Область техники, к которой относится изобретение
Настоящее изобретение относится к способу получения трийодированных ароматических соединений. В частности, изобретение относится к способу введения трех атомов йода с применением активированного молекулярного йода, в молекулы 3,5-дизамещенных фенолов с получением соответствующих 3,5-дизамещенных-2,4,6-трийодфенолов, которые представляют собой интермедиаты, применяемые для синтеза рентгеноконтрастных агентов, а также к общему способу получения самих рентгеноконтрастных агентов.
Уровень техники
Йодированные рентгеноконтрастные средства являются хорошо известными соединениями, широко применяемыми в методиках диагностики, основанных на получении рентгеновских изображений. Подходящие примеры указанных соединений приведены, например, в WO 2009/103666 (Bracco) и цитированной в этой заявке литературе.
Общим признаком подобных соединений является то, что химическая структура большинства из них включает трийодированное ароматическое ядро, которое обеспечивает повышенный эффект контрастирования. Хотя получение этих контрастных агентов может осуществляться целым рядом способов, оно включает в качестве обязательной стадии йодирование ароматического субстрата, главным образом 5-аминоизофталевых групп, которое проходит по свободным положениям 2, 4 и 6 и приводит к соответствующим производным 3,5-дизамещенного-2,4,6-трийоданилина, которые затем подвергают дальнейшей обработке, превращая в конечный продукт, например, как раскрыто в US 5075502.
В качестве альтернативы применяют полийодирование подходящих 3,5-дизамещенных фенолов, которое приводит к получению соответствующих 3,5-дизамещенных-2,4,6-трийодфенолов, которые затем можно подвергнуть дальнейшей обработке с получением ожидаемых конечных агентов, путем так называемой перегруппировки Смайлса.
Для общего ознакомления с описанным выше путем синтеза и перегруппировкой Смайлса смотрите, например, WO 88/09328, WO 97/05097 и WO 00/32561 (Bracco).
Реакцию йодирования можно проводить по различным методикам, известным в технике. При этом, методики, применяемые в настоящее время для получения радиографических контрастирующих агентов, т.е. йодирование ароматического субстрата, как правило, реализуют, используя растворы хлорида йода (ICl) в концентрированной хлористоводородной кислоте (HCl) при высокой температуре или, в качестве альтернативы, с использованием аналогичных йодирующих агентов, например, KICl2 или NaICl2 в водном растворе; для общего ознакомления смотрите US 3914294 (Squibb), WO 92/14695 (Guerbet), US 5013865 (Mallinckrodt), WO 96/37458 и WO 96/37459 (Fructamine).
Основные недостатки описанных выше способов связаны с исключительно кислой средой проведения реакций, причем по ходу проведения реакции кислотность еще больше возрастает, из-за выделения HCl в ходе реакции, коррозионными свойствами йодирующих агентов и ограниченным сроком их хранения.
В качестве примера, йомепрол - хорошо известный радиографический контрастирующий агент, широко применяемый в повседневной диагностической практике, можно получать йодированием ключевого интермедиата формулы
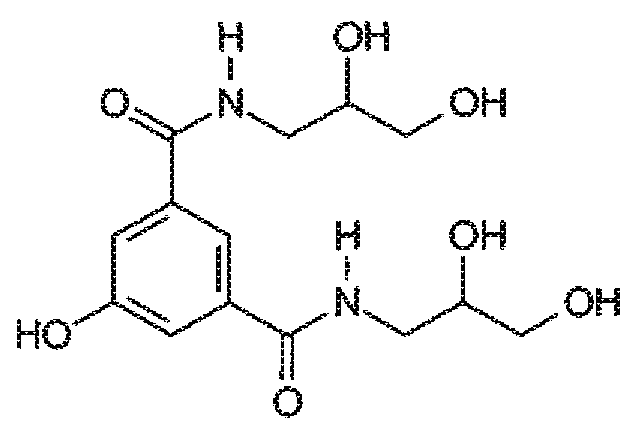
с получением соответствующего йодированного производного формулы
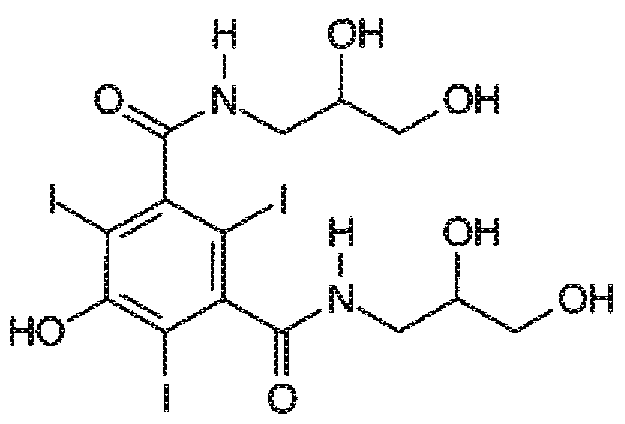
Эту реакцию йодирования, как правило, проводят с использованием водного раствора KICl2 или NaICl2 в качестве йодирующего агента, поддерживая pH реакционной среды примерно 9,5 с помощью подходящего основания, как правило, NaOH, например, как раскрыто в EP 185130.
В качестве альтернативы, йодирование фенольного субстрата проводят с использованием раствора ICl в качестве йодирующего агента (состав: 44,5% I и 14% HCl масс./масс. в H2O), в водной среде, поддерживая значение pH от 6 до 7 добавлением основания, предпочтительно NaOH, при температуре 25°C, например, как описано в WO 00/32561.
При этом очевидно, что при работе в промышленном масштабе основные проблемы появляются из-за необходимости иметь дело и, что более важно, нейтрализовать сильную кислотность используемых йодирующих агентов. С этой целью на практике требуются очень большие количества NaOH для нейтрализации либо HCl, имеющегося в йодирующем растворе, либо HCl, образующегося в ходе реакции.
Кроме того, поскольку нейтрализация такой сильной кислоты является чрезвычайно экзотермической, необходимость поддерживать температуру реакционной смеси около 25°C заставляет осуществлять добавление реагента в течение продолжительного времени, необходимого для предотвращения резкого неуправляемого повышения температуры, хотя реакция йодирования проходит почти мгновенно.
В свете вышеизложенного были предприняты попытки, направленные на разработку методик йодирования, альтернативных применению хлорида йода или его производных. В этом контексте следует отметить способы электрохимического йодирования подходящих ароматических субстратов, например, раскрытые в WO 96/37461, US 3833490 и WO 2009/103666.
В качестве альтернативы Patiel et al., в Tetrahedron Letters 2005, 46, 7179-7181 было предложено монойодирование орто-гидроксизамещенных ароматических карбонильных соединений молекулярным йодом, активированным применением соответствующего сильного окислителя, в т.ч. йодноватой кислоты. Возможность применения такой же йодирующей системы для дальнейшего получения орто/пара дийодированных орто-гидрокси ароматических карбонильных производных была предложена теми же авторами в ARKIVOC 2006, 104-108. В обеих работах в качестве растворителя для проведения реакции применяли имеющийся в продаже 95% водный этанол.
Кроме того, в ES 528109 раскрыто получение 2,4,6-трийодфенолов с применением йода, активированного H2O2 (30%) в метаноле, подкисленном H2SO4 при нагревании до 60°C, причем указанный в источнике выход составил 44%.
Сущность изобретения
Настоящее изобретение относится к способу введения трех атомов йода в молекулы 3,5-дизамещенных фенолов или их солей, которое осуществляют в водной среде с применением молекулярного йода, активированного присутствием соответствующего окислителя, как правило, йодноватой кислоты, а также к улучшенному способу получения рентгеноконтрастных агентов, включающему упомянутую выше стадию йодирования.
Краткое описание иллюстративного материала
Фиг.1: Пример 1: ВЭЖХ реакционного раствора после проведения реакции в течение 6 часов (конечный раствор).
Фиг.2: Пример 6: ВЭЖХ реакционного раствора после проведения реакции в течение 6 часов (конечный раствор).
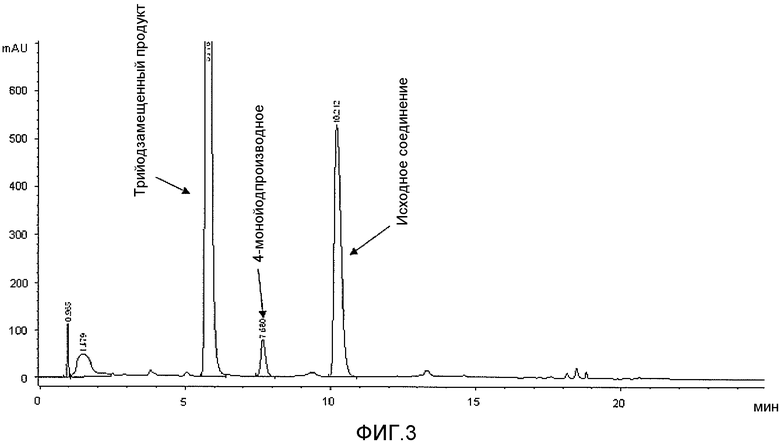
Фиг.3: Сравнительный пример 1: хроматограмма (ВЭЖХ) реакционного раствора через 1,5 часа при 38-40°C.
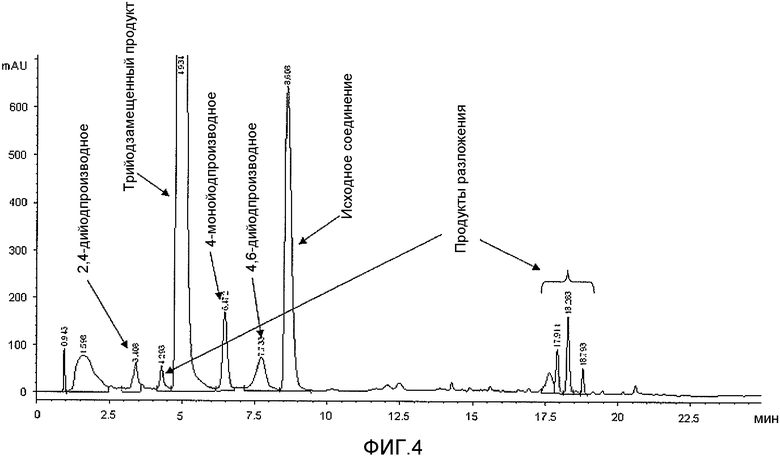
Фиг.4: Сравнительный пример 1: ВЭЖХ реакционного раствора через 3,5 часа при 38-40°C.
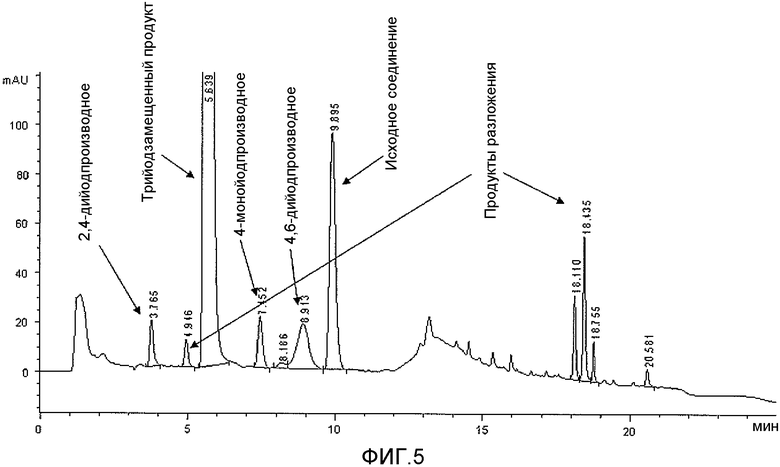
Фиг.5: Сравнительный пример 1: ВЭЖХ маточных растворов после осаждения трийодированного продукта.
Подробное описание изобретения
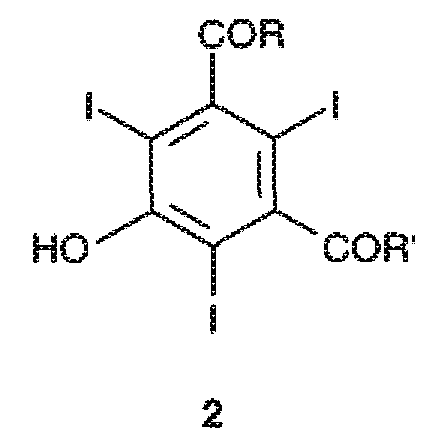
Первым предметом настоящего изобретения является способ получения трийодфенолов формулы 2,
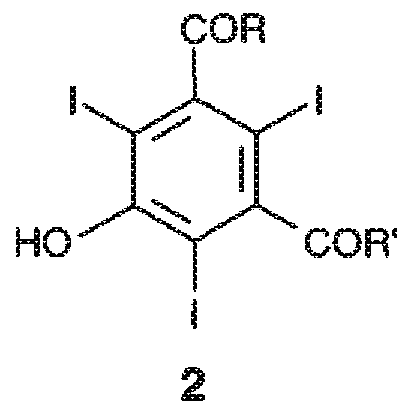
где указанный способ включает йодирование 3,5-дизамещенного фенола формулы 1 или его соли молекулярным йодом в присутствии йодноватой кислоты,
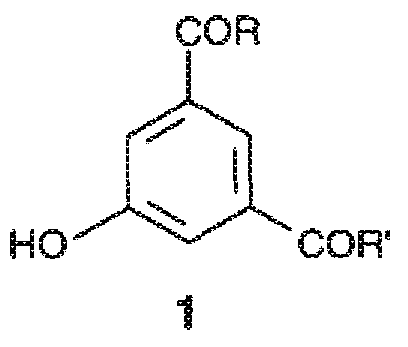
где:
заместители R и R', которые являются одинаковыми или различными, представляют собой группы формулы -NHR1 или формулы -NR2R3, где каждая из групп R1, R2 и R3, независимо от других, представляет собой линейную или разветвленную C1-C6 алкильную группу, которая необязательно замещена одной или несколькими группами, выбранными из гидроксила (-OH), C1-C5 алкокси или гидроксиалкоксигрупп.
Способ йодирования по настоящему изобретению обычно осуществляют в водной среде.
В настоящем описании, если не указано иное, под термином «линейная или разветвленная C1-C6 алкильная группа» подразумевается линейная или разветвленная алкильная цепь, включающая от 1 до 6 атомов углерода. Подходящие примеры алкильных групп включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, н-гексил и т.п.
Перечисленные выше алкильные группы могут быть дополнительно замещены одной или несколькими гидрокси-, алкокси- или гидроксиалкокси-группами, как указано выше.
Под термином «C1-C5 алкокси» подразумевается любая алкилокси-группа, алкильный фрагмент которой представляет собой любую из указанных выше линейных или разветвленных алкильных групп.
Под «гидроксиалкокси группой» подразумевается любая из указанных выше C1-C5 алкоксигрупп, где алкильный фрагмент дополнительно замещен одной или несколькими гидроксильными группами.
Подходящие примеры алкокси или гидроксиалкокси групп по настоящему изобретению включают, например, метокси, этокси, н-пропокси, изопропокси, н-пентокси, 2-гидроксиэтокси, 2,3-дигидроксипропокси, 1,3-дигидроксиизопропокси и т.п.
Согласно предпочтительному варианту осуществления способа по настоящему изобретению, в соединениях формулы 1 и 2, заместители R и R', которые являются одинаковыми или отличаются друг от друга, представляют собой группы, выбранные из -NHR1 или NR2R3, где каждый из заместителей R1, R2 и R3 независимо от других представляет собой линейную или разветвленную C1-C4 алкильную группу, необязательно замещенную одной-тремя гидроксильными группами, например, 1,3-дигидроксиизопропил, 2,3-дигидроксипропил, 1,3-дигидрокси-2-метилизопропил или 2,3,4-тригидроксибутил.
Еще более предпочтительно, в соединениях формул 1 и 2 заместители R и R', которые являются одинаковыми или отличаются друг от друга, представляют собой группы, выбранные из:
-NHCH3,
-NHCH2-CH(OH)-CH2OH,
-NHCH(CH2OH)2 и
-N(CH3)-CH2-CH(OH)-CH2OH.
Поскольку обе группы R и R' не принимают непосредственного участия в реакции, как подробно описано ниже, из изложенного выше специалисту в данной области ясно, что замещающие группы, необязательно присутствующие в заместителях R и R', которые могут вступать в нежелательные побочные реакции, перед проведением реакции следует надлежащим образом защитить.
Введение и последующее снятие защиты указанных групп можно выполнить целым рядом способов, хорошо известных в технике и обычно применяемых в методиках органического синтеза. Для общего ознакомления с применением защитных групп в органической химии см., например, T.W.Green, Protective Groups in Organic Synthesis (Wiley, N.Y. 1981).
Способ по настоящему изобретению является особенно предпочтительным, поскольку он обеспечивает возможность практически полного трийодирования производного фенола формулы 1 или соответствующей соли и позволяет получить трийодированное производное формулы 2, которое, по крайней мере в значительной степени, не загрязнено побочными продуктами, образовавшимися в результате неполного йодирования ароматического цикла, или какими-либо другими примесями.
Таким образом, преимущество способа по настоящему изобретению заключается в возможности избежать очистки трийодированного соединения; фактически продукт уже удовлетворяет аналитическим стандартам для промышленно производимых промежуточных продуктов в реакционном растворе и, следовательно, его можно применять в том виде, в котором он был получен, без выделения и очистки, в следующей стадии реакции для получения целевого йодированного средства.
Как упоминалось выше, в способе по настоящему изобретению, реакция йодирования, приводящая к образованию трийодфенолов формулы 2, происходит при действии молекулярного йода (I2) в присутствии HIO3 по хорошо известному механизму электрофильного замещения.
В описанных выше условиях частицами, осуществляющими эффективное йодирование, вероятно, являются катионы йода (I+), которые образуются при добавлении молекулярного йода (I2), в то время как нереакционноспособные йодид-противоионы (I-) вновь легко окисляются под действием HIO3 до молекулярного йода или даже до катионов йода с более высокой степенью окисления, что позволяет им принимать участие в дальнейшем йодировании ароматического цикла.
Соответственно, в качестве альтернативы йодноватой кислоте, которая, однако, особенно предпочтительна в способе по настоящему изобретению, можно предложить окислители, способные вновь окислять йодид-ионы (I-) до молекулярного йода, включая, например, азотную кислоту, серную кислоту, триоксид серы, пероксид водорода, озон и т.п.
Фактически, если применяется молекулярный йод в присутствии йодноватой кислоты, нереакционноспособные йодид-ионы, образовавшиеся при реакции йодирования, вновь превращаются в молекулярный йод в результате т.н. реакции Дашмана согласно приведенной ниже схеме реакции 1
IO3 - + 5I- + 6H+ → 3I2 + 3H2O
которая, кроме того, ведет к одновременному восстановлению йодат-иона (IO3 -) в молекулярный йод, который затем принимает участие в йодировании ароматического цикла (смотрите, например, Furuichi, R. and Liebhafsky, H.A. Radioactive iodine exchange and the Dushman reaction. Bull. Chem. Soc. Japan 1973, 46, 2008-2010 and Bull. Chem. Soc. Japan 1975, 48, 745-750).
В результате, полное трийодирование 3,5-замещенного фенольного субстрата формулы 1 с получением желаемого трийодзамещенного соединения формулы 2 достигается при полном расходовании стехиометрического количества йодирующих частиц, где указанное количество равно сумме введенных в реакцию I2 и HIO3, при образовании воды в качестве единственного побочного продукта реакции, как показано на следующей общей схеме реакции 2.
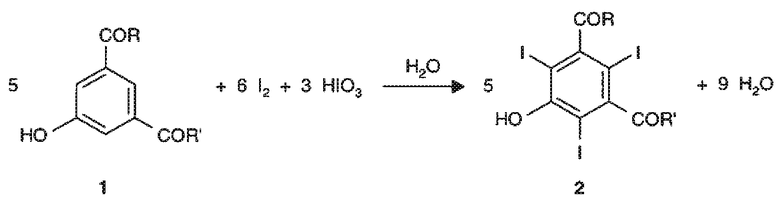
Это означает, что, предпочтительно, совместное применение йода и йодноватой кислоты в способе йодирования по настоящему изобретению обеспечивает возможность исчерпывающего трийодирования ароматического субстрата формулы 1, избегая, с одной стороны, необходимости применения какого-либо избытка йодирующего агента, в частности, молекулярного йода, и, с другой стороны, образования побочных продуктов, в частности, нереакционноспособных полийодид-ионов, например, ионов I3 -, которые, в основном, образуются из комбинации I2 и йодид-ионов.
Следует отметить, что единственной кислотой, входящей в йодирующую смесь по настоящему изобретению, является HIO3, т.е. твердая кислота, которую можно приобрести в свободной продаже в виде готового к применению концентрированного водного раствора, который является значительно менее сильной кислотой и проще для применения, чем соляная кислота (HCl), используемая в современном промышленном способе йодирования.
В этой связи следует отметить, что вся кислотность, связанная со способом йодирования по настоящему изобретению, а именно протоны, поступающие в реакционную смесь из добавленной HIO3, либо образующиеся при реакции йодирования, преимущественно расходуются в упомянутой окислительно-восстановительной реакции Дашмана, протекающей согласно приведенной выше схеме 1. В результате возникает очень благоприятная ситуация, когда pH реакционной смеси самопроизвольно сохраняет желаемое значение в ходе йодирования и нет необходимости добавлять какой-либо нейтрализующий основной раствор, что сопровождается экзотермической реакцией, и, кроме того, удается избежать какого бы то ни было нежелательного разбавления раствора.
Другими словами, способ йодирования по настоящему изобретению позволяет избежать, с одной стороны, применения сильно кислых йодирующих смесей и, с другой стороны, реакция протекает с поглощением всей кислотности, связанной с самим процессом йодирования, как привносимой йодирующим агентом, так и образующейся при реакции йодирования. Следовательно, этот способ дает возможность преодолеть основные недостатки, связанные со способами йодирования, применяемыми в настоящее время, которые требуют регулировать и ограничивать выделение большого количества тепла при реакции нейтрализации с помощью раствора основания, который добавляют совместно с кислотной йодирующей смесью для поддержания pH реакционной среды на желаемом нейтральном уровне.
По причине вышеизложенного, способ по настоящему изобретению дополнительно позволяет, с промышленной точки зрения, значительно сократить общее время процесса йодирования в общей сложности до менее чем 10 часов и, предпочтительно, от 5 до 9 часов.
Кроме того, поскольку не требуется большое количество основного раствора, способ по настоящему изобретению дает возможность добиться высоких концентраций (неочищенной реакционной смеси) и значительно уменьшить количество образующихся солей, а именно NaCl. Этот аспект становится еще более актуальным из-за необходимости обработки и сброса сточных вод, образующихся при промышленном производстве.
Из приведенной ранее общей схемы 2 следует, что способ йодирования по настоящему изобретению требует применения по крайней мере 3 молей йодирующих частиц, под которыми подразумевается, как указано выше, сумма I2 и HIO3, на каждый моль ароматического субстрата 1.
С учетом этого факта, в способе по настоящему изобретению йодирование фенольного субстрата проводят с применением как минимум одного моля молекулярного йода на каждый моль 3,5-дизамещенного фенола формулы 1. Предпочтительно, молярное соотношение между йодом и 3,5-дизамещенным фенольным субстратом 1 [I2/1] должно находиться в пределах от 1,1 до 1,3; еще более предпочтительно, трийодирование 3,5-дизамещенного фенольного субстрата йодом и йодноватой кислотой должно проводиться с применением 1,2 моля йода на моль субстрата 1.
С другой стороны, вследствие стехиометрии указанной реакции, мольное отношение между I2 и йодноватой кислотой должно быть равно по крайней мере 1:0,5, тогда как мольное отношение между йодноватой кислотой и 3,5-дизамещенным фенольным субстратом 1 [HIO3/1] должно находиться в пределах от 0,4 до 0,8.
Соответственно, в особенно предпочтительном варианте осуществления настоящего изобретения, трийодирование 3,5-дизамещенного фенольного субстрата 1 йодом и йодноватой кислотой должно проводиться с применением мольного соотношения 3,5-дизамещенный фенольный субстрат:йод:йодноватая кислота, равного 1:1,2:0,6.
Однако, как указано в экспериментальном разделе, со столь же хорошими результатами можно, необязательно, использовать небольшой избыток, например, 1 мольн.%, йодирующего агента, т.е. либо йода, либо йодноватой кислоты, по сравнению с минимально необходимым стехиометрическим количеством.
При этом к полученной реакционной смеси можно добавить, например, минимальное количество бисульфита натрия для ликвидации возможно присутствующего остатка йодирующего агента. В этом случае оптимальное количество (бисульфита) может быть определено, например, потенциометрически.
Как указано выше, способ йодирования по настоящему изобретению, который включает применение йодирующей системы I2/HIO3, преимущественно реализуют в водной среде, например, в воде или водных растворителях, включая водные солевые растворы или их смеси с органическими растворителями, как, например, низшими спиртами, в т.ч. метанолом или этанолом, диоксаном или гликолями, например, диэтиленгликолем или триэтиленгликолем и их метиловыми эфирами. В последнем случае количество органического растворителя в водной смеси выбирают таким образом, чтобы не менялась общая растворимость как фенольного субстрата или его соли, так и трийодированного продукта в неочищенном растворе.
Предпочтительными растворителями являются вода и водные растворы, например, солевые водные растворы.
При этом применение воды или водного растворителя вместо органического растворителя, применяемого в известном уровне техники, при использовании в качестве йодирующей системы активированного йода, выгодно отличается, в частности, с точки зрения стоимости и воздействия на окружающую среду.
Кроме того, применение водного растворителя благоприятным образом предотвращает необходимость экстракции соединения 1 из водной среды, в которой его, как правило, получают в промышленности, согласно, способам, например, EP 185130 или WO 00/32561, для осуществления его йодирования в органической среде, которая применяется вместо водной среды в указанных литературных источниках.
Аналогично, если получен йодированный продукт 2, использование водной смеси предотвращает необходимость его выделения из органической реакционной смеси для последующего превращения в желаемый радиографический агент в водной среде, которая обычно применяется в промышленных способах, используемых в настоящее время.
Кроме того, неожиданно оказалось, что применение водного растворителя в способе по настоящему изобретению дает возможность решить проблему низкого выхода продукта реакции, который достигается в упомянутых способах известного уровня техники с применением активированного йода в органическом растворителе, что подтверждается в сравнительном примере 1 приведенного ниже экспериментального раздела, причем указанный низкий выход можно объяснить как неполным превращением ароматического субстрата, так и хорошей растворимостью трийодзамещенного продукта в выбранной спиртовой смеси, что предотвращает его полное осаждение или кристаллизацию из реакционного раствора, как показано на фиг.3-5.
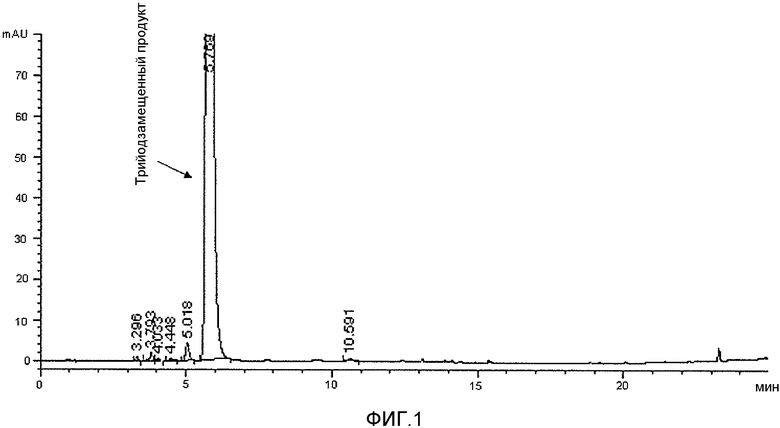
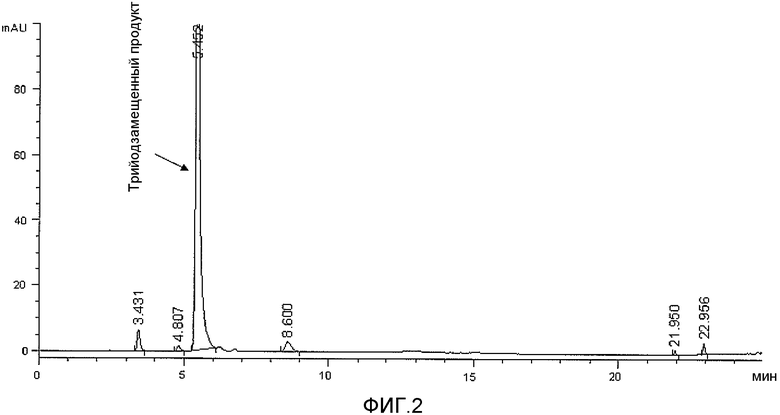
Фактически, применение водного растворителя согласно способу настоящего изобретения обеспечивает возможность почти исчерпывающего трийодирования ароматического субстрата и получения трийодзамещенного продукта, который является практически чистым в реакционном растворе, как видно из фиг.1 и 2. В результате способ по настоящему изобретению не требует проведения каких-либо стадий выделения и очистки йодированного соединения, которое, будучи получено с очень хорошим выходом и высокой чистотой в реакционном растворе, может без дополнительной очистки применяться в следующей стадии реакции для получения целевого йодированного агента. Поэтому с успехом удается избежать любых возможных потерь йодированного продукта при его выделении и/или очистке.
Исходя из сказанного выше, и в соответствии с особенно предпочтительным вариантом осуществления настоящего изобретения, трийодирование 3,5-дизамещенного фенольного субстрата с применением йода и йодноватой кислоты проводят непосредственно в реакционном водном растворе, образующемся в промышленном способе получения желаемого контрастного агента, в котором фенольный субстрат обычно присутствует в виде натриевой соли.
Способ йодирования по настоящему изобретению в основном включает: получение водного раствора 3,5-дизамещенного фенольного субстрата формулы 1 или его соли, используемого в качестве исходного материала, и добавление к указанному раствору твердого I2 и HIO3.
При осуществлении указанного способа, поддерживают температуру ниже 70°C, предпочтительно в пределах от 20 до 70°C и более предпочтительно от 40 до 60°C.
Более конкретно, основные стадии способа по настоящему изобретению включают:
I) получение водного раствора 3,5-дизамещенного фенольного субстрата формулы 1 или его соли, который применяется в качестве исходного материала,
II) добавление твердого I2 к указанному водному раствору, нагретому до температуры от 20 до 70°C, и затем
III) добавление йодноватой кислоты.
В одном из вариантов осуществления настоящего изобретения, стадия I) описанного выше способа включает получение раствора 3,5-дизамещенного фенольного субстрата формулы 1 или его соли, используемых в виде чистого вещества, в водном растворителе, как правило, воде, и применение этого раствора в качестве исходного материала. Предпочтительно, указанный исходный раствор имеет концентрацию в пределах от 24 до 10% (масса/масса) и pH в диапазоне от 9 до 10.
При этом, и если иное не указано в настоящем описании, подходящие соли фенольного субстрата формулы 1 предпочтительно выбирают из солей субстратов со щелочными или щелочноземельными металлами, например, солей натрия, лития, калия, кальция или магния.
Среди этих солей особенно предпочтительными являются натриевые соли 3,5-дизамещенных фенольных субстратов, которые могут применяться сами по себе, т.е. в виде чистых соединений или, в качестве альтернативы, в виде неочищенного раствора, получаемого непосредственно в промышленном способе производства трийодированных контрастных агентов, например, йомепрола, осуществляемого, например, как описано в WO 00/32561. Согласно особенно предпочтительному варианту осуществления настоящего изобретения, водный раствор, применяемый в качестве исходного материала, представляет собой неочищенный водный раствор, получаемый непосредственно в промышленном способе производства желаемого контрастного агента, обычно содержащий исходный 3,5-дизамещенный фенольный субстрат в виде натриевой соли в концентрации от 20 до 25% (масс./масс.).
В этом случае указанный неочищенный раствор, который обычно имеет pH в пределах от 9 до 10, может применяться в неразбавленном состоянии или, необязательно, после разбавления, как правило, водой, например, до половины исходной концентрации.
Затем к раствору фенольного субстрата, предварительно нагретому до температуры менее 70°C, предпочтительно от 20 до 70°C, и более предпочтительно от 30 до 60°C, добавляют твердый I2. При этом практикующему специалисту в данной области должно быть ясно, что как только йод добавлен к нагретому раствору фенольного субстрата, начинается реакция йодирования, протекающая по хорошо известному механизму электрофильного замещения, например, за счет действия ионов I+, образующихся из добавленного йода, которая приводит к образованию ионов H+. В результате pH реакционной смеси снижается с исходных основных значений до величин даже ниже нейтральных.
Затем к реакционной смеси добавляют необходимое количество йодноватой кислоты.
При этом HIO3 предпочтительно добавляют к реакционной смеси, когда pH среды достигает значения в диапазоне от 4,5 до 7 и, предпочтительно, от 5 до 6. В особенно предпочтительном варианте осуществления настоящего изобретения к реакционной смеси добавляют соответствующее количество йодноватой кислоты, когда pH среды достигает значения в пределах от 5 до 5,5.
Интересно отметить, что в данном случае, в противоположность хорошо известному в технике положению, что реакции электрофильного замещения значительно активируются у фенолов, которые находятся в депротонированной (фенатной) форме, и что мольная доля этой фенатной формы возрастает с увеличением pH раствора, авторы изобретения обнаружили, что при указанных выше значениях pH раствора, которые, очевидно, являются неблагоприятными, производные 3,5-дизамещенных-2,4,6-трийодфенолов формулы 2 неожиданно образуются с более высокими выходами и чистотой.
При этом необходимое количество йодной кислоты можно добавить к реакционной смеси сразу или, в качестве альтернативы, постепенно, в течение периода до 4 часов, либо непрерывно, либо порциями с помощью стандартных средств, что приводит к постепенному превращению исходного соединения в соответствующее трийодзамещенное производное. Более конкретно и в соответствии с приведенным ниже экспериментальным разделом, йодноватую кислоту можно добавлять быстро, например, в течение периода времени до двух часов, к исходным растворам, нагретым до температур, например, в диапазоне от 55 до 65°C и предпочтительно до примерно 60°C. Вместо этого, если исходный раствор нагрет до более низкой температуры, например, в диапазоне от 20 до 50°C, предпочтительным является более медленное добавление йодноватой кислоты, причем это добавление можно осуществлять в течение периода времени до 4 часов.
При этом можно с успехом применять водный раствор окислителя, имеющий концентрацию в диапазоне, например, от 30 до 55% (масс./масс.).
Интересно отметить, что при проведении реакции в описанных выше условиях, pH реакционной среды самопроизвольно сохраняется на желаемом значении, а именно в диапазоне от 5 до 5,5, в течение всего периода добавления HIO3 и последующего периода завершения реакции, и не требует какой-либо коррекции с помощью кислотных или основных растворов.
Этот факт дает возможность уменьшить до минимума содержание всех частично йодированных побочных продуктов, а также всех примесей, возникающих например, из-за возможного диспропорционирования йода, которое преимущественно имеет место в щелочной среде, или из-за возможного избытка HIO3, и/или чрезмерно высокой окисляющей способности йодноватой кислоты, которая, напротив, проявляется при более низких значениях pH.
В результате в реакционной смеси образуется трийодзамещенный продукт формулы 2 с хорошими выходами и высокой чистотой, предпочтительно равной или превышающей 98%, который, вследствие этого, может без обработки применяться в следующей стадии получения желаемого радиографического контрастного агента, без необходимости какого-либо выделения и дополнительной очистки.
В данном случае чистоту трийодзамещенного соединения в полученном реакционном растворе можно определить хроматографически, например, с помощью методики ВЭЖХ, либо в виде % площади под кривой, либо относительно стандарта, который, как правило, представляет собой чистый выделенный 3,5-дизамещенный-2,4,6-трийодфенол.
Хотя выделение трийодзамещенного продукта формулы 2, если это желательно, можно осуществить по методикам, известным в органической химии, например, включающим применение ионообменных смол или электродиализ, или фильтрование через мембрану и концентрирование реакционной смеси, согласно особенно предпочтительному варианту осуществления настоящего изобретения, неочищенный раствор трийодзамещенного продукта формулы 2, полученный с помощью способа йодирования по настоящему изобретению, применяется в том виде, в котором он был получен, в следующей стадии синтеза желаемого радиографического агента без какого-либо предварительного выделения или дополнительной очистки йодированного соединения, содержащегося в полученном растворе.
В случае работы при указанных выше температурах, способ по настоящему изобретению не должен приводить к значительным потерям за счет испарения водного растворителя. Напротив, нагревание реакционной смеси при более высоких температурах необязательно могло бы привести к частичной сублимации молекулярного йода. Однако при сохранении температуры реакционной смеси в указанных выше пределах, йодирование обычно протекает без значительных потерь этого реагента. Тем не менее можно также применять обычные охлаждающие или конденсирующие устройства для улавливания сублимированного йода, который затем возвращают в реакционную смесь, необязательно с применением небольшого количества растворителя.
Подробное описание способа по настоящему изобретению приведено в помещенном ниже по тексту экспериментальном разделе, например, в примерах 1-7, относящихся к йодированию 3,5-дизамещенных фенолов по настоящему изобретению.
Однако с точки зрения осуществляемых операций, основные стадии и предпочтительные условия осуществления заявленного способа схематически выглядят следующим образом.
Например, в одном из возможных вариантов осуществления, твердый I2 добавляют к раствору 3,5-дизамещенного фенольного субстрата или его соли, или к основной реакционной смеси, содержащей этот субстрат, полученной непосредственно в промышленном способе получения желаемого радиографического агента, предварительно нагретой до температуры в пределах от 55 до 65°C, предпочтительно, примерно до 60°C.
Затем в полученную смесь вводят водный раствор HIO3 в течение примерно двух часов, начиная введение в момент, когда pH реакционной смеси равно примерно 5. Затем продолжают перемешивать реакционную смесь при указанной выше температуре в течение еще 4 часов (время завершения реакции) и после этого охлаждают до 25°C. Полное время проведения реакции составляет примерно 6 часов.
В качестве альтернативы, в исходный раствор, нагретый примерно до 40°C, можно ввести вначале I2 и затем HIO3 (при указанном выше значении pH реакционной смеси). В этом случае добавление HIO3 предпочтительно осуществляют в течение примерно 3 часов. Затем реакционную смесь нагревают до 50°C и выдерживают при этой температуре примерно 1 час, затем примерно при 60°C в течение еще одного часа и затем охлаждают до 25°C (общее время проведения реакции: 7 часов). Аналогично, оба йодирующих агента (I2 и HIO3) добавляют к реакционной смеси, нагретой примерно до 30°C (время добавления HIO3 составляет примерно 4 часа) и затем температуру реакционной смеси повышают и поддерживают в диапазоне от 55 до 65°C в течение еще 4 часов, после чего охлаждают смесь до комнатной температуры (общее время проведения реакции: 8 часов) или, в еще одном варианте осуществления, I2 вводят в исходный раствор при комнатной температуре (примерно 20°C), затем смесь нагревают до 40°C и в течение примерно 4 часов вводят HIO3, после этого температуру реакционной смеси поднимают до 60°C в течение 2 часов и выдерживают при этой температуре в течение еще 4 часов, после чего охлаждают до комнатной температуры (общее время проведения реакции: 9 часов).
Затем к охлажденной реакционной смеси необязательно можно добавить минимальное количество 18% (масс./масс.) водного раствора бисульфита натрия для устранения всех остатков йодирующих веществ, которые возможно находятся в растворе. Оптимальное количество этого реагента можно определить, например, потенциометрически, как минимальное количество бисульфита, которое позволяет добиться устойчивых отрицательных значений окислительно-восстановительного потенциала конечной смеси (pH которой поддерживают на уровне 5) в пределах от 0 до -20 мВ.
В качестве альтернативы, для облегчения регистрации изменения окислительного потенциала, pH реакционной смеси можно вначале довести до значения 7, добавляя 30% (масс./масс.) водный раствор NaOH, после чего добавлять водный раствор бисульфита натрия до достижения значения окислительно-восстановительного потенциала, в данном случае, в диапазоне от -20 до -50 мВ.
Соединения формулы 1, используемые в качестве исходных веществ в способе по настоящему изобретению, являются известными, и если даже их нельзя приобрести в готовом виде, то все они могут быть получены по известным методикам. Для ознакомления с подобными методиками в качестве основной ссылки смотрите, например, упомянутые выше EP 185130 и WO 00/32561. Аналогично, все остальные реагенты и/или растворители, применяемые в способе по настоящему изобретению, являются известными и легко доступными.
После получения производных 3,5-дизамещенных-2,4,6-трийодфенолов формулы 2, их можно легко превратить в соответствующие радиографические контрастные агенты, представляющие интерес.
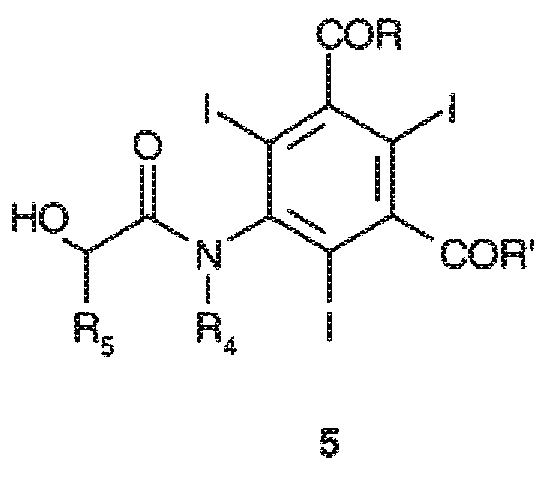
Следовательно, еще одним предметом настоящего изобретения является способ получения соединений формулы 5
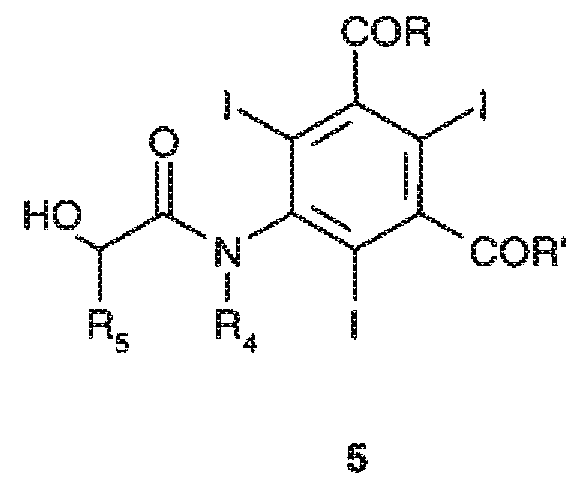
где:
заместители R и R', которые являются одинаковыми или отличаются друг от друга, соответствуют данному ранее определению, и заместители R4 и R5, которые являются одинаковыми или отличаются друг от друга, представляют собой атомы водорода, или линейные или разветвленные C1-C6 алкильные группы, необязательно замещенные одной или несколькими гидроксильными группами или C1-C6 алкоксигруппами,
где указанный способ включает получение производных 3,5-дизамещенных-2,4,6-трийодфенолов формулы 2 путем йодирования 3,5-дизамещенных фенольных субстратов формулы 1 или их солей молекулярным йодом в присутствии HIO3 по способу настоящего изобретения, который в основном описан выше.
Более предпочтительно, указанный способ включает:
a) йодирование 3,5-дизамещенного фенольного субстрата формулы 1 или его соли в водной среде молекулярным йодом в присутствии HIO3 с получением соответствующего 2,4,6-трийодфенольного производного формулы 2; и, кроме этого, указанный способ включает:
b) взаимодействие полученного соединения формулы 2, в котором фенольные группы OH могут необязательно иметь форму соли со щелочным металлом, с соединением формулы 3
где R4 и R5, которые являются одинаковыми или отличаются друг от друга, соответствуют данному выше определению, и Z представляет собой атом галогена, например, Cl, Br, I и, предпочтительно, Cl или Br, или любую подходящую уходящую группу, как, например, остаток сульфоновой кислоты (например, метансульфонилокси (MeSO2O-), бензолсульфонилокси (PhSO2O-), нитробензолсульфонилокси (п-NO2PhSO2O-), толуолсульфонилокси (TsO-) и т.п.) и, предпочтительно, толуолсульфонилокси; с получением соединения формулы 4
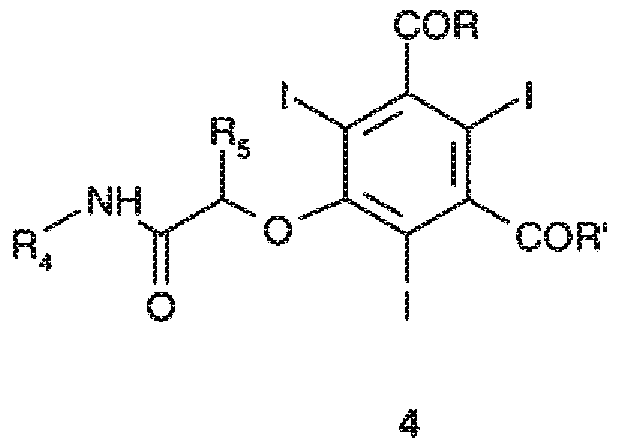
где R, R', R4 и R5 имеют указанные выше значения; и
c) осуществление перегруппировки Смайлса соединения формулы 4 в присутствии основания с получением желаемого конечного соединения формулы 5
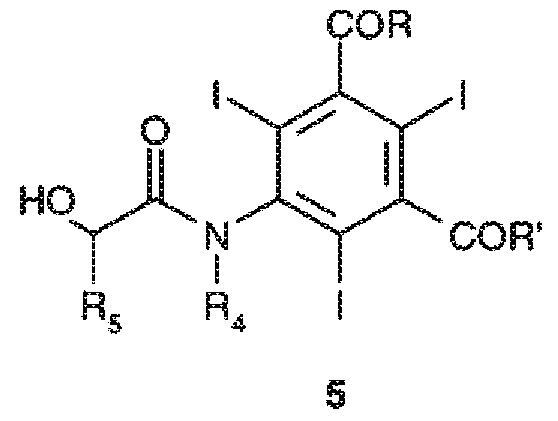
В указанном способе получения рентгеноконтрастных средств стадию a) проводят, как подробно описано выше, способом по настоящему изобретению, тогда как последующие стадии b) и c), условия их осуществления и возможные варианты известны в технике и описаны, например, в заявках на патент WO 97/05097, WO 88/09328, EP 185130 и WO 00/32561.
Способ по настоящему изобретению предпочтительно можно применять для получения радиографических средств, входящих в число соединений формулы 5, в которых R и R' представляют собой одинаковые или отличные друг от друга группы, выбранные из:
-NHCH3,
-NHCH2-CH(OH)-CH2OH,
-NHCH(CH2OH)2 и
-N(CH3)-CH2-CH(OH)-CH2OH,
и заместители R4 и R5, которые являются одинаковыми или отличными друг от друга, представляют собой водород или метильную группу.
Еще более предпочтительно, способ по настоящему изобретению может быть применен для получения широко известных рентгеноконтрастных средств, таких как йопамидол (в котором, соответственно, оба заместителя R и R' представляют собой группы -NH-CH(CH2OH)2, R4 является атомом водорода и R5 является метилом; смотрите The Merck Index, XIII Ed., 2001, No. 5073) или йомепрол (в котором, соответственно, оба заместителя R и R' представляют собой группы -NH-CH2-CH(OH)CH2OH, R4 означает метил, и R5 является атомом водорода; смотрите The Merck Index, XIII Ed., 2001, No. 5071).
Таким образом, еще один вариант осуществления настоящего изобретения представляет собой способ получения йопамидола или йомепрола, который отличается тем, что он включает применение в качестве исходных веществ соединений 2a или 2b, соответственно, полученных йодированием соответствующих субстратов формул 1a и 1b молекулярным йодом в присутствии йодноватой кислоты по способу настоящего изобретения.
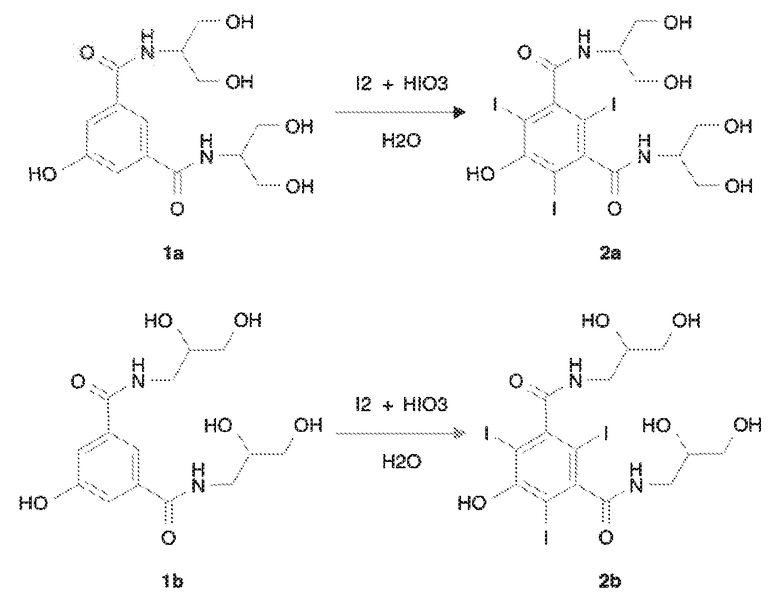
В частности, способ получения йомепрола главным образом включает стадии, представленные на следующей схеме 3:
Схема 3
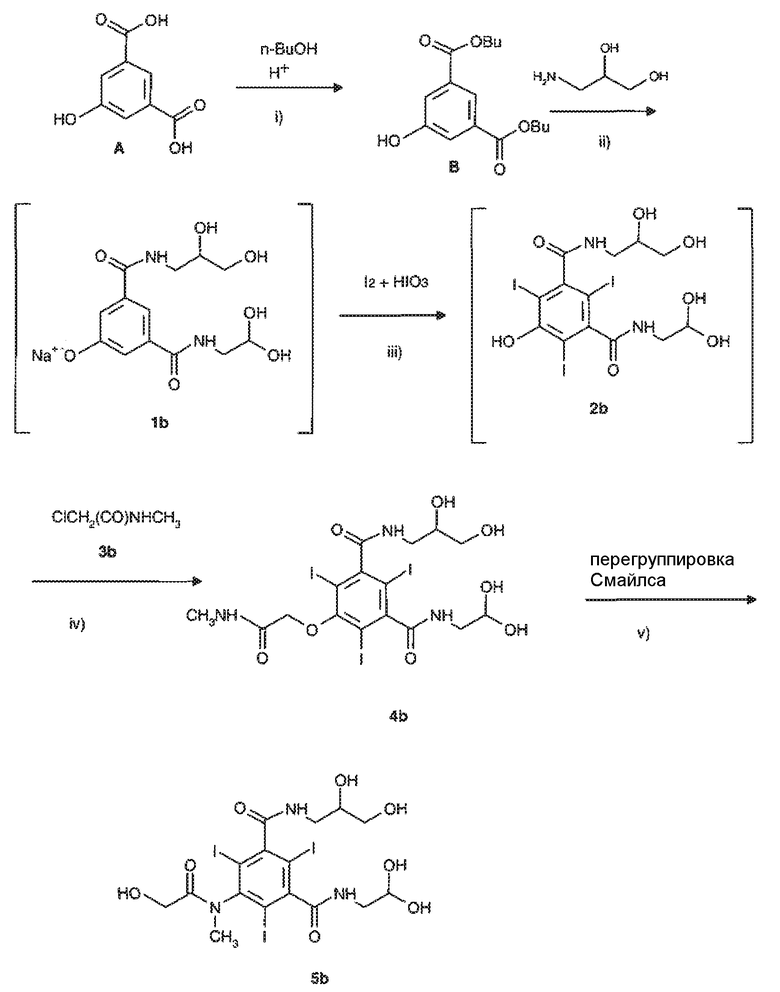
и отличается тем, что стадию йодирования iii) осуществляют с применением молекулярного йода в присутствии йодноватой кислоты и проводят ее в непрерывном режиме, т.е. воздействуя указанными реагентами непосредственно на реакционную смесь соединения формулы 1b, полученную на предыдущей стадии ii) указанного способа, с образованием реакционного раствора йодированного соединения формулы 2b, применяемого в том виде, как он был получен, в следующей стадии алкилирования iv) с получением интермедиата 4b) без выделения или очистки какого либо из упомянутых интермедиатов.
В упомянутом выше способе стадию iii) проводят в соответствии со способом йодирования по настоящему изобретению, который подробно описан выше, в то время, как стадии i), ii), iv) и v), включая экспериментальные условия и необязательные варианты этих стадий, проводят согласно, например, WO 00/32561 и приведенным в этом источнике ссылкам.
При этом предпочтительные условия проведения перегруппировки Смайлса на стадии v) способа, включающие применение основания, например, водного раствора NaOH, и очистку конечного продукта, раскрыты, например, в EP 365541.
Дополнительные подробности, касающиеся способа йодирования по настоящему изобретению, приведены в помещенном ниже экспериментальном разделе, который служит лишь цели лучшей иллюстрации настоящего изобретения, не накладывая никаких ограничений на объем изобретения.
ЭКСПЕРИМЕНТАЛЬНЫЙ РАЗДЕЛ
Определение характеристик полученных соединений
Чистоту полученных 3,5-замещенных-2,4,6-трийодфенолов и их производных определяли с помощью ВЭЖХ, применяя в качестве стандарта чистые соединения.
Общие методики
Методика ВЭЖХ хроматографии
Пример 1
Получение соединения формулы 2, в котором оба заместителя R и R' представляют собой группы -NH-CH2-CH(OH)CH2OH с использованием исходного раствора, нагретого до 60°C.
В 2 л четырехгорлом реакторе с рубашкой, оборудованном механической мешалкой, обратным холодильником и комбинированным электродом для измерения pH/температуры, нагревали до 60°C водный раствор натриевой соли 3,5-дизамещенного фенола 1, соответствующий концентрации фенола 22,8% (масс./масс.) (1175 г раствора; 0,816 моль; pH 9,6) и затем одной порцией добавляли твердый I2 (250,6 г; 0,988 моль). Когда значение pH самопроизвольно понижалось до 5, медленно в течение 2 ч добавляли 50% (масс./масс.) водный раствор HIO3 (173,6 г; 0,494 моль). Реакционную смесь выдерживали при 60°C в течение еще 4 ч, и в течение этого времени значение pH самопроизвольно оставалось в диапазоне 5-5,5. Красный раствор охлаждали до 25°C и гасили добавлением 18% (масс./масс.) водного раствора бисульфита натрия до потери цвета и достижения окислительно-восстановительным потенциалом, измеренным с помощью подходящего электрода, устойчивых отрицательных значений в диапазоне от 0 до -20 мВ.
При гашении реакционной смеси поддерживали pH на уровне 5, добавляя минимальные количества 30% (масс./масс.) водного раствора NaOH.
ВЭЖХ-анализ (результаты которого приведены на фиг.1) показал степень превращения исходного соединения в 3,5-дизамещенный-2,4,6-трийодфенол 2b >98% (по % площади на хроматограмме ВЭЖХ), и полученный раствор использовали в следующей стадии синтеза без какой-либо дополнительной обработки.
Пример 2
Получение соединения формулы 2, в котором оба заместителя R и R' представляют собой группы -NH-CH2-CH(OH)CH2OH с использованием исходного раствора, нагретого до 40°C
В 2 л четырехгорлом реакторе с рубашкой, оборудованном механической мешалкой, обратным холодильником и комбинированным электродом для измерения pH/температуры, твердый I2 (250,6 г; 0,988 моль) одной порцией добавляли к водному раствору натриевой соли 3,5-дизамещенного фенола 1, соответствующий концентрации фенола 22,8% (масс./масс.) (1175 г раствора; 0,816 моль; pH 9,6), нагретому до 40°C. Когда значение pH самопроизвольно понижалось до 5, медленно в течение 3 ч добавляли 50% (масс./масс.) водный раствор HIO3 (173,6 г; 0,494 моль). Затем реакционную смесь нагревали в течение 2 ч при 40°C, 1 ч при 50°C и 1 ч при 60°C, причем в течение всего этого времени значение pH самопроизвольно оставалось в диапазоне 5-5,5. Полученный красный раствор охлаждали до 25°C, доводили pH до 7 и поддерживали его на этом уровне добавлением 30% (масс./масс.) водного раствора NaOH во время гашения, которое осуществляли добавлением 18% (масс./масс.) водного раствора бисульфита натрия до потери цвета и достижения окислительно-восстановительным потенциалом, измеренным с помощью подходящего электрода, устойчивых отрицательных значений в диапазоне от -20 до -50 мВ.
ВЭЖХ-анализ показал степень превращения исходного соединения в 3,5-дизамещенный-2,4,6-трийодфенол 2b >98% (по % площади на хроматограмме ВЭЖХ), и полученный раствор использовали в следующей стадии синтеза без какой-либо дополнительной обработки.
Пример 3
Получение соединения формулы 2, в котором оба заместителя R и R' представляют собой группы -NH-CH2-CH(OH)CH2OH, с использованием исходного раствора, нагретого до 30°C, и гашения реакционной смеси бисульфитом при pH5 на заключительном этапе
В 4 л четырехгорлом реакторе с рубашкой, оборудованном механической мешалкой, обратным холодильником и комбинированным электродом для измерения pH/температуры, разбавляли H2O (1054 г) водный раствор натриевой соли 3,5-дизамещенного фенола 1, соответствующий концентрации фенола 22,8% (масс./масс.) (1175 г раствора; 0,816 моль; pH 9,6) нагревали до 30°C и затем одной порцией добавляли твердый I2 (250,6 г; 0,988 моль). Когда значение pH самопроизвольно понижалось до 5, медленно в течение 4 ч добавляли 50% (масс./масс.) водный раствор HIO3 (173,6 г; 0,494 моль). Температуру реакционной смеси поднимали до 60°C и выдерживали при этой температуре в течение еще 4 ч, причем значение pH самопроизвольно оставалось в диапазоне 5-5,5. Полученный красный раствор охлаждали до 25°C и гасили добавлением 18% (масс./масс.) водного раствора бисульфита натрия, поддерживая pH 5, добавлением 30% (масс./масс.) водного раствора NaOH, до потери цвета и достижения окислительно-восстановительным потенциалом, измеренным с помощью подходящего электрода, устойчивых отрицательных значений в диапазоне от 0 до -20 мВ.
ВЭЖХ-анализ показал степень превращения исходного соединения в 3,5-дизамещенный-2,4,6-трийодфенол 2b >98% (по % площади на хроматограмме ВЭЖХ), и полученный раствор использовали в следующей стадии синтеза без какой-либо дополнительной обработки.
Пример 4
Получение соединения формулы 2, в котором оба заместителя R и R' представляют собой группы -NH-CH2-CH(OH)CH2OH, с использованием исходного раствора, нагретого до 30°C и гашения реакционной смеси бисульфитом при pH7 на заключительном этапе
В 4 л четырехгорлом реакторе с рубашкой, оборудованном механической мешалкой, обратным холодильником и комбинированным электродом для измерения pH/температуры, разбавляли H2O (1054 г) водный раствор натриевой соли 3,5-дизамещенного фенола 1, соответствующий концентрации фенола 22,8% (масс./масс.) (1175 г раствора; 0,816 моль; pH 9,6), нагревали до 30°C и затем одной порцией добавляли твердый I2 (250,6 г; 0,988 моль). Когда значение pH самопроизвольно понижалось до 5, медленно в течение 4 ч добавляли 50% (масс./масс.) водный раствор HIO3 (173,6 г; 0,494 моль). Температуру реакционной смеси поднимали до 60°C и выдерживали при этой температуре в течение еще 4 ч, причем в течение этого времени значение pH самопроизвольно оставалось в диапазоне 5-5,5. Полученный красный раствор охлаждали до 25°C, доводили pH до 7 и поддерживали на этом уровне добавлением 30% (масс./масс.) водного раствора NaOH во время гашения, которое осуществляли добавлением 18% (масс./масс.) водного раствора бисульфита натрия до потери цвета и достижения окислительно-восстановительным потенциалом, измеренным с помощью подходящего электрода, устойчивых отрицательных значений в диапазоне от -20 до -50 мВ.
ВЭЖХ-анализ показал степень превращения исходного соединения в 3,5-дизамещенный-2,4,6-трийодфенол 2b >98% (по % площади на хроматограмме ВЭЖХ), и полученный раствор использовали в следующей стадии синтеза без какой-либо дополнительной обработки.
Пример 5
Получение соединения формулы 2, в котором оба заместителя R и R' представляют собой группы -NH-CH2-CH(OH)CH2OH, с использованием исходного раствора, имеющего комнатную температуру (примерно 20°C)
В 4 л четырехгорлом реакторе с рубашкой, оборудованном механической мешалкой, обратным холодильником и комбинированным электродом для измерения pH/температуры, во-первых, разбавляли H2O (1054 г) водный раствор натриевой соли 3,5-дизамещенного фенола 1, соответствующий концентрации фенола 22,8% (масс./масс.) (1175 г раствора; 0,816 моль; pH 9,6), поддерживая температуру 20°C, и затем одной порцией добавляли твердый I2 (250,6 г; 0,988 моль). Затем полученный раствор нагревали до 40°C, и когда значение pH самопроизвольно понижалось до 5, медленно в течение 4 ч добавляли 50% (масс./масс.) водный раствор HIO3 (173,6 г; 0,494 моль). Температуру реакционной смеси поднимали до 60°C в течение 2 ч и выдерживали при этой температуре в течение еще 3 ч, причем в течение этого времени значение pH самопроизвольно оставалось в диапазоне 5-5,5. Затем красный раствор охлаждали до 25°C, доводили pH до 7 и поддерживали на этом уровне добавлением 30% (масс./масс.) водного раствора NaOH, и гасили бисульфитом натрия (18% (масс./масс.) водный раствор) до потери цвета и достижения устойчивых отрицательных значений (в диапазоне от -20 до -50 мВ) окислительно-восстановительного потенциала, измеренных с помощью подходящих окислительно-восстановительных электродов.
ВЭЖХ-анализ показал степень превращения исходного соединения в 3,5-дизамещенный-2,4,6-трийодфенол 2b >98% (по % площади на хроматограмме ВЭЖХ), и полученный раствор использовали в следующей стадии синтеза без какой-либо дополнительной обработки.
Пример 6
Получение соединения формулы 2, в котором заместитель R представляет собой группу -NH-CH2-CH(OH)CH2OH, и R' представляет собой -NH-CH(CH2OH)2 с использованием исходного раствора, нагретого до 60°C.
В 1 л четырехгорлом реакторе с рубашкой, оборудованном механической мешалкой, обратным холодильником и комбинированным электродом для измерения pH/температуры, N-(2,3-дигидроксипропил)-N'-[2-гидрокси-1-(гидроксиметил)этил]-5-гидрокси-1,3-бензолдикарбоксамид (100,3 г; 0,305 моль) растворяли в H2O (430 г) и превращали в соответствующую натриевую соль добавлением 30% (масс./масс.) NaOH (40,6 г; 0,305 моль) (pH 9,5). Раствор нагревали до 60°C и одной порцией добавляли твердый I2 (93,1 г; 0,367 моль); когда значение pH самопроизвольно понижалось до 5, медленно в течение 2 ч добавляли 50% (масс./масс.) водный раствор HIO3 (64,5 г; 0,183 моль). Температуру реакционной смеси поддерживали на уровне 60°C в течение еще 4 ч, и в это время значение pH реакционной смеси самопроизвольно оставалось в диапазоне 5-5,5. Полученный красный раствор охлаждали до 25°C и гасили добавлением 18% (масс./масс.) водного раствора бисульфита натрия, поддерживая pH равным 5, добавлением 30% (масс./масс.) водного раствора NaOH до обесцвечивания и устойчивого достижения окислительно-восстановительным потенциалом, измеренным с помощью подходящего окислительно-восстановительного электрода, отрицательных значений в диапазоне от 0 до -20 мВ.
ВЭЖХ-анализ (фиг.2) показал степень превращения исходного соединения в N-(2,3-дигидроксипропил)-N'-[2-гидрокси-1-(гидроксиметил)этил]-5-гидрокси-2,4,6-трийод-1,3-бензолдикарбоксамид >98% (по % площади на хроматограмме ВЭЖХ), и полученный раствор использовали в следующей стадии синтеза без какой-либо дополнительной обработки.
Пример 7
Получение соединения формулы 2, в котором оба заместителя R и R' представляют собой группы -NH-CH(CH2OH)2 с использованием исходного раствора, нагретого до 60°C.
В 0,5 л четырехгорлом реакторе с рубашкой, оборудованном механической мешалкой, обратным холодильником и комбинированным электродом для измерения pH/температуры, N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-5-гидрокси-1,3-бензолдикарбоксамид (50 г; 0,152 моль) растворяли в H2O (215 г) и превращали в соответствующую натриевую соль добавлением 30% (масс./масс.) NaOH (20,3 г; 0,152 моль) (pH 9,5). Раствор нагревали до 60°C и одной порцией добавляли твердый I2 (46,4 г; 0,183 моль); когда pH самопроизвольно понижался до 5, медленно в течение 2 ч добавляли 50% (масс./масс.) водный раствор HIO3 (32,2 г; 0,091 моль). Температуру реакционной смеси поддерживали на уровне 60°C в течение еще 4 ч, в течение которых pH реакционной смеси самопроизвольно оставался в диапазоне 5-5,5. Полученный красный раствор охлаждали до 25°C и гасили добавлением 18% (масс./масс.) водного раствора бисульфита натрия, поддерживая pH равным 5, добавлением 30% (масс./масс.) водного раствора NaOH до обесцвечивания и устойчивого достижения окислительно-восстановительным потенциалом, измеренным с помощью подходящего окислительно-восстановительного электрода, отрицательных значений (в диапазоне от -20 до -50 мВ).
ВЭЖХ-анализ показал степень превращения исходного соединения в N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-5-гидрокси-2,4,6-трийод-1,3-бензолдикарбоксамид >98% (по % площади на хроматограмме ВЭЖХ), и полученный раствор использовали в следующей стадии синтеза без какой-либо дополнительной обработки.
Сравнительный пример 1
Этот тест проводили для оценки эффективности реакции йодирования, раскрытой Patil et al., в ARKIVOC, 2006, 104 и Tetrahedron Lett., 2005, 46, 7179.
В 50 мл трехгорлой круглодонной колбе, снабженной термометром и обратным холодильником, в этаноле (30 мл) суспендировали твердый 3,5-дизамещенный фенол 1 (16,4 г; 50 ммоль). Затем к полученной суспензии, нагретой до 38-40°C, добавляли в указанном порядке твердый I2 (15,2 г; 60 ммоль) одной порцией и раствор HIO3 (5,3 г; 30 ммоль) в H2O (3 мл) в течение 5 мин. Полученную темно-коричневую смесь перемешивали при 38-40°C в течение примерно 1 ч и затем регистрировали изменение внешнего вида реакционной смеси, которая превратилась в прозрачный темно-коричневый раствор. Реакционную смесь выдерживали при указанной выше температуре в течение в общей сложности 3,5 ч, затем охлаждали до комнатной температуры, что вызывало кристаллизацию бледно-желтого твердого продукта. Через 15 ч при комнатной температуре это твердое вещество отделяли фильтрованием и высушивали, получая желаемый 3,5-дизамещенный-2,4,6-трийодфенол (12,1 г; 17 ммоль). Выход 34,3%.
По ходу реакции йодирования реакционную смесь анализировали с помощью ВЭЖХ. В частности, первый анализ проводили через 1,5 ч после начала йодирования (время реакции было выбрано на основании указанных литературных источников), причем его результаты показаны на фиг.3, и второй анализ выполняли еще через 2 часа (общее время реакции 3,5 часа), причем его результаты приведены на фиг.4. Полученные результаты показывают, что даже через 3,5 ч превращение не завершается и по-прежнему присутствует значительное количество (13% судя по площади на хроматограмме ВЭЖХ) исходного субстрата. С другой стороны, более продолжительное время реакции ведет к образованию значительного количества примесей, являющихся продуктами распада, которые легко обнаруживаются уже после 3,5 часов реакции (фиг.4). Это, безусловно, является фактором, неблагоприятно влияющим на выходы реакции. Однако невысокие выходы реакции можно также отнести на счет растворимости 3,5-дизамещенного-2,4,6-трийодфенола 2b в спиртовой среде, что подтверждается анализом маточного раствора, показанным на фиг.5, которая мешает количественному выделению продукта йодирования.
В этом отношении, увеличение как выхода реакции, так и чистоты продукта вследствие применения водной среды и условий проведения реакции, выраженное в приведенных выше результатах, очевидно при сравнении фиг.3-5 с фиг.1 и 2, на которых приведены хроматограммы (ВЭЖХ) неочищенных реакционных растворов (примеров 1 и 6 соответственно), полученных с применением способа по настоящему изобретению.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ЙОДИРОВАНИЯ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ | 2009 |
|
RU2469997C2 |
| СПОСОБ ИОДИРОВАНИЯ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ | 2010 |
|
RU2506254C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЙОДИРУЮЩЕГО АГЕНТА | 2010 |
|
RU2528402C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЛЕВОТИРОКСИНА И ЕГО СОЛЕЙ | 2015 |
|
RU2673540C2 |
| СПОСОБ ПОЛУЧЕНИЯ 1-ФЕНИЛ-2,3-ДИМЕТИЛ-4-ИОДПИРАЗОЛОНА-5 (ЙОДАНТИПИРИНА) | 2009 |
|
RU2401830C1 |
| СПОСОБ СИНТЕЗА N,N-ДИЗАМЕЩЕННЫХ АМИНОМЕТИЛСТИРОЛОВ ИЛИ АЛЬФА-АМИНОМЕТИЛСТИРОЛОВ | 2014 |
|
RU2673231C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ МЕТИЛФЕНОЛОВ В ВОДНЫХ СРЕДАХ | 2011 |
|
RU2459203C1 |
| СПОСОБ ПОЛУЧЕНИЯ СУЛЬФАТИРОВАННОГО ПРОИЗВОДНОГО 3,5-ДИЙОД-О-[3-ЙОДФЕНИЛ]-L-ТИРОЗИНА | 2012 |
|
RU2610093C2 |
| СПОСОБ ПОЛУЧЕНИЯ СУЛЬФАТИРОВАННОГО ПРОИЗВОДНОГО 3,5-ДИЙОД-O-[3-ЙОДФЕНИЛ]-L-ТИРОЗИНА | 2018 |
|
RU2708244C1 |
| СПОСОБ ПОЛУЧЕНИЯ СУЛЬФАТИРОВАННОГО ПРОИЗВОДНОГО 3,5-ДИЙОД-O-[3-ЙОДФЕНИЛ]-L-ТИРОЗИНА | 2012 |
|
RU2662826C1 |
Изобретение относится к способу получения производных трийодфенола формулы 2, включающему йодирование 3,5-дизамещенного фенола формулы 1 или его соли, в водной среде с применением молекулярного йода в присутствии йодноватой кислоты, где заместители R и R', которые являются одинаковыми или отличными друг от друга, представляют собой группы формулы -NHR1 или формулы -NR2R3, где каждая из групп R1, R2 и R3, независимо от других, представляет собой линейную или разветвленную C1-C6 алкильную группу, которая необязательно замещена одной или несколькими группами, выбранными из гидроксила (-OH), групп C1-C5 алкокси и гидроксиалкокси. Также изобретение относиться к способу получения соединений формулы 5, где заместители R и R' обозначают заместители, указанные для формулы 2, и заместители R4 и R5, которые являются одинаковыми или отличаются друг от друга, представляют собой атомы водорода, или линейные или разветвленные C1-C6 алкильные группы, необязательно замещенные одной или несколькими гидроксильными группами или C1-C6 алкоксигруппами, включающий (a) получение трийодфенольного соединения формулы 2, причем указанный способ дополнительно включает (b) взаимодействие указанного соединения формулы 2, либо в форме фенола, либо в форме соли фенольной OH-группы с щелочным металлом, с соединением R4HN(C=O)CH(R5)Z (формула 3), где заместители R4 и R5 определены выше и Z представляет собой атом галогена, выбранный из хлора или брома, или уходящую группу, выбранную из групп метансульфонилокси, бензолсульфонилокси, нитробензолсульфонилокси, и толуолсульфонилокси, с получением соединения формулы 4; и (c) введение указанного соединения формулы 4 в перегруппировку Смайлса в присутствии основания с получением соединения формулы 5. Технический результат - получение трийодированных ароматических соединений соответствующей чистоты, подходящей для получения целевого йодированного (рентгеноконтрастного) средства. 2 н. и 13 з.п. ф-лы, 5 ил., 8 пр.
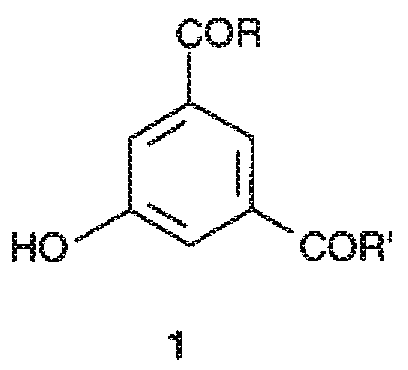
1. Способ получения производных трийодфенола формулы 2
включающий йодирование 3,5-дизамещенного фенола формулы 1
или его соли, в водной среде с применением молекулярного йода в присутствии йодноватой кислоты, где:
заместители R и R', которые являются одинаковыми или отличными друг от друга, представляют собой группы формулы -NHR1 или формулы -NR2R3, где каждая из групп R1, R2 и R3, независимо от других, представляет собой линейную или разветвленную C1-C6 алкильную группу, которая необязательно замещена одной или несколькими группами, выбранными из гидроксила (-OH), групп C1-C5 алкокси и гидроксиалкокси.
2. Способ по п.1, где в соединениях формул 1 и 2 заместители R и R', которые являются одинаковыми или отличаются друг от друга, представляют собой группы формулы -NHR1 или NR2R3, где каждый из заместителей R1, R2 и R3 независимо от других представляет собой линейную или разветвленную C1-C4 алкильную группу, необязательно замещенную одной-тремя гидроксильными группами.
3. Способ по п.2, где в соединениях формул 1 и 2 заместители R и R', которые являются одинаковыми или отличаются друг от друга, представляют собой группы, выбранные из:
-NHCH3,
-NHCH2-CH(OH)-CH2OH,
-NHCH(CH2OH)2 и
-N(CH3)-CH2-CH(OH)-CH2OH.
4. Способ по п.1, где мольное соотношение количества молекулярного йода и 3,5-дизамещенного фенольного субстрата 1 [I2/1] находится в пределах от 1,1 до 1,3, и мольное соотношение количества йодноватой кислоты и 3,5-дизамещенного фенольного субстрата 1 находится в пределах от 0,4 до 0,8.
5. Способ по п.4, где трийодирование 3,5-дизамещенного фенольного субстрата 1 йодом и йодноватой кислотой проводят при мольном соотношении 3,5-дизамещенный фенольный субстрат:йод:йодноватая кислота, равном 1:1,2:0,6.
6. Способ по п.1, где указанная водная среда является водой или водным раствором.
7. Способ по п.6, включающий: получение водного раствора 3,5-дизамещенного фенольного субстрата формулы 1 или его соли и добавление к указанному водному раствору I2 и HIO3.
8. Способ по п.7, где указанный водный раствор 3,5-дизамещенного фенольного субстрата представляет собой реакционный раствор, полученный при осуществлении промышленного способа, и включающий 3,5-дизамещенный фенольный субстрат в виде соли.
9. Способ по п.7, включающий добавление твердого I2 к указанному водному раствору 3,5-дизамещенного фенольного субстрата, нагретому до температуры в диапазоне от 20 до 70°C, с последующим добавлением йодноватой кислоты.
10. Способ по п.9, где йодноватую кислоту добавляют в тот момент, когда реакционная смесь имеет значение pH в пределах от 5 до 6.
11. Способ по любому из пп.1-10, где время проведения реакции находится в пределах от 5 до 9 часов.
12. Способ получения соединений формулы 5
где:
заместители R и R' представляют собой, независимо друг от друга, группы формулы -NHR1 или -NR2R3, где каждая из групп R1, R2 и R3, независимо от других, представляет собой линейную или разветвленную C1-C6 алкильную группу, которая необязательно замещена одной или несколькими группами, выбранными из гидроксила (-OH), групп C1-C5 алкокси и гидроксиалкокси, и
заместители R4 и R5, которые являются одинаковыми или отличаются друг от друга, представляют собой атомы водорода, или линейные или разветвленные C1-C6 алкильные группы, необязательно замещенные одной или несколькими гидроксильными группами или C1-C6 алкоксигруппами, включающий:
a) получение трийодфенольного соединения формулы 2
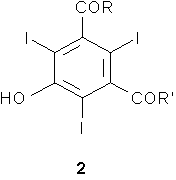
способом по любому из пп.1-11; где указанный способ дополнительно включает:
b) взаимодействие указанного соединения формулы 2, либо в форме фенола, либо в форме соли фенольной OH-группы, с щелочным металлом, с соединением формулы 3
где заместители R4 и R5, которые являются одинаковыми или отличаются друг от друга, соответствуют данному выше определению, и Z представляет собой атом галогена, выбранный из хлора или брома, или уходящую группу, выбранную из групп метансульфонилокси, бензолсульфонилокси, нитробензолсульфонилокси, и толуолсульфонилокси, с получением соединения формулы 4
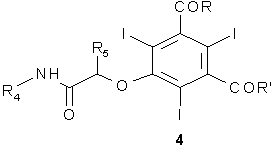
где R, R', R4 и R5 имеют указанные выше значения; и
c) введение указанного соединения формулы 4 в перегруппировку Смайлса в присутствии основания с получением соединения формулы 5.
13. Способ по п.12, где оба заместителя R и R' представляют собой группы -CONH-CH(CH2OH)2, R4 является атомом водорода и R5 является метильной группой.
14. Способ по п.12, где оба заместителя R и R' представляют собой группы -CONH-CH2-CH(OH)CH2OH, R4 означает метил, и R5 является атомом водорода.
15. Способ по п.14, включающий стадии, представленные на схеме 3:
Схема 3
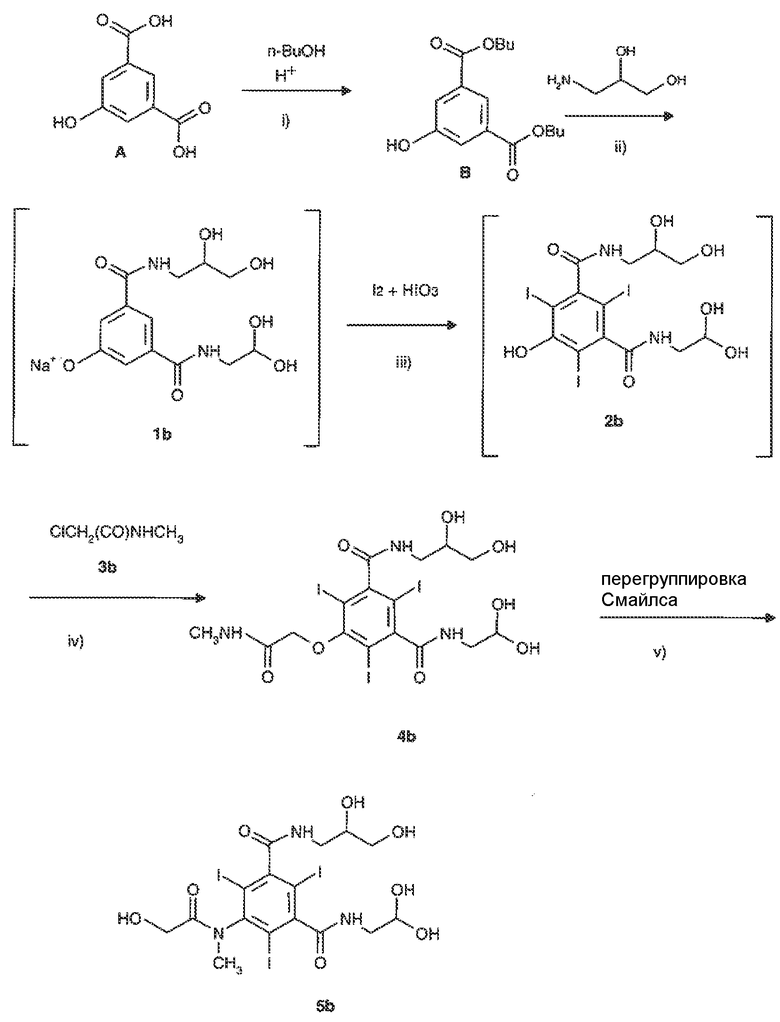
| WO 00/32561 A1, 08.06.2000 | |||
| Колосоуборка | 1923 |
|
SU2009A1 |
| BHAGWAN R.PATIL et al.: "Iodine and iodic acid: an efficient reagent combination for iodination of aryl hydroxy ketones", TETRAHEDRON LETTERS, vol.46, p.7179-7181 | |||
| US 5856596 A1, 05.01.1999 | |||
| RU 99127457 A, 10.09.2001 | |||
| ИЗОМЕРОВ ЙОДФЕНИЛУИДРКАНОВЫХ КИСЛОТ | 0 |
|
SU394355A1 |

