Область техники, к которой относится изобретение
Настоящее изобретение относится к оксидному катализатору, используемому в реакции окисления олефина и/или спирта.
Предпосылки создания изобретения
В качестве способа получения метилакрилата или метилметакрилата в соответствии с реакцией окислительной эстерификации, в которой используются по меньше мере один представитель, выбранный из пропилена, изобутилена и трет-бутилового спирта, в качестве исходного материала, и ненасыщенный альдегид в качестве промежуточного соединения, известны два способа: способ, называемый "прямой мета-способ", содержащий две реакционные стадии; и способ, называемый "способ прямого окисления", содержащий три реакционные стадии. Согласно "Sekiyu Kagaku Purosesu (Petrochemical Processes)" (edited by the Japan Petroleum Institute, pages 172-176, Kodansha Scientific, Ltd.) способ прямого окисления представляет собой способ получения метилакрилата или метилметакрилата в три стадии. Первая стадия окисления представляет собой стадию воздействия на по меньше мере один исходный материал, выбранный из пропилена, изобутилена и трет-бутанола, газофазной каталитической реакции окисления молекулярным кислородом в присутствии катализатора с получением акролеина или метакролеина. Вторая стадия окисления представляет собой стадию воздействия на акролеин или метакролеин, полученные на первой стадии окисления, газофазной каталитической реакции окисления молекулярным кислородом в присутствии катализатора с получением акриловой кислоты или метакриловой кислоты. Стадия эстерификации представляет собой стадию дальнейшей обработки акриловой кислоты или метакриловой кислоты, полученных на второй стадии окисления, эстерификацией при использовании метанола в качестве спирта с получением метилакрилата или метилметакрилата. Между тем прямой мета-способ включает две стадии каталитической реакции, где первая реакционная стадия включает обработку пропилена, изобутилена и/или трет-бутанола в качестве исходного материала (материалов) газофазной каталитической реакцией окисления газом, содержащим молекулярный кислород, с получением акролеина или метакролеина, и вторая стадия окисления включает взаимодействие полученных акролеина или метакролеина, например метанола, в качестве спирта и молекулярного кислорода с получением метилакрилата и метилметакрилата в одну стадию.
В качестве катализатора для получения ненасыщенного альдегида как главного компонента существует катализатор, найденный очень давно фирмой Sohio Company. Затем были представлены многие сложнооксидные катализаторы, содержащие Mo и Bi в качестве основных компонентов. Например, катализатор, фокусирующийся на Mo, Si, Ce, K, Fe, Co, Mg, Cs и Rb в качестве металлов, содержащихся в катализаторе, описан в патентном документе 1. Катализаторы для получения ненасыщенного альдегида и ненасыщенной кислоты описаны также в патентном документе 2. Среди них катализатор, представленный следующей формулой Sb0,5Cs0,5S0,25Ni2,5Co4,5Fe4Bi1Mo12Ox, обеспечивает максимальный выход за один прогон.
Перечень прототипов
Патентный документ
Патентный документ 1: Международная публикация № WO 95/35273
Патентный документ 2: Патент США № 4001317
Краткое описание изобретения
Проблемы, решаемые изобретением
На производительность реакции окисления, описанной выше, значительно влияет концентрация исходного материала и температура реакции. Теоретически считается, что производительность улучшается, когда концентрация исходного материала является высокой, и температура реакции является высокой. Однако, в действительности, когда температура реакции и концентрация исходного материала устанавливаются чрезмерно высокими, возникает проблема заметно сниженной производительности.
Например, когда ненасыщенный альдегид получается реакцией газофазного каталитического окисления, если концентрация исходного материала является высокой, парциальное давление кислорода, требуемое при генерации тепла, увеличивается, что увеличивает образование последующего оксида. Поэтому селективность по ненасыщенному альдегиду значительно снижается, что заметно снижает производительность. Между тем, если температура реакции составляет примерно 350-370°C, селективность по ненасыщенному альдегиду увеличивается. Однако, если температура реакции составляет примерно 370°C или более, селективность по ненасыщенному альдегиду значительно снижается, что заметно снижает производительность. Поэтому требуется катализатор, имеющий высокую производительность требуемого продукта даже при условиях реакции высокой концентрации исходного материала и высокой температуры реакции.
В вышеуказанных наблюдениях был исследован оксидный катализатор системы висмут-молибден (Bi-Mo), используемый в технике, и оксидный катализатор системы, в которой железо, церий или т.п. дополнительно вводятся в систему висмут-молибден, как описано в патентном документе 1. Как результат было установлено, что в таком катализаторе все металлы не образуют сложного оксида и металлы существуют также как Bi2Mo3O12, Ce2Mo3O12, Fe2Mo3O12, Bi2O3, Fe2O3 и СеО2 по результатам рентгеноструктурного анализа или т.п. Указанные однокомпонентные оксиды или двухкомпонентные оксиды имеют сравнительно высокую окислительную способность и поэтому образуют последующий оксид, который находится в состоянии, когда требуемый продукт дополнительно окисляется, создавая проблему сниженной производительности по требуемому продукту.
Тогда авторами настоящего изобретения в результате исследований, посвященных оптимизации окислительной способности катализатора, было установлено, что соотношения Mo, Bi, Fe, Co и Се в катализаторе оптимизируются, и образование не образующих сложный оксид компонентов из указанных компонентов подавляется с подавлением образования последующих оксидов, улучшая производительность требуемого продукта. Таким образом, авторами было сформулировано настоящее изобретение.
Т.е., настоящее изобретение заключается в следующем.
[1] Оксидный катализатор для использования в реакции окисления олефина и/или спирта, который содержит:
молибден, висмут, железо, кобальт и церий; где атомное отношение a висмута к 12 атомам молибдена составляет 2≤a≤6, атомное отношение b железа к 12 атомам молибдена составляет 2,5<b≤5, атомное отношение c кобальта к 12 атомам молибдена составляет 2≤с≤8, атомное отношение d церия к 12 атомам молибдена составляет 0,5≤d≤6, и атомное отношение железо/кобальт составляет 0,4≤b/c≤2,5.
в котором, когда параметр d сложного оксида церия и молибдена, показывающего пик при 33,50° на рентгенограмме, принимается за эталон, уровень изменения d составляет 5000-9000 ч/млн.
[2] Оксидный катализатор по п.[1] выше, где оксидный катализатор имеет состав, представленный следующей формулой состава (1):
Mo12BiaFebCocCedAeBfOg (1),
в которой Мо представляет собой молибден, Bi представляет собой висмут, Fe представляет собой железо, Со представляет собой кобальт, Се представляет собой церий, А представляет собой по меньше мере один элемент, выбранный из группы, состоящей из цезия и рубидия, В представляет собой по меньше мере один элемент, выбранный из группы, состоящей из меди, никеля, магния и свинца, a-f представляют собой атомное отношение каждого из элементов к 12 атомам Мо и удовлетворяют значениям 2≤a≤6, 2,5<b≤5, 2≤с≤8, 0,4≤b/c≤2,5, 0,5≤d≤6, 0,01≤е≤2 и 0≤f<2, и g представляет собой число атомов кислорода, определенное по валентности составляющего элемента, иного, чем кислород.
[3] Способ получения оксидного катализатора по п.[1] или [2], который включает следующие стадии:
созревание суспензии исходного материала, содержащей молибден, висмут, железо, кобальт и церий, при температуре выше, чем комнатная температура;
сушка созревшей суспензии исходного материала;
предварительное прокаливание высушенного продукта при 120°C или выше и 350°C или ниже; и
последующее окончательное прокаливание предварительно прокаленного продукта при температуре 400°C или выше и 700°C или ниже.
[4] Способ получения ненасыщенного альдегида, включающий стадию окисления по меньше мере одного олефина, выбранного из группы, состоящей из пропилена и изобутилена и/или трет-бутилового спирта с использованием оксидного катализатора по п.[1] или [2].
Полезные эффекты изобретения
Настоящее изобретение может предоставить катализатор, обеспечивающий высокую производительность требуемого продукта в условиях реакции высокой концентрации исходного материала и высокой температуры реакции в реакции окисления олефина и/или спирта.
Краткое описание чертежей
На фиг.1 представлены рентгенограммы с соответствующими пиками оксидных катализаторов в примере 2 и сравнительном примере 1.
На фиг.2 представлен увеличенный вид интервала 2θ=32,5-35° пиков рентгенограммы с фиг.1.
Подробное описание изобретения
Вариант осуществления изобретения
Далее подробно описывается вариант осуществления настоящего изобретения (далее называется "настоящий вариант"). Однако настоящее изобретение не ограничивается последующими вариантами и может быть различно модифицировано в объеме сущности изобретения.
[1] Оксидный катализатор для реакции окисления
Оксидный катализатор согласно настоящему изобретению представляет собой оксидный катализатор для использования в реакции окисления олефина и/или спирта, причем оксидный катализатор содержит:
молибден, висмут, железо, кобальт и церий; атомное отношение a висмута к 12 атомам молибдена составляет 2≤a≤6, атомное отношение b железа к 12 атомам молибдена составляет 2,5<b≤5, атомное отношение c кобальта к 12 атомам молибдена составляет 2≤с≤8, атомное отношение d церия к 12 атомам молибдена составляет 0,5≤d≤6, и атомное отношение железо/кобальт составляет 0,4≤b/c≤2,5.
в котором, когда параметр d сложного оксида церия и молибдена, показывающего пик при 33,50° на рентгенограмме, принимается за эталон, уровень изменения d составляет 5000-9000 ч/млн.
Селективность требуемого продукта и подавление последующего окисления вносят вклад в улучшение производительности требуемого продукта. Авторы настоящего изобретения считают, что сложный оксид, имеющий структуру, в которой уровень изменения параметра d составляет 5000-9000 ч/млн, вносит вклад в стабильность промежуточного соединения. Исключение окисляемого водорода образует π-аллильное промежуточное соединение из олефина и/или спирта. Кроме того, реакция присоединения или реакция дегидрирования изменяет π-аллильное промежуточное соединение с получением требуемого продукта. π-Аллильное промежуточное соединение имеет высокую реакционную способность и является нестабильным, и поэтому π-аллильное промежуточное соединение сразу же изменяется или разлагается в другое соединение в обычных обстоятельствах. Однако считается, что оксидный катализатор согласно настоящему изобретению регулирует стабильность π-аллильного промежуточного соединения, и реакция дает предпочтительно получаемый требуемый продукт. Когда кислород вводится и образует π-аллильное промежуточное соединение, последнее изменяется до альдегида или карбоновой кислоты. Когда в π-аллильное промежуточное соединение вводится аммиак, π-аллильное промежуточное соединение изменяется до нитрила. Когда водород исключается из π-аллильного промежуточного соединения, образуется двойная связь, и из спирта получается олефин, и из олефина получается диолефин или т.п. Поэтому различные требуемые продукты могут быть получены с использованием оксидного катализатора согласно настоящему изобретению при соответствующем выборе исходного материала, подаваемого в реактор.
Хотя причина, по которой оксидный катализатор согласно настоящему изобретению подавляет образование последующего оксида, не ясна, авторы настоящего изобретения провели исследование и подтвердили, что последующий оксид снижается, даже если требуемым продуктом является ненасыщенный альдегид. Ненасыщенный альдегид представляет собой соединение, которое является склонным к чрезвычайно легкому окислению. В качестве неожиданного результата последующий оксид, получаемый из ненасыщенного альдегида, снижается даже в условиях высокой концентрации исходного материала и высокой температуры реакции. Если даже ненасыщенный альдегид, который является склонным к чрезвычайно легкому окислению, может быть подавлен, считается, что образование последующего оксида также подавляется для других требуемых продуктов. Последующий оксид является побочным продуктом, который является непредпочтительным во всех реакциях окисления. Если образование последующего оксида может быть подавлено, может ожидаться значительное улучшение производительности.
В настоящем описании последующий оксид означает соединение в состоянии, когда требуемый продукт дополнительно окисляется. Поэтому ненасыщенная карбоновая кислота считается видом последующего оксида, когда требуемым продуктом является ненасыщенный альдегид, так, как на первой стадии реакции прямого мета-способа. Другие примеры последующего оксида могут включать диоксид углерода, пероксиды, дикетоны и эпоксидные соединения.
(1) Состав
В оксидном катализаторе настоящего варианта с точки зрения оксидобразующих элементов металла в Mo-Bi системе оксида металла обязательно существуют Mo, Bi, Ce и Fe. Атомное отношение a висмута к 12 атомам молибдена составляет 2≤a≤6. С точки зрения дополнительного улучшения селективности требуемого продукта атомное отношение a составляет 2≤a≤5 и более предпочтительно 2≤a≤4. С такой же точки зрения атомное отношение d цезия составляет 0,5≤d≤6, предпочтительно 1≤d≤5 и более предпочтительно 1≤d≤4. Bi и Мо аналогично образуют сложные оксиды, такие как Bi2Mo3O12 и Bi2Mo3O6, которые являются активными частицами в реакциях газофазного каталитического окисления и аммоксидирования или т.п. и имеют высокую каталитическую активность. Однако Bi и Мо имеют низкую температуру плавления и низкую теплостойкость. Тем не менее, для Се и Мо трудно образовать сложный оксид, такой как Се2Мо3О12. Однако Се и Мо имеют высокую температуру плавления и высокую теплостойкость. Если оба, соответственно, являются оксидобразующими, образуется система сложного оксида Co-Di-Mo-O, которая имеет сложную структуру, где Bi является твердорастворенным в Се2Мо3О12, имеющем высокую теплостойкость, и имеет высокую активность и теплостойкость.
С точки зрения улучшения каталитической активности без снижения селективности требуемого продукта, Fe является обязательным элементом для того, чтобы промышленно синтезировать как в Мо, так и в Bi. Однако, когда содержание Fe увеличивается, образуется Fe2O3 и последующие оксиды, такие как СО и СО2, имеют тенденцию к увеличению. В результате селективность требуемого продукта снижается. В некоторых случаях, даже если содержание Fe увеличивается, Fe2O3 не образуется. Однако в это время образуется сложный оксид двухкомпонентной системы Fe-Mo-O. Он является неактивным компонентом, не показывающим каталитическую активность. Поэтому обычно атомное отношение железа к 12 атомам молибдена устанавливается обычно при 0<Fe≤2,5, для того чтобы показывать высокий выход. Отсутствует намерение увеличения атомного отношения Fe до 2,5 или более для образования эффективной кристаллической фазы. Между тем, авторами настоящего изобретения установлено, что кристаллическая структура с высокими характеристиками четырехкомпонентной системы Ce-Bi-Fe-Mo-O образуется при увеличении не только содержания Fe, но также содержания Bi и Се в области состава, выходящей за верхний предел атомного отношения Fe, что является традиционными общими знаниями. Авторы настоящего изобретения предполагают, что оксидный катализатор, имеющий кристалл системы Ce-Bi-Fe-Mo-O, дает высокий выход требуемого продукта, потому что энергия связи Мо-О связи Bi-О-Мо сложного оксида, такого как Bi2Mo3O12 и Bi2Mo3O6, в качестве активных частиц, является подходящей. Атомное отношение b железа к 12 атомам молибдена оксидного катализатора согласно настоящему варианту составляет 2,5<b≤5, предпочтительно 2,5<b≤4,5 и более предпочтительно 2,5<b≤4.
В оксидном катализаторе согласно настоящему варианту Со является обязательным элементом для промышленного синтеза требуемого продукта, как в Mo, Bi и Fe. Со образует сложный оксид СоМоО4. Со служит в качестве носителя для высокодиспергированных активных частиц, таких как Bi-Мо-О, и служит для введения кислорода из газовой фазы и подачи его в Bi-Мо-О или т.п. Для того чтобы получить ненасыщенный альдегид с высоким выходом, необходимо объединить Со с Мо с образованием сложного оксида СоМоО4. С точки зрения снижения образования однокомпонентного оксида, такого как Со3О4 и СоО, атомное отношение c кобальта к 12 атомам молибдена составляет 2≤с≤8, предпочтительно 2,5≤с≤6, более предпочтительно 2,5≤с≤4. С точки зрения улучшения активности катализатора, атомное отношение b/c железо/кобальт составляет 0,4≤b/c≤2,5, предпочтительно 0,7≤b/c≤2,0 и более предпочтительно 1≤b/c≤1,5.
Оксидный катализатор согласно настоящему варианту предпочтительно имеет состав, представленный следующей формулой состава (1):
Mo12BiaFebCocCedAeBfOg (1),
в которой Мо представляет собой молибден, Bi представляет собой висмут, Fe представляет собой железо, Со представляет собой кобальт, Се представляет собой церий, А представляет собой по меньше мере один элемент, выбранный из группы, состоящей из цезия и рубидия, В представляет собой по меньше мере один элемент, выбранный из группы, состоящей из меди, никеля, магния и свинца, a-f представляют собой атомное отношение каждого из элементов к 12 атомам Мо; удовлетворяют значениям 2≤a≤6, 2,5<b≤5, 2≤с≤8, 0,4≤b/c≤2,5, 0,5≤d≤6, 0,01≤е≤2 и 0≤f<2, и g представляет собой число атомов кислорода, определенное по валентности составляющего элемента, иного, чем кислород.
В вышеуказанной формуле состава (1) А представляет цезий и/или рубидий, и считается, что А играет роль нейтрализации кислотного участка МоО3 или т.п., который не образует комплекс, катализатором для получения ненасыщенного альдегида. Когда цезий и/или рубидий содержатся/содержится, это не влияет на кристаллическую структуру Ce-Bi-Fe-Mo-O, описанную далее. Атомные отношения указанных элементов к 12 атомам Мо составляют 0,01≤е≤2 с точки зрения каталитической активности. Атомное отношение е элемента А регулируется в указанном числовом интервале, поскольку катализатор является основным, когда количество щелочного элемента увеличивается до интервала или более, и олефин или спирт в качестве исходного материала вряд ли адсорбируется на катализаторе, что имеет тенденцию к блокированию достаточного проявления каталитической активности.
В представляет собой по меньше мере один элемент, выбранный из группы, состоящей из меди, никеля, магния и свинца. Считается, что элемент частично замещает кобальт в оксиде. Медь играет роль улучшения активности катализатора. Однако, с точки зрения поддержания равновесия с образованием кристалла Ce-Bi-Fe-Mo-O, показывающего характеристики катализатора, верхний предел атомного отношения f элемента В составляет предпочтительно f<2. Когда атомное отношение составляет f<2, никель, магний и свинец играют роль стабилизации кристаллической структуры CoMoO4 с подавлением фазового перехода или т.п. в соответствии с давлением и температурой. Поскольку элемент, представленный В, улучшает активность катализатора или стабилизирует кристаллическую структуру CoMoO4 в катализаторе, элемент не влияет на кристаллическую структуру Ce-Bi-Fe-Mo-O и позиционируется как необязательный элемент, чье содержание может быть нулевым (f = 0).
Даже если элементы, представленные А и В, содержатся или не содержатся в катализаторе, образуется кристаллическая структура, кроме кристаллической структуры Ce-Bi-Fe-Mo-O, и, таким образом, элементы не влияют на кристаллическую структуру Ce-Bi-Fe-Mo-O.
Оксидный катализатор, имеющий состав, представленный вышеуказанной формулой состава (1), характеризуется тем, что селективность ненасыщенного альдегида является высокой. Оксидный катализатор подходяще используется на первой стадии реакции прямого мета-способа. Поскольку продуктом поледующего окисления является ненасыщенная карбоновая кислота в способе прямого окисления, способ прямого окисления имеет небольшую характеристику снижения метакриловой кислоты на стадии получения ненасыщенного альдегида в качестве промежуточного соединения. Таким образом, поскольку общий выход метакролеина и метакриловой кислоты является выше, можно сказать, что катализатор является желательным. Тем не менее, поскольку прямой мета-способ образует ненасыщенный альдегид на первой стадии реакции и поэтому образует эфир ненасыщенной карбоновой кислоты из ненасыщенного альдегида на второй стадии реакции, стадия образования ненасыщенной карбоновой кислоты в качестве требуемого продукта не существует. Поэтому в способе окисления со сложнооксидным катализатором желательно образуется только ненасыщенный альдегид, а образование ненасыщенной карбоновой кислоты желательно подавляется насколько возможно. Т.е., когда целью является оптимизация катализатора для первой стадии реакции прямого мета-способа, требуется катализатор, который имеет направленное определенное отличие от катализатора для способа прямого окисления, который обеспечивает высокий выход ненасыщенного альдегида в качестве требуемого продукта и низкий выход ненасыщенной карбоновой кислоты в качестве последующего оксида.
(2) Кристаллическая структура
Когда интервал угла рентгеновской дифракции 2θ=5-60° определяется дифракцией рентгеновских лучей (XRD), оксид, содержащий только церий и молибден, показывает пик при 33,50°. Если железо и висмут дополнительно образуют сложный оксид с оксидом, содержащим только церий и молибден, имеет место сдвиг пика. Поскольку оксидный катализатор согласно настоящему варианту содержит оксид, содержащий церий и молибден, и металл, полученный при образовании комплекса железа с висмутом, оксидный катализатор показывает пик не при 33,50°, а при 33,50°+α° (0<α).
Когда в соответствии с формулой закона Брэгга ("Kotai Hyoumen Kyarakutarizeshon no Jissai (Actual Condinions of Solid Surface Characterization)", edited by Yasuhiro Tanaka and Hiromi Yamashita, pages 13-25, Kodansha Scientific, Ltd.) параметр d между поверхностями кристалла, угол падения и угол отражения θ рентгеновских лучей на поверхность кристалла и длина волны λ имеют зависимость формулы (II):
2dsinθ = nλ (n: целое число) (II),
т.е. когда разность пути между падающими и рассеянными рентгеновскими лучами равна кратному интегралу длины волны падающих рентгеновских лучей, наблюдается явление дифракции. В настоящем варианте устанавливается первичное отражение и устанавливается дифракция, удовлетворяющая условию отражения n=1. Например, когда элемент твердо растворяется при замещении в многокомпонентной системе сложного оксида и угол (2θ) дифракции рентгеновских лучей сдвигается в сторону более низких углов, параметр d находится в состоянии расхождения. Когда угол (2θ) дифракции рентгеновских лучей сдвигается в сторону более высоких углов, параметр d находится в состоянии укорачивания. По указанной причине сдвиг пика, появляющегося при 33,50° в XDR, в сторону более высоких углов (0<α) означает изменение параметра d в состояние параметра d оксида, укорачивающегося при комплексобразовании металлов.
Степень изменения параметра d может быть представлена следующей формулой (III):
Степень изменения d (ч/млн) = (d0-d1)/d0×1000000 (III),
в которой d0 представляет собой параметр сложного оксида церия и молибдена, показывающего пик при 33,50°, и d1 представляет собой параметр оксидного катализатора согласно настоящему варианту. В оксидном катализаторе согласно настоящему варианту степень изменения параметра d составляет 5000-9000 ч/млн. Поскольку оксидный катализатор, близкий к двухкомпонентной системе церия и молибдена, содержится, когда степень изменения параметра d составляет менее 5000, оксидный катализатор имеет высокую окислительную способность, и когда оксидный катализатор используется в качестве катализатора, выход последующего оксида увеличивается. Между тем, когда степень изменения параметра d составляет более 9000, активность снижается. С точки зрения получения требуемого продукта с высокой активностью и высоким выходом, степень изменения параметра d составляет более предпочтительно 5500-8500 ч/млн и еще более предпочтительно 6000-8000 ч/млн.
Механизм, по которому изменяется параметр d, не ясен. Однако считается, что это обусловлено дополнительным твердым растворением Fe в сложном оксиде Ce, Bi и Мо с образованием вновь кристаллической структуры с высокими характеристиками сложнооксидной четырехкомпонентной системы Ce-Bi-Fe-Mo-O. Способ получения катализатора для диспергирования и комплексообразования Bi или т.п. будет описан подробно далее. Однако избыточное соотношение металла является также важным для того, чтобы образовать такую кристаллическую структуру. Когда атомное отношение b железа к 12 атомам Мо удовлетворяет интервалу 2,5<b≤5, образуется кристаллическая структура. Однако, когда атомное отношение b является <2,5, кристаллическая структура не образуется, или даже когда кристаллическая структура образуется, количество кристаллической структуры является чрезвычайно малым, и полученному оксидному катализатору трудно подавлять образование последующего оксида. Т.е., когда атомное отношение b составляет b≤2,5, выход последующего оксида увеличивается. Когда атомное отношение b составляет b>5, выход СОх (СО2 и СО или т.п.) увеличивается. В результате производительность требуемого продукта снижается.
В качестве показателя комплексообразования Ce-Bi-Fe-Mo-O сдвиг 33,50° желательно принимается за эталон. Однако влияние, вызванное комплексообразованием, имеет место также для других пиков. Оксидный катализатор согласно настоящему варианту имеет пики при углах дифракции рентгеновских лучей (2θ) 28,17°±0,05°, 33,50°±α° и 25,44°±0,05° в убывающем порядке интенсивности. Среди них пики при 28,17°±0,05° и 33,50°±α° происходят главным образом от Се-Мо-О, а пик при 25,44°±0,05° происходит главным образом от Со-Мо-О. С точки зрения подавления образования последующего оксида, интенсивность каждого из пиков предпочтительно снижается согласно вышеуказанному порядку.
Так для пика угла дифракции рентгеновских лучей (2θ)=33,50°+α° α представляет собой сдвиг пика от 33,50°. Когда степень сдвига d составляет 5000-9000 ч/млн, устанавливается 0,10°≤α≤0,25°.
(3) Компоненты, иные, чем оксид металла
Оксидный катализатор для реакции окисления согласно настоящему изобретению может содержать носитель для несения оксида металла. Катализатор, содержащий носитель, является предпочтительным тем, что катализатор обеспечивает высокое диспергирование оксида металла и обеспечивает высокую износостойкость оксида металла на носителе. Здесь, когда катализатор формуется способом экструзионного формования, катализатор предпочтительно содержит носитель. Однако, когда метакролеин получается в реакторе с неподвижным слоем и катализатор формуется в таблетки, катализатор может не содержать носитель. Примеры носителя могут включать, но не ограничиваясь этим, диоксид кремния, оксид алюминия, диоксид титана и диоксид циркония. Обычно сам диоксид кремния является более инертным, чем другие носители, и имеет хорошее связующее действие с оксидом металла без снижения селективности к требуемому продукту, который поэтому является предпочтительным носителем. Кроме того, носитель диоксид кремния является предпочтительным, потому что он аналогично обеспечивает высокую износостойкость оксида металла на носителе. Когда катализатор формуется способом экструзионного формования, содержание носителя по отношению ко всему катализатору составляет предпочтительно 5-10% масс.
Также в случае катализатора, используемого в реакторе с псевдоожиженным слоем, с той же точки зрения, как указано выше, в качестве носителя используется диоксид кремния. С точки зрения оптимизации влияния Ce-Bi-Fe-Mo-O на кристаллическую структуру и кажущийся удельный вес с улучшением текучести, содержание носителя в катализаторе составляет предпочтительно 80% масс. или менее по отношению к общей массе катализатора, более предпочтительно 70% масс. или менее и еще более предпочтительно 60% масс. или менее. В случае катализатора, требующего прочность для реактора с псевдоожиженным слоем или т.п., с точки зрения показа практически достаточного сопротивления раздавливанию и износостойкости или т.п., содержание носителя в катализаторе составляет предпочтительно 20% масс. или более по отношению к общей массе катализатора, более предпочтительно 30% масс. или более и еще более предпочтительно 40% масс. или более.
[2] Способ получения катализатора для реакции окисления
Как описано выше, авторы настоящего изобретения сконцентрировались на том, что получается не одно- и/или двухкомпонентная система оксида Ct, Bi, Fe и Мо, но сложный оксид системы Ce-Bi-Fe-Mo-O, получаемый комплексообразованием четырех компонентов, и всесторонне исследовали соотношение состава и способ его получения.
Поскольку Bi является обязательным элементом для образования активных частиц с Мо, также называемый как катализатор системы висмут-молибден (Bi-Mo), с точки зрения активности предпочтительно содержание большого количества Bi. Однако, когда содержание Bi увеличивается, известно, что катализатор является гетерогенным. Например, нитрат висмута в качестве исходного материала Bi, который традиционно используется в промышленности, является плохо водорастворимым веществом. Для растворения нитрата висмута требуется большое количество азотной кислоты. В результате композиция катализатора после прокаливания является гетерогенной, и поэтому традиционная технология получения катализатора имеет ограничение в увеличении содержания Bi. Т.е., образуется однокомпонентный оксид, такой как Bi2O3, и не получается гомогенный катализатор, что неблагоприятно снижает производительность требуемого продукта. С точки зрения улучшения каталитической активности без снижения селективности требуемого продукта, было зафиксировано в течение многих лет, что Fe является обязательным элементом для того, чтобы промышленно синтезировать требуемый продукт, как в случае Mo и Bi. Однако, как указано в Международной публикации № WO 95/35273 (Pamphlet), оптимально вводится небольшое количество Fe. Когда содержание Fe увеличивается, образование последующего оксида, такого как СО и СО2, имеет тенденцию к увеличению, что снижает селективность требуемого продукта.
В результате многих проб и ошибок для того, чтобы решить проблему, авторами настоящего изобретения было установлено, что Се дополнительно вводится в каталитический компонент, содержащий Bi и Fe, более, чем обычно в катализатор, содержащий большое количество азотной кислоты, с подавлением образования единичного оксида в течение первого времени согласно новой технологии получения катализатора, удовлетворяющей трем требованиям (а) специального соотношения компонентов, (b) способа созревания специальной суспензии соли металла и (с) специальному способу прокаливании, и с образованием заново кристалла Ce-Bi-Fe-Mo-O, полученного комплексообразованием четырех компонентов. Требуемое комплексообразование не происходит только при увеличении нитрата висмута и увеличении азотной кислоты. Т.е., авторами настоящего изобретения установлено, что содержание Bi и Fe увеличивается в катализаторе, содержащем большое количество азотной кислоты, и Се дополнительно вводится в катализатор с получением кристаллической структуры, в которой совмещаются четыре компонента, Ce-Bi-Fe-Mo-O в первое время.
Т.е., только когда удовлетворяются все три условия (а) специального соотношения компонентов, (b) способа созревания специальной суспензии соли металла и (с) специального способа прокаливании, образуется кристаллическая структура системы, совмещающей четыре компонента, Ce-Bi-Fe-Mo-O, что дает катализатор, обеспечивающий высокий выход получаемого ненасыщенного альдегида. Когда по меньше мере одно из трех условий не удовлетворяется, образуется сложный оксид двухкомпонентной системы, такой как Ce-Mo-O, Bi-Mo-O и Fe-Mo-O, или одиночный оксид, такой как Fe2O3, Bi2O3, MoO3 и CeO2, и интервал степени изменения параметра d также происходит от интервала 5000-9000 ч/млн, что дает в результате снижение выхода ненасыщенного альдегида.
Оксидный катализатор согласно настоящему варианту может быть получен способом, который содержит первую стадию получения суспензии исходного материала, вторую стадию сушки распылением суспензии исходного материала и третью стадию прокаливания высушенных частиц, полученных на второй стадии. Предпочтительный аспект способа получения оксидного катализатора, имеющего первую-третью стадии, описывается далее.
(1) Получение суспензии исходного материала
На первой стадии исходные материалы элементов металлов, содержащихся в катализаторе, смешиваются с получением суспензии исходного материала. Примеры источников элемента для молибдена, висмута, серия, железа, кобальта, рубидия, цезия, меди, никеля, магния и свинца могут включать соль аммония, соль нитрата, соль гидрохлорида, соль органической кислоты, которые являются растворимыми в воде или азотной кислоте. Источником элемента может быть оксид, гидроксид, карбонатная соль или т.п. В случае оксида предпочтительной является жидкая дисперсия, в которой оксид диспергирован в воде или в органическом растворителе. Оксид, диспергированный в воде, является более предпочтительным. Когда оксид диспергируется в воде, для того чтобы диспергировать оксид, может содержаться стабилизатор дисперсии, такой как полимер. Диаметр частиц оксида составляет предпочтительно 1-500 нм и более предпочтительно 10-80 нм. Когда получают катализатор, содержащий носитель, в суспензию исходного материала предпочтительно вводится силиказоль в качестве исходного материала диоксида кремния.
С точки зрения гомогенного диспергирования суспензии, в суспензию исходного материала могут также, соответственно, вводиться водорастворимые полимеры, такие как полиэтиленгликоль, метилцеллюлоза, поливиниловый спирт, полиакриловая кислота и полиакриламид, многовалентные карбоновые кислоты, такие как амины, аминокарбоновые кислоты, щавелевая кислота, малоновая кислота и янтарная кислота, и органические кислоты, такие как гликолевая кислота, яблочная кислота, винная кислота и лимонная кислота. Количество вводимой органической кислоты специально не ограничивается. Однако, с точки зрения равновесия между гомогенностью и получаемым количеством, органическая кислота предпочтительно вводится в интервале 0-30% масс. по отношению к массе оксида металла.
Способ получения суспензии исходного материала специально не ограничивается, если способ представляет собой способ, который обычно используется. Например, суспензия исходного материала может быть получена смешением раствора, в котором аммониевая соль молибдена растворяется в горячей воде, с раствором, полученным при использовании висмута, церия, железа, кобальта или щелочного металла в качестве нитратной соли, и растворением нитратной соли в воде или в водном растворе азотной кислоты. С точки зрения равновесия между гомогенностью и получаемым количеством, концентрация элемента металла в суспензии после смешения составляет обычно 1-50% масс., предпочтительно 10-40% масс. и более предпочтительно 20-40% масс.
Когда аммониевая соль и нитратная соль смешиваются, получается осадок и, вероятно, с образованием суспензии. Однако суспензия исходного материала предпочтительно созревает в состоянии, когда суспензия исходного материала суспендируется. В настоящем описании "созревание" означает, что суспензия выдерживается в состоянии, когда суспензия исходного материала суспендируется. Предпочтительно, суспензия непрерывно и/или периодически перемешивается, для того чтобы суспендировать суспензию. На данной стадии перемешивания содержащееся твердое вещество измельчается, что способствует образованию предшественника катализатора, который может образовать суспензию, которая является более мелкой и более гомогенной. Когда содержание Bi увеличивается, образуется склонная к образованию суспензия, содержащая большое количество азотной кислоты и имеющая низкую диспергируемость, и поэтому особенно предпочтительно выполняется созревание.
Когда суспензия созреет, суспензия желательно нагревается до температуры, которая выше комнатной температуры и при которой суспензионная среда остается жидкостью, для того чтобы получить желательный комплексный кристалл и/или его предшественник. В частности, температура составляет предпочтительно 20-90°C, более предпочтительно 30-80°C и еще более предпочтительно 50-70°C. Для перемешивания суспензии может использоваться обычное перемешивающее устройство, такое как перемешивающая лопасть и перемешивающий стержень. Скорость перемешивания является различной в зависимости от вязкости суспензии или т.п. и составляет предпочтительно 50-3000 об/мин. Время созревания, требуемое для образования предшественника катализатора, является различным в зависимости от температуры и энергии перемешивания суспензии. Когда температура является высокой или энергия перемешивания является большой, подходящее время созревания имеет тенденцию быть укороченным. Например, когда температура суспензии составляет 20-90°C и операция перемешивания выполняется с использованием мешалки, время созревания составляет предпочтительно 1-24 ч, с точки зрения производительности, более предпочтительно 1-20 ч и еще более предпочтительно 1-10 ч.
В случае суспензии, содержащей большое количество сухого вещества, сухое вещество в суспензии предпочтительно измельчается с использованием гомогенизатора или т.п. перед созреванием. Когда суспензия имеет состав, имеющий увеличенное содержание Bi, как описано выше, содержание азотной кислоты в суспензии имеет тенденцию быть также увеличенным и диспергируемость является склонной к снижению. Поэтому обработка гомогенизатором является особенно эффективной. С точки зрения дальнейшего тонкого измельчения сухого вещества, число оборотов гомогенизатора составляет предпочтительно 5000-30000 об/мин, более предпочтительно 10000-20000 об/мин и еще более предпочтительно 15000-20000 об/мин. Время обработка гомогенизатором является различным в зависимости от числа оборотов или количества сухого вещества. Предпочтительно, время составляет обычно от 5 мин до 2 ч.
Когда суспензия исходного материала является негомогенной, композиция катализатора после прокаливания является гетерогенной и трудно образоваться гомогенной комплекснообразованной кристаллической структуре. Поэтому когда полученный оксид является недостаточно комплексообразованным, стадия получения суспензии предпочтительно оптимизируется. Вышеуказанная стадия получения суспензии исходного материала является примером и не ограничивается. Методика введения источников элементов может быть изменена; концентрация азотной кислоты может быть откорректирована; или аммиачная вода может быть введена в суспензию для изменения рН и вязкости суспензии. Важно образовать гомогенную суспензию для того, чтобы образовать более кристаллическую структуру Ce-Bi-Fe-Mo-O. C этой точки зрения, рН суспензии исходного материала составляет предпочтительно 2,0 или менее. Значение рН суспензии исходного материала составляет более предпочтительно 1,5 или менее и еще более предпочтительно 1,0 или менее. Когда рН суспензии исходного материала является больше 2,0, может образоваться осадок соединения висмута, препятствуя образованию кристаллической структуры Ce-Bi-Fe-Mo-O.
(2) Сушка
На второй стадии суспензия исходного материала, полученная на первой стадии, сушится с получением высушенных частиц. Способ сушки специально не ограничивается, и сушка может выполняться способом, который обычно используется. Способ сушки может осуществляться любым способом, таким как способ сушки выпариванием, способ сушки распылением и способ сушки при пониженном давлении. Способ сушки распылением может осуществляться способом центробежного типа, типа с соплом с двумя жидкостями и типа с соплом высокого давления, который обычно осуществляется в промышленности. Воздух, нагретый водяным паром и электрическим нагревателем или т.п., предпочтительно используется в качестве источника тепла сушки. В данном случае температура на входе сушилки распылением составляет обычно 150-400°C, предпочтительно 180-400°C и более предпочтительно 200-350°C.
(3) Прокаливание
На третьей стадии высушенные частицы, полученные на второй стадии, прокаливаются. Прокаливание может выполняться с использованием такой печи прокаливания, как вращающаяся печь, туннельная печь и муфельная печь. Высушенные частицы предпочтительно прокаливаются в две стадии предварительного прокаливания и окончательного прокаливания. На первой стадии предварительное прокаливание обычно проводится в температурном интервале 120-350°C, предпочтительно 150-350°C и более предпочтительно 200-350°C. Целью предварительного прокаливания является удаление азотной кислоты, оставшейся в высушенных частицах, и постепенное выгорание нитрата аммония и содержащихся органических веществ, происходящих от исходного материала, которым является аммониевая соль, и исходного материала, которым является нитратная соль. Поэтому высушенные частицы могут быть нагреты в такой степени, когда цель может быть достигнута на первой стадии прокаливания. Время предварительного прокаливания составляет обычно 0,1-72 ч, предпочтительно 1-48 ч и более предпочтительно 3-24 ч. В случае низкой температуры 150°C или ниже предварительное прокаливание проводится в течение длительного периода времени. В случае высокой температуры 330°C или выше предварительное прокаливание предпочтительно проводится в течение короткого периода времени 2 ч или менее. Когда температура предварительного прокаливания является слишком высокой или время является слишком длительным, оксид, вероятно, вырастает только в двухкомпонентную систему церия и молибдена на стадии предварительного прокаливания, и как результат затруднено образование кристаллической структуры Ce-Bi-Fe-Mo-O, образуемой на стадии окончательного прокаливания, описываемой далее. Поэтому верхние пределы температуры и времени предварительного прокаливания устанавливаются в такой степени, когда оксид двухкомпонентной системы церия и молибдена не образуется.
В случае предварительного прокаливания скорость повышения температуры является желательно медленной также с точки зрения подавления быстрой реакции горения. Оксидный катализатор в настоящем варианте является многокомпонентной системой. Поэтому, например, когда нитраты металлов используются в качестве исходных материалов, температуры разложения нитратов металлов отличаются друг от друга, и азотная кислота удаляется в процессе прокаливания, и поэтому композиция катализатора после прокаливания является склонной быть гетерогенной. В частности, когда содержание Bi увеличивается, количество нитрата висмута, трудно растворимого в воде, увеличивается, что увеличивает количество азотной кислоты, используемой для растворения. По данной причине, чтобы образовать структуру, которая является более гомогенной, предпочтительно температура повышается медленно для удаления компонентов горения и разложения, таких как азотная кислота и органическое вещество. Скорость повышения температуры обычно составляет 0,1-100°C/мин, более предпочтительно 0,1-75°C/мин и еще более предпочтительно 0,1-50°C/мин.
После предварительного прокаливания предпочтительно осуществляется окончательное прокаливание в виде второй стадии. Однако целью является облегчение образования требуемой кристаллической структуры. Поскольку на кристаллическую структуру влияет произведение температуры прокаливания и времени прокаливания, как установлено авторами настоящего изобретения, предпочтительно температура прокаливания и время прокаливания устанавливаются соответствующим образом. Температура окончательного прокаливания предпочтительно устанавливается выше температуры предварительного прокаливания при 700°C или ниже с точки зрения образования кристалла Ce-Bi-Fe-Mo-O. С точки зрения облегчения образования кристаллической структуры Ce-Bi-Fe-Mo-O температура прокаливания окончательного прокаливания составляет предпочтительно 400-700°C, более предпочтительно 400-650°C и еще более предпочтительно 450-600°C. С точки зрения оптимизации произведения температуры прокаливания и времени прокаливания, чтобы способствовать образованию кристалла, когда прокаливание выполняется при такой температуре, время окончательного прокаливания составляет обычно 0,1-72 ч, предпочтительно 2-48 ч и более предпочтительно 3-24 ч. С точки зрения оптимизации (температура прокаливания)×(время прокаливания) для образования кристаллической структуры в случае низкой температуры 400°C или ниже, например, окончательное прокаливание предпочтительно проводится в течение длительного периода времени примерно 24-72 ч. В случае высокой температуры 600°C или выше, с точки зрения предотвращения снижения активности катализатора, обусловленного чрезмерным снижением площади поверхности, окончательное прокаливание предпочтительно проводится в течение короткого периода времени 1 ч или менее.
Кристаллическая структура комплексной четырехкомпонентной системы Ce-Bi-Fe-Mo-O, вероятно, образуется при выполнении всех вышеуказанных стадий.
Образование кристаллической структуры четырехкомпонентной системы Ce-Bi-Fe-Mo-O на стадии окончательного прокаливания может быть подтверждено осуществлением рентгеноструктурного анализа после окончательного прокаливания. Если образуется кристаллическая структура четырехкомпонентной системы Ce-Bi-Fe-Mo-O, то, когда рентгеноструктурный анализ проводится после окончательного прокаливания, наблюдается пик при 33,50°+α°. Когда образуется кристалл оксида, содержащего только церий и молибден, пик появляется при 33,50°. Однако в случае четырехкомпонентной системы Ce-Bi-Fe-Mo-O пик сдвигается и поэтому образование кристалла четырехкомпонентной системы может быть подтверждено использованием сдвига в качестве указателя.
Была исследована величина сдвига (α°), и параметр d сложного оксида церия и молибдена, показывающего пик при 33,50°, принимается в качестве эталона. Уровень изменения d исследуется с использованием следующих формул:
Если уровень изменения d составляет 5000-9000 ч/млн в настоящем варианте, кристаллическая структура четырехкомпонентной системы Ce-Bi-Fe-Mo-O считается образованной.
[3] Способ получения ненасыщенного альдегида
Ненасыщенный альдегид может быть получен при использовании оксидного катализатора согласно настоящему варианту и при окислении по меньше мере одного олефина, выбранного из группы, состоящей их пропилена и изобутилена и/или трет-бутилового спирта. Далее будет описан его отдельный пример. Однако способ получения согласно настоящему варианту не ограничивается последующим отдельным примером.
(1) Способ получения метакролеина
Метакролеин может быть получен, например, воздействием на изобутилен или трет-бутиловый спирт газофазной каталитической реакции окисления с использованием оксидного катализатора согласно настоящему варианту. В газофазной каталитической реакции окисления газообразный исходный материал, содержащий смешанный газ, к которому добавляются газ, содержащий молекулярный кислород, и газ-разбавитель, вводится в каталитический слой в реактор с неподвижным слоем так, что концентрация молекулярного кислорода в изобутилене, трет-бутиловом спирте или смешанном газе их 1-10% об. составляет 1-20% об. Концентрация изобутилена или трет-бутилового спирта обычно составляет 1-10% об., предпочтительно 6-10% об. и более предпочтительно 7-9% об. Температура реакции составляет 300-480°C, предпочтительно 350-450°C и более предпочтительно 400-450°C. Давление составляет нормальное давление до 5 кг/см2, и газообразный исходный материал может быть введен с пространственной скоростью 400-4000 ч-1 (в условиях нормальной температуры/давления (NTP)). С точки зрения регулирования концентрации кислорода на выходе реактора для того, чтобы улучшить выход ненасыщенного альдегида, мольное соотношение кислорода и изобутилена или трет-бутилового спирта, или их смешанного газа составляет обычно 1,0-2,0, предпочтительно 1,1-1,8 и более предпочтительно 1,2-1,8.
Примеры газа, содержащего молекулярный кислород, могут включать такие газы, содержащие молекулярный кислород, как чистый газообразный кислород, N2O или воздух. С промышленной точки зрения воздух является предпочтительным. Примеры газа-разбавителя могут включать азот, диоксид углерода, водяной пар и их смешанный газ. В качестве соотношения смешения газа, содержащего молекулярный кислород, и газа-разбавителя в смешанном газе предпочтительно удовлетворяется условие 0,01<молекулярный кислород/((газ, содержащий молекулярный кислород) + газ-разбавитель)<0,3 в объемном соотношении. Кроме того, концентрация молекулярного кислорода в исходном материале составляет предпочтительно 1-20% об.
Водяной пар в газообразном исходном материале требуется с точки зрения предотвращения коксования катализатора. Однако концентрация водяного пара в газе-разбавителе предпочтительно снижается насколько возможно для того, чтобы подавлять постобразование карбоновой кислоты, такой как метакриловая кислота и уксусная кислота. Водяной пар в газообразном исходном материале обычно используется в интервале 0-30% об.
(2) Способ получения акролеина
Когда акролеин получается газофазным каталитическим окислением пропилена, условие или т.п. специально не ограничивается. Акролеин может быть получен способом, обычно используемым, когда акролеин получается газофазным каталитическим окислением пропилена. Например, смешанный газ, содержащий 1-15% об. пропилена, 3-30% об. молекулярного кислорода, 0-60% об. водяного пара и 20-80% об. инертного газа, такого как азот и газообразный диоксид углерода или т.п., может быть введен в каталитический слой реактора при 250-450°C, давлении 0,1-1 МПа и пространственной скорости (SV) 300-5000 ч-1. В качестве реактора обычно используется реактор с неподвижным слоем, реактор с псевдоожиженным слоем или реактор с подвижным слоем.
Примеры
Далее настоящий вариант будет дополнительно описан подробно со ссылкой на примеры. Однако настоящий вариант не ограничивается примерами, описанными ниже. Атомное соотношение атомов кислорода в оксидном катализаторе определяется в зависимости от условий валентности других элементов. Поэтому в примерах и сравнительных примерах атомное соотношение атомов кислорода является опущенным в формуле, представляющей каталитическую композицию. Соотношения элементов композиции оксидного катализатора рассчитываются по соотношении зарядов композиции.
Определение угла дифракции рентгеновских лучей
В исследовании методом XRD определяется плоскость (111) и плоскость (200) соединения LaB6, определенного в качестве стандартного эталонного вещества 660 Национальным институтом стандартов и технологии. Значения приводятся к 37,441° и 43,506° соответственно.
В качестве XDR-прибора используют прибор DS ADVANCE (изготовитель - Bruker Corporation). Установленными параметрами XDR-испытаний являются: выход рентгеновских лучей: 40 кВ - 49 мА, щель расхождения (DS): 0,3°, ширина шага: 0,02°/шаг, время счета: 2,0 с и интервал измерения: 2θ=5-60°.
В примерах и сравнительных примерах степень конверсии, селективность и выход, используемые для того, чтобы показать результаты реакции, определяются по следующим формулам:
Степень конверсии = (число молей прореагировавшего исходного материала/число молей подаваемого исходного материала)×100
Селективность = (число молей образовавшегося соединения/число молей прореагировавшего исходного материала)×100
Выход = (число молей образовавшегося соединения/число молей подаваемого исходного материала)×100
Производительность требуемого продукта определяется согласно следующей формуле путем расчета количества образованного требуемого продукта на 1 т каждого из катализаторов, и затем с допущением, что выполняется непрерывная операция с использованием 10 т катализатора в течение 8000 ч.
Производительность (т) = ({число молей (моль/ч) исходного материала, подаваемого в час × выход/количество (т) катализатора}×10(т)×8000(ч)/молекулярная масса требуемого продукта
Пример 1
65,7 г гептамолибдата аммония растворяют в 197,0 г горячей воды с температурой примерно 90°C (жидкость А). 43,8 г нитрата висмута, 25,5 г нитрата церия, 36,4 г нитрата железа, 0,66 г нитрата цезия и 34,5 г нитрата кобальта растворяют в 42,4 г 18% масс. водного раствора азотной кислоты и к этому добавляют 205,0 г горячей воды с температурой примерно 90°C (жидкость В). Как жидкость А, так и жидкость В смешивают и смесь обрабатывают при 20000 об/мин в течение 1 ч с использованием гомогенизатора. Затем обработанную смесь подвергают созреванию при непрерывном перемешивании с использованием магнитной мешалки при температуре примерно 65°C в течение примерно 4 ч с получением суспензии исходного материала. Суспензию исходного материала подают в сушилку распылением и сушат распылением при температуре на входе 250°C и при температуре на выходе примерно 140°C. Температуру полученного высушенного распылением предшественника катализатора дополнительно повышают со скоростью подъема температуры 1,4°C/мин от комнатной температуры и предшественник катализатора предварительно прокаливают при 250°C в течение 3 ч. Полученный предварительно прокаленный предшественник катализатора прокаливают окончательно при 530°C в течение 8 ч. Состав полученного оксидного катализатора показан в таблице 1, а результаты определения дифракции рентгеновских лучей порошка показаны в таблице 2.
Для оценки реакционной способности катализатора трубчатый реактор из нержавеющей стали с рубашкой, имеющий диаметр 14 мм, заполняют 4,0 г катализатора. Смешанный газ, содержащий 8% об. изобутилена, 12,8% об. кислорода, 3,0% об. водяного пара и 76,2% об. азота, насыщают воздухом со скоростью потока 120 мл/мин (NTP) при температуре реакции 430°C с осуществлением реакции синтеза метакролеина. Результаты оценки реакции показаны в таблице 3.
Пример 2
68,8 г гептамолибдата аммония растворяют в 206,3 г горячей воды с температурой примерно 90°C (жидкость А). 33,2 г нитрата висмута, 29,6 г нитрата церия, 44,7 г нитрата железа, 0,57 г нитрата цезия и 32,3 г нитрата кобальта растворяют в 42,6 г 18% масс. водного раствора азотной кислоты и к этому добавляют 196,2 г горячей воды с температурой примерно 90°C (жидкость В). Как жидкость А, так и жидкость В смешивают и смесь обрабатывают при 20000 об/мин в течение 1 ч с использованием гомогенизатора. Затем суспензию подвергают созреванию при перемешивании и смешении при температуре примерно 65°C в течение примерно 4 ч с получением суспензии исходного материала. Суспензию исходного материала подают в сушилку распылением и сушат распылением при температуре на входе 250°C и при температуре на выходе примерно 140°C. Температуру полученного высушенного распылением предшественника катализатора дополнительно повышают со скоростью подъема температуры 1,4°C/мин от комнатной температуры и предшественник катализатора предварительно прокаливают при 250°C в течение 3 ч. Полученный предварительно прокаленный предшественник катализатора прокаливают окончательно при 520°C в течение 14 ч. Состав полученного оксидного катализатора показан в таблице 1, а результаты определения дифракции рентгеновских лучей порошка показаны в таблице 2. Рентгенограммы представлены на фигурах 1 и 2. Проводят анализ методом STEM-EDX (сканирующая просвечивающая электронная микроскопия - энергодисперсионная рентгеновская спектроскопия) Bi-обогащенной фазы катализатора. Как результат, если атомное отношение Bi определяется как 1, атомное отношение Се составляет 0,32, атомное отношение Fe составляет 0,16, атомное отношение Мо составляет 1,1. Ce, Fe и Мо существуют в области, в которой существует большое число Bi, и образуется кристаллическая структура четырехкомпонентной системы Ce-Bi-Fe-Mo-O.
Для оценки реакционной способности катализатора трубчатый реактор заполняют 3,5 г катализатора и реакцию синтеза метакролеина проводят в таких же условиях реакции, как в примере 1. Результаты оценки реакции показаны в таблице 3.
Пример 3
67,4 г гептамолибдата аммония растворяют в 202,3 г горячей воды с температурой примерно 90°C (жидкость А). 40,3 г нитрата висмута, 23,5 г нитрата церия, 50,3 г нитрата железа, 0,56 г нитрата цезия и 28,0 г нитрата кобальта растворяют в 42,7 г 18% масс. водного раствора азотной кислоты и к этому добавляют 201,5 г горячей воды с температурой примерно 90°C (жидкость В). Как жидкость А, так и жидкость В смешивают и смесь обрабатывают при 20000 об/мин в течение 1 ч с использованием гомогенизатора. Затем суспензию подвергают созреванию при перемешивании и смешении при температуре примерно 65°C в течение примерно 4 ч с получением суспензии исходного материала. Суспензию исходного материала подают в сушилку распылением и сушат распылением при температуре на входе 250°C и при температуре на выходе примерно 140°C. Температуру полученного высушенного распылением предшественника катализатора дополнительно повышают со скоростью подъема температуры 1,4°C/мин от комнатной температуры и предшественник катализатора предварительно прокаливают при 250°C в течение 3 ч. Полученный предварительно прокаленный предшественник катализатора прокаливают окончательно при 540°C в течение 3 ч. Состав полученного оксидного катализатора показан в таблице 1, а результаты определения дифракции рентгеновских лучей порошка показаны в таблице 2.
Для оценки реакционной способности катализатора трубчатый реактор заполняют 3,5 г катализатора и реакцию синтеза метакролеина проводят в таких же условиях реакции, как в примере 1. Результаты оценки реакции показаны в таблице 3.
Пример 4
67,4 г гептамолибдата аммония растворяют в 202,2 г горячей воды с температурой примерно 90°C (жидкость А). 40,3 г нитрата висмута, 23,5 г нитрата церия, 55,4 г нитрата железа, 0,56 г нитрата цезия и 24,2 г нитрата кобальта растворяют в 42,9 г 18% масс. водного раствора азотной кислоты и к этому добавляют 202,7 г горячей воды с температурой примерно 90°C (жидкость В). Как жидкость А, так и жидкость В смешивают и смесь обрабатывают при 20000 об/мин в течение 1 ч с использованием гомогенизатора. Затем суспензию подвергают созреванию при перемешивании и смешении при температуре примерно 65°C в течение примерно 4 ч с получением суспензии исходного материала. Суспензию исходного материала подают в сушилку распылением и сушат распылением при температуре на входе 250°C и при температуре на выходе примерно 140°C. Температуру полученного высушенного распылением предшественника катализатора дополнительно повышают со скоростью подъема температуры 1,4°C/мин от комнатной температуры и предшественник катализатора предварительно прокаливают при 250°C в течение 3 ч. Полученный предварительно прокаленный предшественник катализатора прокаливают окончательно при 530°C в течение 8 ч. Состав полученного оксидного катализатора показан в таблице 1, а результаты определения дифракции рентгеновских лучей порошка показаны в таблице 2.
Для оценки реакционной способности катализатора трубчатый реактор заполняют 3,5 г катализатора и реакцию синтеза метакролеина проводят в таких же условиях реакции, как в примере 1. Результаты оценки реакции показаны в таблице 3.
Пример 5
67,2 г гептамолибдата аммония растворяют в 201,6 г горячей воды с температурой примерно 90°C (жидкость А). 40,2 г нитрата висмута, 23,4 г нитрата церия, 60,4 г нитрата железа, 0,55 г нитрата цезия, 18,6 г нитрата кобальта и 1,0 г нитрата свинца растворяют в 37,9 г 18% масс. водного раствора азотной кислоты и к этому добавляют 203,9 г горячей воды с температурой примерно 90°C (жидкость В). Как жидкость А, так и жидкость В смешивают и смесь обрабатывают при 20000 об/мин в течение 1 ч с использованием гомогенизатора. Затем суспензию подвергают созреванию при перемешивании и смешении при температуре примерно 65°C в течение примерно 4 ч с получением суспензии исходного материала. Суспензию исходного материала подают в сушилку распылением и сушат распылением при температуре на входе 250°C и при температуре на выходе примерно 140°C. Температуру полученного высушенного распылением предшественника катализатора дополнительно повышают со скоростью подъема температуры 1,4°C/мин от комнатной температуры и предшественник катализатора предварительно прокаливают при 250°C в течение 3 ч. Полученный предварительно прокаленный предшественник катализатора прокаливают окончательно при 540°C в течение 5 ч. Состав полученного оксидного катализатора показан в таблице 1, а результаты определения дифракции рентгеновских лучей порошка показаны в таблице 2.
Для оценки реакционной способности катализатора трубчатый реактор заполняют 3,6 г катализатора и реакцию синтеза метакролеина проводят в таких же условиях реакции, как в примере 1. Результаты оценки реакции показаны в таблице 3.
Пример 6
66,2 г гептамолибдата аммония растворяют в 198,56 г горячей воды с температурой примерно 90°C (жидкость А). 39,6 г нитрата висмута, 23,0 г нитрата церия, 59,5 г нитрата железа, 0,36 г нитрата цезия, 18,3 г нитрата кобальта и 9,1 г нитрата никеля растворяют в 38,4 г 18% масс. водного раствора азотной кислоты и к этому добавляют 210,0 г горячей воды с температурой примерно 90°C (жидкость В). Как жидкость А, так и жидкость В смешивают и смесь обрабатывают при 20000 об/мин в течение 1 ч с использованием гомогенизатора. Затем суспензию подвергают созреванию при перемешивании и смешении при температуре примерно 65°C в течение примерно 4 ч с получением суспензии исходного материала. Суспензию исходного материала подают в сушилку распылением и сушат распылением при температуре на входе 250°C и при температуре на выходе примерно 140°C. Температуру полученного высушенного распылением предшественника катализатора дополнительно повышают со скоростью подъема температуры 1,4°C/мин от комнатной температуры и предшественник катализатора предварительно прокаливают при 250°C в течение 3 ч. Полученный предварительно прокаленный предшественник катализатора прокаливают окончательно при 520°C в течение 14 ч. Состав полученного оксидного катализатора показан в таблице 1, а результаты определения дифракции рентгеновских лучей порошка показаны в таблице 2.
Для оценки реакционной способности катализатора трубчатый реактор заполняют 3,0 г катализатора и реакцию синтеза метакролеина проводят в таких же условиях реакции, как в примере 1. Результаты оценки реакции показаны в таблице 3.
Пример 7
67,4 г гептамолибдата аммония растворяют в 202,1 г горячей воды с температурой примерно 90°C (жидкость А). 40,3 г нитрата висмута, 23,4 г нитрата церия, 55,4 г нитрата железа, 0,56 г нитрата цезия, 22,3 г нитрата кобальта и 1,5 г нитрата меди растворяют в 37,9 г 18% масс. водного раствора азотной кислоты и к этому добавляют 203,1 г горячей воды с температурой примерно 90°C (жидкость В). Как жидкость А, так и жидкость В смешивают и смесь обрабатывают при 20000 об/мин в течение 1 ч с использованием гомогенизатора. Затем суспензию подвергают созреванию при перемешивании и смешении при температуре примерно 65°C в течение примерно 4 ч с получением суспензии исходного материала. Суспензию исходного материала подают в сушилку распылением и сушат распылением при температуре на входе 250°C и при температуре на выходе примерно 140°C. Температуру полученного высушенного распылением предшественника катализатора дополнительно повышают со скоростью подъема температуры 1,4°C/мин от комнатной температуры и предшественник катализатора предварительно прокаливают при 250°C в течение 3 ч. Полученный предварительно прокаленный предшественник катализатора прокаливают окончательно при 550°C в течение 3 ч. Состав полученного оксидного катализатора показан в таблице 1, а результаты определения дифракции рентгеновских лучей порошка показаны в таблице 2.
Для оценки реакционной способности катализатора трубчатый реактор заполняют 3,2 г катализатора и реакцию синтеза метакролеина проводят в таких же условиях реакции, как в примере 1. Результаты оценки реакции показаны в таблице 3.
Пример 8
67,0 г гептамолибдата аммония растворяют в 202,1 г горячей воды с температурой примерно 90°C (жидкость А). 44,7 г нитрата висмута, 26,1 г нитрата церия, 37,2 г нитрата железа, 0,51 г нитрата рубидия, 18,5 г нитрата кобальта и 14,6 г нитрата магния растворяют в 37,7 г 18% масс. водного раствора азотной кислоты и к этому добавляют 203,1 г горячей воды с температурой примерно 90°C (жидкость В). Как жидкость А, так и жидкость В смешивают и смесь обрабатывают при 20000 об/мин в течение 1 ч с использованием гомогенизатора. Затем суспензию подвергают созреванию при перемешивании и смешении при температуре примерно 65°C в течение примерно 4 ч с получением суспензии исходного материала. Суспензию исходного материала подают в сушилку распылением и сушат распылением при температуре на входе 250°C и при температуре на выходе примерно 140°C. Температуру полученного высушенного распылением предшественника катализатора дополнительно повышают со скоростью подъема температуры 1,4°C/мин от комнатной температуры и предшественник катализатора предварительно прокаливают при 250°C в течение 3 ч. Полученный предварительно прокаленный предшественник катализатора прокаливают окончательно при 540°C в течение 3 ч. Состав полученного оксидного катализатора показан в таблице 1, а результаты определения дифракции рентгеновских лучей порошка показаны в таблице 2.
Для оценки реакционной способности катализатора трубчатый реактор заполняют 3,1 г катализатора и реакцию синтеза метакролеина проводят в таких же условиях реакции, как в примере 1. Результаты оценки реакции показаны в таблице 3.
Пример 9
67,4 г гептамолибдата аммония растворяют в 202,2 г горячей воды с температурой примерно 90°C (жидкость А). 40,3 г нитрата висмута, 23,5 г нитрата церия, 55,4 г нитрата железа, 0,56 г нитрата цезия и 24,2 г нитрата кобальта растворяют в 42,9 г 18% масс. водного раствора азотной кислоты и к этому добавляют 202,7 г горячей воды с температурой примерно 90°C (жидкость В). Как жидкость А, так и жидкость В смешивают и смесь обрабатывают при 20000 об/мин в течение 1 ч с использованием гомогенизатора. Затем суспензию подвергают созреванию при перемешивании и смешении при температуре примерно 65°C в течение примерно 4 ч с получением суспензии исходного материала. Суспензию исходного материала подают в сушилку распылением и сушат распылением при температуре на входе 250°C и при температуре на выходе примерно 140°C. Температуру полученного высушенного распылением предшественника катализатора дополнительно повышают со скоростью подъема температуры 1,4°C/мин от комнатной температуры и предшественник катализатора предварительно прокаливают при 150°C в течение 36 ч. Полученный предварительно прокаленный предшественник катализатора прокаливают окончательно при 520°C в течение 8 ч. Состав полученного оксидного катализатора показан в таблице 1, а результаты определения дифракции рентгеновских лучей порошка показаны в таблице 2.
Для оценки реакционной способности катализатора трубчатый реактор заполняют 3,9 г катализатора и реакцию синтеза метакролеина проводят в таких же условиях реакции, как в примере 1. Результаты оценки реакции показаны в таблице 3.
Пример 10
67,4 г гептамолибдата аммония растворяют в 202,2 г горячей воды с температурой примерно 90°C (жидкость А). 40,3 г нитрата висмута, 23,5 г нитрата церия, 55,4 г нитрата железа, 0,56 г нитрата цезия и 24,2 г нитрата кобальта растворяют в 42,9 г 18% масс. водного раствора азотной кислоты и к этому добавляют 202,7 г горячей воды с температурой примерно 90°C (жидкость В). Как жидкость А, так и жидкость В смешивают и смесь обрабатывают при 20000 об/мин в течение 1 ч с использованием гомогенизатора. Затем суспензию подвергают созреванию при перемешивании и смешении при температуре примерно 65°C в течение примерно 4 ч с получением суспензии исходного материала. Суспензию исходного материала подают в сушилку распылением и сушат распылением при температуре на входе 250°C и при температуре на выходе примерно 140°C. Температуру полученного высушенного распылением предшественника катализатора дополнительно повышают со скоростью подъема температуры 75°C/мин от комнатной температуры и предшественник катализатора предварительно прокаливают при 250°C в течение 3 ч. Полученный предварительно прокаленный предшественник катализатора прокаливают окончательно при 530°C в течение 4 ч. Состав полученного оксидного катализатора показан в таблице 1, а результаты определения дифракции рентгеновских лучей порошка показаны в таблице 2.
Для оценки реакционной способности катализатора трубчатый реактор заполняют 3,9 г катализатора и реакцию синтеза метакролеина проводят в таких же условиях реакции, как в примере 1. Результаты оценки реакции показаны в таблице 3.
Пример 11
67,4 г гептамолибдата аммония растворяют в 202,2 г горячей воды с температурой примерно 90°C (жидкость А). 40,3 г нитрата висмута, 23,5 г нитрата церия, 55,4 г нитрата железа, 0,56 г нитрата цезия и 24,2 г нитрата кобальта растворяют в 42,9 г 18% масс. водного раствора азотной кислоты и к этому добавляют 202,7 г горячей воды с температурой примерно 90°C (жидкость В). Как жидкость А, так и жидкость В смешивают и смесь обрабатывают при 20000 об/мин в течение 1 ч с использованием гомогенизатора. Затем суспензию подвергают созреванию при перемешивании и смешении при температуре примерно 65°C в течение примерно 4 ч с получением суспензии исходного материала. Суспензию исходного материала подают в сушилку распылением и сушат распылением при температуре на входе 250°C и при температуре на выходе примерно 140°C. Температуру полученного высушенного распылением предшественника катализатора дополнительно повышают со скоростью подъема температуры 1,4°C/мин от комнатной температуры и предшественник катализатора предварительно прокаливают при 250°C в течение 3 ч. Полученный предварительно прокаленный предшественник катализатора прокаливают окончательно при 400°C в течение 48 ч. Состав полученного оксидного катализатора показан в таблице 1, а результаты определения дифракции рентгеновских лучей порошка показаны в таблице 2.
Для оценки реакционной способности катализатора трубчатый реактор заполняют 3,2 г катализатора и реакцию синтеза метакролеина проводят в таких же условиях реакции, как в примере 1. Результаты оценки реакции показаны в таблице 3.
Пример 12
67,4 г гептамолибдата аммония растворяют в 202,2 г горячей воды с температурой примерно 90°C (жидкость А). 40,3 г нитрата висмута, 23,5 г нитрата церия, 55,4 г нитрата железа, 0,56 г нитрата цезия и 24,2 г нитрата кобальта растворяют в 42,9 г 18% масс. водного раствора азотной кислоты и к этому добавляют 202,7 г горячей воды с температурой примерно 90°C (жидкость В). Как жидкость А, так и жидкость В смешивают и смесь обрабатывают при 20000 об/мин в течение 1 ч с использованием гомогенизатора. Затем суспензию подвергают созреванию при перемешивании и смешении при температуре примерно 65°C в течение примерно 4 ч с получением суспензии исходного материала. Суспензию исходного материала подают в сушилку распылением и сушат распылением при температуре на входе 250°C и при температуре на выходе примерно 140°C. Температуру полученного высушенного распылением предшественника катализатора дополнительно повышают со скоростью подъема температуры 1,4°C/мин от комнатной температуры и предшественник катализатора предварительно прокаливают при 250°C в течение 3 ч. Полученный предварительно прокаленный предшественник катализатора прокаливают окончательно при 640°C в течение 30 мин. Состав полученного оксидного катализатора показан в таблице 1, а результаты определения дифракции рентгеновских лучей порошка показаны в таблице 2.
Для оценки реакционной способности катализатора трубчатый реактор заполняют 5,4 г катализатора и реакцию синтеза метакролеина проводят в таких же условиях реакции, как в примере 1. Результаты оценки реакции показаны в таблице 3.
Пример 13
Используют такой же катализатор, как в примере 3. Для оценки реакционной способности катализатора трубчатый реактор заполняют 6,4 г катализатора и реакцию синтеза метакролеина проводят в таких же условиях реакции, как в примере 1, за исключением того, что температура реакции была изменена на 400°C. Результаты оценки реакции показаны в таблице 3.
Пример 14
Используют такой же катализатор, как в примере 3. Для оценки реакционной способности катализатора трубчатый реактор заполняют 3,0 г катализатора и реакцию синтеза метакролеина проводят в таких же условиях реакции, как в примере 1, за исключением того, что температура реакции была изменена на 460°C. Результаты оценки реакции показаны в таблице 3.
Пример 15
69,5 г гептамолибдата аммония растворяют в 208,5 г горячей воды с температурой примерно 90°C (жидкость А). 32,0 г нитрата висмута, 7,2 г нитрата церия, 39,9 г нитрата железа, 1,3 г нитрата цезия, 43,2 г нитрата кобальта и 24,2 г нитрата никеля растворяют в 38,3 г 18% масс. водного раствора азотной кислоты и к этому добавляют 208,5 г горячей воды с температурой примерно 90°C (жидкость В). Как жидкость А, так и жидкость В смешивают и смесь обрабатывают при 20000 об/мин в течение 1 ч с использованием гомогенизатора. Затем обработанную смесь подвергают созреванию при перемешивании и смешении при температуре примерно 60°C в течение примерно 4 ч с получением суспензии исходного материала. Суспензию исходного материала подают в сушилку распылением и сушат распылением при температуре на входе 250°C и при температуре на выходе примерно 140°C. Температуру полученного высушенного распылением предшественника катализатора дополнительно повышают со скоростью подъема температуры 1,4°C/мин от комнатной температуры и предшественник катализатора предварительно прокаливают при 280°C в течение 3 ч. Полученный предварительно прокаленный предшественник катализатора прокаливают окончательно при 550°C в течение 10 ч. Состав полученного оксидного катализатора показан в таблице 1, а результаты определения дифракции рентгеновских лучей порошка показаны в таблице 2.
Для оценки реакционной способности катализатора трубчатый реактор заполняют 4,0 г катализатора и реакцию синтеза метакролеина проводят в таких же условиях реакции, как в примере 1. Результаты оценки реакции показаны в таблице 3.
Пример 16
Как жидкость А, так и жидкость В смешивают в такую же композицию, как в примере 1, и смесь подвергают созреванию при непрерывном перемешивании с использованием магнитной мешалки при температуре примерно 65°C в течение примерно 1 ч без выполнения обработки гомогенизатором с получением суспензии исходного материала. Суспензию исходного материала подают в сушилку распылением и сушат распылением при температуре на входе 250°C и при температуре на выходе примерно 140°C. Температуру полученного высушенного распылением предшественника катализатора дополнительно повышают со скоростью подъема температуры 1,4°C/мин от комнатной температуры и предшественник катализатора предварительно прокаливают при 250°C в течение 3 ч. Полученный предварительно прокаленный предшественник катализатора прокаливают окончательно при 530°C в течение 8 ч. Состав полученного оксидного катализатора показан в таблице 1, а результаты определения дифракции рентгеновских лучей порошка показаны в таблице 2.
Для оценки реакционной способности катализатора трубчатый реактор заполняют 4,0 г катализатора и реакцию синтеза метакролеина проводят в таких же условиях реакции, как в примере 1. Результаты оценки реакции показаны в таблице 3.
Пример 17
Как жидкость А, так и жидкость В смешивают в такую же композицию, как в примере 1, и смесь подвергают созреванию при непрерывном перемешивании с использованием магнитной мешалки при температуре примерно 65°C в течение примерно 24 ч без выполнения обработки гомогенизатором с получением суспензии исходного материала. Суспензию исходного материала подают в сушилку распылением и сушат распылением при температуре на входе 250°C и при температуре на выходе примерно 140°C. Температуру полученного высушенного распылением предшественника катализатора дополнительно повышают со скоростью подъема температуры 1,4°C/мин от комнатной температуры и предшественник катализатора предварительно прокаливают при 250°C в течение 3 ч. Полученный предварительно прокаленный предшественник катализатора прокаливают окончательно при 530°C в течение 8 ч. Состав полученного оксидного катализатора показан в таблице 1, а результаты определения дифракции рентгеновских лучей порошка показаны в таблице 2.
Для оценки реакционной способности катализатора трубчатый реактор заполняют 4,0 г катализатора и реакцию синтеза метакролеина проводят в таких же условиях реакции, как в примере 1. Результаты оценки реакции показаны в таблице 3.
Пример 18
Используют такой же катализатор, как в примере 1, и для оценки реакционной способности катализатора трубчатый реактор заполняют 4,5 г катализатора, и реакцию синтеза метакролеина проводят в таких же условиях реакции, как в примере 1, за исключением того, что температуру реакции устанавливают при 350°C. Результаты оценки реакции показаны в таблице 3.
Пример 19
Используют такой же катализатор, как в примере 1, и для оценки реакционной способности катализатора трубчатый реактор заполняют 4,0 г катализатора, и реакцию синтеза метакролеина проводят в таких же условиях реакции, как в примере 1, за исключением того, что температуру реакции устанавливают при 480°C. Результаты оценки реакции показаны в таблице 3.
Пример 20
Используют такой же катализатор, как в примере 1, и для оценки реакционной способности катализатора трубчатый реактор заполняют 4,0 г катализатора. Смешанный газ, содержащий 6% об. изобутилена, 9,6% кислорода, 3,0% об. водяного пара и 81,4% об. азота, насыщают воздухом со скоростью потока 100 мл/мин (NTP) при температуре реакции 430°C с осуществлением реакции синтеза метакролеина. Результаты оценки реакции показаны в таблице 3.
Пример 21
Используют такой же катализатор, как в примере 1, и для оценки реакционной способности катализатора трубчатый реактор заполняют 4,0 г катализатора. Реакцию синтеза метакролеина проводят в таких же условиях реакции, как в примере 20, за исключением того, что температуру реакции устанавливают при 350°C. Результаты оценки реакции показаны в таблице 3.
Сравнительный пример 1
72,8 г гептамолибдата аммония растворяют в 218,4 г горячей воды с температурой примерно 90°C (жидкость А). 26,8 г нитрата висмута, 7,5 г нитрата церия, 19,5 г нитрата железа, 2,0 г нитрата цезия и 79,5 г нитрата кобальта растворяют в 42,1 г 18% масс. водного раствора азотной кислоты и к этому добавляют 177,8 г горячей воды с температурой примерно 90°C (жидкость В). Как жидкость А, так и жидкость В смешивают и смесь обрабатывают при 20000 об/мин в течение 1 ч с использованием гомогенизатора. Затем обработанную смесь подвергают созреванию при перемешивании и смешении при температуре примерно 65°C в течение примерно 4 ч с получением суспензии исходного материала. Суспензию исходного материала подают в сушилку распылением и сушат распылением при температуре на входе 250°C и при температуре на выходе примерно 140°C. Температуру полученного высушенного распылением предшественника катализатора дополнительно повышают со скоростью подъема температуры 1,4°C/мин от комнатной температуры и предшественник катализатора предварительно прокаливают при 250°C в течение 3 ч. Полученный предварительно прокаленный предшественник катализатора прокаливают окончательно при 520°C в течение 5 ч. Состав полученного оксидного катализатора показан в таблице 1, а результаты определения дифракции рентгеновских лучей порошка показаны в таблице 2. Рентгенограммы представлены на фигурах 1 и 2.
Проводят анализ методом STEM-EDX Bi-обогащенной фазы катализатора. Как результат, если атомное отношение Bi определяется как 1, атомное отношение Се составляет 0,07, атомное отношение Fe составляет 0,06, атомное отношение Мо составляет 1,1. Содержание Ce, Fe к Bi является меньше, чем в примере 2, и образуется двухкомпонетная система Bi-Mo-O. Количество образовавшейся четырехкомпонентной кристаллической структуры Ce-Bi-Fe-Mo-O является небольшим.
Для оценки реакционной способности катализатора трубчатый реактор заполняют 4,2 г катализатора и реакцию синтеза метакролеина проводят в таких же условиях реакции, как в примере 1. Результаты оценки реакции показаны в таблице 3.
Сравнительный пример 2
66,0 г гептамолибдата аммония растворяют в 197,9 г горячей воды с температурой примерно 90°C (жидкость А). 39,5 г нитрата висмута, 23,0 г нитрата церия, 75,7 г нитрата железа, 0,54 г нитрата цезия и 15,5 г нитрата кобальта растворяют в 42,1 г 18% масс. водного раствора азотной кислоты и к этому добавляют 214,4 г горячей воды с температурой примерно 90°C (жидкость В). Как жидкость А, так и жидкость В смешивают и смесь обрабатывают при 20000 об/мин в течение 1 ч с использованием гомогенизатора. Затем обработанную смесь подвергают созреванию при перемешивании и смешении при температуре примерно 65°C в течение примерно 4 ч с получением суспензии исходного материала. Суспензию исходного материала подают в сушилку распылением и сушат распылением при температуре на входе 250°C и при температуре на выходе примерно 140°C. Температуру полученного высушенного распылением предшественника катализатора дополнительно повышают со скоростью подъема температуры 1,4°C/мин от комнатной температуры, и предшественник катализатора предварительно прокаливают при 250°C в течение 3 ч. Полученный предварительно прокаленный предшественник катализатора прокаливают окончательно при 540°C в течение 5 ч. Состав полученного оксидного катализатора показан в таблице 1, а результаты определения дифракции рентгеновских лучей порошка показаны в таблице 2.
Для оценки реакционной способности катализатора трубчатый реактор заполняют 4,3 г катализатора и реакцию синтеза метакролеина проводят в таких же условиях реакции, как в примере 1. Результаты оценки реакции показаны в таблице 3.
Сравнительный пример 3
67,4 г гептамолибдата аммония растворяют в 202,2 г горячей воды с температурой примерно 90°C (жидкость А). 40,3 г нитрата висмута, 23,5 г нитрата церия, 55,4 г нитрата железа, 0,56 г нитрата цезия и 24,2 г нитрата кобальта растворяют в 42,9 г 18% масс. водного раствора азотной кислоты и к этому добавляют 202,7 г горячей воды с температурой примерно 90°C (жидкость В). Как жидкость А, так и жидкость В смешивают с получением суспензии исходного материала. Суспензию исходного материала подают в сушилку распылением и сушат распылением при температуре на входе 250°C и при температуре на выходе примерно 140°C. Температуру полученного высушенного распылением предшественника катализатора дополнительно повышают со скоростью подъема температуры 1,4°C/мин от комнатной температуры и предшественник катализатора предварительно прокаливают при 250°C в течение 3 ч. Полученный предварительно прокаленный предшественник катализатора прокаливают окончательно при 540°C в течение 5 ч. Состав полученного оксидного катализатора показан в таблице 1, а результаты определения дифракции рентгеновских лучей порошка показаны в таблице 2.
Для оценки реакционной способности катализатора трубчатый реактор заполняют 4,6 г катализатора и реакцию синтеза метакролеина проводят в таких же условиях реакции, как в примере 1. Результаты оценки реакции показаны в таблице 3.
Сравнительный пример 4
67,4 г гептамолибдата аммония растворяют в 202,2 г горячей воды с температурой примерно 90°C (жидкость А). 40,3 г нитрата висмута, 23,5 г нитрата церия, 55,4 г нитрата железа, 0,56 г нитрата цезия и 24,2 г нитрата кобальта растворяют в 42,9 г 18% масс. водного раствора азотной кислоты и к этому добавляют 202,7 г горячей воды с температурой примерно 90°C (жидкость В). Как жидкость А, так и жидкость В смешивают и смесь обрабатывают при 20000 об/мин в течение 1 ч с использованием гомогенизатора. Затем обработанную смесь подвергают созреванию при перемешивании и смешении при температуре примерно 65°C в течение примерно 4 ч с получением суспензии исходного материала. Суспензию исходного материала подают в сушилку распылением и сушат распылением при температуре на входе 250°C и при температуре на выходе примерно 140°C. Температуру полученного высушенного распылением предшественника катализатора дополнительно повышают со скоростью подъема температуры 1,4°C/мин от комнатной температуры и предшественник катализатора предварительно прокаливают при 105°C в течение 12 ч. Полученный предварительно прокаленный предшественник катализатора прокаливают окончательно при 530°C в течение 8 ч. Состав полученного оксидного катализатора показан в таблице 1, а результаты определения дифракции рентгеновских лучей порошка показаны в таблице 2.
Для оценки реакционной способности катализатора трубчатый реактор заполняют 4,9 г катализатора и реакцию синтеза метакролеина проводят в таких же условиях реакции, как в примере 1. Результаты оценки реакции показаны в таблице 3.
Сравнительный пример 5
67,4 г гептамолибдата аммония растворяют в 202,2 г горячей воды с температурой примерно 90°C (жидкость А). 40,3 г нитрата висмута, 23,5 г нитрата церия, 55,4 г нитрата железа, 0,56 г нитрата цезия и 24,2 г нитрата кобальта растворяют в 42,9 г 18% масс. водного раствора азотной кислоты и к этому добавляют 202,7 г горячей воды с температурой примерно 90°C (жидкость В). Как жидкость А, так и жидкость В смешивают и смесь обрабатывают при 20000 об/мин в течение 1 ч с использованием гомогенизатора. Затем обработанную смесь подвергают созреванию при перемешивании и смешении при температуре примерно 65°C в течение примерно 4 ч с получением суспензии исходного материала. Суспензию исходного материала подают в сушилку распылением и сушат распылением при температуре на входе 250°C и при температуре на выходе примерно 140°C. Температуру полученного высушенного распылением предшественника катализатора дополнительно повышают со скоростью подъема температуры 1,4°C/мин от комнатной температуры и предшественник катализатора предварительно прокаливают при 250°C в течение 3 ч. Полученный предварительно прокаленный предшественник катализатора прокаливают окончательно при 720°C в течение 30 мин. Состав полученного оксидного катализатора показан в таблице 1, а результаты определения дифракции рентгеновских лучей порошка показаны в таблице 2.
Для оценки реакционной способности катализатора трубчатый реактор заполняют 5,9 г катализатора и реакцию синтеза метакролеина проводят в таких же условиях реакции, как в примере 1. Результаты оценки реакции показаны в таблице 3.
Сравнительный пример 6
Катализатор получают таким же образом, как в примере 15, за исключением того, что суспензию исходного материала не подвергают созреванию. Состав полученного оксидного катализатора показан в таблице 1, а результаты определения дифракции рентгеновских лучей порошка показаны в таблице 2.
Для оценки реакционной способности катализатора трубчатый реактор заполняют 4,5 г катализатора и реакцию синтеза метакролеина проводят в таких же условиях реакции, как в примере 1. Результаты оценки реакции показаны в таблице 3.
Сравнительный пример 7
Катализатор получают таким же образом, как в примере 1, за исключением того, что суспензию исходного материала не подвергают созреванию. Используют полученный катализатор и для оценки реакционной способности катализатора трубчатый реактор заполняют 4,0 г катализатора. Реакцию синтеза метакролеина проводят в таких же условиях реакции, как в примере 1. Результаты оценки реакции показаны в таблице 3.
Сравнительный пример 8
Катализатор получают таким же образом, как в примере 1, за исключением того, что суспензию исходного материала не подвергают созреванию. Используют полученный катализатор и для оценки реакционной способности катализатора трубчатый реактор заполняют 5,0 г катализатора. Реакцию синтеза метакролеина проводят в таких же условиях реакции, как в примере 18. Результаты оценки реакции показаны в таблице 3.
Сравнительный пример 9
Катализатор получают таким же образом, как в примере 1, за исключением того, что суспензию исходного материала не подвергают созреванию. Используют полученный катализатор и для оценки реакционной способности катализатора трубчатый реактор заполняют 4,0 г катализатора. Реакцию синтеза метакролеина проводят в таких же условиях реакции, как в примере 19. Результаты оценки реакции показаны в таблице 3.
Сравнительный пример 10
Катализатор получают таким же образом, как в примере 1, за исключением того, что суспензию исходного материала не подвергают созреванию. Используют полученный катализатор и для оценки реакционной способности катализатора трубчатый реактор заполняют 5,0 г катализатора. Реакцию синтеза метакролеина проводят в таких же условиях реакции, как в примере 20. Результаты оценки реакции показаны в таблице 3.
Сравнительный пример 11
Катализатор получают таким же образом, как в примере 1, за исключением того, что суспензию исходного материала не подвергают созреванию. Используют полученный катализатор и для оценки реакционной способности катализатора трубчатый реактор заполняют 6,0 г катализатора. Реакцию синтеза метакролеина проводят в таких же условиях реакции, как в примере 21. Результаты оценки реакции показаны в таблице 3.
Сравнительный пример 12
71,2 г гептамолибдата аммония растворяют в 213,7 г горячей воды с температурой примерно 90°C (жидкость А). 32,8 г нитрата висмута, 14,6 г нитрата церия, 34,1 г нитрата железа, 2,6 г нитрата цезия, 49,2 г нитрата кобальта и 0,35 г нитрата калия растворяют в 37,0 г 18% масс. водного раствора азотной кислоты и к этому добавляют 183,2 г горячей воды с температурой примерно 90°C (жидкость В). Как жидкость А, так и жидкость В смешивают с получением суспензии исходного материала. Суспензию исходного материала подают в сушилку распылением и сушат распылением при температуре на входе 250°C и при температуре на выходе примерно 140°C. Температуру полученного высушенного распылением предшественника катализатора дополнительно повышают со скоростью подъема температуры 1,4°C/мин от комнатной температуры и предшественник катализатора предварительно прокаливают при 250°C в течение 3 ч. Полученный предварительно прокаленный предшественник катализатора прокаливают окончательно при 510°C в течение 3 ч. Состав полученного оксидного катализатора показан в таблице 1, а результаты определения дифракции рентгеновских лучей порошка показаны в таблице 2.
Для оценки реакционной способности катализатора трубчатый реактор заполняют 3,8 г катализатора и реакцию синтеза метакролеина проводят в таких же условиях реакции, как в примере 1. Результаты оценки реакции показаны в таблице 3.
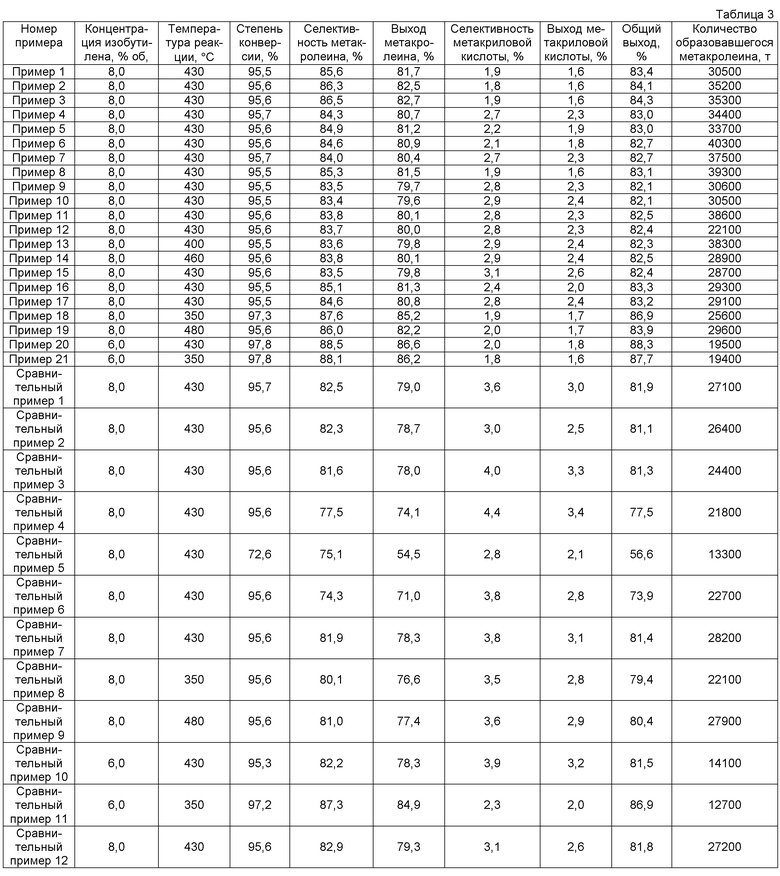
Пример 22
Используют катализатор, полученный в примере 1, и для оценки реакционной способности катализатора трубчатый реактор из нержавеющей стали с рубашкой, имеющий диаметр 14 мм, заполняют 4,0 г катализатора. Смешанный газ, содержащий 8% об. трет-бутилового спирта, 12,8% об. кислорода, 3,0% об. водяного пара и 76,2% об. азота, насыщают воздухом со скоростью потока 120 мл/мин (NTP) при температуре реакции 430°C с осуществлением реакции синтеза метакролеина. Результаты оценки реакции показаны в таблице 4.
Сравнительный пример 12
Используют катализатор, полученный в сравнительном примере 1, и для оценки реакционной способности катализатора трубчатый реактор заполняют 4,2 г катализатора. Реакцию синтеза метакролеина проводят в таких же условиях реакции, как в примере 22. Результаты оценки реакции показаны в таблице 4.
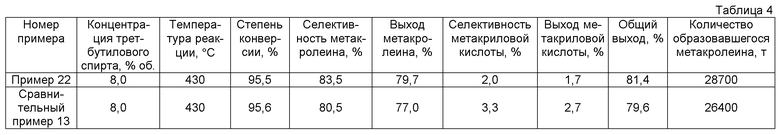
Пример 23
Используют катализатор, полученный в примере 1, и для оценки реакционной способности катализатора трубчатый реактор из нержавеющей стали с рубашкой, имеющий внутренний диаметр 15 мм, заполняют 20 мл катализатора. Исходный материал - газ, имеющий концентрацию пропилена 10% об., концентрацию 17,0% об. водяного пара и концентрацию 73% об. воздуха, пропускают в течение времени контактирования 2,3 сек при нормальном давлении с осуществлением реакции синтеза акролеина при температуре реакции 430°C. Результаты оценки реакции показаны в таблице 5.
Сравнительный пример 13
Используют катализатор, полученный в сравнительном примере 1, и для оценки реакционной способности катализатора трубчатый реактор заполняют 20 мл катализатора. Реакцию синтеза акролеина проводят в таких же условиях реакции, как в примере 23. Результаты оценки реакции показаны в таблице 5.
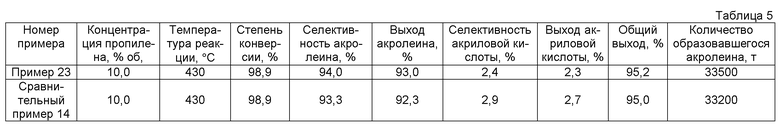
С целью дополнительной иллюстрации отличий настоящего изобретения и обеспечиваемых им технических результатов представляются также следующие справочные примеры.
Справочный пример 1
Катализатор получали таким же образом, как описано в примере 9 документа ЕР 1459803 известного уровня техники. Полученный катализатор подвергали тем же измерениям угла дифракции рентгеновских лучей, которые изложены выше. Результаты измерения показаны в таблице А ниже.
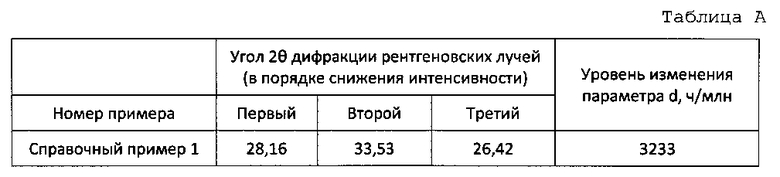
Как видно из таблицы А, в справочном примере 1, где катализатор имеет состав, в котором соотношения компонентов удовлетворяют таковым, указанным в п. 1 формулы настоящего изобретения, но в котором также присутствует сурьма в качестве компонента в составе оксидного катализатора, не удовлетворяется требуемый согласно настоящему изобретению уровень изменения параметра d (шага кристаллической решетки) 5000-9000 ч/млн.
Справочный пример 2
Катализатор получали таким же образом, как описано в примере 4 в JP 2000-237592 (см. JP 2000-237592, абзац [0027]). Полученный катализатор подвергали тем же измерениям угла дифракции рентгеновских лучей, которые изложены выше.
В результате полученный катализатор не имел пиков при углах дифракции 2θ 28,17±0,5°, 33,50+а°, в порядке снижения интенсивности, так что величина уровня изменения параметра d не могла быть определена.
Справочный пример 3
66,8 г гептамолибдата аммония растворяют в 200,41 г горячей воды с температурой примерно 90°С (жидкость А). 44,6 г нитрата висмута, 26,0 г нитрата церия, 28,1 г нитрата железа, 0,67 г нитрата цезия и 35,1 г нитрата кобальта растворяют в 36,8 г 18% масс. водного раствора азотной кислоты и к этому добавляют 197,0 г горячей воды с температурой примерно 90°С (жидкость В). Как жидкость А, так и жидкость В смешивают и смесь обрабатывают при 20000 об/мин в течение 1 ч с использованием гомогенизатора. Затем обработанную смесь подвергают созреванию при непрерывном перемешивании с использованием магнитной мешалки при температуре примерно 65°С в течение примерно 4 ч с получением суспензии исходного материала. Суспензию исходного материала подают в сушилку распылением и сушат распылением при температуре на входе 250°С и при температуре на выходе примерно 140°С. Температуру полученного высушенного распылением предшественника катализатора дополнительно повышают со скоростью подъема температуры 1,4°С/мин от комнатной температуры и предшественник катализатора предварительно прокаливают при 250°С в течение 3 ч. Полученный предварительно прокаленный предшественник катализатора прокаливают окончательно при 530°С в течение 8 ч.
Состав полученного оксидного катализатора определяли на основе масс загруженных исходных компонентов, и он показан в таблице В-1 ниже.
Полученный катализатор подвергали тем же измерениям угла дифракции рентгеновских лучей, которые изложены выше. Результаты данных измерений показаны в таблице В-2.
Для оценки реакционной способности катализатора осуществляли ту же реакцию синтеза метакролеина, что и в примере 1, которая описана выше, но при этом в трубчатый реактор загружали 4,4 г полученного оксидного катализатора. Результаты оценки реакционной способности показаны в таблице В-3.
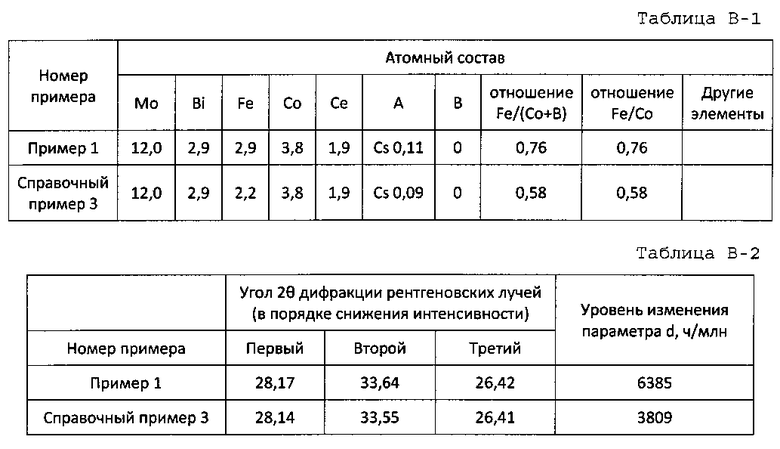
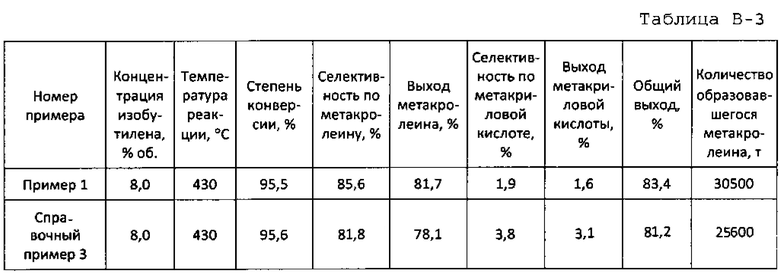
Результаты данного примера подтверждают, что существенным признаком настоящего изобретения является содержание железа в интервале значений b от свыше 2,5 до 5.
Как видно из приведенных выше результатов оценки реакции, оксидный катализатор согласно настоящему варианту вызывает меньшее образование последующего оксида в реакции окисления олефина и/или спирта и может улучшить селективность ненасыщенного альдегида.
Настоящая заявка основана на заявке на Японский патент, поданной 28 июня 2011 г (заявка на Японский патент № 2011-143284), содержание которой поэтому приводится здесь в качестве ссылки.
Промышленная применимость
Оксидный катализатор согласно настоящему изобретению может использоваться в промышленности в качестве катализатора, используемого в реакции окисления олефина и/или спирта.
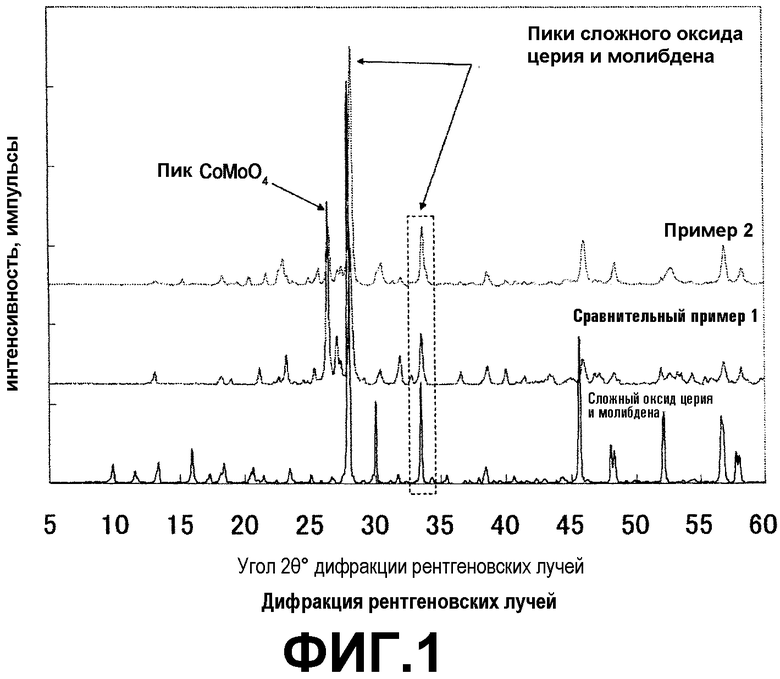
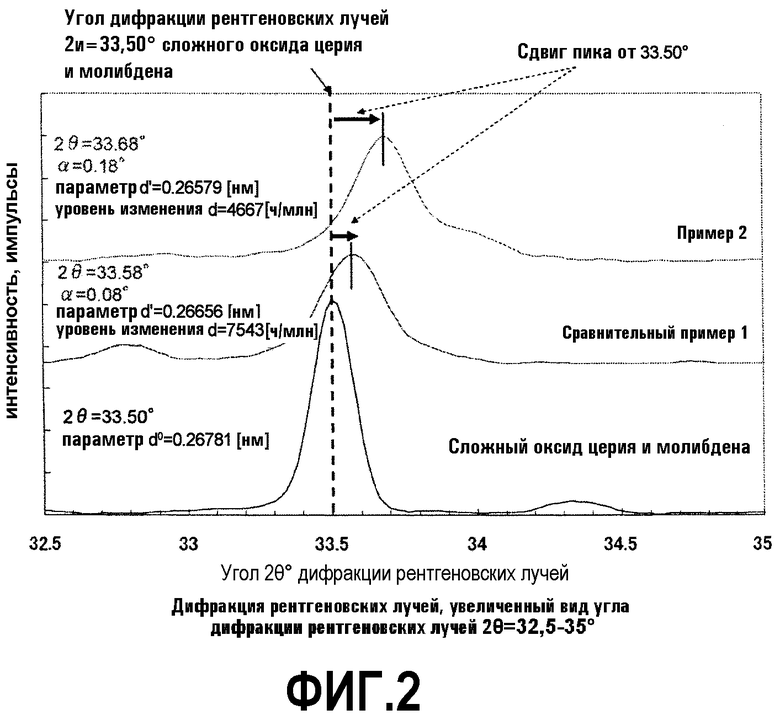
Предложен оксидный катализатор для использования в реакции окисления олефина и/или спирта, способ его получения и способ получения насыщенного альдегида в присутствии указанного катализатора. Катализатор представлен следующей формулой состава (1):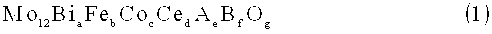
в которой Мо представляет собой молибден, Bi представляет собой висмут, Fe представляет собой железо, Со представляет собой кобальт, Се представляет собой церий, А представляет собой по меньше мере один элемент, выбранный из группы, состоящей из цезия и рубидия, В представляет собой по меньшей мере один элемент, выбранный из группы, состоящей из меди, никеля, магния и свинца, a-f представляют собой атомное отношение каждого из элементов к 12 атомам Мо; 2≤а≤6, 2,5<b≤5, 2≤с≤8, 0,4≤b/c≤2,5, 0,5≤d≤6, 0,01≤е≤2 и 0≤f<2, и g представляет собой число атомов кислорода, определенное по валентности составляющего элемента, иного, чем кислород, в котором, когда параметр d сложного оксида церия и молибдена, показывающего пик при 33,50° на рентгенограмме, принимается за эталон, уровень изменения d составляет 5000-9000 ч/млн. Способ получения катализатора включает следующие стадии: созревание суспензии исходного материала, содержащей молибден, висмут, железо, кобальт и церий, при температуре выше, чем комнатная температура; сушка созревшей суспензии исходного материала; предварительное прокаливание высушенного продукта при 120°С или выше и 350°С или ниже; и последующее окончательное прокаливание предварительно прокаленного продукта при температуре 400°С или выше и 700°С или ниже. Оксидный катализатор вызывает меньшее образование последующего оксида в реакции окисления олефина и/или спирта и может улучшить селективность ненасыщенного альдегида. 3 н.п. ф-лы, 2 ил., 9 табл., 23 пр.
1. Оксидный катализатор для использования в реакции окисления олефина и/или спирта, который имеет состав, представленный следующей формулой состава (1):
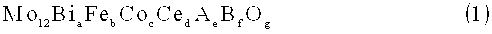
в которой Мо представляет собой молибден, Bi представляет собой висмут, Fe представляет собой железо, Со представляет собой кобальт, Се представляет собой церий, А представляет собой по меньше мере один элемент, выбранный из группы, состоящей из цезия и рубидия, В представляет собой по меньшей мере один элемент, выбранный из группы, состоящей из меди, никеля, магния и свинца, a-f представляют собой атомное отношение каждого из элементов к 12 атомам Мо; при этом удовлетворяются значения 2≤а≤6, 2,5<b≤5, 2≤с≤8, 0,4≤b/c≤2,5, 0,5≤d≤6, 0,01≤е≤2 и 0≤f<2, и g представляет собой число атомов кислорода, определенное по валентности составляющего элемента, иного, чем кислород,
в котором, когда параметр d сложного оксида церия и молибдена, показывающего пик при 33,50° на рентгенограмме, принимается за эталон, уровень изменения d составляет 5000-9000 ч/млн,
причем указанный оксидный катализатор получен способом, включающим стадию созревания суспензии исходного материала, содержащей молибден, висмут, железо, кобальт и церий, при температуре выше, чем комнатная температура.
2. Способ получения оксидного катализатора по п. 1, который включает следующие стадии:
созревание суспензии исходного материала, содержащей молибден, висмут, железо, кобальт и церий, при температуре выше, чем комнатная температура;
сушка созревшей суспензии исходного материала;
предварительное прокаливание высушенного продукта при 120°С или выше и 350°С или ниже; и
последующее окончательное прокаливание предварительно прокаленного продукта при температуре 400°С или выше и 700°С или ниже.
3. Способ получения ненасыщенного альдегида, включающий стадию окисления по меньше мере одного олефина, выбранного из группы, состоящей из пропилена и изобутилена и/или трет-бутилового спирта с использованием оксидного катализатора по п. 1.
| Высокоскоростной молот | 1987 |
|
SU1459803A1 |
| Полирующий состав | 1978 |
|
SU767161A1 |
| ЩИТОВОЙ ДЛЯ ВОДОЕМОВ ЗАТВОР | 1922 |
|
SU2000A1 |
| КАТАЛИТИЧЕСКАЯ КОМПОЗИЦИЯ (ВАРИАНТЫ) И СПОСОБ КОНВЕРСИИ ОЛЕФИНА С ЕЕ ПРИМЕНЕНИЕМ | 2001 |
|
RU2266784C2 |
| Способ приготовления окисного катализатора для окисления пропилена | 1987 |
|
SU1665870A3 |
| US 20110077148 A1, 31.03.2011. | |||

