Область техники, к которой относится изобретение
Настоящее изобретение относится к твердым дисперсиям, содержащим химические соединения, которые ингибируют протеинкиназы, фармацевтическим лекарственным формам, включающим в себя такие дисперсии, способам получения таких дисперсий и лекарственных форм и способам их применения для лечения заболеваний.
Уровень техники изобретения
Митоз представляет собой процесс, при котором полная копия дуплицированного генома при помощи микротрубочек аппарата веретена деления распределяется по двум дочерним клеткам. Было обнаружено, что ключевые регуляторы митоза киназы Аврора, необходимые для стабильности генома, сверхэкспрессируются в опухолях человека. Поэтому в области терапии существует потребность в химических соединениях, ингибирующих киназы Аврора, композициях, содержащих эти ингибиторы, и способах лечения заболеваний, при которых киназы Аврора являются нерегулируемыми или сверхэкспрессируемыми.
Обратимое фосфорилирование белков является одним из основных биохимических механизмов проведения сигнала в эукариотических клетках. Эта реакция катализируется протеинкиназами, которые переносят g-фосфатную группу АТФ на гидроксильные группы целевых белков. В геноме человека существует 518 таких ферментов, из которых приблизительно 90 избирательно катализируют фосфорилирование гидроксильных групп тирозина. Цитозольные тирозинкиназы находятся внутри клетки, в то время как рецепторные тирозинкиназы (RTK) обладают как внеклеточным, так и внутриклеточным доменами и функционируют как трансмембранные рецепторы поверхности клеток. Таким образом, RTK опосредуют клеточные ответы на сигналы внешней среды и обеспечивают протекание широкого круга клеточных процессов, включая пролиферацию, миграцию и выживание.
В норме сигнальные пути с участием RTK строго регулируются, и было показано, что их сверхактивация способствует росту, выживанию и метастазированию раковых клеток. Нарушение регуляции проведения сигнала с участием RTK происходит в результате сверхэкспрессии или мутации генов, и оно коррелирует с развитием различных типов рака у человека.
Семейство рецепторов VEGF (VEGFR) состоит из трех RTK, KDR (рецептор, имеющий домен, содержащий киназу; VEGFR2), FLT1 (Fms-подобная тирозинкиназа; VEGFR1) и FLT4 (VEGFR3). Эти рецепторы опосредуют биологическую функцию факторов роста эндотелия сосудов (VEGF-A, -B, -C, -D, -E и фактора роста плаценты (PlGF)), семейства гомодимерных гликопротеинов, которые связывают рецепторы VEGF с различными аффинностями.
KDR является основным медиатором митогенного, ангиогенного и увеличивающего проницаемость действий VEGF-A, именуемого далее VEGF. Множество различных типов клеток способны продуцировать VEGF, однако его биологическая активность ограничивается преимущественно сосудистой сетью путем избирательной экспрессии KDR в эндотелиальных клетках. Вполне закономерно, что система VEGF/KDR является первичным медиатором ангиогенеза, процесса, при котором новые кровеносные сосуды формируются из предсуществующих сосудов.
FLT1 связывается с VEGF, VEGF-B и фактором роста плаценты. Кроме эндотелиальных клеток, FLT1 экспрессируется на поверхности клеток гладкой мускулатуры, моноцитов и гематопоэтических стволовых клеток. Активация передачи сигнала через FLT1 приводит к мобилизации происходящих из костного мозга эндотелиальных клеток-предшественников, которые рекрутируются в опухоли, где они участвуют в образовании новых кровеносных сосудов.
FLT4 опосредует передачу сигнала VEGF-C и VEGF-D, которые опосредуют образование связанных с опухолью лимфатических сосудов (лимфоангиогенез). Лимфатические сосуды являются одним из путей, по которым раковые клетки распространяются из солидных опухолей при метастазировании.
Семейство рецепторов PDGF (PDGFR) состоит из пяти RTK: PDGFR-a и -b, CSF1R, KIT и FLT3.
CSF-1R кодируется клеточным гомологом ретровирусного онкогена v-fms и является основным регулятором развития макрофагов. Макрофаги являются часто встречающимися компонентами опухолевой стромы, и было показано, что они изменяют внеклеточный матрикс, таким образом, что он становится благоприятным для роста и метастазирования опухоли.
KIT экспрессируется гематопоэтическими клетками-предшественниками, тучными клетками, гаметами и пейсмекерными клетками кишечника (интерстициальными клетками Кахаля). Он способствует развитию опухоли за счет двух основных механизмов, а именно путем его аутокринной стимуляции своим лигандом, фактором стволовых клеток (SCF), и в результате мутаций, приводящих к независимой от лиганда киназной активности.
FLT3 в норме экспрессируется на гематопоэтических стволовых клетках, где его взаимодействие с лигандом FLT3 (FL) стимулирует выживание, пролиферацию и дифференцировку стволовых клеток. Кроме сверхэкспрессии в различных лейкозных клетках, FLT3 часто является мутированным в гематологических злокачественных новообразованиях у приблизительно одной трети пациентов с острым миелоидным лейкозом (AML), несущих активирующие мутации.
Таким образом, желательной является идентификация небольших химических соединений, которые специфично ингибируют передачу сигнала и клеточную пролиферацию путем изменения активности тирозинкиназ, с тем, чтобы регулировать или изменять ненормальную или нарушенную клеточную пролиферацию, дифференцировку или метаболизм. В частности, была бы полезной идентификация способов и химических соединений, которые специфично ингибируют функцию тирозинкиназы, которая является существенной для процессов ангиогенеза или формирования повышенной проницаемости сосудов, приводящей к отеку, асциту, выпоту, экссудату и проникновению макромолекул и накоплению матрикса, а также связанных с этим нарушений.
Были идентифицированы химические соединения, которые ингибируют протеинкиназы, такие как киназы Аврора и киназы семейств VEGFR и PDGFR. Эти химические соединения и способы их получения раскрыты в публикации патента США № 2007-0155776 A1 (далее публикация '776) и в заявке на патент США № 12/632183 (далее заявка '183), включенных в настоящее описание путем ссылки полностью.
Очень низкая растворимость в воде химических соединений, например, описанных в заявке '183, является проблемой для специалистов по разработке рецептур, особенно в тех случаях, когда необходимо поддерживать приемлемую биодоступность при пероральном введении, которая сильно зависит от растворимости в водной среде желудочно-кишечного тракта. Различные решения проблемы низкой биодоступности при пероральном введении известны из уровня техники. Например, в работе Sharma & Joshi (2007) Asian Journal of Pharmaceutics 1(1):9-19 обсуждаются различные стратегии повышения растворимости при приготовлении твердых дисперсий. В ней описан метод испарения растворителя для приготовления твердых дисперсий, с упоминанием в качестве примера твердой дисперсии эторикоксиба, получаемой способом, который включает в себя растворение полиэтиленгликоля (ПЭГ), поливинилпирролидона (ПВП или повидона) и активного ингредиента в 2-пропаноле.
Для расширения клинического применения ингибитора протеинкиназ, например, в качестве химиотерапевтического препарата для онкологических пациентов, очень желательной была бы твердая лекарственная форма с приемлемой биодоступностью при пероральном введении. Такая лекарственная форма и схема ее перорального введения стали бы значительным прогрессом в лечении многих типов рака и сделали бы более легким ее применение в комбинированной терапии с другими химиотерапевтическими препаратами.
Сущность изобретения
Настоящее изобретение относится к продукту в виде твердой дисперсии, содержащему N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевину или ее соль, по меньшей мере, один фармацевтически приемлемый водорастворимый полимерный носитель и, по меньшей мере, одно фармацевтически приемлемое поверхностно-активное вещество.
Кроме того, настоящее изобретение относится к продукту в виде твердой дисперсии, содержащему N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевину или ее соль, по меньшей мере, одну кислоту, по меньшей мере, один фармацевтически приемлемый водорастворимый полимерный носитель и, по меньшей мере, одно фармацевтически приемлемое поверхностно-активное вещество.
Кроме того, настоящее изобретение относится к продукту в виде твердой дисперсии, содержащему N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевину, по меньшей мере, одну кислоту, по меньшей мере, один фармацевтически приемлемый водорастворимый полимерный носитель и, по меньшей мере, одно фармацевтически приемлемое поверхностно-активное вещество.
Также настоящее изобретение относится к твердой лекарственной форме для перорального введения, содержащей такой продукт в виде твердой дисперсии в некоторых случаях вместе с одним или несколькими дополнительными наполнителями.
Кроме того, настоящее изобретение относится к способу получения вышеописанного продукта в виде твердой дисперсии. Этот способ включает в себя:
(a) получение раствора, содержащего (i) N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевину или ее соль, (ii) фармацевтически приемлемую кислоту, (iii), по меньшей мере, один фармацевтически приемлемый водорастворимый полимерный носитель, (iv), по меньшей мере, одно фармацевтически приемлемое поверхностно-активное вещество и (v), по меньшей мере, один подходящий растворитель; и
(b) удаление, по меньшей мере, одного растворителя для получения твердой дисперсии, содержащей, по меньшей мере, один полимерный носитель, по меньшей мере, одно поверхностно-активное вещество, по меньшей мере, одну кислоту, и содержащей N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевину, диспергированную в ней в преимущественно некристаллической форме.
Также настоящее изобретение относится к твердой дисперсии, полученной вышеописанным способом.
Кроме того, настоящее изобретение относится к способу лечения рака, включающему в себя пероральное введение имеющему это заболевание субъекту терапевтически эффективного количества вышеописанной твердой дисперсии, или одной или нескольких твердых лекарственных форм, содержащих такую дисперсию.
Также настоящее изобретение относится к продукту в виде твердой дисперсии, содержащему ингибитор киназы, по меньшей мере, один фармацевтически приемлемый водорастворимый полимерный носитель и, по меньшей мере, одно фармацевтически приемлемое поверхностно-активное вещество, в котором твердая дисперсия (a) остается аморфной в течение, по меньшей мере, 1 месяца в условиях открытого хранения при температуре 25ºС и относительной влажности 75% и (b) имеет температуру стеклования при относительной влажности 75% меньше или равную 15ºС. Предпочтительно, ингибитор киназы представляет собой N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевину или ее фармацевтически приемлемую соль.
Дополнительные варианты осуществления настоящего изобретения, в том числе более подробные аспекты вариантов, приведенных выше, будут раскрыты или будут очевидны из следующего подробного описания.
Подробное описание
Твердая дисперсия в соответствии с настоящим раскрытием включает в себя N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевину или ее фармацевтически приемлемую соль в преимущественно некристаллической или аморфной форме, которая, как правило, является более растворимой, чем кристаллическая форма. Используемый здесь термин «твердая дисперсия» включает в себя системы, имеющие небольшие находящиеся в твердом состоянии частицы одной фазы, диспергированные в другой твердой фазе. В частности, настоящие твердые дисперсии включают в себя N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевину, диспергированную в инертном носителе.
Термин «аморфная форма» относится к частице без определенной структуры, т.е. не имеющей кристаллической структуры.
Используемый здесь термин «преимущественно некристаллический» означает, что не более чем примерно 5%, например, не более чем примерно 2%, или не более чем примерно 1% кристалличности выявляется или рентгеноструктурным анализом, или поляризационной микроскопией, или обоими этими методами.
N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевина, включая ее соли, как правило, имеют очень низкую растворимость в воде, например, менее чем примерно 100 мкг/мл, в большинстве случаев, менее чем примерно 30 мкг/мл. Настоящее изобретение может обеспечить наибольшие преимущества в случае лекарственных препаратов, которые практически нерастворимы в воде, т.е. имеют растворимость менее чем примерно 10 мкг/мл, так как способ настоящего изобретения увеличивает значение условного насыщения раствора такого плохо растворимого активного ингредиента. Примерами таких активных ингредиентов являются, например, лекарственные вещества класса IV Биофармацевтической системы классификации (БСК), которые характеризуются низкой растворимостью и низкой проникающей способностью (смотрите «Waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a biopharmaceutics classification system», U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), August 2000). Признанным является, что водная растворимость многих химических соединений зависит от значения pH; в случае таких химических соединений растворимость в контексте настоящего изобретения представляет интерес при физиологически значимом значении pH, например, значении pH от примерно 1 до примерно 8. Таким образом, в различных вариантах осуществления изобретения лекарственный препарат имеет растворимость в воде, по меньшей мере, при одном из значений pH в диапазоне от примерно 1 до примерно 8, менее чем примерно 100 мкг/мл, например, менее чем примерно 30 мкг/мл, или менее чем примерно 10 мкг/мл. В качестве иллюстрации, N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевина имеет растворимость в воде менее 30 нг/мл при значении pH 7,4.
Твердые дисперсии настоящего изобретения включают в себя в качестве активного ингредиента N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевину или ее фармацевтически приемлемую соль. В некоторых случаях они могут дополнительно включать в себя второй активный ингредиент, например, терапевтический агент, пригодный для применения в комбинированной терапии, как указано ниже в настоящем описании.
Активный ингредиент N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевина для использования в настоящем изобретении в недиспергированном состоянии может находиться в кристаллической или аморфной форме.
Активный ингредиент N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевина для использования в настоящем изобретении может быть в виде соли или в виде несолевого свободного основания.
Например, активный ингредиент N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевина может образовывать соли присоединения кислоты. Соли присоединения кислоты представляют собой соли, полученные в результате реакции N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевины с кислотой. Например, соли, включая ацетат, адипат, альгинат, бикарбонат, цитрат, аспартат, бензоат, бензолсульфонат (безилат), бисульфат, бутират, камфорат, камфорсульфонат, диглюконат, формиат, фумарат, глицерофосфат, глютамат, гемисульфат, гептаноат, гексаноат, гидрохлорид, гидробромид, гидроиодид, лактобионат, лактат, малеат, мезитиленсульфонат, метансульфонат, нафталинсульфонат, никотинат, оксалат, памоат, пектинат, персульфат, фосфат, пикрат, пропионат, сукцинат, тартрат, тиоцианат, трихлорацетат, трифторацетат, пара-толуолсульфонат и ундеканоат N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевины могут быть использованы в твердой дисперсии настоящего изобретения.
N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевину получают, например, так, как описано в примере 1 вышеупомянутой заявки на патент США № 12/632183, полное раскрытие которой включено в настоящее изобретение путем ссылки.
N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевина присутствует в твердой дисперсии настоящего изобретения в количестве, которое может быть терапевтически эффективным при введении этой композиции нуждающемуся в этом субъекту по соответствующей схеме. Величина дозы выражается здесь в виде количества, эквивалентного исходному веществу (эквивалент свободного основания), если иное не следует из контекста. Как правило, разовая доза (количество, вводимое за один раз), которая может вводиться с соответствующей частотой, например, от двух раз в сутки до одного раза в неделю, составляет от примерно 10 до примерно 1000 мг, в зависимости от используемого химического соединения. Когда частота введения составляет один раз в сутки (q.d.), разовая доза равна суточной дозе. В качестве иллюстрации, разовая доза, как правило, составляет от примерно 25 до примерно 1000 мг, более предпочтительно, от примерно 50 до примерно 500 мг, например, примерно 50, примерно 100, примерно 150, примерно 200, примерно 250, примерно 300, примерно 350, примерно 400, примерно 450 или примерно 500 мг. Когда лекарственная форма включает в себя оболочку капсулы, в которую помещена твердая дисперсия, то разовая доза может доставляться в одной капсуле или в нескольких капсулах, в большинстве случаев от 1 до примерно 10 капсул.
Чем выше разовая доза, тем более желательным является получение твердой дисперсии с относительно высокой концентрацией в ней лекарственного препарата. Как правило, концентрация лекарственного препарата в твердой дисперсии составляет, по меньшей мере, примерно 1%, например, от примерно 1% до примерно 50% по массе эквивалента свободного основания, но более низкие и высокие концентрации могут быть приемлемыми или достижимыми в конкретных случаях. Концентрация лекарственного препарата в различных вариантах осуществления изобретения составляет, по меньшей мере, примерно 1%, например, от примерно 1% до примерно 49%, или, по меньшей мере, примерно 5%, например, от примерно 5% до примерно 15%, или примерно 8%, например, от примерно 8% до примерно 12% по массе эквивалента свободного основания.
В некоторых вариантах осуществления изобретения продукт в виде твердой дисперсии настоящего изобретения включает в себя, по меньшей мере, одну кислоту. По меньшей мере, одна кислота выбирается из группы, состоящей из лимонной кислоты, винной кислоты, янтарной кислоты, яблочной кислоты, уксусной кислоты, малеиновой кислоты, малоновой кислоты, аскорбиновой кислоты, молочной кислоты, серной кислоты, фосфорной кислоты, соляной кислоты, бромистоводородной кислоты, метансульфоновой кислоты, бензолсульфоновой кислоты, толуолсульфоновой кислоты, этандисульфоновой кислоты, нафталинсульфоновой кислоты и 1-гидрокси-2-нафтойной кислоты. В предпочтительном варианте осуществления изобретения, по меньшей мере, одна кислота является лимонной кислотой.
По меньшей мере, одна кислота в общей сложности, как правило, составляет от примерно 0,1 до примерно 10 эквивалентов по отношению к N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевине, например, примерно 0,1 или более, примерно 0,5 или более, примерно 1 или более, или примерно 2 или более, или примерно 5 или более эквивалентов.
Основным компонентом матрицы продукта в виде твердой дисперсии является полимер, который является гидрофильным или водорастворимым, по меньшей мере, в части диапазона значений pH, в частности, при значениях pH, существующих в желудочно-кишечном тракте (ЖКТ), или комбинация таких полимеров. Полимер или смесь полимеров, используемые в настоящем изобретении, являются твердыми при комнатной температуре, и, в целях хорошей стабильности при хранении, в диапазоне температур, они должны оставаться твердыми даже при самых высоких температурах, воздействию которых они обычно подвергаются во время хранения, транспортировки или обработки продукта. Поэтому полезным свойством полимера, определяющим его пригодность для использования в настоящем изобретении, является его температура стеклования (Tg). Подходящие водорастворимые полимеры включают в себя без ограничений полимеры, имеющие Tg, по меньшей мере, примерно 50ºС, в частности от примерно 80ºС до примерно 180ºС. Способы определения значений Tg органических полимеров описаны, например, в Sperling, ed. (1992) Introduction To Physical Polymer Science, 2nd edition, John Wiley & Sons, Inc.
Неограничивающие примеры полимерных носителей, используемых в настоящем изобретении, включают в себя:
- гомополимеры и сополимеры N-виниллактамов, особенно гомополимеры и сополимеры N-винилпирролидона, например, гомополимер поливинилпирролидон (ПВП или повидон, например, Kollidon® 12 PF или его эквивалент, Kollidon® 17 PF или его эквивалент, Kollidon® 25 или его эквивалент, Kollidon® 30 PF или его эквивалент, Kollidon® 90 F или его эквивалент) и сополимеры, такие как сополимеры, содержащие в качестве мономеров N-винилпирролидон и винилацетат (коповидон) или мономеров N-винилпирролидон и винилпропионат;
- сложные и простые эфиры целлюлозы, в частности, метилцеллюлозу, этилцеллюлозу, гидроксиалкилцеллюлозы, такие как гидроксипропилцеллюлоза, гидроксиалкилалкилцеллюлозы, такие как гидроксипропилметилцеллюлоза (ГПМЦ или гипромеллоза, например, MethocelTM E3 или его эквивалент, MethocelTM E5 или его эквивалент, MethocelTM E6 или его эквивалент, MethocelTM E15 или его эквивалент, MethocelTM K3 или его эквивалент), фталаты и сукцинаты целлюлозы, такие как ацетатфталат целлюлозы, фталат гидроксипропилметилцеллюлозы, сукцинат гидроксипропилметилцеллюлозы и ацетатсукцинат гидроксипропилметилцеллюлозы (ГПМЦ-АС);
- высокомолекулярные полиалкиленоксиды, такие как полиэтиленоксид, полипропиленоксид и сополимеры этиленоксида и пропиленоксида (полоксамеры);
- полиакрилаты и полиметакрилаты, такие как сополимеры метакриловой кислоты и этилакрилата, сополимеры метакриловой кислоты и метилметакрилата, сополимеры бутилметакрилата и 2-диметиламиноэтилметакрилата, полигидроксиалкилакрилаты, полигидроксиалкилметакрилаты;
- полиакриламиды;
- полимеры винилацетата, такие как сополимеры винилацетата и кротоновой кислоты, частично гидролизованный поливинилацетат (также именуемый частично омыленный «поливиниловый спирт») и поливиниловый спирт;
- олиго- и полисахариды, такие как каррагинаны, галактоманнаны и ксантановая камедь;
и смеси двух или более из них.
В одном варианте осуществления изобретения матрица твердой дисперсии включает в себя один или несколько полимерных носителей, выбранных из группы, состоящей из поливинилпирролидона, гидроксипропилметилцеллюлозы и их смеси. Конкретным примером подходящего коповидона является коповидон, состоящий из примерно 60% мономеров N-винилпирролидона и примерно 40% мономеров винилацетата. Конкретным примером подходящего повидона является повидон, имеющий значение K (показатель вязкости водного раствора повидона), равное примерно 30.
Один или несколько полимерных носителей, как правило, составляют в общей сложности от примерно 20% до примерно 90%, например, от примерно 40% до примерно 85% от массы твердой дисперсии.
Не вдаваясь в теорию, считается, что при пероральном введении и воздействии жидкости ЖКТ, благодаря взаимодействию между полимерным носителем и поверхностно-активным компонентом твердой дисперсии, достигается приемлемая скорость высвобождения и ингибирование кристаллизации или рекристаллизации активного ингредиента, что обеспечивает биоабсорбцию.
Особенно пригодными для использования в качестве поверхностно-активных веществ в настоящем изобретении являются фармацевтически приемлемые неионные поверхностно-активные вещества, особенно те, которые имеют значение гидрофильно-липофильного баланса (ГЛБ) от примерно 12 до примерно 18, например, от примерно 13 до примерно 17, или от примерно 14 до примерно 16. Система ГЛБ (смотрите Fiedler (2002) Encyclopedia of Excipients, 5th edition, Aulendorf: ECV-Editio-Cantor-Verlag) присваивает поверхностно-активным веществам числовые значения, где липофильные вещества получают более низкие значения ГЛБ, а гидрофильные вещества получают более высокие значения ГЛБ.
Неограничивающие примеры неионных поверхностно-активных веществ, используемых в настоящем изобретении, включают в себя:
- полиоксиэтиленовые производные касторового масла, такие как ПЭГ-35 касторовое масло (например, Cremophor® EL от BASF Corp. или эквивалентный продукт), ПЭГ-40 гидрогенизованное касторовое масло (например, Cremophor® RH40 или эквивалентный продукт) и ПЭГ-60 гидрогенизованное касторовое масло (например, Cremophor® RH60 или эквивалентный продукт);
- сложные моноэфиры сорбитана и жирных кислот, например, сорбитан моноолеат (например, Span® 80 или эквивалентный продукт), сорбитан моностеарат (например, Span® 60 или эквивалентный продукт), сорбитан монопальмитат (например, Span® 40 или эквивалентный продукт) и сорбитан монолаурат (например, Span® 20 или эквивалентный продукт);
- полиоксиэтиленовые сложные моноэфиры сорбитана и жирных кислот (полисорбаты), такие как ПЭГ-20 сорбитан моноолеат (полисорбат 80, например, Tween® 80 или эквивалентный продукт), ПЭГ-20 сорбитан моностеарат (полисорбат 60, например, Tween® 60 или эквивалентный продукт), ПЭГ-20 сорбитан монопальмитат (полисорбат 40, например, Tween® 40 или эквивалентный продукт) или ПЭГ-20 сорбитан монолаурат (полисорбат 20, например, Tween® 20 или эквивалентный продукт);
- полоксамеры, такие как полоксамер 124, полоксамер 188, полоксамер 237, полоксамер 388 или полоксамер 407;
- глицериды полиэтиленгликоля, состоящие из моно-, ди- и триглицеридов и сложные моно- и диэфиры полиэтиленгликоля (например, Gelucire® 44/14 или эквивалентный продукт и Gelucire® 50/13 или эквивалентный продукт);
- α-токоферил полиэтиленгликоль сукцинат (ТПГС или витамин Е полиэтиленгликоль сукцинат, смотрите Национальный формуляр США);
и смеси двух или более из них.
Одно или несколько поверхностно-активных веществ, как правило, составляют в общей сложности от примерно 2% до примерно 40%, например, от примерно 5% до примерно 30% от массы твердой дисперсии.
Твердые дисперсии настоящего изобретения являются стабильными, т.е. твердые дисперсии остаются аморфными. Критически важным является, чтобы аморфные твердые дисперсии со временем не изменяли своей формы и потенциально не влияли на свойства лекарственного продукта. Поэтому в еще одном варианте осуществления настоящее изобретение также относится к продукту в виде твердой дисперсии, содержащему ингибитор киназы, по меньшей мере, один фармацевтически приемлемый водорастворимый полимерный носитель и, по меньшей мере, одно фармацевтически приемлемое поверхностно-активное вещество, в котором твердая дисперсия (a) остается аморфной в течение, по меньшей мере, 1 месяца в условиях открытого хранения при температуре 25ºС и относительной влажности 75% и (b) имеет температуру стеклования при относительной влажности 75% меньше или равную 15ºС. Предпочтительно, ингибитор киназы представляет собой N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевину или ее фармацевтически приемлемую соль.
Лекарственная форма настоящего изобретения может состоять или в основном состоит из вышеописанной твердой дисперсии. Однако в некоторых вариантах осуществления изобретения лекарственная форма содержит дополнительные наполнители и требует дополнительной обработки твердой дисперсии. Например, твердая дисперсия может быть измельчена до порошкообразного состояния и помещена в оболочку капсулы или сформована или спрессована в виде таблетки вместе с дополнительными наполнителями, которые могут стандартно использоваться в таких лекарственных формах.
Таким образом, твердая лекарственная форма для перорального введения настоящего изобретения включает в себя без ограничений капсулы, драже, гранулы, пилюли, порошки и таблетки. Наполнители, обычно используемые для приготовления таких лекарственных форм, включают в себя инкапсулирующие материалы или добавки к рецептуре, такие как ускорители всасывания, антиоксиданты, связующие вещества, буферы, покрывающие вещества, красители, разбавители, улучшающие распадаемость вещества, эмульгаторы, объемообразующие вещества, наполнители, ароматизаторы, увлажнители, скользящие вещества, консерванты, пропелленты, разделительные агенты, стерилизующие вещества, подсластители, солюбилизаторы и их смеси. Примеры конкретных наполнителей включают в себя агар, альгиновую кислоту, гидроксид алюминия, бензилбензоат, 1,3-бутиленгликоль, касторовое масло, целлюлозу, ацетат целлюлозы, какао-масло, кукурузный крахмал, кукурузное масло, хлопковое масло, этанол, этилацетат, этилкарбонат, этилцеллюлозу, этиллаурат, этилолеат, желатин, масло проростков, глюкозу, глицерин, масло земляного ореха, изопропанол, изотонический солевой раствор, лактозу, гидроксид магния, стеарат магния, солод, оливковое масло, арахисовое масло, соли фосфата калия, картофельный крахмал, пропиленгликоль, тальк, трагакантовую камедь, воду, сафлоровое масло, кунжутное масло, карбоксиметилцеллюлозу натрия, лаурилсульфат натрия, соли фосфата натрия, соевое масло, сахарозу, тетрагидрофурфуриловый спирт и их смеси.
В одном варианте осуществления твердая лекарственная форма для перорального введения настоящего изобретения представляет собой таблетку, содержащую порошок твердой дисперсии, наполнитель, улучшающее распадаемость вещество, глидант и скользящее вещество. В другом варианте осуществления изобретения таблетка содержит 50% по массе порошка твердой дисперсии.
Способ получения вышеописанной твердой дисперсии включает в себя получение раствора, содержащего N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевину или ее соль, кислоту, полимерный носитель и поверхностно-активное вещество в, по меньшей мере, одном подходящем растворителе; и удаление растворителя для получения твердой дисперсии.
В другом варианте способ получения вышеописанной твердой дисперсии включает в себя получение раствора, содержащего N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевину или ее соль, полимерный носитель и поверхностно-активное вещество в, по меньшей мере, одном подходящем растворителе; и удаление растворителя для получения твердой дисперсии.
В еще одном варианте осуществления изобретения способ получения вышеописанной твердой дисперсии включает в себя получение раствора, содержащего N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевину, кислоту, полимерный носитель и поверхностно-активное вещество в, по меньшей мере, одном подходящем растворителе; и удаление растворителя для получения твердой дисперсии.
На этапе получения раствора различные компоненты могут добавляться в любом порядке. Например, каждый ингредиент может быть добавлен в растворитель по отдельности, и затем растворен в нем. В другом варианте полимерный носитель и/или поверхностно-активное вещество могут быть предварительно смешаны с N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевиной, и полученная смесь затем добавляется в растворитель, или растворитель добавляется к полученной смеси. В другом варианте N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевина и кислота могут быть добавлены в, по меньшей мере, один растворитель, и затем добавляются полимерный носитель и поверхностно-активное вещество.
В принципе, может использоваться любой растворитель, при условии, что он эффективно растворяет активный ингредиент, полимерный носитель и поверхностно-активное вещество. Неограничивающие примеры растворителей, которые могут быть использованы, включают в себя метанол, этанол, ацетон, тетрагидрофуран, воду и их смеси. В предпочтительном варианте осуществления изобретения используется комбинация водного растворителя и смешиваемого с водой органического растворителя. Предпочтительно, компоненты растворяют в смеси воды и ацетона или в смеси воды и тетрагидрофурана.
Удаление растворителя может быть выполнено с использованием тепла, вакуума или их комбинации. Если используется тепло, то, как правило, предпочтительно избегать превышения температуры стеклования (Tg) полимерной матрицы. Для большинства задач подходящим является нагревание при температуре от примерно 50ºС до примерно 80ºС, например, от примерно 55ºС до примерно 75ºС. После удаления растворителя полученный продукт охлаждают (если необходимо) до температуры окружающей среды.
Более подробное описание способа может быть найдено ниже в иллюстративном способе примера 1.
Используемые в настоящем изобретении термины «для перорального введения», «пероральное введение» и «перорально вводимый» относятся к введению субъекту через рот (p.o.), то есть введению, при котором композиция немедленно проглатывается, например, с помощью подходящего объема воды или другой жидкости, пригодной для питья. «Пероральное введение» в соответствии с настоящим изобретением отличается от внутриротового введения, например, сублингвального или трансбуккального введения или местного введения во внутриротовые ткани, такие как периодонтальные ткани, которые не предполагают немедленного проглатывания композиции.
Настоящее изобретение относится к твердой дисперсии или лекарственной форме, обладающей приемлемой биоабсорбцией при пероральном введении. Такая биоабсорбция может быть подтверждена, например, фармакокинетическим (ФК) профилем твердой дисперсии или лекарственной формы, в частности средней максимальной концентрацией (Cmax) или площадью под фармакокинетической кривой (AUC), например, AUC0-24 или AUC0-∞ для конкретной дозы или в диапазоне доз. В качестве иллюстрации, биодоступность может быть выражена в процентах, например, с использованием коэффициента F, который вычисляет AUC для перорального введения тестовой композиции в процентах от AUC для внутривенного введения (i.v.) лекарственного препарата в подходящем растворителе, учитывая любое различие между пероральной и внутривенной дозами.
Биодоступность может быть определена при помощи исследований ФК у человека или у любых подходящих модельных видов. Для целей настоящего собачья модель, в качестве иллюстрации описанная ниже в примере 2, является в целом пригодной. В различных иллюстративных вариантах осуществления изобретения, в которых лекарственным препаратом является N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевина, композиции настоящего изобретения обладают биодоступностью при пероральном введении у собачей модели до, по меньшей мере, примерно 15%, по меньшей мере, примерно 20%, по меньшей мере, примерно 25% или, по меньшей мере, примерно 30% или превосходящей примерно 50%, при введении в виде разовой дозы величиной от примерно 2,5 до примерно 50 мг/кг животным на голодной выдержке или не голодавшим животным.
Упомянутые здесь композиции, включая композиции, описанные в общих чертах или подробно в настоящем изобретении, являются пригодными для перорального введения N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевины субъекту. Таким образом, способ настоящего изобретения по доставке такого лекарственного препарата субъекту включает в себя пероральное введение композиции, как описано выше.
Субъектом может быть человек или животное (например, сельскохозяйственное, содержащееся в зоопарке, рабочее или домашнее животное или лабораторное животное, используемое в качестве модели), но в важном варианте осуществления изобретения субъектом является человек-пациент, нуждающийся в лекарственном препарате, например для лечения рака. Являющийся субъектом человек может быть мужского или женского пола и любого возраста, но, как правило, он является взрослым.
Композицию обычно вводят в количестве, обеспечивающем терапевтически эффективную суточную дозу лекарственного препарата. Используемый здесь термин «суточная доза» означает количество лекарственного препарата, вводимое в течение одних суток, независимо от кратности введения. Например, если субъект получает разовую дозу, равную 150 мг, дважды в сутки, то суточная доза составляет 300 мг. Следует понимать, что использование термина «суточная доза» не предполагает того, что указанная величина дозы обязательно вводится один раз в сутки. Однако в конкретном варианте осуществления изобретения частота введения лекарственного препарата составляет один раз в сутки (q.d.), и в этом варианте осуществления изобретения суточная доза и разовая доза являют одним и тем же количеством.
Размер терапевтически эффективной дозы зависит от конкретного химического соединения, субъекта (включая вид и массу тела субъекта), заболевания (например, конкретного типа рака), которое подлежит лечению, стадии и/или тяжести этого заболевания, индивидуальной переносимости субъектом этого химического соединения, того вводится ли это химическое соединение в режиме монотерапии или в комбинации с одним или несколькими другими лекарственными препаратами, например, другими химиотерапевтическими препаратами для лечения рака, и других факторов. Таким образом, величина суточной дозы может варьировать в широких пределах, например, от примерно 10 до примерно 1000 мг. Большие или меньшие суточные дозы могут быть использованы в конкретных ситуациях. Следует понимать, что использование в настоящем изобретении термина «терапевтически эффективная» доза необязательно подразумевает то, что данный лекарственный препарат является терапевтически эффективным, только если он вводится однократно в такой дозе; как правило, терапевтическая эффективность зависит от композиции, которая вводится неоднократно в соответствии со схемой, включающей в себя необходимую кратность и длительность введения. Крайне предпочтительным является, чтобы выбранная суточная доза была достаточной, для того чтобы обеспечить благоприятный эффект с точки зрения лечения рака, и при этом была не достаточной для того, чтобы вызывать нежелательный побочный эффект в неприемлемой или непереносимой степени. Подходящая терапевтически эффективная доза может быть выбрана обычным врачом без проведения излишних экспериментов на основании настоящего раскрытия и описанного в нем уровня техники с учетом тех факторов, которые были упомянуты выше. Например, врач может начать курс лечения онкологического пациента с назначения относительно низкой суточной дозы и постепенно повышать дозу в течение периода времени продолжительностью несколько дней или недель, чтобы уменьшить риск нежелательных побочных эффектов.
В качестве иллюстрации, подходящие дозы N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевины как правило составляют от примерно 10 до примерно 1000 мг/сутки, более предпочтительно от примерно 50 до примерно 500 мг/сутки или от примерно 200 до примерно 400 мг/сутки, например, примерно 50, примерно 100, примерно 150, примерно 200, примерно 250, примерно 300, примерно 350, примерно 400, примерно 450 или примерно 500 мг/сутки, вводимые со средним интервалом между дозами от 3 до 10 дней или примерно от 4 до 8 дней или примерно 7 дней.
Когда композиция изготовляется в виде капсулы, от одной до нескольких капсул могут быть проглочены целиком, как правило, с помощью воды или другой пригодной для питья жидкости, чтобы помочь процессу глотания. Подходящие материалы оболочки капсулы включают в себя без ограничений желатин (в виде твердых желатиновых капсул или мягких эластичных желатиновых капсул), крахмал, каррагинан и ГПМЦ.
Так как считается, что композиции настоящего изобретения только в незначительной степени обладают свойствами пищи, то в соответствии с настоящим вариантом осуществления изобретения введение можно осуществлять как совместно с приемом пищи, так и отдельно от него, т.е. после приема пищи или натощак. Как правило, предпочтительным является введение настоящих композиций пациенту, принимавшему пищу.
Композиции настоящего изобретения пригодны для применения в монотерапии или в комбинированной терапии, например, с другими химиотерапевтическими препаратами или с ионизирующим излучением. Особым преимуществом настоящего изобретения является то, что оно позволяет осуществлять пероральное введение один раз в сутки - схема, которая является удобной для пациента, проходящего лечение с использованием других перорально принимаемых лекарственных препаратов по схеме один раз в сутки. Пероральное введение легко выполняется самим пациентом или лицом, осуществляющим за ним уход, у пациента дома; также оно является удобным способом введения для пациентов в стационаре или пациентов, получающих медицинскую помощь на дому.
Композиция настоящего изобретения, например, такая композиция, содержащая N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевину, может вводиться в комбинированной терапии вместе с одним или несколькими терапевтическими агентами, которые включают в себя без ограничений алкилирующие агенты, ингибиторы ангиогенеза, антитела, антиметаболиты, антимитотические агенты, антипролиферативные агенты, противовирусные препараты, ингибиторы киназ Аврора, другие индукторы апоптоза (например, ингибиторы Bcl-xL, Bcl-w и Bfl-1), активаторы каскада реакций с участием рецепторов смерти, ингибиторы Bcr-Abl киназы, BiTE-антитела (биспецифичные активаторы Т-клеток), нагруженные лекарственными препаратами антитела, модификаторы биологического отклика, ингибиторы циклинзависимых киназ (CDK), ингибиторы клеточного цикла, ингибиторы циклооксигеназы-2 (ЦОГ-2), связывающие белки с дублированным вариабельным доменом (DVD), ингибиторы рецептора эпидермального фактора роста человека второго типа (ErbB2 или HER/2neu), ингибиторы фактора роста, ингибиторы белка теплового шока (HSP)-90, ингибиторы гистондеацетилазы (HDAC), гормональные терапевтические лекарственные средства, иммунологические лекарственные средства, ингибиторы белков апоптоза (IAP), интеркалирующие антибиотики, ингибиторы киназ, ингибиторы кинезина, ингибиторы JAK2, ингибиторы мишени рапамицина в клетках млекопитающих (mTOR), микроРНК, ингибиторы митогенактивируемой киназы, регулируемой внеклеточными сигналами (MEK), мультивалентные связывающие белки, нестероидные противовоспалительные препараты (НПВП), ингибиторы поли-АДФ (аденозиндифосфат)-рибоза-полимеразы (PARP), химиотерапевтические препараты платины, ингибиторы polo-подобной киназы (Plk), ингибиторы фосфатидилинозитол-3-киназы (PI3K), ингибиторы протеосом, аналоги пурина, аналоги пиримидина, ингибиторы рецепторных тирозинкиназ, ретиноиды, дельтоиды, растительные алкалоиды, малые интерферирующие рибонуклеиновые кислоты (миРНК), ингибиторы топоизомеразы, ингибиторы убиквитин-лигазы и тому подобное.
BiTE-антитела представляют собой биспецифичные антитела, которые направляют Т-клетки атаковать раковые клетки, одновременно связывая две клетки. Затем Т-клетки атакуют раковые клетки-мишени. Примеры BiTE-антител включают в себя без ограничений адекатумумаб (Micromet MT201), блинатумомаб (Micromet MT103) и тому подобное. Не ограничиваясь теорией, одним из механизмов, при помощи которого Т-клетки индуцируют апоптоз целевых раковых клеток, является экзоцитоз компонентов цитолитических гранул, которые включают в себя перфорин и гранзим B. В этой связи было показано, что Bcl-2 ослабляет индукцию апоптоза как перфорином, так и гранзимом B. Эти данные позволяют предположить, что ингибирование Bcl-2 могло бы усилить цитотоксический эффект, вызываемый Т-клетками, когда они нацелены на раковые клетки (Sutton et al. (1997) J. Immunol. 158:5783-5790). миРНК представляют собой молекулы, содержащие эндогенные РНК основания или химически модифицированные нуклеотиды. Эти модификации не устраняют клеточную активность, а скорее придают повышенную стабильность и/или повышенную клеточную эффективность. Примеры химических модификаций включают в себя фосфотиоатные группы, 2'-дезоксинуклеотид, 2'-OCH3-содержащие рибонуклеотиды, 2'-F-рибонуклеотиды, 2'-метоксиэтил рибонуклеотиды, их комбинации и тому подобное. миРНК могут иметь различную длину (например, 10-200 п.н.) и структуру (например, шпильки, одно/двухцепочечную структуру, петли, одно/двухцепочечные разрывы, некомплементарное спаривание оснований) и процессируются в клетках, чтобы обеспечить сайленсинг функционально активных генов. Двухцепочечные миРНК (дцРНК) могут содержать одинаковое количество нуклеотидов в каждой цепи (тупые концы) или иметь асимметричные концы (липкие концы). Липкие концы в 1-2 нуклеотида могут находиться на смысловой и/или антисмысловой цепи, а также они могут находиться на 5'- и/или 3'-концах данной цепи. Например, было показано, что нацеливание миРНК на Mcl-1 повышает активность ABT-236 и ABT-737 в различных линиях опухолевых клеток (Tse et al. (2008) Cancer Res. 68:3421-3428 и ссылки в этой работе).
Мультивалентные связывающие белки представляют собой связывающие белки, содержащие два или более антигенсвязывающих сайта. Мультивалентные связывающие белки сконструированы таким образом, чтобы иметь три или более антигенсвязывающих сайта и, как правило, не являются антителами, существующими в природе. Термин «мультиспецифичный связывающий белок» обозначает связывающий белок, способный связываться с двумя или более взаимосвязанными или независимыми мишенями. Связывающие белки с дублированным вариабельным доменом (DVD) представляют собой тетравалентные или мультивалентные связывающие белки, содержащие два ли более антигенсвязывающих сайта. Такие DVD могут быть моноспецифичными (т.е. способными связываться с одним антигеном) или мультиспецифичными (т.е. способными связываться с двумя или более антигенами). DVD связывающие белки, включающие в себя две тяжелые полипептидные DVD цепи и две легкие полипептидные DVD цепи, называются DVD Ig. Каждая половина DVD Ig содержит тяжелую полипептидную DVD цепь, легкую полипептидную DVD цепь и два антигенсвязывающих сайта. Каждый сайт связывания включает в себя вариабельный домен тяжелой цепи и вариабельный домен легкой цепи с, в общей сложности, 6 определяющими комплементарность областями (CDR), вовлеченными в связывание с антигеном, на один антигенсвязывающий сайт.
Алкилирующие агенты включают в себя алтретамин, AMD-473, AP-5280, апазиквон, бендамустин, бросталлицин, бусульфан, карбоквон, кармустин (БХНМ), хлорамбуцил, CloretazineTM (ларомустин, VNP 30101M), циклофосфамид, дакарбазин, эстрамустин, фотемустин, глуфосфамид, ифосфамид, KW-2170, ломустин (CCNU), мафосфамид, мелфалан, митобронитол, митолактол, нимустин, N-оксида азотистого иприта, ранимустин, темозоломид, тиотепа, треосульфан, трофосфамид и тому подобное.
Ингибиторы ангиогенеза включают в себя ингибиторы рецептора эпидермального фактора роста (EGFR), ингибиторы эндотелиально-специфичной рецепторной тирозинкиназы (Tie-2), ингибиторы рецептора инсулиноподобного фактора роста 2 (IGFR-2), ингибиторы матриксной металлопротеиназы-2 (MMP-2), ингибиторы матриксной металлопротеиназы-9 (MMP-9), ингибиторы рецептора тромбоцитарного фактора роста (PDGFR), аналоги тромбоспондина, ингибиторы рецепторной тирозинкиназы фактора роста эндотелия сосудов (VEGFR) и тому подобное.
Антиметаболиты включают в себя АлимтаТМ (пеметрексед динатрия, LY231514, MTA), 5-азацитидин, КселодаТМ (капецитабин), кармофур, LeustatTM (кладрибин), клофарабин, цитарабин, окфосфат цитарабина, цитозинарабинозид, децитабин, дефероксамин, доксифлуридин, эфлорнитин, EICAR (5-этинил-1-β-D-рибофуранозилимидазол-4-карбоксамид), эноцитабин, этинилцитидин, флударабин, 5-фторурацил (5-ФУ) отдельно или в комбинации с лейковорином, ГемзарТМ (гемцитабин), гидроксикарбамид, АлкеранТМ (мелфалан), меркаптопурин, 6-меркаптопурин рибозид, метотрексат, микофеноловую кислоту, неларабин, нолатрексед, окфосфат, пелитрексол, пентостатин, ралтитрексед, рибавирин, S-1, триапин, триметрексат, TS-1, тиазофурин, тегафур, видарабин, UFT и тому подобное.
Противовирусные препараты включают в себя ритонавир, гидроксихлорохин и тому подобное.
Ингибиторы киназ Аврора включают в себя AZD-1152, MLN-8054, VX-680, ингибиторы, специфичные в отношении киназы Аврора A, ингибиторы, специфичные в отношении киназы Аврора B, ингибиторы всех киназ Аврора и тому подобное.
Ингибиторы белков семейства Bcl-2, отличные от ABT-263 или химических соединений приведенной здесь формулы I, включают в себя AT-101 ((-)госсипол), GenasenseTM - Bcl-2-направленный антисмысловой олигонуклеотид (G3139 или облимерсен), IPI-194, IPI-565, N-(4-(4-((4'-хлор(1,1'-бифенил)-2-ил)метил)пиперазин-1-ил)бензоил)-4-(((1R)-3-(диметиламино)-1-((фенилсульфанил)метил)пропил)амино)-3-нитробензолсульфонамид) (ABT-737), GX-070 (обатоклакс) и тому подобное.
Ингибиторы Bcr-Abl киназы включают в себя дазатиниб (BMS-354825), GleevecTM (иматиниб) и тому подобное.
Ингибиторы CDK включают в себя AZD-5438, BMI-1040, BMS-387032, CVT-2584, флавопиридол, GPC-286199, MCS-5A, PD0332991, PHA-690509, селициклиб (CYC-202 или R-росковитин), ZK-304709 и тому подобное.
Ингибиторы COX-2 включают в себя ABT-963, ArcoxiaTM (эторикоксиб), BextraTM (вальдекоксиб), BMS-347070, CelebrexTM (целекоксиб), COX-189 (люмиракоксиб), CT-3, DeramaxxTM (деракоксиб), JTE-522, 4-метил-2-(3,4-диметилфенил)-1-(4-сульфамоилфенил)-1H-пиррол, MK-663 (эторикоксиб), NS-398, парекоксиб, RS-57067, SC-58125, SD-8381, SVT-2016, S-2474, Т-614, VioxxTM (рофекоксиб) и тому подобное.
Ингибиторы EGFR включают в себя ABX-EGF, анти-EGFR иммунолипосомы, EGF-вакцину, EMD-7200, ErbituxTM (цетуксимаб), HR3, IgA антитела, IressaTM (гефитиниб), TarcevaTM (эрлотиниб или OSI-774), TP-38, EGFR слитый белок, TykerbTM (лапатиниб) и тому подобное. Ингибиторы рецептора ErbB2 включают в себя CP-724714, CI-1033 (канертиниб), HerceptinTM (трастузумаб), TykerbTM (лапатиниб), OmnitargTM (2C4, пертузумаб), TAK-165, GW-572016 (ионафамиб), GW-282974, EKB-569, PI-166, dHER2 (HER2 вакцина), APC-8024 (HER2 вакцина), анти-HER/2neu биспецифичные антитела, B7.her2IgG3, AS HER2 трифункциональные биспецифичные антитела, mAB AR-209, mAB 2B-1 и тому подобное.
Ингибиторы гистондеацетилазы включают в себя депсипептид, LAQ-824, MS-275, трапоксин, субероиланилид гидроксамовой кислоты (САГК), TSA, вальпроевую кислоту и тому подобное. Ингибиторы HSP-90 включают в себя 17AAG, CNF-101, CNF-1010, CNF-2024, 17-DMAG, гелданамицин, IPI-504, КОС-953, MycograbTM (рекомбинантные человеческие антитела к HSP-90), nab-17AAG, NCS-683664, PU24FC1, PU-3, радицикол, SNX-2112, STA-9090, VER-49009 и тому подобное.
Ингибиторы белков апоптоза включают в себя HGS-1029, GDC-0145, GDC-0152, LCL-161, LBW-242 и тому подобное.
Нагруженные лекарственными препаратами антитела включают в себя анти-CD22-MC-MMAF, анти-CD22-MC-MMAE, анти-CD22-MCC-DM1, CR-011-vcMMAE, PSMA-ADC, MEDI-547, SGN-19A, SGN-35, SGN-75 и тому подобное.
Активаторы каскада реакций с участием рецепторов смерти включают в себя ФНО-зависимый апоптоз-индуцирующий лиганд (TRAIL) и антитела или другие агенты, специфичные по отношению к TRAIL или рецепторам смерти (например, DR4 и DR5) такие как апомаб, конатумумаб, ETR2-ST01, GDC0145 (лексатумумаб), HGS-1029, LBY-135, PRO-1762, трастузумаб и тому подобное.
Ингибиторы кинезина включают в себя ингибиторы Eg5, такие как AZD-4877 и ARRY-520, ингибиторы CENPE, такие как GSK-923295A и тому подобное.
Ингибиторы JAK2 включают в себя CEP-701 (лесауртиниб), XL019, INCB-018424 и тому подобное.
Ингибиторы MEK включают в себя ARRY-142886, ARRY-438162, PD-325901, PD-98059 и тому подобное.
Ингибиторы mTOR включают в себя AP-23573, CCI-779, эверолимус, RAD-001, рапамицин, темсиролимус, АТФ-конкурентные ингибиторы TORC1/TORC2, в том числе PI-103, PP242, PP30 и Торин 1, и тому подобное.
Нестероидные противовоспалительные препараты включают в себя AmigesicTM (салсалат), DolobidTM (дифлунизал), MotrinTM (ибупрофен), OrudisTM (кетопрофен), RelafenTM (набуметон), FeldeneTM (пироксикам), ибупрофен мазь, AleveTM и NaprosynTM (напроксен), VoltarenTM (диклофенак), IndocinTM (индометацин), ClinorilTM (сулиндак), TolectinTM (толметин), LodineTM (этодолак), ToradolTM (кеторолак), DayproTM (оксапрозин) и тому подобное.
Ингибиторы PDGFR включают в себя CP-673451, CP-868596 и тому подобное.
Химиотерапевтические препараты платины включают в себя цисплатин, EloxatinTM (оксалиплатин), эптаплатин, лобаплатин, недаплатин, ParaplatinTM (карбоплатин), пикоплатин, сатраплатин и тому подобное.
Ингибиторы polo-подобной киназы включают в себя BI-2536 и тому подобное.
Ингибиторы фосфатидилинозитол-3-киназы включают в себя вортманнин, LY-294002, XL-147, CAL-120, ONC-21, AEZS-127, ETP-45658, PX-866, GDC-0941, BGT226, BEZ235, XL765 и тому подобное.
Аналоги тромбоспондина включают в себя ABT-510, ABT-567, ABT-898, TSP-1 и тому подобное.
Ингибиторы VEGFR включают в себя AvastinTM (бевацизумаб), ABT-869, AEE-788, AngiozymeTM (рибозим, который ингибирует ангиогенез (Ribozyme Pharmaceuticals (Boulder, CO) и Chiron (Emeryville, CA)), акситиниб (AG-13736), AZD-2171, CP-547632, IM-862, MacugenTM (пегаптаниб), NexavarTM (сорафениб, BAY43-9006), пазопаниб (GW-786034), ваталаниб (PTK-787 и ZK-222584), SutentTM (сунитиниб или SU-11248), VEGF-Trap, ZactimaTM (вандетаниб или ZD-6474) и тому подобное.
Антибиотики включают себя интеркалирующие антибиотики, такие как акларубицин, актиномицин D, амрубицин, аннамицин, AdriamycinTM (доксорубицин), BlenoxaneTM (блеомицин), даунорубицин, CaelyxTM и MyocetTM (липосомальный доксорубицин), элсамитруцин, эпирубицин, гларубицин, идарубицин, митомицин С, неморубицин, неокарциностатин, пепломицин, пирарубицин, ребеккамицин, стималамер, стрептозоцин, ValstarTM (валрубицин), зиностатин и тому подобное.
Ингибиторы топоизомеразы включают в себя акларубицин, 9-аминокамптотецин, амонафид, амсакрин, бекатекарин, белотекан, BN-80915, CamptosarTM (иринотекана гидрохлорид), камптотецин, CardioxaneTM (дексразоксан), дифломотекан, эдотекарин, EllenceTM и PharmorubicinTM (эпирубицин), этопозид, экзатекан, 10-гидроксикамптотецин, гиматекан, луртотекан, митоксантрон, оратецин, пирарбуцин, пиксантрон, рубитекан, собузоксан, SN-38, тафлупозид, топотекан и тому подобное.
Антитела включают в себя AvastinTM (бевацизумаб), CD40-специфичные антитела, chTNT-1/B, деносумаб, ErbituxTM (цетуксимаб), Humax-CD4TM (занолимумаб), IGF1R-специфичные антитела, линтузумаб, PanorexTM (эдреколомаб), RencarexTM (WX G250), RituxanTM (ритуксимаб), тицилимумаб, трастузимаб, антитела к CD20 типов I и II и тому подобное.
Гормональные терапевтические лекарственные средства включают в себя ArimidexTM (анастрозол), AromasinTM (эксеместан), арзоксифен, CasodexTM (бикалутамид), CetrotideTM (цетрореликс), дегареликс, деслорелин, DesopanTM (трилостан), дексаметазон, DrogenilTM, (флутамид), EvistaTM (ралоксифен), AfemaTM (фадрозол), FarestonTM (торемифен), FaslodexTM (фулвестрант), FemaraTM (летрозол), форместан, глюкокортикоиды, HectorolTM (доксеркалциферол), RenagelTM (севеламера карбонат), ласофоксифен, леупролид-ацетат, MegaceTM (мегестерол), MifeprexTM (мифепристон), NilandronTM (нилутамид), содержащий тамоксифен NolvadexTM (тамоксифен-цитрат), PlenaxisTM (абареликс), предизон, PropeciaTM (финастерид), рилостан, SuprefactTM (бусерелин), TrelstarTM, содержащий релизинг-фактор лютенизирующего гормона (LHRH), (трипторелин), содержащий гистрелин VantasTM (гистрелиновый имплант), ModrastaneTM (трилостан), ZoladexTM (гозерелин) и тому подобное.
Дельтоиды и ретиноиды включают в себя сеокальцитол (EB1089 или CB1093), лексакальцитрол (KH1060), фенретинид, PanretinTM (алиретиноин), содержащий третиноин AtragenTM (липосомный третиноин), TargretinTM (бексаротен), LGD-1550 и тому подобное.
Ингибиторы PARP включают в себя ABT-888, олапариб, KU-59436, AZD-2281, AG-014699, BSI-201, BGP-15, INO-1001, ONO-2231 и тому подобное.
Растительные алкалоиды включают в себя винкристин, винбластин, виндезин, винорелбин и тому подобное.
Ингибиторы протеосом включают в себя VelcadeTM (бортезомиб), MG132, NPI-0052, PR-171 и тому подобное.
Примеры иммунологических лекарственных средств включают в себя интерфероны и другие лекарственные средства, повышающие иммунитет. Интерфероны включают в себя интерферон-альфа, интерферон-альфа-2a, интерферон-альфа-2b, интерферон-бета, интерферон-гамма-1a, ActimmuneTM (интерферон-гамма-1b), интерферон-гамма-n1, их комбинации и тому подобное. Другие агенты включают в себя AlfaferoneTM (IFN-α), BAM-002 (окисленный глутатион), BeromunTM (тазонермин), BexxarTM (тозитумомаб), CampathTM (алемтузумаб), CTLA4 (цитотоксический лимфоцитный антиген 4), декарбазин, денилейкин, эпратузумаб, GranocyteTM (ленограстим), лентинан, лейкоцитарный альфа-интерферон, имиквимод, MDX-010 (анти-CTLA4), меланомную вакцину, митумомаб, молграмостим, MylotargTM (гемтузумаб озогамицин), NeupogenTM (филграстим), OncoVAC-CL, OvarexTM (ореговомаб), пемтумомаб (Y-muHMFG1), ProvengeTM (сипулеуцел-Т), саргарамостим, сизофилан, тецелейкин, TheracysTM (БЦЖ или Бацилла Кальметта-Герена), убенимекс, VirulizinTM (иммунотерапевтический препарат, Lorus Pharmaceuticals), Z-100 (конкретное вещество Маруяма вакцины или SSM), WF-10 (тетрахлородекаоксид), ProleukinTM (альдеслейкин), ZadaxinTM (тимальфазин), ZenapaxTM (даклизумаб), ZevalinTM (90Y-ибритумомаб тиуксетан) и тому подобное.
Модификаторы биологического отклика представляют собой агенты, которые модифицируют защитные механизмы живых организмов или биологические реакции, такие как выживание, рост или дифференцировку клеток ткани, для направления их на выработку противоопухолевой активности, и включают в себя крестин, лентинан, сизофиран, пицибанил PF-3512676 (CpG-8954), убенимекс и тому подобное.
Аналоги пиримидина включают в себя цитарабин (цитозина арабинозид, ара C или арабинозид C), доксифлуридин, FludaraTM (флударабин), 5-ФУ (5-фторурацил), флоксуридин, GemzarTM (гемцитабин), TomudexTM (ратитрексед), триацетилуридин, TroxatylTM (троксацитабин) и тому подобное.
Аналоги пурина включают в себя LanvisTM (тиогуанин), PurinetholTM (меркаптопурин) и тому подобное.
Антимитотические агенты включают в себя батабулин, эпотилон D (KOS-862), N-(2-((4-гидроксифенил)амино)пиридин-3-ил)-4-метоксибензолсульфонамид, иксабепилон (BMS-247550), паклитаксел, TaxotereTM (доцетаксел), ларотаксел (PNU100940, RPR-109881 или XRP-9881), патупилон, винфлунин, ZK-EPO (синтетический эпотилон) и тому подобное.
Ингибиторы убиквитин-лигазы включают в себя ингибиторы MDM2, такие как нутлины, ингибиторы NEDD8, такие как MLN4924, и тому подобное.
Композиции настоящего изобретения также могут применяться в качестве радиосенсибилизаторов, которые повышают эффективность лучевой терапии. Примеры лучевой терапии включают в себя без ограничений лучевую терапию с внешним пучком излучения (XBRT), дистанционную лучевую терапию, близкофокусную лучевую терапию, лучевую терапию с герметизированным источником излучения, лучевую терапию с негерметизированным источником излучения и тому подобное.
В дополнительном или альтернативном варианте композиции настоящего изобретения могут вводиться в комбинированной терапии с одним или несколькими противоопухолевыми или химиотерапевтическими агентами, выбранными из препаратов AbraxaneTM (ABI-007), ABT-100 (ингибитор фарнезилтрансферазы), AdvexinTM (Ad5CMV-p53 вакцина или контусуген ладеновек), AltocorTM или MevacorTM (ловастатин), AmpligenTM (поли(I)-поли(C12U), синтетическая РНК), AptosynTM (эксисулинд), ArediaTM (памидроновая кислота), арглабин, L-аспарагиназа, атаместан (1-метил-3,17-дион-андроста-1,4-диен), AvageTM (тазаротен), AVE-8062 (производное комбретастатина), BEC2 (митумомаб), кахектин или кахексин (фактор некроза опухоли), CanvaxinTM (противомеланомная вакцина), CeaVacTM (противораковая вакцина), CeleukTM (целмолейкин), гистаминсодержащий CepleneTM (дигидрохлорид гистамина), CervarixTM (адсорбированная вакцина против вируса паллиломы человека (ВПЧ), содержащая адъювант AS04), CHOP (CytoxanTM (циклофосфамид)+AdriamycinTM (доксорубицин)+OncovinTM (винкристин)+преднизон), комбрестатин A4P, CypatTM (ципротерон), DAB(389)EGF (каталитический и транслокационный домены дифтерийного токсина, соединенные через His-Ala линкер с человеческим эпидермальным фактором роста), дакарбазин, дактиномицин, DimericineTM (T4N5 липосомный лосьон), 5,6-диметилксантенон-4-уксусная кислота (DMXAA), дискодермолид, DX-8951f (эксатекан-мезилат), энилурацил (этинилурацил), скваламинсодержащий EvizonTM (скваламинлактат), энзастаурин, EPO-906 (эпотилон B), GardasilTM (квадривалентная рекомбинантная вакцина против вируса папилломы человека (типы 6, 11, 16, 18)), GastrimmuneTM, GenasenseTM (облимерсен), GMK (ганглиозидная конъюгатная вакцина), GVAXTM (вакцина против рака предстательной железы), галофугинон, гистерелин, гидроксикарбамид, ибандроновая кислота, IGN-101, IL-13-PE38, IL-13-PE38QQR (бесудотокс цинтредекина), IL-13-синегнойный экзотоксин, интерферон-α, интерферон-γ, JunovanTM и MepactTM (мифамуртид), лонафарниб, 5,10-метилентетрагидрофолат, милтефозин (гексадецилфосфохолин), NeovastatTM (AE-941), NeutrexinTM (триметрексат-глюкуронат), NipentTM (пентостатин), OnconaseTM (ранпирназа, рибонуклеазный фермент), OncophageTM (витеспен, вакцина для лечения меланомы), OncoVAXTM (IL-2 вакцина), OrathecinTM (рубитекан), OsidemTM (лекарственное средство на основе антитела, действующее на клеточном уровне), OvarexTM MAb (моноклональное мышиное антитело), стабилизированные альбумином наночастицы паклитаксела, паклитаксел, PandimexTM (агликоновые сапонины, выделенные из женьшеня и содержащие 20(S)протопанаксадиол (aPPD) и 20(S)протопанаксатриол (aPPT)), панитумумаб, PanvacTM-VF (вакцина против рака, проходящая клинические испытания), пегаспаргаза, пэгинтерферон альфа (ПЭГ-интерферон A), феноксодиол, прокарбазин, ребимастат, RemovabTM (катумаксомаб), RevlimidTM (леналидомид), RSR13 (эфапроксирал), SomatulineTM LA (ланреотид), SoriataneTM (ацитретин), стауроспорин (Streptomyces staurospores), талабостат (PT100), TargretinTM (бексаротен), TaxoprexinTM (докозогексановая кислота (ДГК)+паклитаксел), TelcytaTM (канфосфамид, TLK-286), TemodaRTM (темозоломид), тесмилифен, тетрандрин, талидомид, TheratopeTM (STn-KLH вакцина), ThymitaqTM (нолатрексед дигидрохлорид), TNFeradeTM (аденовектор: носитель ДНК, содержащий ген фактора некроза опухоли-α), TracleerTM или ZavescaTM (босентан), TransMID-107RTM (KSB-311, дифтерийные токсины), третиноин (ретин-A), TrisenoxTM (триоксид мышьяка), UkrainTM (производное алкалоидов из чистотела большого), VirulizinTM, VitaxinTM (антитело к αvβ3), XcytrinTM (мотексафин гадолиния), XinlayTM (атрасентан), XyotaxTM (паклитаксел полиглумекс), YondelisTM (трабектедин), ZD-6126 (N-ацетилколхинол-O-фосфат), ZinecardTM (дексразоксан), золендроновая кислота, зорубицин и тому подобного.
В одном варианте осуществления композиция настоящего изобретения, например такая композиция, которая содержит N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевину или ее соль, вводится в терапевтически эффективном количестве нуждающемуся в этом субъекту с целью лечения рака.
Примеры включают в себя без ограничений невриному слухового нерва, острый лейкоз, острый лимфоцитарный лейкоз, острый миелолейкоз (моноцитарный, миелобластный, аденокарцинома, ангиосаркома, астроцитома, миеломоноцитарный и промиелоцитарный), острый Т-клеточный лейкоз, базально-клеточную карциному, карциному желчного протока, рак мочевого пузыря, рак мозга, рак молочной железы, бронхогенную карциному, рак шейки матки, хондросаркому, хордому, хориокарциному, хронический лейкоз, хронический лимфоцитарный лейкоз, хронический миелоцитарный (гранулоцитарный) лейкоз, хронический миелобластный лейкоз, рак толстой кишки, колоректальный рак, краниофарингиому, цистаденокарциному, диффузную В-крупноклеточную лимфому, диспролиферативные изменения (дисплазии и метаплазии), эмбриональную карциному, рак эндометрия, эндотелиосаркому, эпендимому, эпителиальную карциному, эритролейкемию, рак пищевода, эстроген-рецептор-положительный рак молочной железы, идиопатическую тромбоцитемию, опухоль Юинга, фибросаркому, фолликулярную лимфому, рак половых клеток яичек, глиому, болезнь тяжелых цепей, гемангиобластому, гепатому, гепатоцеллюлярный рак, гормон-нечувствительны рак предстательной железы, лейомиосаркому, липосаркому, рак легких, лимфагиоэндотелиосаркому, лимфангиосаркому, лимфобластный лейкоз, лимфому (Ходжкина и не-Ходжкина), злокачественные и гиперпролиферативные заболевания мочевого пузыря, молочной железы, толстой кишки, легкого, яичников, поджелудочной железы, предстательной железы, кожи и матки, лимфонеоплазии Т- или В-клеточного происхождения, лейкемию, лимфому, медуллярную карциному, медуллобластому, меланому, менингиому, мезотелиому, множественную миелому, миелолейкоз, миелому, миксосаркому, нейробластому, немелкоклеточный рака легкого, олигодендроглиому, рак ротовой полости, остеогенную саркому, рак яичников, рак поджелудочной железы, папиллярную аденокарциному, папиллярную карциному, пинеалому, полицитемию, рак предстательной железы, рак прямой кишки, почечно-клеточную карциному, ретинобластому, рабдомиосаркому, саркому, карциному сальной железы, семиному, рак кожи, мелкоклеточную карциному легкого, солидные опухоли (карциномы и саркомы), мелкоклеточный рак легкого, рак желудка, плоскоклеточную карциному, синовиому, карциному потовых желез, рак щитовидной железы, макроглобулинемию Вальденстрема, опухоли яичек, рак матки и опухоль Вильмса у млекопитающих.
В более предпочтительном варианте осуществления композиция настоящего изобретения вводится в терапевтически эффективном количестве нуждающемуся в этом субъекту для лечения миелодиспластического синдрома, острого миелолейкоза, колоректального рака, немелкоклеточного рака легкого и рака яичников.
В соответствии с любым из этих вариантов осуществления изобретения композиция вводится в комбинированной терапии с одним или несколькими дополнительными терапевтическими агентами.
Как и в других вариантах осуществления изобретения, введение в соответствии с настоящим вариантом осуществления можно проводить как совместно с приемом пищи, так и отдельно от него, т.е. после приема пищи или натощак. Как правило, предпочтительным является введение настоящих композиций пациенту, принимавшему пищу.
Примеры
Следующие примеры являются только иллюстративными и никаким образом не ограничивают настоящее раскрытие. В этих примерах используются следующие ингредиенты, охраняемые товарными знаками:
Eudragit® L 100-55: сополимер метакриловой кислоты и этилакрилата;
Kollidon® VA64: сополимер винилпирролидона и винилацетата;
Kollidon® SR: поливинилацетат в матрице повидона;
Tween® 20: полисорбат 20 поверхностно-активное вещество;
Cremophor® RH40: полиоксил 40 гидрогенизованное касторовое масло;
Gelucire® 44/14: глицериды полиэтиленгликоля.
Пример 1
Приготовление твердых дисперсий
Свободное основание N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевину (далее API) смешивали в зависимости от рецептуры с кислотой, поверхностно-активным веществом(ами) и водорастворимым полимером(ами) в следующих весовых соотношениях:
Пример 1A: 6% API:3,5% лимонная кислота:20% Tween® 20:10% полоксамер 124:60,5% ПЭГ-1450.
Пример 1B: 10% API:6% лимонная кислота:10% Tween® 20:10% полоксамер 124:24% ГПМЦ-E5:40% ПВП K17.
Пример 1C: 6% API:3% лимонная кислота:20% Tween® 20:71% ПЭГ-1450.
Пример 1D: 7,5% API:4,5% лимонная кислота:10% Tween® 20:10% Cremophor® RH40:25% ГПМЦ-E5:43% ПВП K17.
Пример 1E: 10% API:6% лимонная кислота:10% Tween® 20:10% Cremophor® RH40:24% ГПМЦ-E5:40% ПВП K17.
Пример 1F: 5% API:3% лимонная кислота:20% Tween® 20:27% ГПМЦ-E5:45% ПВП K30.
Пример 1G: 5% API:6% лимонная кислота:20% Tween® 20:24% ГПМЦ-E5:40% ПВП K30.
Пример 1H: 5% API:3% лимонная кислота:20% Tween® 20:72% ГПМЦ-E5.
Пример 1I: 5% API:3% лимонная кислота:20% Tween® 20:67% ПЭГ-8000.
Пример 1J: 10% API:3% лимонная кислота:10% Tween® 20:10% полоксамер 124:64% ГПМЦ-E5.
Пример 1K: 5% API:2% лимонная кислота:10% Tween® 20:83% ПЭГ-3350.
Пример 1L: 10% API:4% лимонная кислота:10% Tween® 20:76% ГПМЦ-E5.
Пример 1M: 10% API:20% Tween® 20:70% ГПМЦ-АС L.
Пример 1N: 5% API:3% лимонная кислота:25% Tween® 20:67% ПЭГ-33500.
Пример 1O: 5% API:2% лимонная кислота:10% Tween® 20:83% ПВП K30.
Пример 1P: 10% API:6% лимонная кислота:10% Tween® 20:10% полоксамер 124:64% ПВП K30.
Пример 1Q: 5% API:3% лимонная кислота:20% Tween® 20:72% ПВП K30.
Пример 1R: 10% API:6% лимонная кислота:10% Tween® 20:30% ГПМЦ-E5:44% ПВП K30.
Пример 1S: 5% API:4% лимонная кислота:Gelucire® 44/14.
Пример 1T: 12,5% API:7,5% лимонная кислота:20% Tween® 20:22,5% ГПМЦ-E5:37,5% ПВП K30.
Пример 1U: 15% API:9% лимонная кислота:20% Tween® 20:21% ГПМЦ-E5:35% ПВП K30.
Пример 1V: 20% API:12% лимонная кислота:20% Tween® 20:18% ГПМЦ-E5:30% ПВП K30.
Пример 1W: 10% API:6% лимонная кислота:10% Tween® 20:10% полоксамер 407:24% ГПМЦ-E5:40% ПВП K17.
Пример 1X: 10% API:6% лимонная кислота:10% Tween® 20:10% полоксамер 188:24% ГПМЦ-E5:40% ПВП K17.
Пример 1Y: 5% API:2% лимонная кислота:10% Tween® 20:83% Eudragit® L 100-55.
Пример 1Z: 10% API:6% лимонная кислота:10% Tween® 20:10% полоксамер 124:24% ГПМЦ-E5:40% ПВП K30.
Пример 1AA: 10% API:6% лимонная кислота:20% Tween® 20:24% ГПМЦ-E3:40% ПВП K30.
Пример 1AB: 10% API:20% Tween® 20:70% ГПМЦ-АС M.
Пример 1AC: 10% API:20% Tween® 20:70% ГПМЦ-АС H.
Пример 1AD: 10% API:6% лимонная кислота:20% Tween® 20:24% ГПМЦ-E3:40% ПВП K17.
Пример 1AE: 10% API:6% лимонная кислота:10% Tween® 20:10% Gelucire® 44/14:24% ГПМЦ-E5:40% ПВП K17.
Пример 1AF: 5% API:4% лимонная кислота:91% витамин Е ТПГС.
Пример 1AG: 10% API:4% лимонная кислота:10% Tween® 20:76% Kollidon® VA64.
Пример 1AH: 10% API:7% лимонная кислота:83% Kollidon® SR.
Пример 1AI: 10% API:6% лимонная кислота:20% Tween® 20:24% ГПМЦ-K3:40% ГПМЦ-K3.
Пример 1AJ: 5% API:2% лимонная кислота:10% Tween® 20:83% Kollidon® VA64.
Пример 1AK: 10% API:6% лимонная кислота:20% Tween® 20:24% ГПМЦ-K3:40% ПВП K30.
Смесь ингредиентов в каждом случае растворяли в водном растворителе и смешиваемом с водой в органическом растворителе, например, ацетоне или тетрагидрофуране, в диапазоне температур от комнатной до 70ºС. Растворитель удаляли или путем ротационного выпаривания при 65ºС в вакууме, или используя распылительную сушилку при температуре 85ºС, и полученную твердую дисперсию охлаждали до комнатной температуры.
Твердую дисперсию в каждом случае просеивали через сито с размером ячеек 30 для получения порошка с уменьшенным размером частиц. Полученную твердую дисперсию сушили под вакуумом при температуре приблизительно 100ºС.
Пример 2
Фармакокинетика твердых дисперсий у собачей модели
Фармакокинетику разовой дозы твердых дисперсий оценивали у собак породы бигль (n=3), подвергнутых голодной выдержке, после того как они перорально получили 10, 25 или 50 мг твердой дисперсии в твердой желатиновой капсуле и затем 10 мл воды. Приблизительно за 30 минут до введения лекарственного препарата каждая собака получила подкожно (sc) гистамин в дозе 100 мг/кг. Приблизительно через 4 часа после введения дозы лекарственного препарата собакам давали корм. Серию образцов гепаринизированной крови получали из яремной вены каждого животного перед введением дозы и через 0,25, 0,5, 1, 1,5, 2, 3, 4, 6, 9, 12, 15 и 24 часа после введения. Плазму крови отделяли центрифугированием (2000 об/мин в течение 10 минут при температуре приблизительно 4ºС), и API выделяли при помощи осаждения белков ацетонитрилом.
Площадь под кривой концентрация в плазме - время в интервале от 0 до t часов (время последнего измерения концентрации в плазме) после введения дозы (AUC0-t) вычисляли с применением линейного метода трапеций для профилей концентрация в плазме - время. Остальную площадь, экстраполированную в бесконечность, определяемую как последняя измеренная концентрация в плазме (Ct), деленная на константу скорости терминальной элиминации (β), прибавляли к AUC0-t, чтобы получить общую площадь под кривой (AUC0-∞). Биодоступность рассчитывали путем деления доза-нормированной AUC0-∞ при пероральном введении на соответствующее значение, полученное при внутривенном (i.v.) введении дозы в виде медленной болюсной инъекции в яремную вену под легкой эфирной анестезией.
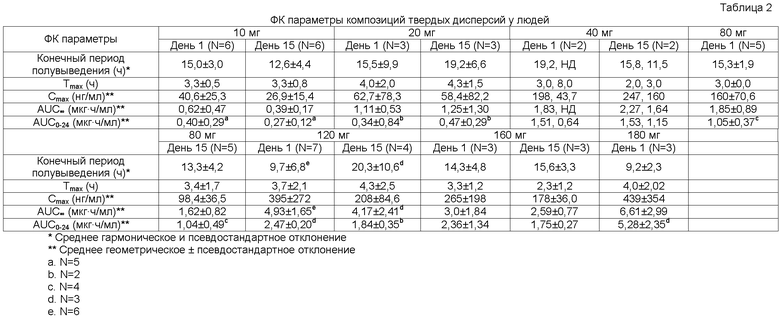
ФК параметры для дисперсий представлены в таблице 1.
ФК параметры композиций твердых дисперсий у собак (n=3)
Пример 3
Фармакокинетика твердых дисперсий у людей
Препараты твердых дисперсий настоящего изобретения использовали в открытой Фазе I исследований на человеке безопасности и фармакокинетики N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевины в качестве монотерапии у пациентов с запущенными солидными опухолями или запущенными гематологическими злокачественными опухолями.
Количество пациентов, которые вошли в исследование и завершили, по меньшей мере, часть исследований было зафиксировано. Пациентам, включенным в исследование, было назначено получение одной из следующих доз: 10 мг, 20 мг, 40 мг, 80 мг, 120 мг, 160 мг и 180 мг. Таблетированный препарат состоял из порошка твердой дисперсии, соответствующей примеру 1E, с наполнителями (50% пример 1E:38,75% микрокристаллическая целлюлоза:10% кросповидон:1% коллоидный диоксид кремния:0,5% стеарат магния).
Дозы вводили в первый, восьмой и пятнадцатый день каждого 28-дневного цикла. В день 1 и день 15 отбирали образцы плазмы крови через 0, 0,5, 1, 2, 3, 4, 6, 8, 10 и 24 часа или через 0, 0,5, 1, 2, 3, 4, 6, 8, 10 или 12, 24, 48 и 72 часа после введения дозы. Определяли концентрацию N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевины в плазме крови, и рассчитывали значения фармакокинетических параметров, которые приведены в таблице 2.
Пример 4
N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1Н-пиразол-4-ил]тиено[3,2-с]пиридин-3-ил}фенил)-N′-(3-фторфенил)мочевина
Пример 4А
3-(4-аминофенил)-7-йодтиено[3,2-с]пиридин-4-амин
Суспензию 3-бромтиено [3,2-c]пиридин-4-амина (13,7 г, 59,7 ммоль), трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенилкарбамата (20 г, 62,7 ммоль), тетракис(трифенилфосфин)палладия(0) (2,5 г, 2,1 ммоль) и Na2CO3 (13,3 г, 125 ммоль) в тетрагидрофуране (150 мл), метаноле (40 мл) и воде (80 мл) дегазировали, затем перемешивали при обратном притоке в течение ночи. Реакционную смесь охлаждали до комнатной температуры, затем распределяли между этилацетатом и водой. Водный слой экстрагировали дополнительным этилацетатом, и комбинированные органические фракции сушили (с использованием MgSO4), фильтровали и фильтрат концентрировали. Остаток очищали посредством хроматографии на силикагеле, элюировали с помощью 50-70% этилацетата-гексанов для получения сырого трет-бутил 4-(4-аминотиено[3,2-с]пиридин-3-ил)фенилкарбамата. Раствор сырого продукта (59,7 ммоль на основании 100% выхода) в N,N-диметилформамиде (80 мл) обрабатывали N-йодсукцинимидом (13,5 г, 59,7 моль - добавленным по частям), и полученный темный раствор перемешивали при комнатной температуре в течение 2 часов, затем распределяли между водой (500 мл) и этилацетатом (100 мл) с добавлением NaCl для облегчения разделения слоев. Водный слой экстрагировали дополнительным этилацетатом (2×75 мл), и комбинированные органические фракции промывали тиосульфатом натрия (3×20 мл) и солевым раствором (50 мл) и затем сушили с использованием MgSO4), фильтровали и концентрировали. Сырой материал обрабатывали TFA (20 мл) и CH2Cl2 (5 мл), перемешивали при комнатной температуре в течение 2 часов, концентрировали под током азота, затем концентрировали в вакууме. Твердые вещества растворяли в воде (100 мл), тщательно обрабатывали твердым Na2CO3 до прекращения выделения газа и фильтровали, промывая дополнительной водой. Собранное твердое вещество сушили для получения указанного в заголовке соединения в виде твердого вещества (загрязненного на прибл. 10% моль PPh3).
Пример 4В
2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол-1-ил)этанол
4-(4,4,5,5,-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол (9,66 г, 49,8 ммоль), 1,3-диоксолан-2-он (21 г, 238 ммоль) и карбонат цезия (16 г, 49,1 ммоль) комбинировали в 100 мл круглодонной колбе. Реакционную смесь нагревали от комнатной температуры до 100°C в масляной бане, в течение указанного времени карбонат таял и служил в качестве растворителя для реакционной смеси, которая оставалась густой суспензией. После нагревания в течение 3,5 часов реакционную смесь охлаждали до комнатной температуры и разводили этилацетатом, затем фильтровали через целит, повторно промывая этилацетатом. Фильтрат концентрировали, затем очищали хроматографией на системе очистки Analogix® Intelliflash™ с использованием колонки SF60-200 g при скорости тока 80 мл/мин, элюируя, как указано далее: 5 минут в 20% этилацетате/гексанах, затем поднимали с 40% до 90% этилацетата/гексанов в течение 35 минут, затем 100% этилацетата в течение еще 20 минут для получения указанного в заголовке соединения.
Пример 4С
2-(4-(4-амино-3-(4-аминофенил)тиено[3,2-c]пиридин-7-ил)-1Н-пиразол-1-ил)этанол
Пример 4А (6 г, 16,34 моль), пример 4В (4,8 г, 20,16 ммоль), PdCl2 (dppf) (1,2 г, 1,640 ммоль) и карбонат натрия (4,6 г, 43,4 ммоль) комбинировали в тетрагидрофуране (400 мл, метаноле (80 мл) и воде (80 мл), и реакционную смесь дегазировали путем барботирования N2 через смесь в течение 1 часа. Затем реакционную смесь нагревали до 80°C в течение 2 часов, затем позволяли остыть и разводили 300 мл этилацетата. Смесь распределяли между H2O (500 мл) и водный слой экстрагировали этилацетатом (2×300 мл). Комбинированные органические экстракты промывали солевым раствором, сушили (Na2SO4), фильтровали через подушечку Целита, концентрировали до общего объема около 200 мл, затем оставляли стоять в течение ночи. Образующееся таким образом твердое вещество собирали фильтрацией, получая указанное в заголовке соединение.
Пример 4D
N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1Н-пиразол-4-ил]тиено[3,2-с]пиридин-3-ил}фенил)-N′-(3-фторфенил)мочевина
Пример 4С (2 г, 5,69 ммоль) растворяли в N,N-диметилформамиде (80 мл) и колбу охлаждали в -20°C бане. 1-фтор-3-изоцианатобензол (0,715 мл, 6,26 ммоль) добавляли по каплям, и реакционной смеси позволяли медленно нагреться до комнатной температуры и перемешивали в течение ночи. Реакционную смесь разводили водой (500 мл) и этилацетатом (75 мл) и перемешивали для переработки в течение 1 часа. Затем смесь помещали в сепараторную воронку. После возможности слоям разделиться, нижний водный слой сливали. Около интерфазы двух слоев находилось существенное количество осадка. Этот материал и органический слой фильтровали, получая твердое вещество. Твердое вещество промывали этилацетатом и сушили для получения указанного в заголовке соединения.
1H ЯМР (ДМСО-d6) δ м.д. 3,80 (кв, J=5,65 Гц, 2Н), 4,23 (т, J=5,59 Гц, 2Н), 4,96 (т, J=5,09 Гц, 1Н), 5,42 (с, 2Н), 6,80 (тд, J=8,31, 2,37 Гц, 1Н), 7,15 (дд, J=8,14, 1,02 Гц, 1Н), 7,32 (тд, J=8,22, 6,95 Гц, 1Н), 7,41 (д, J=8,81 Гц, 2Н), 7,51 (ддд, J=11,70, 2,37, 2,20 Гц, 1Н), 7,50 (с, 1Н), 7,61 (д, J=8,81 Гц, 2Н), 7,91 (д, J=1,02 Гц, 1Н), 8,05 (с, 1Н), 8,15 (д, J=0,68 Гц, 1Н), 8,96 (с, 1Н), 8,99 (с, 1Н); МС (ESI(+)) m/z 489,1 (М+Н)+.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ТИЕНО[3,2-с]ПИРИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ РАКА | 2009 |
|
RU2480472C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ АНАЛОГИ ПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ JAK | 2012 |
|
RU2564419C1 |
| СОЕДИНЕНИЯ ПИРАЗОЛО[3,4-b]ПИРИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ ТАМ И МЕТ | 2019 |
|
RU2773611C1 |
| НОВЫЕ КОНДЕНСИРОВАННЫЕ ПИРИДИНОВЫЕ ПРОИЗВОДНЫЕ, ПРИМЕНИМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ТИРОЗИНКИНАЗЫ с-MET | 2013 |
|
RU2619130C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРИМЕНЕНИЯ В ТЕРАПИИ ЗЛОКАЧЕСТВЕННЫХ НОВООБРАЗОВАНИЙ, ВКЛЮЧАЮЩАЯ КОМБИНАЦИЮ БИФОСФОНАТА, ИНГИБИТОРА СОХ-2 И ТАКСОЛА | 2002 |
|
RU2317819C2 |
| НОВЫЕ СОЕДИНЕНИЯ, КОТОРЫЕ ЯВЛЯЮТСЯ ИНГИБИТОРАМИ ERK | 2013 |
|
RU2660429C2 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ IRAK II ТИПА И ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2810338C2 |
| ИНГИБИТОРЫ СЕРИН/ТРЕОНИНОВЫХ КИНАЗ | 2013 |
|
RU2650501C2 |
| СПОСОБ ПРИМЕНЕНИЯ ИНГИБИТОРОВ ЦИКЛООКСИГЕНАЗЫ-2 ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ НЕОПЛАЗИИ | 1997 |
|
RU2239429C2 |
| ТРИАЗОЛПИРИДИНОВЫЕ СОЕДИНЕНИЯ, ИНГИБИРУЮЩИЕ JAK, И СПОСОБЫ | 2009 |
|
RU2560153C2 |
Изобретение относится к химико-фармацевтической композиции и представляет собой продукт в виде твердой дисперсии, содержащий N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевину или ее фармацевтически приемлемую соль, по меньшей мере, один фармацевтически приемлемый водорастворимый полимерный носитель и, по меньшей мере, одно фармацевтически приемлемое поверхностно-активное вещество. Данная твердая дисперсия пригодна для перорального введения нуждающемуся в этом субъекту для лечения рака. 5 н. и 19 з.п. ф-лы, 4 пр., 2 табл.
1. Продукт в виде твердой дисперсии, содержащий N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевину или ее фармацевтически приемлемую соль, по меньшей мере, один фармацевтически приемлемый водорастворимый полимерный носитель и, по меньшей мере, одно фармацевтически приемлемое поверхностно-активное вещество.
2. Продукт в виде твердой дисперсии по п.1, в котором продукт в виде твердой дисперсии является аморфным.
3. Продукт в виде твердой дисперсии по п.1, содержащий, по меньшей мере, одну кислоту.
4. Продукт в виде твердой дисперсии по п.1, в котором N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевина присутствует в количестве от примерно 1% до примерно 40% по массе эквивалента свободного основания.
5. Продукт в виде твердой дисперсии по п.4, в котором N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевина присутствует в количестве от примерно 5% до примерно 15% по массе эквивалента свободного основания.
6. Продукт в виде твердой дисперсии по п.4, в котором N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевина присутствует в количестве от примерно 8% до примерно 12% по массе эквивалента свободного основания.
7. Продукт в виде твердой дисперсии по п.3, в котором, по меньшей мере, одна кислота составляет от примерно 0,1 до примерно 10 эквивалентов по отношению к N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевине.
8. Продукт в виде твердой дисперсии по п.3, в котором, по меньшей мере, одна кислота выбрана из группы, состоящей из уксусной кислоты, аскорбиновой кислоты, бензолсульфоновой кислоты, лимонной кислоты, этандисульфоновой кислоты, 1-гидрокси-2-нафтойной кислоты, соляной кислоты, бромистоводородной кислоты, молочной кислоты, малеиновой кислоты, яблочной кислоты, малоновой кислоты, метансульфоновой кислоты, фосфорной кислоты, серной кислоты, янтарной кислоты, винной кислоты и толуолсульфоновой кислоты.
9. Продукт в виде твердой дисперсии по п.3, в котором, по меньшей мере, одна кислота выбрана из группы, состоящей из лимонной кислоты, малеиновой кислоты, яблочной кислоты, малоновой кислоты, янтарной кислоты и винной кислоты.
10. Продукт в виде твердой дисперсии по п.3, в котором, по меньшей мере, одна кислота является лимонной кислотой.
11. Продукт в виде твердой дисперсии по п.1, в котором, по меньшей мере, один полимерный носитель присутствует в количестве от примерно 40% до примерно 85% по массе, и, по меньшей мере, одно поверхностно-активное вещество присутствует в количестве от примерно 5% до примерно 30% по массе.
12. Продукт в виде твердой дисперсии по п.1, в котором, по меньшей мере, один полимерный носитель выбран из группы, состоящей из гомополимеров и сополимеров N-виниллактамов, сложных эфиров целлюлозы, простых эфиров целлюлозы, высокомолекулярных полиалкиленоксидов, полиакрилатов, полиметакрилатов, полиакриламидов, полимеров винилацетата, олиго- и полисахаридов, а также их смесей.
13. Продукт в виде твердой дисперсии по п.1, в котором, по меньшей мере, один полимерный носитель выбран из группы, состоящей из поливинилпирролидона, гидроксипропилметилцеллюлозы и их смесей.
14. Продукт в виде твердой дисперсии по п.1, в котором, по меньшей мере, одно поверхностно-активное вещество выбрано из группы, состоящей из производных глицеридов полиэтиленгликоля, полиоксиэтиленовых производных касторового масла, сложных моноэфиров сорбитана и жирных кислот, полисорбатов, полоксамеров, α-токоферил полиэтиленгликоль сукцината и их смесей.
15. Продукт в виде твердой дисперсии по п.1, в котором, по меньшей мере, одно поверхностно-активное вещество выбрано из группы, состоящей из полисорбатов, полиоксиэтиленовых производных касторового масла и их смесей.
16. Способ получения продукта в виде твердой дисперсии, включающий в себя:
(a) получение раствора, содержащего (i) N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевину или ее фармацевтически приемлемую соль, (ii) фармацевтически приемлемую кислоту, (iii), по меньшей мере, один фармацевтически приемлемый водорастворимый полимерный носитель, (iv), по меньшей мере, одно фармацевтически приемлемое поверхностно-активное вещество и (v), по меньшей мере, один подходящий растворитель; и
(b) удаление, по меньшей мере, одного растворителя для получения твердой дисперсии, содержащей, по меньшей мере, один полимерный носитель, по меньшей мере, одно поверхностно-активное вещество, по меньшей мере, одну кислоту, и содержащей N-(4-{4-амино-7-[1-(2-гидроксиэтил)-1H-пиразол-4-ил]тиено[3,2-c]пиридин-3-ил}фенил)-N'-(3-фторфенил)мочевину или ее фармацевтически приемлемую соль, диспергированную в ней в преимущественно некристаллической форме.
17. Способ по п.16, включающий в себя комбинацию водного растворителя и смешиваемого с водой органического растворителя.
18. Способ по п.17, в котором органический растворитель является тетрагидрофураном, метанолом, этанолом или ацетоном.
19. Способ по п.16, в котором растворитель удаляют путем распылительной сушки или в вакууме.
20. Фармацевтическая лекарственная форма для перорального введения, содержащая продукт в виде твердой дисперсии по п.1.
21. Лекарственная форма по п.20, содержащая, по меньшей мере, одну добавку, выбранную из веществ, улучшающих распадаемость, скользящих веществ и объемообразующих агентов.
22. Фармацевтическая лекарственная форма для перорального введения, содержащая продукт в виде твердой дисперсии, полученный способом по п.16.
23. Лекарственная форма по п.22, содержащая, по меньшей мере, одну добавку, выбранную из веществ, улучшающих распадаемость, скользящих веществ и объемообразующих агентов.
24. Способ лечения рака у млекопитающего, включающий в себя пероральное введение имеющему это заболевание субъекту терапевтически эффективного количества вышеописанной твердой дисперсии по п.1.
| WO 2009050291 A2, 23.04.2009 | |||
| WO 2006026500 A1, 09.03.2006 | |||
| WO 2008055966 A1, 15.05.2008 | |||
| СПОСОБ И КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ РАКОВОГО ЗАБОЛЕВАНИЯ, ТОЗИЛАТ И ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ N-(4-ХЛОР-3-(ТРИФТОРМЕТИЛ)ФЕНИЛ)-N'-(4-(2-(N-МЕТИЛКАРБАМОИЛ)-4-ПИРИДИЛОКСИ)ФЕНИЛ)МОЧЕВИНЫ | 2002 |
|
RU2316326C2 |

