Предшествующий уровень техники настоящего изобретения
Фактор XIa представляет собой серинпротеазу плазмы, вовлеченную в регулирование коагуляции крови. При том, что коагуляция крови является необходимой и важной частью регулирования гомеостаза организма, аномальная коагуляция крови также может оказывать вредные эффекты. Например, тромбоз представляет собой образование или наличие кровяного сгустка внутри кровеносного сосуда или в полости сердца. Такой кровяной сгусток может застревать в кровеносном сосуде, блокируя циркуляцию и вызывая сердечный приступ или инсульт. Тромбоэмболические нарушения являются самой частой причиной смертности и инвалидности в промышленно развитом мире.
Свертывание крови представляет собой процесс контроля тока крови, выжный для выживания млекопитающих. Процесс свертывания и последующее растворение сгустка после заживления раны начинаются после повреждения сосудов и могут быть разделены на четыре фазы. Первая фаза вазоконстрикции или вазоконтракции может вызывать снижение потери крови на поврежденном участке. На следующей фазе активации тромбоцитов тромбином тромбоциты прикрепляются к месту повреждения стенки сосуда и образуют тромбоцитарный агрегат. На третьей фазе образование комплексов свертывания приводит к массивному образованию тромбина, который превращает растворимый фибриноген в фибрин путем расщепления двух небольших пептидов. На четвертой фазе после заживления раны тромб растворяется под действием ключевого фермента эндогенной системы фибринолиза - плазмина.
Два альтернативных пути могут приводить к образованию фибринового сгустка - внутренний и внешний пути. Эти пути инициируются различными механизмами, но на более поздней стадии они сходятся с образованием общего конечного пути каскада свертывания. На этом конечном пути свертывания активируется фактор свертывания X. Активированный фактор Х отвечает за образование тромбина из неактивного предшественника протромбина, циркулирующего в крови. Образование тромба на дне аномалии стенки сосуда без раны является результатом внутреннего пути. Образование фибринового сгустка в ответ на повреждение или поражение ткани является результатом внешнего пути. Оба пути включают в себя относительно большое количество белков, которые известны как факторы свертывания. Для внутреннего пути необходимы факторы свертывания V, VIII, IX, X, XI и XII, а также прекалликреин, высокомолекулярный кининоген, ионы кальция и фосфолипиды из тромбоцитов. Активация фактора XIa является центральной точкой пересечения двух путей активации свертывания. Фактор XIa играет важную роль в свертывании крови.
Коагуляция инициируется, когда кровь подвергается воздействию искусственных поверхностей (например, во время гемодиализа, сердечно-сосуцистого хирургического вмешательства с использованием искусственного кровообращения, в сосудистых трансплантатах, при бактериальном сепсисе), клеточных поверхностей, клеточных рецепторов, клеточного дебриса, ДНК, РНК и внеклеточных матриксов. Этот процесс также называется контактной активацией. Поверхностная абсорбция фактора XII приводит к конформационному изменению молекулы фактора XII, тем самым облегчая активацию молекул протеолитического активного фактора XII (фактора XIIa и фактора XIIf). Фактор XIIa (или XIIf) имеет ряд целевых белков, в том числе плазматический прекалликреин и фактор XI. Активный плазматический калликреин далее активирует фактор XII, что приводит к усилению контактной активации. В качестве альтернативы, серинпротеазная поликарбоксипептидаза может активировать плазматический калликреин, входящий в комплекс с высокомолекулярным кининогенином в многокомпонентном комплексе, образованном на поверхности клеток и матриксов (Shariat-Madar et al., Blood, 108:192-199 (2006)). Контактная активация является поверхностно опосредованным процессом, отвечающим частично за регуляцию тромбоза и воспаления, и опосредуется, по меньшей мере частично, фибринолитическим, комплементарным, кининогенным/кининовым и другими гуморальными и клеточными путями (для обзора см. Coleman, R., "Contact Activation Pathway", Hemostasis and Thrombosis, pp. 103-122, Lippincott Williams & Wilkins (2001); Schmaier, A.H., "Contact Activation", Thrombosis and Hemorrhage, pp. 105-128 (1998)). Биологическая значимость системы контактной активации для тромбоэмболических заболеваний подтверждается фенотипом дефицитных по фактору XII мышей. Более конкретно, дефицитные по фактору XII мыши были защищены от тромботической окклюзии сосудов на некоторых моделях тромбоза, а также моделях инсульта, и фенотип дефицитных по фактору XII мышей был идентичным таковому у дефицитных по фактору XI мышей (Renne et al., J Exp. Med., 202:271-281 (2005); Kleinschmitz et al., J Exp.Med., 203:513-518 (2006)). Тот факт, что фактор XI находится в прямом направлении от фактора XIIa в сочетании с идентичным фенотипом дефицитных по факторам XII и XI мышей, свидетельствует о том, что система контактной активации может играть важную роль в активации фактора XI in vivo.
Плазматический калликреин представляет собой зимоген трипсиноподобной серинпротеазы и находится в плазме. Структура гена аналогична структуре фактора XI. В целом, аминокислотная последовательность плазматического калликреина характеризуется 58% гомологией в отношении к фактору XI. Протеолитическая активация с помощью фактора XIIa по внутренней связи I 389-R390 дает тяжелую цепь (371 аминокислота) и легкую цепь (248 аминокислот). Активный сайт плазматического калликреина находится в легкой цепи. Легкая цепь плазматического калликреина реагирует с ингибиторами протеазы, в том числе с альфа-2-макроглобулином и Cl-ингибитором. Интересно, что гепарин значительно ускоряет ингибирование плазматического калликреина антитромбином III в присутствии высокомолекулярного кининогена (HMWK). В крови большая часть плазматического калликреина циркулирует в комплексе с HMWK. Плазматический калликреин расщепляет HMWK с высвобождением брадикинина. Высвобождение брадикинина приводит к повышению проницаемости сосудов и вазодилатации (для обзора см. Coleman, R., "Contact Activation Pathway", Hemostasis and Thrombosis, pp. 103-122, Lippincott Williams & Wilkins (2001); Schmaier A.H., "Contact Activation", Thrombosis and Hemorrhage, pp. 105-128 (1998)).
Больные, демонстрирующие генетический дефицит по C1-эстеразному ингибитору, страдают от наследственной ангиодистрофии (НАЕ), хронического заболевания, которое приводит к периодическому отеку по всему телу, включая руки, ноги, лицо, горло, половые органы и желудочно-кишечный тракт. Анализ показал, что волдыри, возникающие при острых эпизодах, содержат высокие уровни плазматического калликреина, и лечение белковым обратимым ингибитором плазматического калликреина экаллантидом (Kalbitor), одобрено FDA для лечения острых приступов НАЕ (Schneider, L, et al., J. Allergy Clin. Immunol., 120: p. 416 (2007)).
Кроме того, система плазматический калликреин-кинин является аномально избыточной у больных с диагнозом запущенного диабетического отека желтого пятна (DME). Последние публикации показали, что плазматический калликреин способствует обнаружению пропотевания жидкости через сосуды сетчатки и дисфункции в диабетических моделях грызунов (A. Clermont, et al., Diabetes, 60:1590 (2011)), и что лечение с помощью низкомолекулярного ингибитора плазматического калликреина облегчало наблюдаемую проницаемость сосудов в сетчатке и другие аномалии, связанные с кровообращением сетчатки.
Соединения-ингибиторы фактора XIa описаны в WO 2014160592, WO 2013022814, WO 2013022814, WO 2013022818, WO 2013055984, WO 2013056034, WO 2013056060, WO 2013118805, WO 2013093484, WO 2002042273, WO 2002037937, WO 2002060894, WO 2003015715, WO 2004002405, US 20040180855, WO 2004080971, WO 2004094372, US 20050228000, US 20050282805, WO 2005123680, US 20090036438, US 20120088758, US 20060074103, WO 2006062972, WO 2006076246, US 20060154915, US 20090062287, US 20060183771, WO 2007070818, WO 2007070816, WO 2007070826, WO 2008076805, WO 2008157162, WO 2009114677, WO 2011100402 и WO 2011100401.
Краткое описание настоящего изобретения
Настоящее изобретение относится к соединениям формулы I:
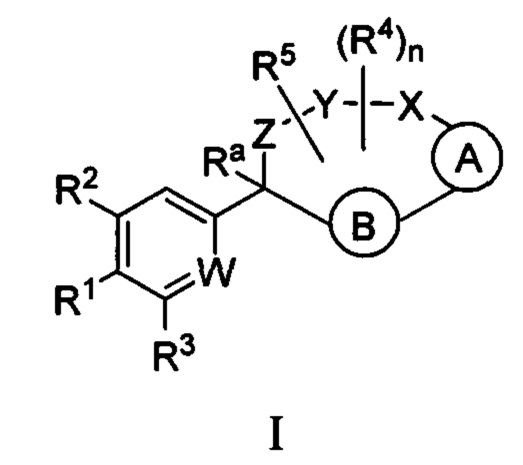
и их фармацевтически приемлемым солям. Соединениями формулы I являются селективные ингибиторы фактора XIa или двойные ингибиторы фактора XIa и плазматического калликреина, и, соответственно, они могут быть применимы при лечении, ингибировании или улучшении одного или нескольких болезненных состояний, на которые может оказывать благоприятное воздействие ингибирование фактора XIa или плазматического калликреина, включая тромбоз, эмболию, гиперкоагуляцию или фиброзные изменения. Соединения по настоящему изобретению дополнительно могут быть использованы в комбинации с другими терапевтически эффективными средствами, включая без ограничения другие лекарственные средства, используемые для лечения тромбоза, эмболии, гиперкоагуляции или фиброзных изменений. Настоящее изобретение дополнительно относится к способам получения соединений I и фармацевтических композиций, которые содержат соединения формулы I, и их фармацевтически приемлемых солей.
Подробное описание настоящего изобретения
Настоящее изобретение относится к соединениям формулы I:
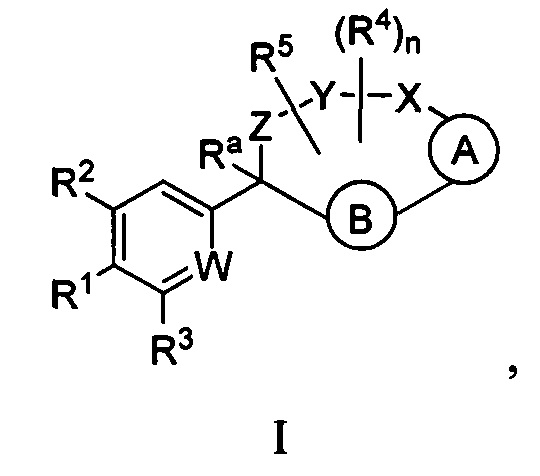
где  представляет собой арил или гетероарил, который необязательно замещен одной-тремя группами, независимо выбранными из группы, состоящей из галогена, оксо, циано, R6, OR6, C(O)OR6, C1-3 алкил-С(O)OR6, NR6R7, NH3+, C1-3 алкил-NR7R8, NHC(O)R6, NHC(O)OR6, NHC(O)OC3-6 циклоалкила, NHC(O)O-C1-3 алкил-OR7, NHC(O)O-C1-3 алкил-C(O)OH, С1-3 алкил-NHC(O)OR7, NHC(O)NR7R8, NHSO2R6, C(O)NR7R8, CH2C(O)NR7R8 и NHCONH-C1-3 алкилгетероциклила;
представляет собой арил или гетероарил, который необязательно замещен одной-тремя группами, независимо выбранными из группы, состоящей из галогена, оксо, циано, R6, OR6, C(O)OR6, C1-3 алкил-С(O)OR6, NR6R7, NH3+, C1-3 алкил-NR7R8, NHC(O)R6, NHC(O)OR6, NHC(O)OC3-6 циклоалкила, NHC(O)O-C1-3 алкил-OR7, NHC(O)O-C1-3 алкил-C(O)OH, С1-3 алкил-NHC(O)OR7, NHC(O)NR7R8, NHSO2R6, C(O)NR7R8, CH2C(O)NR7R8 и NHCONH-C1-3 алкилгетероциклила;
 представляет собой арил или гетероарил, который необязательно замещен одной-тремя группами, независимо выбранными из группы, состоящей из галогена, циано, оксидо, оксо, циклопропила, R6, OR6, C(O)OR6, C1-3 алкил-С(O)OR6, C(O)NR6R7 и NR6R7;
представляет собой арил или гетероарил, который необязательно замещен одной-тремя группами, независимо выбранными из группы, состоящей из галогена, циано, оксидо, оксо, циклопропила, R6, OR6, C(O)OR6, C1-3 алкил-С(O)OR6, C(O)NR6R7 и NR6R7;
W представляет собой N или N+O-;
Y-X представляет собой -C(O)NR6-, -C(O)O-, -CHC(O)OR7-NR6-, -CR6R7-C(O)NR6-, -CHC(O)R7-NR6-, -CHC(O)OR7-CH2-, -CHC(O)NR6R7-NR6-, -CHCR6R7OR8-NR6-, -CHCR6R7-NR6R7-NR6-, -OC(O)NR6-, -NR6C(O)NR6- или -SO2NR6-;
Z представляет собой С3-8 алкилен или С3-8 алкенилен, где один или два атома углерода в указанном алкилене и алкенилене могут быть заменены О, NR6, C=O, C(O)NR6, NR6C(О), S, SO или SO2;
R1 представляет собой арил, гетероарил, С3-6 циклоалкил или гетероалкил, где указанные арильные, гетероарильные, циклоалкильные и гетероциклильные группы необязательно замещены одним-четырьмя заместителями, независимо выбранными из группы, состоящей из галогена, нитро, циано, оксо, R6, OR6, C(O)R6, C(O)OR6, NR6R7, С1-3 алкил-NR6R7, NHC(O)R7, NHC(O)OR7, C(NH)NR6R7, С3-6 циклоалкила и гетероарила (который необязательно замещен галогеном, циано, циклопропилом, С(O)ОН, C(O)NR6R7 или R6);
R2 представляет собой водород, циано, галоген, R6 или OR6;
R3 представляет собой водород, циано, галоген, R6 или OR6;
каждый R4 независимо представляет собой C1-6 алкил, CO2R6, COR6 или CONR7R8, где указанный алкил необязательно замещен одним-тремя атомами галогена;
R5 представляет собой водород, галоген или C1-6 алкил;
или один из R4 и R5 может быть взят вместе с атомами между ними с образованием 3-6-членного кольца;
каждый R6 независимо представляет собой водород или C1-6 алкил, который необязательно замещен одной-тремя группами, независимо выбранными из группы, состоящей из галогена и гидрокси;
каждый R7 независимо представляет собой водород, C1-6 алкил, гетероарил или гетероциклил, где указанная алкильная группа необязательно замещена одной-тремя группами, независимо выбранными из группы, состоящей из галогена и гидрокси;
каждый R8 независимо представляет собой водород или C1-6 алкил;
Ra представляет собой водород, гидрокси или O(C1-6 алкил);
n представляет собой целое число от нуля до трех;
или их фармацевтически приемлемым солям.
Вариант осуществления настоящего изобретения относится к соединениям формулы I:
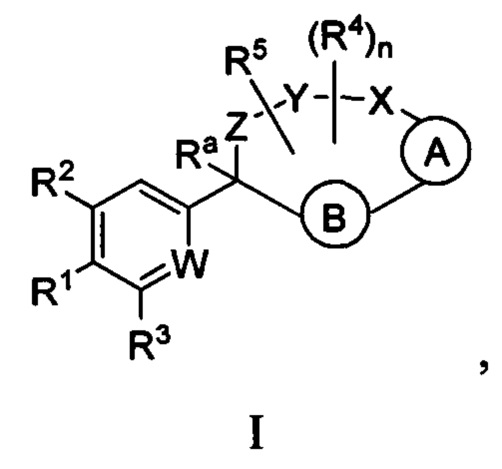
где представляет собой арил или гетероарил, который необязательно замещен одной-тремя группами, независимо выбранными из группы, состоящей из галогена, циано, R6, OR6, C(O)OR6, C1-3 алкил-С(O)OR6, NR6R7, NH3+, C1-3 алкил-NR7R8, NHC(O)R6, NHC(O)OR6, NHC(O)OC3-6 циклоалкила, NHC(O)O-C1-3 алкил-OR7, NHC(O)O-C1-3 алкил-C(O)OH, C1-3 алкил-NHC(O)OR7, NHC(O)NR7R8, NHSO2R6, C(O)NR7R8, CH2C(O)NR7R8 и NHCONH-C1-3 алкилгетероциклила;
представляет собой арил или гетероарил, который необязательно замещен одной-тремя группами, независимо выбранными из группы, состоящей из галогена, циано, оксо, R6, OR6, C(O)OR6, C1-3 алкил-С(O)OR6, C(O)NR6R7 и NR6R7;
W представляет собой N или N+O-;
Y-X представляет собой -C(O)NR6-, -C(O)O-, -CHC(O)OR7-NR6-, -CR6R7-C(O)NR6-, -CHC(O)R7-NR6-, -CHC(O)NR6R7-NR6-, -CHCR6R7OR8-NR6-, -CHCR6R7-NR6R7-NR6-, -OC(O)NR6-, -NR6C(O)NR6- или -SO2NR6-;
Z представляет собой C3-8 алкилен или C3-8 алкенилен, где один или два атома углерода в указанном алкилене и алкенилене могут быть заменены О, NR6, C=O, C(O)NR6, NR6C(O), S, SO или SO2;
R1 представляет собой арил, гетероарил, C3-6 циклоалкил или гетероалкил, где указанные арильные, гетероарильные, циклоалкильные и гетероциклильные группы необязательно замещены одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, нитро, циано, оксо, R6, OR6, C(O)R6, C(O)OR6, NR6R7, C1-3 алкил-NR6R7, NHC(O)R7, NHC(O)OR7, C(NH)NR6R7, С3-6 циклоалкила и гетероарила (который необязательно замещен галогеном, циано, С(O)NR6R7 или R6);
R2 представляет собой водород, циано, галоген, R6 или OR6;
R3 представляет собой водород, циано, галоген, R6 или OR6;
R4 представляет собой C1-6 алкил, CO2R6, COR6 или CONR7R8, где указанный алкил необязательно замещен одним-тремя атомами галогена;
R5 представляет собой водород, галоген или C1-6 алкил;
или R4 и R5 могут быть взяты вместе с атомами между ними с образованием 3-6-членного кольца;
R6 представляет собой водород или C1-6 алкил, который необязательно замещен одной-тремя группами, независимо выбранными из группы, состоящей из галогена и гидрокси;
R7 представляет собой водород, C1-6 алкил, гетероарил или гетероциклил, где указанная алкильная группа необязательно замещена одной-тремя группами, независимо выбранными из группы, состоящей из галогена и гидрокси;
R8 представляет собой водород или C1-6 алкил;
Ra представляет собой водород, гидрокси или O(C1-6 алкил);
n представляет собой целое число от нуля до трех;
или их фармацевтически приемлемым солям.
Вариант осуществления настоящего изобретения относится к соединениям формулы Ia:
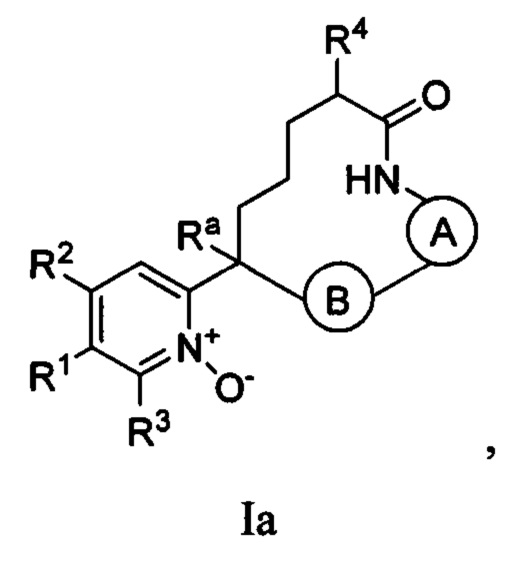
где представляет собой арил или гетероарил, который необязательно замещен одной-тремя группами, независимо выбранными из группы, состоящей из галогена, оксо, циано, R6, OR6, C(O)OR6, C1-3 алкил-С(O)OR6, NR6R7, NH3+, C1-3 алкил-NR7R8, NHC(O)R6, NHC(O)OR6, NHC(O)ОС3-6 циклоалкила, NHC(O)O-C1-3 алкил-OR7, NHC(O)O-C1-3 алкил-C(O)OH, C1-3 алкил-NHC(O)OR7, NHC(O)NR7R8, NHSO2R6, C(O)NR7R8, CH2C(O)NR7R8 и NHCONH-C1-3 алкилгетероциклила;
представляет собой арил или гетероарил, который необязательно замещен одной-тремя группами, независимо выбранными из группы, состоящей из галогена, циано, оксидо, оксо, циклопропила, R6, OR6, C(O)OR6, C1-3 алкил-С(O)OR6, C(O)NR6R7 и NR6R7;
R1 представляет собой арил, гетероарил, C3-6 циклоалкил или гетероалкил, где указанные арильные, гетероарильные, циклоалкильные и гетероциклильные группы необязательно замещены одним-четырьмя заместителями, независимо выбранными из группы, состоящей из галогена, нитро, циано, оксо, R6, OR6, C(O)R6, C(O)OR6, NR6R7, C1-3 алкил-NR6R7, NHC(O)R7, NHC(O)OR7, С(NH)NR6R7, С3-6 циклоалкила и гетероарила (который необязательно замещен галогеном, циано, циклопропилом, С(O)ОН, C(O)NR6R7 или R6);
R2 представляет собой водород, циано, галоген, R6 или OR6;
R3 представляет собой водород, циано, галоген, R6 или OR6;
R4 представляет собой C1-6 алкил, CO2R6, COR6 или CONR7R8, где указанный алкил необязательно замещен одним-тремя атомами галогена;
R6 представляет собой водород или C1-6 алкил, который необязательно замещен одной-тремя группами, независимо выбранными из группы, состоящей из галогена и гидрокси;
R7 представляет собой водород или C1-6 алкил, который необязательно замещен одной-тремя группами, независимо выбранными из группы, состоящей из галогена и гидрокси;
R8 представляет собой водород или C1-6 алкил;
Ra представляет собой водород, гидрокси или O(C1-6 алкил);
n представляет собой целое число от нуля до трех;
или их фармацевтически приемлемым солям.
Вариант осуществления настоящего изобретения относится к соединениям формулы Ib:
где представляет собой арил или гетероарил, который необязательно замещен одной-тремя группами, независимо выбранными из группы, состоящей из галогена, циано, R6, OR6, C(O)OR6, C1-3 алкил-С(O)OR6, NR6R7, NH3+, C1-3 алкил-NR7R8, NHC(O)R6, NHC(O)OR6, NHC(O)OC3-6 циклоалкила, NHC(O)O-C1-3 алкил-OR7, NHC(O)O-C1-3 алкил-C(O)OH, C1-3 алкил-NHC(O)OR7, NHC(O)NR7R8, NHSO2R6, C(O)NR7R8, CH2C(O)NR7R8 и NHCONH-C1-3 алкилгетероциклила;
представляет собой арил или гетероарил, который необязательно замещен одной-тремя группами, независимо выбранными из группы, состоящей из галогена, циано, оксо, R6, OR6, C(O)OR6, C1-3 алкил-С(O)OR6, C(O)NR6R7 и NR6R7;
R1 представляет собой арил, гетероарил, C3-6 циклоалкил или гетероалкил, где указанные арильные, гетероарильные, циклоалкильные и гетероциклильные группы необязательно замещены одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, нитро, циано, оксо, R6, OR6, C(O)R6, C(O)OR6, NR6R7, C1-3 алкил-NR6R7, NHC(O)R7, NHC(O)OR7, C(NH)NR6R7, C3-6 циклоалкила и гетероарила (который необязательно замещен галогеном, циано, C(O)NR6R7 или R6);
R2 представляет собой водород, циано, галоген, R6 или OR6;
R3 представляет собой водород, циано, галоген, R6 или OR6;
R4 представляет собой C1-6 алкил, CO2R6, COR6 или CONR7R8, где указанный алкил необязательно замещен одним-тремя атомами галогена;
R6 представляет собой водород или C1-6 алкил, который необязательно замещен одной-тремя группами, независимо выбранными из группы, состоящей из галогена и гидрокси;
R7 представляет собой водород или C1-6 алкил, который необязательно замещен одной-тремя группами, независимо выбранными из группы, состоящей из галогена и гидрокси;
R8 представляет собой водород или C1-6 алкил;
Ra представляет собой водород, гидрокси или O(C1-6 алкил);
n представляет собой целое число от нуля до трех;
или их фармацевтически приемлемым солям.
Согласно варианту осуществления настоящего изобретения представляет собой фенил, дигидрохинолинин или пиразолил, который необязательно замещен одной-тремя группами, независимо выбранными из группы, состоящей из галогена, оксо, циано, R6, OR6, C(O)OR6, NR6R7, NHC(O)OR6, NHC(O)O-C1-3 алкил-OR7 и NHC(O)O-C1-3 алкил-C(O)OH. В классе настоящего изобретения представляет собой фенил, который необязательно замещен одной-тремя группами, независимо выбранными из группы, состоящей из галогена, C(O)OR6, NHC(O)OR6 и NR6R7. В подклассе настоящего изобретения представляет собой фенил, который необязательно замещен одной-тремя группами, независимо выбранными из группы, состоящей из фтора, С(O)ОН, NHC(O)OH и NHC(O)OCH3.
Согласно варианту осуществления настоящего изобретения выбран из группы, состоящей из фенила, пирролила, пиразолила, имидазолила, триазолила, пиридинила, пиридинил-N-оксида, пиридазинила, пиримидинила, пиразинила и триазинила, где указанные группы необязательно замещены одной-тремя группами, независимо выбранными из группы, состоящей из галогена, оксидо, R6 и циклопропила. В классе настоящего изобретения выбран из группы, состоящей из фенила имидазолила, пиридинила и пиримидинила, где указанные группы необязательно замещены одной-тремя группами, независимо выбранными из группы, состоящей из галогена, оксидо, R6 и циклопропила. В подклассе настоящего изобретения представляет собой фенил, который необязательно замещен галогеном. В другом классе настоящего изобретения представляет собой имидазолил, который необязательно замещен метилом. В другом подклассе настоящего изобретения представляет собой пиридинил, который необязательно замещен галогеном. В другом подклассе настоящего изобретения представляет собой пиридинил, который необязательно замещен оксидо. В другом подклассе настоящего изобретения представляет собой пиримидинил, который необязательно замещен галогеном.
Согласно варианту осуществления настоящего изобретения Y-X представляет собой C(O)NR6. В классе настоящего варианта осуществления Y-X представляет собой C(O)NH. Согласно другому варианту осуществления настоящего изобретения Y-X представляет собой CHC(O)OR7-NR6-. В классе настоящего варианта осуществления Y-X представляет собой CHC(O)OH-NH. Согласно варианту осуществления настоящего изобретения Y-X представляет собой CHC(O)NR6R7-NR6. В классе настоящего варианта осуществления Y-X представляет собой СНС(O)NH(СН3)-NH. Согласно варианту осуществления настоящего изобретения Y-X представляет собой CHC(O)OR7-NR6.
Согласно варианту осуществления настоящего изобретения Z представляет собой C3-8 алкилен.
Согласно варианту осуществления настоящего изобретения W представляет собой N+O-. Согласно другому варианту осуществления настоящего изобретения W представляет собой N.
Согласно варианту осуществления настоящего изобретения R1 представляет собой арил, который необязательно замещен одним-четырьмя заместителями, независимо выбранными из группы, состоящей из хлора, фтора, йода, метила, циклопропила, OCF3, OCF2, CF3, CF2 и гетероарила (который необязательно замещен галогеном, циано, циклопропилом, С(O)ОН, метилом, CF3 или CF2). В классе настоящего варианта осуществления R1 представляет собой фенил, который необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, C3-6 циклоалкила и тетразолила.
Согласно варианту осуществления настоящего изобретения R2 представляет собой водород.
Согласно варианту осуществления настоящего изобретения R3 представляет собой водород.
Согласно варианту осуществления настоящего изобретения R4 представляет собой C1-6 алкил. В классе настоящего изобретения R4 представляет собой метил.
Согласно варианту осуществления настоящего изобретения R5 представляет собой водород. Согласно другому варианту осуществления настоящего изобретения R5 представляет собой галоген. В классе настоящего изобретения R5 представляет собой фтор. Согласно другому варианту осуществления настоящего изобретения R5 представляет собой C1-6 алкил. В классе настоящего изобретения R5 представляет собой метил.
Согласно варианту осуществления настоящего изобретения n равно нулю. Согласно другому варианту осуществления настоящего изобретения n равно одному. Согласно другому варианту осуществления настоящего изобретения n равно двум. Согласно другому варианту осуществления настоящего изобретения n равно трем.
Согласно варианту осуществления настоящего изобретения Ra представляет собой водород или гидрокси. В классе настоящего изобретения Ra представляет собой водород. В другом классе настоящего изобретения Ra представляет собой гидрокси.
Под ссылкой на предпочтительные классы и подклассы, изложенные выше, подразумевается включение всех комбинаций конкретных и предпочтительных групп, если не отмечено иное.
Особые варианты осуществления настоящего изобретения включают в себя без ограничения соединения, определенные в настоящем описании как соединения примеров 1-172 или их фармацевтически приемлемые соли.
Также в объем настоящего изобретения включена фармацевтическая композиция, которая содержит соединение формулы I, формулы Ia или формулы Ib, как описано выше, и фармацевтически приемлемый носитель. Также предусмотрено, что настоящее изобретение охватывает фармацевтическую композицию, которая содержит фармацевтически приемлемый носитель и любое из соединений, особым образом раскрытое в настоящей заявке. Эти и другие аспекты настоящего изобретения будут очевидны из содержащихся в настоящем описании идей.
Настоящее изобретение также относится к композициям для ингибирования потери тромбоцитов, ингибирования образования агрегатов тромбоцитов, ингибирования образования фибрина, ингибирования образования тромба, ингибирования образования эмбола, лечения воспалительных нарушения, лечения диабетической ретинопатии и лечения наследственного ангионевротического отека у млекопитающего, содержащим соединение в соответствии с настоящим изобретением в фармацевтически приемлемом носителе. Эти композиции необязательно могут включать в себя антикоагулянты, противотромбоцитарные средства и тромболитические средства. Композиции могут быть добавлены в кровь, продукты крови или органы млекопитающего для достижения желаемых ингибирований.
Настоящее изобретение также относится к композициям для предупреждения или лечения нестабильной стенокардии, рефракторной стенокардии, инфаркта миокарда, транзиторных ишемических атак, предсердной фибрилляции, тромботического инсульта, эмболического инсульта, тромбоза глубоких вен, диссеминированного внутрисосудистого свертывания, внутриглазного накопления фибрина и реокклюзии или рестеноза реканализированных сосудов у млекопитающего, содержащим соединение в соответствии с настоящим изобретением в фармацевтически приемлемом носителе. Эти композиции необязательно могут включать в себя антикоагулянты, противотромбоцитарные средства и тромболитические средства.
Настоящее изобретение также относится к способу лечения тромбогенности поверхности у млекопитающего путем присоединения к поверхности, либо ковалентно, либо нековалентно, соединения в соответствии с настоящим изобретением.
Соединения в соответствии с настоящим изобретением представляют собой ингибиторы фактора XIa и могут иметь терапевтическое значение, например, в предупреждении заболевания коронарной артерии. Соединения являются селективными ингибиторами фактора XIa или двойными ингибиторами фактора XIa и плазматического калликреина.
Следует учитывать, что, как используется в настоящем документе, упоминание соединений структурной формулы I, формулы Ia и формулы Ib предусматривает фармацевтически приемлемые соли, а также соли, которые не являются фармацевтически приемлемыми, если их используют в качестве предшественников свободных соединений или их фармацевтически приемлемых солей или в других манипуляциях синтеза.
Соединения в соответствии с настоящим изобретением могут быть введены в форме фармацевтически приемлемой соли. Термин «фармацевтически приемлемая соль» относится к солям, полученным из фармацевтически приемлемых нетоксичных оснований или кислот, в том числе из неорганических или органических оснований и неорганических или органических кислот. Соли основных соединений, охватываемые термином «фармацевтически приемлемая соль», относятся к нетоксичным солям соединений в соответствии с настоящим изобретением, которые, как правило, получают путем осуществления реагирования свободного основания с подходящей органической или неорганической кислотой. Типичные соли основных соединений в соответствии с настоящим изобретением включают в себя без ограничения следующие: ацетат, аскорбат, адипат, альгинат, аспират, бензолсульфонат, бензоат, бикарбонат, бисульфат, битартрат, борат, бромид, бутират, камфорат, камфорсульфонат, камсилат, карбонат, хлорид, клавуланат, цитрат, циклопентанпропионат, диэтилуксусную, диглюконат, дигидрохлорид, додецилсульфонат, эдетат, эдизилат, эстолат, эзилат, этансульфонат, муравьиная, фумарат, глюцептат, глюкогептаноат, глюконат, глутамат, глицерофосфат, гликолиларсанилат, гемисульфат, гептаноат, гексаноат, гексилрезорцинат, гидрабамин, гидробромид, гидрохлорид, 2-гидроксиэтансульфонат, гидроксинафтоат, йодид, изоникотиновая, изотионат, лактат, лактобионат, лаурат, малат, малеат, манделат, мезилат, метилбромид, метилнитрат, метилсульфат, метансульфонат, мукат, 2-нафталинсульфонат, напсилат, никотинат, нитрат, N-метилглюкаминаммониевая соль, олеат, оксалат, памоат (эмбонат), пальмитат, пантотенат, пектинат, персульфат, фосфат/дифосфат, пимелиновая, фенилпропионовая, полигалактуронат, пропионат, салицилат, стеарат, сульфат, основный ацетат, сукцинат, таннат, тартрат, теоклат, тиоцианат, тозилат, триэтиодид, трифторацетат, ундеконат, валерат и т.п. Кроме того, если соединения в соответствии с настоящим изобретением несут кислотный фрагмент, то их подходящие фармацевтически приемлемые соли включают в себя без ограничения соли, полученные из неорганических оснований, в том числе алюминия, аммония, кальция, меди, трехвалентного железа, двухвалентного железа, лития, магния, марганца, двухвалентного марганца, калия, натрия, цинка и т.п. Также предусмотрены соли аммония, кальция, магния, калия и натрия. Соли, полученные из фармацевтически приемлемых органических нетоксичных оснований, включают в себя соли первичных, вторичных и третичных аминов, циклических аминов, дициклогексиламинов и основных ионообменных смол, такие как аргинин, бетаин, кофеин, холин, N,N-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этиламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и т.п. Также предусмотрены основные содержащие азот группы, которые могут быть кватернизованы с такими средствами, как галоиды низшего алкила, такие как метил, этил, пропил и бутилхлорид, бромиды и йодиды; диалкилсульфаты, такие как диметил, диэтил, дибутил; и диамилсульфаты, длинноцепочечные галоиды, такие как децил, лаурил, миристил и стеарилхлориды, бромиды и йодиды, аралкилгалоиды, такие как бензил и фенэтилбромиды, и другие.
Такие соли могут быть получены известными способами, например, путем смешивания соединения в соответствии с настоящим изобретением в эквивалентном количестве и раствора, содержащего желаемые кислоту, основание или подобное, а затем сбора желаемой соли путем фильтрования соли или отгонки растворителя. Соединения в соответствии с настоящим изобретением и их соли могут образовывать сольваты с растворителем, таким как вода, этанол или глицерин. Соединения в соответствии с настоящим изобретением могут образовывать соль присоединения кислоты и соль с основанием в одно и то же время в соответствии с типом заместителя боковой цепи.
Если соединения формулы I, формулы Ia или формулы Ib одновременно содержат кислотные и основные группы в молекуле, то настоящее изобретение также предусматривает кроме упомянутых форм солей внутренние соли или бетаины (цвиттерионы).
Настоящее изобретение охватывает все стереоизомерные формы соединений формулы I, формулы Ia и формулы Ib. Если не указана конкретная стереохимия, настоящее изобретение охватывает все такие изомерные формы этих соединений. Все центры асимметрии, которые имеются в соединениях формулы I, формулы Ia и формулы Ib, независимо друг от друга могут иметь (R)-конфигурацию или (S)-конфигурацию. Если связи с хиральным углеродом изображены в виде прямых линий в структурных формулах в соответствии с настоящим изобретением, то следует понимать, что формулой охватываются как (R), так и (S)-конфигурации хирального углерода, а следовательно оба энантиомера и их смеси. Если изображена конкретная конфигурация, то предполагается энантиомер (либо (R), либо (S) в данном центре). Подобным образом, если название соединения упоминается без хирального обозначения для хирального углерода, то следует понимать, что это название охватывает как (R), так и (S)-конфигурации хирального углерода, а, следовательно, и отдельные энантиомеры и их смеси. Получение конкретных стереоизомеров или их смесей может быть определено в примерах, в которых получают такие стереоизомеры или смеси, но это никоим образом не ограничивает включение всех стереоизомеров и их смесей в объем настоящего изобретения.
Настоящее изобретение относится ко всем возможным энантиомерам и диастереомерам, а также к смесям двух или более стереоизомеров, например, к смесям энантиомеров и/или диастереомеров, во всех отношениях. Таким образом, энантиомеры являются предметом настоящего изобретения в энантиомерно чистой форме, как левовращающие, так и правовращающие антиподы, в форме рацематов и в форме смесей двух энантиомеров во всех отношениях. В случае цис/транс-изомерии настоящее изобретение относится как к цис-форме, так и к транс-форме, а также к смесям этих форм во всех отношениях. Получение отдельных стереоизомеров может быть выполнено, при необходимости, путем разделения смеси традиционными способами, например, с помощью хроматографии или кристаллизации, с помощью применения стереохимически однородных исходных материалов для синтеза или с помощью стереоселективного синтеза. Необязательно дериватизация может быть выполнена перед разделением стереоизомеров. Разделение смеси стереоизомеров может быть выполнено при промежуточной стадии в ходе синтеза соединения формулы I, формулы Ia или формулы Ib, или оно может быть выполнено на конечном рацемическом продукте. Абсолютная стереохимия может быть определена с помощью рентгеновской кристаллографии кристаллических продуктов или кристаллических промежуточных соединений, которые дериватизируют, при необходимости, с реагентом, содержащим стереогенный центр известной конфигурации. Если соединения в соответствии с настоящим изобретением способны к таутомеризации, то все отдельные таутомеры, а также их смеси попадают в объем настоящего изобретения. Настоящее изобретение относится ко всем таким изомерам, а также солям, сольватам (в том числе гидратам) и сольватированным солям таких рацематов, энантиомерам, диастереомерам и таутомерам, а также их смесям.
В соединениях в соответствии с настоящим изобретением атомы могут демонстрировать свой природный изотопный состав, или один или несколько из атомов могут быть искусственно обогащены конкретным изотопом, имеющим то же атомное число, но атомную массу или массовое число, отличающиеся от атомной массы или массового числа, преимущественно встречающихся в природе. Настоящее изобретение предусматривает все подходящие изотопные вариации конкретно и в общих чертах описываемых соединений. Например, разные изотопные формы водорода (Н) включают в себя протий (1Н) и дейтерий (2H). Протий является преобладающим изотопом водорода, встречающимся в природе. Обогащение дейтерием может обеспечить определенные терапевтические преимущества, такие как увеличение in vivo время полужизни или снижение необходимых дозировок, или может обеспечить соединение, применимое в качестве стандарта для характеристики биологических образцов. Изотопно обогащенные соединения могут быть получены без излишних экспериментов с помощью обычных методик, хорошо известных специалистам в данной области, или с помощью процессов, аналогичных описываемым в общих схемах процессов и примерах в настоящем документе с использованием соответствующих изотопно обогащенных реагентов и/или промежуточных соединений.
Если какая-либо переменная (например, R4 и т.п.) встречается более чем один раз в какой-либо структурной составляющей, ее определение в каждом случае будет независимым в каждом случае. Также комбинации заместителей и переменных допустимы, только если такие комбинации дают в результате стабильные соединения. Линии, прочерченные в кольцевые системы от заместителей, показывают, что указанная связь может быть присоединена к любому из замещаемых кольцевых атомов. Если кольцевая система является бициклической, то это означает, что связь прикрепляется к любому из подходящих атомов в любом кольце бициклического фрагмента.
Следует учитывать, что один или несколько атомов кремния (Si) могут быть включены в соединения в соответствии с настоящим изобретением вместо одного или нескольких атомов углерода специалистом в данной области для обеспечения соединений, которые являются химически стабильными и которые могут быть легко синтезированы методиками, известными в уровне техники, из легко доступных исходных материалов. Углерод и кремний отличаются своими ковалентными радиусами, что ведет к отличиям в длине связи и в пространственном расположении по сравнению с аналогичными связями С-элемента и Si-элемента. Эти различия приводят к незначительным изменениям размера и формы содержащих кремний соединений по сравнению с содержащими углерод. Рядовому специалисту в данной области будет понятно, что различия в размере и форме могут приводить к незначительным или существенным изменениям в эффективности, растворимости, отсутствии целевой активности, упаковочных свойствах и т.д. (Diass, J.О. et al. Organometallics (2006) 5:1188-1198; Showell, G.A. et al. Bioorganic & Medicinal Chemistry Letters (2006) 16:2555-2558).
Следует учитывать, что заместители и партнеры по замещению в соединениях в соответствии с настоящим изобретением могут быть выбраны специалистом в данной области для обеспечения соединений, которые являются химически стабильными и которые могут быть легко синтезированы согласно методикам, известным в уровне техники, а также согласно способам, изложенным ниже, из легко доступных исходных материалов. Если заместитель сам является замещенным более чем одной группой, то следует учитывать, что эти несколько групп могут быть на одном и том же углероде или на разных углеродах, при том условии, что в результате образуется стабильная структура. Фразу «необязательно замещенный» (одним или несколькими заместителями) следует понимать как означающую, что рассматриваемая группа либо является незамещенной, либо может быть замещена одним или несколькими заместителями.
Кроме того, соединения в соответствии с настоящим изобретением могут существовать в аморфной форме и/или в одной или нескольких кристаллических формах, и, таким образом, все аморфные и кристаллические формы, а также их смеси, соединений формулы I, формулы Ia и формулы Ib охватываются объемом настоящего изобретения. Кроме того, некоторые из соединений в соответствии с настоящим изобретением могут образовывать сольваты с водой (т.е. гидрат) или с традиционными органическими растворителями. Такие сольваты и гидраты, в частности, фармацевтически приемлемые сольваты и гидраты, соединений в соответствии с настоящим изобретением таким же образом охватываются объемом настоящего изобретения наряду с несольватированными и ангидридными формами.
Упоминание соединений в соответствии с настоящим изобретением, как соединений конкретной формулы или варианта осуществления, например, формулы I, формулы Ia или формулы Ib, или какой-либо другой общей структурной формулы, или как конкретного соединения, описываемого или заявляемого в настоящем документе, предусматривает конкретное соединение или соединения, попадающие в объем формулы или варианта осуществления, в том числе их соли, в частности, фармацевтически приемлемые соли, сольваты таких соединений и их сольватированные солевые формы, где такие формы возможны, если не указано иное.
Также в случае, если в соединениях в соответствии с настоящим изобретением имеется карбоновокислотная (-СООН) или спиртовая группа, могут быть использованы фармацевтически приемлемые сложные эфиры производных карбоновых килот, такие как метиловые, этиловые, или пивалоилоксиметиловые, или ацильные производные спиртов, такие как O-ацетильные, O-пивалоильные, O-бензоильные и O-аминоацильные. Такие сложные эфиры и ацильные группы, известные в уровне техники, предусматриваются с целью модификации характеристик растворимости или гидролиза для применения в качестве составов замедленного высвобождения или пролекарственных составов.
Любая модификация фармацевтически приемлемого пролекарства соединения в соответствии с настоящим изобретением, которая обеспечивает в результате превращение in vivo в соединение, охватываемое объемом настоящего изобретения, также попадает в объем настоящего изобретения. Например, сложные эфиры необязательно могут быть получены путем эстерификации доступной карбоновокислотной группы или путем образования сложного эфира на доступной гидроксигруппе в соединении. Подобным образом, могут быть получены лабильные амиды. Фармацевтически приемлемые сложные эфиры или амиды соединений в соответствии с настоящим изобретением могут быть получены для действия в качестве пролекарств, которые могут быть обратно гидролизованы в кислоту (или -СОО- в зависимости от рН жидкости или ткани, в которой происходит превращение) или гидроксиформу, в частности, in vivo, и в таком качестве охватываются объемом настоящего изобретения. Примеры фармацевтически приемлемых модификаций пролекарства включают в себя без ограничения -C1-6алкильные сложные эфиры и -C1-6алкильные, замещенные фенилом, сложные эфиры.
Следовательно, соединения общих структурных формул, вариантов осуществления и конкретные соединения, описываемые и заявляемые в настоящем документе, охватывают соли, все их возможные стереоизомеры и таутомеры, физические формы (например, аморфные и кристаллические формы), сольватные и гидратные формы и любую комбинацию таких форм, а также их соли, их пролекарственные формы и соли их пролекарственных форм, где такие формы возможны, если не указано иное.
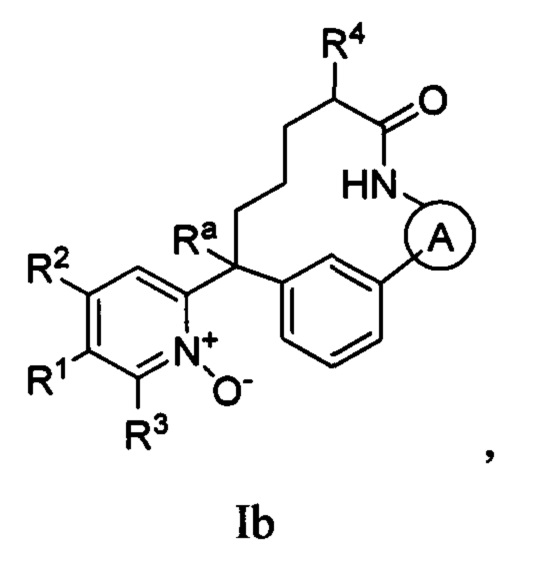
Если в настоящем документе не указано иное, термины «алкил» и «алкилен» предусматривают как разветвленные, так и прямоцепочечные насыщенные алифатические углеводородные группы с определенным числом атомов углерода. По всему настоящему описанию применяются традиционно используемые сокращения для алкильных групп, например, метил, может быть представлен типичными сокращениями, в том числе «Me», или СН3, или символом, который представляет собой продленную связь в качестве концевой группы, например,  , этил может быть обозначен «Et» или СН2СН3, пропил может быть обозначен «Pr» или CH2CH2CH3, бутил может быть обозначен «Bu» или CH2CH2CH2CH3 и т.д. Например, «C1-4алкил» (или «C1-C4алкил») означает линейные или разветвленные алкильные группы, в том числе все изомеры, с определенным числом атомов углерода. Например, структуры
, этил может быть обозначен «Et» или СН2СН3, пропил может быть обозначен «Pr» или CH2CH2CH3, бутил может быть обозначен «Bu» или CH2CH2CH2CH3 и т.д. Например, «C1-4алкил» (или «C1-C4алкил») означает линейные или разветвленные алкильные группы, в том числе все изомеры, с определенным числом атомов углерода. Например, структуры
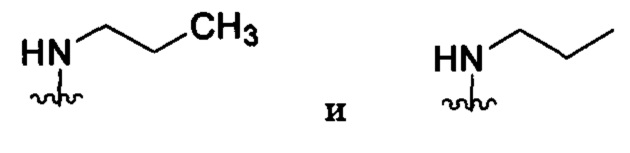
имеют эквивалентные значения. C1-4алкил включает в себя н-, изо-, втор- и трет-бутил, н- и изопропил, этил и метил. Если число не указано, то для линейных или разветвленных алкильных групп используются 1-4 атома углерода.
Если в настоящем документе не указано иное, термин «алкенилен» предусматривает как разветвленные, так и прямые ненасыщенные алифатические углеводородные группы, имеющие определенное число атомов углерода и содержащие по меньшей мере одну углерод-углерод двойную связь.
Если не указано иное, термин «циклоалкил» означает моноциклическую или бициклическую насыщенную алифатическую углеводородную группу с определенным числом атомов углерода. Например, «циклоалкил» включает в себя циклопропил, циклобутил, циклопентил, циклогексил и т.д.
Если не указано иное, термин «галогеновый» или «галоген» означает фтор, хлор, бром или йод.
Если не указано иное, используемый в настоящем документе термин «гетероарил» представляет стабильную моноциклическую или бициклическую кольцевую систему максимум с 10 атомами в каждом кольце, при этом по меньшей мере одно кольцо является ароматическим, и по меньшей мере одно кольцо содержит от 1 до 4 гетероатомов, выбранных из группы, состоящей из О, N и S. Бициклические гетероарильные кольцевые системы включают в себя слитые кольцевые системы, в которых два кольца имеют общие два атома, и спиро кольцевые системы, в которых два кольца имеют общий один атом. Гетероарильные группы в объеме данного определения включают в себя без ограничения бензоимидазолил, бензофуранил, бензофуразанил, бензопиразолил, бензотриазолил, бензотиофенил, бензоксазолил, карбазолил, карболинил, циннолинил, фуранил, индолинил, индолил, индолазинил, индазолил, изобензофуранил, изоиндолил, изохинолил, изотиазолил, изоксазолил, нафтпиридинил, оксадиазолил, оксазолил, оксазолин, изоксазолин, пиранил, пиразинил, пиразолил, пиридазинил, пиридопиридинил, пиридил, пиримидинил, пирролил, хиназолинил, хинолил, хиноксалинил, тетразолил, тетразолопиридил, тиадиазолил, тиазолил, тиенил, триазолил, дигидробензоимидазолил, дигидробензофуранил, дигидробензотиофенил, дигидробензоксазолил, дигидроиндолил, дигидрохинолинил, метилендиоксибензол, бензотиазолил, бензотиенил, хинолинил, изохинолинил, оксазолил, тетра-гидрохинолин и 3-оксо-3,4-дигидро-2N-бензо[b][1,4]тиазин. Если гетероарил содержит атомы азота, то следует учитывать, что соответствующие ему N-оксиды также охватываются данным определением.
Если не указано иное, используемый в настоящем документе термин «гетероцикл» или «гетероциклил» означает стабильную неароматическую моноциклическую или бициклическую кольцевую систему максимум с 10 атомами в каждом кольце, если не указано иное, содержащую от 1 до 4 гетероатомов, выбранных из группы, состоящей из О, N, S, SO или SO2. Бициклические гетероциклические кольцевые системы включают в себя слитые кольцевые системы, в которых два кольца имеют общие два атома, и спиро кольцевые системы, в которых два кольца имеют общий один атом. Термин «гетероциклил», поэтому, включает в себя без ограничения следующее: пиперазинил, пиперидинил, пирролидинил, морфолинил, тиоморфолинил, тетрагидропиранил, дигидропиперидинил, тетрагидротиофенил и т.п. Если гетероцикл содержит азот, то следует учитывать, что соответствующие ему N-оксиды также охватываются данным определением.
Если не указано иное, термин «арил» означает любое стабильное моноциклическое или бициклическое углеродное кольцо максимум с 12 атомами в каждом кольце, при этом по меньшей мере одно кольцо является ароматическим. Примеры таких арильных элементов включают в себя фенил, нафтил, тетрагидронафтил и инданил.
Диатомит Celite® (Fluka) представляет собой диатомовую землю и может называться «Celite».
Если в настоящем документе не указано иное, структуры, содержащие переменные заместителя, такие как приведенная ниже переменная «R»:
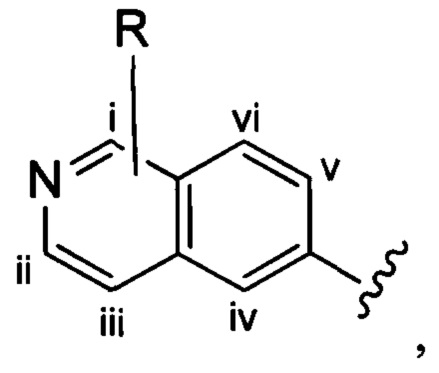
которая изображена как не присоединенная к какому-либо одному конкретному атому углерода бициклического кольца, представляют структуры, в которых переменная необязательно может быть присоединена к любому атому углерода бициклического кольца. Например, переменная R, показанная в приведенной выше структуре, может быть присоединена к любому из 6 атомов углерода бициклического кольца i, ii, iii, iv, v или vi.
Если в настоящем документе не указано иное, бициклические кольцевые системы включают в себя слитые кольцевые системы, в которых два кольца имеют общие два атома, и спиро кольцевые системы, в которых два кольца имеют общий один атом.
Настоящее изобретение также относится к медикаментам, содержащим по меньшей мере одно соединение формулы I, формулы Ia или формулы Ib, и/или фармацевтически приемлемую соль соединения формулы I, формулы Ia или формулы Ib, и/или необязательно стереоизомерную форму соединения формулы I, формулы Ia или формулы Ib, или фармацевтически приемлемую соль стереоизомерной формы соединения формулы I, формулы Ia или формулы Ib, вместе с фармацевтически подходящими и фармацевтически приемлемыми средой-носителем, аддитивными и/или другими активными веществами и вспомогательными средствами.
Антикоагулянтная терапия показана для лечения и предупреждения ряда тромботических состояний, в частности, заболевания коронарной артерии и цереброваскулярного заболевания. Специалистам в данной области безусловно известны обстоятельства, тербующие антикоагулянтной терапии. Используемый в настоящем документе термин «больной» означает млекопитающих, таких как приматы, люди, овцы, лошади, крупный рогатый скот, свиньи, собаки, коты, крысы и мыши.
Ингибирование фактора XIa или двойное ингибирование фактора XIa/плазматического калликреина применимо не только в антикоагулянтной терапии индивидуумов с тромботическими состояниями, но также применимы при необходимости ингибирования коагуляции крови, напимер, для предупреждения коагуляции хранящейся цельной крови и для предупреждения коагуляции в других биологических образцах для тестирования или хранения. Таким образом, ингибиторы фактора XIa или двойные ингибиторы фактора XIa/плазматического калликреина могут быть добавлены в или введены в контакт с какими-либо средами, содержащими или предположительно содержащими тромбин, и для которых желательно, чтобы коагуляция крови была ингибирована, например, при введение крови млекопитающего в контакт с материалом, выбранным из группы, состоящей из сосудистых трансплантатов, стентов, ортопедического протеза, сердечного протеза и систем искусственного кровообращения.
Соединения в соответствии с настоящим изобретением могут быть применимы для лечения или предупреждения венозной тромбоэмболии (например, обструкции или окклюзии вены при отрыве тромба; обструкции или окклюзии легочной артерии при отрыве тромба), кардиогенной тромбоэмболии (например, обструкции или окклюзии сердца при отрыве тромба), артериального тромбоза (например, образования тромба в артерии, что может вызывать инфаркт ткани, питаемой этой артерией), атеросклероза (например, артериосклероза, характеризуемого неравномерно распредиляемыми отложениями липидов) у млекопитающих, а также для снижения тенденции сгущения крови для устройств, контактирующих с кровью.
Примеры венозной тромбоэмболии, которую можно лечить или предупреждать соединениями в соответствии с настоящим изобретением, включают в себя обструкцию вены, обструкцию легочной артерии (легочную эмболию), тромбоз глубоких вен, тромбоз, ассоциированный со злокачественной опухолью и противораковой химиотерапией, тромбоз, присущий тромбофилическим заболеваниям, таким как дефицит белка С, дефицит белка S, дефицит антитромбина III и фактора V Лейдена, а также тромбоз в результате приобретенных тромбофилических нарушений, таких как системная красная волчанка (воспалительное заболевание соединительной ткани). Также в отношении венозной тромбоэмболии соединения в соответствии с настоящим изобретением могут быть применимы для поддержания проходимости постоянных катетеров.
Примеры кардиогенной тромбоэмболии, которую можно лечить или предупреждать соединениями в соответствии с настоящим изобретением, включают в себя тромбоэмболический инсульт (оторванный тромб вызывает неврологическое заболевание, связанное с нарушенным кровоснабжением мозга), кардиогенную тромбоэмболию, ассоциированную с предсердной фибрилляцией (быстрое нерегулярное сокращение мышечных фибрилл верхней камеры сердца), кардиогенную тромбоэмболию, ассоциированную с искусственными клапанами сердца, такими как механические клапаны сердца, и кардиогенную тромбоэмболию, ассоциированную с заболеванием сердца.
Примеры артериального тромбоза включают в себя нестабильную стенокардию (сильная сдавливающая боль в грудной клетке коронарного происхождения), инфаркт миокарда (смерть мышечных клеток сердца в результате недостаточного кровоснабжения), ишемическое заболевание сердца (локальная анемия из-за обструкции (такой как по причине артериального сужения) кровоснабжения), реокклюзию в ходе или после чрескожной транслюминальной коронарной ангиопластики, рестеноз после чрескожной транслюминальной коронарной ангиопластики, окклюзии аортокоронарных шунтирований и окклюзионное цереброваскулярное заболевание. Также при артериальном тромбозе соединения в соответствии с настоящим изобретением могут быть применимы для поддержания проходимости в артериовенозных канюлях.
Примеры атеросклероза включают в себя артериосклероз.
Соединения в соответствии с настоящим изобретением также могут быть ингибиторами калликреина и особенно применимыми для лечения наследственного ангионевротического отека.
Примеры устройств, которые приводятся в контакт с кровью включают в себя сосудистые трансплантаты, стенты, ортопедический протез, сердечный протез и системы искусственного кровообращения.
Медикаменты в соответствии с настоящим изобретением могут быть введены пероральным, ингаляционным, ректальным или чрескожным введением или с помощью подкожной, внутрисуставной, внутрибрюшинной или внутривенной инъекции. Пероральное введение является предпочтительным. Возможно покрытие соединениями формулы (I) стентов и других поверхностей, которые привдятся в контакт с кровью в организме.
Настоящее изобретение также относится к процессу получения медикамента, который предусматривает приведение по меньшей мере одного соединения формулы (I) в подходящую форму введения с использованием фармацевтически подходящего и фармацевтически приемлемого носителя и необязательно дополнительных подходящих активных веществ, добавок или вспомогательных средств.
Подходящими твердыми или галеновыми препаративными формами являются, например, гранулы, порошки, покрытые таблетки, таблетки, (микро)капсулы, суппозитории, сиропы, соки, суспензии, эмульсии, капли или инъекционные растворы и препараты с пролонгированным высвобождением активного вещества, для получения которых используют традиционные вспомогательные ингредиенты, такие как среды-носители, разрыхлители, связующие, покрывающие средства, разбухающие средства, вещества, способствующие скольжению, или смазывающие средства, ароматизаторы, посластители и солюбилизаторы. Обычно используемыми вспомогательными средствами, которые могут быть упомянуты, являются магния карбонат, титана диоксид, лактоза, маннит и другие сахара, тальк, лактоза, желатин, крахмал, целлюлоза и ее производные, животные и растительные масла, такие как жир печени трески, подсолнечное, арахисовое или кунжутное масло, полиэтиленгликоль и растворители, такие как, например, стерильная вода и одно- или многоатомные спирты, такие как глицерин.
Режим введения дозы с использованием ингибиторов фактора XIa или двойных ингибиторов фактора XIa/плазматического калликреина, выбирают в соответствии с рядом факторов, в том числе тип, вид, возраст, масса, пол и состояние здоровья больного; тяжесть состояния, подлежащего лечению; путь введения; почечная и печеночная функции больного; а также конкретное используемое соединение или его соль. Рядовой врач или ветеринар сможет легко определить и назначить эффективное количество лекарственного средства, необходимое для предупреждения, обращения или прекращения прогрессирования состояния.
Пероральные дозировки ингибиторов фактора XIa или двойных ингибиторов фактора XIa/плазматического калликреина при использовании для указанных эффектов будут варьировать от приблизительно 0,01 мг на кг массы тела в сутки (мг/кг/сутки) до приблизительно 30 мг/кг/сутки, предпочтительно 0,025-7,5 мг/кг/сутки, более предпочтительно 0,1-2,5 мг/кг/сутки и наиболее предпочтительно 0,1-0,5 мг/кг/сутки (если не указано иное, количества активных ингредиентов основываются на свободном основании). Например, 80-кг больной может получать от приблизительно 0,8 мг/сутки до 2,4 г/сутки, предпочтительно 2-600 мг/сутки, более предпочтительно 8-200 мг/сутки и наиболее предпочтительно 8-40 мг/кг/сутки. Соответственным образом полученный медикамент для введения один раз в сутки, таким образом, может содержать от 0,8 мг до 2,4 г, предпочтительно от 2 мг до 600 мг, более предпочтительно от 8 мг до 200 мг и наиболее предпочтительно от 8 мг до 40 мг, например, 8 мг, 10 мг, 20 мг и 40 мг. Предпочтительно ингибиторы фактора XIa можно вводить в разделенных дозах два, три или четыре раза в сутки. Для введения дважды в сутки соответственным образом полученный медикамент может сожержать от 0,4 мг до 4 г, предпочтительно от 1 мг до 300 мг, более предпочтительно от 4 мг до 100 мг и наиболее предпочтительно от 4 мг до 20 мг, например, 4 мг, 5 мг, 10 мг и 20 мг.
Внутривенно больной может получать активный ингредиент в количествах, достаточных для доставки 0,025-7,5 мг/кг/сутки, предпочтительно 0,1-2,5 мг/кг/сутки и более предпочтительно 0,1-0,5 мг/кг/сутки. Такие количества можно вводить множеством подходящих способов, например, большие объемы активного ингредиента с низкими концентрациями на протяжении одного длительного периода времени или несколько раз в сутки, низкие объемы активного ингредиента с высокими концентрациями на протяжении короткого периода времени, например, один раз в сутки. Как правило, может быть получен типичный внутривенный состав, который содержит активный ингредиент с концентрацией приблизительно 0,01-1,0 мг/мл, например, 0,1 мг/мл, 0,3 мг/мл и 0,6 мг/мл, и вводится в количествах в сутки от 0,01 мл/кг массы больного до 10,0 мл/кг массы больного, например, 0,1 мл/кг, 0,2 мл/кг, 0,5 мл/кг. В одном примере 80-кг больной, получающий 8 мл два раза в сутки внутривенного состава, содержащего активный ингредиент с концентрацией 0,5 мг/мл, получает 8 мг активного ингредиента в сутки. В качестве буферов может быть использовано сопряженное с глюкуроновой кислотой, L-молочной кислотой, уксусной кислотой, лимонной кислотой или любой фармацевтически приемлемой кислотой основание с достаточной буферной способностью в диапазоне рН, приемлемом для внутривенного введения. Рядовой специалист в данной области без труда сможет выбрать соответствующий буфер и рН состава в зависимости от растворимости лекарственного средства, подлежащего введению.
Соединения формулы I, формулы Ia и формулы Ib могут быть введены как в виде монотерапии, так и в комбинации с другими терапевтическими средствами, в том числе противотромботическими средствами (антикоагулянтами и ингибиторами агрегации тромбоцитов), тромболитическими средствами (активаторами плазминогена), другими профибринолитически активными веществами, гипотензивными средствами, регуляторами сахара в крови, гиполипидемическими средствами и антиаритмическими средствами.
Ингибиторы фактора XIa или двойные ингибиторы фактора XIa/плазматического калликреина также могут быть введены совместно с подходящими антикоагулянтами, в том числе без ограничения с другими ингибиторами фактора XIa, ингибиторами тромбина, антагонистами тромбиновых рецепторов, ингибиторами фактора VIIa, ингибиторами фактора Ха, ингибиторами фактора IXa, ингибиторами фактора XIIa, аденозиндифосфатными антитромбоцитарными средствами (например, антагонистами P2Y12), антагонистами фибриногеновых рецепторов (например, для лечения или предупреждения нестабильной стенокардии или для предупреждения реокклюзии после ангиопластики и рестеноза), другими антикоагулянтами, такими как аспирин, и тромболитическими средствами, такими как активаторы плазминогена или стрептокиназа, для достижения синергических эффектов в лечении различных сосудистых патологий. Такие антикоагулянты включают в себя, например, апиксабен, дабигатран, кангрелор, тикагрелор, ворапаксар, клопидогрел, эдоксабан, мипомерсен, прасугрел, ривароксабан и семулопарин. Например, больные, страдающие заболеванием коронарной артерии, и больные, подвергшиеся ангиопластическим процедурам, получат пользу от совместного введения антагонистов фибриногенных рецепторов и ингибиторов тромбина. Ингибиторы фактора XIa могут быть введены первыми после образования тромба, а затем вводят активатор тканевого плазминогена или активатор другого плазминогена.
В качестве альтернативы или дополннения, одно или несколько дополнительных фармакологически активных средств могут быть введены в комбинации с соединением в соответствии с настоящим изобретением. Дополнительное активное средство (или средства) означает фармацевтически активное средство (или средства), которое является активным в организме, в том числе пролекарства, которые превращаются в фармацевтически активную форму после введения, которое отличается от соединения в соответствии с настоящим изобретением, а также включает в себя свободную кислоту, свободное основание и фармацевтически приемлемые соли указанных дополнительных активных средств, если такие формы имеются в коммерческой продаже или иным образом химически возможны. В целом, любое подходящее дополнительное активное средство или средства, в том числе без ограничения противогипертензивные средства, дополнительные диуретики, противоатеросклеротические средства, такие как липид-модифицирующее соединение, противодиабетические средства и/или средства против ожирения, могут быть использованы в любой комбинации с соединением в соответствии с настоящим изобретением в составе однократной дозировки (комбинации лекарственных средств с фиксированной дозой) или могут быть введены больному в одном или нескольких отдельных дозированных составах, что позволяет одновременное или последовательное введение активных средств (освместное введение отдельных активных средств). Примеры дополнительных активных средств, которые могут быть использованы, включают в себя без ограничения ингибиторы ангиотензин-превращающего фермента (например, алацеприл, беназеприл, каптоприл, церонаприл, цилазаприл, делаприл, эналаприл, эналаприлат, фосиноприл, имидаприл, лизиноприл, мовелтиприл, периндоприл, хинаприл, рамиприл, спираприл, темокаприл или трандолаприл); антагонисты рецептора ангиотензина II, также известные как блокаторы рецептора ангиотензина или ARB, которые могут иметь форму свободного основания, свободной кислоты, соли пролекарства, такие как азилсартан, например, азилсартан медоксомил калия (EDARBI®), кандесартан, например, кандесартан цилексетил (ATACAND®), эпросартан, например, эпросартана мезилат (TEVETAN®), ирбесартан (AVAPRO®), лосартан, например, лосартан калия (COZAAR®), олмесартан, например, олмесартан медоксимил (BENICAR®), телмисартан (MICARDIS®), валсартан (DIOVAN®), и любые из этих лекарственных средств, используемые в комбинации с подобным тиазиду диуретиком, таким как гидрохлортиазид (например, HYZAAR®, DIOVAN HCT®, ATACAND HCT®) и т.д.); калийсберегающие диуретики, такие как амилорид HCl, спиронолактон, эплеранон, триамтерен, какждый с HCTZ или без HCTZ; ингибиторы нейтральной эндопептидазы (например, тиорфан и фосфорамидон); антагонисты альдостерона; ингибиторы альдостеронсинтазы; ингибиторы ренина; эналкреин; RO 42-5892; А 65317; СР 80794; ES 1005; ES 8891; SQ 34017; алискерин (2(S),4(S),5(S),7(S)-N-(2-карбамоил-2-метилпропил)-5-амино-4-гидрокси-2,7-диизопропил-8-[4-метокси-3-(3-метоксипропокси)-фенил]-октанамида гемифумарат) SPP600, SPP630 и SPP635); антагонисты эндотелинового рецептора; вазодилататоры (например, нитропруссид); блокаторы кальциевых каналов (например, млодипин, нифедипин, верапамил, дилтиазем, фелодипин, галлопамил, нилудипин, нимодипин, никардипин); активаторы калиевых каналов (например, никорандил, пинацидил, кромакалим, миноксидил, априлкалим, лопразолам); симпатолитики; бета-адренергические блокирующие лекарственные средства (например, ацебутолол, атенолол, бетаксолол, бисопролол, карведилол, метопролол, метопролола тартрат, надолол, пропранолол, соталол, тимолол); альфа-адренергические блокирующие лекарственные средства (например, доксазозин, празозин или альфа-метилдопа); центральные альфа-адренергические агонисты; периферические вазодилататоры (например, гидралазин); гиполипидемические средства, например, ингибиторы HMG-CoA-редуктазы, такие как симвастатин и ловастатин, которые продаются как ZOCOR® и MEVACOR® в форме лактонового пролекарства и функционируют как ингибиторы после введения, и ингибиторы HMG-CoA-редуктазы в форме фармацевтически приемлемых солей дигидроксикислот с разомкнутым кольцом, такие как аторвастатин (в частности, кальциевая соль, продаваемая под торговой маркой LIPITOR®), розувастатин (в частности, кальциевая соль, продаваемая под торговой маркой CRESTOR®), правастатин (в частности, натриевая соль, продаваемая под торговой маркой PRAVACHOL®) и флувастатин (в частности, натриевая соль, продаваемая под торговой маркой LESCOL®); ингибитор абсорбции холестерина, такой как эзетимиб (ZETIA®), и эзетимиб в комбинации с любыми другими гиполипидемическими средствами, такими как упомянутые выше ингибиторы HMG-CoA-редуктазы и, в частности, с симвастатином (VYTORIN®) или с аторвастатином кальция; ниацин в формах незамедлительного высвобождения или контролируемого высвобождения и, в частности, ниацин в комбинации с DP-антагонистом, таким как ларопипрант, и/или с ингибитором HMG-CoA-редуктазы; агонисты ниациновых рецепторов, такие как аципимокс и ацифран, а также частичные агонисты ниациновых рецепторов; средства, изменяющие метаболизм, в том числе инсулин-сенсибилизирующие средства и родственные соединения для лечения диабета, такие как бигуаниды (например, метформин), меглитиниды (например, репаглинид, натеглинид), сульфонилмочевины (например, хлорпропамид, глимепирид, глипизид, глибурид, толазамид, толбутамид), тиазолидиндионы, также упоминаемые как глитазоны (например, пиоглитазон, розиглитазон), ингибиторы альфа-глюкозидазы (например, акарбоза, миглитол), ингибиторы дипептидилпептидазы (например, ситаглиптин (JANUVIA®), алоглиптин, вилдаглиптин, саксаглиптин, линаглиптин, дутоглиптин, гемиглиптин), алкалоиды спорыньи (например, бромкриптин), комбинированные лекарственные препараты, такие как JANUMET® (ситаглиптин с метформином), и лекарственные препараты, инъецируемые при диабете, такие как эксенатид и прамлинтида ацетат; ингибиторы поглощения глюкозы, такие как ингибиторы натрийзависимого переносчика глюкозы (SGLT) и различные их изоформы, такие как SGLT-1, SGLT-2 (например, ASP-1941, TS-071, BI-10773, тофоглифлозин, LX-4211, канаглифлозин, дапаглифлозин, эртуглифлозин, ипраглифлозин и ремоглифлозин) и SGLT-3; или с другими лекарственными средствами, способствующими предотвращению или лечению упомянутых выше заболеваний, в том числе без ограничения диазоксид и в том числе формы свободной кислоты, свободного основания и фармацевтически приемлемой соли, пролекарственные формы, например, сложные эфиры, и соли пролекарств упомянутых выше лекарственных препаратов, если они химически возможны. Торговые марки фармацевтических лекарственных средств, указанные выше, представлены для иллюстрации поставляемой на рынок формы активного средства(средств); такие фармацевтические лекарственные средства могут применяться в дозированной лекарственной форме для одновременного или последовательного введения с соединением в соответствии с настоящим изобретением, или активное средство(средства) может быть использовано в комбинации лекарственных средств с фиксированной дозой, включающей в себя соединение в соответствии с настоящим изобретением.
Типичные дозы ингибиторов фактора XIa или ингибиторов фактора XIa/плазматического калликреина в соответствии с настоящим изобретением в комбинации с другими подходящими антитромбоцитарными средствами, антикоагулянтными средствами или тромболитическими средствами могут быть такими же, что и дозы ингибиторов фактора XIa, вводимые без совместного введения с дополнительными антитромбоцитарными средствами, антикоагулянтными средствами или тромболитическими средствами, или могут быть значительно меньше доз ингибиторов тромбина, водимых без совместного введения с дополнительными антитромбоцитарными средствами, антикоагулянтными средствами или тромболитическими средствами, в зависимости от терапевтических потребностей больного.
Соединения вводят млекопитающему в терапевтически эффективном количестве. Термин «терапевтически эффективное количество» означает количество соединения в соответствии с настоящим изобретением, которое при введении отдельно или в комбинации с дополнительным терапевтическим средством млекопитающему является эффективным для лечения (т.е. предупреждения, подавления или облегчения) состояния тромбоэмболического и/или воспалительного заболевания или для контроля прогрессирования заболевания у хозяина.
Соединения в соответствии с настоящим изобретением предпочтительно вводить млекопитающему отдельно в терапевтически эффективном количестве. Однако соединения в соответствии с настоящим изобретением также могут быть введены млекопитающему в комбинации с дополнительным терапевтическим средством, как определяется ниже, в терапевтически эффективном количестве. При введении в комбинации комбинация соединений предпочтительно, но не обязательно, является синергической комбинацией. Синергия, как описано, например, в Chou and Talalay, Adv. Enzyme Regul. 1984, 22, 27-55, наблюдается, если эффект (в данном случае ингибирование желаемой цели) соединений при введении в комбинации превышает аддитивный эффект каждого из соединений при отдельном введении в качестве единственного средства. В целом, синергический эффект наиболее четко проявляется при субоптимальных концентрациях соединений. Синергия может выражаться в более низкой цитотоксичности, в повышенном антикоагулянтном эффекте или в каком-либо другом полезном эффекте комбинации по сравнению с отдельными компонентами.
Фразы «введенные в комбинации» или «комбинированная терапия» означают, что соединение в соответствии с настоящим изобретением и одно или несколько дополнительных терапевтических средств вводят одновременно млекопитающему, подлежащему лечению. При введении в комбинации каждый компонент может быть введен в одно и то же время или последовательно в любом порядке в различные моменты времени. Таким образом, каждый компонент может быть введен отдельно, но достаточно близко по времени, чтобы обеспечить желаемый терапевтический эффект.
Объем настоящего изобретения не ограничивается конкретными вариантами осуществления, раскрываемыми в примерах, которые предназначены для иллюстрации некоторых аспектов настоящего изобретения, и любые варианты осуществления, которые являются функционально эквивалентными, входят в объем настоящего изобретения. Действительно, различные модификации настоящего изобретения в дополнение к тем, что показаны и описаны в настоящем документе, будут очевидными для специалистов в данной области и попадают в объем прилагаемой формулы изобретения.
В целях настоящего описания следующие аббревиатуры характеризуются указанными значениями:




Также TLC означает тонкослойная хроматография; Ts означает тозил; UV означает ультрафиолет; Вт означает ватт; масс. % означает процентное содержание по массе; × g означает кратность гравитации; αD означает удельное вращение поляризованного света при 589 нм; °С означает градусы Цельсия; % масса/объем означает процентное содержание по массе предыдущего средства относительно объема последнего средства.
«Человеческий FXIa Ki (нм)» представляет собой человеческий фактор XIa Ki (нм).
Условия аналитической HPLC с обращенной фазой с масс-спектрометрией:
LC1: Колонка: Waters Xterra™ (Waters Technologies Corporation, Wilmington, DE) MS C-18, 3,5 мкм, 3,0×50 мм, температура: 50°С, элюент:10: 90-98:2 объем/объем ацетонитрил/вода + 0,05% TFA в течение 3,75 мин, скорость потока: 1,0 мл/мин, вводимый объем 10 мкл; определение: PDA, 200-600 нм; MS: диапазон 150-750 а.е.м.; ионизация электрораспылением положительных ионов.
LC2: Колонка: Waters Xterra™ (Waters Technologies Corporation, Wilmington, DE) IS C-18, 3,5 мкм, 2,1×20 мм, температура: 50°С, элюент: 5: 95-95: 5 объем/объем ацетонитрил/вода + 0,05% TFA в течение 1,75 мин, скорость потока: 1,5 мл/мин, вводимый объем 5 мкл, определение: PDA, 200-600 нм, MS: диапазон 150-750 а.е.м.; ионизация электрораспылением положительных ионов.
LC3: Колонка: Waters Xterra™ (Waters Technologies Corporation, Wilmington, DE) IS C-18, 3,5 мкм, 2,1×20 мм, температура: 50°С, элюент: 5:95-95:5 объем/объем ацетонитрил/вода + 0,05% TFA в течение 3,00 мин, скорость потока: 1,5 мл/мин, вводимый объем 5 мкл, определение: PDA, 200-600 нм, MS: диапазон 150-750 а.е.м.; ионизация электрораспылением положительных ионов.
LC4: Колонка: Waters Xterra™ (Waters Technologies Corporation, Wilmington, DE) IS C-18, 3,5 мкм, 3,0×50 мм, температура: 50°С, элюент:10: 90-98:2 объем/объем ацетонитрил/вода + 0,05% TFA в течение 1,25 мин, скорость потока: 1,5 мл/мин, вводимый объем 5 мкл, определение: PDA, 200-600 нм, MS: диапазон 150-750 а.е.м.; ионизация электрораспылением положительных ионов.
LC5: Колонка: Sunfire™ (Waters Technologies Corporation, Wilmington, DE) C-18, 5 мкм, 4,6×100 мм, температура: 50°С, элюент:10:90-98:2 объем/объем ацетонитрил/вода + 0,1% муравьиной кислоты в течение 1,25 мин, скорость потока: 1,5 мл/мин, вводимый объем 5 мкл, определение: PDA, 200-600 нм, MS: диапазон 150-750 а.е.м.; ионизация электрораспылением положительных и отрицательных ионов.
LC6: Колонка: Agilent ZORBAX™ (E.I. Du Pont de Nemours and Company, Wilmington, DE) SB-YMC-Actus Pro С18, 3,5 мкм, 2,1×50 мм, температура: 50°С, элюент: 10:90-100:0 объем/объем ацетонитрил/вода + 0,05% TFA в течение 4,00 мин, скорость потока: 0,8 мл/мин, вводимый объем 1 мкл, определение: PDA, 200-400 нм, MS: диапазон 100-1000 а.е.м.; ионизация электрораспылением положительных ионов.
Общие схемы
Соединения в соответствии с настоящим изобретением могут быть получены с использованием типичных методик или согласно методам, изложенным в следующих общих схемах синтеза.
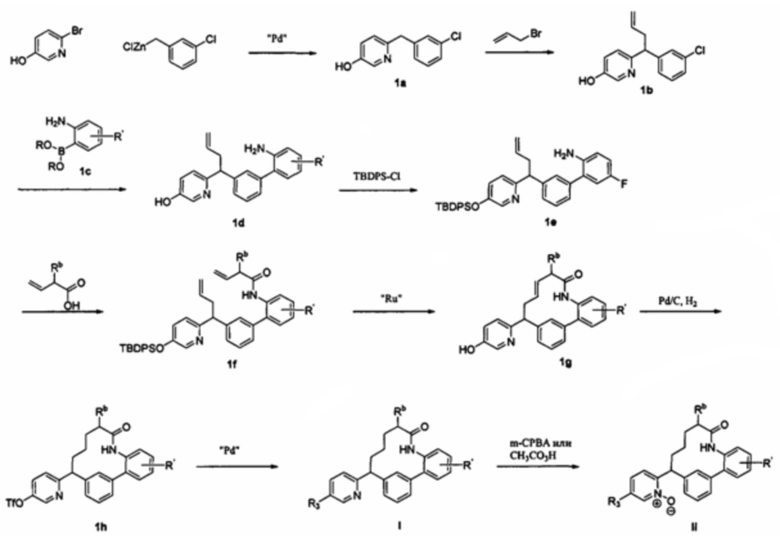
Многочисленные варианты осуществления настоящего изобретения кратко описываются в схеме 1, на которой изображается получение соединений I и II из промежуточного соединения 1а, синтезированного реакцией кросс-сочетания Негиши двух легко доступных соединений в присутствии катализатора, такого как тетракис(трифенилфосфин)палладий(0), в растворителе, таком как тетрагидрофуран, при повышенных температурах. Аллилирование промежуточного соединения 1b с помощью основания, такого как диизопропиламид лития, и аллилбромида дает промежуточное соединение 1b. Промежуточное соединение 1d получают с помощью катализируемого палладием сочетания Сузуки-Мияура промежуточного соединения 1b и бороната или бороновой кислоты 1 с, в присутствии предкатализатора, такого как аддукт хлор-(2-дициклогексилфосфино-2',4',6'-три-изо-пропил-1,1'-бифенил)[2-(2-аминоэтил)фенил]палладия(II) метил-трет-бутиловый эфир, и основания, такого как фосфат калия, в смеси воды и другого растворителя, такого как THF или диоксан, при повышенных температурах. Защита гидроксильной группы промежуточного соединения 1d с помощью защитной группы, такой как трет-бутилдифенилсилильная группа, с последующим амидным сочетанием соединения 1e с 3-бутеновой кислотой или замещенной 3-бутеновой кислотой дает промежуточное соединение 1f. Внутримолекулярный олефиновый метатезис соединения 1f, опосредованный катализатором, таким как катализатор Жана-1B, в растворителе, таком как толуол или дихлорэтан, при окружающих или повышенных температурах закрывает кольцо; снятие защиты in situ или последующая обработка обеспечивает промежуточное соединение 1g. Преобразование фенола в фенилтрифлат 1h с последующим другим катализируемым палладием сочетанием Сузуки-Мияура с боронатом или бороновой кислотой дает соединение I. В качестве альтернативы, соединение I может быть синтезировано с помощью однореакторного двухстадийного преобразования трифлата 1h в бороновую кислоту в присутствии палладиевого катализатора, а затем последующего сочетания Сузуки-Мияура с арилгалоидом или трифлатом. Конечное окисление соединения I оксидантом, таким как mCPBA, перуксусная кислота или оксон, в растворителе, таком как уксусная кислота или дихлорметан, при окружающей температуре дает пиридин-N-оксид II.
Схема 1
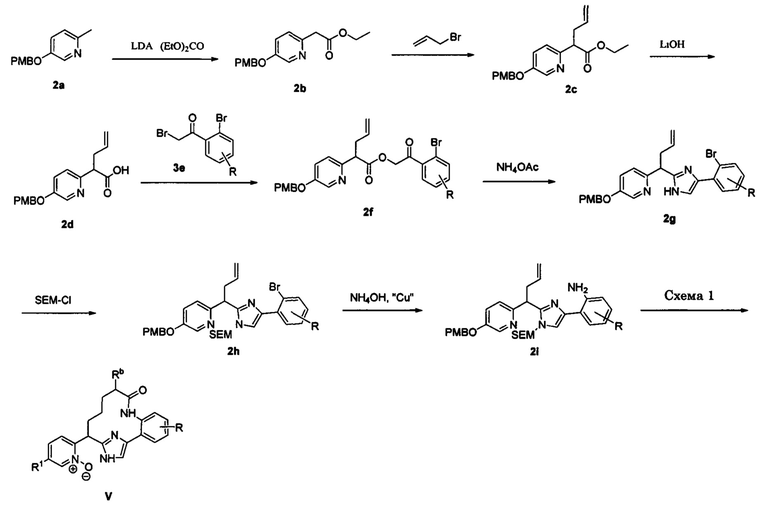
Типичные макроциклы с имидазольным кольцом B в соответствии с настоящим изобретением могут быть получены, как показано на схеме 2. Депротонирование защищенного пара-метоксибензилом 2-метил-5-гидроксипиридина 2а основанием, таким как диизопропиламид лития, с последующим гашением диэтилкарбоната дает сложный эфир 2b. Аллилирование соединения 2b в присутствии основания и аллилбромида с последующей сапонификацией основанием, таким как гидроксид лития, дает карбоновую кислоту 2d. Реакция соединения 2d с альфа-бромкетоном 2е, опосредованная основанием, таким как карбонат цезия, в растворителе, таком как DMF, обеспечивает промежуточное соединение 2f. Конденсация соединения 2f с избыточным количеством ацетата аммония в растворителе, таком как толуол или смешанный растворитель из толуола и уксусной кислоты, при повышенных температурах дает имидазол 2g. Защита NH в имидазоле с помощью группы SEM и превращение брома в аминогруппу с использованием содержащего медь предкатализатора, такого как йодид меди(I), и лиганда, такого как L-пролин, дает промежуточное соединение 2i. Синтез соединения III выполняют с помощью следующих последовательных стадий, описанных на схеме 1.
Схема 2
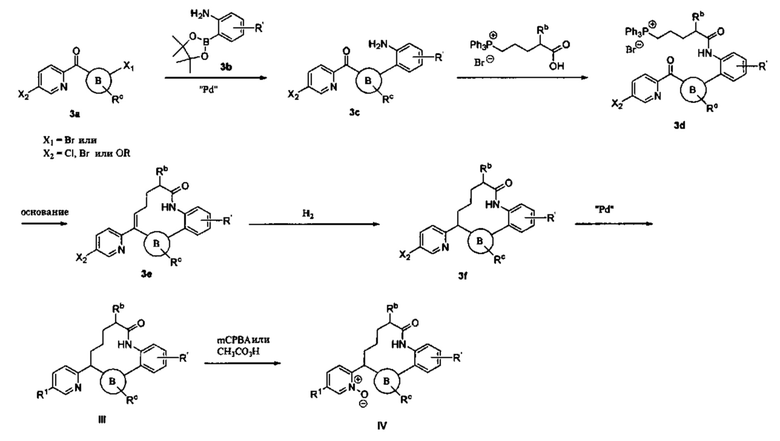
В более широком смысле, типичные макроциклы, содержащие фенильное, замещенное фенильное, 5/6-членное гетероцикльное или замещенное 5/6-членное гетероцикльное кольцо В, могут быть получены, как показано на схеме 3. Сочетание Сузуки-Мияура промежуточного кетона 3а с бороновой кислотой или сложным эфиром 3b в присутствии палладиевого предкатализатора, такого как PdCl2(dppf), и основания, такого как фосфат калия, в смешанном растворителе из воды и диоксана, или в THF при повышенных температурах обечпечивает соединение 3с. Образование амидной связи между соединением 3с и бромидом (4-карбоксибутил)трифенилфосфония, опосредованное реагентом пептидного сочетания, таким как HATU, дает соединение 3d. которое при обработке избыточными эквивалентами основания, такого как LHMDS, в растворителе, таком как THF, при концентрации, равной приблизительно 0,02 М или ниже таковой, при комнатной температуре обеспечивает соединение 3е. Селективная гидрогенизация алкена в присутствии хлорпиридинового фрагмента, катализируемая либо никелем Ренея, либо контаминированной ванадием платиной на угле, дает соединение 3f. Синтез соединения III выполняют путем последующего сочетания Сузуки-Мияура соединения 3f с бороновой кислотой или сложным эфиром. В качестве альтернативы, соединение 3f может быть превращено в бороновую кислоту или сложный эфир, которые реагируют с арилгалоидом или трифлатом в присутствии катализатора с получением соединения III. В случаях, ели Х2 представляет собой алкоксильную группу, то его преобразуют в соответствующий трифлат перед сочетанием Сузуки-Мияура. Окисление соединения IV до N-оксида V описывается на схеме 3.
Схема 3
Промежуточный кетон 3а может быть получен, как показано на схеме 4. Карбоновую кислоту (4а), коммерчески доступную или синтезированную из коммерчески доступных материалов, превращают в соответствующий амид Вайнреба в присутствии реагента пептидного сочетания с получением соединения 4b. В качестве альтернативы, соединение 4b может быть синтезировано реакцией сложного эфира (4а) с N,O-диметилгидроксиламином и подходящим магнийорганическим реагентом или основанием амида лития (Tetrahedron Lett. 1995, 36, 5461-5464). Обработка амида Вайнреба 4b анионом, образуемым при обмене металл-галоген замещенного 2-бромпиридина (4с) и сильного основания, такого как н-BuLi, обеспечивает соответствующий кетон 3а.
Схема 4
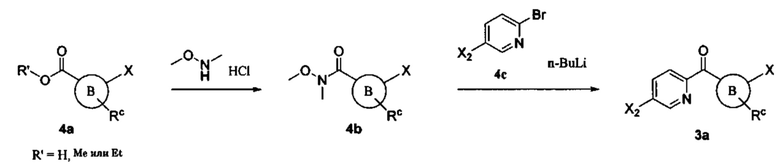
В качестве альтернативы, кетон 3а может быть получен реакцией амида Вайнреба 4-замещенной пиколиновой кислоты (5b) с металлоорганическим нуклеофилом (5а), образованным путем депротонирования или обмена металл-галоген соответствующего арена или гетероарена с металлоорганическим основанием, таким как н-BuLi (схема 5).
Схема 5
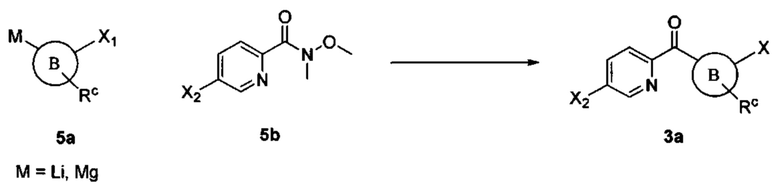
Согласно другому варианту осуществления настоящего изобретения макроциклы с общими структурами V и VI могут быть получены из промежуточного соединения 1d, как показано на схеме 6. Реакция анилина 1d с этил-2-оксоацетатом и аллилтрибутилоловом в присутствии умеренной кислоты, такой как малеиновая кислота (Zhao et. al, Synthesis 2006, 19, 3189), дает соединение 6а. Защита гидроксильной группы с помощью TBDPS с последующей рекцией метатезиса с закрытием кольца, катализируемой рутениевым катализатором, таким как катализатор Жана, и удалением защитной группы TBDPS дает соединение 6с. Гидрогенизация соединения 6с и превращение гидроксильной группы в трифлат дает соединение 6е. Сочетание Сузуки-Мияура соединения 6е с бороновой кислотой или сложным эфиром дает промежуточное соединение 6f. Защита анилина трифторацетильной группой с последующим окислением пиридина оксидантом, таким как мСРВА или перуксусная кислота, дает пиридин-N-оксид 6h. Обрабтка соединения 6h основанием, таким как гидроксид лития, в смешанном растворителе из воды, метанола и THF, при повышенной температуре вызывает гидролиз сложного этилового эфира и удаление трифторацетильной защитной группы на анилине с получением аминокислоты V. Соединение VI может быть получено путем сочетания соединения V с амином в присутствии реагента пептидного сочетания, такого как HATU.
Схема 6
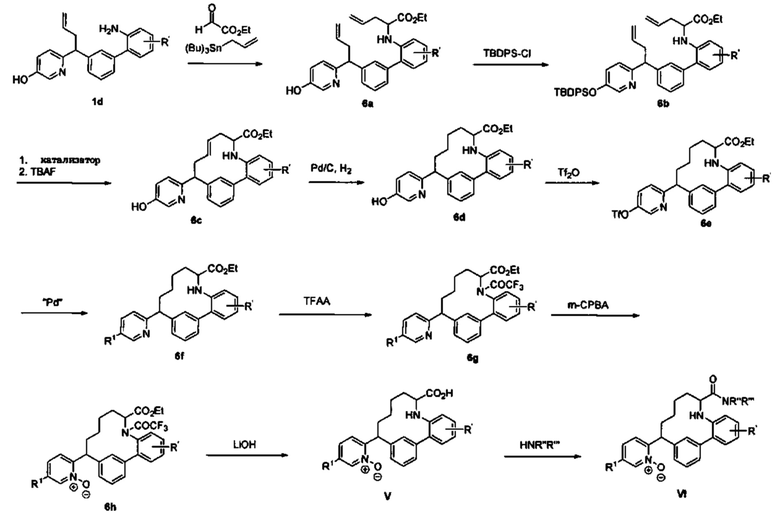
Разделение диастереомеров может быть выполнено на разных стадиях при получении желаемых конечных соединений; однако, как правило, его выполняют с конечными продуктами с помощью хроматографии со сверхкритической подвижной фазой (SFC). Разделения энантиомеров достигают с помощью SFC с использованием различных хиральных колонок. Абсолютную конфигурацию определяют либо непосредственно рентгеновским анализом сокристаллических структур, либо путем сравнения значений Ki FXIa у одной пары энантиомеров исходя из того, что соединения с определенной конфигурацией являются более активными, чем соответствующий им энантиомер.
Препаративную тонкослойную хроматографию (PTLC) проводили на пластинах 20×20 см (силикагель толщиной 500 мкм-1500 мкм). Колоночную флеш-хроматографию проводили на системах флеш-хроматографии ISCO с применением колонок, предварительно заполненных силикагелем, элюировали при помощи градиента гексана/этилацетата или DCM/MeOH, если не отмечено иное.
ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 1
6-(1-(3-ХЛОРФЕНИЛ)БУТ-3-ЕН-1-ИЛ)ПИРИДИН-3-ОЛ
Стадия А: 6-(3-Хлорбензил)пиридин-3-ол: В прозрачную сухую круглодонную колбу объемом 500 мл с магнитной мешалкой добавляли 2-бром-5-гидроксипиридин (4,2 г, 24,14 ммоль) и тетракис(трифенилфосфин)палладий(0) (2,79 г, 2,414 ммоль). Ее закупоривали резиновой септой и трижды продували азотом. В колбу переносили THF (дегазированный, 100 мл) через канюлю и перемешивали с образованием раствора. В раствор переносили 3-хлорбензилцинка хлорид (0,5 М в THF, 100 мл, 50,0 ммоль) через канюлю. Колбу погружали в предварительно нагретую масляную баню при 65°С и перемешивали в течение 4 ч. Ее оставляли охлаждаться до к. т. и выдерживали всю ночь. Реакцию гасили хлоридом аммония (50 мл) и солевым раствором (50 мл). Смесь экстрагировали этилацетатом (100 мл). Органический слой отделяли и сушили над сульфатом натрия, фильтровали и концентрировали. Остаток перетирали в порошок в DCM (50 мл), фильтровали и промывали DCM (2×10 мл) с получением указанного соединения. MS (ES+) m/z: 220 (М+Н).
Стадия В: 5-(2-Хлор-5-нитрофенил)дигидрофуран-2(3H)-он: К раствору 6-(3-хлорбензил)пиридин-3-ола (1,4 г, 6,37 ммоль) в THF (40 мл) при -78°С добавляли раствор LDA (2 M в THF, 8,60 мл, 17,21 ммоль). Реакционную смесь перемешивали в течение 10 мин и по каплям добавляли раствор аллилбромида (0,56 мл, 6,47 ммоль) в THF (5 мл). Полученную смесь перемешивали в течение 30 мин и оставляли нагреваться до к. т. с перемешиванием в течение 3 ч. Ее гасили насыщенным водным хлоридом аммония и смесь экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали солевым раствором и сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-50% этилацетатом в гексане) с получением указанного соединения. MS (ES+) m/z; 260 (М+Н).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 2
(3-БРОМФЕНИЛ)(5-ХЛОРПИРИДИН-2-ИЛ)МЕТАНОН
Стадия А: 3-Бром-N-метокси-N-метилбензамид: К смеси 3-бромбензойной кислоты (40 г, 199 ммоль), N,О-диметилгидроксиламина гидрохлорида (23,29 г, 239 ммоль) и триэтиламина (83 мл, 597 ммоль) в дихлорметане (398 мл) при 0°С добавляли EDC (49,6 г, 259 ммоль). Смесь перемешивали в течение 5 мин и оставляли нагреваться до к. т. с перемешиванием в течение 2 ч. Ее последовательно промывали хлористоводородной кислотой (1 М, 100 мл), водным бикарбонатом натрия (насыщенным, 100 мл) и солевым раствором. Органический слой отделяли и сушили над безводным сульфатом магния. Его фильтровали, а фильтрат концентрировали при пониженном давлении с получением указанного соединения. MS (ES+) m/z: 246, 248 (М+Н).
Стадия В: (3-Бромфенил)(5-хлорпиридин-2-ил)метанон: К перемешиваемому раствору 2-бром-5-хлорпиридина (21,68 г, 113 ммоль) в безводном толуоле (512 мл) при -78°С по каплям добавляли н-бутиллитий (2,5 М в гексане) (49,0 мл, 123 ммоль) в течение 1 ч при помощи шприцевой помпы. Смесь перемешивали при -78°С в течение 90 мин. Раствор 3-бром-N-метокси-N-метилбензамида (25 г, 102 ммоль) в безводном толуоле (80 мл) добавляли к смеси в течение 30 мин при помощи шприцевой помпы. Полученную смесь перемешивали при -78°С еще 2 ч и нагревали до 0°С. Раствор хлористоводородной кислоты (1 М, 100 мл) добавляли к реакционной смеси и перемешивали в течение 20 мин. Значение рН смеси доводили приблизительно до 9 добавлением водного бикарбоната натрия (насыщенного). Смесь экстрагировали этилацетатом (2×200 мл). Объединенные органические слои промывали солевым раствором (100 мл), сушили над сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали EtOAc/гексаном = 9:1 объем/объем) с получением указанного соединения. MS (ES+) m/z: 296, 298 (М+Н).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 3
(5-ХЛОРПИРИДИН-2-ИЛ)(3-(4,4,5,5-ТЕТРАМЕТИЛ-1,3,2-ДИОКСАБОРОЛАН-2-ИЛ)ФЕНИЛ)МЕТАНОН
Смесь (3-бромфенил)(5-хлорпиридин-2-ил)метанона (4,00 г, 13,48 ммоль), бис(пинаколято)диборона (4,10 г, 16,20 ммоль), ацетата калия (2,00 г, 20,00 ммоль) и Pd(dppf)Cl2 (1,00 г, 1,35 ммоль) в диоксане (60 мл) перемешивали при 85°С в течение 4,5 ч. Ее гасили водным хлоридом аммония (насыщенный, 50 мл) и экстрагировали EtOAc (50 мл × 3). Органический слой промывали солевым раствором (100 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (элюировали петролейным эфиром : этилацетатом = 50:1-10:1) с получением указанного соединения. 1Н ЯМР (400 МГц, CDCl3): δ 8.61 (s, 1Н), 8.33 (s, 1H), 8.06-7.95 (m., 3Н), 7.82 (d, J=8.4 Гц, 1H), 7.44 (t, J=7.2 Гц, 1H), 1.28 (s, 12H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 4
(3-БРОМ-5-ФТОРФЕНИЛ)(5-ХЛОРПИРИДИН-2-ИЛ)МЕТАНОН
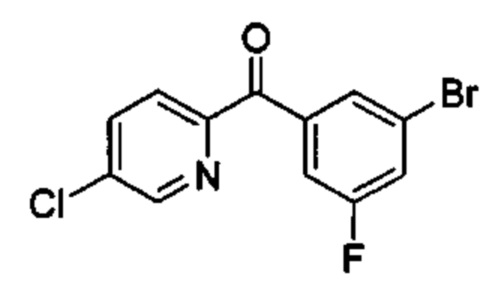
Указанное соединение получали из 3-бром-5-фторбензойной кислоты способом, описанным в синтезе промежуточного соединения 2. MS (ES+) m/z: 314, 316 (М+Н); 1Н ЯМР (400 МГц, CDCl3): δ 8.69 (d, J=1.8 Гц, 1H), 8.02-8.14 (m, 2Н), 7.92 (dd, J=2.2, 8.4 Гц, 1Н), 7.81 (d, J=8.6 Гц, 1H), 7.49 (d, J=7.7 Гц, 1H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 5
2-БРОМ-1-(2-БРОМ-5-ФТОРФЕНИЛ)ЭТАН-1-ОН
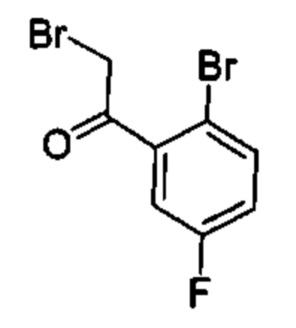
К перемешиваемому раствору 1-(2-бром-5-фторфенил)этанон (5 г, 23,04 ммоль) и HBr (48% масса/масса водный, 0,2 мл, 1,768 ммоль) в диэтиловом эфире (50 мл) при 0°С по каплям добавляли бромин (1,2 мл, 23,29 ммоль). Смесь перемешивали при к. т. в течение 2 ч. Ее переносили в делительную воронку и промывали водой (50 мл). Водный слой экстрагировали диэтиловым эфиром (100 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-20% этилацетатом в гексане) с получением указанного соединения.
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 6
(3-БРОМ-4-ФТОРФЕНИЛ)(5-ХЛОРПИРИДИН-2-ИЛ)МЕТАНОН
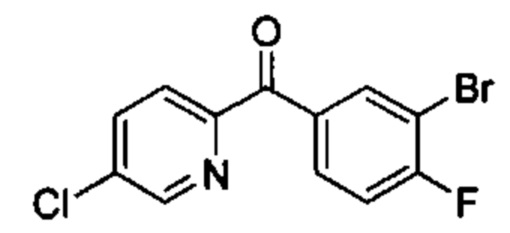
Указанное соединение получали из 3-бром-4-фторбензойной кислоты способом, описанным в синтезе промежуточного соединения 2. MS (ES+) m/z: 314, 316 (М+Н); 1Н ЯМР (400 МГц, CDCl3): δ 8.79 (d, J=2.0 Гц, 1H), 8.29 (dd, J=1.6, 7.0 Гц, 1Н), 8.20 (dd, J=2.2, 8.4 Гц, 1Н), 8.04 (d, J=8.2 Гц, 2H), 7.53 (t, J=8.6 Гц, 1H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 7
(3-БРОМ-4,5-ДИФТОРФЕНИЛ)(5-ХЛОРПИРИДИН-2-ИЛ)МЕТАНОН
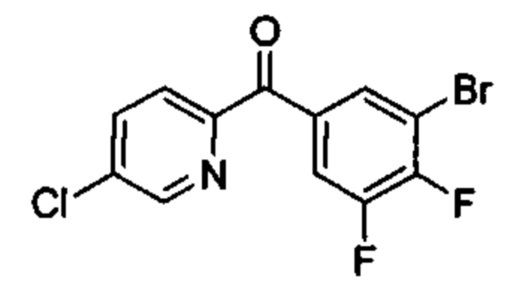
Промежуточное соединение 7 получали из 3-бром-4,5-дифторбензойной кислоты способом, описанным в синтезе промежуточного соединения 2. MS (ES+) m/z: 332, 334 (М+Н).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 8
(2-БРОМПИРИДИН-4-ИЛ)(5-ХЛОРПИРИДИН-2-ИЛ)МЕТАНОН
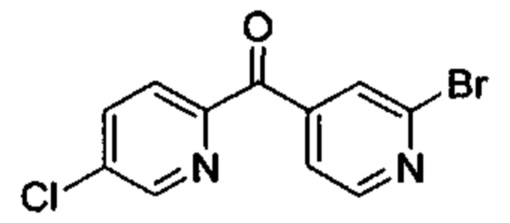
Стадия А: (2-Бромпиридин-4-ил)(5-хлорпиридин-2-ил)метанол: Раствор 2-бром-5-хлорпиридина (3,41 г, 17,74 ммоль) в 10 мл безводного THF при -78°С в атмосфере азота по каплям добавляли к раствору н-бутиллития (2,5 М в гексане, 7,10 мл, 17,74 ммоль) в безводном THF (50 мл). Реакционную смесь перемешивали в течение 15 мин и одной порцией добавляли раствор 2-бромпиридин-4-карбоксальдегида (3,0 г, 16,13 ммоль) в 20 мл THF. Ее перемешивали в течение 10 мин и баню с сухим льдом и ацетоном заменяли баней с ледяной водой. Реакционную смесь перемешивали при 0°С в течение 30 мин, а затем гасили насыщенным водным хлоридом аммония. Смесь экстрагировали этилацетатом. Органический слой отделяли, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-100% этилацетатом в гексане) с получением указанного соединения. MS (ES+) m/z: 299, 301 (М+Н).
Стадия В: (2-Бромпиридин-4-ил)(5-хлорпиридин-2-ил)метанон: Раствор (2-бромпиридин-4-ил)(5-хлорпиридин-2-ил)метанола (1,3 г, 4,34 ммоль) в DCM (40 мл) перемешивали с диоксидом магния (3,77 г, 43,4 ммоль) при к. т. в течение 1 ч. Реакционную смесь фильтровали через слой целита, а твердый кек промывали 10% метанола в DCM (100 мл). Фильтрат концентрировали с получением указанного соединения. MS (ES+) m/z: 297, 299 (М+Н).
Альтернативно, промежуточное соединение 8 получали из 2-бромизоникотиновой кислоты способом, описанным в синтезе промежуточного соединения 2. MS (ES+) m/z: 297, 299 (М+Н); 1Н ЯМР (400 МГц, CDCl3): δ 8.68 (d, J=1.8 Гц, 1Н), 8.56 (d, J=4.9 Гц, 1Н), 8.14 (d, J=8.4 Гц, 1Н), 8.10 (s, 1Н), 7.93 (dd, J=8.4, 2.2 Гц, 1Н), 7.85 (d, J=4.9 Гц, 1Н).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 9
(2-БРОМ-5-ФТОРПИРИДИН-4-ИЛ)(5-ХЛОРПИРИДИН-2-ИЛ)МЕТАНОН
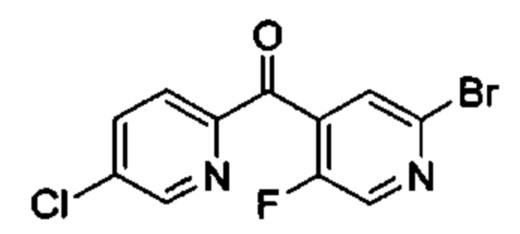
Стадия А: (2-Бром-5-фторпиридин-4-ил)(5-хлорпиридин-2-ил)метанол: К раствору 2,4-дибром-5-фторпиридина (7,53 г, 29,5 ммоль) в 100 мл толуола при -78°С по каплям добавляли раствор н-бутиллития (2,5 М, 7 мл, 17,50 ммоль). Его перемешивали в течение 15 мин и к раствору добавляли раствор 5-хлорпиколинальдегида (3,8 г, 26,8 ммоль) в 30 мл толуола при помощи шприца. Смесь перемешивали в течение 1 ч и ее оставляли нагреваться до 0°С до того, как ее гасили насыщенным водным хлоридом аммония. Смесь экстрагировали этилацетатом. Органический слой промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали с получением указанного соединения (в виде смеси двух региоизомеров в соотношении 9:1 в сторону требуемого изомера). Его использовали без очистки. MS (ES+) m/z: 317, 319 (М+Н).
Стадия В: (2-Бром-5-фторпиридин-4-ил)(5-хлорпиридин-2-ил)метанон: Раствор вышеуказанного продукта (3,8 г, 10,77 ммоль) в DCM (50 мл) перемешивали с диоксидом магния (9,36 г, 108 ммоль) при к.т. в течение 2 ч. Твердые вещества удаляли фильтрацией через слой целита и промывали 10% аммиаком в МеОН (7 н)/DCM (3×50 мл). Фильтрат концентрировали, а остаток растворяли в 20% аммиаке в МеОН (7 н)/DCM и очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-2% аммиака в МеОН (7 н)/DCM) с получением указанного соединения. MS (ES+) m/z: 315, 317 (М+Н); 1Н ЯМР (400 МГц, CDCl3): δ 8.80 (d, J=2.0 Гц, 1Н), 8.63 (s, 1H), 8.26 (dd, J=2.0, 8.5 Гц, 1H), 8.18 (d, J=8.5 Гц, 1H), 7.97 (d, J=4.5 Гц, 1H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 10
(4-БРОМПИРИДИН-2-ИЛ)(5-ХЛОРПИРИДИН-2-ИЛ)МЕТАНОН
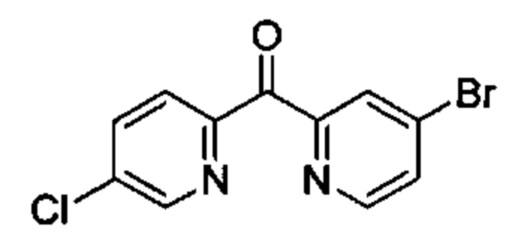
Указанное соединение получали из 2,4-дибромпиридина способом, описанным в синтезе промежуточного соединения 9. Продукт очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-5% NH3 в метаноле (7 н) в DCM) с получением указанного соединения. MS (ES+) m/z: 297, 299 (М+Н).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 11
(5-ХЛОРПИРИДИН-2-ИЛ)(5-ФТОР-4-ЙОДПИРИДИН-2-ИЛ)МЕТАНОН
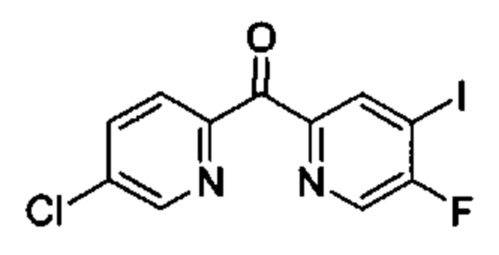
Стадия А: (5-Хлорпиридин-2-ил)(5-фторпиридин-2-ил)метанон: Указанное соединение получали из 2-бром-5-фторпиридина способом, описанным в синтезе промежуточного соединения 9. MS (ES+) m/z: 237.0 [М+Н].
Стадия В: (5-Хлорпиридин-2-ил)(5-фтор-4-йодпиридин-2-ил)метанол: К раствору (5-хлорпиридин-2-ил)(5-фторпиридин-2-ил)метанона (3,00 г, 12,68 ммоль) в THF (30 мл) при -78°С добавляли раствор LDA (2 M в THF, 8,24 мл, 16,48 ммоль). Его перемешивали в течение 30 мин и добавляли одной порцией йод (3,86 г, 15,21 ммоль). Смесь перемешивали при -78°С в течение 2 ч и гасили насыщенным водным хлоридом аммония (20 мл). Ее экстрагировали этилацетатом (100 мл × 3). Объединенные органические слои промывали солевым раствором (200 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали EtOAc : петролейным эфиром = 1:8-1:5 объем/объем) с получением указанного соединения. MS (ES+) m/z: 365.0 [М+Н].
Стадия С: (5-Хлорпиридин-2-ил)(5-фтор-4-йодпиридин-2-ил)метанон: К раствору (5-хлорпиридин-2-ил)(5-фтор-4-йодпиридин-2-ил)метанола (580 мг, 1,591 ммоль) в DCM (6 мл) добавляли оксид марганца (IV) (1383 мг, 15,91 ммоль). Смесь перемешивали при к. т. в течение 12 ч. Твердые вещества удаляли фильтрацией через слой целита и промывали DCM (30 мл). Фильтрат концентрировали в вакууме с получением указанного соединения. MS (ES+) m/z: 363.0 [М+Н].
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 12
(3-БРОМФЕНИЛ)(5-ХЛОР-4-МЕТОКСИПИРИДИН-2-ИЛ)МЕТАНОН
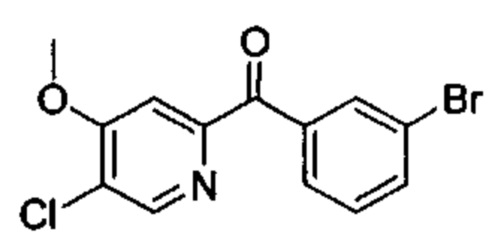
Стадия А: 2-Бром-5-хлор-4-метоксипиридин: К раствору 2-бром-5-хлорпиридин-4-ола (7,80 г, 37,40 ммоль) в DCM (30 мл) и МеОН (3 мл) добавляли TMS-диазометан (94 мл, 187 ммоль, 2 M в гексане) при 0°С. Реакционную смесь перемешивали при 25°С в течение 12 ч. Твердые осадки удаляли фильтрацией через слой целита. Фильтрат концентрировали при пониженном давлении, а остаток очищали методом колоночной флеш-хроматографии (SiO2, петролейный эфир : EtOAc = 1:0-3:1) с получением указанного соединения. 1Н ЯМР (400 МГц, CDCl3): δ 8.19 (s, 1Н), 7.00 (s, 1H), 3.94 (s, 3H).
Стадия B: (3-Бромфенил)(5-хлор-4-метоксипиридин-2-ил)метанон: К перемешиваемому раствору 2-бром-5-хлор-4-метоксипиридина (5,50 г, 24,72 ммоль) в толуоле (200 мл) при -78°С добавляли раствор н-BuLi (10,88 мл, 27,20 ммоль, 2,5 М в гексане). Полученную смесь перемешивали при -78°С в течение 1 ч перед добавлением 3-бром-N-метокси-N-метилбензамида (7,24 г, 29,70 ммоль). Смесь перемешивали в течение 16 ч, при этом температуру оставляли повышаться до к. т. Ее гасили насыщенным водным хлоридом аммония (100 мл) и экстрагировали EtOAc (100 мл × 3). Объединенные органические слои промывали солевым раствором (200 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии (SiO2, петролейный эфир : EtOAc = 50:1-5:1) с получением указанного соединения. MS (ESI) m/z 325.9, 327.9 (М+Н); 1Н ЯМР (400 МГц, CDCl3): δ 8.53 (s, 1H), 8.21 (s, 1Н), 8.01 (d, J=7.8 Гц, 1Н), 7.70 (d, J=8.2 Гц, 1Н), 7.66 (s, 1Н), 7.35 (t, J=7.8 Гц, 1Н), 4.05 (s, 3Н).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 13
(6-БРОМ-5-ФТОРПИРИДИН-2-ИЛ)(5-ХЛОРПИРИДИН-2-ИЛ)МЕТАНОН
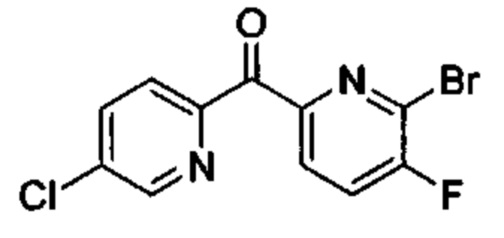
Указанное соединение получали из 6-бром-5-фторпиколиновой кислоты способом, описанным в синтезе промежуточного соединения 2. MS (ES+) m/z: 315, 317 (М+Н); 1H ЯМР (400 МГц, CDCl3): δ 8.79 (d, J=2.0 Гц, 1Н), 8.29 (dd, J=1.6, 7.0 Гц, 1Н), 8.20 (dd, J=2.2, 8.4 Гц, 1H), 8.04 (d, J=8.2 Гц, 2H), 7.53 (t, J=8.6 Гц, 1H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 14
(5-ХЛОРПИРИДИН-2-ИЛ)(6-ХЛОРПИРИМИДИН-4-ИЛ)МЕТАНОН
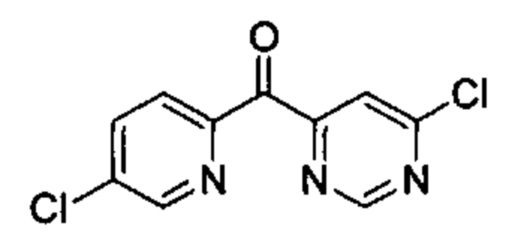
Стадия А: 6-Хлорпиримидин-4-карбонилхлорид: К раствору 6-гидроксипиримидин-4-карбоновой кислоты (2,00 г, 14,28 ммоль) и оксалилдихлорида (1,81 г, 14,28 ммоль) в EtOAc (30 мл) при 0°С добавляли несколько капель DMF (0,5 мл, 6,46 ммоль). Смесь перемешивали при 80°С в течение 16 ч. Ее охлаждали до к. т. и концентрировали при пониженном давлении с получением указанного соединения.
Стадия В: 6-Хлор-N-метокси-N-метилпиримидин-4-карбоксамид: К раствору 6-хлорпиримидин-4-карбонилхлорида (26,60 г, 150 ммоль) и N,O-диметилгидроксиламина гидрохлорида (17,59 г, 180 ммоль) в DCM (300 мл) при 0°С добавляли DIEA (52,50 мл, 301 ммоль). Реакционную смесь оставляли нагреваться до к. т. и перемешивали в течение 16 ч. Ее разбавляли водой (200 мл) и экстрагировали DCM (300 мл × 4). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии (SiO2, петролейный эфир : EtOAc = 5:1) с получением указанного соединения в виде масла. MS (ESI) m/z 202.1 (М+Н); 1Н ЯМР (400 МГц, CDCl3): δ 9.03 (s, 1Н), 7.58 (brs, 1Н), 3.74 (s, 3Н), 3.37 (s, 3Н).
Стадия С: (5-Хлорпиридин-2-ил)(6-хлорпиримидин-4-ил)метанон: Раствор н-BuLi (39,9 мл, 100 ммоль, 2,5 М в гексане) добавляли к перемешиваемому раствору 2-бром-5-хлорпиридина (19,18 г, 100 ммоль) в толуоле (150 мл) при -78°С. Смесь перемешивали в течение 0,5 ч, затем добавляли раствор 5-хлор-N-метокси-N-метилпиколинамида (10 г, 49,80 ммоль) в толуоле (50 мл). Смесь перемешивали при -78°С в течение 1 ч. Ее гасили насыщенным хлоридом аммония (200 мл) и экстрагировали EtOAc (100 мл × 3). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом колоночной флеш-хроматографии (SiO2, петролейный эфир : EtOAc = 12:1-10:1) с получением указанного соединения. MS (ESI) m/z 254.0 (М+Н), 1Н ЯМР (400 МГц, CDCl3): δ 9.16 (s, 1Н), 8.68 (d, J=1.6 Гц, 1Н), 8.18 (d, J=8.6 Гц, 1Н), 7.89-7.99 (m, 2Н).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 15
(5-БРОМПИРИДИН-2-ИЛ)(4-ЙОД-1-((2-(ТРИМЕТИЛСИЛИЛ)ЭТОКСИ)МЕТИЛ)-1Н-ИМИДАЗОЛ-2-ИЛ)МЕТАНОН
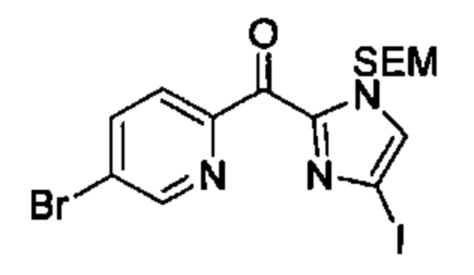
Стадия А: 4-Йод-1-((2-(триметилсилил)этокси)метил)-1Н-имидазол: К суспензии NaH (4,21 г, 105 ммоль, 60% масс.) в DMF (200 мл) при 0°С небольшими частями добавляли 4-йод-1H-имидазол (17 г, 88 ммоль). Ее перемешивали в течение 1 ч и к реакционной смеси добавляли SEM-Cl (16,07 г, 96 ммоль). Смесь перемешивали при к. т. в течение 12 ч и выливали в ледяную воду (200 мл). Смесь экстрагировали EtOAc (100 мл × 3). Объединенные органические слои промывали водой (3×50 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром : EtOAc = 10:1-3:1 объем/объем) с получением указанного соединения. MS (ES+) m/z: 325 (М+Н).
Стадия В: 5-Бром-N-метокси-N-метилпиколинамид: К раствору 5-бромпиколиновой кислоты (15 г, 74,3 ммоль) в DCM (200 мл) при 0°С добавляли EDC (21,35 г, 111 ммоль), N,О-диметилгидроксиламина гидрохлорид (10,86 г, 111 ммоль) и пиридин (15,01 мл, 186 ммоль). Смесь перемешивали при к. т. в течение 16 ч и концентрировали при пониженном давлении. Остаток разбавляли EtOAc (250 мл) и промывали 1 н HCl (50 мл), а затем солевым раствором. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением указанного соединения, которое использовали на следующей стадии без дополнительной очистки. MS (ES+) m/z: 245, 247 (М+Н).
Стадия С: (5-Бромпиридин-2-ил)(4-йод-1-((2-(триметилсилил)этокси)метил)-1H-имидазол-2-ил)метанон: К раствору 4-йод-1-((2-(триметилсилил)этокси)метил)-1H-имидазола (15,88 г, 49,0 ммоль) в THF (100 мл) при -78°С по каплям добавляли раствор LDA (26,5 мл, 53,0 ммоль, 2 M в THF). Реакционную смесь перемешивали в течение 1 ч и по каплям добавляли раствор 5-бром-N-метокси-N-метилпиколинамида (10 г, 40,8 ммоль) в THF (20 мл). Полученную смесь перемешивали при -78°С в течение 1 ч и ее гасили насыщенным водным хлоридом аммония (10 мл). Смесь экстрагировали EtOAc (150 мл). Органический слой промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром : EtOAc = 10:1 объем/объем) с получением указанного соединения. 1Н ЯМР (400 МГц, CDCl3): δ 8.85 (s, 1Н), 8.23 (m, 1H), 8.02 (m, 1H), 7.52 (s, 1H), 5.81 (s, 2H),3.65 (m, 2H), 0.96 (m, 2H), -0.01 (s, 9H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 16
(5-ХЛОРПИРИДИН-2-ИЛ)(4-ЙОД-5-МЕТИЛ-1-((2-(ТРИМЕТИЛСИЛИЛ)ЭТОКСИ)МЕТИЛ)-1Н-ИМИДАЗОЛ-2-ИЛ)МЕТАНОН
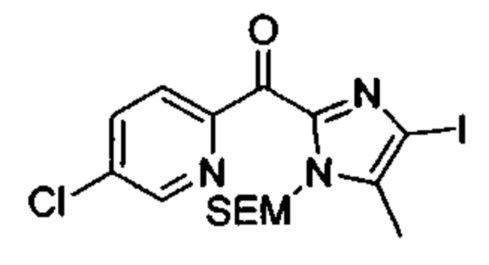
Стадия А: 4-Йод-5-метил-1Н-имидазол: Раствор 4-метил-1Н-имидазола (5,4 г, 65,8 ммоль) и йода (16,69 г, 65,8 ммоль) в диоксане (51 мл) обрабатывали NaOH (132 мл, 132 ммоль) в течение 1 ч. Большую часть органического растворителя удаляли при пониженном давлении. Его нейтрализовали насыщенным хлоридом аммония и экстрагировали этилацетатом (2×100 мл). Объединенные органические слои промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали. Остаток перетирали в порошок в 50 мл диэтилового эфира и выдерживали в течение 10 мин. Белые твердые вещества собирали фильтрацией и сушили под вакуумом с получением указанного соединения.
Стадия В: 4-Йод-5-метил-1-((2-(триметилсилил)этокси)метил)-1Н-имидазол: В раствор 5-йод-4-метил-1H-имидазола (4,42 г, 21,25 ммоль) в DMF (24 мл) при 0°С добавляли NaH (60% масс., 1,020 г, 25,5 ммоль). Его перемешивали в течение 15 мин и добавляли SEM-Cl (4,52 мл, 25,5 ммоль). Смесь перемешивали в течение 30 мин и нагревали до к. т., перемешивали еще 30 мин. Реакцию гасили насыщенным водным хлоридом аммония (50 мл) и экстрагировали этилацетатом (2×100 мл). Объединенные органические слои промывали водой (3×100 мл), солевым раствором (100 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-5% 7 н NH3 в MeOH/DCM) с получением указанного соединения. MS (ESI) m/z 338.9; 1Н ЯМР (CDCl3, 500 МГц): δ 7.47 (s, 1Н), 5.20 (s, 2H), 3.45 (t, J=8.0 Гц, 2H), 2.24 (s, 3H), 0.88 (t, J=8.0 Гц, 2H), -0.03 (s, 9Н). Структуры были подтверждены при помощи ROESY. 5-Иод-4-метил-1-((2-(триметилсилил)этокси)метил)-1H-имидазол выделяли в виде побочного продукта.
Стадия C: (5-Хлорпиридин-2-ил)(4-йод-5-метил-1-((2-(триметилсилил)этокси)метил)-1Н-имидазол-2-ил)метанон: К раствору 4-йод-5-метил-1-((2-(триметилсилил)этокси)метил)-1Н-имидазола (1 г, 2,96 ммоль) в 10 мл безводного THF при -78°С добавляли раствор LDA (2 M в THF/гептане/этилбензоле) (1,8 мл, 3,60 ммоль). Его перемешивали в течение 10 мин и добавляли раствор 5-хлор-N-метокси-N-метилпиколинамида (0,712 г, 3,55 ммоль) в 5 мл THF. Смесь перемешивали в течение 30 мин и нагревали до 0°С с перемешиванием в течение 30 мин. Ее гасили нас. водн. хлоридом аммония и экстрагировали этилацетатом (2×50 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-4% метанола в DCM, градиент) с получением указанного соединения. MS (ESI) m/z 477.8.
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 17
(2-БРОМ-1-((2-(ТРИМЕТИЛСИЛИЛ)ЭТОКСИ)МЕТИЛ)-1Н-ИМИДАЗОЛ-4-ИЛ)(5-ХЛОРПИРИДИН-2-ИЛ)МЕТАНОН
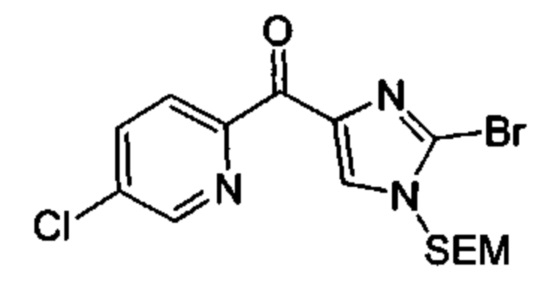
Стадия А: 2-Бром-1-((2-(триметилсилил)этокси)метил)-1Н-имидазол: К перемешиваемому раствору 2-бром-1Н-имидазола (1 г, 6,80 ммоль) в DMF (15 мл) добавляли NaH (0,272 г, 6,80 ммоль, 60% масс.) при 0°С в атмосфере азота. Смесь перемешивали при 0°С в течение 1 ч. К полученной смеси добавляли SEM-Cl (1,207 мл, 6,80 ммоль) и ее перемешивали при 20°С еще 1 ч. Ее гасили осторожно водой (15 мл) при 0°С и смесь экстрагировали EtOAc (20 мл × 4). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром : этилацетатом = 3:1 объем/объем) с получением указанного соединения. MS (ES+) m/z: 277, 279 (М+Н)
Стадия В: (2-Бром-1-((2-(триметилсилил)этокси)метил)-1Н-имидазол-5-ил)(5-хлорпиридин-2-ил)метанол: К перемешиваемому раствору 2-бром-1-((2-(триметилсилил)этокси)метил)-1Н-имидазола (15,53 г, 56,0 ммоль) в THF (100 мл) при -78°С, охлажденному в бане с сухим льдом и ацетоном, добавляли раствор LDA (30,2 мл, 60,3 ммоль, 2 M в THF) в атмосфере азота. Смесь перемешивали при -78°С в течение 1 ч, затем в реакционный раствор по каплям добавляли 5-хлорпиколинальдегид (6,1 г, 43,1 ммоль). После добавления баню удаляли и реакционную смесь оставляли нагреваться до к. т.и перемешивали еще 2 ч. Ее гасили насыщенным водным хлоридом аммония (100 мл) и смесь экстрагировали EtOAc (100 мл × 4). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром : EtOAc = 1:1 объем/объем) с получением указанного соединения. MS (ES+) m/z: 418, 420 (М+Н).
Стадия С: (2-Бром-1-((2-(триметилсилил)этокси)метил)-1H-имидазол-5-ил)(5-хлорпиридин-2-ил)метанон: К перемешиваемому раствору оксалилдихлорида (5,71 г, 45,0 ммоль) в DCM (150 мл) добавляли DMSO (6,76 г, 87 ммоль) при -78°С в атмосфере азота. Смесь перемешивали в течение 1 ч, затем при помощи шприца добавляли (2-бром-1-((2-(триметилсилил)этокси)метил)-1H-имидазол-5-ил)(5-хлорпиридин-2-ил)метанол (14,5 г, 34,6 ммоль). Смесь перемешивали при -78°С в течение 1 ч и добавляли TEA (19,30 мл, 138 ммоль). Реакционную смесь оставляли нагреваться до к. т. и перемешивали в течение 12 ч. Ее гасили ледяной водой (100 мл) и экстрагировали EtOAc (100 мл × 4). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром : EtOAc = 3:1 объем/объем) с получением указанного соединения. MS (ES+) m/z: 416, 418 (М+Н).
Стадия D: (2-Бром-1H-имидазол-5-ил)(5-хлорпиридин-2-ил)метанон: К дегазированному раствору (2-бром-1-((2-(триметилсилил)этокси)метил)-1H-имидазол-5-ил)(5-хлорпиридин-2-ил)метанона (15 г, 36,0 ммоль) в DCM (150 мл) при 0°С добавляли TFA (30 мл, 389 ммоль) в атмосфере азота. Полученную смесь перемешивали при к. т. в течение 2 ч. Ее концентрировали в вакууме с получением указанного соединения, которое использовали на следующей стадии без дополнительной очистки. MS (ES+) m/z: 286, 288 (М+Н).
Стадия Е: (2-Бром-1-((2-(триметилсилил)этокси)метил)-1Н-имидазол-5-ил)(5-хлорпиридин-2-ил)метанон: К дегазированному раствору (2-бром-1H-имидазол-5-ил)(5-хлорпиридин-2-ил)метанона (10,3 г, 35,9 ммоль) и SEM-Cl (8,3 мл, 46,7 ммоль) в DCM (150 мл) добавляли DIEA (18,8 мл, 108 ммоль) при 0°С в атмосфере азота. Полученную смесь перемешивали при 25°С в течение 12 ч. Ее разбавляли водой (100 мл) и экстрагировали EtOAc (200 мл × 4). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром : EtOAc = 3:1 объем/объем) с получением указанного соединения. MS (ES+) m/z: 416, 418 (М+Н).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 18
4-ФТОР-2-(4,4,5,5-ТЕТРАМЕТИЛ-1,3,2-ДИОКСАБОРОЛАН-2-ИЛ)АНИЛИН
Указанное соединение получали модифицированным способом, описанным в литературе (Org. Lett., 2014,16, 2916-2919). Смесь 2-бром-4-фторанилина (20 г, 105 ммоль), PdCl2(PPh3)2 (7,39 г, 10,53 ммоль), 4,4,5,5-тетраметил-1,3,2-диоксаборолана (40,4 г, 316 ммоль) и Et3N (58,7 мл, 421 ммоль) в диоксане (400 мл) перемешивали при 110°С в течение 16 ч в атмосфере азота. Ее охлаждали до к. т. и выливали в насыщенный водный раствор хлорида аммония (300 мл). Смесь экстрагировали DCM (300 мл). Органический слой отделяли, сушили над сульфатом натрия и фильтровали. Фильтрат концентрировали, а остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали EtOAc/петролейным эфиром = 10:90 объем/объем) с получением указанного соединения. 1Н ЯМР (400 МГц, CDCl3): δ 7.25 (m, 1H), 6.92 (m, 1Н), 6.54 (m, 1H), 4.42 (brs, 2H), 1.34 (s, 12H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 19
МЕТИЛ(3-АМИНО-4-(4,4,5,5-ТЕТРАМЕТИЛ-1,3,2-ДИОКСАБОРОЛАН-2-ИЛ)ФЕНИЛ)КАРБАМАТ
Стадия А: Метил(4-бром-3-нитрофенил)карбамат: К перемешиваемой смеси 4-бром-3-нитроанилина (11 г, 50,7 ммоль) в безводном THF (230 мл), охлажденной в бане с ледяной водой, небольшими частями добавляли NaH (60% масс. в минеральном масле, 2,43 г, 60,8 ммоль). Смесь перемешивали в течение 10 мин и по каплям добавляли метилхлорформиат (4,49 мл, 65,9 ммоль). Реакционную смесь перемешивали всю ночь, при этом температуру бани оставляли нагреваться до к. т. Ее охлаждали до 0°С и осторожно гасили водой (8 мл). Смесь разбавляли солевым раствором (100 мл) и экстрагировали этилацетатом (2×100 мл). Органический слой отделяли, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением указанного соединения. 1Н ЯМР (500 МГц, CDCl3): δ 8.05 (d, 1Н), 7.66 (d, 1H), 7.48 (d, 1H), 6.86 (s, 1H), 3.84 (s, 3H).
Стадия В: Метил(3-амино-4-бромфенил)карбамат: К перемешиваемой смеси метил(4-бром-3-нитрофенил)карбамата (14,3 г, 52,0 ммоль) и хлорида аммония (8,34 г, 156 ммоль) в этаноле (195 мл)-воде (65,0 мл) добавляли железный порошок (8,71 г, 156 ммоль). Смесь перемешивали при 80°С в течение 3 ч. Ее охлаждали до к. т. и разбавляли дихлорметаном (300 мл). Смесь фильтровали через слой целита. Фильтрат переносили в делительную воронку и экстрагировали дихлорметаном (2×80 мл). Объединенные органические слои промывали солевым раствором (150 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали EtOAc/гексаном = 1/1 объем/объем) с получением указанного соединения. MS (ES+) m/z: 245, 247 (М+Н).
Стадия C: Метил(3-амино-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)карбамат: Смесь бис(пинаколято)диборона (4,14 г, 16,32 ммоль), метил(3-амино-4-бромфенил)карбамата (2,00 г, 8,16 ммоль), ацетата калия (2,403 г, 24,48 ммоль) и дихлорметанового комплекса 1,1'-бис(дифенилфосфино)ферроцен-палладия (II) дихлорида (0,666 г, 0,816 ммоль) в круглодонной колбе с магнитной мешалкой вакуумировали и снова заполняли трижды азотом. Далее добавляли дегазированный диоксан (40,8 мл) и полученную смесь перемешивали при 100°С в атмосфере азота в течение 4 ч. Ее охлаждали до к. т. и разбавляли этилацетатом (100 мл). Смесь промывали водой (50 мл). Органический слой отделяли и сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали EtOAc/гексаном = 30% объем/объем) с получением указанного соединения. MS (ES+) m/z: 293 (М+Н).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 20
МЕТИЛ-4-АМИНО-3-(4,4,5,5-ТЕТРАМЕТИЛ-1,3,2-ДИОКСАБОРОЛАН-2-ИЛ)БЕНЗОАТ
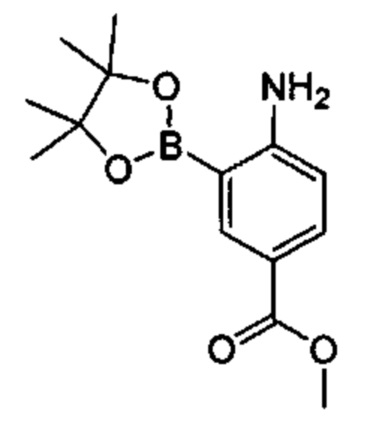
Указанное соединение получали из метил-4-амино-3-бромбензоата способом, описанным в синтезе промежуточного соединения 19, стадия С. MS (ES+) m/z: 278 (М+Н).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 21
ТРЕТ-БУТИЛ(3-АМИНО-4-(4,4,5,5-ТЕТРАМЕТИЛ-1,3,2-ДИОКСАБОРОЛАН-2-ИЛ)ФЕНИЛ)КАРБАМАТ
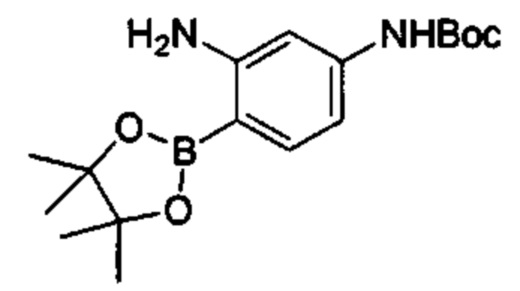
Стадия А: трет-Бутил(4-бром-3-нитрофенил)карбамат: К перемешиваемой смеси 4-бром-3-нитроанилина (20 г, 92 ммоль), DMAP (5,63 г, 46,1 ммоль) и Вос2О (24,14 г, 111 ммоль) в THF (300 мл) при 0°С добавляли DIEA (23,82 г, 184 ммоль). Смесь перемешивали при к. т. в течение 16 ч. Хлористоводородную кислоту (1 М, 200 мл) добавляли к реакционной смеси и смесь экстрагировали этилацетатом (3×200 мл). Объединенные органические фракции промывали водным бикарбонатом натрия (насыщенный, 2×200 мл), сушили над сульфатом натрия, фильтровали и растворитель выпаривали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром : EtOAc = 10:1 объем/объем) с получением указанного соединения.
Стадия В: трет-Бутил(3-амино-4-бромфенил)карбамат: В круглодонную колбу добавляли трет-бутил(4-бром-3-нитрофенил)карбамат (10 г, 31,5 ммоль), EtOH (200 мл), воду (40 мл), гидрохлорид аммония (16,87 г, 315 ммоль) и железный порошок (17,61 г, 315 ммоль). Реакционную смесь перемешивали при 80°С в течение 2 ч и ее оставляли охлаждаться до к. т. Смесь фильтровали через слой целита и фильтрат концентрировали при пониженном давлении. Остаток разбавляли водой (100 мл) и экстрагировали этилацетатом (200 мл × 3). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением указанного соединения, которое использовали без дополнительной очистки.
Стадия С: трет-Бутил(3-амино-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)карбамат: Смесь трет-бутил(3-нитро-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)карбамата (28,50 г, 78,00 ммоль) и Pd-C (2,50 г, 2,35 ммоль) в МеОН (300 мл) перемешивали при 25°С в течение 2 ч в атмосфере Н2. Смесь фильтровали и фильтрационный кек промывали метанолом (500 мл). Фильтрат концентрировали при пониженном давлении с получением указанного соединения. MS (ESI) m/z: 335 [М+Н]. 1Н ЯМР (400 МГц, CDCl3): δ 7.48 (d, J=8.0 Гц, 1Н), 6.92 (brs, 1H), 6.41 (dd, J=2.0, 8.0 Гц, 2H), 4.78 (brs, 2H), 1.50 (s, 9H), 1.32 (s, 12H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 22
ЭТИЛ(3-АМИНО-4-(4,4,5,5-ТЕТРАМЕТИЛ-1,3,2-ДИОКСАБОРОЛАН-2-ИЛ)ФЕНИЛ)КАРБАМАТ
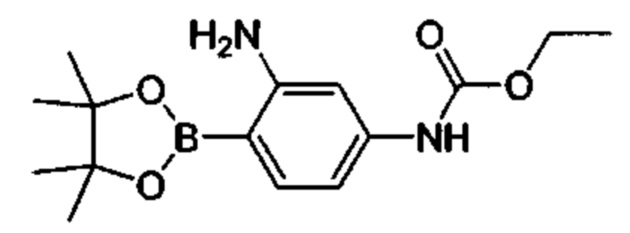
Указанное соединение получали способом, описанным в синтезе промежуточного соединения 19. Продукт очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром : EtOAc = 15:1-5:1) с получением указанного соединения. MS (ESI) m/z 307.1 (М+Н); 1Н ЯМР (400 МГц, CDCl3): δ 7.52 (d, J=8.2 Гц, 1Н), 6.92 (brs, 1H), 6.58-6.41 (m, 2H), 4.79 (brs, 2H), 4.21 (q, J=7.0 Гц, 2H), 1.33 (s, 12H), 1.25 (t, J=7.0 Гц, 3H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 23
МЕТИЛ(5-АМИНО-4-БРОМ-2-ФТОРФЕНИЛ)КАРБАМАТ

Стадия А: Метил(4-бром-2-фтор-5-нитрофенил)карбамат: К раствору 4-бром-2-фтор-5-нитроанилина (4,40 г, 18,72 ммоль) в THF (100 мл) добавляли DIEA (9,81 мл, 56,20 ммоль) и метилхлорформиат (5,31 г, 56,20 ммоль). Смесь перемешивали при 25°С в течение 15 ч. Большую часть растворителя удаляли; к остатку добавляли EtOAc (100 мл) и промывали солевым раствором (50 мл × 2), сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром : EtOAc = 20:1-10:1) с получением указанного соединения. 1Н ЯМР (400 МГц, CDCl3): δ 8.80 (d, J=5.9 Гц, 1Н), 7.46 (d, J=9.8 Гц, 1H), 6.99 (brs, 1H), 3.84 (s, 3H).
Стадия В: Метил(5-амино-4-бром-2-фторфенил)карбамат: К раствору метил(4-бром-2-фтор-5-нитрофенил)карбамата (3,70 г, 12,63 ммоль) в EtOH (60 мл) и воде (20 мл) добавляли железо (1,41 г, 25,30 ммоль) и хлорид аммония (2,70 г, 50,50 ммоль). Смесь перемешивали при 30°С в течение 16 ч. Реакционную смесь фильтровали, а фильтрат концентрировали при пониженном давлении. К остатку добавляли EtOAc (100 мл) и промывали солевым раствором (50 мл × 2), сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром : EtOAc = 15:1) с получением указанного соединения. MS (ESI) m/z 263, 265 (М+Н); 1Н ЯМР (400 МГц, CDCl3): δ 7.62 (brs, 1Н), 7.14 (d, J=10.2 Гц, 1H), 6.77 (brs, 1H), 3.97 (brs, 2H), 3.78 (s, 3H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 24
(4-КАРБОКСИПЕНТИЛ)ТРИФЕНИЛФОСФОНИЯ БРОМИД
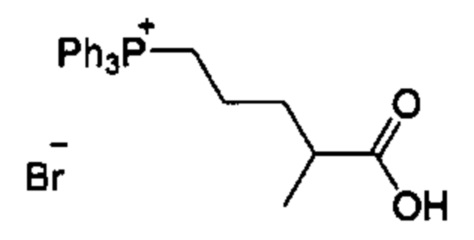
Стадия А: Диэтил-2-(3-бромпропил)-2-метилмалонат: К перемешиваемой суспензии гидрида натрия (12,63 г, 316 ммоль, 60% масс.) в THF (200 мл) в атмосфере азота добавляли раствор диэтил-2-метилмалоната (50 г, 287 ммоль) в THF (50 мл) при 0°С. Ее перемешивали в течение 0,5 ч и добавляли 1,3-дибромпропан (87 г, 431 ммоль). Реакционную смесь нагревали до 70°С и перемешивали еще в течение 3 ч. Ее охлаждали до 0°С и осторожно гасили водой. Ее экстрагировали EtOAc (100 мл × 3). Объединенные органические слои промывали солевым раствором (100 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром : этилацетатом = 100:1-30:1) с получением указанного соединения. MS: (ESI) m/z 295.2, 297.2 (М+Н).
Стадия В: 5-Бром-2-метилпентановая кислота: К перемешиваемому раствору диэтил-2-(3-бромпропил)-2-метилмалоната (48 г, 163 ммоль) в уксусной кислоте (55 мл) добавляли бромид водорода (52,60 г, 650 ммоль). Смесь перемешивали при 120°С в течение 15 ч. Ее охлаждали до к. т. и концентрировали при пониженном давлении. Остаток добавляли воду (50 мл) и экстрагировали DCM (70 мл × 3) и органический слой промывали солевым раствором (30 мл), сушили над сульфатом натрия и фильтровали. Фильтрат концентрировали с получением указанного соединения.
Стадия С: (4-Карбоксипентил)трифенилфосфония бромид: К перемешиваемому раствору 5-бром-2-метилпентановой кислоты (29 г, 149 ммоль) в толуоле (260 мл) добавляли трифенилфосфин (46,80 г, 178 ммоль). Затем смесь перемешивали при 120°С в атмосфере N2 в течение 16 ч. Ее охлаждали до к. т. и переносили в делительную воронку. Слой толуола отделяли, а слой продукт промывали горячим толуолом (30 мл × 5). Его отделяли и очищали методом колоночной флеш-хроматографии на силикагеле (элюировали DCM : МеОН = 20:1) с получением указанного соединения. MS: (ESI) m/z 377.1 (М+Н). 1Н ЯМР (400 МГц, CDCl3): δ 7.81-7.76 (m, 9Н), 7.72-7.69 (m, 6Н), 4.07-3.98 (m, 1Н), 3.32-3.16 (m, 2Н), 2.03-1.98 (m, 1Н), 1.75-1.64 (m, 3Н), 1.09 (d, J=6.4 Гц, 3Н).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 25
5-((1-ФЕНИЛ-1H-ТЕТРАЗОЛ-5-ИЛ)СУЛЬФОНИЛ)ПЕНТАНОВАЯ КИСЛОТА
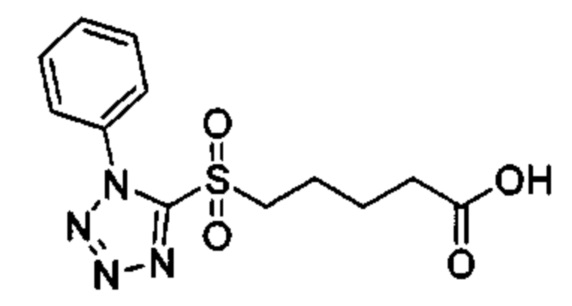
Стадия А: Этил-5-((1-фенил-1H-тетразол-5-ил)тио)пентаноат: К раствору 1-фенил-1H-тетразол-5-тиола (38 г, 213 ммоль) и этил-5-бромпентаноата (49,00 г, 235 ммоль) в ацетоне (40 мл) добавляли K2CO3 (29,50 г, 213 ммоль). Смесь перемешивали в течение 3 ч и ее фильтровали через слой целита. Фильтрат концентрировали при пониженном давлении, а остаток очищали методом колоночной флеш-хроматографии на силикагеле (петролейный эфир : этилацетат от 100:1 до 10:1) с получением указанного соединения. MS: (ESI) m/z 307,0 (М+Н).
Стадия В: 5-((1-Фенил-1H-тетразол-5-ил)тио)пентановая кислота: К перемешиваемому раствору этил-5-((1-фенил-1H-тетразол-5-ил)тио)пентаноата (65 г, 212 ммоль) в THF (200 мл) и воде (160 мл) добавляли LiOH (20,32 г, 849 ммоль). Смесь перемешивали при к. т. в течение 16 ч. Водную HCl (1 н) добавляли для доведения значения рН до 5. Смесь экстрагировали этилацетатом (500 мл × 2). Объединенные органические слои сушили над сульфатом натрия, фильтровали, а фильтрат концентрировали с получением указанного соединения. MS: (ESI) m/z 279,0 (М+Н).
Стадия С: 5-((1-Фенил-1H-тетразол-5-ил)сульфонил)пентановая кислота: К перемешиваемому раствору 5-((1-фенил-1H-тетразол-5-ил)тио)пентановой кислоты (26 г, 93 ммоль) в EtOH (100 мл) добавляли аммоний молибдат тетрагидрат (11,55 г, 9,34 ммоль) при 0°С. Смесь перемешивали в течение 5 мин, затем медленно добавляли пероксид водорода (635 г, 5605 ммоль). Смесь перемешивали при 40°С в течение 16 ч. Ее экстрагировали DCM (200 мл × 2). Объединенные органические слои промывали водным Na2SO3 (0,5 М, 200 мл), сушили над сульфатом натрия и фильтровали. Фильтрат концентрировали с получением указанного соединения. MS: (ESI) m/z 311.0 (М+Н). 1Н ЯМР (400 МГц, CDCl3): δ 7.51-7.68 (m, 5Н), 3.67-3.75 (m, 2Н), 2.39 (t, J=7.2 Гц, 2Н), 1.93-2.04 (m, 2Н), 1.79 (q, J=7.4 Гц, 2Н).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 26
5-(БЕНЗО[D]ТИАЗОЛ-2-ИЛСУЛЬФОНИЛ)ПЕНТАНОВАЯ КИСЛОТА
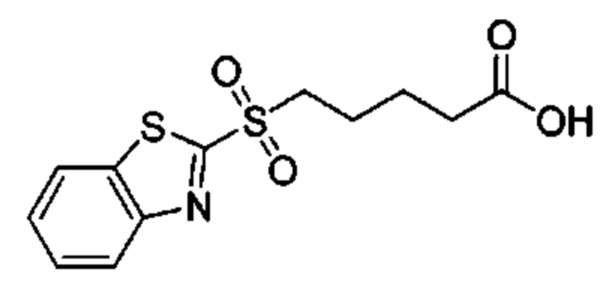
Указанное соединение получали из бензо[d]тиазол-2-тиола способом, описанным в синтезе промежуточного соединения 25. MS: (ESI) m/z 300.0 (М+Н); 1Н ЯМР (400 МГц, CDCl3): δ 8.20 (d, J=7.8 Гц, 1Н), 8.01 (d, J=8.2 Гц, 1Н), 7.55-7.66 (m, 2Н), 3.48-3.58 (m, 2Н), 2.38 (t, J=7.2 Гц, 2Н), 1.89-2.00 (m, 2Н), 1.75-1.84 (m, 2Н).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 27
5-(БЕНЗО[D]ТИАЗОЛ-2-ИЛСУЛЬФОНИЛ)-2,2-ДИМЕТИЛПЕНТАНОВАЯ КИСЛОТА
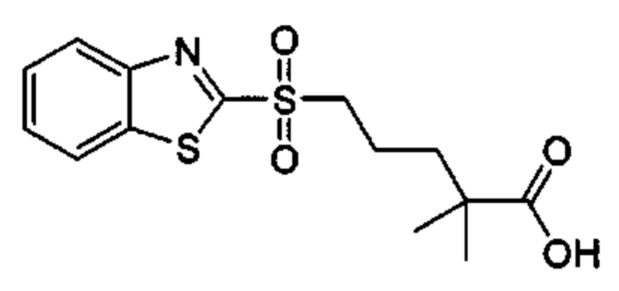
Стадия А: Метил-5-(бензо[d]тиазол-2-илтио)-2-метилпентаноат: К раствору метил-5-(бензо[d]тиазол-2-илтио)пентаноата (120 г, 426 ммоль) в THF (1500 мл) при -78°С по каплям добавляли раствор LDA (299 мл, 597 ммоль, 2 M в THF). Реакционную смесь перемешивали в течение 0,5 ч перед добавлением йодметана (78 мл, 1258 ммоль). Ее перемешивали еще 2 ч и гасили насыщенным водным хлоридом аммония (500 мл). Смесь оставляли нагреваться до к. т. и экстрагировали EtOAc (800 мл × 2). Объединенные органические слои сушили над сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (петролейный эфир : EtOAc, градиент от 20:1 до 10:1) с получением указанного соединения. 1Н ЯМР (400 МГц, CDCl3) δ 7.82 (dd, J=1.1, 8.1 Гц, 1Н), 7.71 (dd, J=1.2, 8.0 Гц, 1H), 7.37 (ddd, J=1.2, 7.2, 8.2 Гц, 1H), 7.23-7.28 (m, 1H), 3.59-3.66 (m, 3H), 3.26-3.34 (m, 2H), 2.46-2.50 (m, 1H), 1.77-1.84 (m, 3H), 1.56-1.61 (m, 1H), 1.14 (d, J=7.0 Гц, 3H).
Стадия В: Метил-5-(бензо[d]тиазол-2-илтио)-2,2-диметилпентаноат: К раствору метил-5-(бензо[d]тиазол-2-илтио)-2-метилпентаноата (3 г, 10,16 ммоль) в THF (45 мл) при -78°С по каплям добавляли раствор LDA (6,60 мл, 13,20 ммоль, 2 M в THF). Реакционную смесь перемешивали в течение 0,5 ч перед добавлением йодметана (2,51 мл, 40,30 ммоль). Ее перемешивали в течение 0,5 ч и гасили насыщенным водным хлоридом аммония (30 мл). Смесь оставляли нагреваться до к. т. и экстрагировали EtOAc (50 мл × 2). Объединенные органические слои сушили над сульфатом натрия и концентрировали при пониженном давлении с получением указанного соединения.
Стадия С: 5-(Бензо[d]тиазол-2-илтио)-2,2-диметилпентановая кислота: К раствору метил-5-(бензо[d]тиазол-2-илтио)-2,2-диметилпентаноата (5,50 г, 17,77 ммоль) в THF (55 мл) и воде (55 мл) добавляли LiOH (3,41 г, 142 ммоль) при 20°С. Смесь перемешивали в течение 72 ч и добавляли HCl (1 М) для доведения значения рН до 5. Смесь экстрагировали EtOAc (50 мл × 2). Объединенные органические слои сушили над сульфатом натрия и фильтровали. Фильтрат концентрировали с получением указанного соединения, которое использовали на следующей стадии сразу без дополнительной очистки. MS (ESI) m/z 296.2 (М+Н).
Стадия D: 5-(Бензо[d]тиазол-2-илсульфонил)-2,2-диметилпентановая кислота: К раствору 5-(бензо[d]тиазол-2-илтио)-2,2-диметилпентановой кислоты (4,90 г, 16,59 ммоль) в EtOH (5 мл) при 0°С добавляли аммоний молибдат тетрагидрат (2,05 г, 1,66 ммоль). Смесь перемешивали в течение 5 мин и медленно добавляли пероксид водорода (113 г, 995 ммоль). Смесь перемешивали при 15°С в течение 12 ч. Добавляли воду (80 мл) и смесь экстрагировали EtOAc (50 мл × 3). Объединенные органические слои промывали насыщенным тиосульфатом натрия (80 мл), сушили над сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного соединения. MS (ESI) m/z 328.0 (М+Н); 1Н ЯМР (400 МГц, CDCl3) δ 8.24-8.19 (m, 1Н), 8.02 (d, J=7.3 Гц, 1H), 7.67-7.58 (m, 2H), 3.55-3.47 (m, 2H), 2.04-1.80 (m, 2H), 1.79-1.62 (m, 2H), 1.25-1.15 (m, 6H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 28
1-(3-(БЕНЗО[D]ТИАЗОЛ-2-ИЛСУЛЬФОНИЛ)ПРОПИЛ)ЦИКЛОПРОПАН-1-КАРБОНОВАЯ КИСЛОТА
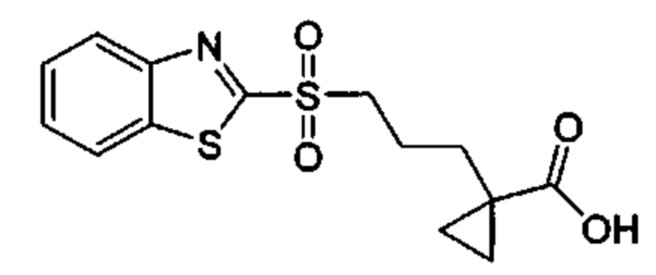
Стадия А: трет-Бутил-1-(3-бромпропил)циклопропанкарбоксилат: К раствору диизопропиламина (12,52 г, 124 ммоль) в THF (300 мл) медленно добавляли н-BuLi (49,5 мл, 124 ммоль, 2,5М в гексане) при -10°С. После завершения добавления смесь перемешивали при -10°С в течение 0,5 ч и охлаждали до -70°С. К реакционной смеси по каплям добавляли трет-бутилциклопропанкарбоксилат (16 г, 113 ммоль), а затем 1,3-дибромпропан (45,4 г, 225 ммоль). После добавления реакционную смесь постепенно нагревали до 25°С в течение 3 ч. Ее гасили насыщенным водным раствором хлорида аммония (150 мл) и экстрагировали EtOAc (150 мл × 3). Объединенные органические фракции промывали солевым раствором (20 мл), сушили над сульфатом натрия, фильтровали и растворитель выпаривали. Неочищенное соединение очищали методом колоночной хроматографии на силикагеле (петролейный эфир : этилацетат = 100:1) с получением указанного соединения. 1Н ЯМР (400 МГц, CDCl3): δ 3.40 (t, J=6.7 Гц, 2Н), 2.25 (d, J=6.7 Гц, 2Н), 1.55-1.60 (m, 2Н), 1.42 (s, 9Н), 0.73-0.76 (m, 2Н), 0.62 (dd, J=1.8, 2.7 Гц, 2Н).
Стадия B: трет-Бутил-1-(3-(бензо[d]тиазол-2-илтио)пропил)циклопропанкарбоксилат: К раствору бензо[d]тиазол-2-тиола (2,67 г, 15,96 ммоль) и трет-бутил-1-(3-бромпропил)циклопропанкарбоксилата (4 г, 6,08 ммоль, 40% чистоты) в ацетоне (30 мл) добавляли K2CO3 (4,20 г, 30,4 ммоль) при 25°С. После добавления смесь перемешивали в течение 2 ч. Твердые вещества удаляли фильтрацией через слой целита. Фильтрат концентрировали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир : EtOAc от 20:1 до 10:1) с получением указанного соединения. MS (ESI) m/z 350.0 (М+Н).
Стадия С: 1-(3-(Бензо[d]тиазол-2-илтио)пропил)циклопропанкарбоновая кислота: К перемешиваемому раствору трет-бутил-1-(3-(бензо[d]тиазол-2-илтио)пропил)циклопропанкарбоксилата (5,5 г, 15,74 ммоль) в EtOAc (40 мл) добавляли раствор HCl в этилацетате (4 М, 40 мл, 40 ммоль). Смесь перемешивали 25°С в течение 2 ч. Ее концентрировали при пониженном давлении. Остаток перетирали в порошок с 80 мл петролейного эфира. Твердые вещества фильтровали и промывали петролейным эфиром (20 мл × 2) с получением указанного соединения, которое использовали на следующей стадии без дополнительной очистки. MS (ESI) m/z 294.0 (М+Н).
Стадия D: 1-(3-(Бензо[d]тиазол-2-илсульфонил)пропил)циклопропанкарбоновая кислота: Раствор 1-(3-(бензо[d]тиазол-2-илтио)пропил)циклопропанкарбоновой кислоты (4 г, 13,63 ммоль) в THF (60 мл) при 0°С добавляли аммоний молибдат тетрагидрат (1,685 г, 1,363 ммоль). Смесь перемешивали в течение 5 мин, затем медленно к смеси добавляли пероксид водорода (15,46 г, 136 ммоль). Смесь перемешивали при 25°С в течение 16 ч и ее гасили водным тиосульфат натрия (100 мл, 50% раствор). Смесь экстрагировали EtOAc (70 мл × 3). Объединенные органические слои промывали солевым раствором (40 мл), сушили над сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного соединения, которое использовали на следующей стадии без дополнительной очистки. MS (ESI) m/z 348.1 (M+Na); 1H ЯМР (400 МГц, CDCl3): δ 8.17-8.23 (m, 1Н), 7.97-8.04 (m, 1H), 7.53-7.66 (m, 2H), 3.40-3.57 (m, 2H), 2.01-2.15 (m, 2H), 1.61-1.73 (m, 2H), 1.24-1.32 (m, 2H), 0.74-0.81 (m, 1H), 0.74-0.81 (m, 1H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 29
5-[БЕНЗО[D]ТИАЗОЛ-2-ИЛСУЛЬФОНИЛ)-4-МЕТИЛПЕНТАНОВАЯ КИСЛОТА
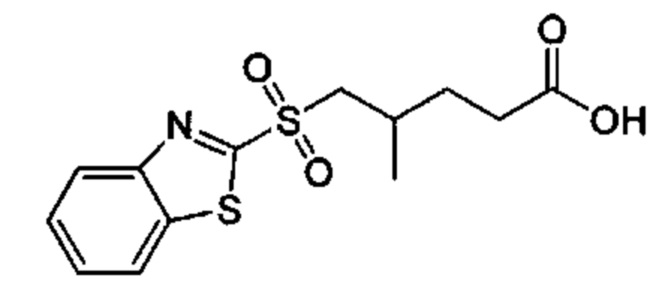
Стадия А: 1,3-Дибром-2-метилпропан: 2-Метилпропан-1,3-диол (10 г, 111 ммоль) медленно добавляли к трибромфосфину (21,08 мл, 222 ммоль) при 0°С. После добавления смесь перемешивали при 80°С в течение 18 ч. Реакционную смесь охлаждали до к. т. и выливали в охлажденный водный раствор карбоната натрия (2,0 л) при 0°С. Смесь экстрагировали DCM (500 мл × 3). Объединенные органические слои промывали солевым раствором (500 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром) с получением указанного соединения. 1Н ЯМР (CDCl3, 400 МГц): δ 3.56-3.49 (m, 2Н), 3.49-3.42 (m, 2Н), 2.24-2.12 (m, 1H), 1.15 (d, J=6.8 Гц, 3Н).
Стадия В: Триэтил-4-бром-3-метилбутан-1,1,1-трикарбоксилат: Смесь триэтилметантрикарбоксилата (12 г, 51,7 ммоль), 1,3-дибром-2-метилпропана (13,39 г, 62,0 ммоль) и K2CO3 (7,86 г, 56,8 ммоль) в DMF (130 мл) перемешивали при 80°С в течение 18 ч. Ее охлаждали до к. т. и гасили водой (300 мл). Смесь экстрагировали EtOAc (150 мл × 2). Объединенные органические слои промывали солевым раствором (150 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (EtOAc в петролейном эфире = 0%-2%, градиент) с получением указанного соединения. 1Н ЯМР (CDCl3, 400 МГц): δ 4.25 (q, J=7.1 Гц, 6Н), 3.51-3.42 (m, 1Н), 3.41-3.32 (m, 1Н), 2.42-2.31 (m, 1Н), 2.22-2.07 (m, 2H), 1.29 (t, J=7.0 Гц, 9H), 1.08 (d, J=6.5 Гц, 3Н).
Стадия С: 5-Бром-4-метилпентановая кислота: К раствору триэтил-4-бром-3-метилбутан-1,1,1-трикарбоксилата (8,5 г, 23,15 ммоль) в АсОН (60 мл) добавляли HBr (18,73 г, 93 ммоль). Смесь перемешивали в течение 18 ч при 120°С в атмосфере азота. Ее охлаждали до к. т. и концентрировали при пониженном давлении. К остатку добавляли EtOAc (150 мл) и промывали солевым раствором (100 мл × 3). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением указанного соединения.
Стадия D: Метил-5-бром-4-метилпентаноат: К раствору 5-бром-4-метилпентановой кислоты (4,7 г, 24,10 ммоль) в МеОН (50 мл) медленно добавляли концентрированную H2SO4 (0,642 мл, 12,05 ммоль). Смесь нагревали до температуры образования флегмы в атмосфере азота в течение 18 ч. Ее охлаждали до к. т. и разбавляли водой (50 мл). Значение рН раствора доводили до 7-8 добавлением водного бикарбоната натрия. Смесь экстрагировали EtOAc (30 мл × 3). Объединенные органические слои промывали солевым раствором (50 мл × 2), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением указанного соединения.
Стадия Е: Метил-5-(бензо[d]тиазол-2-илтио)-4-метилпентаноат: Смесь метил-5-бром-4-метилпентаноата (4,2 г, 20,09 ммоль), бензо[d]тиазол-2-тиола (5,04 г, 30,1 ммоль) и K2CO3 (4,16 г, 30,1 ммоль) в ацетонитриле (100 мл) перемешивали при 25°С в течение 18 ч. Ее гасили водой (300 мл) и экстрагировали EtOAc (100 мл × 3). Объединенные органические слои промывали солевым раствором (150 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (EtOAc в петролейном эфире = 0%-6%, градиент) с получением указанного соединения. MS (ESI) m/z: 296.1 (М+Н);
Стадия F: 5-(Бензо[d]тиазол-2-илтио)-4-метилпентановая кислота: К раствору метил-5-(бензо[d]тиазол-2-илтио)-4-метилпентаноата (5,28 г, 17,87 ммоль) в THF (80 мл) добавляли водный раствор LiOH (2 н, 13,40 мл, 26,8 ммоль). Его перемешивали в течение 3 ч и разбавляли водой (200 мл). Значение рН раствора доводили до 4-5 добавлением водной HCl (1 н). Смесь экстрагировали EtOAc (80 мл × 3). Объединенные органические слои промывали солевым раствором (120 мл), сушили над сульфатом натрия, фильтровали и концентрировали с получением указанного соединения.
Стадия G: 5-(Бензо[d]тиазол-2-илсульфонил)-4-метилпентановая кислота: К перемешиваемой смеси 5-(бензо[d]тиазол-2-илтио)-4-метилпентановой кислоты (5,03 г, 17,88 ммоль) в THF (50 мл) и воде (25 мл) при 0°С добавляли оксон (29,1 г, 47,4 ммоль). Смесь перемешивали при к. т. всю ночь. Ее разбавляли ледяной водой (100 мл) и гасили насыщенным водным тиосульфатом натрия. Смесь экстрагировали DCM (150 мл × 3). Объединенные органические слои промывали солевым раствором (200 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением указанного соединения. MS (ESI) m/z: 314.0 (М+Н); 1Н ЯМР (CDCl3, 400 МГц): δ 8.21 (dd, J=1.3, 7.6 Гц, 1Н), 8.05-7.99 (m, 1Н), 7.68-7.56 (m, 2Н), 3.57 (dd, J=5.0, 14.3 Гц, 1Н), 3.40 (dd, J=7.6,14.4 Гц, 1H), 2.44-2.30 (m, 3Н), 1.96-1.84 (m, 1Н), 1.75-1.61 (m, 1Н), 1.17 (d, J=4.0 Гц, 3Н).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 30
цис-2-(2-((БЕНЗО[D]ТИАЗОЛ-2-ИЛСУЛЬФОНИЛ)МЕТИЛ)ЦИКЛОПРОПИЛ)УКСУСНАЯ КИСЛОТА
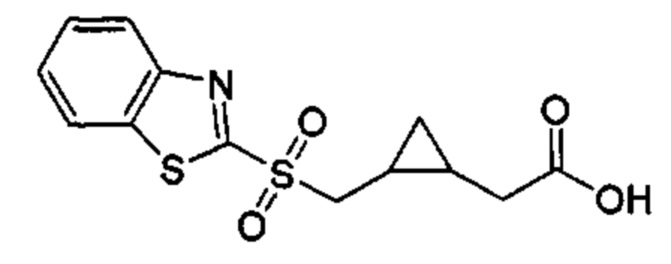
Стадия А: Бензил-2-диазоацетат: К перемешиваемой двухфазной смеси бензил-2-аминоацетата гидрохлорида (10 г, 49,6 ммоль) в 1:1 воде/DCM (166 мл) при 0°С в атмосфере азота по каплям добавляли раствор нитрита натрия (6,84 г, 99 ммоль) в воде (6 мл). Полученную смесь перемешивали в течение 1,5 ч при 0°С, затем нагревали до к. т. всю ночь. Ее разбавляли бикарбонатом натрия (насыщенный, 100 мл) и экстрагировали дихлорметаном (100 мл × 3). Объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 3:1 EtOAc/EtOH и гексаном, 0-30%, градиент) с получением указанного соединения. MS (ES+) m/z: 208.1 (М+32).
Стадия В: цис-Бензил-2-(2-(трет-бутокси)-2-оксоэтил)циклопропан-1-карбоксилат и транс-бензил-2-(2-(трет-бутокси)-2-оксоэтил)циклопропан-1-карбоксилат: К перемешиваемому раствору трет-бутилбут-3-еноата (3,5 г, 24,61 ммоль) и ацетата родия (II) (1,088 г, 4,92 ммоль) в DCM (35,2 мл) в атмосфере азота по каплям добавляли бензил-2-диазоацетат (4,77 г, 27,1 ммоль) и оставляли перемешиваться всю ночь. Полученный раствор фильтровали через слой целита и фильтрат концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 3:1 EtOAc/EtOH и гексаном, 0-20%, градиент) с получением указанных соединений. Изомер А (цис, более быстрое элюирование) MS (ES+) m/z: 313.1 (M+Na). Изомер В (транс, более медленное элюирование) MS (ES+) m/z: 313.1 (M+Na).
Стадия С: цис-2-(2-(трет-Бутокси)-2-оксоэтил)циклопропан-1-карбоновая кислота: Смесь цис-бензил-2-(2-(трет-бутокси)-2-оксоэтил)циклопропан-1-карбоксилата (1,22 г, 4,20 ммоль) и палладия на углеродном катализаторе (0,447 г, 0,420 ммоль) в МеОН (21 мл) перемешивали при атмосферном давлении водорода в течение 7 ч, при этом его фильтровали через слой целита. Фильтрат выпаривали при пониженном давлении с получением указанного соединения. MS (ES+) m/z: 201.1 (М+Н).
Стадия D: цис-трет-Бутил-2-(2-(гидроксиметил)циклопропил)ацетат: К раствору цис-2-(2-(трет-бутокси)-2-оксоэтил)циклопропан-1-карбоновой кислоты (704 мг, 3,52 ммоль) в THF (35,2 мл) с перемешиванием в атмосфере азота медленно добавляли боран (1 M в THF, 8,79 мл, 8,79 ммоль). Реакцию оставляли проходить в течение 6 ч, при этом раствор охлаждали до 0°С и добавляли воду (250 мкл), а затем МеОН (5 мл) до завершения барботирования. Раствор выпаривали при пониженном давлении. Остаток очищали методом нормально-фазовой хроматографии (градиентное элюирование 3:1 EtOAc/EtOH и гексаном, 0-40%, градиент) с получением указанного соединения. MS (ES+) m/z: 187.1 (М+Н).
Стадия Е: цис-Бутил-2-(2-(бромметил)циклопропил)ацетат: К перемешиваемой смеси цис-трет-бутил-2-(2-(гидроксиметил)циклопропил)ацетата (322 мг, 1,729 ммоль) в DCM (14,40 мл) добавляли трифенилфосфин (680 мг, 2,59 ммоль), а затем тетрабромид углерода (860 мг, 2,59 ммоль). Ее перемешивали в течение 24 ч, при этом смесь выпаривали при пониженном давлении. После добавления ацетона (50 мл) твердое вещество отфильтровывали, а фильтрат выпаривали при пониженном давлении с получением указанного соединения, которое использовали без очистки. MS (ES+) m/z: 193.0 (М-56).
Стадия F: цис-трет-Бутил-2-(2-((бензо[d]тиазол-2-илтио)метил)циклопропил)ацетат: Смесь бензо[d]тиазол-2-тиола (434 мг, 2,59 ммоль) и карбоната калия (359 мг, 2,59 ммоль) перемешивали в ацетоне (6,92 мл) в атмосфере азота в течение 10 мин. Добавляли раствор цис-бутил-2-(2-(бромметил)циклопропил)ацетата (431 мг, 1,730 ммоль) в ацетоне (6,9 мл) и реакцию оставляли проходить всю ночь. Твердые вещества отфильтровывали, а фильтрат выпаривали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 3:1 EtOAc/EtOH и гексаном, 0-5%, градиент) с получением указанного соединения. MS (ES+) m/z: 336.1 (М+Н).
Стадия G: цис-2-(2-((Бензо[d]тиазол-2-илтио)метил)циклопропил)уксусная кислота: Раствор цис-трет-бутил-2-(2-((бензо[d]тиазол-2-илтио)метил)циклопропил)ацетата (300 мг, 0,894 ммоль) в 1:1 DCM/TFA (17,88 мл) перемешивали в течение 1 ч. Большую часть растворителя выпаривали при пониженном давлении с получением указанного соединения, которое использовали без очистки. MS (ES+) m/z: 280,0 (М+Н).
Стадия Н: цис-2-(2-((Бензо[d]тиазол-2-илсульфонил)метил)циклопропил)уксусная кислота: К перемешиваемому раствору цис-2-(2-((бензо[d]тиазол-2-илтио)метил)циклопропил)уксусной кислоты (215 мг, 0,770 ммоль) в THF (14,7 мл) добавляли раствор оксона (1419 мг, 2,309 ммоль) в воде (7,3 мл) и реакцию оставляли проходить всю ночь. Смесь гасили сульфитом натрия (насыщенный, 125 мл), а затем экстрагировали EtOAc (100 мл). Водный раствор снова экстрагировали 3:1 хлороформом/IPA (100 мл × 2). Объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученный остаток далее сушили под вакуумом в течение 6 ч с получением указанного соединения. MS (ES+) m/z: 311.9 (М+Н).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 31
транс-2-(2-((БЕНЗО[D]ТИАЗОЛ-2-ИЛСУЛЬФОНИЛ)МЕТИЛ)ЦИКЛОПРОПИЛ)УКСУСНАЯ КИСЛОТА
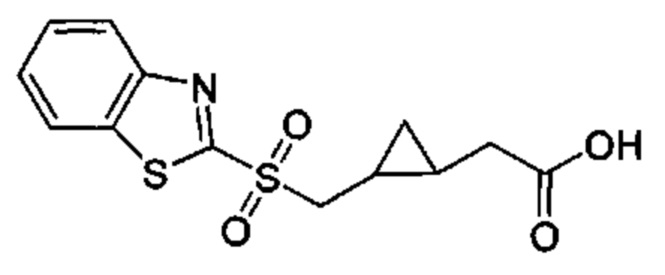
Промежуточное соединение 30 получали из транс-бензил-2-(2-(трет-бутокси)-2-оксоэтил)циклопропан-1-карбоксилата способом, описанным в синтезе промежуточного соединения 29. MS (ES+) m/z: 311.9 (М+Н).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 32
2-(3-ХЛОР-2,6-ДИФТОРФЕНИЛ)-4,4,5,5-ТЕТРАМЕТИЛ-1,3,2-ДИОКСАБОРОЛАН
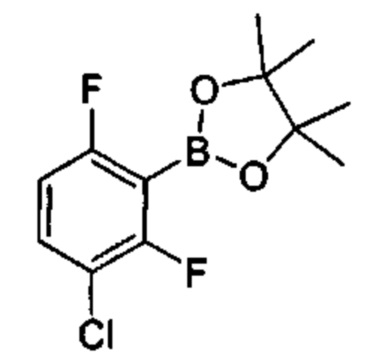
К раствору 1-хлор-2,4-дифторбензола (10 г, 67,3 ммоль) в безводном THF (150 мл) по каплям добавляли н-бутиллитий (2,5 М в гексане, 26,9 мл, 67,3 ммоль) при -78°С. В это время температуру поддерживали ниже -65°С. После добавления смесь перемешивали при -78°С еще 1,5 ч. К смеси по каплям добавляли 2-изопропоксибиспинаколятодиборон (25,05 г, 135 ммоль). Полученную реакционную смесь оставляли нагреваться до к. т. (25°С) и перемешивали в течение 16 ч. Смесь гасили водой (50 мл), фильтровали и фильтрат концентрировали при пониженном давлении. Остаток экстрагировали EtOAc (150 мл × 2). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток перетирали в порошок в петролейном эфире (100 мл) в течение 1 ч. Твердые вещества собирали фильтрацией с получением указанного соединения. 1Н ЯМР (400 МГц, CD3OD): δ 7.02-6.93 (m, 1Н), 6.53 (t, J=8.0 Гц, 1H), 1.09 (s, 12Н).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 33
4-ХЛОР-2-(4,4,5,5-ТЕТРАМЕТИЛ-1,3,2-ДИОКСАБОРОЛАН-2-ИЛ)АНИЛИН
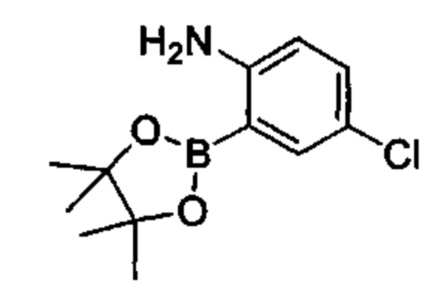
Указанное соединение получали из 2-бром-4-хлоранилина способом, описанным в синтезе промежуточного соединения 18. MS (ESI) m/z: 254 [М+Н].
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 34
2-БРОМ-4-ХЛОР-1-(ТРИФТОРМЕТОКСИ)БЕНЗОЛ
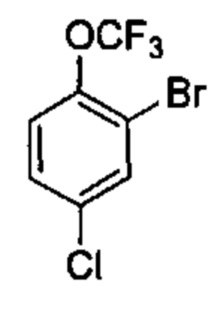
К перемешиваемому раствору 5-хлор-2-(трифторметокси)анилина (1,00 г, 4,73 ммоль) в ацетонитриле (60 мл) и воде (6 мл) добавляли бромид меди (II) (1,48 г, 6,62 ммоль) и изопентилнитрит (0,95 г, 8,13 ммоль). Смесь перемешивали при 70°С в течение 1 ч. Ее охлаждали до к. т. и гасили водой (200 мл). Смесь экстрагировали DCM (60 мл × 2). Объединенные органические слои промывали солевым раствором (60 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром) с получением указанного соединения. 1Н ЯМР (400 МГц, CDCl3): δ 7.58 (d, J=2.2 Гц, 1Н), 7.29-7.22 (m, 1Н), 7.19 (s, 1Н).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 35
2-БРОМ-4-ХЛОР-1-(ДИФТОРМЕТОКСИ)БЕНЗОЛ
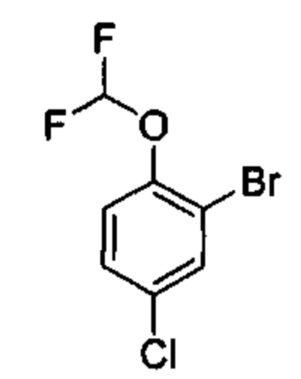
К перемешиваемой смеси 2-бром-4-хлорфенола (500 мг, 2,41 ммоль) и KOH (2,71 г, 48,2 ммоль) в ацетонитриле (15 мл) и воде (15 мл) в атмосфере азота при -78°С добавляли диэтил(бромдифторметил)фосфонат (1,29 г, 4,82 ммоль). Смесь оставляли нагреваться до к.т. и перемешивали в течение 30 мин. Добавляли воду (20 мл) и смесь экстрагировали петролейным эфиром (100 мл). Органическую фракцию промывали солевым раствором (100 мл), сушили над сульфатом натрия, фильтровали и растворитель выпаривали при пониженном давлении с получением указанного соединения. 1Н ЯМР (400 МГц, CDCl3): δ 7.61 (s, 1Н), 7.28 (dd, J=2.3, 9.0 Гц, 1Н), 7.15 (brd, J=9.0 Гц, 1Н), 6.49 (t, J=73.2 Гц, 1Н).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 36
2-БРОМ-1-(ДИФТОРМЕТОКСИ)-3-ФТОРБЕНЗОЛ
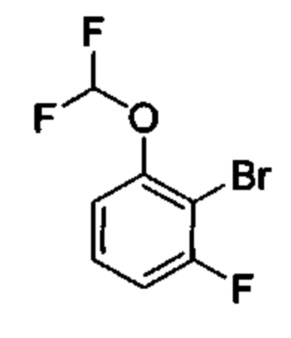
К перемешиваемой смеси 2-бром-3-фторфенола (6,00 г, 31,40 ммоль) в DMF (30 мл) добавляли K2CO3 (5,21 г, 37,70 ммоль) и 2-хлор-2,2-дифторацетат натрия (5,75 г, 37,70 ммоль) одной порцией и смесь перемешивали при 70°С в течение 15 ч в атмосфере N2. Добавляли воду (100 мл) и смесь экстрагировали петролейным эфиром (100 мл × 2). Объединенные органические фракции промывали солевым раствором, сушили над сульфатом натрия, фильтровали и растворитель выпаривали при пониженном давлении с получением указанного соединения, которое использовали сразу. 1Н ЯМР (400 МГц, CDCl3): δ 7.34-7.23 (m, 1Н), 7.07-6.94 (m, 2H), 6.54 (t, J=73.0 Гц, 1H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 37
2-БРОМ-1-(ДИФТОРМЕТИЛ)-3-ФТОРБЕНЗОЛ
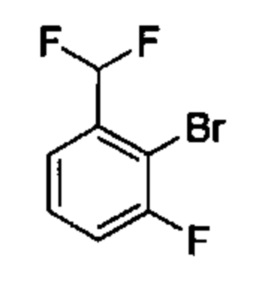
К перемешиваемой смеси 2-бром-3-фторбензальдегида (2,00 г, 9,85 ммоль) в DCM (70 мл) в атмосфере N2 добавляли DAST (1,65 мл, 12,31 ммоль) при 0°С. Смесь перемешивали в течение 1 ч, затем ее оставляли нагреваться до 25°С с перемешиванием в течение еще 1 ч. Добавляли водный насыщенный NaHCO3 (50 мл) и смесь экстрагировали DCM (100 мл × 2). Объединенные органические фракции сушили над сульфатом натрия, фильтровали и растворитель выпаривали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-5% EtOAc в петролейном эфире) с получением указанного соединения. 1Н ЯМР (400 МГц, CDCl3): δ 7.50-7.36 (m, 2Н), 7.25 (t, J=8.2 Гц, 1Н), 6.92 (t, J=60.0 Гц, 1Н).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 38
2-БРОМ-1-(ДИФТОРМЕТОКСИ)-3,4-ДИФТОРБЕНЗОЛ
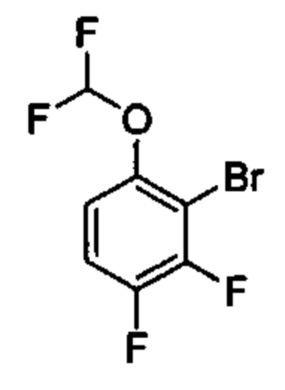
Стадия А: 1.2-Дифтор-4-(метоксиметокси)бензол: К перемешиваемой смеси 3,4-дифторфенола (5,00 г, 38,40 ммоль) в DCM (80 мл) при -10°С добавляли DIEA (13,43 мл, 77 ммоль), затем по каплям MOM-Cl (5,84 мл, 77,00 ммоль). Смесь нагревали до 20°С и перемешивали в течение 16 ч. Добавляли водный хлорид аммония (насыщенный, 100 мл) и смесь экстрагировали DCM (150 мл × 2). Объединенные органические фракции промывали водной НС1 (1М, 100 мл), сушили над сульфатом натрия, фильтровали и растворитель выпаривали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром-петролейным эфиром : EtOAc = 20:1) с получением указанного соединения. 1Н ЯМР (400 МГц, CDCl3): δ 7.10-7.01 (m, 1Н), 6.89 (ddd, J=2.9, 6.7, 11.9 Гц, 1H), 6.74 (dtd, J=1.8, 3.2, 9.1 Гц, 1H), 5.14 (s, 2H), 3.49 (s, 3H).
Стадия В: 2-Бром-3,4-дифтор-1-(метоксиметокси)бензол: К перемешиваемой смеси 1,2-дифтор-4-(метоксиметокси)бензола (5,20 г, 29,90 ммоль) в THF (80 мл) по каплям добавляли н-BuLi (14,33 мл, 35,80 ммоль, 2,5 М в гексане) при -78°С. Смесь перемешивали в течение 60 мин при -78°С, а затем добавляли Br2 (1,85 мл, 35,80 ммоль). Смесь перемешивали еще 1 ч и добавляли водный хлорид аммония (насыщенный, 100 мл). Смесь экстрагировали DCM (200 мл × 2). Объединенные органические фракции промывали солевым раствором (200 мл × 2), сушили над сульфатом натрия, фильтровали и растворитель выпаривали при пониженном давлении с получением указанного соединения.
Стадия С: 2-Бром-3,4-дифторфенол: К перемешиваемой смеси 2-бром-3,4-дифтор-1-(метоксиметокси)бензола (7,00 г, 27,70 ммоль) в THF (40 мл) добавляли водную HCl (46,10 мл, 277 ммоль, 6 М). Смесь перемешивали при 25°С в течение 16 ч, гасили водой (100 мл). Смесь экстрагировали диэтиловым эфиром (200 мл × 2). Объединенные органические фракции промывали солевым раствором (200 мл × 2), сушили над сульфатом натрия, фильтровали и растворитель выпаривали при пониженном давлении с получением указанного соединения.
Стадия D: 2-бром-1-(дифторметокси)-3,4-дифторбензол: К перемешиваемой смеси 2-бром-3,4-дифторфенола (6,30 г, 25,60 ммоль) и гидроксида калия (28,80 г, 512 ммоль) в ацетонитриле (100 мл) и воде (100 мл) добавляли диэтил(бромдифторметил)фосфонат (13,68 г, 51,20 ммоль). Смесь перемешивали при 25°С в течение 30 мин и гасили водой (200 мл). Смесь экстрагировали диэтиловым эфиром (500 мл × 2). Объединенные органические фракции промывали солевым раствором (500 мл × 2), сушили над сульфатом натрия, фильтровали и растворитель выпаривали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром) с получением указанного соединения. 1Н ЯМР (400 МГц, CDCl3): δ 7.17 (dt, J=8.3, 9.2 Гц, 1Н), 7.07-7.00 (m, 1H), 6.51 (t, J=72.0 Гц, 1H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 39
4-ХЛОР-3-ФТОР-2-(4,4,5,5-ТЕТРАМЕТИЛ-1,3,2-ДИОКСАБОРОЛАН-2-ИЛ)АНИЛИН
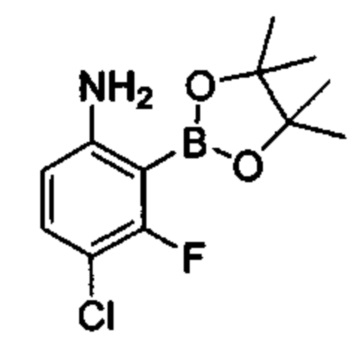
Указанное соединение получали из 2-бром-4-хлор-3-фторанилина способом, описанным в синтезе промежуточного соединения 19, стадия С. MS (ES+) m/z: 272 (М+Н).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 40
(5-ХЛОРПИРИДИН-2-ИЛ)(5-ФТОР-4-ЙОДПИРИДИН-2-ИЛ)МЕТАНОН
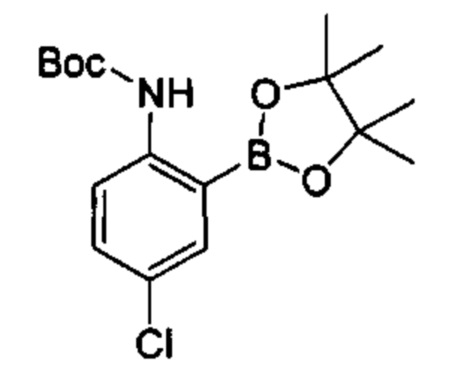
К раствору промежуточного соединения 33 (6,00 г, 23,67 ммоль) в толуоле (80 мл) добавляли Вос2О (16,48 мл, 71,0 ммоль) и TEA (16,49 мл, 118 ммоль). Смесь перемешивали при 80°С в атмосфере азота в течение 13 ч. Ее охлаждали до к. т. и гасили водой (80 мл). Ее экстрагировали EtOAc (3×80 мл). Объединенные органические слои сушили над сульфатом натрия и фильтровали. Фильтрат концентрировали в вакууме. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 10% EtOAc/петролейным эфиром, градиент) с получением указанного соединения. 1Н ЯМР (400 МГц, CDCl3): δ 8.62 (s, 1Н), 8.15 (d, J=8.8 Гц, 1H), 7.66 (d, J=2.4 Гц, 1H), 7.34( dd, J=2.4, 8.8 Гц, 1H), 1.52 (s, 9H), 1.36 (s, 12H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 41
5-(4-ХЛОР-2-(4,4,5,5-ТЕТРАМЕТИЛ-1,3,2-ДИОКСАБОРОЛАН-2-ИЛ)ФЕНИЛ)ОКСАЗОЛ

Стадия А: 2-Бром-4-хлор-N-метокси-N-метилбензамид: В круглодонную колбу добавляли 2-бром-4-хлорбензойную кислоту (10 г, 42,5 ммоль), DCM (150 мл), HATU (19,38 г, 51,0 ммоль), N,O-диметилгидроксиламина гидрохлорид (12,43 г, 127 ммоль) и триэтиламин (29,6 мл, 212 ммоль). Реакционную смесь перемешивали в течение 18 ч при к. т. Смесь гасили водой (200 мл) и экстрагировали DCM (100 мл × 3). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали в вакууме. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром : EtOAc = 10:1, объем/объем) с получением указанного соединения. MS (ES+) m/z: 278, 280 [М+Н].
Стадия В: 2-Бром-4-хлорбензальдегид: К раствору 2-бром-4-хлор-N-метокси-N-метилбензамида (11 г, 39,5 ммоль) в безводном THF (150 мл) добавляли диизобутилалюмогидрид (1 M в толуоле, 71,1 мл, 71,1 ммоль) по каплям при -78°С. Реакционную смесь перемешивали в течение 1 ч и гасили насыщенным раствором виннокислого калия-натрия (300 мл). Смесь перемешивали в течение 20 мин и фильтровали через слой целита. Фильтрат экстрагировали этилацетатом (200 мл × 3). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали в вакууме с получением указанного соединения. Его использовали на следующей стадии без дополнительной очистки.
Стадия С: 5-(2-Бром-4-хлорфенил)оксазол: В круглодонную колбу добавляли 2-бром-4-хлорбензальдегид (8,00 г, 36,5 ммоль), МеОН (200 мл), 1-((изоцианометил)сульфонил)-4-метилбензол (10,68 г, 54,7 ммоль) и K2CO3 (15,11 г, 109 ммоль). Смесь перемешивали при 70°С в течение 3 ч. Смесь концентрировали в вакууме и добавляли воду (200 мл). Смесь экстрагировали этилацетатом (150 мл × 3), Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-10% этилацетатом/петролейным эфиром) с получением указанного соединения. MS (ES+) m/z: 258, 260 [М+Н].
Стадия D: 5-(4-Хлор-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)оксазол: Указанное соединение получали из 5-(2-бром-4-хлорфенил)оксазола способом, описанным в синтезе промежуточного соединения 19. MS (ES+) m/z: 306 [М+Н].
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 42
1-(4-ХЛОР-2-(ТРИМЕТИЛСТАННИЛ)ФЕНИЛ)-4-(ДИФТОРМЕТИЛ)-1Н-1,2,3-ТРИАЗОЛ
Стадия А: 1-Азидо-2-бром-4-хлорбензол: К суспензии 2-бром-4-хлоранилина (5 г, 24,22 ммоль) в HCl (36,5% масс., 30 мл, 365 ммоль) и воде (100 мл) при -5°С по каплям добавляли раствор нитрита натрия (1,838 г, 26,6 ммоль) в воде (10 мл). Смесь перемешивали при -5°С в течение 1 ч и суспензия превращалась в прозрачный раствор. По каплям к реакционной смеси добавляли раствор азида натрия (1,732 г, 26,6 ммоль) в воде (10 мл). В течение добавления из раствора осаждались твердые вещества. Его перемешивали при -5°С в течение 0,5 ч. Смесь экстрагировали этилацетатом (70 мл × 3). Объединенные органические слои последовательно промывали насыщенным водным бикарбонатом натрия (20 мл), водой (40 мл) и солевым раствором (50 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением указанного соединения.
Стадия В: (1-(2-Бром-4-хлорфенил)-1H-1,2,3-триазол-4-ил)метанол: В круглодонную колбу добавляли 1-азидо-2-бром-4-хлорбензол (5 г, 21,51 ммоль), проп-2-ин-1-ол (2,412 г, 43,0 ммоль), (R)-2-((S)-1,2-дигидроксиэтил)-4-гидрокси-5-оксо-2,5-дигидрофуран-3-олят натрия (2,130 г, 10,75 ммоль), сульфат меди (II) (1,716 г, 10,75 ммоль), THF (60 мл) и воду (60 мл). Реакционную смесь перемешивали при 100°С в атмосфере азота в течение 6 ч. Ее охлаждали до к. т. и фильтровали через слой целита. Фильтрат концентрировали в вакууме. К остатку добавляли воду (100 мл) и смесь экстрагировали этилацетатом (3×50 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-40% EtOAc/петролейным эфиром) с получением указанного соединения. MS (ESI) m/z: 288, 290 [М+Н].
Стадия С: 1-(2-Бром-4-хлорфенил)-1H-1,2,3-триазол-4-карбальдегид: В круглодонную колбу добавляли (1-(2-бром-4-хлорфенил)-1H-1,2,3-триазол-4-ил)метанол (5 г, 12,48 ммоль), DCM (100 мл) и оксид марганца (IV) (10,85 г, 125 ммоль). Реакционную смесь перемешивали при 25°С в течение 18 ч. Смесь фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-20% EtOAc/петролейным эфиром, 40 мин, сухая загрузка) с получением указанного соединения. MS (ESI) m/z: 286, 288 [М+Н].
Стадия D: 1-(2-Бром-4-хлорфенил)-4-(дифторметил)-1H-1,2,3-триазол: К раствору 1-(2-бром-4-хлорфенил)-1H-1,2,3-триазол-4-карбальдегида (1,6 г, 5,58 ммоль) в DCM (20 мл) добавляли DAST (1,476 мл, 11,17 ммоль). Полученную смесь перемешивали при 20°С в течение 2 ч и гасили насыщенный водным бикарбонатом натрия (20 мл) и водой (50 мл). Смесь экстрагировали DCM (3×20 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением указанного соединения, которое использовали на следующей стадии без дополнительной очистки. MS (ESI) m/z: 308, 310 [М+Н].
Стадия Е: 1-(4-Хлор-2-(триметилстаннил)фенил)-4-(дифторметил)-1H-1,2,3-триазол: К раствору 1-(2-бром-4-хлорфенил)-4-(дифторметил)-1H-1,2,3-триазола (1,8 г, 4,38 ммоль) и 1,1,1,2,2,2-гексаметилдистаннана (4,30 г, 13,13 ммоль) в толуоле (30 мл) добавляли тетракис(трифенилфосфин)палладий(0) (1,011 г, 0,875 ммоль). Смесь перемешивали при 120°С в атмосфере азота в течение 18 ч. Ее охлаждали до к. т. и фильтровали через слой целита. Фильтрат концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-10% EtOAc/петролейным эфиром) с получением указанного соединения. MS (ESI) m/z: 394 [М+Н].
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 43
1-(2-БРОМ-4-ХЛОРФЕНИЛ)-4-ЦИКЛОПРОПИЛ-1H-1,2,3-ТРИАЗОЛ
К перемешиваемой смеси 1-азидо-2-бром-4-хлорбензола (660 мг, 2,84 ммоль) в толуоле (12 мл) добавляли циклопропилацетилен (0,360 мл, 4,26 ммоль); смесь перемешивали при 110°С всю ночь. Реакционную смесь охлаждали до к. т. и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 20-50% EtOAc/гексан) с получением указанного соединения. MS (ES+) m/z: 298, 300 (М+Н).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 44
1-(4-ХЛОР-2-ЙОДФЕНИЛ)-4-(ДИФТОРМЕТИЛ)-1Н-1,2,3-ТРИАЗОЛ
Стадия А: 1-Азидо-4-хлор-2-йодбензол: К раствору 4-хлор-2-йоданилина (10 г, 39,5 ммоль) в этилацетате (80 мл) и воде (10 мл) в бане с ледяной водой по каплям добавляли HCl (37%, 22,03 мл, 268 ммоль). Полученную смесь перемешивали в течение 10 минут. К раствору добавляли раствор нитрит натрия (2,132 мл, 67,1 ммоль) в воде (15 мл) в течение 10 мин. Смесь перемешивали в течение 30 мин и медленно добавляли раствор азида натрия (4,36 г, 67,1 ммоль) в воде (16 мл). Смесь перемешивали в бане с ледяной водой и оставляли нагреваться до к.т. всю ночь. Смесь разбавляли водой (100 мл) и экстрагировали этилацетатом (2×200 мл). Объединенные органические слои промывали далее водой (150 мл), водным бикарбонатом натрия (насыщенный, 150 мл) и солевым раствором (100 мл), сушили (MgSO4), фильтровали. Растворитель выпаривали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-5% этилацетатом в гексане) с получением указанного соединения.
Стадия В: (1-(4-Хлор-2-йодфенил)-1H-1,2,3-триазол-4-ил)метанол: К смеси 1-азидо-4-хлор-2-йодбензола (5,0 г, 17,9 ммоль), пропаргилового спирта (1,003 г, 17,89 ммоль) в DMF (44,7 мл) добавляли медный купорос (1 М) (3,58 мл, 3,58 ммоль) и далее аскорбат натрия (1 М) (3,58 мл, 3,58 ммоль). Реакционную смесь перемешивали при 50°C всю ночь. Ее охлаждали до к.т., разбавляли водой (50 мл) и экстрагировали этилацетатом (2×100 мл). Объединенные органические слои промывали водой (2×50 мл) и водным бикарбонатом натрия (40 мл). Органический слой отделяли, сушили над MgSO4 и фильтровали. Фильтрат выпаривали при пониженном давлении. Остаток перетирали в порошок в 10% MeOH/DCM и выдерживали в течение 30 мин. Твердые вещества собирали фильтрацией и сушили на воздухе с получением указанного соединения. MS (ES+) m/z: 336 (М+Н).
Стадия С: 1-(4-Хлор-2-йодфенил)-1Н-1,2,3-триазол-4-карбальдегид: Перийодинан Десса-Мартина (6,92 г, 16,31 ммоль) добавляли к перемешиваемой смеси (1-(4-хлор-2-йодфенил)-1H-1,2,3-триазол-4-ил)метанола (4,56 г, 13,59 ммоль) в дихлорметане (68,0 мл) при к.т. Смесь перемешивали в течение 90 мин и гасили смесью водного тиосульфата натрия (насыщенный, 50 мл) и водного бикарбоната натрия (насыщенный, 50 мл). Смесь экстрагировали дихлорметаном (2×50 мл). Объединенные органические слои промывали солевым раствором (насыщенный, 100 мл), сушили (Na2SO4) и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-25% этилацетатом в гексане) с получением указанного соединения. MS (ES+) m/z: 333 (М+Н).
Стадия D: 1-(4-хлор-2-йодфенил)-4-(дифторметил)-1H-1,2,3-триазол: К раствору 1-(4-хлор-2-йодфенил)-1H-1,2,3-триазол-4-карбальдегида (1,5 г, 4,50 ммоль) в дихлорметане (30,0 мл) при 0°C добавляли диэтиламиносеры трифторид (2,90 г, 17,99 ммоль). Реакционную смесь оставляли нагреваться до к.т. и перемешивали в течение 2 ч. Смесь разбавляли дихлорметаном (30 мл) и гасили водным бикарбонатом натрия (насыщенный, 30 мл). Органический слой отделяли и промывали солевым раствором (30 мл), сушили над MgSO4, фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 1:3 этилацетатом/гексаном) с получением указанного соединения. MS (ES+) m/z: 356 (М+Н).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 45
1-(2-БРОМ-4-ХЛОРФЕНИЛ)-4-ХЛОР-1H-1,2,3-ТРИАЗОЛ
Стадия А: 1-(2-Бром-4-хлорфенил)-4-(трибутилстаннил)-1Н-1,2,3-триазол: К перемешиваемому раствору 1-азидо-2-бром-4-хлорбензола (660 мг, 2,84 ммоль) в толуоле (12 мл) добавляли трибутилстаннилацетилен (1073 мг, 3,41 ммоль) и смесь перемешивали при 110°C всю ночь. Реакционную смесь охлаждали до к.т. и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 5-20% этилацетатом в гексане) с получением указанного соединения.
Стадия В: 1-(2-Бром-4-хлорфенил)-4-хлор-1H-1,2,3-триазол: К перемешиваемому раствору 1-(2-бром-4-хлорфенил)-4-(трибутилстаннил)-1H-1,2,3-триазола (1,0 г, 1,826 ммоль) в ацетонитриле (15 мл) добавляли NCS (0,366 г, 2,74 ммоль). Реакционную смесь перемешивали при 60°C всю ночь. Ее охлаждали до к.т. и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 20% этилацетатом в гексане) с получением указанного соединения. MS (ES+) m/z: 292, 294 (М+Н).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 46
1-(2-БРОМ-4-ХЛОРФЕНИЛ)-4-(ТРИФТОРМЕТИЛ)-1H-1,2,3-ТРИАЗОЛ
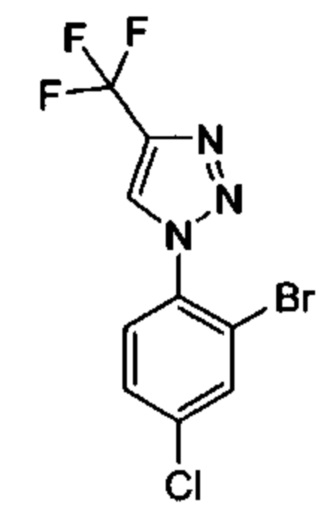
Избыточное количество 3,3,3-трифторпропина барботировали в перемешиваемую смесь 1-азидо-2-бром-4-хлорбензола (4,0 г, 17,21 ммоль) и оксида меди(I) (0,246 г, 1,721 ммоль) в ацетонитриле (43,0 мл) при к.т. в течение 30 мин. Смесь перемешивали всю ночь и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 20% этилацетатом в гексане) с получением указанного соединения. MS (ES+) m/z: 326, 328 (М+Н).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 47
1-(2-БРОМ-4-ХЛОРФЕНИЛ)-1H-ПИРАЗОЛ
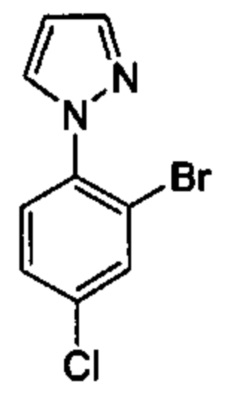
К перемешиваемой смеси 2-бром-4-хлор-1-фторбензола (6,00 г, 28,60 ммоль) в DMF (30 мл) добавляли 1H-пиразол (2,15 г, 31,50 ммоль) и Cs2CO3 (23,33 г, 71,60 ммоль) при к.т. Смесь перемешивали при 100°C в течение 15 ч. Ее охлаждали до к.т. и гасили водным хлоридом аммония (насыщенный, 30 мл). Смесь экстрагировали EtOAc (20 мл × 3). Объединенные органические фракции промывали солевым раствором (насыщенный, 20 мл), сушили над сульфатом натрия, фильтровали и растворитель выпаривали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром : EtOAc = 10:1) с получением указанного соединения. 1H ЯМР (400 МГц, CDCl3): δ 7.83 (d, J=2.5 Гц, 1Н), 7.77-7.65 (m, 2Н), 7.54-7.38 (m, 2Н), 6.55-6.45 (m, 1Н).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 48
1-(2-БРОМ-4-ХЛОРФЕНИЛ)-4-МЕТИЛ-1H-ПИРАЗОЛ
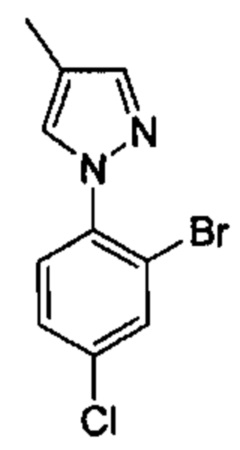
Указанное соединение получали из 4-метил-1H-пиразола способом, описанным в синтезе промежуточного соединения 47. 1H ЯМР (400 МГц, CDCl3): δ 7.69 (d, J=2.2 Гц, 1H), 7.59 (d, J=0.8 Гц, 1Н), 7.55 (s, 1Н), 7.46-7.41 (m, 1Н), 7.40-7.35 (m, 1Н), 2.17 (s, 3H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 49
1-(2-БРОМ-4-ХЛОРФЕНИЛ)-4-(ТРИФТОРМЕТИЛ)-1H-ПИРАЗОЛ
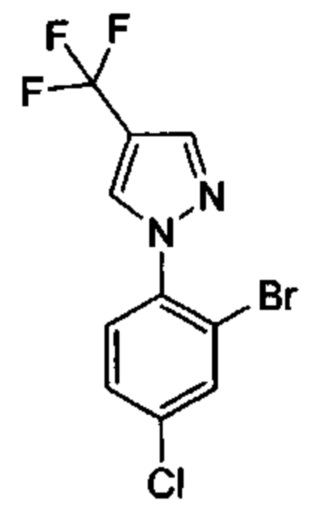
Указанное соединение получали из 4-(трифторметил)-1H-пиразола способом, описанным в синтезе промежуточного соединения 47. MS (ES+) m/z: 325, 327 (М+Н).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 50
1-(4-ХЛОР-3-ФТОР-2-ЙОДФЕНИЛ)-4-(ТРИФТОРМЕТИЛ)-1H-ПИРАЗОЛ
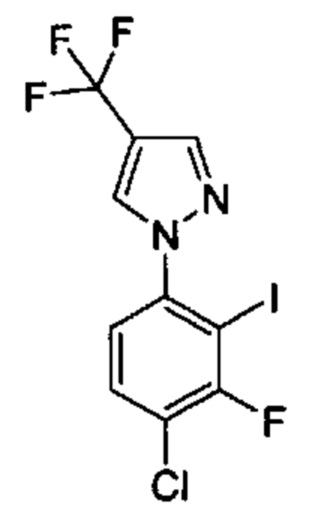
Указанное соединение получали из 4-(трифторметил)-1H-пиразола и 1-хлор-2,4-дифтор-3-йодбензола способом, описанным в синтезе промежуточного соединения 47. MS (ESI) m/z 391.1 (М+Н); 1H ЯМР (400 МГц, CDCl3): δ 7.96-8.03 (m, 1Н), 7.82 (s, 1H), 7.45-7.57 (m, 1H), 7.14-7.26 (m, 1H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 51
1-(2-БРОМ-4-ХЛОРФЕНИЛ)-1H-ПИРАЗОЛ-4-КАРБОНИТРИЛ
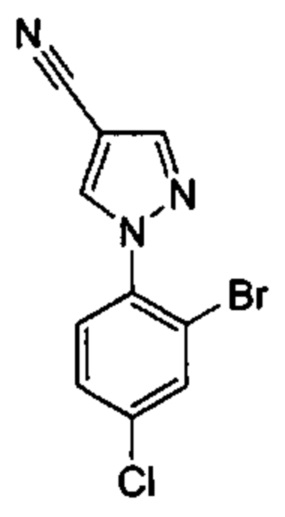
Указанное соединение получали из 1H-пиразол-4-карбонитрила способом, описанным в синтезе промежуточного соединения 47. MS (ES+) m/z: 282, 284 (М+Н).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 52
5-ХЛОР-2-(1-(ДИФТОРМЕТИЛ)-1H-ПИРАЗОЛ-4-ИЛ)ФЕНИЛА ТРИФТОРМЕТАНСУЛЬФОНАТ
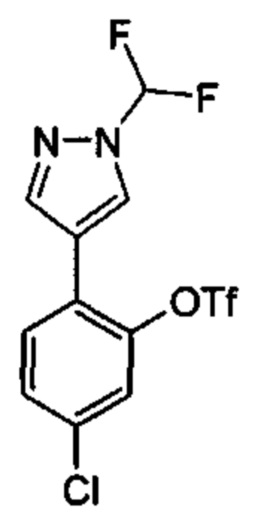
Стадия А: 5-Хлор-2-(1-(дифторметил)-1H-пиразол-4-ил)фенол: 1-(дифторметил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол (282 мг, 1,157 ммоль), 2-бром-5-хлорфенол (200 мг, 0,964 ммоль) и дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцен-палладий(II)дихлорида (79 мг, 0,096 ммоль) смешивали в сосуде с пониженным давлением, дегазировали и повторно заполняли азотом (3×). Далее добавляли диоксан (6 мл) и трехосновный фосфат калия (3М водный) (1 мл). Реакционную смесь перемешивали при 80°C в течение 3 ч. Ее охлаждали до к.т. и очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 50% этилацетатом в гексане) с получением указанного соединения. MS (ES+) m/z: 245 (М+Н).
Стадия В: 5-Хлор-2-(1-(дифторметил)-1H-пиразол-4-ил)фенила трифторметансульфонат: К смеси 5-хлор-2-(1-(дифторметил)-1H-пиразол-4-ил)фенола (90 мг, 0,368 ммоль) и основания Хунига (0,193 мл, 1,104 ммоль) в дихлорметане (2 мл) при 0°C по каплям добавляли трифторметансульфоновый ангидрид (1М в DCM) (0,552 мл, 0,552 ммоль). Смесь перемешивали при к.т. в течение 3 ч. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 50% этилацетатом в гексане) с получением указанного соединения. MS (ES+) m/z: 377 (М+Н).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 53
МЕТИЛ(3-АМИНО-4-БРОМ-5-ФТОРФЕНИЛ)КАРБАМАТ
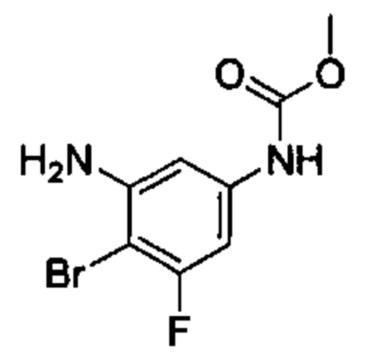
Стадия А: 2-Фтор-4,6-динитрофенол: К перемешиваемому раствору 2-фторфенола (10 г, 89 ммоль) в DCM (100 мл) при 0°C по каплям добавляли азотную кислоту (10,48 мл, 223 ммоль). Полученную смесь перемешивали при 25°C в течение 2 ч. Ее гасили водным NaOH (2 М) для доведения значения pH до 5. Ее разбавляли водой (100 мл), экстрагировали EtOAc (100 мл × 5). Объединенные органические слои промывали солевым раствором (200 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением указанного соединения.
Стадия В: 2-Бром-1-фтор-3,5-динитробензол: К перемешиваемому раствору 2-фтор-4,6-динитрофенола (16 г, 79 ммоль) в DMF (40 мл) и толуоле (300 мл) по каплям добавляли PBr3 (11,20 мл, 119 ммоль). Полученную смесь перемешивали при 110°C в течение 1 ч. Ее охлаждали до к.т., разбавляли EtOAc (200 мл) и переносили в делительную воронку. Ее промывали водой (100 мл × 3), сушили над сульфатом натрия, фильтровали и концентрировали с получением указанного соединения.
Стадия С: 4-Бром-3-фтор-5-нитроанилин: В перемешиваемый раствор 2-бром-1-фтор-3,5-динитробензола (18 г, 67,90 ммоль) в АсОН (400 мл, 6987 ммоль) небольшими частями добавляли железо (11,38 г, 204 ммоль). Смесь перемешивали в течение 1 ч при к.т. и ее гасили водным NaOH (2 М) для доведения pH до 8. Ее разбавляли водой (200 мл), экстрагировали EtOAc (500 мл × 3), промывали солевым раствором (500 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (петролейный эфир : EtOAc = 100:1-3:1) с получением указанного соединения. 1H ЯМР (400 МГц, CDCl3): δ 6.93 (s, 1Н), 6.62 (dd, J=2.7, 9.8 Гц, 1H), 4.15 (brs, 2H).
Стадия D: Метил(4-бром-3-фтор-5-нитрофенил)карбамат: К перемешиваемому раствору 4-бром-3-фтор-5-нитроанилина (12 г, 35,70 ммоль) и DIEA (18,73 мл, 107 ммоль) в DCM (250 мл) при 0°C по каплям добавляли метилхлорформиат (8,44 мл, 107 ммоль). Смесь перемешивали при 25°C в течение 15 ч. Добавляли воду (30 мл) и смесь экстрагировали EtOAc (200 мл × 3). Объединенные органические фракции промывали солевым раствором (200 мл), сушили над сульфатом натрия, фильтровали и растворитель выпаривали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (петролейный эфир : EtOAc = 100:1-3:1) с получением указанного соединения. 1H ЯМР (400 МГц, CDCl3): δ 7.69 (s, 1Н), 7.64 (d, J=9.7 Гц, 1H), 7.02 (brs, 1H), 3.82 (s, 3H).
Стадия E: Метил(3-амино-4-бром-5-фторфенил)карбамат: К раствору метил(4-бром-3-фтор-5-нитрофенил)карбамата (9,50 г, 29,20 ммоль) в EtOH (90 мл) и воде (30 мл) добавляли железо (3,26 г, 58,4 ммоль), хлорид аммония (6,24 г, 117 ммоль) и смесь перемешивали при 90°C в течение 2 ч. Ее охлаждали до к.т. и фильтровали через слой целита. Твердые вещества промывали EtOH (20 мл × 2) и водой (50 мл). Фильтрат экстрагировали EtOAc (100 мл × 3). Объединенные органические фракции промывали солевым раствором (100 мл), сушили над сульфатом натрия, фильтровали и растворитель выпаривали при пониженном давлении с получением указанного соединения. 1H ЯМР (400 МГц, CDCl3): δ 6.74 (brs, 1Н), 6.57 (d, J=11.7 Гц, 2H), 4.25 (brs, 2Н), 3.76 (s, 3H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 54
1-(4-ХЛОР-3-ФТОР-2-ЙОДФЕНИЛ)-4-(ТРИФТОРМЕТИЛ)-1H-1,2,3-ТРИАЗОЛ
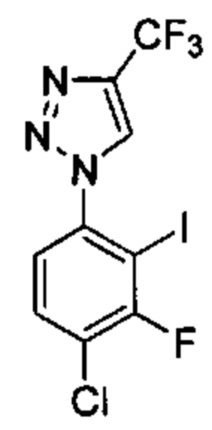
Стадия А: 1-Азидо-4-хлор-3-фтор-2-йодбензол: К раствору 4-хлор-3-фтор-2-йоданилина (300 мг, 1,105 ммоль) в EtOAc (6,9 мл) добавляли раствор HCl (37% масс., 2,3 мл, 27,8 ммоль) в воде (6,9 мл) при 0°C. Полученную смесь перемешивали в течение 10 минут. К этой суспензии добавляли раствор нитрит натрия (84 мг, 1,216 ммоль) в воде (0,5 мл) в течение трех минут. Реакционную смесь перемешивали в течение 30 мин. К вышеуказанной реакционной смеси медленно добавляли раствор азида натрия (79 мг, 1,216 ммоль) в 0,5 мл воды. Смесь затем перемешивали в бане с ледяной водой в атмосфере азота и оставляли нагреваться до к.т. всю ночь. Смесь экстрагировали этилацетатом (2×30 мл). Объединенные органические фракции промывали солевым раствором (30 мл) и сушили над сульфатом натрия, фильтровали и растворитель выпаривали при пониженном давлении с получением указанного соединения.
Стадия В: 1-(4-Хлор-3-фтор-2-йодфенил)-4-(трифторметил)-1H-1,2,3-триазол: 3,3,3-Трифторпропин (104 мг, 1,105 ммоль) барботировали в перемешиваемую смесь 1-азидо-4-хлор-3-фтор-2-йодбензола (329 мг, 1,105 ммоль) и оксида меди(I) (15,81 мг, 0,111 ммоль) в ацетонитриле (2,8 мл) в течение 10 мин. Затем реакционный сосуд закрывали пробкой и запечатывали. Реакционную смесь перемешивали при к.т. всю ночь. Смесь концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-10% этилацетатом в гексане) с получением указанного соединения. MS (ES+) m/z: 391.6 (М+Н).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 55
МЕТИЛ(3-АМИНО-4-(2-(5-БРОМПИКОЛИНОИЛ)1-((2-(ТРИМЕТИЛСИЛИЛ)ЭТОКСИ)МЕТИЛ)-1H-ИМИДАЗОЛ-4-ИЛ)ФЕНИЛ)КАРБАМАТ
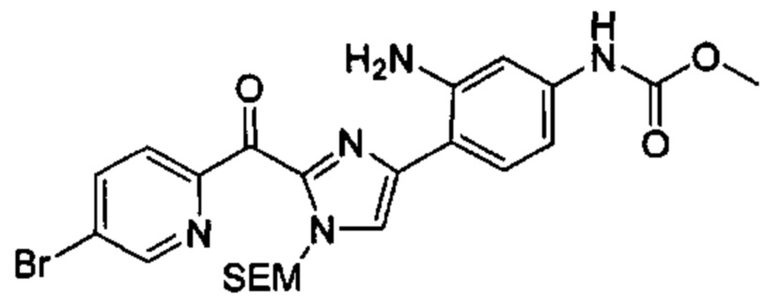
К смеси промежуточного соединения 15 (40 г, 79 ммоль), промежуточного соединения 19 (25,3 г, 87 ммоль), тетракис(трифенилфосфин)палладия(0) (4,55 г, 3,94 ммоль), DMF (354 мл) и трехосновного фосфата калия (50,1 г, 236 ммоль) перемешивали при 60°C в течение 3,5 ч. Ее охлаждали до к.т. и большую часть растворителя удаляли при пониженном давлении. Остаток разбавляли этилацетатом (200 мл) и промывали водой (3×100 мл), затем солевым раствором (100 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-60% EtOAc/гексан, градиент) с получением указанного соединения. MS (ES+) m/z: 546, 548 [М+Н].
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 56
МЕТИЛ(3-АМИНО-4-(2-(5-БРОМПИКОЛИНОИЛ)-1-((2-(ТРИМЕТИЛСИЛИЛ)-ЭТОКСИ)МЕТИЛ)-1H-ИМИДАЗОЛ-4-ИЛ)ФЕНИЛ)КАРБАМАТ
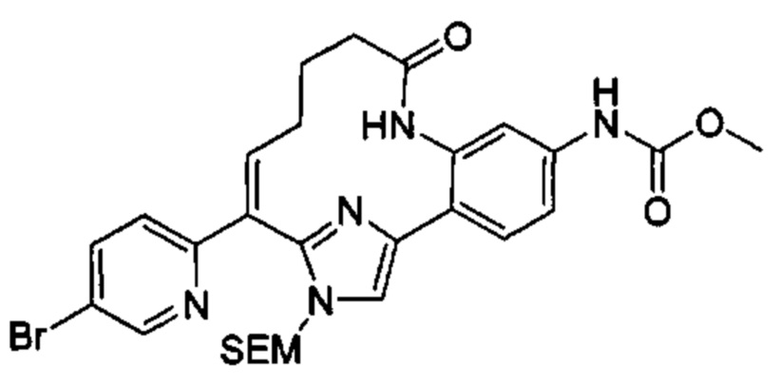
Стадия А: (5-((2-(2-(5-Бромпиколиноил)-1-((2-(триметилсилил)этокси)метил)-1H-имидазол-4-ил)-5-((метоксикарбонил)амино)фенил)амино)-5-оксопентил)трифенилфосфония бромид: К смеси промежуточного соединения 55 (36,5 г, 66,8 ммоль), (4-карбоксибутил)трифенилфосфония бромида (32,6 г, 73,5 ммоль) в DCM (230 мл) добавляли DIEA (35,0 мл, 200 ммоль) и HATU (30,5 г, 80 ммоль). Смесь перемешивали при к.т. в течение 2 ч и реакционную смесь очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-6% MeOH/DCM, градиент) с получением указанного соединения. MS (ES+) m/z: 890, 892 [М+Н].
Стадия В: Метил((12Z,8Z)-9-(5-бромпиридин-2-ил)-4-оксо-11-((2-(триметилсилил)этокси)метил)-11Н-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-8-ен-24-ил)карбамат: К раствору (5-((2-(2-(5-бромпиколиноил)-1-((2-(триметилсилил)этокси)метил)-1H-имидазол-4-ил)-5-((метоксикарбонил)амино)фенил)амино)-5-оксопентил)трифенилфосфония бромида в дегазированном THF (3000 мл) при 0°C добавляли карбонат цезия (50 г, 153 ммоль). Смесь перемешивали при к.т. в течение 16 ч. Твердые вещества удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. К остатку взвеси добавляли 100 мл диэтилового эфира и 20 мл воды и выдерживали в течение 1 ч. Твердые вещества собирали фильтрацией и промывали диэтиловым эфиром (2×50 мл) с получением указанного соединения. MS (ES+) m/z: 612, 614 [М+Н]; 1H ЯМР (CDCl3, 400 МГц): δ 8.65 (d, J=2.0 Гц, 1Н), 7.92 (s, 1Н), 7.84 (dd, J=2.1, 8.2 Гц, 1Н), 7.49 (d, J=8.3 Гц, 1Н), 7.33 (d, J=8.5 Гц, 1H), 7.24 (s, 1H), 6.93 6.84 (m, 1Н), 5.31 (s, 1Н), 3.82-3.71 (m, 5Н), 3.32-3.23 (m, 2Н), 1.62 (m, 4Н), 0.95-0.83 (m, 2Н), 0.77 (t, J=8.2 Гц, 2Н), -0.06 (s, 9Н).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 57
МЕТИЛ((12Z,8Z)-9-(5-БРОМПИРИДИН-2-ИЛ)-5-МЕТИЛ-4-ОКСО-11-((2-(ТРИМЕТИЛСИЛИЛ)ЭТОКСИ)МЕТИЛ)-11H-3-АЗА-1(4,2)-ИМИДАЗОЛ-2(1,2)-БЕНЗОЛАЦИКЛОНОНАФАН-8-ЕН-24-ИЛ)КАРБАМАТ
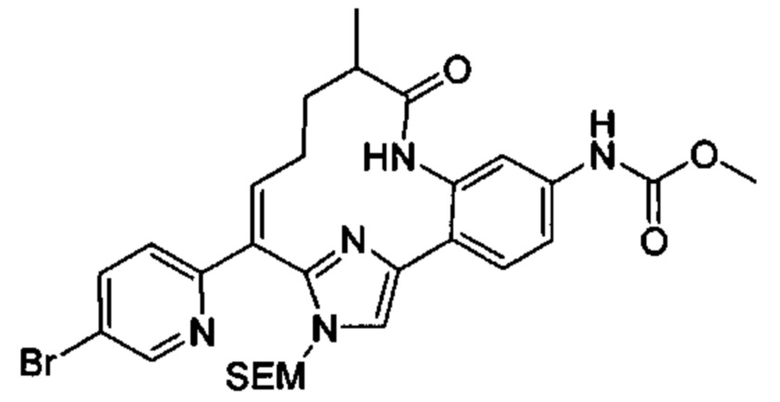
Стадия А: (5-((2-(2-(5-Бромпиколиноил)-1-((2-(триметилсилил)этокси)метил)-1H-имидазол-4-ил)-5-((метоксикарбонил)амино)фенил)амино)-4-метил-5-оксопентил)трифенилфосфония бромид: К перемешиваемой смеси промежуточного соединения 55 (5,00 г, 9,15 ммоль), (4-карбоксипентил)трифенилфосфония бромида (4,18 г, 9,15 ммоль) и DIEA (15,98 мл, 91 ммоль) в DCM (50 мл) добавляли 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфината 2,4,6-триоксид (11,64 г, 18,30 ммоль) при 30°C. Смесь перемешивали при 30°C в течение 16 ч. Ее разбавляли водой и экстрагировали EtOAc (100 мл × 2). Объединенные органические слои промывали солевым раствором (50 мл × 2), сушили над сульфатом натрия, фильтровали и растворитель выпаривали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (DCM : СН3ОН = 50:1) с получением указанного соединения. MS (ESI) m/z 904.2, 906.2 (М-Br).
Стадия B: Метил((12z,8z)-9-(5-бромпиридин-2-ил)-5-метил-4-оксо-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-8-ен-24-ил)карбамат: Указанное соединение получали способом, описанным в синтезе промежуточного соединения 56, стадия В. Продукт очищали методом колоночной флеш-хроматографии на силикагеле (петролейный эфир : EtOAc = 10:1-2:1, градиент) с получением указанного соединения. MS (ESI) m/z 626.2, 628.2 (М+Н).
ПРИМЕРЫ
ПРИМЕР 1 (рацемат), 1-а и 1-b
9-(5-(5-ХЛОР-2-(1H-ТЕТРАЗОЛ-1-ИЛ)ФЕНИЛ)ПИРИДИН-2-ИЛ)-25-ФТОР-3-АЗА-1(1,3),2(1,2)-ДИБЕНЗОЛАЦИКЛОНОНАФАН-4-ОН
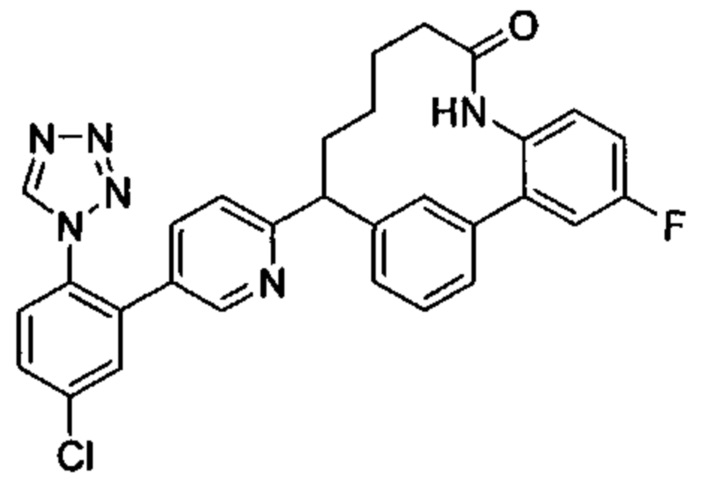
1А: 6-(1-(2'-Амино-5'-фтор-[1,1'-бифенил]-3-ил)бут-3-ен-1-ил)пиридин-3-ол: 6-(3-Хлорбензил)пиридин-3-ол (5,00 г, 19,25 ммоль), метил(3-амино-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)карбамат (5,48 г, 23,10 ммоль), предварительный каталитический нейтрализатор Pd-Xphos (0,796 г, 0,963 ммоль) и дегазированный диоксан (77 мл) добавляли в круглодонную колбу объемом 500 мл с магнитной мешалкой. К смеси добавляли дегазированный водный раствор фосфата калия (3 М, 19,3 мл, 57,8 ммоль). Смесь перемешивали на масляной бане при 80°C в течение 4 ч и охлаждали до к.т. Смесь экстрагировали этилацетатом (2×100 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-100% этилацетатом в гексане) с получением указанного соединения. MS (ES+) m/z: 335 (М+Н).
1В: 3'-(1-(5-((трет-Бутилдифенилсилил)окси)пиридин-2-ил)бут-3-ен-1-ил)-5-фтор-[1,1'-бифенил]-2-амин: К раствору соединения примера 1А (550 мг, 1,645 ммоль) и TEA (0,48 мл, 3,44 ммоль) в DCM (5 мл) при к.т. добавляли TBDPS-C1 (0,5 мл, 1,946 ммоль). Реакционную смесь перемешивали в течение 1 ч и ее сразу очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-50% этилацетатом в гексане) с получением указанного соединения. MS (ES+) m/z: 573 (М+Н).
1С: N-(3'-(1-(5-((трет-Бутилдифенилсилил)окси)пиридин-2-ил)бут-3-ен-1-ил)-5-фтор-[1,1'-бифенил]-2-ил)бут-3-енамид: К перемешиваемой смеси бут-3-еновой кислоты (0,588 мл, 6,91 ммоль), соединения примера 1В (3,3 г, 5,76 ммоль) и основания Хунига (10,06 мл, 57,6 ммоль) в дихлорметане (28,8 мл) добавляли 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфоринан-2,4,6-триоксид (50% в этилацетате, 7,33 г, 11,52 ммоль). Смесь перемешивали при к.т. в течение 1 ч. Ее разбавляли DCM (20 мл), промывали водным бикарбонатом натрия (насыщенный, 15 мл), сушили над MgSO4, фильтровали и растворитель выпаривали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали EtOAc/гексаном = 30% объем/объем) с получением указанного соединения. MS (ES+) m/z: 641 (М+Н).
1D: (E)-9-(5-((трет-Бутилдифенилсилил)окси)пиридин-2-ил)-25-фтор-3-аза-1(1,3),2(1,2)-дибензолациклононафан-6-ен-4-он:
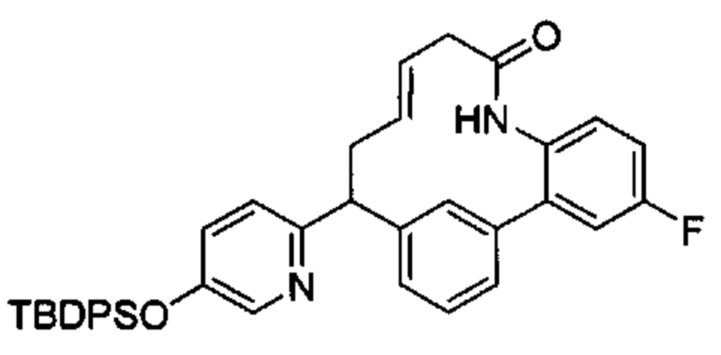
Дегазированный толуол (281 мл) добавляли к смеси соединения примера 1С (3,6 г, 5,62 ммоль), катализатора Чжаня 1В (0,824 г, 1,123 ммоль) и моногидрата пара-толуолсульфоновой кислоты (0,855 г, 4,49 ммоль) в круглодонной колбе объемом 500 мл в атмосфере азота. Смесь перемешивали при 50°C всю ночь. Смесь охлаждали до к.т. и разбавляли дихлорметаном (400 мл). Раствор промывали водным бикарбонатом натрия (насыщенный, 100 мл), сушили (MgSO4), фильтровали и растворитель выпаривали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии (элюировали 0-40% этилацетатом в гексане) с получением указанного соединения. MS (ES+) m/z: 613 (М+Н).
1E: 25-Фтор-9-(5-гидроксипиридин-2-ил)-3-аза-1(1,3),2(1,2)-дибензолациклононафан-4-он:
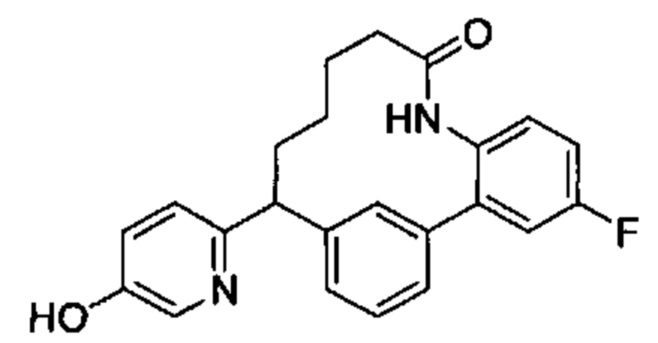
Смесь соединения примера 1D (600 мг, 0,979 ммоль), Pd(OH)2 (20 масс. % на углероде, 68,7 мг, 0,098 ммоль) и метанола (9,8 мл) перемешивали при к.т. в атмосфере водорода в течение 16 ч. Смесь фильтровали через слой целита и фильтрат концентрировали при пониженном давлении. Неочищенный продукт, растворенный в THF (9,4 мл), обрабатывали TBAF (1 M в THF, 1,9 мл, 1,9 ммоль). Смесь перемешивали при к.т. в течение 1 ч, затем ее гасили хлоридом аммония (10 мл) и экстрагировали этилацетатом. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали EtOH-EtOH (3:1) / гексан) с получением указанного соединения. MS (ES+) m/z: 377 (М+Н).
1F: 6-(25-Фтор-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-9-ил)пиридин-3-ила трифторметансульфонат:
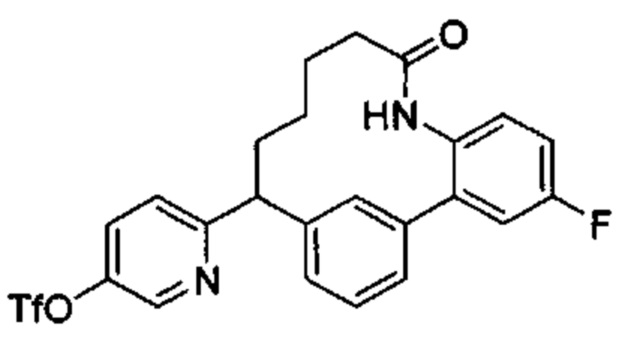
К перемешиваемой смеси соединения примера 1Е (350 мг, 0,930 ммоль) и основания Хунига (487 мкл, 2,79 ммоль) в DCM (9,3 мл) добавляли Tf2O (1 M в DCM, 1,4 мл, 1,4 ммоль). Смесь перемешивали при к.т. в течение 2 ч. Смесь разбавляли дихлорметаном (10 мл), промывали водным бикарбонатом натрия (насыщенный, 10 мл), сушили (MgSO4), фильтровали и растворитель выпаривали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали EtOAc/гексаном) с получением указанного соединения. MS (ES+) m/z: 509 (М+Н).
1G: 9-(5-(2-Амино-5-хлорфенил)пиридин-2-ил)-25-фтор-3-аза-1(1,3),2(1,2)-дибензолациклононафан-4-он:
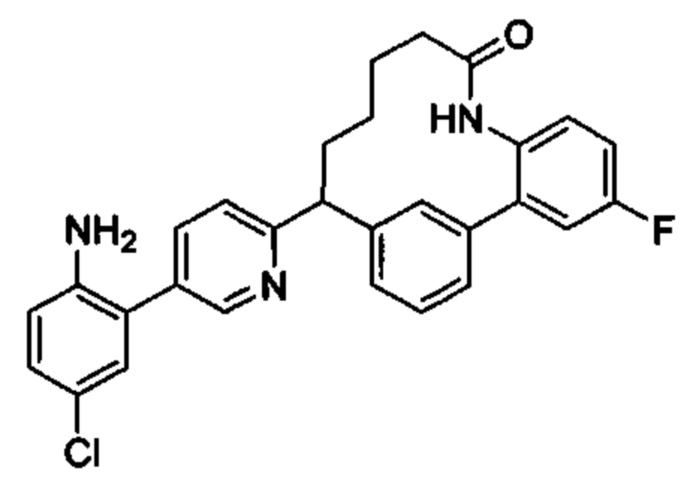
В сосуд для микроволнового облучения с магнитной мешалкой добавляли соединение примера 1F (100 мг, 0,197 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (32,1 мг, 0,039 ммоль), 4-хлор-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин (59,8 мг, 0,236 ммоль), CsF (90,0 мг, 0,590 ммоль) и дегазированный диоксан (2,4 мл). Сосуд закупоривали и смесь перемешивали на масляной бане при 100°C в течение 2 ч. Смесь охлаждали до к.т. и очищали методом колоночной флеш-хроматографии на силикагеле (элюировали EtOAc-EtOH (3:1)/гексан) с получением указанного соединения. MS (ES+) m/z: 486 (М+Н).
Пример 1: 9-(5-(5-Хлор-2-(1H-тетразол-1-ил)фенил)пиридин-2-ил)-25-фтор-3-аза-1(1,3),2(1,2)-дибензолациклононафан-4-он
Пример 1-а: (S)-9-(5-(5-Хлор-2-(1H-тетразол-1-ил)фенил)пиридин-2-ил)-25-фтор-3-аза-1(1,3),2(1,2)-дибензолациклононафан-4-он
Пример 1-b: (R)-9-(5-(5-Хлор-2-(1H-тетразол-1-ил)фенил)пиридин-2-ил)-25-фтор-3-аза-1(1,3),2(1,2)-дибензолациклононафан-4-он
Раствор соединения примера 1G (95 мг, 0,195 ммоль), азида натрия (38,1 мг, 0,586 ммоль) и триметилортоформиата (65 мкл, 0,586 ммоль) в уксусной кислоте (1 мл) перемешивали при 90°C в течение 2 ч и охлаждали до к.т. Его разбавляли этилацетатом (20 мл) и промывали бикарбонатом натрия, а затем солевым раствором. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали EtOAc-EtOH (3:1) в гексане) с получением указанного соединения. MS (ES+) m/z: 539 (М+Н).
Рацемат отделяли методом хиральной SFC с обращенной фазой (OJ, 21×250 мм, 40% MeOH-MeCN (2:1)/СО2, 60 мл/мин, 100 бар, 35°C) с получением соединения примера 1-а (более быстрое элюирование) и соединения примера 1-b (более медленное элюирование). Абсолютную конфигурацию устанавливали на основе биологических данных и рентгеновских структур близких аналогов. MS (ES+) m/z: 539 (М+Н); 1H ЯМР (500 МГц, CDCl3): δ 8.40 (s, 1Н); 8.32 (s, 1Н); 7.55-7.63 (m, 3Н); 7.46-7.50 (m, 2Н); 7.42 (t, J=7.7 Гц, 1Н); 7.17-7.23 (m, 3Н); 7.07-7.12 (m, 3Н); 6.71 (s, 1Н); 4.15 (t, J=7.7 Гц, 1Н); 2.25-2.29 (m, 2H); 2.14 (t, J=6.9 Гц, 2H); 1.72-1.78 (m, 1H); 1.44-1.47 (m, 2H); 0.99 (d, J=6.4 Гц, 1H).
ПРИМЕР 2-a
(S)-5-(5-ХЛОР-2-(1H-ТЕТРАЗОЛ-1-ИЛ)ФЕНИЛ)-2-(25-ФТОР-4-ОКСО-3-АЗА-1(1,3),2(1,2)-ДИБЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
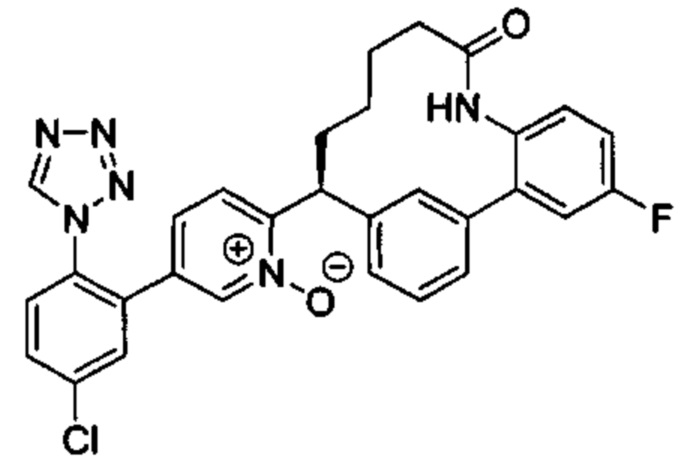
К раствору соединения примера 1-а (35 мг, 0,065 ммоль) в DCM (1 мл) добавляли м-СРВА (22,4 мг, 0,13 ммоль). Реакционную смесь перемешивали при к.т. в течение 3 ч. Ее сразу очищали методом колоночной флеш-хроматографии на силикагеле (элюировали EtOAc-EtOH (3:1) / гексаном) с получением требуемого продукта. MS (ES+) m/z: 555 (М+Н); 1H ЯМР (500 МГц, DMSO-d6): δ 9.68 (s, 1H); 9.53 (s, 1H); 8.20 (s, 1H); 7.89 (s, 1H); 7.83 (s, 2H); 7.60 (s, 1H); 7.50 (d, J=8.3 Гц, 1H); 7.32-7.37 (m, 2H); 7.21-7.28 (m, 3H); 6.95 (d, J=8.3 Гц, 1H); 6.88 (d, J=7.6 Гц, 1H); 4.56 (d, J=12.8 Гц, 1H); 2.27 (t, J=9.6 Гц, 1H); 1.82-2.04 (m, 4H); 1.47 (m, 1H); 1.20 (m, 1H); 1.05 (t, J=7.2 Гц, 1Н).
ПРИМЕР 2-b
(R)-5-(5-ХЛОР-2-(1H-ТЕТРАЗОЛ-1-ИЛ)ФЕНИЛ)-2-(25-ФТОР-4-ОКСО-3-АЗА-1(1,3),2(1,2)-ДИБЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
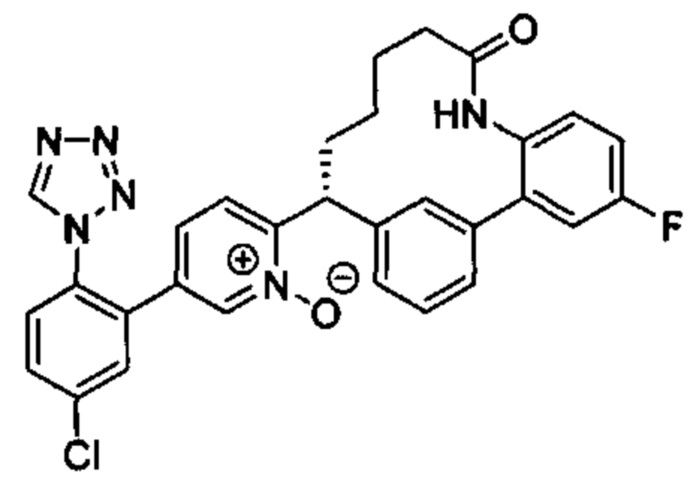
Соединение примера 2-b получали из соединения примера 1-b способом, описанным в примере 2-a. MS (ES+) m/z: 555 (М+Н); 1H ЯМР (500 МГц, DMSO-d6): δ 9.68 (s, 1H); 9.53 (s, 1H); 8.20 (s, 1H); 7.89 (s, 1H); 7.83 (s, 2H); 7.60 (s, 1H); 7.50 (d, J=8.3 Гц, 1H); 7.32-7.37 (m, 2H); 7.21-7.28 (m, 3H); 6.95 (d, J=8.3 Гц, 1H); 6.88 (d, J=7.6 Гц, 1H); 4.56 (d, J=12.8 Гц, 1H); 2.27 (t, J=9.6 Гц, 1H); 1.82-2.04 (m, 4H); 1.47 (m, 1H); 1.20 (m, 1H); 1.05 (t, J=7.2 Гц, 1Н).
ПРИМЕР 3 (рацемат), 3-а, 3-b, 3-е и 3-d
5-(5-ХЛОР-2-(1H-ТЕТРАЗОЛ-1-ИЛ)ФЕНИЛ)-2(25-ФТОР-5-МЕТИЛ-4-ОКСО-3-АЗА-1(1,3),2(1,2)-ДИБЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
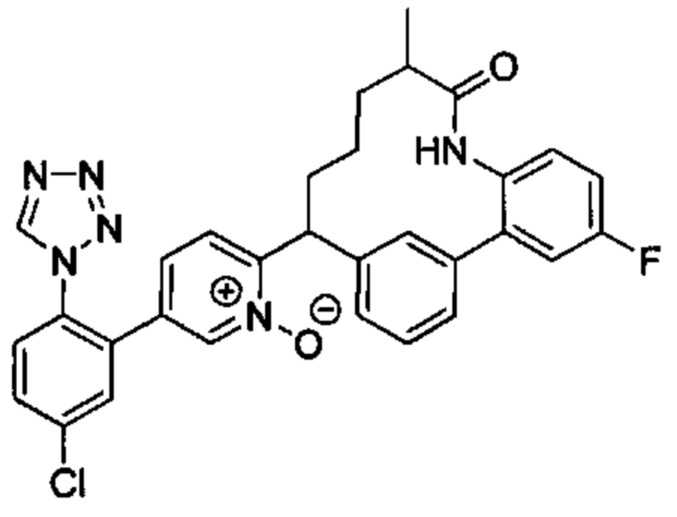
Соединение примера 3 получали способами, описанными в примере 1 и 2, с заменой бут-3-еновой кислоты 2-метилбут-3-еновой кислотой в синтезе соединения 1С.
Четыре диастереомера разделяли методом хиральной HPLC с обращенной фазой (IC-H, 4,6×250 мм, 50% MeOH/СО2, 2,4 мл/мин, 100 бар, 40°C). Абсолютную конфигурацию устанавливали на основе рентгеноспектральных и 1H ЯМР данных.
Пример 3-а: 5-(5-Хлор-2-(1H-тетразол-1-ил)фенил)-2-((5R,9R)-25-фтор-5-метил-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-9-ил)пиридина 1-оксид:
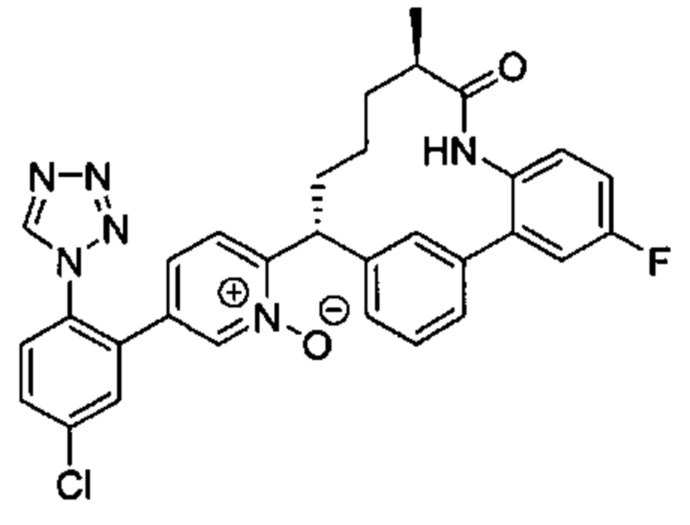
Соединение примера 3-а выделяли в виде пика 1. MS (ES+) m/z: 569 (М+Н); 1H ЯМР (500 МГц, DMSO): δ 9.67 (s, 1Н), 9.40 (s, 1H), 8.20 (s, 1Н), 7.89 (s, 1Н), 7.82-7.84 (m, 2Н), 7.63 (d, J=8.3 Гц, 1Н), 7.56 (s, 1Н), 7.31-7.39 (m, 2Н), 7.19-7.26 (m, 3H), 6.97 (d, J=8.3 Гц, 1Н), 6.78 (d, J=7.6 Гц, 1Н), 4.49 (dd, J=13.0, 3.6 Гц, 1Н), 2.29 (d, J=9.3 Гц, 1Н), 1.94 (m, 1Н); 1.70 (t, J=11.7 Гц, 2Н), 1.25 (m, 3H); 1.09 (d, J=6.8 Гц, 3H).
Пример 3-b: 5-(5-Хлор-2-(1H-тетразол-1-ил)фенил)-2-((5S,9S)-25-фтор-5-метил-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-9-ил)пиридина 1-оксид:
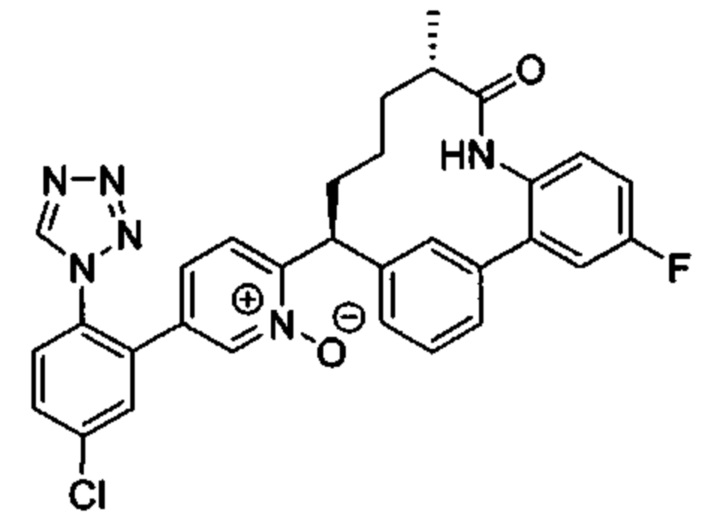
Соединение примера 3-b выделяли в виде пика 2. MS (ES+) m/z: 569 (М+Н); 1H ЯМР (500 МГц, DMSO): δ 9.67 (s, 1Н), 9.40 (s, 1H), 8.20 (s, 1H), 7.89 (s, 1H), 7.82-7.84 (m, 2H), 7.63 (d, J=8.3 Гц, 1H), 7.56 (s, 1H), 7.31-7.39 (m, 2H), 7.19-7.26 (m, 3H), 6.97 (d, J=8.3 Гц, 1H), 6.78 (d, J=7.6 Гц, 1H), 4.49 (dd, J=13.0, 3.6 Гц, 1H), 2.29 (d, J=9.3 Гц, 1H), 1.95 (m, 1H); 1.70 (t, J=11.7 Гц, 2H), 1.25 (m, 3H); 1.09 (d, J=6.8 Гц, 3H).
Пример 3-с: 5-(5-Хлор-2-(1H-тетразол-1-ил)Фенил)-2-((5S,9R)-25-фтор-5-метил-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-9-ил)пиридина 1-оксид:
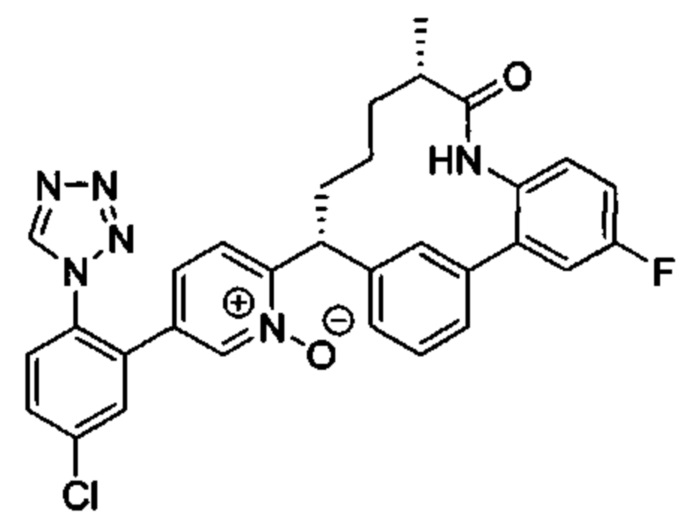
Соединение примера 3-c выделяли в виде пика 3. MS (ES+) m/z: 569 (М+Н); 1H ЯМР (500 МГц, DMSO): δ 9.65 (s, 1Н), 9.51 (s, 1Н), 8.19 (s, 1H), 7.87 (s, 1H), 7.79-7.83 (m, 2Н), 7.63 (s, 1Н), 7.29-7.37 (m, 3H), 7.21 (d, J=6.7 Гц, 2Н), 6.96 (d, J=7.6 Гц, 1H), 6.88 (d, J=8.3 Гц, 1H), 4.60 (m, 1H), 1.85 (br s, 4Н), 1.18-1.36 (m, 3H), 0.92 (d, J=6.8 Гц, 3H).
Пример 3-d: 5-(5-Хлор-2-(1H-тетразол-1-ил)фенил)-2-((5R,9S)-25-фтор-5-метил-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-9-ил)пиридина 1-оксид:
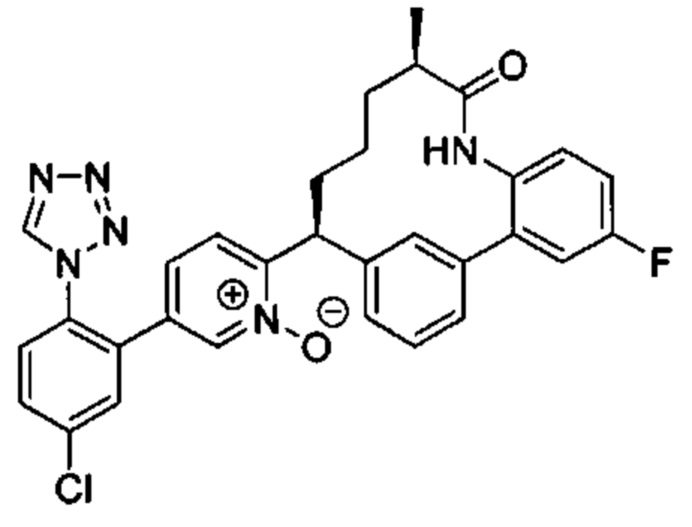
Соединение примера 3-d выделяли в виде пика 4. MS (ES+) m/z: 569 (М+Н); 1H ЯМР (500 МГц, DMSO): δ 9.65 (s, 1H), 9.51 (s, 1H), 8.19 (s, 1H), 7.87 (s, 1H), 7.79-7.83 (m, 2H), 7.63 (s, 1H), 7.29-7.37 (m, 3H), 7.21 (d, J=6.7 Гц, 2H), 6.96 (d, J=7.6 Гц, 1H), 6.88 (d, J=8.3 Гц, 1H), 4.60 (m, 1H), 1.85 (br s, 4H), 1.18-1.36 (m, 3H), 0.92 (d, J=6.8 Гц, 3H).
ПРИМЕР 4 (рацемат), 4-a и 4-b
2-(25-КАРБОКСИ-4-ОКСО-3-АЗА-1(1,3),2(1,2)-УДИБЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)-5-(5-ХЛОР-2-(1H-ТЕТРАЗОЛ-1-ИЛ)ФЕНИЛ)ПИРИДИНА 1-ОКСИД
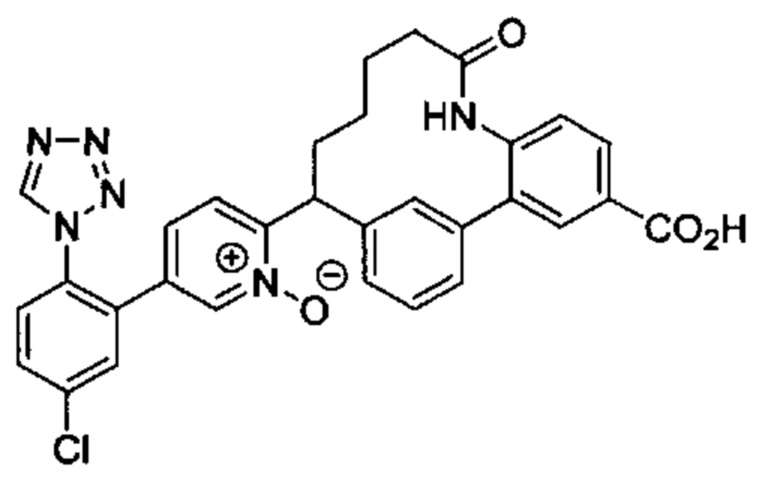
4А: Метил-4-оксо-9-(5-(((трифторметил)сульфонил)окси)пиридин-2-ил)-3-аза-1(1,3),2(1,2)-дибензолациклононафан-25-карбоксилат:
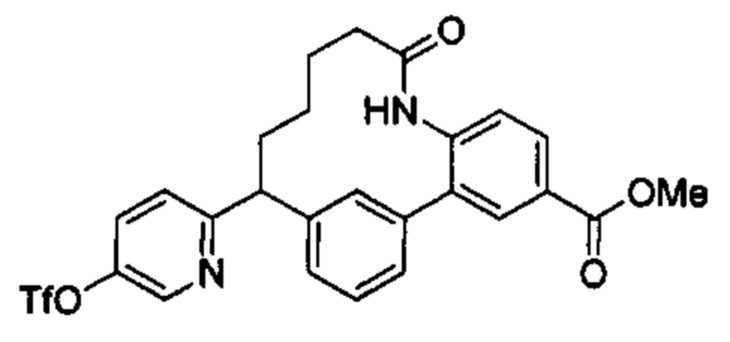
Соединение примера 4А получали таким же путем синтеза, который проводили при синтезе соединения примера 1F с использованием промежуточного соединения 1 и промежуточного соединения 20 в качестве исходных веществ. MS (ES+) m/z: 549 (М+Н).
4В: Метил-9-(5-(2-амино-5-хлорфенил)пиридин-2-ил)-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-25-карбоксилат:
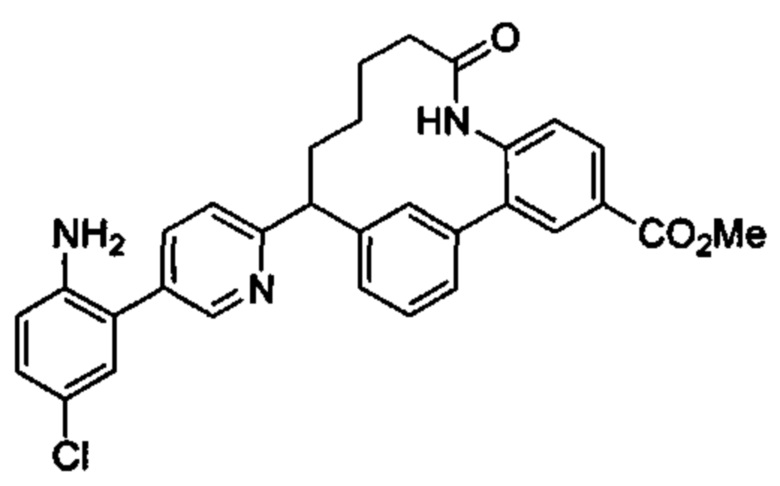
Соединение примера 4В получали из соединения примера 4А способом, описанным в синтезе соединения 1G. MS (ES+) m/z: 526 (М+Н).
4С: 9-(5-(2-Амино-5-хлорфенил)пиридин-2-ил)-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-25-карбоновой кислоты:

К раствору соединения примера 4В (80 мг, 0,152 ммоль) в MeOH (760 мкл) и THF (760 мкл) добавляли водный раствор гидроксида натрия (3 М, 152 мкл, 0,456 ммоль). Смесь перемешивали при к.т. всю ночь и нейтрализовали HCl (1 М) до значения pH 5. Смесь концентрировали при пониженном давлении и сушили под вакуумом. Остаток использовали без дополнительной очистки. MS (ES+) m/z: 512 (М+Н).
4D: 9-(5-(5-Хлор-2-(1H-тетразол-1-ил)фенил)пиридин-2-ил)-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-25-карбоновой кислоты:
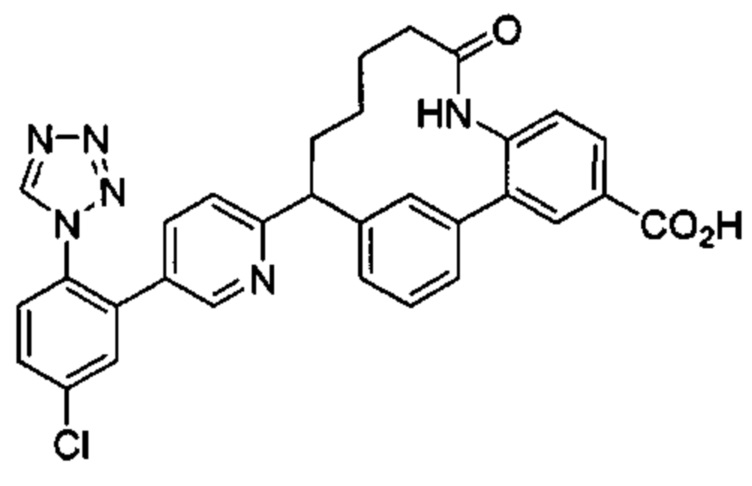
Соединение примера 4D получали из соединения примера 4С способом, описанным в синтезе соединения 1Н. MS (ES+) m/z: 565 (М+Н).
Пример 4:
К раствору соединения примера 4D (73 мг, 0,129 ммоль) в DCM (1 мл) добавляли м-СРВА (96 мг, 0,388 ммоль). Полученный раствор перемешивали при к.т. в течение 2 ч. Твердые вещества осаждали. Его фильтровали и твердые вещества промывали DCM (5 мл) с получением указанного соединения. MS (ES+) m/z: 565 (М+Н).
Рацемический образец соединения примера 4 подвергали хиральному разделению (Whelk 30×250 мм, 60% MeOH (0,2% NH4OH)/CO2, 70 мл/мин, 100 бар, 35°C). Фракции обоих энантиомеров собирали отдельно и концентрировали при пониженном давлении. Остаток более медленного элюента концентрировали и нейтрализовали одной каплей 5% раствора лимонной кислоты и двумя каплями KH2PO4 перед тем, как его очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-20% метанолом в DCM) с получением соединения примера 4-а. MS (ES+) m/z: 581 (М+Н); 1H ЯМР (500 МГц, DMSO-d6): 13.05 (br s, 1Н), 9.73-9.75 (m, 1H), 9.66 (s, 1Н), 8.18 (m, 1Н), 8.03 (s, 1Н), 7.88-7.93 (m, 2Н), 7.78-7.81 (m, 2Н), 7.58 (m, 1H), 7.52 (d, 1H), 7.28-7.39 (m, 3H), 6.94 (t, 1Н), 6.87 (d, 1H), 4.55 (d, 1H), 2.32 (t, 1H), 1.91-1.99 (m, 2H), 1.80 (br s, 1H), 1.47 (br s, 1H), 1.23 (br s, 1H), 1.02 (m, 1H).
Остаток более быстрого элюента растворяли в 2 мл 50% DCM/метанола и добавляли две капли 1 М KH2PO4 и одну каплю 5% раствора лимонной кислоты. Смесь очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-20% метанолом в DCM) с получением соединения примера 4-b. MS (ES+) m/z: 581 (М+Н).
ПРИМЕР 5 (рацемат), 5-а и 5-b
2-(25-КАРБОКСИ-4-ОКСО-3-АЗА-1(1,3),2(1,2)ДИБЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)-5-(3-ХЛОР-2-ФТОР-6-(1H-ТЕТРАЗОЛ-1-ИЛ)ФЕНИЛ)ПИРИДИНА 1-ОКСИД
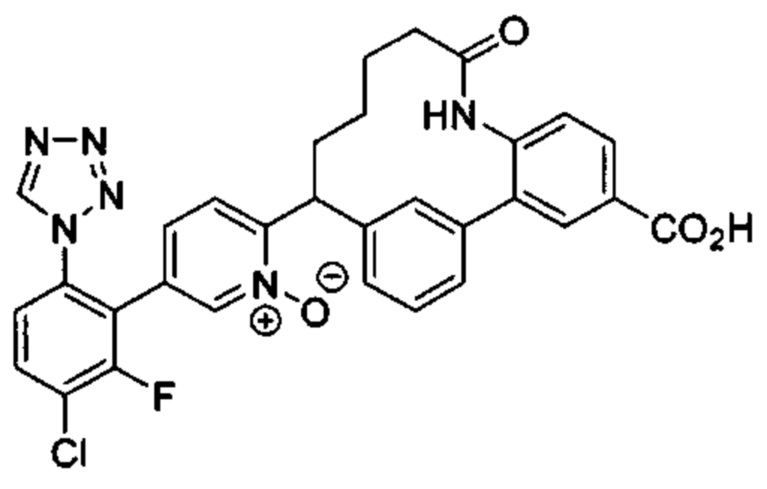
Соединение примера 5 получали способами, описанными в синтезе соединения примера 4. MS (ES+) m/z: 599 (М+Н).
Рацемический образец соединения примера 5 подвергали хиральному разделению методом SFC (OD-H, 2×25 см, 30% MeOH (NH4OH)/CO2, 100 бар, 60 мл/мин, 35°C) с получением соединения примера 5-а (более быстрое элюирование) и соединения примера 5-b (более медленное элюирование). MS (ES+) m/z: 599 (М+Н).
ПРИМЕР 6 (рацемат), 6-а и 6-b
5-(5-ХЛОР-2-(1H-ТЕТРАЗОЛ-1-ИЛ)ФЕНИЛ-2-(24-((МЕТОКСИКАРБОНИЛ)АМИНО)-4-ОКСО-3-АЗА-1(1,3),2(1,2)-ДИБЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
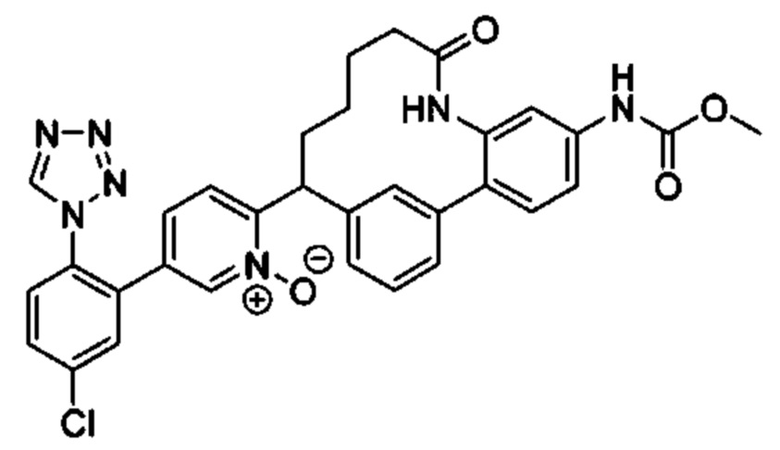
6А: 6-(24-((Метоксикарбонил)амино)-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-9-ил)пиридин-3-илатрифторметансульфонат:
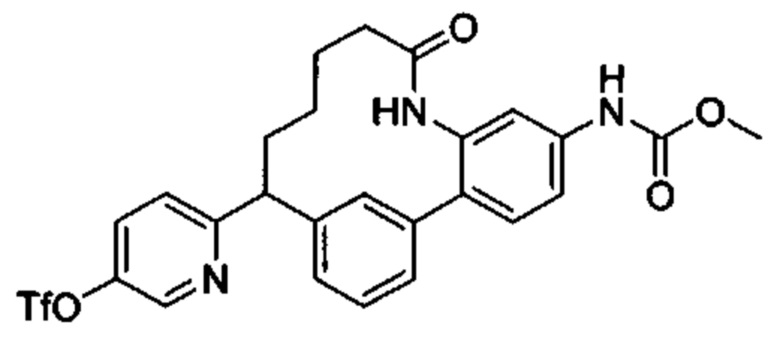
Соединение примера 6А получали таким же способом синтеза, что и в синтезе соединения примера 1F с использованием промежуточного соединения 1 и промежуточного соединения 19 в качестве исходных веществ. MS (ES+) m/z: 564 (М+Н).
Пример 6:
Соединение примера 6 получали из соединения 6А способом синтеза, описанным в синтезе соединения примера 1 и соединения примера 2. MS (ES+) m/z: 610 (М+Н).
Рацемический образец соединения примера 6 подвергали хиральному разделению методом SFC (IC, 30×250 мм, 80% 2:1 MeOH: MeCN/CO2, 70 мл/мин, 100 бар, 35°C) с получением соединения примера 6-а (более медленное элюирование) и соединения примера 6-b (более быстрое элюирование). MS (ES+) m/z: 610 (М+Н); 1H ЯМР (500 МГц, DMSO-d6): δ 9.81 (s, 1Н), 9.66 (s, 1Н), 9.55 (s, 1Н), 8.19 (d, 1H), 7.88 (d, 1Н), 7.79-7.83 (m, 2Н), 7.56 (s, 1Н), 7.47 (d, 2Н), 7.41 (d, 1H), 7.29(m, 2Н), 7.19 (d, 1Н), 6.92 (m, 1Н), 6.79 (d, 1Н), 4.53 (d, 1Н), 3.68 (s, 3H), 2.28 (m, 1Н), 1.94 (m, 2Н), 1.78 (m, 2Н), 1.46 (m, 1Н), 1.21 (m, 1Н), 1.02 (m, 1Н).
ПРИМЕР 7-а и 7-b
5-(5-ХЛОР-3-ФТОР-2-(1H-ТЕТРАЗОЛ-1-ИЛ)ФЕНИЛ)-2-(25-ФТОР-4-ОКСО-3-АЗА-1(1,3),2(1,2)-ДИБЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД

7А: 9-(5-(2-Амино-5-хлор-3-фторфенил)пиридин-2-ил)-25-фтор-3-аза-1(1,3),2(1,2)-дибензолациклононафан-4-он:
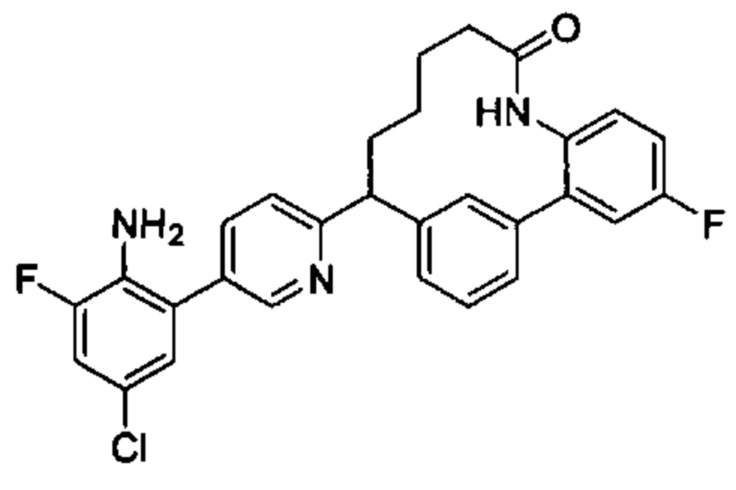
В сосуд добавляли смесь соединения примера 1F (110 мг, 0,216 ммоль), ацетата калия (42,5 мг, 0,433 ммоль), бис(пинаколято)диборона (71,4 мг, 0,281 ммоль) и хлор(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2-аминоэтил)фенил]палладия(II) (15,98 мг, 0,022 ммоль). Ее дегазировали и повторно заполняли азотом трижды. Далее добавляли дегазированный диоксан (2,4 мл) и полученную смесь перемешивали при 100°C в течение 1 ч. Смесь охлаждали до к.т. Добавляли раствор 2-бром-4-хлор-6-фторанилина (72,8 мг, 0,324 ммоль) и хлор(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2-аминоэтил)фенил]палладия(II) (15,98 мг, 0,022 ммоль) в дегазированном диоксане (1 мл), а затем добавляли водный трехосновный фосфат калия (3 М, 0,22 мл, 0,66 ммоль). Реакционную смесь перемешивали при 100°C в течение 1 ч. Ее оставляли охлаждаться до к.т. и сразу очищали методом быстрой колоночной флеш-хроматографии (элюировали EtOAc-EtOH (3:1)/гексаном = 40% объем/объем) с получением указанного соединения. MS (ES+) m/z: 504 (М+Н).
7В: 9-(5-(5-Хлор-3-фтор-2-(1Н-тетразол-1-ил)фенил)пиридин-2-ил)-25-фтор-3-аза-1(1,3),2(1,2)-дибензолациклононафан-4-он:
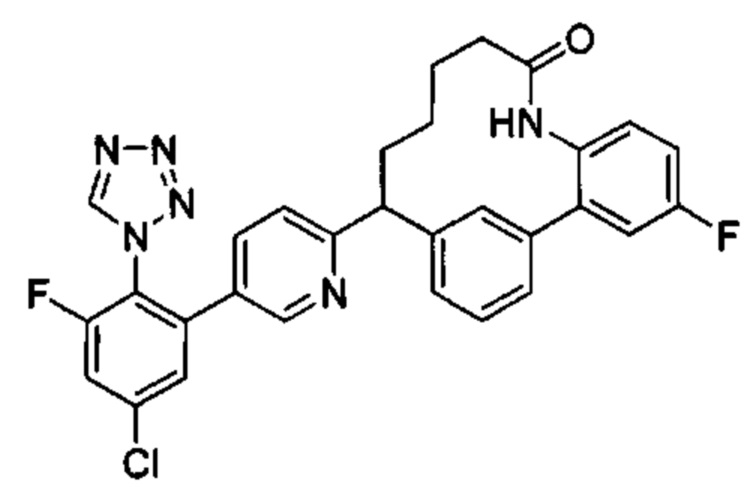
К перемешиваемой смеси соединения примера 7А (36 мг, 0,071 ммоль) и триметилортоформиата (7,58 мг, 0,071 ммоль) в уксусной кислоте (1 мл) добавляли азид натрия (4,64 мг, 0,071 ммоль). Смесь перемешивали при 90°C в течение 2 ч. Ее охлаждали до к.т. и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали EtOAc-EtOH (3:1)/гексаном = 30%-70% объем/объем) с получением указанного соединения. MS (ES+) m/z: 557 (М+Н).
Пример 7:
м-СРВА (11,15 мг, 0,065 ммоль) добавляли к перемешиваемой смеси соединения примера 7В (18 мг, 0,032 ммоль) в дихлорметане (1 мл). Смесь перемешивали при к.т. всю ночь и сразу очищали методом колоночной флеш-хроматографии на силикагеле (элюировали EtOAc-EtOH (3:1) / гексан = 30-100% объем/объем) с получением рацемического соединения. MS (ES+) m/z: 573 (М+Н).
Рацемический образец соединения примера 7 подвергали хиральному разделению методом SFC (IC, 21×250 мм, 50% MeOH : MeCN (2:1)/CO2, 60 мл/мин, 100 бар, 35°C) с получением соединения примера 7-а (более быстрое элюирование) и соединения примера 7-b (более медленное элюирование). MS (ES+) m/z: 573 (М+Н); 1H ЯМР (500 МГц, CH3OH-d4): δ 9.46 (s, 1Н), 8.25 (s, 1H), 7.79 (d, 1H), 7.68 (s, 1Н), 7.64 (s, 1Н), 7.58 (d, 1Н), 7.37 (t, 1Н), 7.21-7.30 (m, 4Н), 7.14 (m, 1H), 6.89 (d, 1H), 4.68 (t, 1Н), 2.38 (t, 1H), 2.11 (t, 1Н), 1.98 (m, 2Н), 1.91 (d, 1Н), 1.56 (m, 1Н), 1.38 (m, 1Н), 1.23 (m, 1Н).
С использованием способов, описанных выше, и соответствующих исходных веществ следующие соединения синтезировали и характеризовали способом LC/MS.
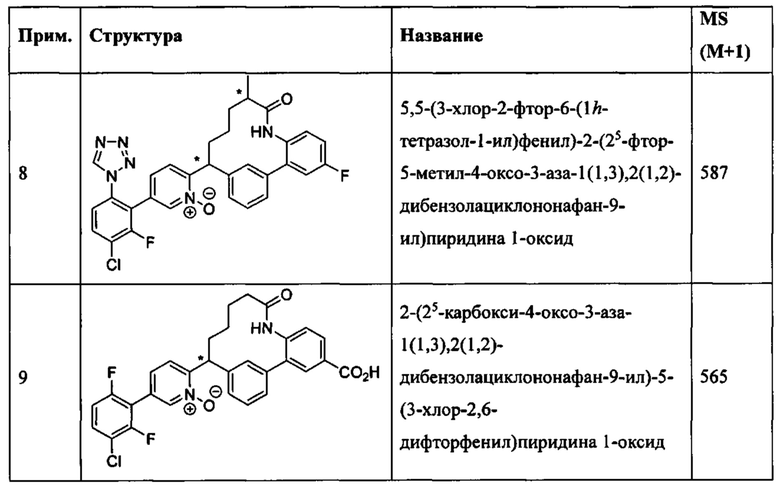
ПРИМЕР 10 (рацемат), 10а и 10b
(Z)-9-(5-(5-ХЛОР-2-(1H-ТЕТРАЗОЛ-1-ИЛ)ФЕНИЛ)ПИРИДИН-2-ИЛ)-25-ФТОР-11H-3-АЗА-1(4,2)-ИМИДАЗОЛ-2(1,2)-БЕНЗОЛАЦИКЛОНОНАФАН-4-ОН
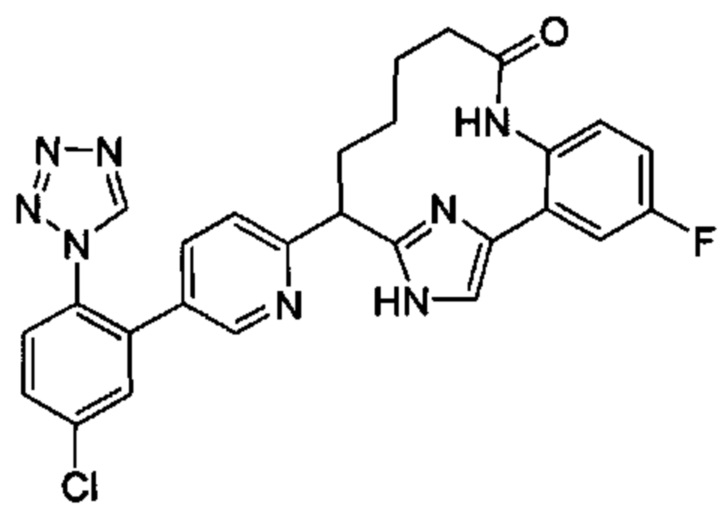
10A: 5-((4-Метоксибензил)окси)-2-метилпиридин: К перемешиваемой смеси 6-метилпиридин-3-ола (10 г, 92 ммоль) в DMF (150 мл) при 0°C небольшими частями добавляли NaH (5,50 г, 137 ммоль, 60% масс.). После добавления смесь перемешивали в атмосфере азота при 0°C в течение 30 мин. К реакционной смеси по каплям добавляли РМВ-Cl (14,86 мл, 110 ммоль) и ее оставляли нагреваться до к.т. с перемешиванием в течение 2 ч. Реакцию гасили насыщенным водным хлоридом аммония (200 мл) и экстрагировали EtOAc (3×100 мл). Объединенные органические слои промывали водой, сушили над сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром : EtOAc = 4:1 объем/объем) с получением указанного соединения. MS (ES+) m/z: 230 (М+Н).
10В: Этил-2-(5-((4-метоксибензил)окси)пиридин-2-ил)ацетат: К перемешиваемой смеси соединения примера 10А (7 г, 30,5 ммоль) в безводном THF (140 мл) при -78°C добавляли раствор LDA (30,5 мл, 61,1 ммоль, 2М в THF). Ее перемешивали в течение 45 мин и к реакционной смеси по каплям добавляли диэтилкарбонат (5,52 мл, 45,8 ммоль). Ее перемешивали еще 1 ч. Реакцию гасили насыщенным водным хлоридом аммония (20 мл) и экстрагировали EtOAc (3×30 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром : EtOAc = 10:1-3:1 объем/объем) с получением указанного соединения. 1H ЯМР (CDCl3, 400 МГц): δ 8.36-8.22 (m, 1Н), 7.35 (d, J=8.6 Гц, 2Н), 7.25-7.17 (m, 2Н), 6.92 (d, J=8.6 Гц, 2Н), 5.09-4.96 (m, 2Н), 4.18 (q, J=7.1 Гц, 2Н), 3.82 (s, 3H), 3.80-3.73 (m, 2Н), 1.26 (t, J=7.1 Гц, 3H).
10С: Этил-2-(5-((4-метоксибензил)окси)пиридин-2-ил)пент-4-еноат: К перемешиваемой смеси соединения примера 10В (6 г, 19,91 ммоль) в THF (100 мл) при -78°C добавляли раствор LDA (11,45 мл, 22,90 ммоль, 2 M в THF). Ее перемешивали при -78°C в течение 30 мин и к реакционной смеси по каплям добавляли аллилбромид (1,9 мл, 21,90 ммоль). Реакционную смесь оставляли нагреваться до 20°C и перемешивали в течение еще 1 ч перед тем, как ее гасили водным насыщенным хлоридом аммония (60 мл). Смесь переносили в делительную воронку и экстрагировали EtOAc (3×60 мл). Объединенные органические слои промывали солевым раствором (насыщенный, 60 мл), сушили (сульфат натрия), фильтровали и концентрировали. Неочищенный продукт очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром : EtOAc = 10:1 объем/объем) с получением указанного соединения. MS (ES+) m/z: 342.1 (М+Н).
10D: 2-(5-((4-Метоксибензил)окси)пиридин-2-ил)пент-4-еновая кислота: Смесь соединения примера 10С (8,6 г, 25,2 ммоль), гидроксида лития моногидрата (5,29 г, 126 ммоль) в сорастворителе MeOH (25 мл)/ТНР (25 мл)/воде (25 мл) перемешивали при 20°C в течение 1 ч. Смесь подкисляли HCl (водная, 1 М) до значения pH 5 и экстрагировали EtOAc (3×50 мл). Объединенные органические слои промывали солевым раствором (насыщенный, 60 мл), сушили (сульфат натрия), фильтровали и концентрировали с получением указанного соединения, которое использовали на следующей стадии без дополнительной очистки. MS (ES+) m/z: 314 (М+Н).
10Е: 2-(2-Бром-5-фторфенил)-2-оксоэтил 2-(5-((4-метоксибензил)окси)пиридин-2-ил)пент-4-еноат: Раствор соединения примера 10D (2,0 г, 6,38 ммоль), 2-бром-1-(2-бром-5-фторфенил)этан-1-она (2,267 г, 7,66 ммоль) и DIEA (2,2 мл, 12,5 ммоль) в ацетонитриле (20 мл) перемешивали при 20°C в течение 2 ч. Смесь концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром : EtOAc = 10:1-3:1 объем/объем) с получением указанного соединения. MS (ES+) m/z: 528, 530 (М+Н);
10F: 2-(1-(4-(2-Бром-5-фторфенил)-1H-имидазол-2-ил)бут-3-ен-1-ил)-5-((4-метоксибензил)окси)пиридин: Смесь соединения примера 10Е (2,2 г, 4,16 ммоль), ацетата аммония (2,74 мл, 41,6 ммоль) и сорастворителя толуола (25 мл)/НОАс (5 мл) в закупоренном сосуде для микроволнового облучения нагревали в микроволновом реакторе при 140°C в течение 30 мин. Смесь экстрагировали EtOAc (3×50 мл). Объединенные органические слои промывали водой (50 мл) и солевым раствором (насыщенный, 50 мл) и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром : EtOAc = 20:1-5:1) с получением указанного соединения. MS (ES+) m/z: 508, 510 (М+Н).
10G: 2-(1-(4-(2-Бром-5-фторфенил)-1-((2-(триметилсилил)этокси)метил)-1Н-имидазол-2-ил)бут-3-ен-1-ил)-5-((4-метоксибензил)окси)пиридин: К суспензии соединения примера 10F (1,7 г, 3,34 ммоль) в DMF (15 мл) добавляли раствор DIEA (0,7 мл, 4,0 ммоль) в DMF (3 мл) при 0°C и смесь перемешивали в течение 30 мин. К раствору реакционной смеси добавляли раствор SEM-Cl (0,65 мл, 3,68 ммоль) в DMF (3 мл). Его перемешивали еще 2 ч при 0°C. Смесь разбавляли EtOAc (200 мл) и промывали водой (100 мл). Органический слой отделяли, сушили над сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром : EtOAc = 30:1-5:1 объем/объем) с получением указанного соединения. MS (ES+) m/z: 638, 640 (М+Н).
10Н: 4-Фтор-2-(2-(1-(5-((4-метоксибензил)окси)пиридин-2-ил)бут-3-ен-1-ил)-1-((2-(триметилсилил)этокси)метил)-1H-имидазол-4-ил)анилин: Соединение примера 10G (1,7 г, 2,66 ммоль), DMSO (17 мл), карбонат калия (1,104 г, 7,99 ммоль), йодида меди(I) (0,051 г, 0,266 ммоль) и L-пролин (0,092 г, 0,799 ммоль) добавляли в сухую закупоренную пробирку. Поток азота барботировали через смесь в течение 2 мин, а затем добавляли гидроксид аммония (0,414 г, 2,95 ммоль). Пробирку закупоривали и перемешивали на масляной бане при 90°C в течение 20 ч. Ее охлаждали до к.т. и реакцию гасили водой (20 мл). Смесь экстрагировали EtOAc (3×50 мл). Объединенные органические слои промывали солевым раствором (2×30 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром : EtOAc = 10:1-3:1 объем/объем) с получением указанного соединения. MS (ES+) m/z: 575 (М+Н).
10I: N-(4-Фтор-2-(2-(1-(5-((4-метоксибензил)окси)пиридин-2-ил)бут-3-ен-1-ил)-1-((2-(триметилсилил)этокси)метил)-1H-имидазол-4-ил)фенил)бут-3-енамид: К раствору соединения примера 10Н (600 мг, 1,044 ммоль), бут-3-еновой кислоты (108 мг, 1,253 ммоль) в DMF (10 мл) добавляли DIEA (0,547 мл, 3,13 ммоль). Реакционную смесь охлаждали до 0°C и по каплям добавляли 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфоринан-2,4,6-триоксид (1329 мг, 2,088 ммоль) (50% масс. в этилацетате). Реакционную смесь перемешивали в течение 5 мин и ее оставляли нагреваться до 30°C в течение 1,5 ч. Смесь охлаждали до 0°C и добавляли раствор водного бикарбоната натрия (насыщенный, 20 мл). Его экстрагировали EtOAc (2×30 мл). Объединенные органические слои промывали солевым раствором (насыщенный, 20 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0~30% этилацетатом/петролейным эфиром, градиент) с получением указанного соединения. MS (ES+) m/z: 643 (М+Н).
10J: (12Z,6Е)-25-Фтор-9-(5-((4-метоксибензил)окси)пиридин-2-ил)-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-6-ен-4-он:
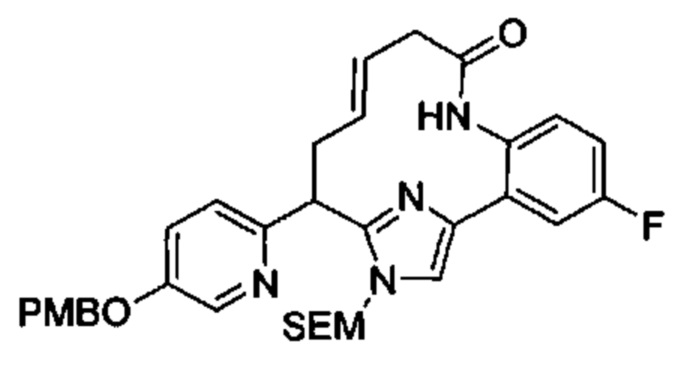
К раствору соединения примера 10I (825 мг, 1,283 ммоль) в DCE (45 мл) (разделенному на 3 пробирки) добавляли Grubbs II ((1,3-бис(2,4,6-триметилфенил)-2-имидазолидинилиден)дихлор(фенилметилен)(трициклогексилфосфин)рутения) (3×109 мг, 0,385 ммоль). Полученную смесь в атмосфере азота перемешивали при 120°C в течение 30 мин в микроволновом реакторе. Объединенные неочищенные продукты очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром : EtOAc = 10:1-2:1 объем/объем) с получением указанного соединения. MS (ES+) m/z: 615 (М+Н).
10K: (Z)-25-Фтор-9-(5-гидроксипиридин-2-ил)-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-4-он:
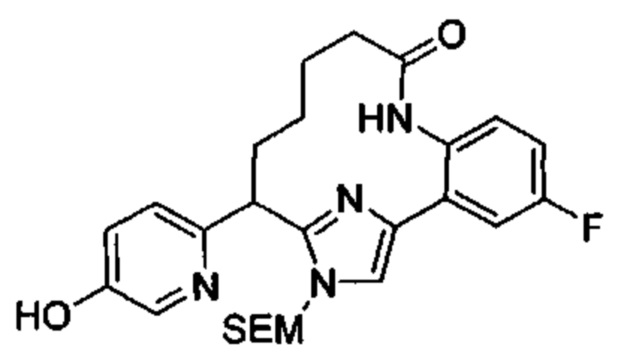
Смесь соединения примера 10J (500 мг, 0,813 ммоль), 10% Pd/C (173 мг, 0,163 ммоль) и MeOH (20 мл) перемешивали при 30°C в атмосфере H2 (1 атм.) в течение 20 ч. Смесь фильтровали через слой целита и фильтрат концентрировали при пониженном давлении с получением указанного соединения, которое использовали на следующей стадии без дополнительной очистки. MS (ES+) m/z: 497 (М+Н)
10L: (Z)-6-(25-Фтор-4-оксо-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-9-ил)пиридин-3-ил трифторметансульфонат:
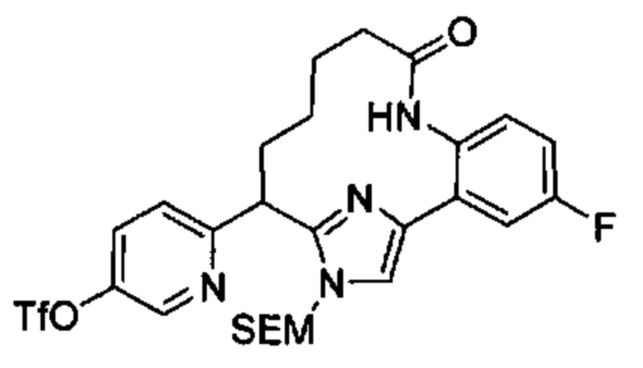
К раствору соединения примера 10K (500 мг, 1,007 ммоль) и Et3N (0,421 мл, 3,02 ммоль) в DCM (8 мл) при 0°C добавляли трифторметансульфоновый ангидрид (1,510 мл, 1,510 ммоль) в атмосфере азота. Смесь перемешивали в течение 20 мин и очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром : EtOAc = 5:1-1:1 объем/объем) с получением указанного соединения. 1H ЯМР (CDCl3, 400 МГц): δ 12.02 (m, 1H), 8.53 (d, J=2.8 Гц, 1Н), 7.94 (dd, J=5.4, 8.9 Гц, 1Н), 7.59 (dd, J=2.8, 8.5 Гц, 1Н), 7.42 (d, J=8.8 Гц, 1Н), 7.17 (dd, J=2.9, 9.2 Гц, 1Н), 7.11 (s, 1Н), 7.00 (dt, J=2.8, 8.5 Гц, 1Н), 5.10 (q, J=11.0 Гц, 2Н), 4.44 (dd, J=3.6, 11.7 Гц, 1H), 3.32 (dt, J=6.7, 10.0 Гц, 2Н), 2.71-2.58 (m, 1Н), 2.56-2.44 (m, 1Н), 2.43-2.31 (m, 1Н), 2.18 (dd, J=3.9, 8.2 Гц, 1Н), 2.08-1.94 (m, 1Н), 1.84-1.62 (m, 2Н), 1.36-1.19 (m, 1Н), 0.83-0.61 (m, 2Н), -0.05 (s, 9Н).
10М: (Z)-9-(5-(2-Амино-5-хлорфенил)пиридин-2-ил)-25-фтор-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-4-он:
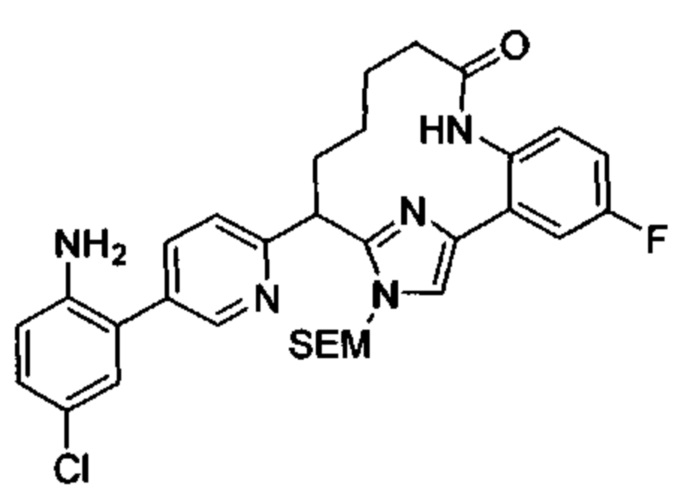
К раствору соединения примера 10L (400 мг, 0,636 ммоль) в DMF (10 мл) в закупоренной пробирке добавляли 4-хлор-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин (323 мг, 1,272 ммоль), K3PO4 (405 мг, 1,909 ммоль) и PdCl2(dppf) (46,6 мг, 0,064 ммоль). Пробирку закупоривали и трижды продували азотом. Смесь перемешивали при 90°C в атмосфере азота в течение 1 ч. Ее охлаждали до к.т. и разбавляли H2O (30 мл). Смесь экстрагировали EtOAc (3×30 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром : EtOAc = 5:1-1:1 объем/объем) с получением указанного соединения. MS (ES+) m/z: 606 (М+Н).
10N: (Z)-9-(5-(5-Хлор-2-(1H-тетразол-1-ил)фенил)пиридин-2-ил)-25-фтор-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-4-он:
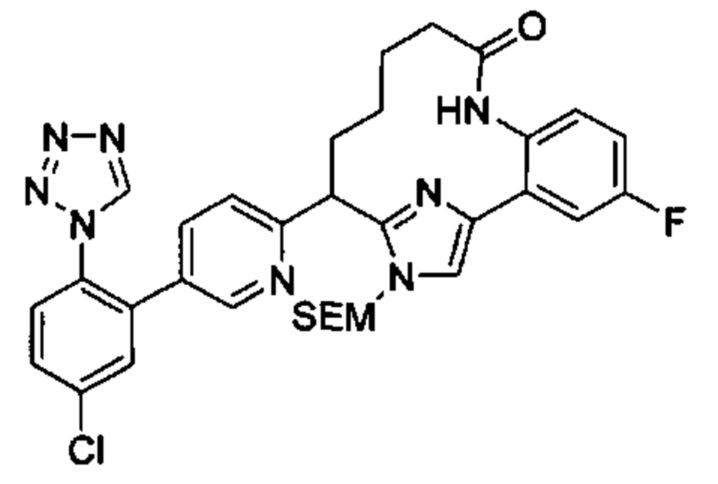
К раствору соединения примера 10М (390 мг, 0,643 ммоль) в НОАс (10 мл) добавляли триметилортоформиат (1365 мг, 12,87 ммоль) и азид натрия (836 мг, 12,87 ммоль). Смесь перемешивали при 30°C в течение 13 ч. Смесь гасили насыщенным NaNO2 (30 мл), значение pH доводили до 7 насыщенным водным бикарбонатом натрия и экстрагировали EtOAc (3×20 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром : EtOAc = 3:1-1:1 объем/объем) с получением указанного соединения. MS (ES+) m/z: 659 (М+Н).
Пример 10:
К раствору соединения примера 10N (250 мг, 0,265 ммоль) в DCM (8 мл) и TFA (4 мл, 51,9 ммоль) добавляли (R)-2-амино-3-меркаптопропановую кислоту (161 мг, 1,327 ммоль) при 25°C. Смесь перемешивали в течение 2 ч. Ее концентрировали при пониженном давлении и остаток очищали методом HPLC с получением рацемического соединения. MS (ES+) m/z: 529 (М+Н).
Образец рацемического соединения примера 10 подвергали хиральному разделению методом SFC (AS, 250 мм × 30 мм, 5 мкм, 40% MeOH/СО2, 60 мл/мин) с получением соединения примера 10-а (более медленное элюирование) и соединения примера 10-b (более быстрое элюирование). MS (ES+) m/z: 529 (М+Н); 1H ЯМР (400 МГц, MeOH-d4): δ 9.28 (s, 1Н), 8.35 (s, 1Н), 7.80-7.70 (m, 3H), 7.56 (m, 1H), 7.50-7.30 (m, 4Н), 7.15 (m, 1Н), 4.59 (m, 1Н), 2.54 (m, 1Н), 2.30-1.90 (m, 4Н), 1.64 (m, 1Н), 1.35 (m, 1Н), 1.05 (m, 1Н).
ПРИМЕР 11 (рацемат), 11а и 11b
(Z)5-(5-ХЛОР-2-(1H-ТЕТРАЗОЛ-1-ИЛ)ФЕНИЛ)-2-(25-ФТОР-4-ОКСО-11H-3-АЗА-1(4,2)-ИМИДАЗОЛ-2(1,2)-БЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД

11А: (Z)-5-(5-Хлор-2-(1H-тетразол-1-ил)фенил)-2-(25-фтор-4-оксо-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-9-ил)пиридина 1-оксид:

Смесь соединения примера 10N (350 мг, 0,531 ммоль) в перуксусной кислоте (10% в уксусной кислоте, 20 мл, 0,531 ммоль) перемешивали при 25°C в течение 13 ч. Реакцию гасили Na2SO3 (нас, 40 мл) и экстрагировали EtOAc (3×30 мл). Объединенные органические слои промывали насыщенным бикарбонатом натрия (30 мл) и солевым раствором (30 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением указанного соединения, которое использовали на следующей стадии без дополнительной очистки. MS (ES+) m/z: 675 (М+Н)
Пример 11:
Раствор соединения примера 11А (25 мг, 0,037 ммоль) и (R)-2-амино-3-меркаптопропановой кислоты (22,43 мг, 0,185 ммоль) в DCM (0,5 мл) и TFA (0,5 мл, 6,49 ммоль) перемешивали при 25°C в течение 3 ч. Смесь концентрировали и очищали методом HPLC с обращенной фазой с получением рацемического продукта. MS (ES+) m/z: 545 (М+Н).
Образец соединения примера 11 подвергали хиральному разделению методом SFC (OJ, 4,6×50 мм, 3 мкм, метанол (0,05% диэтиламин)/СО2, 4 мл/мин, 40°C) с получением соединения примера 11-а (более медленное элюирование) и соединения примера 11-b (более быстрое элюирование). MS (ES+) m/z: 545 (М+Н); 1H ЯМР (CD3OD, 400 МГц): δ 9.42 (s, 1Н), 8.31 (s, 1Н), 7.84-7.77 (m, 2Н), 7.73 (d, J=8.2 Гц, 2Н), 7.52 (s, 1Н), 7.44-7.38 (m, 1Н), 7.36-7.24 (m, 3H), 2.55 (d, J=11.7 Гц, 1Н), 2.38-2.10 (m, 3H), 1.85 (d, J=11.3 Гц, 1H), 1.64 (m, 1H), 1.46 (m, 1H), 1.30-1.20 (m, 1H), 1.09 (m, 1H).
ПРИМЕР 12
МЕТИЛ(9-(5-(3-ХЛОР-2,6-ДИФТОРФЕНИЛ)ПИРИДИН-2-ИЛ)-4-ОКСО-3-АЗА-1(1,3),2(1,2)-ДИБЕНЗОЛАЦИКЛОНОНАФАН-24-ИЛ)КАРБАМАТ
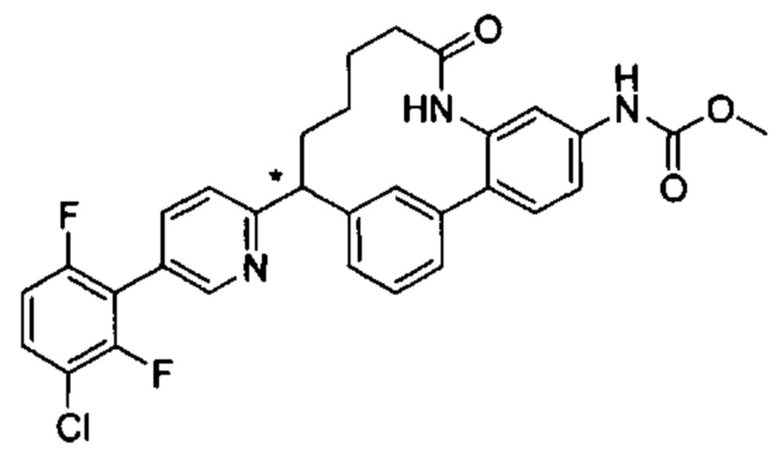
12A: Метил(2-амино-3'-(5-хлорпиколиноил)-[1,1'-бифенил]-4-ил)карбамат: Круглодонную колбу с магнитной мешалкой заполняли промежуточным соединением 19 (2,483 г, 8,50 ммоль), промежуточным соединением 2 (2,1 г, 7,08 ммоль) и дихлорметановым комплексом 1,1'-бис(дифенилфосфино)ферроцен-палладий(II)дихлорида (0,578 г, 0,708 ммоль). Это дегазировали и повторно заполняли азотом трижды. Далее добавляли дегазированный THF (28,3 мл) и водный трехосновный фосфат калия (3 М, 7,08 мл). Смесь перемешивали до 70°C в течение 3 ч и охлаждали до к.т. Ее разбавляли этилацетатом (60 мл) и промывали водой (30 мл). Органический слой отделяли и сушили над безводным сульфатом магния. Его фильтровали, а фильтрат концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали EtOAc/гексаном = 25%-80%) с получением указанного соединения. MS (ES+) m/z: 382 (М+Н).
12В: (5-((3'-(5-Хлорпиколиноил)-4-((метоксикарбонил)амино)-[1,1'-бифенил]-2-ил)амино)-5-оксопентил)трифенилфосфония бромид: К перемешиваемому раствору (4-карбоксибутил)трифенилфосфония бромида (6,94 г, 15,66 ммоль), соединения примера 12А (4,6 г, 12,05 ммоль) и основания Хунига (6,31 мл, 36,1 ммоль) в DMF (58,9 мл) добавляли HATU (5,96 г, 15,66 ммоль). Смесь перемешивали при к.т. всю ночь. Большую часть удаляли при пониженном давлении, а остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали MeOH/CH2Cl2 = 6%) с получением указанного соединения. MS (ES+) m/z: 726 (М).
12С: Метил(E)-(9-(5-хлорпиридин-2-ил)-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-8-ен-24-ил)карбамат
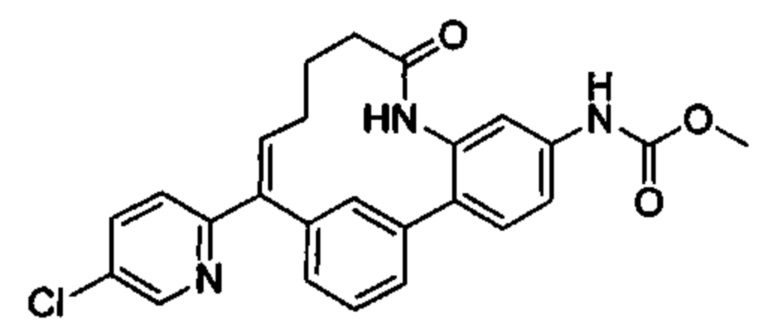
Раствор LiHMDS (1 M в THE, 68,1 мл, 68,1 ммоль) добавляли к перемешиваемой смеси соединения примера 12В (11 г, 13,63 ммоль) в безводном THF (681 мл) при 0°C в течение 1 ч при помощи шприцевой помпы. Полученную смесь перемешивали при к.т. всю ночь и ее гасили водным хлоридом аммония (насыщенный, 100 мл) и водой (100 мл). Смесь экстрагировали этилацетатом (2×200 мл). Объединенные органический слой промывали солевым раствором (150 мл), сушили над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Остаток суспензировали в смеси дихлорметана (20 мл) и метанола (5 мл), а твердые вещества собирали фильтрацией с получением указанного соединения. Фильтрат концентрировали, а остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали MeOH/CH2Cl2 = 5%) с получением другой порции указанного соединения. MS (ES+) m/z: 448 (М+Н).
12D: Метил(9-(5-хлорпиридин-2-ил)-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-24-ил)карбамат:

Смесь соединения примера 12С (3,0 г, 6,70 ммоль), гомогенного Rh катализатора (0,2 г) и 2,2,2-трифторэтанола (80 мл) перемешивали в автоклаве при 50°C в атмосфере водорода (200 фунт/кв. дюйм) в течение 4 ч. Ее охлаждали до к.т. и концентрировали при пониженном давлении. Неочищенный продукт очищали методом колоночной флеш-хроматографии на силикагеле (элюировали MeOH/CH2Cl2 = 7%) с получением указанного соединения. MS (ES+) m/z: 450 (М+Н).
Пример 12:
В сосуд с магнитной мешалкой добавляли бис(пинаколято)диборон (183 мг, 0,720 ммоль), соединение примера 12D (270 мг, 0,600 ммоль), ацетат калия (177 мг, 1,800 ммоль) и предварительный каталитический нейтрализатор Xphos 2-го поколения (47,2 мг, 0,060 ммоль). Это дегазировали и повторно заполняли азотом трижды. В сосуд добавляли дегазированный диоксан (6 мл) и смесь перемешивали при 85°C в течение 1 ч. Ее оставляли охлаждаться до к.т. К смеси добавляли раствор 1-бром-3-хлор-2,6-дифторбензола и дихлорметанового комплекса 1,1'-бис(дифенилфосфино)ферроцен-палладий(II)дихлорида (49,1 мг, 0,060 ммоль) в диоксане (6 мл), а затем добавляли водный трехосновный фосфат калия (3 М, 0,600 мл, 1,80 ммоль). Полученную смесь дегазировали и повторно заполняли азотом трижды, затем перемешивали при 85°C в течение 1 ч. Ее охлаждали до к.т. и большую часть растворителя удаляли при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии (элюировали EtOAc-EtOH (3:1)/гексан = 45%) с получением указанного соединения. MS (ES+) m/z: 562 (М+Н).
ПРИМЕР 13 (рацемат), 13-а и 13-b
5-(3-ХЛОР-2,6-ДИФТОРФЕНИЛ)-2-(24-((МЕТОКСИКАРБОНИЛ)АМИНО)-4-ОКСО-3-АЗА-1(1,3),2(1,2)-ДИБЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
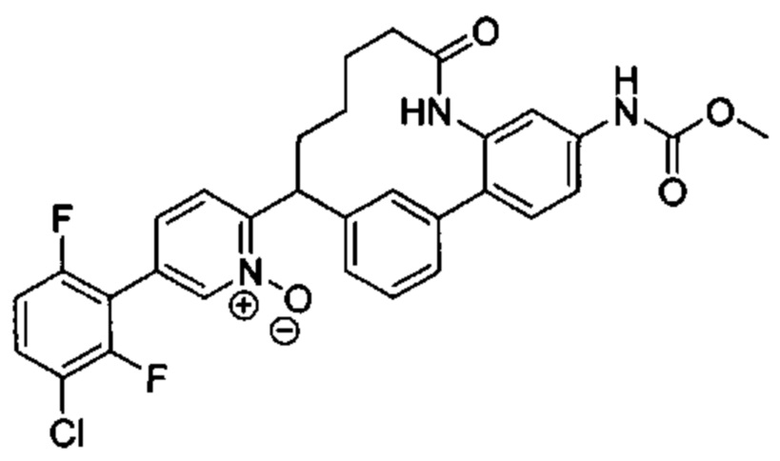
К перемешиваемой смеси соединения примера 12 (198 мг, 0,352 ммоль) в дихлорметане (4 мл) добавляли м-СРВА (237 мг, 1,057 ммоль). Ее перемешивали при к.т. в течение 3 ч. Смесь очищали методом колоночной флеш-хроматографии на силикагеле (элюировали EtOAc-EtOH (3:1) / гексан = 55%) с получением рацемического продукта. MS (ES+) m/z: 578 (М+Н).
Образец рацемического соединения примера 13 подвергали хиральному разделению методом SFC (IC, 21×250 мм, 70% метанол-MeCN (2:1)/CO2, 100 бар, 70 мл/мин, 35°C) с получением соединения примера 13-а (более быстрое элюирование) и соединения примера 13-b (более медленное элюирование). MS (ES+) m/z: 597 (М+Н); 1H ЯМР (500 МГц, СН3ОН-d4): δ 8.50 (s, 1Н), 7.81 (d, 1Н), 7.66-7.73 (m, 2Н), 7.62-7.65 (m, 1Н), 7.43-7.49 (m, 3H), 7.38 (t, 1Н), 7.30 (d, 1Н), 7.20 (t, 1H), 6.97 (d, 1Н), 3.74 (s, 3H), 2.42 (t, 1Н), 2.07-2.15 (m, 3H), 1.98 (d, 1Н), 1.60-1.64 (m, 1Н), 1.40-1.45 (m, 1Н), 1.28-1.30 (m, 1Н).
С использованием методик, описанных в примере 12, и соответствующих исходных веществ, следующие соединения синтезировали и характеризовали способом LC/MS.

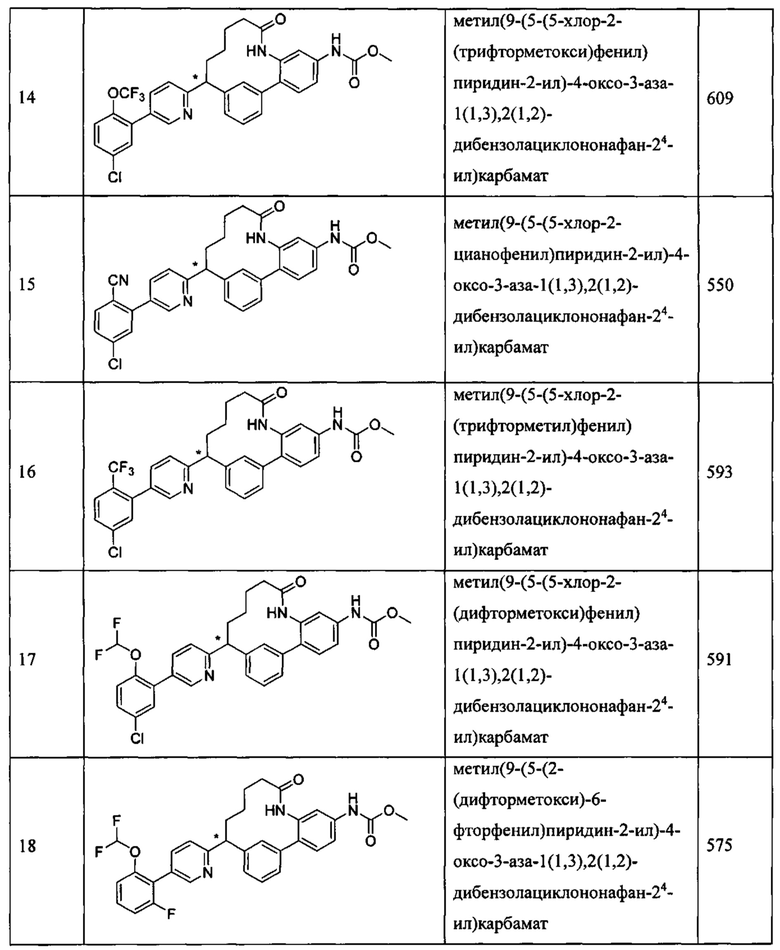
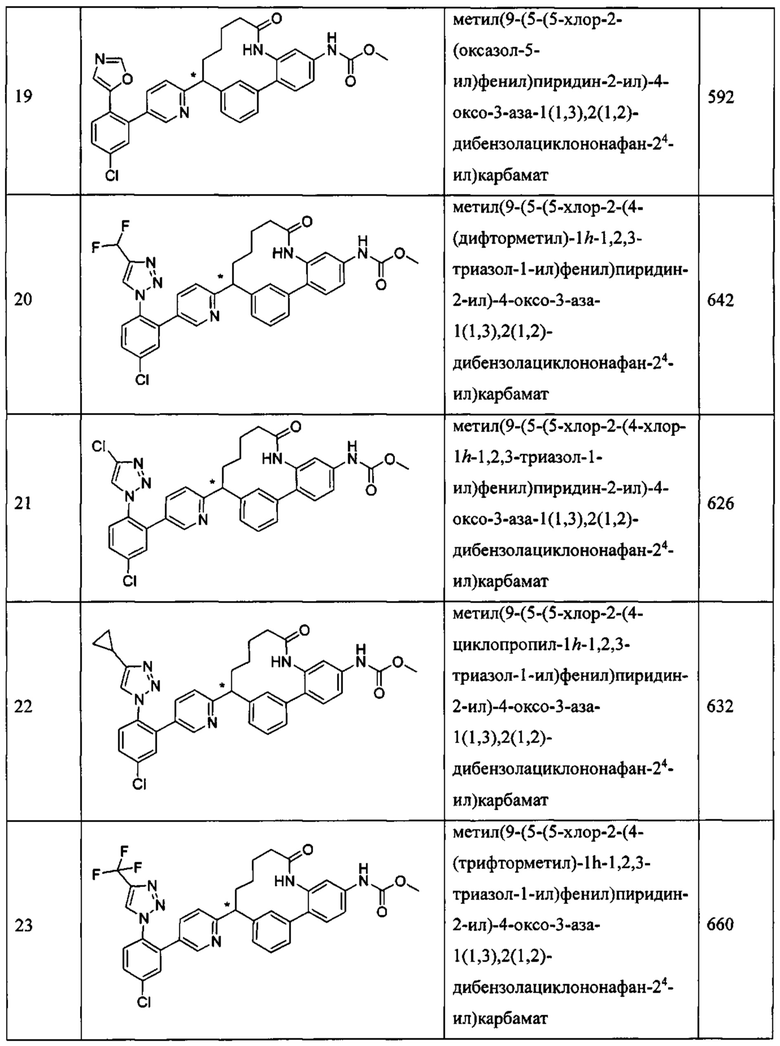
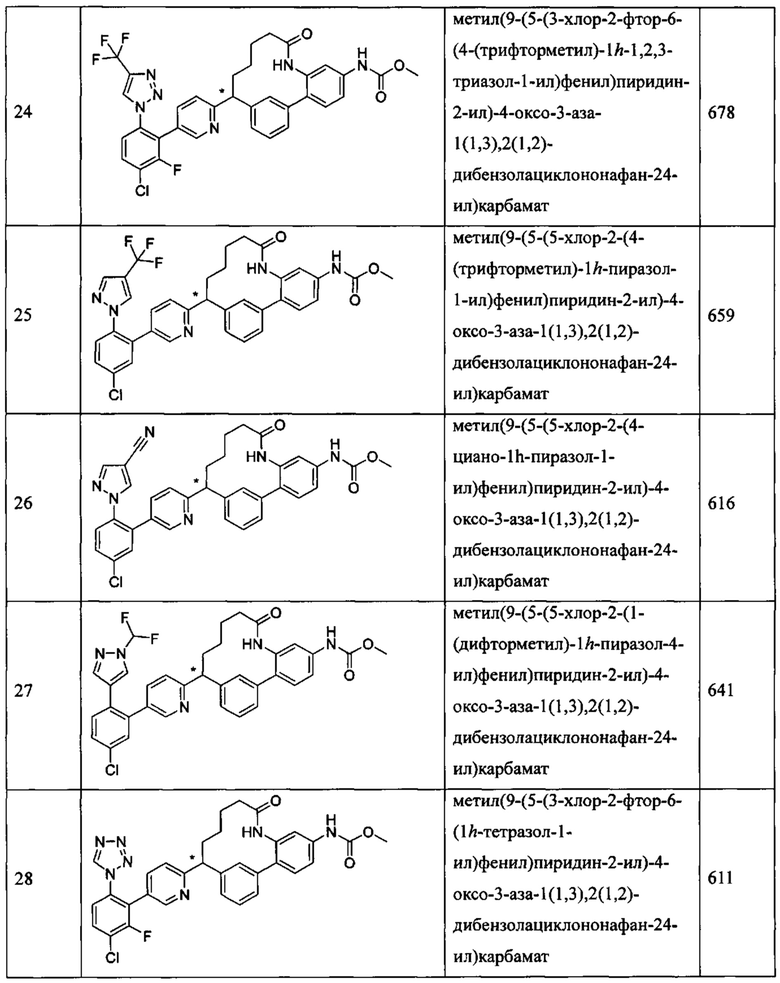
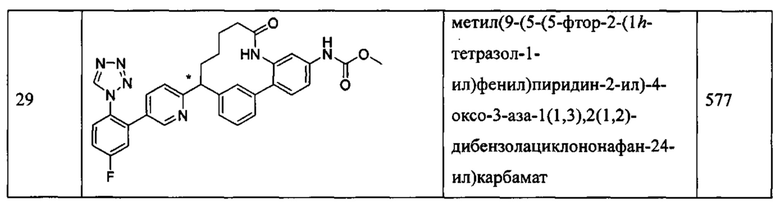
С использованием методик, описанных в примере 13, и соответствующих исходных веществ, следующие соединения синтезировали и характеризовали способом LC/MS.
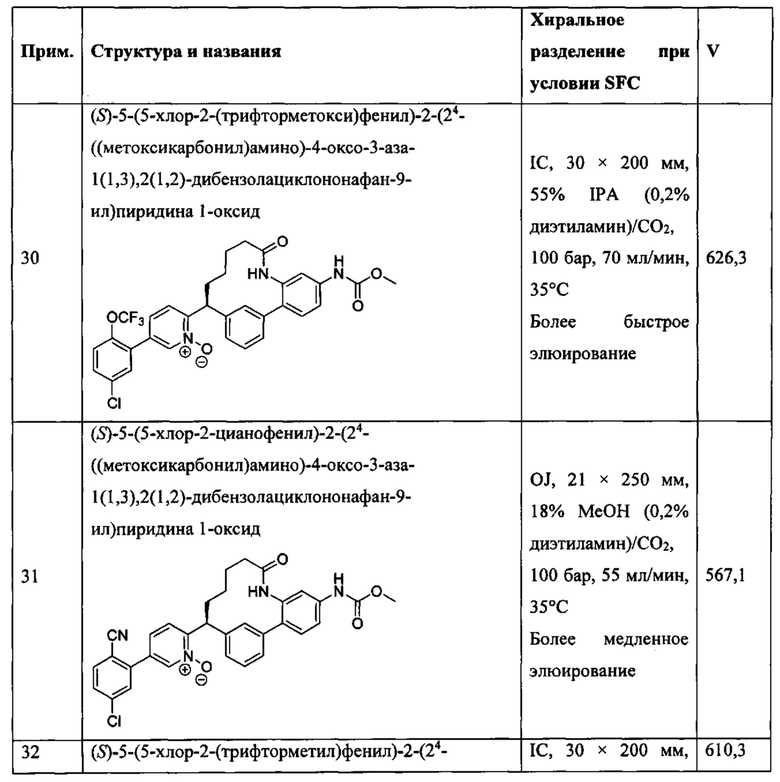
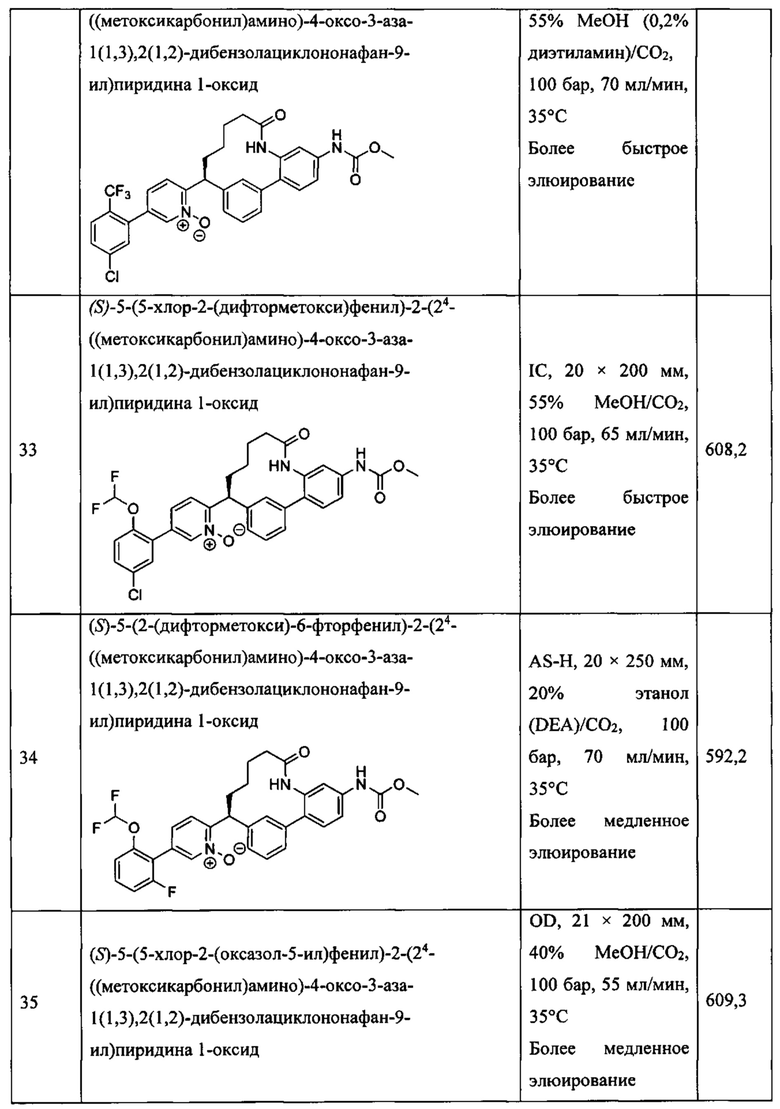
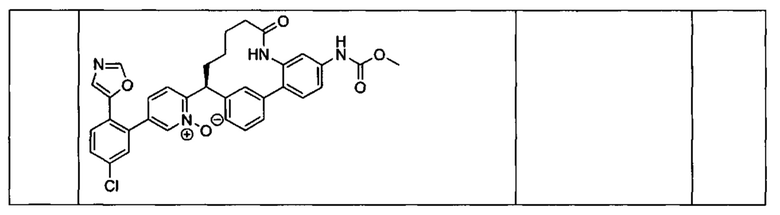
ПРИМЕР 36 (рацемат), 36-а, 36-b
5-(5-ХЛОР-2-(4-(ДИФТОРМЕТИЛ)-1H-1,2,3-ТРИАЗОЛ-1-ИЛ)ФЕНИЛ)-2-(24-((МЕТОКСИКАРБОНИЛ)АМИНО)-4-ОКСО-3-АЗА-1(1,3),2(1,2)-ДИБЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
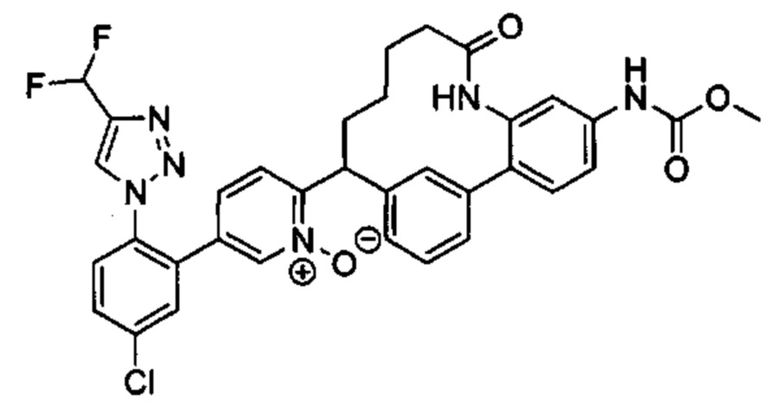
36: 5-Хлор-2-(24-((метоксикарбонил)амино)-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-9-ил)пиридина 1-оксид:
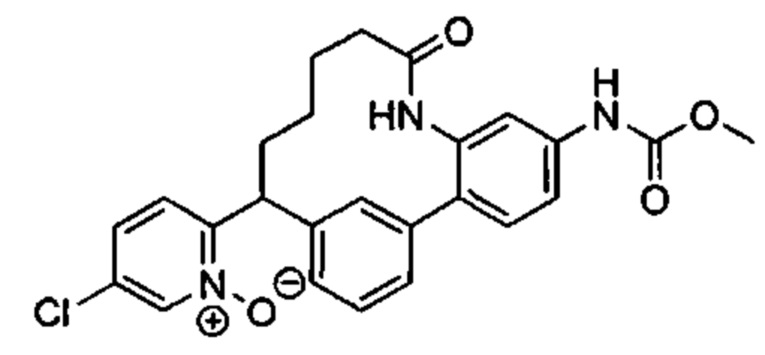
м-СРВА (345 мг, 2,000 ммоль) добавляли к перемешиваемой смеси соединения примера 12D (300 мг, 0.667 ммоль) в дихлорметане (6.668 мл) и смесь перемешивали при к. т.в течение 4 ч. Реакционную смесь очищали методом колоночной флеш-хроматографии на силикагеле (элюировали MeOH/DCM=7%) с получением указанного соединения. MS (ES+) m/z: 466 (М+Н).
36В: 5-(5-Хлор-2-(4-(дифторметил)-1Н-1,2,3-триазол-1-ил)фенил)-2-(24-((метоксикарбонил)амино)-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-9-ил)пиридина 1-оксид: Бис(пинаколято)диборон (65,4 мг, 0,258 ммоль), соединение примера 36А (100 мг, 0,215 ммоль), ацетат калия (63,2 мг, 0,644 ммоль) и предварительный каталитический нейтрализатор XPHOS 2го поколения (16,89 мг, 0,021 ммоль) и диоксан (1,1 мл) смешивали в сосуде пониженного давления, дегазировали и повторно заполняли азотом (3×). Смесь перемешивали при 80°С в течение 30 мин. Ее охлаждали до к. т.К реакционной смеси быстро добавляли 1-(4-хлор-2-йодфенил)-4-(дифторметил)-1H-1,2,3-триазол (99 мг, 0,279 ммоль) и дихлорметановый комплекс 1,1'-бис(дифенилфосфино)-ферроцен-палладий(II)дихлорида (17,57 мг, 0,021 ммоль), а затем добавляли трехосновный фосфат калия (3 М водный) (6,67 мл, 20,01 ммоль). Смесь дегазировали и повторно заполняли N2 (3×) и перемешивали при 80°С в течение 1 ч. Ее охлаждали до к. т.и очищали методом колоночной флеш-хроматографии на силикагеле (элюировали MeOH/DCM=7%) с получением указанного соединения. MS (ES+) m/z: 659 (М+Н).
Образец рацемического соединения примера 36 подвергали хиральному разделению методом SFC (OD, 21×200 мм, 45% МеОН/CO2, 100 бар, 60 мл/мин, 35°С) с получением соединения примера 36-а (более быстрое элюирование) и соединения примера 36-b (более медленное элюирование). MS (ES+) m/z: 659 (М+Н).
С использованием способов, описанных в примере 36, и соответствующих исходных веществ, следующие соединения синтезировали и характеризовали способом LC/MS.
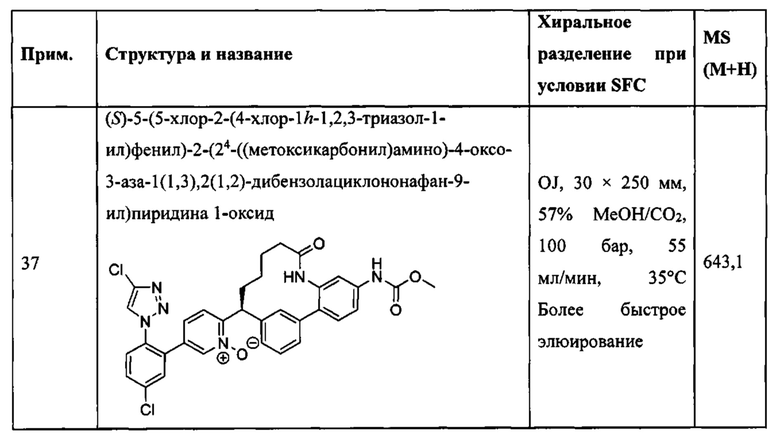
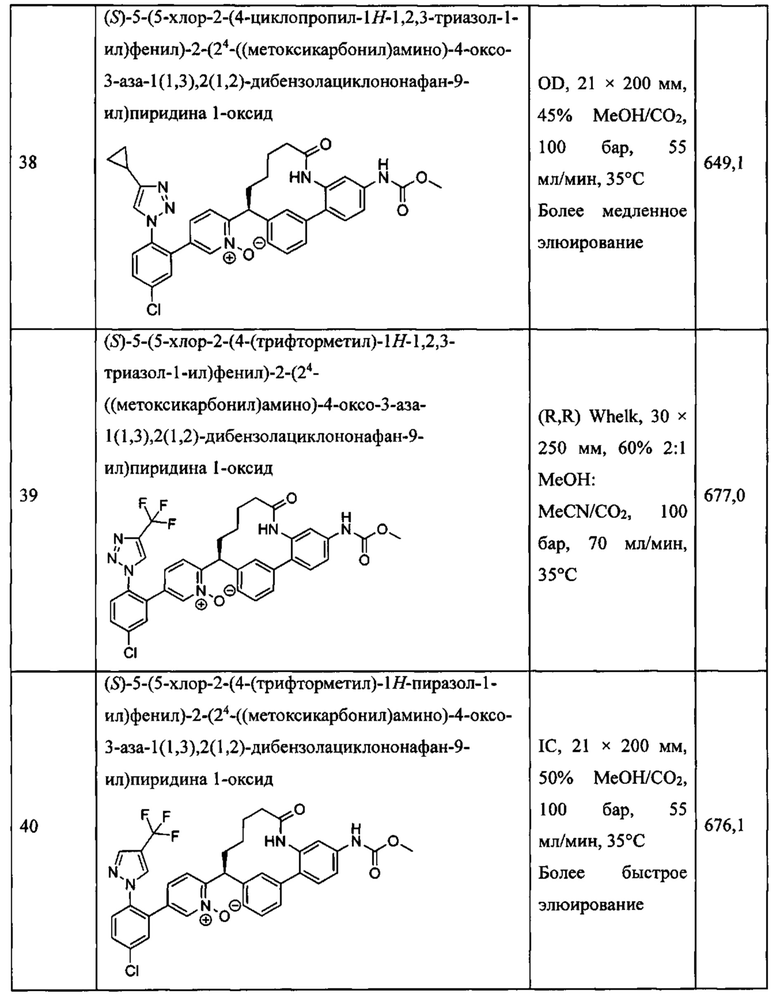
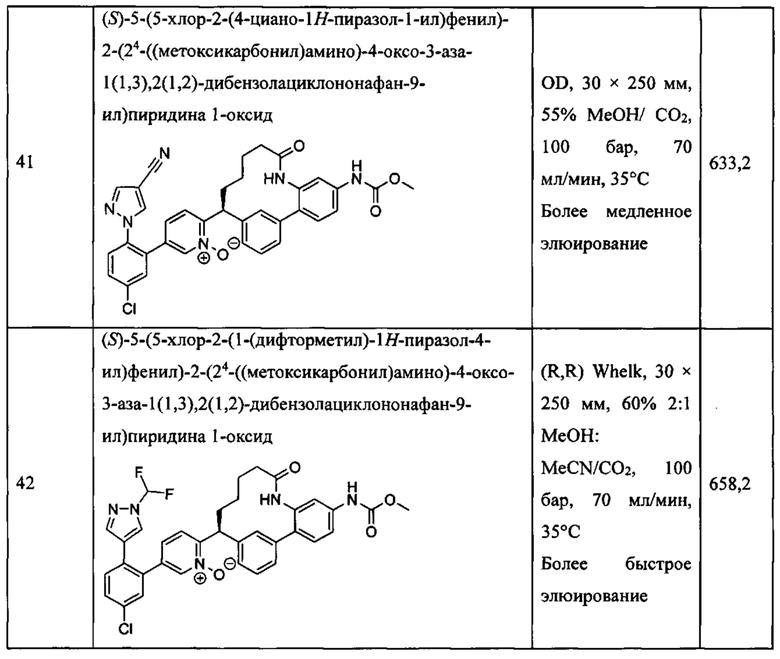
ПРИМЕР 43 (рацемат), 43-а, 43-b
2-(24-АМИНО-4-ОКСО-3-АЗА-1(1,3),2(1,2)-ДИБЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)-5-(3-ХЛОР-2,6-ДИФТОРФЕНИЛ)ПИРИДИНА 1-ОКСИД
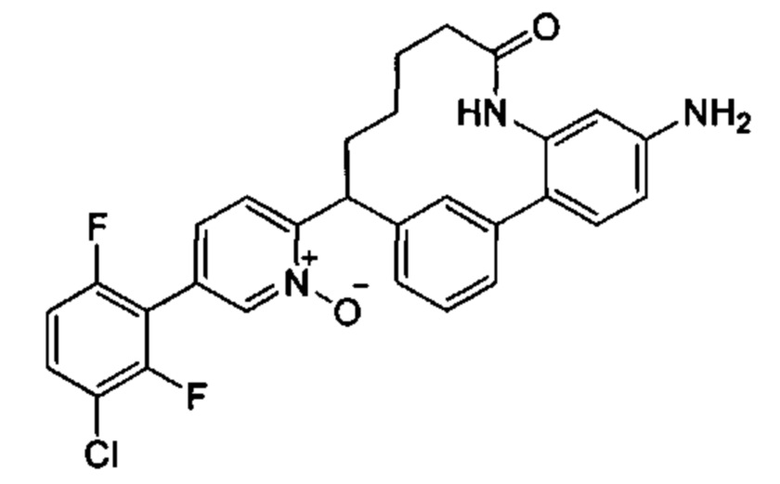
43А: трет-Бутил(Е)-(9-(5-хлорпиридин-2-ил)-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-8-ен-24-ил)карбамат:
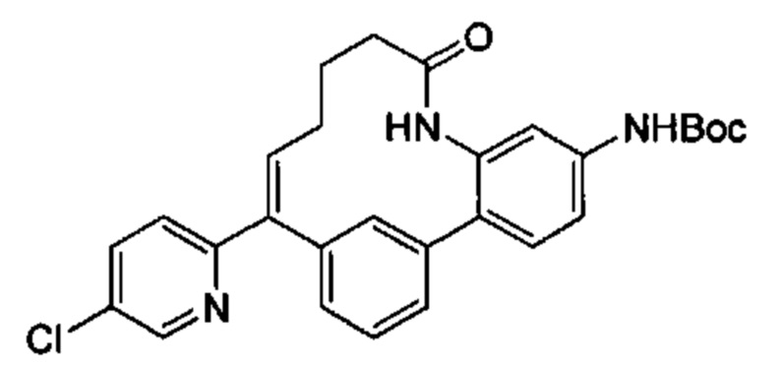
Указанное соединение получали из промежуточного соединения 2 и промежуточного соединения 21 способом, описанным в синтезе соединения 12С. Его очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром : EtOAc = 3:1 объем/объем). MS (ES+) m/z: 490 (М+Н).
43В: трет-Бутил(E)-(9-(5-(3-хлор-2,6-дифторфенил)пиридин-2-ил)-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-8-ен-24-ил)карбамат:
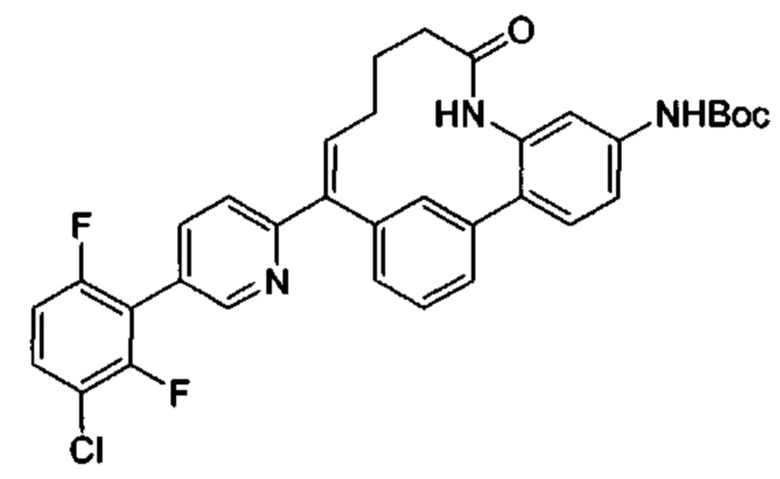
Смесь соединения примера 43А (266 мг, 0,543 ммоль), предварительного каталитического нейтрализатора Xphos второго поколения (42,7 мг, 0,054 ммоль), ацетата калия (107 мг, 1,086 ммоль) и бис(пинаколято)диборона (207 мг, 0,814 ммоль) в диоксане (15 мл) перемешивали при 100°С в атмосфере азота в течение 30 мин, нагревали в микроволновом реакторе. Ее охлаждали до к. т. и добавляли раствор предварительного каталитического нейтрализатора Xphos второго поколения (42,7 мг, 0,054 ммоль), 2-бром-4-хлор-1,3-дифторбензола (185 мг, 0,814 ммоль) в диоксане (1 мл), а затем добавляли водный трехосновный фосфат калия (1 М, 1,086 мл, 1,086 ммоль). Реакционную смесь перемешивали при 80°С в течение 16 ч. Смесь разбавляли EtOAc (200 мл) и промывали солевым раствором (2×100 мл). Органический слой отделяли, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром : EtOAc = 3:1 объем/объем) с получением указанного соединения. MS (ES+) m/z: 602 (М+Н).
43С: трет-Бутил(9-(5-(3-хлор-2,6-дифторфенил)пиридин-2-ил)-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-24-ил)карбамат:
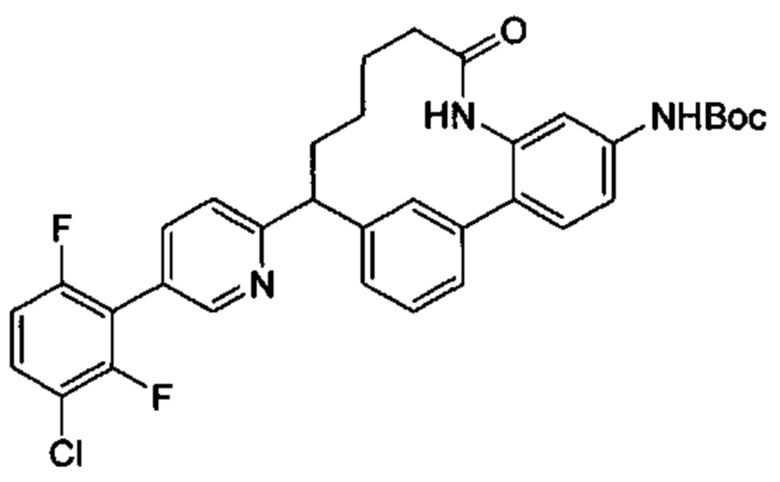
Смесь соединения примера 43В (20 мг, 0,033 ммоль) и никеля Ренея (7,80 мг, 0,133 ммоль) в THF (2 мл) перемешивали при 25°С в атмосфере H2 (1 атм.) в течение 10 мин. Смесь фильтровали через слой целита и фильтрационный кек промывали МеОН (20 мл). Фильтрат концентрировали при пониженном давлении с получением указанного соединения. MS (ES+) m/z: 604 (М+Н).
43D: 2-(24-((трет-Бутоксикарбонил)амино)-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-9-ил)-5-(3-хлор-2,6-дифторфенил)пиридина 1-оксид:
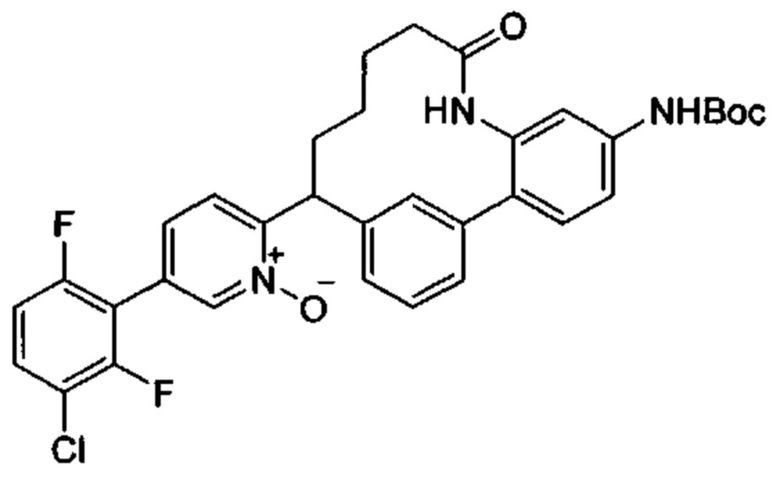
К смеси соединения примера 43С (100 мг, 0,166 ммоль) в CHCl3 (5 мл) добавляли м-СРВА (55,6 мг, 0,248 ммоль) и смесь перемешивали при 25°С в течение 2 ч. Ее гасили водным Na2SO3 (насыщенный, 5 мл) и смесь экстрагировали дихлорметан (2×50 мл). Объединенные органические фракции промывали водным карбонатом натрия (насыщенный, 2×20 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением указанного соединения. MS (ES+) m/z: 620 (М+Н). Неочищенное вещество использовали на следующей стадии без дополнительной очистки.
Пример 43:
К смеси соединения примера 43D (100 мг, 0,161 ммоль) в DCM (5 мл) при 25°С добавляли TFA (0,5 мл, 6,49 ммоль) и смесь перемешивали при 25°С в течение 1 ч. Большую часть растворителя удаляли и остаток очищали методом HPLC с обращенной фазой с получением указанного соединения. MS (ES+) m/z: 520 (М+Н).
Образец рацемического продукта подвергали хиральному разделению методом SFC (OJ, 250×30 мм I.D, 5 мкм; этанол (0,05% диэтиламин)/СО2, градиент, 60 мл/мин, 40°С) с получением соединения примера 43-а (более медленный изомер) и соединения примера 43-b (более быстрый изомер). MS (ES+) m/z: 520 (M+H); 1H ЯМР (CDCl3, 400 МГц): δ 8.49 (s, 1Н), 7.83 (m, 1H), 7.73 (d, J=8.2 Гц, 1H), 7.57-7.69 (m, 3H), 7.46 (m, 1H), 7.31 (d, J=7.7 Гц, 1H), 7.26 (dd, J=2.0, 8.2 Гц, 1H), 7.11-7.23 (m, 2H), 7.00 (d, J=7.5 Гц, 1H), 4.80 (m, 1H), 2.40 (m, 1H), 2.05-2.19 (m, 3H), 1.95 (d, J=7.3 Гц, 1H), 1.43-1.62 (m, 2H), 1.30 (d, J=9.5 Гц, 1H).
ПРИМЕР 44
(S)-5-(3-ХЛОР-2,6-ДИФТОРФЕНИЛ)-2-(24-((ЭТОКСИКАРБОНИЛ)АМИНО)-4-ОКСО-3-АЗА-1(1,3),2(1,2)-ДИБЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
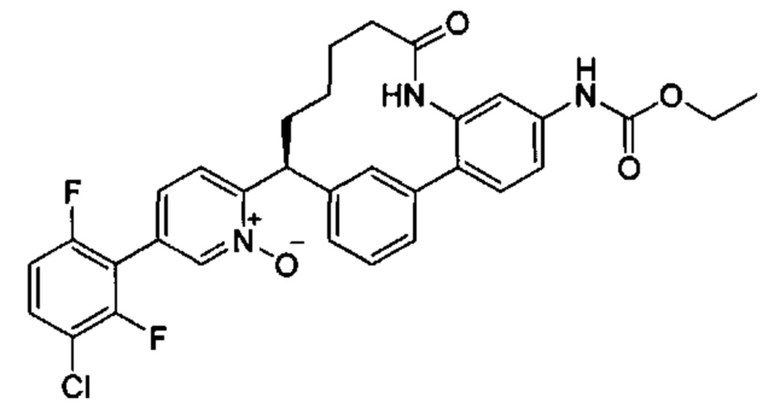
44А: трет-Бутил(S)-(9-(5-(3-хлор-2,6-дифторфенил)пиридин-2-ил)-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-24-ил)карбамат:
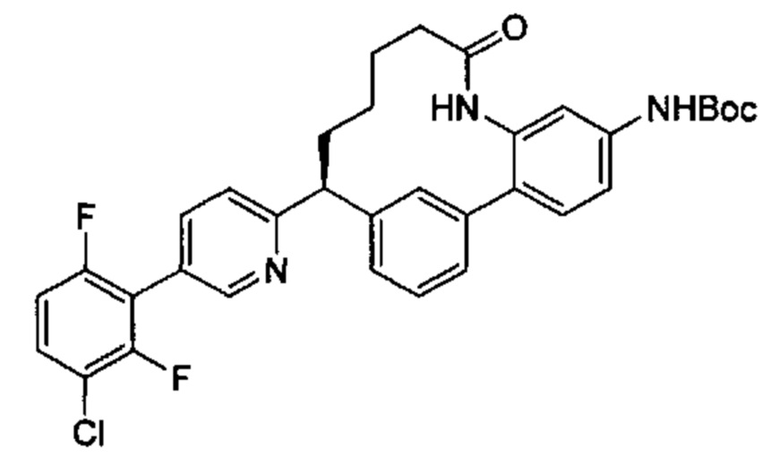
Образец соединения примера 43С подвергали хиральному разделению методом SFC (колонка: Chiralpak AD 250×30 мм I.D, 10 мкм; подвижная фаза: А: СО2, В: этанол (0,05% DEA); градиент: удержание 5% в течение 0,5 мин, затем от 5% до 50% В за 3,5 мин и удержание 50%, скорость потока: 80 мл/мин. Темп. колонки: 40°С) с получением требуемого энантиомера (более быстрое элюирование). MS (ESI) m/z 604.1 (M+H).
44В: (S)-24-амино-9-(5-(3-хлор-2.6-дифторфенил)пиридин-2-ил)-3-аза-1(1,3),2(1,2)-дибензолациклононафан-4-он
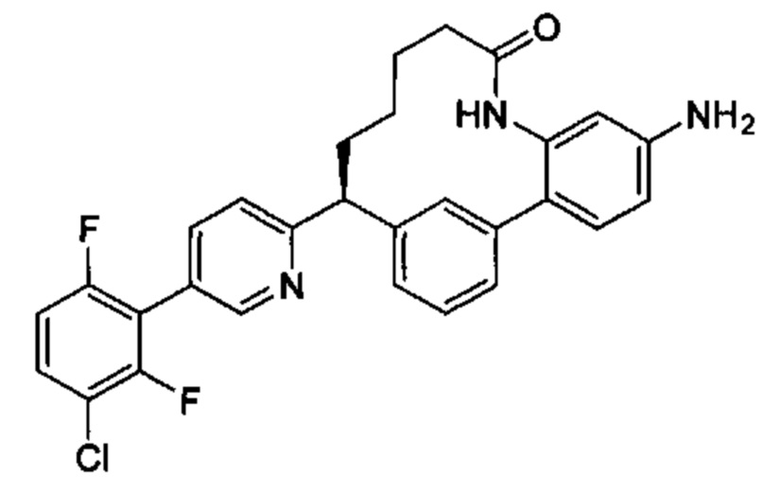
К перемешиваемому раствору соединения примера 44А (100 мг, 0,166 ммоль) в DCM (0,5 мл) добавляли TFA (0,5 мл, 6,49 ммоль) при 25°С. Смесь перемешивали в течение 2 ч и концентрировали при пониженном давлении с получением указанного соединения. MS (ESI) m/z 504.1 (М+Н).
44С: Этил(S)-(9-(5-(3-хлор-2,6-дифторфенил)пиридин-2-ил)-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-24-ил)карбамат:

К перемешиваемому раствору соединения примера 44В (85 мг, 0,169 ммоль) и DIEA (0,236 мл, 1,349 ммоль) в DCM (3 мл) добавляли этилхлорформиат (27,5 мг, 0,253 ммоль) при 25°С. Смесь перемешивали в течение 16 ч. Добавляли водный HCl (1 М, 20 мл) и смесь экстрагировали дихлорметаном (2×50 мл). Объединенные органические слои промывали насыщенным водным NaHCO3 (2×50 мл), сушили над сульфатом натрия, фильтровали. Фильтрат выпаривали при пониженном давлении с получением указанного соединения. MS (ESI) m/z 576.1 (М+Н).
Пример 44:
К перемешиваемому раствору соединения примера 44С (85 мг, 0,125 ммоль) в CHCl3 (3 мл) добавляли м-СРВА (42,2 мг, 0,188 ммоль, 77% чистоты) при 25°С. Смесь перемешивали при 25°С в течение 1 ч и концентрировали при пониженном давлении. Остаток очищали методом преп-HPLC с обращенной фазой с получением указанного соединения. 1Н ЯМР (400 МГц, CD3OD): δ 8.50 (s, 1Н), 7.84-7.78 (m, 1Н), 7.72-7.58 (m, 3Н), 7.50-7.45 (m, 3Н), 7.43-7.29 (m, 2Н), 7.19 (t, J=9.0 Гц, 1Н), 6.96 (d, J=7.3 Гц, 1Н), 4.82-4.78 (m, 1Н), 4.21-4.12 (m, 2Н), 2.49-2.35 (m, 1Н), 2.19-2.04 (m, 3Н), 2.01-1.98 (m, 1Н), 1.69-1.37 (m, 3Н), 1.29 (t, J=6.9 Гц, 3Н).
ПРИМЕР 45
(S)-2-(24-(((2-(ТРЕТ-БУТОКСИ)ЭТОКСИ)КАРБОНИЛ)АМИНО)-4-ОКСО-3-АЗА-1(1,3),2(1,2)-ДИБЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)-5-(3-ХЛОР-2,6-ДИФТОРФЕНИЛ)-ПИРИДИНА 1-ОКСИД

Указанное соединение получали из соединения примера 44В способом, описанным в синтезе соединения примера 44. Его очищали методом HPCL с обращенной фазой. 1Н ЯМР (400 МГц, CD3OD): δ 8.50 (s, 1Н), 7.82-7.59 (m, 4Н), 7.51-7.33 (m, 4Н), 7.31-7.25 (m, 1Н), 7.19 (t, J=8.6 Гц, 1Н), 6.96 (d, J=7.4 Гц, 1Н), 4.83-4.76 (m, 1H), 4.19 (t, J=4.5 Гц, 2Н), 3.62 (t, J=4.9 Гц, 2Н), 2.45-2.32 (m, 1Н), 2.18-1.95 (m, 4Н), 1.59-1.29 (m, 3Н), 1.28 (s, 9Н).
ПРИМЕР 46
(S)-5-(3-ХЛОР-2,6-ДИФТОРФЕНИЛ)-2-(24-(((2-ГИДРОКСИЭТОКСИ)КАРБОНИЛ)АМИНО)-4-ОКСО-3-АЗА-1(1,3),2(1,2)-ДИБЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
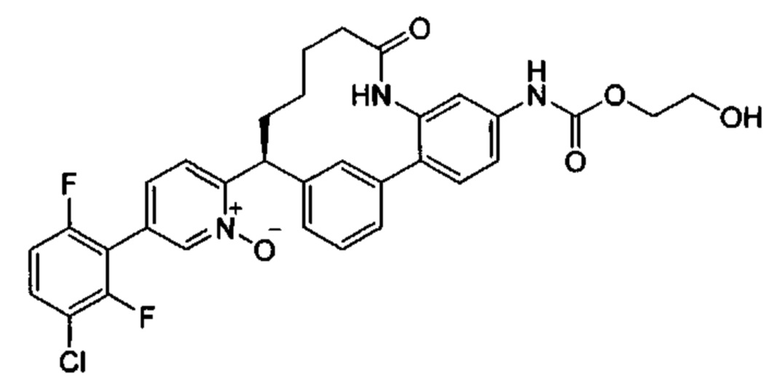
К раствору соединения примера 45 (20 мг, 0,030 ммоль) в CH2Cl2 (5 мл) добавляли TFA (1 мл, 12,97 ммоль). Смесь перемешивали при 26°С в течение 2 ч. Растворитель удаляли при пониженном давлении и остаток очищали методом HPLC с обращенной фазой с получением указанного соединения. 1Н ЯМР (400 МГц, CD3OD): δ 8.47 (s, 1Н), 7.80-7.75 (m, 1Н), 7.72-7.65 (m, 2Н), 7.63-7.57 (m, 1Н), 7.47-7.41 (m, 3Н), 7.38-7.33 (m, 1H), 7.28 (d, J=7.4 Гц, 1Н), 7.17 (t, J=8.6 Гц, 1Н), 6.95 (d, J=7.4 Гц, 1Н), 4.79-4.77 (m, 1Н), 4.19 (t, J=4.5 Гц, 2Н), 3.76 (t, J=4.7 Гц, 2Н), 2.45-2.32 (m, 1Н), 2.18-1.78 (m, 4Н), 1.59-1.49 (m, 1Н), 1.47-1.29 (m, 1Н), 1.27-1.13 (m, 1Н).
ПРИМЕР 47
5-(3-ХЛОР-2,6-ДИФТОРФЕНИЛ)-2-(4-ОКСО-24-(ПИРИДИН-3-ИЛАМИНО)-3-АЗА-1(1,3),2(1,2)-ДИБЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
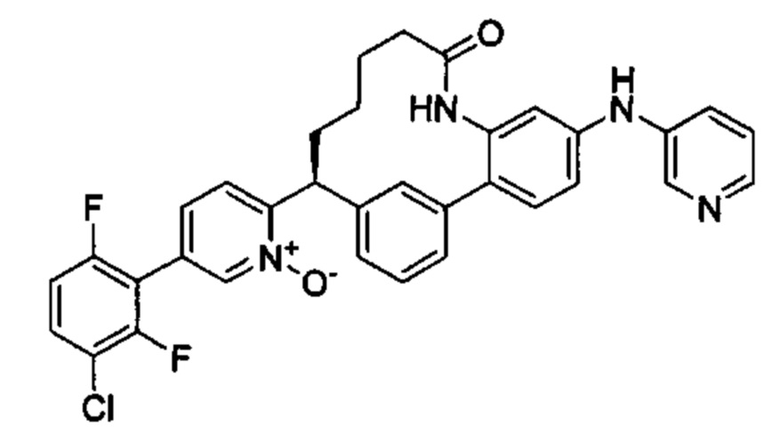
Смесь соединения примера 43 (50 мг, 0,096 ммоль), Pd2(dba)3 (1,761 мг, 1,923 мкмоль), Xantphos (2,226 мг, 3,85 мкмоль), 3-бромпиридина (22,79 мг, 0,144 ммоль) и 2-метилпропан-2-олята натрия (9,24 мг, 0,096 ммоль) в диоксане (5 мл) перемешивали при 80°С в течение 16 ч в закупоренной пробирке в атмосфере азота. Смесь фильтровали и фильтрационный кек промывали дихлорметаном (50 мл). Объединенные органические слои концентрировали, а остаток очищали методом HPLC с обращенной фазой и далее разделяли методом SFC (Chiralpak AS 250×30 мм, подвижная фаза: А: СО2; В: этанол (0,05% DEA); градиент: удержание 5% в течение 0,5 мин, затем от 5% до 40% В за 3,5 мин и удержание 40%, скорость потока: 45 мл/мин. Темп. колонки: 40°С) с получением указанного соединения (более медленное элюирование). 1Н ЯМР (400 МГц, CD3OD): δ 8.48 (s, 1Н), 8.40 (s., 1Н), 8.18-8.03 (m, 2Н), 7.80 (d, J=8.2 Гц, 2Н), 7.74-7.69 (m, 1Н), 7.67-7.59 (m, 2Н), 7.57 (d, J=8.2 Гц, 1Н), 7.42-7.38 (m, 1Н), 7.37-7.25 (m, 2Н), 7.21-7.15 (m, 2Н), 6.97 (d, J=7.4 Гц, 1H), 4.78-4.71 (m, 1Н), 2.45-2.33 (m, 1Н), 2.19-2.04 (m, 3Н), 1.99-1.90 (m, 1Н), 1.68-1.40 (m, 2Н), 1.35-1.27 (m, 1Н).
ПРИМЕР 48
(S)-5-(3-ХЛОР-2,6-ДИФТОРФЕНИЛ)-2-(4-ОКСО-24-(ПИРИМИДИН-2-ИЛАМИНО)-3-АЗА-1(1,3),2(1,2)-ДИБЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
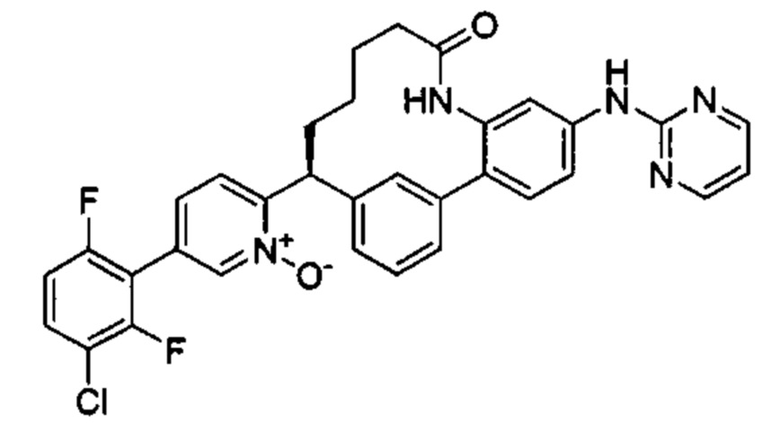
Закупоренную пробирку заполняли соединением примера 43-а (100 мг, 0,135 ммоль), трет-BuOH (1 мл), 2-хлорпиримидином (77 мг, 0,673 ммоль) и HCl/диоксаном (0,4 мл, 1,600 ммоль, 4 М). Смесь нагревали при 85°С в течение 16 ч в атмосфере азота. Ее охлаждали до к. т.и концентрировали при пониженном давлении. Остаток очищали методом HPLC с обращенной фазой с получением указанного соединения. 1Н ЯМР (400 МГц, CD3OD): δ 8.50 (s, 1Н), 8.46 (d, J=5.1 Гц, 2H), 7.83-7.78 (m, 1H), 7.74-7.58 (m, 5H), 7.48 (d, J=8.2 Гц, 1H), 7.40-7.34 (m, 1H), 7.33-7.28 (m, 1H), 7.18 (t, J=8.8 Гц, 1H), 6.95 (d, J=7A Гц, 1H), 6.85 (t, J=4.9 Гц, 1H), 4.81-4.77 (m, 1H), 2.49-2.37 (m, 1H), 2.21-2.05 (m, 3H), 2.02-1.89 (m, 1H), 1.67-1.54 (m, 1H), 1.52-1.39 (m, 1H), 1.32-1.21 (m, 1H).
ПРИМЕР 49
3-(5(3-ХЛОР-2,6-ДИФТОРФЕНИЛ)ПИРИДИН-2-ИЛ)-14-ГИДРОКСИ-11,12-DIHYDRO-9-АЗА-1(6,7)-ХИНОЛИНА-2(1,3)-БЕНЗОЛАЦИКЛОНОНАФАН-12,8-ДИОН
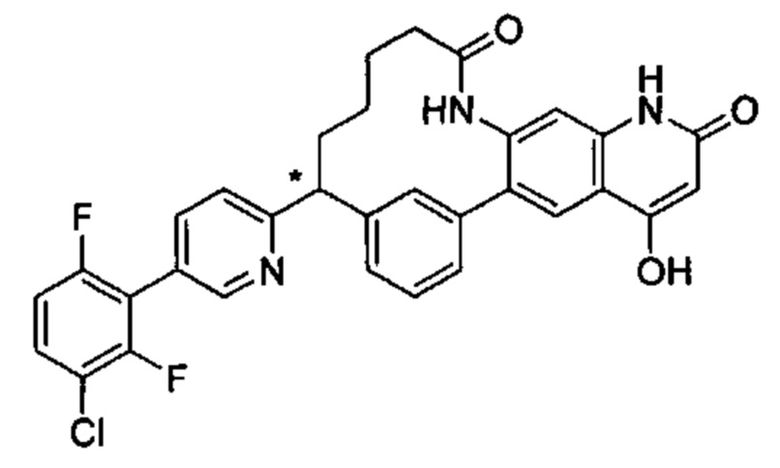
49A: Этил3-((9-(5-(3-хлор-2.6-дифторфенил)пиридин-2-ил)-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-24-ил)амино)-3-оксопропаноат:
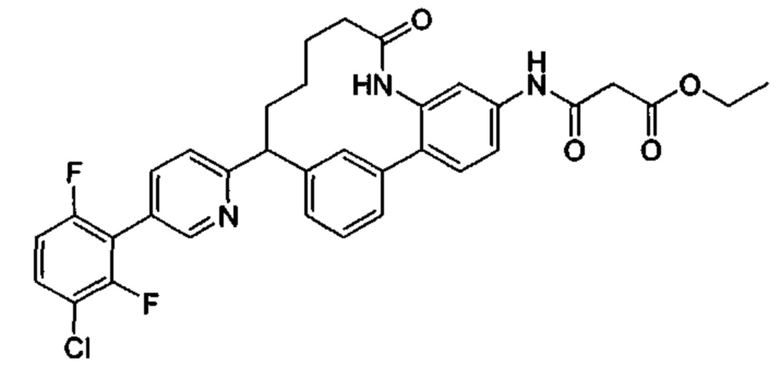
К перемешиваемой смеси соединения примера 44В (40 мг, 0,079 ммоль), N-этил-N-изопропилпропан-2-амина (41,0 мг, 0,317 ммоль) в DCM (2 мл) добавляли этил-3-хлор-3-оксопропаноат (23,90 мг, 0,159 ммоль). Смесь перемешивали при 25°С в течение 16 ч. Ее разбавляли DCM (50 мл), промывали HCl (1М, 20 мл) и водный NaHCO3 (20 мл), сушили над сульфатом натрия, фильтровали и растворитель выпаривали при пониженном давлении. Остаток очищали методом преп-TLC (петролейный эфир : EtOAc = 1:2) с получением указанного соединения. MS (ESI) m/z 618.2 (М+Н).
49В: 3-((9-(5-(3-Хлор-2,6-дифторфенил)пиридин-2-ил)-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-24-ил)амино)-3-оксопропановая кислота:
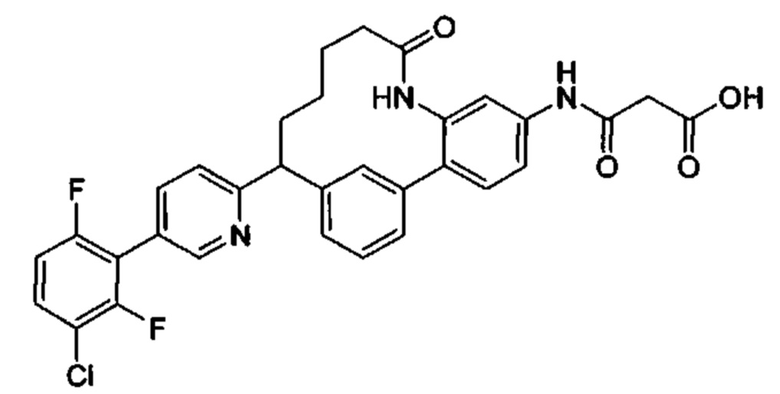
К перемешиваемой смеси соединения примера 49А (30 мг, 0,049 ммоль) в THF (2 мл) и МеОН (0,5 мл) добавляли водный гидроксид лития (0,097 мл, 0,194 ммоль, 2 М); смесь перемешивали при 25°С в течение 1 ч. HCl (1 М) добавляли для доведения значения рН до 3 и смесь экстрагировали EtOAc (20 мл ×2). Объединенные органические слои сушили над сульфатом натрия, фильтровали и растворитель выпаривали при пониженном давлении с получением указанного соединения. MS (ESI) m/z 590.2 (М+Н).
Пример 49:
Смесь соединения примера 49В (200 мг, 0,339 ммоль) в реактиве Итона (1 мл, 0,339 ммоль) перемешивали при 90°С в течение 16 ч в закупоренной пробирке. При помощи LCMS наблюдали завершение реакции. Смесь выливали в воду (10 мл). Добавляли водный NaHCO3 (20 мл) и смесь экстрагировали EtOAc (30 мл ×2). Объединенные органические фракции сушили над сульфатом натрия, фильтровали и растворитель выпаривали при пониженном давлении. Остаток очищали методом HPLC с обращенной фазой (TFA) с получением указанного соединения. MS (ESI) m/z 572.2 (М+Н); 1Н ЯМР (400МГц, DMSO-d6): δ 11.41 (s, 1Н), 9.72 (s, 1Н), 8.64 (s, 1Н), 7.88 (d, J=7.9 Гц, 1Н), 7.83 (s, 1Н), 7.77-7.68 (m, 1Н), 7.58-7.53 (m, 2Н), 7.42-7.30 (m, 3Н), 7.28 (d, J=7.5 Гц, 1Н), 7.18 (d, J=7.5 Гц, 1Н), 7.11 (s, 1Н), 5.76 (s, 1Н), 4.22 (d, J=9.5 Гц, 1H), 2.39-2.17 (m, 2Н), 2.13-1.90 (m, 2Н), 1.88-1.73 (m, 1Н), 1.54-1.49 (m, 1Н), 1.31-1.08 (m, 2Н).
ПРИМЕР 50 (рацемат), 50-а, 50-b, 50-с, 50-d
5-(3-ХЛОР-2.6-ДИФТОРФЕНИЛ)-2-(24-((МЕТОКСИКАРБОНИЛ)АМИНО)-5-МЕТИЛ-4-ОКСО-3-АЗА-1(1,3),2(1,2)-ДИБЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
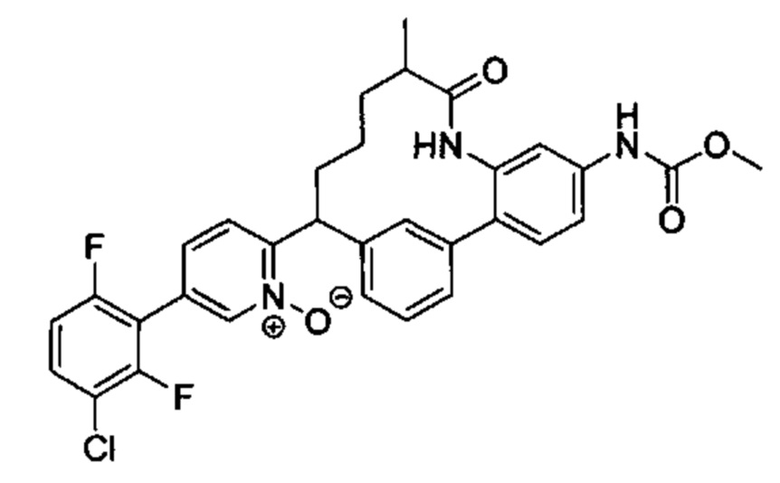
50А: (5-((3'-(5-Хлорпиколиноил)-4-((метоксикарбонил)амино)-[1,1'-бифенил]-2-ил)амино)-4-метил-5-оксопентил)трифенилфосфония бромид: К перемешиваемой смеси соединения примера 12А (6,9 г, 18,07 ммоль), (4-карбоксипентил)трифенилфосфония бромида (10,74 г, 23,49 ммоль) и основания Хунига (31,6 мл, 181 ммоль) в CH2Cl2 (90 мл) при к. т. добавляли 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфоринан-2,4,6-триоксид (50% в EtOAc) (23,00 г, 36,1 ммоль). Смесь перемешивали при к. т.всю ночь. Ее концентрировали; остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали МеОН/CH2Cl2 = 5%) с получением указанного соединения. MS (ES+) m/z: 741 (М).
50В: Метил(E)-(9-(5-хлорпиридин-2-ил)-5-метил-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-8-ен-24-ил)карбамат:
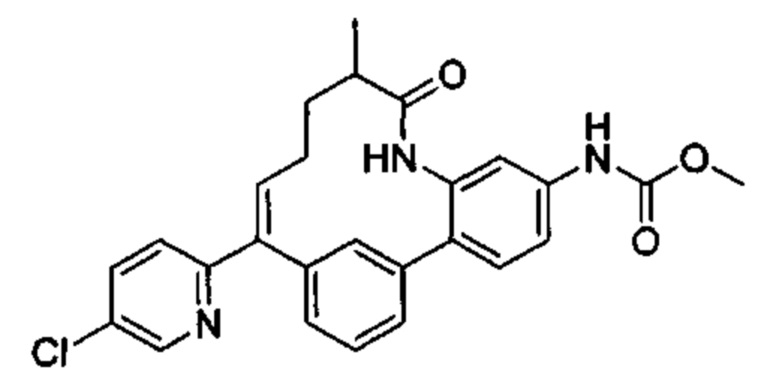
Раствор LiHMDS (1 M в THF) (67,0 мл, 67,0 ммоль) добавляли к перемешиваемому раствору соединения примера 50А (10,0 г, 12,2 ммоль) в тетрагидрофуране (609 мл) при 0°С. Смесь перемешивали при к. т. всю ночь. Ее разбавляли этилацетатом (500 мл), промывали водным хлоридом аммония (насыщенный, 150 мл), водой (150 мл), сушили (MgSCO4), фильтровали. Фильтрат концентрировали при пониженном давлении; остаток перетирали в порошок в дихлорметане (10 мл)-метаноле (5 мл). Осадок фильтровали и сушили с получением указанного соединения. MS (ES+) m/z: 462 (М+Н).
50С: Метил(E)-(9-(5-(3-хлор-2,6-дифторфенил)пиридин-2-ил)-5-метил-4-оксо-3-аза-1(1,3), 2(1,2)-дибензолациклононафан-8-ен-24-ил)карбамат:
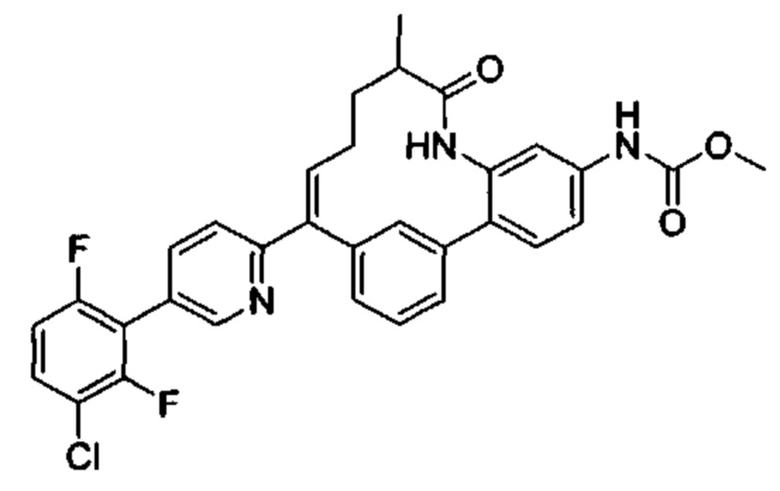
Смесь бис(пинаколято)диборона (198 мг, 0,779 ммоль), соединения примера 50В (300 мг, 0,649 ммоль), ацетата калия (191 мг, 1,948 ммоль), предварительного каталитического нейтрализатора XPHOS 2го поколения (51,1 мг, 0,065 ммоль) и диоксана (3,2 мл) в сосуде с пониженным давлением дегазировали и повторно заполняли азотом (3×).
Смесь нагревали при 80°С в течение 30 мин. Ее охлаждали до к. т.и далее добавляли 1-бром-3-хлор-2,6-дифторбензол (192 мг, 0,844 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)-ферроцен-палладий(II) дихлорида (53,2 мг, 0,065 ммоль) и водного трехосновного фосфата калия (3 М, 0,649 мл, 1,948 ммоль). Смесь дегазировали и повторно заполняли азотом (3×). Ее перемешивали при 80°С в течение 1 ч, охлаждали до к. т. и очищали методом колоночной флеш-хроматографии на силикагеле (элюировали МеОН/CH2Cl2 = 5-8%) с получением указанного соединения. MS (ES+) m/z: 574 (M+H).
50D: Метил(9-(5-(3-хлор-2,6-дифторфенил)пиридин-2-ил)-5-метил-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-24-ил)карбамат:
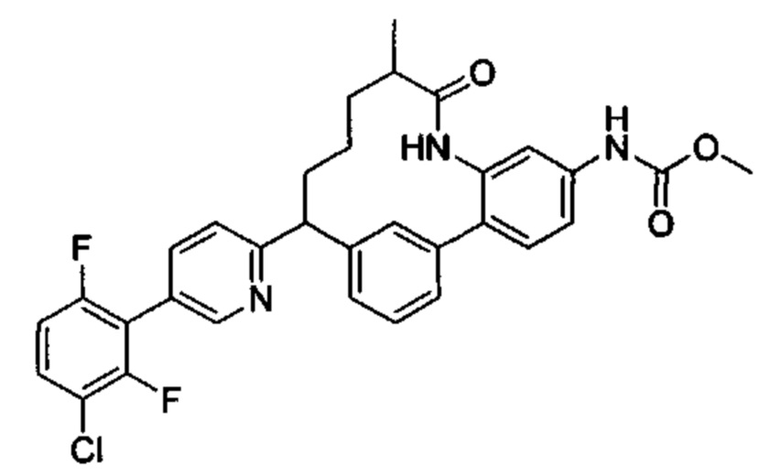
Смесь никеля Ренея (552 мг, 9,41 ммоль) и соединения примера 50С (270 мг, 0,470 ммоль) в тетрагидрофуране (20 мл) встряхивали на шейкере Парра в атмосфере водорода (Р=40 фунт/кв. дюйм) в течение 15 мин. Катализатор удаляли фильтрацией через целит (осторожно). Фильтрат концентрировали при пониженном давлении с получением указанного соединения. MS (ES+) m/z: 576 (M+H).
Пример 50, 50-a, 50-b, 50-c, 50-d:
Соединение примера 50D (165 мг, 0,286 ммоль) в дихлорметане (2,9 мл) перемешивали с м-СРВА (128 мг, 0,573 ммоль) при к. т.в течение 2 ч. Реакционную смесь очищали методом колоночной флеш-хроматографии (элюировали МеОН/CH2Cl2 = 4%) с получением указанного соединения. MS (ES+) m/z: 592 (M+H).
Образец рацемического соединения примера 50 подвергали хиральному разделению методом SFC (IC, 21×200 мм, 48% МеОН/CO2, 100 бар, 58 мл/мин, 35°С) с получением соединения примера 50-а (пик 1), соединения примера 50-b (пик 2), соединения примера 50-с (пик 3) и соединения примера 50-d (пик 4). MS (ES+) m/z: 592 (M+H).
ПРИМЕР 51
МЕТИЛ(9-(5-(5-ХЛОР-2-(1H-ТЕТРАЗОЛ-1-ИЛ)ФЕНИЛ)ПИРИДИН-2-ИЛ)15-ФТОР-4-ОКСО-3-АЗА-1(1,3),2(1,2)-ДИБЕНЗОЛАЦИКЛОНОНАФАН-24-ИЛ)КАРБАМАТ
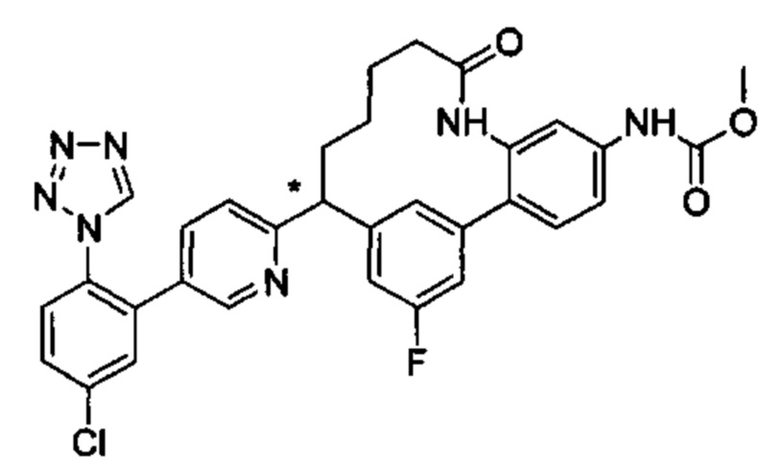
51А: Метил(2-амино-3'-(5-хлорпиколиноил)-5'-фтор-[1,1'-бифенил]-4-ил)карбамат:
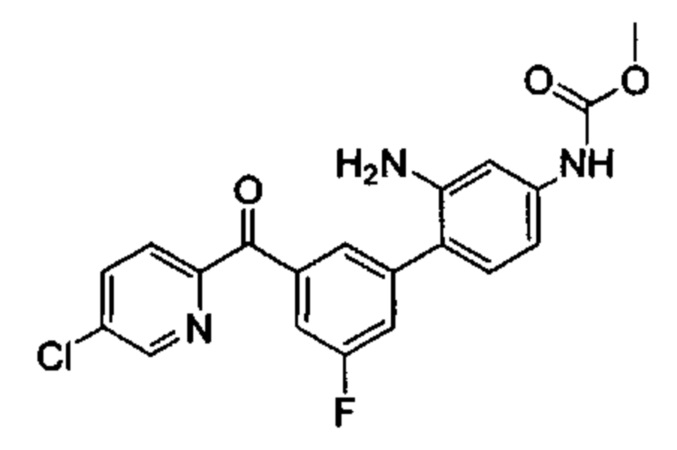
Соединение примера 51А получали реакцией сочетания Сузуки промежуточного соединения 19 и промежуточного соединения 4 способом, описанным в примере 12А. MS (ES+) m/z: 400 (М+Н).
51В: (5-((3'-(5-Хлорпиколиноил)-5'-фтор-4-((метоксикарбонил)амино)-[1,1'-бифенил]-2-ил)амино)-5-оксопентил)трифенилфосфония бромид: К раствору соединения примера 51А (6,0 г, 15,01 ммоль) в DMF (50 мл) при 20°С добавляли (4-карбоксибутил)трифенилфосфония бромид (7,98 г, 18,01 ммоль), 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфината 2,4,6-триоксид (50% масс. в этилацетате, 19,10 г, 30,0 ммоль) и DIEA (7,86 мл, 45,0 ммоль). Смесь перемешивали при 20°С в течение 2 ч и ее гасили насыщенным водным хлоридом аммония (500 мл). Смесь экстрагировали EtOAc (300 мл ×3). Объединенные органические слои промывали солевым раствором (300 мл ×5), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали DCM : МеОН = 30:1 объем/объем) с получением указанного соединения. MS (ES+) m/z: 744 (М).
51С: Метил(E)-(9-(5-хлорпиридин-2-ил)-15-фтор-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-8-ен-24-ил)карбамат:
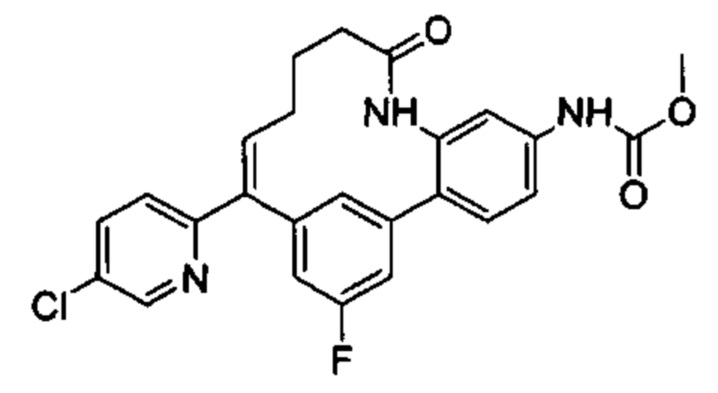
Соединение примера 51С получали из соединения примера 51В способом, описанным в синтезе соединения 12С. MS (ES+) m/z: 466 (М+Н).
51D: Метил(E)-(9-(5-(2-амино-5-хлорфенил)пиридин-2-ил)-15-фтор-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-8-ен-24-ил)карбамат:
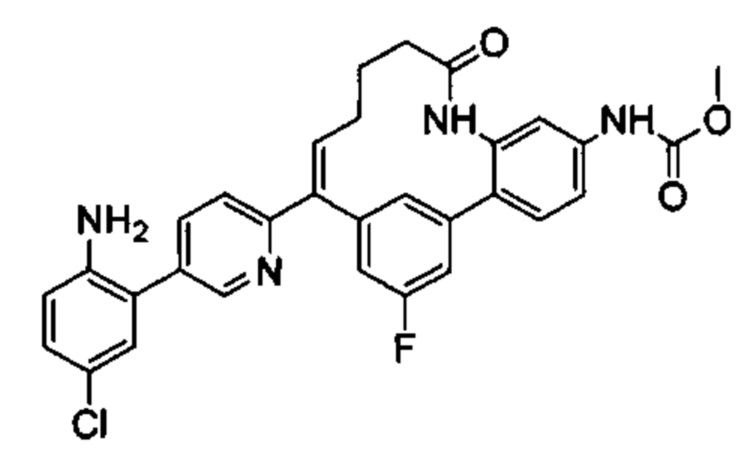
Смесь соединения примера 51С (230 мг, 0,494 ммоль), промежуточного соединения 33 (250 мг, 0,987 ммоль), K2CO3 (205 мг, 1,481 ммоль), 1,1'-бис(ди-трет-бутилфосфино)ферроцена палладий дихлорида (32,2 мг, 0,049 ммоль), THF (3 мл) и воды (1 мл) перемешивали при 120°С в атмосфере азота в течение 40 мин в микроволновом реакторе. Ее охлаждали до к. т.и реакцию гасили водой (20 мл). Смесь экстрагировали EtOAc (10 мл ×3). Объединенные органические слои промывали солевым раствором (10 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением указанного соединения. MS (ES+) m/z: 557 (М+Н).
51Е: Метил(9-(5-(2-амино-5-хлорфенил)пиридин-2-ил)-15-фтор-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-24-ил)карбамат:
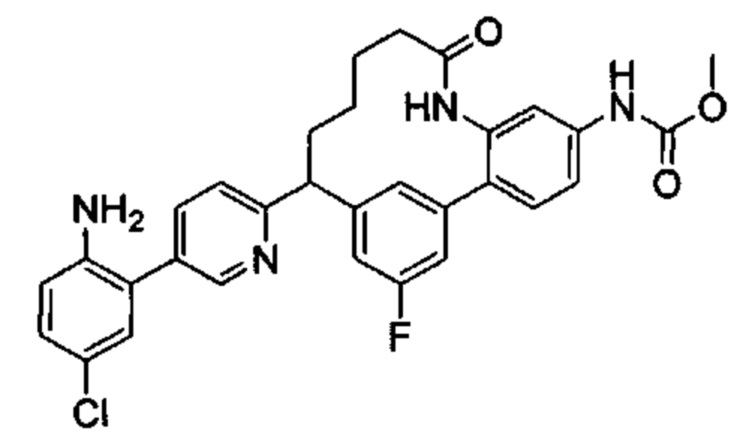
Смесь соединения примера 51D (200 мг, 0,359 ммоль) и никеля Ренея (42,1 мг, 0,718 ммоль) в THF (100 мл) перемешивали при 25°С в атмосфере водорода в течение 10 мин. Реакционную смесь фильтровали, а фильтрат концентрировали с получением указанного соединения, которое использовали на следующей стадии без дополнительной очистки. MS (ES+) m/z: 559 (М+Н).
Пример 51:
К смеси соединения примера 51Е (66 мг, 0,118 ммоль) в НОАс (5.0 мл) добавляли триметилортоформиат (251 мг, 2,361 ммоль) и NaN3 (46,1 мг, 0,708 ммоль). Смесь перемешивали при 30°С в течение 18 ч. Реакцию гасили насыщенным NaNO2 (7 мл) и значение рН доводили до 8 бикарбонатом натрия (насыщенный). Смесь экстрагировали EtOAc (20 мл ×3). Объединенные органические слои промывали солевым раствором (20 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом HPLC с обращенной фазой с получением указанного соединения. MS (ES+) m/z: 612 (М+Н); 1Н ЯМР (CD3OD, 400 МГц): δ 9.29 (s, 1Н), 8.36 (d, J=1.8 Гц, 1Н), 7.64-7.82 (m, 4Н), 7.53 (d, J=8.2 Гц, 1H), 7.39-7.50 (m, 3Н), 7.35 (s, 1Н), 7.02 (d, J=9.3 Гц, 1Н), 6.77 (d, J=9.5 Гц, 1Н), 4.21-4.33 (m, 1Н), 3.75 (s, 3Н), 2.35-2.47 (m, 1Н), 1.96 - 2.23 (m, 3Н), 1.81-1.95 (m, 1Н), 1.60 (m, 1Н), 1.39 (m, 1Н), 1.08-1.25 (m, 1Н).
ПРИМЕР 52
5-(5-ХЛОР-2-(1H-ТЕТРАЗОЛ-1-ИЛ)ФЕНИЛ)-2-(15-ФТОР-24-((МЕТОКСИКАРБОНИЛ)АМИНО)-4-ОКСО-3-АЗА-1(1,3),2(1,2)-ДИБЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
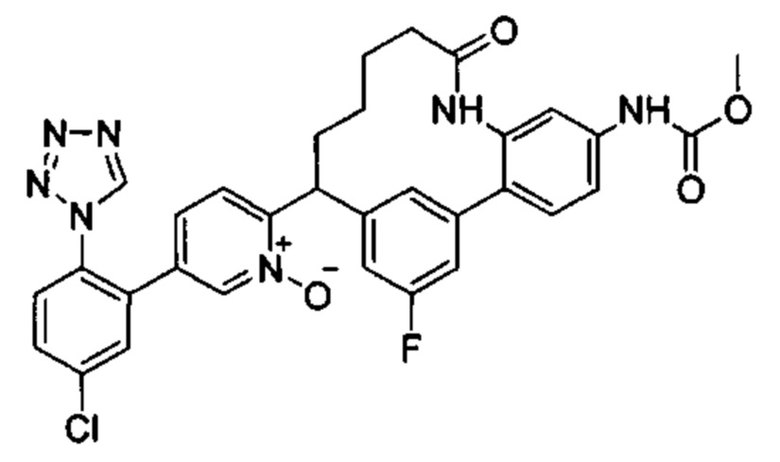
Соединение примера 52 получали из соединения примера 51 способом, описанным в примере 13. MS (ES+) m/z: 628 (М+Н); 1Н ЯМР (DMSO_d6, 400 МГц): δ 9.86 (m, 1Н), 9.65-9.71 (m, 1H), 9.63 (s, 1H), 8.20 (s, 1H), 7.90 (s, 1H), 7.83 (s, 2H), 7.56 (d, J=8.2 Гц, 1H), 7.27-7.51 (m, 4H), 7.03 (d, J=9.4 Гц, 1H), 6.97 (d, J=9.0 Гц, 1H), 6.60 (d, J=9.8 Гц, 1H), 4.56 (d, J=10.6 Гц, 1H), 3.69 (s, 3H), 2.33 (m, 1H), 1.67-2.06 (m, 4H), 1.32-1.56 (m, 1H), 1.11-1.30 (m, 1H), 0.97 (m, 1H).
Следующие соединения синтезировала способами, описанными в примере 51, с соответствующими исходными веществам. Они характеризуются методом LC/MS.

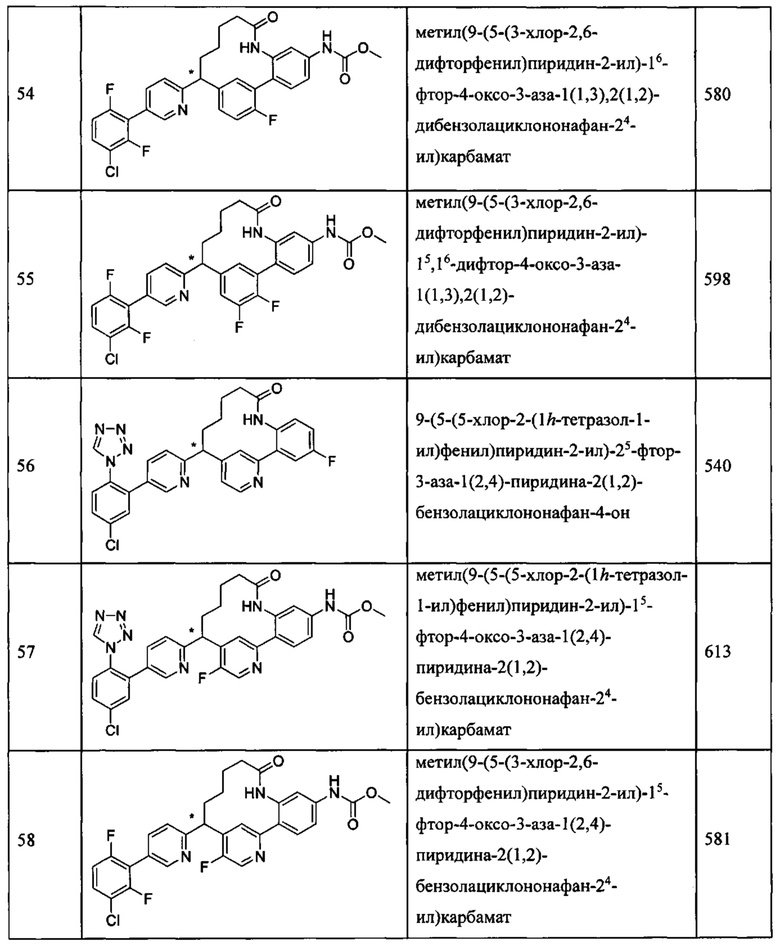
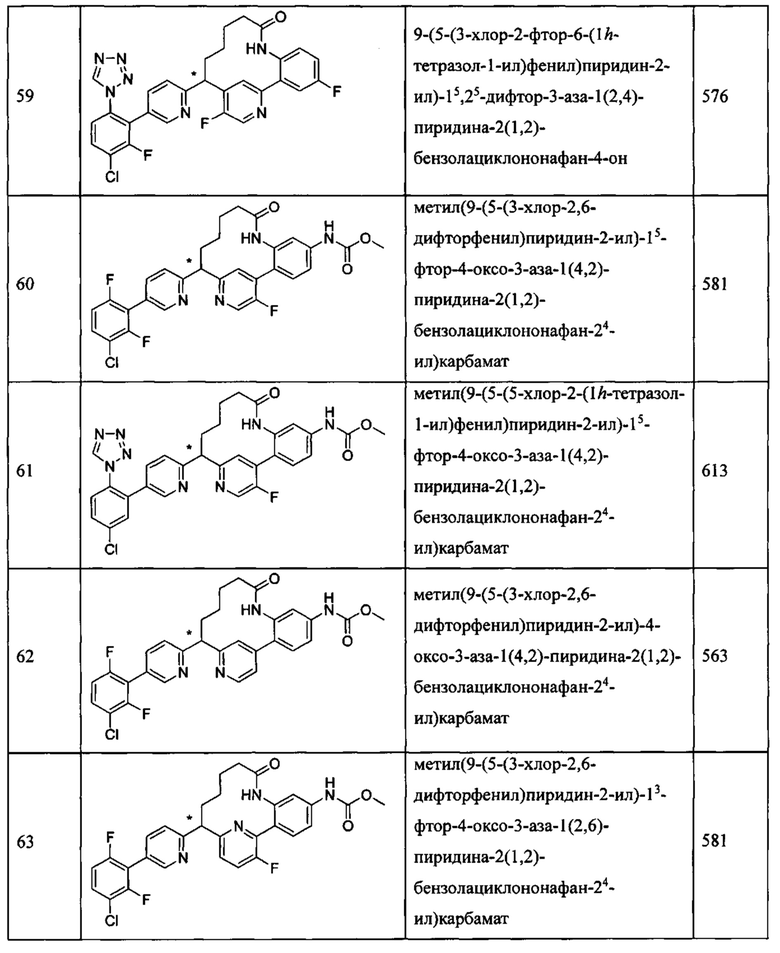
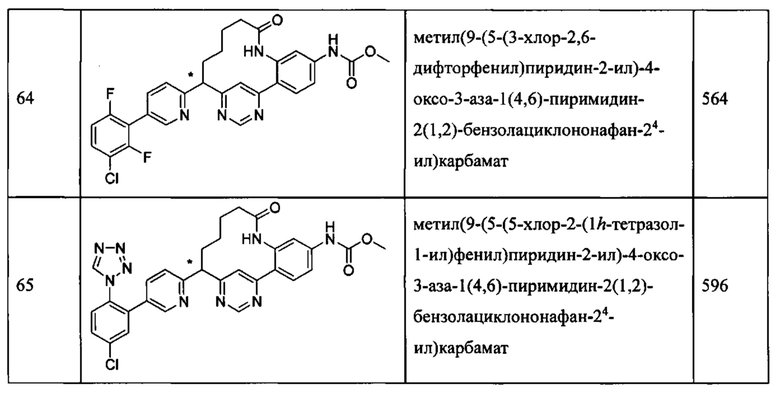
С использованием способов, описанных в примере 13, и соответствующих исходных веществ следующие соединения синтезировали и характеризовали способом LC/MS.
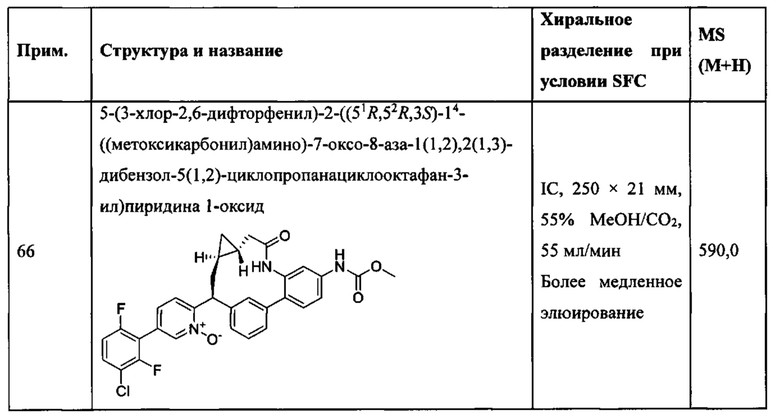
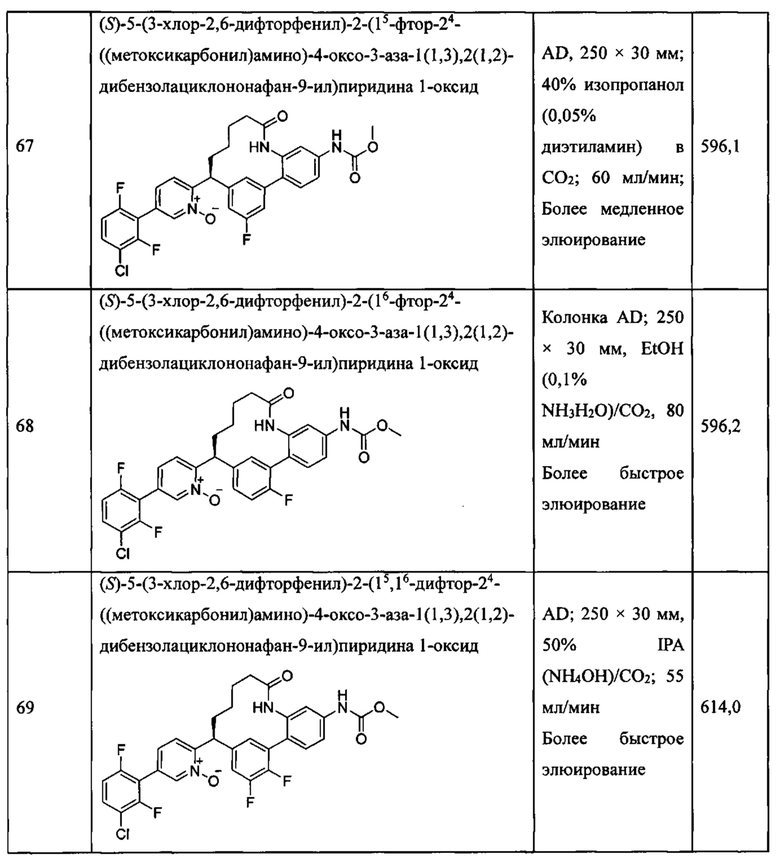
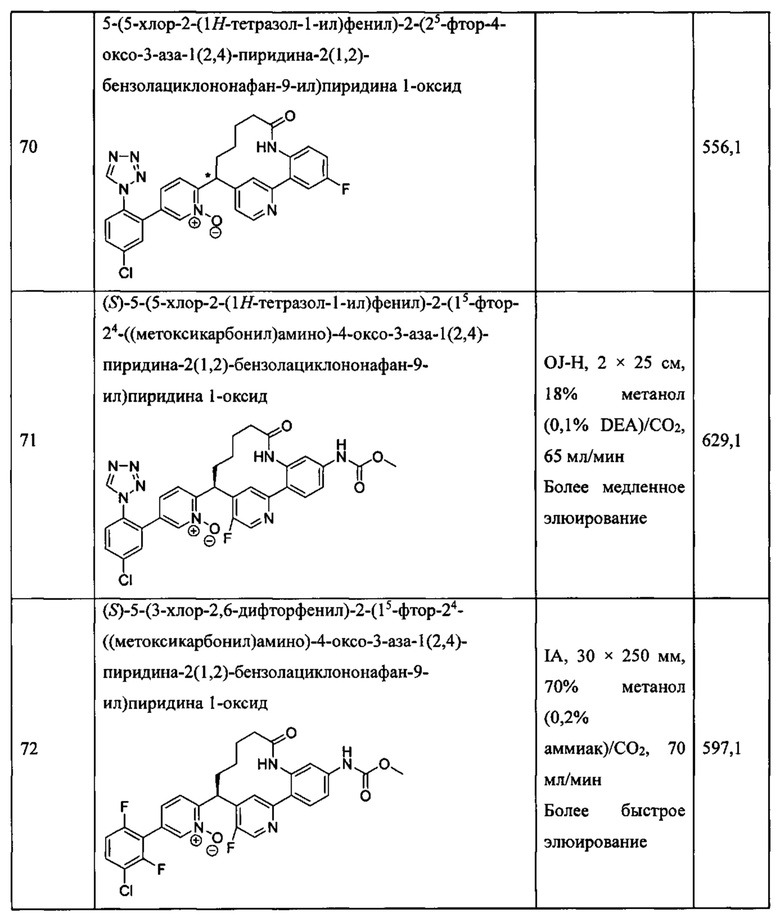
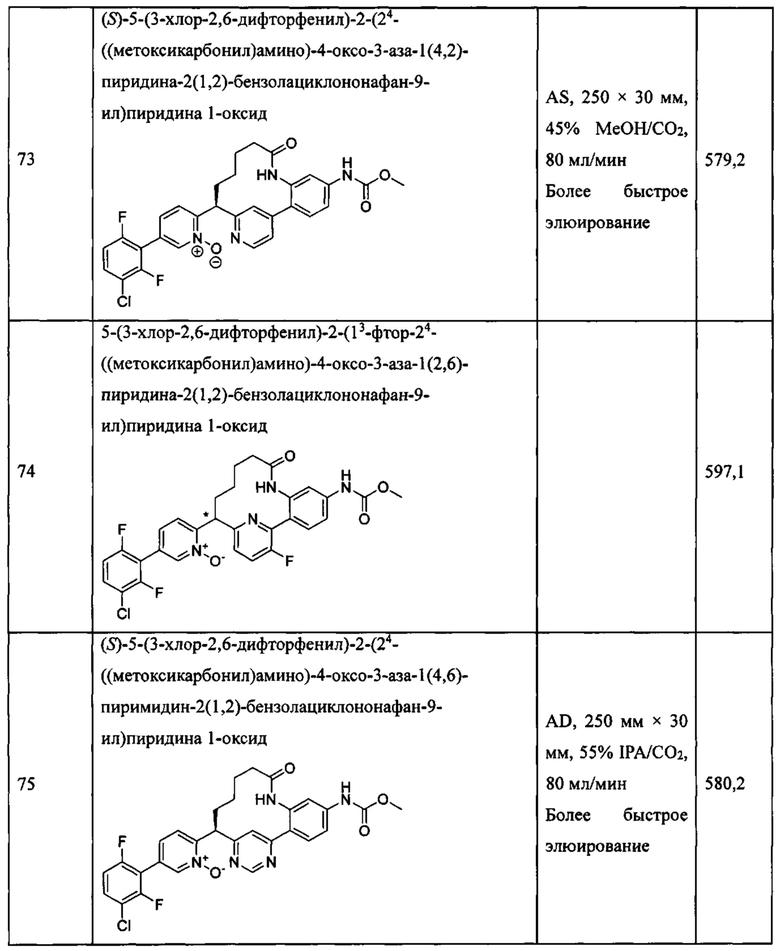
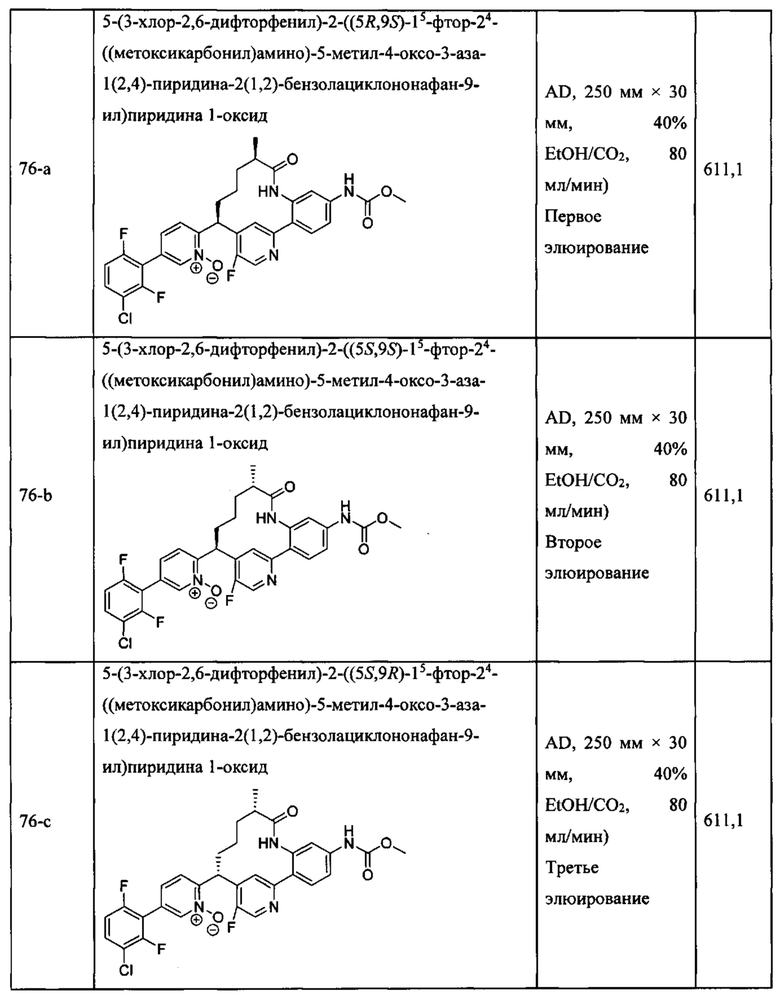
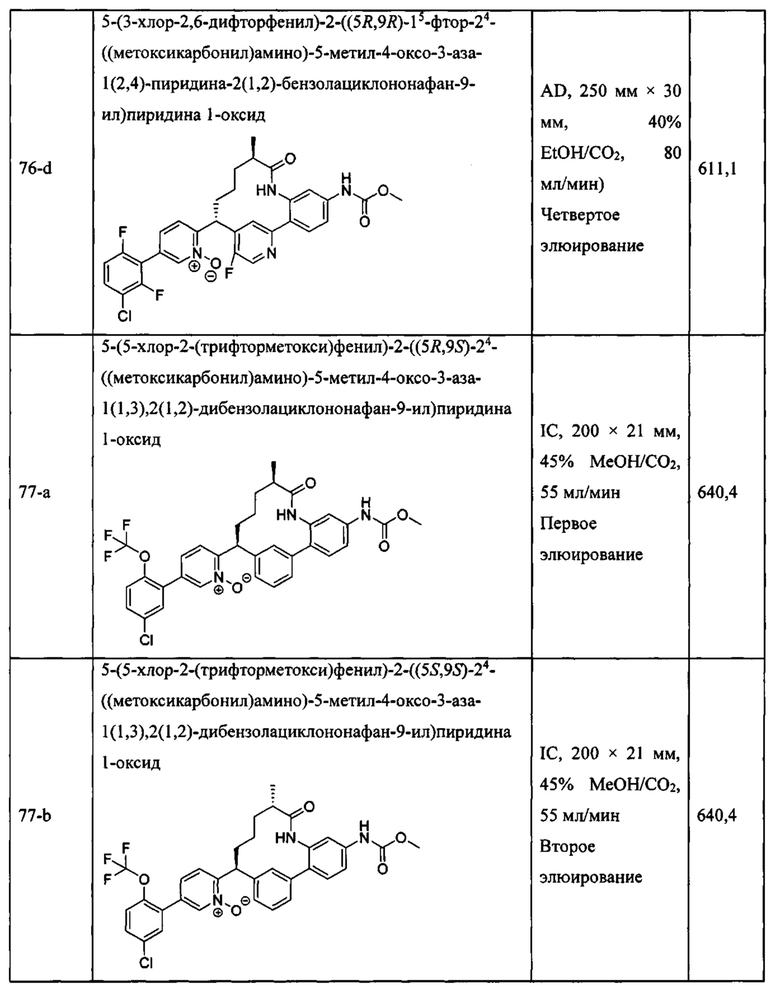
ПРИМЕР 78
2-((3S,8S)-8-КАРБОКСИ-15-ФТОР-1(1,2)2(1,3)-ДИБЕНЗОЛАЦИКЛОНОНАФАН-3-ИЛ)-5-(5-ХЛОР-2-(1Н-ТЕТРАЗОЛ-1-ИЛ)ФЕНИЛ)ПИРИДИНА 1-ОКСИД
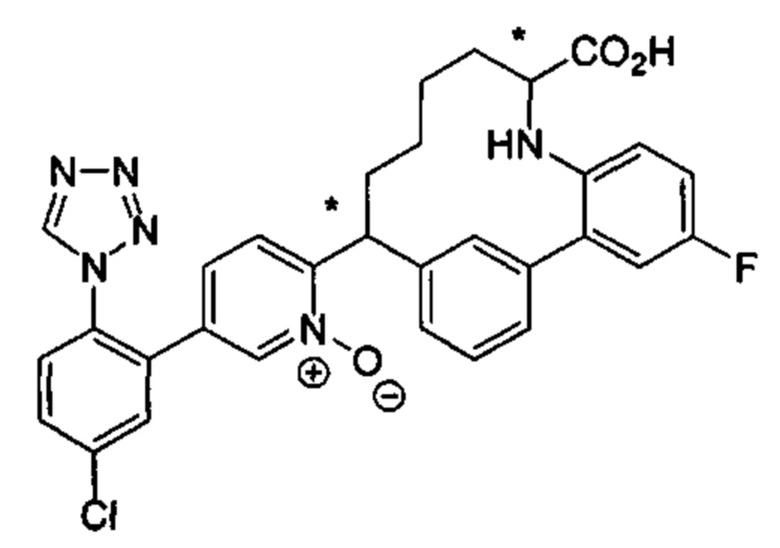
78А: Этил-2-((5-фтор-3'-(1-(5-гидроксипиридин-2-ил)бут-3-ен-1-ил)-[1,1'-бифенил]-2-ил)амино)пент-4-еноат: К раствору соединения примера 1А (5,92 г, 17,70 ммоль) и фумаровой кислоты (2,3 г, 19,83 ммоль) в ацетонитриле (89 мл) добавляли этил-2-оксоацетат (5,42 г, 26,6 ммоль) и аллилтрибутилстаннан (9,88 мл, 31,9 ммоль). Смесь перемешивали при к. т.в течение 2 ч. Ее разбавляли этилацетатом (200 мл) и промывали NaOH (0,1 М, 2×50 мл), затем солевым раствором. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-70% этилацетатом в гексане) с получением указанного соединения. MS (ES+) m/z: 461 (M+H).
78В: Этил-2-((3'-(1-(5-((трет-бутилдифенилсилил)окси)пиридин-2-ил)бут-3-ен-1-ил)-5-фтор-[1,1'-бифенил]-2-ил)амино)пент-4-еноат: К раствору соединения примера 78А (6,17 г, 13,40 ммоль) и TEA (3,73 мл, 26,8 ммоль) в DCM (67,0 мл) добавляли TBDPS-Cl (4,13 мл, 16,08 ммоль). Смесь перемешивали при к. т. всю ночь. Ее концентрировали и очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-30% этилацетатом в гексане) с получением указанного соединения. MS (ES+) m/z: 700 (M+H).
78C: Этил(E)-25-фтор-9-(5-гидроксипиридин-2-ил)-3-аза-1(1,3),2(1,2)-дибензолациклононафан-6-ен-4-карбоксилат:
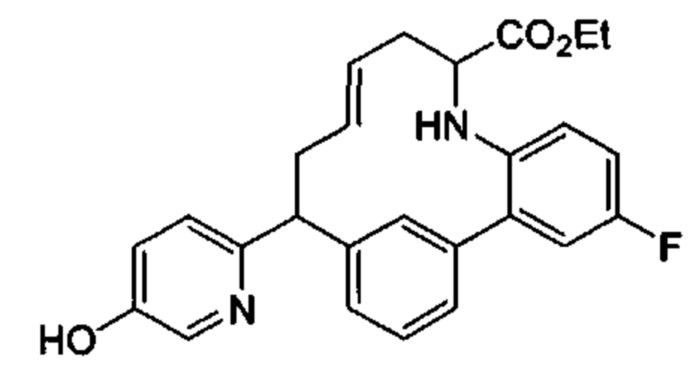
Смесь соединения примера 78В (9,36 г, 13,39 ммоль), катализатора Чжаня-1В (2,95 г, 4,02 ммоль) и пара-толуолсульфоновой кислоты моногидрата (2,1 г, 10,71 ммоль) в толуоле (700 мл) дегазировали барботированием азота в течение 15 мин. Это проводили при 50°С в течение 16 ч. Большую часть растворителя удаляли при пониженном давлении. Остаток растворяли в DCM и промывали насыщенным водным бикарбонатом натрия (30 мл) и солевым раствором. Водный слой экстрагировали DCM (2×200 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-6% МеОН в DCM) с получением указанного соединения. MS (ES+) m/z: 433 (М+Н).
78D: Этил-25-фтор-9-(5-гидроксипиридин-2-ил)-3-аза-1(,3),2(1,2)-дибензолациклононафан-4-карбоксилат:
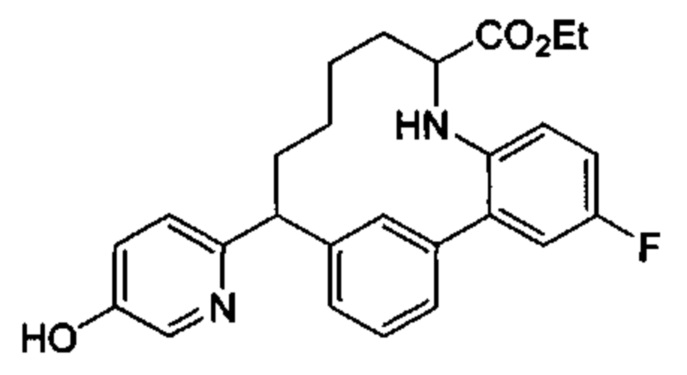
Смесь соединения примера 78С (1,25 г, 2,89 ммоль) и Pd-C (10% масс, 0,615 г, 0,578 ммоль) в МеОН (15 мл) встряхивали при к. т. в атмосфере водороде (45 фунт/кв. дюйм) в течение 16 ч. Катализатор удаляли фильтрацией через слой целита. Фильтрат концентрировали с получением указанного соединения. MS (ES+) m/z: 435 (М+Н).
78Е: Этил-25-фтор-9-(5-(((трифторметил)сульфонил)окси)пиридин-2-ил)-3-аза-1(1,3),2(1,2)-дибензолациклононафан-4-карбоксилат:
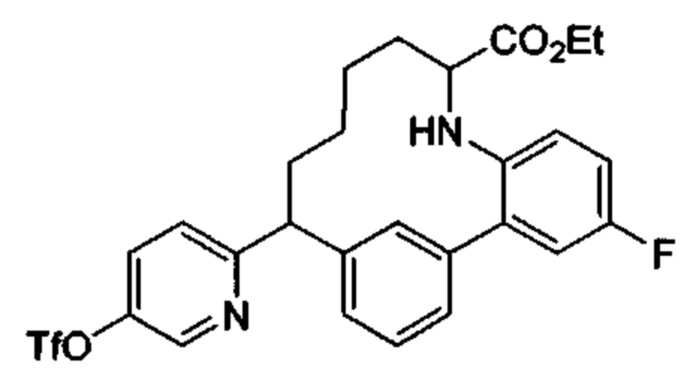
К раствору соединения примера 78D (1,16 г, 2,67 ммоль) в DCM (12 мл) добавляли TEA (1,2 мл, 8,01 ммоль) и Tf2O (3,2 мл, 3,20 ммоль) при 0°С. Смесь перемешивали в течение 0,5 ч. Ее очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-40% этилацетатом в гексане) с получением указанного соединения. MS (ES+) m/z: 567 (М+Н).
78F: Этил-9-(5-(2-амино-5-хлорфенил)пиридин-2-ил)-25-фтор-3-аза-1(1,3),2(1,2)-дибензолациклононафан-4-карбоксилат:
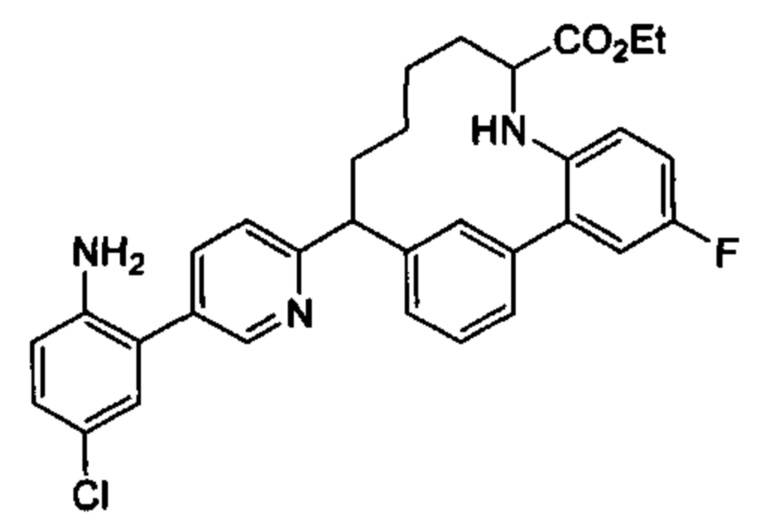
Соединение примера 78Е (1,92 г, 3,39 ммоль), промежуточное соединение 33 (1,117 г, 4,41 ммоль), Pd-Xphos-предварительный каталитический нейтрализатор (0,280 г, 0,339 ммоль) и дегазированный диоксан (13,6 мл) добавляли в колбу с магнитной мешалкой. К смеси добавляли дегазированный водный раствор фосфата калия (3 М, 3,4 мл, 10,2 ммоль). Колбу перемешивали при 80°С в атмосфере азота в течение 3 ч. Ее очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-3% МеОН в DCM) с получением указанного соединения. MS (ES+) m/z: 544 (М+Н).
78G: Этил-9-(5-(5-хлор-2-(2,2,2-трифторацетамидо)фенил)пиридин-2-ил)-25-фтор-3-(2,2,2-трифторацетил)-3-аза-1(1,3),2(1,2)-дибензолациклононафан-4-карбоксилат:
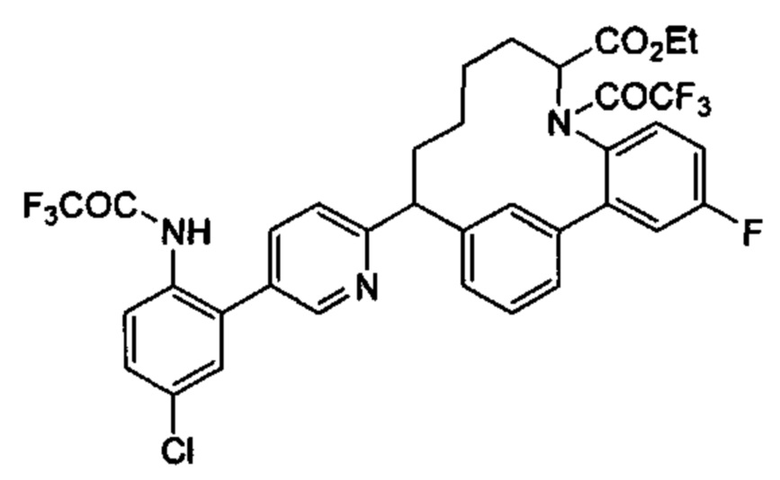
К раствору соединения примера 78F (1,79 г, 3,29 ммоль) в DCM (16,45 мл) и пиридина (1,8 г, 23,03 ммоль) при 0°С добавляли 2,2,2-трифторуксусный ангидрид (2,3 мл, 16,45 ммоль). Его оставляли нагреваться до к. т. и перемешивали всю ночь. Реакционную смесь промывали насыщенным водным бикарбонатом натрия и солевым раствором. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-70% этилацетатом в гексане) с получением указанного соединения. MS (ES+) m/z: 736 (М+Н).
78Н: 5-(5-Хлор-2-(2,2,2-трифторацетамидо)фенил)-2-(4-(этоксикарбонил)-25-фтор-3(2,2,2-трифторацетил)-3-аза-1(1,3),2(1,2)-дибензолациклононафан-9-ил)пиридина 1-оксид:
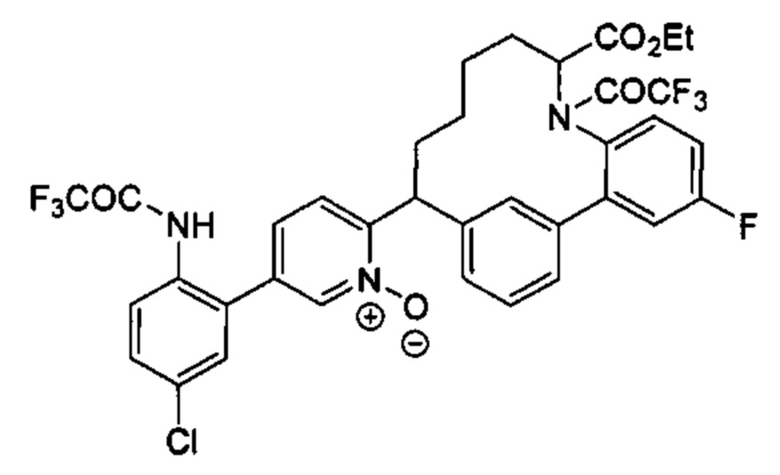
К раствору соединения примера 78G (1,69 г, 2,296 ммоль) в DCM (20 мл) добавляли м-СРВА (1,03 г, 4,59 ммоль). Смесь перемешивали при к. т. в течение 2 ч. Ее очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-7% метанолом в DCM) с получением указанного соединения. MS (ES+) m/z: 752 (М+Н).
78I: 5-(2-Амино-5-хлорфенил)-2-(4-карбокси-25-фтор-3-аза-1(1,3),2(1,2)-дибензолациклононафан-9-ил)пиридина 1-оксид:
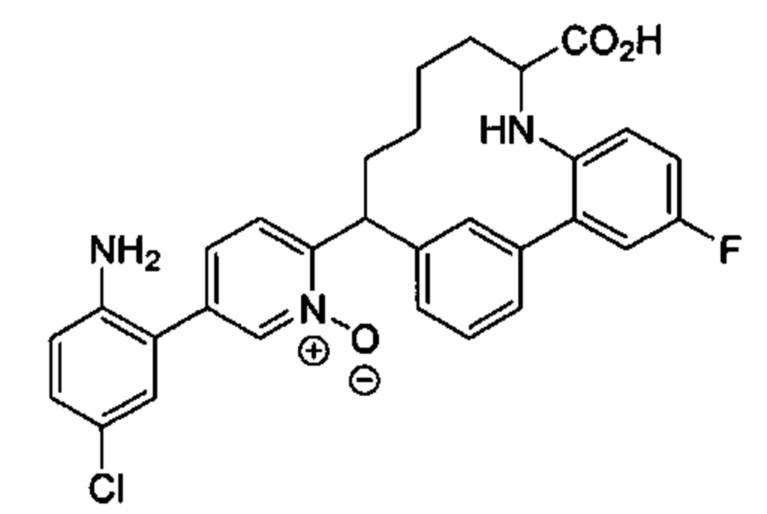
К раствору соединения примера 78Н (1,7 г, 2,260 ммоль) в МеОН (7,5 мл) и THF (7,5 мл) добавляли водный раствор LiOH (1 М, 7,5 мл, 37,5 ммоль). Полученную смесь нагревали при 80°С в закупоренной пробирке в течение 3 ч. Ее подкисляли 4 М HCl до значения рН=5. Смесь дважды экстрагировали CHCl3/IPA (4:1). Объединенные органические слои промывали водой, сушили над сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении. Остаток перетирали в порошок в 10% МеОН в DCM (5 мл) и фильтровали с получением указанного соединения в виде твердого вещества (соотношение 9:1 двух диастереомеров). MS (ES+) m/z: 532 (М+Н).
Пример 78:
К раствору соединения примера 78I (70 мг, 0,132 ммоль) и азида натрия (51,3 мг, 0,789 ммоль) в уксусной кислоте (1,3 мл) добавляли триметилортоформиат (54 мкл, 0,49 ммоль). Смесь перемешивали при 80°С в течение 5 ч и оставляли охлаждаться до к. т всю ночь. Ее разбавляли 3 мл MeCN и подвергали очистке методом HPLC с обращенной фазой (Shimadzu, Sunfire 30×100 мм, 40-50% MeCN в воде с 0,05% TFA, 50 мл/мин, 10 мин градиент). Фракцию требуемого продукта собирали и очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-10% метанолом в DCM) с получением указанного соединения. MS (ES+) m/z: 585 (М+Н).
ПРИМЕР 79
5-(5-ХЛОР-2-(1H-ТЕТРАЗОЛ-1-ИЛ)ФЕНИЛ)-2-(25-ФТОР-4-(ПИРРОЛИДИН-1-КАРБОНИЛ)-3-АЗА-1(1,3),2(1,2)-ДИБЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
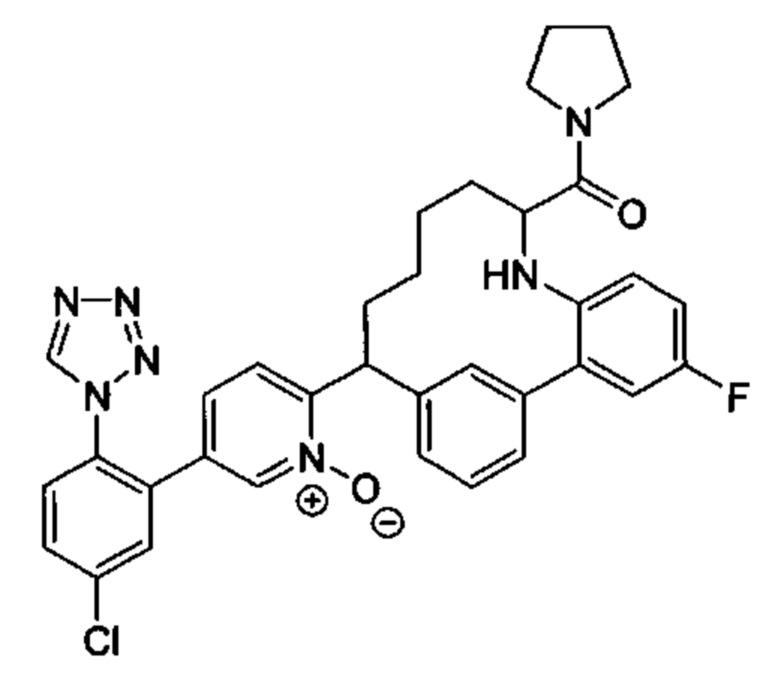
79А: 5-(2-Амино-5-хлорфенил)-2-(25-фтор-4-(пирролидин-1-карбонил)-3-аза-1(1,3),2(1,2)-дибензолациклононафан-9-ил)пиридина 1-оксид:
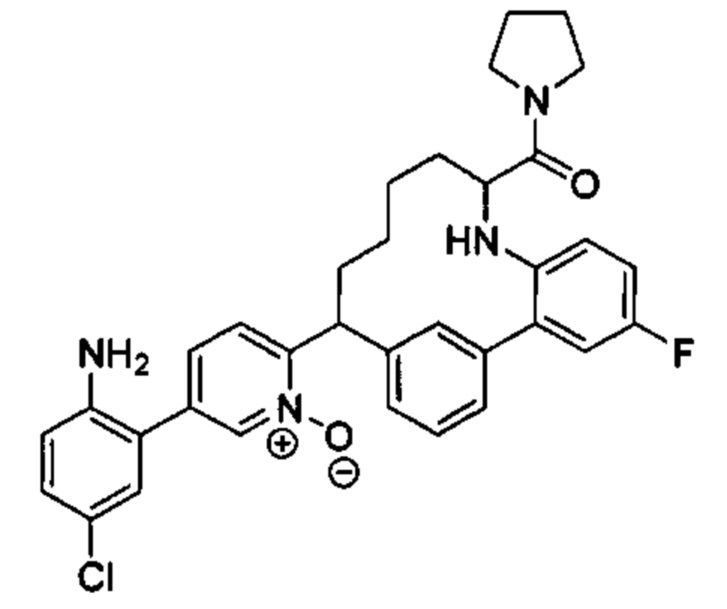
К раствору соединения примера 78I (250 мг, 0,470 ммоль), пирролидина (0,12 мл, 1,451 ммоль) и DIEA (0,16 мл, 0,916 ммоль) в DMF (4 мл) добавляли HATU (214 мг, 0,564 ммоль). Его перемешивали при к. т.в течение 4 ч. Большую часть DMF удаляли при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-5% МеОН в DCM) с получением указанного соединения. MS (ES+) m/z: 585 (М+Н).
Пример 79/79-a/79-b/79-c/79-d:
К смеси соединения примера 79А (208 мг, 0,355 ммоль) добавляли азид натрия (116 мг, 1,777 ммоль) в уксусной кислоте (3,5 мл) TFA (83 мкл, 1,077 ммоль) и триметилортоформиат (0,23 мл, 2,102 ммоль). Ее перемешивали при к. т. всю ночь. При помощи LC-MS наблюдали чистую реакцию. Добавляли TEA (1 мл) и загружали в силикагелевый дозатор, сушили под вакуумом и очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-5% метанолом в DCM) с получением требуемого продукта. MS (ES+) m/z: 638 (М+Н).
Образец рацемического продукта подвергали хиральному разделению методом SFC (OJ, 30×250 мм, 35% МеОН (0,2 NH4OH)/CO2, 70 мл/мин, 100 бар, 35°С) с получением соединения примера 79-а (пик 1), соединения примера 79-b (пик 2), соединения примера 79-с (пик 3) и соединения примера 79-d (пик 4).
ПРИМЕР 80
9-(5-(5-ХЛОР-2-(1H-ТЕТРАЗОЛ-1-ИЛ)ФЕНИЛ)-1-ОКСИДОПИРИДИН-2-ИЛ)-25-ФТОР-4-ОКСО-3-АЗА-1(2,4)-ПИРИДИН-1-ИЯ-2(1,2)-БЕНЗОЛАЦИКЛОНОНАФАН 11-ОКСИД
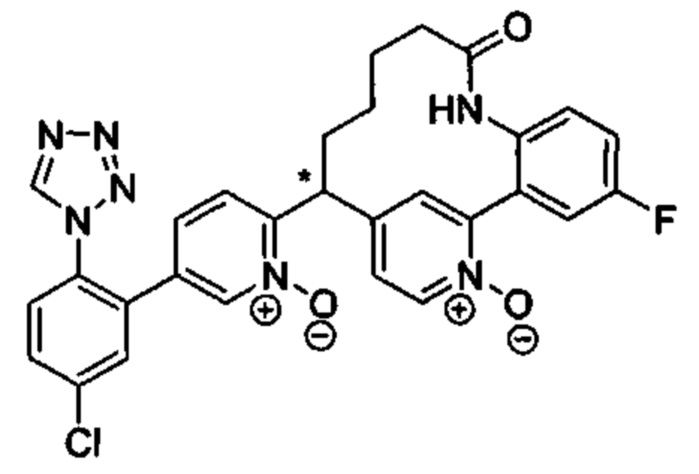
Соединение примера 80 выделяли из соединения примера 70 методом HPLC с обращенной фазой. Фракции, содержащие указанное соединение, концентрировали, а остаток повторно методом колоночной флеш-хроматографии на силикагеле (элюировали 0-10% 7 н аммиаком в метаноле/DCM) с получением указанного соединения. MS (ES+) m/z: 572 (М+Н); 1H ЯМР (500 МГц, DMSO-d6): δ 9.67 (s, 1Н), 9.66 (s, 1H), 8.24 (d, 1H), 8.21 (d, 1H), 7.87 (d, 1H), 7.83 (m, 2H), 7.68 (dd, 1H), 7.52 (d, 1H), 7.28-7.35 (m, 3H), 6.96 (m, 2H), 4.42 (m, 1H), 3.15 (d, 1H), 2.20 (m, 1H), 1.79-2.00 (m, 3H), 1.65 (m, 1H), 1.43 (m, 1H), 1.25 (m, 1H).
ПРИМЕР 81 (рацемат), 81-а и 81-b
9-(5-(5-ХЛОР-2-(1H-ТЕТРАЗОЛ-1-ИЛ)ФЕНИЛ)-1-ОКСИДОПИРИДИН-2-ИЛ)-15-ФТОР-24-((МЕТОКСИКАРБОНИЛ)АМИНО)-4-ОКСО-3-АЗА-1(2,4)-ПИРИДИН-1-ИЯ-2(1,2)-БЕНЗОЛАЦИКЛОНОНАФАН 11-ОКСИД
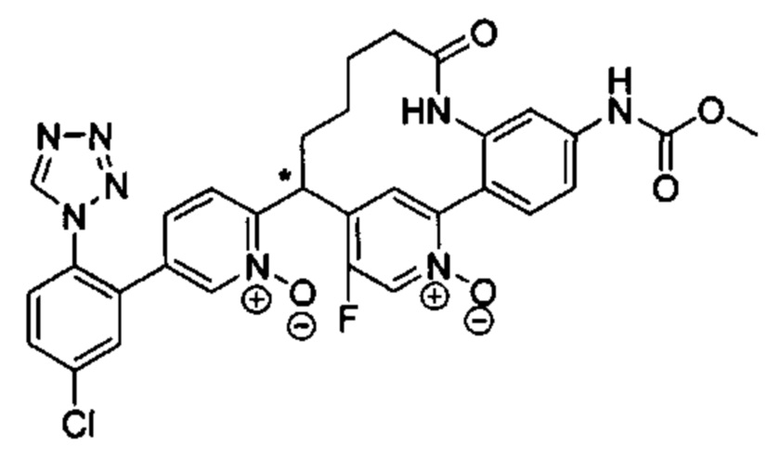
Соединение примера 81 выделяли в форме соединения примера 71 методом HPLC с обращенной фазой (Sunfire 30×100 мм, 20-50% MeCN в воде с 0,05% TFA, 50 мл/мин, 10 мин). MS (ES+) m/z: 645 (М+Н).
Образец рацемического соединения примера 81 подвергали хиральному разделению методом SFC (AD-H, 2×25 см, 35% этанол/СО2, 100 бар, 60 мл/мин, 35°С) с получением соединения примера 81-а (более медленное элюирование) и соединения примера 81-b (более быстрое элюирование). MS (ES+) m/z: 645 (М+Н); 1Н ЯМР (500 МГц, DMSO-d6): δ 9.94 (s, 1Н), 9.74 (s, 1H), 9.66 (s, 1H), 8.54 (d, 1H), 8.24 (s, 1H), 7.90 (s, 1H), 7.83 (t, 2H), 7.75 (d, 1H), 7.35-7.43 (m, 4H), 6.91 (d, 1H), 4.50 (d, 1H), 3.69 (s, 3H), 2.27 (s, 1H), 1.96 (m, 3H), 1.67 (m, 1H), 1.51 (m, 1H), 1.31 (m, 2H).
ПРИМЕР 82
9-(5-(3-ХЛОР-2,6-ДИФТОРФЕНИЛ)-1-ОКСИДОПИРИДИН-2-ИЛ)-15-ФТОР-24-((МЕТОКСИКАРБОНИЛ)АМИНО)-4-ОКСО-3-АЗА-1(2,4)-ПИРИДИН-1-ИЯ-2(1,2)-БЕНЗОЛАЦИКЛОНОНАФАН 11-ОКСИД
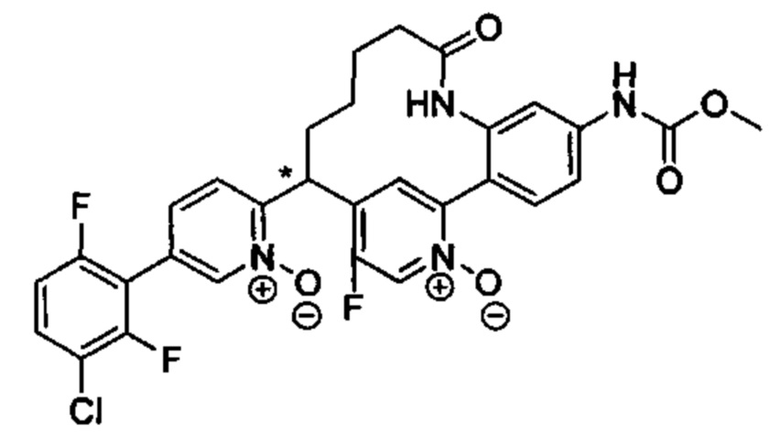
Соединение примера 82 выделяли из соединения примера 72 методом HPLC с обращенной фазой (Sunfire 30×100 мм, 20-50% MeCN в воде с 0,05% TFA, 50 мл/мин, 10 мин). MS (ES+) m/z: 613 (М+Н).
ПРИМЕР 83
9-(5-(3-ХЛОР-2,6-ДИФТОРФЕНИЛ)-1-ОКСИДОПИРИДИН-2-ИЛ)-24-((МЕТОКСИКАРБОНИЛ)АМИНО)-4-ОКСО-3-АЗА-1(4,2)-ПИРИДИН-1-ИЯ-2(1,2)-БЕНЗОЛАЦИКЛОНОНАФАН 11-ОКСИД
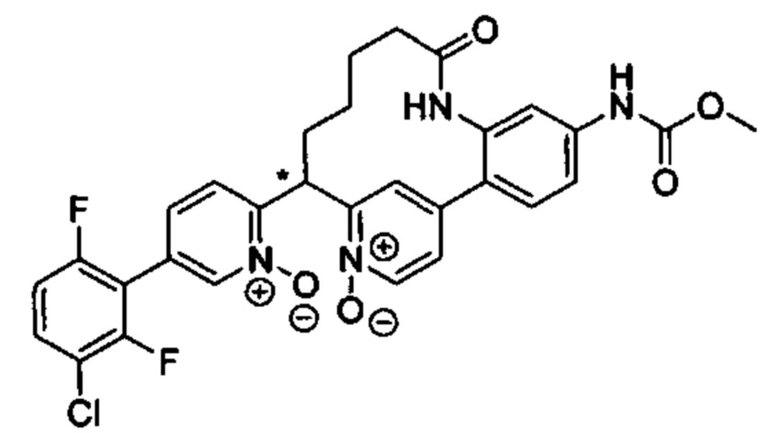
Соединение примера 83 выделяли из соединения примера 73 методом HPLC с обращенной фазой (YMC-Actus Pro С18, 150×30 мм, 20-50% MeCN в воде с 0,5% TFA, 40 мл/мин). MS (ESI) m/z 595.2 (M+H); 1H ЯМР (400МГц, DMSO-d6): δ 9.94 (s, 1H), 9.80 (s, 1H), 8.48 (s, 1H), 8.19 (d, J=6.5 Гц, 1H), 7.82-7.70 (m, 2H), 7.60-7.48 (m, 2H), 7.47-7.22 (m, 5H), 4.84 (d, J=9.6 Гц, 1H), 3.67 (s, 3H), 2.35-2.06 (m, 4H), 1.93-1.78 (m, 1H), 1.59-1.45 (m, 1H), 1.28-1.19 (m, 2H).
ПРИМЕР 84
5-(5-ХЛОР-2-(1H-ТЕТРАЗОЛ-1-ИЛ)ФЕНИЛ)-2-(24-ЦИАНО-5-МЕТИЛ-4-ОКСО-3-АЗА-1(1,3),2(1,2)-ДИБЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
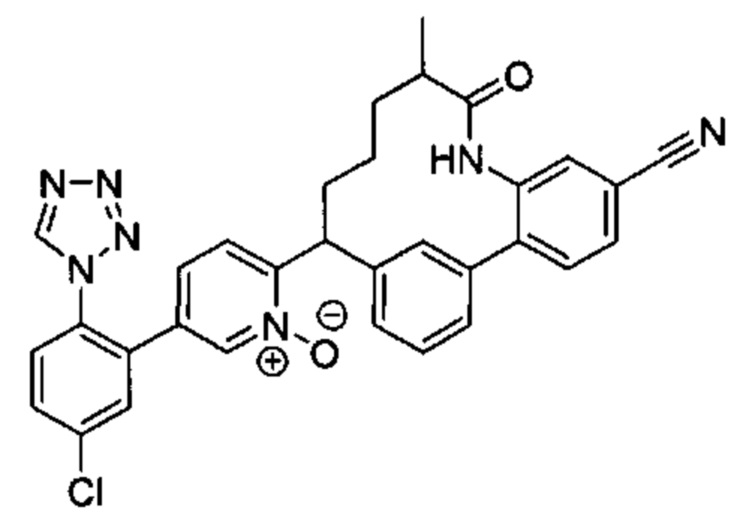
84А: 3'-(5-Хлорпиколиноил)-2-нитро-[1,1'-бифенил]-4-карбонитрил: Смесь (5-хлорпиридин-2-ил)(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)метанона (промежуточное соединение 3) (5,00 г, 14,55 ммоль), 4-бром-3-нитробензонитрила (3,63 г, 16,01 ммоль), Pd(dtbpf)Cl2 (0,95 г, 1,46 ммоль), водного раствора K3PO4 (29,10 мл, 29,10 ммоль, 1 М) и толуола (100 мл) в атмосфере азота перемешивали при 80°С в течение 12 ч. Смесь охлаждали до к. т. и разбавляли водой (80 мл). Ее экстрагировали EtOAc (150 мл ×3). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии (петролейный эфир : EtOAc = 10:1-2:1) с получением указанного соединения. MS (ESI) m/z 364.0 (M+H); 1H ЯМР (400 МГц, CDCl3): δ 8.69 (d, J=1.8 Гц, 1H), 8.18-8.25 (m, 2H), 8.08-8.12 (m, 2H), 7.88-7.97 (m, 2H), 7.67 (d, J=7.9 Гц, 1H), 7.62 (d, J=7.7 Гц, 1H), 7.54-7.59 (m, 1H).
84В: 2-Амино-3'-(5-хлорпиколиноил)-(1,1'-бифенил]-4-карбонитрил: Смесь соединения примера 84А (1,00 г, 2,75 ммоль), железа (1,54 г, 27,50 ммоль) и хлорида аммония (1,47 г, 27,50 ммоль) в EtOH : Н2О=3:1 (20 мл) перемешивали при 80°С в атмосфере азота в течение 1 ч. Ее охлаждали до к. т. и концентрировали при пониженном давлении. Остаток разбавляли водой (20 мл) и экстрагировали смесью DCM/MeOH (10/1, 30 мл ×4). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (петролейный эфир : этилацетат = 3:1) с получением указанного соединения. MS (ESI) m/z 334.1 (M+H); 1H ЯМР (400 МГц, CDCl3): δ 8.66 (d, J=2.0 Гц, 1H), 8.06-8.18 (m, 3H), 7.92 (dd, J=8.2, 2.4 Гц, 1H), 7.58-7.72 (m, 2H), 7.22 (d, J=7.8 Гц, 1H), 7.10 (dd, J=7.8, 0.8 Гц, 1H), 7.02 (s, 1H), 4.04 (brs, 2H).
Пример 84:
Соединение примера 84 получали из соединения примера 84В способом, описанным в синтезе соединения примера 51 и соединения примера 52. Остаток очищали методом преп-HPLC с обращенной фазой с получением указанного соединения. MS (ESI) m/z 576.2 (М+Н); 1Н ЯМР (400 МГц, CD3OD): δ 9.35-9.41 (m, 1Н), 8.22 (s, 1H), 7.58-7.83 (m, 7 H), 7.27-7.47 (m, 3H), 7.12-7.25 (m, 1H), 7.05 (d, J=7.5 Гц, 1H), 4.62-4.75 (m, 1H), 2.55 (brs., 1H), 1.77-2.10 (m, 3H), 1.27-1.53 (m, 3H), 1.05-1.22 (m, 2H).
ПРИМЕР 85-a, 85-b, 85-c, 85-d
5-(3-ХЛОР-2,6-ДИФТОРФЕНИЛ)-2-(26-ФТОР-24-((МЕТОКСИКАРБОНИЛ)АМИНО)-5-МЕТИЛ-4-ОКСО-3-АЗА-1(1,3)2(1,2)-ДИБЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
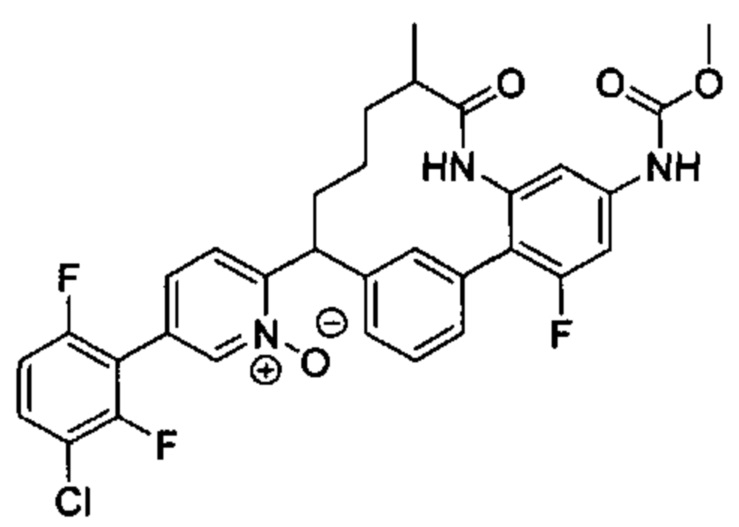
85A: Метил(Е)-(9-(5-(3-хлор-2,6-дифторфенил)пиридин-2-ил)-26-фтор-5-метил-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-8-ен-24-ил)карбамат:
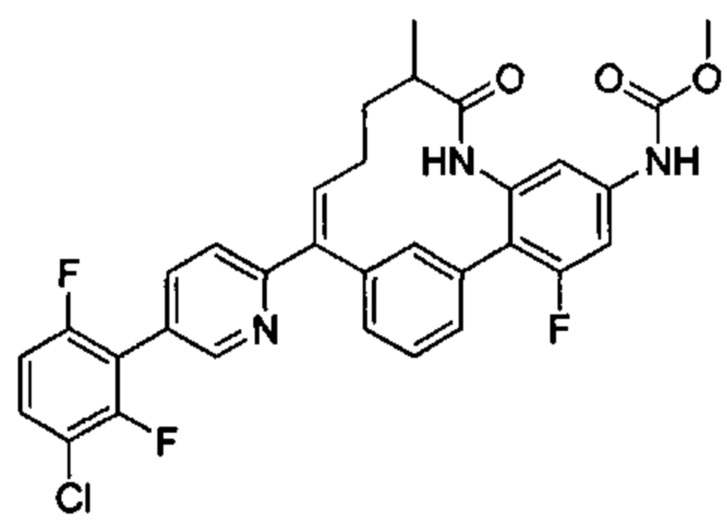
Соединение примера 85А получали из промежуточного соединения 3 и промежуточного соединения 53 способом, подобным описанному в синтезе соединения примера 50С. MS (ESI) m/z 592.2 (М+Н).
Образец рацемического с соединения примера 85А подвергали хиральному разделению методом SFC (OD, 30×250 мм, 50% EtOH/СО2, 100 бар, 80 мл/мин, 35°С) с получением соединения примера 85А-а (более быстрое элюирование) и соединения примера 85А-b (более медленное элюирование). MS (ES+) m/z: 592.2 (М+Н).
85B-a/85B-b: Метил(9-(5-(3-хлор-2,6-дифторфенил)пиридин-2-ил)-26-фтор-5-метил-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-24-ил)карбамат: Смесь соединения примера 85А-а (710 мг, 1,20 ммоль) и никеля Ренея (352 мг, 6,00 ммоль) добавляли в THF (30 мл) и смесь перемешивали при 40°С в атмосфере H2 (1 атм.) в течение 4 ч. Реакционную смесь фильтровали и промывали THF (10 мл × 3) (Осторожно! Никогда не оставляйте катализатор на воздухе). Фильтрат концентрировали с получением смеси двух диастереомеров. MS (ESI) m/z 594.2 (М+Н).
Смесь двух диастереомеров разделяли методом SFC (колонка С2 250 мм × 30 мм, 35% МеОН/СО2, скорость потока: 80 мл/мин) с получением соединения примера 85В-а (более быстрое элюирование) и соединения примера 85В-b (более медленное элюирование). MS (ESI) m/z 594.2 (М+Н).
85B-c/85B-d: Метил(9-(5-(3-хлор-2,6-дифторфенил)пиридин-2-ил)-26-фтор-5-метил-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-24-ил)карбамат: Смесь соединения примера 85А-b (340 мг, 0,554 ммоль) и никеля Ренея (169 мг, 2,87 ммоль) добавляли в THF (30 мл) и смесь перемешивали при 30°С в атмосфере Н2 (1 атм.) в течение 4 ч. Реакционную смесь фильтровали и промывали EtOAc (10 мл × 3). Фильтрат концентрировали, а остаток очищали методом HPLC (YMC-Actus Pro С18, 150 мм × 30 мм, 50-70% MeCN в воде (0,1% TFA), градиент, 25 мл/мин) с получением смеси двух диастереомеров. MS (ESI) m/z 594.2 (М+Н).
Смесь двух диастереомеров разделяли методом SFC (AD, 30×250 мм, 40% МеОН (0,05% DEA)/CO2, 100 бар, 80 мл/мин, 40°С) с получением соединения примера 85В-c (более быстрое элюирование) и соединения примера 85B-d (более медленное элюирование). MS (ESI) m/z 594.2 (М+Н).
Пример 85-a/85-b/85-c/85-d:
Соединение примера 85-a/85-b/85-c/85-d получали из соединений примеров 85В-a/85B-b/85B-c/85B-d, соответственно, способом, описанным в синтезе соединения примера 50.
Соединение примера 85-а: MS (ESI) m/z 610.1 (М+Н); 1Н ЯМР (400 МГц, DMSO_d6): δ 10.01 (s, 1Н), 9.58 (s, 1H), 8.50 (s, 1H), 7.71-7.82 (m, 1H), 7.40-7.58 (m, 4H), 7.31-7.39 (m, 2H), 7.27 (d, J=6.4 Гц, 1H), 7.03-7.18 (m, 2H), 4.58-4.70 (m, 1H), 3.70 (s, 3H), 2.40-2.48 (m, 1H), 2.01-1.91 (m, 2H), 1.81-1.66 (m, 1H), 1.26-1.43 (m, 1H), 0.97-1.25 (m, 2H), 0.92 (d, J=6.6 Гц, 3H).
Соединение примера 85-b: MS (ESI) m/z 610.1 (M+H); 1H ЯМР (400 МГц, DMSO_d6): δ 10.01 (s, 1H), 9.44 (s, 1H), 8.49 (s, 1H), 7.91 (d, J=8.2 Гц, 1H), 7.72-7.84 (m, 1H), 7.59 (d, J=7.8 Гц, 1H), 7.46 (d, J=12.1 Гц, 1H), 7.27-7.40 (m, 3H), 7.08-7.24 (m, 2H), 6.86 (d, J=7.4 Гц, 1H), 4.52 (d, J=10.6 Гц, 1H), 3.70 (s, 3H), 2.31-2.19 (m, 1H), 2.01-1.89 (m, 1H), 1.61-1.86 (m, 2H), 1.47-1.29 (m, 1H), 1.13-1.26 (m, 1H), 1.07 (d, J=7.0 Гц, 3Н), 1.02-0.91 (m, 1H).
Соединение примера 85-c: MS (ESI) m/z 610.1 (M+H); 1H ЯМР (400 МГц, DMSO_d6): δ 10.01 (s, 1H), 9.44 (s, 1H), 8.49 (s, 1H), 7.91 (d, J=8.2 Гц, 1H), 7.72-7.84 (m, 1H), 7.59 (d, J=7.8 Гц, 1H), 7.46 (d, J=12.1 Гц, 1H), 7.27-7.40 (m, 3H), 7.08-7.24 (m, 2H), 6.86 (d, J=7.4 Гц, 1H), 4.52 (d, J=10.6 Гц, 1H), 3.70 (s, 3H), 2.31-2.19 (m, 1H), 2.01-1.89 (m, 1H), 1.61-1.86 (m, 2H), 1.47-1.29 (m, 1H), 1.13-1.26 (m, 1H), 1.07 (d, J=7.0 Гц, 3Н), 1.02-0.91 (m, 1H).
Соединение примера 85-d: MS (ESI) m/z 610.1 (M+H); 1H ЯМР (400 МГц, DMSO_d6): δ 10.01 (s, 1H), 9.58 (s, 1H), 8.50 (s, 1H), 7.71-7.82 (m, 1H), 7.40-7.58 (m, 4H), 7.31-7.39 (m, 2H), 7.27 (d, J=6.4 Гц, 1H), 7.03-7.18 (m, 2H), 4.58-4.70 (m, 1H), 3.70 (s, 3H), 2.40-2.48 (m, 1H), 2.01-1.91 (m, 2H), 1.81-1.66 (m, 1H), 1.26-1.43 (m, 1H), 0.97-1.25 (m, 2H), 0.92 (d, J=6.6 Гц, 3Н).
ПРИМЕР 86 (рацемат), 86-а, 86-b, 86-с, 86-d
5-(3-ХЛОР-2,6-ДИФТОРФЕНИЛ)-2-(16-ФТОР-24-((МЕТОКСИКАРБОНИЛ)АМИНО)-5-МЕТИЛ-4-ОКСО-3-АЗА-1(1,3),2(1,2)-ДИБЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
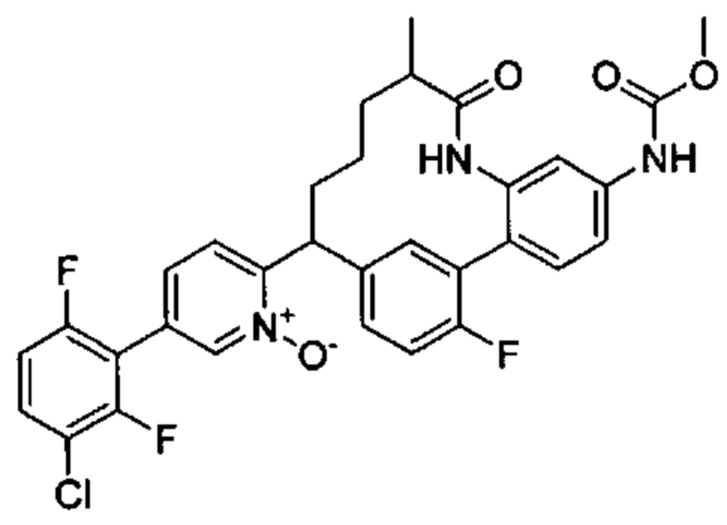
86А: Метил(E)-(9-(5-(3-хлор-2,6-дифторфенил)пиридин-2-ил)-16-фтор-5-метил-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-8-ен-24-ил)карбамат:
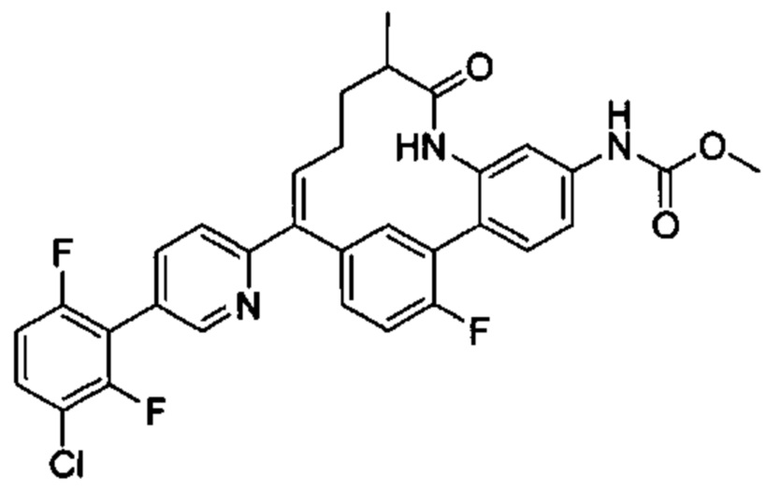
Соединение примера 86А получали из промежуточного соединения 6 и промежуточного соединения 19 способом, подобным описанному в синтезе соединения примера 50С. MS (ESI) m/z 592.1 (M+H).
Образец рацемического соединения примера 86А подвергали хиральному разделению методом SFC (колонка AD, 250 мм × 30 мм, 55% изопропанол/СО2, 70 мл/мин) с получением соединения примера 86А-а (более быстрое элюирование) MS (ESI) m/z 665.3 (М+71) и соединения примера 86А-b (более медленное элюирование).
86В: Метил(9-(5-(3-хлор-2,6-дифторфенил)пиридин-2-ил)-16-фтор-5-метил-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-24-ил)карбамат:
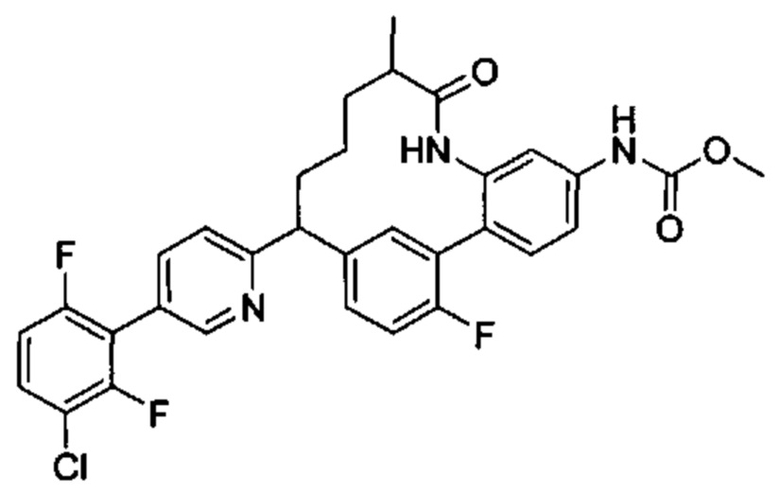
Смесь соединения примера 86А-а (210 мг, 0,355 ммоль) и никеля (100 мг, 1,704 ммоль) в THF (100 мл) перемешивали при 30°С в атмосфере H2 в течение 2 ч. Реакционную смесь фильтровали через слой целита и фильтрат концентрировали с получением указанного соединения в виде смеси двух диастереомеров. MS (ESI) m/z 616.1 (M+Na).
Пример 86-a/86-b:
К раствору соединения примера 86В (100 мг, 0,168 ммоль) в DCM (4 мл) добавляли м-СРВА (83 мг, 0,337 ммоль, 70% чистоты) при 25°С в круглодонной колбе. Смесь перемешивали при 25°С в течение 4 ч. Ее концентрировали и очищали методом HPLC с обращенной фазой (YMC-Actus Pro С18, 150×30 мм, 40-70% MeCN в воде (0,1% TFA), градиент) с получением смеси двух диастереомеров. MS (ESI) m/z 610.2 (M+H).
Образец смеси диастереомеров разделяли методом SFC (колонка AD, 250 мм × 30 мм, 50% IPA/СО2, 70 мл/мин) с получением соединения примера 86-а (более быстрое элюирование) в виде твердого вещества и соединения примера 86-b (более медленное элюирование).
Соединение примера 86-а: MS (ESI) m/z 610.1 (M+H); 1H ЯМР (400 МГц, CD3OD): δ 8.49 (s, 1H), 7.51-7.65 (m, 4Н), 7.39-7.50 (m, 3Н), 7.07-7.20 (m, 3Н), 4.75 (d, J=9.5 Гц, 1Н), 3.73 (s, 3Н), 2.44 (brs, 1Н), 2.12-2.25 (m, 1Н), 2.02 (brs, 1H), 1.76 (brs, 1H), 1.44 (brs, 2Н), 1.19-1.26 (m, 1Н), 1.08 (d, J=6.8 Гц, 3Н).
Соединение примера 86-b: MS (ESI) m/z 610.1 (M+H); 1H ЯМР (400 МГц, CD3OD): δ 8.48 (brs, 1Н), 7.94 (d, J=8.4 Гц, 1H), 7.77 (d, J=8.2 Гц, 1H), 7.62-7.66 (m, 2H), 7.44-7.52 (m, 3Н), 7.17-7.23 (m, 1Н), 7.07-7.13 (m, 1H), 6.89 (brs, 1H), 4.70 (d, J=8.4 Гц, 1H), 3.75 (s, 3H), 2.36 (brs, 1H), 1.98-2.03 (m, 3H), 1.78-1.82 (m, 2H), 1.52 (brs, 1H), 1.20 (d, J=7.1 Гц, 3Н).
Пример 86-c/58-d:
Соединения примеров 86-c/86-d (смесь двух диастереомеров) получали из соединения примера 86А-b способом, как в синтезе соединений примеров 86-а/86-b. Их очищали методом HPLC с обращенной фазой (YMC-Actus Pro С18, 150×30 мм, 40-70% MeCN в воде (0,1% TFA), градиент). MS (ESI) m/z 610.2 (М+Н).
Образец смеси диастереомеров разделяли методом SFC (колонка AD, 250 мм × 30 мм, 50% IPA/CO2, 70 мл/мин) с получением соединения примера 86-с (более быстрое элюирование) в виде твердого вещества и соединения примера 86-d (более медленное элюирование).
Соединение примера 86-с: MS (ESI) m/z 610.1 (М+Н); 1Н ЯМР (400 МГц, CD3OD): δ 8.48 (brs, 1Н), 7.94 (d, J=8.4 Гц, 1Н), 7.77 (d, J=8.2 Гц, 1H), 7.62-7.66 (m, 2Н), 7.44-7.52 (m, 3Н), 7.17-7.23 (m, 1H), 7.07-7.13 (m, 1Н), 6.89 (brs, 1Н), 4.70 (d, J=8.4 Гц, 1Н), 3.75 (s, 3Н), 2.36 (brs, 1Н), 1.98-2.03 (m, 3Н), 1.78-1.82 (m, 2Н), 1.52 (brs, 1Н), 1.20 (d, J=7.1 Гц, 3Н).
Соединение примера 86-b: MS (ESI) m/z 610.1 (М+Н); 1Н ЯМР (400 МГц, CD3OD): δ 8.49 (s, 1Н), 7.51-7.65 (m, 4Н), 7.39-7.50 (m, 3Н), 7.07-7.20 (m, 3Н), 4.75 (d, J=9.5 Гц, 1Н), 3.73 (s, 3Н), 2.44 (brs, 1Н), 2.12-2.25 (m, 1Н), 2.02 (brs, 1Н), 1.76 (brs, 1H), 1.44 (brs, 2Н), 1.19-1.26 (m, 1H), 1.08 (d, J=6.8 Гц, 3Н).
ПРИМЕР 87 (рацемат), 87-а, 87-b
5-(3-ХЛОР-2,6-ДИФТОРФЕНИЛ)-4-МЕТОКСИ-2-(24-((МЕТОКСИКАРБОНИЛ)АМИНО)-4-ОКСО-3-АЗА-1(1,3),2(1,2)-ДИБЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД

87А: Метил(2-амино-3'-(5-хлор-4-метоксипиколиноил)-[1.1'-бифенил]-4-ил)карбамат: Соединение примера 87А получали из промежуточного соединения 19 и промежуточного соединения 12 способом, как описано в синтезе соединения примера 12А. Продукт очищали методом колоночной флеш-хроматографии (SiO2, DCM: EtOAc=50:1-1:1). MS (ESI) m/z 412.1 (M+H).
87B: Метил(2-(5-(бензо[d]тиазол-2-илсульфонил)пентанамидо)-3'-(5-хлор-4-метоксипиколиноил)-[1.1'-бифенил]-4-ил)карбамат: К раствору соединения примера 87А (100 мг, 0,24 ммоль) и промежуточного соединения 26 (104 мг, 0,24 ммоль, 70% чистоты) в пиридине (2 мл) добавляли POCl3 (0,045 мл, 0,49 ммоль) при 0°С. Реакционную смесь перемешивали при 0°С в течение 30 мин, а затем гасили водой (10 мл). Смесь экстрагировали DCM (10 мл × 3). Объединенные органические слои промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом преп-TLC (SiO2, петролейный эфир : EtOAc = 1:1) с получением указанного соединения. MS (ESI) m/z 693.1 (М+Н).
87С: Метил(E)-(9-(5-хлор-4-метоксипиридин-2-ил)-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-8-ен-24-ил)карбамат:
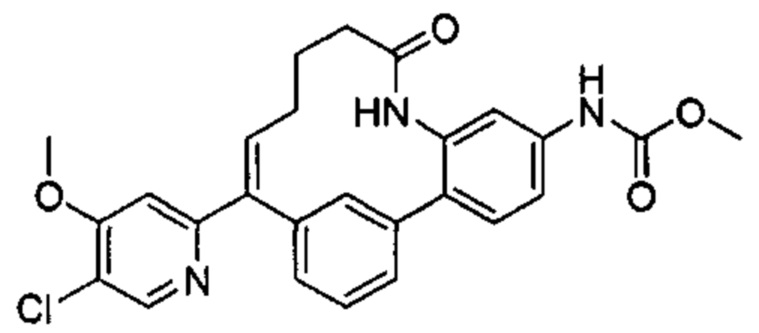
К раствору соединения примера 87В (1,00 г, 1,44 ммоль) в THF (50 мл) при -78°С добавляли раствор LiHMDS (8,66 мл, 8,66 ммоль, 1М в THF). Реакционную смесь перемешивали при -78°С в течение 2 ч и оставляли нагреваться до 25°С. Ее гасили насыщенным водным хлоридом аммония (20 мл) и экстрагировали EtOAc (30 мл × 3). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток перетирали в порошок в EtOAc (100 мл) в течение 0,5 ч. Твердые вещества собирали фильтрацией с получением указанного соединения. MS (ESI) m/z 478.0 (М+Н).
Пример 87/87-a/87-b:
Соединение примера 87 получали из соединения примера 87С способом, описанным в синтезе соединения примера 50. Продукт очищали методом HPLC с обращенной фазой (YMC-Actus Pro С18, 150×30 мм, 29-59% MeCN в воде (0,1% MeCN), 40 мл/мин). MS (ESI) m/z 608.2 (М+Н); 1Н ЯМР (400 МГц, CD3OD): δ 8.52 (s, 1Н), 8.29 (s, 1H), 7.67-7.68 (m, 1H), 7.57-7.63 (m, 1H), 7.44-7.46 (m, 2H), 7.36-7.41 (m, 1H), 7.26-7.33 (m, 2H), 7.11-7.14 (m, 1H), 7.06-7.08 (m, 1H), 4.78-4.82 (m, 1H), 3.94 (s, 3H), 3.73 (s, 3H), 2.35-2.47 (m, 2H), 2.08-2.15 (m, 2H), 1.55-1.65 (m, 2H), 0.86-0.88 (m, 2H).
Рацемический образец соединения примера 87 подвергали хиральному разделению методом SFC (колонка: OD, 250 мм × 30 мм, 50% EtOH/СО2, 70 мл/мин) с получением соединения примера 87-а (более медленное элюирование) и соединения примера 87-b (более быстрое элюирование).
ПРИМЕР 88 (рацемат), 88-а, 88-b, 88-с, 88-d
5-(3-ХЛОР-2,6-ДИФТОРФЕНИЛ)-4-МЕТОКСИ-2-(24-((МЕТОКСИКАРБОНИЛ)АМИНО)-5-МЕТИЛ-4-ОКСО-3-АЗА-1(1,3),2(1,2)-ДИБЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
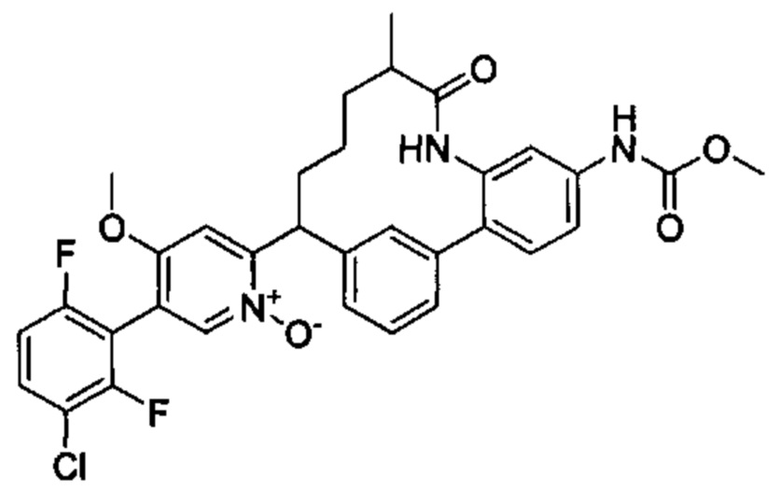
88А: Метил(Е)-(9-(5-(3-хлор-2,6-дифторфенил)-4-метоксипиридин-2-ил)-5-метил-4-оксо-3-аза-1(1,3),2(1,2)-дибензолациклононафан-8-ен-24-ил)карбамат:
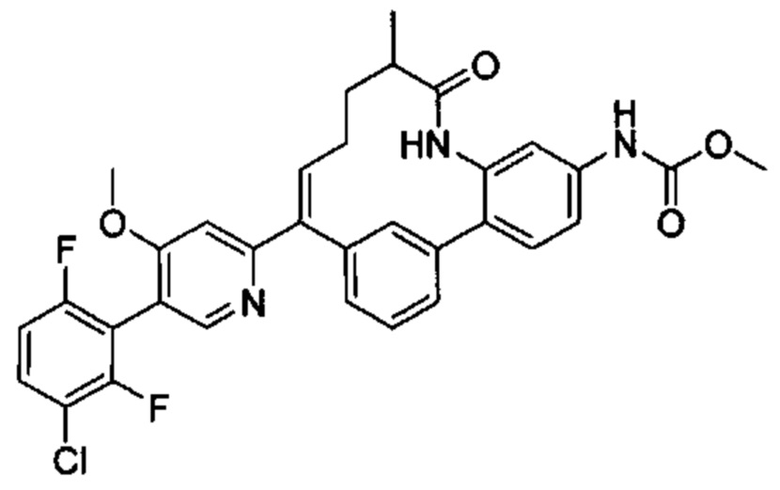
Соединение примера 88А получали из соединения примера 59А способом, описанным в синтезе соединения 50С. MS (ESI) m/z 604.2 (М+Н).
Рацемический образец соединения примера 88А подвергали хиральному разделению методом SFC (колонка AD, 250 мм × 30 мм, 50% изопропанол/СО2, 80 мл/мин) с получением соединения примера 88А-а (более быстрое элюирование) в виде твердого вещества и соединения примера 88А-b (более медленное элюирование).
Пример 88-а/88-b:
Смесь диастереомеров соединений примеров 88-а и 88-b получали из соединения примера 88А-а способом, описанным в синтезе соединения примера 50. Продукт очищали методом HPLC с обращенной фазой (YMC-Actus Pro С18, 150×30 мм, 30-60% MeCN в воде (0,1% TFA), 40 мл/мин). MS (ESI) m/z 622.3 (М+Н).
Образец двух диастереомеров подвергали хиральному разделению методом SFC (колонка AS, 250 мм × 30 м, 50% МеОН/CO2, 80 мл/мин) с получением соединения примера 88-а (более быстрое элюирование) в виде твердого вещества и соединения примера 88-b (более медленное элюирование).
Соединение примера 88-а: MS (ESI) m/z 622.1 (M+H); 1H ЯМР (400 МГц, CD3OD): δ 8.33 (s, 1Н), 7.72 (s, 1H), 7.59-7.64 (m, 1H), 7.38-7.47 (m, 4H), 7.33-7.35 (m, 1H), 7.07-7.19 (m, 3H), 4.78-4.83 (m, 1H), 3.85 (s, 3H), 3.75 (s, 3H), 2.54 (brs, 1H), 2.13-2.15 (m, 2H), 1.85-1.91 (m, 1H), 1.47-1.50 (m, 1H), 1.34-1.37 (m, 2H), 1.10 (d, J=6.6 Гц, 3Н).
Соединение примера 88-b: MS (ESI) m/z 622.1 (M+H); 1H ЯМР (400 МГц, CD3OD): δ 8.30 (s, 1H), 7.69 (s, 1H), 7.62-7.64 (m, 1H), 7.36-7.47 (m, 5H), 7.28-7.30 (m, 1H), 7.14-7.17 (m, 1H), 6.96-6.99 (m, 1H), 4.77-4.79 (m, 1H), 4.03 (s, 3H), 3.74 (s, 3H), 2.37-2.39 (m, 1H), 1.88-2.05 (m, 3H), 1.39-1.51 (m, 2H), 1.24 (d, J=6.8 Гц, 3Н), 1.16-1.18 (m, 1H).
Пример 88-c/88-d:
Смесь диастереомеров соединений примеров 88-с и 88-d получали из соединения примера 88А-b способом, описанным в синтезе соединения примера 50. Продукт очищали методом HPLC с обращенной фазой (YMC-Actus Pro С18, 150×30 мм, 30-60% MeCN в воде (0,1% TFA), 40 мл/мин). MS (ESI) m/z 622.3 (M+H).
Образец двух диастереомеров подвергали хиральному разделению методом SFC (колонка AS, 250 мм × 30 мм, 50% МеОН/CO2, 80 мл/мин) с получением соединения примера 88-с (более быстрое элюирование) в виде твердого вещества и соединения примера 88-d (более медленное элюирование).
Соединение примера 88-с: MS (ESI) m/z 622.3 (M+H); 1H ЯМР (400 МГц, CD3OD): δ 8.30 (s, 1Н), 7.68 (s, 1H), 7.60-7.64 (m, 1H), 7.36-7.48 (m, 5H), 7.27-7.29 (m, 1H), 7.13-7.16 (m, 1H), 6.96-6.99 (m, 1H), 4.79-4.82 (m, 1H), 4.03 (s, 3H), 3.71 (s, 3H), 2.35-2.38 (m, 1H), 1.95-2.11 (m, 3H), 1.39-1.41 (m, 2H), 1.26 (d, J=6.8 Гц, 3Н), 1.18-1.21 (m, 1H).
Соединение примера 88-d: MS (ESI) m/z 622.2 (M+H), 1H ЯМР (400 МГц, CD3OD): δ 8.41 (s, 1H), 7.72 (s, 1H), 7.58-7.66 (m, 1H), 7.39-7.49 (m, 4H), 7.31-7.37 (m, 1H), 7.08-7.20 (m, 3H), 4.81-4.87 (m, 1H), 3.88 (s, 3H), 3.75 (s, 3H), 2.49-2.58 (m, 1H), 2.13-2.17 (m, 2H), 1.84-1.94 (m, 1H), 1.51-1.54 (m, 1H), 1.36 (brs, 2H), 1.10 (d, J=6.8 Гц, 3Н).
ПРИМЕР 89, 89-a, 89-b
5-(5-ФТОР-2-(1Н-ТЕТРАЗОЛ-1-ИЛ)ФЕНИЛ)-2-(25-ФТОР-4-ОКСО-3-АЗА-1(1,3),2(1,2)-ДИБЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
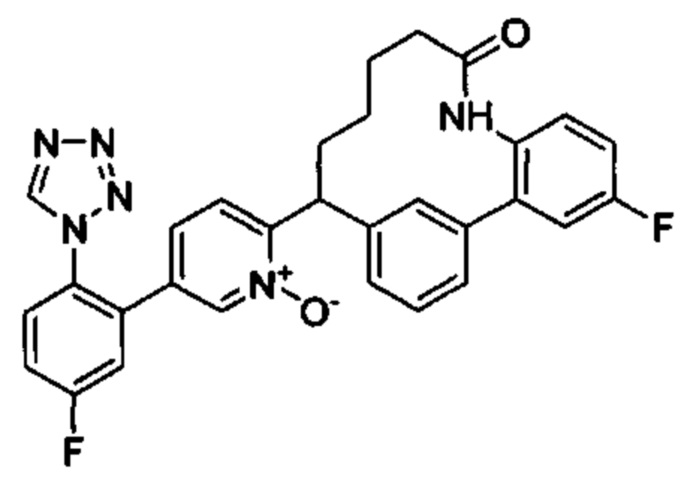
89A: (2'-Амино-5'-фтор-[1.1'-бифенил]-3-ил)(5-хлорпиридин-2-ил)метанон: К раствору промежуточного соединения 2 (9,00 г, 30,30 ммоль) в DMF (150 мл) и воде (20 мл) добавляли промежуточное соединение 18 (7,91 г, 33,40 ммоль), K3PO4 (19,33 г, 91,00 ммоль) и Pd(Ph3P)4 (3,51 г, 3,03 ммоль). Смесь перемешивали при 60°С в атмосфере азота в течение 3 ч. Ее охлаждали до к.т. и фильтровали через слой целита. Твердый кек промывали EtOAc (30 мл × 2). Фильтрат разбавляли водой (100 мл) и экстрагировали EtOAc (100 мл × 3). Объединенные органические слои промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (петролейный эфир : EtOAc = 20:1-5:1) с получением указанного соединения. MS (ESI) m/z 326.8 (М+Н).
89В: N-(3'-(5-Хлорпиколиноил)-5-фтор-[1.1'-бифенил]-2-ил)-5-((1-фенил-1Н-тетразол-5-ил)сульфонил)пентанамид: Раствор соединения примера 89А (11,40 г, 34,90 ммоль), промежуточного соединения 25 (11,91 г, 38,40 ммоль) и EDC (13,38 г, 69,80 ммоль) в пиридине (150 мл) перемешивали при 20°С в течение 16 ч. Его разбавляли EtOAc (200 мл), промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали. Остаток перетирали в порошок с EtOAc (30 мл). Твердое вещество собирали фильтрацией и промывали этилацетатом с получением указанного соединения. MS (ESI) m/z 619.0 (М+Н).
89С: (E)-9-(5-хлорпиридин-2-ил)-25-фтор-3-аза-1(1,3),2(1,2)-дибензолациклононафан-8-ен-4-он:

К раствору соединения примера 89В (5,00 г, 8,08 ммоль) в THF (100 мл) при -78°С добавляли раствор LiHMDS (40,40 мл, 40,40 ммоль, 1 М в THF) и перемешивали в течение 2 ч. Его нагревали до 20°С и перемешивали в течение 16 ч. Реакцию гасили водным хлоридом аммония (нас., 30 мл) и экстрагировали EtOAc (100 мл × 3). Объединенные органические слои промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали. Остаток перетирали в порошок (петролейный эфир : EtOAc = 3:1) с получением указанного соединения. MS (ESI) m/z 393.1 (М+Н).
89D: (Е)-9-(5-(2-Амино-5-фторфенил)пиридин-2-ил)-25-фтор-3-аза-1(1,3),2(1,2)-дибензолациклононафан-8-ен-4-он:
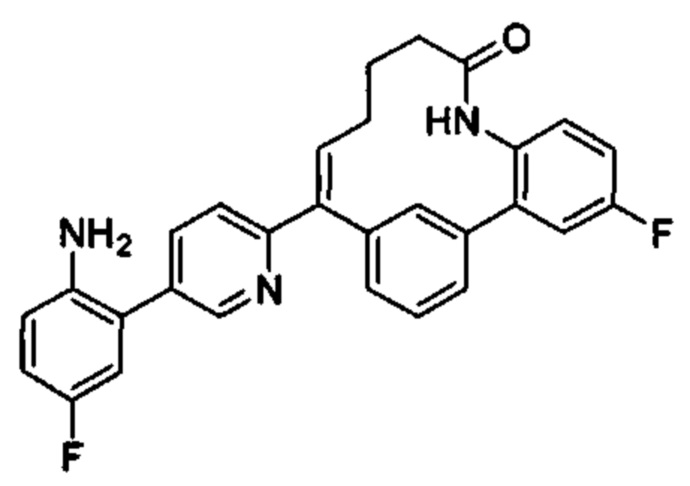
К раствору соединения примера 89С (2,10 г, 4,28 ммоль) в THF (40 мл) добавляли промежуточное соединение 18 (1,83 г, 7,70 ммоль), предварительный каталитический нейтрализатор Xphos 2-го поколения (0,34 г, 0,43 ммоль) и фосфат калия (12,83 мл, 12,83 ммоль, 1M в воде) при 25°С. Смесь перемешивали при 90°С в атмосфере N2 в течение 16 ч. Ее охлаждали до к.т. и гасили Н2О (30 мл). Смесь экстрагировали EtOAc (50 мл × 3), промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (петролейный эфир : EtOAc = 5:1-1:1) с получением указанного соединения. MS (ESI) m/z 468.2 (М+Н).
89Е: 9-(5-(2-Амино-5-фторфенил)пиридин-2-ил)-25-фтор-3-аза-1(1,3),2(1,2)-дибензолациклононафан-4-он:
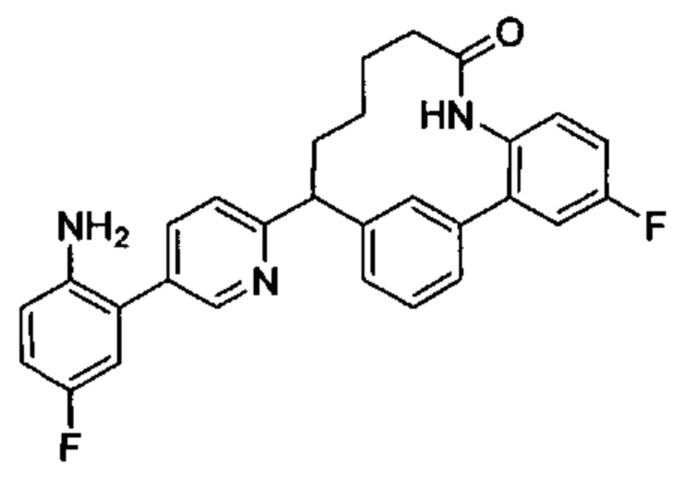
Смесь соединения примера 89D (1,40 г, 2,99 ммоль) и никеля Ренея (3,00 г, 51,10 ммоль) в THF (30 мл) перемешивали при 25°С в течение 4 ч в атмосфере H2 (1 атм.). Катализатор удаляли фильтрацией через слой целита. (Осторожно! Никогда не оставляйте катализатор на воздухе). Фильтрат концентрировали с получением указанного соединения. MS (ESI) m/z 470.2 (М+Н).
Соединение примера 89 получали из соединения примера 89Е способом, описанным в синтезе соединения примера 51. Продукт очищали методом HPLC с обращенной фазой (YMC-Actus Pro С18, 150×30 мм, 27-57% MeCN в воде (0,1% TFA), 40 мл/мин) с получением указанного соединения. MS (ESI) m/z 539.2 (М+Н).
Рацемический образец соединения примера 89 подвергали хиральному разделению методом SFC (колонка: AS, 250 мм × 30 мм, условие: 40% МеОН (0,1% NH3)/CO2) с получением соединения примера 89-а (более медленное элюирование) и соединения примера 89-b (более быстрое элюирование).
ПРИМЕР 90 (рацемат), 90-а, 90-b
5-(2-(4-ХЛОР-1Н-1,2,3-ТРИАЗОЛ-1-ИЛ)-5-ФТОРФЕНИЛ)-2-(25-ФТОР-4-ОКСО-3-АЗА-1(1,3),2(1,2)-ДИБЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
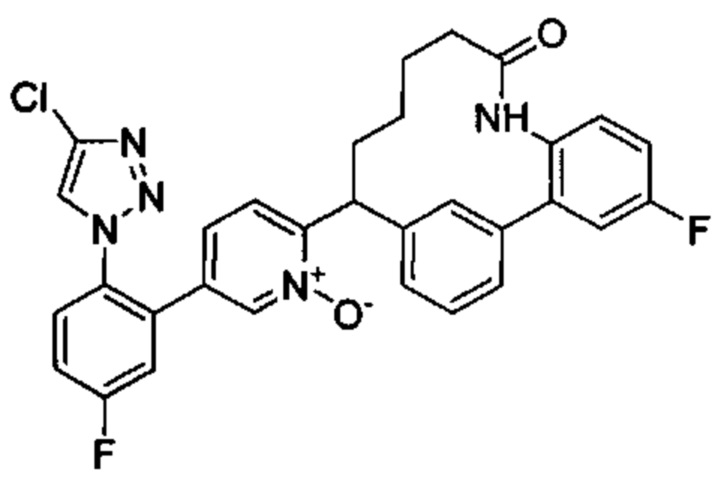
90А: 9-(5-(2-Азидо-5-фторфенил)пиридин-2-ил)-25-фтор-3-аза-1(1,3),2(1,2)-дибензол ациклононафан-4-он:
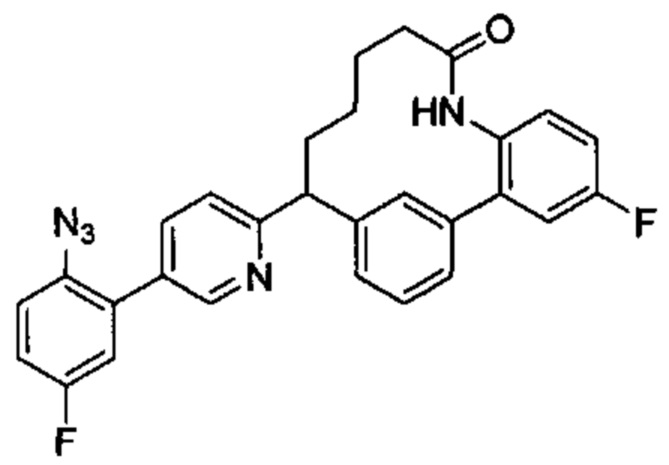
К раствору соединения примера 60D (350 мг, 0,75 ммоль) в хлориде водорода (5 мл) при -5°С добавляли раствор нитрит натрия (103 мг, 1,49 ммоль) в Н2О (15 мл). Его перемешивали при -5°С в течение 1 ч, а затем добавляли раствор азида натрия (97 мг, 1,49 ммоль) в Н2О (15 мл). Смесь перемешивали при 25°С в течение 16 ч. Добавляли водный NaHCO3 (насыщенный) для доведения значения рН до 8. Смесь экстрагировали EtOAc (50 мл × 3). Объединенные органические слои промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали с получением указанного соединения. MS (ESI) m/z 496.2 (М+Н).
90В: 25-Фтор-9-(5-(5-фтор-2-(4-(трибутилстаннил)-1H-1,2,3-триазол-1-ил)фенил)пиридин-2-ил)-3-аза-1(1,3),2(1,2)-дибензолациклононафан-4-он:

К раствору соединения примера 90А (200 мг, 0,40 ммоль) в толуоле (5 мл) добавляли трибутил(этинил)станнан (254 мг, 0,81 ммоль) и смесь перемешивали при 110°С в атмосфере N2 в течение 48 ч. Его охлаждали до к. т.и гасили водным KF (нас., 20 мл). Смесь перемешивали в течение 1 ч, а затем экстрагировали EtOAc (20 мл × 3). Объединенные органические слои промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (петролейный эфир : EtOAc = 10:1-1:1) с получением указанного соединения. MS (ESI) m/z 812.4 (М+Н).
90С: 9-(5-(2-(4-Хлор-1H-1,2,3-триазол-1-ил)-5-фторфенил)пиридин-2-ил)-25-фтор-3-аза-1(1,3),2(1,2)-дибензолациклононафан-4-он:
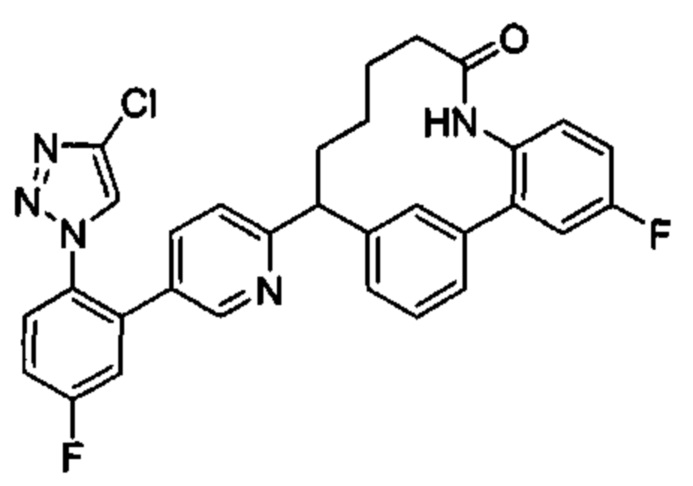
К раствору соединения примера 90В (200 мг, 0,247 ммоль) в ацетонитриле (5 мл) добавляли NCS (65,90 мг, 0,493 ммоль) и смесь перемешивали при 90°С в атмосфере N2 в течение 48 ч. Его охлаждали до к.т. и большую часть растворителя удаляли при пониженном давлении. К остатку добавляли воду (10 мл) и ее экстрагировали EtOAc (10 мл × 3). Объединенные органические слои промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали с получением указанного соединения. MS (ESI) m/z 556.2 (М+Н).
Пример 90/90-a/90-b:
К раствору соединения примера 90С (160 мг, 0,288 ммоль) в АсОН (2 мл) добавляли м-CPBA (93 мг, 0,432 ммоль, 85%) и смесь перемешивали при 25°С в течение 16 ч. Ее гасили водным Na2SO3 (нас., 15 мл). Водный карбонат натрия (насыщенный) добавляли для доведения значения рН до 8. Смесь экстрагировали EtOAc (20 мл × 3). Объединенные органические слои промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом HPLC с обращенной фазой (YMC-Actus Pro С18, 150×30 мм, 33-63% MeCN в воде (0,1% TFA), градиент, 40 мл/мин) с получением указанного соединения. MS (ESI) m/z 572.2 (М+Н). 1Н ЯМР (400МГц, CD3OD) δ 9.53 (s, 1Н), 8.65 (s, 1Н), 8.15 (s, 1Н), 7.83-7.75 (m, 1Н), 7.67 (dd, J=2.8, 9.1 Гц, 1Н), 7.61-7.50 (m, 3Н), 7.39-7.31 (m, 2Н), 7.29-7.19 (m, 3Н), 6.96-6.85 (m, 2Н), 4.61-4.52 (m, 1Н), 2.31-2.22 (m, 1Н), 2.04-1.74 (m, 4Н), 1.51-1.39 (m, 1H), 1.28-1.15 (m, 1Н), 1.10-0.95 (m, 1Н).
Рацемический образец соединения примера 90 подвергали хиральному разделению методом SFC (колонка: OD, 250 мм × 30 м, 40% EtOH (0,1% NH3), 40 мл/мин) с получением соединения примера 90-а (быстрое элюирования) и соединения примера 90-b (более медленное элюирование).
ПРИМЕР 91 (рацемат), 91-а, 91-b
1-(4-ХЛОР-2-(6-(24-((МЕТОКСИКАРБОНИЛ)АМИНО)-4-ОКСО-3-АЗА-1(1,3),2(1,2)-ДИБЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИН-3-ИЛ)ФЕНИЛ)-1H-1,2,3-ТРИАЗОЛ-4-КАРБОНОВОЙ КИСЛОТЫ
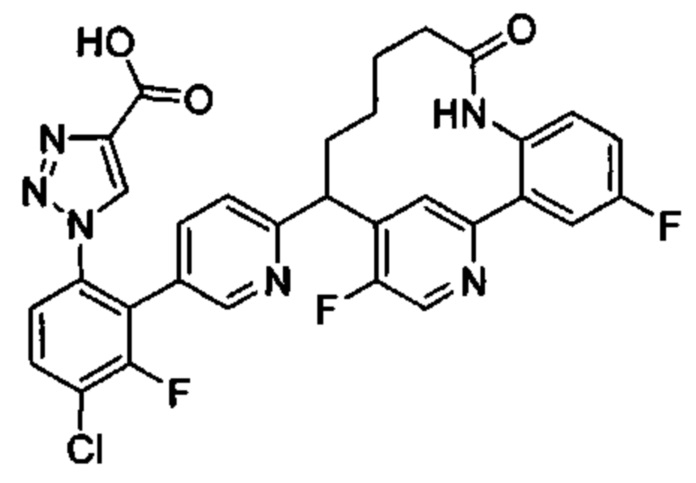
91А. (2-(2-Амино-5-фторфенил)-5-фторпиридин-4-ил)(5-хлорпиридин-2-ил)метанон: В колбу добавляли промежуточное соединение 9 (2,5 г, 7,92 ммоль), промежуточное соединение 18 (2,104 г, 8,87 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцен-палладий(II)дихлорида (0,582 г, 0,713 ммоль) и дегазированный THF (39,6 мл). Полученную смесь дегазировали и трижды повторно заполняли N2. Добавляли дегазированный трехосновный фосфат калия (7,92 мл, 23,77 ммоль). Реакционную смесь нагревали при 60°С в атмосфере N2 в течение 1 ч. Ее охлаждали до к.т. и фильтровали через слой целита. Твердые вещества промывали этилацетатом (50 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-40% этилацетатом в гексане) с получением указанного соединения. MS (ES+) m/z: 346.0 (М+Н).
91В. 5-(Бензо[d]тиазол-2-илсульфонил)-N-(2-(4-(5-хлорпиколиноил)-5-фторпиридин-2-ил)-4-фторфенил)пентанамид: К смеси соединения примера 91А и промежуточного соединения 26 (2,390 г, 7,98 ммоль) в DMF (66,5 мл) добавляли HATU (3,04 г, 7,98 ммоль) и N,N'-диизопропилэтиламин (3,48 мл, 19,96 ммоль). Реакционную смесь перемешивали при к.т. в атмосфере N2 в течение 8 ч. Ее разбавляли водой (50 мл) и экстрагировали этилацетатом (3×75 мл). Объединенные органические слои промывали солевым раствором, сушили над сульфатом натрия, фильтровали. Фильтрат концентрировали при пониженном давлении, а остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-40% этилацетатом в гексане) с получением указанного соединения. MS (ES+) m/z: 626.7 (М).
91С. (Е)-9-(5-Хлорпиридин-2-ил)-15,25-дифтор-3-аза-1(2,4)-пиридина-2(1,2)-бензолациклононафан-8-ен-4-он:
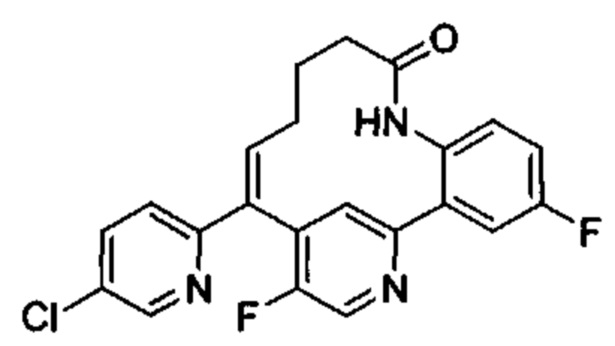
К раствору соединения примера 91В (780 мг, 1,244 ммоль) в THF (25 мл) при -78°С добавляли раствор LHMDS (1,0 М в THF, 8,7 мл, 8,71 ммоль) в атмосфере N2. Реакционную смесь перемешивали при -78°С в течение 1 ч. Ее гасили насыщенным водным хлоридом аммония (15 мл) и водой (30 мл) при 0°С. Водный слой экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали солевым раствором (30 мл) и сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-100% этилацетатом в гексане) с получением указанного соединения. MS (ES+) m/z: 412.2 (M+H).
91D. (E)-9-(5-(6-Амино-3-хлор-2-фторфенил)пиридин-2-ил)-15,25-дифтор-3-аза-1(2,4)-пиридина-2(1,2)-бензолациклононафан-8-ен-4-он:
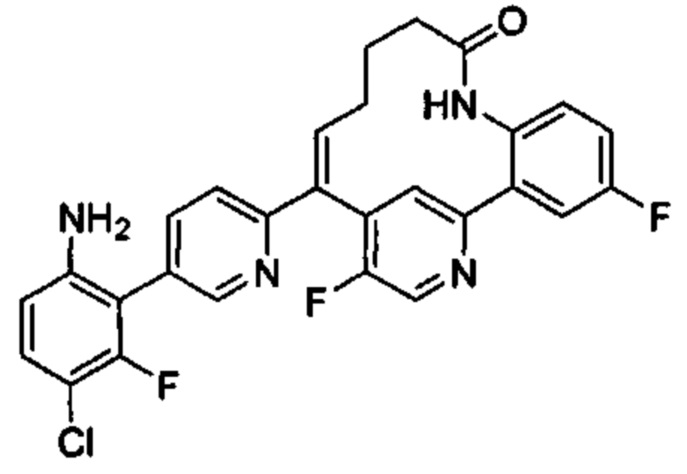
Смесь соединения примера 91С (230 мг, 0,560 ммоль), бис(пинаколято)диборона (171 мг, 0,672 ммоль), Xphos палладиевого (II) комплекса (41,4 мг, 0,056 ммоль) и ацетата калия (165 мг, 1,679 ммоль) и диоксана (5,6 мл) перемешивали при 80°С в атмосфере азота в течение 1,5 ч. Ее охлаждали до к.т., добавляли 4-хлор-3-фтор-2-йоданилин (152 мг, 0,560 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино) ферроцен-палладий(II)дихлорида (45,7 мг, 0,056 ммоль) и трехосновного фосфата калия (3 М водный раствор, 0,56 мл, 1,679 ммоль). Полученную смесь дегазировали и повторно заполняли трижды N2. Ее перемешивали при 80°С в течение 2 ч, охлаждали до к.т. и фильтровали через слой целита. Твердые вещества промывали этилацетатом (50 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали методом преп-TLC (обрабатывали 80% этилацетатом в гексане) с получением указанного продукта. MS (ES+) m/z: 520.9 (М+Н).
91E. трет-Бутил(Е)-1-(4-хлор-2-(6-(15,25-дифтор-4-оксо-3-аза-1(2,4)-пиридина-2(1,2)-бензолациклононафан-8-ен-9-ил)пиридин-3-ил)-3-фторфенил)-1Н-1,2,3-триазол-4-карбоксилат:

К перемешиваемому раствору соединения примера 91D (30 мг, 0,058 ммоль) в ацетонитриле (0,83 мл) при 0°С добавляли раствор изопропилнитрита (24,47 мкл, 0,239 ммоль) в ацетонитриле (0,2 мл), а затем добавляли азидотриметилсилан (31,7 мкл, 0,239 ммоль) в ацетонитриле (0,2 мл). Смесь перемешивали в течение 10 мин при 0°С и ее оставляли нагреваться до к.т., перемешивали в течение 1 ч. Добавляли раствор трет-бутилпропиолята (36,3 мг, 0,288 ммоль) в ацетонитриле (0,2 мл) и оксид меди (I) (0,412 мг, 2,88 мкмоль). Смесь перемешивали в течение 3 ч перед тем, как ее разбавляли DCM, и промывали насыщенным хлоридом аммония и солевым раствором. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-100% этилацетатом в гексане) с получением указанного соединения. MS (ES+) m/z: 673.1 (М+Н).
91F. трет-Бутил-1-(4-хлор-2-(6-(15,25-дифтор-4-оксо-3-аза-1(2,4)-пиридина-2(1,2)-бензолациклононафан-9-ил)пиридин-3-ил)-3-фторфенил)-1Н-1,2,3-триазол-4-карбоксилат:
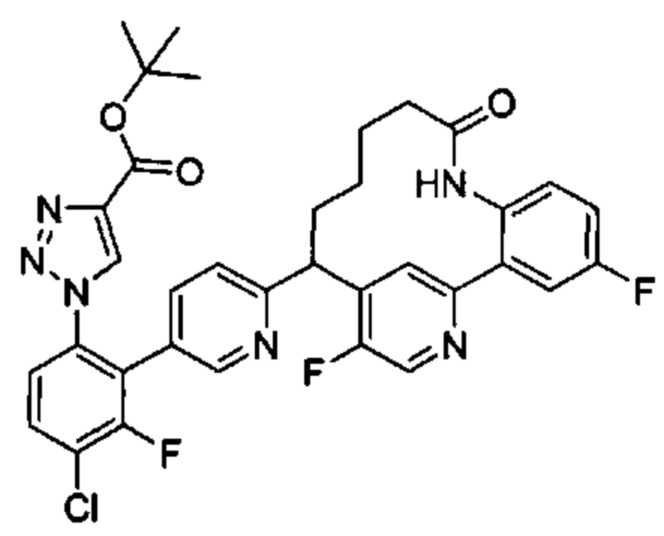
Смесь соединения примера 91Е (23 мг, 0,034 ммоль) и платинового катализатора (1%)/ванадия (2%) на активированном углероде (219 мг, 0,034 ммоль) в этилацетате : этаноле (3:1 объем/объем с 1% АсОН, 7,7 мл) встряхивали в атмосфере водорода (45 фунт/кв. дюйм) в течение 22 ч. Ее разбавляли 10% МеОН в DCM (20 мл) и фильтровали через слой целита. К твердым веществам добавляли МеОН в DCM (30% объем/объем, 30 мл) и аммиак в МеОН (7 н, 1 мл) и обрабатывали ультразвуком в течение нескольких минут. Его фильтровали через слой целита. Фильтрат концентрировали при пониженном давлении с получением указанного соединения. MS (ES+) m/z: 615.4 (М+Н).
Пример 91:
Соединение примера 91F (88 мг, 0,130 ммоль) перемешивали в DCM (10 мл) и TFA (5 мл) при к.т. в течение 1 ч. Большую часть растворителя концентрировали при пониженном давлении. Неочищенное вещество очищали методом преп-TLC (обрабатывали 15% MeOH/DCM с 1% АсОН) с получения указанного соединения. MS (ES+) m/z: 618.9 (М+Н).
Рацемический образец соединения примера 91 подвергали хиральному разделению методом SFC (AS, 21×250 мм, 18% МеОН/CO2, 60 мл/мин, 35°С) с получением соединения примера 91-а (более медленное элюирование) и соединения примера 91-b (более быстрое элюирование).
ПРИМЕР 92, 92-а, 92-b
1-(4-ХЛОР-2-(6-(24-((МЕТОКСИКАРБОНИЛ)АМИНО)-4-ОКСО-3-АЗА-1(1,3),2(1,2)-ДИБЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИН-3-ИЛ)ФЕНИЛ)-1Н-1,2,3-ТРИАЗОЛ-4-КАРБОНОВОЙ КИСЛОТЫ
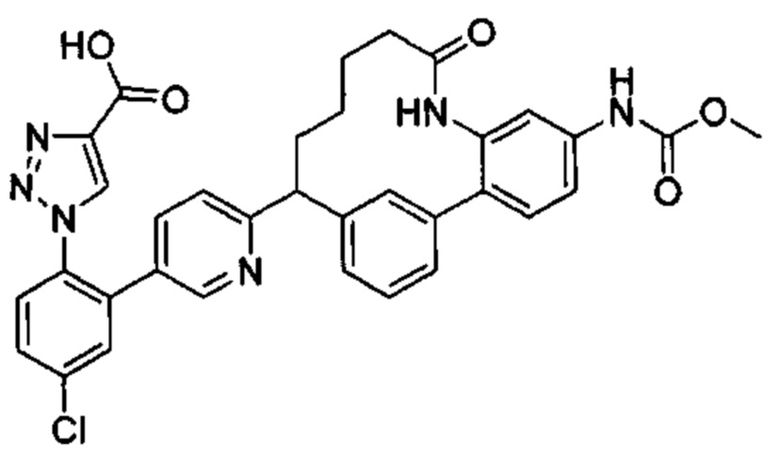
Соединение примера 92 получали из 12С способом, подобным описанному в синтезе соединения примера 91. MS (ES+) m/z: 637 (М+Н).
Рацемический образец соединения примера 92 подвергали хиральному разделению методом SFC (IC, 21×200 мм, 50% МеОН/CO2, 60 мл/мин, 35°С) с получением соединения примера 92-а (более медленное элюирование) и соединения примера 92-b (более быстрое элюирование).
ПРИМЕР 93, 93-а, 93-b
5-(3-ХЛОР-2,6-ДИФТОРФЕНИЛ)-2-(24-((МЕТОКСИКАРБОНИЛ)АМИНО)-4-ОКСО-3-АЗА-1(2,4)-ПИРИДИНА-2(1,2)-БЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
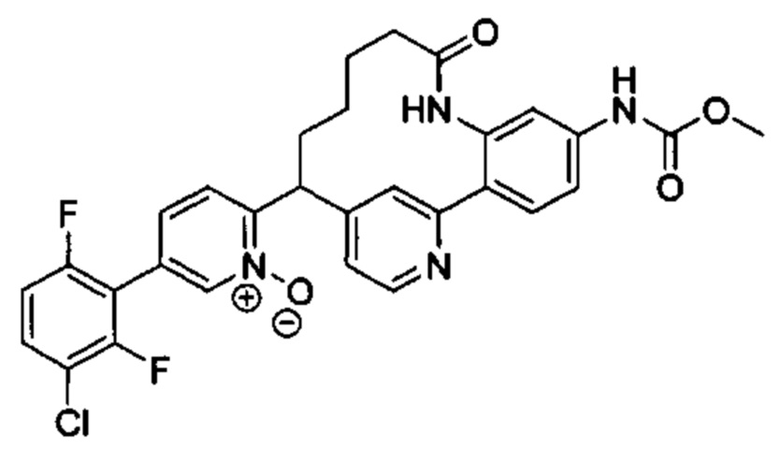
93А: Метил(3-амино-4-(4-(5-хлорпиколиноил)пиридин-2-ил)фенил)карбамат:
Указанное соединение получали из промежуточного соединения 8 способом, подобным описанному в синтезе соединения примера 12А. Продукт очищали методом колоночной флеш-хроматографии (SiO2, DCM: EtOAc=100:l-5:l). MS (ESI) m/z 383.0 (M+H).
93B: Метил(3-(5-бромпентанамидо)-4-(4-(5-хлорпиколиноил)пиридин-2-ил)фенил)карбамат: К смеси соединения примера 93А (4,00 г, 10,45 ммоль) и 5-бромпентаноилхлорида (2,29 г, 11,49 ммоль) в DCM (100 мл) при 0°С добавляли Et3N (4,37 мл, 31,30 ммоль). Смесь перемешивали при 25°С в течение 1 ч. Ее разбавляли водой (50 мл) и экстрагировали DCM (50 мл × 3). Объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии (SiO2, петролейный эфир : EtOAc = 5:1-1:1) с получением указанного соединения. MS (ESI) m/z 545.7, 546.7 (M+H).
93C: Метил(3-(5-бромпентанамидо)-4-(4-((5-хлорпиридин-2-ил)(гидрокси)метил)пиридин-2-ил)фенил)карбамат: К раствору соединения примера 93В (4,00 г, 7,33 ммоль) в DCM/MeOH (3:1, 100 мл) при 0°С добавляли NaBH4 (1,39 г, 36,60 ммоль). Полученную смесь перемешивали при 25°С в течение 1 ч перед тем, как ее гасили насыщенным водным раствором хлорида аммония (50 мл). Ее экстрагировали DCM (50 мл × 3). Объединенные органические слои промывали водой (50 мл) и солевым раствором (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением указанного соединения. MS (ESI) m/z 547.1.7, 549.1 (M+H).
93D: S-(5-((2-(4-((5-Хлорпиридин-2-ил)(гидрокси)метил)пиридин-2-ил)-5-((метоксикарбонил)амино)фенил)амино)-5-оксопентил)этантиоат: К раствору соединения примера 93С (5,00 г, 9,13 ммоль) и DIEA (4,78 мл, 27,40 ммоль) в DMF (100 мл) при 0°С добавляли тиоуксусную кислоту (1,957 мл, 27,40 ммоль). Полученную смесь перемешивали при 25°С в течение 1 ч. Ее разбавляли водой (300 мл) и экстрагировали DCM (200 мл × 3). Объединенные органические слои промывали водой (200 мл) и солевым раствором (200 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали методом колоночной флеш-хроматографии (SiO2, петролейный эфир : EtOAc = 5:1-1:1) с получением указанного соединения. MS (ESI) m/z 543.2 (M+H).
93E: S-(5-((2-(4-((5-Хлорпиридин-2-ил)((метилсульфонил)окси)метил)пиридин-2-ил)-5-((метоксикарбонил)амино)фенил)амино)-5-оксопентил)этантиоат: К раствору соединения примера 93D (4,70 г, 8,66 ммоль) и TEA (3,62 мл, 26,00 ммоль) в DCM (120 мл) при 0°С добавляли метансульфонилхлорид (1,38 мл, 17,31 ммоль). Полученную смесь перемешивали при 25°С в течение 3 ч. Ее разбавляли водой (100 мл) и экстрагировали DCM (100 мл × 3). Объединенные органические слои промывали водой (100 мл) и солевым раствором (100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением указанного соединения. MS (ESI) m/z 621.1 (М+Н).
93F: Метил(10-(5-хлорпиридин-2-ил)-4-оксо-9-тиа-3-аза-1(2,4)-пиридина-2(1,2)-бензолациклодекафан-24-ил)карбамат:
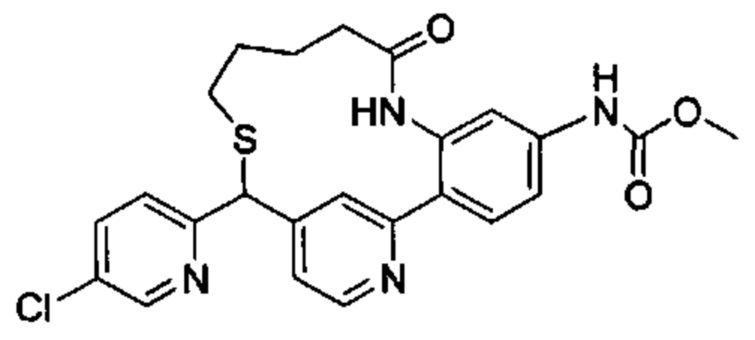
К смеси KOH (0,542 г, 9,66 ммоль) в EtOH (300 мл) при 0°С добавляли раствор соединения примера 93Е (4,00 г, 6,44 ммоль) в DCM (10 мл). Полученную смесь перемешивали при 25°С в течение 3 ч. Ее разбавляли водой (50 мл) и экстрагировали DCM (100 мл × 3). Объединенные органические слои промывали солевым раствором (100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением указанного соединения. MS (ESI) m/z 483.1 (М+Н).
93G: Метил(10-(5-хлорпиридин-2-ил)-9,9-диоксидо-4-оксо-9-тиа-3-аза-1(2,4)-пиридина-2(1,2-)-бензолациклодекафан-24-ил)карбамат:
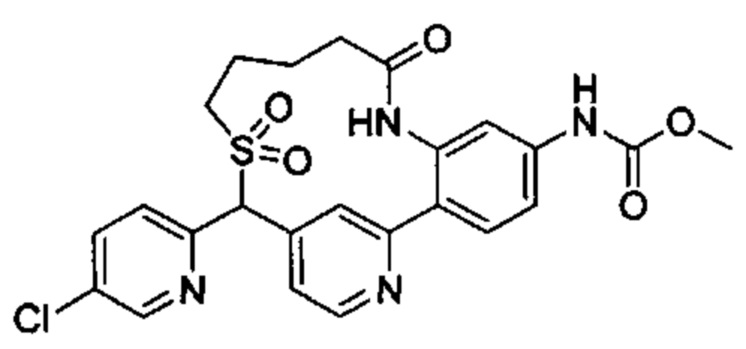
К раствору пероксимоносульфата калия (4,84 г, 7,87 ммоль) в воде (75 мл) при 0°С добавляли раствор соединения примера 93F (3,80 г, 7,87 ммоль) в МеОН (75 мл). Смесь перемешивали при 20°С в течение 13 ч. Ее разбавляли насыщенным раствором водного NaHCO3 (200 мл) и экстрагировали DCM (200 мл × 3). Объединенные органические слои промывали водой (200 мл) и солевым раствором (200 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением указанного соединения. MS (ESI) m/z 515.1 (M+H).
93H: Метил(E)-(9-(5-хлорпиридин-2-ил)-4-оксо-3-аза-1(2,4)-пиридина-2(1,2)-бензолациклононафан-8-ен-24-ил)карбамат:
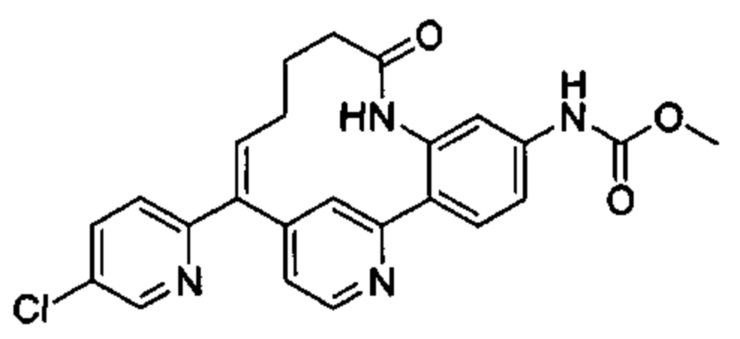
К суспензии KOH (5,88 г, 105 ммоль) и соединения примера 93G (2,70 г, 5,24 ммоль) в DCM (10 мл) и трет-BuOH (30 мл) добавляли CCl4 (2,024 мл, 20,97 ммоль) при 40°С. Смесь перемешивали в течение 4 ч при 40°С. Ее разбавляли водой (100 мл) и экстрагировали DCM (100 мл × 3). Органические слои сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии (SiO2, петролейный эфир : EtOAc = 3:1-1:3) с получением указанного соединения в виде твердого вещества. MS (ESI) m/z 449.1 (М+Н).
Пример 93:
Соединение примера 93 получали из соединения примера 93Н способом, подобным описанному в синтезе соединения примера 50. Продукт очищали методом HPLC с обращенной фазой (YMC-Actus Pro С18, 150×30 мм, MeCN/вода (0,1% TFA), 40 мл/мин) с получением указанного соединения в виде твердого вещества. MS (ESI) m/z 579.2 (М+Н). 1Н ЯМР (400МГц, CD3OD): δ 8.51 (s, 1H), 8.47 (d, J=5.1 Гц, 1Н), 7.92 (d, J=8.4 Гц, 1Н), 7.82 (s, 1Н), 7.77 (d, J=8.2 Гц, 1Н), 7.60-7.69 (m, 2Н), 7.43-7.51 (m, 2Н), 7.18-7.22 (m, 1Н), 6.93 (d, J=4.4 Гц, 1H), 4.80 (dd, J=12.1, 4.2 Гц, 1Н), 3.72 (s, 3Н), 2.46-2.56 (m, 1Н), 1.98-2.19 (m, 4Н), 1.68-1.72 (m, 1H), 1.50 (brs, 1Н), 1.10-1.18 (m, 1Н).
Рацемический образец соединения примера 93 подвергали хиральному разделению методом SFC (колонка AD, 250 мм × 30 мм, 55% EtOH 80 мл/мин) с получением соединения примера 93-а (быстрое элюирование) и соединения примера 93-b (более медленное элюирование).
ПРИМЕР 94
9-(5-(3-ХЛОР-2,6-ДИФТОРФЕНИЛ)-1-ОКСИДОПИРИДИН-2-ИЛ)-24-((МЕТОКСИКАРБОНИЛ)АМИНО)-4-ОКСО-3-АЗА-1(2,4)-ПИРИДИН-1-ИЯ-2(1,2)-БЕНЗОЛАЦИКЛОНОНАФАН 11-ОКСИД
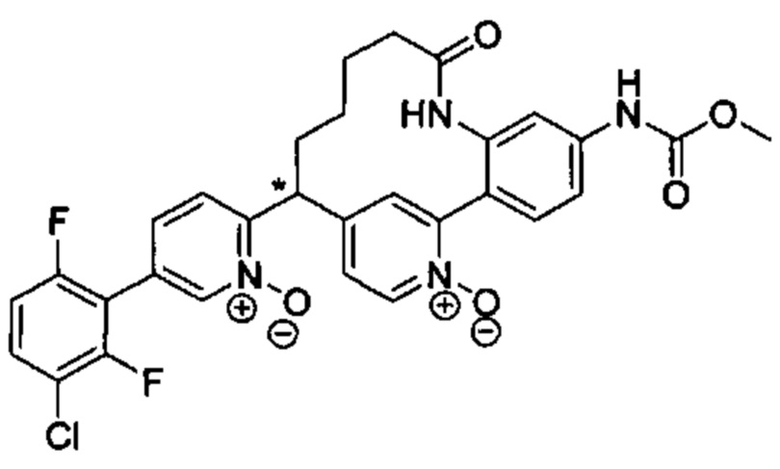
Соединение примера 94 выделяли из соединения примера 93 методом HPLC с обращенной фазой (YMC-Actus Pro С18, 150×30 мм, 20-50% MeCN в воде с 0,5% TFA, 40 мл/мин). MS (ES+) m/z: 595 (М+Н); 1Н ЯМР: (CD3OD, 400 МГц): δ 9.60 (br. s., 1Н), 8.59-8.71 (m, 1Н), 8.49 (br. s., 1H), 8.36 (d, J=6.7 Гц, 1H), 7.89 (d, J=7.8 Гц, 1H), 7.69-7.83 (m, 2H), 7.58-7.67 (m, 2H), 7.43 (d, J=8.6 Гц, 1H), 7.16-7.23 (m, 1H), 4.67-4.75 (m, 1H), 3.69-3.81 (m, 3H), 2.31-2.43 (m, 1H), 2.03-2.24 (m, 3H), 1.80 (br. s., 1H), 1.53 (br. s., 2H), 1.35 (br. s., 1H).
ПРИМЕР 95-a, 95-b, 95-c, 95-d
МЕТИЛ(9-(5-(3-ХЛОР-2,6-ДИФТОРФЕНИЛ)ПИРИДИН-2-ИЛ)-5-МЕТИЛ-4-ОКСО-3-АЗА-1(4,2)-ПИРИДИНА-2(1,2)-БЕНЗОЛАЦИКЛОНОНАФАН-24-ИЛ)КАРБАМАТ
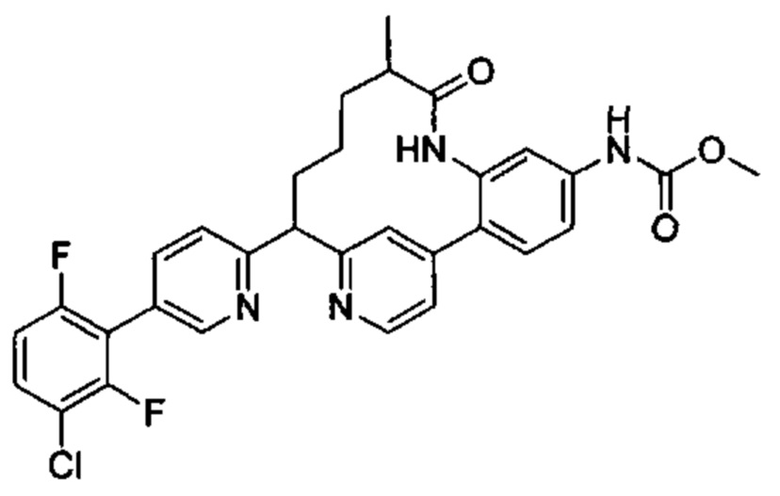
95A: Метил(Z)-(9-(5-(3-хлор-2,6-дифторфенил)пиридин-2-ил)-5-метил-4-оксо-3-аза-1(4,2)-пиридина-2(1,2)-бензолациклононафан-8-ен-24-ил)карбамат:
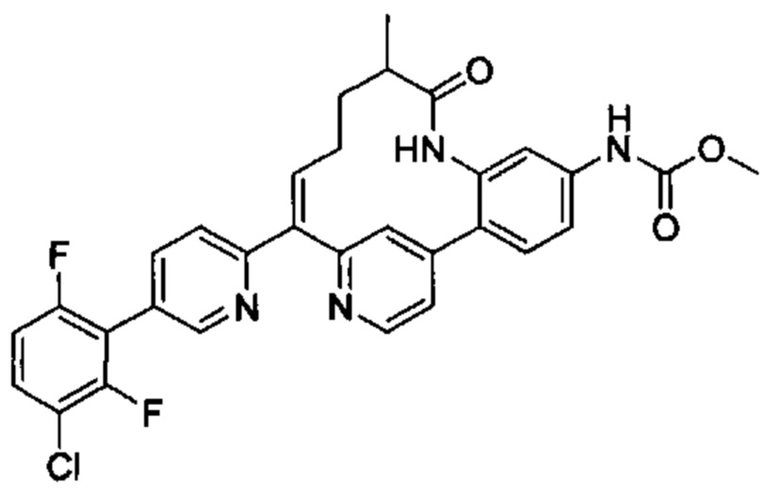
Указанное соединение получали из промежуточного соединения 10 способом, подобным описанному в синтезе соединения примера 50С. Продукт очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром : EtOAc = 1:1). MS (ESI) m/z 575.1 (М+Н).
Рацемический образец соединения примера 95А подвергали хиральному разделению методом SFC (Chiralpak AD, 250×30 мм, 50% EtOH (0,05% DEA)/CO2, 80 мл/мин) с получением соединения примера 95А-а (более быстрое элюирование) и соединения примера 95А-b (более медленное элюирование).
95В-а/95В-b: Метил(9-(5-(3-хлор-2,6-дифторфенил)пиридин-2-ил)-5-метил-4-оксо-3-аза-1(4,2)-пиридина-2(1,2)-бензолациклононафан-24-ил)карбамат:
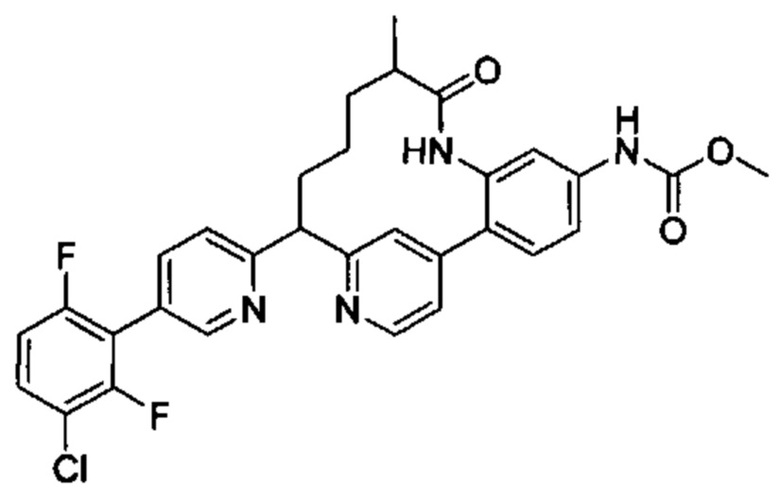
Смесь соединения примера 95А-а (120 мг, 0,209 ммоль) и никеля Ренея (12,25 мг, 0,209 ммоль) в THF (2 мл) перемешивали при 25°С в атмосфере водород (1 атм.) в течение 10 мин. Смесь фильтровали через слой целита (осторожно, воспламеняющееся вещество) и фильтрационный кек промывали метанолом (50 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали методом HPLC с обращенной фазой (YMC-Actus Pro С18, 150×30 мм, MeCN/вода (0,1% TFA), 40 мл/мин) с получением соединения примера 95В-а, MS (ESI) m/z 577.2 (М+Н) и соединения примера 95B-b, MS (ESI) m/z 577.2 (M+H).
95B-c/95B-d: Метил(9-(5-(3-хлор-2,6-дифторфенил)пиридин-2-ил)-5-метил-4-оксо-3-аза-1(4,2)-пиридина-2(1,2)-бензолациклононафан-24-ил)карбамат:
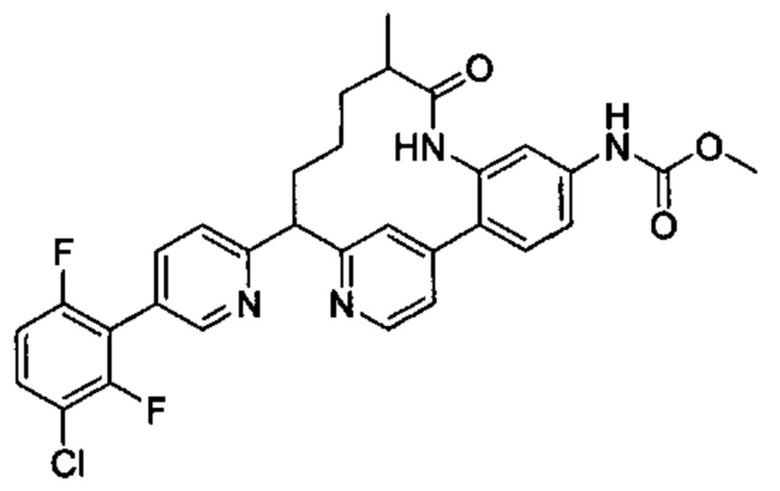
Смесь двух диастереомеров примеров 95B-c/95B-d получали из соединения примера 95А-b способом, описанным в синтезе соединения примеров 95B-a/95B-d. Продукт очищали методом HPLC с обращенной фазой (YMC-Actus Pro С18, 150×30 мм, MeCN/вода (0,1% TFA), 40 мл/мин) с получением соединения примера 95В-С (более быстрое элюирование), MS (ESI) m/z 577.2 (М+Н), и соединения примера 95B-d (более медленное элюирование), MS (ESI) m/z 577.2 (М+Н).
Пример 95-a/95-b/95-c/95-d:
Соединение примера 95-a/95-b/95-c/95-d получали из соединения примера 95В-a/95B-b/95B-c/95B-d, соответственно, способом, описанным в синтезе соединения примера 13. Продукты очищали методом HPLC с обращенной фазой (YMC-Actus Pro С18, 100×21 мм, MeCN/вода (0,1% TFA), 25 мл/мин).
Соединение примера 95-а: MS (ESI) m/z 593.2 (М+Н); 1Н ЯМР (400МГц, CD3OD): δ 8.61 (d, J=5.9 Гц, 1Н), 8.54 (s, 1Н), 8.01 (d, J=8.2 Гц, 1Н), 7.97 (s, 1Н), 7.81 (d, J=8.2 Гц, 1Н), 7.71 (d, J=5.5 Гц, 1Н), 7.69-7.60 (m, 2Н), 7.56 (d, J=2.7 Гц, 2Н), 7.21 (t, J=9.2 Гц, 1Н), 5.12-5.02 (m, 1Н), 3.76 (s, 3Н), 2.55-2.47 (m, 1Н), 2.40-2.27 (m, 1Н), 2.25-2.13 (m, 1Н), 1.82 (q, J=10.7 Гц, 1Н), 1.65-1.51 (m, 1H), 1.27 (d, J=6.1 Гц, 4Н), 1.15-1.03 (m, 1Н).
Соединение примера 95-b: MS (ESI) m/z 593.2 (М+Н); 1Н ЯМР (400МГц, CD3OD): δ 8.61-8.54 (m, 2Н), 8.11 (s, 1H), 7.98 (d, J=8.2 Гц, 1Н), 7.82 (d, J=8.2 Гц, 1Н), 7.71-7.64 (m, 2Н), 7.60-7.56 (m, 2Н), 7.52-7.47 (m, 1Н), 7.22 (t, J=8.6 Гц, 1Н), 4.99-4.95 (m, 1Н), 3.77 (s, 3Н), 2.91-2.75 (m, 1Н), 2.44 (t, J=12.7 Гц, 1H), 2.11 (t, J=11.2 Гц, 2Н), 1.71-1.54 (m, 2Н), 0.99 (d, J=7.0 Гц, 3Н), 0.63-0.47 (m, 1Н).
Соединение примера 95-с: MS (ESI) m/z 593.2 (М+Н); 1Н ЯМР (400МГц, CD3OD): δ 8.61 (d, J=5.9 Гц, 1Н), 8.54 (s, 1Н), 8.01 (d, J=8.2 Гц, 1Н), 7.97 (s, 1Н), 7.81 (d, J=8.2 Гц, 1Н), 7.71 (d, J=5.5 Гц, 1Н), 7.69-7.60 (m, 2Н), 7.56 (d, J=2.7 Гц, 2Н), 7.21 (t, J=9.2 Гц, 1Н), 5.12-5.02 (m, 1Н), 3.76 (s, 3Н), 2.55-2.47 (m, 1Н), 2.40-2.27 (m, 1H), 2.25-2.13 (m, 1Н), 1.82 (q, J=10.7 Гц, 1H), 1.65-1.51 (m, 1Н), 1.27 (d, J=6.7 Гц, 4Н), 1.15-1.03 (m, 1Н).
Соединение примера 95-d: MS (ESI) m/z 593.2 (М+Н); 1H ЯМР (400МГц, CD3OD): δ 8.61 -8.54 (m, 2Н), 8.11 (s, 1Н), 7.98 (d, J=8.2 Гц, 1Н), 7.82 (d, J=8.2 Гц, 1Н), 7.71-7.64 (m, 2Н), 7.60-7.56 (m, 2Н), 7.52-7.47 (m, 1Н), 7.22 (t, J=8.6 Гц, 1H), 4.99-4.95 (m, 1Н), 3.77 (s, 3Н), 2.91-2.75 (m, 1Н), 2.44 (t, J=12.7 Гц, 1Н), 2.11 (t, J=11.2 Гц, 2Н), 1.71-1.54 (m, 2Н), 0.99 (d, J=7.0 Гц, 3Н), 0.63-0.47 (m, 1Н).
ПРИМЕР 96 (рацемат), 96-а и 96-b
(Z)-5-(3-ХЛОР-2,6-ДИФТОРФЕНИЛ)-2-(24-((МЕТОКСИКАРБОНИЛ)АМИНО)-4-ОКСО-11H-3-АЗА-1(4,2)-ИМИДАЗОЛ-2(1,2)-БЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
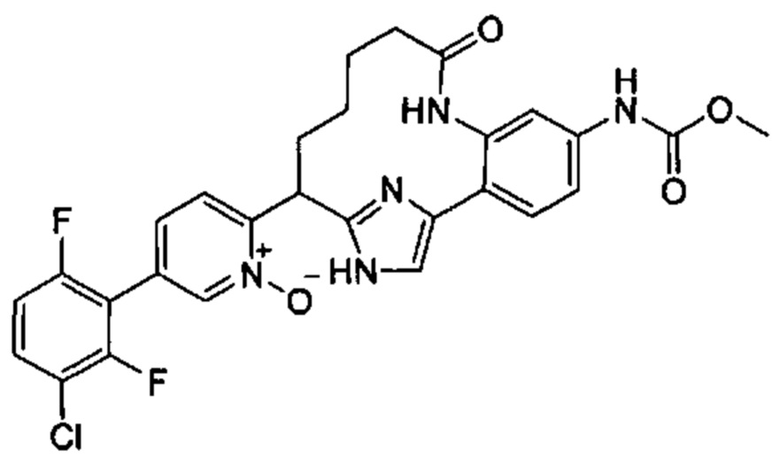
96А: Метил((12Z,8Z)-9-(5-(3-хлор-2,6-дифторфенил)пиридин-2-ил)-4-оксо-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-8-ен-24-ил)карбамат:
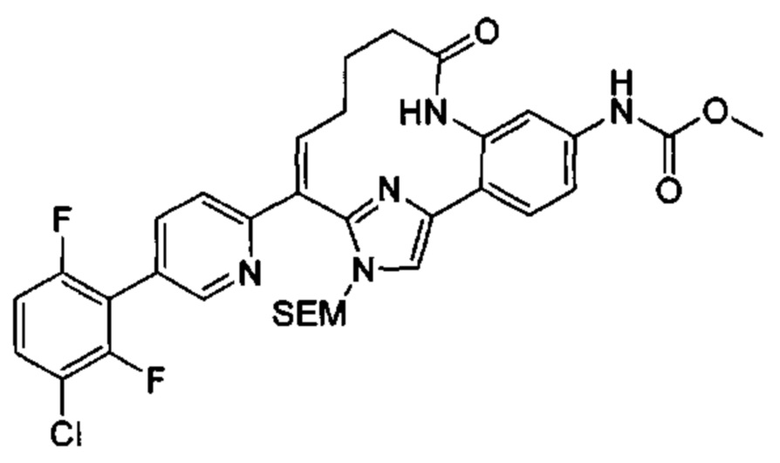
Указанное соединение получали из промежуточного соединения 56 способом, описанным в примере 50С. Продукт очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром : EtOAc = 1:1 объем/объем). MS (ES+) m/z: 680 (М+Н.
96В: Метил(Z)-(9-(5-(3-хлор-2,6-дифторфенил)пиридин-2-ил)-4-оксо-11-((2-(триметилсилил)этокси)метил-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-24-ил)карбамат:
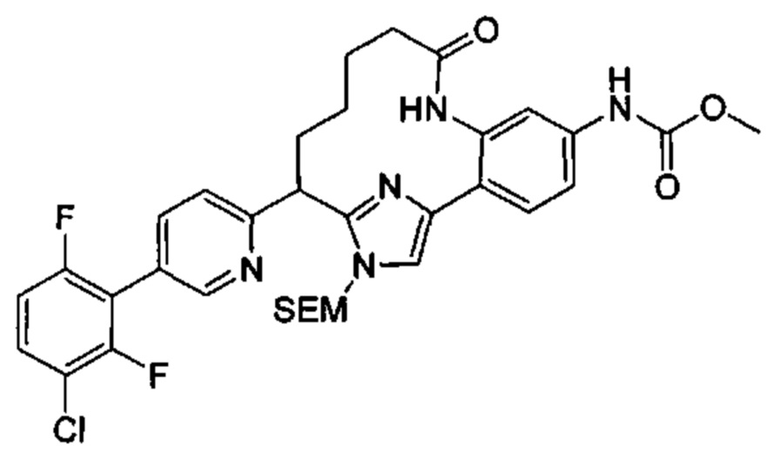
Мутный раствор соединения примера 96А (1,1 г, 1,455 ммоль) в THF (40 мл) добавляли большое избыточное количество никеля Ренея. Смесь встряхивали в атмосфере водорода (40 фунт/кв. дюйм) при к.т. в течение 1 ч. В сосуд добавляли 200 мл 10% метанола в DCM и обрабатывали ультразвуком в течение 10 мин. Катализаторы удаляли фильтрацией через слой целита и промывали 200 мл 10% МеОН в DCM, а затем 50 мл метанола (Осторожно! Никогда не оставляйте катализатор на воздухе). Катализатор покрывали водой (10 мл) и размещали в контейнер, покрытый водой. Фильтрат концентрировали при пониженном давлении. Остаток суспендировали в метаноле (50 мл) и фильтровали. Твердые вещества промывали метанолом, затем диэтиловым эфиром. Их сушили на воздухе всю ночь с получением указанного соединения. MS (ES+) m/z: 683 (М+Н).
96С: (Z)-5-(3-Хлор-2,6-дифторфенил)-2-(24-((метоксикарбонил)амино)-4-оксо-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-9-ил)пиридина 1-оксид:
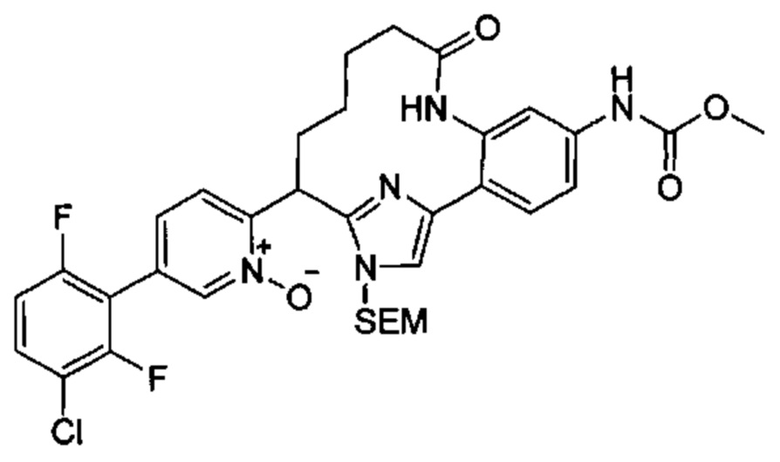
В круглодонную колбу объемом 100 мл с магнитной мешалкой добавляли соединение примера 96В (7 г, 10,26 ммоль) и уксусную кислоту (35,0 мл). Ее помещали в водяную баню при к.т. и к перемешиваемой суспензионной смеси добавляли охлажденную перуксусную кислоту (39% масс, в уксусной кислоте, 51,1 мл, 308 ммоль). Ее перемешивали в течение 5 ч. Реакционную смесь переносили в делительную воронку и медленно вливали по каплям в больший химический стакан с перемешиваемой смесью льда (1000 г), тиосульфата натрия (81 г, 513 ммоль) и карбоната натрия (98 г, 923 ммоль). После вливания смесь перемешивали в течение 30 мин, пока не растаял лед. Осадок собирали фильтрацией. Твердый кек промывали водой и сушили на воздухе. Неочищенный продукт повторно растворяли в DCM/MeOH (5:1) и очищали методом колоночной хроматографии на силикагеле (элюировали 0-85% EtOAc/DCM и 0-6% MeOH/DCM, соответственно) с получением указанного соединения. MS (ES+) m/z: 698 (М+Н).
Пример 96:
В колбу с соединением примера 96С (2,4 г, 3,44 ммоль) и DCM (12,00 мл) добавляли TFA (13,24 мл, 172 ммоль). Смесь перемешивали при к.т. в течение 6 ч. Ее переносили в делительную воронку и медленно вливали по каплям к перемешиваемой смеси льда (200 г) и карбоната натрия (25,5 г, 241 ммоль). Смесь экстрагировали DCM (150 мл × 3). Объединенные органические слои промывали солевым раствором, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-6% MeOH/DCM) с получением указанного соединения. MS (ES+) m/z: 568 (М+Н).
Образец рацемического соединения примера 96 подвергали хиральному разделению методом SFC (AS-H, 250×30 мм; 50% метанол (0,05% диэтиламин)/CO2, 80 мл/мин) с получением соединения примера 96-а (более медленное элюирование) и соединения примера 96-b (более быстрое элюирование). MS (ES+) m/z: 568 (М+Н); 1Н ЯМР (CD3OD, 400 МГц): δ 8.57 (s, 1Н), 7.96 (d, J=8.4 Гц, 1Н), 7.81 (d, J=8.4 Гц, 1H), 7.68 (dt, J=5.7, 8.6 Гц, 1Н), 7.61 (s, 1Н), 7.54-7.48 (m, 1Н), 7.44-7.40 (m, 2Н), 7.27-7.18 (m, 1Н), 4.97 (dd, J=6.9, 10.7 Гц, 1Н), 3.77 (s, 3Н), 2.57 (d, J=12.8 Гц, 1H), 2.44-2.28 (m, 2Н), 2.11 (m, 1Н), 1.95-1.80 (m, 1Н), 1.76-1.51 (m, 2Н), 1.12 (m, 1Н).
ПРИМЕР 97
МЕТИЛ(Z)-(9-(5-(5-ХЛОР-2-(1H-ТЕТРАЗОЛ-1-ИЛ)ФЕНИЛ)ПИРИДИН-2-ИЛ)-4-ОКСО-11H-3-АЗА-1(4,2)-ИМИДАЗОЛ-2(1,2)-БЕНЗОЛАЦИКЛОНОНАФАН-24-ИЛ)КАРБАМАТ

97А: Метил((12Z,8Z)-9-(5-(2-амино-5-хлорфенил)пиридин-2-ил)-4-оксо-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-8-ен-24-ил)карбамат:

В реакционный сосуд для микроволнового облучения с магнитной мешалкой добавляли промежуточное соединение 56 (200 мг, 0,326 ммоль), промежуточное соединение 33 (166 мг, 0,653 ммоль), 1,1'-бис(ди-трет-бутилфосфино)ферроцен палладия дихлорид (21,28 мг, 0,033 ммоль), K2CO3 (135 мг, 0,979 ммоль), THF (9 мл) и воду (3 мл). Смесь нагревали при 120°С в микроволновом реакторе в течение 0,5 ч. Ее охлаждали до к.т. и разбавляли Н2О (10 мл). Смесь экстрагировали DCM (3×20 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали методом TLC (1000 мкм, петролейный эфир : EtOAc = 1:2) с получением указанного соединения. MS (ES+) m/z: 659 (М+Н).
97В: Метил(Z)-(9-(5-(2-амино-5-хлорфенил)пиридин-2-ил)-4-оксо-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-24-ил)карбамат:

Указанное соединение получали из соединения примера 97А способом, описанным в пример 102В. Его использовали без очистки. MS (ES+) m/z: 661 (М+Н).
97С: Метил(Z)-(9-(5-(5-хлор-2-(1H-тетразол-1-ил)фенил)пиридин-2-ил)-4-оксо-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-24-ил)карбамат:
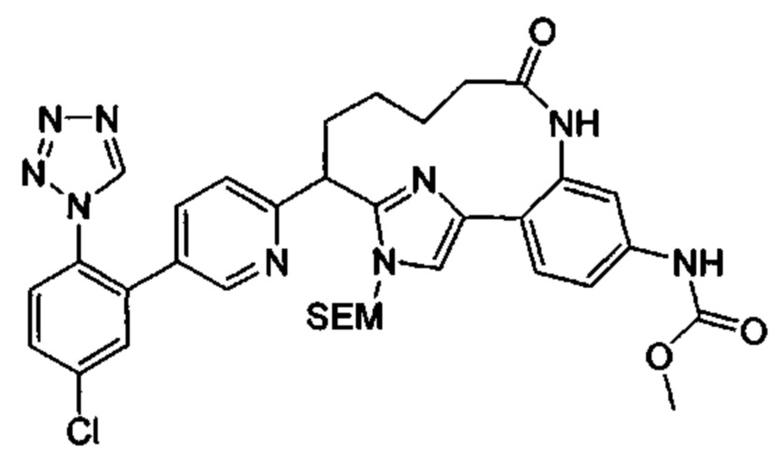
К раствору соединения примера 97В (120 мг, 0,127 ммоль) в НОАс (5 мл) добавляли триметилортоформиат (135 мг, 1,270 ммоль) и азид натрия (49,5 мг, 0,762 ммоль). Смесь перемешивали при 30°С в течение 15 ч. Смесь гасили насыщенным NaNO2 (30 мл) и насыщенным водным бикарбонатом натрия. Ее экстрагировали EtOAc (3×20 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением указанного соединения, которое использовали на следующей стадии без дополнительной очистки. MS (ES+) m/z: 714 (М+Н).
Пример 97:
Соединение примера 97 получали из соединения примера 97С способом, описанным в примере 11. Остаток очищали методом HPLC с обращенной фазой (YMC-Actus Pro С18, 150×30 мм, MeCN/вода (0,1% TFA), 40 мл/мин) с получением указанного соединения. MS (ES+) m/z: 584 (М+Н).
ПРИМЕР 98 (рацемат), 98-а и 98-b
(Z)-5-(5-ХЛОР-2-(1H-ТЕТРАЗОЛ-1-ИЛ)ФЕНИЛ)-2-(24-((МЕТОКСИКАРБОНИЛ)АМИНО)-4-OKCO-11H-3-АЗА-1(4,2)-ИМИДАЗОЛ-2(1,2)-БЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
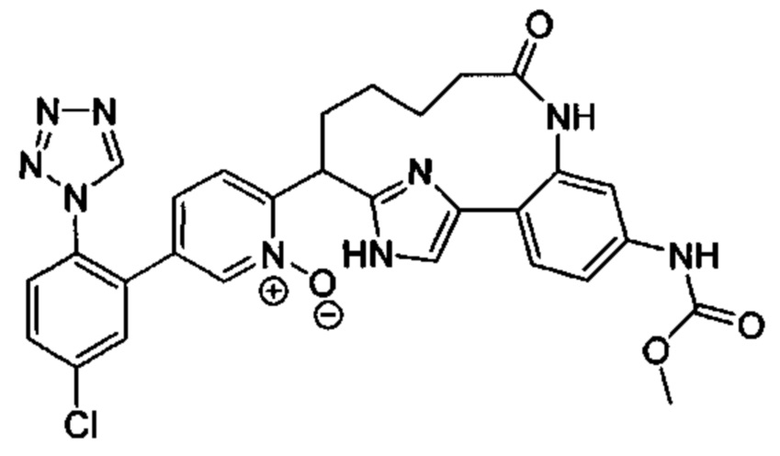
Соединение примера 98 получали из соединения примера 97С способами, описанными в примере 96. Его очищали методом HPLC с обращенной фазой с получением рацемического продукта. MS (ES+) m/z: 600 (М+Н).
Образец соединения примера 98 подвергали хиральному разделению методом SFC (OD-3,4.6×50 мм, 40% метанол (0,05% диэтиламин) в СО2; 4 мл/мин; 40°С) с получением соединения примера 98-а (более быстрое элюирование) и соединения примера 98-b (более медленное элюирование). MS (ES+) m/z: 600 (М+Н); 1Н ЯМР: (CD3OD, 400 МГц): δ 9.42 (s, 1Н), 8.31 (s, 1Н), 7.78-7.83 (m, 2Н), 7.67-7.76 (m, 2Н), 7.61 (s, 1Н), 7.49 (d, J=8.6 Гц, 1Н), 7.41 (m, 1Н), 7.35 (s, 1H), 7.30 (d, J=7.8 Гц, 1Н), 3.76 (s, 3Н), 2.53 (d, J=11.7 Гц, 1Н), 2.11-2.35 (m, 3Н), 1.87 (d, J=11.0 Гц, 1Н), 1.63 (m, 1Н), 1.45 (m, 1H), 1.29 (q, J=6.9 Гц, 1Н), 1.13 (m, 1Н).
ПРИМЕР 99, 99-а, 99-b
(Z)-5-(5-ХЛОР-2-(ТРИФТОРМЕТОКСИ)ФЕНИЛ)-2-(24-((МЕТОКСИКАРБОНИЛ)АМИНО)-4-ОКСО-11H-3-АЗА-1(4,2)-ИМИДАЗОЛ-2(1,2)-БЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
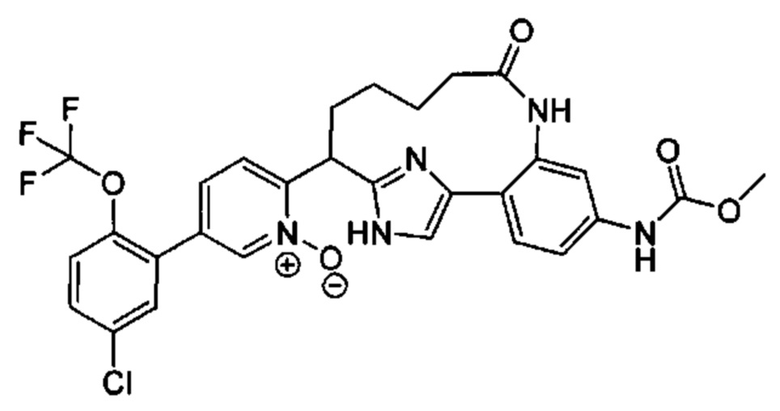
Соединение примера 99 получали способом, описанным в синтезе соединения примера 96. Продукт очищали методом HPLC с обращенной фазой (YMC-Actus Pro С18, 150×30 мм, MeCN/вода (0,1% TFA), 40 мл/мин) с получением указанного соединения. MS (ESI) m/z 616.2 (М+Н).
Рацемический образец соединения примера 99 подвергали хиральному разделению методом SFC (AS-H, 250×21 мм, 40% этанол (0,05% DEA)/CO2, 60 мл/мин) с получением соединения примера 99-а (более быстрое элюирование) и соединения примера 99-b (второе элюирование). MS (ESI) m/z 616.1 (М+Н); 1Н ЯМР (400МГц, CD3OD): δ 8.53 (s, 1Н), 7.74-7.54 (m, 5Н), 7.53-7.43 (m, 2Н), 7.38 (d, J=7.9 Гц, 1H), 7.14 (s, 1H), 4.95 (d, J=5.1 Гц, 1Н), 3.74 (s, 3Н), 2.56-2.24 (m, 3Н), 2.23-2.09 (m, 1Н), 2.08-1.93 (m, 1Н), 1.73-1.57 (m, 1H), 1.31-1.14 (m, 2Н).
ПРИМЕР 100, 100-а, 100-b
(Z)-5-(3-ХЛОР-6-(ДИФТОРМЕТОКСИ)-2-ФТОРФЕНИЛ)-2-(24-((МЕТОКСИКАРБОНИЛ)АМИНО)-4-ОКСО-11H-3-АЗА-1(4,2)-ИМИДАЗОЛ-2(1,2)-БЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
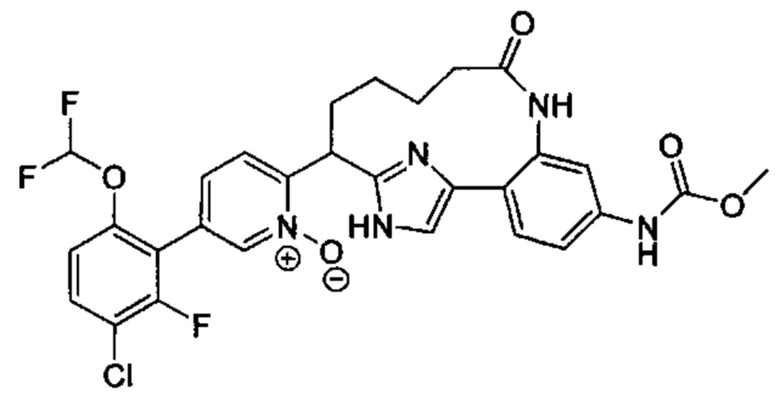
100А: (Z)-5-(3-Хлор-6-(дифторметокси)-2-фторфенил)-2-(24-((метоксикарбонил)амино)-4-оксо-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-9-ил)пиридина 1-оксид:
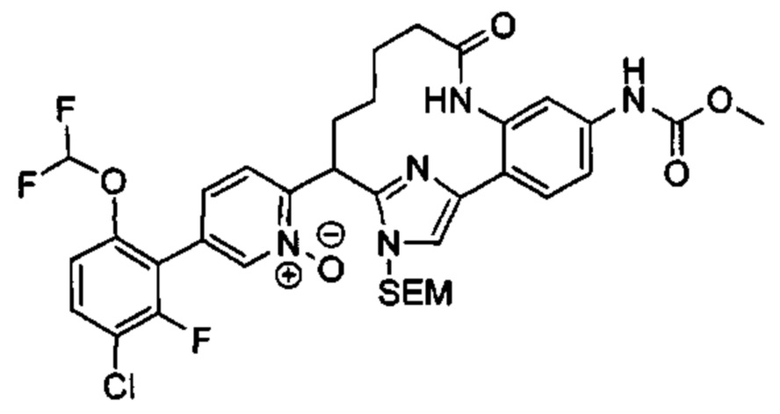
Указанное соединение получали способом, описанным в синтезе соединения примера 96С. Продукт очищали методом колоночной флеш-хроматографии (элюировали МеОН/CH2Cl2=5%). MS (ES+) m/z: 746 (М+Н).
Пример 100, 100-а и 100-b
Смесь соединения примера 100А (89 мг, 0,119 ммоль), HCl (4 н в диоксане) (1,2 мл, 4,77 ммоль) и воды (0,298 мл) перемешивали при 50°С в течение 2 ч. Ее охлаждали до к.т. и большую часть растворителя удаляли при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии (элюировали МеОН/CH2Cl2=7%) с получением указанного соединения. MS (ES+) m/z: 616 (М+Н).
Рацемический образец соединения примера 100 подвергали хиральному разделению методом SFC (IC, 21×250 мм, 48% МеОН/CO2, 100 бар, 60 мл/мин, 35°С) с получением соединения примера 100-а (более быстрое элюирование) и соединения примера 100-b (более медленное элюирование). MS (ES+) m/z: 616(М+Н).
ПРИМЕР 101, 101-а, 101-b
(Z)-5-(2-ДИФТОРМЕТОКСИ)-6-ФТОРФЕНИЛ)-2-(24-((МЕТОКСИКАРБОНИЛ)АМИНО)-4-ОКСО-11H-3-АЗА-1(4,2)-ИМИДАЗОЛ-2(1,2)-БЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
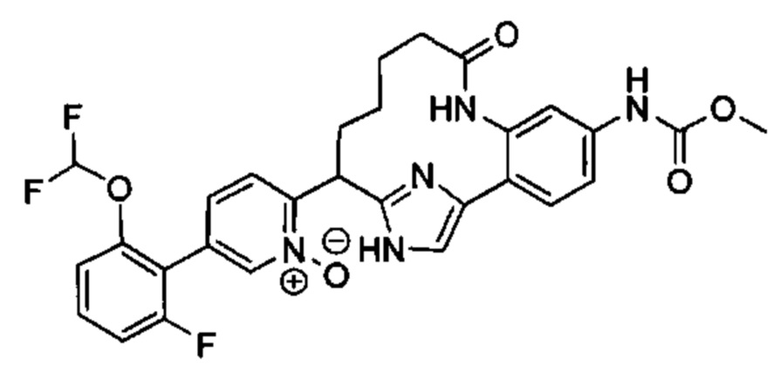
Соединение примера 101 получали способом, описанным в синтезе соединения примера 100. MS (ES+) m/z: 582(М+Н).
Рацемический образец соединения примера 101 подвергали хиральному разделению методом SFC (OJ, 30×250 мм, 45% МеОН+0,2%DEA/CO2, 100 бар, 70 мл/мин, 35°С) с получением соединения примера 101-а (более быстрое элюирование) и соединения примера 101-b (более медленное элюирование). MS (ES+) m/z: 582 (М+Н).
ПРИМЕР 102, 102-а, 102-b
(Z)-5-(5-ХЛОР-2-(ДИФТОРМЕТОКСИ)ФЕНИЛ)-2-(24-((МЕТОКСИКАРБОНИЛ)АМИНО)-4-ОКСО-11H-3-АЗА-1(4,2)-ИМИДАЗОЛ-2(1,2)-БЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
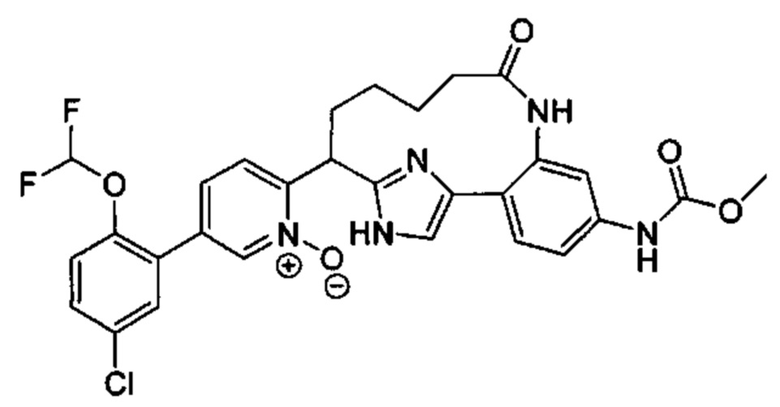
102А: Метил(Z)-(9-(5-(5-хлор-2-(дифторметокси)фенил)пиридин-2-ил)-4-оксо-11-((2-(триметилсилил)этокси)метил-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-24-ил)карбамат:
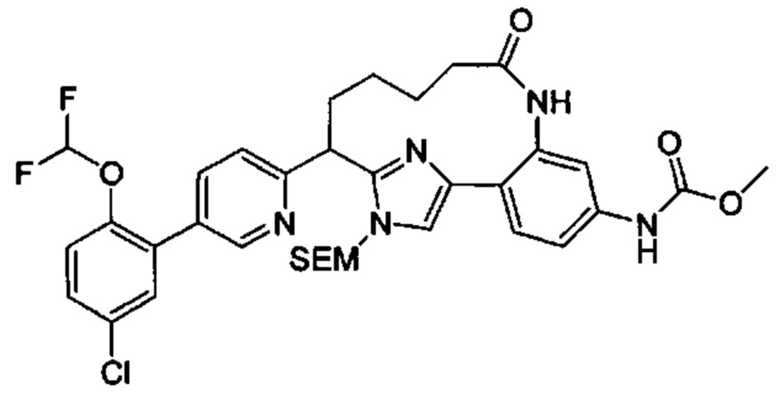
Указанное соединение получали способом, описанным в синтезе соединения примера 96В. MS (ESI) m/z 712 (М+Н).
102В: Метил(Z)-(9-(5-(5-хлор-2-(дифторметокси)фенил)пиридин-2-ил)-4-оксо-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-24-ил)карбамат:
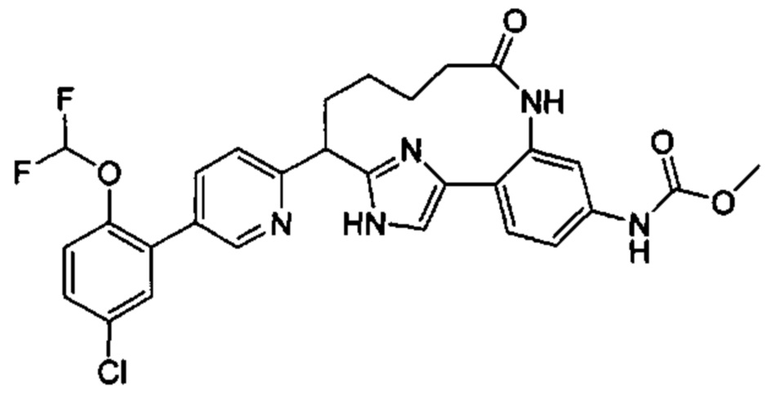
Соединение примера 102А (2,82 г, 3,96 ммоль) растворяли в HCl (4M в диоксане) (36 мл, 144 ммоль) и воде (3,60 мл). Раствор перемешивали при 50°С в течение 3 ч. Его охлаждали до к.т. и большую часть растворителя удаляли при пониженном давлении. Остаток медленно добавляли к перемешиваемой смеси льда (30 г) и насыщенного водного карбоната натрия (30 мл). Твердые вещества осаждались и смесь выдерживали всю ночь. Твердое вещество собирали фильтрацией и промывали водой (2×20 мл) и диэтиловым эфиром (20 мл). Его сушили на воздухе с получением указанного соединения. MS (ESI) m/z 581.9 (М+Н).
Пример 102:
К раствору соединения примера 102В (1,85 г, 3,18 ммоль) в 16 мл уксусной кислоты добавляли перуксусную кислоту (39% раствор в уксусной кислоте) (16 мл, 96 ммоль). Раствор перемешивали при к.т. в течение 6 ч. Его медленно добавляли к перемешиваемой смеси льда (500 г), насыщенного карбоната натрия (100 мл) и насыщенного тиосульфата натрия (100 мл). Образовывались осадки и их выдерживали в течение 1 ч перед фильтрацией. Серые твердые вещества собирали и очищали методом колоночной флеш-хроматографии (0-10% метанола в DCM) с получением указанного соединения. MS (ES+) m/z: 598.0 (M+H).
Рацемический образец соединения примера 102 подвергали хиральному разделению методом SFC (Kromasil-5, 30×250 мм, 60% MeOH-MeCN (2:1)/СО2, 100 бар, 70 мл/мин, 35°С) с получением соединения примера 102-а (более медленное элюирование) и соединения примера 102-b (более быстрое элюирование). MS (ES+) m/z: 582 (М+Н). 1Н ЯМР (500 МГц, DMSO-d6): δ 12.13 (s, 1Н), 11.85 (s, 1H), 9.66 (s, 1H), 8.51 (s, 1H), 7.77 (s, 1H), 7.63 (m, 1H), 7.57 (m, 1H), 7.50-7.40 (m, 2H), 7.38-7.25 (m, 4H), 7.22 (t, J=73.5 Гц, 1 H), 4.82 (t, J=8.0 Гц, 1 H), 3.64 (s, 3 H), 2.47 (m, 1H), 2.25 (m, 2H), 2.01 (m, 1H), 1.87 (m, 1H), 1.57 (m, 1H), 1.46 (m, 1H), 1.14 (m, 1H).
ПРИМЕР 103, 103-a, 103-b
(Z)-5-(3-ХЛОР-2,6-ДИФТОР-4-ЙОДФЕНИЛ)-2-(24-((МЕТОКСИКАРБОНИЛ)АМИНО)-4-ОКСО-11H-3-АЗА-1(4,2)-ИМИДАЗОЛ-2(1,2)-БЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
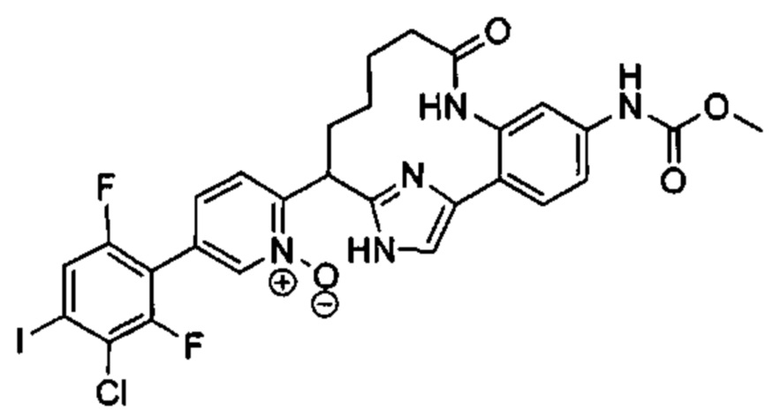
103А: 4-Бром-2-хлор-3,5-дифторанилин: Смесь 4-бром-3,5-дифторанилина (400 мг, 1,923 ммоль) и NCS (257 мг, 1,923 ммоль) в DMF (3,8 мл) перемешивали при 60°С в течение 90 мин. Ее разбавляли диэтиловым эфиром (40 мл), промывали водой (20 мл) и солевым раствором (20 мл). Органический слой сушили над сульфатом магния, фильтровали и растворитель выпаривали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии (элюировали EtOAc/гексаном=50%) с получением указанного соединения. MS (ES+) m/z: 242, 244 (М+Н).
103В: Метил(Z)-(9-(5-(4-амино-3-хлор-2,6-дифторфенил)пиридин-2-ил)-4-оксо-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-24-ил)карбамат:
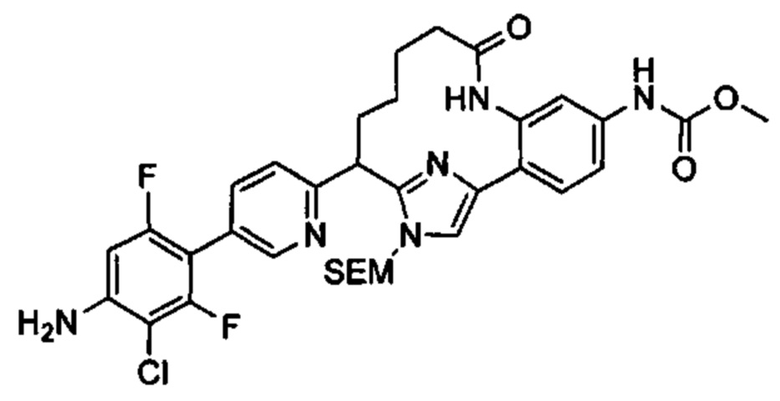
Указанное соединение получали из промежуточного соединения 55 и соединения примера 109А способом, описанным в синтезе соединения 102В. MS (ES+) m/z: 698 (М+Н).
103С: Метил(Z)-(9-(5-(3-хлор-2,6-дифтор-4-йодфенил)пиридин-2-ил)-4-оксо-11-((2-(триметилсилил)этокси)метил)-11Н-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-24-ил)карбамат:
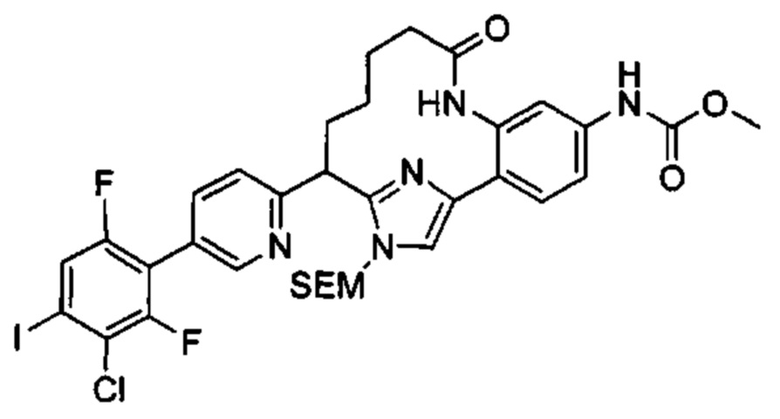
Смесь нитрита натрия (0,252 мл, 0,252 ммоль), соединения примера 103В (160 мг, 0,229 ммоль), хлористоводородной кислоты (37% масс., 1,5 мл), воды (3,00 мл) и ацетонитрила (3 мл) перемешивали при 0°С в течение 10 мин. К смеси добавляли водный KI (1 М, 0,241 мл, 0,241 ммоль) и перемешивали при к.т. в течение 1 ч. Добавляли водный карбонат калия (10% масс., 1 мл) и водный тиосульфат натрия (насыщенный, 1 мл). Значение рН раствора доводили до 8 добавлением водного карбоната натрия (10%). Образовывался желтый осадок и его выдерживали в течение 1 ч. Твердые вещества собирали фильтрацией и сушили на воздухе. Неочищенный продукт очищали методом колоночной флеш-хроматографии (элюировали МеОН/CH2Cl2=7%) с получением указанного соединения. MS (ES+) m/z: 808 (М+Н).
Пример 103. 103-а и 103-b:
Соединение примера 103 получали из соединения примера 103С способами, описанными в синтезе соединения примера 102. Продукт очищали методом колоночной флеш-хроматографии (элюировали МеОН/CH2Cl2=5%) с получением указанного соединения. MS (ES+) m/z: 694 (М+Н).
Образец рацемического соединения примера 103 подвергали хиральному разделению методом SFC (OJ, 21×250 мм, 25% МеОН (0,2% NH4OH)/CO2, 100 бар, 60 мл/мин, 35°С) с получением соединения примера 103-а (более быстрое элюирование) и соединения примера 103-b (более медленное элюирование). MS (ES+) m/z: 694 (М+Н).
ПРИМЕР 104, 104-а и 104-b
((Z)-9-(5-(3-ХЛОР-2,6-ДИФТОРФЕНИЛ)ПИРИДИН-2-ИЛ)-25-ФТОР-11H-3-АЗА-1(4,2)-ИМИДАЗОЛ-2(1,2)-БЕНЗОЛАЦИКЛОНОНАФАН-4-ОН
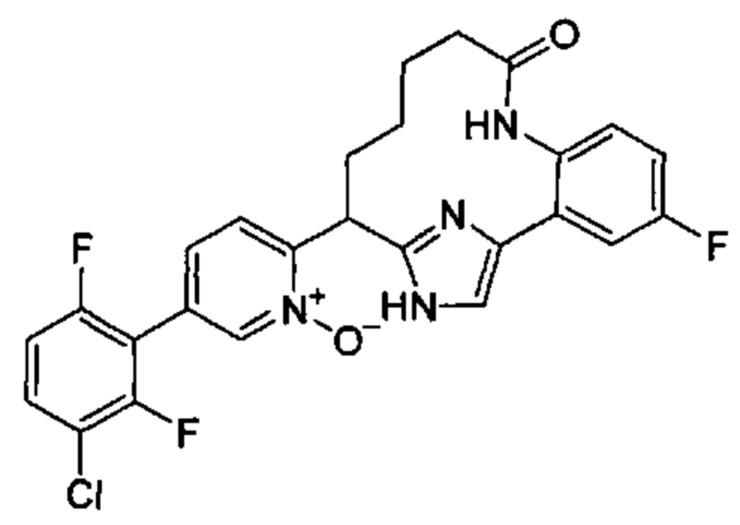
104А: (12Z,8Z)-9-(5-Бромпиридин-2-ил)-25-фтор-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-8-ен-4-он:
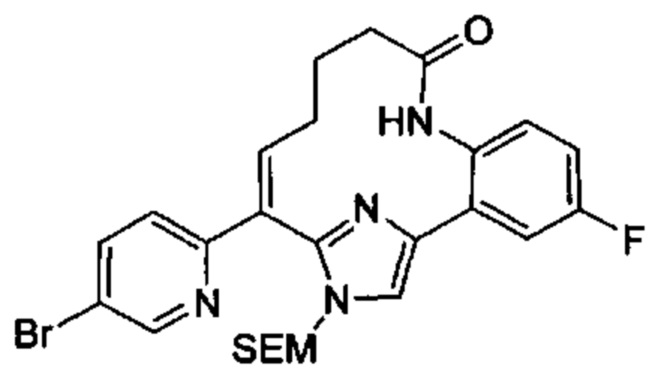
Указанное соединение получали из промежуточного соединения 15 и промежуточного соединения 18 способом, описанным в синтезе соединения примера 12С. Его очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром : EtOAc = 5:1 объем/объем). MS (ES+) m/z: 557, 559 (М+Н).
Пример 104:
Соединение примера 104 получали из соединения примера 104А способом, описанным в синтезе соединения примера 96. Продукт очищали методом HPLC с обращенной фазой. MS (ES+) m/z: 513 (М+Н).
Образец рацемического продукта подвергали хиральному разделению методом SFC (AS-H, 4,6×150 мм, 5 мкм; этанол (0,05% диэтиламин)/СО2 градиент; 3 мл/мин, 40°С) с получением соединения примера 104-а (более быстрое элюирование) и соединения примера 104-b (более медленное элюирование). MS (ES+) m/z: 513 (М+Н); 1Н ЯМР (CD3OD, 400 МГц): δ 8.57 (s, 1Н), 7.97 (d, J=8.4 Гц, 1Н), 7.81 (d, J=8.4 Гц, 1Н), 7.68 (dt, J=6.0, 8.6 Гц, 1Н), 7.54 (s, 1Н), 7.43 (dd, J=2.3, 8.5 Гц, 1Н), 7.38-7.27 (m, 2Н), 7.23 (t, J=8.5 Гц, 1Н), 5.00-4.96 (m, 1Н), 2.57 (d, J=12.8 Гц, 1Н), 2.36 (d, J=3.7 Гц, 2Н), 2.16 (t, J=12.1 Гц, 1Н), 1.88 (q, J=12.1 Гц, 1Н), 1.75-1.50 (m, 2Н), 1.12 (m, 1Н).
ПРИМЕР 105, 105-а, 105-b
(Z)-5-(5-ХЛОР-2-(4-(ДИФТОРМЕТИЛ)-1H-1,2,3-ТРИАЗОЛ-1-ИЛ)ФЕНИЛ)-2-(25-ФТОР-4-ОКСО-11H-3-АЗА-1(4,2)-ИМИДАЗОЛ-2(1,2)-БЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
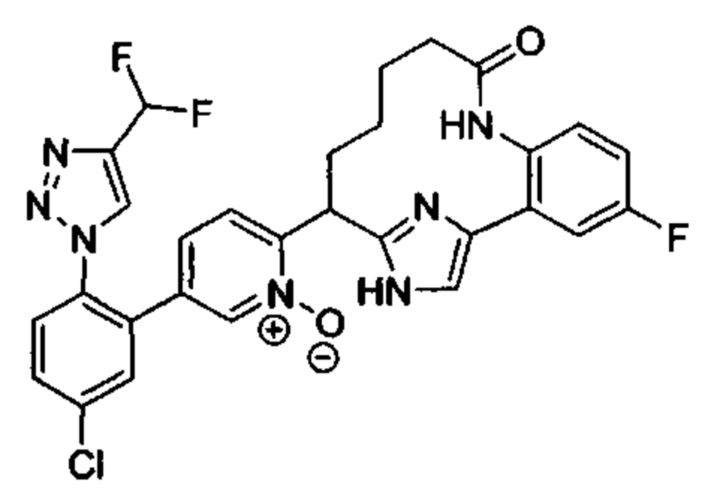
105А: (12Z,8Z)-9-(5-(5-Хлор-2-(4-(дифторметил)-1H-1,2,3-триазол-1-ил)фенил)пиридин-2-ил)-25-фтор-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-8-ен-4-он:
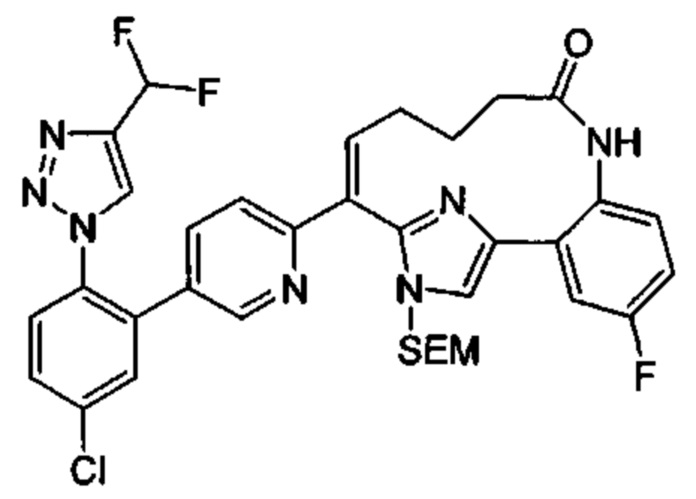
К раствору соединения примера 104А (230 мг, 0,413 ммоль) и промежуточного соединения 46 (194 мг, 0,495 ммоль) в толуоле (10 мл) в перчаточный бокс добавляли K3PO4 (175 мг, 0,835 ммоль) и бифенильный предварительный каталитический нейтрализатор Ad2nBuP (55,2 мг, 0,083 ммоль) в атмосфере азота. Полученную смесь перемешивали при 80°С в атмосфере азота в течение 18 ч. Ее оставляли охлаждаться до к. т и смесь экстрагировали EtOAc (3×10 мл). Объединенные органические слои промывали солевым раствором (10 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом TLC (петролейный эфир : EtOAc=2:1) с получением указанного соединения. MS (ES+) m/z: 706 (М+Н).
Пример 105:
Соединение примера 105 получали из соединения примера 105А способами, описанными в синтезе примера 96. Продукт очищали методом HPLC с обращенной фазой с получением указанного соединения. MS (ES+) m/z: 594 (М+Н).
Образец соединения примера 105 подвергали хиральному разделению методом SFC (Lux Cellulose-2, 30×250 мм, 45% EtOH/СО2; 80 мл/мин) с получением соединения примера 105-а (более быстрое элюирование) и соединения примера 105-b (более медленное элюирование). MS (ES+) m/z: 594 (М+Н); 1Н ЯМР: (CD3OD, 400 МГц): δ 8.54 (s, 1Н), 8.26 (m, 1Н), 7.72-7.81 (m, 2Н), 7.65-7.72 (m, 1Н), 7.50 (m, 2Н), 7.27-7.38 (m, 2Н), 7.20 (d, J=7.0 Гц, 1Н), 6.81-7.13 (m, 2Н), 2.58 (m, 1Н), 2.24-2.50 (m, 2Н), 1.87-2.15 (m, 2Н), 1.45-1.69 (m, 2Н), 1.12 (m, 1Н), 0.79-1.01 (m, 1H).
ПРИМЕР 106, 106-a, 106-b
(Z)-2-(25-КАРБОКСИ-4-ОКСО-11H-3-АЗА-1(4,2)-ИМИДАЗОЛ-2(1,2)-БЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)-5-(3-ХЛОР-2,6-ДИФТОРФЕНИЛ)ПИРИДИНА 1-ОКСИД
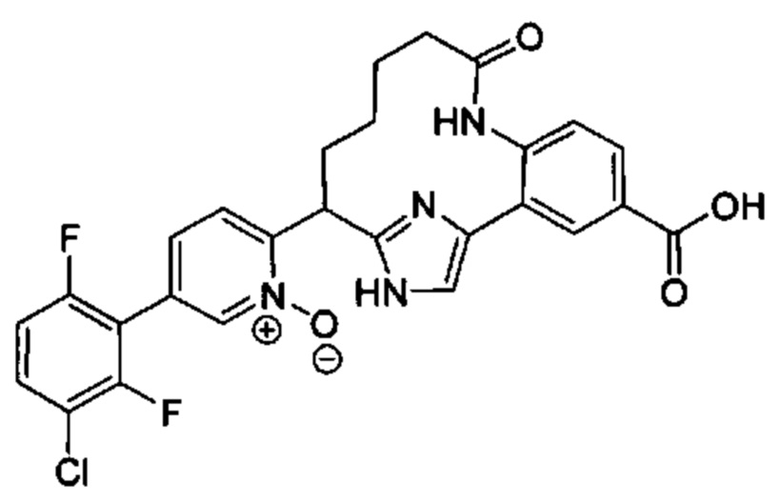
106A: (Z)-(3-Хлор-2.6-дифторфенил)-2-(25-(метоксикарбонил)-4-оксо-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-9-ил)пиридина 1-оксид:
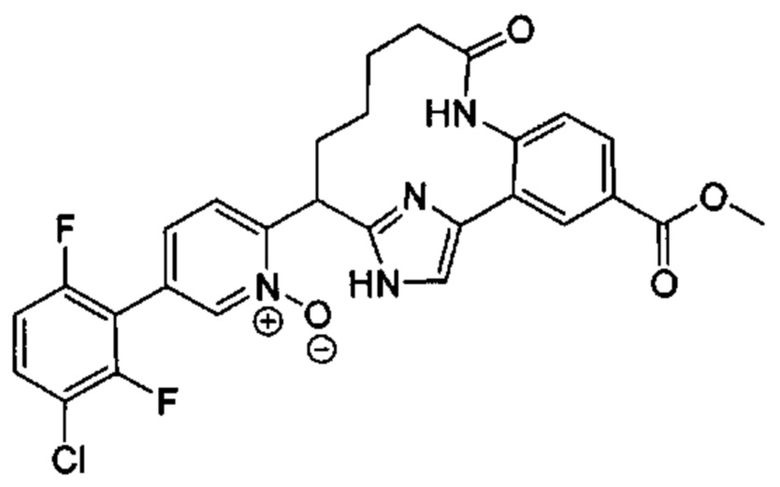
Указанное соединение получали из промежуточного соединения 15 и промежуточного соединения 20 способом, описанным в синтезе соединения примера 96. MS (ES+) m/z: 553 (М+Н).
Пример 106:
К раствору соединения примера 106А (100 мг, 0,181 ммоль) в THF/H2O (5:1, 5 мл) добавляли LiOH (13 мг, 0,543 ммоль) при 25°С. Смесь перемешивали при 25°С в течение 12 ч и ее концентрировали при пониженном давлении. Остаток очищали методом HPLC с обращенной фазой с получением указанного соединения. MS (ES+) m/z: 539 (М+Н).
Образец соединения примера 106 подвергали хиральному разделению методом SFC (AD, 30 мм × 250 м, 10 мкм; 45% IPA/CO2, 100 бар, 40°С) с получением соединения примера 106-а (более медленное элюирование) и соединения примера 106-b (более быстрое элюирование). MS (ES+) m/z: 539 (М+Н); 1Н ЯМР: (CD3OD, 400 МГц): δ 8.57 (s, 1Н), 8.22 (s, 1H), 7.83-7.96 (m, 2H), 7.57-7.70 (m, 3H), 7.35 (s, 1H), 7.18 (t, J=8.6 Гц, 1H), 5.02 (dd, J=3.7, 11.2 Гц, 1H), 3.90 (td, J=6.2, 12.2 Гц, 1H), 2.33-2.61 (m, 3H), 2.06-2.28 (m, 2H), 1.76 (m, 1H), 1.57 (m, 1H), 1.38 (m, 1H), 1.26 (m, 2H), 1.13 (d, J=6.3 Гц, 3Н).
ПРИМЕР 107 (рацемат), 107-а и 107-b
(Z)-5-(5-ХЛОР-2-(1H-ТЕТРАЗОЛ-1-ИЛ)ФЕНИЛ)-2-(25-ФТОР-9-ГИДРОКСИ-4-ОКСО-11H-3-АЗА-1(4,2)-ИМИДАЗОЛ-2(1,2)-БЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
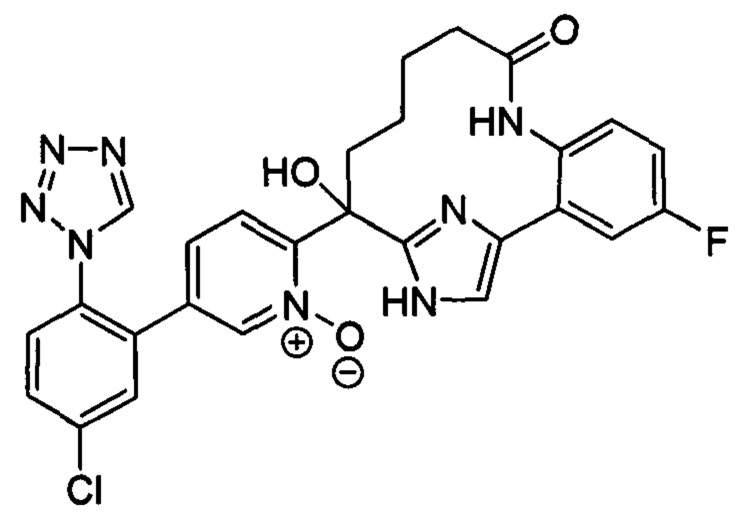
107А: 1-(5-Бромпиридин-2-ил)-1-(4-йод-1-((2-(триметилсилил)этокси)метил)-1Н-имидазол-2-ил)бут-3-ен-1-ол: К раствору промежуточного соединения 15 (3,2 г, 6,30 ммоль) в THF (40 мл) при -40°С по каплям добавляли раствор аллилмагния бромида (1 М в THF, 12,59 мл, 12,59 ммоль). Смесь перемешивали в течение 10 мин и ее нагревали до 0°C с перемешиванием в течение 30 мин. Ее гасили насыщенным водным хлоридом аммония (20 мл) и экстрагировали EtOAc (100 мл). Органический слой промывали солевым раствором, сушили над сульфатом натрия и фильтровали. Фильтрат концентрировали, а остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром/этилацетат = 20:1 объем/объем) с получением указанного соединения. MS (ES+) m/z: 550, 552 [M+H].
107B: 1-(4-(2-Амино-5-фторфенил)-1-((2-(триметилсилил)этокси)метил)-1Н-имидазол-2-ил)-1-(5-бромпиридин-2-ил)бут-3-ен-1-ол: Указанное соединение получали из соединения примера 107А способом, описанным в синтезе соединения примера 12А. Его очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром : EtOAc = 30:1-5:1 объем/объем). MS (ES+) m/z: 533, 535 [M+H].
107C: N-(2-(2-(1-(5-Бромпиридин-2-ил)-1-гидроксибут-3-ен-1-ил)-1-((2-(триметилсилил)этокси)метил)-1H-имидазол-4-ил)-4-фторфенил)бут-3-енамид: Указанное соединение получали из соединения примера 107В способом, описанным в синтезе соединения примера 1C. Неочищенный продукт очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-30% этилацетата в петролейный эфире, градиент). MS (ES+) m/z: 601, 603 [M+H].
107D: (12Z,6E)-9-(5-Бромпиридин-2-ил)-25-фтор-9-гидрокси-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-6-ен-4-он:
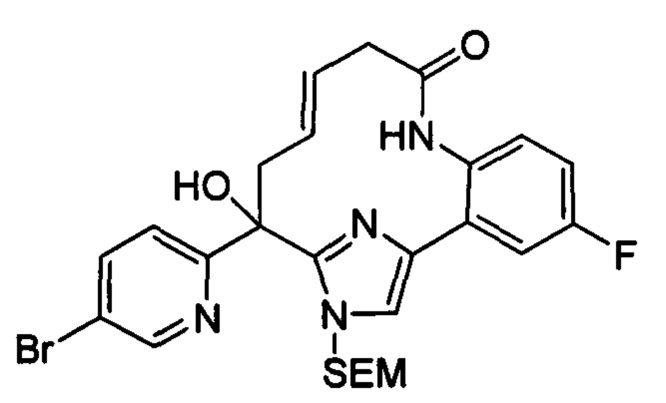
Раствор соединения примера 107С (280 мг, 0,465 ммоль) и катализатора Grubbs II ((1,3-бис(2,4,6-триметилфенил)-2-имидазолидинилиден)дихлор(фенилметилен)(трициклогексилфосфин)рутений) (158 мг, 0,186 ммоль) в DCE (дегазированный, 15 мл) перемешивали при 120°С в атмосфере азота в течение 30 мин в микроволновом реакторе. Большую часть растворителя удаляли при пониженном давлении и остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром : этилацетатом = 10:1-2:1 объем/объем) с получением указанного соединения. MS (ES+) m/z: 573, 575 [M+H].
107Е: трет-Бутил(4-хлор-2-(6-((12Z,6E)-25-фтор-9-гидрокси-4-оксо-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-6-ен-9-ил)пиридин-3-ил)фенил)карбамат:
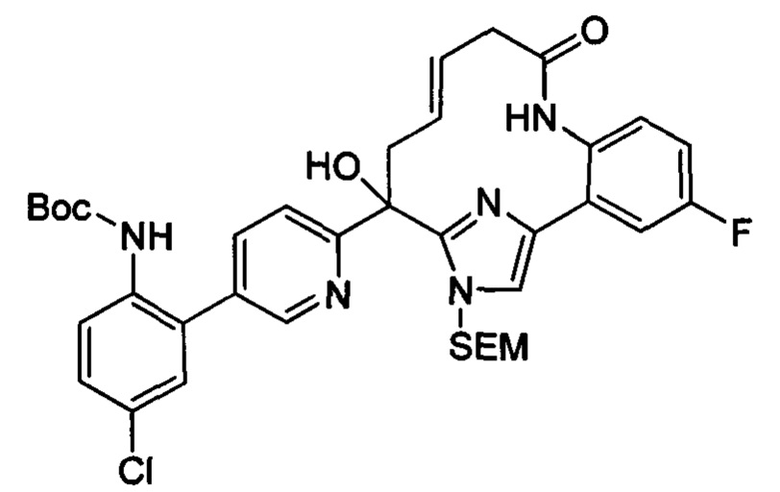
Смесь соединения примера 107D (120 мг, 0,209 ммоль), промежуточного соединения 40 (89 мг, 0,251 ммоль), PdCl2(dppf) (15,3 мг, 0,021 ммоль), K2CO3 (72,3 мг, 0,523 ммоль) в THF (8 мл) и воде (2 мл) перемешивали при 65°С в атмосфере азота в течение 2 ч. Ее охлаждали до к. т. и разбавляли водой (10 мл). Смесь экстрагировали EtOAc (30 мл × 3). Объединенные органические слои промывали водой (20 мл) и солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом преп-TLC (силикагель, петролейный эфир : EtOAc = 2:1 объем/объем) с получением указанного соединения. MS (ES+) m/z: 720 [M+H].
107F: трет-Бутил(Z)-(4-хлор-2-(6-(25-фтор-9-гидрокси-4-оксо-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-9-ил)пиридин-3-ил)фенил)карбамат:
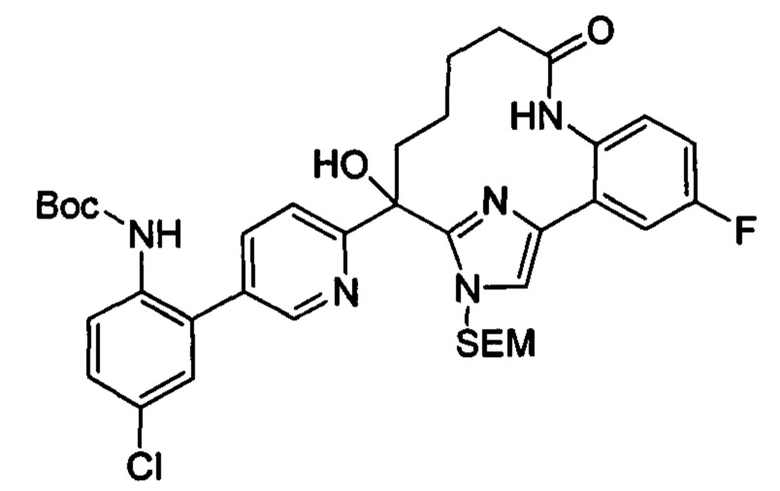
Смесь соединения примера 107Е (450 мг, 0,625 ммоль) и никеля Ренея (3,67 мг, 0,062 ммоль) в THF (5 мл) перемешивали в атмосфере водорода (1 атм.) в течение 18 ч. Его фильтровали через слой целита и твердые вещества промывали DCM. Фильтрат концентрировали с получением указанного соединения. Его использовали на следующей стадии без дополнительной очистки. MS (ES+) m/z: 722 [М+Н].
107G: (Z)-9-(5-(2-Амино-5-хлорфенил)пиридин-2-ил)-25-фтор-9-гидрокси-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-4-он:
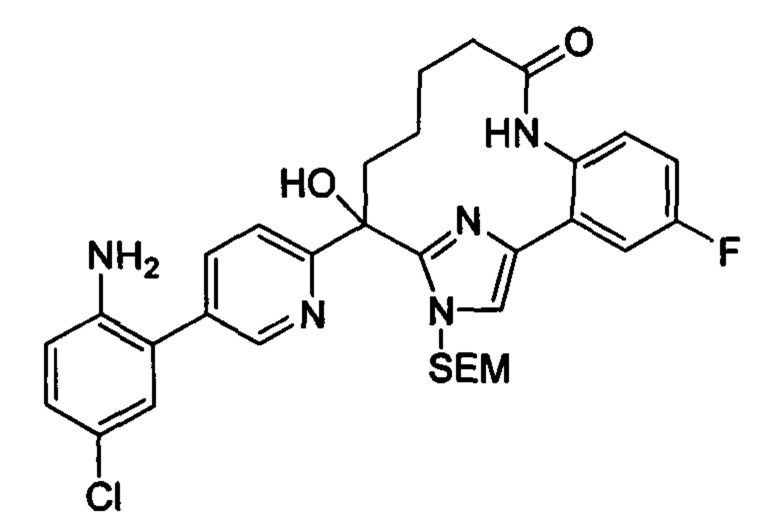
Соединение примера 107F (400 мг, 0,554 ммоль) обрабатывали трифторуксусной кислоты (1 мл, 12,98 ммоль) в DCM (5 мл) при 20°С в течение 3 ч. Большую часть растворителя удаляли при пониженном давлении и остаток разбавляли DCM. Раствор осторожно гасили насыщенным водным карбонатом натрия (20 мл). Смесь экстрагировали DCM (20 мл × 3). Объединенные органические слои промывали солевым раствором, сушили над сульфатом натрия и фильтровали. Фильтрат концентрировали с получением указанного соединения. Его использовали на следующей стадии без дополнительной очистки. MS (ES+) m/z: 622 [М+Н].
107Н: (Z)-9-(5-(5-Хлор-2-(1H-тетразол-1-ил)фенил)пиридин-2-ил)-25-фтор-9-гидрокси-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-4-он:
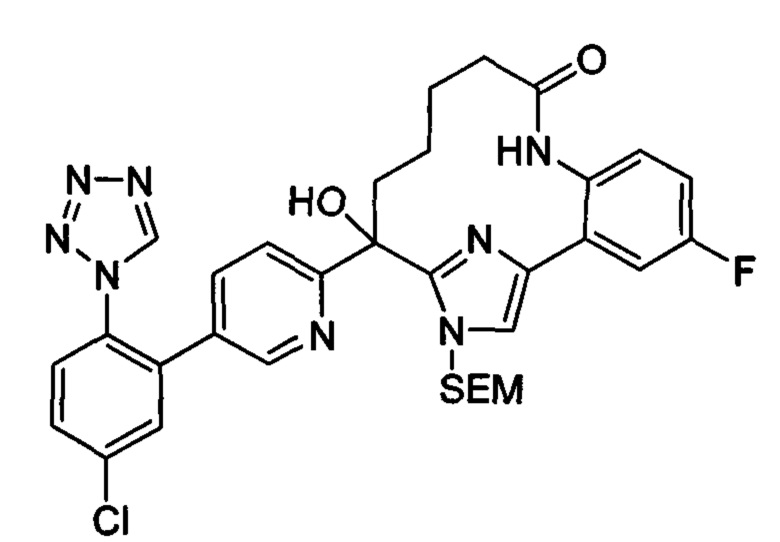
К раствору соединения примера 107G (345 мг, 0,554 ммоль) в АсОН (3 мл) добавляли триметилортоформиат (1,23 мл, 11,09 ммоль) и азид натрия (721 мг, 11,09 ммоль) при к. т. Смесь перемешивали в течение 12 ч. Ее гасили насыщенным водным карбонатом натрия (30 мл) и экстрагировали EtOAc (40 мл × 3). Объединенные органические слои промывали солевым раствором, сушили над сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, а остаток очищали методом преп-TLC (силикагель, петролейный эфир : этилацетат = 1:2 объем/объем) с получением указанного соединения. MS (ES+) m/z: 675 [M+H].
107I: (Z)-5-(5-Хлор-2-(1H-тетразол-1-ил)фенил)-2-(25-фтор-9-гидрокси-4-оксо-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-9-ил)пиридина 1-оксид:
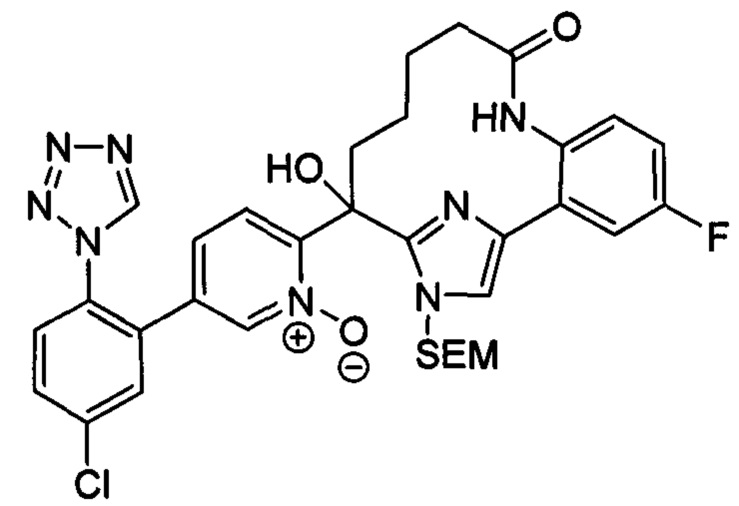
Смесь соединения примера 107Н (90 мг, 0,133 ммоль) в перуксусной кислоте (10% в уксусной кислоте, 2 мл, 0,133 ммоль) перемешивали при 25°С в течение 16 ч. Смесь гасили насыщенным водным Na2SO3 и значение рН смеси доводили приблизительно до 7 насыщенным водным Na2CO3. Смесь экстрагировали EtOAc (10 мл × 3). Объединенную органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением указанного соединения. Ее использовали на следующей стадии без дополнительной очистки. MS (ES+) m/z: 691 [M+H].
Пример 107:
К раствору соединения примера 107I (110 мг, 0,064 ммоль) в DCM (2 мл) добавляли (R)-2-амино-3-меркаптопропановую кислоту (38,6 мг, 0,318 ммоль) и TFA (2 мл, 26,0 ммоль) при 20°С. Смесь перемешивали в течение 4 ч. Ее нейтрализовали 30% водным аммиаком (5 мл) и перемешивали в течение еще 30 мин при 0°С. Смесь экстрагировали EtOAc (20 мл × 3). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом HPLC с получением рацемического соединения. MS (ES+) m/z: 561 [M+H].
Образец рацемического соединения примера 107 подвергали хиральному разделению методом SFC (OD, 50×4,6 мм, 40% метанола (0,05% диэтиламина) в CO2, 4 мл/мин, 40°С) с получением соединения примера 107-а (более быстрое элюирование) и соединения примера 107-b (более медленное элюирование). LCMS (ESI) m/z: 561.2 [М+Н+]; 1Н ЯМР: (CD3OD, 400МГц): δ 9.43 (s, 1Н), 8.35 (s, 1H), 7.91 (d, J=5.2 Гц, 1Н), 7.80-7.70 (m, 3Н), 7.54 (s, 1Н), 7.45-7.30 (m, 4H), 2.66 (m, 1Н), 2.47 (m, 1Н), 2.33 (m, 2H), 1.95 (m, 1H), 1.50-1.11 (m, 3H).
ПРИМЕР 108, 108-a, 108-b
(Z)-2-(24-АМИНО-4-ОКСО-11H-3-АЗА-1(4,2)-ИМИДАЗОЛ-2(1,2)-БЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)-5-(3-ХЛОР-2,6-ДИФТОРФЕНИЛ)ПИРИДИНА 1-ОКСИД
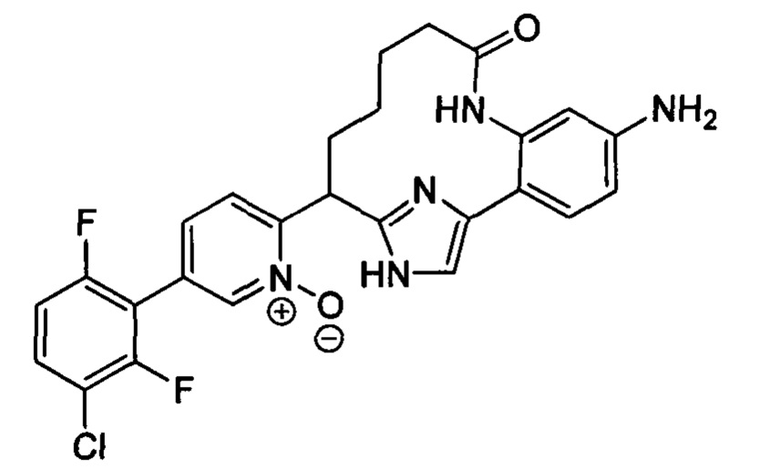
108А: трет-Бутил(Z)-(9-(5-(3-хлор-2,6-дифторфенил)пиридин-2-ил)-4-оксо-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-24-ил)карбамат:
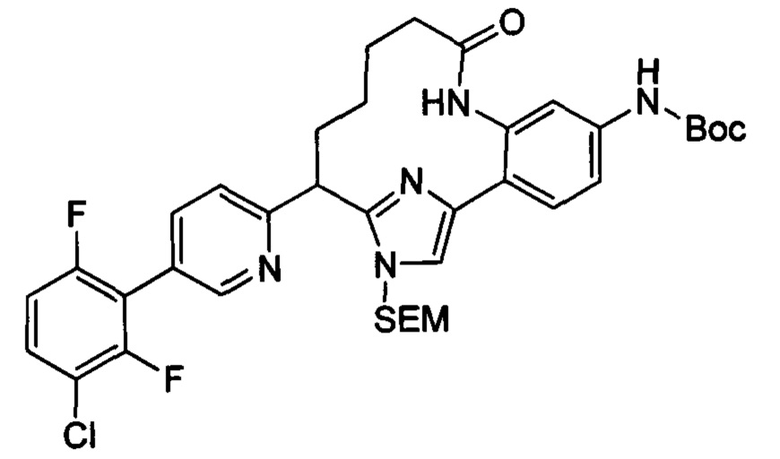
Указанное соединение получали из промежуточного соединения 15 и промежуточного соединение 40 способом, описанным в синтезе соединения примера 102В. MS (ESI) m/z 724.3 (M+H).
108В: (Z)-2-(24-((трет-Бутоксикарбонил)амино)-4-оксо-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-9-ил)-5-(3-хлор-2,6-дифторфенил)пиридина 1-оксид:
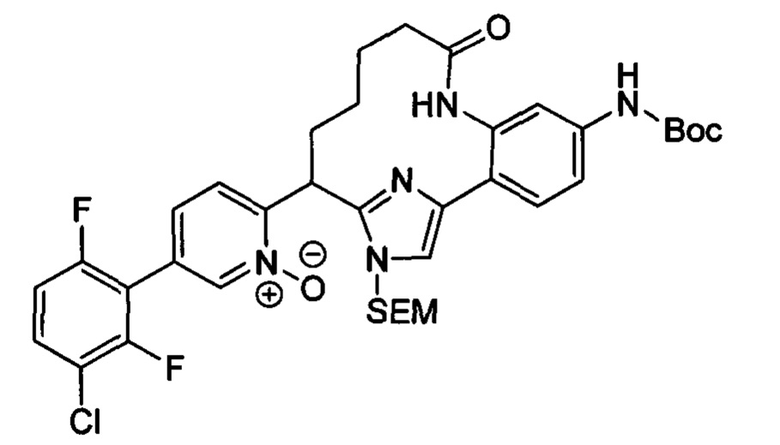
К раствору соединения примера 108А (4 г, 5,52 ммоль), NaHCO3 (1,39 г, 16,57 ммоль) и комплекса мочевины и пероксида водорода (1:1) (1,56 г, 16,57 ммоль) в DCM (50 мл) добавляли 2,2,2-трифторуксусный ангидрид (3,48 г, 16,57 ммоль). Смесь перемешивали при 25°С в течение 1 ч. Реакционную смесь выливали в смесь водного бикарбоната натрия (насыщенный, 100 мл) и водного Na2SO3 (насыщенный, 100 мл) и экстрагировали EtOAc (100 мл × 3). Объединенные органические слои промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом HPLC с обращенной фазой с получением указанного соединения. MS (ESI) m/z 740.2 (М+Н).
Рацемический образец соединения примера 108 В подвергали хиральному разделению методом SFC (колонка AD, 250 мм × 50 мм, 45% EtOH/CO2, 200 мл/мин) с получением соединения примера 108В-а (более быстрое элюирование) и соединения примера 108В-b (более медленное элюирование).
Пример 108-а: (R,Z)-2-(24-амино-4-оксо-11H-3-аза-1(4,2)-имидазол-2(1,2))-бензолациклононафан-9-ил)-5-(3-хлор-2,6-дифторфенил)пиридина 1-оксид: К перемешиваемой смеси соединения примера 108В-а (1,5 г, 2,026 ммоль) и (R)-2-амино-3-меркаптопропановой кислоты (1,23 г, 10,13 ммоль) в DCM (15 мл) добавляли TFA (10 мл, 130 ммоль). Смесь перемешивали при 40°С в течение 2 ч. Растворитель удаляли при пониженном давлении и остаток очищали методом HPLC с обращенной фазой с получением указанного соединения. MS (ESI) m/z 509.9 (М+Н).
Пример 108-b: (S,Z)-2-(24-амино-4-оксо-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-9-ил)-5-(3-хлор-2,6-дифторфенил)пиридина 1-оксид: Соединение примера 108-b получали из соединения примера 108В-b способом, описанным в синтезе соединения примера 108-а. Продукт очищали методом HPLC с обращенной фазой с получением указанного соединения. MS (ESI) m/z 509.9 (М+Н); 1Н ЯМР (400МГц, CDCl3): δ 12.71 (brs, 1H), 11.42 (brs, 1H), 8.49 (s, 1H), 7.59-7.46 (m, 4H), 7.28 (s, 1H), 7.06 (dt, J=1.5, 9.0 Гц, 1Н), 6.95 (s, 1Н), 6.45 (dd, J=2.4, 8.2 Гц, 1Н), 4.89 (dd, J=3.4, 12.7 Гц, 1Н), 3.78 (s, 2H), 2.83-2.62 (m, 1Н), 2.66-2.44 (m, 2H), 2.26 (d, J=8.2 Гц, 1Н), 2.15-1.97 (m, 1Н), 1.93-1.75 (m, 1Н), 1.52-1.42 (m, 2H).
ПРИМЕР 109, 109-a, 109-b
(Z)-5-(3-ХЛОР-2,6-ДИФТОРФЕНИЛ)-2-(24-ЭТОКСИКАРБОНИЛ)АМИНО)-4-ОКСО-11H-3-АЗА-1(4,2)-ИМИДАЗОЛ-2(1,2)-БЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
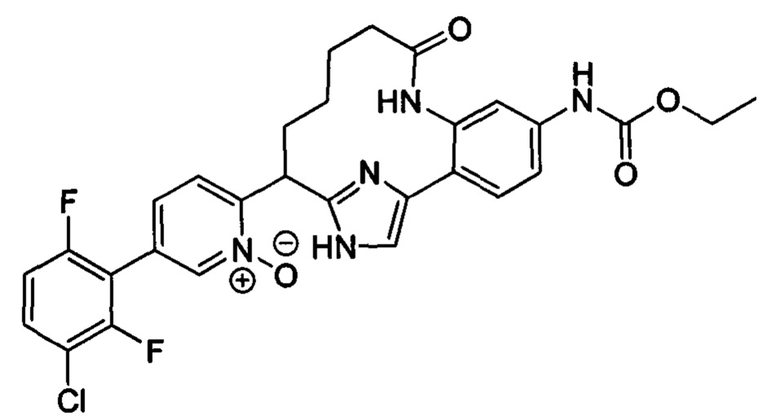
109А: (Z)-5-(3-Хлор-2,6-дифторфенил)-2-(24-нитро-4-оксо-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-9-ил)пиридина 1-оксид:
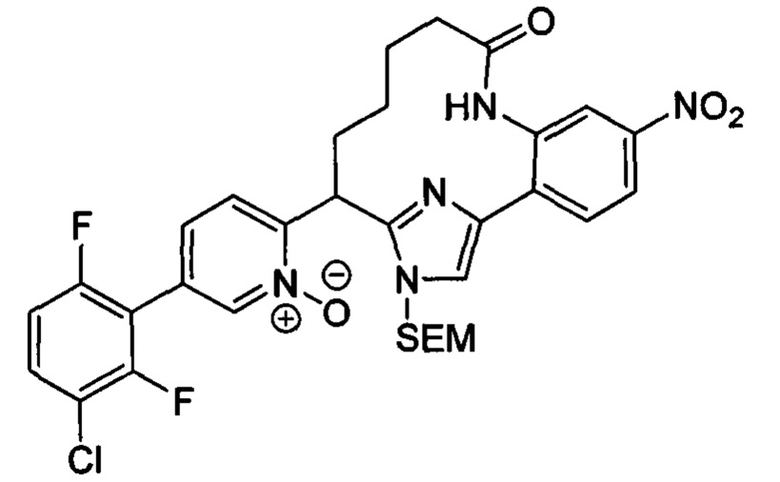
Смесь соединения примера 118А (400 мг, 0,55 ммоль, 50%) и перуксусной кислоты (15 мл, 8% масс. в уксусной кислоте) перемешивали при 30°С в течение 16 ч. Реакционную смесь выливали в ледяную воду (200 мл) и добавляли насыщенный водный NaHCO3 для доведения значения рН до 8. Добавляли водный Na2SO3 (нас., 400 мл) и смесь экстрагировали EtOAc (800 мл × 2). Объединенные органические слои промывали солевым раствором, сушили над сульфатом натрия, фильтровали и растворитель удаляли при пониженном давлении с получением указанного соединения. MS (ESI) m/z 670.3 (М+Н).
109В: (Z)-2-(24-амино-4-оксо-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-9-ил)-5-(3-хлор-2,6-дифторфенил)пиридина 1-оксид):
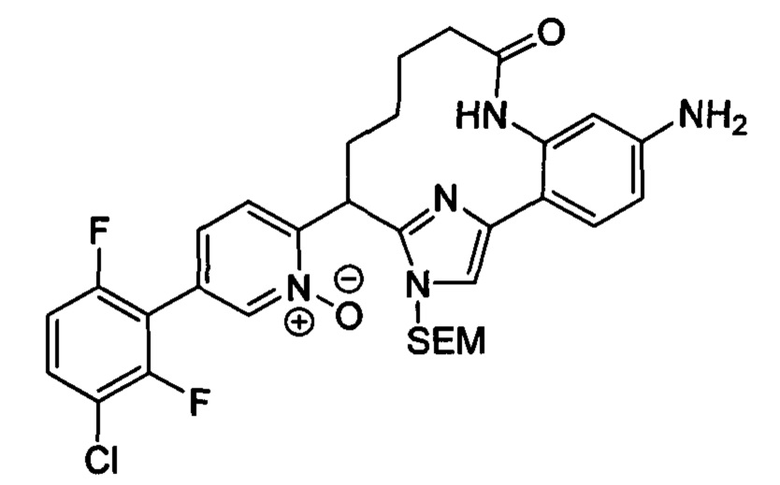
Смесь соединения примера 109А (400 мг, 0,30 ммоль, 40%), катализатора Адамса (100 мг, 0,44 ммоль), EtOAc (10 мл) и МеОН (2 мл) перемешивали при 30°С в атмосфере Н2 в течение 1 ч. Катализатор удаляли фильтрацией через слой целита. Фильтрат концентрировали с получением указанного соединения. MS (ESI) m/z 640.3 (М+Н).
109С: (Z)-5-(3-Хлор-2,6-дифторфенил)-2-(24-((этоксикарбонил)амино)-4-оксо-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-9-ил)пиридина 1-оксид:
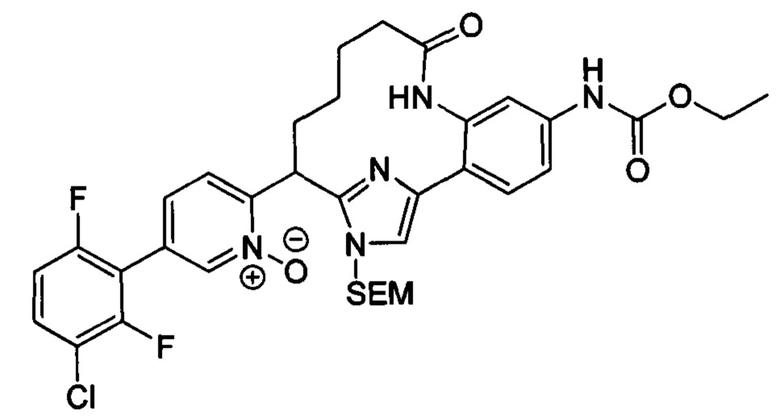
К перемешиваемой смеси соединения примера 109В (350 мг, неочищенный) и DIEA (0,12 мл, 0,66 ммоль) в DCM (10 мл) при 0°С по каплям добавляли этилхлорформиат (47,50 мг, 0,44 ммоль). Смесь перемешивали при 25°С в течение 1 ч. К ней добавляли H2O (20 мл) и экстрагировали EtOAc (30 мл × 3). Органические слои сушили над сульфатом натрия, фильтровали и концентрировали с получением указанного соединения. MS (ESI) m/z 712.2 (M+H).
Пример 109:
К перемешиваемой смеси соединения примера 109С (140 мг, неочищенный) и (R)-2-амино-3-меркаптопропановой кислоты (71,60 мг, 0,59 ммоль) в DCM (3 мл) добавляли TFA (3 мл). Смесь перемешивали при 40°С в течение 2 ч. Большую часть растворителя удаляли при пониженном давлении и остаток очищали методом HPLC с обращенной фазой с получением указанного соединения в виде твердого вещества. MS (ESI) m/z 582.3 (М+Н).
Рацемический образец соединения примера 109 подвергали хиральному разделению методом SFC (колонка: AS, 250×30 мм, 50% EtOH/CO2, 70 мл/мин) и дополнительно очищали методом HPLC с обращенной фазой с получением соединения примера 109-а (более быстрое элюирование) и соединения примера 109-b (более медленное элюирование). MS (ESI) m/z 581.9 (М+Н); 1Н ЯМР (400МГц, CD3OD): δ 8.58 (s, 1H), 7.96 (d, J=8.4 Гц, 1H), 7.81 (d, J=8.8 Гц, 1Н), 7.68 (dt, J=5.9, 8.7 Гц, 1Н), 7.60-7.55 (m, 1H), 7.53-7.48 (m, 1H), 7.46-7.39 (m, 2H), 7.27-7.20 (m, 1H), 4.97 (dd, J=6.4, 11.2 Гц, 1Н), 4.22 (q, J=7.0 Гц, 2H), 2.57 (d, J=13.7 Гц, 1H), 2.34 (brs, 2H), 2.26-2.09 (m, 1H), 1.90-1.74 (m, 1H), 1.71-1.49 (m, 2H), 1.32 (t, J=7.1 Гц, 3Н), 1.22-0.97 (m, 1H).
ПРИМЕР 110, 110-a, 110-b
(Z)-5-(3-ХЛОР-2,6-ДИФТОРФЕНИЛ)-2-(15-ХЛОР-24-((МЕТОКСИКАРБОНИЛ)АМИНОМ)-4-ОКСО-11H-3-АЗА-1(4,2)-ИМИДАЗОЛ-2(1,2)-БЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
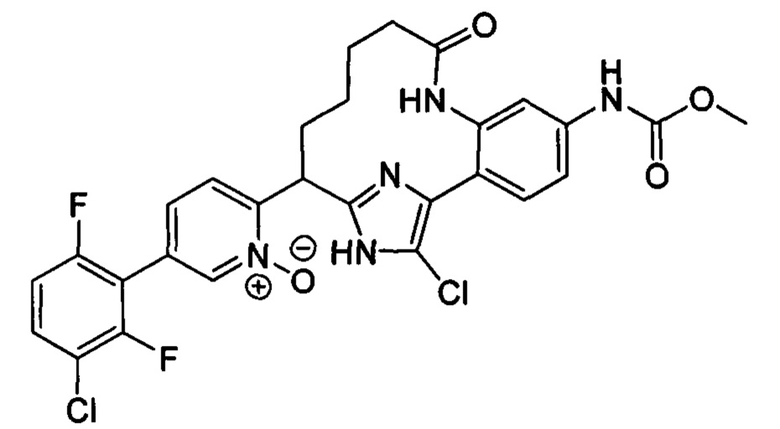
110А: Метил(Z)-(15-хлор-9(5-(3-хлор-2,6-дифторфенил)пиридин-2-ил)-4-оксо-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-24-ил)карбамат:
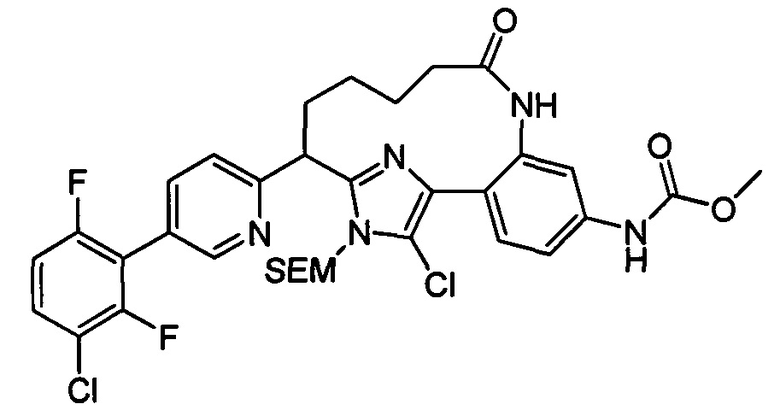
Раствор соединения примера 96В (100 мг, 0,15 ммоль) и 2-хлор-1,3-бис(метоксикарбонил)гуанидина (33,8 мг, 0,16 ммоль) в CHCl3 (4 мл) перемешивали при 20°С в течение 0,5 ч. К смеси добавляли воду (10 мл) и экстрагировали DCM (10 мл × 3). Объединенные органические слои промывали солевым раствором, сушили над сульфатом натрия, фильтровали и растворитель выпаривали при пониженном давлении. Остаток очищали методом HPLC с обращенной фазой с получением указанного соединения. MS (ESI) m/z 716.1 (М+Н).
Пример 110:
Соединение примера 110 получали из соединения примера 110А способом, описанным в синтезе соединения примера 96. MS (ESI) m/z 602.1 (M+H).
Рацемический образец соединения примера 110 (55 мг, 0,091 ммоль) подвергали хиральному разделению методом SFC (колонка AS (250×30 мм), 45% EtOH/CO2, 80 мл/мин) с получением соединения примера 110-а (более быстрое элюирование) и соединения примера 110-b (более медленное элюирование). MS (ESI) m/z 602.1 (M+H); 1H ЯМР (400МГц, CD3OD): δ 8.49 (s, 1Н), 8.05 (d, J=8.2 Гц, 1Н). 7.75 (d, J=8.2 Гц, 1Н), 7.59-7.69 (m, 1Н), 7.45-7.56 (m, 2H), 7.39 (dd, J=1.7, 8.3 Гц, 1Н), 7.20 (t, J=9.0 Гц, 1Н), 3.75 (s, 3Н), 2.39-2.48 (m, 1Н), 2.18-2.03 (m, 3H), 1.98-1.86 (m, 1Н), 1.61-1.42 (m, 2H), 1.26-1.08 (m, 1H).
ПРИМЕР 111, 111-a, 111-b
(Z)-5-(3-ХЛОР-2,6-ДИФТОРФЕНИЛ)-2-(24-((МЕТОКСИКАРБОНИЛ)АМИНО)-15-МЕТИЛ-4-ОКСО-11H-3-АЗА-1(4,2)-ИМИДАЗОЛ-2(1,2)-БЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
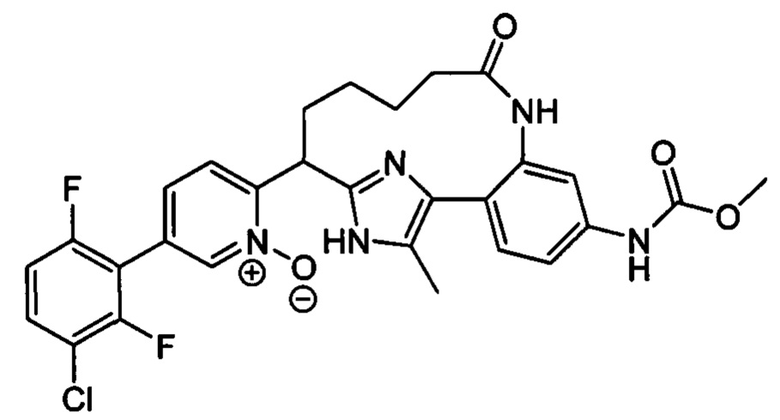
111А: Метил(Z)-(15-бром-9-(5-(3-хлор-2,6-дифторфенил)пиридин-2-ил)-4-оксо-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-24-ил)карбамат:
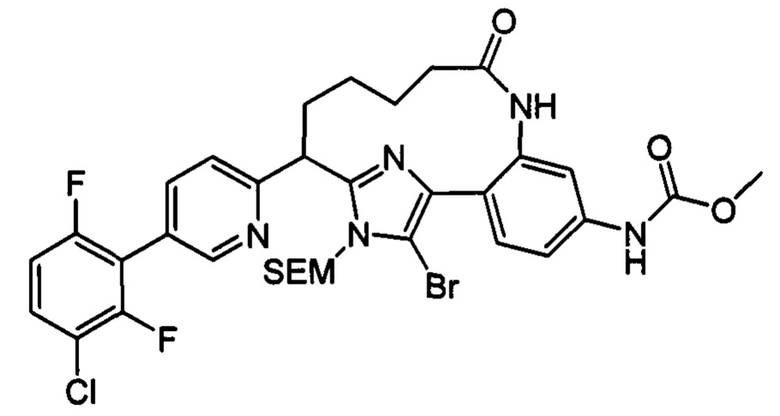
К перемешиваемой смеси соединения примера 102В (650 мг, 0,857 ммоль) в CHCl3 (10 мл) добавляли пиридиний трибромид (233 мг, 0,729 ммоль) и смесь перемешивали при 20°С в течение 1 ч. Ее добавляли к водному бикарбонату натрия (насыщенный, 10 мл) и смесь экстрагировали дихлорметаном (3×20 мл). Объединенные органические слои промывали солевым раствором (2×15 мл), сушили над сульфатом натрия, фильтровали и растворитель выпаривали при пониженном давлении с получением указанного соединения. MS (ES+) m/z: 760, 762 (M+H).
111В: Метил(Z)-(9-(5-(3-хлор-2,6-дифторфенил)пиридин-2-ил)-15-метил-4-оксо-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-24-ил)карбамат:

Смесь соединения примера 111А (570 мг, 0,599 ммоль), 2,4,6-триметил-1,3,5,2,4,6-триоксатриборинана (226 мг, 1,797 ммоль), карбоната калия (331 мг, 2,396 ммоль) и Pd(Ph3P)4 (208 мг, 0,180 ммоль) в диоксане (10 мл) перемешивали при 90°С в атмосфере азота в течение 15 ч. Добавляли водный хлорид аммония (насыщенный, 10 мл) и смесь экстрагировали этилацетатом (3×20 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над сульфатом натрия, фильтровали и растворитель выпаривали при пониженном давлении. Остаток очищали методом препаративной HPLC (YMC-Actus Pro С18, 150×30 мм, 34-64% MeCN в воде (0,1% TFA), 40 мл/мин) с получением указанного соединения. MS (ES+) m/z: 696.3 (M+H).
Пример 111:
Соединение примера 111 получали из соединения примера 111В способом, описанным в синтезе соединения примера 96, Продукт очищали методом HPLC с обращенной фазой (YMC-Actus Pro С18, 150×30 мм, 17-47% MeCN в воде (0,1% TFA), 40 мл/мин) с получением указанного соединения. MS (ES+) m/z: 582.2 (M+H).
Рацемический образец соединения примера 111 подвергали хиральному разделению методом SFC (колонка AS, 250×30 мм, 50% МеОН/CO2, 80 мл/мин) с получением соединения примера 111-а (более медленное элюирование) и соединения примера 111-b (более быстрое элюирование). MS (ES+) m/z: 582.2 (M+H).
ПРИМЕР 112, 112-а, 112-b
(Z)-5-(3-ХЛОР-2,6-ДИФТОРФЕНИЛ)-2-(15-ЭТИЛ-24-((МЕТОКСИКАРБОНИЛ)АМИНО)-4-ОКСО-11H-3-АЗА-1(4,2)-ИМИДАЗОЛ-2(1,2)-БЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
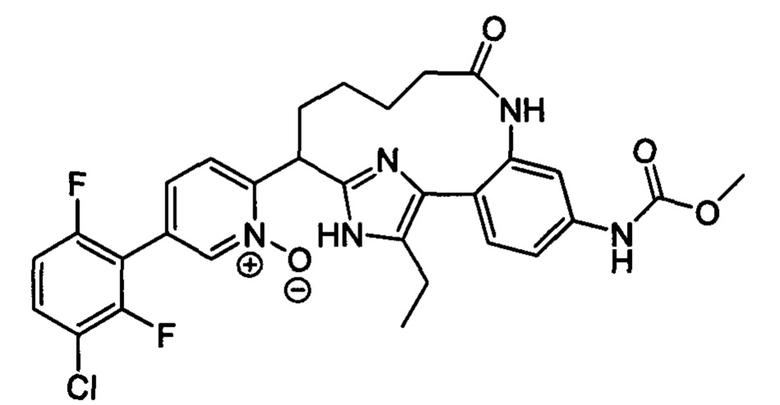
112А: Метил(Z)-(9-(5-(3-хлор-2,6-дифторфенил)пиридин-2-ил)-4-оксо-11-((2-(триметилсилил)этокси)метил)-15-винил-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-24-ил)карбамат:
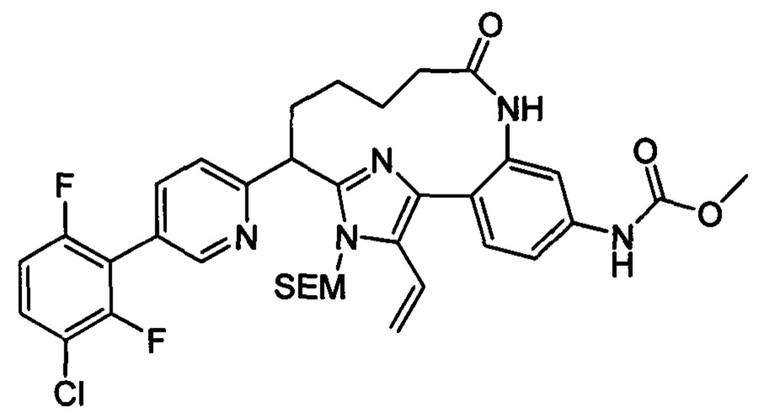
В закупоренную пробирку для микроволнового облучения с магнитной мешалкой добавляли соединение примера 102В (150 мг, 0,197 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцен-палладий(II) дихлорида (32,2 мг, 0,039 ммоль), винилтрифторборат калия (52,8 мг, 0,394 ммоль). Ее закупоривали и трижды продували азотом. К смеси добавляли дегазированный этанол (1 мл) и TEA (0,082 мл, 0,591 ммоль). Смесь перемешивали при 80°С в течение 2 ч. Ее охлаждали до к. т.и очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-5% метанолом в DCM) с получением указанного соединения. MS (ES+) m/z: 708.3 (M+H).
112В: Метил(Z)-(9-(5-(3-хлор-2,6-дифторфенил)пиридин-2-ил)-15-этил-4-оксо-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-24-ил)карбамат:
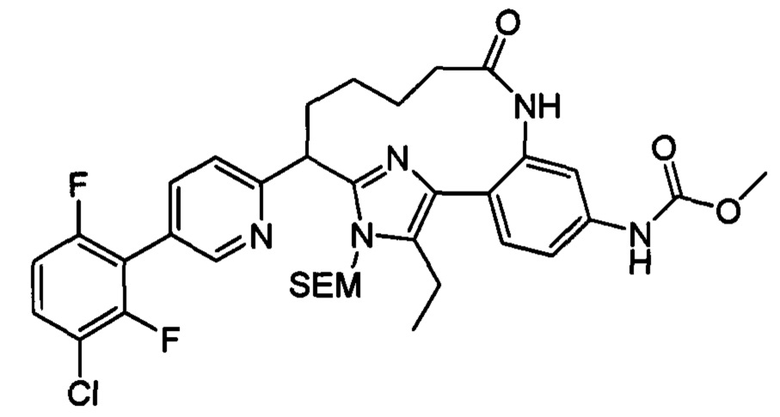
Смесь соединения примера 112А (210 мг, 0,296 ммоль) и никеля Ренея (348 мг, 5,93 ммоль) в THF (5 мл) встряхивали в атмосфере водорода (40 фунт/кв. дюйм) в течение 30 мин. Его фильтровали через слой целита, промывали 10% метанолом в DCM (2×10 мл). Фильтрат концентрировали, а остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-5% метанолом в DCM) с получением указанного соединения. MS (ES+) m/z: 710.3 (M+H).
Пример 112:
Соединение примера 112 получали из соединения примера 112В способом синтеза соединения примера 108. Продукт очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-7% метанолом в DCM). MS (ES+) m/z: 596.2 (M+H).
Рацемический образец соединения примера 112 подвергали хиральному разделению методом SFC (RR Whelk, 30×250 мм, 65% МеОН (0,2% NH4OH)/CO2, 70 мл/мин, 100 бар, 35°С) с получением соединения примера 112-а (более быстрое элюирование) и соединения примера 112-b (более медленное элюирование). Два энантиомера повторно очищали методом колоночной флеш-хроматографии на силикагеле (0-7% метанола в DCM). MS (ES+) m/z: 596.2 (M+H). 1H ЯМР (500 МГц, DMSO-d6): δ 12.00 (s, 1H), 11.75 (s, 1H), 9.66 (s, 1H), 8.59 (s, 1H), 7.84 (s, 1H), 7.71 (m, 1H), 7.62 (d, J=8.5 Гц, 1H), 7.49 (d, J=8.5 Гц, 1H), 7.40-7.20 (m, 3Н), 4.84 (d, J=4.0 Гц, 1Н),3.64 (s, 3Н), 2.62 (q, J=7.5 Гц, 2Н), 2.40 (m, 1 H), 2.26 (m, 1 H), 2.18 (brs, 1 H), 2.04 (brs, 1H), 1.85 (brs, 1H), 1.57 (brs, 1H). 1.33 (brs, 1H), 1.15 (t, J=7.5 Гц, 3Н), 1.04 (brs, 1H).
ПРИМЕР 113, 113-a, 113-b
(Z)-2-(15-ЦИКЛОПРОПИЛ-24-((МЕТОКСИКАРБОНИЛ)АМИНО)-4-ОКСО-11H-3-АЗА-1(4,2)-ИМИДАЗОЛ-2(1,2)-БЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)-5-(2-(ДИФТОРМЕТОКСИ)-6-ФТОРФЕНИЛ)ПИРИДИНА 1-ОКСИД
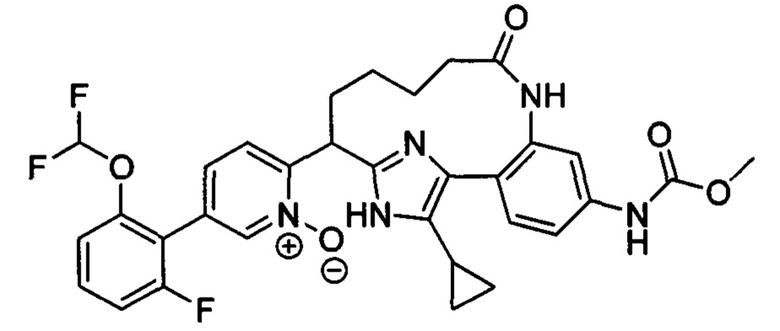
113А: Метил(Z)-(15-бром-9-(5-(2-(дифторметокси)-6-фторфенил)пиридин-2-ил)-4-оксо-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-24-ил)карбамат:
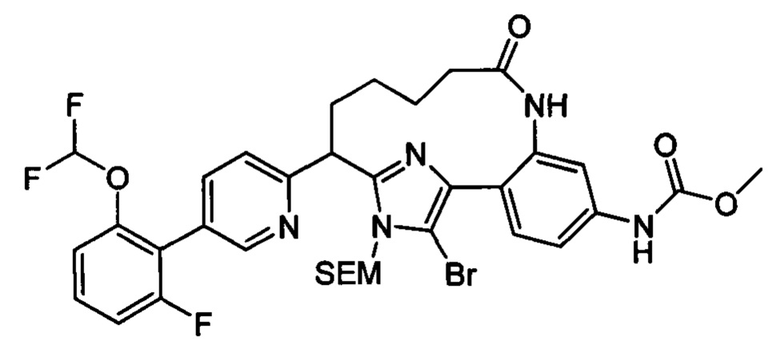
Указанное соединение получали из промежуточного соединения 56 и промежуточного соединения 36 способом, описанным в синтезе соединения примера 111А. Продукт очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-4,5% метанолом в DCM). MS (ES+) m/z: 774.1, 776.1 (M+H).
113В: Метил(Z)-(15-циклопропил-9-(5-(2-(дифторметокси)-6-фторфенил)пиридин-2-ил)-4-оксо-11-((2-(триметилсилил)этокси)метил)-11Н-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-24-ил)карбамат:
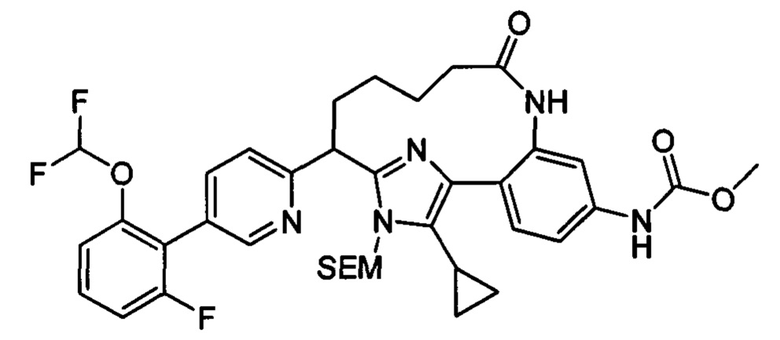
Сосуд, заполненный соединением примера 113А (420 мг, 0,542 ммоль), циклопропилбороновой кислотой (69,9 мг, 0,813 ммоль), тетракис (188 мг, 0,163 ммоль) и карбонатом калия (300 мг, 2,169 ммоль) дегазировали и повторно заполняли азотом (3×). Далее добавляли диоксан (5,4 мл); полученную смесь нагревали при 80°С всю ночь. Ее охлаждали до к. т. и очищали методом колоночной флеш-хроматографии (элюировали MeOH/CH2Cl2 = 4,5%) с получением указанного соединения. MS (ES+) m/z: 736 (M+H).
Пример 113:
Соединение примера 113 получали из соединения примера 113В способом, описанным в синтезе соединения примера 102. Продукт очищали методом колоночной флеш-хроматографии на силикагеле (элюировали MeOH/CH2Cl2 = 7%). MS (ES+) m/z: 622.3 (M+H).
Рацемический образец соединения примера 113 подвергали хиральному разделению методом SFC (RR Whelk, 30×250 мм, 45% МеОН (0,2% NH4OH)/CO2, 70 мл/мин, 100 бар, 35°С) с получением соединения примера 113-а (более быстрое элюирование) и соединения примера 113-b (более медленное элюирование). MS (ES+) m/z: 622.3 (M+H).
ПРИМЕР 114, 114-а, 114-b, 114-c, 114-d
(Z)-5-(3-ХЛОР-2,6-ДИФТОРФЕНИЛ)-2-(24-((МЕТОКСИКАРБОНИЛ)АМИНО)-5-МЕТИЛ-4-ОКСО-11H-3-АЗА-1(4,2)-ИМИДАЗОЛ-2(1,2)-БЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
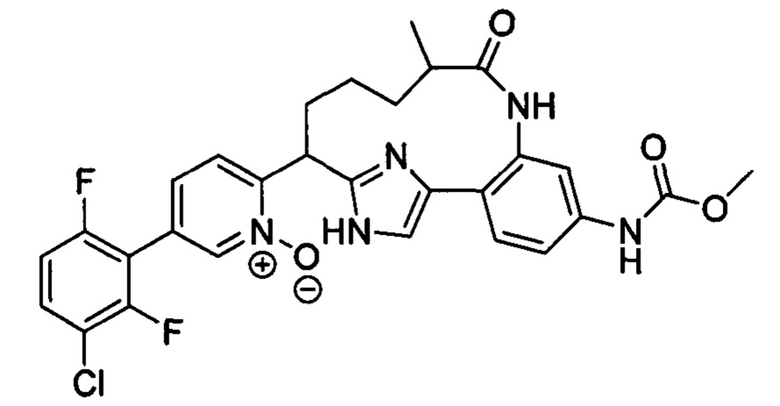
114А: Метил(Z)-(9-(5-(3-хлор-2,6-дифторфенил)пиридин-2-ил)-5-метил-4-оксо-11-((2-(триметилсилил)этокси)метил)-11H-3-аза-1(4,2)-имидазол-2(1,2)-бензолациклононафан-24-ил)карбамат:
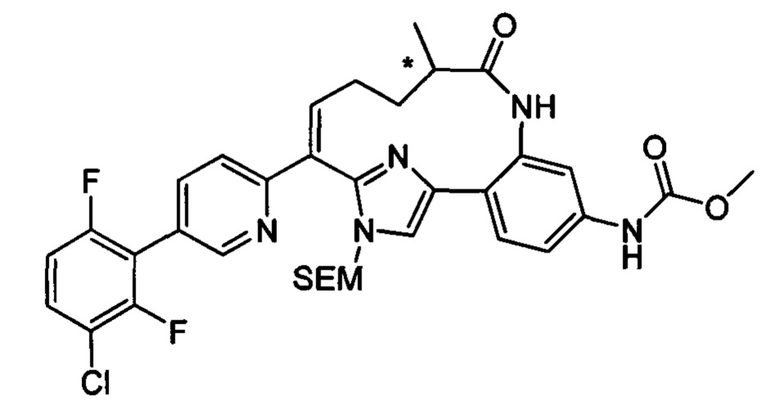
Смесь промежуточного соединения 57 (1,50 г, 2,39 ммоль), 2-(3-хлор-2,6-дифторфенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (1,31 г, 4,79 ммоль), Pd(dtbpf)Cl2 (0,16 г, 0,24 ммоль), K2CO3 (0,99 г, 7,18 ммоль), THF (24 мл) и воды (8 мл) в закупоренном сосуде перемешивали при 120°С в течение 16 ч. Ее охлаждали до к. т. и разбавляли водой (10 мл). Смесь экстрагировали EtOAc (100 мл × 2). Объединенные органические слои промывали солевым раствором, сушили над сульфатом натрия, фильтровали и растворитель выпаривали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (0-6% метанола в DCM, градиент) с получением указанного соединения. MS (ESI) m/z 694.3 (М+Н).
Рацемический образец вышеуказанного продукта подвергали хиральному разделению методом SFC (Chiralpak AD 250×50 мм, 45% EtOH (0,05% DEA)/CO2, 200 мл/мин) с получением соединения примера 114А-а (более быстрое элюирование) и соединения примера 114A-b (более медленное элюирование). MS (ESI) m/z 694.3 (М+Н).
Пример 114-а/114-b:
Смесь соединения примера 114-а/114-b получали из соединения примера 114А-а способом, описанным в синтезе соединения примера 102.
Образец двух диастереомеров подвергали хиральному разделению методом SFC (Chiralpak AS-H 250×30 мм, 40% этанола (0,05% DEA)/CO2, 70 мл/мин) с получением соединения примера 114-а (более быстрое элюирование) и соединения примера 114-b (более медленное элюирование).
Соединение примера 114-а: MS (ESI) m/z 582.2 (М+Н); 1Н ЯМР (400МГц, CD3OD): δ 8.56 (s, 1Н), 7.96 (d, J=8.4 Гц, 1Н), 7.81 (d, J=8.4 Гц, 1Н), 7.67 (dt, J=5.8, 8.5 Гц, 1Н), 7.58 (s, 1Н), 7.50-7.46 (m, 1Н), 7.43-7.38 (m, 2H), 7.22 (t, J=8.6 Гц, 1Н), 4.98 (dd, J=6.0, 11.9 Гц, 1Н), 3.76 (s, 3H), 2.83-2.75 (m, 1Н), 2.42 (t, J=12.6 Гц, 1Н), 2.25-2.21 (m, 1H), 2.00-1.91 (m, 1Н), 1.78-1.59 (m, 2H), 1.05 (d, J=6.8 Гц, 3H), 0.78-0.65 (m, 1H).
Соединение примера 114-b: MS (ESI) m/z 582.1 (М+Н); 1Н ЯМР (400МГц, CD3OD): δ 8.56 (s, 1H), 7.85 (d, J=8.4 Гц, 1H), 7.72 (d, J=8.4 Гц, 1H), 7.65 (dt, J=5.7, 8.5 Гц, 1Н), 7.56 (s, 1H), 7.51-7.47 (m, 1H), 7.43-7.38 (m, 1H), 7.25 (s, 1H), 7.20 (t, J=8.6 Гц, 1H), 4.88-4.85 (m, 1H), 3.74 (s, 3H), 2.66-2.57 (m, 1H), 2.34-2.18 (m, 2H), 1.76 (d, J=7.7 Гц, 1H), 1.44 -1.32 (m, 2H), 1.22 (brd, J=7.1 Гц, 3H), 1.16-1.04 (m, 1H).
Пример 114-c/114-d:
Смесь соединения примера 114-c/114-d получали из соединения примера 114А-b способом, описанным в синтезе соединения примера 102.
Образец двух диастереомеров подвергали хиральному разделению методом SFC (Chiralpak AS-H 250×30 мм, 40% этанола (0,05% DEA)/CO2, 70 мл/мин) с получением соединения примера 114-с (более быстрое элюирование) и соединения примера 114-d (более медленное элюирование).
Соединение примера 114-с: MS (ESI) m/z 582.1 (М+Н); 1Н ЯМР (400МГц, CD3OD): δ 8.56 (s, 1H), 7.85 (d, J=8.4 Гц, 1H), 7.72 (d, J=8.4 Гц, 1H), 7.65 (dt, J=5.7, 8.5 Гц, 1Н), 7.56 (s, 1H), 7.51-7.47 (m, 1H), 7.43-7.38 (m, 1H), 7.25 (s, 1H), 7.20 (t, J=8.6 Гц, 1H), 4.88-4.85 (m, 1H), 3.74 (s, 3H), 2.66-2.57 (m, 1H), 2.34-2.18 (m, 2H), 1.76 (d, J=7.7 Гц, 1H), 1.44-1.32 (m, 2H), 1.22 (brd, J=7.1 Гц, 3H), 1.16-1.04 (m, 1H).
Соединение примера 114-d: MS (ESI) m/z 582.2 (М+Н); 1H ЯМР (400МГц, CD3OD): δ 8.56 (s, 1H), 7.96 (d, J=8.4 Гц, 1H), 7.81 (d, J=8.4 Гц, 1H), 7.67 (dt, J=5.8, 8.5 Гц, 1Н), 7.58 (s, 1H), 7.50-7.46 (m, 1H), 7.43-7.38 (m, 2H), 7.22 (t, J=8.6 Гц, 1Н), 4.98 (dd, J=6.0,11.9 Гц, 1H), 3.76 (s, 3H), 2.83-2.75 (m, 1H), 2.42 (t, J=12.6 Гц, 1H), 2.25-2.21 (m, 1H), 2.00-1.91 (m, 1H), 1.78-1.59 (m, 2H), 1.05 (d, J=6.8 Гц, 3H), 0.78-0.65 (m, 1H).
Следующие соединения синтезировала способами, описанными в примере 96 или примере 102 с соответствующими исходными веществам. Они характеризуются методом LC/MS.
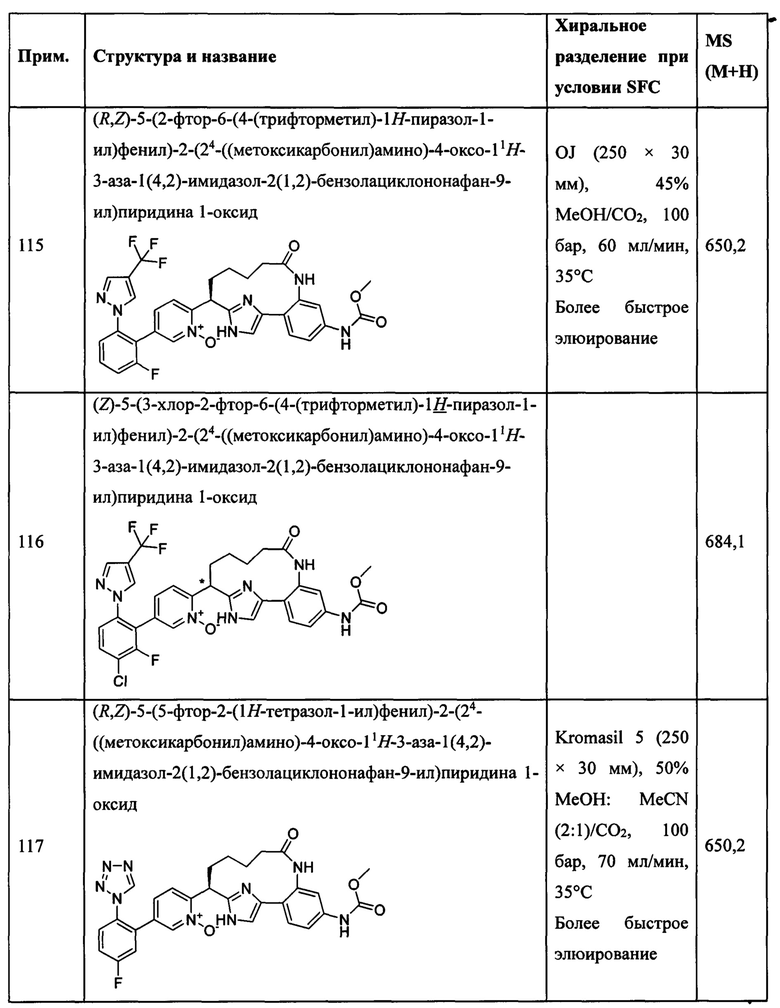
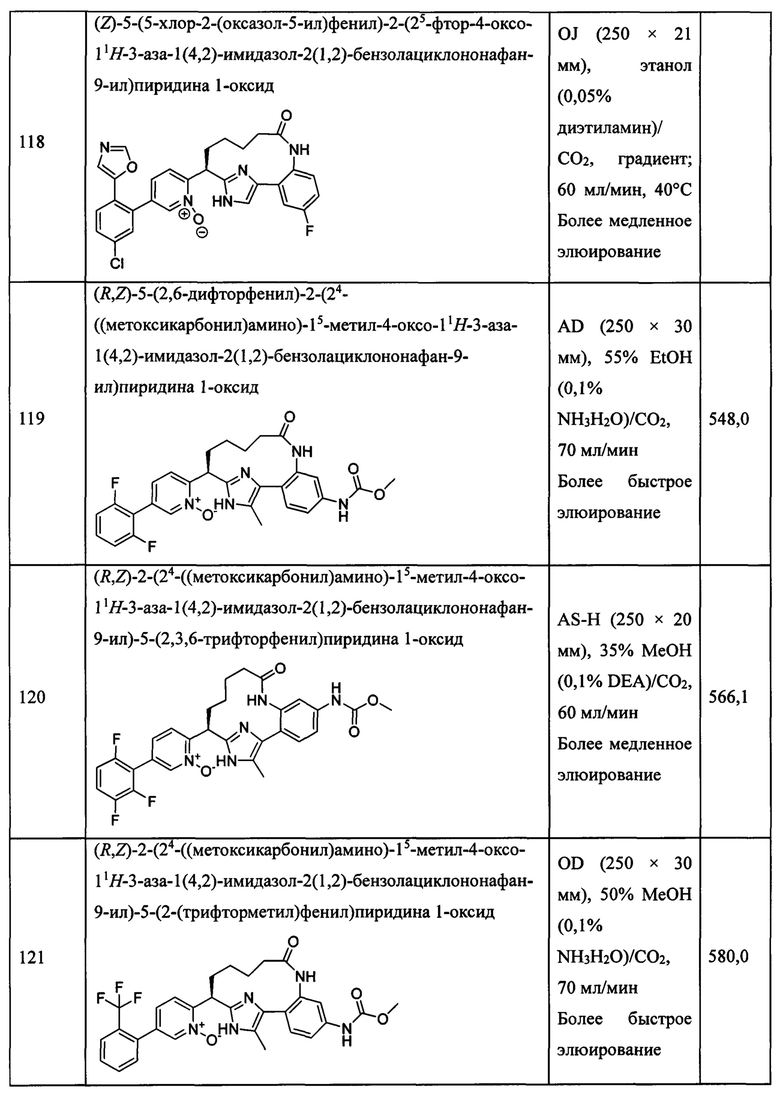
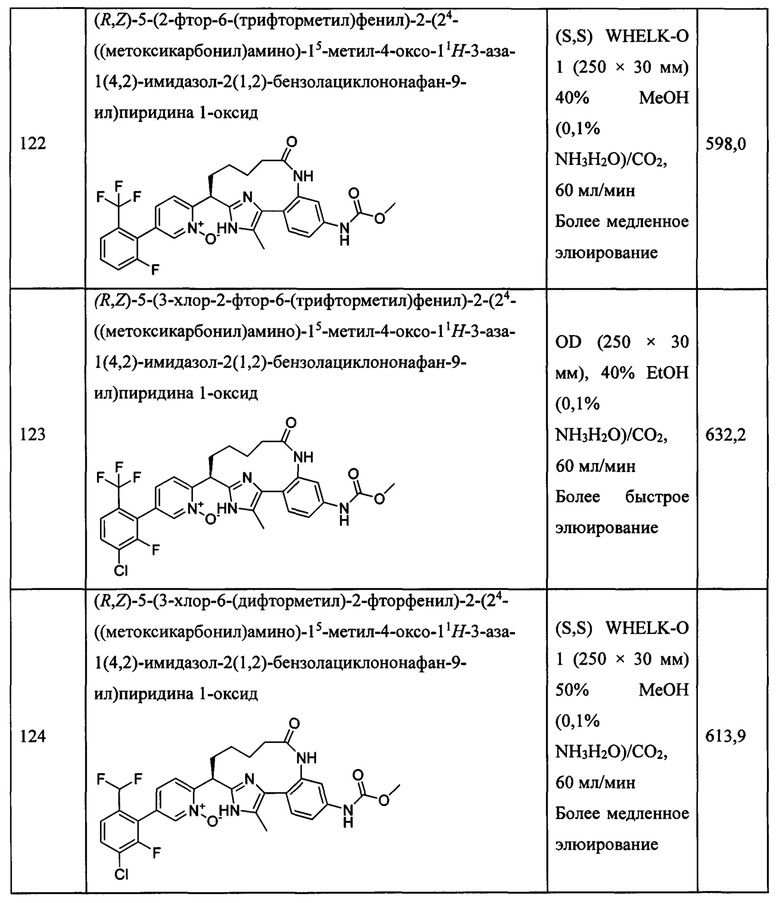
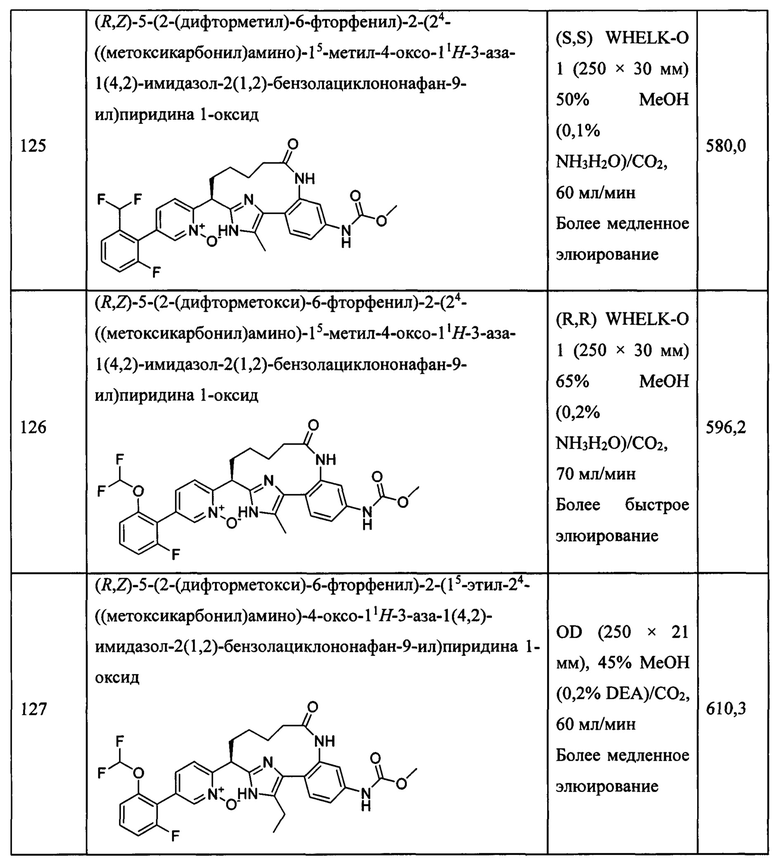
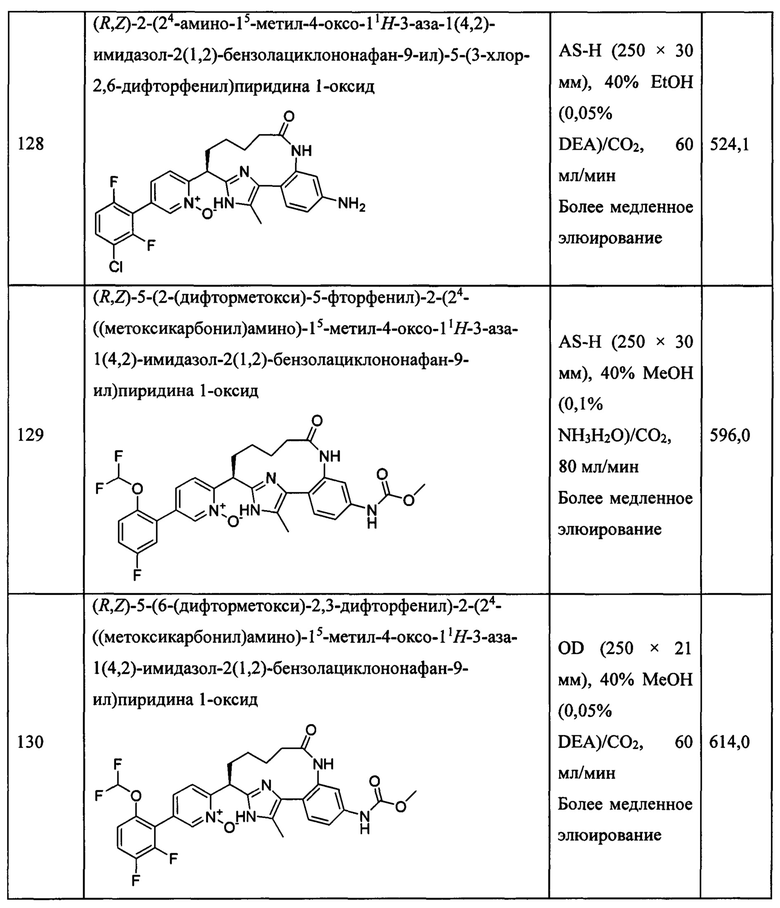
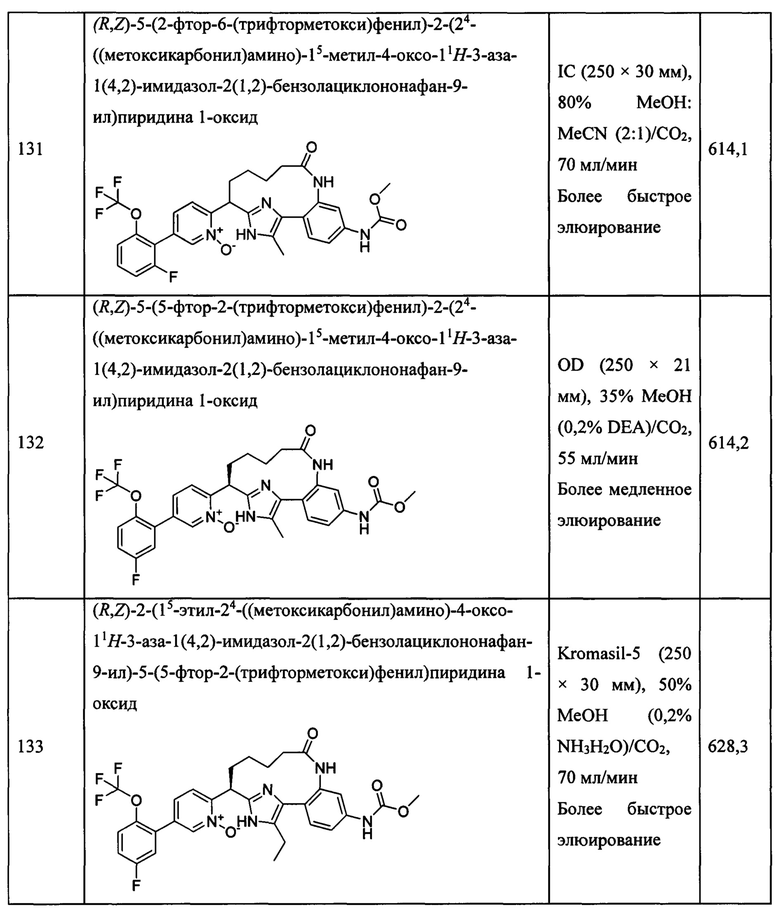
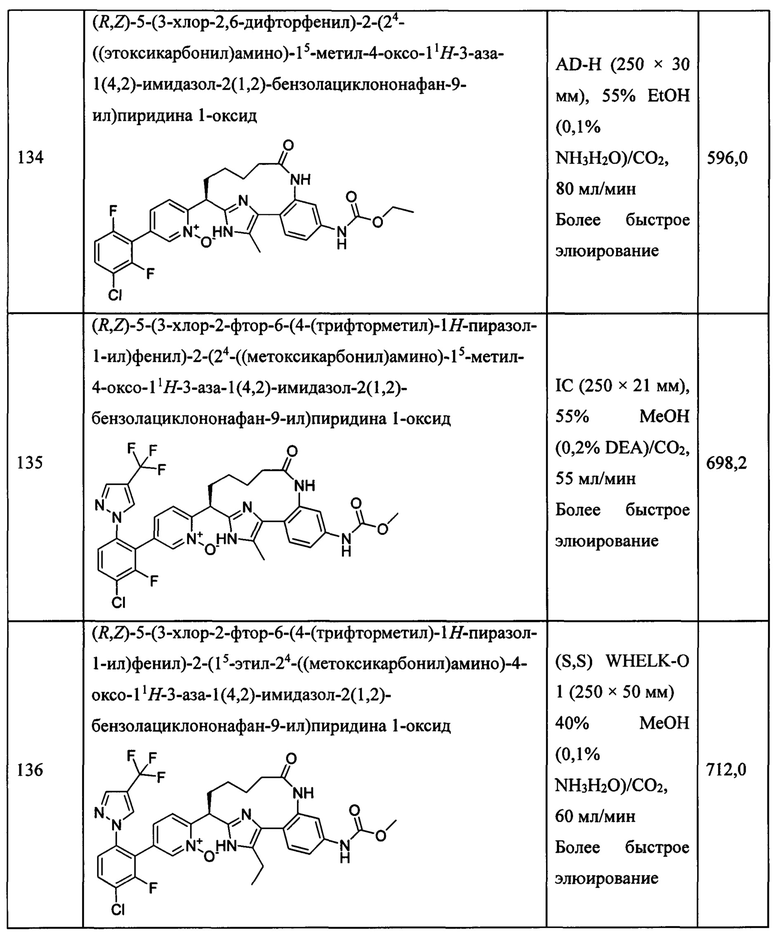
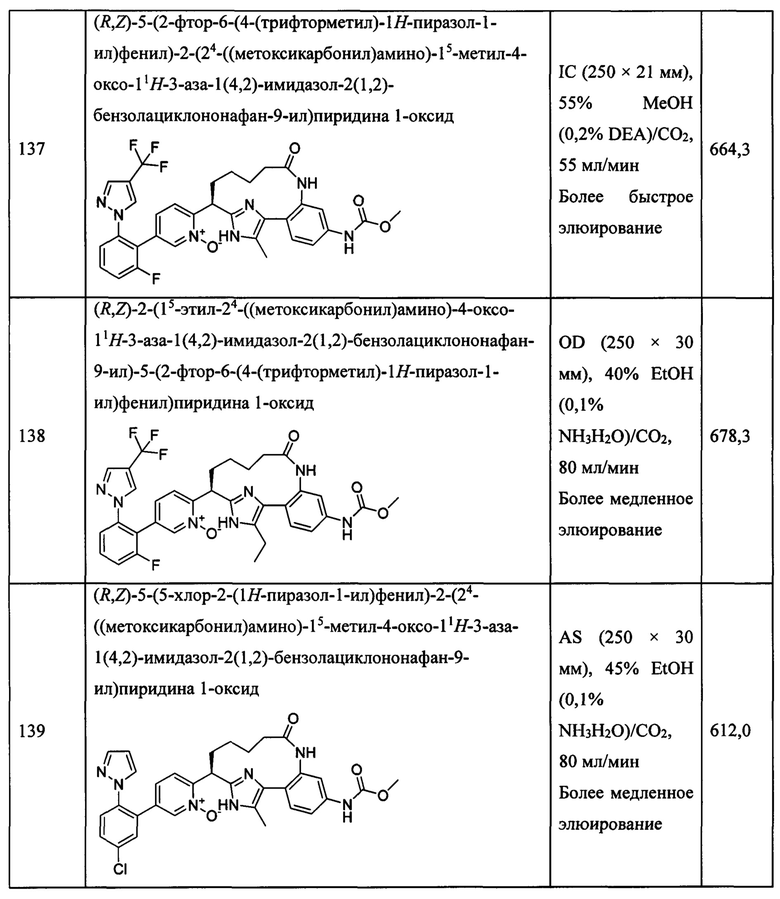
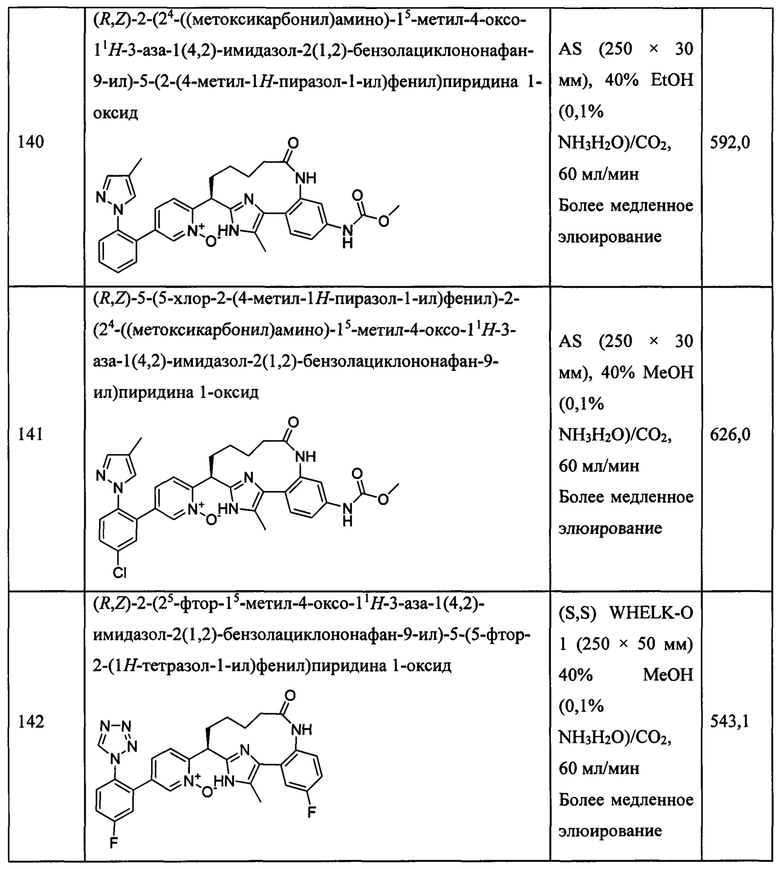
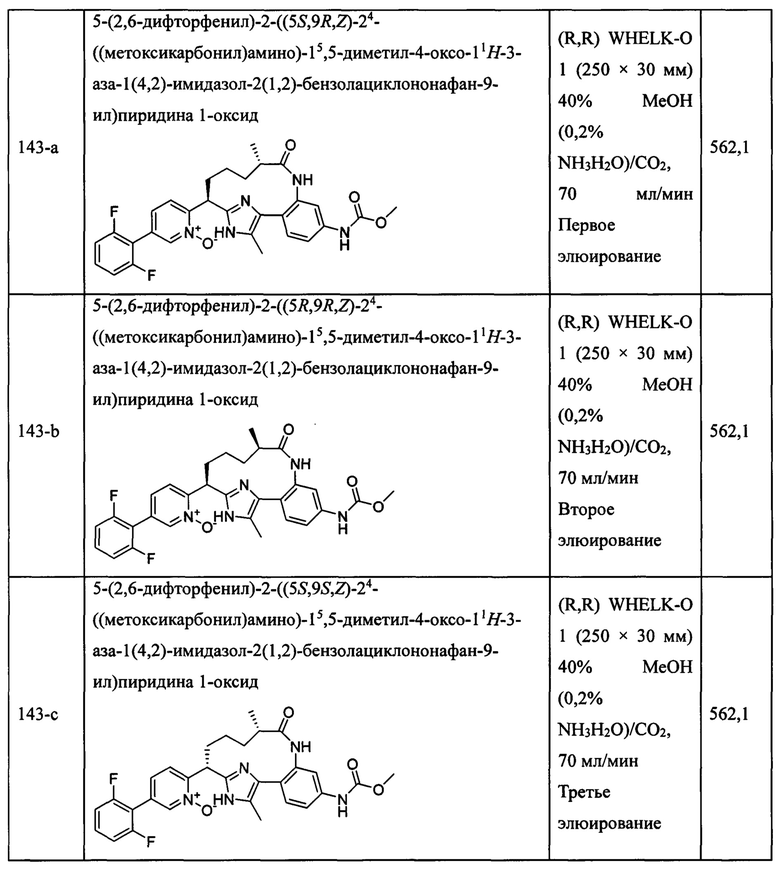
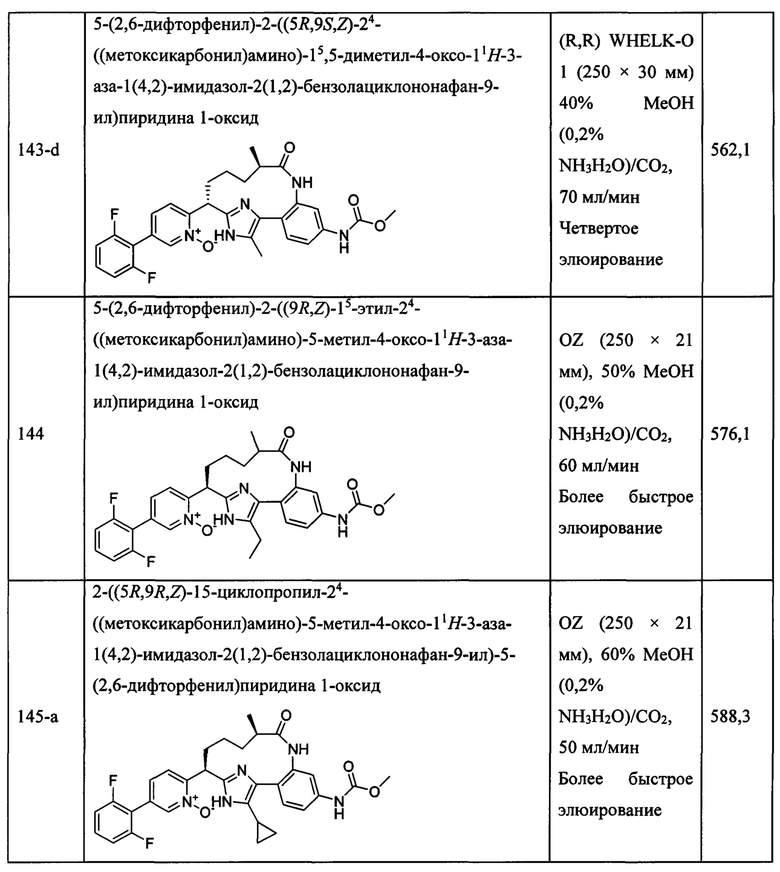
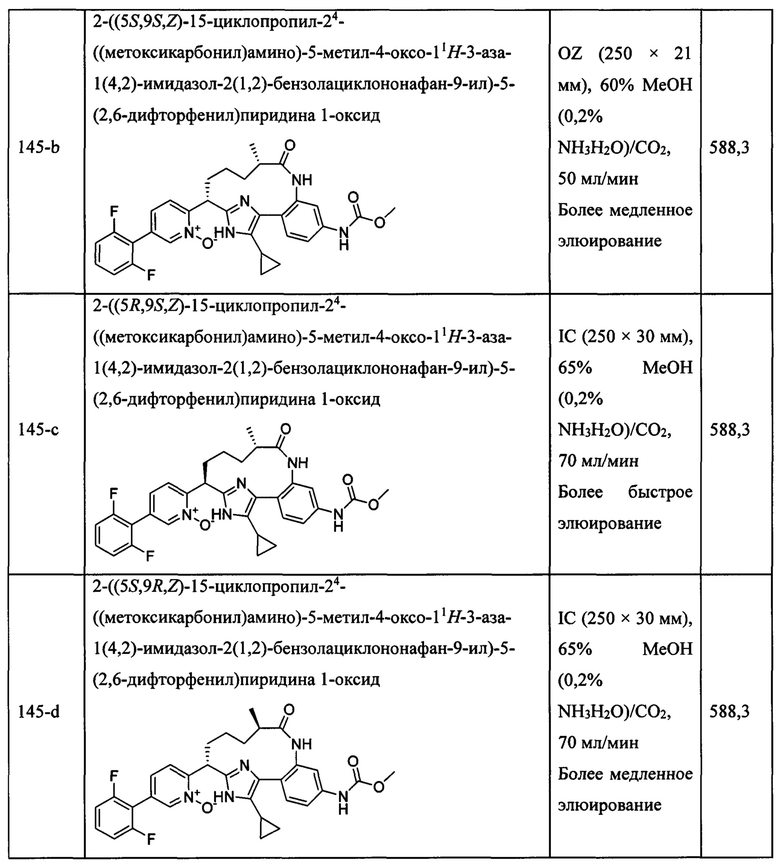
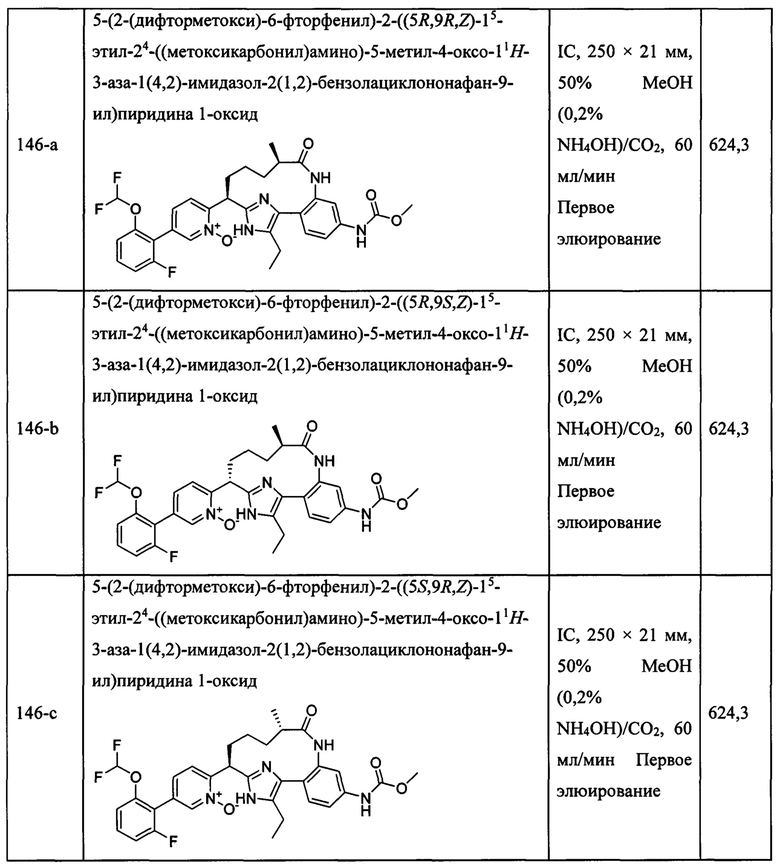
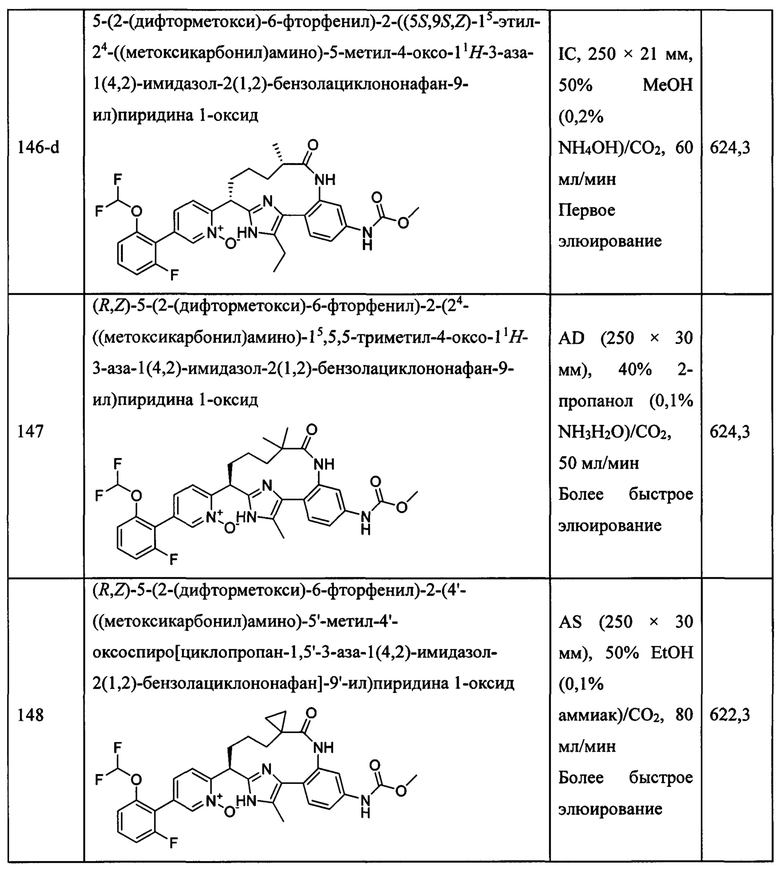
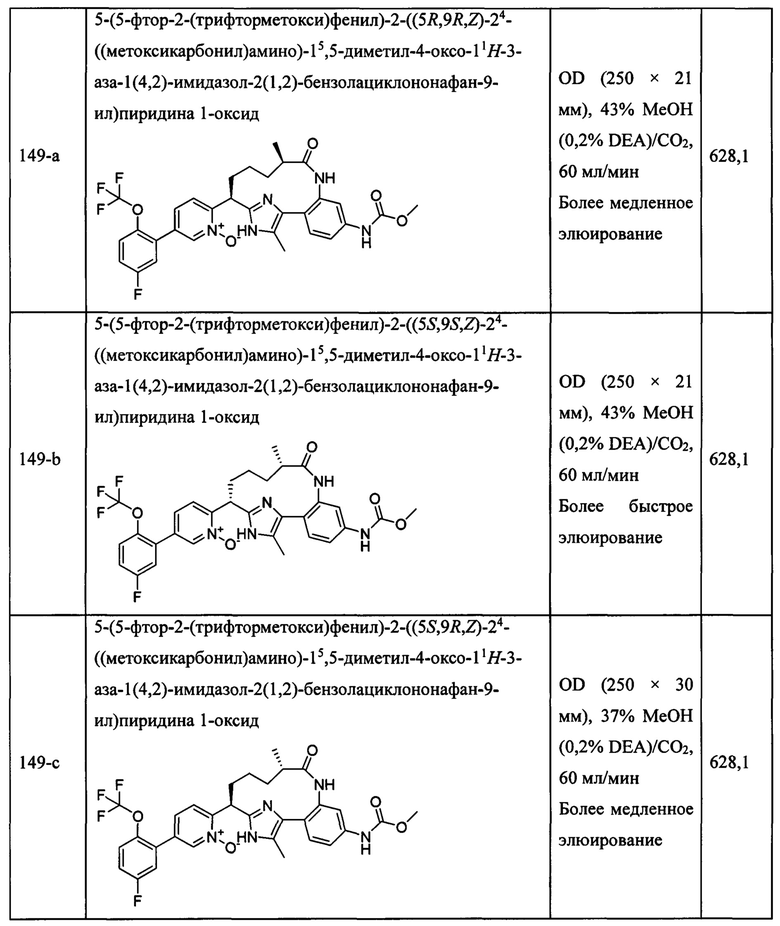
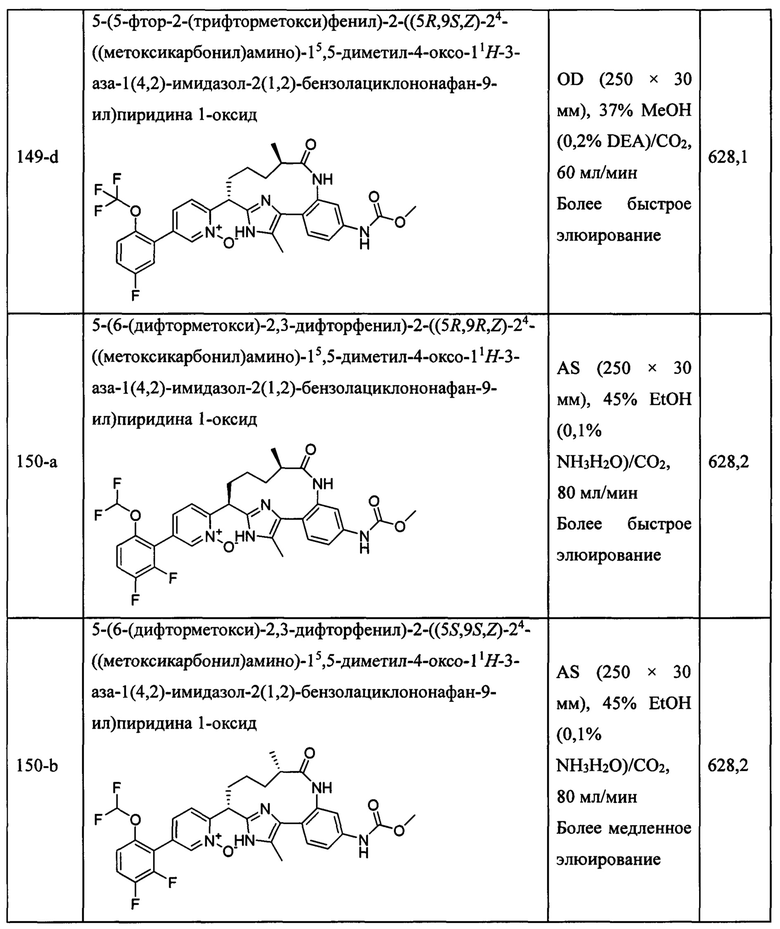
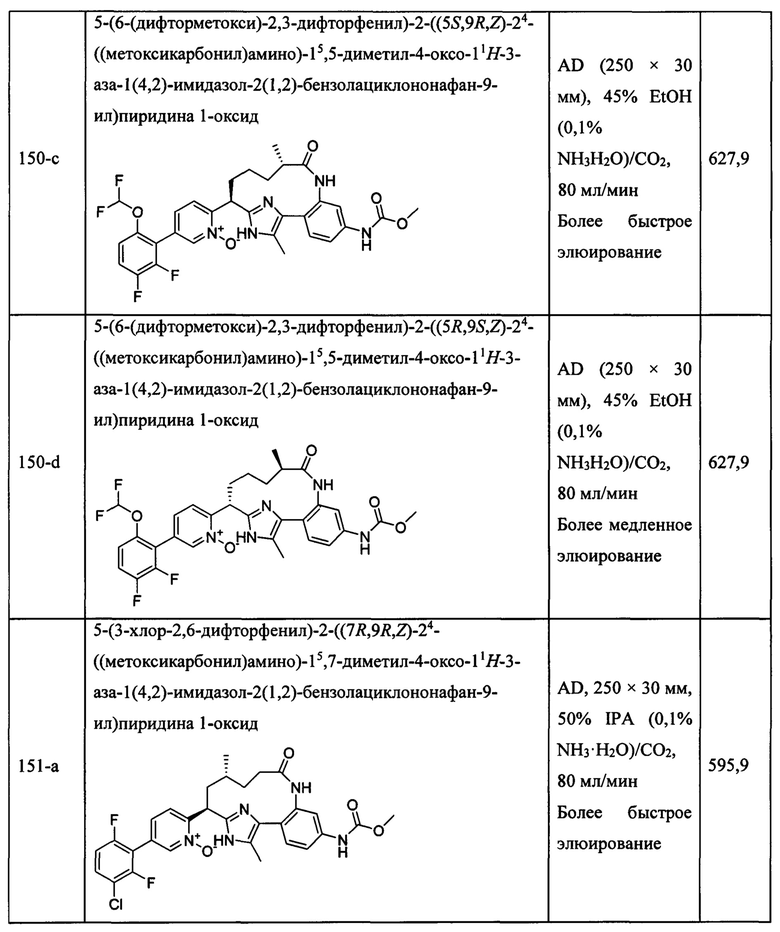
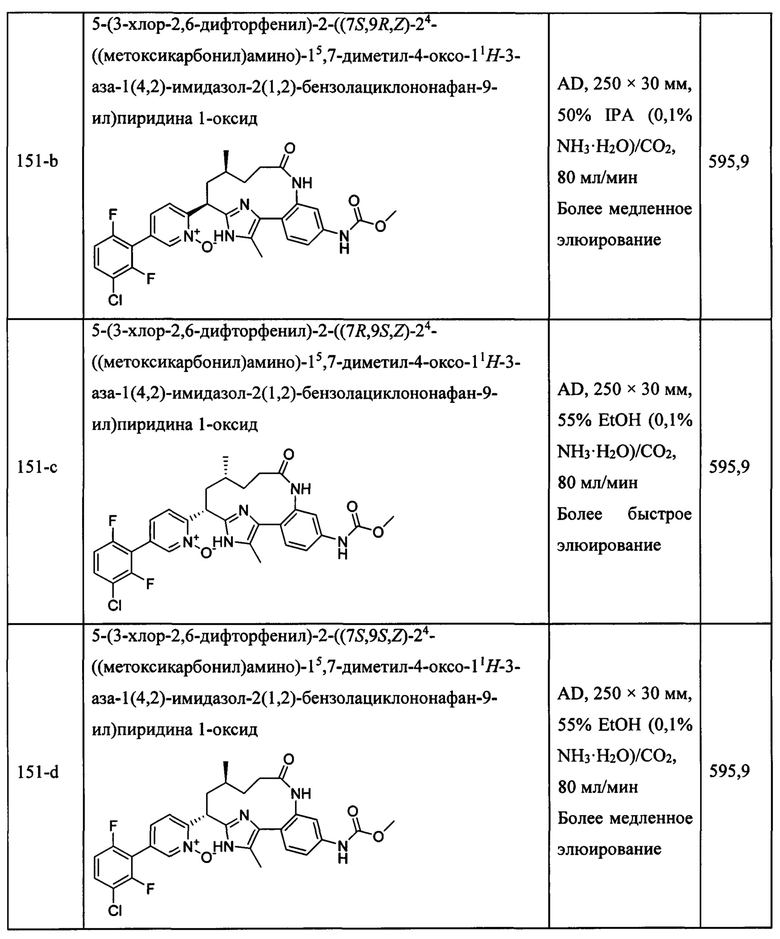
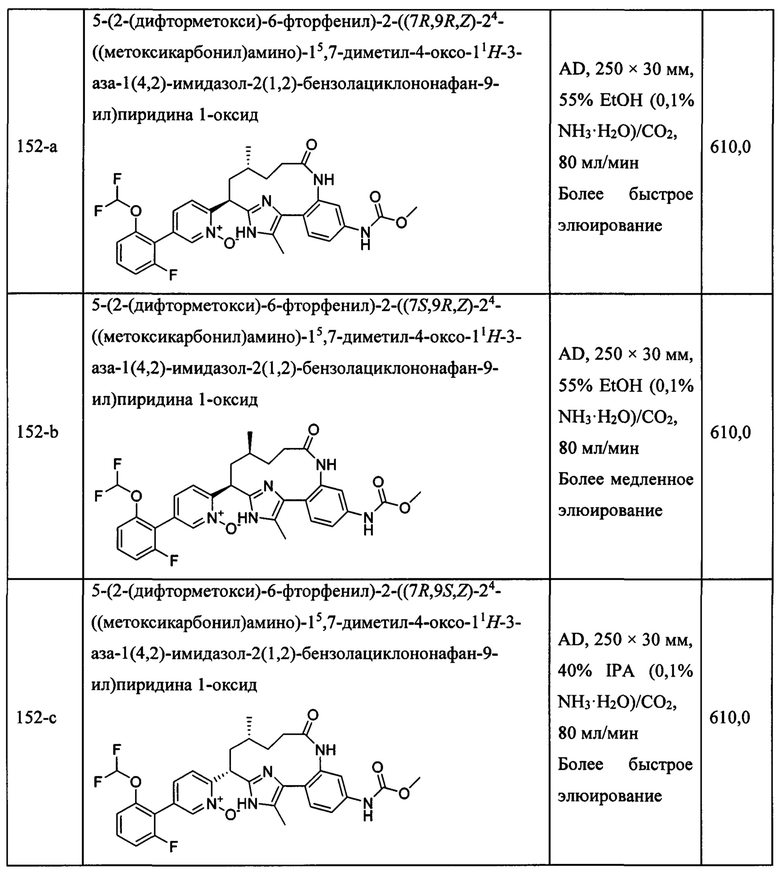
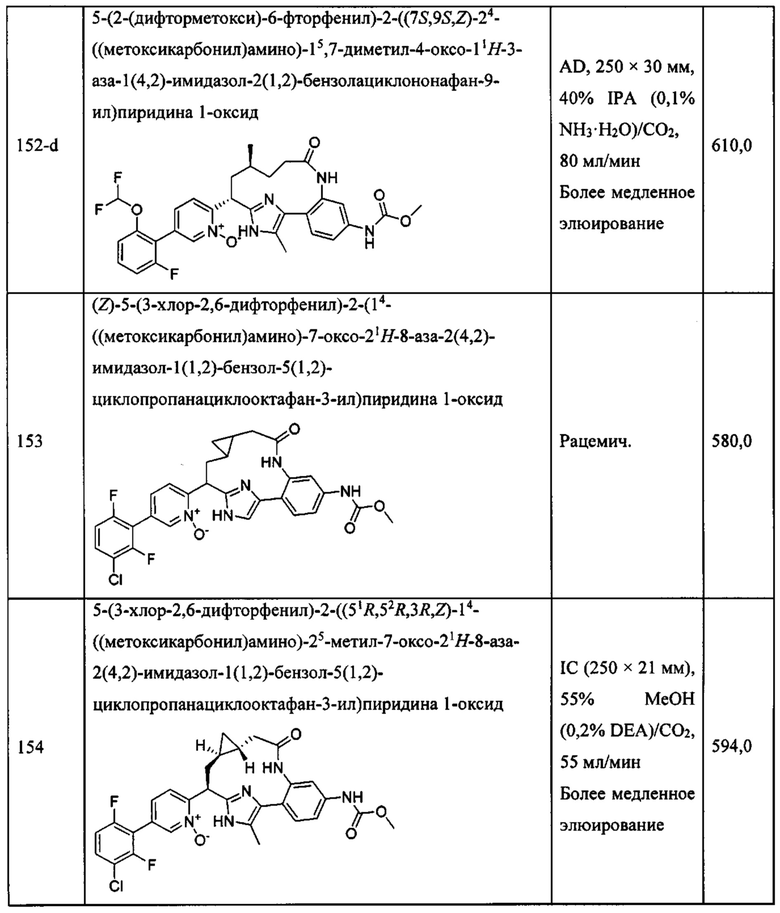
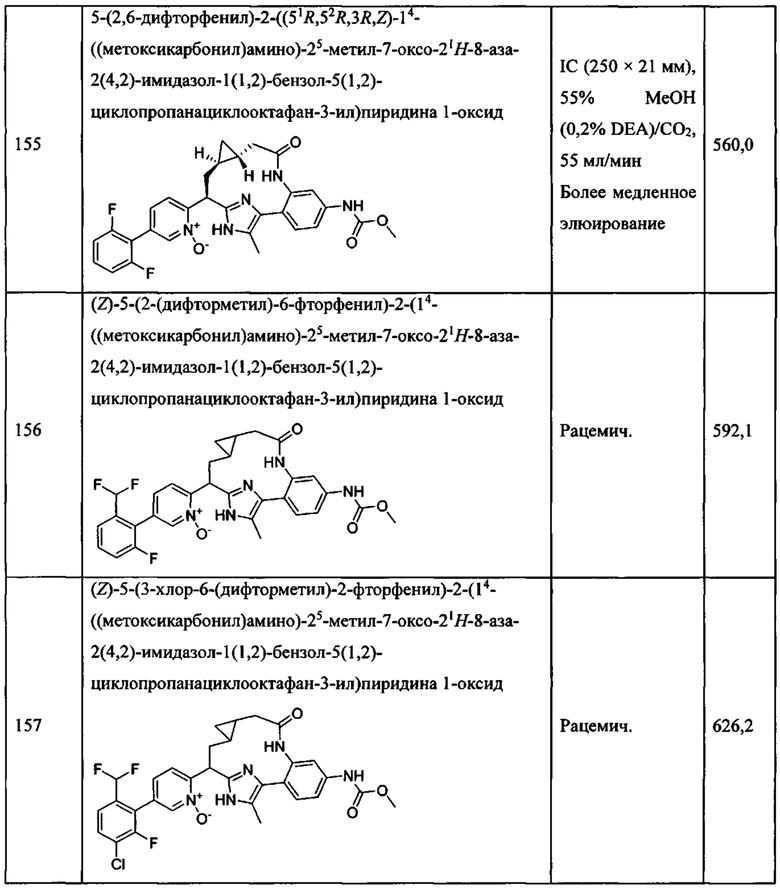
ПРИМЕР 160 (рацемат), 160-а и 160-b
(Z)-5-(5-ХЛОР-2-(1H-ТЕТРАЗОЛ-1-ИЛ)ФЕНИЛ)-2-(25-ФТОР-4-ОКСО-11H-3-АЗА-1(2,4)-ИМИДАЗОЛ-2(1,2)-БЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
Соединение примера 160 получали из промежуточного соединения 17 и промежуточного соединения 18 способами, описанными в синтезе примера 97 и примера 98. Продукт очищали методом HPLC с обращенной фазой. MS (ES+) m/z: 545 (М+Н).
Образец рацемического соединения примера 160 подвергали хиральному разделению методом SFC (OJ, 50×4,6 мм; метанол (0,05% диэтиламин)/СО2; 40 мл/мин, 40°С) с получением соединения примера 160-а (более медленное элюирование) и соединения примера 160-b (более быстрое элюирование). MS (ES+) m/z: 545 (М+Н).
ПРИМЕР 161 (рацемат), 161-а и 161-b
(Z)-5-(3-ХЛОР-2-ФТОР-6-(1H-ТЕТРАЗОЛ-1-ИЛ)ФЕНИЛ)-2-(25-ФТОР-4-ОКСО-11H-3-АЗА-1(2,4)-ИМИДАЗОЛ-2(1,2)-БЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
Соединение примера 161 получали способами, описанными в примере 160. Его очищали методом HPLC с обращенной фазой. MS (ES+) m/z: 563 (М+Н).
Образец рацемического соединения примера 161 подвергали хиральному разделению методом SFC (OJ, 50×4,6 мм; этанол (0,05% диэтиламин)/CO2; 4 мл/мин, 40°С) с получением соединения примера 161-а (более медленное элюирование) и соединения примера 161-b (более быстрое элюирование). MS (ES+) m/z: 545 (М+Н).
ПРИМЕР 162, 162-а, 162-b
(Z)-5-(3-ХЛОР-2,6-ДИФТОРФЕНИЛ)-2-(25-((МЕТОКСИКАРБОНИЛ)АМИНО)-4-ОКСО-11H-3-АЗА-1(1,4)-ПИРАЗОЛ-2(1,2)-БЕНЗОЛАЦИКЛОНОНАФАН-9-ИЛ)ПИРИДИНА 1-ОКСИД
162А: Этил-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-4-карбоксилат
К раствору этил-4-пиразолкарбоксилата (5 г, 35,7 ммоль) в DMF (10 мл) при к.т. добавляли SEM-Cl (7,6 мл, 42,8 ммоль). Смесь перемешивали в течение 10 мин. Ее разбавляли этилацетатом (50 мл) и промывали 10% водным карбонатом натрия (10 мл), водой (4×30 мл) и солевым раствором (20 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-30% этилацетатом в гексане) с получением указанного соединения. MS (ES+) m/z: 271 (М+Н).
162В: N-Метокси-N-метил-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-4-карбоксамид: К смеси соединения примера 162А (4,8 г, 17,75 ммоль) и N,O-диметилгидроксиламина гидрохлорида (2,60 г, 26,6 ммоль) в THF (40 мл) при 0°С добавляли раствор изопропилмагния бромида (2,9 М в 2-метилтетрагидрофуране, 18 мл, 52,2 ммоль). Ее перемешивали всю ночь и оставляли нагреваться до к.т. Ее гасили насыщенным водным хлоридом аммония (20 мл) и солевым раствором (50 мл). Смесь экстрагировали этилацетатом (2×50 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии (элюировали 0-100% этилацетатом в гексане) с получением указанного соединения. MS (ES+) m/z: 286 (М+Н).
162С: (5-Хлорпиридин-2-ил)(1-((2-(триметилсилил)этокси)метил)-1H-пиразол-4-ил)метанон: К раствору 2-бром-5-хлорпиридина (7,2 г, 37,4 ммоль) в безводном толуоле (100 мл) при -78°С медленно добавляли н-бутиллитий (2,5 М в гексане, 15 мл, 37,5 ммоль). Его перемешивали в течение 1 ч и к раствору добавляли раствор соединения примера 162В (8,7 г, 30,5 ммоль) в безводном толуоле (20 мл). Реакционную смесь перемешивали в течение 15 мин и нагревали до 0°С. Ее перемешивали в течение 1 ч и гасили насыщенным водным хлоридом аммония. Смесь экстрагировали этилацетатом (2×50 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии (элюировали 0-30% этилацетатом в гексане) с получением указанного соединения. MS (ES+) m/z: 338 (М+Н).
162D: (5-Хлорпиридин-2-ил)(1H-пиразол-4-ил)метанон: Соединение примера 162С (5,6 г, 16,57 ммоль) обрабатывали HCl в диоксане (50 мл, 200 ммоль) при к.т. в течение 1 ч. Это концентрировали при пониженном давлении и к остатку добавляли 100 мл смеси CHCl3/IPA (5:1) и 100 мл насыщенного водного бикарбоната натрия. Смесь переносили в делительную воронку и органический слой отделяли. Водный слой дважды экстрагировали CHCl3/IPA (5:1) (2×50 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-3% метанолом в DCM) с получением указанного соединения. MS (ES+) m/z: 208 (М+Н).
162Е: Метил(4-фтор-3-нитрофенил)карбамат: К суспензии гидрида натрия (60% масс. в минеральном масле) (5,5 г, 138 ммоль) в безводном DMA (60 мл) при 0°С добавляли раствор 4-фтор-3-нитроанилина (10 г, 64,1 ммоль) в DMA (40 мл). Ее перемешивали в течение 0,5 ч и добавляли метилхлорформиат (11 мл, 142 ммоль). Реакционную смесь перемешивали в течение 15 мин и оставляли нагреваться до к.т. в течение 1 ч. Ее гасили водным NaOH (3 н, 50 мл) и смесь перемешивали в течение 1 ч при к.т. Ее разбавляли водой (300 мл) и экстрагировали этилацетатом (3×200 мл). Объединенные органические слои промывали водой (4×100 мл), солевым раствором (50 мл), сушили над сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, а остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-5% метанолом в DCM) с получением указанного соединения. MS (ES+) m/z: 215 (М+Н).
162F: Метил(4-(4-(5-хлорпиколиноил)-1H-пиразол-1-ил)-3-нитрофенил)карбамат: Смесь соединения примера 162D (11,3 г, 46,3 ммоль), соединения примера 162Е (11,3 г, 46,3 ммоль) и карбоната калия (14 г, 101 ммоль) в DMA (100 мл) перемешивали при 100°С всю ночь. Ее охлаждали до к.т. и разбавляли этилацетатом (500 мл), промывали водой (4×200 мл). Объединенные водные слои экстрагировали этилацетатом (300 мл). Объединенные органические слои промывали солевым раствором (2×100 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-7% метанолом в DCM) с получением смеси требуемого продукта и побочного продукта гидролиза (1-(4-амино-2-нитрофенил)-1H-пиразол-4-ил)(5-хлорпиридин-2-ил)метанона.
К раствору вышеуказанной смеси в пиридине (50 мл) при 0°С добавляли метилхлорформиат (5,68 г, 60,1 ммоль). Ее перемешивали в течение 30 мин. Большую часть растворителя удаляли при пониженном давлении. Остаток растворяли в этилацетате (200 мл) и промывали 1 н HCl (2×50 мл) и солевым раствором (100 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-5% метанолом в DCM) с получением указанного соединения. MS (ES+) m/z: 402 (М+Н).
162G: Метил(3-амино-4-(4-(5-хлорпиколиноил)-1H-пиразол-1-ил)фенил)карбамат: Смесь соединения примера 162F (1,68 г, 4,18 ммоль), железа (0,934 г, 16,73 ммоль) и хлорида аммония (0,447 г, 8,36 ммоль) в изопропаноле (15 мл) и воде (5,00 мл) перемешивали при 80°С в течение 1 ч. Ее сразу загружали в силикагелевый дозатор и очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-5% метанолом в DCM) с получением указанного соединения. MS (ES+) m/z: 372 (М+Н).
162Н: (5-((2-(4-(5-Хлорпиколиноил)-1H-пиразол-1-ил)-5-((метоксикарбонил)амино)фенил)амино)-5-оксопентил)трифенилфосфония бромид: К смеси соединения примера 162G (1,87 г, 5,03 ммоль), (4-карбоксибутил)трифенилфосфония бромида (2,5 г, 5,64 ммоль) в DCM (50,3 мл) добавляли DIEA (2,5 мл, 14,31 ммоль) и HATU (2,3 г, 6,05 ммоль). Ее перемешивали в течение 16 ч. Ее концентрировали при пониженном давлении и остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-10% метанолом в DCM) с получением указанного соединения. MS (ES+) m/z: 716 (M-Br).
162I: Метил((14Z,8Е)-9-(5-хлорпиридин-2-ил)-4-оксо-11H-3-аза-1(1,4)-пиразол-2(1,2)-бензолациклононафан-8-ен-24-ил)карбамат:
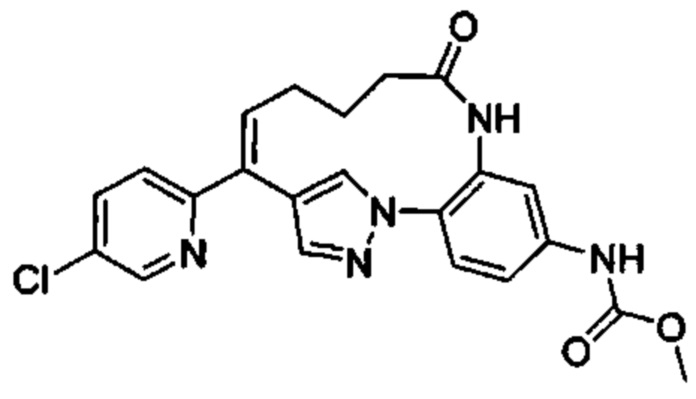
К раствору соединения примера 162Н (4 г, 5,02 ммоль) в THF (500 мл) добавляли трет-бутилат калия (1 M в THF, 21 мл, 21,00 ммоль). Смесь перемешивали при к.т. в течение 16 ч. Ее гасили насыщенным водным хлоридом аммония, экстрагировали этилацетатом (200 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали. Его очищали методом колоночной флеш-хроматографии на силикагеле (элюировали 0-6% метанолом в DCM) с получением указанного соединения. MS (ES+) m/z: 438 (М+Н).
Пример 162:
Соединение примера 162 получали из соединения примера 162I способами, описанными в примере 50. Его очищали методом HPLC с обращенной фазой с получением рацемического продукта. MS (ES+) m/z: 568.3 (М+Н).
Образец соединения примера 162 подвергали хиральному разделению методом SFC (Kromasil, 250×30 мм, 50% (2:1 метанол/MeCN) в CO2; 70 мл/мин; 100 бар, 35°С) с получением соединения примера 162-а (более медленное элюирование) и соединения примера 162-b (более быстрое элюирование). MS (ES+) m/z: 568 (М+Н); 1Н ЯМР: (500 МГц, DMSO-d6): δ 9.96 (s, 1Н), 9.21 (s, 1Н), 8.53 (s, 1Н), 8.07 (s, 1Н), 7.80 (m, 2Н), 7.63 (s, 1Н), 7.49 (m, 2Н), 7.34 (t, J=9.0 Гц, 1Н), 7.16 (d, J=9.0 Гц, 1Н), 4.61 (m, 1Н), 3.68 (s, 3Н), 2.18 (brs, 2Н), 2.04 (m, 1Н), 1.80-1.60 (m, 2Н), 1.40-1.00 (m, 3Н).
ПРИМЕР 163
5-(3-ХЛОР-2,6-ДИФТОРФЕНИЛ)-2-(11-МЕТИЛ-8-ОКСО-11H-9-АЗА-1(3,4)-ПИРАЗОЛ-2(1,3)-БЕНЗОЛАЦИКЛОНОНАФАН-3-ИЛ)ПИРИДИНА 1-ОКСИД
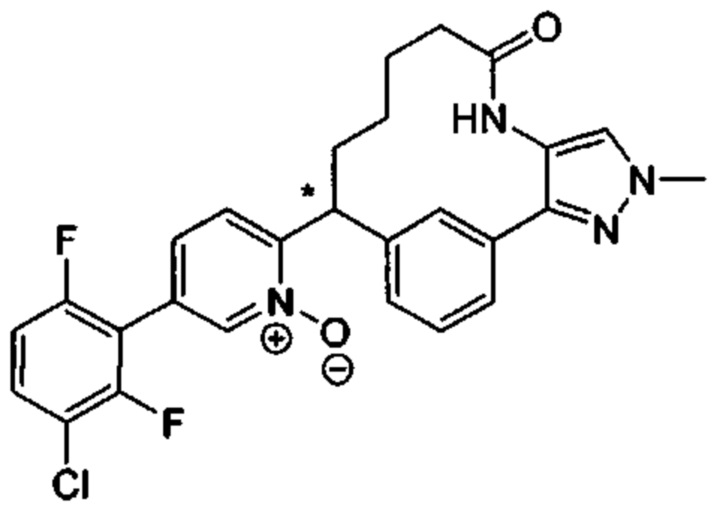
163А: 4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразол: К суспензии гидрида натрия (7,07 г, 177 ммоль, 60% масс. в масле) в THF (100 мл) при 0°С в атмосфере N2 добавляли 4-нитро-1H-пиразол (10 г, 88 ммоль). Смесь перемешивали в течение 0,5 ч перед добавлением SEM-Cl (17,25 мл, 97 ммоль). Реакционную смесь перемешивали при к.т. еще 1,5 ч. Ее гасили водным хлоридом аммония (нас., 50 мл) и экстрагировали EtOAc (50 мл×4). Объединенные органические слои промывали солевым раствором (100 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом колоночной флеш-хроматографии (SiO2, петролейный эфир : этилацетат = 100:1-10:1, градиент) с получением указанного соединения. 1Н ЯМР (400 МГц, CDCl3): δ 8.31 (s, 1Н), 8.11 (s, 1H), 5.46 (s, 2H), 3.59-3.68 (m, 2H), 0.99-0.91 (m, 2H), 0.00 (s, 9H).
163B: (5-Хлорпиридин-2-ил)(3-(4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-5-ил)фенил)метанон: Смесь промежуточного соединения 2 (975 мг, 3,29 ммоль), соединения примера 163А (500 мг, 2,055 ммоль), бутилди-1-адамантилфосфина (111 мг, 0,308 ммоль), K2CO3 (852 мг, 6,16 ммоль), пивалиновой кислоты (0,036 мл, 0,308 ммоль) и Pd(OAc)2 (46,1 мг, 0,205 ммоль) в DMF (10 мл) в атмосфере азота перемешивали при 120°С в течение 6 ч. Ее охлаждали до к.т. и разбавляли водой (20 мл). Смесь экстрагировали EtOAc (20 мл×3). Объединенные органические слои промывали солевым раствором (10 мл×4), сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом колоночной флеш-хроматографии (SiO2, петролейный эфир : этилацетат = 50:1-10:1) с получением указанного соединения. MS (ES+) m/z: 459.0 (М+Н); 1Н ЯМР (400 МГц, CDCl3): δ 8.67 (d, J=2.0 Гц, 1Н), 8.25-8.34 (m, 3Н), 8.10 (d, J=8.2 Гц, 1Н), 7.91 (dd, J=2.2, 8.4 Гц, 1Н), 7.81 (d, J=7.8 Гц, 1Н), 7.65-7.73 (m, 1Н), 5.33 (s, 2Н), 3.66-3.75 (m, 2Н), 0.84-0.93 (m, 2Н), 0.04 (s, 9Н).
163С: (3-(4-Амино-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-5-ил)фенил)(5-хлорпиридин-2-ил)метанон: К раствору соединения примера 163В (200 мг, 0,436 ммоль) в EtOH (15 мл) добавляли железо (97 мг, 1,743 ммоль) и хлорид аммония (46,6 мг, 0,872 ммоль) в воде (5 мл). Смесь перемешивали при 90°С в течение 1 ч. Ее охлаждали до к.т. и разбавляли водой (20 мл). Смесь экстрагировали EtOAc (20 мл×3). Объединенные органические слои промывали солевым раствором (10 мл×4), сушили над сульфатом натрия, фильтровали и концентрировали с получением указанного соединения. MS (ES+) m/z: 429.1 (М+Н); 1Н ЯМР (400 МГц, CDCl3): δ 8.66 (d, J=1.8 Гц, 1Н), 8.21-8.27 (m, 1Н), 8.03-8.12 (m, 2Н), 7.84-7.93 (m, 2Н), 7.61-7.68 (m, 1Н), 7.31 (s, 1H), 5.30-5.41 (m, 2Н), 3.61-3.68 (m, 2Н), 0.83-0.90 (m, 2Н), 0.02-0.06 (m, 9Н).
163D: 5-(3-Хлор-2,6-дифторфенил)-2-(8-оксо-11H-9-аза-1(5,4)-пиразол-2(1,3)-бензолациклононафан-3-ил)пиридина 1-оксид:
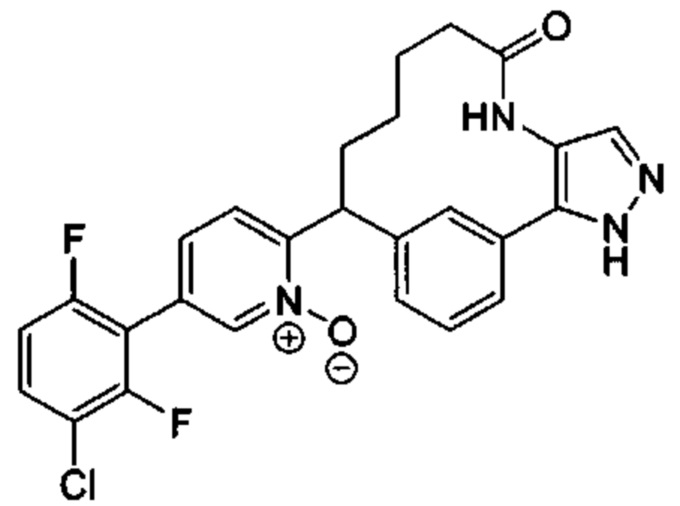
Указанное соединение получали из соединения примера 163С способом, описанным в синтезе соединения примера 50. MS (ES+) m/z: 495.1 (М+Н).
Пример 163:
К смеси соединения примера 163D и K2CO3 (8,38 мг, 0,061 ммоль) в DMF (2 мл) добавляли йодметан (5,74 мг, 0,040 ммоль). Ее перемешивали при 40°С в течение 14 ч. К реакционной смеси добавляли воду (10 мл) и ее экстрагировали EtOAc (20 мл×2). Объединенные органические слои промывали солевым раствором (20 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом HPLC с обращенной фазой с получением указанных соединений. MS (ES+) m/z: 509.1 (М+Н); 1Н ЯМР (400 МГц, CD3CN): δ 8.29-8.38 (m, 1Н), 7.81 (s, 1Н), 7.49-7.73 (m, 4Н), 7.41-7.48 (m, 1Н), 7.33-7.40 (m, 2Н), 7.17 (t, J=9.2 Гц, 1H), 7.04 (d, J=7.5 Гц, 1Н), 4.65-4.84 (m, 1Н), 3.98 (s, 3Н), 2.24-2.28 (m, 2Н), 2.03-2.15 (m, 2Н), 1.56-1.70 (m, 1Н), 1.21-1.33 (m, 2Н), 1.02-1.16 (m, 1Н).
ПРИМЕР 164
5-(3-ХЛОР-2,6-ДИФТОРФЕНИЛ)-2-(11-МЕТИЛ-8-ОКСО-11H-9-АЗА-1(5,4)-ПИРАЗОЛ-2(1,3)-БЕНЗОЛАЦИКЛОНОНАФАН-3-ИЛ)ПИРИДИНА 1-ОКСИД
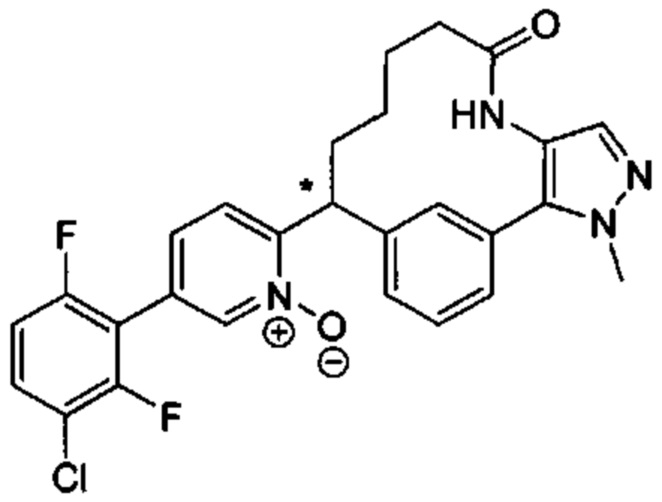
Соединение примера 164 отделяли от соединения примера 163 методом HPLC. MS (ES+) m/z: 509.1 (М+Н); 1Н ЯМР (400 МГц, CD3CN): δ 8.36 (s, 1Н), 7.98 (br. s., 2H), 7.61 (dt, J=6.0, 8.6 Гц, 1H), 7.44-7.57 (m, 4H), 7.34 (t, J=7.7 Гц, 1H), 7.17 (t, J=9.0 Гц, 1H), 6.95 (d, J=7.5 Гц, 1H), 4.81 (dd, J=4.4, 11.2 Гц, 1H), 3.90 (s, 3H), 2.23-2.29 (m, 2H), 2.07-2.15 (m, 2H), 1.52-1.68 (m, 1H), 0.80-1.49 (m, 3H).
ПРИМЕР 165, 465-a, 165-b
5-(5-ХЛОР-2-(4-(ТРИФТОРМЕТИЛ)-1H-1,2,3-ТРИАЗОЛ-1-ИЛ)ФЕНИЛ)-2-(11-(ДИФТОРМЕТИЛ)-8-ОКСО-11H-9-АЗА-1(5,4)-ПИРАЗОЛ-2(1,3)-БЕНЗОЛАЦИКЛОНОНАФАН-3-ИЛ)ПИРИДИНА 1-ОКСИД
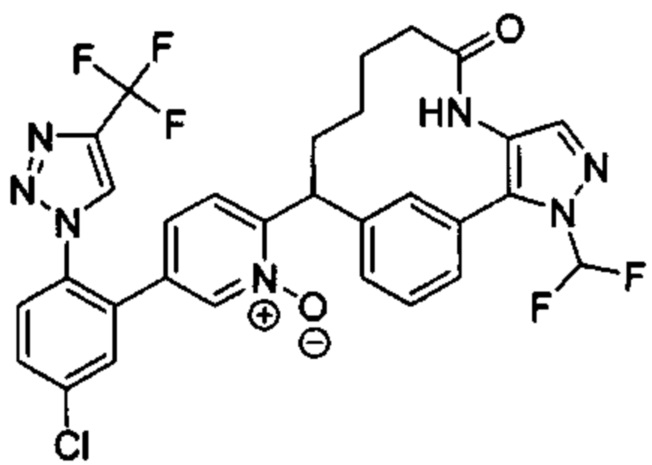
165А: 1-(Дифторметил)-4-нитро-1H-пиразол: Смесь 4-нитро-1H-пиразола (2 г, 17,69 ммоль), Cs2CO3 (5,76 г, 17,69 ммоль) и 2-хлор-2,2-дифторацетата натрия (5,39 г, 35,4 ммоль) в DMF (10 мл) перемешивали ппри 120°С в атмосфере азота в течение 2 ч. Ее охлаждали до к.т. и разбавляли водой (20 мл). Смесь экстрагировали EtOAc (70 мл×3). Объединенные органические слои промывали водой (40 мл) и солевым раствором (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом колоночной флеш-хроматографии на силикагеле (элюировали петролейным эфиром : этилацетатом = 100:1-10:1, градиент) с получением указанного соединения.
Пример 165:
Соединение примера 165 получали из соединения примера 165А способом, описанным в синтезе соединения 164D. MS (ES+) m/z: 509.1 (М+Н); 1Н ЯМР (CD3OD, 400 МГц): δ 8.75 (s, 1Н), 8.24 (s, 1Н), 7.69-7.81 (m, 4Н), 7.58-7.67 (m, 3Н), 7.37-7.64 (m, 2Н), 7.22 (d, J=8.2 Гц, 1H), 6.98 (d, J=6.8 Гц, 1H), 4.75 (d, J=10.4 Гц, 1H), 2.39-2.52 (m, 1H), 1.95-2.14 (m, 4H), 1.67 (br. s., 1H), 1.24-1.37 (m, 1H), 1.10 (brs, 1H).
Рацемический образец соединения примера 165 подвергали хиральному разделению методом SFC (AS, 250×30 мм, 30% МеОН (0,1% NH3⋅H2O)/CO2, 50 мл/мин) с получением соединения примера 165-а (более медленное элюирование) и соединения примера 165-b (более быстрое элюирование). MS (ES+) m/z: 644.2 (М+Н).
С применением методик, подобных описанным выше, следующие соединения синтезировали и характеризовали способом LCMS.
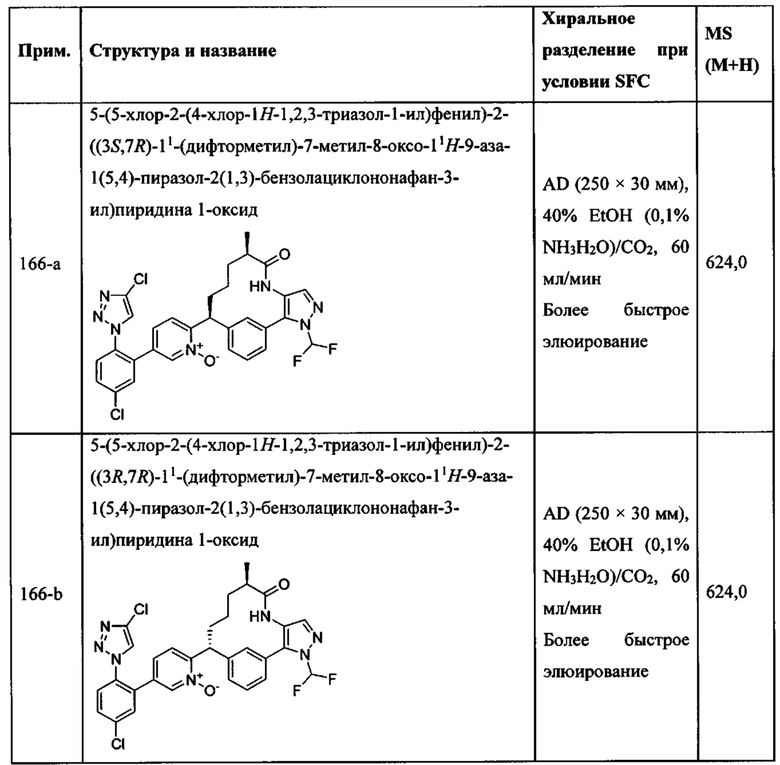
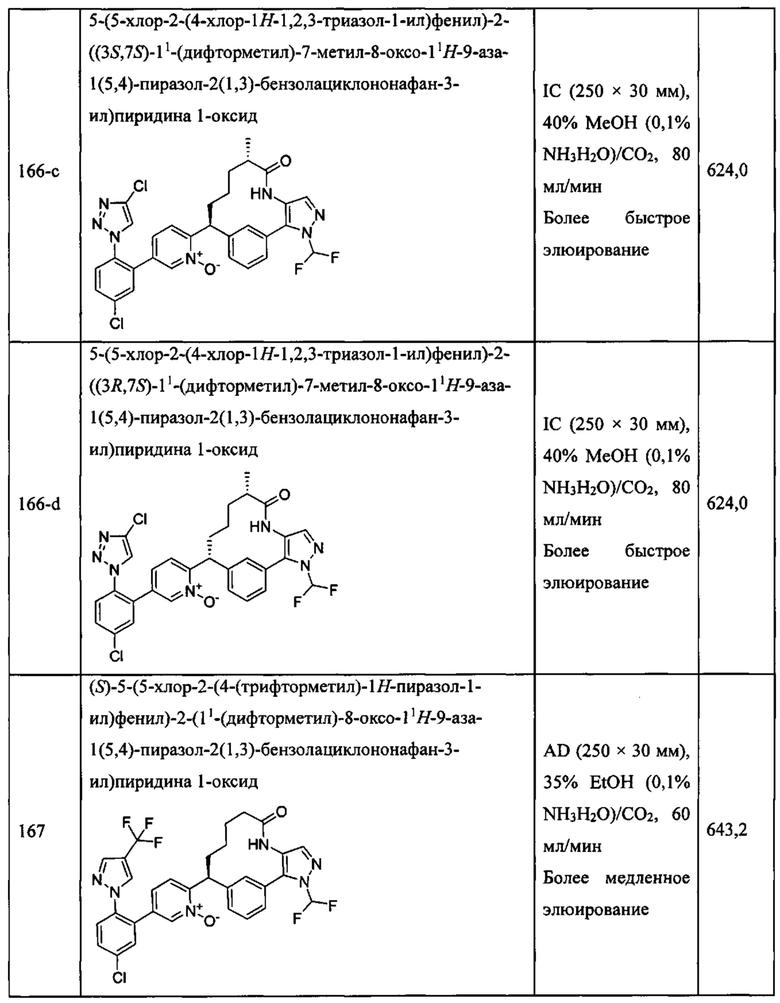
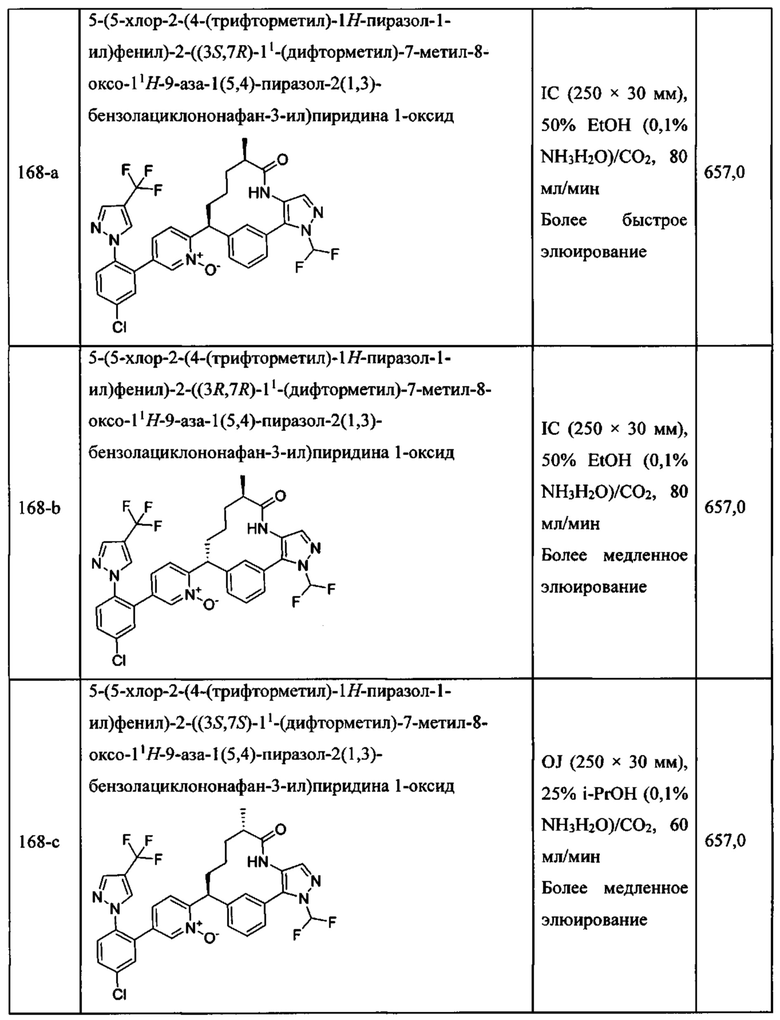
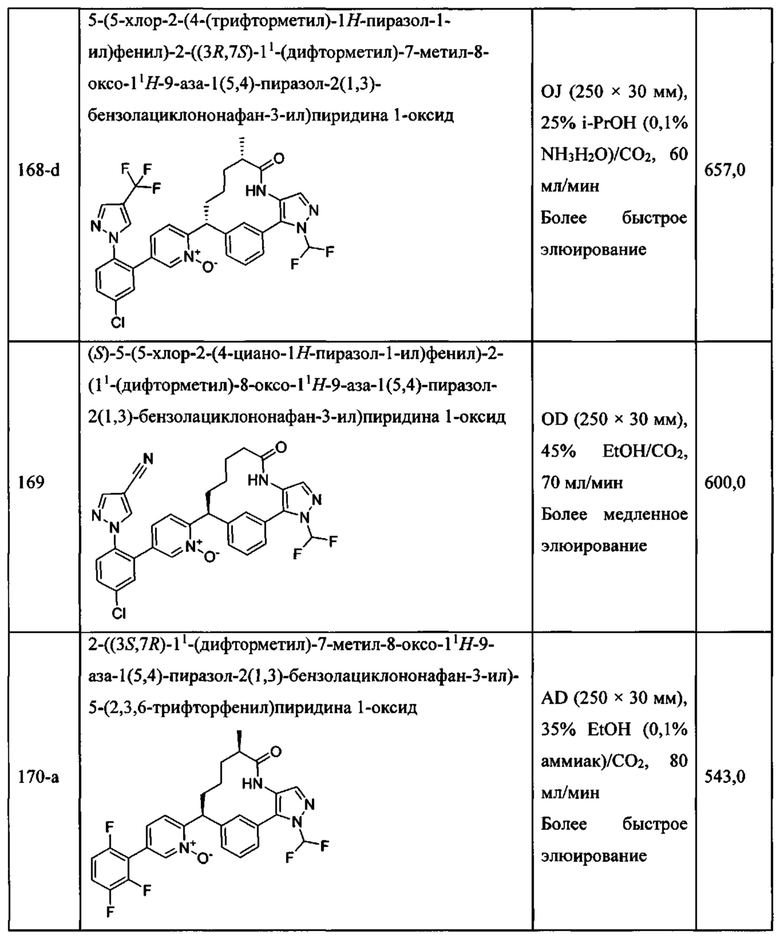
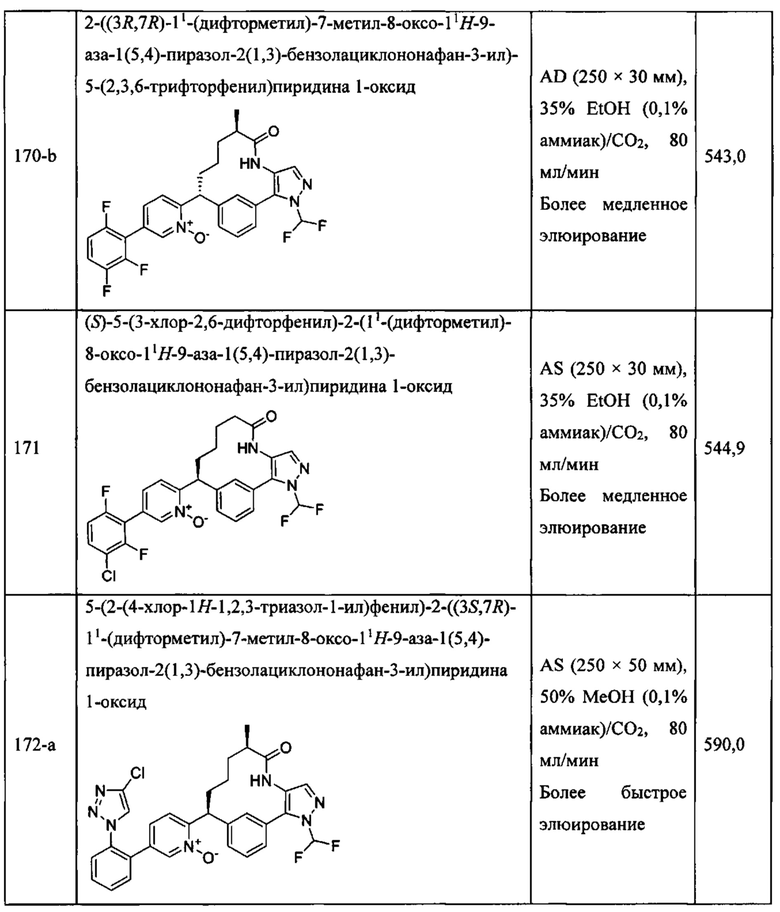
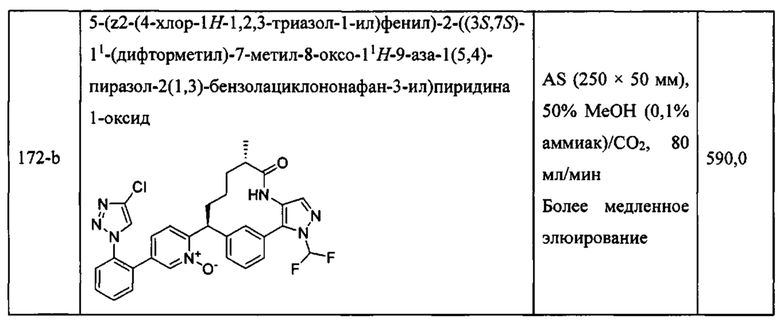
Анализ фактора XIa
Эффективность соединения в соответствии с настоящим изобретением в качестве ингибитора фактора коагуляции XIa может быть определена с использованием соответствующей очищенной серинпротеазы и подходящего синтетического субстрата. Скорость гидролиза хромогенного или флуорогенного субстрата соответствующей серинпротеазой измеряли как при отсутствии, так и в присутствии соединений в соответствии с настоящим изобретением. Анализы выполняли при комнатной температуре или при 37°С. Гидролиз субстрата приводил к высвобождению аминотрифторметилкумарина (AFC), который контролировали спектрофотометрически путем измерения повышения эмиссии при 510 нм с возбуждением при 405 нм. Снижение скорости изменения флуоресценции в присутствии ингибитора указывает на ингибирование фермента. Такие способы известны специалисту в данной области. Результаты данного анализа выражены в виде константы ингибирования Ki.
Определения фактора XIa осуществляли в 50 мМ буфере HEPES при рН 7,4, содержащем 150 мМ NaCl, 5 мМ CaCl2 и 0,1% PEG 8000 (полиэтиленгликоль; JT Baker или Fisher Scientific). Определения осуществляли с использованием очищенного человеческого фактора XIa при конечной концентрации 40 пМ (Sekisui Diagnostics) и синтетического субстрата Z-Gly-Pro-Arg-AFC, соль TFA (Sigma, № С0980) при концентрации 100 мкМ.
Анализы активности выполняли путем разбавления маточного раствора субстрата по меньшей мере в десять раз до конечной концентрации ≤0,1 Km раствором, содержащим фермент или фермент, уравновешенный ингибитором. Время, необходимое для достижения равновесия между ферментом и ингибитором, определяли в контрольных экспериментах. Измеряли начальные скорости образования продукта в отсутствии (Vo) или в присутствии ингибитора (Vi). Принимая во внимание конкурентное ингибирование и то, что величина пренебрежимо мала по сравнению с Km/[S], [I]/е и [I]/е (где [S], [I] и е, соответственно, представляют собой общие концентрации субстрата, ингибитора и фермента), константа равновесия (Ki) для диссоциации ингибитора от фермента может быть получена из зависимости Vo/Vi при [I], показанной в следующем уравнении:
Vo/Vi=1+[I]/Ki.
Активности, показанные в данном анализе, указывают на то, что соединения в соответствии с настоящим изобретением, могут быть терапевтически применимы для лечения или предупреждения различных сердечно-сосудистых и/или цереброваскулярных тромбоэмболических состояний у больных, страдающих нестабильной стенокардией, острым коронарным синдромом, рефрактерной стенокардией, инфарктом миокарда, транзиторными ишемическими атаками, предсердной фибрилляцией, инсультом, таким как тромботический инсульт или эмболический инсульт, тромбозом вен, тромбозом коронарных и церебральных артерий, церебральной и легочной эмболией, атеросклерозом, тромбозом глубоких вен, диссеминированным внутрисосудистым свертыванием и реокклюзией или рестенозом реканализированных сосудов.
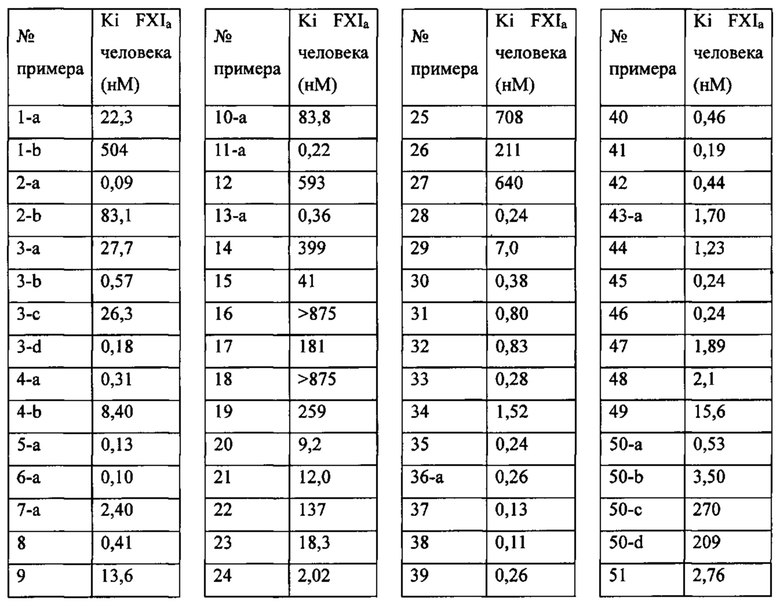
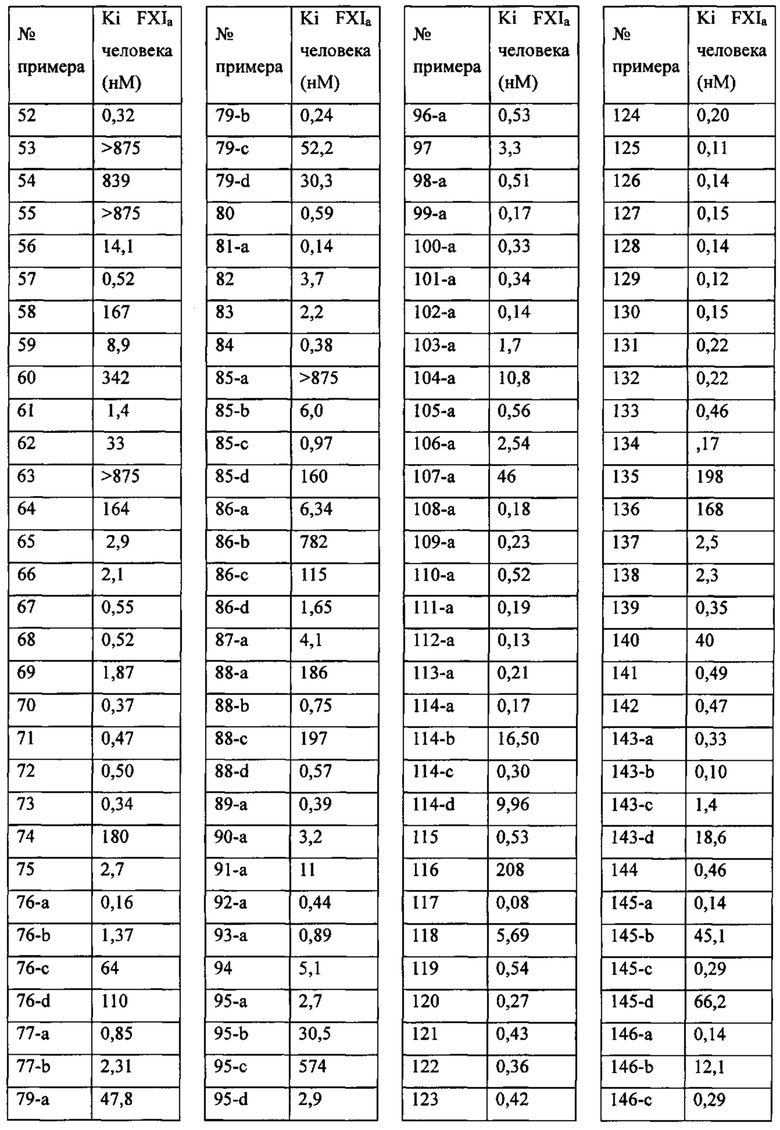
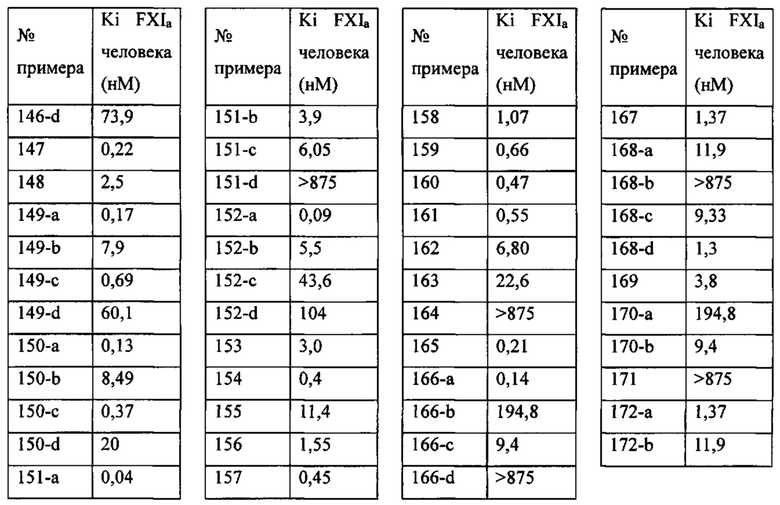
Анализ калликреина
Эффективность соединения в соответствии с настоящим изобретением в качестве ингибитора калликреина может быть определена с использованием соответствующей очищенной серинпротеазы и подходящего синтетического субстрата. Скорость гидролиза хромогенного или флуорогенного субстрата соответствующей серинпротеазой измеряли как при отсутствии, так и в присутствии соединений в соответствии с настоящим изобретением. Анализы выполняли при комнатной температуре или при 37°С. Гидролиз субстрата приводил к высвобождению аминотрифторметилкумарина (AFC), который контролировали спектрофотометрически путем измерения повышения эмиссии при 510 нм с возбуждением при 405 нм. Снижение скорости изменения флуоресценции в присутствии ингибитора указывает на ингибирование фермента. Такие способы известны специалисту в данной области. Результаты данного анализа выражены в виде коонстанты ингибирования Ki.
Определения калликреина осуществляли в 50 мМ буфере HEPES при рН 7,4, содержащем 150 мМ NaCl, 5 мМ CaCl2 и 0,1% PEG 8000 (полиэтиленгликоль; Fisher Scientific). Определения осуществляли с использованием очищенного человеческого плазматического калликреина при конечной концентрации 0,5 нМ (Enzyme Research Laboratories) и синтетического субстрата ацетил-K-P-R-AFC (Sigma, № С6608) при концентрации 100 мМ.
Анализы активности выполняли путем разбавления маточного раствора субстрата по меньшей мере в десять раз до конечной концентрации ≤0,2 Km раствором, содержащим фермент или фермент, уравновешенный ингибитором. Время, необходимое для достижения равновесия между ферментом и ингибитором, определяли в контрольных экспериментах. Реакции выполняли при условиях кривой линейной прогрессии и усиления флуоресценции, измеряемого при возбуждении 405 нм/эмиссии 510 нм. Значения преобразовывали в процентное ингибирование контрольной реакции (после вычитания значения 100% ингибирования). IC50 определяли по точке перегиба на четырехпараметрической логистической кривой. Ki вычисляли с использованием уравнения Ченга-Прусоффа: Ki=IC50/(1+([S]/Km)).
Активности, показанные в данном анализе, указывают на то, что соединения в соответствии с настоящим изобретением, могут быть терапевтически применимы для лечения или предупреждения различных сердечно-сосудистых и/или цереброваскулярных тромбоэмболических состояний у больных, страдающих нестабильной стенокардией, острым коронарным синдромом, рефрактерной стенокардией, инфарктом миокарда, транзиторными ишемическими атаками, предсердной фибрилляцией, инсультом, таким как тромботический инсульт или эмболический инсульт, тромбозом вен, тромбозом коронарных и церебральных артерий, церебральной и легочной эмболией, атеросклерозом, тромбозом глубоких вен, диссеминированным внутрисосудистым свертыванием и реокклюзией или рестенозом реканализированных сосудов.
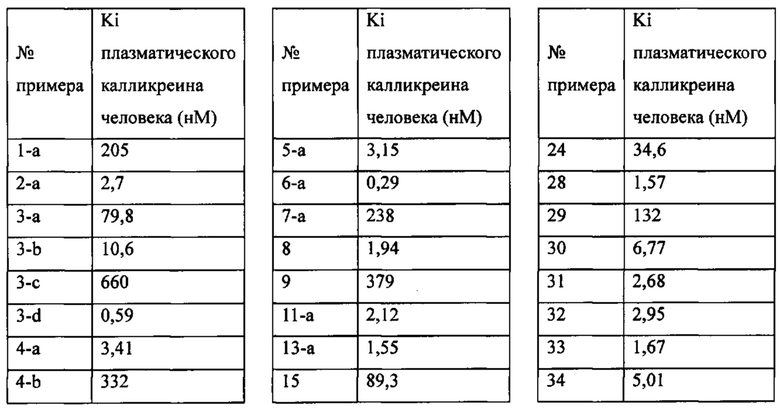
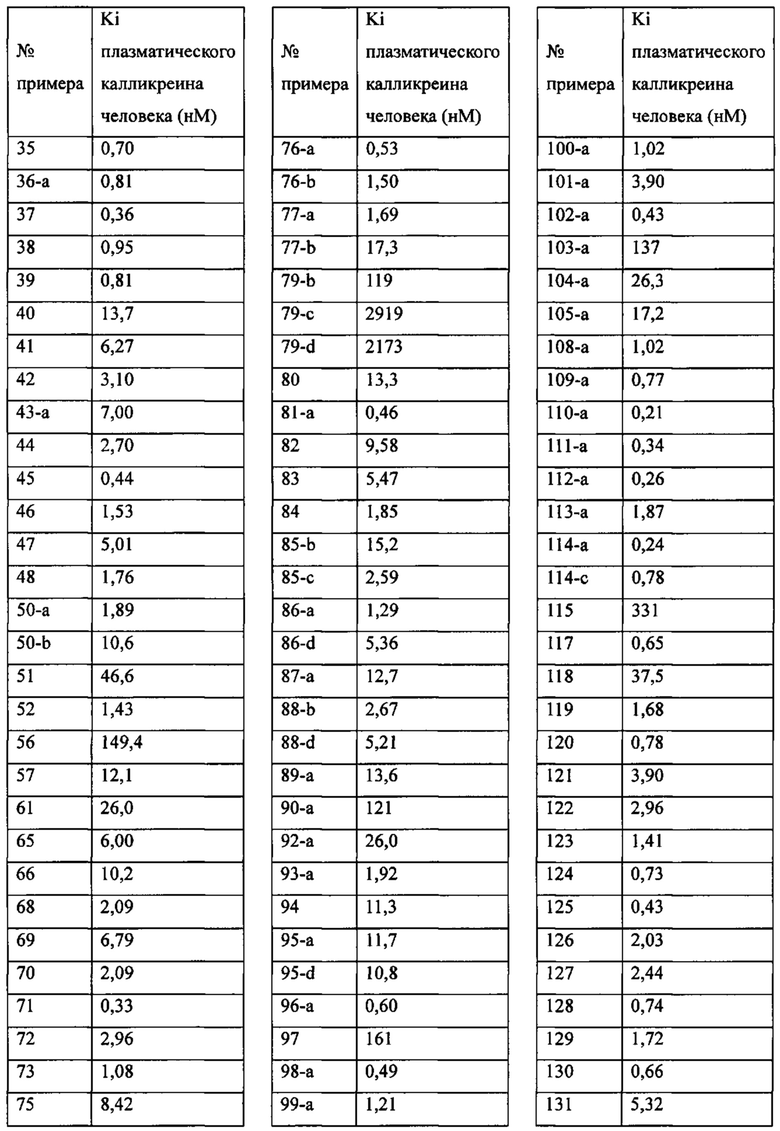
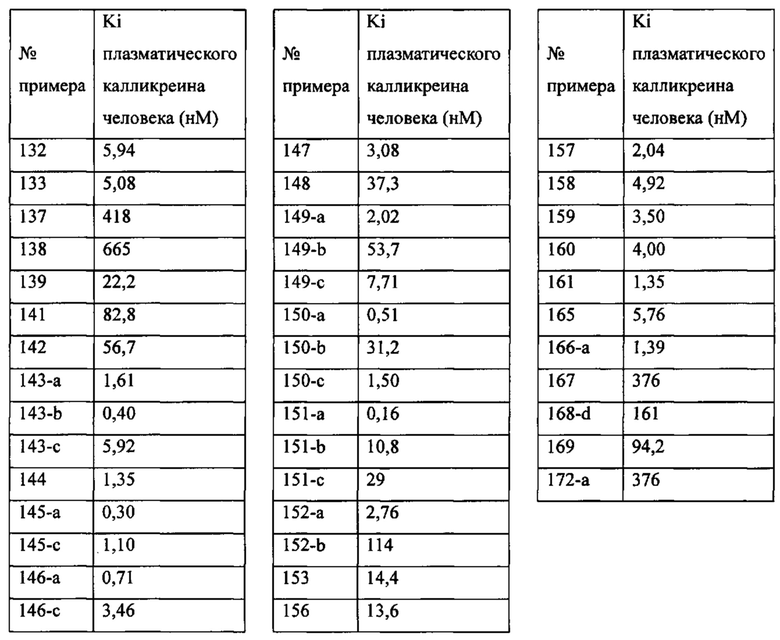
| название | год | авторы | номер документа |
|---|---|---|---|
| Соединения 1-циано-пирролидинов в качестве ингибиторов USP30 | 2016 |
|
RU2717238C2 |
| НОВЫЕ СОЕДИНЕНИЯ | 2018 |
|
RU2767652C2 |
| СПОСОБ ЛЕЧЕНИЯ | 2012 |
|
RU2621148C2 |
| АЗАИНДОЛИЛПИРИДОНЫ И ДИАЗАИНДОЛИЛПИРИДОНЫ | 2018 |
|
RU2788659C2 |
| ЗАМЕЩЕННОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ КОНДЕНСИРОВАННОЕ ЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2815814C1 |
| ПИРИДИЛПИРИДОНЫ | 2018 |
|
RU2805334C2 |
| ХИНОЛИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ SMO | 2015 |
|
RU2695815C2 |
| Бензотиофены и родственные соединения в качестве агонистов STING | 2019 |
|
RU2806274C2 |
| ХИМИЧЕСКИЕ СОСТАВЫ, КОМПОЗИЦИИ И СПОСОБЫ ИХ ИСПОЛЬЗОВАНИЯ | 2005 |
|
RU2413720C2 |
| ИНГИБИТОРЫ ФУРИНА | 2019 |
|
RU2799824C2 |
Изобретение относится к соединению формулы I, обладающему свойствами ингибитора фактора коагуляции XIa, фармацевтической композиции на его основе, способу ингибирования и предупреждения образования тромба в крови, способу лечения и применению для лечения венозной тромбоэмболии и легочной эмболии, тромбоза глубоких вен, тромботического инсульта. В общей формуле I  представляет собой фенил или 5-10-членный гетероарил с 1-2 атомами азота, который необязательно замещен одной-двумя группами, независимо выбранными из группы, состоящей из галогена, оксо, циано, R6, OR6, C(O)OR6, NR6R7, NHC(O)O-C1-3 алкил-OR7;
представляет собой фенил или 5-10-членный гетероарил с 1-2 атомами азота, который необязательно замещен одной-двумя группами, независимо выбранными из группы, состоящей из галогена, оксо, циано, R6, OR6, C(O)OR6, NR6R7, NHC(O)O-C1-3 алкил-OR7;  представляет собой фенил или 5-6-членный гетероарил с 1-2 атомами азота, который необязательно замещен одной-двумя группами, независимо выбранными из группы, состоящей из галогена, оксидо, циклопропила, R6; W представляет собой N или N+O-; Y-X представляет собой -CHC(O)OR7-NR6-, -CR6R7-C(O)NR6-, -CHC(O)R7-NR6 -; Z представляет собой C3-4 алкилен; R1 представляет собой фенил, необязательно замещенный одним-четырьмя заместителями, независимо выбранными из группы, состоящей из галогена, циано, R6, OR6, C(O)R6, NR6R7, и 5-членного гетероарила с 2-4 гетероатомами, выбранными из азота и кислорода (который необязательно замещен галогеном, циано, циклопропилом, C(O)OH, или R6); R2 представляет собой водород, или OR6; R3 представляет собой водород; каждый R4 независимо представляет собой C1-6 алкил; R5 представляет собой водород, галоген или C1-6 алкил; или один из R4 и R5 может быть взят вместе с атомами между ними с образованием 3-6-членного алкильного кольца; каждый R6 независимо представляет собой водород или C1-6 алкил, который необязательно замещен одной-тремя группами, независимо выбранными из группы, состоящей из галогена; каждый R7 независимо представляет собой водород, C1-6 алкил, 6-членный гетероарил с 1-2 атомами азота или 5-членный гетероциклил с 1 атмом азота, где указанная алкильная группа необязательно замещена одной-тремя группами, независимо выбранными из группы, состоящей из галогена; Ra представляет собой водород или гидрокси; n представляет собой целое число от нуля до одного. 8 н. и 7 з.п. ф-лы, 172 пр.
представляет собой фенил или 5-6-членный гетероарил с 1-2 атомами азота, который необязательно замещен одной-двумя группами, независимо выбранными из группы, состоящей из галогена, оксидо, циклопропила, R6; W представляет собой N или N+O-; Y-X представляет собой -CHC(O)OR7-NR6-, -CR6R7-C(O)NR6-, -CHC(O)R7-NR6 -; Z представляет собой C3-4 алкилен; R1 представляет собой фенил, необязательно замещенный одним-четырьмя заместителями, независимо выбранными из группы, состоящей из галогена, циано, R6, OR6, C(O)R6, NR6R7, и 5-членного гетероарила с 2-4 гетероатомами, выбранными из азота и кислорода (который необязательно замещен галогеном, циано, циклопропилом, C(O)OH, или R6); R2 представляет собой водород, или OR6; R3 представляет собой водород; каждый R4 независимо представляет собой C1-6 алкил; R5 представляет собой водород, галоген или C1-6 алкил; или один из R4 и R5 может быть взят вместе с атомами между ними с образованием 3-6-членного алкильного кольца; каждый R6 независимо представляет собой водород или C1-6 алкил, который необязательно замещен одной-тремя группами, независимо выбранными из группы, состоящей из галогена; каждый R7 независимо представляет собой водород, C1-6 алкил, 6-членный гетероарил с 1-2 атомами азота или 5-членный гетероциклил с 1 атмом азота, где указанная алкильная группа необязательно замещена одной-тремя группами, независимо выбранными из группы, состоящей из галогена; Ra представляет собой водород или гидрокси; n представляет собой целое число от нуля до одного. 8 н. и 7 з.п. ф-лы, 172 пр.
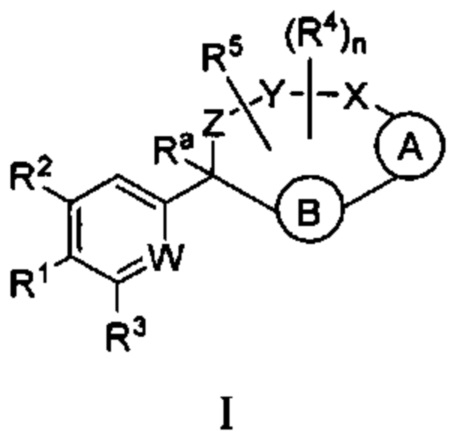
1. Соединение формулы:
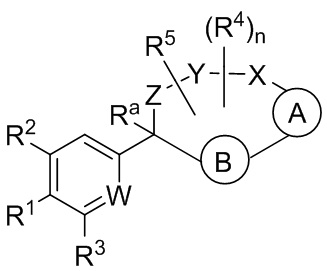 ,
,
где  представляет собой фенил или 5-10-членный гетероарил с 1-2 атомами азота, который необязательно замещен одной-двумя группами, независимо выбранными из группы, состоящей из галогена, оксо, циано, R6, OR6, C(O)OR6, NR6R7, NHC(O)O-C1-3 алкил-OR7;
представляет собой фенил или 5-10-членный гетероарил с 1-2 атомами азота, который необязательно замещен одной-двумя группами, независимо выбранными из группы, состоящей из галогена, оксо, циано, R6, OR6, C(O)OR6, NR6R7, NHC(O)O-C1-3 алкил-OR7;
 представляет собой фенил или 5-6-членный гетероарил с 1-2 атомами азота, который необязательно замещен одной-двумя группами, независимо выбранными из группы, состоящей из галогена, оксидо, циклопропила, R6;
представляет собой фенил или 5-6-членный гетероарил с 1-2 атомами азота, который необязательно замещен одной-двумя группами, независимо выбранными из группы, состоящей из галогена, оксидо, циклопропила, R6;
W представляет собой N или N+O-;
Y-X представляет собой -CHC(O)OR7-NR6-, -CR6R7-C(O)NR6-, -CHC(O)R7-NR6 -;
Z представляет собой C3-4 алкилен;
R1 представляет собой фенил, необязательно замещенный одним-четырьмя заместителями, независимо выбранными из группы, состоящей из галогена, циано, R6, OR6, C(O)R6, NR6R7, и 5-членного гетероарила с 2-4 гетероатомами, выбранными из азота и кислорода (который необязательно замещен галогеном, циано, циклопропилом, C(O)OH, или R6);
R2 представляет собой водород, или OR6;
R3 представляет собой водород;
каждый R4 независимо представляет собой C1-6 алкил;
R5 представляет собой водород, галоген или C1-6 алкил;
или один из R4 и R5 может быть взят вместе с атомами между ними с образованием 3-6-членного алкильного кольца;
каждый R6 независимо представляет собой водород или C1-6 алкил, который необязательно замещен одной-тремя группами, независимо выбранными из группы, состоящей из галогена;
каждый R7 независимо представляет собой водород, C1-6 алкил, 6-членный гетероарил с 1-2 атомами азота или 5-членный гетероциклил с 1 атмом азота, где указанная алкильная группа необязательно замещена одной-тремя группами, независимо выбранными из группы, состоящей из галогена;
Ra представляет собой водород или гидрокси;
n представляет собой целое число от нуля до одного;
или его фармацевтически приемлемая соль.
2. Соединение по п. 1 формулы
 ,
,
где представляет собой фенил или 5-10-членный гетероарил с 1-2 атомами азота, который необязательно замещен одной-двумя группами, независимо выбранными из группы, состоящей из галогена, оксо, циано, R6, OR6, C(O)OR6, NR6R7, NHC(O)O-C1-3 алкил-OR7;
представляет собой фенил или 5-6-членный гетероарил с 1-2 атомами азота, который необязательно замещен одной-двумя группами, независимо выбранными из группы, состоящей из галогена, оксидо, циклопропила, R6;
R1 представляет собой фенил, необязательно замещенный одним-четырьмя заместителями, независимо выбранными из группы, состоящей из галогена, циано, R6, OR6, NR6R7, и 5-членного гетероарила с 2-4 гетероатомами, выбранными из азота и кислорода (который необязательно замещен галогеном, циано, циклопропилом, C(O)OH или R6);
R2 представляет собой водород, или OR6;
R3 представляет собой водород;
R4 представляет собой C1-6 алкил;
R6 представляет собой водород или C1-6 алкил, который необязательно замещен одной-тремя группами, независимо выбранными из группы, состоящей из галогена;
R7 представляет собой водород или C1-6 алкил, который необязательно замещен одной-тремя группами, независимо выбранными из группы, состоящей из галогена;
Ra представляет собой водород или гидрокси;
n представляет собой целое число от нуля до одного;
или его фармацевтически приемлемая соль.
3. Соединение по п. 1 или 2, где выбран из группы, состоящей из фенила имидазолила, пиридинила и пиримидинила, где указанные группы необязательно замещены одной-двумя группами, независимо выбранными из группы, состоящей из галогена, оксидо, R6 и циклопропила; или его фармацевтически приемлемая соль.
4. Соединение по любому из пп. 1-3, где представляет собой фенил, который необязательно замещен одной-двумя группами, независимо выбранными из группы, состоящей из галогена, и C(O)OR6; или его фармацевтически приемлемая соль.
5. Соединение по любому из пп. 1-4, где R1 представляет собой фенил, который необязательно замещен одним-четырьмя заместителями, независимо выбранными из группы, состоящей из хлора, фтора, йода, и метила OCF3, OCHF2, CF3, CHF2 и 5-членного гетероарила с 2-4 гетероатомами, выбранными из азота и кислорода (который необязательно замещен галогеном, циано, циклопропилом, C(O)OH, метилом, CF3 или CHF2); или его фармацевтически приемлемая соль.
6. Соединение по любому из пп. 1-5, где R1 представляет собой фенил, который необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, и тетразолил; или его фармацевтически приемлемая соль.
7. Соединение по п. 1, выбранное из:
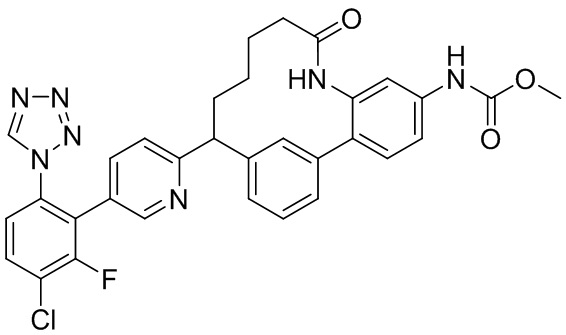 ,
,  ,
, 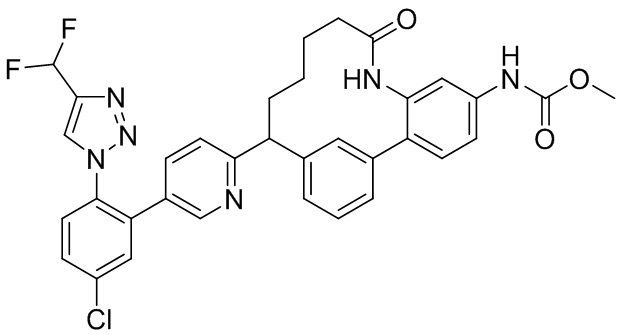 ,
, 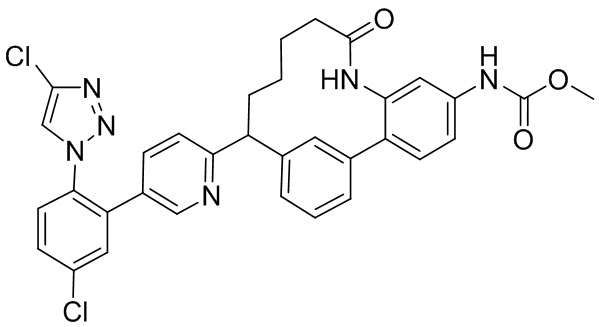 ,
,  ,
, 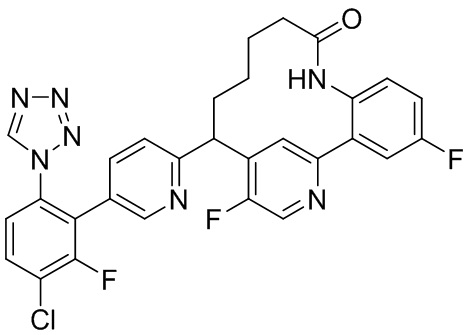 ,
, 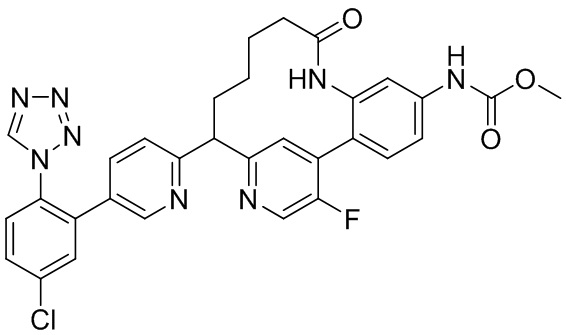 ,
, 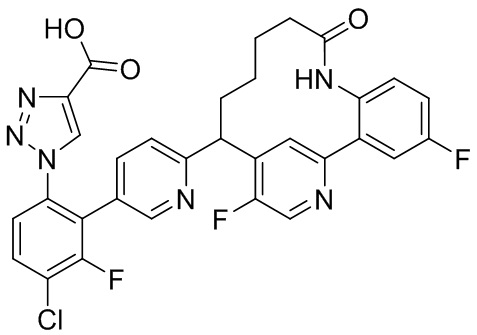 ,
, 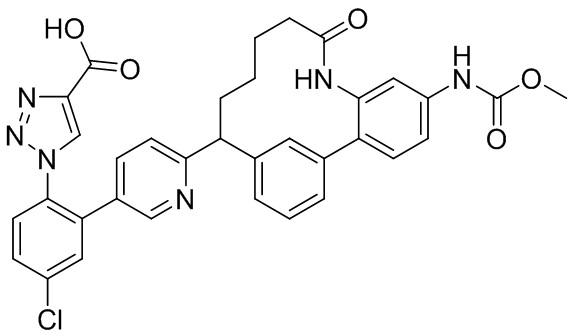 ,
, 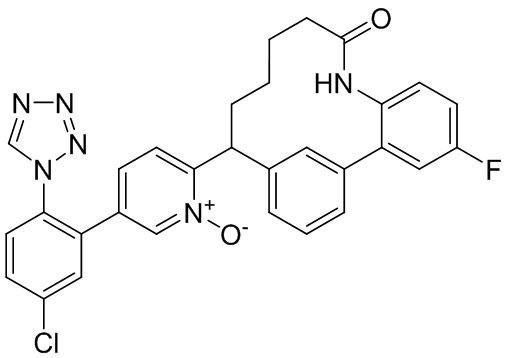 ,
, 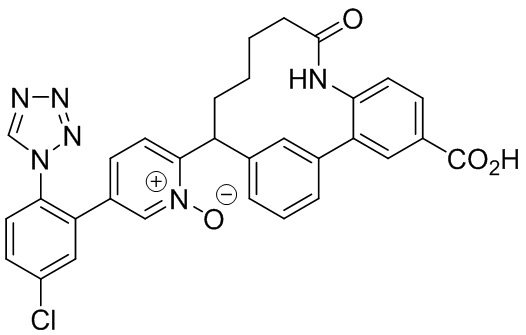 ,
,  ,
, 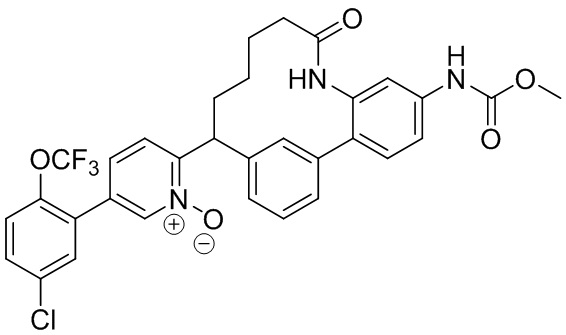 ,
,  ,
, 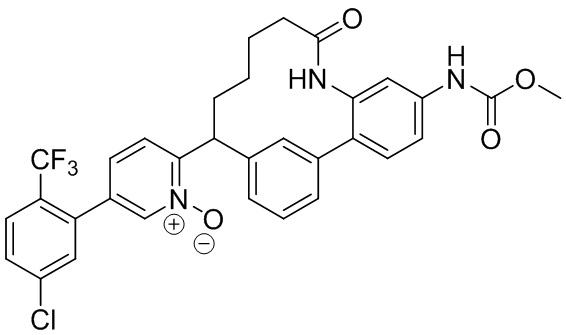 ,
, 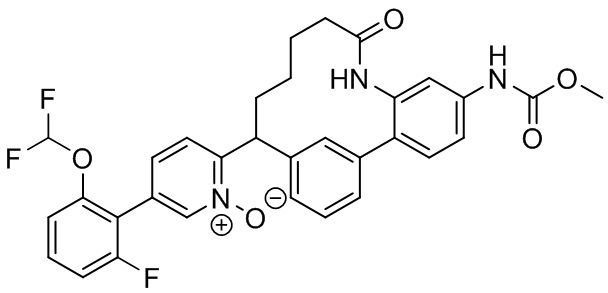 ,
, 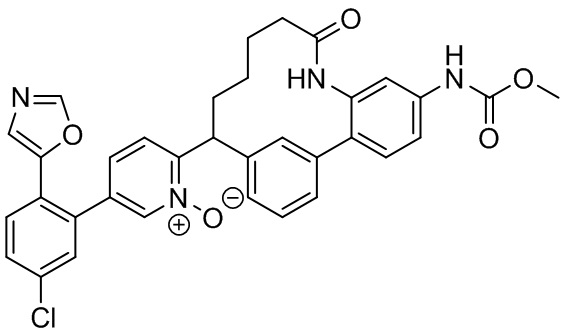 ,
, 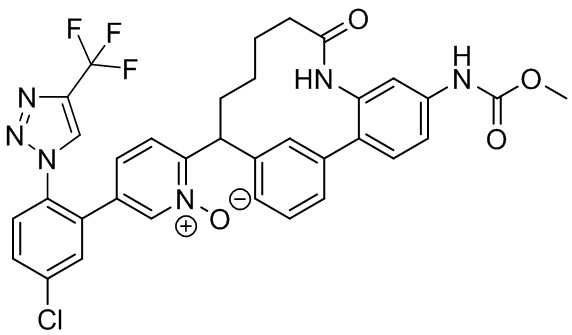 ,
, 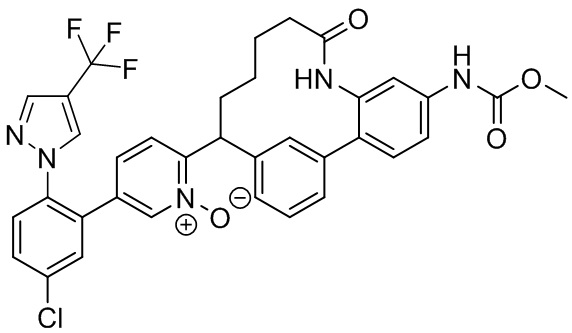 ,
, 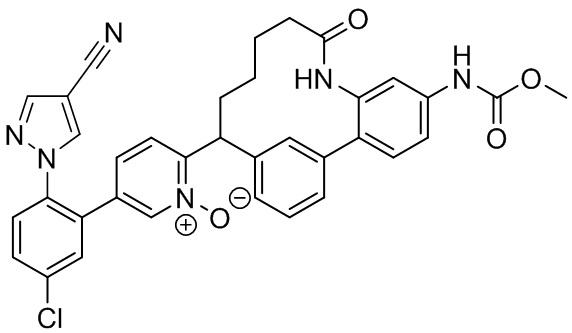 ,
, 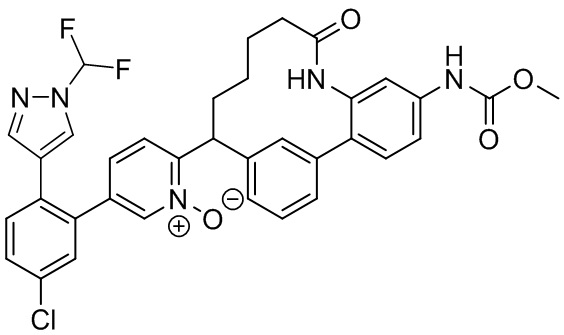 ,
, 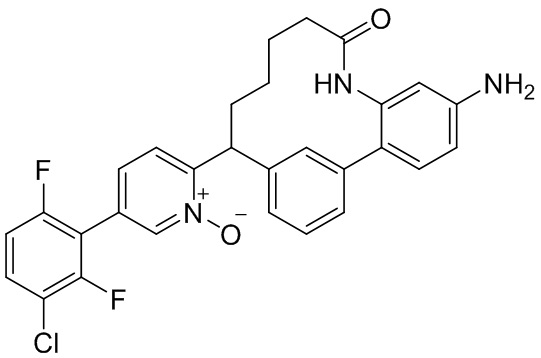 ,
, 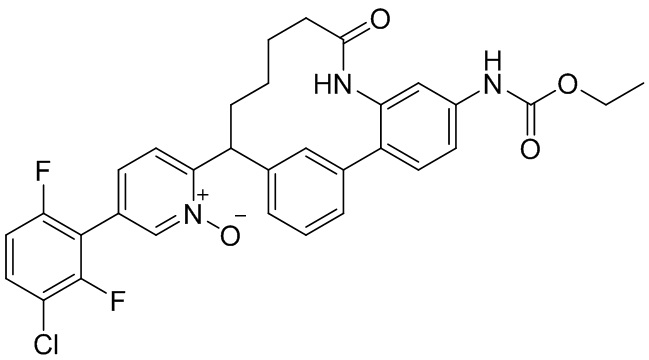 ,
, 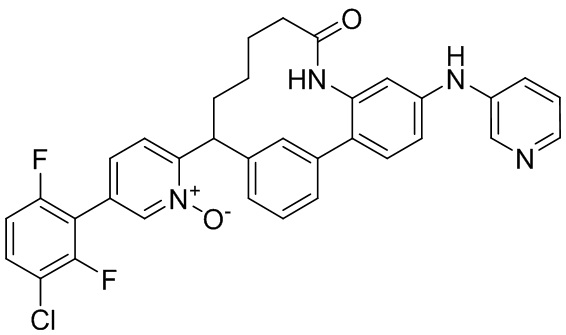 ,
, 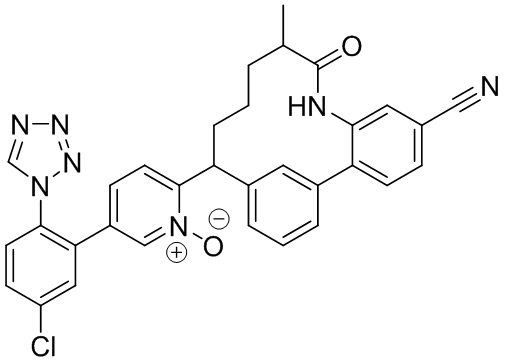 ,
, 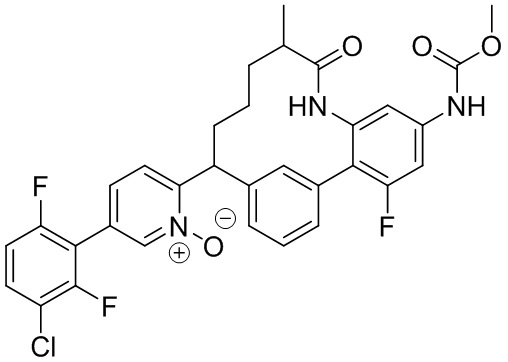 ,
, 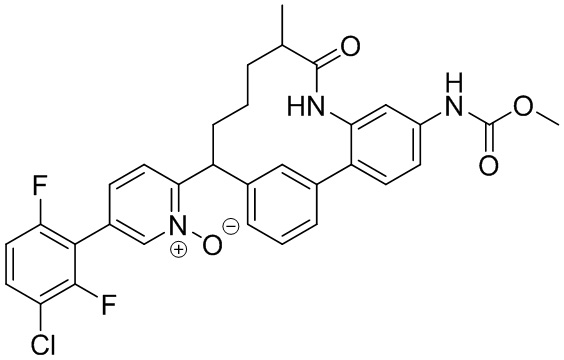 ,
, 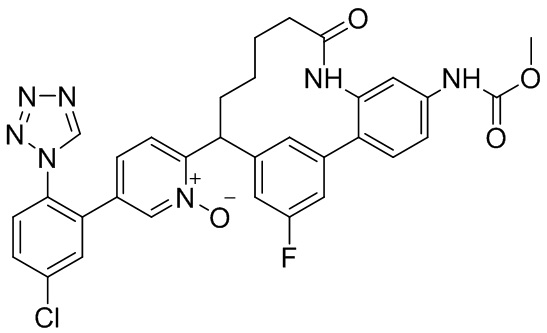 ,
, 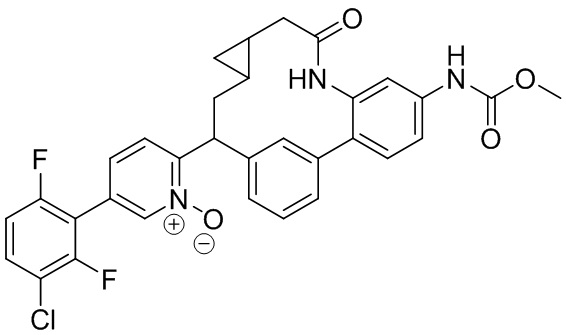 ,
, 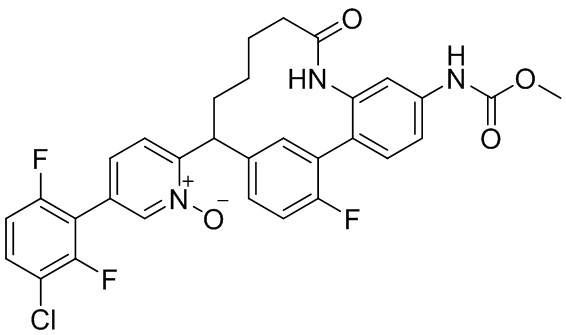 , ,
, , 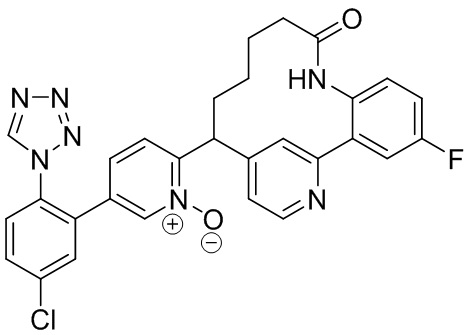 ,
, 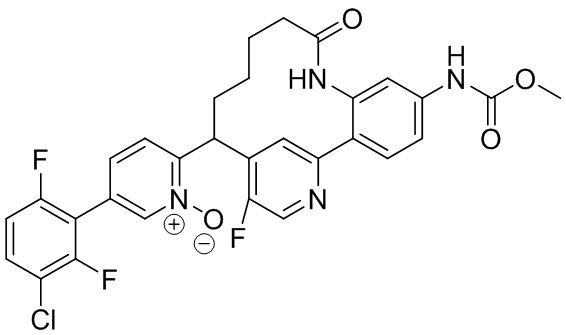 ,
, 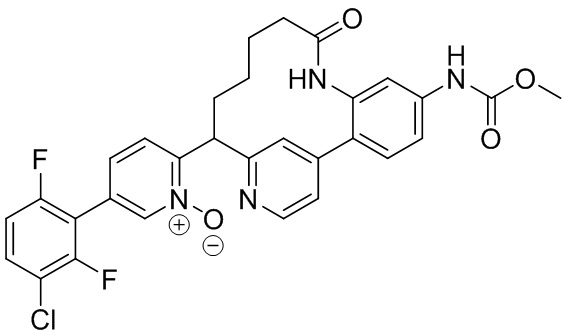 ,
, 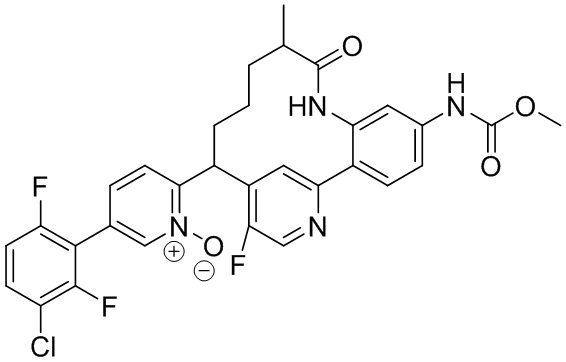 ,
, 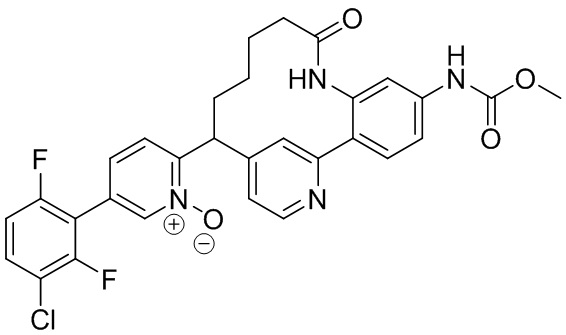 ,
, 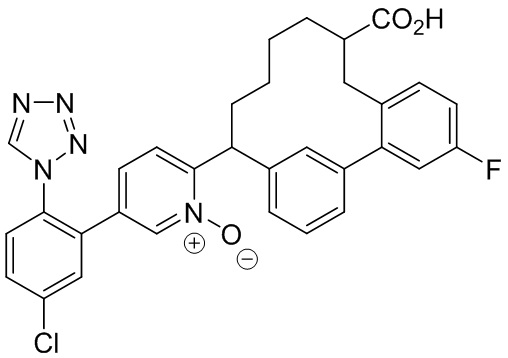 ,
, 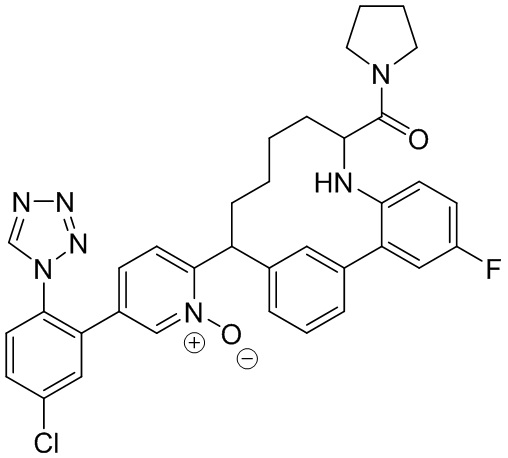 ,
, 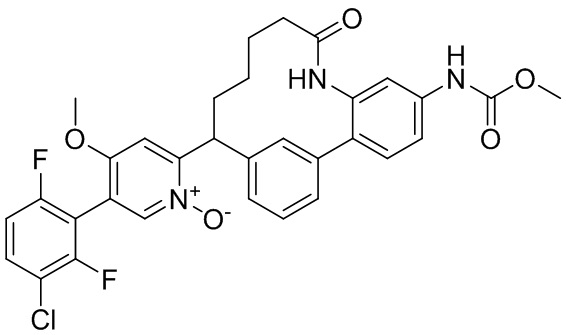 ,
, 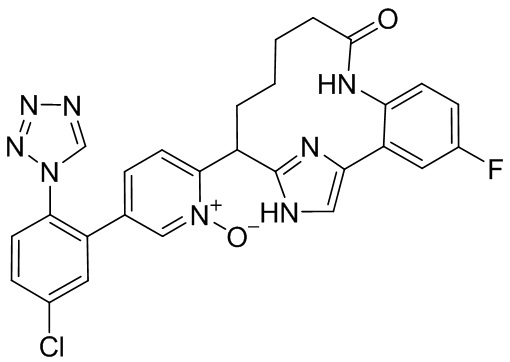 ,
, 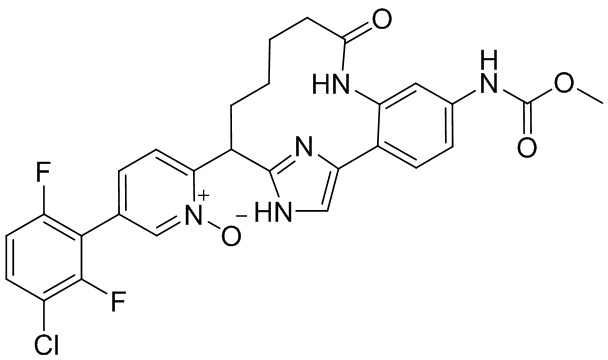 ,
, 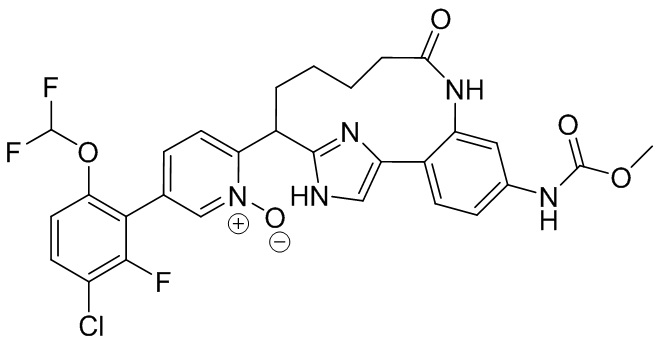 ,
, 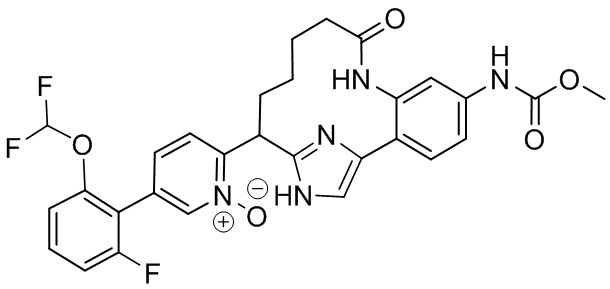 ,
, 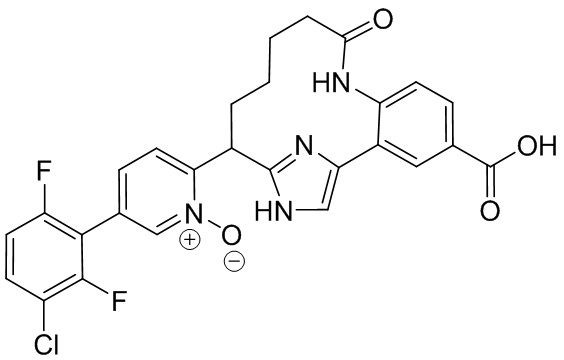 ,
, 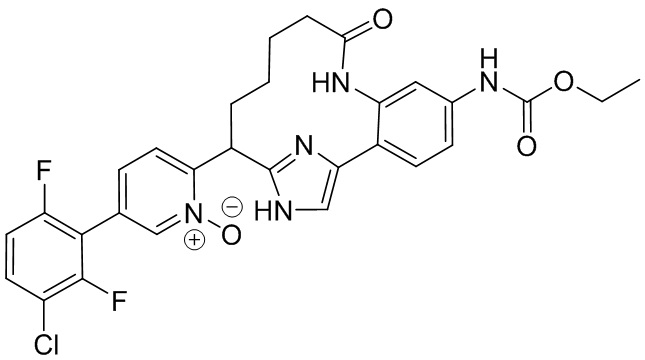 ,
, 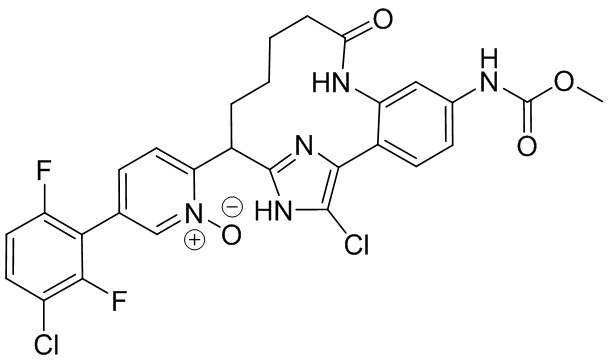 ,
, 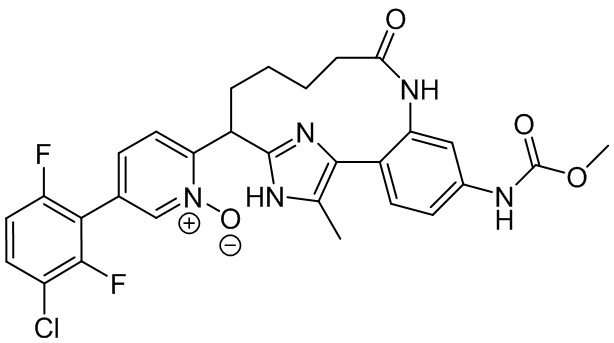 ,
, 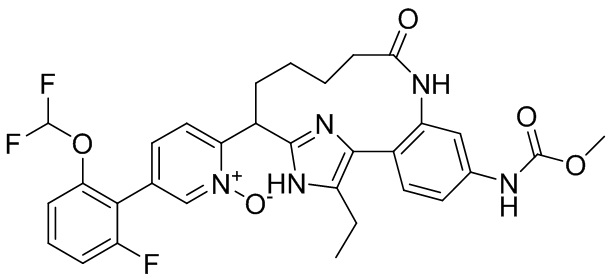 ,
, 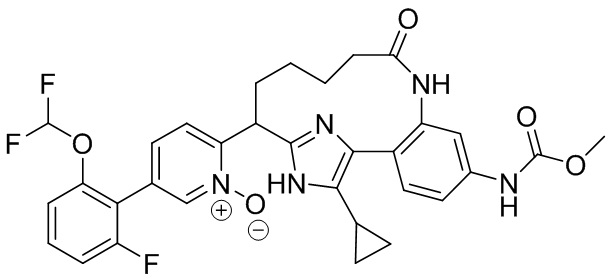 ,
, 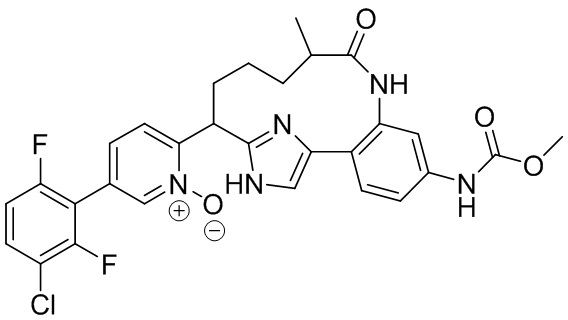 ,
, 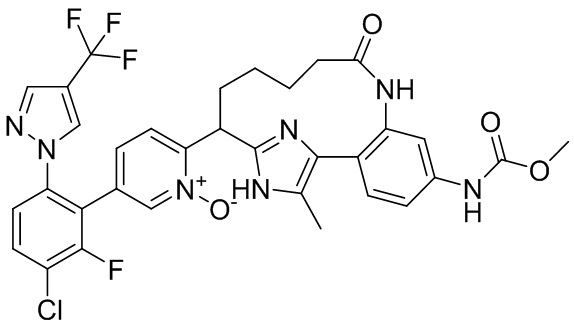 ,
, 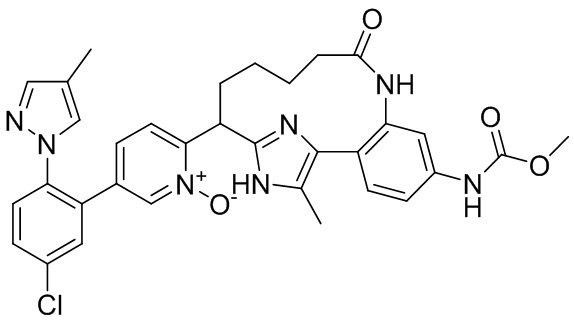 ,
,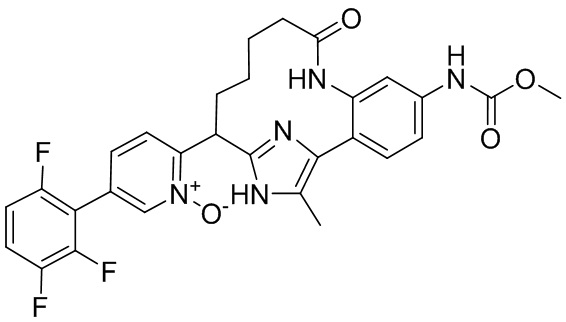 ,
, 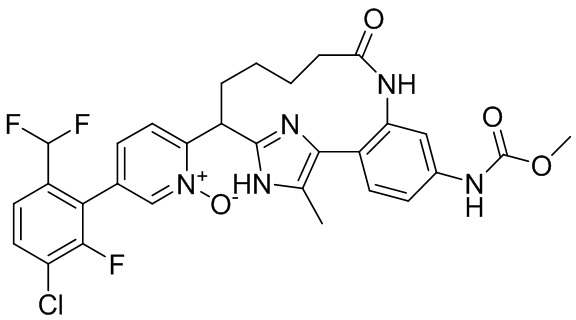 ,
, 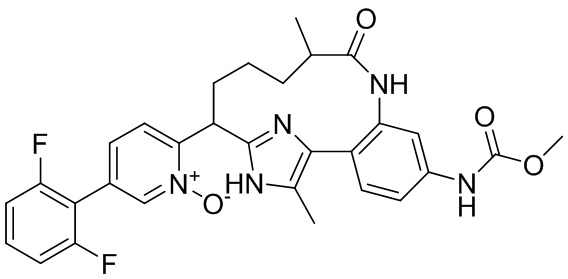 ,
, 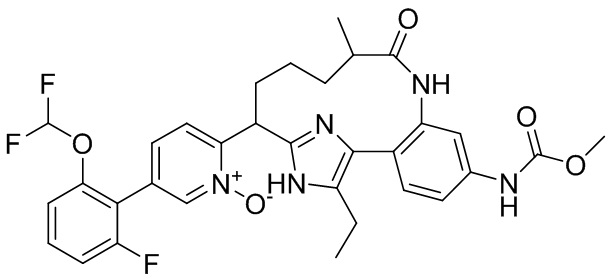 ,
, 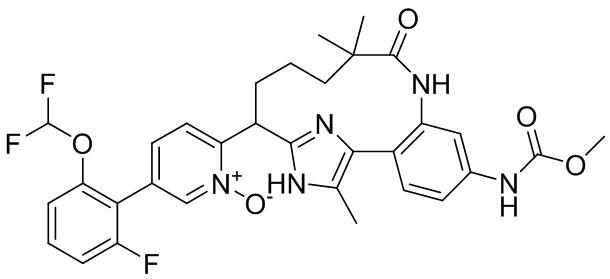 ,
, 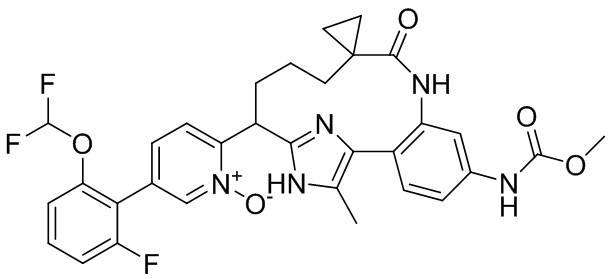 ,
, 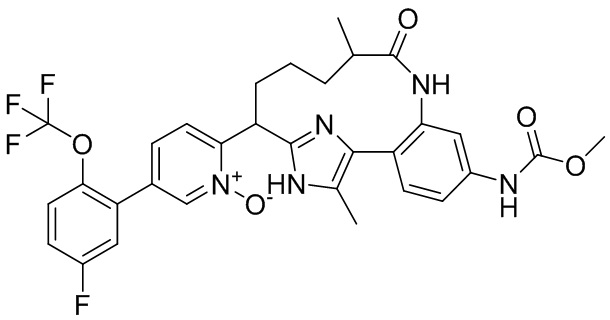 ,
, 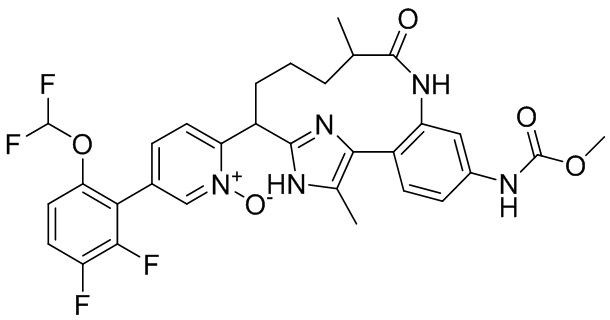 ,
, 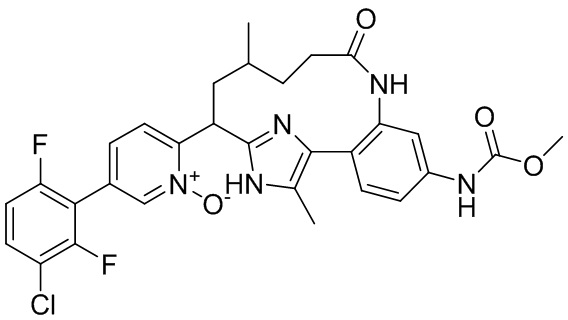 ,
, 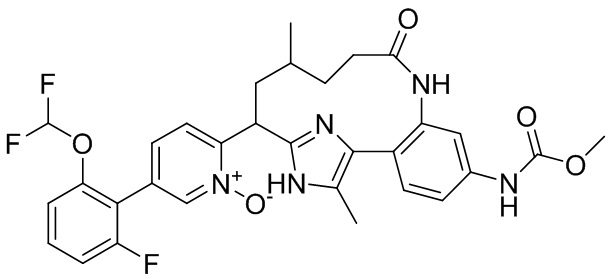 ,
, 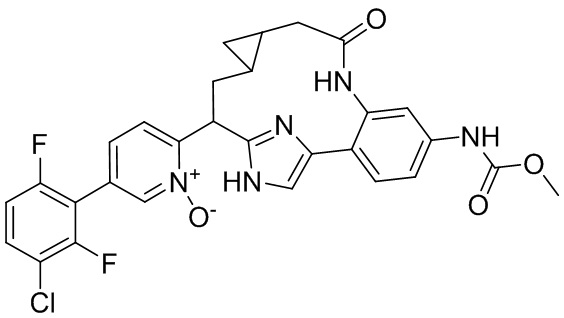 ,
, 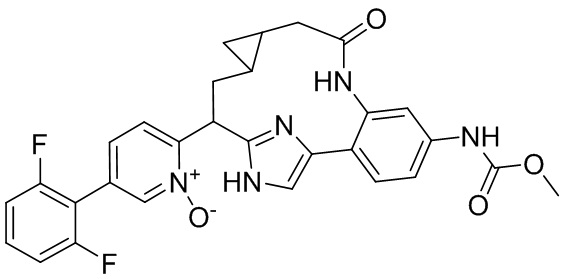 ,
, 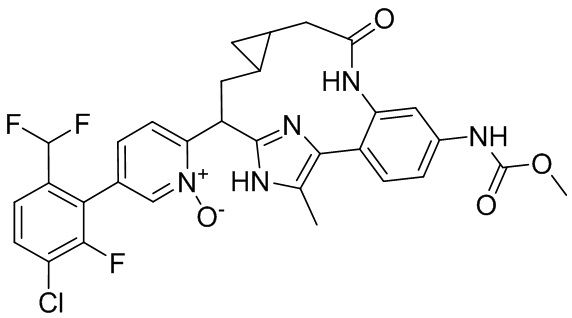 ,
, 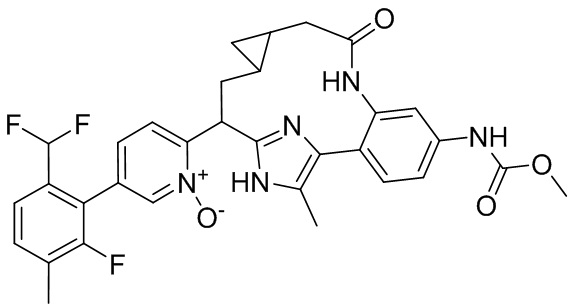 ,
, 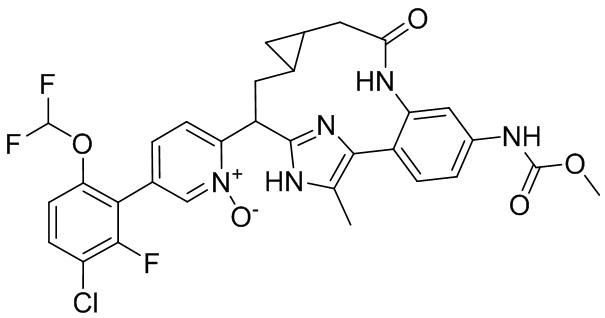 ,
, 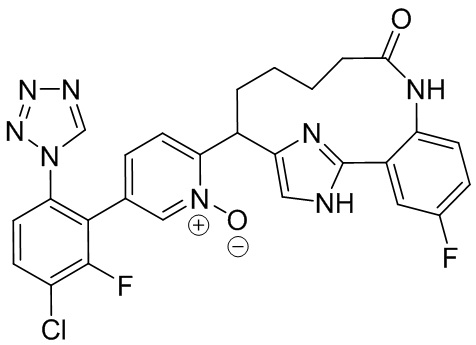 ,
, 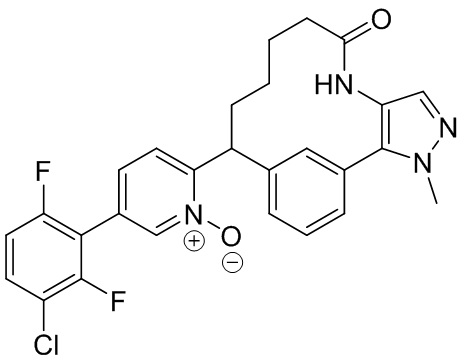 ,
, 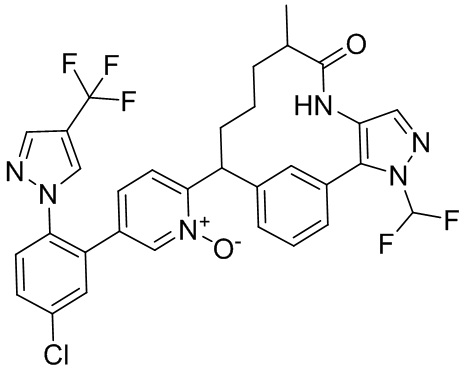 ,
, 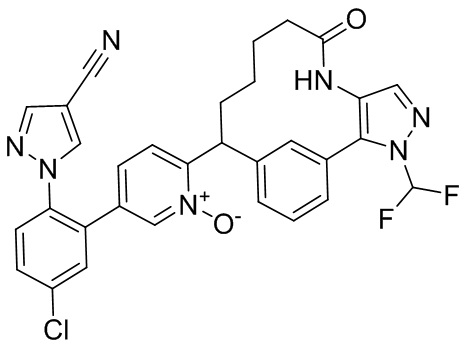 ,
, 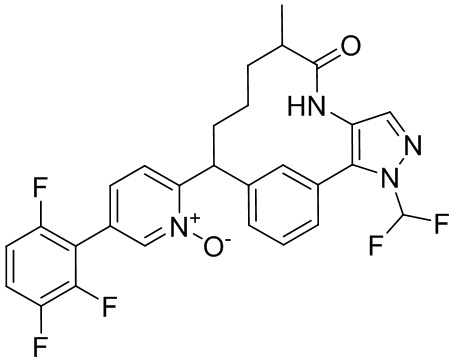 ,
, 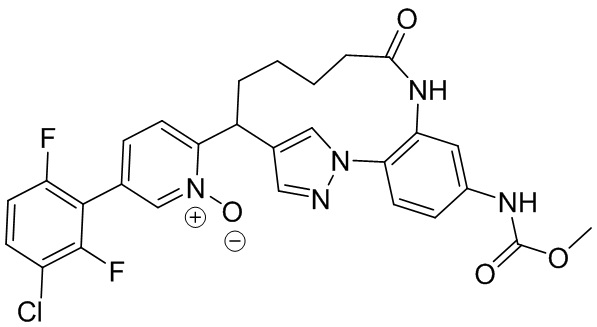
или его фармацевтически приемлемая соль.
8. Фармацевтическая композиция, обладающая свойствами ингибитора фактора коагуляции XIa, содержащая соединение по любому из пп. 1-7 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
9. Способ ингибирования образования тромба в крови или лечения образования тромба в крови, причем способ предусматривает введение композиции по п. 8 млекопитающему, нуждающемуся в этом.
10. Способ предупреждения образования тромба в крови, причем способ предусматривает введение композиции по п. 8 млекопитающему, нуждающемуся в этом.
11. Способ лечения венозной тромбоэмболии и легочной эмболии у млекопитающего, причем способ предусматривает введение композиции по п. 8 млекопитающему, нуждающемуся в этом.
12. Способ лечения тромбоза глубоких вен у млекопитающего, причем способ предусматривает введение композиции по п. 8 млекопитающему, нуждающемуся в этом.
13. Способ лечения тромботического инсульта у человека, причем способ предусматривает введение композиции по п. 8 млекопитающему, нуждающемуся в этом.
14. Применение соединения по любому из пп. 1-7 или его фармацевтически приемлемой соли при изготовлении лекарственного препарата для ингибирования тромбина, ингибирования образования тромба, лечения образования тромба или предупреждения образования тромба у млекопитающего.
15. Соединение по любому из пп. 1-7, обладающее свойствами ингибитора фактора коагуляции XIa.
| WO 2014022767 A1, 06.02.2014 | |||
| US 20150290194 A1, 15.10.2015 | |||
| Устройство для передачи электрической энергии | 1931 |
|
SU28581A1 |

