Область техники, к которой относится изобретение
Настоящее изобретение относится к области органического синтеза, в частности, к способу синтеза N,N-дизамещенных аминометилстиролов или α-аминометилстиролов (I)
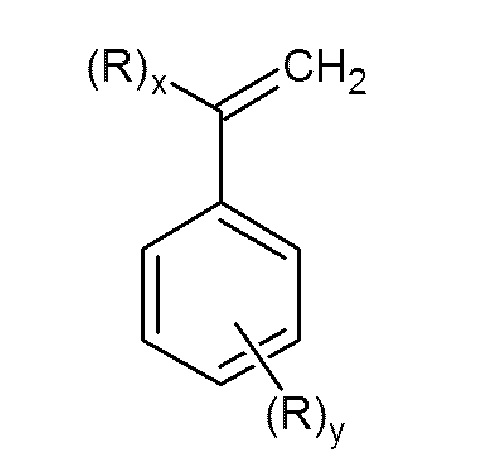
которые являются перспективными мономерами для получения функционализированных каучуков и конструкционных пластиков.
Уровень техники
Взаимодействие алкилгалогенидов с вторичными аминами является общим методом синтеза третичных аминов. Например, для синтеза N,N-диэтиламинометистирола по указанному методу проводят реакцию между винилбензилхлоридом и N,N-диэтиламином в смеси полярных растворителей, а именно, в растворе диоксана в воде [Giffin D.Jonens, James R.Runyon, Josephine Ong, Ind. Eng. Chem. 53 volume 297-8 (1961)] или этанола в воде [JPS58181038A]. Известные способы характеризуются высоким расходом N,N-диэтиламина и необходимостью регенерации гидрохлорида амина, вследствие его образования из исходного амина при протекании реакции и образовании соляной кислоты, а также необходимостью дополнительной стадии очистки от полярных растворителей и препаративно неудобной стадией очистки конечных соединений.
Известно также получение N,N-дизамещенных аминометилстиролов взаимодействием винилбензилгалогенида и вторичного амина в двухфазной системе (JPS6379855). N,N-диэтиламинометистирол получают в эмульсии винилбензилхлорида и N,N-диэтиламина в органическом растворителе, например, в бензоле. Данный способ имеет те же недостатки, что и представленные выше методы, а именно, высокий расход N,N-диэтиламина, необходимость регенерации гидрохлорида амина, дополнительные стадии очистки конечных соединений.
В патенте JPS529092A, также описано применение двухфазной системы для получения N,N-диэтиламинометистирола, при этом проводят взаимодействие 40% водного раствора N,N диэтиламина с винилбензилхлоридом в присутствии гидроксида калия. Данный метод характеризуется длительностью протекания процесса и невысоким выходом целевого соединения, вследствие инициирования побочного процесса полимеризации исходного и конечного соединений гидроксидом калия.
В заявке на патент JP2004123559 описан способ получения N,N-диалкилзамещенных аминометилстиролов, который является наиболее близким техническим решением к заявленному изобретению. Способ осуществляют в двухфазной системе, образуемой водным раствором N,N-диалкиламина и винилбензилхлоридом без использования основания, но с применением бромида тетраалкиламмония в качестве катализатора межфазного переноса. Это наиболее современный способ получения N,N-диалкилзамещенных аминометилстиролов. Из всех представленных способов он характеризуется наивысшими значениями конверсий исходных продуктов и выходов конечных соединений, быстрым протеканием процесса получения конечных соединений и простым способом их очистки. К недостаткам данного способа можно отнести высокий расход применяемого катализатора для обеспечения необходимой скорости процесса и конверсии. Более того, вследствие склонности N,N-дизамещенных аминометилстиролов, как и самого стирола к радикальной полимеризации, особенно усиливающейся при нагревании (Александров И.А., Перегонка и ректификация в нефтепереработке, 1981, М.: Химия, 352 с.), в ходе синтеза и, особенно, в ходе очистки целевых продуктов вакуумной перегонкой, согласно данному способу, неизбежно будет происходить частичное осмоление целевого продукта и уменьшение его выхода. Кроме того, растворимость целевых соединений в воде также приводит к снижению выхода за счет выведения части продукта с водной фракцией при промывке органической фракции водой. Еще одним недостатком известного способа является применение большого избытка амина, часть которого используется для нейтрализации соляной кислоты, образующейся, в ходе протекания реакции нуклеофильного замещения атома хлора аминогруппой. Вследствие этого возникает необходимость дополнительной стадии регенерации амина из образовавшегося гидрохлорида, что усложняет процесс.
В патенте SU233680 описано получение α-диэтиламинометилстирола применением уже описанного выше приема с использованием двухфазной системы, состоящей из водного раствора диэтиламина и α-хлорметилстирола, и дополнительно содержащей добавку эмульгатора ОП-7. При этом достигается 100% конверсия. Основными недостатками известного способа являются использование большого избытка диэтиламина, необходимость регенерации амина из образующегося гидрохлорида, как следствие препаративно неудобный способ выделения и очистки конечных соединений и длительное время проведения синтеза.
Общим недостатком известных способов также является узкий спектр получаемых соединений, ограничивающийся N,N-диалкиламинометилстиролами и N,N-диалкил-α-аминометилстиролами.
Задачей данного изобретения является получение N,N-дизамещенных аминометилстиролов или α-аминометилстиролов общей формулы (I)
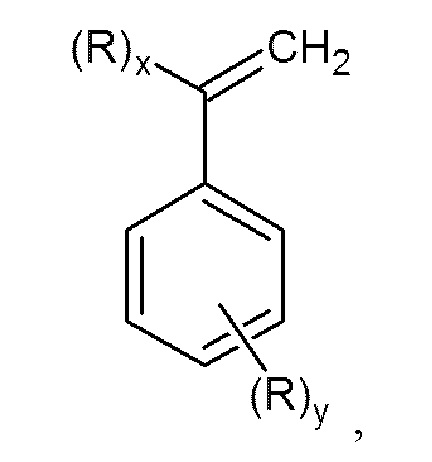
с высокой конверсией исходных продуктов, а также высоким выходом N,N-дизамещенных аминометилстиролов или α-аминометилстиролов без использования большого избытка вторичного амина, дорогостоящих катализаторов и трудоемких способов выделения и очистки конечных продуктов.
Техническим результатом заявленного изобретения является получение N,N-дизамещенных аминометилстиролов или α-аминометилстиролов общей формулы (I) с высокими выходами, высокой конверсией исходных продуктов, а также сокращение времени процесса. Кроме того, заявленный способ обеспечивает исключение дорогостоящих катализаторов и оборудования, трудоемких и ресурсозатратных методов выделения и очистки целевых соединений и уменьшение расхода катализатора и реагентов.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способу получения N,N-дизамещенных аминометилстиролов или α-аминометилстиролов общей формулы (I):
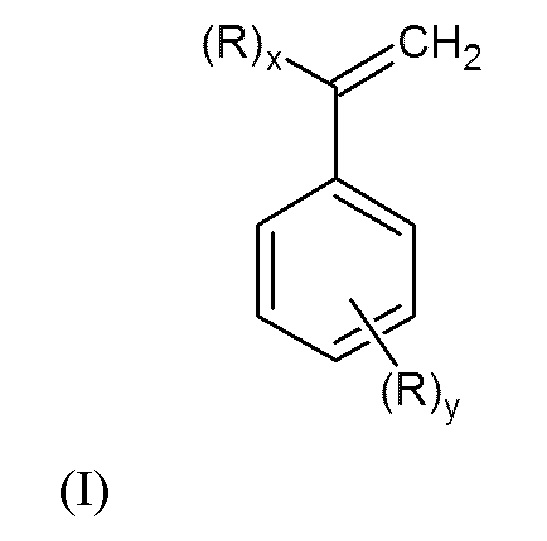
или их смеси
где x=1, y=0 или x=0, y=1;
R представляет собой группу -СН2N(R 1)(R2), в которой R1 и R2 могут быть одинаковыми или различными, и независимо представляют собой алкил или циклоалкил необязательно замещенные одним или более заместителями, выбранными из гидроксигруппы, диалкиламиногруппы, алкоксигруппы, арилоксигруппы, алкилсульфанильной группы, арилсульфанильной группы, арила;
или арил, необязательно замещенный одним или более заместителями, выбранными из галогена, алкила, диалкиламиногруппы, алкоксигруппы, арилоксигруппы, алкилсульфанильной группы, арилсульфанильной группы, арила;
или гетероарил, содержащий один или более атомов азота, где гетероарил необязательно замещен одним или более заместителями, выбранными из галогена, алкила, диалкиламиногруппы, алкоксигруппы, арилоксигруппы, алкилсульфанильной группы, арилсульфанильной группы, арила;
или R1 и R2, взятые вместе с азотом, образуют 5-6-членное гетероциклическое или 5-членное гетероароматическое кольцо, необязательно содержащие один или более дополнительных гетероатомов, выбранных из азота, кислорода или серы;
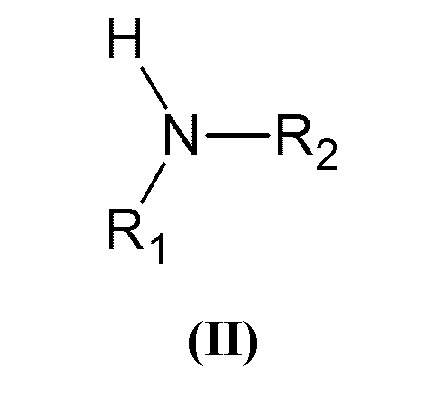
который включает взаимодействие вторичного амина общей формулы (II):
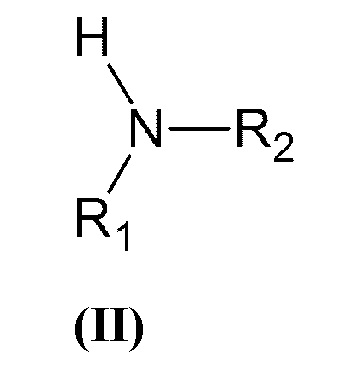
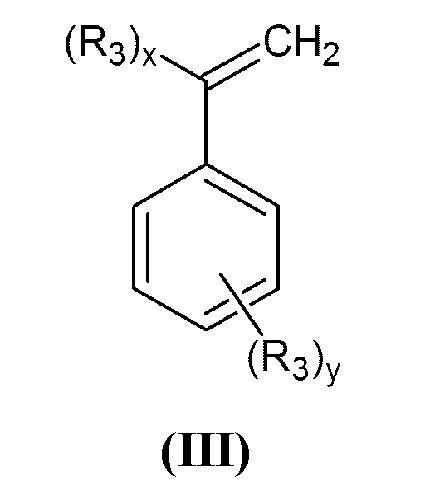
где R1 и R2 принимают значения, определенные выше, с винилбензилгалогенидом, или α-галогенметилстиролом общей формулы (III):

в которой R3 представляет собой группу -СН2-Hal, а x=1, y=0 или x=0, y=1;
в двухфазной системе, в щелочной среде, в атмосфере инертного газа в присутствии катализатора, представляющего собой иодиды аммониевых или фосфониевых соединений и антиоксиданта.
В контексте данного описания алкил представляет собой насыщенный или ненасыщенный С1-С6 алкил с линейной или разветвленной цепью; циклоалкил представляет собой С3-С7 циклоалкил; арил представляет собой 6-членную арильную группу; гетероарил представляет собой ненасыщенное 6-членное кольцо, содержащее один или более атомов азота.
Заявленный способ проводят в присутствии катализатора межфазного переноса йодида аммониевых или фосфониевых соединений, которые предпочтительно выбраны из группы, состоящей из тетраалкиламмоний или тетраалкилфосфоний иодидов, тетраариламмоний или тетраарилфосфоний иодидов, арилтриалкиламмоний или арилтриалкилфосфоний иодидов, в количестве 0,01-10 мольных %, предпочтительно 0,1-5 мольных %, более предпочтительно 0,5-1 мольных % на количество используемого соединения III.
В качестве щелочной среды в способе по настоящему изобретению используют водный раствор карбоната щелочного металла или аммония с концентрацией 10-50 массовых %, предпочтительно 25-40 массовых %, более предпочтительно 30-35 массовых %. Эти же концентрации предпочтительны при применении карбоната аммония, при использовании карбоната натрия предпочтительно использовать водные растворы с концентрацией 10-14 массовых %.
Используемый в способе антиоксидант представляет собой фенольный или тиофенольный антиоксидант, в количестве 0,01-10 мольных %, предпочтительно 0,1-5 мольных %, наиболее предпочтительно 0,5-1 мольных % на количество используемого соединения III.
Заявленный способ проводят при температуре менее 150 °С, более предпочтительно при температуре 20-130°С, более предпочтительно 80-120°С и давлении 0,5-2 атм., предпочтительно 1-1,5 атм. Время реакции согласно заявленному способу составляет 1-3 ч, более предпочтительно 1,5- 2 ч.
Мольное соотношение вторичный амин: винилбензилгалогенид или α-галогенметилстирол, в заявленном способе составляет 1,0÷1,5/1, более предпочтительно 1,01÷1,1/1.
Дополнительно в способе могут быть использованы органические растворители, выбранные из группы, включающей: алифатические углеводородные растворители, ароматические углеводородные растворители, хлорсодержащие углеводородные растворители, в частности бензол, толуол, гексан, гептан, хлорбензол, тетрахлорметилметан, трихлорметан, хлористый метилен, или любые их смеси.
Способ настоящего изобретения дополнительно включает стадию экстракции соединений формулы (I) из водного раствора указанного карбоната щелочного металла или аммония органическим растворителем, выбранным из группы, включающей: алифатические углеводородные растворители, ароматические углеводородные растворители, хлорсодержащие углеводородные растворители, а также использование вакуумной перегонки для выделения целевого соединения формулы (I).
В предпочтительном варианте способ осуществляют в течение 2 часов в атмосфере азота при температуре 80-120°C в двухфазной системе при взаимодействии соединений общей формулы II и III в водном растворе карбоната калия в присутствии катализатора тетрабутиламмоний йодида (1 мольных %) с применением антиоксиданта 2,2-метилен-бис(4-метил-6-третбутилфенол) (агидола-2). Дальнейшее разделение фаз и очистка вакуумной перегонкой обеспечивают высокие выходы целевых соединений общей формулы I.
Настоящее изобретение также относится к новым соединениям общей формулы (I), выбранным из:
3-[(N,N-диизопропиламино)метил]стирола;
N-(3-этенилбензил)-N-метиланилина;
N-(4-этенилбензил)-N-метиланилина.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В контексте данного изобретения в соединениях общей формулы (I) алкил предпочтительно представляет собой метил, этил, пропил, изопропил, бутил, втор-бутил, трет-бутил, изобутил, гексил, бут-3-енил, бут-3-инил; циклоалкил предпочтительно представляет собой циклогексил, циклогептил; необязательно замещенный арил предпочтительно представляет собой фенил, 2-хлорфенил, 2-бромфенил, 2-иодфенил, 2-N,N-диметиламинофенил, 2-метоксифенил, 2-феноксифенил, 2-(метилсульфанил)фенил, 2-метилсульфанилфенил; 6- членный гетероарил предпочтительно представляет собой пиридинил, пиримидинил.
Заместители R1 и R2 предпочтительно выбирают из:
- алкила или циклоалкила необязательно замещенных одной или несколькими гидроксигруппами, в частности гидроксиметила, 2-гидроксиэтанила, 2-гидроксипропила, 2-гидроксиизопропила, 3-гидроксипропила, 2,3-дигидроксипропила, 2-гидроксибутила 2-гидроксициклогексила;
- алкила или циклоалкила необязательно замещенных диалкиламиногруппой, в частности N,N-диметиламинометила N,N-дифениламинометила, N-метил-N-фениламинометила, N,N-диметиламиноэтила, N,N-диизопропиламинометила, N,N-дициклогексиламинометила, 2-(N,N-диметиламино)изопропила, 2-(N,N-диметиламино)циклогексила;
- алкила или циклоалкила необязательно замещенных алкокси- и/или арилоксигруппой, в частности метоксиметила, изопропоксиметила, феноксиметила, 2-(метокси)изопропила, 2-(метокси)циклогексила.
- алкила или циклоалкила необязательно замещенных алкил- и/или арилсульфанильной группой, в частности (метилсульфанил)метила, (изопропилсульфанил)метила, 2-(метилсульфанил)циклогексила, (фенилсульфанил)метила.
R1 и R2, взятые вместе с азотом, образуют 5-6-членное гетероциклическое кольцо, которое необязательно содержит один или более дополнительных гетероатомов, выбранных из азота, кислорода или серы, в частности пирролидинил, морфолинил, тиоморфолинил, пиперидинил, 1,3-оксазолидинил, 1,3-тиозолидинил, N-метилпиперазинил, N-фенилпиперазинил.
R1 и R2, взятые вместе с азотом, образуют 5-членное гетероароматическое кольцо, необязательно содержащее один или два дополнительных гетероатома, выбранных из азота, кислорода, серы, в частности имидазолил, пиразолил, фуранил, пирролил, оксазолил, тиазолил.
В качестве галогена используют фтор, хлор, йод или бром.
В способе настоящего изобретения в качестве вторичных аминов могут быть использованы соединения общей формулы (II), выбранные из группы, включающей: N,N-диалкиламины, N,N-диариламины, N-(алкил)-N-(арил)амины, гетероциклические и гетероароматические соединения, содержащие в кольце один атом азота, имеющий в качестве заместителя один атом водорода, в частности N,N-диалкиламины, N,N-бис(алкоксиалкил)амины, N,N-бис(арилоксиалкил)амины, N,N-бис(N',N'-диалкиламиноалкил)амины, N,N-бис(N',N'-диариламиноалкил)амины, N-(алкил)-N-(алкилоксиалкил)амины, N-(алкил)-N-(арилоксиалкил)амины, N-(алкил)-N-(N',N'-диалкиламиноалкил)амины, N-(алкил)-N-(N',N'-диариламиноалкил)амины, N,N-диалкениламины, N-(алкенил)-N-(алкилоксиалкил)амины, N-(алкенил)-N-(алкилоксиалкенил)амины, N-(алкенил)-N-(алкилоксиалкинил)амины, N-(алкенил)-N-(арилоксиалкил)амины, N-(алкенил)-N-(N',N'-диалкениламиноалкил)амины, N-(алкенил)-N-(N',N'-диалкиламиноалкил)амины, N-(алкенил)-N-(N',N'-диариламиноалкил)амины, N,N-диалкиниламины, N-(алкинил)-N-(алкинилоксиалкил)амины, N-(алкинил)-N-(алкилоксиалкил)амины, N-(алкинил)-N-(арилоксиалкил)амины, N-(алкинил)- N-(N',N'-диалкиламиноалкил)амины, N-(алкинил)-N-(N',N'-диалкиниламиноалкил)амины, N-(алкинил)-N-(N',N'-диариламиноалкил)амины, N-(алкоксиалкил)-N-(арилоксиалкил)амины, N-(алкоксиалкил)-N-(N',N'-диалкиламиноалкил)амины, N-(алкоксиалкил)-N-(N',N'-диариламиноалкил)амины, N-(арилоксиалкил)-N-(N',N'-диалкиламиноалкил)амины, N-(арилоксиалкил)-N-(N',N'-диариламиноалкил)амины, N,N-бис(N',N'-алкилариламиноалкил)амины, N-(алкил)-N-(N',N'-алкилариламиноалкил)амины, N-(алкоксиалкил)-N-(N',N'-алкилариламиноалкил)амины, N-(арилоксиалкил)-N-(N',N'-алкилариламиноалкил)амины. В способе по настоящему изобретению в качестве исходных соединений общей формулы (III) могут быть использованы о-, м-, п-винилбензилгалогениды (галогенметилстиролы) или α-галогенметилстиролы, или их смеси с различным соотношением изомеров. Атом галогена в таких соединениях может быть выбран из хлора, брома или йода, при этом от хлора к йоду скорость реакции и достигаемая конверсия увеличиваются.
Карбонат щелочного металла или аммония предпочтительно выбирают из группы, включающей карбонат калия, карбонат натрия или карбонат аммония, предпочтительно использование карбоната калия, концентрация которого в водном растворе составляет 10-50 массовых %, предпочтительно 25-40 массовых %, более предпочтительно 30-35 массовых %. Эти же концентрации предпочтительны при применении карбоната аммония, при использовании карбоната натрия предпочтительно использовать водные растворы с концентрацией 10-14 массовых %. Карбонат щелочного металла или аммония применяют для регенерации вторичного амина из образующегося в ходе синтеза гидрохлорида за счет нейтрализации соляной кислоты, кроме того, карбонат не инициирует побочных реакций полимеризации, в результате которых уменьшается выход.
Образующиеся в ходе синтеза хлориды щелочных металлов или аммония, обладают меньшей растворимостью по сравнению с карбонатом щелочного металла или аммония, поэтому концентрация водного раствора карбоната подбирается таким образом, чтобы по окончании синтеза и охлаждении реакционной массы водный раствор был насыщенным, но выпадения осадка хлорида не происходило, так как это приведет к затруднению выделения конечных соединений. Для синтеза всех заявленных соединений концентрация водного раствора карбоната составляет 10-35 массовых %. Предпочтительно использование карбоната калия, но также могут быть использованы карбонат натрия или карбонат аммония.
Применение катализатора межфазного переноса в количестве 0,01-10 мольных %, предпочтительно 0,1-5 мольных %, более предпочтительно 0,5-1 мольных % на количество используемого соединения III обеспечивает увеличение конверсии и скорости образования соединений общей формулы (I). Наиболее эффективными катализаторами межфазного переноса являются соединения, содержащие атом йода, такие как йодид тетразамещенного аммония, вследствие ускорения фазового переноса и протекания реакции Финкельштейна в реакционной массе, например тетраалкиламмоний йодид, тетраариламмоний йодид, арилтриалкиламмоний йодид, в которых алкил представляет собой С1-С6 алкильную группу, а арил представляет собой фенил, предпочтительно бензилтриэтиламмоний йодид, тетрабутиламмоний йодид, тетраэтилфосфоний йодид, тетрафенилфосфоний йодид, бензилтриэтилфосфоний йодид, тетрафенилфосфоний йодид и другие.
Реакция Финкельштейна представляет собой синтез алкилйодидов либо алкилфторидов взаимодействием алкилхлоридов либо алкилбромидов с йодидами или фторидами щелочных металлов. Реакция Финкельштейна протекает по механизму SN2 (бимолекулярного нуклеофильного замещения) и является равновесной:
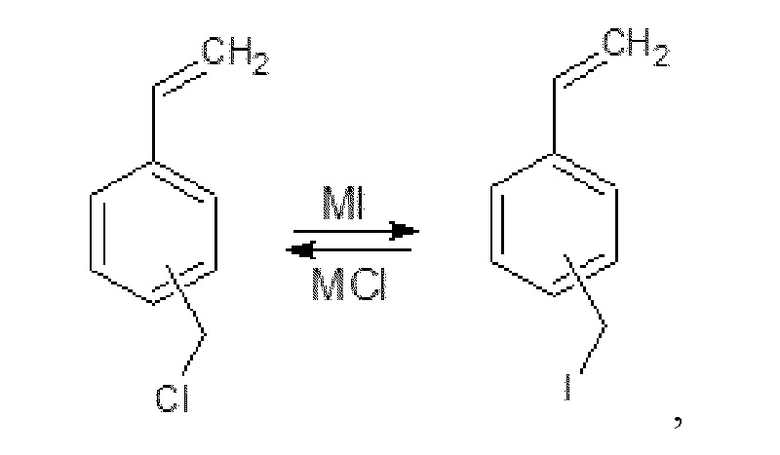
где М представляет собой ион калия или ион тетрабутиламмония.
В классическом варианте реакции Финкельштейна для повышения выходов алкилйодидов синтез проводится в растворителях, в которых йодиды натрия или калия хорошо растворимы, а образующиеся в ходе реакции хлориды или бромиды щелочных металлов малорастворимы.
В настоящем изобретении меньшая растворимость образующегося в ходе реакции хлорида тетразамещенного аммония по сравнению с йодидом, как в воде, так и в диэтиламине и в органической фазе приводит к тому, что один из продуктов реакции выводится из реакционной среды, выпадая в осадок, и равновесие реакции смещается по правилу Ле-Шателье в сторону образования целевых продуктов. В свою очередь атом йода в субстрате является лучшей уходящей группой по сравнению с атомом хлора, что увеличивает, как общую скорость процесса получения соединений формулы (I), так и конверсию исходных соединений.
Содержащие в качестве атома галогена бром или хлор катализаторы межфазного переноса, например, тетрабутиламмоний хлорид, тетраэтиламмоний хлорид, бензилтриэтиламмоний хлорид, тетрафенилфосфония хлорид, тетрабутиламмоний бромид, тетраэтиламмоний бромид, бензилтриэтиламмоний бромид, тетрафенилфосфония бромид и им подобные, являются менее эффективными, так как в реакционной массе реакция Финкельштейна не протекает. Наименьшей каталитической эффективностью обладают йодиды щелочных металлов, так как они слаборастворимы в органической фазе, и каталитическое действие основано только на протекании реакции Финкельштейна. В качестве подтверждения указанной тенденции эффективности катализаторов в таблице 1 представлены данные серии синтезов N,N-диэтиаминометилстирола с применением различных катализаторов.
Таблица 1. Серия синтезов N,N-диэтиаминометилстирола с различными катализаторами.
В способе настоящего изобретения в качестве ингибитора радикальной полимеризаиции используют фенольный или тиофенольный антиоксидант, в частности бензол-1,4-диол, 4-метил-2,6-дитретбутилфенол, 4-гидрокси-3,5-дитретбутоксибензол, 4-третбутилбен-1,2-диол, 4,6-бис-октилтиометил-о-крезол, 2,2'-тиобис(4-метил-6-трет-бутил-фенол), 4,4'-тиобис(6-трет-бутил-м-крезол), катехины, 1,3,5-триметил-2,4,6-трис(3,5-ди-трет-бутил-4-гидроксибензил)бензол, 2,5-диметил-4-(4-бутилбензилтио)фенол, 2-диметилбензил-4,4(гексилтио)фенол, 2,4-дибутил-6-(бутилтио)фенол, 2,6-бис(1,1-диметилбутил)-4-(1,1-диметилбутилтио)фенол, 2,2-метилен-бис(4-метил-6-третбутилфенол), предпочтительно 2,2-метилен-бис-4-метил-6-третбутилфенол (агидол-2), так как он хорошо растворим в органических растворителях и не растворим в воде, что важно для стабилизации соединений именно в органической фазе. Кроме того, агидол-2 не подвергается совместной перегонке с соединениями общей формулы (I), что облегчает очистку конечных соединений. В заявленном способе также возможно применение в качестве ингибиторов радикальной полимеризации бензол-1,4-диола (гидрохинона) или 4-метил-2,6-ди-третбутилфенола (агидол-1), но только в случаях перегонки соединений с температурой кипения не выше 100°C при 1-2 мбар, так как они имеют тенденцию к совместной перегонке с высококипящими соединениями (I) при последующей очистке. Также могут быть использованы другие ингибиторы радикальной полимеризации фенольной или тиофенольной природы, имеющие высокие температуры кипения или разложения (выше 300 оС), то есть не подвергающиеся совместной перегонке при очистке продуктов, например, бензол-1,4-диол, 4-метил-2,6-дитретбутилфенол, 4-гидрокси-3,5-дитретбутоксибензол, 4-третбутилбен-1,2-диол, 4,6-бис-октилтиометил-о-крезол, 2,2'-тиобис(4-метил-6-трет-бутил-фенол), 4,4'-тиобис(6-трет-бутил-м-крезол), катехины, 1,3,5-триметил-2,4,6-трис(3,5-ди-трет-бутил-4-гидроксибензил)бензол, 2,5-диметил-4-(4-бутилбензилтио)фенол, 2-диметилбензил-4,4(гексилтио)фенол, 2,4-дибутил-6-(бутилтио)фенол, 2,6-бис(1,1-диметилбутил)-4-(1,1-диметилбутилтио)фенол, 2,2-метилен-бис(4-метил-6-третбутилфенол), предпочтительно 2,2-метилен-бис(4-метил-6-третбутилфенол) и подобные соединения.
Фенольные или тиофенольные антиоксиданты используются в количестве 0,01-10 мольных %, предпочтительно 0,1-5 мольных %, наиболее предпочтительно 0,5-1 мольных % на количество используемого соединения III.
Предпочтительно, заявленный способ получения соединений (I) осуществляют в атмосфере инертного газа при температуре 80-120°C при атмосферном давлении, но также можно использовать повышенное давление и проводить процесс при температурах до 150 оС. Нагрев реакционной массы выше 150°C не допустим и приводит к уменьшению выхода за счет осмоления реакционной массы. Применение повышенного давления, обуславливающее усложнение применяемого оборудования, нежелательно, так как это ведет к усложнению разработанного метода синтеза.
Предпочтительная длительность способа согласно настоящему изобретению составляет от 1 до 3 часов, более предпочтительно 1,5-2 часа.
Предпочтительно синтез проводят без использования органических растворителей, тем не менее, допускается применение таких органических растворителей, как алифатические углеводородные растворители, ароматические углеводородные растворители, хлорсодержащие углеводородные растворители, в частности бензол, толуол, гексан, гептан, хлорбензол, тетрахлорметилметан, трихлорметан, хлористый метилен или любые их смеси.
Согласно способу настоящего изобретения синтез предпочтительно проводят в эмульсии водного раствора карбоната калия и соединений (II), (III) при 80-120°C в течение 2 часов с использованием антиоксиданта 2,2-метилен-бис(4-метил-6-третбутилфенола) (агидол-2) и катализатора йодида тетрабутиламмония.
Для наиболее полного выделения соединений формулы (I) необходимо использовать экстракцию водного раствора карбоната, после проведения синтеза, что дает увеличение выхода конечных соединений на 1-4%. Предпочтительно в качестве экстрагента использовать углеводородные растворители, например, пентан или гексан, так как:
- они обладают невысокой температурой кипения, что облегчает последующее удаление экстрагента отгонкой;
- они обладают низкой растворимостью в воде, что препятствует переходу экстрагента в водную фазу и увеличению растворимости конечных соединений в водной фазе;
- они являются хорошими растворителями по отношению к соединениям общей формулы (I).
После объединения органических фракций, полученных при разделении фаз и экстракции водной фазы, растворитель удаляют из органической фазы. Далее проводят вакуумную перегонку остатка после упаривания, при этом недопустимо нагревать перегоняемую массу выше 150оС, так как происходит полимеризация очищаемого соединения. В качестве ректификационного оборудования возможно использование пленочных и роторно-пленочных колонн.
Таким образом, заявляемый способ получения соединений общей формулы (I) характеризуется широким спектром получаемых соединений, высокими конверсией исходных соединений и выходом соединений формулы (I), является приемлемым для крупномасшатабного производства вследствие препаративного удобства проведения и обработки синтеза, отсутствия затруднений на стадии выделения конечного вещества, а также малых количеств используемых катализаторов.
Осуществление изобретения ниже иллюстрируется экспериментальными примерами. Заявленный способ не ограничивается представленными примерами и может быть распространен на получение любых соединений заявленных структур.
ПРИМЕРЫ
Строение и чистоту полученных соединений анализировали методами ИК-спектрометрии, 1H, 13С ЯМР-спектроскопии, газовой хроматографией и определением температуры кипения.
Пример 1
Получение смеси 3-[(N,N-диэтиламино)метил]стирола и 4-[(N,N-диэтиламино)метил]стирола.

Синтез проводят под атмосферой азота, с добавкой агидола-2 (0,085 г, 0,5 мольных % по отношению к винилбензил хлориду). В реактор помещают 10,366 г (0,075 моль) карбоната калия в 24,0 мл (1,33 моль) дистиллированной воды, 7,0 мл (0,05 моль) смеси м- и п-винилбензилхлоридов (53% мета-изомера, 47% пара-изомера) и 0,185 г (0,0005 моль) тетрабутиламмоний йодида, к полученной эмульсии при перемешивании добавляют 7,7 мл (0,075 моль) N,N-диэтиламина. Затем реакционную массу перемешивают при кипячении в течение 2 часов. Далее фракции делят на делительной воронке, водный слой экстрагируют гексаном (2×5 мл), после чего органические фазы объединяют и упаривают растворитель. Остаток после упаривания, очищают перегонкой под вакуумом, собирают основную фракцию кипящую при 90-92°C и давлении 3-4 мБар и сушат над молекулярными ситами. Выход смеси изомеров конечного соединения (53% мета-изомера, 47% пара-изомера) 91% от теоретического количества, степень конверсии- 99%.
ИК- спектр (см-1):С-Н 2797-3087 (сильное), С=С (обертон) 1813 (слабое), С=С 1602-1630 (слабое).
1Н ЯМР (400 МГц, хлороформ-D) δ, м. д., J (Гц),
Спектр ЯМР 1Н (400 МГц) смеси изомеров, δ, м. д., J (Гц), (СDCl3): 1.07-1.22 (12H, м, СH3); 2.61 (8H, м, 4CH2-N); 3.63 (1.88H, c, п-St-CH2-), 3.64 (2.12H, c, м-St-CH2-), 5.30 (2H, тд, 2J=11.09, 4J=0.94 =CH e к заместителю); 5.83 (2H, ддд, 2J=17.61, 3J=15.68, 4J=0.94 =CH z к заместителю); 6.80 (2H, ддд, 3J=17.61, 3J=10.90, 3J=8.6, Ph-CH=); 7.31-7.51 (8H, м, Н-St)
Спектр ЯМР 13С (100 МГц) м- изомера, δ, м. д., (СDCl3): 11,74 (2СН3); 46,70 (N(СН2)2); 57.51 (St-CH2) 112.96 (=CH2); 124.48 (2(С) Ph); 126.66 (4(С) Ph); 128.22 (5(С) Ph); 128.22 (6(С) Ph) 136.03 (3(С) Ph); 136.98 (Ph-CH=); 139.75 (1(С) Ph)
Спектр ЯМР 13С (100 МГц) п- изомера, δ, м. д., (СDCl3): 11,74 (2СН3); 46,70 (N(СН2)2); 57.28 (St-CH2) 113.42 (=CH2); 125.94 (3,5(С) Ph); 128.90 (2,6(С) Ph); 136.69 (Ph-CH=); 137.36 (4(С) Ph); 140.30 (1(С) Ph)
Пример 2.
Получение смеси 3-[(N,N-диизопропиламино)метил]стирола и 4 [(N,N диизопропиламино)метил]стирола.
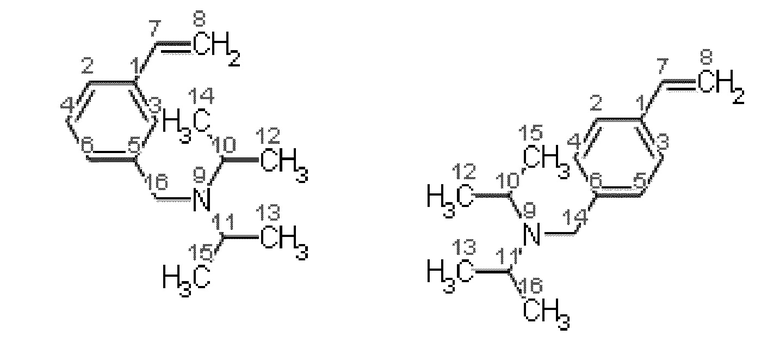
Синтез проводят под атмосферой азота, с добавкой агидола-2 (0,085 г, 0,5 мольных % по отношению к винилбензил хлориду). В реактор помещают 10,366 г (0,075 моль) карбоната калия и 24,0 мл (1,33 моль) дистиллированной воды, 7,0 мл (0,05 моль) смеси м- и п-винилбензилхлоридов (53% мета-изомера, 47% пара-изомера) и 0,185 г (0,0005 моль) тетрабутиламмоний иодида, к полученной эмульсии при перемешивании добавляют 10,5 мл (0,075 моль) N,N-диизопропиламина. После чего реакционную массу перемешивают при кипячении в течение 2 часов. Далее фракции делят на делительной воронке, водный слой экстрагируют гексаном (2×5 мл), после чего органические фазы объединяют и упаривают растворитель. Остаток после упаривания, очищают перегонкой под вакуумом, собирают фракцию кипящую при 114 118°C и давлении 5-7 мБар и сушат над молекулярными ситами. Выход смеси изомеров конечного соединения (53% мета-изомера, 47% пара-изомера) 92% от теоретического количества, степень конверсии- 99%. Нагрев перегоняемой массы выше 150°C приводит к осмолению.
ИК- спектр (см-1):С-Н 2811-3086 (сильное), С=С (обертон) 1811 (слабое), С=С 1582-1630 (слабое).
Спектр ЯМР 1Н (400 МГц) смеси изомеров, δ, м. д., J (Гц), (СDCl3): 1.21-1.23 (12H, м, 4СH3); 3.21 (1H, спт, 3J=6.60 м-N(CH)2); 3.22 (1H, спт, 3J=6.60 п-N(CH)2); 3.81 (1,87H, c, п-St-CH2-), 3.83 (2,13H, c, м-St-CH2-), 5.37 (2H, ддд, 2J=14.09, 3J=10.90 3J=1.10 =CH e к заместителю); 5.91 (2H, ддд, 2J=17.70, 3J=16.90, 4J=1.10 =CH z к заместителю); 6.90 (2H, ддд, 2J=17.61, 3J=10.90, 4J=9.40, Ph-CH=); 7.41-7.63 (8H, м, Н-St)Спектр ЯМР 13С (100 МГц) м- изомера, δ, м. д., (СDCl3): 20,70 (4СН3); 47,71 (N(СН2)2); 48.79 (St-CH2) 113.14 (=CH2); 124.10 (2(С) Ph); 125.68 (4(С) Ph); 127.42 (5(С) Ph); 127.95 (6(С) Ph) 136.84 (3(С) Ph); 137.22 (Ph-CH=); 143.38 (1(С) Ph)
Спектр ЯМР 13С (100 МГц) п- изомера, δ, м. д., (СDCl3): 20,70 (2СН3); 47,73 (N(СН2)2); 48.579 (St-CH2) 112.58 (=CH2); 125.86 (3,5(С) Ph); 127.73 (2,6(С) Ph); 135.63 (4(С) Ph); 137.19 (Ph-CH=); 142.99 (1(С) Ph);
Пример 3.
Получение смеси 4-(3-этенилбензил)морфолина и 4-(4-этенилбензил)морфолина.
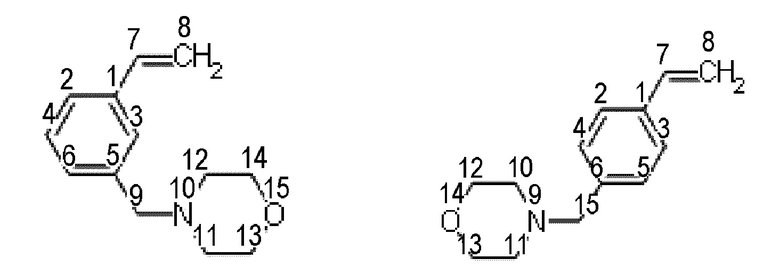
Синтез проводят под атмосферой азота, с добавкой агидола-2 (0,085 г, 0,5 мольных % по отношению к винилбензил хлориду). В реактор помещают 10,366 г (0,075 моль) карбоната калия в 24,0 мл (1,33 моль) дистиллированной воды, 7,0 мл (0,05 моль) смеси м- и п-винилбензилхлоридов (53% мета-изомера, 47% пара-изомера) и 0,185 г (0,0005 моль) тетрабутиламмоний йодида, к полученной эмульсии при перемешивании добавляют 4,8 мл (0,055 моль) морфолина. После этого реакционную массу перемешивают при 100°C в течение 2 часов. Далее фракции делят на делительной воронке, водный слой экстрагируют гексаном (2×5 мл), после чего органические фазы объединяют и упаривают растворитель. Остаток после упаривания, очищают перегонкой под вакуумом, собирают фракцию кипящую при 126-128°C и давлении 3 4 мБар. Выход смеси изомеров конечного соединения (53% мета-изомера, 47% пара-изомера) 94% от теоретического количества, степень конверсии- 99%. Нагрев перегоняемой массы выше 150°C приводит к осмолению.
ИК спектр (см-1):С-Н 2690-2958 (среднее), С=С (обертон) 1817 (слабое), С=С 1581-1629 (слабое).
Спектр ЯМР 1Н (400 МГц) смеси изомеров, δ, м. д., J (Гц), (СDCl3): 2.39-2.42 (8H, м, 2N(СH2)2); 3.44 (1,89H, c, п-St-CH2-); 3.45 (2,11H, c, м-St-CH2-); 3.67 3.70 (8H, м, 2О(CH2)2); 5.23 (2H, тд, 2J=11.00, 4J=1.00 =CH e к заместителю); 5.74 (2H, ддд, 2J=17.60, 3J=13.20, 4J=1.00 =CH z к заместителю); 6.75 (2H, ддд, 3J=17.50, 3J=11.00, 3J=6.40, Ph-CH=); 7,20-7,37 (8H, м, Н-St)
Спектр ЯМР 13С (100 МГц) м- изомера, δ, м. д., (СDCl3): 53,43 (N(СН2)2); 63.16 (St-CH2); 66,74 (O(СН2)2); 113.63 (=CH2); 124.79 (2(С) Ph); 126.83 (4(С) Ph); 128.24 (5(С) Ph); 128.46 (6(С) Ph) 136.30 (3(С) Ph); 136.66 (Ph-CH=); 137.96 (1(С) Ph)
Спектр ЯМР 13С (100 МГц) п- изомера, δ, м. д., (СDCl3): 53,46 (N(СН2)2); 62.92 (St-CH2); 66,74 (O(СН2)2); 113.28 (=CH2); 125.92 (3,5(С) Ph); 129.09 (2,6(С) Ph); 136.41 (Ph-CH=); 137.35 (4(С) Ph); 137.36 (1(С) Ph).
Пример 4.
Получение смеси 1-(3-этенилбензил)-1Н-имидазола и 1-(4-этенилбензил)-1Н-имидазола
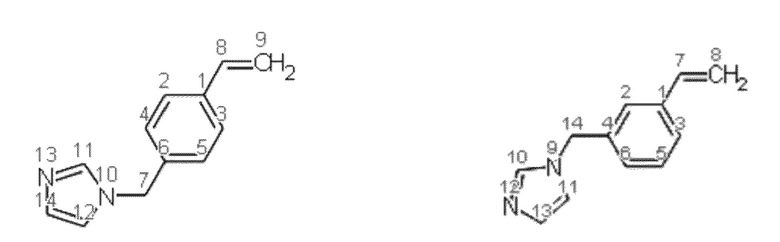
Синтез проводят под атмосферой азота, с добавкой агидола 2 (0,085 г, 0,5 мольных % по отношению к винилбензил хлориду). В реактор помещают 20.732 г (0,15 моль) карбоната калия, 48,5 мл (2,684 моль) дистиллированной воды, 7,148 г (0,055 моль) имидазола и 0,3694 г (0,0010 моль) тетрабутиламмоний йодида, при перемешивании добавляют 7,0 мл (0,5 моль) винилбензилхлорид, полученную эмульсию перемешивают при 100°C в течение 2 часов. Далее фракции делят на делительной воронке, водный слой экстрагируют гексаном (2×5 мл), после чего органические фазы объединяют и упаривают растворитель. Остаток после упаривания, очищают перегонкой под вакуумом, собирают фракцию кипящую при 130-132 °С и давлении 0,5-1 мБар. Выход смеси изомеров конечного соединения (53% мета-изомера, 47% пара-изомера) 90% от теоретического количества, степень конверсии - 99%. Нагрев перегоняемой массы выше 150°C приводит к осмолению.
ИК спектр (см-1):С-Н δ 2828-3088 (сильное), С=С (обертон) 1827 (слабое), С=С δ 1603-1629 (слабое), 1560 (слабое), 1504 (среднее) колебания имидазольного кольца.
Спектр ЯМР 1Н (400 МГц) смеси изомеров, δ, м. д., J (Гц), (СDCl3): 4.98 (2H, c, п-St-CH2-), 5.15 (2H, тд, 2J=11.09, 4J=0.94 =CH e к заместителю); 5.38 (2H, c, м-St-CH2-), 5.62 (2H, ддд, 2J=17.61, 3J=15.68, 4J=0.94 =CH z к заместителю); 6.55 (2H, ддд, 3J=17.61, 3J=10.90, 3J=8.6, Ph-CH=); 6.77-7.54 (8H, м, Н-St, H-Im)
Спектр ЯМР 13С (100 МГц) м-изомера, δ, м. д., (СDCl3): 51.11 (St-CH2); 115.52 (=CH2); 120.14 (11(С) Ph); 122.73 (2(С) Ph); 122,81 (3(С) Ph); 127.37 (5(С) Ph); 127.78 (6(С) Ph); 132.88 (13(С) Ph); 135.85 (Ph-CH=); 136.20 (1(С) Ph); 137.14 (10(С) Ph); 137.75 (4(С) Ph).
Спектр ЯМР 13С (100 МГц) п- изомера, δ, м. д., (СDCl3): 51.32 (St-CH2); 116.13 (=CH2); 120.05 (12(С) Ph); 125.75 (2,3(С) Ph); 130.22 (4.5(С) Ph); 133.95 (14(С) Ph); 136.03 (Ph-CH=); 136.61 (6(С) Ph); 136.66 (1(С) Ph); 137.42 (11(С) Ph);
Пример 5.
Получение смеси N-(3-этенилбензил)-N-метиланилина и N-(4-этенилбензил)-N-метиланилина.
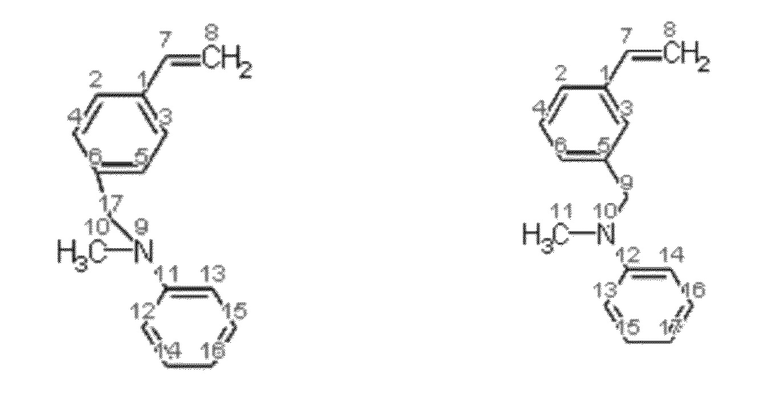
Синтез проводят в токе азота, с добавкой агидола-2 (0,085 г, 0,5 мольных % по отношению к винилбензил хлориду). В реактор помещают 10,366 г (0,075 моль) карбоната калия в 24,0 мл (1,33 моль) дистиллированной воды, 7,0 мл (0,05 моль) смеси м- и п-винилбензилхлоридов (53% мета-изомера, 47% пара-изомера) и 0,185 г (0,0005 моль) тетрабутиламмоний иодида, к полученной эмульсии при перемешивании добавляют 5,9 мл (0,055 моль) N-метиланилина. Затем реакционную массу перемешивают при 100°C в течение 2 часов. Далее фракции делят на делительной воронке, водный слой экстрагируют гексаном (2×5 мл), после чего органические фазы объединяют и упаривают растворитель. Остаток после упаривания, очищают перегонкой под вакуумом, собирают фракцию кипящую при 130-135°C и давлении 1 мБар. Выход смеси изомеров конечного соединения (53% мета-изомера, 47% пара-изомера) 87% от теоретического количества, степень конверсии- 99%. Нагрев перегоняемой массы выше 150°C приводит к осмолению.
ИК спектр (см-1):С-Н 2800-3090 (среднее), С=С (обертон) 1821 (слабое), С=С 1590 (сильное), ароматическое кольцо 1505 (сильное).
Спектр ЯМР 1Н (400 МГц) смеси изомеров, δ, м. д., J (Гц), (СDCl3): 1.54 (6H, c, СH3); 4.70 (4H, c, м,п-St-CH2-), 5.44 (2H, ддд, 2J=14.09, 3J=10.90 3J=1.10 =CH e к заместителю); 5.95 (2H, ддд, 2J=17.61, 3J=15.68, 4J=0.94 =CH z к заместителю); 6.85-7,05 (8H, м, Ph-CH=, 3Н-An); 7.31-7.62 (12H, м, 4Н-St, 2Н-An)
Спектр ЯМР 13С (100 МГц) м- изомера, δ, м. д., (СDCl3): 39,35 (СН3); 57,60 (St-CH2-N) 113.37 (=CH2); 113.42 (13(C), 14(C)-An) 117.60 (17(C)-An) 125.65 (3(С) Ph); 125.67 (2(С) Ph); 127.39 (4(С) Ph); 129.73 (6(С) Ph); 130.15 (15,16(С) An); 137.28 (Ph-CH=); 137.52 (1(С) Ph); 138.80 (5(С) Ph);150.77 (12(С) Ph)
Спектр ЯМР 13С (100 МГц) п- изомера, δ, м. д., (СDCl3): 39,35 (СН3); 57,35 (St-CH2-N) 113.42 (12(C), 13(C)-An) 114.87 (=CH2); 117.65 (16(C)-An); 127.19 (2,3(С) Ph); 127.90 (4,5(С) Ph); 130.15 (14,15(С) Ph); 137.83 (Ph-CH=); 139.67 (1(С) Ph); 140.41 (1(С) Ph); 150.66 (11(С) Ph).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ТРЕТИЧНЫХ АМИНОВ, СОДЕРЖАЩИХ ЭТЕНИЛБЕНЗИЛЬНЫЕ ЗАМЕСТИТЕЛИ | 2015 |
|
RU2732296C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2-(N,N-ДИАЛКИЛАМИНОМЕТИЛ)СТИРОЛОВ, СОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИЙ ФРАГМЕНТ | 2015 |
|
RU2575176C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-АМИНОМЕТИЛСТИРОЛОВ, СОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИЙ ФРАГМЕНТ | 2014 |
|
RU2562775C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-АМИНОМЕТИЛСТИРОЛОВ, СОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИЙ ФРАГМЕНТ | 2015 |
|
RU2596198C1 |
| СПОСОБ ПОЛУЧЕНИЯ МОДИФИЦИРОВАННОГО КАУЧУКА МЕТОДОМ РАСТВОРНОЙ АНИОННОЙ ПОЛИМЕРИЗАЦИИ, РЕЗИНОВАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ТАКОЙ КАУЧУК, И ЕЕ ПРИМЕНЕНИЕ | 2016 |
|
RU2707102C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-(N,N-ДИАЛКИЛАМИНОМЕТИЛ)СТИРОЛОВ | 2015 |
|
RU2579116C1 |
| НОВЫЕ ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ | 2002 |
|
RU2294326C2 |
| ПОЛУЧЕНИЕ N-МОНОФТОРАЛКИЛТРОПАНОВ | 2010 |
|
RU2552355C2 |
| ПРОИЗВОДНЫЕ АМИДА ФЕНИЛЦИКЛОГЕКСИЛКАРБОНОВОЙ КИСЛОТЫ, СМЕСЬ ИХ ИЗОМЕРОВ ИЛИ ОТДЕЛЬНЫЕ ИЗОМЕРЫ И ИХ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ С АНТИАРТЕРИОСКЛЕРОТИЧЕСКОЙ И АНТИРЕСТЕНОЗНОЙ АКТИВНОСТЬЮ | 1996 |
|
RU2158261C2 |
| СПОСОБ ПОЛУЧЕНИЯ (-)-N-МЕТИЛ-N-[4-(ФЕНИЛ-4-АЦЕТАМИНОПИПЕРИДИН-1-ИЛ)-2-(3,4-ДИХЛОРФЕНИЛ)БУТИЛ]БЕНЗАМИДА И ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ | 1995 |
|
RU2097376C1 |
Изобретение относится к области органического синтеза, в частности к способу получения N,N-дизамещенных аминометилстиролов или α-аминометилстиролов общей формулы (I) или их смеси, где x=1, y=0 или x=0, y=1; R представляет собой группу -СН2N(R1)(R2), в которой R1 и R2 могут быть одинаковыми или различными и независимо представляют собой алкил или циклоалкил, необязательно замещенные одним или более заместителями, выбранными из гидроксигруппы, диалкиламиногруппы, алкоксигруппы, арилоксигруппы, алкилсульфанильной группы, арилсульфанильной группы, арила; или арил, необязательно замещенный одним или более заместителями, выбранными из галогена, алкила, диалкиламиногруппы, алкоксигруппы, арилоксигруппы, алкилсульфанильной группы, арилсульфанильной группы, арила; или гетероарил, содержащий один или более атомов азота, где гетероарил необязательно замещен одним или более заместителями, выбранными из галогена, алкила, диалкиламиногруппы, алкоксигруппы, арилоксигруппы, алкилсульфанильной группы, арилсульфанильной группы, арила; или R1 и R2, взятые вместе с азотом, образуют 5-6-членное гетероциклическое или 5-членное гетероароматическое кольцо, необязательно содержащее один или более дополнительных гетероатомов, выбранных из азота, кислорода или серы. Способ включает взаимодействие вторичного амина общей формулы (II), где R1 и R2 принимают значения, определенные выше, с винилбензилгалогенидом или α-галогенметилстиролом общей формулы (III), в которой R3 представляет собой группу -СН2-Hal, а x=1, y=0 или x=0, y=1, в двухфазной системе в щелочной среде в атмосфере инертного газа в присутствии катализатора, представляющего собой иодиды аммониевых или фосфониевых соединений, и антиоксиданта. Предлагаемый способ позволяет получить соединения общей формулы (I) с высокими выходами при высокой конверсии исходных реагентов, а также сократить время проведения процесса. Изобретение относится также к конкретным соединениям формулы (I), выбранным из 3-[(N,N-диизопропиламино)метил]стирола, N-(3-этенилбензил)-N-метиланилина или N-(4-этенилбензил)-N-метиланилина. 2 н. и 23 з.п. ф-лы, 1 табл., 5 пр.
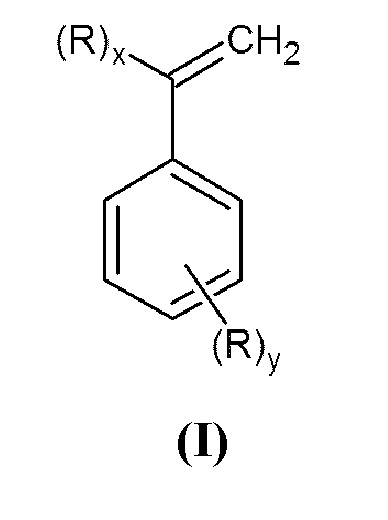
1. Способ получения N,N-дизамещенных аминометилстиролов или α-аминометилстиролов общей формулы (I)
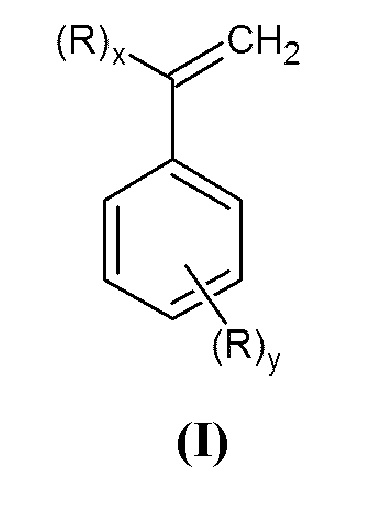
или их смеси,
где x=1, y=0 или x=0, y=1; R представляет собой группу -СН2N(R1)(R2), в которой R1 и R2 могут быть одинаковыми или различными и независимо представляют собой алкил или циклоалкил, необязательно замещенные одним или более заместителями, выбранными из гидроксигруппы, диалкиламиногруппы, алкоксигруппы, арилоксигруппы, алкилсульфанильной группы, арилсульфанильной группы, арила;
или арил, необязательно замещенный одним или более заместителями, выбранными из галогена, алкила, диалкиламиногруппы, алкоксигруппы, арилоксигруппы, алкилсульфанильной группы, арилсульфанильной группы, арила;
или гетероарил, содержащий один или более атомов азота, где гетероарил необязательно замещен одним или более заместителями, выбранными из галогена, алкила, диалкиламиногруппы, алкоксигруппы, арилоксигруппы, алкилсульфанильной группы, арилсульфанильной группы, арила;
или R1 и R2, взятые вместе с азотом, образуют 5-6-членное гетероциклическое или 5-членное гетероароматическое кольцо, необязательно содержащее один или более дополнительных гетероатомов, выбранных из азота, кислорода или серы;
который включает взаимодействие вторичного амина общей формулы (II)
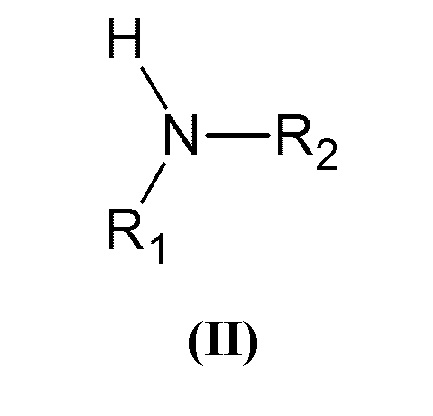 ,
,
где R1 и R2 принимают значения, определенные выше, с винилбензилгалогенидом или α-галогенметилстиролом общей формулы (III)
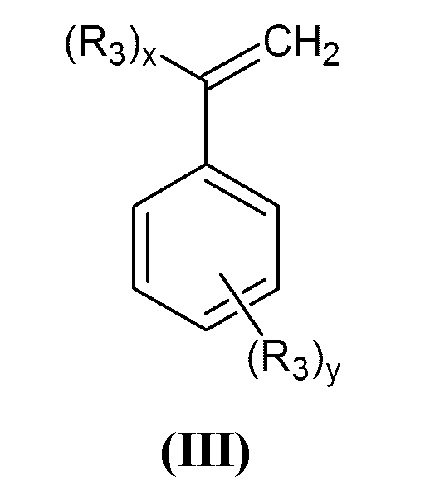 ,
,
в которой R3 представляет собой группу -СН2-Hal, а x=1, y=0 или x=0, y=1,
в двухфазной системе в щелочной среде в атмосфере инертного газа в присутствии катализатора, представляющего собой иодиды аммониевых или фосфониевых соединений, и антиоксиданта.
2. Способ по п.1, в котором алкил представляет собой насыщенный или ненасыщенный С1-С6 алкил с линейной или разветвленной цепью, предпочтительно метил, этил, пропил, изопропил, бутил, втор-бутил, трет-бутил, изобутил, гексил, бут-3-енил, бут-3-инил.
3. Способ по п.1, в котором циклоалкил представляет собой С3-С7 циклоалкил, предпочтительно циклогексил, циклогептил.
4. Способ по п.1, в котором арил представляет собой 6-членную арильную группу, предпочтительно фенил, 2-хлорфенил, 2-бромфенил, 2-йодфенил, 2-N,N-диметиламинофенил, 2-метоксифенил, 2-феноксифенил, 2-метилсульфанилфенил.
5. Способ по п.1, в котором гетероарил представляет собой 6-членную арильную группу, содержащую один или более атомов азота, предпочтительно пиридинил, пиримидинил.
6. Способ по п.1, в котором R1 и R2 независимо представляют собой гидроксиметил, 2-гидроксиэтанил, 2-гидроксипропил, 2-гидроксиизопропил, 3-гидроксипропил, 2,3-дигидроксипропил, 2-гидроксибутил, 2-гидроксициклогексил.
7. Способ по п.1, в котором R представляет собой N,N-диметиламинометил N,N-дифениламинометил, N-метил-N-фениламинометил, N,N-диметиламиноэтил, N,N-диизопропиламинометил, N,N-дициклогексиламинометил, 2-(N,N-диметиламино)изопропил, 2-(N,N-диметиламино)циклогексил.
8. Способ по п.1, в котором R1 и R2 независимо представляют собой метоксиметил, изопропоксиметил, феноксиметил, 2-(метокси)изопропил, 2-(метокси)циклогексил.
9. Способ по п.1, в котором R1 и R2 независимо представляют собой (метилсульфанил)метил, (изопропилсульфанил)метил, 2-(метилсульфанил)циклогексил, (фенилсульфанил)метил.
10. Способ по п.1, в котором R1 и R2, взятые вместе с азотом, образуют 5-6-членное гетероциклическое кольцо, которое необязательно содержит один или несколько дополнительных гетероатомов, выбранных из азота, кислорода, серы, представляют собой соединение, выбранное из группы пирролидинил, морфолинил, тиоморфолинил, пиперидинил, 1,3-оксазолидинил;
или R1 и R2, взятые вместе с азотом, образуют 5-членное гетероароматическое кольцо, необязательно содержащее один или два дополнительных гетероатома, выбранных из азота, кислорода, серы, в частности имидазолил, пиразолил, фуранил, пирролил, оксазолил, тиазолил.
11. Способ по п.1, в котором галоген представляет собой фтор, хлор, йод или бром.
12. Способ по п.1, в котором катализатор представляет собой йодиды аммониевых или фосфониевых соединений, выбранных из группы, включающей тетраалкиламмоний или тетраалкилфосфоний йодиды, тетраариламмоний или тетраарилфосфоний йодиды, арилтриалкиламмоний или арилтриалкилфосфоний йодиды, в частности бензилтриэтиламмоний йодид, тетрабутиламмоний йодид, тетраэтилфосфоний йодид, тетрафенилфосфоний йодид, бензилтриэтилфосфоний йодид, тетрафенилфосфоний йодид.
13. Способ по п.1, в котором количество йодида аммониевого или фосфониевого соединения составляет 0,01-10 мол.%, предпочтительно 0,1-5 мол.%, более предпочтительно 0,5-1 мол.% на количество используемого соединения III.
14. Способ по п.1, в котором в качестве щелочной среды используют раствор соединения карбоната щелочного металла или аммония, выбранного из группы, включающей карбонат калия, карбонат натрия или карбонат аммония.
15. Способ по п.14, в котором концентрация карбоната щелочного металла или аммония в водном растворе составляет 10-50 мас.%, предпочтительно 25-40 мас.%, более предпочтительно 30-35 мас.%.
16. Способ по п.1, в котором антиоксидант представляет собой фенольный или тиофенольный антиоксидант, выбранный из группы, включающей бензол-1,4-диол, 4-метил-2,6-ди-трет-бутилфенол, 4-гидрокси-3,5-ди-трет-бутоксибензол, 4-трет-бутилбен-1,2-диол, 4,6-бис-октилтиометил-о-крезол, 2,2'-тиобис(4-метил-6-трет-бутилфенол), 4,4'-тиобис(6-трет-бутил-м-крезол), катехины, 1,3,5-триметил-2,4,6-трис(3,5-ди-трет-бутил-4-гидроксибензил)бензол, 2,5-диметил-4-(4-бутилбензилтио)фенол, 2-диметилбензил-4,4(гексилтио)фенол, 2,4-дибутил-6-(бутилтио)фенол, 2,6-бис(1,1-диметилбутил)-4-(1,1-диметилбутилтио)фенол, 2,2-метилен-бис(4-метил-6-трет-бутилфенол), предпочтительно 2,2-метилен-бис(4-метил-6-трет-бутилфенол).
17. Способ по п.1, в котором антиоксидант используют в количестве 0,01-10 мол.%, предпочтительно 0,1-5 мол.%, наиболее предпочтительно 0,5-1 мол.% на количество используемого соединения III.
18. Способ по п.1, в котором взаимодействие осуществляют при температуре менее 150°С, предпочтительно при температуре 20-130°С, более предпочтительно 80-120°С.
19. Способ по п.1, в котором взаимодействие осуществляют при давлении 0,5-2 атм, предпочтительно 1-1,5 атм.
20. Способ по п.1, в котором время реакции составляет 1-3 ч, более предпочтительно 1,5- 2 ч.
21. Способ по п.1, в котором мольное соотношение вторичный амин:винилбензилгалогенид или α-галогенметилстирол составляет 1,0÷1,5/1, более предпочтительно 1,01÷1,1/1.
22. Способ по п.1, в котором дополнительно могут быть использованы органические растворители, выбранные из группы, включающей алифатические углеводородные растворители, ароматические углеводородные растворители, хлорсодержащие углеводородные растворители, в частности бензол, толуол, гексан, гептан, хлорбензол, тетрахлорметилметан, трихлорметан, хлористый метилен или любые их смеси.
23. Способ по п.1, который дополнительно включает стадию экстракции соединений формулы (I) из водного раствора указанного карбоната щелочного металла или аммония органическим растворителем, выбранным из группы, включающей алифатические углеводородные растворители, ароматические углеводородные растворители, хлорсодержащие углеводородные растворители.
24. Способ по п.1, в котором для выделения соединения формулы (I) применяют вакуумную перегонку.
25. Соединение общей формулы (I), выбранное из 3-[(N,N-диизопропиламино)метил]стирола, N-(3-этенилбензил)-N-метиланилина или N-(4-этенилбензил)-N-метиланилина.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Г.М | |||
| ПОГОСЯН и др., Производные стирола | |||
| XXI | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| ИГРУШКА С ПЛАВАЮЩЕЙ ФИГУРОЙ | 1922 |
|
SU451A1 |
| СПОСОБ ПОЛУЧЕНИЯ о;-ДИЭТИЛАМИНОМЕТИЛСТИРОЛА | 0 |
|
SU233680A1 |

