1. ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Изобретение относится к доставке и конъюгированию биологически-активных веществ, включая главным образом использование повышенной биологической доступности и клеточного поглощения для воздействия на такую доставку и конъюгирование.
2. ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Существенной проблемой клинической фармакологии является избирательная доставка определенных биологически-активных соединений к клеткам-мишеням организма. В большинстве случаев требуемые вещества только пассивно и частично диффундируют в клетки-мишени, а концентрации в плазме крови, необходимые для достижения значительного внутриклеточного уровня, труднодостижимы из-за токсичности, выведения и расщепления печенью, почками и другими органами или жидкостями. В случае центральной нервной системы проблема часто обостряется из-за наличия гематоэнцефалического барьера (ВВВ), а в опухолях может обостряться дополнительно из-за низкой или неэффективной васкуляризации. Дополнительные трудности могут возникнуть из-за усвоения и расщепления биологически-активных соединений в кишечнике, и/или плохого проникновения большинства таких соединений через стенку кишечника. Также трудности возникают вследствие случайной доставки биологически-активных соединений к нецелевым клеткам.
Проблемы, касающиеся точной доставки, имели место в случае многих биологически-активных соединений, включая высокотоксичные противоопухолевые лекарственные препараты, такие как доксорубицин и метотрексат, противовирусные лекарственные препараты, такие как арабинозилцитозин и арабинозиладенозин, и противопаразитарные лекарственные препараты, такие как хлорокин и пириметамин, для которых указанная специфичность особенно важна. Проблемы доставки имели место также в отношении веществ, которые лекарственными препаратами не считаются, такие как радиоактивные маркеры и контрастные вещества, но которые хотелось бы отнести к биологически-активным веществам вследствие того, что они оказывают воздействие на живой организм или могут в нем использоваться.
Известно, что углеводная часть гликопротеинов играет огромную роль в абсорбции, транспорте и последующем распределении по тканям в теле, и ранее некоторые исследователи пытались решить проблему транспорта путем введения биологически-активных материалов, связанных с сахарами. В этой области были получены значительные результаты (Mizuma T., et al, Intestinal Active Absorption of Sugar-Conjugated Compounds by Glucose Transport System: Implication of Improvement of Poorly Absorbable Drugs, Biochem. Pharma., 43 (9) 2037-2039).
В другой работе, где исследовалось дифференциальное конъюгирование различных конъюгатов сахарид-поли-L-лизин, сообщается, что конъюгаты галактозы предпочтительно доставляются к печени, коньюгаты маннозы и фруктозы предпочтительно доставляются в ретикулоэндотелиальную систему, а конъюгаты ксилозы предпочтительно доставляются к печени и легким (Gonsho, A., et.al, Tissue-Targeting Ability of Saccharide-Poly (L-Lysine) Conjugates, Biol. Pharm. Bull., 17 (2) 275-282 (1994).
Исследование в этой области подразделяется на три основных направления:
(1) терапия, при которой сахар никоим образом не связан с биологически-активным соединением;
(2) композиции, в которых сахар ковалентно присоединен либо непосредственно, либо опосредованно, через гликозидную связь (связывая C1 атом сахара), к биологически-активному соединению;
(3) композиции, в которых лекарственный препарат присоединен к амфифилу, например в виде включения внутри липосомы, покрытой оболочкой из сахара.
Исследование по первому направлению проводилось с использованием маннита для преодоления гематоэнцефалического барьера. Концентрированные растворы маннита, например, вводили в сонную артерию для разрушения гематоэнцефалического барьера на срок, достаточно продолжительный для того, чтобы дать возможность метотрексату или другим сильным химиотерапевтическим агентам проникать в опухоли головного мозга (Angier, N. Discover, pp. 67-72). Хотя по этому направлению и были получены значительные результаты, оно имеет некоторые недостатки. В частности, открытие гематоэнцефалического барьера является неспецифичным и дает возможность проникать в мозг многим опасным веществам вместе с необходимым соединением. Кроме того, эта методика главным образом применима для центральной нервной системы, а в широком масштабе для других систем неприменима.
Исследования по второму направлению, непосредственное или опосредованное гликозидное присоединение cахаров к лекарственным препаратам, имеют гораздо большее значение. Так, цитотоксический лекарственный препарат, метотрексат (МТС), был сконъюгирован с маннозилированным бычьим сывороточным альбумином (BSA) и соединение, полученное в итоге, узнавалось рецепторами маннозы, имеющимися на поверхности макрофагов (Chakraborty, P. et al, Sugar Receptors Mediated Drug Delivery to Macrophages in the Therapy of Experimental Visceral Leishmaniasis, Biochem. Biophys. Res. Comm., 166(1) 404-410 (1990)). Инсулин был гликозилирован для получения преимущества в конкурентном связывании между глюкозой и гликозилированным инсулином (Seminoff, L.A. et al, A Selfregulating insulin delivery system. Characterization of a synthetic glycosylated insulin derivative, Int'l J. Pharm, 54(1989) 241-249)). Анти-ВИЧ препарат 3'-азидо-3'-дезокситимидин (AZT) также был посредством глюкозидирования сшит с сывороточным альбумином человека (HSA) и остатками различных сахаров с получением маннозо-, фруктозо-, галактозо- и глюкозо-неогликопротеинов (Molema, G., Targeting of Antiviral Drugs To T4-Lymphocytes, Biochem. Pharm, 40(12) 2603-2610 (1990)). Противовоспалительный агент, напроксин, также был посредством глюкозидирования присоединен к сывороточному альбумину человека (HSA) с сахарным остатком (Franssen, E.J.F., Hepatic and Intrahepatic Targeting of an Anti-inflammatory Agent With Human Serum Albumin and Neoglycoproteins as Carrier Molecules, Biochem. Pharm., 45(6) 1215-1226 (1993)). В последних двух случаях гликозидная связь формируется с сывороточным альбумином человека (HSA) и, таким образом, только опосредованно - с лекарственными препаратами - AZT или напроксином. В 1994 г. группа японских ученых сообщила о способе радиойодирования дигоксина с использованием гликозидов (Takemuru, Y., et al, Development of Glycoside-Bound Radiopharmaceuticals: Novel Radioiodination Method for Digoxin, Biol. Pharm. Bull., 17(1) 97-101 (1974)). Сахара также были ковалентно присоединены к поли-L-лизину посредством гликозидной связи (Monsigny, М., et al, Sugar Specific Delivery of Drugs, Oligonucleotides and Genes, Targeting of Drugs, 4 31-50 (ed. by G. Gregoriadis et al., Putnam Press, N.Y., 1994)).
В дополнение к этим синтетическим конъюгатам сахаров в природе существуют многочисленные примеры ковалентного присоединения сахара к биологически-активной части. Гликозиды, например, являются продуктом конденсации сахаров с различными органическими гидрокси- (или изредка амино- или тиол-) соединениями, в которых OH гемиацетальной части углеводорода участвует в конденсации (Remington, The Science and Practice of Pharmacy, 19th ed. 386-387 (Mack Publ., Co., Easton, PA 1995)). Действительно, многие широко известные биологически-активные соединения являются гликозидами, включая амигдалин, цимарин, дигитоксин, кубаин, рутин и салицин. Существуют также эндогенные гликозиды, включая ганглиозиды, сахара нуклеотидов и адгезивные молекулы нервных клеток. Другие встречающиеся в природе соединения, обычно не классифицируемые как гликозиды, на самом деле имеют гликозидные связи в своей структуре. Примеры включают антибиотики - гентамицин, амикацин, нетилмицин, тобрамицин, новобиоцин и стрептомицин, глюкоалкалоиды, такие как соланин, и нуклеозиды, которые состоят из пуринового или пиримидинового основания, присоединенного к D-рибозе D-2-дезоксирибозе.
Исследование как природных, так и синтетических соединений показывает, что ковалентное прямое или опосредованное гликозидное присоединение сахаров к лекарственным веществам является успешной стратегией доставки лекарственных веществ. Несмотря на то, что биологически-активные соединения с присоединенными сахарами встречаются достаточно часто и хорошо изучены, количество соединений, которые могут быть получены посредством использования стандартной гликозидной связи, ограничено. Например, в некоторых случаях такое присоединение полностью невозможно, поскольку из-за величины и пространственных затруднений получившееся в результате сконъюгированное соединение не может быть распознано и точно доставлено соответствующими клеточными переносчиками. В других случаях гликозидные связи являются очень неустойчивыми и легко гидролизуются гликозидазами.
В отношении третьего направления известно, что гликозид-несущие липосомы могут использоваться in vivo для доставки лекарственных веществ специфически к макрофагам (Medda, S. et al., Sugar-coated Liposomes: a Novel Delivery System for Increasing Drug Efficacy and Reduced Drug Toxicity, Biotechnol. Appl, Biochem, 17 37-47 (1993)). Также были синтезированы анионные амфифилы с эфирной связью, образованной фосфорной кислотой между фторофильно-липофильной хвостовой частью и гидрофильной головной частью на основе сахара (Guillod, F. , et al., Amphiphilic Sugar Phosphates with Single or Double Perfluoroalkylated Hidrophobic Chains for Use in Oxigen and Drug Delivery Systems, Art. Cells, Blood Subs. , and Immob. Biotech., 22(4)1273-1279 (1994)). В 1995 г. был опубликован научный анализ функций липосом с поверхностными гликолипидами (Jones, М., The Surface Properties of Phospholipid Liposome Systems and Their Characterization, Advanced in Colloid and Interface Science, 54 93-128 (1995)). Однако, эта третья категория соединений, поначалу казавшаяся многообещающей, имеет те же недостатки, что и гликозиды и неогликопротеины, о которых говорилось выше. Стандартная гликозидная связь, на основе которой образованы эти соединения, является слишком ограничивающей. Кроме того, существуют постоянные проблемы синтеза и/или очистки при получении стереохимически чистых или α или β-гликозидных связей.
Учитывая недостатки доступных в настоящее время способов и составов для улучшения клеточного поглощения и конъюгирования, обеспечивающего направленную доставку, например тканеспецифического поглощения, представляется перспективным создание новых способов и составов для присоединения сахаров к биологически активным соединениям.
3. РЕЗЮМЕ
Предлагаются способы и составы, которые повышают клеточное поглощение биологически-активных материалов посредством ковалентного присоединения таких соединений к углеводной части через химические линкеры с использованием не-гликозидных связей. Многочисленные углеводороды, линкеры и биологически-активные материалы можно объединить, образовав новые составы, которые здесь в совокупности именуются глинкозидами. Предпочтительные глинкозиды предпочтительно захватываются рецепторами для глюкозы и/или другими клеточными рецепторами для моносахаридов и, уже попав внутрь клетки, глинкозиды расщепляются на производное сахара, линкер или линкерные фрагменты и биологически-активное соединение. Различные аспекты данного изобретения включают в себя глинкозидные составы, способы синтеза глинкозидов и способы лечения заболеваний с применением глинкозидов.
4. КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Фиг.1 - схема, отображающая пример синтеза производных линкера.
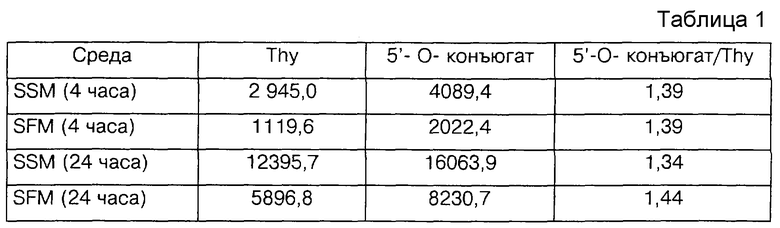
Фиг.2 - схема, отображающая пример синтеза 3-O- и 6-O-D- галактозо-линкера в виде промежуточных соединений.
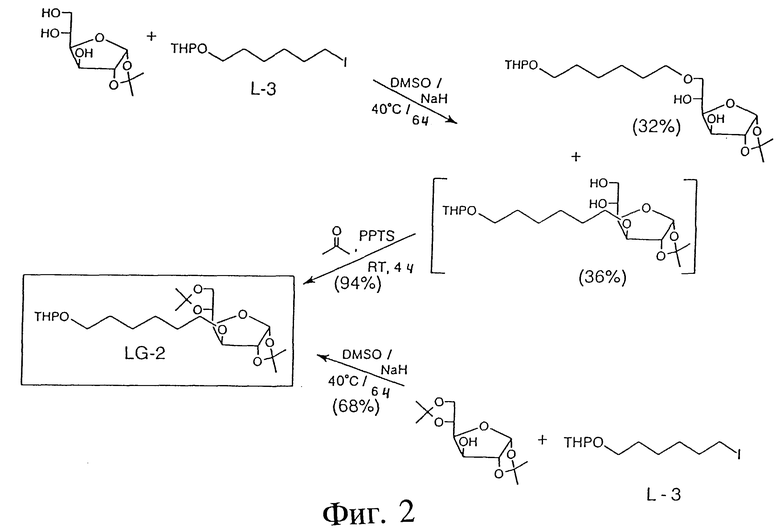
Фиг. 3 - схема, отображающая пример синтеза 3-O- и 6-O-D-галактозо-линкера в виде реактивных промежуточных соединений.
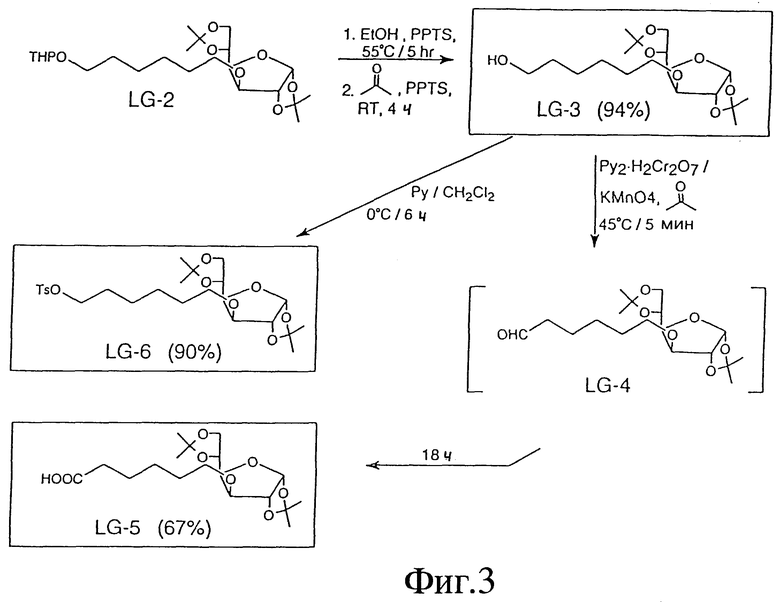
Фиг. 4 - схема, отображающая пример синтеза конъюгатов тимидина с LG-5 посредством сложно-эфирной связи.
Фиг. 5 - схема, отображающая пример конъюгации виразола (VirazoleTM) с LG-5 посредством сложно-эфирной связи.
Фиг. 6 - схема, отображающая пример конъюгации виразола (VirazoleTM) с LG-6 посредством эфирной связи.
Фиг. 7 - схема, отображающая пример синтеза 6-O-D-галактозо-линкера в виде промежуточных соединений.
Фиг.8 - схема, отображающая пример синтеза 4-O- и 6-O-D-маннозо-линкеров в виде промежуточных соединений.
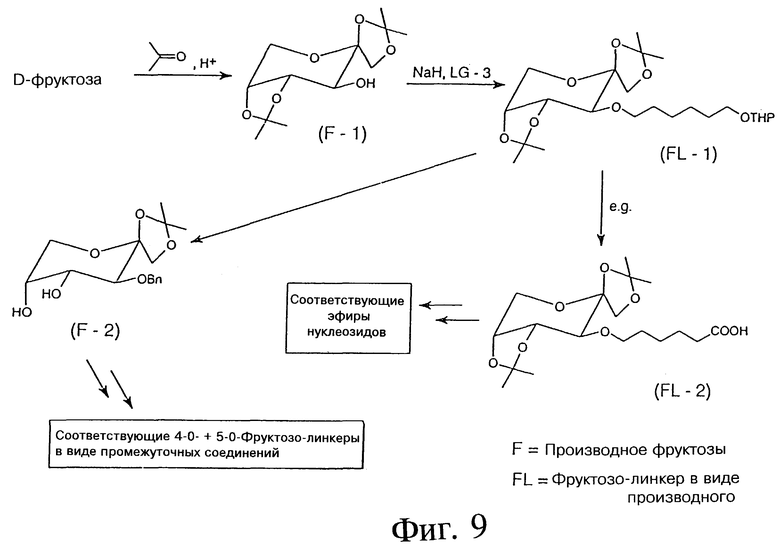
Фиг. 9 - схема, отображающая пример синтеза 3-O-, 4-O- и 5-O-D-фруктозо-линкеров в виде промежуточных соединений.
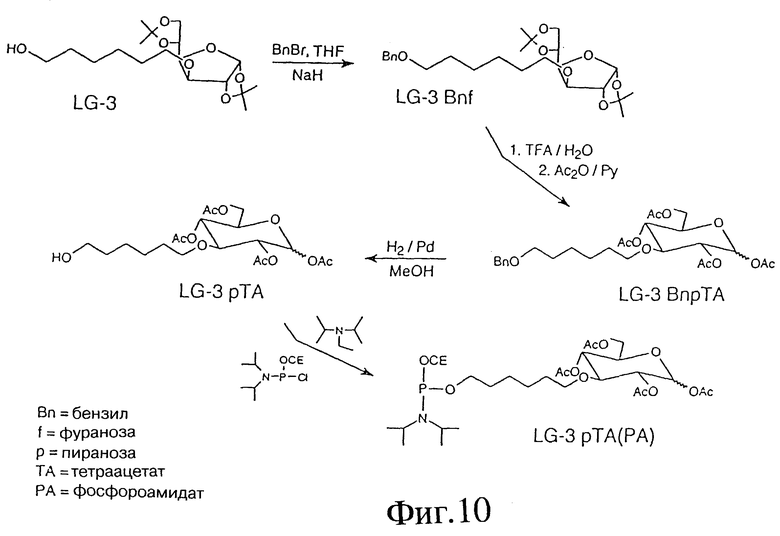
Фиг. 10 - схема, отображающая пример синтеза глюкозо-линкера в виде фосфорамидатных промежуточных соединений для автоматизированного синтеза олигодезоксинуклеотидов.
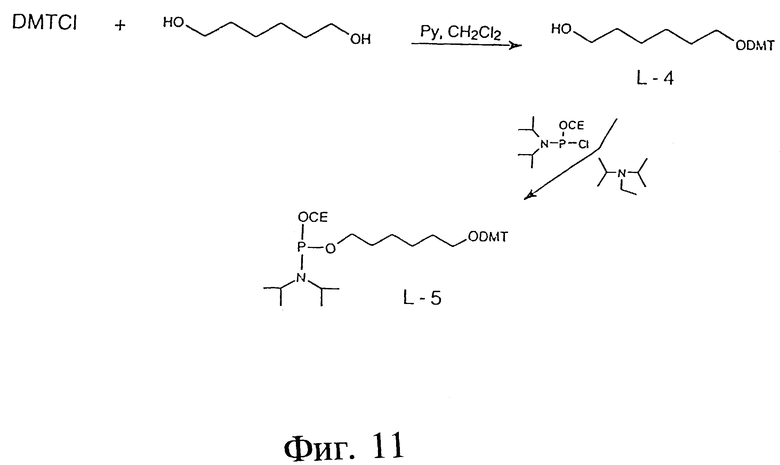
Фиг. 11 - схема, отображающая пример синтеза фосфорамидатов производных линкера для автоматизированного синтеза олигодезоксинуклеотидов.
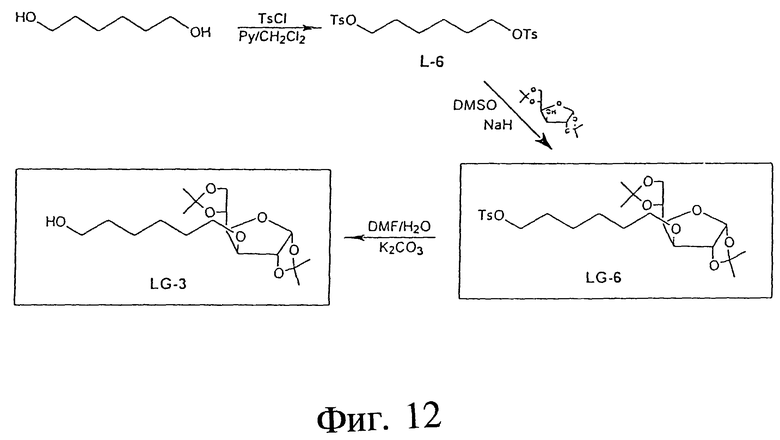
Фиг. 12 - схема, отображающая альтернативный синтез 1,2:5,6- ди-O-изопропилиден-3-O-(6'-гидрокси)-гексил- α -D-глюкофуранозы (LG-3).
Фиг. 13 - схема, отображающая пример синтеза олигодезоксинуклеотидов (ODN), модифицированных в 3' и 5' положениях (ICN 16967).
Фиг. 14 - кривые абсорбции TM' и УФ для модифицированных (14 а,в) (ICN 16967) и немодифицированных (14 б,г) олигонуклеотидов.
5. ПОДРОБНОЕ ОПИСАНИЕ
Согласно новой терминологии, применяемой здесь, глинкозиды имеют общую структуру MS - LINK - BAM, где MS - моносахарид, LINK - линкер, а ВАМ - биологически-активный материал, MS ковалентно присоединен к LINK по другому положению, чем MS C1, a LINK ковалентно связан с BAM. Существует большое количество вариаций глинкозидов. Например, в глинкозиде может быть использован любой моносахарид, включая моносахарид, имеющий от трех до восьми и более атомов углерода. LINK-часть глинкозида также может варьироваться и может являться прямым или разветвленным алкилом, замещенным алкилом, аралкилом, замещенным аралкилом, арилом и замещенным арилом. BAM-часть может состоять из различных биологически-активных молекул, включая следующие:
желудочно-кишечные лекарственные средства и лекарственные препараты, улучшающие функцию печени
гематологические лекарственные средства
сердечно-сосудистые лекарственные средства лекарственные средства, улучшающие дыхание
симпатикомиметики
холиномиметики
адренергические лекарственные средства и адренергические нейроноблокаторы
противосудорожные и спазмолитические средства
костно-мышечные релаксанты
маточные средства и средства от мигрени
гормоны
анестетики общего и местного действия
седативные и снотворные средства
противоэпилептические средства
психотропные средства
противовоспалительные средства
стимуляторы ЦНС
Соответствующие MS-LINK и LINK-BAM связи также широко варьируются и как пример могут независимо состоять из простых эфирных, сложных эфирных, амидных, дисульфидных гемиацетальных, гемикетальных, ацетальных и кетальных связей.
В представленных предпочтительных глинкозидах использованы встречающиеся в природе сахара - рибоза (5 атомов углерода) или гексоза (6 атомов углерода), линкеры с прямой цепью, содержащие от четырех до шестнадцати атомов углерода, бифункциональные α,ω линкеры, такие как гексан, и известные биологически-активные лекарственные препараты. Каждая MS-LINK и LINK-BAM связь предпочтительно расщепляется в физиологических условиях таким образом, что глинкозид действует как про-препарат, доставляя активный или активируемый лекарственный препарат в позицию-мишень на поверхности или внутри клетки-мишени. Представленные предпочтительные воплощения включают глинкозиды, в которых связь LINK-BAM прочнее, чем связь MS-LINK.
Глинкозиды имеют много преимуществ по сравнению с гликозидами, включая следующие:
(1) Моносахарид может быть химически присоединен посредством специально сконструированного α,ω бифункционального линкера к ВАМ по любому из различных OH заместителей (кроме C1), находящихся на моносахариде.
(2) Использование линкера может исключить синтетические стереохимические проблемы.
(3) В отношении гликозидов, и в частности гликозидов, в которых ВАМ прямо присоединена к моносахариду, пространственные затруднения значительно или полностью устранены. Это облегчает прохождение глинкозида через специфические мембранные белковые транспортные пути, такие как одна из систем глюкозного транспорта. Глутаминовая транспортная система (GLUT-1) является примером такой системы.
(4) Относительная прочность связей моносахарид-линкер и линкер-BAM может изменяться таким образом, что ВАМ может быть освобожден в определенной точке при транспорте.
(5) Глинкозиды также обеспечивают улучшенное прохождение через гематоэнцефалический барьер.
Синтез глинкозидов
Для того, чтобы химически присоединить углеводы к биологически-активному соединению (соединениям), выбранные функциональные группы обоих химических соединений могут быть использованы для их ковалентного связывания посредством специфического химического линкера. Весьма часто функциональные группы в углеводах и биологически-активных веществах включают гидроксильную, амино-, меркапто-, карбонильную, карбоксильную и амидо-функции. Химические линкеры в большинстве случаев имеют альфа, омега бифункциональные алкильные цепочки. Очевидно, существует гораздо больше функциональных групп и химических связей, которые можно использовать для этих целей.
В общем, глинкозиды могут быть получены по следующей схеме:
(1) Подготовить соответствующий линкер. В случае бифункционального α,ω линкера, α-конец будет реактивным, а ω-конец будет защищенным.
(2) Выбрать моносахарид и химически связать одну из его не- С1-ОН групп с линкером, и
(3) Химически связать линкер с биологически-активным материалом. Типичные способы синтеза и глинкозидные соединения описываются в примерах, приведенных ниже. Естественно, подразумевается, что приведенные примеры являются только иллюстративными и не предназначены для ограничения объема изобретения.
ПРИМЕРЫ 1-3 - СИНТЕЗ ПРОИЗВОДНЫХ ЛИНКЕРОВ
На фиг.1 первый ряд показывает пример синтеза 1-O-п-толуол- сульфонил-1,6-гександиола (L-1) с использованием следующих реактивов: 1,6-гександиола= 1,6-HD; п-толуолсульфонилхлорида=TsCl; пиридина=Py; и дихлорметана CH2Cl2. В данной процедуре 1,6 HD (44 г, 0,372 моль) растворяли в смеси (80 мл) Py/CH2Cl2 (1:1). Получившийся раствор охлаждали до 0oC и добавляли TsCl (20 г, 0,105 моль). Полученную в результате реакции смесь сохраняли в течение 5 часов при 4oC. Затем раствор нейтрализовали 10%-ной водной HCl и экстрагировали CH2Cl2 (3x100 мл). Органический слой промывали рассолом, высушивали и выпаривали. Сырой продукт очищали с помощью флэш-хроматографии на силикагелевой колонне (с использованием ТСХ системы растворителей). L-1 (масло); выход: 20,6 г (72%). ТСХ: этилацетат/гексан (1:1); Rf: 0,56. Структуру и чистоту L-1 проверяли посредством 1H-ПМР-спектра (протонный магнитный резонанс).
Второй ряд показывает пример синтеза 1-O-п-толуолсульфонил-6-O-(2'-тетрагидропиранил)-1,6-гександиола (L-2) с использованием следующих реактивов: L-1; дигидропирана=DHP; п-толуолсульфоната пиридиния=PPTS (приготовленного из пиридиния (Py) и п-толуолсульфокислоты=TsOH); и CH2Cl2. В этой процедуре L-1 (10 г, 36,7 мМ), DHP (3,4 г, 40,3 мМ) и PPTS (1,0 г, 3,7 мМ) добавляли друг за другом к безводному CH2Cl2 (40 мл). Реакционную смесь перемешивали при комнатной температуре до завершения реакции (6-8 часов). Реакционную смесь промывали рассолом (3x20 мл), сушили и выпаривали. Сырой продукт очищался методом хроматографии на силикагелевой колонке (с использованием ТСХ системы растворителей). L-2 (масло); выход: 9,42 г (72%). ТСХ: 15% этилацетат, 1% триэтиламин в гексане; Rf=0,9: структуру и чистоту L-2 проверяли посредством 1H-ПМР-спектра.
Третий ряд показывает пример синтеза 1-O-(2'-тетрагидропиранил)-6-йодо-1-гексанола (L-3) с использованием следующих реактивов: L-2; Nal и ацетона. В этой процедуре смесь из L-2 (9,0 г, 25,2 мМ), Nal (11,3 г, 75 мМ) и сухого ацетона (100 мл) перемешивали при температуре 50oC до завершения реакции (8-12 часов). Ацетон выпаривали, а остаток обрабатывали водой (100 мл). Продукт эстрагировали с помощью CH2Cl2 (3x50 мл). Органический слой промывали рассолом (3 x на 50 мл), сушили и выпаривали. Продукт собирали посредством колоночной хроматографии на силикагеле (ТСХ система растворителей). L-3 (масло), выход: 5.0 г (63%). ТСХ: 5% триэтиламин в гексане, Rf: 0,50; 1H-ПМР - спектр (CDCl3) подтвердил структуру и чистоту L-3.
ПРИМЕР 4 - СИНТЕЗ 3-O- и 6-O-D-ГЛЮКОЗО-ЛИНКЕРА В ВИДЕ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ
Фиг.2 показывает пример синтеза 1,2-O-изопропилиден-3-O- (6'-тетрагидропиранилокси)-гексил-альфа-D-глюкофуранозы (LG-1a) и 1,2-O-изопропилиден-6-O-(6'-тетрагидропиранилокси)-гексил-альфа-D- глюкофуранозы (LB-1б) с использованием следующих реактивов: L-3; 1,2-O-изопропилиден-альфа-D-глюкофуранозы= IPGF; NaH; и диметилсульфоксида=DMSO. В этой процедуре IPGF (2,0 г, 9,08 мМ) растворяли в безводном DMSO (7 мл) и добавляли NaH (всего 545 мг, 13,6 мМ) несколькими небольшими порциями при комнатной температуре. Реакционную смесь выдерживали при 40oC в течение 0,5 часов и затем добавляли L-3 (2,27 г, 7,26 мМ). Получившуюся в результате смесь выдерживали при комнатной температуре в течение ночи и затем выливали в этилацетат (200 мл). Органический слой промывали рассолом (3x50 мл), сушили (безвод. Na2SO4) и выпаривали. Сырой продукт очищали и изомеры разделялись флэш-хроматографией на силикагелевой колонке (ТСХ система растворителей). LG-1a (масло); выход: 1,06 г, (36%) и LG-1б (масло); выход: 0,94 г, (32%). ТСХ: 50% этилацетат, 2,5% триэтиламин, 47,5% гексан: Rf(LG-1a): 0,34; Rf(LG-1б):0,47. 1H-ПМР - спектры обоих изомеров в CDCl3 соответствовали предполагаемым структурам (образцы были химически чистыми).
ПРИМЕР 5 - 1,2:5,6-ДИ-O-ИЗОПРОПИЛИДЕН-3-O- (6'-ТЕТРАГИДРОПИРАНИЛОКСИ)-ГЕКСИЛ-АЛЬФА-D-ГЛЮКОФУРАНОЗА (LG-2)
Фиг. 2 показывает также пример синтеза 1,2:5,6-ди-O-изопропилиден-3-O-(6'-тетрагидропиранилокси)-гексил-альфа-D- глюкофуранозы (LG-2) с использованием следующих реагентов: LG-1a; ацетона и PPTS (приготовленного из TsOH и Py). В этой процедуре смесь LG-1a (500 мг, 1,233 мМ), PPTS (67 мг, 0,247 мМ) и ацетона перемешивали при комнатной температуре до завершения реакции (6 часов). Ацетон выпаривали, а остаточное вещество подвергали хроматографии. LG-2 (масло). Выход; 490 мг (91%). ТСХ: 15% этилацетат, 5% триэтиламин, гексан 80%. Rf: 0.66. 1H-ПМР -спектр (CDCl3) подтверждает чистоту и структуру LG-2.
ПРИМЕР 6 АЛЬТЕРНАТИВНЫЙ СИНТЕЗ 1,2:5,6-ДИ-O-ИЗОПРОПИЛИДЕН-3- O-(6'-ТЕТРАГИДРО-ПИРАНИЛОКСИ)-ГЕКСИЛ-АЛЬФА-D-ГЛЮКОФУРАНОЗЫ
Альтернативный прямой синтез LG-2 может быть осуществлен при использовании следующих реактивов: 1,2:5,6-ди-O- изопропилиден-альфа-D-глюкофураноза= DIPGF L-3; NaH; и DMSO. В этой процедуре DIPGF (2,5 г, 9,6 мМ) растворяли в безводном DMSO (7 мл) и добавляли NaH (576 мг, 14,40 мМ) несколькими небольшими порциями при комнатной температуре. Реакционную смесь выдерживали при 40oC в течение 30 минут и затем добавляли L-3 (3,6 г, 11,52 мМ). Реакционную смесь выдерживали в течение ночи при комнатной температуре и затем выливали в этилацетат (200 мл). Органический слой промывали рассолом (3x50 мл), сушили (Na2SO4) и выпаривали. Продукт очищали посредством колоночной хроматографии на селикагеле. LG-2 (масло); выход; 2,42 г (58%). ТСХ: 15% этилацетат, 5% триэтиламин, 80% гексан. Rf: 0,66.
ПРИМЕР 7 - СИНТЕЗ 3-O-D-ГЛЮКОЗО-ЛИНКЕРА В ВИДЕ РЕАКТИВНЫХ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ
Фиг. 3 показывает пример синтеза 1,2:5,6-ди-O-изопропилиден-3-O-(6'-гидрокси)-гексил-альфа-D-глюкофуранозы (LG-3) с использованием следующих реактивов: LG-2; этанола (96%); PPTS и ацетона. В этой процедуре LG-2 (2,0 г, 4,49 мМ) и PPTS (245 мг, 0,90 мМ) растворяли в этаноле (100 мл) и реакционную смесь нагревали в течение 5 часов при 55oC. Затем этанол выпаривали. Оставшийся материал растворяли в ацетоне (100 мл) и перемешивали при комнатной температуре в течении 4 часов. После удаления ацетона под вакуумом осадок растворяли в этилацетате (50 мл), промывали рассолом (3x50 мл), высушивали (Na2SO4) и выпаривали. Продукт окончательно очищался методом флэш-хромотографии на двуокиси кремния. LG-3 (масло); выход: 1,52 г (94%). ТСХ: этилацетат/гексан (1:1): Rf:0,48: 1H-ПМР-спектр подтвердил чистоту и структуру LG-3.
ПРИМЕР 8 - 1,2:5,6-ДИ-O-ИЗОПРОПИЛИДЕН-3-O-(5'-ГИДРОКСИ-КАРБОНИЛ)-ПЕНТИЛ-АЛЬФА-D- ГЛЮКОФУРАНОЗА (LG-5)
Фиг. 3 также показывает пример синтеза 1,2:5,6-ди-O-изопропилиден-3-O-(5'-гидроксикарбонил)-пентил-альфа-D- глюкофуранозы (LG-5) с использованием следующих реактивов: LG-3; Py2H2Cr2O7 (бихромат пиридиния) = PDCh; KMnO4 и ацетона. В этой процедуре смесь LG-3 (1,0 г, 2,77 мМ), PDCh (2,13 г, 5,65 мМ), KMnO4 (0,89 г, 5,65 мМ) и ацетона (50 мл) перемешивали при 50oC до завершения реакции (7-8 часов). Мониторинг реакции посредством ТСХ показал, что почти полностью трансформация исходного LG-3 в промежуточный альдегид (LG-4) происходила всего через 5 минут (в отдельном опыте выделяли LG-4, и его чистота и структура подтверждалась соответствующим 1H-ПМР - спектром). После окончания окисления LG-5 до кислоты реакционную смесь фильтровали и фильтрат выпаривали. Продукт очищали методом флэш-хромотографии (система растворителей ТСХ). LG-5 (масло); выход: 700 мг (67%). ТСХ: этилацетат/гексан (1: 1); Rf: 0,23; 1H-ПМР - спектр (CDCl3) подтвердил чистоту и структуру LG-5.
ПРИМЕР 9 - 1,2: 5,6-ДИ-O-ИЗОПРОПИЛИДЕН-3-O-(6'-П-ТОЛУОЛ- СУЛЬФОНИЛОКСИ)-ГЕКСИЛ-АЛЬФА-D-ГЛЮКОФУРАНОЗА (LG-6)
Фиг. 3 также показывает пример синтеза 1,2:5,6-ди-O-изопропилиден-3-O-(6'-п-толуолсульфонилокси)-гексил-альфа-D- глюкофуранозы (LG-6) с использованием следующих реактивов: LG-3; TsCl; Py и CH2Cl2. В этой процедуре LG-3 (1,40 г, 3,884 мМ) растворяли в смеси пиридина (5 мл) и CH2Cl2 (10 мл). Реакционную смесь охлаждали до 0oC и добавляли TsCl (0,89 г, 4,668 мМ). Реакционную смесь выдерживали в течение 5 часов при 4oC, затем нейтрализовали водной 10%-ной HCl. Органический слой промывали рассолом, высушивали и выпаривали. Продукт очищали посредством колоночной хроматографии (система растворителей ТСХ). LG-6 (масло); выход: 1,8 г (90%). ТСХ: 33% этилацетат, 67% гексан. 1H-ПМР-спектр (CDCl3) подтвердил чистоту и структуру LG-6.
ПРИМЕР 10 - КОНЪЮГАТЫ ТИМИДИНА С LG-5 (ПОСРЕДСТВОМ СЛОЖНОЭФИРНОЙ СВЯЗИ)
Фиг. 4 показывает пример синтеза 3'-O- и 5'-O-эфиров тимидина с LG-5 с использованием следующих реактивов: тимидина: LG-5; бициклогексилкарбодиимида = DCC и диметиламинопиридина = DMAP. В этой процедуре смесь тимидина (100 мг, 0,413 мМ), LG-5 (233 мг, 0,620 мМ), DCC (128 мг, 0,620 мМ), DMAP (15,1 мг, 0,124 мМ) и Py (3 мл) выдерживали при 60oC в течение ночи, что было достаточно для завершения реакции. Py выпаривали в вакууме, а осадочное вещество подвергали хроматографии на силикагелевой колонке (система растворителей ТСХ). 5'-O- и 3'-O-эфиры тимидина (масла); выход: 68,7 мг (28%) и 59,0 мг (24%). ТСХ: 25% гексан, 75% этилацетат; Rf (5):0,40 и Rf (3): 0,25. 1H-ПМР - спектры (CDCl3) обоих изомеров соответствовали предполагаемым структурам.
ПРИМЕР 11 ОТЩЕПЛЕНИЕ ИЗОПРОПИЛИДЕНА (СНЯТИЕ ЗАЩИТЫ) ОТ РАЗДЕЛЕННЫХ СЛОЖНЫХ ЭФИРОВ ТИМИДИНА
Фиг. 4 показывает пример снятия защиты у разделенных сложных эфиров тимидина с использованием трифторуксусной кислоты= TFA. В этой процедуре разделенный (отдельный) сложный эфир тимидина (из предыдущего опыта), 30 мг, 0,05 мМ обрабатывали смесью TFA/вода (9/1, об/об) при комнатной температуре в течение 5 минут. Затем реакционную смесь выпаривали в вакууме, снова растворяли в 1 мл воды, фильтровали и снова выпаривали. Эфиры после снятия защиты (масла); выходы: 81-89%; ТСХ: 10% метанол, 2% уксусная кислота, 88% этилацетат: Rf: 0,17 (свободный 5'-ОН) и 0,20 (свободный 3'-ОН). 1H-ПМР-спектры согласовались с предполагаемой структурой.
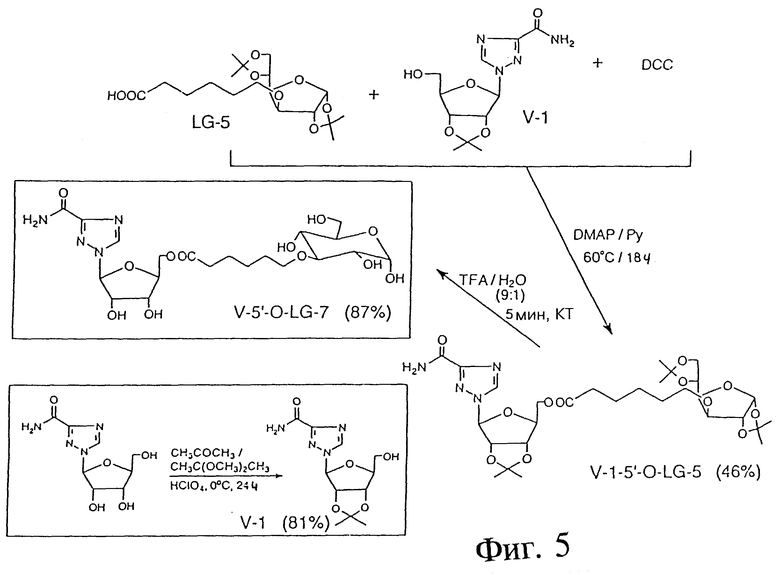
ПРИМЕР 12 - КОНЪЮГАЦИЯ VIRAZOLE С LG-5 (ПОСРЕДСТВОМ СЛОЖНОЭФИРНОЙ СВЯЗИ)
Фиг. 5 показывает пример синтеза 2',3'-O-изопропилиден-виразола (V-1) с использованием следующих реактивов: VirazoleTM (товарный знак ICN для рибавирина (Ribavirin USP)); ацетона; 2,2-диметоксипропана=DMP и (пер)хлорной кислоты (70%). В этой процедуре VirazoleTM (370 г, 12,28 мМ) суспендировали в смеси ацетона (40 мл) и DMP (20 мл). Смесь охлаждали на ледяной бане и добавляли (пер)хлорную кислоту (600 мкл). Смесь выдерживали при комнатной температуре в течение 3 часов, а затем при 5oC в течение ночи. Получившийся в результате раствор оранжевого цвета нейтрализовали 2 молями водного КОН, фильтровали и выпаривали до сухости. Твердый остаток обрабатывали метанолом (5 мл) и нерастворимый продукт удаляли фильтрацией. Метанольный фильтрат выпаривали до сухости и твердый остаток перекристаллизовывали из ацетона. Выход: 2,95 г (81%); ТСХ: 15% метанол, 85% этилацетат; Rf: 0,54. 1H-ПМР-спектр (ацетон - d6) подтвердил чистоту и структуру V-1.
ПРИМЕР 13 - 2',3'-O-ИЗОПРОПИЛИДЕН-VIRAZOLE-5'-O-ЭФИР
Фиг. 5 также показывает пример синтеза 2',3'-O- изопропилиден-Virazole-5'-O-эфира с LG-5 (V-1-5'-O-LG-5) с использованием следующих реактивов: V-1; LG-5; DCC; DMAP; пиридина. В этой процедуре смесь V-1 (236 мг, 0,796 мМ), LG-5 (200 мг, 0,531 мМ), DCC (108 мг, 0,531 мМ), DMAP (13 мг, 0,106 мМ) и Py (5 мл) выдерживали при 60oC в течение ночи. Пиридин удаляли в вакууме, а осадок очищали посредством колоночной хроматографии на силикагеле. Выход: 160 мг (46%); ТСХ: 25% гексан, 75% этилацетат; Rf: 0,21. 1H-ПМР - спектр (ацетон - d6) подтвердил чистоту и структуру V-1-5'-O-LG-5.
ПРИМЕР 14 - СНЯТИЕ ЗАЩИТЫ С V-1-5'-O-LG-5 И СИНТЕЗ V-5'-O-LG-7
Фиг. 5 показывает пример снятия защиты с V-1-5'-O-LG-5 и синтеза V-5'-O-LG-7 с использованием защищенного сложного эфира VirazoleTM из предыдущего опыта (V-1-сложный эфир)TFA. В этой процедуре сложный эфир V-1 (50 мг, 0,076 мМ) обрабатывали смесью TFA/вода (9/1, об/об) при комнатной температуре в течение 5 минут. Реакционную смесь выпаривали до сухости, снова растворяли в 1 мл воды, фильтровали и снова выпаривали. Выход: 35 мг (87%); ТСХ: 10% метанол, 2% уксусная кислота, 88% этилацетат; Rf: 0,19. 1H-ПМР-спектр (D2O) подтвердил чистоту и структуру V-5'-O-LG-7.
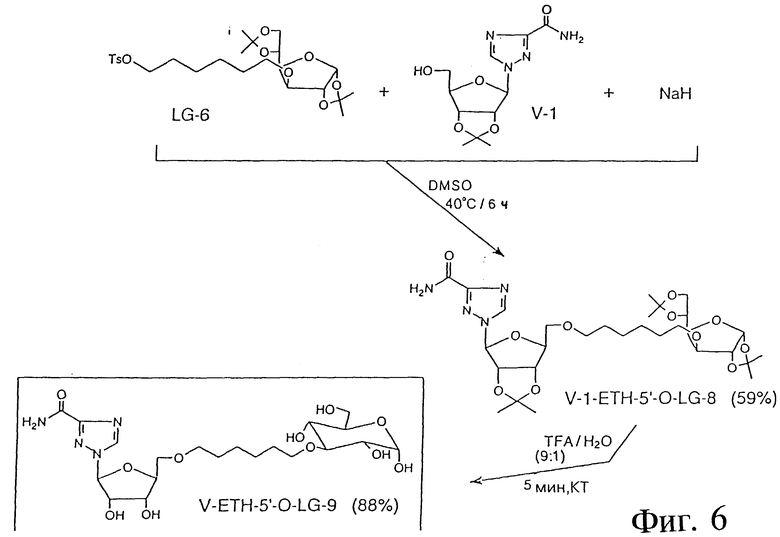
ПРИМЕР 15 - КОНЪЮГАЦИЯ VIRAZOLETM С LG-6 (ПОСРЕДСТВОМ СЛОЖНОЭФИРНОЙ СВЯЗИ)
Фиг. 6 показывает пример синтеза V-1-ETH-5'-O-: LG-8 и LG-9 с использованием следующих реактивов: V-1; LG-6; NaH и DMSO. В этой процедуре V-1 (250 мг, 0,843 мМ) растворяли в безводном DMSO (5 мл) и добавляли небольшими порциями NaH (118 мг, 2,95 мМ) в течение 30 минут. Реакционную смесь нагревали 30 минут при 40oC и затем добавляли LG-6. Смесь выдерживали при комнатной температуре в течение ночи, а затем разбавляли 100 мл этилацетата. Раствор промывали рассолом (3x30 мл), высушивали (Na2SO4), выпаривали и очищали посредством колоночной хроматографии на силикагеле. Выход: 220 мг (49%); ТСХ: 10% гексан, 90% этилацетат: Rf: 0,53; 1H-ПМР-спектр (ацетон-d6) подтвердил предполагаемую структуру.
ПРИМЕР 16 - КОНЪЮГАЦИЯ VIRAZOLETM С LG-6 (ПОСРЕДСТВОМ СЛОЖНОЭФИРНОЙ СВЯЗИ)
В другом примере синтеза предыдущее соединение (50 мг, 0,078 мМ) обрабатывали смесью TFA/вода (9/1, об/об) при комнатной температуре в течение 5 минут. Реакционную смесь выпаривали до сухости, снова растворяли в 1 мл воды, фильтровали и снова выпаривали. Выход V-1 ETH-5'-O-LG-9: 36 мг (88%); TLC: 10% метанол, 2% уксусная кислота, 88% этилацетат.
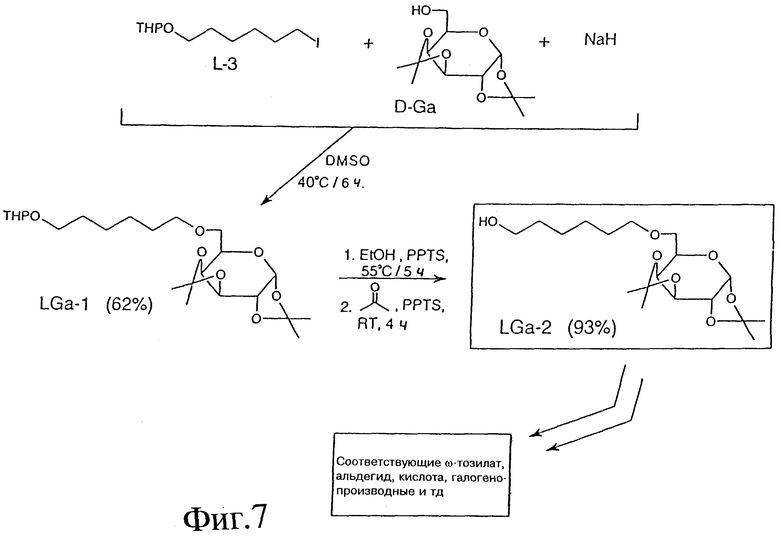
ПРИМЕР 17 - СИНТЕЗ 6-О-D-ГАЛАКТОЗО-ЛИНКЕРА В ВИДЕ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ
Фиг. 7 показывает пример синтеза 1,2:3,4-ди-O-изопропилиден-6-O- (6'-тетрагидропиранилокси)-гексил-альфа-D-галактопиранозы (LGa-2) с использованием следующих реактивов: L-3; 1,2:3,4-ди-O- изопропилиден-альфа-D-галактопиранозы=DIPGap; NaH и DMSO. В этой процедуре DIPGap (500 мг, 1,92 мМ) растворяли в безводном DMSO (5 мл) и добавляли небольшими порциями NaH (96 мг, 2,40 мМ) при комнатной температуре. Реакционную смесь нагревали при 40oC в течение 0,5 часов и добавляли L-3 (660 мг, 2,11 мМ) при комнатной температуре. Смесь выдерживали при комнатной температуре в течение ночи и затем вливали в 100 мл этилацетата. Органический слой промывали рассолом (3x25 мл), высушивали (Na2SO4) и выпаривали. Вещество очищали методом флэш-хроматографии на двуокиси кремния (система растворителей: 10% этилацетат, 2,5% триэтиламин, 87,5% гексан). Выход: 500 мг (62%); ТСХ: 15% этилацетат, 2,5% триэтиламин, 82.5% гексан; Rf: 0,44. 1H-ПМР-спектр (CDCl3) подтвердил чистоту и структуру LGa-2. LGa-1 был получен подобной процедурой как и LG-3 (см. фиг.3).
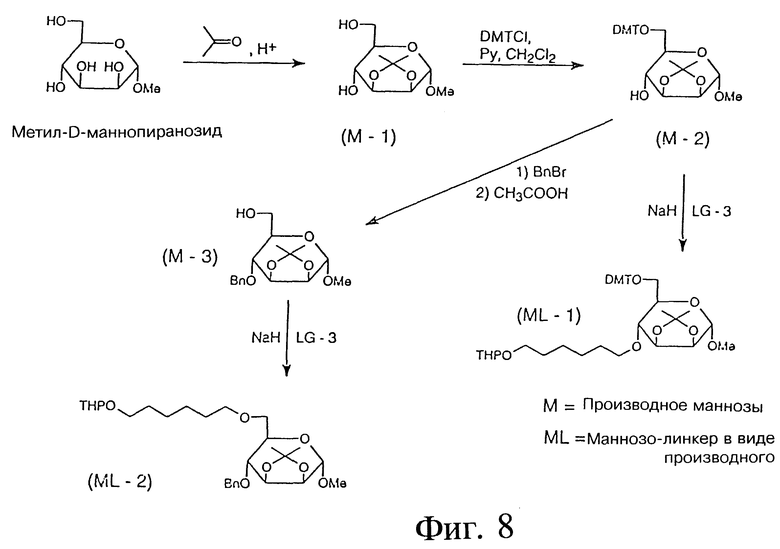
ПРИМЕР 18 - СИНТЕЗ 4-O- И 6-O-D-МАННОЗО-ЛИНКЕРОВ В ВИДЕ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ
Фиг. 8 показывает синтез 4-O и 6-О-D-маннозо-линкеров в виде промежуточных соединений. Синтез проводится с использованием химических реактивов подобно тому, как описано ранее для галактозы и глюкозы.
ПРИМЕР 19 - СИНТЕЗ 3-O-, 4-O- И 5-O-D-ФРУКТОЗО-ЛИНКЕРА В ВИДЕ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ
Фиг. 9 показывает синтез 4-O и 6-O-D-маннозо-линкеров в виде промежуточных соединений. Синтез проводится с использованием химических реактивов подобно тому, как описано ранее для галактозы и глюкозы.
ПРИМЕР 20 - ГЛЮКОЗО-ЛИНКЕР В ВИДЕ ФОСФОРАМИДАТНЫХ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ ДЛЯ АВТОМАТИЧЕСКОГО СИНТЕЗА ОЛИГОДЕЗОКСИНУКЛЕОТИДОВ (ODN)
Фиг. 10 показывает пример синтеза LG-3 Bnf с использованием следующих реактивов: LG-3; бензилбромида=BnBr; гидрида натрия=NaH и тетрагидрофурана= THF. В этой процедуре смесь LG-3 (1,00 г, 2,77 мМ) и NaH (150 мг, 3,75 мМ) в THF (10 мл) перемешивали при 40oC в течение 1 часа с последующим добавлением BnBr (1,3 мл, 11,08 мМ). Реакционную смесь перемешивали далее при 40oC до завершения реакции (6 часов), затем добавляли этилацетат (100 мл) и полученный раствор промывали рассолом (3x50 мл). Органический слой высушивали (Na2SO4) и выпаривали. Продукт очищали на флэш-хроматографией на силикагеле (система растворителей ТСХ). Выход 1,25 г (94%); ТСХ: 15% этилацетат, 85% гексан; Rf: 0,45. 1H-ПМР-спектр (ацетон-d6) соответствовал предполагаемой структуре
ПРИМЕР 21- LG-3BnpTA
Фиг. 10 также показывает пример синтеза LG-3BnpTA с использованием следующих реактивов: LG-3Bnf; трифторуксусной кислоты=TFA; ангидрида уксусной кислоты= Ac20 и пиридина=Py. В этой процедуре LG-3Bnf (1,00 г, 2,22 мМ) обрабатывали 4 мл смеси TFA/вода (9/1, об/об) при комнатной температуре в течение 7 минут. Реакционную смесь выпаривали в вакууме до сухости, снова растворяли в смеси Ac20/Py (10 мл, 1:1, об/об) и оставляли при комнатной температуре на ночь. После нейтрализации (5%-ная водн. HCl, 50 мл) продукт экстрагировали этилацетатом (3x50 мл). Органический слой промывали рассолом (3x50 мл), высушивали (Na2SO4) и выпаривали. Продукт окончательно очищали посредством флэш-хроматографии на колонке с силикагелем (система растворителей ТСХ). Выход 1,15 г (96%); ТСХ: 33% этилацетат, 67% гексан; Rf: 0,44. 1H-ПМР-спектр (ацетон-d6) подтвердил чистоту и структуру LG-3BnpTA.
ПРИМЕР 22- LG-3pTA
Фиг. 10 также показывает пример синтеза LG-3pTA с использованием следующих реактивов: LG-3BnpTA; 10% Pd/C и метанола. В этой процедуре LG-3pTA (1,00 г, 1,86 мМ) растворяли в метаноле (50 мл) и затем добавляли катализатор (Pd/C, 200 мг). Реакционную смесь взбалтывали в атмосфере H2 до завершения реакции (18-24 часа). Затем катализатор удаляли фильтрацией и метаноловый раствор выпаривали до сухости. Продукт очищали методом флэш-хроматографии на диоксиде кремния. Выход: 815 мг (98,1%); ТСХ: этилацетат/гексан(1:1); Rf: 0,21. 1H-ПМР-спектр (CDCl3) соответствовал предполагаемой структуре.
ПРИМЕР 23 - LG-3pTAPA
Фиг. 10 также показывает пример синтеза фосфорамидатного промежуточного соединения, LG-3pTAPA, с использованием следующих реактивов: LG-3pTA; хлоро-циано-этилокси-диизопропиламино-фосфина= PAP (предшественник фосфорамидата); диизопропилэтиламина=DIPEA и CH2Cl2. В этой процедуре LG-3pTA (300 мг, 0,669 мМ) растворяли в CH2Cl2 (3 мл) под аргоном. Получившийся раствор охлаждали до 0oC и добавляли DIPEA (450 мкл, 2,62 мМ). Через 15 минут добавляли также PAP (315 мкл, 1,408 мМ, 0oC). Полученную реакционную смесь выдерживали при комнатной температуре в течение последующих двух часов, затем охлаждали (0oC), разбавляли холодным CP2Cl2 (100 мл), промывалась холодным 5%-ным водн. NaHCO3, высушивали (Na2SO4) и выпаривали в вакууме. Сырой продукт очищали методом флэш-хроматографии на диоксиде кремния. Чистый продукт сохраняли под аргоном до его использования в автоматическом синтезе олигонуклеотидов. Для автоматического ODN синтеза LG-3pTAPA использовали в виде 0,1 М раствора в безводном ацетонитриле. Выход: 257 мг (62,2%); ТСХ: 32% этилацетат, 5% триэтиламин, 63% гексан; Rf: 0,53. 31P-ПМР-спектр соответствовал предполагаемой структуре.
ПРИМЕР 24 - ФОСФОРАМИДАТНЫЕ ПРОИЗВОДНЫЕ ЛИНКЕРА ДЛЯ АВТОМАТИЧЕСКОГО СИНТЕЗА ОЛИГОНУКЛЕОТИДОВ
Фиг. 11 показывает пример синтеза L-4 с использованием следующих реактивов: 1,6-гександиол=HD и диметокситритилхлорид=DMT-Cl. В этой процедуре HD (4,00 г, 29,52 мМ) растворяли в смеси Py/CH2Cl2 (1:1, об/об) (8,0 мл). Получившийся раствор охлаждали до 0oC и добавляли DMT-Cl (2,50 г, 7,38 мМ). Полученную реакционную смесь выдерживали в течение 6 часов при 4oC. Реактивный раствор затем тщательно нейтрализовали (pH 8) 5%-ной водн. HCl и продукт экстрагировали CH2Cl2 (3x50 мл). Органический слой промывали рассолом (3x30 мл), высушивали (Na2SO4) и выпаривали. Сырой продукт очищали методом флэш-хроматографии на диоксиде кремния. Выход: 2,53 г (81,5%); ТСХ: 32% этилацетат, 3% триэтиламин, 65% гексан; Rf: 0,38; 1H-ПМР-спектр соответствовал предполагаемой структуре
ПРИМЕР 25 - L-5
Фиг. 11 также показывает пример синтеза L-5 с использованием следующих реактивов: L-4; PAP и DIPEA. В этой процедуре L-4 (500 мг, 1,19 мМ) растворяли в CH2Cl2 (5 мл) под аргоном. Получившийся раствор охлаждали до 0oC и добавляли DIPEA (800 мкл, 4,65 мМ). Через 15 минут добавляли PAP (550 мкл, 2,46 мМ). Полученную реакционную смесь выдерживали при комнатной температуре в течение последующих двух часов, затем охлаждали (0oC), разбавляли холодным CH2Cl2 (100 мл), промывали холодным 5%-ным водн. NaHCO3, высушивали (Na2SO4, NaHCO3) и выпаривали в вакууме. Сырой продукт очищали флэш-хроматографией на диоксиде кремния. Чистый продукт (бесцветное масло) сохранялся до использования под аргоном при -180oC. Фосфорамидат L-5 использовали в виде 0,1 М раствора в безводном ацетонитриле. Выход: 430 мг (58,3%); ТСХ: 16% этилацетат, 4% триэтиламин, 80% гексан; Rf: 0,37. 1H и 31P-ПМР- спектр соответствовал предполагаемой структуре.
ПРИМЕР 26 - L-6
На фиг. 12 первый ряд показывает пример синтеза 1,6-O-п-дитолуол-сульфонил-1,6-гександиола (L-6) с использованием следующих реактивов: 1,6-гександиола= 1,6-HD; п-толуолсульфонилхлорида=TsCl; пиридина=Py и дихлорметана= CH2Cl2. В этой процедуре 6-HD (70,9 г, 0,60 моль) растворяли в 150 мл Py и 250 мл CH2Cl2. Получившийся раствор охлаждали до 0oC и добавляли TsCl (252 г, 1,32 моль). Полученную реакционную смесь выдерживали при 4oC в течение 5 часов. Реактивный раствор затем нейтрализовали 10%-ной HCl и продукт экстрагировали CH2Cl2 (3x250 мл). Органический слой промывали рассолом, высушивали и выпаривали. Сырой продукт суспендировали в этаноле (300 мл), фильтровали и промывали холодным этанолом. L-6 (белые кристаллы); выход: 219,1 г (85,6%); ТСХ: этилацетат/гексан (1: 2); Rf: 0,48. Чистоту и структуру L-6 проверяли и подтверждали посредством 1H-ПМР-спектра и FAB (бомбардировка быстрыми атомами) масс-спектроскопии высокого разрешения.
ПРИМЕРЫ 27 И 28 - АЛЬТЕРНАТИВНЫЙ СИНТЕЗ LG-6 И LG-3
Фиг. 12 во втором ряду также показывает альтернативный прямой синтез LG-6, который может быть осуществлен с использованием следующих реактивов: 1,2: 5,6-ди-O-изопропилиден- α -D-глюкофуранозы= DIPGF, L-6; NaH и DMSO. В этой процедуре DIPGF (11,68 г, 44,88 мМ) растворяли в безводном DMSO (50 мл) и добавляли небольшими порциями NaH (2,334 г, 58,34 мМ) при комнатной температуре. Реакционную смесь выдерживали при 40oC в течение 30 минут и затем добавляли L-6 (67,0 г, 157,0 мМ) растворенного в 150 мл теплого (40oC) DMSO. Спустя 30 минут реакционную смесь вливали в этилацетат (800 мл). Органический слой промывали рассолом (3x50 мл), высушивали (Na2SO4) и выпаривали. Продукт очищали методом хроматографии на силикагелевой колонке (система растворителей ТСХ). LG-6 (бесцветное масло); выход: 16,0 г (69,0%); ТСХ: 33% этилацетат, 67% гексан; Rf: 0,53. Чистоту и структуру L-6 проверяли и подтверждали 1H-ПМР-спектром и FAB масс-спектроскопией высокого разрешения.
В дополнение, фиг. 12 показывает пример синтеза 1,2:5,6-ди-O- изопропилиден-3-O-(6'-гидрокси)-гексил- α -D-глюкофуранозы (LG-3) с использованием следующих реактивов: LG-6; DMF; K2CO3; H2O. В этой процедуре LG-6 (16,0 г, 31,03 мМ) и K2CO3 растворяли в DMF (200 мл) и воде (20 мл) и реакционную смесь нагревали 5 часов при 90oC. После удаления растворителя (выпаривание под высоким вакуумом) остаток растворяли в этилацетате (600 мл), промывали рассолом (3x200 мл), высушивали (Na2SO4) и выпаривали. Продукт окончательно очищали методом флэш-хроматографии на диоксиде кремния. LG-3 (бесцветное масло); выход: 7,85 r (70,2%); ТСХ: этилацетат/гексан; Rf: 0,48; чистоту и структуру LG-3 проверяли и подтвердили 1H-ПМР-спектром и FAB масс-спектроскопией высокого разрешения.
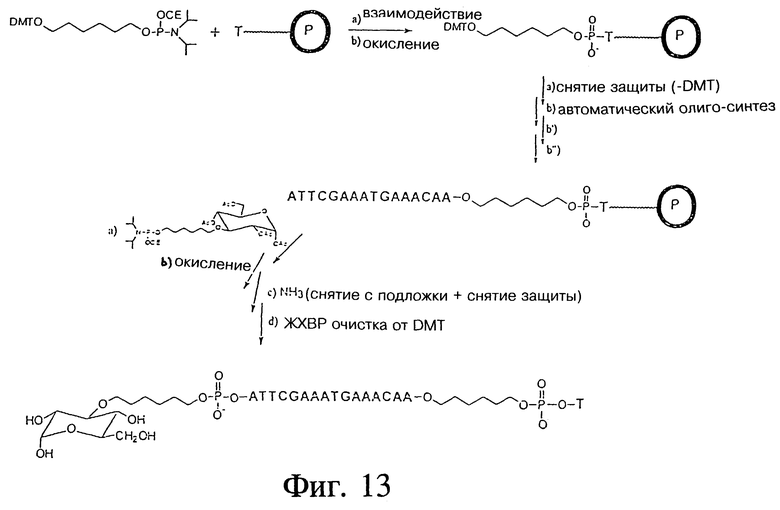
ПРИМЕР 29 - СИНТЕЗ ОЛИГОДЕЗОКСИНУКЛЕОТИДОВ (ODN), МОДИФИЦИРОВАННЫХ В ПОЛОЖЕНИЯХ 3' И 5', И ИХ БИОФИЗИЧЕСКИЕ СВОЙСТВА
Фиг. 13 показывает пример синтеза олигодезоксинуклеотидов (ODN), модифицированных в положениях 3' и 5' (ICN 16967) с использованием автоматического синтезатора ДНК (Applied Biosystems, model 394) и обычной фосфорамидатной химии; β -цианоэтилфосфорамидатов, реактивов для синтеза и CPG полистироловых колонок (1 мкМ и 10 мкМ T, размер пор 500 Ao, ABI). После синтеза от олигонуклеотида отщепляли защитные группы с помощью концентрированного гидроксида аммония в течение 8 часов при 55oC. Первичный и окончательный анализ проводили с использованием аналитической колонки C8 HPLC (Beckman; ультрасферический октил).
Очистку проводили на полупрепаративной колонке с обращенной фазой C8 HPLC (Beckman C8 ультрасферический ODS 5 мкМ, 10 мм x 25 см) со скоростью потока 3 мл/мин с использованием 0,1М TEAA и 5% ацетонитрила. После очистки и кондиционирования чистое вещество осаждали 2-пропанолом, растворяли в воде и лиофилизировали. Выход: 2,23 мг, (36% для 1 мкмольного синтеза). Структуру и чистоту ICN 16967 проверяли и подтвердили HPLC (высокоэффективная жидкостная хроматография) анализом, капиллярным электрофорезом и масс-спектроскопией с облучением электронами.
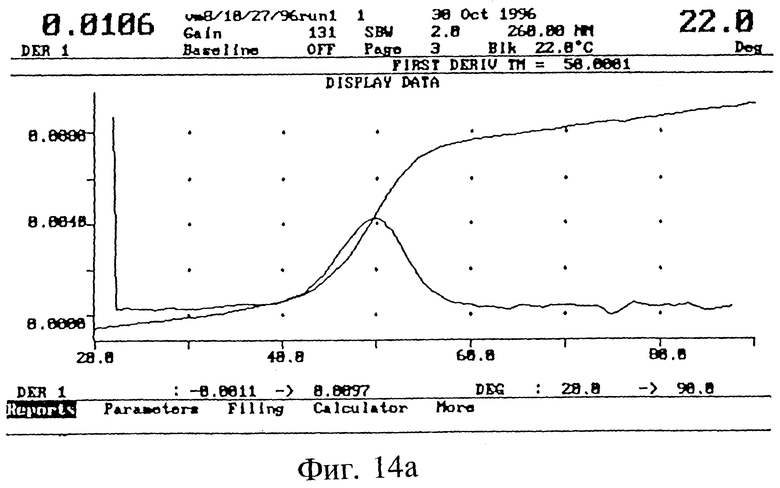
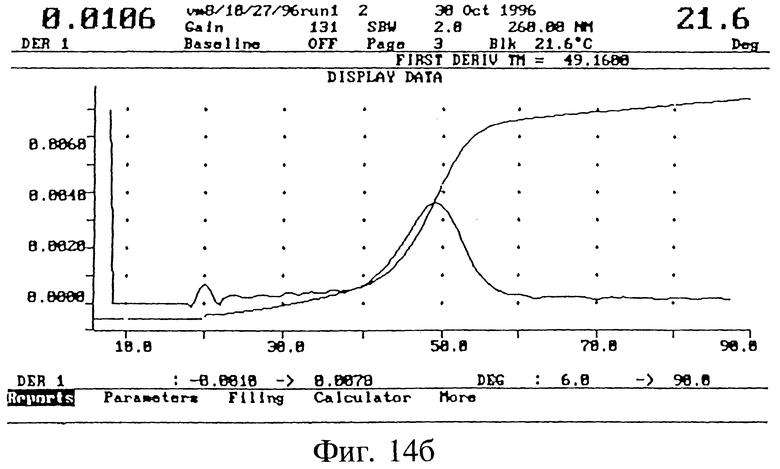
БИОФИЗИЧЕСКИЕ СВОЙСТВА ICN 16967
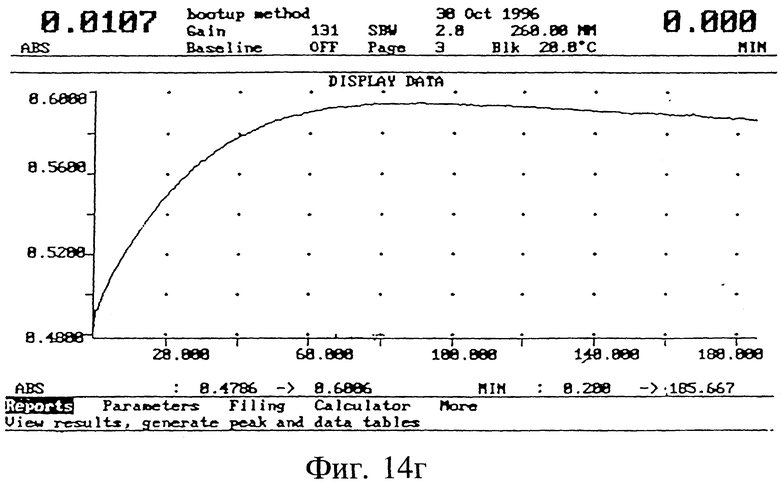
Гибридизационные свойства полученных 3', 5'-модифицированных олигонуклеотидов изучали в экспериментах по термодинамическому плавлению (Tm) (Methods in Fuglisi, 3. D.; Tinoco, I.Jr. Methods Enzymol 1989, 180, 304). Опыты проводили на спектрометре Varian UV, оснащенном электронным терморегулятором и программным обеспечением для Cary гибридизации. Как показано на фиг. 14 (а), эта модифицированная последовательность гибридизуется с комплементарной РНК практически также, как и немодифицированная последовательность (б).
Образцы для Tm измерений содержали 2 мкМ модифицированных олигонуклеотидов и 2.0 мкМ комплементарной РНК в буфере (10 мМ фосфата натрия, 0,1 мМ EDTA и 0,1 М хлорида натрия, pH 7,0).
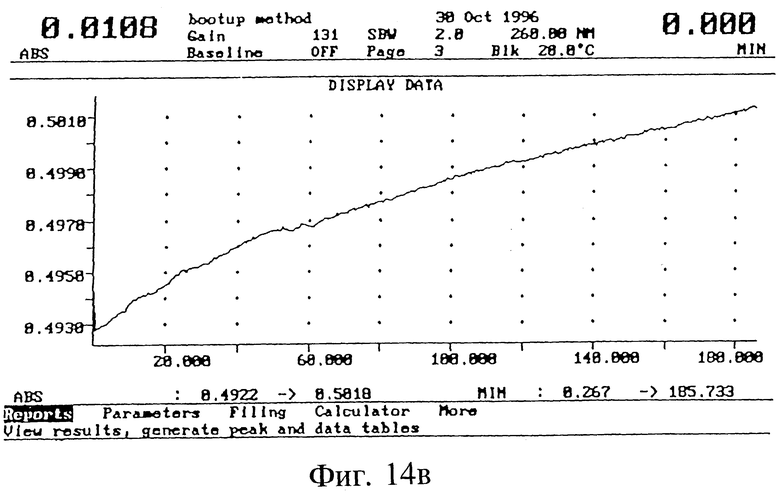
Стабильность и период полураспада модифицированного (фиг. 14, в) и немодифицированного (г) олигонуклеотида вычислялись по кривым спектрального поглощения ультрафиолета образцов олигонуклеотида в процессе расщепления фосфодиэстеразой из змеиного яда при 37oC, используя известный метод (Svedsen, M. L; Wengel, 3.; Dahl, O.; Kirpekar, F.; Roepstorff, P. Tetrahedron 1993, 49, 11341.) Олигонуклеотиды (0,75 OD (оптическая плотность)) инкубировали с фосфодиэстеразой из змеиного яда (1,2 единиц) в 1,5 мл буфера (0,1 М трис HCl, pH 7,5: 0,1 М NaCl; 14 мМ MgCl2) в кювете Varian UV спектрометра при 25oC, и в процессе расщепления снимали зависимость возрастания спектрального поглощения при 260 нм от времени. По этим кривым спектрального поглощения вычисляли период полураспада олигонуклеотидов. При этих условиях ICN 16976 (глинкозид) олигонуклеотид был примерно в 20 раз более стабилен, чем соответствующий немодифицированный олигонуклеотид.
ДОКАЗАТЕЛЬСТВО ВОЗРОСШЕЙ БИОЛОГИЧЕСКОЙ ДОСТУПНОСТИ
Мы провели испытание биологической доступности типичного глинкозида, меченного тритием 5'-O-глюкозо-тимидина (D-G-L-Thy), путем сравнения включения свободного тимидина с включением D-G-L-Thy в простом анализе in vitro. В этом опыте мы использовали клетки линии 177 Lung Cancer Cell (немелкоклеточная карцинома легкого человека), растущие в среде, обогащенной 10% фетальной телячьей сывороткой (SSM) и в бессывороточной основной среде (SFM). Клетки обрабатывали свободным тимидином и конъюгированным тимидином (D-G-L-Thy) в течение 4 и 24 часов. D-G-L-*Thy был синтезирован методом изотопного разбавления (1 часть высокорадиоактивного (Hot) Thy: 100 частей низкорадиоактивного (Cold) Thy). Исходный раствор был приготовлен со следующими средними значениями cpm (число импульсов в минуту): Thy: 481660 и 5'-O-конъюгат: 467242 (поправочный коэффициент 1,03). Были получены результаты (средние значения) (см. табл.1).
Во всех условиях включение тимидина, высвобожденного из соединения D-G-L-Thy, было выше по сравнению с контролем. Эти данные показывают, что тимидин может быть высвобожден из комплекса D-G-L-Thy в клетках благодаря наличию фосфодиэстераз, и что высвобожденный тимидин может быть затем включен в ДНК. Маловероятно, что тимидин мог быть высвобожден ферментативно из внеклеточного комплекса, так как эта среда не содержит фосфодиэстеразы.
Таким образом, наши данные показывают, что глинкозидное соединение D-G-L-Thy поглощается клетками как пролекарство и внутри клетки превращается в свободный тимидин и свободную глюкозу. Поскольку поглощение совместно с параллельным и последовательным (вне- и внутриклеточным) гидролизом пролекарства представляют собой кинетический комплекс, испытание на этой стадии, может иметь следующие пояснения:
- Поглощение глинкозида очень высокое, весьма вероятно, что оно намного выше, чем поглощение самого тимидина.
- Внутриклеточный гидролиз пролекарства происходит достаточно быстро для того, чтобы высвобождать достаточное количество свободного лекарства внутриклеточно.
- Это является очевидным признаком возможного поглощения глинкозида посредством GLUT системы.
ДОКАЗАТЕЛЬСТВО ВОЗРОСШЕЙ БИОЛОГИЧЕСКОЙ ДОСТУПНОСТИ
Клетки меланомы человека ATТC HTB140 (HS294T) культивировали в DMEM (модифицированная по способу Дульбекко среда Игла) с добавлением 10% бычьей сыворотки (культуральная среда). Для опыта эти клетки были обработаны трипсином и собраны в виде суспензии в культуральной среде. После посева на 96-ти луночные планшеты с примерной плотностью 1000/клеток/лунку/200 мкл культуральной среды, клетки обрабатывали 0,5 мкМ олигонуклеотидов в течение 72 часов. После этого клетки подсчитывали с использованием гемоцитометра. Каждый подсчет проводили три раза для каждого опыта. Указанные результаты вычисляли как среднее ингибирование роста клеток против необработанных клеток (контроль) и основаны на трех независимых опытах (см. табл.2).
Опыты показали, что немодифицированный олигонуклеотид эффективен в концентрациях 0,5 мкМ в течение 72 часов клеточного роста, а модифицированный олигонуклеотид более эффективен, чем немодифицированный олигонуклеотид, при тех же концентрациях и условиях опыта.
ЛЕЧЕНИЕ С ПРИМЕНЕНИЕМ ГЛИНКОЗИДОВ
Глинкозиды могут применяться для увеличения абсорбции, распространения и клеточного поглощения как уже существующих, так и разрабатываемых фармацевтических препаратов. Например, глинкозиды могут использоваться для контроля и воздействия in vitro, in vivo и ex vivo на нормальный, физиологически стареющий и патологический рост клеток, для индукции/предотвращения апоптоза (утраты клеток), индукции/подавления дифференциации клеток, для контроля метаболизма и реакций клеток, активности и репродукции вирусов в инфицированных клетках и функционирования вирус-инфицированных клеток, а также для лечения нейро-дегенеративных заболеваний.
Изобретение имеет определенное отношение к транспорту различных моносахаридов и особенно к функционированию переносчиков глюкозы в различных тканях и клетках. Переносчики глюкозы напрямую задействованы в транспорте различных моносахаридов в клетки (глюкозы, маннозы, галактозы и др.). Некоторые из этих переносчиков глюкозы являются характерными для определенных тканей (как пример) или типа клеток (как пример), или биологического функционирования (например, GLUT-1-ион быстрорастущих нормальных и раковых клеток). Тот факт, что различные типы переносчиков глюкозы по-разному экспрессируются в различных клетках, обеспечивает возможность специфического клеточного поглощения или доставки с лечебной целью биологически-активных соединений, сконъюгированных с сахаридом, как описывается ниже.
1. Переносчики глюкозы в быстрорастущих клетках. Нерастущие (неактивные) нормальные клетки, активированные фактором роста, вступают в клеточный цикл и делятся. Ростовые факторы вызывают каскад событий, которые можно наблюдать по экспрессии генов, мРНК, белка и уровню клеточной структурной организации. Например, краткосрочное (0,5 - 1 час) повышение экспрессии GLUT-1 мРНК и белка наблюдали в клетках, активированных фактором роста (Endocrinology, 1990, 127, 2025). Ванадат натрия, известный как неспецифический стимулятор роста, был способен индуцировать экспрессию GLUT-1 в нормальных ЗТЗ фибробластах и, кроме того, продлевал период полураспада GLUT-1 мРНК в 2-3 раза (Endocrinology, 1990, 126, 2778).
Был установлен аутокринный механизм неконтролируемой пролиферации раковых клеток. Раковые клетки растут быстро и имеют очень высокий уровень GLUT-1. Таким образом, раковые клетки поглощают значительно больше глюкозы, чем нормальные клетки и используют ее для кликолиза. Также было доказано, что для роста раковых клеток необходим высокий уровень экспрессии функционального GLUT-1 белка. Любое ингибирование функционирования GLUT-1 в раковых клетках, вызванное связыванием GLUT-1-специфичных антител или связыванием сурамина, значительно снижает поглощение глюкозы и скорость роста обработанных клеток. Эти данные показывают на необходимость высокого поглощения глюкозы для поддержания большой скорости пролиферации раковых клеток, что специфически опосредовано зависимостью GLUT от фактора роста. Таким образом, раковые клетки молочной железы, легкого, простаты, глиомы и другие показывают значительно повышенный уровень GLUT.
2. Переносчики глюкозы в гематоэнцефалическом барьере. Чтобы преодолеть гематоэнцефалический барьер (BBB), лекарственные препараты должны проникнуть через луминальные и базальные мембраны эндотелиальных клеток. Это возможно путем использования преимущества опосредованного рецептором поглощения, как например, в случае препарата от болезни Паркинсона L-Dopa. Допамин сам не может преодолеть гематоэнцефалический барьер. Однако, его естественный предшественник L-Dopa транспортируется через гематоэнцефалический барьер посредством системы аминокислотного переноса.
Десенсибилизирующие олигонуклеотиды - это высокозаряженные молекулы, обладающие низкой проницаемостью через стенки капилляров мозга. Введя олигонуклеотиды в глинкозиды, как описано здесь, транспорт этих потенциально эффективных десенсибилизирующих препаратов можно значительно облегчить за счет поглощения посредством переносчиков глюкозы эндотелиальных клеток, составляющих BBB.
Транспорт глюкозы из крови к нейронным и глиальным клеткам мозга обеспечивается двумя специфическими переносчиками, находящимися в эндотелии капилляров мозга, которые составляют ВВВ (M.W. Brightman 1977, "Morphology of Blood-brain Interfaces" Exp.Eye Res. 25, 1-25). Два этих переносчика глюкозы были идентифицированы как два различных члена супергенного семейства натрий-независимых переносчиков глюкозы. В то время как изоформа переносчика глюкозы типа 3 (GLUT-3) локализована на мембране нейронной клетки (S. Nagamatsu et al, J. Biol. Chem. 267, 467-472), изоформа переносчика глюкозы типа 1 (GLUT-1) локализована на BBB (R.J. Boado and W.M. Partridge, Biochem. Biophys. Res. Commun. 166, 174-179 (1990)). Глинкозид должен быть способен проникать через BBB благодаря наличию GLUT-1 транспортной системы. Это значительно увеличит возможность применения десенсибилизирующих олигонуклеидов для нейродегенеративных заболеваний ЦНС, таких как, например, болезнь Паркинсона (Parkinson's) и болезнь Альцгеймера (Aizheimer's). Лекарственный препарат может вводиться перорально или внутривенно, а затем распространяться посредством циркуляции крови к мозгу, или он может быть введен в сонную артерию для улучшения локального поступления в мозг.
3. Другие цели, которые подходят для применения глинкозидов благодаря наличию специфичных переносчиков глюкозы, включают нервные клетки, ткани печени, желудка, жировые и мышечные ткани и стареющие клетки.
ФАРМАЦЕВТИЧЕСКИЕ ПРЕПАРАТЫ С ИСПОЛЬЗОВАНИЕМ ГЛИНКОЗИДОВ
Как и другие фармацевтические препараты, глинкозиды могут быть приспособлены для введения в тело пациента различными способами, подходящими для выбранного способа введения, включая пероральное, внутривенное, внутримышечное, внутрибрюшное, местное и др. В дополнение к содержанию одного или большего числа различных глинкозидов, предлагаемые фармацевтические препараты могут содержать одно или большее число не-биологически-активных соединений, например, эксципиентов, таких как стабилизаторы (для увеличения срока хранения), эмульгаторы, связывающие вещества, уплотняющие вещества, соли, консерванты и т.п.
Глинкозиды могут употребляться в дозах и количествах, которые являются традиционными в уровне техники для основных биологически-активных соединений, но приспособленных для более эффективной абсорбции, транспорта и поглощения клеткой. Так, для рибавирина (Ribavirin), который назначается перорально по 1200 мг ежедневно, доза соответствующего глинкозида будет составлять примерно 600 мг рибовирина ежедневно или меньше. Доза может вводиться в один прием или может быть разделена на несколько меньших доз, которые затем вводятся через определенные интервалы времени.
Схема приема лекарственного средства может быть адаптирована для обеспечения оптимальной терапевтической реакции. Например, наиболее предпочтительная доза может измениться в соответствии с определенным выбранным агентом, и в ходе курса лечения доза может быть пропорционально увеличена или уменьшена в зависимости от терапевтической ситуации.
Глинкозиды могут вводиться любым удобным способом - перорально, внутривенно, внутрибрюшинно, внутримышечно, подкожно и другими известными путями. При пероральном применении глинкозиды могут вводиться с инертным растворителем или с усваивающимся пищевым носителем, либо глинкозиды могут включаться непосредственно в продукты диеты. Перорально вводимые глинкозиды могут быть объединены с эксципиентами и применяться в виде таблеток для проглатывания, трансбуккальных (щечных) таблеток, пастилок, капсул, эликсиров, суспензий - сиропов, облаток и т.п.
Таблетки, пастилки, драже, капсулы и т.п. могут содержать также следующие компоненты: связующие вещества, такие как трагакант, аравийская камедь, кукурузный крахмал или желатин; эксципиенты, такие как дикальцийфосфат; разрыхлители, такие как кукурузный и картофельный крахмал, альгиновая кислота и т. п.; смазывающие вещества, такие как стеарат магния, и подслащивающие вещества, такие как сахароза, лактоза или сахарин; ароматические вещества, такие как мятный ароматизатор, масло грушанки, вишневый ароматизатор. Когда единицей дозы является капсула, она может содержать в дополнение к веществам, указанным выше, жидкий носитель. Различные другие вещества могут также присутствовать в качестве оболочки или как либо по-другому для изменения физической формы единицы дозы. Например, таблетки, облатки, капсулы могут быть покрыты оболочкой, сахаром или и тем и другим. Сироп или эликсир может содержать сахарозу как подслащивающее вещество, метил и пропилпарабены как консерванты, краситель и ароматизатор, например вишневый или апельсиновый ароматизатор. Эти дополнительные вещества в применяемых количествах должны быть совершенно нетоксичны. Кроме того, глинкозиды могут быть включены в препараты и составы задержанного высвобождения.
Препараты для парентерального введения могут включать стерильные водные растворы или дисперсии, стерильные порошки для спонтанного приготовления стерильных препаратов, растворы и дисперсии для инъекций. Растворы и дисперсии могут также содержать буферы, разбавители и другие необходимые добавки, подобранные для улучшения поглощения клеткой глинкозидов в этом составе, например, глинкозиды могут быть инкапсулированы в соответствующие липосомы. Растворы и дисперсии для парентерального введения предпочтительно являются стерильными и достаточно текучими для удобства применения, достаточно стабильными для производства и использования и защищенными от инфицирования микроорганизмами, такими как бактерии и грибы. Носитель может быть растворителем или дисперсионной средой, содержащей, например, воду, этанол, полиол (например глицерин, пропиленгликоль, жидкий полиэтиленгликоль и подобное), смеси из них и растительные масла. Необходимая текучесть может поддерживаться, например, применением покрытия, такого как лецитин, выдерживанием требуемых размеров частиц в случае дисперсий и использованием поверхностно-активных веществ.
Стерильные растворы для инъекций готовят внесением активных соединений в необходимом количестве в соответствующий растворитель с одним или большим числом других различных ингредиентов, описанных выше, с последующей стерилизацией. Дисперсии могут быть приготовлены введением различных стерилизованных активных ингредиентов в стерильный растворитель, содержащий основную дисперсионную среду и другие необходимые компоненты из описанных выше. В случае стерильных порошков, используемых для приготовления стерильных растворов для инъекций, предпочтительными способами приготовления являются вакуумная сушка и сушка замораживанием, в результате которых получают порошок активного ингредиента плюс любые дополнительные желаемые компоненты из описанных выше.
Фармацевтические препараты для местного введения могут быть особенно удобны для локального воздействия определенными биологически-активными соединениями. Препараты для местного воздействия включают мази, аэрозоли, гели, суспензии, примочки, кремы и т.п. Препараты для местного введения могут содержать, в дополнение к глинкозидам, известные носители, такие как изопропанол, глицерин, парафин, стеариловый спирт, полиэтиленгликоль и т.д. Фармацевтически приемлемый носитель может также содержать известный химический активатор абсорбции. Примером активатора абсорбции является, например, диметилацетамид (патент США N 3, 472, 931), трихлороэтанол или трифтороэтанол (патент США N 3, 891, 757), определенные спирты и их смеси (патент Великобритании N 1, 001, 949 и описание к патенту Великобритании N 1, 464, 975).
Растворы глинкозидов могут сохраняться и/или вводиться в виде свободного основания или фармакологически приемлемых солей и могут быть приготовлены в смеси воды с поверхностно-активным веществом, таким как гидроксипропилцеллюлоза. Дисперсии могут быть также приготовлены в глицерине, жидких полиэтиленгликолях, их смесях и в маслах. Эти составы и препараты могут содержать консервант для предупреждения роста микроорганизмов.
Предупреждение воздействия микроорганизмов может быть осуществлено различными антимикробными и противогрибковыми агентами, например парабеном, хлоробутанолом, фенолом, сорбиновой кислотой, тимерозалом и т.п. В большинстве случаев желательно включать изотонические агенты, такие как хлорид натрия. Пролонгирование введения составов для инъекций может быть осуществлено путем использования агентов, замедляющих абсорбцию, таких как моностеарат алюминия и желатин.
Описанные составы и препараты предпочтительно содержат по меньшей мере 0.1% активного глинкозида. Процентный состав препаратов может варьироваться и может содержать примерно от 2 мас.% до 60 мас.% вводимого количества. Количество активных соединений в терапевтических составах и препаратах таково, чтобы была получена подходящая доза.
Здесь подразумевается, что "фармацевтически приемлемый носитель" включает любой из растворителей, дисперсионные среды, оболочки, антимикробные и противогрибковые агенты, изотонические и замедляющие абсорбцию агенты и т.п. Использование подобных сред и агентов для фармацевтически активных веществ хорошо известно из уровня техники. За исключением случаев, когда какая-то обычная среда или агент несовместимы с терапевтически активными ингредиентами, их использование в терапевтических составах рассматривается. В составы и препараты могут также быть включены дополнительные активные ингредиенты.
В дополнение к терапевтическому применению глинкозидов, они могут также использоваться как лабораторный инструмент для изучения абсорбции, распределения, клеточного поглощения и эффективности
ЭКВИВАЛЕНТЫ
Вышеизложенное подробное описание является достаточным, чтобы опытные специалисты могли осуществить изобретение на практике. Действительно, различные модификации вышеизложенного, дающие возможность для осуществления изобретения, которые очевидны для специалистов в области органической химии и близких областях науки, попадают в объем изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ТИАЗОФУРИНА И ДРУГИХ С-НУКЛЕОЗИДОВ | 1998 |
|
RU2185385C2 |
| НУКЛЕОЗИДЫ С МОДИФИЦИРОВАННЫМИ САХАРАМИ И ОЛИГОНУКЛЕОТИДЫ | 1995 |
|
RU2145964C1 |
| МОНОЦИКЛИЧЕСКИЕ L-НУКЛЕОЗИДЫ, ИХ АНАЛОГИ И ПРИМЕНЕНИЯ | 1997 |
|
RU2188828C2 |
| НОВЫЕ НУКЛЕОЗИДЫ, ИМЕЮЩИЕ БИЦИКЛИЧЕСКУЮ САХАРНУЮ ГРУППИРОВКУ, И СОДЕРЖАЩИЕ ИХ ОЛИГОНУКЛЕОТИДЫ | 1999 |
|
RU2211223C2 |
| ПУРИНОВЫЕ L-НУКЛЕОЗИДЫ, ИХ АНАЛОГИ И ПРИМЕНЕНИЕ | 1997 |
|
RU2183639C2 |
| НУКЛЕИНОВЫЕ КИСЛОТЫ, МОДИФИЦИРОВАННЫЕ АМИНОКИСЛОТАМИ | 1995 |
|
RU2154638C2 |
| МОДУЛЯЦИЯ ЭКСПРЕССИИ ТН1/ТН2 ЦИТОКИНОВ РИБАВИРИНОМ И АНАЛОГАМИ РИБАВИРИНА В АКТИВИРОВАННЫХ Т-ЛИМФОЦИТАХ | 1997 |
|
RU2186569C2 |
| МОДУЛЯЦИЯ ИММУННОГО ОТВЕТА С ПОМОЩЬЮ РИБАВИРИНА | 1999 |
|
RU2211698C2 |
| Способ получения гетероциклических кетонов | 1988 |
|
SU1678207A3 |
| Способ получения производного цинолина или его кислотно-аддитивных солей | 1986 |
|
SU1500158A3 |

Описываются новые соединения - глинкозиды формулы MS-LINK-BAM, где MS моносахарид; LINK - линкер; BAM - биологически активный материал; MS ковалентно присоединен к LINR в ином положении, чем С, углерод MS, и LINR ковалентно присоединен к BAM. Они повышают клеточное поглощение биологически активных материалов посредством ковалентного присоединения таких соединений к углеводной части через химические линкеры с использованием не-гликозидных связей. Описывается также способ повышения биологической доступности биологически активного материала и способ воздействия на живой организм биологически активного материала. 5 с. и 10 з.п.ф-лы, 2 табл., 14 ил.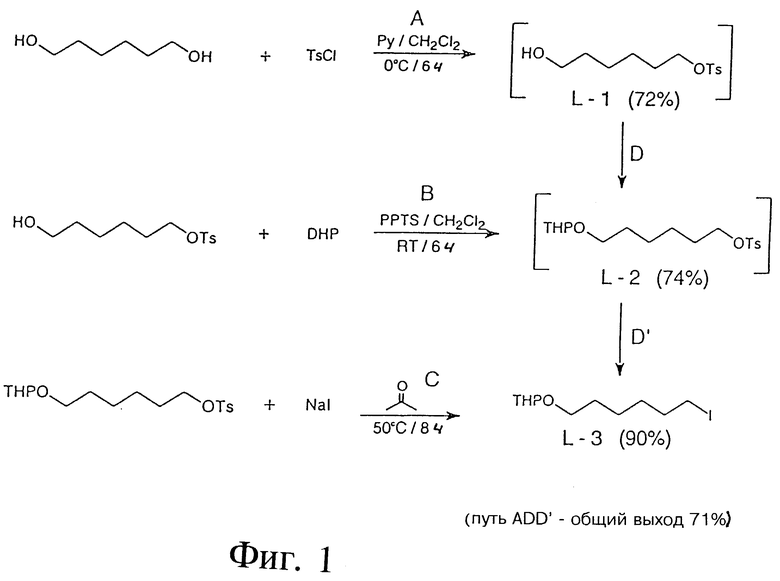
| СПОСОБ ПОЛУЧЕНИЯ НИЗКОАЛКИЛ-2-0-(р- | 0 |
|
SU298117A1 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Molema et al "TARGETING OF ANTIVIRAL DRUG | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| COPROTEIN - AZTMP CONJUGATES IN VITRO" in BIOCHEMICAL PHARMACOLOGY, 1990, N 12, v.40, p.2603 - 2610 | |||
| Gousho A | |||
| et al - in Biol | |||
| Pharma Bull., 1994, 17(2), pp.275 - 282 | |||
| Mizuma T | |||
| et al - in Biochem | |||
| Pharma, 1994, 43(9), pp.2037 - 2039. | |||

