Настоящее изобретение относится к способу получения иодированных ароматических соединений. В частности, оно относится к способу, включающему электрохимическое йодирование 3,5-дизамещенных фенолов до соответствующих 3,5-дизамещенных-2,4,6-трийодфенолов, которые представляют собой интермедиаты, применимые в синтезе рентгеноконтрастных веществ, и к получению самих контрастных веществ.
Уровень техники
Йодированные контрастные вещества представляют собой хорошо известные соединения, широко используемые в методах диагностики, основанных на получении рентгеновских изображений. Подходящие примеры указанных соединений включают, например, диатризоат, йоталамат, йокситаламат, метризоат, йогексол, йомепрол, йопамидол, йопентол, йопромид, йоверсол, йоксилан, йодиксанол, йосаркол, йогуламид, йоглунид, йоглуамид, ацетризоат, йодамид, йоцетамид и метризамид, которые все являются мономерами, тогда как, например, йоксаглат, йотролан, йотазул, йодипамид, йокармат, йодоксамат, йотроксат, йотролан и тому подобное являются димерами. Другие примеры йодированных контрастных веществ описаны, например, в WO 94/14478 (Bracco).
Общим признаком этих химических структур является наличие трийодированного ароматического ядра, которое обеспечивает усиленный эффект контрастности.
Указанные соединения могут быть получены множеством способов, некоторые из которых включают йодирование ароматических составляющих данных субстратов, в частности пригодных анилинов, с тем чтобы получить соответствующие производные 2,4,6-трийоданилина, чтобы преобразовать их и переработать в конечные соединения.
Применимыми предшественниками получения указанных выше соединений также являются 3,5-дизамещенные фенолы, которые сначала вступают в реакцию трийодирования в свободные 2, 4 и 6 позиции, с тем чтобы привести к получению соответствующих 3,5-дизамещенных-2,4,6-трийодфенолов. Эти последние, в свою очередь, могут быть далее преобразованы и переработаны через так называемую перегруппировку Смайлса в требуемые конечные соединения.
Для получения общей справочной информации, касающейся указанных выше путей синтеза и перегруппировки Смайлса, смотрите, например, WO 88/09328, WO 97/05097 и WO 00/32561 (Bracco).
Йодирование фенольного кольца может происходить по хорошо известному механизму электрофильного замещения в свободные орто- и пара-позиции, то есть в позиции 2, 4 и 6, приводя таким образом к трийодированному кольцу.
Стадию йодирования, в частности, можно осуществлять согласно нескольким методам, известным в данной области техники.
Например, ее можно осуществлять, используя растворы хлорида йода (ICl) в концентрированной хлористоводородной кислоте (HCl) или, альтернативно, посредством аналогичных йодирующих агентов, таких как, например, KICl2 or NaICl2 в водном растворе. Для получения общей справочной информации смотрите WO 92/14695 (Guerbet) или US 5013865 (Mallinckrodt).
Описанные выше методы страдают от значительных недостатков, обусловленных ограниченным сроком годности хранения йодирующих агентов и их корродирующими свойствами. В дополнение, присутствие атомов хлора может привести к побочным реакциям и, таким образом, к нежелательному образованию хлорсодержащих побочных продуктов, которые могут оказывать влияние на выходы реакции и чистоту конечных соединений.
Указанные выше проблемы могут быть вполне решены при альтернативном пути, включающем электрохимическое йодирование подходящих ароматических ядер. Электрохимическое йодирование включает, в частности, анодное образование ионов I+ из источника йода, например самого I2, в электролитической ячейке, по существу согласно нижеследующей схеме:

и затем катионы I+, образованные таким образом, могут действовать как йодирующие агенты на ароматическое ядро заданного соединения.
Кроме молекулярного йода также можно использовать множество йодидов, таких как йодиды щелочных металлов и даже йодистоводородная кислота или их смеси, чтобы электрохимически инициировать ионы I+.
Примечательно, однако, что электрохимическое йодирование из источника I2 преимущественно обеспечивает из 1 моля источника йода возникновение двух молей йодирующих ионов. В дополнение, еще одно главное преимущество, которое предлагают в указанном методе, заключается в том, что йодирующие частицы можно генерировать по мере необходимости, избегая таким образом хранения корродирующих реагентов. Более того, вследствие отсутствия источника хлора не могут быть получены побочные продукты хлорирования, влияющие на чистоту конечного соединения.
Электрохимическое йодирование ароматических субстратов известно в данной области техники, о чем сообщали, например, в J. Am. Chem. Soc, Vol. 98, No. 6, 1976, pages 1515-1519; и EP 828705 (Nycomed Imaging As).
Согласно EP 828705, в частности, описан способ получения мономерных или двумерных соединений трийоданилина, включающий электрохимическое йодирование 3,5-дизамещенных анилинов или 3,3'-дизамещенных-5,5'-связанных бисанилинов. Когда вместо этого речь идет о фенолах и их производных, то в US 3833490 описывают способ получения гербицидов, включающий электрохимическое йодирование 4-гидроксибензонитрила в присутствии ионов IO-, с тем чтобы получить соответствующие моно- или дийодированные производные, содержащие атомы йода в позициях 3 и/или 5.
Примечательно, что описанный выше способ йодирования непосредственно осуществляют внутри электролитической ячейки, которая находится в окислительных условиях, имеющихся в анодном отделении, тогда как наличие заместителя (например, цианогруппы), находящегося в пара-положении по отношению к фенольной гидроксигруппе, может уменьшить склонность вышеописанного субстрата к окислительной деструкции.
Однако, даже осуществляя стадию йодирования вне электролитической ячейки, йодирование фенола с помощью электрохимически генерированных йодирующих частиц не давало ожидаемых результатов, так как вместо ожидаемых продуктов получали смеси побочных продуктов. Экспериментальное подтверждение этого можно найти в сравнительных примерах 1 и 2 нижеследующего экспериментального раздела.
Несмотря на эти значительные недостатки, касающиеся электрохимического йодирования фенолов, авторы, однако, неожиданно обнаружили, что электрохимическое йодирование данных 3,5-дизамещенных фенолов позволяет получить соответствующие трийодированные соединения.
Цель изобретения
Таким образом, в настоящем изобретении предлагают способ электрохимического йодирования 3,5-дизамещенных фенолов и, кроме того, способ получения рентгеноконтрастных веществ, который включает стадию электрохимического йодирования, описанного выше.
Поэтому первой целью настоящего изобретения является способ получения 3,5-дизамещенных-2,4,6-трийодфенолов формулы (2). Этот способ включает:
(a) электрохимическое генерирование I+ катионов из пригодного источника йода; и
(b) йодирование 3,5-дизамещенных фенолов формулы (1) в присутствии указанных выше катионов I+, получаемых на стадии (a):
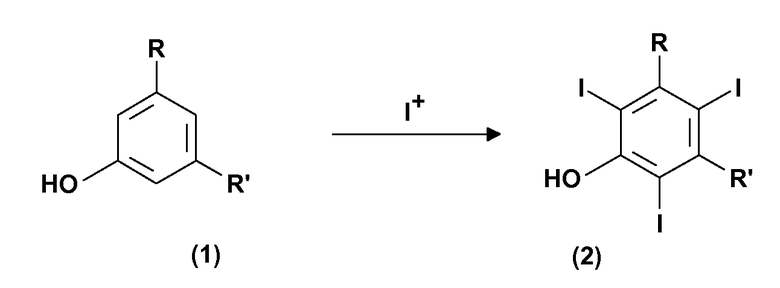
где
R и R', одинаковые или отличающиеся друг от друга, представляют группу, выбираемую из карбокси-COOH, карбоксиэфира -COOR1 и карбоксиамидо -CONH2, -CONHR1 или -CONR2R3; где R1, R2 и R3, одинаковые или отличающиеся друг от друга, представляют прямую или разветвленную C1-C6 алкильную группу, которая необязательно замещена одной или несколькими группами, выбираемыми из гидрокси, амино, сульфгидрила, C1-C6 алкокси или карбокси, и/или необязательно прерывается одной или несколькими двухвалентными группами, выбираемыми из -NH-, -O-, >C=О, -(C=О)О-, -О(C=О)-, -NH(C=О)-, -(C=О)NH-, >SO или >SO2 групп.
Настоящий способ преимущественно дает возможность регенерации трийодированных 3,5-дизамещенных фенолов формулы (2) с высокими выходами и чистотой.
В настоящем описании, если не оговорено иначе, под термином «прямая или разветвленная C1-C6 алкильная группа» авторы подразумевают линейную или разветвленную алкильную цепь, содержащую от 1 до 6 атомов углерода. Таким образом, пригодные примеры указанных алкильных групп могут включать метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, н-гексил и тому подобное.
Указанные выше алкильные группы могут быть дополнительно замещены и/или прерываться разнообразными частями, определенными выше.
В пределах этих групп, под термином «C1-C6 алкокси» авторы подразумевают любую алкилоксигруппу, в которой алкильная часть как раз представляет любую из указанных выше прямых или разветвленных C1-C6 алкильных групп.
Пригодные примеры алкоксигрупп по изобретению могут, таким образом, включать в себя метокси, этокси, н-пропокси, изопропокси, н-пентилокси и тому подобное.
В соответствии с предпочтительным вариантом осуществления способа по изобретению в пределах соединений формул (1) и (2), R и R', одинаковые или отличающиеся друг от друга, представляют собой группу, выбираемую из карбокси (-COOH), карбоксиэфира (-COOR1) и карбоксамидо (-CONH2, -CONHR1 или -CONR2R3), где R1, R2 и R3, одинаковые или отличающиеся друг от друга, представляют собой прямую или разветвленную C1-C4 алкильную группу, необязательно замещенную одной или несколькими гидроксигруппами.
Даже еще более предпочтительно, когда в соединениях формул (1) и (2) R и R', одинаковые или отличающиеся друг от друга, представляют собой группу, выбираемую из:
-COOH,
-COOCH3,
-COO(CH2)3-CH3,
-CONH2,
-CONHCH3,
-CONHCH2-CH(OH)-CH2(OH),
-CONHCH[CH2OH]2.
Из всего сказанного выше, поскольку обе группы R и R', согласно приведенным ниже подробностям, не принимают непосредственного участия в реакционной стадии, специалисту в данной области техники очевидно, что необязательные заместители или группы, так или иначе присутствующие среди значений R и R', которые могут подвергаться нежелательным побочным реакциям, необходимо нуждаются в соответствующей защите перед осуществлением реакции.
Защита указанных групп и последующее удаление защиты могут быть выполнены множеством способов, широко известных в данной области техники и обычно принятых в методах органического синтеза. Для получения общей справочной информации, касающейся защитных групп в органической химии, смотрите, например, T. W. Green, Protective Groups in Organic Synthesis (Wiley, N.Y. 1981). Электрохимическое генерирование I+ согласно стадии (a) способа по изобретению осуществляют в электролитической ячейке, следовательно, в присутствии анода и катода, иначе обозначаемых как анодное и катодное отделения.
Пригодными анодами являются аноды, традиционно известные в данной области техники, и они могут включать углеграфитовые аноды, стеклоуглеродные аноды, платиновые аноды или даже аноды из платиновых сплавов, эти сплавы могут содержать другие металлы, такие как, например, иридий, титан или обладающий электропроводностью диоксид титана. В частности, предпочтительными являются платиновый и углеграфитовый аноды.
В качестве катода могут быть использованы общепринятые катоды, включая также, например, угольный, платиновый, палладиевый, свинцовый и медный катоды или катоды из нержавеющей стали, а также катоды из их смесей. В частности, угольные и стальные катоды являются наиболее предпочтительными. Катод и анод электрохимически взаимосвязаны обычным способом, посредством пористой перегородки или мембраны, например, состоящей из пористой фритты или любого другого проницаемого мембранного материала, известного в данной области техники как подходящий для заданной цели.
Согласно способу по изобретению йодирующие частицы представлены катионами йода (I+), которые на месте генерируют от любого пригодного источника йода, например, включая молекулярный I2 или подходящие производные йодидов, такие как соли щелочных металлов NaI или KI или даже йодистоводородная кислота, или любая их смесь.
В рамках настоящего изобретения предпочтительным, в частности, является молекулярный йод (I2).
Согласно стехиометрии реакции, которую используют в настоящем способе, для трийодирования каждого моля ароматического субстрата формулы (1) до соответствующего соединения формулы (2) необходимо, по крайней мере, 3 моля реакционно-способных ионов (I+).
Предпочтительно использовать незначительный избыток йодирующего агента по отношению к фенольному субстрату, с тем чтобы соотношение эквивалентов между ними могло варьировать от 1:1 до 3:1 или даже более предпочтительно от 1:1 до 2:1.
Анодную реакцию осуществляют в присутствии органического растворителя, предпочтительно полярного и/или протонного растворителя, такого как, например, ацетонитрил, низшие спирты С1-С4 и их смеси.
Предпочтительно, когда способ осуществляют в присутствии низших спиртов, где метанол является даже более предпочтительным.
Предпочтительно следует избегать водно-спиртовых систем растворителей, содержащих значительные количества воды, например смеси вода/метанол при массовом соотношении 1:1 или даже при более низком содержании воды, например 1:4.
В этих пределах, когда, в частности, осуществляют генерирование реакционно-способных ионов (I+) на стадии (a) из молекулярного йода, самого по себе или совместно с любым соответствующим йодидом, присутствие количеств воды, которые могут уменьшить его растворимость в среде растворителя, по-видимому, негативно влияет на выход реакции.
Фактически, согласно Сравнительному примеру 3, когда электрохимическое окисление йода, согласно стадии (a) способа по изобретению, осуществляют с помощью данной смеси вода/спирт, значительные количества непрореагировавшего йода регенерируют по завершении способа.
Вместо этого, в том что касается катодного отделения, отсутствует необходимость в указанных выше ограничениях выбора системы растворителей, и, следовательно, можно успешно использовать варьирование количества воды.
Обычно могут быть использованы полярный и/или протонный растворители, такие как ацетонитрил, низшие спирты, и их смесь с водой.
Среди них смеси метанол/вода являются особенно предпочтительными.
Обычно pH реакционной среды поддерживают кислотным, то есть ниже 7, добавляя кислоту, например концентрированную азотную или серную кислоту, например 97% H2SO4. Предпочтительно, когда pH поддерживают между 0 и 4, и даже более предпочтителен pH между 1 и 2.
Альтернативно стадию (a) можно осуществлять в присутствии известных соединений, пригодных для поддержания электропроводности внутри электролитической ячейки, и таким образом использовать для указанной выше цели.
Для электрохимического генерирования катионов I+ у анода можно использовать такое электрическое напряжение на электродах, чтобы анодный потенциал составлял от приблизительно 1 до приблизительно 3 В по отношению к SCE (насыщенному каломельному электроду, используемому в качестве электрода сравнения).
Предпочтительно, когда реакцию осуществляют при анодном потенциале, охватывающем от приблизительно 1,2 до приблизительно 1,8 В по отношению к SCE.
Альтернативно электролитический способ по настоящему изобретению можно осуществлять в гальваностатическом режиме, то есть во время осуществления процесса через раствор пропускают постоянный ток, например, соответствующий 1-500 мА/см2.
Температуру во время осуществления способа поддерживают постоянной, например, между 15°C и 25°C, действуя согласно общепринятым методикам. Обычно, когда тепло, генерируемое во время осуществления способа, может привести к частичному испарению растворителя, в частности, при использовании низкокипящих растворителей, таких как метанол, можно добавлять дополнительные количества свежего растворителя, с тем чтобы поддерживать неизменными и температуру, и объем растворителя.
Получающийся раствор, содержащий йодирующие частицы, затем приводят в контакт с ароматическим субстратом (1) или его пригодным раствором, как было указано ранее.
Обычно, как сообщают в экспериментальном разделе, йодирование ароматического субстрата формулы (1) осуществляют в отдельном реакторе, в котором и йодирующий раствор со стадии (a), и субстрат (1), или как таковой, или растворенный в выбранном пригодном растворителе, включая, например, растворители, указанные ранее, приводят в контакт надлежащим образом.
Предпочтительно, когда используют раствор субстрата (1) в таком низшем спирте, как метанол.
Последующая обработка реакционной смеси, с тем чтобы выделить и получить конечное соединение формулы (2), может быть выполнена согласно общепринятым методикам, широко известным в данной области техники.
В качестве примера, при использовании молекулярного йода для генерирования (I+) катионов, любой избыток (I+) или I2 в конце реакции можно было бы легко удалить согласно общепринятым способам, добавляя пригодные количества йодида щелочного металла, например KI, с тем чтобы получить I2 и затем комплекс KI3. Аналогично, избыток йода также можно было бы удалить, добавляя сульфит натрия или бисульфит до обесцвечивания реакционной среды.
Соединения формулы (1) как исходные вещества для способа по изобретению известны и, если они не являются сами по себе, по существу коммерчески доступны, все они могут быть получены по известным методикам.
Для получения общей справочной информации о соединениях формулы (1) смотрите, например, упомянутые выше WO 88/09328, WO 97/05097 и WO 00/32561.
Аналогично, любой другой реагент и/или растворитель, используемый в настоящем способе, является известным и легкодоступным.
Подробности, касающиеся способа по изобретению, изложены в нижеследующем экспериментальном разделе по электрохимическому йодированию 3,5-дизамещенных фенолов по настоящему изобретению, который, однако, кратко приводится ниже.
Плавильный котел, иначе называемый тиглем, помещают внутри кристаллизатора, подключенного к системе магнитной мешалки. Затем подвешивают платиновый анод и размещают между пористой фриттой, составляющей дно тигля, и внутренним дном кристаллизатора. Пригодное количество спиртового раствора выбранного источника йода, например раствора I2 в метаноле, заливают в кристаллизатор, так чтобы его объем был таким, что обеспечивал электрохимический контакт с пористой фриттой, и затем добавляют нужное количество кислоты. Затем в тигель вливают соответствующее количество смеси спирт/вода с последующим добавлением кислоты. Когда тигель вместе с его содержимым опускают в спиртовой раствор йода, в тигель вводят графитовый катод; и тогда система оказывается электрически связанной. Поддерживают постоянный электрический поток, затем на катоде образуется водород, и раствор йода из коричневого становится желто-оранжевым. Во время реакции в анодное отделение порциями добавляют соответствующее количество свежего метанола, с тем чтобы компенсировать испарение и поддерживать постоянный контакт между стеклообразной фриттой тигля и окружающим раствором йода. После соответствующего периода времени, например приблизительно трех часов, раствор становится бледно-желтым. Затем электроэнергию отключают и йодирующий раствор из кристаллизатора перемещают в одногорлую колбу. Затем добавляют соответствующее количество выбранного соединения формулы (1) или его раствора, или как такового или порциями, и смесь нагревают с обратным холодильником. Реакцию контролируют с помощью ТСХ (TLC) и нагревание продолжают до количественного превращения. Полученный таким образом конечный раствор охлаждают до комнатной температуры.
Затем любой избыток I+ и I2 можно устранить добавлением соответствующих растворов KI и насыщенного раствора Na2SO3, добавляемого до обесцвечивания. Растворитель затем выпаривают и остаток экстрагируют пригодным растворителем, например этилацетатом. Раствор сушат, фильтруют и упаривают до остатка. Неочищенный продукт очищают флэш-хроматографией на силикагеле или элюированием на колонке с гидрофобной полимерной смолой, сначала элюируя водой, чтобы исключить соли йода, и затем пригодным растворителем, например ацетоном, чтобы вымыть продукт.
После получения соединения формулы (2) можно легко превратить в представляющие интерес соответствующие рентгеноконтрастные вещества, имеющие формулу (5), приведенную ниже.
Следовательно, следующей целью настоящего изобретения является способ получения соединений формулы (5), приведенной ниже, включающий:
(a) электрохимическое генерирование I+ катионов из соответствующего источника йода; и
(b) йодирование 3,5-дизамещенных фенолов формулы (1) в присутствии указанных выше I+ катионов стадии (a):
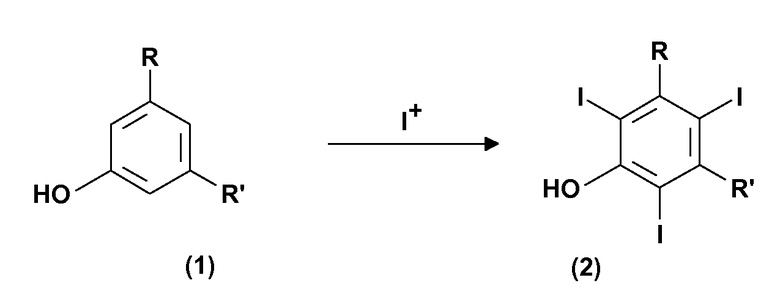
где R и R' определены выше, с тем чтобы получить соединения формулы (2);
(с) взаимодействие соединения формулы (2), или как такового, или такого, в котором гидроксигруппа в бензольном кольце присутствует в форме соли щелочного или щелочноземельного металла, с соединением формулы (3)

где R4 и R5, одинаковые или отличающиеся друг от друга, представляют собой водород или прямую или разветвленную C1-C6 алкильную группу, необязательно замещенную одной или несколькими гидрокси или C1-C6 алкоксигруппами, а Z представляет собой атом галогена или любую подходящую уходящую группу; с тем чтобы получить соединение формулы (4)
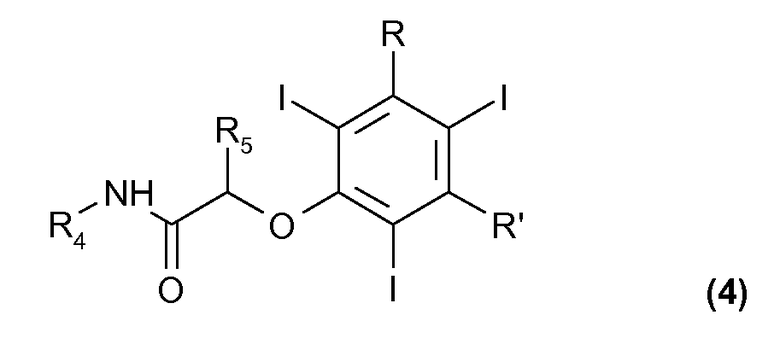
где R, R', R4 и R5 были определены выше; и
(d) осуществление перегруппировки Смайлса в присутствии оснований для соединения формулы (4), с тем чтобы получить конечное соединение формулы (5)
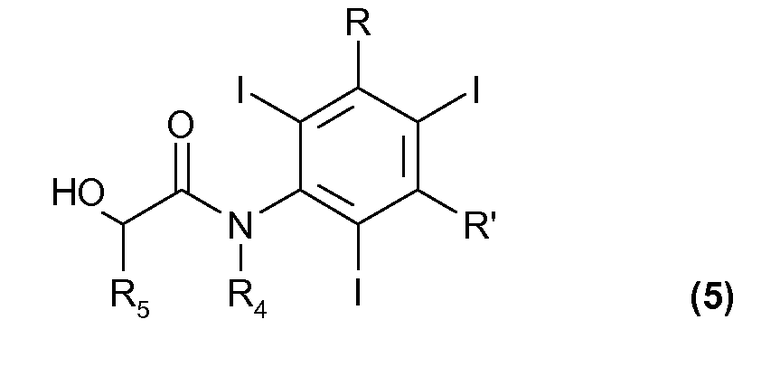
Согласно настоящему способу получения рентгеноконтрастных веществ электрохимическое йодирование стадий (a) и (b) осуществляют, как сообщали ранее, тогда как стадии (c) и (d), исчерпывающе охватывающие экспериментальные условия и необязательные их варианты, известны в данной области техники, как, например, сообщали в упомянутых выше заявках на патенты WO 88/09328, WO 97/05097 и WO 00/32561.
Предпочтительно, когда в соединениях формулы (3) Z представляет собой атом брома или хлора.
Даже более предпочтительно, когда настоящий способ может быть применен к получению широкоизвестных рентгеноконтрастных веществ, таких как йопамидол (где соответственно и R, и R' представляют собой группу -CONH-CH(CH2OH)2, R4 является водородом, а R5 представляют собой метил; см., The Merck Index, XIII Ed., 2001, No. 5073) или йомепрол (где соответственно и R, и R' представляют собой группу -CONH-CH2-CH(OH)CH2OH, R4 представляют собой метил, а R5 является атомом водорода; (см., The Merck Index, XIII Ed., 2001, No. 5071).
Поэтому дополнительной целью изобретения является способ получения йопамидола или йомепрола, исходя из соединений формулы (2a) или (2b) соответственно. Указанные соединения формулы (2a) и (2b) получают электрохимическим йодированием соединений (1a) или (1b) соответственно согласно стадиям (a) и (b) способа по изобретению.
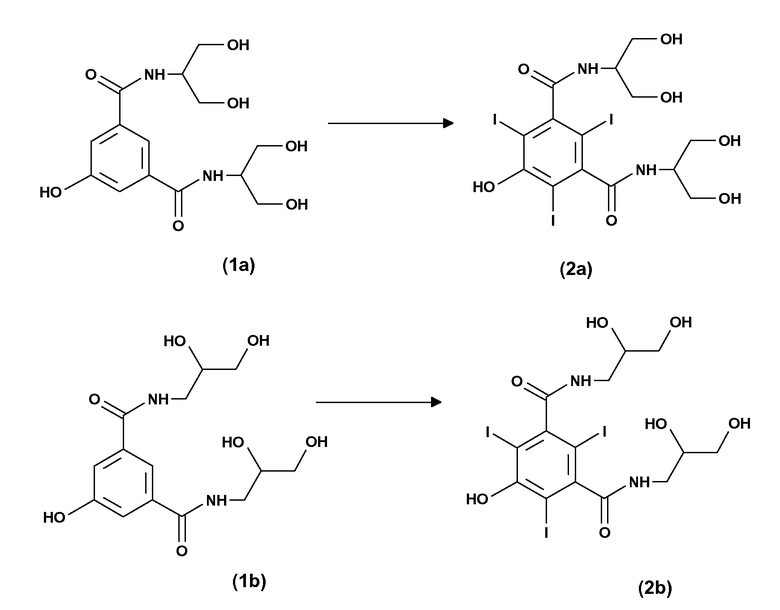
Согласно дополнительному аспекту изобретения настоящий способ также можно легко использовать для получения соответствующим образом замещенных анилинов, содержащих данные карбокси- или карбоксиэфирные группы, как по существу изложено ниже.
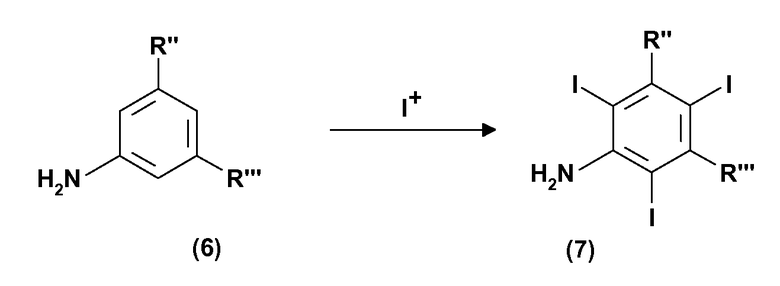
Следовательно, согласно дополнительному варианту осуществления изобретения настоящим обеспечивают способ получения 3,5-дизамещенных-2,4,6-трийоданилинов формулы (7), включающий:
(a) электрохимическое генерирование I+ катионов из пригодного источника йода; и
(b) йодирование 3,5-дизамещенных анилинов формулы (6) в присутствии указанных выше I+ катионов стадии (a):
где
R” и R”', одинаковые или отличные друг от друга, представляют карбокси -COOH или карбоксиэфирную группу -COOR1, где R1 представляет собой прямую или разветвленную С1-С6 алкильную группу, которая необязательно замещена одной или несколькими группами, выбираемыми из гидрокси, амино, сульфгидрила, С1-С6 алкокси или карбокси и/или необязательно прерывается одной или несколькими двухвалентными группами, выбираемыми из -NH-, -О-, >C=О, -(C=О)О-, -О(C=О)-, -NH(C=О)-, -(C=О)NH-, >SO или >SO2 групп.
В настоящем описании, если не оговорено иначе, в рамках формул (6) и (7) определения R” и R'” и, следовательно, R1 являются такими, как изложено ранее.
Предпочтительно, когда в рамках указанного выше способа получения соединений формулы (7) R” и R'” являются одинаковыми, и оба представляют собой группу -COOH или -COOR1, где R1 является прямой или разветвленной С1-C4 алкильной группой.
Указанный выше способ осуществляют посредством первоначальной генерации источника йодирования I+, что, как сообщали ранее, дает возможность полного трийодирования ароматического кольца с высокими выходом и чистотой на последующей стадии (b).
К тому же, в этом случае известны исходные вещества формулы (6) или же легко могут быть получены по известным методикам, например, гидрируя соответствующим образом или, так или иначе, восстанавливая соответствующую нитроизофталевую кислоту, с последующим необязательным преобразованием типа функциональности по общепринятым методикам превращения карбоксигрупп в карбоксиэфирные группы.
Полученные таким образом соединения формулы (7) являются применимыми интермедиатами для синтеза рентгеноконтрастных веществ, среди которых находятся упомянутые выше йопамидол и йомепрол, как используемые в методиках, широкоизвестных в данной области техники.
Нижеследующие примеры приведены для того, чтобы лучше проиллюстрировать настоящее изобретение, не накладывая на него каких-либо ограничений.
Пример 1
Генерирование раствора I +
Концентрированный раствор йода в метаноле (0,55 М, 10 мл) вносили в 100-мл химический стакан, разбавляли метанолом (20 мл) и подкисляли 0,2 мл 97% H2SO4. Пористый тигель (с диаметром пор 6 мкм) частично заполняли метанолом (2 мл), водой (6 мл) и 97% H2SO4 (0,2 мл) и вставляли в химический стакан. Анод в виде платиновой пластины вставляли в химический стакан и в тигель вводили графитовый катод. Электролиз осуществляли в гальваностатическом режиме (200 мА/см2) до тех пор, пока анолит не обесцвечивался до бледно-желтого цвета. Полученный раствор использовали как таковой в последующих реакциях йодирования.
Пример 2
Синтез 3,5-бис(2,3-дигидроксипропиламинокарбонил)-2,4,6-трийодфенола

3,5-Бис(2,3-дигидроксипропиламинокарбонил)фенол (56,5 мг, 0,17 ммоль) добавляли в круглодонную колбу, содержащую раствор I+ (полученный из 1,55 ммоль I2 и 50 мл MeOH, действуя согласно описанному в примере 1). Реакционную смесь кипятили с обратным холодильником в течение 1 часа. Затем добавляли дополнительное количество 3,5-бис(2,3-дигидроксипропиламинокарбонил)фенола (56,5 мг, 0,17 ммоль) и раствор 1 час кипятили с обратным холодильником. В заключение добавляли третью порцию 3,5-бис(2,3-дигидроксипропиламинокарбонил)фенола (56,5 мг, 0,17 ммоль) и раствор кипятили с обратным холодильником до тех пор, пока не наблюдали полного превращения. После охлаждения добавляли KI (257 мг, 1,55 ммоль) и по каплям добавляли насыщенный водный раствор Na2SO3 до тех пор, пока раствор не сделался бледно-желтым. Метанол удаляли при пониженном давлении и желтый остаток очищали на колонке Amberlite® XAD 1600, элюируя водой (100 мл), пока соли не были удалены полностью, и затем смесью 8/2 вода/ацетон (100 мл). Выход: 300 мг, 82%.
Спектральные данные
Пример 3
Синтез 3,5-бис(1-гидроксиметил-2-гидроксиэтиламинокарбонил)-2,4,6-трийодфенола
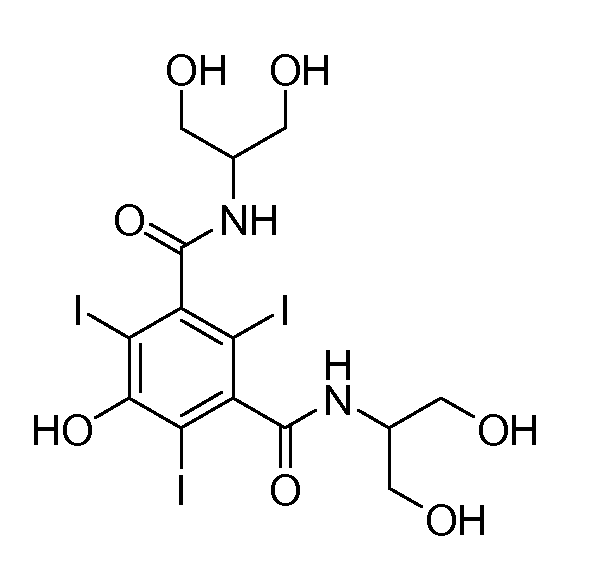
В круглодонную колбу, содержащую раствор I+ (полученный из 1,55 ммоль I2 и 50 мл MeOH, действуя согласно описанному в примере 1), добавляли 3,5-бис(1-гидроксиметил-2-гидроксиэтиламинокарбонил)фенол (56,5 мг, 0,17 ммоль).
Реакционную смесь кипятили с обратным холодильником в течение 1 часа. Затем добавляли дополнительное количество указанного выше производного фенола (56,5 мг, 0,17 ммоль) и раствор кипятили с обратным холодильником в течение 1 часа. В заключение добавляли третью порцию указанного выше производного фенола (56,5 мг, 0,17 ммоль) и раствор кипятили с обратным холодильником, пока не наблюдали полного превращения. После охлаждения добавляли KI (257 мг, 1,55 ммоль) и по каплям добавляли насыщенный водный раствор Na2SO3, пока раствор не становился бледно-желтым.
Метанол удаляли при пониженном давлении и желтый остаток очищали на колонке Amberlite® XAD 1600, элюируя водой (100 мл) до полного удаления солей и затем смесью 8/2 вода/ацетон (100 мл). Выход: 200 мг, 55%.
Спектральные данные
Пример 4
Синтез 3,5-бис(н-бутоксикарбонил)-2,4,6-трийодфенола
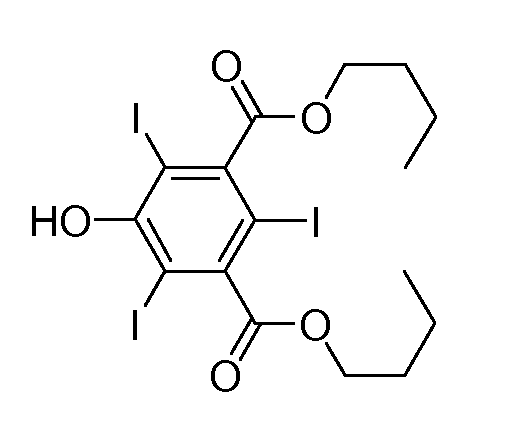
В круглодонную колбу, содержащую раствор I+ (полученный из 0,78 ммоль I2 и 50 мл MeOH, действуя согласно описанному в примере 1), добавляли 3,5-бис(н-бутоксикарбонил)фенол (25,0 мг, 0,085 ммоль). Полученный раствор 1 час кипятили с обратным холодильником. Затем добавляли дополнительное количество указанного выше производного фенола (25,0 мг, 0,085 ммоль) и раствор 1 час кипятили с обратным холодильником. В заключение добавляли третью порцию указанного выше производного фенола (25,0 мг, 0,085 ммоль) и раствор 7 ч кипятили с обратным холодильником, пока не наблюдали полного превращения.
После охлаждения добавляли KI (125 мг, 0,76 ммоль) и по каплям добавляли насыщенный водный раствор Na2SO3, пока раствор не принимал бледно-желтую окраску.
Метанол удаляли при пониженном давлении и полученный таким образом желтый раствор разбавляли водой и экстрагировали AcOEt (3x20 мл). Органическую фазу сушили над Na2SO4 и упаривали при пониженном давлении. Черный остаток очищали флэш-хроматографией на силикагеле (элюент: смесь 9 петролейный эфир/0,5 изопропанол/0,5 AcOEt). Выход: 100 мг, 58%.
Спектральные данные
Пример 5
Синтез 3,5-бис(метоксикарбонил)-2,4,6-трийодфенола
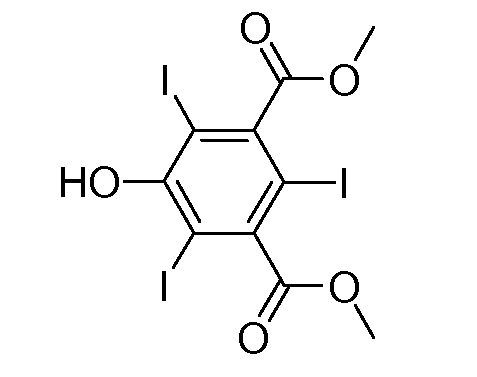
В круглодонную колбу, содержащую раствор I+ (полученный из 0,78 ммоль I2 и 50 мл MeOH, действуя согласно описанному в примере 1), добавляли 3,5-бис(метоксикарбонил)фенол (53,0 мг, 0,085 ммоль). Полученный раствор 1 ч кипятили с обратным холодильником. Затем добавляли дополнительное количество указанного выше производного фенола (53,0 мг, 0,085 ммоль) и раствор 1 ч кипятили с обратным холодильником. В заключение добавляли третью порцию указанного выше производного фенола (53,0 мг, 0,085 ммоль) и раствор 10 ч кипятили с обратным холодильником, пока не наблюдали полного превращения.
После охлаждения добавляли KI (125 мг, 0,76 ммоль) и по каплям добавляли насыщенный водный раствор Na2SO3, пока раствор не принимал бледно-желтую окраску.
Метанол удаляли при пониженном давлении, и полученный таким образом желтый раствор разбавляли водой и экстрагировали AcOEt (3×20 мл). Органическую фазу сушили над Na2SO4 и упаривали при пониженном давлении. Черный остаток очищали флэш-хроматографией на силикагеле (элюент: смесь 9 петролейный эфир/0,5 изопропанол/0,5 AcOEt).
Выход: 90 мг, 60%.
Спектральные данные
Сравнительный Пример 1
Йодирование фенола
В круглодонную колбу, содержащую раствор I+ (полученный в примере 1 из 0,78 ммоль I2 и 50 мл MeOH,), добавляли фенол (24,4 мг, 0,26 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 5 ч. После охлаждения добавляли KI (131 мг, 0,79 ммоль) и по каплям добавляли насыщенный водный раствор Na2SO3.
Метанол удаляли при пониженном давлении, и полученный таким образом желтый раствор разбавляли водой и экстрагировали AcOEt (3×20 мл). Органическую фазу сушили над Na2SO4 и упаривали при пониженном давлении. Коричневый остаток загружали в колонку для флэш-хроматографии и элюировали смесью 9 петролейный эфир/1 AcOEt.
Выход: 25 мг, 21%.
Спектральные данные
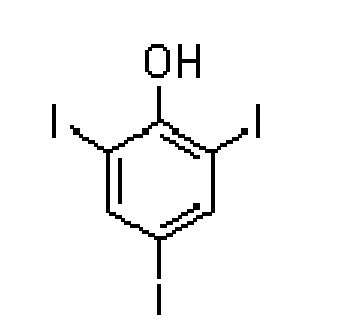
ESI-MS (масс-спектры при ионизации электрораспылением) (в режиме отрицательных ионов)
Рассчитано для C6H3I3O: 471,7 u.m.a. (единиц атомной массы)
Найдено: 471,1 (M-H+), 964,4 (M+Na+-2H+)
В последующих фракциях собирали более полярные побочные продукты, среди которых были 2-йодгидрохинон и дийодбифенол.
Сравнительный Пример 2
Йодирование пирогаллола
В круглодонную колбу, содержащую раствор I+ (полученный из 0,78 ммоль I2 и 50 мл MeOH аналогично примеру 1), добавляли пирогаллол (32 мг, 0,26 ммоль). Реакционную смесь 3 ч кипятили с обратным холодильником. После охлаждения добавляли KI (131 мг, 0,79 ммоль) и по каплям добавляли насыщенный водный раствор Na2SO3.
Метанол удаляли при пониженном давлении и желтый раствор, полученный таким образом, разбавляли водой и экстрагировали AcOEt (3×20 мл). Органическую фазу сушили над Na2SO4 и упаривали при пониженном давлении. Хроматографический анализ коричневого остатка, полученного таким образом, показал наличие комплекса и неразделяемой смеси окисленных и йодированных побочных продуктов.
Сравнительный Пример 3
Электрохимическое генерирование I+ в водно-спиртовой среде
Аппарат электрохимического йодирования состоял из ячейки, сформированной из:
анодного отделения с платиновым анодом (фольга, 3 см2) и магнитной мешалкой;
катодного отделения с катодом из нержавеющей стали (пластина, 8 см2).
Два отделения разделяли пористой стеклообразной фриттой (средний размер пор 6 мкм). В анодное отделение помещали 0,15 М раствор NaBF4 в смеси метанол/вода (50/50, 40 мл) и туда при интенсивном перемешивании добавляли йод (762 мг, 3 ммоль). Вследствие низкой растворимости йода в смеси большая часть его все же присутствовала в виде осадка на дне анодного отделения. Смесь была бледно-оранжевого цвета. Устанавливали pH, равный 1,5, используя 40% водный раствор HBF4. В катодное отделение помещали 0,15 М раствора NaBF4 в смеси метанол/вода (50/50, 15 мл).
Использовали постоянный ток в 400 мА. Приблизительно через 30 минут ток начинал уменьшаться, и через приблизительно 40 минут ток падал до ~60 мА при максимальном напряжении блока питания, составляющем 31,6 В. Раствор был желтого цвета, и значительная часть исходного йода все еще находилась в нижней части отделения анолита. Твердый йод регенерировали фильтрованием, промывали водой и сушили, получая массу 678 мг (регенерировали 89% непрореагировавшего йода).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ЙОДИРОВАНИЯ ПРОИЗВОДНЫХ ФЕНОЛА | 2011 |
|
RU2563645C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЙОДИРУЮЩЕГО АГЕНТА | 2010 |
|
RU2528402C2 |
| КОНТРАСТНЫЕ АГЕНТЫ | 2008 |
|
RU2469021C2 |
| СПОСОБ ИОДИРОВАНИЯ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ | 2010 |
|
RU2506254C2 |
| 2,4-Диарил-6-алкил-1,3,5-триазины и способ их получения | 2023 |
|
RU2812149C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ МЕТИЛФЕНОЛОВ В ВОДНЫХ СРЕДАХ | 2011 |
|
RU2459203C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЛЕВОТИРОКСИНА И ЕГО СОЛЕЙ | 2015 |
|
RU2673540C2 |
| СИСТЕМА АРИЛФЕНОКСИКАТАЛИЗАТОРА ДЛЯ ПОЛУЧЕНИЯ ГОМОПОЛИМЕРА ЭТИЛЕНА ИЛИ СОПОЛИМЕРОВ ЭТИЛЕНА И АЛЬФА-ОЛЕФИНОВ | 2005 |
|
RU2374267C2 |
| ИОДИРОВАННЫЕ АРИЛЬНЫЕ СОЕДИНЕНИЯ И ДИАГНОСТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1995 |
|
RU2145955C1 |
| СПОСОБ ПОЛУЧЕНИЯ СУЛЬФАТИРОВАННОГО ПРОИЗВОДНОГО 3,5-ДИЙОД-О-[3-ЙОДФЕНИЛ]-L-ТИРОЗИНА | 2012 |
|
RU2610093C2 |
Изобретение относится к способу получения 3,5-дизамещенных-2,4,6-трийодфенолов формулы (2) (где значения радикалов R и R' определены в п.1 формулы изобретения). Способ осуществляют путем электрохимического генерирования катионов I+ из природного источника йода в присутствии протонного полярного растворителя в отдельном реакторе. Затем проводят йодирование 3,5-дизамещенных фенолов в присутствии катионов I+, полученных на предыдущей стадии. Также изобретение относится к способам получения соединений формулы (5), йопамидола и йомепрола. Технический результат - усовершенствованные способы получения 3,5-дизамещенных-2,4,6 - трийодфенолов. 3 н. и 12 з.п. ф-лы, 8 пр.

1. Способ получения 3,5-дизамещенных-2,4,6-трийодфенолов формулы (2), включающий:
(a) электрохимическое генерирование катионов I+ из пригодного источника йода в растворителе в анодном отделении, выбранном из ацетонитрила, низших спиртов С1-С4 или их смеси; и
(b) йодирование 3,5-дизамещенных фенолов формулы (1) в присутствии катионов I+, полученных выше на стадии (а):
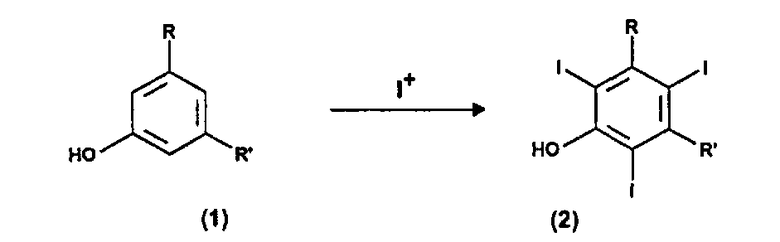
где R и R', одинаковые или отличные друг от друга, представляют собой группу, выбираемую из карбокси -СООН, карбоксиэфира -COOR1 и карбоксамидо -CONH2, -CONHR1 или -CONR2R3; где R1, R2 и R3, одинаковые или отличные друг от друга, представляют прямую или разветвленную C1-C6 алкильную группу, которая необязательно замещена одной или несколькими группами, выбираемыми из гидрокси, амино, сульфгидрил, С1-С6алкокси или карбокси, и/или необязательно прерывается одной или несколькими двухвалентными группами, выбираемыми из -NH-, -O-, >С=O, -(С=O)O-, -O(С=O)-, -NH(C=O)-, -(C=O)NH-, >SO или >SO2 групп, и где стадию (b) осуществляют в отдельном реакторе.
2. Способ по п.1, в котором в соединениях формулы (1) и (2)
R и R', одинаковые или отличные друг от друга, представляют собой группу, выбираемую из карбокси -СООН, карбоксиэфира -COOR1 и карбоксамидо -CONH2, -CONHR1 или -CONR2R3, где R1, R2 и R3, одинаковые или отличные друг от друга, представляют собой прямую или разветвленную С1-С4 алкильную группу, необязательно замещенную одной или несколькими гидроксигруппами.
3. Способ по п.2, в котором в соединениях формулы (1) и (2) R и R', одинаковые или отличные друг от друга, представляют собой группу, выбираемую из:
-СООН,
-СООСН3,
-СОО(СН2)3-СН3,
-CONH2,
-CONHCH3,
-CONHCH2-CH(OH)-CH2(OH),
-CONHCH[CH2OH]2.
4. Способ по любому из пп.1-3, в котором катионы йода, I+, генерируют in situ из пригодного источника йода, включая молекулярный I2, йодид натрия или калия или йодистоводородную кислоту, или их смесь.
5. Способ по п.4, в котором катионы йода, I+, генерируют in situ из молекулярного I2.
6. Способ по п.1, осуществляемый в присутствии полярного и/или протонного органического растворителя, одинакового или различающегося для стадий (а) и (b).
7. Способ по п.1, в котором растворителем является низший спирт С1-С4.
8. Способ по п.1, в котором растворителем является метанол.
9. Способ по любому из пп.1-3, 5-8, в котором получающийся раствор стадии (а) перемещают в отдельный реактор и подвергают контакту с соединением формулы (1) или его раствором.
10. Способ по п.4, в котором получающийся раствор стадии (а) перемещают в отдельный реактор и подвергают контакту с соединением формулы (1) или его раствором.
11. Способ получения соединения формулы (5), включающий:
(a) электрохимическое генерирование катионов I+ из пригодного источника йода; и
(b) йодирование 3,5-дизамещенных фенолов формулы (1) в присутствии катионов I+, получаемых выше на стадии (а):
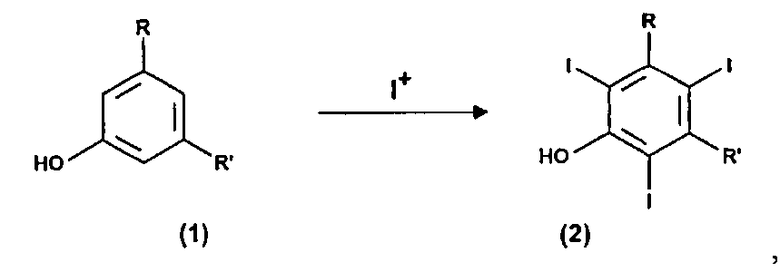
где R и R' определены в п.1, с тем чтобы получить соединение формулы (2);
с) взаимодействие соединения формулы (2), или как такового, или в котором гидроксигруппа бензольного кольца присутствует в форме соли щелочного или щелочноземельного металла, с соединением формулы (3) 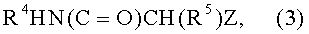
в котором R4 и R5, одинаковые или отличные друг от друга, представляют собой водород или прямую или разветвленную С1-С6 алкильную группу, необязательно замещенную одной или несколькими гидрокси- или С1-С6 алкоксигруппами, a Z представляет собой атом галогена или любую пригодную уходящую группу, с тем чтобы получить соединение формулы (4)
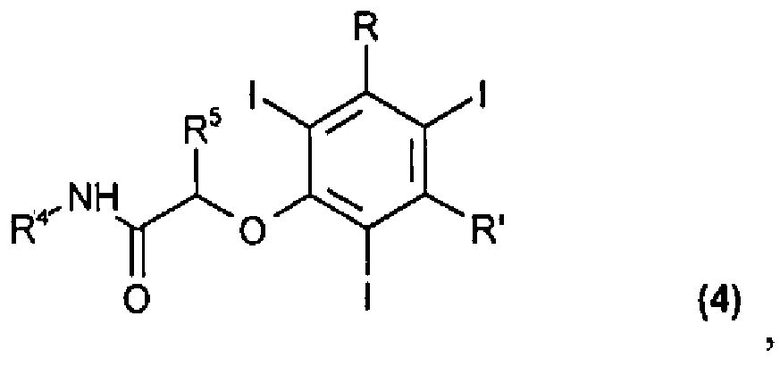
в котором R, R', R4 и R5 определены выше; и
(d) подвергание соединения формулы (4) перегруппировке Смайлса в присутствии оснований, с тем чтобы получить конечное соединение формулы (5)

12. Способ по п.11, в котором в соединениях формулы (3) Z представляет собой атом брома или хлора.
13. Способ получения соединения формулы (5) по п.11 или 12, в котором и R, и R' представляют собой -CONH-CH(CH2OH) 2 группу, R4 является водородом, a R5 является метилом.
14. Способ для получения соединения формулы (5) по п.11 или 12, в котором и R, и R' представляют группу -CONH-CH2-CH(OH)CH2OH, R4 является метилом, a R5 является водородом.
15. Способ получения йопамидола или йомепрола, исходя из соответствующих соединений формулы (2), согласно способу по любому из пп.1-11.
| WO 00/32561 А1, 08.06.2000 | |||
| US 3833490 А, 03.09.1974 | |||
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| вСЕСОЮЗНАЯ | 0 |
|
SU376858A1 |
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| US 3647864 A, 07.05.1972 | |||
| Larry l | |||
| Планшайба для точной расточки лекал и выработок | 1922 |
|
SU1976A1 |
| RU 2060246 C1, 20.05.1996. | |||

