Область техники, к которой относится изобретение
Настоящее изобретение относится к новым фармацевтическим соединениям, фармацевтическим композициям, содержащим такие соединения, и применению соединений в качестве антагонистов рецепторов меланокортина 4 (MC4R).
Предпосылки создания изобретения
Меланокортины представляют собой пептиды, происходящие из проопиомеланокортинов (РОМС), которые связываются и активируют рецепторы, связанные с G-белком (GPCR) семейства рецепторов меланокортинов. Меланокортины регулируют широкий спектр физиологических процессов, включая сексуальную функцию и сексуальное поведение, прием пищи и метаболизм. На сегодняшний день у млекопитающих идентифицировано пять рецепторов меланокортина (MCR): MC1R, MC2R, MC3R, MC4R и MC5R, которые экспрессируются в различных тканях. MC1R специфически экспрессируется в меланоцитах и клетках меланомы, MC2R представляет собой рецептор АСТН и экспрессируется в ткани надпочечников, MC3R преимущественно экспрессируется в головном мозге и лимбической системе, MC4R широко экспрессируется в головном и спинном мозге, a MC5R экспрессируется в головном мозге и многих периферических тканях, включая кожу, жировую ткань, скелетные мышцы и лимфоидную ткань. См., например, патент США №8138188 и Saleh et al., Front. Pharmacol., 2018, 9:560.
MC4R представляет собой связанный с G-белком семи-трансмембранный рецептор, преимущественно экспрессируемый в гипоталамусе, гиппокампе и таламусе (Gantz et al. 1993 J. Biol. Chem. 2 68:15174-15179). Рецептор участвует в центральной регуляции массы тела: MC4R активируется α-меланоцит-стимулирующим гормоном (MSH), который образуется из проопиомеланокортина и инактивируется агути-родственным белком, (AGRP). α-MSH вызывает потерю массы тела, тогда как эктопическая экспрессия белка агути приводит к ожирению у агути-мышей (Fan et al. 1993 Nature 385:165-168; Lu et al. 1994 Nature 371:799-802). Дополнительные доказательства роли MC4R в регуляции массы тела вытекают из модели нокаута у мышей (Huszar et al. 1997 Cell 88:131-141), а также из мутаций, связанных с гаплонедостаточностью, у людей (Vaisse et al. 1998 Nat. Genet. 20:113-114; Yeo et al. 1998 Nat. Genet. 20:111-112; Hinney et al. 1999 J. Clin. Endocrinol. Metab. 8 4:1483-1486). У мышей с MC4R-нокаутом увеличение массы тела было заметно к возрасту 5 недель. К 15-недельному возрасту гомозиготные мутантные самки были в среднем в два раза тяжелее своих однопометников дикого типа, тогда как гомозиготные мутантные самцы были на ~50% тяжелее, чем контрольные особи дикого типа. Мыши, гетерозиготные по MC4R-нокауту, показали прибавку массы тела, промежуточную по сравнению с наблюдаемой у однопометных мышей дикого типа и гомозиготных мутантов, таким образом демонстрируя дозовый эффект абляции MC4R на регуляцию массы тела. Потребление пищи гомозиготными мутантами было увеличено на ~50% по сравнению с сибсами дикого типа (Huszar et al. 1997 Cell 88:131-141). [Из Am. J. Hum. Genet., 65:1501-1507,1999]. Было показано, что активация MC4R индуцирует эрекцию полового члена у грызунов, а инактивация MC4R вызывает ожирение (см. Hadley, 1999, Ann. NY Acad. Sci., 885:1-21; Wikberg et al. 2000, Pharmacol. Res., 42(5), 393-420; и Saleh et al., Front. Pharmacol., 2018, 9:560).
В последние годы в литературе и патентных заявках сообщалось о нескольких видах низкомолекулярных антагонистов MC4R [см., например, WO 2010052256; WO 2010081666; патент США №8044068; Chaki et al., Current Topics in Medicinal Chemistry, 2007, 7, 1145-1151; Foster et al., Current Topics in Medicinal Chemistry, 2007, 7, 1131-1136; Pontillo et al., Bioorganic & Medicinal Chemistry Letters 15 (2005) 2541-46; Vos et al., Bioorganic & Medicinal Chemistry Letters 16(2006) 2302-2305; Tao, Endocrine Reviews, 2010, 31(4):506-543; и Saleh et al., Front. Pharmacol., 2018, 9: 560]. Эти антагонисты MC4R полезны для лечения и/или профилактики связанных с MC4R состояний, заболеваний или расстройств, например кахексии [включая, например, кахексию, связанную с хроническим заболеванием, такую как кахексия, связанная с раком, кахексия, связанная с синдромом приобретенного иммунодефицита (СПИД), кахексия, связанная с сердечной недостаточностью, например кахексия, связанная с застойной сердечной недостаточностью (CHF), кахексия, связанная с хроническим заболеванием почек (CKD); кахексия, связанная с лечением хронического заболевания, такая как кахексия, связанная с лечением рака, или кахексия, связанная с лечением сердечной недостаточности (например, CHF)]; анорексии или нервной анорексии (например, анорексии у пожилых людей, анорексии, связанной с химиотерапией и/или лучевой терапией); тошноты; рвоты; потери массы тела (например, непреднамеренной потери массы тела); задержки развития; саркопении; мышечной атрофии; мышечной слабости; немощности; остеопороза; заболеваний костей (например, потери костной массы); боли; невропатической боли; беспокойства (например, посттравматического стрессового расстройства, или ПТСР); депрессии; гипертензии; нарушения питания; ожирения (например, саркопении в результате хронического ожирения); сексуальной дисфункции; и воспалительного заболевания (например, воспалительного заболевания, связанного с анорексией, или кахексией, или саркопенией, или атрофией мышц).
По-прежнему существует потребность в альтернативных антагонистах MC4R, например, для разработки новых и/или улучшенных фармацевтических препаратов (например, более эффективных, более селективных, менее токсичных и/или обладающих улучшенными биофармацевтическими свойствами, такими как физическая стабильность, растворимость, пероральная биодоступность; соответствующая метаболическая стабильность; клиренс; период полувыведения) для лечения или профилактики связанных с MC4R состояний, заболеваний или расстройств, таких как описанные в настоящей заявке. Настоящее изобретение направлено на эти и другие важные цели.
Сущность изобретения
В одном варианте осуществления (Вариант осуществления А1) настоящее изобретение предоставляет соединение формулы I:
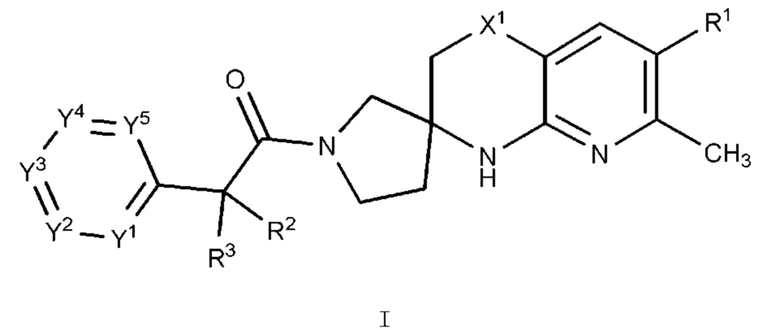
или его фармацевтически приемлемую соль, где:
R1 представляет собой Н, галоген, С1-4 алкил, С1-4 галогеналкил, С3-6 циклоалкил, 4-7-членный гетероциклоалкил, фенил или R1a, где каждый из С3-6 циклоалкила и 4-7-членного гетероциклоалкила необязательно замещен 1, 2, 3 или 4 независимо выбранными С1-4 алкильными группами, и где фенил необязательно замещен 1, 2, 3 или 4 независимо выбранными группами RB, где каждый RB представляет собой галоген, -ОН, -CN, алкил, галогеналкил, алкокси, С1-4 галогеналкокси, С3-4 циклоалкил или RB1, или два смежных RB вместе с двумя образующими кольцо атомами фенила, к которым они присоединены, образуют конденсированный 5- или 6-членный гетероарил, каждый из которых необязательно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из галогена, -ОН, -ON, алкила, галогеналкила, С1-4 алкокси и С1-4 галогеналкокси;
R1a представляет собой 5- или 6-членный гетероарил, необязательно замещенный 1, 2, 3 или 4 независимо выбранными группами RA, где каждый RA представляет собой галоген, -ОН, -CN, C1-4 алкил, C1-4 галогеналкил, C1-4 алкокси, C1-4 галогеналкокси, С3-4 циклоалкил, -N (С1-4 алкил) 2, RA1 или (С3-4 циклоалкил) -С1-4 алкил-, где каждый из C1-4 алкила, С3-4 циклоалкила и (С3-4 циклоалкил) -С1-4 алкил- необязательно замещен 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена, -ОН, -CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси и C1-4 галогеналкокси; или два смежных RA вместе с двумя образующими кольцо атомами 5- или 6-членного гетероарила, к которым они присоединены, образуют конденсированное бензольное кольцо, или конденсированный 5- или 6-членный гетероарил, или конденсированный 5- или 6-членный гетероциклоалкил, или конденсированный 5- или 6-членный циклоалкил, каждый из которых необязательно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из галогена, -ОН, -CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси и C1-4 галогеналкокси;
RA1 представляет собой 5- или 6-членный гетероарил или 5-или 6-членный гетероциклоалкил, каждый из которых необязательно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из галогена, -ОН, -CN, алкила, галогеналкила, алкокси и Cl-4 галогеналкокси;
RB1 представляет собой 5- или 6-членный гетероарил, каждый из которых необязательно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из галогена, -ОН, -CN, алкила, С1-4 галогеналкила, С1-4 алкокси и С1-4 галогеналкокси;
X1 представляет собой C(RX)2, где каждый RX независимо представляет собой Н или С1-4 алкил;
каждый из R2 и R3 независимо представляет собой Н, галоген, C1-4 алкил, С1-4 гидроксиалкил, С1-4 галогеналкил, (C1-4 алкокси)-С1-4 алкил-, С3-4 циклоалкил или (С3-4 циклоалкил) -С1-4 алкил, где каждый из С3-4 циклоалкила и (С3-4 циклоалкил) -С1-4 алкил-необязательно замещен 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена, -ОН, C1-4 алкила, C1-4 галогеналкила, С1-4 алкокси и С1-4 галогеналкокси;
или R2 и R3 вместе с атомом углерода, с которым они связаны, образуют С3-6 циклоалкил, необязательно замещенный 1, 2, 3, 4 или
5 заместителями, каждый из которых независимо выбран из галогена, -ОН, С1-4 алкила, С1-4 галогеналкила, C1-4 алкокси и С1-4 галогеналкокси;
каждый из Y1, Y2, Y3, Y4, и Y5 независимо представляет собой CR4 или N, при условии, что не более чем 3 из Y1, Y2, Y3, Y4 и Y5 представляют собой N; и
каждый R4 независимо представляет собой Н, галоген, -ОН, CN, C1-4 алкил, C1-4 галогеналкил, C1-4 алкокси, C1-4 галогеналкокси, -N(C1-2 алкил)2, С3-4 циклоалкил или (С3-4 циклоалкил)-C1-4 алкил-, где каждый из C1-4 алкила, С3-4 циклоалкила и (С3-4 циклоалкил) -C1-4 алкил- необязательно замещен 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена, -ОН, -CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси и C1-4 галогеналкокси.
Настоящее изобретение также предоставляет фармацевтическую композицию, содержащую терапевтически эффективное количество соединения формулы I или фармацевтически приемлемой соли такого соединения и фармацевтически приемлемый носитель.
Настоящее изобретение также предоставляет способ для лечения MC4R-связанного состояния, заболевания или расстройства у млекопитающего (например, человека), нуждающегося в таком лечении, при этом способ включает введение млекопитающу (например, человеку) соединения формулы I или фармацевтически приемлемой соли такого соединения.
Настоящее изобретение также предоставляет соединение формулы I или фармацевтически приемлемую соль такого соединения для применения в лечении MC4R-связанного состояния, заболевания или расстройства.
MC4R-связанное состояние, заболевание или расстройство включает такое, которое выбрано из кахексии [включая, например, кахексию, связанную с хроническим заболеванием, такую как кахексия, связанная с раком, кахексия, связанная с синдромом приобретенного иммунодефицита (СПИДом), кахексия, связанная с сердечной недостаточностью, например кахексия, связанная с застойной сердечной недостаточностью (CHF), кахексия, связанная с хроническим заболеванием почек (CKD); кахексия, связанная с лечением хронического заболевания, такая как кахексия, связанная с лечением рака, или кахексия, связанная с лечением сердечной недостаточности (например, CHF)]; анорексии или нервной анорексии (например, анорексии у пожилых людей, анорексии, связанной с химиотерапией и/или лучевой терапией); тошноты; рвоты; потери массы тела (например, непреднамеренной потери массы тела); задержки развития; саркопении; мышечной атрофии; мышечной слабости [например, мышечной слабости, связанной с хронической обструктивной болезнью легких (COPD)]; немощности; остеопороза; заболеваний костей (например, потери костной массы); боли; невропатической боли; беспокойства (например, посттравматического стрессового расстройства, или ПТСР); депрессии; гипертензии; нарушения питания; ожирения (например, саркопении в результате хронического ожирения); сексуальной дисфункции; и воспалительного заболевания (например, воспалительного заболевания, связанного с анорексией, или кахексией, или саркопенией, или мышечной атрофией).
Настоящее изобретение также предоставляет способ для лечения состояния, заболевания или расстройства у млекопитающего (например, человека), нуждающегося в таком лечении, при этом способ включает введение млекопитающу (например, человеку) соединения формулы I или фармацевтически приемлемой соли такого соединения, где состояние, заболевание или расстройство выбрано из кахексии [включая, например, кахексию, связанную с хроническим заболеванием, такую как кахексия, связанная с раком, кахексия, связанная с синдромом приобретенного иммунодефицита (СПИДом), кахексия, связанная с сердечной недостаточностью, например кахексия, связанная с застойной сердечной недостаточностью (CHF), кахексия, связанная с хроническим заболеванием почек (CKD); кахексия, связанная с лечением хронического заболевания, такая как кахексия, связанная с лечением рака, или кахексия, связанная с лечением сердечной недостаточности (например, CHF)]; анорексии или нервной анорексии (например, анорексии у пожилых людей, анорексии, связанной с химиотерапией и/или лучевой терапией); тошноты; рвоты; потери массы тела (например, непреднамеренной потери массы тела); задержки развития; саркопении; мышечной атрофии; мышечной слабости; немощности; остеопороза; заболеваний костей (например, потери костной массы); боли; невропатической боли; беспокойства (например, посттравматического стрессового расстройства, или ПТСР); депрессии; гипертензии; нарушения питания; ожирения (например, саркопении в результате хронического ожирения); сексуальной дисфункции; и воспалительного заболевания (например, воспалительного заболевания, связанного с анорексией, или кахексией, или саркопенией, или мышечной атрофией).
Настоящее изобретение также предоставляет способ антагонизирования рецептора меланокортина-4 (MC4R), при этом способ включает контактирование MC4R с соединением формулы I или фармацевтически приемлемой солью такого соединения.
Должно быть понятно, что как представленное выше общее описание, так и следующее далее подробное описание представлены только для иллюстрации и объяснения, а не для ограничения заявленного изобретения.
Краткое описание чертежей
Фиг. 1 показывает иллюстративную монокристаллическую структуру соединения Примера 14.
Фиг. 2 показывает иллюстративную монокристаллическую структуру соединения Примера 15.
Фиг. 3 показывает наблюдаемую репрезентативную порошковую рентгеновскую дифрактограмму кристаллической формы Р23.
Фиг. 4 показывает наблюдаемую репрезентативную порошковую рентгеновскую дифрактограмму кристаллической формы С69.
Фиг. 5 показывает наблюдаемую репрезентативную порошковую рентгеновскую дифрактограмму кристаллической формы Р28.
Фиг. 6 показывает наблюдаемую репрезентативную порошковую рентгеновскую дифрактограмму кристаллической формы Примера 14.
Подробное описание изобретения
Настоящее изобретение можно будет легче понять, обратившись к следующему подробному описанию иллюстративных вариантов осуществления изобретения и включенных в них примеров.
Должно быть понятно, что настоящее изобретение не ограничивается конкретными способами получения путем синтеза, которые, конечно, могут варьироваться. Также должно быть понятно, что терминология, используемая в настоящей заявке, предназначена для описания только конкретных вариантов осуществления и не предназначена для ограничения. В настоящем описании и следующей далее формуле изобретения используется ряд терминов, которые должны определяться как имеющие следующие значения:
Как используется в описании настоящего изобретения, формы единственного числа могут означать "один или более". Как используется в формуле изобретения, при использовании в сочетании со словом "включающий", формы единственного числа могут означать "один или более чем один". Как используется в настоящей заявке, "другой" могут означать по меньшей мере "еще один или более".
Термин "около" относится к относительному понятию, означающему значение близкое к указанному, находящееся в пределах плюс или минус 10% от указанного значения, к которому этот термин относится, в одном в варианте осуществления означает плюс или минус 5%, в другом в варианте осуществления означает плюс или минус 2%. Для области настоящего изобретения этот уровень приближения является подходящим, если только специально не указано, что требуются более узкие пределы значения.
"Соединение" в контексте настоящей заявки включает любое фармацевтически приемлемое производное или вариант, включая конформационные изомеры (например, цис и транс изомеры) и все оптические изомеры (например, энантиомеры и диастереомеры), рацемические, диастереомерные и другие смеси таких изомеров, а также сольваты, гидраты, изоморфы, полиморфы, таутомеры, сложные эфиры, солевые формы и пролекарства. Выражение "пролекарство" относится к соединениям, которые являются предшественниками лекарственных средств, которые после введения высвобождают лекарственное средство in vivo в результате некоторого химического или физиологического процесса (например,
пролекарство в условиях физиологического рН или при воздействии фермента преобразуется в желаемую форму, представляющую собой лекарственное средство).
Термин "алкил" означает ациклическую насыщенную алифатическую углеводородную группу, которая может быть с прямой цепью/линейной или разветвленной. Примеры таких групп включают, но не ограничиваются этим, метил, этил, н-пропил, изопропил, бутил, втор-бутил, изобутил и трет-бутил. Количество атомов углерода в алкиле и других различных углеводородсодержащих группах указано префиксом, обозначающим нижний и верхний предел количества атомов углерода в группе, т.е. префикс Ci-j означает группу с количеством атомов углерода от целого числа "i" до целого числа "j" включительно. Таким образом, например, алкил относится к алкилу, содержащему от одного до четырех атомов углерода включительно. Репрезентативные примеры алкила включают метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил и трет-бутил. В качестве другого примера, алкил относится к алкилу, содержащему от одного до двух атомов углерода включительно (т.е. метил или этил). Алкильная группа необязательно может быть замещена 1 или несколькими (например, 1-5) подходящими заместителями, если это указано.
В различных местах настоящего описания заместители соединений по изобретению раскрыты в виде групп или диапазонов. В частности, предполагается, что изобретение включает каждую отдельную подкомбинацию членов таких групп и диапазонов. Например, термин "C1-4 алкил" специально предназначен для включения C1 алкила (метила), С2 алкила (этила), С3 алкила и С4 алкила. В другом примере термин "4-7-членный гетероциклоалкил" специально предназначен для включения любой 4-, 5-, 6- или 7-членной гетероциклоалкильной группы.
В контексте настоящей заявки термин "n-членный", где n представляет собой целое число, обычно описывает количество образующих кольцо атомов в группе, где число образующих кольцо атомов равно п. Например, пиридинил является примером 6-членного гетероарильного кольца, а пиразолил является примером 5-членной гетероарильной группы.
В контексте настоящей заявки термин "алкокси" или "алкилокси" относится к -О-алкильной группе. Например, термин "C1-4 алкокси" или алкилокси" относится к группе -О-(C1-4 алкил); в качестве другого примера, термин "C1-2 алкокси" или "С1-2 алкилокси" относится к группе -О-(С1-2 алкил). Примеры алкокси включают метокси, этокси, пропокси (например, н-пропокси и изопропокси), трет-бутокси и т.п. Алкокси или алкилокси группа необязательно может быть замещена 1 или несколькими (например, 1-5) подходящими заместителями, если это указано.
Термин "гало" или "галоген" в контексте настоящей заявки означает -F, -Cl, -Br или -I.
В контексте настоящей заявки термин "галогеналкил" относится к алкильной группе, содержащей один или несколько галогеновых заместителей (вплоть до пергалогеналкила, т.е. каждый атом водорода алкильной группы замещен атомом галогена). Например, термин галогеналкил" относится к С1-4 алкильной группе, содержащей один или несколько галогеновых заместителей (вплоть до пергалогеналкила, т.е. каждый атом водорода алкильной группы замещен атомом галогена); и термин "С1-2 галогеналкил" относится к С1-2 алкильной группе (т.е. метилу или этилу), содержащей один или несколько галогеновых заместителей (вплоть до пергалогеналкила, т.е. каждый атом водорода алкильной группы замещен атомом галогена). Примеры галогеналкильных групп включают -CF3, -CHF2, -CH2F, -CH2CF3, -C2F5, -CH2Cl и т.п.
"Фторалкил" означает алкил, как определено выше, замещенный одним или несколькими фтор (-F)-заместителями (вплоть до перфторалкила, т.е. каждый атом водорода алкильной группы замещен атомом фтора). Термин "C1-2 фторалкил" относится к C1-2 алкильной группе (т.е. метилу или этилу), содержащей один или несколько фтор-заместителей (вплоть до перфторалкила, т.е. каждый атом водорода алкильной группы замещен атомом фтора); и термин "C1 фторалкил" относится к метилу, содержащему 1, 2 или 3 фтор-заместителя. Примеры C1 фторалкила включают фторметил, дифторметил и трифторметил; некоторые примеры С2 фторалкила включают 1-фторэтил, 2-фторэтил, 2,2-дифторэтил, 1,2-дифторэтил, 2,2,2-трифторэтил, 1,1,2-трифторэтил и т.п.
В контексте настоящей заявки термин "галогеналкокси" относится к -О-галогеналкильной группе. Например, термин "C1-4 галогеналкокси" относится к группе -O-(C1-4 галогеналкил); и термин "C1-2 галогеналкокси" относится к группе -O-(C1-2 галогеналкил). В качестве еще одного примера, термин "C1 галогеналкокси" относится к метокси группе, содержащей один, два или три галогеновых заместителя. Примером галогеналкокси является -OCF3 или -OCHF2.
В контексте настоящей заявки термин "фторалкокси" относится к -О-фторалкильной группе. Например, термин "С1-2 фторалкокси" относится к группе -O-(С1-2 фторалкил); и термин "С1-2 фторалкокси" относится к группе -О-(С1 фторалкил). Примеры С1 фторалкокси включают -O-CH2F, -O-CHF2 и -O-CF3. Некоторые примеры С2 фторалкокси включают -O-CH2CHF2, -O-CH2-CHF2, -O-CH2CF3, -O-CF2CH3 и -О-CF2CF3.
В контексте настоящей заявки термин "гидроксилалкил" или "гидроксиалкил" относится к алкильной группе, содержащей один или несколько (например, 1, 2 или 3) ОН заместителей. Термин "С1-4 гидроксилалкил" или "С1-4 гидроксиалкил" относится к C1-4 алкильной группе, содержащей один или несколько (например, 1, 2 или 3) ОН заместителей; и термин "C1-2 гидроксилалкил" или "С1-2 гидроксиалкил" относится к С1-2 алкильной группе, содержащей один или несколько (например, 1, 2 или 3) ОН заместителей. Примером гидроксилалкила является -СН2ОН или -СН2СН2ОН.
В контексте настоящей заявки термин "циклоалкил" относится к насыщенным или ненасыщенным неароматическим моноциклическим или полициклическим (таким как бициклические) углеводородным кольцам (например, моноциклическим, таким как циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклононил, или бициклическим, включая спиро, конденсированные или связанные мостиковой связью системы (такие как бицикло[1.1.1]пентанил, бицикло[2.2.1]гептанил, бицикло[3.2.1]октанил или бицикло[5.2.О]нонанил, декагидронафталенил и т.д.).
Циклоалкильная группа содержит 3-15 (например, 3-14, 3-10, 3-6, 3-4 или 4-6) атомов углерода. В некоторых вариантах осуществления циклоалкил необязательно может содержать один, два или более некумулированных неароматических двойных или тройных связей и/или одну-три оксо группы. В некоторых вариантах осуществления бициклоалкильная группа содержит от 6 до 14 атомов углерода. Термин "С3-4 циклоалкил" в контексте настоящей заявки означает насыщенную циклическую углеводородную группу, содержащую от 3 до 4 атомов углерода. Примеры С3-4 циклоалкила включают циклопропил и циклобутил. Также в определение циклоалкила включены группы, которые содержат одно или несколько ароматических колец (включая арил и гетероарил), конденсированных с циклоалкильным кольцом, например, бензо или пиридинильные производные циклопентана (5-членный циклоалкил), циклопентена, циклогексана (6-членный циклоалкил) и т.п., например, 6,7-дигидро-5H-циклопента[b]пиридинил, 5, 6,7,8-тетрагидрохинолинил или 1 5,6,7,8-тетрагидроизохинолинил, каждый из которых включает 5-членную или 6-членную циклоалкильную группу, которая является конденсированной с гетероарильным кольцом (т.е. пиридинильным кольцом). Циклоалкил или С3-4 циклоалкильная группа необязательно могут быть замещены 1 или несколькими (например, 1-5) подходящими заместителями, если это указано.
Термин "С3-4 циклоалкил-С1-4 алкил-" в контексте настоящей заявки означает С3-4 циклоалкил, как определено в настоящей заявке, присоединенный к исходной молекулярной группе через С3-4 алкильную группу, определенную в настоящей заявке. Некоторые примеры группы С3-4 циклоалкил-С1-4 алкил- включают циклопропилметил, 2-циклопропилэтил, 2-циклопропилпропил, 3-циклопропилпропил, циклобутилметил, 2-циклобутилэтил, 2-циклобутилпропил и 3-циклобутилпропил.
В контексте настоящей заявки термин "гетероциклоалкил" относится к моноциклической или полициклической [включая 2 или более колец, которые конденсированы вместе, включая спиро, конденсированные или связанные мостиковой связью системы, например, бициклическую кольцевую систему] насыщенной или ненасыщенной неароматической 4-15-членной кольцевой системе (такой как 4-14-членная кольцевая система, 4-12-членная кольцевая система, 5-10-членная кольцевая система, 4-7-членная кольцевая система, 4-6-членная кольцевая система или 5-6-членная кольцевая система), включающей 1-14 образующих кольцо атомов углерода и 1-10 образующих кольцо гетероатомов, каждый из которых независимо выбран из О, S и N (и необязательно Р или В, если присутствуют). Гетероциклоалкильная группа также необязательно может содержать одну или несколько оксо (т.е. =O) или тионо (т.е. =S) групп. Например, термин "4-7-членный гетероциклоалкил" относится к моноциклической или полициклической, насыщенной или ненасыщенной, неароматической 4-7-членной кольцевой системе, которая включает один или несколько образующих кольцо гетероатомов, каждый из которых независимо выбран из О, S и N. В качестве другого примера, термин "5- или 6-членный гетероциклоалкил" относится к моноциклической или полициклической, насыщенной или ненасыщенной, неароматической 5-или 6-членной кольцевой системе, которая включает один или несколько образующих кольцо гетероатомов, каждый из которых независимо выбран из О, S и N. Гетероциклоалкильная группа необязательно может быть замещена 1 или несколькими (например, 1-5) подходящими заместителями, когда это указано.
Некоторые примеры 4-7-членного гетероциклоалкила включают азетидинил, оксетанил, тетрагидрофуранил, имидазолидинил, пирролидинил, пиперидинил, пиперазинил, оксазолидинил, тиазолидинил, пиразолидинил, тиоморфолинил, тетрагидротиазинил, тетрагидротиадиазинил, морфолинил, тетрагидродиазинил и тетрагидропиранил (также известный как оксанил). Некоторые другие примеры 4-7-гетероциклоалкила включают тетрагидрофуран-2-ил, тетрагидрофуран-3-ил, тетрагидропиранил (например, тетрагидро-2H-пиран-4-ил), имидазолидин-1-ил, имидазолидин-2-ил, имидазолидин-4-ил, пирролидин-1-ил, пирролидин-2-ил, пирролидин-3-ил, пиперидин-1-ил, пиперидин-2-ил, пиперидин-3-ил, пиперидин-4-ил, пиперазин-1-ил, пиперазин-2-ил, 1, 3-оксазолидин-3-ил, 1,4-оксазепан-2-ил, изотиазолидинил, 1, 3-тиазолидин-3-ил, 1,2-пиразолидин-2-ил, 1,2-тетрагидротиазин-2-ил, 1,3-тиазинан-3-ил, 1,2-тетрагидродиазин-2-ил, 1,3-тетрагидродиазин-1-ил, 1,4-оксазин-4-ил, оксазолидинонил, 2-оксо-пиперидинил (например, 2-оксо-пиперидин-1-ил), 2-оксоазепан-3-ил, и т.п.
В контексте настоящей заявки термин "гетероарил" относится к моноциклическим или конденсированным полициклическим ароматическим гетероциклическим группам с одним или несколькими гетероатомами в качестве кольцевых членов (образующими кольцо атомами), каждый из которых независимо выбран из О, S и N, в по меньшей мере одном кольце. Гетероарильная группа содержит 5-14 образующих кольцо атомов, в том числе 1-13 атомов углерода и 1-8 гетероатомов, выбранных из О, S, и N. В некоторых вариантах осуществления гетероарильная группа содержит 5-10 образующих кольцо атомов, в том числе от одного до четырех гетероатомов. Гетероарильная групп также может содержать от одной до трех оксо или тионо (т.е. =S) групп. В некоторых вариантах осуществления гетероарильная группа содержит 5-8 образующих кольцо атомов, в том числе один, два или три гетероатома. Например, термин "5-членный гетероарил" относится к моноциклической гетероарильной группе, определенной выше, с 5 образующими кольцо атомами в моноциклическом гетероарильном кольце; термин "6-членный гетероарил" относится к моноциклической гетероарильной группе, определенной выше, с 6 образующими кольцо атомами в моноциклическом гетероарильном кольце; и термин "5- или 6-членный гетероарил" относится к моноциклической гетероарильной группе, определенной выше, с 5 или 6 образующими кольцо атомами в моноциклическом гетероарильном кольце. Гетероарильная группа необязательно может быть замещена 1 или несколькими (например, 1-5) подходящими заместителями, когда это указано. Примеры моноциклических гетероарилов включают группы с 5 образующими кольцо атомами, в том числе с одним-тремя гетероатомами, или группы с 6 образующими кольцо атомами, в том числе с одним, двумя или тремя гетероатомами азота. Примеры конденсированных бициклических гетероарилов включают два конденсированных 5-и/или 6-членных моноциклических кольца, включающих от одного до четырех гетероатомов.
Некоторые примеры гетероарильных групп включают пиридинил (например, пиридин-2-ил, пиридин-3-ил, пиридин-4-ил), пиразинил, пиримидинил (например, пиримидин-2-ил, пиримидин-4-ил или пиримидин-5-ил), пиридазинил (например, пиридазин-3-ил или пиридазин-4-ил), тиенил, фурил, имидазолил (например, 1H-имидазол-4-ил), пирролил, оксазолил (например, 1,3-оксазолил, 1, 2-оксазолил), тиазолил (например, 1,2-тиазолил, 1,3-тиазолил), пиразолил (например, пиразол-1-ил, пиразол-3-ил, пиразол-4-ил), тетразолил (например, 2H-тетразол-5-ил), триазолил (например, 1,2,3-триазолил, 1,2,4-триазолил), оксадиазолил (например, 1,2,3-оксадиазолил, 1, 2, 4-оксадиазолил или 1,3,4-оксадиазолил), тиадиазолил (например, 1,3,4-тиадиазолил или 1,2,4-тиадиазолил), хинолил, изохнолил, бензотиенил, бензофурил, индолил, бензотиазолил, 1, 2-бензоксазолил, 1H-имидазо[4,5-с]пиридинил, имидазо[1,2-а]пиридинил, 1H-пирроло[3,2-с]пиридинил, имидазо[1,2-а]пиразинил, имидазо[2,1-е][1,2,4]триазинил, имидазо[1,5-а]пиразинил, имидазо[1,2-а]пиримидинил, 1H-индазолил, 9H-пуринил, имидазо[1,2-а]пиримидинил, [1,2,4]триазоло[1,5-а]пиридинил, [1,2,4]триазоло[1,5-а]пиримидинил, [1,2,4]триазоло[4,3-b]пиридазинил, изоксазоло[5,4-е]пиридазинил, изоксазоло[3,4-е]пиридазинил, пиразоло[1,5-а]пиримидинил, 6,7-дигидро-5H-пирроло[1,2-b] [1,2,4]триазолил, пиридон, пиримидон, пиразинон, пиримидинон, 1H-имидазол-2(3H) -он, 1H-пиррол-2,5-дион, 3-оксо-2H-пиридазинил, 1H-2-оксо-пиримидинил, 1H-2-оксо-пиридинил, 2, 4 (1H, 3H)-диоксо-пиримидинил, 1H-2-оксо-пиразинил, и т.п.
В контексте настоящей заявки соединение формулы I, описанное в настоящей заявке, включает необязательные замещения и переменные. Должно быть понятно, что нормальная валентность каждого из указанных (необязательно замещенных) атомов или групп, не превышается, и что любое из необязательных замещений приводит к стабильному соединению. Также должно быть понятно, что комбинации необязательных заместителей и/или переменных допустимы, только если такие комбинации приводят к стабильному соединению.
В контексте настоящей заявки, если не указано иное, точка присоединения заместителя может быть из любого подходящего положения заместителя. Например, пиперидинил может представлять собой пиперидин-1-ил (присоединенный через N атом пиперидинила), пиперидин-2-ил (присоединенный через С атом в 2-положении пиперидинила), пиперидин-3-ил (присоединенный через С атом в 3-положении пиперидинила) или пиперидин-4-ил (присоединенный через С атом в 4-положении пиперидинила). В качестве другого примера, пиридинил (или пиридил) может представлять собой 2-пиридинил (или пиридин-2-ил), 3-пиридинил (или пиридин-3-ил) или 4-пиридинил (или пиридин-4-ил).
В контексте настоящей заявки точка присоединения заместителя может быть определена для указания положения, где заместитель присоединен к другой группе. Например, " (С3-4 циклоалкил)-С1-4 алкил-" означает, что точка присоединения находится в "С1-4 алкильной" части группы " (С3-4 циклоалкил) -C1-4 алкил-".
Когда замещенная или необязательно замещенная группа описана без указания атома, через который такая группа связана с заместителем, тогда заместитель может быть связан через любой подходящий атом в такой группе. Например в замещенном " (С3-4 циклоалкил)-C1-4 алкиле-", заместитель на циклоалкилалкиле [т.е. (С3-4 циклоалкил) -C1-4 алкил-] может быть связан с любым атомом углерода алкильной части или циклоалкильной части циклоалкилалкила. Комбинации заместителей и/или переменных допустимы, только если такие комбинации приводят к стабильным соединениям.
В контексте настоящей заявки термин "смежный" при описании относительных положений двух групп заместителей на кольцевой структуре относится к двум группам заместителей, которые соответственно присоединены к двум образующим кольцо атомам одного и того же кольца, где два образующих кольцо атома непосредственно через химическую связь. Например, в каждой из следующих структур:
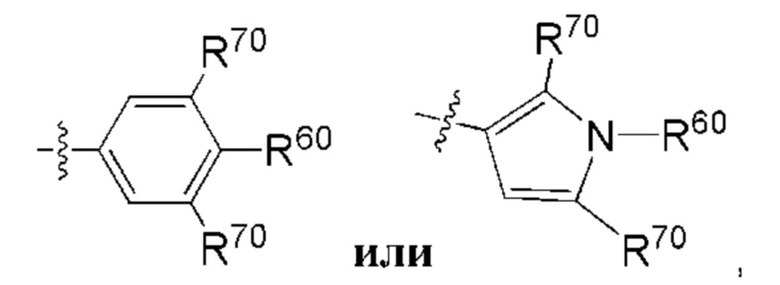
любая из двух групп R70 является смежной с группой R60.
"Млекопитающие" относятся к теплокровным позвоночным животным, характеризующимся секрецией молока самками для выкармливания детенышей, таким как морские свинки, мыши, крысы, песчанки, кошки, кролики, собаки, крупный рогатый скот, козы, овцы, лошади, обезьяны, шимпанзе и люди.
Термин «фармацевтически приемлемый» означает вещество (например, соединения по изобретению) и любую его соль или композицию, содержащую вещество или соль по изобретению, которые подходят для введения пациенту.
В контексте настоящей заявки выражения «реакционно-инертный растворитель» и «инертный растворитель» относятся к растворителю или смеси растворителей, которые не взаимодействуют с исходными веществами, реагентами, промежуточными соединениями или продуктами так, чтобы оказывать неблагоприятное влияние на выход желаемого продукта.
В контексте настоящей заявки термин «селективность» или «селективный» относится к большему эффекту соединения в первом анализе по сравнению с эффектом того же соединения во втором анализе. Например, в «кишечно-селективных» соединениях первый анализ предназначен для определения периода полужизни соединения в кишечнике, а второй анализ предназначен для определения периода полужизни соединения в печени.
«Терапевтически эффективное количество» означает количество соединения по настоящему изобретению, которое (i) лечит или предотвращает конкретное заболевание, состояние или
расстройство; (ii) ослабляет, улучшает или устраняет один или несколько симптомов конкретного заболевания, состояния или расстройства; или (iii) предотвращает или задерживает появление одного или нескольких симптомов конкретного заболевания, состояния или расстройства, описанных в настоящей заявке.
Термин «осуществлять лечение», «лечить» или «лечение», используемый в настоящей заявке, охватывает как превентивное, то есть профилактическое, так и паллиативное лечение, включая реверсию развития, облегчение, уменьшение тяжести или замедление прогрессирования заболевания (или расстройства или состояния) или любого повреждения ткани, связанного с одним или несколькими симптомами заболевания (или расстройства, или состояния).
В контексте настоящей заявки термин «контактирование» относится к объединению указанных частей в системе in vitro или системе in vivo. Например, «контактирование» MC4R с соединением по изобретению включает введение соединения по настоящему изобретению млекопитающему, такому как человек, имеющему MC4R, а также, например, введение соединения по настоящему изобретению в образец, содержащий клеточный или очищенный препарат, содержащий MC4R.
Вариант осуществления А2 представляет собой дополнительный вариант осуществления Варианта осуществления А1, где соединение формулы I или его фармацевтически приемлемая соль представляет собой соединение формулы Ia:
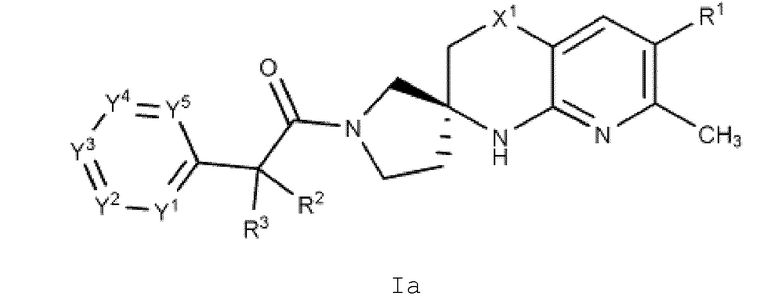
или его фармацевтически приемлемую соль, и где переменные R1, R2, R3, X1, Y1, Y2, Y3, Y4 и Y5 имеют значение, определенное в Варианте осуществления А1.
Вариант осуществления A3 представляет собой дополнительный вариант осуществления Варианта осуществления А1 или А2, где соединение или его фармацевтически приемлемая соль представляет собой соединение формулы II:
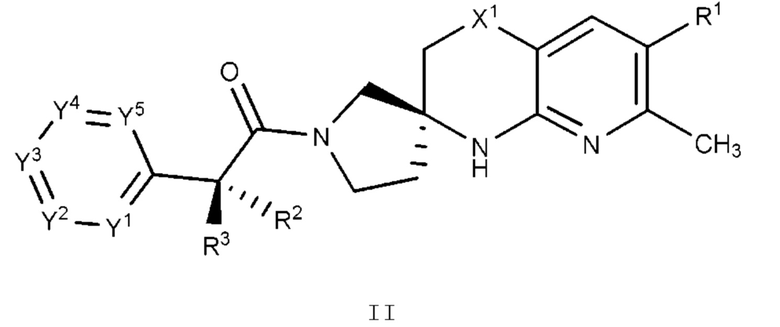
или его фармацевтически приемлемую соль, и где переменные R1, R2, R3, X1, Y1, Y2, Y3, Y4 и Y5 имеют значение, определенное в Варианте осуществления А1.
Вариант осуществления А4 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А1 - A3, где соединение или его фармацевтически приемлемая соль представляет собой соединение формулы III:
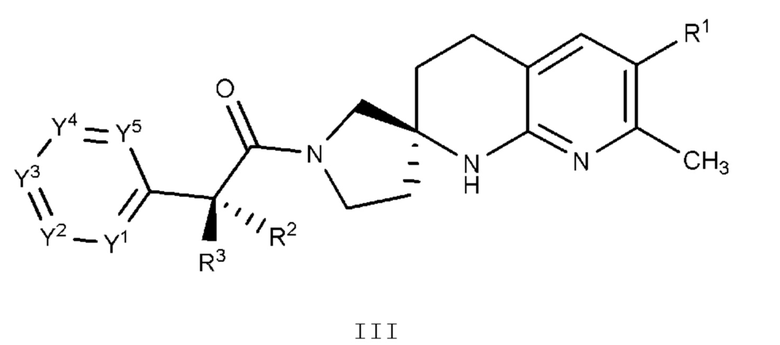
или его фармацевтически приемлемую соль, и где переменные R1, R2, R3, Y1, Y2, Y3, Y4 и Y5 имеют значение, определенное в Варианте осуществления А1.
Вариант осуществления А5 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления A1 - А4, где:
R1 представляет собой Н, галоген, C1-4 алкил, C1-4 галогеналкил, С3-6 циклоалкил, 4-7-членный гетероциклоалкил, необязательно замещенный 1-4 С1-4 алкильными группами, или R1a;
R1a представляет собой 5- или 6-членный гетероарил, необязательно замещенный 1, 2, 3 или 4 независимо выбранными группами RA, где каждый RA представляет собой галоген, -ОН, -CN, C1-4 алкил, C1-4 галогеналкил, С1-4 алкокси, С1-4 галогеналкокси, С3-4 циклоалкил или (С3-4 циклоалкил) -C1-4 алкил-, где каждый из С1-4 алкила, С3-4 циклоалкила и (С3-4 циклоалкил) -C1-4 алкил-необязательно замещен 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена, -ОН, -CN, С1-4 алкила, С1-4 галогеналкила, С1-4 алкокси и С1-4 галогеналкокси; или два смежных RA вместе с двумя кольцевыми атомами 5- или 6-членного гетероарила, к которым они присоединены, образуют конденсированное бензольное кольцо или конденсированный 5- или 6-членный гетероарил, каждый из которых необязательно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из галогена, -ОН, -CN, алкила, С1-4 галогеналкила, С1-4 алкокси и C1-4 галогеналкокси.
Вариант осуществления Аб представляет собой дополнительный вариант осуществления любого из Вариантов осуществления A1 - А4, где R1 представляет собой Н, галоген или 4-7-членный гетероциклоалкил.
Вариант осуществления А7 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления A1 - А4, где R1 представляет собой Н или галоген.
Вариант осуществления А8 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления A1 - А4, где R1 представляет собой Н.
Вариант осуществления А9 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления A1 - А4, где R1 представляет собой галоген (например, С1).
Вариант осуществления А10 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления A1 - А4, где R1 представляет собой 4-7-членный гетероциклоалкил (например, тетрагидрофуранил, тетрагидропиранил, пирролидинил, пиперидинил, пиперазинил или морфолино), необязательно замещенный 1-4 C1-4 алкильными группами.
Вариант осуществления A11 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления A1 - А4, где R1 представляет собой R1a.
Вариант осуществления А12 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления A1 - А4, где:
R1 представляет собой R1a; и
R1a представляет собой 5-членный гетероарил, необязательно замещенный 1, 2, 3 или 4 независимо выбранными группами RA, где каждый RA представляет собой галоген, -ОН, -CN, C1-4 алкил, C1-4 галогеналкил, C1-4 алкокси, C1-4 галогеналкокси, С3-4 циклоалкил или (С3-4 циклоалкил)-C1-4 алкил-, где каждый из C1-4 алкила, С3-4 циклоалкила и (С3-4 циклоалкил) -C1-4 алкил- необязательно замещен 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена, -ОН, -CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси и С1-4 галогеналкокси; или два смежных RA вместе с двумя кольцевыми атомами 5-членного гетероарила, к которым они присоединены, образуют конденсированный 5- или 6-членный гетероарил, каждый из которых необязательно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из галогена, -ОН, -CN, С1-4 алкила, C1-4 галогеналкила, С1-4 алкокси и С1-4 галогеналкокси.
Вариант осуществления А13 представляет собой дополнительный вариант осуществления Варианта осуществления A11 или А12, где R1a представляет собой 5-членный гетероарил, необязательно замещенный 1, 2, 3 или 4 независимо выбранными группами RA, где каждый RA представляет собой галоген, -ОН, -CN, С1-4 алкил, С1-4 галогеналкил, C1-4 алкокси, C1-4 галогеналкокси, С3-4 циклоалкил или (С3-4 циклоалкил)-С1-4 алкил-, где каждый из С1-4 алкила, С3-4 циклоалкила и (С3-4 циклоалкил) -С1-4 алкил- необязательно замещен 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена, -ОН, -CN, алкила, галогеналкила, алкокси и C1-4 галогеналкокси.
Вариант осуществления А14 представляет собой дополнительный вариант осуществления Варианта осуществления A11 или А12, где R1a представляет собой 5-членный гетероарил, необязательно замещенный 1, 2, 3 или 4 независимо выбранными группами RA, где каждый RA представляет собой галоген, -ОН, -CN, алкил, галогеналкил, алкокси, галогеналкокси или С3-4 циклоалкил.
Вариант осуществления А15 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А12 А14, где каждый из образующих кольцо атомов 5-членного гетероарила R1a представляет собой атом углерода или азота.
Вариант осуществления А16 представляет собой дополнительный вариант осуществления Варианта осуществления A11, где R1a представляет собой пиразолил, 1,2,4-триазолил, 1,2,3-триазолил, тетразолил, 1,2-тиазолил, 1,3,4-тиадиазолил, 1,2,4-тиадиазолил, 1,3,4-оксадиазолил, 1,2,4-оксадиазолил, 1,3-тиазолил, имидазолил, пиразоло[1,5-а]пиримидинил или [1,2,4]триазоло[1,5-а]пиридинил-, каждый из которых необязательно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из галогена, -ОН, -CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и С3-4 циклоалкил.
Вариант осуществления А17 представляет собой дополнительный вариант осуществления Варианта осуществления A11, где R1a представляет собой 1Н-пиразол-4-ил, 1Н-1, 2, 4-триазол-3-ил, 2Н-1,2,3-триазол-4-ил, 2Н-тетразол-5-ил, 1,2-тиазол-5-ил, 1,3,4-тиадиазол-2-ил, 1,2,4-тиадиазол-5-ил, 1,3,4-оксадиазол-2-ил, 1, 2, 4-оксадиазол-3-ил, 1, 3-тиазол-2-ил, 1, 3-тиазол-4-ил, 1Н-имидазол-4-ил, пиразоло[1,5-а]пиримидин-3-ил или [1,2,4]триазоло[1,5-а]пиридин-2-ил, каждый из которых необязательно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из галогена, -ОН, -CN, алкил, галогеналкила, алкокси, галогеналкокси и С3-4 циклоалкила.
Вариант осуществления А18 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления A1 - А4, где:
R1 представляет собой R1a; и
R1a представляет собой 5-членный гетероарил, замещенный 2, 3 или 4 независимо выбранными RA, где каждый RA представляет собой галоген, -ОН, -CN, С1-4 алкил, галогеналкил, C1-4 алкокси, C1-4 галогеналкокси, С3-4 циклоалкил или (С3-4 циклоалкил)-С1-4 алкил-, где каждый из C1-4 алкила, С3-4 циклоалкила и (С3-4 циклоалкил) -C1-4 алкил- необязательно замещен 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена, -ОН, -CN, С1-4 алкила, C1-4 галогеналкила, С1-4 алкокси и C1-4 галогеналкокси; или два смежных RA вместе с двумя кольцевыми атомами 5-членного гетероарила, к которым они присоединены, образуют конденсированный 5- или 6-членный гетероарил или конденсированный 5- или 6-членный гетероциклоалкил, каждый из которых необязательно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из галогена, -ОН, -CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси и C1-4 галогеналкокси.
Вариант осуществления А19 представляет собой дополнительный вариант осуществления Варианта осуществления А18, где два RA являются смежными и они, вместе с двумя кольцевыми атомами 5-членного гетероарила, к которым они присоединены, образуют конденсированный 5- или 6-членный гетероарил, который необязательно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из галогена, -ОН, -CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси и C1-4 галогеналкокси; и где каждый из остальных RA, если присутствует, независимо представляет собой галоген, -ОН, -CN, C1-4 алкил, C1-4 галогеналкил, C1-4 алкокси, C1-4 галогеналкокси, С3-4 циклоалкил или (С3-4 циклоалкил)-С1-4 алкил-, где каждый из C1-4 алкила, С3-4 циклоалкила и (С3-4 циклоалкил) -C1-4 алкил- необязательно замещен 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена, -ОН, -CN, С1-4 алкила, С1-4 галогеналкила, С1-4 алкокси и С1-4 галогеналкокси.
Вариант осуществления А20 представляет собой дополнительный вариант осуществления Варианта осуществления А18, где два RA являются смежными и они, вместе с двумя кольцевыми атомами 5-членного гетероарила, к которым они присоединены, образуют конденсированный 5- или 6-членный гетероциклоалкил, который необязательно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из галогена, -ОН, -CN, С1-4 алкила, С1-4 галогеналкила, C1-4 алкокси и C1-4 галогеналкокси; и где каждый из остальных RA, если присутствует, независимо представляет собой галоген, -ОН, -CN, С1-4 алкил, галогеналкил, C1-4 алкокси, C1-4 галогеналкокси, С3-4 циклоалкил или (С3-4 циклоалкил) -С1-4 алкил-, где каждый из С1-4 алкила, С3-4 циклоалкила и (С3-4 циклоалкил) -C1-4 алкила- необязательно замещен 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена, -ОН, -CN, С1-4 алкила, С1-4 галогеналкила, С1-4 алкокси и С1-4 галогеналкокси.
Вариант осуществления А21 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления A1 - А4, где:
R1 представляет собой R1a; и
R1a представляет собой 6-членный гетероарил, необязательно замещенный 1, 2, 3 или 4 независимо выбранными RA, где каждый RA представляет собой галоген, -ОН, -CN, C1-4 алкил, C1-4 галогеналкил, C1-4 алкокси, C1-4 галогеналкокси, С3-4 циклоалкил или (С3-4 циклоалкил)-C1-4 алкил-, где каждый из C1-4 алкила, С3-4 циклоалкила и (С3-4 циклоалкил) -C1-4 алкила- необязательно замещен 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена, -ОН, -CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси и C1-4 галогеналкокси; или два смежных RA вместе с двумя кольцевыми атомами 6-членного гетероарила, к которым они присоединены, образуют конденсированное бензольное кольцо или конденсированный 5- или 6-членный гетероарил, каждый из которых необязательно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из галогена, -ОН, -CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси и C1-4 галогеналкокси.
Вариант осуществления А22 представляет собой дополнительный вариант осуществления Варианта осуществления А21, где R1a представляет собой 6-членный гетероарил, необязательно замещенный 1, 2, 3 или 4 независимо выбранными RA, где каждый RA представляет собой галоген, -ОН, -CN, С1-4 алкил, С1-4 галогеналкил, С1-4 алкокси, С1-4 галогеналкокси, С3-4 циклоалкил или (С3-4 циклоалкил)-С1-4 алкил-, где каждый из С1-4 алкила, С3-4 циклоалкила и (С3-4 циклоалкил) -С1-4 алкил- необязательно замещен 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена, -ОН, -CN, С1-4 алкила, С1-4 галогеналкила, С1-4 алкокси и C1-4 галогеналкокси.
Вариант осуществления А23 представляет собой дополнительный вариант осуществления Варианта осуществления А21, где R1a представляет собой 6-членный гетероарил, необязательно замещенный 1, 2, 3 или 4 независимо выбранными RA, и где каждый RA представляет собой галоген, -ОН, -CN, С1-4 алкил, галогеналкил, С1-4 алкокси, С1-4 галогеналкокси или С3-4 циклоалкил.
Вариант осуществления А24 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А21 - А23, где каждый из образующих кольцо атомов 6-членного гетероарила R1a представляет собой атом углерода или азота. Еще в одном варианте осуществления 1, 2 или 3 образующих кольцо атома 6-членного гетероарила R1a являются атомами азота (а остальные из образующих кольцо атомов являются атомами углерода).
Вариант осуществления А25 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А21 - А23, где R1a представляет собой пиридинил, пиридазинил, пиразинил или пиримидинил, каждый из которых необязательно замещен 1, 2 или 3 независимо выбранными RA, где каждый RA представляет собой галоген, -ОН, -CN, C1-4 алкил, C1-4 галогеналкил, C1-4 алкокси, C1-4 галогеналкокси или С3-4 циклоалкил.
Вариант осуществления А26 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А21 - А23, где R1a представляет собой пиридин-2-ил, пиридин-3-ил, пиридазин-3-ил, пиридазин-4-ил, пиразин-2-ил, пиримидин-2-ил, пиримидин-4-ил или пиримидин-5-ил, каждый из которых необязательно замещен 1, 2 или 3 независимо выбранными RA, где каждый RA представляет собой галоген, -ОН, -CN, C1-4 алкил, C1-4 галогеналкил, C1-4 алкокси, C1-4 галогеналкокси или С3-4 циклоалкил.
Вариант осуществления А27 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А21 - А23, где R1a представляет собой пиримидинил, необязательно замещенный 1, 2 или 3 независимо выбранными RA, где каждый RA представляет собой галоген, -ОН, -CN, С1-4 алкил, С1-4 галогеналкил, С1-4 алкокси, С1-4 галогеналкокси или С3-4 циклоалкил.
Вариант осуществления А28 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А21 - А23, где R1a представляет собой пиримидин-2-ил, необязательно замещенный 1 или 2 независимо выбранными RA, где каждый RA представляет собой галоген, -ОН, -CN, С1-4 алкил, С1-4 галогеналкил, С1-4 алкокси, С1-4 галогеналкокси или С3-4 циклоалкил.
Вариант осуществления А29 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А21 - А23, где R1a представляет собой пиримидин-2-ил.
Вариант осуществления А30 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления A1 - А4, где R1 представляет собой фенил, где фенил замещен 3 или 4 независимо выбранными RB, где два смежных RB вместе с двумя образующими кольцо атомами фенила, к которым они присоединены, образуют конденсированный 5- или 6-членный гетероарил, каждый из которых необязательно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из галогена, -ОН, -CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси и C1-4 галогеналкокси; и где каждый из остальных RB независимо представляет собой галоген, ОН, -CN, С1-4 алкил, C1-4 галогеналкил, C1-4 алкокси, C1-4 галогеналкокси или С3-4 циклоалкил;
Вариант осуществления А31 представляет собой дополнительный вариант осуществления Варианта осуществления А30, где R1 представляет собой 1,2-бензоксазолил (например, 2-бензоксазол-6-ил) или 1,3-бензотиазолил (например, 1,3-бензотиазол-5-ил), каждый из которых необязательно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из галогена, -ОН, -CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси и C1-4 галогеналкокси.
Вариант осуществления А32 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления A1 - А4, где R1 представляет собой фенил, где фенил замещен группой RB1 и необязательно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из галогена, -ОН, -CN, С1-4 алкила, С1-4 галогеналкила, С1-4 алкокси, C1-4 галогеналкокси и С3-4 циклоалкила.
Вариант осуществления А33 представляет собой дополнительный вариант осуществления Варианта осуществления А32, где RB1 представляет собой 1,3,4-оксадиазолил (например, 1,3,4-оксадиазол-2-ил), 1,2,4-оксадиазолил (например, 1,2,4-оксадиазол-3-ил) или 1,3-оксазолил (например, 1,3-оксазол-5-ил), каждый из которых необязательно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из галогена, -ОН, -CN, C1-4 алкила, C1-4 галогеналкила, С1-4 алкокси, С1-4 галогеналкокси и С3-4 циклоалкила.
Вариант осуществления А34 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А1 - A3 и А5 - А33, где X1 представляет собой СН2.
Вариант осуществления А35 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А1 - A3 и А5 - А33, где X1 представляет собой СН(СН3).
Вариант осуществления А36 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А1 - А35, где каждый из R2 и R3 независимо представляет собой Н, F или С1-4 алкил.
Вариант осуществления А37 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А1 - А33, где каждый из R2 и R3 независимо представляет собой Н, F или C1-2 алкил.
Вариант осуществления А38 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А1 - А33, где каждый из R2 и R3 независимо представляет собой Н или C1-4 алкил.
Вариант осуществления А39 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А1 - А33, где каждый из R2 и R3 независимо представляет собой Н или C1-2 алкил.
Вариант осуществления А40 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А1 - А33, где каждый из R2 и R3 независимо представляет собой Н или метил.
Вариант осуществления А41 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А1 - А35, где R2 представляет собой С1-4 алкил и R3 представляет собой Н.
Вариант осуществления А42 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А1 - А35, где R2 представляет собой С1-2 алкил и R3 представляет собой Н.
Вариант осуществления А43 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А1 - А35, где R2 представляет собой метил, и R3 представляет собой Н.
Вариант осуществления А44 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А1 - А43, где каждый из Y1, Y2, Y3, Y4 и Y5 независимо представляет собой CR4.
Вариант осуществления А45 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А1 - А43, где один из Y1, Y2, Y3, Y4 и Y5 представляет собой N, а каждый из остальных независимо представляет собой CR4.
Вариант осуществления А46 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А1 - А43, где Y3 представляет собой N и каждый из Y1, Y2, Y4 и Y5 независимо представляет собой CR4.
Вариант осуществления А47 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А1 - А43, где два из Y1, Y2, Y3, Y4 и Y5 представляют собой N, а каждый из остальных из Y1, Y2, Y3, Y4 и Y5 независимо представляет собой CR4.
Вариант осуществления А48 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А1 - А43, где Y1 представляет собой N, Y3 представляет собой N, а каждый из Y2, Y4 и Y5 независимо представляет собой CR4.
Вариант осуществления А49 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А1 - А48, где каждый R4 независимо представляет собой Н, галоген, C1-2 алкил, C1-2 галогеналкил, -N(C1-4 алкил)2, С1-2 алкокси или C1-2 галогеналкокси.
Вариант осуществления А50 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А1 - А48, где каждый R4 независимо представляет собой Н, F, Cl, -СН3, C1 фторалкил, -ОСН3 или C1 фторалкокси.
Вариант осуществления А51 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А1 - А48, где каждый R4 независимо представляет собой Н, галоген или С1-2 алкокси.
Вариант осуществления А52 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А1 - А48, где каждый R4 независимо представляет собой Н, F, Cl или -ОСН3.
Вариант осуществления А53 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А1 - А48, где каждый R4 независимо представляет собой Н, F или -ОСН3.
Вариант осуществления А54 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А1 - A3, где:
R1 представляет собой R1a;
R1a представляет собой пиразолил, 1, 2,4-триазолил, 1,2,3-триазолил, тетразолил, 1,2-тиазолил, 1, 3, 4-тиадиазолил, 1,2,4-тиадиазолил, 1, 3,4-оксадиазолил, 1, 2, 4-оксадиазолил, 1,3-тиазолил, имидазолил, пиразоло[1,5-а]пиримидинил или [1,2,4]триазоло[1,5-а]пиридинил-, каждый из которых необязательно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из галогена, -ОН, -CN, C1-4 алкила, C1-4 галогеналкила, алкокси, С1-4 галогеналкокси и С3-4 циклоалкила;
X1 представляет собой СН2 или СН(СН3);
R2 представляет собой C1-2 алкил и R3 представляет собой Н;
один из Y1, Y2, Y3, Y4 и Y5 представляет собой N и каждый из остальных из Y1, Y2, Y3, Y4 и Y5 независимо представляет собой CR4; и
каждый R4 независимо представляет собой Н, F, Cl, -СН3, C1 фторалкил, -ОСН3 или C1 фторалкокси.
Вариант осуществления А33 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А1 - A3, где:
R1 представляет собой R1a;
R1a представляет собой 1,2,4-триазолил, 1, 2,3-триазолил или тетразолил (например, 2H-тетразол-5-ил), каждый из которых необязательно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из галогена, -ОН, -CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и С3-4 циклоалкила;
X1 представляет собой СН2;
R2 представляет собой С1-2 алкил и R3 представляет собой Н;
Y3 представляет собой N и каждый из Y1, Y2, Y4 и Y5 независимо представляет собой CR4; и
каждый R4 независимо представляет собой Н, F, Cl, -СН3, С1 фторалкил, -ОСН3 или С1 фторалкокси.
Вариант осуществления А56 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А1 - A3, где:
R1 представляет собой R1a;
R1a представляет собой тетразолил (например, 2H-тетразол-5-ил), необязательно замещенный 1, 2 или 3 независимо выбранными RA, где каждый RA представляет собой галоген, -ОН, -CN, алкил, C1-4 галогеналкил, алкокси, С1-4 галогеналкокси или С3-4 циклоалкил (например, R1a представляет собой 2H-тетразол-5-ил, замещенный С1-4 алкилом, таким как метил);
X1 представляет собой СН2;
R2 представляет собой метил, и R3 представляет собой Н;
Y3 представляет собой N и каждый из Y1, Y2, Y4 и Y5 независимо представляет собой CR4; и
каждый R4 независимо представляет собой Н, F, Cl или -ОСН3.
Вариант осуществления А57 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А1 - A3, где:
R1 представляет собой R1a;
R1a представляет собой пиразолил (например, 1Н-пиразол-4-ил), необязательно замещенный 1, 2 или 3 независимо выбранными RA, где каждый RA представляет собой галоген, -ОН, -CN, C1-4 алкил, C1-4 галогеналкил, C1-4 алкокси, C1-4 галогеналкокси или С3-4 циклоалкил (например, R1a представляет собой 1H-пиразол-4-ил, замещенный C1-4 алкилом, таким как метил);
X1 представляет собой СН2;
R2 представляет собой метил, и R3 представляет собой Н;
Y3 представляет собой N и каждый из Y1, Y2, Y4 и Y5 независимо представляет собой CR4; и
каждый R4 независимо представляет собой Н, F, Cl или -ОСН3 (например, каждый R4 независимо представляет собой Н, F или -ОСН3).
Вариант осуществления А58 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А1 - A3, где:
R1 представляет собой R1a;
R1a представляет собой пиридинил, пиридазинил, пиразинил или пиримидинил, каждый из которых необязательно замещен 1, 2 или 3 независимо выбранными RA, где каждый RA представляет собой галоген, -ОН, -CN, C1-4 алкил, С1-4 галогеналкил, С1-4 алкокси, С1-4 галогеналкокси или С3-4 циклоалкил;
X1 представляет собой СН2 или СН(СН3);
R2 представляет собой С1-2 алкил и R3 представляет собой Н; один из Y1, Y2, Y3, Y4 и Y5 представляет собой N, и каждый из остальных из Y1, Y2, Y3, Y4 и Y5 независимо представляет собой CR4; и
каждый R4 независимо представляет собой Н, F, Cl, -CHF2 или
-осн3.
Вариант осуществления А59 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А1 - A3, где:
R1 представляет собой R1a;
R1a представляет собой пиридинил, пиридазинил, пиразинил или пиримидинил, каждый из которых необязательно замещен 1, 2 или 3 независимо выбранными RA, где каждый RA представляет собой галоген, -ОН, -CN, С1-4 алкил, С1-4 галогеналкил, С1-4 алкокси, С1-4 галогеналкокси или С3-4 циклоалкил;
X1 представляет собой СН2;
R2 представляет собой C1-2 алкил и R3 представляет собой Н;
Y3 представляет собой N и каждый из Y1, Y2, Y4 и Y5 независимо представляет собой CR4; и
каждый R4 независимо представляет собой Н, F или -ОСН3.
Вариант осуществления А60 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А1 - A3, где:
R1 представляет собой R1a;
R1a представляет собой пиримидинил (например, пиримидин-2-ил), необязательно замещенный 1, 2 или 3 независимо выбранными RA, где каждый RA представляет собой галоген, -ОН, -CN, C1-4 алкил, C1-4 галогеналкил, C1-4 алкокси, C1-4 галогеналкокси или С3-4 циклоалкил (например, R1a представляет собой незамещенный пиримидин-2-ил);
X1 представляет собой СН2;
R2 представляет собой метил, и R3 представляет собой Н;
Y3 представляет собой N и каждый из Y1, Y2, Y4 и Y5 независимо представляет собой CR4; и
каждый R4 независимо представляет собой Н, F, Cl или -ОСН3 (например, каждый R4 независимо представляет собой Н, F или -ОСН3).
Вариант осуществления А61 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А1 - A3, где:
R1 представляет собой R1a;
R1a представляет собой [1, 2, 4]триазоло[1,5-а]пиридин-2-ил, необязательно замещенный 1, 2 или 3 независимо выбранными RA, где каждый RA представляет собой галоген, -ОН, -CN, алкил, C1-4 галогеналкил, алкокси, С1-4 галогеналкокси или С3-4 циклоалкил (например, R1a представляет собой незамещенный [1,2,4]триазоло[1,5-а]пиридин-2-ил);
X1 представляет собой СН2;
R2 представляет собой метил, и R3 представляет собой Н;
Y3 представляет собой N и каждый из Y1, Y2, Y4 и Y5 независимо представляет собой CR4; и
каждый R4 независимо представляет собой Н, F, Cl или -ОСН3 (например, каждый R4 независимо представляет собой Н, F или -ОСН3).
Вариант осуществления А62 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А1 - A3, где:
R1 представляет собой R1a;
R1a представляет собой пиридинил, пиридазинил, пиразинил или пиримидинил, каждый из которых необязательно замещен 1, 2 или 3 независимо выбранными RA, где каждый RA представляет собой галоген, -ОН, -CN, C1-4 алкил, C1-4 галогеналкил, C1-4 алкокси, C1-4 галогеналкокси или С3-4 циклоалкил;
X1 представляет собой СН2 или СН(СН3);
R2 представляет собой C1-2 алкил и R3 представляет собой Н;
каждый из Y1, Y2, Y3, Y4 и Y5 независимо представляет собой CR4; и
каждый R4 независимо представляет собой Н, F, Cl, -СН3, CF3, -CHF2 или -OCH3.
Вариант осуществления А63 представляет собой дополнительный вариант осуществления любого из Вариантов осуществления А1 - A3, где:
R1 представляет собой Н;
X1 представляет собой СН2 или СН(СН3);
каждый из R2 и R3 независимо представляет собой Н или С1-2 алкил (например, каждый из R2 и R3 представляет собой Н);
каждый из Y1, Y2, Y3, Y4 и Y5 независимо представляет собой CR4; и
каждый R4 независимо представляет собой Н, F, Cl, -СН3, CF3, -CHF2 или -ОСН3 (например, каждый R4 независимо представляет собой Н или F, например, один из R4 представляет собой F и каждый из остальных R4 представляет собой Н).
Вариант осуществления А64 (дополнительный вариант осуществления Варианта осуществления А1) предоставляет соединение, выбранное из Примеров 1-201 раздела "ПРИМЕРЫ", или его фармацевтически приемлемую соль (или его исходное соединение, когда примером соединения, например, является соль), описанные в настоящей заявке.
Вариант осуществления А65 (дополнительный вариант осуществления Варианта осуществления А1) предоставляет соединение, выбранное из
2-(5-хлор-2-метоксипиридин-4-ил)-1-[7-метил-6-(2-метил-2H-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она;
2-(б-метокси-2-метилпиримидин-4-ил)-1-[7-метил-6-(2-метил-2H-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она;
2-[6-(дифторметокси)пиридин-3-ил]-1-[7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она;
1-[7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]-2-[4-(трифторметил)фенил]пропан-1-она;
1-(4,7-диметил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил)-2-(4-фторфенил)этан-1-она;
2-(5-фтор-2-метоксипиридин-4-ил)-1-[7-метил-6-(2-метил-2H-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она;
2-(5-фтор-2-метоксипиридин-4-ил)-1-[7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она;
2-(5-хлор-2-метоксипиридин-4-ил)-1-[7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она;
2-(5-фтор-2-метоксипиридин-4-ил)-1-[7-метил-6-(1-метил-1H-пиразол-4-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она; и
2-(5-фтор-2-метоксипиридин-4-ил)-1-{7-метил-6-[(4,6-2Н2)пиримидин-2-ил]-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил}пропан-1-она,
или его фармацевтически приемлемую соль.
Вариант осуществления А66 (дополнительный вариант осуществления Варианта осуществления 1) предоставляет соединение, выбранное из
(2R)-2-(5-хлор-2-метоксипиридин-4-ил)-1-[7-метил-6-(2-метил-2H-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она;
2-(6-метокси-2-метилпиримидин-4-ил)-1-[{2S)-7-метил-6-(2-метил-2H-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она;
2-[б-(дифторметокси)пиридин-3-ил]-1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она;
1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]-2-[4 -(трифторметил)фенил]пропан-1-она;
1-(4,7-диметил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил)-2-(4-фторфенил)этан-1-она;
(2R)-2-(5-фтор-2-метоксипиридин-4-ил)-1-[(2S)-7-метил-6-(2-метил-2H-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она;
(2R)-2-(5-фтор-2-метоксипиридин-4-ил)-1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она;
(2R)-2-(5-хлор-2-метоксипиридин-4-ил)-1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2, 3'-пирролидин]-1'-ил]пропан-1-она;
(2R)-2-(5-фтор-2-метоксипиридин-4-ил)-1-[7-метил-6-(1-метил-1H-пиразол-4-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она; и
(2R)-2-(5-фтор-2-метоксипиридин-4-ил)-1-{(2S)-7-метил-6-[(4,6-2Н2)пиримидин-2-ил]-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил}пропан-1-она,
или его фармацевтически приемлемую соль.
Вариант осуществления А67 (дополнительный вариант осуществления Варианта осуществления А1) предоставляет соединение, выбранное из:
(2R)-2-(5-хлор-2-метоксипиридин-4-ил)-1-[7-метил-6-(2-метил-2H-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она, DIAST-1;
2-(6-метокси-2-метилпиримидин-4-ил)-1-[(2S)-7-метил-6-(2-метил-2H-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она, DIAST-1;
2-[6-(дифторметокси)пиридин-3-ил]-1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она, DIAST-2;
1-[(25)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]-2-[4-(трифторметил)фенил]пропан-1-она, DIAST-1;
1-(4,7-диметил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил)-2-(4-фторфенил)этан-1-она, DIAST-1;
(2R)-2-(5-фтор-2-метоксипиридин-4-ил)-1-[{2S)-7-метил-6-(2-метил-2H-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она;
(2R)-2-(5-фтор-2-метоксипиридин-4-ил)-1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она;
(2R)-2-(5-хлор-2-метоксипиридин-4-ил)-1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она;
(2R)-2-(5-фтор-2-метоксипиридин-4-ил)-1-[7-метил-6-(1-метил-1H-пиразол-4-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она, DIAST-1; и
(2R)-2-(5-фтор-2-метоксипиридин-4-ил)-1-{(25)-7-метил-6-[(4, 6-2Н2)пиримидин-2-ил]-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил}пропан-1-она,
или его фармацевтически приемлемую соль.
Вариант осуществления А68 (дополнительный вариант осуществления Варианта осуществления А1) предоставляет соединение, которое представляет собой (2R)-2-(5-Фтор-2-метоксипиридин-4-ил)-1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-он или его фармацевтически приемлемую соль.
Вариант осуществления А69 (дополнительный вариант осуществления Варианта осуществления А1) предоставляет соединение, которое представляет собой (2R)-2-(5-Фтор-2-метоксипиридин-4-ил)-1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-он.
Вариант осуществления А70 (дополнительный вариант осуществления Варианта осуществления А69) предоставляет кристаллическую форму (2R)-2-(5-Фтор-2-метоксипиридин-4-ил)-1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она. Еще в одном варианте осуществления Варианта осуществления А70 кристаллическая форма демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере один характеристический пик, выраженный в градусах 2θ, выбранный из 8,7±0,2°; 11,1±0,2°; и 13,3±0,2°.
Еще в одном варианте осуществления Варианта осуществления А70, кристаллическая форма представляет собой Форму I, описанную в настоящей заявке в Примере 14. В некоторых вариантах осуществления Форма I демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере один характеристический пик, выраженный в градусах 2θ, при 8,7±0,2°. В некоторых вариантах осуществления Форма I демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере один характеристический пик, выраженный в градусах 2θ, при 11,1±0,2°. В некоторых вариантах осуществления Форма I демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере один характеристический пик, выраженный в градусах 2θ, при 13,3±0,2°. В некоторых вариантах осуществления Форма I демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере два характеристических пика, выраженных в градусах 2θ, выбранных из 8,7±0,2°; 11,1±0,2°; и 13,3±0,2°. В некоторых вариантах осуществления Форма I демонстрирует порошковую рентгеновскую дифрактограмму, включающую два характеристических пика, выраженных в градусах 2θ, выбранных из 8,7±0,2°; и 11,1±0,2. В некоторых вариантах осуществления Форма I демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере три характеристических пика, выраженных в градусах 2θ, выбранных из 8,7±0,2°; 11,1±0,2°; и 13,3±0,2°.
В некоторых вариантах осуществления Форма I демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере два характеристических пика, выраженных в градусах 2θ, выбранных из 8,7±0,2°; 11,1±0,2°; и 26,0±0,2. В некоторых вариантах осуществления Форма I демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере два характеристических пика, выраженных в градусах 2θ, выбранных из 8,7±0,2°; и 26,0±0,2°. В некоторых вариантах осуществления Форма I демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере два характеристических пика, выраженных в градусах 2θ, выбранных из 11,1±0,2°; и 26,0±0,2°. В некоторых вариантах осуществления Форма I демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере три характеристических пика, выраженных в градусах 2θ, выбранных из 8,7±0,2°; 11,1±0,2°; и 26,0±0,2°.
В некоторых вариантах осуществления Форма I демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере два характеристических пика, выраженных в градусах 2θ, которые представлены в Таблице X1. В некоторых вариантах осуществления Форма I демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере три характеристических пика, выраженных в градусах 2θ, которые представлены в Таблице X1. В некоторых вариантах осуществления Форма I демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере четыре (например, 4, 5, 6, 7, 8, 9 или 10) характеристических пика, выраженных в градусах 2θ, которые представлены в Таблице X1.
В некоторых вариантах осуществления Форма I демонстрирует порошковую рентгеновскую дифрактограмму, по существу такую, как показано на Фиг. 1.
Каждый вариант осуществления, Пример или его фармацевтически приемлемая соль могут быть заявлены индивидуально или сгруппированными вместе в любой комбинации с любым количеством каждых отдельных вариантов осуществления, описанных в настоящей заявке.
Спироциклическое соединение формулы I (также включающее соединение формулы la, II или III) по изобретению можно использовать в любых из фармацевтических композиций, применений и способов по изобретению, описанных в настоящей заявке.
Соединение формулы I или его фармацевтически приемлемая соль по настоящему изобретению является антагонистом MC4R. Таким образом, настоящее изобретение также предоставляет способ антагонизирования MC4R (либо in vitro, либо in vivo), включающий контактирование (включая инкубацию) MC4R с соединением формулы I или его фармацевтически приемлемой солью (например, соединением, выбранным из Вариантов осуществления A1 - А70 или Примеров 1-201, представленных в настоящей заявке), описанными в настоящей заявке.
Количество соединения формулы I или его фармацевтически приемлемой соли, используемое в любом из способов (или применений) по настоящему изобретению, является эффективным для антагонизирования MC4R.
Еще один вариант осуществления настоящего изобретения включает применение соединения формулы I или фармацевтически приемлемой соли соединения (такого как соединение, выбранное из Вариантов осуществления A1 - А70 или Примеров 1-201, представленных в настоящей заявке) в качестве лекарственного средства, в частности, когда лекарственное средство предназначено для применения в лечении MC4R-связанного состояния, заболевания или расстройства, включающее введение млекопитающему, такому как человек, нуждающемуся в таком лечении, терапевтически эффективного количества.
Еще один вариант осуществления настоящего изобретения включает применение соединения формулы I или фармацевтически приемлемой соли соединения (такого как соединение, выбранное из Вариантов осуществления A1 - А70 или Примеров 1-201, представленных в настоящей заявке) для получения лекарственного средства для лечения MC4R-связанного состояния, заболевания или расстройства, которое включает введение млекопитающему, такому как человек, нуждающемуся в таком лечении, терапевтически эффективного количества.
Еще один вариант осуществления настоящего изобретения включает применение соединения формулы I или фармацевтически приемлемой соли соединения (такого как соединение, выбранное из Вариантов осуществления A1 - А70 или Примеров 1-201, представленных в настоящей заявке) в качестве лекарственного средства, в частности, когда лекарственное средство предназначено для применения в лечении состояния, заболевания или расстройства, выбранного из кахексии [включая, например, кахексию, связанную с хроническим заболеванием, такую как кахексия, связанная с раком, кахексия, связанная с синдромом приобретенного иммунодефицита (СПИДом), кахексия, связанная с сердечной недостаточностью, например кахексия, связанная с застойной сердечной недостаточностью (CHF), кахексия, связанная с хроническим заболеванием почек (CKD); кахексия, связанная с лечением хронического заболевания, такая как кахексия, связанная с лечением рака, или кахексия, связанная с лечением сердечной недостаточности (например, CHF)]; анорексии или нервной анорексии (например, анорексии у пожилых людей, анорексии, связанной с химиотерапией и/или лучевой терапией); тошноты; рвоты; потери массы тела (например, непреднамеренной потери массы тела); задержки развития; саркопении; мышечной атрофии; мышечной слабости; немощности; остеопороза; заболеваний костей (например, потери костной массы); боли; невропатической боли; беспокойства (например, посттравматического стрессового расстройства, или ПТСР); депрессии; гипертензии; нарушения питания; ожирения (например, саркопении в результате хронического ожирения); сексуальной дисфункции; и воспалительного заболевания (например, воспалительного заболевания, связанного с анорексией, или кахексией, или саркопенией, или мышечной атрофией), которое включает введение млекопитающему, такому как человек, нуждающемуся в таком лечении, терапевтически эффективного количества соединения или его фармацевтически приемлемой соли.
Еще один вариант осуществления настоящего изобретения включает применение соединения формулы I или фармацевтически приемлемой соли соединения (такого как соединение, выбранное из Вариантов осуществления A1 - А70 или Примеров 1-201, представленных в настоящей заявке) для получения лекарственного средства для лечения состояния, заболевания или расстройства, выбранного из кахексии [включая, например, кахексию, связанную с хроническим заболеванием, такую как кахексия, связанная с раком, кахексия, связанная с синдромом приобретенного иммунодефицита (СПИДом), кахексия, связанная с сердечной недостаточностью, например кахексия, связанная с застойной сердечной недостаточностью (CHF), кахексия, связанная с хроническим заболеванием почек (CKD); кахексия, связанная с лечением хронического заболевания, такая как кахексия, связанная с лечением рака, или кахексия, связанная с лечением сердечной недостаточности (например, CHF)]; анорексии или нервной анорексии (например, анорексии у пожилых людей, анорексии, связанной с химиотерапией и/или лучевой терапией); тошноты; рвоты; потери массы тела (например, непреднамеренной потери массы тела); задержки развития; саркопении; мышечной атрофии; мышечной слабости; немощности; остеопороза; заболеваний костей (например, потери костной массы); боли; невропатической боли; беспокойства (например, посттравматического стрессового расстройства, или ПТСР); депрессии; гипертензии; нарушения питания; ожирения (например, саркопении в результате хронического ожирения); сексуальной дисфункции; и воспалительного заболевания (например, воспалительного заболевания, связанного с анорексией, или кахексией, или саркопенией, или мышечной атрофией), которое включает введение млекопитающему, такому как человек, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы I или фармацевтически приемлемой соли такого соединения.
Соединения по настоящему изобретению могут содержать асимметрические или хиральные центры, и поэтому существуют в различных стереоизомерных формах. Если не указано иное, предполагается, что все стереоизомерные формы соединений по настоящему изобретению, а также их смеси, включая рацемические смеси, составляют часть настоящего изобретения. Кроме того, настоящее изобретение охватывает все геометрические и позиционные изомеры. Например, если соединение по настоящему изобретению включает двойную связь или конденсированное кольцо, обе формы цис- и транс-, а также смеси охватываются объемом изобретения.
Хиральные соединения по изобретению (и их хиральные предшественники) могут быть получены в энантиомерно-обогащенной форме с использованием хроматографии, типично высокоэффективной жидкостной хроматографии (ВЭЖХ) или сверхкритической флюидной хроматографии (СФХ), на смоле с асимметрической неподвижной фазой и с подвижной фазой, состоящей из углеводорода, типично гептана или гексана, содержащего от 0 до 50% изопропанола, типично от 2 до 20%, и от 0 до 5% алкиламина, типично 0,1% диэтиламина (DEA) или изопропиламина. Концентрация элюента дает обогащенную смесь. В случае использования СФХ подвижная фаза может состоять из сверхкритической жидкости, типично диоксида углерода, содержащей 2-50% спирта, такого как метанол, этанол или изопропанол.
Диастереомерные смеси можно разделить на их индивидуальные диастереоизомеры на основе их физико-химических различий способами, хорошо известными специалистам в данной области, такими как хроматография и/или фракционная кристаллизация. Энантиомеры можно разделить путем преобразования энантиомерной смеси в диастереомерную смесь путем взаимодействия с подходящим оптически активным соединением (например, хиральным вспомогательным веществом, таким как хиральный спирт или хлорангидрид кислоты Мошера), разделения диастереоизомеров и преобразования (например, путем гидролиза) отдельных диастереоизомеров в соответствующие чистые энантиомеры. Энантиомеры также можно разделить на хиральной ВЭЖХ колонке. Альтернативно, определенные стереоизомеры могут быть синтезированы с использованием оптически активного исходного вещества, путем асимметричного синтеза с использованием оптически активных реагентов, субстратов, катализаторов или растворителей или путем преобразования одного стереоизомера в другой путем асимметрической трансформации.
В некоторых вариантах осуществления соединения по изобретению могут содержать асимметричные атомы углерода. Углерод-углеродные связи соединений формулы I могут быть показаны в настоящей заявке с использованием сплошной линии ( ), волнистой линии
), волнистой линии  , сплошного клина
, сплошного клина  или штрихованного клина
или штрихованного клина  . Использование сплошной линии для представления связей с асимметричными атомами углерода означает, что включены все возможные стереоизомеры (например, конкретные энантиомеры, рацемические смеси и т.д.) по этому атому углерода. Использование сплошного или штрихованного клина для представления связей с асимметричными атомами углерода означает, что только показанный стереоизомер предназначен для включения. Использование волнистой линии для представления связей с асимметричными атомами углерода означает, что стереохимия неизвестна (если не указано иное). Возможны случаи, когда соединения по изобретению могут содержать более одного асимметричного атома углерода. Для таких соединений использование сплошной линии для представления связей с асимметричными атомами углерода означает, что предполагается включение всех возможных стереоизомеров. Например, если не указано иное, предполагается, что соединения по изобретению могут существовать в виде энантиомеров и диастереомеров или в виде рацематов и их смесей. Использование сплошной линии для представления связей с одним или несколькими асимметричными атомами углерода в соединении по изобретению и использование сплошного или штрихованного клина для представления связей с другими асимметричными атомами углерода в том же соединении означает, что присутствует смесь диастереомеров.
. Использование сплошной линии для представления связей с асимметричными атомами углерода означает, что включены все возможные стереоизомеры (например, конкретные энантиомеры, рацемические смеси и т.д.) по этому атому углерода. Использование сплошного или штрихованного клина для представления связей с асимметричными атомами углерода означает, что только показанный стереоизомер предназначен для включения. Использование волнистой линии для представления связей с асимметричными атомами углерода означает, что стереохимия неизвестна (если не указано иное). Возможны случаи, когда соединения по изобретению могут содержать более одного асимметричного атома углерода. Для таких соединений использование сплошной линии для представления связей с асимметричными атомами углерода означает, что предполагается включение всех возможных стереоизомеров. Например, если не указано иное, предполагается, что соединения по изобретению могут существовать в виде энантиомеров и диастереомеров или в виде рацематов и их смесей. Использование сплошной линии для представления связей с одним или несколькими асимметричными атомами углерода в соединении по изобретению и использование сплошного или штрихованного клина для представления связей с другими асимметричными атомами углерода в том же соединении означает, что присутствует смесь диастереомеров.
Если соединения по настоящему изобретению обладают двумя или более стереогенными центрами и в названии указана абсолютная или относительная стереохимия, обозначения R и S относятся соответственно к каждому стереогенному центру в порядке возрастания номеров (1, 2, 3 и т.д.) в соответствии с общепринятыми системами нумерации IUPAC для каждой молекулы. Когда соединения по настоящему изобретению обладают одним или несколькими стереогенными центрами и стереохимия не указана ни в названии, ни в структуре, подразумевается, что название или структура предназначены для охвата всех форм соединения, включая рацемическую форму.
Соединения по настоящему изобретению могут содержать олефиноподобные двойные связи. Когда такие связи присутствуют, соединения по изобретению существуют в виде цис- и трансконфигураций и в виде их смесей. Термин «цис» относится к ориентации двух заместителей относительно друг друга и плоскости кольца (либо оба «вверх», либо оба «вниз»). Аналогично, термин «транс» относится к ориентации двух заместителей относительно друг друга и плоскости кольца (заместители находятся на противоположных сторонах кольца).
Также возможно, что промежуточные соединения и соединения по настоящему изобретению могут существовать в различных таутомерных формах, и все такие формы охватываются объемом изобретения. Термин «таутомер» или «таутомерная форма» относится к структурным изомерам с различной энергией, которые являются взаимопревращаемыми через низкий энергетический барьер. Например, протонные таутомеры (также известные как прототропные таутомеры) включают взаимные превращения посредством миграции протона, такие как кето-енольная и имин-енаминовая изомеризация.
Валентные таутомеры включают взаимные превращения за счет реорганизации некоторых связывающих электронов.
В объем заявленных соединений по настоящему изобретению включены все стереоизомеры, геометрические изомеры и таутомерные формы соединений формулы I, включая соединения, демонстрирующие более одного типа изомерии, и смеси одного или нескольких из них. Также включены соли присоединения кислоты или основания, в которых противоион является оптически активным, например, D-лактат или L-лизин, или рацемические, например, DL-тартрат или DL-аргинин.
Настоящее изобретение включает все фармацевтически приемлемые изотопно-меченые соединения формулы I, в которых один или несколько атомов заменены атомами, имеющими такой же атомный номер, но атомную массу или массовое число, отличающееся от атомной массы или массового числа, обычно встречающихся в природе.
Примеры изотопов, подходящих для включения в соединения по изобретению, включают изотопы водорода, такие как 2Н и 3Н, углерода, такие как 11С, 13С и 14С, хлора, такие как 36Cl, фтора, такие как 18F, йода, такие как 123I, 124I и 125I, азота, такие как 13N и 15N, кислорода, такие как 15O, 17О и 18О, фосфора, такие как 32Р, и серы, такие как 35S.
Некоторые изотопно-меченые соединения формулы I, например, соединения, включающие радиоактивный изотоп, полезны в исследованиях распределения лекарственного средства и/или субстрата в тканях. Радиоактивные изотопы тритий, т.е. 3Н, и углерод-14, т.е. 14С, особенно полезны для этой цели ввиду легкости их введения и удобных способов детекции.
Замещение более тяжелыми изотопами, такими как дейтерий, т.е. 2Н, может обеспечить определенные терапевтические преимущества, обусловленные большей метаболической стабильностью, например, увеличенным периодом полувыведения in vivo или уменьшенными требованиями к дозировке, и, следовательно, может быть предпочтительным в некоторых обстоятельствах.
Замещение изотопами, излучающими позитроны, такими как 11С, 18F, 15О и 13N, может быть полезно в исследованиях позитронно-эмиссионной томографии (ПЭТ) для изучения степени занятости рецепторов субстратом.
Изотопно-меченные соединения формулы I, как правило, могут быть получены обычными способами, известными специалистам в данной области, или способами, аналогичными тем, которые описаны в прилагаемых Примерах и Получениях, с использованием соответствующих изотопно-меченных реагентов вместо использовавшегося ранее немеченого реагента.
Соединения по настоящему изобретению можно выделить и использовать как таковые или, когда это возможно, в форме их фармацевтически приемлемой соли. Термин «соли» относится к неорганическим и органическим солям соединения по настоящему изобретению. Эти соли могут быть получены in situ во время окончательного выделения и очистки соединения или путем отдельной обработки соединения подходящей органической или неорганической кислотой и выделения образованной таким образом соли.
Соли, охватываемые термином «фармацевтически приемлемые соли», относятся к соединениям по настоящему изобретению, которые обычно получают путем взаимодействия свободного основания с подходящей органической или неорганической кислотой с получением соли соединения по настоящему изобретению, подходящей для введения пациенту. Подходящие кислотно-аддитивные соли образуются из кислот, образующих нетоксичные соли. Примеры солей включают ацетат, адипат, аспартат, бензоат, безилат, бикарбонат/карбонат, бисульфат/сульфат, борат, камзилат, цитрат, цикламат, эдисилат, эзилат, формиат, фумарат, глуцептат, глюконат, глюкуронат, гексафторфосфат, гибензат, гидрохлорид/хлорид, гидробромид/бромид, гидройодид/йодид, изетионат, лактат, малат, малеат, малонат, мезилат, метилсульфат, нафтилат, 2-напсилат, никотинат, нитрат, оротат, оксалат, пальмитат, памоат, фосфат/гидрофосфат/дигидрофосфат, пироглутамат, сахарат, стеарат, сукцинат, таннат, тартрат, тозилат, трифторацетат и ксинофоат. См., например, Berge, et al. J. Pharm. Sci. 66, 1-19 (1977); Handbook of Pharmaceutical Salts: Properties, Selection and Use by Stahl and Wermuth (Wiley-VCH, 2002).
Соединения формулы I и их фармацевтически приемлемые соли могут существовать в несольватированных и сольватированных формах. Термин «сольват» используется в настоящей заявке для описания молекулярного комплекса, включающего соединение формулы I или его фармацевтически приемлемую соль и одну или несколько молекул фармацевтически приемлемого растворителя, например, этанола. Термин «гидрат» используется, когда указанный растворитель представляет собой воду.
В настоящее время принятой системой классификации органических гидратов является система, которая определяет гидраты с изолированными участками, канальные гидраты или координированные с ионами металлов гидраты - см. Polymorphism in Pharmaceutical Solids by K.R. Morris (Ed. HG Brittain, Marcel Dekker, 1995). Гидраты с изолированными участками представляют собой гидраты, в которых молекулы воды изолированы от прямого контакта друг с другом за счет промежуточных органических молекул. В канальных гидратах молекулы воды находятся в каналах решетки, где они находятся рядом с другими молекулами воды. В координированных с ионами металлов гидратах молекулы воды связаны с ионами металлов.
Когда растворитель или вода прочно связаны, комплекс может иметь четко определенную стехиометрию, не зависящую от влажности. Однако, когда растворитель или вода слабо связаны, как в канальных сольватах и гигроскопичных соединениях, содержание воды/растворителя может зависеть от влажности и условий сушки. В таких случаях нестехиометрия будет нормой.
Также в объем изобретения включены многокомпонентные комплексы (отличные от солей и сольватов), в которых лекарственное средство и по меньшей мере один другой компонент присутствуют в стехиометрических или нестехиометрических количествах. Комплексы этого типа включают клатраты (комплексы включения лекарственное средство-хозяин) и сокристаллы. Последние обычно определяются как кристаллические комплексы нейтральных молекулярных составляющих, которые связаны друг с другом посредством нековалентных взаимодействий, но также могут представлять собой комплекс нейтральной молекулы с солью. Сокристаллы могут быть получены кристаллизацией из расплава, перекристаллизацией из растворителей или физическим измельчением компонентов вместе - см. О. Almarsson and М.J. Zaworotko, Chem. Commun.f 17, 1889-1896 (2004). Общий обзор многокомпонентных комплексов см. в Haleblian, J. Pharm. Sci., 64 (8), 1269-1288 (1975).
Соединения по изобретению включают соединения формулы I или их фармацевтически приемлемые соли, как определено выше, их полиморфы и изомеры (включая оптические, геометрические и таутомерные изомеры), как определено ниже, и изотопно-меченные соединения формулы I или их фармацевтически приемлемые соли.
Соединения по настоящему изобретению можно вводить в виде пролекарств. Таким образом, некоторые производные соединений формулы I или их фармацевтически приемлемые соли, которые сами могут иметь небольшую фармакологическую активность или не иметь ее, могут при введении внутрь или нанесении на поверхность тела превращаться в соединения формулы I или их фармацевтически приемлемые соли, обладающие желаемой активностью, например, в результате гидролитического расщепления. Такие производные называются «пролекарствами». [Дополнительную информацию об использовании пролекарств можно найти в Prodrugs as Novel Delivery Systems, Vol. 14, ACS Symposium Series (T. Higuchi and W. Stella) и «Bioreversible Carriers in Drug Design», Pergamon Press, 1987 (ed. E.B. Roche, American Pharmaceutical As soci ation).]
Пролекарства можно получить, например, путем замены соответствующих функциональных групп, присутствующих в соединениях формулы I или их фармацевтически приемлемых солях, определенными группами, известными специалистам в данной области как лпро-группы, как описано, например, в "Design of Prodrugs" Н. Bundgaard (Elsevier, 1985).
Некоторые примеры таких пролекарств включают:
(i) когда соединение формулы I или его фармацевтически приемлемая соль содержит спиртовую функциональную группу (-ОН) -эфир такого соединения, например, замещение водорода (C1-С6)алканоилоксиметилом; или фосфатный эфир (-O-PO3H2) или сульфатный эфир (-O-SO3H) или их фармацевтически приемлемые соли; и
(ii) амид или карбамат функциональной аминогруппы, присутствующей в формуле (I) или (II), где водород амино NH группы замещен (C1-С10)алканоилом или (C1-С10)алкоксикарбонилом, соответственно.
Также включены в объем изобретения активные метаболиты соединений формулы I (включая пролекарства) или их фармацевтически приемлемых солей, т.е. соединения, образуемые in vivo при введении лекарственного средства, часто в результате окисления или дезалкилирования. Некоторые примеры метаболитов в соответствии с изобретением включают:
(i) когда соединение формулы I или его фармацевтически приемлемая соль содержит метильную группу - гидроксиметильное производное такого соединения (-СН3 → -СН2ОН), и
(ii) когда соединение формулы I или его фармацевтически приемлемая соль содержит алкокси группу - гидрокси-производное такого соединения (-OR → -ОН).
Некоторые соединения по настоящему изобретению могут существовать более чем в одной кристаллической форме (обычно называются «полиморфами»). Полиморфы могут быть получены кристаллизацией в различных условиях, например, с использованием различных растворителей или различных смесей растворителей для перекристаллизации; кристаллизацией при разных температурах; и/или различных режимах охлаждения, от очень быстрого до очень медленного охлаждения в процессе кристаллизации. Полиморфы также могут быть получены нагреванием или плавлением соединения по настоящему изобретению с последующим постепенным или быстрым охлаждением. Присутствие полиморфов можно определить методами ЯМР-спектроскопии с твердым зондом, ИК-спектроскопии, дифференциальной сканирующей калориметрии, порошковой рентгеновской дифракции или другими подобными методами.
Как правило, соединения по настоящему изобретению можно получить способами, которые включают способы, аналогичные известным в области химии, особенно в свете описания, содержащегося в настоящей заявке. Некоторые способы получения соединений по настоящему изобретению представлены в качестве дополнительных признаков изобретения и проиллюстрированы следующими схемами реакций. Другие способы могут быть описаны в экспериментальной части. Конкретные схемы синтеза соединений формулы I или их фармацевтически приемлемых солей приведены ниже. Следует отметить, что тетразолы обычно представляют собой высокоэнергетическую функциональную группу, и следует соблюдать осторожность при синтезе и обращении с тетразол-содержащими молекулами.
Для синтеза соединений по настоящему изобретению можно использовать пути синтеза, которые включают способы, аналогичные тем, которые хорошо известны в области химии, особенно в свете содержащегося в настоящей заявке описания. Исходные вещества, как правило, доступны из коммерческих источников, таких как MilliporeSigma (Milwaukee, WI), или легко могут быть получены способами, хорошо известными специалистам в данной области [например, могут быть получены способами, в общем виде описанными в Louis F. Fieser and Mary Fieser, Reagents for Organic Synthesis, v. 1-19, Wiley, New York (1967-1999 ed.), или Beilsteins Handbuch der Organischen Chemie, 4, Aufl. ed. Springer-Verlag, Berlin, включая приложения (также доступны в онлайн-базе данных Beilstein)]. Многие из используемых в настоящей заявке соединений родственны или получены из соединений, представляющих большой научный интерес и коммерческую потребность, и, соответственно, многие такие соединения коммерчески доступны, или о них сообщается в литературе, или их легко получить из других общедоступных соединений методами, описанными в литературе.
Следует отметить, что при получении соединений формулы I или их солей некоторые способы получения, описанные в настоящей заявке, могут потребовать защиты отдаленной функциональной группы (например, первичный амин, вторичный амин, карбоксил в предшественниках формулы I). Необходимость такой защиты будет варьироваться в зависимости от характера отдаленной функциональной группы и условий способов получения. Необходимость такой защиты легко может определить специалист в данной области техники. Использование таких способов защиты/снятия защиты также находится в пределах компетенции специалистов в данной области. Общее описание защитных групп и их использования см. в T.W. Greene, Protective Groups in Organic Synthesis, 5th Edition, John Wiley & Sons, New York, 2014. Например, некоторые соединения содержат функциональные группы первичных аминов или карбоновых кислот, которые могут мешать реакциям на других участках молекулы, если их оставить незащищенными. Соответственно, такие функциональные группы могут быть защищены соответствующей защитной группой, которую можно удалить на последующей стадии. Подходящие защитные группы для защиты аминов и карбоновых кислот включают те защитные группы, которые обычно используются в синтезе пептидов (такие как N-трет-бутоксикарбонил (Boc), бензилоксикарбонил (Cbz) и 9-флуоренилметоксикарбонил (Fmoc) для аминов и низшие алкиловые или бензиловые эфиры для карбоновых кислот), которые обычно не являются химически реакционноспособными в описанных условиях реакции и обычно могут быть удалены без химического изменения других функциональных групп в соединениях формулы I.
Реакции можно отслеживать любым подходящим способом, известным в данной области. Например, образование продукта можно контролировать спектроскопическими методами, такими как спектроскопия ядерного магнитного резонанса (например, ХН или 13С), инфракрасная спектроскопия, спектрофотометрия (например, в УФ- и видимой области), масс-спектрометрия, или
хроматографическими методами, такими как высокоэффективная жидкостная хроматография (ВЭЖХ) или тонкослойная хроматография (ТСХ).
Соединения формулы I, их соли и промежуточные соединения могут быть получены в соответствии со следующими схемами реакций и сопутствующим обсуждением. Схемы реакций, описанные ниже, предназначены для предоставления общего описания методологии, используемой при получении соединений по настоящему изобретению.
Некоторые из соединений по настоящему изобретению содержат один хиральный центр со стереохимическим обозначением (R или S), а другие будут содержать два отдельных хиральных центра со стереохимическим обозначением (R или S). Специалистам в данной области будет очевидно, что большинство синтетических преобразований можно осуществить подобным образом, независимо от того, являются ли вещества энантиообогащенными или рацемическими. Кроме того, разделение на желаемые оптически активные вещества может происходить в любой желаемой точке последовательности с использованием хорошо известных способов, таких как описанные в настоящей заявке и в литературе по химии.
Если не указано иное, R1, R2, R3, X1, Y1, Y2, Y3, Y4, Y5 и структурная Формула I (включая, например, Ia, II) в схемах реакций и обсуждении, которые представлены ниже, имеют такое же значение, как определено в настоящей заявке, или соответствуют описанному в настоящей заявке. Как правило, соединения по настоящему изобретению можно получить способами, которые включают способы, аналогичные тем, которые известны в области химии, в частности, в свете описания, содержащегося в настоящей заявке. Некоторые способы получения соединений по настоящему изобретению и их промежуточных соединений представлены в качестве дополнительных признаков изобретения и проиллюстрированы следующими схемами реакций. Другие способы могут быть описаны в экспериментальной части. Схемы и примеры, представленные в настоящей заявке (включая соответствующее описание) представлены только для иллюстрации и не предназначены для ограничения объема настоящего изобретения.
На Схемах реакций, которые представлены ниже, переменные Xc, X2, X3, X4, X5, X6, X7, X8, X9, R5, R6, R7, R8, R9, R10, R11, R12, R13, R14, LG1, LG2, LG3, LG4, PG1, PG2, PG3, PG4, PG5, PG6, PG7 и PG8 имеют такое же значение, как определено в настоящей заявке, или соответствуют описанному в настоящей заявке в формуле изобретения для формулы I, если не указано иное. Для каждой из переменных ее значение остается таким же, как описано выше, если в дальнейшем не указано иное.
Схема 1 относится к синтезу соединений формулы I, Ia и II. Кислоты формулы 1-1 можно подвергнуть взаимодействию с аминами формулы 1-2 (или их солями) с использованием стандартных условий амидирования, используя реагенты для реакций сочетания, такие как 1,1'-карбонилдиимидазол, 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинан 2,4,6-триоксид (Т3Р), О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфат (HATU) или другие, с получением соединений формулы I. Альтернативно, кислоты формулы 1-1 можно подвергнуть взаимодействию с аминами формулы 1-3 аналогичным способом с получением соединений формулы Ia. Кислоты формулы 1-1 могут быть приобретены коммерческим путем, синтезированы, как описано в Org. Process Res. Dev. 1997, 1, 72, или синтезированы, как описано в настоящей заявке. Амины формулы 1-2 могут быть синтезированы, как описано в настоящей заявке. Соединения формулы I, которые содержат смеси энантиомеров или диастереомеров, можно разделить с использованием сверхкритической флюидной хроматографии или обращеннофазовой хроматографии с хиральной колонкой, когда необходимо разделить их на индивидуальные диастереомеры или энантиомеры, если это требуется для получения соединений формулы Ia или II.
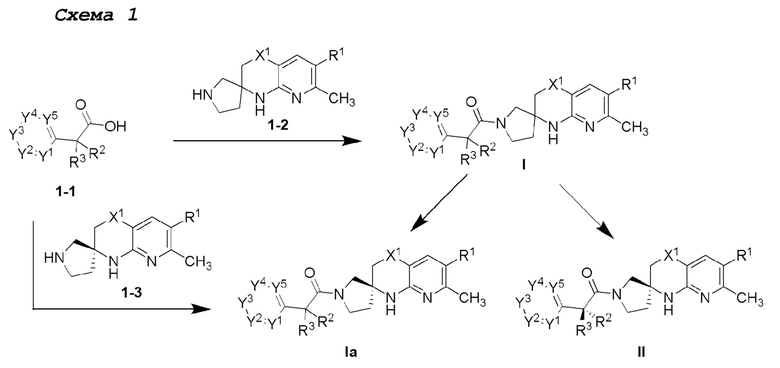
Схема 2 описывает альтернативный синтез соединений формулы
II, когда используют один энантиомер кислот формулы 2-1 (подтип кислот формулы 1-1). Некоторые кислоты формулы 2-1 могут быть рацемизированы или эпимеризированы в некоторых общих условиях амидирования, как описано на Схеме 1. Вместо этого, путем использования более низких температур, с использованием растворителей, которые способствуют растворению взаимодействующих веществ, с использованием добавок, таких как соли имидазолия или пиридиния, или других способов, описанных в Org. Process Res. Dev. 2016, 20, 140; Org. Lett. 2012, 14, 1970; или Org. Process Res. Dev. 2009, 13, 106, или с использованием формы свободного основания аминов формулы 1-3, высокий энантиомерный избыток можно сохранить в ходе осуществления реакции. Альтернативно, если используются общие условия или если происходит эпимеризация или рацемизация, смесь образовавшихся диастереомеров можно разделить с использованием сверхкритической флюидной или обращеннофазовой хроматографии с хиральной колонкой, или они могут быть разделены в виде диастереомерной соли с соответствующей хиральной кислотой в обычных условиях разделения с получением соединений формулы II.
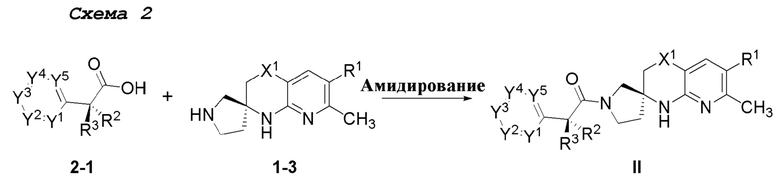
Схема 3 описывает способ синтеза кислоты формулы 2-1 селективно в виде отдельного энантиомера. Кислоты формулы 3-1 могут быть приобретены коммерческим путем или синтезированы с использованием способов, описанных в литературе или в настоящей заявке, и их можно подвергнуть взаимодействию с хорошо известным хиральным вспомогательным агентом (Xe), например агентом типа агента Эванса (оптически чистые оксазолидиноны), агента Майерса (производное псевдоэфедрина) или другими, описанными в литературе, с получением промежуточных соединений формулы 3-2. Обработка соединений формулы 3-2 сильным основанием, таким как диизопропиламид лития, бис(триметилсилил)амид лития,
бис(триметилсилил)амид натрия, бис(триметилсилил)амид калия или т.п., и алкилгалогенидом (когда R2 и/или R3 представляет собой алкильную группу) или другими электрофилами, такими как N-фторбензолсульфонимид (когда R2 и/или R3 представляет собой фтор), может дать соединения формулы 3-3 с высоким диастереомерным избытком. Условия гидролиза Xc группы в соединениях формулы 3-3 зависят от индивидуальных свойств, но часто для получения соединений формулы 2-1 можно использовать реагенты (неорганические основания), такие как используемый в чистом виде или водный гидроксид калия, натрия гидроксид, гидроксид лития, с использованием или без пероксида водорода, и протонные растворители, такие как метанол, этанол или другие, или апротонные растворители, такие как тетрагидрофуран, среди прочих.
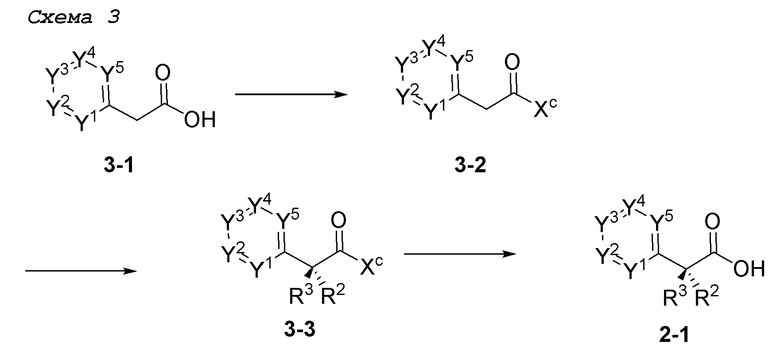
Схема 4 описывает способы, которые можно использовать для синтеза кислот формулы 4-9 (подтип соединений формулы 1-1), 4-7 (подтип соединений формулы 2-1) или 4-8 (подтип соединений формулы 2-1). Арильные или гетероарильные соединения формулы 4-1 [где X2 представляет собой галогенид (например, F, Cl, Br или I) или удаляемую группу, такую как -OTf] можно подвергнуть взаимодействию с дизащищенными малонатами формулы 4-2 (где PG1 может представлять собой метил, этил, трет-бутил, бензил, п-метоксибензил или др., и PG2 может представлять собой ортогонально удаляемую защитную группу, которая выбрана из вышеуказанных или может быть такой же защитной группой) с использованием условий SNAr или путем перекрестного сочетания с использованием палладиевого катализатора, такого как ацетат палладия(II) или трис(дибензилиденацетон)дипалладий(0) [Pd2(dba)3] или другие, с рядом доступных фосфиновых лигандов, или медного катализатора, такого как иодид меди (I), или кислотного лиганда, такого как 2-пиколиновая кислота, как описано в Org. Lett. 2007, 9, 3469, с получением промежуточных соединений формулы 4-3. Соединения формулы 4-3 можно обработать подходящим основанием, таким как гидрид натрия, диизопропиламид лития, бис(триметилсилил)амид лития, бис(триметилсилил)амид натрия, бис(триметилсилил)амид калия, карбонат калия, карбонат цезия или т.п., а затем алкилировать алкилирующими реагентами, такими как метилиодид, этилиодид или др., фторирующими агентами, такими как N-фторбензолсульфонимид, или другими электрофилами с получением соединений формулы 4-4. Альтернативно, соединения формулы 4-1 можно подвергнуть взаимодействию с соединениями формулы 4-6 для непосредственного образования соединений формулы 4-4 (в таких же условиях, как преобразование соединений формулы 4-1 в соединения формулы 4-3). Удаление защитных групп соединений формулы 4-4 можно осуществить с использованием стандартных способов (щелочного или кислотного гидролиза); или, когда PG1 или PG2 представляет собой бензил, путем использования палладиевых катализаторов с водородом или источниками восстановителей, такими как формиат, триалкилсиланы или др., с получением промежуточных соединений формулы 4-5. Альтернативно, соединения формулы 4-4 могут непосредственно образовывать кислоты формулы 4-9 в таких же условиях (как условия для получения соединений формулы 4-5), или в тех, где могут требоваться повышенные температуры. Ди-кислоты формулы 4-5 также могут быть моно-декарбоксилированы с получением рацемических кислот формулы 4-9 с использованием основания, кислоты, оксида меди(I), нагревания или других подходящих условий. Кислоты формулы 4-9, которые содержат смеси энантиомеров, можно разделить с использованием сверхкритической флюидной или обращеннофазовой хроматографии с хиральной колонкой, или их можно разделить и выделить в виде диастереомерной соли с соответствующей хиральной кислотой в классических условиях разделения, таких, которые описаны в Org. Process Res. Dev. 2011, 15, 53, или один энантиомер можно селективно преобразовать в сложный эфир с использованием биокатализа, как описано в Adv. Synth. Catal., 2009, 351, 2333 (см. также J. Org. Chem. 2003, 68, 7234), и разделить с получением кислот формулы 4-7 или 4-8 с высоким энантиомерным избытком. Ди-кислоты формулы 4-5 также могут быть моно-декарбоксилированы с использованием биокатализа, например, ферментов арилмалонатдекарбоксилаз (АМДаза) с получением единственного энантиомера соединений формулы 4-7 или 4-8 с высоким энантиомерным избытком. См., например, (a) J. Am. Chem. Soc. 1990, 112, 4077; (b) () Eur J. Biochem. 1992, 210, 475; (c) Appl. Environ. Microbiol. 2007, 73, 5676; (d) Appl. Microbiol. Biotechnol. 2016, 100, 8621.
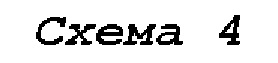
Схема 5 относится к синтезу кислот формулы 5-6 (подтип соединений формулы 1-1), 5-7 (подтип соединений формулы 2-1) и 5-8 (подтип соединений формулы 2-1), где R2c может представлять собой, например, Н, алкил, С3-4 циклоалкил и т.д. (см. определения R2 или R3). Арил- или гетероарилгалогениды формулы 5-1 (где X3 представляет собой I, Br или в некоторых случаях Cl) могут быть приобретены коммерческим путем или синтезированы с использованием способов, известных специалистам в области синтеза. Арил- или гетероарилгалогениды формулы 5-1 можно подвергнуть взаимодействию с соответствующим реагентом для осуществления обмена металл-галоген, таким как н-бутиллитий, изопропилмагний хлорид, или подобные металл-содержащие основания, или металлический магний, и осуществить гашение реакции дикарбонильным соединением формулы 5-3 с получением соединений формулы 5-4. Альтернативно, арены или гетероарены формулы 5-2 могут быть непосредственно депротонированы подобным сильным основанием или реагентами, такими как диизопропиламид лития, 2,2,6,6-тетраметилпиперидид лития, бис(2,2,6,6-тетраметилпиперидинил)цинк или другие их варианты, и их можно подвергнуть взаимодействию с дикарбонильными соединениями формулы 5-3 с получением соединений формулы 5-4. Соединения формулы 5-4 можно обработать сильной кислотой, такой как хлористоводородная кислота, серная кислота, диэтилэфират трифторида бора или другие кислоты Бренстеда или Льюиса, с получением соединений формулы 5-5. Соединения формулы 5-5 можно обработать восстановителями, такими как силаны, в присутствии кислот или водородом и металлическим катализатором, таким как палладий, с получением соединений формулы 5-6. Альтернативно, соединения формулы 5-4 также можно обработать подобными кислотами в присутствии восстановителя, такого как силаны, или водородом и металлическим катализатором, таким как палладий, с получением кислот формулы 5-6. Альтернативно, соединения формулы 5-5 можно обработать водородом и металлами, такими как рутений или родий или др., и хиральным лигандом или многими другими способами, такими, как описанные в Org. Chem. Front. 2014, 1, 155, для селективного образования кислот формулы 5-7 или 5-8 с высоким энантиомерным избытком. Альтернативно, соединения формулы 5-5 могут быть преобразованы с использованием биокатализатора, такого как ENE-редуктаза (как описано в ACS Catal. 2018, 8, 3532), или другими способами для селективного образования соединений формулы 5-7 или 5-8 с высоким энантиомерным избытком. Альтернативно, соединения формулы 5-6, которые содержат смеси энантиомеров, можно разделить с использованием сверхкритической флюидной или обращеннофазовой хроматографии с хиральной колонкой, или их можно разделить и выделить в виде диастереомерной соли с соответствующей хиральной кислотой в обычных условиях разделения с получением соединений общей формулы 5-7 или 5-8.

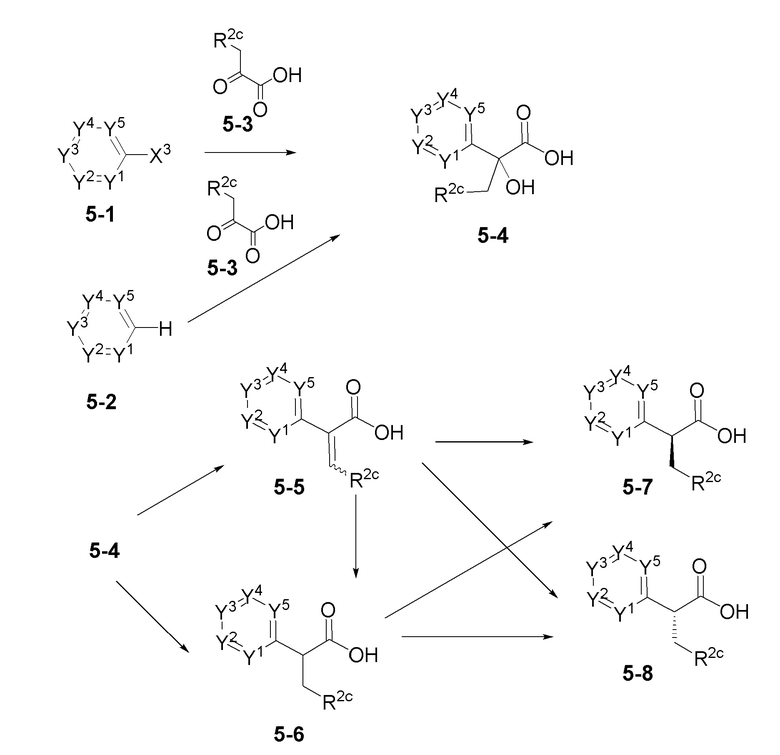
Схема 6 описывает некоторые другие способы для синтеза кислот формул 1-1 и 2-1. Соединения формулы 6-1 можно депротонировать с использованием сильных оснований и ацилировать диоксидом углерода или карбонильным соединением формулы 6-2 (где LG1 представляет собой удаляемую группу, такую как хлорид или алкоксид, a PG1 представляет собой защитную группу, как описано выше) с получением соединений формулы 6-3. Соединения формулы 6-3 можно депротонировать с использованием сильных оснований и обработать алкилирующими агентами с получением соединений формулы 6-4 способом, аналогичным описанному выше для преобразования соединений формулы 3-2 в соединения формулы 3-3. Соединения 6-4 можно обработать в условиях гидролиза с получением соединений формулы 1-1 способом, аналогичным описанному выше для преобразования соединений формулы 3-3 в соединения формулы 2-1, или металлическим катализатором, таким как палладий на углероде, и водородом, когда PG1 представляет собой бензильную группу, или кислотой, когда PG1 может отделяться в виде стабильного катиона или может удаляться с получением соединений формулы 1-1. Альтернативно, можно изменить порядок стадий таким образом, чтобы соединения формулы 6-3 подвергались гидролизу с образованием соединений формулы 6-5 с последующим алкилированием с использованием условий, аналогичных описанным, с получением соединений формулы 1-1. Альтернативно, соединения формулы 6-4 можно обработать в условиях биокатализа, таких как ферменты эстеразы, с получением кислот формулы 2-1 с высоким энантиомерным избытком. Альтернативно, соединения формулы 6-6 можно обработать сильной кислотой, такой как хлористоводородная кислота или серная кислота, в присутствии спирта, такого как метанол или этанол, с получением соединений формулы 6-3. Альтернативно, соединения формулы 6-6 можно подвергнуть алкилированию с получением соединений формулы 6-7 с использованием способа, аналогичного описанному для преобразовани соединений формулы 6-3 в соединения формулы 6-4. Альтернативно, соединения формулы 6-7 могут быть непосредственно гидролизованы с образованием кислот формулы 1-1 с использованием либо сильной кислоты, такой как хлористоводородная кислота или серная кислота, либо сильного основания, такого как гидроксид натрия, гидроксид калия или гидроксид лития, в присутствии воды. Альтернативно, соединения формулы 6-8 (где LG2 представляет собой удаляемую группу, такую как Cl, Br, I, OMs, OTs или др.) можно обработать источниками цианида, такими как цианид натрия, триметилсилилцианид или др., с получением соединений формулы 6-6. Соединения формулы 6-8 могут быть приобретены коммерческим путем или синтезированы различными способами, как описано в литературе, или, когда LG2 представляет собой, например, Br или Cl, путем взаимодействия соединений формулы 6-1 с галогенирующим электрофилом, таким как N-бромсукцинимид, бром или др., с использованием радикал-инициирующего активатора, такого как 2,2'-азобисизобутиронитрил, свет или другие реагенты. Соединения формулы 6-12 (подтип соединений формулы 6-8, где R2 представляет собой Н) могут быть преобразованы во все из аналогичных промежуточных соединений и соединений, являющихся производными 6-8, с использованием аналогичных способов. Соединения формулы 6-14 можно обработать основанием и алкилирующими агентами, как описано для преобразования 6-3 в 6-4, с получением соединений формулы 1-1. Соединения формулы 6-11 могут быть приобретены коммерческим путем или синтезированы с использованием способов, описанных в литературе. Соединения формулы 6-9 могут быть окислены с использованием окисляющего реагента, такого как перманганат калия, также с образованием соединений формулы 6-11. Соединения формулы 6-11 можно подвергнуть реакции гомологенизации с использованием любого количества способов, описанных в литературе, таких как реакция Арндта-Айстерта (с использованием активирующего реагента, такого как тионилхлорид, этилхлорформиат или др.; затем диазометанового реагента; соли серебра, такой как бензоат серебра, оксид серебра или др.; и нуклеофила, такого как вода или спирт), или других способов, описанных в литературе, таких как описанные в J. Org. Chem. 2001, 66, 5606, с получением соединений формулы 6-10. Кислоты формулы 1-1, которые содержат смеси энантиомеров, можно разделить с использованием сверхкритической флюидной или обращеннофазовой хроматографии с хиральной колонкой, или они могут быть разделены в виде диастереомерной соли с соответствующей хиральной кислотой в классических условиях разделения, как описано в Org. Process Res. Dev. 2011, 15, 53, или нежелательный энантиомер можно преобразовать в сложный эфир с использованием биокатализа, как описано в Adv. Synth. Cat al., 2009, 351, 2333 (см. также J. Org. Chem. 2003, 68, 7234), и разделить с получением кислот формулы 2-1 с высоким энантиомерным избытком.

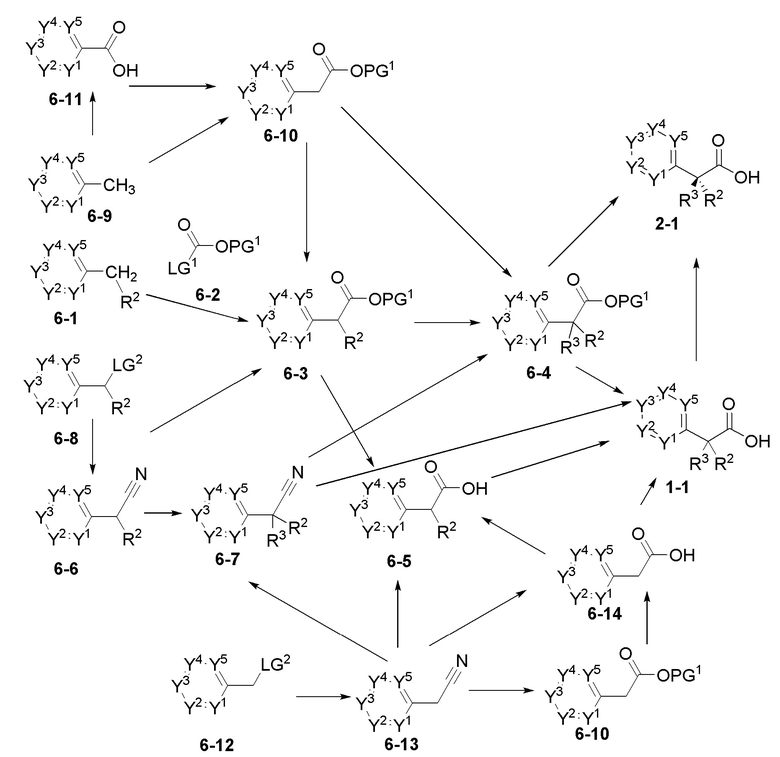
Схема 7 описывает синтез соединений формулы 7-3, 7-4, 7-7, 7-8, 7-9 и 7-10 (где Q1 могут представлять собой любые фрагменты соединений, которые описаны в формуле I, Ia, II или на Схемах 3-б), которые можно использовать в качестве любого из промежуточных соединений, уже описанных выше, при необходимости, и эти заместители могуть быть введены во многих точках во время синтеза, описанного на Схемах 3-6. Соединения формулы 7-1 (где PG1 имеет значение, описанное выше) можно подвергнуть процедуре снятия защиты с использованием деалкилирующих условий, таких как триметилсилилиодид, метантиолат натрия или др., сильных кислот таких как бромистоводородная кислота, трибромид бора, или, когда PG1 представляет собой бензильную группу, можно использовать палладий или соответствующие металлы и газообразный водород для образования соединений формулы 7-2. Соединения формулы 7-2 можно подвергнуть взаимодействию с источниками дифторметила, такими как дифторгалогенацетаты или (бромдифторметил)триметилсилан, с получением соединений формулы 7-3. Соединения формулы 7-2 также можно подвергнуть взаимодействию с источниками трифторметила, такими как дифторгалогенацетаты, с добавлением источника электрофильного фтора, такого как Selectfluor™, трифторметилгалогениды, или через промежуточный ксантан, который можно обработать XtalFluor® и источником электрофильного фтора, таким как N-фторбензолсульфонимид или 1, 3, 5-трихлор-1,3,5-триазинан-2,4,6-трион (ТССА) (как описано в J. Org. Chem. 2019, 84, 15776) или др., с получением соединений формулы 7-4. Соединения формулы 7-5 (где LG3 может представлять собой Cl, Br, I, OTf или др.) можно обработать источником нуклеофильного винила, таким как винилборонат, винилстаннан или др., с использованием условий палладий-катализируемого перекрестного сочетания, описанных в литературе, с получением соединений формулы 7-6. Соединения формулы 7-6 можно подвергнуть окислительному расщеплению до альдегида с использованием реагентов, таких как озон с трифенилфосфином или диметилсульфидом, тетроксид осмия (или трихлорид рутения) и периодат натрия или др., с получением соединений формулы 7-7. Соединение формулы 7-7 можно подвергнуть взаимодействию с источниками нуклеофильного дифторметилирования, такими как Deoxo-Fluor® или XtalFluor® и соответствующие реагенты, с получением соединений формулы 7-8. Соединения формулы 7-5 можно обработать спиртами в условиях SNAr или перекрестного сочетания с использованием палладия и различных лигандов с получением соединений формулы 7-9 [где R8 представляет собой, например, C1-4 алкил (такой как метил) или C1-4 галогеналкил (такой как C1 фторалкил)]. Соединения формулы 7-5 также можно подвергнуть взаимодействию с аминами в таких же условиях с получением соединений формулы 7-10 [где каждый из R6 и R7 независимо представляет собой C1-4 алкил (такой как метил), или R6 и R7 вместе с атомом углерода, с которым они связаны, образуют циклопропил].
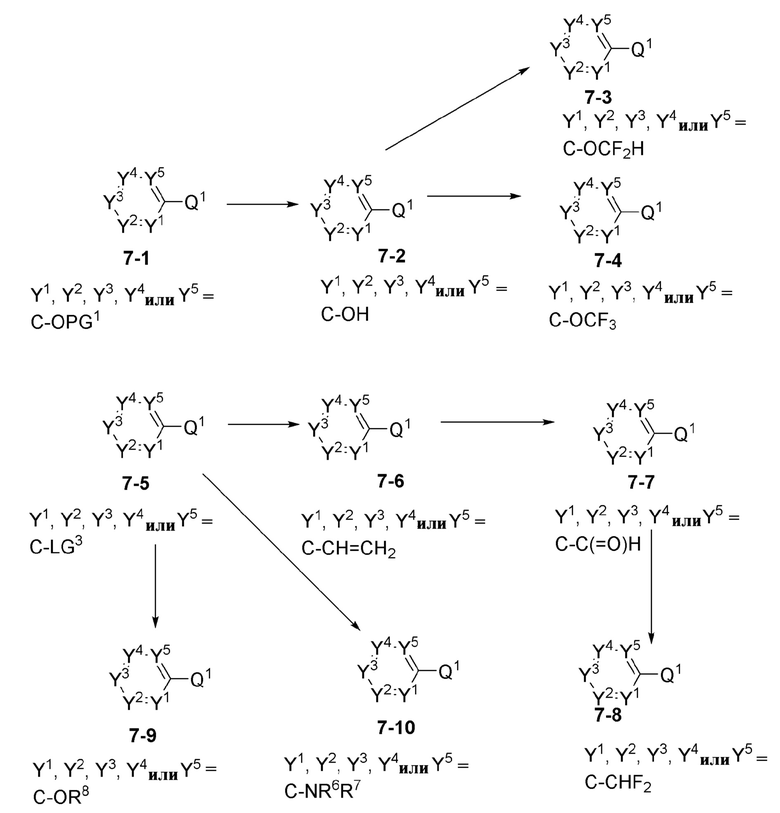
Схема 8 описывает синтез соединений формул 1-2 и 1-3. Соединения формулы 8-1 (где PG4 может представлять собой бензил, л-метоксибензил, трет-бутоксикарбонил, бензилоксикарбонил, ацетил, бензоил или другие обычные азот-защитные группы; PG5 может быть таким же, как PG4, или может представлять собой любую из подобных защитных групп, которые могут удаляться ортогонально) можно подвергнуть галогенированию с использованием электрофильных галогенирующих реагентов, таких как дибромгидантоин, N-бромсукцинимид, N-хлорсукцинимид, бром, иод или др., с получением соединений формулы 8-3 (где X4 может представлять собой Cl, Br или I). Соединения формулы 8-3 можно подвергнуть взаимодействию с источником дибора [таким как тетрагидроксидибор, бис (неопентилгликолято)дибор (5,5,5',5'-тетраметил-2,2'-би-1,3,2-диоксаборинан) или бис(пинаколято)дибор (4,4,4',4',5,5,5',5'-октаметил-2,2'-би-1,3,2-диоксаборолан)], источником ди-олова, таким как бис(трибутилолово) или др., сильным металл-содержащим основанием, таким как изопропилмагний хлорид, затем с источником цинка, таким как дихлорид цинка или др., с получением соединений формулы 8-2 (где М1 может представлять собой бороновую кислоту, боронат, оловоорганическое соединение, цинкорганическое соединение или другой металл, способный взаимодействовать в условиях С-С перекрестного сочетания) и выделением, если они стабильны, или направлением в другую реакцию, если желательно. Соединения формулы 8-2 можно подвергнуть взаимодействию с соединениями формулы 8-9 (где X5 представляет собой Cl, Br, I, OTf или др.) в условиях С-С перекрестного сочетания, таких как реакции Сузуки (М1=бор), Стилле (М1=олово), Негиши (М1=галогенид цинка), Кумада (М1=галогенид магния) или др., с получением соединений формулы 8-4. Альтернативно, соединения формулы 8-3 и 8-10 можно подвергнуть взаимодействию таким же способом, как 8-2 и 8-9, с нуклеофилом и электрофилом, возвращенным в реакцию перекрестного сочетания, с получением соединений формулы 8-4. Альтернативно, в некоторых случаях соединения формулы 8-1 также можно подвергнуть взаимодействию в условиях СН активации/прямого арилирования с соединениями формулы 8-9 для непосредственного образования соединений формулы 8-4. PG4 и PG5 группу соединений формулы 8-4 можно удалить с использованием подходящих условий удаления защиты, таких как кислота или гидрогенолиз или др., с получением соединений формулы 1-2. Необходимо отметить, что в случае, когда соединения формулы 8-1, 8-2, 8-3, 8-4 или 1-2 содержат смеси стереохимии или являются рацемическими, их можно разделить на отдельные энантиомеры с использованием сверхкритической флюидной или обращеннофазовой хроматографии с хиральной колонкой или в виде диастереомерной соли с соответствующей хиральной кислотой в классических условиях разделения и выделить с получением соединений формулы 8-5, 8-7, 8-6, 8-8 или 1-3, соответственно, с высоким энантиомерным избытком. Альтернативно, соединения формулы 8-5, 8-7, 8-6 и 8-8 можно подвергнуть взаимодействию в таких же условиях, как их аналогичные промежуточные соединения из этой схемы, без модификации условий, приводящих к образованию соединений формулы 1-3.
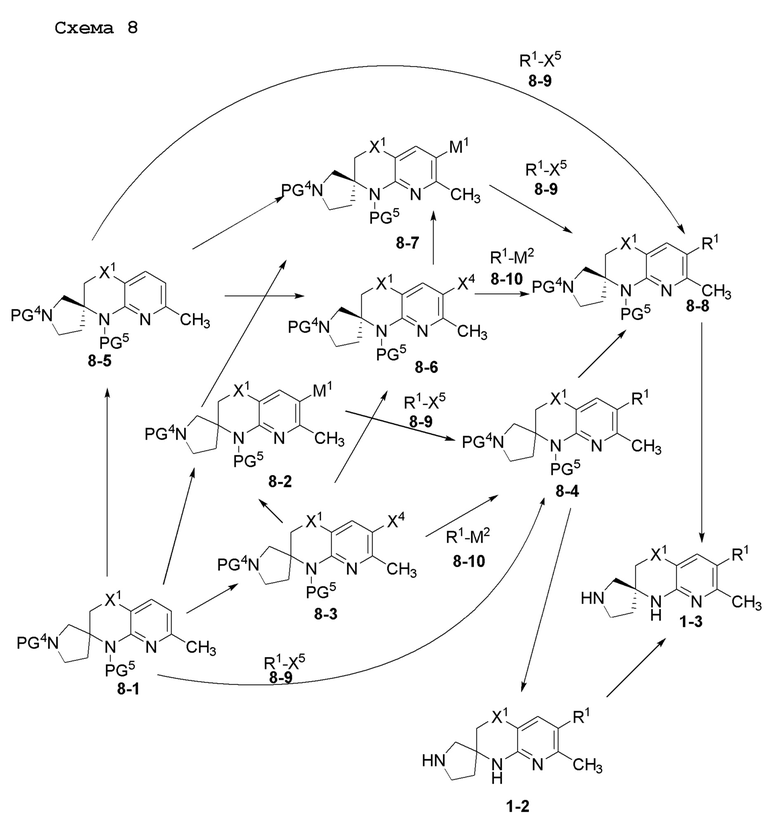
Схема 9 относится к синтезу соединения формулы 9-13. Соединения формулы 9-1 (где X7 является более реакционноспособным, чем X6; например, Х7=Br, Х6=Cl, или Х7=I, Х6=Br или Cl, или другие подобные комбинации) можно подвергнуть взаимодействию с соединением формулы 9-2 в условиях реакции Соногаширы с использованием медного и палладиевого катализатора с получением соединений формулы 9-3. Соединения формулы 9-3 можно обработать различными палладиевыми, платиновыми или родиевыми катализаторами (на углероде или оксиде алюминия или свободными) и водородом, триалкилсиланами, муравьиной кислотой с получением соединений формулы 9-8. Соединения 9-8 можно обработать с использованием условий SNAr типа или палладия или меди с подходящими лигандами в обычных условиях C-N перекрестного сочетания с получением соединений формул 9-4. В таких же условиях с определенными PG5 группами, такими как карбамат и более сильное основание, такое как трет-бутоксид натрия или калия, соединения формулы 9-8 могут быть непосредственно преобразованы в соединения формулы 9-6. Альтернативно, соединения формулы 9-9 (где R11 может представлять собой арен, такой как фенил, или алкильную группу, такую как этил или бутил, или спирт, такой как этанол, или др.; и (Xs)" может представлять собой OMs-, OTs-, OTf-, Cl-, Br-, I- или т.п.) и соединения формулы 9-10 можно подвергнуть взаимодействию вместе в условиях реакции Виттига (как описано в Tetrahedron Lett. 2007, 48, 3359) или подобных условиях с основаниями, такими как карбонат калия, трет-бутоксид натрия, н-бутиллитий или подобные основания, с получением соединений формулы 9-11. Соединения формулы 9-11 могут быть преобразованы в соединения формулы 9-8 с использованием условий, аналогичных условиям преобразования, описанным для соединений формулы 9-3, с получением соединений формулы 9-8. Альтернативно, соединения формулы 9-10 могут быть преобразованы в соединения формулы 9-12 с использованием подходящей метилен-содержащей соли Виттига способом, аналогичным преобразованию 9-10 в 9-11. Альтернативно, соединения формулы 9-11 можно подвергнуть взаимодействию в условиях окислительно-восстановительной изомеризации под действием света с использованием катализаторов, таких как иридий или др., подходящих лигандов и синего светодиода с получением соединений формулы 9-7. Альтернативно, преобразование соединений формулы 9-11 можно осуществить путем взаимодействия в таких же окислительно-восстановительных условиях под действием света с вторым катализатором, обычно палладиевым, добавляемым для осуществления циклизации после изомеризации, с получением соединений формулы 9-5. Альтернативно, соединения формулы 9-3 можно обработать отравленными катализаторами, такими как катализатор Линдлара (например, способзами, описанными в J. Org. Chem. 2001, 66, 3634) или палладий на сульфате бария с источником водорода, или способами, описанными в Tetrahedron Lett. 2008, 49, 2839, с получением соединений формулы 9-7. Альтернативно, соединения формулы 9-7 можно обработать в условиях гидрирования, аналогичных описанным для преобразования соединений формулы 9-11 в 9-8, с получением соединений формулы 9-8. Альтернативно, соединения формул 9-10 и 9-2 могут быть взаимопреобразованы с использованием условий образования алкина, таких как реакция Кори-Фукса или др., с получением соединений формулы 9-2, или обработаны в условиях окислительного расщепления с получением соединений формулы 9-10. Альтернативно, соединения формулы 9-7 можно подвергнуть взаимодействию в условиях, аналогичных описанным для преобразования соединений формулы 9-8 в соединения формулы 9-6, с получением соединений формулы 9-5. Соединения формулы 9-5 можно подвергнуть взаимодействию в условиях, аналогичных условиям преобразования соединений формулы 9-11 в соединения формулы 9-8, с получением соединений формулы 9-4, 9-13 или 9-6 в зависимости от выбора используемых защитных групп.Альтернативно, соединения формулы 9-5 можно подвергнуть взаимодействию в стандартных условиях для удаления PG5 с получением соединений формулы 9-14. Соединения формулы 9-14 затем можно подвергнуть взаимодействию в условиях, аналогичных условиям преобразования соединений формулы 9-5 в соединения формулы 9-6, с получением соединений формулы 9-6 или 9-13 в зависимости от выбора защитной группы. Альтернативно, соединения формулы 9-12 могут быть преобразованы в соединения формулы 9-11 с использованием условий реакции перекрестного сочетания Гека с соединениями формулы 9-1. Альтернативно, соединения формулы 9-12 и 9-1 могут быть преобразованы в соединения формулы 9-15 с использованием условий SNAr или C-N перекрестного сочетания (когда X6 является более реакционноспособным, чем X7, например, Х7=Cl, Х6=Cl или Вг, или Х7=Br, Х6=Br или I, или другие подобные комбинации). Соединения формулы 9-15 можно подвергнуть взаимодействию в условиях, аналогичных описанным для преобразования соединений формулы 9-12 в соединения формулы 9-11, с получением соединений формулы 9-5.
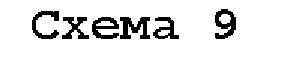
Схема 10 описывает синтез соединения формулы 10-11, подтипа соединения формулы 9-13, где стереохимия определена. Все из показанных преобразований можно осуществить, как описано для аналогичных соединений и промежуточных соединений, описанных на Схеме 9, и они не требуют модификаций или использования других условий.

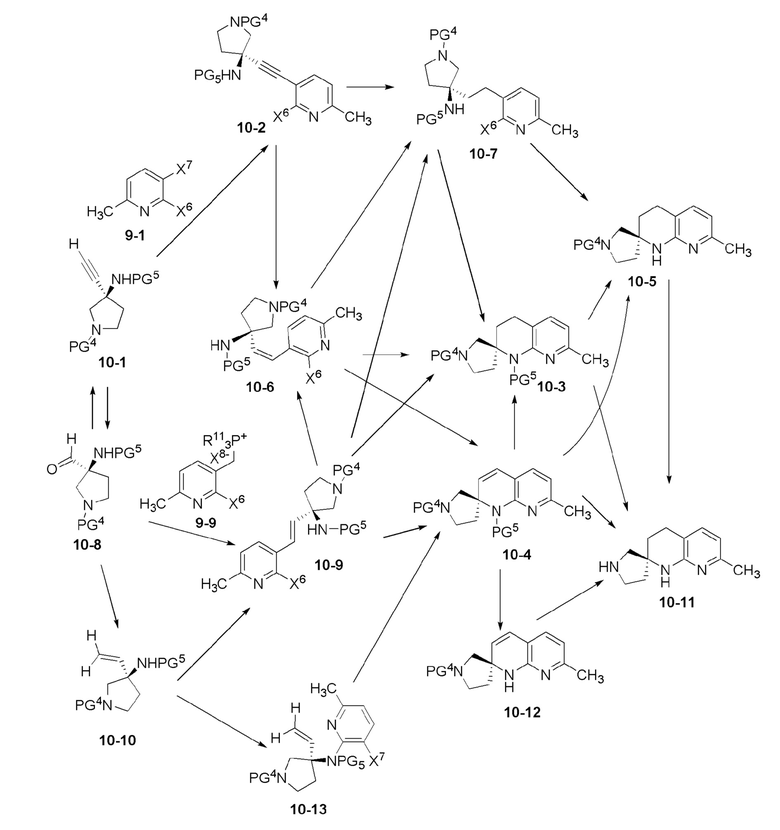
Схема 11 описывает синтез соединений формулы 11-6 и 11-7 (подтип соединений формул 9-2 и 10-1, соответственно, где PG7 представляет собой защитную группу, которая позволяет смежному азоту оставаться нуклеофильным, такую как бензил, п-метоксибензил или др., или возможно отсутствие защитной группы). Соединения формулы 11-1 могут быть приобретены коммерческим путем или синтезированы в соответствии со способами, описанными в литературе, и их можно подвергнуть взаимодействию с соединениями формулы 11-2 (где PG6 представляет собой триметилсилил или другую подходящую алкин-защитную группу), которые были депротонированы под действием основания, такого как трет-бутоксид калия, диизопропиламид лития, гидрид натрия, н-бутиллитий, металлический цинк или магний, с получением соединения формулы 11-3. Соединения формулы 11-3 затем можно подвергнуть процедуре удаления защиты для получения терминального алкина с использованием реагентов, таких как фторид тетрабутиламмония, карбонат калия, гидроксид калия или др., с получением соединения формулы 11-4. Гидроксил соединений формулы 11-4 может быть активирован для превращения в удаляемую группу OR9 (где R9 представляет собой ацетил, бензоил, трет-бутоксикарбонил, диалкилфосфат [Р(O)(OAlk)2] или т.п.} с использованием ацетилхлорида, бензоилхлорида, других ацилгалогенидов, других подходяще активированных кислот или других активирующих групп, таких как галогенформиаты, диалкилгалогенфосфаты или др., и основания, такого как триэтиламин, N, iV-диизопропилэтиламин, пиридин, 4-(диметиламино)пиридин или т.п., с получением активированных соединений формулы 11-5. Соединения формулы 11-5 можно подвергнуть взаимодействию с аминами, защищенными п-метоксибензилом, бензилом или др., в реакции, катализируемой хлоридом меди(I), бромидом меди(I) (см. J. Org. Chem. 2013, 78, 5647), рутениевыми катализаторами (см. New J. Chem. 2011, 35, 2427) и т.п., с получением соединений формулы 11-6. Если соединения формулы 11-6 содержат смеси стереохимии или являются рацемическими, их можно разделить на отдельные энантиомеры с использованием сверхкритической флюидной или обращеннофазовой хроматографии с хиральной колонкой или в виде диастереомерной соли с соответствующей хиральной кислотой в классических условиях разделения и выделить с получением соединений формулы 11-7 с высоким энантиомерным избытком.
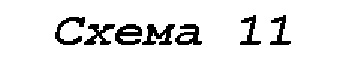
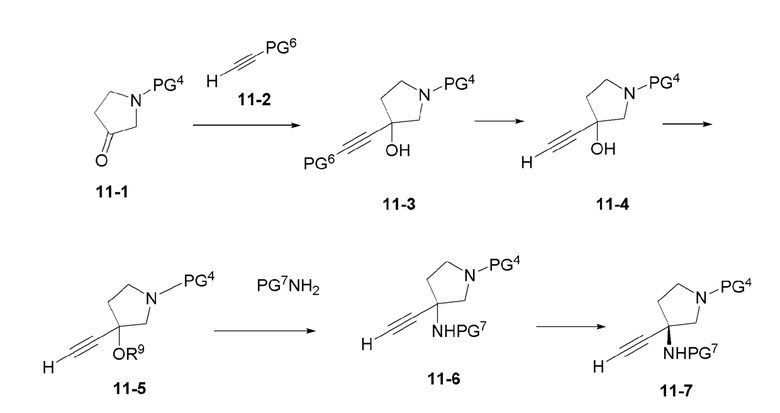
Схема 12 описывает синтез соединений формулы 9-1 и 9-9 (где R11 может представлять собой алкильные группы, арильные группы, алкокси группы, или их комбинации, или их оксидный вариант). Соединения формулы 12-1 (которые могут быть приобретены коммерческим путем или синтезированы из 12-6 или другими способами, описанными в литературе) можно обработать восстановителем, таким как гидриды на основе алюминия (литийалюминийгидрид, натрия бис(2-метоксиэтокси)алюминия дигидрид, диизобутилалюминийгидрид или др.) или борогидриды (такие как борогидрид лития, борогидрид натрия или др.), с получением соединений формулы 12-3. Альтернативно, соединения формулы 12-2 (которые могут быть приобретены коммерческим путем или образованы из соединений формулы 12-1 с использованием стандартных условий гидролиза или другими способами, описанными в литературе) могут быть преобразованы с использованием таких же реагентов, но также с использованием борановых реагентов или производных борана, с получением соединений формулы 12-3. ОН группа соединений формулы 12-3 может быть активирована, становясь удаляемой группой (LG4, которая может представлять собой OMs, OTs, OTf, Cl, Br, I или др.), с использованием подходящего реагента, такого как метансульфонилхлорид, п-толуолсульфонилхлорид, трифторметансульфоновый ангидрид, оксихлорид фосфора или тионилхлорид, оксибромид фосфора или трибромид фосфора, иод с трифенилфосфином или имидазолом, кислоты такие как бромистоводородная кислота или хлористоводородная кислота, или с использованием комбинации способов, такой как метансульфонилхлорид с последующим иодидом натрия, хлоридом натрия, бромидом натрия, иодидом калия, хлоридом калия, бромидом калия или др., с получением соединений формулы 12-4. Альтернативно, соединения формулы 12-5 могут быть галогенированы в условиях радикального галогенирования, таких как условия, описанные для преобразования соединений 6-1 в 6-8, с получением соединений формулы 12-4. Соединение формулы 12-4 можно подвергнуть взаимодействию с соединением формулы 12-9 (например, трифенилфосфином, триэтилфосфином, триэтилфосфитом или другими фосфорными нуклеофилами) для получения соединений формулы 9-9. Соединения формулы 12-6 можно подвергнуть нитрованию в стандартных условиях нитрования, таких как дымящая азотная кислота, для получения соединений формулы 12-7. Нитро группа соединений формулы 12-7 может быть восстановлена до амина с использованием различных условий, таких как палладий на углероде с водородом, цинк или железо с уксусной или хлористоводородной кислотой, хлорид олова (II) или др., с получением соединений формулы 12-8. Соединения формулы 12-8 можно обработать нитритом натрия или изоамилнитритом с бромистоводородной кислотой, бромидом калия, иодидом калия или с использованием других стандартных условий реакции Зандмейера с получением соединений формулы 9-1. Соединения формулы 9-1 можно обработать с использованием условий карбонилирования, таких как палладий с подходящим лигандом, таким как 1,1'-бис(дифенилфосфино)ферроцен, источник оксида углерода и спирт, такой как этанол или метанол, или можно обработать с использованием условий обмена металл-галоген и гашения источником ацила, таким как диэтилкарбонат, диоксид углерода, этилхлорформиат или др., с получением соединений формулы 12-1.

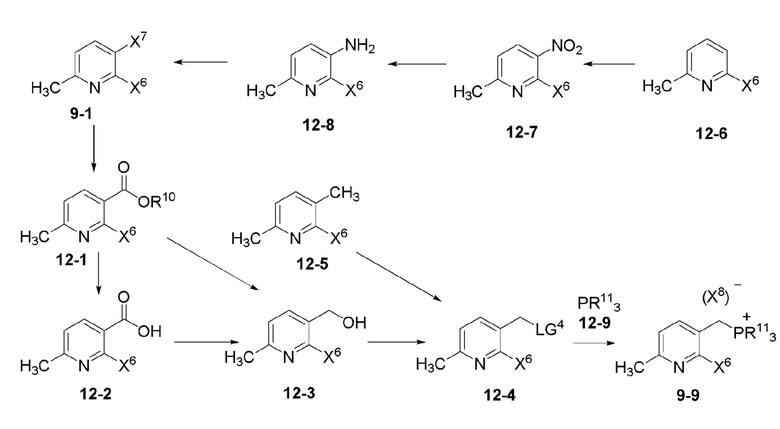
Схема 13 описывает способ синтеза соединений формулы 9-10. Соединения формулы 13-1 (где PG8 может представлять собой любой С-связанный алкил или арил, и которые могут быть приобретены коммерческим путем или синтезированы с использованием способов, описанных в литературе) можно обработать в условиях удаления гидроксильной группы, например, с использованием метансульфонилхлорида или л-толуолсульфонилхлорида, как описано в Org. Lett. 2016, 18, 1812, или с использованием других стандартных условий с получением соединений формулы 13-2. Соединения формулы 13-2 можно подвергнуть взаимодействию в реакции 3+2 циклоприсоединения с коммерчески доступными соединениями формулы 13-3 с получением соединений формулы 13-4. Соединения формулы 13-4 могут быть восстановлены непосредственно до соединений формулы 9-10 с использованием условий, таких как диизобутилалюминийгидрид, или других восстановителей, с которыми можно избежаать чрезмерного восстановления. Альтернативно, соединения формулы 13-4 могут быть восстановлены до соединений формулы 13-5 с использованием условий, аналогичных тем, которые описаны для преобразования соединений формулы 12-1 в 12-3. Соединения формулы 13-5 затем могут быть окислены с использованием ряда хорошо известных реагентов, таких как хромовый реагент Коллинза, периодинановые реагенты Десса-Мартина, реагент Парика-Деринга, другие активированные реагенты на основе DMS0 типа реагентов Сверна или многие другие, с получением соединений формулы 9-10. Альтернативно, соединения формулы 13-8 можно обработать диизопропиламидом лития, бис(триметилсилил)амидом лития, н-бутиллитием или многими другими подобными сильными основаниями и подходящими ацилирующими реагентами, такими как этилхлорформиат, этилцианоформиат или диэтилкарбонат, с получением соединений формулы 13-9 (где PG9 может быть таким же, как PG8, или может представлять собой другую алкильную или арильную группу, чтобы таким образом было возможно ортогональное удаление этой группы в селективных условиях). Соединения формулы 13-9 могут быть селективно гидролизованы с использованием одного эквивалента основания, такого как гидроксид натрия или гидроксид лития или многие другие, или с использованием селективной PG9, такой как бензил, которую можно удалить обработкой палладием на углероде и водородом, с получением соединений формулы 13-10. Соединения формулы 13-10 могут быть преобразованы в соединения формулы 13-4 с использованием реакций, таких как перегруппировка Курциуса (как описано в Org. Biomol. Chem. 2018, 16, 2006), или других подобных реакций, которые обеспечивают перегруппировку кислоты или соответствующей ацильной группы в дегомологированный амин или защищенный амин, таких как перегруппировка Гофмана, перегруппировка Лоссена или реакция Шмидта.

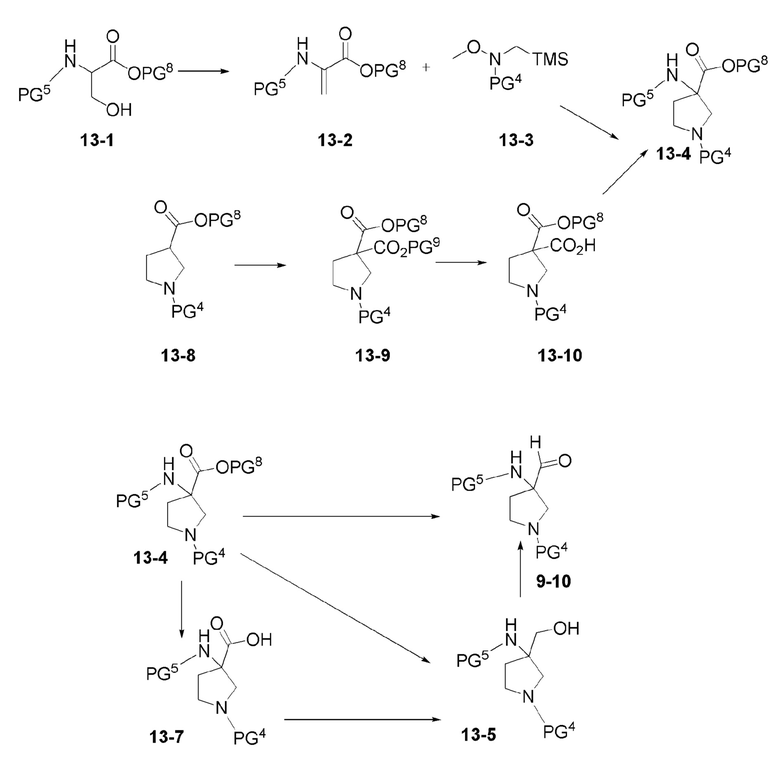
Схема 14 описывает способ синтеза соединений формулы 10-8 (подтипа 9-10, где стереохимия определена) и соответствующих промежуточных соединений. Рацемические варианты или смеси соединений формулы 13-4 можно разделить на отдельные энантиомеры с использованием сверхкритической флюидной хроматографии или обращеннофазовой хроматографии с хиралвной колонкой, или, когда PG4 или PG5 представляет собой защитную группу, которая не устраняет основность азота, к которому она присоединена, также можно разделить с использованием хиральной кислоты в классических условиях разделения с получением соединений формулы 14-4. Альтернативно, соединения формулы 13-9 могут подвергатвся воздействию различных биокаталитических условий, таких как Эстераза ECS03 (АВ 5035 74) от Enzymlcals, или условий, описанных в Tetrahedron: Asymmetry 1998, 9, 2 663, с образованием соединений общей формулы 14-1 (подтипа соединений формулы 13-10). Соединения общей формулы 14-1 и 14-4 могут быть преобразованы в аналогичные им соединения, описанные на Схеме 13, с использованием описанных условий, без какой-либо необходимости в модификациях или изменениях реакционных условий.
Схема 15 описывает синтез соединения 15-4 (подтипа соединений формулы 8-4, где X1 представляет собой СН-СН3). Соединения формулы 11-1 можно подвергнуть взаимодействию с аммиаком и аллилборным реагентом, как описано в Chem. Commun. 2005, 44, 5551, с получением соединений формулы 15-1. Соединения формулы 15-1 можно подвергнуть взаимодействию способом, аналогичным тому, как описано для преобразования соединений формулы 9-12 в 9-15, с получением соединений формулы 15-2. Соединения 15-2 можно подвергнуть взаимодействию способом, аналогичным способу преобразования соединений формулы 9-15 в 9-5, с получением соединений формулы 15-3 с одновременной перегруппировкой во внутренний олефин, как показано. Соединения формулы 15-3 могут быть преобразованы в соединения формулы 15-4 с использованием способов, аналогичным описанным для преобразования соединений формулы 9-5 в 9-4. Альтернативно, соединения формулы 15-2 можно подвергнуть взаимодействию с использованием условий озонолиза в присутствии борогидрида натрия или других условий окислительного расщепления с получением соединений формулы 15-7. Соединения формулы 15-7 можно подвергнуть взаимодействию в условиях удаления гидроксильной группы до олефина, таких как использование метансульфонилхлорида или л-толуолсульфонилхлорида, а затем сильного основания, или использование условий элиминирования по Грико с арилселеноцианатом в присутствии триалкилфосфина, с получением соединений формулы 15-8 (которые представляет собой подтип соединений формулы 9-15, где PG5 представляет собой Н).
Схема 16 описывает синтез соединений формулы 16-3 (где Y7 представляет собой N или СН), 16-7, 16-11 и 16-14, которые все являются подтипами 8-9, которые могут быть коммерчески недоступны. Соединения формулы 16-1 можно подвергнуть взаимодействию с источником электрофильного галогена способом, аналогичным преобразованию соединений формулы 8-1 в 8-3, с получением соединений формулы 16-2. Соединения формулы 16-2 можно подвергнуть взаимодействию в условиях, аналогичных условиям преобразования соединений формулы 7-2 в 7-3 (когда R12 представляет собой Н) или соединений формулы 7-2 в 7-4 (когда R12 представляет собой F), с получением соединений формулы 16-3. Соединения формулы 16-4 (где X9 может представлять собой галоген, который является таким же, как X5, или может быть более реакционноспособным, таким образом, X9 представляет собой Br, когда Xs представляет собой Cl, или X9 представляет собой I, когда X5 представляет собой Br или Cl) можно подвергнуть взаимодействию в условиях по типу реакции Чан-Лама с циклопропилбороновой кислотой (когда R13 представляет собой циклопропил) и источником меди, таким как ацетат меди(II) или др., и подходящими лигандами, такими как 2,2'-бипиридин, и любым количеством оснований, таких как карбонаты или амины, и соокислителем, таким как кислород, который может быть добавлен или может поступать из воздуха, с получением соединений формулы 16-5. Соединения формулы 16-5 (которые могут быть приобретены коммерческим путем, например, когда R13 представляет собой метил, или могут быть получены, как описано в настоящей заявке или в литературе) можно подвергнуть взаимодействию в условиях обмена металл-галоген, таких как изопропилмагнийхлорид, н-бутиллитий, магний или др., и осуществить гашение источником карбонила, таким как N,N-диметилформамид, морфолин-4-карбальдегид или др., с получением соединений формулы 16-6. Соединения формулы 16-6 можно подвергнуть взаимодействию в условиях, аналогичных тем, которые использовали для преобразования соединений формулы 7-7 в 7-8, с получением соединений формулы 16-7. Соединения формулы 16-4 можно подвергнуть взаимодействию с 4-галоген-1-бутеном в присутствии основания, такого как карбонат калия, бикарбонат натрия, гидрид натрия, диизопропиламид лития или др., с получением соединений формулы 16-8. Соединения формулы 16-8 можно подвергнуть взаимодействию в условиях по типу реакции Гека, как описано для преобразования соединений формулы 9-15 в 9-5, с получением соединений формулы 16-9. Соединения формулы 16-9 можно подвергнуть взаимодействию в несколько стадий таким же способом, как описано для последовательности преобразования соединений формулы 7-6 в 7-7, затем в 7-8, с получением соединений формулы 16-10 и 16-11. Соединения формулы 16-12 (где X10 может представлять собой Cl, Br или I, OTf или др.) можно подвергнуть взаимодействию с палладием на углероде или другими обычными катализаторами и газообразным дейтерием (или другим источником дейтеридов, таким как дейтерировэнный формиат, в условиях трансферной гирогенизации) с получением соединений формулы 16-13. Соединения формулы 16-13 можно подвергнуть взаимодействию в условиях по типу реакции Зандмейера, как описано для преобразования соединений формулы 12-8 в 9-1, с получением соединений формулы 16-14. Альтернативно, соединения формулы 16-15 можно подвергнуть взаимодействию в условиях по типу реакции Зандмейера, как описано для преобразования соединений формулы 12-8 в 9-1.
Схема 17 описывает способ синтеза соединений формулы 17-5, которые являются подтипом соединений формулы 8-4, где R1 представляет собой тетразол, замещенный группой R14, которая может представлять собой необязательно замещенную алкильную или арильную группу. Соединения формулы 17-1 можно подвергнуть взаимодействию с использованием способа преобразования, аналогичного описанному для синтеза соединений формулы 7-5 до 7-6 и 7-7, с получением соединений формулы 17-2 и 17-3. Альтернативно, соединения формулы 17-1 могут быть непосредственно преобразованы в соединения формулы 17-3 способом, аналогичным способу преобразования соединений формулы 16-5 в соединения формулы 16-6. Соединения формулы 17-3 можно подвергнуть взаимодействию с замещенными гидразинами в стандартных условиях конденсации с использованием кислотного или щелочного катализа с получением соединений формулы 17-4. Соединения формулы 17-4 можно подвергнуть взаимодействию с диазо-соединениями, такими как диэтил- или ди-трет-бутилазодикарбоксилат, в присутствии источников гипервалентного иода, таких как [бис(трифторацетокси)иод]бензол или др., с получением соединений формулы 17-5.

Подробное описание отдельных стадий реакции приведено ниже в разделе «Примеры». Специалистам в данной области техники должно быть понятно, что для синтеза соединений можно использовать другие пути синтеза. Хотя конкретные исходные вещества и реагенты обсуждаются ниже, их можно легко заменить другими исходными веществами и реагентами для получения различных производных и/или условий реакции. Кроме того, многие из соединений, полученных способами, описанными ниже, могут быть дополнительно модифицированы в свете настоящего раскрытия с использованием обычных химических методов, хорошо известных специалистам в данной области.
Комбинированные средства
Соединения по настоящему изобретению можно вводить отдельно или в комбинации с одним или несколькими дополнительными терапевтическими средствами. Под «вводимым в комбинации» или «комбинированной терапией» подразумевается, что соединение по настоящему изобретению и одно или несколько дополнительных терапевтических средств вводят одновременно млекопитающему, подвергаемому лечению. При введении в комбинации каждый компонент можно вводить одновременно или последовательно в любом порядке в разные моменты времени. Таким образом, каждый компонент можно вводить отдельно, но достаточно близко по времени, чтобы обеспечить желаемый терапевтический эффект.Фразы «совместно вводимые», «совместное введение», «одновременное введение» и «вводимые одновременно» означают, что соединения вводят в комбинации. Таким образом, описанные в настоящей заявке способы профилактики и лечения включают использование комбинированных средств.
Комбинированные средства вводят млекопитающему в терапевтически эффективном количестве. Под «терапевтически эффективным количеством» подразумевается количество соединения по настоящему изобретению, которое при введении млекопитающему отдельно или в комбинации с дополнительным терапевтическим средством является эффективным для лечения целевого заболевания/расстройства/состояния (например, кахексии, анорексии, нервной анорексии, тошноты, рвоты, задержки развития, саркопении, мышечной атрофии, слабости, остеопороза, потери костной массы, боли, беспокойства, депрессии или гипертензии).
В некоторых вариантах осуществления соединение по настоящему изобретению можно вводить совместно с одним или несколькими другими средствами, такими как орлистат, TZD и другие средства, повышающие чувствительность к инсулину, аналоги FGF21, метформин, этиловые эфиры омега-3-кислоты (например, ловаза), фибраты, ингибиторы ГМГ-KoA-редуктазы, эзетимиб, пробукол, урсодезоксихолевая кислота, агонисты TGR5, агонисты FXR, витамин Е, бетаин, пентоксифиллин, антагонисты СВ1, карнитин, N-ацетилцистеин, восстановленный глутатион, лоркасерин, комбинация налтрексона с бупроприоном, ингибиторы SGLT2 (включая дапаглифлозин, канаглифлозин, эмпаглифлозин, тофоглифлозин, эртуглифлозин, ASP-1941, THR1474, TS-071, ISIS388626 и LX4211, а также описанные в WO 2010023594), фентермин, топирамат, агонисты рецепторов GLP-1, агонисты рецепторов GIP, двойные агонисты рецептора GLP-1/рецептора глюкагона (например, OPK88003, MEDI0382, JNJ-64565111, NN9277, BI 456906), двойные агонисты рецептора GLP-1/рецептора GIP [например, тирзепатид (LY3298176), NN9423], блокаторы рецепторов ангиотензина, ингибитор ацетил-СоА карбоксилазы (АСС), ингибитор BCKDK, ингибитор кетогексокиназы (КНК), ингибиторы ASK1, ингибиторы киназы дегидрогеназы альфа-кетокислот с разветвленной цепью (ингибиторы BCKDK), ингибиторы CCR2 и/или CCR5, ингибиторы PNPLA3, ингибиторы DGAT1, ингибиторы DGAT2, аналог FGF21, аналоги FGF19, агонисты PPAR, агонисты FXR, активаторы АМРК [например, ЕТС-1002 (бемпедоевая кислота)], ингибиторы SCD1 или ингибиторы МРО.
Примеры агонистов рецепторов GLP-1 включают лираглутид, албиглутид, эксенатид, албиглутид, ликсисенатид, дулаглутид, семаглутид, НМ15211, LY3298176, Medi-0382, NN-9924, ТТР-054, ТТР-273, эфпегленатид, средства, описанные в WO 2018109607, описанные в РСТ/IB 2019/054867, поданной 11 июня 2019 г., и описанные в РСТ/IB2019/054961, поданной 13 июня 2019 г., включая следующие соединения:
2-({4-[2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1Н-бензимидазол-6-карбоновая кислота;
2-({4-[2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-7-фтор-1-[(2S)-оксетан-2-илметил]-1Н-бензимидазол-6-карбоновая кислота;
2-({4-[(2S)-2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1Н-бензимидазол-6-карбоновая кислота;
2-({4-[(2S)-2-(4-хлор-2-фторфенил)-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-7-фтор-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-({4-[2-(4-циано-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-({4-[2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-3-(1,3-оксазол-2-илметил)-3N-имидазо[4,5-b]пиридин-5-карбоновая кислота;
2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(1-этил-1H-имидазол-5-ил)метил]-1Н-бензимидазол-6-карбоновая кислота;
2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-(1,3-оксазол-4-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-(пиридин-3-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-(1, 3-о кса зол-5-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(1-этил-1H-1,2,3-триазол-бил)метил]-1Н-бензимидазол-6-карбоновая кислота;
2-({4-[2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-(1,3-оксазол-2-илметил)-1Н-бензимидазол-6-карбоновая кислота;
2-({4-[2-(4-хлор-2-фторфенил)-7-фтор-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1Н-бензимидазол-6-карбоновая кислота;
2-({4-[2-(4-циано-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-(1,3-оксазол-2-илметил)-1Н-бензимидазол-6-карбоновая кислота;
2-({4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-7-фтор-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-({4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-({4-[(2S)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-7-фтор-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-({4-[(2S)-2-(4-циано-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-({4-[(2S)-2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-({4-[(2S)-2(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(1-этил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоновая кислота;
2-({4-[(2R)-2-(4-хлор-2-фторфенил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(1-этил-1H-имидазол-5-ил)метил]-1H-бензимидазол-6-карбоновая кислота;
2-({4-[2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2 -илметил]-1H-бензимидазол-6-карбоновая кислота;
2-({4-[(2S)-2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1Н-бензимидазол-6-карбоновая кислота;
2-({4-[(2R)-2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1Н-бензимидазол-6-карбоновая кислота;
2-({4-[2-(5-хлорпиридин-2-ил)-2-метил-1,3-бензодиоксол-4-ил]пиперидин-1-ил}метил)-1-[(2S)-оксетан-2-илметил]-1Н-бензимидазол-6-карбоновая кислота, DIAST-X2;
2-[(4-{2-[(4-хлор-2-фторбензил)окси]пиридин-3-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1Н-бензимидазол-6-карбоновая кислота;
2-[(4-{2-[(4-хлор-2-фторбензил)окси]пиридин-3-ил}пиперидин-1-ил)метил]-1-(1,3-оксазол-2-илметил)-1Н-бензимидазол-6-карбоновая кислота;
2-[(4-{2-[(4-циано-2-фторбензил)окси]пиридин-3-ил}пиперидин-1-ил)метил]-1-(1,3-оксазол-2-илметил)-1Н-бензимидазол-6-карбоновая кислота;
2-[(4-{2-[(4-циано-2-фторбензил)окси]пиридин-3-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{3-[(4-хлор-2-фторбензил)окси]пиразин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1Н-бензимидазол-6-карбоновая кислота;
2-(6-{6-[(4-циано-2-фторбензил)окси]пиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-H-бензимидазол-6-карбоновая кислота;
2-(6-{2-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-4-ил}-6-азаспиро [2.5] окт-1 -ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-(6-{2-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-4-ил}-6-азаспиро[2.5]окт-1-ил)-1-(1,3-оксазол-2-илметил)-1H-бензимидазол-6-карбоновая кислота;
2-(6-{6-[(4-циано-2-фторбензил)окси]-5-фторпиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-(6-{6-[(4-циано-2-фторбензил)окси]-3-фторпиридин-2-ил}-6-азаспиро[2.5]окт-1-ил)-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-[(4-{2-[(4-хлор-2-фторбензил)окси]пиримидин-4-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-{[(2S)-4-{2-[(4-хлор-2-фторбензил)окси]-5-фторпиримидин-4-ил}-2-метилпиперазин-1-ил]метил}-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота;
2-{[(2S)-4-{2-[(4-хлор-2-фторбензил)окси]пиримидин-4-ил}-2-метилпиперазин-1-ил]метил}-1-[(2S)-оксетан-2-илметил]-1Н-бензимидазол-6-карбоновая кислота; и
2-[(4-{6-[(4-циано-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1Н-бензимидазол-6-карбоновая кислота, и их фармацевтически приемлемые соли.
Примеры ингибиторов АСС включают 4-(4-[(1-изопропил-7-оксо-1,4,6,7-тетрагидро-1'H-спиро[индазол-5,4'-пиперидин]-1'-ил)карбонил]-6-метоксипиридин-2-ил)бензойную кислоту, гемкабен и фирсокостат (GS-0976) и их фармацевтически приемлемые соли.
Примеры агонистов FXR включают тропифексор (2-[(1R,3R,55)-3-({5-циклопропил-3-[2-(трифторметокси)фенил]-1,2-оксазол-4-ил}метокси)-8-азабицикло[3,2,1]октан-8-ил]-4-фтор-1,3-бензотиазол-6-карбоновая кислота), цилофексор (GS-9674), обетихолевую кислоту, LY2562175, Met409, TERN-101 и EDP-305, и их фармацевтически приемлемые соли.
Примеры ингибиторов КНК включают [(1R,5S,6R)-3-{2-[(2S)-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3.1.0]гекс-6-ил]уксусную кислоту и ее фармацевтически приемлемые соли.
Примеры ингибиторов DGAT2 включают (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид [включая его кристаллические твердые формы (Форму 1 и Форму 2)]. См. патент США №10071992.
Примеры ингибиторов BCKDK включают ингибиторы, описанные в US Serial No. 62/868057, поданной 28 июня 2019 г., и US Serial No. 62/868,542 поданной 28 июня 2019 г., включая следующие соединения:
5-(5-хлор-4-фтор 3-метилтиофен-2-ил)-1H-тетразол;
5-(5-хлор-3-дифторметилтиофен-2-ил)-1H-тетразол;
5-(5-фтор-3-метилтиофен-2-ил)-1H-тетразол;
5-(5-хлор-3-метилтиофен-2-ил)-1H-тетразол;
5-(3,5-дихлортиофен-2-ил)-1H-тетразол;
5-(4-бром-3-метилтиофен-2-ил)-1H-тетразол;
5-(4-бром-3-этилтиофен-2-ил)-1H-тетразол;
5-(4-хлор-3-этилтиофен-2-ил)-1H-тетразол;
3-хлор-5-фтортиено[3,2-b]тиофен-2-карбоновая кислота;
3-бром-5-фтортиено[3,2-b]тиофен-2-карбоновая кислота;
3-(дифторметил)-5-фтортиено[3,2-b]тиофен-2-карбоновая кислота;
5,6-дифтортиено[3,2-b]тиофен-2-карбоновая кислота; и
3,5-дифтортиено[3,2-b]тиофен-2-карбоновая кислота;
или их фармацевтически приемлемые соли.
В некоторых вариантах осуществления соединение по настоящему изобретению можно вводить совместно с одним или несколькими противодиабетическими средствами. Подходящие противодиабетические средства включают инсулин, метформин, агонисты рецептора GLP-1 (описанные в настоящей заявке выше), ингибитор ацетил-СоА-карбоксилазы (АСС) (описанный в настоящей заявке выше), ингибиторы SGLT2 (описанные в настоящей заявке выше), ингибиторы моноацилглицерол-O-ацилтрансферазы, ингибиторы фосфодиэстеразы (PDE)-10, активаторы AMPK [например, ЕТС-1002 (бемпедоевая кислота)], средства на основе сульфонилмочевины (например, ацетогексамид, хлорпропамид, диабинез, глибенкламид, глипизид, глибурид, глимепирид, гликлазид, глипентид, гликвидон, глизоламид, толазамид, и толбутамид), меглитиниды, ингибиторы α-амилазы (например, тендамистат, трестатин и AL-3688), ингибитор α-глюкозидгидролазы (например, акарбоза), ингибиторы α-глюкозидазы (например, адипозин, камиглибоза, эмиглитат, миглитол, воглибоза, прадимицин-Q и сальбостатин), агонисты PPARγ (например, балаглитазон, циглитазон, дарглитазон, энглитазон, изаглитазон, пиоглитазон и розиглитазон), агонисты PPAR α/γ (например, CLX-0940, GW-1536, GW-1929, GW-2433, KRP-297, L-796449, LR-90, MK-0767 и SB-219994), ингибиторы протеинтирозинфосфатазы-1B (РТР-1В) [например, тродускемин, экстракт гиртиозала и соединения, раскрытые в Zhang, S. et al., Drug Discovery Today, 12(9/10), 373-381 (2007)], активаторы SIRT-1 (например, ресвератрол, GSK2245840 или GSK184072), ингибиторы дипептидилпептидазы IV (DPP-IV) (например, описанные в WO 2005116014, ситаглиптин, вилдаглиптин, алоглиптин, дутоглиптин, линаглиптин и саксаглиптин), средства, стимулирующие секрецию инсулина, ингибиторы окисления жирных кислот, антагонисты А2, ингибиторы c-jun амино-концевой киназы (JNK), активаторы глюкокиназы (GKa), такие как описанные в WO 2010103437, WO 2010103438, WO 2010013161, WO 2007122482, ТТР-399, ТТР-355, ТТР-547, AZD1656, ARRY1403, MK-0599, TAK-329, AZD5658 или GKM-001, инсулин, миметики инсулина, ингибиторы гликогенфосфорилазы (например, GSK1362885), агонисты рецептора VPAC2, модуляторы рецептора глюкагона, такие как описанные в Demong, D.E. et al., Annual Reports in Medicinal Chemistry 2008, 43, 119-137, модуляторы GPR119, особенно агонисты, такие как описанные в WO 2010140092, WO 2010128425, WO 2010128414, WO 2010106457, Jones, R.M. et al., Annual Reports in Medicinal Chemistry 2009, 44, 149-170 (например, МВХ-2982, GSK1292263, APD597 и PSN821), производные или аналоги FGF21, например описанные в Kharitonenkov, А. et al., Current Opinion in Investigational Drugs 2009, 10(4)359-364, модуляторы рецепторов TGR5 (также называемые GPBAR1), особенно агонисты, такие как описанные в Zhong, M., Current Topics in Medicinal Chemistry, 2010, 10(4), 386-396 и INT777, агонисты GPR40, такие как описанные в Medina, J.C., Annual Reports in Medicinal Chemistry, 2008, 43, 75-85, включая но не ограничиваясь этим, модуляторы TAK-875, GPR120, особенно агонисты, активаторы высокоаффинного рецептора никотиновой кислоты (НМ74А) и ингибиторы SGLT1, такие как GSK1614235. Дополнительный репрезентативный перечень антидиабетических средств, которые можно комбинировать с соединениями по настоящему изобретению, можно найти, например, на странице 28, строка 35 - страница 30, строка 19 WO 2011005611.
Другие противодиабетические средства могут включать ингибиторы или модуляторы ферментов карнитинпальмитоилтрансферазы, ингибиторы фруктозо-1,6-дифосфатазы, ингибиторы альдозоредуктазы, ингибиторы минералокортикоидных рецепторов, ингибиторы TORC2, ингибиторы CCR2 и/или CCR5, ингибиторы изоформ PKC (например,, PKCα, PKCβ, PKCγ), ингибиторы синтетазы жирных кислот, ингибиторы серинпальмитоилтрансферазы, модуляторы GPR81, GPR39, GPR43, GPR41, GPR105, Kv1.3, ретинолсвязывающего белка 4, глюкокортикоидного рецептора, рецепторов соматостатина (например, SSTR1, SSTR2, SSTR3 и SSTR5), ингибиторы или модуляторы PDHK2 или PDHK4, ингибиторы MAP4K4, модуляторы семейства IL1, включая IL1бета, модуляторы RХRальфа. Кроме того, подходящие антидиабетические средства включают механизмы, перечисленные в Carpino, P.A., Goodwin, В. Expert Opin. Ther. Pat., 2010, 20(12), 1627-51.
Соединения по настоящему изобретению можно вводить совместно со средствами против сердечной недостаточности, такими как ингибиторы АСЕ (например, каптоприл, эналаприл, фозиноприл, лизиноприл, периндоприл, хинаприл, рамиприл, трандолаприл), блокаторы рецепторов ангиотензина II (например, кандесартан, лозартан, валсартан), ингибиторы неприлизина ангиотензиновых рецепторов (сакубитрил/валсартан), блокатор If-каналов ивабрадин, бета-адреноблокаторы (например, бисопролол, метопролола сукцинат, карведилол), антагонисты альдостерона (например, спиронолактон, эплеренон), гидралазин и изосорбид динитрат, диуретики (например, фуросемид, буметанид, торасемид, хлоротиазид, амилорид, гидрохлоротиазид, индапамид, метолазон, триамтерен) или дигоксин.
Соединения по настоящему изобретению можно также вводить совместно со средствами, снижающими уровень холестерина или липидов, включая следующие иллюстративные средства: ингибиторы HMG СоА-редуктазы (например, правастатин, питавастатин, ловастатин, аторвастатин, симвастатин, флувастатин, NK-104 (также известный как итавастатин, или нисвастатин, или нисбастатин) и ZD-4522 (также известный как розувастатин, или атавастатин, или висастатин); ингибиторы скваленсинтетазы; фибраты (например, гемфиброзил, пемафибрат, фенофибрат, клофибрат); секвестранты желчных кислот (такие как квестран, колестипол, колесевелам); ингибиторы АСАТ; ингибиторы МТР, ингибиторы липооксигеназы, ингибиторы абсорбции холестерина (например, эзетимиб), препараты никотиновой кислоты (например, ниацин, ниакор, сло-ниацин), омега-3 жирные кислоты (например, эпанова, рыбий жир, эйкозапентаеновая кислота), ингибиторы белков-переносчиков эфира холестерина (например, обицетрапиб) и модуляторы PCSK9 [например, алирокумаб, эволокумаб, бокоцизумаб, ALN-PCS (инклизиран)].
Соединения по настоящему изобретению также можно использовать в комбинации с антигипертензивными средствами, и такую антигипертензивную активность легко могут определить специалисты в данной области на основании стандартных анализов (например, измерения кровяного давления). Примеры подходящих антигипертензивных средств включают: блокаторы альфа-адренорецепторов; бета-адреноблокаторы; блокаторы кальциевых каналов (например, дилтиазем, верапамил, нифедипин и амлодипин); вазодилататоры (например, гидралазин), диуретики (например, хлоротиазид, гидрохлоротиазид, флуметиазид, гидрофлуметиазид, бендрофлуметиазид, метилхлоротиазид, трихлорметиазид, политиазид, бензтиазид, этакриновая кислота, трикринафен, хлорталидон, торасемид, фуросемид, мусолимин, буметанид, триамтренен, амилорид, спиронолактон); ингибиторы ренина; ингибиторы АСЕ (например, каптоприл, зофеноприл, фозиноприл, эналаприл, цераноприл, цилазоприл, делаприл, пентоприл, хинаприл, рамиприл, лизиноприл); антагонисты рецептора АТ-1 (например, лозартан, ирбесартан, валсартан); антагонисты рецептора ЕТ (например, ситакссентан, атрсентан и соединения, раскрытые в патентах США №№5612359 и 6043265); двойной антагонист ET/AII (например, соединения, раскрытые в WO 00/01389); ингибиторы нейтральной эндопептидазы (NEP); ингибиторы вазопептидазы (двойные ингибиторы NEP-ACE) (например, гемопатрилат и нитраты). Иллюстративным антиангинальным средством является ивабрадин.
Примеры подходящих блокаторов кальциевых каналов (L-типа или Т-типа) включают дилтиазем, верапамил, нифедипин и амлодипин и мибефрадил.
Примеры подходящих сердечных гликозидов включают дигиталис и уабаин.
В одном варианте осуществления соединение по изобретению можно вводить совместно с одним или несколькими диуретиками. Примеры подходящих диуретиков включают (а) петлевые диуретики, такие как фуросемид (такой как LASIX™), торасемид (такой как DEMADEX™), беметанид (такой как BUMEX™) и этакриновую кислоту (такую как EDECRIN™); (b) диуретики тиазидного типа, такие как хлоротиазид (например, DIURIL™, ESIDRIX™ или HYDRODIURIL™), гидрохлоротиазид (например, MICROZIDE™ или ORETIC™), бензтиазид, гидрофлуметиазид (например, SALURON™), бендрофлуметиазид, метилхлортиазид, политиазид, трихлорметиазид и индапамид (такой как LOZOL™); (с) диуретики фталимидинового типа, такие как хлорталидон (такой как HYGROTON™) и метолазон (такой как ZAROXOLYN™); (d) диуретики хиназолинового типа, такие как хинетазон; и (е) калийсберегающие диуретики, такие как триамтерен (например, DYRENIUM™) и амилорид (например, MIDAMOR™ или MODURETIC™).
В другом варианте осуществления соединение по изобретению можно вводить совместно с петлевым диуретиком. В еще одном варианте осуществления петлевой диуретик выбирают из фуросемида и торасемида. В еще одном варианте осуществления одно или несколько соединений формулы I или их фармацевтически приемлемых солей можно вводить совместно с фуросемидом. В еще одном варианте осуществления одно или несколько соединений формулы I или их фармацевтически приемлемых солей можно вводить совместно с торасемидом, который необязательно может представлять собой форму торасемида с контролируемым или модифицированным высвобождением.
В другом варианте осуществления соединение по изобретению можно вводить совместно с диуретиком тиазидного типа. В еще одном варианте осуществления диуретик тиазидного типа выбирают из группы, состоящей из хлоротиазида и гидрохлоротиазида. В еще одном варианте осуществления одно или несколько соединений формулы I или их фармацевтически приемлемых солей можно вводить совместно с хлоротиазидом. В еще одном варианте осуществления одно или несколько соединений формулы I или их фармацевтически приемлемых солей можно вводить совместно с гидрохлоротиазидом.
В другом варианте осуществления одно или несколько соединений формулы I или их фармацевтически приемлемых солей можно вводить совместно с диуретиком фталимидинового типа. В еще одном варианте осуществления диуретик фталимидинового типа представляет собой хлорталидон.
Примеры подходящих антагонистов минералокортикоидных рецепторов включают сприонолактон и эплеренон.
Примеры подходящих ингибиторов фосфодиэстеразы включают: ингибиторы PDE III (такие как цилостазол); и ингибиторы PDE V (такие как силденафил).
Специалистам в данной области должно быть понятно, что соединения по настоящему изобретению можно также использовать в сочетании с другими видами лечения сердечно-сосудистых или цереброваскулярных заболеваний, включая чрескожное коронарное вмешательство, стентирование, стенты с лекарственным покрытием, терапию стволовыми клетками и медицинские устройства, такие как имплантированные кардиостимуляторы, дефибрилляторы или сердечная ресинхронизирующая терапия.
В частности, при предоставлении в виде единой дозированной лекарственной формы существует вероятность химического взаимодействия между комбинированными активными ингредиентами. По этой причине, когда соединение по настоящему изобретению и второе терапевтическое средство объединяют в одной дозированной лекарственной форме, они сформулированы таким образом, что, хотя активные ингредиенты объединены в одной дозированной лекарственной форме, физический контакт между активными ингредиентами сведен к минимуму (т.е. уменьшен). Например, один активный ингредиент может иметь энтеросолюбильное покрытие. Когда один из активных ингредиентов имеет энтеросолюбильное покрытие, можно не только минимизировать контакт между комбинированными активными ингредиентами, но также можно контролировать высвобождение одного из этих компонентов в желудочно-кишечном тракте таким образом, чтобы один из этих компонентов не высвобождался в желудке, а высвобождался в кишечнике. Один из активных ингредиентов также может быть покрыт материалом, который предоставляет замедленное высвобождение в желудочно-кишечном тракте, а также служит для сведения к минимуму физического контакта между комбинированными активными ингредиентами. Кроме того, компонент с замедленным высвобождением может быть дополнительно покрыт энтеросолюбильным покрытием, так что высвобождение этого компонента происходит только в кишечнике. Еще один подход предполагает формулирование комбинированного продукта, в котором один компонент покрыт полимером, обеспечивающим замедленное высвобождение и/или высвобождение в кишечнике, а другой компонент также покрыт полимером, таким как гидроксипропилметилцеллюлоза с низкой вязкостью (НРМС) или другие подходящие вещества, известные в данной области техники, для дальнейшего разделения активных компонентов. Полимерное покрытие служит дополнительным барьером для взаимодействия с другим компонентом.
Эти, а также другие способы сведения к минимуму контакта между компонентами комбинированных продуктов по настоящему изобретению, независимо от того, вводятся ли они в одной лекарственной форме или вводятся в отдельных формах, но в то же время одним и тем же способом, будут очевидны специалистам в данной области техники, когда они ознакомятся с настоящим раскрытием.
При лечении комбинированной терапией соединения по настоящему изобретению, а также другие лекарственные терапии вводят млекопитающим (например, людям, мужчинам или женщинам) обычными способами. Соединение формулы I или его соль адаптированы для терапевтического применения в качестве средств, которые проявляют антагонистическое действие (включая ингибирование) в отношении MC4R у млекопитающих, в частности у людей, и, таким образом, применимы для лечения различных состояний (например, описанных в настоящей заявке), при которых подразумевается такое действие.
Заболевание/расстройство/состояние, которое можно лечить в соответствии с настоящим изобретением, включает, но не ограничивается этим, кахексию (например, кахексию, связанную с раком, СПИДом, застойной сердечной недостаточностью и/или хронической болезнью почек); анорексию/нервную анорексию (например, гериатрическую анорексию, анорексию, связанную с химиотерапией и/или лучевой терапией); тошноту; рвоту; потерю массы тела (например, непреднамеренную потерю массы тела); задержку развития; саркопению; мышечную атрофию; немощность; остеопороз; заболевания костей (например, потерю костной массы); боль; невропатическую боль; беспокойство; депрессию; гипертензию; нарушение питания; ожирение; сексуальную дисфункцию; и воспалительное заболевание.
Введение соединений по настоящему изобретению можно осуществлять любым способом, при котором соединение по настоящему изобретению доставляется системно и/или локально. Эти способы включают пероральный, парентеральный, интрадуоденальный пути, буккальный, интраназальный и т.д. Обычно соединения по настоящему изобретению вводят перорально, но можно использовать парентеральное введение (например, внутривенное, внутримышечное, подкожное или интрамедуллярное), например, когда пероральное введение не подходит для мишени или когда пациент не может проглотить препарат.
Для введения пациентам-людям пероральная суточная доза соединений по настоящему изобретению может составлять, например, от 0,01 мг до 5000 мг в зависимости, конечно, от способа и частоты введения, состояния болезни, возраста и состояния пациента и т.д. Можно использовать пероральную суточную дозу в диапазоне от 1 мг до 2000 мг (например, от 3 мг до 2000 мг). В качестве еще одного примера, пероральная суточная доза составляет от 5 мг до 1000 мг. Для удобства соединения по настоящему изобретению можно вводить в стандартной лекарственной форме. При желании можно использовать несколько доз стандартной лекарственной формы в день для увеличения общей суточной дозы. Стандартная лекарственная форма, например, может представлять собой таблетку или капсулу, содержащую около 0,1, 0,5, 1, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 125, 150, 175, 200, 250, 300, 500 или 1000 мг соединения по настоящему изобретению. Общую суточную дозу можно вводить в виде разовой дозы или в несколько приемов, и она может, по усмотрению врача, выходить за пределы типичных диапазонов, указанных в настоящей заявке.
Для введения пациентам-людям суточная доза соединений по настоящему изобретению может находиться в диапазоне от 1 мг до 2000 мг, в зависимости, конечно, от способа и частоты введения, болезненного состояния, а также возраста и состояния пациента и т.д. В качестве еще одного примера, суточная доза инфузии находится в диапазоне от 5 мг до 1000 мг. Общую суточную дозу можно вводить в виде разовой дозы или в несколько приемов, и она может, по усмотрению врача, выходить за пределы типичных диапазонов, указанных в настоящей заявке.
В соответствии со способами лечения по изобретению соединение по настоящему изобретению или комбинацию соединения по настоящему изобретению и по меньшей мере одного дополнительного лекарственного средства (называемого в настоящей заявке «комбинацией») вводят субъекту, нуждающемуся в таком лечении, предпочтительно в виде фармацевтической композиции. В аспекте изобретения, относящемся к комбинации, соединение по настоящему изобретению и по меньшей мере одно другое лекарственное средство (например, другое средство против кахексии или анорексии) можно вводить либо по отдельности, либо в фармацевтической композиции, включающей оба средства. Обычно предпочтительно, чтобы такое введение было пероральным.
Когда комбинацию соединения по настоящему изобретению и по меньшей мере одного другого лекарственного средства вводят вместе, такое введение может быть последовательным по времени или одновременным. В некоторых вариантах осуществления используют одновременное введение комбинаций лекарственных средств. Для раздельного или последовательного введения соединение по настоящему изобретению и дополнительное лекарственное средство можно вводить в любом порядке, и каждое из них можно вводить с независимой частотой или режимом дозирования. В некоторых вариантах осуществления такое введение может быть пероральным. В некоторых вариантах осуществления такое введение может быть пероральным и одновременным. Когда соединение по настоящему изобретению и дополнительное лекарственное средство вводят последовательно, введение каждого из них можно осуществлять одним и тем же или разными способами.
В соответствии со способами по изобретению соединение по настоящему изобретению или комбинацию можно вводить в виде фармацевтической композиции. Соответственно, соединение по настоящему изобретению или комбинацию можно вводить пациенту по отдельности или вместе в любой обычной лекарственной форме для перорального, ректального, чрескожного, парентерального (например, внутривенного, внутримышечного или подкожного), интрацистернального, интравагинального, интраперитонеального, местного (например, порошок, мазь, крем, спрей или лосьон), буккального или назального (например, спрей, капли или средство для ингаляции) введения.
Соединения по изобретению или их комбинации можно вводить отдельно или в смеси с одним или несколькими подходящими фармацевтическими эксципиентами, адъювантами, разбавителями или носителями, известными в данной области и выбранными с учетом предполагаемого пути введения и стандартной фармацевтической практики. Соединение по изобретению или комбинацию можно формулировать для получения лекарственных форм с немедленным, отсроченным, модифицированным, пролонгированным, импульсным или контролируемым высвобождением в зависимости от желаемого пути введения и специфики профиля высвобождения, соизмеримо с терапевтическими потребностями.
Фармацевтическая композиция включает соединение по изобретению или комбинацию в количестве, как правило, в диапазоне от около 1% до около 75%, 80%, 85%, 90% или даже 95% (по массе) композиции, обычно в диапазоне от около 1%, 2% или 3% до около 50%, 60% или 70%, чаще в диапазоне от около 1%, 2% или 3% до менее чем 50%, например, около 25%, 30% или 35%.
Способы получения различных фармацевтических композиций с определенным количеством активного соединения известны специалистам в данной области. Например, см. Remington, J.P., The Science and Practice of Pharmacy, Lippincott Williams and Wilkins, Baltimore, Md. 20th ed., 2000.
Композиции, подходящие для парентеральной инъекции, обычно включают фармацевтически приемлемые стерильные водные или неводные растворы, дисперсии, суспензии или эмульсии и стерильные порошки для восстановления в стерильные растворы или дисперсии для инъекций. Примеры подходящих водных и неводных носителей или разбавителей (включая растворители и наполнители) включают воду, этанол, полиолы (пропиленгликоль, полиэтиленгликоль, глицерин и т.п.), их подходящие смеси, триглицериды, включая растительные масла, такие как оливковое масло, и инъекционные органические сложные эфиры, такие как этилолеат. Предпочтительным носителем является Miglyol.RTM, фирменный эфир каприловой/каприновой кислоты с глицерином или пропиленгликолем (например, Miglyol.RTM. 812, Miglyol.RTM. 829, Miglyol.RTM. 840), доступный от Condea Vista Co., Cranford, N.J. Надлежащую текучесть можно поддерживать, например, за счет использования покрытия, такого как лецитин, за счет поддержания требуемого размера частиц в случае дисперсий и за счет использования поверхностно-активных веществ.
Эти композиции для парентерального введения могут также содержать эксципиенты, такие как консерванты, смачивающие, эмульгирующие и диспергирующие агенты. Загрязнение композиций микроорганизмами можно предотвратить с использованием различных антибактериальных и противогрибковых средств, например, парабенов, хлорбутанола, фенола, сорбиновой кислоты и т.п. Также может быть желательно включить изотонические агенты, например, сахара, хлорид натрия и т.п. Пролонгированную абсорбцию инъецируемых фармацевтических композиций можно обеспечить с использованием агентов, способных замедлять абсорбцию, например, моностеарата алюминия и желатина.
Твердые лекарственные формы для перорального введения включают капсулы, таблетки, жевательные таблетки, леденцы, пилюли, порошки и препараты, состоящие из множества частиц (гранулы). В таких твердых лекарственных формах соединение по настоящему изобретению или их комбинацию смешивают по меньшей мере с одним инертным эксципиентом, разбавителем или носителем. Подходящие эксципиенты, разбавители или носители включают такие вещества, как цитрат натрия или дикальцийфосфат и/или (а) один или несколько наполнителей или объемообразующих веществ (например, микрокристаллическую целлюлозу (доступную как Avicel ТМ от FMC Corp.), крахмалы, лактозу, сахарозу, маннит, кремниевую кислоту, ксилит, сорбит, декстрозу, гидрофосфат кальция, декстрин, альфа-циклодекстрин, бета-циклодекстрин, полиэтиленгликоль, жирные кислоты со средней длиной цепи, оксид титана, оксид магния, оксид алюминия и т.п.); (b) одно или несколько связующих веществ (например, карбоксиметилцеллюлозу, метилцеллюлозу, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, желатин, гуммиарабик, этилцеллюлозу, поливиниловый спирт, пуллулан, прежелатинизированный крахмал, агар, трагакант, альгинаты, желатин, поливинилпирролидон, сахарозу, аравийскую камедь и т.п.); (с) один или несколько увлажнителей (например, глицерин и т.п.); (d) один или несколько разрыхлителей (например, агар-агар, карбонат кальция, картофельный или маниоковый крахмал, альгиновую кислоту, некоторые комплексные силикаты, карбонат натрия, лаурилсульфат натрия, натрия крахмалгликолят (доступны как Explotab™ от Edward Mendell Co.), сшитый поливинилпирролидон, натрия кроскармеллозу типа А (доступна как Ac-di-sol™), полиакрилин калия (ионообменная смола) и т.п.); (е) один или несколько замедлителей растворения (например, парафин и т.п.); (f) один или несколько ускорителей абсорбции (например, соединения четвертичного аммония и т.п.); (д) один или несколько смачивающих агентов (например, цетиловый спирт, моностеарат глицерина и т.п.); (h) один или несколько адсорбентов (например, каолин, бентонит и т.п.); и/или одно или несколько смазывающих веществ (например, тальк, стеарат кальция, стеарат магния, стеариновую кислоту, полиоксилстеарат, цетанол, гидрогенизированное касторовое масло, эфиры сахарозы и жирных кислот, диметилполисилоксан, микрокристаллический воск, желтый пчелиный воск, белый пчелиный воск, твердые полиэтиленгликоли, лаурилсульфат натрия и т.п.). В случае капсул и таблеток лекарственные формы могут также содержать буферные агенты.
Твердые композиции аналогичного типа можно также использовать в качестве наполнителей в мягких или твердых желатиновых капсулах с использованием таких эксципиентов, как лактоза или молочный сахар, а также высокомолекулярных полиэтиленгликолей и т.п.
Твердые лекарственные формы, такие как таблетки, драже, капсулы и гранулы, могут быть получены с покрытиями и оболочками, такими как энтеросолюбильные покрытия и другие, хорошо известные в данной области техники. Они также могут содержать агенты, придающие непрозрачность, и могут также иметь такую композицию, чтобы они могли высвобождать соединение по настоящему изобретению и/или дополнительное лекарственное средство замедленным образом. Примерами заливочных композиций, которые можно использовать, являются полимерные вещества и воски. Лекарственное средство также может быть в микрокапсулированной форме, при необходимости, с одним или несколькими из вышеуказанных эксципиентов.
В случае таблеток, активное вещество обычно будет составлять менее 50% (по массе) композиции, например, менее примерно 10%, например, 5% или 2,5% по массе. Преобладающую часть композиции составляют наполнители, разбавители, разрыхлители, смазывающие вещества и, необязательно, ароматизаторы. Смеси таких эксципиентов хорошо известны в данной области. Часто наполнители/разбавители включают смеси двух или более из следующих компонентов: микрокристаллическая целлюлоза, маннит, лактоза (все виды), крахмал и дикальцийфосфат. Смеси наполнитель/разбавитель обычно составляют менее 98% композиции и предпочтительно менее 95%, например, 93,5%. Предпочтительные разрыхлители включают Ac-di-sol™, Explotab™, крахмал и лаурилсульфат натрия. Разрыхлитель, если он присутствует, обычно составляет менее 10% композиции или менее 5%, например, около 3%. Предпочтительным смазывающим веществом является стеарат магния. Смазывающее вещество, если оно присутствует, обычно составляет менее 5% композиции или менее 3%, например, около 1%.
Таблетки могут быть получены стандартными способами таблетирования, например, прямым прессованием или влажным, сухим гранулированием или гранулированием из расплава, способом отверждения расплава и экструзией. Ядра таблеток могут быть моно- или многослойными и могут иметь соответствующие покрытия, известные в данной области техники.
Жидкие лекарственные формы для перорального введения включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры. В дополнение к соединению по настоящему изобретению или комбинации жидкая лекарственная форма может содержать инертные разбавители, обычно используемые в данной области техники, такие как вода или другие растворители, солюбилизирующие агенты и эмульгаторы, как, например, этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (например, масло семян хлопчатника, арахисовое масло, масло зародышей кукурузы, оливковое масло, касторовое масло, кунжутное масло и т.п.), Miglyol.RTM. (доступный от CONDEA Vista Co., Cranford, N.J.), глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и сложные эфиры сорбитана и жирных кислот или смеси этих веществ и т.п.
Помимо таких инертных разбавителей композиция может также включать эксципиенты, такие как смачивающие агенты, эмульгаторы и суспендирующие агенты, подсластители, ароматизаторы и отдушки.
Пероральные жидкие формы соединений по изобретению или комбинаций включают растворы, в которых активное соединение полностью растворено. Примеры растворителей включают все известные фармацевтические растворители, подходящие для перорального введения, в частности те, в которых соединения по изобретению проявляют хорошую растворимость, например, полиэтиленгликоль, полипропиленгликоль, пищевые масла и системы на основе глицерила и глицерида. Системы на основе глицерила и глицерида могут включать, например, следующие фирменные продукты (и соответствующие непатентованные продукты): Captex.TM. 355 ЕР (глицерилтрикаприлат/капрат, от Abitec, Columbus Ohio), Crodamol.ТМ. (триглицерид со средней длиной цепи, от Croda, Cowick Hall, UK) или Labrafac.TM. CC (триглииды со средней длиной цепи от Gattefosse), Captex.TM. 500P (глицерилтриацетат, т.е. триацетин, от Abitec), Capmnl.TM. MCM (моно- и диглицериды со средней длиной цепи от Abitec), Migyol.TM. 812 (каприловый/каприновый триглицерид, от Condea, Cranford N.J.), Migyol.TM. 829 (каприловый/каприновый/янтарный триглицерид от Condea), Migyol.TM. 840 (пропиленгликоль дикаприлат/дикапрат, от Condea), Labrafil.TM. M1944CS (глицериды олеоилмакрогола-6, от Gattefosse), Peceol.TM. (глицерилмоноолеат, от Gattefosse) и Maisine.TM. 35-1 (глицерилмоноолеат, от Gattefosse). Особый интерес представляют триглицеридные масла со средней длиной цепи (от C8 до С10). Эти растворители часто составляют преобладающую часть композиции, т.е. больше чем около 50%, обычно больше чем около 80%, например, около 95% или 99%. Адъюванты и добавки также могут быть включены в состав растворителей, главным образом, в качестве агентов, маскирующих вкус, улучшающих вкусовые качества и ароматизирующих агентов, антиоксидантов, стабилизаторов, модификаторов текстуры и вязкости и солюбилизаторов.
Суспензии, в дополнение к соединению по настоящему изобретению или комбинации, могут также включать носители, такие как суспендирующие агенты, например, этоксилированные изостеариловые спирты, полиоксиэтиленсорбит и сложные эфиры сорбитана, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, или смеси этих веществ и т.п.
Композиции для ректального или вагинального введения предпочтительно включают суппозитории, которые можно получить путем смешивания соединения по настоящему изобретению или комбинации с подходящими нераздражающими эксципиентами или носителями, такими как масло какао, полиэтиленгликоль или воск для суппозиториев, которые являются твердыми при обычной комнатной температуре, но жидкими при температуре тела и, следовательно, расплавляются в прямой кишке или вагинальной полости, высвобождая таким образом активный компонент(компоненты).
Лекарственные формы для местного введения соединений по настоящему изобретению или их комбинаций включают мази, кремы, лосьоны, порошки и спреи. Лекарственные средства смешивают с фармацевтически приемлемым эксципиентом, разбавителем или носителем и любыми консервантами, буферами или пропеллентами, которые могут потребоваться.
Некоторые из соединений по настоящему изобретению могут быть плохо растворимы в воде, например, меньше чем около 1 мкг/мл. Следовательно, жидкие композиции в солюбилизирующих неводных растворителях, таких как триглицеридные масла со средней длиной цепи, обсуждавшиеся выше, являются предпочтительной лекарственной формой для этих соединений.
Твердые аморфные дисперсии, включая дисперсии, образованные в процессе распылительной сушки, также являются предпочтительной лекарственной формой для плохо растворимых соединений по изобретению. Под «твердой аморфной дисперсией» понимают твердое вещество, в котором по меньшей мере часть плохо растворимого соединения находится в аморфной форме и диспергирована в водорастворимом полимере. Под «аморфным» подразумевается, что плохо растворимое соединение не является кристаллическим. Под «кристаллическим» подразумевается, что соединение демонстрирует дальний порядок в трех измерениях, по меньшей мере, 100 повторяющихся единиц в каждом измерении. Таким образом, термин «аморфный» предназначен для включения не только вещества, которое по существу не имеет порядка, но также вещества, которое может иметь некоторую небольшую степень порядка, но порядок менее чем в трех измерениях и/или только на коротких расстояниях. Аморфное вещество может быть охарактеризовано методами, известными в данной области техники, такими как кристаллографический метод порошковой рентгеновской дифракции (PXRD), твердотельный ЯМР или термические методы, такие как дифференциальная сканирующая калориметрия (DSC).
Предпочтительно, чтобы по меньшей мере большая часть (т.е. по меньшей мере около 60% масс.) плохо растворимого соединения в твердой аморфной дисперсии была аморфной. Соединение может существовать в твердой аморфной дисперсии в относительно чистых аморфных доменах или областях, в виде твердого раствора соединения, гомогенно распределенного по всему полимеру, или в любой комбинации этих состояний или тех состояний, которые являются промежуточными. Твердая аморфная дисперсия предпочтительно является по существу гомогенной, таким образом, аморфное соединение диспергировано как можно однороднее в полимере. Термин «по существу гомогенный» означает, что доля соединения, которое присутствует в относительно чистых аморфных доменах или областях в твердой аморфной дисперсии, относительно мала, порядка менее 20% масс. и предпочтительно менее 10% масс. от общего количества лекарственного средства.
Водорастворимые полимеры, подходящие для использования в твердых аморфных дисперсиях, должны быть инертными в том смысле, что они не вступают в неблагоприятные химические реакции с плохо растворимым соединением, являются фармацевтически приемлемыми и обладают по меньшей мере некоторой растворимостью в водном растворе при физиологически релевантных значениях рН (например, 1-8). Полимер может быть нейтральным или ионизируемым и должен иметь растворимость в воде по меньшей мере 0,1 мг/мл по меньшей мере в части диапазона рН 1-8.
Водорастворимые полимеры, подходящие для использования в настоящем изобретении, могут быть целлюлозными или нецеллюлозными. Полимеры могут быть нейтральными или ионизируемыми в водном растворе. Из них предпочтительными являются ионизируемые и целлюлозные полимеры, при этом более предпочтительными являются ионизируемые целлюлозные полимеры.
Примеры водорастворимых полимеров включают сукцинат ацетата гидроксипропилметилцеллюлозы (HPMCAS), гидроксипропилметилцеллюлозу (НРМС), фталат гидроксипропилметилцеллюлозы (НРМСР), карбоксиметилэтилцеллюлозу (СМЕС), фталат ацетата целлюлозы (CAP), тримеллитат ацетата целлюлозы (CAT), поливинилпирролидон (PVP), гидроксипропилцеллюлозу (НРС), метилцеллюлозу (МС), блок-сополимеры этиленоксида и пропиленоксида (РЕО/РРО, также известные как полоксамеры) и их смеси. Особенно предпочтительные полимеры включают HPMCAS, НРМС, НРМСР, СМЕС, CAP, CAT, PVP, полоксамеры и их смеси. Наиболее предпочтительным является HPMCAS. См. публикацию заявки на европейский патент №0901786 А2, описание которой включено в настоящую заявку посредством ссылки.
Твердые аморфные дисперсии могут быть получены любым способом, который предоставляет получение твердых аморфных дисперсий, в которых по меньшей мере большая часть (по меньшей мере 60%) слабо растворимого соединения находится в аморфном состоянии. Такие способы включают механические, термические способы и способы с использованием растворителей. Примеры механических способов включают измельчение и экструзию; способы плавления, включая высокотемпературное плавление, модифицированное растворителем плавление и методы отверждения расплава; и способы с использованием растворителей, включая осаждение без растворителя, нанесение покрытия распылением и сушку распылением. См., например, следующие патенты США, соответствующие раскрытия которых включены в настоящую заявку посредством ссылки: №№5456923 и 5939099, которые описывают получение дисперсий методами экструзии; 5340591 и 4673564, которые описывают получение дисперсий способами измельчения; и №№5707646 и 4894235, которые описывают образование дисперсий способами отверждения расплава. В предпочтительном способе твердую аморфную дисперсию получают распылительной сушкой, как описано в публикации заявки на европейский патент №0901786 А2. В этом способе соединение и полимер растворяют в растворителе, таком как ацетон или метанол, после чего растворитель быстро удаляют из раствора методом распылительной сушки с образованием твердой аморфной дисперсии. Твердые аморфные дисперсии могут быть получены таким образом, чтобы они содержали до около 99% масс. соединения, например, 1% масс., 5% масс., 10% масс., 25% масс., 50% масс., 75% масс., 95% масс. или 98% масс., по желанию.
Твердую дисперсию можно использовать как таковую в качестве лекарственной формы, или она может служить продуктом, используемым в производстве (MUP) для получения других лекарственных форм, таких как капсулы, таблетки, растворы или суспензии. Примером водной суспензии является водная суспензия высушенной распылением дисперсии 1:1 (масс./масс.) соединение/HPMCAS-HF, содержащей 2,5 мг/мл соединения в 2% полисорбата-80. Твердые дисперсии для использования в таблетках или капсулах обычно смешивают с другими эксципиентами или адъювантами, обычно присутствующими в таких лекарственных формах. Например, иллюстративный наполнитель для капсул содержит 2:1 (масс./масс.) соединение/HPMCAS-MF высушенную распылением дисперсию (60%), лактозу (быстротекущая) (15%), микрокристаллическую целлюлозу (например, Avicel.sup. (RO-102) (15,8%), натрий крахмал (7%), лаурилсульфат натрия (2%) и стеарат магния (1%).
Полимеры HPMCAS доступны с низкой, средней и высокой степенью чистоты как Aqoa(R)-LF, Aqoat(R)-MF и Aqoat(R)-HF, соответственно, от Shin-Etsu Chemical Co., LTD, Tokyo, Japan. Как правило, предпочтительны более высокие сорта MF и HF.
Для удобства соединение по настоящему изобретению (или комбинация) может доставляться с питьевой водой, так что терапевтическая доза соединения попадает внутрь с ежедневным водообеспечением. Соединение можно добавлять непосредственно в питьевую воду, предпочтительно в виде жидкого водорастворимого концентрата (такого как водный раствор водорастворимой соли).
Эти соединения можно также вводить животным, отличным от человека, например, по показаниям, подробно описанным выше. Точная вводимая доза каждого активного ингредиента будет варьироваться в зависимости от ряда факторов, включая, но не ограничиваясь этим, тип животного и тип болезненного состояния, возраст животного и способ(способы) введения.
Лекарственные средства, используемые в комбинации с соединениями формулы I или их солями, используют в дозе, которая эффективна при показаниях, подлежащих лечению. Такие дозы могут быть определены стандартными анализами, такими как указанные выше и представленные в настоящей заявке. Комбинированные средства можно вводить одновременно или последовательно в любом порядке.
Эти дозировки расчитаны на среднего субъекта-человека, имеющего массу тела примерно от 60 до 70 кг. Лечащий врач без труда сможет определить дозы для субъектов, масса тела которых выходит за пределы этого диапазона, таких как младенцы и пожилые люди.
Схемы введения можно скорректировать для обеспечения оптимального желаемого ответа. Например, можно вводить один болюс, можно вводить несколько дробных доз в течение определенного времени, или доза может быть пропорционально уменьшена или увеличена в соответствии с терапевтической ситуацией. Особенно выгодно формулирование парентеральных композиций в виде стандартной лекарственной формы для простоты введения и однородности дозировки. Стандартная лекарственная форма, в контексте настоящей заявки, относится к физически дискретным единицам, подходящим в качестве единичных доз для млекопитающих, подлежащих лечению; каждая единица содержит заранее определенное количество активного соединения, рассчитанное для получения желаемого терапевтического эффекта, в сочетании с требуемым фармацевтическим носителем. Спецификация для стандартных лекарственных форм по изобретению диктуется и напрямую зависит от (а) уникальных характеристик химиотерапевтического средства и конкретного терапевтического или профилактического эффекта, который должен быть достигнут, и (b) ограничений, существующих в области составления композиций такого активного соединения, для лечения чувствительности у индивидуумов.
Таким образом, специалистам в данной области техники будет понятно, основываясь на представленном раскрытии, что доза и схема введения корректируются в соответствии со способами, хорошо известными в терапии. То есть, максимально переносимая доза может быть легко установлена, а также может быть определено эффективное количество, обеспечивающее определяемый терапевтический эффект у пациента, а также временные требования для введения каждого средства для обеспечения определяемого терапевтического эффекта у пациента. Соответственно, хотя в настоящей заявке приведены примеры определенных доз и схем введения, эти примеры никоим образом не ограничивают дозы и схемы введения, которые могут предоставляться пациенту при осуществлении настоящего изобретения.
Следует отметить, что дозы могут варьироваться в зависимости от типа и тяжести состояния, подлежащего облегчению, и могут включать разовые или многократные дозы. Кроме того, должно быть понятно, что для любого конкретного субъекта конкретные схемы введения должны корректироваться с течением времени в соответствии с индивидуальными потребностями и профессиональным мнением лица, осуществляющего введение или контролирующего введение композиций, и что диапазоны доз, указанные в настоящей заявке, являются только иллюстративными и не предназначены для ограничения объема или применения заявленной композиции. Например, дозы можно корректировать на основе фармакокинетических или фармакодинамических параметров, которые могут включать клинические эффекты, такие как токсические эффекты и/или лабораторные показатели. Таким образом, настоящее изобретение охватывает повышение дозы для индивидуального пациента, определяемое специалистом в данной области. Определение подходящих доз и схем введения химиотерапевтического средства хорошо известно в соответствующей области техники, и специалистам в данной области будет понятно, что они охвачены, когда они ознакомятся с раскрытием, представленным в настоящей заявке.
Настоящее изобретение также включает применение соединения формулы I или его фармацевтически приемлемой соли в качестве лекарственного средства (такого как таблетка с стандартной дозировкой или капсула с стандартной дозировкой). В другом варианте осуществления настоящее изобретение включает применение соединения формулы I или его фармацевтически приемлемой соли для получения лекарственного средства (такого как таблетка с стандартной дозировкой или капсула с стандартной дозировкой) для лечения одного или нескольких состояний, определенных в разделах, описанных выше, обсуждающих способы лечения.
Фармацевтическая композиция по настоящему изобретению может быть получена, упакована или может продаваться в нерасфасованном виде, в виде разовой стандартной дозы или в виде множества разовых стандартных доз. В контексте настоящей заявки термин «разовая доза» представляет собой дискретное количество фармацевтической композиции, содержащее заданное количество активного ингредиента. Количество активного ингредиента обычно равно дозе активного ингредиента, которая должна вводиться субъекту, или подходящей части такой дозы, такой как, например, половина или одна треть такой дозы.
Эти средства и соединения по изобретению можно комбинировать с фармацевтически приемлемыми носителями, такими как физиологический раствор, раствор Рингера, раствор декстрозы и т.п. Конкретная схема введения, т.е. доза, интервалы между введением доз и повторение, будет зависеть от конкретного субъекта и истории болезни этого субъекта.
Приемлемые носители, эксципиенты или стабилизаторы нетоксичны для реципиентов в используемых дозировках и концентрациях и могут включать буферы, такие как фосфат, цитрат и другие органические кислоты; соли, такие как хлорид натрия; антиоксиданты, включая аскорбиновую кислоту и метионин; консерванты (такие как октадецилдиметилбензиламмоний хлорид, гексаметоний хлорид, бензалконий хлорид, бензетоний хлорид, фенол, бутиловый или бензиловый спирт, алкилпарабены, такие как метил- или пропилпарабен, катехол, резорцин, циклогексанол, 3-пентанол и м-крезол); низкомолекулярные (менее примерно 10 остатков) полипептиды; белки, такие как сывороточный альбумин, желатин или иммуноглобулины; гидрофильные полимеры, такие как поливинилпирролидон; аминокислоты, такие как глицин, глутамин, аспарагин, гистидин, аргинин или лизин; моносахариды, дисахариды и другие углеводы, включая глюкозу, маннозу или декстрины; хелатирующие агенты, такие как EDTA; сахара, такие как сахароза, маннит, трегалоза или сорбит; солеобразующие противоионы, такие как натрий; комплексы с металлами (например, комплексы Zn-белок); и/или неионогенные поверхностно-активные вещества, такие как TWEENT™, PLURONICS™ или полиэтиленгликоль (PEG).
Липосомы, содержащие эти средства и/или соединения по изобретению, получают способами, известными в данной области, такими как описанные в патентах США №№4485045 и 4544545. Липосомы с увеличенным временем циркуляции раскрыты в патенте США №5013556. Особенно полезные липосомы могут быть получены методом обращенно-фазового испарения с липидной композицией, включающей фосфатидилхолин, холестерин и PEG-дериватизированный фосфатидилэтаноламин (PEG-PE). Липосомы экструдируют через фильтры с определенным размером пор, чтобы получить липосомы желаемого диаметра.
Эти средства и/или соединения по изобретению также могут быть заключены в микрокапсулы, полученные, например, методами коацервации или межфазной полимеризации, например, гидроксиметилцеллюлозные или желатиновые микрокапсулы и поли(метилметакрилатные) микрокапсулы, соответственно, в коллоидных системах доставки лекарственных средств (например, липосомы, альбуминовые микросферы, микроэмульсии, наночастицы и нанокапсулы) или в макроэмульсиях. Такие методы раскрыты в Remington, The Science and Practice of Pharmacy, 20th Ed., Mack Publishing (2000).
Можно использовать препараты пролонгированного действия. Подходящие примеры препаратов с замедленным высвобождением включают полупроницаемые матрицы из твердых гидрофобных полимеров, содержащие соединение по изобретению, при этом матрицы имеют форму формованных изделий, например, пленок или микрокапсул. Примеры матриц с пролонгированным высвобождением включают сложные полиэфиры, гидрогели [например, поли(2-гидроксиэтилметакрилат) или поли(виниловый спирт)], полилактиды (патент США №3773919), сополимеры L-глутаминовой кислоты и этил-L-глутамата, неразлагаемый этилен-винилацетат, разлагаемые сополимеры молочной и гликолевой кислот, такие как используемые в LUPRON DEPOT™ (инъекционные микросферы, состоящие из сополимера молочной и гликолевой кислот и лейпролид ацетата), изобутират ацетата сахарозы и поли-D-(-)-3-гидроксимасляную кислоту.
Композиции, используемые для внутривенного введения, должны быть стерильными. Это легко осуществить, например, путем фильтрации через стерильные фильтрационные мембраны. Соединения по изобретению обычно помещают в контейнер со стерильным входным отверстием, например, пакет для внутривенного раствора или флакон с пробкой, прокалываемой иглой для подкожных инъекций.
Подходящие эмульсии можно получить с использованием коммерчески доступных жировых эмульсий, таких как Intralipid™, Liposyn™, Infonutrol™, Lipofundin™ и Lipiphysan™. Активный ингредиент можно растворить в предварительно смешанной эмульсионной композиции, либо, альтернативно, его можно растворить в масле (например, соевом масле, сафлоровом масле, масле скмян хлопчатника, кунжутном масле, кукурузном масле или миндальном масле), и эмульсию получают при смешивании с фосфолипидом (например, яичными фосфолипидами, соевыми фосфолипидами или соевым лецитином) и водой. Следует понимать, что могут быть добавлены другие ингредиенты, например, глицерин или глюкоза, для регулирования тоничности эмульсии. Подходящие эмульсии обычно содержат до 20% масла, например от 5 до 20%. Жировая эмульсия может включать капли жира размером от 0,1 до 1,0 мкм, в частности от 0,1 до 0,5 мкм, и иметь рН в диапазоне от 5,5 до 8,0.
Эмульсионные композиции могут быть получены путем смешивания соединения по изобретению с Intralipid™ или его компонентами (соевым маслом, яичными фосфолипидами, глицерином и водой).
Композиции для ингаляции или инсуффляции включают растворы и суспензии в фармацевтически приемлемых водных или органических растворителях или их смесях, а также порошки. Жидкие или твердые композиции могут содержать подходящие фармацевтически приемлемые эксципиенты, как указано выше. В некоторых вариантах осуществления композиции вводят пероральным или назальным путем для местного или системного действия. Композиции в предпочтительно стерильных фармацевтически приемлемых растворителях можно распылять с использованием газов. Распыляемые растворы можно вдыхать непосредственно из распылителя, или распылитель можно прикрепить к лицевой маске, палатке или дыхательному аппарату прерывистого действия с положительным давлением. Растворы, суспензии или порошковые композиции можно вводить, предпочтительно перорально или назально, из устройств, которые доставляют композицию соответствующим образом.
Соединения по настоящему изобретению могут быть сформулированы для перорального, трансбуккального, интраназального, парентерального (например, внутривенного, внутримышечного или подкожного) или ректального введения или в форме, подходящей для введения путем ингаляции. Соединения по изобретению также могут быть сформулированы для пролонгированной доставки.
Способы получения различных фармацевтических композиций с определенным количеством активного ингредиента известны или станут очевидными для специалистов в данной области в свете настоящего раскрытия. Примеры способов получения фармацевтических композиций см. в Remington, The Science and Practice of Pharmacy, 20th Edition (Lippincott Williams & Wilkins, 2000).
Фармацевтические композиции по изобретению могут содержать от 0,1% до 95% соединения(соединений) по изобретению, предпочтительно от 1% до 70%. В любом случае вводимая композиция будет содержать соединение(соединения) по изобретению в количестве, эффективном для лечения заболевания/состояния субъекта, которого лечат.
Поскольку настоящее изобретение имеет аспект, который относится к лечению заболеваний/состояний, описанных в настоящей заявке, комбинацией активных ингредиентов, которые можно вводить по отдельности, изобретение также относится к объединению отдельных фармацевтических композиций в форме набора. Набор включает две отдельные фармацевтические композиции: соединение формулы I, или его фармацевтически приемлемую соль, или его пролекарство, или соль такого соединения или пролекарства, и второе соединение, как описано выше. Набор включает средства для содержания отдельных композиций, такие как контейнер, разделенная бутыль или разделенный пакет из фольги. Обычно набор включает инструкции по применению отдельных компонентов. Форма набора является особенно предпочтительной, когда отдельные компоненты предпочтительно вводят в разных лекарственных формах (например, перорально и парентерально), вводят с разными интервалами между введениями или когда по усмотрению лечащего врача желательно титрование отдельных компонентов комбинации.
Примером такого набора является так называемая блистерная упаковка. Блистерные упаковки хорошо известны в упаковочной промышленности и широко используются для упаковки стандартных лекарственных форм (таблетки, капсулы и т.п.). Блистерная упаковка обычно состоит из листа относительно жесткого материала, покрытого фольгой, предпочтительно из прозрачного пластика. В процессе упаковки в пластиковой пленке образуются углубления. Углубления имеют размер и форму упаковываемых таблеток или капсул. Затем таблетки или капсулы помещают в углубления и лист относительно жесткого материала запечатывают с пластиковой фольгой на лицевой стороне фольги, противоположной направлению, в котором были сформированы углубления. В результате таблетки или капсулы запаиваются в углублениях между пластиковой пленкой и листом. Предпочтительно прочность листа такова, что таблетки или капсулы могут быть извлечены из блистерной упаковки путем нажатия рукой на углубления, в результате чего в листе образуется отверстие в месте углубления. Затем таблетку или капсулу можно извлечь через указанное отверстие.
Может быть желательным предусмотреть в наборе памятку, например, в виде чисел рядом с таблетками или капсулами, при этом числа соответствуют определенным дням схемы введения, когда указанные таким образом таблетки или капсулы следует принимать. Другим примером такой памятки является календарь, напечатанный на карточке, например, следующим образом: «Первая неделя, понедельник, вторник и т.д… Вторая неделя, понедельник, вторник… и т.д.» Другие варианты памяток будут весьма очевидны. «Суточная доза» может представлять собой одну таблетку или капсулу, или несколько таблеток или капсул, принимаемых в определенный день. Кроме того, суточная доза соединения по настоящему изобретению может состоять из одной таблетки или капсулы, тогда как суточная доза второго соединения может состоять из нескольких таблеток или капсул, и наоборот. Памятка должна отражать это.
В другом конкретном варианте осуществления изобретения предусмотрен дозатор, предназначенный для отмеривания суточных доз по одной за раз в порядке их предполагаемого использования. Предпочтительно дозатор снабжен запоминающим устройством, чтобы еще больше облегчить соблюдение режима. Примером такого запоминающего устройства является механический счетчик, который показывает количество отмеренных суточных доз. Другим примером такого средства запоминания является микрочип-память с питанием от батареи в сочетании с жидкокристаллическим считывающим устройством или звуковым сигналом напоминания, который, например, считывает дату приема последней суточной дозы и/или напоминает субъекту, когда следует принять следующую дозу.
Кроме того, поскольку настоящее изобретение имеет аспект, который относится к лечению описанных в настоящей заявке заболеваний/состояний комбинацией активных ингредиентов, которые можно вводить совместно, изобретение также относится к объединению отдельных фармацевтических композиций в одной лекарственной форме, такой как (но не ограничиваясь этим) одна таблетка или капсула, двухслойная или многослойная таблетка или капсула, или за счет использования сегрегированных компонентов или перегородок в таблетке или капсуле.
Активный ингредиент может доставляться в виде раствора в водном или неводном носителе, с использованием или без дополнительных растворителей, сорастворителей, эксципиентов или комплексообразователей, выбранных из фармацевтически приемлемых разбавителей, эксципиентов, растворителей или носителей.
Активный ингредиент может быть сформулирован в виде твердой дисперсии или в виде самоэмульгирующейся системы доставки лекарственного средства (SEDDS) с фармацевтически приемлемыми эксципиентами.
Активный ингредиент может быть сформулирован в виде таблетки или капсулы с немедленным или контролируемым (например, отсроченным, замедленным или пролонгированным) высвобождением. Альтернативно, активный ингредиент может доставляться в виде только активного ингредиента в оболочке капсулы без дополнительных эксципиентов.
ПРИМЕРЫ
Ниже проиллюстрирован синтез различных соединений по настоящему изобретению. Дополнительные соединения, охватываемые объемом настоящего изобретения, могут быть получены с использованием способов, проиллюстрированных в этих Примерах, либо отдельно, либо в сочетании с методами, общеизвестными в данной области. Все исходные вещества в этих Получениях и Примерах либо коммерчески доступны, либо могут быть получены способами, известными в данной области, или описанными в настоящей заявке.
Реакции осуществляли на воздухе или, при использовании чувствительных к кислороду или влаге реагентов или промежуточных продуктов, в инертной атмосфере (азот или аргон). При необходимости реакционные аппараты сушили в динамическом вакууме с использованием тепловой пушки и использовали безводные растворители (продукты Sure-Seal™ от Aldrich Chemical Company, Milwaukee, Wisconsin, или продукты DriSolv™ от EMD Chemicals, Gibbstown, NJ). В некоторых случаях коммерческие растворители пропускали через колонки, заполненные молекулярными ситами 4Å, до тех пор, пока не были достигнуты следующие стандарты контроля качества для воды: а) <100 ч/млн для дихлорметана, толуола, N,N-диметилформамида и тетрагидрофурана; b) <180 ч/млн для метанола, этанола, 1,4-диоксана и диизопропиламина. Для очень чувствительных реакций растворители дополнительно обрабатывали металлическим натрием, гидридом кальция или молекулярными ситами и подвергали перегонке непосредственно перед использованием. Другие коммерческие растворители и реагенты использовали без дополнительной очистки. Для способов синтеза со ссылкой на процедуры в других Примерах или Методах, условия реакции (время реакции и температура) могут варьироваться. Продукты, как правило, сушили в вакууме перед их использованием в дальнейших реакциях или биологических испытаниях.
Когда указано, реакционные смеси нагревали в условиях микроволнового облучения с использованием микроволнового устройства Biotage Initiator или Personal Chemistry Emrys Optimizer. Ход реакции контролировали методами тонкослойной хроматографии (ТСХ), жидкостной хроматографии/масс-спектрометрии (ЖХ-МС), высокоэффективной жидкостной хроматографии (ВЭЖХ) и/или газовой хроматографии/масс-спектрометрии (ГХ-МС). ТСХ осуществляли на пластинах с силикагелем, предварительно покрытых флуоресцентным индикатором (длина волны возбуждения 254 нм), и визуализировали в УФ-свете и/или при помощи окрашивания йодом, перманганатом калия, хлоридом кобальта (II), фосфомолибденовой кислотой и/или молибдатом церия-аммония. Данные ЖХ-МС получали на устройстве Agilent серии 1100 с автоматическим пробоотборником Leap Technologies, колонками Gemini C18, градиентами ацетонитрил/вода и модификаторами, такими как трифторуксусная кислота, муравьиная кислота или гидроксид аммония. Элюент из колонки анализировали с использованием масс-спектрометра Waters ZQ, сканирующего в режиме как положительной, так и отрицательной ионизации в диапазоне от 100 до 1200 Да. Также использовали другие подобные устройства. Данные ВЭЖХ обычно получали на устройстве Agilent серии 1100 с использованием указанных колонок, градиентов ацетонитрил/вода и трифторуксусной кислоты или гидроксида аммония в качестве модификаторов. Данные ГХМС получали с использованием печи Hewlett Packard 6890 с инжектором HP 6890, колонкой HP-1 (12 м × 0,2 мм × 0,33 мкм) и газом-носителем гелием. Образец анализировали на масс-селективном детекторе HP 5973, сканирующем от 50 до 550 Да, с использованием электронной ионизации. Очистку осуществляли жидкостной хроматографией среднего давления (ЖХСД) с использованием устройства Isco CombiFlash Companion, AnaLogix IntelliFlash 280, Biotage SP1 или Biotage Isolera One и предварительно упакованных картриджей с силикагелем Isco RediSep или Biotage Snap. Хиральную очистку осуществляли методом хиральной сверхкритической флюидной хроматографии (СФХ), обычно с использованием аппаратов Berger или Thar; колонок, таких как колонки ChiralPAK-AD, -AS, -IC, Chiralcel-OD или -OJ; и смесей CO2 с метанолом, этанолом, 2-пропанолом или ацетонитрилом, используемых как таковые или модифицированных с использованием трифторуксусной кислоты или пропан-2-амина. УФ-детекцию использовали для запуска сбора фракций. Для способов синтеза со ссылкой на процедуры в других Примерах или Методах очистки могут варьироваться: как правило, растворители и соотношения растворителей, используемые для элюентов/градиентов, выбирались для обеспечения соответствующих значений Rf или времени удерживания.
Данные масс-спектрометрии были получены из ЖХМС анализов. Масс-спектрометрию (МС) осуществляли методом химической ионизации при атмосферном давлении (APCI), ионизации электрораспылением (ESI), ионизации электронным ударом (EI) или ионизации рассеянием электронов (ES). Химические сдвиги протонной ядерно-магнитной спектроскопии (1H ЯМР) указаны в миллионных долях в более слабом поле от тетраметилсилана, и они были получены на спектрометрах Varian, Bruker или Jeol с частотой 300, 400, 500 или 600 МГц. Химические сдвиги выражены в миллионных долях (м.д., δ) относительно остаточных пиков дейтерированного растворителя (хлороформ, 7,26 м.д.; CD2HOD, 3,31 м.д.; ацетонитрил-d2, 1,94 м.д.; диметилсульфоксид-d5, 2,50 м.д.; DHO, 4,79 м.д.). Формы пиков описаны следующим образом: с синглет; д - дублет; т - триплет; кв. - квартет; квинт. - квинтет; м - мультиплет; ушир.с - уширенный синглет; каж. - кажущийся. Аналитические данные СФХ обычно получали на аналитическом устройстве Berger, как описано выше. Данные оптического вращения были получены на поляриметре PerkinElmer модели 343 с использованием кюветы 1 дм. Микроанализы были выполнены Quantitative Technologies Inc. и были в пределах 0,4% от расчетных значений.
Если не указано иное, химические реакции осуществляли при комнатной температуре (около 23 градусов Цельсия).
Если не указано иное, все реагенты были получены коммерческим путем и использовались без дополнительной очистки, или были получены с использованием методов, известных из литературы.
Термины «концентрировали», «упаривали» и «концентрировали в вакууме» относятся к удалению растворителя при пониженном давлении на роторном испарителе с температурой бани менее 60°С. Сокращения «мин» и «ч» означают «минуты» и «часы», соответственно. Термин «ТСХ» относится к тонкослойной хроматографии, «комнатная температура или температура окружающей среды» означает температуру от 18 до 25°С, «ГХМС» относится к газовой хроматографии/масс-спектрометрии, «ЖХМС» относится к жидкостной хроматографии/масс-спектрометрии, «СВЭЖХ» относится к сверхэффективной жидкостной хроматографии, а «ВЭЖХ» относится к высокоэффективной жидкостной хроматографии, «СФХ» относится к сверхкритической флюидной хроматографии.
Гидрирование можно осуществить в шейкере Парра под давлением газообразного водорода или в проточном реакторе гидрирования H-Cube от Thales-nano при полном использовании водорода и скорости потока 1-2 мл/мин при заданной температуре.
Время удерживания ВЭЖХ, СВЭЖХ, ЖХМС, ГХМС и СФХ измеряли с использованием методов, указанных в процедурах.
В некоторых примерах хиральное разделение осуществляли для разделения энантиомеров или диастереомеров некоторых соединений по изобретению (в некоторых примерах разделенные энантиомеры обозначаются как ENT-1 и ENT-2 в соответствии с порядком их элюирования; аналогичным образом, разделенные диастереомеры обозначаются как DIAST-1 и DIAST-2 в соответствии с порядком их элюирования). В некоторых примерах оптическое вращение энантиомера измеряли с использованием поляриметра. В соответствии с его наблюдаемыми данными вращения (или его данными удельного вращения), энантиомер с вращением по часовой стрелке был обозначен как (+)-энантиомер, а энантиомер с вращением против часовой стрелки был обозначен как (-)-энантиомер. Рацемические соединения обозначаются либо отсутствием показанной или описанной стереохимии, либо присутствием (+/-) рядом со структурой; в этом последнем случае указанная стереохимия представляет собой только один из двух энантиомеров, составляющих рацемическую смесь.
Соединения и промежуточные соединения, описанные ниже, были названы с использованием соглашения о наименовании, предоставленного ACD/ChemSketch 2017.2.1, File Version C40H41, Build 99535 (Advanced Chemistry Development, Inc., Toronto, Ontario, Canada). Соглашение о наименовании, представленное в ACD/ChemSketch 2017.2.1, хорошо известно специалистам в данной области, и считается, что соглашение о наименовании, предоставленное в ACD/ChemSketch 2017.2.1, в целом соответствует рекомендациям IUPAC (International Union for Pure and Applied Chemistry) по номенклатуре органической химии и правилам индекса CAS.
Спектры 1H ЯМР многих соединений в настоящей заявке указывают на смесь ротамеров из-за присутствия амидной и/или карбаматной функциональной группы, и они были сведены в таблицу, чтобы отразить присутствие более чем одного ротамера.
ПОЛУЧЕНИЯ
Получения Р1 - Р33 описывают получение некоторых исходных веществ или промежуточных соединений, используемых для получения некоторых соединений по изобретению.
Получение Р1
2-(5-Хлор-2-метоксипиридин-4-ил)пропановая кислота (Р1)
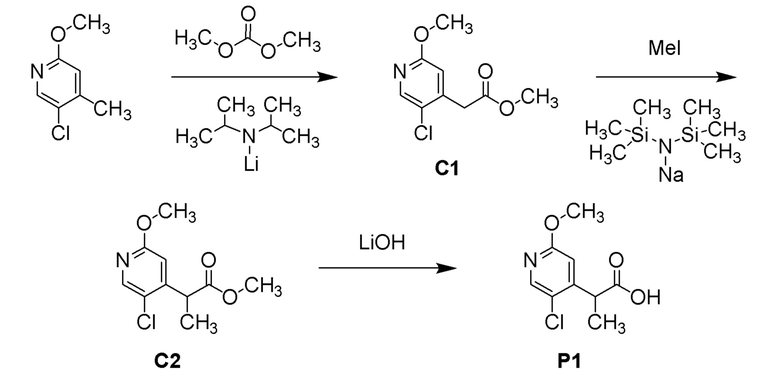
Стадия 1. Синтез метил(5-Хлор-2-метоксипиридин-4-ил)ацетата (С1).
Раствор диизопропиламида лития в тетрагидрофуране (2 М; 1,9 л, 3,8 моль) добавляли по каплям к -30°С раствору 5-хлор-2-метокси-4-метилпиридина (197 г, 1,25 моль) в тетрагидрофуране (1,4 л). После перемешивания реакционной смеси при -30°С в течение 1 часа добавляли по каплям диметилкарбонат (338 г, 3,75 моль); по завершении добавления реакционную смесь нагревали до 25°С и перемешивали в течение 1 часа. Затем смесь выливали в хлористоводородную кислоту (0,5 М, 7 л, 3,5 моль) и экстрагировали этилацетатом (2 × 1,5 л); объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле (градиент: 0% до 20% этилацетата в петролейном эфире) давала С1 в виде желтого масла. Выход: 203 г, 0,941 моль, 75%. ЖХМС m/z 216,1 (наблюдаемый изотопный паттерн хлора) [М+Н]+. 1H ЯМР (400 МГц, метанол-d4) δ 8,10 (с, 1Н), 6,82 (с, 1Н), 3,90 (с, 3Н), 3,79 (с, 2Н), 3,71 (с, 3Н).
Стадия 2. Синтез метил-2-(5-хлор-2-метоксипиридин-4-ил)пропаноата (С2).
К -78°С раствору С1 (175 г, 0,812 моль) в тетрагидрофуране (1,2 л) добавляли раствор бис(триметилсилил)амида натрия в тетрагидрофуране (2 М; 455 мл, 0,910 моль) по каплям. Реакционную смесь перемешивали при -78°С в течение 1 часа, затем добавляли по каплям раствор иодметана (172,6 г, 1,216 моль) в тетрагидрофуране (100 мл) при -78°С и перемешивание продолжали при этой температуре в течение 2 часов. Реакционную смесь затем выливали в насыщенный водный раствор хлорида аммония (500 мл) и экстрагировали этилацетатом (2 × 100 мл); объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением С2 в виде коричневого масла. По данным ЯМР и ЖХМС анализа это вещество было загрязнено некоторым количеством диметилированного побочного продукта метил-2-(5-хлор-2-метоксипиридин-4-ил)-2-метилпропаноата. Выход: 136 г, ≤0,592 моль, ≤73%. ЖХМС m/z 230,1 (наблюдаемый изотопный паттерн хлора) [М+Н]+. 1H ЯМР (400 МГц, метанол-d4), только пик продукта: δ 8,10 (с, 1Н), 6,76 (с, 1Н), 4,10 (кв., J=7,2 Гц, 1Н), 3,89 (с, 3Н), 3,69 (с, 3Н), 1,48 (д, J=7,2 Гц, 3Н).
Стадия 3. Синтез 2-(5-хлор-2-метоксипиридин-4-ил)пропановой кислоты (Р1).
К 25°С раствору С2 (168 г, 0,732 моль) в тетрагидрофуране (1 л) добавляли по каплям раствор гидроксида лития моногидрата (61,4 г, 0,146 моль) в воде (300 мл) при 25°С. Смесь перемешивали в течение 2 часов, затем концентрировали в вакууме. Водный остаток выливали в воду (500 мл) и промывали трет-бутилметиловым эфиром (2 × 500 мл). Водный слой затем доводили до рН 4 добавлением 3 М хлористоводородной кислоты и экстрагировали этилацетатом (2 × 500 мл); объединенные этилацетатные слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением Р1 в виде белого твердого вещества. Выход: 122 г, 0,566 моль, 77%. ЖХМС m/z 216,1 (наблюдаемый изотопный паттерн хлора) [М+Н]+. 1H ЯМР (400 МГц, метанол-d4) δ 8,10 (с, 1Н), 6,79 (с, 1Н), 4,06 (кв., J=7,1 Гц, 1Н), 3,89 (с, 3Н), 1,48 (д, J=7,2 Гц, 3Н).
Получения Р2 и Р3
(2R)-2-(5-Хлор-2-метоксипиридин-4-ил)пропановая кислота (Р2) и (2S)-2-(5-Хлор-2-метоксипиридин-4-ил)пропановая кислота (Р3)
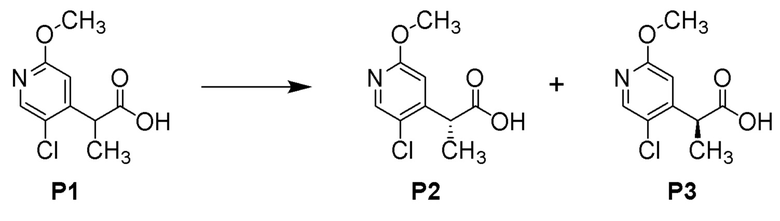
Разделение Р1 (5,00 г, 23,2 ммоль) на составляющие его энантиомеры осуществляли сверхкритической флюидной хроматографией (Колонка: Chiral Technologies Chiralpak IG, 30 × 250 мм, 5 мкм; Подвижная фаза: 95:5 диоксид углерода/метанол; Скорость потока: 80 мл/мин; Обратное давление: 120 бар). Первый элюирующий энантиомер, масло, которое отверждалось при выстаивании, был указан как Р2, а второй элюирующий энантиомер как Р3.
Указанная абсолютная стереохимия была присвоена путем определения при помощи рентгеновских лучей кристаллической структуры соединения 15, которое было синтезировано с использованием этой партии Р2 (см. ниже. Пример 15, Альтернативная Стадия 3).
Р2 - Выход: 2,4 г, 11,1 ммоль, 48%. 1H ЯМР (400 МГц, хлороформ-d) δ 8,13 (с, 1Н), 6,75 (с, 1Н), 4,12 (кв., J=7,2 Гц, 1Н), 3,91 (с, 3Н), 1,53 (д, J=7,2 Гц, 3Н). Время удерживания: 3,98 минут (Аналитические условия. Колонка: Chiral Technologies Chiralpak IG, 4,6 × 250 мм, 5 мкм; Подвижная фаза А: диоксид углерода; Подвижная фаза В: метанол; Градиент: 5% В в течение 1,00 минуты, затем 5% до 60% В в течение 8 минут; Скорость потока: 3,0 мл/мин; Обратное давление: 120 бар).
Р3 - Выход: 2,4 г, 11,1 ммоль, 48%. Время удерживания: 4,22 минут (Аналитические условия идентичны используемым для Р2).
Получение Р4
2-(6-Метокси-2-метилпиримидин-4-ил)пропаноат лития (Р4)
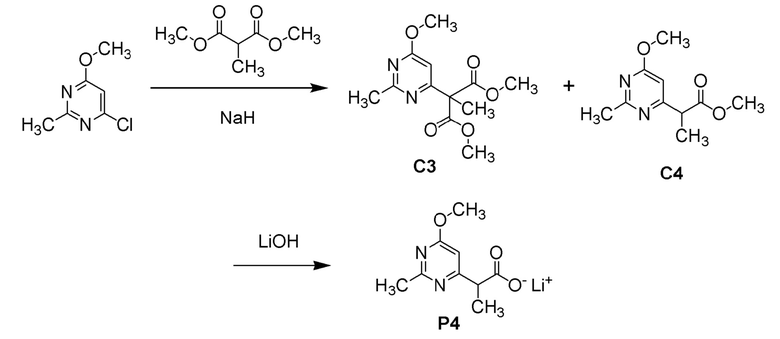
Стадия 1. Синтез диметил(6-метокси-2-метилпиримидин-4-ил) (метил)пропандиоата (С3) и метил-2-(6-метокси-2-метилпиримидин-4-ил)пропаноата (С4).
Гидрид натрия (60% в минеральном масле; 1,14 г, 28,5 ммоль) добавляли к раствору диметилметилпропандиоата (5,53 г, 37,8 ммоль) в N,N-диметилформамиде (25 мл). Через 30 минут добавляли 4-хлор-6-метокси-2-метилпиримидин (3,00 г, 18,9 ммоль), затем реакционную смесь нагревали при 100°С в течение 16 часов. Затем смесь разбавляли водой (150 мл) и экстрагировали этилацетатом (3 × 50 мл); объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали, концентрировали в вакууме и очищали хроматографией на силикагеле (градиент: 0% до 10% этилацетата в петролейном эфире) с получением продукта (2,60 г) в виде желтого масла. На основании ЯМР и ЖХМС анализа считалось, что это содержащая примеси смесь С3 и С4, которую непосредственно использовали на следующей стадии. ЖХМС m/z 211,1 и 269,2 [М+Н]+. 1H ЯМР (400 МГц, метанол-d4), характеристические пики: δ 6,68 (с), 6,60 (с), 3,96 (с), 3,94 (с), 3,81 (кв., J=7,2 Гц), 3,75 (с), 3,68 (с), 2,54 (с), 2,52 (с), 1,79 (с), 1,47 (д, J=7,3 Гц).
Стадия 2. Синтез 2-(6-метокси-2-метилпиримидин-4-ил)пропаноата лития (Р4).
Раствор С3 и С4 (с предыдущей стадии; 2,60 г, ≤18,9 ммоль) и гидроксид лития моногидрат (1,22 г, 29,1 ммоль) в смеси тетрагидрофурана (45 мл) и воды (15 мл) перемешивали при 45°С в течение 3 часов. Реакционную смесь концентрировали в вакууме, затем остаток подвергали лиофилизации с получением Р4 в виде белого твердого вещества. Выход: 2,3 г, 11 ммоль, 58% за 2 стадии. ЖХМС m/z 197,1 [М+Н]+. 1H ЯМР (400 МГц, метанол-d4) δ 6,66 (с, 1Н), 3,94 (с, 3Н), 3,61 (кв., J=7,3 Гц, 1Н), 2,53 (с, 3Н), 1,44 (д, J=7,2 Гц, 3Н).
Получение Р5
2-[6-(Дифторметокси)пиридин-3-ил]пропановая кислота (Р5)
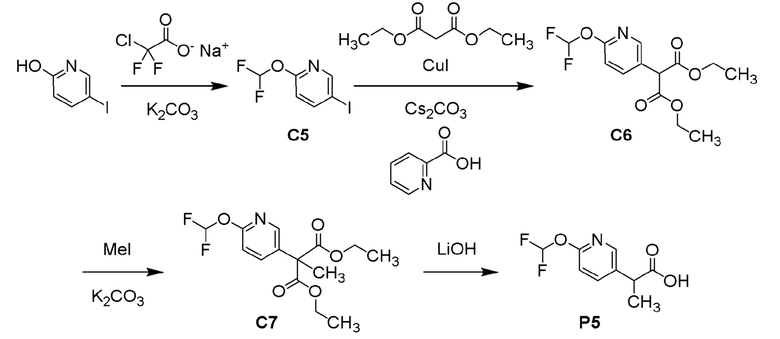
Стадия 1. Синтез 2-(дифторметокси)-5-иодпиридина (С5).
Хлор(дифтор)ацетат натрия (4,62 г, 30,3 ммоль) и карбонат калия (5,58 г, 40,4 ммоль) добавляли к 25°С раствору 5-иодпиридин-2-ола (4,46 г, 20,2 ммоль) в N,N-диметилформамиде (100 мл) и реакционную смесь перемешивали при 50°С в течение 16 часов. Затем смесь разбавляли водой (500 мл) и экстрагировали этилацетатом (3 × 100 мл) и объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (градиент: 0% до 8% этилацетата в петролейном эфире) давала С5 в виде масла. Выход: 2,10 г, 7,75 ммоль, 38%. 1H ЯМР (400 МГц, хлороформ-d) δ 8,39 (ушир.д, J=2,2 Гц, 1Н), 7,97 (дд, J=8,6, 2,3 Гц, 1Н), 7,40 (т, JHF=72,6 Гц, 1Н), 6,74 (ушир.д, J=8,6 Гц, 1Н).
Стадия 2. Синтез диэтил[6-(дифторметокси)пиридин-3-ил]пропандиоата (С6).
Смесь С5 (1,9 г, 7,0 ммоль), диэтилпропандиоата (1,68 г, 10,5 ммоль), иодида меди(I) (133 мг, 0,698 ммоль), пиридин-2-карбоновой кислоты (172 мг, 1,40 ммоль) и карбоната цезия (7,42 г, 22,8 ммоль) в тетрагидрофуране (50 мл) перемешивали при 80°С в течение 16 часов, затем реакционную смесь разбавляли этилацетатом (100 мл) и промывали водным раствором хлорида аммония (100 мл). Водный слой экстрагировали этилацетатом (2 × 50 мл) и объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле (градиент: 0% до 15% этилацетата в петролейном эфире) давала С6 в виде бесцветного масла (2,4 г). По данным 1H ЯМР анализа это вещество содержало остаточный диэтилпропандиоат; часть этого образца использовали непосредственно на следующей стадии. ЖХМС m/z 304,0 [М+Н]+. lH ЯМР (400 МГц, хлороформ-d), только пики продукта: δ 8,14 (ушир.с, 1Н), 7,90 (ушир.д, J=8,4 Гц, 1Н), 7,45 (т, JHF=72,9 Гц, 1Н), 6,92 (д, J=8,4 Гц, 1Н), 4,59 (с, 1Н), 4,26-4,17 (м, 4Н, предположительно; частично скрыт остаточным диэтилпропандиоатом), 1,31-1,24 (м, 6Н, предположительно; частично скрыт остаточным диэтилпропандиоатом).
Стадия 3. Синтез диэтил[6-(дифторметокси)пиридин-3-ил](метил)пропандиоата (С7).
К раствору С6 (с предыдущей стадии; 750 мг, ≤2,2 ммоль) в N,N-диметилформамиде (15 мл) добавляли карбонат калия (1,03 г, 7,45 ммоль). Добавляли по каплям иодметан (527 мг, 3,71 ммоль) и реакционную смесь перемешивали при 25°С в течение 4 часов. Затем смесь объединяли с подобной реакцией, осуществляемой с использованием С6 (250 мг, ≤0,73 ммоль), выливали в воду (200 мл) и экстрагировали этилацетатом (2 × 50 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия и концентрировали в вакууме; хроматография на силикагеле (градиент: 0% до 15% этилацетата в петролейном эфире) давала С7 в виде масла. Объединенный выход: 738 мг, 2,33 ммоль, 80% за 2 стадии. ЖХМС m/z 318,2 [М+Н]+. 1H ЯМР (400 МГц, хлороформ-d) δ 8,20 (ушир.с, 1Н), 7,81 (ушир.д, J=8,7 Гц, 1Н), 7,45 (т, JHF=72,9 Гц, 1Н), 6,90 (д, J=8,7 Гц, 1Н), 4,30-4,18 (м, 4Н), 1,87 (с, 3Н), 1,27 (т, J=7,1 Гц, 6Н).
Стадия 4. Синтез 2-[6-(дифторметокси)пиридин-3-ил]пропановая кислота (Р5).
К 25°С раствору С7 (738 мг, 2,33 ммоль) в тетрагидрофуране (10 мл) добавляли раствор гидроксида лития (279 мг, 11,6 ммоль) в воде (3 мл). Реакционную смесь перемешивали при 25°С в течение 16 часов, затем разбавляли водой (100 мл) и промывали дихлорметаном (3 × 50 мл). Эти органические слои сливали. Водный слой доводили до рН 5 добавлением 5 М хлористоводородной кислоты, затем экстрагировали дихлорметаном (3 × 50 мл); объединенные органические слои концентрировали в вакууме с получением Р5 в виде твердого вещества. Выход: 337 мг, 1,55 ммоль, 67%. ЖХМС m/z 218,1 [М+Н]+. 1H ЯМР (400 МГц, метанол-d4) δ 8,15 (д, J=2,5 Гц, 1Н), 7,83 (дд, J=8,5, 2,5 Гц, 1Н), 7,51 (т, JHF=73,2 Гц, 1Н), 6,94 (д, J=8,5 Гц, 1Н), 3,78 (кв., J=7,2 Гц, 1Н), 1,49 (д, J=7,2 Гц, 3Н).
Получение Р6
2-(5-Фтор-2-метоксипиридин-4-ил)пропановая кислота (Р6)
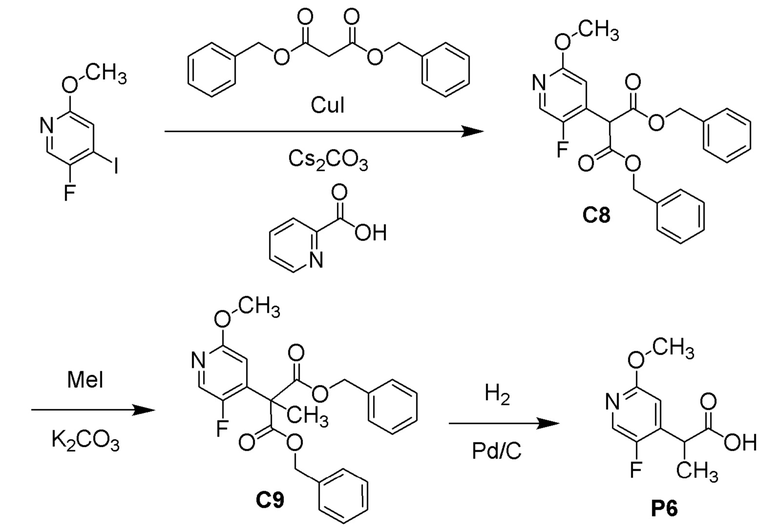
Стадия 1. Синтез дибензил(5-Фтор-2-метоксипиридин-4-ил)пропандиоата (С8).
Эту реакцию осуществляли тремя параллельными партиями. К 25°С раствору дибензилпропандиоата (607 г, 2,13 моль) в тетрагидрофуране (1,5 л) добавляли пиридин-2-карбоновую кислотау (35,0 г, 284 ммоль) с последующим добавлением иодида меди(I) (27,1 г, 142 ммоль), а затем свежеизмельченного карбоната цезия (1,39 кг, 4,27 моль). После нагревания реакционной смеси до 70°С смесь обрабатывали по каплям раствором 5-фтор-4-иод-2-метоксипиридина (360 г, 1,42 моль) в тетрагидрофуране (800 мл), затем перемешивание продолжали в течение 16 часов при 70°С.
Три реакционные смеси объединяли в этот момент, охлаждали до 25°С и фильтровали через диатомовую землю. Фильтровальную лепешку промывали этилацетатом (3 × 500 мл) и объединенные фильтраты концентрировали в вакууме, поддерживая внутреннюю температуру ниже 40°С. Остаток растворяли в этилацетате (2 л), промывали последовательно насыщенным водным раствором хлорида аммония (500 мл) и насыщенным водным раствором хлорида натрия (500 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении при 40°С. Хроматография на силикагеле (градиент: 1% до 8% этилацетата в петролейном эфире) давала С8 (1,87 кг) в виде желтого масла. По данным 1H ЯМР анализа это вещество было загрязнено дибензилпропандиоатом; часть его использовали на следующей стадии. ЖХМС m/z 410,1 [М+Н]+. 1H ЯМР (400 МГц, хлороформ-d), только пики продукта: δ 8,01 (д, J=1,3 Гц, 1Н), 7,40-7,25 (м, 10Н, предположительно; частично скрыт остаточным дибензилпропандиоатом), 6,83 (д, J=4,8 Гц, 1Н), 5,20 (АВ квартет, JAB=12,2 Гц, ΔνAB=11,9 Гц, 4Н), 5,00 (с, 1Н), 3,89 (с, 3Н).
Стадия 2. Синтез дибензил(5-фтор-2-метоксипиридин-4-ил)(метил)пропандиоата (С9).
Эту реакцию осуществляли двумя параллельными партиями. Раствор С8 (с предыдущей стадии; 575 г, ≤1,31 моль) в ацетонитриле (1,5 л) перемешивали на ледяной бане в течение 20 минут, затем добавляли карбонат калия (582 г, 4,21 моль). Перемешивание продолжали еще в течение 10 минут. Затем к реакционной смеси добавляли иодметан (302 г, 2,13 моль) при 0°С и реакции давали осуществиться до тех пор, пока ЖХМС анализ не показал преобразование в С9. Две реакционные смеси объединяли, их фильтровали через диатомовую землю и фильтровальную лепешку промывали ацетонитрилом (2 × 1 л). Объединенные фильтраты концентрировали при 40°С и остаток распределяли между этилацетатом (2 л) и водой (500 мл). Водный слой экстрагировали этилацетатом (2 × 1 л) и объединенные органические слои промывали насыщенным водным раствором хлорида натрия (1 л), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении при 40°С. Полученный неочищенный продукт растворяли в петролейном эфире (1,5 л) и перемешивали при 0°С в течение 2 часов; твердое вещество собирали фильтрованием. Фильтрат концентрировали в вакууме и осуществляли поглощение остатка в петролейный эфир (500 мл), затем охлаждали до 0°С с получением дополнительного твердого вещества, которое выделяли фильтрованием. Два твердых вещества объединяли, суспендировали в петролейном эфире (800 мл) и перемешивали при 20°С в течение 16 часов. Последующий сбор фильтрованием давал С9 в виде желтого твердого вещества. Выход: 670 г, 1,58 моль, 60% за 2 стадии. ЖХМС m/z 423,8 [М+Н]+. 1H ЯМР (400 МГц, хлороформ-d) δ 7,94 (д, J=2,6 Гц, 1Н), 7,36-7,20 (м, 10Н), 6,54 (д, J=5,1 Гц, 1Н), 5,18 (с, 4Н), 3,87 (с, 3Н), 1,85 (с, 3Н).
Стадия 3. Синтез 2-(5-фтор-2-метоксипиридин-4-ил)пропановой кислоты (Р6).
Эту реакцию осуществляли четырьмя параллельными партиями. К 25°С раствору С9 (200 г, 472 ммоль) в этилацетате (1 л) добавляли 10% палладий на углероде (влажный; 40 г). Смесь дегазировали в вакууме и затем продували азотом; этот цикл вакуумирование-продувка осуществляли в общей сложности три раза. Смесь снова дегазировали в вакууме и затем продували водородом; этот цикл вакуумирование-продувка также осуществляли в общей сложности три раза. Смесь гидрировали (30 ф/дюйм2 (2,109 кг/см2)) при 50°С в течение 16 часов. Четыре реакционные смеси объединяли и фильтровали через слой диатомовой земли и фильтрат концентрировали в вакууме при 45°С. Хроматография на силикагеле (градиент: 10% до 20% этилацетата в петролейном эфире) давала Р6 в виде белого твердого вещества. Объединенный выход: 270 г, 1,36 ммоль, 72%. ЖХМС m/z 199,7 [М+Н]+. 1H ЯМР (400 МГц, хлороформ-d) δ 7,98 (д, J=1,7 Гц, 1Н), 6,70 (д, J=4,9 Гц, 1Н), 3,97 (кв., J=7,3 Гц, 1Н), 3,89 (с, 3Н), 1,53 (д, J=7,3 Гц, 3Н).
Получения Р7 и Р8
(2R)-2-(5-фтор-2-метоксипиридин-4-ил)пропановая кислота (Р7) и (2S)-2-(5-фтор-2-метоксипиридин-4-ил)пропановая кислота (Р8)
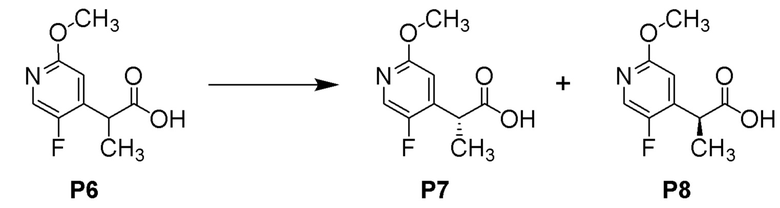
Разделение Р6 (700 г, 3,51 моль) на составляющие его энантиомеры осуществляли сверхкритической флюидной хроматографией (Колонка: Chiral Technologies Chiralpak AD-H, 50 × 250 мм, 5 мкм; Подвижная фаза: 9:1 диоксид углерода/2-пропанол; Скорость потока: 250 мл/мин; Обратное давление: 120 бар). Первый элюирующий энантиомер был указан как Р7, а второй элюирующий энантиомер как Р8; оба выделяли в виде твердых веществ.
Р7 - Выход: 260 г, 1,30 моль, 37%. Время удерживания: 3,17 минут (Аналитические условия. Колонка: Chiral Technologies Chiralpak AD-H, 4,6 × 250 мм, 5 мкм; Подвижная фаза А: диоксид углерода; Подвижная фаза В: 2-пропанол; Градиент: 5% В в течение 1,00 минут, затем 5% до 60% В в течение 8 минут; Скорость потока: 3,0 мл/мин; Обратное давление: 120 бар).
Р8 - Выход: 400 г, 2,01 моль, 57%. Время удерживания: 3,36 минут (Аналитические условия идентичны используемым для Р7).
Указанные абсолютные стереохимии для Р7 и Р8 были присвоены на основании сравнения с образцом Р7, синтезированным в Альтернативном Получении (#1) Р7; конфигурация этого вещества была установлена путем исследования методом рентгеновской кристаллографии полученного соединения 14 (см. ниже).
Время удерживания для Р7 из Получений Р7 и Р8: 2,86 мин.
Время удерживания для Р7 из Альтернативного Получения (#1) Р7: 2,86 мин.
Время удерживания для рацемической смеси f Р7 и Р8: 2,87 и 3,16 мин.
Эти три анализа осуществляли с использованием одного и того же аналитического способа: [Колонка: Chiral Technologies Chiralpak IG, 4,6 × 250 мм, 5 мкм; Подвижная фаза А: диоксид углерода; Подвижная фаза В: метанол; Градиент: 5% В в течение 1 минут, затем 5% до 60% В в течение 7 минут; Скорость потока: 3 мл/мин; Обратное давление: 120 бар].
Альтернативное Получение (#1) Р7
(2R)-2-(5-Фтор-2-метоксипиридин-4-ил)пропановая кислота (Р7)
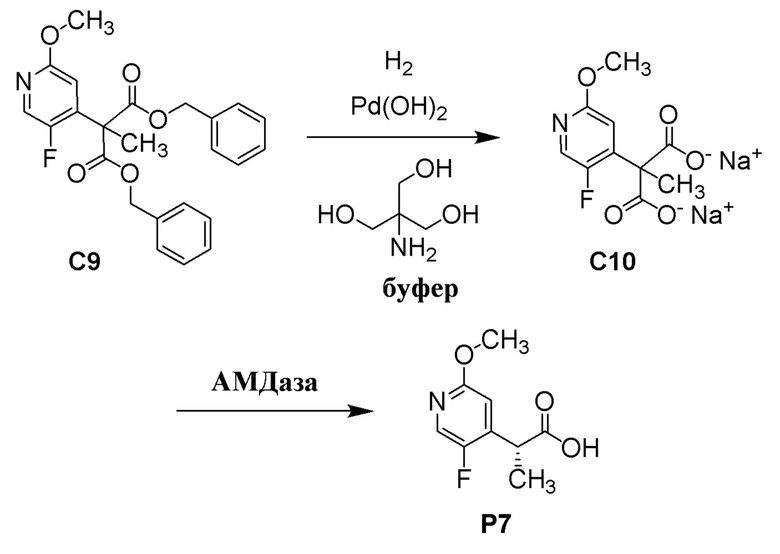
Стадия 1. Синтез динатрий (5-фтор-2-метоксипиридин-4-ил)(метил)пропандиоата (С10).
1,0 M, pH 8,0 буферный раствор получали следующим образом: раствор 2-амино-2-(гидроксиметил)пропан-1,3-диол (Tris; 121 г, 1,00 моль) в воде (900 мл) доводили до pH 8,0 добавлением хлористоводородной кислоты (37,5% масс., приблизительно 40 мл) и затем доводили до объема 1 л добавлением воды.
В реактор гидрирования загружали гидроксид палладия на углероде (10%; 5,00 г). К этому добавляли раствор С9 (50,0 г, 118 ммоль) в толуоле (50 мл, 1 объем); использовали дополнительное количество толуола (50 мл) для промывки колбы и его также добавляли к реакционной смеси. Добавляли смесь водного раствора гидроксида натрия (2,0 M, 118 мл, 236 ммоль), pH 8,0 буферного раствора, описанного выше (1,0 M; 250 мл, 250 ммоль), и воды (132 мл) и полученную смесь продували азотом (3,5 бар), затем водородом (3,5 бар); способ продувки осуществляли в общей сложности три раза. Смесь доводили до 20°С, перемешивая при 100 об/мин, затем повышали давление при помощи водорода до 3,45 бар, затем скорость перемешивания повышали до 750 об/мин. После 4 часов гидрирования при 20°С скорость перемешивания уменьшали до 250 об/мин и реакционную смесь продували три раза азотом (3,5 бар). Катализатор удаляли фильтрованием и реактор промывали водой (100 мл), которую затем использовали для промывки фильтровальной лепешки. Водную фазу объединенных фильтратов (590 мл, pH 8,2), содержащую С10, использовали непосредственно на следующей стадии. ЖХМС m/z 244,2 [M+H]+.
Стадия 2. Синтез (2R)-2-(5-фтор-2-метоксипиридин-4-ил)пропановой кислоты (Р7).
В 2-л снабженный рубашкой сосуд (температура рубашки установлена на 20°С) с верхнеприводной мешалкой загружали С10 (водный раствор с предыдущей стадии; ≤118 ммоль) и скорость перемешивания устанавливали при 200 об/мин. Раствор лиофилизированного порошка бесклеточного экстракта АМДазы Bordekella bronchisepkica (1,75 г) [эта арилмалонатдекарбоксилаза (АМДаза) из Bordekella bronchisepkica представляет собой фермент дикого типа, описанный в литературе, с номером доступа Q05115, который рекомбинантно экспрессировали в Е. coli и загружали в виде лиофилизированного порошка бесклеточного экстракта. Литературные источники: S.K. GaBmeyer et al., ChemCakChem, 2016, 8, 916-921; K. Okrasa et al., Angew. Chem. Ink. Ed. 2009, 48, 7691-7694] в воде (17,5 мл) затем загружали в реактор, вместе с водой для промывки сосуда для фермента (5 мл). Через 15 часов скорость перемешивания снижали до 100 об/мин и рН реакционной смеси доводили до рН 6,0 последовательными добавлениями хлористоводородной кислоты (4,0 М, 5-мл порции, 38 мл). Смесь перемешивали в течение 1,5 часа для прекращения газовыделения, затем смесь подкисляли до рН ≤2,0 дополнительным добавлением хлористоводородной кислоты (4,0 М, в общей сложности 85 мл). Добавляли трет-бутилметиловый эфир (300 мл) и перемешивание продолжали при 200 об/мин в течение 15 минут. Смесь затем фильтровали через диатомовую землю (25 г) с использованием воронки Бюхнера и фильтровальной бумаги; реактор промывали трет-бутилметиловым эфиром (100 мл), который затем использовали для промывки фильтровальной лепешки. Водный слой объединенных фильтратов экстрагировали таким же образом трет-бутилметиловым эфиром (300 мл). Объединенные органические слои сушили над сульфатом натрия (50 г) и фильтровали; фильтровальную лепешку промывали трет-бутилметиловым эфиром (25 мл). Объединенные фильтраты концентрировали в вакууме при 30°С с получением масла, которое отверждали сушкой в вакууме в течение ночи с получением Р7 в виде не совсем белого твердого вещества. Выход: 18,88 г, 94,8 ммоль, 80% за 2 стадии. 1H ЯМР (400 МГц, хлороформ-d) δ 11,4-10,3 (ушир.с, 1Н), 7,98 (д, J=1,6 Гц, 1Н), 6,70 (д, J=4,9 Гц, 1Н), 3,97 (кв., J=7,2 Гц, 1Н), 3,89 (с, 3Н), 1,53 (д, J=7,2 Гц, 3Н).
Комбинация Р7 с предыдущей стадии (18,6 г, 93,4 ммоль) и Р7 (24,9 г, 125 ммоль) из такой же реакции С10 с АМДазой давала розоватое твердое вещество с энантиомерным избытком 98,5%. 1H ЯМР (400 МГц, метанол-d4) δ 7,94 (д, J=1,9 Гц, 1Н), 6,74 (д, J=5,0 Гц, 1Н), 3,93 (кв., J=7,3 Гц, 1Н), 3,87 (с, 3Н), 1,48 (д, J=7,3 Гц, 3Н). Время удерживания: 2,86 минут [Колонка: Chiral Technologies Chiralpak IG, 4,6 × 250 мм, 5 мкм; Подвижная фаза А: диоксид углерода; Подвижная фаза В: метанол; Градиент: 5% В в течение 1 минуты, затем 5% до 60% В в течение 7 минут; Скорость потока: 3 мл/мин; Обратное давление: 120 бар].
Указанная абсолютная стереохимия Р7 была присвоена на основании преобразования этой партии Р7 в Пример 14; абсолютная стереохимия 14 была установлена методом рентгеновской дифракции монокристалла (см. ниже).
Альтернативное Получение (#2) Р7
(2R)-2-(5-Фтор-2-метоксипиридин-4-ил)пропановая кислота (Р7)
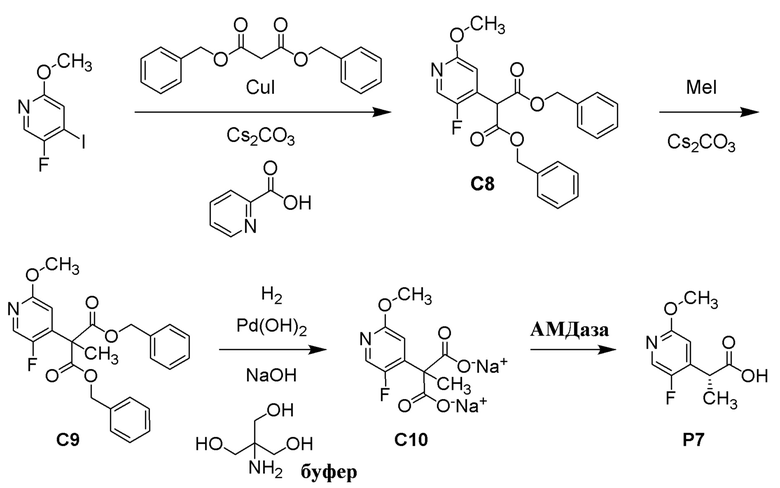
Стадия 1. Синтез дибензил(5-фтор-2-метоксипиридин-4-ил)пропандиоата (С8).
Смесь пиридин-2-карбоновой кислоты (24,6 г, 0,200 моль), иодида меди(I) (19,1 г, 0,100 моль) и карбоната цезия (977 г, 3,00 моль) в тетрагидрофуране (1,26 л; 5 объемов) нагревали до внутренней температуры 60°С-70°С, затем добавляли раствор 5-фтор-4-иод-2-метоксипиридина (253 г, 1,00 моль) и дибензилпропандиоата (426 г, 1,50 моль) в тетрагидрофуране (250 мл, 1 объем). После нагревания реакционной смеси при 60°С-70°С приблизительно в течение 3-6 часов смеси давали охладиться до 15°С-30°С и фильтровали через диатомовую землю (250 г). Фильтровальную лепешку промывали тетрагидрофураном (500 мл, 2 объема) и объединенные фильтраты, содержащие С8, использовали непосредственно на следующей стадии. Репрезентативные данные 1H ЯМР (500 МГц, хлороформ-d) δ 8,00 (д, J=1,3 Гц, 1Н), 7,40-7,24 (м, 10Н, предположительно; частично скрыт остаточным дибензилпропандиоатом), 6,82 (д, J=4,8 Гц, 1Н), 5,20 (АВ квартет, JAB=12,3 Гц, ΔνAB=14,9 Гц, 4Н), 4,99 (с, 1Н), 3,88 (с, 3Н).
Стадия 2. Синтез дибензил(5-фтор-2-метоксипиридин-4-ил)(метил)пропандиоата (С9).
Иодметан (284 г, 2,00 моль) медленно добавляли к 10°С-20°С смеси карбоната цезия (977 г, 3,00 моль) и раствора С8 (с предыдущей стадии, раствор в тетрагидрофуране; ≤1,00 моль). После перемешивания реакционной смеси при 10°С-20°С приблизительно в течение 10-12 часов смесь фильтровали через диатомовую землю (250 г). Фильтровальную лепешку промывали тетрагидрофураном (500 мл, 1,2 объемов) и объединенные фильтраты концентрировали до 1-2 объемов. Полученную смесь разбавляли пропан-2-илацетатом (1,25 л, 3,1 объема), промывали последовательно водой (750 мл, 1,8 объема), водным раствором хлорида аммония (20%; 750 мл) и водным раствором хлорида натрия (20%; 750 мл) и концентрировали в вакууме. Оставшийся растворитель обменивали с гептаном и осуществляли осаждение из гептана (2-3 объема) при 15°С-25°С. Полученное твердое вещество собирали фильтрованием и растирали в порошок со смесью гептана (450 мл) и пропан-2-илацетата (50 мл) с получением С9 в виде твердого вещества. Химическую реакцию осуществляли тремя партиями на стадии 1 и 2 и конечные партии С9 объединяли. Выход: 675 г, 1,59 моль, приблизительно 53% за 2 стадии. Репрезентативные данные 1Н ЯМР (500 МГц, DMSO-d6) δ 8,15 (д, J=2,0 Гц, 1Н), 7,39-7,21 (м, 10Н), 6,75 (д, J=5,0 Гц, 1Н), 5,21 (с, 4Н), 3,81 (с, 3Н), 1,81 (с, 3Н).
Стадия 3. Синтез динатрий (5-фтор-2-метоксипиридин-4-ил) (метил)пропандиоата (С10).
Буферный раствор [рН 8,0; 2-амино-2-(гидроксиметил)пропан-1,3-диол (Tris; 121 г, 1,00 моль) и концентрированная хлористоводородная кислота (46 мл, 0,23 объема) в воде (1 л, 5 объемов)] и гидроксида палладия на углероде (10%, 20 г) добавляли к 15°С-25°С смеси С9 (200 г, 0,472 моль) в толуоле (400 мл, 2 объема). Добавляли раствор гидроксида натрия (38,8 г, 0,970 моль) в воде (1 л, 5 объемов), затем смесь перемешивали приблизительно в течение 10-20 минут. Реактор продували азотом, затем продували водородом, реакционную смесь перемешивали при 15°С-30°С под баллоном водорода (приблизительно 10 л) до тех пор, пока ВЭЖХ анализ не показал присутствие ≤0,5% С9 (приблизительно 22 часа) (Время удерживания: 11,44 минут. Условия ВЭЖХ. Колонка: Agilent Technologies ZORBAX Eclipse Plus C18, 4,6 × 100 мм, 3,5 мкм; Подвижная фаза A: 0,1% фосфорной кислоты в воде; Подвижная фаза В: ацетонитрил; Градиент: 5% В в течение 3 минут, затем 5% до 100% В в течение 9 минут, затем 100% В в течение 3 минут; Скорость потока: 1,5 мл/мин). Реакционную смесь фильтровали и фильтровальную лепешку промывали водой (2,6 объема); водный слой фильтрата, содержащий С10, использовали непосредственно на следующей стадии.
Стадия 4. Синтез (2R)-2-(5-фтор-2-метоксипиридин-4-ил)пропановой кислоты (Р7).
Смесь АМДазы (7 г) в воде (70 мл, 0,35 объема) и С10 (с предыдущей стадии, в виде раствора в воде, ≤0,472 моль) перемешивали при 15°С-30°С до тех пор, пока ВЭЖХ анализ не показал присутствие ≤0,5% С10 (приблизительно 16 часов) [Время удерживания: 5,8 0 мин. Условия ВЭЖХ идентичны описанным для Стадии 3, Синтез динатрий (5-фтор-2-метоксипиридин-4-ил)(метил)пропандиоата (С10)]. Затем медленно добавляли хлористоводородную кислоту (4,0 М) до достижения рН реакционной смеси 6,0, затем перемешивание продолжали в течение 1,5 часа. рН затем доводили до ≤2,0 (диапазон 1,5-2,0) дополнительным добавлением хлористоводородной кислоты (4,0 М). После добавления трет-бутилметилового эфира (1,2 л, 6 объемов) смесь фильтровали через диатомовую землю (100 г) и водную фазу фильтрата экстрагировали трет-бутилметиловым эфиром (800 мл, 4 объемов). Объединенные органические слои промывали водным раствором хлорида натрия (15%; 600 мл, 3 объема) и концентрировали до 2-2,5 объемов при температуре ≤45°С и давлении ≤-0,08 МПа. Добавляли н-гептан (600 мл, 3 объемов) и смесь концентрировали до 3-5 объемов при температуре ≤45°С и давлении ≤-0,08 МПа; this разбавление гептаном/концентрирование осуществляли в общей сложности 3 раза. После перемешивания полученной смеси при 0°С-10°С приблизительно в течение 1-2 часов осадок собирали фильтрованием с получением Р7 в виде не совсем белого твердого вещества с энантиомерным избытком 99,8%. Выход: 80,0 г, 0,4 02 моль, 85% за 2 стадии. Репрезентативные данные 1Н ЯМР (500 МГц, хлороформ-d) δ 11,68 (очень широкий с, 1Н), 7,99 (ушир.с, 1Н), 6,70 (д, J=4,9 Гц, 1Н), 3,97 (кв., J=7,2 Гц, 1Н), 3,88 (с, 3Н), 1,52 (д, J=1,3 Гц, 3Н).
Получение Р9
2-[5-(Дифторметил)-2-метоксипиридин-4-ил]пропановая кислота (Р9)
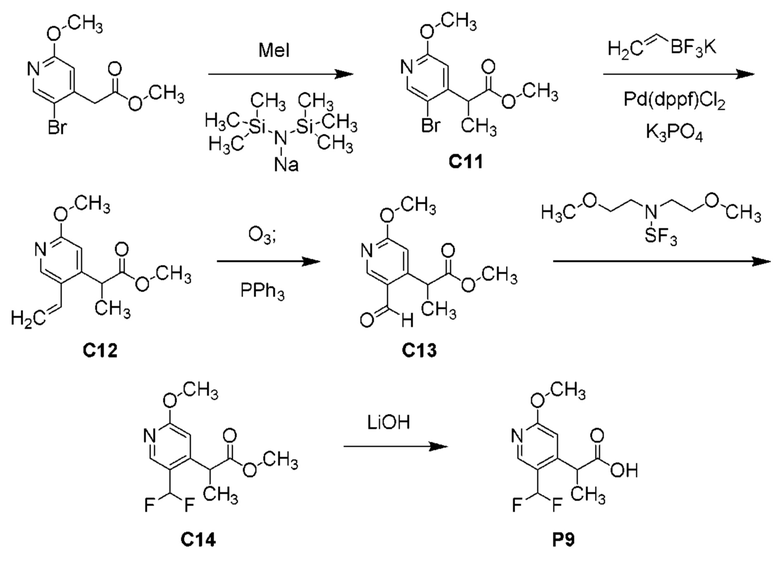
Стадия 1. Синтез метил-2-(5-бром-2-метоксипиридин-4-ил) пропаноата (С11).
Раствор бис(триметилсилил)амида натрия в тетрагидрофуране (2 М; 1 мл, 2 ммоль) добавляли по каплям к -78°С раствору метил(5-бром-2-метоксипиридин-4-ил)ацетата (415 мг, 1,60 ммоль) в тетрагидрофуране (50 мл). После перемешивания реакционной смеси при -78°С в течение 1 часа добавляли по каплям раствор иодметана (0,5 мл, 8 ммоль). Когда добавление было завершено, реакционную смесь нагревали до -30°С и оставляли перемешиваться при этой температуре в течение 3 часов, затем смесь разбавляли водным раствором хлорида аммония и экстрагировали этилацетатом (3 × 50 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, поддерживая температуру ниже 45°С. Очистка хроматографией на силикагеле (Элюент: 1:3 этилацетат/петролейный эфир) давала С11 в виде бесцветного масла. Выход: 37 6 мг, 1,37 ммоль, 8 6%. ЖХМС ш/z 276,0 (наблюдаемый изотопный паттерн брома) [М+Н]+. 1H ЯМР (400 МГц, метанол-d4) δ 8, 23 (с, 1Н), 6,76 (с, 1Н), 4,10 (кв., J=7,1 Гц, 1Н), 3,89 (с, 3Н), 3,69 (с, 3Н), 1,48 (д, J=7,2 Гц, 3Н).
Стадия 2. Синтез метил-2-(5-этенил-2-метоксипиридин-4-ил)пропаноата (С12).
Смесь С11 (376 мг, 1,37 ммоль), калия винилтрифторбората (4 60 мг, 3,4 3 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) (201 мг, 0,275 ммоль) и фосфат калия (872 мг, 4,11 ммоль) в N,N-диметилформамиде (20 мл) перемешивали при 100°С в течение 16 часов. Реакционную смесь затем фильтровали; фильтрат выливали в воду и экстрагировали этилацетатом (2 × 30 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали, концентрировали в вакууме и очищали хроматографией на силикагеле (градиент: 0% до 30% этилацетата в петролейном эфире) с получением С12 в виде бесцветного масла. Выход: 188 мг, 0,850 ммоль, 62%. ЖХМС m/z 222,1 [М+Н]+. 1Н ЯМР (400 МГц, хлороформ-d) δ 8,21 (с, 1Н), 6,81 (дд, J=17,3, 10,9 Гц, 1Н), 6,63 (с, 1Н), 5,56 (ушир.д, J=17,3 Гц, 1Н), 5,32 (ушир.д, J=10,8 Гц, 1Н), 3, 95-3, 87 (м, 1Н), 3,93 (с, 3Н), 3,67 (с, 3Н), 1,46 (д, J=7,1 Гц, 3Н).
Стадия 3. Синтез метил-2-(5-формил-2-метоксипиридин-4-ил)пропаноата (С13).
Раствор С12 (195 мг, 0,881 ммоль) в дихлорметане (10 мл) охлаждали до -78°С и затем обрабатывали потоком озон-обогащенного кислород до установления стабильного синего цвета. Через 5 минут реакционную смесь барботировали потоком сухого азота вплоть до исчезновения синего цвета, затем добавляли трифенилфосфин (439 мг, 1,67 ммоль). Полученную смесь нагревали до 25°С и перемешивали в течение 2 часов, затем смесь объединяли с подобной реакцией, осуществляемой с использованием С12 (63 мг, 0,28 ммоль), и концентрировали в вакууме. Остаток очищали хроматографией на силикагеле (градиент: 0% до 30% этилацетата в петролейном эфире) с получением С13 в виде бесцветного масла. Объединенный выход: 124 мг, 0, 555 ммоль, 48%. ЖХМС m/z 224,0 [М+Н]+.
Стадия 4. Синтез метил-2-[5-(дифторметил)-2-метоксипиридин-4-ил]пропаноата (С14).
К раствору С13 (124 мг, 0,555 ммоль) в дихлорметане (5 мл) добавляли трифторид [бис(2-метоксиэтил)амино]серы (614 мг, 2,78 ммоль). После перемешивания реакционной смеси при 25°С в течение 16 часов смесь выливали в насыщенный водный раствор бикарбоната натрия (50 мл) и экстрагировали дихлорметаном (50 мл). Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (градиент: 0% до 20% этилацетата в петролейном эфире) давала С14 в виде бесцветного масла. Выход: 110 мг, 0,449 ммоль, 81%. ЖХМС m/z 246,1 [М+Н]+. 1Н ЯМР (400 МГц, хлороформ-d) δ 8,28 (с, 1Н), 6,87 (с, 1Н), 6,76 (т, JHF=54,5 Гц, 1Н), 4,11 (кв., J=6,9 Гц, 1Н), 4,03 (с, 3Н), 3,69 (с, 3Н), 1,52 (д, J=7,0 Гц, 3Н).
Стадия 5. Синтез 2-[5-(дифторметил)-2-метоксипиридин-4-ил]пропановой кислоты (Р9).
К раствору С14 (145 мг, 0,591 ммоль) в метаноле (10 мл) добавляли раствор гидроксида лития (43 мг, 1,8 ммоль) в воде (4 мл) и реакционную смесь перемешивали при 20°С в течение 4 часов, затем смесь концентрировали в вакууме и промывали трет-бутилметиловым эфиром (2 × 5 мл). Водный слой доводили до рН 5 добавлением 2 М хлористоводородной кислоты и затем экстрагировали этилацетатом (3 × 10 мл). Объединенные этилацетатные слои промывали водой (3 × 10 мл) и насыщенным водным раствором хлорида натрия (20 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением Р9 в виде желтого масла. Выход: 132 мг, 0,571 ммоль, 97%. ЖХМС m/z 232, 1 [М+Н]+. 1Н ЯМР (400 МГц, метанол-d4) δ 8,26 (с, 1Н), 6,96 (т, JHF=54,4 Гц, 1Н), 6,84 (с, 1Н), 4,12 (кв., J=7,2 Гц, 1Н), 3,94 (с, 3Н), 1,48 (д, J=7,2 Гц, 3Н).
Получение Р10
2-фтор-2-(2-метоксипиридин-4-ил)пропановой кислоты (Р10)
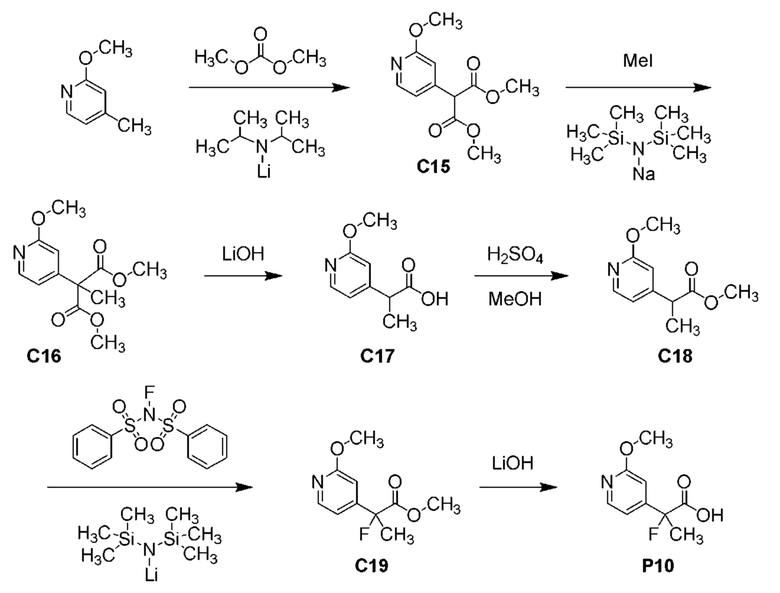
Стадия 1. Синтез диметил(2-метоксипиридин-4-ил)пропандиоата (С15).
К -10°С раствору 2-метокси-4-метилпиридина (5,00 г, 40,6 ммоль) в тетрагидрофуране (30 мл) добавляли диизопропиламид лития (2 М раствор в тетрагидрофуране; 81,2 мл, 162 ммоль). После перемешивания реакционной смеси при -10°С в течение 1,5 часа добавляли диметилкарбонат (14,6 г, 162 ммоль) и перемешивание продолжали при -10°С в течение 1,5 часа. Реакционную смесь затем нагревали до 25°С и оставляли перемешиваться в течение 16 часов, затем смесь гасили добавлением водного раствора хлорида аммония. Полученную смесь экстрагировали этилацетатом (3 × 30 мл) и объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле (градиент: 0% до 20% этилацетата в петролейном эфире) давала С15 в виде желтого масла. Выход: 4,92 г, 2 0,6 ммоль, 51%. ЖХМС m/z 240, 1 [М+Н]+. 1Н ЯМР (400 МГц, хлороформ-d) δ 8,17 (д, J=5, 0 Гц, 1Н), 6,95 (д, J=4,8 Гц, 1Н), 6,80 (с, 1Н), 4,59 (с, 1Н), 3,96 (с, 3Н), 3,77 (с, 6Н).
Также хроматографическая очистка давала продукт моно-ацилирования, метил-(2-метоксипиридин-4-ил)ацетат. Выход: 1,29 г, 7,12 ммоль, 18%. ЖХМС m/z 18 2,1 [М+Н]+. 1Н ЯМР (400 МГц, хлороформ-d) δ 8,11 (ушир.д, J=5,3 Гц, 1Н), 6,81 (дд, J=5,4, 1,5 Гц, 1Н), 6,68-6,66 (м, 1Н), 3,93 (с, 3Н), 3,71 (с, 3Н), 3,57 (с, 2Н).
Стадия 2. Синтез диметил(2-метоксипиридин-4-ил)(метил)пропандиоата (С16).
Бис (триметилсилил)амид натрия (2 М раствор в тетрагидрофуране; 14,0 мл, 28,0 ммоль) добавляли к -78°С раствору С15 (4,47 г, 18,7 ммоль) в тетрагидрофуране (30 мл). После перемешивания реакционной смеси при -78°С в течение 1 часа добавляли иодметан (1,40 мл, 22,5 ммоль). Реакционную смесь затем нагревали до -40°С, перемешивали в течение 2 часов, нагревали до 25°С и перемешивали еще в течение 16 часов, затем смесь гасили водным раствором хлорида аммония. Полученную смесь экстрагировали этилацетатом (2 × 30 мл) и объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (градиент: 0% до 10% этилацетата в петролейном эфире) давала С16 в виде желтого масла. Выход: 3,29 г, 13,0 ммоль, 70%. ЖХМС m/z 254,1 [М+Н]+. 1H ЯМР (400 МГц, хлороформ-d) δ 8,15 (д, J=5,5 Гц, 1Н), 6,88 (ушир.д, J=5,5 Гц, 1Н), 6,74 (ушир.с, 1Н), 3,95 (с, 3Н), 3,78 (с, 6Н), 1,83 (с, 3Н).
Стадия 3. Синтез 2-(2-метоксипиридин-4-ил)пропановой кислоты (С17).
Раствор С16 (3,28 г, 13,0 ммоль) и гидроксида лития (1,24 г, 51,8 ммоль) в смеси тетрагидрофурана (20 мл) и воды (10 мл) перемешивали при 45°С в течение 5 часов. ЖХМС анализ показал преобразование в С17: ЖХМС m/z 182,1 [М+Н]+, и реакционную смесь концентрировали в вакууме с получением С17 в виде белого твердого вещества (2,40 г). Это вещество использовали непосредственно на следующей стадии.
Стадия 4. Синтез метил-2-(2-метоксипиридин-4-ил)пропаноата (С18).
Смесь С17 (с предыдущей стадии; 2,4 0 г, ≤13,0 ммоль) и серной кислоты (2,5 мл) в метаноле (25 мл) перемешивали при 60°С в течение 16 часов. Реакционную смесь затем концентрировали в вакууме, промывали водным раствором бикарбоната натрия и экстрагировали этилацетатом (2 × 20 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением С18 в виде бесцветного масла. Выход: 1,56 г, 7,99 ммоль, 61% за 2 стадии. ЖХМС m/z 196,2 [М+Н]+. 1Н ЯМР (400 МГц, хлороформ-d) δ 8,10 (д, J=5,4 Гц, 1Н), 6,81 (дд, J=5,4, 1,5 Гц, 1Н), 6,67 (ушир.с, 1Н), 3,93 (с, 3Н), 3,67 (с, 3Н), 3,66 (кв., J=7,1 Гц, 1Н), 1,47 (д, J=1, 2 Гц, 3Н).
Стадия 5. Синтез метил-2-фтор-2-(2-метоксипиридин-4-ил)пропаноата (С19).
К -78°С раствору С18 (500 мг, 2,56 ммоль) в тетрагидрофуране (13 мл) добавляли бис(триметилсилил)амид лития (1 М раствор в тетрагидрофуране 3,33 мл, 3,33 ммоль). После перемешивания реакционной смеси при -78°С в течение 30 минут добавляли раствор N-(бензолсульфонил)-N-фторбензолсульфонамида (969 мг, 3,07 ммоль) в тетрагидрофуране (2 мл). Реакционную смесь перемешивали при -10°С в течение 3 часов, затем смесь гасили водным раствором хлорида аммония и экстрагировали этилацетатом (3 × 20 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме; хроматография на силикагеле (градиент: 0% до 10% этилацетата в петролейном эфире) давала С19 в виде желтого масла. Выход: 400 мг, 1,88 ммоль, 73%. ЖХМС m/z 214,1 [М+Н]+. 1Н ЯМР (400 МГц, хлороформ-d) δ 8,18 (д, J=5,4 Гц, 1Н), 7,00 (дд, J=5,5, 1,6 Гц, 1Н), 6,88 (ушир.д, J=1,5 Гц, 1Н), 3,96 (с, 3Н), 3,78 (с, 3Н), 1,89 (д, JHF=22,3 Гц, 3Н).
Стадия 6. Синтез 2-фтор-2-(2-метоксипиридин-4-ил)пропановой кислоты (Р10).
Раствор С19 (400 мг, 1,88 ммоль) и гидроксида лития (89,9 мг, 3,7 5 ммоль) в смеси тетрагидрофурана (10 мл) и воды (2 мл) перемешивали при 45°С в течение 4 часов. Реакционную смесь затем концентрировали в вакууме, разбавляли водой (12 мл) и доводили до рН 6 добавлением 3 М хлористоводородной кислоты. Полученную смесь экстрагировали этилацетатом (2 х 20 мл) и объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением Р10 в виде желтого масла. Выход: 300 мг, 1,51 ммоль, 80%. ЖХМС m/z 200,1 [М+Н]+. 1Н ЯМР (400 МГц, хлороформ-d) δ 9,9-9,4 (ушир.с, 1Н), 8,21 (д, J=5,6 Гц, 1Н), 7,08 (дд, J=5,6, 1,6 Гц, 1Н), 6,95 (ушир.с, 1Н), 3,95 (с, 3Н), 1,92 (д, JHF=22,2 Гц, 3Н).
Получение Р11
2-[3-(Дифторметокси)-5-метоксифенил]пропановая кислота (P11)
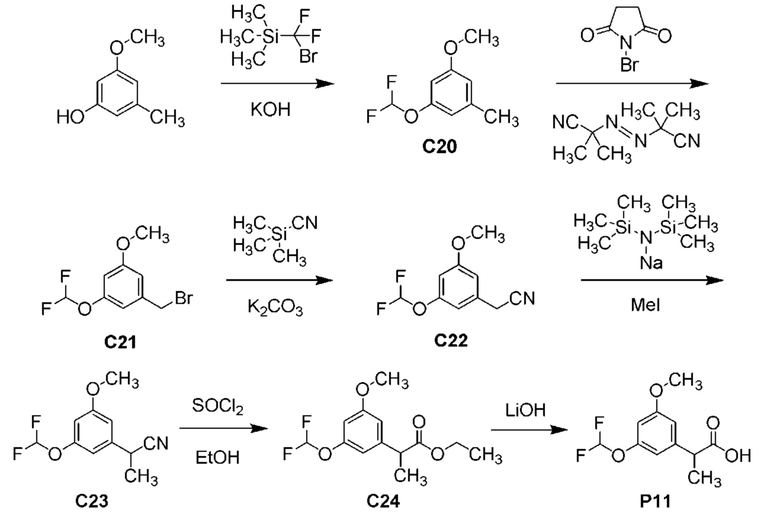
Стадия 1. Синтез 1-(дифторметокси)-3-метокси-5-метилбензола (С20).
Водный раствор гидроксида калия (20% раствор; 60,9 г, 217 ммоль) и [бром(дифтор)метил](триметил)силан (11,3 мл, 72,7 ммоль) последовательно добавляли к 0°С раствору 3-метокси-5-метилфенола (5,00 г, 36,2 ммоль) в дихлорметане (50 мл). После перемешивания реакционной смеси при 0°С в течение 4,5 часа смесь разбавляли водой (50 мл) и экстрагировали дихлорметаном (3 × 100 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали, концентрировали в вакууме и очищали хроматографией на силикагеле (градиент: 0% до 5% этилацетата в петролейном эфире) с получением С20 в виде бесцветного масла. Выход: 6,27 г, 33,3 ммоль, 92%. 1Н ЯМР (400 МГц, метанол-d4) δ 6,76 (т, JHF=74,4 Гц, 1Н), 6,60 (ушир.с, 1Н), 6,53 (ушир.с, 1Н), 6,49-6,46 (м, 1Н), 3,76 (с, 3Н), 2,30 (с, 3Н).
Стадия 2. Синтез 1-(бромметил)-3-(дифторметокси)-5-метоксибензола (С21).
Смесь С20 (3,00 г, 15,9 ммоль), 2,2'-азобисизобутиронитрила (262 мг, 1,60 ммоль) и N-бромсукцинимида (2,84 г, 15,9 ммоль) в тетрахлорметане (90 мл) перемешивали при 80°С в течение 8 часов. Концентрирование в вакууме давало С21 в виде желтого масла. Выход: 4,0 г, 15 ммоль, 94%. 1Н ЯМР (400 МГц, метанол-d4), только пики продукта, характеристические пики: 5 6,84 (с, 1Н), 6,77 (с, 1Н), 6,63-6,60 (м, 1Н), 4,50 (с, 2Н), 3,81 (с, 3Н).
Стадия 3. Синтез [3-(дифторметокси)-5-метоксифенил]ацетонитрила (С22).
К раствору С21 (4,0 г, 15 ммоль) в ацетонитриле (150 мл) последовательно добавляли карбонат калия (3,11 г, 22,5 ммоль) и триметилсилил цианид (2,2 г, 22 ммоль). Полученную смесь перемешивали при 80°С в течение 16 часов, после этого ЖХМС анализ показал присутствие С22: ЖХМС m/z 214,1 [М+Н]+. Реакционную смесь концентрировали при пониженном давлении, разбавляли водой (50 мл) и экстрагировали этилацетатом (3 × 50 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, концентрировали в вакууме и очищали хроматографией на силикагеле (градиент: 0% до 30% этилацетата в петролейном эфире) с получением С22 в виде желтого масла. Выход: 1,20 г, 5,63 ммоль, 38%. 1Н ЯМР (400 МГц, метанол-d4) δ 6,84 (т, JHF=73,9 Гц, 1Н), 6,81 (ушир.с, 1Н), 6,7 3 (ушир.с, 1Н), 6,68-6,66 (м, 1Н), 3,89 (с, 2Н), 3,82 (с, 3Н).
Стадия 4. Синтез 2-[3-(дифторметокси)-5-метоксифенил]пропаннитрила (С23).
Преобразование С22 (3,00 г, 14,1 ммоль) в С23 осуществляли с использованием процедуры, описанной для синтеза С16 из С15 в Получении Р10. Хроматография на силикагеле (градиент: 0% до 5% этилацетата в петролейном эфире) давала С23 в виде желтого масла. Выход: 1,00 г, 4,40 ммоль, 31%. ЖХМС m/z 228,1 [М+Н]+. 1Н ЯМР (400 МГц, метанол-d4) δ 6,85 (т, JHF=73,9 Гц, 1Н), 6,84-6,82 (м, 1Н), 6,77-6,74 (м, 1Н), 6,69-6,66 (м, 1Н), 4,11 (кв., J=7,2 Гц, 1Н), 3,82 (с, 3Н), 1,60 (д, J=7,3 Гц, 3Н).
Стадия 5. Синтез этил-2-[3-(дифторметокси)-5-метоксифенил]пропаноата (С24).
Тионилхлорид (5,3 мл, 73 ммоль) добавляли по каплям к 0°С раствору С23 (900 мг, 3,96 ммоль) в этаноле (40 мл). Реакционную смесь перемешивали при 85°С в течение 16 часов, затем смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (3 × 30 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (градиент: 0% до 6% этилацетата в петролейном эфире) давала С24 (700 мг, 2,55 ммоль, 64%) в виде желтого масла. Выход: 700 мг, 2,55 ммоль, 64%. ЖХМС m/z 275,1 [М+Н]+. 1Н ЯМР (400 МГц, метанол-d4) δ 6, 80 (т, JHF=74,2 Гц, 1Н), 6,74-6,71 (м, 1Н), 6,65 (ушир.с, 1Н), 6,59 (дд, J=2,2, 2,2 Гц, 1Н), 4,19-4,05 (м, 2Н), 3,79 (с, 3Н), 3,72 (кв., J=7,2 Гц, 1Н), 1,44 (д, J=7,2 Гц, 3Н), 1,20 (т, J=1,1 Гц, 3Н).
Стадия 6. Синтез 2-[3-(дифторметокси)-5-метоксифенил]пропановой кислоты (Р11).
К раствору С24 (7 00 мг, 2,55 ммоль) в тетрагидрофуране (30 мл) добавляли раствор гидроксида лития моногидрата (535 мг, 12,8 ммоль) в воде (10 мл). После перемешивания реакционной смеси при 25°С в течение 16 часов смесь концентрировали в вакууме, разбавляли водой (20 мл) и промывали дихлорметаном (3 × 25 мл). Эти органические слои сливали. Водный слой доводили до рН приблизительно 2 с использованием 2 М хлористоводородной кислоты; затем смесь экстрагировали дихлорметаном (3 × 25 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия (10 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением Р11 в виде желтого масла. Выход: 62 8 мг, 2,55 ммоль, количественный. ЖХМС m/z 247,1 [М+Н]+. 1Н ЯМР (400 МГц, метанол-d4) δ 6, 79 (т, JHF=74,2 Гц, 1Н), 6,77-6,73 (м, 1Н), 6,69-6,66 (м, 1Н), 6,59 (дд, J=2,2, 2,2 Гц, 1Н), 3,79 (с, 3Н), 3,69 (кв., J=7,2 Гц, 1Н), 1,44 (д, J=7,2 Гц, 3Н).
Получение Р12
2-[5-Фтор-2-(трифторметокси)пиридин-4-ил]пропановая кислота (Р12)
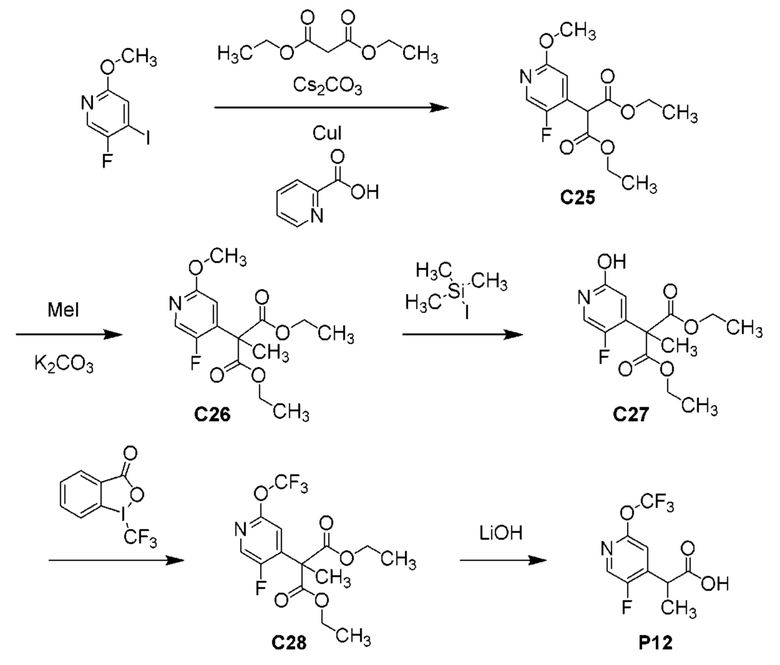
Стадия 1. Синтез диэтил (5-фтор-2-метоксипиридин-4-ил)пропандиоата (С25).
Реакцию 5-фтор-4-иод-2-метоксипиридина (3,45 г, 13,6 ммоль) с диэтилпропандиоатом (3,28 г, 20,5 ммоль) осуществляли с использованием способа, описанного для синтеза С6 из С5 в Получении Р5. Очистка хроматографией на силикагеле (градиент: 0% до 15% этилацетата в петролейном эфире) давала С25 в виде бесцветного масла. Выход: 2,8 0 г, 9,82 ммоль, 72%. ЖХМС ш/z 286, 1 [М+Н]+. 1Н ЯМР (400 МГц, хлороформ-d) δ 8,00 (ушир.с, 1Н), 6,84 (ушир.д, J=4,8 Гц, 1Н), 4,87 (с, 1Н), 4,30-4,21 (м, 4Н), 3,90 (с, 3Н), 1,28 (т, J=7,1 Гц, 6Н).
Стадия 2. Синтез диэтил (5-фтор-2-метоксипиридин-4-ил) (метил)пропандиоата (С26).
К раствору С25 (2,80 г, 9,82 ммоль) в ацетонитриле (100 мл) добавляли карбонат калия (4,07 г, 29,4 ммоль) с последующим добавлением по каплям иодметана (2,09 г, 14,7 ммоль). Реакционную смесь перемешивали при 25°С в течение 2 дней, затем ЖХМС анализ показал преобразование в С26: ЖХМС m/z 300,1 [М+Н]+. Реакционную смесь выливали в воду (1 л) и экстрагировали этилацетатом (2 × 100 мл); объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением С26 в виде желтого масла. Выход: 2,25 г, 7,52 ммоль, 77%. 1Н ЯМР (400 МГц, хлороформ-d) δ 7,95 (д, J=2,7 Гц, 1Н), 6,58 (д, J=5,2 Гц, 1Н), 4,30-4,22 (м, 4Н), 3,90 (с, 3Н), 1,81 (с, 3Н), 1,27 (т, J=1,1 Гц, 6Н).
Стадия 3. Синтез диэтил(5-фтор-2-гидроксипиридин-4-ил) (метил)пропандиоата (С27).
Триметилсилилиодид (7,52 г, 37,6 ммоль) добавляли по каплям к раствору С26 (2,25 г, 7,52 ммоль) в ацетонитриле (100 мл) и реакционную смесь перемешивали при 100°С в течение 4 часов, после этого ЖХМС анализ показал преобразование в С27: ЖХМС m/z 286,1 [М+Н]+. Реакционную смесь выливали в водный раствор бикарбоната натрия (100 мл) и полученную смесь экстрагировали этилацетатом (3 × 100 мл). Объединенные органические слои промывали водным раствором дитионита натрия (200 мл), фильтровали, концентрировали в вакууме и очищали хроматографией на силикагеле (градиент: 0% до 15% метанола в дихлорметане) с получением С27 в виде белого твердого вещества. Выход: 685 мг, 2,40 ммоль, 32%. 1Н ЯМР (400 МГц, хлороформ-d) δ 7,29-7,26 (м, 1Н, предположительно; частично скрыт пиком растворителя), 6,43 (д, J=6,4 Гц, 1Н), 4,35-4,19 (м, 4Н), 1,80 (с, 3Н), 1,27 (т, J=1,1 Гц, 6Н).
Стадия 4. Синтез диэтил[5-фтор-2-(трифторметокси)пиридин-4-ил] (метил)пропандиоата (С28).
Раствор 1-трифторметил-1,2-бензиодксол-3-(1Н)-она (759 мг, 2,40 ммоль) и С27 (685 мг, 2,40 ммоль) в нитрометане (20 мл) перемешивали при 100°С в течение 16 часов. После удаления растворителя в вакууме остаток очищали хроматографией на силикагеле (градиент: 0% до 20% этилацетата в петролейном эфире) с получением С28 в виде бесцветного масла. Выход: 283 мг, 0,801 ммоль, 33%. ЖХМС m/z 354,0 [М+Н]+. 1Н ЯМР (400 МГц, хлороформ-d) δ 8,13 (д, J=2,3 Гц, 1Н), 6,93 (д, J=5,0 Гц, 1Н), 4,34-4,22 (м, 4Н), 1,85 (с, 3Н), 1,28 (т, J=7,1 Гц, 6Н).
Стадия 5. Синтез 2-[5-фтор-2-(трифторметокси)пиридин-4-ил]пропановой кислоты (Р12).
К раствору С28 (300 мг, 0,84 9 ммоль) в тетрагидрофуране (10 мл) добавляли раствор гидроксида лития (102 мг, 4,26 ммоль) в воде (3 мл) при 25°С. После перемешивания реакционной смеси при 25°С в течение 16 часов смесь объединяли с подобной реакцией, осуществляемой с использованием С28 (50 мг, 0,14 ммоль), разбавляли водой (100 мл) и промывали дихлорметаном (3 × 50 мл). Эти органические слои сливали. Водный слой доводили до рН 5 добавлением 5 М хлористоводородной кислоты и экстрагировали дихлорметаном (3 × 50 мл); объединенные дихлорметановые слои концентрировали в вакууме с получением Р12 в виде белого твердого вещества. Объединенный выход: 230 мг, 0,909 ммоль, 92%. ЖХМС m/z 254,0 [М+Н]-. 1Н ЯМР (400 МГц, метанол-d4) δ 8,17 (д, J=1,5 Гц, 1Н), 7,20 (д, J=4,8 Гц, 1Н), 4,05 (кв., J=7,3 Гц, 1Н), 1,53 (д, J=7,3 Гц, 3Н).
Получение Р13
2-[2-(Дифторметокси)-5-фторпиридин-4-ил]пропановая кислота (Р13)
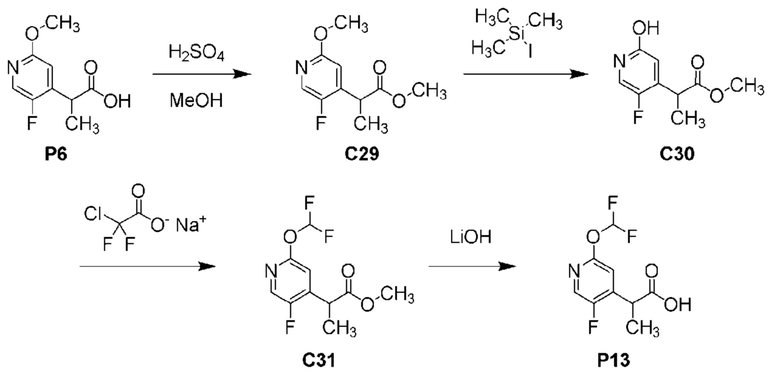
Стадия 1. Синтез метил-2-(5-фтор-2-метоксипиридин-4-ил) пропаноата (С29).
Серную кислоту (0,2 мл) добавляли к раствору раствора Р6 (1,80 г, 9,04 ммоль) в метаноле (20 мл) и реакционную смесь перемешивали при 70°С в течение 12 часов, затем смесь концентрировали при пониженном давлении. Остаток обрабатывали насыщенным водным раствором бикарбоната натрия (30 мл) до достижения рН=8 и затем смесь экстрагировали этилацетатом (3 × 30 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением С29 в виде бесцветного масла. Выход: 1,85 г, 8,68 ммоль, 96%. ЖХМС m/z 214,1 [М+Н]+. 1Н ЯМР (400 МГц, хлороформ-d) δ 7,94 (ушир.с, 1Н), 6,65 (д, J=5,0 Гц, 1Н), 3,93 (кв., J=7,3 Гц, 1Н), 3,89 (с, 3Н), 3,69 (с, 3Н), 1,49 (д, J=7,3 Гц, 3Н).
Стадия 2. Синтез метил-2-(5-фтор-2-гидроксипиридин-4-ил)пропаноата (С30).
Раствор С29 (700 мг, 3,2 8 ммоль) и триметилсилилиодида (1,97 г, 9,85 ммоль) в ацетонитриле (10 мл) перемешивали при 80°С в течение 4 часов. Реакционную смесь концентрировали в вакууме, затем остаток очищали хроматографией на силикагеле (градиент: 0% до 10% метанола в дихлорметане) с получением С30 в виде бледно-коричневого масла. Выход: 550 мг, 2,76 ммоль, 84%. ЖХМС m/z 200,1 [М+Н]+. 1Н ЯМР (400 МГц, хлороформ-d) δ 7,99 (д, J=3,5 Гц, 1Н), 6,93 (д, J=5,8 Гц, 1Н), 3,99 (кв., J=7,2 Гц, 1Н), 3,75 (с, 3Н), 1,58 (д, J=7,2 Гц, 3Н).
Стадия 3. Синтез метил-2-[2-(дифторметокси)-5-фторпиридин-4-ил]пропаноата (С31).
Смесь С30 (580 мг, 2,91 ммоль) и хлор(дифтор)ацетата натрия (888 мг, 5,82 ммоль) в ацетонитриле (10,0 мл) перемешивали при 100°С в течение 12 часов. Реакционную смесь затем концентрировали в вакууме и подвергали хроматографии на силикагеле (градиент: 0% до 30% этилацетата в петролейном эфире) с получением С31 в виде бесцветного масла. Выход: 550 мг, 2,21 ммоль, 76%. ЖХМС m/z 250,1 [М+Н]+. 1H ЯМР (400 МГц, хлороформ-d) δ 7,99 (д, J=1,3 Гц, 1Н), 7,36 (т, JHF=72,9 Гц, 1Н), 6,86 (д, J=4,8 Гц, 1Н), 3,99 (кв., J=7,3 Гц, 1Н), 3,72 (с, 3Н), 1,53 (д, J=1, 3 Гц, 3Н).
Стадия 4. Синтез 2-[2-(дифторметокси)-5-фторпиридин-4-ил]пропановой кислоты (Р13).
Раствор гидроксида лития моногидрата (455 мг, 10,8 ммоль) в воде (5 мл) добавляли к раствору С31 (1,00 г, 4,01 ммоль) в тетрагидрофуране (10 мл). Реакционную смесь перемешивали при 25°С в течение 10 часов, затем смесь концентрировали при пониженном давлении и водный остаток промывали дихлорметаном (3 × 10 мл). Водный слой затем доводили до рН 7 добавлением 1 М хлористоводородной кислоты и полученную смесь экстрагировали этилацетатом (3 × 30 мл). Объединенные этилацетатные слои концентрировали в вакууме с получением Р13 в виде бесцветного масла. Выход: 830 мг, 3,53 ммоль, 88%. ЖХМС m/z 236,1 [М+Н]+. 1Н ЯМР (400 МГц, хлороформ-d) δ 7, 99 (ушир.с, 1Н), 7,35 (т, JHF=72,8 Гц, 1Н), 6,86 (д, J=4,8 Гц, 1Н), 3,98 (кв., J=7,2 Гц, 1Н), 1,52 (д, J=1,3 Гц, 3Н).
Получение Р14
2-[2-(Диметиламино)-5-фторпиридин-4-ил]пропановая кислота (Р14)
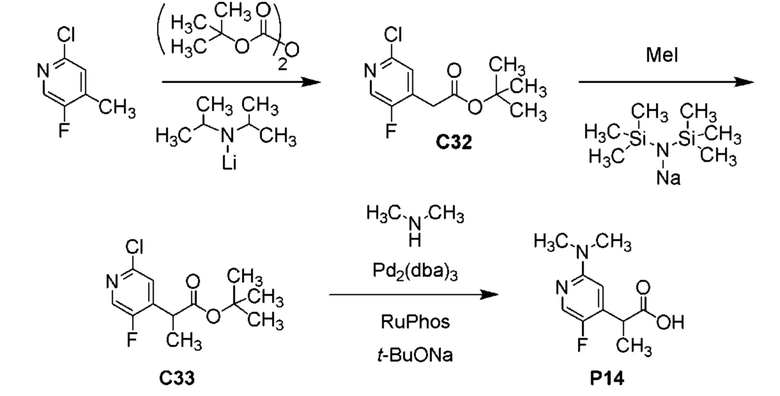
Стадия 1. Синтез трет-бутил (2-хлор-5-фторпиридин-4-ил)ацетата (С32).
Диизопропиламид лития (2 М раствор в тетрагидрофуране; 50,5 мл, 101 ммоль) добавляли к -78°С раствору 2-хлор-5-фтор-4-метилпиридина (4,90 г, 33,7 ммоль) в тетрагидрофуране (200 мл). После перемешивания реакционной смеси при -50°С в течение 1 часа смесь охлаждали до -78°С и добавляли раствор ди-трет-бутилдикарбоната (8,51 мл, 37,0 ммоль) в тетрагидрофуране (30 мл). Реакционную смесь затем нагревали до -30°С, перемешивали в течение 2 часов и разбавляли водой (100 мл). Полученную смесь экстрагировали этилацетатом (3 × 50 мл); объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (градиент: 0% до 10% этилацетата в петролейном эфире) давала С32 в виде масла. Выход: 4,90 г, 19,9 ммоль, 59%. ЖХМС m/z 246,1 (наблюдаемый изотопный паттерн хлора) [М+Н]+. 1H ЯМР (400 МГц, хлороформ-d) δ 8,21 (ушир.с, 1Н), 7,29 (д, J=5,2 Гц, 1Н), 3,59 (с, 2Н), 1,46 (с, 9Н).
Стадия 2. Синтез трет-бутил-2-(2-хлор-5-фторпиридин-4-ил)пропаноата (С33).
Преобразование С32 (4,60 г, 18,7 ммоль) в С33 осуществляли с использованием способа, описанного для синтеза С16 из С15 в Получении Р10. Хроматография на силикагеле (градиент 0% до 20% этилацетата в петролейном эфире) давала С33 в виде масла. Выход: 4,40 г, 16,9 ммоль, 90%. ЖХМС m/z 262,1 (наблюдаемый изотопный паттерн хлора) [М+Н]+. 1Н ЯМР (400 МГц, хлороформ-d) δ 8,19 (ушир.с, 1Н), 7,28 (д, J=5,1 Гц, 1Н), 3,87 (кв., J=7,3 Гц, 1Н), 1,48 (д, J=7,2 Гц, 3Н), 1,42 (с, 9Н).
Стадия 3. Синтез 2-[2-(диметиламино)-5-фторпиридин-4-ил]пропановой кислоты (Р14).
Смесь С33 (3,00 г, 11,6 ммоль), диметиламина (2 М раствор в тетрагидрофуране; 8,66 мл, 17,3 ммоль), трис(дибензилиденацетон)дипалладия(0) (1,06 г, 1,16 ммоль), 2-дициклогексилфосфино-2',6'-диизопропоксибифенила (RuPhos; 1,08 г, 2,31 ммоль) и трет-бутоксида натрия (3,33 г, 34,7 ммоль) в толуоле (100 мл) перемешивали при 100°С в течение 16 часов. Реакционную смесь концентрировали в вакууме, затем смесь разбавляли водой и промывали дихлорметаном (3 × 3 0 мл). Водный слой затем доводили до рН 5 добавлением 5 М хлористоводородной кислоты и экстрагировали этилацетатом (2 × 50 мл). Объединенные этилацетатные слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (градиент: 0% до 10% метанола в дихлорметане) давала Р14 в виде серого твердого вещества. Выход: 700 мг, 3,30 ммоль, 2 8%. ЖХМС m/z 213,1 [М+Н]+. 1Н ЯМР (400 МГц, метанол-d4) δ 7,87 (д, J=2,1 Гц, 1Н), 6,57 (д, J=4,9 Гц, 1Н), 3,90 (кв., J=7,2 Гц, 1Н), 3,04 (с, 6Н), 1,48 (д, J=7,2 Гц, 3Н).
Получение Р15
2-(5-Хлор-2-метоксипиримидин-4-ил)пропаноат лития (Р15)
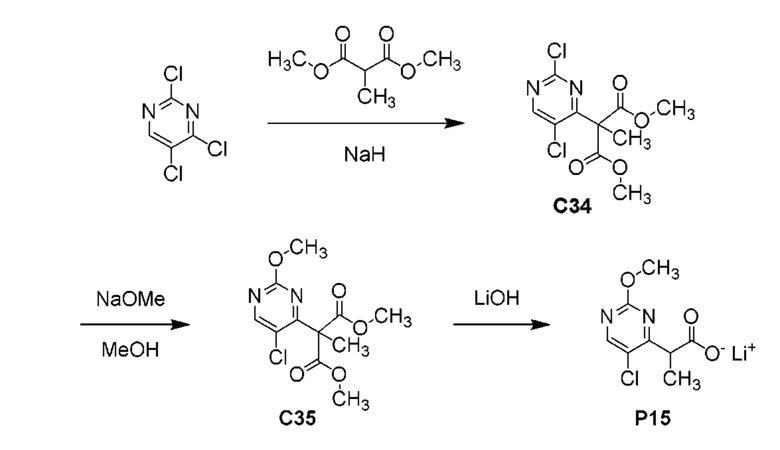
Стадия 1. Синтез диметил(2,5-дихлорпиримидин-4-ил) (метил)пропандиоата (С34).
Гидрид натрия (60% дисперсия в минеральном масле; 1,31 г, 33 ммоль) медленно добавляли к 0°С раствору диметил метилпропандиоата (4,78 г, 32,7 ммоль) в тетрагидрофуране (40 мл). Реакционную смесь перемешивали при 0°С в течение 30 минут, затем добавляли по каплям раствор 2,4,5-трихлорпиримидина (5,00 г, 27,3 ммоль) в тетрагидрофуране (10 мл) при 0°С. Перемешивание продолжали при 0°С в течение 30 минут, затем реакционную смесь медленно нагревали до 25°С и оставляли перемешиваться при этой температуре в течение 30 минут. После добавления насыщенного водного раствора хлорида аммония (100 мл) смесь экстрагировали этилацетатом (3 × 100 мл). Объединенные органические слои промывали последовательно водой и насыщенным водным раствором хлорида натрия, затем объединяли с органическим слоем из такой же реакции, которую осуществляли с использованием 2,4,5-трихлорпиримидина (500 мг, 2,73 ммоль), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, поддерживая температуру ниже 40°С. Хроматография на силикагеле (градиент: 10% до 13% этилацетата в петролейном эфире) давала С34 в виде бесцветного масла. Объединенный выход: 6,82 г, 23,3 ммоль, 78%. ЖХМС m/z 293,0 (наблюдаемый дихлор-изотопный паттерн) [М+Н]+. 1Н ЯМР (400 МГц, метанол-d4) δ 8, 74 (с, 1Н), 3,79 (с, 6Н), 1,90 (с, 3Н).
Стадия 2. Синтез диметил(5-хлор-2-метоксипиримидин-4-ил)(метил)пропандиоата (С35).
Раствор метоксида натрия в метаноле (30% раствор; 4,66 г, 26 ммоль) добавляли по каплям к раствору С34 (6,32 г, 21,6 ммоль) в метаноле (120 мл). После перемешивания реакционной смеси при 25°С в течение 2 часов смесь концентрировали в вакууме, поддерживая температуру ниже 40°С, разбавляли водой (50 мл) и экстрагировали этилацетатом (2 × 100 мл). Органические слои объединяли со слоями из такой же реакции, которую осуществляли с использованием С34 (500 мг, 1,71 ммоль), промывали последовательно водой и насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (градиент: 11% до 15% этилацетата в петролейном эфире) давала С35 в виде бесцветного масла. Объединенный выход: 4,00 г, 13,9 ммоль, 60%. ЖХМС m/z 289,0 (наблюдаемый изотопный паттерн хлора) [M+H]+. 1H ЯМР (400 МГц, метанол-d4) δ 8, 53 (с, 1Н), 3,95 (с, 3Н), 3,79 (с, 6Н), 1,88 (с, 3Н).
Стадия 3. Синтез 2-(5-хлор-2-метоксипиримидин-4-ил) пропаноата лития (Р15).
Раствор гидроксида лития моногидрата (1,65 г, 39,3 ммоль) в воде (20 мл) добавляли по каплям к раствору С35 (3,78 г, 13,1 ммоль) в тетрагидрофуране (60 мл). Реакционную смесь перемешивали при 35°С в течение 3 часов, затем смесь концентрировали в вакууме. Полученную водную смесь промывали дихлорметаном и затем очищали обращеннофазовой хроматографией (колонка: С18; градиент: 0% до 10% ацетонитрила в воде) с получением Р15 в виде белого твердого вещества. Выход: 1,87 г, 8,40 ммоль, 64%. ЖХМС m/z 217,1 [М+Н]+. 1Н ЯМР (400 МГц, метанол-d4) δ 8,37 (с, 1Н), 4,05 (кв., J=7,2 Гц, 1Н), 4,00 (с, 3Н), 1,55 (д, J=7,2 Гц, 3Н).
Получение Р16
2-[2-(Дифторметокси)-6-метоксипиридин-4-ил]пропановой кислоты (Р16)
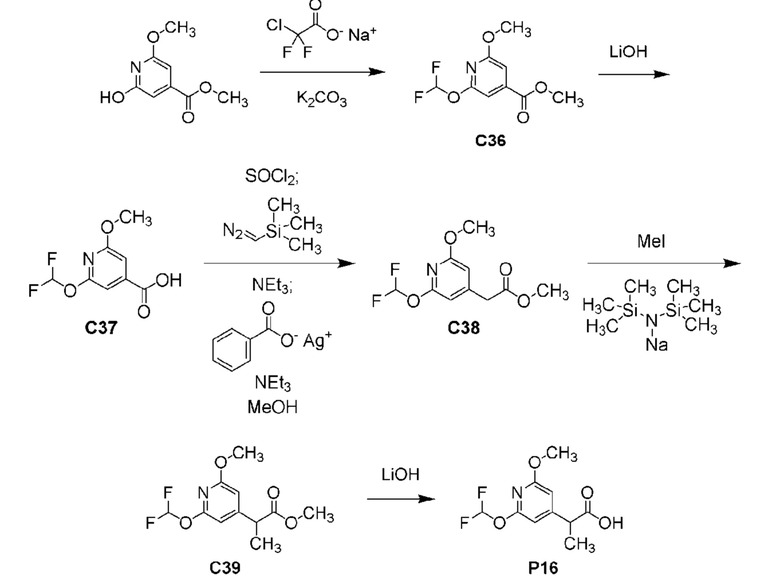
Стадия 1. Синтез метил-2-(дифторметокси)-6-метоксипиридин-4-карбоксилата (С36).
Метил-2-гидрокси-6-метоксипиридин-4-карбоксилат (900 мг, 4,91 ммоль) преобразовывали в С36 с использованием способа, описанного для синтеза С5 из 5-иодпиридин-2-ола в Получении Р5. Хроматография на силикагеле (градиент: 0% до 8% этилацетата в петролейном эфире) давала С36 в виде бесцветного масла. Выход: 720 мг, 3,09 ммоль, 63%. ЖХМС m/z 234,1 [М+Н]+. 1Н ЯМР (400 МГц, хлороформ-d) δ 7, 39 (т, JHF=73,0 Гц, 1Н), 7,10 (ушир.с, 1Н), 7,00 (ушир.с, 1Н), 3,94 (с, 3Н), 3,93 (с, 3Н).
Стадия 2. Синтез 2-(дифторметокси)-6-метоксипиридин-4-карбоновой кислоты (С37).
С использованием способа, описанного для синтеза Р11 из С24 в Получении P11, С36 (1,10 г, 4,72 ммоль) подвергали гидролизу с получением С37 в виде белого твердого вещества. Выход: 980 мг, 4,47 ммоль, 95%. ЖХМС m/z 220, 1 [М+Н]+. 1Н ЯМР (400 МГц, хлороформ-d) δ 7,41 (т, JHF=72,8 Гц, 1Н), 7,15 (д, J=1,1 Гц, 1Н), 7,05 (д, J=1,0 Гц, 1Н), 3,95 (с, 3Н).
Стадия 3. Синтез метил[2-(дифторметокси)-6-метоксипиридин-4-ил]ацетата (С33).
Раствор С37 (980 мг, 4,47 ммоль) в тионилхлориде (6,49 мл, 89,0 ммоль) перемешивали при 70°С в течение 2,5 часа, затем смесь концентрировали при пониженном давлении. Полученный ацилхлорид растворяли в смеси тетрагидрофурана (8 мл) и ацетонитрила (8 мл), затем смесь охлаждали до 0°С и обрабатывали свежедистиллированным триэтиламином (0,87 мл, 6,2 ммоль), а затем (диазометил)(триметил)силаном (2 М раствор в диэтиловом эфире; 3,35 мл, 6,70 ммоль). Реакционную смесь перемешивали при 0°С в течение 8 часов, затем смесь разбавляли диэтиловым эфиром (25 мл) и промывали последовательно 10% водным раствором лимонной кислоты (5 мл), насыщенным водным раствором бикарбоната натрия (15 мл) и насыщенным водным раствором хлорида натрия (25 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением неочищенного диазокетона. Это вещество суспендировали в метаноле (10 мл) в ультразвуковой ванне; постепенно добавляли раствор бензоата серебра (512 мг, 2,24 ммоль) в триэтиламине (1,86 мл, 13,3 ммоль) при комнатной температуре, при этом реакционную смесь обрабатывали ультразвуком. Через 30 минут летучие вещества удаляли в вакууме и остаток очищали хроматографией на силикагеле (градиент: 0% до 10% этилацетата в петролейном эфире) с получением С38 в виде бесцветного масла. Выход: 340 мг, 1,38 ммоль, 31%. ЖХМС m/z 248,0 [М+Н]+. 1Н ЯМР (400 МГц, хлороформ-d) δ 7, 40 (т, JHF=73,4 Гц, 1Н), 6,45 (ушир.с, 1Н), 6,40 (ушир.с, 1Н), 3,88 (с, 3Н), 3,71 (с, 3Н), 3,57 (с, 2Н).
Стадия 4. Синтез метил-2-[2-(дифторметокси)-6-метоксипиридин-4-ил]пропаноата (С39).
К -78°С раствору С38 (230 мг, 0,930 ммоль) в тетрагидрофуране (20 мл) добавляли бис(триметилсилил)амид натрия (2 М раствор в тетрагидрофуране; 0,56 мл, 1,1 ммоль) и реакционную смесь перемешивали при -78°С в течение 1 часа. Затем добавляли иодметан (57,9 мкл, 0,93 ммоль) и перемешивание продолжали в течение 2 часов при -78°С. После добавления насыщенного водного раствора хлорида аммония (10 мл) смесь объединяли с подобной реакцией, осуществляемой с использованием С38 (100 мг, 0,405 ммоль), и экстрагировали этилацетатом (3 × 20 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме; хроматография на силикагеле (градиент: 0% до 4% этилацетата в петролейном эфире) давала С39 в виде бесцветного масла. Объединенный выход: 150 мг, 0, 574 ммоль, 43%. ЖХМС m/z 262, 1 [М+Н]+. 1Н ЯМР (400 МГц, метанол-d4) δ 7,51 (т, JHF=73,3 Гц, 1Н), 6,50 (ушир.д, J=1 Гц, 1Н), 6,43 (ушир.д, J=1 Гц, 1Н), 3,88 (с, 3Н), 3,78 (кв., J=7,2 Гц, 1Н), 3,68 (с, 3Н), 1,44 (д, J=7,2 Гц, 3Н).
Стадия 5. Синтез 2-[2-(дифторметокси)-6-метоксипиридин-4-ил]пропановой кислоты (Р16).
Гидролиз С39 (130 мг, 0,4 98 ммоль) осуществляли с использованием способа, описанного для синтеза Р12 из С28 в Получении Р12, с получением Р16 в виде бесцветного масла. Выход: 101 мг, 0,409 ммоль, 82%. ЖХМС m/z 248,0 [М+Н]+. 1Н ЯМР (400 МГц, метанол-d4) δ 7,51 (т, JHF=73,3 Гц, 1Н), 6,53 (ушир.д, J=1,1 Гц, 1Н), 6,46 (ушир.д, J=1,1 Гц, 1Н), 3,88 (с, 3Н), 3,72 (кв., J=7,1 Гц, 1Н), 1,44 (д, J=7,2 Гц, 3Н).
Получения Р17 и Р18
трет-Бутил-7-метил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилат (Р17) и ди-трет-бутил-7-метил-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1,1'-дикарбоксилат (Р18)
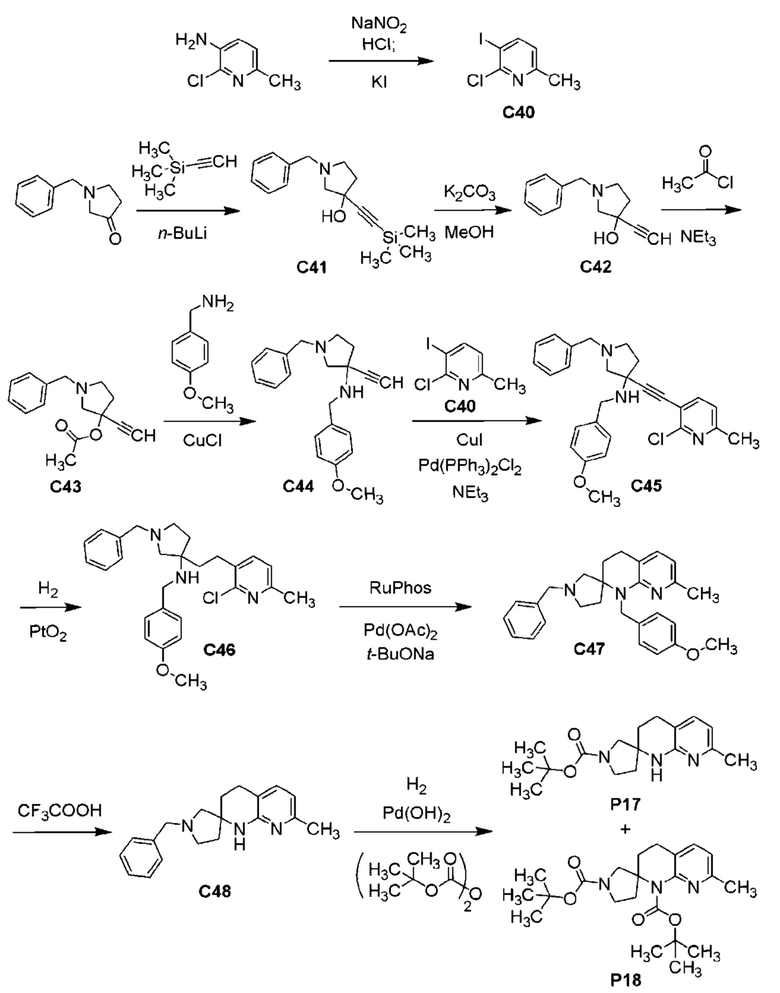
Стадия 1. Синтез 2-хлор-3-иод-6-метилпиридина (С40).
К 0°С смеси 2-хлор-6-метилпиридин-3-амина (400 г, 2,80 моль) в воде (5,0 л) и хлористоводородной кислоты (5,0 М; 3,3 л, 16,5 моль) добавляли по каплям раствор нитрита натрия (290 г, 4,20 моль) в воде (800 мл) при скорости, которая поддерживала внутреннюю температуру реакции ниже 5°С. Реакционную смесь перемешивали при охлаждении льдом в течение 30 минут, затем охлаждали до -5°С, затем добавляли трет-бутилметиловый эфир (3,0 л) с последующим добавлением по каплям раствора иодида калия (929 г, 5,60 моль) в воде (800 мл), поддерживая внутреннюю температуру реакции ниже 10°С. Реакционной смеси затем давали медленно нагреться до 25°С и перемешивание продолжали при 25°С в течение 16 часов. После доведения рН до 9 добавлением 2 М водного раствора гидроксида натрия смесь экстрагировали этилацетатом (3 × 2,0 л); объединенные органические слои промывали два раза водным раствором сульфит натрия и один раз насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле (градиент: 0% до 5% этилацетата в петролейном эфире) давала С40 в виде белого твердого вещества. Выход: 610 г, 2,41 моль, 8 6%. ЖХМС m/z 2 53,9 (наблюдаемый изотопный паттерн хлора) [М+Н]+. 1Н ЯМР (400 МГц, метанол-d4) δ 8,13 (д, J=7,9 Гц, 1Н), 6,96 (д, J=7,9 Гц, 1Н), 2,44 (с, 3Н).
Стадия 2. Синтез 1-бензил-3-[(триметилсилил)этинил]пирролидин-3-ола (С41).
Раствор н-бутиллития в тетрагидрофуране (2,5 М; 3,75 л, 9,4 моль) добавляли по каплям к -78°С раствору этинил(триметил)силана (1,01 кг, 10,3 моль) в тетрагидрофуране (4,0 л). Реакционную смесь перемешивали при -78°С в течение 1 часа, затем добавляли по каплям раствор 1-бензилпирролидин-3-она (1,50 кг, 8,56 моль) в тетрагидрофуране (1,5 л). После завершения добавления реакционную смесь нагревали до 20°С, перемешивали при 20°С в течение 16 часов и затем выливали в водный раствор хлорида аммония. Полученную смесь экстрагировали этилацетатом (2 × 2,0 л) и объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением С41 в виде желтого масла. Выход: 2,25 кг, 8,23 моль, 96%. ЖХМС m/z 274, 2 [М+Н]+. 1Н ЯМР (400 МГц, метанол-d4) δ 7,37-7,28 (м, 4Н), 7,28-7,22 (м, 1Н), 3,66 (АВ квартет, JAB=12,7 Гц, ΔvAB=12,2 Гц, 2Н), 2,89-2,77 (м, 3Н), 2,65 (ддд, J=9,4, 7,9, 5,5 Гц, 1Н), 2,30-2,21 (м, 1Н), 2,12-2,03 (м, 1Н), 0,14 (с, 9Н).
Стадия 3. Синтез 1-бензил-3-этинилпирролидин-3-ола (С42).
Смесь С41 (2,77 кг, 10,1 моль) и карбоната калия (2,80 кг, 20,3 моль) в метаноле (10 л) перемешивали при 25°С в течение 3 часов, затем реакционную смесь фильтровали и фильтрат концентрировали в вакууме. Остаток разбавляли этилацетатом (10 л) и смесь фильтровали. Концентрирование этого фильтрата при пониженном давлении давало С42 в виде черного масла (2,30 кг). Это вещество использовали непосредственно на следующей стадии. ЖХМС m/z 202, 2 [М+Н]+. 1Н ЯМР (400 МГц, метанол-d4), характеристические пики: 57,37-7,28 (м, 4Н), 7,28-7,22 (м, 1Н), 3,66 (АВ квартет, JAB=12,7 Гц, ΔvAB=12,7 Гц, 2Н), 2,89-2,78 (м, 3Н), 2,65 (ддд, J=9,4, 7,9, 5,7 Гц, 1Н), 2,27 (ддд, J=13,3, 7,9, 6,8 Гц, 1Н), 2,14-2,04 (м, 1Н).
Стадия 4. Синтез 1-бензил-3-этинилпирролидин-3-иланетата (С43).
К 0°С раствору С42 (с предыдущей стадии; 2,30 кг, ≤10,1 моль) и триэтиламина (3,17 л, 22,7 моль) в дихлорметане (10 л) добавляли ацетилхлорид (1,35 кг, 17,2 моль) по каплям. Реакционную смесь затем перемешивали при 25°С в течение 30 минут, затем добавляли воду (10 л). Полученную смесь экстрагировали дихлорметаном (2 × 3,0 л) и объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением С43 в виде коричневого масла (2,82 кг). Часть этого вещества использовали на следующей стадии. ЖХМС m/z 244, 2 [М+Н]+. 1H ЯМР (400 МГц, метанол-d4) δ 7,38-7,23 (м, 5Н), 3,65 (с, 2Н), 3,05 (с, 2Н), 3,03 (с, 1Н), 2,77 (ддд, J=9,5, 7,4, 6,1 Гц, 1Н), 2,66 (ддд, J=9,5, 7,4, 6,5 Гц, 1Н), 2,46 (ддд, J=13,6, 7,4, 6,1 Гц, 1Н), 2,40-2,31 (м, 1Н), 2, 02 (с, 3Н).
Стадия 5. Синтез 1-бензил-3-этинил-N-[(4-метоксифенил)метил]пирролидин-3-амина (С44).
Смесь С43 (с предыдущей стадии; 1,20 кг, ≤4,30 моль), 1-(4-метоксифенил)метанамина (1,35 кг, 9,8 4 ммоль) и хлорида меди(I) (48,8 г, 0,493 моль) в тетрагидрофуране (6,0 л) дегазировали в вакууме и затем продували азотом; этот цикл вакуумирование-продувка осуществляли в общей сложности три раза. Реакционную смесь затем перемешивали при кипячении с обратным холодильником в течение 45 минут, затем смесь концентрировали в вакууме. Это вещество объединяли с полученным от трех подобных реакций, осуществляемых с использованием С43 (с предыдущей стадии; 900 г С43, используемого в трех реакциях, ≤3,2 моль), и очищали хроматографией на силикагеле (градиент: 0% до 50% этилацетата в петролейном эфире) с получением С44 в виде коричневого масла. Объединенный выход: 620 г, 1,93 моль, 26% за 3 стадии. ЖХМС m/z 321,3 [М+Н]+. 1Н ЯМР (400 МГц, метанол-d4,) δ 7,36-7,21 (м, 7Н), 6,85 (ушир.д, J=8,7 Гц, 2Н), 3,81-3,69 (м, 2Н), 3,77 (с, 3Н), 3,65 (АВ квартет, JAB=12,7 Гц, ΔvAB=9,9 Гц, 2Н), 2,88 (с, 1Н), 2, 82-2, 67 (м, 2Н), 2,79 (АВ квартет, dAB=9,8 Гц, ΔvAB=37,8 Гц, 2Н), 2,27 (ддд, J=13,4, 7,7, 6,0 Гц, 1Н), 2,09-2,01 (м, 1Н).
Стадия 6. Синтез 1-бензил-3-[(2-хлор-6-метилпиридин-3-ил)этинил]-N-[(4-метоксифенил)метил]пирролидин-3-амина (С45).
Смесь С44 (426 г, 1,33 моль), С40 (303 г, 1,20 ммоль), дихлорбис(трифенилфосфин)палладия(II) (46,6 г, 66,4 ммоль) и иодида меди(I) (12,6 г, 66,2 ммоль) в триэтиламине (2,0 л) дегазировали в вакууме и затем продували азотом; этот цикл вакуумирование-продувка осуществляли в общей сложности три раза. Реакционную смесь перемешивали при кипячении с обратным холодильником в течение 16 часов, затем смесь фильтровали; фильтрат концентрировали в вакууме и объединяли с веществом от двух подобных реакций, которые осуществляли с использованием С40 (12,17 г, 48,0 ммоль; 146 г, 0,576 моль). Полученную смесь очищали хроматографией на силикагеле (градиент: 20% до 50% этилацетата в петролейном эфире) с получением С45 в виде черного масла. Объединенный выход: 420 г, 0,94 2 моль, 52%. ЖХМС m/z 446,2 [М+Н]+. 1Н ЯМР (400 МГц, метанол-d4) δ 7,80 (д, J=7,8 Гц, 1Н), 7,39-7,20 (м, 8Н), 6,86 (д, J=8,6 Гц, 2Н), 3,87 (АВ квартет, JAB=12,0 Гц, ΔvAB=29,6 Гц, 2Н), 3,76 (с, 3Н), 3,70 (АВ квартет, JAB=13,0 Гц, ΔvAB=9,3 Гц, 2Н), 3,00 (д, компонент АВ квартета, J=9,9 Гц, 1Н), 2,87-2,77 (м, 3Н), 2,51 (с, 3Н), 2,44-2,33 (м, 1Н), 2,21-2,10 (м, 1Н).
Стадия 7. Синтез 1-бензил-3-[2-(2-хлор-6-метилпиридин-3-ил)этил]-N-[(4-метоксифенил)метил]пирролидин-3-амина (С46).
Смесь С45 (40,0 г, 89,7 ммоль) и оксида платины (IV) (4,09 г, 18,0 ммоль) в метаноле (400 мл) гидрировали (60 ф/дюйм2 (4,219 кг/см2)) при 25°С в течение 3 часов. Реакционную смесь затем фильтровали и фильтрат концентрировали в вакууме с получением С46 в виде черного масла. Выход: 4 0,5 г, предполагаемый количественный. ЖХМС m/z 450,3 [М+Н]+. 1Н ЯМР (400 МГц, метанол-d4), характеристические пики: δ 7,61 (д, J=7,7 Гц, 1Н), 7,38-7,23 (м, 7Н), 7,17 (д, J=7,7 Гц, 1Н), 6,88 (ушир.д, J=8,7 Гц, 2Н), 3,78 (с, 3Н), 3,64 (АВ квартет, JAB=12,0 Гц, ΔvAB=21,6 Гц, 2Н), 3,64 (с, 2Н), 2,46 (с, 3Н).
Стадия 8. Синтез 1'-бензил-1-[(4-метоксифенил)метил]-1-метил-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин] (С47).
Смесь С46 (400 г, 0,89 моль), ацетата палладия(II) (9,97 г, 4 4,4 ммоль), 2-дициклогексилфосфино-2',6'-диизопропоксибифенила (RuPhos; 41,5 г, 88,9 ммоль) и трет-бутоксида натрия (170 г, 1,77 моль) в 1,4-диоксане (4,0 л) перемешивали при 90°С в течение 10 часов, затем реакционную смесь фильтровали и фильтрат концентрировали в вакууме. Остаток распределяли между этилацетатом (2 л) и водой (2 л) и водный слой экстрагировали этилацетатом (1 л). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали, концентрировали в вакууме и подвергали хроматографии на силикагеле (градиент: 0% до 10% этилацетата в петролейном эфире) с получением С47 в виде белого твердого вещества. Выход: 195 г, 0,472 моль, 53%. ЖХМС m/z 414,3 [М+Н]+. 1Н ЯМР (400 МГц, хлороформ-d) δ 7,38-7,21 (м, 5Н, предположительно; частично скрыт пиком растворителя), 7,17 (д, J=8,2 Гц, 2Н), 7,03 (д, J=7,2 Гц, 1Н), 6,77 (д, J=8,2 Гц, 2Н), 6,32 (д, J=7,3 Гц, 1Н), 5,07-4,92 (м, 2Н), 3,77 (с, 3Н), 3,54 (ушир. АВ квартет, JAB=13 Гц, ΔvAB=40 Гц, 2Н), 2,95 (д, J=10,2 Гц, 1Н), 2,92-2,83 (м, 1Н), 2,83-2,73 (м, 1Н), 2,73-2,63 (м, 1Н), 2,43-2,31 (м, 1Н), 2,29-2,08 (м, 2Н), 2,23 (с, 3Н), 2,03-1,73 (м, 3Н).
Стадия 9. Синтез 1'-бензил-1'-метил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин] (С48).
К 0°С раствору С47 (190 г, 0,459 моль) в дихлорметане (1,5 л) добавляли трифторуксусную кислоту (523 г, 4,59 моль) и реакционную смесь перемешивали при 25°С в течение 3 часов. Затем смесь концентрировали в вакууме; остаток разбавляли этилацетатом (1,5 л) и промывали насыщенным водным раствором карбоната натрия (1,0 л) и этот водный слой экстрагировали этилацетатом (2 × 300 мл). Объединенные органические слои концентрировали в вакууме и очищали хроматографией на силикагеле (градиент: 0% до 10% метанола в дихлорметане) с получением С48 в виде коричневого масла (17 9 г). Это вещество использовали непосредственно на следующей стадии. ЖХМС m/z 294, 3 [М+Н]+. 1Н ЯМР (400 МГц, хлороформ-d) δ 9,1-8,3 (ушир.с, 1Н), 7,41-7,35 (м, 2Н), 7,35-7,28 (м, 2Н), 7,28-7,22 (м, 2Н, предположительно; частично скрыт пиком растворителя), 6,35 (д, J=1,3 Гц, 1Н), 3,72 (с, 2Н), 2,96-2,85 (м, 1Н), 2,80-2,62 (м, 5Н), 2,42 (с, 3Н), 2,05 (ддд, J=13,1 8,1, 5,0 Гц, 1H), 1,98-1,81 (м, 3Н).
Стадия 10. Синтез трет-бутил-7-метил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-11-карбоксилата (Р17) и ди-трет-бутил-7'-метил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1,1'-дикарбоксилата (Р18).
Смесь С48 (с предыдущей стадии; 179 г, ≤0, 459 моль), ди-трет-бутилдикарбоната (199,7 г, 915 ммоль) и гидроксида палладия (17,9 г, 127 ммоль) в метаноле (2,0 л) и этилацетате (2,0 л) гидрировали при 55 ф/дюйм2 (3, 8 67 кг/см2) и 25°С в течение 18 часов. Реакционную смесь затем фильтровали через слой диатомовой земли и фильтрат концентрировали в вакууме. Хроматография на силикагеле (градиент: 0% до 50% дихлорметана в этилацетате) давала Р17 и Р18, оба в виде белого твердого вещества.
Р17 - Выход: 101 г, 0, 333 моль, 73% за 2 стадии. ЖХМС m/z 304, 3 [М+Н]+. 1Н ЯМР (400 МГц, метанол-d4) δ 7, 20 (д, J=7,3 Гц, 1Н), 6,44 (д, J=7,4 Гц, 1Н), 3,62-3,44 (м, 2Н), 3,37-3,3 (м, 2Н, предположительно; частично скрыт пиком растворителя), 2,84-2,66 (м, 2Н), 2,27 (с, 3Н), 2,05-1,92 (м, 2Н), 1,92-1,76 (м, 2Н), [1,48 (с) и 1,46 (с), всего 9Н].
Р18 - Выход: 21,3 г, 52,8 ммоль, 12% за 2 стадии. ЖХМС m/z 404, 3 [М+Н]+. 1Н ЯМР (400 МГц, метанол-d4) δ 7,49 (д, J=7,7 Гц, 1Н), 7,02 (ушир.д, J=7,7 Гц, 1Н), [3,85 (д, J=11,3 Гц) и 3,75 (д, J=11,2 Гц), всего 1Н), 3,62-3,47 (м, 2Н), 3,40-3,24 (м, 1Н, предположительно; частично скрыт пиком растворителя), 2,89-2,73 (м, 2Н), 2,54-2,27 (м, 1Н), 2,44 (с, 3Н), 2,13-1,82 (м, 3Н), 1,46 (с, 9Н), [1,43 (с) и 1,43 (с), всего 9Н].
Получения Р19 и Р20
трет-Бутил-(2S)-7-метил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилат (Р19) и трет-Бутил-(2R)-7-метил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилат (Р20)
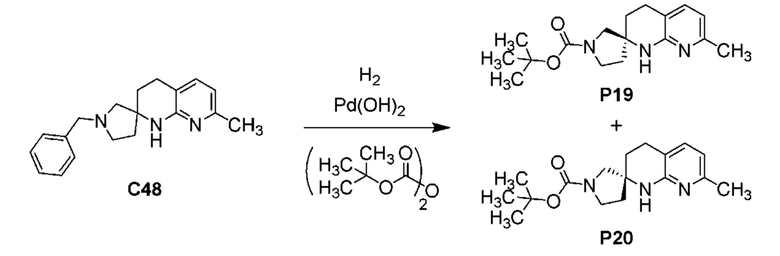
Ди-трет-бутилдикарбонат (3,97 г, 18,2 ммоль) добавляли к раствору С48 (4,45 г, 15,2 ммоль) в смеси метанола (20 мл) и этилацетата (25 мл). После добавления гидроксида палладия на углероде (900 мг) реакционную смесь гидрировали при 80 ф/дюйм2 (5,625 кг/см2) в течение 18 часов, после этого ЖХМС анализ показал полное преобразование в Р19/Р20: ЖХМС m/z 304,2 [М+Н]+. Реакционную смесь фильтровали и фильтрат концентрировали при пониженном давлении; остаток растворяли в этилацетате, промывали последовательно насыщенным раствором бикарбоната натрия и насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Разделение компонентов-энантиомеров осуществляли сверхкритической флюидной хроматографией {Колонка: Chiral Technologies Chiralpak IB, 30 × 250 мм, 5 мкм; Подвижная фаза 9:1 диоксид углерода / [этанол, содержащий 0,2% (7 М аммиака в метаноле)]; Скорость потока: 80 мл/мин; Обратное давление: 100 бар). Первый элюирующий энантиомер был указан как Р19, а второй элюирующий энантиомер как Р20. Оба выделяли в виде твердых веществ.
Р19 - Выход: 1,60 г, 5,27 ммоль, 35%. Время удерживания: 3,75 минут [Аналитические условия. Колонка: Chiral Technologies Chiralpak IB, 4, 6 × 250 мм, 5 мкм; Подвижная фаза А: диоксид углерода; Подвижная фаза В: этанол, содержащий 0,2% (7 М аммиака в метаноле); Градиент: 5% В в течение 1 минуты, затем 5% до 60% В в течение 8 минут; Скорость потока: 3,0 мл/мин; Обратное давление: 120 бар].
Р20 - Выход: 1,50 г, 4,94 ммоль, 32%. Время удерживания: 3,96 минут (Аналитические условия идентичны используемым для Р19).
Указанная абсолютная стереохимия была присвоена на основании преобразования этой партии Р19 в Р23 в Альтернативном Получении (#1) Р23 ниже. Абсолютная конфигурация Р23 была установлена при его использовании в синтезе 14, который анализировали при помощи рентгеноструктурного анализа монокристаллов (см. ниже).
Получение Р21
Ди-трет-бутил-6-бром-7-метил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1,1'-дикарбоксилат (Р21)
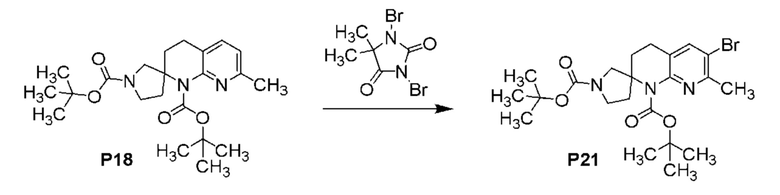
К 0°С раствору Р18 (20 г, 50 ммоль) в дихлорметане (200 мл) добавляли 1,3-дибром-5,5-диметилимидазолидин-2,4-дион (7,09 г, 2 4,8 ммоль) шестью порциями в течение 3 0 минут. Реакционную смесь перемешивали при 0°С в течение 1 часа, затем смесь обрабатывали насыщенным водным раствором сульфита натрия (2 00 мл) и экстрагировали дихлорметаном (3 × 100 мл). Объединенные органические слои промывали насыщенным водным раствором бикарбоната натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (градиент: 0% до 40% этилацетата в петролейном эфире) давала Р21 в виде белого твердого вещества. Выход: 22,8 г, 47,2 ммоль, 94%. ЖХМС m/z 384,1 (наблюдаемый изотопный паттерн брома) {[М-(2-метилпроп-1-ен и CO2)]+Н}+. 1Н ЯМР (400 МГц, хлороформ-d) δ 7,51 (ушир.с, 1Н), [3,89 (д, J=11,0 Гц) и 3,73 (д, J=11,0 Гц), всего 1Н], 3,65-3,51 (м, 1Н), 3,46 (д, J=11,0 Гц, 1Н), 3,38-3,26 (м, 1Н), [2,87-2,56 (м) и 2,15-1,70 (м), всего 6Н], 2,57 (с, 3Н), [1,46 (с) и 1,45 (с), всего 18Н].
Получение Р22
трет-Бутил-6-бром-7-метил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилат (Р22)
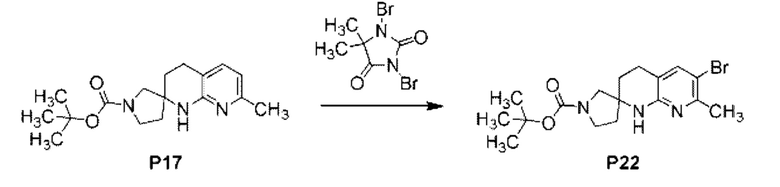
1,3-Дибром-5,5-диметилимидазолидин-2,4-дион (2,47 г, 8,64 ммоль) добавляли по порциям в течение 2 0 минут к 0°С раствору Р17 (5,25 г, 17,3 ммоль) в дихлорметане (69 мл). После перемешивания реакционной смеси при 0°С в течение 4 5 минут ЖХМС анализ показал преобразование в Р22: ЖХМС m/z 384,3 (наблюдаемый изотопный паттерн брома) [М+Н]+. Через 1 час при 0°С реакционную смесь обрабатывали насыщенным водным раствором сульфита натрия (100 мл) и смесь экстрагировали дихлорметаном. Органический слой промывали последовательно насыщенным водным раствором бикарбоната натрия и насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и концентрировали в вакууме с получением Р22 в виде твердого вещества. Выход: 6,60 г, 17,3 ммоль, количественный. 1H ЯМР (400 МГц, метанол-d4) δ 7,40 (ушир.с, 1Н), 3,61-3,43 (м, 2Н), 3,37-3,3 (м, 2Н, предположительно; почти полностью скрыт пиком воды), 2,85-2,67 (м, 2Н), 2,37 (с, 3Н), 2,06-1,92 (м, 2Н), 1,92-1,75 (м, 2Н), [1,47 (с) и 1,46 (с), всего 9Н].
Получения Р23 и Р24
трет-Бутил-(2S)-6-бром-1-метил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилат (Р23) и трет-бутил-(2R)-6-бром-7-метил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилат (Р24)
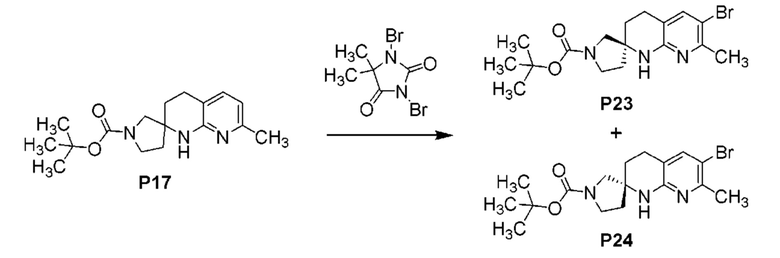
1,3-Дибром-5,5-диметилимидазолидин-2,4-дион (5,65 г, 19,8 ммоль) добавляли по порциям к 0°С раствору Р17 (10,0 г, 32,9 ммоль) в дихлорметане (150 мл) и реакционную смесь перемешивали при 0°С-5°С в течение 1 часа, после этого ЖХМС анализ показал, что произошло бромирование: ЖХМС m/z 382,3 [М+Н]+. Добавляли насыщенный водный раствор сульфита натрия (20 мл) с последующим добавлением воды (50 мл); полученный водный слой экстрагировали дихлорметаном (2 × 50 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и концентрировали в вакууме; хроматография на силикагеле (градиент: 0% до 100% этилацетата в гептане) давала рацемическую смесь Р23 и Р24 в виде светло-желтого пенистого вещества (11,8 г). Это вещество объединяли с продуктом подобной реакции, которую осуществляли с использованием Р17 (7,40 г, 24,4 ммоль), с получением светло-желтого пенистого вещества (20,9 г, 54,6 ммоль, объединенный выход 95%), и его разделяли на составляющие его энантиомеры сверхкритической флюидной хроматографией [Колонка: Chiral Technologies Chiralcel OJ, 50 × 250 мм, 5 мкм; Подвижная фаза 4:1 диоксид углерода/ (1:1 метанол/ацетонитрил); Скорость потока: 250 мл/мин; Обратное давление: 120 бар]. Первый элюирующий энантиомер был указан как Р23, а второй элюирующий энантиомер был указан как Р24.
Указанная абсолютная стереохимия была присвоена на основании преобразования этой партии Р23 в Р28 (см. Получение Р28) и затем в Пример 14; абсолютная стереохими 14 была установлена методом рентгеновской дифракции монокристалла (см. ниже).
Р23, выделенный в виде желтого масла, которое отверждалось при выстаивании - Объединенный выход: 9,37 г, 24,5 ммоль, 43%. 1Н ЯМР (400 МГц, DMSO-d6) δ 7,37 (с, 1Н), 7,02-6,96 (м, 1Н), [3,55-3,40 (м), 3,36-3,26 (м, предположительно; частично скрыт пиком воды) и 3,24-3,13 (м), всего 4Н], 2,75-2,55 (м, 2Н), 2,31 (с, 3Н), 1,95-1,78 (м, 2Н), 1,76-1,60 (м, 2Н), [1,40 (с) и 1,38 (с), всего 9Н]. Время удерживания: 4,01 мин [Аналитические условия. Колонка: Chiral Technologies Chiralcel OJ-H, 4, 6 × 250 мм, 5 мкм; Подвижная фаза А: диоксид углерода; Подвижная фаза В: метанол, содержащий 0,2% (7 М аммиака в метаноле); Градиент: 5% В в течение 1 минуты, затем 5% до 60% В в течение 8 минут; Скорость потока: 3,0 мл/мин; Обратное давление: 120 бар].
Р24 - Объединенный выход: 11,8 г, который содержал этанол; скорректированная оценка: 28,4 ммоль, 50%. 1Н ЯМР (400 МГц, DMSO-d6), характеристические пики: δ 7,37 (с, 1Н), 7,01-6,96 (м, 1Н), 2,75-2,55 (м, 2Н), 2,31 (с, 3Н), 1,95-1,78 (м, 2Н), 1,76-1,60 (м, 2Н), [1,40 (с) и 1,38 (с), всего 9Н]. Время удерживания: 4,32 мин (Аналитические условия идентичны используемым для Р23).
Альтернативное Получение (#1) Р23
трет-Бутил-(2S)-6-бром-7-метил-3б4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилат (Р23)
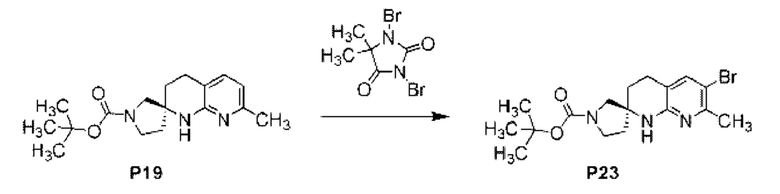
1,3-Дибром-5,5-диметилимидазолидин-2,4-дион (625 мг, 2,19 ммоль) добавляли по порциям к 0°С раствору Р19 (вещество из Получения Р19 и Р20; 1,10 г, 3,63 ммоль) в дихлорметане (20 мл). После перемешивания реакционной смеси при комнатной температуре в течение 1 часа ЖХМС анализ показал преобразование в Р23: ЖХМС m/z 384,2 (наблюдаемый изотопный паттерн брома) [М+Н]+. Затем добавляли насыщенный водный раствор сульфита натрия и полученную смесь экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали, концентрировали в вакууме и очищали хроматографией на силикагеле (градиент: 10% до 40% этилацетата в гептане) с получением Р23 в виде белого твердого вещества. Выход: 1,25 г, 3,27 ммоль, 90%. 1Н ЯМР (400 МГц, хлороформ-d) δ 7,33 (с, 1Н), 5,15-5,01 (ушир.с, 1Н), 3,59-3,45 (м, 2Н), 3,43-3,25 (м, 2Н), 2,81-2,66 (м, 2Н), 2,43 (с, 3Н), 2,01-1,74 (м, 4Н), 1,48-1,43 (ушир.с, 9Н).
Абсолютная стереохимия этого образца Р23 была присвоена как указано путем сравнения с образцами их Получения Р23 и Р24:
Время удерживания Р23 из Альтернативного Получения (#1) Р23: 4,08 мин
Время удерживания рацемической смеси Р23 и Р24: 4,07 и 4,36 мин.
Время удерживания Р23 из Получения Р23 и Р24: 4,01 мин
Время удерживания Р24 из Получения Р23 и Р24: 4,32 мин
Эти четыре анализа осуществляли с использованием одного и того же аналитического способа: [Колонка: Chiral Technologies Chiralcel OJ-H, 4,6 × 100 мм, 5 мкм; Подвижная фаза А: диоксид углерода; Подвижная фаза В: метанол, содержащий 0,2% (7 М аммиака в метаноле); Градиент: 5% В в течение 1 минуты, затем 5% до 60% В в течение 8 минут; Скорость потока: 3,0 мл/мин].
Альтернативное Получение (#2) Р23
трет-Бутил-(2S)-6-бром-7-метил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилат (Р23)
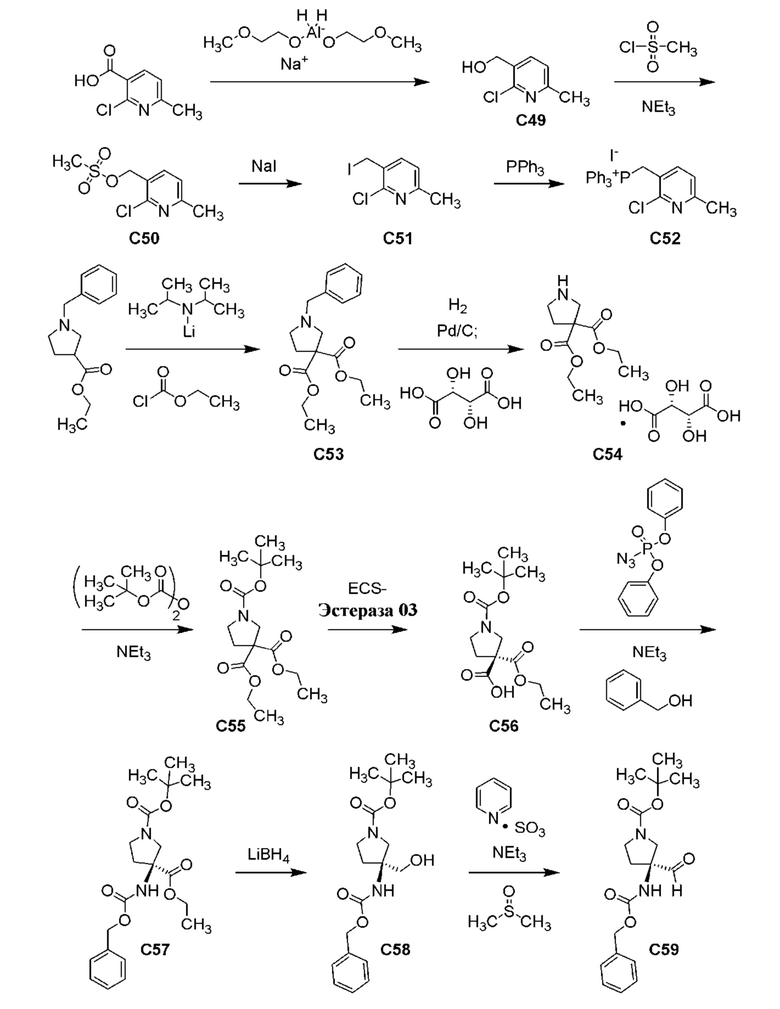
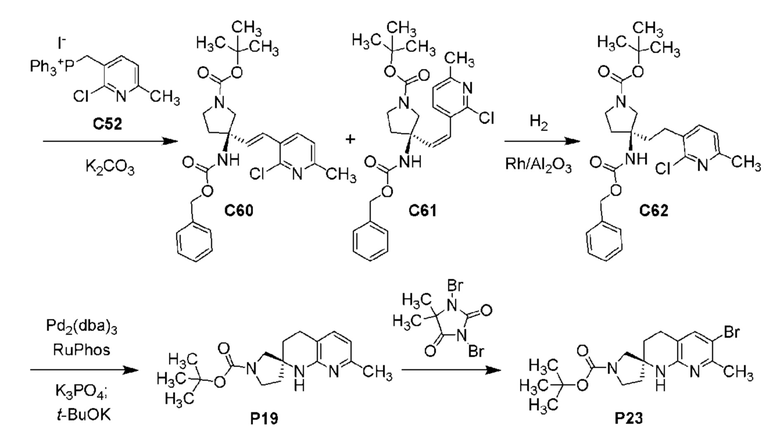
Стадия 1. Синтез (2-хлор-6-метилпиридин-3-ил)метанола (С49).
Раствор натрий бис(2-метоксиэтокси)алюминийгидрида (70%; 1,05 кг, 2,5 экв.) добавляли к раствору, при от -5°С до 5°С, 2-хлор-6-метилпиридин-3-карбоновой кислоты (250 г, 1,4 6 моль) в толуоле (2,5 л). После перемешивания реакционной смеси при температуре от -5°С до 5°С в течение 19 часов смесь обрабатывали раствором гидроксида натрия (145,7 г, 3,642 моль, 2,50 экв.) в воде (1,25 л), поддерживая внутреннюю температуру ниже от 0°С до 10°С. Полученную смесь затем нагревали до 25°С; через 15 минут водный слой экстрагировали пропан-2-илацетатом (2 × 1,25 л). Эти два экстракта объединяли с толуольным слоем и фильтровали через силикагель (125 г). Фильтровальную лепешку промывали пропан-2-илацетатом (125 мл) и объединенные фильтраты концентрировали до 8 объемов при температуре 40°С-45°С с получением С49 в виде раствора в толуоле (1,602 кг, 11,2% С49 по массе); основной объем этого раствора использовали на следующей стадии. Расчетный выход: 179,4 г, 1,138 моль, 78%. 1Н ЯМР (400 МГц, DMSO-d6) δ 7,81 (д, J=7,8 Гц, 1Н), 7,29 (д, J=7,7 Гц, 1Н), 5,48 (т, J=5,6 Гц, 1Н), 4,50 (д, J=5,6 Гц, 2Н), 2,43 (с, 3Н).
Стадия 2. Синтез (2-хлор-6-метилпиридин-3-ил)метилметансульфоната (С50).
Триэтиламин (134,2 г, 1,32 6 моль) добавляли к раствору С49 в толуоле (с предыдущей стадии; 1,537 кг, содержащий 11,2% С49, 172,1 г, 1,09 моль). Раствор охлаждали до температуры от -5°С до 5°С и затем обрабатывали по каплям метансульфонилхлоридом (128,5 г, 1,122 моль), поддерживая внутреннюю температуру при -5°С-5°С. После перемешивания реакционной смеси при этой температуре в течение 2 часов снова добавляли триэтиламин (22,7 г, 0,224 моль) с последующим добавлением по каплям метансульфонилхлорида (25,7 г, 0, 224 моль). Перемешивание продолжали при -5°С-5°С в течение 1 часа, затем реакционную смесь обрабатывали водой (805 мл), поддерживая внутреннюю температуру ниже 25°С, и затем перемешивали в течение 15 минут при 25°С. Органический слой промывали водой (805 мл) и концентрировали с получением С50 в виде раствора в толуоле (861 г). Этот раствор использовали непосредственно на следующей стадии. 1H ЯМР (400 МГц, DMSO-d6) δ 7,92 (д, J=7,7 Гц, 1Н), 7,36 (д, J=1,7 Гц, 1Н), 5,30 (с, 2Н), 3,29 (с, 3Н), 2,48 (с, 3Н).
Стадия 3. Синтез 2-хлор-3-(подметил)-6-метилпиридина (С51).
Иодид натрия (230 г, 1,53 моль) растворяли в ацетоне (1,13 кг) при 25°С; к этому раствору добавляли раствор С50 в толуоле (с предыдущей стадии; 861 г, ≤1,09 моль С50). После перемешивания реакционной смеси при 25°С в течение 1 часа добавляли раствор метабисульфита натрия (57,86 г, 0,3044 моль) в воде (1,45 л) и перемешивание продолжали в течение 30 минут. Органический слой отделяли, разбавляли толуолом (417 мл) и концентрировали до 5 объемов с получением С51 в виде раствора в толуоле (1,110 кг, 22,93% С51 по массе). Этот раствор использовали непосредственно на следующей стадии. Расчетный выход: 254,5 г, 0,9514 моль, 87% за 2 стадии. 1Н ЯМР (400 МГц, DMSO-d6) δ 7,90 (д, J=7,7 Гц, 1Н), 7,26 (д, J=7, 8 Гц, 1Н), 4,55 (с, 2Н), 2,41 (с, 3Н).
Стадия 4. Синтез [(2-хлор-6-метилпиридин-3-ил)метил](трифенил)фосфоний иодида (С52).
Раствор С51 в толуоле (с предыдущей стадии; 1,110 кг, 22,93% С51 по массе, 254,5 г, 0,9514 моль) разбавляли ацетонитрилом (1,29 л) и обрабатывали трифенилфосфином (262 г, 0,999 моль). После перемешивания реакционной смеси в течение 4 часов при 25°С смесь охлаждали до 10°С, перемешивали при этой температуре в течение 16 часов и фильтровали. Фильтровальную лепешку промывали толуолом (255 мл) и сушили при 45°С в течение 4 часов с получением С52 в виде твердого вещества. Выход: 412,6 г, 0,7788 моль, 56% за 4 стадии. Чистота: 99,7% по данным ВЭЖХ. 1Н ЯМР (400 МГц, DMSO-d6) δ 7,97-7,90 (м, 3Н), 7,80-7,71 (м, 8Н), 7,71-7,66 (м, 4Н), 7,44 (дд, J=1,8, 2,4 Гц, 1Н), 7,20 (д, J=1,8 Гц, 1Н), 5,15 (д, JHP=15,0 Гц, 2Н), 2,40 (д, J=2,4 Гц, 3Н).
Стадия 5. Синтез диэтил-1-бензилпирролидин-3,3-дикарбоксилата (С53).
Раствор этил-1-бензилпирролидин-3-карбоксилата (700 г, 3,00 моль) в тетрагидрофуране (4,20 л) добавляли по каплям в течение 5 часов к раствору, при температуре от -80°С до -70°С, диизопропиламида лития (2,0 М, 2,40 л, 4,80 моль). Перемешивание продолжали при температуре от -80°С до -70°С в течение 2 часов, затем добавляли этилхлорформиат (423,5 г, 3,90 моль) в течение 3 часов, поддерживая температуру реакции от -80°С до -70°С. После перемешивания реакционной смеси в течение 2 часов при температуре от -80°С до -70°С температуру доводили до температуры от -50°С до -40°С и реакционную смесь гасили добавлением раствора уксусной кислоты (288 г, 4,80 моль) в тетрагидрофуране (1,40 л), поддерживая температуру от -50°С до -40°С. Полученную смесь нагревали до 15°С-25°С и распределяли между водой (3,50 л) и 2-метилтетрагидрофураном (7,0 л). После перемешивания смеси в течение 30 минут при 15°С-25°С, водный слой экстрагировали 2-метилтетрагидрофураном (7,0 л) и объединенные органические слои промывали раствором уксусной кислоты (288 г, 4,80 моль) в воде (4,2 л), а затем водным раствором сульфата натрия (10%; 2 × 3,50 кг). Органические слои концентрировали в вакууме ло 2-3 объемов, поддерживая температуру ниже 50°С. Добавляли этанол (4,90 л, 7 объемов) и раствор снова концентрировали в вакууме до 2-3 объемов, поддерживая температуру ниже 50°С. Это добавление этанола/концентрирование осуществляли в общей сложности три раза, используя для конечного цикла 2,80 л этанола, с последующим концентрированием до 4-5 объемов. Это давало С53 в виде раствора в этаноле (3,148 кг, 24, 23% С53 по массе). Часть этого раствора использовали на следующей стадии. Расчетный выход: 762, 8 г, 2,498 моль, 83%. 1Н ЯМР (400 МГц, DMSO-d6) δ 7,35-7,20 (м, 5Н), 4,12 (кв., J=7,1 Гц, 4Н), 3,57 (с, 2Н), 2,90 (с, 2Н), 2,55 (т, J=6,9 Гц, 2Н), 2,29 (т, J=6,8 Гц, 2Н), 1,14 (т, J=1,1 Гц, 6Н).
Стадия 6. Синтез диэтилпирролидин-3,3-дикарбоксилата, L-тартратной соли (С54).
Этанол (720 мл, 6 объемов) добавляли к раствору С53 (120 г, 0,393 моль) в этаноле (с предыдущей стадии; приблизительно 500 мл). После добавления влажного палладия на углероде (10%; 12 г) реакционный сосуд вакуумировали и загружали аргоном три раза, а затем вакуумировали и загружали водородом три раза. Затем осуществляли гидрирование при 40-50 ф/дюйм2 (2,812-3,515 кг/см2) и 40°С-50°С в течение 24 часов. Полученную смесь фильтровали через диатомовую землю (50 г); фильтровальную лепешку промывали этанолом (240 мл, 2 объемов) и объединенные фильтраты концентрировали в вакууме до 2,5-3,5 объемов, поддерживая температуру при 45°С или ниже. Этот раствор добавляли, в течение 2 часов, к 40°С-50°С раствору L-винной кислоты (16,1 г, 0,511 моль) в воде (85 мл, 0,7 объемов) и этанола (465 мл). После перемешивания смеси при 4 0°С-50°С в течение 1 часа добавляли затравку С54 (0,4 г; см. ниже) при 45°С. Смесь охлаждали до 10°С в течение 6 часов и затем перемешивали при 10°С в течение 4 часов; фильтрование давало фильтровальную лепешку, которую промывали этанолом (2 объема) и сушили при 40°С в течение 20 часов с получением С54 в виде твердого вещества. Выход: 127,4 г, 0,3487 моль, 89%. ВЭЖХ чистота: 99,1%. 1Н ЯМР (400 МГц, DMSO-d6) δ 4,16 (кв., J=7,1 Гц, 4Н), 4,03 (с, 2Н), 3,49 (с, 2Н), 3,08 (т, J=7,1 Гц, 2Н), 2,32 (т, J=7,1 Гц, 2Н), 1,18 (т, J=7,1 Гц, 6Н).
Затравочный материал, используемый выше, получали из другого цикла этого же синтеза С54, в котором твердое вещество С54 образовывалось непосредственно при охлаждении.
Стадия 7. Синтез 1-трет-бутил-3,3-диэтилпирролидин-1,3,3-трикарбоксилата (С55).
Ди-трет-бутилдикарбонат (19,7 г, 90,3 ммоль) добавляли по каплям к 20°С-30°С смеси С54 (88,12 г, 0,2412 моль) и триэтиламина (73,33 г, 0,7247 моль) в дихлорметане (881 мл, 10 объемов). Добавляли дополнительное количество ди-трет-бутилдикарбоната (19,2 г, 88,0 ммоль и 19,3 г, 88,4 ммоль) по каплям, периодически осуществляя ВЭЖХ-анализ. После перемешивания реакционной смеси при 20°С-30°С в течение 18 часов рН доводили до 7 добавлением хлористоводородной кислоты (1 М; 309 г) и перемешивание продолжали в течение 15 минут. Органический слой перемешивали с водным раствором сульфата натрия (10%; 485,30 г) при 20°С-30°С в течение 15 минут и затем органический слой концентрировали в вакууме до 1-2 объемов поддерживая температуру ниже 40°С. Добавляли диметилсульфоксид (71,7 г) с получением С55 в виде раствора в диметилсульфоксиде (154,2 г, 48,9% С55 по массе). Основной объем этого вещества использовали на следующей стадии. Расчетный выход: 75,4 г, 0,239 моль, 99%. 1Н ЯМР (400 МГц, DMSO-d6) δ 4,16 (кв., J=1,1 Гц, 4Н), 3,67 (ушир.с, 2Н), 3,34-3,26 (м, 2Н), 2,37-2,28 (м, 2Н), 1,39 (с, 9Н), 1,17 (ушир.т, J=1,1 Гц, 6Н).
Стадия 8. Синтез (3PJ -1- (трет-бутоксикарбонил)-3-(этоксикарбонил)пирролидин-3-карбоновой кислоты (С56).
ECS-Эстераза 03 фермент [Bacillus stearothermophilus, рекомбинантный из Escherichia coli, (ЕС 3.1.1.1); 0, 540 г] добавляли в фосфатный буфер (0,1 М; рН=6,92, 580 мл, 8,2 объемов) при 20°С-30°С. Добавляли раствор С55 (72,2 г, 0,229 моль) в диметилсульфоксиде (с предыдущей стадии; приблизительно 148 г); дополнительный диметилсульфоксид (9 мл) использовали для промывки первоначального сосуда и его также добавляли к реакционной смеси. Исходное значение рН реакции было 7,08; после перемешивания при 20°С-30°С в течение 1 часа рН снижался до 6,58. Автотитратор рН использовали для поддержания рН при 7,5 добавлением водного раствора гидроксида натрия (2 М; 121 мл, 0,242 моль) в течение 24 часов. Добавляли хлористоводородную кислоту (6 М; 52 мл, 0,312 моль) с доведением рН до 2,39; затем добавляли этилацетат (435 мл, 6,0 объемов) и смесь перемешивали в течение 30 минут при 20°С-30°С. Фильтрование через диатомовую землю (18,0 г) давало фильтровальную лепешку, которую промывали этилацетатом (2 × 7 5 мл). Объединенные фильтраты перемешивали при 20°С-30°С в течение 30 минут и затем водный слой перемешивали с этилацетатом (217 мл, 3,0 объема) в течение 30 минут. Объединенные органические слои промывали два раза водой (360 мл, 5,0 объемов) при перемешивании в течение 30 минут. Полученный раствор концентрировали в вакууме до 1-2 объемов, поддерживая температуру ниже 40°С, затем разбавляли толуолом (360 мл); эту процедуру концентрирования/разбавления осуществляли в общей сложности три раза с получением С56 в виде раствора в толуоле (418,3 г, 15,67% С56 по массе). Расчетный выход: 65,6 г, 0, 228 моль, количественный. 1Н ЯМР (400 МГц, хлороформ-d) δ 7,99 (очень широкий с, 1Н), 4,22 (кв., J=1,1 Гц, 2Н), [3,88 (ушир. с) и 3,83 (ушир. с), всего 2Н), 3,51-3,38 (м, 2Н), 2,41 (т, J=7,1 Гц, 2Н), 1,44 (с, 9Н), 1,26 (ушир.т, J=1 Гц, 3Н).
Стадия 9. Синтез 1-трет-бутил-3-этил-(3S)-3-{[(бензилокси)карбонил]амино}пирролидин-1,3-дикарбоксилата (С57).
Толуол (17 0 мл, 1,2 объемов) добавляли к раствору С56 в толуоле (3,8 объемов, содержащий 2 8,9% по массе С56, 14 6,4 г, 0,5096 моль); раствор нагревали до 80°С-90°С. К этому медленно добавляли, в течение 2 часов, смесь триэтиламина (77,4 г, 0,765 моль) и дифенилфосфоразидата (140,3 г, 0,5098 моль) в толуоле (732 мл, 5 объемов). Реакционную смесь перемешивали при 80°С-90°С в течение 3 часов, затем смесь охлаждали до 50°С и обрабатывали по каплям, в течение 2 часов, раствором бензилового спирта (55,12 г, 0,5097 моль) в толуоле (290 мл, 2 объема). После перемешивания реакционной смеси при 100°С в течение 16 часов смесь охлаждали до 15°С-25°С и распределяли между толуолом (1,46 л, 10 объемов) и водой (2,20 л, 15 объемов) при перемешивании в течение 30 минут. Органический слой промывали последовательно водным раствором карбоната калия (10%; 3 × 1,46 л) и водой (2 × 750 мл). Затем смесь концентрировали в вакууме до 1-2 объемов, поддерживая температуру ниже 50°С, и разбавляли тетрагидрофураном (1,0 л); эту процедуру концентрирования/разбавления осуществляли в общей сложности три раза, затем смесь концентрировали в вакууме до 4-5 объемов, поддерживая температуру ниже 50°С. Это давало С57 в виде раствора в тетрагидрофуране (595,8 г, 19,14% С57 по массе). Расчетный выход: 114 г, 0, 290 моль, 57%. 1Н ЯМР (400 МГц, хлороформ-d) δ 7,40-7,28 (м, 5Н), 5,25 (очень широкий с, 1Н), 5,10 (ушир.с, 2Н), 4,28-4,12 (м, 2Н), 3,91-3,76 (м, 1Н), 3,71-3,38 (м, 3Н), 2,54-2,15 (м, 2Н), 1,45 (с, 9Н), 1,27-1,16 (м, 3Н).
Стадия 10. Синтез трет-бутил-(3S)-3-{[(бензилокси)карбонил]амино}-3-(гидроксиметил)пирролидин-1-карбоксилата (С58).
Раствор борогидрида лития в тетрагидрофуране (2 М; 511 мл, 1,02 моль) добавляли в течение 2 часов к 0°С-10°С раствору С57 в тетрагидрофуране (835,6 г, содержащий 19,20% С57 по массе, 160,4 г, 0,4 087 моль). После перемешивания реакционной смеси при 0°С-10°С в течение 15 часов смесь охлаждали до температуры от -5°С до 5°С и обрабатывали по каплям хлористоводородной кислотой (0,5 М; 2,08 л, 1,04 моль, 13 объемов) до рН 7. Смесь затем нагревали до 20°С-30°С, разбавляли этилацетатом (1,60 л, 10 объемов) и перемешивали в течение 10 минут, затем органический слой концентрировали в вакууме до 2-3 объемов, поддерживая температуру при 50°С или ниже. Полученную смесь разбавляли ацетонитрилом (880 мл) и концентрировали в вакууме до 2-3 объемов, поддерживая температуру при 50°С или ниже; эту процедуру разбавления/концентрирования осуществляли в общей сложности три раза. Смесь затем нагревали до 40°С-50°С и перемешивали в течение 1 часа, затем смесь охлаждали в течение 4 часов до 15°С-25°С. Добавляли воду (164 мл) по каплям в течение 2 часов при 15°С-25°С и смесь перемешивали при 15°С-25°С в течение 12 часов. Полученное твердое вещество собирали фильтрованием и сушили в вакууме в течение 4 0 часов при температуре при 50°С или ниже с получением С58 в виде твердого вещества. Выход: 123,2 г, 0,3516 моль, 86%. ВЭЖХ чистота: 99,8%. 1Н ЯМР (400 МГц, DMSO-d6) δ 7,40-7,27 (м, 5Н), 4,99 (с, 2Н), 4,93 (т, J=5,8 Гц, 1Н), 3,58-3,45 (м, 3Н), 3,31-3,21 (м, 3Н), 2,10-1,85 (м, 2Н), 1,38 (с, 9Н).
Стадия 11. Синтез трет-бутил-(3S)-3-{[(бензилокси)карбонил]амино}-3-формилпирролидин-1-карбоксила та (С59).
Раствор С58 (125 г, 0,357 моль) и диметилсульфоксида (144,5 г, 1,84 9 моль) в дихлорметане (2,02 л) перемешивали в течение 2 часов при 35°С-45°С; анализ по методу Карла Фишера показал содержание воды 0,029%. Раствор концентрировали в вакууме до 3-4 объемов при 35°С-45°С и затем разбавляли дихлорметаном (1,80 л). Еще один анализ по методу Карла Фишера показал содержание воды 0,034%. Раствор концентрировали в вакууме при 35°С-45°С до 6-7 объемов, затем добавляли триэтиламин (112,3 г, 1,110 моль) при 20°С-30°С и реакционную смесь охлаждали до температуры от -5°С до 0°С и перемешивали при этой температуре в течение 15 минут. Добавляли комплекс триоксида серы с пиридином (141,3 г, 0,8878 моль) по порциям в течение 2 часов; перемешивание затем продолжали при температуре от -5°С до 0°С в течение 16 часов, после этого реакционную смесь нагревали до 35°С-45°С и концентрировали до 2-3 объемов. Смесь охлаждали до 20°С-30°С, затем распределяли между этилацетатом (945 мл) и водой (675 мл) и водный слой экстрагировали этилацетатом (675 мл). Объединенные органические слои промывали последователвно хлористоводородной кислотой (1 М; 675 мл), водой (675 мл) и насыщенным водным раствором бикарбоната натрия (675 мл), затем концентрировали досуха при 30°С-40°С с получением С59 в виде масла. Выход: 118,7 г, 0, 3407 моль, 95%. ВЭЖХ чистота: 91,2%. 1Н ЯМР (400 МГц, хлороформ-d) δ 9,59 (с, 1Н), 7,42-7,29 (м, 5Н), 5,39 (ушир.с, 1Н), 5,12 (с, 2H), 3,85-3,70 (м, 1Н), 3,63-3,43 (м, 3Н), 2,44-2,04 (м, 2Н), 1,45 (с, 9Н).
Стадия 12. Синтез трет-бутил-(3R)-3-{[(бензилокси)карбонил]амино}-3-[(Е)-2-(2-хлор-6-метилпиридин-3-ил)этенил]пирролидин-1-карбоксилата (С60) и трет-бутил-(3R)-3-{[(бензилокси)карбонил]амино}-3-[(Z)-2-(2-хлор-6-метилпиридин-3-ил)этенил]пирролидин-1-карбоксилата (С61).
Смесь С59 (237,1 г, 0,6805 моль) и С52 (393, 6 г, 0,7429 моль) в диметилсульфоксиде (2,40 л, 10 объемов) обрабатывали карбонатом калия (188,7 г, 1,365 моль) и нагревали при 60°С в течение 2 часов. Затем добавляли пропан-2-илацетат (1,54 л, 6,5 объемов), воду (6,40 л, 27 объемов) и водный раствор сульфата натрия (10%; 710 мл, 3,0 объема) и смесь перемешивали в течение 20 минут при 25°С. Органический слой промывали три раза водным раствором сульфата натрия (10%; 1,30 л, 5,5 объемов) при перемешивании каждой смеси в течение 20 минут перед разделением. Затем смесь промывали водным раствором бикарбоната натрия (7%; 1,30 л, 5,5 объемов) таким же образом и концентрировали в вакууме до 1-2 объемов при температуре 50°С или ниже. Добавляли пропан-2-илацетат (1,06 л) и смесь концентрировали в вакууме до 1-2 объемов при температуре 50°С или ниже. Добавляли пропан-2-илацетат (480 мл) с последующим добавлением по каплям метилциклогексана (1,66 л) при 20°С-30°С. Полученную смесь перемешивали при 20°С-30°С в течение 1 часа, затем смесь охлаждали до температуры от -15°С до -5°С и перемешивали при этой температуре в течение 16 часов. Фильтрование суспензии осуществляли при -15°С до -5°С и фильтровальную лепешку промывали смесью пропан-2-илацетата и метилциклогексана (соотношение 3:7, 710 мл) при температуре от -15°С до -5°С. Объединенные фильтраты концентрировали в вакууме, разбавляли метилциклогексаном (20 объемов) и подвергали хроматографии на силикагеле (градиент: 14% до 25% этилацетата в метилциклогексане) с получением смеси С60 и С61 в виде масла. Это вещество, как считали на основании 1Н ЯМР анализа, состояло из 3-4 изомеров/ротамеров. Выход: 268,1 г, 0,5680 моль, 83%. ВЭЖХ чистота: ≥99,7%. 1Н ЯМР (400 МГц, DMSO-d6), характеристические пики: δ [8,00 (д, J=1,9 Гц) и 7,59 (д, J=1,6 Гц), всего 1Н], [7,85 (с) и 7,41 (с), всего 1Н], [7,38-7,25 (м), 7,22 (ушир. д, J=7,2 Гц) и 7,16 (д, J=1,1 Гц), всего 6Н], [6,62 (д, компонент АВ квартета, J=16,1 Гц) и 6,34 (д, J=12,3 Гц), всего 1Н], [6,49 (ушир. д, компонент АВ квартета, J=16,0 Гц), 5,88 (д, J=12,3 Гц) и 5,87 (д, J=12,3 Гц), всего 1Н], [5,04 (АВ квартет, JAB=12,7 Гц, ΔνAB=16,4 Гц), 4,74 (д, компонент АВ квартета, J=12,4 Гц) и 4,70-4,62 (м), всего 2Н], 3,83-3,68 (м, 1Н), 3,32-3,16 (м, 3Н), [2,43 (с) и 2,36 (с), всего 3Н], 2,27-2,12 (м, 1Н), 2,00-1,85 (м, 1Н), [1,40 (с), 1,39 (с), 1,37 (с) и 1,34 (с), всего 9Н].
Стадия 13. Синтез трет-бутил-(3S)-3-{[(бензилокси)карбонил]амино}-3-[2-(2-хлор-6-метилпиридин-3-ил)этил]пирролидин-1-карбоксилата (С62).
Реакционный сосуд, содержащий смесь С60 и С61 (283,0 г, 0,5996 моль) и родия на оксиде алюминия (5%; 14,15 г) в метаноле (1,98 л), вакуумировали и загружали аргоном три раза, затем вакуумировали и загружали водородом три раза. Затем осуществляли гидрирование в течение 40 часов при 30-40 ф/дюйм2 (2,109-2,812 кг/см2) и 20°С-25°С. Реакционную смесь фильтровали через диатомовую землю (424 г), фильтровальную лепешку промывали метанолом (5 объемов); объединенные фильтраты концентрировали в вакууме при 35°С-45°С. Полученное вещество обрабатывали толуолом (5 объемов) и концентрировали в вакууме при 50°С-60°С; эту процедуру добавление толуола/концентрирование осуществляли в общей сложности три раза с получением С62. Выход: 254,4 г, 0,5367 моль, 90%. ВЭЖХ чистота: 97,1%. 1H ЯМР (400 МГц, хлороформ-d) δ 7,41-7,28 (м, 6Н), 6,99 (ушир. д, J=7,6 Гц, 1Н), 5,06 (с, 2Н), 4,91-4,79 (м, 1Н), 3,62 (д, J=11,7 Гц, 1Н), 3,57-3,36 (м, 2Н), 3,36-3,26 (м, 1Н), 2,74-2,55 (м, 2Н), 2,48 (с, 3Н), [2,48-2,40 (м), 2,39-2,07 (м) и 2,05-1,82 (м), всего 4Н], 1,45 (ушир. с, 9Н).
Стадия 14. Синтез трет-бутил-(2S)-7-метил-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилата (Р19).
Раствор С62 в толуоле (947,73 г, содержащий 19% С62 по массе, 180 г, 0,380 моль) разбавляли толуолом (1,17 л, 6,5 объемов) и обрабатывали последовательно 2-дициклогексилфосфино-2',6'-диизопропоксибифенилом (RuPhos; 35,44 г, 75,95 ммоль) и фосфатом калия (145,1 г, 0,6836 моль). Полученную смесь продували три раза азотом, затем добавляли трис(дибензилиденацетон)дипалладий(0) (34,78 г, 37,98 ммоль) и осуществляли три дополнительных цикла продувки азотом. Реакционную смесь перемешивали в течение 24 часов при 75°С-85°С. Добавляли фосфат калия (16,2 г, 0,117 моль) и перемешивание продолжали при 75°С-85°С еще в течение 16 часов. Реакционную смесь охлаждали до 20°С-30°С, затем добавляли трет-бутоксид калия (76,7 г, 0,684 моль) и реакционную смесь перемешивали в течение 2 часов при 75°С-85°С. Затем смесь охлаждали и распределяли между водой (2,25 л) и этилацетатом (2,25 л); после перемешивания в течение 30 минут при 20°С-30°С смесь фильтровали через диатомовую землю (180 г) и фильтровальную лепешку промывали этилацетатом (1,80 л). Органический слой объединенных фильтратов промывали последовательно водой (2×2,25 л) и водным раствором сульфата натрия (10%; 2,25 л), затем экстрагировали три раза водным раствором лимонной кислоты (0,5 М; 1,072 кг, 1,4 экв.). Объединенные слои лимонной кислоты промывали этилацетатом (2×1,07 л), затем доводили до рН 7 добавлением водного раствора гидроксида натрия (30%; 596 г) при 20°С-30°С. Экстрагирование водного слоя этилацетатом (3×1,07 л) и последующее добавление комбинации этих трех органических слоев давали Р19 в виде раствора в этилацетате (3,218 кг, 2,7% Р19 по массе); основной объем этого вещества использовали на следующей стадии. Расчетный выход: 86,9 г, 0,286 моль, 75%. ВЭЖХ чистота: 98,9%. 1Н ЯМР (400 МГц, хлороформ-d) δ 7,11 (д, J=7,3 Гц, 1Н), 6,41 (д, J=7,4 Гц, 1Н), 4,90 (ушир. с, 1Н), 3,59-3,43 (м, 2Н), [3,40 (д, компонент АВ квартета, J=11,1 Гц) и 3,36-3,25 (м), всего 2Н], 2,80-2,65 (м, 2Н), 2,31 (с, 3Н), 2,00-1,75 (м, 4Н), [1,45 (с) и 1,44 (с), всего 9Н].
Стадия 15. Синтез трет-бутил-(2S)-6-бром-7-метил-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксила та(Р23).
1,3-Дибром-5,5-диметилимидазолидин-2,4-дион (45,60 г, 0,1595 моль) добавляли к 0°С-5°С раствору Р19 в этилацетате (с предыдущей стадии; 2986 г, содержащий 2,7% Р19, 80,6 г, 0,266 моль). После перемешивания реакционной смеси в течение 1 часа при 0°С-5°С смесь гасили добавлением водного раствора сульфита натрия (10%; 203 г) и воды (456 мл) и полученную смесь перемешивали при 10°С-20°С в течение 20 минут. Водный слой экстрагировали два раза этилацетатом (415 мл, 5,1 объемов) при перемешивании при 10°С-20°С в течение 20 минут; объединенные органические слои затем перемешивали в течение 20 минут с водным раствором сульфата натрия (10%; 456 г). Органический слой концентрировали до 1-2 объемов в вакууме при ниже 50°С, затем разбавляли метанолом (480 мл, 6 объемов). Эту процедуру концентрирования/разбавления осуществляли в общей сложности три раза и конечный раствор концентрировали в вакууме, при ниже 50°С, до 5-6 объемов. Полученный раствор охлаждали до 15°С-25°С и медленно добавляли воду (415 мл) в течение 2 часов при 15°С-25°С и затем перемешивали в течение 14 часов при 15°С-25°С. Фильтрование давало фильтровальную лепешку, которую промывали смесью метанола и воды (1:1, 2×200 мл), а затем сушили в вакууме при 45°С в течение 48 часов с получением Р23 в виде твердого вещества. Выход: 99,50 г, 0,2603 моль, 98%. ВЭЖХ чистота: 99,7%. ЖХМС m/z 384,1 (наблюдаемый изотопный паттерн брома) [М+Н]+. 1Н ЯМР (400 МГц, DMSO-d6) δ 7,37 (с, 1Н), 7,03-6,97 (м, 1Н), 3,55-3,43 (м, 1Н), 3,3-3,25 (м, 1Н, предположительно; частично скрыт пиком воды), 3,24-3,13 (м, 2Н), 2,75-2,55 (м, 2Н), 2,30 (с, 3Н), 1,95-1,77 (м, 2Н), 1,76-1,59 (м, 2Н), [1,40 (с) и 1,38 (с), всего 9Н].
Получение данных рентгеновской порошковой дифракции (PXRD) для кристаллического Р23
Образец Р23 [полученный, как описано на стадии 15 Альтернативного Получения (#2) выше] был представлен для порошкового рентгеностуктурного анализа (PXRD), и было обнаружено, что он представляет собой кристаллическое вещество.
Порошковый рентгеностуктурный анализ осуществляли с использованием дифрактометра Bruker AXS D8 Endeavour, оснащенного медным источником излучения (Cu). Щель расходимости устанавливали на постоянное освещение 15 мм. Дифрагированное излучение регистрировалось детектором PSD-Lynx Eye с щелью PSD детектора, установленной на 4,123 градуса. Напряжение и сила тока рентгеновской трубки были установлены на 40 кВ и 4 0 мА, соответственно. Кроме того, использовали энергодисперсионный детектор, никелевый фильтр. Данные собирали в гониометре Theta-Theta при длине волны Cu от 3,0 до 40,0 градусов 2-тета с использованием размера шага 0,0100 градуса и времени шага 1,0 секунды. Противорассеивающий экран был установлен на фиксированное расстояние 1,5 мм. Образцы подготавливали, помещая их в силиконовый держатель для образцов с низким фоном и вращая их со скоростью 15 об/мин во время сбора. Данные собирали с использованием программного обеспечения Bruker DIFFRAC Plus, а анализ осуществляли с использованием программного обеспечения EVA diffract plus. Файл данных PXRD не обрабатывали перед поиском пиков. Используя алгоритм поиска пиков в программном обеспечении EVA, пики, выбранные с пороговым значением 1, использовали для предварительного отнесения пиков. Для обеспечения достоверности корректировки делались вручную; результаты отнесений, выполненных автоматически, проверяли визуально, а положения пиков были скорректированы по максимуму пиков. Обычно выбирались пики с относительной интенсивностью ≥3%. Обычно не выбирали пики, которые не были разрешены или согласовывались с шумом. Типичная ошибка, связанная с положением пика из PXRD, как указано в USP, до +/-0,2° 2-тета (USP-941).
Один репрезентативный паттерн дифракции наблюдали для кристаллической формы Р23, и он представлен на Фиг. 3. Перечень дифракционных пиков в градусах 20 и относительных интенсивностей с относительной интенсивностью ≥3,0% PXRD от образца кристаллического Р23, показан в Таблице Х-Р23 ниже.
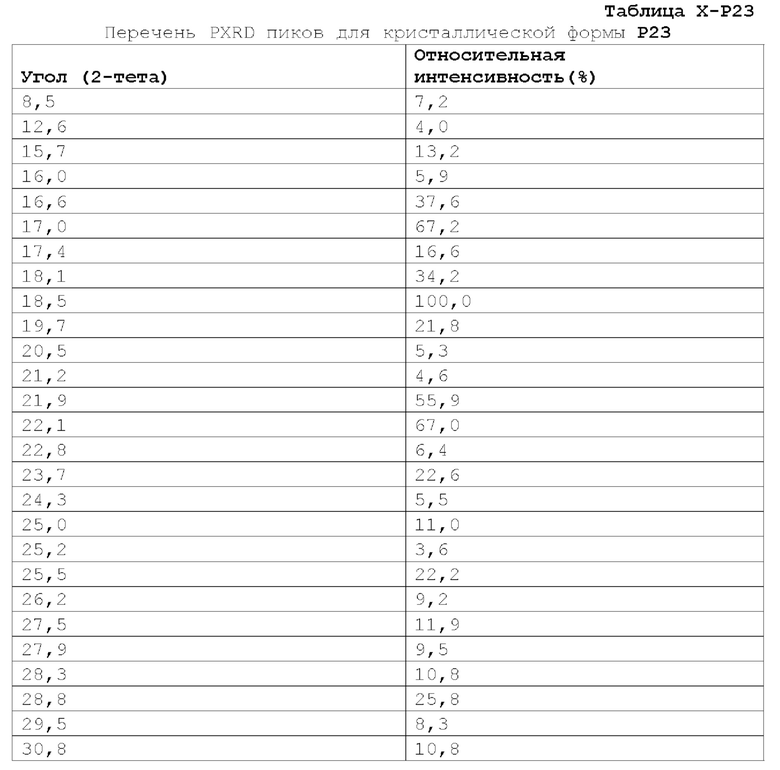
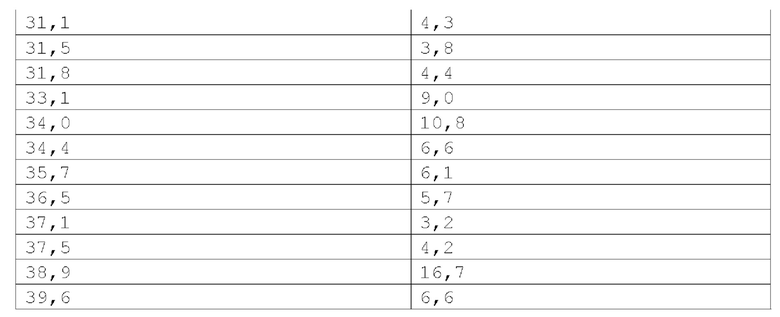
В некоторых вариантах осуществления настоящее изобретение предоставляет соединение, которое представляет собой трет-бутил-(2S)-6-бром-7-метил-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилат или его соль. В некоторых вариантах осуществления настоящее изобретение предоставляет соединение, которое представляет собой трет-бутил-(2S)-6-бром-7-метил-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилат. В некоторых других вариантах осуществления настоящее изобретение предоставляет кристаллическую форму трет-бутил-(2S)-6-бром-7-метил-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилата. В некоторых других вариантах осуществления кристаллическая форма трет-бутил-(2S)-6-бром-7-метил-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилата демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере один характеристический пик, выраженный в градусах 20, как показано в Таблице Х-Р23.
В некоторых вариантах осуществления кристаллическая форма трет-бутил-(2S)-6-бром-7-метил-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилата демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере два характеристических пика, выраженных в градусах 20, которые представлены в Таблице Х-Р23. В некоторых вариантах осуществления кристаллическая форма трет-бутил-(2S)-6-бром-7-метил-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилата демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере три характеристических пика, выраженных в градусах 20, которые представлены в Таблице Х-Р23. В некоторых вариантах осуществления кристаллическая форма трет-бутил-(2S)-6-бром-7-метил-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилата демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере четыре (например, 4, 5, 6, 7, 8, 9 или 10) характеристических пика, выраженных в градусах 20, которые представлены в Таблице Х-Р23. В некоторых вариантах осуществления кристаллическая форма трет-бутил-(2S)-6-бром-7-метил-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилата демонстрирует порошковую рентгеновскую дифрактограмму, по существу такую, как показано на Фиг. 3.
Получение Р25
Ди-трет-бутил-7-метил-6-(2-метил-2Е-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1,1'-дикарбоксилат (Р25)
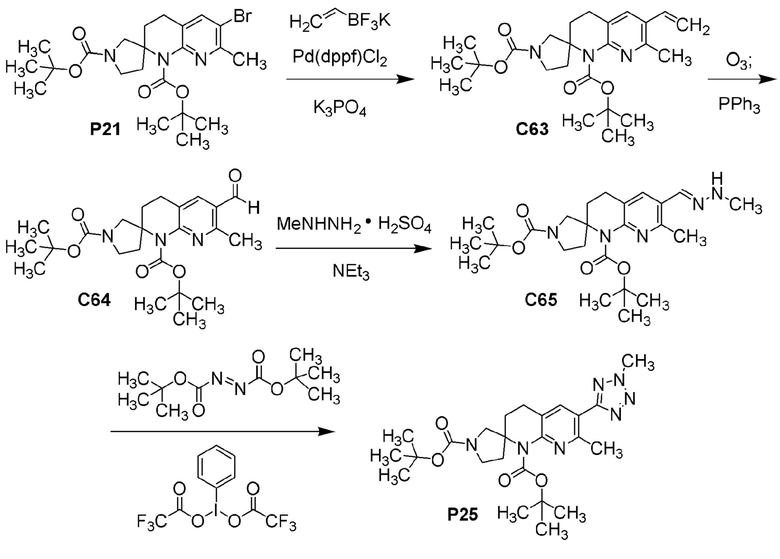
Стадия 1. Синтез ди-трет-бутил-6-этенил-7-метил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1,1'-дикарбоксилата (С63).
Смесь Р21 (15,0 г, 31,1 ммоль), винилтрифторбората калия (10,4 г, 77,6 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) (2,27 г, 3,10 ммоль) и фосфата калия (19,8 г, 93,3 ммоль) в N,N-диметилформамиде (500 мл) перемешивали при 95°С в течение 16 часов, затем реакционную смесь фильтровали; фильтрат выливали в воду (4 л) и экстрагировали этилацетатом (2×800 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток объединяли с продуктом подобной реакции, которую осуществляли с использованием Р21 (5,00 г, 10,4 ммоль), смесь очищали хроматографией на силикагеле (градиент: 0% до 20% этилацетата в петролейном эфире) с получением С63 в виде белого твердого вещества. Объединенный выход: 17,1 г, 38,9 ммоль, 94%. ЖХМС m/z 430,3 [М+Н]+. 1Н ЯМР (400 МГц, хлороформ-d) δ 7,46 (ушир. с, 1Н), 6,83 (дд, J=11,4, 11,1 Гц, 1Н), 5,59 (ушир. д, J=11,4 Гц, 1Н), 5,37-5,24 (м, 1Н), [3,90 (д, J=11,0 Гц) и 3,72 (д, J=11,0 Гц), всего 1Н], 3,64-3,41 (м, 2Н), 3,38-3,24 (м, 1Н), [2,86-2,64 (м), 2,62-2,39 (м) и 2,16-1,72 (м), всего 9Н], 1,45 (с, 18Н).
Стадия 2. Синтез ди-трет-бутил-6-формил-7-метил-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1,1'-дикарбоксилата (С64).
Раствор С63 (17,0 г, 39,6 ммоль) в дихлорметане (200 мл) охлаждали до температуры -78°С и вводили поток озон-обогащенного кислорода до установления стабильного синего цвета. Еще через 5 минут реакционную смесь барботировали потоком сухого азота вплоть до исчезновения синего цвета, затем добавляли трифенилфосфин (20,7 г, 78,9 ммоль). Полученную смесь нагревали до 25°С и перемешивали в течение 2 часов, после этого ЖХМС анализ показал присутствие С64: ЖХМС m/z 454, 3 [M+Na+]. Реакционную смесь концентрировали в вакууме, затем остаток очищали хроматографией на силикагеле (градиент: 0% до 50% этилацетата в петролейном эфире) с получением С64 в виде бесцветного смолистого вещества. Выход: 9,98 г, 23,1 ммоль, 58%.
Стадия 3. Синтез ди-трет-бутил-7-метил-6-[(2-метилгидразинилиден)метил]-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1,1'-дикарбоксилата (С65).
Раствор метилгидразинсульфата (3,20 г, 22,2 ммоль) и триэтиламина (7,78 мл, 55,8 ммоль) в метаноле (50 мл) перемешивали при 25°С в течение 5 минут, затем добавляли раствор С64 (7,98 г, 18,5 ммоль) в метаноле (20 мл). После перемешивания реакционной смеси при 25°С в течение 1 часа сбор осадка фильтрованием давал С65 в виде белого твердого вещества. Выход: 7,60 г, 16,5 ммоль, 89%. ЖХМС m/z 460,3 [М+Н]+.
Стадия 4. Синтез ди-трет-бутил-7-метил-6-(2-метил-2H-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1,1'-дикарбоксилата (Р25).
К раствору С65 (6,70 г, 14,6 ммоль) в смеси 2,2,2-трифторэтанола (35 мл) и дихлорметана (35 мл) добавляли ди-трет-бутилазодикарбоксилат (4,36 г, 18,9 ммоль) с последующим добавлением [бис(трифторацетокси)иод]бензола (33,2 г, 77,2 ммоль). Реакционную смесь перемешивали при 25°С в течение 30 минут, затем смесь выливали в насыщенный водный раствор сульфита натрия (300 мл) и экстрагировали дихлорметаном (2×100 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме; хроматография на силикагеле (градиент: 0% до 20% этанола в дихлорметане) давала Р25 в виде белого твердого вещества. Выход: 2,10 г, 4,32 ммоль, 30%. ЖХМС m/z 508,3 [M+Na+]. 1H ЯМР (400 МГц, метанол-d4) δ 8,11 (с, 1Н), 4,44 (с, 3Н), [3,93 (д, J=11,3 Гц) и 3,86 (д, J=11,1 Гц), всего 1Н], 3,68-3,56 (м, 1Н), 3,56-3,46 (м, 1Н), 3,46-3,3 (м, 1Н, предположительно; частично скрыт пиком растворителя), 2,92-2,81 (м, 2Н), 2,73 (с, 3Н), [2,69-2,58 (м) и 2,58-2,47 (м), всего 1Н], 2,15-1,88 (м, 3Н), 1,48 (ушир. с, 18Н).
Получение Р26
(2S)-7-Метил-6-(2-метил-2Н-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин], дигидрохлоридной соли (Р26)
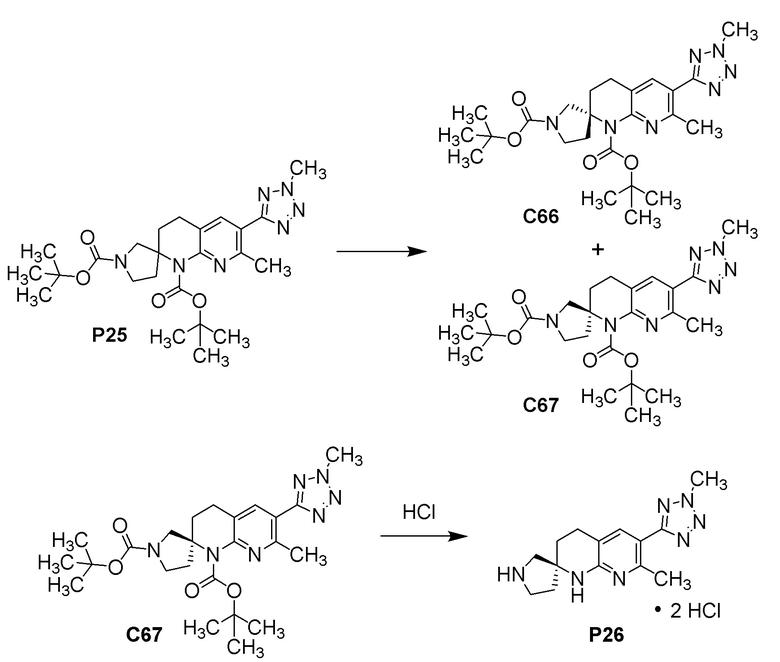
Стадия 1. Разделение ди-трет-бутил-(2R)-7-метил-6-(2-метил-2H-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1,1'-дикарбоксилата (С66) и ди-трет-бутил-(2S)-7-метил-6-(2-метил-2H-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1,1'-дикарбоксилата (С67).
Разделение Р25 (2,37 г, 4,88 ммоль) на составляющие его диастереомеры осуществляли с использованием сверхкритической флюидной хроматографии {Колонка: Chiral Technologies Chiralcel OJ-H, 21,2×250 мм, 5 мкм; Подвижная фаза 9:1 диоксид углерода/[метанол, содержащий 0,2% (7 М аммиака в метаноле)]; Скорость потока: 80 мл/мин; Обратное давление: 120 бар). Первый элюирующий диастереомер был указан как С66, а второй элюирующий диастереомер был указан как С67.
С66 выделяли в виде твердого вещества. Выход: 1,01 г, 2,0 8 ммоль, 43%. Время удерживания: 2,68 мин [Аналитические условия. Колонка: Chiral Technologies Chiralcel OJ-H, 4,6×250 мм, 5 мкм; Подвижная фаза А: диоксид углерода; Подвижная фаза В: метанол, содержащий 0,2% (7 М аммиака в метаноле); Градиент: 5% В в течение 1,0 минут, затем 5% до 60% В в течение 8,0 минут; Скорость потока: 3,0 мл/мин; Обратное давление: 120 бар].
С67 выделяли в виде масла. Выход: 1,00 г, 2,06 ммоль, 42%. Время удерживания: 3,33 мин (Аналитические условия идентичны используемым для С66).
Присвоение абсолютной стереохимии см. ниже.
Стадия 2. Синтез (2S)-7-метил-6-(2-метил-2H-тетразол-5-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин], дигидрохлоридной соли (Р26).
Раствор С67 (150 мг, 0,309 ммоль) в смеси дихлорметана (1,0 мл) и 1,1,1,3,3,3-гексафторпропан-2-ола (1,0 мл) обрабатывали раствором хлористого водорода в 1,4-диоксане (4 М; 0,309 мл, 1,24 ммоль). После перемешивания реакционной смеси при комнатной температуре в течение 2 часов ЖХМС анализ показал преобразование в Р26: ЖХМС m/z 286,3 [М+Н]+. Концентрирование реакционной смеси в вакууме давало Р26 в виде твердого вещества. Выход: 105 мг, 0,293 ммоль, 95%.
Указанная абсолютная стереохимия была установлена следующим образом. Эту партию Р26 использовали для получения 3 и 4 в Альтернативном синтезе Примеров 3 и 4. Корреляция между этими партиями 3 и 4 с такими же соединениями, полученными из предшественника с известной абсолютной стереохимией (см. Примеры 3 и 4), показана в Альтернативном синтезе Примеров 3 и 4.
Альтернативное Получение Р26
(2S)-7-Метил-6-(2-метил-2H-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин], дигидрохлоридная соль (Р26)
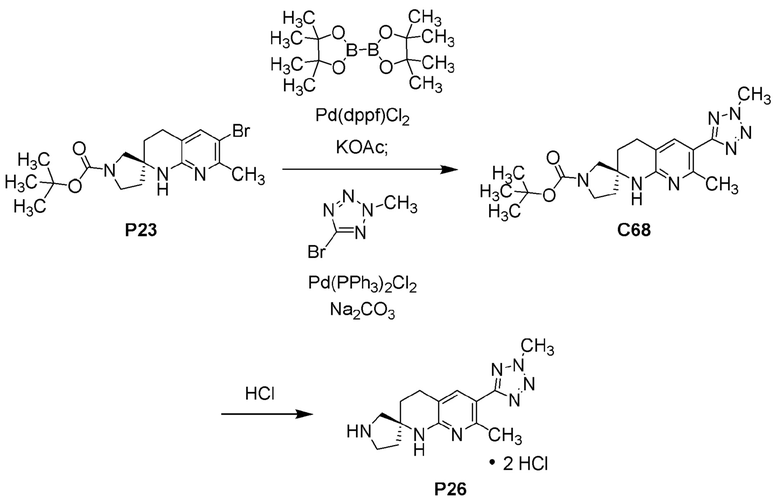
Стадия 1. Синтез трет-бутил-(2S)-7-метил-6-(2-метил-2H-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилата (С68).
Смесь 4,4,4',4',5,5,5',5'-октаметил-2,2'-би-1,3,2-диоксаборолана (299 мг, 1,18 ммоль), Р23 (300 мг, 0,785 ммоль), комплекса [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) с дихлорметаном (32,0 мг, 39,2 мкмоль) и высушенного в печи ацетата калия (308 мг, 3,14 ммоль) в 1,4-диоксане (10 мл) дегазировали путем барботирования смеси азотом в течение 5 минут. Реакционный сосуд затем герметично закрывали и нагревали при 100°С в алюминиевом блоке в течение 2 часов, затем смеси давали охладиться до комнатной температуры. Добавляли 5-бром-2-метил-2H-тетразол (134 мг, 0,822 ммоль), дихлорбис(трифенилфосфин)палладий(II) (27,5 мг, 39,2 мкмоль) и дегазированный водный раствор карбоната натрия (2,0 М; 0,981 мл, 1,96 ммоль) и реакционную смесь снова дегазировали путем барботирования азотом в течение 5 минут. Затем смесь перемешивали при 90°С в течение 18 часов, охлаждали до комнатной температуры, разбавляли этилацетатом и фильтровали через диатомовую землю. Органический слой фильтрата промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и концентрировали в вакууме; ЖХМС анализ показал присутствие С68: ЖХМС m/z 38 6,3 [М+Н]+. Хроматография на силикагеле (градиент: 20% до 50% этилацетата в гептане) давала С68 в виде светло-желтого масла. Выход: 280 мг, 0,726 ммоль, 92%. Эту партию С68 использовали в Примерах 3 и 4 ниже.
Стадия 2. Синтез (2S)-7-метил-6-(2-метил-2H-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин], дигидрохлоридной соли (Р26).
Смесь С68 (185 мг, 0,480 ммоль) и раствора хлористого водорода в 2-пропаноле (1,25 М; 1,9 мл, 2,4 ммоль) нагревали до 50°С в течение 1 часа. Концентрирование реакционной смеси в вакууме давало Р26 в виде твердого вещества, которое использовали без дополнительной очистки. Выход: 170 мг, 0,47 ммоль, 98%.
Получение Р27
[(2S)-1'-(трет-Бутоксикарбонил)-7-метил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-6-ил]бороновая кислота (Р27)
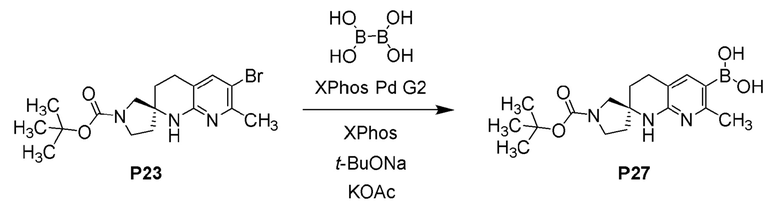
Реакционный сосуд, содержащий смесь Р23 (19,5 г, 51,0 ммоль), ацетата калия (12,5 г, 127 ммоль), трет-бутоксида натрия (49,0 мг, 0,510 ммоль), хлор(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладия(II) [XPhos Pd G2; 401 мг, 0,510 ммоль) и 2-дициклогексилфосфино-2',4',6'-триизопропилбифенила (XPhos; 729 мг, 1,53 ммоль) продували азотом. Затем добавляли метанол (200 мл), этан-1,2-диол (20 мл) и тетрагидроксидибор (11,4 г, 127 ммоль), затем реакционную смесь барботировали азотом в течение 10 минут. Реакционную смесь нагревали до внутренней температуры 50°С в течение 2 часов, охлаждали до комнатной температуры и затем до 0°С и доводили до рН 14 добавлением водного раствора гидроксида натрия (4 М; 80 мл) {Осторожно: выделение газа}. Полученную смесь перемешивали при комнатной температуре в течение 30 минут и фильтровали; фильтрат концентрировали в вакууме и экстрагировали два раза трет-бутилметиловым эфиром. Объединенные органические слои затем экстрагировали водным раствором гидроксида натрия (2 М; 2×100 мл). Все водные слои объединяли и перемешиваемую смесь обрабатывали по каплям хлористоводородной кислотой (4 М; приблизительно 20 мл) до осаждения твердых частиц (это происходило при рН приблизительно 9). После перемешивания смеси при комнатной температуре еще в течение 30 минут смесь экстрагировали четыре раза этилацетатом. Объединенные этилацетатные слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением Р27 в виде светло-желтого порошка. Выход: 12,5 г, 36,0 ммоль, 71%. ЖХМС m/z 348,4 [М+Н]+. 1Н ЯМР (400 МГц, метанол-d4), характеристические пики: δ 7,74 (ушир. с, 1Н), 3,46-3,35 (м, 2Н), 2,92-2,72 (м, 2Н), 2,48 (с, 3Н), 2,12-1,83 (м, 4Н), [1,47 (с) и 1,46 (с), всего 9Н].
Получение Р28
(2S)-7-Метил-6-(пиримидин-2-мл)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин], дигидрохлоридная соль (Р28)
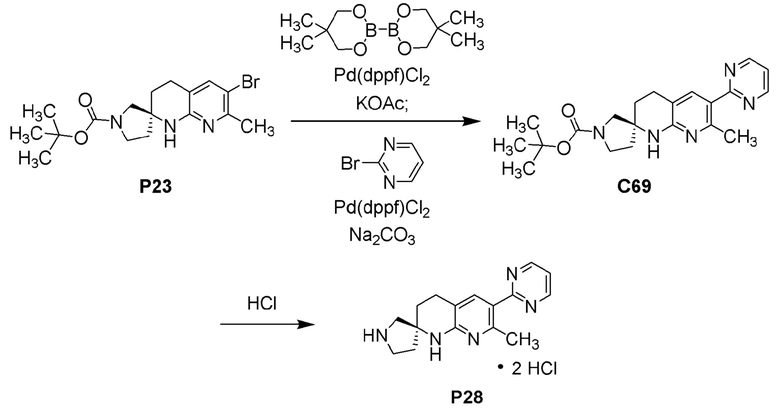
Стадия 1. Синтез трет-бутил-(2S)-7'-метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилата (С69).
Осуществляли барботирование азотом смеси высушенного в печи ацетата калия (2,07 г, 21,1 ммоль), Р23 (вещество из Получения Р23 и Р24; 2,02 г, 5,28 ммоль), 5,5,5',5'-тетраметил-2,2'-би-1,3,2-диоксаборинана (1,79 г, 7,92 ммоль) и комплекса [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) с дихлорметаном (216 мг, 0,264 ммоль) в 1,4-диоксане (20 мл) в течение 5 минут. Реакционную смесь затем нагревали в 105°С алюминиевом блоке в течение 2 часов, затем смеси давали охладиться до комнатной температуры и затем обрабатывали 2-бромпиримидином (840 мг, 5,28 ммоль), дополнительным количеством комплекса [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) с дихлорметаном (216 мг, 0,264 ммоль) и водным раствором карбоната натрия (2,0 М; 7,93 мл, 15,9 ммоль). Реакционную смесь продували азотом, затем смесь нагревали до 100°С в течение 18 часов, после этого ЖХМС анализ показал преобразование в С69: ЖХМС m/z 382,4 [М+Н]+. Реакционную смесь охлаждали, распределяли между водным раствором хлорида аммония и этилацетатом и затем всю смесь фильтровали через слой диатомовой земли. Фильтровальную лепешку промывали водой и этилацетатом и водный слой объединенного фильтрата экстрагировали этилацетатом (2×30 мл). Все органические слои объединяли и промывали последовательно водой (100 мл) и насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Остаток (2,9 г) растворяли в этилацетате (10 мл) и обрабатывали при помощи SiliaMetS Thiol (SiliCycle, R51030B; 2 г); полученную смесь нагревали при кипячении с обратным холодильником в течение 10 минут и затем охлаждали до комнатной температуры. Фильтрование через слой диатомовой земли давало фильтрат, который концентрировали при пониженном давлении с получением С69 в виде коричневого смолистого вещества (2 г). Это вещество использовали на следующей стадии без дополнительной очистки.
Стадия 2. Синтез (2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин], дигидрохлоридная соль (Р28).
Раствор хлористого водорода получали медленным добавлением ацетилхлорида (1,50 мл, 21,1 ммоль) к 2-пропанолу (4 мл). В отдельной колбе, С69 (с предыдущей стадии; 2 г; ≤5,28 ммоль) растворяли в смеси пропан-2-илацетата (15 мл) и 2-пропанола (15 мл); это требовало нагревания при 50°С. Как только раствор был получен, к нему медленно добавляли раствор хлористого водорода и реакционную смесь нагревали при 50°С в течение 2 часов. Затем смеси давали медленно охладиться до комнатной температуры при перемешивании; перемешивание продолжали при комнатной температуре в течение 18 часов. Полученное твердое вещество собирали вакуумным фильтрованием в атмосфере азота с получением Р28 в виде гигроскопичного твердого вещества. Выход: 750 мг, 2,12 ммоль, 40% за 2 стадии. ЖХМС m/z 282, 3 [М+Н]+. 1Н ЯМР (400 МГц, метанол-d4) δ 8,91 (д, J=4,9 Гц, 2Н), 8,58 (с, 1Н), 7,43 (т, J=4,9 Гц, 1Н), 3,76-3,66 (м, 1Н), 3,66-3,52 (м, 2Н), 3,46 (д, компонент АВ квартета, J=12,5 Гц, 1Н), 3,12-2,95 (м, 2Н), 2,90 (с, 3Н), 2,49-2,38 (м, 1Н), 2,37-2,25 (м, 1Н), 2,24-2,06 (м, 2Н).
Указанная абсолютная стереохимия была присвоена на основании преобразования этой партии Р28 в Пример 14; абсолютная стереохими 14 была установлена методом рентгеновской дифракции монокристалла (см. Пример 14 ниже).
Получение Р29
трет-Бутил-4,7-диметил-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилат (Р29)
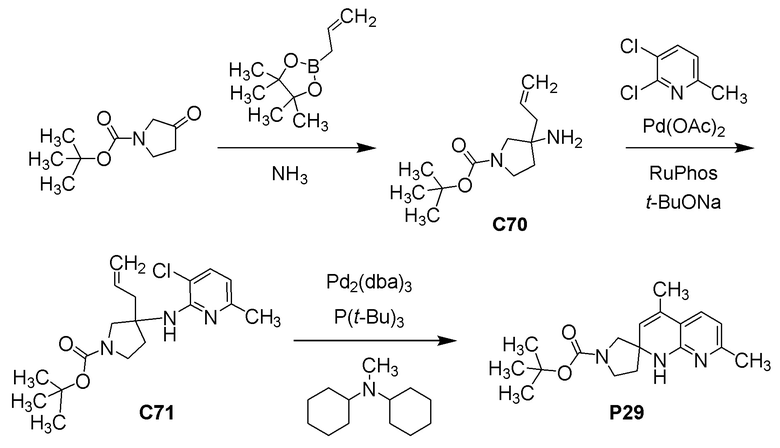
Стадия 1. Синтез трет-бутил-3-амино-3-(проп-2-ен-1-ил)пирролидин-1-карбоксилата (С70).
Смесь трет-бутил-3-оксопирролидин-1-карбоксилата (500 мг, 2,70 ммоль) и раствора аммиак в метаноле (7 М; 3,9 мл, 27 ммоль) перемешивали при комнатной температуре в течение 30 минут. К этому затем добавляли по каплям раствор 4,4,5,5-тетраметил-2-(проп-2-ен-1-ил)-1,3,2-диоксаборолана (907 мг, 5,40 ммоль) в метаноле и реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Летучие вещества удаляли в вакууме и остаток подвергали хроматографии на силикагеле (градиент: 0% до 10% метанола в дихлорметане) с получением С70. Выход: 200 мг, 0,884 ммоль, 33%. 1Н ЯМР (400 МГц, хлороформ-d) δ 5,91-5,76 (м, 1Н), 5,20 (м, 1Н), 5,15 (ушир. д, J=11 Гц, 1Н), 3,53-3,38 (м, 2Н), 3,32-3,08 (м, 2Н), 2,28 (д, J=7,5 Гц, 2Н), 1,89-1,79 (м, 1Н), 1,73-1,63 (м, 1Н), 1,46 (с, 9Н).
Стадия 2. Синтез трет-бутил-3-[(3-хлор-6-метилпиридин-2-ил)амино]-3-(проп-2-ен-1-ил)пирролидин-1-карбоксилата (С71).
Сосуд, содержащий смесь 2,3-дихлор-6-метилпиридина (100 мг, 0,617 ммоль), С70 (168 мг, 0,742 ммоль), ацетата палладия(II) (6,93 мг, 30,9 мкмоль), 2-дициклогексилфосфино-2',6'-диизопропоксибифенила (RuPhos; 28,8 мг, 61,7 мкмоль) и трет-бутоксида натрия (119 мг, 1,24 ммоль) в 1,4-диоксане (8 мл) продували азотом, герметично закрывали и нагревали при 80°С в течение ночи. ЖХМС анализ показал преобразование в С71: ЖХМС m/z 352,3 (наблюдаемый изотопный паттерн хлора) [М+Н]+, затем реакционную смесь охлаждали до комнатной температуры и распределяли между водой и дихлорметаном. Водный слой экстрагировали дихлорметаном и объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали, концентрировали в вакууме и подвергали хроматографии на силикагеле (градиент: 0% до 50% этилацетата в гептане) с получением С71 в виде масла, которое отверждалось при выстаивании. Выход: 121 мг, 0,344 ммоль, 56%. 1Н ЯМР (400 МГц, хлороформ-d) δ 7,29 (д, J=8,1 Гц, 1Н), 6,42-6,34 (м, 1Н), 5,82-5,68 (м, 1Н), 5,11-5,03 (м, 1Н), 5,03-4,93 (м, 1Н), 3,79-3,69 (м, 1Н), [3,62 (д, компонент АВ квартета, J=11,6 Гц), 3,56 (д, компонент АВ квартета, J=11,4 Гц) и 3,54-3,36 (м), всего 3Н], 2,95-2,83 (м, 1Н), 2,76-2,63 (м, 1Н), 2,45-2,28 (м, 1Н), 2,34 (с, 3Н), 2,08-1,96 (м, 1Н), 1,50-1,41 (ушир. с, 9Н).
Стадия 3. Синтез трет-бутил-4,7'-диметил-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилата (Р29).
Смесь С71 (40 мг, 0,11 ммоль), трис (дибензилиденацетон) дипалладия (0) (5,20 мг, 5,68 мкмоль), N-циклогексил-N-метилциклогексанамина (111 мг, 0, 568 ммоль) и три-трет-бутилфосфина (1,15 мг, 5,68 мкмоль) в N,N-диметилформамиде (1,0 мл) дегазировали и затем нагревали при 80°С в течение 2 часов. Температуру повышали до 120°С и реакционную смесь поддерживали при этой температуре в течение 3 дней. ЖХМС анализ показал преобразование в Р29: ЖХМС m/z 316,3 [М+Н]+. Реакционную смесь охлаждали до комнатной температуры, затем смесь распределяли между этилацетатом и водой и водный слой экстрагировали два раза этилацетатом. Объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и концентрировали в вакууме; хроматографии на силикагеле (градиент: 0% до 100% этилацетата в гептане) давала Р29. Выход: 30 мг, 95 мкмоль, 86%. 1H ЯМР (400 МГц, хлороформ-d), характеристические пики: δ 7,13 (д, J=7,5 Гц, 1Н), 6,40 (д, J=7,5 Гц, 1Н), 5,27 (ушир. с, 1Н), 5,06-4,99 (ушир. с, 1Н), 3,58-3,40 (м, 3Н), 3,31-3,21 (м, 1Н), 2,31 (с, 3Н), [1,96 (с) и 1,96 (с), всего 3Н], [1,46 (с) и 1,44 (с), всего 9Н].
Получение Р30
трет-Бутил-1-бензил-7-метил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилат (Р30)
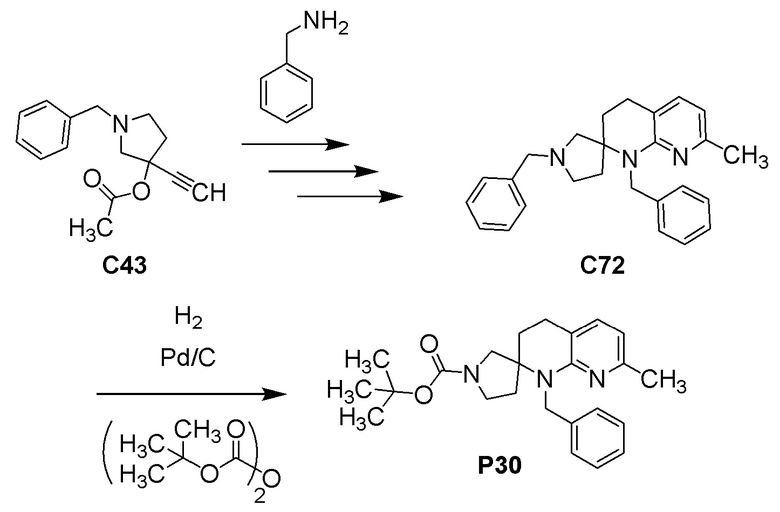
Стадия 1. Синтез 1,1'-дибензил-7-метил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин] (С72).
Преобразование С43 в С72 осуществляли с использованием способа, описанного для синтеза С47 из С43 в Получениях Р17 и Р18, с использованием 1-фенилметанамина вместо 1-(4-метоксифенил)метанамина. Хроматография на силикагеле (градиент: 0% до 10% метанола в дихлорметане) давала С72. Выход для стадии циклизации с получением С72: 580 мг, 1,51 ммоль, 69%.
Стадия 2. Синтез трет-бутил-1-бензил-7-метил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилата (Р30).
Смесь С72 (550 мг, 1,43 ммоль), палладия на углероде (50 мг, 0,143 ммоль) и ди-трет-бутилдикарбоната (376 мг, 1,72 ммоль) в метаноле (20 мл) гидрировали в течение ночи при 75 ф/дюйм2 (5,273 кг/см2). Реакционную смесь фильтровали через диатомовую землю и фильтрат концентрировали в вакууме; хроматография на силикагеле (градиент: 0% до 100% этилацетата в гептане) давала Р30 в виде белого полутвердого вещества. Выход: 482 мг, 1,22 ммоль, 85%. 1Н ЯМР (400 МГц, хлороформ-d) δ 7,29-7,07 (м, 6Н, предположительно; частично скрыт пиком растворителя), 6,39 (д, J=7,2 Гц, 1Н), 5,15-4,99 (м, 1Н), 4,97-4,78 (м, 1Н), 3,58-3,19 (м, 4Н), 2,87-2,71 (м, 2Н), 2,31-2,16 (м, 1Н), 2,24 (с, 3Н), 2,07-1,95 (м, 1Н), 1,92-1,79 (м, 1Н), 1,75-1,63 (м, 1Н), [1,45 (с) и 1,43 (с), всего 9Н].
Получение Р31
6-[5-(Дифторметил)-1-метил-1H-1,2,4-триазол-3-ил]-1-метил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин] (Р31)
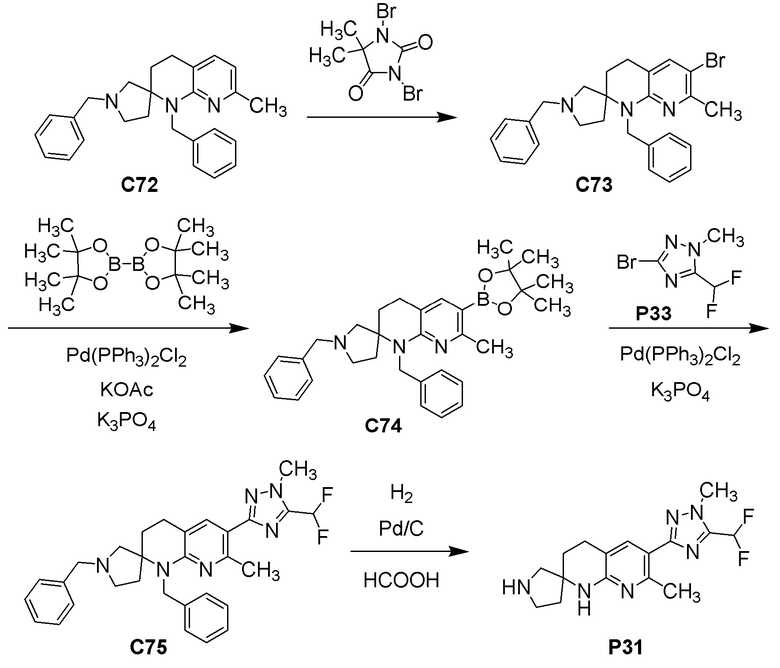
Стадия 1. Синтез 1,1'-дибензил-6-бром-7-метил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин] (С73).
1,3-Дибром-5,5-диметилимидазолидин-2,4-дион (532 мг, 1,86 ммоль) добавляли по порциям к 0°С раствору С72 (1,19 г, 3,10 ммоль) в дихлорметане (16 мл). ЖХМС анализ через 1 час показал преобразование в С73: ЖХМС m/z 462,2 (наблюдаемый изотопный паттерн брома) [М+Н]+. Реакционную смесь разбавляли дихлорметаном (20 мл), промывали последовательно насыщенным водным раствором сульфита натрия, насыщенным водным раствором бикарбоната натрия и насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (градиент: 0% до 50% этилацетата в гептане) давала С73 в виде масла. Выход: 980 мг, 2,12 ммоль, 68%. 1Н ЯМР (400 МГц, хлороформ-d) δ 7,32-7,12 (м, 11Н, предположительно; частично скрыт пиком растворителя), 5,03 (АВ квартет, JAB=16,3 Гц, ΔνАВ=2б,б Гц, 2Н), 3,54 (АВ квартет, JAB=13,1 Гц, ΔνАВ=41,8 Гц, 2Н), 2,93 (д, J=10,2 Гц, 1Н), 2,88 (ддд, J=8,5, 8,5, 3,4 Гц, 1Н), 2, 84-2, 75 (м, 1Н), 2, 74-2, 65 (м, 1Н), 2,40-2,32 (м, 1Н), 2,29 (с, 3Н), 2,19 (д, d=10,2 Гц, 1Н), 2,12 (ддд, d=13,4, 8,3, 8,3 Гц, 1Н), 1,99 (ддд, d=13,7, 8,8, 5,2 Гц, 1Н), 1,93-1,85 (м, 1Н), 1,81 (ддд, d=13,4, 7,3, 3,5 Гц, 1Н).
Стадия 2. Синтез 1,1'-дибензил-7-метил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)3,4-дигидро-1H-спиро [1,8-нафтиридин-2,3'-пирролидин] (С74).
В реакционный сосуд загружали 4,4,4',4',5,5,5',5'-октаметил-2,2'-би-1,3,2-диоксаборолан (148 мг, 0,583 ммоль), С73 (180 мг, 0,38 9 ммоль), комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) с дихлорметаном (31,8 мг, 38,9 мкмоль) и высушенный в печи ацетат калия (153 мг, 1,56 ммоль) в 1,4-диоксане (5 мл). Реакционную смесь барботировали азотом в течение 5 минут, затем сосуд герметично закрывали и нагревали при 100°С в алюминиевом блоке в течение 2 часов. ЖХМС анализ показал присутствие С74: ЖХМС m/z 510,4 [М+Н]+. Реакционную смесь охлаждали до комнатной температуры, затем смесь разбавляли этилацетатом и фильтровали через слой диатомовой земли. Фильтрат концентрировали в вакууме с получением соединения С74, которое использовали непосредственно на следующей стадии.
Стадия 3. Синтез 1,1'-дибензил-6-[5-(дифторметил)-1-метил-1H-1,2,4-триазол-3-ил]-7-метил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин] (С75).
Дихлорбис(трифенилфосфин)палладий(II) (5,24 мг, 7,46 мкмоль), водный раствор фосфата калия (2,0 М; 0,466 мл, 0,932 ммоль) и 3-бром-5-(дифторметил)-1-метил-1H-1,2,4-триазол (Р33; 79,1 мг, 0,373 ммоль) добавляли к раствору С74 (с предыдущей стадии; ≤0,389 ммоль) в тетрагидрофуране (5 мл). Реакционную смесь продували азотом, затем реакционный сосуд герметично закрывали и нагревали при 70°С в алюминиевом блоке в течение 1 часа. Температуру затем повышали до 100°С и нагревание продолжали в течение ночи. Снова добавляли 3-бром-5-(дифторметил)-1-метил-1H-1,2,4-триазол (Р33; 79,1 мг, 0,373 ммоль) и нагревание осуществляли еще в течение 6 часов, затем реакционную смесь охлаждали и распределяли между этилацетатом и водой. Водный слой экстрагировали два раза этилацетатом и объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (градиент: 0% до 100% этилацетата в гептане) давала С75. Выход: 105 мг, 0,204 ммоль, 52% за 2 стадии. ЖХМС m/z 515,3 [М+Н]+. 1Н ЯМР (400 МГц, хлороформ-d) δ 7,69 (с, 1Н), 7,33-7,19 (м, 9Н, предположительно; частично скрыт пиком растворителя), 7,18-7,12 (м, 1Н), 6,85 (т, JHF=52,6 Гц, 1Н), 5,14 (АВ квартет, JAB=16,3 Гц, ΔνAB=17,6 Гц, 2Н), 4,05 (с, 3Н), 3,54 (АВ квартет, JAB=13,0 Гц, ΔνAB=38,8 Гц, 2Н), 2,97 (д, J=10,2 Гц, 1Н), 2,93-2,81 (м, 2Н), 2,81-2,72 (м, 1Н), 2,53 (с, 3Н), 2,41-2,32 (м, 1Н), 2,22 (д, J=10,2 Гц, 1Н), 2,15 (ддд, J=13,5, 8,3, 8,2 Гц, 1Н), 2,06-1,97 (м, 1Н), 1,96-1,88 (м, 1Н), 1,84 (ддд, J=13,4, 7,3, 3,4 Гц, 1Н).
Стадия 4. Синтез 6-[5-(дифторметил)-1-метил-1H-1,2,4-триазол-3-ил]-7'-метил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин] (P31).
Палладий на углероде (10%, влажный, содержащий воду; 20 мг) добавляли к раствору С75 (105 мг, 0,204 ммоль) в метаноле (5 мл), содержащем каплю муравьиной кислоты, и полученную смесь гидрировали в течение ночи при комнатной температуре и 70 ф/дюйм2 (4,922 кг/см2). После фильтрования фильтрат концентрировали в вакууме с получением Р31 в виде светло-желтого твердого вещества. Выход: 63 мг, 0,19 ммоль, 93%. ЖХМС m/z 335,2 [М+Н]+. 1Н ЯМР (400 МГц, хлороформ-d) δ 8,20 (с, 1Н), 6,86 (т, JHF=52,4 Гц, 1Н), 4,10 (с, 3Н), 3,78-3,51 (м, 3Н), 3,41 (д, J=12,3 Гц, 1Н), 2,99-2,85 (м, 2Н), 2,83 (с, 3Н), 2,29-2,19 (м, 2Н), 2,19-2,01 (м, 2Н).
Получение Р32
(2S)-6-[5-(Дифторметил)-1-метил-1H-1,2,4-триазол-3-ил]-7-метил-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин], дигидрохлоридная соль (Р32)
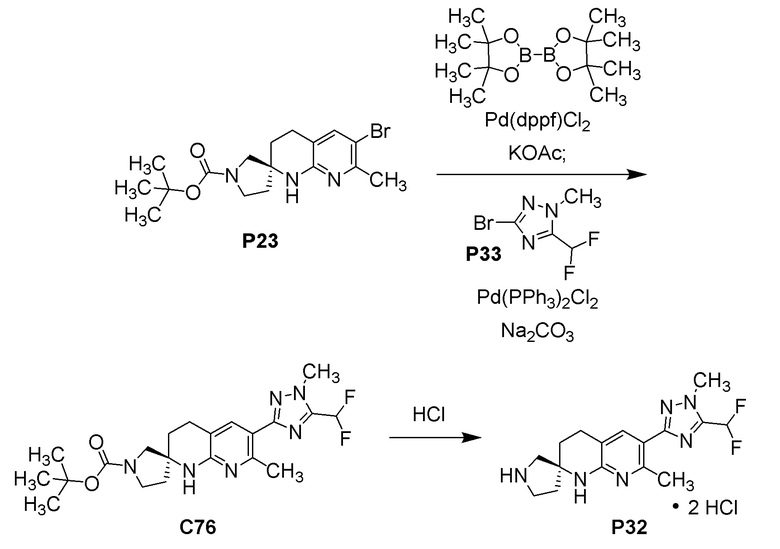
Стадия 1. Синтез трет-бутил-(2S)-6-[5-(дифторметил)-1-метил-1H-1,2,4-триазол-3-ил]-7-метил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилата (С76).
С использованием способа, описанного для синтеза С68 из Р23 в Альтернативном Получении Р26, Р23 (220 мг, 0, 575 ммоль) и 3-бром-5-(дифторметил)-1-метил-1H-1,2,4-триазол (Р33; 128 мг, 0,604 ммоль) использовали для получения С76. Хроматография на силикагеле (градиент: 20% до 50% этилацетата в гептане) давала С76 в виде белого твердого вещества. Выход: 110 мг, 0,253 ммоль, 44%. ЖХМС m/z 435,4 [М+Н]+.
Стадия 2. Синтез (2S)-6-[5-(дифторметил)-1-метил-1H-1,2,4-триазол-3-ил]-7-метил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин], дигидрохлоридной соли (Р32).
Смесь С76 (110 мг, 0,253 ммоль) в растворе хлористого водорода в 2-пропаноле (1,25 М, 1,0 мл, 1,2 ммоль) нагревали при 50°С в течение 1 часа. ЖХМС анализ показал образование Р32: ЖХМС m/z 335,3 [М+Н]+. Концентрирование в вакууме давало Р32 в виде твердого вещества. Выход: 74 мг, 0,182 ммоль, 72%.
Получение Р33
3-Бром-5-(дифторметил)-1-метил-1H-1,2,4-триазол (Р33)
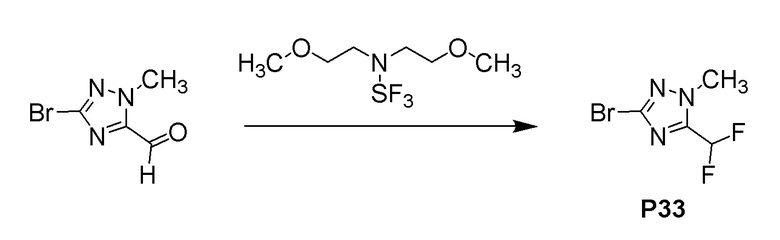
Трифторид [бис(2-метоксиэтил)амино]серы (47,0 мл, 255 ммоль) добавляли по каплям к 0°С смеси 3-бром-1-метил-1H-1,2,4-триазол-5-карбальдегида (24,2 г, 127 ммоль) в дихлорметане (400 мл); реакционной смеси давали нагреться до 20°С и перемешивали при 20°С в течение 16 часов. После добавления по каплям водного раствора бикарбоната натрия полученную смесь экстрагировали дихлорметаном (3×300 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (градиент: 50% до 70% дихлорметана в петролейном эфире) давала 3-бром-5-(дифторметил)-1-метил-1H-1,2,4-триазол (Р33) в виде светло-желтого масла (17,7 г). Это вещество объединяли с продуктом подобной реакции, которую осуществляли с использованием 3-бром-1-метил-1H-1,2,4-триазол-5-карбальдегида (12,0 г, 63,2 ммоль); концентрирование при пониженном давлении давало Р33 в виде белого твердого вещества. Объединенный выход: 2 5,2 г, 119 ммоль, 63%. ЖХМС m/z 212 (наблюдаемый изотопный паттерн брома) [М+Н]+. 1H ЯМР (400 МГц, метанол-d4) δ 7,06 (т, JHF=52,2 Гц, 1Н), 4,01 (с, 3Н).
Примеры 1 и 2
(2R)-2-(5-Хлор-2-метоксипиридин-4-ил)-1-[7-метил-6-(2-метил-2H-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-он, DIAST-1 (1) и (2R)-2-(5-Хлор-2-метоксипиридин-4-ил)-1-[7-метил-6-(2-метил-2Е-тетразол-5-ил)-3,4-дигидро-1Е-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-он, DIAST-2 (2)
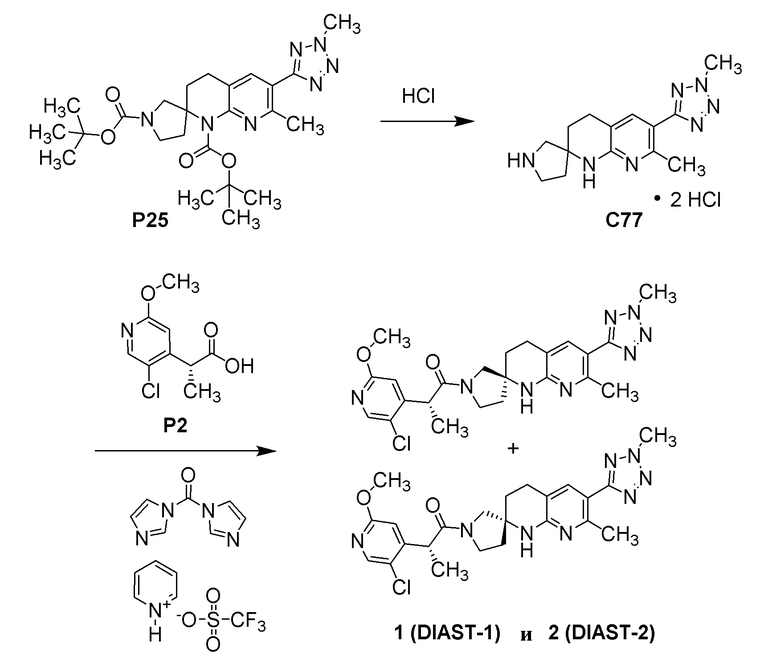
Стадия 1. Синтез 7'-метил-6-(2-метил-2H-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин], дигидрохлоридной соли (С77).
Раствор хлористого водорода в 1,4-диоксане (4,0 М; 0,587 мл, 2,35 ммоль) добавляли к раствору Р25 (285 мг, 0,587 ммоль) в смеси дихлорметана (1 мл) и 1, 1, 1, 3, 3, 3-гексафторпропан-2-ола (1 мл). После перемешивания реакционной смеси при комнатной температуре в течение 2 часов ЖХМС анализ показал присутствие С77: ЖХМС m/z 286,3 [М+Н]+. Удаление летучих веществ в вакууме давало С77 в виде белого твердого вещества. Выход: 210 мг, 0,586 ммоль, количественный.
Стадия 2. Синтез (2R)-2-(5-хлор-2-метоксипиридин-4-ил)-1-[7-метил-6-(2-метил-2H-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она, DIAST-1 (1) и (2R)-2-(5-хлор-2-метоксипиридин-4-ил)-1-[7-метил-6-(2-метил-2H-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она, DIAST-2 (2).
К раствору Р2 (65,7 мг, 0,305 ммоль) в ацетонитриле (1 мл) добавляли трифторметансульфонат пиридиния (140 мг, 0,611 ммоль) и смесь перемешивали до образования раствора. Добавляли 1,1'-карбонилдиимидазол (49,4 мг, 0,305 ммоль) одной порцией и реакционную смесь перемешивали при комнатной температуре в течение 45 минут, затем вводили раствор С77 (104 мг, 0,290 ммоль) в ацетонитриле (2 мл). После перемешивания реакционной смеси при комнатной температуре в течение 3 часов смесь разбавляли водным раствором хлорида аммония и полученную смесь экстрагировали три раза этилацетатом. Объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (градиент: 20% до 100% этилацетата в гептане) давала смесь 1 и 2 в виде белого твердого вещества (105 мг), ЖХМС m/z 483,3 (наблюдаемый изотопный паттерн хлора) [М+Н]+. Разделение диастереомеров осуществляли сверхкритической флюидной хроматографией [Колонка: Chiral Technologies Chiralpak IB, 21×250 мм, 5 мкм; Подвижная фаза 85:15 диоксид углерода/(0,2% гидроксида аммония в метаноле); Скорость потока: 75 мл/мин; Обратное давление: 200 бар]; первый элюирующий диастереомер был указан как 1 {(2R)-2-(5-хлор-2-метоксипиридин-4-ил)-1-[7-метил-6-(2-метил-2H-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-он, DIAST-1}, а второй элюирующий диастереомер как 2 {(2R)-2-(5-хлор-2-метоксипиридин-4-ил)-1-[7-метил-6-(2-метил-2Н-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-он, DIAST-2}.
1 - Выход: 7,2 мг, 15 мкмоль, 5%. ЖХМС m/z 483,2 [М+Н]+. 1Н ЯМР (400 МГц, метанол-d4) δ [8,15 (с) и 8,14 (с), всего 1Н], [7,87 (с) и 7,83 (с), всего 1Н], [6,81 (с) и 6,75 (с), всего 1Н], [4,39 (с) и 4,39 (с), всего 3Н], [4,31 (кв., J=6,8 Гц) и 4,22 (кв., J=6,9 Гц), всего 1Н], 3,90 (с, 3Н), [3,9-3,81 (м) и 3,76-3,52 (м), всего 3Н], [3,48 (д, компонент АВ квартета, J=12,3 Гц) и 3,35 (д, J=10,7 Гц), всего 1Н], [2,93-2,72 (м) и 2,6-2,46 (м), всего 2Н], [2,60 (с) и 2,58 (с), всего 3Н], 2,16-1,84 (м, 3Н), 1,80-1,72 (м, 1Н), [1,43 (д, J=6,8 Гц) и 1,42 (д, J=6,9 Гц), всего 3Н]. Время удерживания: 2,32 минут [Аналитические условия. Колонка: Chiral Technologies Chiralpak IB, 4,6×100 мм, 5 мкм; Подвижная фаза 3:2 диоксид углерода/(0,2% гидроксида аммония в метаноле); Скорость потока: 1,5 мл/мин; Обратное давление: 120 бар].
2 - Выход: 7,9 мг, 16 мкмоль, 6%. ЖХМС m/z 483,2 [М+Н]+. Время удерживания: 2,53 минут (Аналитические условия идентичны условиям, используемым для 1).
Примеры 3 и 4
2-(6-Метокси-2-метилпиримидин-4-ил)-1-[(2S)-7-метил-6-(2-метил-2Н-тетразол-5-ил)-3,4-дигидро-2Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-он, DIAST-1 (3) и 2-(6-Метокси-2-метилпиримидин-4-ил)-1-[(2S)-7-метил-6-(2-метил-2Н-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-он, DIAST-2 (4)
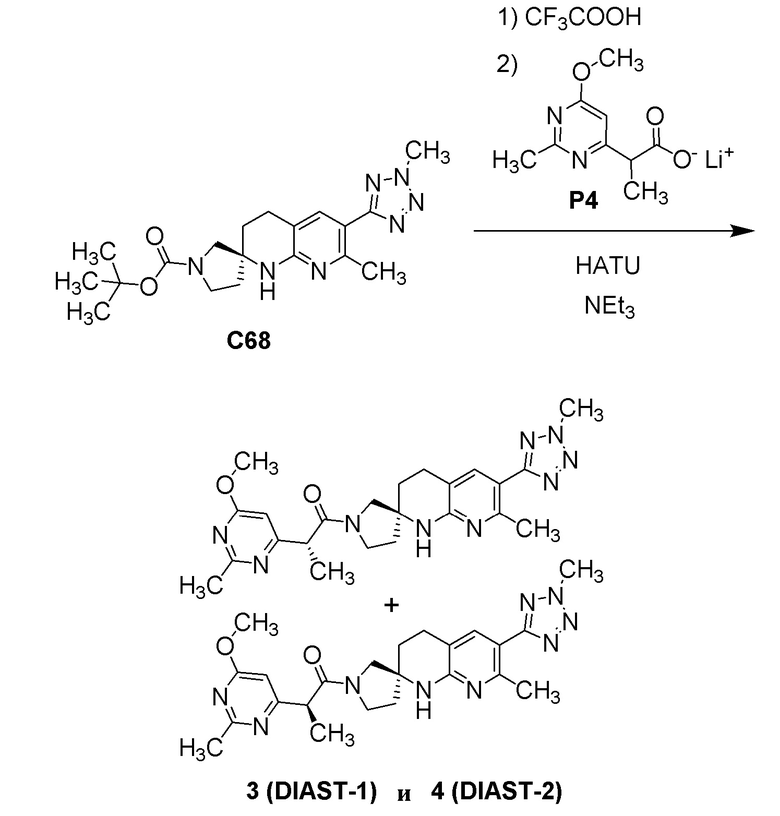
Трифторуксусную кислоту (2 мл) добавляли к раствору С68 (280 мг, 0,72 6 ммоль) в дихлорметане (10 мл) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Затем смесь концентрировали в вакууме и упаривали два раза с этилацетатом с получением вещества, у которого удалена защита, в виде темно-коричневого масла (200 мг), ЖХМС m/z 286,3 [М+Н]+. Часть этого масла (35 мг) и Р4 (24,9 мг, 0,123 ммоль) растворяли в дихлорметане (3 мл) и обрабатывали О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфатом (HATU; 70,0 мг, 0,184 ммоль) и триэтиламином (51,3 мкл, 0,368 ммоль), а затем N,N-диметилформамидом (2 капли) для облегчения растворимости. После перемешивания реакционной смеси при комнатной температуре в течение ночи смесь разбавляли дихлорметаном, промывали последовательно водным раствором бикарбоната натрия и насыщенным водным раствором хлорида натрия, фильтровали, сушили и концентрировали при пониженном давлении. Разделение составляющих диастереомеров осуществляли с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralpak IA, 21×250 мм, 5 мкм; Подвижная фаза: 7:3 диоксид углерода/(0,5% гидроксида аммония в метаноле); Скорость потока: 75 мл/мин; Обратное давление: 120 бар]; первый элюирующий диастереомер был указан как 3 {2-(6-метокси-2-метилпиримидин-4-ил)-1-[{2S)-7-метил-6-(2-метил-2H-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-он, DIAST-1}, а второй элюирующий диастереомер как 4 {2-(6-метокси-2-метилпиримидин-4-ил)-1-[(2S)-7-метил-6-(2-метил-2H-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-он, DIAST-2}.
3 - Выход: 3,1 мг, 6,7 мкмоль, 5%. ЖХМС m/z 464,3 [М+Н]+. 1Н ЯМР (400 МГц, метанол-d4) δ [7,86 (с) и 7,85 (с), всего 1Н], [6,65 (с) и 6,61 (с), всего 1Н], [4,39 (с) и 4,39 (с), всего 3Н], [4,05 (кв., J=7,0 Гц), 4,01-3,89 (м), 3,88-3,55 (м), 3,59 (с) и 3,53 (с), всего 5Н], [3,98 (с) и 3,96 (с), всего 3Н], 2,95-2,75 (м, 2Н), [2,60 (с), 2,58 (с) и 2,55 (с), всего 6Н], 2,19-1,71 (м, 4Н), [1,46 (д, J=7,1 Гц) и 1,44 (д, J=7,1 Гц), всего 3Н]. Время удерживания: 2,47 мин [Аналитические условия. Колонка: Chiral Technologies Chiralpak IA, 4,6×100 мм, 5 мкм; Подвижная фаза: 65:35 диоксид углерода/(метанол, содержащий 0,5% гидроксида аммония); Скорость потока: 1,5 мл/мин; Обратное давление: 120 бар].
4 - Выход: 3,6 мг, 7,8 мкмоль, 6%. ЖХМС m/z 486, 3 [M+Na+]. Время удерживания: 2,92 мин (Аналитические условия идентичны условиям, используемым для 3).
Альтернативный синтез Примеров 3 и 4
2-(6-Метокси-2-метилпиримидин-4-ил)-1-[(2S)-7'-метил-6-(2-метил-2Н-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-он, DIAST-1 (3) и 2-(6-Метокси-2-метилпиримидин-4-ил)-1-[(2S)-7-метил-6-(2-метил-2Н-тетразол-5-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-он, DIAST-2 (4)
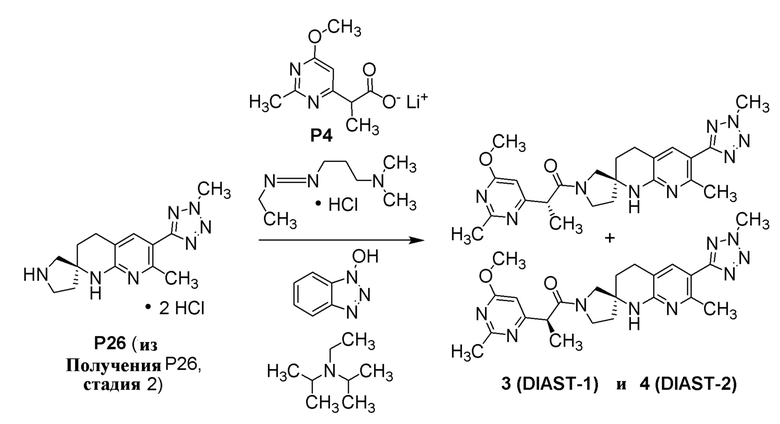
Раствор Р26 (вещество из Получения Р26; 105 мг, 0,293 ммоль), Р4 (69,0 мг, 0,352 ммоль), гидрохлорида 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (EDCI; 169 мг, 0,882 ммоль), 1H-бензотриазол-1-ола (119 мг, 0,881 ммоль) и N,N-диизопропилэтиламина (0,255 мл, 1,46 ммоль) в N,N-диметилформамиде (3 мл) перемешивали при 25°С в течение 16 часов. Реакционную смесь затем разбавляли водой (40 мл) и экстрагировали этилацетатом (3×30 мл) и объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле (градиент: 0% до 10% метанола в дихлорметане) осуществляли разделение двух диастереомеров с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralcel OJ, 30×250 мм, 5 мкм; Подвижная фаза: 85:15 диоксид углерода/(2-пропанол, содержащий 0,2% пропан-2-амина); Скорость потока: 80 мл/мин; Обратное давление: 100 бар]. Первый элюирующий диастереомер был указан как 3 {2-(6-метокси-2-метилпиримидин-4-ил)-1-[(2S)-7-метил-6-(2-метил-2H-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-он, DIAST-1}, а второй элюирующий диастереомер как 4 {2-(6-метокси-2-метилпиримидин-4-ил)-1-[(2S)-7-метил-6-(2-метил-2H-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-он, DIAST-2}.
3 - Выход: 30 мг, 65 мкмоль, 22%. ЖХМС m/z 464,2 [М+Н]+. 1Н ЯМР (400 МГц, метанол-d4) δ [7,86 (с) и 7,84 (с), всего 1Н], [6,65 (с) и 6,61 (с), всего 1Н], [4,39 (с) и 4,39 (с), всего 3Н], [4,04 (кв., J=7,0 Гц), 4,00-3,89 (м), 3,88-3,60 (м), 3,59 (с) и 3,53 (с), всего 5Н], [3,97 (с) и 3,96 (с), всего 3Н], [2,94-2,74 (м) и 2,67-2,59 (м), всего 2Н], [2,60 (с), 2,58 (с) и 2.55 (с), всего 6Н], [2,16-2,06 (м) и 2,06-1,71 (м), всего 4Н], [1,46 (д, J=7,1 Гц) и 1,44 (д, J=7,1 Гц), всего 3Н]. Время удерживания: 4,92 мин (Аналитические условия. Колонка: Chiral Technologies Chiralcel OJ, 4,6×250 мм, 5 мкм; Подвижная фаза А: диоксид углерода; Подвижная фаза В: 2-пропанол, содержащий 0,2% пропан-2-амина; Градиент: 5% В в течение 1,00 минут, затем 5% до 60% В в течение 8,00 минут; Скорость потока: 3,0 мл/мин; Обратное давление: 120 бар).
4 - Выход: 30 мг, 65 мкмоль, 22%. ЖХМС m/z 464,2 [М+Н]+. 1Н ЯМР (400 МГц, метанол-d4) δ 7,85 (с, 1Н), [6,62 (с) и 6,59 (с), всего 1Н], [4,40 (с) и 4,39 (с), всего 3Н], [4,04 (кв., J=7,1 Гц), 3,98-3,85 (м), 3,77-3,60 (м), 3,58 (д, компонент АВ квартета, J=10,6 Гц) и 3, 55-3, 48 (м), всего 5Н], [3,96 (с) и 3,91 (с), всего 3Н], 2,92-2,76 (м, 2Н), [2,59 (с), 2,57 (с), 2,56 (с) и 2,37 (с), всего 6Н], [2,21-2,09 (м), 2,08-2,01 (м) и 2,01-1,78 (м), всего 4Н], [1,47 (д, J=6,9 Гц) и 1,42 (д, J=7,0 Гц), всего 3Н]. Время удерживания: 5,05 мин (Аналитические условия идентичны условиям, используемым для 3).
Отнесение двух диастереомеров к соединениям 3 и 4 осуществляли на основании схожести спектров 1Н ЯМР этого первого элюирующего энантиомера (3) с образцом 3 из Примеров 3 и 4, описанных выше. Дополнительное подтверждение получали путем сравнения хроматографического времени удерживания для этой партии 3 с продуктами из Примеров 3 и 4, описанных выше:
Время удерживания 3 из Альтернативного синтеза Примеров 3 и 4: 2,28 мин
Время удерживания 3 из Примеров 3 и 4: 2,4 6 мин
Время удерживания 4 из Примеров 3 и 4: 2,91 мин
Эти анализы осуществляли с использованием одного и того же аналитического способа: [Колонка: Chiral Technologies Chiralpak IA, 4,6×100 мм, 5 мкм; Подвижная фаза: 65:35 диоксид углерода/(метанол, содержащий 0,5% гидроксида аммония); Скорость потока: 1,5 мл/мин; Обратное давление: 120 бар].
Биологическая активность (Ki) соответствующих примеров из этих двух экспериментов также соответствовала указанным отнесениям (данные от индивидуальных партий, которые представлены в Таблице 2):
Пример 3 из Примеров 3 и 4: 0,36 нМ
Пример 3 из Алвтернативного синтеза Примеров 3 и 4: 1,2 нМ
Пример 4 из Примеров 3 и 4: 25 нМ
Пример 4 из Алвтернативного синтеза Примеров 3 и 4: 34 нМ
Примеры 5 и 6
2-[6-(Дифторметокси)пиридин-3-ил]-1-[(2S)-1-метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-он, DIAST-1 (5) и 2-[6-(Дифторметокси)пиридин-3-ил]-1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-он, DIAST-2 (6)
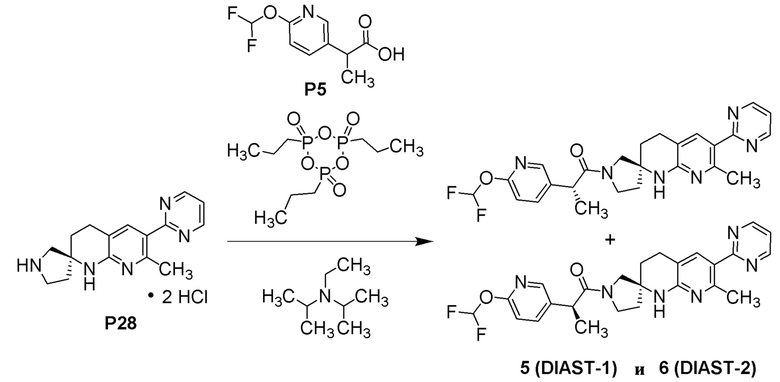
Смесь Р28 (50 мг, 0,14 ммоль), Р5 (30,6 мг, 0,141 ммоль), N,N-диизопропилэтиламина (0,12 мл, 0,69 ммоль) и 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинан 2,4,6-триоксида (50% раствор в этилацетате; 0,25 мл, 0,42 ммоль) в дихлорметане (10 мл) перемешивали при 25°С в течение 16 часов, затем смесь разбавляли водой (20 мл) и экстрагировали дихлорметаном (2×20 мл). Объединенные органические слои последовательно промывали водным раствором бикарбоната натрия (30 мл) и насыщенным водным раствором хлорида натрия (30 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (градиент: 0% до 10% метанола в дихлорметане) давала смесь 5 и 6; эти диастереомеры разделяли с использованием обращеннофазовой ВЭЖХ (Колонка: Chiral Technologies Chiralpak IE; 50×250 мм; 10 мкм; Подвижная фаза: 95:5 этанол/ацетонитрил; Скорость потока: 60 мл/мин). Первый элюирующий диастереомер был указан как 5 {2-[6-(дифторметокси)пиридин-3-ил]-1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-он, DIAST-1}, а второй элюирующий диастереомер был указан как 6 {2-[6-(дифторметокси)пиридин-3-ил]-1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-он, DIAST-2}; оба были выделены в виде белого твердого вещества.
5 - Выход: 10 мг, 21 мкмоль, 15%. ЖХМС m/z 481,3 [М+Н]+. 1H ЯМР (400 МГц, метанол-d4) δ [8,82 (д, J=4,9 Гц) и 8,81 (д, J=4,9 Гц), всего 2Н], [8,19 (д, J=2,5 Гц) и 8,12 (д, J=2,5 Гц), всего 1Н], 7,88-7,76 (м, 2Н), [7,52 (т, JHF=73,2 Гц) и 7,43 (т, JHF=13,1 Гц), всего 1Н], [7,31 (т, J=4,9 Гц) и 7,31 (т, J=4,9 Гц), всего 1Н], [6,96 (д, J=8,5 Гц) и 6,89 (д, J=8,5 Гц), всего 1Н], [4,07 (кв., J=6,9 Гц), 4,03-3,91 (м), 3,74-3,63 (м), 3,60 (д, компонент АВ квартета, J=12,1 Гц), 3,58-3,51 (м), 3,44 (д, J=12,4 Гц) и 3,40 (д, J=10,6 Гц), всего 5Н], 2,92-2,77 (м, 2Н), [2,58 (с) и 2,54 (с), всего 3Н], [2,22-2,10 (м), 2,08-1,93 (м) и 1,93-1,77 (м), всего 4Н], [1,46 (д, J=6,9 Гц) и 1,42 (д, J=6,9 Гц), всего 3Н]. Время удерживания: 7,12 мин (Аналитические условия. Колонка: Chiral Technologies Chiralpak AY-H; 4,6×250 мм; Подвижная фаза: 95:5:0,1 этанол/ацетонитрил/диэтиламин; Скорость потока: 0,6 мл/мин).
6 - Выход: 9,8 мг, 2 0 мкмоль, 14%. ЖХМС m/z 481,3 [М+Н]+. 1H ЯМР (400 МГц, метанол-d4) δ [8,81 (д, J=4,9 Гц) и 8,80 (д, J=4,9 Гц), всего 2Н], [8,21 (д, J=2,5 Гц) и 8,16 (д, J=2,5 Гц), всего 1Н], 7,90-7,78 (м, 2Н), [7,54 (т, JHF=73,2 Гц) и 7,53 (т, JHF=73,2 Гц), всего 1Н], [7,31 (т, J=4,9 Гц) и 7,30 (т, J=4,9 Гц), всего 1Н], [6,98 (д, J=8,5 Гц) и 6,96 (д, J=8,5 Гц), всего 1Н], [4,08 (кв., J=6,9 Гц) и 4,00 (кв., J=6,9 Гц), всего 1Н], [3,95-3,87 (м), 3,78-3,54 (м), 3,51 (АВ квартет, JAB=12,3 Гц, ΔνAB=33,2 Гц) и 3,39 (д, J=10,7 Гц), всего 4Н], [2,94-2,71 (м) и 2,62-2,49 (м), всего 2Н], [2,57 (с) и 2,54 (с), всего 3Н], [2,16-2,04 (м) и 2,02-1,84 (м), всего 3Н], 1,78-1,70 (м, 1Н), [1,45 (д, J=7,0 Гц) и 1,42 (д, J=7,0 Гц), всего 3Н]. Время удерживания: 10,66 мин (Аналитические условия идентичны условиям, используемым для 5).
Примеры 7 и 8
1-[(2S)-7'-Метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]-2-[4-(трифторметил)фенил]пропан-1-он, DIAST-1 (7) и 1-[(2S)-7-Метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]-2-[4-(трифторметил)фенил]пропан-1-он, DIAST-2 (8)
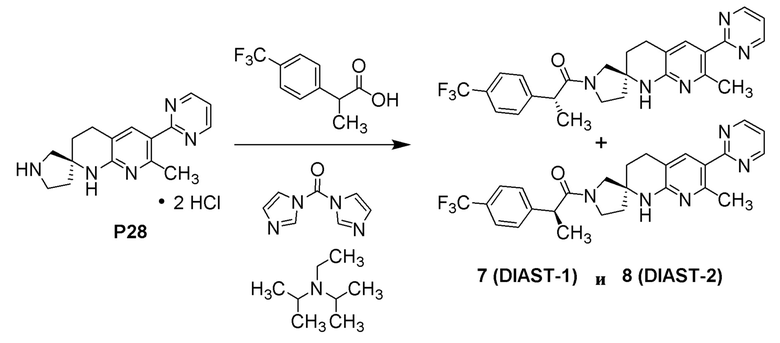
1,1'-Карбонилдиимидазол (24 0 мг, 1,4 8 ммоль) добавляли по порциям к раствору 2-[4-(трифторметил)фенил]пропановой кислоты (323 мг, 1,48 ммоль) в ацетонитриле (5 мл). После перемешивания реакционной смеси при комнатной температуре в течение 45 минут добавляли смесь Р28 (500 мг, 1,41 ммоль) и N,N-диизопропилэтиламина (0,504 мл, 2,89 ммоль) в ацетонитриле (2 мл). Перемешивание продолжали при комнатной температуре в течение 18 часов, затем реакционную смесь экстрагировали этилацетатом. Объединенные органические слои промывали последовательно насыщенным водным раствором бикарбоната натрия и насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Остаток разделяли на составляющие его диастереомеры сверхкритической флюидной хроматографией {Колонка: Chiral Technologies Chiralcel OJ, 30×250 мм, 5 мкм; Подвижная фаза 85:15 диоксид углерода/[метанол, содержащий 0,2% (7 М аммиака в метаноле)]; Скорость потока: 80 мл/мин; Обратное давление: 100 бар). Первый элюирующий диастереомер был указан как 7 {1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]-2-[4-(трифторметил)фенил]пропан-1-он, DIAST-1}, а второй элюирующий диастереомер был указан как 8 (1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]-2-[4-(трифторметил)фенил]пропан-1-он, DIAST-2}; оба были выделены в виде твердых веществ.
7 - Выход: 250 мг, 0,519 ммоль, 3%. ЖХМС m/z 482,4 [М+Н]+. 1H ЯМР (400 МГц, метанол-d4) δ [8,80 (д, J=4,9 Гц) и 8,79 (д, J=4,9 Гц), всего 2Н], [7,83 (с) и 7,75 (с), всего 1Н], 7,68-7,62 (м, 2Н), [7,54 (д, компонент АВ квартета, J=8, 1 Гц) и 7,49 (д, компонент АВ квартета, J=8,1 Гц), всего 2Н], [7,28 (т, J=4,9 Гц) и 7,28 (т, J=4,9 Гц), всего 1Н], [4,10 (кв., J=6,9 Гц) и 4,00 (кв., J=6,9 Гц), всего 1Н], [3,92-3,83 (м) и 3,71 (ддд, J=12,5, 8,5, 6,2 Гц), всего 1Н], [3,62-3,46 (м), 3,46 (д, компонент АВ квартета, J=12,3 Гц) и 3,26 (д, J=10,7 Гц), всего 3Н], [2,91-2,75 (м), 2,68-2,58 (м) и 2,35-2,25 (м), всего 2Н], [2,56 (с) и 2,53 (с), всего 3Н], [2,13-1,99 (м) и 1,99-1,81 (м), всего 3Н], 1,66-1,58 (м, 1Н), [1,45 (д, J=6,9 Гц) и 1,42 (д, J=6,9 Гц), всего 3Н]. Время удерживания: 4,28 мин [Колонка: Chiral Technologies Chiralcel OJ, 4,6×250 мм, 5 мкм; Подвижная фаза А: диоксид углерода; Подвижная фаза В: метанол, содержащий 0,2% (7 М аммиака в метаноле); Градиент: 5% В в течение 1,0 минуты, затем 5% до 60% В в течение 8,0 минут; Скорость потока: 3,0 мл/мин; Обратное давление: 120 бар].
8 - Выход: 260 мг, 0,540 ммоль, 38%. ЖХМС m/z 482,4 [М+Н]+. 1H ЯМР (400 МГц, метанол-d4) δ [8,80 (д, J=4,9 Гц) и 8,79 (д, J=4,9 Гц), всего 2Н], [7,82 (с) и 7,81 (с), всего 1Н], [7,64 (д, компонент АВ квартета, J=8,1 Гц) и 7,57 (д, компонент АВ квартета, J=8,2 Гц), всего 2Н], [7,52 (д, компонент АВ квартета, J=8, 1 Гц) и 7,47 (д, компонент АВ квартета, J=8,2 Гц), всего 2Н], [7,29 (т, J=4,9 Гц) и 7,28 (т, J=4,9 Гц), всего 1Н], [4,09 (кв., J=6,9 Гц) и 4,03 (кв., J=6,9 Гц), всего 1Н], [3,96-3,87 (м) и 3,46-3,37 (м), всего 1Н], [3,73-3,63 (м), 3,52 (АВ квартет, JAB=12,3 Гц, ΔνAB=62,6 Гц) и 3,27 (д, J=10,6 Гц), всего 3Н]. 2,90-2,71 (м, 2Н), [2,57 (с) и 2,53 (с), всего 3Н], [2,15-2,05 (м), 2,04-1,90 (м) и 1,89-1,70 (м), всего 4Н], [1,45 (д, J=6,9 Гц) и 1,43 (д, J=6,9 Гц), всего 3Н]. Время удерживания: 4,74 мин (Аналитические условия идентичны условиям, используемым для 7).
Примеры 9, 10, 11 и 12
1-(4,7-Диметил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил)-2-(4-фторфенил)этан-1-он, DIAST-1 (9), 1-(4,7-Диметил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил)-2-(4-фторфенил)этан-1-он, DIAST-2 (10), 1-(4,7-Диметил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил)-2-(4-фторфенил)этан-1-он, DIAST-3 (11) и 1-(4,7-Диметил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил)-2-(4-фторфенил)этан-1-он, DIAST-4 (12)
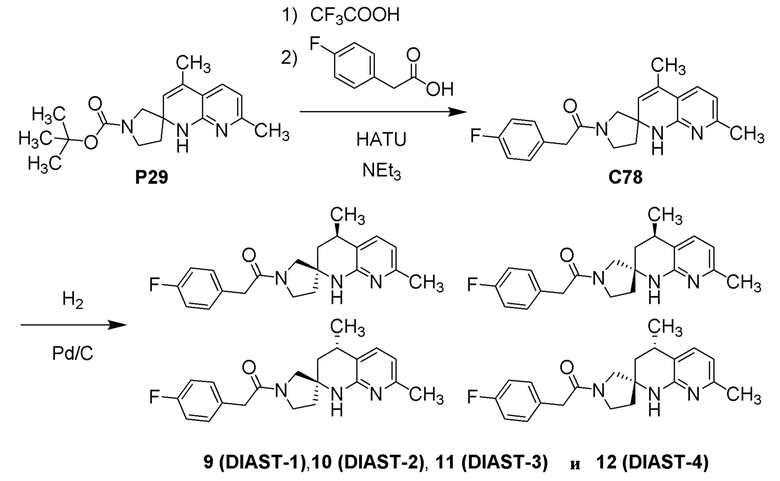
Стадия 1. Синтез 1-(4,7-диметил-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил)-2-(4-фторфенил)этан-1-она (С78).
Трифторуксусную кислоту (0,5 мл) добавляли к раствору Р29 (30 мг, 95 мкмоль) в дихлорметане (3 мл) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. После удаления летучих веществ концентрированием в вакууме остаток два раза совместно упаривали с этилацетатом и гептаном, затем растворяли в дихлорметане (5 мл). К этому раствору добавляли триэтиламин (13,3 мкл, 95,4 мкмоль), (4-фторфенил)уксусную кислоту (14,7 мг, 95,4 мкмоль) и О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфат (HATU; 36,2 мг, 95,2 мкмоль). После перемешивания реакционной смеси при комнатной температуре в течение 1 часа смесь концентрировали в вакууме и очищали хроматографией на силикагеле (градиент: 0% до 10% метанола в дихлорметане) с получением С78 в виде не совсем белого порошка. Выход: 34 мг, количественный. ЖХМС m/z 352,2 [М+Н]+.
Стадия 2. Синтез 1-(4,1'-диметил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил)-2-(4-фторфенил)этан-1-она, DIAST-1 (9), 1-(4,1-диметил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил)-2-(4-фторфенил)этан-1-она, DIAST-2 (10), 1-(4,7-диметил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил)-2-(4-фторфенил)этан-1-она, DIAST-3 (11) и 1-(4,7-диметил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил)-2-(4-фторфенил)этан-1-она, DIAST-4 (12).
Раствор С78 (22 мг, 63 мкмоль) в метаноле (3 мл) обрабатывали палладием на углероде (10%; 5 мг) и гидрировали в течение ночи при 50 ф/дюйм2 (3,515 кг/см2). Реакционную смесь затем фильтровали, концентрировали в вакууме и подвергали сверхкритической флюидной хроматографии (Колонка: Chiral Technologies Chiralcel OJ-H, 5 мкм; Подвижная фаза А: диоксид углерода; Подвижная фаза В: метанол, содержащий 0,2% гидроксида аммония; Градиент: 3% до 5% В; Скорость потока: 75 мл/мин; Обратное давление: 200 бар) для разделения четырех диастереомеров. Первый элюирующий диастереомер был указан как 9 {1-(4,7-диметил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил)-2-(4-фторфенил)этан-1-он, DIAST-1}, элюирующий вторым - как 10 {1-(4,7-диметил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил)-2-(4-фторфенил)этан-1-он, DIAST-2}, элюирующий третьим - как 11 {1-(4,7-диметил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил)-2-(4-фторфенил)этан-1-он, DIAST-3} и элюирующий четвертым - как 12 {1-(4,7-диметил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил)-2-(4-фторфенил)этан-1-он, DIAST-4}.
9 - Выход: 1,2 мг, 3,4 мкмоль, 5%. ЖХМС m/z 354,3 [М+Н]+. 1Н ЯМР (400 МГц, хлороформ-d) δ 7,31-7,21 (м, 2Н, предположительно; частично скрыт пиком растворителя), 7,21-7,16 (м, 1Н), 7,05-6,94 (м, 2Н), [6,48 (д, J=7,5 Гц) и 6,47 (д, J=7,5 Гц), всего 1Н], [3,77-3,52 (м) и 3,44 (д, компонент АВ квартета, J=12,1 Гц), всего 4Н], [3,62 (с) и 3,39 (с), всего 2Н], [2,90-2,77 (м) и 2,61-2,48 (м), всего 1Н], 2,33 (с, 3Н), 2,13-2,03 (м, 1Н), 2,02-1,94 (м, 1Н), 1,89-1,74 (м, 1Н), [1,33 (д, d=6,7 Гц) и 1,28 (д, J=6,1 Гц), всего 3Н]. Время удерживания: 2,77 мин (Аналитические условия. Колонка: Chiral Technologies Chiralcel OJ-H, 4,6×100 мм, 5 мкм; Подвижная фаза: 85:15 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 1,5 мл/мин; Обратное давление: 120 бар).
10 - Выход: 1,3 мг, 3,7 мкмоль, 6%. ЖХМС m/z 354,3 [М+Н]+. 1Н ЯМР (400 МГц, хлороформ-d) δ 7,30-7,19 (м, 3Н, предположительно; частично скрыт пиком растворителя), 7,06-6,98 (м, 2Н), [6,48 (д, d=7,4 Гц) и 6,47 (д, J=7,4 Гц), всего 1Н], [3,75-3,55 (м) и 3,50-3,40 (м), всего 6Н], 2,95-2,82 (м, 1Н), 2,33 (с, 3Н), [2,13-1,79 (м) и 1,74-1,66 (м, предположительно; частично скрыт пиком воды), всего 4Н], 1,36-1,30 (м, 3Н). Время удерживания: 2,92 мин (Аналитические условия идентичны условиям, используемым для 9).
11 - Выход: 1,3 мг, 3,7 мкмоль, 6%. ЖХМС m/z 354,3 [М+Н]+. 1H ЯМР (400 МГц, хлороформ-d) δ 7,29-7,21 (м, 2Н, предположительно; частично скрыт пиком растворителя), 7,21-7,15 (м, 1Н), 7,05-6,93 (м, 2Н), [6,48 (д, d=7,5 Гц) и 6,47 (д, J=7,5 Гц), всего 1Н], [3,74-3,52 (м) и 3,45 (д, компонент АВ квартета, J=12,0 Гц), всего 4Н], [3,62 (с) и 3,39 (с), всего 2Н], [2,90-2,78 (м) и 2,61-2,49 (м), всего 1Н], [2,33 (с) и 2,32 (с), всего 3Н], 2,10-2,04 (м, 1Н), 2,00-1,94 (м, 1Н), 1,88-1,74 (м, 1Н), [1,32 (д, J=6,7 Гц) и 1,28 (д, J=6,7 Гц), всего 3Н]. Время удерживания: 3,48 мин (Аналитические условия идентичны условиям, используемым для 9).
12 - Выход: 2,1 мг, 5,9 мкмоль, 9%. ЖХМС m/z 354,3 [М+Н]+. 1H ЯМР (400 МГц, хлороформ-d) δ 7,29-7,20 (м, 3Н, предположительно; частично скрыт пиком растворителя), 7,06-6,98 (м, 2Н), [6,48 (д, J=7,4 Гц) и 6,46 (д, J=7,4 Гц), всего 1Н], [3,74-3,55 (м) и 3,50-3,40 (м), всего 6Н], 2,95-2,82 (м, 1Н), 2,32 (с, 3Н), [2,12-1,78 (м) и 1,74-1,66 (м, предположительно; частично скрыт пиком воды), всего 4Н], 1,36-1,30 (м, 3Н). Время удерживания: 4,14 мин (Аналитические условия идентичны условиям, используемым для 9).
На основании сравнения данных 1Н ЯМР, 9 и 11 являются энантиомерами друг друга. Подобным образом, 10 и 12 включают пару энантиомеров.
Пример 13
(2R)-2-(5-Фтор-2-метоксипиридин-4-ил)-1-[(2S)-7-метил-6-(2-метил-2H-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-он (13)
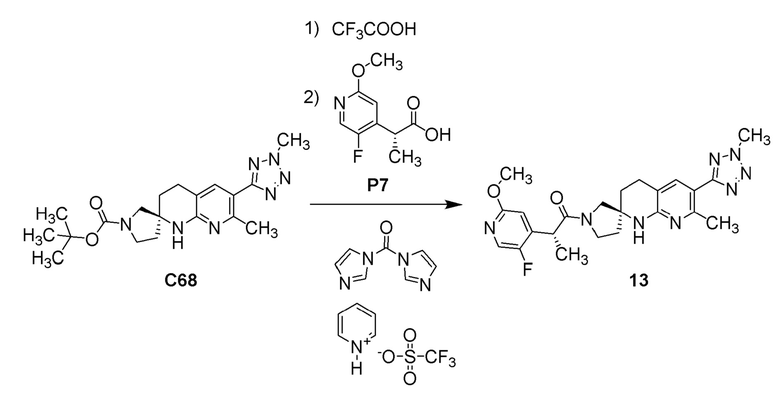
Раствор С68 (280 мг, 0,726 ммоль) в дихлорметане (10 мл) обрабатывали трифторуксусной кислотой (2 мл) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Затем смесь концентрировали в вакууме и выпаривали два раза из этилацетата с получением незащищенного субстрата в виде темно-коричневого масла (200 мг); часть этого вещества использовали в последующей реакции сочетания.
К раствору Р7 (36,4 мг, 0,183 ммоль) в ацетонитриле (3 мл) добавляли трифторметансульфонат пиридиния (88,0 мг, 0,384 ммоль) с последующим добавлением 1,1'-карбонилдиимидазола (31,1 мг, 0,192 ммоль). После перемешивания смеси при комнатной температуре в течение 45 минут добавляли часть незащищенного вещества из полученного выше (73 мг, ≤0,18 ммоль), в виде раствора в ацетонитриле (3 мл), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем смесь распределяли между дихлорметаном и разбавленным водным раствором хлорида аммония; органический слой промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (градиент: 0% до 10% метанола в дихлорметане) с последующей сверхкритической флюидной хроматографией [Колонка: Chiral Technologies Chiralpak IA, 21×250 мм, 5 мкм; Подвижная фаза: 7:3 диоксид углерода/(0,5% гидроксида аммония в метаноле); Скорость потока: 75 мл/мин; Обратное давление: 120 бар] давали (2R)-2-(5-фтор-2-метоксипиридин-4-ил)-1-[(2S)-7-метил-6-(2-метил-2H-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-он (13). Выход: 13,6 мг, 29,1 мкмоль, приблизительно 16%. ЖХМС m/z 489,3 [M+Na+]. Время удерживания: 2,6 мин [Аналитические условия. Колонка: Chiral Technologies Chiralpak IA, 4,6×100 мм, 5 мкм; Подвижная фаза: 65:35 диоксид углерода/(метанол, содержащий 0,5% гидроксида аммония); Скорость потока: 1,5 мл/мин; Обратное давление: 120 бар].
Пример 14
(2R)-2-(5-Фтор-2-метоксипиридин-4-ил)-1-[(2S)-1-метил-6-(пиримидин-2-ил)-3,4- дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-он (14)
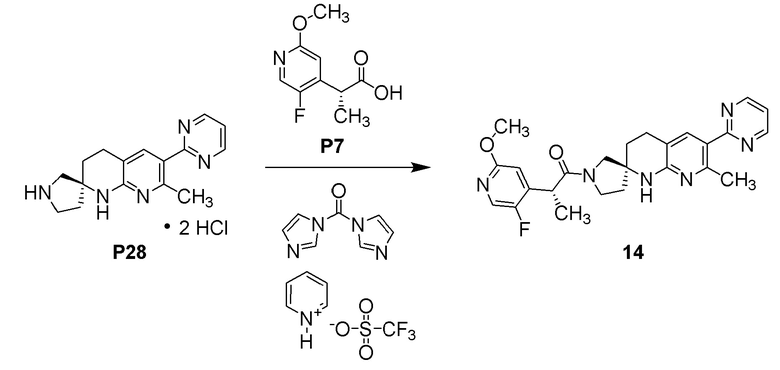
Трифторметансульфонат пиридиния (1,02 г, 4,45 ммоль) добавляли к раствору Р7 (вещество со стадии 2 Альтернативного Получения (#1) Р7; 422 мг, 2,12 ммоль) в ацетонитриле (10 мл). К полученному раствору добавляли 1,1'-карбонилдиимидазол (360 мг, 2,22 ммоль) одной порцией и реакционную смесь оставляли перемешиваться при комнатной температуре в течение 45 минут, затем добавляли раствор Р28 (вещество со стадии 2 Получения Р28; 750 мг, 2,12 ммоль) в ацетонитриле (5 мл) одной порцией. Реакционную смесь перемешивали при комнатной температуре еще в течение 3 часов, затем смесь разбавляли насыщенным водным раствором хлорида аммония и экстрагировали три раза этилацетатом. Объединенные органические слои промывали последовательно насыщенным водным раствором бикарбоната натрия и насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и концентрировали в вакууме; хроматография на силикагеле (градиент: 30% до 100% этилацетата в гептане) давала (2R)-2-(5-фтор-2-метоксипиридин-4-ил)-1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-он (14) в виде белого твердого вещества.
Указанная абсолютная стереохимия была присвоена на основании рентгеноструктурного анализа монокристалла, который осуществляли на соединении 14, полученном в результате кристаллизации этой партии (см. ниже).
Выход: 670 мг, 1,45 ммоль, 68%. ЖХМС m/z 463,4 [М+Н]+. 1Н ЯМР (400 МГц, метанол-d4) δ [8,81 (д, J=4,9 Гц) и 8,80 (д, J=4,9 Гц), всего 2Н], [7,99 (д, J=1,6 Гц) и 7,98 (д, J=1,7 Гц), всего 1Н], [7,84 (с) и 7,81 (с), всего 1Н], [7,30 (т, J=4,9 Гц) и 7,29 (т, J=4,9 Гц), всего 1Н], [6,78 (д, J=4,9 Гц) и 6,73 (д, J=4,9 Гц), всего 1Н], [4,27 (кв., d=6,9 Гц) и 4,19 (кв., d=6,9 Гц), всего 1Н], [3,93-3,83 (м) и 3,76-3,67 (м), всего 1Н], [3,88 (с) и 3,88 (с), всего 3Н], [3,67-3,57 (м), 3,53 (АВ квартет, JAB=12,3 Гц, ΔνAB=34,7 Гц) и 3,39 (д, компонент АВ квартета, J=10,6 Гц), всего 3Н], [2,94-2,72 (м) и 2,63-2,54 (м), всего 2Н], [2,57 (с) и 2,55 (с), всего 3Н], 2,15-1,83 (м, 3Н), 1,83-1,74 (м, 1Н), [1,45 (д, J=6,8 Гц) и 1,43 (д, J=6,8 Гц), всего 3Н].
Перекристаллизация из 3:2 смеси этилацетата и гептана давала вещество с диастереомерным избытком 99,1%; дальнейшая перекристаллизация из ацетонитрила давала монокристалл, который использовали для рентгеноструктурного анализа.
Определение структуры 14 методом рентгеновской дифракции монокристаллов
Рентгеноструктурный анализ монокристалла
Сбор данных осуществляли на дифрактометре D8 Quest Bruker при комнатной температуре. Сбор данных состоял из омега- и фи-сканов.
Структура была разрешена методом внутреннего фазирования с использованием пакета программ SHELX в группе Р1 триклинного класса. Структуру затем уточняли методом наименьших квадратов в полноматричном приближении. Все не-водородные атомы были обнаружены и уточнены с использованием параметров анизотропного смещения.
Атомы водорода, находящиеся на азоте, были обнаружены из разностной карты Фурье и уточнены с ограничением расстояний. Остальные атомы водорода были размещены в рассчитанных положениях, и им давали перемещаться на их атомах-носителях. Конечное уточнение включало параметры изотропного смещения для всех атомов водорода.
Анализ абсолютной структуры с использованием методов вероятности (Hooft, 2008) осуществляли с использованием PLATON (Spek). Результаты показывают, что абсолютная структура присвоена корректно. Этим методом рассчитано, что вероятность того, что структура присвоена корректно, составляет 100%. Параметр Хофта указан как 0,05 с esd (расчетное стандартное отклонение), составляющим (10), а параметр Парсона указан как 0,04 с esd, составляющим (10).
Конечный R-индекс был равен 4,5%. Конечная разностная карта Фурье показала, что нет недостающей или смещенной электронной плотности.
Соответствующая информация, качающаяся кристалла, сбора данных и уточнения структуры, обобщенно представлена в Таблице А. Атомные координаты, длина связей, углы связей и параметры смещения перечислены в Таблицах В - D.
Программа и ссылки
SHELXTL, Version 5.1, Bruker AXS, 1997.
PLATON, A.L. Spek, J. Appl. Cryst. 2003, 36, 7-13.
MERCURY, C.F. Macrae, P.R. Edington, P. McCabe, E. Pidcock, G.P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Cryst. 2006, 39, 453-457.
OLEX2, О.V. Dolomanov, L.J. Bourhis, R.J. Gildea, J.A.K. Howard and H. Puschmann, J. Appl. Cryst. 2009, 42, 339-341.
R.W.W. Hooft, L.H. Straver and A.L. Spek, J. Appl. Cryst. 2008, 41, 96-103.
H.D. Flack, Acta Cryst. 1983, A39, 867-881.


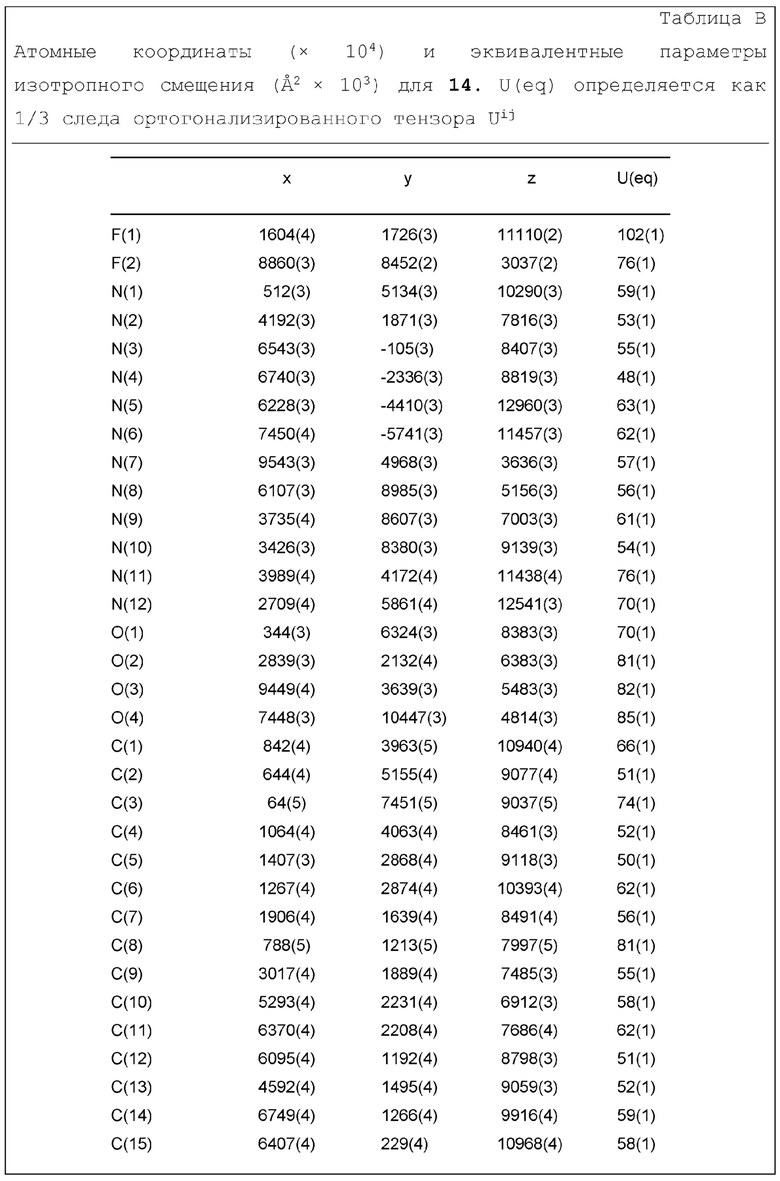
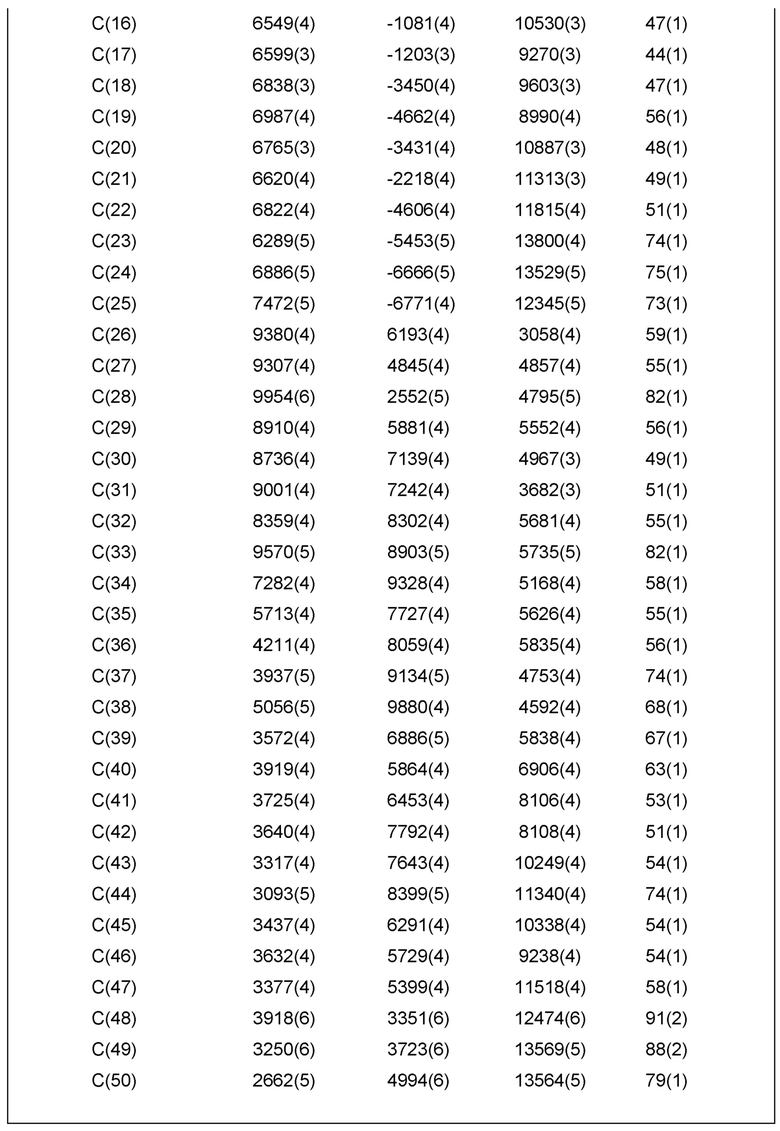









Таким образом, абсолютную стереохимию соединения Примера 14 определяли рентгеноструктурным анализом монокристалла. Фиг. 1 представляет полученную методом рентгенографии монокристаллическую структуру (ORTEP изображение) кристаллического соединения Примера 14: (2R)-2-(5-фтор-2-метоксипиридин-4-ил)-1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она.
В некоторых вариантах осуществления настоящее изобретение предоставляет кристаллическую форму (2R)-2-(5-фтор-2-метоксипиридин-4-ил)-1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она.
В некоторых других вариантах осуществления кристаллическая форма (2R)-2-(5-фтор-2-метоксипиридин-4-ил)-1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она представляет собой соединение, описанное (или полученное) в Примере 14.
Альтернативный синтез Примера 14
(2R)-2-(5-Фтор-2-метоксипиридин-4-ил)-1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-он (14)
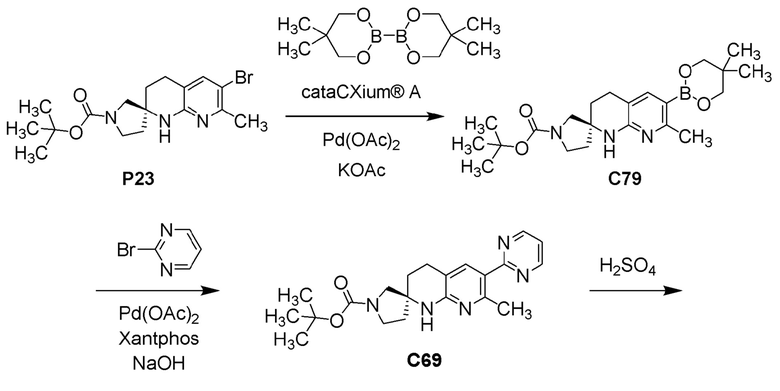
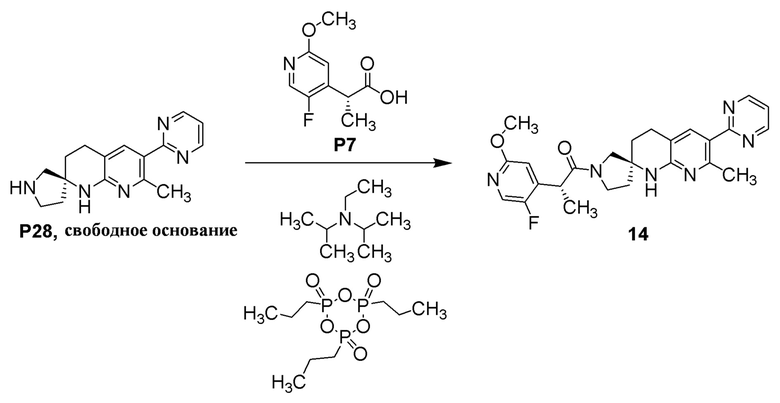
Стадия 1. Синтез трет-бутил-(2S)-6-(5,5-диметил-1,3,2-диоксаборинан-2-ил)-1-метил-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилата (С79).
Ди(1-адамантил)-н-бутилфосфин (cataCXium® А; 2,21 г, 6,16 ммоль), затем ацетат палладия(II) (0,461 мг, 2,05 ммоль), добавляли к 2-метилтетрагидрофурану (170 мл); смесь катализаторов продували аргоном в течение 10-20 минут между каждыми процедурами. Смесь нагревали при кипячении с обратным холодильником в течение 1 часа, затем охлаждали до ≤50°С.
В отдельном реакторе, Р23 (98,2% по массе; 80,0 г, 205 ммоль), 5,5,5',5'-тетраметил-2,2'-би-1,3,2-диоксаборинан (60,3 г, 267 ммоль), ацетат калия (97% по массе; 62,4 г, 617 ммоль) и воду (2,37 мл, 132 ммоль) добавляли к 2-метилтетрагидрофурану (220 мл). Стенки реактора промывали 2-метилтетрагидрофураном (100 мл) и полученную смесь продували аргоном приблизительно в течение 1 часа. Затем добавляли через канюлю смесь катализаторов, меньше чем за 2 минуты, и реакционную смесь нагревали до температуры кипения с обратным холодильником при скорости 1°С/мин. После 4 часов кипячения с обратным холодильником смесь охлаждали до 10°С, выдерживали при этой температуре в течение ночи и быстро обрабатывали по каплям, в течение 15 минут, водным раствором гидроксида натрия (1,0 М; 410 мл, 410 ммоль). Внутреннюю температуру в процессе добавления поддерживали ниже 17°С. Полученную смесь нагревали до 20°С, разбавляли трет-бутилметиловым эфиром (180 мл) и тщательно смешивали в течение 5 минут, после чего было подтверждено, что водный слой имел рН 10. К органическому слою добавляли водный раствор гидроксида натрия (1,0 М; 480 мл, 480 ммоль) четырьмя порциями в течение 4 минут; после перемешивания в течение 5 минут органический слой отделяли и аналогичным образом экстрагировали водным раствором гидроксида натрия (1,0 М; 480 мл, 480 ммоль). Объединенные экстракты гидроксида натрия смешивали с толуолом (240 мл) и обрабатывали по порциям хлористоводородной кислотой (12,2 М; 62,3 мл, 760 ммоль) при скорости, которая поддерживала температуру ниже 30°С. Полученная смесь имела рН 14; добавляли дополнительное количество хлористоводородной кислоты (12,2 М; 34 мл, 415 ммоль) для доведения рН до 10. После перемешивания смеси в течение 5 минут водный слой экстрагировали толуолом (2×240 мл) и толуольные слои объединяли, с получением С79 в виде раствора в толуоле. Основной объем этого вещества использовали на следующей стадии. Расчетный выход: 73,2 г (по данным количественного анализа ЯМР), 176 ммоль, выход 86%, в виде раствора в толуоле.
Стадия 2. Синтез трет-бутил-(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилата (С69).
К раствору С79 в толуоле (с предыдущей стадии; 509 мл, содержащий 72,7 г, 175 ммоль С79) добавляли водный раствор гидроксида натрия (1 М; 530 мл, 530 ммоль) с последующим добавлением 2-бромпиримидина (39,0 г, 245 ммоль). Полученную смесь продували аргоном в течение 30 минут, затем добавляли 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos; 1,27 г, 2,19 ммоль) и ацетат палладия(II) (394 мг, 1,76 ммоль). После нагревания реакционной смеси при 50°С в течение 3,5 часа смесь охлаждали до 20°С, оставляли перемешиваться в течение ночи и фильтровали. Фильтровальную лепешку промывали толуолом (150 мл) и органический слой объединенных фильтратов промывали водой при перемешивании в течение 5 минут и затем смеси давали выстояться в течение 30 минут; твердые вещества в смеси поддерживали в органическом слое, который подвергали молекулярной перегонке при 100 мбар и 60°С. Смесь перегоняли до тех пор, пока не осталось приблизительно 275 мл, затем смесь охлаждали до 20°С при скорости 1°С/мин. После перемешивания смеси в течение 30 минут, в течение которых были обнаружены твердые веществы, медленно добавляли гептан (727 мл) по каплям в течение 30 минут. Полученный раствор перемешивали в течение 10 минут, нагревали до 60°С при скорости 1°С/мин и перемешивали при 60°С в течение 90 минут, затем смесь охлаждали до 20°С при скорости 1°С/мин и оставляли перемешиваться в течение 3 дней. Фильтрование с последующей промывкой твердой фильтровальной лепешки два раза фильтратом и один раз гептаном (220 мл) давали С69 в виде твердого вещества. Выход: 63,85 г, 167,4 ммоль, 96%. ВЭЖХ чистота: 99,4%. 1Н ЯМР (600 МГц, DMSO-d6) δ 8,80 (д, J=4,8 Гц, 2Н), 7,90 (с, 1Н), 7,27 (т, J=4,8 Гц, 1Н), [7,25 (ушир. с) и 7,24 (ушир. с), всего 1Н], 3,56-3,49 (м, 1Н), 3,37-3,30 (м, 1Н), 3,28-3,21 (м, 2Н), 2,80-2,73 (м, 1Н), 2,73-2,65 (м, 1Н), 2,59 (с, 3Н), 1,99-1,84 (м, 2Н), 1,82-1,69 (м, 2Н), [1,41 (с) и 1,39 (с), всего 9Н].
Получение данных рентгеновской порошковой дифракции (PXRD) для кристаллического С69
Образец С69 (полученный, как описано на стадии 2 выше) подвергали порошковому рентгеновскому дифракционному анализу (PXRD), и было обнаружено, что это кристаллическое вещество.
Порошковый рентгеновский дифракционный анализ осуществляли с использованием дифрактометра Bruker AXS D8 Endeavour, оснащенного медным источником излучения (Cu). Щель расходимости устанавливали на постоянное освещение 15 мм. Дифрагированное излучение регистрировалось детектором PSD-Lynx Eye с щелью PSD детектора, установленной на 4,123 градуса. Напряжение и сила тока рентгеновской трубки были установлены на 40 кВ и 40 мА, соответственно. Кроме того, использовали энергодисперсионный детектор, никелевый фильтр. Данные собирали в гониометре Theta-Theta при длине волны Cu от 3,0 до 40,0 градусов 2-тета с использованием размера шага 0,0100 градуса и времени шага 1,0 секунда. Противорассеивающий экран был установлен на фиксированное расстояние 1,5 мм. Образцы подготавливали, помещая их в силиконовый держатель для образцов с низким фоном и вращая их со скоростью 15 об/мин во время сбора. Данные собирали с использованием программного обеспечения Bruker DIFFRAC Plus, а анализ осуществляли с использованием программного обеспечения EVA diffract plus. Файл данных PXRD не обрабатывали перед поиском пиков. Используя алгоритм поиска пиков в программном обеспечении EVA, пики, выбранные с пороговым значением 1, использовали для предварительного отнесения пиков. Для обеспечения достоверности корректировки делались вручную; результаты отнесений, выполненных автоматически, проверяли визуально, а положения пиков были скорректированы по максимуму пиков. Обычно выбирались пики с относительной интенсивностью ≥3%. Обычно не выбирали пики, которые не были разрешены или согласовывались с шумом. Типичная ошибка, связанная с положением пика из PXRD, как указано в USP, до +/- 0,2° 2-тета (USP-941).
Один репрезентативный паттерн дифракции наблюдали для кристаллической формы С69, и он представлен на Фиг. 4. Перечень дифракционных пиков в градусах 2θ и относительных интенсивностей с относительной интенсивностью ≥3,0% PXRD от образца кристаллического С69, показан в Таблице Х-С69 ниже.
В некоторых вариантах осуществления настоящее изобретение предоставляет соединение, которое представляет собой трет-бутил-(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилат или его соль. В некоторых вариантах осуществления настоящее изобретение предоставляет соединение, которое представляет собой трет-бутил-(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилат. В некоторых других вариантах осуществления настоящее изобретение предоставляет кристаллическую форму трет-бутил-(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилата. В некоторых других вариантах осуществления кристаллическая форма трет-бутил-(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилата демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере один характеристический пик, выраженный в градусах 2θ, такой как показано в Таблице Х-С69.
В некоторых вариантах осуществления кристаллическая форма трет-бутил-(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилата демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере два характеристических пика, выраженных в градусах 2θ, которые представлены в Таблице Х-С69. В некоторых вариантах осуществления кристаллическая форма трет-бутил-(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилата демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере три характеристических пика, выраженных в градусах 2θ, которые представлены в Таблице Х-С69. В некоторых вариантах осуществления кристаллическая форма трет-бутил-(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилата демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере четыре (например, 4, 5, 6, 7, 8, 9 или 10) характеристических пика, выраженных в градусах 2θ, которые представлены в Таблице Х-С69. В некоторых вариантах осуществления кристаллическая форма трет-бутил-(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилата демонстрирует порошковую рентгеновскую дифрактограмму, по существу такую, как показано на Фиг. 4.
Стадия 3. Синтез (2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин] (Р28, свободное основание).
Раствор С69 (96% по массе; 50,0 г, 126 ммоль) в воде (100 мл) и 2-пропаноле (150 мл) добавляли в течение 10 минут к 80°С смеси воды (150 мл) и концентрированной серной кислоты (14,5 мл, 272 ммоль). После перемешивания реакционной смеси при 80°С в течение 4 часов смесь охлаждали до 25°С и затем подвергали молекулярной перегонке при 120°С и атмосферном давлении. При достижении в результате перегонки смеси объема приблизительно 200 мл температуру снижали до 50°С, добавляли активированный уголь (Darco G-60; 10 г) и перемешивание продолжали в течение 1,5 часа при 50°С. Смесь затем охлаждали до 25°С и фильтровали с использованием 10 мкм фильтра. Фильтровальную лепешку промывали водой (100 мл) и объединенные фильтраты разбавляли 2-пропанолом (20 мл); полученную смесь, рН 0,86, подщелачивали до точки помутнения, затем осветляли добавлением 6 М водного раствора гидроксида натрия (приблизительно 75 мл). Получали рН=9,32. Смесь обрабатывали по каплям дополнительным количеством 6 М водного раствора гидроксида натрия (приблизительно 20 капель) до рН 9,6-9,7, в этот момент мутность сохранялась. Перемешивание продолжали в течение 45 минут, затем добавляли дополнительное количество 6 М водного раствора гидроксида натрия (всего приблизительно 80 мл, 480 ммоль) и перемешивание продолжали при 20°С в течение 30 минут. Смесь затем нагревали до 50°С при скорости 1°С/мин, перемешивали в течение 1,5 часа и охлаждали до 20°С при скорости 1°С/мин. После перемешивания в течение 1,5 часа смесь фильтровали; фильтровальную лепешку промывали водным раствором гидроксида натрия (1 М; 100 мл, 100 ммоль) и сушили в течение ночи в вакууме при 50°С с получением Р28, свободного основания. Выход: 30,87 г, 98,1% Р28 по данным количественного анализа ЯМР, 108 ммоль, 86%. 1Н ЯМР (600 МГц, DMSO-d6) δ 8,79 (д, J=4,8 Гц, 2Н), 7,88 (с, 1Н), 7,25 (т, J=4,8 Гц, 1Н), 7,01 (с, 1Н), 2,99 (ддд, J=11,0, 8,4, 6,4 Гц, 1Н), 2,79 (ддд, J=10,9, 8,6, 5,6 Гц, 1Н), 2,75-2,68 (м, 3Н), 2,61 (д, J=11,3 Гц, 1Н), 2,58 (с, 3Н), 1,80-1,68 (м, 3Н), 1,65 (ддд, J=12,7, 8,6, 6,4 Гц, 1Н).
Получение данных рентгеновской порошковой дифракции (PXRD) для кристаллического Р28
Образец Р28 (полученный, как описано на стадии 3 выше) подвергали порошковому рентгеновскому дифракционному анализу (PXRD), и было обнаружено, что это кристаллическое вещество.
Порошковый рентгеновский дифракционный анализ осуществляли с использованием дифрактометра Bruker AXS D8 Endeavour, оснащенного медным источником излучения (Cu). Щель расходимости устанавливали на постоянное освещение 15 мм. Дифрагированное излучение регистрировалось детектором PSD-Lynx Eye с щелью PSD детектора, установленной на 4,123 градуса. Напряжение и сила тока рентгеновской трубки были установлены на 40 кВ и 40 мА, соответственно. Кроме того, использовали энергодисперсионный детектор, никелевый фильтр. Данные собирали в гониометре Theta-Theta при длине волны Cu от 3,0 до 40,0 градусов 2-тета с использованием размера шага 0,0100 градуса и времени шага 1,0 секунда. Противорассеивающий экран был установлен на фиксированное расстояние 1,5 мм. Образцы подготавливали, помещая их в силиконовый держатель для образцов с низким фоном и вращая их со скоростью 15 об/мин во время сбора. Данные собирали с использованием программного обеспечения Bruker DIFFRAC Plus, а анализ осуществляли с использованием программного обеспечения EVA diffract plus. Файл данных PXRD не обрабатывали перед поиском пиков. Используя алгоритм поиска пиков в программном обеспечении EVA, пики, выбранные с пороговым значением 1, использовали для предварительного отнесения пиков. Для обеспечения достоверности корректировки делались вручную; результаты отнесений, выполненных автоматически, проверяли визуально, а положения пиков были скорректированы по максимуму пиков. Обычно выбирались пики с относительной интенсивностью ≥3%. Обычно не выбирали пики, которые не были разрешены или согласовывались с шумом. Типичная ошибка, связанная с положением пика из PXRD, как указано в USP, до +/- 0,2° 2-тета (USP-941).
Один репрезентативный паттерн дифракции наблюдали для кристаллической формы Р28, и он представлен на Фиг. 5. Перечень дифракционных пиков в градусах 2θ и относительных интенсивностей с относительной интенсивностью ≥3,0% PXRD от образца кристаллического Р28 показаны в Таблице Х-Р28 ниже.

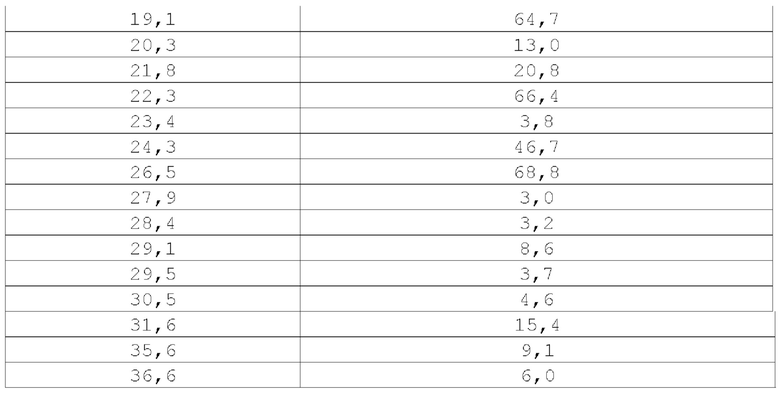
В некоторых вариантах осуществления настоящее изобретение предоставляет соединение, которое представляет собой (2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин] или его соль. В некоторых вариантах осуществления настоящее изобретение предоставляет соединение, которое представляет собой (2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]. В некоторых других вариантах осуществления настоящее изобретение предоставляет кристаллическую форму (2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]. В некоторых других вариантах осуществления кристаллическая форма (2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин] демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере один характеристический пик, выраженный в градусах 2θ, такой как показано в Таблице Х-Р28.
В некоторых вариантах осуществления кристаллическая форма (2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин] демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере два характеристических пика, выраженных в градусах 2θ, которые представлены в Таблице Х-Р28. В некоторых вариантах осуществления кристаллическая форма (2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин] демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере три характеристических пика, выраженных в градусах 2θ, которые представлены в Таблице Х-Р28.
В некоторых вариантах осуществления кристаллическая форма (2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин] демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере четыре (например, 4, 5, 6, 7, 8, 9 или 10) характеристических пика, выраженных в градусах 2θ, которые представлены в Таблице Х-Р28. В некоторых вариантах осуществления кристаллическая форма (2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин] демонстрирует порошковую рентгеновскую дифрактограмму, по существу такую, как показано на Фиг. 5.
Стадия 4. Синтез (2R)-2-(5-фтор-2-метоксипиридин-4-ил)-1-[(2S)-7'-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она (14).
Суспензию Р7 (19,1 г, 95,9 ммоль) в 2-метилтетрагидрофуране (200 мл) обрабатывали соединением Р28 в форме свободного основания (98,1% по массе, 25 г, 87,2 ммоль), затем N,N-диизопропилэтиламином (19 мл, 110 ммоль). Добавляли 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинан 2,4,6-триоксид (раствор 50% по массе в этилацетате; 65 мл, 110 ммоль) в течение 15 минут, при скорости, которая поддерживала внутреннюю температуру реакции ниже 30°С. После перемешивания реакционной смеси в течение 100 минут добавляли водный раствор бикарбоната натрия (1,14 М; 250 мл, 285 ммоль) {Осторожно: выделение газа} и перемешивание продолжали в течение 10 минут при 20°С. Полученную смесь нагревали до 40°С, перемешивали в течение 30 минут и снова обрабатывали водным раствором бикарбоната натрия (1,14 М; 125 мл, 142 ммоль). После перемешивания смеси в течение 80 минут, добавляли воду (75 мл) и перемешивание продолжали в течение 10 минут. Органический слой подвергали перегонке при 60°С и 500 мбар до уменьшения смеси до 5 объемов. Добавляли 2-метилтетрагидрофуран (125 мл), температуру доводили до 45°С-50°С и смесь фильтровали через диатомовую землю. Дополнительное количество 2-метилтетрагидрофурана (50 мл) использовали для промывки фильтровальной лепешки и объединенные фильтраты подвергали перегонке при 60°С и 500 мбар до приблизительно 3 объемов. Температуру повышали до 80°С до осаждения твердых частиц на дно реактора, затем снижали до 50°С. Полученное вещество обрабатывали при 50°С, в течение 15 минут, гептаном (250 мл) и оставляли перемешиваться при 50°С в течение 90 минут. Затем смесь охлаждали до 20°С при скорости 1°С/мин и оставляли перемешиваться в течение 3 дней, затем смесь разбавляли до объема 600 мл добавлением 10 моль% 2-метилтетрагидрофурана в гептане. Фильтрование давало фильтровальную лепешку, которую промывали гептаном (75 мл) и сушили в течение ночи при 50°С в вакууме с получением (2R)-2-(5-фтор-2-метоксипиридин-4-ил)-1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она (14) в виде твердого вещества. Выход: 29,63 г, 64,06 ммоль, 73%. ВЭЖХ чистота: 100%. ЖХМС m/z 463,3 [М+Н]+. 1Н ЯМР (600 МГц, DMSO-d6) δ [8,81 (д, J=4,8 Гц) и 8,80 (д, J=4,7 Гц), всего 2Н], [8,12 (с) и 8,10 (с), всего 1Н], [7,90 (с) и 7,87 (с), всего 1Н], [7,33 (с) и 7,23 (с), всего 1Н], 7,30-7,26 (м, 1Н), [6,75 (д, J=4,8 Гц) и 6,69 (д, J=4,8 Гц), всего 1Н], [4,15 (кв., J=6,9 Гц) и 4,10 (кв., J=6,9 Гц), всего 1Н], [3,83 (с) и 3,82 (с), всего 3Н], [3,78-3,71 (м), 3,61-3,49 (м), 3,47-3,41 (м), 3,42 (д, J=11,2 Гц), 3,32-3,28 (м, предположительно; частично скрыт пиком воды) и 3,25 (д, J=10,4 Гц), всего 4Н], [2,80-2,65 (м) и 2,5-2,43 (м, предположительно; частично скрыт пиком растворителя), всего 2Н], [2,59 (с) и 2,57 (с), всего 3Н], [2,03-1,94 (м) и 1,87-1,72 (м), всего 3Н], 1,67-1,60 (м, 1Н), 1,36-1,30 (м, 3Н).
Получение данных рентгеновской порошковой дифракции (PXRD) для кристаллического Примера 14
Образец Примера 14 (полученный по существу так, как описано в этом Альтернативном способе синтеза, за исключением того, что на стадии 4 фильтрование через диатомовую землю заменяли обработкой смеси веществом SiliaMetS® Thiol с последующим фильтрованием. SiliaMetS® Thiol: Silicycle Inc., Продукт номер R51030B) измельчали и предоставляли для порошкового рентгеновского дифракционного анализа (PXRD), и было обнаружено, что это кристаллическое вещество (указано как Форма I).
Порошковый рентгеновский дифракционный анализ осуществляли с использованием дифрактометра Bruker AXS D8 Endeavour, оснащенного медным источником излучения (Cu). Щель расходимости устанавливали на постоянное освещение 15 мм. Дифрагированное излучение регистрировалось детектором PSD-Lynx Eye с щелью PSD детектора, установленной на 4,123 градуса. Напряжение и сила тока рентгеновской трубки были установлены на 40 кВ и 40 мА, соответственно. Кроме того, использовали энергодисперсионный детектор, никелевый фильтр. Данные собирали в гониометре Theta-Theta при длине волны Cu от 3,0 до 40,0 градусов 2-тета с использованием размера шага 0,0100 градуса и времени шага 1,0 секунда. Противорассеивающий экран был установлен на фиксированное расстояние 1,5 мм. Образцы подготавливали, помещая их в силиконовый держатель для образцов с низким фоном и вращая их со скоростью 15 об/мин во время сбора. Данные собирали с использованием программного обеспечения Bruker DIFFRAC Plus, а анализ осуществляли с использованием программного обеспечения EVA diffract plus. Файл данных PXRD не обрабатывали перед поиском пиков. Используя алгоритм поиска пиков в программном обеспечении EVA, пики, выбранные с пороговым значением 1, использовали для предварительного отнесения пиков. Для обеспечения достоверности корректировки делались вручную; результаты отнесений, выполненных автоматически, проверяли визуально, а положения пиков были скорректированы по максимуму пиков. Обычно выбирались пики с относительной интенсивностью ≥3%. Обычно не выбирали пики, которые не были разрешены или согласовывались с шумом. Типичная ошибка, связанная с положением пика из PXRD, как указано в USP, до +/- 0,2° 2-тета (USP-941).
Один репрезентативный паттерн дифракции наблюдали для Формы I Примера 14, и он представлен на Фиг. 1. Перечень дифракционных пиков в градусах 2θ и относительных интенсивностей с относительной интенсивностью ≥3,0% PXRD от образца кристаллического Примера 14 показаны в Таблице XI ниже.
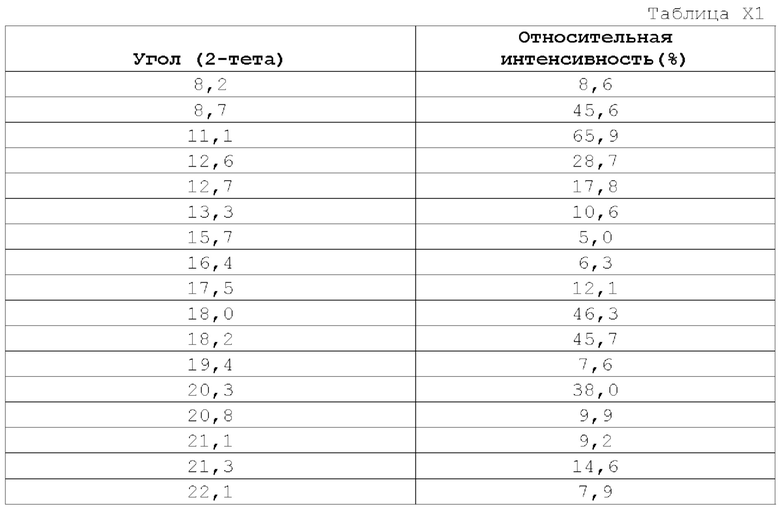
В некоторых вариантах осуществления настоящее изобретение предоставляет кристаллическую форму (2R)-2-(5-Фтор-2-метоксипиридин-4-ил)-1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она. В некоторых других вариантах осуществления настоящее изобретение предоставляет кристаллическую форму (2R)-2-(5-Фтор-2-метоксипиридин-4-ил)-1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она, которая указана как Форма I.
В некоторых вариантах осуществления Форма I (Примера 14) демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере один характеристический пик, выраженный в градусах 2θ, выбранный из 8,7±0,2°; 11,1±0,2°; и 13,3±0,2°. В некоторых вариантах осуществления Форма I демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере один характеристический пик, выраженный в градусах 2θ, при 8,7±0,2°. В некоторых вариантах осуществления Форма I демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере один характеристический пик, выраженный в градусах 2θ, при 11,1±0,2°. В некоторых вариантах осуществления Форма I демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере один характеристический пик, выраженный в градусах 2θ, при 13,3±0,2°. В некоторых вариантах осуществления Форма I демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере два характеристических пика, выраженных в градусах 2θ, выбранных из 8,7±0,2°; 11,1±0,2°; и 13,3±0,2°. В некоторых вариантах осуществления Форма I демонстрирует порошковую рентгеновскую дифрактограмму, включающую два характеристических пика, выраженных в градусах 2θ, выбранных из 8,7±0,2°; и 11,1±0,2. В некоторых вариантах осуществления Форма I демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере три характеристических пика, выраженных в градусах 2θ, выбранных из 8,7±0,2°; 11,1±0,2°; и 13,3±0,2°.
В некоторых вариантах осуществления Форма I демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере два характеристических пика, выраженных в градусах 2θ, выбранных из 8,7±0,2°; 11,1±0,2°; и 26,0±0,2. В некоторых вариантах осуществления Форма I демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере два характеристических пика, выраженных в градусах 2θ, выбранных из 8,7±0,2°; и 26,0±0,2°. В некоторых вариантах осуществления Форма I демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере два характеристических пика, выраженных в градусах 2θ, выбранных из 11,1±0,2°; и 26,0±0,2°. В некоторых вариантах осуществления Форма I демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере три характеристических пика, выраженных в градусах 2θ, выбранных из 8,7±0,2°; 11,1±0,2°; и 26,0±0,2°.
В некоторых вариантах осуществления Форма I демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере два характеристических пика, выраженных в градусах 2θ, которые представлены в Таблице XI. В некоторых вариантах осуществления Форма I демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере три характеристических пика, выраженных в градусах 2θ, которые представлены в Таблице XI. В некоторых вариантах осуществления Форма I демонстрирует порошковую рентгеновскую дифрактограмму, включающую по меньшей мере четыре (например, 4, 5, 6, 7, 8, 9 или 10) характеристических пика, выраженных в градусах 2θ, которые представлены в Таблице XI.
В некоторых вариантах осуществления Форма I демонстрирует порошковую рентгеновскую дифрактограмму, по существу такую, как показано на Фиг. 1.
Пример 15
(2R)-2-(5-Хлор-2-метоксипиридин-4-ил)-1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-он (15)
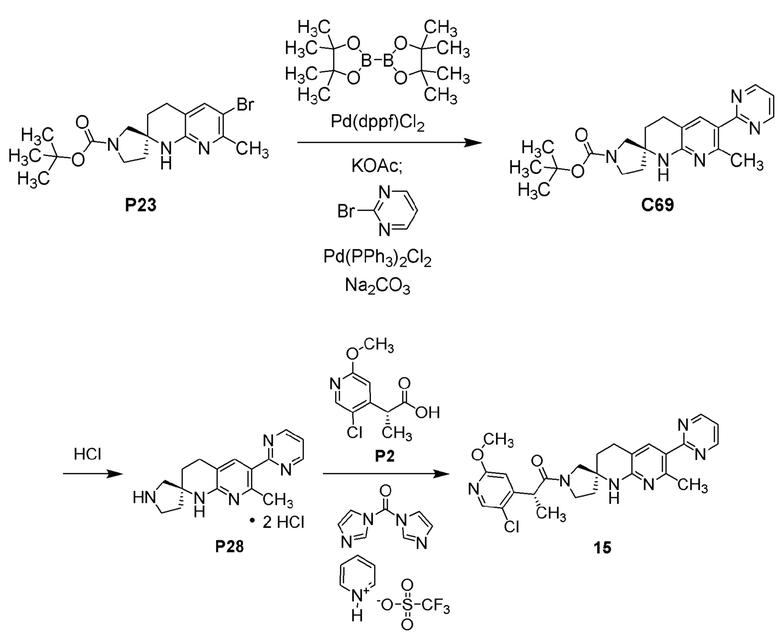
Стадия 1. Синтез трет-бутил-(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилата (С69).
Смесь 4,4,4',4',5,5,5',5'-октаметил-2,2'-би-1,3,2-диоксаборолана (249 мг, 0,981 ммоль), Р23 (250 мг, 0,654 ммоль), комплекса бис(дифенилфосфино)ферроцен]дихлорпалладия(II) с дихлорметаном (26,7 мг, 32,7 мкмоль) и высушенного в печи ацетата калия (257 мг, 2,62 ммоль) в 1,4-диоксане (12 мл) дегазировали путем барботирования смеси азотом в течение 5 минут. Реакционный сосуд герметично закрывали и смесь нагревали до 100°С в алюминиевом блоке в течение 2 часов, затем давали охладиться до комнатной температуры. Затем к реакционной смеси добавляли 2-бромпиримидин (109 мг, 0,686 ммоль), дихлорбис(трифенилфосфин)палладий(II) (22,9 мг, 32,6 мкмоль) и дегазированный водный раствор карбоната натрия (2,0 М; 0,817 мл, 1,63 ммоль) и смесь нагревали при 90°С в течение 18 часов. После охлаждения до комнатной температуры реакционную смесь разбавляли этилацетатом и фильтровали через диатомовую землю. Органический слой фильтрата промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и концентрировали в вакууме; хроматография на силикагеле (элюенты: 20%, затем 50%, затем 100% этилацетата в гептане) давала С69 в виде белого твердого вещества. Выход: 55,0 мг, 0,144 ммоль, 22%. ЖХМС m/z 382,3 [М+Н]+. 1Н ЯМР (400 МГц, хлороформ-d) δ 8,76 (д, J=4,8 Гц, 2Н), 7,92 (с, 1Н), 7,11 (т, J=4,9 Гц, 1Н), 5,37 (ушир.с, 1Н), 3,62-3,26 (м, 4Н), 2,88-2,76 (м, 2Н), 2,68 (с, 3Н), 2,06-1,77 (м, 4Н), 1,46 (ушир.с, 9Н).
Стадия 2. Синтез (2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин], дигидрохлоридной соли (Р28).
Раствор хлористого водорода в 1,4-диоксане (4,0 М; 0,144 мл, 0,576 ммоль) добавляли к раствору С69 (55,0 мг, 0,144 ммоль) в смеси дихлорметана (0,5 мл) и 1,1,1,3,3,3-гексафторпропан-2-ола (0,5 мл) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов, после этого ЖХМС анализ показал преобразование в Р28: ЖХМС m/z 282,3 [М+Н]+. Реакционную смесь концентрировали в вакууме, с получением Р28 в виде желтого смолистого вещества. Выход: 50 мг, 0,141 ммоль, 98%.
Стадия 3. Синтез (2R)-2-(5-хлор-2-метоксипиридин-4-ил)-1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она (15).
Трифторметансульфонат пиридиния (35,6 мг, 0,155 ммоль) добавляли к раствору Р2 (16,7 мг, 77,4 мкмоль) в ацетонитриле (1 мл). Полученный раствор обрабатывали 1,1'-карбонилдиимидазолом (12,6 мг, 77,7 мкмоль) одной порцией и реакционную смесь перемешивали при комнатной температуре в течение 45 минут. Затем добавляли раствор Р28 (25,0 мг, 70,6 мкмоль) в ацетонитриле (2 мл) одной порцией и перемешивание продолжали при комнатной температуре в течение 3 часов, затем реакционную смесь разбавляли водным раствором хлорида аммония и экстрагировали три раза этилацетатом. Объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле (градиент: 20% до 100% этилацетата в гептане), затем сверхкритическая флюидная хроматографией (Колонка: Chiral Technologies Chiralcel OJ-H, 21×250 мм, 5 мкм; Подвижная фаза 9:1 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 75 мл/мин; Обратное давление: 150 бар) давала (2R)-2-(5-хлор-2-метоксипиридин-4-ил)-1-[{2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-он (15). Выход: 5,9 мг, 12 мкмоль, 17%. ЖХМС m/z 479,3 (наблюдаемый изотопный паттерн хлора) [М+Н]+. 1Н ЯМР (4 00 МГц, метанол-d4) δ [8,81 (д, J=5,0 Гц) и 8,81 (д, J=5,0 Гц), всего 2Н], [8,15 (с) и 8,14 (с), всего 1Н], [7,85 (с) и 7,81 (с), всего 1Н], [7,31 (т, J=4,9 Гц) и 7,30 (т, J=4,9 Гц), всего 1Н], [6,81 (с) и 6,76 (с), всего 1Н], [4,32 (кв., J=7,0 Гц) и 4,23 (кв., J=6,9 Гц), всего 1Н], 3,91 (ушир.с, 3Н), [3,9-3,83 (м) и 3,76-3,52 (м), всего 3Н], [3,49 (д, J=12,2 Гц) и 3,38-3,3 (м, предположительно; частично скрыт пиком растворителя), всего 1Н], [2,93-2,72 (м) и 2,56-2,47 (м), всего 2Н], [2,57 (с) и 2,56 (с), всего 3Н], [2,16-2,07 (м) и 2,05-1,84 (м), всего 3Н], 1,80-1,73 (м, 1Н), [1,43 (д, J=6,9 Гц) и 1,42 (д, J=6, 9 Гц), всего 3Н].
Альтернативная стадия 3. Синтез (2R)-2-(5-хлор-2-метоксипиридин-4-ил)-1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она (15) для определения кристаллической структуры методом рентгенографии.
Трифторметансульфонат пиридиния (112 мг, 0,487 ммоль) добавляли к раствору Р2 (вещество из Получения Р2 и Р3; 50,0 мг, 0,232 ммоль) в ацетонитриле (3 мл). Полученный раствор обрабатывали 1,1'-карбонилдиимидазолом (39,5 мг, 0,244 ммоль) одной порцией и реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Затем добавляли раствор Р28 (82,1 мг, 0,232 ммоль) одной порцией; через 1 час добавляли каплю воды с получением раствора. После перемешивания реакционной смеси при комнатной температуре в течение 2 часов ЖХМС анализ показал преобразование в 15: ЖХМС m/z 479,3 (наблюдаемый изотопный паттерн хлора) [М+Н]+. Реакционную смесь затем распределяли между этилацетатом и водным раствором бикарбоната натрия; органический слой промывали последовательно водой и насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (градиент: 0% до 10% метанола в дихлорметане) давала (2R)-2-(5-хлор-2-метоксипиридин-4-ил)-1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-он (15) в виде твердого вещества. Выход: 102 мг, 0,213 ммоль, 92%.
Это вещество растворяли в смеси (приблизительно 12 мл) 10% этилацетата в гептане путем нагревания. Раствору давали охладиться и затем выстояться, частично закрытым, при комнатной температуре в течение 3 дней. Полученное твердое вещество обеспечивало а кристалл для рентгеноструктурного анализа (см. ниже).
Определение структуры 15 методом рентгеновской дифракции монокристаллов
Рентгеноструктурный анализ монокристалла
Сбор данных осуществляли на дифрактометре Bruker D8 Venture при комнатной температуре. Сбор данных состоял из омега- и фи-сканов. Используемое кристаллическое вещество микроразмерного и мультидоменного типа показало тета-дифракцию выше области разрешения 0,90-0,94 А.
Структура была разрешена методом внутреннего фазирования с использованием пакета программ SHELX в пространственной группе Р1 триклинного класса. Структуру затем уточняли методом наименьших квадратов в полноматричном приближении. Все неводородные атомы были обнаружены и уточнены с использованием параметров анизотропного смещения.
Атомы водорода, находящиеся на азоте, были обнаружены из разностной карты Фурье и уточнены с ограничением расстояний. Остальные атомы водорода были размещены в рассчитанных положениях, и им давали перемещаться на их атомах-носителях. Конечное уточнение включало параметры изотропного смещения для всех атомов водорода.
Анализ абсолютной структуры с использованием методов вероятности (Hooft, 2008) осуществляли с использованием PLATON (Spek). Результаты показывают, что абсолютная структура присвоена корректно. Этим методом рассчитано, что вероятность того, что структура присвоена корректно, составляет 100%. Параметр Хофта указан как 0,04 с esd (расчетное стандартное отклонение), составляющим (3), а параметр Парсона указан как 0,05 с esd, составляющим (3).
Конечный R-индекс был равен 6,9%. Конечная разностная карта Фурье показала, что нет недостающей или смещенной электронной плотности.
Соответствующая информация, качающаяся кристалла, сбора данных и уточнения структуры, обобщенно представлена в Таблице Е. Атомные координаты, длина связей и параметры смещения перечислены в Таблицах F-Н.
Программа и ссылки
См. перечень, представленный выше для Определения структуры 14 методом рентгеновской дифракции монокристаллов.

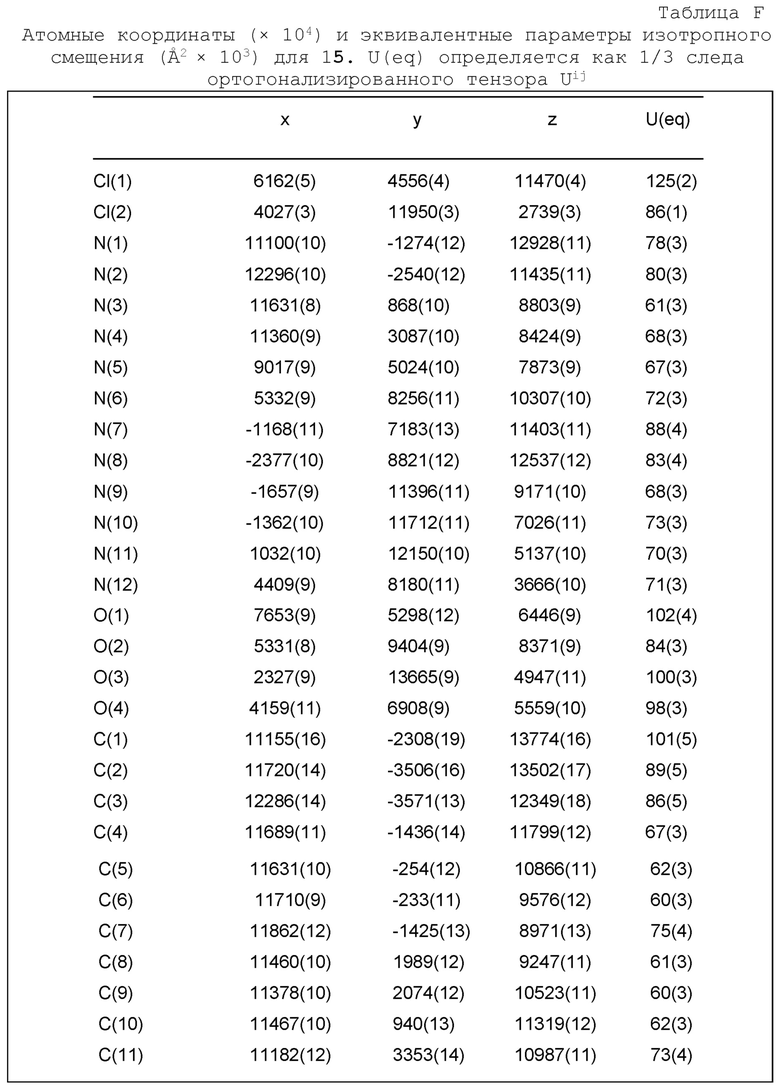

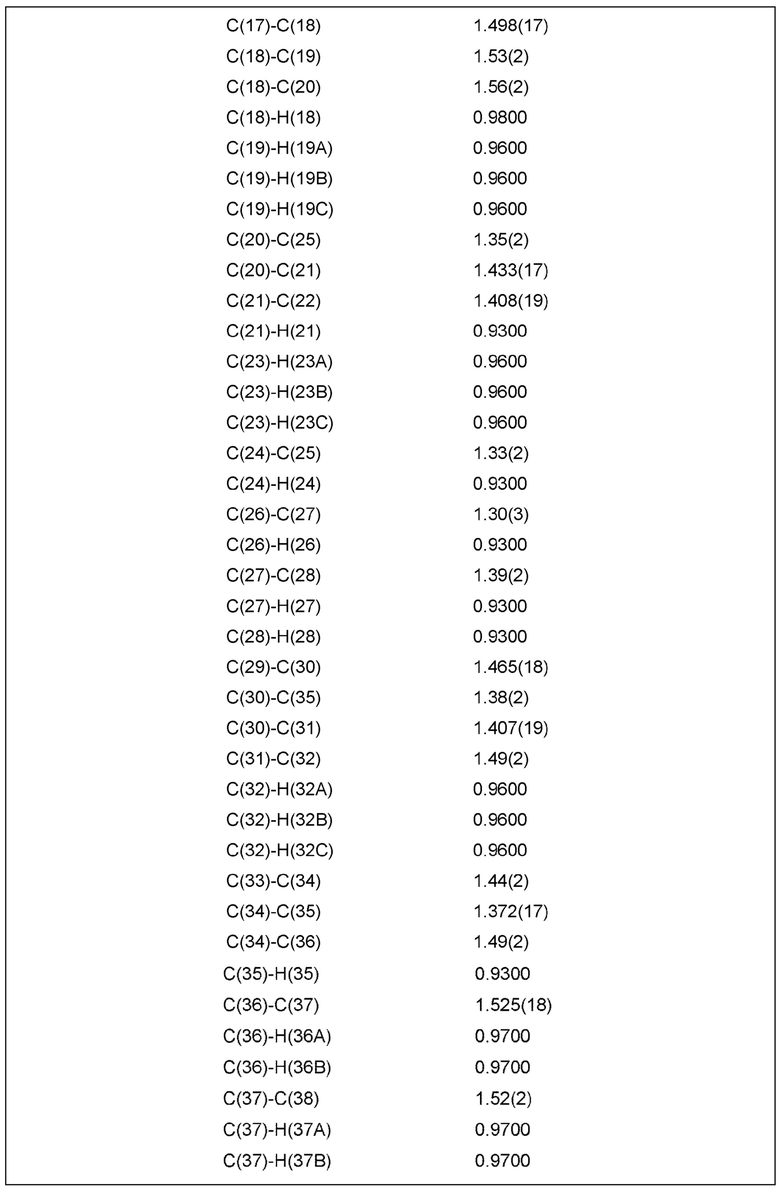
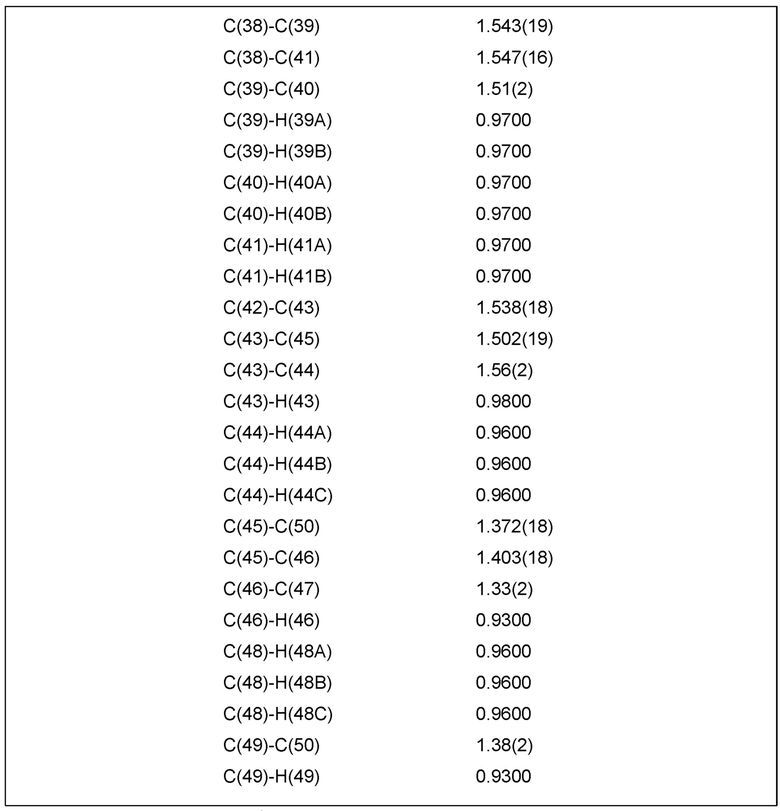
Использовали преобразования симметрии для получения эквивалентных атомов
Таким образом, абсолютную стереохимию соединения Примера 15 определяли рентгеноструктурным анализом монокристалла. Фиг. 2 представляет полученную методом рентгенографии монокристаллическую структуру (ORTEP изображение) кристаллического соединения Примера 15: (2R)-2-(5-Хлор-2-метоксипиридин-4-ил)-1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она.
В некоторых вариантах осуществления настоящее изобретение предоставляет кристаллическую форму (2R)-2-(5-хлор-2-метоксипиридин-4-ил)-1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она. В некоторых других вариантах осуществления кристаллическая форма (2R)-2-(5-хлор-2-метоксипиридин-4-ил)-1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она представляет собой соединение, описанное (или полученное) в Примере 15.
Примеры 16 и 17
(2R)-2-(5-Фтор-2-метоксипиридин-4-ил)-1-[7-метил-6-(1-метил-1Н-пиразол-4-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-он, DIAST-1 (16) и (2R)-2-(5-Фтор-2-метоксипиридин-4-ил)-1-[7-метил-6-(1-метил-1H-пиразол-4-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-он, DIAST-2 (17)
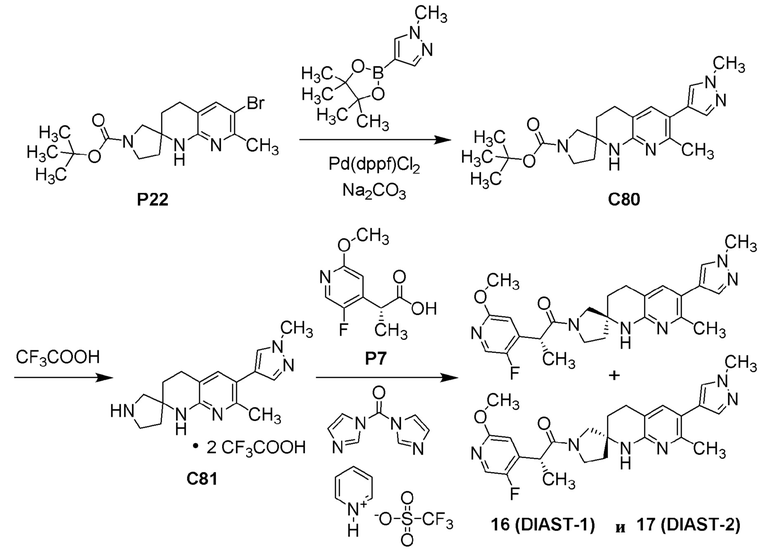
Стадия 1. Синтез трет-бутил-7-метил-6-(1-метил-1Н-пиразол-4-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилата (С80).
Смесь Р22 (100 мг, 0,262 ммоль), 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (109 мг, 0,524 ммоль), комплекс бис(дифенилфосфино)ферроцен]дихлорпалладия(II) с дихлорметаном (10,7 мг, 13,1 мкмоль) и водного раствора карбоната натрия (2,0 М; 0,33 мл, 0,66 ммоль) в 1,4-диоксане (3 мл) продували азотом. Реакционный сосуд герметично закрывали и нагревали до 80°С в течение ночи, затем ЖХМС анализ показал преобразование в С80: ЖХМС m/z 384,3 [М+Н]+. Реакционную смесь охлаждали до комнатной температуры, затем смесь распределяли между этилацетатом и водой и водный слой экстрагировали два раза этилацетатом. Объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и очищали хроматографией на силикагеле (градиент: 0% до 100% этилацетата в гептане) с получением С80 в виде твердого вещества. Выход: 93 мг, 0,24 ммоль, 92%. 1H ЯМР (400 МГц, хлороформ-d), характеристические пики: δ 7,52 (с, 1Н), 7,36 (с, 1Н), 7,19 (с, 1Н), 3,95 (с, 3Н), 3,62-3,26 (м, 4Н), 2,85-2,68 (м, 2Н), 2,41 (с, 3Н), [1,47 (с) и 1,45 (с), всего 9Н].
Стадия 2. Синтез 7-метил-6-(1-метил-1Н-пиразол-4-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин], бис-трифторацетатной соли (С81).
Трифторуксусную кислоту (1,0 мл) добавляли к раствору С80 (92 мг, 0,24 ммоль) в дихлорметане (3 мл) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Затем смесь концентрировали в вакууме и остаток совместно упаривали два раза с этилацетатом/гептаном с получением С81 в виде смолистого вещества. Выход: 128 мг, предполагаемый количественный. ЖХМС m/z 284,2 [М+Н]+.
Стадия 3. Синтез (2R)-2-(5-фтор-2-метоксипиридин-4-ил)-1-[7-метил-6-(1-метил-1H-пиразол-4-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она, DIAST-1 (16) и (2R)-2-(5-фтор-2-метоксипиридин-4-ил)-1-[7-метил-6-(1-метил-1H-пиразол-4-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она, DIAST-2 (17).
К раствору Р7 (23,9 мг, 0,120 ммоль) в ацетонитриле (1 мл) добавляли трифторметансульфонат пиридиния (57,8 мг, 0,252 ммоль), с последующим добавлением 1,1'-карбонилдиимидазола (20,4 мг, 0,126 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 45 минут, затем добавляли раствор С81 (34,0 мг, 66,5 мкмоль) в ацетонитриле и перемешивание продолжали в течение ночи при комнатной температуре. ЖХМС анализ в этот момент показал присутствие продукта сочетания: ЖХМС m/z 465,3 [М+Н]+. Реакционную смесь затем распределяли между дихлорметаном и разбавленным водным раствором хлорида аммония; органический слой промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистку осуществляли хроматографией на силикагеле (градиент: 0% до 10% метанола в дихлорметане), затем сверхкритической флюидной хроматографией (Колонка: Phenomenex Lux Cellulose-1, 21×250 мм, 5 мкм; Подвижная фаза: 4:1 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 75 мл/мин; Обратное давление: 120 бар]. Первый элюирующий диастереомер был указан как 16 {(2R)-2-(5-фтор-2-метоксипиридин-4-ил)-1-[7-метил-6-(1-метил-1H-пиразол-4-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-он, DIAST-1}, а второй элюирующий диастереомер как 17 {(2R)-2-(5-фтор-2-метоксипиридин-4-ил)-1-[7-метил-6-(1-метил-1H-пиразол-4-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-он, DIAST-2}.
16 - Выход: 7,3 мг, 15,7 мкмоль, 24%. ЖХМС m/z 465,5 [М+Н]+. 1H ЯМР (400 МГц, метанол-d4) δ [7,99 (д, J=1,6 Гц) и 7,97 (д, J=1,7 Гц), всего 1Н], [7,67 (с) и 7,67 (с), всего 1Н], 7,55-7,52 (м, 1Н), [7,29 (с) и 7,27 (с), всего 1Н], [6,78 (д, J=4,9 Гц) и 6,72 (д, J=4,9 Гц), всего 1Н], [4,27 (кв., J=6,9 Гц) и 4,18 (кв., J=6,9 Гц), всего 1Н], [3,92 (с) и 3,92 (с), всего 3Н], [3,88 (с) и 3,88 (с), всего 3Н], [3,88-3,83 (м), 3,75-3,56 (м) и 3,54 (д, компонент АВ квартета, J=12,1 Гц), всего 3Н], [3,45 (д, компонент АВ квартета, J=12,3 Гц) и 3,36 (д, J=10,6 Гц), всего 1Н], [2,89-2,70 (м) и 2,59-2,49 (м), всего 2Н], [2,37 (с) и 2,34 (с), всего 3Н], 2,13-1,81 (м, 3Н), 1,80-1,71 (м, 1Н), 1,47-1,40 (м, 3Н). Время удерживания: 3,71 минут [Аналитические условия. Колонка: Phenomenex Lux Cellulose-1, 4,6×100 мм, 5 мкм; Подвижная фаза: 3:1 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 1,5 мл/мин; Обратное давление: 200 бар].
17 - Выход: 6,2 мг, 13,3 мкмоль, 20%. ЖХМС m/z 466,6 [М+Н]+. Время удерживания: 4,64 мин (Аналитические условия идентичны условиям, используемым для 16).
Пример 18
(2R)-2-(5-Фтор-2-метоксипиридин-4-ил)-1-{(2S)-7-метил-6-[(4,6-2H2)пиримидин-2-ил]-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил}пропан-1-он (18)
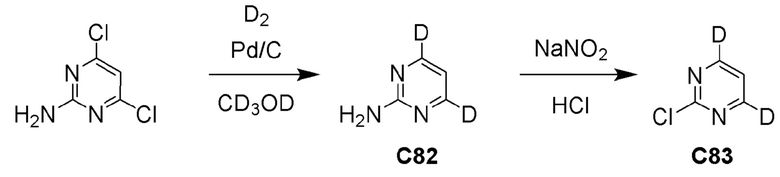
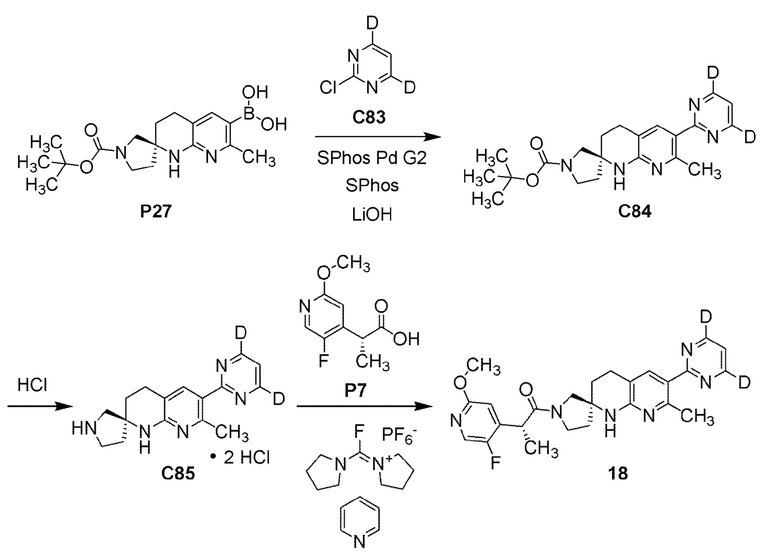
Стадия 1. Синтез (4,6-2Н2)пиримидин-2-амина (С82).
К раствору 4,6-дихлорпиримидин-2-амина (500 мг, 3,05 ммоль) в метаноле-d4 (10 мл) добавляли палладий на углероде (100 мг) и триэтиламин (1,3 мл, 9,3 ммоль). Реакционную смесь перемешивали под газообразным дейтерием при 20°С в течение 6 часов, затем смесь фильтровали для удаления катализатора. Собранный катализатор промывали метанолом (2×10 мл), затем объединенные фильтраты концентрировали в вакууме, затем подвергали хроматографии на силикагеле (градиент: 0% до 80% этилацетата в петролейном эфире) с получением С82 в виде белого твердого вещества. Выход: 210 мг, 2,16 ммоль, 71%. ЖХМС m/z 98,2 [М+Н]+. 1H ЯМР (400 МГц, DMSO-d6) δ 6,60-6,52 (ушир.с, 2Н), 6,53 (с, 1Н).
Стадия 2. Синтез 2-хлор (4,6-2H2)пиримидина (С83).
Промежуточное соединение С82 (210 мг, 2,16 ммоль) добавляли по порциям к концентрированной хлористоводородной кислоты (0,7 мл) при 0°С и полученную смесь перемешивали до тех пор, пока она не стала гомогенной. Раствор затем охлаждали до около -15°С, затем добавляли по каплям охлажденный раствор нитрита натрия (298 мг, 4,32 ммоль) в воде (0,5 мл) в течение 1 часа, поддерживая температуру реакции между -15°С и -10°С. Реакционную смесь перемешивали в течение 1 часа и температуре давали повыситься до около -5°С; затем смесь осторожно нейтрализовали до рН 7 добавлением 30% водного раствора гидроксида натрия, поддерживая температуру реакции ниже 0°С. Полученную смесь экстрагировали диэтиловым эфиром (3×5 мл) и объединенные органические слои промывали насыщенным водным раствором хлорида натрия (10 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением С83 в виде белого твердого вещества. Выход: 115 мг, 0,987 ммоль, 46%. 1H ЯМР (400 МГц, DMSO-d6) δ 7,60 (с, 1Н).
Стадия 3. Синтез трет-бутил-(2S)-7-метил-6-[(4,6-2H2)пиримидин-2-ил]-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилата (С84).
Смесь С83 (40 мг, 0,34 ммоль), Р27 (119 мг, 0,34 ммоль), 2-дициклогексилфосфино-2',6'-диметоксибифенила (SPhos; 5,6 мг, 14 мкмоль), хлор(2-дициклогексилфосфино-2',6'-диметокси-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладия(II) (SPhos Pd G2; 4,9 мг, 6,8 мкмоль) и водного раствора гидроксида лития (2 М; 0,4 мл, 0,8 ммоль) в тетрагидрофуране (5 мл) продували азотом в течение 3 минут, затем реакционную смесь перемешивали при 60°С в течение 4 часов. Затем смесь концентрировали в вакууме; остаток очищали хроматографией на силикагеле (градиент: 0% до 50% этилацетата в петролейном эфире) с получением С84 в виде желтого твердого вещества. Выход: 116 мг, 0,302 ммоль, 89%. ЖХМС m/z 384,3 [М+Н]+. 1H ЯМР (400 МГц, хлороформ-d) δ [8,04 (с) и 8,01 (с), всего 1Н], 7,14 (с, 1Н), 3,67-3,30 (м, 4Н), 2,92-2,76 (м, 2Н), [2,74 (с) и 2,73 (с), всего 3Н], 2,12-1,79 (м, 4Н), [1,47 (с) и 1,46 (с), всего 9Н].
Стадия 4. Синтез (2S)-7-метил-6-[(4,6-2Н2)пиримидин-2-ил]-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин], дигидрохлориднои соли (С85).
Раствор хлористого водорода в 1,4-диоксане (4 М; 3 мл) добавляли к раствору С84 (116 мг, 0,302 ммоль) в дихлорметане (3 мл) и реакционную смесь перемешивали при 20°С в течение 2 часов. Концентрирование в вакууме давало С85 в виде желтого твердого вещества. Выход: 108 мг, 0,303 ммоль, количественный. ЖХМС m/z 284,2 [М+Н]+. 1H ЯМР (400 МГц, DMSO-d6) δ 10,09-9,93 (ушир.с, 1Н), 9,82-9,67 (ушир.с, 1Н), 9,01 (с, 1Н), 8,45 (с, 1Н), 7,50 (с, 1Н), 3,50-3,34 (м, 2Н), 3,34-3,27 (м, 2Н), 3,01-2,84 (м, 2Н), 2,82 (с, 3Н), 2,26-2,07 (м, 3Н), 1,99-1,87 (м, 1Н).
Стадия 5. Синтез (2R)-2-(5-фтор-2-метоксипиридин-4-ил)-1-{(2S)-7-метил-6-[(4,6-2H2)пиримидин-2-ил]-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил}пропан-1-она (18).
Раствор С85 (80 мг, 0,22 ммоль), Р7 (45 мг, 0,23 ммоль), фтор-N,N,N',N'-бис(тетраметилен) формамидиний гексафторфосфата (BTFFH; 85 мг, 0,27 ммоль) и пиридина (71 мг, 0,890 ммоль) в дихлорметане (10 мл) перемешивали при 25°С в течение 16 часов. Реакционную смесь выливали в водный раствор бикарбоната натрия (10 мл), затем смесь экстрагировали этилацетатом (2×20 мл); объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (градиент: 0% до 10% метанола в дихлорметане) давала (2R)-2-(5-фтор-2-метоксипиридин-4-ил)-1-{(2S)-7-метил-6-[(4,6-2H2)пиримидин-2-ил]-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил}пропан-1-он (18) в виде белого твердого вещества. Выход: 27 мг, 58 мкмоль, 26%. ЖХМС m/z 465,3 [М+Н]+. 1H ЯМР (400 МГц, метанол-d4) δ [8,00 (д, J=1,7 Гц) и 7,98 (д, J=1,7 Гц), всего 1Н], [7,85 (с) и 7,82 (с), всего 1Н], [7,31 (с) и 7,30 (с), всего 1Н], [6,78 (д, J=4,9 Гц) и 6,73 (д, J=4,9 Гц), всего 1Н], [4,28 (кв., J=6,9 Гц) и 4,20 (кв., J=6,9 Гц), всего 1Н], [3,93-3,85 (м), 3,77-3,67 (м), 3,67-3,57 (м), 3,53 (АВ квартет, JAB=12,2 Гц, ΔνAB=35,5 Гц) и 3,39 (д, J=10,6 Гц), всего 4Н], [3,89 (с) и 3,88 (с), всего 3Н], [2,95-2,75 (м) и 2,64-2,55 (м), всего 2Н], [2,58 (с) и 2,55 (с), всего 3Н], [2,16-2,06 (м) и 2,05-1,85 (м), всего 3Н], 1,84-1,75 (м, 1Н), [1,45 (д, J=6,9 Гц) и 1,44 (д, J=6,9 Гц), всего 3Н].
Примеры 19 и 20
2-(4-Фторфенил)-1-[7-метил-6-(1-метил-1H-пиразол-4-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]этан-1-он, ENT-1 (19) и 2-(4-Фторфенил)-1-[7-метил-6-(1-метил-1H-пиразол-4-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]этан-1-он, ENT-2 (20)
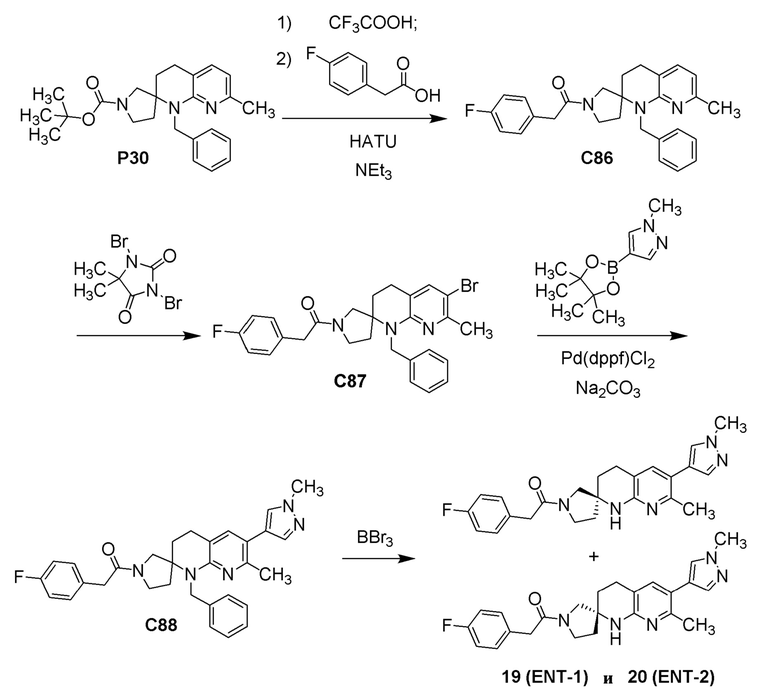
Стадия 1. Синтез 1-(1-бензил-7-метил-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил)-2-(4-фторфенил)этан-1-она (С86).
Трифторуксусную кислоту (319 мг, 2,80 ммоль) добавляли к раствору Р30 (110,0 мг, 0,280 ммоль) в дихлорметане (4 мл) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Затем смесь концентрировали в вакууме, совместно упаривали с этилацетатом несколько раз и растворяли в дихлорметане (4 мл). Полученный раствор обрабатывали триэтиламином (84,9 мг, 0,839 ммоль). O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфатом (HATU; 117 мг, 0,308 ммоль) и (4-фторфенил)уксусной кислотой (43,1 мг, 0,280 ммоль) и перемешивали в течение ночи при комнатной температуре. После удаления растворителя в вакууме остаток очищали хроматографией на силикагеле (градиент: 0% до 10% метанола в дихлорметане) с получением С86 в виде светлого желтовато-коричневого твердого вещества. Выход: 120 мг, 0,279 ммоль, количественный. 1H ЯМР (400 МГц, хлороформ-d) δ [7,31-7,13 (м), 7,13-7,06 (м) и 7,05-6,91 (м), всего 10Н], 6,44-6,38 (м, 1Н), 5,11-4,97 (м, 1Н), [4,88 (д, компонент АВ квартета, J=16,8 Гц) и 4,77 (д, компонент АВ квартета, J=16,7 Гц), всего 1Н], [3,71-3,60 (м), 3,59-3,37 (м) и 3,33-3,24 (м), всего 6Н], [2,83-2,69 (м) и 2,62-2,51 (м), всего 2Н], 2,35-2,17 (м, 1Н), [2,24 (с) и 2,23 (с), всего 3Н], 2,01-1,68 (м, 3Н).
Стадия 2. Синтез 1-(1-бензил-6-бром-7-метил-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил)-2-(4-фторфенил)этан-1-она (С87).
1,3-Дибром-5,5-диметилимидазолидин-2,4-дион (47,9 мг, 0,168 ммоль) добавляли несколькими небольшими порциями к 0°С раствору С86 (120 мг, 0,279 ммоль) в дихлорметане (5 мл). Реакционной смеси давали нагреться до комнатной температуры; через 30 минут ЖХМС анализ показал присутствие С87: ЖХМС m/z 508,3 (наблюдаемый изотопный паттерн брома) [М+Н]+. Через 1 час реакционную смесь концентрировали в вакууме и подвергали хроматографии на силикагеле (градиент: 0% до 100% этилацетата в гептане) с получением С87. Выход: 65,0 мг, 0,128 ммоль, 46%. 1H ЯМР (400 МГц, хлороформ-d) δ [7,35-7,17 (м), 7,16-7,06 (м) и 7,05-6,91 (м), всего 10Н], [4,98 (д, компонент АВ квартета, J=16,7 Гц) и 4,98 (д, компонент АВ квартета, J=16, 8 Гц), всего 1Н], [4,80 (д, компонент АВ квартета, J=16,8 Гц) и 4,72 (д, компонент АВ квартета, J=16,7 Гц), всего 1Н], [3,72-3,62 (м), 3,58-3,43 (м) и 3,33-3,24 (м), всего 6Н], [2,85-2,69 (м) и 2,62-2,48 (м), всего 2Н], 2,38-2,15 (м, 1Н), [2,34 (с) и 2,32 (с), всего 3Н], 2,01-1,70 (м, 3Н).
Стадия 3. Синтез 1-[1-бензил-7-метил-6-(1-метил-1H-пиразол-4-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]-2-(4-фторфенил)этан-1-она (С88).
В реакционный сосуд загружали С87 (65,0 мг, 0,128 ммоль), 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол (33,1 мг, 0,159 ммоль), комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) с дихлорметаном (5,20 мг, 6,37 мкмоль), водный раствор карбоната натрия (2,0 М; 0,127 мл, 0,254 ммоль) и 1,4-диоксан (3 мл). Сосуд продували азотом, затем смесь герметично закрывали и нагревали при 90°С в течение ночи, после этого ЖХМС анализ показал преобразование в С88: ЖХМС m/z 510,4 [М+Н]+. Реакционную смесь затем охлаждали до комнатной температуры и распределяли между этилацетатом и водным раствором хлорида аммония. Водный слой экстрагировали два раза этилацетатом и объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (градиент: 0% до 10% метанола в дихлорметане) давала С88 в виде твердого вещества. Выход: 65,0 мг, 0,128 ммоль, количественный.
Стадия 4. Синтез 2-(4-фторфенил)-1-[7-метил-6-(1-метил-1H-пиразол-4-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]этан-1-она, ENT-1 (19) и 2-(4-фторфенил)-1-[7-метил-6-(1-метил-1H-пиразол-4-ил)-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]этан-1-она, ENT-2 (20).
Раствор трибромида бора в дихлорметане (1,0 М; 0,765 мл, 0,765 ммоль) добавляли к -78°С раствору С88 (65,0 мг, 0,128 ммоль) в дихлорметане (3 мл), затем реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 15 часов. Затем смесь обрабатывали метанолом (0,5 мл) и концентрировали в вакууме. Разделение на составляющие энантиомеры осуществляли с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralpak AS-H, 21×250 мм, 5 мкм; Подвижная фаза: 83:7 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 75 мл/мин; Обратное давление: 200 бар]. Первый элюирующий диастереомер был указан как 19 {2-(4-фторфенил)-1-[7-метил-6-(1-метил-1H-пиразол-4-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]этан-1-он, ENT-1}, а второй элюирующий диастереомер как 20 {2-(4-фторфенил)-1-[7-метил-6-(1-метил-1H-пиразол-4-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]этан-1-он, ENT-2}.
19 - Выход: 3,8 мг, 9,1 мкмоль, 7%. ЖХМС m/z 442,5 [M+Na+]. Время удерживания: 2,88 мин [Аналитические условия. Колонка: Chiral Technologies Chiralpak AS-H, 4,6×100 мм, 5 мкм; Подвижная фаза: 4:1 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 1,5 мл/мин; Обратное давление: 200 бар].
20 - Выход: 2,0 мг, 4,8 мкмоль, 4%. ЖХМС m/z 442,5 [M+Na+]. Время удерживания: 4,01 мин (Аналитические условия идентичны условиям, используемым для 19).
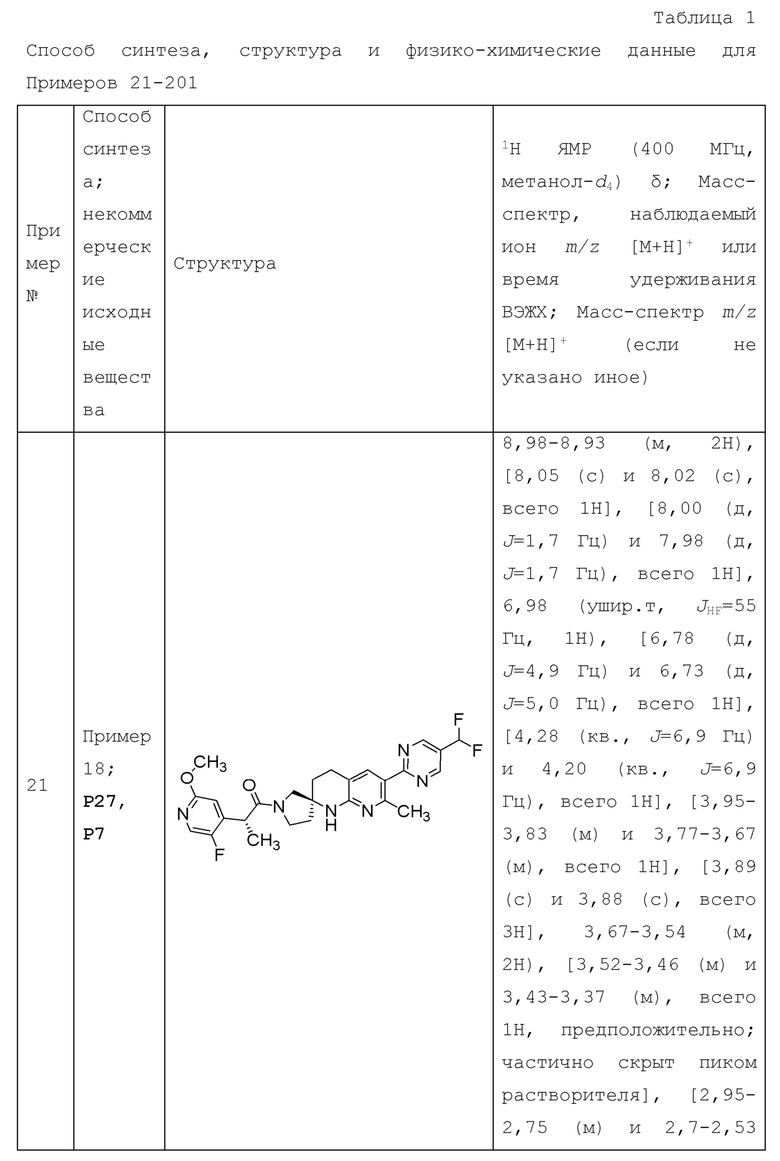
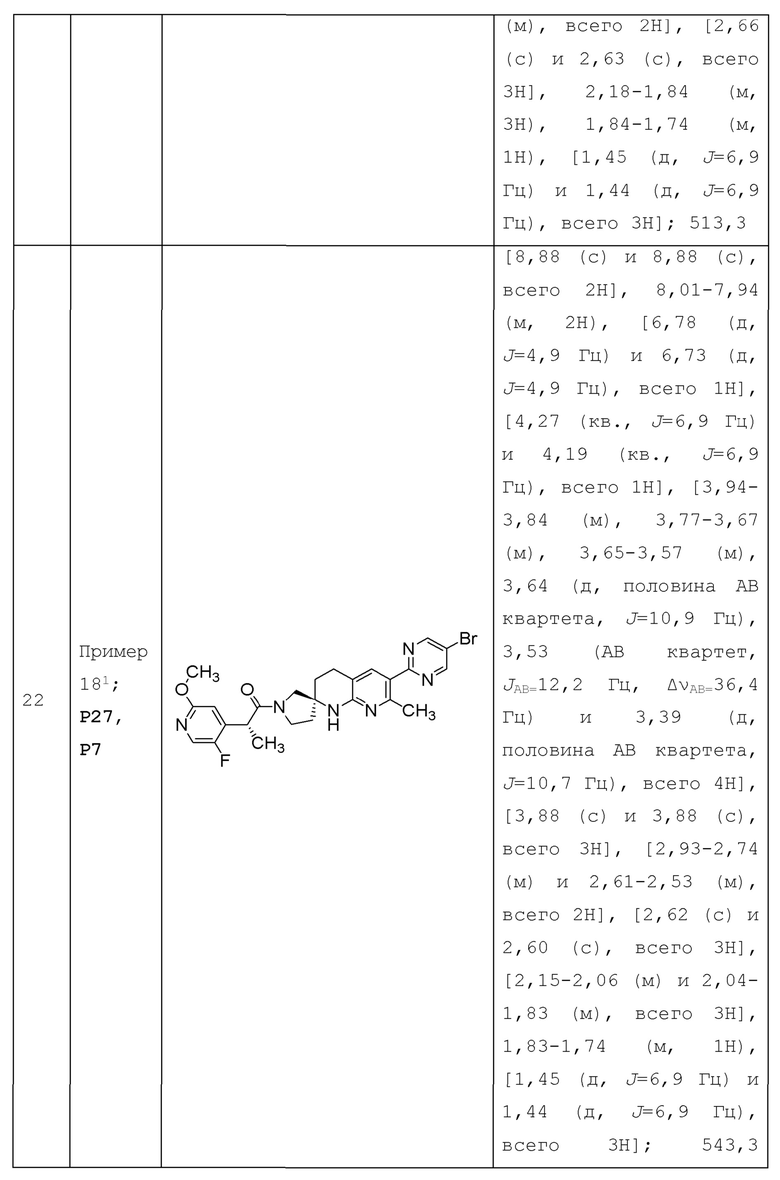
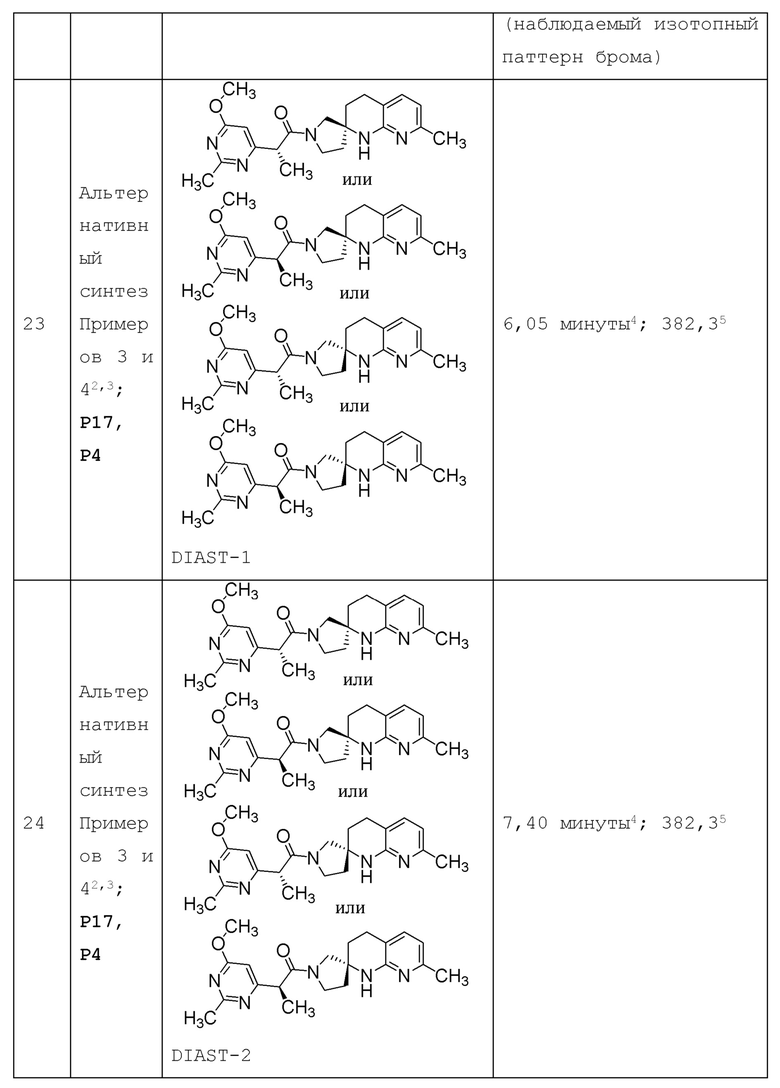
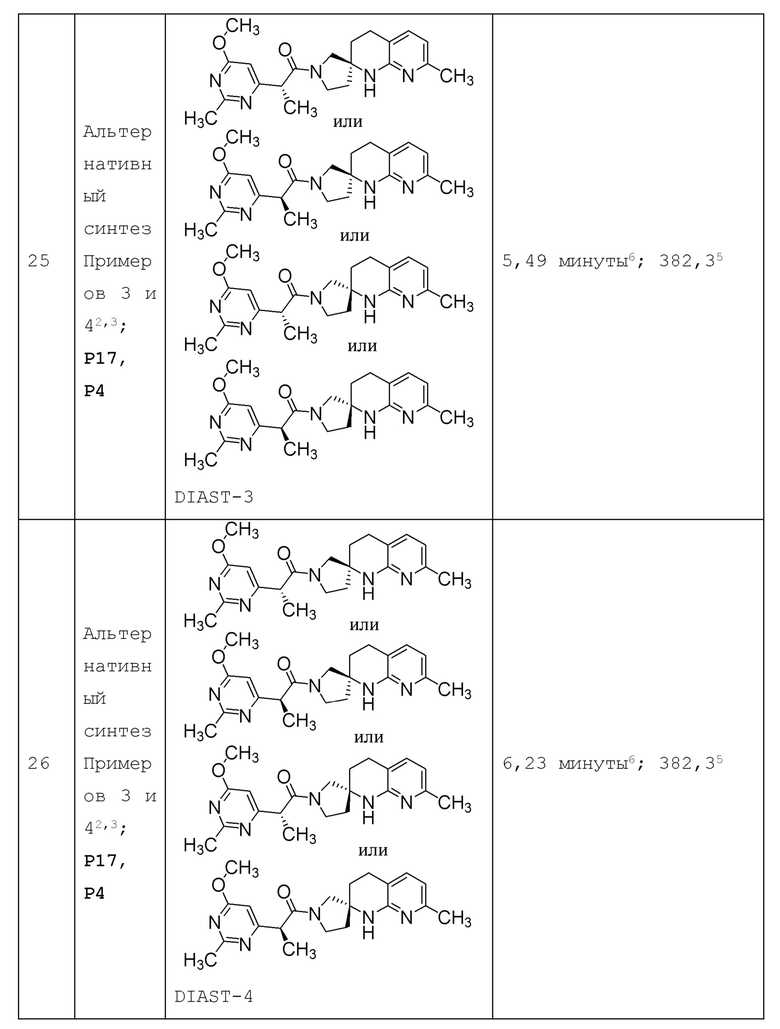
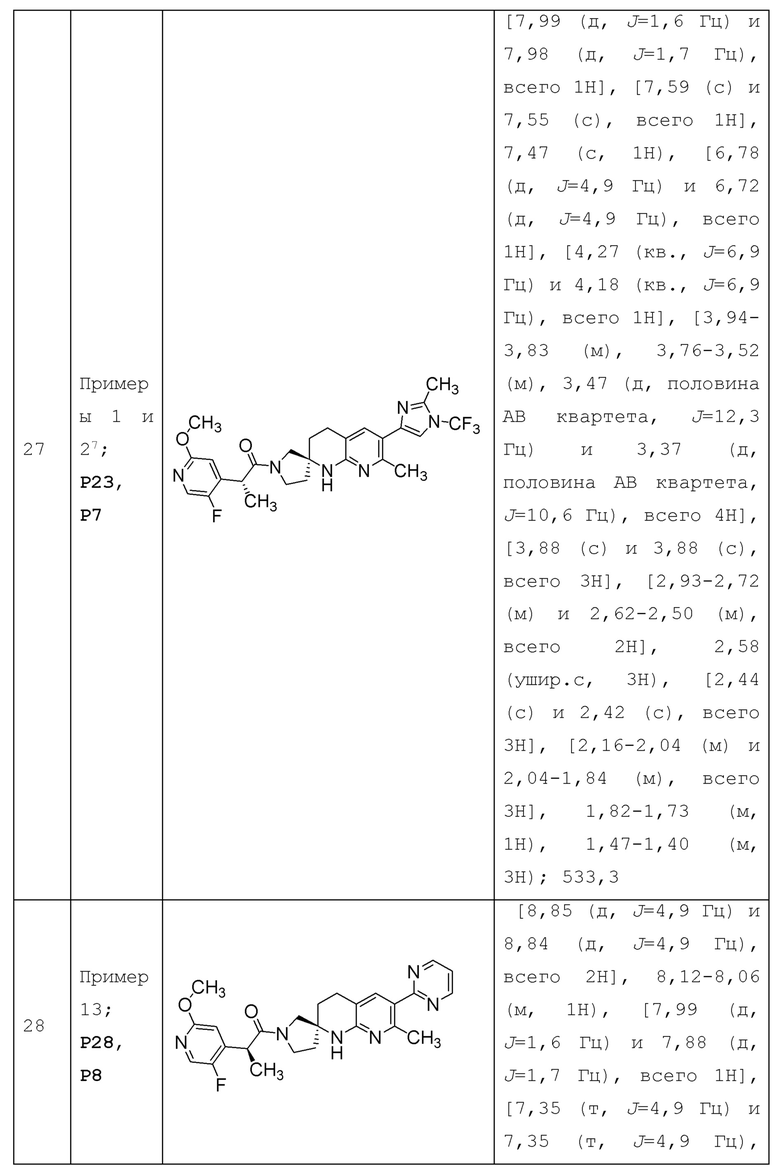
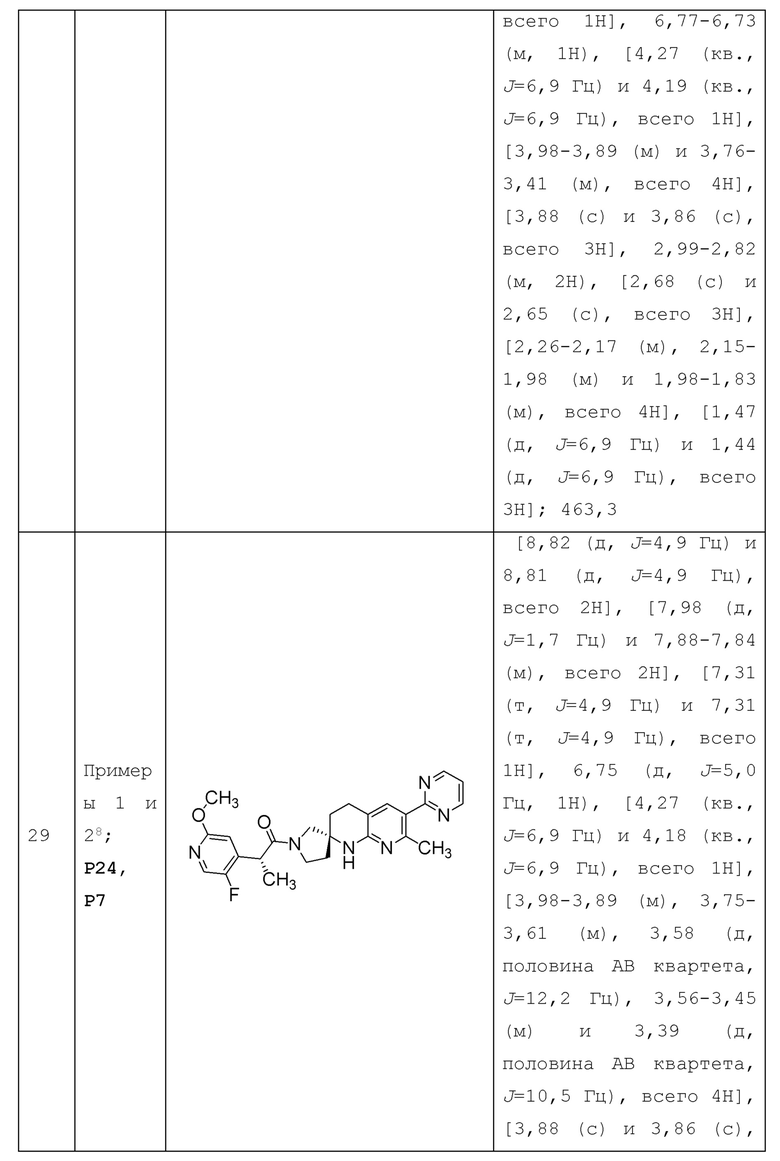
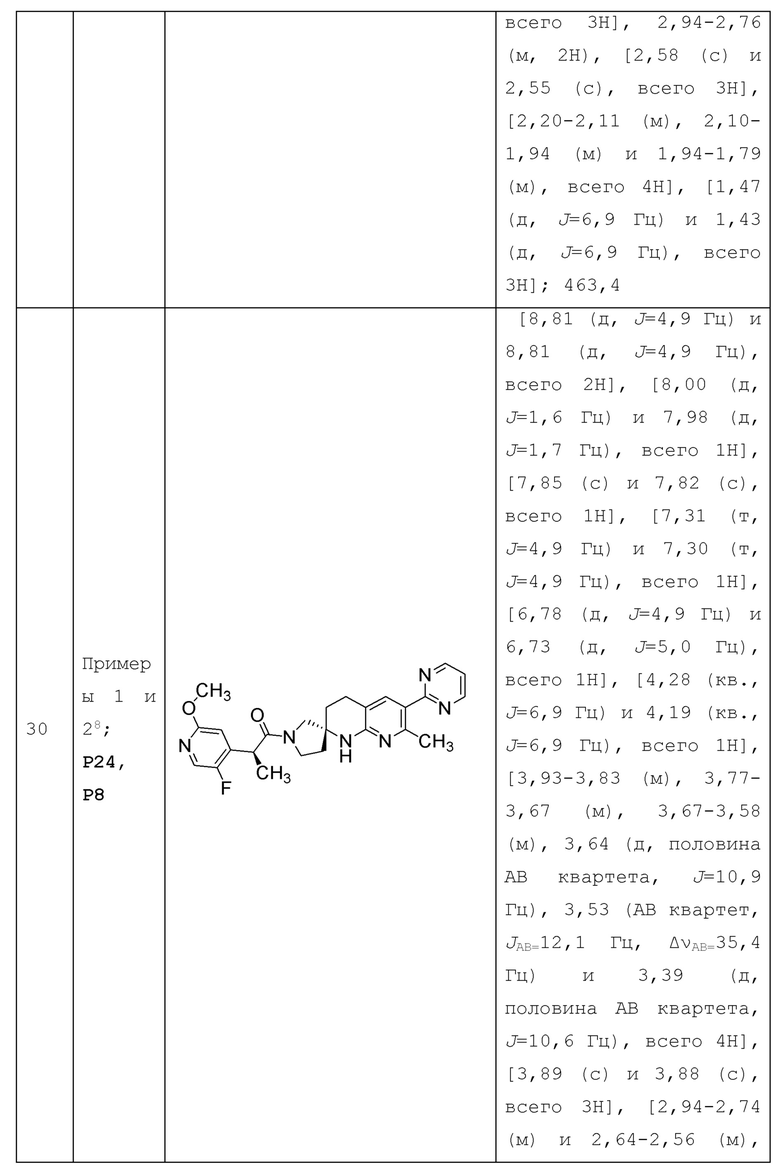
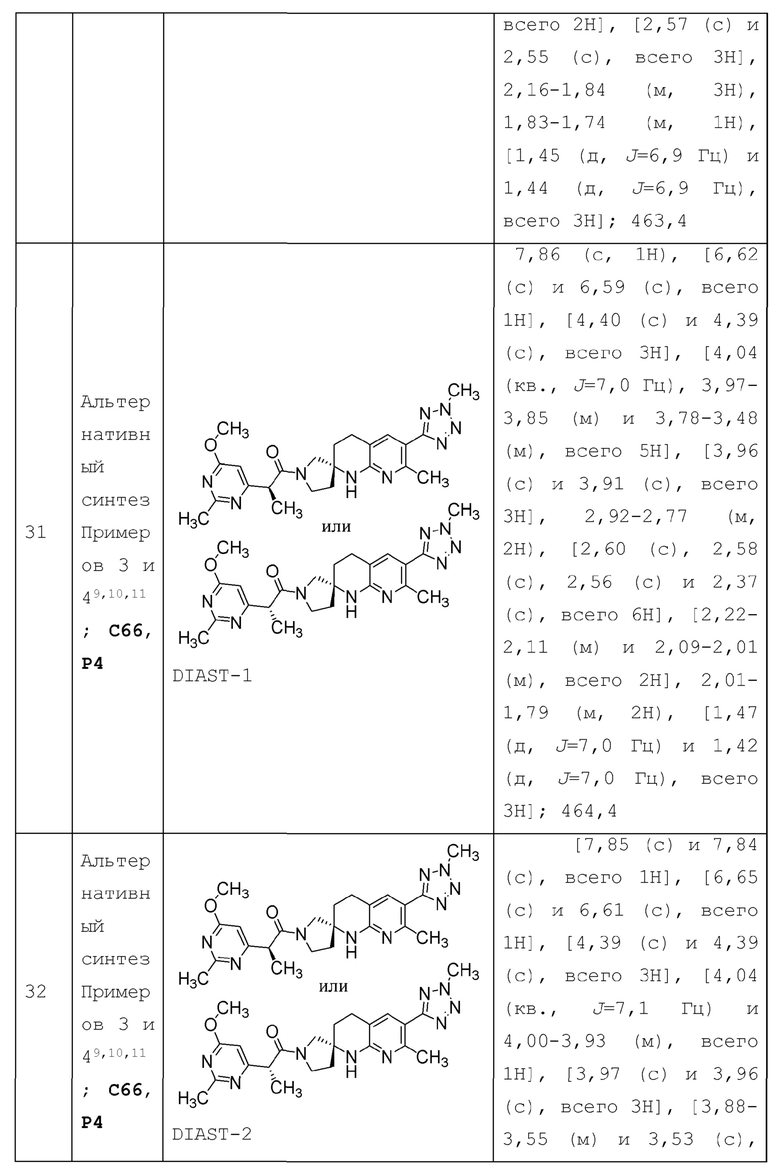
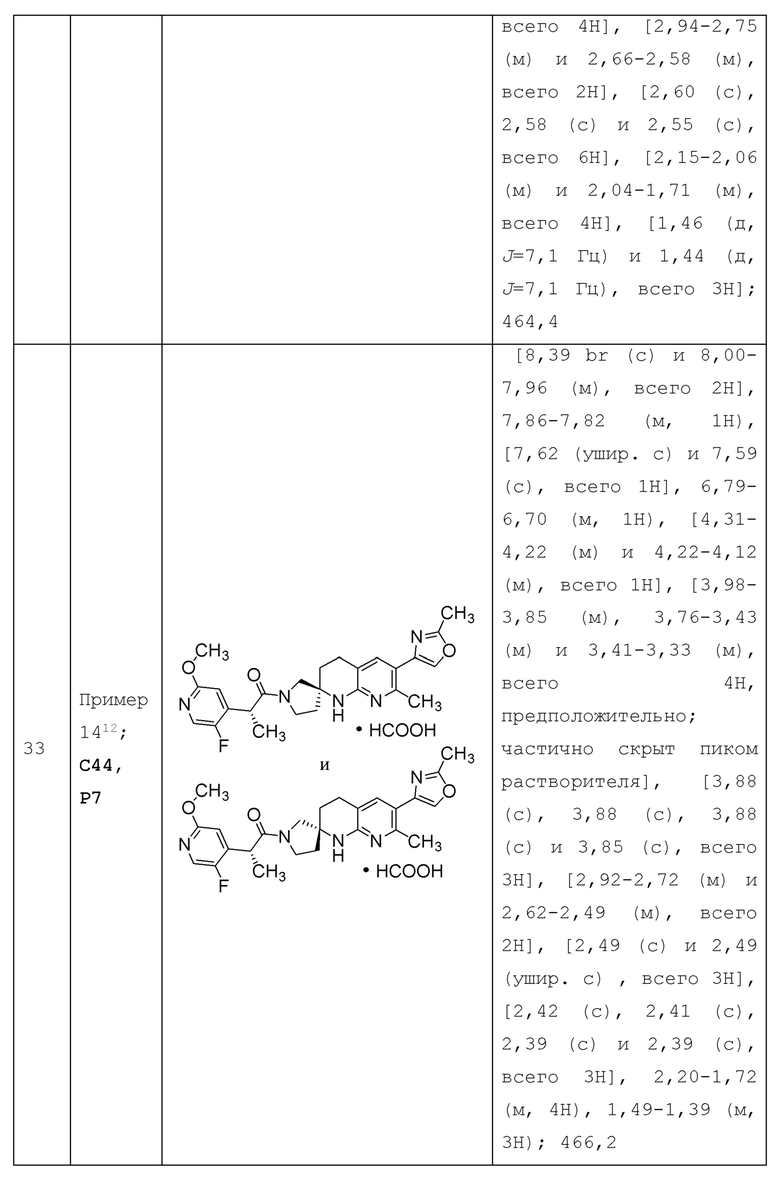
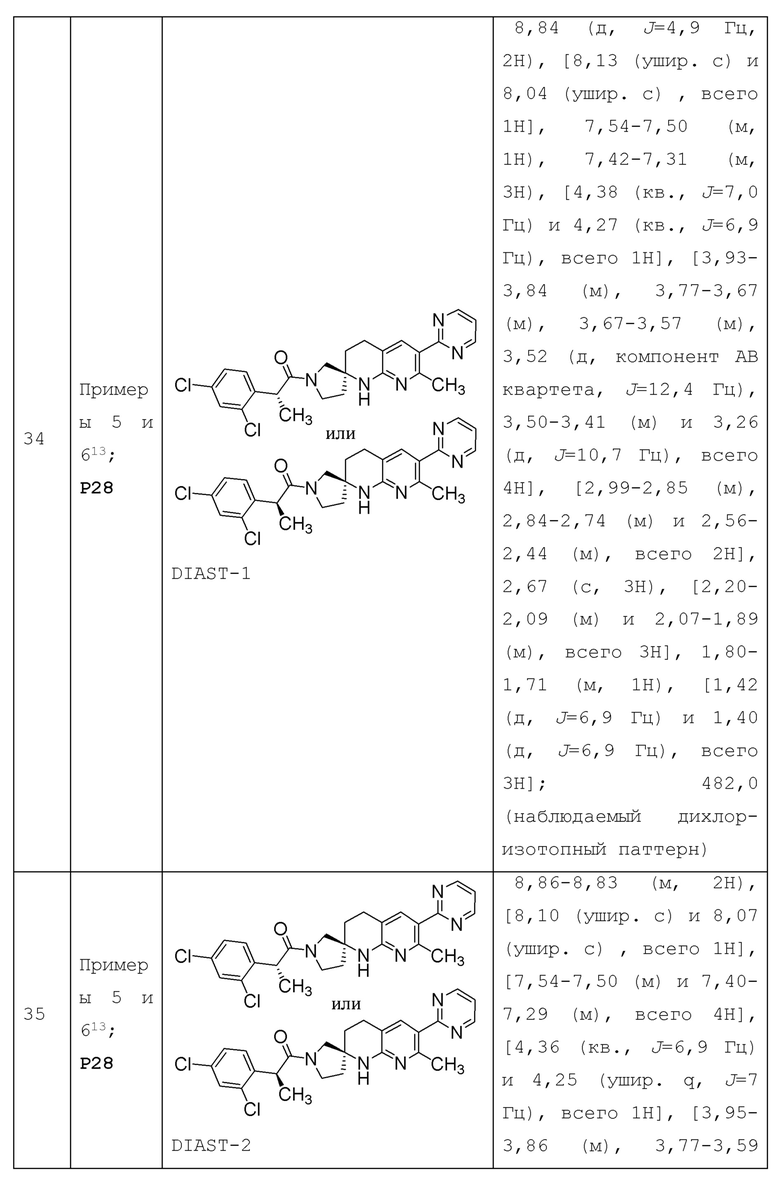
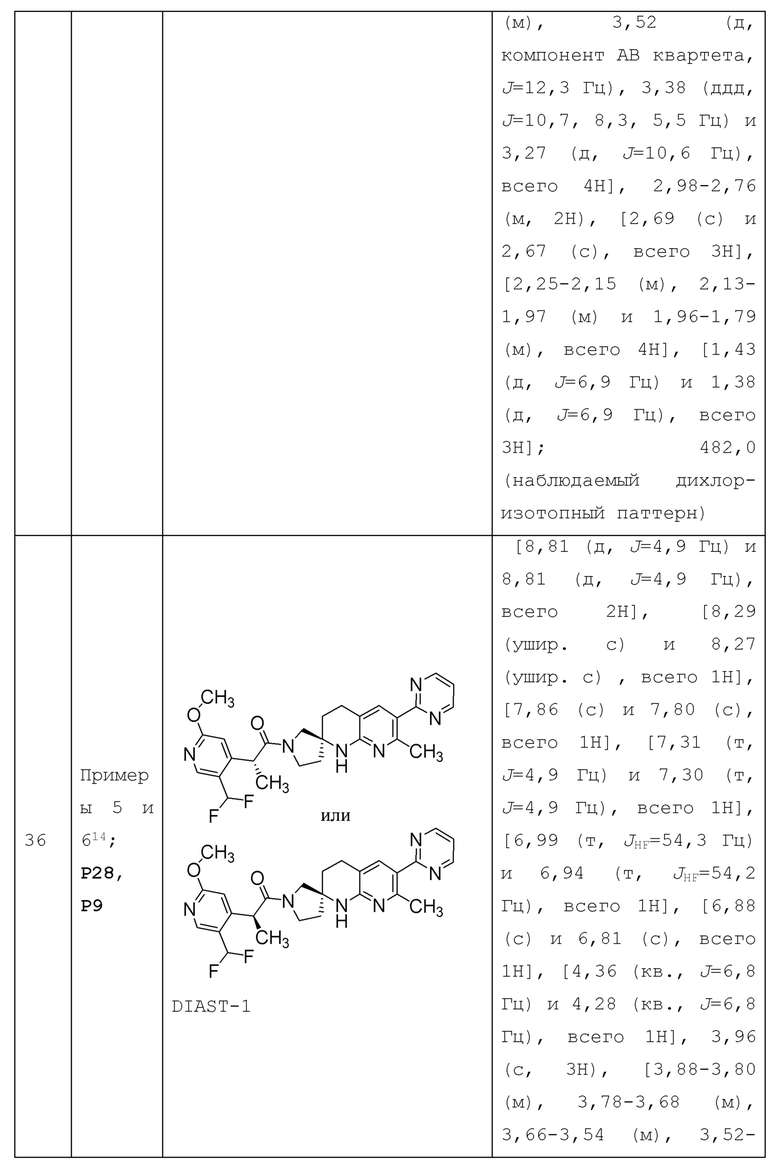
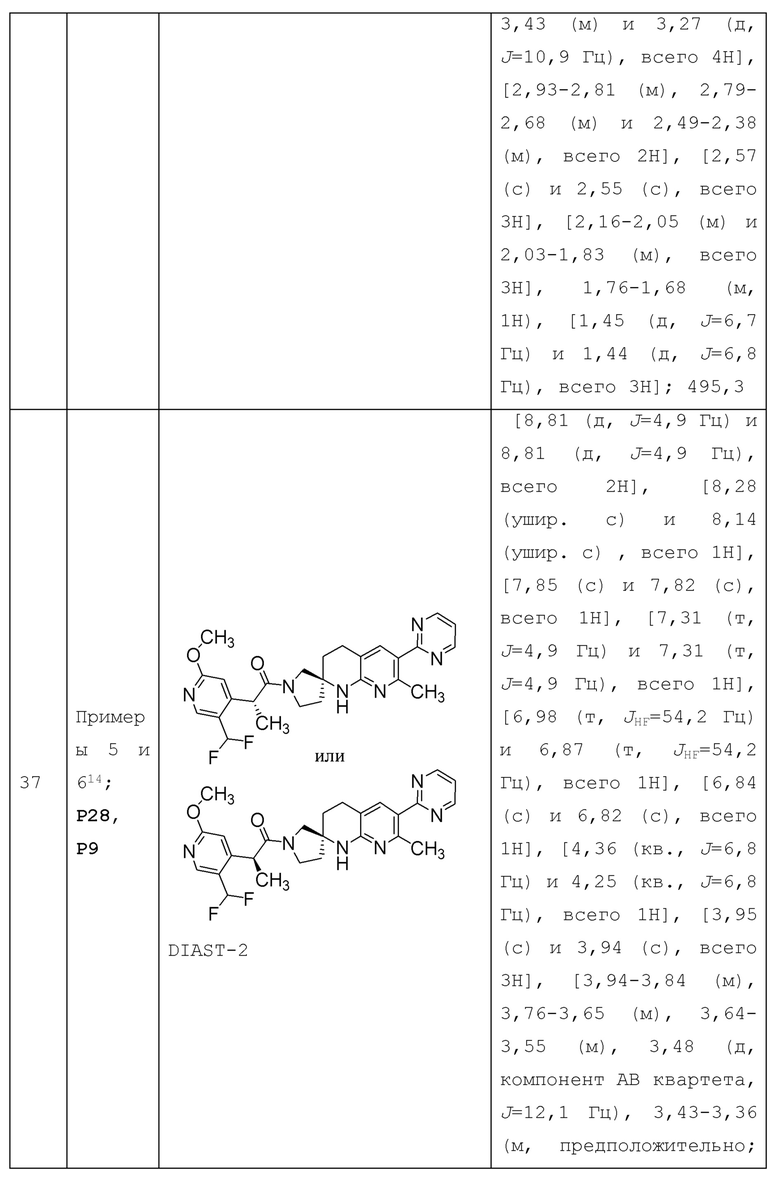
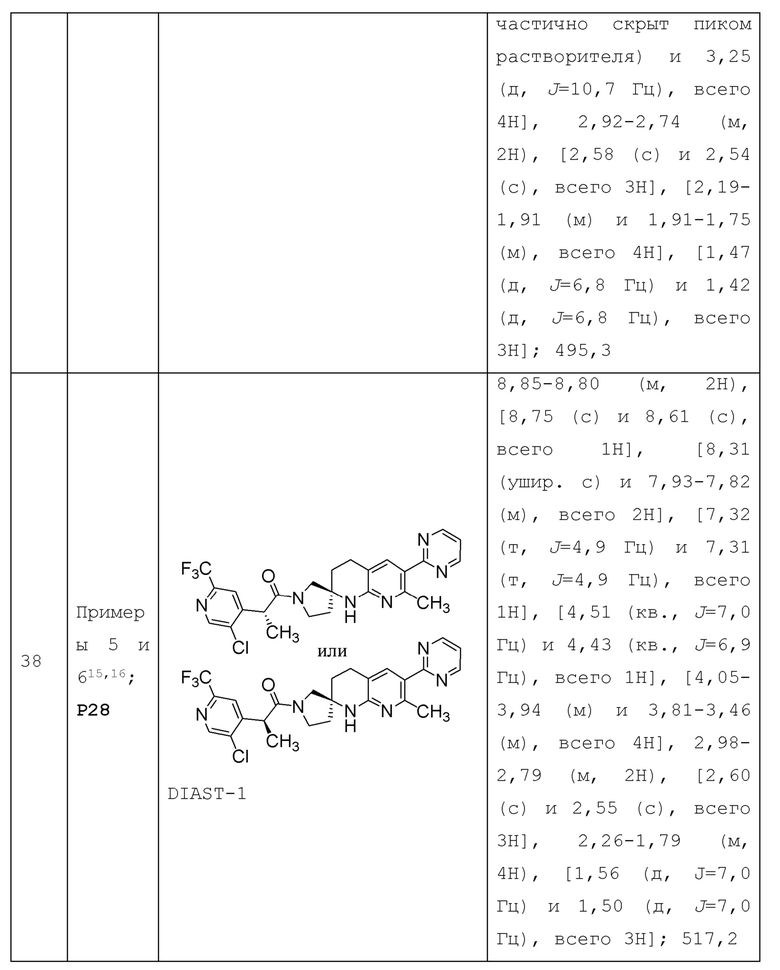
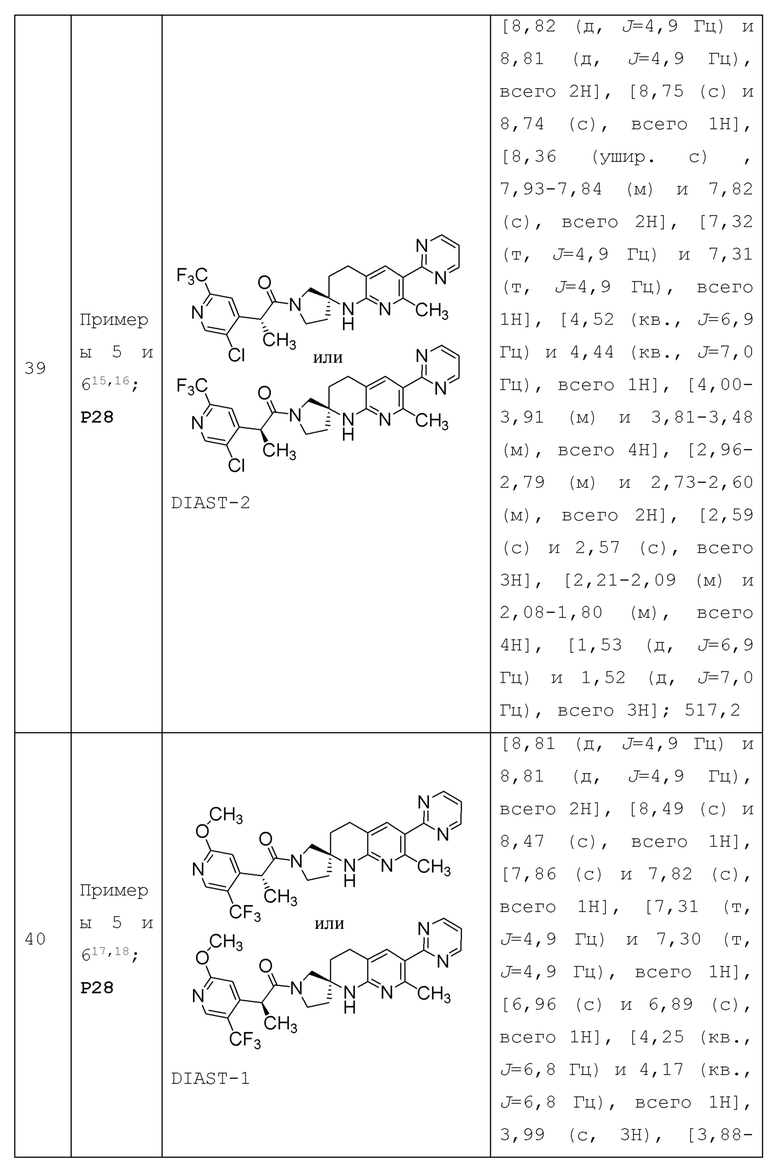
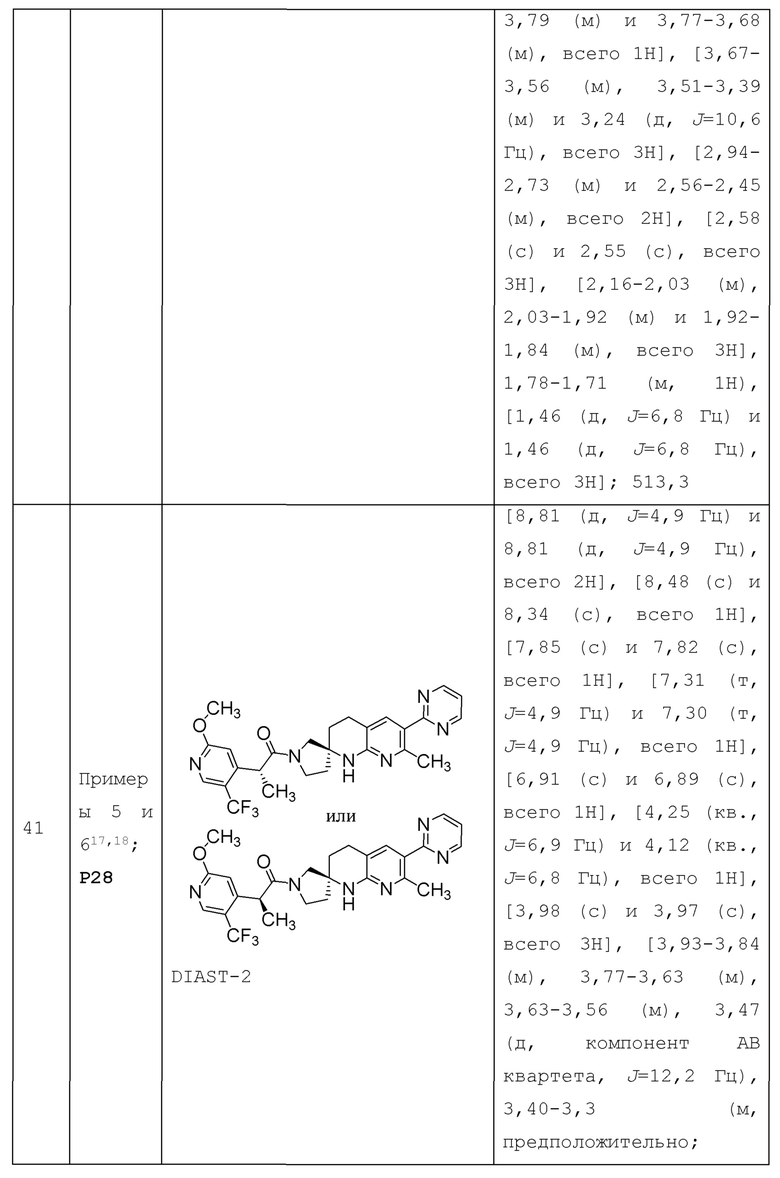
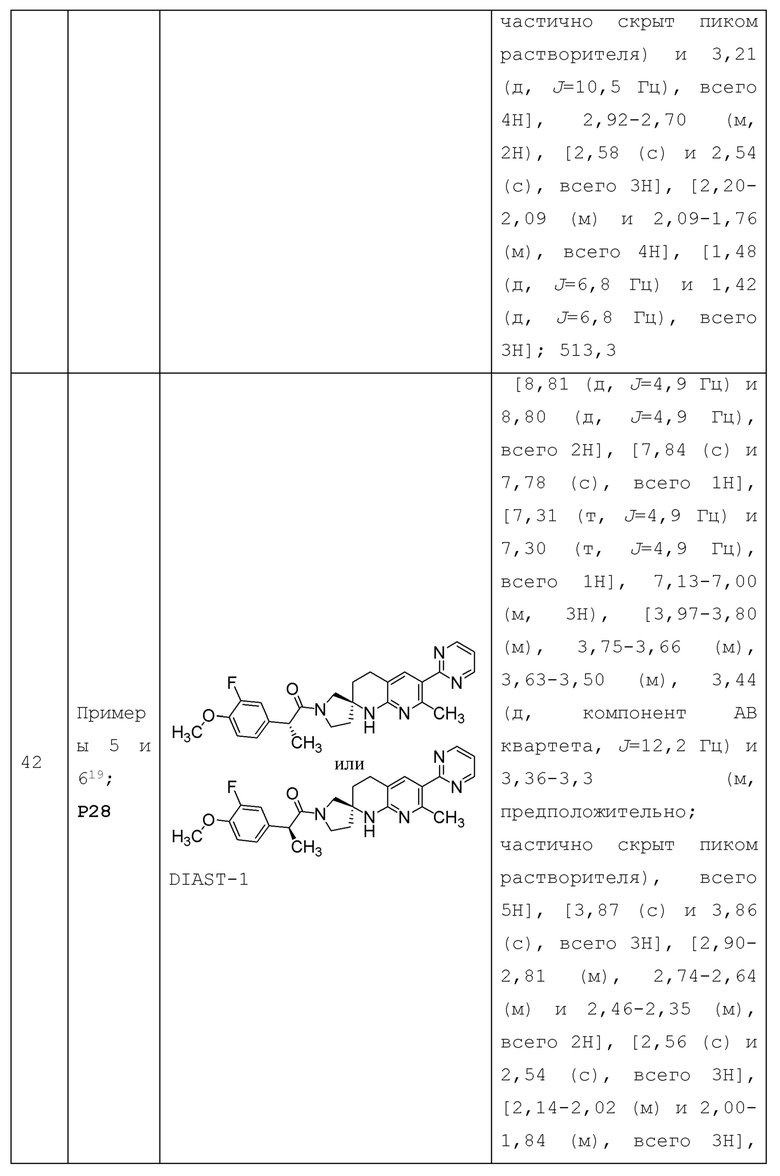
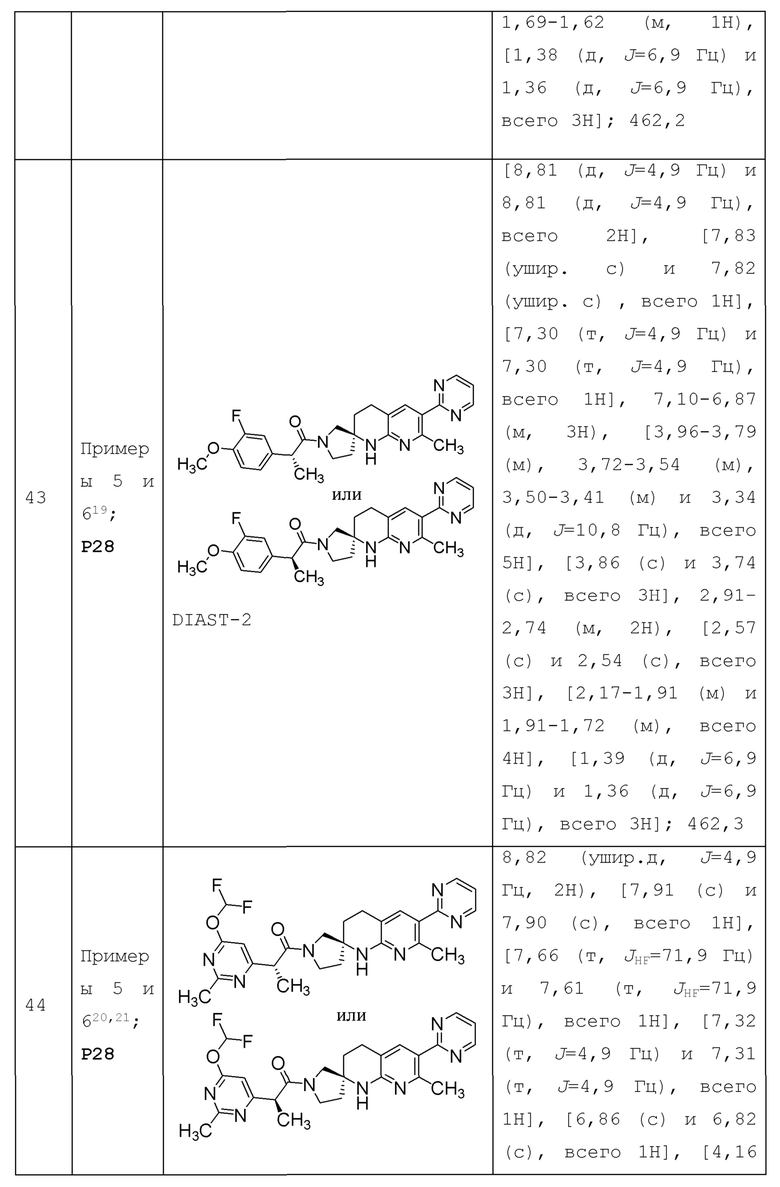
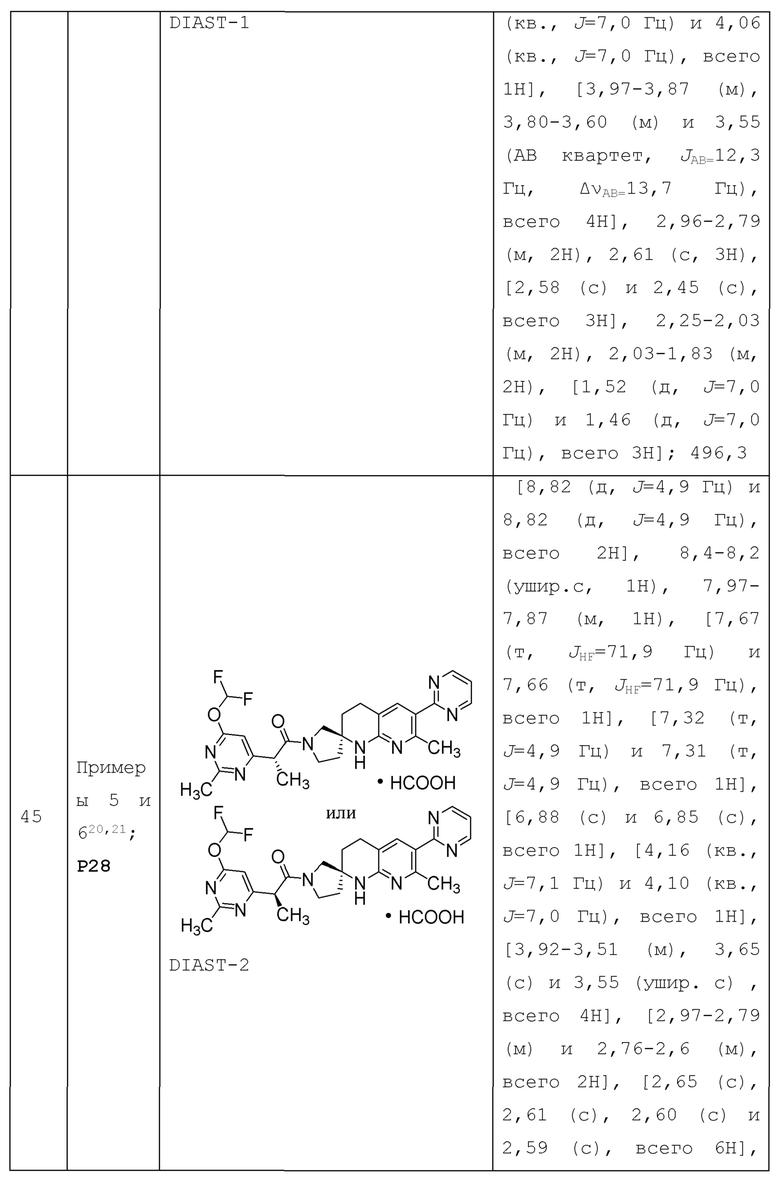
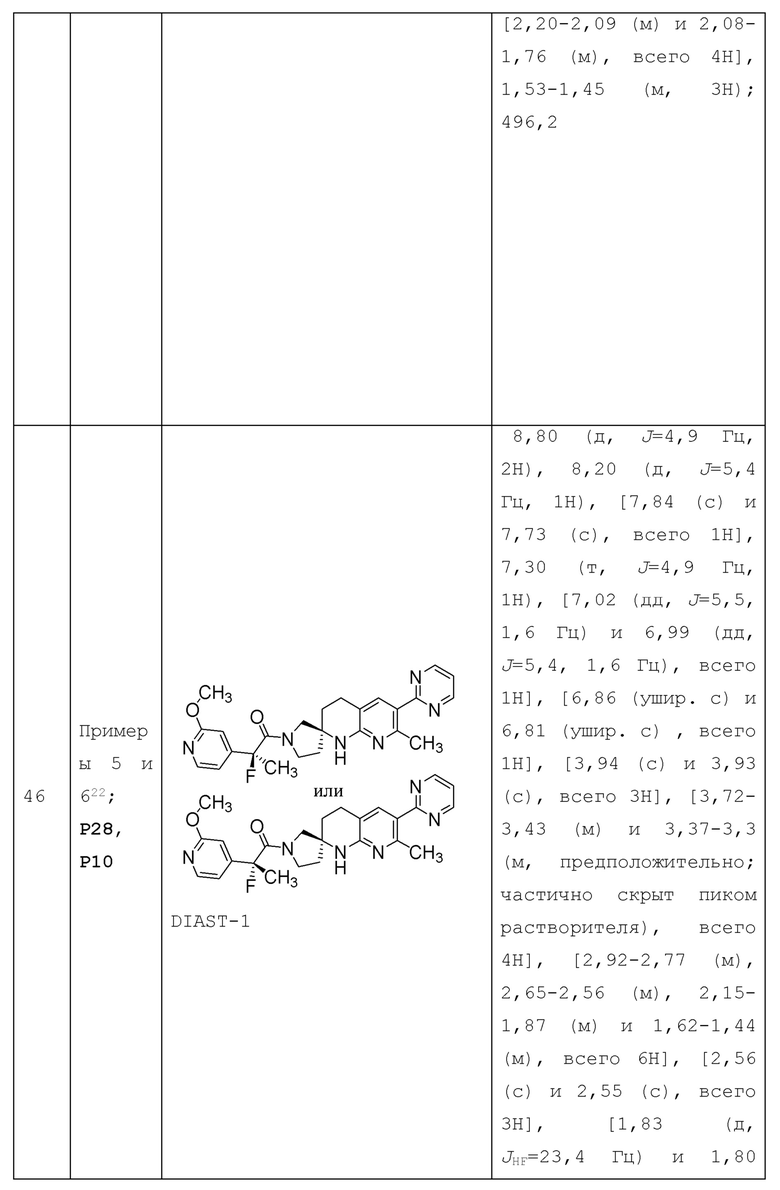
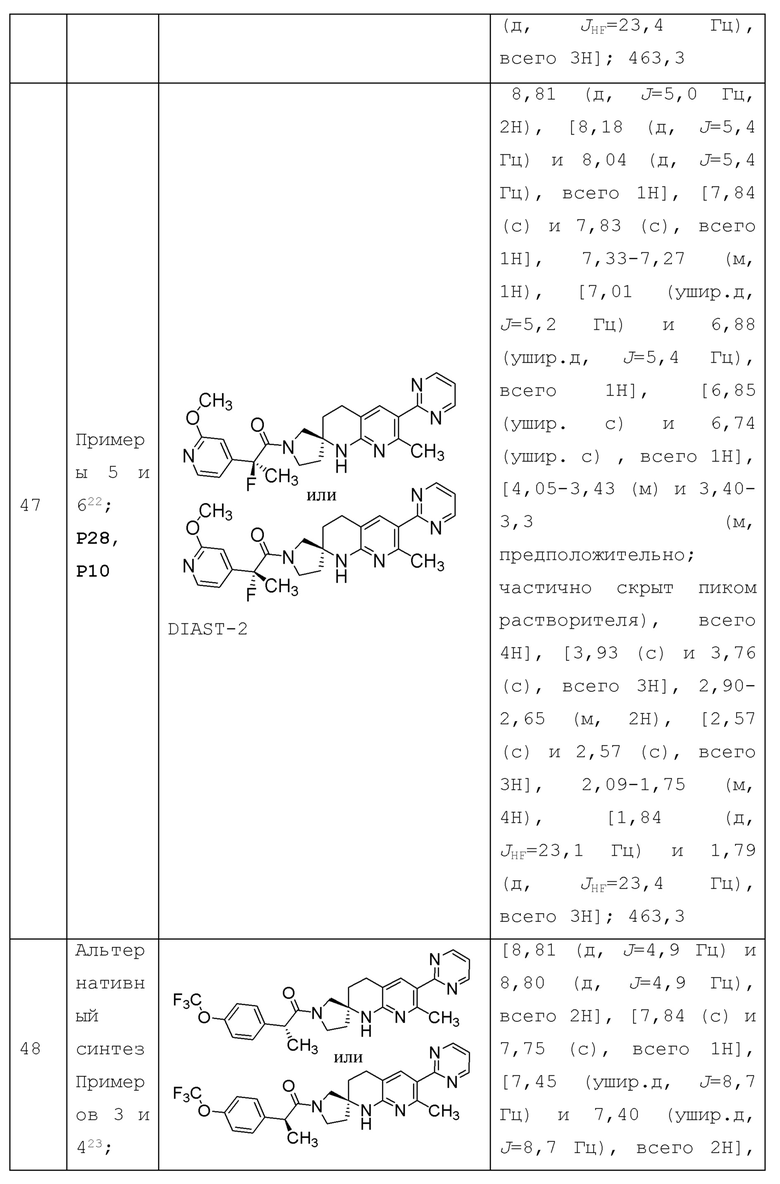
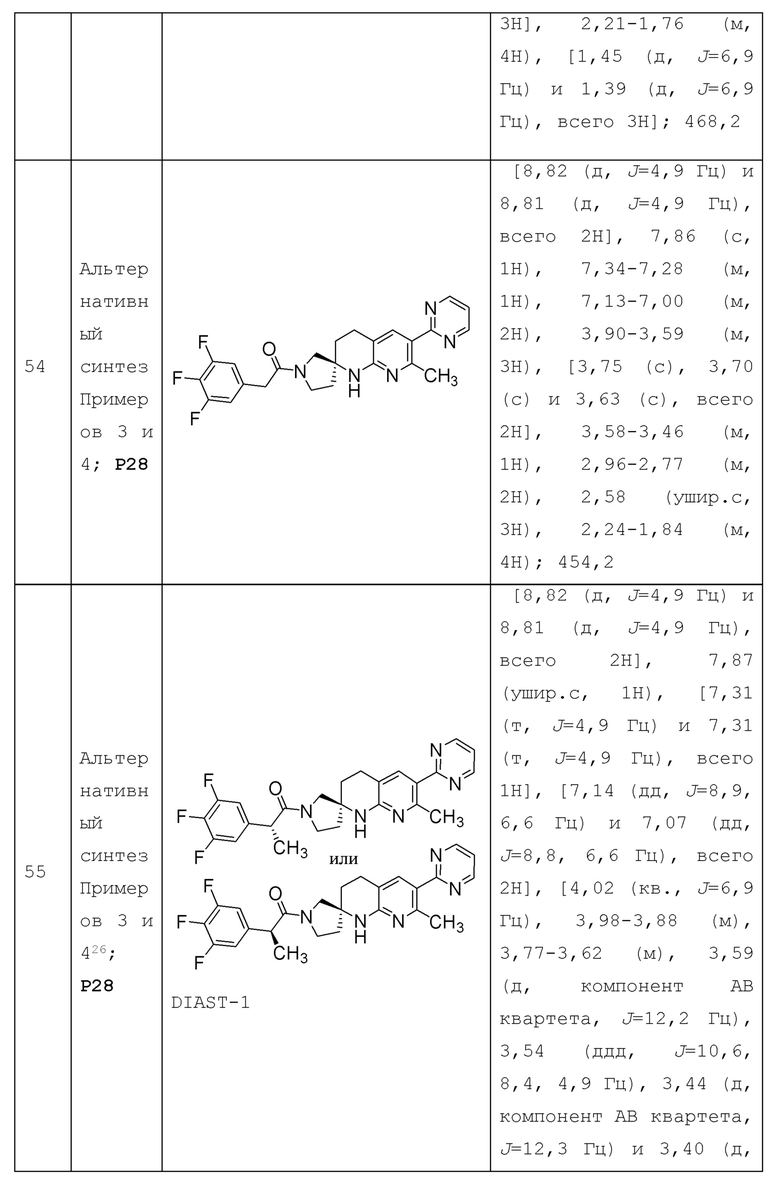
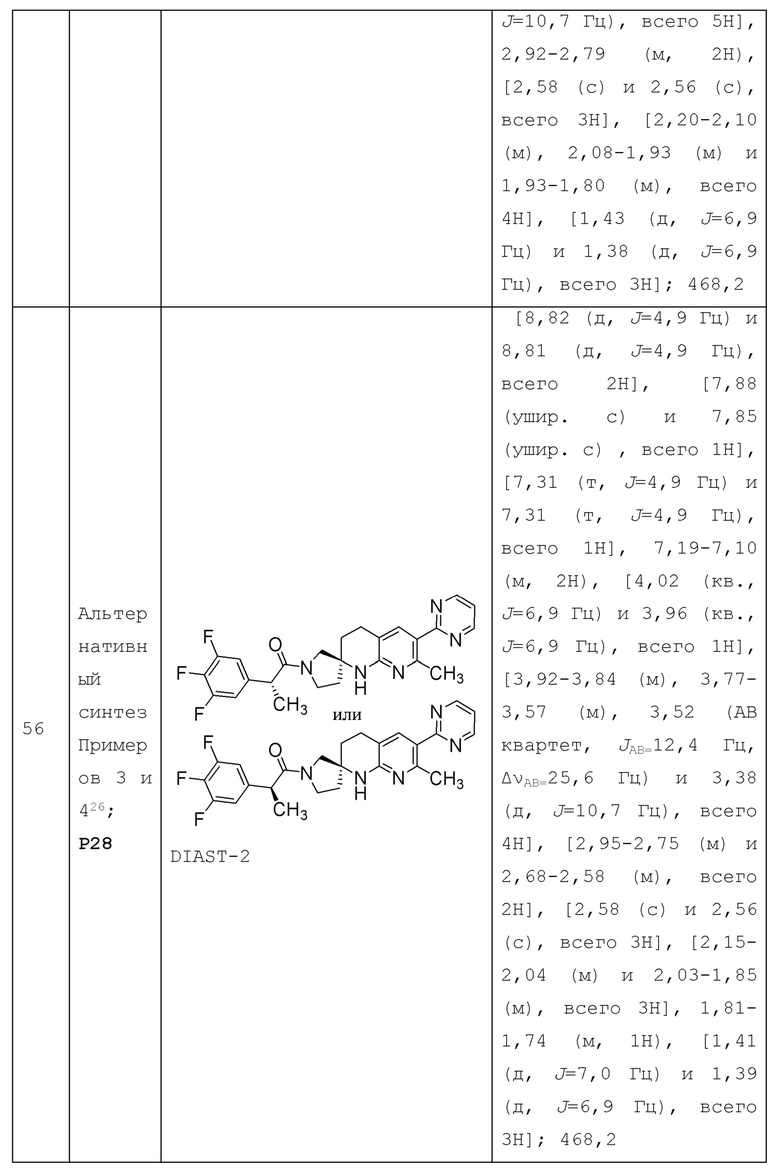
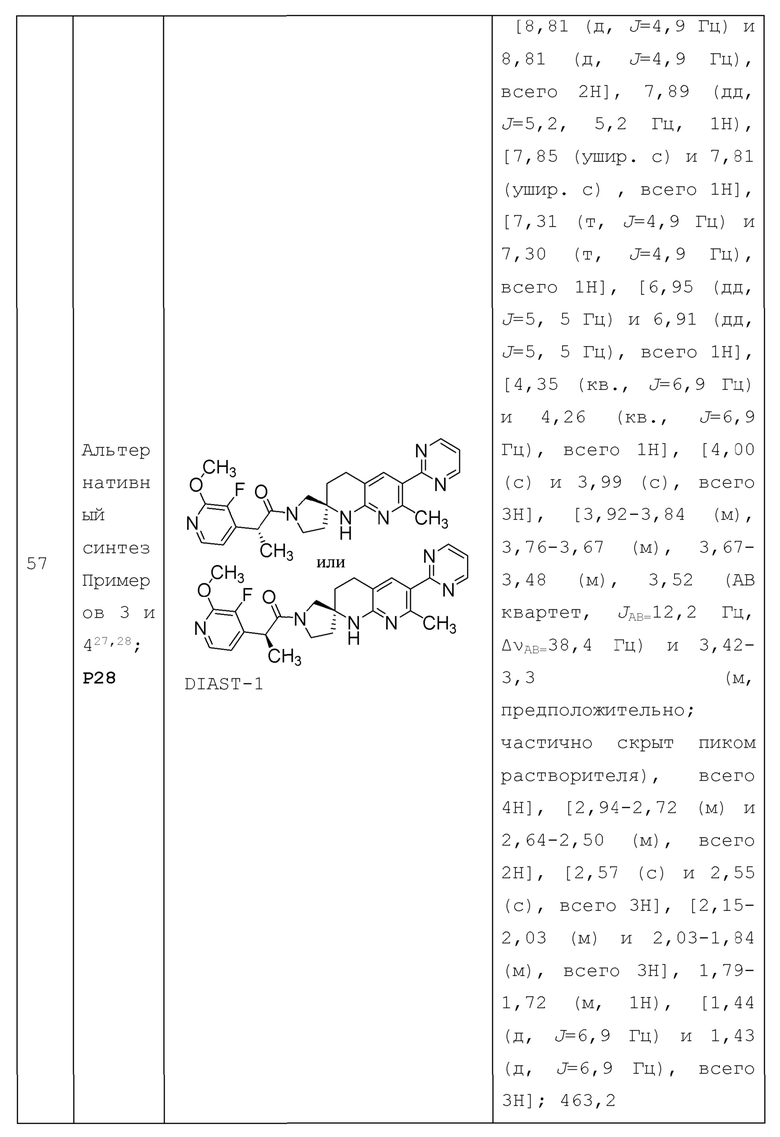
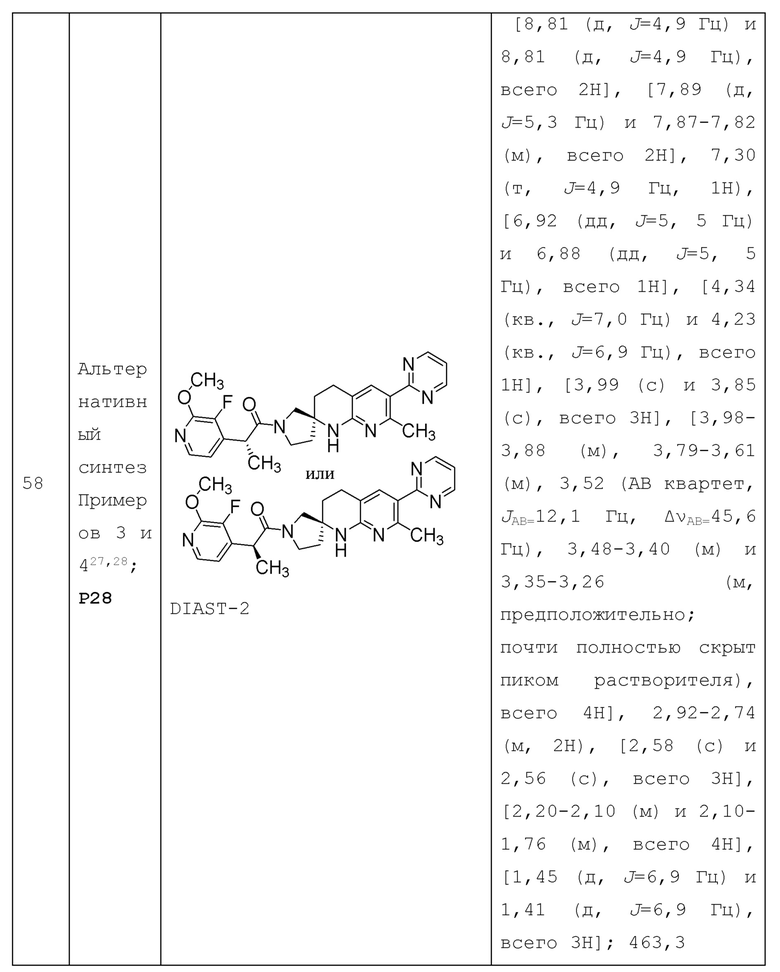
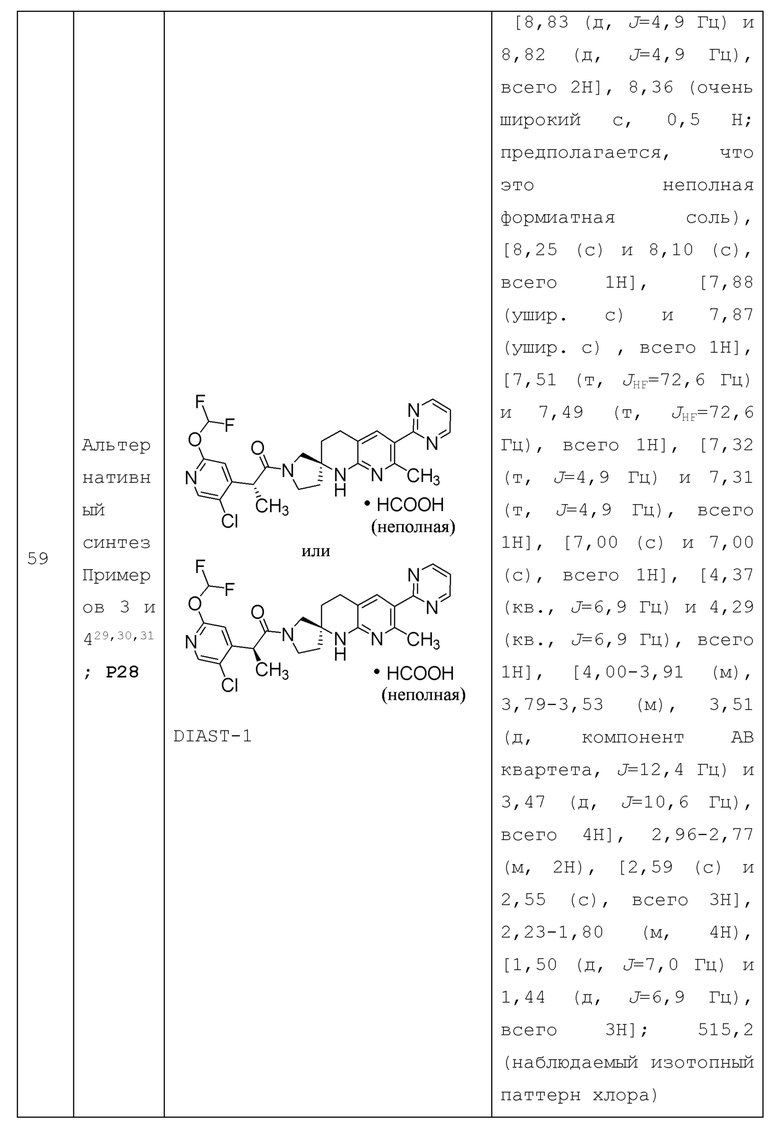
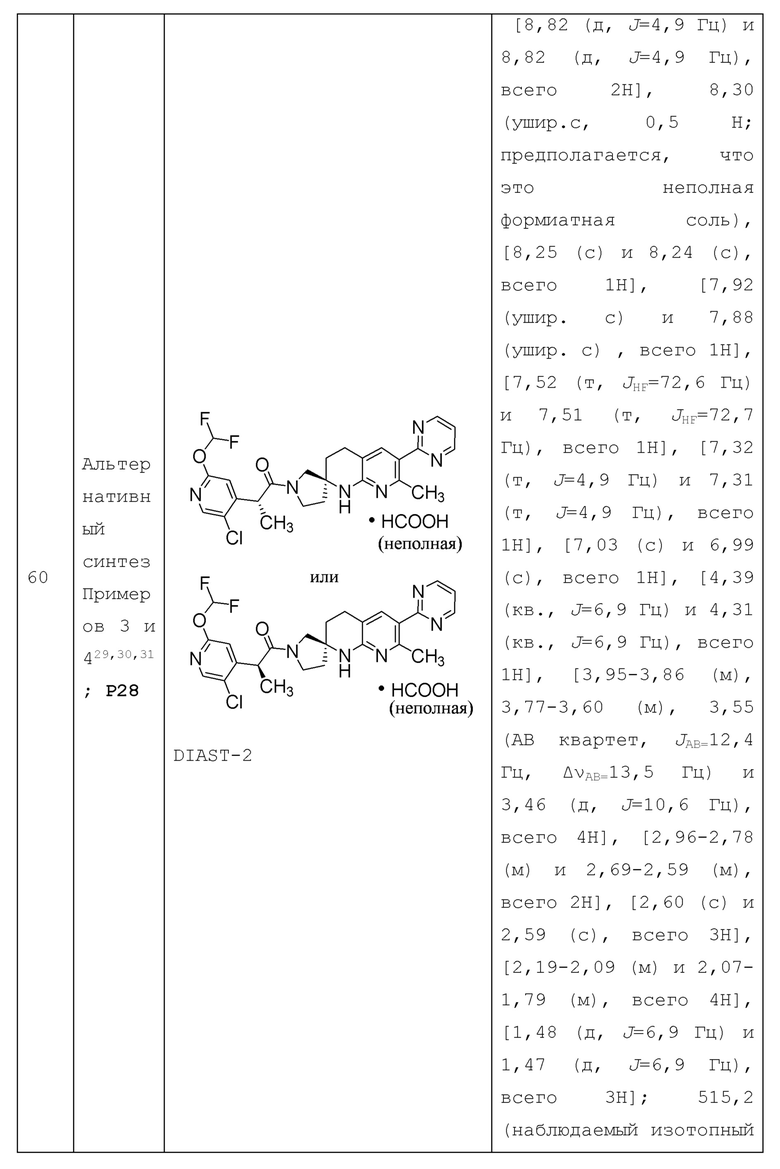
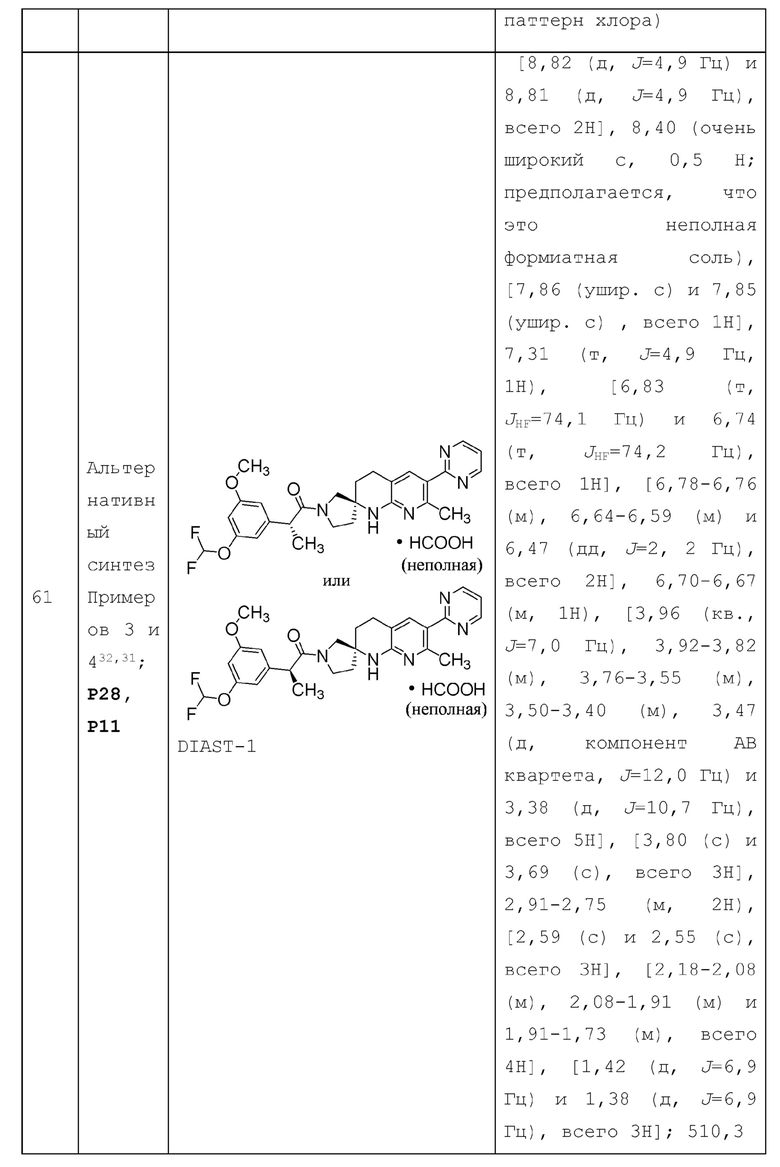
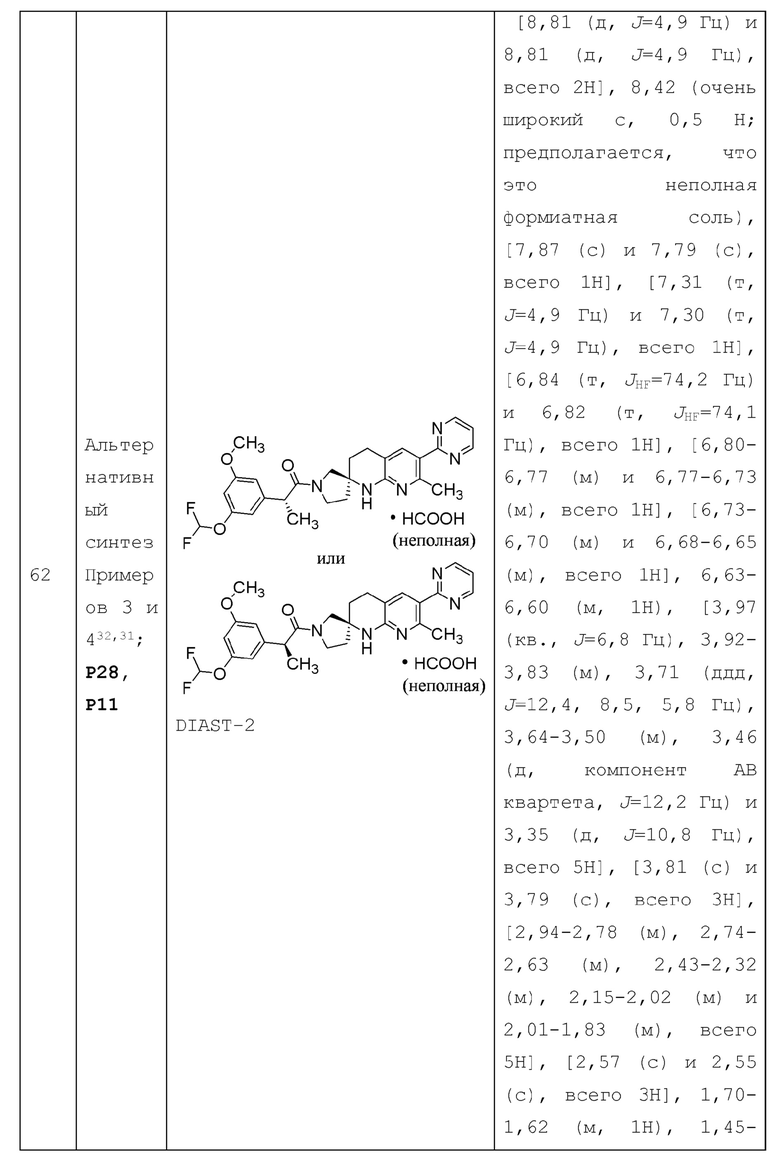
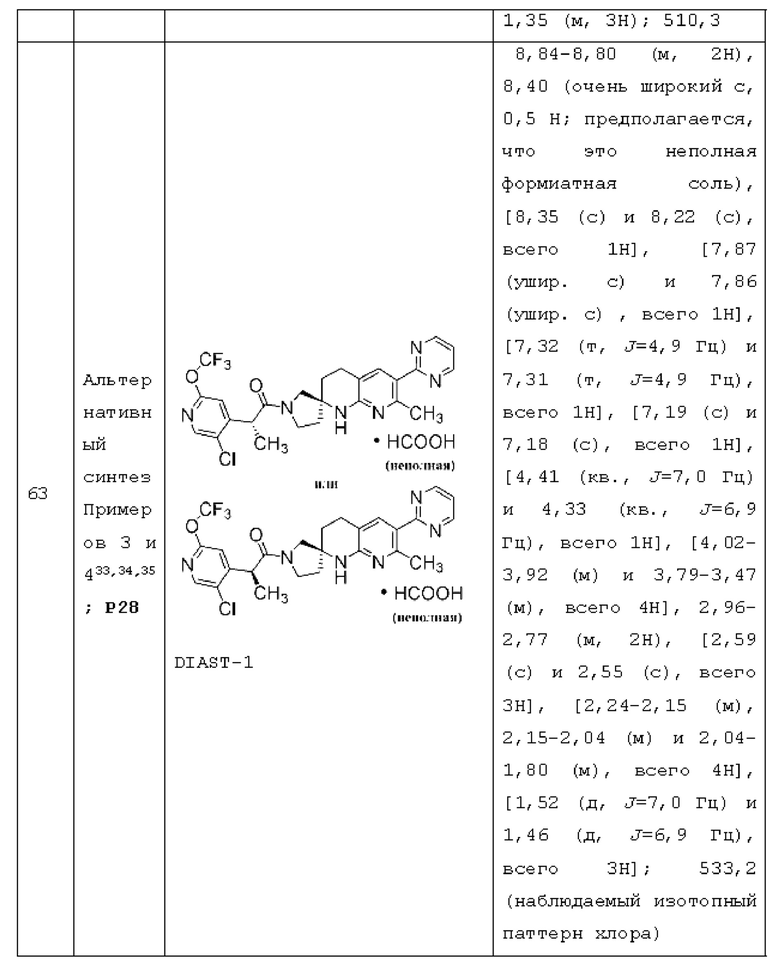
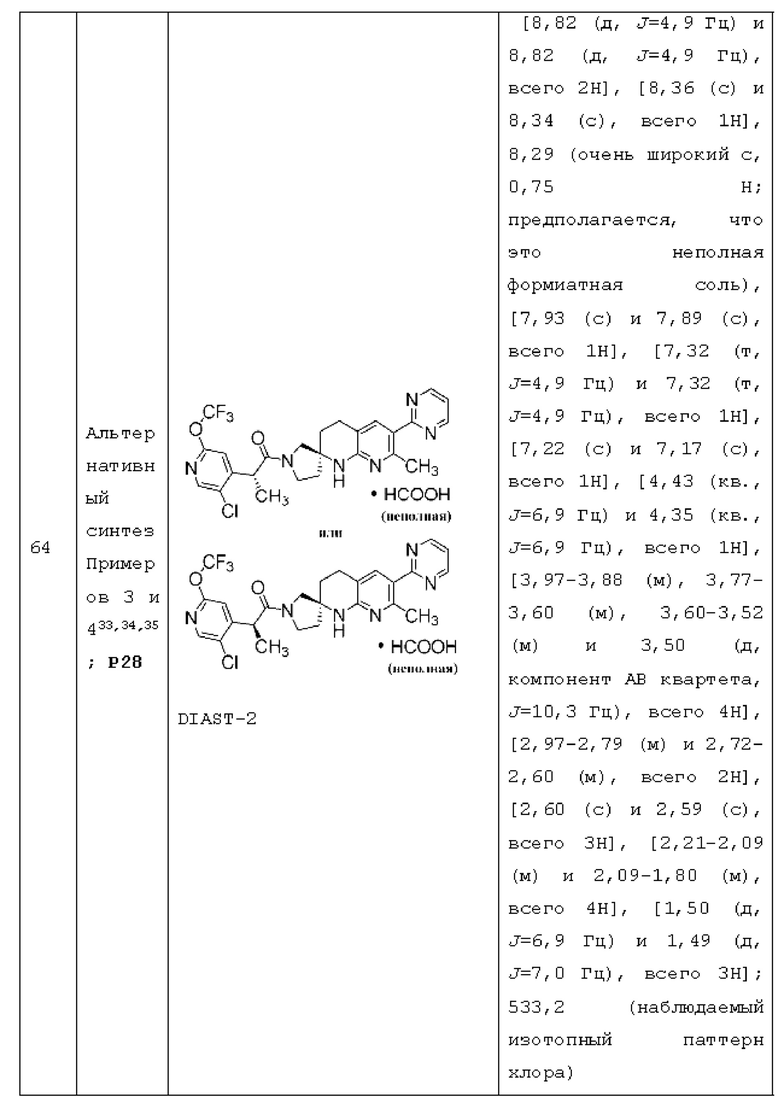
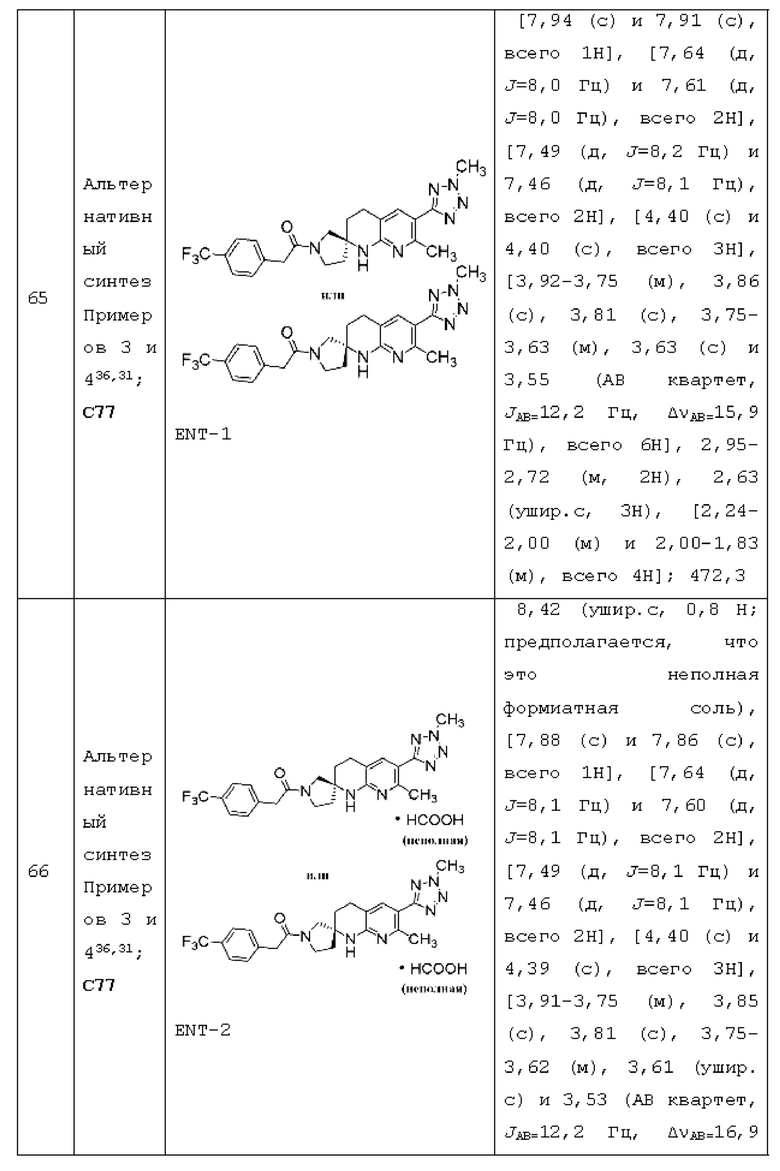
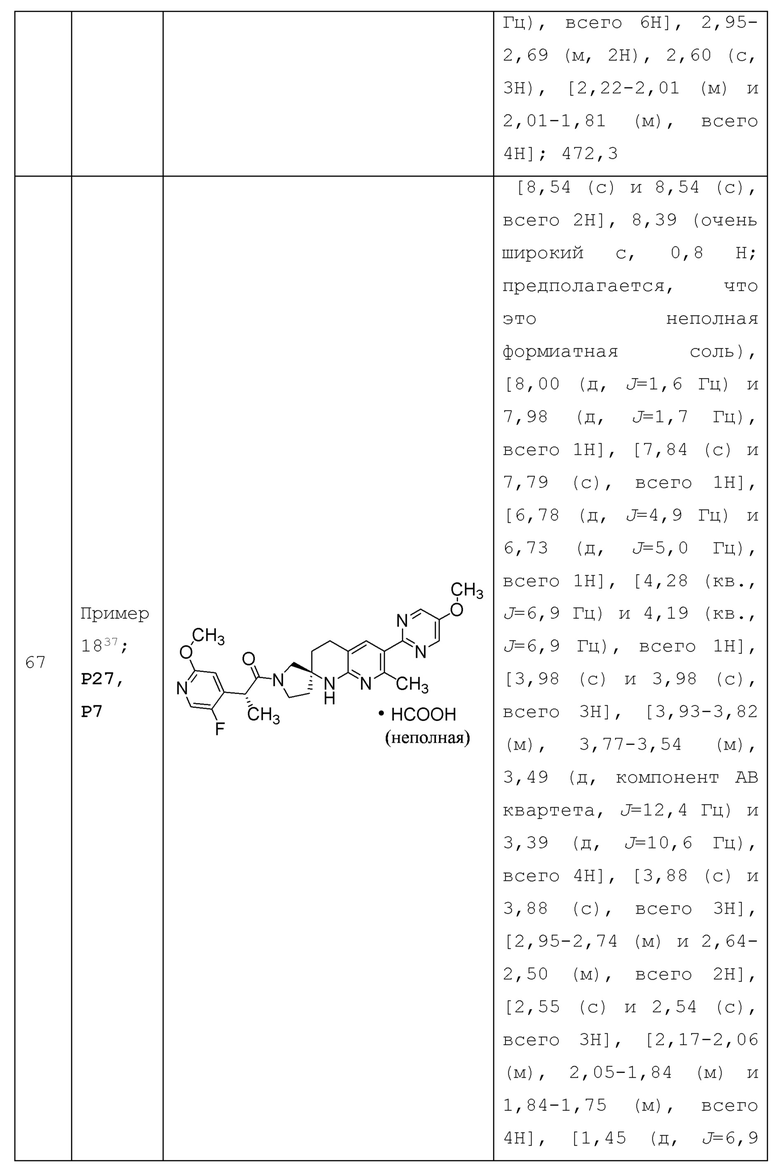
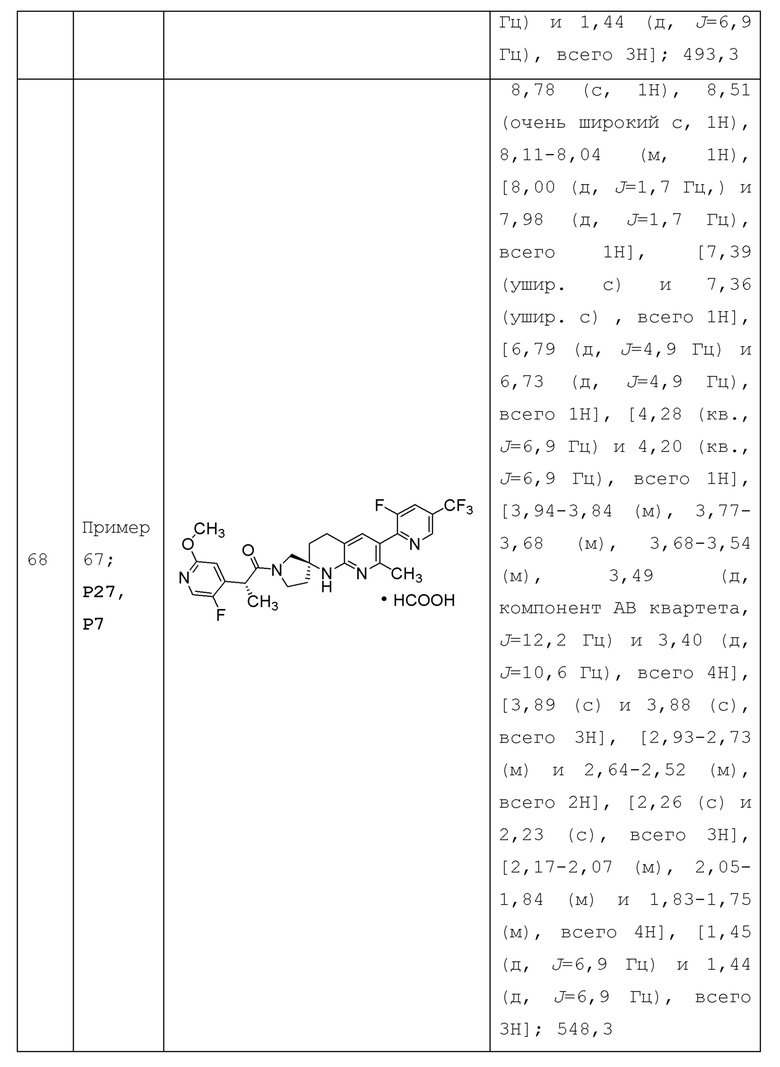
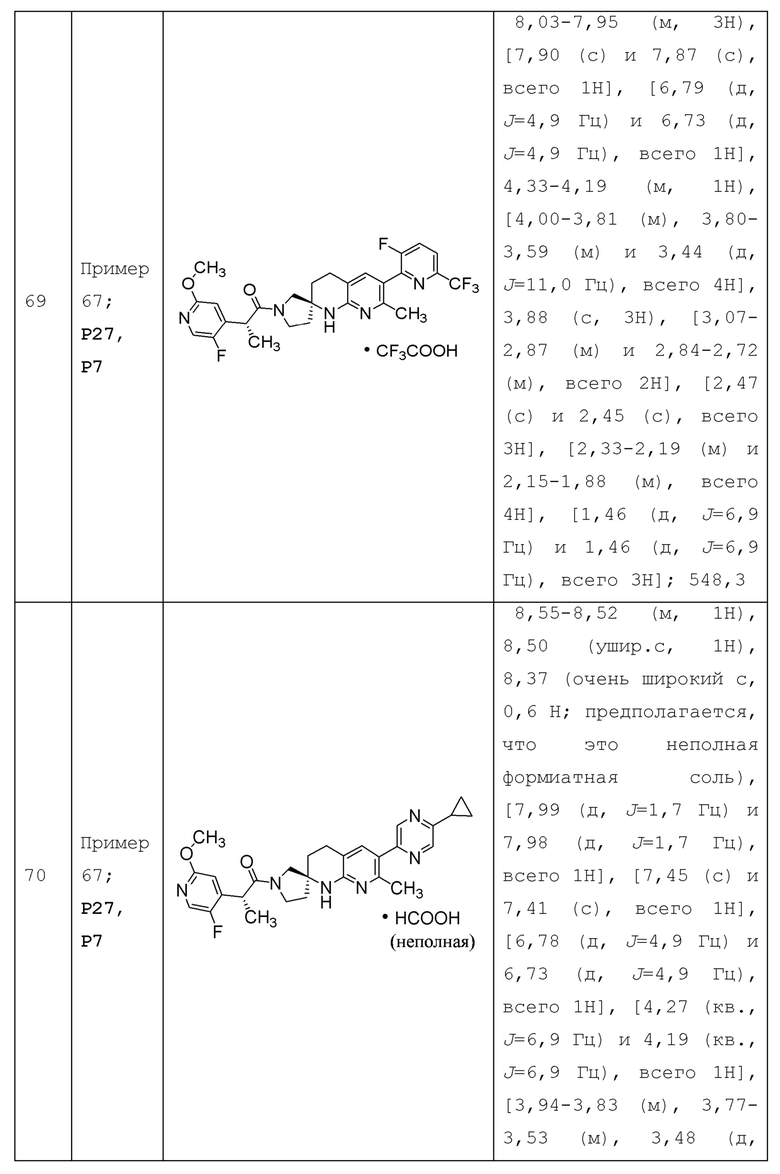
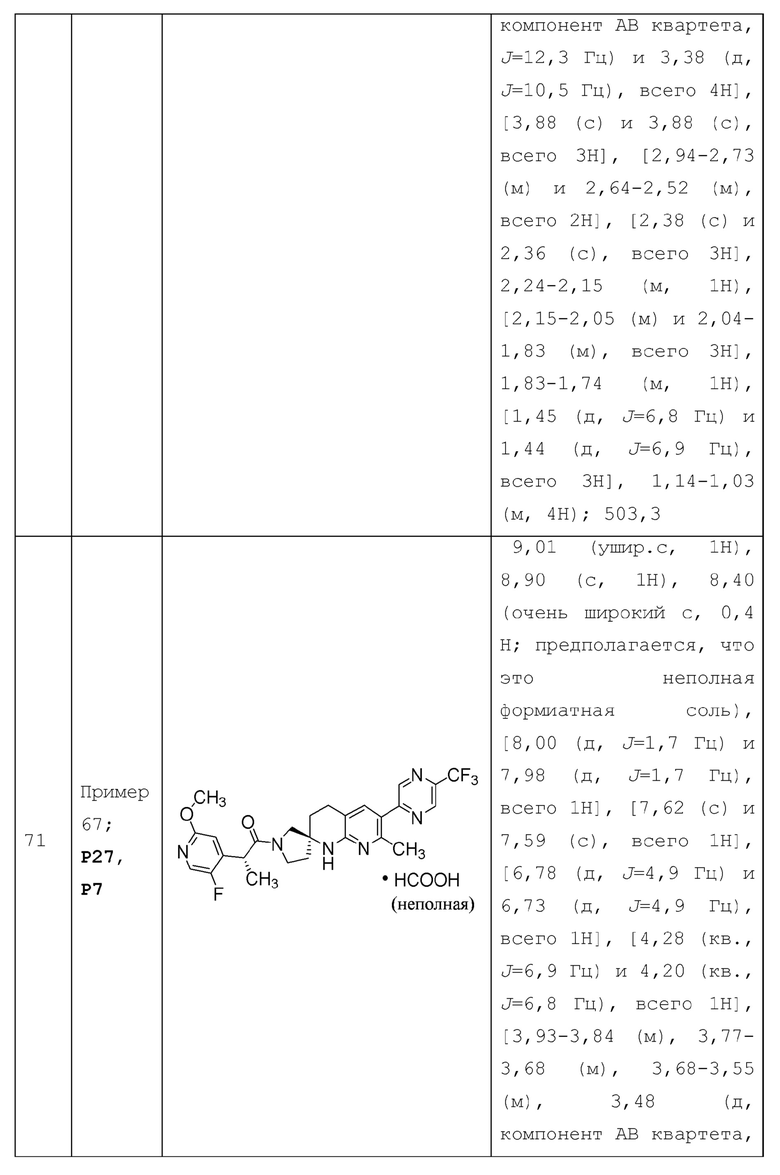
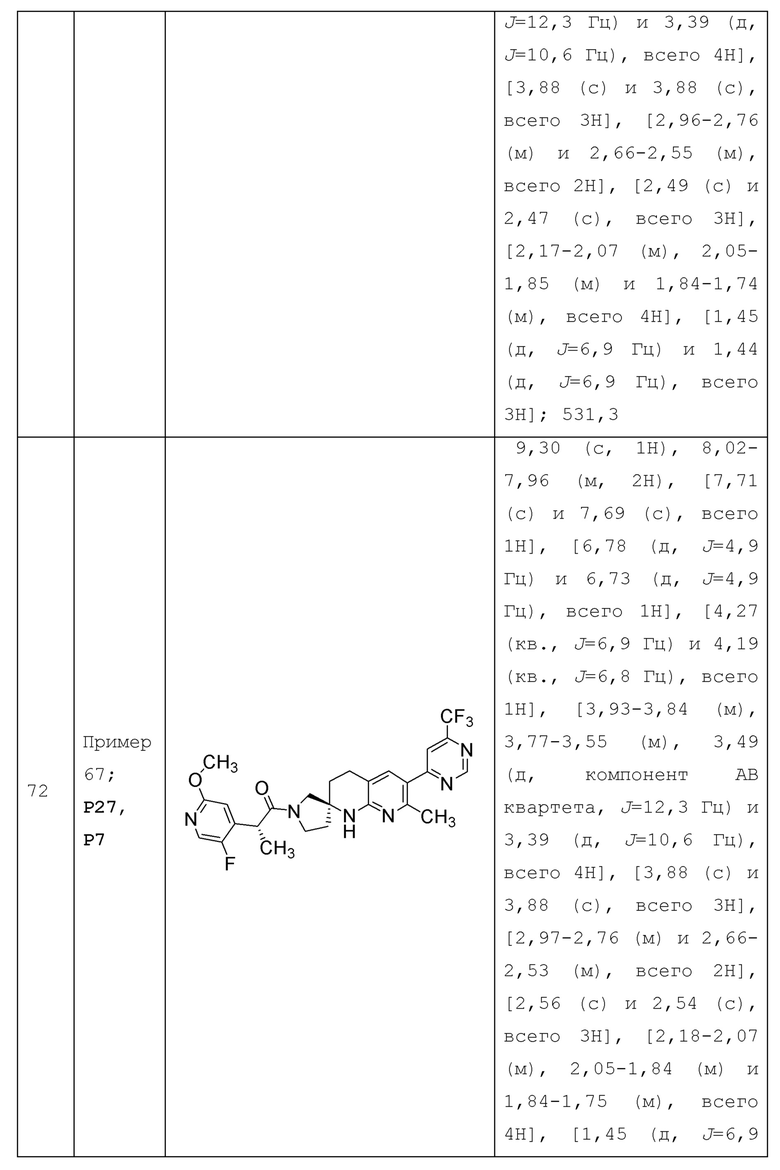
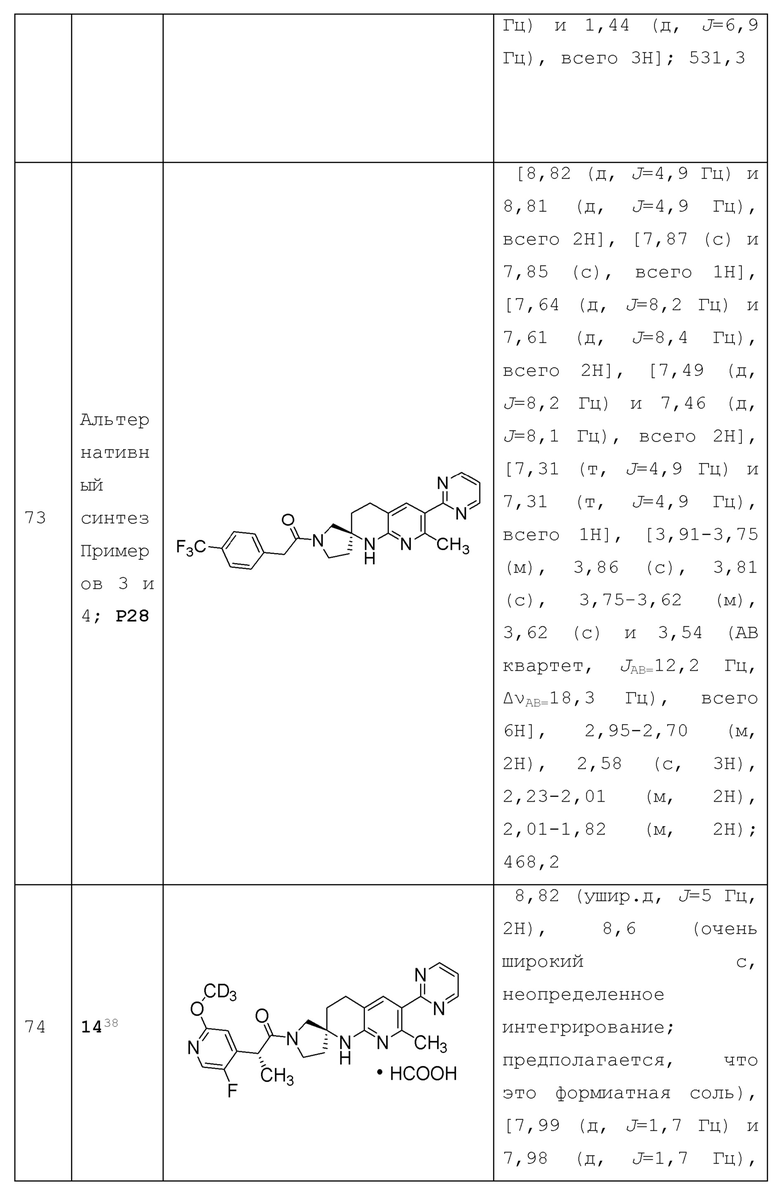
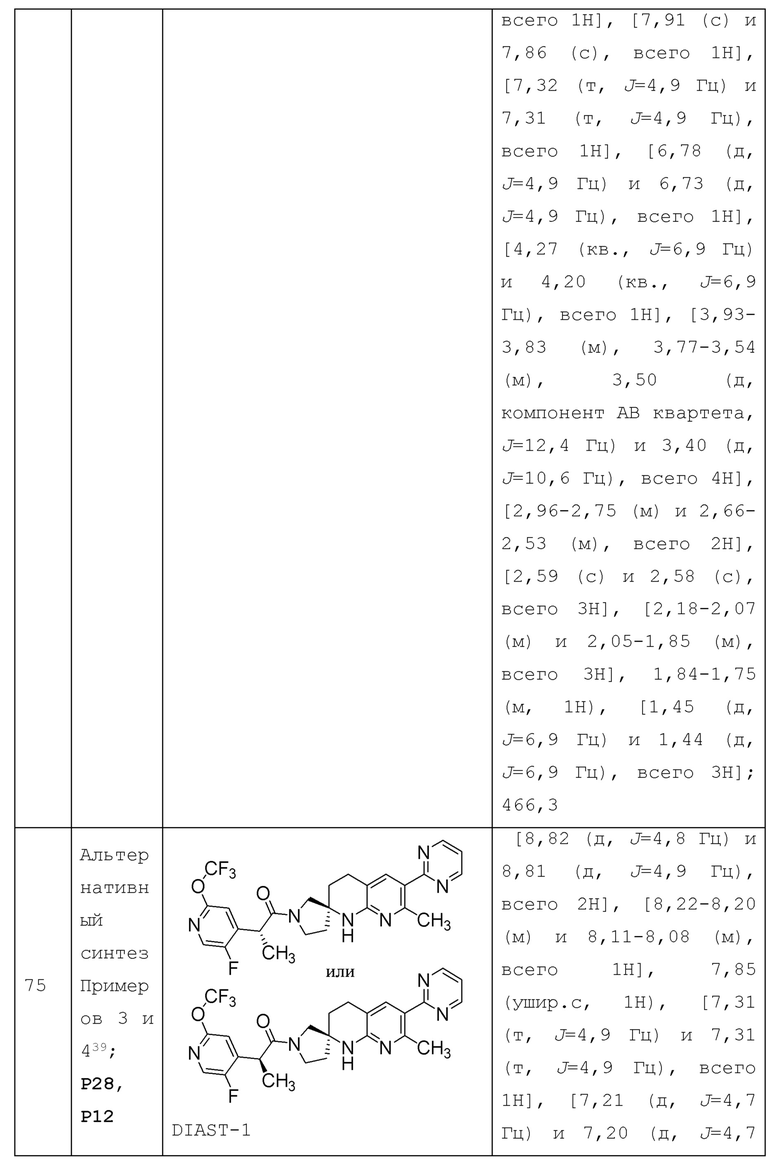
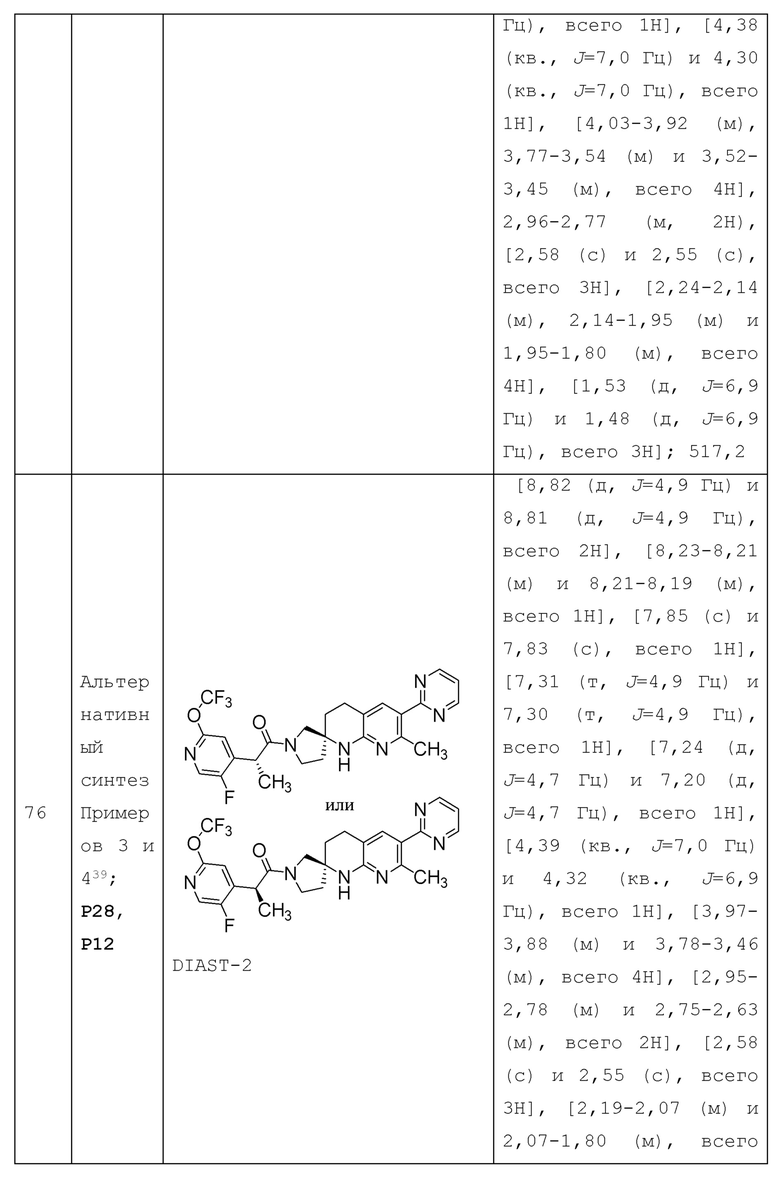
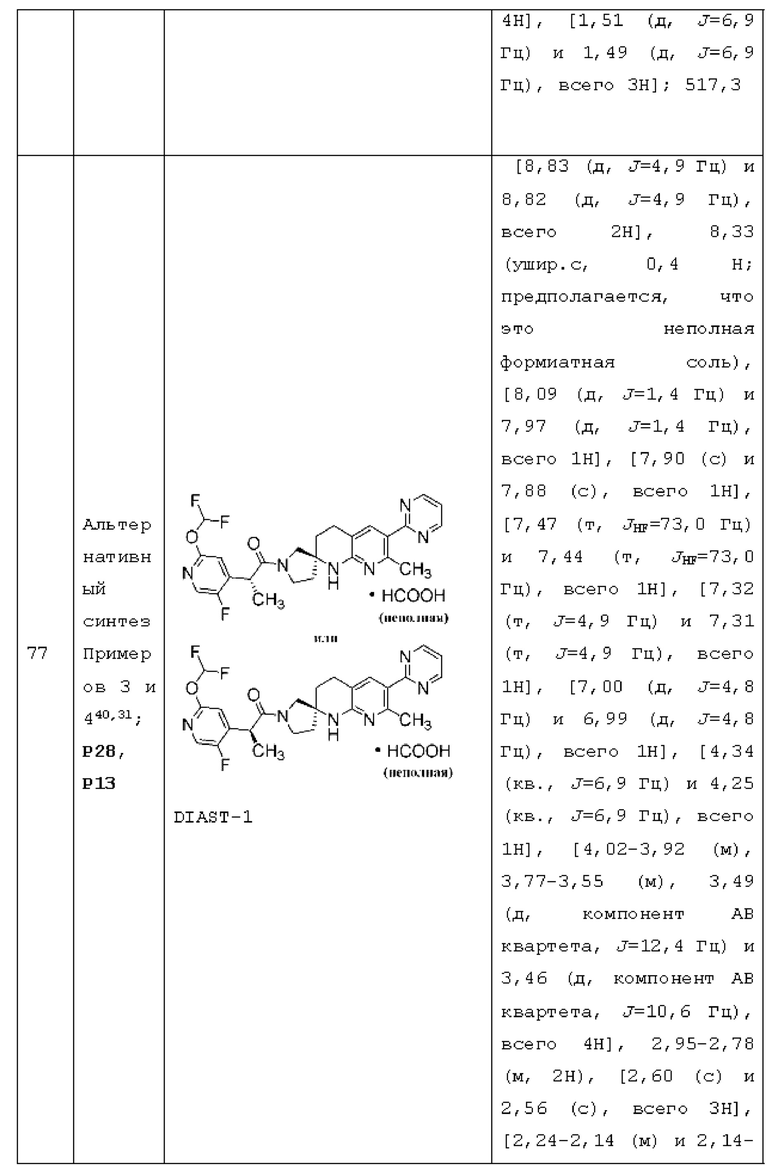
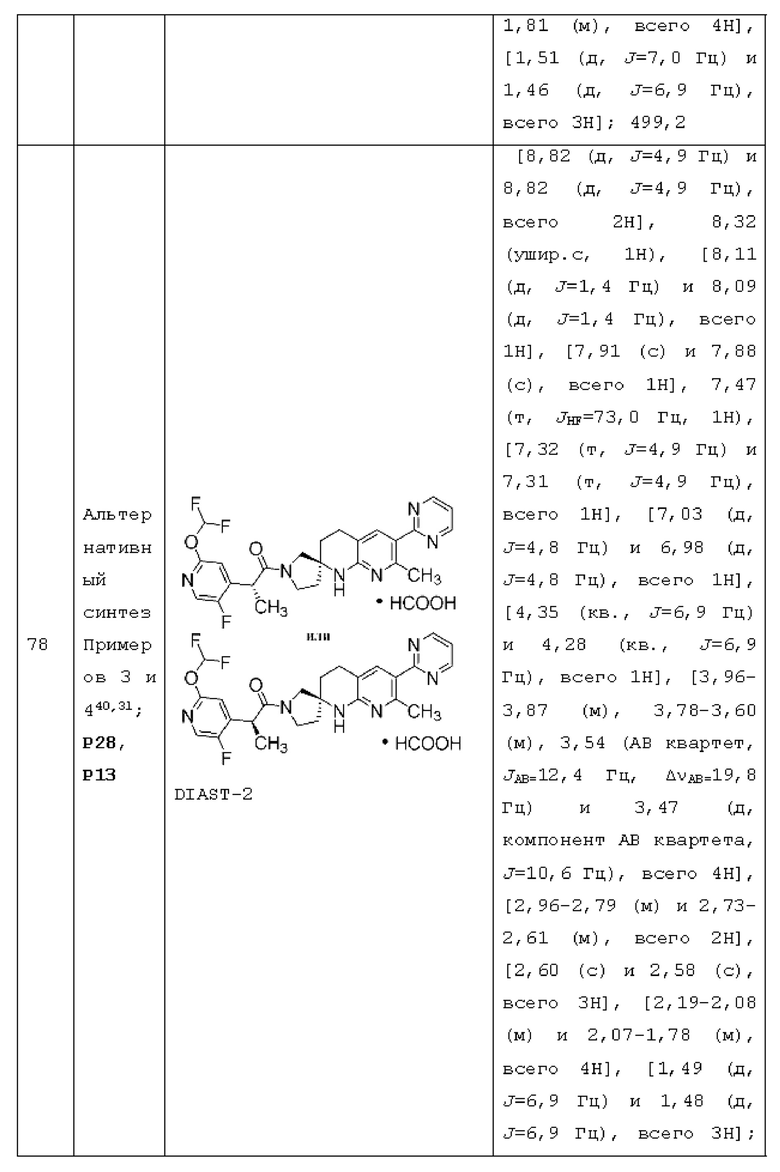
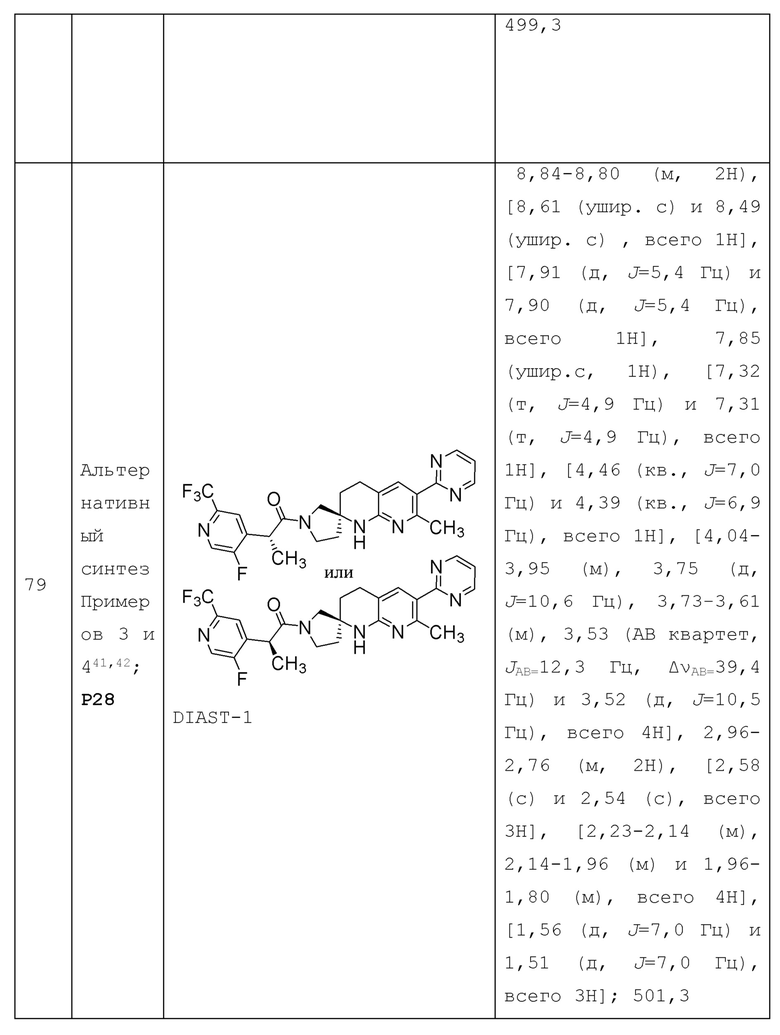
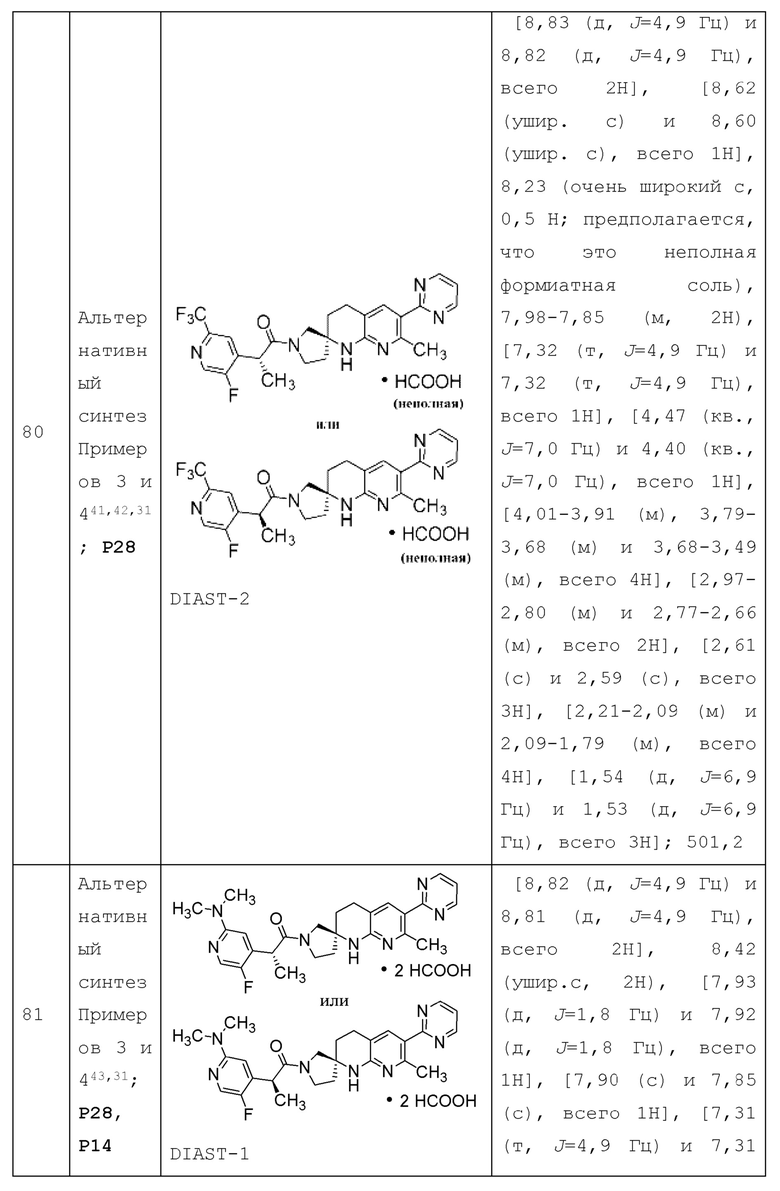
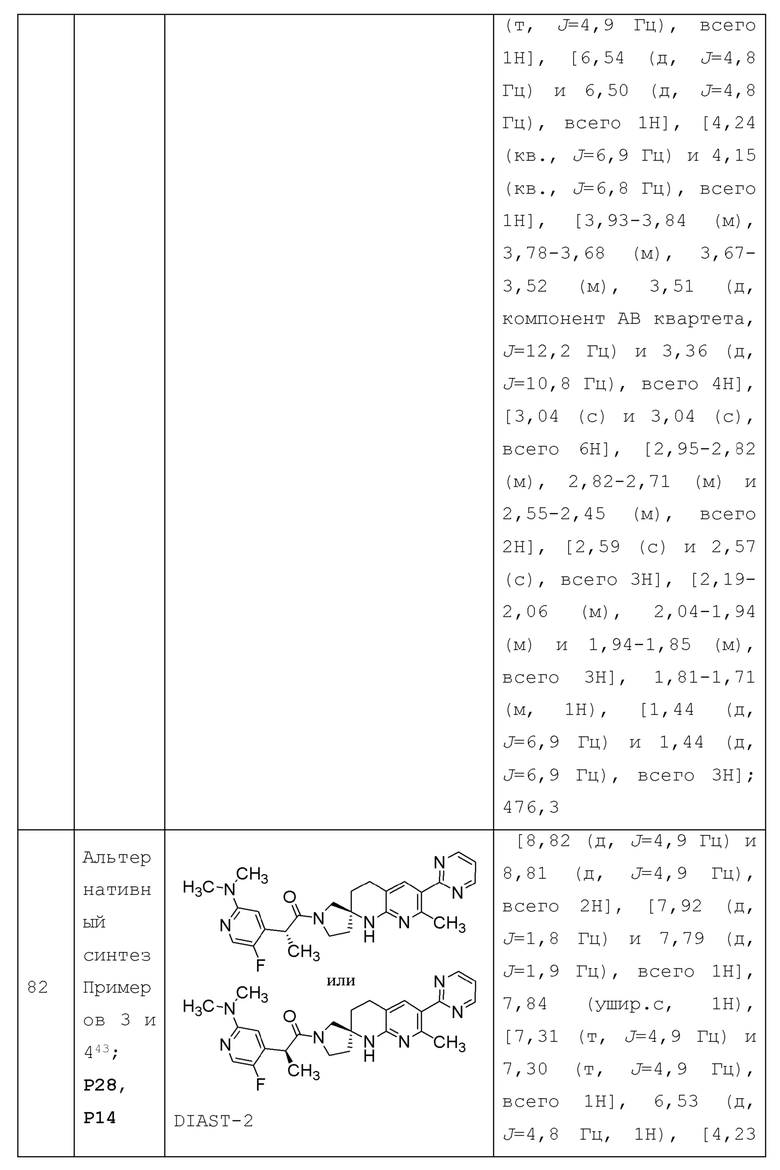
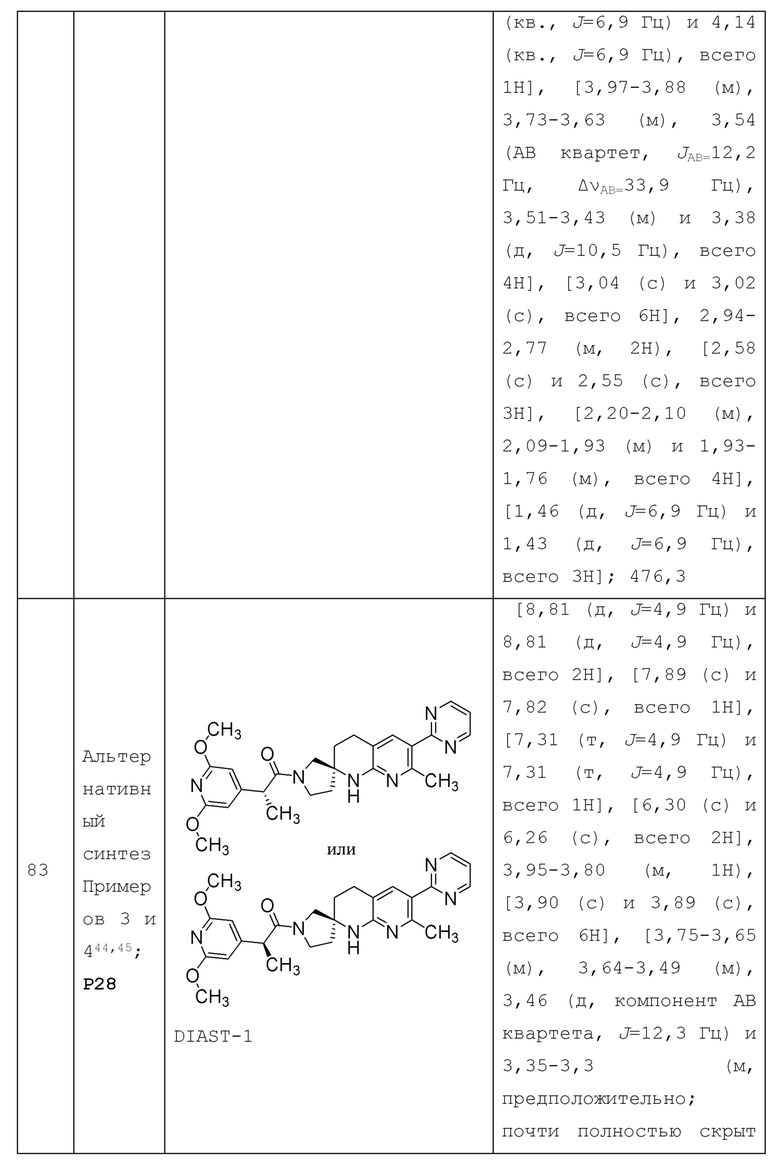
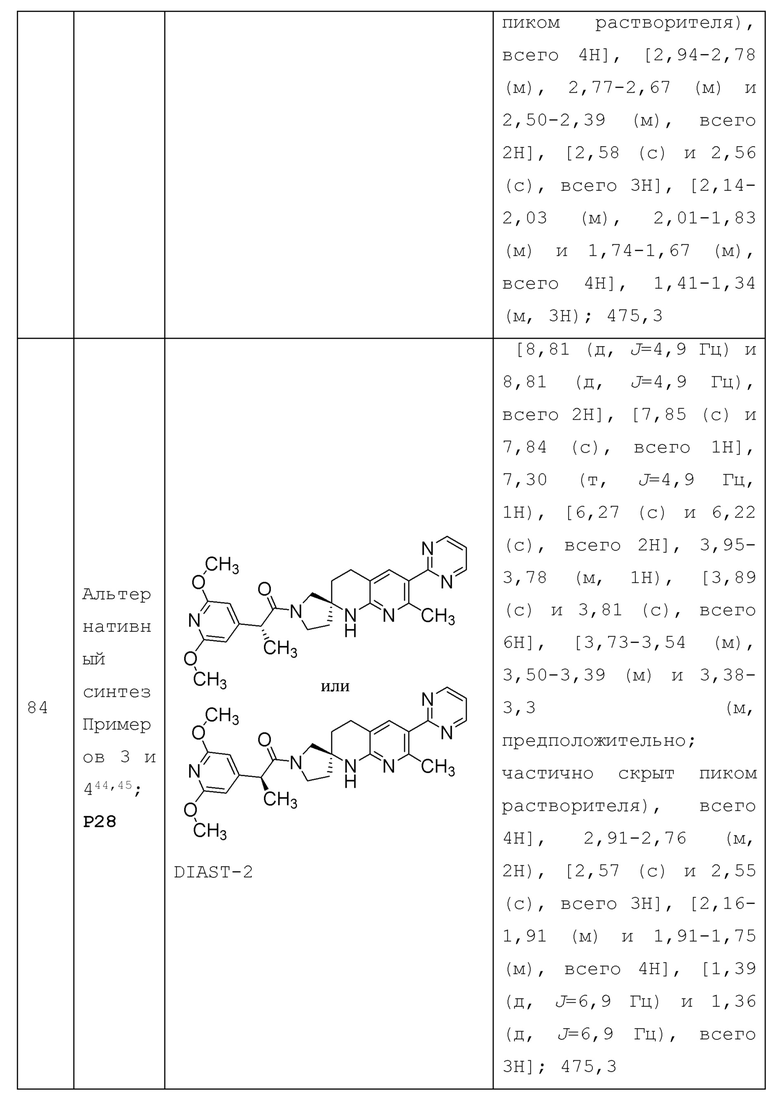
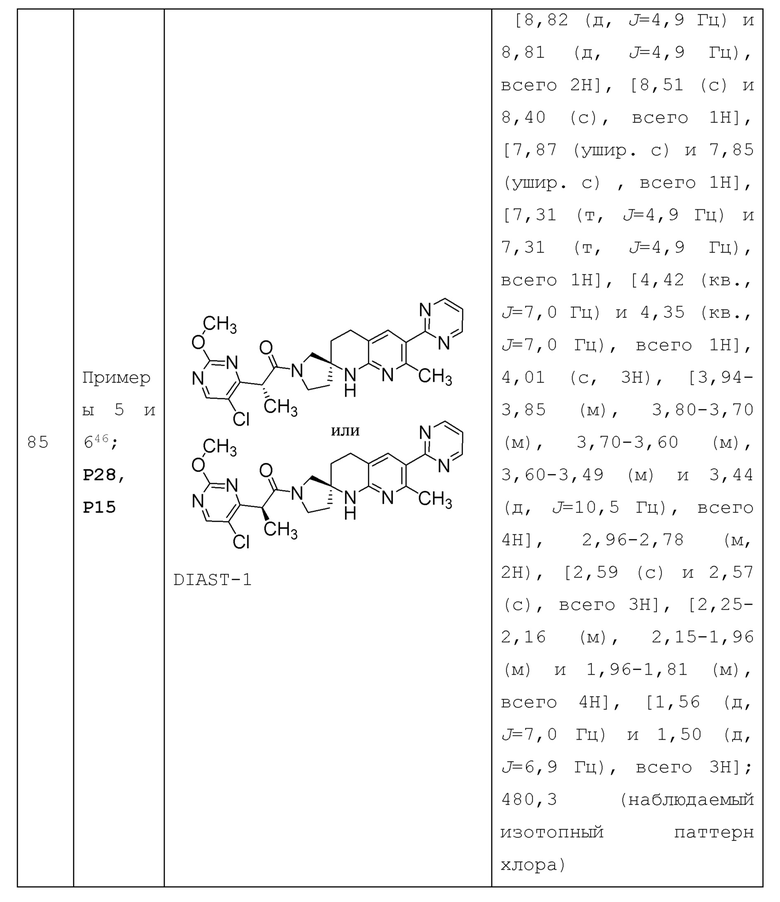
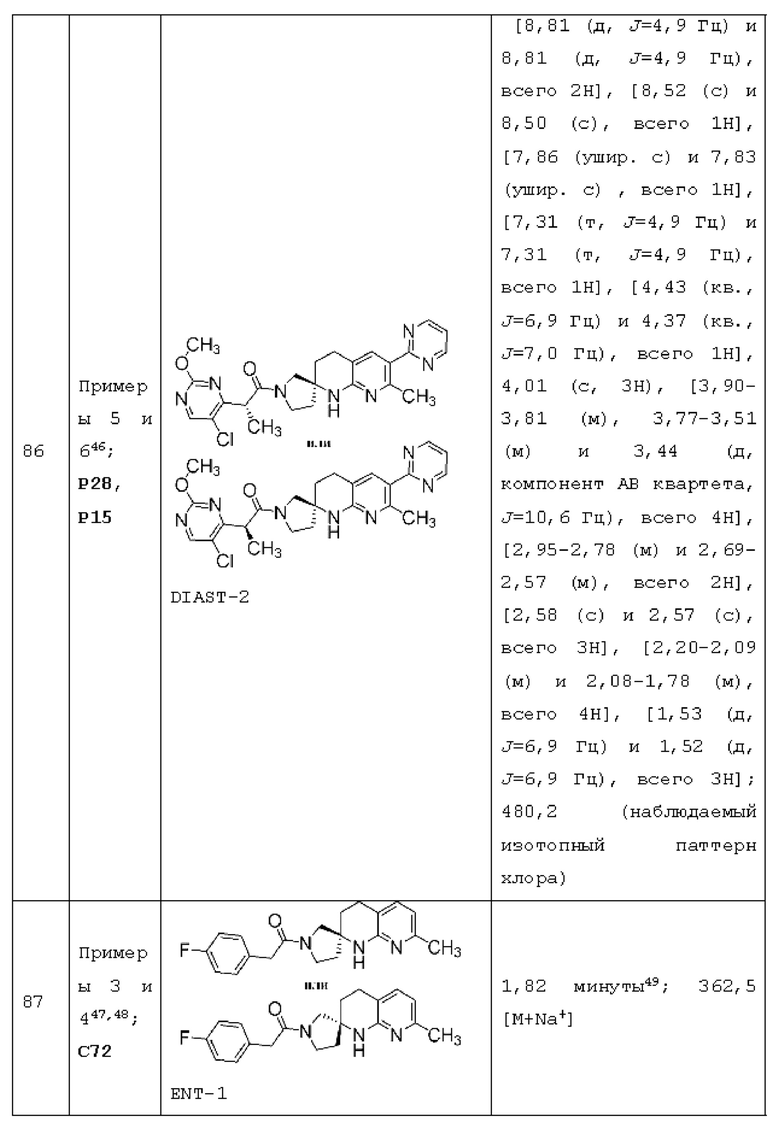
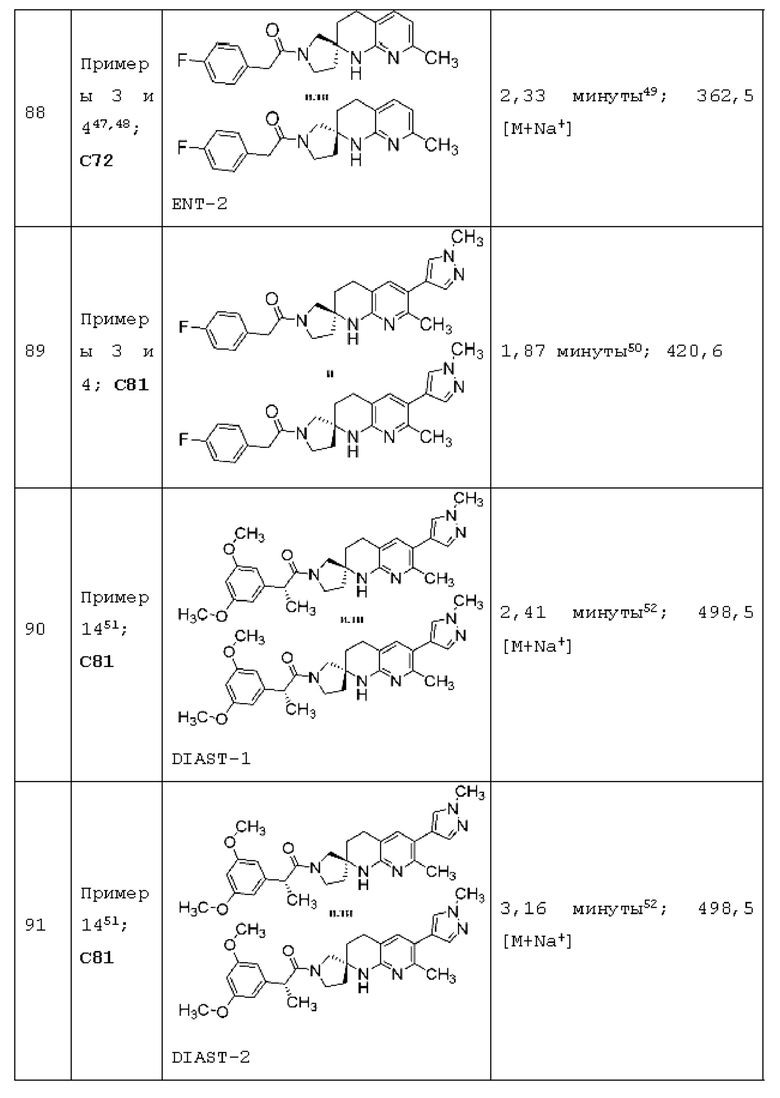
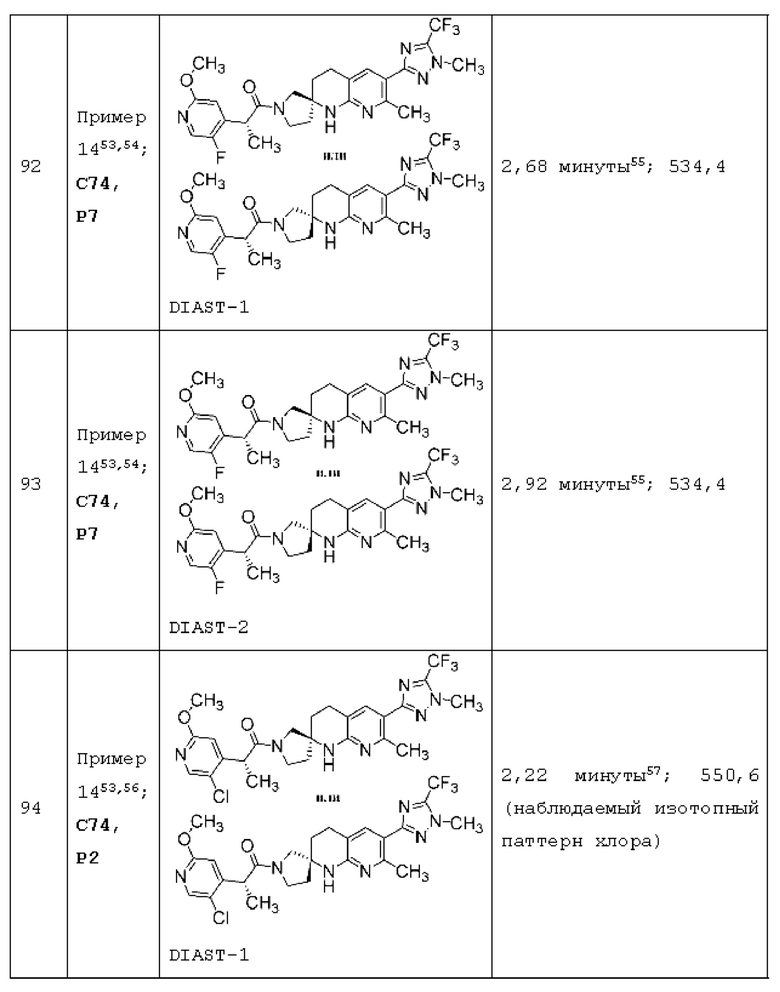
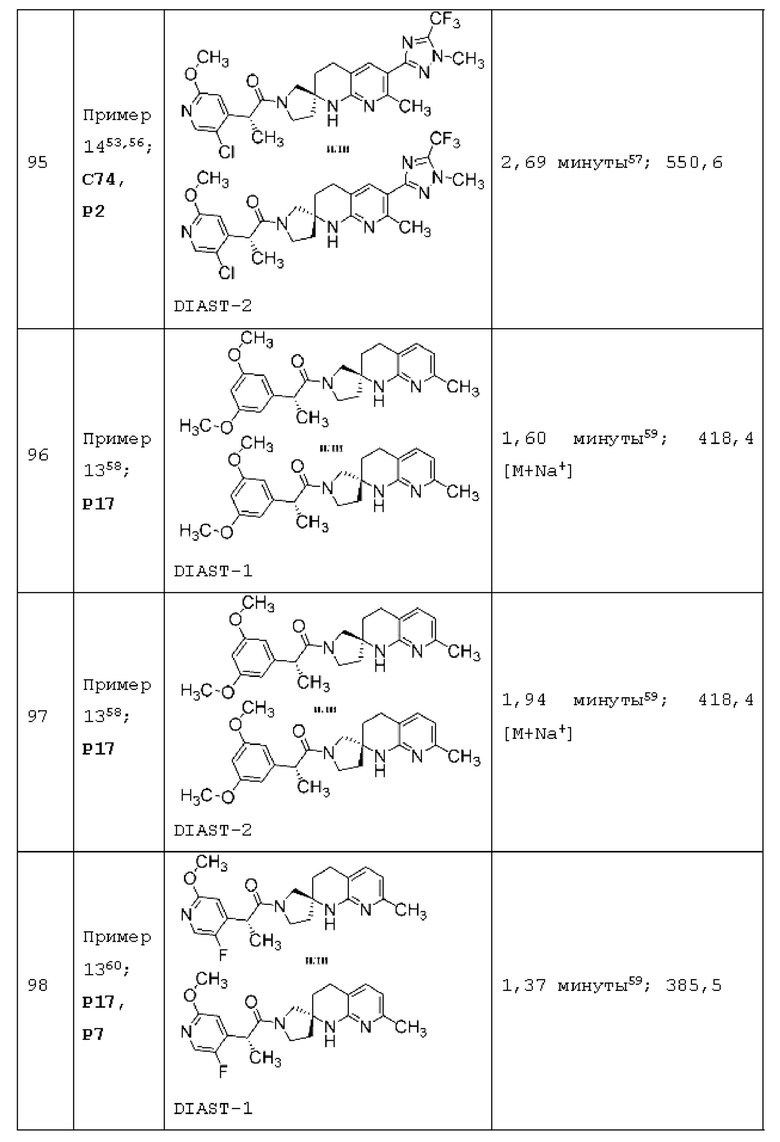
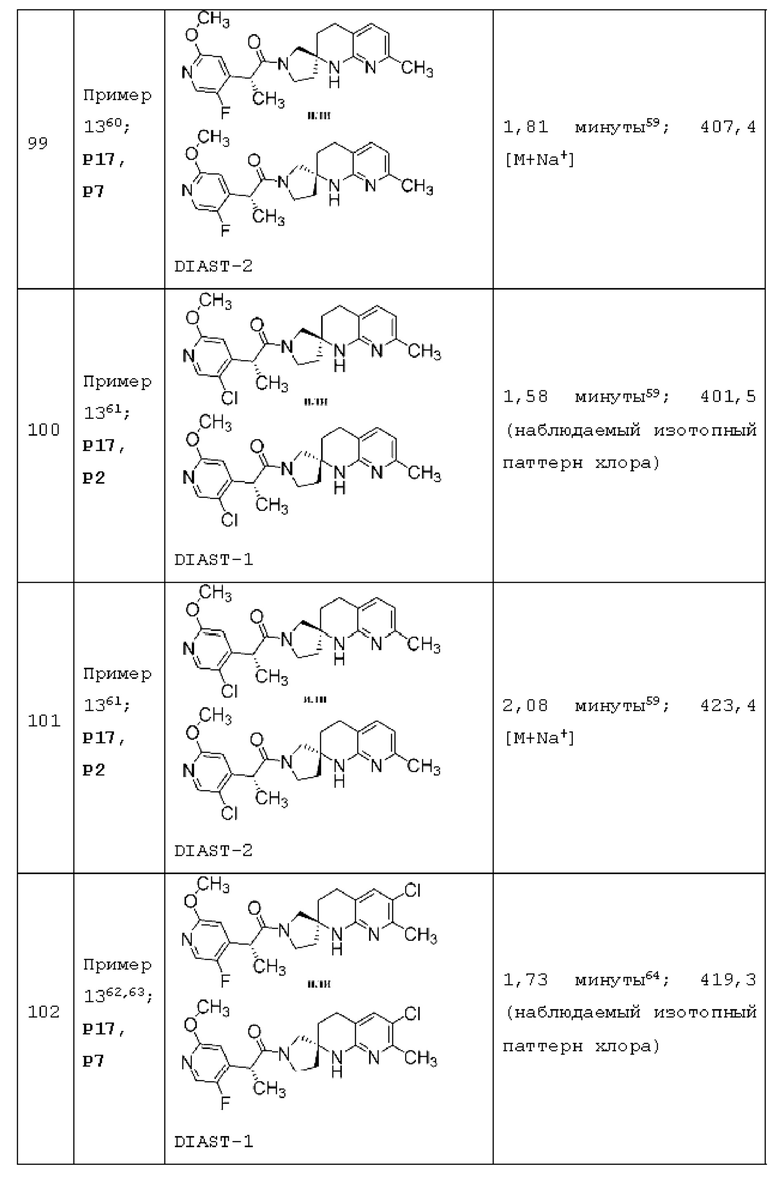
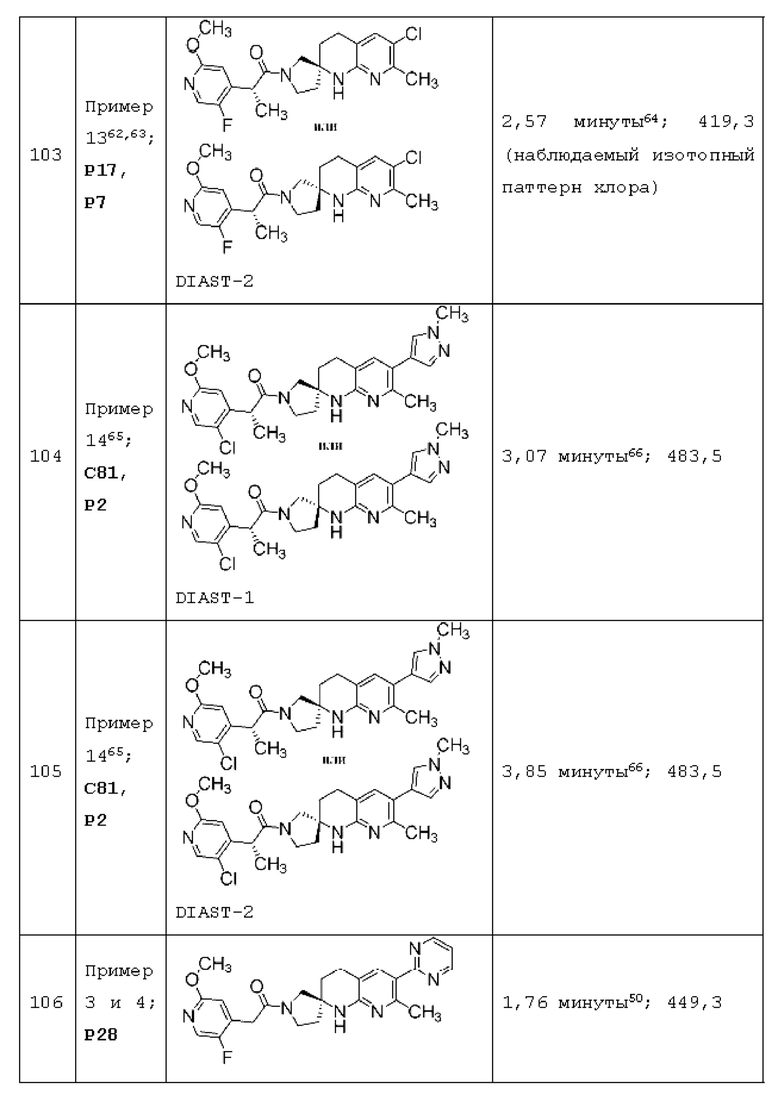
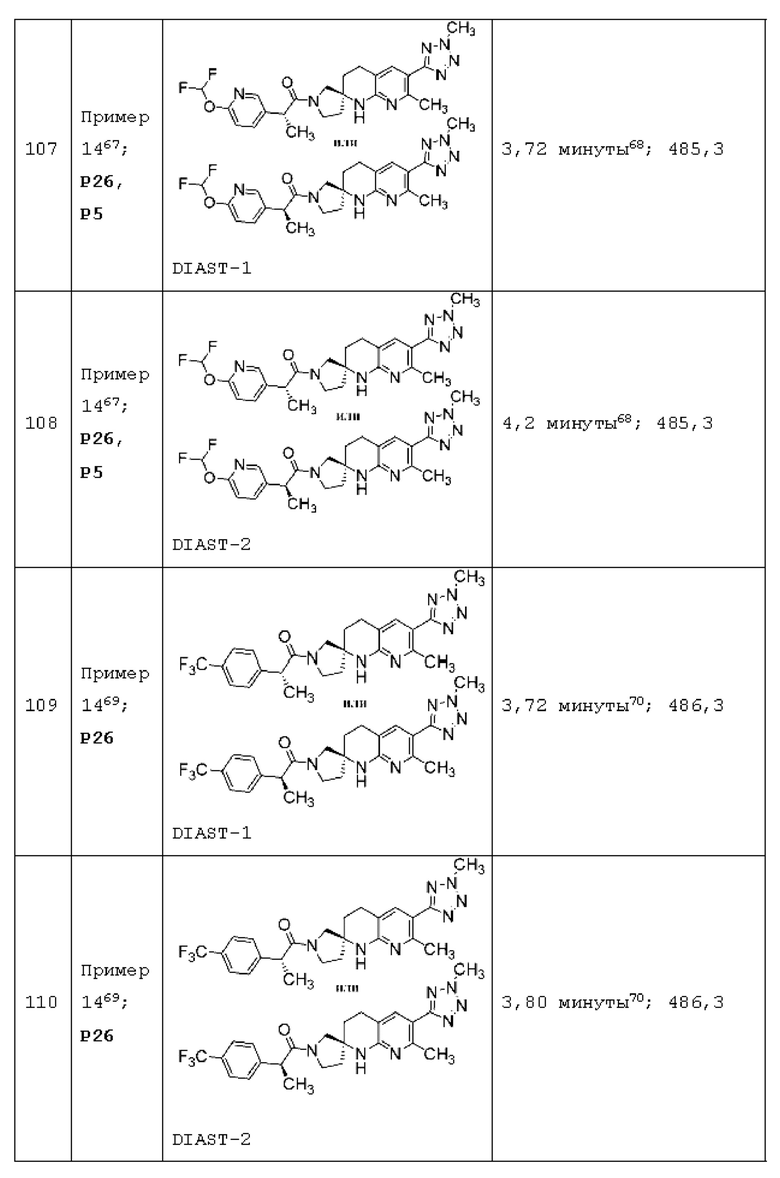
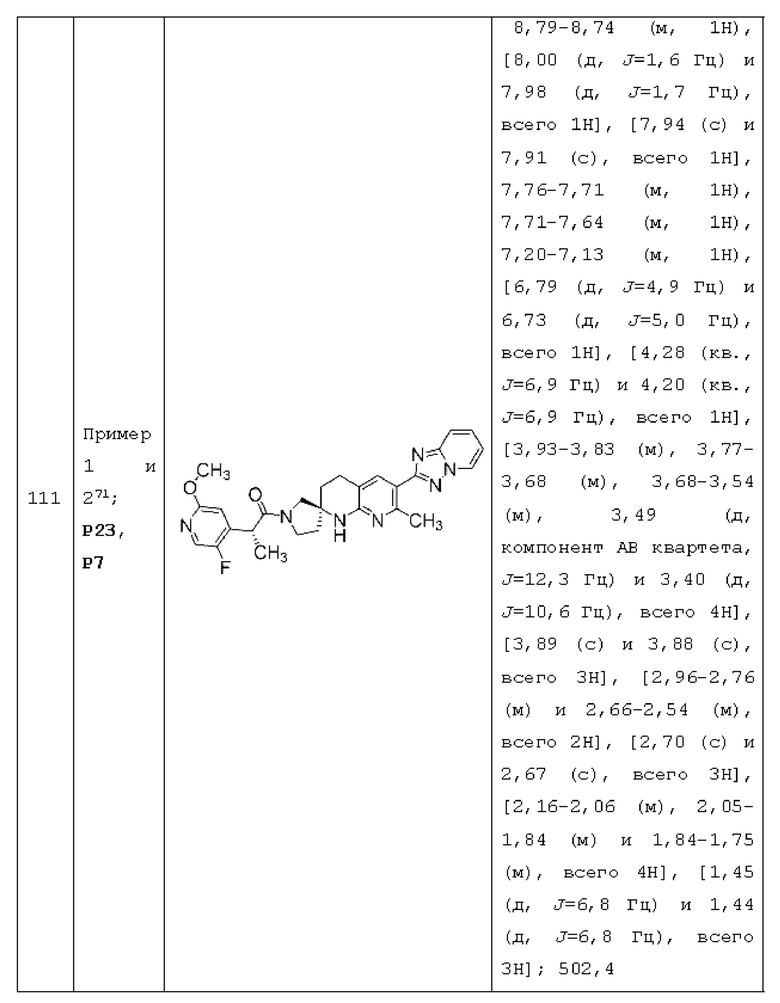
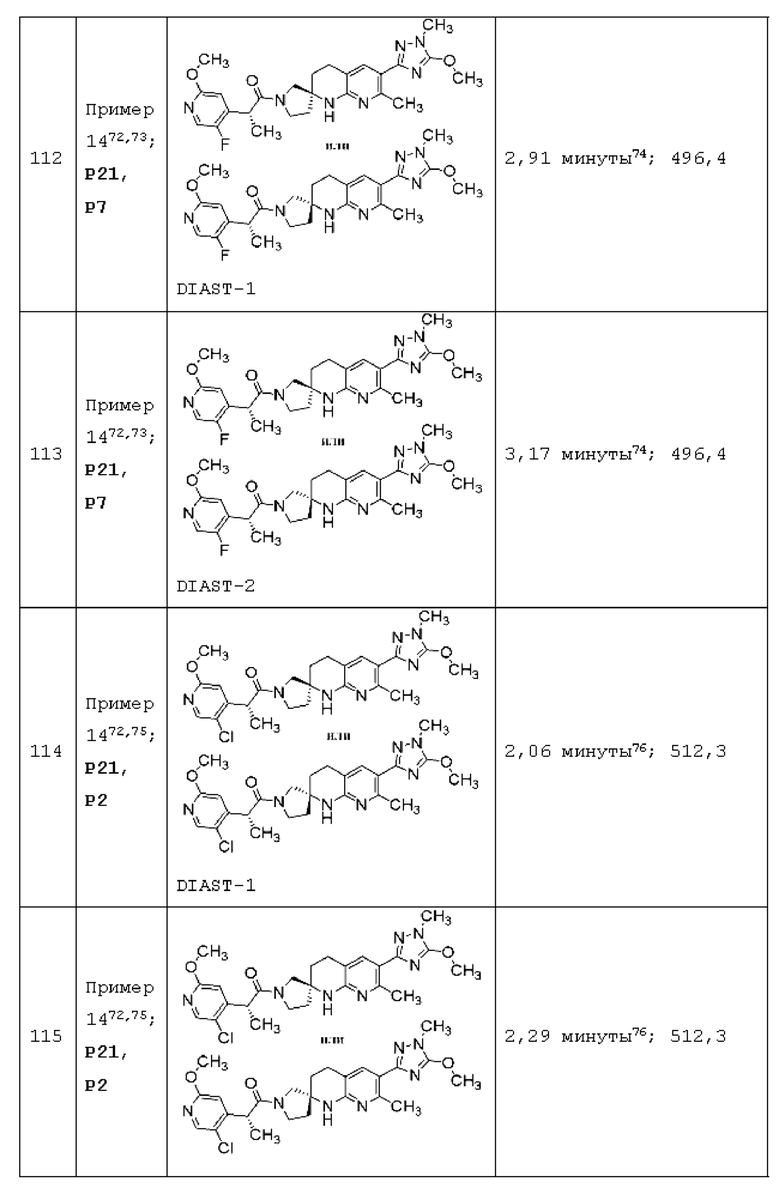
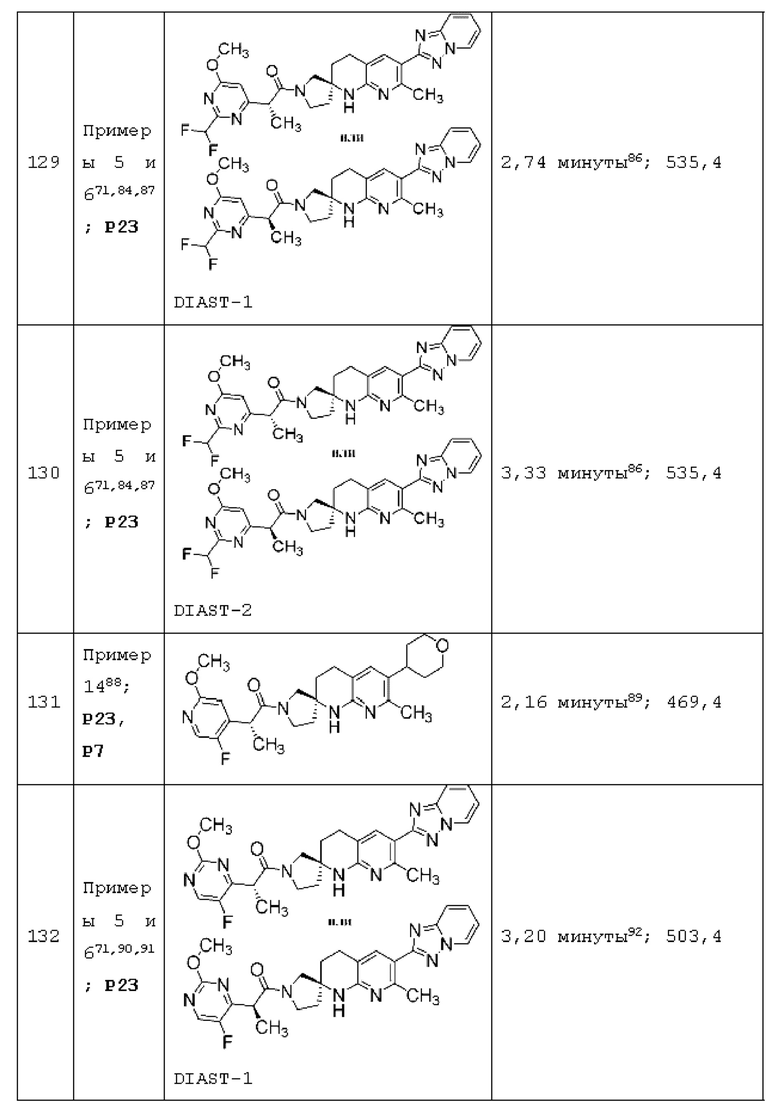
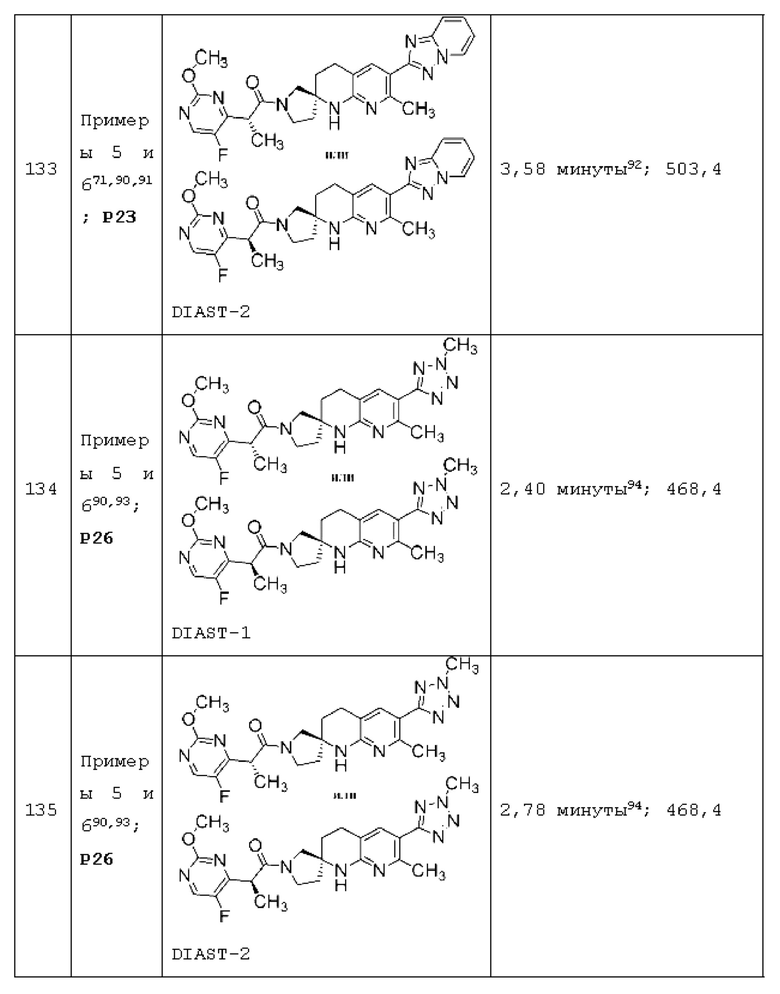
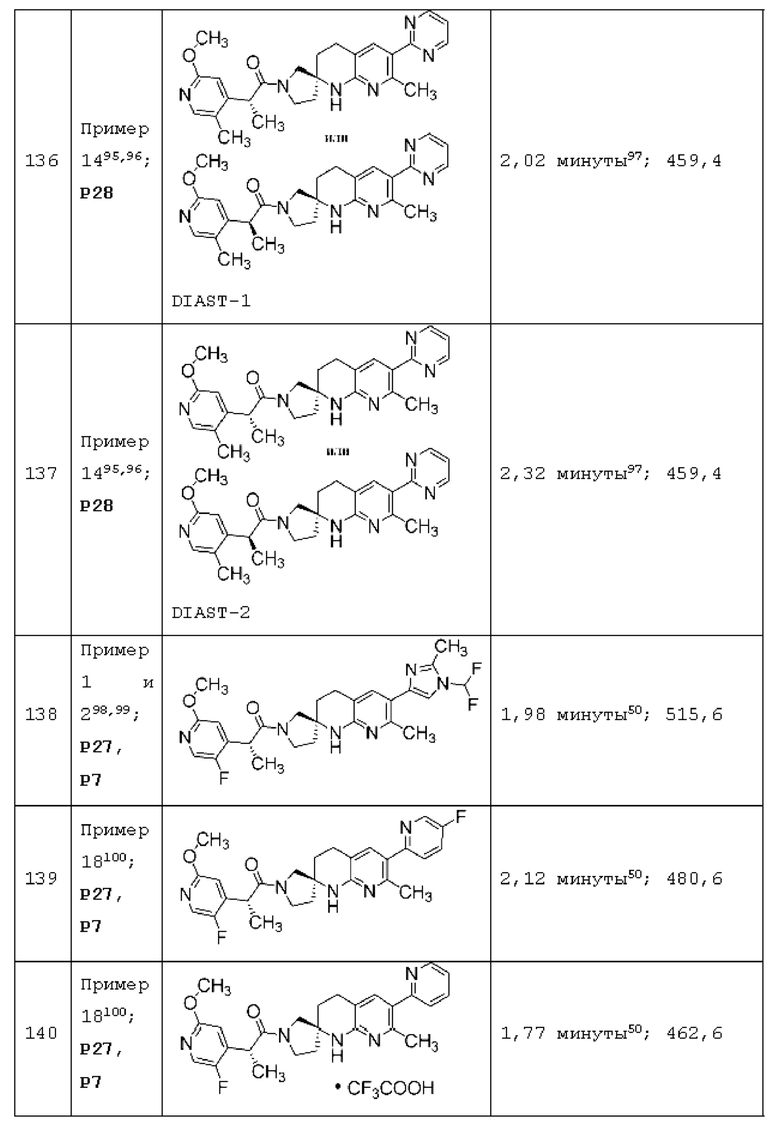
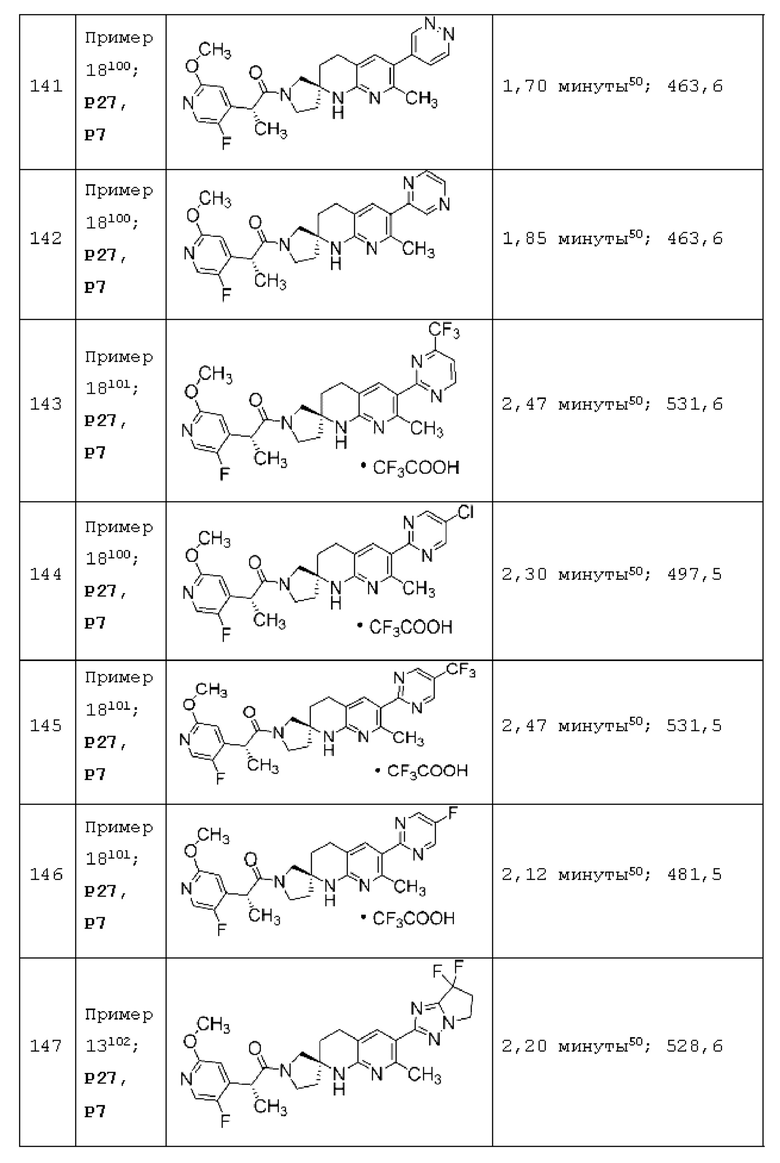
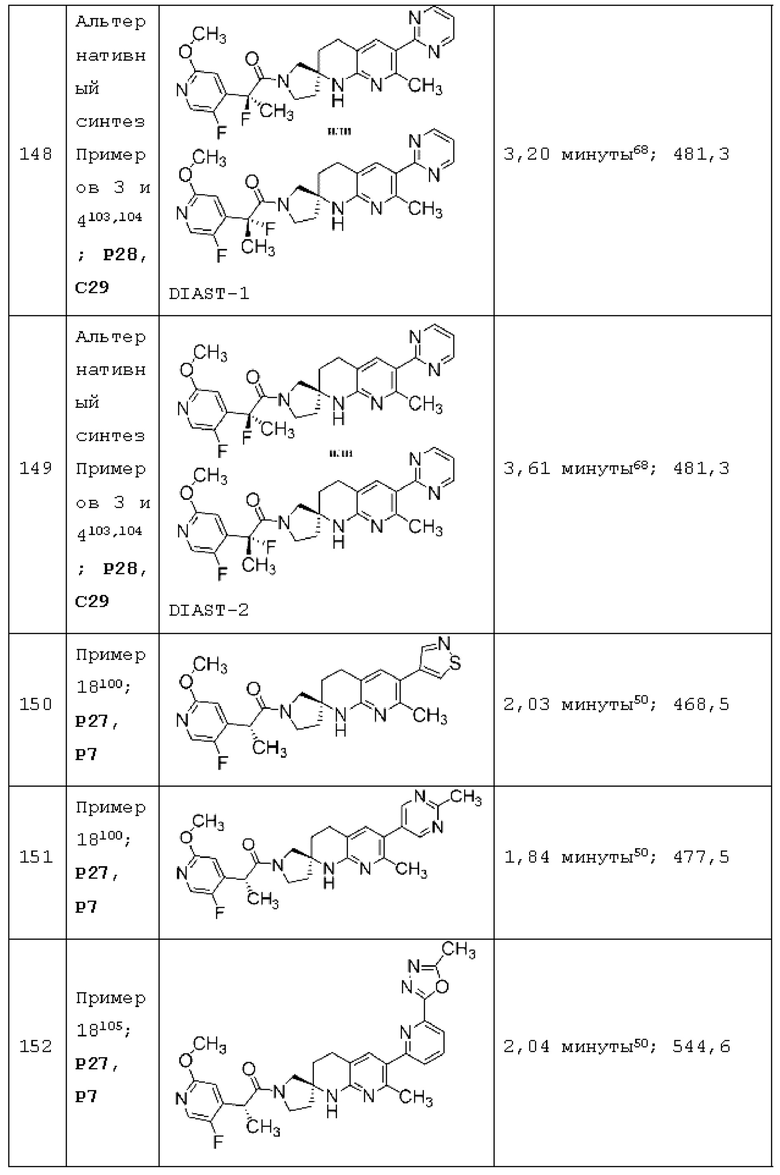
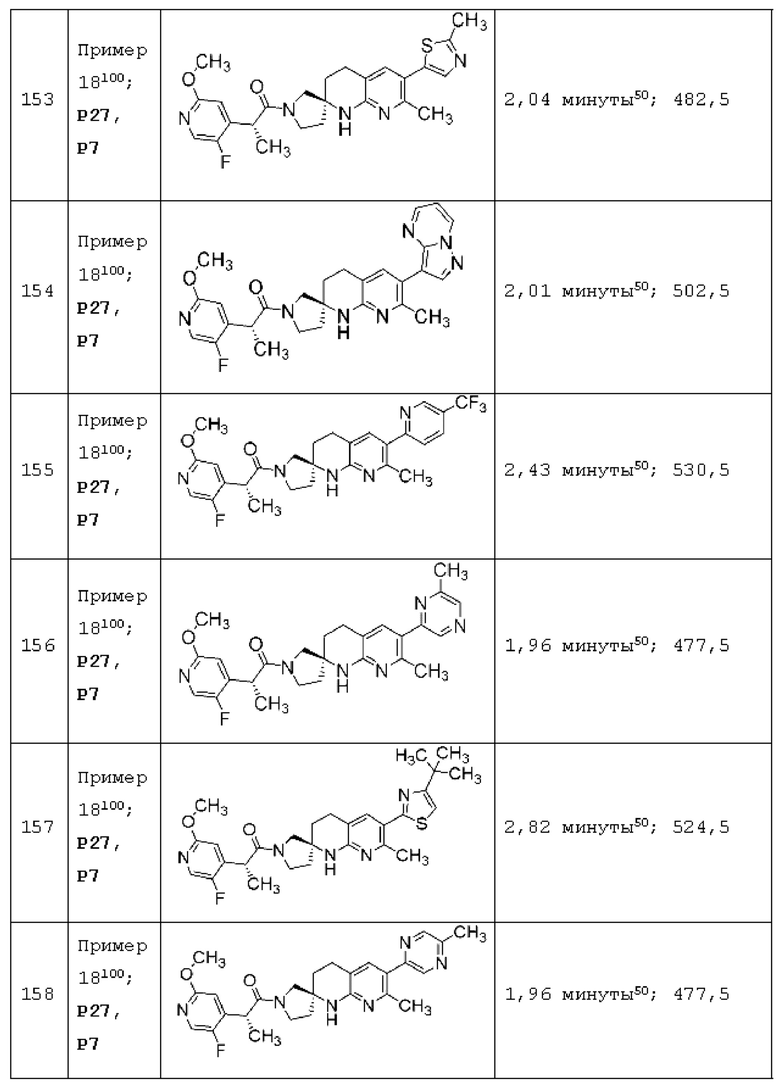
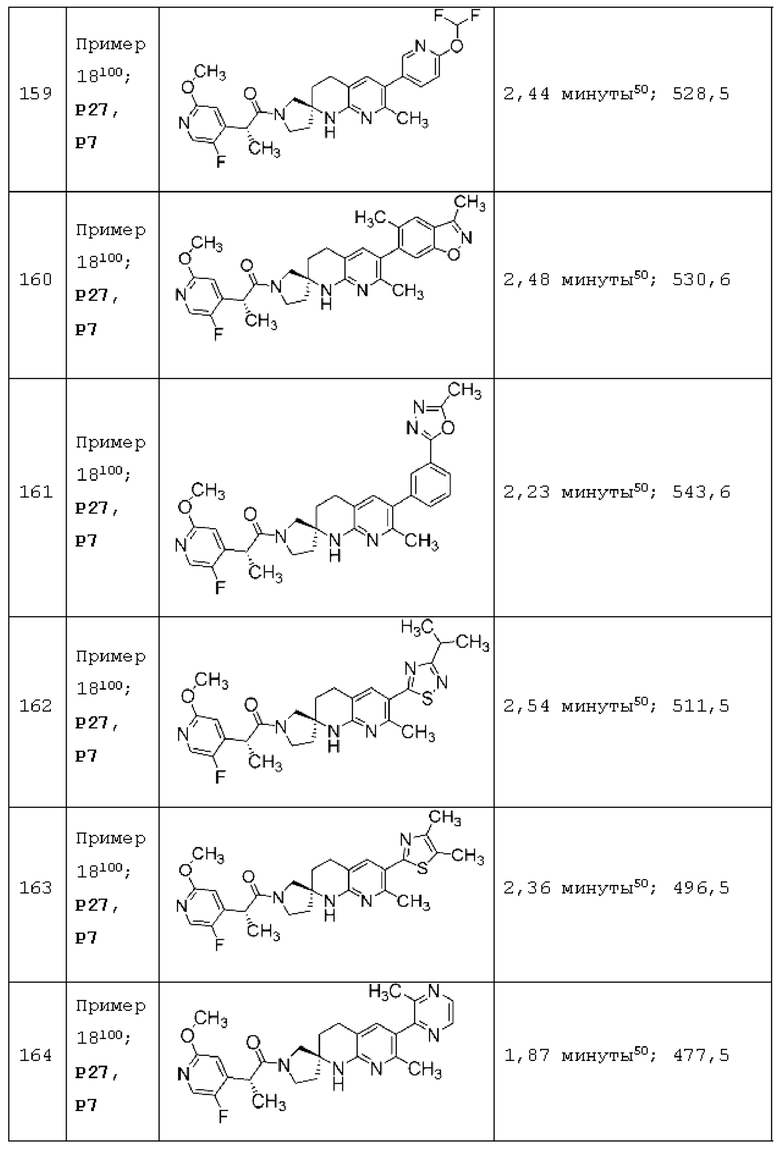
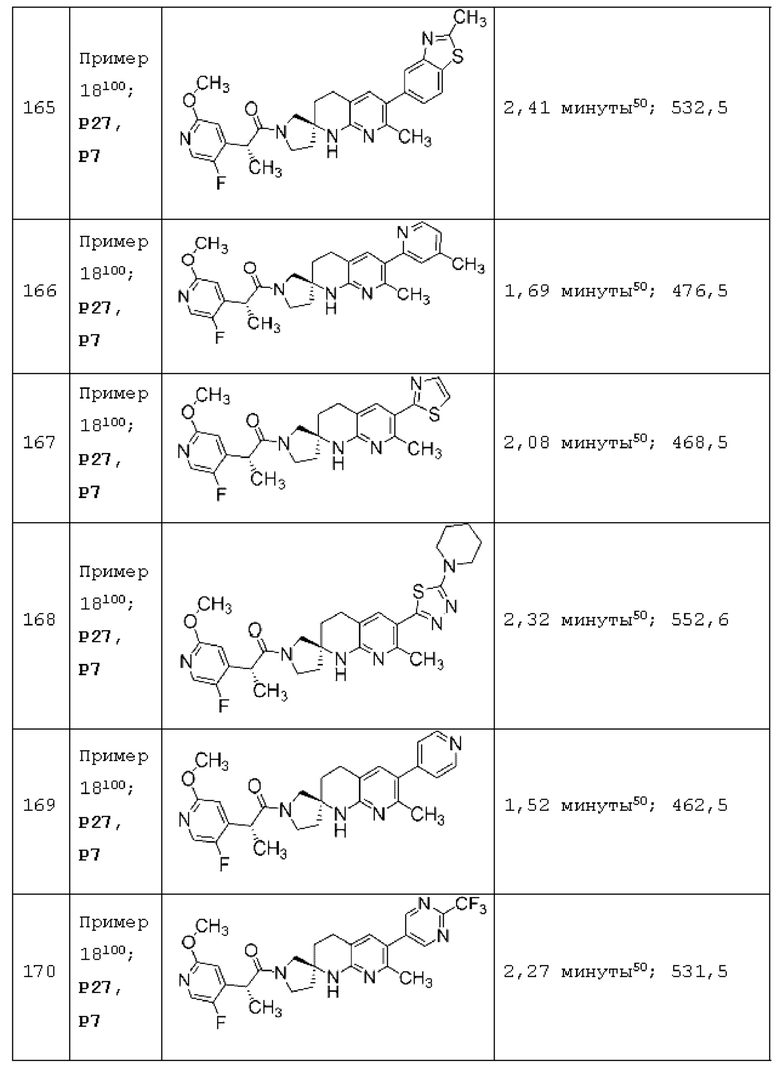
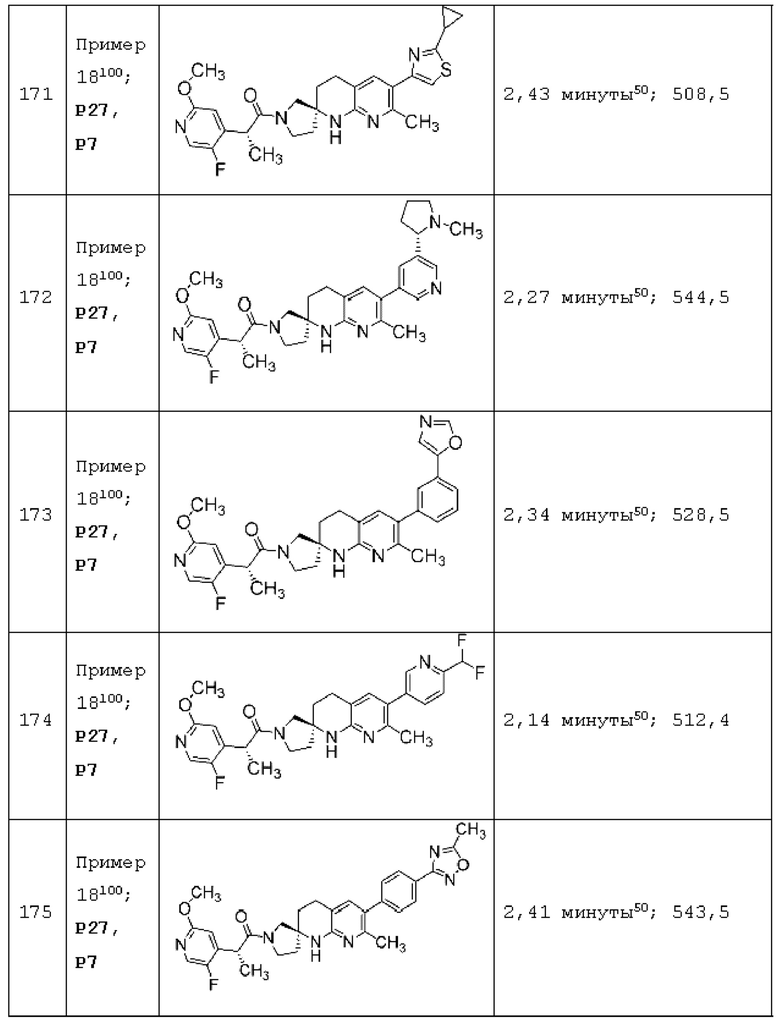
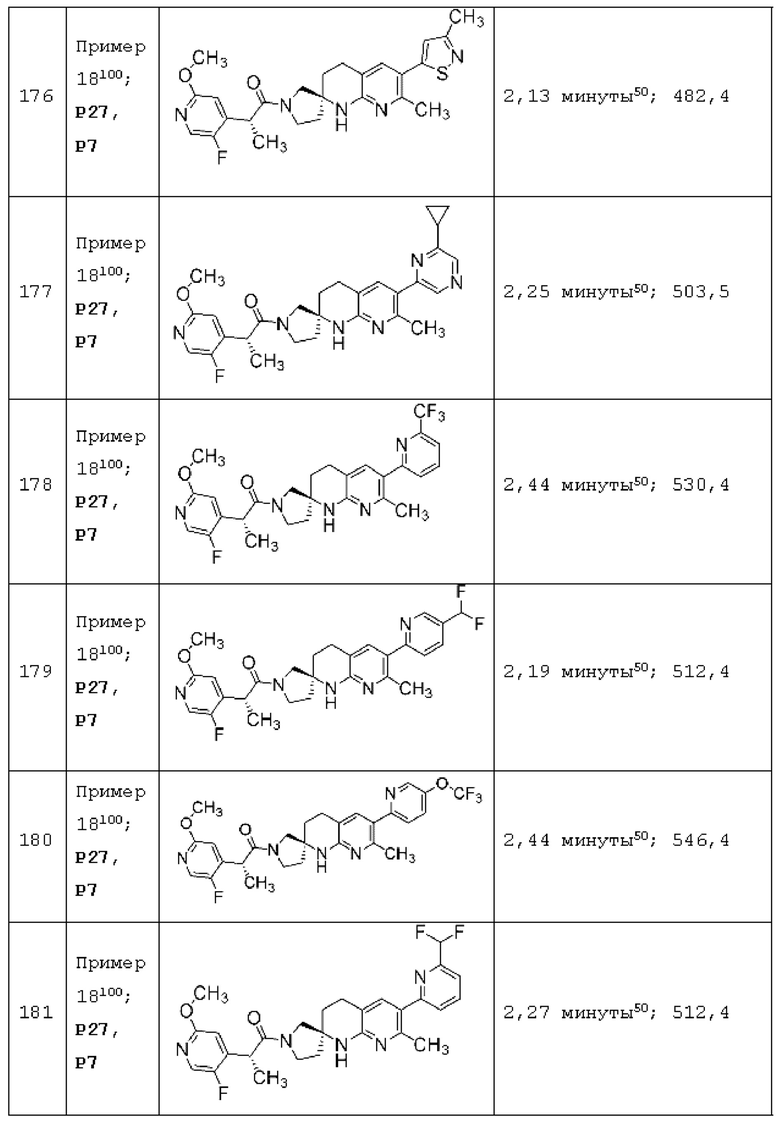
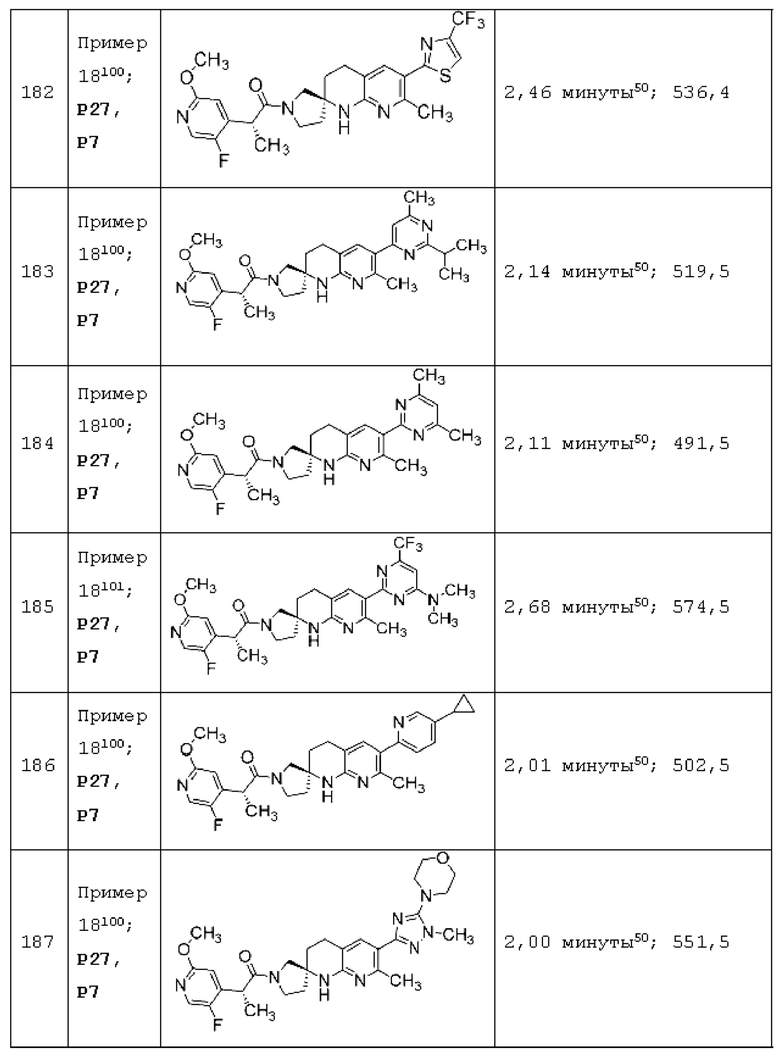
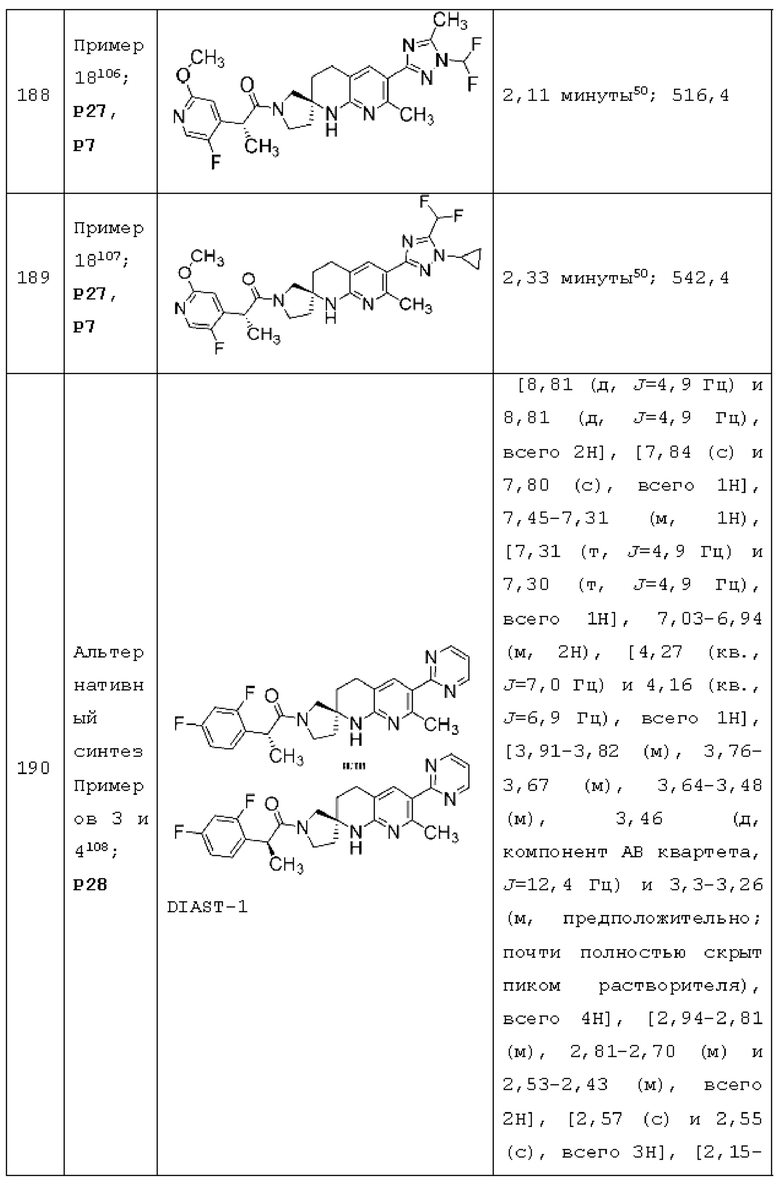
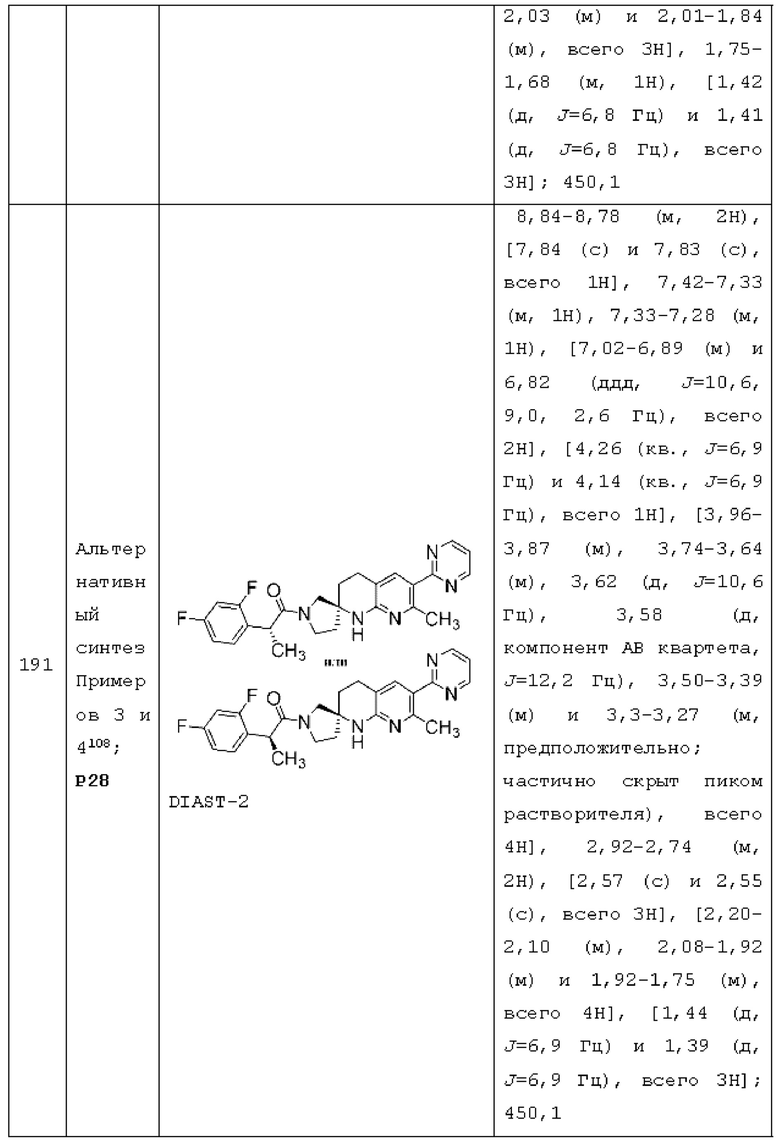
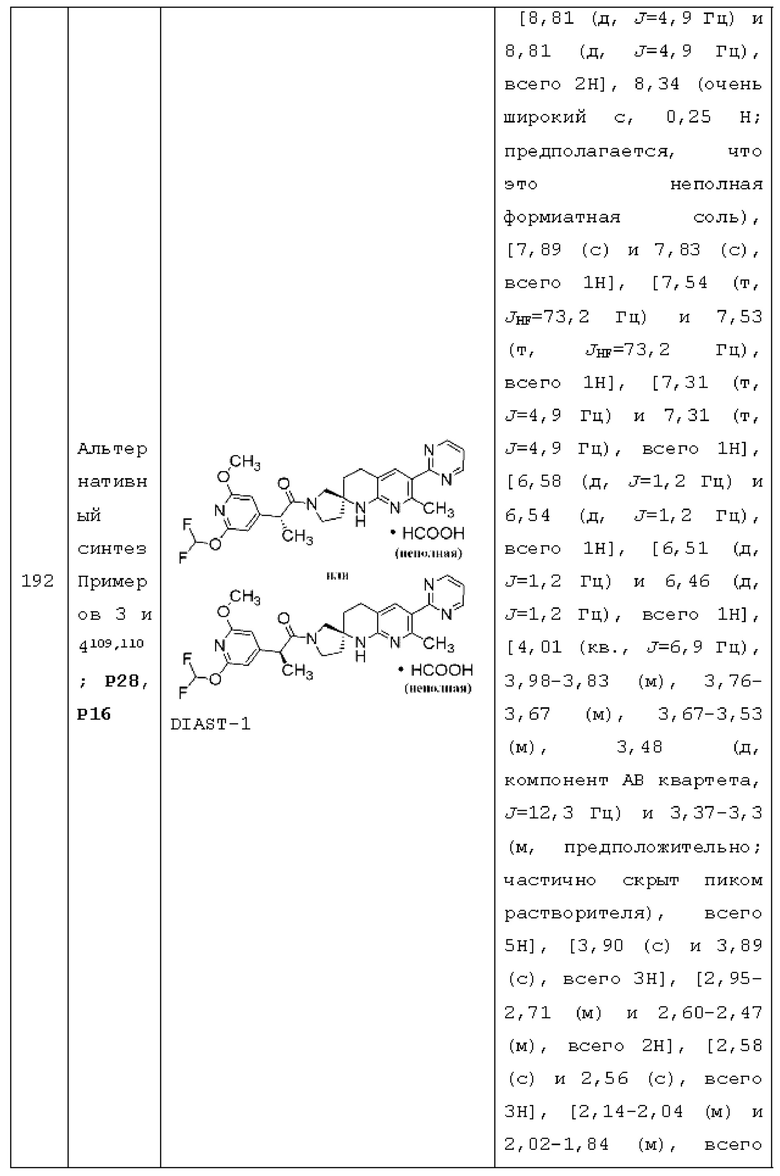
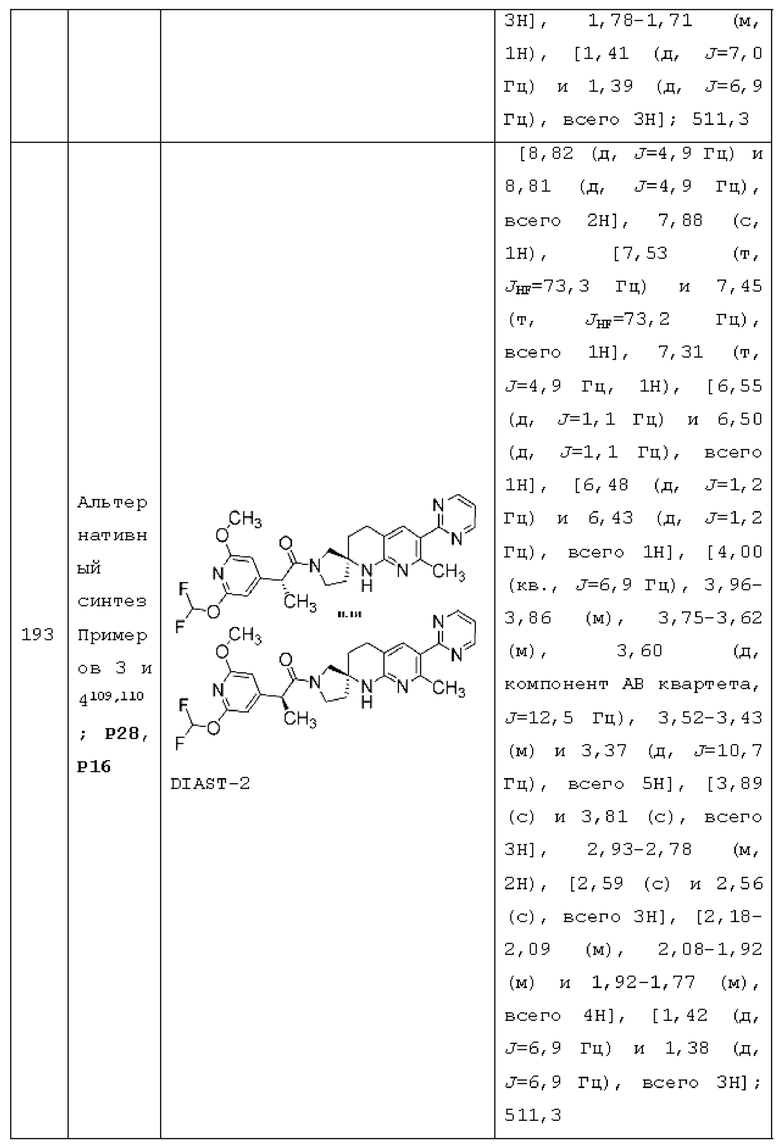
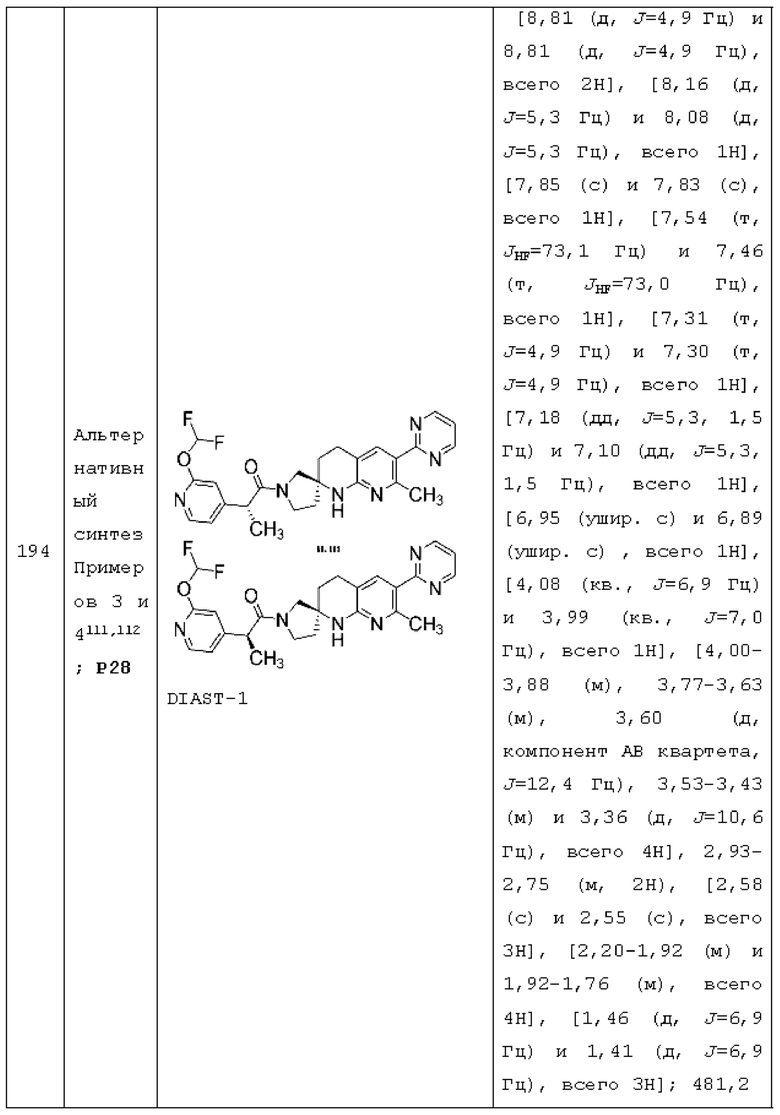
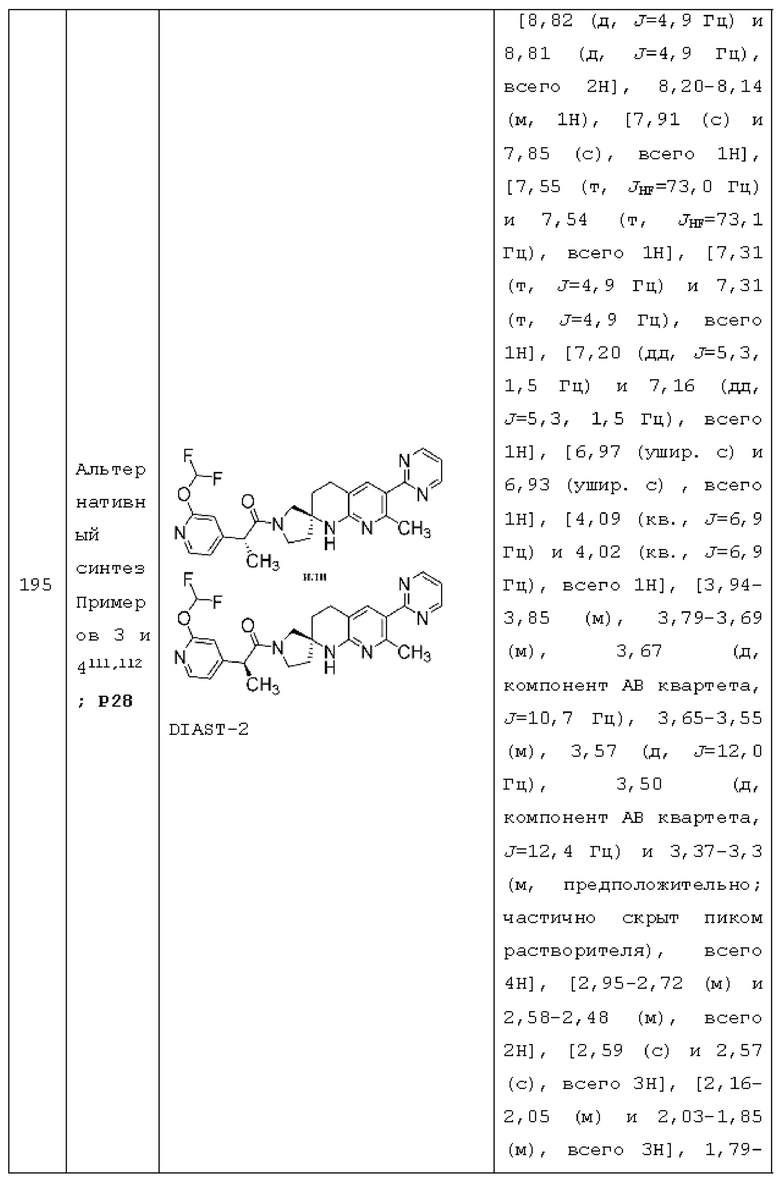
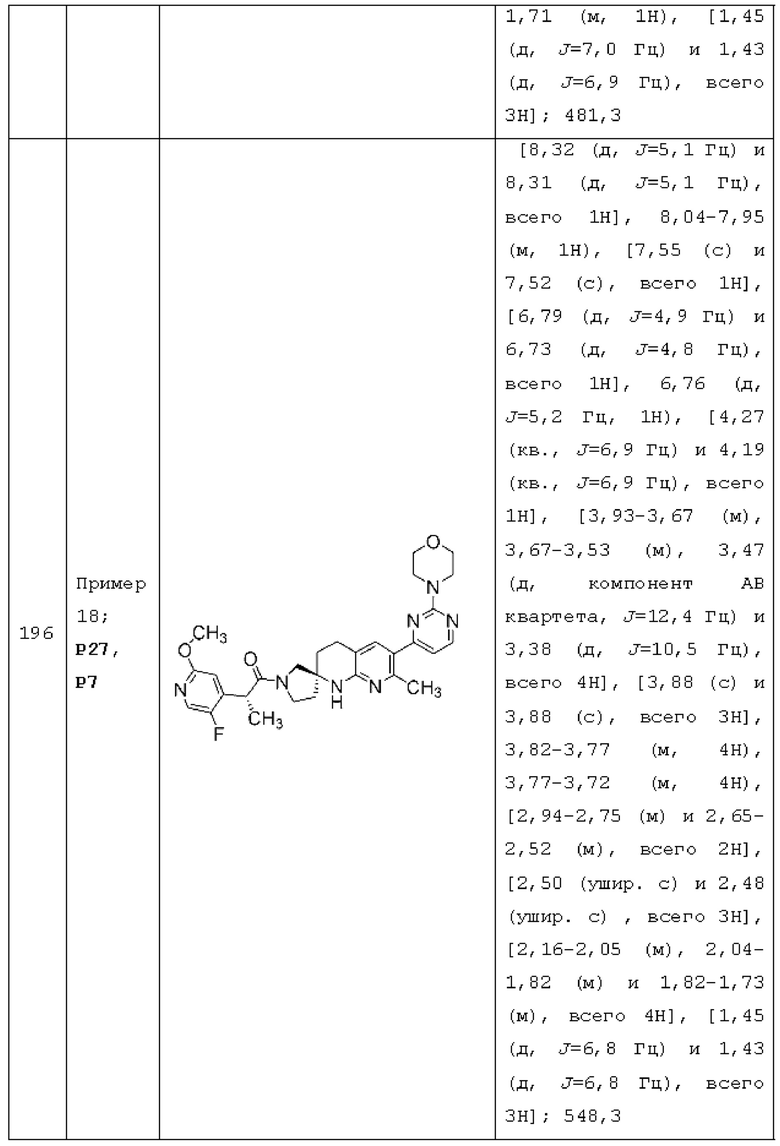
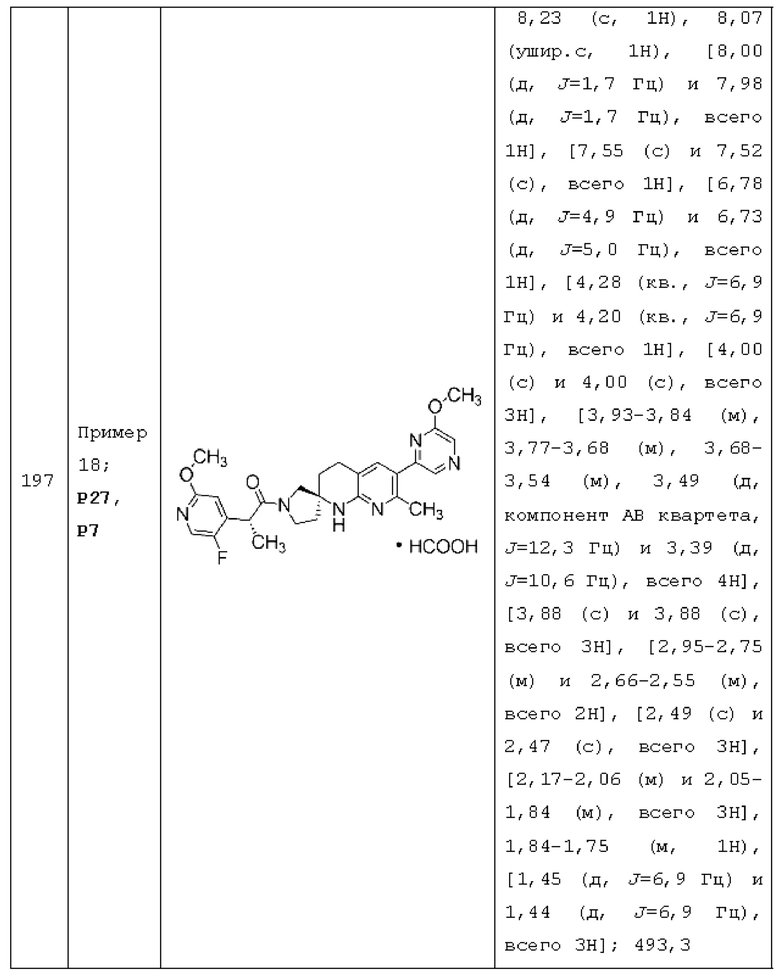
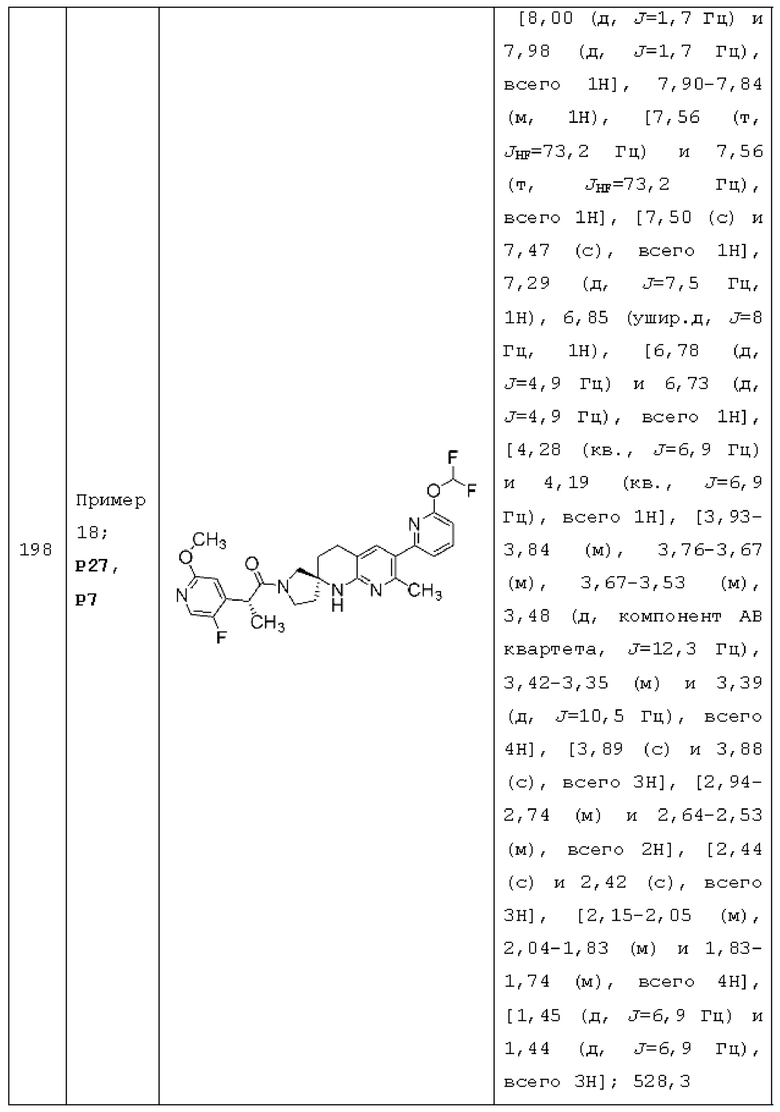
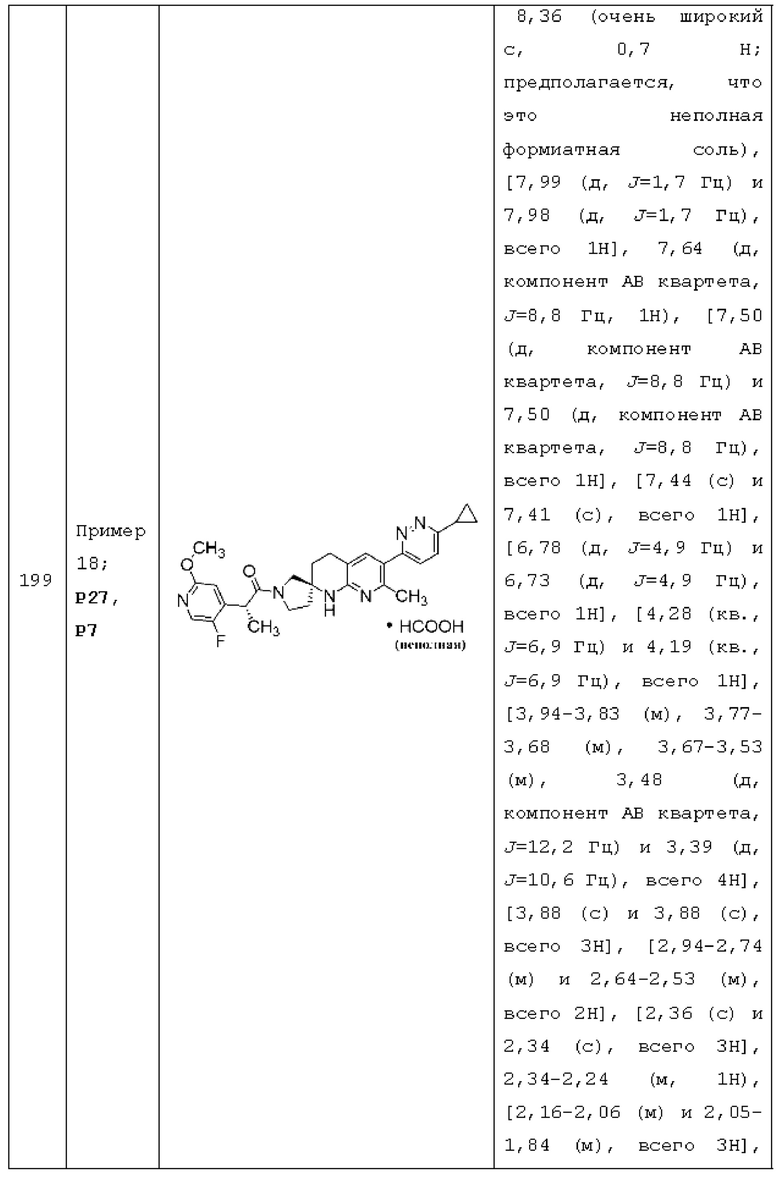
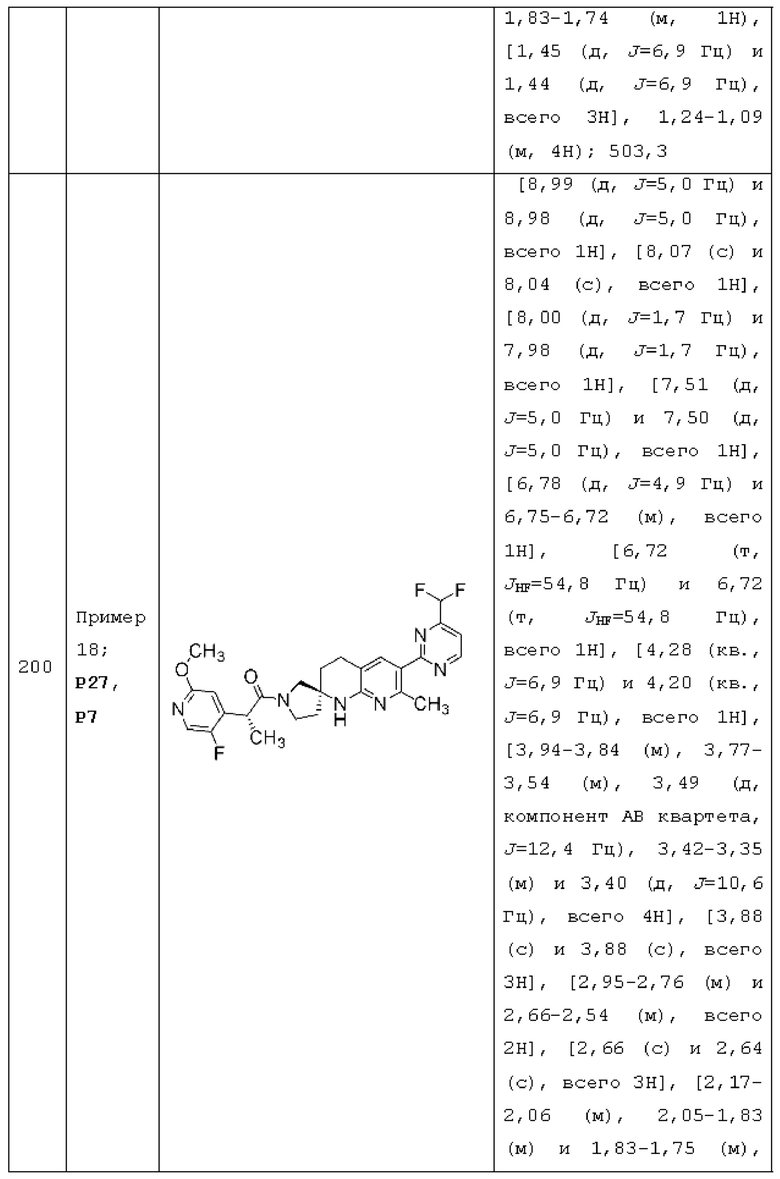
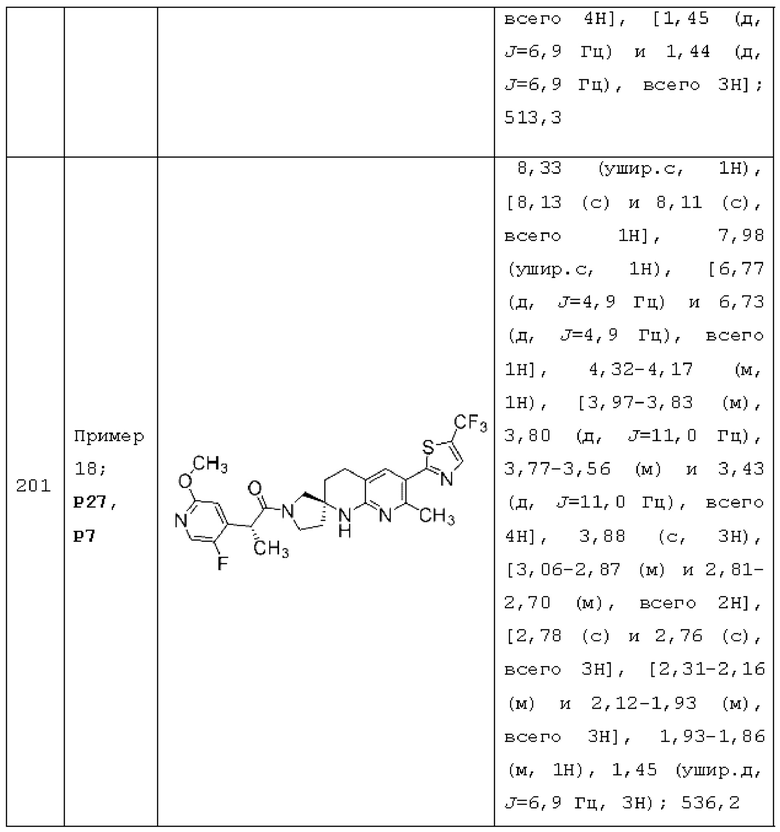
1. Взаимодействие 5-бром-2-иодпиримидина с P27 осуществляли с использованием [1,1'-бис (дифенилфосфино) ферроцен] дихлорпалладия (II) и карбоната натрия с получением требуемого трет-бутил-(2S)-6-(5-бромпиримидин-2-ил)-7-метил-3,4-дигидро-1Н-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилата.
2. Удаление защиты соединения P17 с использованием хлористого водорода в 2-пропаноле давало требуемый 7-метил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин].
3. Разделение продукта на четыре составляющих его диастереомера осуществляли сверхкритической флюидной хроматографией следующим образом: Осуществляли первоначальное разделение [Колонка: Chiral Technologies Chiralcel OJ, 30×250 мм, 5 мкм; Подвижная фаза: 9:1 диоксид углерода/(2-пропанол, содержащий 0,2% 1-аминопропан-2-ола); Скорость потока: 80 мл/мин; Обратное давление: 100 бар] с получением первой элюирующей смеси (Pdt1) и второй элюирующей смеси (Pdt2). Время удерживания для Pdtl: 3,8 0 мин [Аналитические условия. Колонка: Chiral Technologies Chiralcel OJ, 4,6×250 мм, 5 мкм; Подвижная фаза А: диоксид углерода; Подвижная фаза В: 2-пропанол, содержащий 0,2% 1-аминопропан-2-ола; Градиент: 5% В в течение 1,00 минуты, затем 5% до 60% В в течение 8,00 минут; Скорость потока: 3,0 мл/мин; Обратное давление: 120 бар]. Время удерживания для Pdt2: 4,08 мин (Аналитические условия идентичны используемым для Pdt1).
Затем разделяли Pdt1 {Колонка: Phenomenex Lux Amylose-1, 30×250 мм, 5 мкм; Подвижная фаза: 3:2 диоксид углерода/[(1:1 ацетонитрил:метанол), содержащая 0,2% (7 М аммиака в метаноле)]; Скорость потока: 80 мл/мин; Обратное давление: 100 бар). Первый элюирующий диастереомер был указан как Пример 23, а второй элюирующий диастереомер как Пример 24.
Разделяли Pdt2 {Колонка: Phenomenex Lux Amylose-1, 30×250 мм, 5 мкм; Подвижная фаза: 7:3 диоксид углерода/[этанол, содержащий 0,2% (7 М аммиака в метаноле)]; Скорость потока: 80 мл/мин; Обратное давление: 100 бар) с получением первого элюирующего диастереомера Примера 25 и второго элюирующего диастереомера Примера 26.
4. Аналитические условия. Колонка: Phenomenex Lux Amylose-1, 4,6×250 мм, 5 мкм; Подвижная фаза А: диоксид углерода; Подвижная фаза В: (1:1 ацетонитрил/метанол), содержащая 0,2% (7 М аммиака в метаноле); Градиент: 5% В в течение 1,00 минуты, затем 5% до 60% В в течение 8,00 минут; Скорость потока: 3,0 мл/мин; Обратное давление: 120 бар.
5. Эти ЖХМС данные были получены из анализа реакционной смеси.
6. Аналитические условия. Колонка: Phenomenex Lux Amylose-1, 4,6×250 мм, 5 мкм; Подвижная фаза А: диоксид углерода; Подвижная фаза В: этанол, содержащий 0,2% (7 М аммиака в метаноле)]; Градиент: 5% В в течение 1,00 минуты, затем 5% до 60% В в течение 8,00 минут; Скорость потока: 3,0 мл/мин; Обратное давление: 120 бар.
7. Требуемый трет-бутил-(2S)-7-метил-6-[2-метил-1-(трифторметил)-1H-имидазол-4-ил]-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилат синтезировали из P23 с использованием способа, описанного в Получении Р28 для синтеза C69; 4-иод-2-метил-1-(трифторметил)-1H-имидазол использовали вместо 2-бромпиримидина.
8. [(2R)-1'-(трет-Бутоксикарбонил)-7-метил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-6-ил]бороновую кислоту получали из р24 с использованием способа, описанного в Получении Р27. Последующее преобразование в требуемый трет-бутил-(2R)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилат осуществляли так же, как синтез C84 из P27 в Примере 18.
9. Удаление защиты соединения с66 с использованием хлористого водорода в дихлорметане давало требуемый {2R)-1-метил-6-(2-метил-2H-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин].
10. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralcel OJ-H, 30×250 мм, 5 мкм; Подвижная фаза: 4:1 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 80 мл/мин; Обратное давление: 120 бар]. Первый элюирующий диастереомер был указан как Пример 31, а второй элюирующий диастереомер как Пример 32. При осуществлении аналитической сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralcel OJ-H, 4,6×250 мм, 5 мкм; Подвижная фаза А: диоксид углерода; Подвижная фаза В: метанол, содержащий 0,2% (7 М аммиака в метаноле); Градиент: 5% В в течение 1,00 минуты, затем 5% до 60% В в течение 8,00 минут; Скорость потока: 3,0 мл/мин; Обратное давление: 120 бар] Пример 31 показал время удерживания 4,22 мин. Пример 32 имел время удерживания 4,4 8 минут в таких же условиях.
11. Сравнение спектров 1Н ЯМР показало, что Пример 31 представляет собой энантиомер Примера 4, а Пример 32 представляет собой энантиомер Примера 3.
12. Преобразование C44 в 1'-бензил-7-метил-6-(2-метил-1,3-оксазол-4-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин] осуществляли с использованием способа, описанного для синтеза C48 в Получениях Р17 и Р18, за исключением того, что использовали 2-хлор-3-иод-6-метил-5-(2-метил-1,3-оксазол-4-ил)пиридин вместо C40. Последующее гидрирование давало требуемый 7-метил-6-(2-метил-1,3-оксазол-4-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин].
13. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralcel OJ-H, 20×250 мм, 5 мкм; Подвижная фаза: 78:22 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 50 мл/мин]. Первый элюирующий диастереомер был указан как Пример 34, а второй элюирующий диастереомер как Пример 35. При осуществлении аналитической сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralcel OJ-3R, 3,0×150 мм, 3 мкм; Подвижная фаза: 3:1 диоксид углерода/(метанол, содержащий 0,1% диэтиламин); Скорость потока: 2,5 мл/мин] Пример 34 показал время удерживания 2,63 мин. Пример 35 имел время удерживания 3,36 мин в таких же условиях.
14. Продукт разделяли на составляющие его диастереомеры с использованием ВЭЖХ (Колонка: Chiral Technologies Chiralpak AD, 50×250 мм, 10 мкм; Подвижная фаза: 95:5:0,1 этанол/ацетонитрил/диэтиламин; Скорость потока: 60 мл/мин). Первый элюирующий диастереомер был указан как Пример 36, а второй элюирующий диастереомер как Пример 37. При осуществлении аналитической ВЭЖХ (Колонка: Chiral Technologies Chiralpak AD-H, 4,6×150 мм, 5 мкм; Подвижная фаза: 95:5:0,1 этанол/ацетонитрил/диэтиламин; Скорость потока: 0,8 мл/мин), Пример 36 показал время удерживания 5,04 мин. Пример 37 имел время удерживания 7,8 9 мин в таких же условиях.
15. Преобразование 5-хлор-4-иод-2-(трифторметил)пиридина в требуемую 2-[5-хлор-2-(трифторметил)пиридин-4-ил]пропановую кислоту осуществляли с использованием способа, описанного в Получении Р5. ЖХМС m/z 254, 0 (наблюдаемый изотопный паттерн хлора) [М+Н]+. 1Н ЯМР (400 МГц, метанол-d4) δ 8, 72 (с, 1Н), 7,83 (с, 1Н), 4,27 (кв., J=7,3 Гц, 1Н), 1,57 (д, J=7,3 Гц, 3Н).
16. Продукт разделяли на составляющие его диастереомеры с использованием ВЭЖХ (Колонка: Chiral Technologies Chiralpak AY, 50×250 мм, 10 мкм; Подвижная фаза: 60:40:0,1 гексан/этанол/диэтиламин; Скорость потока: 60 мл/мин). Первый элюирующий диастереомер был указан как Пример 38, а второй элюирующий диастереомер как Пример 39. При осуществлении аналитической ВЭЖХ (Колонка: Chiral Technologies Chiralpak AD-H, 4,6×150 мм, 5 мкм; Подвижная фаза: метанол; Скорость потока: 1,0 мл/мин), Пример 38 показал время удерживания 3,53 минут. Пример 39 имел время удерживания 5,4 6 минут в таких же условиях.
17. Преобразование 4-иод-2-метокси-5-(трифторметил)пиридина в требуемую 2-[2-метокси-5-(трифторметил)пиридин-4-ил]пропановую кислоту осуществляли с использованием способа, описанного в Получении Р5. ЖХМС m/z 250,1 [М+Н]+. 1Н ЯМР (4 00 МГц, хлороформ-d) δ 8,46 (с, 1Н), 6,88 (с, 1Н), 4,10 (кв., J=7,1 Гц, 1Н), 4,00 (с, 3Н), 1,54 (д, J=7,0 Гц, 3Н).
18. Продукт разделяли на составляющие его диастереомеры с использованием ВЭЖХ (Колонка: Chiral Technologies Chiralpak IA, 50×250 мм, 10 мкм; Подвижная фаза: 70:30:0,1 гексан/этанол/диэтиламин; Скорость потока: 60 мл/мин). Первый элюирующий диастереомер был указан как Пример 40, а второй элюирующий диастереомер как Пример 41. При осуществлении аналитической ВЭЖХ (Колонка: Chiral Technologies Chiralpak IA-3, 4,6×250 мм, 3 мкм; Подвижная фаза: 70:30:0,1 гексан/этанол/диэтиламин; Скорость потока: 1,0 мл/мин), Пример 40 показал время удерживания 6,49 минут. Пример 41 имел время удерживания 8,3 9 мин в таких же условиях.
19. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralcel OJ-H, 20×250 мм, 5 мкм; Подвижная фаза: 7:3 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 50 мл/мин]. Первый элюирующий диастереомер был указан как Пример 42, а второй элюирующий диастереомер как Пример 43. При осуществлении аналитической сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralcel OJ-3R, 3,0×150 мм, 3 мкм; Подвижная фаза: 7:3 диоксид углерода/(метанол, содержащий 0,1% диэтиламина); Скорость потока: 2,5 мл/мин], Пример 42 показал время удерживания 1,81 мин. Пример 43 имел время удерживания 2,54 мин в таких же условиях.
20. 6-Хлор-2-метилпиримидин-4-ол преобразовывали в 4-хлор-6-(дифторметокси)-2-метилпиримидин с использованием способа, описанного в Получении Р11, Стадия 1. Это вещество затем использовали для получения требуемого 2-[6-(дифторметокси)-2-метилпиримидин-4-ил]пропаноата лития в соответствии со способом, представленным в Получении Р4. ЖХМС m/z 233, 1 [М+Н]+. 1Н ЯМР (4 00 МГц, метанол-d4) δ 7,62 (т, JHF=72,1 Гц, 1Н), 6,83 (с, 1Н), 3,70 (кв., J=7,2 Гц, 1Н), 2,58 (с, 3Н), 1,48 (д, J=7,2 Гц, 3Н).
21. Продукт разделяли на составляющие его диастереомеры с использованием ВЭЖХ (Колонка: Chiral Technologies Chiralpak IA 4,6 см × 25 мм; Подвижная фаза: этанол; Скорость потока: 1 мл/мин). Первый элюирующий диастереомер был указан как Пример 44, а второй элюирующий диастереомер как Пример 45. каждый из этих диастереомеров подвергали конечной очистке методом обращеннофазовой ВЭЖХ (Колонка: Nouryon Kromasil 100-5 С18, 21,5×100 мм, 5 мкм; Подвижная фаза А: вода, содержащая 0,1% муравьиной кислоты; Подвижная фаза В: ацетонитрил; Градиент: 20% до 4 0% В). При осуществлении аналитической ВЭЖХ (Колонка: Колонка: Chiral Technologies Chiralpak IA, 4,6×250 мм; Подвижная фаза: этанол; Скорость потока: 1,0 мл/мин) Пример 44 показал время удерживания 9,78 мин. Пример 45 имел время удерживания 13,69 мин в таких же условиях.
22. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии (Колонка: Chiral Technologies Chiralcel OJ-H, 20×250 мм, 5 мкм; Подвижная фаза: 72:28 диоксид углерода/этанол; Скорость потока: 50 мл/мин). Первый элюирующий диастереомер был указан как Пример 46, а второй элюирующий диастереомер как Пример 47. При осуществлении аналитической сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralcel OJ-3R, 3,0×150 мм, 3 мкм; Подвижная фаза: 3:1 диоксид углерода/(метанол, содержащий 0,1% диэтиламина); Скорость потока: 2,5 мл/мин] Пример 46 показал время удерживания 1,62 мин. Пример 4 7 имел время удерживания 2,77 мин в таких же условиях.
23. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralcel OJ-H, 20×250 мм, 5 мкм; Подвижная фаза: 88:12 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 50 мл/мин]. Первый элюирующий диастереомер был указан как Пример 48, а второй элюирующий диастереомер как Пример 49. При осуществлении аналитической сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralcel OJ-3R, 3,0×150 мм, 3 мкм; Подвижная фаза: 4:1 диоксид углерода/(метанол, содержащий 0,1% диэтиламин); Скорость потока: 2,0 мл/мин] Пример 48 показал время удерживания 2,43 мин. Пример 49 имел время удерживания 3,04 мин в таких же условиях.
24. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralcel OJ-H, 20×250 мм, 5 мкм; Подвижная фаза: 84:16 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 50 мл/мин]. Первый элюирующий диастереомер был указан как Пример 50, а второй элюирующий диастереомер как Пример 51. При осуществлении аналитической сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralcel OJ-3R, 3,0×150 мм, 3 мкм; Подвижная фаза: 3:1 диоксид углерода/(метанол, содержащий 0,1% диэтиламин); Скорость потока: 2,5 мл/мин] Пример 50 показал время удерживания 1,77 мин. Пример 51 имел время удерживания 2,16 мин в таких же условиях.
25. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralcel OJ-H, 20×250 мм, 5 мкм; Подвижная фаза: 86:14 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 50 мл/мин]. Первый элюирующий диастереомер был указан как Пример 52, а второй элюирующий диастереомер как Пример 53. При осуществлении аналитической сверхкритической флюидной хроматографии (Колонка: Chiral Technologies Chiralcel 0J-3R, 3,0×150 мм, 3 мкм; Подвижная фаза: 4:1 диоксид углерода/(метанол, содержащий 0,1% диэтиламина); Скорость потока: 2,0 мл/мин) Пример 52 показал время удерживания 2,2 4 мин. Пример 53 имел время удерживания 2,58 мин в таких же условиях.
26. Продукт разделяли на составляющие его диастереомеры с использованием ВЭЖХ (Колонка: Chiral Technologies Chiralpak IG, 25×250 мм, 10 мкм; Подвижная фаза: этанол; Скорость потока: 30 мл/мин). Первый элюирующий диастереомер был указан как Пример 55, а второй элюирующий диастереомер как Пример 56. При осуществлении аналитической ВЭЖХ (Колонка: Chiral Technologies Chiralpak IG-3, 4,6×150 мм, 3 мкм; Подвижная фаза: этанол; Скорость потока: 0,5 мл/мин) Пример 55 показал время удерживания 9,7 9 мин. Пример 56 имел время удерживания 12,37 мин в таких же условиях.
27. Преобразование 3-фтор-4-иод-2-метоксипиридина в требуемую 2-(3-фтор-2-метоксипиридин-4-ил)пропановую кислоту осуществляли с использованием способа, описанного в Получении Р5. ЖХМС m/z 200,1 [М+Н]+. 1Н ЯМР (400 МГц, хлороформ-d) δ 7,89 (д, J=5,3 Гц, 1Н), 6,83 (дд, J=5, 5 Гц, 1Н), 4,09 (кв., J=7,2 Гц, 1Н), 4,02 (с, 3Н), 1,52 (д, J=7,4 Гц, 3Н).
28. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralcel OJ-H, 20×250 мм, 5 мкм; Подвижная фаза: 4:1 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 50 мл/мин]. Первый элюирующий диастереомер был указан как Пример 57, а второй элюирующий диастереомер как Пример 58. При осуществлении аналитической сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralcel OJ-3R, 3,0×150 мм, 3 мкм; Подвижная фаза: 3:1 диоксид углерода/(метанол, содержащий 0,1% диэтиламина); Скорость потока: 2,5 мл/мин] Пример 57 показал время удерживания 1,7 7 мин. Пример 58 имел время удерживания 2,92 мин в таких же условиях.
29. Преобразование 5-хлор-4-иодпиридин-2-ола в требуемую 2-[5-хлор-2-(дифторметокси)пиридин-4-ил]пропановую кислоту осуществляли с использованием способа, описанного в Получении Р5. ЖХМС m/z 252, 1 (наблюдаемый изотопный паттерн хлора) [М+Н]+. 1Н ЯМР (400 МГц, метанол-d4) δ 8,21 (с, 1Н), 7,50 (т, JHF=72,8 Гц, 1Н), 7,01 (с, 1Н), 4,13 (кв., J=7,2 Гц, 1Н), 1,52 (д, J=7,2 Гц, 3Н).
30. Продукт разделяли на составляющие его диастереомеры с использованием ВЭЖХ (Колонка: Chiral Technologies Chiralpak AD, 50×250 мм, 10 мкм; Подвижная фаза: 70:30:0,1 метанол/ацетонитрил/диэтиламин; Скорость потока: 50 мл/мин). Первый элюирующий диастереомер был указан как Пример 59, а второй элюирующий диастереомер как Пример 60. При осуществлении аналитической ВЭЖХ (Колонка: Chiral Technologies Chiralpak AD-H, 4,6×150 мм, 5 мкм; Подвижная фаза: 4:1 метанол/ацетонитрил; Скорость потока: 1,0 мл/мин) Пример 59 показал время удерживания 3,16 мин. Пример 60 имел время удерживания 6,23 мин в таких же условиях.
31. Конечную очистку осуществляли с использованием обращеннофазовой С18 хроматографии (Подвижная фаза А: вода, содержащая 0,1% муравьиной кислоты; Подвижная фаза В: ацетонитрил; Градиент: 0% до 50% В).
32. Продукт разделяли на составляющие его диастереомеры с использованием ВЭЖХ (Колонка: Chiral Technologies Chiralpak AY, 50×250 мм, 10 мкм; Подвижная фаза: 70:30:0,1 гексан/этанол/диэтиламин; Скорость потока: 60 мл/мин). Первый элюирующий диастереомер был указан как Пример 61, а второй элюирующий диастереомер как Пример 62. При осуществлении аналитической ВЭЖХ (Колонка: Chiral Technologies Chiralpak AY-3, 4,6×150 мм, 3 мкм; Подвижная фаза: 70:30:0,1 гексан/этанол/диэтиламин; Скорость потока: 1,0 мл/мин) Пример 61 показал время удерживания 4,41 мин. Пример 62 имел время удерживания 5,60 мин в таких же условиях.
33. Взаимодействие 5-хлор-4-иодпиридин-2-ола с 1-трифторметил-1,2-бензиодксол-3-(1H)-оном в нитрометане при 100°С давало 5-хлор-4-иод-2-(трифторметокси)пиридин. Это вещество преобразовывали в требуемую 2-[5-хлор-2-(трифторметокси)пиридин-4-ил]пропановую кислоту с использованием способа, описанного в Получении Р5. ЖХМС m/z 270,0 (наблюдаемый изотопный паттерн хлора) [М+Н]+. 1Н ЯМР (400 МГц, метанол-d4) δ 8,32 (с, 1Н), 7,20 (с, 1Н), 4,17 (кв., J=7,3 Гц, 1Н), 1,54 (д, J=7,3 Гц, 3Н).
34. Продукт разделяли на составляющие его диастереомеры с использованием ВЭЖХ (Колонка: Chiral Technologies Chiralpak AD, 50×250 мм, 10 мкм; Подвижная фаза: 100:0,1 метанол/диэтиламин; Скорость потока: 60 мл/мин). Первый элюирующий диастереомер был указан как Пример 63, а второй элюирующий диастереомер как Пример 64. При осуществлении аналитической ВЭЖХ (Колонка: Chiral Technologies Chiralpak AD-H, 4,6×250 мм, 5 мкм; Подвижная фаза: 100:0,1 метанол/диэтиламин; Скорость потока: 1,0 мл/мин) Пример 63 показал время удерживания 7,22 мин. Пример 64 имел время удерживания 10,44 мин в таких же условиях.
35. Конечную очистку осуществляли с использованием обращеннофазовой С18 хроматографии (Подвижная фаза А: вода, содержащая 0,1% муравьиной кислоты; Подвижная фаза В: ацетонитрил; Градиент: 0% до 40% В).
36. Продукт разделяли на составляющие его энантиомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralpak AD-H, 20×250 мм, 5 мкм; Подвижная фаза: 64:36 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 50 мл/мин]. Первый элюирующий энантиомер был указан как Пример 65, а второй элюирующий энантиомер как Пример 66. При осуществлении аналитической сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralpak AD-3R, 3,0×150 мм, 3 мкм; Подвижная фаза: 65:35 диоксид углерода/(метанол, содержащий 0,1% диэтиламина); Скорость потока: 2,0 мл/мин] Пример 65 показал время удерживания 1,32 мин. Пример 66 имел время удерживания 1,79 мин в таких же условиях.
37. В реакции использовали бром-содержащее гетероциклическое соединение (2-бром-5-метоксипиримидин), а не хлор-содержащее производное.
38. Деметилирование Примера 14 осуществляли с использованием триметилсилилиодида в ацетонитриле при кипячении с обратным холодильником с получением (2R)-2-(5-фтор-2-гидроксипиридин-4-ил)-1-[{2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она. Это вещество можно подвергнуть взаимодействию с иод (2Н3)метаном и карбонатом калия в N, N-диметилформамиде с получением Примера 74.
39. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralpak IG-H, 20×250 мм, 5 мкм; Подвижная фаза: 54:46 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 45 мл/мин]. Первый элюирующий диастереомер был указан как Пример 75, а второй элюирующий диастереомер как Пример 76. При осуществлении аналитической сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralpak IG-3R, 3,0×150 мм, 3 мкм; Подвижная фаза: 1:1 диоксид углерода/(метанол, содержащий 0,1% диэтиламина); Скорость потока: 1,5 мл/мин] Пример 75 показал время удерживания 1,93 мин. Пример 76 имел время удерживания 2,9 9 мин в таких же условиях.
40. Продукт разделяли на составляющие его диастереомеры с использованием ВЭЖХ (Колонка: Chiral Technologies Chiralpak AD, 50×250 мм, 10 мкм; Подвижная фаза: 70/30/0,1 метанол/ацетонитрил/диэтиламин; Скорость потока: 60 мл/мин). Первый элюирующий диастереомер был указан как Пример 77, а второй элюирующий диастереомер как Пример 78. При осуществлении аналитической ВЭЖХ (Колонка: Chiral Technologies Chiralpak AD-3, 4,6×250 мм, 3 мкм; Подвижная фаза: 70/30/0,1 метанол/ацетонитрил/диэтиламин; Скорость потока: 1,0 мл/мин) Пример 77 показал время удерживания 2,58 мин. Пример 78 имел время удерживания 4,41 мин в таких же условиях.
41. Преобразование 5-фтор-4-иод-2-(трифторметил)пиридина в требуемую 2-[5-фтор-2-(трифторметил)пиридин-4-ил]пропановую кислоту осуществляли с использованием способа, описанного в Получении Р5. ЖХМС m/z 238,1 [М+Н]+. 1Н ЯМР (400 МГц, хлороформ-d) δ 8,55 (с, 1Н), 7,70 (д, J=5,4 Гц, 1Н), 4,14 (кв., J=l,3 Гц, 1Н), 1,61 (д, J=7, 3 Гц, 3Н).
42. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralpak IG-H, 20×250 мм, 5 мкм; Подвижная фаза: 54:46 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 45 мл/мин]. Первый элюирующий диастереомер был указан как Пример 79, а второй элюирующий диастереомер как Пример 80. При осуществлении аналитической сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralpak IG-3R, 3,0×150 мм, 3 мкм; Подвижная фаза: 1:1 диоксид углерода/(метанол, содержащий 0,1% диэтиламина); Скорость потока: 1,5 мл/мин] Пример 79 показал время удерживания 1,97 мин. Пример 80 имел время удерживания 3,41 мин в таких же условиях.
43. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralcel OJ-H, 20×250 мм, 5 мкм; Подвижная фаза: 86:14 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 50 мл/мин]. Первый элюирующий диастереомер был указан как Пример 81, а второй элюирующий диастереомер как Пример 82. При осуществлении аналитической сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralcel OJ-3R, 3,0×150 мм, 3 мкм; Подвижная фаза: 3:1 диоксид углерода/(метанол, содержащий 0,1% диэтиламина); Скорость потока: 2,5 мл/мин] Пример 81 показал время удерживания 1,55 мин. Пример 82 имел время удерживания 1,79 мин в таких же условиях.
44. 2,6-Диметокси-4-метилпиридин преобразовывали в требуемую 2-(2,6-диметоксипиридин-4-ил)пропановую кислоту с использованием способа, описанного в Получении Р1. ЖХМС m/z 212,1 [М+Н]+. 1Н ЯМР (400 МГц, хлороформ-d) δ 6,26 (с, 2Н), 3,90 (с, 6Н), 3,63 (кв., J=7,2 Гц, 1Н), 1,47 (д, J=7,1 Гц, 3Н).
45. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralcel OJ-H, 20×250 мм, 5 мкм; Подвижная фаза: 86:14 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 50 мл/мин]. Первый элюирующий диастереомер был указан как Пример 83, а второй элюирующий диастереомер как Пример 84. При осуществлении аналитической сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralcel 0J-3R, 3,0×150 мм, 3 мкм; Подвижная фаза: 3:1 диоксид углерода/(метанол, содержащий 0,1% диэтиламина); Скорость потока: 2,5 мл/мин] Пример 83 показал время удерживания 1,75 мин. Пример 84 имел время удерживания 2,11 мин в таких же условиях.
46. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralcel OD-H, 20×250 мм, 5 мкм; Подвижная фаза: 68:32 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 50 мл/мин]. Первый элюирующий диастереомер был указан как Пример 85, а второй элюирующий диастереомер как Пример 86. При осуществлении аналитической сверхкритической флюидной хроматографии [Колонка: Chiral Technologies ChiralCel OD-3R, 3,0×150 мм, 3 мкм; Подвижная фаза: 7:3 диоксид углерода/(метанол, содержащий 0,1% диэтиламина); Скорость потока: 2,0 мл/мин] Пример 85 показал время удерживания 1,70 мин. Пример 8 6 имел время удерживания 2,14 мин в таких же условиях.
47. Гидрирование C72 над гидроксидом палладия давало требуемый 7-метил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин].
48. Продукт разделяли на составляющие его энантиомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralcel OJ-H, 21×250 мм, 5 мкм; Подвижная фаза: 9:1 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 75 мл/мин; Обратное давление: 200 бар]. Первый элюирующий энантиомер был указан как Пример 87, а второй элюирующий энантиомер как Пример 88.
49. Условия для аналитической ВЭЖХ. Колонка: Phenomenex Lux Cellulose-3, 4,6×100 мм, 5 мкм; Подвижная фаза: 4:1 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 1,5 мл/мин; Обратное давление: 200 бар.
50. Условия для аналитической ВЭЖХ. Колонка: Waters Atlantis dC18, 4,6×50 мм, 5 мкм; Подвижная фаза А: 0,05% трифторуксусной кислоты в воде (об/об); Подвижная фаза В: 0,05% трифторуксусной кислоты в ацетонитриле (об/об); Градиент: 5,0% до 95% В, линейный в течение 4,0 минут, затем 95% В в течение 1,0 минут; Скорость потока: 2 мл/мин.
51. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralcel OD-H, 21×250 мм, 5 мкм; Подвижная фаза: 3:1 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 75 мл/мин; Обратное давление: 200 бар]. Первый элюирующий диастереомер был указан как Пример 90, а второй элюирующий диастереомер как Пример 91.
52. Условия для аналитической ВЭЖХ. Колонка: Phenomenex Lux Cellulose-1, 4,6×100 мм, 5 мкм; Подвижная фаза: 3:2 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 1,5 мл/мин; Обратное давление: 200 бар.
53. Преобразование C74 в требуемый 7-метил-6-[1-метил-5-(трифторметил)-1H-1,2,4-триазол-3-ил]-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин] осуществляли с использованием способа, описанного для синтеза P31 из C74 в Получении Р31.
54. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralpak IB, 21×250 мм, 5 мкм; Подвижная фаза: 9:1 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 75 мл/мин; Обратное давление: 200 бар]. Первый элюирующий диастереомер был указан как Пример 92, а второй элюирующий диастереомер как Пример 93.
55. Условия для аналитической ВЭЖХ. Колонка: Chiral Technologies Chiralpak IB, 4,6×100 мм, 5 мкм; Подвижная фаза: 85:15 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 1,5 мл/мин; Обратное давление: 200 бар.
56. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Phenomenex Lux Cellulose-1, 21×250 мм, 5 мкм; Подвижная фаза: 3:1 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 75 мл/мин; Обратное давление: 120 бар]. Первый элюирующий диастереомер был указан как Пример 94, а второй элюирующий диастереомер как Пример 95.
57. Условия для аналитической ВЭЖХ. Колонка: Phenomenex Lux Cellulose-1, 4,6×100 мм, 5 мкм; Подвижная фаза: 7:3 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 1,5 мл/мин; Обратное давление: 200 бар.
58. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralcel OJ-H, 21×250 мм, 5 мкм; Подвижная фаза: 92:8 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 75 мл/мин; Обратное давление: 200 бар]. Первый элюирующий диастереомер был указан как Пример 96, а второй элюирующий диастереомер как Пример 97.
59. Условия для аналитической ВЭЖХ. Колонка: Phenomenex Lux Cellulose-3, 4,6×100 мм, 5 мкм; Подвижная фаза: 4:1 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 1,5 мл/мин; Обратное давление: 120 бар.
60. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralcel OJ-H, 21×250 мм, 5 мкм; Подвижная фаза: 92:8 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 75 мл/мин; Обратное давление: 200 бар]. Первый элюирующий диастереомер был указан как Пример 98, а второй элюирующий диастереомер как Пример 99.
61. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralcel OJ-H, 21×250 мм, 5 мкм; Подвижная фаза: 92:8 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 75 мл/мин; Обратное давление: 200 бар]. Первый элюирующий диастереомер был указан как Пример 100, а второй элюирующий диастереомер как Пример 101.
62. Взаимодействие P17 с 1,3-дихлор-5,5-диметилимидазолидин-2,4-дионом давало требуемый трет-бутил-6-хлор-7-метил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилат.
63. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralcel OJ-H, 21×250 мм, 5 мкм; Подвижная фаза: 4:1 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 75 мл/мин; Обратное давление: 120 бар]. Первый элюирующий диастереомер был указан как Пример 102, а второй элюирующий диастереомер как Пример 103.
64. Условия для аналитической ВЭЖХ. Колонка: Chiral Technologies Chiralcel OJ-H, 4,6×100 мм, 5 мкм; Подвижная фаза: 7:3 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 1,5 мл/мин; Обратное давление: 120 бар.
65. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Phenomenex Lux Cellulose-1, 21×250 мм, 5 мкм; Подвижная фаза: 4:1 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 75 мл/мин; Обратное давление: 120 бар]. Первый элюирующий диастереомер был указан как Пример 104, а второй элюирующий диастереомер как Пример 105.
66. Условия для аналитической ВЭЖХ. Колонка: Phenomenex Lux Cellulose-1, 4,6×100 мм, 5 мкм; Подвижная фаза: 65:35 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 1,5 мл/мин; Обратное давление: 200 бар.
67. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralpak AS-H, 21×250 мм, 5 мкм; Подвижная фаза: 9:1 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 75 мл/мин; Обратное давление: 120 бар]. Первый элюирующий диастереомер был указан как Пример 107, а второй элюирующий диастереомер как Пример 108.
68. Условия для аналитической ВЭЖХ. Колонка: Chiral Technologies Chiralpak AS-H, 4,6×100 мм, 5 мкм; Подвижная фаза: 85:15 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 1,5 мл/мин; Обратное давление: 120 бар.
69. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralpak IB, 21×250 мм, 5 мкм; Подвижная фаза: 85:15 диоксид углерода/ (метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 75 мл/мин; Обратное давление: 120 бар]. Первый элюирующий диастереомер был указан как Пример 109, а второй элюирующий диастереомер как Пример 110.
70. Условия для аналитической ВЭЖХ. Колонка: Chiral Technologies Chiralpak IB, 4,6×100 мм, 5 мкм; Подвижная фаза: 4:1 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 1,5 мл/мин; Обратное давление: 120 бар.
71. Преобразование P23 в требуемое соединение (2S)-7-метил-6-([1,2,4]триазоло[1,5-а]пиридин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин], дигидрохлоридную соль, осуществляли с использованием способа, описанного в Альтернативном Получении Р26; в этом случае использовали 2-бром[1,2,4]триазоло[1,5-а]пиридин вместо 5-бром-2-метил-2H-тетразола.
72. Преобразование P21 в требуемое соединение 6-(5-метокси-1-метил-1H-1,2,4-триазол-3-ил)-7-метил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин], дигидрохлоридную соль, осуществляли с использованием способа, описанного в Альтернативном Получении Р26; в этом случае использовали 3-бром-5-метокси-1-метил-1Н-1,2,4-триазол вместо 5-бром-2-метил-2H-тетразола.
73. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralpak IB, 21×250 мм, 5 мкм; Подвижная фаза: 85:15 диоксид углерода/ (метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 75 мл/мин; Обратное давление: 200 бар]. Первый элюирующий диастереомер был указан как Пример 112, а второй элюирующий диастереомер как Пример 113.
74. Условия для аналитической ВЭЖХ. Колонка: Chiral Technologies Chiralpak IB, 4,6×100 мм, 5 мкм; Подвижная фаза: 3:1 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 1,5 мл/мин; Обратное давление: 120 бар.
75. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralpak IB, 21×250 мм, 5 мкм; Подвижная фаза: 4:1 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 75 мл/мин; Обратное давление: 200 бар]. Первый элюирующий диастереомер был указан как Пример 114, а второй элюирующий диастереомер как Пример 115.
76. Условия для аналитической ВЭЖХ. Колонка: Chiral Technologies Chiralpak IB, 4,6×100 мм, 5 мкм; Подвижная фаза: 3:2 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 1,5 мл/мин; Обратное давление: 120 бар.
77. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralpak IB, 21×250 мм, 5 мкм; Подвижная фаза: 85:15 диоксид углерода/ (метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 75 мл/мин; Обратное давление: 200 бар]. Первый элюирующий диастереомер представлял собой Пример 13, а второй элюирующий диастереомер был указан как Пример 116. Пример 13 показал время удерживания 3,2 6 мин. Пример 116 имел время удерживания 3,52 мин в таких же условиях (см. примечание 74).
78. Преобразование P23 в требуемое соединение (2S)-7-метил-6-(2-метил-2H-1,2,3-триазол-4-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин], дигидрохлоридную соль, осуществляли с использованием способа, описанного в Альтернативном Получении Р26; в этом случае использовали 4-бром-2-метил-2H-1,2,3-триазол вместо 5-бром-2-метил-2H-тетразола.
79. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralpak IA, 21×250 мм, 5 мкм; Подвижная фаза: 4:1 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 75 мл/мин; Обратное давление: 150 бар]. Первый элюирующий диастереомер был указан как Пример 123, а второй элюирующий диастереомер как Пример 124.
80. Условия для аналитической ВЭЖХ. Колонка: Chiral Technologies Chiralpak IA, 4,6×100 мм, 5 мкм; Подвижная фаза: 3:1 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 1,5 мл/мин; Обратное давление: 200 бар.
81. Метил(2-метоксипиридин-4-ил)ацетат преобразовывали в требуемую 2-(2-метоксипиридин-4-ил)пропановую кислоту с использованием способа, описанного в Получении Р1. ЖХМС m/z 182,1 [М+Н]+. 1H ЯМР (400 МГц, DMSO-d6) δ 12,53 (ушир.с, 1Н), 8,09 (д, J=5,3 Гц, 1Н), 6,90 (д, J=5,3 Гц, 1Н), 6,71 (с, 1Н), 3,83 (с, 3Н), 3,69 (кв., J=7,l Гц, 1Н), 1,34 (д, J=7,l Гц, 3Н).
82. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralcel OJ-H, 21×250 мм, 5 мкм; Подвижная фаза: 3:1 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 75 мл/мин; Обратное давление: 120 бар]. Первый элюирующий диастереомер был указан как Пример 125, а второй элюирующий диастереомер как Пример 126.
83. Условия для аналитической ВЭЖХ. Колонка: Chiral Technologies Chiralcel OJ-H, 4,6×100 мм, 5 мкм; Подвижная фаза: 3:2 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 1,5 мл/мин; Обратное давление: 120 бар.
84. 2,4-Дихлор-6-метоксипиримидин преобразовывали в диметил(2-хлор-6-метоксипиримидин-4-ил)(метил)пропандиоат в соответствии со способом, описанным на стадии 1 Получения Р4. Это вещество затем использовали для синтеза требуемого 2-[2-(дифторметил)-6-метоксипиримидин-4-ил]пропаноата лития с использованием способа, описанного в Получении Р9. ЖХМС m/z 233,1 [М+Н]+. 1Н ЯМР (400 МГц, метанол-d4) δ 6,91 (с, 1Н), 6,54 (т, JHF=54,7 Гц, 1Н), 4,01 (с, 3Н), 3,71 (кв., J=7,2 Гц, 1Н), 1,48 (д, J=l,2 Гц, 3Н).
85. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralpak IA, 21×250 мм, 5 мкм; Подвижная фаза: 7:3 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 75 мл/мин; Обратное давление: 120 бар]. Первый элюирующий диастереомер был указан как Пример 127, а второй элюирующий диастереомер как Пример 128.
86. Условия для аналитической ВЭЖХ. Колонка: Chiral Technologies Chiralpak IA, 4,6×100 мм, 5 мкм; Подвижная фаза: 3:2 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 1,5 мл/мин; Обратное давление: 120 бар.
87. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralpak IA, 21×250 мм, 5 мкм; Подвижная фаза: 3:2 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 75 мл/мин; Обратное давление: 120 бар]. Первый элюирующий диастереомер был указан как Пример 129, а второй элюирующий диастереомер как Пример 130.
88. С использованием способа, описанного в D. W. С. MacMillan et al., J. Amer. Chem. Soc. 2016, 138, 8084-8087, p23 можно подвергнуть взаимодействию с 4-бромтетрагидро-2H-пираном, в присутствии фотохимического катализатора [Ir{dF(CF3)рру}2(dtbpy)]PF6, с получением трет-бутил-(2S)-7-метил-6-(оксан-4-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилата. Удаление защиты с использованием хлористого водорода давало требуемое соединение (2S)-7-метил-6-(оксан-4-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин] дигидрохлоридную соль.
89. Условия для аналитической ВЭЖХ. Колонка: Chiral Technologies Chiralpak IA, 4,6×100 мм, 5 мкм; Подвижная фаза: 65:35 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 1,5 мл/мин; Обратное давление: 120 бар.
90. Преобразование 4-хлор-5-фтор-2-метоксипиримидина в требуемую 2-(5-фтор-2-метоксипиримидин-4-ил)пропановую кислоту осуществляли с использованием способа, описанного в Получении Р4. Полученная кислота содержала 4-этил-5-фтор-2-метоксипиримидин в виде примеси. ЖХМС m/z 201,1 [М+Н]+. 1Н ЯМР (400 МГц, DMSO-d6), только пики продукта: δ 8,32 (с, 1Н), 3,85 (с, 3Н), 3,56 (кв., J=7,3 Гц, 1Н), 1,34 (д, J=7,3 Гц, 3Н).
91. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralcel OJ-H, 21×250 мм, 5 мкм; Подвижная фаза: 4:1 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 75 мл/мин; Обратное давление: 12 0 бар]. Первый элюирующий диастереомер был указан как Пример 132, а второй элюирующий диастереомер как Пример 133.
92. Условия для аналитической ВЭЖХ. Колонка: Chiral Technologies Chiralcel OJ-H, 4,6×100 мм, 5 мкм; Подвижная фаза: 3:1 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Обратное давление: 120 бар.
93. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralpak AS-H, 21×250 мм, 5 мкм; Подвижная фаза: 85:15 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 75 мл/мин; Обратное давление: 120 бар]. Первый элюирующий диастереомер был указан как Пример 134, а второй элюирующий диастереомер как Пример 135.
94. Условия для аналитической ВЭЖХ. Колонка: Chiral Technologies Chiralpak AS-H, 4,6×100 мм, 5 мкм; Подвижная фаза: 4:1 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 1,5 мл/мин; Обратное давление: 120 бар.
95. Преобразование 2-метокси-4,5-диметилпиридина в требуемую 2-(2-метокси-5-метилпиридин-4-ил)пропановую кислоту осуществляли с использованием способа, описанного в Получении Р1. ЖХМС m/z 196,1 [М+Н]+. 1Н ЯМР (400 МГц, метанол-d4) δ 7,89 (ушир.с, 1Н), 6,69 (с, 1Н), 3,90 (кв., J=7,1 Гц, 1Н), 3,86 (с, 3Н), 2,26 (ушир.с, 3Н), 1,44 (д, J=1,2 Гц, 3Н).
96. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralpak IA, 5 мкм; Подвижная фаза: 1:1 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 75 мл/мин; Обратное давление: 120 бар]. Первый элюирующий диастереомер был указан как Пример 136, а второй элюирующий диастереомер как Пример 137.
97. Условия для аналитической ВЭЖХ. Колонка: Chiral Technologies Chiralpak IA, 4,6×100 мм, 5 мкм; Подвижная фаза: 1:1 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 1,5 мл/мин; Обратное давление: 120 бар.
98. Взаимодействие р27 с 1-(дифторметил)-4-иод-2-метил-1Л-имидазолом с использованием способа, описанного для преобразования р27 в с84 в Примере 18, давало требуемый трет-бутил-(2S)-6-[1-(дифторметил)-2-метил-1H-имидазол-4-ил]-7-метил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-карбоксилат.
99. Требуемый 1-(дифторметил)-4-иод-2-метил-1H-имидазол получали из 4-иод-2-метил-1H-имидазола с использованием способа, описанного для синтеза C5 в Получении Р5.
100. Подходящее бром-замещенное гетероароматическое соединение использовали в катализируемом палладием сочетании.
101. Подходящее хлор-замещенное гетероароматическое соединение использовали в катализируемом палладием сочетании.
102. Взаимодействие 3,5-дибром-1H-1,2,4-триазола с 4-бромбут-1-еном в присутствии карбоната калия давало 3,5-дибром-1-(бут-3-ен-1-ил)-1H-1,2,4-триазол; это вещество подвергали циклизации до 2-бром-7-метилиден-6,7-дигидро-5H-пирроло[1,2-b][1,2,4]триазола с использованием 2-дициклогексилфосфино-2',6'-диметоксибифенила (SPhos), хлор(2-дициклогексилфосфино-2',6'-диметокси-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладия(II) (SPhos Pd G2) и триэтиламина при повышенной температуре. После озонолиза до кетона осуществляли взаимодействие с трифторидом (диэтиламино)серы с получением требуемого 2-бром-7,7-дифтор-6,7-дигидро-5H-пирроло[1,2-b] [1,2,4] триазола.
103. Преобразование с29 в требуемую 2-фтор-2-(5-фтор-2-метоксипиридин-4-ил)пропановую кислоту осуществляли с использованием способа, описанного для стадий 5 и 6 Получения Р10. ЖХМС m/z 218,1 [М+Н]+. 1Н ЯМР (400 МГц, метанол-d4) δ 8,01 (д, J=2,6 Гц, 1Н), 6,92 (д, J=5,0 Гц, 1Н), 3,90 (с, 3Н), 1,91 (ушир.д, JHF=23,1 Гц, 3Н).
104. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralpak AS-H, 21×250 мм, 5 мкм; Подвижная фаза: 9:1 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 75 мл/мин; Обратное давление: 120 бар]. Первый элюирующий диастереомер был указан как Пример 14 8, а второй элюирующий диастереомер как Пример 149.
105. 3-Бромбензогидразид ацилировали ацетилхлоридом в присутствии триэтиламина и затем подвергали циклизации путем взаимодействия с п-толуолсульфонилхлоридом и триэтиламином с получением требуемого 2-бром-6-(5-метил-1,3,4-оксадиазол-2-ил)пиридина.
106. Иодирование 5-метил-1H-1,2,4-триазола с использованием N-иодсукцинимида в N, N-диметилформамиде при повышенной температуре давало 3-иод-5-метил-1H-1,2,4-триазол; это вещество преобразовывали в требуемый 1-(дифторметил)-3-иод-5-метил-1H-1,2,4-триазол с использованием способа, описанного для синтеза C31 из C30 в Получении Р13.
107. Взаимодействие 3,5-дибром-1H-1,2,4-триазола с циклопропилбороновой кислотой в присутствии ацетата меди(II), 2,2'-бипиридина и карбоната натрия в 1,2-дихлорэтане при 70°С давало 3,5-дибром-1-циклопропил-1H-1,2,4-триазол. Затем осуществляли обмен литий-галоген с использованием н-бутиллития (1,1 эквивалента) при -78°С и полученный анион подвергали взаимодействию с N,N-диметилформамидом с получением 3-бром-1-циклопропил-1H-1,2,4-триазол-5-карбальдегида. Это вещество подвергали взаимодействию с трифторидом [бис(2-метоксиэтил)амино]серы в дихлорметане с получением требуемого 3-бром-1-циклопропил-5-(дифторметил)-1H-1,2,4-триазола.
108. Продукт разделяли на составляющие его диастереомеры с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralcel OJ-H, 20×250 мм, 5 мкм; Подвижная фаза: 86:14 диоксид углерода/(метанол, содержащий 0,2% гидроксида аммония); Скорость потока: 50 мл/мин]. Первый элюирующий диастереомер был указан как Пример 190, а второй элюирующий диастереомер как Пример 191. При осуществлении аналитической сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralcel OJ-3R, 3,0×150 мм, 3 мкм; Подвижная фаза: 4:1 диоксид углерода/(метанол, содержащий 0,1% диэтиламина); Скорость потока: 2,0 мл/мин] Пример 190 показал время удерживания 2,7 0 мин. Пример 191 имел время удерживания 3,33 мин в таких же условиях.
109. Продукт разделяли на составляющие его диастереомеры с использованием ВЭЖХ (Колонка: Chiral Technologies Chiralcel OD, 25×250 мм, 10 мкм; Подвижная фаза: 4:1 гексан/этанол; Скорость потока: 25 мл/мин). Первый элюирующий диастереомер был указан как Пример 192, а второй элюирующий диастереомер как Пример 193. При осуществлении аналитической ВЭЖХ (Колонка: Chiral Technologies Chiralcel OD-H, 4,6×150 мм, 5 мкм; Подвижная фаза: 85:15 гексан/этанол; Скорость потока: 1,0 мл/мин) Пример 192 показал время удерживания 6,2 0 мин. Пример 193 имел время удерживания 7,06 мин в таких же условиях.
110. Конечную очистку осуществляли с использованием обращеннофазовой С18 хроматографии (Подвижная фаза А: вода, содержащая 0,1% муравьиной кислоты; Подвижная фаза В: ацетонитрил; Градиент: 0% до 45% В).
111. С использованием способа, описанного для синтеза P13 из C30 в Получении Р13, метил-2-(2-гидроксипиридин-4-ил)пропаноат преобразовывали в требуемую 2-[2-(дифторметокси)пиридин-4-ил]пропановую кислоту. ЖХМС m/z 218,0 [М+Н]+. 1H ЯМР (400 МГц, метанол-d4) δ 8,14 (ушир.д, J=5,3 Гц, 1Н), 7,53 (т, JHF=73,1 Гц, 1Н), 7,16 (дд, J=5,3, 1,5 Гц, 1Н), 6,93-6,91 (м, 1Н), 3,80 (кв., J=7,2 Гц, 1Н), 1,48 (д, J=7,2 Гц, 3Н).
112. Продукт разделяли на составляющие его диастереомеры с использованием ВЭЖХ (Колонка: Chiral Technologies Chiralpak IG, 50×250 мм, 10 мкм; Подвижная фаза: 4:1 этанол/ацетонитрил; Скорость потока: 30 мл/мин). Первый элюирующий диастереомер был указан как Пример 194, а второй элюирующий диастереомер как Пример 195. При осуществлении аналитической ВЭЖХ (Колонка: Chiral Technologies Chiralpak IG, 4,6×150 мм, 5 мкм; Подвижная фаза: 4:1 этанол/ацетонитрил; Скорость потока: 0,5 мл/мин), Пример 194 показал время удерживания 6,48 мин. Пример 195 имел время удерживания 8,08 мин в таких же условиях.
Пример АА. Анализ аффинности связывания in vitro с использованием hMC4R
Аффинность связывания испытываемых соединений с рецептором α-меланоцитстимулирующего гормона (hMC4R) оценивали с использованием анализа конкурентного связывания радиолиганда. Рекомбинантные клетки яичников китайского хомячка (СНО), стабильно экспрессирующие hMC4R (PerkinElmer # ES-191-C), использовали для конкурентного связывания. Мембраны hMC4R выращивали в модифицированной по способу Дульбекко эссенциальной среде и среде F-12 Хэма (DMEM/F12), с 10% инактивированной нагреванием эмбриональной бычьей сыворотки (FBS), 0,4 мг/мл генетицина и 2 мМ L-глутамина. Клеточные мембраны собирали и замораживали до осуществления анализа.
Соединения солюбилизировали в 100% диметилсульфоксиде (DMSO) до концентрации 30 мМ. 10-точечную серию промежуточных разведений с использованием полулогарифмических разведений получали в 100% DMSO с максимальной концентрацией 0,03 мМ. Серийно разведенные соединения наносили по 1 мкл/лунка в 96-луночные планшеты Costar 3363. Конечный диапазон соединений в анализе составлял от 300 нМ до 0,01 нМ с конечной концентрацией DMSO 1%. Контрольные лунки, содержащие 1 мкл 2 мМ (конечная концентрация 2 мкМ) альфа-меланоцитстимулирующего гормона (сх-MSH-Tocris # 2584), добавляли в лунки для неспецифического связывания и 1 мкл 100% DMSO для всех контрольных лунок связывания. После этого добавляли 80 мкл буфера для анализа [25 мМ НЕ PES, 5 мМ MgCl2, 2,5 мМ CaCl2, 150 мМ NaCl, полная таблетка ингибитора протеазы без EDTA (Thermo Scientific #11873580001) и 0,25% BSA]. 10 мкл [125I] - (Nle4, D-Phe7)-α-MSH (PerkinElmer #NEX3520) добавляли во все лунки при 10-кратной конечной концентрации 0,5 нМ. Используемая концентрация радиолиганда была ниже равновесной константы диссоциации (Kd) 2,59 нМ. Точную концентрацию радиолиганда, используемого для каждого эксперимента, определяли методом жидкостного сцинтилляционного счета и при необходимости корректировали.
Замороженные клеточные мембраны hMC4R оттаивали и гомогенизировали в гомогенизаторе Даунса. Гомогенаты ресуспендировали в буфере для анализа до концентрации 2 мкг на лунку. Реакцию конкурентного связывания инициировали добавлением 10 мкл раствора мембран MC4R в готовые для анализа планшеты, содержащие испытываемое соединение и [125I]-(Nle4, D-Phe7)-α-MSH. Планшеты инкубировали в течение 2 часов при комнатной температуре. Затем образцы для анализа быстро фильтровали через фильтровальные планшеты Unifilter-96 GF/B, покрытые PEI, с использованием устройства для сбора образцов фильтровальных планшетов (PerkinElmer) и промывали охлажденным льдом промывочным буфером [25 мМ (4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота (HEPES), 1 мМ MgCl2, 2,5 мМ CaCl2 и 500 мМ NaCl]. Фильтровальные планшеты высушивали в течение ночи при комнатной температуре. Затем планшеты герметично закрывали снизу перед добавлением 50 мкл/лунка сцинтилляционной жидкости Ultima Gold XR (PerkinElmer 6013111). Затем планшеты герметично закрывали сверху, инкубировали в течение 60 минут при комнатной температуре, а затем определяли количество присутствующей радиоактивности при помощи жидкостного сцинтилляционного счета на Microbeta Trilux (PerkinElmer # 2450-0060).
Необработанные данные (выраженные как количество импульсов в минуту) анализировали с использованием ActivityBase (программное обеспечение для управления данными IDBS). Выраженный в процентах эффект при каждой концентрации соединения рассчитывали с использованием ActivityBase на основании значений для лунок без ингибирования (контроли общего связывания) и лунок с полным ингибированием (контроли неспецифического связывания) на каждом планшете для анализа. Концентрацию, необходимую для 50% ингибирования (IC50), определяли из этих данных с использованием 4-параметрической логистической модели. Равновесную константу диссоциации для ингибитора взаимодействия лиганда и рецептора (Ki) затем рассчитывали из значений IC50 с использованием уравнения Ченга-Прусоффа: Ki=IC50/(1+([L]/Kd)), где [L] представляет собой концентрацию радиолиганда, используемого в эксперименте, a Kd представляет собой аффинность радиолиганда (определенную в отдельных экспериментах с насыщением).
Пример ВВ. Анализ функциональной активности антагониста MC4R in vitro
Функциональную антагонистическую активность испытываемых соединений in vitro определяли путем мониторинга уровней внутриклеточного циклического аденозинмонофосфата (цАМФ) в клетках яичника китайского хомячка (СНО-), стабильно экспрессирующих рецептор меланокортина-4 человека (MC4R). После активации агонистом человеческий MC4R связывается с G-белковым комплексом, заставляя субъединицу Gα обменивать связанный GDP на GTP с последующей диссоциацией комплекса Gα-GTP. Активированная субъединица Gα может соединяться с находящимися на пути ниже эффекторами для регулирования уровней вторичных мессенджеров или цАМФ внутри клетки. Таким образом, определение внутриклеточных уровней цАМФ предоставляет возможность определения фармакологических характеристик. Уровни внутриклеточного цАМФ количественно определяют с использованием гомогенного анализа, используя метод гомогенной флуоресценции с временным разрешением (HTRF) от CisBio. Этот метод представляет собой конкурентный иммуноанализ между нативным цАМФ, продуцируемым клетками, и цАМФ, меченным акцепторным красителем d2. Затем эти две молекулы конкурируют за связывание с моноклональным анти-цАМФ антителом, меченным криптатом. Специфический сигнал обратно пропорционален концентрации цАМФ в клетках.
Испытываемые соединения солюбилизировали до концентрации 30 мМ в 100% диметилсульфоксиде (DMSO) и хранили. Серию 11-точечных разведений с использованием 1 к 3 162-кратных серийных разведений осуществляли в 100% DMSO с максимальной концентрацией 800 мкМ. Серийно разведенное соединение вводили в 384-луночный планшет (Greiner, № по каталогу 781280) в количестве 40 нл/лунка с дублирующими точками для каждой концентрации, а затем разбавляли 1 к 1000 с использованием 40 мкл буфера для анализа, содержащего HBSS, 20 мМ HEPES (Invitrogen), 0,1% BSA и 250 мкМ IBMX (Sigma Aldrich), для создания промежуточного планшета с 2х конечной анализируемой концентрацией (FAC). Конечный диапазон концентраций соединения в анализе составлял от 400 нМ до 4 пМ с конечной концентрацией DMSO 0,1%.
Полученные в собственной лаборатории СНО-клетки, стабильно экспрессирующие Gs-связанный рецептор MC4R человека, высевали в 384-луночные планшеты для анализа (Corning, № по каталогу 3570) в 50 мкл/лунка среды Хэма F-12, содержащей 10% термоинактивированной FBS, 1х пенициллина/стрептомицина, 1 мМ Glutamax (Invitrogen), при плотности 2500 клеток на лунку и инкубировали при 37°С (95% O2: 5% CO2) в течение ночи, используя крышки micro-clime (Labcyte, № по каталогу LLS-0310). В день анализа среду удаляли из планшета для анализа путем осторожного постукивания и промокания планшета бумажным полотенцем и заменяли на 5 мкл 2х соединения-антагониста в буфере для анализа (HBSS, 20 мМ HEPES, 0,1% BSA, 250 мкМ IBMX) и 0,1% DMSO. Клетки инкубировали с соединением в течение 30 минут при 37°С (95% 02: 5% CO2) перед стимуляцией путем добавления 5 мкл агониста ЕС80 (200 нМ α-меланоцитостимулирующего гормона, aMSH, Bachem) и еще одной 30-минутной инкубацией при 37°С (95% O2: 5% CO2). Уровни внутриклеточного цАМФ определяли количественно в соответствии с протоколом Cisbio (5 мкл D2, а затем 5 мкл криптата, инкубация в течение 1-2 часов при комнатной температуре). Образцы измеряли на планшет-ридере Envision (PerkinElmer Life and Analytical Sciences; возбуждение при 320 нм; эмиссия при 665 нм/620 нм).
Данные анализировали, используя соотношение интенсивности флуоресценции при 620 и 665 нм для каждой лунки, экстраполировали из стандартной кривой цАМФ для выражения данных в виде нМ цАМФ для каждой лунки. Затем данные, выраженные как нМ цАМФ, нормализовали к контрольным лункам с использованием Activity Base (IDBS). Нулевой процентный эффект (ZPE) определяли как нМ цАМФ, образующегося при стимуляции агонистом ЕС80 (200 нМ aMSH). В отсутствие контрольного соединения-антагониста стопроцентный эффект (НРЕ) определяли как нМ цАМФ, образующегося только из буфера/носителя для анализа. Значения концентрации и % эффекта для каждого соединения наносили на график при помощи Activity Base с использованием четырехпараметрического логистического уравнения доза-ответ и определяли концентрацию, необходимую для 50% ингибирования (IC50). Затем рассчитывали значения равновесной константы диссоциации (Кь) по уравнению Леффа-Дугалла: Kb=[IC50]/((2+([А]/[ЕС50])n)1/n-1), где А=концентрация анализируемого агониста, используемая в эксперименте (200 нМ), и n = угол наклона.
Таблица 2 представляет биологическую активность (Ki значения, см. Пример АА; и Kb значения, см. Пример ВВ) и названия соединений для Примеров 1-201.
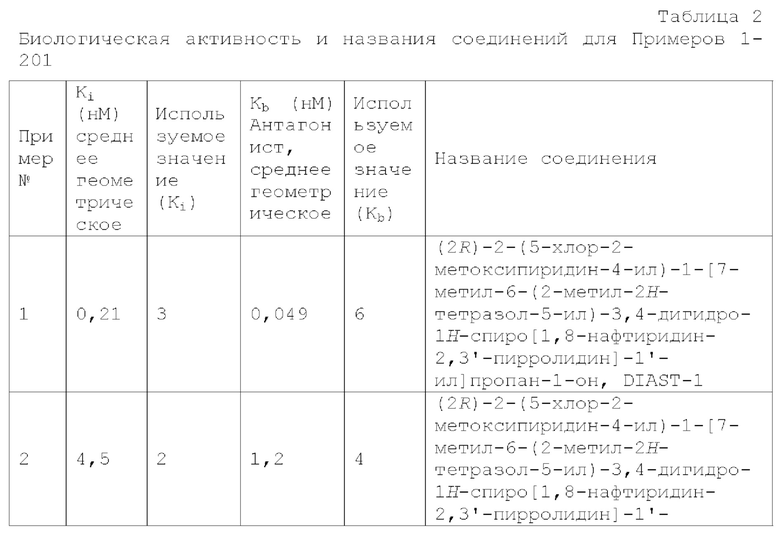
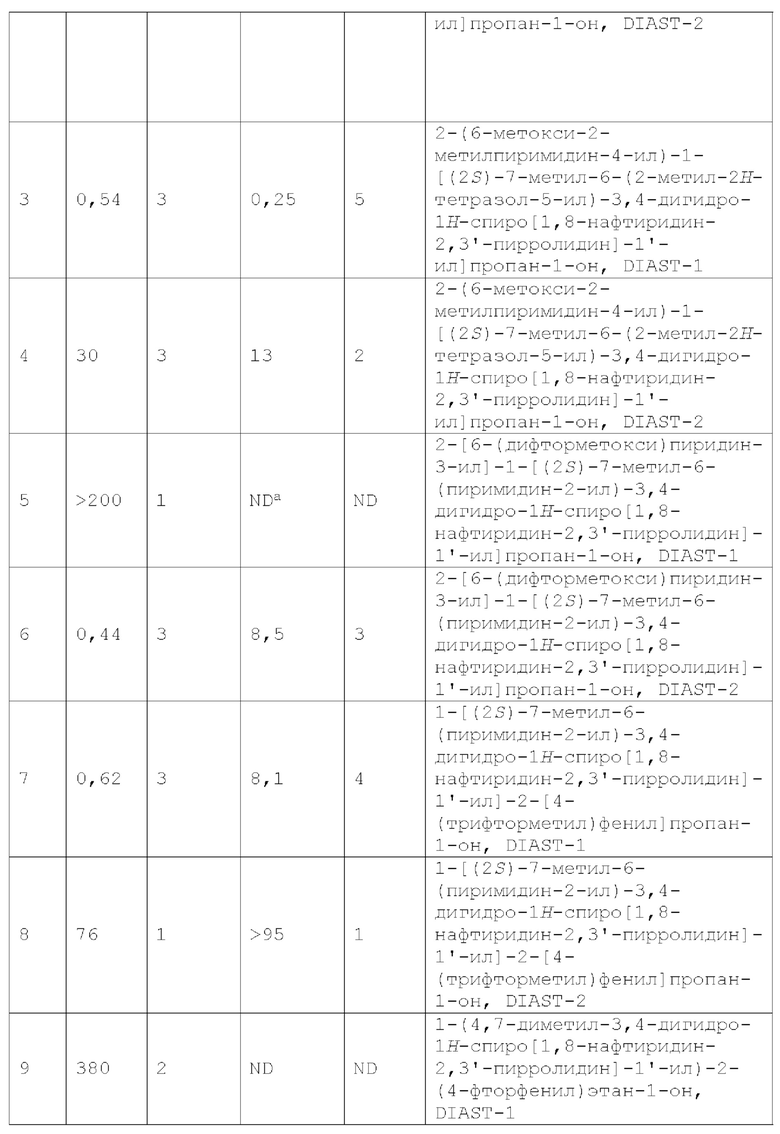
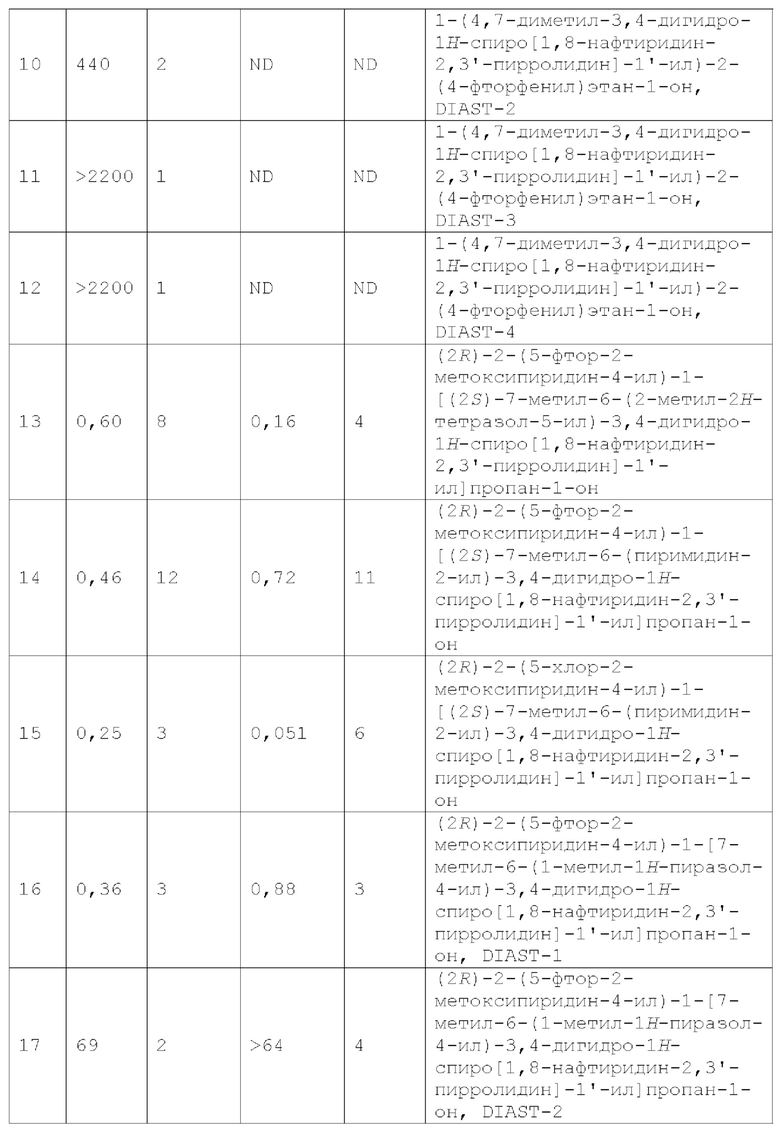
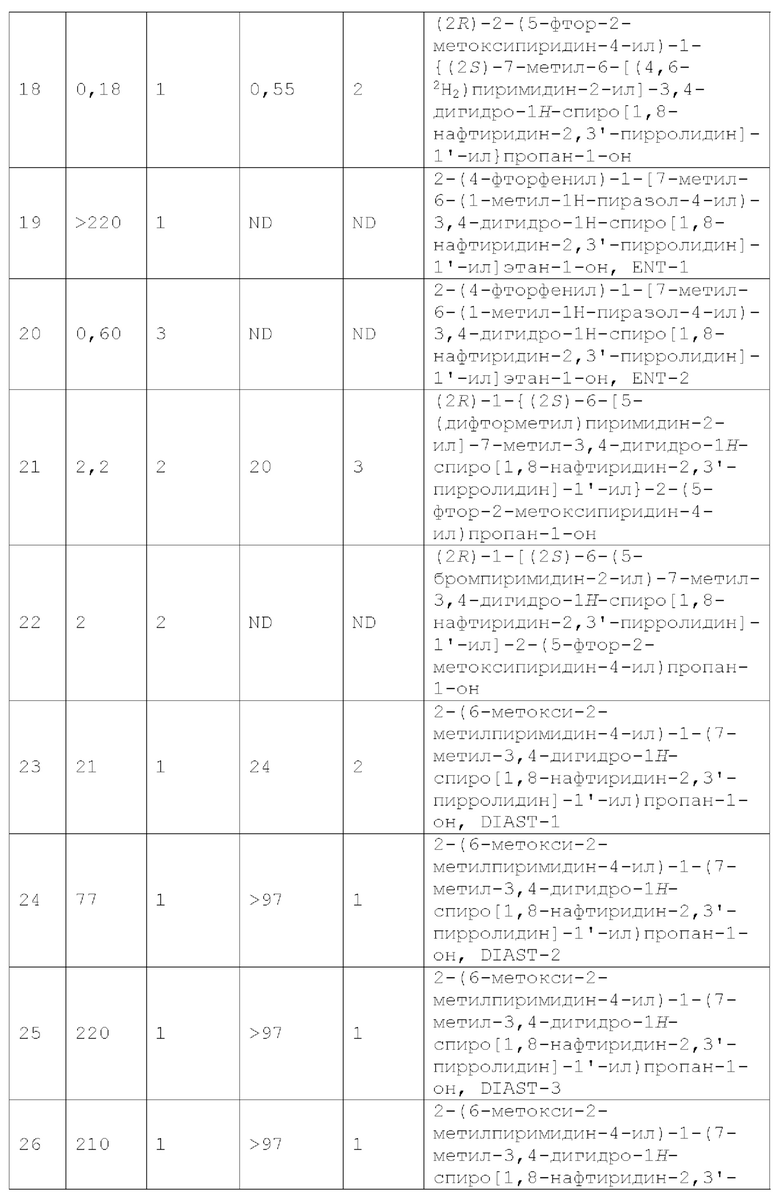
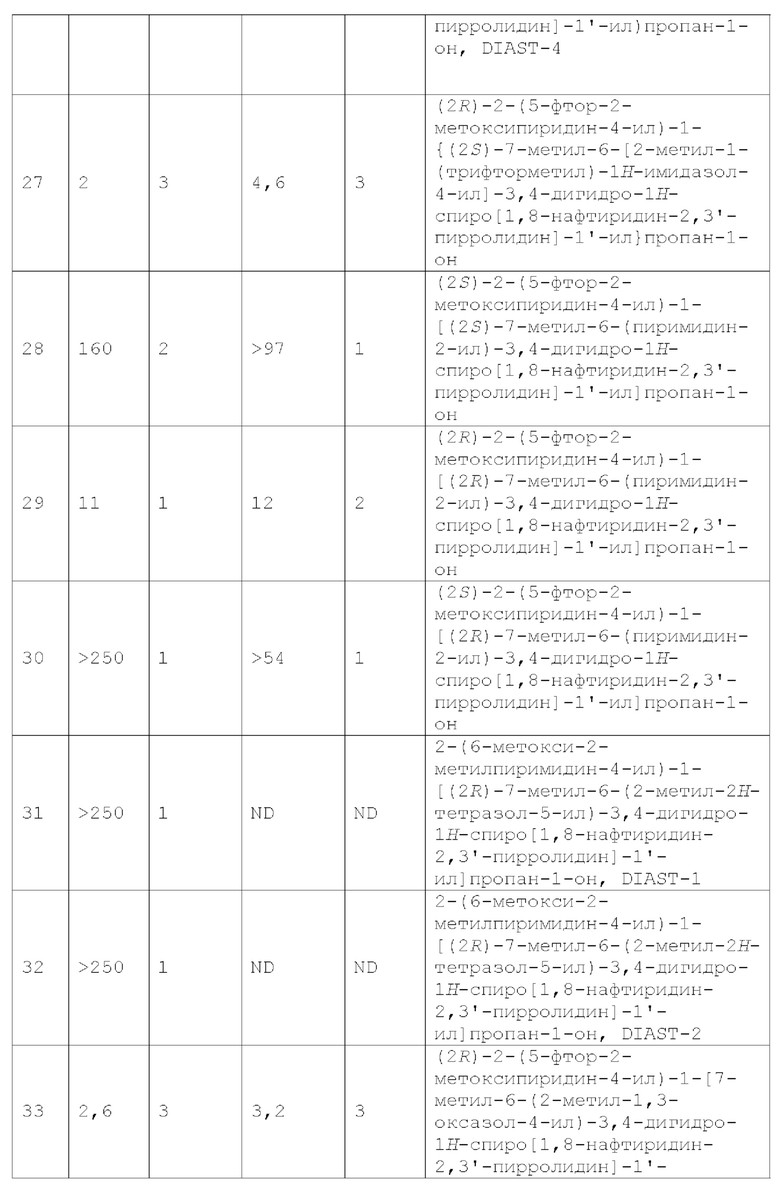
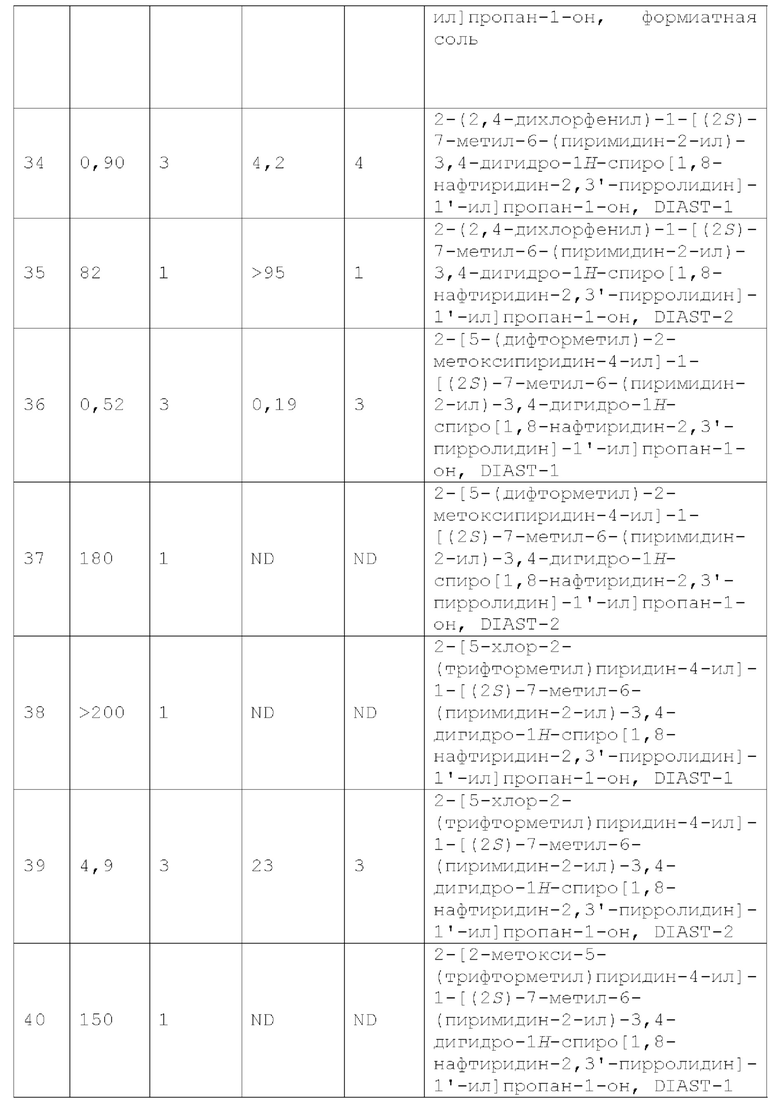
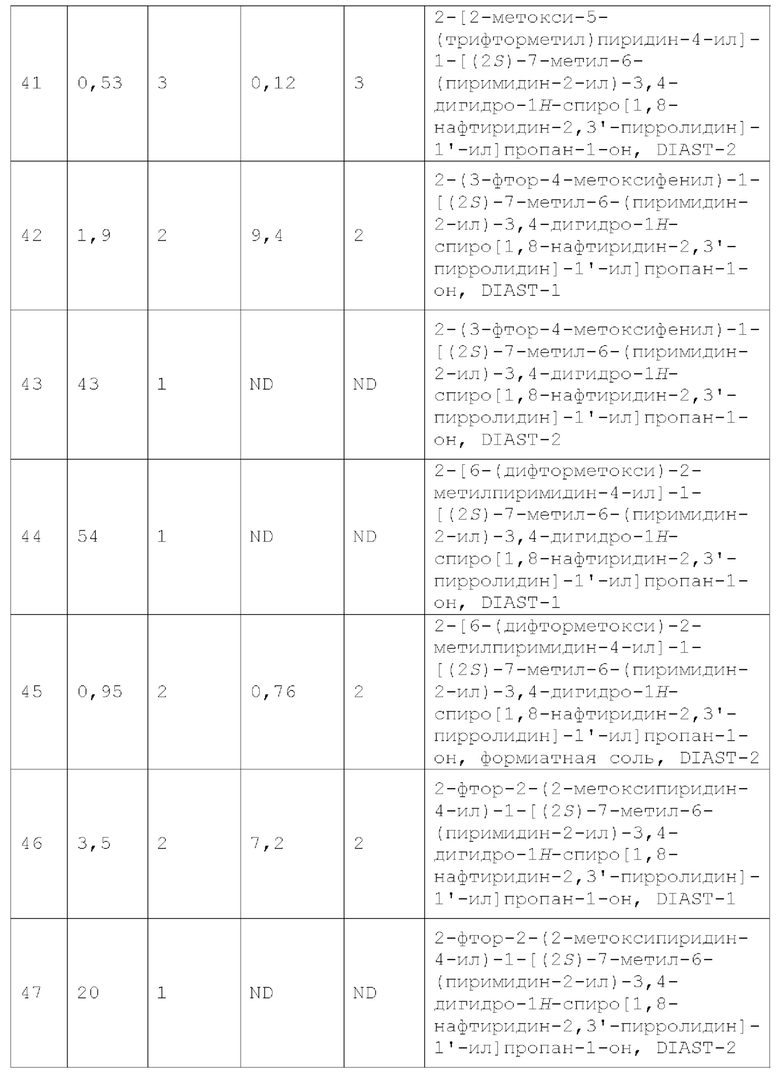
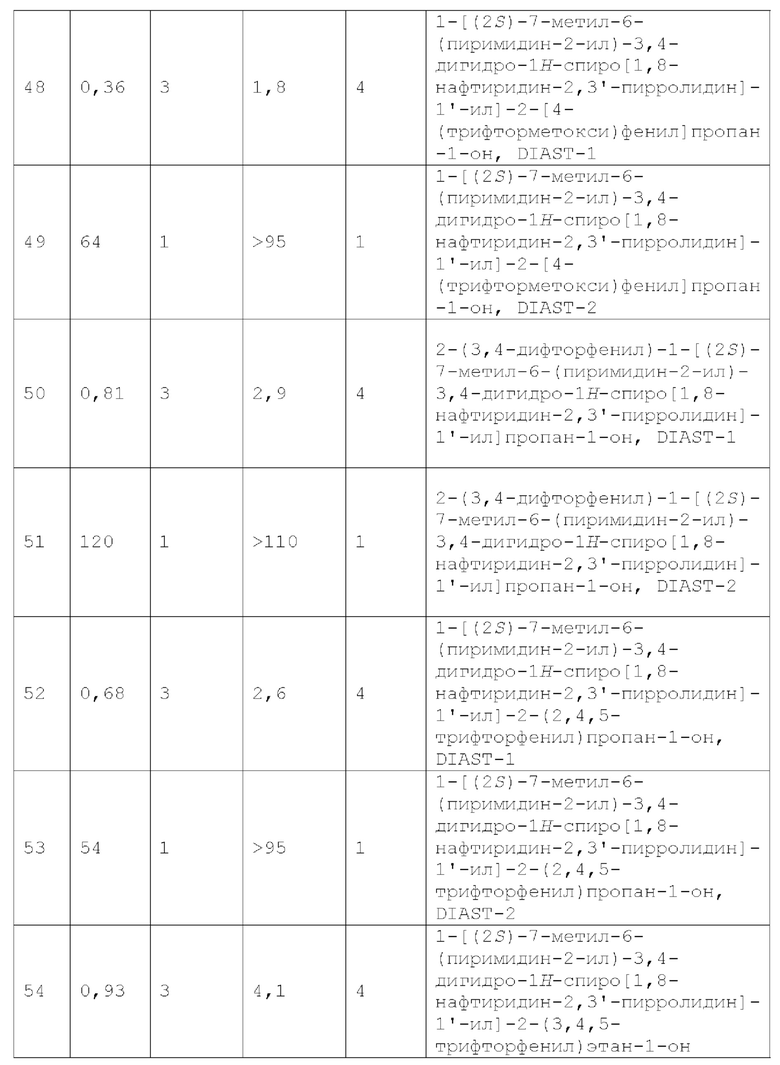
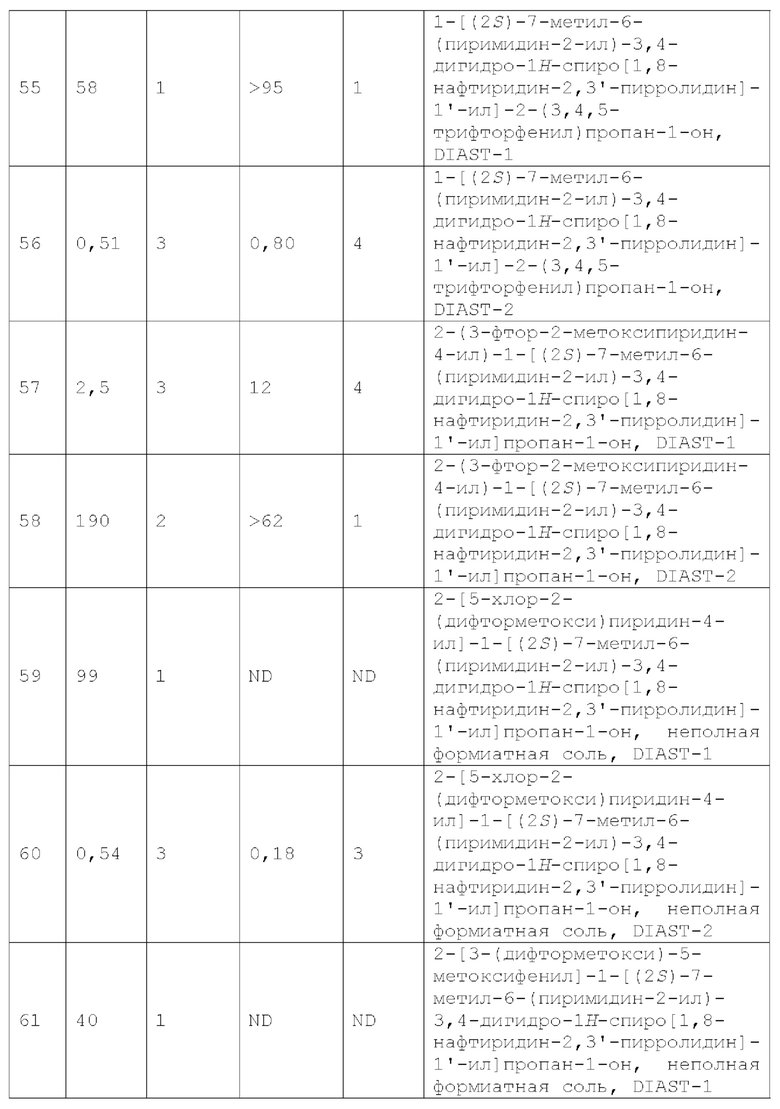
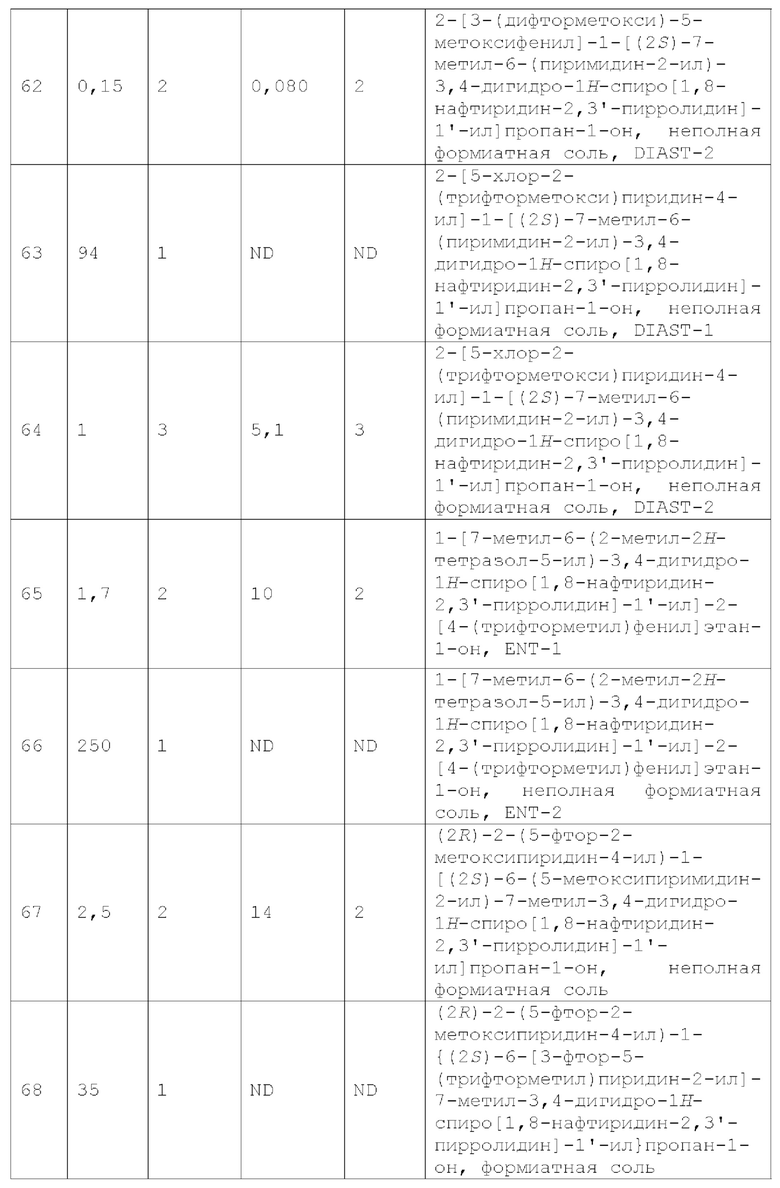
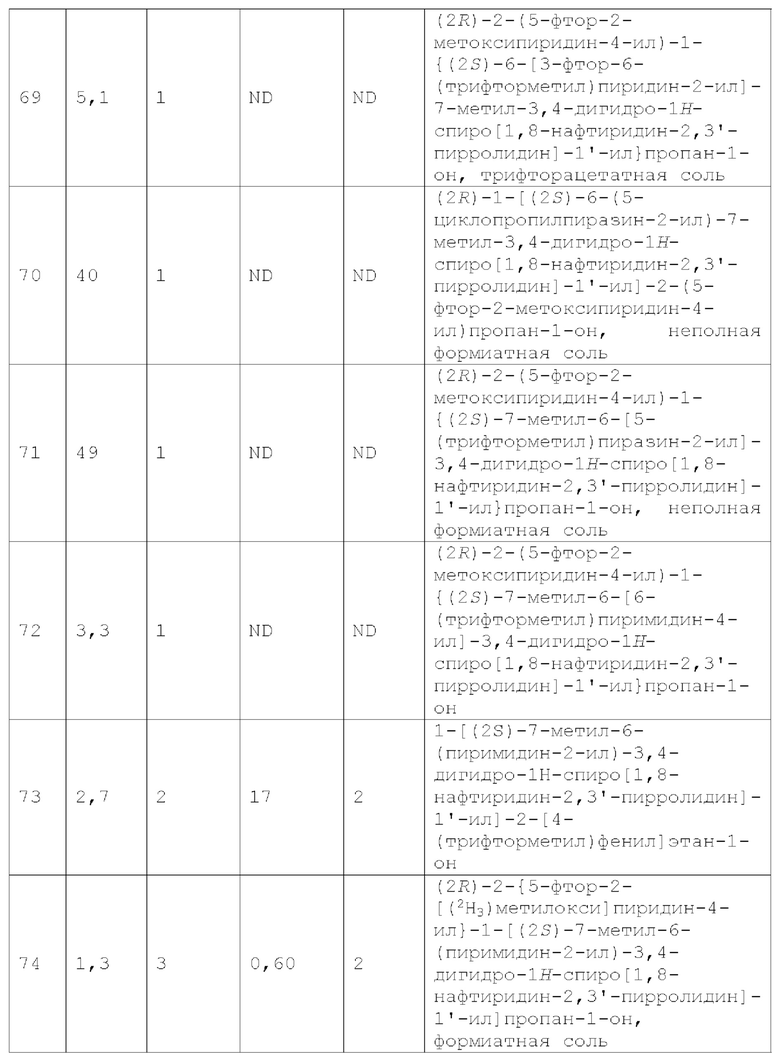
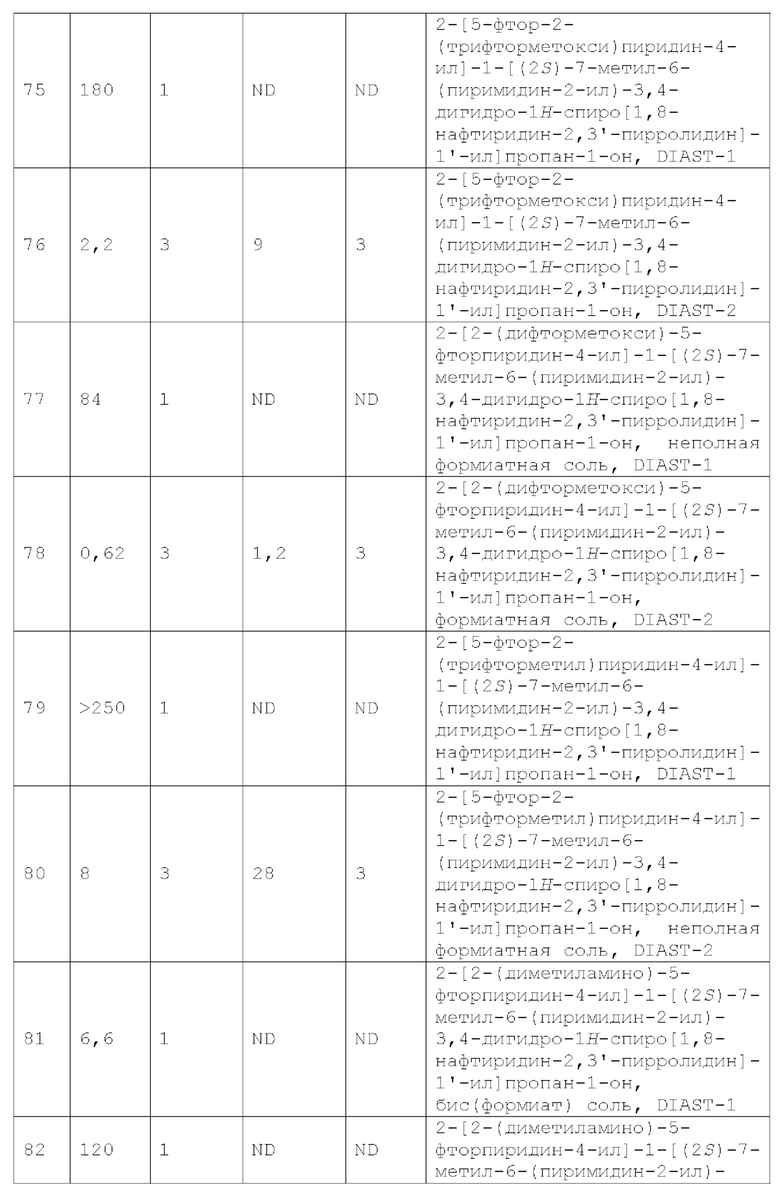
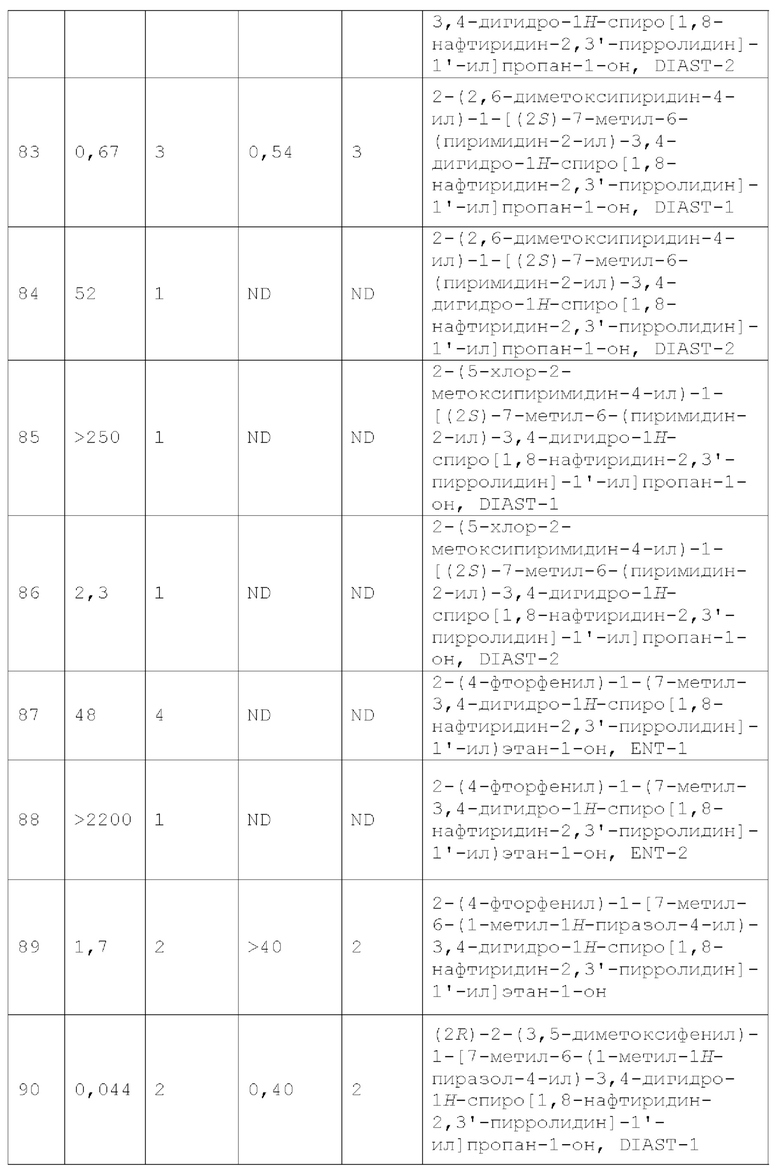
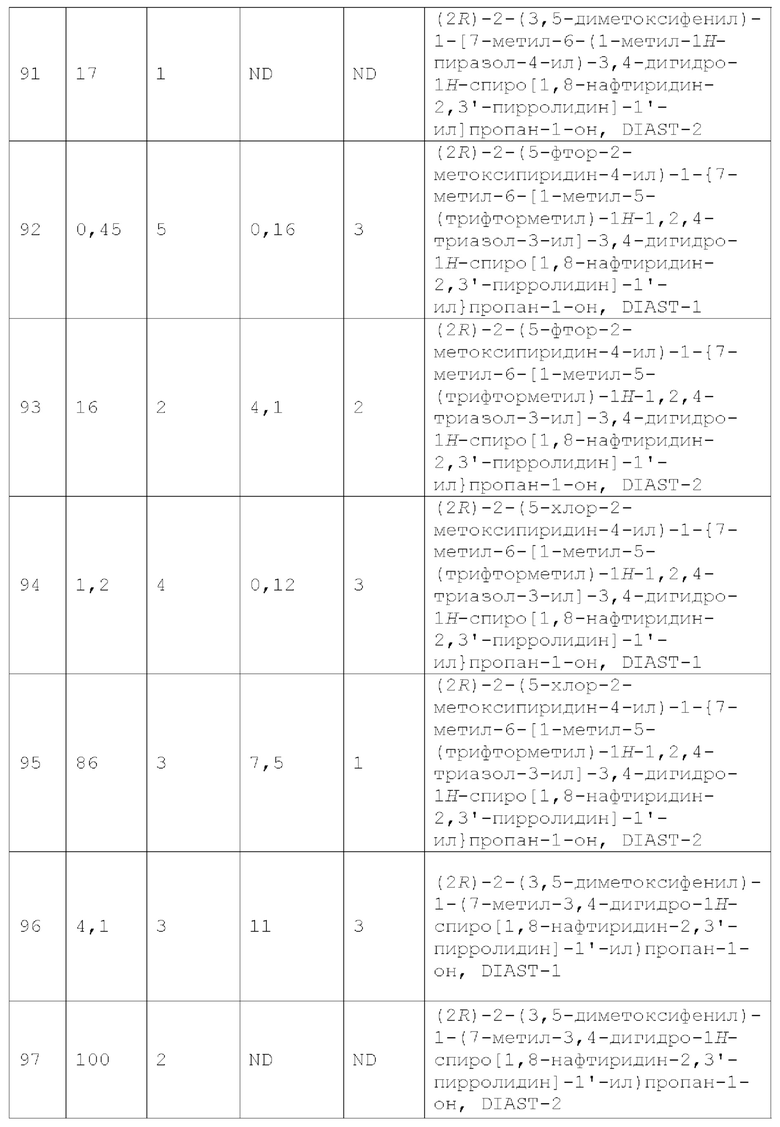
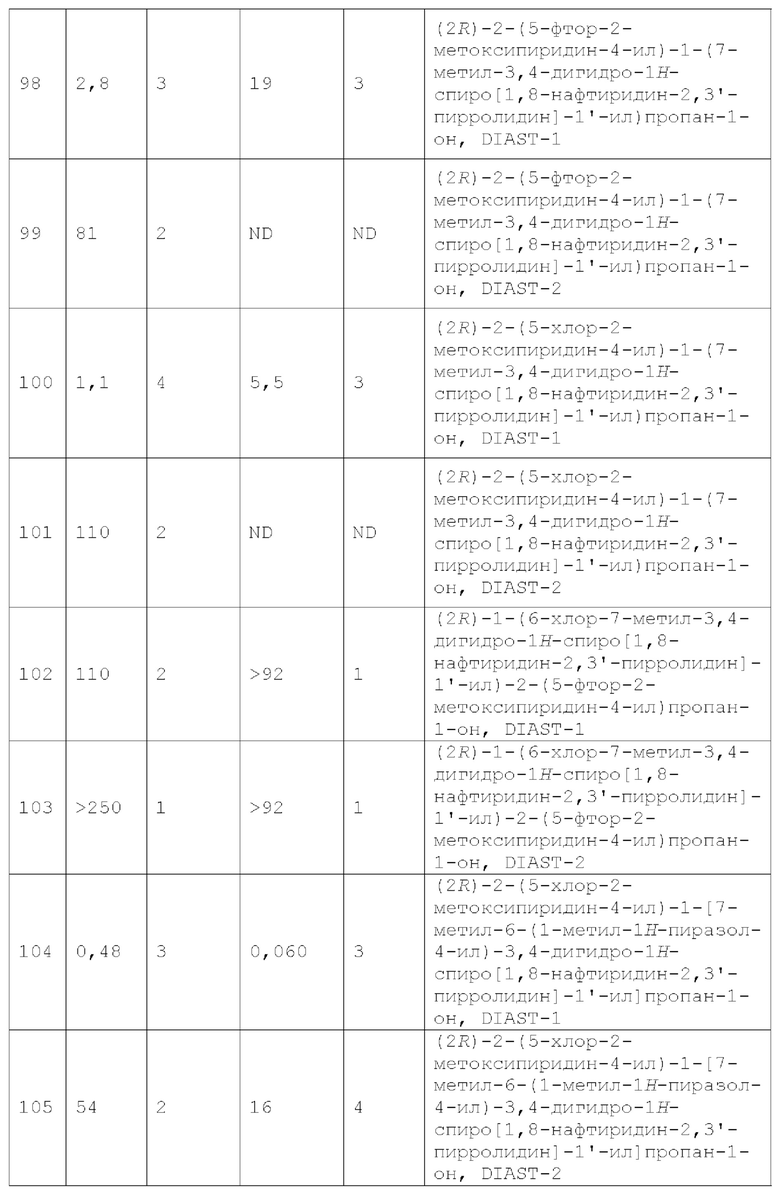
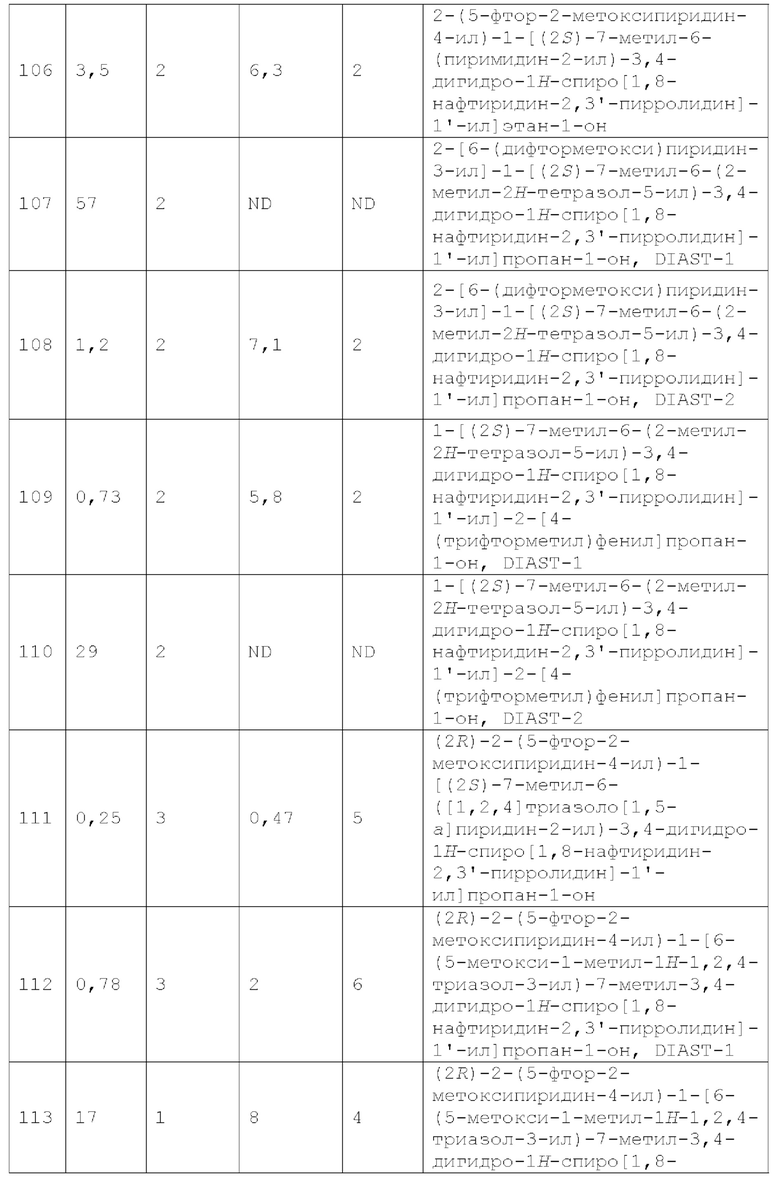
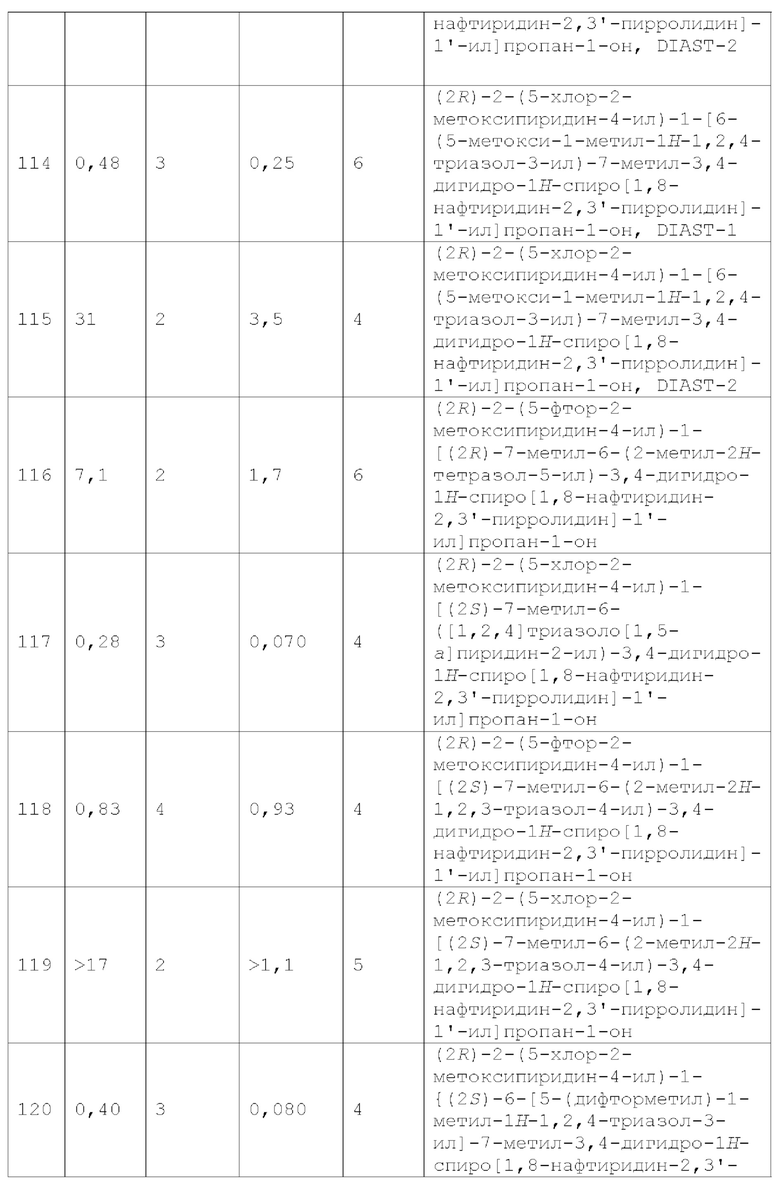
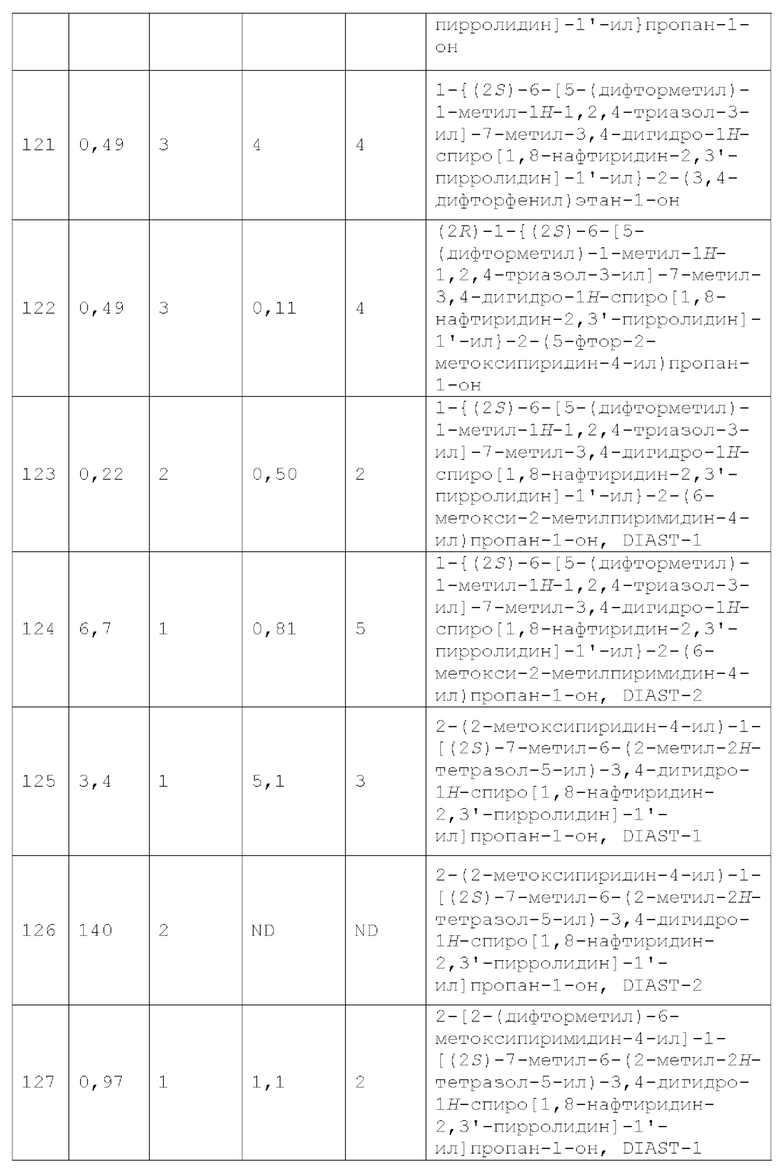
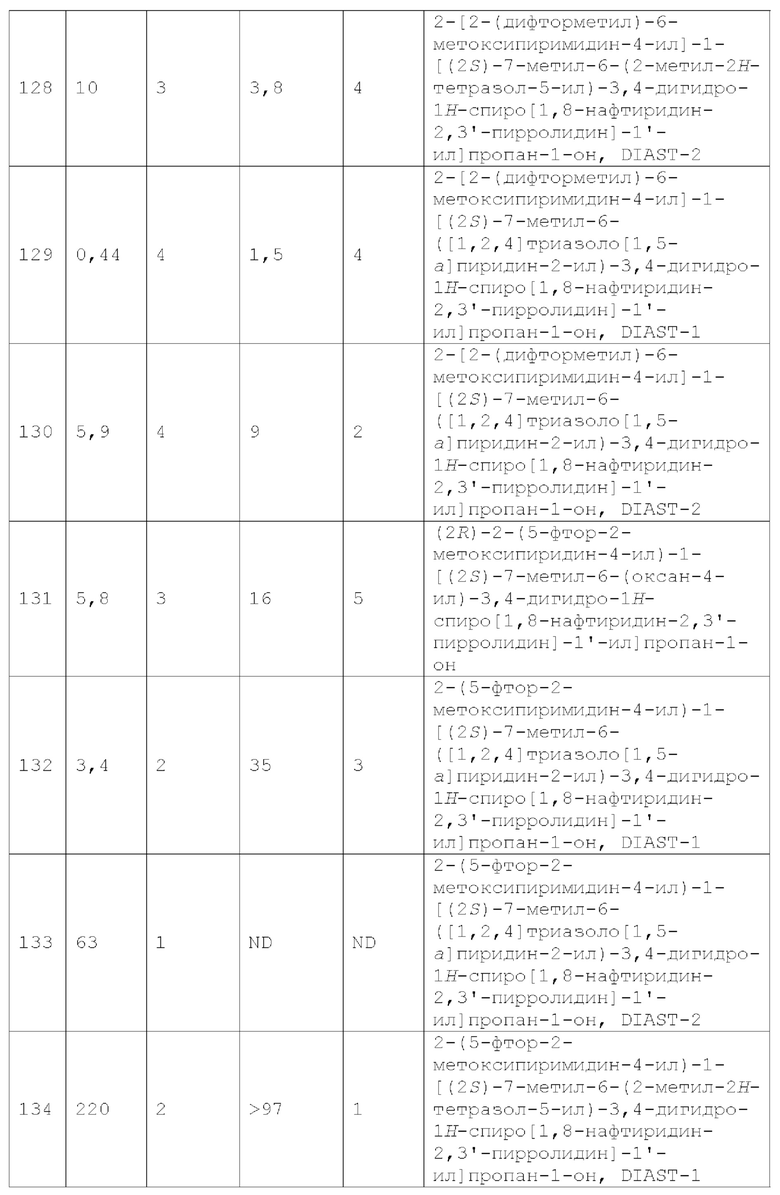
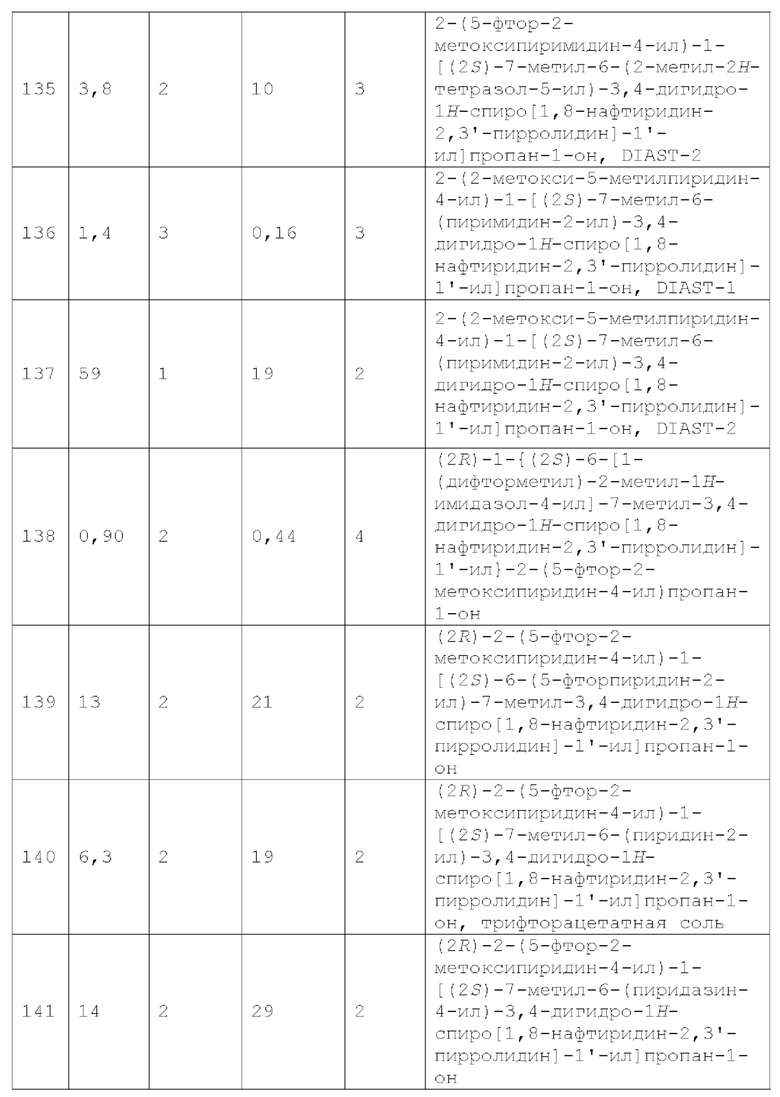
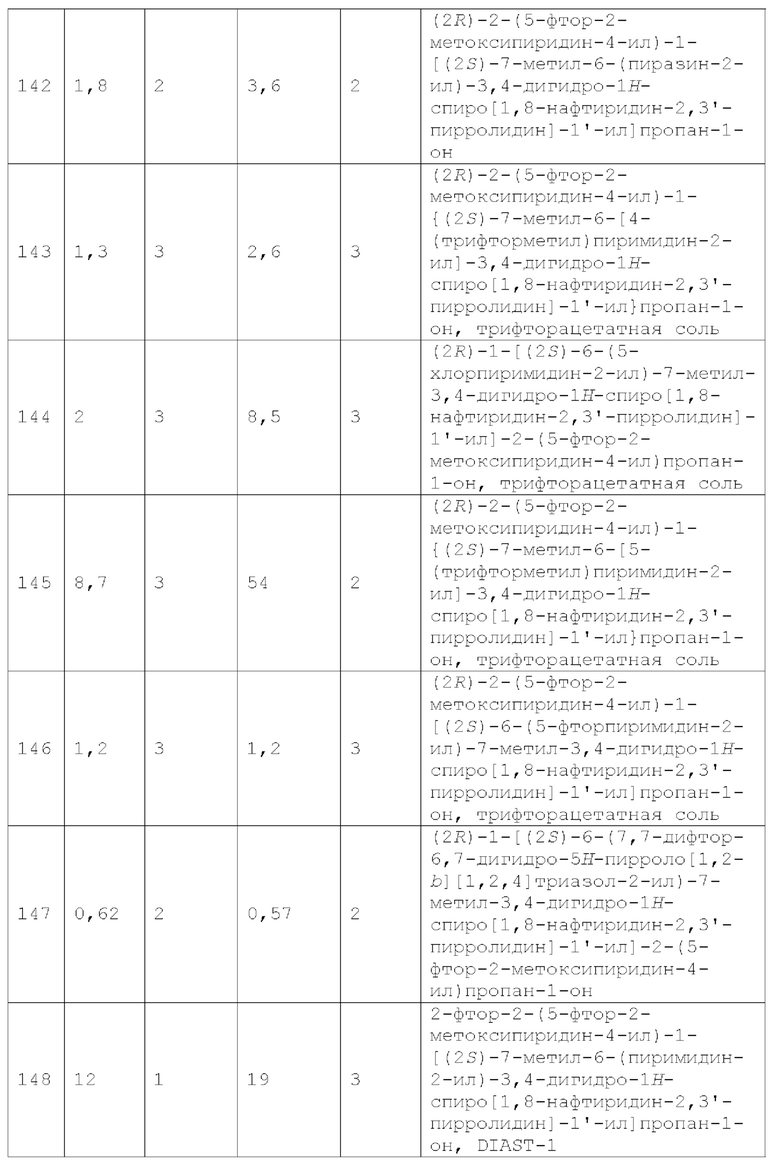
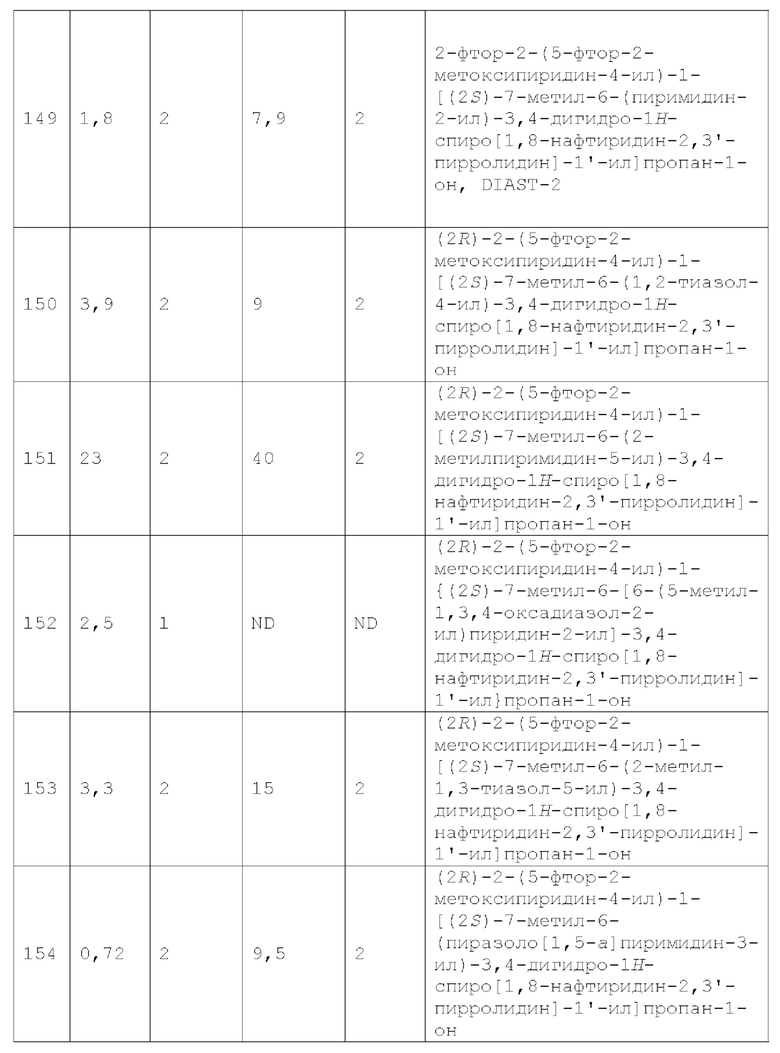
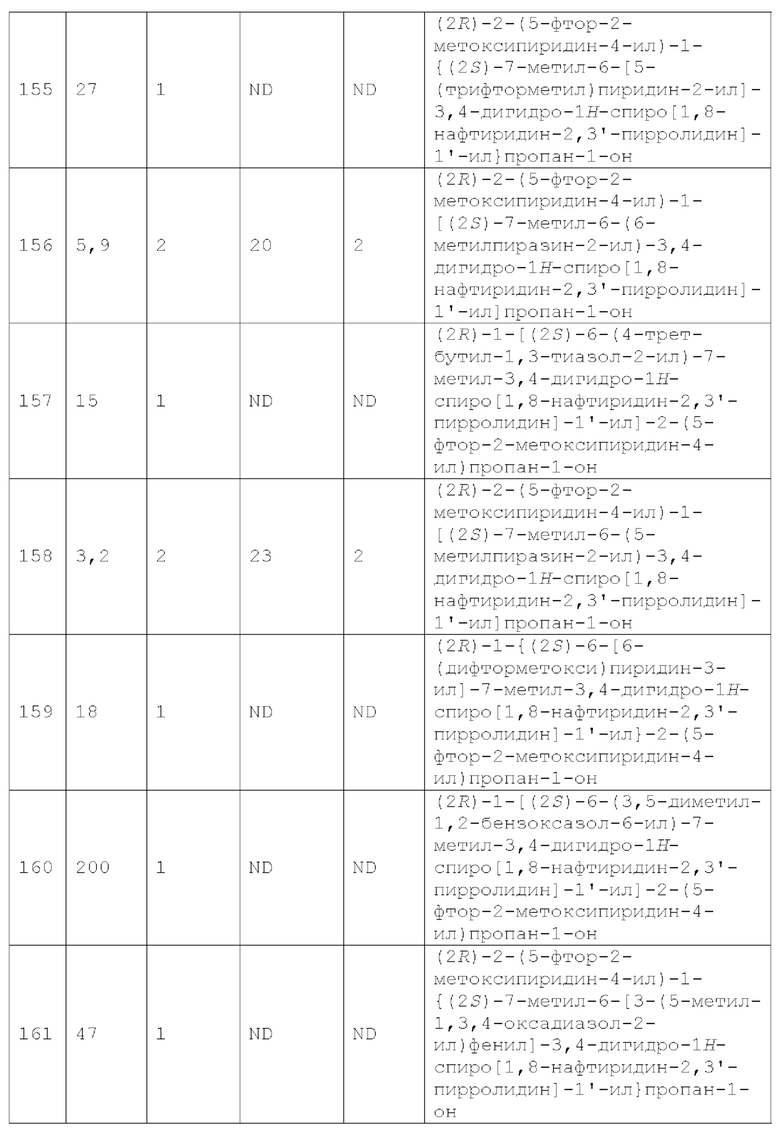
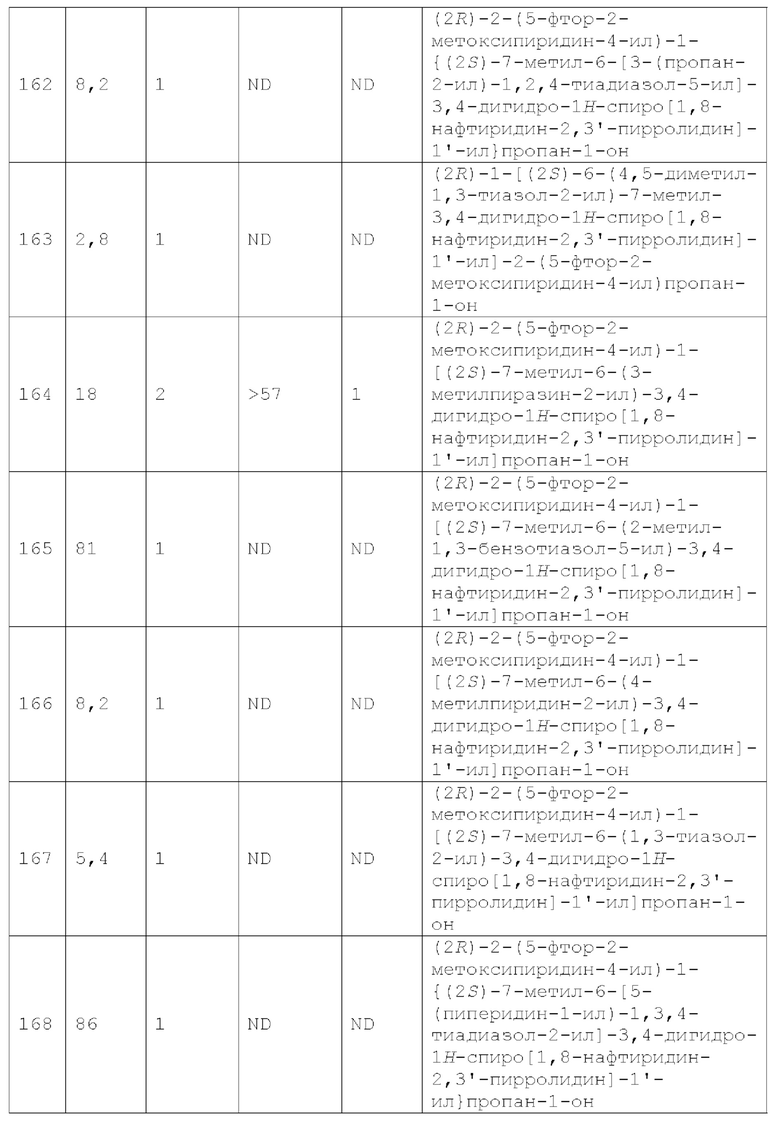
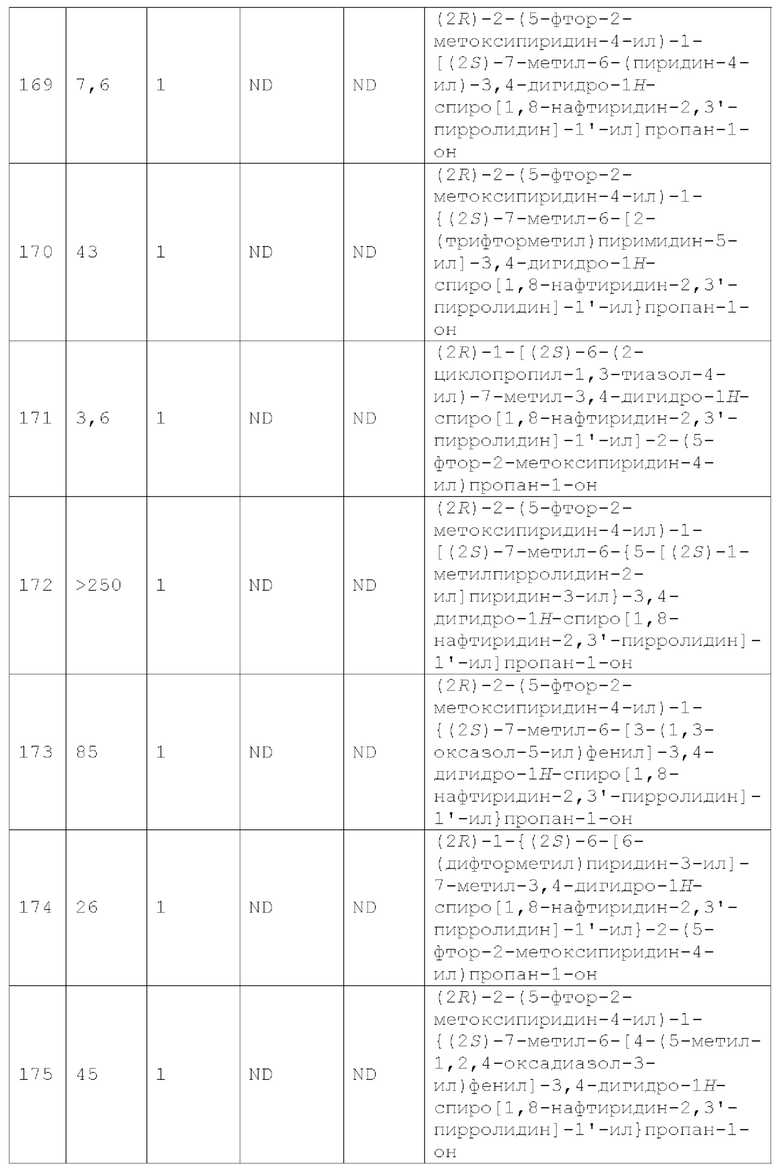

В настоящей заявке имеются ссылки на различные публикации. Полное раскрытие этих публикаций включено в настоящую заявку посредством ссылки для всех целей.
Специалистам в данной области техники будет очевидно, что в настоящее изобретение могут быть внесены различные модификации и изменения без отклонения от объема или сущности изобретения. Другие варианты осуществления изобретения будут очевидны специалистам в данной области техники из рассмотрения описания и практического применения изобретения, раскрытого в настоящей заявке. Предполагается, что описание и примеры должны рассматриваться только как иллюстративные, а истинный объем и сущность изобретения указаны в следующей формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| СОЕДИНЕНИЯ С КОНДЕНСИРОВАННЫМИ КОЛЬЦАМИ | 2019 |
|
RU2783414C2 |
| БЕНЗОКСАЗЕПИНОВЫЕ ИНГИБИТОРЫ PI3K И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2600927C2 |
| СОЕДИНЕНИЯ ПИРИДАЗИНАМИДА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ТИРОЗИНКИНАЗЫ СЕЛЕЗЕНКИ (SYK) | 2013 |
|
RU2627661C2 |
| БЕНЗОКСАЗЕПИНОВЫЕ ИНГИБИТОРЫ PI3 И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2654068C1 |
| ЗАМЕЩЕННЫЕ ИМИДАЗОПИРИДИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ HDM2 | 2013 |
|
RU2690663C2 |
| ЦИКЛОАЛКИЛНИТРИЛПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС-КИНАЗЫ | 2014 |
|
RU2655380C2 |
| Спироконденсированные пирролидиновые производные в качестве ингибиторов деубиквитилирующих ферментов (DUB) | 2017 |
|
RU2730552C2 |
| ПРОИЗВОДНЫЕ ПИРИДИНА И ИХ ПРИМЕНЕНИЕ ПРОТИВ МИКОБАКТЕРИЙ | 2015 |
|
RU2664587C1 |
| ПРОИЗВОДНЫЕ ХИНОЛИН-2-ОНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ C-KIT | 2016 |
|
RU2754858C2 |
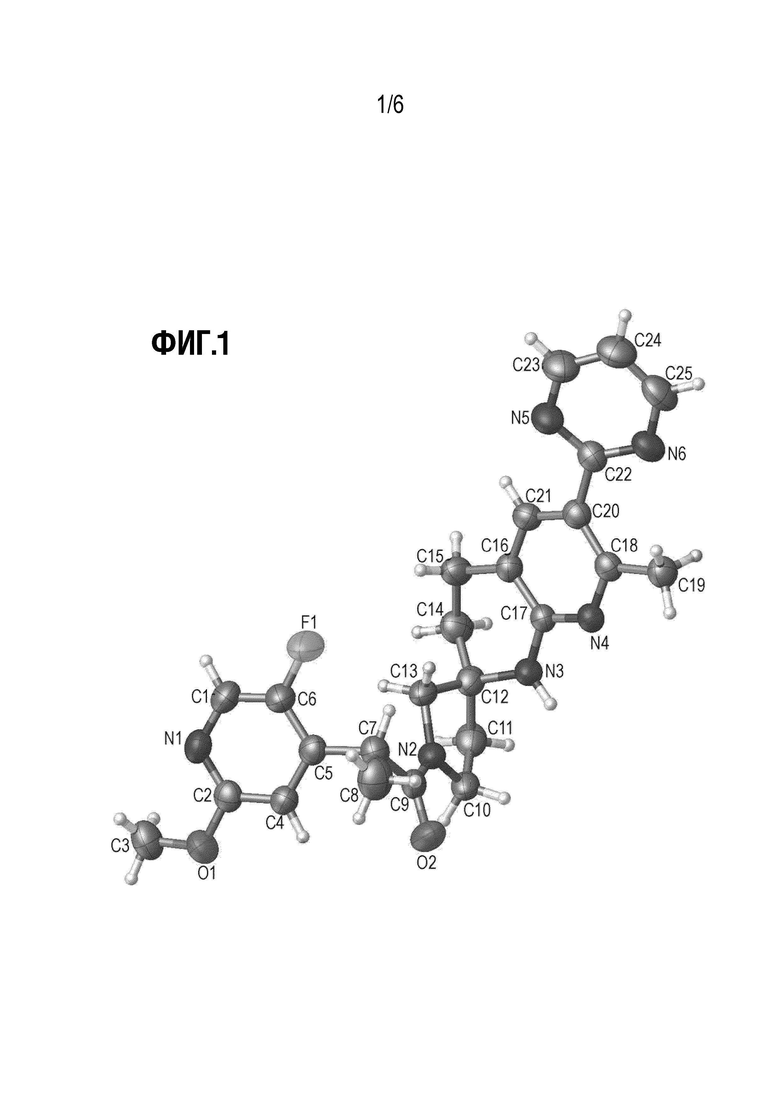
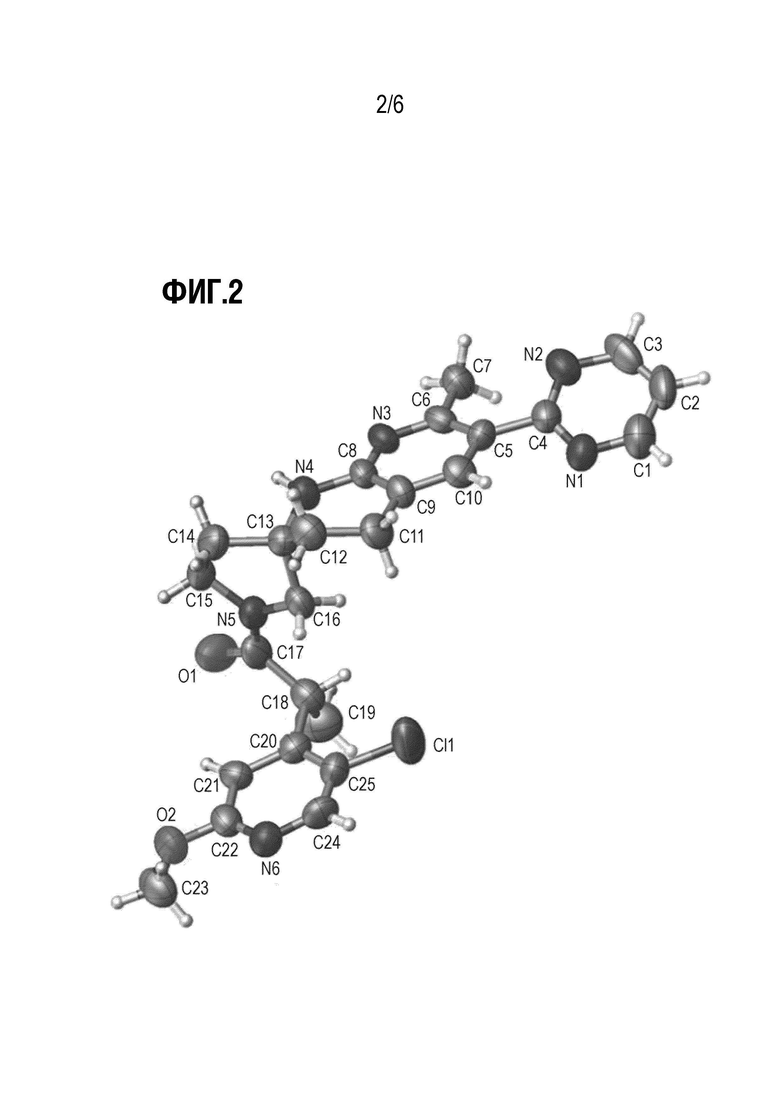
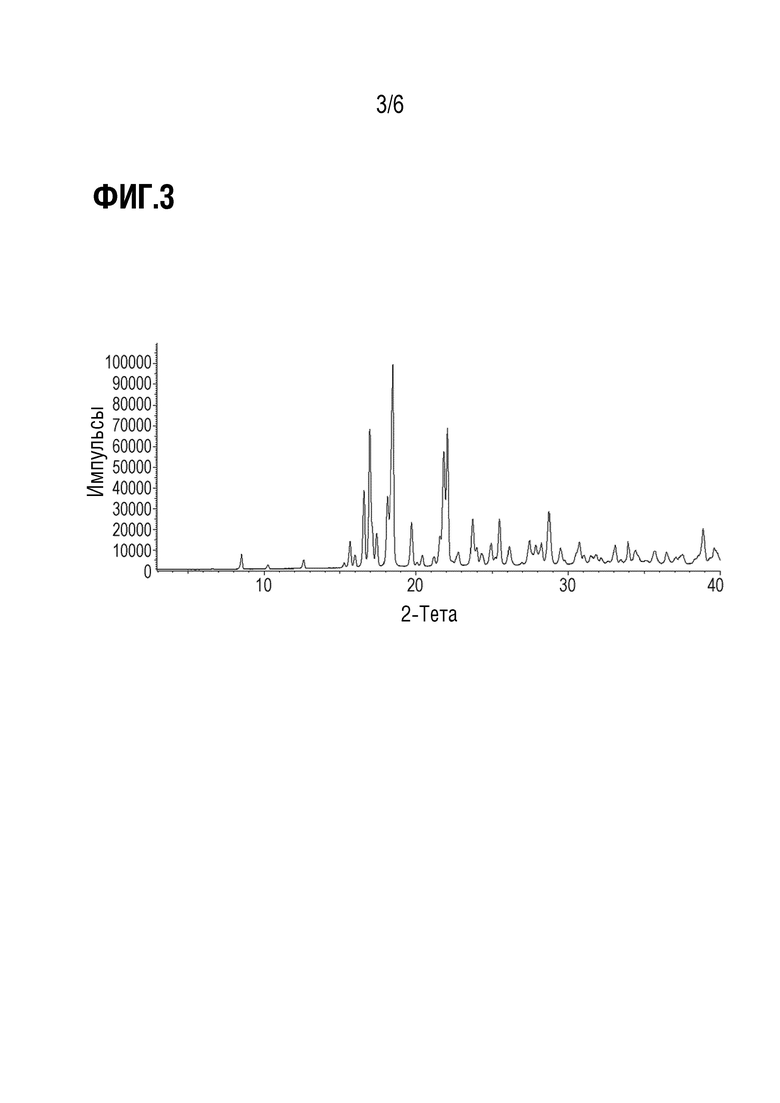
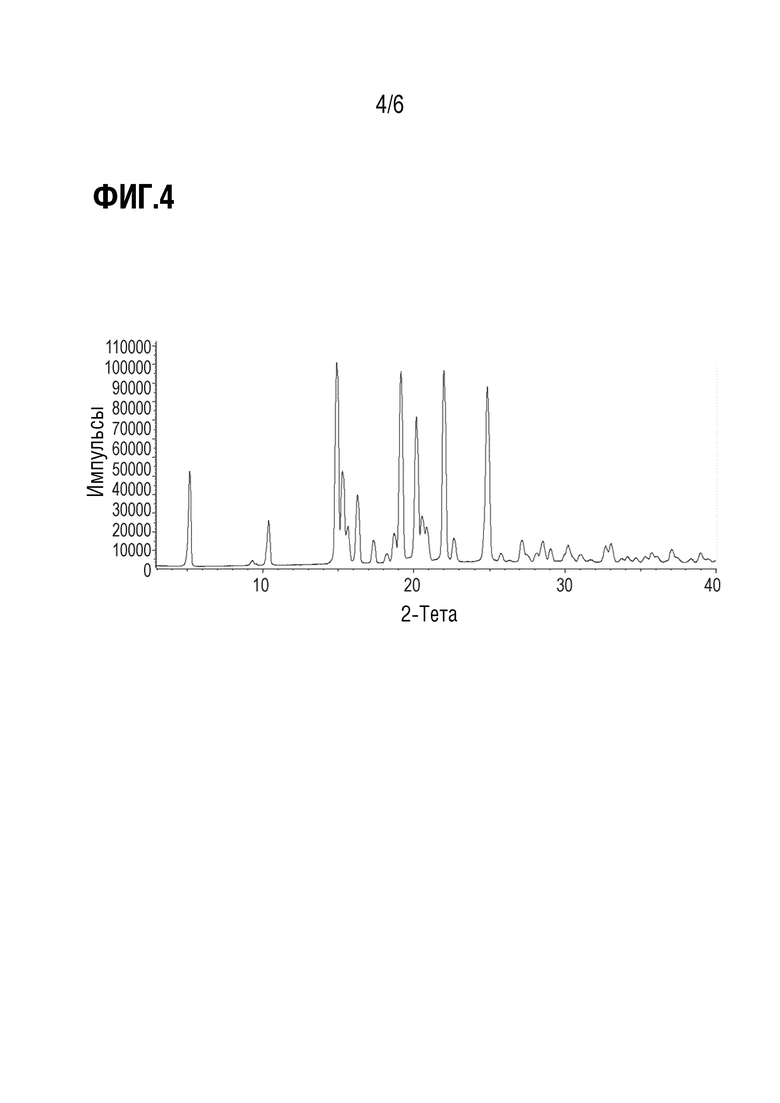
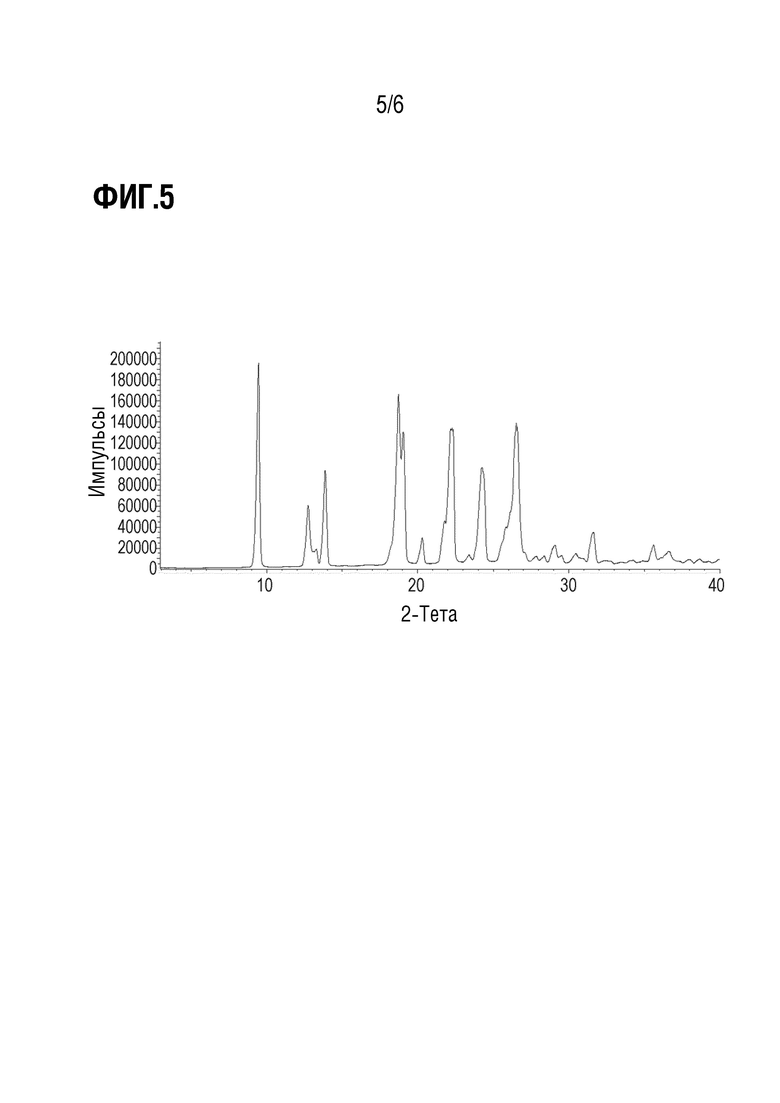
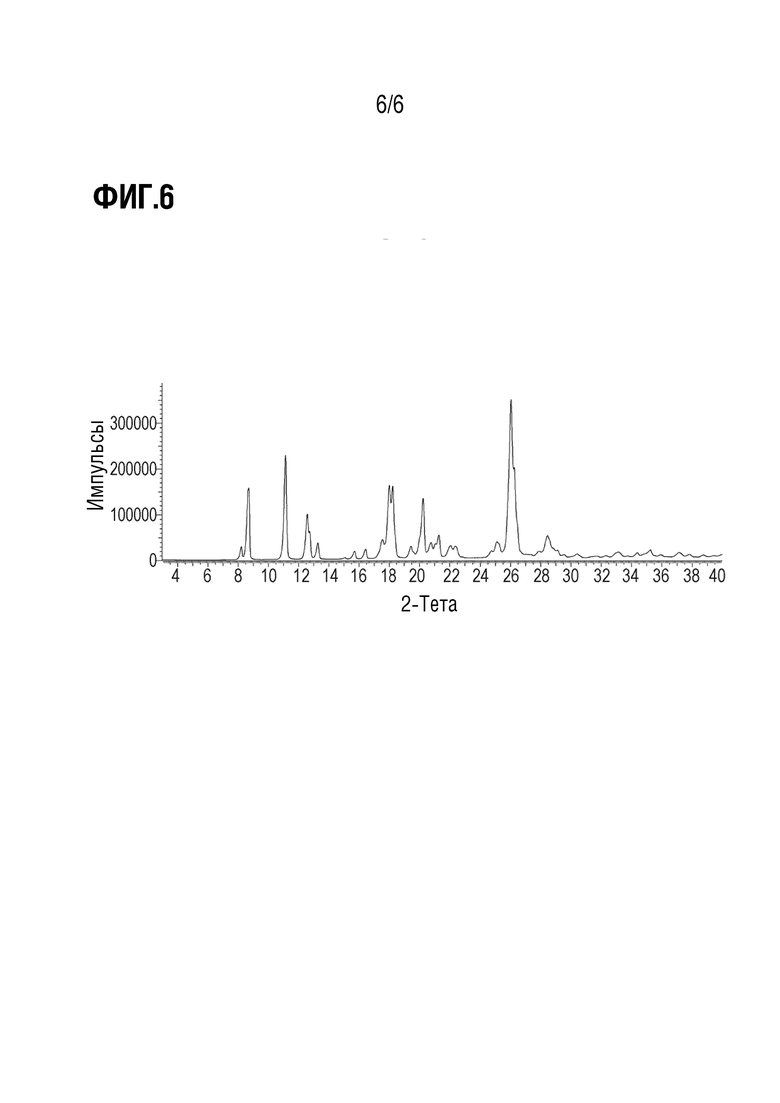
Изобретение относится к соединению формулы I, где R1, R2, R3, X1, Y1, Y2, Y3, Y4 и Y5 имеют значения, определенные в настоящей заявке, и его фармацевтически приемлемой соли, в качестве антагониста MC4R. Также предложены фармацевтические композиции, содержащие такие соединения и соли, и применение таких соединений и солей для лечения, например, кахексии, анорексии или нервной анорексии. 6 н. и 14 з.п. ф-лы, 6 ил., 14 табл., 201 пр.
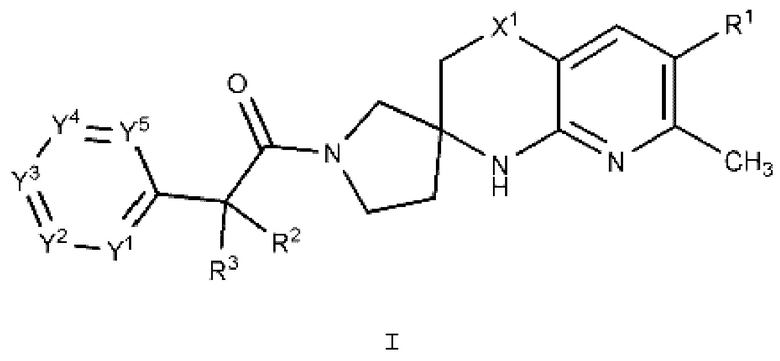
1. Соединение формулы I:
или его фармацевтически приемлемая соль, где:
R1 представляет собой Н, галоген, 4-7-членный гетероциклоалкил, фенил или R1a, где 4-7-членный гетероциклоалкил необязательно замещен 1, 2, 3 или 4 независимо выбранными С1-4 алкильными группами, и где фенил необязательно замещен 1 или 2 независимо выбранными RB1;
R1a представляет собой 5- или 6-членный гетероарил, необязательно замещенный 1 или 2 независимо выбранными RA, где каждый RA представляет собой галоген, C1-4 алкил, C1-4 галогеналкил, C1-4 алкокси, C1-4 галогеналкокси, С3-4 циклоалкил, -N(С1-4 алкил)2 или RA1, где два смежных RA вместе с двумя образующими кольцо атомами 5- или 6-членного гетероарила, к которым они присоединены, образуют конденсированный 5- или 6-членный гетероарил или конденсированный 5- или 6-членный гетероциклоалкил, каждый из которых необязательно замещен 1 или 2 заместителями, каждый из которых независимо выбран из галогена или С1-4 алкила;
RA1 представляет собой 5-или 6-членный гетероциклоалкил, который необязательно замещен C1-4 алкилом;
RB1 представляет собой 5- или 6-членный гетероарил, который необязательно замещен C1-4 алкилом;
X1 представляет собой C(RX)2, где каждый RX независимо представляет собой Н или C1-4 алкил;
каждый из R2 и R3 независимо представляет собой Н, галоген или C1-4 алкил;
каждый из Y1, Y2, Y3, Y4 и Y5 независимо представляет собой CR4 или N, при условии, что не более чем 3 из Y1, Y2, Y3, Y4 и Y5 представляют собой N; и
каждый R4 независимо представляет собой Н, галоген, C1-4 алкил, С1-4 галогеналкил, С1-4 алкокси или C1-4 галогеналкокси;
где каждый гетероатом независимо выбран из N, O и S.
2. Соединение или фармацевтически приемлемая соль по п. 1, где соединение представляет собой соединение формулы Ia:
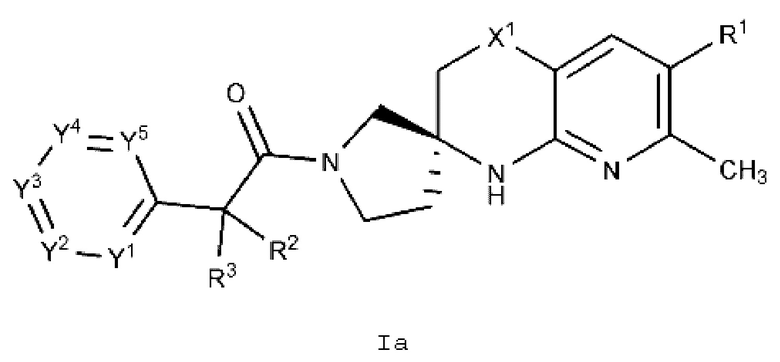
или его фармацевтически приемлемую соль.
3. Соединение или фармацевтически приемлемая соль по п. 1 или 2, где соединение представляет собой соединение формулы II:
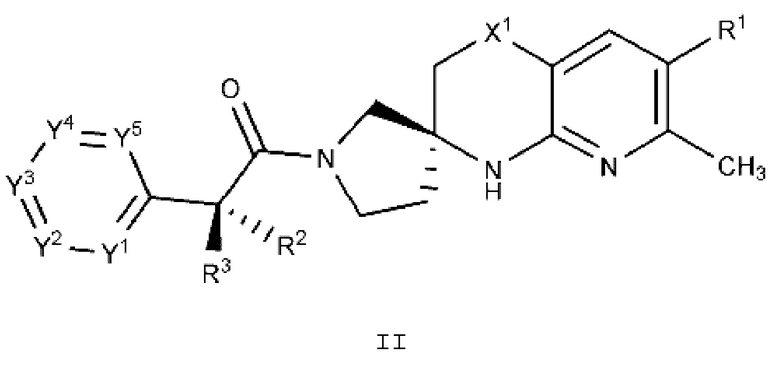
или его фармацевтически приемлемую соль.
4. Соединение или фармацевтически приемлемая соль по любому из пп. 1-3, где соединение представляет собой соединение формулы III:
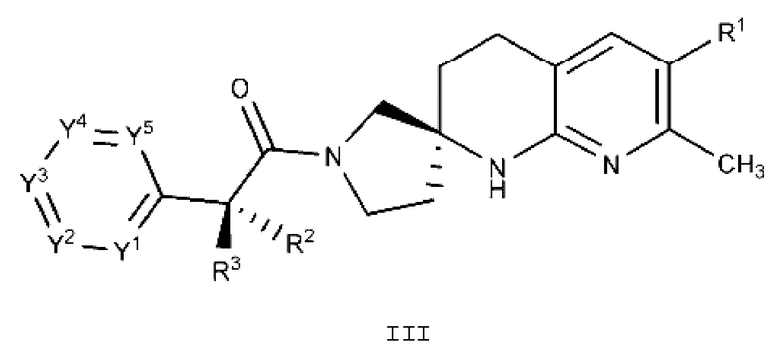
или его фармацевтически приемлемую соль.
5. Соединение или фармацевтически приемлемая соль по любому из пп. 1-4, где:
R1 представляет собой R1a; и
R1a представляет собой 6-членный гетероарил, необязательно замещенный 1 или 2 независимо выбранными RA, где каждый RA представляет собой галоген, С1-4 алкил, С1-4 галогеналкил, С1-4 алкокси, С1-4 галогеналкокси или С3-4 циклоалкил.
6. Соединение или фармацевтически приемлемая соль по п. 5, где R1a представляет собой пиримидинил, необязательно замещенный 1 или 2 независимо выбранными RA, где каждый RA представляет собой галоген, C1-4 алкил, C1-4 галогеналкил, C1-4 алкокси, C1-4 галогеналкокси или С3-4 циклоалкил.
7. Соединение или фармацевтически приемлемая соль по п. 6, где R1a представляет собой пиримидин-2-ил.
8. Соединение или фармацевтически приемлемая соль по любому из пп. 1-3 и 5-7, где X1 представляет собой СН2.
9. Соединение или фармацевтически приемлемая соль по любому из пп. 1-8, где каждый из R2 и R3 независимо представляет собой Н или C1-4 алкил.
10. Соединение или фармацевтически приемлемая соль по любому из пп. 1-9, где R2 представляет собой метил, a R3 представляет собой Н.
11. Соединение или фармацевтически приемлемая соль по любому из пп. 1-10, где Y3 представляет собой N, а каждый из Y1, Y2, Y4 и Y5 независимо представляет собой CR4.
12. Соединение или фармацевтически приемлемая соль по любому из пп. 1-11, где каждый R4 независимо представляет собой Н, галоген или C1-2 алкокси.
13. Соединение по п. 1, выбранное из:
(2R)-2-(5-хлор-2-метоксипиридин-4-ил)-1-[7-метил-6-(2-метил-2H-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она, DIAST-1;
2-(6-метокси-2-метилпиримидин-4-ил)-1-[(2S)-7-метил-6-(2-метил-2H-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она, DIAST-1;
2-[6-(дифторметокси)пиридин-3-ил]-1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она, DIAST-2;
1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]-2-[4-(трифторметил)фенил]пропан-1-она, DIAST-1;
1-(4,7-диметил-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил)-2-(4-фторфенил)этан-1-она, DIAST-1;
(2R)-2-(5-фтор-2-метоксипиридин-4-ил)-1-[(2S)-7-метил-6-(2-метил-2H-тетразол-5-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она;
(2R)-2-(5-фтор-2-метоксипиридин-4-ил)-1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она;
(2R)-2-(5-хлор-2-метоксипиридин-4-ил)-1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она;
(2R)-2-(5-фтор-2-метоксипиридин-4-ил)-1-[7-метил-6-(1-метил-1H-пиразол-4-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она, DIAST-1; и
(2R)-2-(5-фтор-2-метоксипиридин-4-ил)-1-{(2S)-7-метил-6-[(4,6-2Н2)пиримидин-2-ил]-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил}пропан-1-она,
или его фармацевтически приемлемая соль.
14. Соединение по п. 1, которое представляет собой (2R)-2-(5-фтор-2-метоксипиридин-4-ил)-1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-он или его фармацевтически приемлемую соль.
15. Соединение по п. 1, которое представляет собой кристаллическую форму (2R)-2-(5-фтор-2-метоксипиридин-4-ил)-1-[(2S)-7-метил-6-(пиримидин-2-ил)-3,4-дигидро-1H-спиро[1,8-нафтиридин-2,3'-пирролидин]-1'-ил]пропан-1-она.
16. Фармацевтическая композиция, содержащая терапевтически эффективное количество соединения или фармацевтически приемлемой соли по любому из пп. 1-15 и фармацевтически приемлемый носитель, где соединение обладает антагонистической активностью в отношении MC4R.
17. Способ лечения MC4R-связанного состояния, заболевания или расстройства у человека, где способ включает введение человеку, нуждающемуся в этом, соединения или фармацевтически приемлемой соли по любому из пп. 1-15 в терапевтически эффективном количестве.
18. Способ лечения MC4R-связанного состояния, заболевания или расстройства у человека, где способ включает введение человеку, нуждающемуся в этом, соединения или фармацевтически приемлемой соли по любому из пп. 1-15 в терапевтически эффективном количестве, где состояние, заболевание или расстройство выбрано из кахексии [включая, например, кахексию, связанную с хроническим заболеванием, такую как кахексия, связанная с раком, кахексия, связанная с синдромом приобретенного иммунодефицита (СПИДом), кахексия, связанная с сердечной недостаточностью, например кахексия, связанная с застойной сердечной недостаточностью (CHF), кахексия, связанная с хроническим заболеванием почек (CKD); кахексия, связанная с лечением хронического заболевания, такая как кахексия, связанная с лечением рака, или кахексия, связанная с лечением сердечной недостаточности (например, СНГ)]; анорексии или нервной анорексии (например, анорексии у пожилых людей, анорексии, связанной с химиотерапией и/или лучевой терапией); тошноты; рвоты; потери массы тела (например, непреднамеренной потери массы тела); задержки развития; саркопении; мышечной атрофии; мышечной слабости; немощности; остеопороза; заболеваний костей (например, потери костной массы); боли; невропатической боли; беспокойства (например, посттравматического стрессового расстройства, или ПТСР); депрессии; гипертензии; нарушения питания; ожирения (например, саркопении в результате хронического ожирения); сексуальной дисфункции; и воспалительного заболевания (например, воспалительного заболевания, связанного с анорексией, или кахексией, или саркопенией, или мышечной атрофией).
19. Применение соединения или фармацевтически приемлемой соли по любому из пп. 1-15 в терапевтически эффективном количестве в лечении MC4R-связанного состояния, заболевания или расстройства, выбранного из кахексии [включая, например, кахексию, связанную с хроническим заболеванием, такую как кахексия, связанная с раком, кахексия, связанная с синдромом приобретенного иммунодефицита (СПИДом), кахексия, связанная с сердечной недостаточностью, например кахексия, связанная с застойной сердечной недостаточностью (CHF), кахексия, связанная с хроническим заболеванием почек (CKD); кахексия, связанная с лечением хронического заболевания, такая как кахексия, связанная с лечением рака, или кахексия, связанная с лечением сердечной недостаточности (например, CHF)]; анорексии или нервной анорексии (например, анорексии у пожилых людей, анорексии, связанной с химиотерапией и/или лучевой терапией); тошноты; рвоты; потери массы тела (например, непреднамеренной потери массы тела); задержки развития; саркопении; мышечной атрофии; мышечной слабости; немощности; остеопороза; заболеваний костей (например, потери костной массы); боли; невропатической боли; беспокойства (например, посттравматического стрессового расстройства, или ПТСР); депрессии; гипертензии; нарушения питания; ожирения (например, саркопении в результате хронического ожирения); сексуальной дисфункции; и воспалительного заболевания (например, воспалительного заболевания, связанного с анорексией, или кахексией, или саркопенией, или мышечной атрофией).
20. Способ антагонизирования рецептора меланокортина-4 (MC4R), включающий контактирование MC4R с соединением или фармацевтически приемлемой солью по любому из пп. 1-15.
| EP 2003131 A1, 17.12.2008 | |||
| RU 2008116844 A, 10.11.2009 | |||
| ALAN C | |||
| FOSTER et al | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Походная разборная печь для варки пищи и печения хлеба | 1920 |
|
SU11A1 |

