Заявки, связанные с данным изобретением
Данная заявка основана на предварительной заявке США №61/019,770, поданной 8 января 2008 в соответствии с Разделом 35 Кодекса законов США, статья 119(е), и любыми другими применимыми законами. Таким образом, предварительная заявка США №61/019,770 во всей своей полноте включена в данную заявку в виде ссылки.
Область техники
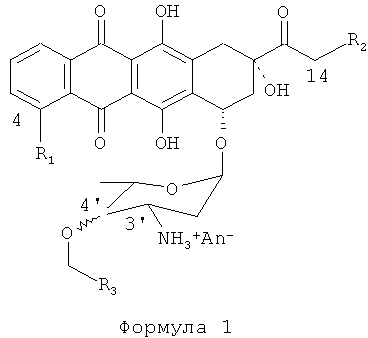
Изобретение относится к химическим способам, используемым для производства антрациклинов. Более конкретно, изобретение относится к способам и процессам, используемым для производства антрациклинов Формулы (I):
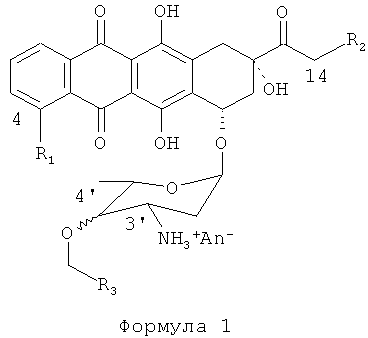
где: R1=H, ОН, OMe;
R2=Н, ОН, OCOAлк1; Алк1=линейный или разветвленный алкил, алкенил или алкинил C1-C12,
4'-ОСН2-R3эк[ваториальный] или акс[иальный]; R3=Н, Алк1, Ar, где
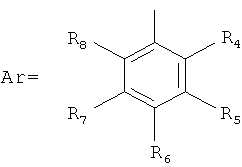
An- - анион сильной кислоты,
в частности (4-R1=ОМе, 14-R2=ОН, акс[иальный]4'-BnO).
Уровень техники
Антрациклины образуют одно из самых больших семейств природных биологически активных соединений. Было установлено, что некоторые члены этого семейства являются клинически эффективными противоопухолевыми агентами. Примерами таких соединений являются даунорубицин, доксорубицин, идарубицин, эпирубицин, пирарубицин, зорубицин, акларубицин и карминомицин. Например, было показано, что эти соединения полезны при лечении карциномы молочной железы, острого лимфоцитарного и нелимфоцитарного лейкоза, хронического лимфолейкоза, лимфомы, не являющейся лимфомой Ходжкина, и других твердых злокачественных опухолей.
Однако во многих случаях проблема трансмембранного транспорта или переноса через гематоэнцефалический барьер является первостепенной для повышения биодоступности лекарства. Поиск новых производных антрациклинов, особенно соединений, способных легко преодолевать гематоэнцефалический барьер, продолжается вплоть до сегодняшнего дня. Такие свойства позволят расширить показания к применению антрациклинов и включить в них как первичные, так и метастатические опухоли центральной нервной системы. Выше перечислены лишь некоторые причины постоянно увеличивающегося интереса к синтезу новых антрациклиновых антибиотиков с варьируемой лиофильностью, таким как описаны в патентах США №№5,625,043 и 6,673,907. Изменение лиофильности может быть достигнуто путем модификации гликозидного остатка молекулы, в частности, путем алкилирования 3'-N и/или 4'-О атомов сахара.
Согласно способу, раскрытому в патенте США №6,673,907, ряд 3'-N замещенных соединений получают прямым алкилированием антрациклинов бензилбромидами в диметилформамиде (ДМФА). Традиционно считается, что введение аралкильных заместителей (замещенных бензильных радикалов) в положении 4'-О антрациклинов является существенно менее доступным. Такой синтез является сложным вследствие следующих трудностей:
(a) функциональные группы и агликона, и сахара должны быть защищены защитными группами;
(b) получение 3'-азидо гликозидного фрагмента осложняется возникновением экваториального и аксиального изомеров, которые затем должны быть разделены с помощью стереоспецифического гидролиза;
(c) стадия соединения требует использования, как минимум, двойного избытка синтона сахара, который, в свою очередь, получают 5-6-стадийным синтезом. Стадия соединения протекает с менее чем 100% стереоспецифичностью, что приводит к получению нежелательного стереоизомера, который в дальнейшем необходимо удалить;
(d) общее количество стадий синтеза и стадий хроматографической очистки превышает 10, что препятствует высокому выходу желаемого продукта.
Современные представления об относительной реакционно-способности нуклеофильных групп позволяют расположить их в следующем порядке: NH2 > ароматический ОН > алифатический ОН, и исключают возможность селективного алкилирования алифатических ОН групп на фоне незащищенных NH2 или ароматических ОН групп. Это приводит к сложному способу синтеза производных антрациклинов, замещенных в 4'-О положении, как описано выше.
Бензилирование сахара в 4' положение даунорубицина или его аналогов с использованием общепринятых бензилирующих агентов, таких как бензилгалогениды + NaH; +BuLi; +t-BuOK, является невозможным вследствие прямого предпочтительного бензилирования азота в отсутствие защитных групп на 3'-NH2 или возникновения реакционного центра на 3'-N защищенном азоте. Кроме того, бензилирование сахара в 4' положении препятствует удалению защитной группы с 3'-NH группы.
Комбинация этих факторов приводит к тому, что одновременно протекают различные реакции, результатом чего является получение сложной, плохо разделяемой смеси множества продуктов.
Ранее, согласно общепринятым способам алкилирования 4' гидроксильной группы сахаров, в качестве исходного продукта использовали 3,4-ди-O-ацетил-Rhamnal. Сначала его превращали в 3-азид (рацемат); затем нужный оптический изомер выделяли и бензилировали BnBr в присутствии NaH. Полученный таким образом синтон затем соединяли с независимо синтезированным агликоном.
Дальнейшие модификации и удаление защитных групп приводили к получению конечного продукта.
Упрощение получения этого класса соединений за счет модификации микробиологически получаемых предшественников антрациклинов без выделения агликона и сахара дает такому процессу существенное преимущество. Например, один такой подход к синтезу идарубицина раскрыт в патенте США №7,053,191. Способ, описанный в патенте США №7,053,191, позволяет уменьшить количество стадий синтеза с 11 или 12 до всего 5.
Способ превращения 3'-NH2 в 3'-N3 группу в гликозидной части молекулы антрациклина ранее описан в Journal of Medicinal Chemistry 2006 VoI 49, No 5, pp.1792-1799. Этот способ позволяет получить соответствующий азид, сохраняя незатронутой молекулу антрациклина.
Раскрытие изобретения
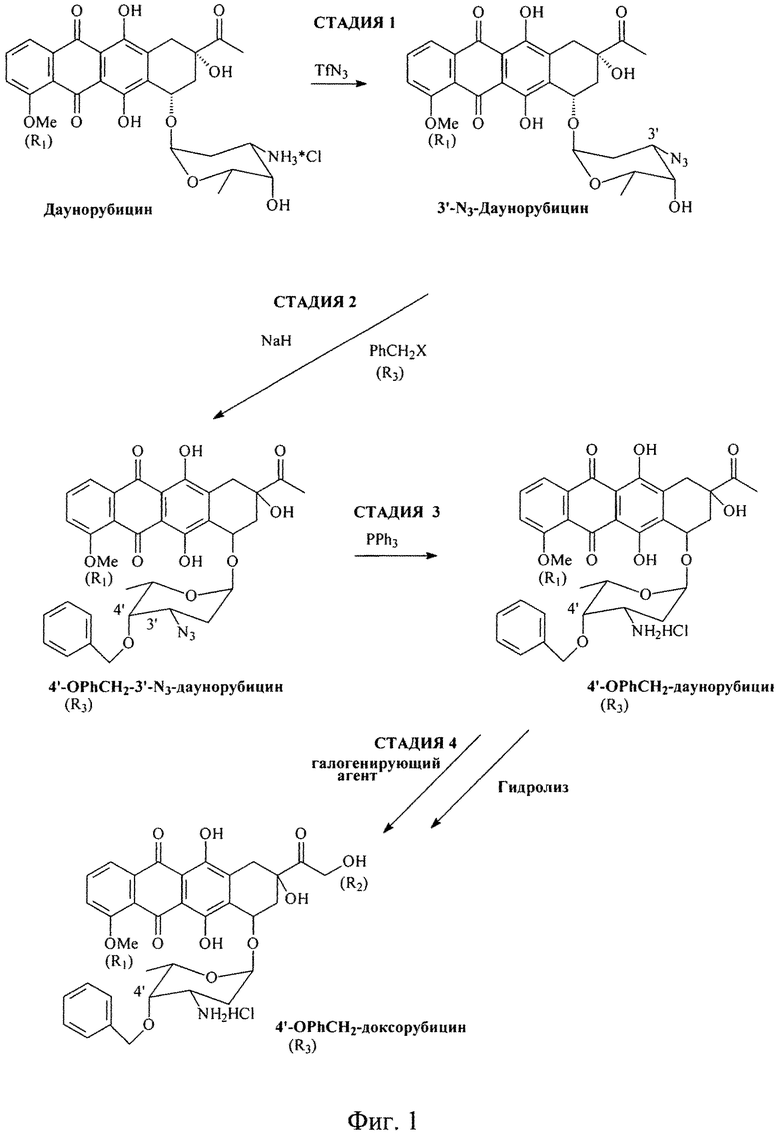
В данном изобретении раскрыт инновационный способ аралкилирования антрациклинов с использованием аралкилирующего агента R3-CH2X (например, BnBr) в соответствии со схемой реакции, приведенной на Фиг.1. Согласно данному изобретению 4-R1, 3'-N3-дауномицины являются подходящими субстратами для селективного 4'-O-бензилирования с получением 4-R1, 3'-N3-4'-O-аралкил-даунорубицинов (в частности, 4'-O-Bn-дауномицинов). Таким образом, данное изобретение раскрывает путь для простого получения 4'-O-аралкилированных производных антрациклинов.
Краткое описание чертежей (рисунков)
Фиг.1 представляет собой схему последовательностей химических реакций согласно одному из вариантов выполнения изобретения.
Детальное раскрытие изобретения
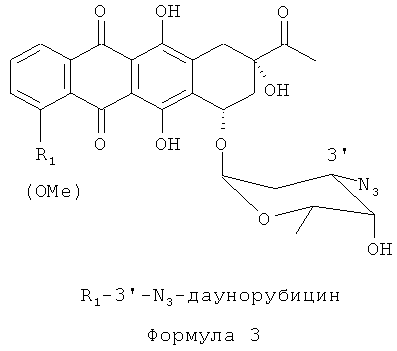
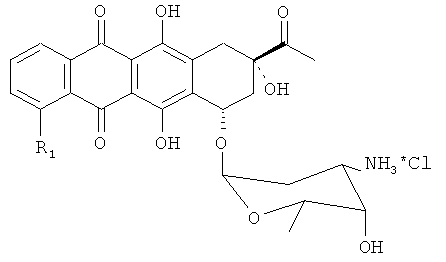
Способ аралкилирования антрациклинов с использованием аралкилирующего агента R3.CH2X (например, ВпВг) согласно данному изобретению включает стадии, схематически показанные на Фиг.1. Исходным материалом является производное антрациклина в виде соли в спирте (предпочтительно, в метаноле). Раствор TfN3 в дихлорметане добавляют к раствору соли производного антрациклина в спирте (предпочтительно, в метаноле), и инкубируют смесь в течение 4-24 часов до тех пор, пока исходное соединение полностью не прореагирует. В результате получают азидное производное, представленное Формулой 3:
Азид-производное, представленное Формулой 3, растворяют в апротонном растворителе, устойчивом к действию сильных оснований и алкилирующих агентов, таком как диалкиламиды, простые эфиры (линейные эфиры (например, диэтиловый эфир, метил-t-бутиловый эфир), циклические эфиры (например, ТГФ), и эфиры этиленгликоля) или смесь таких растворителей (предпочтительно, ДМФА). При перемешивании в смесь добавляют избыток сильного основания (предпочтительно NaH) в соотношении 1.2-10 М на 1 М антрациклина. Затем добавляют алкилирующий агент R3CH2X (например, BnBr) в избытке, составляющем 1.2-10 М на 1 М антрациклина при температуре от 0 до 90°С или при температуре кипения растворителя. Длительность реакции сильно зависит от реакционно-способности алкилирующего агента и может варьироваться от часов до дней. Степень завершенности реакции контролируют тонкослойной хроматографией. («ТСХ»).
После завершения реакции реакционную смесь упаривают при пониженном давлении и промывают диэтиловым эфиром. Продукт экстрагируют дихлорметаном из водно-органической эмульсии реакционной смеси в дистиллированной воде. Экстракт в дихлорметане промывают дистиллированной водой и удаляют дихлорметан упариванием при пониженном давлении.
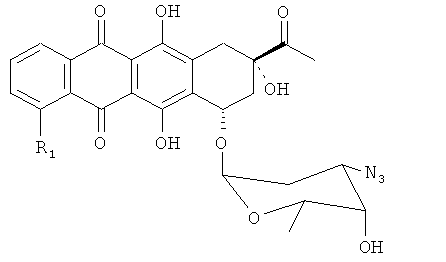
Полученный алкилированный антрациклин-азид растворяют в ТГФ, к раствору добавляют 2 М избыток трифенилфосфина. Длительность реакции варьируется от часов до дней. Завершенность реакции снова контролируют ТСХ. В результате получают аралкилированный антрациклин, представленный Формулой 4:
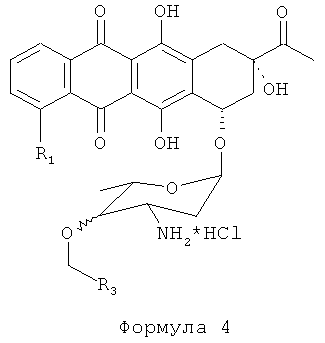
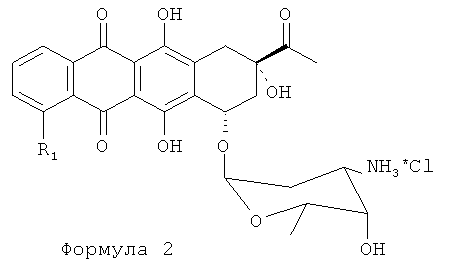
Затем проводят галогенирование аралкилированного антрациклина Формулы 4 комплексным галогенидом, представленным Формулой 2:
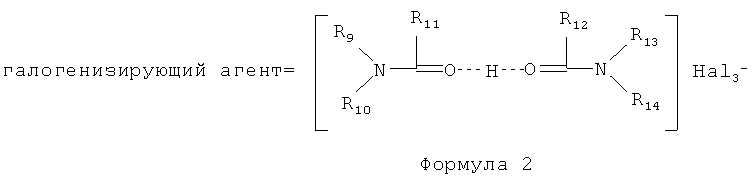
где заместители R9-R14 являются Н или углеводородным радикалом, содержащим от 1 до 4 атомов углерода в цепи (C1-C4); Hal является Cl, Br, I.
Растворителями, используемыми для этой реакции, являются апротонные растворители средней основности, способные связывать галогеноводород, получающийся в процессе галогенирования, например амиды, простые эфиры и их смеси, предпочтительно диметилформамид и тетрагеидрофуран. Реакцию проводят при температуре 20-60°С в течение 2-20 часов, предпочтительно при 50°С в течение примерно 3 часов. В результате получают производное, галогенированное в 14 положении. Это 14-галогенированное производное затем осаждают добавлением холодного ацетона или ацетонитрила и гидролизуют в водно-ацетоновом растворе в присутствии солей карбоновых кислот, предпочтительно формиата натрия, при рН=2.5-5.5 или, более предпочтительно, при рН=3.5-4.0. Если желательно получить 14-0 эфиры (R2=OCOАлк1; Алк1=линейный или разветвленный алкил, алкенил или алкинил C1-C12), используют соль соответствующей карбоновой кислоты.
ПРИМЕР
Сначала 20 г даунорубицин гидрохлорида растворяют в 125 мл МеОН. Добавляют раствор 7.5 г K2CO3 в 20 мл воды и интенсивно перемешивают в течение 1 минуты. Затем к смеси добавляют раствор ТfNз в дихлорметане. Смесь продолжают перемешивать на магнитной мешалке до тех пор, пока не будет достигнута полная конверсия исходного антрациклина (подтверждается ТСХ). Полученную реакционную массу выливают в 300 мл воды. Органический слой отделяют, водный экстрагируют дихлорметаном. Дихлорметан удаляют из раствора на роторном испарителе. В результате получают 3'-N3-дауномицин.
Затем 3'-N3-дауномицин растворяют в 100 мл диметилформамида и добавляют 2 г 60% суспензии NaH в парафине. Смесь перемешивают при комнатной температуре в течение 30 минут, после чего к ней добавляют 4 мл бензилбромида. Перемешивание продолжают до тех пор, пока концентрация исходного азида дауномицина не уменьшается в 8-10 раз. Затем полученную реакционную смесь выливают в подкисленную дистиллированную воду и экстрагируют дихлорметаном. Дихлорметан удаляют из раствора на роторном испарителе.
Полученный полутвердый остаток растворяют в 100 мл тетрагидрофурана, и к раствору добавляют 7 г трифенилфосфина. Полученный раствор оставляют при комнатной температуре вплоть по полной конверсии 3'-N3-4'-OBn-дауномицина. Полученную реакционную массу полностью высушивают выпариванием, избыток трифенилфосфина удаляют хроматографией. В результате получают 4'-OBn-дауномицин.
Полученный в результате 4'-OBn-дауномицин растворяют в 100 мл диметилформамида и добавляют к смеси 5 г дибромбромата водорода бис диметилформамида ((ДМФ)2·HBr·Br2). Смесь инкубируют при 40°С в течение 2 часов. Затем реакционную смесь выливают в 350 мл ацетонитрила. Полученный осадок отделяют фильтрованием и промывают ацетонитрилом, растворитель удаляют.
Твердый осадок растворяют в смеси 80 мл ацетона, 80 мл 0.25 М водного раствора бромистого водорода и 8 грамм формиата натрия. Реакционную смесь инкубируют в течение 30 часов при 35°С.
Затем ацетон удаляют из реакционной смеси, а полученный остаток очищают с помощью хроматографии. Выход 4'-OBn-доксорубицина составляет 3.1 г.
Настоящее изобретение относится к способу получения антрациклинов формулы 1 и может быть использовано в химической промышленности,
 ,
,
где: R1=H, ОН, ОМе; R2=Н, ОН, или OCOАлк1; где Алк1 представляет собой алкил, алкенил или алкинил C1-C12, 4'-ОСН2-R3; R3 представляет собой H, Алк1 или необзательно замещенный арил; An- является анионом сильной кислоты; получают алкилированием с помощью R3-CH2X, где Х выбран из Cl-, Br-, I-, Ts, CH3SO2O-, CF3SO2O, через стадии: (a) получения соли формулы 2, (b) инкубирования ее с раствором TfN3 в дихлорметане до образования соответствующего 3'-N3-даунорубицина, (c) растворения продукта стадии (b) в апротонном растворителе; (d) взаимодействия продукта стадии (с) с избытком R3-CH2X и основания до получения соответствующего 4'ОR3-3'-N3-даунорубицина, (e) взаимодействия продукта стадии (d) в ТГФ с трифенилфосфином до получения соответствующего 4'-OR3-даунорубицина, (f) взаимодействия продукта стадии (е) в апротонном растворителе с галогенирующим агентом до получения производного, галогенированного в 14 положении, (g) гидролиза продукта стадии (f). Предложен новый эффективный способ получения производных антрациклинов. 13 з.п. ф-лы, 1 ил., 1 пр.
1. Способ получения 4'-O-аралкил производных антрациклинов формулы 1
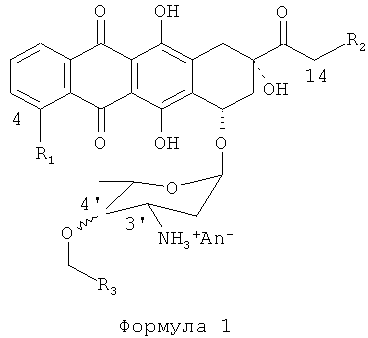
где: R1=H, ОН, ОМе;
R2=Н, ОН, или OCOАлк1; Алк1 = линейный или разветвленный алкил, алкенил или алкинил C1-C12,
4'-ОСН2-R3экваториальный или аксиальный; R3=H, Алк1, или Ar, где
An- - анион сильной кислоты;
с использованием в качестве алкилирующих агентов R3-CH2X, где Х выбран из группы, включающей Cl-, Br-, I-, Ts, CH3SO2O-, CF3SO2O,включающий стадии:
(a) получение исходного материала, представляющего собой производное антрациклина в виде соли даунорубицин гидрохлорида в спирте в соответствии со следующей формулой, где R1 определен, как в формуле 1;
(b) инкубирование указанного исходного материала с раствором TfN3 в дихлорметане до тех пор, пока исходный материал полностью не прореагирует, с образованием 3'-N3-даунорубицина в соответствии со следующей формулой, где R1,
как определено в формуле 1,
(c) растворение продукта, полученного на стадии (b), в апротонном растворителе;
(d) взаимодействие продукта, полученного на стадии (с), с избытком алкилирующего агента R3-CH2X и сильным основанием для получения 4'ОR3-3'-N3-даунорубицина, в соответствии со следующей формулой, где R3 и Х, как они определены в формуле 1;
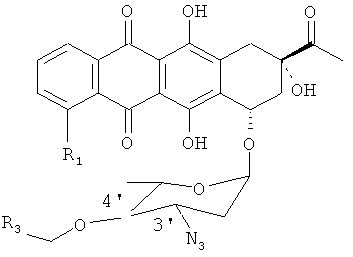
(e) растворение продукта, полученного на стадии (d), в тетрагидрофуране (ТГФ) и взаимодействие его с трифенилфосфином для получения 4'-OR3-даунорубицина в соответствии со следующей формулой, где R3, как определено в формуле 1;
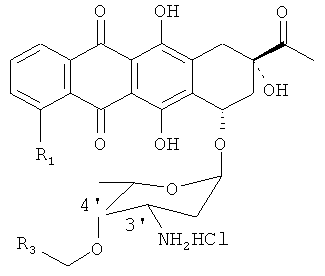
(f) взаимодействие продукта, полученного на стадии (е), с комплексным галогенидом формулы 2
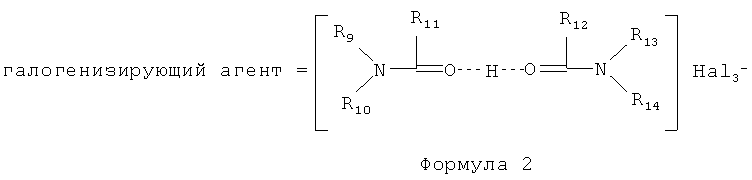
где заместители R9-R14 являются H или углеводородным радикалом, содержащим от 1 до 4 атомов углерода в цепи (C1-C4); Hal является Cl, Br, I
в апротонном растворителе средней основности для получения производного, галогенированного в 14 положении;
(g) гидролиз продукта, полученного на стадии (f), для получения 4'-OR3-доксорубицина в соответствии со следующей формулой, где R1 и R2 определены для формулы 1:
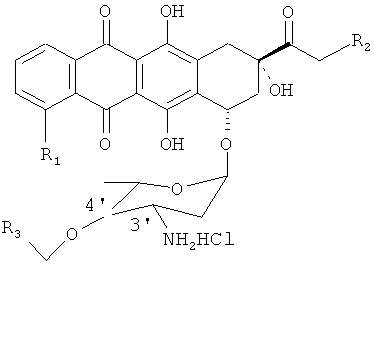
2. Способ по п.1, отличающийся тем, что алкилирующий агент, используемый на стадии (d), является R3-CH2X, где R3 = Ar;
причем на стадии (d) реакцию с указанным алкилирующим агентом проводят при соотношении 1.2-10 М алкилирующего агента на 1 М антрациклина, реакцию на стадии (d) проводят в одном из диалкиламидов или эфиров и реакцию на стадии (d) проводят при температуре от 0°С до 90°С или при температуре кипения апротонного растворителя.
3. Способ по п.2, отличающийся тем, что указанный растворитель средней основности выбран из группы, включающей амиды, простые эфиры и их смеси, и реакцию на стадии (f) проводят при температуре от 20°С до 60°С в течение от 2 до 20 часов.
4. Способ по п.3, отличающийся тем, что реакцию на стадии (f) проводят при температуре 50°С в течение 3 часов.
5. Способ по п.4, отличающийся тем, что указанный растворитель средней основности выбран из группы, включающей диметилформамид и тетрагидрофуран.
6. Способ по п.5, отличающийся тем, что инкубацию на стадии (b) проводят в течение от 4 до 24 часов до тех пор, пока исходное соединение полностью не прореагирует.
7. Способ по п.1, отличающийся тем, что указанный апротонный растворитель выбран из группы, включающей диалкиламиды, простые эфиры, линейные эфиры, циклические эфиры и их смеси.
8. Способ по п.6, отличающийся тем, что указанный апротонный растворитель выбран из группы, включающей диметилформамид и тетрагидрофуран.
9. Способ по п.1, отличающийся тем, что алкилирующим агентом является бензилбромид.
10. Способ по п.2, отличающийся тем, что алкилирующим агентом является бензилбромид, а сильным основанием является NaH.
11. Способ по п.8, отличающийся тем, что алкилирующим агентом является бензилбромид, а сильным основанием является NaH.
12. Способ по п.8, отличающийся тем, что спиртом является метанол.
13. Способ по п.1, отличающийся тем, что R1=OMe; R2=OH и 4'-BnO находится в аксиальном положении.
| US 20070037758 A1, 15.02.2007 | |||
| WO 2007076345 A2, 05.07.2007 | |||
| US 20060223766 A1, 05.10.2006 | |||
| WO 2005021565 A1, 10.03.2005 | |||
| 8-ФТОРАНТРАЦИКЛИНГЛИКОЗИДЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ ПРИСОЕДИНЕНИЯ КИСЛОТ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2095365C1 |

