для обработанного солевым раствором животного (ромбы) и для двух животных, обработанных испытуемым соединением (квадраты, треугольники). Эти данные показывают, что соединение Ac-crrrrrr-NH2 (SEQ ID NO:6) можно доставлять системно в достаточных количествах трансдермальным путем (в данном случае облегченной микропорацией трансдермальной доставкой), применяя дерма-роллер для того, чтобы эффективно и заметно снижать концентрацию PTH по сравнению с исходной концентрацией в течение ~4 часов, в течение которых проходило исследование. Следует отметить, что было показано, что Ac-crrrrrr-NH2 (SEQ ID NO:6) эффективно снижает концентрацию PTH по сравнению с исходной концентрацией в 1K1C крысиной модели острой почечной недостаточности при введении коротким IV вливанием, а также у нормальных крыс (данные не показаны).
В другом исследовании, описанном в примере 10, соединение Ac-c(C)arrrar-NH2 (SEQ ID NO:3) вводился облегченной микропорацией трансдермальной доставкой нормальным крысам, применяя трансдермальный пластырь. Трансдермальную пластырную систему, содержащую 10% раствор (по массе) Ac-c(C)arrrar-NH2 (SEQ ID NO:3) в солевом растворе, помещали на микропорированную площадь и оставляли на этом месте в течение ~30 часов. Образцы крови периодически отбирали у крыс в течение 30 часов, и образцы плазмы анализировали на PTH концентрацию ELISA. Результаты показаны на фигуре 12. Неожиданно, эти данные демонстрируют, что облегченная микропорацией трансдермальная доставка может осуществлять достаточную замедленную доставку Ac-c(C)arrrar-NH2 (SEQ ID NO:3) для оказания заметного и продолжительного снижения концентрации PTH в течение >30 часов у крыс с нормальной почечной функцией. Эти данные демонстрируют, что облегченная микропорацией трансдермальная доставка конъюгата Ac-c(C)arrrar-NH2 (SEQ ID NO:3), применяя пластырь, может обеспечивать достаточным воздействием пептида в крови в процессе курса лечения для получения значительного и длительного снижения концентрации PTH по сравнению с исходной концентрацией в течение >30 часов. Эти данные демонстрируют, что трансдермальная доставка с помощь пластыря на ежедневной или более продолжительной основе делает возможным лечение диализных и негемодиализных пациентов, нуждающихся в лечении. Например, CKD (стадия 4), первичный гиперпаратиреоз и вторичный гиперпаратиреоз (SHPT) у пациентов с почечным трансплантатом, которых обычно не лечат IV лекарственными средствами, но которых можно легко лечить трансдермальным пластырем на ежедневной основе с помощью облегченной микропорацией трансдермальной доставки.
Другие исследования проводили для дополнительной оценки пути введения соединений. Как описано в примере 11, соединение Ac-c(C)arrrar-NH2 (SEQ ID NO:3) вводили IV вливанием с очень низкой дозой нормальным крысам и крысам с почечной недостаточностью для определения наименьшей дозы, необходимой для введения вливанием, трансдермальной пластырной системой или другим долговременным способом доставки для достижения значительного снижения концентрации PTH. Здоровым крысам вливали внутривенно в течение шести часов очень низкие дозы (1 мкг/кг/час, 3 мкг/кг/час и 10 мкг/кг/час) Ac-c(C)arrrar-NH2 (SEQ ID NO:3). Образцы крови отбирали до введения дозы (перед введением) и через 2 часа, 4 часа, 6 часов (непосредственно перед завершением вливания; EOI) и 8 часов (2 часа после EOI) после начала вливания, и плазму анализировали на концентрацию PTH ELISA. Неожиданно, данные, показанные на фигуре 13, демонстрируют, что вливание очень низких доз Ac-c(C)arrrar-NH2 (SEQ ID NO:3) (1 мкг/кг/час (квадраты), 3 мкг/кг/час (окружности) и 10 мкг/кг/час (треугольники)) в течение 6 часов является эффективным для достижения значительного снижения концентрации PTH по сравнению с исходной концентрацией в процессе вливания. Эти данные показывают, что низкие дозы, доставляемые непрерывно, могут быть такими же эффективными (или даже более эффективными, чем) как доставка с гораздо большими дозами в виде болюса.
PTH-снижающий эффект дополнительно оценивали на крысиной 1K1C модели острой почечной недостаточности. Крысам 1K1C модели внутривенно вливали низкие дозы Ac-c(C)arrrar-NH2 (SEQ ID NO:3) (30 мкг/кг/час и 100 мкг/кг/час) в течение 6 часов. Образцы крови отбирали до введения дозы (перед) и через 2 часа, 4 часа, 6 часов (непосредственно перед завершением вливания; EOI), 8 часов (2 часа после EOI) и 24 часа после начала вливания, и плазму анализировали на уровень PTH ELISA. Данные, показанные на фигуре 14A, демонстрируют, что IV вливание низких доз Ac-c(C)arrrar-NH2 (SEQ ID NO:3) заметно снижает концентрацию PTH по сравнению с исходной концентрацией в 1K1C модели, модели почечной недостаточности, когда исходную концентрацию PTH можно наблюдать от 400 до >1100 пг/мл. Неожиданно, 6-часовое IV вливание низкой дозы (ромбы, 30 мкг/кг/час, и квадраты, 100 мкг/кг/час) Ac-c(C)arrrar-NH2 (SEQ ID NO:3) было способно снижать концентрацию PTH по сравнению с исходной концентрацией в течение ~24 часов. В соответствии с данным резким снижением PTH в 1K1C крысиной модели, фигура 14B показывает гистограмму, содержащую данные о концентрации кальция в сыворотке, при данной острой почечной недостаточности, и показывает соответствующее снижение концентрации кальция в плазме после IV вливания с низкой дозой Ac-c(C)arrrar-NH2 (SEQ ID NO:3). Эти данные демонстрируют, что Ac-c(C)arrrar-NH2 (SEQ ID NO:3) представляет собой очень эффективное кальциймиметическое соединение, которое способно снижать концентрацию PTH и кальция после вливания или доставки низких доз (например, трансдермальной доставкой) в течение ~24 часов. Эти данные дополнительно подтверждают вывод о том, что длительная доставка низкой дозы кальциймиметического агента IV вливанием или облегченной микропорацией трансдермальной доставкой может представлять собой эффективное лечение для пациентов на ежедневной и менее частой основе.
Комбинированная терапия
Как описано выше, способы применения можно применять отдельно или в комбинации с другими подходами для лечения гиперкальциемии и/или болезни костей. Данные другие подходы включают, но не ограничиваются ими, лечение агентами, такими как бисфосфонатные агенты, блокаторы интегрина, гормонозаместительную терапию, селективные модуляторы рецепторов эстрогена, ингибиторы катепсина K, терапию с применением витамина D, аналоги витамина D, такие как ZEMPLAR™ (парикальцитол), CALCIJEX® (кальцитриол), ONE-ALPHA® (альфакальцидол) и HECTOROL® (доксеркальциферол), противовоспалительные агенты, низкодозовую PTH терапию (с или без эстрогена), кальциймиметики, связывающие фосфат агенты, кальцитонин, ингибиторы RANK лиганда, антитела к RANK лиганду, остеопротегерин, антагонисты аденозина и ингибиторы ATP протонного насоса.
В одном варианте осуществления в комбинированной терапии применяют витамин D или аналог витамина D в комбинации с кальциймиметическим агентом. Витамин D способствует абсорбции кальция и функционирует, поддерживая нормальные уровни кальция и фосфора в крови. PTH функционирует, усиливая абсорбцию кальция в кишечнике увеличением синтеза 1,25-(OH)2 витамина D, активной формы витамина D. PTH также стимулирует выведение фосфора из почек и усиливает высвобождение из кости.
Как обсуждалось выше, вторичный гиперпаратиреоз характеризуется повышением концентрации паратиреоидного гормона (PTH), связанным с недостаточной концентрацией активного гормона витамина D. Витамин D или аналог витамина D можно применять для снижения повышенной концентрации PTH при лечении вторичного гиперпаратиреоза. В одном варианте осуществления настоящее изобретение включает фармацевтическую композицию, содержащую кальциймиметический агент и аналог витамина D.
В одном варианте осуществления настоящее изобретение включает фармацевтическую композицию, содержащую кальциймиметический агент и ZEMPLAR™ (парикальцитол). Парикальцитол представляет собой синтетический аналог кальцитриола, метаболически активной формы витамина D. Рекомендованная первоначальная доза Zemplar зависит от исходной концентрации интактного паратиреоидного гормона (iPTH). Если исходная iPTH концентрация является меньшей, чем или равной 500 пг/мл, ежедневная доза составляет 1 мкг, и доза "три раза в неделю" (вводимая не чаще, чем каждый второй день) составляет 2 мкг. Если исходная iPTH концентрация является большей, чем 500 пг/мл, ежедневная доза составляет 2 мкг, и доза "три раза в неделю" (вводимая не чаще, чем каждый второй день) составляет 4 мкг. Соответственно, доза должна быть индивидуальной и основываться на концентрации iPTH в сыворотке при контроле концентрации кальция и фосфора в сыворотке. Парикальцитол описывают в патенте США № 5246925 и патенте США № 5587497.
В другом варианте осуществления настоящее изобретение включает фармацевтическую композицию, содержащую кальциймиметический агент и CALCIJEX® (кальцитриол). Кальцитриол представляет собой метаболически активную форму витамина D. Рекомендованная первоначальная доза CALCIJEX® (пероральная) составляет 0,25 мкг/день. Данное количество можно увеличивать до 0,25 мкг/день при 4-8-недельных интервалах. Нормальная или только слегка пониженная концентрация кальция может отвечать дозам 0,25 мкг каждый второй день. Для пациентов на диализе, рекомендованная первоначальная доза CALCIJEX® (IV) составляет 0,02 мкг/кг (1-2 мкг) 3 раза/неделю, каждый второй день. Данное количество можно увеличить до 0,5-1 мкг, каждые 2-4 недели. Кальцитриол описывают в патенте США № 6051567, патенте США № 6265392 и патенте США № 6274169.
В одном варианте осуществления обеспечивают фармацевтической композицией, содержащей кальциймиметический агент и HECTOROL® (доксеркальциферол). Доксеркальциферол представляет собой синтетический аналог витамина D, который подвергается метаболической активации in vivo, образуя 1α,25-дигидроксивитамин D2, встречающийся в природе, биологически активная форма витамина D. Рекомендованная первоначальная доза HECTOROL® составляет 10 мкг, вводимая три раза в неделю при диализе (приблизительно каждый второй день). Первоначальную дозу следует регулировать, при необходимости, для того чтобы снизить iPTH в крови до диапазона 150-300 пг/мл. Дозу можно повышать при 8-недельных интервалах до 2,5 мкг, если iPTH не снижается на 50% и не достигает требуемого диапазона. Максимальная рекомендованная доза HECTOROL составляет 20 мкг, вводимая три раза в неделю при диализе в суммарном количестве 60 мкг в неделю. Доксеркальциферол описывают в патенте США № 5602116, патенте США № 5861386, патенте США № 5869473 и патенте США № 6903083.
Конкретная комбинация терапий (терапевтических средств или способов) для применения в комбинированном режиме будет учитывать совместимость требуемых терапевтических средств и/или способов и требуемого терапевтического эффекта, который надо достигнуть. Также ясно, что применяемые терапии могут достигать требуемого эффекта для того же заболевания (например, соединение по настоящему изобретению можно вводить одновременно с другим агентом, применяемым для лечения того же заболевания), или они могут достигать различных эффектов (например, контроль любых побочных эффектов). Как применяют в настоящем изобретении, дополнительные терапевтические агенты, которые обычно вводят для лечения или предотвращения конкретного заболевания или состояния, являются известными как "подходящие для заболевания или состояния, которые подвергают лечению".
Комбинированное лечение по настоящему изобретению, как определено в настоящем изобретении, можно достигнуть посредством одновременного, последовательного или раздельного введения отдельных компонентов упомянутого лечения.
Соединения или их фармацевтически приемлемые композиции можно также вводить в композиции для нанесения покрытия на имплантируемые медицинские изделия, биоразлагаемые полимеры, имплантируемый насос и суппозитории. Соответственно, в другом аспекте предполагается композиция для нанесения покрытия на имплантируемое устройство, содержащая описанное соединение, как описано в общем выше, и носитель, подходящий для нанесения покрытия на имплантируемое устройство. В еще другом аспекте, включенным является имплантируемое устройство, покрытое композицией, содержащей соединение, как описано в общем выше, и носитель, подходящий для нанесения покрытия на имплантируемое устройство.
Подходящие покрытия и общий способ получения покрытых имплантируемых устройств описывают в патентах США №№ 6099562, 5886026 и 5304121. Покрытия обычно представляют собой биосовместимые полимерные материалы, такие как полимерные гидрогели, полиметилдисилоксан, поликапролактон, полиэтиленгликоль, полимолочная кислота, этиленвинилацетат и их смеси. Покрытия можно необязательно дополнительно покрывать подходящим верхним покрытием фторсиликона, полисахаридов, полиэтиленгликоля, фосфолипидов или их комбинаций для придания композиции свойств контролируемого высвобождения.
Потенциальные клинические маркеры для определения эффективности лечения
Определение эффективности описанного способа лечения можно осуществить рядом способов.
Нормальные концентрации кальция в сыворотке находятся в диапазоне от 8,8 мг/дл до 0,4 мг/дл (от 2,2 ммоль/л до 2,6 ммоль/л). В определенных случаях эффективность лечения можно определить измерением концентрации маркера в сыворотке и моче, связанного с кальцием, включая, но не ограничиваясь ими, суммарный и ионизированный кальций в сыворотке, альбумин, PTH в плазме, PTHrP, фосфат, витамин D и магний.
В других случаях эффективность можно определить измерением минеральной плотности костной ткани (BMD) или измерением концентрации биохимических маркеров для образования кости и/или резорбции кости в сыворотке или моче. Потенциальные маркеры образования кости включают, но не ограничиваются ими, суммарное количество щелочной фосфатазы, щелочную фосфатазу в костях, остеокальцин, не полностью карбоксилированный остеокальцин, C-концевой пропептид проколлагена I типа и N-концевой пропептид проколлагена I типа. Потенциальные маркеры резорбции кости включают, но не ограничиваются ими, гидроксипролин, гидроксилизин, гликозил-галактозилгидроксилизин, галактозилгидроксилизин, пиридинолин, дезоксипиридинолин, N-концевой сшитый телопептид коллагена I типа, C-концевой сшитый телопептид коллагена I типа, C-концевой сшитый телопептид коллагена I типа, полученный MMP, сиалопротеин кости, кислую фосфатазу и устойчивую к тартрату кислую фосфатазу.
В других случаях эффективность можно определить процентным снижением концентрации PTH относительно концентрации до введения дозы (исходная концентрация) и/или достижением требуемой концентрации PTH, которая, как общепринято, является полезной для пациентов (например, руководство, составленное Национальным Почечным Фондом). Также в других случаях эффективность можно определить измерением ослабления гиперплазии паратиреоидной железы, связанной с гиперпаратиреозным заболеванием.
Ожидается, что когда описанный способ лечения применяют для нуждающегося в нем субъекта, способ лечения будет оказывать эффект, как измерено, например, одним или более из суммарного количества кальция в сыворотке, количества ионизированного кальция в сыворотке, суммарного количества кальция в крови, количества ионизированного кальция в крови, альбумина, PTH в плазме, PTH в крови, PTHrP, фосфата, витамина D, магния, минеральной плотности костной ткани (BMD), суммарного количества щелочной фосфатазы, щелочной фосфатазы в костях, остеокальцина, не полностью карбоксилированного остеокальцина, C-концевого пропептида проколлагена I типа, N-концевого пропептида проколлагена I типа, гидроксипролина, гидроксилизина, гликозил-галактозилгидроксилизина, галактозилгидроксилизина, пиридинолина, дезоксипиридинолина, N-концевого сшитого телопептида коллагена I типа, C-концевого сшитого телопептида коллагена I типа, C-концевого сшитого телопептида коллагена I типа, полученного MMP, сиалопротеина кости, кислой фосфатазы и устойчивой к тартрату кислой фосфатазы. Эффекты включают профилактическое лечение, а также лечение имеющегося заболевания.
Биологически эффективная молекула может быть функционально связана с описанным пептидом ковалентной связью или нековалентным взаимодействием. В конкретных вариантах осуществления функционально связанные биологически эффективные молекулы могут изменять фармакокинетические свойства описанных соединений посредством придания соединению свойств как части соединенной молекулы. Некоторые свойства, которые биологически эффективные молекулы могут придавать описанным соединениям, включают, но не ограничиваются ими, доставку соединения в отдельное определенное место в теле; концентрирование активности соединения в требуемом месте в теле и снижение его эффектов в других местах; ослабление побочных эффектов лечения соединением; изменение проникающей способности соединения; изменение биодоступности или скорости доставки в тело соединения; изменение длительности эффекта лечения соединением; изменение in vitro химической стабильности соединения; изменение in vivo стабильности соединения, периода полувыведения, клиренса, абсорбции, распределения и/или выведения; изменение скорости возникновения и ослабления эффектов соединения; обеспечение позволительным действием, допуская то, чтобы соединение обладало эффектом.
В следующем аспекте описанное соединение можно конъюгировать с полиэтиленгликолем (PEG). Выбранный PEG может иметь любую подходящую молекулярную массу, и может быть линейным или разветвленным, и может быть необязательно конъюгирован через линкер. Средняя молекулярная масса PEG будет предпочтительно находиться в диапазоне от приблизительно 2 килодальтон (кДа) до приблизительно 100 кДа, более предпочтительно от приблизительно 5 кДа до приблизительно 40 кДа. Альтернативно, применяемый PEG-фрагмент может иметь молекулярную массу 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 или 20 кДа.
Описанные соединения можно конъюгировать с PEG через подходящий аминокислотный остаток, расположенный в любом положении в соединениях. Описанные соединения могут необязательно содержать дополнительный аминокислотный остаток, с которым конъюгируют PEG, включая, например, дополнительный аминосодержащий остаток, такой как лизин.
В данной области техники известно, что пегилированные пептиды увеличивают время полувыведения конъюгированного пептида из сыворотки. В данной области техники известен ряд способов для получения пегилированных пептидов. Например, PEG-фрагмент может быть соединен с амино концом, карбокси концом или через боковую цепь заявленного пептида, необязательно благодаря присутствию линкерной группы. В других вариантах осуществления PEG-фрагмент может быть связан с атомом серы тиолсодержащей аминокислоты, такой как цистеин, или может быть соединен с боковой цепью аминосодержащей аминокислоты, такой как лизин.
PEG-группы, как правило, будут присоединять к описанному соединению ацилированием или алкилированием через реакционноспособную группу в PEG-фрагменте (например, альдегидную, аминовую, оксимную, гидразиновую, тиольную, эфирную или карбоксильную группу) к реакционноспособной группе в описанном соединении (например, альдегидной, аминовой, оксимной, гидразиновой, эфирной, карбоксильной или тиольной группе), которая может располагаться на амино конце, карбокси конце или в боковой цепи описанного соединения. Один подход для осуществления пегилирования синтетических пептидов включает соединение через конъюгатную связь в растворе пептида и PEG-фрагмента, причем каждый содержит функциональную группу, которая является реакционноспособной по отношению к другой. Пептиды можно легко получить, применяя общепринятые способы синтеза в растворе или на твердой фазе. Конъюгирование пептида и PEG обычно осуществляют в водной фазе, и его можно контролировать обращенно-фазовой ВЭЖХ. Пегилированные пептиды можно легко очистить и охарактеризовать, применяя стандартные способы, известные специалистам в данной области техники.
Одну или более отдельных субъединиц описанных соединений можно также модифицировать различными агентами для получения производных, о которых известно, что они реагируют с конкретными боковыми цепями или концевыми остатками. Например, лизиновые остатки и аминоконцевые остатки могут реагировать с янтарным ангидридом или другими аналогичными ангидридами карбоновых кислот, которые меняют заряд лизинового или аминоостатка. Другие подходящие реагенты включают, например, имидоэфиры, такие как метилпиколинимидат; пиридоксаль; пиридоксальфосфат; хлорборогидрид; тринитробензолсульфокислоту; O-метилизомочевину; 2,4-пентандион, и катализируемую трансаминазой реакцию с глиоксалатом. Остатки аргинина можно модифицировать реакцией с общепринятыми агентами, такими как фенилглиоксаль, 2,3-бутандион, 1,2-циклогександион и нингидрин.
Кроме того, описанные соединения можно модифицировать так, чтобы они содержали некатионные остатки, которые обеспечивают иммуногенными остатками, пригодными для получения антител для биоаналитических ELISA измерений, а также для оценки иммуногенности. Например, описанные соединения можно модифицировать введением тирозинового и/или глицинового остатка. Специфические модификации тирозиновых остатков являются особенно интересными для введения спектральных меток в тирозиновые остатки. Неограничивающие примеры включают реакцию с ароматическими диазониевыми соединениями или тетранитрометаном. Обычно N-ацетилимидазол и тетранитрометан применяют для получения O-ацетилтирозинового и 3-нитро-производных, соответственно.
Наборы, содержащие описанные соединения
Настоящее изобретение также относится к наборам для осуществления терапевтических режимов по настоящему изобретению. Данные наборы содержат терапевтически эффективные количества описанных соединений, обладающих активностью в качестве CaSR модулятора, в фармацевтически приемлемой форме, отдельно или в комбинации с другими агентами, в фармацевтически приемлемой форме.
Предпочтительные фармацевтические формы включают описанные соединения в комбинации со стерильным солевым раствором, раствором декстрозы, буферным раствором, стерильной водой или другой фармацевтически приемлемой стерильной жидкостью. Альтернативно, композиция может содержать противомикробный или бактериостатический агент. Альтернативно, композицию можно лиофилизировать или сушить. В этом случае набор может дополнительно содержать фармацевтически приемлемый раствор, предпочтительно стерильный, для получения раствора для целей инъекции. В другом варианте осуществления набор может дополнительно содержать иглу или шприц, предпочтительно упакованный в стерильной форме, для инъекции композиции. В других вариантах осуществления набор дополнительно содержит инструкцию по введению композиции субъекту. Инструкция может представлять собой вкладыш с текстом, аудиозапись, аудиовизуальную запись, или любое другое средство для инструктирования по введению композиции субъекту.
В одном варианте осуществления набор содержит (i) первую емкость, содержащую описанное соединение, обладающее активностью в качестве CaSR модулятора; и (ii) инструкцию по применению. В другом варианте осуществления набор содержит (i) первую емкость, содержащую соединение, как описано в настоящем изобретении, и (ii) вторую емкость, содержащую фармацевтически приемлемую среду для разбавления или растворения.
В другом варианте осуществления набор содержит (i) первую емкость, содержащую описанное соединение, обладающее активностью в качестве CaSR модулятора; (ii) вторую емкость, содержащую антикальциемический агент; и (iii) инструкцию по применению.
В одном варианте осуществления антикальциемический агент представляет собой агент, выбранный из группы, состоящей из бисфосфонатных агентов, гормонзаместительных терапевтических агентов, терапии с применением витамина D, аналогов витамина D, таких как ZEMPLAR™ (парикальцитол); CALCIJEX® (кальцитриол), ONE-ALPHA® (альфакальцидол) и HECTOROL® (доксеркальциферол), низкодозового PTH (с или без эстрогена) и кальцитонина.
В родственных аспектах настоящее изобретение обеспечивает изделиями, которые содержат составляющие наборов, описанные выше. Например, настоящее изобретение обеспечивает изделием, содержащим эффективное количество описанного пептида, отдельно или в комбинации с другими агентами, и инструкцию с указанием применения для лечения заболеваний, описанных в настоящем изобретении.
ПРИМЕРЫ
Следующие примеры предлагаются для иллюстрации, но не для ограничения соединений и способов, описанных в настоящем изобретении. Специалисты могут осуществлять различные модификации, не выходя за пределы объема и сущности объекта изобретения, описанного в настоящем изобретении.
Пример 1
Катионные соединения с PTH понижающей активностью
Модель почечной недостаточности
Крысиная модель острой почечной недостаточности (также называемая 1K1C модель) разрабатывалась для симуляции патологии SHPT, связанной с болезнью почек последней стадии. Модель обладает патологическими характеристиками гиперпаратиреоза, связанного с недостаточным функционированием почек, в частности, с заметным увеличением концентрации PTH в плазме и снижением концентрации кальция в сыворотке. Разработка данной модели позволяет дополнительно охарактеризовать описанные соединения в отношении субъекта с почечной дисфункцией и повышенной концентрацией PTH. Обычные исходные уровни PTH в данной модели равнялись в среднем ~450 пг/мл.
1K1C модель острой почечной недостаточности включает удаление одной почки, с последующим воздействием на оставшуюся почку в течение 45 минут ишемией и 48 часов реперфузией. Последующее ишемия/реперфузия (I/R) повреждение оставшейся почки приводит в результате к существенному некрозу и почечной недостаточности. Уровни креатитина в сыворотке оценивали в течение более 24-48 часов после I/R повреждения (данные не показаны). Также, в связи с возникшей в результате почечной дисфункцией, суммарная концентрация PTH резко увеличивалась по сравнению с концентрацией до I/R повреждения ~100 пг/мл. Через 48 часа после I/R, уровень PTH в плазме оценивали равной ~450 пг/мл (~5-кратное увеличение), и, в некоторых случаях, он достигал величины вплоть до ~1200 пг/мл.
Данное воспроизводимое увеличение концентрации креатинина и PTH в сыворотке обеспечивало надежной моделью, которая имитирует физиологию, наблюдаемую у ESRD пациентов.
Гидрохлорид цинакальцета (SENSIPAR®), одобренный кальциймиметический агент, который применяют для снижения PTH при лечении SHPT, испытывали в 1K1C модели острой почечной недостаточности. Пероральное введение цинакальцета при 30 мг/кг заметно снижало PTH на приблизительно 50% в течение вплоть до 6 часов. Данный результат согласуется с опубликованными преклиническими данными для цинакальцета (Nemeth et al., J. Pharmacol. Exp. Ther, 308(2):627-35 (2004)) и подтверждает, что 1K1C модель острой почечной недостаточности представляет собой подходящую модель для оценки активности кальциймиметических агентов для данного заболевания.
Протокол, применяемый в данном исследовании, был следующим. Мужские особи крыс Спрага-Доули получали из лаборатории Charles River (Hollister, CA; требуемая масса 250-275 г). Для исследований с применением испытуемых изделий, животных предварительно канюлировали в бедренную и яремную вены для введения лекарственного средства и отбора крови, соответственно. Животных держали в среде с контролируемой температурой с постоянным циклом 12 часов свет/12 часов темнота и свободным доступом к пище и воде в течение всего времени. Все экспериментальные методики с животными проводили согласно руководству IACUC.
Общую анестезию вызывали и поддерживали внутрибрюшинной (IP) инъекцией пентобарбитала натрия (5,2%, 0,4 мл/крыса). Животным, которые подвергались 45-минутной почечной ишемии, вводили дополнительную IP инъекцию пентобарбитала натрия (5,2%, 0,1 мл/крыса) для поддержания уровня наркоза. Отбор образцов крови для PTH измерений после введения соединений нормальным крысам осуществляли при непрерывной анестезии изофлураном.
Чистую, асептическую методику применяли для всего способа. После анестезирования крыс брюшной отдел брили электростригальной машиной перед операцией, и кожу очищали 70% спиртовым раствором.
Что касается исследований по разработке модели, левую бедренную вену канюлировали PE-10 канюлей для отбора крови. Обе почки обнажали лапаротомией. Правую нефректомию осуществляли после того, как ножку правой почки и мочеточник лигировали двойной 2-0 шелковой нитью. После подтверждения отсутствия кровотечения в правой ножке, левую почечную артерию осторожно разрезали и зажимали микрососудистой клипсой, для того чтобы вызвать глобальную ишемию левой почки. Почечную ишемию подтверждали наблюдением повсеместного изменения цвета с серого на белый (побледнение). Разрез брюшной стенки временно закрывали марлей, для того чтобы способствовать поддержанию температуры абдоминального органа. Через 45 минут указанного периода ишемии клипсу удаляли, и ток крови через артерию левой почки считали восстановленным при наблюдении повсеместного восстановления красного цвета. Разрез брюшной стенки закрывали слоями 2-0 шелковой нитью. Затем животного выводили из наркоза. Физиологические параметры, включая температуру тела (36-37,5°C) и массу тела, измеряли в течение всей методики. Температуру тела контролировали и поддерживали, применяя систему обратной связи с ректальным зондом с нагревательным элементом.
Приблизительно через 48 часов после 1K1C операции (I/R повреждения), животным вводили различные соединения для оценки влияния на концентрации PTH и кальция в плазме. В большинстве случаев испытуемые изделия вводили IV вливанием (время вливания 5, 10 или 30 минут), хотя в некоторых исследованиях соединения вводили IV болюсом или подкожной (SC) болюсной инъекцией. Для введения лекарственного средства и отбора крови животных анестезировали изофлураном.
Образцы крови собирали периодически в течение всего времени исследования. Образцы сыворотки анализировали на концентрацию кальция, и образцы плазмы анализировали на PTH. Из-за диапазона исходных величин PTH для отдельных крыс, все данные нормализовали по отношению к концентрации до введения дозы (исходной). Концентрацию креатинина в крови измеряли, применяя имеющийся в продаже набор от BioAssay Systems (Hayward, Ca), номер по каталогу #DICT-500. Анализы осуществляли согласно инструкции производителя.
B. Испытание соединений в модели почечной недостаточности
Соединения со следующими последовательностями получали для испытания в модели почечной недостаточности: Ac-crrrr-NH2 (SEQ ID NO:4), n=4, Ac-crrrrr-NH2 (SEQ ID NO:5), n=4, Ac-crrrrrr-NH2 (SEQ ID NO:6), n=7, Ac-crrrrrrr-NH2 (SEQ ID NO:7), n=4, и солевой контроль, n=2. Пептиды вводили животным при дозе 3 мг/кг 30-минутным IV вливанием. До введения дозы отбирали образец крови для определения исходной концентрации PTH в плазме до введения дозы. Результаты показаны на фигуре 1 следующим образом: Ac-crrrr-NH2 (SEQ ID NO:4, ромбы), Ac-crrrrr-NH2 (SEQ ID NO:5, квадраты), Ac-crrrrrr-NH2 (SEQ ID NO:6, треугольники) и Ac-crrrrrrr-NH2 (SEQ ID NO:7, пустые квадраты).
Пример 2
In vitro клеточный анализ в HEK-293, экспрессирующих человеческий кальцийчувствительный рецептор
Человеческие эмбриональные клетки почек (HEK) 293T высевали в T25 колбе с 2 миллионами клеток на колбу и инкубировали при 37°C в 5% CO2 в течение ночи. Через день данные клетки трансицировали человеческим CaSR рецептором, применяя реагент для трансфекции на основе липофектамина 2000. Через 24 часа после трансфекции клетки высевали в 384-луночные планшеты при 8000 клеток/лунка. Анализы осуществляли через 48 часов после трансфекции. В некоторых случаях величины EC50 определяли измерением синтеза инозитолмонофосфата в HEK293 клетках, стабильно трансфицированных человеческим кальцийчувствительным рецептором (см. таблицу 1).
Культуральную среду отсасывали из лунок и заменяли 28 мкл 1X буфером для стимуляции (Hepes 10 мМ, CaCl2 1 мМ, MgCl2 0,5 мМ, KCl 4,2 мМ, NaCl 146 мМ, глюкоза 5,5 мМ, LiCl 50 мМ, pH 7,4). Клетки инкубировали с соединениями при различных концентрациях (1 мМ или 300 мкМ, в качестве самой большой, и дополнительно 1/2 log серийное разбавление) при 37°C в течение 1,5 часов перед прекращением реакции. Концентрацию синтезированного IP1 определяли в клетках, применяя Cisbio IP-One Tb набор (621 PAPEC) согласно инструкции производителя. Вкратце, инкубирование с соединением прекращали последовательным добавлением D2 меченного-IP1 и криптат-меченного анти-IP1 в буфере для лизиса и дополнительным инкубированием при комнатной температуре в течение 60 минут. Пластины считывали при 620 нм и 668 нм с 314 нм возбуждением. Нетрансфицированные клетки 293 применяли в качестве отрицательного контроля.
Определяли отношение флуоресценции при 668 нм и 620 нм, IP1 концентрацию рассчитывали на основе стандартных кривых (полученных с помощью Graph Pad Prism версия 4), применяя известные концентрации IP1 стандартов. EC50 рассчитывали, исходя из величин отношения флуоресценции OD(668 нм)/OD(620 нм), применяя аппроксимацию кривых с нелинейной регрессией в программном обеспечении Prism.
Пептиды и конъюгаты получали твердофазным способом получения в 0,25-ммольном масштабе на ABI автоматизированном синтезаторе. Последовательную конденсацию Fmoc-аминокислот (4 экв., Anaspec) с амидной смолой Ринка (NovaBiochem) осуществляли, применяя HBTU/DIEA активацию. Полученный пептид отщепляли, применяя TFA коктейль (фенол (5%), триизопропилсилан (2,5%) и вода (2,5%); 10 мл на грамм смолы) и выделяли осаждением в диэтиловом эфире. После очистки C18 ВЭЖХ конечный продукт выделяли в виде TFA соли лиофилизацией подходящих фракций и характеризовали ВЭЖХ (>95% чистота) и LC-MS (подтвержденная MW).
Пример 3
In vivo Введение соединений с катионными субъединицами
Пептиды вводили внутривенно с дозой 0,5 мг/кг анестезированным изофлураном нормальным крысам Спрага-Доули. Контрольную группу крыс обрабатывали солевым раствором. Кровь отбирали до введения дозы и каждый час в течение 4 часов. Крыс держали с анестезией изофлураном в течение всего исследования. Концентрацию PTH в плазме измеряли ELISA, обнаруживающим биоактивный интактный PTH 1-84 (Immutopics international номер в каталоге 60-2700), и суммарную площадь под кривой для AUC рассчитывали для данных точек, включающих 1-4 часа. Процентное снижение концентрации PTH рассчитывали согласно следующей формуле: AUCобрабатываемое соединение/AUCсолевой контроль*100.
Пример 4
Исследования связи структура-активность: In vivo активность
Испытуемые пептиды, показанные в настоящем изобретении как SEQ ID NO:26 (Ac-carrrar-NH2) и SEQ ID NO:29 (Ac-arrrar-NH2), испытывали in vitro, применяя HEK293 клетки, трансфицированные CaSR, согласно способу в примере 2. Пептиды также испытывали in vivo введением в виде IV болюса нормальным крысам Спрага-Доули при дозах 9 мг/кг для SEQ ID NO:29 и при 0,5 мг/кг для SEQ ID NO:26. Внутривенный (IV) болюс солевого раствора применяли в качестве контроля. Концентрацию PTH в плазме (K2EDTA) определяли до введения дозы и через 1, 2, 3 и 4 часа после введения дозы. Крыс поддерживали при анестезии изофлураном в течение всего исследования. Результаты показаны на фигурах 2A-2B, представленные как среднее по группе ± стандартное отклонение (SD). На фигуре 2B PTH показано как процент от исходной величины до введения дозы.
Пример 5
Исследования связи структура-активность: D- и L-аминокислотные субъединицы
Получали ряд соединений, содержащих L-аминокислотный остаток, используемый вместо D-аминокислотного остатка. Соединения вводились в виде IV болюса нормальным крысам Спрага-Доули при дозе 0,5 мг/кг. Внутривенный (IV) болюс солевого раствора применяли в качестве контроля. Концентрации PTH в плазме (K2EDTA) определяли до введения дозы и через 1, 2, 3 и 4 часа после введения дозы, и AUC рассчитывали, как описано выше. Крыс поддерживали при анестезии изофлураном в течение всего исследования. Результаты показаны в таблице 4 выше.
Пример 6
Исследования связи структура-активность: высвобождение гистамина
Для оценки влияния общего положительного заряда на высвобождение гистамина, связанного с соединением, получали пептиды, содержащие 4-7 катионных (аргинин) остатков, и испытывали их на способность вызывать высвобождение гистамина in vivo. Испытуемые пептиды включали (i) Ac-crrrr-NH2 (SEQ ID NO:4), (ii) Ac-crrrrr-NH2 (SEQ ID NO:5), (iii) Ac-crrrrrr-NH2 (SEQ ID NO:6) и (iv) Ac-crrrrrrrr-NH2 (SEQ ID NO:41).
Получали мужские особи крыс Спрага-Доули (Charles River) и предварительно канюлировали в бедренную и яремную вены для вливания лекарственного средства и отбора крови, соответственно. Все IV обработки лекарственными средствами осуществляли при анестезии (изофлуран). Животным вводили дозы 1-минутным IV введением в суммарном объеме 0,5 мл. Образцы крови отбирали через 5, 15 и 30 минут после IV болюса для получения образцов плазмы (K2EDTA) для анализа на гистамин. Что касается исследования с 30-минутным IV вливанием, образцы отбирали в конце вливания. В некоторых случаях применяли крыс в 1K1C модели острой почечной ишемии.
Равный объем солевого раствора вводили после каждого отбора крови для восстановления потерянного объема. Приблизительно 0,2 мл крови отбирали в каждый момент времени, применяя предварительно покрытые EDTA шприцы для облегчения отбора сыворотки.
ELISA на гистамин проводили на разбавленной плазме, применяя иммуноферментный набор для определения гистамина (EIA) (номер в каталоге # A05890, SPI-BIO, Montigny le Bretonneux, France). EIA набор для определения гистамина представляет собой усиленный дериватизацией конкурентный иммуноферментный анализ, который обнаруживает гистамин в диапазоне от 40 пг/мл до 5500 пг/мл. Образцы анализировали в двух экземплярах согласно протоколу производителя.
Лиофилизированные пептиды (TFA соли) взвешивали, и зарегистрированную массу приводили в соответствие с содержанием пептида. Растворы получали растворением веществ в изотоническом растворе хлорида натрия для получения требуемых концентраций пептидов. В некоторых случаях молярность пептида регулировали для того, чтобы позволить осуществлять сравнение пептидов между собой. Пептиды вводили IV болюсом в эквивалентной дозе в пересчете на один моль как SEQ ID NO:41 (т.е. 0,7 мкмоль/крыса) 1-минутным IV болюсом, и концентрацию гистамина в плазме измеряли до введения дозы (pre), через 5, 15 и 30 минут после введения дозы. Данные представлены как средние величины для группы (n=2) ± SD. Высвобождение гистамина показано как кратное изменение относительно (исходной) концентрации до введения дозы. Результаты показаны на фигуре 3. Данные представлены как средние для группы (n=2) ± SD.
Пример 7
Исследования связи структура-активность: высвобождение гистамина
Что касается in vitro оценки высвобождения гистамина, выделенные перитонеальные тучные клетки крыс выделяли проведением перитонеального лаважа, применяя холодный HBSS+25 мМ HEPES pH 7,4, содержащий гепарин (5 мкл/мл). Клетки промывали дважды буфером для стимулирования (HBSS+25 мМ HEPES pH 7,4) и инкубировали с 10 мкМ соединения в буфере для стимулирования (HBSS+25 мМ HEPES pH 7,4) в течение 15 минут в 96-луночном планшете (106/лунка) при 37°C. Клеточный супернатант анализировали на гистамин, применяя набор EIA для определения гистамина (Cayman # 589651). Данные показаны в таблице 10.
Что касается in vivo оценки высвобождения гистамина, соединения вводили анестезированным изофлураном нормальным крысам при 2 мг/кг IV болюсом (введенный менее чем за одну минуту). Концентрацию гистамина в плазме измеряли через 5 минут после введения соединения (Cayman EIA на гистамин # 589651). Данные показаны в таблице 11.
Сокращения, применяемые в настоящем изобретении и в частности в таблицах 10-11 приведены здесь.
Пример 8
Исследования связи структура-активность: in vivo активность
Соединение Ac-c(C)arrrar-NH2 (SEQ ID NO:3) получали для сравнения с соединением Ac-carrrar-NH2 (SEQ ID NO:26). В соединении Ac-c(C)arrrar-NH2 (SEQ ID NO:3) тиолсодержащую субъединицу в положении X1 конъюгировали через дисульфидную связь с L-Cys остатком. Данные два соединения вводили IV болюсом животным с 1K1C моделью острой почечной недостаточности при дозах 0,3 и 0,5 мг/кг. Концентрацию PTH в плазме определяли до введения дозы и периодически в течение 24 часов после введения дозы. Результаты показаны на фигуре 10, на которой показанные данные представляют собой средние значения для группы ± SEM, в виде функции времени, в часах, у крыс с 1K1C моделью острой почечной недостаточности, соединение Ac-c(C)arrrar-NH2 (SEQ ID NO:3) представлено квадратами (0,3 мг/кг, n=5) и * символами (0,5 мг/кг, n=6) и соединение Ac-с(Ac-C)arrrar-NH2 (SEQ ID NO:141) треугольниками (0,3 мг/кг, n=8) и ромбами (0,5 мг/кг, n=7).
Пример 9
Облегченная микропорацией трансдермальная доставка кальциймиметических агентов
Для оценки системной доставки кальциймиметического агента, Ac-crrrrrr-NH2 (SEQ ID NO:6) вводили CD безволосым крысам трансдермально, применяя резервуар. Ac-crrrrrr-NH2 (SEQ ID NO:6) наносили в виде 10% раствора в солевом растворе на приблизительно 1 см2 площади на спину CD® безволосым крысам, которые были микропорированы 5 проходами 1,0-мм дерма-роллера при умеренном давлении. Полистирольную камеру (внутренний диаметр 9,5 мм) наклеивали сверху микропорированной площади для создания резервуара с лекарственным средством, в которой помещали раствор Ac-crrrrrr-NH2 (SEQ ID NO:6) или солевой раствор. 10% раствор Ac-crrrrrr-NH2 (SEQ ID NO:6) вводили в камеру-резервуар на двух крысах, только солевой раствор вводили в камеру-резервуар на одной крысе. Резервуары закрывали крышкой для предотвращения испарения. Образцы крови отбирали в течение 4 часов, и плазму анализировали на PTH-концентрацию ELISA. Результаты показаны на фигуре 11.
Пример 10
Продолжительная доставка кальциймиметических агентов с помощью облегченного микропорацией трансдермального пластыря
Для дополнительной оценки системной доставки кальциймиметического агента, Ac-c(C)arrrar-NH2 (SEQ ID NO:3) вводился трансдермально нормальным крысам, применяя трансдермальный пластырь. Нормальных крыс обрабатывали матрицей с микроиглами и трансдермальной пластырной системой. Небольшую площадь на спинах крыс Спрага-Доули (~350 г), покрытую мехом, срезали, применяя ножницы, и данную площадь кожи микропорировали, применяя 14×14 матрицу (~1 см2) микроигл (~0,5 мм). Трансдермальную пластырную систему, содержащую 10% раствор (по массе) Ac-c(C)arrrar-NH2 (SEQ ID NO:3) в солевом растворе, помещали на микропорированную площадь и оставляли в течение ~30 часов. Образцы крови отбирали у крыс периодически в течение 30 часов, и образцы плазмы анализировали на PTH-концентрацию ELISA. Результаты показаны на фигуре 12.
Пример 11
Вливание кальциймиметических агентов
Для дополнительной оценки снижающего PTH эффекта кальцимиметического соединения, Ac-c(C)arrrar-NH2 (SEQ ID NO:3) вводили IV вливанием с очень низкой дозой нормальным крысам и крысам с почечной недостаточностью для определения наименьшей дозы, необходимой для введения вливанием, трансдермальной пластырной системой или другим продолжительным способом доставки для достижения заметного снижения концентрации PTH. Мужским особям нормальных крыс Спрага-Доули (250-300 г) вливали внутривенно в течение 6 часов Ac-c(C)arrrar-NH2 (SEQ ID NO:3) при скорости дозирования 1 мкг/кг/час, 3 мкг/кг/час и 10 мкг/кг/час. Образцы крови отбирали до введения дозы, через 2 часа, 4 часа, 6 часов (непосредственно перед окончанием вливания; EOI) и 8 часов (2 часа после EOI) после введения дозы, и плазму анализировали на PTH-концентрацию ELISA. Данные показаны на фигуре 13, где обработки 1 мкг/кг/час (квадраты), 3 мкг/кг/час (ромбы) и 10 мкг/кг/час (треугольники) в течение 6 часов были эффективными для обеспечения заметного снижения концентрации PTH по сравнению с исходной концентрацией в течение вливания.
Аналогичное исследование проводили на крысах с 1K1C моделью острой почечной недостаточности. Крысам с 1K1C моделью внутривенно вливали Ac-c(C)arrrar-NH2 (SEQ ID NO:273) при скоростях дозирования 30 мкг/кг/час и 100 мкг/кг/час в течение 6 часов. Образцы крови отбирали до введения дозы (pre), через 2 часа, 4 часа, 6 часов (непосредственно перед окончанием вливания; EOI), 8 часов (2 часа после EOI) и 24 часа, и плазму анализировали на PTH-концентрацию ELISA. Данные показаны на фигуре 14A (30 мкг/кг/час, ромбы, и 100 мкг/кг/час, квадраты), и концентрация кальция в сыворотке у животных показана на фигуре 14B.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТЕРАПЕВТИЧЕСКИЕ СРЕДСТВА ДЛЯ РЕГУЛЯЦИИ УРОВНЯ ФОСФОРА В СЫВОРОТКЕ | 2012 |
|
RU2612912C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ИСПОЛЬЗОВАНИЯ ПЕПТИДОВ НЕОГЕНЕЗА ОСТРОВКОВ И ИХ АНАЛОГОВ | 2014 |
|
RU2685958C2 |
| МОДУЛЯЦИЯ АКТИВНОСТИ КОМПЛЕМЕНТА | 2015 |
|
RU2670988C2 |
| ПЕПТИДЫ И ИХ ПРОИЗВОДНЫЕ, ВЗАИМОДЕЙСТВУЮЩИЕ С НИКОТИНОВЫМ АЦЕТИЛХОЛИНОВЫМ РЕЦЕПТОРОМ И ПРИГОДНЫЕ ДЛЯ ИСПОЛЬЗОВАНИЯ В КОСМЕТОЛОГИИ ПРОТИВ МИМИЧЕСКИХ И ВОЗРАСТНЫХ МОРЩИН | 2013 |
|
RU2524428C1 |
| АНАЛОГИ АЛЬФА- и ГАММА-MSH | 2013 |
|
RU2668791C2 |
| Соединение, представляющее собой агонист кальций-чувствительного рецептора, и его применение | 2020 |
|
RU2827834C1 |
| НОВЫЕ ПОЛИПЕПТИДЫ | 2014 |
|
RU2674604C2 |
| ПЕПТИДОМИМЕТИЧЕСКИЕ МАКРОЦИКЛЫ | 2011 |
|
RU2582678C2 |
| НОВЫЕ АГОНИСТЫ NPR-B | 2010 |
|
RU2636738C2 |
| МОДУЛЯЦИЯ АКТИВНОСТИ КОМПЛЕМЕНТА | 2015 |
|
RU2778514C2 |





































































































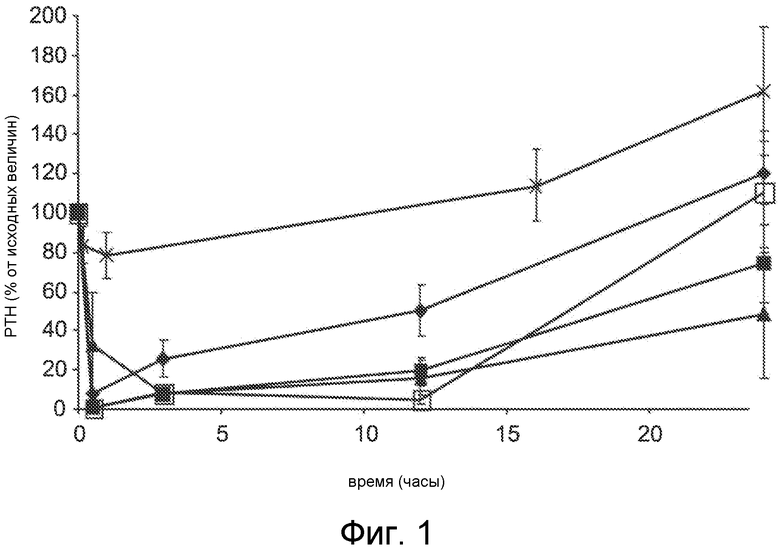
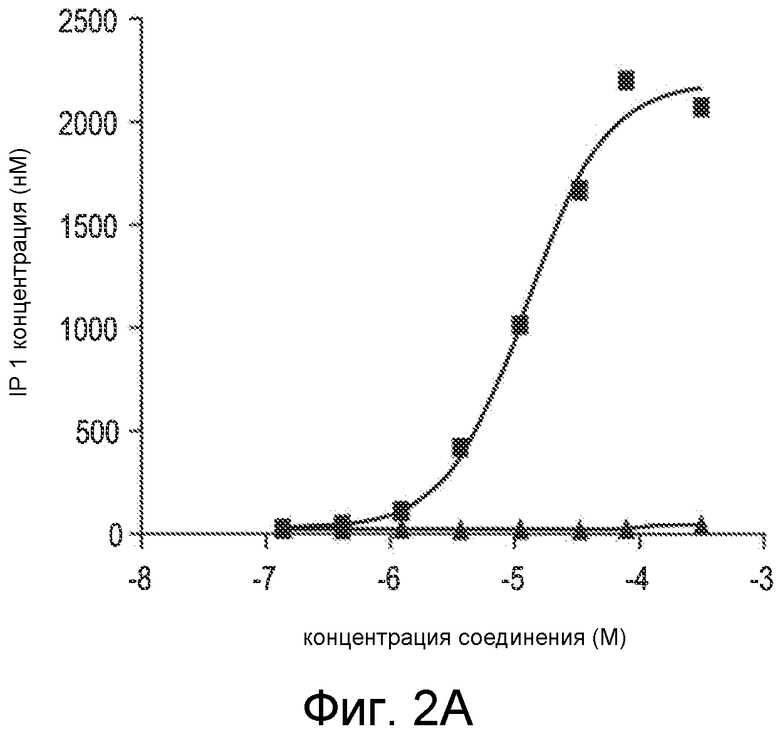
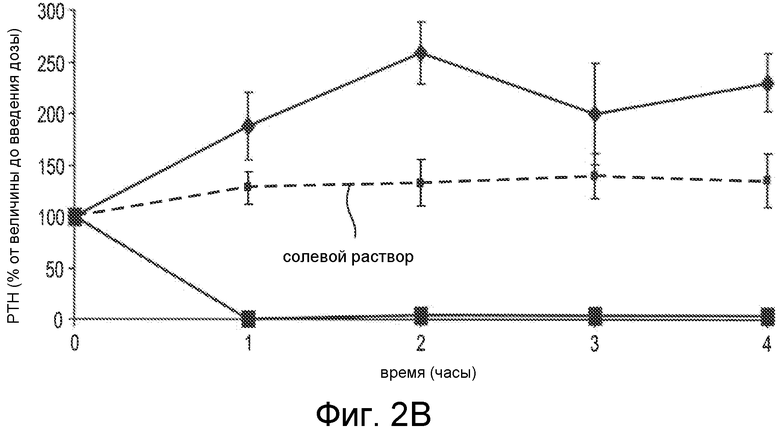
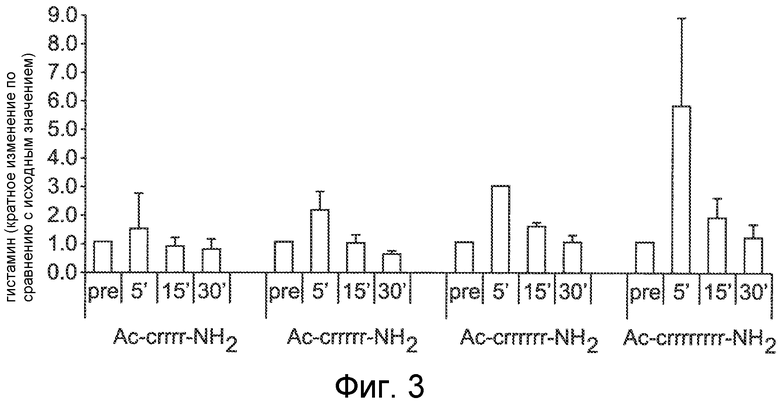
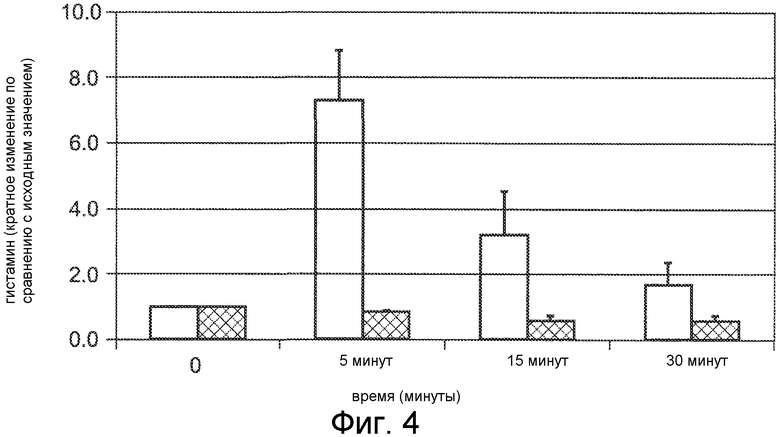

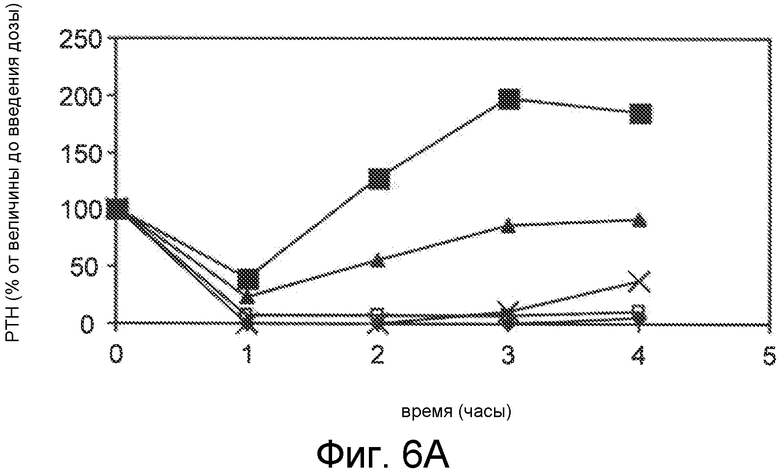
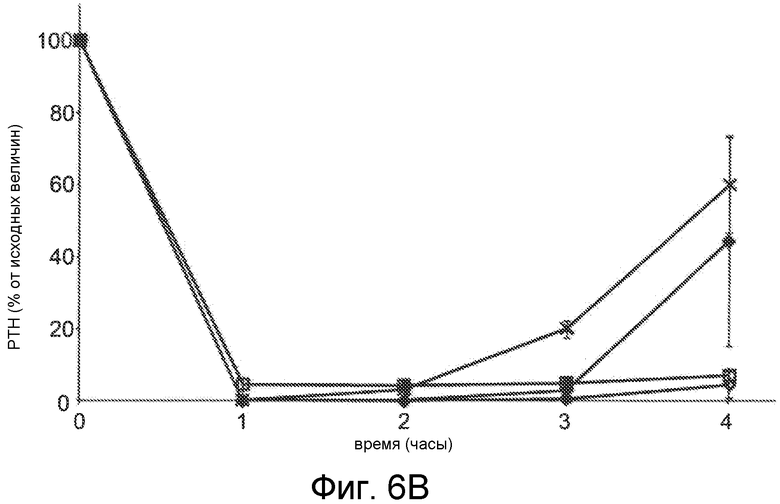
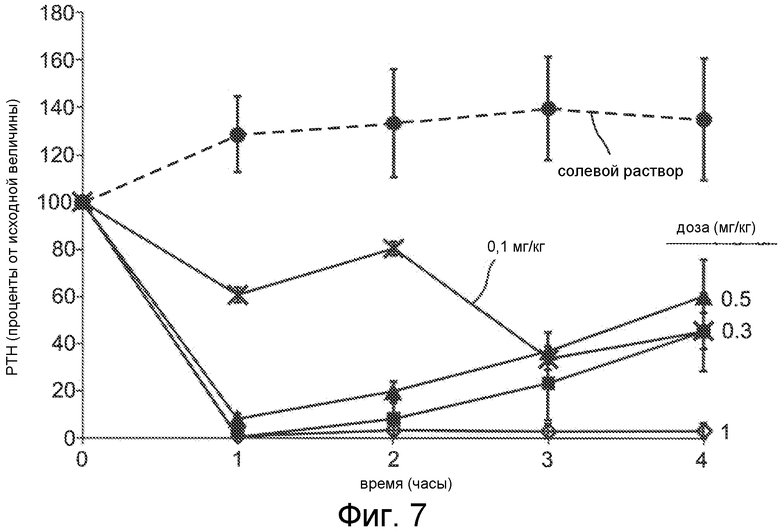
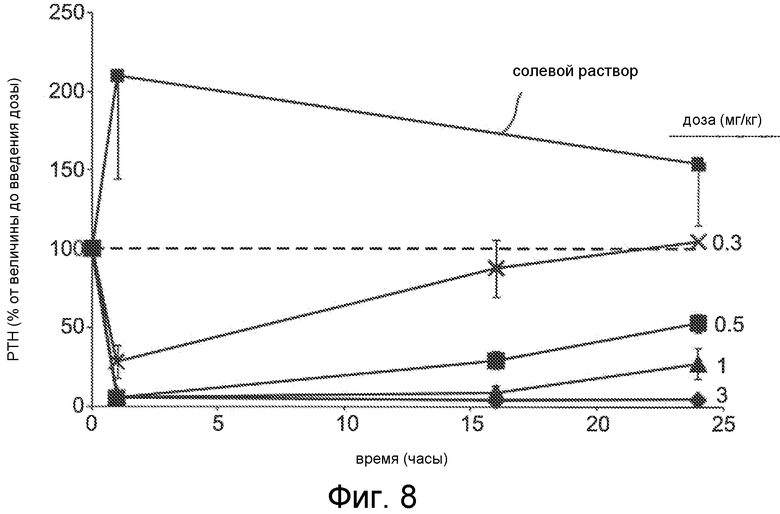
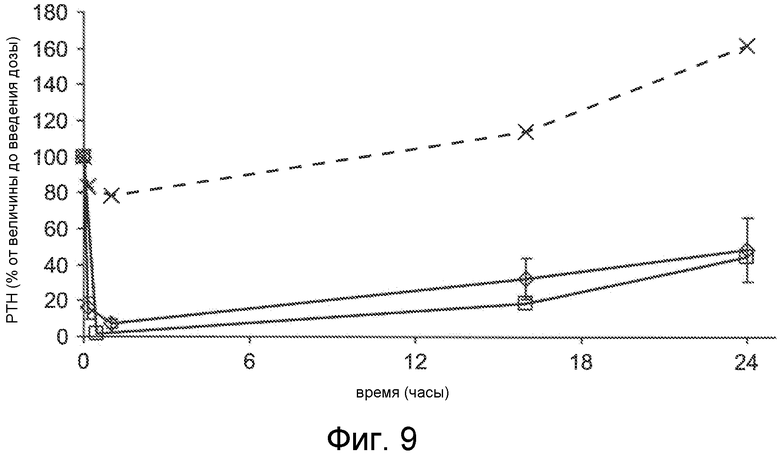
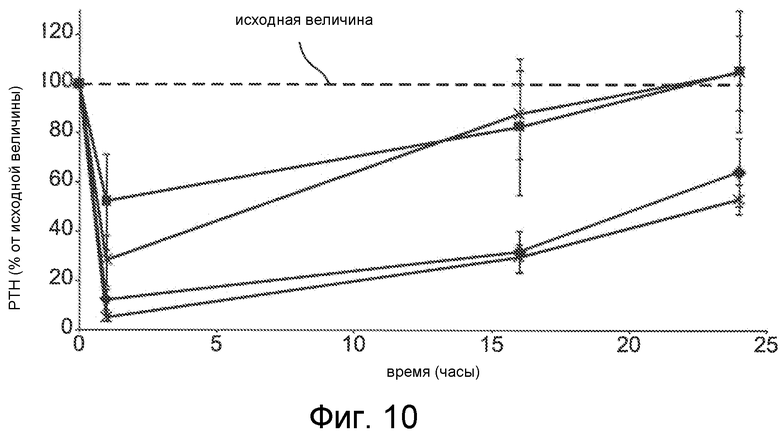
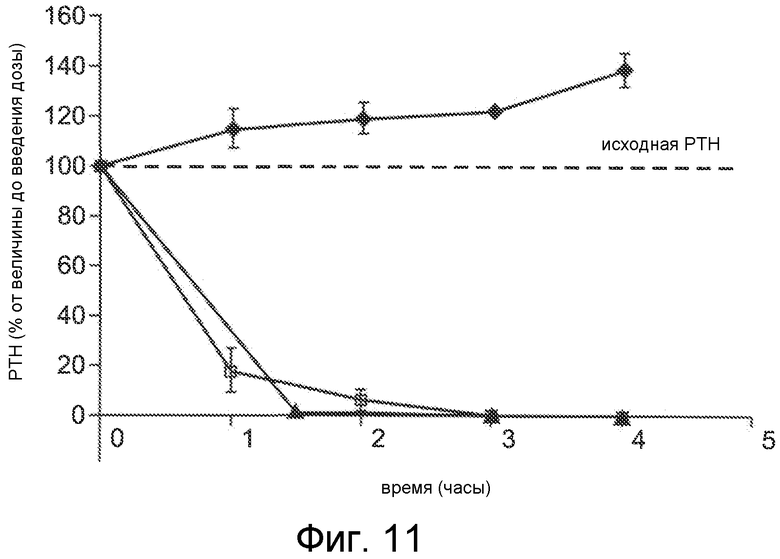
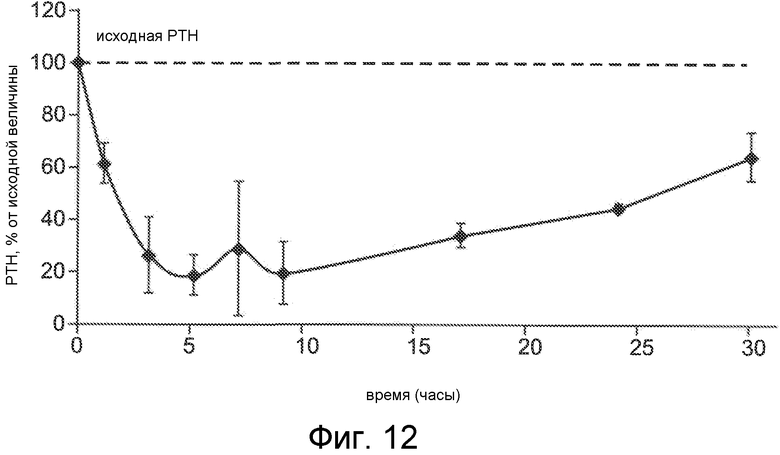
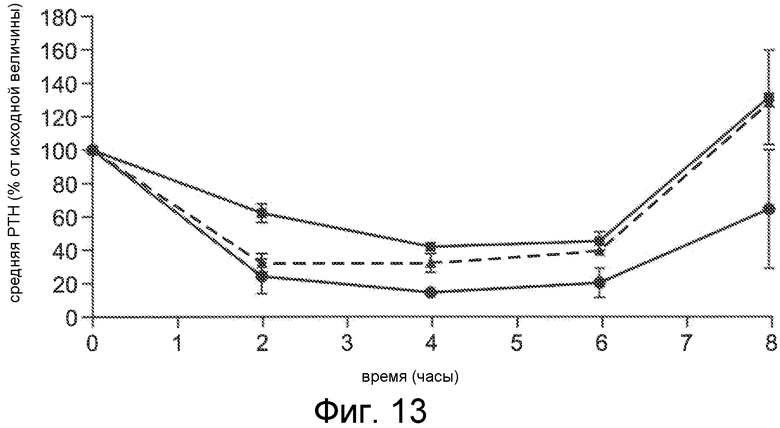
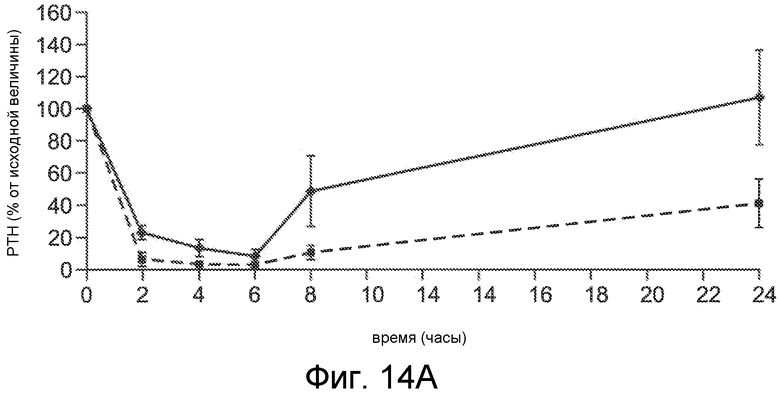
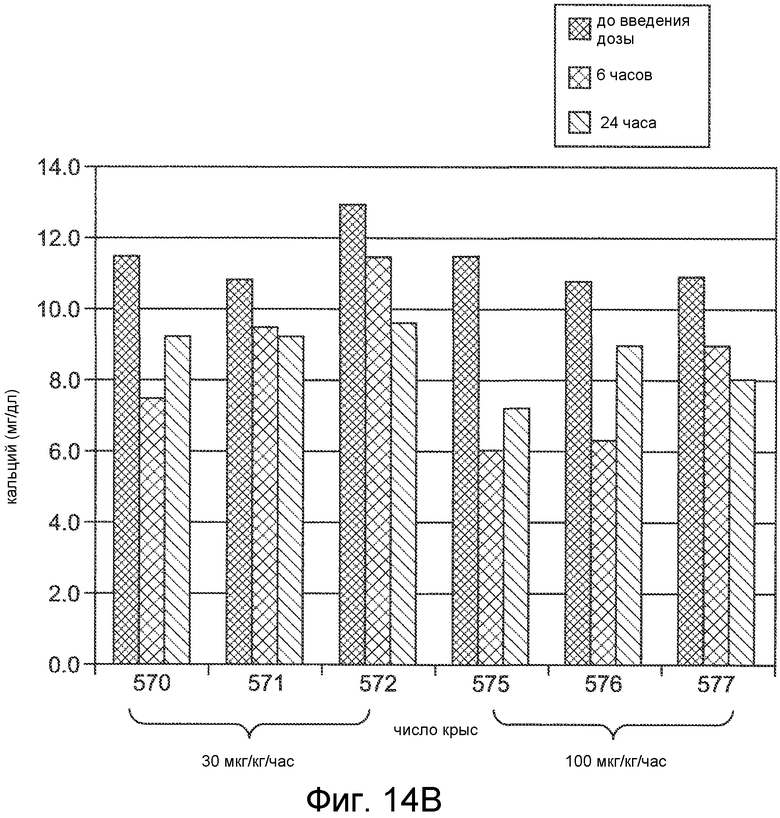
Изобретение относится к соединениям, обладающим активностью, снижающей уровни паратиреоидного гормона. Соединение содержит пептид и конъюгирующую группу, где пептид имеет аминокислотную последовательность формулы: Х1-Х2-Х3-Х4-Х5-X6 _Х7, в которой X1 представляет собой D-цистеин; Х2 представляет собой аминокислоту, выбранную из группы, состоящей из D-аргинина и некатионной аминокислоты; Х3 представляет собой D-аргинин; Х4 представляет собой аминокислоту, выбранную из группы, состоящей из D-аргинина и некатионной аминокислоты; Х5 представляет собой D-аргинин; Х6 представляет собой некатионную аминокислоту; Х7 представляет собой D-аргинин; и где пептид связан с конъюгирующей группой посредством дисульфидной связи. Соединения можно применять для лечения субъектов, страдающих, например, от первичного, вторичного или третичного гиперпаратиреоза, гиперкальциемии злокачественной опухоли, метастатической болезни кости или остеопороза. 12 н. и 48 з.п. ф-лы, 14 ил., 11 табл., 11 пр.
1. Соединение, содержащее пептид и конъюгирующую группу, где пептид имеет аминокислотную последовательность, имеющую формулу:
Х1-Х2-Х3-Х4-Х5-X6 _Х7,
в которой
X1 представляет собой D-цистеин;
Х2 представляет собой аминокислоту, выбранную из группы, состоящей из D-аргинина и некатионной аминокислоты;
Х3 представляет собой D-аргинин;
Х4 представляет собой аминокислоту, выбранную из группы, состоящей из D-аргинина и некатионной аминокислоты;
Х5 представляет собой D-аргинин;
Х6 представляет собой некатионную аминокислоту;
Х7 представляет собой D-аргинин; и
где пептид связан с конъюгирующей группой посредством дисульфидной связи.
2. Соединение по п. 1, где N-конец пептида является ацетилированным.
3. Соединение по п. 1, где С-конец является амидированным.
4. Соединение по п. 2, где С-конец является амидированным.
5. Соединение по п. 1, где конъюгирующая группа выбрана из группы, состоящей из цистеина, гомоцистеина, глутатиона, пегилированного цистеина и тиолсодержащего полипептида.
6. Соединение по п. 5, где конъюгирующая группа является ацетилированной.
7. Соединение по п. 1, где конъюгирующая группа представляет собой цистеин.
8. Соединение по п. 7, где конъюгирующая группа является ацетилированной.
9. Соединение по п. 1, где соединение представляет собой гомодимер.
10. Соединение по п. 1, где Х2 представляет собой D-аргинин.
11. Соединение по п. 10, где N-конец пептида является ацетилированным.
12. Соединение по п. 11, где С-конец пептида является амидированным.
13. Соединение по п. 10, где соединение представляет собой гомодимер.
14. Соединение по п. 5, где Х4 представляет собой D-аргинин.
15. Соединение по п. 14, где конъюгирующая группа представляет собой цистеин.
16. Соединение по п. 15, где конъюгирующая группа является ацетилированной.
17. Соединение по п. 14, где N-конец пептида является ацетилированным.
18. Соединение по п. 17, где С-конец пептида является амидированным.
19. Соединение по п. 5, где конъюгирующая группа представляет собой цистеин.
20. Соединение по п. 19, где конъюгирующая группа является ацетилированной.
21. Соединение по п. 10, где Х4 представляет собой D-аланин.
22. Соединение по п. 21, где конъюгирующая группа представляет собой L-цистеин.
23. Соединение по п. 21, где конъюгирующая группа представляет собой D-цистеин.
24. Соединение по п. 22, где конъюгирующая группа является ацетилированной.
25. Соединение по п. 23, где конъюгирующая группа является ацетилированной.
26. Соединение по п. 21, где соединение представляет собой гомодимер.
27. Соединение по п. 21, где N-конец пептида является ацетилированным.
28. Соединение по п. 27, где С-конец пептида является амидированным.
29. Соединение по п. 14, где соединение является гомодимером.
30. Соединение по п. 14, где Х2 представляет собой D-аланин.
31. Соединение по п. 30, где конъюгирующая группа представляет собой L-цистеин.
32. Соединение по п. 30, где конъюгирующая группа представляет собой D-цистеин.
33. Соединение по п. 31, где конъюгирующая группа является ацетилированной.
34. Соединение по п. 32, где конъюгирующая группа является ацетилированной.
35. Соединение по п. 30, где соединение является гомодимером.
36. Соединение по п. 30, где N-конец пептида является ацетилированным.
37. Соединение по п. 36, где С-конец пептида является амидированным.
38. Соединение по п. 1, где пептид представляет собой от 8 до 15 аминокислотных остатков в длину.
39. Соединение по п. 38, где пептид представляет собой от 8 до 11 аминокислотных остатков в длину.
40. Соединение по п. 39, где пептид представляет собой от 8 до 9 аминокислотных остатков в длину.
41. Соединение по п. 1, где пептид состоит из аминокислотной последовательности, имеющей формулу:
X1-Х2-Х3-Х4-Х5-Х6-Х7.
42. Соединение, содержащее Ас-с(С)arrrar-NH2 (SEQ ID NO: 3).
43. Соединение, содержащее Ас-с(С)arrrar-NH2 (SEQ ID NO: 3), где конъюгирующая группа является ацетилированной.
44. Соединение, содержащее Ас-с(с)arrrar-NH2 (SEQ ID NO: 142).
45. Соединение, содержащее Ас-с(С)rrarar-NH2 (SEQ ID NO: 28).
46. Соединение, содержащее Ас-с(С)rrarar-NH2 (SEQ ID NO: 28), где конъюгирующая группа является ацетилированной.
47. Соединение, содержащее:
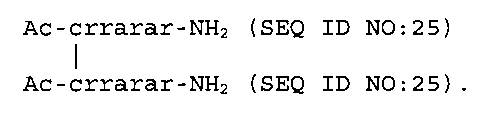
48. Соединение, содержащее:

49. Фармацевтическая композиция для лечения вторичного гиперпаратиреоза (SHPT) у субъекта, содержащая соединение по любому из пп. 1-48 в эффективном количестве и фармацевтически приемлемое вспомогательное вещество или носитель.
50. Фармацевтическая композиция для снижения уровней паратиреоидного гормона у субъекта, содержащая соединение по любому из пп. 1-48 в эффективном количестве и фармацевтически приемлемое вспомогательное вещество или носитель.
51. Фармацевтическая композиция по п. 49, которую вводят внутривенно.
52. Фармацевтическая композиция по п. 49, которую вводят трансдермально.
53. Фармацевтическая композиция по п. 50, которую вводят трансдермально.
54. Фармацевтическая композиция по п. 50, которую вводят внутривенно.
55. Применение композиции, содержащей соединение по любому из пп. 1-48 для изготовления лекарственного средства для лечения вторичного гиперпаратиреоза (SHPT) у субъекта.
56. Применение композиции, содержащей соединение по любому из пп. 1-48 для изготовления лекарственного средства для снижения уровней паратиреоидного гормона у субъекта.
57. Применение по п. 55, дополнительно включающее второй терапевтический агент.
58. Применение по п. 57, где второй терапевтический агент представляет собой витамин D, аналог витамина D или гидрохлорид цинакальцета.
59. Применение по п. 56, дополнительно включающее второй терапевтический агент.
60. Применение по п. 59, где второй терапевтический агент представляет собой витамин D, аналог витамина D или гидрохлорид цинакальцета.
| US 2009023652 A1, 22.01.2009 | |||
| US 2008249016 A1, 09.10.2008 | |||
| WO 1995011988 A1, 04.05.1995 | |||
| EA 199901025 A1, 26.06.2000 | |||
| CHATTOPADHYAY, N | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| J | |||
| PHYSIOL | |||
| CELL PHYSIOL., 2000, vol | |||
| АППАРАТ ДЛЯ ОБОГАЩЕНИЯ РУД ПО МЕТОДУ ВСПЛЫВАНИЯ | 1915 |
|
SU279A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |

