Заявки, имеющие отношение к данному изобретению
В соответствии с разделом 119(е) Кодекса законов США (35 U.S.С.) и другими применимыми законами данная заявка включает преимущества предварительной заявки США №60/751765, поданной 20 декабря 2005. Таким образом, предварительная заявка США №60/751765 во всей своей полноте включена в данную заявку в виде ссылки.
Область техники
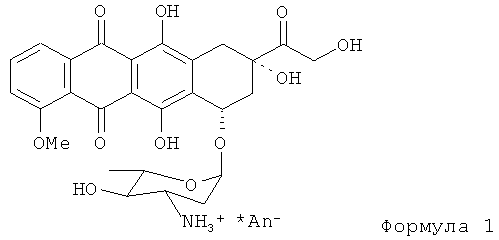
Данное изобретение относится к химическим способам, используемыми для получения антрациклинов, соединений, используемых в качестве противоракового лекарства при химиотерапии. Более конкретно, данное изобретение относится к способам получения антрациклинов Формулы (1) (где An- является анионом любой сильной кислоты, например, в случае 4'-эпирубицина An- является Cl-).
Уровень техники
Антрациклины образуют одну из крупнейших групп (семейств) природных биологически активных соединений. Было показано, что несколько членов этого семейства обладают терапевтической (клинической) эффективностью в качестве противоопухолевых агентов. Эти соединения включают, например, даунорубицин, доксорубицин, идарубицин, эпирубицин, пирарубицин, зорубицин, акларубицин и карминомицин. Было показано, в частности, что эти соединения полезны при лечении карциномы молочной железы, острого лимфолейкоза и нелимфоцитарной лейкемии (non-lymphocytic leukemia), хронического лимфолейкоза, лимфомы, не являющейся лимфомой Ходжкина (non-Hodgkin's lymphoma), и других солидных злокачественных опухолей.
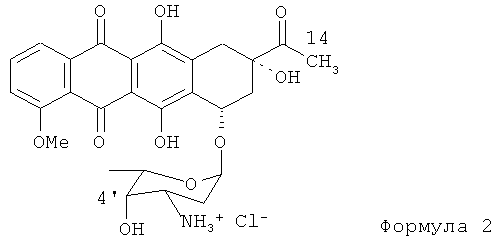
Антрациклиновые антибиотики обладают очень высокой противоопухолевой активностью, что позволяет эффективно использовать их для лечения широкого спектра опухолей. Исходным материалом для синтеза большинства антрациклиновых антибиотиков является даунорубицин в форме, представленной Формулой (2). Эпирубицин Формулы (1) отличается от даунорубицина, получаемого микробиологическим способом, наличием 14-оксиметильной группы и экваториальной ориентацией НО-4'-С.
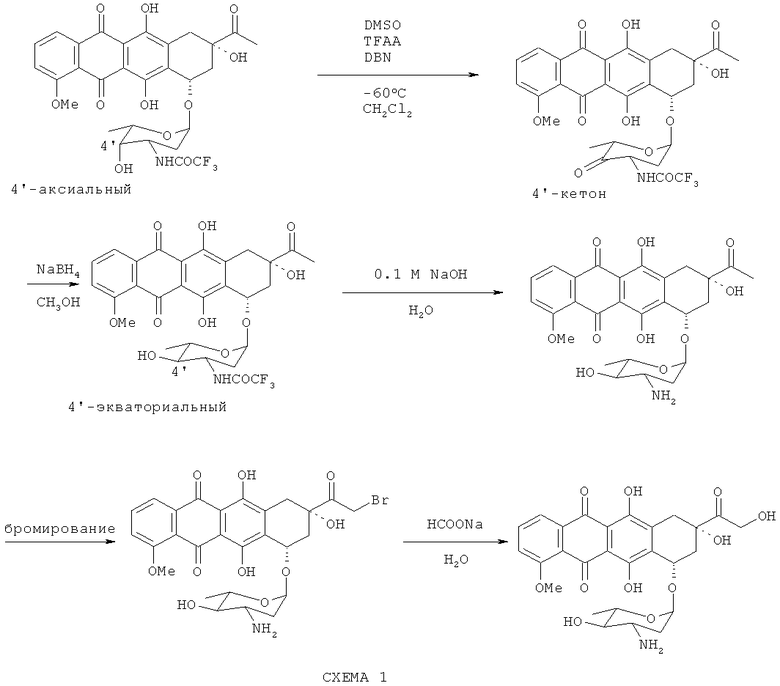
Превращение даунорубицина в эпирубицин осуществляют путем окисления 4'-гидроксильного фрагмента в кетон, что сопровождается потерей оптического центра, и дополнительного стереоспецифического восстановления (в требуемой конформации) с последующим превращением эпи-даунорубицина в эпи-рубицин бромированием 14-СНз-(СО)- фрагмента и гидролизом полученного 14-CH2Br-фрагмента в -(СО)-СН2ОН радикал. Этот процесс схематически показан на Схеме 1 ниже.
Частично этот синтез был разработан Фармиталия (Farmitalia), как описано в патенте США №4345068 на имя Suarato и др. Ранее были описаны и другие способы синтеза эпи-даунорубицина, см., например, патент США №5945518 на имя Bigatti и др., патент США №5874550 на имя van der Rijst и др. Однако во всех существующих способах синтеза эпи-даунорубицина используется один и тот же исходный материал, а именно, даунорубицин Формулы (2).
Раскрытие изобретения
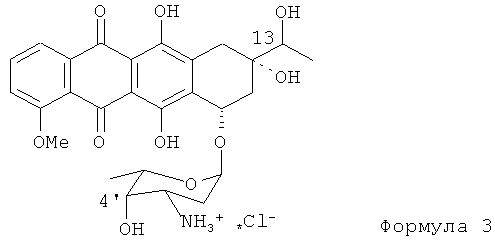
Данное изобретение направлено на создание новаторского способа получения эпирубицина с использованием нового исходного материала для синтеза. Конкретнее, новым исходным материалом является 13-даунорубицинол (13-дигидродаунорубицин, представленный Формулой (3)). Ключевое различие между даунорубицинолом и даунорубицином состоит в наличии гидроксильной группы в положении 13 антрациклинового ядра в отличие от 13-кето группы.
Согласно данному изобретению новый способ включает следующие стадии:
(1) первой стадией нового процесса является селективное введение защитной группы по амино-группе гликозидной части антибиотика, как показано Формулой (4); 13-ОН и 4'-ОН группы предпочтительно остаются немодифицированными.
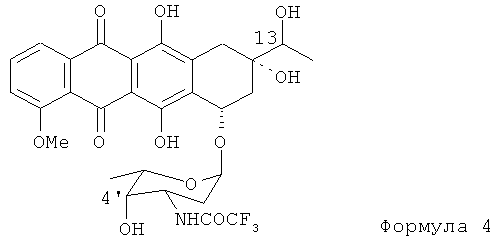
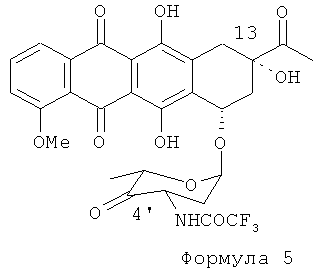
(2) вторая стадия процесса включает окисление 13-ОН и 4'-ОН групп в соответствующие кетоны путем обработки N-трифторацетил-13-даунорубицинола Формулы (4) диметилсульфоксидом, активированным различными ацилирующими агентами (АсХ), для получения соединения Формулы (5):
АсХ = PySO3, SOCl2, PHal3, POHal3; Hal=Cl, Br;
Ac = AlkCO; OC-(CH2)nCO, n=0-4; AlkSO2; ArCO; ArSO2,
Alk = алкил или галогеналкильный радикал;
Ar = фенил или замещенный фенильный радикал;
Х=Cl, Br, I, ОАс.

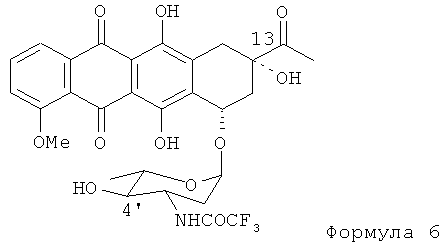
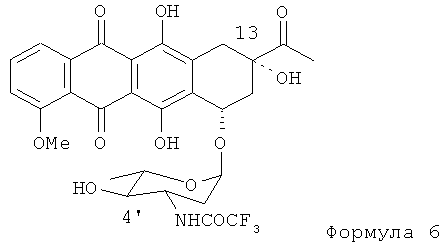
(3) в ходе третьей стадии 4'-кето группу 4'-кето-N-трифторацетил-даунорубицина Формулы (5) восстанавливают для получения экваториальной 4'-ОН группы без модификации 13-кето группы. Реакцию осуществляют путем взаимодействия 4'-кето-N-трифторацетил-даунорубицина с восстановителем, таким как производное боргидрида щелочного металла MHBL3, где М=Li, Na, К; L=AlkO, AlkCOO, ArCOO (Alk = Me, Et, n-Pr, все, -(CH2)n, n=0-4; Ar=Ph или замещенный Ph=Ph-Alk, с получением N-трифторацетил-4'-эпи-даунорубицина Формулы (6).
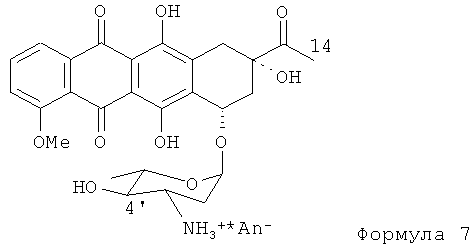
(4) гидролиз N-трифторацетил-4'-эпи-даунорубицина в щелочном растворе для получения производного Формулы (7).
(5) галогенирование 4'-эпи-даунорубицина Формулы (7) в положение С14 осуществляют реакцией с комплексными галогенидами Формулы (8):
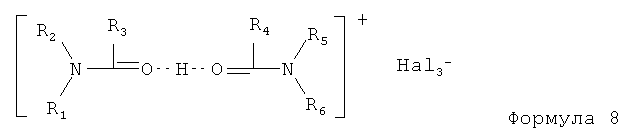
где радикалы от R1 до R6 являются Н или углеводородными радикалами, содержащими от 1 до 4 углеродных атомов в цепи (C1-C4); Hal является Cl, Br, I.
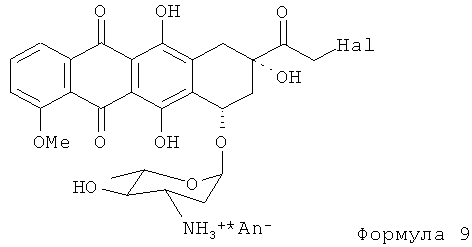
(6) полученное 14-Hal-производное Формулы (9) (где Hal является Cl, Br или I; и An- представляет собой анион сильной кислоты) гидролизуют хорошо известными способами в присутствии формиата щелочного металла с получением в конечном итоге продукта Формулы (1).
Детальное раскрытие изобретения
Согласно данному изобретению способ получения эпирубицина с использованием даунорубицинола в качестве исходного соединения включает следующие стадии.
I. Синтез N-трифторацетил-13-даунорубицинола
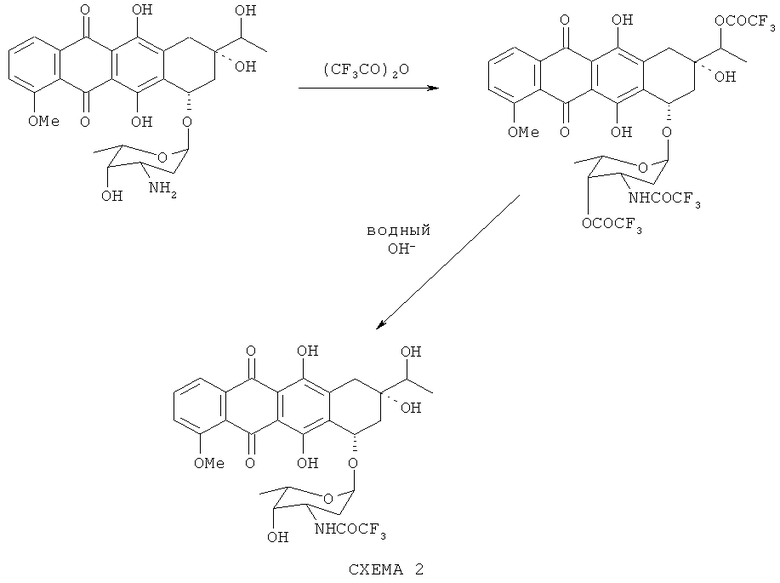
N-TFA-13-даунорубицинол получают из 13-даунорубицинола путем ацилирования последнего ангидридом трифторуксусной кислоты в сухих апротонных растворителях, не смешивающихся с водой, предпочтительно в дихлорметане, с последующим мягким гидролизом полученного амидоэфира в двухфазной системе, содержащей водный раствор основания и органический раствор амидоэфира, с получением N-TFA-даунорубицинола (как показано на схеме 2 ниже).
II. Синтез 4'-кето-N-трифторацетилдаунорубицина
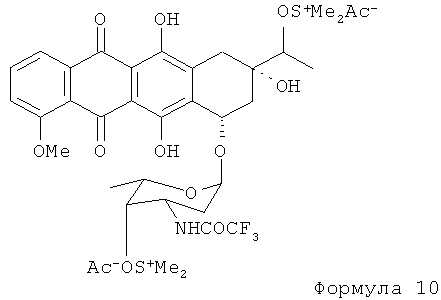
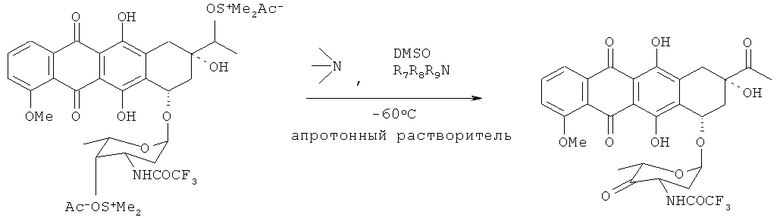
4'-кето-N-TFA-даунорубицин получают путем взаимодействия N-TFA-13-даунорубицинола с диметилсульфоксидом, активированным различными ацилирующими агентами (АсХ). N-TFA-13-даунорубицинол превращают в его сульфокси соль (4), которая в дальнейшем расщепляется на 4'-кето-N-TFA-даунорубицин, представленный Формулой (10), помимо других продуктов.
При определенных условиях выход искомого кетона может превышать 85% (см. Схему 3).
Подпись к Схеме 3:
АсХ = PySO3, SOCl2, PHal3, POHal3; Hal = Cl, Br;
Ac = AlkCO, OC-(CH2)n-CO n=0÷4, AlkSO2, ArCO Ar SO2
Alk = алкильный или галогеналкильный радикал,
Ar = фенильный или замещенный фенильный радикал.
Х=Cl, Br, I, ОАс.
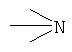
Апротонный растворитель = неводный апротонный растворитель, например, диметилсульфоксид (DMSO), диметилацетамид (DMAA), гексаметилфосфорамид (НМРА), дихлорметан (DCM) и другие галогеноалканы, ароматические углеводороды и их смесь.
Реакцию проводят при температурах от -80°С до 0°С; более оптимально при
-70±5°С. Увеличение температуры, при которой проводят реакцию, значительно увеличивает количество побочных продуктов (примесей).
III. Синтез 4'-эпи-N-трифторацетилдаунорубицина
4'-эпи-N-трифторацетилдаунорубицин синтезируют с помощью стереоспецифического восстановления 4'-кето-N-TFA-даунорубицина в экваториальной конформации боргидридом натрия (L=H).
Эта реакция (см. Схему 4) повышает выход желаемого эпимера до более чем 90%. Однако использование этого восстановителя также ведет к восстановлению 13-кето-группы агликонового фрагмента молекулы с образованием N-TFA-даунорубицинола.
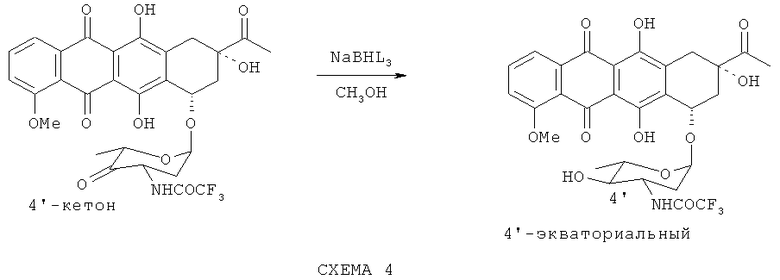
Альтернативно, восстановителем может быть боргидрид натрия с L≠Н; в частности L=AlkO (Alk = Me, Et, n-Pr или все); АсО (Ас = CR3CO, R=H, Hal). Использование такого боргидрида снижает его восстановительную способность, улучшая таким образом как регио-, так и стереоселективность реакции.
Реакцию проводят в растворителе, не подверженном восстановлению, таком как спирты, эфиры, углеводороды и галогенированные углеводороды, а также их смеси, предпочтительно в метаноле. Реакцию проводят при температуре от -35°С до 10°С и более предпочтительно при -20±5°С.
Превращение 4'-эпи-N-ТТА-даунорубицина в 4'-эпи-даунорубицин при удалении трифторацетильной защитной группы у 4'-эпи-N-TFA производных антрациклинов достигается путем обработки водной щелочью, имеющей рН 10-13, при температуре от 0°С до 40°С, предпочтительно при 20±5°С.
IV. Превращение 14-СН3 радикала агликонового фрагмента 4'-эпи-даунорубицина в 14-CH2OH
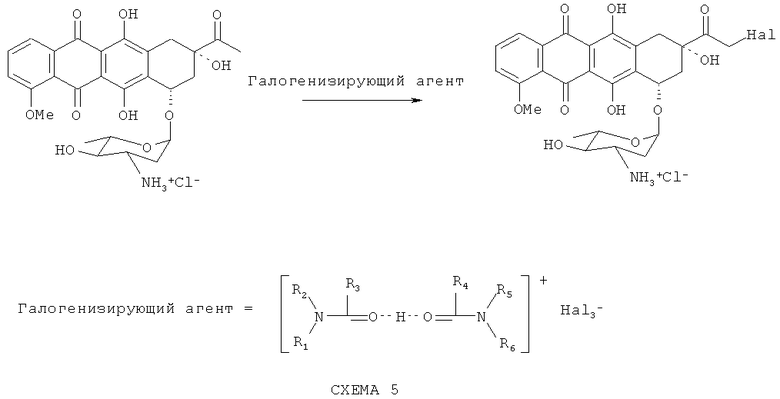
Галогенирование 4'-эпи-даунорубицина с получением продукта Формулы (6), показанное на Схеме (5), осуществляют с использованием комплексных галогенидов в качестве галогенизирующих агентов. Такой подход позволяет снизить количество стадий синтеза, а также увеличивает выход и чистоту конечного продукта.
В качестве растворителей для этой реакции используются амиды, простые эфиры и их смеси, предпочтительно диметилформамид и тетрагидрофуран.
Реакцию проводят при температуре 20-60°С в течение 2-20 часов; предпочтительно при 50°С в течение 3 часов. Полученное 14-галоген производное Формулы (9) гидролизуют в водном растворе ацетона в присутствии солей карбоновых кислот, предпочтительно формиата натрия, рН 2.5-5.5. В результате получают конечный продукт Формулы (1).
ПРИМЕР 1
(a) 5 грамм 13-дигидродаунорубицина Формулы (3) суспендируют в 200 мл дихлорметана (DCM) и охлаждают до 0°С. Ангидрид трифторуксусной кислоты в DCM (8 мл: 15 мл) добавляют медленно, по каплям в течение 1 часа при интенсивном перемешивании суспензии.
(b) Полученную смесь выдерживают при 0°С еще в течение 0.5 часа, после чего выливают в 250 мл дистиллированной воды, перемешивают и отделяют органический слой.
(c) К полученному органическому слою добавляют 200 мл насыщенного раствора бикарбоната натрия и оставляют смесь при интенсивном перемешивании при комнатной температуре на 24 часа для осуществления гидролиза и получения раствора 3'-N, 4',13-ди-О-три-трифторацетилдаунорубицинола.
(d) После завершения гидролиза (контролируется жидкостной хроматографией высокого давления, HPLC) органический слой отделяют и упаривают при пониженном давлении до полного высыхания.
(e) После упаривания получают 5 грамм N-трифторацетил-13-даунорубицинола с чистотой примерно 90% (подтверждено HPLC).
(f) N-трифторацетил-13-даунорубицинол, полученный на стадии (е) ПРИМЕРА 1, используют на следующей стадии синтеза, описанной в ПРИМЕРЕ 2, без дополнительной очистки.
ПРИМЕР 2
(a) 8 мл DMSO растворяют в 100 мл DCM и охлаждают до -60°С при перемешивании. Затем к раствору добавляют 2 мл оксалилхлорида в 5 мл DCM и инкубируют при -60°С в течение 40 минут для получения реакционной смеси.
(b) 5 грамм N-трифторацетил-13-даунорубицинола растворяют в 50 мл DCM и добавляют в реакционную смесь в течение 20 минут, поддерживая температуру в интервале -60±5°С. После этого реакционную смесь инкубируют в течение 1 часа.
(c) В реакционную смесь добавляют 10 мл триэтиламина при температуре не выше -60°С. Общее время взаимодействия реакционной смеси с триэтиламином составляет 10 минут.
(d) В реакционную смесь добавляют раствор 5 мл уксусной кислоты в 10 мл DCM и перемешивают в течение 2 минут.
(e) После этого реакционную смесь выливают в 300 мл дистиллированной воды, перемешивают и отделяют органический слой. Эту стадию повторяют трижды.
(f) Органический слой упаривают на роторном испарителе при пониженном давлении.
(g) После упаривания получают 4.7 грамма 4'кето-N-трифторацетилдаунорубицина с чистотой около 85% (подтверждено HPLC).
(h) 4'кето-N-трифторацетилдаунорубицин, полученный на стадии (g) ПРИМЕРА 2, используют на следующей стадии синтеза, описанной в ПРИМЕРЕ 3. без дополнительной очистки.
ПРИМЕР 3
(a) 4.7 грамма 4'кето-N-трифторацетилдаунорубицина растворяют в 180 мл тетрагидрофурана и, при перемешивании, в течение 40 минут добавляют 2.1 грамма тетраацетилборгидрида натрия. Реакционную смесь инкубируют при встряхивании в течение 1 часа при температуре в интервале 20±2°С.
(b) Реакционную массу переносят в смесь 150 мл DCM + 300 мл дистиллированной воды + 2 мл 1 М соляной кислоты и перемешивают. Органический слой отделяют и дважды промывают аликвотами по 300 мл дистиллированной воды.
(c) После упаривания получают 4.6 г 4'эпи-N-трифторацетилдаунорубицина с чистотой примерно 79% (подтверждено HPLC).
(d) Полученный неочищенный продукт подвергают очистке на препаративном хроматографе. После упаривания элюата получают 3.0 грамма 4'эпи-N-трифторацетилдаунорубицина с чистотой примерно 95% (подтверждено HPLC).
ПРИМЕР 4
3.0 грамма 4'эпи-N-трифторацетилдаунорубицина суспендируют в 200 мл дистиллированной воды при температуре 30°С, после чего добавляют 10 мл раствора 1.0 N NaOH. Смесь инкубируют в течение 30 минут, затем нейтрализуют до рН 7 раствором соляной кислоты и подвергают очистке с использованием препаративной хроматографии. После выпаривания элюата получают 2.1 грамм гидрохлорида 4'эпи-даунорубицина с чистотой около 96% (подтверждено HPLC).
ПРИМЕР 5
(a) 2.1 грамма гидрохлорида 4'эпи-даунорубицина растворяют в 70 мл диметилформамида и добавляют к смеси 2.8 грамм дибромбромата бис(диметилформамид) водорода. Смесь инкубируют при 40°С в течение 2 часов.
(b) Реакционную смесь выливают в 350 мл ацетонитрила. Образовавшийся осадок отфильтровывают и промывают ацетонитрилом, растворитель удаляют.
(c) Твердый осадок растворяют в смеси 80 мл ацетона + 80 мл 0.25 М водного раствора бромистого водорода + 8 грамм формиата натрия. Реакционную смесь инкубируют в течение 30 часов при 35°С.
(d) Реакционную смесь подвергают препаративной хроматографии, выделяя фракцию, содержащую эпирубицин.
(e) Элюат упаривают, и остаток кристаллизуют путем добавления ацетона.
(f) Выход на этой стадии составляет 1.3 г. гидрохлорида эпирубицина с чистотой 99,8% (подтверждено HPLC).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 4-ДЕМЕТИЛДАУНОРУБИЦИНА | 2006 |
|
RU2450013C2 |
| СПОСОБ АРАЛКИЛИРОВАНИЯ 4'-ГИДРОКСИЛЬНОЙ ГРУППЫ АНТРАЦИКЛИНОВ | 2008 |
|
RU2563453C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4-R-ЗАМЕЩЕННОГО 4-ДЕМЕТОКСИДАУНОРУБИЦИНА | 2004 |
|
RU2336277C2 |
| ПРОИЗВОДНЫЕ АНТРАЦИКЛИНА | 1995 |
|
RU2159619C2 |
| ПРОИЗВОДНЫЕ АНТРАЦИКЛИНОВ, СПОСОБ ПОЛУЧЕНИЯ 13-БЕНЗЕНИДСУЛЬФОНИЛГИДРАЗОНОВЫХ ПРОИЗВОДНЫХ АНТРАЦИКЛИНОВ, СПОСОБ ПОЛУЧЕНИЯ 13-ДЕОКСИАНТРАЦИКЛИНОВ, СПОСОБ ПОЛУЧЕНИЯ 5-ИМИНО-13-ДЕОКСИАНТРАЦИКЛИНОВ | 2005 |
|
RU2404188C9 |
| СПОСОБ ПОЛУЧЕНИЯ 4-ДЕМЕТОКСИДАУНОРУБИЦИНА | 2012 |
|
RU2540968C2 |
| Способ получения хлоргидратов замещенных антрациклинов | 1979 |
|
SU867315A3 |
| Способ получения антрациклинов | 1977 |
|
SU776562A3 |
| Способ получения 4 @ -галоид-антрациклингликозидов | 1984 |
|
SU1579465A3 |
| Способ получения 4-деокси-даунорубицина или 4-деокси-доксорубицина | 1981 |
|
SU1277902A3 |
Изобретение описывает способ получения антрациклина, представленного Формулой (1)
из 13-даунорубицинола путем ацилирования. N-трифторацетил-13-даунорубицинол взаимодействует с апротонным растворителем и ацилирующим агентом с образованием промежуточной сульфокси соли, которую обрабатывают сильным основанием для получения 4'-кето-N-трифторацетилдаунорубицина. 4'-кето-N-трифторацетил-даунорубицин взаимодействует с восстановителем, таким как боргидрид щелочного металла, для получения N-трифторацетил-4'-эпидаунорубицина. N-трифторацетил-4'-эпидаунорубицин гидролизуют в щелочном растворе для получения промежуточного соединения. Промежуточное соединение вступает в реакцию с галогенизирующим агентом для получения 14-Hal-производного. 14-Hal производное гидролизуют в присутствии формиата щелочного металла для получения требуемого конечного продукта. Технический результат: описан новый способ получения эпирубицина, который отличается высоким выходом конечного продукта. 2 н. и 15 з.п. ф-лы.
1. Способ получения антрациклина, представленного формулой (1)
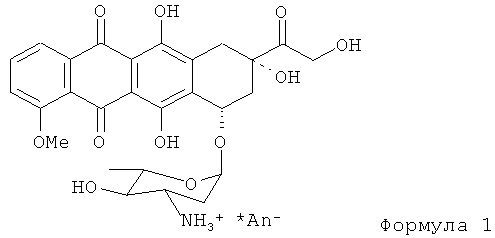
из 13-дигидродаунорубицина(даунорубицинола), включающий:
(а) получение N-трифторацетил-13-даунорубицинола, представленного формулой (4)
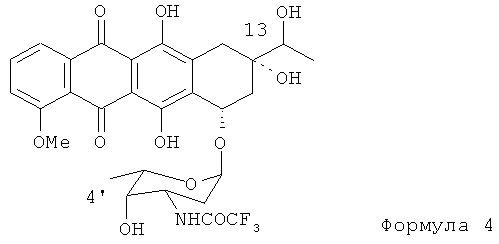
(b) взаимодействие N-трифторацетил-13-даунорубицинола с апротонным растворителем и ацилирующим агентом для получения промежуточной сульфоксисоли и обработку промежуточной сульфоксисоли сильным основанием для получения 4'-кето-N-трифторацетилдаунорубицина формулы (5)
(с) взаимодействие 4'-кето-N-трифторацетилдаунорубицина с боргидридом щелочного металла для получения N-трифторацетил-4'-эпи-даунорубицина, представленного формулой (6)
(d) гидролиз N-трифторацетил-4'-эпи-даунорубицина в щелочном растворе для получения соединения, представленного формулой (7) 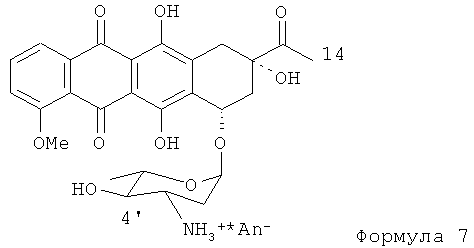
где An- является анионом сильной кислоты;
(е) взаимодействие соединения формулой (7) с галогенизирующим агентом, представленным формулой (8)
где хотя бы один из радикалов от R1 до R6 является Н или углеводородным радикалом, содержащим от 1 до 4 атомов углерода в цепи (C1-C4), и Hal является Cl, Br или I, для получения соединения, представленного формулой (9)
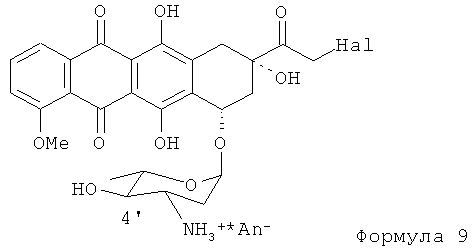
где Hal является Cl, Br или I, и An- является анионом сильной кислоты; и
(f) гидролиз соединения формулы (9) в присутствии формиата щелочного металла для получения антрациклина формулы (1)
2. Способ по п.1, отличающийся тем, что апротонный растворитель на стадии (b) выбран из группы, включающей DMSO (диметилсульфоксид), диметилацетамид (DMAA), гексаметилфосфорамид (НМРА), ацетонитрил, DCM (дихлорметан), галогеноалканы, ароматические углеводороды и их смеси.
3. Способ по п.1, отличающийся тем, что ацилирующим агентом на стадии (b) является соединение АсХ,
где АсХ - PySO3, SOCl2, PHal3, POHal3, Hal - Cl, Br;
Ac - AlkCO, OC-(CH2)n-CO, n=0-4, AlkSO2, ArCO, ArSO2;
Alk - алкил или галогеналкильный радикал;
Ar - фенил или замещенный фенильный радикал; и
Х - Cl, Br, I, ОАс.
4. Способ по п.1, отличающийся тем, что боргидрид щелочного металла на стадии (с) представляет собой соединение общей формулы MHBL3, где М является одним из Li, Na или К, и L является одним из AlkO, AlkCOO, ArCOO, где Alk является одним из Me, Et, n-Pr, все, -(СН2)n, n=0-4, и где Ar - Ph или замещенный Ph - Ph-Alk.
5. Способ по п.1, отличающийся тем, что сильным основанием на стадии (b) является третичный амин общей формулы NR7R8R9, где N является циклическим или полициклическим третичным амином, R7, R8, R9 - алкил или циклоалкил, и R8, R9 - -(СН2)n, где n=3-6.
6. Способ по п.1, отличающийся тем, что стадию (b) проводят при температуре в интервале от примерно -80°С до примерно 0°С.
7. Способ по п.1, отличающийся тем, что реакцию с галогенизирующим агентом проводят при температуре от примерно 20°С до примерно 60°С в течение от 2 до 20 ч.
8. Способ по п.1, отличающийся тем, что стадия (а) включает: получение 13-даунорубицинола формулы (3), и ацилирование 13-даунорубицинола ангидридом трифторуксусной кислоты в растворителе для получения N-трифторацетил-13-даунорубицинола формулы (4), предусмотренного стадией (а).
9. Способ по п.8, отличающийся тем, что растворителем является дихлорметан.
10. Способ получения антрациклина, представленного формулой (1)
из 13-дигидродаунорубицина, включающий:
(а) получение 13-даунорубицинола, представленного формулой (3)
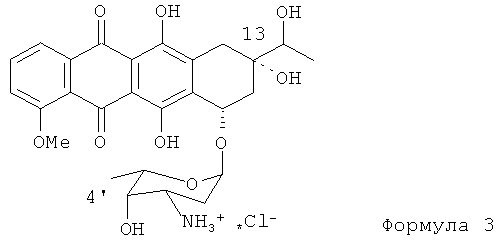
(b) ацилирование 13-даунорубицинола, полученного на стадии (а), ангидридом трифторуксусной кислоты в растворителе для получения N-трифторацетил-13-даунорубицинола, представленного формулой (4)
(d) взаимодействие N-трифторацетил-13-даунорубицинола с апротонным растворителем и ацилирующим агентом для получения промежуточной сульфоксисоли и обработка промежуточной сульфоксисоли сильным основанием для получения 4'-кето-N-трифторацетилдаунорубицина формулы (5)
(е) взаимодействие 4'-кето-N-трифторацетилдаунорубицина с боргидридом щелочного металла для получения N-трифторацетил-4'-эпи-даунорубицина, представленного формулой (6)
(f) гидролиз N-трифторацетил-4'-эпи-даунорубицина в щелочном растворе для получения соединения формулы (7)
где An- является анионом сильной кислоты;
(g) взаимодействие соединения формулы (7) с галогенизирующим агентом, представленным формулой (8)
где хотя бы один из радикалов от R1 до R6 является Н или углеводородным радикалом, содержащим от 1 до 4 атомов углерода в цепи (C1-C4), и Hal является Cl, Br или I, для получения соединения, представленного формулой (9)
где Hal является Cl, Br или I, и An- является анионом сильной кислоты; и
(h) гидролиз соединения формулы (9) в присутствии формиата щелочного металла для получения антрациклина формулы (1)
11. Способ по п.10, отличающийся тем, что апротонный растворитель на стадии (d) выбран из группы, включающей DMSO (диметилсульфоксид), диметилацетамид (DMAA), гексаметилфосфорамид (НМРА), ацетонитрил, DCM (дихлорметан), галогеноалканы, ароматические углеводороды и их смеси.
12. Способ по п.10, отличающийся тем, что ацилирующим агентом на стадии (d) является соединение общей формулы АсХ,
где AcX - PySO3, SOCl2, PHal3, POHal3, Hal - Cl, Br;
Ac - AlkCO, ОС-(СН2)n-СО, n=0-4, AlkSO2, ArCO, ArSO2;
Alk - алкил или галогеналкильный радикал;
Ar - фенил или замещенный фенильный радикал; и
Х - Cl, Br, I, ОАс.
13. Способ по п.10, отличающийся тем, что боргидрид щелочного металла на стадии (е) представляет собой соединение общей формулы MHBL3, где М является одним из Li, Na или К, и L является одним из AlkO, AlkCOO, ArCOO, где Alk является одним из Me, Et, n-Pr, -(CH2)n, n=0-4, и где Ar - Ph или замещенный Ph - Ph-Alk.
14. Способ по п.10, отличающийся тем, что сильным основанием на стадии (d) является третичный амин общей формулы NR7R8R9, где N является циклическим или полициклическим третичным амином, R7, R8, R9 - алкил или циклоалкил, и R8, R9 - -(CH2)n, где n=3-6.
15. Способ по п.10, отличающийся тем, что стадию (d) проводят при температуре в интервале от примерно -80°С до примерно 0°С.
16. Способ по п.10, отличающийся тем, что реакцию с галогенизирующим агентом проводят при температуре от примерно 20°С до примерно 60°С в течение от 2 до 20 ч.
17. Способ по п.10, отличающийся тем, что растворителем является дихлорметан.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
| RU 2003117429 А, 10.12.2004. | |||

