Область техники, к которой относится изобретение
Настоящая заявка раскрывает улучшенные способы синтеза производных дигидропиридофталазинона, например, таких как 5-фтор-8-(4-фторфенил)-9-(1-метил-1Н-1,2,4-триазол-5-ил)-8,9-дигидро-2Н-пиридо[4,3,2-де]фталазин-3(7Н)-он и его стереоизомеров, которые являются эффективными ингибиторами поли(АДФ-рибоза)полимеразы (PARP), и новые синтезированные промежуточные соединения.
Уровень техники
Семейство поли(АДФ-рибоза)полимераз (PARP) включает в себя приблизительно 18 белков, которые демонстрируют определенный уровень гомологии в их каталитическом домене, но различаются по их клеточным функциям (Ame et al., BioEssays., 26 (8), 882-893 (2004)). PARP-1 и PARP-2 являются уникальными членами семейства, в том отношении, что их каталитическая активность стимулируется образованием разрывов нитей ДНК.
PARP вовлечена в сигнализацию повреждения ДНК, благодаря своей способности распознавать и быстро связываться с одинарными или двойными разрывами нитей ДНК (D'Amours et al., Biochem. J., 342, 249-268 (1999)). Она принимает участие в различных ДНК-связывающих функциях, включая генную амплификацию, клеточное деление, дифференцировку, апоптоз, эксцизионную репарацию оснований ДНК, а также влияние на длину теломер и стабильность хромосом (d´Adda di Fagagna et al., Natura Gen., 23(1), 76-80 (1999)).
Синтез производных дигидропиридофталазинона был раскрыт в патентной публикации США № US 2010/0035883 A1, содержание которой включено сюда в качестве ссылки в полном объеме. Тем не менее у методов синтеза, раскрытых в US 2010/0035883 A1, имеются, в частности, проблемы масштабирования. Соответственно, существует необходимость в создании улучшенных способов синтеза этого класса соединений, и в частности 5-фтор-8-(4-фторфенил)-9-(1-метил-1Н-1,2,4-триазол-5-ил)-8,9-дигидро-2Н-пиридо[4,3,2-де]фталазин-3(7Н)-она и его стереоизомеров.
Сущность изобретения
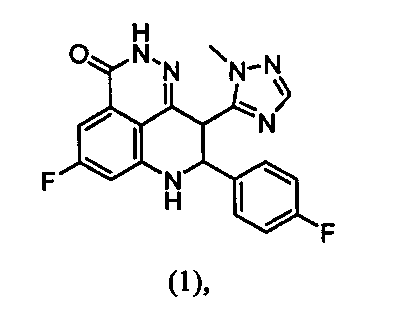
В настоящем документе представлены улучшенные способы синтеза и новые промежуточные соединения, которые представляют собой масштабируемые и эффективные способы получения 5-фтор-8-(4-фторфенил)-9-(1-метил-1Н-1,2,4-триазол-5-ил)-8,9-дигидро-2Н-пиридо[4,3,2-де]фталазин-3(7Н)-она, как показано в формуле (1), и его энантиомеров, как показано в формулах (1а) и (1b):

или их солей. В частности, общий выход для синтезов, включенных в настоящий документ, является улучшенным по сравнению с имеющимся в уровне техники.
В первом варианте осуществления этот способ включает следующие этапы:
Этап а) - взаимодействие соединения формулы (2):

с 1-метил-1H-1,2,4-триазол-5-карбальдегидом для получения соединения формулы (3):

или его соли;
Этап b) - обработка соединения формулы (3) низшим алифатическим спиртом, например, таким как HO-R, где R=C1-C4 алкил, предпочтительно метанол, для получения соединения формулы (4):

или его соли;
Этап с) - взаимодействие соединения формулы (4) или его соли с 4-фторбензальдегидом в присутствии восстанавливающего реагента и кислоты или кислоты Льюиса для получения соединения формулы (5):

или его соли;
Этап d) - осуществление разделения, например, хирального хроматографического разделения соединения формулы (5) до выхода двух энантиомеров формул (6а) и (6b):

и
Этап e) - взаимодействие энантиомера формулы (6а) или (6b) с гидразином для получения энантиомера формул (1а) или (1b).
Во втором варианте осуществления энантиомер формулы (1а) или (1b) получают первой обработкой соединения формулы (5) гидразином, чтобы получить рацемат формулы (1), а затем осуществлением хирального хроматографического разделения до выхода двух энантиомеров формул (1а) и (1b).
В третьем варианте осуществления альтернативный метод включает следующие этапы:
Этап а) - взаимодействие соединения формулы (3) или его соли с гидразин-гидратом для получения соединения формулы (7):

или его соли;
Этап b) - взаимодействие соединения формулы (7) с 4-фторбензальдегидом в инертном растворителе при повышенной температуре для получения соединения формулы (8):

или его соли;
Этап с) - обработка соединения формулы (8) основанием в инертном растворителе для получения соединения формулы (1); и
Этап d) - осуществление хирального хроматографического разделения соединения формулы (1) до выхода двух энантиомеров формул (1а) и (1b).
В четвертом варианте осуществления еще один альтернативный метод включает следующие этапы:
Этап а) - взаимодействие соединения формулы (4) с 4-фторбензальдегидом в присутствии основания для получения соединения формулы (9):

или его соли, в котором R представляет собой C1-C6 алкил (низший алкил).
Этап b) - взаимодействие соединения формулы (9) с восстанавливающим реагентом для получения соединения формулы (5) или соединения формулы (10):

или его соли, в котором R представляет собой C1-C6 алкил;
Этап c) - восстановление соединения формулы (10) с помощью восстанавливающего реагента для получения соединения формулы (5);
Этап d) - осуществление хирального хроматографического разделения соединения формулы (5) для получения энантиомеров формул (6а) и (6b); и
Этап e) - взаимодействие энантиомера формулы (6а) или (6b) с гидразин-гидратом для получения энантиомера формулы (1а) или (1b).
В пятом варианте осуществления еще один способ включает взаимодействие соединения формулы (9) с гидразином для получения соединения формулы (1) и разделение с помощью хирального хроматографического разделения до выхода двух энантиомеров (1а) и (1b).
Подробное описание изобретения
PARP играет важную роль в облегчении репарации ДНК, в регуляции транскрипции РНК, в медиации клеточной гибели и в регуляции иммунного ответа. Ингибиторы PARP демонстрируют эффективность на многочисленных моделях болезни, в частности на моделях ишемического реперфузионного повреждения, воспалительного заболевания, дегенеративных заболеваний, защиту от вышеуказанных побочных эффектов цитотоксических соединений и потенцирование цитотоксической терапии рака. Ингибиторы PARP эффективны в профилактике ишемического реперфузионного повреждения на моделях инфаркта миокарда, инсульта, другой нейрональной травмы, при трансплантации органов, а также при реперфузии глаз, почек, кишечника и скелетных мышц. Ингибиторы PARP эффективны при воспалительных заболеваниях, таких как артрит, подагра, воспалительное заболевание кишечника, при воспалениях ЦНС, таких как рассеянный склероз и аллергический энцефалит, при сепсисе, септическом шоке, геморрагическом шоке, легочном фиброзе и увеите. Ингибиторы PARP также оказывают благоприятное воздействие на некоторые модели дегенеративных заболеваний, включая диабет и болезнь Паркинсона. Ингибиторы PARP уменьшают печеночную токсичность вследствие передозировки ацетаминофена, кардиотоксичность и почечную токсичность из-за доксорубицина и противоопухолевых средств на основе платины, а также вторичные поражения кожи в результате воздействия иприта. В различных моделях рака ингибиторы PARP демонстрируют потенцирование лучевой и химиотерапии, увеличивая апоптоз раковых клеток, ограничивая рост опухоли, уменьшая метастазы, а также продлевая выживаемость животных с опухолями.
В настоящем документе представлены улучшенные способы и новые промежуточные вещества для получения производных дигидропиридофталазинона и, в частности, соединения формулы (1) или его энантиомеров формулы (1а) и (1b):

или их солей.
Одним из вариантов осуществления изобретения является способ получения соединения формулы (1), который включает взаимодействие соединения формулы (5):

или его соли с моногидратом гидразина. В некоторых вариантах осуществления реакция протекает в инертном растворителе при температуре от приблизительно 0 до приблизительно 140°С в течение от приблизительно 1 до приблизительно 24 часов.
В некоторых вариантах осуществления инертный растворитель может включать воду, спирт, такой как метанол, этанол, н-пропанол, изопропанол, н-бутанол, изобутанол, трет-бутанол или этиленгликоль; простой эфир, такой как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, 2-метил-тетрагидрофуран или диоксан; амид, такой как диметилформамид или диметилацетамид; сложный эфир, такой как этилацетат, метилацетат или этилформиат; хлористый углеводород, такой как дихлорметан, хлороформ или дихлорэтан; углеводород, такой как гексан, гептаны, бензол, толуол или ксилол; или нитрил, такой как ацетонитрил, или их комбинации. В некоторых вариантах осуществления растворитель представляет собой спирт. В некоторых вариантах осуществления растворитель представляет собой метанол или этанол.
В некоторых вариантах осуществления температура реакции может иметь любое значение или диапазон и включает от приблизительно 0°C до приблизительно 140°C. Например, в некоторых вариантах осуществления температура реакции может быть от приблизительно 0°C до приблизительно 25°C; от приблизительно 0°C до приблизительно 100°C; от приблизительно 0 до приблизительно 120°C; от приблизительно 20°C до приблизительно 40°C; от приблизительно 25°C до приблизительно 60°C; от приблизительно 100°C до приблизительно 140°C или приблизительно при 25°С; приблизительно при 40°С; приблизительно при 50°C или приблизительно при 60°C.
Другим вариантом осуществления изобретения является способ получения соединения формулы (5) или его соли, где R представляет собой C1-C6 алкил. В некоторых вариантах осуществления способ включает взаимодействие соединения формулы (4):

или его соли с 4-фторбензальдегидом в присутствии кислоты или кислоты Льюиса и восстанавливающего реагента в инертном растворителе при температуре от приблизительно 0 до приблизительно 80°С в течение приблизительно от 1 до приблизительно 5 часов.
В некоторых вариантах осуществления кислота может быть подходящей неорганической или органической кислотой. Подходящие неорганические кислоты включают соляную, бромистоводородную, иодистоводородную, азотную, угольную, серную и фосфорную кислоты. В других вариантах осуществления подходящие органические кислоты могут быть выбраны из алифатических, циклоалифатических, ароматических, аралифатических, карбоновых и сульфоновых классов кислот. Примерами таких кислот являются муравьиная, уксусная, пропионовая, янтарная, гликолевая, глюконовая, молочная, яблочная, винная, лимонная, аскорбиновая, глюкуроновая, малеиновая, фумаровая, пировиноградная, аспарагиновая, глутаминовая, бензойная, антраниловая, 4-гидроксибензойная, фенилуксусная, миндальная, эмбоновая, метилсульфоновая, трифторметансульфоновая, этансульфоновая, 2-гидроксиэтансульфоновая, камфорсульфоновая, бензолсульфоновая, пантотеновая, трифторуксусная, п-толуолсульфоновая, сульфаниловая, циклогексиламиносульфоновая, стеариновая, альгиновая, β-гидроксимасляная, салициловая, галактаровая и галактуроновая кислоты. В некоторых вариантах осуществления кислотой является неорганическая кислота. В некоторых вариантах осуществления кислотой является HCl. В других вариантах осуществления кислотой является органическая кислота. В некоторых вариантах осуществления органической кислотой является уксусная кислота. В некоторых вариантах осуществления органической кислотой является трифторуксусная кислота. В некоторых вариантах осуществления кислотой является п-толуолсульфоновая кислота.
В некоторых вариантах осуществления кислоту Льюиса выбирают из группы, состоящей из трифлата металла, галогенида металла, перхлората металла или трифторбората металла. Примеры включают трифлаты металлов Li(OTf), Sn(OTf)2, Cu(OTf)2, Bi(OTf)3, Ca(OTf)2, Al(OTf)3, Sm(OTf)3, Yb(OTf)3 и Sc(OTf)3.
Примеры галогенидов металлов включают CeCl3, WC13, ZrCl4, RuCl3, AlCl3, SbCl3, CoCl2, CdCl2, ZnCl2, TaCl5, InCl3, BiCl3, VCl3, SnCl4, TiCl4, ZrCl4, InBr3, MgBr2, SmI2 и SmCl3. Примеры перхлоратов включают LiClO4, NaClO4, Zn(ClO4)2 и Cu(ClO4)2. В некоторых вариантах осуществления кислотой Льюиса является AlCl3.
В некоторых вариантах осуществления восстанавливающий реагент выбирают из группы, состоящей из борогидрида натрия, цианоборогидрида натрия, порошкового Fe, TiCl3, SnCl2, гидразина и водорода в присутствии катализатора на основе переходного металла. В некоторых вариантах осуществления восстанавливающий реагент представляет собой TiCl3. В некоторых вариантах осуществления восстанавливающий реагент представляет собой порошковое Fe. В некоторых вариантах осуществления восстанавливающий реагент представляет собой водород в присутствии катализатора на основе переходного металла. В некоторых вариантах осуществления восстанавливающий реагент представляет собой SnCl2.
В некоторых вариантах катализатор на основе переходного металла выбран из группы, состоящей из палладия, никеля и платины.
Другим вариантом осуществления изобретения является способ получения соединения формулы (4) или его соли, где R представляет собой C1-C6 алкил. В некоторых вариантах осуществления способ включает взаимодействие соединения формулы (3):
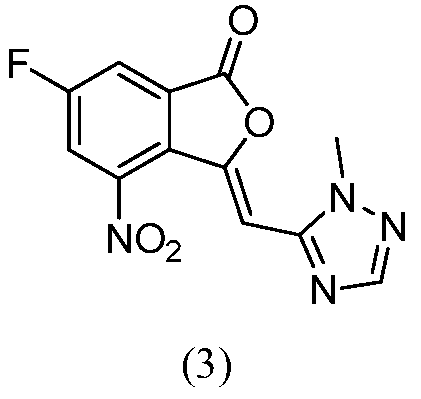
или его соли со спиртом либо в присутствии, либо в отсутствии кислоты при температуре от приблизительно 0 до приблизительно 140°С в течение от примерно 1 до примерно 24 часов.
В некоторых вариантах осуществления спирт выбран из группы, состоящей из низших алифатических спиртов, таких как метанол, этанол, н-пропанол, изопропанол, н-бутанол, изобутанол, трет-бутанол. В некоторых вариантах осуществления спиртом является метанол. В некоторых вариантах осуществления используются либо неорганическая кислота, такая как HCl, либо органическая кислота, такая как уксусная или трифторуксусная кислоты. В некоторых вариантах осуществления используется уксусная кислота.
Другим вариантом осуществления изобретения является способ получения соединения формулы (3). В некоторых вариантах осуществления способ включает взаимодействие соединения формулы (2):

с 1-метил-1H-1,2,4-триазол-5-карбальдегидом в присутствии основания и нейтрализатора воды в инертном растворителе при температуре от приблизительно 0°C до приблизительно 140°С в течение от приблизительно 1 до приблизительно 24 часов.
В некоторых вариантах осуществления основание выбирается либо из неорганического основания, такого как карбонат натрия, карбонат калия, карбонат цезия, гидрид калия и гидрид натрия; либо из органического основания, такого как триметиламин, триэтиламин, диизопропилэтиламин, пиридин, N,N-диметиламинопиридин, лутидин, имидазол и пиперидин. В некоторых вариантах осуществления основание представляет собой органическое основание. В некоторых вариантах осуществления основание представляет собой триэтиламин.
В некоторых вариантах осуществления нейтрализатор воды выбирается из группы, состоящей из натрия сульфата, магния сульфата, цеолита и ангидридов кислот. В некоторых вариантах осуществления нейтрализатор воды представляет собой уксусный ангидрид.
Другим вариантом осуществления изобретения является способ получения соединения формулы (5). Способ включает в себя проведение превращения соединения формулы (9):

или его соли в присутствии восстанавливающего реагента в инертном растворителе. В некоторых вариантах осуществления восстанавливающий реагент представляет собой порошковое железо. В некоторых вариантах осуществления восстанавливающий реагент представляет собой борогидрид натрия. В некоторых вариантах осуществления восстанавливающий реагент представляет собой водород в присутствии катализатора на основе переходного металла. В некоторых вариантах осуществления катализатор на основе переходного металла представляет собой палладий на угле.
В некоторых вариантах осуществления соединение формулы (9) превращается в соединение формулы (10):

или его соль, в котором R представляет собой C1-C6 алкил. В этом случае соединение формулы (10) дополнительно восстанавливается восстанавливающим реагентом до соединения формулы (5).
Другим вариантом осуществления изобретения является способ получения соединения формулы (9). В некоторых вариантах осуществления способ включает в себя взаимодействие соединения формулы (4):

или его соли с 1-метил-1H-1,2,4-триазол-5-карбальдегидом в инертном растворителе при условиях протекания альдольной конденсации дегидратации, которые включают, но не ограничиваясь этим, использование основания, выбранного из алкоксида лития, алкоксида натрия, лития диизопропиламина, триэтиламина, диизопропилэтиламина, пиперидина, пирролидина и пролина при температуре от приблизительно 0 до приблизительно 140°С в течение от приблизительно 1 до приблизительно 60 часов. В некоторых вариантах осуществления основание представляет собой L-пролин, и инертный растворитель представляет собой смесь метанола и дихлорметана. В некоторых вариантах осуществления растворителем является ДМСО.
Другим вариантом осуществления изобретения является альтернативный способ получения соединения формулы (1), который включает взаимодействие соединения формулы (8):

или его соли с основанием в инертном растворителе при температуре от приблизительно 0°C до приблизительно 140°С в течение от приблизительно 1 до приблизительно 24 часов. В некоторых вариантах осуществления основанием является гидрид натрия. В некоторых вариантах осуществления основанием является карбонат цезия, и растворителем - тетрагидрофуран. В некоторых вариантах осуществления температура реакции составляет от приблизительно 50 до приблизительно 60°C.
Другим вариантом осуществления изобретения является способ получения соединения формулы (8) или его соли. В некоторых вариантах осуществления способ включает взаимодействие соединения формулы (7):

или его соли с 4-фторбензальдегидом в инертном растворителе при температуре от приблизительно 0 до приблизительно 140°С в течение от приблизительно 1 до приблизительно 48 часов. В некоторых вариантах осуществления растворителем является ацетонитрил.
Другим вариантом осуществления изобретения является способ получения соединения формулы (7) или его соли, включающий взаимодействие соединения формулы (3):

или его соли с моногидратом гидразина в присутствии кислоты в инертном растворителе при температуре реакции от приблизительно 0 до приблизительно 140°С в течение от приблизительно 1 до приблизительно 48 часов. В некоторых вариантах осуществления растворителем является тетрагидрофуран, и кислотой является уксусная кислота.
Другим вариантом осуществления изобретения является альтернативный способ получения соединения формулы (1), который включает в себя проведение реакции соединения формулы (9):

или его соли с моногидратом гидразина в инертном растворителе при температуре от приблизительно 0 до приблизительно 140°С в течение от приблизительно 1 до приблизительно 48 часов. В некоторых вариантах осуществления инертным растворителем является спирт. В некоторых вариантах осуществления спиртом является метанол.
Другим вариантом осуществления изобретения является способ получения энантиомера формулы (1а) или (1b), включающий осуществление хирального хроматографического разделения соединения формулы (1).
В некоторых вариантах осуществления хиральное хроматографическое разделение включает использование препаративной ВЭЖХ, препаративной сверхкритической флюидной хроматографии (SFC) или хроматографии псевдоподвижного слоя (SMB) на хиральной неподвижной фазе. В некоторых вариантах осуществления хиральное разделение достигается с помощью SFC с использованием колонки CHIRALPAK AD или IA и CO2/MeOH или CO2/EtOH в качестве подвижной фазы.
Также вариантом осуществления является способ получения энантиомера формулы (1а) или (1b), включающий обработку энантиомера формулы (6а) или (6b):

моногидратом гидразина, где R представляет собой C1-C6 алкил. В некоторых вариантах осуществления реакция протекает в инертном растворителе при температуре от приблизительно 0 до приблизительно 140°С в течение от приблизительно 1 до приблизительно 24 часов. В некоторых вариантах осуществления инертным растворителем является спирт. В некоторых вариантах осуществления растворителем является метанол или этанол.
Также вариантом осуществления является способ получения энантиомера формулы (6а) или (6b), включающий разделение соединения формулы (5), используя хиральное хроматографическое разделение:

В некоторых вариантах осуществления хиральное хроматографическое разделение соединения формулы (5) включает в себя использование препаративной ВЭЖХ, препаративной сверхкритической флюидной хроматографии (SFC) или хроматографии псевдоподвижного слоя (SMB) на хиральной неподвижной фазе. В некоторых вариантах осуществления хиральное разделение достигается с помощью SFC с использованием колонки CHIRALPAK IC и CO2/MeOH в качестве подвижной фазы.
В некоторых вариантах хиральное разделение достигается с помощью SMB с использованием колонки CHIRALPAK IC и ацетонитрила в качестве подвижной фазы.
Примеры
Следующие примеры предназначены для иллюстрации различных вариантов, описанных здесь. В некоторых вариантах осуществления соединения получают с помощью различных схем синтеза. Эти примеры не предназначены и не должны толковаться как ограничивающие объем изобретения. Все публикации, патенты и патентные заявки, цитированные в настоящей заявке, приведены здесь в качестве ссылки во всей их полноте для любых целей.
Пример 1
(Z)-6-Фтор-3-((1-метил-1H-1,2,4-триазол-5-ил)метилен)-4-нитроизобензофуран-1(3Н)-он (3)

В 80 л стеклянный реактор с рубашкой, оборудованный холодильником, механической мешалкой, термопарой и азотом на входе/выходе, при 15-25°С загружали один за другим безводный 2-метил-тетрагидрофуран (22,7 кг), 6-фтор-4-нитроизобензофуран-1(3Н)-он (2) (2,4 кг, 12,2 моль, 1,00 экв.) и 2-метил-2Н-1,2,4-триазол-3-карбальдегид (49,6-52,6% концентрации в дихлорметане по данным ГХ, 3,59-3,38 кг, 16,0 моль, 1,31 экв.). Затем в вышеупомянутую реакционную смесь загружали триэтиламин (1,50 кг, 14,8 моль, 1,21 экв.). Реакционную смесь перемешивали в течение еще 10 минут. Ангидрид уксусной кислоты (9,09-9,10 кг, 89,0-89,1 моль, 7,30 экв.) загружали в вышеуказанную реакционную смесь при комнатной температуре в течение 20-30 минут. Реакционную смесь нагревали от температуры окружающей среды до температуры флегмы (85-95°С) в течение 80-90 минут, и смесь нагревали с обратным холодильником еще 70-90 минут. Реакционную смесь анализировали с помощью ВЭЖХ, определяя, что количество соединения (2) было снижено до ≤5%. Полученную суспензию охлаждали до 5-15°C в течении 150-250 минут. Суспензию выдерживали при 5-15°C в течение еще 80-90 минут. Суспензию фильтровали и влажный осадок промывали этилацетатом (2 л × 3). Влажный осадок высушивали в вакууме при температуре 40-50°C в течение 8 часов до получения 2,65-2,76 кг (Z)-6-фтор-3-((1-метил-1H-1,2,4-триазол-3-ил)метилен)-4-нитроизобензофуран-1(3Н)-она (3) в виде желтого твердого вещества (2,66 кг, выход: 75,3%, чистота: 98,6-98,8% по данным ВЭЖХ). LC-MS (ESI) m/z: 291 (M+1)+. 1H-ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 3,94 (с, 3Н), 7,15 (с, 1H), 8,10 (с, 1H), 8,40-8,42 (дд, J1=6,4 Гц, J2=2,4 Гц, 1H), 8,58-8,61 (дд, J1=8,8 Гц, J2=2,4 Гц, 1H).
Пример 2
Метил-5-фтор-2-(2-(1-метил-1Н-1,2,4-триазол-5-ил)ацетил)-3-нитробензоат (4)

Пример 2А
(Z)-6-Фтор-3-((1-метил-1H-1,2,4-триазол-3-ил)метилен-4-нитроизобензофуран-1(3Н)-он (3) (177 г, 0,6 моль, 1,0 экв.) и HCl (2 н. в метаноле, 3 л, 6 моль, 10 экв.) загружали в 5 л трехгорлую колбу, снабженную механической мешалкой, термометром и азотом на входе/выходе. Реакционную смесь перемешивали при комнатной температуре в течение 25 часов. Реакционную смесь анализировали с помощью ВЭЖХ, определяя, что осталось 0,8% соединения (3). Реакционную смесь концентрировали под вакуумом при 40°C досуха и получали метил 5-фтор-2-(2-(1-метил-1H-1,2,4-триазол-3-ил)ацетил)-3-нитробензоата гидрохлорид (4) в виде желтого твердого вещества (201 г, выход: 93,4%). Это вещество было использовано для следующего этапа без дополнительной очистки. LC-MS (ESI) m/z: 323 (M+1)+. 1Н-ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 3,89 (с, 3Н), 3,92 (с, 3Н), 4,60 (с, 2Н), 7,85 (с, 1H), 8,25-8,28 (дд, J1=8,4 Гц, J2=2,8 Гц, 2H), 8,52-8,54 (дд, J1=8,4 Гц, J2=2,8 Гц, 2H).
Пример 2B
Альтернативная процедура обработки, показанная в примере 2А, является следующей. Вместо того, чтобы выпаривать реакционную смесь досуха, ее конденсировали до 2 объемов, после чего растворитель замещали 12 объемами ТГФ, а затем 12 объемами гептана. Суспензионную смесь концентрировали в 2-х объемах и фильтровали, до получения продукта. Таким образом, 1,8 кг (Z)-6-фтор-3-((1-метил-1H-1,2,4-триазол-3-ил)метилен)-4-нитроизобензофуран-1(3Н)-она (3) дают 2,15 кг (выход 96,4%) продукта метил-5-фтор-2-(2-(1-метил-1H-1,2,4-триазол-3-ил)ацетил)-3-нитробензоата гидрохлорида (4).
Пример 3
Метил-7-фтор-2-(4-фторфенил)-3-(1-метил-1Н-1,2,4-триазол-5-ил)-4-оксо-1,2,3,4-тетрагидрохинолина-5-карбоксилат (5)

Пример 3A
К суспензии метил-5-фтор-2-(2-(1-метил-1H-1,2,4-триазол-5-ил)ацетил)-3-нитробензоата (4) (5 г, 15,5 ммоль, 1 экв.) и 4-фторбензальдегида (3,6 г, 29 ммоль, 1,87 экв.) в смеси растворителей тетрагидрофурана (30 мл) и метанола (5 мл) добавляли хлорид титана(III) (20% по массе раствора в 2 н. соляной кислоте) (80 мл, 6 экв.) по каплям при перемешивании при комнатной температуре. Реакционную смесь перемешивали при 30-50°C в течение 2 часов. Затем смесь разбавляли водой (160 мл) и полученный раствор экстрагировали этилацетатом (100 мл × 4). Объединенные органические слои промывали насыщенным NaHCO3 (50 мл × 3) и водным NaHSO3 (100 мл × 3), сушили с помощью Na2SO4 и концентрировали досуха. Таким образом была получена сырая твердая масса, которую промывали петролейным эфиром (120 мл) до получения указанного в заголовке соединения в виде твердого вещества желтого цвета (5,9 г, выход: 95%, чистота 97%). LC-MS (ESI) m/z: 399 (M+1)+. 1Н-ЯМР (400 МГц, CDCl3) δ (м.д.): 3,58 (с, 3Н), 3,87 (с, 3Н), 4,16-4,19 (д, J2=13,2 Гц, 1H), 4,88 (с, 1H), 5,37-5,40 (д, J2=13,2 Гц, 1H), 6,47-6,53 (м, 2H), 6,97-7,01 (м, 2H), 7,37-7,41 (м, 2H), 7,80 (с, 1H).
Пример 3B
Альтернативная процедура обработки, показанная в примере 3А, является следующей. После завершения реакции смесь экстрагировали изопропилацетатом (20 объемов × 4) без разбавления водой. Продукт изолировали растворителем с замещением изопропилацетата на гептан с последующим ресуспендированием МТБЭ и фильтрацией. Таким образом, из 3 килограммов метил-5-фтор-2-(2-(1-метил-1H-1,2,4-триазол-5-ил)ацетил)-3-нитробензоата (4) получили 2,822 кг указанного в заголовке соединения (5) (выход 81%).
Пример 3C
К перемешиваемому раствору метил-5-фтор-2-(2-(1-метил-1H-1,2,4-триазол-5-ил)ацетил)-3-нитробензоата (4) (580 мг, 2 ммоль) и 4-фторбензальдегида (488 мг, 4 ммоль) в метаноле (0,75 мл) и тетрагидрофуране (4,5 мл) добавляли концентрированный раствор HCl (по массе 37%, 6 мл), затем в реакционную систему медленно добавляли восстанавливающее порошковое Fe (672 мг, 12 ммоль). После того как добавление было завершено, полученную смесь нагревали до 60°C и выдерживали при этой температуре в течение 3 часов. После исчезновения исходного материала (4), как это было показано LC-MS, реакционную смесь распределяли между этилацетатом (30 мл) и водой (30 мл) и водную фазу экстрагировали этилацетатом (20 мл × 3). Объединенную органическую фазу высушивали Na2SO4, концентрировали в вакууме и очищали с помощью колоночной хроматографии (этилацетат:петролейный эфир=1:1) с получением указанного в заголовке соединения (5) в виде бледно-желтого твердого вещества (300 мг, выход 40%). LC-MS (ESI) m/z: 399 (M+1)+. 1Н-ЯМР (400 МГц, CDCl3) δ (м.д.): 3,58 (с, 3Н), 3,87 (с, 3Н), 4,17 (д, 1H), 4,87 (с, 1H), 5,38 (д, 1H), 6,50 (дд, 2Н), 6,99 (дд, 2Н), 7,38 (дд, 2Н), 7,80 (с, 1H).
Пример 3D
К перемешиваемому раствору метил-5-фтор-2-(2-(1-метил-1H-1,2,4-триазол-5-ил)ацетил)-3-нитробензоата (4) (580 мг, 2 ммоль) и 4-фторбензальдегида (488 мг, 4 ммоль) в метаноле (0,75 мл) и тетрагидрофуране (4,5 мл) добавляли SnCl2 (2,28 г, 12 ммоль) и концентрированную HCl (по массе 37%, 6 мл), в полученной смеси реакция шла при 45°C в течение 3 часов, до тех пор пока LC-MS не определит исчезновение исходного материала (4) и формирование продукта около 50%. Затем смесь распределяли между этилацетатом (30 мл) и водой (30 мл) и водную фазу экстрагировали этилацетатом (20 мл × 3). Объединенную органическую фазу высушивали Na2SO4, концентрировали в вакууме и очищали с помощью колоночной хроматографии (этилацетат:петролейный эфир=1:1) с получением указанного в заголовке соединения (5) в виде бледно-желтого твердого вещества (10 мг, выход 1,3%). LC-MS (ESI) m/z: 399 (M+1)+. 1Н-ЯМР (400 МГц, CDCl3) δ (м.д.): 3,58 (с, 3Н), 3,87 (с, 3Н), 4,17 (д, 1H), 4,87 (с, 1H), 5,38 (д, 1H), 6,50 (дд, 2Н), 6,99 (дд, 2Н), 7,38 (дд, 2Н), 7,80 (с, 1H).
Пример 3E
Раствор метил-5-фтор-2-(2-(1-метил-1H-1,2,4-триазол-5-ил)ацетил)-3-нитробензоата (4) (580 мг, 2 ммоль) и 4-фторбензальдегида (488 мг, 4 ммоль) в метаноле (20 мл) и уксусной кислоте (1 мл) перемешивали при комнатной температуре в течение 24 часов в атмосфере водорода (1 бар) в присутствии каталитического количества 10% Pd/C (212 мг, 0,2 ммоль). После завершения реакции, катализатор удаляли фильтрованием через слой Celit, растворитель удаляли в вакууме, а остаток очищали с помощью колоночной хроматографии (этилацетат:петролейный эфир=1:1) с получением указанного в заголовке соединения (5) в виде бледно-желтого твердого вещества (63 мг, выход 8%). LC-MS (ESI) m/z: 399 (M+1)+. 1Н-ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 3,56 (с, 3Н), 3,86 (с, 3Н), 7,02 (дд, 2H), 7,21 (дд, 2H), 7,90 (с, 1H), 8,08 (с, 1Н), 8,26 (дд, 1Н), 8,56 (дд, 1Н).
Пример 4
5-Фтор-8-(4-фторфенил)-9-(1-метил-1Н-1,2,4-триазол-5-ил)-8,9-дигидро-2Н-пиридо[4,3,2-де]фталазин-3(7Н)-он (1)

Метил-7-фтор-2-(4-фторфенил)-3-(1-метил-1H-1,2,4-триазол-5-ил)-4-оксо-1,2,3,4-тетрагидрохинолина-5-карбоксилат (5) (150 г, 0,38 мол, 1,0 экв.) и метанол (1,7 л) загружали в 3 л трехгорлую колбу, снабженную механической мешалкой, термометром и азотом на входе/выходе. Полученную суспензию перемешивали при комнатной температуре в течение 15 минут. Гидразина гидрат (85% чистоты, 78,1 г, 1,33 мол, 3,5 экв.) загружали по каплям в вышеуказанную реакционную смесь в течение 30 минут при температуре окружающей среды. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакцию анализировали с помощью ВЭЖХ, показывающей, что оставалось примерно 2% соединения (5). Полученную суспензию фильтровали. Влажный осадок суспендировали в метаноле (2 л) и перемешивали при комнатной температуре в течение 3 часов. Вышеуказанную суспензию фильтровали и влажный осадок промывали метанолом (0,5 л). Влажный осадок затем высушивали в вакууме при 45-55°C в течение 12 часов. Таким образом было получено указанное в заголовке соединение в виде бледно-желтого твердого вещества (112 г, выход: 78,1%, чистота: 95,98% по данным ВЭЖХ). LC-MS (ESI) m/z: 381 (M+1)+. 1H-ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 3,66 (с, 3Н), 4,97-5,04 (м, 2H), 6,91-6,94 (дд, J1=2,4, J2=11,2 Гц, 1H), 7,06-7,09 (дд, J1=2,4, J2=8,8 Гц, 1H), 7,14-7,18 (м, 3H), 7,47-7,51 (м, 2H), 7,72 (с, 1H), 7,80 (с, 1H), 12,35 (с, 1H).
Пример 5
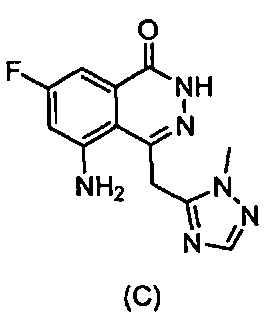
5-Амино-7-фтор-4-((1-метил-1Н-1,2,4-триазол-5-ил)метил)фталазин-1(2Н)-он

К раствору 6-фтор-3-((1-метил-1H-1,2,4-триазол-3-ил)метилен)-4-нитрозобензофуран-1(3Н)-она (3) (4,0 г, 135 ммоль) в ТГФ (100 мл) добавляли моногидрат гидразина (85%) (6 мл) при комнатной температуре в атмосфере азота. Смесь перемешивали в течение 2 часов, затем добавляли уксусную кислоту (6 мл) и смесь нагревали и выдерживали при температуре 60°C в течение 18 часов. Полученную смесь разбавляли водой (100 мл) и экстрагировали этилацетатом (100 мл × 3). Органический слой сушили над безводным Na2SO4 и выпаривали досуха, получая указанное в заголовке соединение в виде твердого вещества желтого цвета (1,6 г, выход 42%). LC-MS (ESI) m/z: 275 (M +1)+.
Пример 6
(Е)-7-Фтор-5-(4-фторбензилиденамино)-4-((1-метил-1H-1,2,4-триазол-5-ил)метил)фталазин-1(2Н)-он
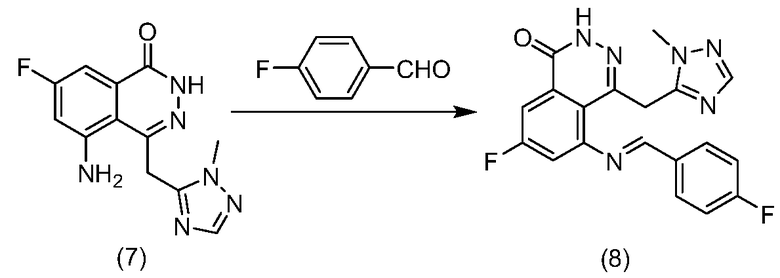
К суспендированному 5-амино-7-фтор-4-((1-метил-1H-1,2,4-триазол-3-ил)метил)фталазин-1(2Н)-он (7) (1,6 г, 5,8 ммоль) в ацетонитриле (50 мл) добавляли 4-фторбензальдегид (2,2 г, 17,5 ммоль). Смесь перемешивали при нагревании с обратным холодильником в атмосфере азота в течение 48 часов. Осадок отфильтровывали и промывали смесью растворителей (этилацетат/гексан, 1:1, 10 мл). После сушки в вакууме, получили указанное в заголовке соединение в виде твердого вещества желтого цвета (1,2 г, выход 52%). LC-MS (ESI) m/z: 381 (M+1)+.
Пример 7
5-Фтор-8-(4-фторфенил)-9-(1-метил-1Н-1,2,4-триазол-5-ил)-8,9-дигидро-2Н-пиридо[4,3,2-де]фталазин-3(7Н)-он (1)

К суспензии (E)-7-фтор-5-(4-фторбензилиденамино)-4-((1-метил-1Н-1,2,4-триазол-5-ил)метил)фталазин-1(2Н)-она (8) (2,0 г, 5,3 ммоль) в ТГФ (80 мл) добавляли карбонат цезия (3,4 г, 10,6 ммоль). Реакционную смесь перемешивали при 55°С в течение 4 часов и охлаждали до комнатной температуры. Полученную смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (50 мл × 3). Объединенные органические слои сушили над безводным Na2SO4 и выпаривали досуха, получая указанное в заголовке соединение в виде белого твердого вещества (1,6 г, выход 80%). LC-MS (ESI) m/z: 381 (M+1)+. 1H-ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 3,66 (с, 3Н), 4,97-5,04 (м, 2H), 6,91-6,94 (дд, J1=2,4, J2=11,2 Гц, 1H), 7,06-7,09 (дд, J1=2,4, J2=8,8 Гц, 1H), 7,14-7,18 (м, 3Н), 7,47-7,51 (м, 2H), 7,72 (с, 1H), 7,80 (с, 1H), 12,35 (с, 1H).
Пример 8
(Е)-Метил-5-фтор-2-(3-(4-фторфенил)-2-(1-метил-1Н-1,2,4-триазол-5-ил)акрилоил)-3-нитробензоат (9)

К перемешиваемому раствору метил-5-фтор-2-(2-(1-метил-1H-1,2,4-триазол-5-ил)ацетил)-3-нитробензоата (4) (580 мг, 2 ммоль) и 4-фторбензальдегида (488 мг, 4 ммоль) в диметилсульфоксиде (2 мл) добавляли L-пролин (230 мг, 2 ммоль). Полученную смесь выдерживали при перемешивании при 45°C в течение 48 часов. Затем реакционную систему распределяли между этилацетатом (50 мл) и водой (30 мл) и органическую фазу промывали водой (20 мл × 3), сушили Na2SO4, концентрировали в вакууме и очищали с помощью колоночной хроматографии (этилацетат:петролейный эфир=1:3) с получением указанного в заголовке соединения (9) в виде бледно-желтой пены (340 мг, выход 40%). LC-MS (ESI) m/z: 429 (M+1)+. 1H-ЯМР (400 МГц, ДМСО-d6), δ (м.д.): 3,56 (с, 3Н), 3,86 (с, 3Н), 7,02 (дд, 2Н), 7,21 (дд, 2Н), 7,90 (с, 1H), 8,08 (с, 1H), 8,26 (дд, 1H), 8,56 (дд, 1H).
Пример 9
Метил-7-фтор-2-(4-фторфенил)-1-гидрокси-3-(1-метил-1H-1,2,4-триазол-5-ил)-4-оксо-1,2,3,4-тетрагидрохинолина-5-карбоксилат (10)

К раствору (Е)-метил-5-фтор-2-(3-(4-фторфенил)-2-(1-метил-1H-1,2,4-триазол-5-ил)акрилоил)-3-нитробензоата (9) (200 мг, 0,467 ммоль) в метаноле (20 мл) добавляли 10% Pd/C (24 мг). После добавления смесь перемешивали в H2 (1 атм) при комнатной температуре в течение 0,5 часа. Затем реакционную систему фильтровали и выпаривали при пониженном давлении. Остаток очищали с помощью хроматографии (этилацетат:петролейный эфир=1:1) с получением указанного в заголовке соединения (10) (110 мг, выход 57%) в виде не совсем белой пены. LC-MS (ESI) m/z: 415 (М+Н)+. 1H-ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 3,53 (с, 3Н), 3,73 (с, 3Н), 5,08 (д, 2H), 5,27 (д, 2H), 6,95 (дд, 1H), 7,08 (дд, 2Н), 7,15 (дд, 1H), 7,42 (дд, 2Н), 7,77 (с, 1H), 9,92 (с, 1H).
Пример 10
Метил-7-фтор-2-(4-фторфенил)-3-(1-метил-1Н-1,2,4-триазол-5-ил)-4-оксо-1,2,3,4-тетрагидрохинолина-5-карбоксилат (5)

К перемешиваемому раствору метил-7-фтор-2-(4-фторфенил)-1-гидрокси-3-(1-метил-1H-1,2,4-триазол-5-ил)-4-оксо-1,2,3,4-тетрагидрохинолина-5-карбоксилата (10) (41,4 мг, 0,1 ммоль) в метаноле (5 мл) добавляли концентрированную HCl (по массе 37%, 1 мл) и восстанавливающее порошковое Fe (56 мг, 1 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 3 часов. После исчезновения соединения (10), как это было показано LC-MS, реакционная система была разделена между этилацетатом (20 мл) и водой (20 мл), а затем водную фазу экстрагировали этилацетатом (10 мл × 3). Объединенную органическую фазу сушили Na2SO4, концентрировали в вакууме и очищали с помощью колоночной хроматографии (этилацетат:петролейный эфир=1:1) с получением указанного в заголовке соединения (5) в виде бледно-желтого твердого вещества (12 мг, выход 30%). LC-MS (ESI) m/z: 399 (M+1)+. 1Н-ЯМР (400 МГц, CDCl3) δ (м.д.): 3,58 (с, 3Н), 3,87 (с, 3Н), 4,17 (д, 1H), 4,87 (с, 1H), 5,38 (д, 1H), 6,50 (дд, 2Н), 6,99 (дд, 2Н), 7,38 (дд, 2Н), 7,80 (с, 1H).
Пример 11
Метил-7-фтор-2-(4-фторфенил)-3-(1-метил-1Н-1,2,4-триазол-5-ил)-4-оксо-1,2,3,4-тетрагидрохинолина-5-карбоксилат (5)

К раствору (Е)-метил-5-фтор-2-(3-(4-фторфенил)-2-(1-метил-1H-1,2,4-триазол-5-ил)акрилоил)-3-нитробензоата (9) (214 мг, 0,5 ммоль) в метаноле (5 мл) добавляли концентрированную HCl (по массе 37%, 1 мл), затем в реакционную систему медленно добавляли восстанавливающий порошок Fe (140 мг, 2,5 ммоль). После добавления полученную смесь нагревали с обратным холодильником в течение 24 часов. Затем реакционную смесь фильтровали, концентрировали, нейтрализовали насыщенным NaHCO3 (20 мл) и экстрагировали этилацетатом (10 мл × 3). Остаток очищали с помощью хроматографии (этилацетат:петролейный эфир=1:1) с получением указанного в заголовке соединения (5) (30 мг, выход 15%) в виде не совсем белой пены. LC-MS (ESI) m/z: 399 (М + Н)+. 1Н-ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 3,56 (с, 3Н), 3,86 (с, 3Н), 7,02 (дд, 2Н), 7,21 (дд, 2Н), 7,90 (с, 1H), 8,08 (с, 1H), 8,26 (дд, 1H), 8,56 (дд, 1H).
Пример 12
(8R,9S)-5-Фтор-8-(4-фторфенил)-9-(1-метил-1Н-1,2,4-триазол-5-ил)-8,9-дигидро-2Н-пиридо[4,3,2-де]фталазин-3(7H)-он (1а) и (8S,9R)-5-фтор-8-(4-фторфенил)-9-(1-метил-1H-1,2,4-триазол-5-ил)-8,9-дигидро-2Н-пиридо[4,3,2-де]фталазин-3(7Н)-он (1b)

Хиральное разделение 5-фтор-8-(4-фторфенил)-9-(1-метил-1Н-1,2,4-триазол-5-ил)-8,9-дигидро-2Н-пиридо[4,3,2-де]фталазин-3 (7H)-она (1) (52,5 г) осуществляли на устройстве для сверхкритической флюидной хроматографии (SFC) с использованием колонки CHIRALPAK IA и CO2/метанола/диэтиламина (80/30/0,1) в качестве подвижной фазы. Таким образом были получены два энантиомера с временем удерживания 7,9 мин (23,6 г, восстановление 90%, >98% энантиомерного избытка (эи)) и 9,5 минуты (20,4 г, восстановление 78%, >98% эи), как проанализировано с помощью колонки CHIRALPAK IA 0,46 см × 15 см и СО2/метанола/диэтиламина (80/30/0,1) в качестве подвижной фазы при скорости потока 2 г/мин.
Пример 13
(2R,3R)-Метил-7-фтор-2-(4-фторфенил)-3-(1-метил-1Н-1,2,4-триазол-5-ил)-4-оксо-1,2,3,4-тетрагидрохинолина-5-карбоксилат (6а) и (2S,3S)-метил-7-фтор-2-(4-фторфенил)-3-(1-метил-1H-1,2,4-триазол-5-ил)-4-оксо-1,2,3,4-тетрагидрохинолина-5-карбоксилат (6b)

Пример 13A
Хиральное разделение соединения (5) проводили на устройстве SFC с 5 мкм колонкой CHIRALPAK®IC 3 см (внутренний диаметр) × 25 см, используя СО2/MeOH (80/20) в качестве подвижной фазы при скорости потока 65 г/мин, поддерживая при этом температуру колонки 35°C и с детекцией длины волны УФ 254 нм. Таким образом, рацемат соединения (5) (5 г) в растворе метанола разделили, в результате чего получили два энантиомера с временем удерживания 2,35 мин (2,2 г, 88% восстановление, >98% эи) и 4,25 минуты (2,3 г, 92% на восстановление, >98% эи), соответственно, когда анализировали с помощью колонки CHIRALPAK®IC 0,46 см × 15 см и CO2/MeOH (80/20) в качестве подвижной фазы при скорости потока скоростью 2 мл/мин.
Пример 13B
Хиральное разделение соединения (5) проводили на устройстве SFC с 5 мкм колонкой CHIRALPAK®IC 5 см (внутренний диаметр) × 25 см, используя СО2/MeOH (75/25) в качестве подвижной фазы при скорости потока 200 мл/мин, поддерживая при этом температуру колонки 40°C и с детекцией длины волны УФ 255 нм. Таким образом, рацемат соединения (5) (1,25 кг) в растворе метанола разделили, в результате чего получили два энантиомера с выходом приблизительно 83% и 97,4% чистоты.
Пример 13C
Альтернативно, разделение также может быть достигнуто на устройстве для хроматографии псевдоподвижного слоя (SMB), с колонкой CHIRALPAK®IC и ацетонитрилом в качестве подвижной фазы. Время удерживания для двух энантиомеров составляет 3,3 и 4,1 минуты соответственно. В некоторых вариантах осуществления производительность может быть больше, чем 6 кг сырья/сут/кг конечного продукта.
Пример 14
(8R,9S)-5-Фтор-8-(4-фторфенил)-9-(1-метил-1Н-1,2,4-триазол-5-ил)-8,9-дигидро-2Н-пиридо[4,3,2-де]фталазин-3(7Н)-он (1а) и (8S,9R)-5-фтор-8-(4-фторфенил)-9-(1-метил-1H-1,2,4-триазол-5-ил)-8,9-дигидро-2Н-пиридо[4,3,2-де]фталазин-3(7Н)-он (1b)

Пример 14A
К раствору (2R,3R)-метил-7-фтор-2-(4-фторфенил)-3-(1-метил-1Н-1,2,4-триазол-5-ил)-4-оксо-1,2,3,4-тетрагидрохинолин-5-карбоксилата (6а) или (2S,3S)-метил-7-фтор-2-(4-фторфенил)-3-(1-метил-1Н-1,2,4-триазол-5-ил)-4-оксо-1,2,3,4-тетрагидрохинолин-5-карбоксилата (6b) (400 мг, 1,0 ммоль) в этаноле (8,0 мл) добавляли гидразина моногидрат (85%, 2,0 мл) и раствор перемешивали при комнатной температуре в течение 2 часов. Затем полученный раствор концентрировали до объема 2 мл и фильтровали и полученный осадок промывали этанолом (1 мл). После сушки в вакууме при 50°C было получено указанное в заголовке соединение в виде белого твердого вещества (209 мг, выход 55%). LC-MS (ESI) m/z: 381 (M+1)+. 1Н-ЯМР (400 МГц, ДМСО-d6): δ (м.д.): 3,681 (с, 3Н), 4,99-5,06 (м, 2H), 6,92-6,96 (м, 1H), 7,08-7,11 (м, 1H), 7,16-7,21 (т, J=8,8 Гц, 2H), 7,49-7,53 (м, 2H), 7,75 (с, 1H), 7,83 (с, 1H), 12,35 (с, 1H).
Пример 14B
К раствору (2R,3R)-метил-7-фтор-2-(4-фторфенил)-3-(1-метил-1Н-1,2,4-триазол-5-ил)-4-оксо-1,2,3,4-тетрагидрохинолин-5-карбоксилата (6а) или (2S,3S)-метил-7-фтор-2-(4-фторфенил)-3-(1-метил-1Н-1,2,4-триазол-5-ил)-4-оксо-1,2,3,4-тетрагидрохинолин-5-карбоксилата (6b) (446 г) в ацетонитриле (10 объемов) добавляли моногидрат гидразина (2,9 экв.) и раствор перемешивали при комнатной температуре в течение 2 часов. Затем полученный раствор концентрировали до объема 2 мл и фильтровали. Сырой продукт суспендировали с водой (3-5 объемов) при 15-16°C. После сушки в вакууме при 50°C было получено указанное в заголовке соединение в виде белого твердого вещества (329 г, выход 77%, 99,93% чистоты). LC-MS (ESI) m/z: 381 (M+1)+; 1Н-ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 3,681 (с, 3Н), 4,99-5,06 (м, 2H), 6,92-6,96 (м, 1H), 7,08-7,11 (м, 1H), 7,16-7,21 (т, J=8,8 Гц, 2H), 7,49-7,53 (м, 2H), 7,75 (с, 1H), 7,83 (с, 1H), 12,35 (с, 1Н).
Множество вариантов было описано в настоящем документе. Однако следует понимать, что могут быть сделаны различные модификации без отступления от сущности и объема настоящего изобретения. Соответственно, другие варианты входят в объем следующей формулы.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПИРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2016 |
|
RU2745069C2 |
| ЗАМЕЩЕННЫЕ ПИРИДИНОВЫЕ И ПИРАЗИНОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE4 | 2014 |
|
RU2668073C2 |
| КРИСТАЛЛИЧЕСКАЯ ТОЗИЛАТНАЯ СОЛЬ (8S,9R)-5-ФТОР-8-(4-ФТОРФЕНИЛ)-9-(1-МЕТИЛ-1Н-1,2,4-ТРИАЗОЛ-5-ИЛ)-8-9-ДИГИДРО-2Н-ПИРИДО[4,3,2-de]ФТАЛАЗИН-3(7Н)-ОНА | 2011 |
|
RU2598606C2 |
| Соединения формул (I) и (A), фармацевтическая композиция, лекарственное средство, применение и способ получения соединения формулы (I) | 2018 |
|
RU2822758C2 |
| ДИАМИНОТРИАЗОЛЫ, ПРИГОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗ | 2003 |
|
RU2350606C2 |
| ДИГИДРОПИРИДОФТАЛАЗИНОНОВЫЕ ИНГИБИТОРЫ ПОЛИ(АДФ-РИБОЗА)ПОЛИМЕРАЗЫ | 2009 |
|
RU2514937C2 |
| ХИНОЛИНКАРБОКСАМИДНЫЕ И ХИНОЛИНКАРБОНИТРИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ mGLuR2-НЕГАТИВНЫХ АЛЛОСТЕРИЧЕСКИХ МОДУЛЯТОРОВ, КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2610262C2 |
| СПИРОСОЕДИНЕНИЯ В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРОВ МЕЛАНОКОРТИНА 4 И ИХ ПРИМЕНЕНИЯ | 2021 |
|
RU2813541C1 |
| ОКТАГИДРО КОНДЕНСИРОВАННЫЕ АЗАДЕКАЛИНОВЫЕ МОДУЛЯТОРЫ ГЛЮКОКОРТИКОИДНОГО РЕЦЕПТОРА | 2014 |
|
RU2674983C1 |
| ИНГИБИТОРЫ КАНАЛОВ С ТРАНЗИТОРНЫМ ПОТЕНЦИАЛОМ НА ОСНОВЕ ОКСАДИАЗОЛОВ | 2019 |
|
RU2818244C2 |
Изобретение относится к способу получения соединения формулы (1):

или его соли, включающий взаимодействие соединения формулы (5) 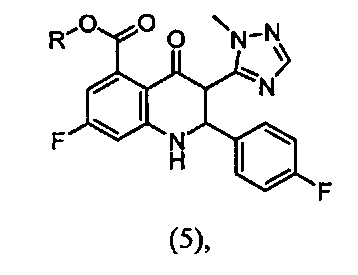 или его соли с моногидратом гидразина, где R представляет собой C1-C6 алкил; где соединение формулы (5) или его соль получают взаимодействием соединения формулы (4)
или его соли с моногидратом гидразина, где R представляет собой C1-C6 алкил; где соединение формулы (5) или его соль получают взаимодействием соединения формулы (4)  или его соли с 4-фторбензальдегидом в присутствии восстанавливающего реагента и кислоты или кислоты Льюиса, где R представляет собой C1-C6 алкил. Изобретение также относится к альтернативным способам получения соединения формулы (1) и к промежуточным соединениям, структурные формулы которых представлены в формуле изобретения. Технический результат: получение соединения формулы (1) новыми способами с улучшенными выходами и получение новых промежуточных соединений. 6 н. и 41 з.п. ф-лы, 14 пр.
или его соли с 4-фторбензальдегидом в присутствии восстанавливающего реагента и кислоты или кислоты Льюиса, где R представляет собой C1-C6 алкил. Изобретение также относится к альтернативным способам получения соединения формулы (1) и к промежуточным соединениям, структурные формулы которых представлены в формуле изобретения. Технический результат: получение соединения формулы (1) новыми способами с улучшенными выходами и получение новых промежуточных соединений. 6 н. и 41 з.п. ф-лы, 14 пр.
1. Способ получения соединения формулы (1):
или его соли, включающий:
взаимодействие соединения формулы (5)
или его соли с моногидратом гидразина, где R представляет собой C1-C6 алкил;
где соединение формулы (5) или его соль получают взаимодействием соединения формулы (4):
или его соли с 4-фторбензальдегидом в присутствии восстанавливающего реагента и кислоты или кислоты Льюиса, где R представляет собой C1-C6 алкил.
2. Способ по п.1, в котором восстанавливающий реагент представляет собой галогенид металла.
3. Способ по п.2, в котором галогенид металла представляет собой TiCl3 или SnCl2.
4. Способ по п.1, в котором восстанавливающий реагент представляет собой порошковое Fe.
5. Способ по п.1, в котором восстанавливающий реагент представляет собой водород в присутствии металлического катализатора.
6. Способ по п.5, в котором металлический катализатор выбирают из группы, состоящей из палладия, никеля и платины.
7. Способ по п.1, в котором восстанавливающий реагент представляет собой MCNBH3, где M выбирают из группы, состоящей из Li, Na, K, и Bu4N.
8. Способ по п.1, в котором кислота представляет собой неорганическую кислоту, выбранную из группы, состоящей из HCl, HBr, HI, H2SO4 и H3PO4.
9. Способ по п.1, в котором кислота представляет собой органическую кислоту, выбранную из группы, состоящей из муравьиной кислоты, уксусной кислоты, пропионовой кислоты, масляной кислоты, трифторуксусной кислоты, п-толуолсульфоновой кислоты и метилсульфоновой кислоты.
10. Способ по п.1, в котором кислоту Льюиса выбирают из группы, состоящей из AlCl3, TiCl4, SnCl4 и ZnCl2.
11. Способ по п.1, в котором соединение формулы (4) или его соль получают обработкой соединения формулы (3):

или его соли соединением R-OH в присутствии кислоты, где R выбран из группы, состоящей из метила, этила, пропила, изопропила, бутила и изобутила; и кислоту выбирают из группы, состоящей из HCl, уксусной кислоты, п-толуолсульфоновой кислоты и трифторуксусной кислоты.
12. Способ по п.11, в котором соединение формулы (3) или его соль получают взаимодействием соединения формулы (2):

с 1-метил-1H-1,2,4-триазол-5-карбальдегидом в условиях конденсации.
13. Способ по п.12, в котором условия конденсации включают использование основания в присутствии нейтрализатора воды при температуре от приблизительно 80 до приблизительно 90°C.
14. Способ по п.13, в котором основание представляет собой триэтиламин, и нейтрализатор воды представляет собой уксусный ангидрид.
15. Способ получения соединения формулы (1):
или его соли, включающий:
взаимодействие соединения формулы (8):

или его соли с основанием.
16. Способ по п.15, в котором основание представляет собой карбонат металла или гидрид металла.
17. Способ по п.16, в котором карбонат металла выбирают из группы, состоящей из Na2CO3, K2CO3 и Cs2CO3.
18. Способ по п.16, в котором гидрид металла представляет собой NaH или KH.
19. Способ по п.16, в котором соединение (8) или его соль получают взаимодействием соединения формулы (7):

или его соли с 4-фторбензальдегидом.
20. Способ по п.19, в котором соединение формулы (7) или его соль получают взаимодействием соединения формулы (3):

или его соли с моногидратом гидразина в присутствии кислоты.
21. Способ по п.20, в котором кислоту выбирают из группы, состоящей из уксусной кислоты, трифторуксусной кислоты, HCl и п-толуолсульфоновой кислоты.
22. Способ получения соединения формулы (1):

или его соли, включающий: взаимодействие соединения формулы (5)

или его соли с моногидратом гидразина, где R представляет собой C1-C6 алкил;
где соединение формулы (5) или его соль получают взаимодействием соединения формулы (9):

или его соли с восстанавливающим реагентом в инертном растворителе при температуре от приблизительно 0 до приблизительно 140°C в течение от приблизительно 1 до приблизительно 48 часов, где R представляет собой C1-C6 алкил.
23. Способ по п.22, в котором восстанавливающий реагент представляет собой боргидрид металла.
24. Способ по п.22, в котором боргидрид металла представляет собой NaBH4 или NaCNBH3.
25. Способ по п.22, в котором восстанавливающий реагент представляет собой порошковое Fe.
26. Способ по п.22, в котором восстанавливающий реагент представляет собой TiCl3 или SnCl2.
27. Способ по п.22, в котором восстанавливающий реагент представляет собой водород в присутствии переходного металла.
28. Способ по п.27, в котором переходный металл выбирают из группы, состоящей из палладия, никеля и платины.
29. Способ получения соединения формулы (1):

или его соли, включающий:
взаимодействие соединения формулы (5)

или его соли с моногидратом гидразина, где R представляет собой C1-C6 алкил;
где соединение формулы (5) или его соль получают взаимодействием соединения формулы (10):

или его соли с восстанавливающим реагентом, где R представляет собой C1-C6 алкил.
30. Способ по п.29, в котором восстанавливающий реагент представляет собой порошковое Fe.
31. Способ по п.29, в котором восстанавливающий реагент представляет собой боргидрид металла.
32. Способ по п.31, в котором боргидрид металла представляет собой NaBH4.
33. Способ по п.29, в котором восстанавливающий реагент представляет собой водород в присутствии переходного металла.
34. Способ по п.33, в котором переходный металл выбирают из группы, состоящей из палладия, никеля и платины.
35. Способ по п.29, в котором соединение формулы (10) или его соль получают взаимодействием соединения формулы (9):

или его соли с восстанавливающим реагентом, где R представляет собой C1-C6 алкил.
36. Способ по п.35, в котором восстанавливающий реагент представляет собой водород при атмосферном давлении при комнатной температуре.
37. Способ получения соединения формулы (1a) или (1b):

или его соли, включающий взаимодействие соединения формулы (6a) или (6b):

или его соли с гидразином в спирте, где R представляет собой C1-C6 алкил.
38. Способ по п.37, в котором соединение формулы (6a) или (6b) получают, осуществляя хиральное хроматографическое разделение соединения формулы (5):

39. Способ по п.38, в котором хиральное разделение достигается с помощью устройства для сверхкритической флюидной хроматографии (SFC).
40. Способ по п.39, дополнительно включающий использование хиральной неподвижной фазы, в котором хиральная неподвижная фаза представляет собой колонку CHIRALPAK.
41. Способ по п.40, в котором колонка CHIRALPAK представляет собой колонку IC или IA.
42. Способ по п.39, дополнительно включающий использование подвижной фазы, где подвижная фаза содержит CO2 и MeOH.
43. Способ по п.38, в котором хиральное разделение достигается с помощью устройства для хроматографии псевдоподвижного слоя (SMB).
44. Способ по п.43, дополнительно включающий использование хиральной неподвижной фазы, в котором хиральная неподвижная фаза представляет собой колонку CHIRALPAK.
45. Способ по п.44, в котором колонка CHIRALPAK представляет собой колонку IC.
46. Способ по п.43, дополнительно включающий использование подвижной фазы, где подвижная фаза представляет собой ацетонитрил.
47. Соединение, выбранное из группы, состоящей из:
 ,
, 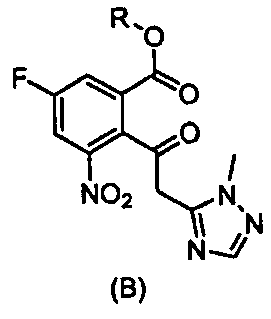 ,
, 
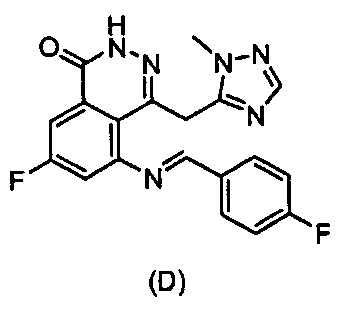 ,
, 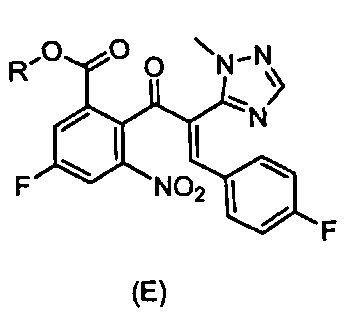 и
и 
где R представляет собой C1-C6 алкил.
| RU 2004127932 A, 27.05.2005 | |||
| EP 0778275 A1, 11.06.1997 | |||
| WO 2004080976 A1, 23.09.2004 | |||
| US 20060004028 A1, 05.01.2005 | |||
| US 20090098084 A1, 16.04.2009 |

