В настоящей заявке испрашивается приоритет в соответствии 35 U.S.C. § 119 предварительных заявок США под номером 60/426681, зарегистрированной 15 ноября 2002 и озаглавленной «Compositions Useful as Inhibitors of Protein Kinase», и номером 60/447705, зарегистрированной 11 февраля 2003 и озаглавленной «Compositions Useful as Inhibitors of Protein Kinase», причем полное содержание каждой из этих заявок, таким образом, включено в качестве ссылки.
Настоящее изобретение относится к ингибиторам протеинкиназ. Изобретение относится также к фармацевтическим композициям, содержащим соединения изобретения и способам применения композиций при лечении различных заболеваний.
Поиску новых терапевтических агентов значительно помогло в последние годы лучшее понимание структуры ферментов и других биомолекул, ассоциированных с заболеваниями. Одним важным классом ферментов, которые были предметом экстенсивного исследования, являются протеинкиназы.
Протеинкиназы составляют большое семейство структурно близких ферментов, которые являются ответственными за контроль различных процессов сигнальной трансдукции в клетке. (См. Hardie G. and Hanks, S. The Protein Kinase Facts Book, I and II, Academic Press, San Diego, CA: 1995). Считается, что протеинкиназы возникли из обычного предкового гена вследствие консервации их структуры и каталитической функции. Почти все киназы содержат похожий каталитический домен из 250-300 аминокислот. Киназы могут быть группированы на классы по субстратам, которые они фосфорилируют (например, протеинтирозин, протеинсерин/треонин, липиды и тому подобное). Идентифицированы мотивы последовательности, которые обычно соответствуют каждому из этих семейств киназ (см., например, Hanks, S.K. Hunter, T., FASEB J. 1995, 9, 576-596; Knighton et al., Science, 1991, 253, 407-414; Hiles et al., Cell 1992, 70, 419-429, Kunz et al., Cell 1993, 73, 585-596; Carcia-Bustos et al., EMBO J. 1994, 13, 2352-2361).
В общем, протеинкиназы опосредуют внутриклеточную передачу сигнала действием переноса фосфорила от нуклеозидтрифосфата к акцептору-белку, который принимает участие в пути переноса сигнала. Эти явления фосфорилирования действуют в качестве молекулярных включений/выключений, которые могут модулировать или регулировать биологическую функцию целевого белка. Эти события фосфорилирования в конце концов запускаются в ответ на различные внеклеточные и другие стимулы. Примеры таких стимулов включают в себя сигналы стрессов окружающей среды и химических стрессов (например, осмотического шока, теплового шока, ультрафиолетового излучения, бактериального эндотоксина и Н2О2), цитокинов (например, интерлейкина-1 (IL-1) и фактора α некроза опухолей (TNF-α)) и факторов роста (например, кофактора колониеобразования гранулоцитов-макрофагов (GM-CSF) и фактора роста фибробластов (FGF)). Внеклеточные стимулы могут действовать на одну или несколько клеточных реакций, относящихся к росту, миграции, дифференциации клеток, секреции гормонов, активации факторов транскрипции, сокращению мышц, метаболизму глюкозы, регуляции синтеза белков и регуляции клеточного цикла.
Многие заболевания ассоциируются с аномальными клеточными реакциями, запускаемыми опосредованными протеинкиназами событиями, как описано выше. Эти заболевания включают в себя, но не ограничиваются перечисленным, аутоиммунные заболевания, воспалительные заболевания, костные заболевания, метаболические заболевания, неврологические и нейродегенеративные заболевания, рак, сердечно-сосудистые заболевания, аллергии и астму, болезнь Альцгеймера и гормон-связанные заболевания. В соответствии с этим значительные усилия в лекарственной химии были направлены на нахождение ингибиторов протеинкиназ, которые являются эффективными в качестве терапевтических агентов.
Семейство рецепторов типа III тирозинкиназ, включающее в себя Flt3, c-Kit, PDGF-рецептор и c-Fms, играет важную роль в сохранении, росте и развитии гемопоэтических и негемопоэтических клеток [Scheijen, B, Criffin JD, Oncogene, 2002, 21, 3314-3333 and Reille, JT, British Journal of Haematology, 2002, 116, 744-757]. FLT-3 и с-Kit регулируют сохранение пулов стволовых клеток/ранних клеток-предшественников, а также развитие зрелых лимфоидных и миелоидных клеток [Lyman, S. Jacobsen, S, Blood, 1998, 91, 1101-1134]. Оба рецептора содержат присущий домен киназы, который активируется при опосредованной лигандом димеризации рецепторов. При активации домен киназы индуцирует аутофосфорилирование рецептора, а также фосфорилирование различных цитоплазмических белков, которые помогают распространяться сигналу активации, приводящему к росту, дифференциации и выживанию. Некоторые передающие сигнал негативные регуляторы FLT-3 и с-Kit включают в себя PLCγ, PI3-киназу, Grb-2, SHIP и родственные Sre киназы [Scheijen, B. Griffin, JD, Oncogene, 2002, 21, 3314-3333]. Обнаружено, что обе рецепторные тирозинкиназы играют роль в различных гемопоэтических и негемопоэтических злокачественных образованиях. Мутации, которые индуцируют лиганднезависимую активацию FLT-3 и c-Kit, участвуют в остром миелогенном лейкозе (AML), остром лимфоцитном лейкозе (ALL), мастоцитозе и опухоли желудочно-кишечной стромы (GIST). Эти мутации включают в себя одно аминокислотное изменение в домене киназы или внутренние тандемные дупликации, точечные мутации или делеции в рамке считывания, расположенные рядом с регионом мембраны рецепторов. Кроме активирующих мутаций лигандзависимая (аутокринная или паракринная) стимуляция сверхэкспрессированной FLT3 дикого типа или c-Kit может способствовать злокачественному фенотипу [Scheijen, B. Griffin JD, Oncogene, 2002, 21, 3314-3333].
С-fms кодирует рецептор колониестимулирующего фактора макрофагов (M-CSF-1R), который экспрессируется преимущественно в линии дифференцировки моноцитов/марофагов [Dai, XM et al., Blood, 2002, 99, 111-120]. M-CSF-1R и его лиганд регулируют линию роста и дифференцировки макрофага. Подобно другим членам семейства M-CSF-1R содержит присущий домен киназы, который активируется при лиганд-индуцированной димеризации рецептора. M-CSF-1R экспрессируется также в негемопоэтических клетках, включающих эпителиальные клетки молочной железы и нейроны. Мутации в этом рецепторе потенциально связаны с миелоидным лейкозом и его экспрессия коррелирует с метастатической карциномой молочной железы, яичников и эндометрия [Reilly, JT, British Journal of Haematology, 2002, 116, 744-757 and Kacinski, BM, Mol. Reprod. and Devel., 1997, 46, 71-74]. Другим возможным показанием для антагонистов M-CSF-1R является остеопороз [Teitelbaum, S., Science 2000, 289, 1504-1508].
PDGF-рецептор (PDGFR) имеет две субъединицы, PDGFR-α и PDGFR-β, которые могут образовывать гомо- или гетеродимеры при связывании лиганда. Имеется несколько лигандов PDGFR: АВ, ВВ, СС и DD. PDGFR экспрессируется на ранних стволовых клетках, тучных клетках, миелоидных клетках, мезенхимальных клетках и клетках гладких мышц [Scheijen, B. Griffin, JD, Oncogene, 2002, 21, 3314-3333]. Только PDGFR-β участвует в миелоидных лейкозах, обычно в качестве партнера транслокации с Tel, белком взаимодействия Хантингтона (HIP1) или рабаптином 5. Недавно было обнаружено, что активация мутации в домене киназы PDGFR-α происходит в опухолях желудочно-кишечной стромы (GIST) [Heinrich, MC et al., Sciencexpress, 2003].
Циклинзависимые киназы (CDKs) являются серин/треонинпротеинкиназами, состоящими из обогащенной β-складкой аминоконцевой долей и большой карбоксиконцевой долей, которая в основном является α-спиральной. CDKs обнаруживает 11 субдоменов, участвующих во всех протеинкиназах и имеющих диапазон молекулярных масс от 33 до 44 кД. Это семейство киназ, которое включает в себя CDK1, CDK2, CDK4 и CDK6, требует фосфорилирования у остатка, соответствующего Thr 160 CDK2, чтобы быть полностью активным [Meijer, L., Drug Resistance Updates 2000, 3, 83-88].
Каждый комплекс CDK образован из регуляторной субъединицы циклина (например, циклина А, В1, В2, D1, D2, D3 и Е) и субъединицы каталитической киназы (например, CDK1, CDK2, CDK4, CDK5 и CDK6). Каждая разная пара киназа/циклин функционирует для регуляции разных и специфических фаз клеточного цикла, известных как фазы G1, S, G2 и М [Nigg, E., Nature Reviews 2001, 2, 21-32; Flatt, P., Pietenpol, J. Drug Metabolism Reviews 2000, 32, 283-305].
CDKs принимают участие в нарушениях пролиферации клеток, особенно при раке. Клеточная пролиферация является результатом прямой или косвенной дерегуляции цикла деления клетки, и CDKs играет критическую роль в регуляции различных фаз этого цикла. Например, сверхэкспрессия циклина D1 обычно ассоциируется с различными раковыми заболеваниями человека, включающими карциномы и глиомы молочной железы, толстой кишки и гепатоклеточные карциномы и глиомы [Flatt, P., Pietenpol, J., Drug Metabolism Rewiews 2000, 32, 283-305]. Комплекс CDK2/циклин Е играет ключевую роль в развитии от ранней фазы G1 до фазы S клеточного цикла, и сверхэкспрессия циклина Е была ассоциирована с различными солидными опухолями. Следовательно, ингибиторы циклинов D1, E или ассоциированных с ними CDKs являются пригодными целями для терапии рака [Kaubisch, A. Schwartz, G., The Cancer Journal 2000, 6, 192-212].
CDKs, особенно CDK2, также играет роль в апоптозе и развитии Т-клеток. CDK2 был индентифицирован как ключевой регулятор апоптоза тимоцита [Williams, O., et al., European Journal of Immunology 2000, 709-713]. Стимуляция активности CDK2-киназы ассоциируется с развитием апоптоза в тимоцитах в ответ на специфические стимулы. Ингибирование активности CDK2-киназы блокирует этот апоптоз, что приводит к защите тимоцитов.
Кроме регуляции клеточного цикла и апоптоза CDKs непосредственно принимают участие в процессе транскрипции. Различные вирусы требуют CDKs для их процесса репликации. Примеры, в которых ингибиторы CDK ограничивают вирусную репликацию, включают в себя цитомегаловирусы человека, вирус герпеса и вирус ветряной оспы [Meijer, L., Drug Resistance Updates 2000, 3, 83-88].
Ингибирование CDK также является пригодным для лечения невродегенеративных нарушений, таких как болезнь Альцгеймера. Появление спаренных спиральных нитей (PHF), связанное с болезнью Альцгеймера, вызывается гиперфосфорилированием белка Tau CDK5/р25 [Meijer, L., Drug Resistance Updates, 2000, 3, 83-88].
Другим особенно интересным семейством киназ является семейство киназ Src. Эти киназы принимают участие в раковом заболевании, дисфункции иммунной системы и ремоделирующих кости заболеваниях. Для общих обзоров см. Thomas and Brugge, Annu. Rtv. Cell Dev. Biol. 1997, 13, 513; Lawrence and Niu, Pharmacol. Ther. 1998, 77, 81; Tatosyan and Mizenina, Biochemistry (Moscow) 2000, 65, 49; Boschelli et al., Drugs of The Future 2000, 25(7), 717, (2000).
Члены семейства Src включают в себя следующие восемь киназ в организме млекопитающих: Src, Fyn, Yes, Fgr, Lyn, Hck, Lck и Blk. Эти нерецепторные протеинкиназы имеют диапазон молекулярной массы от 52 до 62 кД. Все они характеризуются общей структурной организацией, которая состоит из шести определенных функциональных доменов: домен гомологии Src 4 (SH4), уникальный домен, домен SH3, домен SH2, каталитический домен (SH1) и С-концевая регуляторная область. Tatosyan et al. Biochemistry (Moscow) 2000, 65, 49-58.
На основе опубликованных исследований киназы Src рассматривают в качестве потенциальных терапевтических мишеней для различных заболеваний человека. Мыши, которые имеют дефицит в Src, характеризуются развитием остеопороза или построением костей вследствие пониженной резорбции костей остеокластами. Это позволяет предположить, что остеопороз, возникающий вследствие аномально высокой резорбции костей, можно лечить ингибированием Src. Soriano et al., Cell 1992, 69, 551 and Soriano et al., Cell 1991, 64, 693.
Подавление деструкции артритных костей достигалось сверхэкспрессией CSK в ревматоидных синовиоцитах и остеокластах. Takayanagi et al., J. Clin Invest. 1999, 104, 137. CSK или С-концевая Src-киназа фосфорилирует и тем самым ингибирует каталитическую активность Src. Это значит, что ингибирование Src может предотвратить деструкцию сустава, которая является характеристикой у пациентов, страдающих ревматоидным артритом. Boschelli et al., Drugs of Future 2000, 25(7), 717.
Src играет также роль в репликации вируса гепатита В. Кодированный вирусом фактор транскрипции НВх активирует Src в стадии, требуемой для размножения вируса. Klein et al., EMBO J. 1999, 18, 5019 and Klein et al., Mol. Cell. Biol. 1997, 17, 6427.
В ряде исследований связывали экспрессию Src с раковыми заболеваниями, такими как рак толстой кишки, молочной железы, печени и поджелудочной железы, некоторые В-клеточные лейкозы и лимфомы. Talamonti et al., J. Clin. Invest. 1993, 91, 53; Lutz et al., Biochem. Biophys. Res. 1998 243, 503; Rosen et al., J. Biol. Chem. 1986, 261, 13754; Bolen et al., Proc. Natl. Acad. Sci. USA 1987, 84, 2251; Masaki et al., Hepatology 1998, 27, 1257; Biscardi et al., Adv. Cancer Res. 1999, 76, 61; Lynch et al., Leukemia, 1993, 7, 1416. Кроме того, было обнаружено, что антисмысловая Src, экспрессированная в опухолевых клетках яичников и прямой кишки, ингибирует рост опухоли. Wiener et al., Clin Cancer Res., 1999, 5, 2164; Staley et al., Cell Growth Diff., 1997, 8, 269.
Другие киназы семейства Src также являются потенциальными терапевтическими целями. Lck играет роль в передаче сигнала Т-клеток. Мыши, у которых отсутствует ген Lck, обладают слабой способностью к развитию тиомоцитов. Функция Lck в качестве позитивного активатора передачи сигнала Т-клеток подтверждает, что ингибиторы Lck могут быть применимыми для лечения аутоиммунного заболевания, такого как ревматоидный артрит. Molina et al., Nature, 1992, 357, 161. Hck, Fgr и Lyn были идентифицированы в качестве важных медиаторов передачи сигнала интегрина в миелоидных лейкоцитах. Lowell et al., J. Leukoc. Biol., 1999, 65, 313. Ингибирование этих медиаторов киназ может быть, следовательно, полезным для лечения воспаления. Boschelli et al., Drugs of the Future 2000, 25(7), 717.
Syk является тирозинкиназой, которая играет критическую роль в опосредованной FcεRI дегрануляции тучных клеток и активации эозинофилов. В соответствии с этим киназа Syk принимает участие в различных аллергических нарушениях, в частности, астме. Было обнаружено, что Syk связывается с фосфорилированной гамма-цепью рецептора FcεRI через N-концевые домены SH2 и является существенным для передачи последующего сигнала [Taylor et al., Mol. Cell. Biol. 1995, 15, 4149].
Ингибирование апоптоза эозинофилов было предложено в качестве ключевого механизма развития эозинофилии крови и тканей при астме. IL-5 и GM-CSF позитивно регулируются при астме и предлагаются для индуцирования эозинофилии крови и тканей ингибированием апоптоза эозинофилов. Ингибирование апоптоза эозинофилов было предложено в качестве ключевого механизма для развития эозинофилии крови и тканей при астме. Указывается, что киназа Syk требуется для предотвращения апоптоза эозинофилов цитокинами (использование антисмысловых последовательностей) [Yousefi et al., J. Exp Med 1996, 183, 1407].
Роль Syk в FcγR-зависимой и -независимой реакции в полученных из костного мозга макрофагов определяли с использованием облученных мышиных химер, реконструированных клетками фетальной печени из Syk-/-эмбрионов. SYK-дефицитные макрофаги были дефективными при фагоцитозе, индуцированном FcγR, но проявили нормальный фагоцитоз в ответ на комплемент [Kiefer et al., Mol Cell Biol. 1998, 18, 4209]. Указывалось также, что антисмысловая последовательность аэролизированного Syk подавляет экспрессию Syk и высвобождает медиатор из макрофагов (Stenton et al., Immunology 2000, 164, 3790].
Киназы Janus (JAK) являются семейством, состоящим из JAK1, JAK2, JAK3 и TYK2. JAK играют критическую роль в передаче сигнала цитокинов. Последующие по ходу событий субстраты семейства киназ JAK включают в себя трансдуктор сигнала и активатор белков транскрипции (STAT). Передача сигнала JAK/STAT принимает участие в опосредовании многих аномальных иммунных реакций, таких как аллергии, астма, аутоиммунные заболевания, такие как отторжение трансплантата, ревматоидный артрит, боковой амиотрофический склероз и рассеянный склероз, а также солидные и гематологические злокачественности, такие как лейкозы и лимфомы. Фармацевтическое вмешательство в путь JAK/STAT указывается в обзорах [Frank Mol. Med. 5, 432-456 (1999) & Seidel et al., Oncogene 19, 2646-2656 (2000)].
JAK1, JAK2 и TYK2 экспрессируются повсеместно, тогда как Jak3 преимущественно экспрессируется в гемопоэтических клетках. JAK3 связывается эксклюзивно с гамма-цепью (γc) обычного рецептора цитокина и активируется IL-2, IL-4, IL-7, IL-9 и IL-15. Пролиферация и выживаемость мышиных тучных клеток, индуцированных IL-4 и IL-9, фактически, как было показано, зависит от подачи сигнала JAK3 и γc [Suzuki et al., Blood 96, 2172-2180 (2000)].
Связывание рецепторов иммуноглобулина (Ig) Е с высокой аффинностью сенсибилизированных тучных клеток приводит к высвобождению провоспалительных медиаторов, включающих ряд вазоактивных цитокинов, являющихся результатом острых аллергических или промежуточных (типа I) реакций гиперчувствительности [Gordon et al., Nature 346, 274-276 (1990) & Galli, N. Engl. J. Med., 328, 257-265 (1993)]. Была установлена критическая роль JAK3 в опосредованных рецептором IgE реакциях тучных клеток in vitro и in vivo [Malaviya, et al., Biochem. Biophys. Res. Commun. 257, 807-813 (1999)]. Кроме того, описано также предотвращение реакций гиперчувствительности типа I, в том числе анафилаксии, опосредованной активацией тучных клеток посредством ингибирования JAK3 [Malaviya et al., J. Biol. Chem. 274, 27028-27038 (1999)]. Нацеливание на тучные клетки ингибиторов JAK3 модулирует дегрануляцию тучных клеток in vitro и предотвращает опосредованные IgE-рецептором/антигеном анафилактические реакции in vivo.
В недавнем исследовании описано удачное «нацеливание» JAK3 для иммунной супрессии и переносимости аллотрансплантата. В исследовании показана доза-зависимая выживаемость сердечного аллотрансплантата буйвола у реципиентов Wistar Furth при введении ингибиторов JAK3, что указывает на возможность регуляции нежелательных иммунных реакций при гомологичной болезни [Kirken Transpl. Proc. 33, 3268-3270 (2001)].
IL-4-опосредованное STAT-фосфорилирование принимало участие в качестве механизма, включенного в ранние и поздние стадии ревматоидного артрита (RA). Позитивная регуляция провоспалительных цитокинов в синовиальной оболочке и синовиальной жидкости при RA является характеристикой данного заболевания. Было показано, что IL-4-опосредованная активация пути IL-4/STAT опосредуется через киназы Janus (JAK 1 & 3) и что ассоциированные с IL-4 киназы JAK экспрессируются в синовиальной оболочке RA [Muller-Ladner, et al., J. Immunol. 164, 3894-3901 (2000)].
Семейный боковой амиотрофический склероз (FALS) является фатальным нейродегенеративным нарушением, поражающим приблизительно 10% пациентов ALS. Степени выживаемости мышей с FALS повышались при лечении JAK3-специфичного ингибитора. Это подтверждает, что JAK3 играет роль в FALS [Trieu, et al., Biochem. Biophys. Res. Commun. 267, 22-25 (2000)].
Белки, являющиеся трансдуктором сигнала и активатором транскрипции (STAT), активируются среди прочих киназами семейства JAK. Результаты недавнего исследования подтвердили возможность вмешательства в путь передачи сигнала JAK/STAT посредством «прицеливающихся» в киназы семейства JAK специфических ингибиторов для лечения лейкоза [Sudbeck, et al., Clin. Cancer Res. 5, 2569-1582 (1999)]. Обнаружено, что JAR3-специфические соединения ингибируют клоногенный рост JAK3-экспрессирующих клеточных линий DAUDI, RAMOS, LC1; 19, NALM-6, MOLT-3 и HL-60.
На животных моделях белки слияния TEL/JAK2 вызывали миелопролиферативные нарушения и в гематопоэтических клеточных линиях введение TEL/JAK2 приводило к активации STAT1, STAT3, STAT5 и цитокин-независимому росту [Schwaller, et al., EMBO, J. 17, 5321-5333 (1998)].
Ингибирование JAK3 и TYK 2 отменяет фосфорилирование тирозина STAT3 и ингибирует рост клеток при mycosis fungoides, форме кожной лимфомы Т-клеток. Эти результаты подразумевают участие киназ семейства JAK в существенно активированном пути JAK/STAT, который присутствует при mycosis fungoides [Neilsen, et al., Proc. Nat Acad. Sci. U.S.A. 94, 6764-6769 (1997)]. Аналогично показано, что STAT3, STAT5, JAK1 и JAK2 существенно активируются в лимфоме Т-клеток мышей, характеризующейся первоначально сверхэкспрессией LCK, таким образом далее вовлекая путь JAK/STAT в аномальный рост клеток [Yu, et al., J. Immunol. 159, 5206-5210 (1997)]. Кроме того, IL-6 опосредованная STAT3-активация блокируется ингибитором JAK, что приводит к сенсибилизации клеток миеломы к апоптозу [Catlett-Falcone, et al., Immunity 10, 105-115 (1999)].
Одним представляющим интерес семейством киназ является Rho-ассоциированная образующая суперспираль протеинсерин/треонинкиназа (ROCK), которая, как считается, является эффектором Ras-связанной небольшой GTPазой Rho. Семейство ROCK включает в себя p160ROCK (ROCK-1) (Ishizaki et al., EMBO J 1996, 15, 1885-1893) и ROKα/Rho-киназа/ROCK-II (Leung et al., J. Biol. Chem. 1995, 270, 29051-29054; Matsui et al., EMBO J. 1996, 15, 2208-2216; Nakagawa et al., FEBS Lett. 1996, 392, 189-193), протеинкиназа PKN (Amano et al., Science 1996, 271, 648-650; Watanabe et al., Science 1996, 271, 645-648), и цитрон и цитронкиназа (Madaule et al. Nature, 1998, 394, 491-494; Madaule et al., FEBS Lett. 1995, 377, 243-248). Обнаружено, что киназы семейства ROCK принимают участие в осуществлении различных функций, включая Rho-индуцированное образование актиновых стрессовых волокон и фокальных адгезий, (Leung et al., Mol. Cell Biol. 1996, 16, 5313-5327; Amano et al., Science, 1997, 275, 1308-1311; Ishizaki et al., FEBS Lett. 1997, 404, 118-124) и в регуляции по типу обратной связи миозинфосфатазы (Kimura et al., Science, 1996, 273, 245-248), активации тромбоцитов (Klages et al., J. Cell Biol., 1999, 144, 745-754), сокращении гладких мышц аорты под действием различных стимулов, (Fu et al., FEBS Lett., 1998, 440, 183-187), индуцированные тромбином реакции клеток гладких мышц аорты, (Seasholtz et al., Cir. Res., 1999, 84, 1186-1193), гипертрофию кардиомиоцитов (Kuwahara et al., FEBS Lett., 1999, 452, 314-318), сокращение бронхиальных гладких мышц (Yoshii et al., Am. J. Respir. Cell Mol. Biol, 1999, 20, 1190-1200), сокращение гладких мышц и цитоскелетной реорганизации немышечных клеток (Fukata et al., Trends in Pharm. Sci 2001, 22, 32-39), активацию регулируемых объемом анионных каналов (Nilius et al., J. Physiol., 1999, 516, 67-74), невритную ретракцию (Hirose et al., J. Cell. Biol., 1998, 141, 1625-1636), хемотаксис нейтрофилов (Niggli, FEBS Lett., 1999, 445, 69-72), заживление ран (Nobes and Hall, J. Cell. Biol., 144, 1235-1244), инвазии опухоли (Itoh et. al., Nat. Med., 1999, 5, 221-225) и клеточной трансформации (Sahai et al., Curr. Biol, 1999, 9, 136-145)). Более конкретно, ROCK принимает участие в различных заболеваниях и нарушениях, включающих в себя гипертензию (Satoh et al., J. Clin. Invest. 1994, 94, 1397-1403; Mukai et al., FASEB J. 2001, 75, 1062-1064; Uehata et al., Nature 1997, 389, 990-994; Masumoto et al., Hypertension, 2001, 38, 1307-1310), церебральный вазоспазм (Sato et al., Circ. Res. 2000, 87,195-200; Miyagi et al., J. Neurosurg. 2000, 93, 471-476; Tachibana et al., Acta Neurochir (Wien) 1999, 141, 13-19), коронарный вазоспазм (Shimokawa et al., Jpn. Cir. J. 2000, 64, 1-12; Kandabashi et al., Circulation 2000, 101, 1319-1323; Katsumata et al., Circulation 1997, 96, 4357-4363; Shimokawa et al., Cardiovasc. Res. 2001, 51, 169-177; Utsunomiya et al., J. Pharmacol. 2001, 134, 1724-1730; Masumoto et al., Circulation 2002, 105, 1545-154), бронхиальную астму (Chiba et al., Comp. Biochem. Physiol. C Pharmacol. Toxicol. Endocrinol. 1995, 11, 351-357; Chiba et al., Br. J. Pharmacol. 1999, 127, 597-600; Chiba et al., Br. J. Pharmacol. 2001, 133, 886-890; Iizuka et al., Eur. J. Pharmacol. 2000, 406, 273-279), преждевременные роды (Niro et al., Biochem. Biophys. Res. Commun. 1997, 230, 356-359; Tahara et al., Endocrinology 2002, 143, 920-929; Kupittayanant et al., Pflugers Arch. 2001, 443, 112-114), эректильную дисфункцию (Chitaley et al., Nat. Med. 2001, 7, 119-122; Mills et al., J. Appl. Physiol. 2001, 91, 1269-1273), глаукому (Honjo et al., Arch. Ophthalmol. 2001, 1171-1178; Rao et al. Invest. Ophthalmol. Vis. Sci. 2001, 42, 1029-1037), пролиферацию клеток васкулярных гладких мышц (Shimokawa et al., Cardiovasc. Res. 2001, 51, 169-177; Morishige et al., Arterioscler. Thromb. Vase. Biol. 2001, 21, 548-554; Eto et al., Am. J. Physiol. Heart Circ. Physiol 2000, 278, H1744-H1750; Sawada et al., Circulation 2000, 101, 2030-2023; Shibata et al., Circulation 2001, 103, 284-289), гипертрофию миокарда (Hoshijima et al., J. Biol. Chem. 1998, 273, 7725-77230; Sah et al., J. Biol. Chem. 1996, 271, 31185-31190; Kuwahara et al. FEBS Lett. 1999, 452, 314-318; Yanazume et al. J. Biol. Chem. 2002, 277, 8618-8625), малигному (Itoh et al. Nat. Med. 1999, 5, 221-225; Genda et al. Hepatology 1999, 30, 1027-1036; Somlyo et al. Biochem., Biophys. Res. Commun. 2000, 269, 652-659), повреждение, вызванное ишемией/реперфузией (Ikeda et al. J. of Surgical Res. 2003, 109, 155-160; Miznuma et al. Transplantation 2003, 75, 579-586), эндотелиальную дисфункцию (Hernandez-Perera et al. Circ. Res. 2000, 87, 616-622; Laufs et al., J. Biol. Chem. 1998, 273, 24266-24271; Eto et al., Circ. Res. 2001, 89, 583-590), болезнь Крона и колит (Segain et al. Gastroenterology 2003, 124(5), 1180-1187), отрастание нейритов (Fournier et al. J. Neurosci. 2003, 23, 1416-1423), болезнь Рейно (Shimokawa et al. J. Cardiovasc. Pharmacol. 2002, 39, 319-327) и атеросклероз (Retzer et al. FEBS Lett. 2000, 466, 70-74; Ishibashi et al. Biochim. Biophys. Acta 2002, 1590, 123-130). В соответствии с этим разработка ингибиторов киназ ROCK может быть применима в качестве терапевтических агентов для лечения нарушений, принимающих участие в пути киназы ROCK.
ERK2 (киназа, регулирующая внеклеточный сигнал) является членом семейства митоген-активированных протеинкиназ (МАР)1 млекопитающих. Киназы (МАР)1 являются серин/треонинкиназами, которые опосредуют пути внутриклеточной сигнальной трансдукции (Cobb and Goldsmith, J Biol. Chem., 1995, 270, 14843; Davis, Mol. Reprod. Dev. 1995, 42, 459) и активируются митогенами и факторами роста (Bokemeyer et al. Kidney Int. 1996, 49, 1187). Члены семейства киназ МАР подобным образом имеют общую последовательность и консервативные структурные домены и помимо ERK2 включают в себя киназы JNK (N-концевая киназа Jun) и р38. Киназы JNKs и р38 активируются в ответ на провоспалительные цитокины TNF-альфа и интерлейкин-1 и клеточным стрессом, таким как тепловой шок, гиперосомолярность, ультрафиолетовое излучение, липополисахарид и ингибиторы синтеза белка (Derijard et al., Cell 1994, 76, 1025; Han et al., Science 1994, 265, 808; Raingeaud et al., J Biol. Chem. 1995, 270, 7420; Shapiro and Dinarello, Proc. Natl. Acad. Sci. USA 1995, 92, 12230). В противоположность этому ERK активируются митогенами и факторами роста (Bokemeyer et al., Kidney Int. 1996, 49, 1187).
ERK2 является широко распространенной протеинкиназой, которая достигает максимальную активность, когда Thr183 и Tyr185 фосфорилируются киназой МАР предшествующего события, МЕК1 (Anderson et al., Nature 1990, 343, 651; Crews et al., Science 1992, 255, 478). При активации ERK2 фосфорилирует многие регуляторные белки, включая протеинкиназы Rsk90 (Bjorbaek et al., J. Biol. Chem. 1995, 270, 18848) и МАРКАР2 (Rouse et al., Cell 1994, 78, 1027), и факторы транскрипции, такие как ATF2 (Raingeaud et al., Mol Cell Biol. 1996, 16, 1247), Elk-1 (Raingeaud et al., Mol. Cell Biol. 1996, 16, 1247), c-Fos (Chen et al., Proc. Natl Acad. Sci. USA 1993, 90, 10952), и с-Мус (Oliver et al., Proc. Soc. Exp. Biol. Med. 1995, 210, 162). ERK2 является также мишенью в последующем событии Ras/Raf-зависимых путей (Moodie et al., Science 1993, 260, 1658) и может помочь заменить сигналы от этих потенциально онкогенных белков. Обнаружено, что ERK2 играет роль в негативном контроле роста раковых клеток молочной железы (Frey and Mulder, Cancer Res. 1993, 57, 628), и описана сверхэкспрессия ERK2 в раковой опухоли молочной железы человека (Sivaraman et al., J Clin. Invest. 1997, 99, 1478). Активированный ERK2 участвует также в пролиферации эндотелин-стимулированных клеток гладких мышц дыхательных путей, что подтверждает роль этой киназы при заболевании астмой (Whelchel et al., Am. J. Respir. Cell Mol. Biol. 1997, 16, 589).
Гликогенсинтаза киназа-3 (GSK-3) является серин/треонинпротеинкиназой, состоящей из α- и β-изоформ, каждая из которых кодируется определенными генами (Coghlan et al., Chemistry & Biology 2000, 7, 793-803; and Kim and Kimmel, Curr. Opinion Genetics Dev., 2000 10, 508-514). GSK-3 принимает участие в различных заболеваниях, включающих в себя болезнь Альцгеймера, нарушения деятельности ЦНС, такие как маниакальное депрессивное нарушение и невродегенеративные заболевания, и кардиомиоцитная гипертрофия [PCT Application Nos.: WO 99/65897 and WO 00/38675; and Haq et al., J. Cell Biol. 2000, 151, 117-130]. Эти заболевания ассоциируются с аномальной операцией некоторых путей передачи сигналов клеток, в которых GSK-3 играет роль. Обнаружено, что GSK-3 фосфорилирует и моделирует активность ряда ругуляторных белков. Эти белки включают в себя гликогенсинтазу, которая является ограничивающим скорость ферментом, необходимым для синтеза гликогена, связанным с микротрубочками белком Tau, фактором транскрипции гена β-катенином, фактор инициации транскрипции е1F2B, а также цитратлиазой АТФ, аксином, фактором-1 теплового шока, с-Jun, c-myc, c-myb, CREB и СЕРВАα. Эти разнообразные белковые цели вовлекают GSK-3 во многие аспекты клеточного метаболизма, пролиферации, дифференциации и развития.
В опосредованном GSK-3 пути, который является приемлемым для лечения диабета типа II, индуцированная инсулином передача сигнала приводит к клеточному поглощению глюкозы и синтезу гликогена. Наряду с этим путем GSK-3 является негативным регулятором инсулин-индуцированного сигнала. Обычно присутствие инсулина вызывает ингибирование опосредованного GSK-3 фосфорилирования и деактивации гликогенсинтазы. Ингибирование GSK-3 приводит к повышенному синтезу гликогена и поглощению глюкозы [Klein et al., PNAS 1996, 93, 8455-8459; Cross et al., Biochem. J. 1994, 303, 21-26; Cohen, Biochem. Soc. Trans. 1993, 21, 555-567; and Massillon et al., Biochem J. 1994, 299, 123-128]. Однако у диабетического пациента, у которого инсулиновая реакция ослаблена, синтез гликогена и поглощение глюкозы не повышается несмотря на присутствие относительно высоких уровней инсулина в крови. Это приводит к аномально высоким уровням в крови глюкозы с острыми и продолжительными эффектами, которые могут в конце концов привести к сердечно-сосудистому заболеванию, почечной недостаточности и слепоте. У таких пациентов нормальное, индуцированное инсулином ингибирование GSK-3 не имеет места. Описано также, что у пациентов с диабетом типа II GSK-3 сверхэкспрессируется [см. заявку РСТ: WO 00/38675]. Терапевтические ингибирования GSK-3, следовательно, являются потенциально применимыми для лечения диабетических пациентов, страдающих ослабленной реакцией на инсулин.
Активность GSK-3 связана также с болезнью Альцгеймера. Это заболевание характеризуется хорошо известным β-амилоидным пептидом и образованием внутриклеточных нейрофибриллярных клубков. Пептиды Аβ образуются из амилоидного белка-предшественника (АРР) последовательным протеолизом, катализируемым аспартилпротеазой ВАСЕ2, с последующим расщеплением презенелин-зависимой γ-секретазой. Было показано, что антитела против β-амилоидных бляшек могут медленно снижать познавательную способность у пациентов с болезнью Альцгеймера (Hock et al., Neuron, 2003, 38, 547-554) и, таким образом, другие снижающие β-амилоид стратегии (например, разработка агентов, способных ингибировать β-амилоидный пептид) могут быть применимыми при лечении болезни Альцгеймера и других психотических и нейродегенеративных нарушений. Кроме того, нейрофибриллярные клубки содержат гиперфосфорилированный белок Tau, который фосфорилируется на аномальных сайтах, и эти агенты, способные ингибировать гиперфосфорилирование белка Tau, могут быть применимыми при лечении болезни Альцгеймера и других психотических и нейродегенеративных нарушений.
Известно, что GSK-3 фосфорилирует эти аномальные сайты в клетках на моделях животных. Кроме того, обнаружено, что ингибирование GSK-3 предотвращает гиперфосфорилирование Tau в клетках [Lovestone et al., Current Biology 1994, 4, 1077-86; and Brownlees et al., Neuroreport 1997, 8, 3251-55]. Следовательно, активность GSK-3 стимулирует генерацию нейрофибриллярных клубков и развитие болезни Альцгеймера. Обнаружено также, что GSK-3 облегчает процессинг АРР и что ингибитор GSK-3 (литий) ингибирует генерацию Аβ-пептидов посредством ингибирования GSK-3 (Phiel et al. Nature 2003, 423, 435-439). Таким образом, разработка ингибиторов GSK-3 может быть применимой для снижения образования амилоидных бляшек и нейрофибриллярных клубков, патологических отличительных признаков болезни Альцгеймера, и может быть применимой для лечения других психотических и нейродегенеративных нарушений.
Другим субстратом GSK-3 является β-катенин, который деградирует после фосфорилирования GSK-3. Пониженные уровни β-катенина обнаружены у шизофренических пациентов и были ассоциированы с другими заболеваниями, относящимися к увеличению гибели нейронных клеток [Zhong et al., Nature 1998, 395, 698-702; Takashima et al., PNAS 1993, 90, 7789-93; and Pei et al., J. Neuropathol. Exp 1997, 56, 70-78].
Активность GSK-3 ассоциируется также с ударом [Wang et al., Brain Res 2000, 859, 381-5; Sasaki et al., Neurol Res 2001, 23, 588-92; Hashimoto et al., J. Biol. Chem 2002, 277, 32985-32991].
Подсемейство киназ AGC фосфорилирует их субстраты у остатков серина и треонина и принимает участие в различных хорошо известных процессах передачи сигналов, включающих в себя, но не ограничивающихся перечисленным, передачу сигнала циклического АТФ, реакцию на инсулин, защиту апоптоза, передачу сигнала диацилглицерина и регулирование трансляции белка (Peterson et al., Curr. Biol. 1999, 9, R521). Это подсемейство включает в себя РКА, РКВ (с-Akt), PCK, PRK1, 2, p70S6K и PDK.
Обнаружено, что АКТ (известная также как РКВ или Rac-PK бета), серин/треонинпротеинкиназа, сверхэкспрессируется в нескольких типах раковых заболеваний и является медиатором функций нормальных клеток [(Khwaja, A., Nature 1999, 401, 33-34); (Yuan, Z.Q., et al., Oncogene 2000, 19, 2324-2330); (Namikawa, K., et al., J Neurosci. 2000, 20, 2875-2886)]. АКТ содержит домен N-концевой гомологии плекскина (РН), домен киназы и С-концевую «хвостовую» область. До сих пор обнаружены три изоформы АКТ-киназы человека (АКТ-1, -2 и -3) [(Cheng, J.Q., Proc. Natl. Acad. Sci. USA 1992, 89, 9267-9271); (Brodbeck, D. et al., J. Biol. Chem. 1999, 274, 9133-9136)]. Домен РН связывает 3-фосфоинозитиды, которые синтезируются фосфатидилинозит-3-киназой (PI3K) при стимуляции факторами роста, такими как полученный из тромбоцитов фактор роста (PDGF), фактор роста нервов (NGF) и инсулиноподобный фактор роста (IFG-1) [(Kulik et al., Mol Cell. Biol, 1997, 17, 1595-1606,); (Hemmings, B.A., Science, 1997, 275, 628-630)]. Связывание липида с доменом РН стимулирует транслокацию АКТ на мембранах плазмы и облегчает фосфорилирование другими РН-доменсодержащими протеинкиназами, PDK1 у Thr308, Thr309 и Thr305 для изоформ АКТ 1, 2 и 3 соответственно. Вторая до сих пор неизвестная киназа требуется для фосфорилирования Ser473, Ser474 или Ser472 в С-концевых «хвостах» АКТ-1, -2 и -3 соответственно для создания полностью активированного фермента АКТ.
После расположения на мембране АКТ опосредует несколько функций в клетке, включающих метаболические действия инсулина (Calera, M.R. et al., J. Biol. Chem. 1998, 273, 7201-7204), индуцирование дифференциации и/или пролиферации, синтез белка и стрессовые реакции (Alessi, D.R. et al., Curr. Opin. Genet. Dev. 1998, 8, 55-62).
Проявления регуляций измененной АКТ появляются как при повреждении, так и при заболевании, причем наиболее важным является проявление его при раке. Первая оценка АКТ была связана с карциномой яичников человека, когда было обнаружено, что экспрессия АКТ амплифицирует в 15% случаев (Cheng, J.Q. et al., Proc. Natl. Acad. Sci. U.S.A. 1992, 89, 9267-9271). Было также обнаружено, что она сверхэкспрессируется в 12% случаев панкреатических раковых заболеваний (Cheng, J. Q. et al., Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 3636-3641). Было показано, что АКТ-2 сверхэкспрессируется в 12% карцином яичников и что амплификация АКТ особенно частой была у 50% недифференцированных опухолей, подтверждая, что АКТ может также быть ассоциирована с агрессивностью опухоли (Bellacosa, et al., Int. J. Cancer 1995, 64, 280-285).
Обнаружено, что РКА (известная также как цАМФ-зависимая протеинкиназа) регулирует многие жизненные функции, включая метаболизм энергии, генную транскрипцию, пролиферацию, дифференциацию, репродуктивную функцию, секрецию, нейронную активность, память, сократительную способность и двигательную функцию (Beebe, S.J., Semin. Cancer Biol. 1994, 5, 285-294). РКА представляет собой тетрамерный гомофермент, который содержит две каталитические субъединицы, связанные с гомодимерной регуляторной субъединицей (которая действует для ингибирования каталитических субъединиц). При связывании цАМФ (активация фермента) каталитические субъединицы диссоциируют из регуляторных субъединиц с образованием активной серин/треонинкиназы (McKnight, G.S. et al., Recent Prog. Horm. Res. 1988,44, pp. 307). На сегодняшний день обнаружены три изоформы каталитической субъединицы (С-α, С-β и С-γ) (Beebe, S.J. et al., J. Biol. Chem. 1992, 267, 25505-25512), причем С-α-субъединица является наиболее экстенсивно изученной в основном благодаря ее повышенной экспрессии в первичных и метастатических меланомах (Becker, D. et al., Oncogene 1990, J, 1133). На сегодняшний день стратегии для модуляции активности С-α-субъединицы включают в себя использование антител, молекул, которые блокируют активность РКА посредством «нацеливания» на регуляторные димеры и экспрессии антисмысловых олигонуклеотидов.
Рибосомные протеинкиназы р70S6K -1 и -2 также являются членами подсемейства AGC протеинкиназ и катализируют фосфорилирование и последующую активацию рибосомного белка S6, который принимал участие в трансляционной позитивной регуляции мРНК, кодирующих компоненты аппаратуры синтеза белков. Эти мРНК содержат олигопиримидиновый тракт у их 5'-транскрипционного начального сайта, названного 5ТОР, который, как было обнаружено, является существенным для их регуляции на трансляционном уровне (Volarevic, S. et al., Prog. Nucleic Acid Res. Mol. Biol. 2001, 65, 101-186). Р70S6K-зависимое фосфорилирование S6 стимулируется в ответ на различные гормоны и факторы роста в основном через путь PI3K (Coffer, P.J. et al., Biochem. Biophys. Res. Commun, 1994 198, 780-786), который может быть под регуляцией mTOR, поскольку рапамицин действует с ингибированием активности Р70S6K и блокированием протеинсинтазы, особенно в результате регуляции по типу обратной связи трансляции этих мРНК, кодирующих рибосомные белки (Kuo, C.J. et al., Nature 1992, 555, 70-73).
In vitro PDK1 катализирует фосфорилирование Thr252 в петле активации каталитического домена р70, который является недиспенсируемым для активности р70 (Alessi, D.R., Curr. Biol, 1998, 8, 69-81). Использование исследования рапамицина и делеции гена dp70S6K из дрозофил и Р70S6K из мышей установило центральную роль р70 как в росте клеток, так и в передаче сигнала пролиферации.
3-Фосфоинозитид-зависимая протеинкиназа-1 (PDK1) играет ключевую роль в регуляции активности ряда киназ, принадлежащих к подсемейству AGC протеинкиназ (Alessi, D. et al., Biochem. Soc. Trans 2001, 29, 1). Они включают в себя изоформы протеинкиназы В (РКВ, известной также как АКТ), рибосомную S6-киназу р70 (S6K) (Avruch, J. et al., Prog. Mol. Subcell. Biol. 2001, 26, 115) и рибосомную S6-киназу р90 (Frödin, M. et al., EMBO J. 2000, 19, 2924-2934). PDK1, опосредующая передачу сигнала, активируется в ответ на инсулин и факторы роста и в результате присоединения клетки к межклеточной матрице (передача сигнала интегрина). После активации эти ферменты опосредуют многие другие клеточные события фосфорилированием ключевых регуляторных белков, которые играют важную роль в регуляции процессов, таких как выживаемость, рост, пролиферация клеток и регуляция глюкозы [(Lawlor, M.A. et al., J. Cell Sci. 2001, 114, 2903-2910), (Lawlor, M.A. et al., EMBO J. 2002, 21, 3728-3738)]. PDK1 представляет собой белок из 556 аминокислот с N-концевым каталитическим доменом и C-концевым доменом гомологии плекстрина (РН), который активирует свой субстрат фосфорилированием этих киназ у их петли активации (Belham, C. et al., Curr. Biol. 1999, 9, R93-R96). Многие раковые заболевания человека, в том числе простаты и NSCL, имеют повышенную функцию пути передачи сигнала PDK1 вследствие ряда характерных генетических событий, таких как мутации PTEN и сверхэкспрессия некоторых ключевых регуляторных белков [(Graff, J.R., Expert Opin. Ther. Targets 2002, 6, 103-113), (Brognard, J., et al., Cancer Res. 2001, 61, 3986-3997)]. Ингибирование PDK1 как потенциальный механизм лечения рака было демонстрировано трансфицированием PTEN-отрицательной раковой клеточной линии человека (U87MG) с антисмысловыми олигонуклеотидами, направленными против PDK1. Образовавшееся понижение в уровнях белка PDK1 приводит к снижению клеточной пролиферации и выживаемости (Flynn, P., et al., Curr. Biol. 2000, 10, 1439-1442). Следовательно, разработка ингибиторов сайта связывания АТФ PDK1 предоставляет среди других лечений цель для химиотерапии рака.
Иной диапазон генотипов раковых клеток относится к проявлению следующих шести существенных чередований в клеточной физиологии: самодостаточность в подаче сигнала роста, уклонение от апоптоза, интенсивность для подачи ингибирующего рост сигнала, безграничный репликативный потенциал, непрерывный ангиогенез и инвазия ткани, приводящая к метастазам (Hanahan, D. et al., Cell 2000, 100, 57-70). PDK1 является критическим медиатором пути подачи сигнала PI3K, который регулирует мультитуду клеточной функции, включающей рост, пролиферацию и выживание. Следовательно, ингибирование этого пути может влиять на четыре из шести определенных условий для развития рака. Предполагается, что как таковой ингибитор PDK1 может оказывать действие на рост очень широкого диапазона раковых заболеваний человека.
В частности, повышенные уровни активности пути PI3K непосредственно связаны с развитием ряда раковых заболеваний человека, развитием до агрессивного резистентного состояния (приобретенная резистентность к химиотерапиям) и плохим прогнозом. Такую повышенную активность относили к ряду ключевых событий, включающих в себя пониженную активность негативных регуляторов пути, таких как фосфатаза PTEN, активирующие мутации позитивных регуляторов пути, таких как Ras, и сверхэкспрессия компонентов самого пути, такого как РКВ, причем примеры их включают головной мозг (глиома), молочную железу, голову и шею, почки, легкие, печень, меланому, яичники, поджелудочную железу, простату, саркому, щитовидную железу [(Teng, D.H. et al., Cancer Res., 1997 57, 5221-5225), (Brognard, J. et al., Cancer Res., 2001, 61, 3986-3997), (Cheng, J.Q. et al., Proc. Natl. Acad.Sci. 1996, 93, 3636-3641), (Int. J. Cancer 1995, 64, 280), (Graff, J.R., Expert Opin. Ther. Targets 2002, 6, 103-113), (Am. J. Pathol. 2001, 159, 431)].
Кроме того, понижение функции пути посредством генного "knockout", генного "knockdown", доминантные отрицательные исследования и ингибиторы в виде маленьких молекул пути продемонстрировали реверсию многих фенотипов рака in vitro (некоторые исследования демонстрировали также аналогичное действие in vivo), такую как блокирование пролиферации, снижали выживание и сенсибилизировали раковые клетки к воздействию химиотерапии различных типов в ряде клеточных линий, относящихся к раковым заболеваниям следующих органов: поджелудочной железы [(Cheng, J.Q. et al., Proc. Natl. Acad. Sci. 1996, 93, 3636-3641), (Neoplasia 2001, 3, 278)], легких [(Brognard, J. et al., Cancer Res. 2001, 61, 3986-3997), (Neoplasia 2001, 3, 278)], яичников [(Hayakawa, J. et al., Cancer Res. 2000, 60, 5988-5994), (Neoplasia 2001, 3, 278)],молочной железы (Mol. Cancer Ther. 2002, 1, 707), прямой кишки [(Neoplasia 2001, 3, 278), (Arico, S. et al., J. Biol. Chem. 2002, 277, 27613-27621)], шеи (Neoplasia 2001, 3, 278), простаты [(Endocrinology 2001, 142, 4795), (Thakkar, H. et al. J. Biol. Chem. 2001, 276, 38361-38369), (Chen, X. et al., Oncogene 2001, 20, 6073-6083)] и головного мозга (глиобластома) [(Flynn, P. et al., Curr. Biol. 2000, 10,1439-1442)].
В соответствии с этим существует потребность в разработке ингибиторов подсемейств протеинкиназ FLT-3, FMS, c-KIT, PDGFR, JAK, AGC (например, PKA, PDK, p70S6K-1 и -2 и РКВ), протеинкиназ CDK, GSK, SRC, ROCK и/или SYK, которые являются применимыми при лечении различных заболеваний или состояний, связанных с активацией подсемейства протеинкиназ FLT-3, FMS, c-KIT, PDGFR, JAK, AGC (например, PKA, PDK, p70S6K-1 и -2 и РКВ), CDK, GSK, SRC, ROCK и/или SYK, особенно приверженных неадекватным лечениям, в настоящее время пригодным для большей части этих заболеваний.
Теперь обнаружено, что соединения данного изобретения и их фармацевтически приемлемые композиции являются эффективными в качестве ингибиторов подсемейства протеинкиназ FLT-3, FMS, c-KIT, PDGFR, JAK, AGC (например, PKA, PDK, p70S6K-1 и -2 и РКВ), протеинкиназ CDK, GSK, SRC, ROCK и/или SYK. В некоторых вариантах осуществления эти соединения являются эффективными в качестве ингибиторов протеинкиназ FLT-3, JAK-3, PDK-1 и/или SYK. Эти соединения имеют общую формулу I:
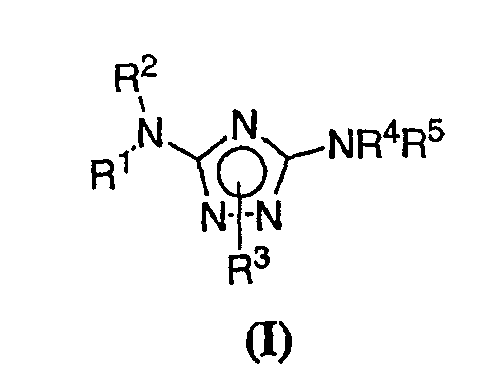
или являются их фармацевтически приемлемыми солями, где R1, R2, R3, R4 и R5 имеют значения, указанные ниже.
Указанные соединения и их фармацевтические композиции являются применимыми для лечения или профилактики различных заболеваний, включающих в себя, но не ограничивающихся перечисленным, болезнь сердца, диабет, болезнь Альцгеймера, нарушения иммунодефицита, воспалительные заболевания, гипертензию, аллергические заболевания, аутоиммунные заболевания, деструктивные костные нарушения, такие как остеопороз, пролиферативные заболевания, инфекционные заболевания, заболевания, иммунологически опосредованные, и вирусные заболевания. Эти композиции являются также применимыми в способах предупреждения гибели клеток и гиперплазии и, следовательно могут применяться для лечения или профилактики реперфузии/ишемии при ударе, сердечных приступов и гипоксии органа. Композиции являются также применимыми в способах профилактики индуцированной тромбином агрегации тромбоцитов. Композиции являются особенно применимыми для лечения таких нарушений, как хронический миелогенный лейкоз (CML), острый миелоидный лейкоз (AML), острый промиелоцитный лейкоз (APL), ревматоидный артрит, астма, остеоартрит, ишемия, рак (включающий, но не ограничивающийся перечисленным, рак яичников, рак молочной железы и эндометриальный рак), заболевание печени, включая печеночную ишемию, заболевание сердца, такое как инфаркт миокарда и застойная сердечная недостаточность, патогенные иммунные состояния, включающие в себя активацию Т-клеток, и нейродегенеративные нарушения.
1. Общее описание соединений изобретения:
Настоящее изобретение относится к соединению формулы I:
или его фармацевтически приемлемой соли,
где R1 представляет собой водород или Y-R', где Y представляет собой необязательно замещенную С1-6-алкилиденовую цепь, в которой до двух метиленовых звеньев необязательно и независимо заменены на -О-, -S-, -NR-, -OCO-, -СОО- или -СО;
в каждом случае R независимо представляет собой водород или необязательно замещенную С1-6алифатическую группу, в каждом случае R' независимо представляет собой водород или необязательно замещенную группу, выбранную из С1-6алифатической группы, 3-8-членного насыщенного, частично ненасыщенного или полностью ненасыщенного моноциклического кольца, имеющего 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, или 8-12-членной насыщенной, частично ненасыщенной или полностью ненасыщенной бициклической системы колец, имеющей 0-5 гетероатомов, независимо выбранных из азота, кислорода или серы, или R и R', два случая R или два случая R' взяты вместе с атомом(ами), с которым они связаны, с образованием необязательно замещенного 3-12-членного насыщенного, частично ненасыщенного или полностью ненасыщенного моноциклического или бициклического кольца, имеющего 0-4 гетероатома, независимо выбранных из азота, кислорода или серы;
R2 представляет собой -(T)nAr1 или -(Т)nCy1, где Т представляет собой необязательно замещенную С1-4алкилиденовую цепь, в которой одно метиленовое звено Т необязательно заменено на -NR-, -S-, -O-, -CS-, -CO2-, -OCO-, -CO-, -СОСО-, -CONR-, -NRCO-, -NRCO2-, -SO2NR-, -NRSO2-, -CONRNR-, -NRCONR-, -OCONR-, -NRNR-, -NRSO2NR-, -SO-, -SO2-, -PO-, -PO2- или -POR-; n равно 0 или 1; Ar1 представляет собой необязательно замещенную арильную группу, выбранную из 5-6-членного моноциклического или 8-12-членного бициклического кольца, имеющего 0-5 гетероатомов, независимо выбранных из азота, кислорода или серы; и Cy1 представляет собой необязательно замещенную группу, выбранную из 3-7-членного насыщенного или частично ненасыщенного моноциклического кольца, имеющего 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, или 8-12-членной насыщенной или частично ненасыщенной бициклической системы колец, имеющей 0-5 гетероатомов, независимо выбранных из азота, кислорода или серы;
или R1 и R2, взятые вместе с атомом азота, образуют необязательно замещенную 5-8-членное моноциклическое или 8-12-членное бициклическое насыщенное, частично ненасыщенное или полностью ненасыщенное кольцо, имеющее 0-3 дополнительных гетероатомов, независимо выбранных из азота, кислорода или серы;
где Ar1, Cy1 или любое кольцо, образованное R1 и R2, взятых вместе, являются, каждое независимо, необязательно замещенными х независимыми группами Q-Rx, где х равно 0-5, Q представляет собой связь или С1-6-алкилиденовую цепь, в которой до двух метиленовых звеньев Q необязательно заменены на -NR-, -S-, -O-, -CS-, -CO2-, -OCO-, -CO-, -СОСО-, -CONR-, -NRCO-, -NRCO2-, -SO2NR-, -NRSO2-, -CONRNR-, -NRCONR-, -OCONR-, -NRNR-, -NRSO2NR-, -SO-, -SO2-, -PO-, -PO2- или -POR-; в каждом случае Rх представляет собой, независимо, R', галоген, NO2, CN, OR', SR', N(R')2, NR'COR', NR'CONR'2, NR'CO2R', COR', CO2R', OCOR', CON(R')2, OCON(R')2, SOR', SO2R', SO2N(R')2, NR'SO2R', NR'SO2N(R')2, COCOR' или СОСН2COR';
R3 связан с атомом азота либо в 1-, либо в 2-положении кольца и представляет собой (L)mAr2 или (L)mCy2, где L представляет собой необязательно замещенную С1-4алкилиденовую цепь, в которой одно метиленовое звено L необязательно заменено на -NR-, -S-, -O-, -CS-, -CO2-, -OCO-, -CO-, -COCO-,-CONR-, -NRCO-, -NRCO2-, -SO2NR-, -NRSO2-, -CONRNR-, -NRCONR-, -OCONR-, -NRNR-, -NRSO2NR-, -SO-, -SO2-, -PO-, -PO2- или -POR-; m равно 0 или 1; Ar2 представляет собой необязательно замещенную арильную группу, выбранную из 5-6-членного моноциклического или 8-12-членного бициклического кольца, имеющего 0-5 гетероатомов, независимо выбранных из азота, кислорода или серы; и Cy2 представляет собой необязательно замещенную группу, выбранную из 3-7-членного насыщенного или частично ненасыщенного моноциклического кольца, имеющего 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, или 8-12-членной насыщенной или частично ненасыщенной бициклической системы колец, имеющей 0-5 гетероатомов, независимо выбранных из азота, кислорода или серы; где Ar2 и Cy2, каждый независимо, необязательно замещены группами Z-Ry, где y равно 0-5, Z представляет собой связь или С1-6-алкилиденовую цепь, в которой до двух метиленовых звеньев Z необязательно заменены на -NR-, -S-, -O-, -CS-, -CO2-, -OCO-, -CO-, -СОСО-, -CONR-, -NRCO-, -NRCO2-, -SO2NR-, -NRSO2-, -CONRNR-, -NRCONR-, -OCONR-, -NRNR-, -NRSO2NR-, -SO-, -SO2-, -PO-, -PO2- или -POR-; в каждом случае Ry представляет собой, независимо, R', галоген, NO2, CN, OR', SR', N(R')2, NR'COR', NR'CONR'2, NR'CO2R', COR', CO2R', OCOR', CON(R')2, OCON(R')2, SOR', SO2R', SO2N(R')2, NR'SO2R', NR'SO2N(R')2, COCOR' или СОСН2COR';
R4 представляет собой водород или С1-6алкил, при условии что, когда R5 представляет собой водород, тогда R4 также представляет собой водород;
R5 представляет собой водород или R3 и R5, взятые вместе, образуют необязательно замещенную группу, выбранную из 5-7-членного насыщенного, частично ненасыщенного или полностью ненасыщенного моноциклического кольца, имеющего 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, или 8-10-членной насыщенной, частично ненасыщенной или полностью ненасыщенной бициклической системы колец, имеющей 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, и
где любое кольцо, образованное R3 и R5, взятыми вместе, является необязательно замещенным до пяти заместителями, выбранными из W-Rw, где W представляет собой связь или С1-6-алкилиденовую цепь, в которой до двух метиленовых звеньев W необязательно заменены на -NR-, -S-, -O-, -CS-, -CO2-, -OCO-, -CO-, -СОСО, -CONR-, -NRCO-, -NRCO2-, -SO2NR-, -NRSO2-, -CONRNR-, -NRCONR-, -OCONR-, -NRNR-, -NRSO2NR-, -SO-, -SO2-, -PO-, -PO2- или -POR-; в каждом случае RW представляет собой, независимо, R', галоген, NO2, CN, OR', SR', N(R')2, NR'COR', NR'CONR'2, NRCO2R', COR', CO2R', OCOR', CON(R')2, OCON(R')2, SOR', SO2R', SO2N(R')2, NR'SO2R', NR'SO2N(R')2, COCOR' или СОСН2COR'.
В некоторых вариантах осуществления для соединений формулы I соблюдается одно или несколько или все из следующих условий:
а) когда R3 представляет собой незамещенный фенил и R1 представляет собой водород, тогда R2 не является
i) незамещенным фенилом;
ii) незамещенным пиридилом;
iii) бензилом, замещенным о-OMe;
iv) -(C=S)NH(C=O)фенилом;
v) 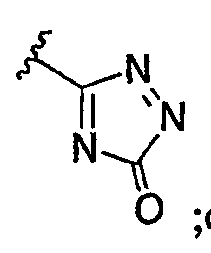 ; или
; или
vi) -(С=S)NH-нафтилом или -(С=О)нафтилом, или
b) когда R3 представляет собой замещенный или незамещенный фенил, тогда R2 не является фенилом, замещенным в параположении оксазолом, тиазолом, тиадиазолом, оксадиазолом, тетразолом, триазолом, диазолом или пиразолом;
с) когда R3 представляет собой фенил, пиридил, пиримидиндион или циклогексил и R1 представляет собой водород, тогда R2 не представляет собой фенил, одновременно замещенный одной группой OMe в метаположении и одной группой оксазола в параположении;
d) когда R3 представляет собой 4-Cl-фенил или 3,4-ди-Cl-фенил, тогда R2 не представляет собой п-Cl-фенил;
е) когда R3 представляет собой незамещенный пиримидинил, тогда R2 не является незамещенным фенилом, п-ОМе-замещенным фенилом, п-OEt-замещенным фенилом или о-ОМе-замещенным фенилом, или, когда R3 представляет собой 4-Ме-пиримидинил или 4,6-диметилпиримидинил, тогда R2 не является незамещенным фенилом;
f) исключаются соединения формулы I:
g) когда R2 представляет собой 3-пиридинил и R1 представляет собой водород, тогда R3 не является триметоксибензоилом;
h) когда R3 представляет собой необязательно замещенный фенил и R1 представляет собой водород, тогда R2 не может быть -(C=S)NH(C=O)фенилом, -(С=О)NHфенилом, -(С=S)NHфенилом или -(С=О)СН2(С=О)фенилом;
i) когда R1 представляет собой водород, R2 представляет собой незамещенный бензил, тогда R3 не может быть тиадиазолом, замещенным необязательно замещенным фенилом;
j) когда R1 представляет собой водород, R2 представляет собой пиридил и R3 представляет собой пиридил, тогда R2 не может быть замещен одним или несколькими CF3, Me, OMe, Br или Cl;
k) когда R1 представляет собой водород, R2 представляет собой пиридил, тогда R3 не является незамещенным пиридилом, незамещенным хинолином, незамещенным фенилом или незамещенным изохинолином;
l) когда R1 представляет собой водород и R2 представляет собой незамещенный хинолин, тогда R3 не может быть незамещенным пиридилом или незамещенным хинолином;
m) когда R1 представляет собой водород, R2 представляет собой незамещенный изохинолин или незамещенный нафтил, тогда R3 не является незамещенным пиридилом;
n) исключены соединения формулы I, имеющие общую структуру:
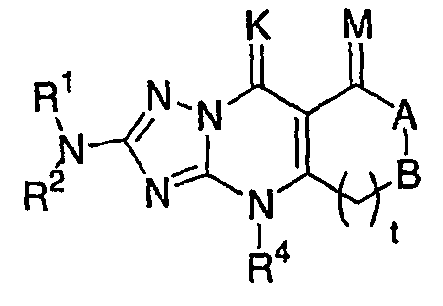 ,
,
в которой R1,R2 и R3 имеют значения, указанные выше, М и К представляют собой О или Н2, при условии что К и М являются разными, А и В представляют собой, каждый, -СН2-, -NH-, -N-алкил, N-аралкил, -NCORa, -NCONHRb или -NCSNHRb, где Ra представляет собой низший алкил или аралкил и Rb представляет собой алкил с неразветвленной или разветвленной цепью, аралкил или арил, который может быть либо незамещенным либо замещенным одним или несколькими алкильными и/или галогеналкильными заместителями;
о) исключены соединения формулы I, имеющие общую структуру:
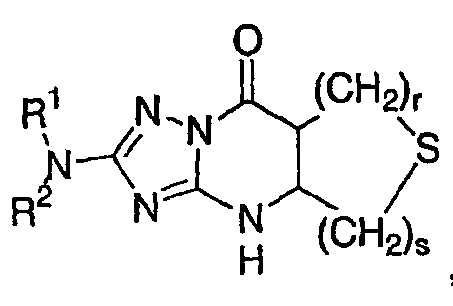 ,
,
в которой R1 и R2 имеют значения, указанные выше, и r и s равны, каждый независимо, 0, 1, 2, 3 или 4, при условии что сумма s и r равна, по меньшей мере, 1;
р) исключено любое одно или несколько или все из соединений формулы I, имеющих следующие структуры:
где R2 представляет собой NH(CH)(Ph)C=O(Ph);
где R2 представляет собой незамещенный фенил или фенил, замещенный ОМе, Cl или Ме;
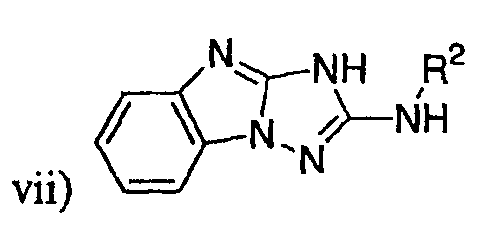 ,
,
где R2 представляет собой незамещенный фенил или фенил, замещенный ОМе, Cl, Ме, ОМе, или R2 представляет собой незамещенный бензил;
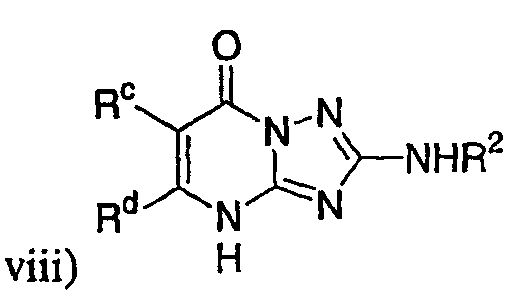 ,
,
где R2 представляет собой необязательно замещенный аралкил и Rc и Rd представляют собой, каждый независимо, Ме, водород, СН2Cl или Cl;
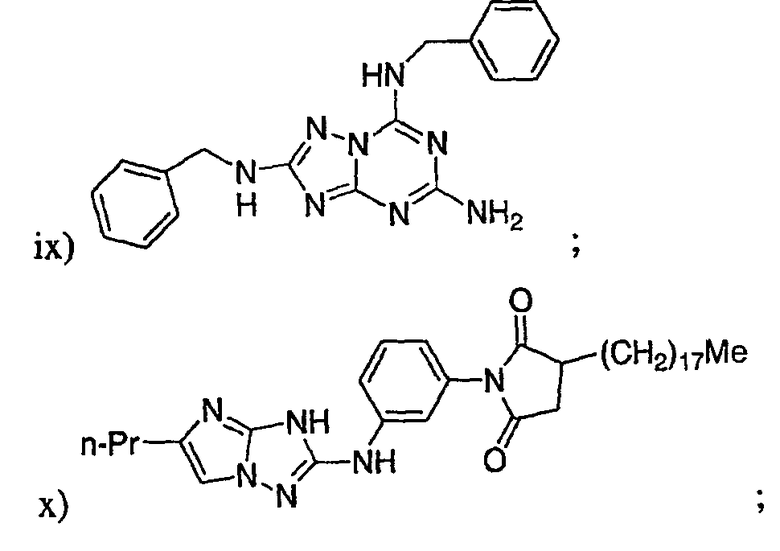
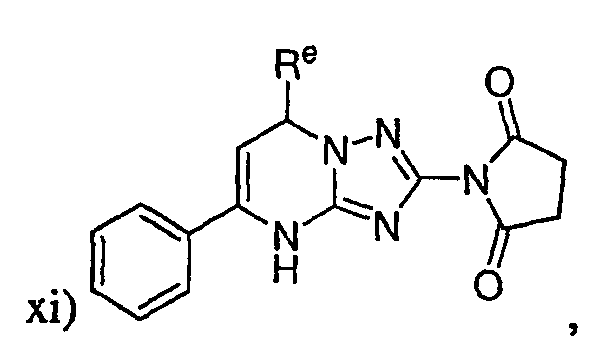
где Re представляет собой необязательно замещенный фенил;
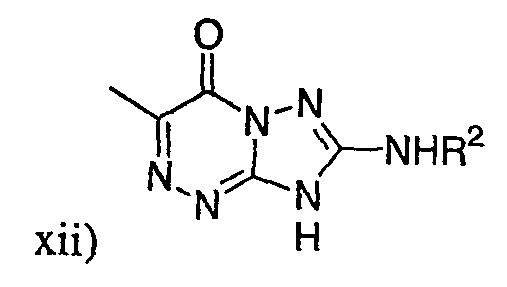 ,
,
где R2 представляет собой фенил, необязательно замещенный Ме, ОМе, Br или Cl; или
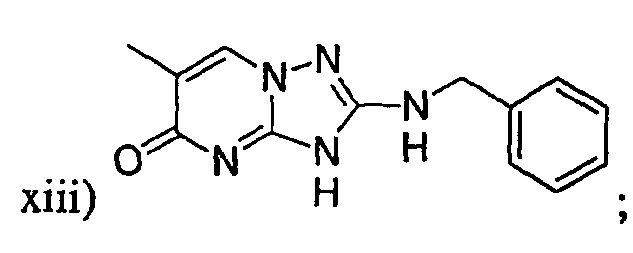
q) когда R1 представляет собой водород и R2 представляет собой фенил или необязательно замещенный фенил и m равно 1, тогда L не может быть -СО-, -СОСН2- или -СОСН=СН-.
В некоторых других вариантах осуществления для соединений формулы I, когда R1 представляет собой водород и R2 представляет собой фенил или необязательно замещенный фенил и m равно 1, тогда L не может быть -СО-, -CS-, -CONR-, -CSNR-, -SO2-, -SO2NR-, -COSO2- или -CSSO2-.
Соединения данного изобретения включают в себя соединения, в общем описанные выше, и дополнительно иллюстрированы описанными здесь классами, подклассами и видами. Будут применяться используемые здесь следующие определения, если не указано особо. Для целей данного изобретения химические элементы идентифицируют в соответствии с Периодической таблицей элементов, версии CAS, Handbook of Chemistry and Physics, 75th Ed. Кроме того, общие принципы органической химии описываются в "Organic Chemistry", Thomas Sorrell, University Science Books, Sausalito: 1999, and "March's Advanced Organic Chemistry", 5th Ed., Ed.: Smith, M.B. and March, J., John Wiley & Sons, New York: 2001, полное содержание которой таким образом включается в качестве ссылки.
Как описано здесь, соединения изобретения могут быть необязательно замещены одним или несколькими заместителями, такими, как иллюстрированы в общем выше или приводятся в качестве примеров конкретными классами, подклассами и видами изобретения. Должно быть принято, что фразу «необязательно замещенный» используют взаимозаменяемым образом с фразой «замещенный или незамещенный». В общем термин «замещенный» вне зависимости от того, предшествует ли ему термин «необязательно» или нет, относится к замене радикалов водорода в данной структуре радикалом определенного заместителя. Если не оговорено особо, необязательно замещенная группа может иметь заместитель у каждого замещаемого положения группы, и когда более чем одно положение в любой данной структуре может быть замещено более чем одним заместителем, выбранным из определенной группы, заместители могут быть одинаковыми или различными в разных положениях. Комбинации заместителей, рассматриваемые данным изобретением, являются предпочтительно такими, чтобы они привели к образованию стабильных или химически возможных соединений. Термин «стабильный», используемый здесь, относится к соединениям, которые по существу не изменяются, когда их подвергают обработке в условиях их получения, определения и предпочтительно их выделения, очистки и применения для одной или нескольких описанных здесь целей. В некоторых вариантах осуществления стабильное соединение или химически возможное соединение является соединением, которое по существу не изменяется при температуре 40°С или ниже, в отсутствие влаги или других химически реакционных условий в течение, по меньшей мере, недели.
Термин «алифатический» или «алифатическая группа», используемый здесь, означает прямую (т.е. неразветвленную) или разветвленную, замещенную или незамещенную углеводородную цепь, которая является полностью насыщенной или которая содержит одно или несколько ненасыщенных звеньев, или моноциклический углеводород или бициклический углеводород, который является полностью насыщенным или который содержит одно или несколько ненасыщенных звеньев, но который не является ароматическим (это относится также здесь к «карбоциклу», «циклоалифатическому» или «циклоалкилу»), который имеет одну точку присоединения к остальной части молекулы. Если не оговорено особо, алифатические группы содержат 1-20 алифатических атомов углерода. В некоторых вариантах осуществления алифатические группы содержат 1-10 алифатических атомов углерода. В других вариантах осуществления алифатические группы содержат 1-8 алифатических атомов углерода. Еще в других вариантах осуществления алифатические группы содержат 1-6 алифатических атомов углерода и в других вариантах осуществления алифатические группы содержат 1-4 алифатических атома углерода. В некоторых вариантах осуществления термин «циклоалифатический» (или «карбоцикл» или «циклоалкил») относится к моноциклическому С3-С8-углеводороду или бициклическому С8-С12-углеводороду, который является полностью насыщенным или который содержит одно или несколько ненасыщенных звеньев, но который не является ароматическим и который имеет одну точку присоединения к остальной части молекулы и в котором любое отдельное кольцо в указанной бициклической системе колец имеет 3-7 членов. Подходящие алифатические группы включают в себя, но не ограничиваются перечисленным, неразветвленные или разветвленные, замещенные или незамещенные алкильные, алкенильные, алкинильные группы и их сочетания (гибриды), такие как (циклоалкил)алкил, (циклоалкенил)алкил или (циклоалкил)алкенил.
Термин «гетероалифатический», используемый здесь, означает алифатические группы, в которых один или два атома углерода независимо заменены одним или несколькими атомами кислорода, серы, азота или кремния. Гетероалифатические группы могут быть замещенными или незамещенными, разветвленными или неразветвленными, циклическими или ациклическими и включают в себя «гетероцикл», «гетероциклил», «гетероциклоалифатические» или «гетероциклические» группы.
Термин «гетероцикл», «гетероциклил», «гетероциклоалифатическая» или «гетероциклическая группа», используемый здесь, означает неароматические моноциклические, бициклические или трициклические системы колец, в которых один или несколько членов колец являются независимо выбранными гетероатомами. В некоторых вариантах осуществления «гетероцикл», «гетероциклил», «гетероциклоалифатическая» или «гетероциклическая» группа имеет от трех до четырнадцати членов, где один или несколько членов кольца являются гетероатомами, независимо выбранными из кислорода, серы, азота или фосфора, и где каждое кольцо является 3-7-ми членным кольцом.
Термин «гетероатом» означает один или несколько атомов кислорода, серы, азота, фосфора или кремния (в том числе любую окисленную форму азота, серы, фосфора или кремния, кватернизованную форму любого азотистого основания или замещаемый атом азота гетероциклического кольца, например, N (как в 3,4-дигидро-2Н-пирролиле), NH (как в пирролидиниле) или NR+ (как в N-замещенном пирролидиниле)).
Термин «ненасыщенный», используемый здесь, означает, что группа имеет одно или несколько ненасыщенных звеньев.
Термин «алкокси» или «тиоалкил», используемый здесь, относится к алкильной группе, как указано ранее, присоединенной к основной углеродной цепи через атом кислорода («алкокси») или серы («тиоалкил»).
Термин «галогеналкил», «галогеналкенил» и «галогеналкокси» означает алкил, алкенил или алкокси в зависимости от того, какой случай может быть, замещенный одним или несколькими атомами галогена. Термин «галоген» означает F, Cl, Br или I.
Термин «арил», используемый отдельно или как часть большей группы, как в «аралкиле», «аралкокси» или «арилоксиалкиле», относится в моноциклическим, бициклическим и трициклическим системам колец, имеющим всего от пяти до четырнадцати членов колец, где, по меньшей мере, одно кольцо в системе является ароматическим и где каждое кольцо в системе содержит от 3 до 7 членов. Термин «арил» может быть использован взаимозаменяемым образом с термином «арильное кольцо». Термин «арил» относится также к гетероарильным кольцевым системам, как указано здесь выше.
Термин «гетероарил», используемый отдельно или как часть большей группы, как в «гетероаралкиле» или «гетероарилалкокси», относится в моноциклическим, бициклическим и трициклическим системам колец, имеющим всего от пяти до четырнадцати членов в кольце, где, по меньшей мере, одно кольцо в системе является ароматическим, по меньшей мере, одно кольцо в системе содержит одно или несколько гетероатомов и где каждое кольцо в системе содержит от 3 до 7 членов. Термин «гетероарил» может быть использован взаимозаменяемым образом с термином «гетероароматическое кольцо» или термином «гетероароматический».
Арильная (в том числе аралкильная, аралкокси, арилоксиалкильная и тому подобное) или гетероарильная (в том числе гетероаралкильная и гетероарилалкокси и тому подобное) группа может содержать один или несколько заместителей и, таким образом, может быть «необязательно замещенной». Если не оговорено особо выше и здесь, подходящие заместители на ненасыщенном атоме углерода арильной или гетероарильной группы обычно выбраны из галогена; -R°; -OR°; -SR°; фенила (Ph), необязательно замещенного -R°; -O(Ph), необязательно замещенного -R°; -(СН2)1-2(Ph), необязательно замещенного -R°; -СН=СН(Ph), необязательно замещенного -R°; -NO2; -CN; -N(R°)2, -NR°C(O)R°; -NR°C(S)R°; -NR°C(O)N(R°)2; -NR°C(S)N(R°)2; -NR°CO2R°; -NR°NR°C(O)R°; -NR°NR°C(O)N(R°)2; -NR°NR°CO2R°; -С(О)С(О)Ro; -C(O)CH2C(O)R°; -CO2R°; -C(O)R°; -C(S)R°; -C(O)N(R°)2; -C(S)N(R°)2; -OC(O)N(R°)2; -OC(O)R°; -C(O)N(OR°)R°; -C(NOR°)R°; -S(O)2R°; -S(O)3R°; -SO2N(R°)2; -S(O)R°; -NR°SO2N(R°)2, -NR°SO2R°; -N(ORo)Ro, -C(=NH)-N(R°)2, -P(O)2R°; -PO(R°)2, -OPO(R°)2; -(CH2)0-2NHC(O)R°; фенила (Ph), необязательно замещенного R°; -О(Ph), необязательно замещенного R°; -(СН2)1-2(Ph), необязательно замещенного R°; или -СН=СН(Ph), необязательно замещенного R°, где в каждом независимом случае R° выбран из водорода, необязательно замещенной С1-6алифатической группы, ненасыщенного 5-6-членного гетероарильного или гетероциклического кольца, фенила, -О(Ph) или -СН2(Ph) или, несмотря на приведенные выше значения, два независимых R° на одном и том же заместителе или на разных заместителях, взятые вместе с атомом(ами), с которым каждый R° связан, образуют необязательно замещенное 3-12-членное насыщенное, частично ненасыщенное или полностью ненасыщенное моноциклическое или бициклическое кольцо, имеющее 0-4 гетероатома, выбранные из азота, кислорода или серы.
Необязательные заместители на алифатической группе R° выбраны из NH2, NH(С1-4алифатической группы), N(С1-4алифатической)2 группы, галогена, С1-4алифатической группы, ОН, О(С1-4алифатической) группы, NO2, CN, CO2Н, СО2(С1-4алифатической) группы, О(галогенС1-4алифатической) или галоген(С1-4алифатической) группы, где каждая из вышеуказанных С1-4алифатических групп R° является незамещенной.
Алифатическая или гетероалифатическая группа или неароматическое гетероциклическое кольцо может содержать один или несколько заместителей и, таким образом, может быть «необязательно замещенной». Если не оговорено особо выше и здесь, подходящие заместители на насыщенном атоме углерода алифатической или гетероалифатической группы или неароматического гетероциклического кольца выбраны из заместителей, перечисленных выше для ненасыщенного атома углерода арильной или гетероарильной группы и дополнительно включают в себя следующие заместители: =О, =S, =NNHR*, =NN(R*)2, =NNHC(O)R*, =NNHCO2(алкил), =NNHSO2(алкил) или =NR*, где каждый R* независимо выбран из водорода или необязательно замещенной С1-6алифатической группы.
Если не оговорено особо выше и здесь, необязательные заместители на атоме азота неароматического гетероциклического кольца обычно выбраны из -R+, -N(R+)2, -C(O)R+, -CO2R+, -C(O)C(O)R+, -C(O)CH2C(O)R+, -SO2R+, -SO2N(R+)2, -C(=S)N(R+1)2, -C(=NH)N(R+)2 или -NR+SO2R+, где R+ представляет собой водород, необязательно замещенную С1-6алифатическую группу, необязательно замещенный фенил, необязательно замещенный -О(Ph), необязательно замещенный -СН2(Ph), необязательно замещенный -(СН2)1-2(Ph); необязательно замещенный -СН=СН(Ph) или незамещенный 5-6-членный гетероарил или гетероциклическое кольцо, имеющее от одного до четырех гетероатомов, независимо выбранных из кислорода, азота или серы, или несмотря на приведенные выше значения две независимых группы R+ на одном и том же заместителе или на разных заместителях, взятые вместе с атомом(ами), с которым каждый R+ связан, образуют необязательно замещенное 3-12-членное насыщенное, частично ненасыщенное или полностью ненасыщенное моноциклическое или бициклическое кольцо, имеющее 0-4 гетероатома, выбранные из азота, кислорода или серы.
Необязательные заместители на алифатической группе или фенильном кольце R+ выбраны из -NH2, -NH(С1-4алифатической), N(С1-4алифатической)2 групп, галогена, С1-4алифатической группы, -ОН, -О(С1-4алифатической) группы, -NO2, -CN, -CO2Н, -СО2(С1-4алифатической), -О(галогенС1-4алифатической) или галоген(С1-4алифатической) группы, где каждая из вышеуказанных С1-4алифатических групп R+ является незамещенной.
Термин «алкиленовая цепь» относится к неразветвленной или разветвленной углеродной цепи, которая может быть полностью насыщенной или имеет одно или несколько ненасыщенных звеньев и имеет две точки присоединения к остальной части молекулы.
Как подробно указано выше, в некоторых вариантах осуществления два независимых R° (или R+, R, R' или любых других символов, аналогично указанных здесь) взяты вместе с атомом(ами), к которому они присоединены, с образованием необязательно замещенного 3-12-членного насыщенного, частично ненасыщенного или полностью ненасыщенного моноциклического или бициклического кольца, имеющего 0-4 гетероатома, независимо выбранных из азота, кислорода или серы.
Примеры колец, которые образуются, когда два независимых R° (или R+, R, R' или любых других символов, аналогично указанных здесь) взяты вместе с атомом(ами), с которым они связаны, включают в себя кольца, но не ограничиваются перечисленными: а) когда два независимых R° (или R+, R, R' или любых других символа, аналогично указанных здесь) связываются с одним и тем же атомом и взяты с этим атомом с образованием кольца, например, N(R°)2, где оба R° взяты вместе с атомом азота с образованием пиперидин-1-ильной, пиперазин-1-ильной или морфолин-4-ильной группы; и b) два независимых R° (или R+, R, R' или любых других символов, аналогично указанных здесь) связываются с различными атомами и взяты вместе с этими атомами с образованием кольца, например, когда фенильная группа замещена двумя OR°  эти два R° взяты вместе с атомами кислорода, к которым они присоединены, с образованием конденсированного 6-членного кислородсодержащего кольца
эти два R° взяты вместе с атомами кислорода, к которым они присоединены, с образованием конденсированного 6-членного кислородсодержащего кольца  . Должно быть понятно, что различные другие кольца могут быть образованы, когда два независимых R° (или R+, R, R' или любых других символа, аналогично указанных здесь) взяты вместе с атомом(ами), к которому каждый символ присоединен, и примеры таких колец, указанных подробно выше, не предназначены для ограничения.
. Должно быть понятно, что различные другие кольца могут быть образованы, когда два независимых R° (или R+, R, R' или любых других символа, аналогично указанных здесь) взяты вместе с атомом(ами), к которому каждый символ присоединен, и примеры таких колец, указанных подробно выше, не предназначены для ограничения.
Имеется в виду, что если не оговорено особо, структуры, приведенные здесь, включают в себя все изомерные (например, энантиомерные, диастереомерные и геометрические (или конформационные) формы структуры; например, R- и S-конфигурации для каждого асимметричного центра, (Z)- (Е)-изомеры по двойной связи и (Z)- и (Е)-конформационные изомеры. Таким образом, индивидуальные стереохимические изомеры, а также смеси энантиомерных, диастереомерных и геометрических (или конформационных) изомеров настоящих соединений находятся в пределах объема изобретения. Если не оговорено особо, все таутомерные формы соединений изобретения находятся в пределах объема изобретения. Кроме того, если не оговорено особо, имеется также в виду, что структуры, изображенные здесь, включают в себя соединения, которые отличаются только присутствием одного или нескольких обогащенных изотопами атомов. Например, соединения, имеющие указанные структуры, за исключением замены водорода дейтерием или тритием или замены углерода углеродом, обогащенным 13С или 14С, находятся в пределах объема изобретения. Такие соединения являются применимыми, например, в качестве аналитических приспособлений или зондов в биологических анализах.
Как описано в общем выше для соединений формулы I, в некоторых вариантах осуществления R2 представляет собой -(Т)nAr1. В некоторых вариантах осуществления Ar1 выбран из одной из следующих групп:
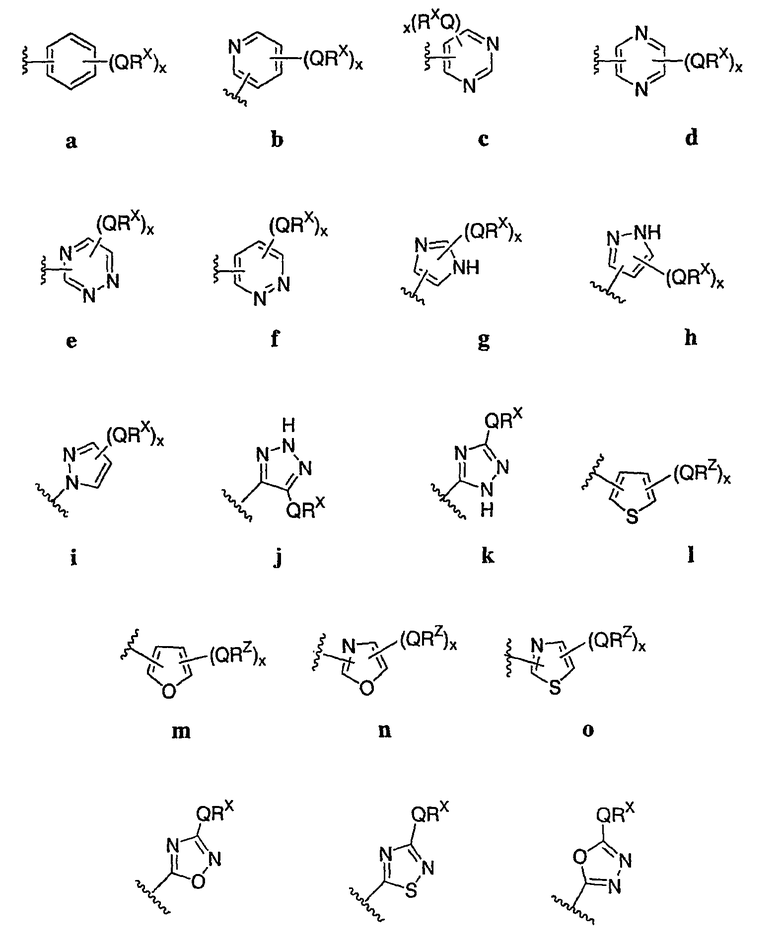
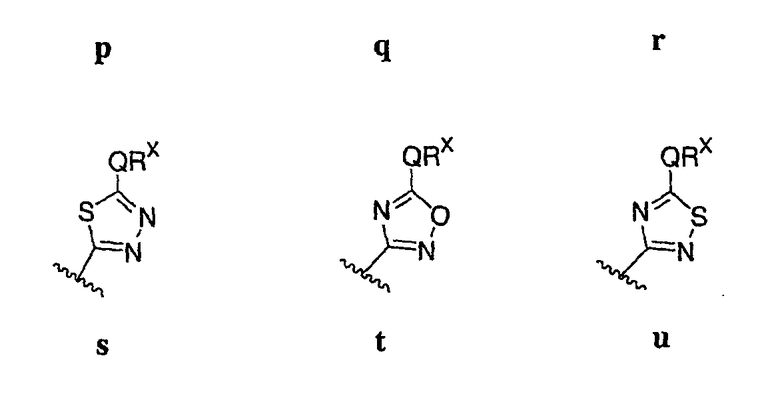
где значения Q и Rx указаны в общем выше и указаны здесь в классах и подклассах, и х равно 0-5.
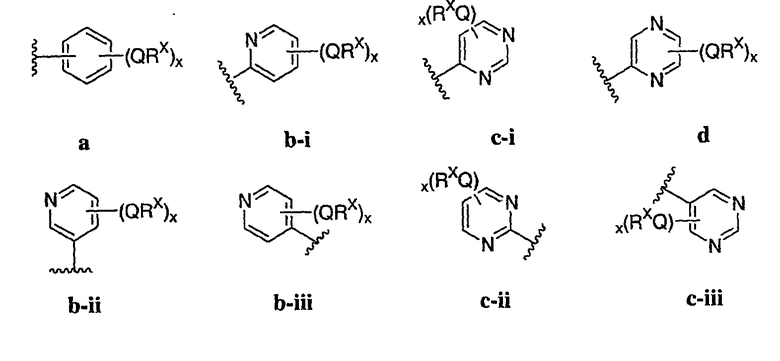
В других вариантах осуществления Ar1 представляет собой фенил (а), пиридил (b) (предпочтительно присоединенный в 2-, 3- или 4-положении, как показано в b-i, b-ii и b-iii), пиримидинил (с) (предпочтительно присоединенный в 2-, 4- или 5-положении, как показано в с-i, с-ii и с-iii)
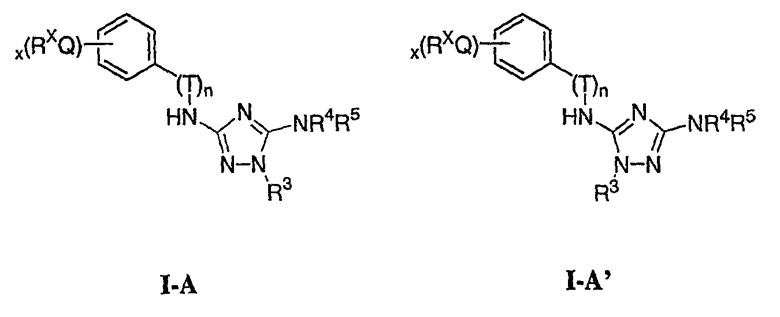
В следующих вариантах осуществления R1 представляет собой водород, Ar1 представляет собой фенил (а) и соединения имеют формулу I-A или I-A':
В других вариантах осуществления R2 представляет собой (Т)nCy1. В предпочтительных вариантах осуществления Cy1 выбран из одной из следующих групп:
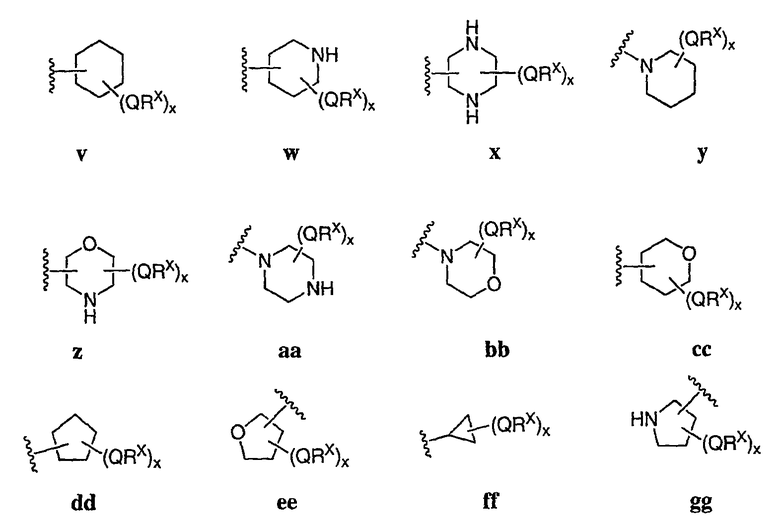
где любой замещаемый атом углерода или азота необязательно замещен QRx и где Q и Rx имеют значения, в общем указанные выше и указанные здесь в классах и подклассах, и х равно 0-5.
В следующих вариантах осуществления Cy1 выбран из одной из следующих групп:
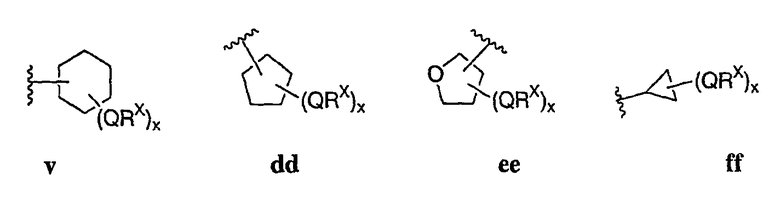
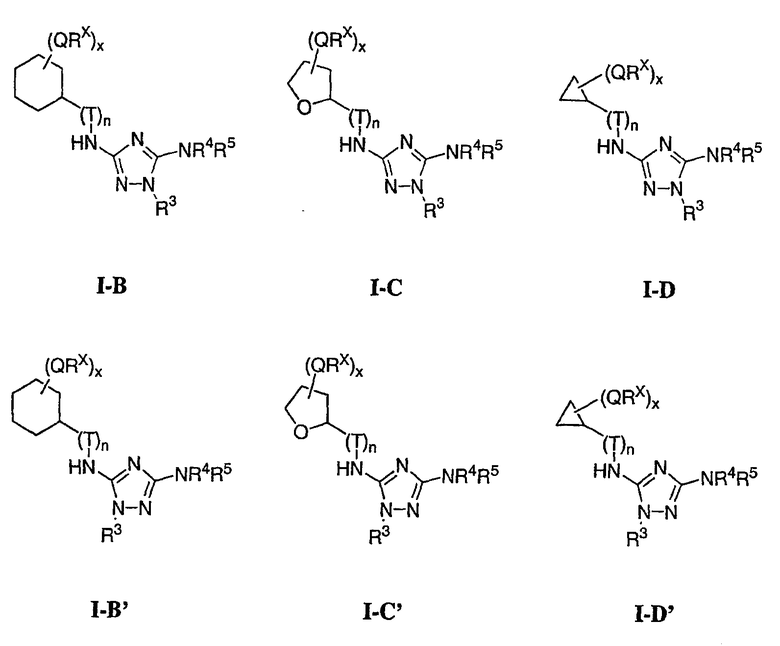
В других вариантах осуществления R1 представляет собой водород, Cy1 представляет собой циклогексил (v), тетрагидрофуранил (ее) (предпочтительно присоединенный в 2-положении) или циклопропил (ff) и соединения имеют одну из следующих формул I-B, I-C, I-D, I-B', I-C' или I-D':
В некоторых вариантах осуществления R1 представляет собой водород, С1-4алкил, -СН2OCOR', -СН2OCOCHRNRR', COOR', -COCHROCOR', COR', -CO(CH2)3NHR', где R' представляет собой С1-6алкил или алкилдиоксолон. В наиболее предпочтительных вариантах осуществления R1 представляет собой водород.
Примеры групп Т, когда они присутствуют, включают в себя СН2 и -СН2СН2-. В некоторых других вариантах осуществления n равно 0 и Т отсутствует.
Как подробно указано выше, Ar1 или Cy1 может быть необязательно замещен у одного или нескольких замещаемых атомов углерода или азота до 5 группами QRx. В некоторых вариантах осуществления х равно 0-3 и Ar1 или Cy1, каждый независимо, замещен 0-3 группами QRx. В следующих вариантах осуществления х равно 0 и Ar1 или Cy1 является незамещенным.
В некоторых вариантах осуществления группы QRx представляют собой, каждый независимо, R', галоген, CN, NO2, -N(R')2, -CH2N(R')2, -OR', CH2OR', -SR', -CH2SR', -COOR', -NRCOR', -CON(R')2, -SO2N(R')2, -CONR(CH2)2N(R')2, -CONR(CH2)3N(R')2, -CONR(CH2)4N(R')2, -O(CH2)2OR', -O(CH2)3OR', -O(CH2)4OR', -O(CH2)2N(R')2, -O(CH2)3N(R')3 или -O(CH2)4N(R')2. В других вариантах осуществления группы QRx представляют собой, каждая независимо, Cl, Br, F, CF3, Me, Et, CN, -COOH, -N(CH3)2, -N(Et)2, -N(iPr)2, -О(СН2)ОСН3, -CONH2, -COOCH3, -ОН, -СН2ОН, -NHCOCH3, -SO2NH2, метилендиокси, этилендиокси, -О(СН2)2N-морфолино, -О(СН2)3N-морфолино, -О(СН2)4N-морфолино, -О(СН2)2N-пиперазинил, -О(СН2)3N-пиперидинил, -О(СН2)4N-пиперидинил, -NHCH(CH2OH)фенил, -CONH(CH2)2N-морфолино, -CONH(CH2)2N-пиперазинил, -CONH(CH2)3N-морфолино, -CONH(CH2)3N-пиперазинил, -CONH(CH2)4N-морфолино, -CONH(CH2)4N-пиперазинил, -SO2NH(CH2)2N-морфолино, -SO2NH(CH2)2N-пиперазинил, -SO2NH(CH2)3N-морфолино, -SO2NH(CH2)3N-пиперазинил, -SO2NH(CH2)4N-морфолино, -SO2NH(CH2)4N-пиперазинил, где каждая из указанных выше фенильных, морфолино, пиперазинильных или пиперидинильных групп является необязательно замещенной или необязательно замещенной группой, выбранной из С1-4алкокси, фенила, фенилокси, бензила, пиперидинила, пиперазинила, морфолино или бензилокси. В некоторых вариантах осуществления атом азота пиперидинильной или пиперазинильной группы необязательно замещен -COR'. В некоторых других вариантах осуществления атом азота пиперидинильной или пиперазинильной группы необязательно замещен -СОСН2CN или -СОСН3. Примеры групп QRx также включают в себя группы, показанные ниже в таблице 1.
Должно быть понятно, что некоторые классы соединений общей формулы I являются особенно интересными. В некоторых вариантах осуществления настоящее изобретение предлагает моноциклические производные триазола, у которых R4 и R5 представляют собой, каждый, водород, эти соединения имеют общую формулу II или II':
где R1 и R2 имеют значения, указанные выше в общем, и указываются здесь в классах и подклассах; R3 представляет собой (L)mAr2 или (L)mCy2, где L представляет собой необязательно замещенную C1-4алкилиденовую цепь, в которой одно метиленовое звено L необязательно заменено на -NR-, -S-, -O-, -CS-, -CO2-, -OCO-, -CO-, -COCO-, -CONR-, -NRCO-, -NRCO2-, -SO2NR-, -NRSO2-, -CONRNR-, -NRCONR-, -OCONR-, -NRNR-, -NRSO2NR-, -SO-, -SO2-, -PO-, -PO2- или -POR-; m равно 0 или 1; Ar2 представляет собой необязательно замещенную арильную группу, выбранную из 5-6-членного моноциклического или 8-12-членного бициклического кольца, имеющего 0-5 гетероатомов, независимо выбранных из азота, кислорода или серы; и Cy2 представляет собой необязательно замещенную группу, выбранную из 3-7-членного насыщенного или частично ненасыщенного моноциклического кольца, имеющего 0-3 гетероатома, независимо выбранные из азота, кислорода или серы, или 8-12-членной насыщенной или частично ненасыщенной бициклической системы колец, имеющей 0-5 гетероатомов, независимо выбранных из азота, кислорода или серы; где Ar1 и Cy2, каждый независимо, необязательно замещены y группами Z-Ry, где y равно 0-5, Z представляет собой связь или С1-6-алкилиденовую цепь, в которой до двух метиленовых звеньев Z необязательно заменены на -NR-, -S-, -O-, -CS-, -CO2-, -OCO-, -CO-, -СОСО-, -CONR-, -NRCO-, -NRCO2-, -SO2NR-, -NRSO2-, -CONRNR-, -NRCONR-, -OCONR-, -NRNR-, -NRSO2NR-, -SO-, -SO2-, -PO-, -PO2- или -POR-; в каждом случае Ry представляет собой, независимо, R', галоген, NO2, CN, OR', SR', N(R')2, NR'COR', NR'CONR'2, NR'CO2R', COR', CO2R', OCOR', CON(R')2, OCON(R')2, SOR', SO2R', SO2N(R')2, NR'SO2R', NR'SO2(R')2, COCOR' или СОСН2COR';
в каждом случае R независимо представляет собой водород или необязательно замещенную С1-6алифатическую группу, в каждом случае R' независимо представляет собой водород или необязательно замещенную группу, выбранную из С1-6алифатической группы, 3-8-членного насыщенного, частично ненасыщенного или полностью ненасыщенного моноциклического кольца, имеющего 0-3 гетероатома, независимо выбранные из азота, кислорода или серы, или 8-12-членной насыщенной, частично ненасыщенной или полностью ненасыщенной бициклической системы колец, имеющей 0-5 гетероатомов, независимо выбранных из азота, кислорода или серы, или R и R', R в двух случаях или R' в двух случаях взяты вместе с атомом(ами), с которым они связаны, с образованием необязательно замещенного 3-12-членного насыщенного, частично ненасыщенного или полностью ненасыщенного моноциклического или бициклического кольца, имеющего 0-4 гетероатома, независимо выбранных из азота, кислорода или серы.
В некоторых вариантах осуществления для соединений формулы II, описанных непосредственно выше, учитывается одно или несколько или все из следующих условий:
а) когда R3 представляет собой незамещенный фенил и R1 представляет собой водород, тогда R2 не является
i) незамещенным фенилом;
ii) незамещенным пиридилом;
iii) бензилом, замещенным о-ОМе;
iv) -(C=S)NH(C=O)фенилом;
v) 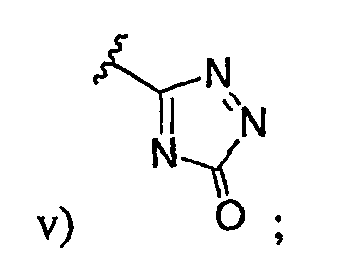 или
или
vi) -(С=S)NH-нафтилом или -(С=О)NHнафтилом, или
b) когда R3 представляет собой замещенный или незамещенный фенил, тогда R2 не является фенилом, замещенным в параположении оксазолом, тиазолом, тиадиазолом, оксадиазолом, тетразолом, триазолом, диазолом или пирролом;
с) когда R3 представляет собой фенил, пиридил, пиримидиндион или циклогексил и R1 представляет собой водород, тогда R2 не представляет собой фенил, одновременно замещенный одной группой ОМе в метаположении и одной группой оксазола в параположении;
d) когда R3 представляет собой 4-Cl-фенил или 3,4-Cl-фенил, тогда R2 не представляет собой п-Cl-фенил;
е) когда R3 представляет собой незамещенный пиримидинил, тогда R2 не является незамещенным фенилом, п-ОМе-замещенным фенилом, п-OEt-замещенным фенилом или о-ОМе-замещенным фенилом, или когда R3 представляет собой 4-Ме-пиримидинил или 4,6-диметилпиримидинил, тогда R2 не является незамещенным фенилом;
f) исключаются соединения формулы I:
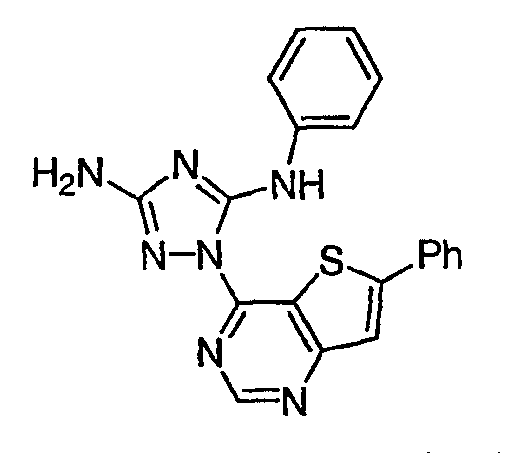
g) когда R2 представляет собой 3-пиридинил и R1 представляет собой водород, тогда R3 не является триметоксибензоилом;
h) когда R3 представляет собой необязательно замещенный фенил и R1 представляет собой водород, тогда R2 не может быть -(C=S)NH(C=O)фенилом, -(С=О)NHфенилом, -(С=S)NHфенилом или -(С=О)СН2(С=О)фенилом;
i) когда R1 представляет собой водород, R2 представляет собой незамещенный бензил, тогда R3 не может быть тиадиазолом, замещенным необязательно замещенным фенилом;
j) когда R1 представляет собой водород, R2 представляет собой пиридил и R3 представляет собой пиридил, тогда R2 не может быть замещен одним или несколькими CF3, Me, OMe, Br или Cl;
k) когда R1 представляет собой водород, R2 представляет собой пиридил, тогда R3 не является незамещенным пиридилом, незамещенным хинолином, незамещенным фенилом или незамещенным изохинолином;
l) когда R1 представляет собой водород и R2 представляет собой незамещенный хинолин, тогда R3 не может быть незамещенным пиридилом или незамещенным хинолином;
m) когда R1 представляет собой водород и R2 представляет собой незамещенный изохинолин или незамещенный нафтил, тогда R3 не является незамещенным пиридилом; или
n) когда R1 представляет собой водород и R2 представляет собой фенил или необязательно замещенный фенил и m равно 1, тогда L не может быть -СО-, -СОСН2- или -СОСН=СН-.
В некоторых других вариантах осуществления для соединений формулы I, когда R1 представляет собой водород и R2 представляет собой фенил или необязательно замещенный фенил и m равно 1, тогда L не может быть -СО-, -CS-, -CONR-, -CSNR-, -SO2-, -SO2NR-, -COSO2 или -CSSO2-.
Как описано в общем выше, в некоторых вариантах осуществления Ar1 выбран из любой одной из формул от а до u, изображенных выше (включая некоторые подгруппы b-i, c-i, b-ii, b-iii, c-ii или c-iii) и в некоторых других вариантах осуществления Cy1 выбран из любой одной формулы от v до ff, изображенных выше. Однако должно быть понятно, что для соединений формулы II, описанных выше, некоторые дополнительные соединения представляют особый интерес. Например, в некоторых приведенных в качестве примеров вариантах осуществления для приведенных выше соединений общей формулы II или II' имеющие определенный интерес соединения включают в себя те соединения, у которых R1 представляет собой водород, Ar1 представляет собой необязательно замещенный фенил и R3 представляет собой -(L)mAr2 или -(L)mCy2, эти соединения имеют формулу II-A-(i), II-A-(ii), II-A-(i)' или II-A-(ii)':

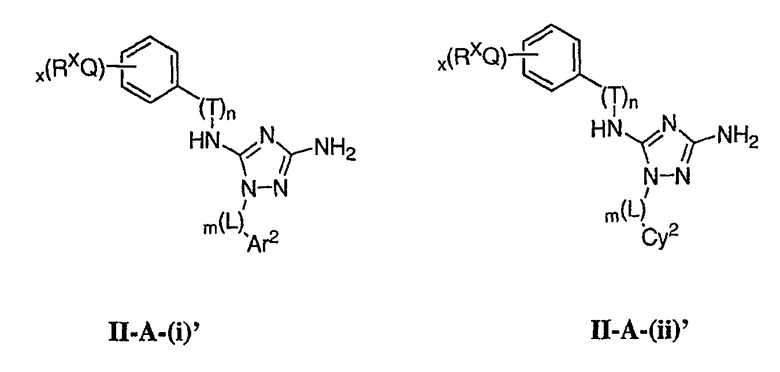
В некоторых вариантах осуществления для соединений, описанных непосредственно выше, m и n, оба, равны 0.
В некоторых других приведенных в качестве примеров вариантов осуществления R1 представляет собой водород, Cy1 представляет собой необязательно замещенный циклогексил, тетрагидрофуранил или циклопропил и R3 представляет собой -(L)mAr2 или -(L)mCy2, эти соединения имеют формулу II-B-(i), II-B-(ii), II-C-(i), II-c-(ii), II-D-(i), II-D-(ii)', II-B-(i)', II-B-(ii)', II-C-(i)', II-C-(ii)', II-D-(i)' или II-D-(ii)'.
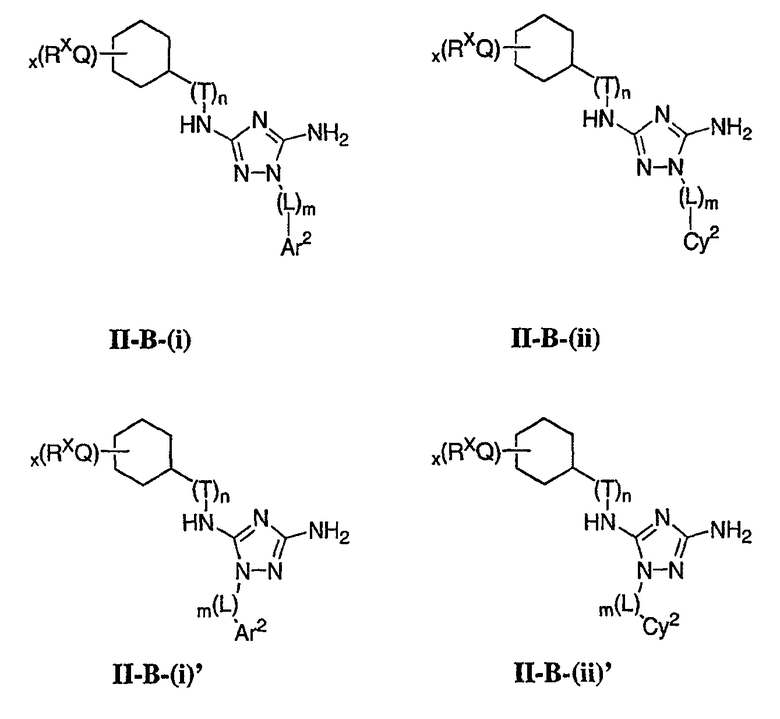
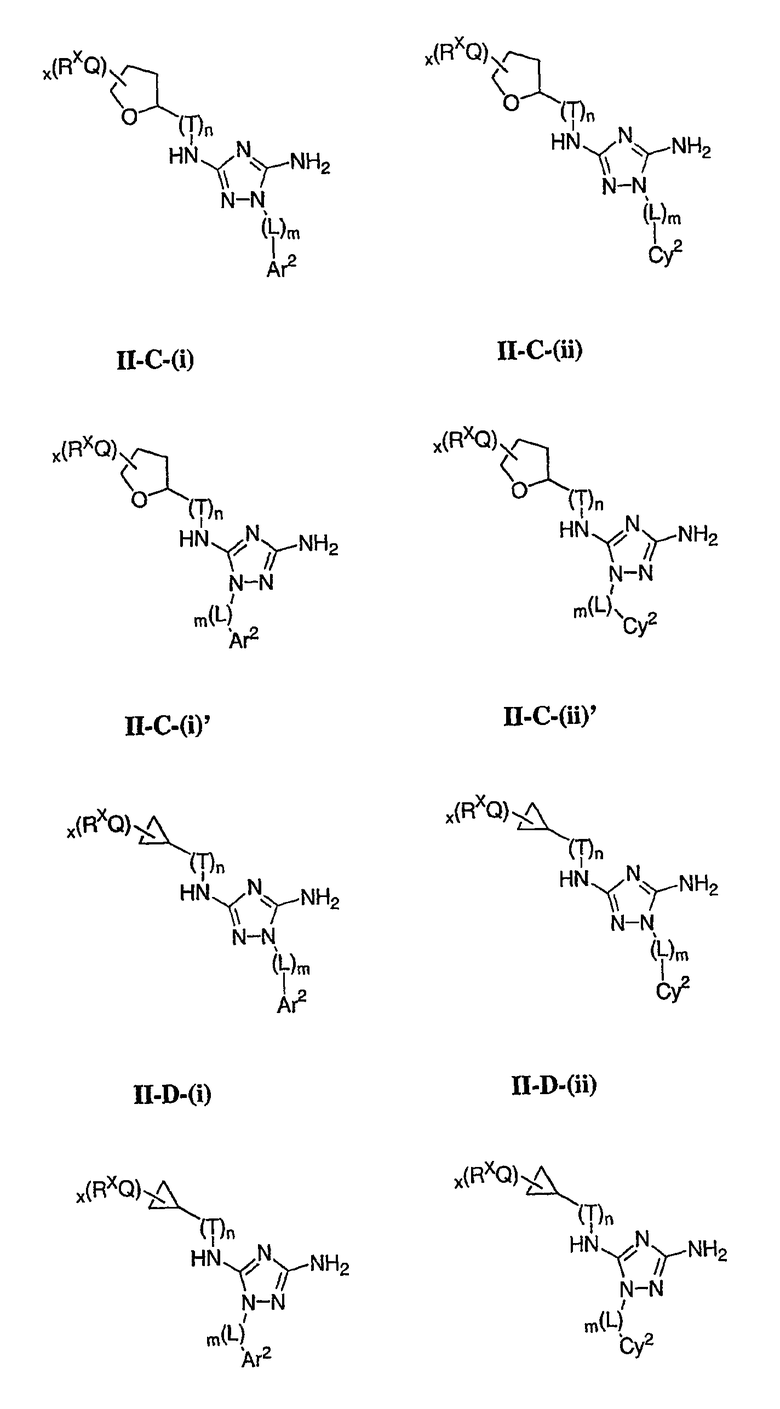

В некоторых вариантах осуществления для соединений, описанных непосредственно выше, m и n, оба, равны 0.
В некоторых других вариантах осуществления для соединений общей формулы II и подгрупп II-A-(i), II-B-(i), II-C-(i), II-D-(i), II-A-(i)', II-B-(i)', II-C-(i)', II-D-(i)', описанных непосредственно выше, Ar2 выбран из одной из следующих групп:
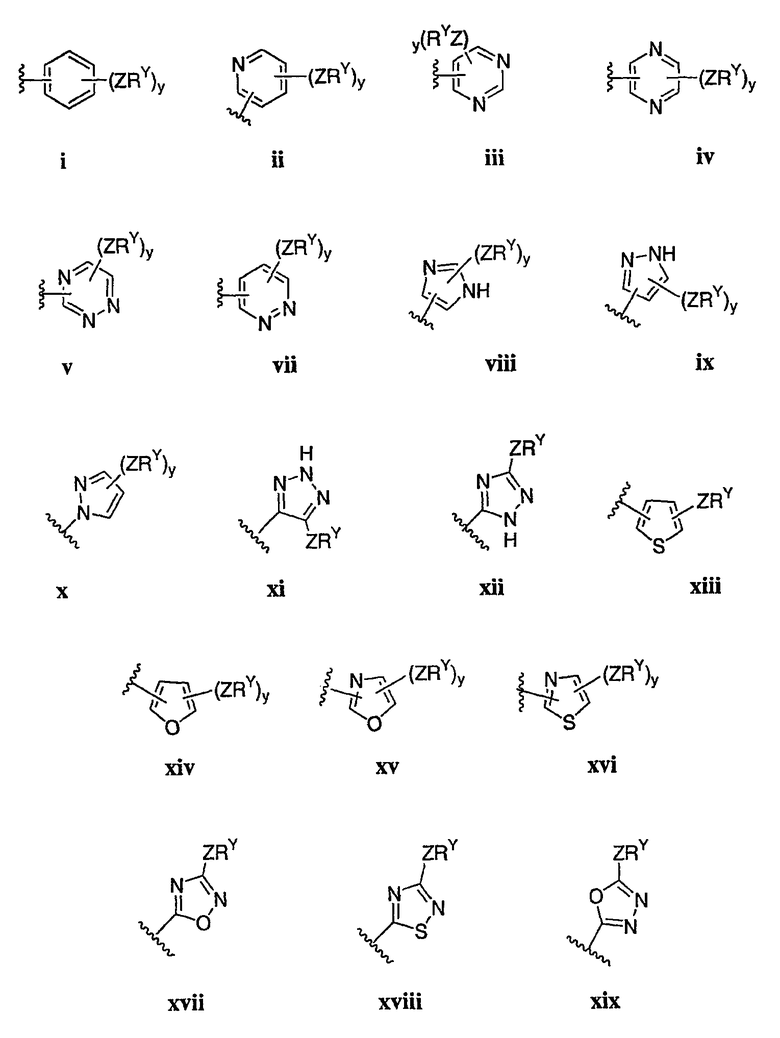
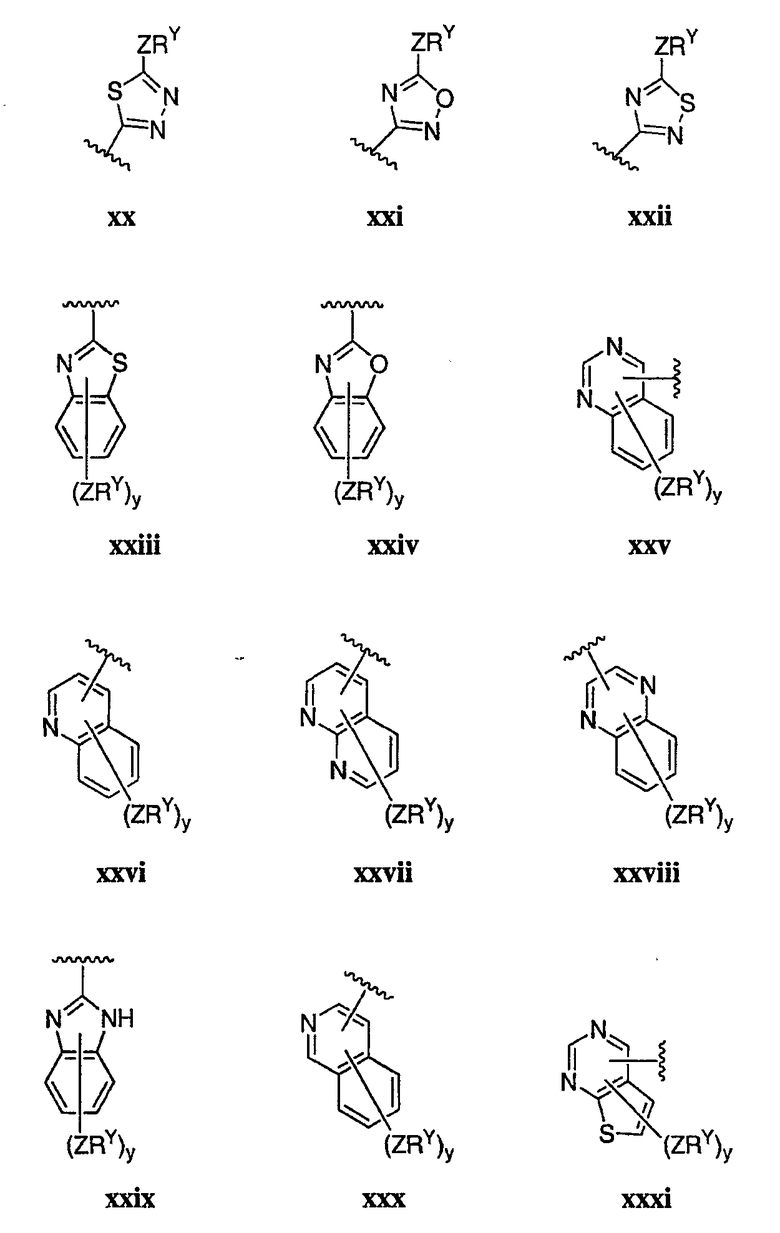
где любой замещаемый атом углерода или азота необязательно замещен ZRY, где Z и RY имеют значения, описанные в общем выше и описанные здесь в классах и подклассах, и y равно 0-5.
В более предпочтительных вариантах осуществления Ar2 выбран из одной из следующих групп:
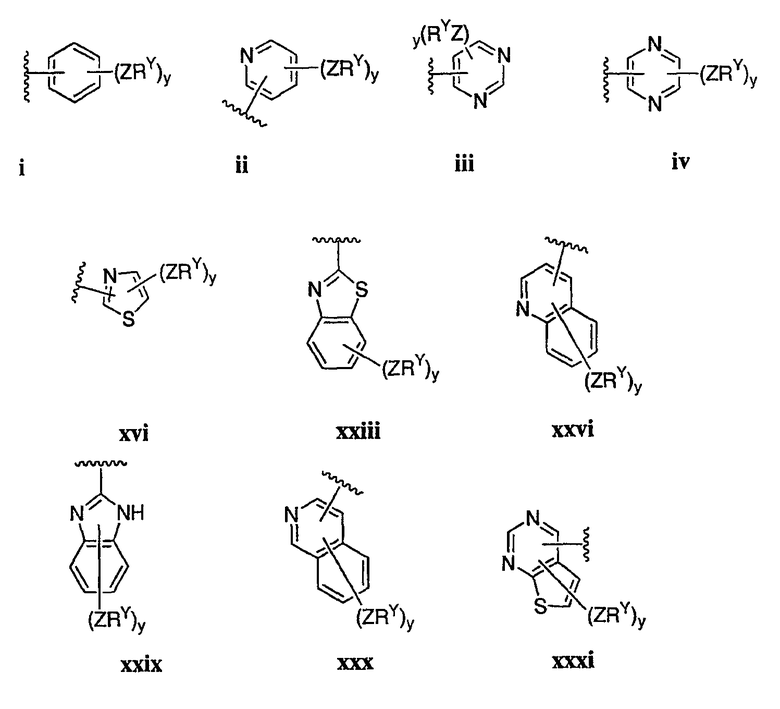
Еще в одних вариантах осуществления для соединений формулы II и подгрупп II-A-(ii), II-B-(ii), II-C-(ii), II-D-(ii), II-A-(ii)', II-B-(ii)', II-C-(ii)', II-D-(ii)' R3 представляет собой -(L)mCy2 и Cy2 выбран из одной из следующих групп:
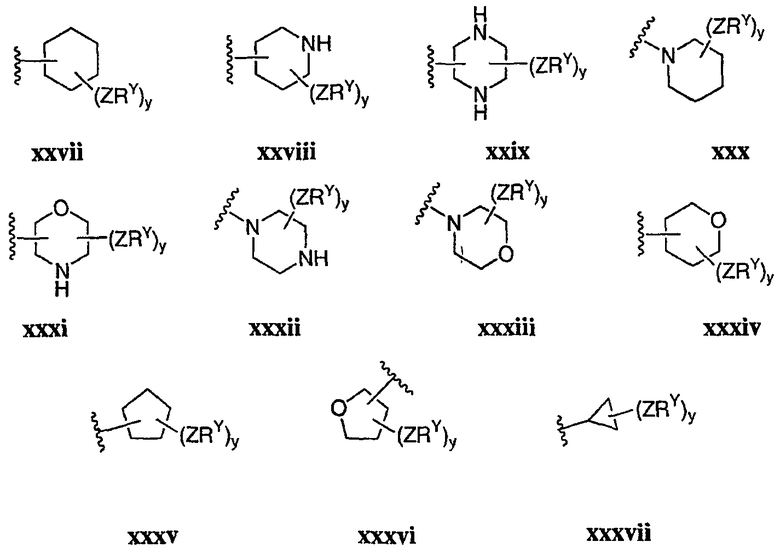
где любой замещаемый атом углерода или азота необязательно замещен ZRY, где Z и RY имеют значения, описанные в общем выше и описанные здесь в классах и подклассах, и y равно 0-5.
В следующих вариантах осуществления Cy2 выбран из одной из следующих групп i-b или viii-b:

В некоторых вариантах осуществления m равно 1 и L представляет собой -О-, -NR- или -СН2-. В других вариантах осуществления m равно 0.
В некоторых других вариантах осуществления для соединений общей формулы II R1 и R2 имеют значения, описанные в общем выше и указанные в классах и подклассах здесь, m равно 0 и Ar2 представляет собой необязательно замещенный фенил, 2-пиридил, 2-тиазолил, 2-пиримидинил, 6-пиримидинил, 4-пиридил, бензотиазолил или 2-хинолинил, эти соединения имеют одну из структур II-E-(i), II-E-(ii), II-F-(i), II-F-(ii), II-G-(i), II-G-(ii), II-G'-(i), II-G'-(ii), II-H-(i), II-H-(ii), II-I-(i), II-I-(ii), II-I'-(i), II-I'-(ii), II-J-(i), II-J-(ii), II-E-(i)', II-E-(ii)', II-F-(i)', II-F-(ii)', II-G-(i)', II-G-(ii)', II-G'-(i)', II-G'-(ii)', II-H-(i)', II-H-(ii)', II-I-(i)', II-I-(ii)', II-I'-(i)', II-I'-(ii)', II-J-(i)', II-J-(ii)':


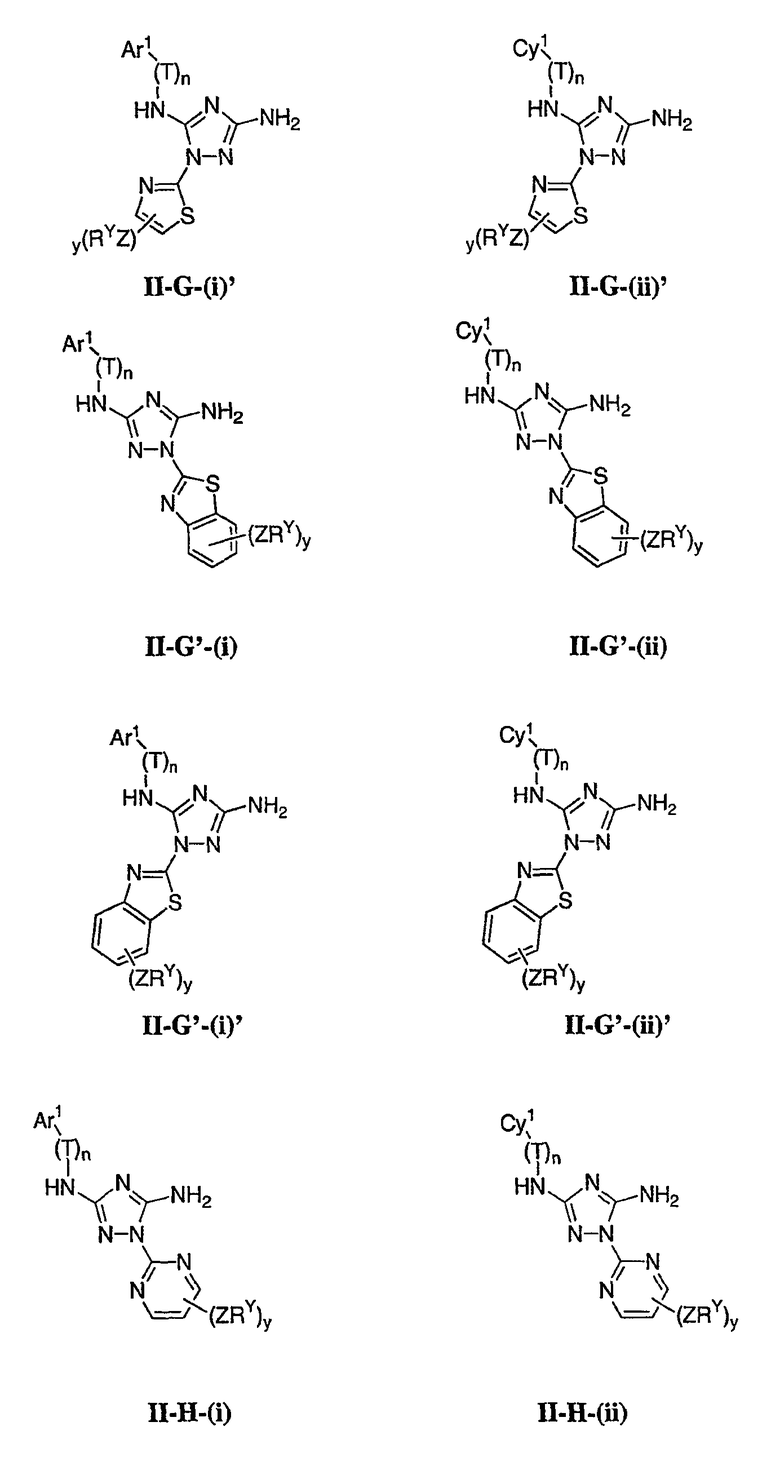
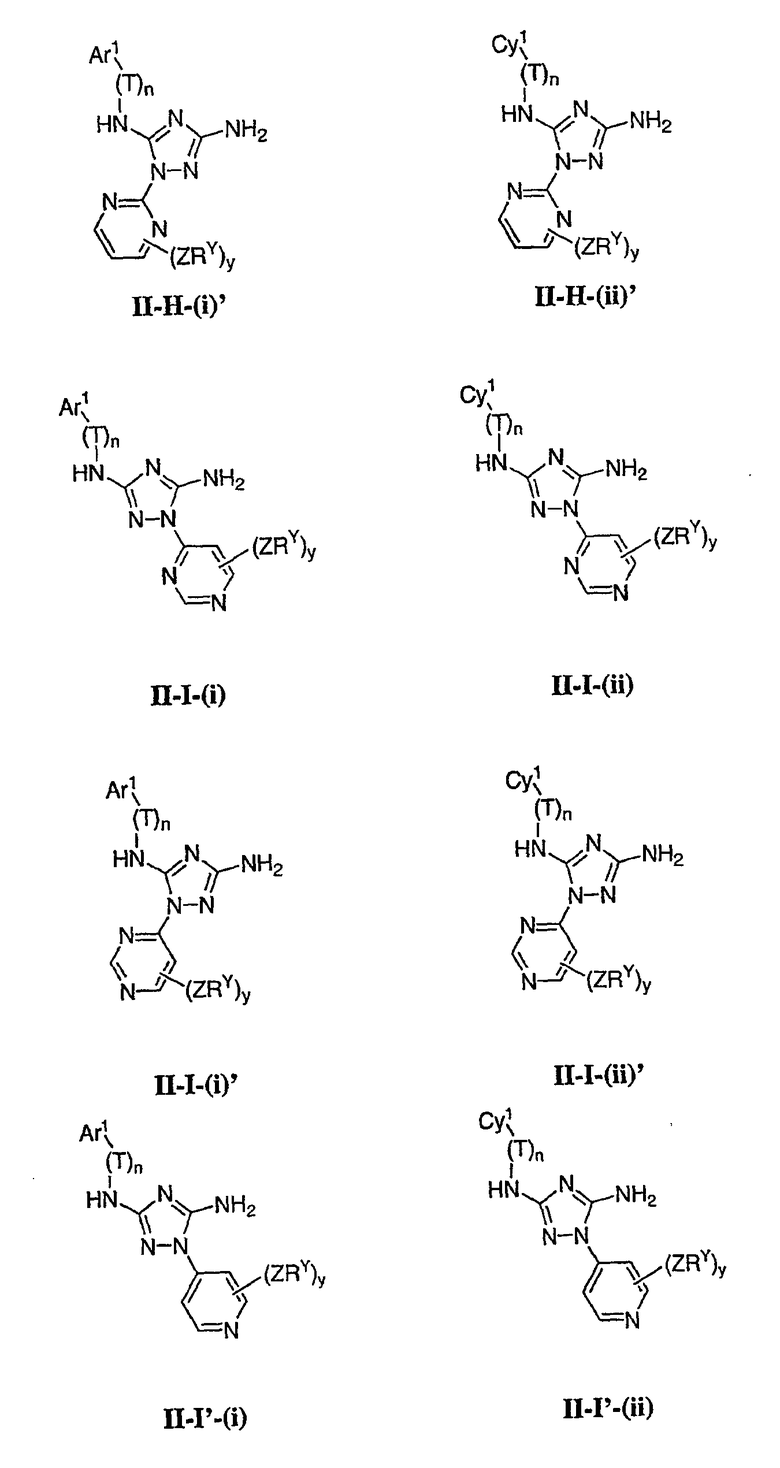
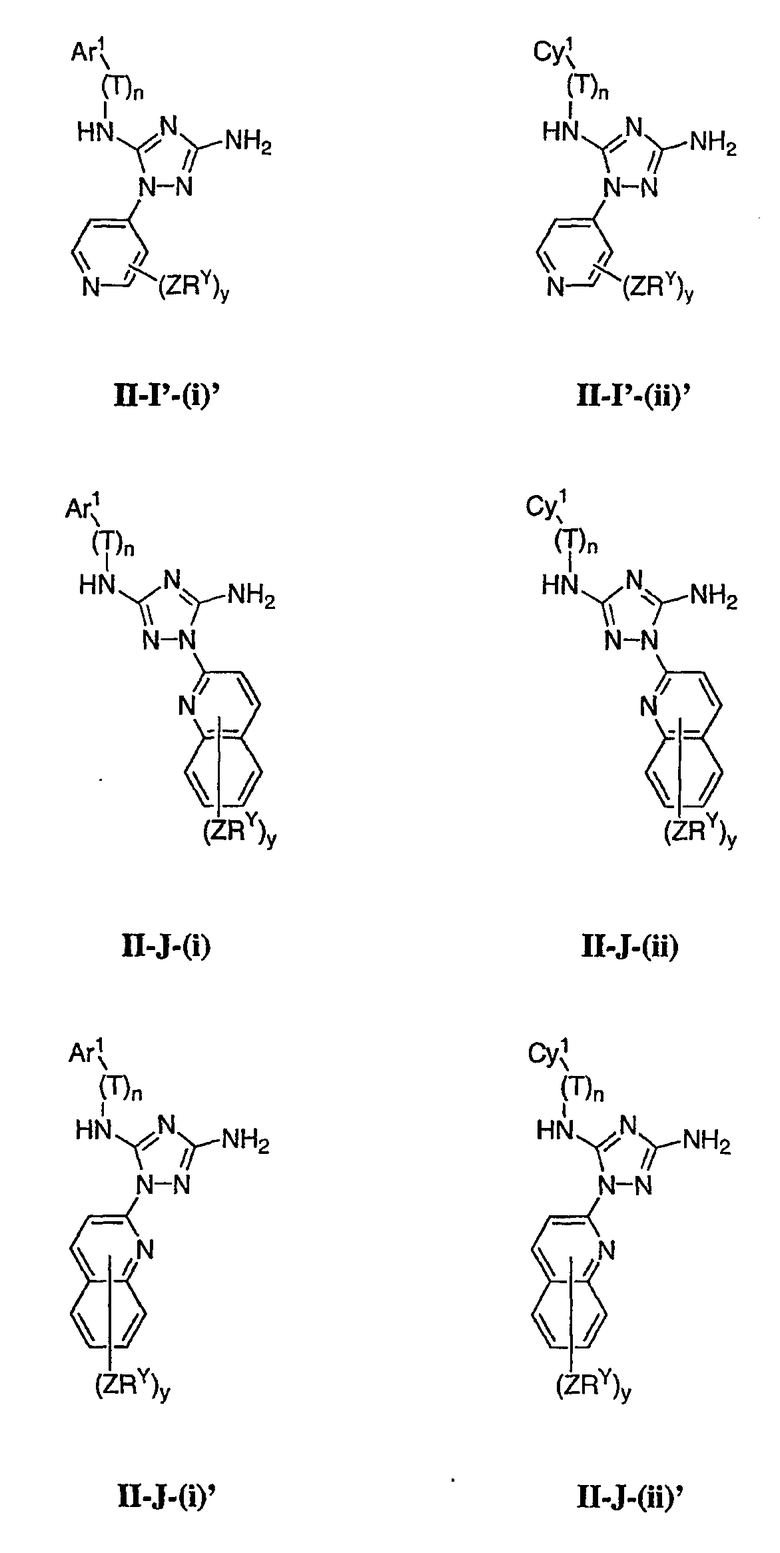
В следующих предпочтительных вариантах осуществления Cy2 представляет собой циклогексил и соединения имеют формулу II-K-(i), II-K-(ii), II-K-(i)' или II-K-(ii)':
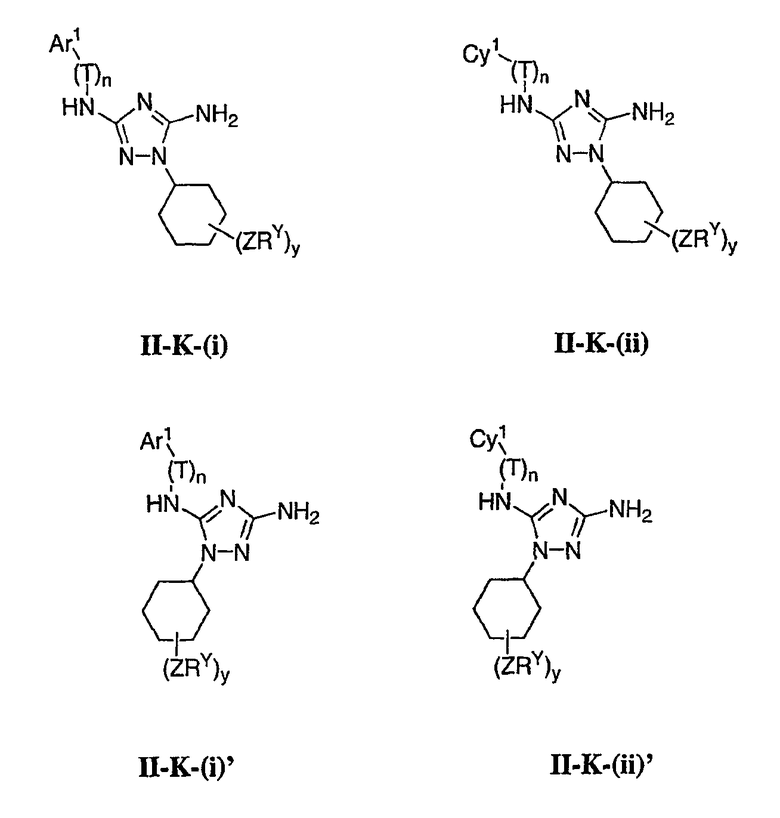
Как описано выше, Ar2 или Cy2 может быть необязательно замещен у любого замещаемого атома углерода или азота до 5 заместителями ZRy. В некоторых предпочтительных вариантах осуществления y равно 0-3, и поэтому Ar2 или Cy2, каждый независимо, замещен 0-3 заместителями ZRy. В других предпочтительных вариантах осуществления y равно 0 и Ar2 или Cy2 является незамещенным.
В предпочтительных вариантах осуществления каждая из групп ZRY независимо представляет собой R', галоген, CN, NO2, N(R')2, -CH2N(R')2, -OR', -CH2OR', -SR', -CH2SR', -COOR', -NRCOR', -CON(R')2, -SO2N(R')2, -CONR(CH2)2N(R')2, -CONR(CH2)3N(R')2, -CONR(CH2)4N(R')2, -O(CH2)2OR', -O(CH2)3OR', -O(CH2)4OR', -O(CH2)2N(R')2, -O(CH2)3N(R')2 или -O(CH2)4N(R')2. В других вариантах осуществления группы ZRY представляют собой, каждый независимо, Cl, Br, F, CF3, Me, Et, CN, -COOH, -N(CH3)2, -N(Et)2, -N(iPr)2, -О(СН2)2ОСН3, -CONH2, -COOCH3, -ОН, -СН2ОН, -NHCOCH3, -SO2NH2, метилендиокси, этилендиокси, -О(СН2)2N-морфолино, -О(СН2)3N-морфолино, -О(СН2)4N-морфолино, -О(СН2)2N-пиперазинил, -О(СН2)3N-пиперазинил, -О(СН2)4N-пиперазинил, -NHCH(CH2OH)фенил, -CONH(CH2)2N-морфолино, -CONH(CH2)2N-пиперазинил, -CONH(CH2)3N-морфолино, -CONH(CH2)3N-пиперазинил, -CONH(CH2)4N-морфолино, -CONH(CH2)4N-пиперазинил, -SO2NH(CH2)2N-морфолино, -SO2NH(CH2)2N-пиперазинил, -SO2NH(CH2)3N-морфолино, -SO2NH(CH2)3N-пиперазинил, -SO2NH(CH2)4N-морфолино, -SO2NH(CH2)4N-пиперазинил, где каждая из указанных выше фенильных, морфолино, пиперазинильных и пиперидильных групп является необязательно замещенной или необязательно замещенной группой, выбранной из С1-4алкокси, фенила, фенилокси, бензила, пиперидинила, пиперазинила, морфолино или бензилокси. В некоторых вариантах осуществления атом азота пиперидинильной или пиперазинильной группы необязательно замещен -COR'. В некоторых других вариантах осуществления атом азота пиперидинильной или пиперазинильной группы необязательно замещен -СОСН2CN или -СОСН3. Примеры групп ZRY включают в себя также группы, показанные ниже в таблице 1.
Должно быть понятно, что некоторые подклассы вышеуказанных соединений формул II, II-A-(i), II-A-(ii), II-B-(i), II-B-(ii), II-C-(i), II-C-(ii), II-D-(i), II-D-(ii), II-E-(i), II-E-(ii), II-F-(i), II-F-(ii), II-G-(i), II-G-(ii), II-G'-(i), II-G'-(ii), II-H-(i), II-H-(ii), II-I-(i), II-I-(ii), II-I'-(i), II-I'-(ii), II-J-(i), II-J-(ii), II-K-(i), II-K-(ii), II', II-A-(i)', II-A-(ii)' II-B-(i)' II-B-(ii)' II-C-(i)' II-C-(ii)' II-D-(i)', II-D-(ii)', II-E-(i)', II-E-(ii)', II-F-(i)', II-F-(ii)', II-G-(i)', II-G-(ii)', II-G'-(i)', II-G'-(ii)', II-H-(i)', II-H-(ii)', II-I-(i)', II-I-(ii)', II-I'-(i)', II-I'-(ii)', II-J-(i)', II-J-(ii)', II-K-(i)', II-K-(ii)' представляют особый интерес.
Например, в некоторых предпочтительных вариантах осуществления для соединений формул II-A-(i), II-A-(ii), II-B-(i), II-B-(ii), II-C-(i), II-C-(ii), II-D-(i), II-D-(ii), II-A-(i)', II-A-(ii)', II-B-(i)', II-B-(ii)', II-C-(i)', II-C-(ii)', II-D-(i)' или II-D-(ii)' Ar2 представляет собой фенил, пиридил, пиримидинил (более предпочтительно 2- иди 6-пиримидинил), хинолинил, тиазолил или бензтиазолил, каждый из которых необязательно замещен 0-3 группами ZRY, и Cy2 представляет собой циклогексил, необязательно замещенный 0-3 группами ZRY. В более предпочтительных вариантах осуществления для соединений, описанных выше, n равно 0 или n равно 1 и Т представляет собой СН2; m равно 0, х равно 0-3; y равно 0-3 и в каждом из случаев QRx или ZRY независимо представляет собой R', галоген, CN, NO2, N(R')2, -CH2N(R')2, -OR', -CH2OR', -SR', -CH2SR', -COOR', -NRCOR', -CON(R')2, -SO2N(R')2, -CONR(CH2)2N(R')2, -CONR(CH2)3N(R')2, -CONR(CH2)4N(R')2, -O(CH2)2OR', -O(CH2)3OR', -O(CH2)4OR', -O(CH2)2N(R')2, -O(CH2)2N(R')3 или -O(CH2)4N(R')2. В более предпочтительных вариантах осуществления группы QRX или ZRY представляют собой, каждая независимо, Cl, Br, F, CF3, Me, Et, CN, -COOH, -N(CH3)2, -N(Et)2, -N(iPr)2, -О(СН2)ОСН3, -CONH2, -COOCH3, -ОН, -СН2ОН, -NHCOCH3, -SO2NH2, метилендиокси, этилендиокси, -О(СН2)2N-морфолино, -О(СН2)3N-морфолино, -О(СН2)4N-морфолино, -О(СН2)2N-пиперазинил, -О(СН2)3N-пиперазинил, -О(СН2)4N-пиперазинил, -NHCH(CH2OH)фенил, -CONH(CH2)2N-морфолино, -CONH(CH2)2N-пиперазинил, -CONH(CH2)3N-морфолино, -CONH(CH2)3N-пиперазинил, -CONH(CH2)4N-морфолино, -CONH(CH2)4N-пиперазинил, -SO2NH(CH2)2N-морфолино, -SO2NH(CH2)2N-пиперазинил, -SO2NH(CH2)3N-морфолино, -SO2NH(CH2)3N-пиперазинил, -SO2NH(CH2)4N-морфолино, -SO2NH(CH2)4N-пиперазинил, где каждая из указанных выше фенильных, морфолино, пиперазинильных и пиперидинильных групп является необязательно замещенной или необязательно замещенной группой, выбранной из С1-4алкокси, фенила, фенилокси, бензила, пиперидинила, пиперазинила, морфолино или бензилокси. В некоторых вариантах осуществления атом азота пиперидинильной или пиперазинильной группы необязательно замещен -COR'. В некоторых других вариантах осуществления атом азота пиперидинильной или пиперазинильной группы необязательно замещен -СОСН2CN или -СОСН3.
Для соединений формул II-E-(i), II-E-(ii), II-F-(i), II-F-(ii), II-G-(i), II-G-(ii), II-G'-(i), II-G'-(ii), II-H-(i), II-H-(ii), II-I-(i), II-I-(ii), II-I'-(i), II-I'-(ii), II-J-(i), II-J-(ii), II-K-(i), II-K-(ii), II-E-(i)', II-E-(ii)', II-F-(i)', II-F-(ii)', II-G-(i)', II-G-(ii)', II-G'-(i)', II-G'-(ii)', II-H-(i)', II-H-(ii)', II-I-(i)', II-I-(ii)', II-I'-(i)', II-I'-(ii)', II-J-(i)', II-J-(ii)', II-K-(i)' или II-K-(ii)' Ar1 представляет собой необязательно замещенную группу, выбранную из фенила, и Cy1 выбран из циклогексила, фуранила или циклопропила, необязательно замещенного 0-3 группами QRX. В более предпочтительных вариантах осуществления для соединений, описанных выше, n равно 0 или n равно 1 и Т представляет собой СН2; х равно 0-3, y равно 0-3 и в каждом случае QRX или ZRY представляет собой независимо R', галоген, CN, NO2, -N(R')2, -CH2N(R')2, -OR', -CH2OR', -SR', -CH2SR', -COOR', -NRCOR', -CON(R')2, -SO2N(R')2, -CONR(CH2)2N(R')2, -CONR(CH2)3N(R')2, -CONR(CH2)4N(R')2, -O(CH2)2OR', -O(CH2)3OR', -O(CH2)4OR', -O(CH2)2N(R')2, -O(CH2)3N(R')2 или -O(CH2)4N(R')2. В более предпочтительных вариантах осуществления группы QRX или ZRY представляют собой, каждый независимо, Cl, Br, F, CF3, Me, Et, CN, -COOH, N(CH3)2, -N(Et)2, -N(iPr)2, -О(СН2)2ОСН3, -CONH2, COOCH3, -ОН, -СН2ОН, -NHCOCH3, SO2NH2, метилендиокси, этилендиокси, -О(СН2)2N-морфолино, -О(СН2)3N-морфолино, -О(СН2)4N-морфолино, -О(СН2)2N-пиперазинил, -О(СН2)3N-пиперазинил, -О(СН2)4N-пиперазинил, -NHCH(CH2OH)фенил, -CONH(CH2)2N-морфолино, -CONH(CH2)2N-пиперазинил, -CONH(CH2)3N-морфолино, -CONH(CH2)3N-пиперазинил, -CONH(CH2)4N-морфолино, -CONH(CH2)4N-пиперазинил, -SO2NH(CH2)2N-морфолино, -SO2NH(CH2)2N-пиперазинил, -SO2NH(CH2)3N-морфолино, -SO2NH(CH2)3N-пиперазинил, -SO2NH(CH2)4N-морфолино, -SO2NH(CH2)4N-пиперазинил, где каждая из указанных выше фенильных, морфолино, пиперазинильных и пиперидинильных групп является необязательно замещенной или необязательно замещенной группой, выбранной из С1-4алкокси, фенила, фенилокси, бензила, пиперидинила, пиперазинила, морфолино или бензилокси. В некоторых вариантах осуществления атом азота пиперидинильной или пиперазинильной группы необязательно замещен -COR'. В некоторых других вариантах осуществления атом азота пиперидинильной или пиперазинильной группы необязательно замещен -СОСН2CN или -СОСН3.
В следующих вариантах осуществления предложены соединения, имеющие одну из формул:
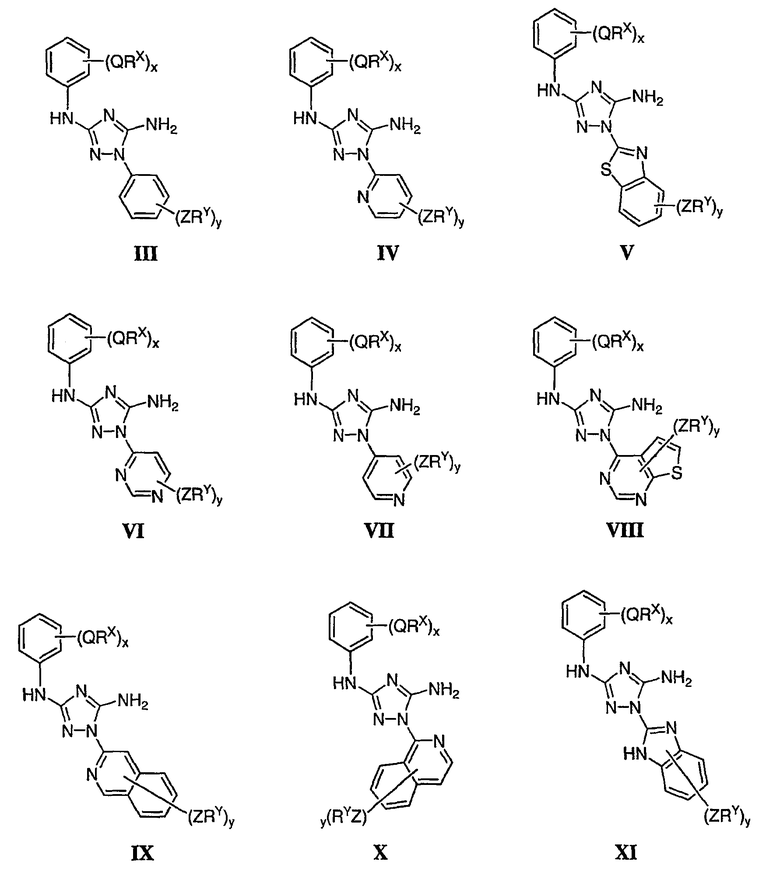
В некоторых предпочтительных вариантах осуществления для соединений формул III, IV, V, VI, VII, VIII, IX, X или XI х равно 0-3, y равно 0-3 и в каждом случае QRX или ZRY представляют собой независимо R', галоген, CN, NO2, -N(R')2, -CH2N(R')2, -OR', -CH2OR', -SR', -CH2SR', -COOR', -NRCOR', -CON(R')2, -SO2N(R')2, -CONR(CH2)2N(R')2, -CONR(CH2)3N(R')2, -CONR(CH2)4N(R')2, -O(CH2)2OR', -O(CH2)3OR', -O(CH2)4OR', -O(CH2)2N(R')2, -O(CH2)3N(R')2 или -O(CH2)4N(R')2. В более предпочтительных вариантах осуществления группы QRX или ZRY представляют собой, каждый независимо, Cl, Br, F, CF3, Me, Et, CN, -COOH, N(CH3)2, -N(Et)2, -N(iPr)2, -О(СН2)2ОСН3, -CONH2, COOCH3, -ОН, -СН2ОН, -NHCOCH3, -SO2NH2, метилендиокси, этилендиокси, -О(СН2)2N-морфолино, -О(СН2)3N-морфолино, -О(СН2)4N-морфолино, -О(СН2)2N-пиперазинил, -О(СН2)3N-пиперазинил, -О(СН2)4N-пиперазинил, -NHCH(CH2OH)фенил, -CONH(CH2)2N-морфолино, -CONH(CH2)2N-пиперазинил, -CONH(CH2)3N-морфолино, -CONH(CH2)3N-пиперазинил, -CONH(CH2)4N-морфолино, -CONH(CH2)4N-пиперазинил, -SO2NH(CH2)2N-морфолино, -SO2NH(CH2)2N-пиперазинил, -SO2NH(CH2)3N-морфолино, -SO2NH(CH2)3N-пиперазинил, -SO2NH(CH2)4N-морфолино, -SO2NH(CH2)4N-пиперазинил, где каждая из указанных выше фенильных, морфолино, пиперазинильных и пиперидинильных групп является необязательно замещенной или необязательно замещенной группой, выбранной из С1-4алкокси, фенила, фенилокси, бензила, пиперидинила, пиперазинила, морфолино или бензилокси. В некоторых вариантах осуществления атом азота пиперидинильной или пиперазинильной группы необязательно замещен -COR'. В некоторых других вариантах осуществления атом азота пиперидинильной или пиперазинильной группы необязательно замещен -СОСН2CN или -СОСН3.
В следующих вариантах осуществления соединения формул III, VI, VIII, IX, X и XI (включая их подгруппы) являются предпочтительными в качестве ингибиторов PDK-1.
В следующих вариантах осуществления соединения формул VI и VII (включая их подгруппы) являются предпочтительными в качестве ингибиторов JAC-3. В некоторых предпочтительных вариантах осуществления формул VI и VII, применимые в качестве ингибиторов JAC-3, являются замещенными, по меньшей мере, одной группой ZRy, где ZRY представляет собой N(R'), и соединения имеют структуру
В следующих вариантах осуществления соединения формул III, IV, VI и VII (включая их подгруппы) являются предпочтительными в качестве ингибиторов FLT-3.
Некоторые другие классы соединений формулы I, имеющие особый интерес, включают в себя би- или трициклические производные триазола, у которых
R1 и R2 имеют значения, указанные в общем выше и приведенные в классах и подклассах здесь;
R4 представляет собой водород или С1-4алкил;
R3 и R5, взятые вместе, образуют необязательно замещенную группу, выбранную из 5-7-членного насыщенного, частично ненасыщенного или полностью ненасыщенного моноциклического кольца, имеющего 0-3 гетероатома, независимо выбранного из азота, кислорода или серы, или 8-10-членной насыщенной, частично ненасыщенной, или полностью ненасыщенной бициклической системы колец, имеющей 0-3 гетероатома, независимо выбранного из азота, кислорода или серы;
R3 и R5, взятые вместе, образуют необязательно замещенную группу, выбранную из 5-7-членного насыщенного, частично ненасыщенного или полностью ненасыщенного моноциклического кольца, имеющего 0-3 гетероатома, независимо выбранного из азота, кислорода или серы, или 8-10-членной насыщенной, частично ненасыщенной, или полностью ненасыщенной бициклической системы колец, имеющей 0-3 гетероатома, независимо выбранного из азота, кислорода или серы;
где любое кольцо, образованное R3 и R5, взятых вместе, необязательно замещено до пяти заместителями, выбранными из W-RW, где W представляет собой связь или С1-6-алкилиденовую цепь, в которой до двух метиленовых звеньев W необязательно и независимо заменены на -NR-, -S-, -O-, -CS-, -CO2-, -OCO-, -CO-, -СОСО-, -CONR-, -NRCO-, -NRCO2-, -SO2NR-, -NRSO2-, -CONRNR-, -NRCONR-, -OCONR-, -NRNR-, -NRSO2NR-, -SO-, -SO2-, -PO-, -PO2- или -POR-; и в каждом случае Rw представляет собой, независимо R', галоген. NO2, CN, OR', SR', N(R')2, NR'COR', NR'CONR'2, NR'CO2R', COR', CO2R', OCOR', CON(R')2, OCON(R')2, SOR', SO2R', SO2N(R')2, NR'SO2R', NR'SO2N(R')2, COCOR' или COCH2COR'.
В некоторых вариантах осуществления соединения, описанные непосредственно выше, включают в себя одно или несколько или все из следующих ограничений:
а) соединения формулы I исключают соединения, имеющие общую структуру:
в которой R1, R2 и R3 имеют указанные выше значения, М и К представляют собой О или Н2, при условии что K и М являются разными, А и В представляют собой, каждый, -CH2-, -NH-, -N-алкил-, -N-аралкил-, -NCORa, -NCONHRb или -NCSNHRb, где Ra представляет собой низший алкил или аралкил и Rb представляет собой алкил с неразветвленной или разветвленной цепью, аралкил или арил, который может быть либо незамещенным, либо замещенным одним или несколькими алкильными и/или галогеналкильными заместителями;
b) соединения формулы I исключают соединения, имеющие общую структуру:
в которой R1 и R2 имеют указанные выше значения и r и s равны, каждый независимо, 0, 1, 2, 3 или 4, при условии что сумма s и r равна, по меньшей мере, 1;
с) соединения формулы I исключают любое одно или несколько или все из следующих соединений:
 ,
,
в которой R2 представляет собой NH(CH)(Ph)C=O(Ph);
 ,
,
в которой R2 представляет собой незамещенный фенил или фенил, замещенный ОМе, Cl или Ме;
 ,
,
в которой R2 представляет собой незамещенный фенил или фенил, замещенный ОМе, Cl, Ме, ОМе, или R2 представляет собой незамещенный бензил;
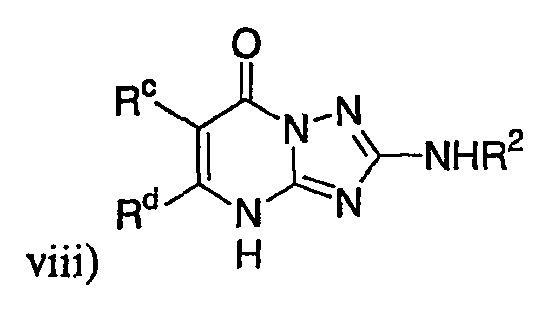 ,
,
в которой R2 представляет собой необязательно замещенный аралкил и Rc и Rd представляют собой, каждый независимо, Ме, водород, СН2Cl или Cl;
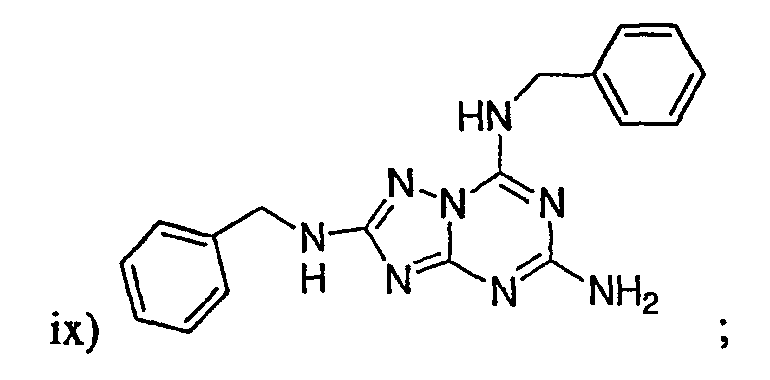
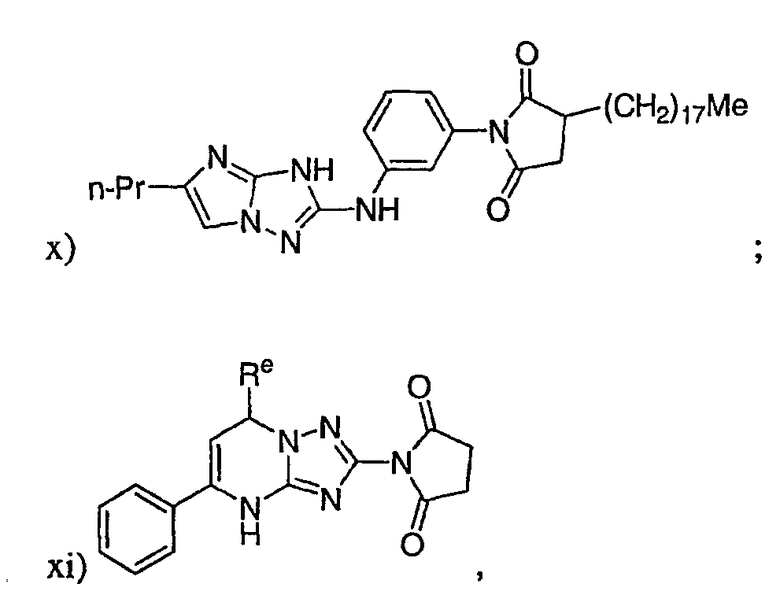
в которой Re представляет собой необязательно замещенный фенил;
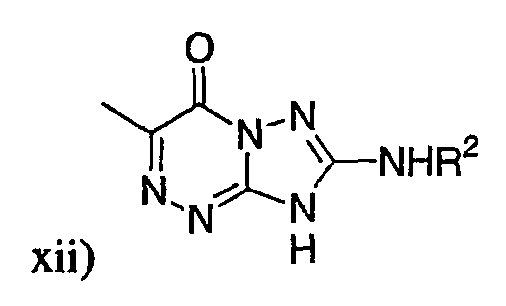 ,
,
в которой R2 представляет собой фенил, необязательно замещенный Ме, ОМе, Br или Cl, или
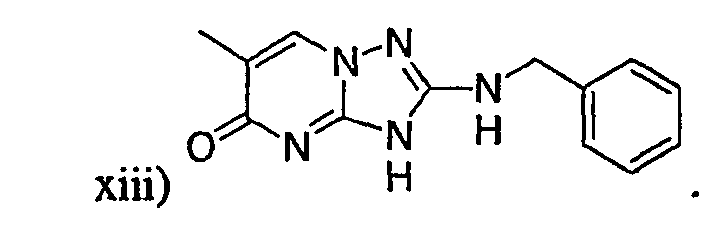
В некоторых предпочтительных вариантах осуществления соединения имеют одну из следующих формул:
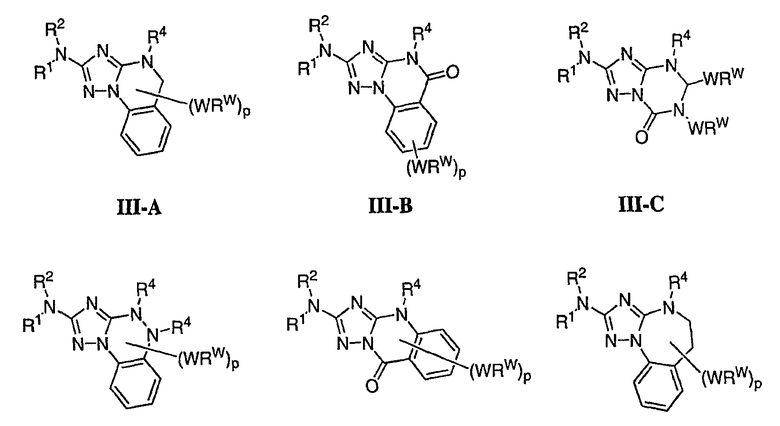
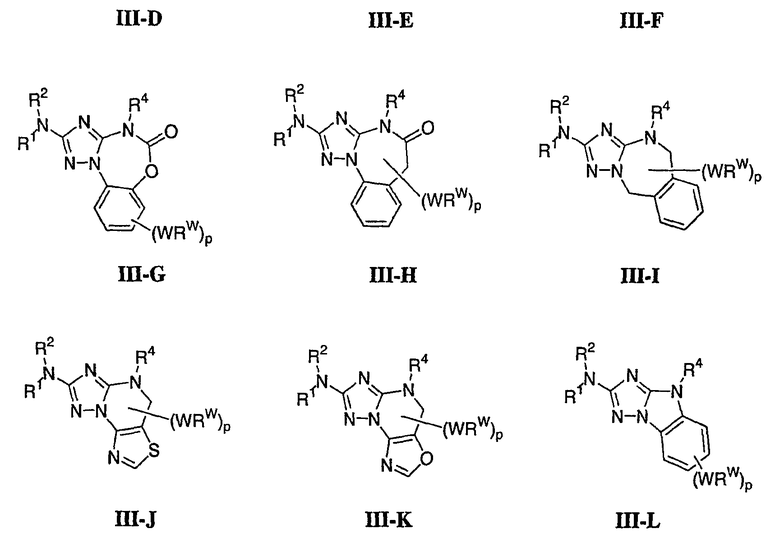
где W и RW имеют значения, указанные в общем выше и описанные здесь в классах и подклассах, и р равно 0-5.
Как описано в общем выше, в некоторых предпочтительных вариантах осуществления Ar1 выбран из любой одной из формул от а до u, приведенных выше (включая некоторые подгруппы b-i, c-i, b-ii, b-iii, c-ii или c-iii), и в некоторых других вариантах осуществления Cy1 выбран из любой одной формулы от v до ff, приведенных выше. Должно быть понятно, однако, что для соединений, описанных непосредственно выше, некоторые дополнительные соединения являются соединениями особого интереса. Например, в некоторых приведенных в качестве примеров вариантах осуществления особый интерес представляют те соединения, у которых R1 представляет собой водород и Ar1 представляет собой необязательно замещенный фенил.
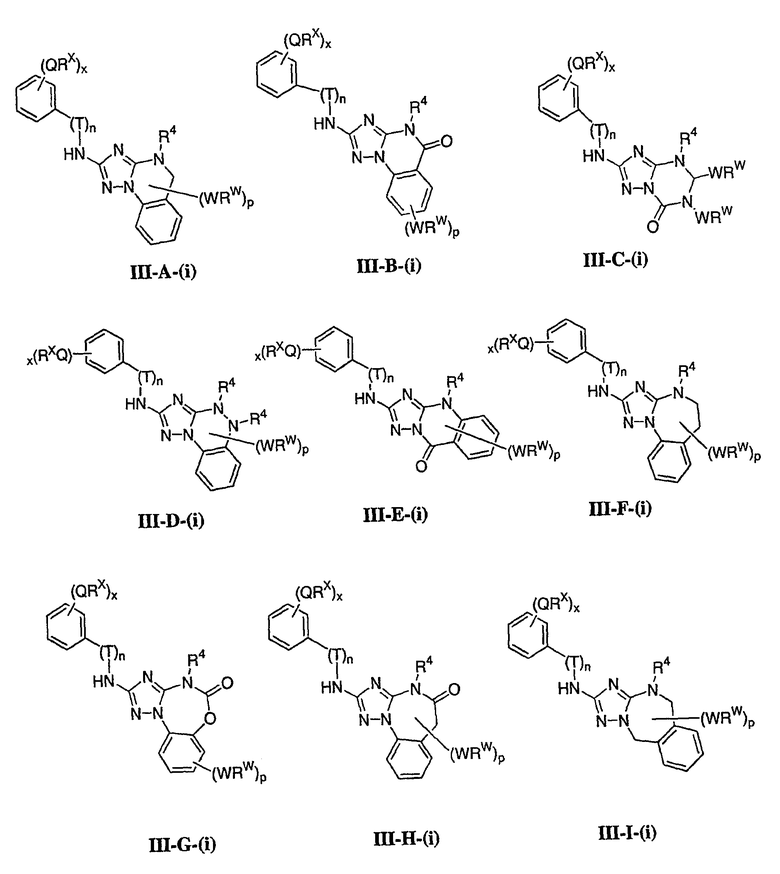

Должно быть понятно, что для соединений, описанных выше, некоторые дополнительные соединения, представляют особый интерес. Например, в некоторых других приведенных в качестве примеров вариантов осуществления особый интерес представляют те соединения, у которых R1 представляет собой водород и Ar1 представляет собой необязательно замещенный пиридил.
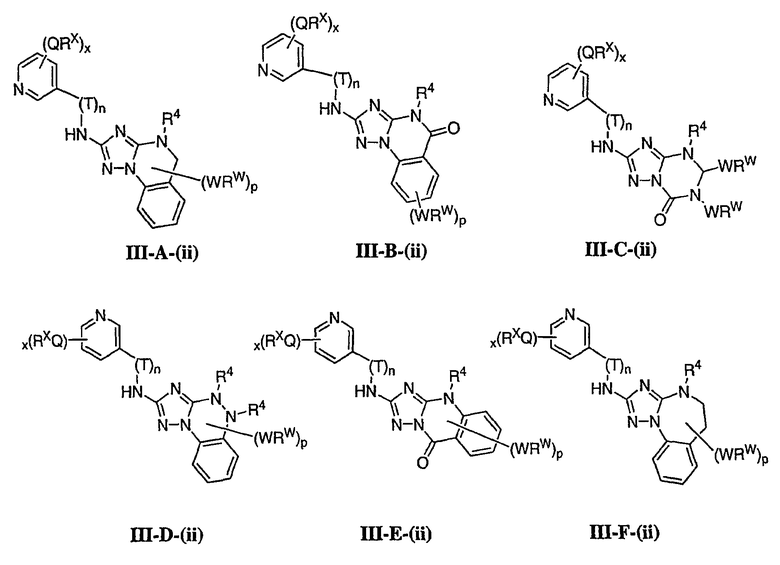
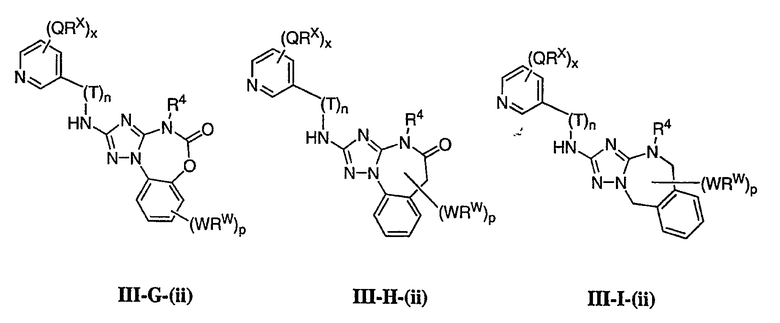
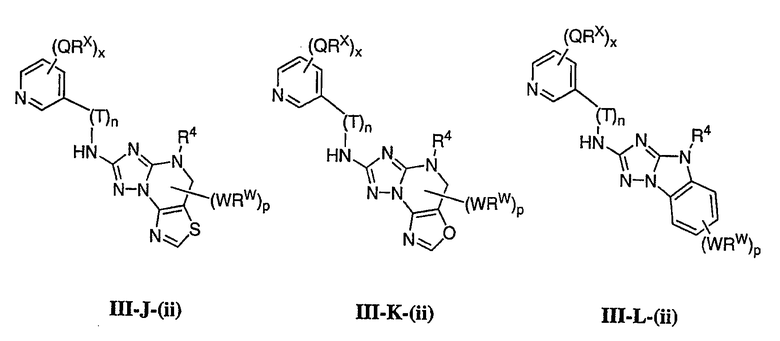
Должно быть понятно, что для соединений, описанных выше, некоторые дополнительные соединения представляют особый интерес. Например, в некоторых других приведенных в качестве примеров вариантах осуществления особый интерес представляют те соединения, у которых R1 представляет собой водород и Ar1 представляет собой необязательно замещенный циклогексил.
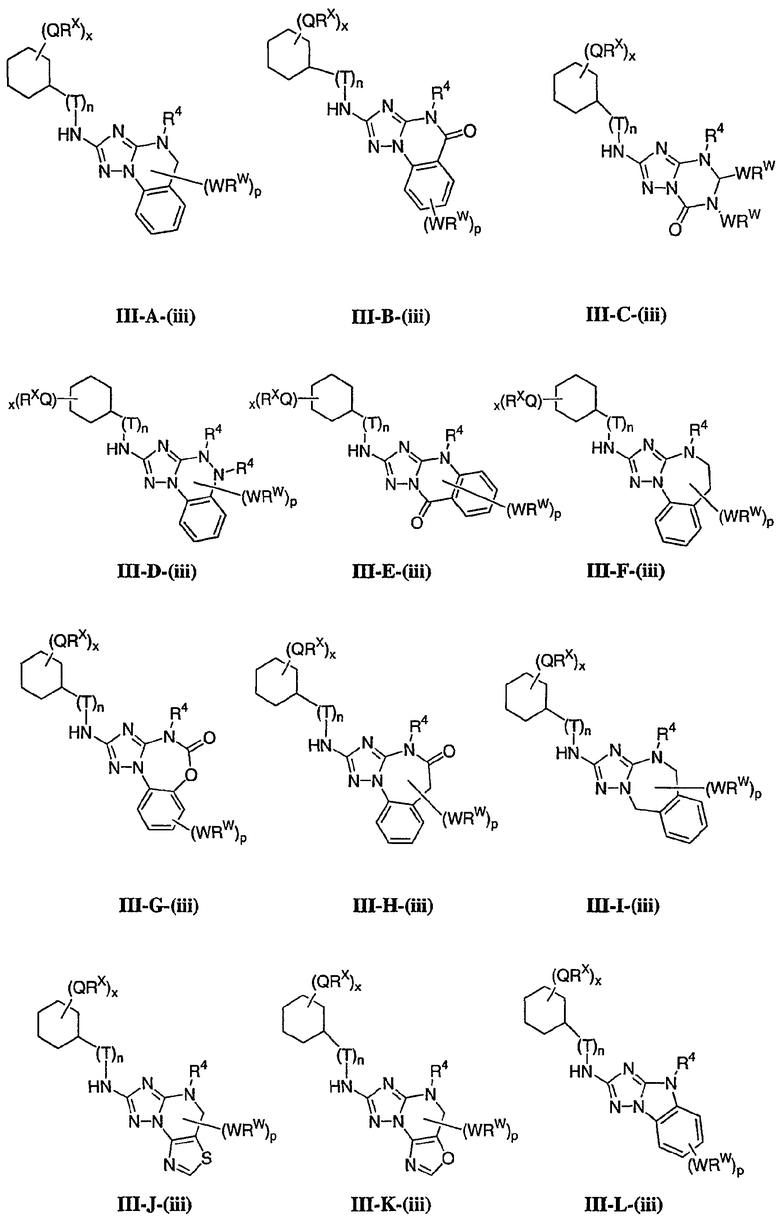
Должно быть понятно, что для соединений, описанных выше, некоторые дополнительные соединения представляют особый интерес. Например, в других приведенных в качестве примеров вариантах осуществления соединения особого интереса включают в себя те соединения, у которых R1 представляет собой водород и Ar1 представляет собой необязательно замещенный тетрагидрофурил.
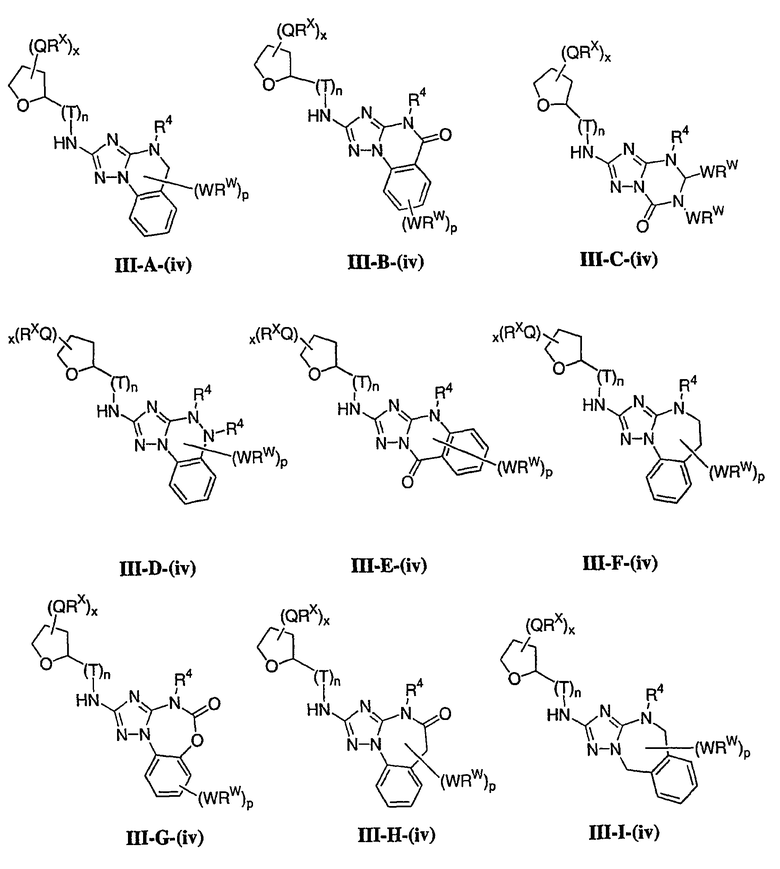
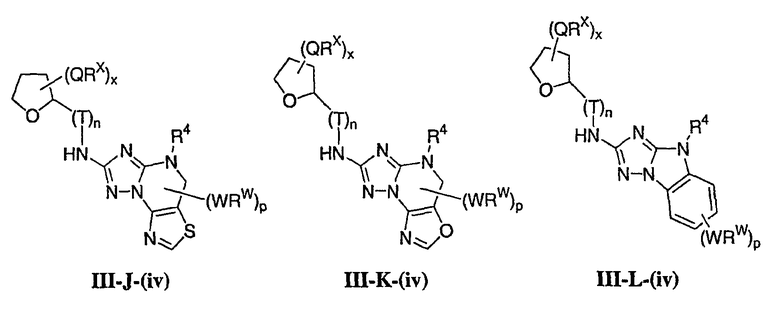
Должно быть понятно, что для соединений, описанных выше, некоторые дополнительные соединения представляют особый интерес. Например, в других приведенных в качестве примеров вариантах осуществления соединения особого интереса включают в себя те соединения, у которых R1 представляет собой водород и Ar1 представляет собой необязательно замещенный циклопропил.
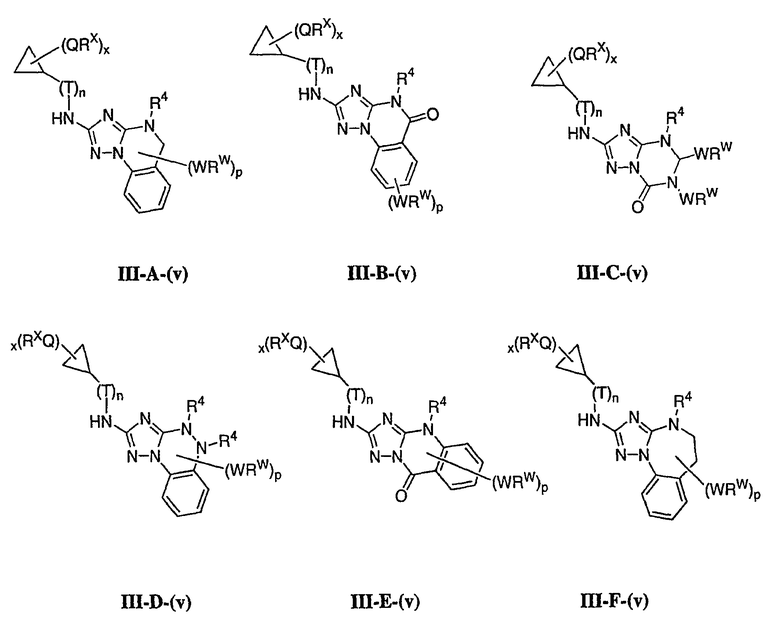
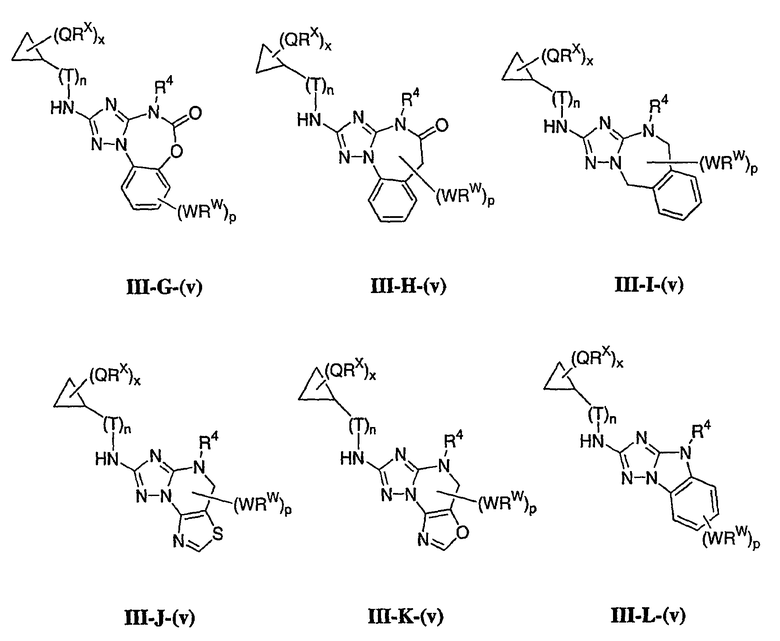
Как описано выше, любое кольцо, образованное R3 и R5, взятыми вместе, может быть необязательно замещено до 5 группами WRW. В некоторых предпочтительных вариантах осуществления р равно 0-3. В других предпочтительных вариантах осуществления р равно 0 и кольцо, образованное R3 и R5, является незамещенным.
В предпочтительных вариантах осуществления группы WRW представляют собой, каждая независимо, R', галоген, CN, NO2, N(R')2, -CH2N(R')2, -OR', -CH2OR', -SR', -CH2SR', -COOR', -NRCOR', -CON(R')2, -SO2N(R')2, -CONR(CH2)2N(R')2, -CONR(CH2)3N(R')2, -CONR(CH2)4N(R')2, -O(CH2)2OR', -O(CH2)3OR', -O(CH2)4OR', -O(CH2)2N(R')2, -O(CH2)3N(R')2 или -O(CH2)4N(R')2. В других вариантах осуществления группы WRW представляют собой, каждая независимо, Cl, Br, F, CF3, Me, Et, CN, -COOH, N(CH3)2, -N(Et)2, -N(iPr)2, -О(СН2)ОСН3, -CONH2, COOCH3, -ОН, -СН2ОН, -NHCOCH3, -SO2NH2, метилендиокси, этилендиокси, -О(СН2)2N-морфолино, -О(СН2)3N-морфолино, -О(СН2)4N-морфолино, -О(СН2)2N-пиперазинил, -О(СН2)3N-пиперазинил, -О(СН2)4N-пиперазинил, -NHCH(CH2OH)фенил, -CONH(CH2)2N-морфолино, -CONH(CH2)2N-пиперазинил, -CONH(CH2)3N-морфолино, -CONH(CH2)3N-пиперазинил, -CONH(CH2)4N-морфолино, -CONH(CH2)4N-пиперазинил, -SO2NH(CH2)2N-морфолино, -SO2NH(CH2)2N-пиперазинил, -SO2NH(CH2)3N-морфолино, -SO2NH(CH2)3N-пиперазинил, -SO2NH(CH2)4N-морфолино, -SO2NH(CH2)4N-пиперазинил, где каждая из указанных выше фенильных, морфолино, пиперазинильных и пиперидинильных групп является необязательно замещенной или необязательно замещенной группой, выбранной из С1-4алкокси, фенила, фенилокси, бензила, пиперидинила, пиперазинила, морфолино или бензилокси. В некоторых вариантах осуществления атом азота пиперидинильной или пиперазинильной группы необязательно замещен -COR'. В некоторых других вариантах осуществления атом азота пиперидинильной или пиперазинильной группы необязательно замещен -СОСН2CN или -СОСН3. Примеры групп ZRW включают также группы, показанные ниже в таблице 1.
В более предпочтительных вариантах осуществления для соединений, описанных выше, n равно 0 или n равно 1 и Т представляет собой СН2; р=0-3, y равно 0-3; и в каждом случае WRW или ZRY представляет собой, независимо, R', галоген, CN, NO2, -N(R')2, -CH2N(R')2, -OR', CH2OR', -SR', -CH2SR', -COOR', -NRCOR', -CON(R')2, -SO2N(R')2, -CONR(CH2)2N(R')2, -CONR(CH2)3N(R')2, -CONR(CH2)4N(R')2, -O(CH2)2OR', -O(CH2)3OR', -O(CH2)4OR', -O(CH2)2N(R')2, -O(CH2)3N(R')2 или -O(CH2)4N(R')2.
В более предпочтительных вариантах осуществления группы WRW или ZRY представляют собой, каждая независимо, Cl, Br, F, CF3, Me, Et, CN, -COOH, N(CH3)2, -N(Et)2, -N(iPr)2, -О(СН2)2ОСН3, -CONH2, COOCH3, -ОН, -СН2ОН, -NHCOCH3, -SO2NH2, метилендиокси, этилендиокси, -О(СН2)2N-морфолино, -О(СН2)3N-морфолино, -О(СН2)4N-морфолино, -О(СН2)2N-пиперазинил, -О(СН2)3N-пиперазинил, -О(СН2)4N-пиперазинил, -NHCH(CH2OH)фенил, -CONH(CH2)2N-морфолино, -CONH(CH2)2N-пиперазинил, -CONH(CH2)3N-морфолино, -CONH(CH2)3N-пиперазинил, -CONH(CH2)4N-морфолино, -CONH(CH2)4N-пиперазинил, -SO2NH(CH2)2N-морфолино, -SO2NH(CH2)2N-пиперазинил, -SO2NH(CH2)3N-морфолино, -SO2NH(CH2)3N-пиперазинил, -SO2NH(CH2)4N-морфолино, -SO2NH(CH2)4N-пиперазинил, где каждая из указанных выше фенильных, морфолино, пиперазинильных и пиперидинильных групп является необязательно замещенной, или необязательно замещенной группой, выбранной из С1-4алкокси, фенила, фенилокси, бензила, пиперидинила, пиперазинила, морфолино или бензилокси. В некоторых вариантах осуществления атом азота пиперидинильной или пиперазинильной группы необязательно замещен -COR'. В некоторых других вариантах осуществления атом азота пиперидинильной или пиперазинильной группы необязательно замещен -СОСН2CN или -СОСН3.
В других предпочтительных вариантах осуществления R4 представляет собой водород или С1-4алкил. В более предпочтительных вариантах осуществления R4 представляет собой водород или метил. В наиболее предпочтительных вариантах осуществления R4 представляет собой водород.
Репрезентативные примеры соединений формулы I представлены ниже в таблице 1.
Таблица 1. Примеры соединений формулы I:
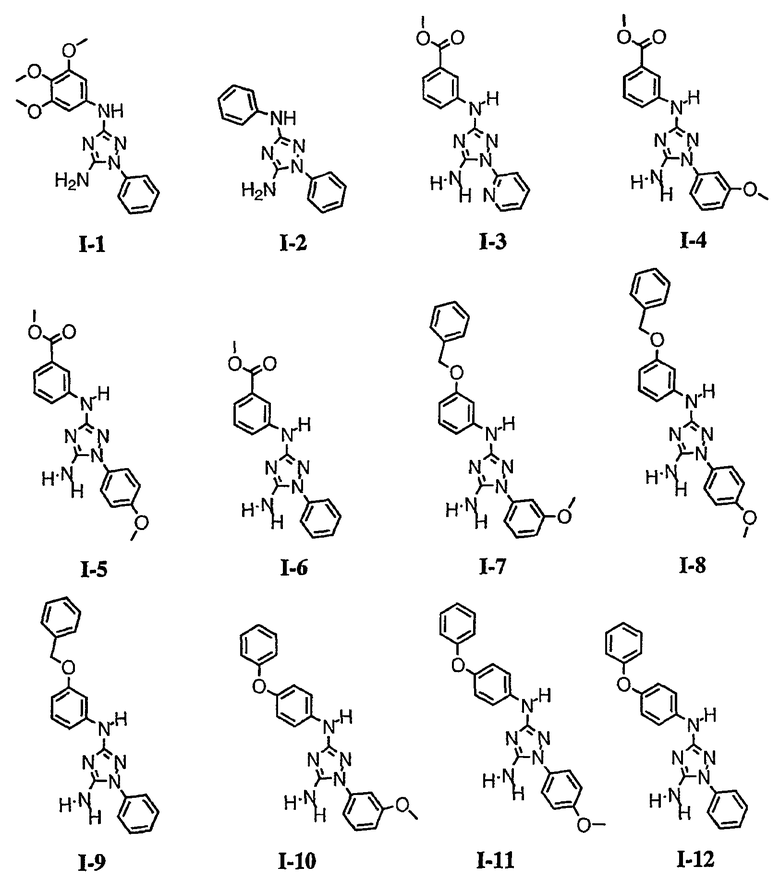
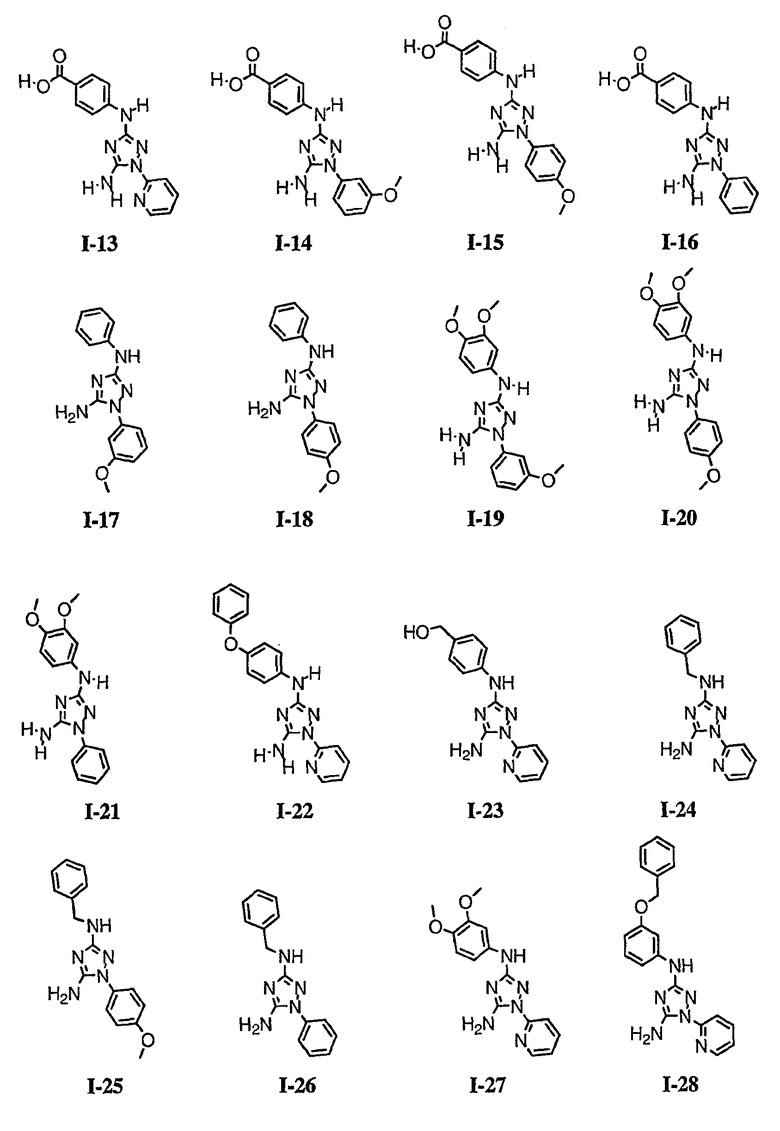
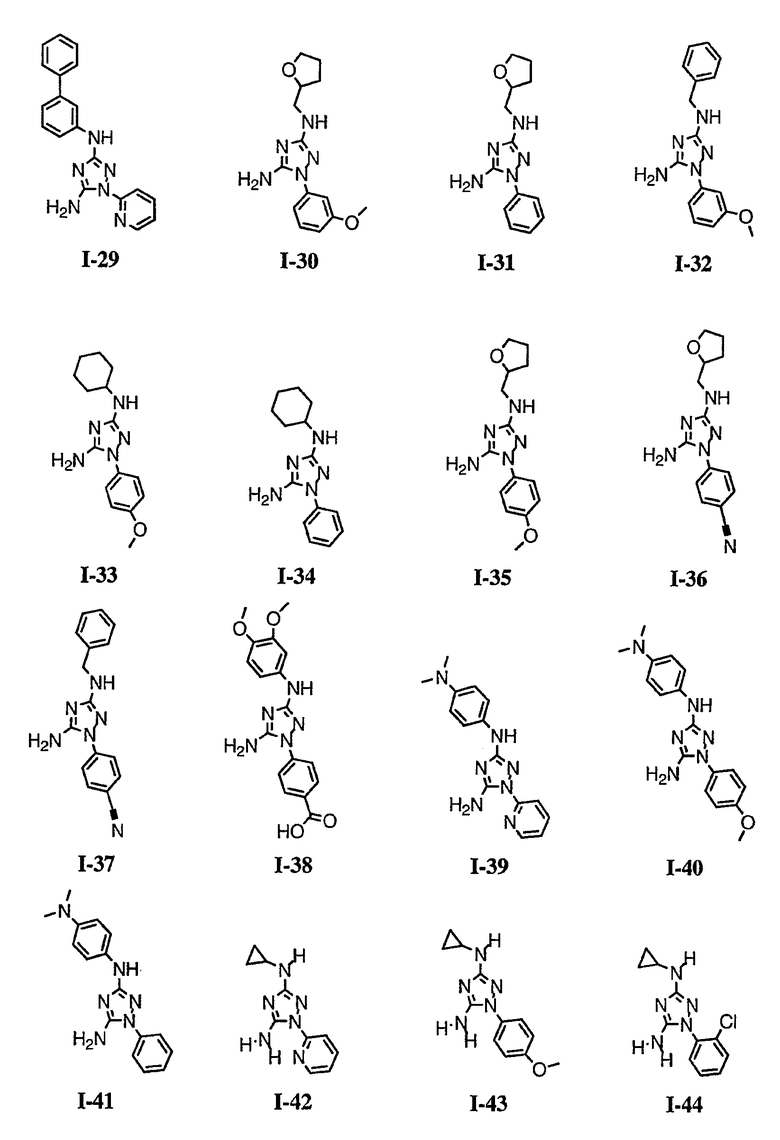
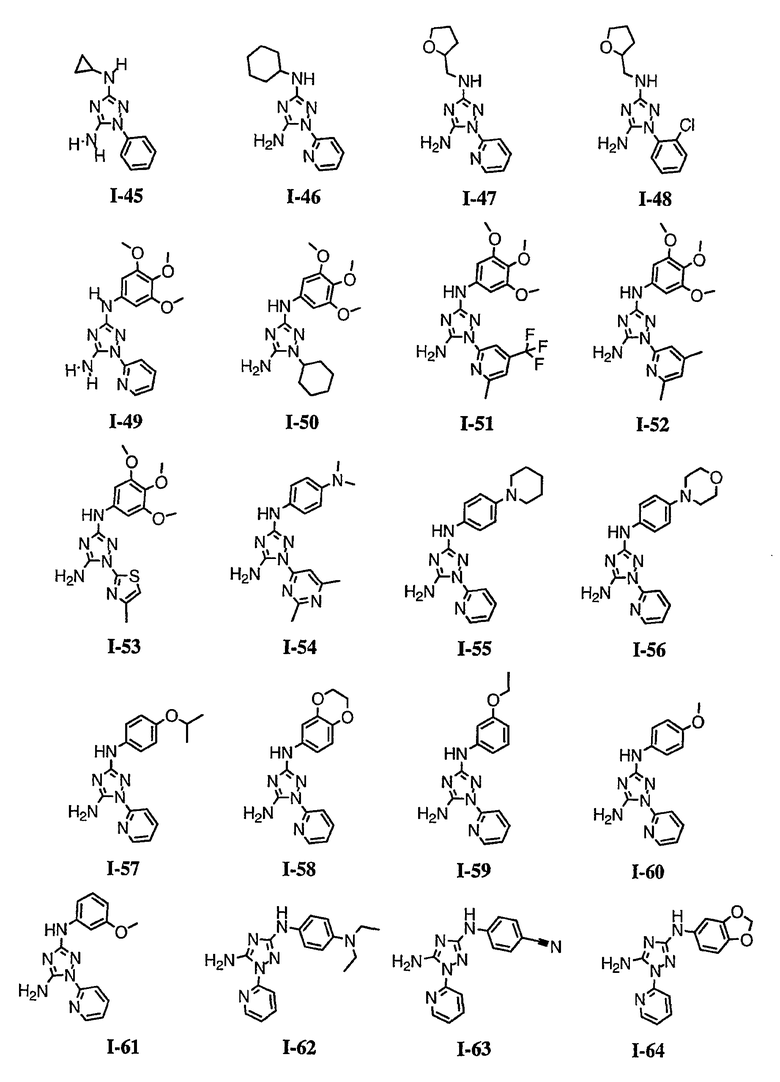
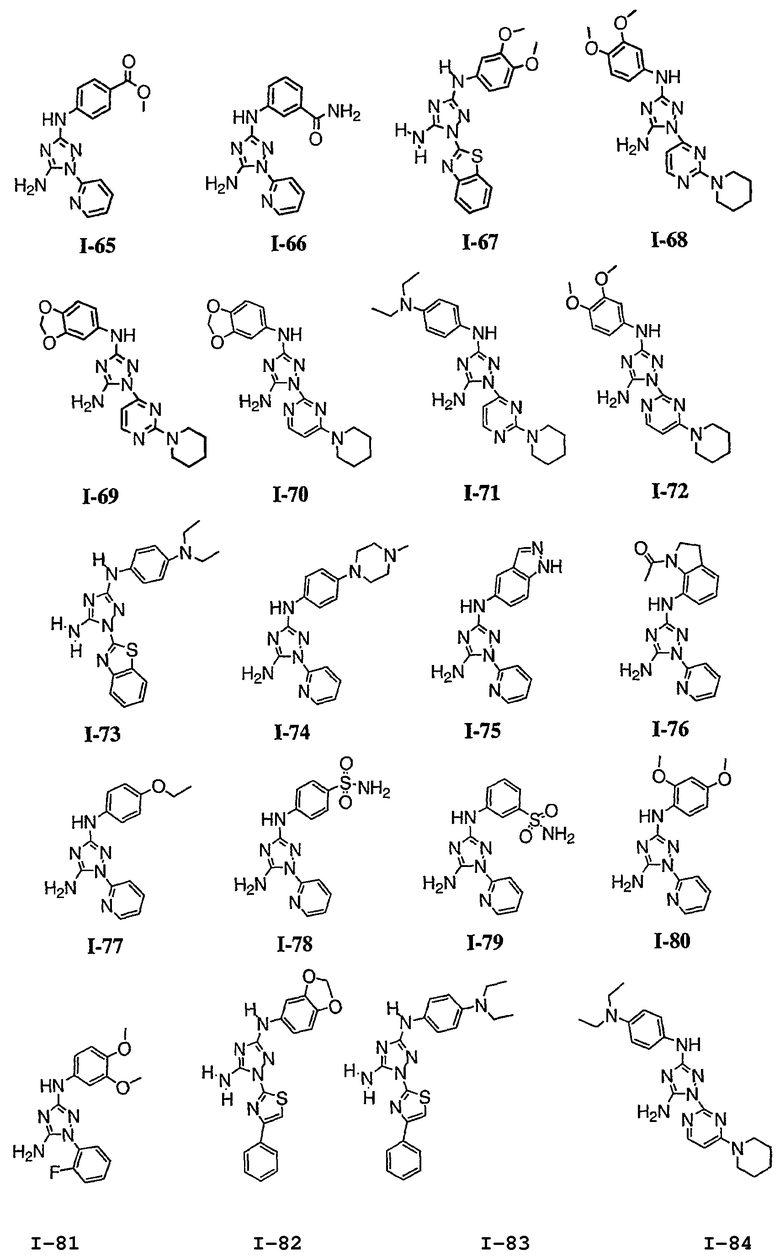
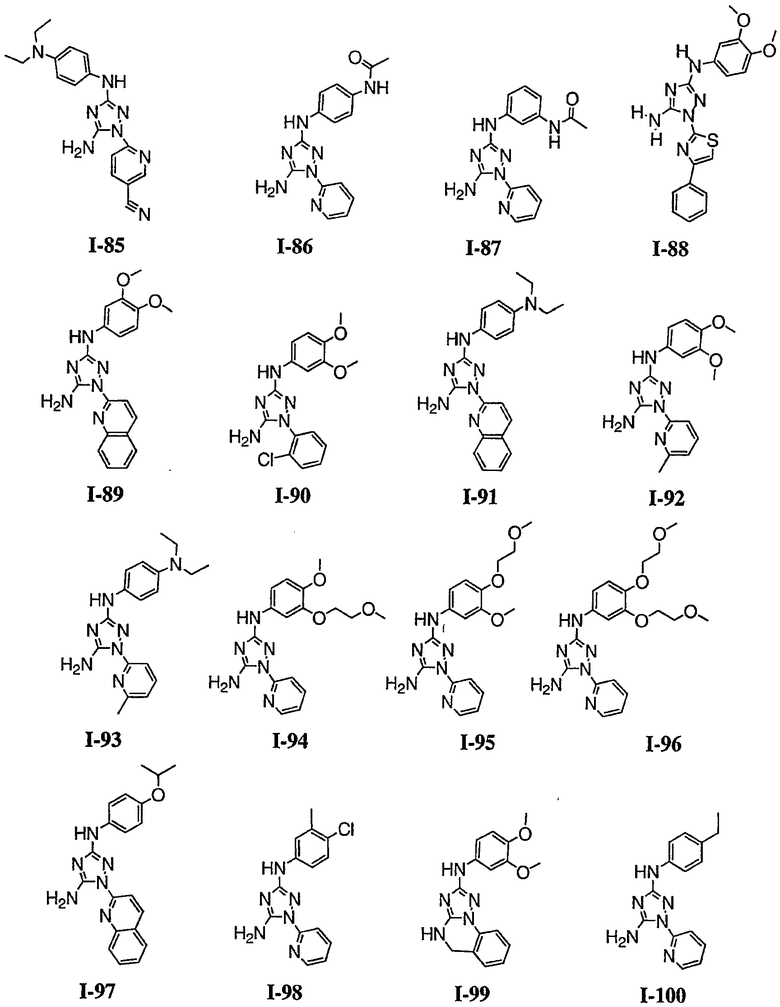
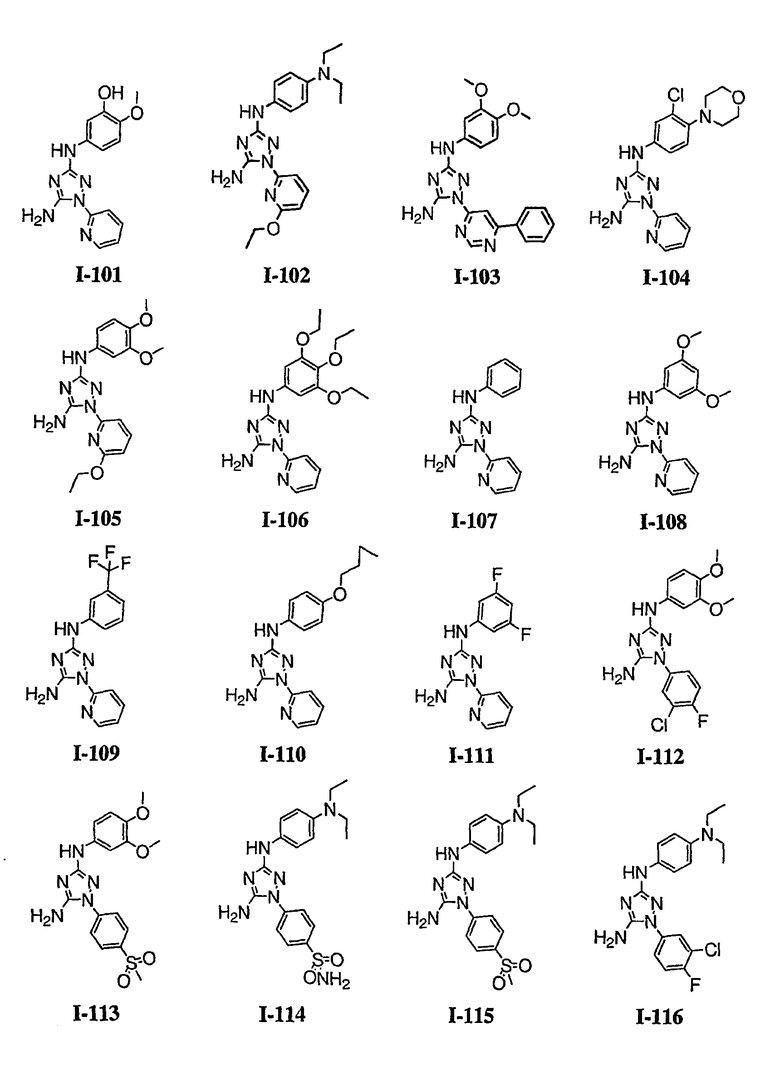
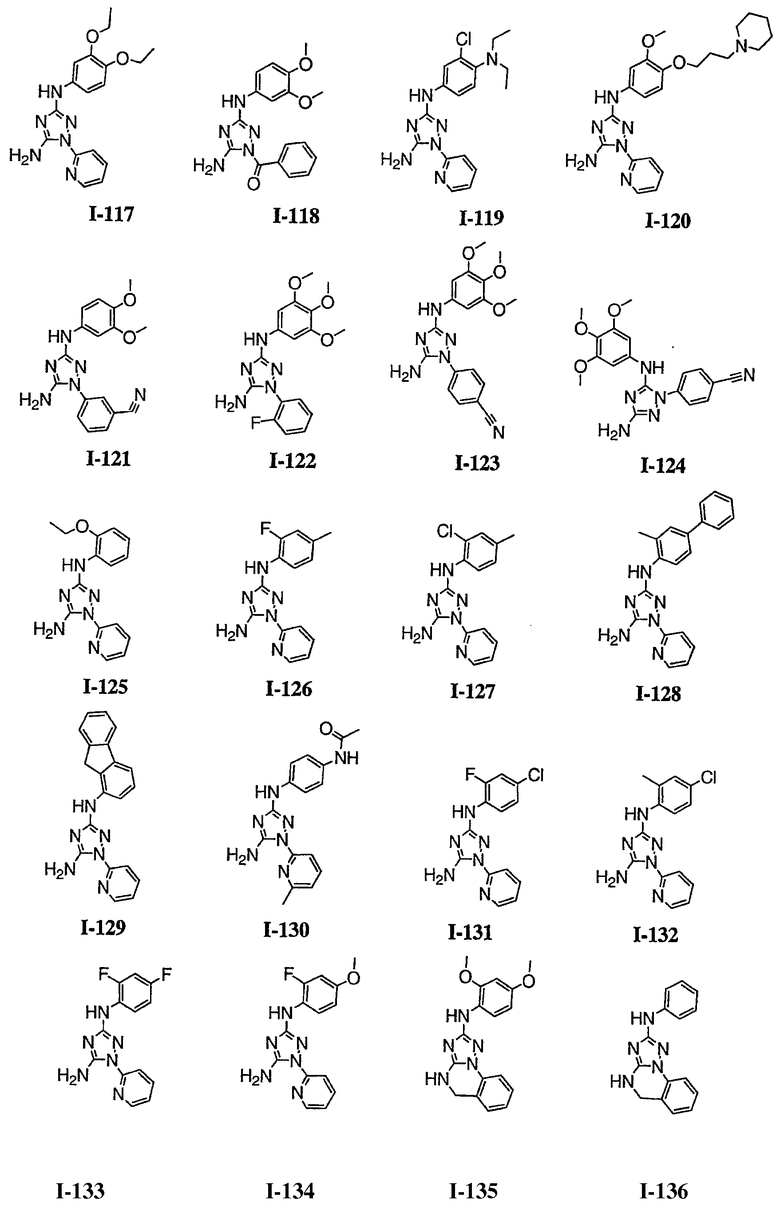
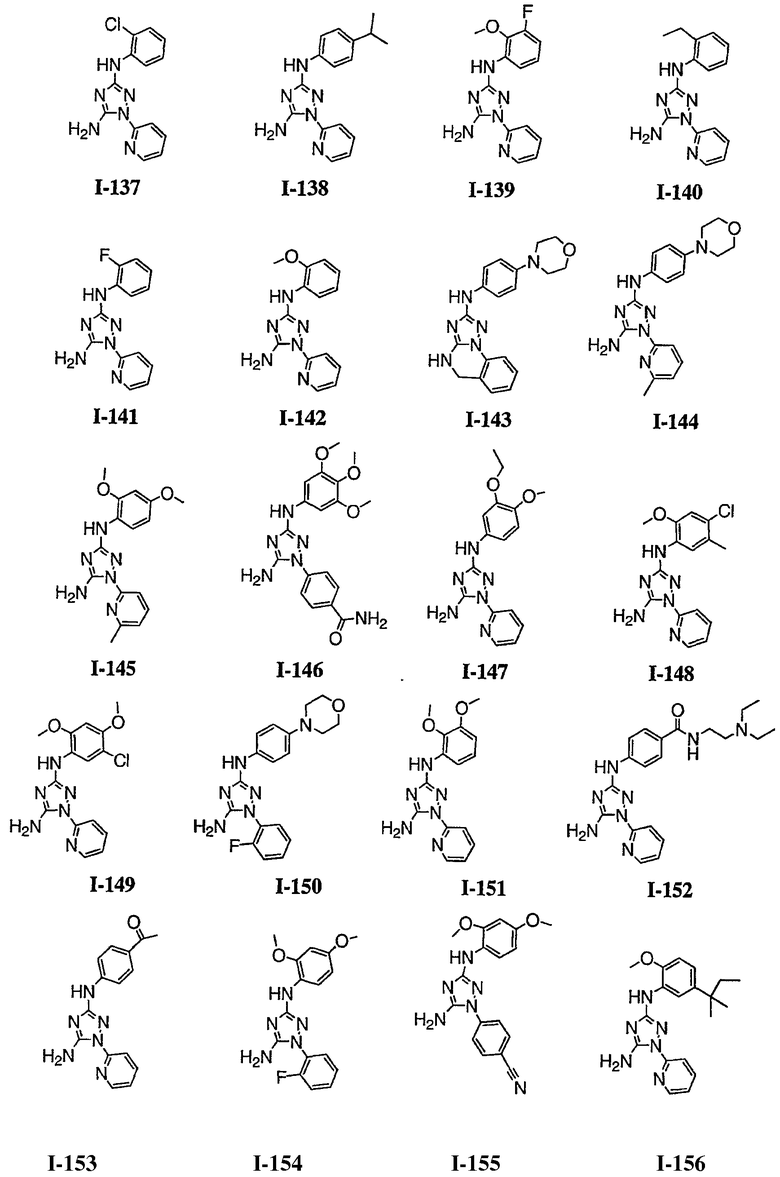
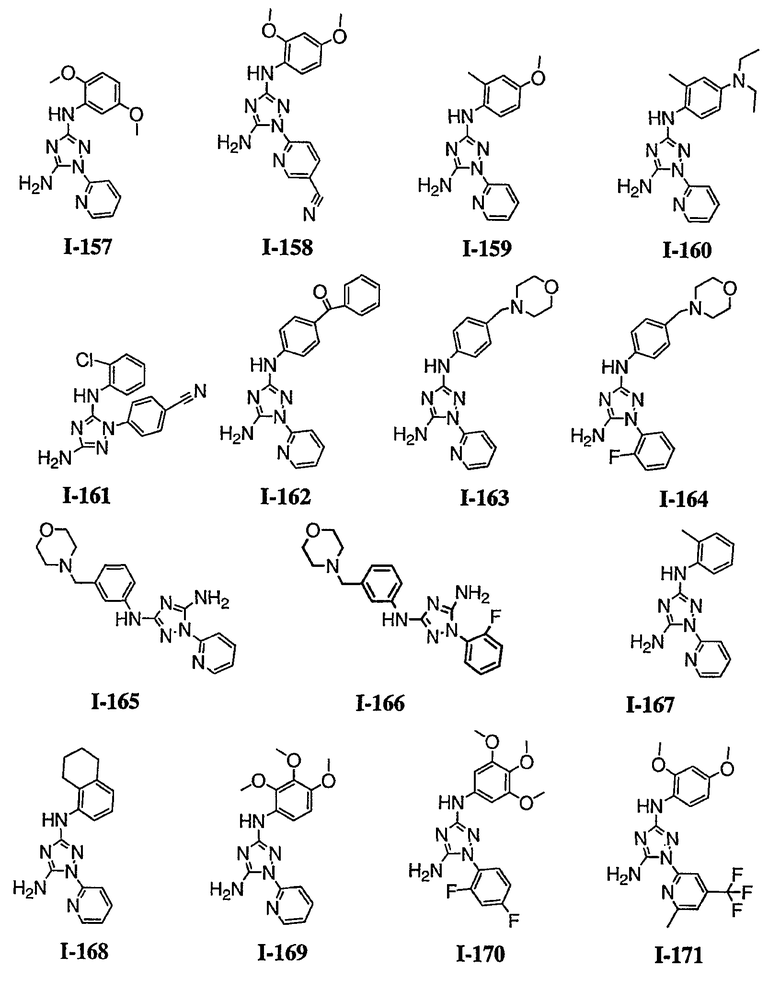
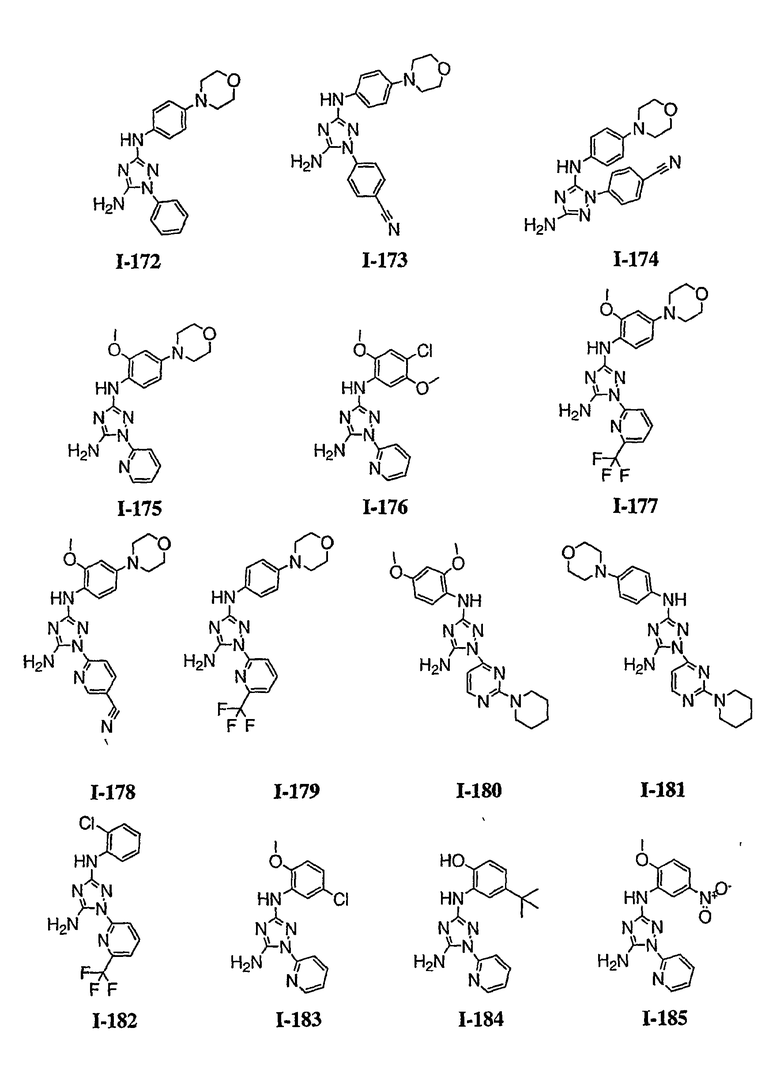
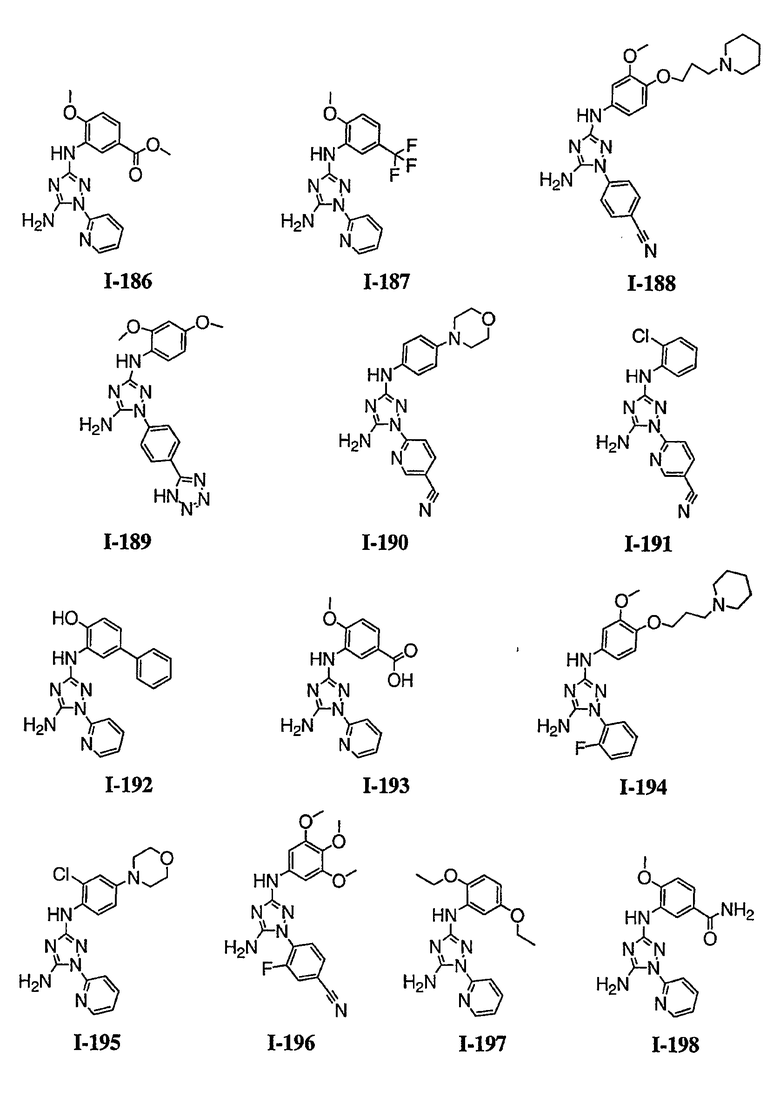
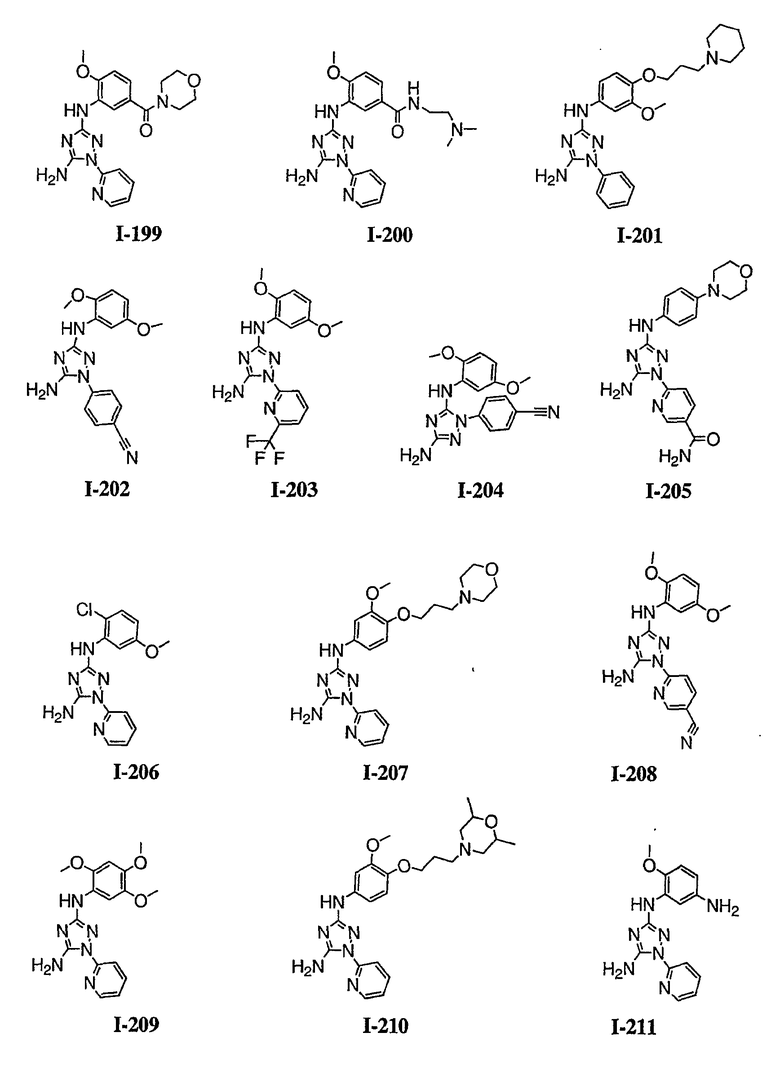
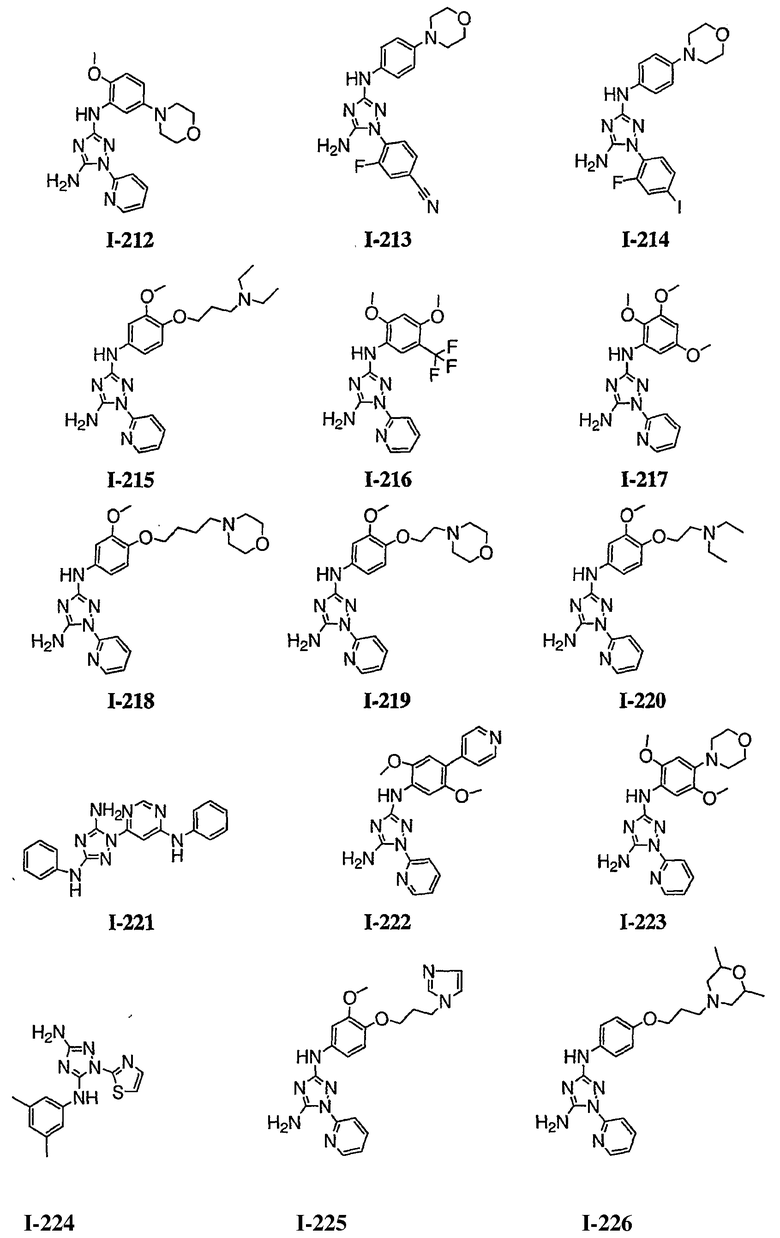

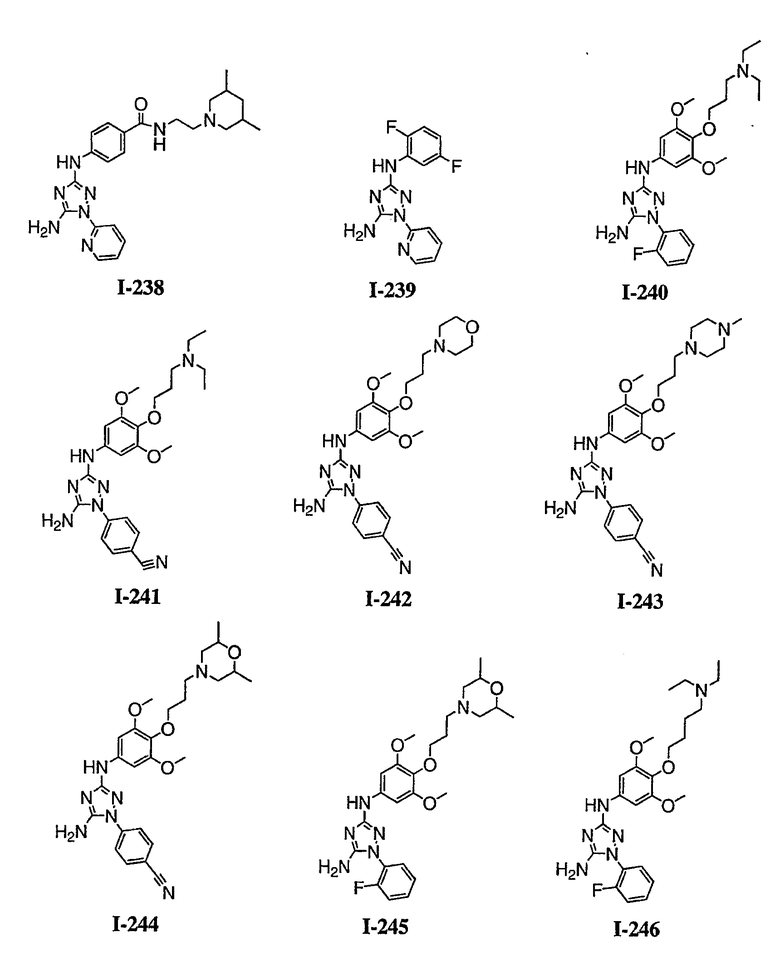
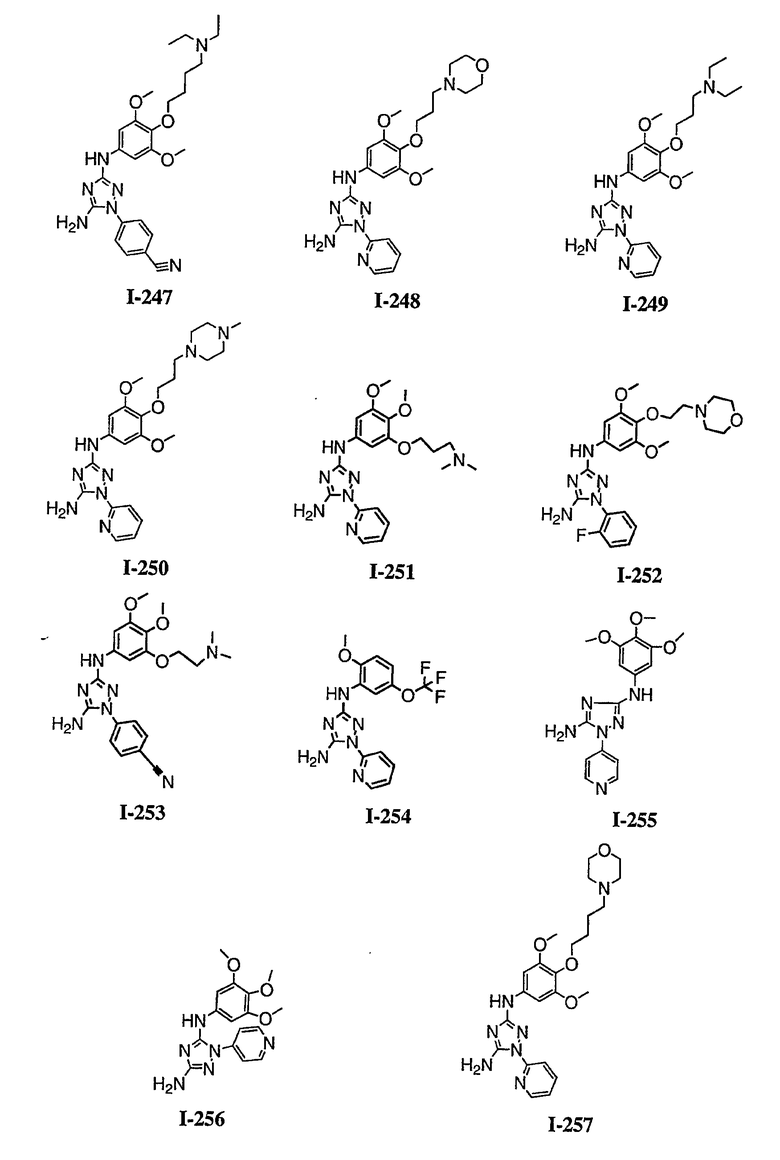
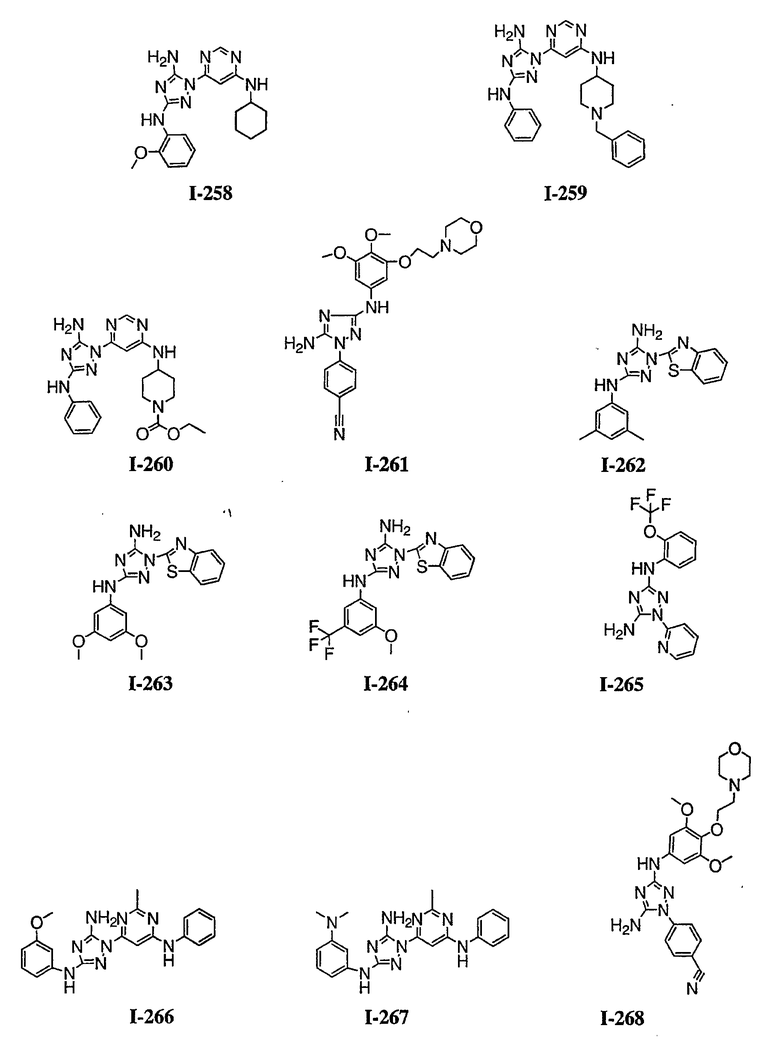


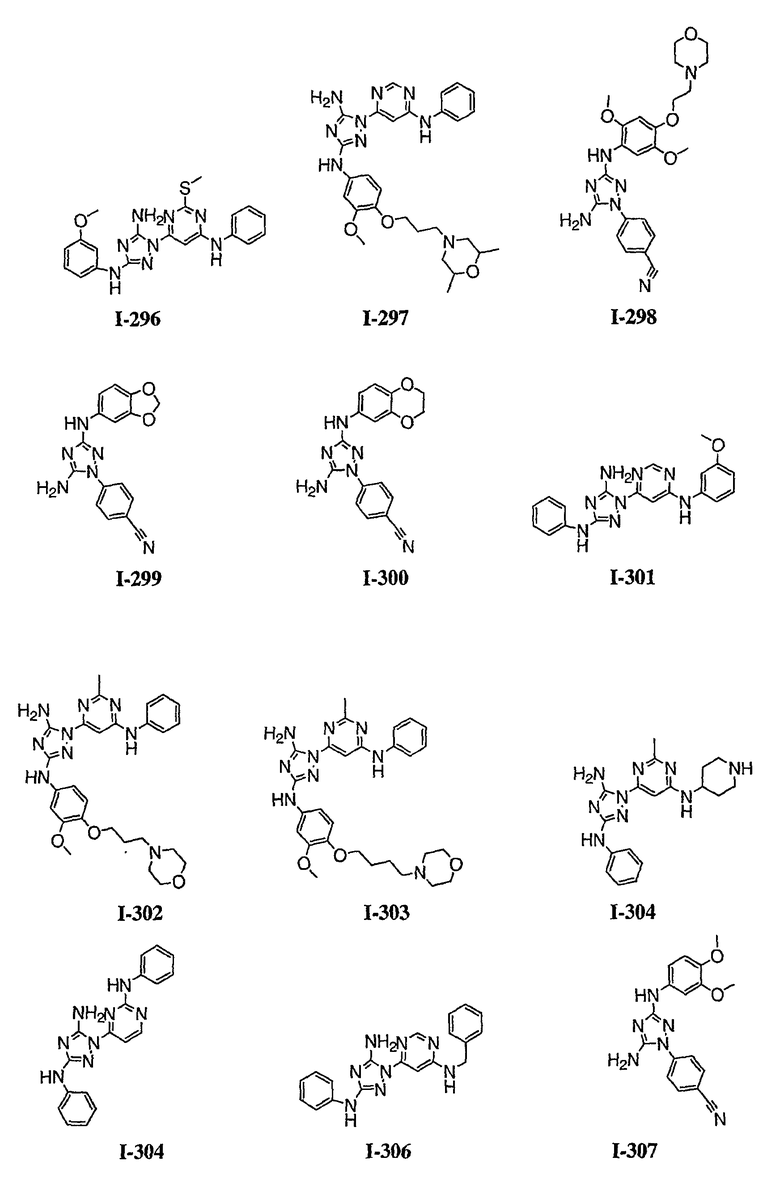
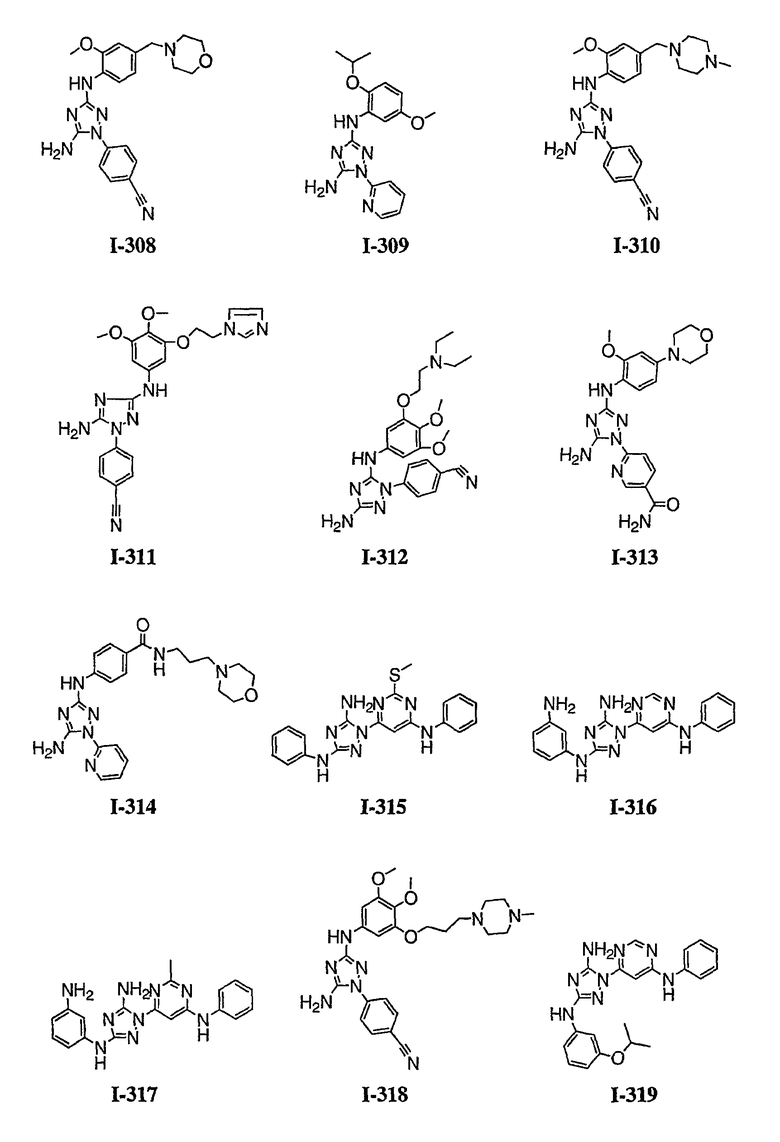
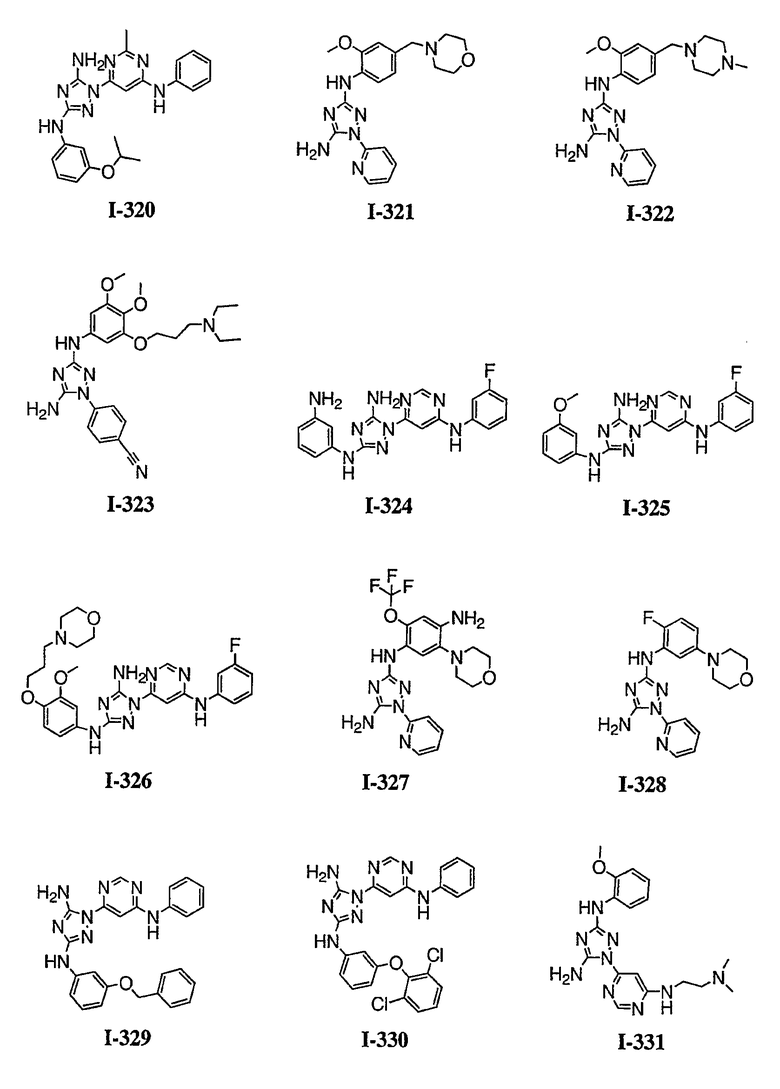
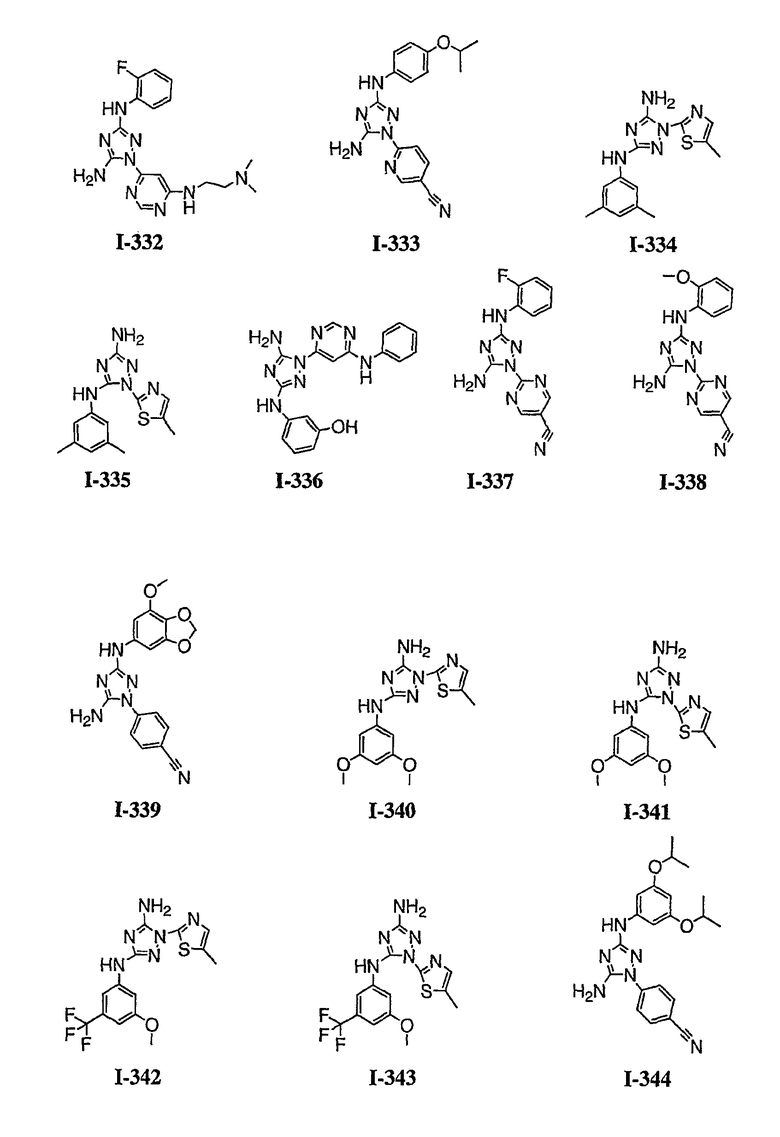
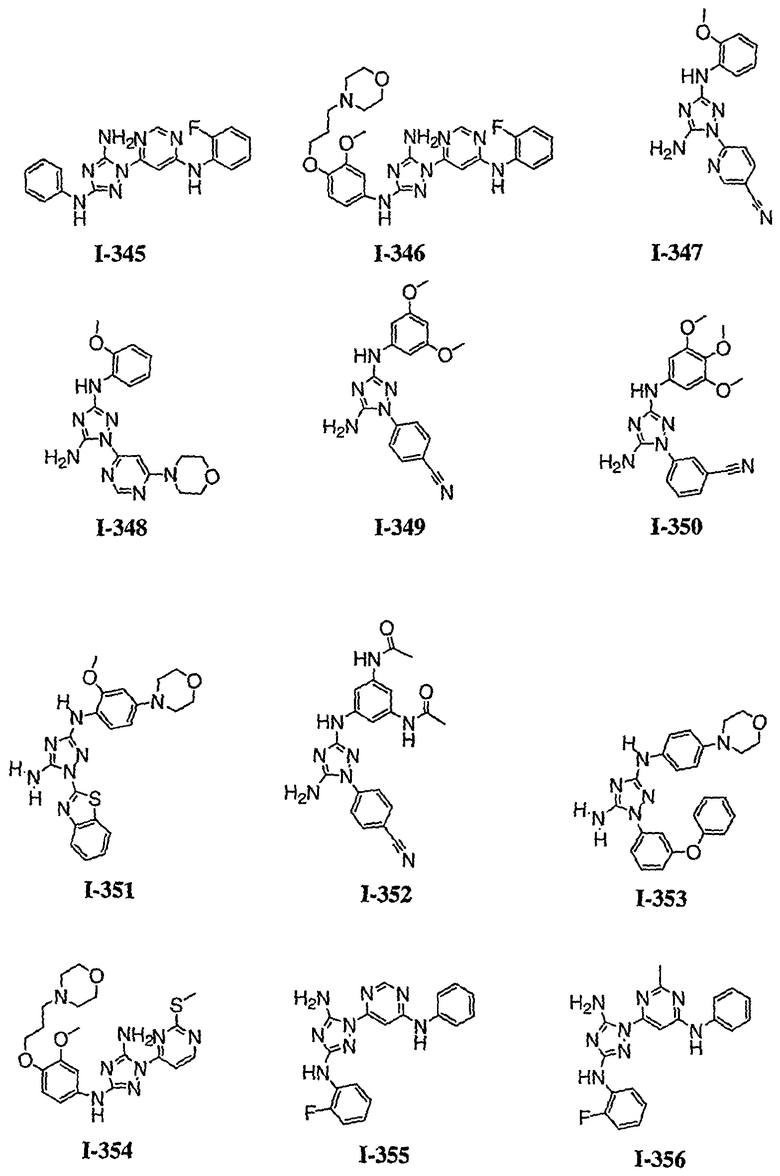
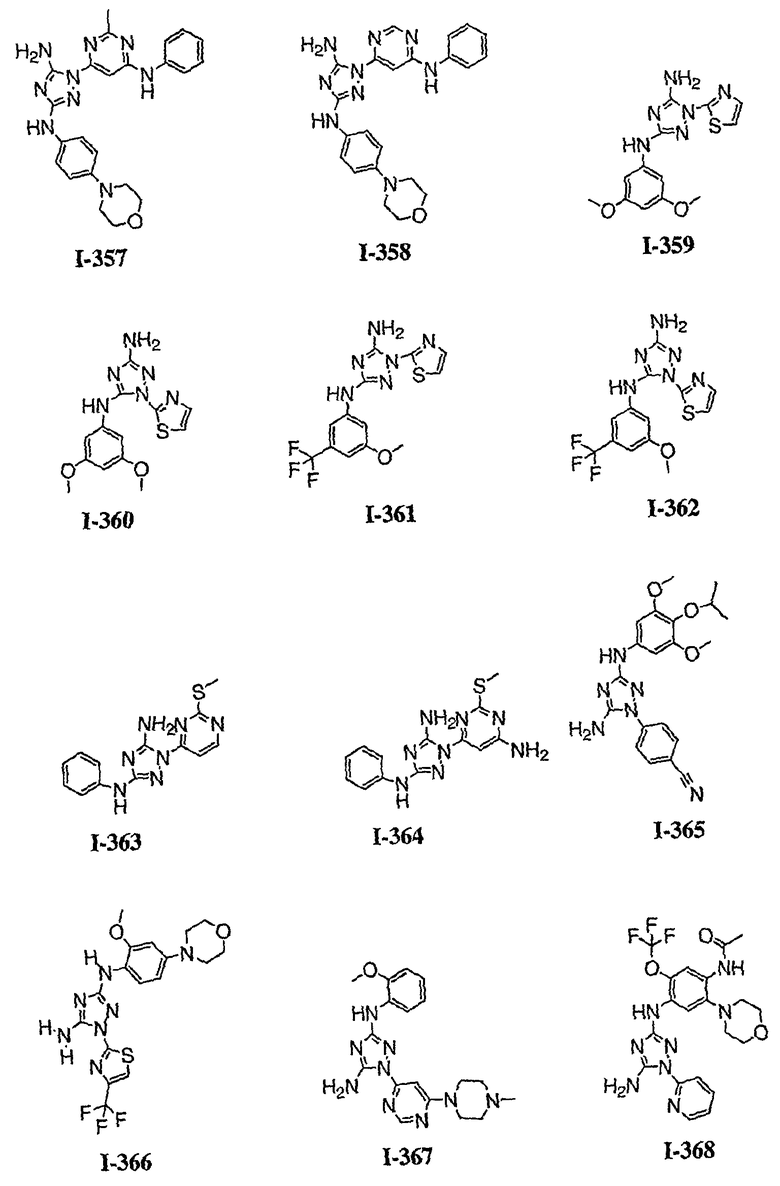
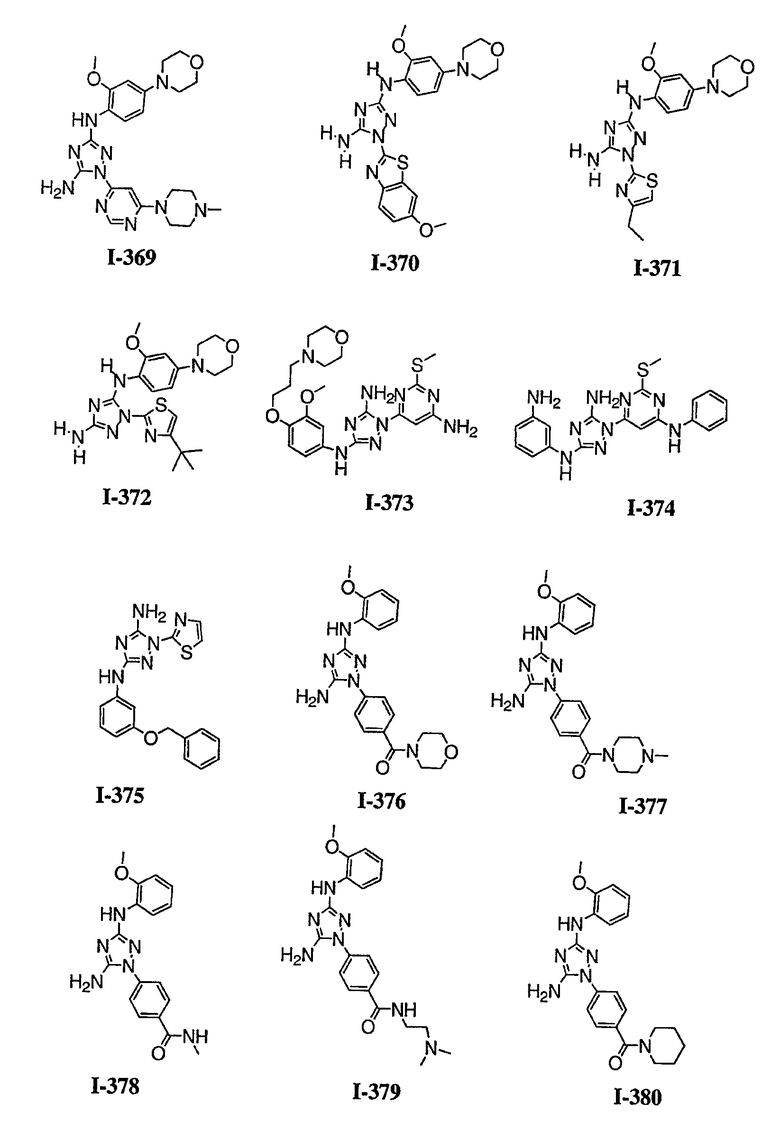
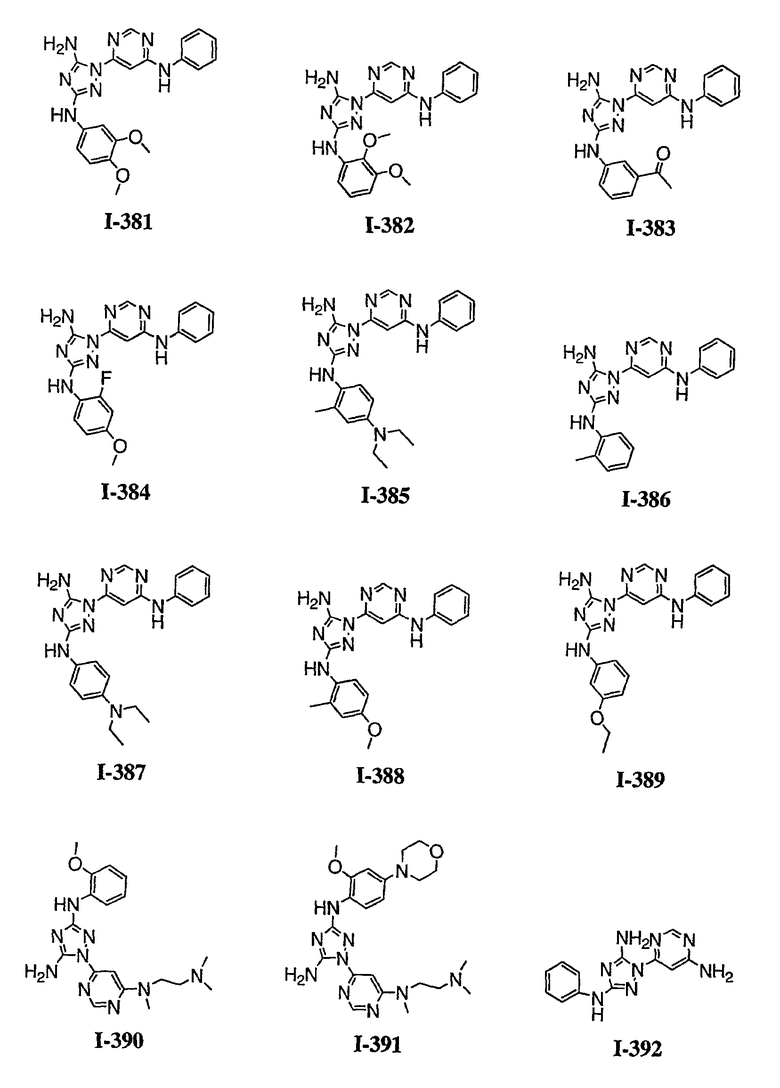

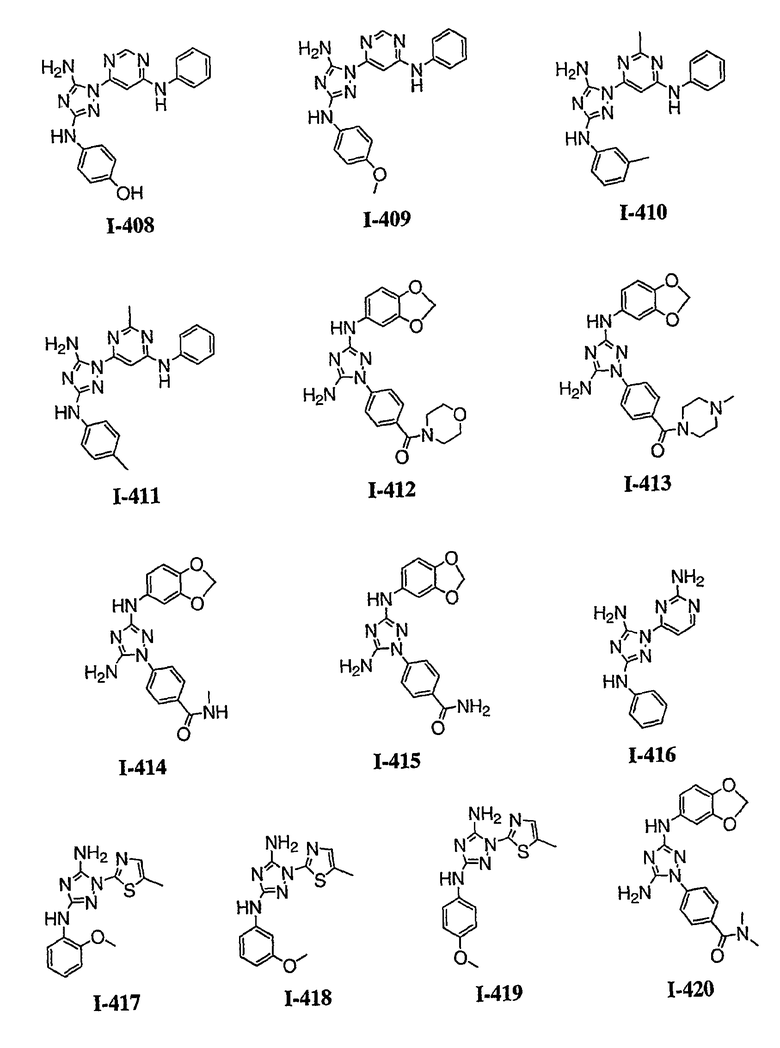
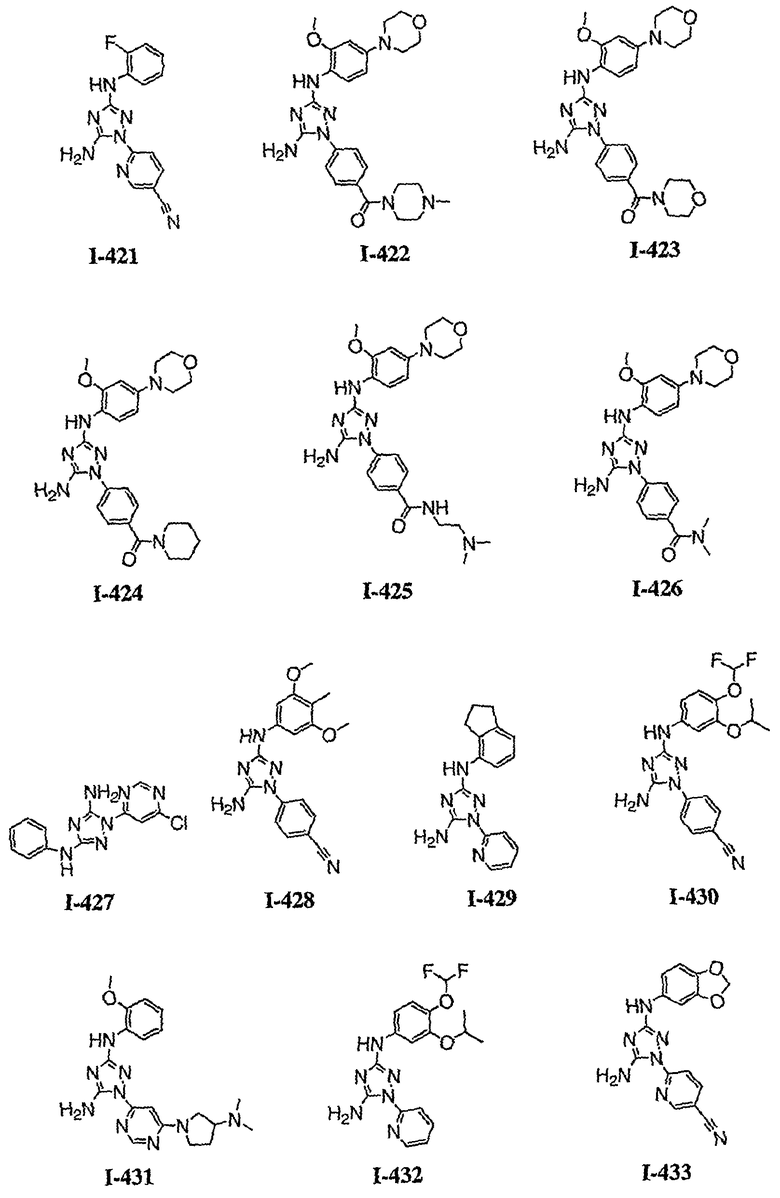
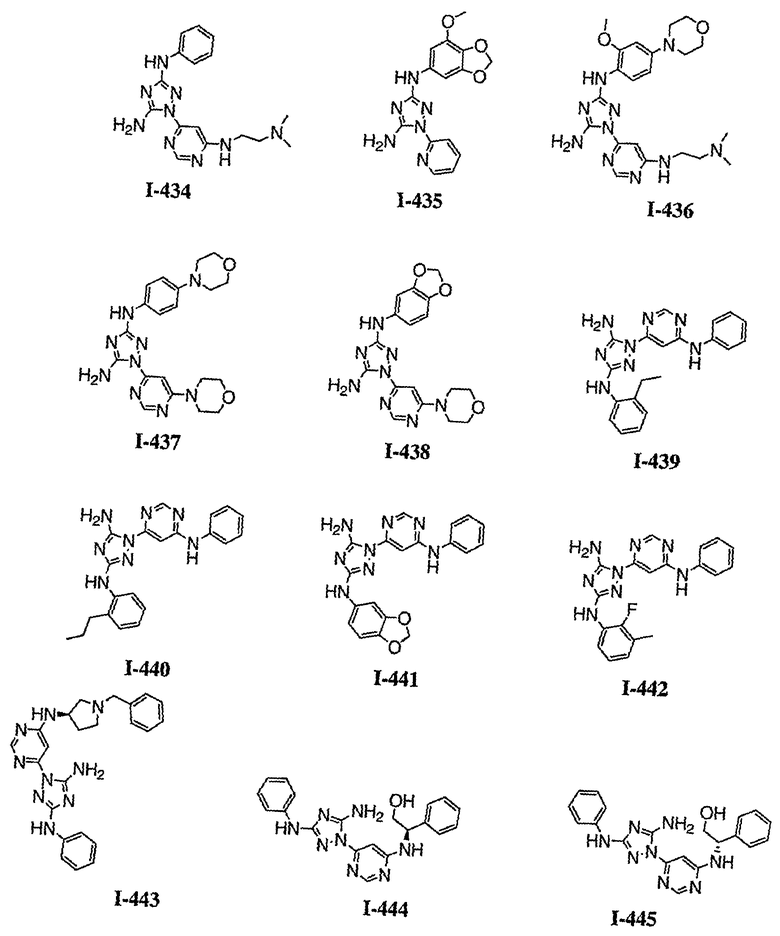
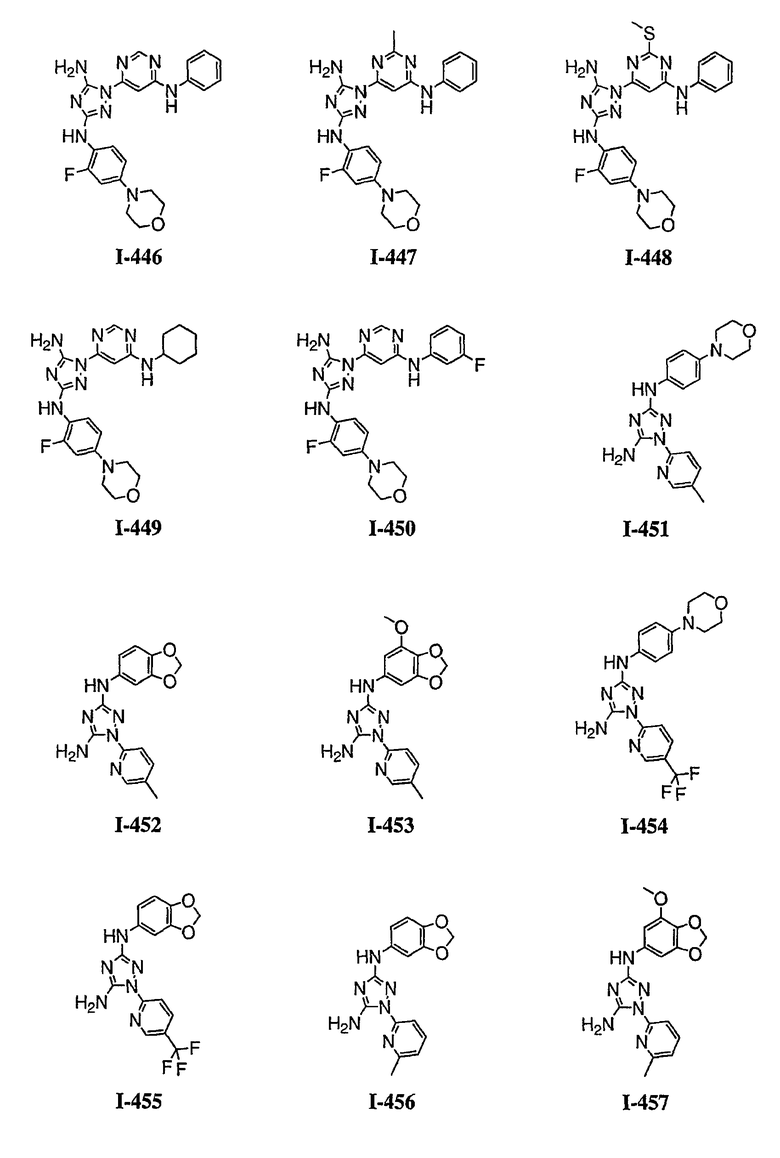

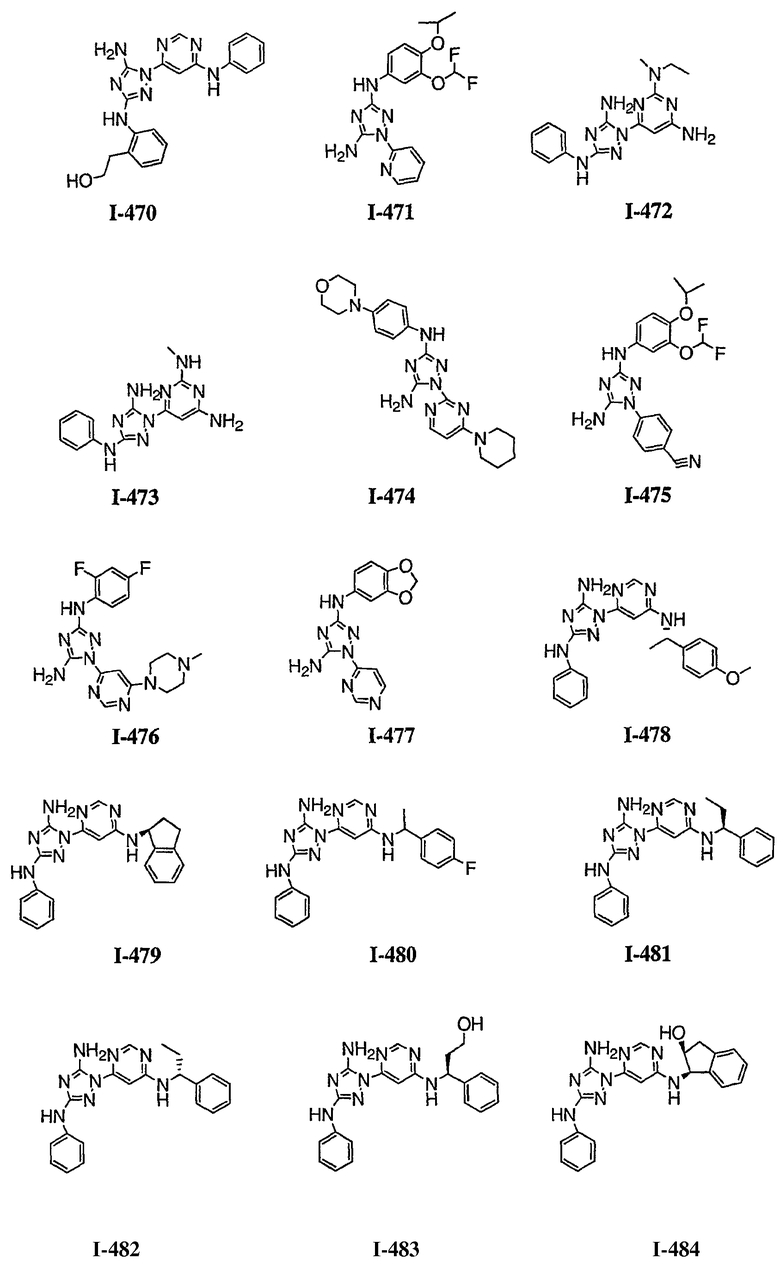
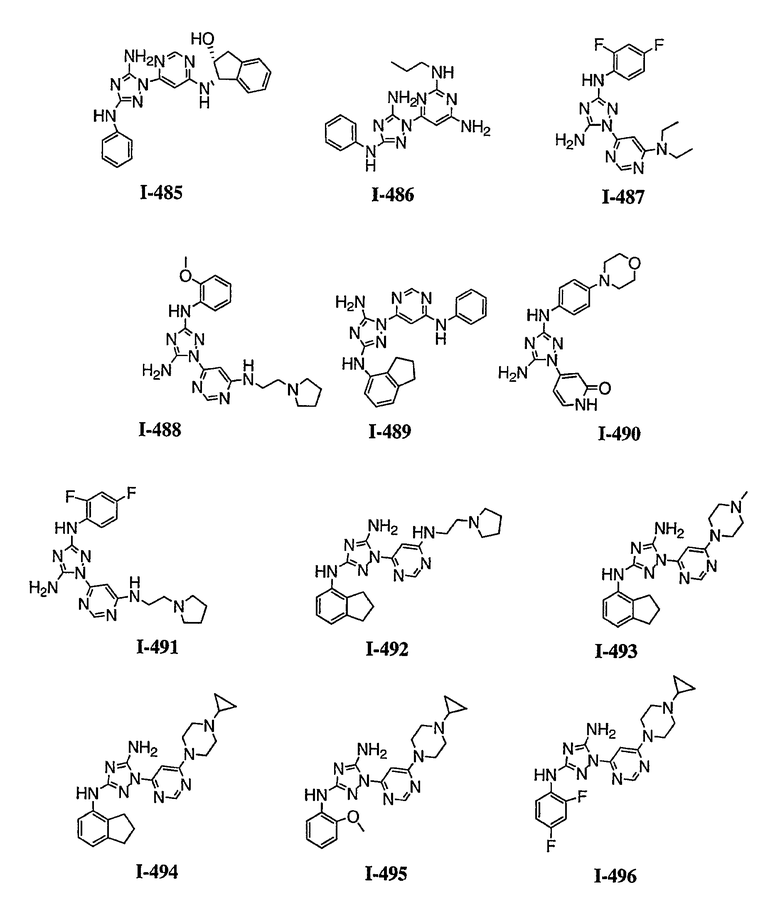
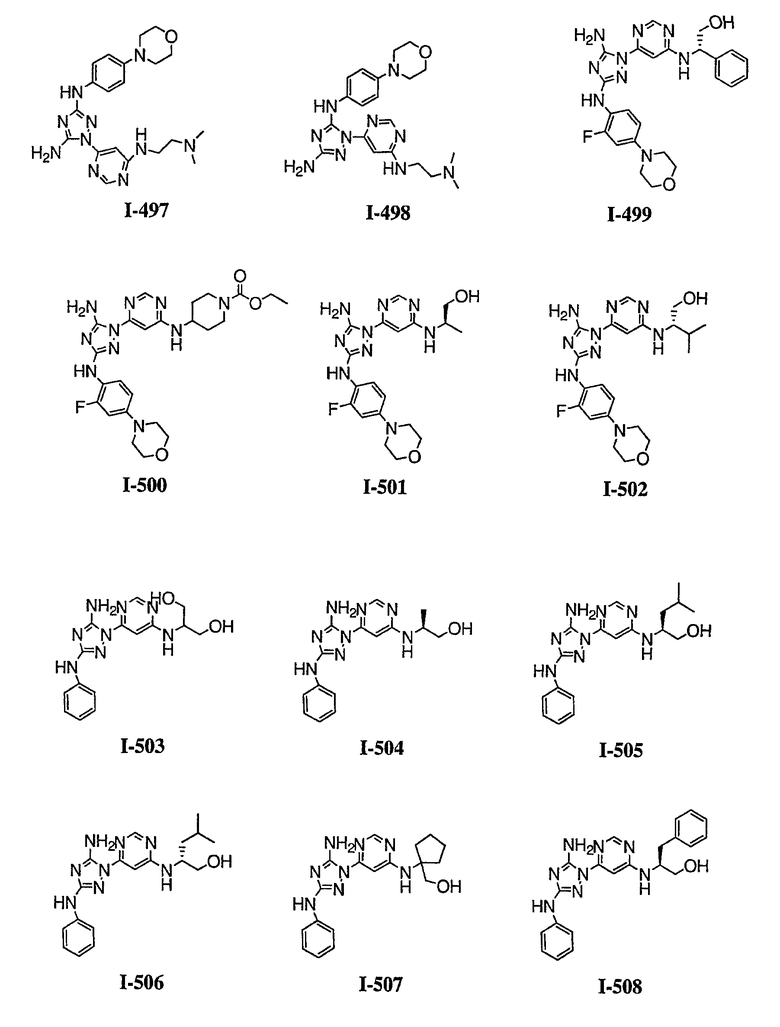
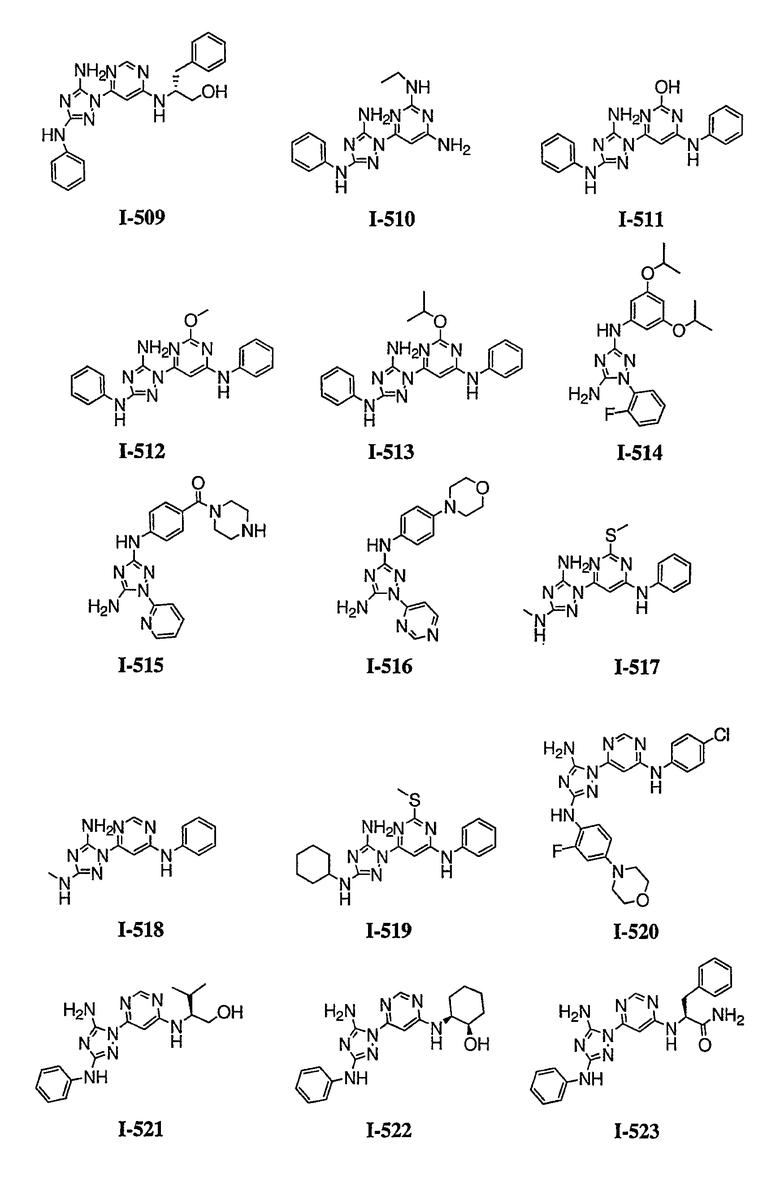
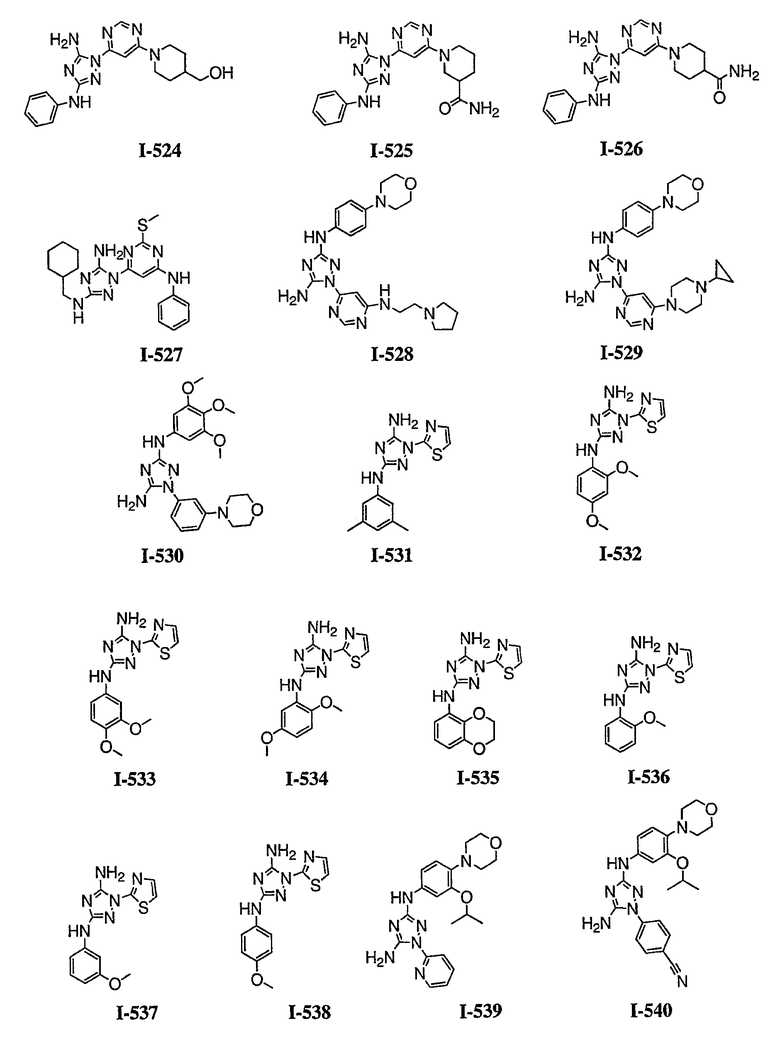
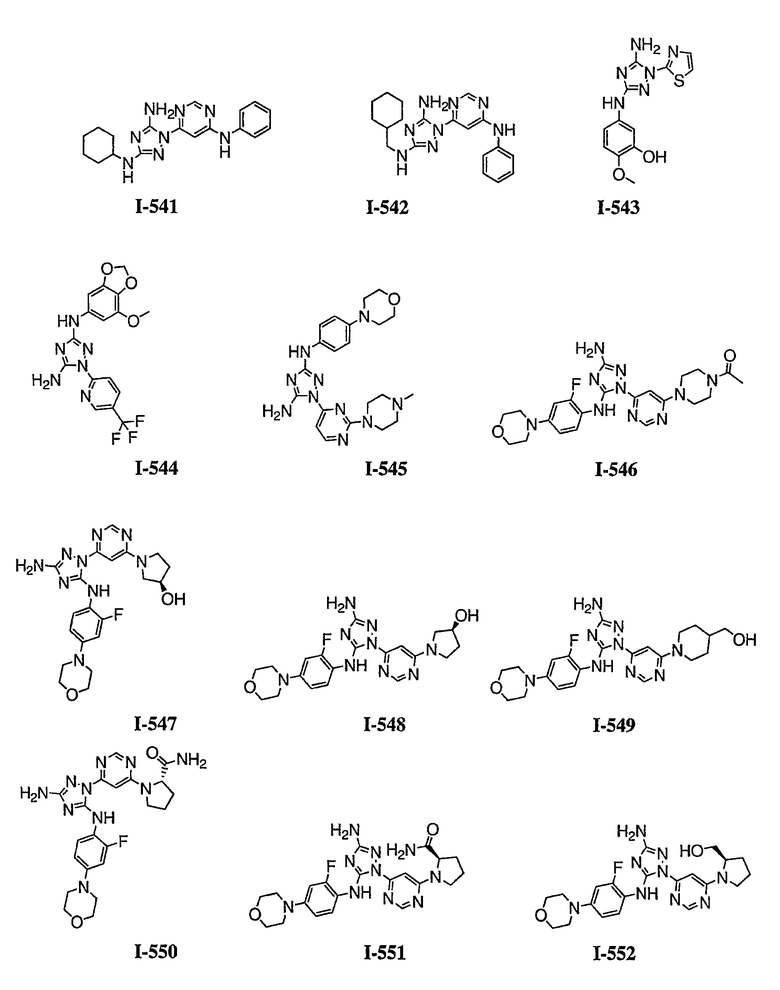
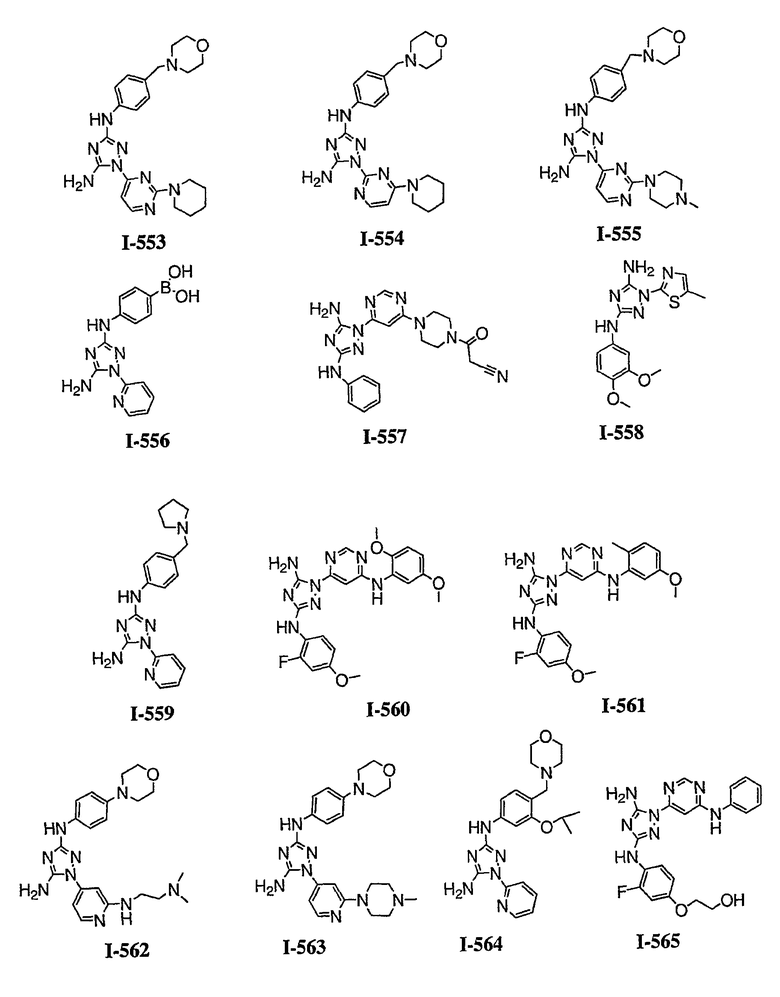
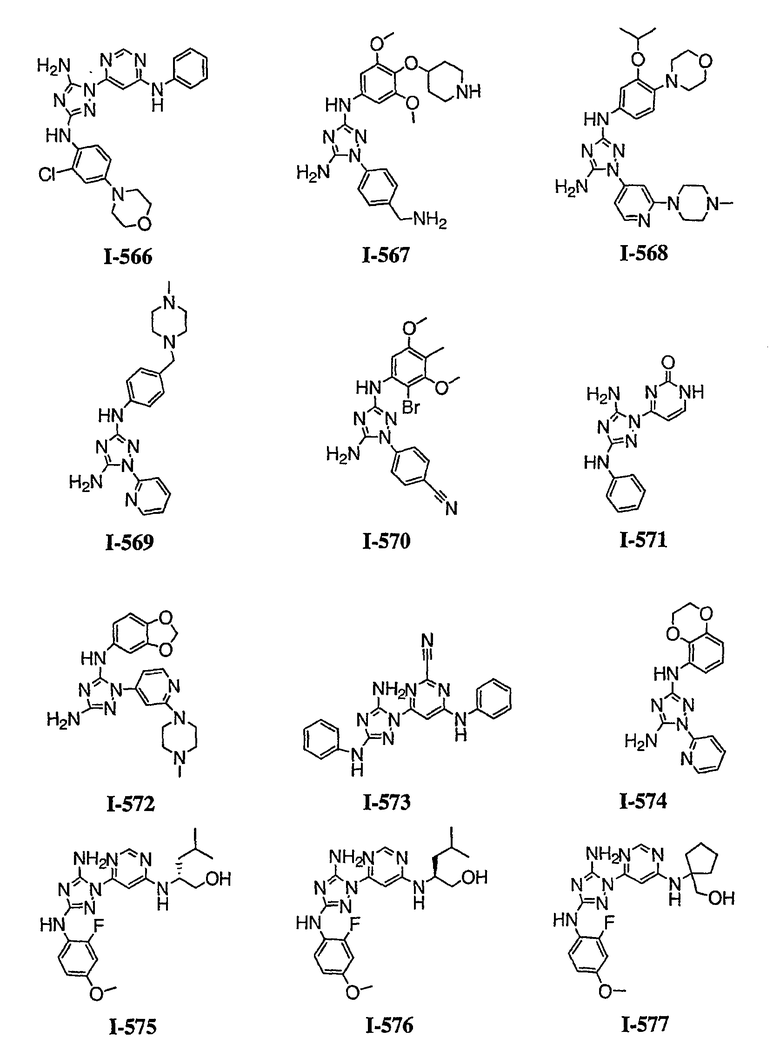

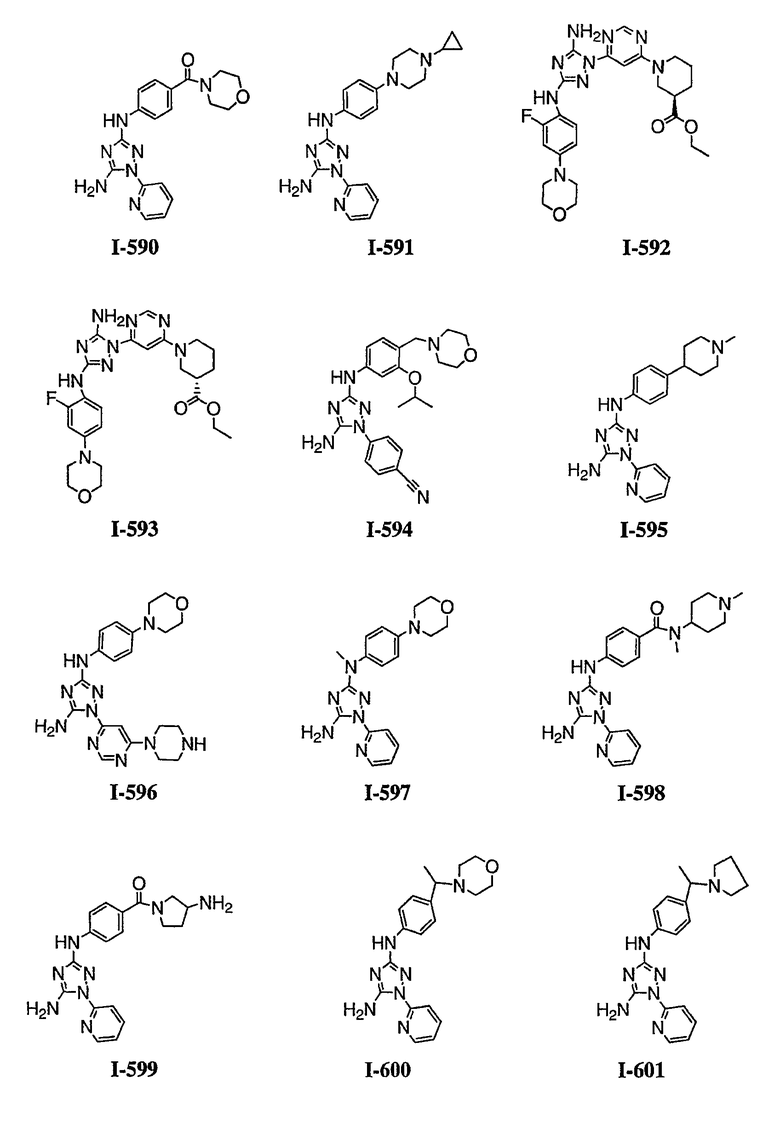
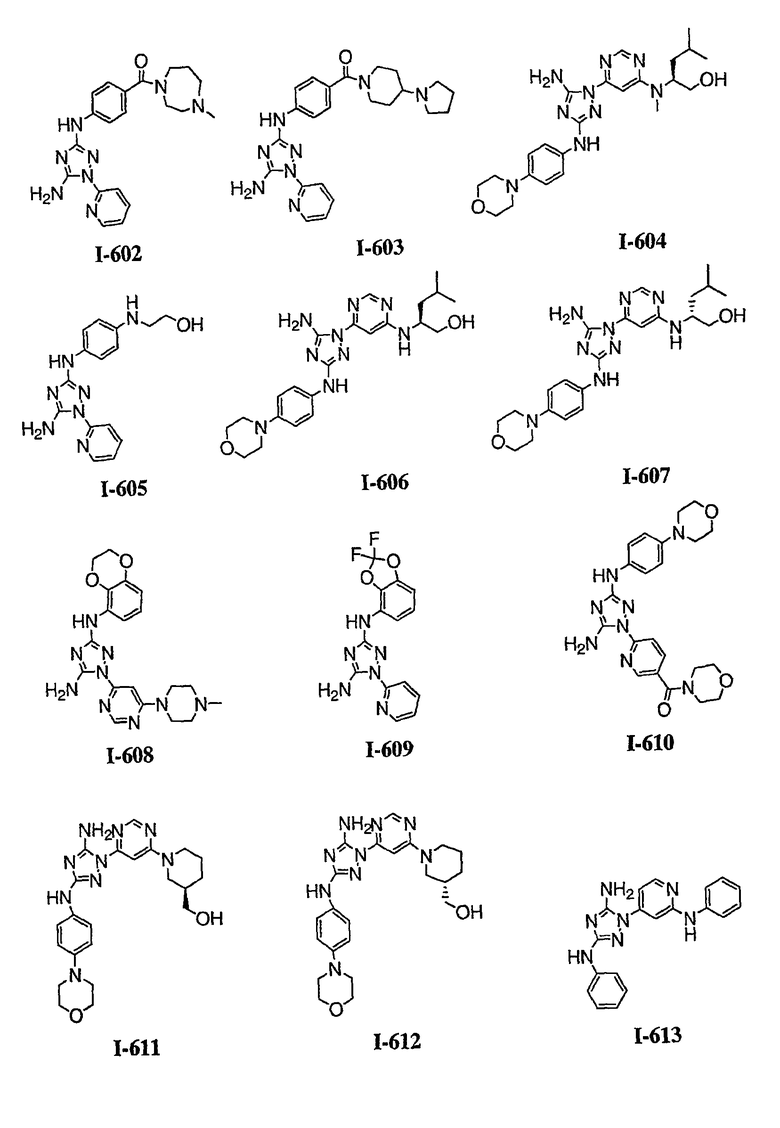
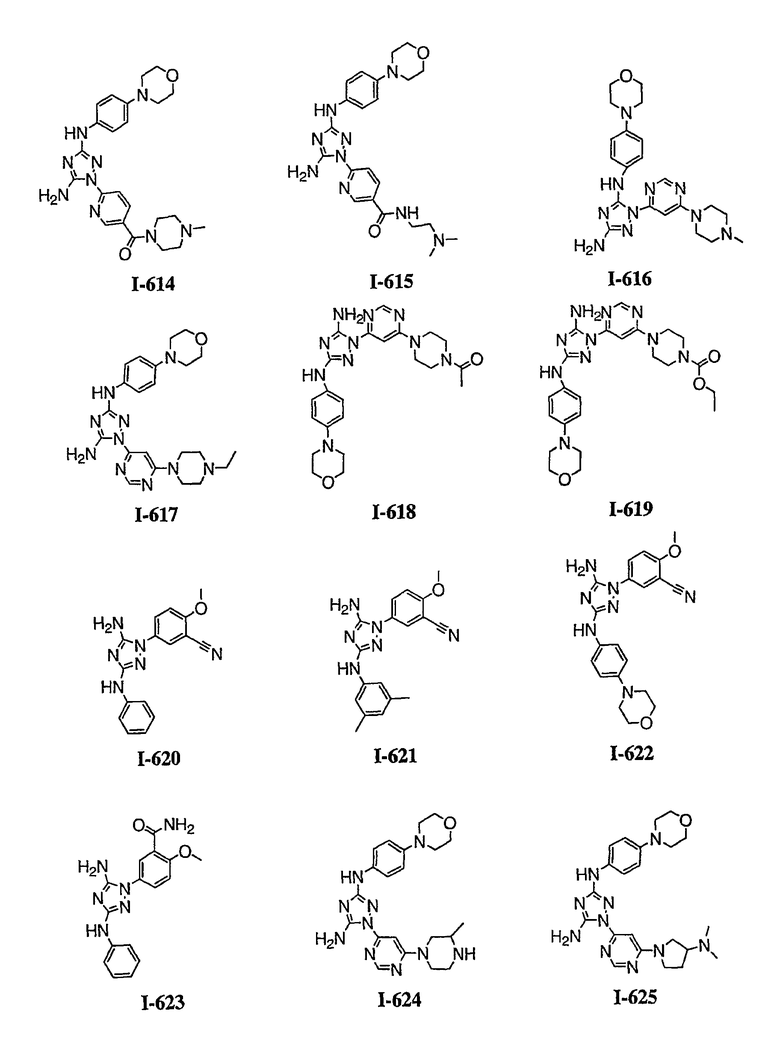

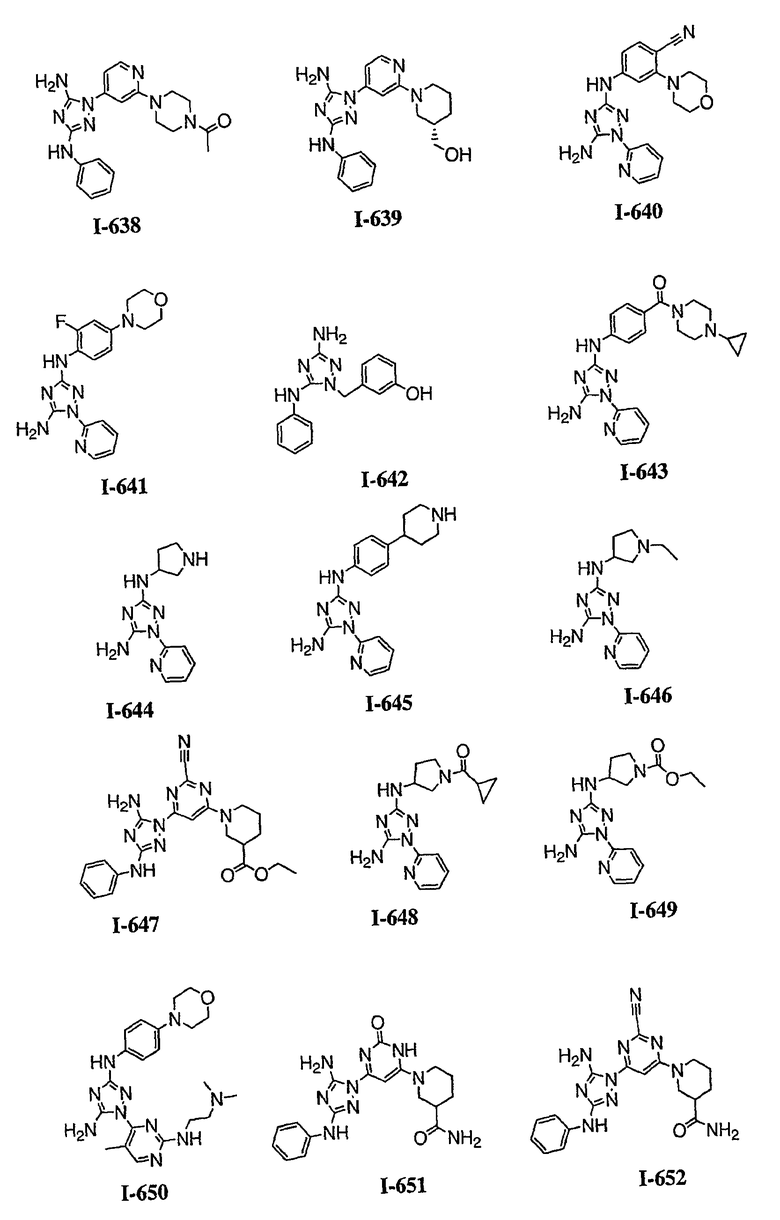
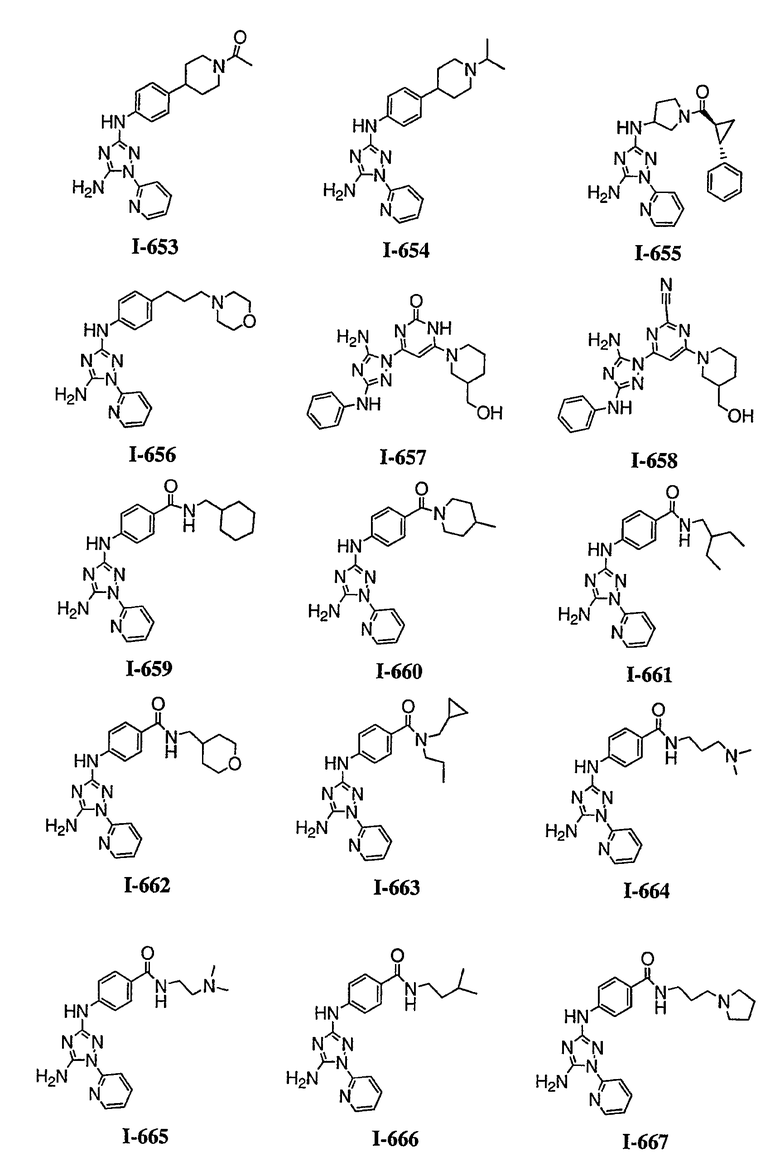
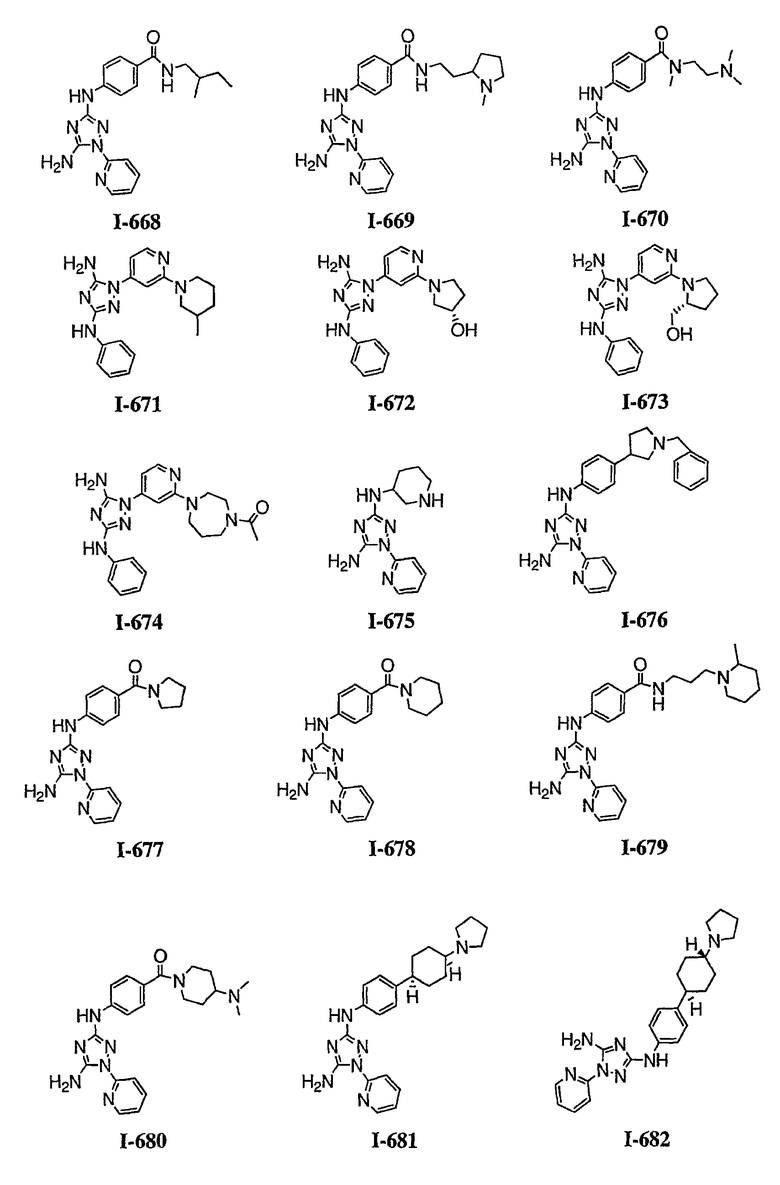
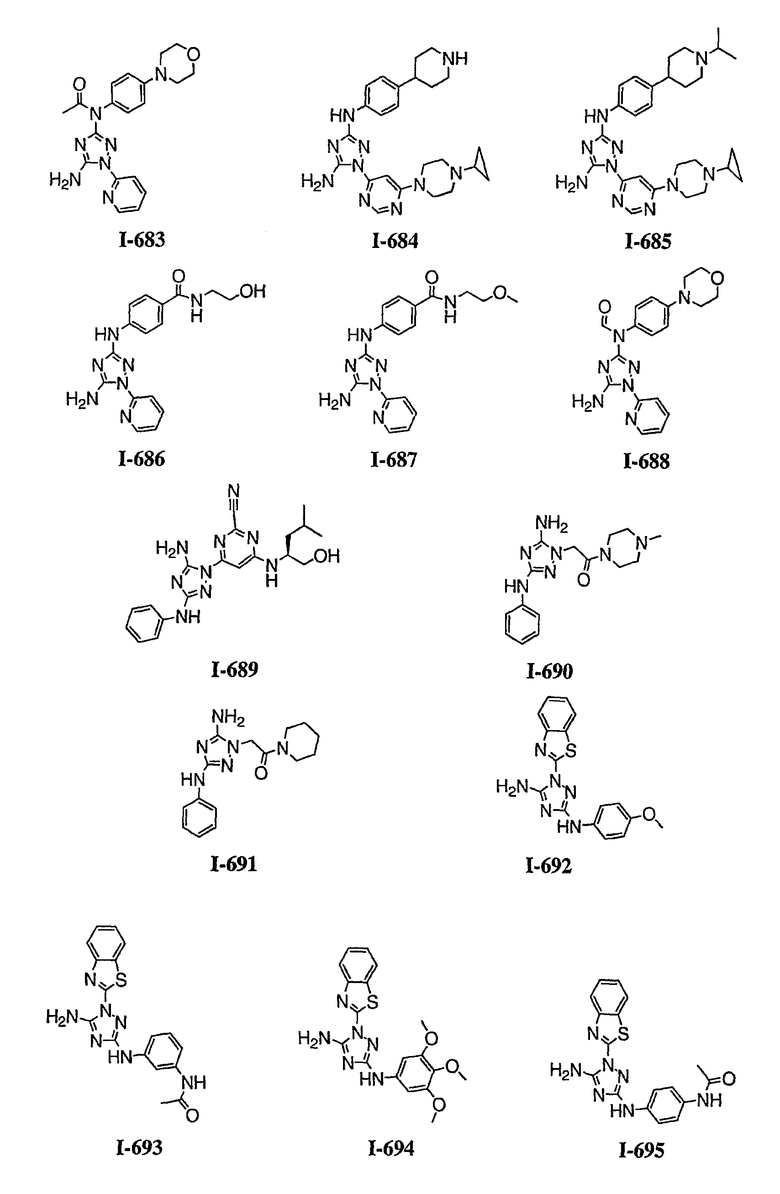
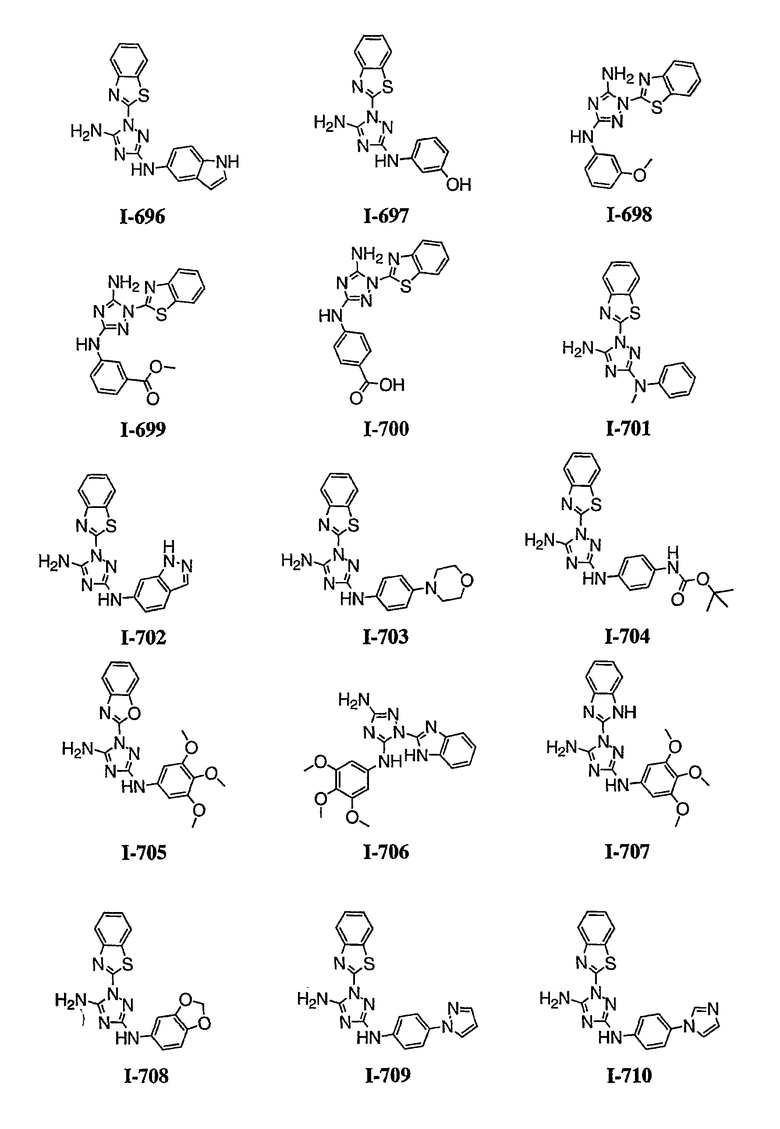
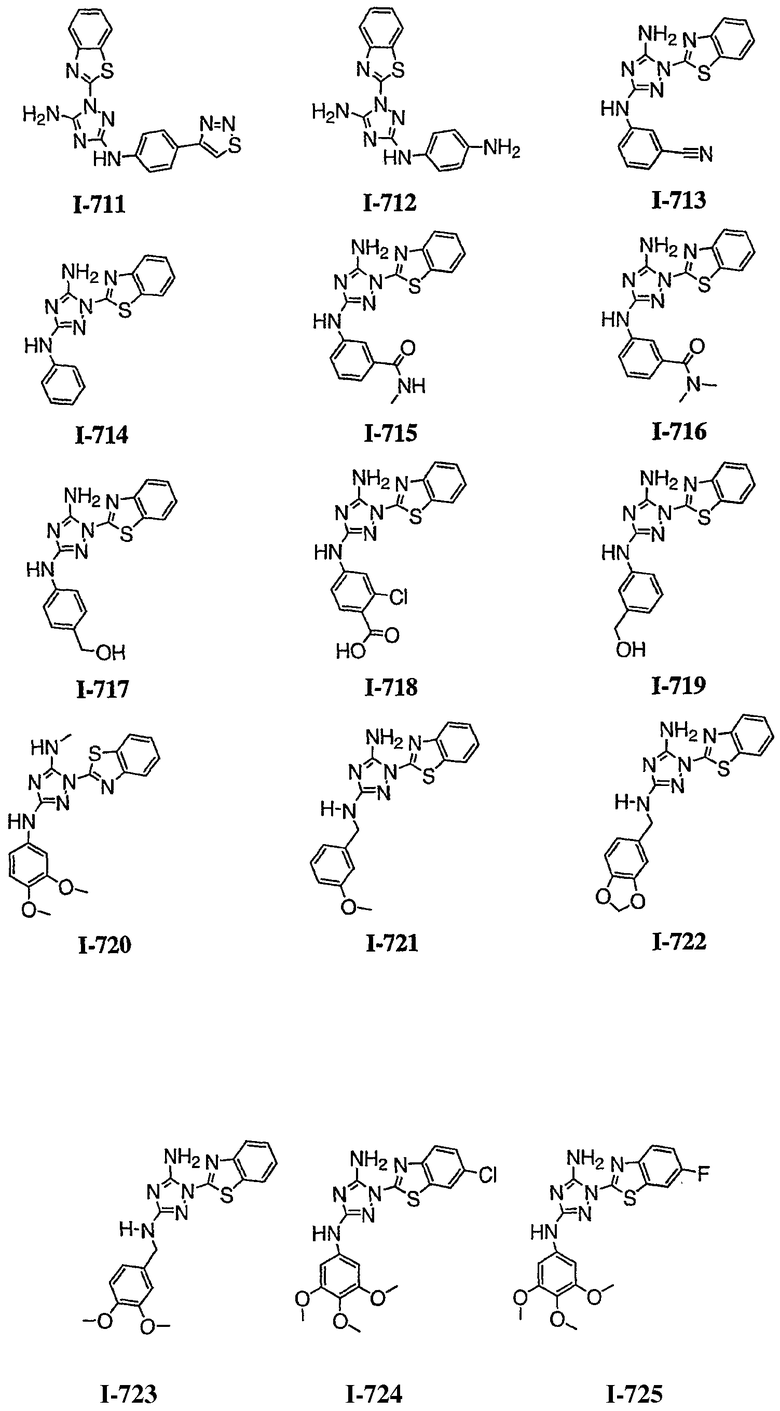
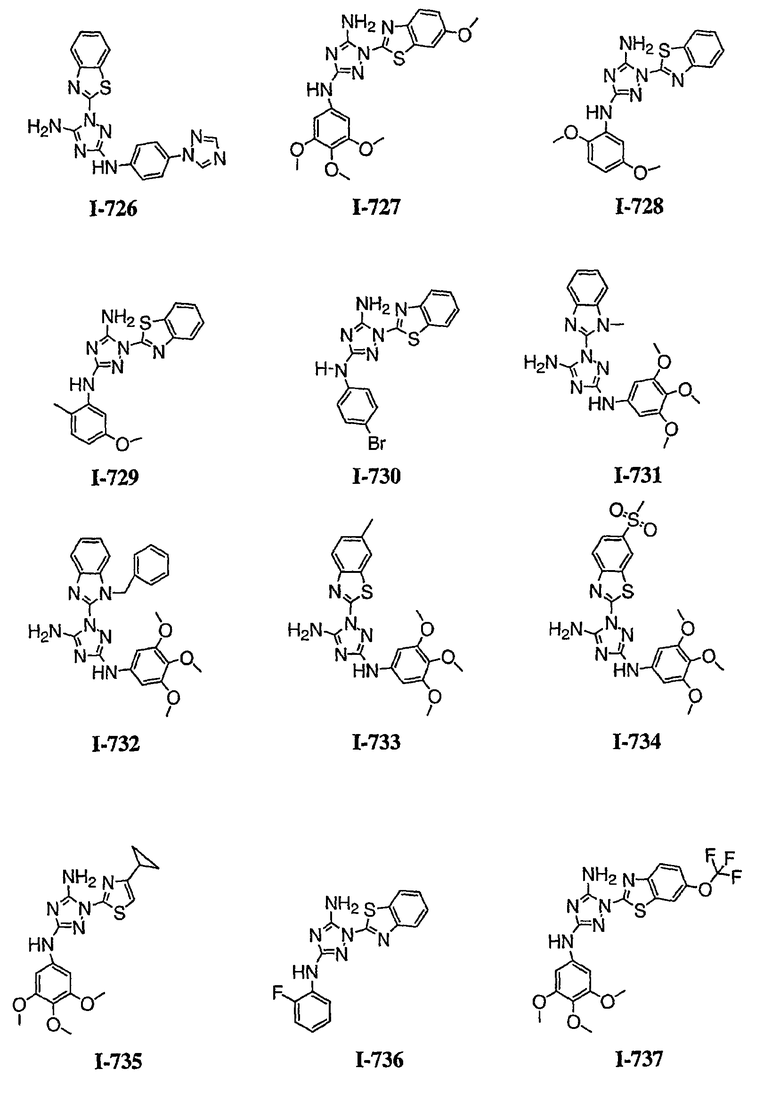
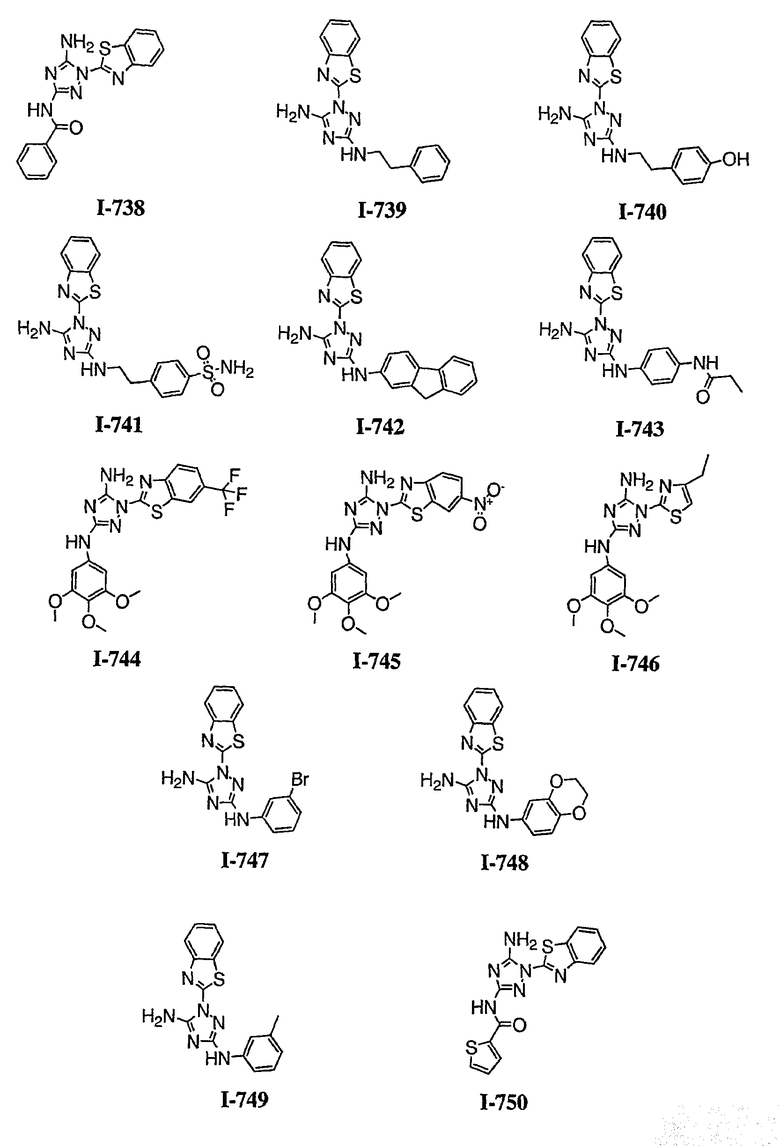
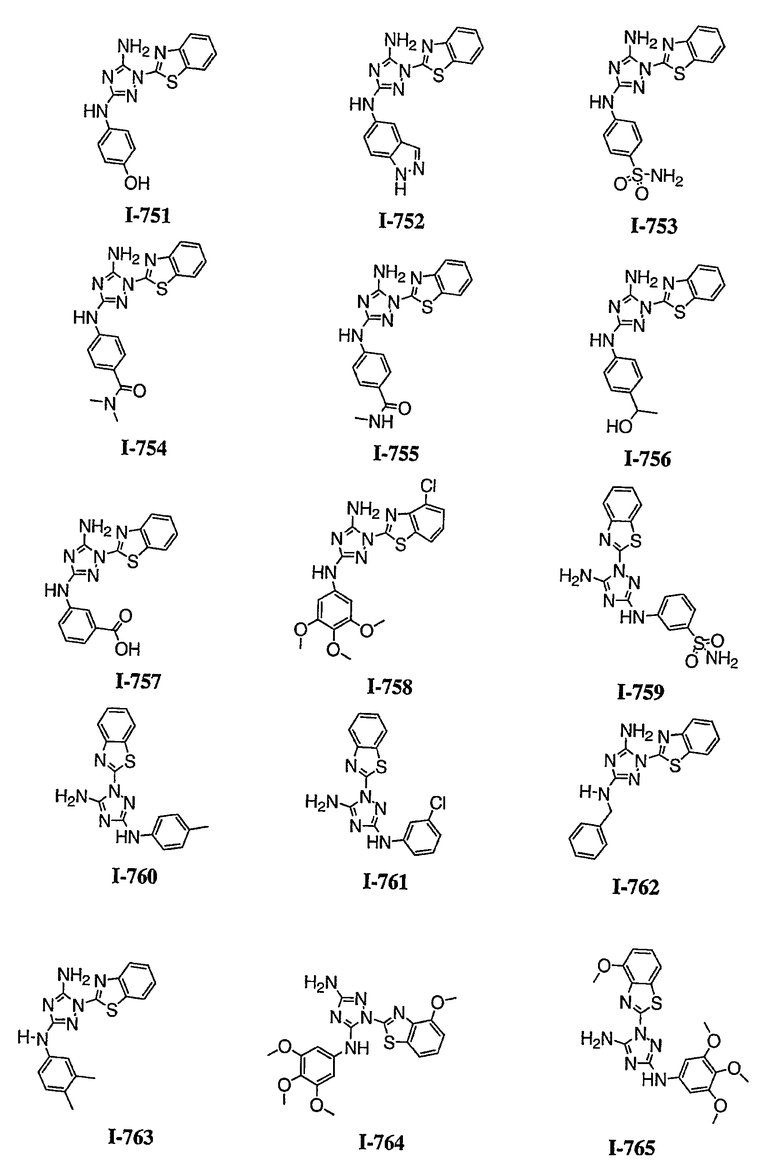
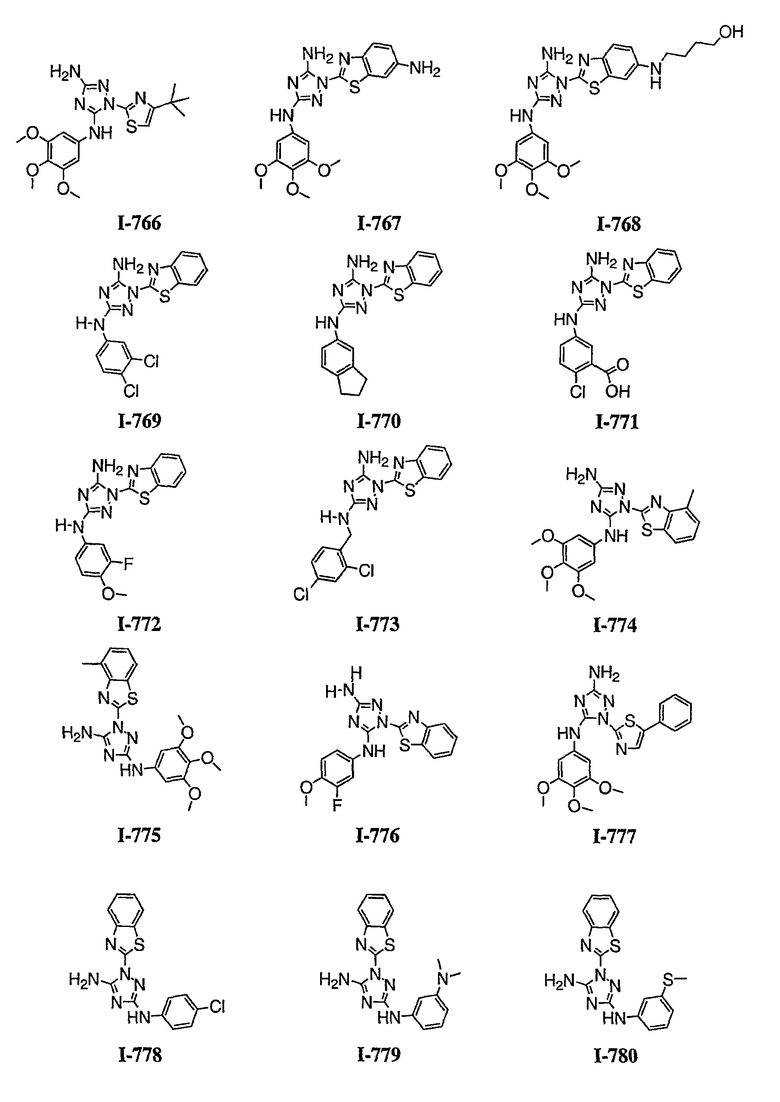
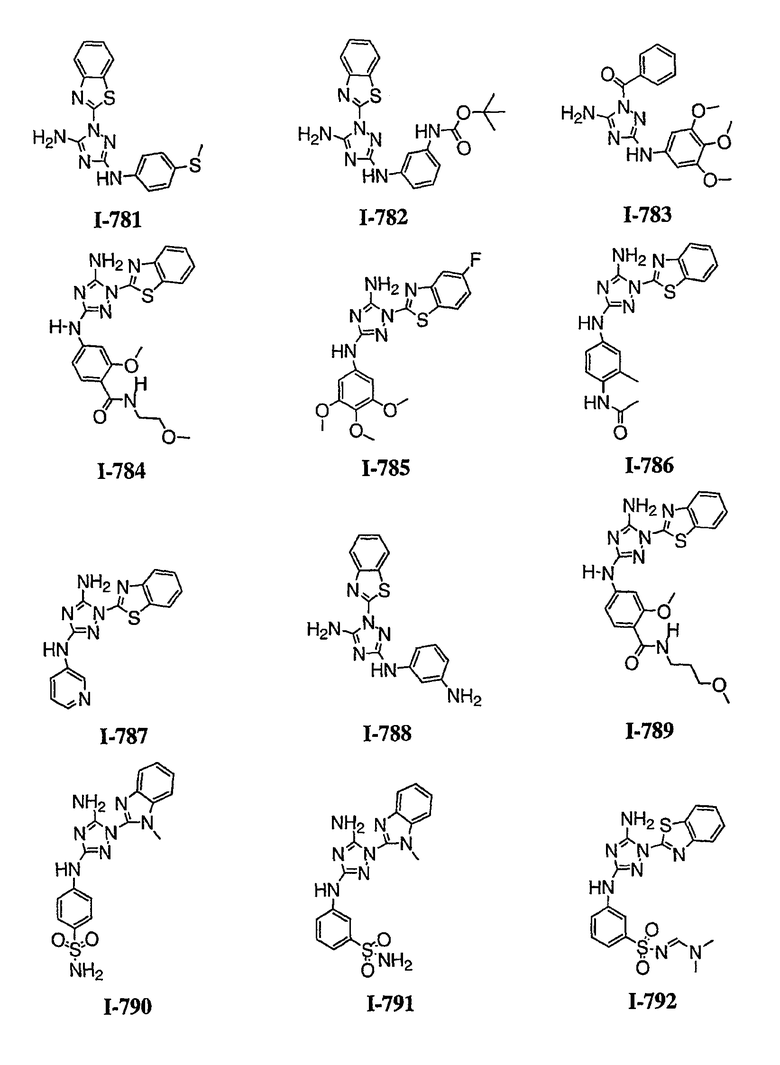
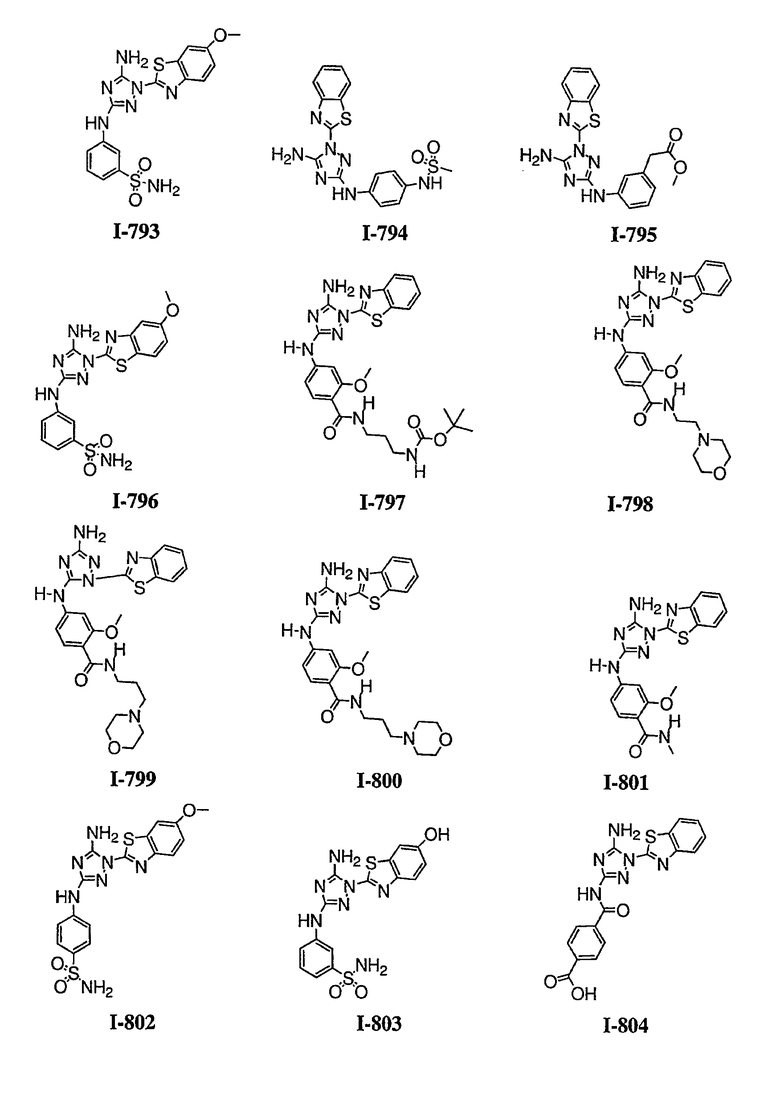
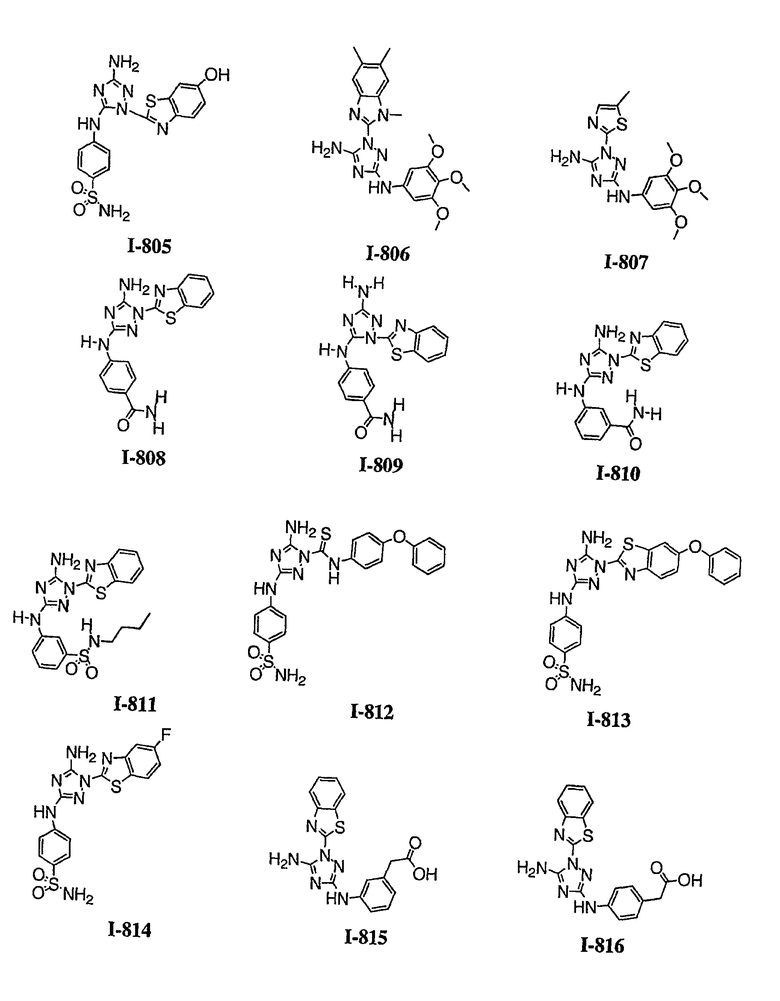
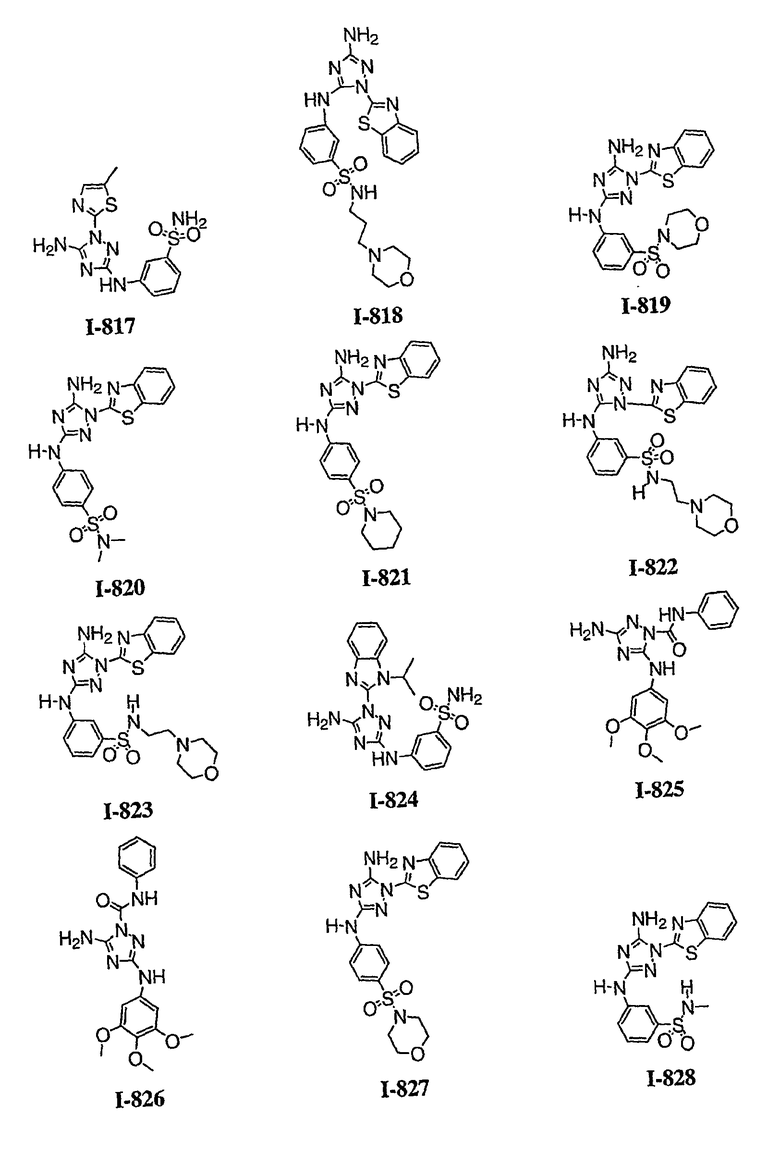
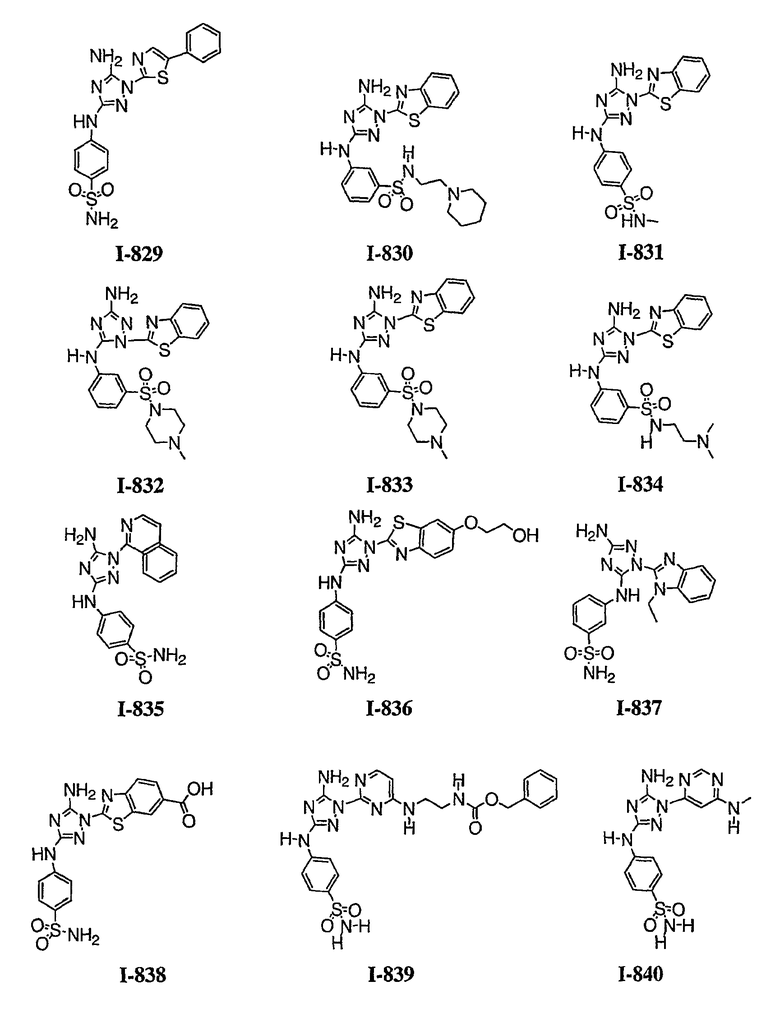
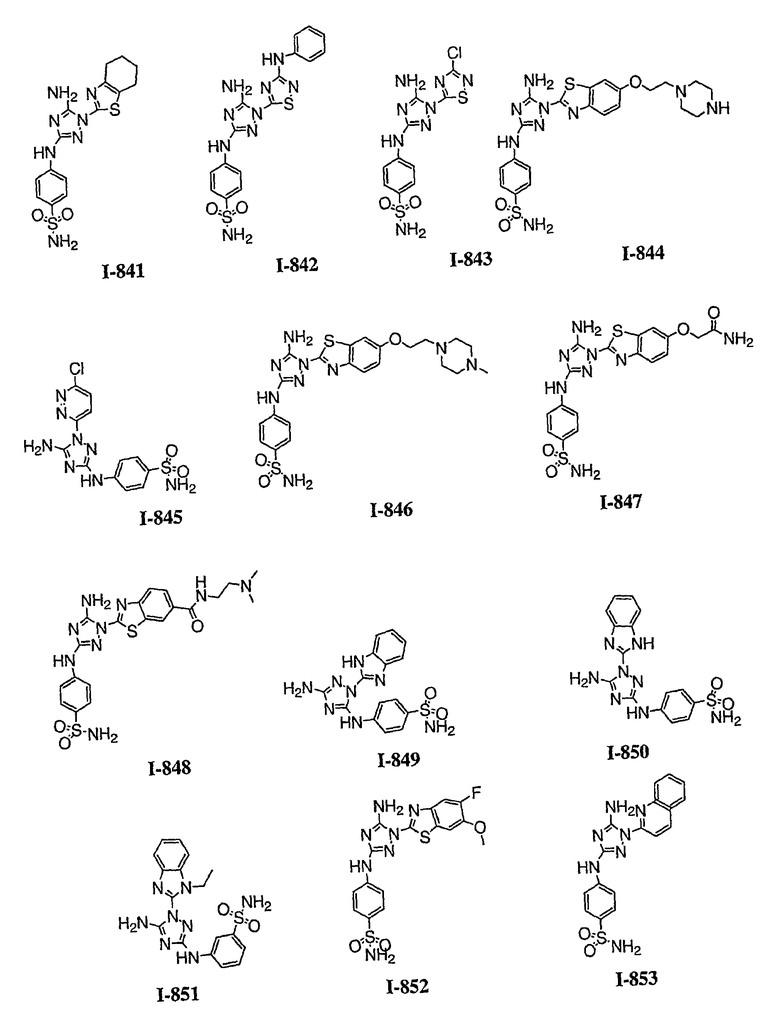
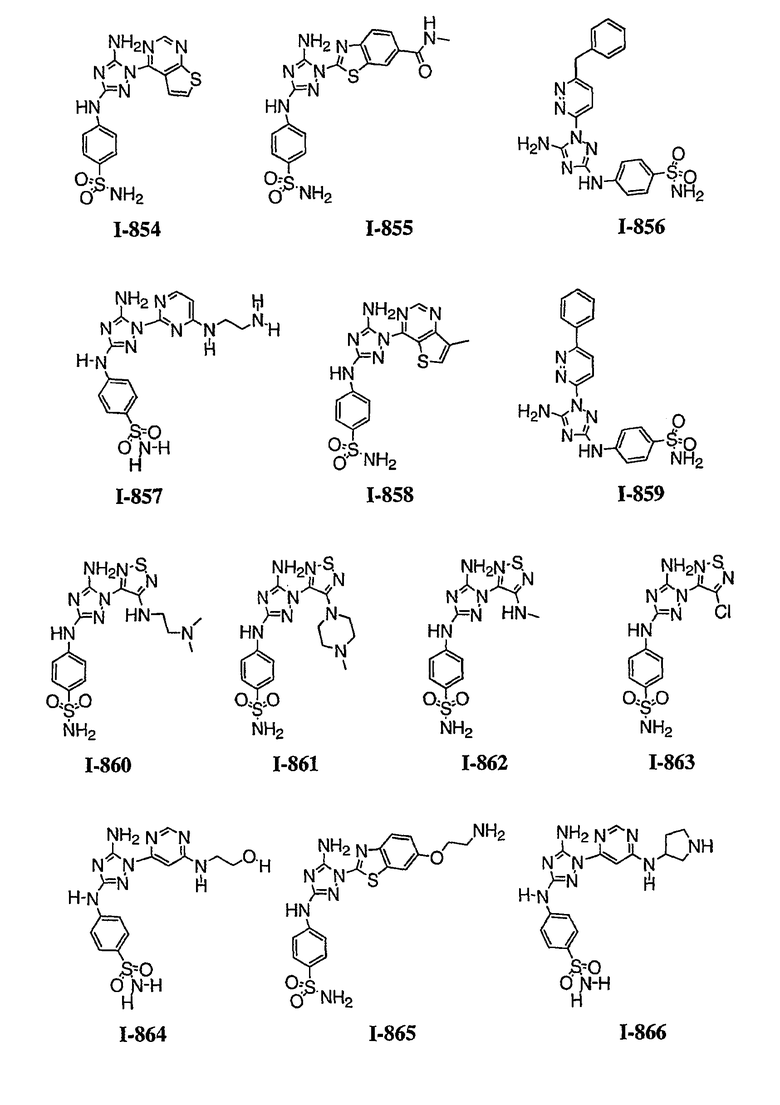
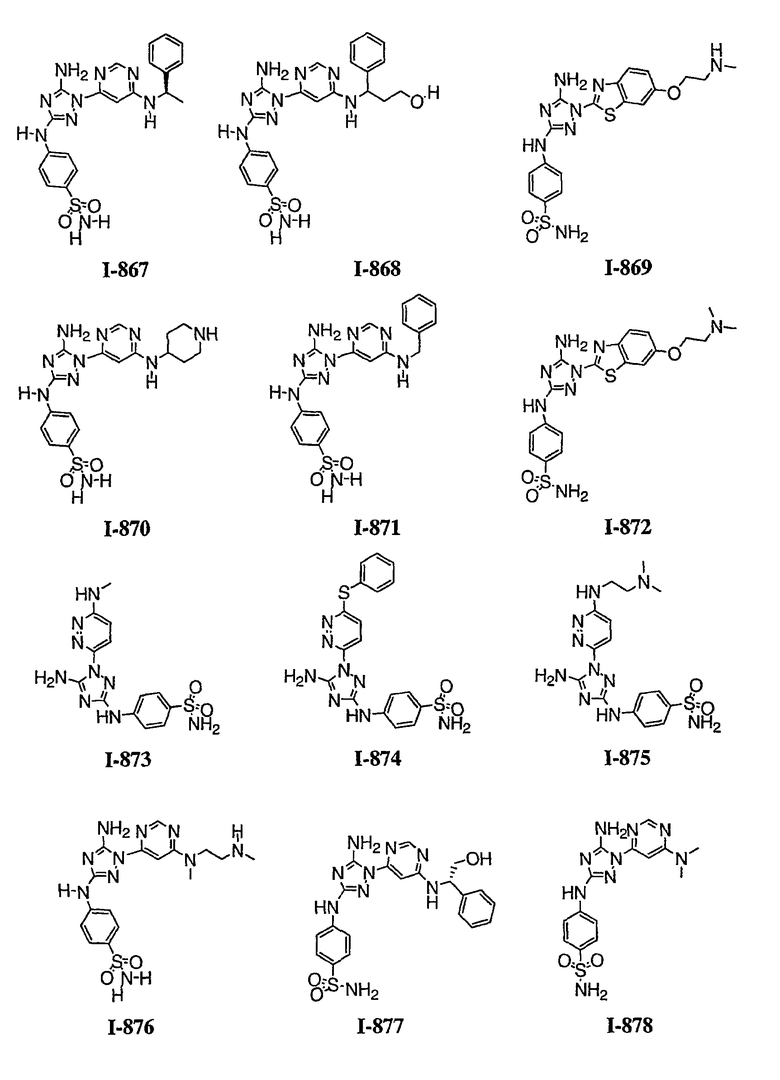
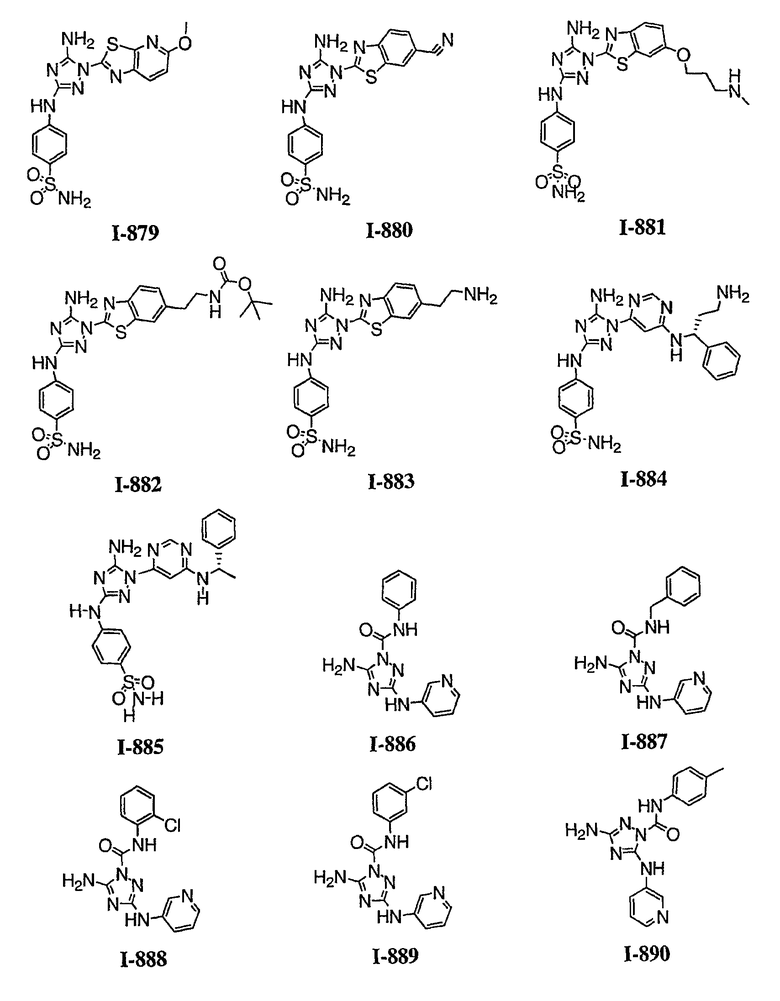
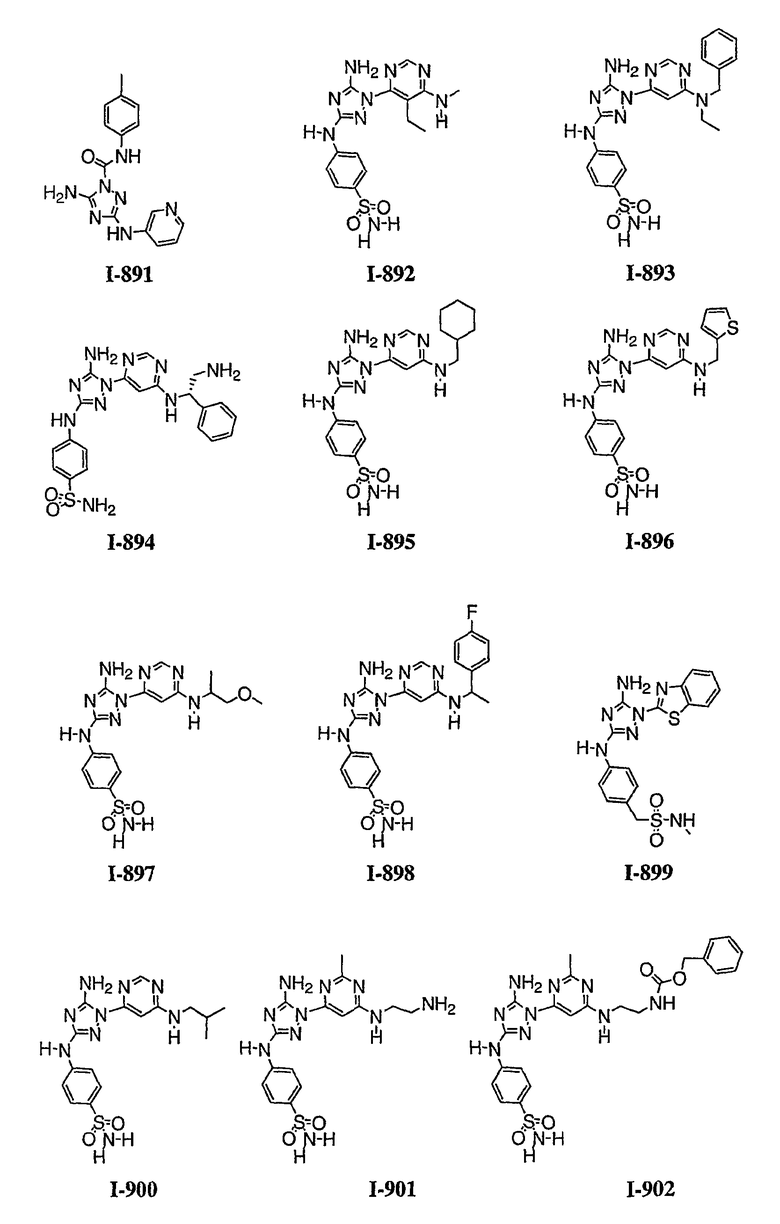
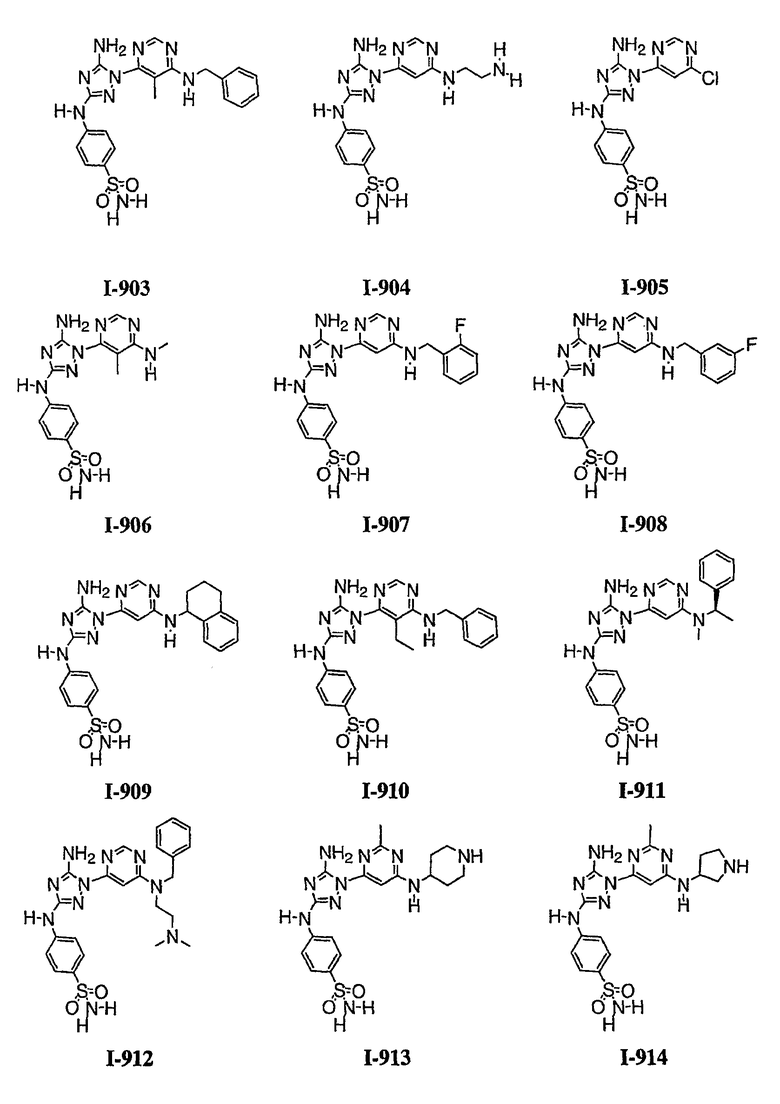
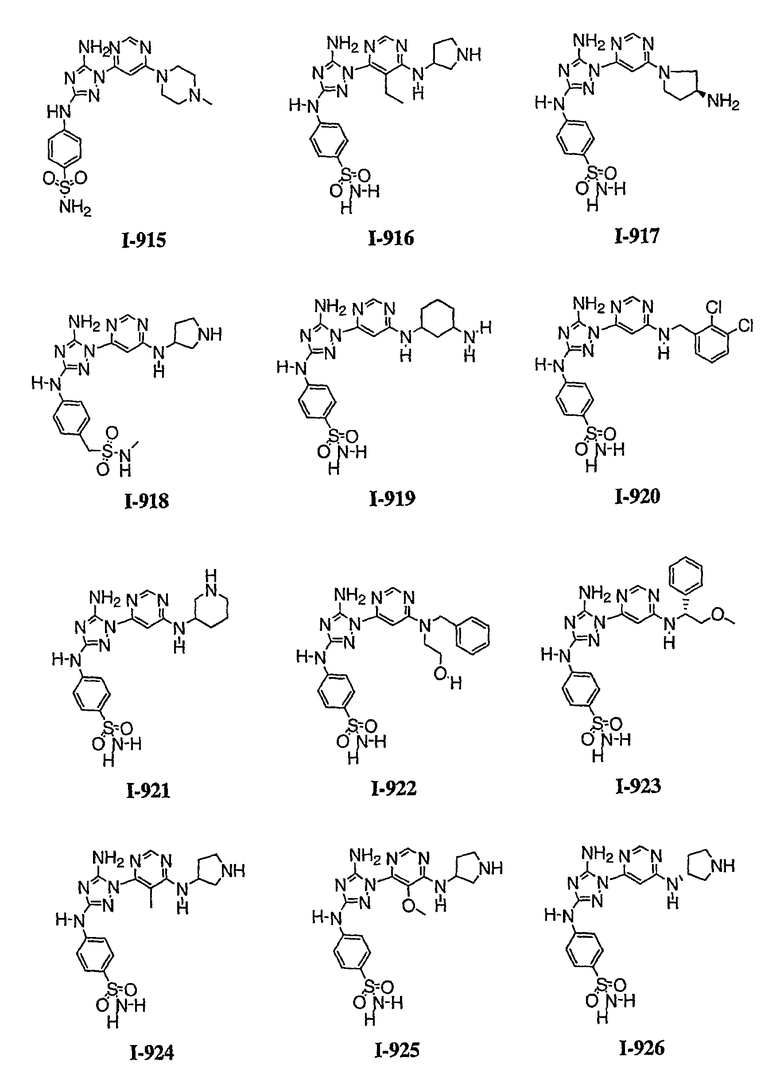
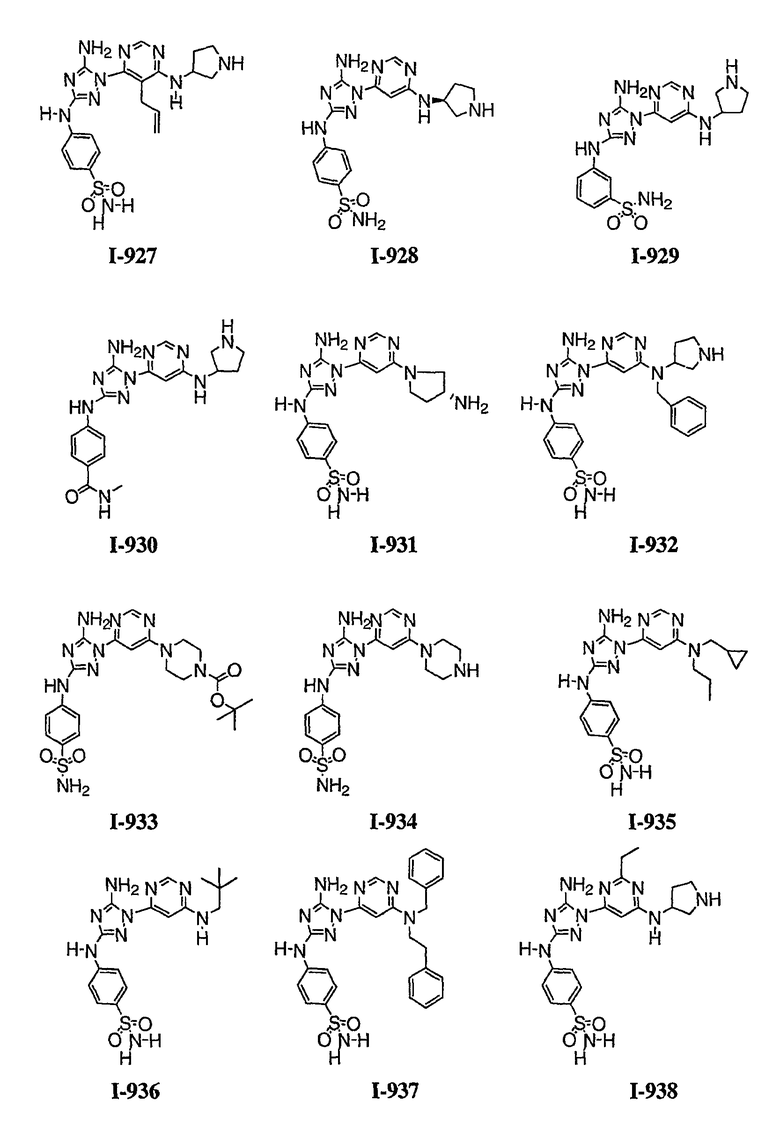
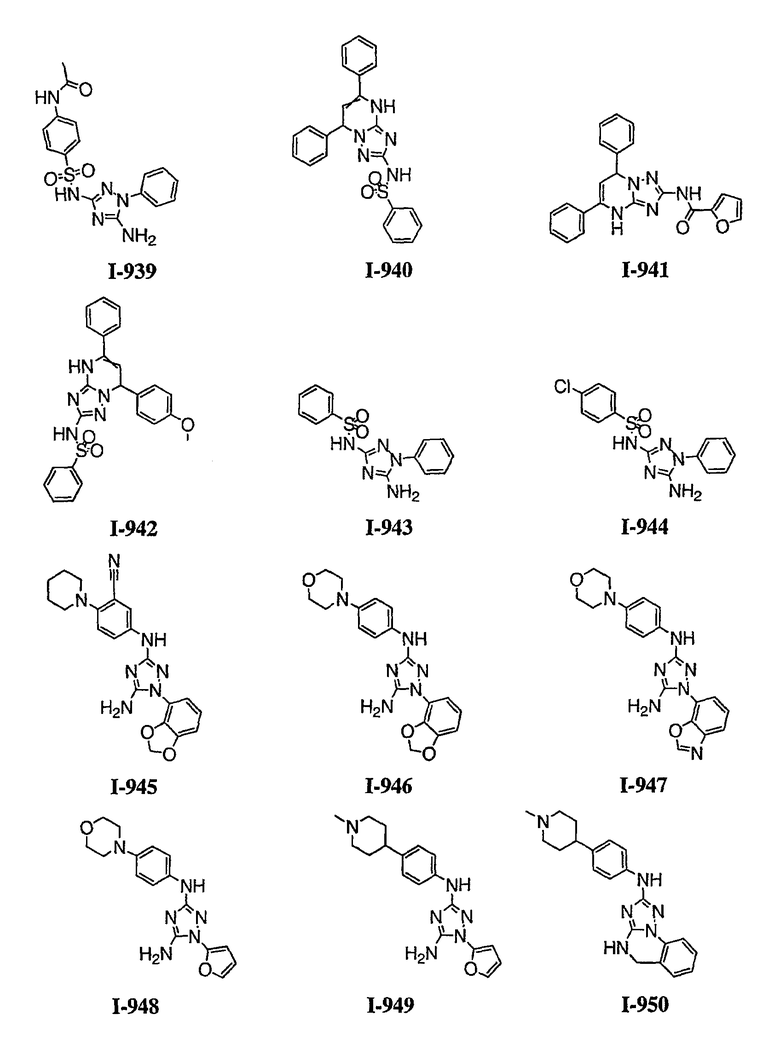
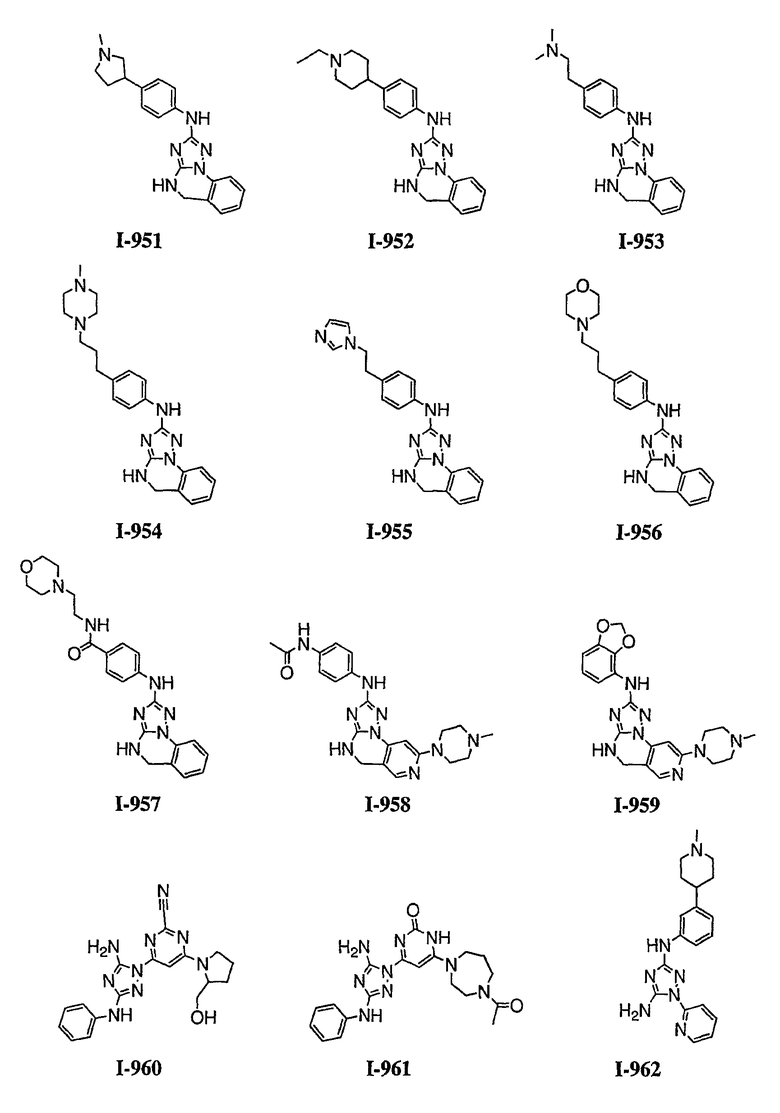
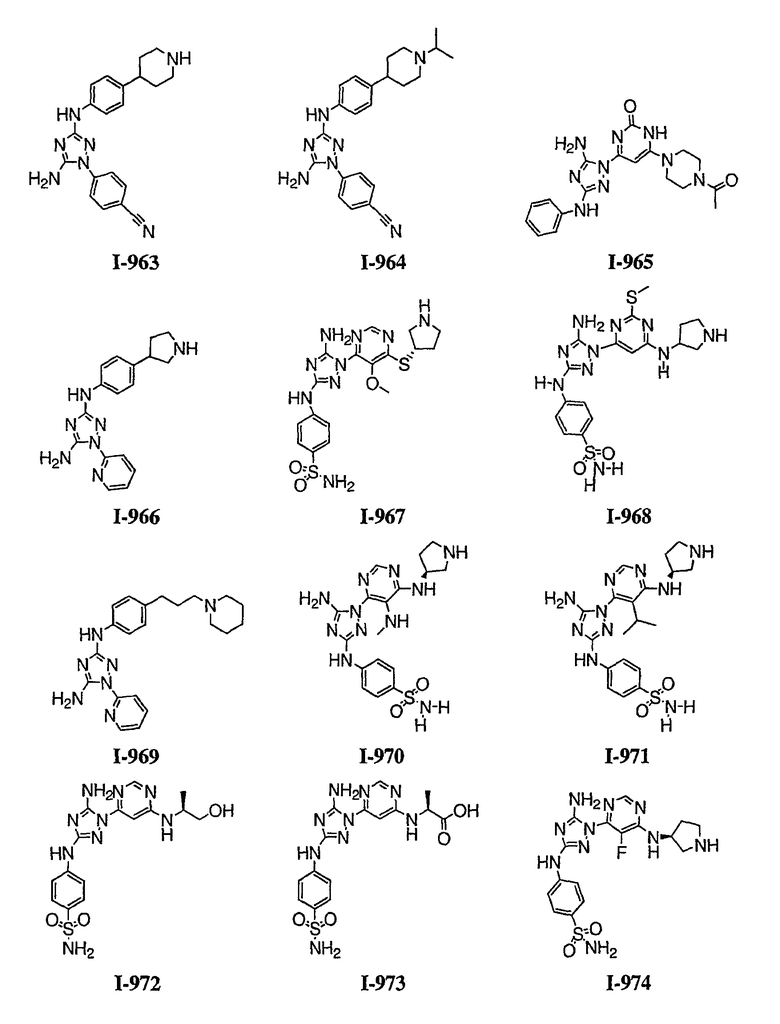
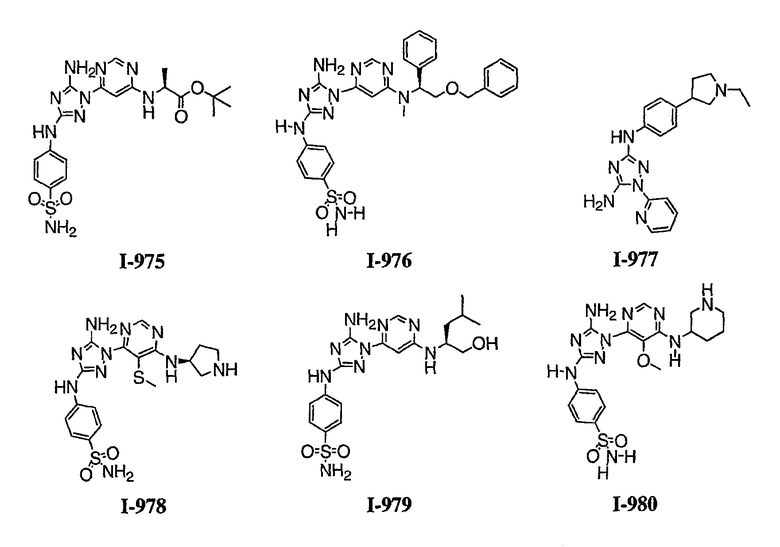
III. Общая синтетическая методология:
Соединения данного изобретения могут быть получены в общем способами, известными специалистам в данной области для аналогичных соединений, как показано на приведенной ниже общей схеме и указанных ниже примерах.
Схема 1:

(I)=изопропанол при 100 градусах Цельсия в течение 1 часа.
(II)=изопропанол при 100 градусах Цельсия на протяжении ночи.
Приведенная выше схема I показывает общий способ получения соединений формулы I. Например, соединения изобретения могут быть получены взаимодействием исходного материала (Q) с подходящим амином с образованием промежуточного соединения (А). Последующим взаимодействием (А) с подходящим гидразином получают описанные соединения формулы I.
Приведенная ниже схема 2 показывает синтез некоторых приведенных в качестве примеров соединений, у которых R3 представляет собой -(L)mAr2, эти соединения получают также общими процедурами, описанными выше.
Схема 2:
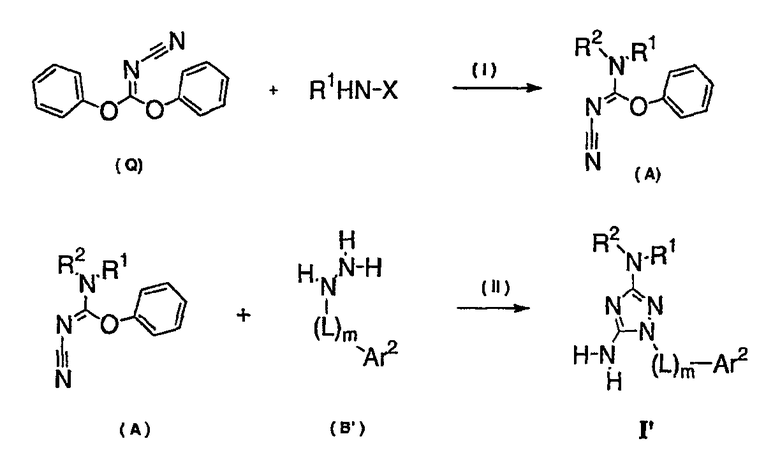
(I)=изопропанол при 100 градусах Цельсия в течение 1 часа.
(II)=изопропанол при 100 градусах Цельсия на протяжении ночи.
На схемах 3, 4 и 5 изображен синтез некоторых приведенных в качестве примеров соединений, у которых R3 и R5, взятые вместе, образуют необязательно замещенное кольцо, как здесь указано. Хотя ниже приведен синтез некоторых соединений, должно быть понятно, что другие би- и трициклические соединения, определяемые здесь в общем, могут быть также получены описываемым здесь способом.
Схема 3:
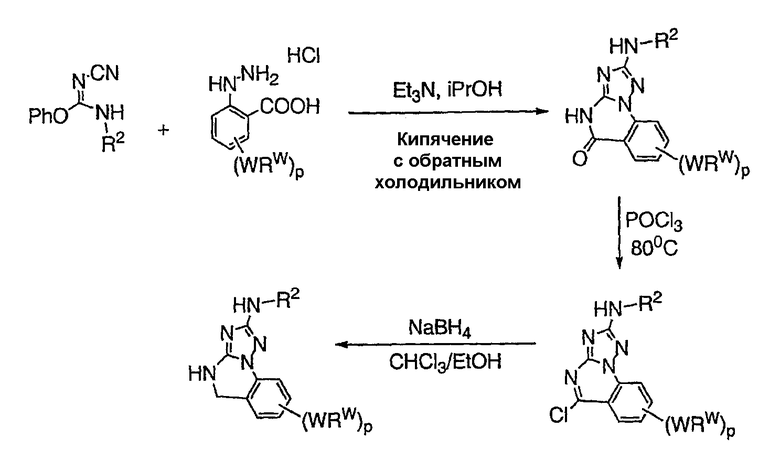
На схемах 4 и 5 изображены общие синтезы соединений, имеющих общую формулу:
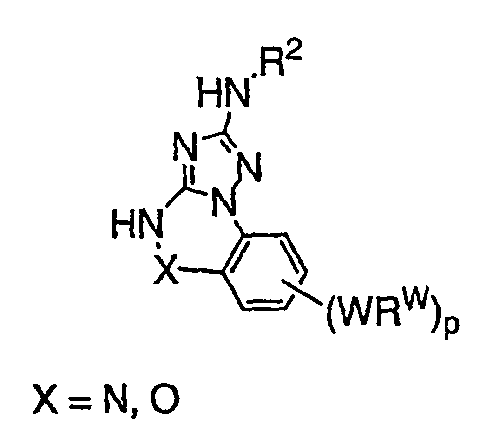
Схема 4:
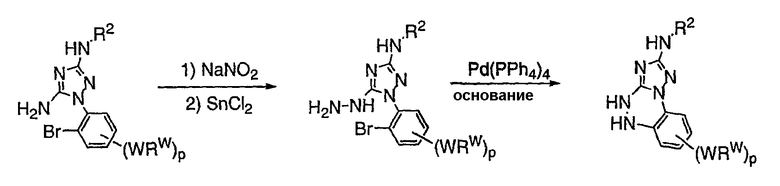
Схема 5:
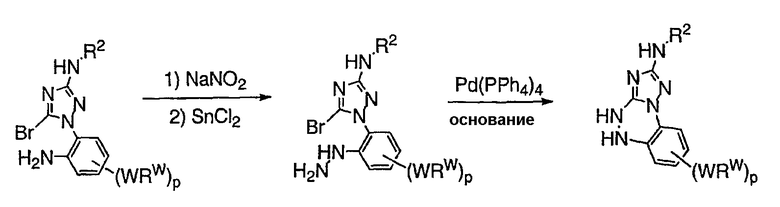
Хотя некоторые приведенные в качестве примеров варианты осуществления изображены и описаны выше и здесь, должно быть понятно, что соединения изобретения могут быть получены по способам, описанным в общем виде выше, с использованием подходящих исходных материалов.
Фармацевтически приемлемые композиции
Как описано выше, настоящее изобретение предлагает соединения, которые являются ингибиторами протеинкиназ, и поэтому настоящие соединения являются пригодными для лечения заболеваний, нарушений и состояний, включающих в себя, но не ограничивающихся перечисленным, сердечное нарушение, нейродегенеративное нарушение, психотические нарушения, аутоиммунное нарушение, состояние, связанное с трансплантацией органа, воспалительное нарушение, иммунологически опосредованное нарушение, вирусную болезнь или костное нарушение. В предпочтительных вариантах осуществления соединения являются полезными для лечения аллергии, астмы, диабета, болезни Альцгеймера, болезни Хантингтона, болезни Паркинсона, СПИД-ассоциированной деменции, бокового амиотрофического склероза (AML, болезнь Lou Gehrig), рассеянного склероза (MS), шизофрении, кардиомиоцитной гипертрофии, реперфузии/ишемии (например, удара), алопеции, рака, гепатомегалии, сердечно-сосудистого заболевания, включая кардиомегалию, муковисцидоз, вирусного заболевания, аутоиммунного заболевания, атеросклероза, рестеноза, псориаза, воспаления, гипертензии, стенокардии, цереброваскулярного сокращения, нарушения периферического кровообращения, преждевременных родов, артериосклероза, вазоспазма (церебрального вазоспазма, коронарного вазоспазма), ретинопатии, эректильной дисфункции (ED), СПИДА, остеопороза, болезни Крона и колита, выроста нейрита и болезни Рено. В предпочтительных вариантах осуществления заболеванием, состоянием или нарушением является атеросклероз, гипертензия, эректильная дисфункция (ED), реперфузия/ишемия (например, удар) или вазоспазм (церебральный вазоспазм, коронарный вазоспазм).
В соответствии с этим в другом аспекте настоящего изобретения предложены фармацевтически приемлемые композиции, которые содержат любое из описываемых здесь соединений и необязательно фармацевтически приемлемый носитель, вспомогательное вещество или наполнитель. В некоторых вариантах осуществления указанные композиции необязательно далее включают один или несколько дополнительных терапевтических агентов.
Должно быть также известно, что некоторые из соединений настоящего изобретения могут существовать для лечения в свободной форме или, если требуется, в виде их фармацевтически приемлемых производных. В соответствии с настоящим изобретением фармацевтически приемлемые производные включают в себя, но не ограничиваются перечисленным, фармацевтически приемлемые пролекарства, соли, сложные эфиры, соли таких эфиров или любой другой аддукт или производное, которое при введении пациенту, нуждающемуся в этом, способен обеспечить прямым или косвенным образом образование соединения, в противном случае описываемого здесь, или его метаболит или остаток.
Используемый здесь термин «фармацевтически приемлемая соль» относится к тем солям, которые находятся в пределах объема здравого медицинского суждения, являются подходящими для применения в контакте с тканями человека и низших животных без чрезмерной токсичности, раздражения, аллергической реакции и тому подобное и имеют соразмерное отношение приемлемая польза/риск. Термин «фармацевтически приемлемая соль» означает любую нетоксичную соль или соль сложного эфира соединения данного изобретения, которая при введении реципиенту способна обеспечивать образование либо прямым, либо косвенным образом, соединения данного изобретения или его ингибирующего активного метаболита или остатка. Используемый здесь термин «его ингибирующий активный метаболит или остаток» означает, что метаболит или его остаток также является ингибитором подсемейств протеинкиназ FLT-3, FMS, c-KIT, PDGFR, JAK, AGC (например, PKA, PDK, p70S6K-1 и -2 и РКВ), протеинкиназ CDK, GSK, SRC, ROCK и/или SYK.
Фармацевтически приемлемые соли являются хорошо известными в данной области. Например, S.M Berge et al. описали подробно фармацевтически приемлемые соли в публикации Pharmaceutical Sciences, 1977, 66, 1-19, включенной здесь в качестве ссылки. Фармацевтически приемлемые соли соединений данного изобретения включают в себя соли, полученные из подходящих неорганических и органических кислот и оснований. Примерами фармацевтически приемлемых, нетоксичных кислотно-аддитивных солей являются соли аминогруппы, образованной с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, фосфорная кислота, серная кислота и перхлорная кислота, или с органическими кислотами, такими как уксусная кислота, щавелевая кислота, малеиновая кислота, винная кислота, лимонная кислота, янтарная кислота или малоновая кислота, или с использованием других способов, известных в данной области, таких как ионообмен. Другие фармацевтически приемлемые соли включают в себя адипат, альгинат, аскорбат, аспартат, бензолсульфонат, бензоат, бисульфат, борат, бутират, камфорат, камфорасульфонат, цитрат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептаноат, глицерофосфат, глюконат, полусульфат, гептаноат, гексаноат, гидроиодид, 2-гидроксиэтансульфонат, лактобионат, лактат, лаурат, лаурилсульфат, малат, малеат, малонат, метансульфонат, 2-нафталинсульфонат, никотинат, нитрат, олеат, оксалат, пальмитат, памоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, стеарат, сукцинат, сульфат, тартрат, тиоцианат, п-тоуолсульфонат, ундеканоат, валерат и тому подобное. Соли, полученные из подходящих оснований, включают в себя соли щелочного металла, щелочноземельного металла, аммония и N+(C1-4алкил)4. Данное изобретение рассматривает также кватернизацию любой из основных азотсодержащих групп описанных здесь соединений. Растворимые или диспергируемые в воде или масле продукты могут быть получены такой кватернизацией. Репрезентативные соли щелочных или щелочноземельных металлов включают в себя соли натрия, лития, калия, кальция, магния и тому подобное. Следующие фармацевтически приемлемые соли включают в себя, если являются подходящими, нетоксичные катионы аммония, четвертичного аммония и амина, образованные с применением противоионов, таких как галогенид, гидроксид, карбоксилат, сульфат, фосфат, нитрат, низший алкилсульфонат и арилсульфонат.
Как описано выше, фармацевтически приемлемые композиции настоящего изобретения дополнительно содержат фармацевтически приемлемый носитель, вспомогательное вещество или наполнитель, который при использовании здесь включает в себя любой и все растворители, разбавители или другие жидкие наполнители, вспомогательные средства для диспергирования или суспендирования, поверхностно-активные вещества, изотонические агенты, загущающие или эмульгирующие агенты, консерванты, твердые связывающие вещества, смазывающие вещества и тому подобное, что является подходящим для требуемой конкретной лекарственной формы. В Remington's Pharmaceutical Sciences, Sixteenth Edition, E. W. Martin (Mack Publishing Co., Easton, Pa., 1980) описал различные носители, используемые при изготовлении фармацевтически приемлемых композиций, и различные способы их получении. За исключением того, что любая общепринятая среда-носитель является несовместимой с соединениями изобретения, например, вследствие продуцирования любого нежелательного биологического действия или, иначе, вредного взаимодействия с любым другим компонентом(ами) фармацевтически приемлемой композиции, предполагается, что ее применение находится в пределах объема данного изобретения. Некоторые примеры материалов, которые могут служить в качестве фармацевтически приемлемых носителей, включают в себя, но не ограничиваются перечисленным, ионообменники, оксид алюминия, стеарат алюминия, лецитин, белки сыворотки, такие как альбумин сыворотки человека, буферные вещества, такие как фосфаты, глицин, сорбиновая кислота или сорбат калия, смеси неполных глицеридных эфиров насыщенных растительных жирных кислот, вода, соли или электролиты, такие как сульфат протамина, гидрофосфат натрия, гидрофосфат калия, хлорид натрия, соли цинка, коллоидальный диоксид кремния, трисиликат магния, поливинилпирролидон, полиакрилаты, воски, блок-сополимеры полиэтилен-полиоксипропилен, ланолин, сахара, такие как лактоза, глюкоза и сахароза; крахмалы, такие как кукурузный крахмал и картофельный крахмал; целлюлоза и ее производные, такие как натрийкарбоксиметилцеллюлоза, этилцеллюлоза и ацетат целлюлозы, порошкообразный трагакант; мальт; желатин; тальк, эксципиенты, такие как какао-масло и воски суппозиториев; масла, такие как арахисовое масло, хлопковое масло; подсолнечное масло; кунжутное масло; оливковое масло; кукурузное масло и соевое масло; гликоли, такие как пропиленгликоль или полиэтиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные агенты, такие как гидроксид магния и гидроксид алюминия; альгиновая кислота; вода без пирогена; изотонический солевой раствор; раствор Рингера; этиловый спирт и фосфатные буферные растворы, а также другие нетоксичные совместимые смазывающие вещества, такие как лаурилсульфат натрия и стеарат магния, а также красящие агенты, высвобождающие агенты, агенты для получения покрытий, подслащивающие, придающие вкус агенты и отдушки, консерванты и антиокислители могут также присутствовать в композиции в соответствии с решением изготовителя.
Применение соединений и фармацевтически приемлемых композиций
Еще в одном аспекте предложен способ лечения или снижения тяжести пролиферативного нарушения, сердечного нарушения, нейродегенеративного нарушения, психотического нарушения, аутоиммунного нарушения, состояния, связанного с трансплантацией органа, воспалительного нарушения, иммунологически опосредованного нарушения, вирусной болезни и костного нарушения, содержащий введение эффективного количества соединения или фармацевтически приемлемой композиции, содержащей соединение, в котором нуждается субъект. В некоторых вариантах осуществления настоящего изобретения «эффективным количеством» соединения или фармацевтически приемлемой композиции является такое количество, которое эффективно для лечения или снижения тяжести пролиферативного нарушения, сердечного нарушения, нейродегенеративного нарушения, психотического нарушения, аутоиммунного нарушения, состояния, связанного с трансплантацией органа, воспалительного нарушения, иммунологически опосредованного нарушения, вирусной болезни и костного нарушения. Соединения и композиции по способу настоящего изобретения могут быть введены с применением любого количества и любого пути введения, эффективного для лечения или снижения тяжести пролиферативного нарушения, сердечного нарушения, нейродегенеративного нарушения, аутоиммунного нарушения, состояния, связанного с трансплантацией органа, воспалительного нарушения, иммунологически опосредованного нарушения, вирусной болезни и костного нарушения. Точное требуемое количество будет варьировать от субъекта к субъекту в зависимости от вида, возраста и общего состояния субъекта, тяжести инфекции, конкретного агента, способа его введения и тому подобного. Соединения изобретения предпочтительно изготовляют в унифицированной лекарственной форме для легкости введения и однородности дозы. Выражение «унифицированная лекарственная форма», используемое здесь, относится к физически дискретной единице агента, подходящей для подвергаемого лечению пациента. Должно быть понятно, однако, что общая суточная доза, необходимая для приема соединений или композиций настоящего изобретения, решается штатным врачом в пределах объема здравого медицинского суждения. Определенный уровень эффективной дозы для любого конкретного пациента или организма будет зависеть от различных факторов, включающих в себя подвергаемое лечению нарушение и серьезность нарушения; активности определенного применяемого соединения; определенной применяемой композиции; возраста, массы тела, общего здоровья, пола и диеты пациента; времени введения, пути введения и скорости экскреции определенного применяемого соединения; продолжительности лечения; лекарственных средств, применяемых в комбинации или совместно с определенным применяемым соединением и подобных факторов, хорошо известных в медицинской области. Термин «пациент», используемый здесь, означает животное, предпочтительно млекопитающее, наиболее предпочтительно человек.
Фармацевтически приемлемые композиции данного изобретения могут быть введены человеку и другим животным перорально, ректально, парентарально, внутриполостным путем, интравагинально, внутрибрюшинно, местно (например, в виде порошков, мазей или капель), трансбуккально, в виде орального или назального спрея или тому подобное, в зависимости от тяжести подвергаемой лечению инфекции. В некоторых вариантах осуществления соединения изобретения могут быть введены перорально или парентерально при уровнях дозирования приблизительно от 0,01 мг/кг до приблизительно 50 мг/кг и предпочтительно от 1 мг/кг до приблизительно 25 мг/кг массы тела субъекта в день один или несколько раз в день для получения требуемого терапевтического действия.
Жидкие лекарственные формы для перорального введения включают в себя, но не ограничиваются перечисленным, фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. Кроме активных соединений жидкие лекарственные формы могут содержать инертные разбавители, обычно используемые в данной области, такие как, например, вода или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (в частности, хлопковое, арахисовое, кукурузное, зародышевое, оливковое, касторовое и кунжутное масла), глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и эфиры жирных кислот и сорбитана и их смеси. Кроме инертных разбавителей пероральные композиции могут также включать в себя вспомогательные средства, такие как смачивающие агенты, эмульгирующие и суспендирующие агенты, подслащивающие вещества, вкусовые добавки и отдушки.
Инъецируемые препараты, например стерильные инъецируемые водные или масляные суспензии, могут быть изготовлены известными в данной области способами с использованием подходящих диспергирующих или смачивающих агентов и диспергирующих агентов. Стерильный инъецируемый препарат может быть также стерильным инъецируемым раствором, суспензией или эмульсией в нетоксичном парентерально приемлемом разбавителе или растворителе, например в виде раствора в 1,3-бутандиоле. В качестве приемлемых наполнителей и растворителей может быть использована вода, раствор Рингера, U.S.P. (фармакопея США) и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды обычно применяют стерильный жидкий жир. Для этой цели можно применять любое смешанное жирное масло, включая синтетические моно- и диглицериды. Кроме того, при получении инъецируемых препаратов применяют жирные кислоты, такие как олеиновая кислота.
Инъецируемые композиции могут быть стерилизованы, например, фильтрованием через удерживающий бактерии фильтр или включением стерилизующих агентов в форме стерильных твердых композиций, которые могут быть растворены или диспергированы в стерильной воде или другой стерильной инъецируемой среде перед применением.
Чтобы пролонгировать действие соединения настоящего изобретения, часто желательно замедлить абсорбцию соединения из подкожной или внутримышечной инъекции. Это можно выполнить посредством применения жидкой суспензии кристаллического или аморфного материала с плохой растворимостью в воде. Скорость абсорбции соединения тогда зависит от его скорости растворения, которая, в свою очередь, зависит от размера кристаллов и кристаллической формы. В альтернативном варианте замедленная абсорбция парентерально введенной формы соединения достигается растворением или суспендированием соединения в масляном наполнителе. Инъецируемые формы депо получают образованием микрокапсульных матриц соединения в биоразрушаемых полимерах, таких как полилактид-полигликолид. Скорость высвобождения соединения можно регулировать в зависимости от отношения соединения к полимеру и природы применяемого конкретного полимера. Примеры других биоразрушаемых полимеров включают в себя поли(ортоэфиры) и поли(ангидриды). Инъецируемые препараты типа депо также получают улавливанием соединения в липосомах или микроэмульсиях, которые являются совместимыми с тканями тела.
Композициями для ректального или вагинального введения предпочтительно являются суппозитории, которые могут быть получены смешиванием соединений данного изобретения с подходящими нераздражающими эксципиентами или носителями, такими как какао-масло, полиэтиленгликоль или воски для суппозиториев, которые являются твердыми при температуре окружающей среды, но становятся жидкими при температуре тела и, следовательно, плавятся в прямой кишке или вагинальной полости и высвобождают активное соединение.
Твердые лекарственные формы для перорального введения включают в себя капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых лекарственных формах активное соединение смешивают, по меньшей мере, с одним инертным, фармацевтически приемлемым эксципиентом или носителем, таким как цитрат натрия или фосфат кальция, и/или а) наполнителями или заполнителями, такими как крахмалы, лактоза, сахароза, глюкоза, маннит и кремниевая кислота, b) связывающими веществами, такими как, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидинон, сахароза и аравийская камедь, с) увлажнителями, такими как глицерин, d) дезинтегрирующими агентами, такими как агар-агар, карбонат кальция, картофельный крахмал или крахмал тапиока, альгиновая кислота, некоторые силикаты и карбонат натрия, е) агентами, замедляющими растворение, такими как парафин, f) ускорителями растворения, такими как соединения четвертичного аммония, g) смачивающими агентами, такими как, например, цетиловый спирт и моностеарат глицерина, h) абсорбентами, такими как каолиновая и бентонитовая глина, и i) смазывающими веществами, такими как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и их смеси. В случае капсул, таблеток и пилюль лекарственная форма может содержать также буферные агенты.
Твердые композиции подобного типа могут также применяться в качестве наполнителей в мягкие и твердые желатиновые капсулы с использованием таких эксципиентов, как лактоза или молочный сахар, а также полиэтиленгликоли с высокой молекулярной массой и тому подобное. Твердые лекарственные формы таблеток, драже, капсул, пилюль и гранул могут быть получены с покрытиями и оболочками, такими как энтеросолюбильные покрытия и другие покрытия, хорошо известные в области изготовления фармацевтических препаратов. Они могут необязательно содержать агенты, придающие непрозрачность, и могут также быть композицией, которая высвобождает активный ингредиент(ы) только или преимущественно в некоторой части кишечного тракта, необязательно, замедленным образом. Примеры заделывающих композиций, которые можно применять, включают в себя полимерные вещества и воски. Твердые композиции подобного типа могут также применяться в качестве наполнителей в мягких и твердых желатиновых капсулах с использованием таких эксципиентов, как лактоза или молочный сахар, а также полиэтиленгликоли с высокой молекулярной массой и тому подобное.
Активные соединения могут быть также в микрокапсулированной форме с одним или несколькими эксципиентами, как указано выше. Твердые лекарственные формы в виде таблеток, драже, капсул, пилюль и гранул могут быть получены с покрытиями и оболочками, такими как энтеросолюбильные покрытия, регулирующие высвобождения покрытия, и другие покрытия, хорошо известные в области изготовления фармацевтических препаратов. В таких твердых лекарственных формах активное соединение может быть смешано, по меньшей мере, с одним инертным разбавителем, таким как сахароза, лактоза или крахмал. Такие лекарственные формы могут содержать также, как это бывает в обычной практике, дополнительные вещества, другие, чем инертные разбавители, например смазывающие вещества для таблетирования и другие вспомогательные средства для таблетирования, такие как стеарат магния и микрокристаллическая целлюлоза. В случае капсул, таблеток и пилюль лекарственные формы могут также содержать буферные агенты. Они могут необязательно содержать агенты, придающие непрозрачность, и могут также быть композицией, которая высвобождает активный ингредиент(ы) только или преимущественно в некоторой части кишечного тракта, необязательно, замедленным образом. Примеры заделывающих композиций, которые можно применять, включают в себя полимерные вещества и воски.
Лекарственные формы для местного или чрескожного введения соединения данного изобретения включают в себя мази, пасты, кремы, лосьоны, гели, порошки, растворы, спреи, формы для ингаляции или пластыри. Активный компонент смешивают в стерильных условиях с фармацевтически приемлемым носителем и, когда требуется, с любыми необходимыми консервантами или буферами. Офтальмический препарат, глазные капли, ушные капли также рассматриваются как находящиеся в пределах объема данного изобретения. Кроме того, настоящее изобретение рассматривает применение чрескожных пластырей, которые имеют дополнительное преимущество в обеспечении регулируемой доставки соединения в организм. Такие лекарственные формы могут быть изготовлены растворением или распределением соединения в подходящей среде. Можно также использовать усилители абсорбции для повышения потока соединения, протекающего через кожу. Скорость можно регулировать либо обеспечением регулирующей скорости мембраны, либо распределением соединения в полимерной матрице или геле.
Как описано в общем выше, соединения изобретения могут быть исследованы в качестве ингибиторов протеинкиназы. В одном варианте осуществления соединения и композиции изобретения являются ингибиторами одного или нескольких из подсемейств протеинкиназ FLT-3, FMS, c-KIT, PDGFR, JAK, AGC (например, PKA, PDK, p70S6K-1 и -2 и РКВ), CDK, GSK, SRC, ROCK и/или SYK, и, таким образом, без связи с какой-либо конкретной теорией соединения, и композиции являются особенно применимыми для лечения или снижения тяжести заболевания, состояния или нарушения, когда при заболевании, состоянии или нарушении принимает участие активация одного или нескольких подсемейств протеинкиназ из FLT-3, FMS, c-KIT, PDGFR, JAK, AGC (например, PKA, PDK, p70S6K-1 и -2 и РКВ), протеинкиназ CDK, GSK, SRC, ROCK и/или SYK. Когда активация подсемейством протеинкиназ FLT-3, FMS, c-KIT, PDGFR, JAK, AGC (например, PKA, PDK, p70S6K-1 и -2 и РКВ), CDK, GSK, SRC, ROCK и/или SYK имеет место при определенном заболевании, состоянии или нарушении, такое заболевание, состояние или нарушение может называться «опосредованным подсемейством протеинкиназ FLT-3, FMS, c-KIT, PDGFR, JAK, AGC (например, PKA, PDK, p70S6K-1 и -2 и РКВ), CDK, GSK, SRC, ROCK и/или SYK заболеванием» или симптомом заболевания. В соответствии с этим в другом аспекте настоящего изобретения предложен способ лечения или снижения тяжести заболевания, состояния или нарушения, где в данном патологическом состоянии имеет место активация подсемейством протеинкиназ FLT-3, FMS, c-KIT, PDGFR, JAK, AGC (например, PKA, PDK, p70S6K-1 и -2 и РКВ), CDK, GSK, SRC, ROCK и/или SYK. Активность соединения, применяемого в данном изобретении в качестве ингибитора подсемейства протеинкиназ FLT-3, FMS, c-KIT, PDGFR, JAK, AGC (например, PKA, PDK, p70S6K-1 и -2 и РКВ), CDK, GSK, SRC, ROCK и/или SYK, можно анализировать in vitro, in vivo или в клеточной линии. Анализы in vitro включают в себя анализы, которые определяют либо активность фосфорилирования, либо активность АТФазы, активированного подсемейством протеинкиназ FLT-3, FMS, c-KIT, PDGFR, JAK, AGC (например, PKA, PDK, p70S6K-1 и -2 и РКВ), CDK, GSK, SRC, ROCK и/или SYK. Другие анализы in vitro количественно определяют способность ингибитора связываться с подсемейством протеинкиназ FLT-3, FMS, c-KIT, PDGFR, JAK, AGC (например, PKA, PDK, p70S6K-1 и -2 и РКВ), CDK, GSK, SRC, ROCK и/или SYK. Связывание ингибитора можно измерить мечением радиоактивным изотопом ингибитора перед связыванием, выделением комплекса ингибитор подсемейства протеинкиназ/FLT-3, FMS, c-KIT, PDGFR, JAK, AGC (например, PKA, PDK, p70S6K-1 и -2 и РКВ), CDK, GSK, SRC, ROCK и/или SYK и определением количества связанной радиоактивной метки. В альтернативном случае связывание ингибитора может быть определено проведением эксперимента конкуренции, в котором новые ингибиторы инкубируют с подсемейством протеинкиназ FLT-3, FMS, c-KIT, PDGFR, JAK, AGC (например, PKA, PDK, p70S6K-1 и -2 и РКВ), CDK, GSK, SRC, ROCK и/или SYK, связанной с известными радиоактивными метками.
Термин «ингибирует измеряемым образом», используемый здесь, означает поддающееся измерению изменение в активности подсемейства протеинкиназ FLT-3, FMS, c-KIT, PDGFR, JAK, AGC (например, PKA, PDK, p70S6K-1 и -2 и РКВ), CDK, GSK, SRC, ROCK и/или SYK между образцом, содержащим указанную композицию и киназу FLT-3, FMS, c-KIT, PDGFR, JAK, AGC (например, PKA, PDK, p70S6K-1 и -2 и РКВ), CDK, GSK, SRC, ROCK и/или SYK, и эквивалентным образцом, содержащим киназу FLT-3, FMS, c-KIT, PDGFR, JAK, подсемейство протеинкиназ AGC (например, PKA, PDK, p70S6K-1 и -2 и РКВ), CDK, GSK, SRC, ROCK и/или SYK в отсутствие указанной композиции.
Термин «FLT-3-опосредованное заболевание», применяемый здесь, означает любое заболевание или ухудшенное состояние, в котором, как известно, киназа семейства FLT-3 играет роль. Такие состояния включают, без ограничения, гемопоэтитические нарушения, в частности острый миелогенный лейкоз (AML), острый промиелоцитный лейкоз (APL) и острый лимфоцитный лейкоз (ALL).
В соответствии с другим вариантом осуществления изобретение предлагает лечение или снижение тяжести FMS-опосредованного заболевания или состояния у пациента, содержащий стадию введения указанному пациенту композиции по настоящему изобретению.
Термин «FMS-опосредованное заболевание», применяемый здесь, означает любое заболевание или ухудшенное состояние, в котором, как известно, киназа семейства FLT-3 играет роль. Такие состояния включают, без ограничения, рак (включающий в себя, но не ограничивающийся перечисленным, рак яичников, эндометрия, молочной железы), воспалительные нарушения и гипертензию.
В соответствии с другим вариантом осуществления изобретение предлагает лечение или снижение тяжести с-KIT-опосредованного заболевания или состояния у пациента, содержащее стадию введения указанному пациенту композиции по настоящему изобретению.
Термин «с-KIT-опосредованное заболевание», применяемый здесь, означает любое заболевание или ухудшенное состояние, в котором, как известно, киназа семейства c-KIT играет роль. Такие состояния включают, без ограничения, AML, хронический миелогенный лейкоз (CML), мастоцитоз, анапластическую крупноклеточную лимфому, ALL, опухоль желудочно-кишечной стромы (GIST), Т-клеточную лимфому, аденокистозную карциному, ангиосаркому, эндометриальную карциному, мелкоклеточный рак легких, рак простаты, рак яичников, карциному молочной железы, карциному щитовидной железы, злокачественную меланому и карциному толстой кишки.
В соответствии с другим вариантом осуществления изобретение предлагает способ лечения или снижения тяжести CDK-2-опосредованного заболевания или состояния у пациента, содержащий стадию введения указанному пациенту композиции по настоящему изобретению.
Термин «CDK-2-опосредованное заболевание», применяемый здесь, означает любое заболевание или ухудшенное состояние, в котором, как известно, киназа семейства CDK-2 играет роль. В соответствии с этим эти соединения являются применимыми для лечения заболеваний или состояний, на которые, как известно, влияет активность CDK-2-киназы. Такие заболевания и состояния включают в себя рак, болезнь Альцгеймера, рестеноз, ангиогенез, гломерулонефрит, цитомегаловирусное заболевание, ВИЧ-заболевание, герпес, псориаз, атеросклероз, алопецию и аутоиммунные заболевания, такие как ревматоидный артрит, вирусные инфекции, нейродегенеративные заболевания, заболевания, связанные с тимоцитным апоптозом, или пролиферативные нарушения, являющиеся результатом дерегуляции клеточного цикла, особенно прогрессирования от фазы G1 до фазы S.
В соответствии с другим вариантом осуществления изобретение предлагает способ лечения или снижения тяжести GSK-3-опосредованного заболевания или состояния у пациента, содержащий стадию введения указанному пациенту композиции по настоящему изобретению.
В соответствии с другим вариантом осуществления изобретение предлагает способ лечения или снижения тяжести Src-опосредованного заболевания или состояния у пациента, содержащий стадию введения указанному пациенту композиции по настоящему изобретению.
Термин «Src-опосредованное заболевание», применяемый здесь, означает любое заболевание или ухудшенное состояние, в котором, как известно, играет роль Src. Такие состояния включают, без ограничения, раковые заболевания, такие как рак толстой кишки, молочной железы, печени и поджелудочной железы, аутоиммунные заболевания, такие как отторжение трансплантата, аллергии, ревматоидный артрит, лейкоз, заболевания, заключающиеся в реконструкции костей, такие как остеопороз, и вирусные заболевания, такие как инфекция гепатита В.
В соответствии с другим вариантом осуществления изобретение предлагает способ лечения или снижения тяжести Syk-опосредованного заболевания или состояния у пациента, содержащий стадию введения указанному пациенту композиции по настоящему изобретению.
Термин «Syk-опосредованное заболевание» или «Syk-опосредованное состояние», применяемый здесь, означает любое заболевание или другое ухудшенное состояние, в котором, как известно, играет роль Syk. Такие состояния включают, без ограничения, аллергические нарушения, особенно астму.
Термин «JAK-опосредованное заболевание», применяемый здесь, означает любое заболевание или другое ухудшенное состояние, в котором, как известно, играет роль JAK. Такие состояния включают, без ограничения, иммунные реакции, такие как аллергические реакции или реакции повышенной чувствительности типа I, астма, аутоиммунные заболевания, такие как отторжение трансплантата, гомологичная болезнь, ревматоидный артрит, боковой латеральный склероз и рассеянный склероз, нейродегенеративные нарушения, такие как семейный боковой латеральный склероз (FALS), а также солидные и гематологические злокачественности, такие как лейкозы и лимфомы.
Термин «PDK1-опосредованное состояние» или «заболевание», применяемый здесь, означает любое заболевание или другое ухудшенное состояние, в котором, как известно, играет роль PDK1. Термин «PDK1-опосредованное состояние» или «заболевание» означает те заболевания или состояния, которые облегчают лечением ингибитором PDK1. PDK1-опосредованные заболевания или состояния включают в себя, но не ограничиваются перечисленным, пролиферативные нарушения и рак. Указанный рак предпочтительно выбран из панкреатического рака, рака простаты или яичников.
Термин «РКА-опосредованное состояние» или «заболевание», применяемый здесь, означает любое заболевание или другое ухудшенное состояние, в котором, как известно, играет роль PKA. Термин «PKA-опосредованное состояние» или «заболевание» означает те заболевания или состояния, которые облегчают лечением ингибитором PKA. PKA-опосредованные заболевания или состояния включают в себя, но не ограничиваются перечисленным, пролиферативные нарушения и рак.
Термин «p70S6K-опосредованное состояние» или «заболевание», применяемый здесь, означает любое заболевание или другое ухудшенное состояние, в котором, как известно, играет роль p70S6K. Термин «p70S6K-опосредованное состояние» или «заболевание» означает те заболевания или состояния, которые облегчают лечением ингибитором p70S6K. p70S6K-опосредованные заболевания или состояния включают в себя, но не ограничиваются перечисленным, пролиферативные нарушения, такие как рак и туберозный склероз.
Термин «GSK-3-опосредованное заболевание», применяемый здесь, означает любое заболевание или другое ухудшенное состояние, в котором, как известно, играет роль GSK-3. Такие заболевания или состояния включают в себя, без ограничения, аутоиммунные заболевания, воспалительные заболевания, метаболические, неврологические и нейродегенеративные заболевания (например, болезнь Альцгеймера болезнь Хантингтона, болезнь Паркинсона и нарушения движения базального ядра, хорею, дистонию, болезнь Вилсона, болезнь Пика, дегенерация фронтальной части (головного мозга), прогрессивный супрануклеарный паралич (PSP), болезнь Крейтфелда-Якоба, таупатологию и кортикобазальную дегенерацию (CBD)), психотические нарушения (например, шизофрению, СПИД-ассоциированную деменцию, депрессию, биполярное нарушение и тревожные состояния), сердечно-сосудистые заболевания, аллергию, астму, диабет, боковой амиотрофический склероз (AML, болезнь Lou Gehrig), рассеянный склероз (MS), кардиомиоцитную гипертрофию, реперфузию/ишемию, удар и алопецию.
Термин «ROCK-опосредованное состояние» или «заболевание», применяемый здесь, означает любое заболевание или другое ухудшенное состояние, в котором, как известно, играет роль ROCK. Термин «ROCK-опосредованное состояние» или «заболевание» означает те заболевания или состояния, которые облегчают лечением ингибитором ROCK. ROCK-опосредованные заболевания или состояния включают в себя, но не ограничиваются перечисленным, гипертензию, стенокардию, цереброваскулярное сокращение, астму, нарушение периферического кровообращения, преждевременные роды, рак, эректильную дисфункцию, атеросклероз, спазм (церебральный вазоспазм и коронарный вазоспазм), ретинопатию (например, глаукому), воспалительные нарушения, иммунные нарушения, СПИД, остеопороз, гипертрофию миокарда, повреждение, индуцированное ишемией/реперфузией, и эндотелиальную дисфункцию.
В других вариантах осуществления изобретение относится к способу усиления синтеза гликогена и/или понижения уровней глюкозы в крови у пациента, нуждающегося в этом, содержащему введение указанному пациенту терапевтически эффективного количества композиции, содержащей соединение формулы I. Этот способ особенно применим для диабетических пациентов.
Еще в одном варианте осуществления изобретение относится к способу ингибирования продуцирования гиперфосфорилированного Tau-белка у пациента, нуждающегося в этом, содержащему введение указанному пациенту терапевтически эффективного количества композиции, содержащей соединение формулы I. Этот способ особенно применим при остановке развития или замедлении прогресса болезни Альцгеймера.
Еще в одном варианте осуществления изобретение относится к способу ингибирования фосфорилирования β-катенина у пациента, нуждающегося в этом, содержащему введение указанному пациенту терапевтически эффективного количества композиции, содержащей соединение формулы I. Этот способ особенно применим для лечения шизофрении.
Должно быть понятно, что соединения и фармацевтически приемлемые композиции настоящего изобретения можно применять в комбинационных терапиях, то есть соединения и фармацевтически приемлемые композиции могут быть введены одновременно с одной или несколькими другими требуемыми терапиями или медицинскими процедурами, до или после таких терапий или процедур. В конкретной комбинаций лечений (терапий или процедур), которую применяют в комбинационной схеме таких лечений следует принимать во внимание совместимость требуемых терапий и/или процедур и требуемого терапевтического эффекта, который должен быть достигнут. Должно быть понятно, что применяемые лечения могут достичь требуемого действия для одного и того же нарушения (например, соединение изобретения может быть введено с другим агентом, применяемым для лечения такого же нарушения) или они могут привести к различным действиям (например, подавление любых отрицательных действий). Используемые здесь дополнительные терапевтические агенты, которые обычно вводят для лечения или профилактики конкретного заболевания или состояния, являются известными как «подходящие для подвергаемого лечению заболевания или состояния».
Например, химиотерапевтические агенты или другие антипролиферативные агенты могут быть комбинированы с соединениями данного изобретения для лечения пролиферативных заболеваний и рака. Примеры известных химиотерапевтических агентов включают в себя, но не ограничиваются перечисленным, например, другие лечения или противораковые агенты, которые могут применяться в комбинации с противораковыми патентоспособными агентами настоящего изобретения, может применяться также хирургия, радиотерапия (несколькими примерами ее являются гамма-излучение, нейтронная лучевая радиотерапия, электронная лучевая радиотерапия, протонная терапия, брахитерапия и системное лечение радиоактивными изотопами, не говоря уже о других терапиях), эндокринная терапия, лечение модификаторами биологических реакций (интерферонами, интерлейкинами и фактором некроза тканей (TNF), не говоря уже о других), гипертермия и криотерапия, лечение агентами для ослабления отрицательных действий (например, противорвотными средствами) и другими разрешенными химиотерапевтическими лекарственными средствами, включающими в себя, но не ограничивающимися перечисленным, алкилирующие лекарственные средства (меторетамин, хлорамбуцил, циклофосфамид, мелфалан, ифосфамид), антиметаболиты(метотрексат), пуриновые антагонисты и пиримидиновые антагонисты (6-меркаптопурин, 5-фторурацил, цитарабил, гемицитабин), веретенообразные яды (винбластин, винкристин, винорелбин, паклитаксел), подофиллотоксины (этопозид, иринотекан, топотекан), антибиотики (доксорубицин, блеомицин, митомицин), нитрозомочевины (кармустин, ломустин), неорганические ионы (цисплатин, карбоплатин), ферменты (аспарагиназа) и гормоны (тамоксифен, лейпролид, флутамид и мегестрол), гливекТМ, адриамицин, дексаметазон и циклофосфамид. Для более подробного обсуждения уточненных терапий рака см. http://www.nci.nih.gov/, список разрешенных FDA лекарственно средств для онкологии в . и The Merck Manual, Seventeenth ed. 1999, полное содержание которых включено таким образом в качестве ссылки.
Другие примеры агентов, которые могут также быть комбинированы с ингибиторами данного изобретения, включают в себя, без ограничения, агенты для лечения болезни Альцгеймера, такие как арицепт® и эксцелон®; агенты для лечения болезни Паркинсона, такие как L-DOPA/карбидопа, энтакапон, ропинрол, прамипексол, бромокриптин, перголид, тригексефендил и амантадин; агенты для лечения рассеянного склероза (MS), такие как бета-интерферон (например, авонекс® и ребиф®), копаксон® и митоксантрон; агенты для лечения астмы, такие как албутерол и сингулаир®; агенты для лечения шизофрении, такие как зупрекса, риспердал, сероквел и галоперидол; противовоспалительные агенты, такие как кортикостероиды, блокаторы TNF, IL-1 RA, азатиоприн, циклофосфамид и сульфазалазин; иммуномодуляторные и иммунодепрессивные агенты, такие как циклоспорин, такролимус, рапамицин, микрофенолят мофетил, интерфероны, кортикостероиды, циклофосфамид, азатиоприн и сульфазалазин; нейротрофные факторы, такие как ингибиторы ацетилхолинэстеразы, ингибиторы МАО, интерфероны, противосудорожные средства, блокаторы ионных каналов, рилузол и агенты против паркинсонизма; агенты для лечения сердечно-сосудистого заболевания, такие как бета-блокаторы, ингибиторы АСЕ, диуретики, нитраты, блокаторы кальциевых каналов и статины; агенты для лечения заболевания печени, такие как кортикостероиды, холестериламин, интерфероны и противовирусные агенты; агенты для лечения заболеваний крови, такие как кортикостероиды, антилейкозные агенты и факторы роста, и агенты для лечения нарушений типа иммунодефицита, такие как гамма-глобулин.
Количество дополнительного терапевтического агента, присутствующего в композициях данного изобретения, должно быть не больше, чем количество, которое обычно вводят в композиции, содержащей такой терапевтический агент в качестве единственного активного агента. Количество дополнительного терапевтического агента в настоящее время в предложенных композициях будет находиться в диапазоне приблизительно от 50% до 100% количества, обычно присутствующего в композиции, содержащей этот агент в качестве единственного терапевтически активного агента.
Соединения данного изобретения или их фармацевтически приемлемые композиции могут быть также включены в композиции для покрытия имплантируемых медицинских устройств, таких как протезы, искусственные клапаны, васкулярные трансплантаты, стенты и катетеры. В соответствии с этим настоящее изобретение в другом аспекте включает в себя композицию для покрытия имплантируемого устройства, содержащую соединение настоящего изобретения, как описано в общем выше и описано в классах и подклассах здесь, и носитель, подходящий для покрытия имплантируемого устройства. Еще в одном аспекте настоящее изобретение включает в себя имплантируемое устройство, покрытое композицией, содержащей соединение настоящего изобретения, как описано в общем выше и здесь в классах и подклассах, и носитель, подходящий для покрытия имплантируемого устройства.
Васкулярные стенты, например, использовали для преодоления рестеноза (повторного сужения стенок сосудов после повреждения). Однако пациенты, использующие стенты или другие имплантируемые устройства, испытывают риск образования тромба и активации тромбоцитов. Эти нежелательные действия могут быть предотвращены или ослаблены предварительным покрытием устройства фармацевтически приемлемой композицией, содержащей ингибитор киназы. Подходящие покрытия и общее получение покрытых имплантируемых устройств описаны в патентах США 6099562, 5886026 и 5304121. Покрытия обычно являются биосовместимыми полимерными материалами, такими как полимер гидрогель, полиметилдисилоксан, поликапролактон, полиэтиленгликоль, полимолочная кислота, этиленвинилацетат и их смеси. Покрытия могут быть необязательно дополнительно покрыты подходящим верхним слоем фторсиликона, полисахаридов, полиэтиленгликоля, фосфолипидов или их комбинаций для придания композиции характеристик регулируемого высвобождения.
Другой аспект изобретения относится к способу ингибирования активности подсемейства протеинкиназ FLT-3, FMS, c-KIT, PDGFR, JAK, AGC (например, PKA, PDK, p70S6K-1 и -2 и РКВ), CDK, GSK, SRC, ROCK и/или SYK в биологическом образце или в организме пациента, который содержит введение пациенту или контактирование указанного биологического образца с соединением формулы I или композицией, содержащей указанное соединение. Термин «биологический образец», используемый здесь, включает в себя, без ограничения, культуры клеток или их экстракты, материал биопсии, полученный из организма млекопитающего, или его экстракты; кровь, слюна, моча, фекалии, сперму, слезы или другие жидкости тела или их экстракты.
Ингибирование активности подсемейства протеинкиназ FLT-3, FMS, c-KIT, PDGFR, JAK, AGC (например, PKA, PDK, p70S6K-1 и -2 и РКВ), CDK, GSK, SRC, ROCK и/или SYK в биологическом образце является применимым для различных целей, которые известны специалисту в данной области. Примеры таких целей включают в себя, но не ограничиваются перечисленным, трансфузию крови, трансплантацию органа, хранение биологических образцов и биологические анализы.
ПРИМЕРЫ
А) Синтез соединений изобретения
Соединения общей формулы I получали по общей процедуре следующим образом (по схемам 1 и 2).
Исходный материал (Q) растворяли в 2-пропаноле до достижения 1 М раствора и раствор нагревали до 100 градусов Цельсия. К горячей смеси затем добавляли амин и смесь перемешивали в течение 1 часа в запаянной трубке. ВЭЖХ показывала, что реакция была завершенной и реакционную смесь концентрировали досуха. Образец затем очищали на системе нормальной фазы комбифлэш. Системой растворителей была смесь дихлорметан:метанол. Элюирование начинали с 0% метанола смеси и повышали содержание в ней метанола максимум до 10% в зависимости от свойств соединений.
Исходный материал (А) растворяли в 2-пропаноле до достижения 1 М раствора. К этому раствору затем добавляли 1 эквивалент (В). Реакцию проводили в запаянной трубке при 100 градусах Цельсия в течение ночи. В случае когда гидразином была его соль HCl, добавляли 1 эквивалент триэтиламина.
Реакцию проводили следующим образом. Реакционную смесь концентрировали досуха, проводили ЖХ-МС для определения полноты реакции. Остаток продукта растворяли в метаноле и промывали через предварительно кондиционированную колонку SCX. Продукт затем элюировали раствором метанол/аммиак. Продукт элюирования концентрировали досуха и затем очищали препаративной хроматографией с обращенной фазой Гилсона.
Следующие примеры описывают в качестве примеров синтез различных исходных материалов и соединений изобретения. После каждой серии процедур приводится список некоторых приведенных в качестве примеров соединений, полученных способами изобретения.
Пример 1
4-(3-Иодфенил)морфолин. К раствору 1,3-дииодбензола (5,05 г, 15,3 ммоль) в изопропаноле (16 мл) в атмосфере азота добавляют морфолин (1,33 г, 1,33 мл, 15,3 ммоль), фосфат калия (6,50 г, 30,6 моль), этиленгликоль (1,90 г, 1,70 мл, 30,6 ммоль) и иодид меди (I) (146 мг, 0,765 ммоль). Смесь нагревают при 80°С в течение 15 час и затем охлаждают до комнатной температуры. Твердые вещества удаляют фильтрованием и раствор концентрируют. Остаток очищают колоночной хроматографией на силикагеле, элюируя смесью EtOAc:гексаны (от 5 до 25% EtOAc), получая при этом 4-(3-иодфенил)морфолин (1,81 г, 41%) в виде бесцветного масла. МС (ES+): m/z=290,0; 1Н ЯМР (CDCl3, 500 МГц): δ 3,15 (т, 4Н), 3,85 (т, 4Н), 6,87 (дд, 1Н), 6,96-7,00 (м, 1Н), 7,20 (д, 1Н), 7,22-7,25 (м, 1Н).
Пример 2
Трет-Бутиловый эфир N-(3-морфолин-4-илфенил)гидразинкарбоновой кислоты. К раствору 4-(3-иодфенил)морфолина (1,81 г, 6,26 ммоль) в ДМФ (6,5 мл) в атмосфере азота добавляют трет-бутилкарбазат (993 мг, 7,52 ммоль), иодид меди (I) (59,5 мг, 0,313 ммоль), 1,10-фенантролин (113 мг, 0,626 ммоль) и карбонат цезия (2,85 г, 8,77 ммоль). Смесь нагревают при 80°С в течение 18 час и затем охлаждают до комнатной температуры. Добавляют воду (100 мл) и смесь экстрагируют этилацетатом. Органический слой сушат над MgSO4, концентрируют. Остаток очищают колоночной хроматографией на силикагеле с элюированием смесью EtOAc:гексаны (от 25 до 50% EtOAc), получая при этом трет-бутиловый эфир N-(3-морфолин-4-илфенил)гидразинкарбоновой кислоты (1,14 г, 62%) в виде желтого масла. МС (ES+): m/z=294,2; 1Н ЯМР (ДМСО-d6, 500 МГц): δ 1,44 (с, 9Н), 3,06 (т, 4Н), 3,73 (т, 4Н), 4,97 (с, 2Н), 6,66 (дд, 1Н), 6,91 (д, 1Н), 7,01-7,05 (м, 1Н), 7,09-7,14 (м, 1Н).
Пример 3
Соль с HCl (3-морфолин-4-илфенил)гидразина. К раствору трет-бутилового эфира N-(3-морфолин-4-илфенил)гидразинкарбоновой кислоты (468 мг, 1,60 ммоль) в метаноле (20 мл) добавляют раствор 4 н. HCl в диоксане (10 мл). Смесь перемешивают при комнатной температуре на протяжении ночи, затем концентрируют, получая при этом соль с 3 HCl (3-морфолин-4-илфенил)гидразина (484 мг, 100%) в виде желтого твердого вещества.
МС (ES+): m/z=194,1; 1Н ЯМР (CD3OD, 500 МГц): δ 3,60-3,71 (м, 4Н), 4,06-4,15 (м, 4Н), 7,06 (д, 1Н), 7,26-7,35 (м, 2Н), 7,50-7,58 (м, 1Н).
Пример 4
(6-Хлорпиридин-3-ил)морфолин-4-илметанон. К 6-хлорникотиноилхлориду (0,540 г, 3,07 ммоль) в дихлорметане (10 мл) добавляют морфолин (0,294 г, 1,1 эквивалента) с последующим добавлением триэтиламина (940 мкл, 2,2 эквивалента). Реакционную смесь перемешивают на протяжении ночи при комнатной температуре, получая при этом (6-хлорпиридин-3-ил)морфолин-4-илметанон. Масс-спектрометрия не показала присутствия исходного материала, 6-хлорникотиноилхлорида и показала точный ион М+. Проводят водную обработку продукта и сырой материал переносят для следующей стадии. Сырой остаток, (6-хлорпиридин-3-ил)морфолин-4-илметанон, имеет массу 0,610 г (выход 88%) после водной обработки.
Аналогично получают следующие соединения:
Пример 5
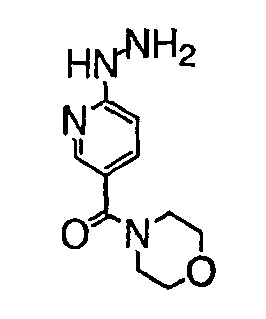
(6-Гидразинопиридин-3-ил)морфолин-4-илметанон. К (6-хлорпиридин-3-ил)морфолин-4-илметанону (0,649 г, 2,86 ммоль) в этаноле (6 мл) добавляют 0,270 мкл (3,0 эквивалента) гидразина с последующим добавлением 438,9 мкл триэтиламина (1,1 эквивалента). Реакционную смесь перемешивают на протяжении ночи при 100°С, получая при этом (6-гидразинопиридин-3-ил)морфолин-4-илметанон. Реакционную смесь фильтруют и затем концентрируют досуха. Сырой остаток, (6-гидразинопиридин-3-ил)морфолин-4-илметанон, имеет массу 0,372 г (выход 76%).
Аналогично получают следующие соединения:
Пример 6
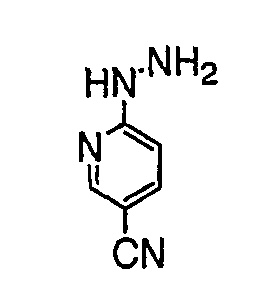
6-Гидразиноникотинонитрил (CF#H-1). Смесь 6-хлорникотинонитрила (2,77 г, 20 ммоль) и гидрата гидразина (15 мл) перемешивают при 100°С в течение 3 час и упаривают. Остаток суспендируют в эфире и фильтруют, затем суспендируют в растворе бикарбоната натрия и фильтруют, промывая водой, и сушат, получая при этом 6-гидразиноникотинонитрил (1,25 г, выход 46%) в виде рыжевато-коричневого твердого вещества.
1Н ЯМР (ДМСО-d6, 500 МГц): 8,59 (с, 1Н), 8,35 (с, 1Н), 7,74 (д, 1Н), 6,75 (с, 1Н), 4,44 (с, 2Н) м.д.; MS (FIA) 135,1 (M+H).
Аналогично получают следующие соединения
Пример 7
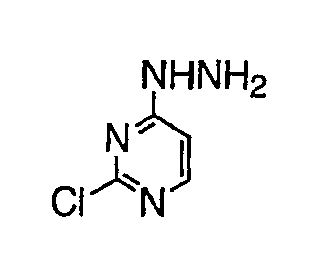
(2-Хлорпиримидин-4-ил)гидразин. К раствору 2,4-дихлорпиримидина (1,49 г, 10,0 ммоль) в этаноле (25 мл) добавляют триэтиламин (2,02 г, 2,78 мл, 20,0 моль) и гидразин (321 мг 0,321 мл, 10,0 ммоль). Смесь перемешивают при комнатной температуре в течение 2 час. Добавляют воду и смесь экстрагируют дихлорметаном. Органический слой сушат над MgSO4, концентрируют. Остаток очищают колоночной хроматографией на силикагеле при элюировании смесью метанол:дихлорметан (от 2 до 5% метанола), получая при этом (2-хлорпиримидин-4-ил)гидразин (330 мг, 23%) в виде белого твердого вещества. МС (ES+): m/z=144,9.
Пример 8
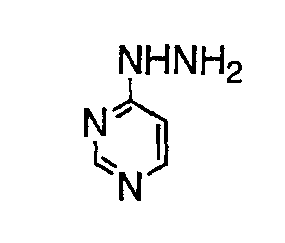
Пиримидин-4-илгидразин. К раствору (2-хлорпиримидин-4-ил)гидразина в метаноле добавляют формиат аммония и Pd/C (10%). Смесь нагревают при 55°С в течение 15 час. Смесь охлаждают до комнатной температуры и фильтруют и фильтрат концентрируют. Добавляют воду и смесь экстрагируют дихлорметаном. Органический слой сушат над MgSO4 и концентрируют, получая при этом пиримидин-4-илгидразин (62,0 мг, 25%) в виде желтого твердого вещества.
МС (ES+): m/z=111,3; 1Н ЯМР (CD3OD, 500 МГц): (с, шир., 1Н), 8,06 (д, 1Н), 8,39 (с, 1Н)..
Схема 6
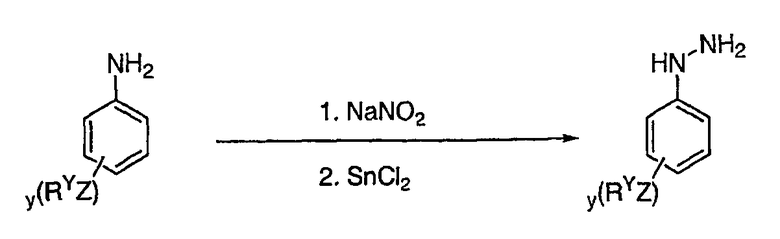
Пример 9
(3-Феноксифенил)гидразин. К раствору 3-феноксифениланилина (2,32 г, 12,5 ммоль) в метаноле (5 мл), воде (10 мл) и концентрированной HCl (3 мл) при 0°С добавляют в виде быстрых капель раствор нитрита натрия (0,87 г, 12,7 ммоль) в воде (2 мл). Реакционную смесь перемешивают 10 мин, затем обрабатывают добавлением в виде быстрых капель находящегося при 0°С раствора дигидрата хлорида олова (6,77 г, 30 ммоль) в концентрированной HCl (25 мл). Реакционную смесь перемешивают в течение 1 часа затем устанавливают рН до ˜7 при помощи 6 н. NaOH и бикарбоната натрия, затем фильтруют через целит, промывают смесью 1:3 метанол:дихлорметан. Фильтрат отделяют, водную фазу экстрагируют смесью 1:3 метанол:дихлорметан (2х). Объединенный органический слой сушат над сульфатом натрия, упаривают, затем очищают флэш-хроматографией (SiO2) с элюированием смесью 35:65 этилацетат:гексаны, получая при этом (3-феноксифенил)гидразин (1,78 г, выход 71%) в виде оранжевого масла. 1Н-ЯМР (CDCl3, 500 МГц) 7,30 (м, 2Н), 7,14 (т, 1Н), 7,08 (т, 1Н), 7,00 (м, 2Н), 6,52 (м, 1Н), 6,47 (м, 1Н), 6,45 (м, 1Н), 5,2 (шир. 1Н), 3,5 (шир. 2Н) м.д.; МС (FIA) 201,1 (М+Н); ВЭЖХ (способ А) 2,887 мин.
Аналогично получают следующее соединение:
Схема 7
Пример 10

4-Хлор-2-пиперидин-1-илпиримидин и 2-хлор-4-пиперидин-1-илпиримидин
Эти промежуточные соединения получают по процедуре K. Yoshida et al., J. Chem. Soc. Perkins Transactions I., (1992), 919-922. Раствор 2,4-дихлорпиримидина (4,00 г, 26,8 ммоль) и 1-метилпиперидина (3,25 мл, 29,5 ммоль) в 1,4-диоксане (60 мл) перемешивают при 100оС в течение 3 час, затем охлаждают и упаривают. Очистка флэш-хроматографией (SiO2) с элюированием смесью 15:85 этилацетат:гексаны дает 4-хлор-2-пиперидин-1-илпиримидин (0,58 г, выход 12%) в виде светло-желтого твердого вещества: 1Н-ЯМР (CDCl3, 500 МГц) 8,06 (д, 1Н), 6,37 (д, 1Н), 3,70 (м, 4Н), 1,61 (м, 2Н), 1,53 (м, 4Н) м.д.; МС (FIA) 198,1(М+Н);ВЭЖХ (способ А) 3,550 мин и 2-хлор-4-пиперидин-1-илпиримидин (1,87 г, выход 38%) в виде белого твердого вещества: 1Н-ЯМР (CDCl3, 500 МГц) 8,01 (д, 1Н), 6,41 (д, 1Н), 3,65 (м, 4Н), 1,73 (м, 2Н), 1,65 (м, 4Н) м.д.; МС (FIA) 198 (М+Н); ВЭЖХ (способ А) 2,583 мин.
Аналогично получают следующее соединение:
Схема 8
4-Хлор-6-(4-метилпиперазин-1-ил)пиримидин
4,6-Дихлорпиримидин (2 г, 13,4 ммоль) и N-метилпиперазин (1,5 мл, 13,4 ммоль) растворяют в 20 мл ТГФ вместе с ТЕА (1,9 мл, 13,4 ммоль) и перемешивают в течение 18 час. ТГФ выпаривают и добавляют 10 мл воды и смесь затем экстрагируют DCM. Слой DCM сушат над сульфатом натрия, фильтруют и упаривают, получая при этом 2,4 г 4-хлор-6-(4-метилпиперазин-1-ил)пиримидина в виде желтого воскообразного твердого вещества, который применяют без дополнительной очистки.
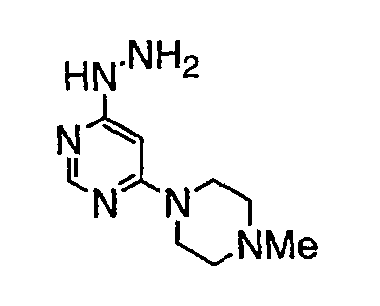
[6-(4-Метилпиперазин-1-ил)пиримидин-4-ил]гидразин
4-Хлор-6-(4-метилпиперазин-1-ил)пиримидин (200 мг, 0,94 ммоль) и гидрат гидразина (200 мкл, 4 ммоль) нагревают до 180оС в течение 6 мин в персональной химической микроволновой печке. Растворитель выпаривают, получая при этом [6-(4-метилпиперазин-1-ил)пиримидин-4-ил]гидразин в виде желто-коричневых кристаллов, которые используют без дополнительной очистки.
Аналогично получают следующие соединения:
Пример 11
[6-(4-Этилпиперазин-1-ил)пиримидин-4-ил]гидразин
4,6-Дихлорпиримидин (1,1 г, 7,4 ммоль) растворяют в 20 мл изопропанола, добавляют карбонат калия (2 г, 15 ммоль) и N-этилпиперазин (843 мг, 7,4 ммоль). Смесь перемешивают при комнатной температуре в течение 18 час, затем добавляют гидразин (1,6 г, 50 ммоль) и нагревают для кипячения с обратным холодильником в течение 22 час. Смесь охлаждают и фильтруют, затем из фильтрата выпаривают растворитель. Остаток растворяют в 20 мл кипящего ацетонитрила и фильтруют. При охлаждении образуется белый осадок. Осадок отфильтровывают, получая при этом приблизительно 1 г [6-(4-этилпиперазин-1-ил)пиримидин-4-ил]гидразина в виде белого твердого вещества (60%). МС ES+ 223,2.
Схема 9
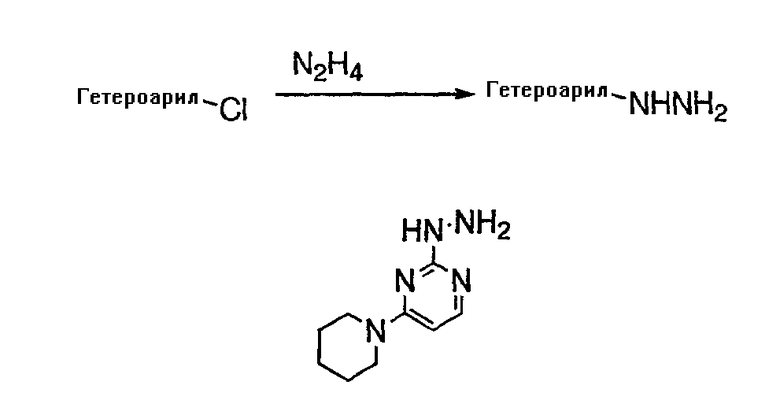
(4-Пиперидин-1-илпиримидин-2-ил)гидразин. Указанное в заголовке соединение получают, как описано в примере 10.
1Н-ЯМР (ДМСО-d6, 500 МГц) 7,82 (шир., 1Н), 7,74 (шир., 1Н), 5,88 (с, 1Н), 4,17 (с, 2Н), 3,65 (м, 4Н), 1,58 (м, 2Н), 1,45 (м, 4Н) м.д.; МС (FIA) 194,2 (М+Н);ВЭЖХ (способ А) 0,648 мин.
Аналогично получают следующие соединения:
Схема 10
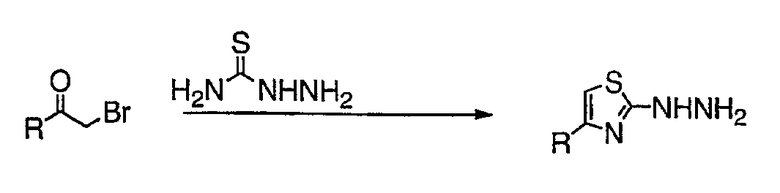
Пример 12
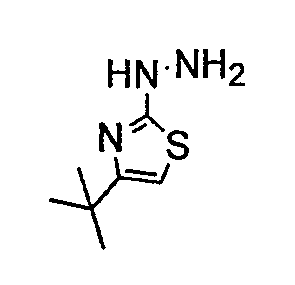
4-(трет-Бутилтиазол-2-ил)гидразин. Смесь 1-бром-3,3-диметилбутан-2-она (1,35 мл, 10 ммоль) и тиосемикарбазида (0,91 г, 10 ммоль) в этаноле (35 мл) кипятят с обратным холодильником в течение 1,5 часа и упаривают. Очистка флэш-хроматографией (SiO2) дает 4-(трет-бутилтиазол-2-ил)гидразин (1,01 г, выход 59%) в виде оранжевого твердого вещества. 1Н-ЯМР (ДМСО-d6, 500 МГц) 9,0 (шир., 1Н), 7,3 (шир., 2Н), 6,37 (с, 1Н), 1,22 (с, 9Н) м.д.; МС (ЖХ-МС) 172,1 (М+Н); (способ А) 2,520 мин.
Аналогично получают следующие соединения:
Пример 13
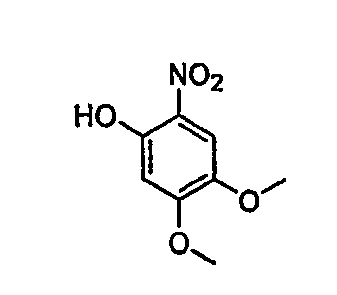
4,5-Диметокси-2-нитрофенол. К раствору 4,5-диметокси-2-нитробензальдегида (3,75 г, 14,2 ммоль) в дихлорметане (75 мл) при 0°С в атмосфере азота добавляют метахлорпероксибензойную кислоту (чистота 75%, 4,90 г, 28,4 ммоль), затем трифторуксусную кислоту (1,05 мл, 14,2 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 18 час, затем снова охлаждают до 0°С. Избыточный реагент гасят 5% раствором бисульфита натрия и осадок удаляют фильтрованием, промывают дихлорметаном. Органическую фазу фильтрата промывают бикарбонатом натрия и насыщенным раствором соли, сушат (сульфатом натрия) и упаривают, получая при этом желтое твердое вещество. Это промежуточное соединение суспендируют в метаноле (50 мл), обрабатывают 2 н. NaOH (16 мл, 32 ммоль) и перемешивают при комнатной температуре в течение 1 часа. Реакционную смесь подкисляют 1 н. HCl и фильтруют, промывая метанолом, получая при этом 4,5-диметокси-2-нитрофенол (2,00 г, выход 71%) в виде ярко-желтого твердого вещества.
1Н-ЯМР (CDCl3, 500 МГц) 11,0 (с, 1Н), 7,39 (с, 1Н), 6,48 (с, 1Н), 3,90 (с, 3Н), 3,83 (с, 3Н) м.д.; МС (FIA) 197,9 (М-Н); ВЭЖХ (способ А) 3,357 мин
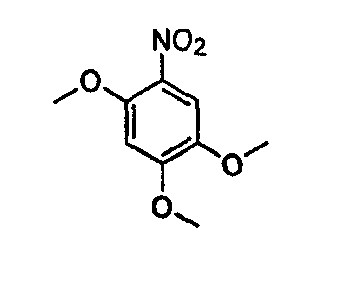
1,2,4-Триметокси-5-нитробензол. Смесь 4,5-диметокси-2-нитрофенола (2,00 г, 10 ммоль), карбоната калия (2,76 г, 20 ммоль) и иодметана (0,75 мл, 12 ммоль) в ДМФ помещают в запаянную трубку и нагревают при 75-80°С в течение 20 час. Реакционную смесь охлаждают и фильтруют через целит, промывая этилацетатом. Фильтрат промывают водой (воду первой промывки обратно экстрагируют этилацетатом), бикарбонатом натрия и насыщенным раствором соли, сушат (сульфатом натрия) и упаривают. Очистка флэш-хроматографией (SiO2) с элюированием смесью 1:1 этилацетат:гексаны дает 1,2,4-триметокси-5-нитробензол (1,20 г, выход 57%) в виде желтого твердого вещества.
1Н-ЯМР (CDCl3, 500 МГц) 7,53 (с, 1Н), 6,50 (с, 1Н), 3,92 (с, 3Н), 3,91 (с, 3Н), 3,84 (с, 3Н) м.д.; МС (FIA) 214,1 (М+Н); ВЭЖХ (способ А) 3,253 мин
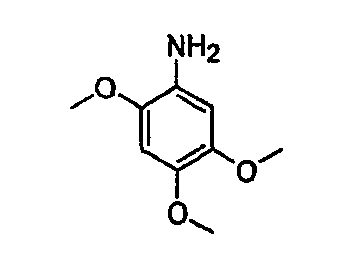
2,4,5-Триметоксифениламин. 1,2,4-Триметокси-5-нитробензол (1,20 г, 5,63 ммоль) и дигидрат хлорида олова (3,81 г, 16,9 ммоль) в этилацетате (50 мл) перемешивают при 65-70°С в течение 20 час. Реакционную смесь охлаждают, осторожно нейтрализуют бикарбонатом натрия и фильтруют через целит. Органическую фазу промывают насыщенным раствором соли, сушат (сульфатом натрия) и упаривают. Очистка флэш-хроматографией (SiO2) при элюировании смесью 3:7 этилацетат:гексаны дает 2,4,5-триметоксифениламин (0,56 г, выход 54%) в виде рыжевато-коричневого твердого вещества. 1Н-ЯМР (ДМСО-d6, 500 МГц) 6,57 (с, 1Н), 6,37 (с, 1Н), 3,70 (с, 3Н), 3,65 (с, 3Н), 3,63 (с, 3Н) м.д.; ВЭЖХ (способ А) 2,163 мин
Схема 11
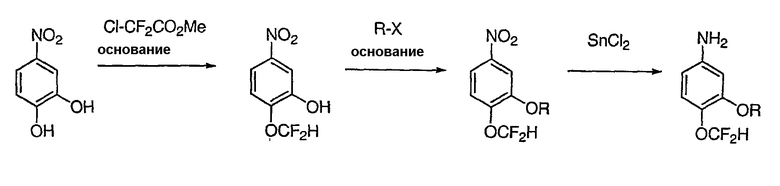
Пример 14
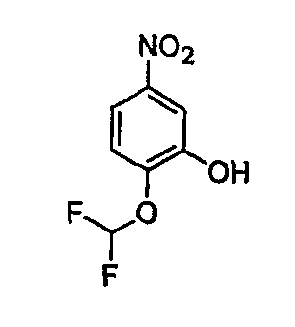
2-Дифторметокси-5-нитрофенол. Раствор 4-нитробензол-1,2-диола (4,18 г, 27,1 ммоль), метилхлордифторацетата (3,0 мл, 28,5 ммоль) и карбоната цезия (11,05 г, 33,9 ммоль) в ДМФ (75 мл) нагревают при 90°С в течение 24 час. Реакционную смесь охлаждают, выпаривают и разбавляют этилацетатом. Продукт экстрагируют два раза в 1 н. NaOH, объединенную водную фазу подкисляют, экстрагируют этилацетатом (дважды) и органические слои промывают водой (дважды) и насыщенным раствором соли, сушат (сульфатом натрия) и упаривают. Очистка флэш-хроматографией (SiO2) при элюировании смесью 2:8 этилацетат:гексаны дает 2-дифторметокси-5-нитрофенол (1,50 г, выход 27%) в виде ярко-желтого твердого вещества.
1Н-ЯМР (ДМСО-d6, 500 МГц) 10,9 (с, 1Н), 7,76 (д, 1Н), 7,73 (дд, 1Н), 7,36 (д, 1Н), 7,28 (т, 1Н) м.д.; МС (FIA) 204,1 (М-Н); ВЭЖХ (способ А) 3,307 мин
Аналогично получают следующее соединение.
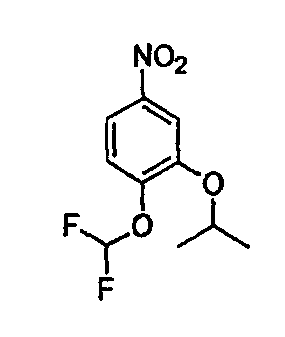
1-Дифторметокси-2-изопропокси-4-нитробензол. 2-Дифторметокси-5-нитрофенол (1,49 г, 7,26 ммоль), иодизопропан (0,87 мл, 8,72 ммоль) и карбонат цезия (3,55 г, 10,9 ммоль) в ДМФ (20 мл) в запаянной трубке нагревают при 90°С в течение 20 час. Реакционную смесь охлаждают и упаривают, разбавляют водой и экстрагируют (два раза) этилацетатом. Объединенную органическую фазу промывают водой (три раза) и насыщенным раствором соли, сушат (сульфатом натрия) и упаривают, получая при этом 1-дифторметокси-2-изопропокси-4-нитробензол (1,712 г, выход 95%) в виде оранжевого масла.
1Н-ЯМР (CDCl3, 500 МГц) 7,95 (м, 2Н), 7,38 (д, 1Н), 7,37 (т, 1Н), 4,80 (м, 1Н), 1,54 (д, 6Н) м.д.; МС (FIA) 216,1 (М-Н); ВЭЖХ (способ А) 4,108 мин
Аналогично получают следующее соединение.
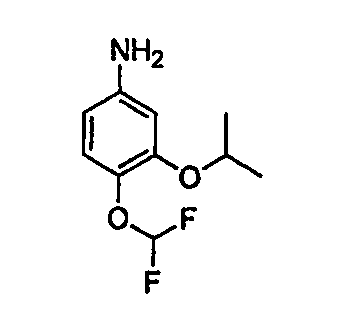
4-Дифторметокси-3-изопропоксифениламин. К дигидрату хлорида олова (5,46 г, 24,2 ммоль) в концентрированной HCl (7 мл) при 0°С добавляют 1-дифторметокси-2-изопропокси-4-нитробензол (1,712 г, 6,92 ммоль) в этилацетате (7 мл) и реакционную смесь перемешивают в течение 1 часа. Реакционную смесь нейтрализуют до рН ˜7 NaOH и фильтруют через целит, промывая этилацетатом. Фильтрат разделяют и водную фазу обратно экстрагируют этилацетатом. Объединенную органическую фазу промывают насыщенным раствором соли, сушат (сульфатом натрия) и упаривают. Очистка флэш-хроматографией (SiO2) при элюировании смесью 2:8 этилацетат:гексаны дает 4-дифторметокси-3-изопропоксифениламин (0,55 г, выход 37%, выход 57% на основе выделенного исходного материала) в виде оранжевого масла.
1Н-ЯМР (CDCl3, 500 МГц) 6,94 (д, 1Н), 6,41 (т, 1Н), 6,31 (дд, 1Н), 6,23 (д, 1Н), 4,47 (м, 1Н), 1,33 (д, 6Н) м.д.; МС (FIA) 218,2 (М+Н); ВЭЖХ (способ А) 2,853 мин
Аналогично получают следующее соединение.
Схема 12

Пример 15
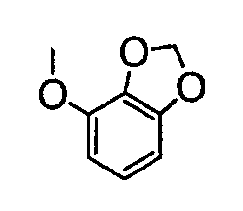
4-Метоксибензо[1,3]диоксол. Смесь 3-метоксибензол-1,2-диола (1,161 г, 8,28 ммоль) в ДМФ (10 мл) добавляют к бромхлорметану (611 мкл, 1,1 эквивалента) и смесь перемешивают при 90 градусах Цельсия в течение 4 часов. Смесь выливают в воду и экстрагируют дихлорметаном. Органический слой выливают через картридж разделения фаз и концентрируют досуха. Сырой продукт представляет собой желтую жидкость. Жидкость очищают колоночной хроматографией, получая при этом 1,21 г (96%).
1Н-ЯМР (ДМСО, 500 МГц) 6,7 (т, 1Н), 6,63 (д, 1Н), 6,58 (д, 1Н), 5,97 (с, 2Н), 3,83 (с, 3Н), ВЭЖХ (способ А) 2,86 мин
Схема 13
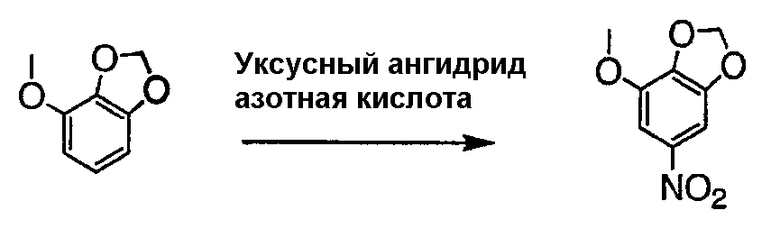
Пример 16
4-Метокси-6-нитробензо[1,3]диоксол. 4-Метоксибензо[1,3]диоксол (2,03 г, 13,34 ммоль) растворяют в уксусном ангидриде (20 мл) и охлаждают на ледяной бане при перемешивании. Делительной воронкой по каплям добавляют азотную кислоту (1,5 мл) на протяжении 30 минут. Ледяную баню убирают и смесь перемешивают на протяжении ночи, позволяя реакционной смеси нагреться до комнатной температуры. Смесь выливают в ледяную воду и продукт измельчают и фильтруют и промывают водой. Осадок сушат в вакууме в дессикаторе, получая при этом (1,21 г, выход 46%) 4-метокси-6-нитробензо[1,3]диоксол.
ЯМР-ДМСО-d6: 7,63 (с, 1Н), 7,52 (с, 1Н), 6,25 (с, 2Н), 3,95 (с, 3Н)
Схема 14
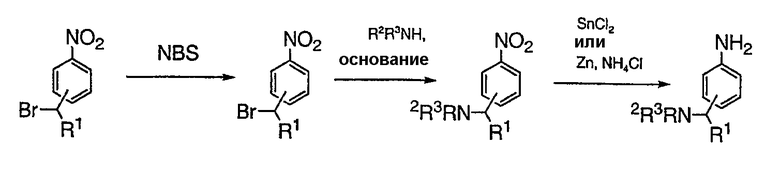
Пример 17
4-(4-Нитробензил)морфолин. К смеси 1-бромметил-4-нитробензола (5,0 г, 29,1 ммоль) и карбоната калия (12,0 г, 87 ммоль) в ТГФ (100 мл) добавляют медленный поток морфолина (6,35 мл, 73 ммоль). Реакционную смесь перемешивают 24 час при комнатной температуре, фильтруют через целит и упаривают. Очистка флэш-хроматографией (SiO2) дает 4-(4-нитробензил)морфолин (5,27 г, выход 81%) в виде светло-желтого твердого вещества.
1Н-ЯМР (ДМСО-d6, 500 МГц) 8,20 (д, 2Н), 7,60 (д, 2Н), 3,61 (м, 6Н), 2,38 (м, 4Н) м.д.; МС (FIA) 223,1(М+Н); ВЭЖХ (способ А) 1,577 мин
Аналогично получают следующие соединения:

4-Морфолин-4-илметилфениламин. По процедуре, описанной в примере 1.С, указанное в заголовке соединение получают (1,70 г, выход 98%) в виде оранжевого твердого вещества.
1Н-ЯМР (ДМСО-d6, 500 МГц) 6,91 (д, 2Н), 6,49 (д, 2Н), 4,95 (с, 2Н), 3,53 (м, 4Н), 2,28 (м, 4Н) м.д.; МС (FIA) 193,2 (М+Н); ВЭЖХ (способ А) 1,038 мин.
Аналогично получают следующее соединение:
Пример 18
4-Пирролидин-1-илметилфениламин. Указанное в заголовке соединение (0,37 г, выход 20%) получают по процедурам, описанным выше, в виде желтого масла.
1Н-ЯМР (ДМСО-d6, 500 МГц) 6,92 (д, 2Н), 6,49 (д, 2Н), 4,88 (с, 2Н), 3,38 (с, 2Н), 2,38 (м, 4Н), 1,66 (м, 4Н) м.д.; МС (FIA) 177,2 (М+Н); ВЭЖХ (способ А) 1,162 мин.
Пример 19
4-(4-Метилпиперазин-1-илметил)фениламин (CF#А-5). Смесь 1-метил-4-(4-нитробензил)пиперазина (3,09 г, 13,1 ммоль), цинковой пыли (4,29 г, 65,6 ммоль) и хлорида аммония (2,81 г, 52,5 ммоль) в метаноле (100 мл) кипятят с обратным холодильником 1 час, охлаждают, фильтруют через целит (промывка метанолом) и упаривают, получая при этом 4-(4-метилпиперазин-1-илметил)фениламин (2,67 г, выход 99%) а виде светло-желтого твердого вещества.
1Н-ЯМР (ДМСО-d6, 500 МГц) 6,89 (д, 2Н), 6,49 (д, 2Н), 4,89 (с, 2Н), 3,24 (с, 2Н), 2,3 (шир. м, 8Н) м.д.; МС (FIA) 206,2 (М+Н); ВЭЖХ (способ А), продукт элюируется с фронтом растворителя.
Пример 20

1-(1-Бромэтил)-4-нитробензол. Смесь 1-этил-4-нитробензола (3,4 мл, 25 ммоль), N-бромсукцинимида (4,38 г, 24,6 ммоль) и бензоилпероксида (0,04 г, 0,18 ммоль) в тетрахлориде углерода (30 мл) кипятят с обратным холодильником 1 час, охлаждают и фильтруют, промывая смесью 1:1 этилацетат:гексаны. Фильтрат упаривают и очищают флэш-хроматографией (SiO2) с элюированием смесью 2:98 этилацетат:гексаны, получая при этом 1-(1-бромэтил)-4-нитробензол (5,18 г, выход 90%) в виде желтого масла.
1Н-ЯМР (CDCl3, 500 МГц) 8,22 (д, 2Н), 7,62 (д, 2Н), 5,22 (кв., 1Н), 2,08 (д, 3Н) м.д.; ВЭЖХ (способ А) 3,837 мин.
4-[1-(4-Нитрофенил)этил]морфолин. Смесь 1-(1-бромэтил)-4-нитробензола (1,24 г, 5,43 ммоль), карбоната калия (2,25 г, 16,3 ммоль) и морфолина (1,2 мл, 13,6 ммоль) в ДМФ (10 мл) перемешивают при комнатной температуре в течение 16 час, затем упаривают. Остаток суспендируют в этилацетате, промывают водой и насыщенным раствором соли, сушат (сульфатом натрия) и упаривают, получая при этом 4-[1-(4-нитрофенил)этил]морфолин (1,225 г, выход 95%) в виде желтого масла. 1Н-ЯМР (ДМСО-d6, 500 МГц) 8,19 (д, 2Н), 7,16 (д, 2Н), 3,56 (м, 5Н), 2,41 (м, 2Н), 2,26 (м, 2Н), 1,29 (д, 3Н) м.д.; МС (FIA) 237, 2 (М+Н); ВЭЖХ (способ А) 2,248 мин.
Аналогично получают следующее соединение:
4-(1-Морфолин-4-илэтил)фениламин. Указанное в заголовке соединение получают способом, описанным выше.
1Н-ЯМР (ДМСО-d6, 500 МГц) 6,90 (д, 2Н), 6,49 (д, 2Н), 4,87 (с, 2Н), 3,51 (м, 4Н), 3,14 (кв., 1Н), 2,30 (м, 2Н), 2,25 (м, 2Н), 1,21 (д, 3Н) м.д.; МС (FIA) 207,3 (М+Н).
Аналогично получают следующее соединение:
Пример 21
2-Метокси-4-морфолино-4-илфениламин. К суспензии 5-морфолино-2-нитроанизола (0,76 г, 3,21 ммоль) в МеОН (20 мл) в атмосфере N2 добавляют 5% Pd/C. Реакционную смесь перемешивают в атмосфере Н2 при комнатной температуре в течение 4 час, фильтруют через целит, который промывают МеОН. Фильтрат концентрируют в вакууме, получая при этом продукт в виде клейкого красного твердого вещества (0,63 г, выход 95%).
(способ А)
Схема 15
Пример 22
Метиловый эфир 2-хлор-4-фторбензойной кислоты. Смесь 2-хлор-4-фторбензойной кислоты (6,5 г, 37 ммоль) в метаноле (100 мл) обрабатывают хлортриметилсиланом (14,0 мл, 111 ммоль), перемешивают 24 часа при комнатной температуре и упаривают. Остаток растворяют в дихлорметане, промывают бикарбонатом натрия, сушат (сульфатом натрия) и упаривают, получая при этом метиловый эфир 2-хлор-4-фторбензойной кислоты (7,01 г, выход 99%) в виде светло-желтого масла.
1Н-ЯМР (CDCl3, 500 МГц) 7,93 (м, 1Н), 7,22 (м, 1Н), 7,06 (м, 1Н), 3,95 (с, 3Н) м.д.; МС (FIA) 189,1 (М+Н); ВЭЖХ (способ А) 3,37 мин.

Метиловый эфир 3,5-диметокси-4-(2-морфолин-4-илэтокси)бензойной кислоты. К раствору метил-3,5-диметокси-4-гидроксибензоата (3,0 г, 14 ммоль) в ДМФ (10 мл) добавляют гидрохлорид 4-(2-хлорэтил)морфолина (3,99 г, 21 ммоль) и твердый К2СО3 (8,4 г, 60 ммоль). Смесь нагревают при 60°С в атмосфере N2 в течение 30 час. Смесь разбавляют EtOAc (100 мл) и промывают Н2О (2 × 50 мл), обратно экстрагируют водную фазу и объединенные органические фазы промывают насыщенным раствором соли. Органический слой сушат над Na2SO4, фильтруют и упаривают, получая при этом продукт в виде коричневого твердого вещества (4,79 г, выход количественный).
1Н-ЯМР (CDCl3, 500 МГц) 7,28 (с, 2Н), 4,15 (т, 2Н), 3,91 (с, 3Н), 3,88 (с, 6Н), 3,75-3,71 (м, 4Н), 2,78 (т, 2Н), 2,59 (шир. с, 4Н) м.д.; МС (FIA) 326,17 (М+Н)
Аналогично получают следующее соединение:
Метиловый эфир 2-хлор-4-морфолин-4-илбензойной кислоты. Смесь метилового эфира 2-хлор-4-фторбензойной кислоты (3,51 г, 18,6 ммоль), морфолина (1,95 мл, 22,3 ммоль) и карбоната калия (5,12 г, 37,1 ммоль) в N-метилпирролидоне (20 мл) перемешивают при 120°С в течение 5 час. Реакционную смесь охлаждают, разбавляют этилацетатом и фильтруют через целит. Фильтрат промывают четыре раза водой, один раз насыщенным раствором соли, сушат (сульфатом натрия) и упаривают. Очистка флэш-хроматографией (SiO2) с элюированием смесью 2:8 этилацетат:гексаны дает метиловый эфир 2-хлор-4-морфолин-4-илбензойной кислоты (3,08 г, выход 65%) в виде белого твердого вещества.
1Н-ЯМР (CDCl3, 500 МГц) 7,79 (д, 1Н), 6,81 (д, 1Н), 6,67 (дд, 1Н), 3,81 (с, 3Н), 3,78 (м, 4Н), 3,20 (м, 4Н) м.д.; МС (FIA) 256,1 (М+Н); ВЭЖХ (способ А) 3,275 мин.
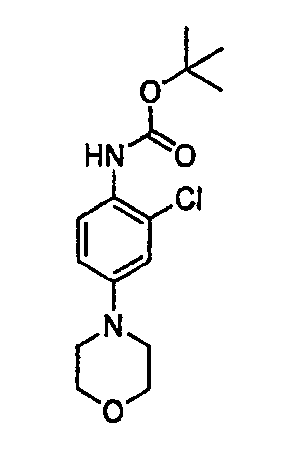
Трет-Бутиловый эфир (2-хлор-4-морфолин-4-илфенил)карбаминовой кислоты. Смесь метилового эфира 2-хлор-4-морфолин-4-илбензойной кислоты (3,08 г, 12,0 ммоль) и 6 н. NaOH (2,5 мл, 15 ммоль) в метаноле (50 мл) и воде (7,5 мл) перемешивают 24 час при комнатной температуре, затем подкисляют 2 н. HCl. Осадок отфильтровывают, промывают водой и сушат, получая при этом 2-хлор-4-морфолин-4-илбензойную кислоту (2,56 г, выход 88%) в виде белого твердого вещества. Это твердое вещество (10,6 ммоль) суспендируют в трет-бутаноле (20 мл), обрабатывают дифенилфосфорилазидом (2,30 мл, 10,6 ммоль), затем триэтиламином (1,45 мл, 10,6 ммоль), перемешивают при кипячении с обратным холодильником в течение 20 час и упаривают. Очистка флэш-хроматографией (SiO2) при элюировании смесью 2:8 этилацетат:гексаны дает трет-бутиловый эфир (2-хлор-4-морфолин-4-илфенил)карбаминовой кислоты (2,84 г) в виде смеси (˜ 1:1) с трет-бутиловым эфиром 2-хлор-4-морфолин-4-илбензойной кислоты. Эту смесь применяют без дополнительной очистки.
1Н-ЯМР (CDCl3, 500 МГц) 8,0 (шир., 1Н), 7,80 (д, 1Н), 7,4 (д, 1Н), 6,90 (д, 1Н), 6,87 (д, 1Н), 6,84 (дд, 1Н), 6,75 (дд, 1Н), 3,87 (м, 8Н), 3,26 (м, 4Н), 3,11 (м, 4Н), 1,61 (с, 9Н), 1,55 (с, 9Н) м.д.; МС (FIA) 313,1 (М+Н); ВЭЖХ (способ А) 3,70 мин.
Аналогичным способом получают следующие соединения:
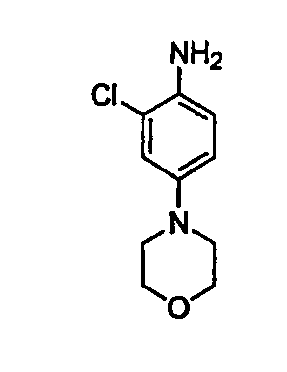
2-Хлор-4-морфолин-4-илфениламин. Раствор нечистого трет-бутилового эфира (2-хлор-4-морфолин-4-илфенил)карбаминовой кислоты (2,84 г) в дихлорметане (30 мл) обрабатывают трифторуксусной кислотой (3,5 мл), перемешивают при комнатной температуре в течение 24 час и упаривают. Остаток растворяют в этилацетате, промывают 1 н. NaOH, водой и насыщенным раствором соли, сушат (сульфатом натрия) и упаривают. Очистка флэш-хроматографией (SiO2) при элюировании смесью 35:65 этилацетат:гексаны дает 2-хлор-4-морфолин-4-илфениламин (0,77 г, выход 40%) в виде не совсем белого твердого вещества.
1Н-ЯМР (CDCl3, 500 МГц) 6,92 (шир., 1Н), 6,77 (шир., 2Н), 3,89 (м, 6Н), 3,05 (м, 4Н) м.д.; МС (FIA) 213,1 (М+Н); ВЭЖХ (способ А) 1,975 мин.
Пример 23
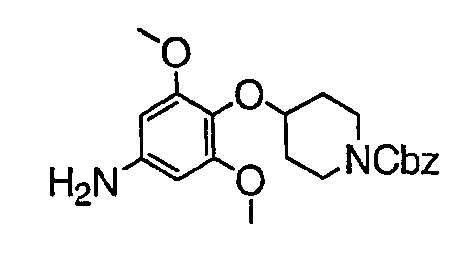
Бензиловый эфир 4-(4-амино-2,6-диметоксифенокси)пиперидин-1-карбоновой кислоты. Указанное в заголовке соединение получают из бензилового эфира 4-(4-трет-бутоксикарбониламино-2,6-диметоксифенокси)пиперидин-1-карбоновой кислоты по процедуре, описанной в примере ST-3. МС (ES+): m/z=387,2.
Пример 24
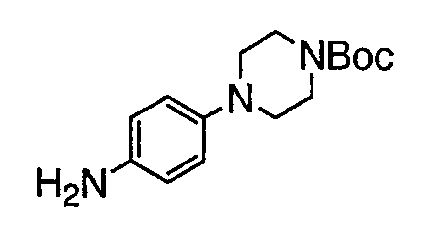
трет-Бутиловый эфир 4-(4-аминофенил)пиперазин-1-карбоновой кислоты. Указанное в заголовке соединение получают из 4-иоданилина и трет-бутилпиперазинкарбоксилата по процедуре, описанной в примере ST-1. МС (ES+): m/z=278,2.
Пример 25
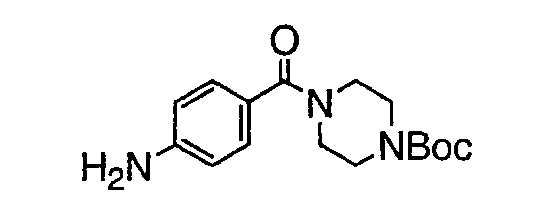
трет-Бутиловый эфир 4-(4-аминобензоил)пиперазин-1-карбоновой кислоты. К раствору 4-аминобензойной кислоты (411 мг, 3,00 ммоль) в ДМФ (3,0 мл) при комнатной температуре добавляют EDC (862 мг, 4,50 ммоль), HOBt (608 мг, 4,50 ммоль), триэтиламин (606 мг; 0,835 мл, 6,00 ммоль) и трет-бутилпиперазинкарбоксилат (671 мг, 3,60 ммоль). Смесь перемешивают в течение 22 час, затем добавляют 2 н. водный NaOH до установления рН>10. Смесь экстрагируют этилацетатом и органический слой сушат над MgSO4, концентрируют. Остаток очищают колоночной хроматографией на силикагеле при элюировании смесью EtOAc:гексаны (от 50 до 90% EtOAc), получая при этом трет-бутиловый эфир 4-(4-аминобензоил)пиперазин-1-карбоновой кислоты (796 мг, 87%) в виде бесцветного масла. МС (ES+): m/z=306,2.
Пример 26
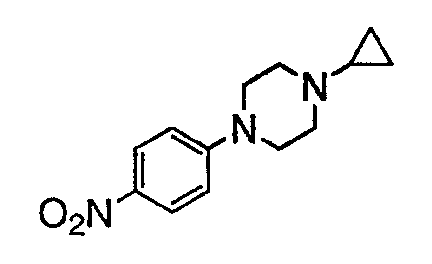
1-Циклопропил-4-(4-нитрофенил)пиперазин. К раствору 1-(4-нитрофенил)пиперазина (1,04 г, 5,00 ммоль) в метаноле (25 мл) в атмосфере азота добавляют молекулярные сита (1,0 г), уксусную кислоту (3,00 г, 2,86 мл, 50,0 ммоль) [(1-этоксициклопропил)окси]триметилсилан (5,22 г, 5,99 мл, 30,0 ммоль), цианоборогидрид натрия (1,41 г, 22,5 ммоль). Смесь перемешивают при комнатной температуре а течение 2,5 час, фильтруют и концентрируют. К остатку добавляют воду и 1 н. водную NaOH для регулирования рН до >11. Смесь экстрагируют этилацетатом и органический слой сушат над Na2SO4, концентрируют, получая при этом указанное в заголовке соединение (1,24 г, 100%) в виде желтого твердого вещества. МС (ES+): m/z=247,8.
Пример 27
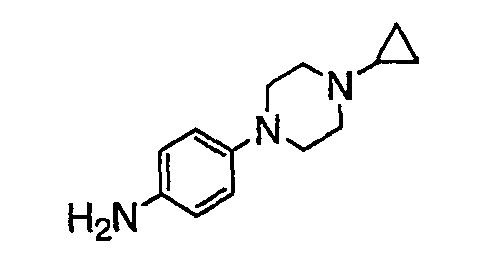
4-(4-Циклопропилпиперазин-1-ил)фениламин. К раствору 1-циклопропил-4-(4-нитрофенил)пиперазина (1,24 г, 5,00 ммоль) в метаноле (25 мл) добавляют Pd/C (10%, 100 мг) и трифторуксусную кислоту (1,0 мл). Смесь перемешивают под давлением водорода (из баллона) 1 атм на протяжении ночи, фильтруют и концентрируют. К остатку добавляют воду и 1 н. водный NaOH для регулирования рН >11. Смесь экстрагируют этилацетатом и органический слой сушат над Na2SO4, концентрируют, получая при этом указанное в заголовке соединение (1,08 г, 100%) в виде коричневого масла. МС (ES+) m/z=218,1
Пример 28
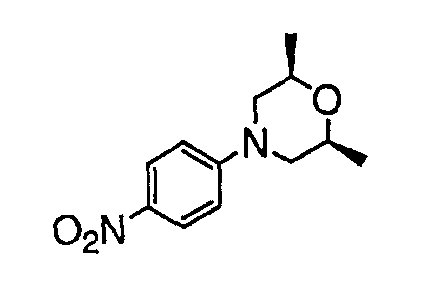
цис-2,6-Диметил-4-(4-нитрофенил)мофолин. Смесь 1-фтор-4-нитробензола, 2,6-диметилморфолина (куплен у Aldrich) и диизопропиламина нагревают при 110°С в течение 16 час. После охлаждения до комнатной температуры в смесь добавляют воду и экстрагируют EtOAc. Органическую фазу сушат над MgSO4, концентрируют. Остаток очищают колоночной хроматографией на силикагеле с элюированием смесью EtOAc:гексаны (от 5 до 35% EtOAc), получая при этом транс-2,6-диметил-4-(4-нитрофенил)мофолин (258 мг) и цис-2,6-диметил-4-(4-нитрофенил)мофолин (838 мг), оба в виде желтых твердых веществ. Транс-изомер: МС (ES+): m/z=237,2; 1Н-ЯМР (CDCl3, 500 МГц): δ 1,30 (д, 6Н), 3,17 (дд, 2Н), 3,46 (дд, 2Н), 4,15-4,22 (м, 2Н), 6,77 (д, 2Н), 8,14 (д, 2Н).Цис-изомер: МС (ES+): m/z=237,2; 1Н-ЯМР (CDCl3, 500 МГц): δ 1,29 (д, 6Н), 2,62 (дд, 2Н), 3,67 (дд, 2Н), 3,73-3,81 (м, 2Н), 6,84 (д, 2Н), 8,14 (д, 2Н).
Пример 29
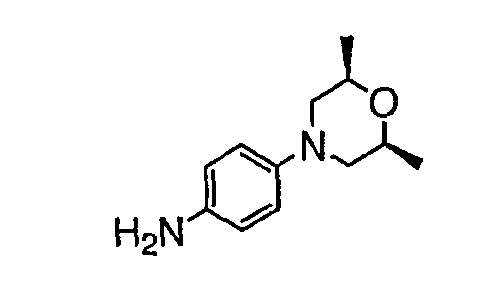
4-(2,6-Диметилморфолин-4-ил)фениламин. Указанное в заголовке соединение получают из цис-2,6-диметил-4-(4-нитрофенил)морфолина по процедуре, описанной выше. МС (ES+): m/z=207,3
Пример 30
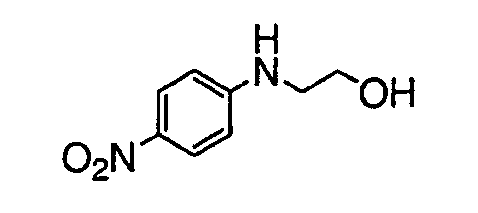
2-(4-Нитрофениламино)этанол. Указанное в заголовке соединение получают из 1-фтор-4-нитробензола и 2-аминоэтанола по процедуре, описанной выше.
МС (ES+): m/z=183,0; 1Н-ЯМР (CDCl3, 500 МГц): δ 3,41 (т, 2Н), 3,92 (т, 2Н), 6,59 (д, 2Н), 8,11 (д, 2Н).
Пример 31
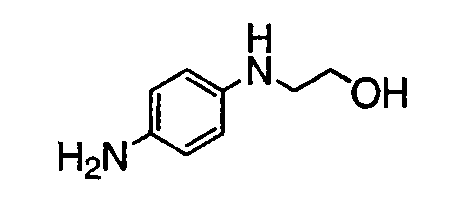
2-(4-Аминофениламино)этанол. Указанное в заголовке соединение получают из 2-(4-нитрофениламино)этанола по процедуре, описанной выше. МС (ES+): m/z=153,0.
Схема 16

Пример 32

1-(2-Хлорэтокси)-2-метокси-4-нитробензол. Смесь 2-метокси-4-нитрофенола (2,50 г, 14,8 ммоль), 1-бром-2-хлорэтана (1,35 мл, 16,3 ммоль) и карбонат калия (4,08 г, 19,6 ммоь) в ДМФ (50 мл) в запаянной трубке нагревают при 90°С в течение 18 час. Реакционную смесь охлаждают и фильтруют, промывая этилацетатом. Фильтрат промывают бикарбонатом натрия, водой (4 раза) и насыщенным раствором соли и сушат (сульфатом натрия) и упаривают. Очистка флэш-хроматографией (SiO2) с элюированием смесью 35:65 этилацетат:гексаны дает 1-(2-хлорэтокси)-2-метокси-4-нитробензол (1,93 г, выход 56%) в виде не совсем белого твердого вещества.
1Н-ЯМР (ДМСО, 500 МГц): 7,89 (дд, 1Н), 7,76 (д, 1Н), 7,21 (д, 1Н), 4,41 (т, 2Н), 4,00 (т, 2Н), 3,90 (с, 3Н) м.д.; ВЭЖХ (способ А) 3,781 мин.
Аналогичным способом получают следующие соединения:
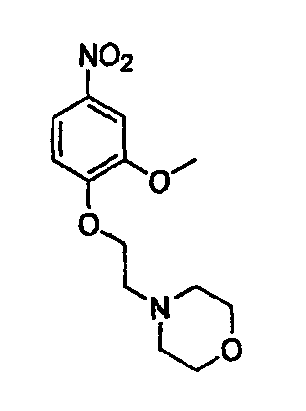
4-[2-(2-Метокси-4-нитрофенокси)этил]морфолин. Смесь 1-(2-хлорэтокси)-2-метокси-4-нитробензол (0,60 г, 2,59 ммоль), морфолина (0,28 мл, 3,11 ммоль), иодида натрия (0,39 г, 2,59 ммоль) и карбоната калия (0,71 г, 5,18 ммоль) в этаноле (5 мл) нагревают в запаянной трубке при 90°С в течение 18 час, охлаждают, фильтруют и упаривают. Очистка флэш-хроматографией (SiO2) с элюированием смесью 2:98 метанол:дихлорметан дает 4-[2-(2-метокси-4-нитрофенокси)этил]морфолин (0,37 г, выход 51%) в виде светло-желтого твердого вещества. 1Н-ЯМР (ДМСО-d6, 500 МГц) 7,89 (дд, 1Н), 7,74 (д, 1Н), 7,21 (д, 1Н), 4,23 (т, 2Н), 3,88 (с, 3Н), 3,88 (м, 4Н), 2,73 (т, 2Н), 2,50 (м, 4Н) м.д.; МС (FIA) 283,2 (М+Н); ВЭЖХ (способ А) 2,652 мин.
Аналогично получают следующие соединения:
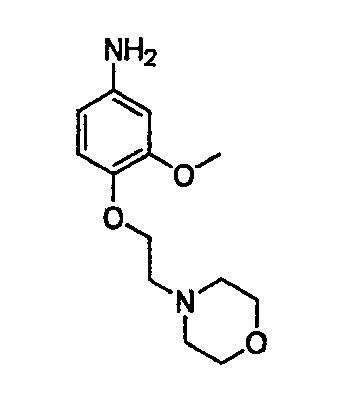
3-Метокси-4-(2-морфолин-4-илэтокси)фениламин. Указанное в заголовке соединение получают по процедуре, описанной выше, в виде красного масла (0,14 г, выход 43%).
1Н-ЯМР (ДМСО-d6, 500 МГц) 6,65 (д, 1Н), 6,24 (д, 1Н), 6,03 (дд, 1Н), 4,70 (с, 2Н), 3,87 (т, 2Н), 3,66 (с, 3Н), 3,56 (м, 4Н), 2,58 (т, 2Н), 2,44 (м, 4Н) м.д.; МС (FIA) 253,2 (М+Н); ВЭЖХ (способ А) продукт элюируется с фронтом растворителя.
Аналогично получают следующие соединения:
Схема 17:
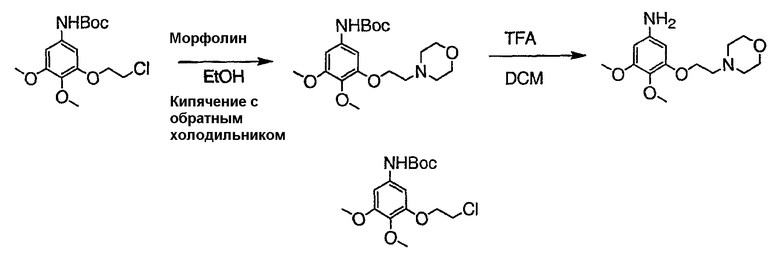
Трет-Бутиловый эфир [3-(2-хлорэтокси)-4,5-диметоксифенил]карбаминовой кислоты. 3-(2-Хлорэтокси)-4,5-диметоксибензойную кислоту (500 мг, 1,9 ммоль), DPPA (550 мг, 2 ммоль) и ТЕА (2 ммоль) смешивают в 5 мл трет-бутанола и кипятят с обратным холодильником в течение 5 час. трет-бутанол удаляют в вакууме и остаток очищают хроматографией на силикагеле (5% метанол/DCM), получая при этом трет-бутиловый эфир [3-(2-хлорэтокси)-4,5-диметоксифенил]карбаминовой кислоты.
ЯМР CDCl3: 6,7 (д, 2Н), 6,4 (шир.с, 1Н), 4,3 (т, 3Н), 3,85 (с, 3Н), 3,80 (с, 3Н), 3,82 (м, 2Н), 1,5 (с, 9Н).
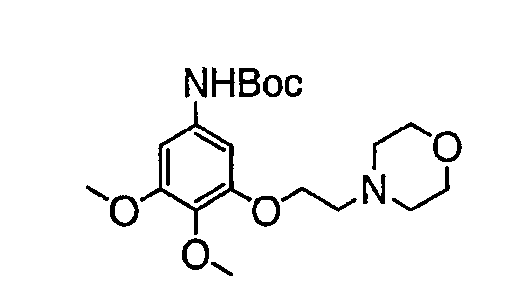
трет-Бутиловый эфир [3,4-диметокси-5-(2-морфолин-4-илэтокси)фенил]карбаминовой кислоты. трет-Бутиловый эфир [3-(2-хлорэтокси)-4,5-диметоксифенил]карбаминовой кислоты (90 мг, 0,27 ммоль) в 1 мл этанола обрабатывают 200 мкл морфолина и кипятят с обратным холодильником в течение 18 час. Растворитель и избыток морфолина выпаривают и остаток очищают препаративной ТСХ, получая при этом 79 мг трет-бутиловый эфир [3,4-диметокси-5-(2-морфолин-4-илэтокси)фенил]карбаминовой кислоты. МС ES+ 383.
3,4-Диметокси-5-(2-морфолин-4-илэтокси)фениламин.
трет-Бутиловый эфир [3,4-диметокси-5-(2-морфолин-4-илэтокси)фенил]карбаминовой кислоты (97 мг, 0,24 ммоль) перемешивают в 1 мл DCM и 1 мл TFA. Спустя ˜1 час TFA и DCM выпаривают и добавляют 2 мл насыщенного водного раствора бикарбоната натрия. Водный слой экстрагируют DCM, который выпаривают, получая при этом 60 мг 3,4-диметокси-5-(2-морфолин-4-илэтокси)фениламин. МС ES+ 283,1.
Аналогичными способами получили следующие анилины:
Пример 33
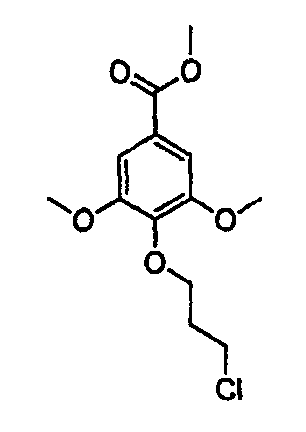
Метиловый эфир 4-(3-хлорпропокси)-3,5-диметоксибензойной кислоты. Указанное в заголовке соединение получают по процедуре, описанной выше.
1Н-ЯМР (CDCl3, 500 МГц) 7,22 (с, 2Н), 4,11 (т, 2Н), 3,84 (с, 3Н), 3,83 (с, 6Н), 3,78 (т, 2Н), 2,11 (м, 2Н) м.д.; ВЭЖХ (способ А) 4,060 мин.
Аналогично получают следующие соединения:
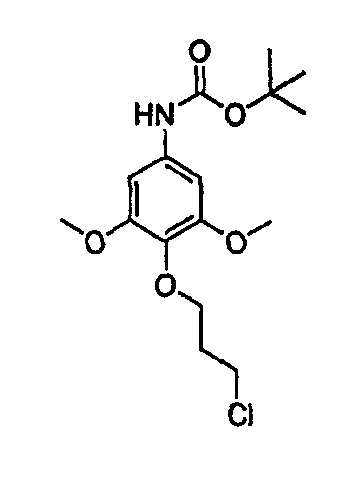
трет-Бутиловый эфир [4-(3-хлорпропокси)-3,5-диметоксифенил]карбаминовой кислоты. Указанное в заголовке соединение получают по процедуре, описанной в примере 7.С.
1Н-ЯМР (CDCl3, 500 МГц) 6,58 (с,2Н), 6,35 (с, 1Н), 3,98 (т, 2Н), 3,77 (т, 2Н), 3,75 (с, 6Н), 2,08 (м,2Н), 1,44 (с, 9Н) м.д.; МС (FIA) 246,1 (М+Н-ВОС); ВЭЖХ (способ А) 4,301 мин.
Аналогично получают следующие соединения:
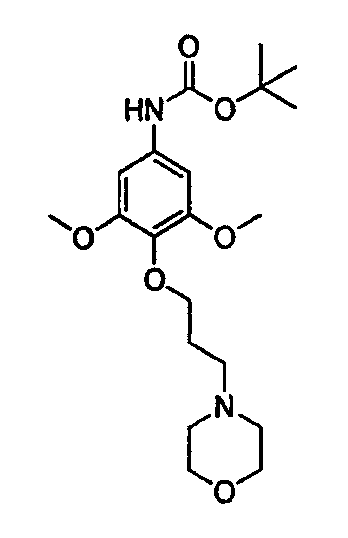
трет-Бутиловый эфир [3,5-диметокси-4-(3-морфолин-4-илпропокси)фенил]карбаминовой кислоты. Указанное в заголовке соединение получают по процедуре, описанной выше.
1Н-ЯМР (ДМСО-d6, 500 МГц) 9,2 (с, 1Н), 6,82 (с, 2Н), 3,80 (т, 2Н), 3,69 (с, 6Н), 3,56 (м, 4Н), 2,42 (т, 2Н), 2,33 (м, 4Н), 1,72 (м, 2Н), 1,46 (с, 9Н) м.д.; МС (FIA) 397,2 (М+Н); ВЭЖХ (способ А) 3,049 мин.
Аналогично получают следующие соединения:
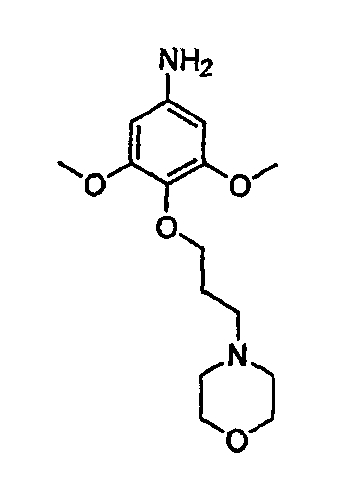
3,5-Диметокси-4-(3-морфолин-4-илпропокси)фениламин. Указанное в заголовке соединение получают по процедуре, описанной выше. Указанное в заголовке соединение используют в неочищенной форме и не анализируют.
Аналогично получают следующие соединения:
Схема 18:
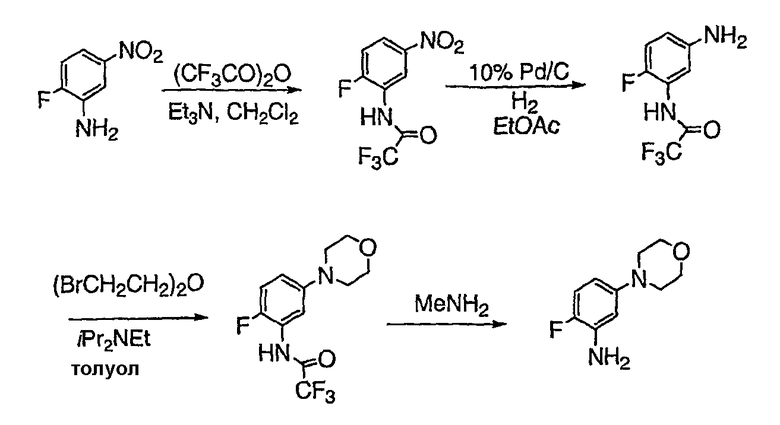
2-Фтор-5-морфолин-4-илфениламин
К раствору 2-фтор-5-нитрофениламина (4,68 г, 30 ммоль), триэтиламина (13,8 мл, 100 ммоль) в дихлорметане (100 мл) добавляют по каплям при 0°С трифторуксусный ангидрид (5,7 мл, 40 ммоль). Спустя 1 час реакционную смесь промывают водой, разбавленной HCl (рН 2) и водой, концентрируют, получая при этом сырой 2,2,2-трифтор-N-(2-фтор-5-нитрофенил)ацетамид (12,9 г).
Сырой ацетамид растворяют в EtOAc (50 мл), встряхивают с 10% Pd/C (450 мг) в атмосфере Н2 (50 фунт/кв. дюйм). Фильтрование дает N-(5-амино-2-фторфенил)-2,2,2-трифторацетамид (8,42 г).
Смесь анилина (1,5 г, 6,7 ммоль), бис-(2-бромэтилового) эфира (1,5 г, 6,7 ммоль), диизопропилэтиламина (4,7 мл, 31 ммоль) в смеси толуола (100 мл) диметилацетамида (ДМА, 5 мл) кипятят с обратным холодильником в течение 5 дней. Концентрирование и колоночная хроматография (гексан/EtOAc, 7:3) дают 2,2,2-трифтор-N-(2-фтор-5-морфолин-4-илфенил)ацетамид (1,45 г). ЖХ-МС: m/e=291,1 (М-Н), 293,2 (М+Н). 1Н-ЯМР (ДМСО-d6, 500 МГц) 11,19 (с, 1Н), 7,20 (т, 1Н), 6,98 (дд, 1Н), 6,94 (дт, 1Н), 3,72 (т, 4Н), 3,06 (т, 4Н).
Схема 19
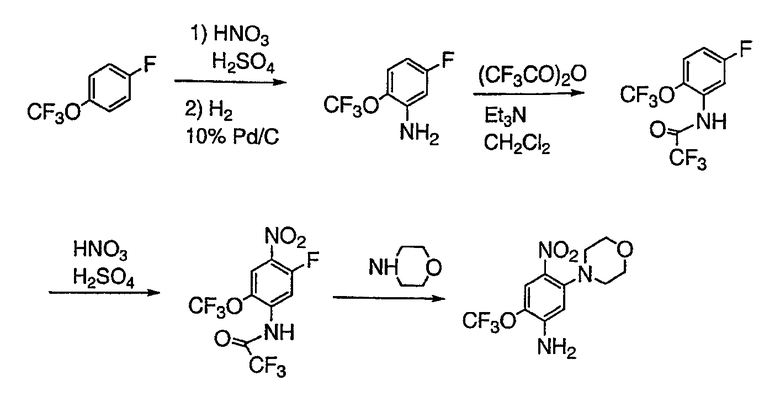
5-Морфолин-4-ил-4-нитро-2-трифторметоксифениламин. При -10°С 90% дымящую азотную кислоту (25 мл) медленно добавляют к концентрированной серной кислоте (50 мл), чтобы поддерживать температуру ниже 0°С. При -20°С добавляют порциями 1-фтор-4-трифторметоксибензол (5 г, 27,7 ммоль), чтобы поддерживать температуру реакционной смеси ниже 0°С. После добавления реакционную смесь выдерживают при 0°С в течение 30 мин, выливают в смесь лед-вода, экстрагируют EtOAc. Экстракты концентрируют, получая при этом смесь (6,05 г) 4-фтор-2-нитро-1-трифторметоксибензола и 1-фтор-2-нитро-4-трифторметоксибензола в отношении 3:1. Сырой продукт нитрования (6,05 г) растворяют в этаноле (40 мл) и встряхивают с 10% Pd/C (310 мг) и концентрированной HCl (2,8 мл) в атмосфере Н2 (50 фунт/кв. дюйм) в течение 3,5 часа. Фильтрование и концентрирование дают смесь (8,0 г) 5-фтор-2-трифторметоксифениламина и его изомера 2-фтор-5-трифторметоксифениламина в отношении 3:1. Без очистки фениламины (7 г, 35,8 ммоль) суспендируют в дихлорметане (100 мл), обрабатывают трифторуксусным ангидридом (71 ммоль) и триэтиламином (20 мл) в течение 16 час. Реакционную смесь промывают насыщенным NaHCO3 и насыщенным раствором соли. Органическую фазу далее очищают хроматографией на силикагеле (гексан/EtOAc, 9:1), получая при этом смесь (5,29 г) 2,2,2-трифтор-N-(5-фтор-2-трифторметоксифенил)ацетамида и его изомера в отношении 1:4, которую нитруют такой же процедурой, как в первой стадии. Нитрат (3 г) нагревают при кипячения с обратным холодильником с морфолином (10 мл) в 1,2-дихлорэтане (30 мл) в течение 3 час, смесь упаривают для удаления морфолина. Остаток разбавляют дихлорметаном, промывают HCl (0,5 н., 100 мл). Органическую фазу очищают флэш-хроматографией (гексан/EtOAc, от 7:3 до 1:1), получая при этом указанное в заголовке соединение 5-морфолин-4-ил-4-нитро-2-трифторметоксифениламин (2,48 г), ЖХ-МС: m/e=306,1 (М-Н), 308,2 (М+Н). 1Н-ЯМР (500 МГц, CDCl3): 8,04 (с, 1Н), 6,35 (с, 1Н), 4,54 (шир.с, 1Н), 3,90 (т,4Н), 3,07 (т, 4Н).
Раствор ацетамида (210 мг) в метаноле (2 мл) обрабатывают 40% водным раствором метиламина (0,5 мл) в течение 16 час. После выпаривания остаток суспендируют в воде, и фильтрование суспензии дает указанный в заголовке продукт (90 мг). FIA-МС: m/e=197,1 (М-Н).
Пример 34
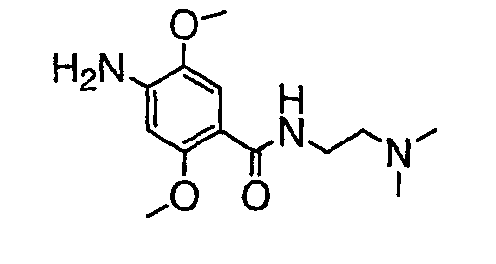
4-Амино-N-(2-диметиламиноэтил)-2,5-диметоксибензамид (DC-1787-162). К раствору 4-амино-5-хлор-2-метоксибензойной кислоты (300 мг, 1,49 ммоль) в CH2Cl2 (8 мл) добавляют N,N-диметилэтилендиамин (263 мг, 2,98 ммоль), EDCI (428 мг, 2,23 ммоль), гидрат НОВТ (201 мг, 1,49 ммоль) и диизопропилэтилендиамин (777 мкл, 4,47 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 48 час. Реакционную смесь разбавляют CH2Cl2, промывают насыщенным NaHCO3 и насыщенным раствором соли. Органическую фазу сушат над MgSO4, фильтруют и упаривают, получая при этом белое твердое вещество (423 мг, содержит небольшое количество примесей).
1Н-ЯМР (500 МГц, ДМСО-d6) 8,08 (т, 1Н), 7,70 (с, 1Н), 6,47 (с, 1Н), 5,96 (с, 2Н), 3,82 (с, 1Н), 3,34 (кв, 2Н), 2,45 (т, 2Н), 2,25 (с, 6Н) м.д.; МС (FIA) 227,1 (М+Н).
Пример 35

N-Циано-N'-((4-(4-трет-бутоксикарбонил)пиперазинокарбонил)фенил)-О-фенилизомочевина. Смесь трет-бутилового эфира 4-(4-аминобензоил)пиперазин-1-карбоновой кислоты (520 мг, 1,70 ммоль) и дифенилцианокарбонимид (406 мг, 1,70 ммоль) в диметилацетонитриле (3,0 мл) нагревают при 150°С в течение 30 мин. Смесь концентрируют и очищают колоночной хроматографией на силикагеле с элюированием смесью EtOAc:гексаны (от 5 до 35% EtOAc), получая при этом указанное в заголовке соединение (177 мг, 23%) в виде белого твердого вещества.
МС (ES+): m/z=450,1; 1Н-ЯМР (CDCl3, 500 МГц): δ 1,48 (с, 9Н), 3,26-3,83 (м, 8Н), 7,16 (д, 2Н), 7,34 (т, 1Н), 7,42-7,49 (м, 6Н).
Пример 36
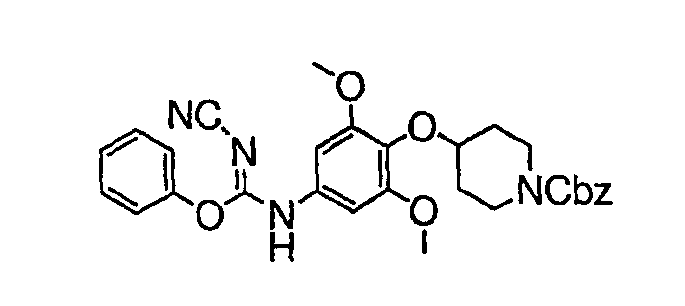
Смесь бензилового эфира 4-(4-амино-2,6-диметоксифенокси)пиперидин-1-карбоновой кислоты (57,8 мг, 0,180 ммоль) и дифенилцианокарбонимида (42,4 мг, 0,180 ммоль) в толуоле (1,0 мл) нагревают при 100°С на протяжении ночи. Смесь концентрируют и очищают колоночной хроматографией на силикагеле с элюированием смесью EtOAc:гексаны (от 40 до 60% EtOAc), получая при этом указанное в заголовке соединение (65,3 мг, 82%) в виде бесцветного масла. МС (ES+): m/z=531,2.
Пример 37
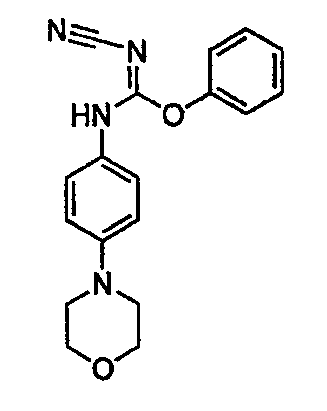
N-Циано-N'-(4-морфолинофенил)-О-фенилизомочевина. К 4-морфолиноанилину (196 г, <1,10 моль) в изопропаноле (2 л) добавляют на протяжении 0,5 часа дифенилцианокарбонимидат (250 г, 1,05 моль) и смесь перемешивают 22 часа. Твердое вещество отфильтровывают, промывают изопропанолом до получения бесцветной промывной жидкости, затем суспендируют в МТВЕ и снова фильтруют. Соединение сушат, получая при этом N-циано-N'-(4-морфолинофенил)-О-фенилизомочевину (319 г, выход 95%) в виде не совсем белого твердого вещества.
1Н-ЯМР (ДМСО-d6, 500 МГц) 10,6 (с, 1Н), 7,43 (т, 2Н), 7,29 (м, 5Н), 6,95 (д, 2Н), 3,73 (м, 4Н), 3,10 (м, 4Н) м.д.; МС (FIA) 323,2 (М+Н); ВЭЖХ (способ А) 3,126 мин.
Аналогично получают следующие соединения:
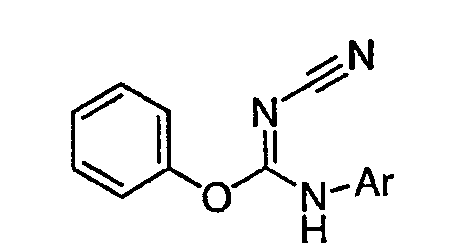
500 МГц
(растворитель)
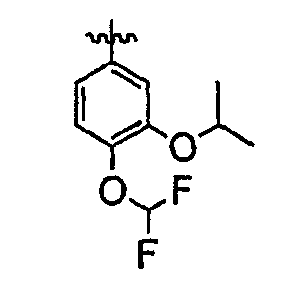
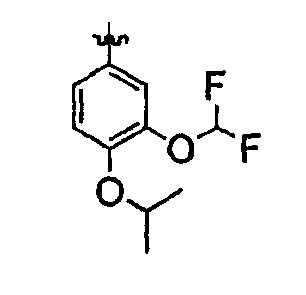
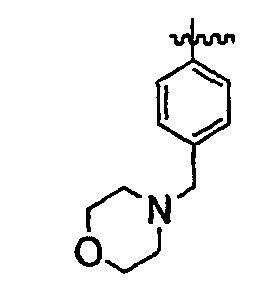
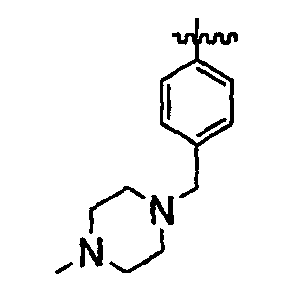


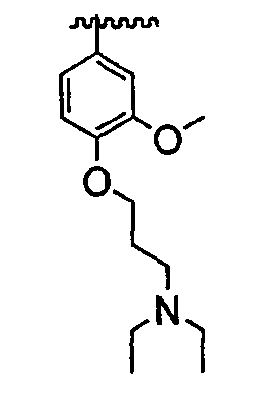
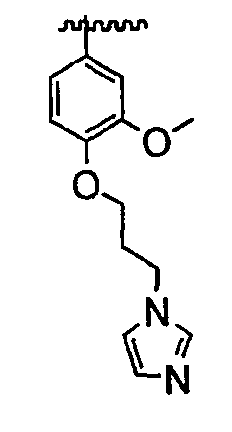
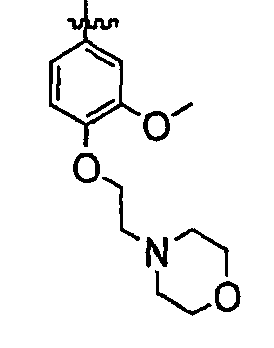
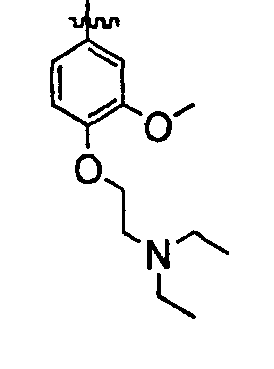
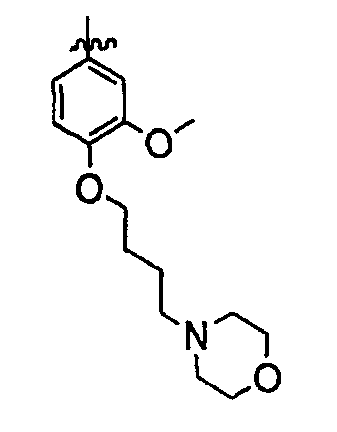
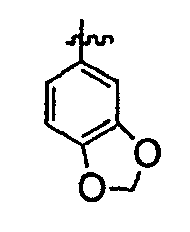
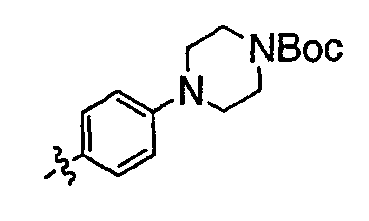
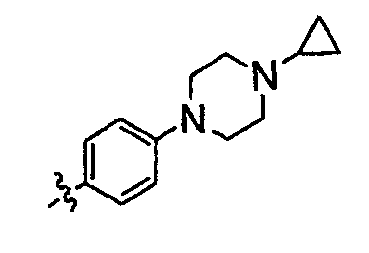
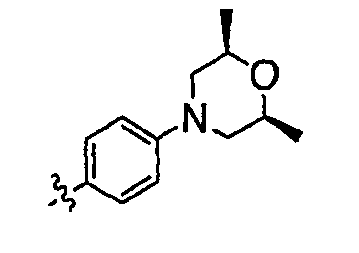
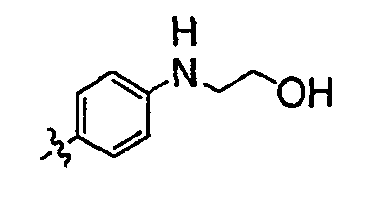

Пример 38
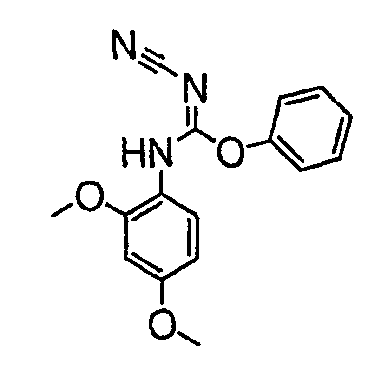
N-Циано-N'-(2,4-диметоксифенил)-О-фенилизомочевина. К суспензии дифенилцианокарбонимидата (3,0 г, 12,59 ммоль) в изопропаноле (15 мл) добавляют 2,4-диметоксианилин (2,02 г, 13,22 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 24-48 час. Твердое вещество отделяют фильтрованием, промывают изопропанолом и сушат в вакууме, получая при этом указанное в заголовке соединение в виде коричневого твердого вещества (3,55 г, выход 95%).
1Н-ЯМР (500 МГц, ДМСО-d6) 10,60 (шир.с, 1Н), 7,52-7,40 (м, 3Н), 7,35-7,07 (м, 3Н), 7,00 (д,д, 1Н), 6,85 (дд, 1Н), 3,81 (с, 3Н) м.д.; ЖХ-МС 289,12 (М+Н); ВЭЖХ (способ А) 3,32 мин.
Аналогично получают следующие соединения:
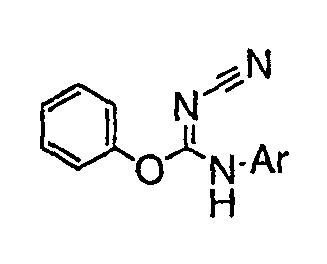
500 МГц
(растворитель)
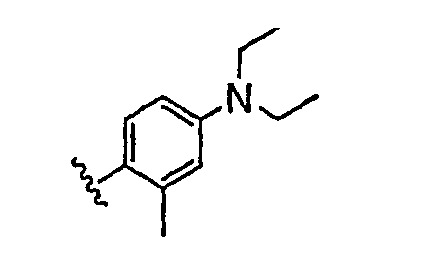
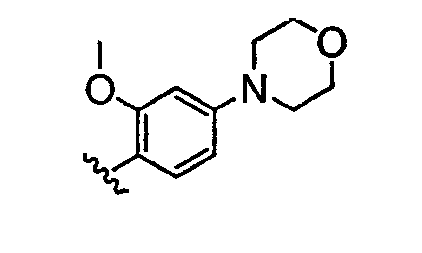
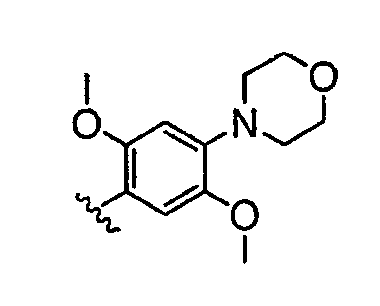
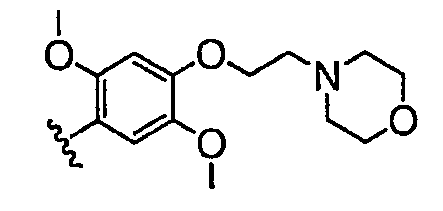
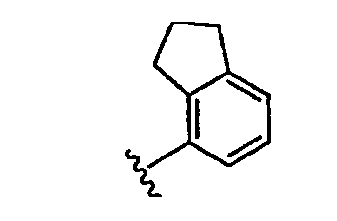

Пример 39
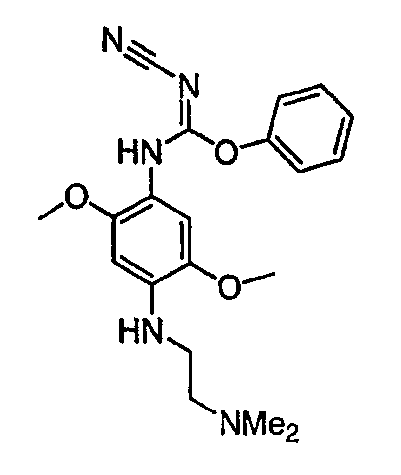
N-Циано-1-[4-(2-диметиламиноэтиламино)-2,5-диметоксифенил]-2-фенилизомочевина. К раствору гидрохлорида N-(2-диметиламиноэтил)-2,5-диметоксибензол-1,4-диамина (0,05 г, 0,143 ммоль) в дистиллированной воде (1 мл) добавляют К2СО3 (0,065 г, 0,47 ммоль). Смесь разбавляют EtOAc (1 мл) и добавляют дифенилцианокарбонимидат (0,032 г, 0,136 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 18 час. Осадок отделяют фильтрованием и промывают минимальным количеством EtOAc, получая при этом указанное в заголовке соединение в виде светло-красного твердого вещества (0,015 г, выход 29%).
1Н-ЯМР (500 МГц, ДМСО-d6) 10,11 (с, 1Н), 7,51-7,33 (м, 2Н), 7,31-6,98 (м, 3Н), 6,81-6,64 (м, 1Н), 6,28 (шир.с, 1Н), 4,87 (шир.с, 1Н), 3,85-3,62 (м, 5Н), 3,12 (с, 3Н), 2,17 (с, 6Н) м.д.; ЖХ-МС 384,3 (М+Н); ВЭЖХ 1,9 мин (способ А).
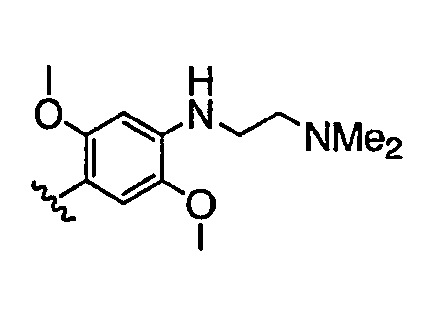
Пример 40
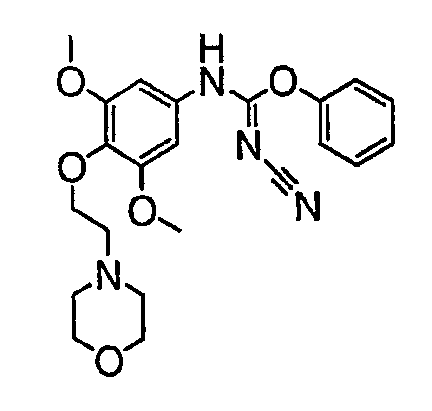
N-Циано-1-[3,5-диметокси-4-(2-морфолин-4-илэтокси)фенил]-2-фенилизомочевина. Раствор 3,5-диметокси-4-(2-морфолин-4-илэтокси)фениламина (0,62 ммоль) в изопропаноле (5 мл) и триэтиламине (0,25 мл, 1,79 ммоль) перемешивают при комнатной температуре в течение 10 мин. Добавляют раствор дифенилцианокарбонимидат (163 мг, 0,68 ммоль) в изопропаноле (1 мл) и реакционную смесь нагревают при 60°С в течение 3 час. Растворитель выпаривают и остаток очищают колоночной хроматографией с элюированием от 2% до 5% МеОН в CH2Cl2, получая при этом указанное в заголовке соединение в виде желтого твердого вещества (0,275 г, выход количественный).
1Н-ЯМР (500 МГц, ДМСО-d6) 10,9-10,49 (шир., 1Н), 7,45 (т, 2Н), 7,35-7,25 (м, 3Н), 5,76 (с, 2Н), 4,0-3,94 (м, 2Н), 3,76 (с, 6Н), 3,68-3,50 (шир. с, 4Н), 2,80-2,35 (м, 6Н) м.д.; ЖХ-МС 427,18 (М+Н); ВЭЖХ 1,78 мин.
500 МГц
(растворитель)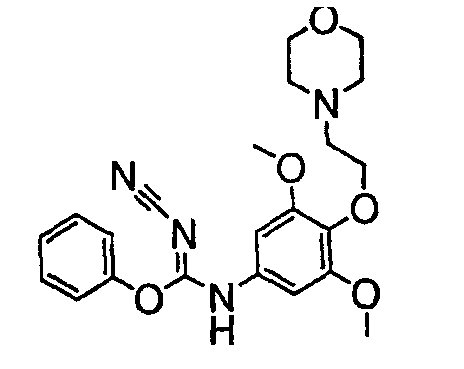
Пример 41
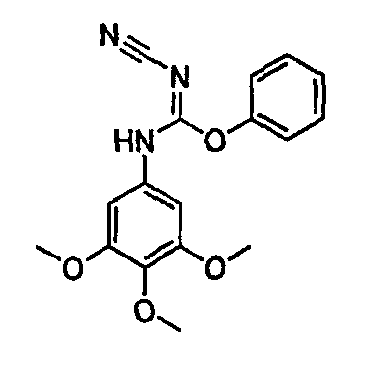
N-Циано-N'-(3,4,5-триметоксифенил)-О-фенилизомочевина. Смесь 3,4,5-триметоксианилина (1,83 г, 10 ммоль) и дифенилцианокарбонимидата (2,62 г, 11 ммоль) в изопропаноле (30 мл) перемешивают при 100-110°С в течение 1 часа. Реакционную смесь охлаждают и фильтруют при промывании эфиром, получая при этом указанное в заголовке соединение (2,79 г, выход 85%) в виде белого твердого вещества.
1Н-ЯМР (500 МГц, ДМСО-d6) 10,8 (с, 1Н), 7,45 (т, 2Н), 7,31 (м, 3Н), 6,83 (с, 2Н), 3,76 (с, 6Н), 3,65 (с, 3Н) м.д.; МС (FIA) 328,1 (М+Н); ВЭЖХ (способ А) 3,211 мин.
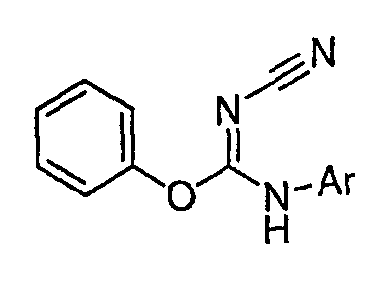
Аналогично получают следующие соединения:
500 МГц
(растворитель)
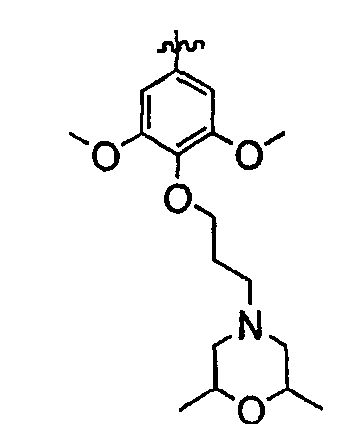
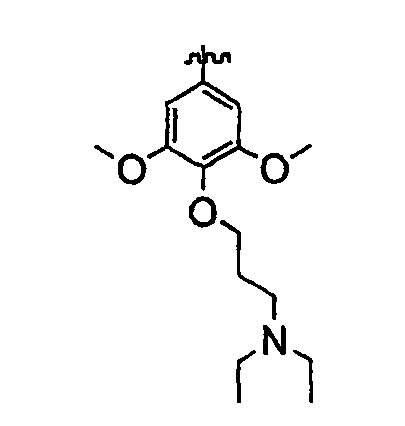
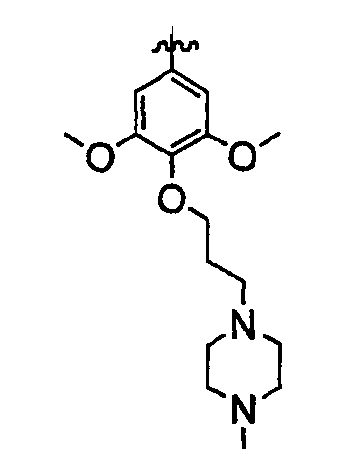
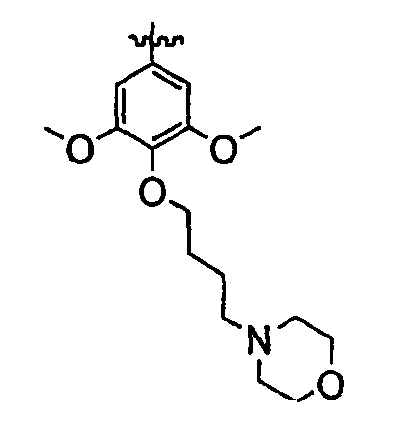
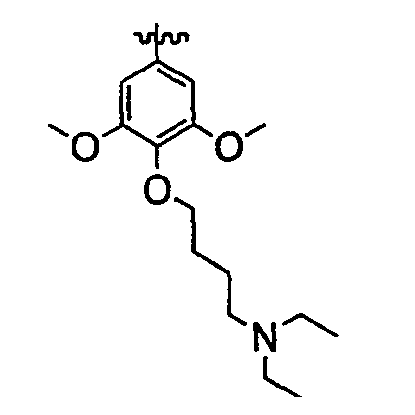
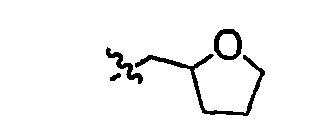
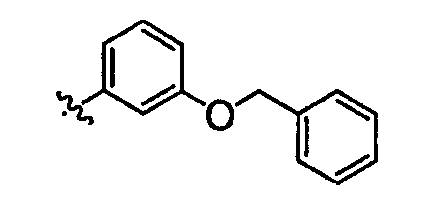
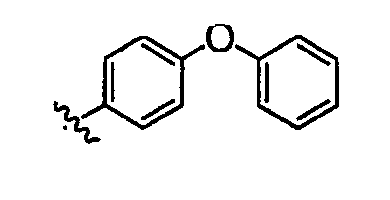
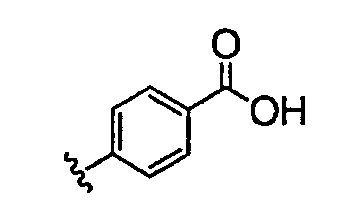
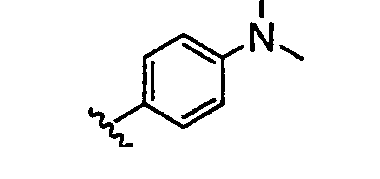
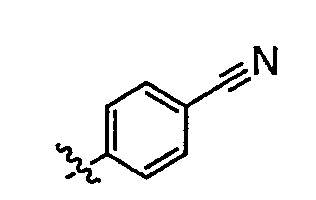
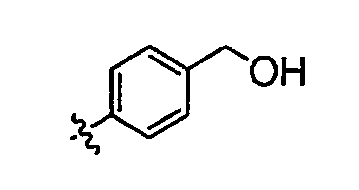
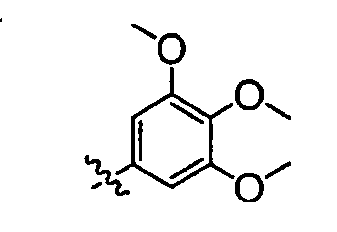
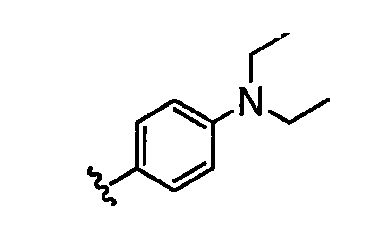
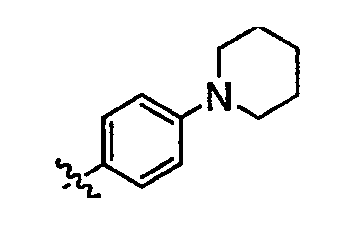
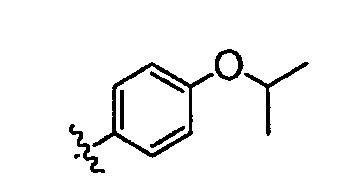
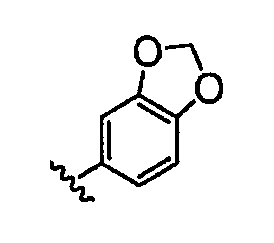
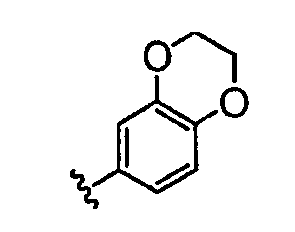

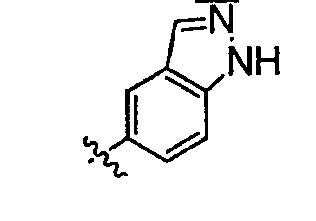
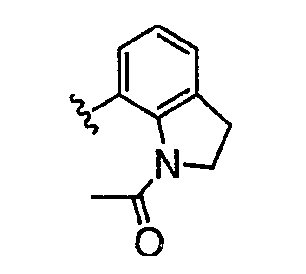




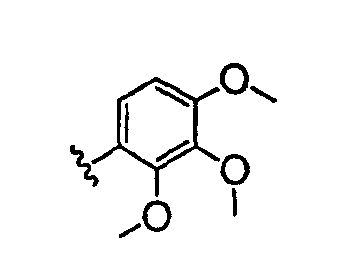
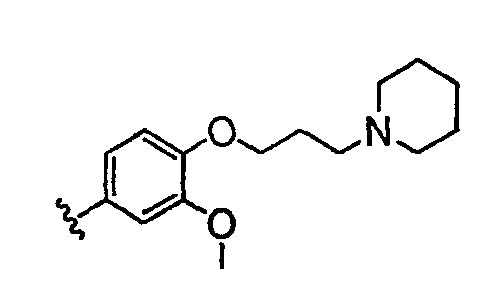
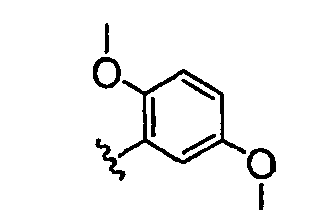
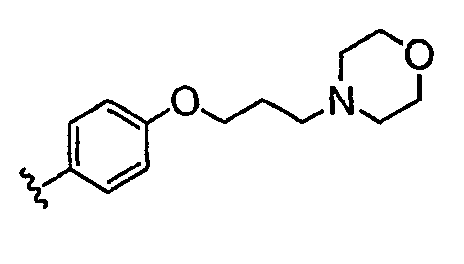
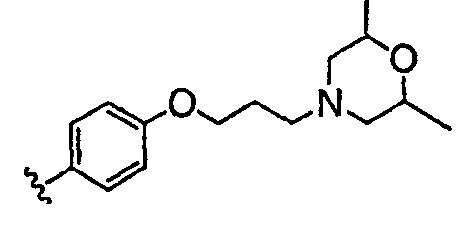
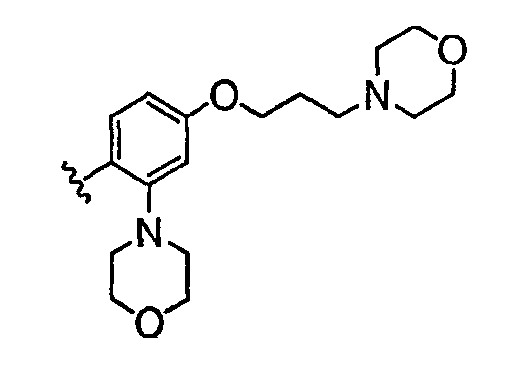
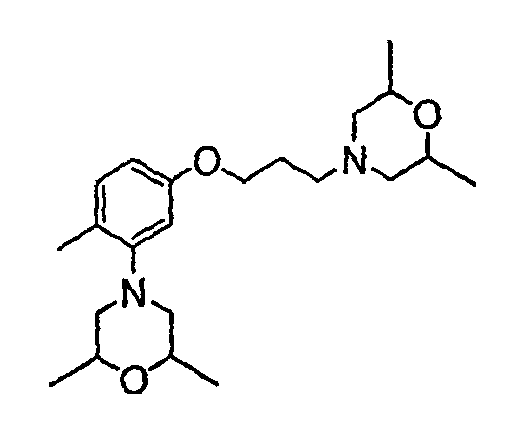
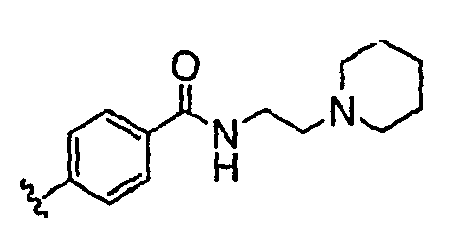
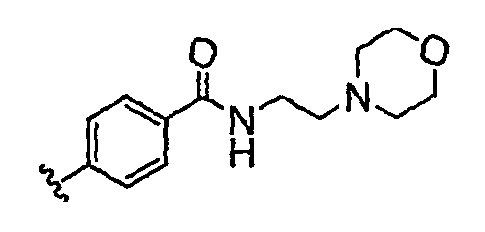
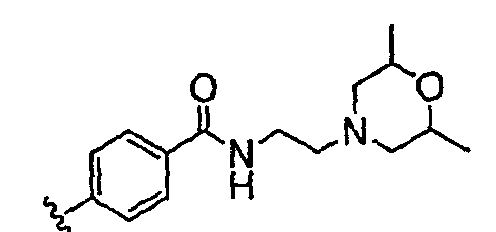
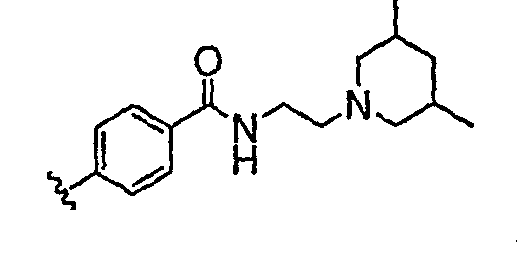
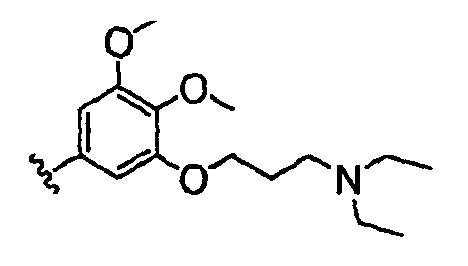
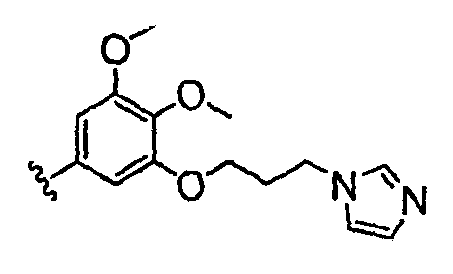
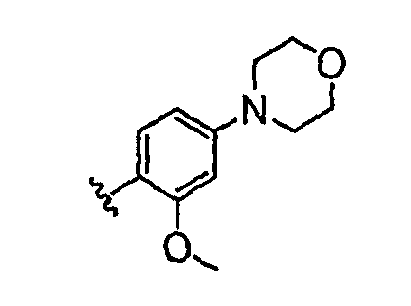
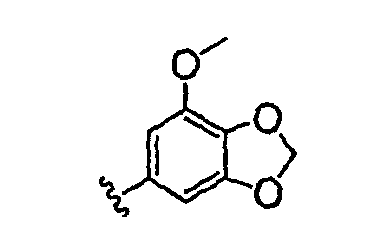
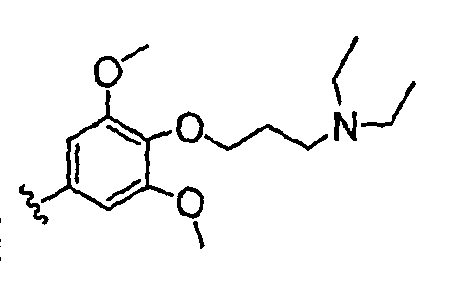


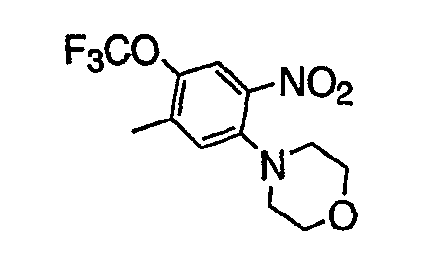

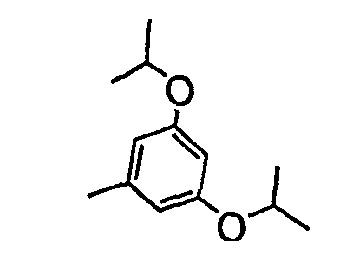
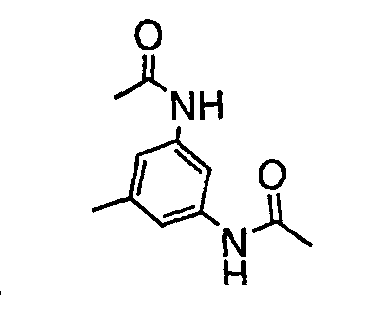
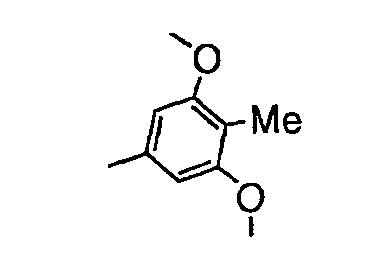

Ниже приводятся примеры способов, используемых при получении диаминотриазолов. Примеры служат также для описания нескольких способов очистки. Данные для указанных соединений содержатся в приведенной ниже таблице.
Пример 42
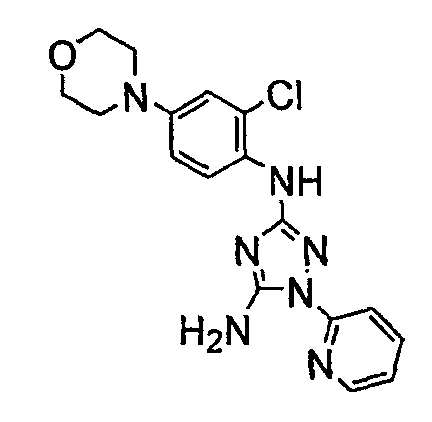
Способ А
N3-(2-Хлор-4-морфолин-4-илфенил)-1-пиридин-2-ил-1Н-[1,2,4]триазол-3,5-диамин. Смесь N-циано-N'-(2-хлор-4-морфолинфенил)-О-фенилизомочевины (0,10 г, 0,28 моль) и 2-гидразинопиридина (0,046 г, 0,42 ммоль) в изопропаноле (3 мл) нагревают при 115°С в течение 20 час. Осадок отфильтровывают, промывают изопропанолом и очищают флэш-хроматографией (SiO2) при элюировании смесью 2:98 метанол:дихлорметан, получая при этом указанное в заголовке соединение (0,080 г, выход 79%) в виде белого твердого вещества.
Пример 43
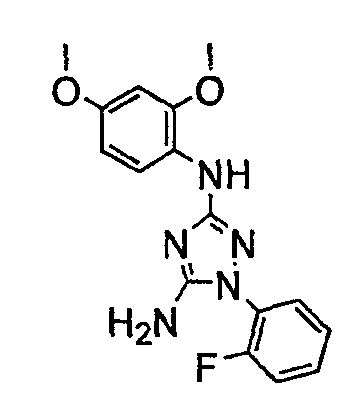
Способ А*
N3-(2,4-Диметоксифенил)-1-(2-фторфенил)-1Н-[1,2,4]триазол-3,5-диамин. Смесь N-циано-N'-(2,4-диметоксифенил)-О-фенилизомочевины (0,10 г, 0,34 ммоль), гидрохлорида 2-фторфенилгидразина (0,08 г, 0,50 ммоль) и триэтиламина (0,01 мл, 0,68 ммоль) в изопропаноле (3 мл) нагревают при 100°С в течение 18 час и упаривают. Очистка флэш-хроматографией (SiO2) с элюированием смесью 1:1 этилацетат:гексаны дает указанное в заголовке соединение (0,10 г, выход 85%).
Пример 44
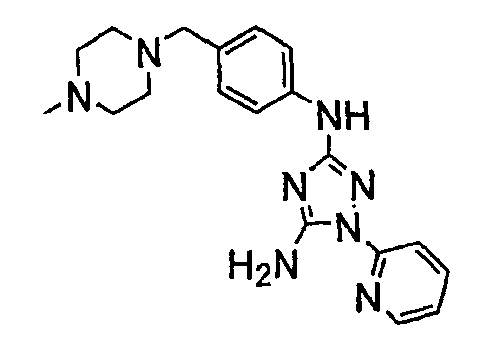
Способ А**
4-[5-Амино-3-(3,4-диметоксифениламино)[1,2,4]триазол-1-ил]бензонитрил. К N-циано-N'-(3,4-диметоксифенил)-О-фенилизомочевине (0,215 г, 0,723 ммоль) в изопропаноле (3 мл) добавляют 0,184 г (1,5 эквивалента) гидрохлорида 4-гидразинобензонитрила с последующим добавлением 152 мкл (1,5 эквивалента) триэтиламина и каталитического количества (0,2 эквивалента) 4-диметиламинопиридина. Реакционную смесь перемешивают на протяжении ночи при 100 градусах Цельсия. Реакционную смесь концентрируют досуха и очищают колоночной хроматографией с обращенной фазой. Чистые фракции сушат, получая при этом 4-[5-амино-3-(3,4-диметоксифениламино)[1,2,4]триазол-1-ил]бензонитрила в виде рыжевато-коричневого твердого вещества (18,3 мг, выход 7,5%).
Способ В
N3-[4-(4-Метилпиперазин-1-илметил)фенил]-1-пиридин-2-ил-1Н-[1,2,4]триазол-3,5-диамин. Смесь N-циано-N'-(4-(4-метил)пиперазинилметилфенил)-О-фенилизомочевины (0,60 г, 1,72 ммоль) и 2-гидразинопиридина (0,23 г, 2,06 ммоль) в изопропаноле (8 мл) нагревают микроволновой печкой (прибор Emrys) при 180°С в течение 10 мин, затем упаривают. Очистка флэш-хроматографией (SiO2) с элюированием смесью 0,2:2:98 NH4OH:метанол:дихлорметан дает указанное в заголовке соединение (0,54 г, выход 87%) в виде светло-рыжевато-коричневого твердого вещества.
Пример 45
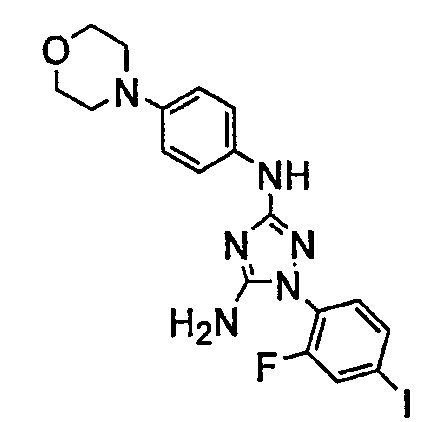
Способ С
1-(2-фтор-4-иодфенил)-N3-(4-морфолин-4-илфенил)-1Н-[1,2,4]триазол-3,5-диамин. Смесь N-циано-N'-(4-морфолинофенил)-О-фенилизомочевины (0,69 г, 2,14 ммоль) и 2-фтор-4-иодфенилгидразина (0,65 г, 2,57 ммоль) в диметилацетамиде (4 мл) нагревают при 120°С в течение 24 час. Реакционную смесь разбавляют этилацетатом, промывают водой (3 раза) и насыщенным раствором соли, сушат (сульфатом натрия) и упаривают. Очисткой флэш-хроматографией (SiO2) с элюированием смесью 4:96 метанол:дихлорметан с последующей полупрепаративной ВЭЖХ получают указанное в заголовке соединение (0,03 г, выход 5%) в виде белого лиофилизата.
Пример 46
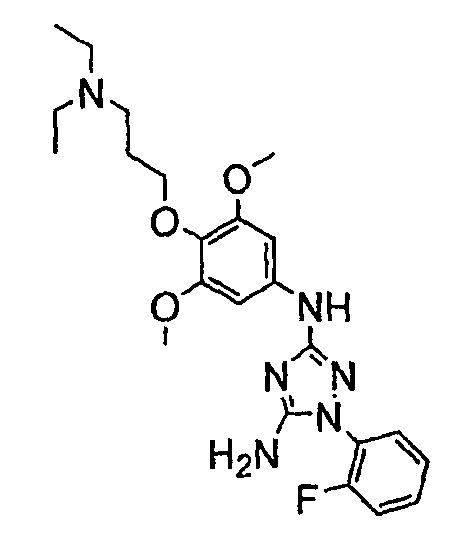
Способ С*
N3-[4-(3-Диэтиламинопропокси)-3,5-диметоксифенил]-1-(2-фторфенил)-1Н-[1,2,4]триазол-3,5-диамин. Раствор гидрохлорида 2-фторфенилгидразина (0,15 г, 0,91 ммоль) и триэтиламина (0,13 мл, 0,91 ммоль) в диметилацетамиде (1 мл) перемешивают 0,5 часа при комнатной температуре. Добавляют N-циано-N'-(4-(3-диэтиламинопропокси)-3,5-диметоксифенил)-О-фенилизомочевину (0,30 г в смеси с солью триэтиламина и трифторуксусной кислоты, 0,45 ммоль) в диметилацетамиде (1 мл) и реакционную смесь перемешивают при 120°С в течение 20 час. Осадок удаляют и фильтрат промывают эфиром. Оставшуюся водную фазу упаривают и очищают полупрепаративной ВЭЖХ, получая при этом указанное в заголовке соединение (0,01 г, выход 4%) в виде белого лиофилизата.
Пример 47
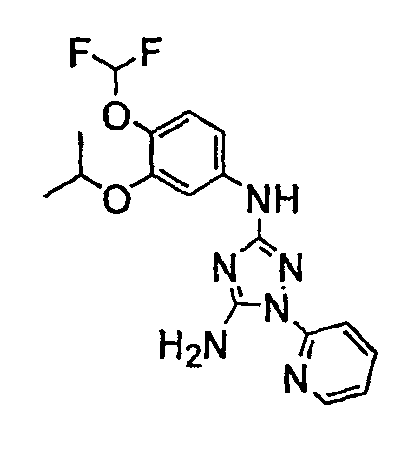
Способ D
N-(4-Дифторметокси-3-изопропоксифенил)-1-пиридин-2-ил-1Н-[1,2,4]триазол-3,5-диамин. Смесь N-циано-N'-(дифторметокси-3-изопропилоксифенил)-О-фенилизомочевины (0,10 г, 0,28 ммоль) и 2-гидразинопиридина (0,06 г, 0,56 ммоль) в диметилацетамиде (3 мл) нагревают на микроволновом приборе при 220°С в течение 15 мин. Реакционную смесь охлаждают, выливают в смесь лед-вода и перемешивают в течение 0,5 часа. Осадок отделяют фильтрованием, промывают холодной водой и сушат, получая при этом указанное в заголовке соединение (0,10 г, выход 97%) в виде светло-розового твердого вещества.
Пример 48
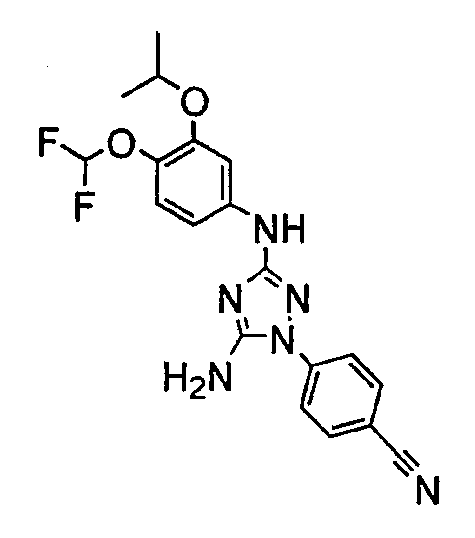
Способ D*
4-[5-Амино-3-(4-дифторметокси-3-изопропоксифениламино)-[1,2,4]триазол-1-ил]бензонитрил. Смесь N-циано-N'-(3-дифторметокси-4-изопропилоксифенил)-О-фенилизомочевины (0,10 г, 0,28 ммоль), 4-цианофенилгидразина (0,09 г, 0,55 ммоль) и триэтиламина (0,08 мл, 0,55 ммоль) в диметилацетамиде (3 мл) нагревают в микроволновом приборе при 220°С в течение 5 мин. Охлажденную реакционную смесь выливают в смесь лед-вода и сырой продукт выделяют фильтрованием. Очистка полупрепаративной ВЭЖХ дает указанное в заголовке соединение (0,07 г, выход 51%) в виде светло-оранжевого твердого вещества.
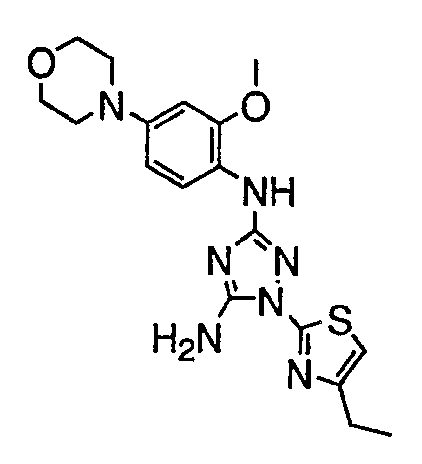
Пример 49
Способ Е
1-(4-Этилтиазол-2-ил)-N3-(2-метокси-4-морфолин-4-илфенил-1Н-[1,2,4]триазол-3,5-диамин. Смесь N-циано-N'-(2-метоксифенил)-О-фенилизомочевины (0,10 г, 0,28 ммоль), (4-этилтиазол-2-ил)гидразина (0,060 г, 0,56 ммоль) и DMAP (несколько кристаллов) в диметилацетамиде (3 мл) нагревают на микроволновой аппаратуре при 220°С в течение 7 мин. Очистка полупрепаративной ВЭЖХ дает указанное в заголовке соединение (0,01 г, выход 8%).
Пример 50
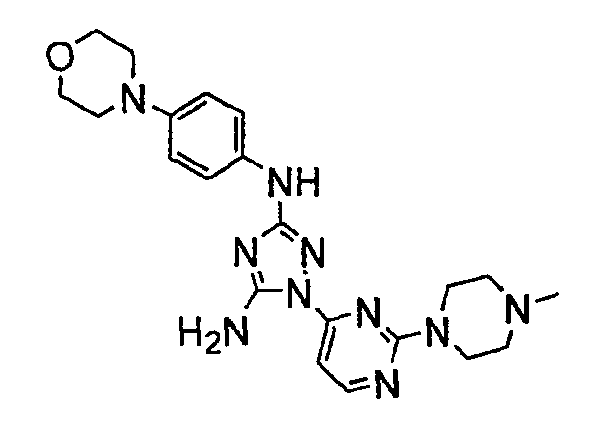
Способ G
1-[2-(4-Метилпиперазин-1-ил)пиримидин-4-ил]-N3-(4-морфолин-4-илфенил)-1Н-[1,2,4]триазол-3,5-диамин. Смесь N-циано-N'-(4-морфолинофенил)-О-фенилизомочевины (0,10 г, 0,31 ммоль) и [2-(4-метилпиперазин-1-ил)пиримидин-4-ил]гидразина (0,08 г, 0,34 ммоль) в N-метилпирролидиноне (3 мл) нагревают в микроволновом аппарате при 220°С в течение 5 мин. Очистка полупрепаративной ВЭЖХ дает указанное в заголовке соединение (0,09 г, выход 2%).
Пример 51
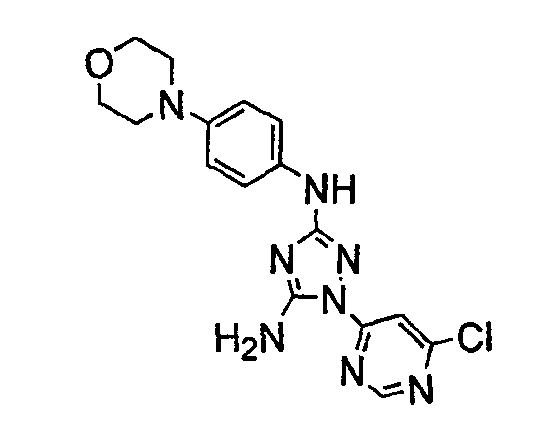
Способ G*
1-(6-Хлорпиримидин-4-ил)-N3-(4-морфолин-4-илфенил)-1Н-[1,2,4]триазол-3,5-диамин. Смесь N-циано-N'-(4-морфолинофенил)-О-фенилизомочевины (8,76 г, 27,2 ммоль, 4-хлор-6-гидразинопиримидина (4,12 г, 28,5 ммоль) и диизопропилэтиламина (18,9 мл, 109 ммоль) в N-метилпирролидиноне (50 мл) нагревают в микроволновом аппарате при 220°С в течение 5 мин, охлаждают, выливают в смесь лед-вода, перемешивают 0,5 часа, фильтруют для получения сырого продукта (9,30 г). Очистка флэш-хроматографией (SiO2) с элюированием смесью 2:98 метанол:дихлорметан дает указанное в заголовке соединение (3,91 г, выход 39%) в виде желтого порошка.
Пример 52
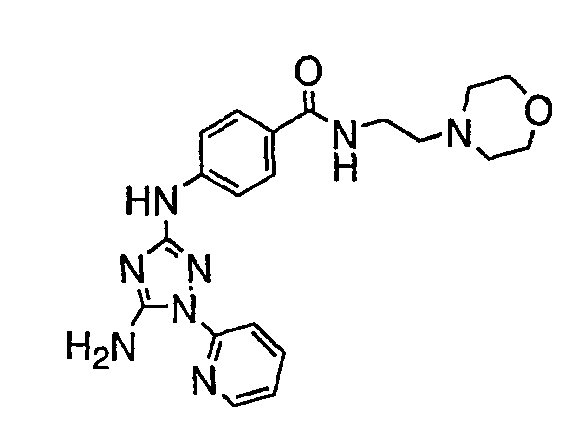
Способ Н
4-(5-Амино-1-пиридин-2-ил-1Н-[1,2,4]триазол-3-иламино)-N-(2-морфолин-4-илэтил)бензамид. К 4-(5-амино-1-пиридин-2-ил-1Н-[1,2,4]триазол-3-иламино)бензойной кислоте (4,635 г, 15,643 ммоль) в тетрагидрофуране (50 мл) добавляют (7,12 г, 1,2 эквивалента) HBTU с последующим добавлением 2-морфолин-4-илэтиламина (2,24 г, 1,1 эквивалента) и триэтиламина (5,45 мл, 2,5 эквивалента). Реакционную смесь перемешивают на протяжении ночи при комнатной температуре. Твердое вещество отфильтровывают, промывают тетрагидрофураном, холодным этанолом, водой, этанолом и, наконец, эфиром. Соединение сушат, получая при этом 4-(5-амино-1-пиридин-2-ил-1Н-[1,2,4]триазол-3-иламино)-N-(2-морфолин-4-илэтил)бензамид (1,17 г, выход 18%) в виде белого твердого вещества. Органические фазы содержали продукт, который дополнительно не выделяли.
Процедуры очистки:
1. Осаждение из растворителя
2. Силикагель
3. Полупрепаративная ВЭЖХ
Время удерживания определяли по ЖХ-МС, если не указано иначе.
(М+Н)
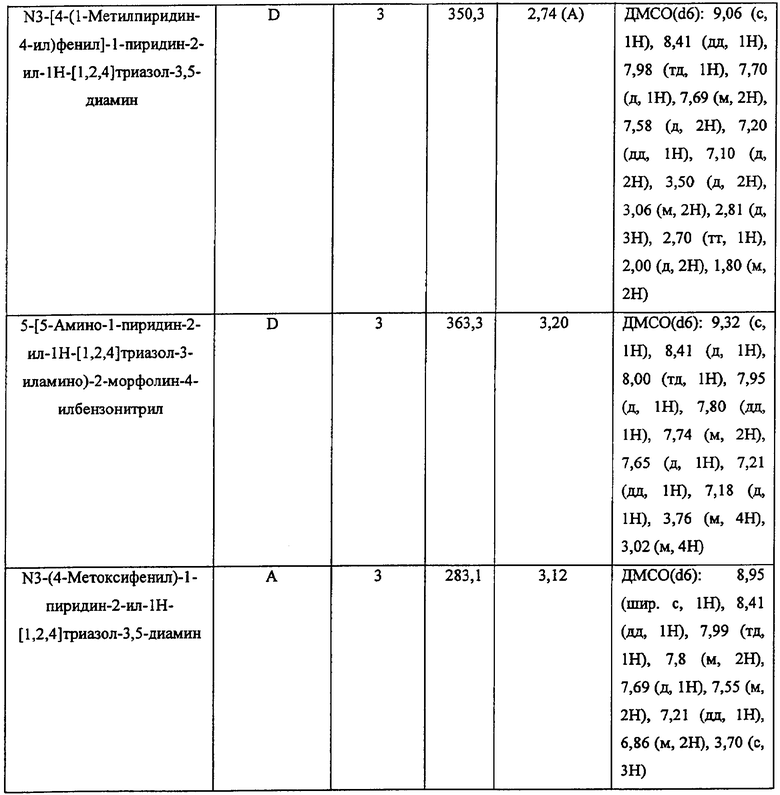
ДМСО(d6): 8,43 (дд, 1H), 8,02 (тд, 1H), 7,93 (шир. с, 2H), 7,86 (д, 1H), 7,65 (д, 1H), 7,54
ДМСО: 8,9 (с, 1H), 8,46-8,26 (д,д, 1H), 8,06-7,88 (м, 1Н), 7,72-7,52 (м, 3H), 7,41-7,24 (д, 1H), 7,24-7,10 (д,д,
(ДМСО-d6): 9,0 (с, 1H), 8,42 (д, 1H), 7,98 (т, 1H), 7,70 (с, 2H), 7,66 (д, 1H), 7,21 (т, 1H), 7,05 (с, 2H), 3,79 (с,
Пример 53
Схема 20
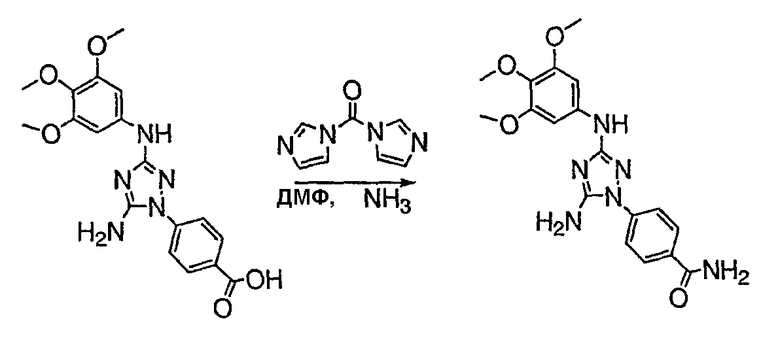
4-[5-Амино-3-(3,4,5-триметоксифениламино)-[1,2,4]триазол-1-ил]бензамид. При 0°С 1,1'-карбонилдиимидазол (83 мг, 0,5 ммоль) добавляют к раствору 4-[5-амино-3-(3,4,5-триметоксифениламино)-[1,2,4]триазол-1-ил]бензойной кислоты (100 мг, 0,25 ммоль) в ДМФ (3 мл). Реакционную смесь перемешивают при комнатной температуре в течение 1 часа и добавляют аммиак (7,0 М в МеОН, 1 мл). Реакционную смесь перемешивают при комнатной температуре в течение 2 дней. Смесь затем очищают полупрепаративной ВЭЖХ. FIA-МС: m/e=385,1 (М+Н), 381,1 (ES-), Rt=3,60 мин (способ А). 1Н ЯМР (500 МГц, ДМСО(d6)): 8,87 (с, 1Н), 8,02 (шир.с, 1Н), 7,99 (д, 2Н), 7,67 (д, 2Н), 7,39 (шир.с, 1Н), 7,00 (с, 2Н), 6,62 (с, 2Н), 3,78 (с, 6Н), 3,65 (с, 3Н).
Схема 21
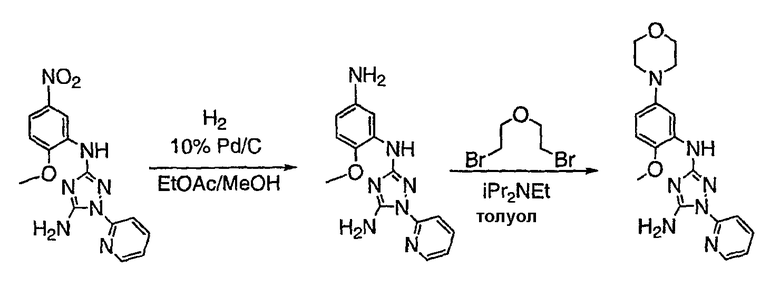
N3-(5-Амино-2-метоксифенил)-1-пиридин-2-ил-1Н-[1,2,4]триазол-3,5-диамин и N3-(2-метокси-5-морфолин-4-илфенил)-1-пиридин-2-ил-1Н-[1,2,4]триазол-3,5-диамин
Гидрирование N3-(5-нитро-2-метоксифенил)-1-пиридин-2-ил-1Н-[1,2,4]триазол-3,5-диамин 10% Pd/C в смешанном растворителе EtOAc/МеОН (1:1) дает N3-(5-амино-2-метоксифенил)-1-пиридин-2-ил-1Н-[1,2,4]триазол-3,5-диамин.
FIA-МС: m/e=298,2 (М+Н). Rt=2,40 мин (способ А). 1Н ЯМР (500 МГц, ДМСОd-6): 9,93 (шир.с, 2Н), 8,45 (дд, 1Н), 8,33 (д, 1Н), 8,00 (тд, 1Н), 7,88 (д, 1Н), 7,82 (шир.с, 2Н), 7,99 (с, 1Н), 7,26 (дд, 1Н), 7,08 (д, 1Н), 6,87 (дд, 1Н), 3,90 (с, 3Н).
Раствор N3-(5-Амино-2-метоксифенил)-1-пиридин-2-ил-1Н-[1,2,4]триазол-3,5-диамина (160 мг, 0,5 ммоль) и бис-(2-бромэтилового эфира (140 мг, 0,6 ммоль) и изопропилэтиламина (258 мг, 2 ммоль) в смеси толуола (30 мл) и DMAC (3 мл) нагревают при 110°С в течение 70 час. Смесь концентрируют. Остаток очищают ВЭЖХ, получая при этом указанное в заголовке соединение (26 мг).
FIA-МС: m/e=368,2 (М+Н). Rt=2,13 мин (способ А). 1Н ЯМР (500 МГц, ДМСОd-6): 8,43 (дд, 1Н), 8,03 (м, 2Н), 7,90 (м, 1Н), 7,68 (д, 1Н), 7,56 (шир.с, 1Н), 7,26 (дд, 1Н), 6,94 (д, 1Н), 6,64 (дд, 1Н), 3,80 (с, 3Н), 3,28 (шир.с, 4Н).
Схема 22
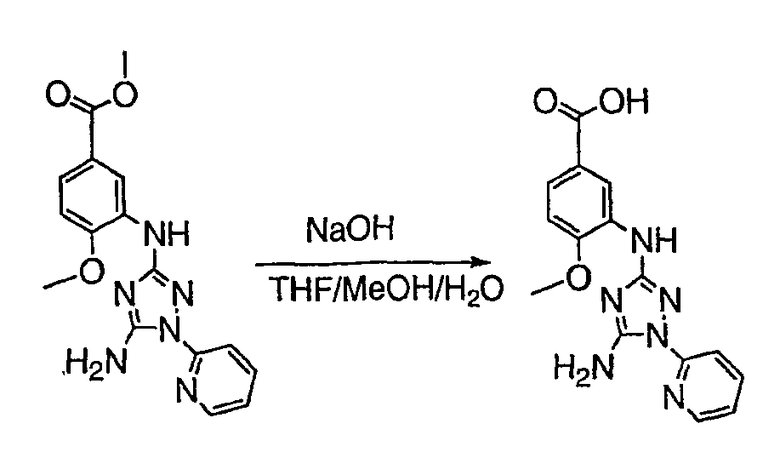
3-(5-Амино-1-пиридин-2-ил-1Н-[1,2,4]триазол-3-иламино)-4-метоксибензойная кислота
Суспензию метилового эфира 3-(5-амино-1-пиридин-2-ил-1Н-[1,2,4]триазол-3-иламино)-4-метоксибензойной кислоты (1,31 г, 3,85 ммоль) в смешанном растворителе ТГФ (40 мл), МеОН (5 мл) и воды (10 мл) обрабатывают 2 н. NaOH (8 мл) при 50°С в течение 1 часа. Реакционную смесь охлаждают до комнатной температуры, нейтрализуют 6 н. HCl. Образуется осадок, который собирают фильтрованием, получая при этом указанное в заголовке соединение (1,20 г) с выходом 95%. Небольшое количество дополнительно очищают ВЭЖХ.
FIA-МС: m/e=327,1 (М+Н), 325,0 (М-Н). Rt=3,09 мин (способ А). 1Н ЯМР (500 МГц, ДМСОd-6): 12,53 (с, 1Н), 8,81 (д, 1Н), 8,43 (дд, 1Н), 8,01 (тд, 1Н), 7,78 (с, 2Н), 7,65 (д, 1Н), 7,56 (с, 1Н), 7,353 (дд, 1Н), 7,23 (дд, 1Н), 7,07 (дд, 1Н), 3,92 (с, 3Н).
Схема 23

3-(5-Амино-1-пиридин-2-ил-1Н-[1,2,4]триазол-3-иламино)-4-метоксибензамид. Суспензию 3-(5-Амино-1-пиридин-2-ил-1Н-[1,2,4]триазол-3-иламино)-4-метоксибензойной кислоты (108 мг, 0,8 ммоль) в смешанном растворителе из ТГФ (50 мл) и ДМФ (15 мл) обрабатывают 1,1'- карбонилдиимидазолом (194 мг, 1,2 ммоль) при комнатной температуре. Спустя 1 час добавляют аммиак в МеОН (7,0 М, 1 мл). Реакционную смесь перемешивают при 50°С в течение 16 час, выливают в воду. Осадок собирают фильтрованием и далее очищают ВЭЖХ, получая при этом 3-(5-Амино-1-пиридин-2-ил-1Н-[1,2,4]триазол-3-иламино)-4-метоксибензамид (112 мг).
FIA-МС: m/e=326,1 (М+1). Rt=2,65 мин (способ А). 1Н ЯМР (500 МГц, ДМСОd-6): 8,65 (д, 1Н), 8,43 (дд, 1Н), 8,02 (тд, 1Н), 7,98 (м, 1Н), 7,83 (м, 1Н), 7,73 (д, 1Н), 7,70 (м, 1Н), 7,47 (дд, 1Н), 7,27 (ff, 1Н), 7,14 (м, 1Н), 7,04 (д, 1Н), 3,90 (с, 1Н).
Аналогично получают следующие соединения:
Способ А
Схема 24
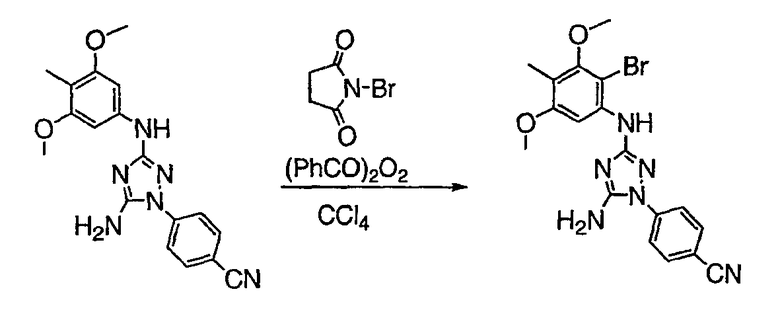
4-[5-Амино-3-(2-бром-3,5-диметокси-4-метилфениламино)-[1,2,4]триазол-1-ил]бензонитрил
К суспензии 4-(5-Амино-3-(3,5-диметокси-4-метилфениламино)-[1,2,4]триазол-1-ил]бензонитрила (155 мг, 0,44 ммоль) в CCl4 (10 мл) и бензоле (10 мл) добавляют N-бромсукцимид (100 г, 0,56 ммоль) и пероксид бензоила (10 мг). Реакционную смесь кипятят с обратным холодильником в течение 16 час. Концентрируют. Остаток очищают ВЭЖХ, получая при этом указанное в заголовке соединение (80 мг).
FIA-МС: m/e=429,1 и 431,1 (М+Н), 427,1 и 429,1 (М-Н). Rt=3,89 мин (способ А). 1Н ЯМР (500 МГц, ДМСО(d6)): 7,95 (д, 2Н), 7,82 (с, 1Н), 7,80 (д, 2Н), 7,37 (с, 1Н), 6,88 (шир.с, 2Н), 3,80 (с, 3Н), 3,69 (с, 3Н), 2,08 (с, 3Н).
Схема 25

N3-(4-Амино-5-морфолин-4-ил-2-трифторметоксифенил)-1-пиридин-2-ил-1Н-[1,2,4]триазол-3,5-диамин и N-[4-(5-амино-1-пиридин-2-ил-1Н-[1,2,4]триазол-3-иламино)-2-морфолин-4-ил-5-трифторметоксифенил]ацетамид
Раствор 1-циано-3-(5-морфолин-4-ил-4-нитро-2-трифторметоксифенил)-2-фенилизомочевины (3,60 г, 8 ммоль), 2-гидразинопиридина (2,0 г, 18,3 ммоль) в DMA (50 мл) перемешивают при 110°С в течение 18 час. Смесь упаривают в высоком вакууме и остаток суспендируют в воде (200 мл) и фильтруют. Твердое вещество суспендируют в смеси EtOH (50 мл) и EtOAc (30 мл), встряхивают с 10% Pd/C (835 мг) и 6 н. HCl (2 мл) в атмосфере Н2 (50 фунт/кв. дюйм) в течение 18 час. Реакционную смесь фильтруют через целит и целит промывают ДМФ. Фильтрат и промывные жидкости объединяют и перегоняют в высоком вакууме и затем лиофилизуют, получая при этом указанное в заголовке соединение (2,35 г). Небольшое количество дополнительно очищают ВЭЖХ для биологического анализа.
FIA-МС: m/e=437,2 (М+Н). Rt=3,14 мин (способ А). 1Н ЯМР (500 МГц, ДМСО(d6)): 8,41 (д, 1Н), 8,31 (м, 1Н), 8,0 (т, 1Н), 7,96 (м, 1Н), 7,75 (шир.с, 2Н), 7,61 (д, 1Н), 7,22 (дд, 1Н), 6,93 (м, 1Н), 5,0 (шир.с, 2Н), 3,79 (м, 4Н), 2,86 (м, 4Н).
Раствор N3-(4-амино-5-морфолин-4-ил-2-трифторметоксифенил)-1-пиридин-2-ил-1Н-[1,2,4]триазол-3,5-диамина (150 мг, 0,34 ммоль) в ДМФ (6 мл) обрабатывают пиридином (0,1 мл) и уксусным ангидридом (0,040 мл) при 23°С в течение 4 час. Смесь концентрируют и остаток очищают ВЭЖХ, получая при этом указанное в заголовке соединение (43 мг).
FIA-МС: m/e=479,2 (М+Н). Rt=3,30 мин (способ А). 1Н ЯМР (500 МГц, ДМСО(d6)): ДМСО(d6): 8,89 (с, 1Н), 8,56 (с, 1Н), 8,43 (дд, 1Н), 8,19 (с, 1Н), 8,02 (тд, 1Н), 7,88 (с, 1Н), 7,75 (м, 2Н), 7,65 (д, 1Н), 7,23 (дд, 1Н), 3,82 (м, 4Н), 2,88 (м, 4Н), 2,11 (с, 3Н).
Пример 54
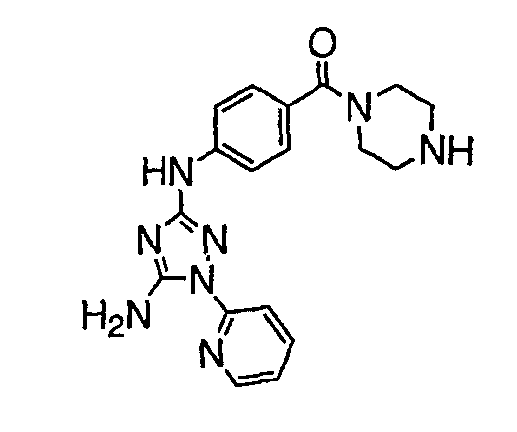
[4-(5-Амино-1-пиридин-2-ил-1Н-[1,2,4]триазол-3-иламино)фенил]пиперазин-1-илметанон. Смесь трет-бутилового эфира 4-[4-(5-амино-1-пиридин-2-ил-1Н-[1,2,4]триазол-3-иламино)бензоил]пиперазин-1-карбоновой кислоты (22,1 мг) и трифторуксусной кислоты (0,50 ил) перемешивают при комнатной температуре в течение 1 часа. Реакционную смесь концентрируют, получая при этом указанное в заголовке соединение (13,6 мг) в виде белого твердого вещества.
МС (ES+): m/z=365,1. 1Н ЯМР (CD3SOCD3, 500 МГц): δ 3,09-3,23 (м, 4Н), 3,63-3,76 (м, 4Н), 7,20-7,25 (м, 1Н), 7,41 (д, 2Н), 7,64-7,74 (м,4Н), 7,95-8,02 (м, 1Н), 8,39-8,44 (м, 1Н), 8,66-8,95 (м, 2Н), 9,44-9,48 (м, 1Н).
Пример 55
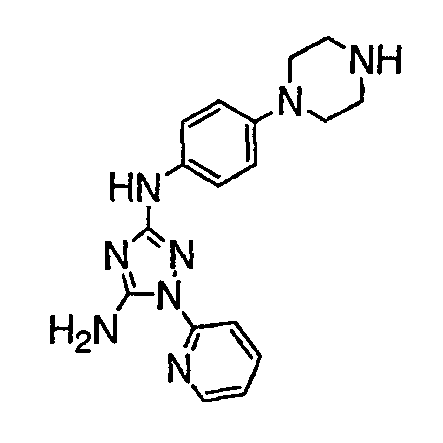
N3-(4-Пиперазин-1-илфенил)-1-пиридин-2-ил-1Н-[1,2,4]триазол-3,5-диамин. Указанное в заголовке соединение получают из трет-бутилового эфира 4-[4-(5-амино-1-пиридин-2-ил-1Н-[1,2,4]триазол-3-иламино)фенил]пиперазин-1-карбоновой кислоты по процедуре, описанной выше.
МС (ES+): m/z=337,20. 1Н ЯМР (CD3OD, 500 МГц): δ 3,34-3,41 (м, 8Н), 7,06 (д, 2Н), 7,30 (дд, 1Н), 7,53 (д, 2Н), 7,82 (д, 1Н), 7,97-8,02 (м, 1Н), 8,45-8,48 (м, 1Н).
Пример 56

1-(4-Аминометилфенил)-N3-[3,5-диметокси-4-(пиперидин-4-илокси)фенил]-1Н-[1,2,4]триазол-3,5-диамин. Указанное в заголовке соединение получают из бутилового эфира 4-{4-[5-амино-1-(4-цианофенил)-1Н-[1,2,4]триазол-3-иламино]-2,6-диметоксифенокси}пиперидин-1-карбоновой кислоты по процедуре, описанной выше.
МС (ES+): m/z=440,20. 1Н ЯМР (CD3OD, 500 МГц): δ 1,90-2,09 (м, 4Н), 3,13-3,21 (м, 2Н), 3,50-3,59 (м, 2Н), 3,82 (с, 6Н), 4,19 (с, 2Н), 4,29-4,35 (м, 1Н), 6,93 (с, 2Н), 7,61 (д, 2Н), 7,70 (д, 2Н).
Схема 26

Пример 57

4-[5-Амино-3-(4-морфлин-4-ил)фениламино-[1,2,4]триазол-1-ил]-3-фторбензонитрил. Смесь 1-(2-фтор-4-иодфенил)-N3-(4-морфолинофенил)-1Н-[1,2,4]триазол-3,5-диамина (0,48 г, 0,99 моль) и цианида меди (I) (0,09 г, 0,99 ммоль) в НМРА (3 мл) и нагревают при 55°С в течение 2 час, затем выливают в воду (75 мл) и фильтруют, промывая водой. Фильтровальный осадок суспендируют в хлороформе (100 мл) и метаноле (5 мл), кипятят с обратным холодильником в течение 2 час, охлаждают, фильтруют и выпаривают. Очистка полупрепаративной ВЭЖХ дает указанное в заголовке соединение (0,04 г, выход 9%) в виде светло-рыжевато-коричневого лиофилизата.
Аналогичным способом получают следующие соединения:
Пример 58
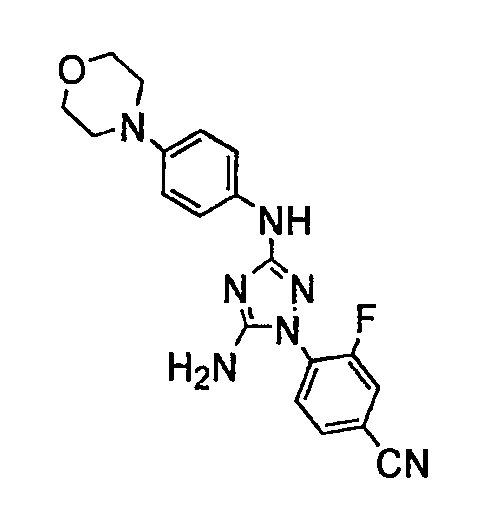
4-[5-Амино-3-(4-морфолин-4-илфениламино)-[1,2,4]триазол-1-ил]-3-фторбензонитрил
Смесь 1-(2-фтор-4-иодфенил)-N3-(4-морфолинофенил)-1Н-[1,2,4]триазол-3,5-диамина (0,48 г, 0,99 ммоль) и цианида меди (I) (0,09 г 0,99 ммоль) в НМРА (3 мл) нагревают при 55°С, затем выливают в воду (75 мл) и фильтруют, промывая водой. Фильтровальный осадок суспендируют в хлороформе (100 мл) и метаноле (5 мл), кипятят с обратным холодильником в течение 2 час, охлаждают, фильтруют и упаривают. Очистка полупрепаративной ВЭЖХ дает указанное в заголовке соединение (0,04 г, выход 9%) в виде светло-рыжевато-коричневого лиофилизата.
Схема 27
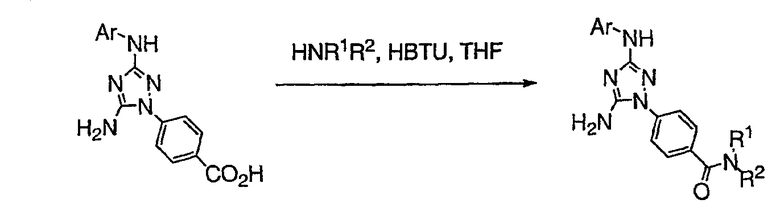
Пример 59
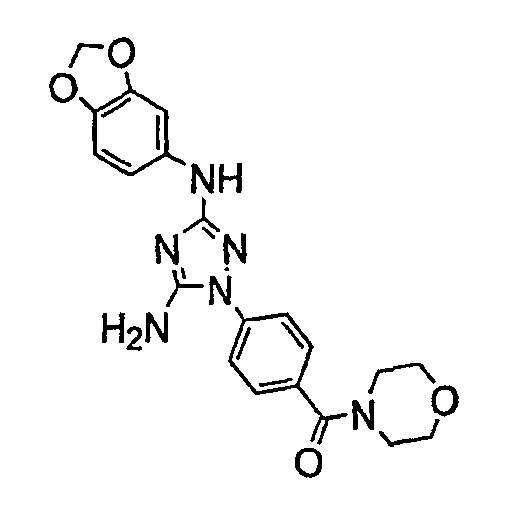
{4-[5-Амино-3-(бензо[1,3]диоксол-5-иламино)-[1,2,4]триазол-1-ил]фенил}морфолин-4-илметанон. Смесь 4-[5-амино-3-бензо[1,3]диоксол-5-иламино)[1,2,4]триазол-1-ил]бензойной кислоты (0,15 г, 0,44 ммоль), морфолина (0,05 мл, 0,55 ммоль) и HBTU (0,21 г, 0,55 ммоль) в ТГФ (5 мл) перемешивают при комнатной температуре в течение 4 час. Реакционную смесь разбавляют водой, экстрагируют смесью метанол/дихлорметан, сушат (сульфатом натрия) и упаривают. Очистка 2 последовательными полупрепаративными ВЭЖХ дает указанное в заголовке соединение (0,008 г, выход 5%) в виде светло-розового лиофилизата.
Аналогичным способом получают следующие соединения:
Пример 60
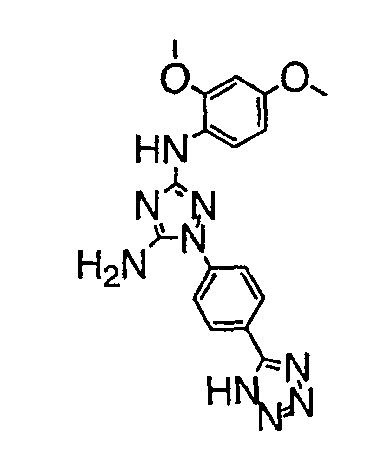
N3-(2,4-диметоксифенил)-1-[4-1Н-тетразол-5-ил)фенил]-1Н-[1,2,4]триазол-3,5-диамин
4-[5-Амино-3-(2,4-диметоксифениламино)-[1,2,4]триазол-1-ил]бензонитрил (52 мг, 0,15 ммоль) и триметилсилилазид (20 мг, 0,165 моль) суспендируют в 1 мл толуола с каталитическим количеством оксида дибутилолова и нагревают до 110°С в течение 18 часов. Толуол выпаривают и остаток очищают препаративной ВЭЖХ, получая при этом 13 мг продукта в виде соли с TFA.
Схема 28
(а) NMP, 220°С, микроволновая аппаратура, (b) Метод замещения А: амин, ТГФ, DIEA, кипячение с обратным холодильником; способ замещения В: амин, NMP, 220°С, микроволновая аппаратура.
Пример 61
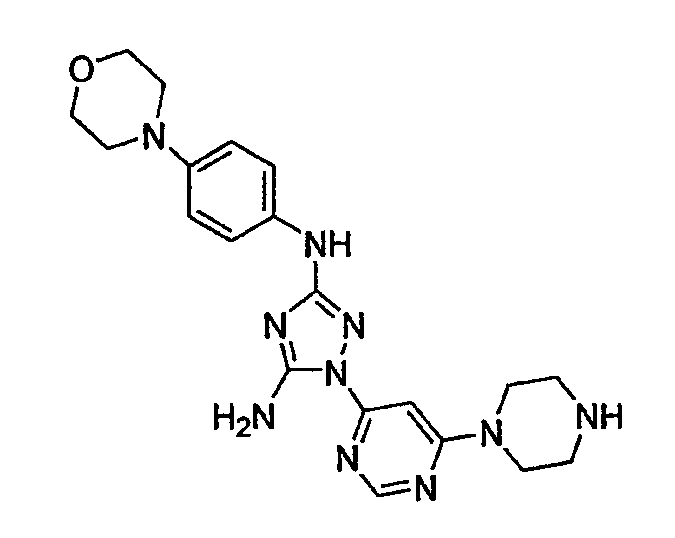
N3-(4-Морфолин-4-илфенил)-1-(6-пиперазин-1-илпиримидин-4-ил)-1Н-[1,2,4]триазол-3,5-диамин. Смесь 1-(6-хлорпиримидин-4-ил)-N3-(4-морфолин-4-илфенил)-1Н-[1,2,4]триазол-3,5-диамина (0,11 г, 0,30 ммоль), пиперазина (0,26 г, 3,0 ммоль) и диизопропилэтиламина (0,21 мл) в ТГФ (100 мл) перемешивают при кипячении с обратным холодильником в течение 3 час, затем охлаждают и упаривают. Очистка полупрепаративной ВЭЖХ дает указанное в заголовке соединение (0,13 г, выход 64%) в виде светло-желтого твердого вещества.
Следующие соединения получают с использованием указываемых аналогичных способов (схема 28).
Схема 29
(2) амин, DIEA, п-диоксан, 100°С, (b) гидразин, ТГФ, кипячение с обратным холодильником, (с) 4, ДМФ, 220°С (микроволновая аппаратура)
Следующие соединения получены, как указано и описано на схеме 29:
Пример 62
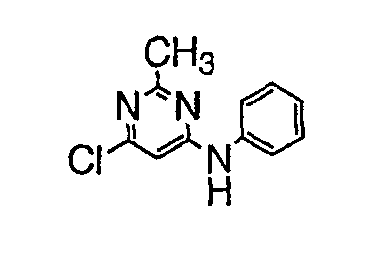
(6-Хлор-2-метилпиримидин-4-ил)фениламин
2-Метил-4,6-дихлорпиримидин (400 мг; 2,64 ммоль) кипятят с обратным холодильником в 10 мл п-диоксана с DIEA (575 мкл; 426 мг; 3,3 ммоль) и анилином (239 мкл; 247 мг; 2,65 ммоль) в течение 48 час в атмосфере N2. Растворитель удаляют при пониженном давлении и остаток распределяют между этилацетатом и 0,5 н. HCl. Органическую фракцию промывают водой, насыщенным раствором соли и сушат над Na2SO4 и растворитель удаляют при пониженном давлении. Сырой материал растирают с МТВЕ, фильтруют с отсасыванием для выделения и промывают еще раз МТВЕ и сушат на воздухе, получая при этом 375 мг белого твердого вещества, выход 64,5%. 1Н ЯМР (500 МГц, ДМСО-d6) d 8,9 (с, 1Н), 7,7 (с, 1Н), 7,55 (м, 2Н), 7,27 (т, 2Н), 6,95 (т, 1Н), 6,0 (с, 1Н), 2,25 (с, 3Н) м.д.
Пример 63
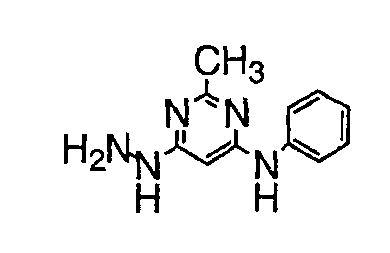
(6-Гидразино-2-метилпиримидин-4-ил)фениламин. (6-Хлор-2-метилпиримидин-4-ил)фениламин (2,83 г; 12,9 ммоль) кипятят с обратным холодильником в ТГФ (35 мл) с безводным гидразином (5 мл; 4,9 г; 153 ммоль) в атмосфере N2 в течение 26 час. Реакционную смесь охлаждают и растворитель удаляют при пониженном давлении. Остаток распределяют между этилацетатом и водой, причем органическую фазу промывают снова водой, насыщенным раствором соли и сушат (Na2SO4) и растворитель удаляют при пониженном давлении. Сырой материал растворяют в минимальном количестве метиленхлорида (горячего) и добавляют 100 мл горячих гексанов и материал перемешивают, в то время пока он охлаждается до температуры окружающей среды. Твердое вещество выделяют посредством фильтрования с отсосом и промывают еще раз гексанами и сушат на воздухе, получая при этом 2,5 г белого порошка, выход 89%.
1Н ЯМР (500 МГц, ДМСО-d6) d 8,9 (с, 1Н), 7,7 (с, 1Н), 7,5 (д, 2Н), 7,26 (т, 2Н), 6,9 (т, 1Н), 6,0 (с, 1Н), 4,15 (с, 2Н), 2,3 (с, 3Н), м.д.; МС (М+Н) 216.
Пример 64
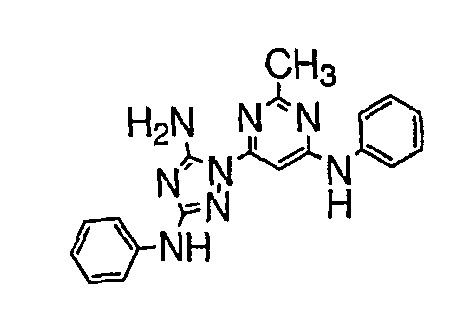
1-(2-Метил-6-фениламинопиримидин-4-ил)-N3-фенил-1Н-[1,2,4]триазол-3,5-диамин
(6-Гидразино-2-метилпиримидин-4-ил)фениламин (50 мг; 0,232 ммоль) нагревают в запаянном сосуде в 2 мл 2-пропанола и 60 мг N-циано-N'-фенил-О-фенилизомочевины в течение 8 час. Реакционную смесь охлаждают и гасят водой и фильтруют с отсасыванием для выделения твердого вещества. Сырой материал растирают с теплым 2-пропанолоом и собирают посредством фильтрования с отсасыванием, получая при этом 15 мг белого материала, выход 18%.
1Н ЯМР (500 МГц, ДМСО-d6) d 9,73 (с, 1Н), 9,17 (с, 1Н), 7,8 (с, 2Н), 7,7 (д, 2Н), 7,62 (д,2Н), 7,35 (т, 2Н), 7,25 (т, 2Н), 7,05 (т, 1Н), 6,83 (т, 1Н), 6,81 (с, 1Н), 2,53 (с, 3Н); МС (М+Н) 359.
Схема 30
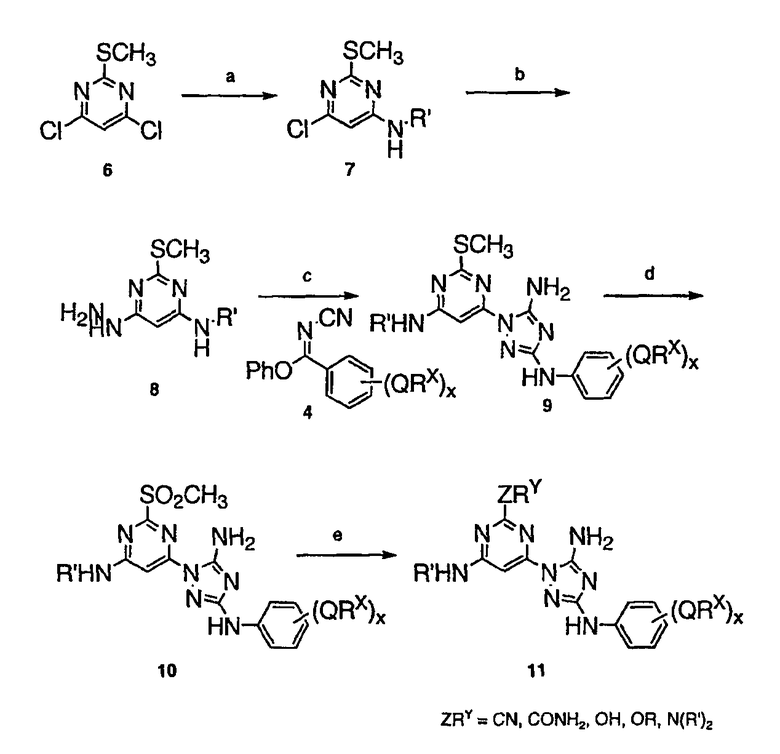
(а) Амин, DIEA, п-диоксан, 100°С, (b) гидразин, ТГФ, кипячение с обратным холодильником, (с) 4, ДМФ, 220°С (микроволновая аппаратура), (d) mCPBA ТГФ/п-диоксан, (е) для ZRY = CN, CONH2: i) KCN, ДМСО, ii) К2СО3, 30% Н2О2, ДМСО; для ZRY=OR: iii) NaOR, ДМФ; для ZRY=ОН; iv) NaOH, ДМФ; для ZRY=N(R')2, v) NH(R')2, ТГФ, 80°С.
Как описано в схеме 29, получают следующие соединения:
Пример 65
(6-Хлор-2-метилсульфанилпиримидин-4-ил)фениламин
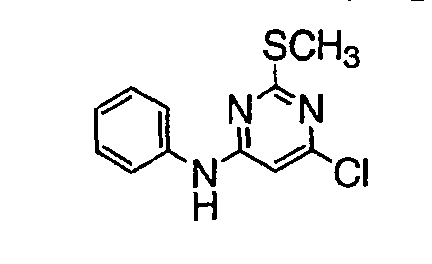
Смесь 2,00 г (10,2 ммоль) 2-тиометил-4,6-дихлорпиримидина нагревают до 100°С в 20 мл п-диоксана вместе с (1,8 мл; 1,35 г; 10,2 ммоль) DIEA и (0,93 мл; 0,96 г; 10,3 ммоль) анилина в атмосфере N2 в течение 24 час. Реакционную смесь охлаждают и растворитель удаляют при пониженном давлении. Остаток распределяют между водой и этилацетатом и органический слой промывают 0,1 н. HCl, водой, насыщенным раствором соли и сушат (Na2SO4); растворитель удаляют при пониженном давлении. Материал отверждается при стоянии с получением 2,57 г белого порошка, выход 88%. MS m/e (FIA+) 250/252.
(6-Гидразино-2-метилсульфанилпиримидин-4-ил)фениламин
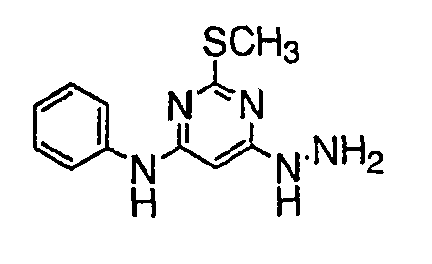
6-Хлор-2-метилсульфанилпиримидин-4-ил)фениламин (2,7 г; 11 ммоль) нагревают при кипячении с обратным холодильником в 25 мл ТГФ с безводным гидразином (2,5 мл; 2,44 г; 76 ммоль) в течение 5 час в атмосфере N2. Реакционную смесь охлаждают и растворитель удаляют при пониженном давлении. Смесь перемешивают с 100 мл воды, вследствие чего образуется белое твердое вещество. Материал собирают фильтрованием с отсасыванием, промывают еще раз водой и сушат на воздухе, получая при этом 2,69 г белого порошка, выход 95%.
МС m/e (FIA+) 246; 1Н ЯМР (500 МГц ДМСО-d6) d 9,05 (с, 1Н), 7,8 (с, 1Н), 7,5 (д, 2Н), 7,25 (2Н), 6,9 (т, 1Н), 5,85 (с, 1Н), 4,26 (шир. с, 2Н), 2,45 (с, 3Н) м.д.
1-(2-Метилсульфанил-6-фениламинопиримидин-4-ил)-N3-фенил-1Н-[1,2,4]триазол-3,5-диамин
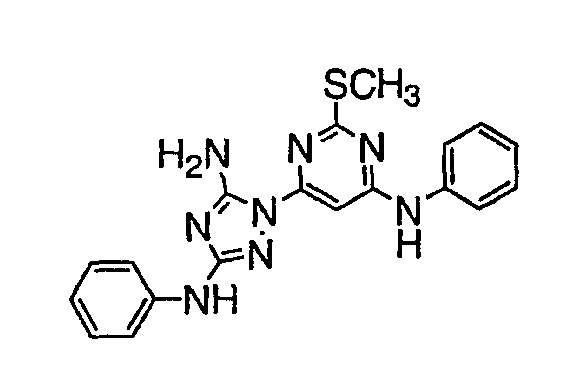
(6-Гидразино-2-метилсульфанилпиримидин-4-ил)фениламин (100 мг 0,40 ммоль) нагревают с N-циано-N'-фенил-О-фенилизомочевиной (96 мг, 0,44 ммоль) в 0,5 мл ДМСО в запаянной трубке в течение 4 часов при 100°С. Реакцию гасят водой и образовавшееся твердое вещество выделяют посредством фильтрования с отсасыванием. Твердое вещество промывают водой и переносят в круглодонную колбу с ацетонитрилом и растворитель удаляют при пониженном давлении. ВЭЖХ (градиент элюента: вода-ацетонитрил, 0,1% TFA) дает 15 мг бежевого порошка, выход 11%.
МС m/e (FIA+) 391, m/e (FIA-) 389; 1Н ЯМР (500 МГц ДМСО-d6) d 10,0 (шир. с, 1Н), 9,38 (шир. м, 1Н), 7,7 (д, 2Н), 7,63 (д, 2Н), 7,63 (д, 2Н), 7,36 (т, 2Н), 7,27 (т, 2Н), 7,08 (т, 1Н), 6,88 (т, 1Н), 6,77 (с, 1Н), 2,6 (с, 3Н) м.д.
1-(2-Метансульфонил-6-фениламинопиримидин-4-ил)-N3-фенил-1Н-[1,2,4]триазол-3,5-диамин
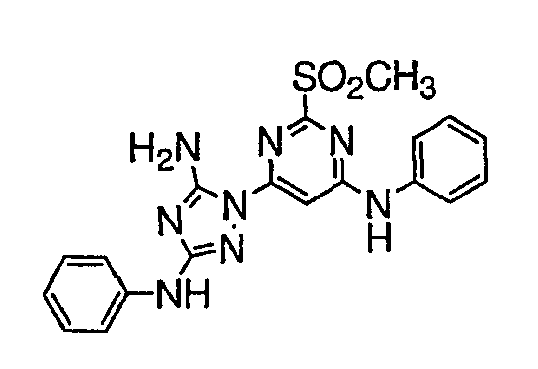
К 1-(2-метилсульфанил-6-фениламинопиримидин-4-ил)-N3-фенил-1Н-[1,2,4]триазол-3,5-диамину (240 мг; 0,615 ммоль) в 10 мл п-диоксана и 10 мл ТГФ добавляют 77% mCPBA (413 мг; 2,4 ммоль). Реакционную смесь перемешивают при температуре окружающей среды в течение 2 час. Реакционную смесь гасят в воде. Образовавшееся твердое вещество выделяют посредством фильтрования с отсасыванием и твердое вещество промывают еще раз водой. Твердое вещество переносят в круглодонную колбу с ацетонитрилом и растворитель удаляют при пониженном давлении. Образовавшееся твердое вещество растирают с МТВЕ и твердое вещество собирают посредством фильтрования с отсасыванием и сушат на воздухе, получая при этом 214 г не совсем белого порошка (выход 82,3%).
МС m/e (FIA+) 423, m/e (FIA-) 421; 1Н ЯМР (500 МГц ДМСО-d6) δ 10,4 (с, 1Н), 9,25 (с, 1Н), 7,72 (с, 1Н), 7,13 (т, 4Н), 7,43 (т, 2Н), 7,25 (т, 2Н), 7,15 (т, 1Н), 7,03 (с, 1Н), 6,85 (т, 1Н), 3,3 (с, 3Н) м.д.
4-(5-Амино-3-фениламино-[1,2,4]триазол-1-ил)-6-фениламинопиримидин-2-карбонитрил
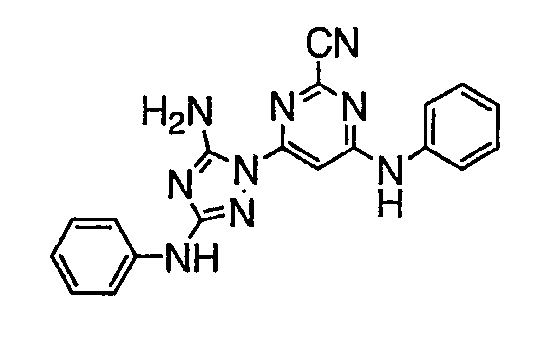
К раствору 1-(2-метансульфонил-6-фениламинопиримидин-4-ил)-N3-фенил-1Н-[1,2,4]триазол-3,5-диамина (50 мг; 0,12 ммоль) в 2 мл ДМСО добавляют 8 мг (0,13 ммоль) цианида калия и реакционную смесь перемешивают в течение 1 часа при температуре окружающей среды. Растворитель удаляют при пониженном давлении. Остаток растирают в воде и твердое вещество выделяют посредством фильтрования с отсасыванием. Колоночная хроматография (SiO2, 5% EtOH-CH2Cl2) с последующим растиранием с диэтиловым эфиром дает 5 мг не совсем белого порошка (выход 11%).
МС m/e (FIA+) 370, m/e (FIA-) 368; 1Н ЯМР (500 МГц ДМСО-d6) δ 10,25 (с, 1Н), 9,25 (с, 1Н), 7,63 (м, 6Н), 7,4 (т, 2Н), 7,25 (т, 2Н), 7,15 (т, 1Н), 7,07 (с, 1Н), 6,88 (т, 1Н) м.д.
Амид 4-(5-Амино-3-фениламино-[1,2,4]триазол-1-ил)-6-фениламинопиримидин-2-карбоновой кислоты
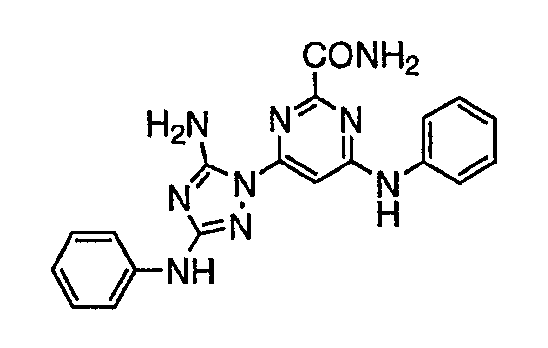
К 4-(5-амино-3-фениламино-[1,2,4]триазол-1-ил)-6-фениламинопиримидин-2-карбонитрилу (20 мг; 0,054 ммоль) в 500 мкл ДМСО добавляют при температуре окружающей среды безводный К2СО3 (5 мг; 0,036 ммоль) с последующим добавлением 10 капель 30% водного Н2О2. Реакционную смесь перемешивают в течение 10 мин и гасят водой. Водный слой экстрагируют EtOAc (2х). Органические фракции объединяют и промывают водой, насыщенным раствором соли и затем сушат (Na2SO4), фильтруют и концентрируют. Твердое вещество растирают с горячим 2-пропанолом, оставляют для охлаждения и твердое вещество собирают посредством фильтрования с отсасыванием, промывают 2-пропанолом и МТВЕ и сушат на воздухе, получая при этом не совсем белый порошок (выход 61%).
МС m/e (FIA+) 388, 410 (М+Na), m/e (FIA-) 386; 1Н ЯМР (500 МГц ДМСО-d6) δ 10,0 (шир. с, 1Н), 9,2 (с, 1Н), 7,83 (д, 4Н), 7,68 (д, 2Н), 7,63 (д, 2Н), 7,4 (т, 2Н), 7,23 (т, 2Н), 7,1 (т, 1Н), 6,85 (т, 1Н) м.д.
4-(5-Амино-3-фениламино-[1,2,4]триазол-1-ил)-6-фениламино-1Н-пиримидин-2-он
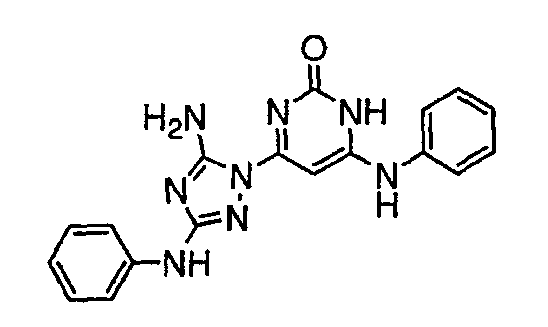
1-(2-Метансульфонил-6-фениламинопиримидин-4-ил)-N3-фенил-1Н-[1,2,4]триазол-3,5-диамин (12 мг, 0,028 ммоль) перемешивают при температуре окружающей среды в 500 мл ДМФ с 200 мкл 1 н. NaOH в течение 2 час. Реакционную смесь гасят водой и раствору дают возможность образовать осадок. Фильтрат декантируют и твердое вещество снова суспендируют в воде, дают ему возможность осесть и снова декантируют. Твердое вещество переносят в круглодонную колбу с ацетонитрилом и растворители удаляют при пониженном давлении. Образовавшийся материал, бежево-желтое твердое вещество, получают с выходом 90%.
МС m/e (FIA-) 359; 1Н ЯМР (500 МГц ДМСО-d6) δ 10,5 (шир.с, 1Н), 9,15 (с, 1Н), 7,9 (шир.с, 2Н), 7,66 (д, 4Н), 7,4 (т, 2Н), 7,2 (т, 2Н), 7,18 (шир.с, 1Н), 6,8 (т, 1Н), 6,35 (шир.с, 1Н) м.д.
1-(2-Метокси-6-фениламинопиримидин-4-ил)-N3-фенил-1Н-[1,2,4]триазол-3,5-диамин
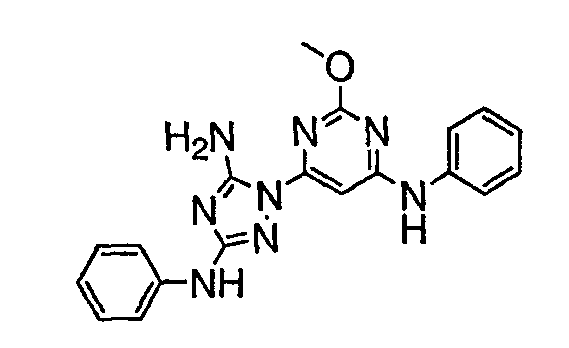
1-(2-Метансульфонил-6-фениламинопиримидин-4-ил)-N3-фенил-1Н-[1,2,4]триазол-3,5-диамин (12 мг, 0,028 ммоль) перемешивают при температуре окружающей среды в 500 мкл ДМФ с 200 мкл свежеполученного раствора метоксида натрия в течение 2 час. Реакционную смесь гасят водой и осадку дают возможность высадиться из раствора. Фильтрат декантируют и материал промывают водой, снова декантируют и твердое вещество переносят в круглодонную колбу с ацетонитрилом и растворители удаляют при пониженном давлении. Препаративная ВЭЖХ (градиент элюента: ацетонитрил-вода, 0,1% TFA) дает 12 мг бежевого твердого вещества, выход 90%.
МС m/e (FIA+) 375; 1Н ЯМР (500 МГц ДМСО-d6) δ 9,8 (с, 1Н), 9,15 (с, 1Н), 7,7 (м, 7Н), 7,37 (т, 2Н), 7,25 (т, 2Н), 7,02 (т, 1Н), 6,83 (т, 1Н), 6,64 (с, 1Н) м.д.
6-[5-Амино-3-фениламино-[1,2,4]триазол-1-ил)-N2-этил-N2-метил-N4-фенилпиримидин-2,4-диамин
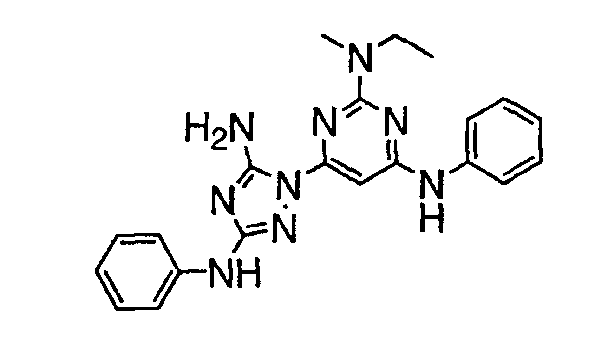
1-(2-Метансульфонил-6-фениламинопиримидин-4-ил)-N3-фенил-1Н-[1,2,4]триазол-3,5-диамин (75 мг, 0,18 ммоль) перемешивают с N-этилметиламином (200 мкл; 1,5 ммоль) в 1 мл ТГФ в запаянной трубке в течение 10 час при 80°С. Реакционную смесь гасят 1 н. HCl и образовавшийся осадок центрифугируют до получения осадка. Осадок суспендируют в воде и снова центрифугируют до получения осадка. Препаративная ВЭЖХ (градиент элюента: ацетонитрил-вода, 0,1% TFA) дает указанное в заголовке соединение. Очищенный материал превращают в соль HCl добавлением 1 н. HCl, причем растворитель удаляют при пониженном давлении. Материал получают в виде не совсем белого твердого вещества (5 мг, выход 6%).
МС m/e (FIA+) 402, m/e (FIA-) 400; 1Н ЯМР (500 МГц ДМСО-d6) δ 9,45 (шир.с, 1Н), 9,05 (шир.с, 1Н), 7,7 (д, 2Н), 7,65 (шир.с, 2Н), 7,61 (д, 2Н), 7,35 (т, 2Н), 7,25 (т, 2Н), 6,97 (т, 1Н), 6,85 (т, 1Н), 6,33 (с, 1Н), 3,6 (шир. кварт., 2Н), 3,1 (шир. с, 3Н), 1,15 (т, 3Н) м.д.
Схема 30
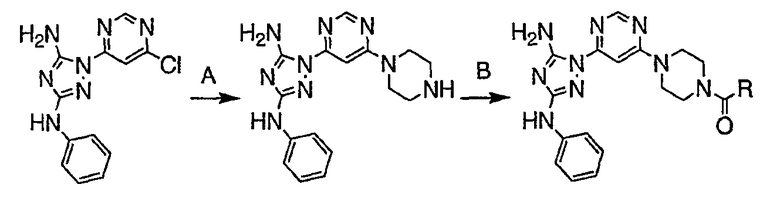
Условия реакции: А. пиперазин, NMP, 220°С, 6 мин; В. хлорангидрид кислоты, основание Хунига, CH2Cl2.
Пример 66
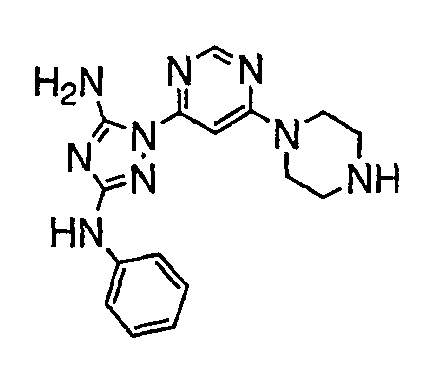
N3-Фенил-1-(6-пиперазин-1-илпиримидин-4-ил)-1Н-[1,2,4]триазол-3,5-диамин
К раствору 200 мг 1-(6-хлорпиримидин-4-ил)-N3-фенил-1Н-[1,2,4]триазол-3,5-диамина (0,69 ммоль, 1 экв.) в 5 мл NMP добавляют 200 мг пиперазина (2,32 ммоль, 3,3 экв.). Реакционный сосуд запаивают и нагревают до 220°С посредством облучения микроволновой печью в течение 6 мин и оставляют для охлаждения. Образовавшийся раствор выливают в 50 мл воды и осадок отфильтровывают и промывают водой (3 х 20 мл). Образовавшееся воскообразное твердое вещество (150 мг) используют без дополнительной очистки.
ЖХ-МС: Rf=1,35 мин, 338,24 (М+Н).
Пример 67

3-{4-[6-(5-Амино-3-фениламино-[1,2,4]триазол-1-ил)пиримидин-4-ил]пиперазин-1-ил}-3-оксопропионитрил. К перемешиваемому раствору 100 мг 2-цианоуксусной кислоты (1,2 ммоль, 7,9 экв.) в 10 мл CH2Cl2 добавляют последовательно 300 мкл оксалилхлорида (435 мг, 3,45 ммоль, 23 экв.) и 1 каплю ДМФ. Реакционную смесь оставляют для перемешивания при 25°С до прекращения выделения пузырьков. Реакционную смесь концентрируют и подвергают азеотропной перегонке с CH2Cl2 (3 × 10 мл) перед повторным растворением в 10 мл CH2Cl2. К этому раствору добавляют 50 мг N3-фенил-1-(6-пиперазин-1-илпиримидин-4-ил)-1Н-[1,2,4]триазол-3,5-диамина (0,148 ммоль, 1 экв.) в 5 мл CH2Cl2. После последующего добавления 200 мкл основания Хунига реакционную смесь перемешивают в течение 12 час при 25°С. Реакционную смесь затем концентрируют и очищают хроматографией на силикагеле, получая при этом 2,9 мг (0,007 ммоль, выход 5%).
ЖХ-МС: 2,72 мин/405,2 (М+Н). 1Н ЯМР (500 МГц, CDCl3) δ 8,31 (1Н, с), 7,40 (2Н, д), 7,25 (2Н, т), 6,89 (1Н, т), 6,77 (1Н, с), 6,65 (2Н, шир.с), 6,45 (1Н, с), 3,82 (2Н, м), 3,70 (4Н, м), 3,52 (2Н, м), 3,40 (2Н, с) м.д.
Схема 31
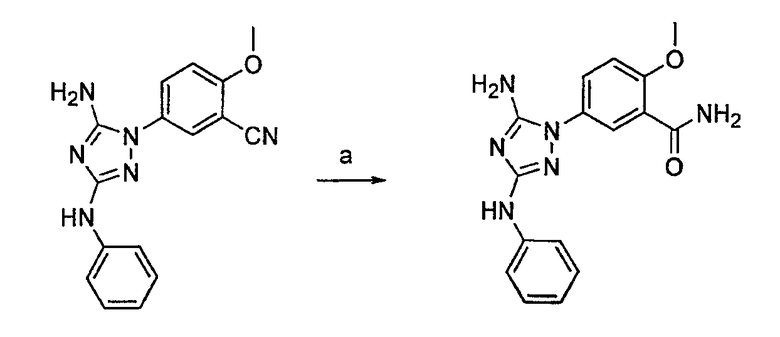
(а) К2СО3, 30% Н2О2, ДМСО
5-(5-Амино-3-фениламино-[1,2,4]триазол-1-ил)-2-метоксибензамид. К перемешиваемому раствору 5-(5-амино-3-фениламино)-[1,2,4]триазол-1-ил)-2-метоксибензонитрила (20 мг, 0,065 ммоль) и К2СО3 (2 мг) в ДМСО (0,3 мл) при комнатной температуре добавляют 30% водный раствор Н2О2 (0,3 мл). Спустя 1 час добавляют дополнительный 30% водный Н2О2 (0,15 мл). Спустя 40 мин добавляют воду вместе с 5% водным Na2CO3 и смесь перемешивают в течение нескольких минут. Твердое вещество собирают и промывают несколькими порциями воды и сушат в вакууме, получая при этом белое твердое вещество (18 мг, 0,055 ммоль, выход 85%).
1Н ЯМР 500 МГц (ДМСО) 8,82 (с, 1Н), 7,93 (д, 1Н), 7,71 (шир. с, 1Н), 7,63 (дд, 1Н), 7,60 (шир. с, 1Н), 7,52 (д, 2Н), 7,25 (д, 1Н), 7,19 (т, 2Н), 6,75 (т, 1Н), 6,28 (шир. с, 2Н), 3,93 (с, 3Н), м.д. ЖХ/МС: Rt=2,27 мин, (М+Н)= 325,2.
Схема 32
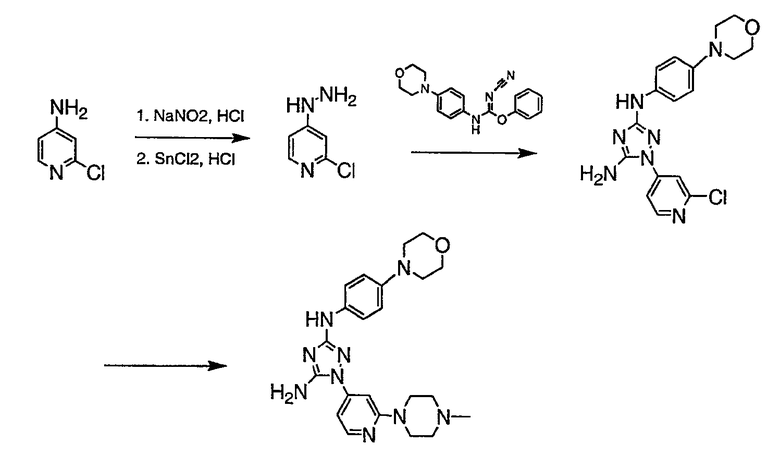
(2-Хлорпиридин-4-ил)гидразин
2-Хлорпиридин-4-иламин (2 г, 15,6 ммоль) растворяют в 20 мл 1 М HCl и 4 мл конц. HCl и охлаждают до 0°С. Нитрит натрия (1 г, 17 ммоль) растворяют в 2 мл воды и раствор добавляют по каплям к раствору пиридина. Смесь перемешивают при 0-5°С в течение 2 час и затем добавляют по каплям к суспензии SnCl2 в 35 мл конц. HCl при 0°С. Смесь перемешивают при 0°С в течение 1 часа, затем рН осторожно повышают до рН 9-10 NaOH при использовании эффективного охлаждения и перемешивания. Водную смесь экстрагируют 10% раствором МеОН/хлороформ и органический слой отделяют, сушат (сульфатом натрия) и упаривают. Сырой продукт в виде смеси очищают колоночной хроматографией (диоксид кремния, 5% МеОН/DCM), получая при этом 400 мг (2-хлорпиридин-4-ил)гидразина. МС ES+ 144,0, 146,3.
Диаминотриазол, 1-(2-хлорпиридин-4-ил)-N3-(4-морфолин-4-илфенил)-1Н-[1,2,4]триазол-3,5-диамин и другие диаминотриазолы, описанные в схеме и таблице, получают из (2-хлорпиридин-4-ил)гидразина и имидатных сложных эфиров с использованием процедур, описанных в другом месте данного изобретения.
5-[2-(4-Метилпиперазин-1-ил)пиридин-4-ил]-N2-(4-морфолин-4-илфенил)-5Н-имидазол-2,4-диамин. 1-(2-хлорпиридин-4-ил)-N3-(4-морфолин-4-илфенил)-5Н-имидазол-2,4-диамин (100 мг, 0,27 ммоль) и N-метилпиперазин (101 мг, 1 ммоль) смешивают в 1 мл н-бутанола и нагревают до 230°С в течение 360 сек в персональном микроволновом инструменте. Растворитель выпаривают в вакууме и остаток очищают препаративной ТСХ (1% NH4OH/10% МеОН/DCM), получая при этом 45 мг 5-[2-(4-метилпиперазин-1-ил)пиридин-4-ил]-N2-(4-морфолин-4-илфенил)-5Н-имидазол-2,4-диамина.
Аналогичным способом получают следующие соединения.
Пример 68
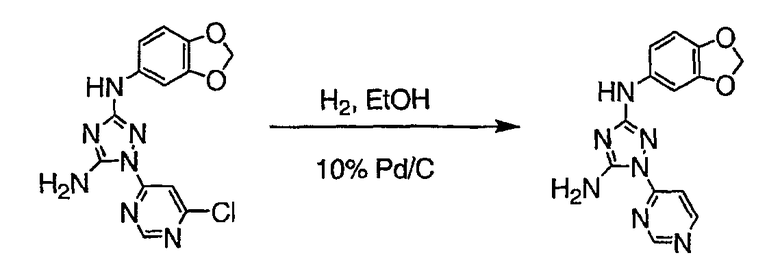
N3-Бензо[1,3]диоксол-5-ил-1-пиримидин-4-ил-1Н-[1,2,4]триазол-3,5-диамин. N3-Бензо[1,3]диоксол-5-ил-1-(6-хлорпиримидин-4-ил)-1Н-[1,2,4]триазол-3,5-диамин (40 мг, 0,12 ммоль) перемешивают с 10% Pd/C в 1 мл этанола при 1 атм водорода в течение 18 час. Катализатор удаляют фильтрованием и растворитель выпаривают. Сырой продукт очищают препаративной ВЭЖХ, получая при этом 11 мг N3-бензо[1,3]диоксол-5-ил-1-пиримидин-4-ил-1Н-[1,2,4]триазол-3,5-диамина в виде соли TFA.
1Н ЯМР ацетон-d6: 8,9 (с, 1Н), 8,8 (д, 1Н), 8,1 (шир.с, 1Н), 7,4 (м, 3Н), 7,1 (д, 1Н), 6,8 (д, 1Н), 5,9 (с, 2Н).
Аналогичным способом получают следующее соединение:
2-(3,4-Диметоксифениламино)-4Н-[1,2,4]триазоло[1,5-a]хиназолин-5-он и 2-амино-4-(3,4-диметоксифенил)-4Н-[1,2,4]триазоло[1,5-a]хиназолин-5-он
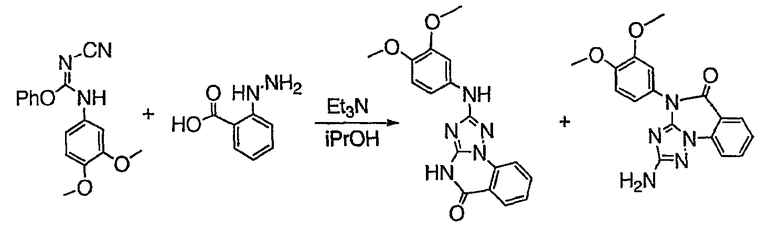
Раствор 1-(3,4-диметоксифенил)-3-циано-2-фенилизомочевины (600 мг, 2 ммоль), гидрохлорида 2-гидразинобензойной кислоты (760 мг, 4 ммоль) и триэтиламина (1,6 мл) в изопропаноле (20 мл) и нагревают при кипячении с обратным холодильником в течение 24 час. Смесь упаривают. Остаток суспендируют в воде (50 мл), фильтруют. Твердое вещество очищают ВЭЖХ, получая при этом 2-(3,4-диметоксифениламино)-4Н-[1,2,4]триазоло[1,5-a]хиназолин-5-он (263 мг) и его изомер (72 мг). Данные 2-(3,4-диметоксифениламино)-4Н-[1,2,4]триазоло[1,5-a]хиназолин-5-она: FIA-MC; m/e=338,2 (M+H), 336,1 (M-H). Rt=3,09 мин (способ А). 1Н ЯМР (500 МГц, ДМСО-d6): 12,90 (с, 1Н), 9,36 (с, 1Н), 8,15 (д, 1Н), 7,91 (т, 1Н), 7,86 (д, 1Н), 7,45 (т, 1Н), 7,41 (д, 1Н), 7,18 (дд, 1Н), 6,90 (д, 1Н), 3,80 (с, 3Н), 3,71 (с, 3Н). Данные2-амино-4-(3,4-диметоксифенил)-4Н-[1,2,4]триазоло[1,5-a]хиназолин-5-она: ЖХ-МС; m/e=338,2 (M+H), 336,1 (M-H). Rt=2,34 мин. 1Н ЯМР (500 МГц, ДМСО-d6): 8,17 (д, 1Н), 7,90 (т, 1Н), 7,76 (д, 1Н), 7,45 (т, 1Н), 7,16 (д, 1Н), 7,09 (д, 1Н), 7,02 (дд, 1Н), 6,04 (с, 2Н), 3,82 (с, 3Н), 3,70 (с, 3Н).
(4,5-Дигидро-[1,2,4]триазоло[1,5-a]хиназолин-2-ил)-(3,4-диметоксифенил)амин
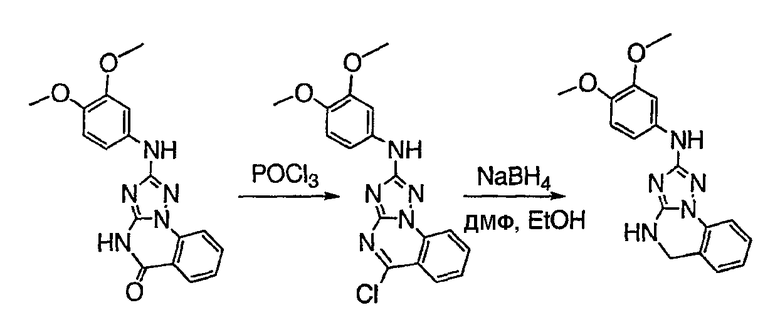
Суспензию 2-(3,4-диметоксифениламино)-4Н-[1,2,4]триазоло[1,5-a]хиназолин-5-она (57 мг) в оксихлориде фосфора (5 мл) нагревают при 90°С в течение 2 час. Смесь упаривают. Остаток суспендируют в дихлорметане, промывают холодным бикарбонатом натрия, насыщенным раствором соли и сушат (Na2SO4). Фильтрование и концентрирование дает сырой (5-хлор-[1,2,4]триазоло[1,5-a]хиназолин-2-ил)-(3,4-диметоксифенил)амин (68 мг). ЖХ-МС: m/e=356,1 (М+Н).
При 0°С раствор борогидрида натрия (2,0 М, 0,25 мл) добавляют к раствору вышеуказанного хлорида (38 мг, 0,107 ммоль) в хлороформе (5 мл) и этаноле (2 мл) (или ДМФ и МеОН). Реакционную смесь выдерживают при комнатной температуре в течение 1 часа (мониторинг проводят аналитической ВЭЖХ), подкисляют трифторуксусной кислотой и очищают ВЭЖХ, получая при этом указанное в заголовке соединение (12 мг). FIA-MC: m/e=324,1 (M+H). Rt=3,08 (способ А). 1Н ЯМР (500 МГц, ДМСО-d6): 8,90 (с, 1Н), 7,71 (с, 1Н), 7,39 (д, 1Н), 7,34 (т, 1Н), 7,31 (дд, 1Н), 7,25 (д, 1Н), 7,12 (дд, 1Н), 7,08 (тд, 1Н), 6,84 (д, 1Н), 4,50 (с, 2Н), 3,77 (с, 3Н), 3,68 (с, 3Н).
Аналогично получают следующие соединения:
Схема 33
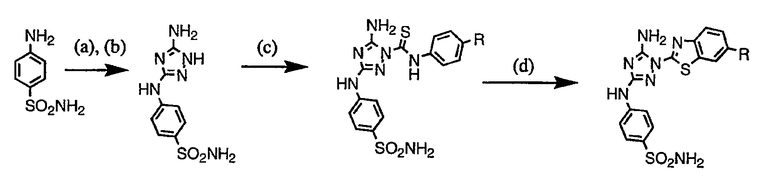
Реагенты: (а) (PhO)2CN(CN), iPrOH; 75°С; (b) моногидрат гидразина, iPrOH, 70°С; (c) Im2CS, имидазол, ДМФ, комнатная температура, затем анилин, 0°С; (d) Br2, ДМФ, комнатная температура.
На указанной выше схеме 33 показан общий способ получения некоторых 6-замещенных триазолилбензотиазолов, где R представляет собой простую эфирную группу или аминогруппу.
Пример 69
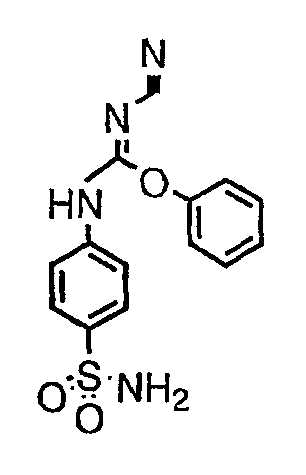
4-(3-Циано-2-фенилизоуреидо)бензолсульфонамид. 4-Аминобензолсульфонамид (2 ммоль) добавляют в одну порцию к раствору дифеноксицианоимидата (2 ммоль) в изопропаноле. Реакционную смесь перемешивают при 75°С в течение 4 часов. Реакционной смеси затем дают возможность охладиться до комнатной температуры. Образованный твердый осадок отфильтровывают. Его промывают изопропанолом, получая при этом указанное в заголовке соединение (0,33 г; выход 70%).
1Н ЯМР (400 МГц, ДМСО-d6) δ 7,30-7,40 (5Н, м), 7,45-7,50 (2Н, т), 7,65-7,70 (2Н, д), 7,80-7,85 (2Н, д), 11,20 (1Н, с); МС (ES+) m/e=317.
Пример 70
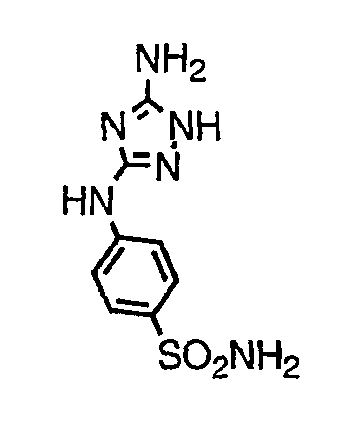
4-(5-Амино-1Н-[1,2,4]триазол-3-иламино)бензолсульфонамид. К раствору 4-(3-циано-2-фенилизоуреидо)бензолсульфонамида (1,35 ммоль) в ТГФ (5 мл) добавляют раствор моногидрата гидразина (2 ммоль; 1,5 эквивалентов). Раствор перемешивают при 70°С в течение 16 часов. Реакционной смеси дают возможность охладиться до комнатной температуры. Осаждается белое твердое вещество. Его отделяют фильтрованием и промывают этанолом, получая при этом указанное в заголовке соединение (300 мг; выход 87%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 5,95-6,00 (2Н, с), 7,00-7,05 (2Н, с), 7,55-7,65 (4Н, м), 9,15-9,20 (1Н, с); МС (ES+) m/e=255.
Пример 71

(4-Феноксифенил)амид 5-амино-3-(4-сульфамоилфениламино)-[1,2,4]триазол-1-тиоугольной кислоты. Тиокарбонилдиимидазол (0,56 ммоль, 1,5 эквивалента) добавляют к раствору 4-феноксианилина (0,6 ммоль, 1,6 эквивалента) и имидазол (0,07 ммоль, 0,2 эквивалента) в ацетонитриле (5 мл). Реакционную смесь перемешивают при комнатной температуре в течение трех часов. В виде одной порции добавляют 4-(5-амино-1Н-[1,2,4]триазол-3-иламино)бензолсульфонамид (0,37 мМ, 1 эквивалент). Реакционную смесь перемешивают при 50°С в течение 16 часов. После добавления ДМФ (1 мл) реакционную смесь перемешивают при 80°С в течение одного часа. Реакционную смесь концентрируют в вакууме. Остаток очищают хроматографией на силикагеле при элюировании смесью EtOAc:пентан (от 30:70 до 0:100), получая при этом указанное в заголовке соединение (40 мг; выход 40%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,05-7,18 (6Н, м), 7,20-7,23 (1Н, т), 7,40-7,50 (4Н, м), 7,70-7,75 (2Н, д), 7,80-7,85 (2Н, д), 8,45-8,50 (2Н, с), 9,80-9,85 (1Н, с), 10,90 (1Н, с); МС (ES+) m/e=482.
Пример 72
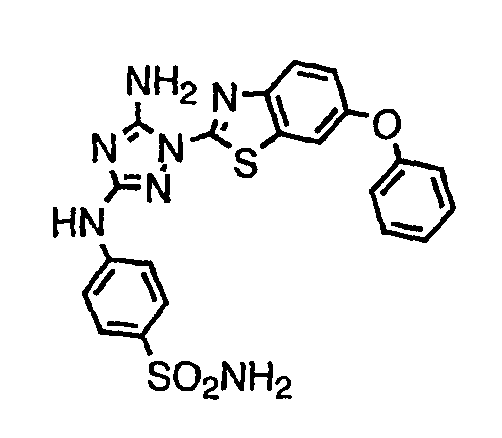
4-[5-Амино-1-(6-феноксибензотиазол-2-ил)-1Н-[1,2,4]триазол-3-иламино]бензолсульфонамид. Бром (7 мкл, 1 эквивалент) добавляют к перемешиваемой суспензии (4-феноксифенил)амида 5-амино-3-(4-сульфамоилфениламино)-[1,2,4]триазол-1-тиоугольной кислоты в дихлорметане (3 мл). Реакционную смесь перемешивают при комнатной температуре в течение 18 часов. Затем добавляют раствор брома (7 мкл) в уксусной кислоте (1 мл) для доведения реакции до завершения: реакционную смесь перемешивают в течение дополнительных четырех часов. Осажденное белое твердое вещество отделяют фильтрованием, получая при этом указанное в заголовке соединение (5 мг; выход 7%).
1Н ЯМР (400 МГц, ДМСО-d6) δ 7,05-7,20 (8Н, м), 7,35-7,50 (4Н, м), 7,70-7,75 (2Н, д), 7,80-7,85 (2Н, д), 9,90-9,95 (1Н, с); МС (ES+) m/e=480.
Схема 34
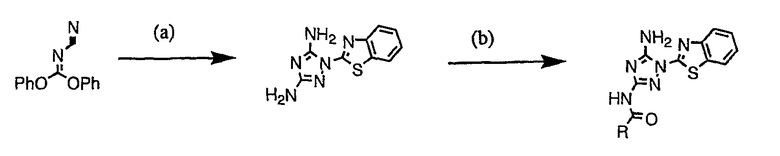
Реагенты: (а) NH3, EtOH, 90°С, затем бензотиазол-2-илгидразин, NMM, 110°С; RCOCl, пиридин.
На приведенной выше схеме 34 показан общий способ получения N-(5-амино-1-бензотиазол-2-ил-1Н-[1,2,4]триазол-3-ил)амидов.
Пример 73
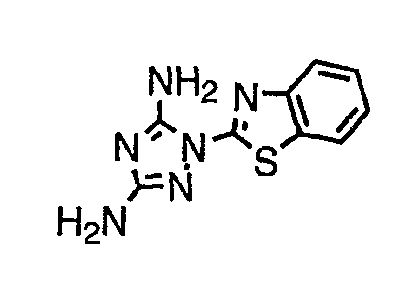
1-Бензотиазол-2-ил-1Н-[1,2,4]триазол-3,5-диамин. К суспензии дифеноксицианоимидата (2 ммоль) в этаноле (3 мл) в запаянной колбе добавляют 2 М раствор аммиака в этаноле (4 ммоль). Реакционную смесь перемешивают при 90°С в течение 48 часов, затем концентрируют в вакууме. Остаток растворяют в N-метилморфолине (5 мл). К этому раствору добавляют бензотиазол-2-илгидразин (2 ммоль). Реакционную смесь перемешивают при 110°С в течение 24 часов. После охлаждения к реакционной смеси добавляют дистиллированную воду (20 мл) и смесь распределяют между этилацетатом и насыщенным раствором соли (100 мл/100 мл). На этой стадии твердое вещество удаляют фильтрованием. Это белое твердое вещество промывают еще этилацетатом и сушат в вакууме, получая при этом чистое указанное в заголовке соединение (170 мг; выход 37%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 5,85 (2Н, с), 7,30-7,35 (1Н, т), 7,45-7,50 (1Н, т), 7,60 (2Н, с), 7,80-7,85 (1Н, д), 7,98-8,02 (1Н, д). МС (ES+) m/e=233.
Пример 74
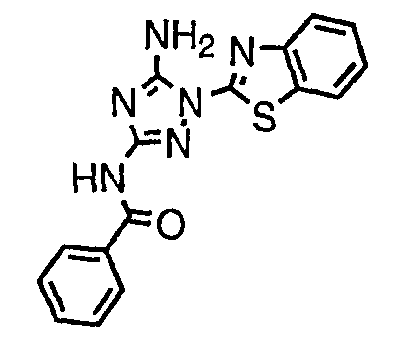
N-(5-Амино-1-бензотиазол-2-ил-1Н-[1,2,4]триазол-3-ил)бензамид. Бензоилхлорид (0,34 ммоль) добавляют по каплям к перемешиваемому раствору 1-бензтиазол-2-ил-1Н-[1,2,4]триазол-3,5-диамина (0,34 ммоль) в пиридине (3 мл). Реакционную смесь перемешивают при комнатной температуре в течение 2 часов, затем концентрируют в вакууме. Остаток распределяют между этилацетатом и 10% водной лимонной кислотой. На этой стадии отфильтровывают белое твердое вещество. Твердое вещество сушат в вакууме, получая при этом чистое указанное в заголовке соединение (10 мг; выход 15%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,40-7,45 (1Н, т), 7,50-7,70 (3Н, м), 7,85-8,00 (6Н, м), 8,1 (1Н, д), 10,95 (1Н, с). МС (ES+) m/e=337.
Схема 35
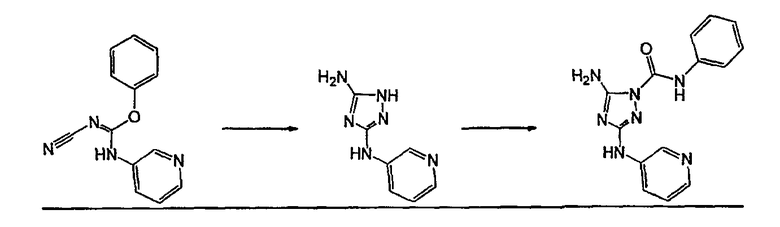
Пример 75
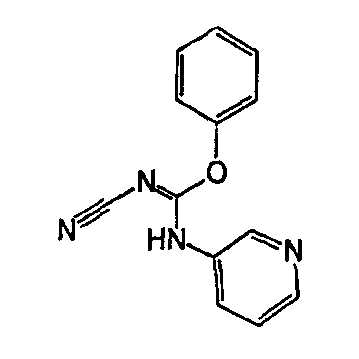
Фенил-(3-пиридил)-N-цианокарбонимидат. К раствору дифенил-N-цианокарбонимидата (1,85 ммоль) в трет-бутаноле (4 мл) добавляют 3-аминопиридин (1,85 ммоль) и смесь нагревают при кипячении с обратным холодильником в течение 3 часов. После охлаждения до комнатной температуры образовавшийся белый осадок собирают фильтрованием при пониженном давлении и промывают небольшим количеством холодного Et2O, затем сушат в вакууме при 40°С в течение 3 часов, получая при этом указанное в заголовке соединение (0,23 г, 54%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,33 (3Н, м), 7,91 (1Н, д), 8,43 (1Н, д), 8,67 (1Н, с), 10,99 (1Н, с); МС (ES+) m/e=239,2 (100%).
Пример 76
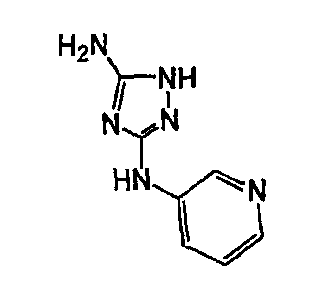
N3-(3-Пиридил)-1Н-[1,2,4]триазол-3,5-диамин. К раствору фенил-(3-пиридил)-N-цианокарбинимидата (1,00 ммоль) в изопропаноле (10 мл) добавляют гидрат гидразина (1 ммоль) и смесь нагревают для кипячения с обратным холодильником в течение 2,5 часа. После охлаждения до комнатной температуры образовавшийся белый осадок собирают фильтрованием при пониженном давлении и промывают небольшим количеством холодного изопропанола, затем сушат в вакууме при 40°С в течение 6 часов, получая при этом указанное в заголовке соединение (0,15 г, выход 87,5%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 5,93 (2Н, с), 7,17 (1Н, м), 7,94 (2Н, м), 8,64 (1Н, д), 8,88 (1Н, с), 11,24 (1Н, с); МС (ES+) m/e=177,2 (100%).
Пример 77
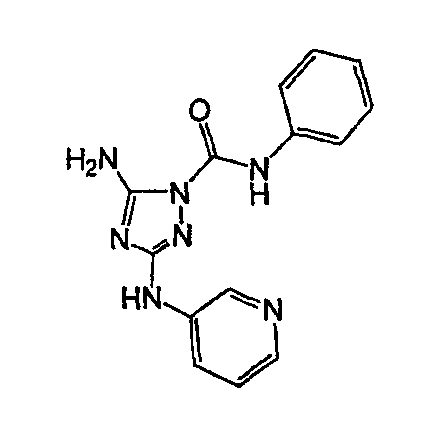
Фениламид 5-амино-3-(3-пиридиламино-[1,2,4]триазол-1-карбоновой кислоты. К раствору N3-(3-пиридил)-1Н-[1,2,4]триазол-3,5-диамина (0,43 ммоль) в сухом ТГФ (3 мл) и сухом DCM (3 мл) добавляют по каплям фенилизоцианат (0,43 ммоль) на протяжении 1 минуты и смесь перемешивают при комнатной температуре в течение 2 часов. Реакционную смесь концентрируют в вакууме и остаток очищают колоночной хроматографией [диоксид кремния Merck, элюирование EtOAc и гексанами (4:1)], получая при этом указанное в заголовке соединение (22,0 мг, 17%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,17 (1Н, т), 7,28 (1Н, м), 7,39 (4Н, м), 7,65 (2Н, д), 8,09 (1Н, м), 8,23 (1Н, м), 8,79 (1Н, д), 9,44 (1Н, с), 9,64 (1Н, с); МС (ES+) m/e=296,3 (100%).
Дополнительные данные для соединений изобретения:
В) Биологические данные:
Пример 1: ингибирование FLT-3:
Скрининг соединений проводили на их способность ингибировать активность FLT-3 с использованием анализа связывания с радиометрическим фильтром. Анализ проводил контроль включения 33Р в субстрат поли(Glu, Tyr) 4:1 (pE4Y). Реакции проводили в растворе, содержащем 100 мМ HEPES (pH 7,5), 10 мМ MgCl2, 25 мМ NaCl, 1 мМ DTT, 0,01% BSA и 2,5% ДМСО. Конечные концентрации субстратов в анализе были 90 мкМ АТФ и 0,5 мг/мл рЕ4Y (оба из Sigma Chemicals, St Louis MO). Конечные концентрации соединений обычно находятся между 0,01 и 5 мкМ. Обычно титрование по 12 точкам проводили приготовлением серийных разведений от 10 мМ испытуемого соединения в исходном ДМСО. Реакции проводили при комнатной температуре.
Получали два раствора для анализа. Раствор 1 содержит 100 мМ HEPES (рН 7,5), 10 мМ MgCl2, 25 мМ NaCl, 1 мг/мл рЕ4Y и 180 мкМ АТФ (содержащий 0,3 мкКи [γ-33P]АТФ для каждой реакции). Раствор 2 содержит 100 мМ HEPES (рН 7,5), 10 мМ MgCl2, 25 мМ NaCl, 2 мМ DTT, 0,02% BSA и 3 нМ FLT-3. Анализ проводили на 96-луночном планшете смешиванием 50 мкл каждого раствора 1 с 2,5 мл испытуемого соединения. Реакцию инициировали раствором 2. После инкубации в течение 20 минут при комнатной температуре реакцию останавливали 50 мкл 20% ТСА, содержащего 0,4 мМ АТФ. Все объемы реакций затем переносили в планшет для фильтрования и промывали 5% ТСА при помощи Harvester 9600 из ТОМТЕС (Hamden, CT). Количество включения 33Р в рЕ4y анализировали сцинтилляционным счетчиком Packard TopCount Microplate (Meriden, CT). Данные подбирали с применением программного обеспечения Prism, получая при этом IC50 или Ki.
В общем соединения изобретения, включающие соединения таблицы 1, являются эффективными для ингибирования FLT-3.
Пример 2: ингибирование c-KIT
Скрининг соединений проводили на их способность ингибировать активность c-KIT с использованием анализа связывания с радиометрическим фильтром. Анализ проводил контроль включения 33Р в субстрат поли(Glu, Tyr) 4:1 (pE4Y). Реакции проводили в растворе, содержащем 100 мМ HEPES (pH 7,5), 10 мМ MgCl2, 25 мМ NaCl, 1 мМ DTT, 0,01% BSA и 2,5% ДМСО. Конечные концентрации субстратов в анализе были 700 мкМ АТФ и 0,5 мг/мл рЕ4Y (оба из Sigma Chemicals, St Louis MO). Конечные концентрации соединений обычно находятся между 0,01 и 5 мкМ. Обычно титрование по 12 точкам проводили приготовлением серийных разведений от 10 мМ испытуемого соединения в исходном ДМСО. Реакции проводили при комнатной температуре.
Получали два раствора для анализа. Раствор 1 содержит 100 мМ HEPES (рН 7,5), 10 мМ MgCl2, 25 мМ NaCl, 1 мг/мл рЕ4Y и 1,4 мкМ АТФ (содержащий 0,5 мкКи [γ-33P]АТФ для каждой реакции). Раствор 2 содержит 100 мМ HEPES (рН 7,5), 10 мМ MgCl2, 25 мМ NaCl, 2 мМ DTT, 0,02% BSA и 25 нМ c-KIT. Анализ проводили на 96-луночном планшете смешиванием 33 мкл раствора 1 и 1,65 мкл испытуемых соединений. Реакцию инициировали 33 мкл раствора 2. После инкубации в течение 20 минут при комнатной температуре реакцию останавливали 50 мкл 10% ТСА, содержащей 0,2 мМ АТФ. Все объемы реакций затем переносили в планшет для фильтрования и промывали 5% ТСА при помощи Harvester 9600 из ТОМТЕС (Hamden, CT). Количество включения 33Р в рЕ4y анализировали сцинтилляционным счетчиком Packard TopCount Microplate (Meriden, CT). Данные подбирали с применением программного обеспечения Prism, получая при этом IC50 или Ki.
В общем соединения изобретения, включающие соединения таблицы 1, являются эффективными для ингибирования c-KIT.
Пример 3: Ингибирование GSK-3
Скрининг соединений проводили на их способность ингибировать активность GSK-3β (АА 1-420) с использованием стандартной системы связанного фермента (Fox et al. (1998) Protein Sci. 7, 2249). Реакции проводили в растворе, содержащем 100 мМ HEPES (pH 7,5), 10 мМ MgCl2, 25 мМ NaCl, 300 мкМ NADH, 1 мМ DTT и 1,5% ДМСО. Конечные концентрации субстратов в анализе были 20 мкМ АТФ (Sigma Chemicsls, St. Louis, MO) и 300 мкМ пептида (American Peptide, Sunnyvale, CA). Реакции проводили при 30°С и 20 нМ GSK-3β. Конечные концентрации компонентов системы связанного фермента были 2,5 мМ фосфоенолпирувата, 300 мкМ NADH, 30 мкг/мл пируваткиназы и 10 мкг/мл лактатдегидрогеназы.
Получали исходный буферный раствор для анализа, содержащий все реагенты, перечисленные выше, за исключением АТР и представляющего интерес испытуемого соединения. Исходный буферный раствор для анализа (175 мкл) инкубировали в 96-луночном планшете с 5 мкл представляющего интерес испытуемого соединения при конечных диапазонах концентраций от 0,002 мкМ до 30 мкМ при 30°С в течение 10 мин. Обычно титрование по 12 точкам проводили приготовлением серийных разведений (от 10 мМ исходных растворов соединения) ДМСО испытуемых соединений в «дочерних» планшетах. Реакцию инициировали добавлением 20 мкл АТФ (конечная концентрация 20 мкМ). Скорости реакции получали с использованием аппарата для прочтения планшетов Molecular Devices Spectramax (Sunnyvale, CA) на протяжении 10 мин при 30°С. Величины Ki определяли из данных скоростей как функцию концентрации ингибитора.
В общем соединения изобретения, включающие соединения таблицы 1, являются эффективными для ингибирования GSK-3.
Пример 4: ингибирование CDK-2
Скрининг соединений проводили на их способность ингибировать активность CDK-2/циклина А с использованием стандартной системы связанного фермента (Fox et al. (1998) Protein Sci. 7, 2249). Реакции проводили в 100 мМ HEPES (pH 7,5), 10 мМ MgCl2, 25 мМ NaCl, 1 мМ DTT и 1,5% ДМСО. Конечные концентрации субстратов в анализе были 100 мкМ АТФ (Sigma chemicals) и 100 мкМ пептида (American Peptide, Sunnyvale, CA). Анализ проводили при 30°С и 25 нМ CDK-2/циклина А. Конечные концентрации компонентов системы связанных ферментов были 2,5 мМ фосфоенолпирувата, 350 мкМ NADH, 30 мкг/мл пируваткиназы и 10 мкг/мл лактатдегидрогеназы.
Получали исходный буферный раствор для анализа, содержащий все реагенты, перечисленные выше, за исключением CDK-2/циклина А, DTT и представляющего интерес испытуемого соединения. 56 мкл испытуемой реакционной смеси помещали в 384-луночный планшет с последующим добавлением 1 мкл 2 мМ исходного раствора ДМСО, содержащего испытуемое соединение (конечная концентрация соединения 30 мкМ). Планшет предварительно инкубировали в течение ˜ 10 минут при 30°С и реакцию инициировали добавлением 10 мкл фермента (конечная концентрация 25 нМ). Скорости реакции получали с использованием аппарата для прочтения планшетов BioRad Ultramark (Hercules, CA) на протяжении 5 минут при 30°С. Величины Ki определяли по стандартным методам.
В общем соединения изобретения, включающие соединения таблицы 1, являются эффективными для ингибирования CDK-2.
Пример 5: ингибирование SRC
Соединения оценивали в качестве ингибиторов Src-киназы человека с использованием либо анализа на основе радиоактивности, либо спектрофотометрическим анализом.
Анализ A ингибирования Src: анализ на основе радиоактивности
Соединения анализировали в качестве ингибиторов полноразмерной рекомбинантной Src-киназы человека (из Upstate Biotechnology, cat. № 14-117), экспрессированной и очищенной из бакуловирусных клеток. Мониторинг активности Src проводили после включения 33Р из АТФ в тирозин произвольного полимера поли-Glu-Tyr композиции, Glu:Tyr=4:1 (Sigma, cat. № P-0275). Далее представлены конечные концентрации компонентов анализа: 0,05 М HEPES, pH 7,6, 10 мМ MgCl2, 2 мМ DTT, 0,25 мг/мл BSA, 10 мкМ АТФ (1-3 мкКи 33Р-АТФ на реакцию), 5 мг/мл поли-Glu-Tyr и 1-2 единицы рекомбинантной Src-киназы человека. В типичном анализе компоненты реакции, за исключением АТФ, предварительно смешивали и аликвоты отбирали для анализа в лунки планшета. Растворенные в ДМСО ингибиторы добавляли к лункам для получения конечной концентрации ДМСО 2,5%. Аналитические планшеты инкубировали при 30°С в течение 10 мин перед инициацией реакции 33Р-АТФ. После 20 минутной реакции реакции гасили 150 мкл 10% трифторуксусной кислоты (ТСА), содержащей 20 мМ Na3РО4. Потушенные образцы затем переносили в 69-луночный планшет для фильтрования (Whatman, UNI-Filter GF/F Glass Fiber Filter, cat. № 7700-3310), установленный на вакуумную систему для планшета для фильтрования. Планшеты для фильтрования промывали четыре раза 10% ТСА, содержащей 20 мМ Na3PO4, и затем 4 раза метанолом. К каждой лунке затем добавляли 200 мкл сцинтилляционной жидкости. Планшеты герметизировали и количество радиоактивности, связанной с фильтрами, количественно определяли на сцинтилляционном счетчике TopCount. Включенную радиоактивность наносили на график как функцию концентрации ингибитора. Данные подбирали для модели кинетик конкурирующего ингибирования, чтобы получить Ki для соединения.
Анализ В ингибирования Src: спектрофотометрический анализ
ADP, продуцированный из АТФ посредством катализируемого рекомбинантной Src-киназой человека фосфорилирования поли-Glu-Tyr-субстрата, количественно определяли с использованием анализа связанного фермента (Fox et al. (1998) Protein Sci 7, 2249). В этом анализе одна молекула NADH окисляется в NAD на каждую молекулу ADP, продуцированную в реакции киназы. Исчезновение NADH обычно прослеживается при 340 нм.
Конечные концентрации компонентов анализа являются следующими: 0,025 М HEPES, рН 7,6, 10 мМ MgCl2, 2 мМ DTT, 0,25 мг/мл поли-Glu-Tyr и 25 нМ рекомбинантной Src-киназы человека. Конечными концентрациями компонентов системы связанного фермента являются 2,5 мМ фосфоенолпируват, 200 мкМ NADH, 30 мкг/мл пируваткиназы и 10 мкг/мл лактатдегидрогеназы.
В типичном анализе все компоненты, за исключением АТФ, предварительно смешивали и аликвоты добавляли в аналитический планшет. аликвоты отбирали для анализа в лунки планшета. Растворенные в ДМСО ингибиторы добавляли к лункам для получения конечной концентрации ДМСО 2,5%. Аналитический планшет инкубировали при 30°С в течение 10 мин перед инициацией реакции 100 мкМ АТФ. Мониторинг изменения поглощение при 340 нм со временем, скорость реакции проводили на ридер-планшете молекулярным способом. Данные скорости как функцию концентрации ингибитора подбирали по модели кинетики конкурирующего ингибирования для получения Ki для соединения.
В общем соединения изобретения, включающие соединения таблицы 1, являются эффективными для ингибирования SRC.
Пример 6: ингибирование SYK
Скрининг соединений проводили на их способность ингибировать активность Syk с использованием стандартной системы связанного фермента (Fox et al. (1998) Protein Sci. 7, 2249). Реакции проводили в 100 мМ HEPES, pH 7,5, 10 мМ MgCl2, 25 мМ NaCl, 1 мМ DTT и 1,5% ДМСО. Конечные концентрации в анализе были 200 мкМ АТФ (Sigma Chemical, Co.) и 4 мкМ поли Gly-Tyr пептида (Sigma Chemical, Co.). Анализ проводили при 30°С и 200 нМ Syk. Конечные концентрации компонентов системы связанного фермента были 2,5 мМ фосфоенолпирувата, 300 мкМ NADH, 30 мкг/мл пируваткиназы и 10 мкг/мл лактатдегидрогеназы.
Получали исходный буферный раствор для анализа, содержащий все реагенты, перечисленные выше, за исключением Syk, DTT и представляющего интерес испытуемого соединения. 56 мкл испытуемой реакционной смеси помещали в 96-луночный планшет с последующим добавлением 1 мкл 2 мМ исходного раствора ДМСО, содержащего испытуемое соединение (конечная концентрация соединения 30 мкМ). Планшет предварительно инкубировали в течение ˜ 10 минут при 30°С и реакцию инициировали добавлением 10 мкл фермента (конечная концентрация 25 нМ). Скорости реакции получали с использованием ридер-планшета (Hercules, CA) на протяжении 5 минут при 30°С и величины Ki определяли по стандартным методам.
В общем соединения изобретения, включающие соединения таблицы 1, являются эффективными для ингибирования SYK.
Пример 7: ингибирование FMS
Скрининг соединений проводили на их способность ингибировать активность FMS с использованием анализа связывания радиометрического фильтра. Анализ проводит контроль включения 33Р в субстрат поли(Glu, Tyr) 4:1 (pE4Y). Реакции проводили в растворе, содержащем 100 мМ HEPES (pH 7,5), 10 мМ MgCl2, 25 мМ NaCl, 1 мМ DTT, 0,01% BSA и 2,5% ДМСО. Конечные концентрации субстратов в анализе были 90 мкМ АТФ и 0,5 мг/мл рЕ4Y (оба из Sigma Chemicals, St Louis MO). Конечные концентрации соединений обычно находятся между 0,01 и 5 мкМ. Обычно титрование по 12 точкам проводили приготовлением серийных разведений от 10 мМ испытуемого соединения в исходном ДМСО. Реакции проводили при комнатной температуре.
Получали два раствора для анализа. Раствор 1 содержит 100 мМ HEPES (рН 7,5), 10 мМ MgCl2, 25 мМ NaCl, 1 мг/мл рЕ4Y и 180 мкМ АТФ (содержащий 0,3 мкКи [γ-33P]АТФ для каждой реакции). Раствор 2 содержит 100 мМ HEPES (рН 7,5), 10 мМ MgCl2, 25 мМ NaCl, 2 мМ DTT, 0,02% BSA и 3 нМ FMS. Анализ проводили на 96-луночном планшете смешиванием 50 мкл каждого раствора 1 и 2,5 мл испытуемых соединений. Реакцию инициировали раствором 2. После инкубации в течение 20 минут при комнатной температуре реакцию останавливали 50 мкл 20% ТСА, содержащей 0,4 мМ АТФ. Все объемы реакций затем переносили в планшет для фильтрования и промывали 5% ТСА при помощи Harvester 9600 от ТОМТЕС (Hamden, CT). Количество включения 33Р в рЕ4y анализировали сцинтилляционным счетчиком Packard TopCount Microplate (Meriden, CT). Данные подбирали с применением программного обеспечения Prism, получая при этом IC50 или Ki.
В общем соединения изобретения, включающие соединения таблицы 1, являются эффективными для ингибирования FMS.
Пример 8: анализ ингибирования Rock
Скрининг соединений проводили на их способность ингибировать активность ROCK I (АА 6-553) c использованием стандартной системы связанного фермента (Fox et al. (1998) Protein Sci, 7, 2249). Реакции проводили в растворе, содержащем 100 мМ HEPES (pH 7,5), 10 мМ MgCl2, 25 мМ NaCl, 2 мМ DTT и 1,5% ДМСО. Конечные концентрации субстратов в анализе были 45 мкМ АТФ (Sigma Chemicals, St Louis MO) и 200 мкМ пептида (American peptide, Sunnyvale, CA). Реакции проводили при 30°С и 45 нМ ROCK I. Конечными концентрациями компонентов системы связанного фермента были 2,5 мМ фосфоенолпирувата, 350 мкМ NADH, 30 мкг/мл пируваткиназы и 10 мкг/мл лактатдегидрогеназы.
Обнаружено, что некоторые соединения изобретения ингибируют ROCK.
Пример 9: анализ ингибирования JAK3
Ингибирование соединениями JAK анализировали способом, описанным G.R. Brown, et al., Bioorg. Med. Chem. Lett. 2000, vol. 10, pp. 575-579, следующим образом. В планшеты Maxisorb, предварительно покрытые при 4°С поли (Glu, Ala, Tyr), 6:3:1, затем промытые 0,05% фосфатным забуференным солевым раствором и твином (PBST), добавляют 2 мкМ АТФ, 5 мМ MgCl2 и раствором соединения в ДМСО. Реакцию начинают ферментом JAK и планшеты инкубируют в течение 60 минут при 30°С. Планшеты затем промывают PBST, 100 мкл HRP-сопряженным антителом 4G10 и планшет инкубируют в течение 90 минут при 30°С. Планшет снова промывают PBST, добавляют 100 мкл раствора ТМВ и планшеты инкубируют в течение еще 30 минут при 30°С. Для остановки реакции добавляют серную кислоту (100 мкл 1 М) и планшет считывают при 450 нм для получения оптических плотностей для анализа с целью определения величин Ki.
Обнаружено, что соединения ингибируют JAK-3.
Анализ ингибирования PDK-1
Скрининг соединений проводили на их способность ингибировать PDK-1 с использованием анализа включения радиоактивного фосфата (Pitt and Lee, J. Biomol. Screen., (1996), 1, 47). Анализ проводили в смеси 100 мМ HEPES (pH 7,5), 10 мМ MgCl2, 25 мМ NaCl, 2 мМ DTT. Конечные концентрации субстратов в анализе были 40 мкМ АТФ (Sigma Chemicals) и 65 мкМ пептида (PDKtide, Upstate, Lake Placid, NY). Анализ проводили при 30°С и 25 нМ PDK-1 в присутствии ˜27,5 нКи/мкл [γ-32P]АТФ (Amersham Pharmacia Biotech, Amersham UK). Получали исходный буферный раствор для анализа, содержащий все реагенты, перечисленные выше, за исключением АТФ и представляющего интерес испытуемого соединения. 15 мкл исходного раствора помещали в 96-луночный планшет с последующим добавлением 1 мкл 0,5 мМ исходного раствора ДМСО, содержащем испытуемое соединение (конечная концентрация соединения 25 мкМ, конечная концентрация ДМСО 5%). Планшет предварительно инкубировали в течение 10 минут при 30°С и реакцию инициировали добавлением 4 мкл АТФ (конечная концентрация 40 мкМ).
Реакцию останавливали спустя 10 минут добавлением 100 мкл 100 мМ фосфорной кислоты, 0,01% твина-20. Фосфоцеллюлозный 96-луночный планшет (Millipore, Cat. № NAPHNOB50) предварительно обрабатывали 100 мкл 100 мМ фосфорной кислоты, 0,01 твина-20 перед добавлением реакционной смеси (100 мкл). Пятна оставляли для пропитки в течение, по меньшей мере, 5 минут перед стадией промывки (4 × 200 мкм 100 мМ фосфорной кислоты, 0,01% твина-20). После сушки к лунке добавляли 20 мкл жидкого сцинтилляционного коктейля Optiphase "SuperMix" (Перкин Эльмер) перед сцинтилляционным подсчетом (1450 Microbeta Liquid Scintillation Counter, Wallac).
Соединения, обнаруживающие более чем 50% ингибирование по сравнению со стандартными лунками, содержащими смесь для анализа и ДМСО без испытуемого соединения, титровали для определения величин IC50.
Соединения изобретения испытывали, и было обнаружено, что они ингибируют PDK-1.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ Mcl-1 НА ОСНОВЕ 7-ЗАМЕЩЕННЫХ ИНДОЛОВ | 2008 |
|
RU2473542C2 |
| ПРОИЗВОДНЫЕ ЗАМЕЩЕННОГО ТРИАЗОЛДИАМИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2001 |
|
RU2274639C2 |
| АМИНОТРИАЗОЛОПИРИДИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2009 |
|
RU2552642C2 |
| ПРОИЗВОДНЫЕ ДИАМИНОВ | 2002 |
|
RU2319699C2 |
| ПРОИЗВОДНЫЕ ДИАМИНА | 2002 |
|
RU2314303C2 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ПРИМЕНЕНИЕМ ЭТИХ СОЕДИНЕНИЙ | 2007 |
|
RU2478635C2 |
| ПРОИЗВОДНОЕ 1,2,4-ТРИАЗОЛОНА | 2011 |
|
RU2566754C2 |
| BCL-2-СЕЛЕКТИВНЫЕ АПОПТОЗ-ИНДУЦИРУЮЩИЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ РАКА И ИММУННЫХ ЗАБОЛЕВАНИЙ | 2010 |
|
RU2542994C2 |
| ПРОИЗВОДНЫЕ ПИРИМИДИНА | 2002 |
|
RU2320658C2 |
| КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ПИРИДАЗИНА И ЛЕКАРСТВЕННЫЕ ПРЕПАРАТЫ, СОДЕРЖАЩИЕ ДАННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2003 |
|
RU2292337C2 |
Описывается соединение формулы (II), (II') или его фармацевтически приемлемая соль, где значения радикалов указаны в формуле изобретения. Соединения являются эффективными в качестве ингибиторов протеинкиназ FLT-3 или KIT. Описываются также способ ингибирования активности киназ FLT-3 или KIT в биологическом образце in vitro и применение соединений для производства лекарственного средства, пригодного для лечения или облегчения тяжести заболевания или состояния выбранного острого миелогенного лейкоза, острого промиелоцитного лейкоза или острого лимфоцитного лейкоза или рака яичников. 3 н. и 8 з.п. ф-лы.

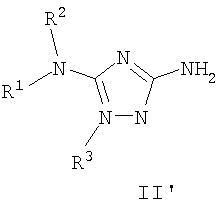
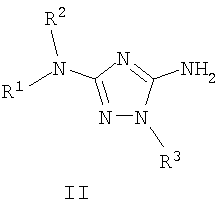
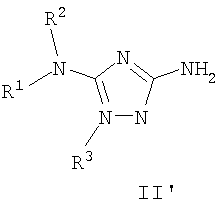
или его фармацевтически приемлемая соль,
где R1 представляет собой водород;
R2 представляет собой -(Т)nAr1, где n равно 0; Ar1 представляет собой замещенный фенил;
где Ar1 являются замещенными х независимыми группами Q-Rx, где х равно 1-3, Q представляет собой связь или C1-6-алкилиденовую цепь, в которой до двух метиленовых звеньев Q необязательно заменены на -NR-, -O-, -СО-, -CONR-, -NRCO-, -SO2NR-; в каждом случае Rx представляет собой, независимо, R', галоген, NO2, CN, OR', N(R')2, COR', CO2R', CON(R')2, SO2R' или SO2N(R')2, где по меньшей мере один Q-Rx не является водородом;
R3 связан с атомом азота либо в 1-, либо в 2-положении кольца и представляет собой -(L)mAr2; m равно 0; Ar2 представляет собой необязательно замещенную арильную группу, выбранную из пиридина, пиримидина, тиазола или 9-10-членного бициклического кольца, имеющего 1-2 гетероатомов, независимо выбранных из азота или серы; где Ar2 независимо, необязательно замещен группами Z-Ry, где y равно 0-3, Z представляет собой связь или C1-6-алкилиденовую цепь, в которой до двух метиленовых звеньев Z необязательно заменены на -NR-, -O-, -СО- или -CONR-; в каждом случае Ry представляет собой независимо R', галоген, CN, OR', SR', N(R')2, CO2R', CON(R')2 или SO2R';
в каждом случае R независимо представляет собой водород или С1-6алкильную группу, или бензильную группу;
в каждом случае R' независимо представляет собой водород или группу, выбранную из С1-6алкильной группы, 3-7-членного насыщенного или полностью ненасыщенного моноциклического кольца, имеющего 0-2 гетероатома, независимо выбранных из азота или кислорода, где указанная алкильная группа или моноциклическое кольцо необязательно замещено C1-3алкильной группой, бензильной группой, СН2ОН, ОН, NH2, N(CH3)2, CN, F, Cl, циклопропилом, ацетилом, С(O)ОС1-2алкильной группой, пирролидинилом или морфолино или два случая R' взяты вместе с атомом, с которым они связаны, с образованием 5-6-членного насыщенного или полностью ненасыщенного моноциклического кольца, имеющего 0-2 гетероатома, независимо выбранных из азота или кислорода, где указанное моноциклическое кольцо является необязательно замещенным галогеном;
при условии, что
a) когда R3 представляет собой пиридил, тогда R2 не является фенилом, одновременно замещенным одной группой ОМе в метаположении и одной группой оксазола в параположении;
b) когда R3 представляет собой незамещенный пиримидинил, тогда R2 не является незамещенным фенилом, пара-ОМе-замещенным фенилом, пара-OEt-замещенным фенилом или орто-ОМе-замещенным фенилом.

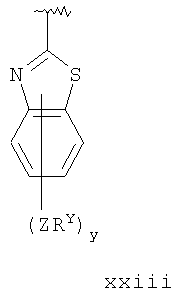
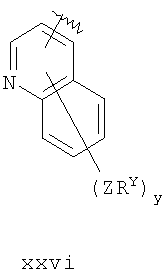
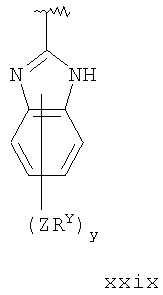
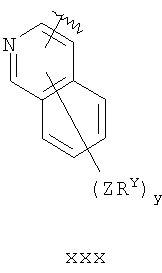
где любой замещаемый атом углерода или азота является необязательно замещенным ZRy.
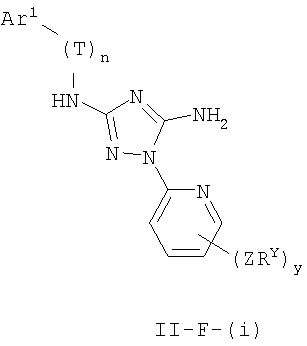
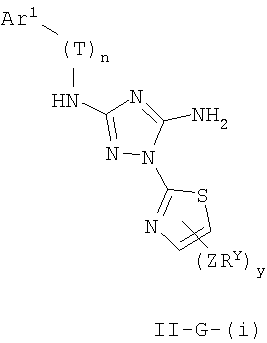
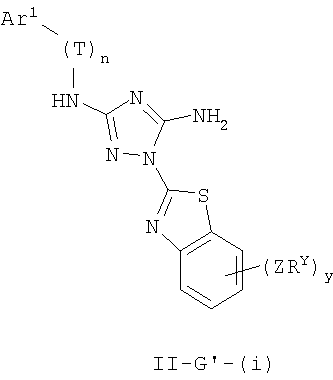
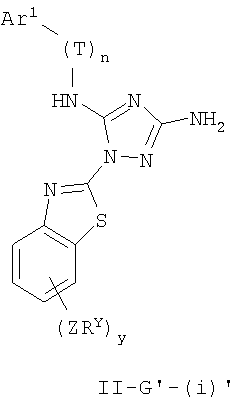
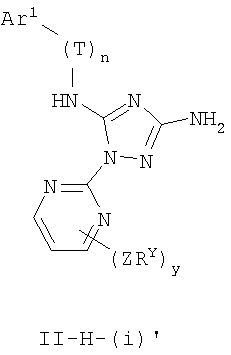

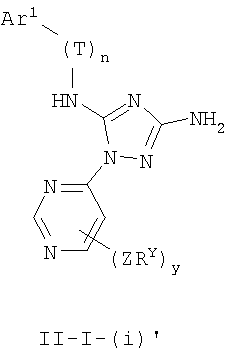
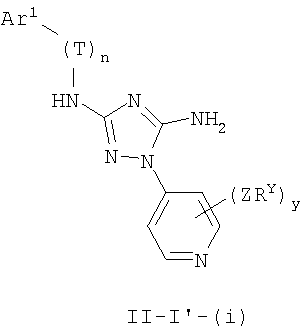
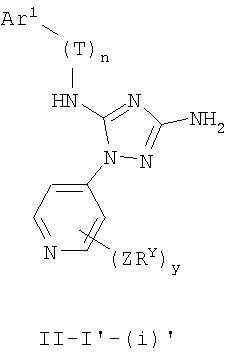
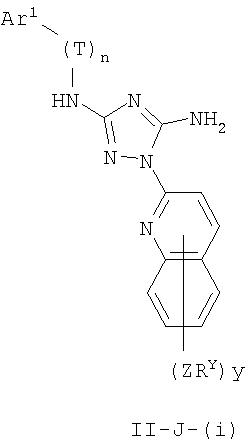
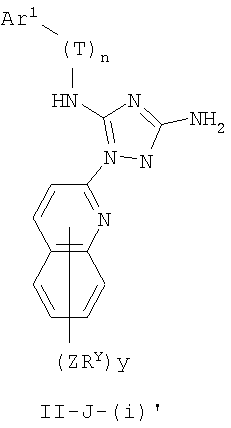
где n равно 0.
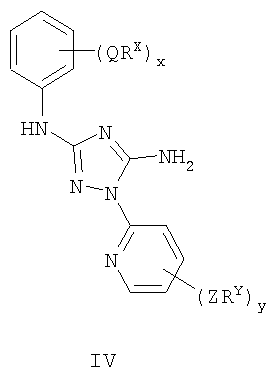
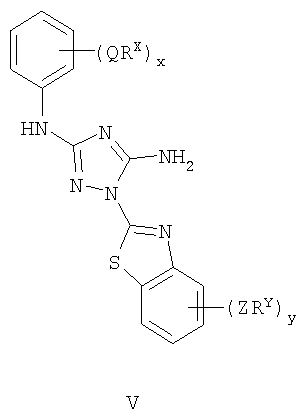
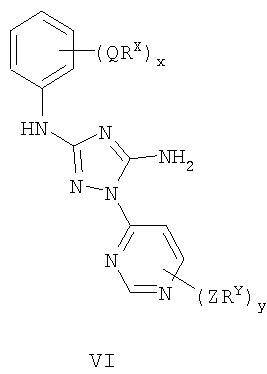
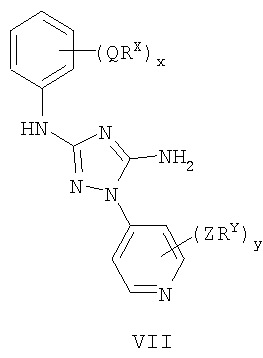
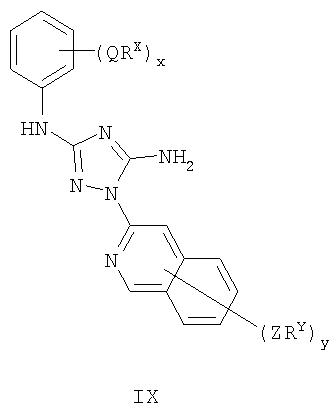
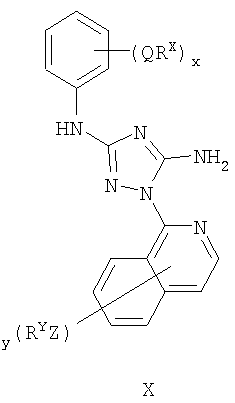 или
или 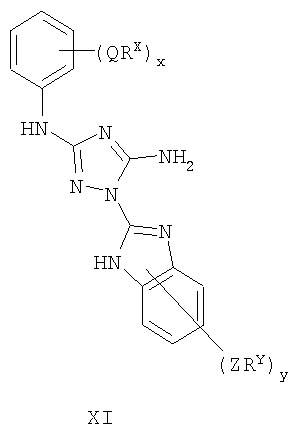
| АВТОМОБИЛЬНОЕ УСТРОЙСТВО ОБНАРУЖЕНИЯ СИГНАЛОВ РАДАРНЫХ УСТАНОВОК КОНТРОЛЯ СКОРОСТИ ДВИЖЕНИЯ НА АВТОТРАССАХ | 1993 |
|
RU2094814C1 |
| ДВЕРНАЯ ЗАЩЕЛКА | 1992 |
|
RU2057240C1 |
| Контактное приспособление к карманным или ручным электрическим лампам | 1927 |
|
SU10563A1 |
| ПРОИЗВОДНОЕ 2-(1,2,4-ТРИАЗОЛ-1-ИЛ)-1,3,4-ТИАДИАЗОЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ | 1998 |
|
RU2180903C2 |

