Изобретение относится к области биоорганической химии, в частности к производным углеводов и аминокислот, содержащих четыре терминальных углеводных остатка.
Воспаление является иммунным ответом на повреждение ткани и вторжения патогенов, в котором проникновение лейкоцитов из крови в ткани является ключевым процессом. Однако чрезмерный приток лейкоцитов при определенных патологических состояниях может быть результатом нежелательных реакций. Например, локализация лейкоцитов в месте повреждения ткани может вызвать ее дальнейшие повреждение, что ведет к углублению, расширению и ухудшению поражения ткани, а также необратимому некрозу и окончательной потери функций ткани [Kaila N. / N. Kaila, K. Janz, S. DeBernardo, P.W. Bedard, R.T. Camphausen, S. Tam, D. H. H. Tsao, J.C. Keith Jr., C. Nickerson-Nutter, A. Shilling, R. Young-Sciame, Q.J. Wang // Med. Chem. - 2007. - V. 50. - P. 21-39].
Важную роль в прочном креплении лейкоцитов к эндотелию играют интегрины лейкоцитов CD11b/CD18. Исследования антиадгезионного биологического механизма с использованием флуоресцентно меченных мультивалентных лактозидов показали, что они являются мишенями CD11b на поверхности лейкоцитов. Кроме того, при исследовании лечения тяжелого ожогового шока, клинический синдром которого связан с недостаточным притоком крови к жизненно важным органам и тканям, было продемонстрировано, что мультивалентные лактозиды могут эффективно ингибировать адгезию лейкоцитов в эндотелиальных клетках [Watterson S.Н. / S.Н. Watterson, Z. Xiao, D.S. Dodd, D.R. Tortolani, W. Vaccaro, D. Potin, M. Launay, J. Stetsko // Med. Chem. - 2010. - V. 53. - P. 3814-3830].
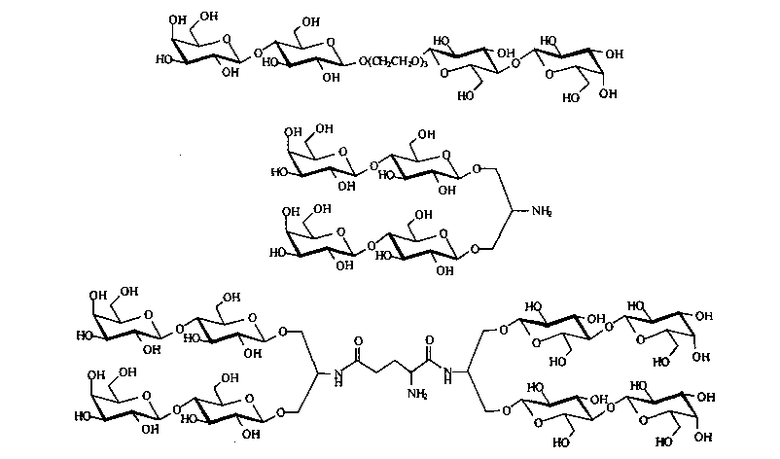
Наиболее близким техническим решением к заявленному изобретению является гликоконъюгат с двумя терминальными остатками лактозы, который, обладая двумя гидрофобными цепями, способен встраиваться в бислой липосом и эффективно связываться с лектинами.
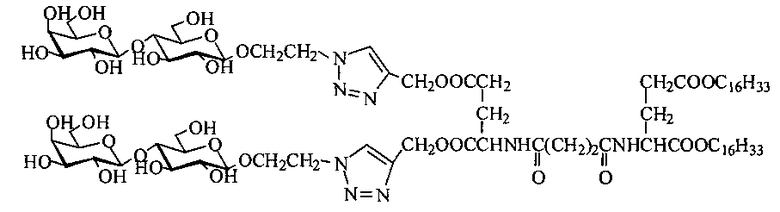
Техническим результатом предлагаемого изобретения является повышение аффинности углеводных остатков к рецепторам благодаря созданию новых тетравалентных неогликоконъюгатов с D-глюкозой в качестве разветвляющего ядра и производным L-глутаминовой кислоты в гидрофобном блоке, специфичных к соответствующим рецепторам и содержащих в своем составе остатки различных углеводов (например, лактозы, D-маннозы и других), имеющих следующую общую формулу:
Для достижения указанного технического результата разработан способ получения неогликоконъюгатов, включающий синтез производного тетрапропаргилового эфира D-глюкозы с дигексадециловым эфиром L-глутаминовой кислоты, синтез азидоэтильного производного углевода и конъюгацию этих компонентов с образованием целевых соединений.
Реализация данного изобретения подтверждается следующими примерами.
Пример 1
Синтез N-(23,4,6-тетра-O-ацетил-β-D-глюкопиранозилокси)-сукцинимида. К раствору 2,00 г (5,13 ммоль) 1,2,3,4,6-пента-O-ацетил-β-D-глюкопиранозида в 22 мл этилацетата прибавляли 0,450 мл (5,64 ммоль) эфиратного комплекса трехфтористого бора. Через 15 мин к реакционной смеси прибавляли 1,18 г (10,3 ммоль) N-гидроксисукцинимида и выдерживали 12 ч при температуре 40°C. Затем реакционную массу нейтрализовывали 25%-ным раствором аммиака до pH=7. Раствор промывали 5×100 мл воды, органический слой сушили, растворитель удаляли в вакууме. Продукт очищали при помощи колоночной хроматографии в системе толуол-ацетонитрил 5:1. Получали 1,03 г (45%) N-(2,3,4,6-тетра-O-ацетил-β-D-глюкопиранозил)-сукцинимида в виде белых кристаллов, Rf (толуол-ацетонитрил 2:1) 0,45. Tпл=177-178°C (из изопропилового спирта). ИК-спектр (вазелиновое масло, vmax, см-1): 2915, 2850, 1410, (C-H), 1750 (C=O), 1232 (C-O), 1168-1048 (C-O, 4 полосы, углеводный скелет).
Пример 2
Синтез N-(β-D-глюкопиранозил)-3-(карбоксиметил)-пропанамида. К раствору 100 мг (0,224 ммоль) N-(2,3,4,6-тетра-O-ацетил-β-D-глюкопиранозид)-сукцинимида в 5 мл метанола прикапывали 0,1 М раствор метилата натрия в метаноле до pH=8. Реакционную смесь оставляли на 24 ч. Затем реакционную смесь обрабатывали ионообменной смолой КУ-2 (H+) до pH=7. Смолу отфильтровывали, остаток растворителя удаляли в вакууме. ИК-спектр (в пленке, vmax, см-1): 3370 (ОН), 2895, 1470, 1354 (С-Н), 1750 (С=О), 1671 (С=O, I амидная полоса), 1632 (NH, II амидная полоса), 1225 (С-О), 1110-1044 (С-О, 4 полосы, углеводный скелет).
Пример 3
Синез N-(β-D-глюкопиранозил)-3-(карбокси)-пропанамида. 62,0 мг N-(β-D-глюкопиранозид)-3-(карбоксиметил)-пропанамида перемешивали в 0,05 М растворе NaOH в метаноле при комнатной температуре 24 ч. Реакционную смесь нейтрализовывали до pH=7 ионообменной смолой КУ-2 (Н+), смолу отфильтровывали. Остаток растворителя удаляли в вакууме и высушивали над P2O5 в вакуум-эксикаторе. Получали 50,0 г (67%) N-(β-D-глюкопиранозил)-3-(карбокси)-пропанамида в виде аморфного вещества, Rf (хлороформ-метанол-вода 18:6:1) 0,67. ИК-спектр (vmax, см-1): 3332 (ОН), 2890, 1476, 1350 (С-Н), 1715 (С=O), 1671 (С=O, I амидная полоса), 1632 (NH, II амидная полоса), 1280 (С-О), 1118-1041 (С-О, 4 полосы, углеводный скелет).
Пример 4
Синтез бисгексадецил-N-({3-[N′-β-D-глюкопиранозилокси]-карбамоил}-пропионил)-L-глутамата. К раствору 50 мг (0,169 ммоль) N-(β-D-глюкопиранозил)-3-(карбокси)-пропанамида в ДМФА добавляли DCC и DMAP в качестве катализатора. Смесь охлаждали до 0° в бане со льдом в течение 15 мин. Добавляли 100 мг (0,169 ммоль) дигексадецилового эфира L-глутаминовой кислоты в ДМФА. Реакцию проводили в инертной атмосфере в течение 3-х дней. Выпавший осадок отфильтровывали, остаток растворителя удаляли в вакууме. Продукт выделяли при помощи препаративной хроматографии в системах хлороформ-метанол 5:1, толуол-ацетонитрил 1:1. Получали 33 мг (21%) продукта, Rf (хлороформ-метанол 1:1) 0,4. ИК-спектр (в пленке, vmax, см-1): 3322 (ОН), 2917, 2848 (С-Н), 1731 (С=O), 1695 (I амидная полоса, С=O), 1650 (II амидная полоса, NH), 1467 (С-О), 1197-1035 (С-О, 4 полосы, углеводный скелет). Масс-спектр, m/z (Iотн, %): 895.6 [М+Na]+(100), 429.2 (18.9), 256.7 (25), 163.4 (21.8), 147.2 (36), 71.7 (34).
Пример 5
Синтез бисгексадецил-N-({3-[N′-2,3,4,6-тетра-O-пропинил-β-D-глюкопиранозилокси]-карбамоил}-пропионил)-L-глутамата. К 33 мг (0,0368 ммоль) бисгексадецил-N-({3-[N′-β-D-глюкопиранозилокси]-карбамоил}-пропионил)-L-глутамата в 3 мл ДМФА прибавляли порциями гидрид натрия 88 мг (0,368 ммоль) при температуре 0°С. Через 20 мин по каплям добавляли пропаргилбромид. Оставляли на магнитной мешалке при 0°C на 12 ч. Растворитель удаляли в вакууме. Остаток растворяли в этилацетате и промывали водой. Органический слой сушили над безводным Na2SO4. Остаток растворителя удаляли в вакууме. Очистку проводили при помощи препаративной хроматографии в системе гексан-этилацетат 1:1. Получали 20 мг (53%) соединения в виде аморфного вещества, Rf (гексан-этилацетат 2:1) 0,8. ИК-спектр (вазелиновое масло, см-1): 3324 (О-Н), 2920, 2850 (С-Н), 2130 (С≡С), 1700 (С=O), 1619 (C=O, I амидная полоса), 1580 (NH, II амидная полоса), 1420 (С-О), 1189-1070 (С-О, углеводный скелет). Масс-спектр, m/z (Iотн, %): 1025.497 (100) [М]+, 989.933 (11), 978.933 (9), 816.801 (10), 716.341 (12).
Пример 6
Синтез 2,3,4,6-тетра-O-ацетил-1-O-(2-бромэтил)-α-D-маннопиранозида. К раствору 3 г (7,69 ммоль) 1,2,3,4,6-пента-O-ацетил-α-D-маннопиранозида в 22 мл безводного хлористого метилена прибавляли 0,710 мл (8,84 ммоль) эфиратного комплекса трехфтористого бора. Через 15 мин к реакционной смеси прибавляли 0,985 мл (8,84 ммоль) 2-бромэтанола и выдерживали 12 ч при 20°C. Реакционную массу перемешивали при нагревании до 40°C. Затем реакционную массу нейтрализовывали 25%-ным раствором аммиака до pH=7. Раствор промывали 3×100 мл воды, органический слой сушили через складчатый фильтр, смоченный хлористым метиленом, растворитель удаляли в вакууме.
Образовавшееся после обработки реакционной массы масло хроматографировали с помощью колоночной хроматографии, элюируя системой растворителей толуол-ацетонитрил 9:1. Получали 2,2 г (59%) чистого соединения. Rf (хлороформ-метанол 9:1) 0,45, Tпл=56-58°C.
ИК- спектр (vmax, см-1): 2836 (С-Н), 1755 (С=O), 1460, 1380 (С-Н), 1218 (С-O), 1158-1035 (С-О, 4 полосы, углеводный скелет).
1H-ЯМР-спектр (CDCl3, δ, м.д.): 1.99, 2.05, 2.10, 2.16 (с, 12Н, COCH3), 3.50-3.53 (т, 2Н, OCH2CH2), 3.86-4.0 (м, 2Н, OCH2CH2), 4.10-4.16 (м, 2Н, Н-6), 4.24-4.30 (дд, 1H, Н-5), 4.87 (д, 1H, Н-1, J12 1,88), 5.26-5.31 (м, 2Н, Н-2, Н-4), 5.34-5.37 (дд, 1Н, Н-3).
Пример 7
Синтез 1-O-(2-азидоэтил)-α-D-маннопиранозида. К раствору 200 мг (0,40 ммоль) 2,3,4,6-тетра-O-ацетил-1-O-(2-бромэтил)-α-D-маннопиранозида в 5 мл безводного диметилформамида добавляли 80 мг (1,20 ммоль) азида натрия и перемешивали при комнатной температуре в течение 20 ч. В раствор добавляли избыток безводного диэтилового эфира. Образовавшийся осадок отфильтровывали, растворитель удаляли в вакууме.
После удаления растворителя получали 157 мг (85,5%) (2,3,4,6-тетра-O-ацетил-1-O-(2-азидоэтил)-α-D-маннопиранозида в виде аморфного вещества, Rf (толуол-ацетонитрил 2:1) 0,65.
ИК-спектр (vmax, см-1): 2831 (С-Н), 2099 (N=N), 1742 (С=O), 1460, 1380 (С-Н), 1250 (С-N), 1210 (С-О), 1217-1040 (С-O, 4 полосы, углеводный скелет).
1H-ЯМР-спектр (CDCl3, δ, м.д.): 1.99, 2.05, 2.10, 2.16 (с, 12Н, СОСН3), 3.40-3.52 (м, 2Н, OCH2CH2), 3.62-3.9 (м, 2Н, OCH2CH2) 4.0-4.15 (м, 2Н, Н-6), 4.24-4.32 (дд, 1Н, Н-5), 4.85-4.87 (д, 1H, Н-1, J12 1,88), 5.26-5.30 (м, 2Н, Н-2, Н-4), 5.34-5.38 (дд, 1Н, Н-3).
К раствору 200 мг 2,3,4,6-тетра-O-ацетил-1-O-(2-азидоэтил)-β-D-маннопиранозида в 5 мл безводного метанола при перемешивании при комнатной температуре прибавляли 0,1 мл свежеприготовленного 0,1 М раствора метилата натрия в метаноле до достижения pH 8 и нагревали смесь на водяной бане. Обессоливали раствор ионообменной смолой КУ-2 (Н+-форма), отфильтровывали и удаляли растворитель в вакууме.
Получали 0,125 г (82%) 1-O-(2-азидоэтил)-α-D-маннопиранозида в виде аморфного вещества, Rf (хлороформ-метанол 2:1) 0,4.
ИК-спектр: (vmax, см-1): 3325 (О-Н), 2900, 1435, 1340 (С-Н), 1215 (С-О), 1140-1030 (С-О, 4 полосы, углеводный скелет).
Пример 8
Получение гликоконъюгата с четырьмя терминальными остатками лактозы. К 20 мг (0,323 ммоль) 1-O-(2-азидоэтил)-лактозида в ацетонитриле добавляли 80 мг (0,094 ммоль) производного бисгексадецил-N-({3-[N′-2,3,4,6-тетра-O-пропинил-β-D-глюкопиранозилокси]-карбамоил}-пропионил)-L-глутамата, растворенного в смеси ацетонитрил-ДМФА (2:1), каталитическое количество CuI, DIPEA и перемешивали при комнатной температуре в течение 7 суток в атмосфере аргона. Реакционную массу отфильтровывали от осадка. Растворитель удаляли в вакууме. Очистку проводили при помощи препаративной хроматографии в системе хлороформ-метанол 9:1. Получали 30 мг (35%) продукта в виде аморфного вещества, Rf (В) 0,8. ИК-спектр (в пленке, vmax,см-1): 3374 (ОН), 2929, 2854, 1477, 1444 (С-Н), 2700 (C=N), 1749 (С=O), 1683 (C=O, I амидная полоса), 1627 (NH, II амидная полоса), 1400 (N=N), 1280 (С-О), 1255 (C-N), 1249-1074 (С-О, 4 полосы, углеводный скелет). Масс-спектр (Iотн, %) m/z: 2670,550 (29) [М]+, 531.63 (39), 530.21 (100), 259.74 (56.1), 199.01 (10), 149.1 (51.9).
Аналогично из 50 мг (0.43 ммоль) 2,3,4,6-тетра-O-ацетил-1-O-(2-азидоэтил)-α-D-маннопиранозида и 90 мг (0,11 ммоль) производного бисгексадецил-N-({3-[N′-2,3,4,6-тетра-O-пропинил-β-D-глюкопиранозилокси]-карбамоил}-пропионил)-L-глутамата получали 48 мг (39%) тетравалентного гликоконъюгата с терминальными остатками D-маннозы. Масс-спектр (Iотн, %) m/z: 1949,950 (100) [М]+, 480.63 (39), 351.70 (32.9).
Для изучения возможности применения синтезированных соединений как маркерных молекул в составе липосом было исследовано взаимодействие модифицированных тетравалентным неогликоконъюгатом с остатками лактозы везикул с галактозосвязывающим лектином клещевины Ricinus communis (RCA1) с использованием спектрофотометрического метода анализа.
Известно, что гликопротеины с концевыми галактозильными остатками, дающими положительную реакцию преципитации с лектином RCA1, обладают также сродством и к другим галактозоспецифичным лектинам, в частности к асиалогликопротеиновым рецепторам гепатоцитов человека и животных. В связи с этим логично предположить, что гликозилированные липосомы, преципитирующие в присутствии RCA1, будут также характеризоваться повышенной аффинностью к гепатоцитам.
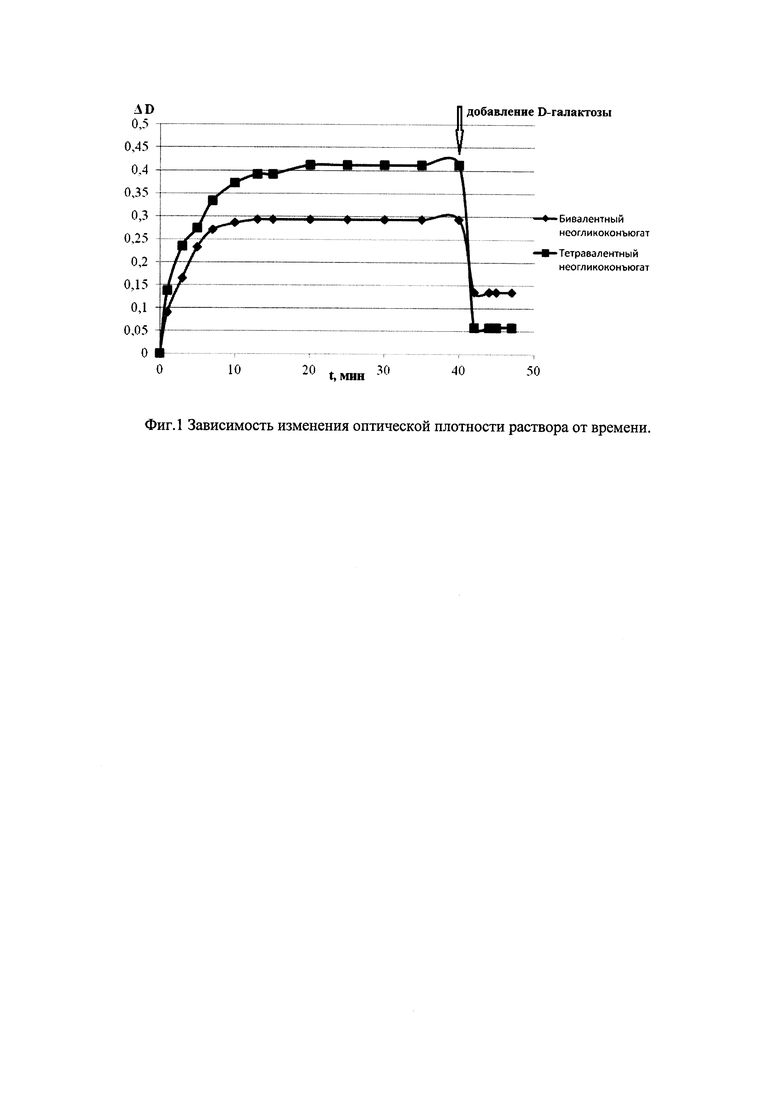
Было исследовано взаимодействие липосом, модифицированных би- и тетравалентными гликоконъюгатами, содержащими производные лактозы в качестве терминальных остатков, с лектином клещевины RCA1. Тетравалентное соединение показало большую эффективность связывания, чем бивалентное соединение. Это подтверждает литературные данные об увеличении связывания с рецептором с ростом числа углеводных остатков. Добавление D-галактозы приводило к снижению оптической плотности, что свидетельствует о специфическом взаимодействии конъюгатов с лектином RCA1.
Таким образом, полученные данные свидетельствуют о наличии углеводной специфичности лектина RCA1 к терминальной галактозе сформированных модельных систем. При этом важным фактором, влияющим на эффективность связывания маркер-рецептор, является количество терминальных углеводных остатков. Например, с увеличением числа остатков галактозы увеличилось сродство модифицированной липосомы к лектину RCA1.
| название | год | авторы | номер документа |
|---|---|---|---|
| БИВАЛЕНТНЫЕ НЕОГЛИКОКОНЪЮГАТЫ НА ОСНОВЕ ДИЭФИРА L-ГЛУТАМИНОВОЙ КИСЛОТЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2011 |
|
RU2462471C1 |
| УГЛЕВОДСОДЕРЖАЩИЕ КАТИОННЫЕ АМФИФИЛЫ, ОБЛАДАЮЩИЕ СПОСОБНОСТЬЮ ДОСТАВЛЯТЬ НУКЛЕИНОВЫЕ КИСЛОТЫ В КЛЕТКИ МЛЕКОПИТАЮЩИХ | 2009 |
|
RU2394834C1 |
| ЧУВСТВИТЕЛЬНЫЕ К ГЛЮКОЗЕ КОНЪЮГАТЫ ИНСУЛИНА | 2014 |
|
RU2676307C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА, МЕДИЦИНСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ, ИХ ПРИМЕНЕНИЕ В МЕДИЦИНЕ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ | 2003 |
|
RU2369613C2 |
| СПОСОБ ПОЛУЧЕНИЯ 6-O-[β-D-(2,3,4,6-ТЕТРА-O-АЦЕТИЛ)ГЛИКОПИРАНОЗИЛ]-d,l-α-ТОКОФЕРОЛОВ | 2007 |
|
RU2350620C1 |
| ПРОИЗВОДНЫЕ ГЛЮКОПИРАНОЗИЛОКСИПИРАЗОЛА И ИХ ПРИМЕНЕНИЕ В ЛЕКАРСТВЕННЫХ СРЕДСТВАХ | 2001 |
|
RU2317302C2 |
| ПРИМЕНЕНИЕ 5-МЕТИЛ-1,3-БЕНЗДИОЛА ИЛИ ЕГО ПРОИЗВОДНЫХ В ИЗГОТОВЛЕНИИ ЛЕКАРСТВА И ФУНКЦИОНАЛЬНОГО ПРОДУКТА ПИТАНИЯ ДЛЯ ЛЕЧЕНИЯ ИЛИ ПРЕДУПРЕЖДЕНИЯ ДЕПРЕССИИ | 2008 |
|
RU2447887C2 |
| ГЛЮКОПИРАНОЗИЛОКСИПИРАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ЭТИ ПРОИЗВОДНЫЕ, И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ | 2000 |
|
RU2232767C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА, ЛЕКАРСТВЕННЫЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ЭТИ ПРОИЗВОДНЫЕ, ИХ ПРИМЕНЕНИЕ В МЕДИЦИНЕ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ | 2003 |
|
RU2356906C2 |
| Средство, обладающее противовоспалительным и анальгезирующим действием | 2017 |
|
RU2657803C1 |
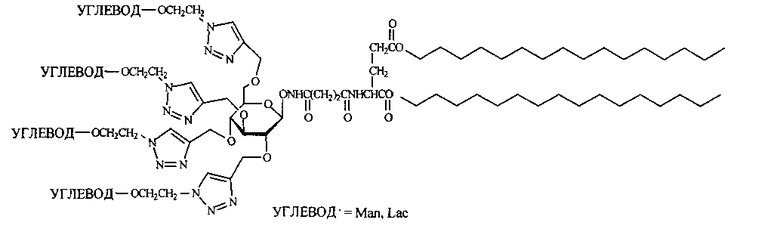
Изобретение относится к получению тетравалентных неогликоконъюгатов с углеводным разветвляющим ядром и производным L-глутаминовой кислоты в гидрофобном блоке, пригодных для приготовления липосом, формулы:
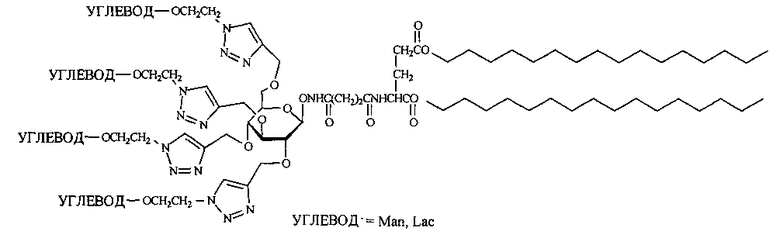
Предложены новые тетравалентные неогликоконъюгаты, обеспечивающие повышение аффинности углеводных остатков к рецепторам, а также эффективный способ их получения, включающий конъюгацию азидоэтильного производного углевода, выбранного из 1-O-(2-азидоэтил)-α-D-маннопиранозида или 1-O-(2-азидоэтил)лактозида, с бисгексадецил-N-({3-[N′-2,3,4,6-тетра-O-пропинил-β-D-глюкопиранозилокси]-карбамоил}-пропионил)-L-глутаматом. 2 н.п. ф-лы, 1 ил., 8 пр.
1. Тетравалентные неогликоконъюгаты с углеводным разветвляющим ядром и производным L-глутаминовой кислоты в гидрофобном блоке:
2. Способ получения тетравалентных неогликоконъюгатов, охарактеризованных в п. 1, включающий конъюгацию азидоэтильного производного углевода, выбранного из 1-O-(2-азидоэтил)-α-D-маннопиранозида или 1-O-(2-азидоэтил) лактозида, с бисгексадецил-N-({3-[N′-2,3,4,6-тетра-O-пропинил-β-D-глюкопиранозилокси]-карбамоил}-пропионил)-L-глутаматом.
| НОСИТЕЛИ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2008 |
|
RU2505315C2 |
| WO 1997023494 A1, 03.07.1997 | |||
| ГЛИНКОЗИДЫ, СПОСОБ ИХ СИНТЕЗИРОВАНИЯ, СПОСОБ ПОВЫШЕНИЯ БИОЛОГИЧЕСКОЙ ДОСТУПНОСТИ БИОЛОГИЧЕСКИ-АКТИВНОГО МАТЕРИАЛА, СПОСОБ ВОЗДЕЙСТВИЯ НА ЖИВОЙ ОРГАНИЗМ БИОЛОГИЧЕСКИ-АКТИВНОГО МАТЕРИАЛА | 1996 |
|
RU2141963C1 |

