Область техники, к которой относится изобретение
Настоящее изобретение относится к способу получения (6R)-3-гексил-4-гидрокси-6-ундецил-5,6-дигидропиран-2-она и (5R)-2-гексил-5-гидрокси-3-иминогексадеканоатного производного, применяемого в указанном способе в качестве промежуточного соединения.
Описание предшествующего уровня техники
(6R)-3-гексил-4-гидрокси-6-ундецил-5,6-дигидропиран-2-он формулы (I), который может быть в форме своего таутомера, структура формулы (Ia), является важным промежуточным соединением, применяемым при получении химических соединений тонкого органического синтеза и фармацевтических веществ(API):

(6R)-3-гексил-4-гидрокси-6-ундецил-5,6-дигидропиран-2-он является промежуточным соединением, применяемым в синтезе оксетанонов, таких как тетрагидролипстатин (Orlistat™), и способы его получения описывают в патентах США № 4983746, 5245056, 5399720, 5274143 и 5420305. В патенте США № 4983746 описывают получение 3,6-диалкил-5,6-дигидро-4-гидроксипиран-2-она окислением диалкил-3,4,5,6-тетрагидро-4-гидроксипиран-2-она, применяя реагент Джонса. Однако, сильное основание, такое как н-бутиллитий и LDA (литийдиизопропиламид), которые не подходят для промышленного применения, применяют при получении диалкил-3,4,5,6-тетрагидро-4-гидроксипиран-2-она, и требуется дополнительное окисление 3,6-диалкил-5,6-дигидро-4-гидроксипиран-2-она для получения требуемого оптического изомера.
В патенте США № 5245056 и 5399720 описывают способ, включающий стадию получения (6R)-3-гексил-4-гидрокси-6-ундецил-5,6-дигидропиран-2-она активацией 3-гидрокситетрадекановой кислоты карбонилдиимидом, но выход данной реакции был низким, только 34%.
Для увеличения выхода (6R)-3-гексил-4-гидрокси-6-ундецил-5,6-дигидропиран-2-она в патенте США № 5274143 и 5420,305 описывают способ, включающий внутримолекулярную циклизацию (R)-3-[2-бром-1-оксооктилоксо]тетрадеканоата в присутствии цинка для получения (6R)-3-гексил-4-гидрокси-6-ундецил-5,6-дигидропиран-2-она с выходом 61-67%, тогда как в патенте США № 6545165 описывают внутримолекулярную циклизацию (R)-3-[2-бром-1- оксооктилоксо]тетрадеканоата в присутствии трет-бутилмагний хлорида для получения (6R)-3-гексил-4-гидрокси-6-ундецил-5,6-дигидропиран-2-она с выходом 78%.
Кроме того, в патенте США № 6552204 описывают способ получения 3,6-диалкил-5,6-дигидро-4-гидрокси-пиран-2-она, который включает: (i) реакцию защищенного β-гидроксиацилгалогенида с кетенацеталем для получения эфира δ-гидрокси-β-енольного эфира или защищенного δ-гидрокси-β-енольного эфира, или (ii) реакцию β-гидроксиацилгалогенида с мономалоновой кислотой для получения δ-гидрокси-β-кетоэфира и, затем, обработку δ-гидрокси-β-кетоэфира кислотой. Однако исходное соединение должно быть получено при высоком давлении для получения требуемого оптического изомера, что приводит к низкой эффективности процесса.
СУЩНОСТЬ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Цель настоящего изобретения заключается в обеспечении улучшенным способом получения (6R)-3-гексил-4-гидрокси-6-ундецил-5,6-дигидропиран-2-она формулы (I) с высокой чистотой и высоким выходом.
Другая цель настоящего изобретения заключается в обеспечении промежуточным соединением, применяемым в получении (6R)-3-гексил-4-гидрокси-6-ундецил-5,6-дигидропиран-2-она формулы (I).
Согласно одному аспекту настоящего изобретения, обеспечивают способом получения (6R)-3-гексил-4-гидрокси-6-ундецил-5,6-дигидропиран-2-она формулы (I), который включает:
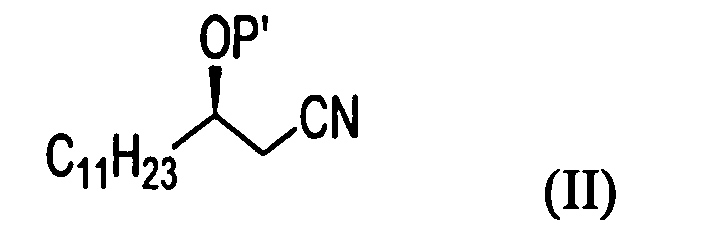
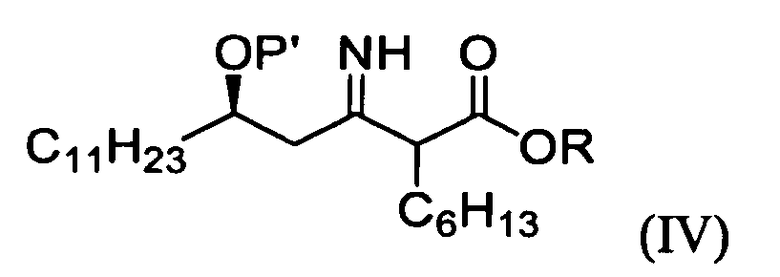
1) взаимодействие (R)-3-гидрокситетрадеканнитрильного производного формулы (II) с алкил 2-бромоктаноатом формулы (III) в присутствии цинка для получения (5R)-2-гексил-5-гидрокси-3-иминогексадеканоатного производного формулы (IV);
2) обработку соединения формулы (IV) водной кислотой для получения (5R)-2-гексил-5-гидрокси-3-оксогексадеканоатного производного формулы (V); и
3) циклизацию соединения формулы (V) в кислых или основных условиях:
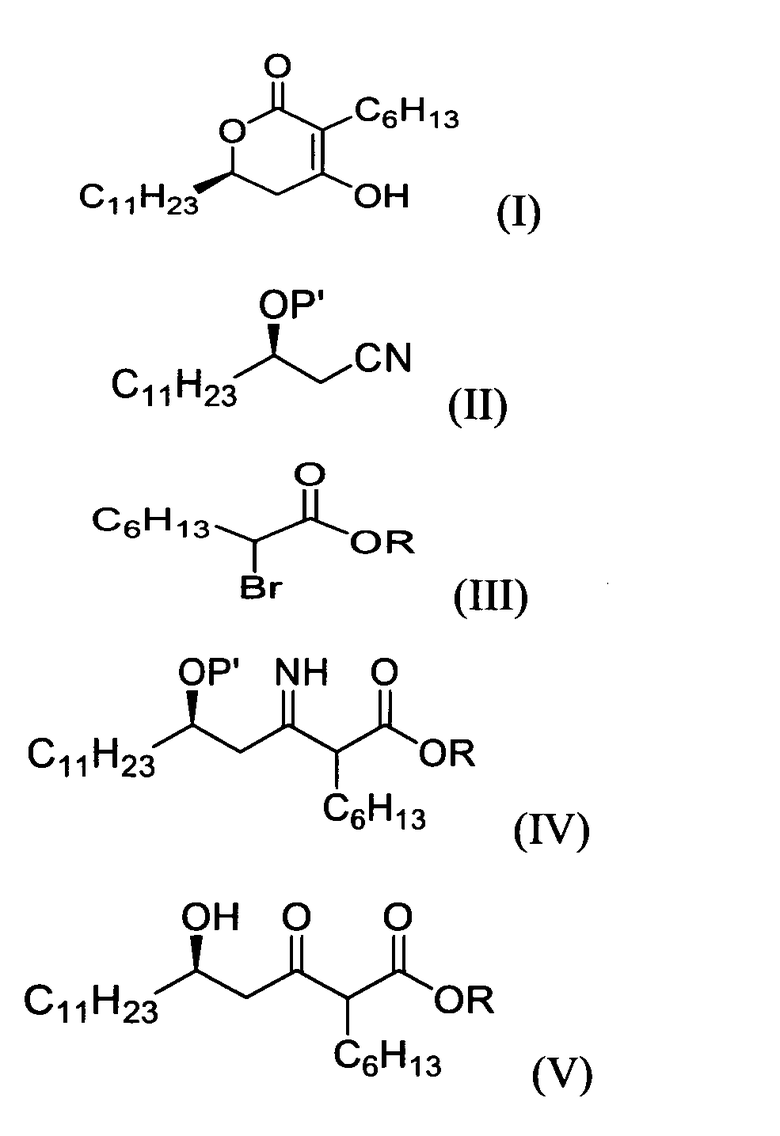
в которых
P' представляет собой водород или защитную группу, выбранную из триалкилсилила и алкоксиалкила, которые можно удалить в кислых условиях, и
R представляет собой метил, этил или пропил.
Согласно другому аспекту настоящего изобретения обеспечивают соединением формулы (IV), применяемым в получении соединения формулы (I).
ПОДРОБНОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
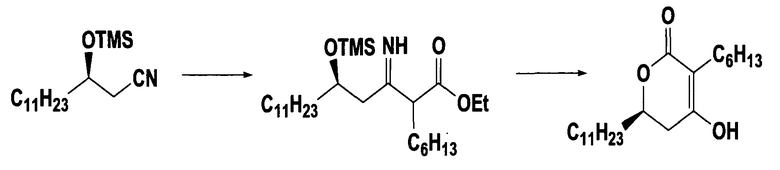
(R)-3-гидрокситетрадеканнитрильное производное формулы (II), применяемое в качестве исходного соединения в способе настоящего изобретения, можно получить способом, описанным в Tetrahedron Asymmetry 1999, 2945.
Согласно настоящему изобретению соединение формулы (I) можно получить реакцией Блейза (R)-3-гидрокситетрадеканнитрила или (R)-3-гидрокситетрадеканнитрила, содержащего защищенную гидроксильную группу, с соединением формулы (III) в присутствии цинка для получения соединения формулы (IVa) или соединения формулы (IVb); обработкой соединения формулы (IVa) или (IVb) водной кислотой для получения соединения формулы (V); и циклизацией соединения формулы (V).
Конкретно, способ настоящего изобретения получения соединения формулы (I) можно осуществить, как показано на реакционной схеме 1.
Реакционная схема 1
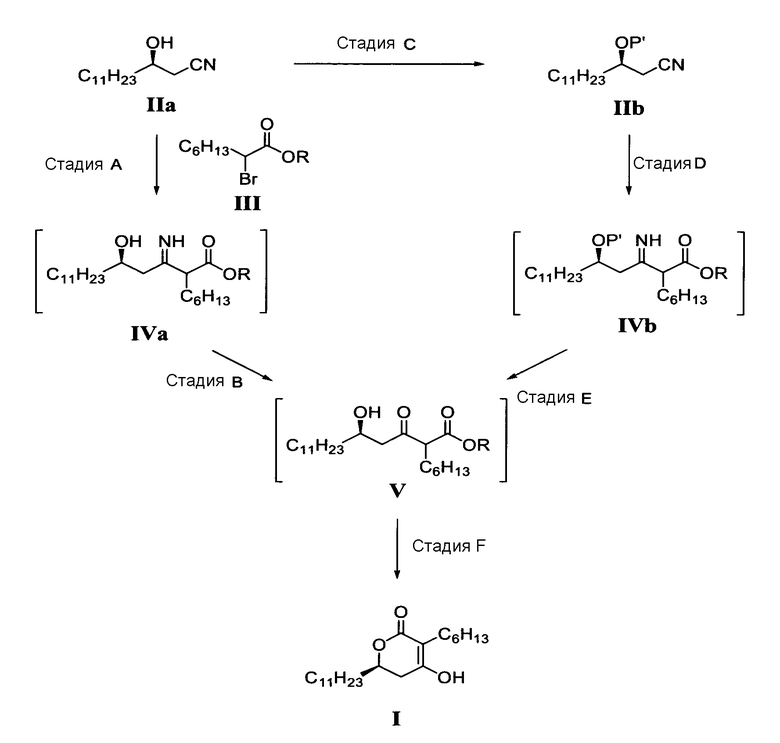
в которой
P' представляет собой водород или защитную группу, выбранную из триалкилсилила и алкоксиалкила, которые можно удалить в кислых условиях, и
R представляет собой метил, этил или пропил.
На стадии A реакционной схемы 1, (R)-3-гидрокситетрадеканнитрил формулы (IIa) подвергают взаимодействию с алкил 2-бромоктаноатом формулы (III) в присутствии цинка в подходящем растворителе для получения (5R)-2-гексил-5-гидрокси-3-иминогексадеканоатного производного формулы (IVa). Примеры алкил 2-бромоктаноата формулы (III) включают метил 2-бромоктаноат, этил 2-бромоктаноат и пропил 2-бромоктаноат. Алкил 2-бромоктаноат применяют в количестве в диапазоне 1-10 молярных эквивалентов, предпочтительно 1,5-3 молярных эквивалента на 1 моль (R)-3-гидрокситетрадеканнитрила формулы (IIa).
Цинк может быть в форме порошка или пыли, которая ускоряет реакционную стадию A, и активированный цинковый порошок является самым предпочтительным. Цинк применяют в количестве в диапазоне 1-10 молярных эквивалентов, предпочтительно 3-5 молярных эквивалента на 1 моль (R)-3-гидрокситетрадеканнитрила формулы (IIa). Кроме того, можно применять каталитическую добавку для активации цинка в количестве 0,01-0,2 молярных эквивалентов на 1 моль (R)-3-гидрокситетрадеканнитрила формулы (IIa). Примеры данных добавок включают I2, 1,2-диброметан, метансульфокислоту и тиметилсилилхлорид. Триметилсилилхлоридную добавку можно также применять в качестве реакционного растворителя.
Кроме того, для увеличения воспроизводимости стадии A вместе с цинком можно применять небольшое количество свинца. Например, свинцовый порошок можно применять в количестве в диапазоне 2-10% по весу, предпочтительно 3-5% по весу относительно суммарного веса цинка. Пример применения свинца в реакции с применением цинка описывают в Organic Process Research & Development 2001, 28.
Растворитель, который можно применять на стадии A, включает апротонный растворитель, такой как эфир, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, ацетонитрил, метилацетат, этилацетат, бензол, толуол и их смеси. Среди них этиловый эфир, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан и толуол являются предпочтительными.
Стадию A можно проводить при температуре в диапазоне от 0°C до температуры кипения применяемого растворителя, предпочтительно 30-100°C.
На стадии B, (5R)-2-гексил-5-гидрокси-3-иминогексадеканоатное производное формулы (IVa) обрабатывают водной кислотой в подходящем растворителе для получения (5R)-2-гексил-5-гидрокси-3-оксогексадеканоатного производного формулы (V). Примеры кислоты включают неорганические кислоты, такие как хлороводородная кислота, серная кислота и бромоводородная кислота, а также органические кислоты, такие как толуолсульфокислота, метансульфокислота и камфорсульфокислота, и кислота находится в водном растворе в количестве в диапазоне 0,01-5 молярных эквивалентов, предпочтительно 1-3 молярных эквивалента на 1 моль (5R)-2-гексил-5-гидрокси-3-иминогексадеканоатного производного формулы (IVa).
Растворитель, который можно применять на стадии B, включает протонные растворители, такие как вода, метанол и этанол; апротонные растворители, такие как этиловый эфир, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, ацетонитрил, метилацетат, этилацетат, бензол и толуол; и их смеси.
Таким образом полученное (5R)-2-гексил-5-гидрокси-3-оксогексадеканоатное производное формулы (V) можно непосредственно применять на стадии F без дополнительной очистки.
В течение стадии B, небольшое количество (5R)-2-гексил-5-гидрокси-3-оксогексадеканоатного производного формулы (V) можно подвергнуть циклизации для получения целевого соединения, (6R)-3-гексил-4-гидрокси-6-ундецил-5,6-дигидропиран-2-она формулы (I).
Стадию B можно осуществлять при температуре в диапазоне от 0°C до температуры кипения применяемого растворителя, предпочтительно 10-60°C. На стадии C гидроксильную группу (R)-3-гидрокситетрадеканнитрила формулы (IIa) защищают защитной группой для гидроксила, которую можно легко удалить. Примеры защитной группы включают триалкилсилил, такой как триметилсилил (TMS) и триэтилсилил, алкоксиалкил, такой как тетрагидропиранил (THP), 4-метокситетрагидропиранил (MTHP), 2-метокси-2-пропил (MOP) и 1-этокси-1-этил (EE), которые можно легко удалить в кислых условиях.
Триметилсилилирование гидроксильной группы (R)-3-гидрокситетрадеканнитрила можно осуществлять общепринятым способом (см. Protective groups in organic synthesis 4th Ed., pp 171-178), и введение тетрагидропиранильной защитной группы можно осуществлять общепринятым способом, применяя дигидрофуран и п-толуолсульфонат пиридиния (см. Protective groups in organic synthesis 4th Ed., pp 59-68, Protecting groups).
Защищенный (R)-3-гидрокситетрадеканнитрил формулы (IIb) можно применять в стадии D без выделения или очистки. На стадии D защищенное (5R)-2-гексил-5-гидрокси-3-иминогексадеканоатное производное формулы (IVb) получают способом, аналогичным способу, применяемому на стадии A. На стадии E из защищенного (5R)-2-гексил-5-гидрокси-3-иминогексадеканоатного производного формулы (IVb) удаляют защитную группу в кислых условиях способом, аналогичным способу на стадии B, для получения (5R)-2-гексил-5-гидрокси-3-оксогексадеканоатного производного формулы (V). Таким образом полученное (5R)-2-гексил-5-гидрокси-3-оксогексадеканоатное производное формулы (V) можно непосредственно применять в стадии F без дополнительной очистки.
В течение стадии E небольшое количество (5R)-2-гексил-5-гидрокси-3-оксогексадеканоатного производного формулы (V) можно подвергнуть циклизации для получения целевого соединения, (6R)-3-гексил-4-гидрокси-6-ундецил-5,6-дигидропиран-2-она формулы (I).
На стадии F (5R)-2-гексил-5-гидрокси-3-оксогексадеканоатное производное формулы (V), полученное на стадии B или E, циклизуют для получения целевого соединения, (6R)-3-гексил-4-гидрокси-6-ундецил-5,6-дигидропиран-2-она формулы (I).
Циклизацию можно осуществлять в подходящем растворителе в присутствии кислоты или основания, как описано в патенте США № 6552204.
Кислоту или основание можно применять в количестве в диапазоне 0,01-10 молярных эквивалентов, предпочтительно 0,1-3 молярных эквивалента на 1 моль (5R)-2-гексил-5-гидрокси-3-оксогексадеканоатного производного формулы (V).
Примеры кислоты включают неорганические кислоты, такие как хлороводородная кислота и серная кислота; органические кислоты, такие как толуолсульфокислота, метансульфокислота и камфорсульфокислота; и их смеси, и метансульфокислота является предпочтительной. Примеры оснований включают гидроксид натрия и гидроксид калия.
Растворитель, который можно применять на стадии B, включает протонные растворители, такие как вода, метанол и этанол; апротонные растворители, такие как этиловый эфир, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, ацетонитрил, метилацетат, этилацетат, бензол и толуол; и их смеси, среди которых ацетонитрил является предпочтительным.
Настоящее изобретение будет описано более подробно с ссылкой на примеры. Однако ясно, что настоящее изобретение не ограничивается конкретными примерами.
Получение (6R)-3-гексил-4-гидрокси-6-ундецил-5,6-дигидропиран-2-она
Пример 1

4,35 г активированного цинкового порошка, 5 г (R)-3-гидрокситетрадеканнитрила и 30 мл тетрагидрофурана помещали в реактор и перемешивали, добавляли 0,14 мл метансульфокислоты, и полученную в результате смесь кипятили с обратным холодильником в течение 1 часа, к которой добавляли по каплям в течение приблизительно одного часа разбавленный раствор 9,53 мл этил 2-бромоктаноата в 10 мл толуола и кипятили с обратным холодильником в течение 1 часа. Полученную в результате смесь охлаждали до комнатной температуры, и добавляли по каплям 20 мл 3N HCl в течение 30 минут и перемешивали в течение приблизительно 1 часа. Смесь нагревали до приблизительно 50°C и перемешивали в течение приблизительно 2 часов.
Полученный в результате твердый остаток фильтровали, применяя воронку Бюхнера со слоем целита, и промывали 25 мл этилацетата. Органический слой объединяли, промывали 50 мл воды, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Таким образом полученный концентрат растворяли в 20 мл ацетонитрила, добавляли 4,22 г метансульфокислоты, перемешивали в течение приблизительно 12 часов, охлаждали до 5°C и дополнительно перемешивали в течение приблизительно 2 часов. Образовавшийся твердый остаток выделяли фильтрацией, промывали 5 мл холодного ацетонитрила и сушили, получая 4,85 г заявленного в заголовке соединения (выход: 62%) в виде грязно-белого твердого остатка.
Тпл: 110-112°C.
1H-ЯМР (CDCl3, смесь кето- и эндоформ, м.д.): δ 0,80-0,99 (м, 6H), 1,16-2,03 (м, 30H), 2,16-2,80 (м, 2H), 3,22 (т, 0,1H, J=5,5 Гц), 3,41 (т, 0,9H, J=5,5 Гц), 4,25-4,40 (т, 0,1H), 4,48-4,62 (т, 0,1H), 4,62-4,75 (т, 0,8H, 6,75-6,96 (ушир.с, 0,1H).
Пример 2

2,9 г активированного цинкового порошка и 10 мл тетрагидрофурана помещали в реактор и перемешивали, добавляли 6 мл хлортриметилсилана, и полученную в результате смесь кипятили с обратным холодильником в течение 1 часа, к которой медленно добавляли в течение приблизительно 5 минут разбавленный раствор 2 г (R)-3-гидрокситетрадеканнитрила в 4 мл тетрагидрофурана и перемешивали в течение приблизительно 5 минут. Затем добавляли по каплям в течение 30 минут разбавленный раствор 5,6 мл этил 2-бромоктаноата в 4 мл тетрагидрофурана и кипятили с обратным холодильником в течение 1 часа. Полученную в результате смесь охлаждали до приблизительно 5°C, добавляли по каплям в течение 30 минут 6 мл 3N HCl. Смесь нагревали до приблизительно 50°C и перемешивали в течение 1 часа.
Полученный в результате твердый остаток фильтровали, применяя воронку Бюхнера со слоем целита, и промывали 20 мл этилацетата. Органический слой объединяли, промывали 20 мл воды и 20 мл соляного раствора, сушили над безводным сульфатом магния и концентрировали при пониженном давлении.
Таким образом полученный маслянистый остаток растворяли в 10 мл ацетонитрила, добавляли 1,5 г метансульфокислоты, перемешивали в течение 12 часов при комнатной температуре. Образовавшийся твердый остаток выделяли фильтрацией, промывали 2 мл холодного ацетонитрила и сушили, получая 1,0 г заявленного в заголовке соединения (выход: 32%) в виде грязно-белого твердого остатка.
1H-ЯМР (CDCl3, смесь кето- и эндоформ, м.д.): δ 0,80-0,99 (м, 6H), 1,16-2,03 (м, 30H), 2,16-2,80 (м, 2H), 3,22 (т, 0,1H, J=5,5 Гц), 3,41 (т, 0,9H, J=5,5 Гц), 4,25-4,40 (м, 0,1H), 4,48-4,62 (м, 0,1H), 4,62-4,75 (м, 0,8H, 6,75-6,96 (ушир.с, 0,1H).
Пример 3
Стадия 1:
Получение (R)-3-[тетрагидропиран-2-илокси]тетрадеканнитрила
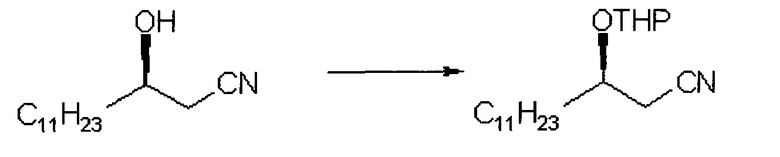
2,16 г (R)-3-гидрокси-тетрадеканнитрила, 1,93 мл дигидропирана и 20 мл тетрагидрофурана помещали в реактор и перемешивали, и добавляли 266 мг п-толуолсульфат пиридиния, нагревали до 50°C и перемешивали в течение приблизительно 2 часов. Смесь охлаждали до комнатной температуры, добавляли 30 мл воды и 30 мл гексана и энергично перемешивали в течение 5 минут. Полученная в результате смесь разделялась на две фазы, и водный слой удаляли. Органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении, получая 2,97 г заявленного в заголовке соединение (выход: 100%) в виде масла.
1H-ЯМР (CDCl3, смесь кето- и эндоформ, м.д.): δ 0,80-0,99 (м, 6H), 1,16-2,03 (м, 30H), 2,16-2,80 (м, 2H), 3,22 (т, 0,1H, J=5,5 Гц), 3,41 (т, 0,9H, J=5,5 Гц), 4,25-4,40 (м, 0,1H), 4,48-4,62 (м, 0,1H), 4,62-4,75 (м, 0,8H, 6,75-6,96 (ушир.с, 0,1H).
Стадия 2:
Получение 5,6-дигидро-3-гексил-4-гидрокси-6-ундецил-2H- пиран-2-она
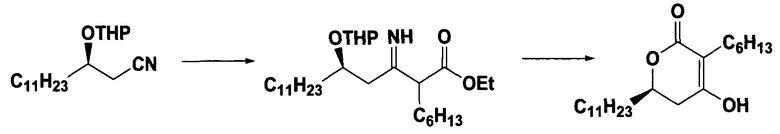
1,27 г активированного цинкового порошка, 2,0 г (R)-3-[тетрагидропиран-2-илокси]тетрадеканнитрила, полученного на стадии 1, и 8 мл тетрагидрофурана помещали в реактор и перемешивали, и добавляли 0,07 мл метансульфокислоты. Затем смесь кипятили с обратным холодильником в течение 1 часа, добавляли по каплям в течение приблизительно 1 часа раствор 2,77 мл этил 2-бромоктаноата, растворенного в 4 мл тетрагидрофурана, и дополнительно кипятили с обратным холодильником в течение 1 часа.
Полученную в результате смесь охлаждали до приблизительно 5 °C, добавляли по каплям в течение 30 минут 8 мл 3N HCl и перемешивали в течение приблизительно 2 часов при 40°C. Полученный в результате твердый остаток фильтровали, применяя воронку Бюхнера со слоем целита, и промывали 5 мл этилацетата. Органический слой объединяли, промывали 50 мл воды, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Таким образом полученный маслянистый остаток растворяли в 8 мл ацетонитрила, добавляли 0,12 г метансульфокислоты, перемешивали в течение 5 часов при комнатной температуре, охлаждали до 5°C и дополнительно перемешивали в течение приблизительно 1 часа. Образовавшийся твердый остаток выделяли фильтрацией, получая 1,46 г заявленного в заголовке соединения (выход: 64%) в виде грязно-белого твердого остатка.
1H-ЯМР (CDCl3, смесь кето- и эндоформ, м.д.): δ 0,80-0,99 (м, 6H), 1,16-2,03 (м, 30H), 2,16-2,80 (м, 2H), 3,22 (т, 0,1H, J=5,5 Гц), 3,41 (т, 0,9H, J=5,5 Гц), 4,25-4,40 (м, 0,1 H), 4,48-4,62 (м, 0,1H), 4,62-4,75 (м, 0,8H, 6,75-6,96 (ушир.с, 0,1H).
Пример 4
Стадия 1:
Получение (R)-3-[триметилсилилокси]тетрадеканнитрила

5 г (R)-3-гидрокситетрадеканнитрила и 35 мл хлористого метилена помещали в реактор и перемешивали. Смесь охлаждали до 0°C, и добавляли 3,87 мл триэтиламина, к которой добавляли в течение приблизительно 30 минут 3,38 мл триметилсилилхлорида, и перемешивали в течение приблизительно 1 часа. Затем полученную в результате смесь медленно нагревали до комнатной температуры и перемешивали в течение приблизительно 1 часа, добавляли 50 мл воды и энергично перемешивали в течение 5 минут. Водный слой удаляли. Органический слой объединяли с 50 мл воды, сушили над безводным сульфатом магния и концентрировали при пониженном давлении, получая 6,66 г заявленного в заголовке соединения (выход: 100%) в виде масла.
1H-ЯМР (CDCl3, м.д.): δ 0,15 (с, 9H), 0,88(т, 3H, J=6,7 Гц), 1,18-1,43(м, 18H), 1,18-1,65(м, 2H), 2,44(дд, 2H, J=5,7 Гц), 3,82-4,00 (м, 1H).
Стадия 2: Получение 5,6-дигидро-3-гексил-4-гидрокси-6-ундецил-2H- пиран-2-она
2.2 г активированного цинкового порошка, 10 мл тетрагидрофурана и 0,02 мл метансульфокислоты помещали в реактор, и полученную в результате смесь кипятили с обратным холодильником в течение 1 часа, к которой медленно добавляли в течение приблизительно 5 минут разбавленный раствор 2 г (R)-3-триметилсилилоокситетрадеканнитрила, полученного на стадии 1, в 4 мл тетрагидрофурана и дополнительно перемешивали в течение приблизительно 5 минут. Добавляли по каплям в течение 30 минут разбавленный раствор 3,6 мл этил 2-бромоктаноата в 4 мл тетрагидрофурана и дополнительно кипятили с обратным холодильником в течение 1 часа.
Полученную в результате смесь охлаждали до приблизительно 5°C, добавляли по каплям в течение 30 минут 6 мл 3N HCl и перемешивали в течение 2 часов при 40°C. Полученный в результате твердый остаток фильтровали, применяя воронку Бюхнера со слоем целита, и промывали 20 мл этилацетата. Органический слой объединяли, промывали 20 мл воды и 20 мл соляного раствора, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Таким образом полученный маслянистый остаток растворяли в 10 мл ацетонитрила, добавляли 1,1 г метансульфокислоты и перемешивали в течение 12 часов при комнатной температуре. Образовавшийся твердый остаток выделяли фильтрацией, получая 1,54 г заявленного в заголовке соединения (выход: 65%) в виде грязно-белого твердого остатка.
1H-ЯМР (CDCl3, смесь кето- и эндоформ, м.д.): δ 0,80-0,99 (м, 6H), 1,16-2,03 (м, 30H), 2,16-2,80 (м, 2H), 3,22 (т, 0,1H, J=5,5 Гц), 3,41 (т, 0,9H, J=5,5 Гц), 4,25-4,40 (м, 0,1H), 4,48-4,62 (м, 0,1H), 4,62-4,75 (м, 0,8H, 6,75-6,96 (ушир.с, 0,1H).
Тогда как настоящее изобретение описано с учетом конкретных вариантов осуществления, ясно, что различные модификации и изменения могут быть сделаны специалистами в данной области техники в настоящем изобретении, которые также включены в объем настоящего изобретения, как описано в формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ (3S,4S)-4-((R)-2-(БЕНЗИЛОКСИ)ТРИДЕЦИЛ)-3-ГЕКСИЛ-2-ОКСЕТАНОНА И ИСПОЛЬЗУЕМОГО ДЛЯ ЭТОГО НОВОГО ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ | 2009 |
|
RU2471790C1 |
| СПОСОБ ПОЛУЧЕНИЯ ВОРИКОНАЗОЛА | 2008 |
|
RU2434009C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГИДРОХЛОРИДА 1-(3,4-ДИХЛОР-2-ФТОРФЕНИЛАМИНО)-7-МЕТОКСИХИНАЗОЛИН-6-ИЛОКСИ)ПИПЕРИДИН-1-ИЛ)ПРОП-2-ЕН-1-ОНА И ИСПОЛЬЗУЕМЫХ В НЕМ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2012 |
|
RU2563630C1 |
| РЕАКЦИИ МАКРОЦИКЛИЗАЦИИ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И ДРУГИЕ ФРАГМЕНТЫ, ПРИГОДНЫЕ В ПОЛУЧЕНИИ АНАЛОГОВ ХАЛИХОНДРИНА B | 2014 |
|
RU2710545C2 |
| С-ГЛИКОЗИДНЫЕ ПРОИЗВОДНЫЕ, СОДЕРЖАЩИЕ КОНДЕНСИРОВАННОЕ ФЕНИЛЬНОЕ КОЛЬЦО, ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ПОЛУЧЕНИЯ ТАКОВЫХ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ТАКОВЫЕ | 2017 |
|
RU2739024C2 |
| ИНГИБИТОР DPP-IV, ВКЛЮЧАЮЩИЙ БЕТА-АМИНОГРУППУ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРЕДУПРЕЖДЕНИЯ И ЛЕЧЕНИЯ ДИАБЕТА ИЛИ ОЖИРЕНИЯ | 2008 |
|
RU2419615C1 |
| НОВОЕ ТРИЦИКЛИЧЕСКОЕ ПРОИЗВОДНОЕ ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2009 |
|
RU2470934C1 |
| СПОСОБ ПОЛУЧЕНИЯ КИСЛОТЫ МОНТЕЛУКАСТ В ИОННОЙ ЖИДКОЙ СРЕДЕ | 2008 |
|
RU2436774C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИФЕНИЛМЕТАНА | 2017 |
|
RU2797392C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИФЕНИЛМЕТАНА | 2023 |
|
RU2814846C1 |
Настоящее изобретения относится к способу получения (6R)-3-гексил-4-гидрокси-6-ундецил-5,6-дигидропиран-2-она ,включающему:
1) взаимодействие соединения формулы (II) с соединением формулы (III) в присутствии цинка для получения соединения формулы (IV);
2) обработку соединения формулы (IV) водной кислотой для получения соединения формулы (V); и 3) циклизацию соединения формулы (V) в
кислых или основных условиях:

где Р' представляет собой водород или защитную группу, выбранную из триалкилсилила и алкоксиалкила, которые легко удаляются в кислых условиях, и R представляет собой метил, этил или пропил, а также к промежуточному (5R)-2-гексил-5-гидрокси-3-иминогексадеканоатному производному (IV), применяемому в этом способе, и способу его получения. (6R)-3-гексил-4-гидрокси-6-ундецил-5,6-дигидропиран-2-он применяется в синтезе оксетанов, таких как тетрагидролипстатин. 3 н. и 10 з.п. ф-лы.
1. Способ получения (6R)-3-гексил-4-гидрокси-6-ундецил-5,6-дигидропиран-2-она формулы (I), включающий:
1) взаимодействие соединения формулы (II) с соединением формулы (III) в присутствии цинка для получения соединения формулы (IV);
2) обработку соединения формулы (IV) водной кислотой для получения соединения формулы (V); и
3) циклизацию соединения формулы (V) в кислых или основных условиях:



в которых Р' представляет собой водород или защитную группу, выбранную из триалкилсилила и алкоксиалкила, которая легко удаляется в кислых условиях, и R представляет собой метил, этил или пропил.
2. Способ по п.1, в котором Р' представляет собой водород, и R представляет собой метил, этил или изопропил.
3. Способ по п.1, в котором алкоксиалкильный остаток представляет собой тетрагидропиранил, 4-метокситетрагидропиранил, 2-метокси-2-пропил или 1-этокси-1-этил.
4. Способ по п.1, в котором соединение формулы (III) представляет собой метил 2-бромоктаноат, этил 2-бромоктаноат или пропил 2-бромоктаноат.
5. Способ по п.1, в котором цинк используют в количестве в диапазоне 1-10 молярных эквивалентов на 1 моль соединения формулы (II), применяемого на стадии 1).
6. Способ по п.1, в котором стадия 1) дополнительно включает добавление добавки, выбранной из группы, состоящей из I2, 1,2-этандибромида, метансульфокислоты и триметилсилилхлорида.
7. Способ по п.1, в котором стадия 1) дополнительно включает добавление свинца.
8. Способ по п.1, в котором стадию 1) осуществляют в растворителе, выбранном из группы, состоящей из этилового эфира, тетрагидрофурана, 1,4-диоксана, 1,2-диметоксиэтана, ацетонитрила, метилацетата, этилацетата, бензола, толуола и их смеси.
9. Способ по п.1, в котором кислота, используемая на стадии 2) представляет собой неорганическую кислоту, выбранную из группы, состоящей из хлороводородной кислоты, серной кислоты и бромоводородной кислоты; органическую кислоту, выбранную из группы, состоящей из толуолсульфокислоты, метансульфокислоты и камфорсульфокислоты; или их смеси.
10. Способ по п.1, в котором количество кислоты, используемой на стадии 2) находится в диапазоне 0,01-5 молярных эквивалентов на 1 моль соединения формулы (IV).
11. Способ по п.1, в котором стадию 2) осуществляют в протонном растворителе, выбранном из группы, состоящей из воды, метанола и этанола; в апротонном растворителе, выбранном из группы, состоящей из этилового эфира, тетрагидрофурана, 1,4-диоксана, 1,2-диметоксиэтана, ацетонитрила, метилацетата, этилацетата, бензола и толуола; или их смеси.
12. (5R)-2-гексил-5-гидрокси-3-иминогексадеканоатное производное формулы (IV):
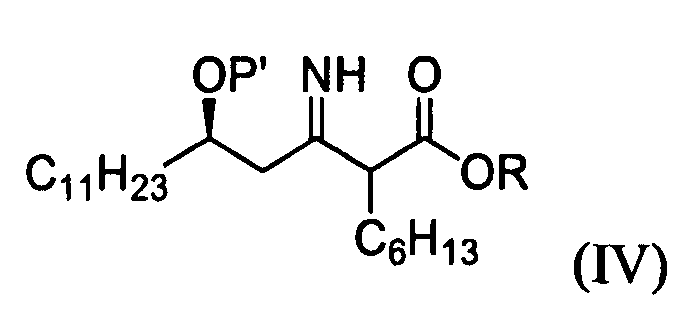
в котором Р' представляет собой водород или защитную группу, выбранную из триалкилсилила и алкоксиалкила, которые легко удалить в кислых условиях, и R представляет собой метил, этил или пропил.
13. Способ получения (5R)-2-гексил-5-гидрокси-3-иминогексадеканоатного производного формулы (IV), включающий взаимодействие соединения формулы (II) с соединением формулы (III) в присутствии активированного цинка:


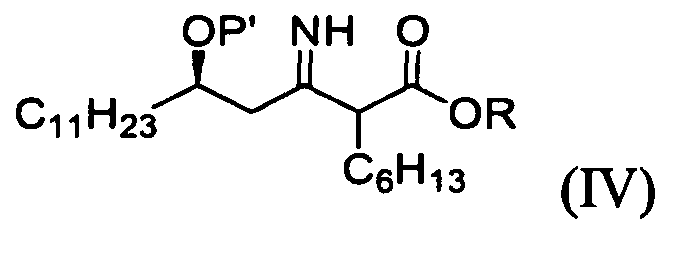
в которых Р' представляет собой водород или защитную группу, выбранную из триалкилсилила и алкоксиалкила, которые легко удалить в кислых условиях, и R представляет собой метил, этил или пропил.
| John J.Landi et al, Tetrahedron Lttters, 1993, 34(2), c.277-280 | |||
| US 6545165 В1, 08.04.2003 | |||
| 4-ГИДРОКСИПИРАН-2-ОНЫ, ЦИКЛООКТИЛ-ИЛИ БЕНЗОПИРАН-2-ОНЫ, 4-ГИДРОКСИ-2Н-ПИРАН-2-ОНЫ И 4-ГИДРОКСИ-ЦИКЛООКТАПИРАН-2-ОНЫ | 1995 |
|
RU2139284C1 |
| Способ получения производных 2н-пиран-2,6(3н)-диона или их солей | 1976 |
|
SU679142A3 |
| 3-Циано-4,6,6-триметил-5,6-дигидро-2-пирон, проявляющий противовоспалительную активность | 1983 |
|
SU1197412A1 |

