Область техники, к которой относится изобретение
Настоящее изобретение относится к новым конденсированным гетероциклическим производным, обладающим аффинностью к рецепторам S1P, к фармацевтической композиции, содержащей указанные соединения, а также к применению указанных соединений для получения лекарственного средства для лечения, смягчения или предупреждения заболеваний и состояний, в которые вовлечен любой из рецепторов S1P или в которые вовлечено модулирование эндогенной системы передачи сигнала S1P через любой из рецепторов S1P.
Уровень техники, к которому относится изобретение
Сфингозин-1-фосфат (S1P) является биоактивным сфинголипидом, который опосредует широкое множество клеточных ответов, таких как пролиферация, организация и миграция цитоскелета, сборка адгезионных соединений и прочных контактов и морфогенез. S1P может связываться с представителями семейства генов дифференцировки эндотелиальных клеток (рецепторы EDG), включающего локализованные на плазматической мембране сопряженные с G-белком рецепторы. На сегодняшний день пять представителей этого семейства идентифицировано в качестве рецепторов S1P в различных типах клеток, S1P1 (EDG-1), S1P2 (EDG-5), S1P3 (EDG-3), S1P4 (EDG-6) и S1P5 (EDG-8). S1P может вызывать перестройку цитоскелета во многих типах клеток для регуляции транспорта иммунных клеток, гомеостаза сосудов и сообщения клеток в центральной нервной системе (ЦНС) и в периферических системах органов.
Известно, что S1P секретируется эндотелием сосудов и присутствует в крови в концентрации 200-900 наномоль и связан с альбумином и другими белками плазмы. Это обеспечивает как стабильный резервуар во внутриклеточных жидкостях, так и эффективную доставку к высокоаффинным рецепторам клеточной поверхности. S1P связывается с низкой наномолярной аффинностью с пятью рецепторами S1P1-5. Кроме того, тромбоциты также содержат S1P, и он может локально высвобождаться, вызывая, например, сужение сосудов. Подтипы рецепторов S1P1, S1P2 и S1P3 широко экспрессируются и представляют собой преобладающие рецепторы в сердечно-сосудистой системе. Кроме того, S1P1 также является рецептором на лимфоцитах. Рецепторы S1P4 расположены практически исключительно в гемопоэтической и лимфоидной системах. S1P5 в основном (хотя и не исключительно) экспрессируется в центральной нервной системе. Экспрессия S1P5, по-видимому, ограничивается олигодендроцитами у мышей, миелинизирующими клетками головного мозга, в то время как у крыс и человека была выявлена экспрессия на уровне астроцитов и эндотелиальных клеток, но не на олигодендроцитах.
Модуляторы рецепторов S1P представляют собой соединения, которые передают сигнал в качестве (ант)агонистов на один или несколько рецепторов S1P. Настоящее изобретение относится к модуляторам рецептора S1P5, в частности, к агонистам, и предпочтительно к агонистам с селективностью в отношении рецепторов S1P1 и/или S1P3, ввиду нежелательных сердечно-сосудистых/или иммуномодулирующих эффектов. В настоящее время установлено, что агонисты S1P5 можно использовать для лечения когнитивных нарушений, в частности, связанного со старением снижения когнитивной способности.
Хотя продолжаются исследования по разработке лекарственных средств, которые можно использовать для лечения связанного со старением снижения когнитивной способности и деменции, это еще не привело к множеству успешных кандидатов. Таким образом, существует потребность в новых терапевтических средствах с желаемыми свойствами.
Описание изобретения
В рамках изобретения было выявлено, что конденсированные гетероциклические производные формулы (I):
Конденсированное гетероциклическое производное формулы (I)
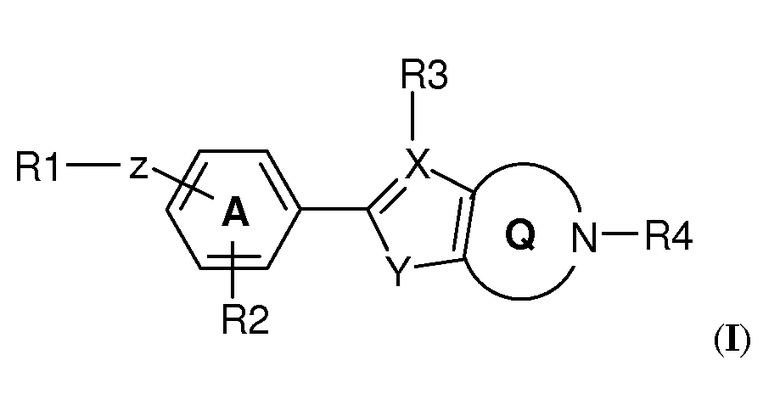 ,
,
где
R1 выбран из
циано,
(2-4C)алкенила, (2-4C)алкинила, (1-4C)алкила, каждый из которых необязательно замещен CN или одним или несколькими атомами фтора,
(3-6C)циклоалкила, (4-6C)циклоалкенила или (8-10C)бициклической группы, каждый из которых необязательно замещен галогеном или (1-4C)алкилом, необязательно замещенным одним или несколькими атомами фтора,
фенила, бифенила, нафтила, каждый из которых необязательно замещен одним или несколькими заместителями, независимо выбранными из галогена, циано, (1-4C)алкила, необязательно замещенного одним или несколькими атомами фтора, (1-4C)алкокси, необязательно замещенного одним или несколькими атомами фтора, амино, диметиламино, и (3-6C)циклоалкила, необязательно замещенного фенилом, который может быть замещен (1-4C)алкилом или галогеном, и
фенила, замещенного фенокси, бензила, бензилокси, фенилэтила или моноциклического гетероцикла, каждый из которых необязательно замещен (1-4C)алкилом,
Z представляет собой линкерную группу-W-(Cn-алкилен)-T-, где
W связан с R1 и выбран из связи, -O-, -CO-, -S-, -SO-, -SO2-, -NH-, -CH=CH-, -C(CF3)=CH-, -C≡C-, -CH2-O-, -O-CO-, -CO-O-, -CO-NH-, -NH-CO- и транс-циклопропилена;
n представляет собой целое число от 0 до 10; и
T связан с частью фенилена/пиридила и выбран из связи, -O-, -S-, -SO-, -SO2-, -NH-, -CO-, -C=C-, -C≡C- и транс-циклопропилена;
R2 представляет собой H или один или несколько заместителей, независимо выбранных из циано, галогена, (1-4C)алкила, необязательно замещенного одним или несколькими атомами галогена, или (1-4C)алкокси, необязательно замещенного одним или несколькими атомами галогена;
кольцевая структура A может содержать один атом азота;
X выбран из C или N; если X представляет собой C, R3 выбран из H и (1-4C)алкила, в ином случае R3 отсутствует;
Y выбран из NH, O и S;
структура Q представляет собой 5-, 6- или 7-членный циклический амин;
и
R4 представляет собой (1-4C)алкилен-R5, где один или несколько атомов углерода в алкиленовой группе могут быть независимо замещены одним или несколькими атомами галогена или (CH2)2, образуя циклопропильную часть, или R4 представляет собой (3-6C)циклоалкилен-R5, -CH2-(3-6C)циклоалкилен-R5, (3-6C)циклоалкилен-CH2-R5 или -CO-CH2-R5, где R5 представляет собой -OH, -PO3H2, -OPO3H2, -COOH, -COO(1-4C)алкил или тетразол-5-ил;
или его фармацевтически приемлемая соль, сольват или гидрат, или один или несколько их N-оксидов, проявляют аффинность к рецепторам S1P. В частности, соединения по изобретению демонстрируют селективную аффинность к рецептору S1P5 относительно рецептора(ов) S1P1 и/или S1P3.
В WO 2008/012010 некоторые из описанных соединений имеют некоторое структурное сходство с соединениями по настоящему изобретению; однако они описаны в качестве лигандов H3-рецепторов гистамина.
Соединения по изобретению являются модуляторами рецептора S1P, в частности, рецептора S1P5. Более конкретно, соединения по изобретению представляют собой агонисты рецептора S1P5. Соединения по изобретению пригодны для лечения, смягчения и профилактики заболеваний и состояний, в которые вовлечены (любые) рецептор(ы) S1P, в частности S1P5, или в которые вовлечено модулирование эндогенной системы передачи сигнала S1P через любой из рецепторов S1P. В частности, соединения по настоящему изобретению можно использовать для лечения, смягчения или профилактики нарушений ЦНС (центральная нервная система), таких как нейродегенеративные нарушения, в частности, но не ограничиваясь ими, когнитивные нарушения (в частности, связанное со старением снижение когнитивной способности) и родственные состояния, болезнь Альцгеймера, (сосудистая) деменция, болезнь Ниманна-Пика, и нарушение когнитивной способности при шизофрении, обсессивно-компульсивное поведение, большая депрессия и аутизм, рассеянный склероз, боль и т.д. Предпочтительно, соединения по настоящему изобретению можно использовать для лечения, смягчения или профилактики когнитивных нарушений (в частности, связанного со старением снижения когнитивной способности) и родственных состояний.
В предпочтительном варианте осуществления изобретения, соединения имеют формулу (I), где
R1 выбран из
(3-6C)циклоалкила или (8-10C)бициклической группы, необязательно замещенных галогеном, (1-4C)алкила, и
фенила, необязательно замещенного одним или несколькими заместителями, независимо выбранными из галогена, циано, (1-4C)алкила, (1-4C)алкокси, трифторметила и трифторметокси;
W выбран из связи, -O-, -CO-, -S-, -SO-, -SO2-, -NH-, -CH=CH-, -C≡C- и транс-циклопропилена; и
n представляет собой целое число от 0 до 4; и предпочтительно n выбран из 0, 1 и 2; и
R2 представляет собой H или один или несколько заместителей, независимо выбранных из галогена, (1-4C)алкила, необязательно замещенного одним или несколькими атомами фтора, или (1-4C)алкокси, необязательно замещенного одним или несколькими атомами фтора;
и где определение других групп/символов является таким, как определено выше.
В другом варианте осуществления соединение по изобретению имеет структуру (Ia)
 .
.
В одном варианте осуществления изобретения кольцевая структура Q представляет собой 6-членное кольцо. В частности, соединение по изобретению имеет структуру (Ib)
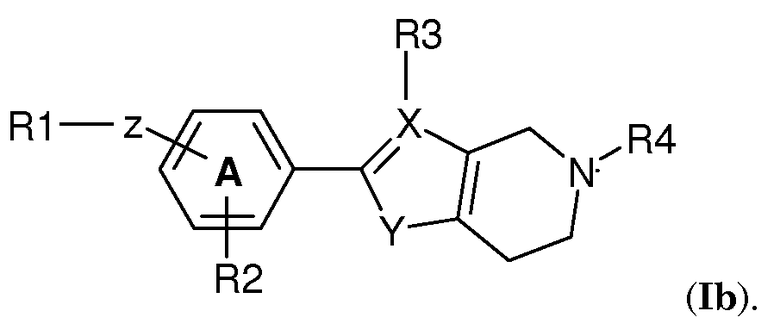
В следующем варианте осуществления изобретения R4 выбран из -(CH2)2-OH, -CH2-COOH, -(CH2)2-COOH, -(CH2)3-COOH, -CH2-CHCH3-COOH, -CH2-C(CH3)2-COOH, -CHCH3-CH2-COOH, -CH2-CF2-COOH, -CO-CH2-COOH,  , 1,3-циклобутилен-COOH, -(CH2)2-PO3H2, -(CH2)3-PO3H2, -(CH2)2-OPO3H2, -(CH2)3-OPO3H2, -CH2-тетразол-5-ила, -(CH2)2-тетразол-5-ила и -(CH2)3-тетразол-5-ила. Предпочтительные группы R4 выбраны из -(CH2)2-COOH, -(CH2)3-COOH, -CH2-CHCH3-COOH, -CH2-C(CH3)2-COOH, -CHCH3-CH2-COOH, -CH2-CF2-COOH. Высоко предпочтительными являются -(CH2)2-COOH, -CHCH3-CH2-COOH, -CH2-CHCH3-COOH и 1,3-циклобутилен-COOH. Особенно предпочтительным является -CH2-CHCH3-COOH.
, 1,3-циклобутилен-COOH, -(CH2)2-PO3H2, -(CH2)3-PO3H2, -(CH2)2-OPO3H2, -(CH2)3-OPO3H2, -CH2-тетразол-5-ила, -(CH2)2-тетразол-5-ила и -(CH2)3-тетразол-5-ила. Предпочтительные группы R4 выбраны из -(CH2)2-COOH, -(CH2)3-COOH, -CH2-CHCH3-COOH, -CH2-C(CH3)2-COOH, -CHCH3-CH2-COOH, -CH2-CF2-COOH. Высоко предпочтительными являются -(CH2)2-COOH, -CHCH3-CH2-COOH, -CH2-CHCH3-COOH и 1,3-циклобутилен-COOH. Особенно предпочтительным является -CH2-CHCH3-COOH.
В другом предпочтительном варианте осуществления соединения имеют формулу (I), где Y представляет собой O.
Кроме того, в предпочтительном варианте осуществления изобретения, X представляет собой N.
В предпочтительных вариантах осуществления изобретения R1 представляет собой инданил, необязательно замещенный галогеном, (1-4C)алкилом, или более предпочтительный R1 представляет собой необязательно замещенный фенил, где необязательные заместители выбраны из любого из ранее определенных заместителей, однако, в частности, необязательные заместители представляют собой один или несколько заместителей, независимо выбранных из галогена, циано, (1-4C)алкила, (1-4C)алкокси, трифторметила и трифторметокси. В высоко предпочтительных вариантах осуществления R1 представляет собой 4Cl-фенил или 4CF3-фенил.
В одном из вариантов осуществления изобретения R2 представляет собой H или один или несколько заместителей, независимо выбранных из метила, метокси, хлора или фтора. В предпочтительном варианте осуществления R2 представляет собой H, или R2 представляет собой один метил, один метокси, один атом хлора, один атом хлора, или один или два атома фтора.
В вариантах осуществления изобретения, где X представляет собой CR3, R3 представляет собой предпочтительно H или метил, и, в частности, R3 представляет собой H.
Кроме того, в одном варианте осуществления изобретения Z представляет собой линкерную группу -W-(CH2)n-T-, значение которой выбрано из связи, -O-, -CO-, -S-, -SO2-, -NH-, -CH2-, -(CH2)2-, -CCH3-O-, -CH=CH-, -C≡C-, -CH2-O-, -O-CH2-, -CH2-S-, -S-CH2-, -CH2-SO2-, -SO2-CH2-, -CH2-NH-, -NH-CH2- и транс-циклопропилена. В предпочтительных вариантах осуществления Z представляет собой -O-, -CH2-O- или транс-циклопропилен. В частности, Z представляет собой -CH2-O-.
Термин галоген относится к фтору, хлору, брому или йоду. Предпочтительными галогенами являются фтор и хлор, и в частности хлор.
Термин (1-4C)алкил означает разветвленную или неразветвленную алкильную группу, имеющую 1-4 атомов углерода, например, метил, этил, пропил, изопропил и бутил. Предпочтительной алкильной группой является метил.
Термин (1-4C)алкокси означает алкоксигруппу, имеющую 1-4 атомов углерода, где алкильная часть является такой, как определено выше. Предпочтительной алкоксигруппой является метокси.
Термины (1-4C)алкилен и (Cn-алкилен) означают разветвленную или неразветвленную алкиленовую группу, имеющую 1-4 или n атомов углерода, соответственно, например, метилен, -CHCH3-, -C(CH3)2-, -CHCH3CH2- и т.п. В определении R4, который представляет собой (1-4C)алкилен-R5, один или несколько атомов углерода в алкиленовой группе (среди прочих) может быть независимо замещен (CH2)2, образуя циклопропильную часть, т.е. образуя группу R4, такую как  .
.
Термин (2-4C)алкинил означает разветвленную или неразветвленную алкинильную группу, имеющую 2-4 атома углерода, где тройная связь может присутствовать в различных положениях в группе, например, этинил, пропаргил, 1-бутинил, 2-бутинил и т.д.
Термин (3-6C)циклоалкил означает циклическую группу, имеющую 3-6 атомов углерода, соответственно, циклопропил, циклобутил, циклопентил или циклогексил. Предпочтительными являются циклопентил и циклогексил.
Термин (4-6C)циклоалкенил означает циклическую алкенильную группу, имеющую 4-6 атомов углерода и содержащую одну или две двойных связи, например, циклогексенил.
Термин (3-6C)циклоалкилен означает циклическую алкильную группу, имеющую две точки присоединения. Предпочтительным является 1,3-циклобутилен, имеющий структуру
 .
.
Термин (8-10C)бициклическая группа означает конденсированную кольцевую систему из двух групп, выбранную из ароматических и неароматических кольцевых структур, имеющих вместе 8-10 атомов углерода, например, индановую группу.
Что касается заместителей, термин "независимо" означает, что заместители могут быть одинаковыми или могут отличаться друг от друга в одной и той же молекуле.
Соединения по изобретению можно пригодным образом получать способами, доступными в данной области, и как проиллюстрировано в экспериментальном разделе этого описания.
Соединения по настоящему изобретению могут содержать один или несколько асимметричных центров и, таким образом, могут встречаться в качестве рацематов и рацемических смесей, единичных энантиомеров, диастереомерных смесей и отдельных диастереомеров. Могут присутствовать дополнительные центры асимметрии, в зависимости от природы различных заместителей на молекуле. Каждый такой центр асимметрии независимо обеспечивает два оптических изомера, и подразумевается, что в объем этого изобретения входят все возможные изомеры и диастереомеры в смесях и в качестве чистых или частично очищенных соединений. Подразумевается, что настоящее изобретение охватывает все такие изомерные формы этих соединений. Можно осуществлять независимый синтез диастереомеров или их хроматографическое разделение, как известно в данной области, путем соответствующей модификации методологии, описанной в настоящем описании. Их абсолютную стехиометрию можно определять с помощью рентгеновской кристаллографии кристаллических продуктов или кристаллических промежуточных соединений, которые являются преобразованными, при необходимости, реагентом, содержащим центр асимметрии с известной абсолютной конфигурацией. Если желательно, можно разделять рацемические смеси соединений, так чтобы выделять отдельные энантиомеры. Разделение можно проводить способами, хорошо известными в данной области, такими как присоединение соединений в рацемической смеси к энантиомерно чистому соединению с образованием диастереомерной смеси, с последующим разделением отдельных диастереомеров стандартными способами, такими как фракционная кристаллизация или хроматография.
Соединения могут существовать в качестве полиморфов, и подразумевается, что они как таковые включены в настоящее изобретение. Кроме того, соединения могут образовывать сольваты с водой (т.е. гидраты) или обычными органическими растворителями, и подразумевается, что такие сольваты также охватываются объемом настоящего изобретения.
Также в объем изобретения входят изотопно меченное соединение формулы (I) или его фармацевтически приемлемые соли, включая соединения формулы (I), изотопно меченные, чтобы они поддавались детекции с помощью PET или SPECT. То же самое справедливо для соединений формулы (I), меченных [13C]-, [14C]-, [3H]-, [18F]-, [125I]- или другими изотопно обогащенными атомами, пригодными для исследований связывания рецепторов или метаболизма.
Термин "фармацевтически приемлемая соль" относится к солям, которые, по мнению медицинского специалиста, пригодны для применения в контакте с тканями человека и низших животных без чрезмерной токсичности, раздражения, аллергического ответа и т.п., и имеют приемлемое соотношение польза/риск. Фармацевтически приемлемые соли хорошо известны в данной области. Их можно получать in situ при выделении и очистке соединений по изобретению, или отдельно путем их реакции с фармацевтически приемлемыми нетоксичными основаниями или кислотами, включая неорганические или органические основания и неорганические или органические кислоты.
Соединения по изобретению можно вводить энтерально или парентерально. Точная доза и режим для этих соединений и их композиций будут зависеть от биологической активности самого соединения, возраста, массы тела и пола пациента, потребности отдельного индивидуума, которому вводят лекарственное средства, степени тяжести или потребности, и мнения практикующего медицинского специалиста. Как правило, парентеральное введение требует более низких дозировок, чем другие способы введения, которые более зависят от всасывания. Однако дозировки для человека предпочтительно составляют 0,001-10 мг на кг массы тела. Как правило, энтеральные и парентеральные дозировки находятся в диапазоне от 0,1 до 1000 мг всех активных ингредиентов в сутки.
Смешанные с фармацевтически приемлемыми добавками, например, как описано в стандартном справочнике “Remington, The Science и Practice of Pharmacy” (21st edition, Lippincott Williams & Wilkins, 2005, см. особенно, Part 5: Pharmaceutical Manufacturing), соединения можно прессовать в твердые дозированные единицы, такие как пилюли или таблетки, или их можно преобразовывать в капсулы или суппозитории. С помощью фармацевтически приемлемых жидкостей соединения также можно применять в форме раствора, суспензии или эмульсии.
Для получения дозированных единиц, например таблеток, предусмотрено использование общепринятых добавок, таких как наполнители, красители, полимерные связующие агенты и т.п. Как правило, можно использовать любую фармацевтически приемлемую добавку, которая не препятствует функции активных соединений.
Пригодные носители, с которым можно вводить соединения по изобретению, включают, например, лактозу, крахмал, производные целлюлозы и т.п., или их смеси, используемые в подходящих количествах. Композиции для внутривенного введения могут представлять собой, например, растворы соединений по изобретению в стерильном изотоническом водном буфере. Когда это необходимо, внутривенные композиции могут включать, например, солюбилизирующие агенты, стабилизаторы и/или местный анестетик для облегчения боли в области инъекции.
Фармацевтические композиции по изобретению можно изготавливать для любого пути введения, и они содержат по меньшей мере одно соединение по настоящему изобретению и его фармацевтически приемлемые соли с любым фармацевтически приемлемым ингредиентом, эксципиентом, носителем, адъювантом или наполнителем.
Под "фармацевтически приемлемым" понимают, что носитель, разбавитель или эксципиент должны быть совместимы с другими ингредиентами состава и не должны быть вредоносными для их реципиента.
В одном варианте осуществления изобретения предусмотрена фармацевтическая упаковка или набор, содержащие один или несколько контейнеров, заполненных одной или несколькими фармацевтическими композициями по изобретению. К такому контейнеру(ам) могут прилагаться различные письменные материалы, такие как инструкции по применению, или указание в форме, предписанной правительственным учреждением, регулирующим изготовление, применение или продажу фармацевтических продуктов, которое отражает одобрение учреждением изготовления, применения или продажи для введения человеку или ветеринарного введения.
Если не определено иначе, все технические и научные термины, используемые в настоящем описании, обладают тем же значением, которое обычно подразумевает специалист в области, к которой относится это изобретение. Хотя при применении на практике или исследовании настоящего изобретения можно использовать способы и материалы, сходные или эквивалентные способам и материалам, описанным в настоящем описании, пригодные способы и материалы описаны в настоящем описании.
ПЕРЕВОД НАДПИСЕЙ НА ЧЕРТЕЖАХ:
Фиг. 1. Процентное изменение у молодых и пожилых самцов мышей C57BL/6J в T-лабиринте либо в случае носителя (контрольные группы), либо в случае соединения по изобретению (доза мг/кг; п. о.)
Представленные ниже примеры предназначены для дальнейшей более подробной иллюстрации изобретения.
Любое новое промежуточное соединение, описанное в настоящем описании, является дополнительным вариантом осуществления настоящего изобретения.
ПРИМЕРЫ
§1. АНАЛИТИЧЕСКИЕ СПОСОБЫ
Спектры ядерного магнитного резонанса (1H-ЯМР и 13C-ЯМР, APT) определяли в указанном растворителе с использованием Bruker ARX 400 (1H: 400 МГц, 13C: 100 МГц) при 300 K, если нет иных указаний. Эксперименты 19F-ЯМР и 13C-ЯМР проводили на спектрометре Varian Inova 500 при 11,74 T (499,9 МГц для 1H; 125,7 МГц для 13C; 50,7 МГц, 470,4 МГЦ для 19F) с использованием 5-мм зонда SW. Спектры определяли в дейтерированном хлороформе или DCM, полученных от Cambridge Isotope Laboratories Ltd. Химический сдвиг (δ) приведен в м.д. в сторону слабого поля относительно тетраметилсилана (1H, 13C) или CCl3F (19F). Константы взаимодействия J приведены в Гц. Формы пиков в спектрах ЯМР обозначены с помощью символов “кв” (квартет), “дкв” (двойной квартет), “т” (триплет), “дт” (двойной триплет), “д” (дублет), “дд” (двойной дублет), “с” (синглет), “уширенный спектр” (уш.с) и “м” (мультиплет). Сигналы NH и OH идентифицировали после смешения образца с каплей D2O.
Флэш-хроматография относится к очистке с использованием указанного элюента и силикагеля (силикагель либо Acros: 0,030-0,075 мм, либо Merck 60: 0,040-0,063 мм).
Колоночную хроматографию проводили с использованием силикагеля 60 (0,063-0,200 мм, Merck).
Мониторинг реакций проводили с использованием тонкослойной хроматографии (TLC) на покрытых диоксидом кремния пластмассовых листах (предварительно покрытый силикагель 60 F254, Merck) с указанным элюентом. Пятна визуализировали с помощью УФ-света (254 нм) или I2.
Температуры плавления регистрировали на устройстве для определения температуры плавления Büchi B-545.
Жидкостная хроматография-масс-спектрометрия (LC-MS):
- Способ A.
Система LC-MS состоит из µ-насоса Waters 1525. Насос соединен с автоматическим пробоотборником Waters 2777.
Способ LC представляет собой:
B=100% ACN с 0,2% HCOOH
Автоматический пробоотборник имеет 10-мкл инжекционную петлю; объем инжекции составляет 10 мкл. Автоматический пробоотборник соединен с колонкой Waters Sunfire C18, 30*4,6 мм, с 2,5-мкм частицами. Колонка термостатируется при комнатной температуре +/- 23°C.
Колонка соединена с Waters 2996 PDA. Сканирование длины волны происходит от 240 до 320 нм. Разрешение составляет 1,2 нм и частота взятия образца составляет 20 Гц. После PDA поток разделяется 1:1 и соединяется с Waters 2424 ELSD.
ELSD имеет следующие параметры:
Давление газа: 40 фунт/кв. дюйм
Скорость передачи данных: 20 точек/с
Усиление: 500
Константа времени: 0,2 с
Режим распылителя: охлаждение
Пролетная трубка: 50°C
Образцы также исследовали с помощью детектора масс Waters ZQ.
Масс-спектрометр имеет следующие параметры:
Диапазон сканирования: 117-900 а.е.м.
Полярность: положительная
Формат данных: центроидный
Время на сканирование: 0,500 с
Время между сканированиями: 0,05 с
Капилляры: 2,5 кВ
Конус: 25 В
Экстрактор: 2 В
RF-линза: 0,5 В
Температура источника: 125°C
Температура десольватации: 400°C
Газ в конусе: 100 л/ч
Газ десольватации: 800 л/ч
Разрешение LM 1: 15
Разрешение HM 1: 15
Энергия ионов: 0,5
Усилитель: 500 В
Вся система контролируется Masslynx 4.1.
- Способ B.
Система LC-MS состоит из 2 микронасосов Perkin Elmer серии 200. Насосы соединены друг с другом с помощью 50-мкл T-образного смесителя. Смеситель соединен с автоматическим пробоотборником Gilson 215.
Способ LC представляет собой:
B=100% ацетонитрил с 0,1% HCOOH
Автоматический пробоотборник имеет 2-мкл инжекционную петлю. Автоматический пробоотборник соединен с колонкой Waters Sunfire C18, 30*4,6 мм, с 2,5-мкм частицами. Колонка термостатируется в термостате колонки Perkin Elmer серии 200 при 23°C. Колонка соединена с измерительным устройством Perkin Elmer 785 UV/VIS meter с 2,7-мкл проточной ячейкой. Длина волны установлена на 254 нм. Измерительное устройство в УФ-диапазоне соединено с масс-спектрометром Sciex API 150EX. Масс-спектрометр имеет следующие параметры:
Диапазон сканирования: 100-900 а.е.м.
Полярность: положительная
Режим сканирования: профильный
Разрешение Q1: единицы
Размер шага: 0,10 а.е.м.
Время на сканирование: 0,500 с
NEB: 10
CUR: 10
IS: 5200
TEM: 325
DF: 30
FP: 225
EP: 10
Детектор рассеяния света соединен с Sciex API 150. Детектор рассеяния света представляет собой Polymer Labs PL-ELS 2100, действующий при 70°C и давлении N2 1,7 бар.
Вся система контролируется компьютером Dell precision GX370, работающем на Windows 2000.
Описанное время удержания в таблице 1 (Rt) приведено для пика на хроматограмме общего ионного тока (TIC), которая показывала массу для [M+H]+ с точностью в пределах 0,5 а.е.м. от вычисленной точной ММ и имела ассоциированный пик в хроматограмме испарительного рассеяния света (ELS) с относительной площадью % (чистотой) >85%.
§2. СОКРАЩЕНИЯ
§3. ОСНОВНЫЕ АСПЕКТЫ СИНТЕЗА
Пригодные способы синтеза заявленных соединений описаны ниже.
Для синтеза соединений 1 описаны два пути, соответственно, на схемах 2 и 3. Оба пути начинаются с соединения 6, синтез которого представлен на схеме 1. Альфа-алкилирование пирролидин-енамина 4 альфа-бром-ацетофенонами (3), тем самым, вводящее Rb-группу в молекулу, дает соединение 5. Последующие замыкание кольца 5 в кислотных условиях дало соединение 6 с достаточным выходом.
Схема 1. Синтез ключевого промежуточного соединения 6

Путь A (см. схему 2) начинается алкилированием пиперидиновой части в 6 путем либо стандартного алкилирования, либо восстановительного алкилирования, либо реакции присоединения Майкла, с получением соединений 7 с защищенной карбоновой кислотой. Часть бензилового простого эфира может быть введена двумя путями. Во-первых, бром в 7 можно прямо превращать в производное бензилового эфира 9 с помощью катализируемой палладием реакции. Кроме того, бромид 7 можно конвертировать в производное фенола 8 через опосредуемую палладием реакцию. Соединение 8 можно конвертировать в желаемые производные бензилового эфира 9 в условиях фазового переноса с бензилбромидами или через реакцию Мицунобу с бензиловыми спиртами. Наконец, из соединений 9 можно удалять защитную группу с получением конечных продуктов 1.
Схема 2. Путь A к соединениям 1

Альтернативно, для синтеза соединений 1 можно следовать пути B (см. схему 3). Пиперидин в соединении 6 защищали группой BOC. После этого, сначала часть бензилового простого эфира вводили либо путем прямой опосредуемой палладием реакции брома в 10 с 12, либо через преобразование брома в фенольное производное 11, которое можно было конвертировать в 12 в условиях алкилирования или Мицунобу. Наконец, соединение 12 можно было конвертировать в 9 удалением кислотой группы BOC и последующим введением защищенных концевых частей карбоновых кислот.
Схема 3. Путь B к соединениям 1

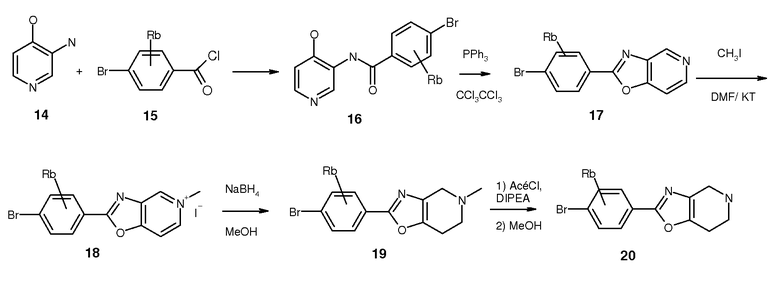
Для синтеза оксазолопроизводных 2 было разработано три пути. Синтез ключевого промежуточного соединения 20 представлен на схеме 4. Ацилирование коммерчески доступного 14 надлежащим образом защищенным бензоилхлоридом (15) дало 16, которое затем подвергали замыканию кольца до 17 с использованием трифенилфосфина и гексахлорэтана. Метилирование пиридина в 17 в четвертичную соль 18 и последующее восстановление 18 боргидридом натрия дали соединение 19. Соединение 19 деметилировали 1-хлорэтилхлорформиатом с получением ключевого промежуточного соединения 20.
Схема 4. Синтез соединений 20
Первый путь (путь C) к соединениям 2 представлен на схеме 5, и он начинается с соединения 20. Аналогично тому, как описано для синтеза соединений 7 в фуранильной серии, в 20 можно вводить концевые части в виде защищенных трет-бутилом карбоновых кислот с получением 21. Начиная с 21, производные бензилового эфира 23 можно получать либо путем прямой опосредуемой палладием реакции сочетания (от 21 к 23) с бензиловыми спиртами, либо путем первоначального преобразования бромида в 21 в фенол 22 и последующего бензилирования 22 (до 23) в условиях фазового перехода или Мицунобу. Наконец удаление защитной группы кислотой из карбоновой кислоты в 23 давало соединения 2.
Схема 5. Путь к соединениям 2
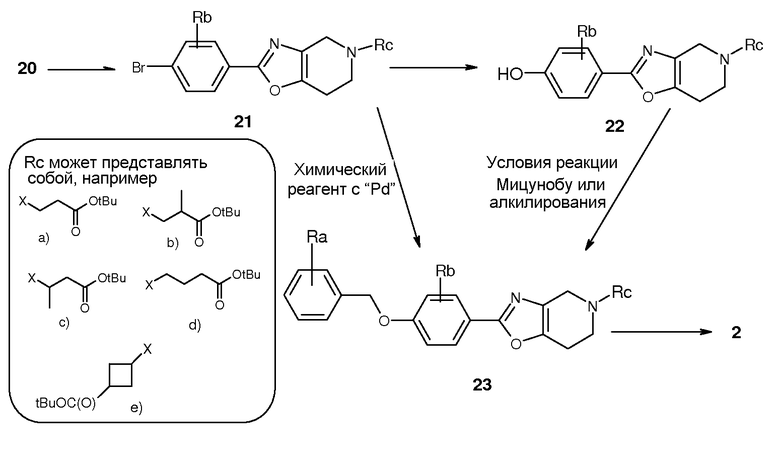
Альтернативно, можно следовать пути D, как представлено на схеме 6. Соединения 25 можно получать, начиная с 14 и надлежащим образом замещенного производного 4-бензилоксибензойной кислоты (24) под действием трифенилфосфина и трихлорацетонитрила. Соединение 25 можно конвертировать в бензилоксипроизводные 23 аналогично тому, как описано выше на схемах 4 и 5 для синтеза соединений 21. Таким образом, метилирование 25 и последующее восстановление NaBH4 дали 26, которое деметилировали с помощью ACE-Cl с получением 27. Наконец, в 27 вводили концевые части с получением соединения 23. После этого, трет-бутильную группу в 23 можно было удалить в кислотных условиях с получением соединения 2. С другой стороны, бензил в 23 можно удалять гидрогенизацией с получением фенольных производных 22.
Схема 6. Путь D к соединениям 2
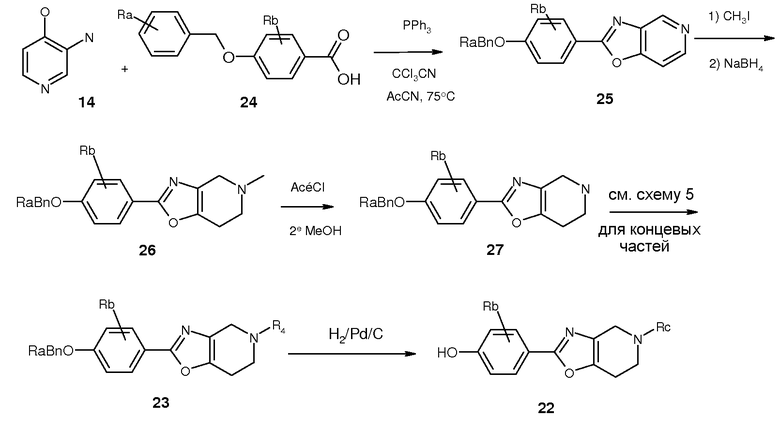
И, наконец, третий путь (путь E) к соединениям 2 представлен на схеме 7. Соединение 20 защищали трет-бутилоксикарбонильной группой с получением 28, которое можно было конвертировать в соответствующий фенол (29) в стандартных условиях с палладием. Алкилирование 29 в условиях фазового переноса или Мицунобу дало 30. С другой стороны, соединение 30 также можно было получить непосредственно из бромида 28 в условиях присутствия химического реагента с палладием. Удаление кислотой группы BOC в 30 привело к образованию соединения 27, которое можно было алкилировать в 23, как описано на схеме 5.
Схема 7. Путь E для синтеза соединений 2
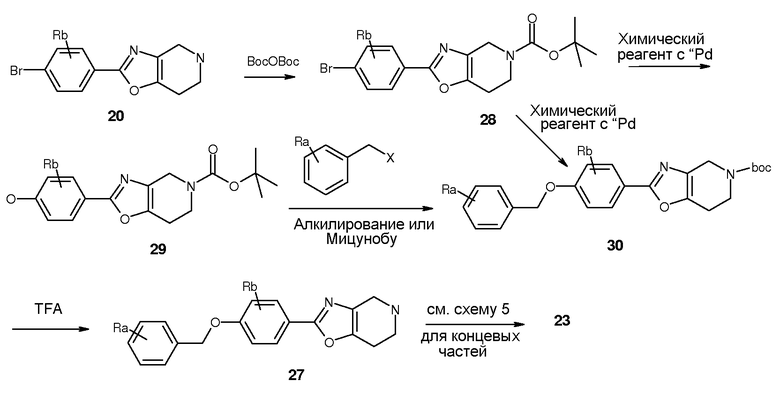
Схема 8
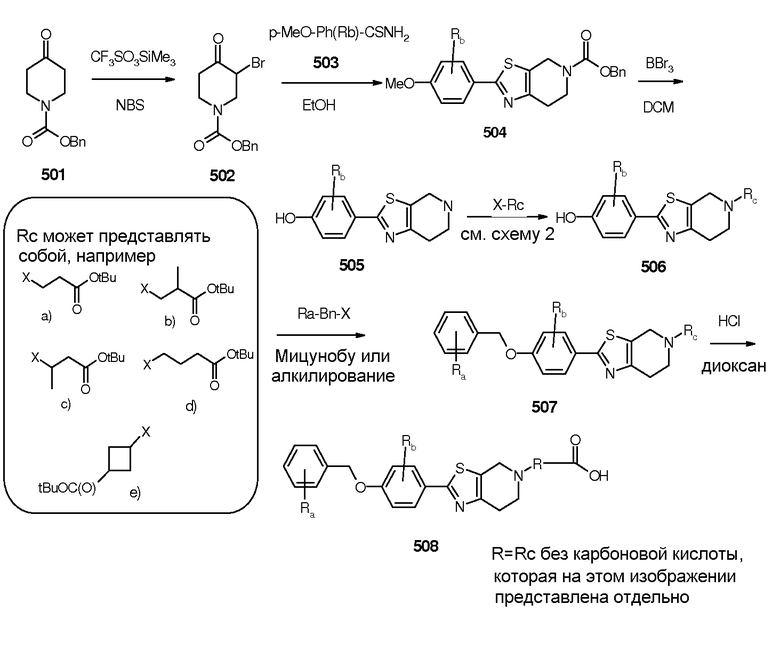
Тиазолопроизводные 508 и 520 синтезировали, как описано на схеме 8 и 9. Корректирование R-групп в реагентах приводит к введению Ra, Rb и Rc.
Схема 9

Путь синтеза в направлении ряда альтернативных концевых частей и линкеров представлен на схеме 10.
Схема 10

Для специалистов в данной области очевидно, что выбор определенного пути может быть основан на доступности реагентов. Кроме того, пути B, D и E в высокой степени подходят для внесения разнообразия в Rc-концевую часть соединений 1 и 2. В путях A и C введение части Ra-Bn происходит в последней части синтеза, что делает их более подходящими для исследования разнообразия в этой части молекулы.
§4. СИНТЕЗ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ
Общая методика синтеза соединений 5. К раствору трет-бутилового эфира 4-оксопиперидин-1-карбоновой кислоты в толуоле (2 мл/ммоль) добавляли каталитическое количество пара-толуолсульфоновой кислоты моногидрата (0,1 экв.) и пирролидин (4 г. э.). Смесь нагревали до температуры кипения с обратным холодильником в условиях Дина-Старка в течение 18 часов. Смесь концентрировали при пониженном давлении, и осадок перерастворяли в толуоле. К этому раствору медленно добавляли (в течение 25 минут) раствор надлежащим образом замещенного 2-бром-1-(4-бром-фенил)этанона (1,05 экв.) в толуоле/DCM (2 мл/ммоль, ½, об./об.). Смесь перемешивали в течение ночи при комнатной температуре и полученную белую взвесь выливали в воду. Водный слой экстрагировали DCM (3 раза), и объединенные органические слои сушили (MgSO4), а затем концентрировали при пониженном давлении. Полученное масло очищали хроматографией на силикагеле с получением соединения 5 с выходом 50-90%.
Общая методика синтеза соединений 6. Соединение 5 суспендировали в концентрированной хлористоводородной кислоте (10 экв., 12 Н). Смесь нагревали (с шагом 10°C в 30 минут) до 80°C. Смесь начинает интенсивно пениться, следовательно, следует обеспечить достаточно объема в исходной реакционной емкости. Через 45 минут смесь охлаждали до 0°C и нейтрализовывали 50 масс. % раствором NaOH (экзотермическая реакция). После перемешивания в течение ночи при комнатной температуре полученный твердый материал собирали фильтрацией и промывали 0,1 M NaOH. Светло-коричневый материал очищали путем экстракции с помощью Soxhlet в EtOAc с получением 6 в виде бежевого твердого вещества, которое использовали на следующей стадии без дальнейшей очистки.
Общая методика введения концевых частей в виде защищенных карбоновых кислот (7).
a) Введение трет-бутилового эфира пропионовой кислоты. Соединение 6 суспендировали в метаноле (4 мл/ммоль) и добавляли DIPEA (1,05 г. э.). К смеси добавляли трет-бутилакрилат (1,2 экв.) и смесь кипятили с обратным холодильником в течение 16 ч. Преобразование проверяли с помощью анализа TLC. Растворители выпаривали, и осадок перерастворяли в EtOAc и экстрагировали 5% раствором NaHCO3. Органический слой сушили (MgSO4), концентрировали в вакууме, и осадок очищали колоночной хроматографией на силикагеле с получением чистого 7a.
b) Введение трет-бутилового эфира 2-метилпропионовой кислоты. Соединение 6 суспендировали в DMF (6 мл/ммоль). К этой суспензии добавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (3 г. э.) и трет-бутилметакрилат (2 экв.). Смесь нагревали при 125°C в течение 16-100 ч. Раствор охлаждали и добавляли 5% NaHCO3 (15 мл/ммоль), и экстрагировали EtOAc. Органический слой промывали водой (4x), сушили над MgSO4, концентрировали в вакууме, и осадок очищали колоночной хроматографией на силикагеле с получением чистого 7b.
c) Введение трет-бутилового эфира 3-масляной кислоты. Соединение 6 суспендировали в 1,2-дихлорэтане (6 мл/ммоль). К этой суспензии добавляли трет-бутилацетоацетат (1 экв.) и триацетоксиборгидрид натрия (1,4 экв.). Смесь перемешивали при комнатной температуре в течение 16 ч. Если реакция не завершалась, добавляли другую часть трет-бутилацетоацетата (1 экв.) и триацетоксиборгидрида натрия (1,4 экв.). К раствору добавляли 5% NaHCO3 (15 мл/ммоль) и смесь экстрагировали DCM. Объединенные органические слои сушили над Na2SO4, концентрировали в вакууме, и осадок очищали колоночной хроматографией на силикагеле с получением чистого 7c.
d) Введение трет-бутилового эфира 4-масляной кислоты. Соединение 6 суспендировали в ацетонитриле (3 мл/ммоль). К этой суспензии добавляли карбонат калия (2 экв.), трет-бутил-4-бромбутаноат (1,1 экв.) и йодид калия (1,1 экв.). Смесь перемешивали при комнатной температуре в течение 16 ч, после чего анализ TLC выявил завершение реакции. Смесь концентрировали в вакууме, и осадок растворяли в EtOAc, промывали 5% NaHCO3 (15 мл/ммоль). Органический слой сушили над Na2SO4, концентрировали в вакууме, и осадок очищали колоночной хроматографией на силикагеле с получением 7d.
Общая методика для введения части бензилового эфира на 7 в соединение 9. Раствор соединения 7, надлежащим образом замещенного бензилового спирта (1,1 экв.), ацетата палладия(II) (0,02 экв.), 2-ди-трет-бутилфосфино-3,4,5,6-тетраметил-2',4',6'-триизопропил-1,1'-бифенила (0,02 экв.), карбоната цезия (1,5 экв.) в дегазированном толуоле (4 мл/ммоль) нагревали при 75°C в течение 16 ч. Преобразование проверяли с помощью анализа TLC. Раствор охлаждали до комнатной температуры, разбавляли DCM, фильтровали и концентрировали в вакууме. Осадок очищали колоночной хроматографией на силикагеле с получением соединения 9 с выходами, варьирующими в диапазоне 30-80%.
Общая методика для преобразования бромсодержащих производных 7 в фенольные производные 8. Соединение 7 растворяли в толуоле (8 мл/ммоль) и к раствору добавляли гидроксид калия (2 экв., 11,7 Н), и раствор дегазировали. К раствору добавляли 2-ди-трет-бутилфосфино-2',4',6'-триизопропилбифенил (0,06 экв.) и трис-(дибензилиденацетон)дипалладий(0) (0,03 экв.). Смесь перемешивали при 60°C в течение 1,25 ч. Смеси позволяли достигнуть комнатной температуры, разбавляли EtOAc и промывали 5% раствором NaHCO3 (10 мл/ммоль). Органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме. Осадок очищали колоночной хроматографией на силикагеле с получением соединения 8 с выходами, варьирующими в диапазоне 25-85%.
Общая методика для преобразования фенола 8 в бензиловые простые эфиры 9.
Способ A). Соединение 8 растворяли в смеси DCM/вода, 2/1, об./об. (4 мл/ммоль) и к этому раствору добавляли гидроксид натрия (2 Н, 3 экв.). К этой смеси добавляли бромид тетрабутиламмония (0,1 экв.) и надлежащим образом замещенный бензилбромид (1,1 экв.). Смесь перемешивали в течение 16 ч при комнатной температуре, после чего анализ TLC показал завершение реакции. Смесь разбавляли DCM (15 мл/ммоль), слои разделяли и водный слой экстрагировали DCM. Органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме. Осадок очищали колоночной хроматографией на силикагеле с получением чистого соединения 9 с выходами, варьирующими в диапазоне 80-90%.
Способ B). Соединение 8 растворяли в N,N'-диметилацетамиде (4 мл/ммоль) и к этому раствору добавляли трифенилфосфин (1,25 экв.) и диизопропилазодикарбоксилат (1,25 экв.) и надлежащим образом замещенный бензиловый спирт (1,2 экв.). Смесь перемешивали при комнатной температуре в течение 16 ч, после чего анализ TLC показал завершение реакции. Смесь разбавляли диэтиловым эфиром и промывали водой (3x). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме. Осадок очищали колоночной хроматографией на силикагеле с получением соединения 9 с выходами, варьирующими в диапазоне 70-90%.
Общая методика для удаления защитной группы кислотой из соединений 9 до 1. Соединение 9 растворяли в растворе HCl в 1,4-диоксане (4 Н, 45 экв.) и смесь перемешивали при комнатной температуре в течение 24 ч. При необходимости применяли нагревание при 50°C для направления реакции к завершению. Растворители выпаривали и добавляли диизопропиловый эфир для осаждения продукта. Белый твердый материал фильтровали и сушили в вакууме с получением соединения 1 с выходом, варьирующим в диапазоне 80-100%.
Общая методика для синтеза BOC-защищенных производных соединения 6. К суспензии соединения 6 в DCM (6 мл/ммоль) добавляли DIPEA (1 экв.), диметиламинопиридин (DMAP, 0,05 экв.) и ди-трет-бутилдикарбонат (1,1 экв.). Смесь перемешивали при комнатной температуре в течение 16 ч, после чего анализ TLC выявил завершение реакции. Реакционную смесь промывали 5% водн. раствором NaHCO3, и полученные водные слои экстрагировали DCM. Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме. Осадок очищали колоночной хроматографией на силикагеле с получением соединения 10 с выходами, варьирующими в диапазоне 70-90%.
Общая методика синтеза соединений 11. Соединение 10 растворяли в смеси 1,4-диоксан/вода, 1/1, об./об. (2 мл/ммоль) и к раствору добавляли гидроксид калия (4 экв., 11,7 Н) и раствор дегазировали. К раствору добавляли 2-ди-трет-бутилфосфино-2',4',6'-триизопропилбифенил (0,04 экв.) и трис-(дибензилиденацетон)дипалладий(0) (0,02 экв.). Смесь перемешивали при 80°C в течение 16 ч. Смесь охлаждали до комнатной температуры, разбавляли EtOAc, подкисляли до pH 6 с помощью 0,1 Н HCl и экстрагировали EtOAc. Органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме. Осадок очищали колоночной хроматографией на силикагеле с получением чистого соединения 11 с выходами, варьирующими в диапазоне 60-95%.
Общая методика для синтеза производных бензилового эфира 12.
Способ A). Соединение 11 растворяли в смеси DCM/вода, 2/1, об./об. (4 мл/ммоль) и к этому раствору добавляли гидроксид натрия (2 Н, 3 экв.). К этой смеси добавляли бромид тетрабутиламмония (0,1 экв.) и надлежащим образом замещенный бензилбромид (1,1 экв.). Смесь перемешивали в течение 16 ч при комнатной температуре, после чего анализ TLC показал завершение реакции. Смесь разбавляли DCM (15 мл/ммоль), слои разделяли, и водный слой экстрагировали DCM. Органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме. Осадок очищали колоночной хроматографией на силикагеле с получением соединения 12 с выходами, варьирующими в диапазоне 80-90%.
Способ B). Соединение 11 растворяли в N,N'-диметилацетамиде (4 мл/ммоль) и к этому раствору добавляли трифенилфосфин (1,25 экв.), диизопропилазодикарбоксилат (DIAD, 1,25 экв.) и надлежащим образом замещенный бензиловый спирт (1,2 экв.). Смесь перемешивали в течение 16 ч при комнатной температуре, после чего анализ TLC показал завершение реакции. Смесь разбавляли диэтиловым эфиром и промывали водой (3x). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме. Осадок очищали колоночной хроматографией на силикагеле с получением соединения 12 с выходами, варьирующими в диапазоне 70-90%.
Способ C). Соединение 10 растворяли в толуоле (8 мл/ммоль) и к раствору добавляли гидроксид калия (2 экв., 11,7 Н) и раствор дегазировали. К раствору добавляли надлежащим образом замещенный бензилбромид (1,1 экв.), 2-ди-трет-бутилфосфино-2',4',6'-триизопропилбифенил (0,06 экв.) и трис-(дибензилиденацетон)дипалладий(0) (0,03 экв.). Смесь перемешивали при 60°C в течение 1,25 ч. Смесь охлаждали до комнатной температуры, разбавляли EtOAc и промывали 5% раствором NaHCO3 (10 мл/ммоль). Органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме. Осадок очищали колоночной хроматографией на силикагеле с получением соединения 12 с выходами, варьирующими в диапазоне 30-80%.
Общая методика для удаления защитной группы из соединений 12-13.
Соединение 12 растворяли в DCM (10 мл/ммоль) и добавляли трифторуксусную кислоту (10 экв.). Раствор кипятили с обратным холодильником в течение 16 ч, после чего анализ TLC показал завершение реакции. Смесь нейтрализовывали 5% водн. NaHCO3. Смесь экстрагировали DCM (3x), и объединенные органические слои промывали рассолом, сушили над Na2SO4 и концентрировали в вакууме с получением соединения 13, которое использовали на следующей стадии без дальнейшей очистки.
Общая методика для введения концевых частей в виде защищенной карбоновой кислоты (9), начиная с соединения 13.
a) Введение трет-бутилового эфира пропионовой кислоты. Соединение 13 суспендировали в метаноле (4 мл/ммоль) и добавляли DIPEA (1,05 г. э.). К смеси добавляли 1,2 г. э. трет-бутилакрилата и смесь кипятили с обратным холодильником в течение 16 ч. Преобразование проверяли с помощью анализа TLC. Растворители выпаривали, и осадок перерастворяли в EtOAc и экстрагировали 5% раствором NaHCO3. Органический слой сушили (MgSO4), концентрировали в вакууме, и осадок очищали колоночной хроматографией на силикагеле с получением чистого 9a.
b) Введение трет-бутилового эфира 2-метилпропионовой кислоты. К раствору соединения 13 в DMF (6 мл/ммоль) в бутыли из пирекса добавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (3 г. э.) и трет-бутилметакрилат (2 экв.). Смесь нагревали при 125°C в течение 100 ч. Раствор охлаждали и добавляли 5% NaHCO3 (15 мл/ммоль) и экстрагировали смесью диэтиловый эфир/EtOAc, 1/1, об./об. Органический слой промывали водой (4x), сушили над MgSO4, концентрировали в вакууме, и осадок очищали колоночной хроматографией на силикагеле с получением чистого 9b.
c) Введение трет-бутилового эфира 3-масляной кислоты. Соединение 13 суспендировали в 1,2-дихлорэтане (6 мл/ммоль). К этой суспензии добавляли трет-бутилацетоацетат (1 экв.) и триацетоксиборгидрид натрия (1,4 экв.). Смесь перемешивали при комнатной температуре в течение 16 ч. Если реакция не завершалась, добавляли другую порцию трет-бутилацетоацетата (1 экв.) и триацетоксиборгидрида натрия (1,4 экв.). После завершения реакции раствор разбавляли 5% NaHCO3 (15 мл/ммоль) и смесь экстрагировали DCM. Объединенные органические слои сушили над Na2SO4, концентрировали в вакууме, и осадок очищали колоночной хроматографией на силикагеле с получением чистого соединения 9c.
d) Введение трет-бутилового эфира 4-масляной кислоты. Соединение 13 суспендировали в ацетонитриле (3 мл/ммоль). К этой суспензии добавляли карбонат калия (2 экв.), трет-бутил-4-бромбутаноат (1,1 экв.) и йодид калия (1,1 экв.). Смесь нагревали при комнатной температуре в течение 16 ч, после чего анализ TLC выявил завершение реакции. Смесь концентрировали в вакууме, и осадок растворяли в EtOAc, промывали 5% NaHCO3 (15 мл/ммоль). Органический слой сушили над Na2SO4, концентрировали в вакууме, и осадок очищали колоночной хроматографией на силикагеле с получением соединения 9d.
Общая методика для синтеза 2-(4-бром-фенил)-оксазоло[4,5-c]пиридина 17. К охлажденной (0°C) суспензии коммерчески доступного 4-гидрокси-3-аминопиридина в DCM (14, 6 мл/ммоль) добавляли триэтиламин (1,25 экв.) и раствор надлежащим образом замещенного бензоилхлорида 15 (1 экв., 0,3 М в DCM). Реакционной смеси позволяли достигнуть комнатной температуры, и смесь перемешивали в течение от 16 до 64 ч, после чего анализ TLC показал завершение реакции. Смесь фильтровали, промывали DCM и простым эфиром с получением 16 в качестве твердого материала (выход 50-80%), который использовали на следующей стадии без дальнейшей очистки. Гексахлорэтан (2,5 экв.) растворяли в DCM и добавляли трифенилфосфин (3 экв.) и триэтиламин (8 экв.). Смесь перемешивали в течение 10 минут при комнатной температуре и медленно добавляли соединение 16 5-ью равными порциями. Смесь перемешивали при комнатной температуре в течение 64 ч, после чего анализ TLC (DCM/MeOH, 97/3, об./об.) показал завершение реакции. Раствор концентрировали, и осадок суспендировали в DCM. Смесь фильтровали, и осадок промывали DCM и диэтиловым эфиром с получением 17 с выходом 30-80%.
Общая методика синтеза соединений 19. К раствору соединений 17 в DMF добавляли йодметан (4 г. э.) и смесь перемешивали в течение 16 ч. Смесь концентрировали в вакууме, и осадок перемешивали в EtOAc с получением 18 в виде белого твердого вещества. Соединение 18 растворяли в метаноле (10 мл/ммоль) и раствор охлаждали до 0°C. Добавляли боргидрид натрия (2 г. э.) и смесь перемешивали при 0°C в течение 2 ч, после чего ей позволяли достигнуть комнатной температуры, и перемешивание продолжали в течение 16 ч. Добавляли воду (1 мл/ммоль) и смесь перемешивали в течение 5 минут. Смесь совместно упаривали с ацетонитрилом, и осадок очищали колоночной хроматографией на силикагеле с получением соединения 19 с выходом 50-90%.
Общая методика синтеза соединений 20. К охлажденному (0°C) раствору соединения 19 в 1,2-дихлорэтане (10 мл/ммоль) добавляли DIPEA (2 экв.). При 0°C добавляли 1-хлорэтилхлорформиат (3 экв.) и смесь перемешивали в течение 10 минут при 0°C, после чего температуру повышали до температуры кипячения с обратным холодильником. Через 2 ч смесь концентрировали в вакууме, и осадок растворяли в метаноле (10 мл/ммоль). Раствор перемешивали в течение 48 ч при комнатной температуре. Удаление растворителя приводило к выделению соединения 20 с выходом 70-90%.
Общая методика синтеза соединений 21.
a) Введение трет-бутилового эфира пропионовой кислоты.
Соединение 20 суспендировали в метаноле (10 мл/ммоль) и добавляли DIPEA (2,05 г. э.). К смеси добавляли 1,2 г. э. трет-бутилакрилат и кипятили с обратным холодильником в течение 16-120 ч. Преобразование проверяли с помощью анализа TLC и при необходимости добавляли дополнительные реагенты для направления реакции к завершению. Растворители выпаривали, и осадок перерастворяли в EtOAc и экстрагировали 5% раствором NaHCO3. Органический слой сушили (MgSO4), концентрировали в вакууме, и осадок очищали колоночной хроматографией на силикагеле с получением 21a с выходами, варьирующими в диапазоне 50-90%.
b) Введение трет-бутилового эфира 2-метилпропионовой кислоты.
К раствору соединения 20 в DMF (6 мл/ммоль) добавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU, 3 г. э.) и трет-бутилметакрилат (5 экв.). Смесь нагревали при 125°C в течение 100 ч. Раствор охлаждали и добавляли 5% NaHCO3 (15 мл/ммоль) и экстрагировали смесью диэтиловый эфир/EtOAc, 1/1, об./об. Органический слой промывали водой (4x), сушили над MgSO4, концентрировали в вакууме, и осадок очищали колоночной хроматографией на силикагеле с получением чистого 21b.
c) Введение трет-бутилового эфира 3-масляной кислоты.
Соединение 20 суспендировали в 1,2-дихлорэтане (8 мл/ммоль). К этой суспензии добавляли трет-бутилацетоацетат (1,4 экв.), уксусную кислоту (1 г. э.) и триацетоксиборгидрид натрия (1,8 экв.). Смесь перемешивали при комнатной температуре в течение 16 ч. Если реакция не завершалась, добавляли другую порцию трет-бутилацетоацетата (1 экв.) и триацетоксиборгидрида натрия (1,4 экв.). После завершения реакции раствор разбавляли 5% NaHCO3 (15 мл/ммоль) и смесь экстрагировали DCM. Объединенные органические слои сушили над Na2SO4, концентрировали в вакууме, и осадок очищали колоночной хроматографией на силикагеле с получением чистого соединения 21c.
d) Введение трет-бутилового эфира 4-масляной кислоты.
Соединение 20 суспендировали в DMF (5 мл/ммоль). К этой суспензии добавляли карбонат калия (3 экв.) и трет-бутил-4-бромбутаноат (3 экв.). Смесь нагревали при 80°C в течение 16 ч, после чего анализ TLC выявил завершение реакции. Смесь концентрировали в вакууме, и осадок очищали колоночной хроматографией на силикагеле с получением 21d.
Общая методика синтеза соединений 22. Соединение 21 растворяли в ацетонитриле (25 мл/ммоль) и к раствору добавляли порошковый гидроксид калия (2 экв.), и раствор дегазировали. К раствору добавляли 2-ди-трет-бутилфосфино-3,4,5,6-тетраметил-2',4',6'-триизопропил-1,1-бифенил (0,06 экв.) и трис-(дибензилиденацетон)дипалладий(0) (0,03 экв.). Смесь перемешивали при 60°C в течение 4 ч. Смесь охлаждали до комнатной температуры и концентрировали в вакууме. Осадок растворяли в DCM и промывали 0,1 M HCl и водой. Водные слои экстрагировали DCM, и объединенные органические слои сушили над MgSO4. Соединение 22 получали после колоночной хроматографии на силикагеле с выходами, варьирующими в диапазоне 30-70%.
Общая методика введения части бензилового эфира в 21 для получения соединения 23. Раствор соединения 21, надлежащим образом замещенный бензиловый спирт (2 экв.), палладия(II) ацетат (0,02 экв.), 2-ди-трет-бутилфосфино-3,4,5,6-тетраметил-2',4',6'-триизопропил-1,1'бифенил (0,02 экв.), карбонат цезия (1,5 экв.) в дегазированном толуоле (3 мл/ммоль) нагревали при 75°C в течение 16 ч. Преобразование проверяли с помощью анализа TLC. Раствор охлаждали до комнатной температуры, разбавляли DCM, фильтровали и концентрировали в вакууме. Осадок очищали колоночной хроматографией на силикагеле с получением чистого соединения 23 с выходами, варьирующими в диапазоне 30-80%.
Общая методика для преобразования фенола 22 в бензиловые эфиры 23.
Способ A). Соединение 22 растворяли в смеси DCM/вода, 2/1, об./об. (4 мл/ммоль) и к этому раствору добавляли гидроксид натрия (2 Н, 3 экв.). К этой смеси добавляли бромид тетрабутиламмония (0,1 экв.) и надлежащим образом замещенный бензилбромид (1,1 экв.). Смесь перемешивали в течение 16 ч при комнатной температуре, после чего анализ TLC показал завершение реакции. Смесь разбавляли DCM (15 мл/ммоль), слои разделяли, и водный слой экстрагировали DCM. Органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме. Осадок очищали колоночной хроматографией на силикагеле с получением чистого соединения 23 с выходами, варьирующими в диапазоне 80-90%.
Способ B). Соединение 22 растворяли в сухом DCM (15 мл/ммоль) и к этому раствору добавляли трифенилфосфин (1,8 экв.) и надлежащим образом замещенный бензиловый спирт (1,8 экв.). К этой смеси добавляли диизопропилазодикарбоксилат (1,8 экв.) и перемешивали в течение 16 ч при комнатной температуре, после чего анализ TLC показал завершение реакции. Смесь концентрировали в вакууме. Осадок очищали колоночной хроматографией на силикагеле с получением чистого соединения 23 с выходами, варьирующими в диапазоне 70-90%.
Способ C). Смолу PS-TBD (3,7 экв.) инкубировали с раствором 22 (1,1 г. э.) в 1 мл ацетонитрила в течение 1,5 ч при 50°C. После этого добавляли надлежащим образом замещенный бензилбромид (1,10 экв.) в ацетонитриле. После этого реакционную смесь встряхивали и нагревали при 75°C в течение 16 ч. Затем растворитель удаляли фильтрацией, и смолу промывали 3×2,5 мл ACN. Объединенные органические слои концентрировали в вакууме с последующей колоночной флэш-хроматографией на диоксиде кремния с получением соединения 23 с выходами, варьирующими в диапазоне 60-95%.
Общая методика для удаления защитной группы из 23 для получения 2. Соединение 23 растворяли в растворе HCl в 1,4-диоксане (4 Н, 100 экв.) и смесь перемешивали в течение 16 ч при комнатной температуре. При необходимости применяли нагревание при 50°C для направления реакции к завершению. Растворители выпаривали и добавляли диизопропиловый эфир для осаждения продукта. Белый твердый материал фильтровали и сушили в вакууме с получением соединения 2 с выходом, варьирующим в диапазоне 70-100%.
Общая методика синтеза соединений 25. К охлажденной (0°C) суспензии коммерчески доступного 4-гидрокси-3-аминопиридина (14) в ацетонитриле (15 мл/ммоль) добавляли надлежащим образом замещенную 4-бензилоксибензойную кислоту (24, 1 экв.), трифенилфосфин (3 экв.) и трихлорацетонитрил (3 экв.). Реакционной смеси позволяли достигнуть комнатной температуры и смесь перемешивали в течение 16-64 ч при 80°C. Смесь концентрировали в вакууме, и осадок растворяли в DCM и промывали 2 Н NaOH (3x). Объединенные водные слои экстрагировали DCM, и органические слои сушили (Na2SO4) с получением неочищенного 25 в качестве масла, которое использовали на следующей стадии без дальнейшей очистки.
Общая методика синтеза соединений 26. К раствору соединения 25 в DMF (5 мл/ммоль) добавляли йодметан (4 г. э.) и смесь перемешивали в течение 16 ч. Смесь концентрировали в вакууме, и осадок перемешивали в EtOAc с получением четвертичной соли 25 в виде белого твердого вещества. Неочищенный материал растворяли в метаноле (10 мл/ммоль) и раствор охлаждали до 0°C. Добавляли боргидрид натрия (2,5 г. э.) и смесь перемешивали при 0°C в течение 2 ч, после чего ей позволяли достигнуть комнатной температуры и перемешивание продолжали в течение 16-64 ч. Добавляли воду (1 мл/ммоль) и смесь перемешивали в течение 5 минут. Смесь концентрировали в вакууме, осадок суспендировали в 2 Н NaOH (5 мл/ммоль) и экстрагировали DCM (3x). Объединенные органические слои сушили (Na2SO4) и концентрировали с получением неочищенного 26 в виде желтого твердого вещества, которое использовали на следующей стадии без дальнейшей очистки.
Общая методика для получения соединений 27. К охлажденному (0°C) раствору соединения 26 в 1,2-дихлорэтане (10 мл/ммоль) добавляли DIPEA (2 экв.) и добавляли 1-хлорэтилхлорформиат (3 экв.). Смесь перемешивали в течение 10 минут при 0°C, после чего температуру повышали до температуры кипячения с обратным холодильником. Через 4 ч смеси позволяли достигнуть комнатной температуры, и перемешивание продолжали в течение 16 ч. Смесь концентрировали в вакууме, и осадок растворяли в метаноле (10 мл/ммоль). Раствор перемешивали в течение 16-48 ч при комнатной температуре. Удаление растворителя приводило к выделению неочищенного 27 с общим выходом 20-40% в расчете на 25.
Общая методика введения концевых частей в виде защищенной карбоновой кислоты (для получения 23), начиная с соединения 27.
a) Введение трет-бутилового эфира пропионовой кислоты.
Соединение 27 суспендировали в метаноле (4 мл/ммоль) и добавляли DIPEA (1,05 г. э.). К смеси добавляли 1,2 г. э. трет-бутилакрилата и смесь кипятили с обратным холодильником в течение 16 ч. Преобразование проверяли с помощью анализа TLC. Растворители выпаривали, и осадок перерастворяли в EtOAc и экстрагировали 5% раствором NaHCO3.Органический слой сушили (MgSO4), концентрировали в вакууме, и осадок очищали колоночной хроматографией на силикагеле с получением чистого 23a.
b) Введение трет-бутилового эфира 2-метилпропионовой кислоты.
К раствору соединения 27 в DMF (6 мл/ммоль) добавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (3 г. э.) и трет-бутилметакрилат (4 экв.). Смесь нагревали при 125°C в течение 100 ч. Раствор охлаждали и добавляли 5% NaHCO3 (15 мл/ммоль) и экстрагировали смесью диэтиловый эфир/EtOAc, 1/1, об./об. Органический слой промывали водой (4×), сушили над MgSO4, концентрировали в вакууме, и осадок очищали колоночной хроматографией на силикагеле с получением чистого 23b.
c) Введение трет-бутилового эфира 3-масляной кислоты.
Соединение 27 суспендировали в 1,2-дихлорэтане (6 мл/ммоль). К этой суспензии добавляли трет-бутилацетоацетат (1 экв.) и триацетоксиборгидрид натрия (1,4 экв.). Смесь перемешивали при комнатной температуре в течение 16 ч. Если реакция не завершалась, добавляли другую порцию трет-бутилацетоацетата (1 экв.) и триацетоксиборгидрида натрия (1,4 экв.). После завершения реакции раствор разбавляли 5% NaHCO3 (15 мл/ммоль) и смесь экстрагировали DCM. Объединенные органические слои сушили над Na2SO4, концентрировали в вакууме, и осадок очищали колоночной хроматографией на силикагеле с получением чистого соединения 23c.
d) Введение трет-бутилового эфира 4-масляной кислоты.
Соединение 27 суспендировали в ацетонитриле (3 мл/ммоль). К этой суспензии добавляли карбонат калия (2 экв.), трет-бутил-4-бромбутаноат (1,1 экв.) и йодид калия (1,1 экв.). Смесь нагревали при комнатной температуре в течение 16 ч, после чего анализ TLC выявил завершение реакции. Смесь концентрировали в вакууме, и осадок растворяли в EtOAc, промывали 5% NaHCO3 (15 мл/ммоль). Органический слой сушили над Na2SO4, концентрировали в вакууме, и осадок очищали колоночной хроматографией на силикагеле с получением 23d.
e) Введение 3-циклобутанкарбоновой кислоты.
Соединение 27 суспендировали в 1,2-дихлорэтане (20 мл/ммоль). К этой суспензии добавляли 3-оксоциклобутанкарбоновую кислоту (1,3 экв.) и триацетоксиборгидрид натрия (1,6 экв.). Смесь перемешивали при комнатной температуре в течение 16 ч. Если реакция не завершалась, добавляли другую порцию 3-оксоциклобутанкарбоновой кислоты (1,3 экв.) и триацетоксиборгидрида натрия (1,6 экв.). После завершения реакции раствор разбавляли 5% NaHCO3 (15 мл/ммоль) и смесь экстрагировали DCM. Объединенные органические слои сушили над Na2SO4, концентрировали в вакууме, и осадок очищали колоночной хроматографией на силикагеле с получением чистого соединения 2e.
Общая методика для гидрогенизации 23 для получения соединения 22. К раствору соединения 23 в этаноле (10 мл/ммоль) добавляли гидроксид палладия на угле (20%, 0,22 г. э.). Гидрогенизацию начинали при атмосферном давлении водорода. Перемешивание продолжали в течение 16 ч при комнатной температуре. Смесь фильтровали над Hyflo, и осадок промывали этанолом. Фильтрат концентрировали в вакууме с получением соединения 22.
Общая методика синтеза соединений 28. К суспензии соединения 20 в DCM (6 мл/ммоль) добавляли DIPEA (1 экв.), диметиламинопиридин (DMAP, 0,05 экв.) и ди-трет-бутилдикарбонат (1,1 экв.). Смесь перемешивали при комнатной температуре в течение 16 ч, после чего анализ TLC выявил завершение реакции. Смесь концентрировали в вакууме. Осадок очищали колоночной хроматографией на силикагеле с получением чистого соединения 28 с выходами, варьирующими в диапазоне 70-90%.
Общая методика синтеза соединений 29. Соединение 28 растворяли в смеси 1,4-диоксан/вода, 1/1, об./об. (10 мл/ммоль) и к раствору добавляли гидроксид калия (4 экв., 11,7 Н), и раствор дегазировали. К раствору добавляли 2-ди-трет-бутилфосфино-2',4',6'-триизопропилбифенил (0,04 экв.) и трис-(дибензилиденацетон)дипалладий(0) (0,02 экв.). Смесь перемешивали при 80°C в течение 16 ч. Смесь охлаждали до комнатной температуры, разбавляли EtOAc, подкисляли до pH 6 с помощью 0,1 Н HCl и экстрагировали EtOAc. Органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме. Осадок очищали колоночной хроматографией на силикагеле с получением чистого соединения 29 с выходами, варьирующими в диапазоне 60-95%.
Общая методика для синтеза производных бензилового эфира 30.
Способ A). Соединение 29 растворяли в смеси DCM/вода, 2/1, об./об. (4 мл/ммоль) и к этому раствору добавляли гидроксид натрия (2 Н, 3 экв.). К этой смеси добавляли бромид тетрабутиламмония (0,1 экв.) и надлежащим образом замещенный бензилбромид (1,1 экв.). Смесь перемешивали в течение 16 ч при комнатной температуре, после чего анализ TLC показал завершение реакции. Смесь разбавляли DCM (15 мл/ммоль), слои разделяли, и водный слой экстрагировали DCM. Органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме. Осадок очищали колоночной хроматографией на силикагеле с получением чистого соединения 30 с выходами, варьирующими в диапазоне 80-90%.
Способ B) Соединение 29 растворяли в N,N'-диметилацетамиде (4 мл/ммоль) и к этому раствору добавляли трифенилфосфин (1,25 экв.), диизопропилазодикарбоксилат (1,25 экв.) и надлежащим образом замещенный бензиловый спирт (1,2 экв.). Смесь перемешивали в течение 16 ч при комнатной температуре, после чего анализ TLC показал завершение реакции. Смесь разбавляли диэтиловым эфиром и промывали водой (3×). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме. Осадок очищали колоночной хроматографией на силикагеле с получением чистого соединения 30 с выходами, варьирующими в диапазоне 70-90%.
Способ C). Соединение 28 растворяли в толуоле (8 мл/ммоль) и к раствору добавляли гидроксид калия (2 экв., 11,7 Н), и раствор дегазировали. К раствору добавляли надлежащим образом замещенный бензилбромид (1,1 экв.), 2-ди-трет-бутилфосфино-2',4',6'-триизопропилбифенил (0,06 экв.) и трис-(дибензилиденацетон)дипалладий(0) (0,03 экв.). Смесь перемешивали при 60°C в течение 1,25 ч. Смесь охлаждали до комнатной температуры, разбавляли EtOAc и промывали 5% раствором NaHCO3 (10 мл/ммоль). Органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме. Осадок очищали колоночной хроматографией на силикагеле с получением чистого соединения 30 с выходами, варьирующими в диапазоне 30-80%.
Общая методика для удаления защитной группы из соединений 30-27.
Соединение 30 растворяли в DCM (10 мл/ммоль) и добавляли трифторуксусную кислоту (6 экв.). Раствор кипятили с обратным холодильником в течение 16 ч, после чего анализ TLC показал завершение реакции. Смесь нейтрализовывали 5% водн. NaHCO3. Смесь экстрагировали DCM (3×), и объединенные органические слои промывали рассолом, сушили над Na2SO4 и концентрировали в вакууме с получением соединения 27, которое использовали на следующей стадии без дальнейшей очистки.
§5. СИНТЕЗ КОНКРЕТНЫХ СОЕДИНЕНИЙ
(См. таблицу 1)
Все фуранильные производные из таблицы 1 можно получать, следуя либо пути A, либо пути B, выбирая соответствующие реагенты. Представленные ниже соединения являются типичными примерами.
Все оксазолопроизводные из таблицы 1 можно получать, следуя любому из путей C, D или E, выбирая соответствующие реагенты. Представленные ниже соединения являются типичными примерами.
Трет-бутиловый эфир 3-[2-(4-бромфенил)-2-оксоэтил]-4-оксопиперидин-1-карбоновой кислоты (5, Rb=H). К раствору трет-бутилового эфира 4-оксо-пиперидин-1-карбоновой кислоты (4, 104,1 г, 522 ммоль) в толуоле (800 мл) добавляли каталитическое количество пара-толуолсульфоновой кислоты моногидрата (0,5 г, 2,6 ммоль) и пирролидин (172,8 мл, 2090 ммоль). Смесь нагревали до температуры кипения с обратным холодильником в условиях Дина-Старка в течение 18 часов. Смесь концентрировали при пониженном давлении, и осадок перерастворяли в толуоле (600 мл). К этому раствору медленно добавляли (в течение 25 минут раствор 2-бром-1-(4-бромфенил)этанона (3, Rb=H, 145,2 г, 522 ммоль) в толуоле/DCM (900 мл, ½, об./об.). Смесь перемешивали в течение ночи при комнатной температуре, и полученную белую взвесь выливали в воду (1,5 л). Водный слой экстрагировали DCM (3×300 мл), и объединенные органические слои сушили (MgSO4), а затем концентрировали при пониженном давлении. Полученное масло очищали хроматографией на силикагеле (от диэтиловый эфир/петролейный эфир, 2/3, об./об. до 100% диэтилового эфира) с получением соединения 5 (Rb=H, 166,6 г, 87%) в виде желтого твердого вещества. Анализ TLC, Rf 0,3 в смеси диэтиловый эфир/петролейный эфир, 1/1, об./об.
2-[4-бромфенил]-4,5,6,7-тетрагидрофуро[3,2-c]пиридин (6, Rb=H). Соединение 5 (Rb=H, 166 г, 456 ммоль) суспендировали в концентрированной хлористоводородной кислоте (500 мл, 12 Н, 6 моль). Смесь нагревали со скоростью 10°C за 30 минут до 80°C. Смесь начинает интенсивно пениться, следовательно, следует обеспечить достаточно объема в исходной реакционной емкости. Через 45 минут смесь охлаждали до 0°C и нейтрализовывали 50 масс. % раствором NaOH (экзотермическая реакция). После перемешивания в течение ночи при комнатной температуре полученный твердый материал собирали фильтрацией и промывали 250 мл 0,1 М NaOH. Светло-коричневый материал очищали экстракцией с помощью Soxhlet в EtOAc с получением 6 (Rb=H, 51 г, 38%) в виде белого твердого вещества, которое использовали на следующей стадии без дальнейшей очистки. LC-MS (способ A): Rt 1,19, [M+H] 278.
Трет-бутиловый эфир 3-[2-(4-бромфенил)-6,7-дигидро-4H-фуро[3,2-c]пиридин-5-ил]пропионовой кислоты (7a, Rb=H). Соединение 6 (Rb=H, 1,46 г, 5 ммоль) суспендировали в метаноле (30 мл) и добавляли DIPEA (0,91 мл, 1,05 г. э.). К смеси добавляли трет-бутилакрилат (0,88 мл, 1,2 экв.) и кипятили с обратным холодильником в течение 16 ч. Преобразование проверяли с помощью анализа TLC (диэтиловый эфир/петролейный эфир, 1/1, об./об.). Растворители выпаривали, и осадок перерастворяли в EtOAc и экстрагировали с помощью 5% раствора NaHCO3. Органический слой сушили (MgSO4), концентрировали в вакууме, и осадок очищали колоночной хроматографией на силикагеле (диэтиловый эфир/петролейный эфир, от 2/3 до 1/1, об./об.) с получением чистого 7a (Rb=H, 1,75 г, 86%) в виде белого твердого вещества. LC-MS (способ A): Rt 1,38, [M+H] 407.
Трет-бутиловый эфир 3-[2-(4-гидроксифенил)-6,7-дигидро-4H-фуро[3,2-c]пиридин-5-ил]пропионовой кислоты (8a, Rb=H). Соединение 7a (Rb=H, 3,85 г, 9,5 ммоль) растворяли в толуоле (80 мл) и к раствору добавляли гидроксид калия (2 экв., 11,7 Н), и раствор дегазировали. К раствору добавляли 2-ди-трет-бутилфосфино-2',4',6'-триизопропилбифенил (0,24 г, 0,57 ммоль, 0,06 экв.) и трис-(дибензилиденацетон)дипалладий(0) (0,26 г, 0,28 ммоль, 0,03 экв.). Смесь перемешивали при 60°C в течение 1,25 ч. Смесь охлаждали до комнатной температуры, разбавляли EtOAc и промывали 5% раствором NaHCO3 (10 мл/ммоль). Органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме. Осадок очищали колоночной хроматографией на силикагеле (диэтиловый эфир/петролейный эфир, 1/1, об./об., Rf 0,1) с получением чистого соединения 8a (Rb=H, 1,86 г, 57%) в виде желтого твердого вещества. LC-MS (способ A): Rt 1,14, [M+H] 344.
Трет-бутиловый эфир 3-{2-[4-(2-фторбензилокси)фенил]-6,7-дигидро-4H-фуро[3,2-c]пиридин-5-ил}пропионовой кислоты (9a, Ra=2F, Rb=H). Соединение 8a (Rb=H, 1,24 г, 3,61 ммоль) растворяли в N,N-диметилацетамиде (10 мл) и к этому раствору добавляли трифенилфосфин (1,33 г, 5,06 ммоль, 1,4 экв.), диизопропилазодикарбоксилат (1 мл, 5,05 ммоль, 1,4 экв.) и 2-фторбензиловый спирт (0,46 мл, 4,33 ммоль, 1,2 экв.). Смесь перемешивали в течение 16 ч при комнатной температуре, после чего анализ TLC (диэтиловый эфир, Rf 0,3) показал завершение реакции. Смесь разбавляли диэтиловым эфиром и промывали водой (3×). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме. Осадок очищали колоночной хроматографией на силикагеле (диэтиловый эфир/петролейный эфир, от 1/1, об./об. до 2/1, об./об.) с получением чистого соединения 9a (Ra=2F, Rb=H, 1,38 г, 84%) в виде масла. LC-MS (способ A): Rt 1,46, [M+H] 452.
3-{2-[4-(2-фторбензилокси)фенил]-6,7-дигидро-4H-фуро[3,2-c]пиридин-5-ил}пропионовая кислота (33). Соединение 9a (Ra=2F, Rb=H, 1,38 г, 3,1 ммоль) растворяли в растворе HCl в 1,4-диоксане (4 Н, 30 мл) и смесь перемешивали в течение 2 ч при 35°C. Растворители выпаривали и добавляли диизопропиловый эфир (30 мл) для осаждения продукта в качестве соли хлористоводородной кислоты. Белый твердый материал фильтровали и сушили в вакууме с получением соединения 33 (0,75 г, 54%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6): δ ч/млн 2,93 (т, J=7,6 Гц, 2H), 3,07 (уш.с, 2H), 3,28-3,55 (уш.с, 2H), 3,44 (т, J=7,6 Гц, 2H), 3,60-3,90 (уш.с, 2H), 4,06-4,56 (уш.с, 2H), 5,17 (с, 2H), 6,76 (с, 1H), 7,10 (д, J=8,8 Гц, 2H), 7,21-7,31 (м, 2H), 7,39-7,48 (м, 1H), 7,57 (т, J=7,5 Гц, 1H), 7,63 (д, J=8,8 Гц, 2H), 10,7-11,5 (уш.с, 1H), 12,3-13,2 (уш.с, 1H); LC-MS (способ A): Rt 1,39, [M+H] 396.
Трет-бутиловый эфир 2-(4-бромфенил)-6,7-дигидро-4H-фуро[3,2-c]пиридин-5-карбоновой кислоты 10 (Rb=H). К суспензии соединения 6 (Rb=H, 5 г, 17 ммоль) в DCM (100 мл) добавляли DIPEA (2,92 мл, 1 экв.), DMAP (0,1 г, 0,05 экв.) и ди-трет-бутилдикарбонат (4,1 г, 18,8 ммоль, 1,1 экв.). Смесь перемешивали при комнатной температуре в течение 16 ч, после чего анализ TLC (DCM, Rf 0,40) показал полное завершение реакции. Реакционную смесь промывали 5% водн. раствором NaHCO3, и полученные водные слои экстрагировали DCM. Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме. Осадок очищали колоночной хроматографией на силикагеле (элюент: 100% DCM) с получением соединения 10 (Rb=H, 5,99 г, 92%) в качестве масла.
Трет-бутиловый эфир 2-(4-гидроксифенил)-6,7-дигидро-4H-фуро[3,2-c]пиридин-5-карбоновой кислоты 11a (Rb=H). Соединение 10 (Rb=H, 11,77 г, 31 ммоль) растворяли в смеси 1,4-диоксан/вода, 1/1, об./об. (200 мл) и к раствору добавляли гидроксид калия (6,98 г, 124,5 ммоль, 4 экв.) и дегазировали. К раствору добавляли 2-ди-трет-бутилфосфино-2',4',6'-триизопропилбифенил (0,53 г, 1,24 ммоль, 0,04 экв.) и трис-(дибензилиденацетон)дипалладий(0) (0,57 г, 0,62 ммоль, 0,02 экв.). Смесь перемешивали при 80°C в течение 16 ч. Смесь охлаждали до комнатной температуры, разбавляли EtOAc, подкисляли до pH 6 с помощью 0,1 Н HCl и экстрагировали EtOAc. Органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме. Осадок очищали колоночной хроматографией на силикагеле (элюент: DCM/MeOH, от 1/0 до 99,5/0,5) с получением соединения 11 (Rb=H, 9 г, 90%) в виде белого твердого вещества.
Трет-бутиловый эфир 2-[4-(4-хлорбензилокси)фенил]-6,7-дигидро-4H-фуро[3,2-c]пиридин-5-карбоновой кислоты (12, Ra=4Cl, Rb=H). Соединение 11a (Rb=H, 2,0 г, 6,34 ммоль) растворяли в смеси DCM/вода, 2/1, об./об. (30 мл) и к этому раствору добавляли гидроксид натрия (2 Н, 10 мл). К этой смеси добавляли бромид тетрабутиламмония (0,2 г, 0,63 ммоль, 0,1 экв.) и 4-хлорбензилбромид (1,43 г, 6,98 ммоль, 1,1 г. э.). Смесь перемешивали в течение 16 ч при комнатной температуре, после чего анализ TLC (100% DCM, Rf 0,55) показал полное завершение реакции. Смесь разбавляли DCM (15 мл/ммоль), слои разделяли, и водный слой экстрагировали DCM. Органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме. Осадок очищали колоночной хроматографией на силикагеле (DCM/петролейный эфир, от 3/1 до 1/0, об./об.) с получением соединения 12 (Ra=4Cl, Rb=H, 2,3 г, 82%) в виде желтого масла. 1H ЯМР (400 МГц, CDCl3) δ ч/млн 1,4 (с, 9H); 2,75 (уш.с, 2H); 3,75 (уш.с, 2H); 4,35 (уш.с, 2H); 5,05 (с, 2H); 6,4 (с, 1H), 6,94 (д, 1H); 7,30-7,55 (м, 7H).
2-[4-(4-Хлорбензилокси)фенил]-6,7-дигидро-4H-фуро[3,2-c]пиридин (13, Ra=4Cl, Rb=H). Соединение 12 (Ra=4Cl, Rb=H, 2,3 г, 5,2 ммоль) растворяли в DCM (50 мл) и добавляли трифторуксусную кислоту (4 мл, 10 экв.). Раствор кипятили с обратным холодильником в течение 16 ч, после чего анализ TLC (100% DCM, Rf 0,05) показал завершение реакции. Смесь нейтрализовывали 5% водн. NaHCO3. Смесь экстрагировали DCM (3x), и объединенные органические слои промывали рассолом, сушили над Na2SO4 и концентрировали в вакууме с получением неочищенного 13 (Ra=4Cl, Rb=H, 1,79 г), которое использовали на следующей стадии без дальнейшей очистки. LC-MS (способ A): Rt 1,49, [M+H] 340.
Трет-бутиловый эфир 3-{2-[4-(4-хлорбензилокси)фенил]-6,7-дигидро-4H-фуро[3,2-c]пиридин-5-ил}-2-метилпропионовой кислоты (9b, Ra=4Cl, Rb=H). К раствору соединения 13a (0,25 г, 0,74 ммоль) в DMF (5 мл) в 25-мл бутыли из пирекса добавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (0,33 мл, 2,21 ммоль) и трет-бутилметакрилат (0,24 мл, 1,47 ммоль). Смесь нагревали при 140°C в течение 16 ч. Раствор охлаждали и добавляли 5% NaHCO3 (10 мл), и экстрагировали смесью диэтиловый эфир/EtOAc, 1/1, об./об. Органический слой промывали водой (4×20 мл), сушили над MgSO4, концентрировали в вакууме, и осадок очищали колоночной хроматографией на силикагеле (диэтиловый эфир/петролейный эфир, от 9/1 до 4/1, об./об., Rf 0,65) с получением чистого 9b (Ra=4Cl, Rb=H, 0,1 г, 28%) в виде бесцветного масла. LC-MS (способ A): Rt 1,88, [M+H] 482.
3-{2-[4-(4-Хлорбензилокси)фенил]-6,7-дигидро-4H-фуро[3,2-c]пиридин-5-ил}-2-метилпропионовая кислота (77). Соединение 9b (Ra=4Cl, Rb=H, 0,12 г, 0,25 ммоль) растворяли в растворе HCl в 1,4-диоксане (4 Н, 2,8 мл) и смесь перемешивали в течение 16 ч при комнатной температуре. Растворитель выпаривали и добавляли диизопропиловый эфир (30 мл) для осаждения продукта в качестве соли хлористоводородной кислоты. Белый твердый материал фильтровали и сушили в вакууме с получением соединения 77 (0,09 г, 74%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6): δ ч/млн 1,29 (д, J=7,2 Гц, 3Н), 3,05-3,17 (м, 3Н), 3,23 (дд, J=5,4, 13,3 Гц, 1Н), 3,51-3,68 (м, 3Н), 4,24 (уш.с, 2Н), 5,16 (с, 2Н), 6,73 (с, 1Н), 7,08 (д, J=8,9 Гц, 2Н), 7,42-7,53 (м, 4Н), 7,61 (д, J=8,9 Гц, 2Н), 10,4-13,1 (уш.с, 2H); 13C ЯМР (101 МГц, ДМСО-d6): δ ч/млн 16,42 (кв., 1C), 20,36 (т, 1C), 35,17 (д, 1C), 48,74 (т, 1C), 49,57 (т, 1C), 56,96 (т, 1C), 68,60 (т, 1C), 102,92 (д, 1C), 113,06 (с, 1C), 115,41 (д, 1C), 123,20 (с, 1C), 124,87 (д, 1C), 128,41 (д, 1C), 129,42 (д, 1C), 132,46 (с, 1C), 136,01 (с, 1C), 145,10 (с, 1C), 152,99 (с, 1C), 157,85 (с, 1C), 174,95 (с, 1C). LC-MS (способ A): Rt 1,56, [M+H] 426.
Трет-бутиловый эфир 2-[4-(бензилокси)фенил]-6,7-дигидро-4H-фуро[3,2-c]пиридин-5-карбоновой кислоты 12 (Ra=Rb=H). Соединение 11 (Rb=H, 0,84 г, 2,66 ммоль) растворяли в смеси DCM/вода, 2/1, об./об. (30 мл) и к этому раствору добавляли гидроксид натрия (2 Н, 4,2 мл). К этой смеси добавляли бромид тетрабутиламмония (0,09 г, 0,27 ммоль, 0,1 экв.) и бензилбромид (0,35 мл, 2,93 ммоль, 1,1 г. э.). Смесь перемешивали в течение 16 ч при комнатной температуре, после чего анализ TLC (DCM/MeOH, 98/2, об./об., Rf 0,8) показал завершение реакции. Смесь разбавляли DCM (100 мл), слои разделяли, и водный слой экстрагировали DCM. Органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме. Осадок очищали колоночной хроматографией на силикагеле (DCM/петролейный эфир, от 3/1 до 1/0, об./об.) с получением соединения 12 (Ra=Rb=H, 1,03 г, 95%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ ч/млн 1,4 (с, 9Н); 2,75 (уш.с, 2Н); 3,75 (уш.с, 2H); 4,35 (уш.с, 2H); 5,05 (с, 2Н); 6,35 (с, 1H); 6,98 (д, 2Н); 7,30-7,55 (м, 7Н).
2-[4-Бензилоксифенил]-6,7-дигидро-4H-фуро[3,2-c]пиридин (13, Ra=Rb=H). Соединение 12 (Ra=Rb=H, 1,03 г, 2,54 ммоль) растворяли в DCM (20 мл) и добавляли трифторуксусную кислоту (1,5 мл). Раствор кипятили с обратным холодильником в течение 16 ч, после чего анализ TLC (100% DCM, Rf 0,05) показал завершение реакции. Смесь нейтрализовывали 5% водн. NaHCO3 (40 мл) и экстрагировали DCM (3×50 мл), и объединенные органические слои промывали рассолом, сушили над Na2SO4 и концентрировали в вакууме с получением соединения 13 (Ra=Rb=H, 0,67 г, 86%), которое использовали на следующей стадии без дальнейшей очистки. LC-MS (способ A): Rt 1,50, [M+H] 306.
Трет-бутиловый эфир 3-{2-[4-бензилоксифенил]-6,7-дигидро-4H-фуро[3,2-c]пиридин-5-ил}масляной кислоты (9c, Ra=Rb=H). Соединение 13 (Ra=Rb=H, 0,16 г, 0,52 ммоль) суспендировали в 1,2-дихлорэтане (3,2 мл). К этой суспензии добавляли трет-бутилацетоацетат (0,09 мл, 0,52 ммоль) и триацетоксиборгидрид натрия (0,16 г, 0,73 ммоль). Смесь перемешивали при комнатной температуре в течение 16 ч, после чего добавляли другую порцию трет-бутилацетоацетата (1 экв.) и триацетоксиборгидрида натрия (1,4 экв.) вместе с каплей уксусной кислоты. После перемешивания в течение дополнительных 60 ч вновь добавляли порцию трет-бутилацетоацетата (1 экв.) и триацетоксиборгидрида натрия (1,4 экв.), и перемешивание продолжали в течение 36 ч. Раствор разбавляли 5% NaHCO3 (10 мл), и смесь экстрагировали DCM (3×100 мл). Объединенные органические слои сушили над Na2SO4, концентрировали в вакууме, и осадок очищали колоночной хроматографией на силикагеле (диэтиловый эфир/петролейный эфир, от 9/1 до 4/1, об./об.) с получением чистого соединения 9c (Ra=Rb=H, 0,06 г, 25%) в виде белого твердого вещества. LC-MS (способ A): Rt 1,68, [M+H] 448.
3-{2-[4-Бензилоксифенил]-6,7-дигидро-4H-фуро[3,2-c]пиридин-5-ил}масляная кислота (76). Соединение 9c (Ra=Rb=H, 0,08 г, 0,18 ммоль) растворяли в растворе HCl в 1,4-диоксане (4 мл, 2 Н) и смесь перемешивали в течение 16 ч при комнатной температуре. Растворители выпаривали, и осадок совместно упаривали с циклогексаном. Добавляли диизопропиловый эфир (30 мл) для осаждения продукта в виде соли хлористоводородной кислоты, белый твердый материал фильтровали и сушили в вакууме с получением соединения 76 (0,06 г, 80%). 1H ЯМР (400 МГц, ДМСО-d6), δ ч/млн: 1,38 (д, J=6,6 Гц, 3Н), 2,58-2,75 (м, 1Н), 2,90-3,18 (м, 3Н), 3,39-3,54 (м, 1Н), 3,61-3,78 (м, 1Н), 3,80-3,93 (м, 1Н), 4,13-4,32 (м, 2Н), 5,14 (с, 2Н), 6,76 (с, 1Н), 7,07 (д, J=8,8 Гц, 2Н), 7,30-7,36 (м, 1Н), 7,40 (с, 2Н), 7,45 (с, 2Н), 7,62 (д, J=8,8 Гц, 2Н), 10,15-10,80 (м, 1Н), 12,54-13,10 (м, 1Н);). LC-MS (способ A): Rt 1,46, [M+H] 392.
Трет-бутиловый эфир 4-[2-(4-бромфенил)-6,7-дигидро-4H-фуро[3,2-c]пиридин-5-ил]масляной кислоты (7d). Соединение 6 (Rb=H, 4,55 г, 16,3 ммоль) суспендировали в ацетонитриле (55 мл). К этой суспензии добавляли карбонат калия (4,52 г, 32,7 ммоль), трет-бутил-4-бромбутаноат (4,38 г, 19,6 ммоль, 1,2 экв.) и йодид калия (3,2 г, 19,6 ммоль, 1,2 экв.). Смесь нагревали до температуры кипения с обратным холодильникомв в течение 16 ч, после чего анализ TLC (диэтиловый эфир/петролейный эфир, 1/1, об./об., Rf 0,1) показал завершение реакции. Смесь концентрировали в вакууме, и осадок растворяли в EtOAc и промывали 5% NaHCO3 (2×60 мл). Органический слой сушили над Na2SO4, концентрировали в вакууме, и осадок очищали колоночной хроматографией на силикагеле (диэтиловый эфир/петролейный эфир, 1/1, об./об.) с получением 7d (Rb=H, 4,94 г, 71%) в виде желтого твердого вещества. LC-MS (способ A): Rt 1,37, [M+H] 420.
Трет-бутиловый эфир 4-[2-(4-гидроксифенил)-6,7-дигидро-4H-фуро[3,2-c]пиридин-5-ил]масляной кислоты (8d). Соединение 7d (Rb=H, 4,91 г, 11,68 ммоль) растворяли в толуоле (100 мл) и к раствору добавляли гидроксид калия (2 мл, 11,7 Н) и дегазировали. К раствору добавляли 2-ди-трет-бутилфосфино-2',4',6'-триизопропилбифенил (0,27 г, 0,64 ммоль, 0,06 экв.) и трис-(дибензилиденацетон)дипалладий(0) (0,29 г, 0,32 ммоль, 0,03 экв.). Смесь перемешивали при 60°C в течение 1,25 ч. Смесь охлаждали до комнатной температуры, разбавляли EtOAc и промывали 5% раствором NaHCO3 (100 мл). Органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме. Осадок очищали колоночной хроматографией на силикагеле (диэтиловый эфир/петролейный эфир, от 1/1 до 1/0, об./об., Rf 0,1) с получением чистого соединения 8d (Rb=H, 4,0 г) в виде желтого твердого вещества. LC-MS (способ A): Rt 1,21, [M+H] 358.
Трет-бутиловый эфир 4-{2-[4-(2-фторбензилокси)фенил]-6,7-дигидро-4H-фуро[3,2-c]пиридин-5-ил}масляной кислоты (9d). Соединение 8d (Rb=H, 0,43 г, 1,2 ммоль) растворяли в смеси DCM/вода, 2/1, об./об. (5 мл) и к этому раствору добавляли гидроксид натрия (1,8 мл, 2 Н, 3 экв.). К этой смеси добавляли бромид тетрабутиламмония (0,1 экв.) и 2F-бензилбромид (1,32 ммоль, 250 мг). Смесь перемешивали в течение 16 ч при комнатной температуре, после чего анализ TLC (диэтиловый эфир, Rf 0,5) показал завершение реакции. Смесь разбавляли DCM (15 мл), слои разделяли, и водный слой экстрагировали DCM. Органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме. Осадок очищали колоночной хроматографией на силикагеле (диэтиловый эфир/петролейный эфир, от 1/1 до 1/0, об./об.) с получением соединения 9d (Ra=2F, Rb=H) с выходом 80%. LC-MS (способ A): Rt 1,49, [M+H] 466.
4-{2-[4-(2-Фторбензилокси)фенил]-6,7-дигидро-4H-фуро[3,2-c]пиридин-5-ил}масляная кислота (35). Соединение 9d (Ra=2F, Rb=H, 0,3 г, 0,64 ммоль) растворяли в растворе HCl в 1,4-диоксане (4 Н, 2,8 мл) и смесь перемешивали в течение 16 ч при комнатной температуре. Растворители выпаривали и добавляли диизопропиловый эфир (30 мл) для осаждения продукта в виде соли хлористоводородной кислоты. Белый твердый материал фильтровали и сушили в вакууме с получением соединения 35 (0,32 г, 95%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ ч/млн: 1,69-1,83 (м, 2Н), 2,18 (т, J=7,2 Гц, 2Н), 2,81-2,91 (м, 2Н), 3,02-3,13 (м, 2Н), 3,23-3,36 (уш.с, 1Н), 3,50-3,68 (уш.с, 1Н), 3,89-4,05 (м, 1Н), 4,15-4,29 (м, 1Н), 4,98 (с, 2Н), 6,58 (с, 1Н), 6,91 (д, J=8,7 Гц, 2Н), 7,02-7,12 (м, 2Н), 7,21-7,29 (м, 1Н), 7,38 (дт, J=7,7, 1,5 Гц, 1Н), 7,44 (д, J=8,7 Гц, 2Н), 9,80 (уш.с, 1Н), 11,37-13,06 (уш.с, 1Н); LC-MS (способ A): Rt 1,37, [M+H] 410.
4-{2-[4-(2-Фторбензилокси)-2-фторфенил]-6,7-дигидро-4H-фуро[3,2-c]-пиридин-5-ил}масляная кислота (73). Соединение 73 получали аналогично тому, как описано для 35, начиная с 2-бром-1-(4-бром-2-фторфенил)этанона. Соединение 73: 1H ЯМР (400 МГц, ДМСО-d6), δ ч/млн: 1,92-2,04 (м, 2Н), 2,38 (т, J=7,1 Гц, 2Н), 3,02-3,13 (м, 2Н), 3,17-3,28 (м, 2Н), 3,44-3,92 (уш.с, 2Н), 3,98-4,54 (уш.с, 2Н), 5,21 (с, 2Н), 6,70 (д, J=2,8 Гц, 1Н), 7,00 (дд, J=8,9, 1,9 Гц, 1Н), 7,12 (дд, J=13,4, 1,9 Гц, 1Н), 7,23-7,32 (м, 2Н), 7,41-7,50 (м, 1Н), 7,56-7,63 (м, 1Н), 7,69 (т, J=9,0 Гц, 1Н), 10,06-10,93 (уш.с, 1Н), 12,07-12,85 (уш.с, 1Н); LC-MS (способ A): Rt 1,35, [M+H] 428.
2-(4-Бромфенил)-оксазоло[4,5-c]пиридин (17, Rb=H). К охлажденной (0°C) суспензии коммерчески доступного 4-гидрокси-3-аминопиридина 14 (4 г, 36 ммоль) в DCM (200 мл) добавляли триэтиламин (6,3 мл, 1,25 экв.) и раствор 4-бромбензоилхлорида (15, Rb=H, 8 г, 36 ммоль, 1 экв., 0,3 M в DCM). Реакционной смеси позволяли достигнуть комнатной температуры и перемешивали в течение 16 ч. Смесь фильтровали, промывали DCM и простым эфиром с получением неочищенного 16 (Rb=H) в виде твердого материала, который использовали на следующей стадии без дальнейшей очистки. Гексахлорэтан (10,2 г, 43 ммоль, 2,5 экв.) растворяли в DCM (150 мл) и добавляли трифенилфосфин (13,56 г, 51,69 ммоль, 3 экв.) и триэтиламин (19,2 мл, 137,8 ммоль, 8 экв.). Смесь перемешивали в течение 10 минут при комнатной температуре и медленно добавляли неочищенное соединение 16 (Rb=H) 5-ью равными частями. Смесь перемешивали при комнатной температуре в течение 64 ч, после чего анализ TLC (DCM/MeOH, 97/3, об./об., Rf 0,3) показал завершение реакции. Раствор концентрировали, и осадок суспендировали в DCM. Смесь фильтровали, и осадок промывали DCM и диэтиловым эфиром с получением неочищенного 17 (Rb=H), которое использовали на следующей стадии без дальнейшей очистки.
2-(4-Бромфенил)-5-метил-4,5,6,7-тетрагидрооксазоло[4,5-c]пиридин (19, Rb=H). К раствору соединения 17 (Rb=H, 11,4 ммоль) в DMF (95 мл) добавляли йодметан (2,84 мл, 45,58 ммоль, 4 г. э.) и смесь перемешивали в течение 16 ч. Смесь концентрировали в вакууме, и осадок перемешивали в EtOAc с получением неочищенного 18 (Rb=H, 3,3 г, 69%) в виде белого твердого вещества. Соединение 18 (Rb=H, 2,3 г, 5,5 ммоль) растворяли в метаноле (55 мл), и раствор охлаждали до 0°C. Добавляли боргидрид натрия (0,42 г, 11 ммоль, 2 г. э.) и смесь перемешивали при 0°C в течение 2 ч, после чего ей позволяли достигнуть комнатной температуры и перемешивание продолжали в течение 16 ч. Добавляли воду (4 мл) и смесь перемешивали в течение 5 минут. Смесь совместно упаривали с ацетонитрилом, и осадок очищали колоночной хроматографией на силикагеле (DMA 0,5) с получением соединения 19 (Rb=H) с выходом 61%.
2-(4-Бромфенил)-4,5,6,7-тетрагидрооксазоло[4,5-c]пиридин (20, Rb=H). К охлажденному (0°C) раствору соединения 19 (Rb=H, 0,95 г, 3,2 ммоль) в 1,2-дихлорэтане (32 мл) добавляли DIPEA (1,1 мл, 6,4 ммоль, 2 экв.). При 0°C добавляли 1-хлорэтилхлорформиат (1,05 мл, 9,72 ммоль, 3 экв.) и смесь перемешивали в течение 10 минут при 0°C, после чего температуру повышали до температуры кипячения с обратным холодильником. Через 2 ч смесь концентрировали в вакууме, и осадок растворяли в метаноле (35 мл). Раствор перемешивали в течение 48 ч при комнатной температуре. Осадок отфильтровывали, твердый продукт промывали диэтиловым эфиром с получением соединения 20 (Rb=H, 0,9 г, 88%). LC-MS (способ A): Rt 1,1, [M+H] 280.
Трет-бутиловый эфир 3-[2-(4-бромфенил)-6,7-дигидро-H-оксазоло[4,5-c]пиридин-5-ил]пропионовая кислота (21, Rb=H). Соединение 20 (Rb=H, 8 г, 22,8 ммоль) суспендировали в метаноле (200 мл) и добавляли DIPEA (8,15 мл, 46,8 ммоль, 2,05 г. э.). К смеси добавляли трет-бутилакрилат (3,97 мл, 27,4 ммоль, 1,2 экв.) и смесь кипятили с обратным холодильником в течение 120 ч. Преобразование проверяли с помощью анализа TLC, и через 16 и 64 ч добавляли дополнительный трет-бутилакрилат (3,97 мл, 27,4 ммоль, 1,2 экв.) для направления реакции к завершению. Растворители выпаривали, и осадок перерастворяли в EtOAc и экстрагировали 5% раствором NaHCO3. Органический слой сушили (MgSO4), концентрировали в вакууме, и осадок очищали колоночной хроматографией на силикагеле (элюент: диэтиловый эфир/петролейный эфир, 1/1, об./об.) с получением 21a (Rb=H, 8,8 г, 93%). LC-MS (способ A): Rt 1,38, [M+H] 408.
Трет-бутиловый эфир 3-{2-[4-(бензилокси)фенил)]-6,7-дигидро-H-оксазоло[4,5-c]пиридин-5-ил}пропионовой кислоты (23a, Ra=Rb=H). Соединение 21a (Rb=H, 0,9 г, 2,21 ммоль) растворяли в дегазированном толуоле (7 мл) и к раствору добавляли карбонат цезия (1,08 г, 3,3 ммоль), бензиловый спирт (0,46 мл, 4,42 ммоль, 2 экв.), 2-ди-трет-бутилфосфино-3,4,5,6-тетраметил-2',4',6'-триизопропил-1,1-бифенил (25,5 мг, 0,05 ммоль, 0,02 экв.) и ацетат палладия(II) (9,92 мг, 0,04 ммоль, 0,02 экв.). Смесь перемешивали при 70°C в течение 16 ч. Смесь охлаждали до комнатной температуры, концентрировали в вакууме, и осадок очищали колоночной хроматографией на силикагеле (диэтиловый эфир/петролейный эфир, 3/1, об./об.) с получением чистого 23a (Ra=Rb=H, 0,71 г, 74%) в виде белого твердого вещества. LC-MS (способ A): Rt 1,46, [M+H] 435.
3-{2-[4-(Бензилокси)фенил)]-6,7-дигидро-H-оксазоло[4,5-c]пиридин-5-ил}пропионовая кислота (47). Соединение 23a (Ra=Rb=H, 0,71 г, 1,63 ммоль) растворяли в растворе HCl в 1,4-диоксане (4 Н, 12 мл, 30 экв.) и смесь перемешивали в течение 16 ч при 50°C. Растворители выпаривали и добавляли диэтиловый эфир для осаждения продукта. Белый твердый материал фильтровали и сушили в вакууме с получением соединения 47 (0,67 г, 93%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6): δ ч/млн, 2,92 (т, J=7,1 Гц, 2Н), 3,06-3,31 (уш.с, 2Н), 3,57 (т, J=7,1 Гц, 2Н), 3,50-3,61 (уш.с, 1Н), 3,79-4,04 (уш.с, 1Н), 4,21-4,42 (уш.с, 1Н), 4,42-4,60 (уш.с, 1Н), 5,20 (с, 2Н), 7,16 (д, J=8,6 Гц, 2Н), 7,35 (т, J=7,5 Гц, 1Н), 7,41 (т, J=7,5 Гц, 2Н), 7,49 (д, J=7,5 Гц, 2Н), 7,93 (д, J=8,6 Гц, 2Н), 10,31-10,84 (уш.с, 1Н). LC-MS (способ A): Rt 1,32, [M+H] 379.
Трет-бутиловый эфир 4-[2-(4-бромфенил)-6,7-дигидро-H-оксазоло[4,5-c]пиридин-5-ил]масляной кислоты (21d, Rb=H). Соединение 20 (Rb=H, 2,5 г, 7,92 ммоль) суспендировали в DMF (40 мл). К этой суспензии добавляли карбонат калия (3,8 г, 27,7 ммоль, 3,5 экв.) и трет-бутил-4-бромбутаноат (5,3 г, 23,7 ммоль, 3 экв.). Смесь нагревали при 80°C в течение 16 ч, после чего анализ TLC выявил завершение реакции. Смесь концентрировали в вакууме, и осадок очищали колоночной хроматографией на силикагеле (элюент: диэтиловый эфир/петролейный эфир, от 1/1, об./об. до 100% диэтиловый эфир) с получением 21d (Rb=H, 3,15 г, 94%) в виде белого твердого вещества.
Трет-бутиловый эфир 4-{2-[4-(2,3-дифторбензилокси)фенил)]-6,7-дигидро-H-оксазоло[4,5-c]пиридин-5-ил}масляной кислоты (23d, Ra=2,3-diF, Rb=H). Соединение 21d (Rb=H, 0,6 г, 1,42 ммоль) растворяли в дегазированном толуоле (5 мл) и к раствору добавляли карбонат цезия (0,7 г, 2,14 ммоль), 2,3-дифторбензиловый спирт (0,32 мл, 2,85 ммоль, 2 экв.), 2-ди-трет-бутилфосфино-3,4,5,6-тетраметил-2',4',6'-триизопропил-1,1-бифенил (16,43 мг, 0,03 ммоль, 0,02 экв.) и ацетат палладия(II) (6,39 мг, 0,03 ммоль, 0,02 экв.). Смесь перемешивали при 70°C в течение 16 ч. Смесь охлаждали до комнатной температуры, концентрировали в вакууме, и осадок очищали колоночной хроматографией на силикагеле (диэтиловый эфир/петролейный эфир, от 2/1, об./об. до 100% диэтиловый эфир) с получением чистого 23d (Ra=2,3-ди-F, Rb=H, 0,48 г, 70%) в качестве масла.
4-{2-[4-(2,3-Дифторбензилокси)фенил)]-6,7-дигидро-H-оксазоло[4,5-c]пиридин-5-ил}масляная кислота (57). Соединение 23d (Ra=2,3-ди-F, Rb=H, 0,46 г, 0,95 ммоль) растворяли в растворе HCl в 1,4-диоксане (4 Н, 14 мл, 60 экв.) и смесь перемешивали в течение 16 ч при комнатной температуре. Растворители выпаривали и добавляли диэтиловый эфир для осаждения продукта. Белый твердый материал фильтровали и сушили в вакууме с получением соединения 57 (0,45 г, 99%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6): δ ч/млн 2,08 (м, 2Н), 2,41 (т, J=7,0 Гц, 2Н), 3,00-3,29 (м, 2Н), 3,31-3,40 (м, 2Н), 3,48-3,70 (уш.с, 1Н), 3,70-3,96 (уш.с, 1Н), 4,18-4,38 (м, 1Н), 4,38-4,61 (м, 1Н), 5,26 (с, 2Н), 7,16 (д, J=8,8 Гц, 2Н), 7,19-7,25 (м, 1Н), 7,28-7,39 (м, 2Н), 7,94 (д, J=8,8 Гц, 2Н), 10,37-10,88 (уш.с, 1Н); LC-MS (способ A): Rt 1,16, [M+H] 429.
4-{2-[4-Бензилоксифенил]-6,7-дигидро-H-оксазоло[4,5-c]пиридин-5-ил}масляная кислота (53). Соединение 53 получали аналогично тому, как описано для синтеза 57, начиная с 23d (Ra=Rb=H). 1H ЯМР (400 МГц, ДМСО-d6): δ ч/млн 2,01-2,17 (м, 2Н), 2,42 (т, J=6,9 Гц, 2Н), 3,00-3,30 (м, 2Н), 3,32-3,41 (м, 2Н), 3,50-3,69 (м, 1Н), 3,78-3,96 (м, 1Н), 4,18-4,37 (м, 1Н), 4,43-4,59 (м, 1Н), 5,18 (с, 2Н), 7,14 (д, J=8,8 Гц, 2Н), 7,34 (т, J=7,9 Гц, 1Н), 7,40 (т, J=7,9 Гц, 2Н), 7,47 (д, J=7,9 Гц, 2Н), 7,93 (д, J=8,8 Гц, 2Н), 10,45-10,89 (уш.с, 1Н); LC-MS (способ A): Rt 1,17, [M+H] 393.
4-{2-[4-(4-Трифторметилбензилокси)-2-фторфенил]-6,7-дигидро-H-оксазоло[4,5-c]пиридин-5-ил}масляная кислота (85). Соединение 85 получали, следуя пути C, начиная с 2-фтор-4-бромбензоилхлорида. 1H ЯМР (400 МГц, ДМСО-d6), δ ч/млн: 1,98-2,11 (м, 2Н), 2,40 (т, J=7,1 Гц, 2Н), 3,10-3,24 (м, 2Н), 3,30-3,42 (м, 2Н), 3,51-3,66 (уш.с, 1Н), 3,79-3,97 (уш.с, 1Н), 4,25-4,39 (м, 1Н), 4,49-4,65 (м, 1Н), 5,32 (с, 2Н), 6,98-7,13 (м, 2Н), 7,66-7,78 (м, 4Н), 7,93 (т, J=8,6 Гц, 1Н), 9,91-10,46 (уш.с, 1Н); LC-MS (Способ A): Rt 1,77, [M+H] 479.
4-{2-[4-(2-Фторбензилокси)-2-метилфенил]-6,7-дигидро-H-оксазоло[4,5-c]пиридин-5-ил}масляная кислота (89). Соединение 89 получали, следуя пути C, начиная с 2-метил-4-бромбензоилхлорида. 1H ЯМР (400 МГц, ДМСО-d6), δ ч/млн: 2,00-2,12 (м, 2Н), 2,41 (т, J=7,1 Гц, 2Н), 2,64 (с, 3Н), 3,05-3,26 (м, 2Н), 3,32-3,42 (м, 2Н), 3,50-3,70 (уш.с, 1Н), 3,79-3,95 (уш.с, 1Н), 4,22-4,39 (м, 1Н), 4,48-4,65 (м, 1Н), 5,21 (с, 2Н), 6,97-7,04 (м, 2Н), 7,17-7,27 (м, 2Н), 7,37-7,46 (м, 1Н), 7,56 (дт, J=7,4, 1,4 Гц, 1Н), 7,89 (д, J=8,5 Гц, 1Н), 9,97-10,46 (уш.с, 1Н); LC-MS (способ A): Rt 1,61, [M+H] 425.
4-{2-[4-(3,4-Дихлорбензилокси)-2-фторфенил]-6,7-дигидро-H-оксазоло[4,5-c]пиридин-5-ил}масляная кислота (227). Соединение 227 получали, следуя пути C, начиная с 2-фтор-4-бромбензоилхлорида. 1H ЯМР (600 МГц, ДМСО-d6), δ ч/млн: 1,74 (кв., J=7,1 Гц, 2Н), 2,26 (т, J=7,1 Гц, 2Н), 2,55 (т, J=7,1 Гц, 2Н), 2,73-2,78 (м, 2Н), 2,81 (т, J=5,3 Гц, 2Н), 3,43 (уш.с, 2Н), 5,21 (с, 2Н), 7,00 (дд, J=8,8, 2,5 Гц, 1Н), 7,11 (дд, J=13,0, 2,5 Гц, 1Н), 7,47 (дд, J=8,4, 2,0 Гц, 1Н), 7,69 (д, J=8,3 Гц, 1Н), 7,76 (д, J=1,9 Гц, 1Н), 7,89 (т, J=8,3 Гц, 1Н), 11,30-12,80 (уш.с, 1Н); LC-MS (способ A): Rt 1,42, [M+H] 479.
4-{2-[4-(2-Фторбензилокси)-3-хлорфенил]-6,7-дигидро-H-оксазоло[4,5-c]пиридин-5-ил}масляная кислота (283). Соединение 283 получали, следуя пути C, начиная с 3-хлор-4-бром-бензоилхлорида. 1H ЯМР (400 МГц, ДМСО-d6): δ ч/млн 1,69-1,80 1,74 (м, 2Н), 2,27 (т, J=7,2 Гц, 2Н), 2,55 (т, J=7,0 Гц, 2Н), 2,73-2,79 (м, 2Н), 2,81 (т, J=4,6 Гц, 2Н), 3,43 (с, 2Н), 5,32 (с, 2Н), 7,25-7,33 (м, 2Н), 7,42-7,50 (м, 2Н), 7,58-7,65 (м, 1Н), 7,88 (дд, J=8,7, 2,1 Гц, 1Н), 7,93 (д, J=2,1 Гц, 1Н), 11,0-13,0 (уш.с, 1Н); LC-MS (способ B): Rt 1,99*, [M+H] 445.
4-{2-[4-(4-Хлорбензилокси)-3-фторфенил]-6,7-дигидро-H-оксазоло[4,5-c]пиридин-5-ил}масляная кислота (211). Соединение 211 получали, следуя пути C, начиная с 3-фтор-4-бромбензоилхлорида. 1H ЯМР (400 МГц, ДМСО-d6) δ ч/млн: 1,95-2,06 (м, 2Н), 2,38 (т, J=7,2 Гц, 2Н), 3,09-3,20 (м, 2Н), 3,23-3,32 (м, 2Н), 3,44-3,53 (м, 1Н), 3,76-3,86 (м, 1Н), 4,20-4,32 (м, 1Н), 4,43-4,52 (м, 1Н), 5,28 (с, 2Н), 7,43 (т, J=8,6 Гц, 1Н), 7,51 (м, 4Н), 7,76 (м, 2Н), 10,65-11,02 (уш.с, 1Н), 12,03-12,77 (уш.с, 1Н); LC-MS (способ B): Rt 2,03*, [M+H] 445.
2-(4-Бензилоксифенил)оксазоло[4,5-c]пиридин (25, Ra=Rb=H). К охлажденной (0°C) суспензии коммерчески доступного 4-гидрокси-3-аминопиридина (14, 19,3 г, 175 ммоль) в ацетонитриле (1500 мл) добавляли 4-бензилоксибензойную кислоту (24, Ra=Rb=H, 40 г, 175 ммоль), трифенилфосфин (142,5 г, 543 ммоль, 3,1 экв.) и трихлорацетонитрил (54,5 мл, 543 ммоль, 3,1 экв.). Реакционной смеси позволяли достигнуть комнатной температуры и перемешивали в течение 16 ч при 80°C. Смесь концентрировали в вакууме, и остатки растворяли в DCM и промывали 2 Н NaOH (3x). Объединенные водные слои экстрагировали DCM, и органические слои сушили (Na2SO4) с получением неочищенного 25 (Ra=Rb=H) в виде масла, которое использовали на следующей стадии без дальнейшей очистки.
2-(4-Бензилоксифенил)-5-метил-4,5,6,7-тетрагидрооксазоло[4,5-c]пиридин (26, Ra=Rb=H). К раствору неочищенного 25 (Ra=Rb=H, 117 ммоль) в DMF (540 мл) добавляли йодметан (29,35 мл, 471 ммоль, 4 г. э.) и смесь перемешивали в течение 16 ч. Смесь концентрировали в вакууме, и осадок перемешивали в EtOAc с получением четвертичной соли 25 в виде белого твердого вещества, которое растворяли в метаноле (950 мл), и раствор охлаждали до 0°C. Добавляли боргидрид натрия (10,2 г, 268 ммоль, 2,5 г. э.) и смесь перемешивали при 0°C в течение 2 ч, после чего ей позволяли достигнуть комнатной температуры, и перемешивание продолжали в течение 64 ч. Добавляли воду (117 мл) и смесь перемешивали в течение 5 минут. Смесь концентрировали в вакууме, осадок суспендировали в 2 Н NaOH (5 мл/ммоль) и экстрагировали DCM (3x). Объединенные органические слои сушили (Na2SO4) и концентрировали с получением неочищенного 26 (Ra=Rb=H) в виде желтого твердого вещества, которое использовали на следующей стадии без дальнейшей очистки.
2-(4-Бензилоксифенил)-4,5,6,7-тетрагидрооксазоло[4,5-c]пиридин (27, Ra=Rb=H). К охлажденному (0°C) раствору соединения 26 (Ra=Rb=H, 35,2 г, 109,8 ммоль) в 1,2-дихлорэтане (880 мл) добавляли DIPEA (37,61 мл, 219,7 ммоль, 2 экв.) и добавляли 1-хлорэтилхлорформиат (35,56 мл, 329,6 ммоль, 3 экв.). Смесь перемешивали в течение 10 минут при 0°C, после чего температуру повышали до температуры кипячения с обратным холодильником. Через 4 ч смеси позволяли достигнуть комнатной температуры, и перемешивание продолжали в течение 16 ч. Смесь концентрировали в вакууме, и осадок растворяли в метаноле (880 мл). Раствор перемешивали в течение 16 ч при комнатной температуре, после чего анализ TLC показал завершение реакции. Удаление растворителя привело к выделению неочищенного 27 (Ra=Rb=H) с общим выходом 20% в расчете на 25 (Ra=Rb=H).
Трет-бутиловый эфир 3-[2-(4-бензилоксифенил)-4,5,6,7-тетрагидрооксазоло[4,5-c]пиридин]-2-метилпропионовой кислоты (28b). К раствору соединения 27 (Ra=Rb=H, 13,45 г, 43,9 ммоль) в DMF (270 мл) добавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (3 г. э.) и трет-бутилметакрилат (28,54 мл, 175,6 ммоль, 4 экв.). Смесь нагревали при 125°C в течение 100 ч. Раствор охлаждали и добавляли 5% NaHCO3 (15 мл/ммоль), и экстрагировали смесью диэтиловый эфир/EtOAc, 1/1, об./об. Органический слой промывали водой (4x), сушили над MgSO4, концентрировали в вакууме, и осадок очищали колоночной хроматографией на силикагеле (элюент: диэтиловый эфир/петролейный эфир, от 1/2 до 3/1, об./об.) с получением 23b (Ra=Rb=H, 14,58 г, 74%) в качестве масла.
3-[2-(4-Бензилоксифенил)-6,7-дигидро-H-оксазоло[4,5-c]пиридин-5-ил]-2-метилпропионовая кислота (146). Соединение 23b (Ra=Rb=H, 0,45 г, 1 ммоль) растворяли в растворе HCl в 1,4-диоксане (4 Н, 14 мл, 60 экв.) и смесь перемешивали в течение 16 ч при комнатной температуре. Растворители выпаривали и добавляли диэтиловый эфир для осаждения продукта. Белый твердый материал фильтровали и сушили в вакууме с получением соединения 146 (0,43 г, 99%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6), δ ч/млн: 1,28 (д, J=7,3 Гц, 3Н), 3,07-3,20 (м, 3Н), 3,26 (дд, J=13,2, 4,9 Гц, 1Н), 3,60 (дд, J=13,2, 7,9 Гц, 1Н), 3,66 (уш.с, 2Н), 4,34 (уш.с, 2Н), 5,18 (с, 2Н), 7,11-7,17 (м, 2Н), 7,31-7,37 (м, 1Н), 7,37-7,43 (м, 2Н), 7,44-7,49 (м, 2Н), 7,85-7,93 (м, 2Н), 10,17-12,85 (уш.с, 1Н); LC-MS (способ A): Rt 1,48, [M+H] 393.
3-{2-[4-(4-Трифторметилбензилокси)фенил]-6,7-дигидро-H-оксазоло[4,5-c]пиридин-5-ил}-2-метилпропионовая кислота (156). Соединение 156 получали, следуя пути C. 1H ЯМР (400 МГц, ДМСО-d6), δ ч/млн: 1,29 (д, J=7,2 Гц, 3Н), 3,09-3,21 (уш.с, 3Н), 3,27 (дд, J=13,1, 5,3 Гц, 1Н), 3,57-3,72 (дд, J=13,1, 7,3 Гц, 1Н), 3,65-3,72 (уш.с, 2Н), 4,34 (уш.с, 2Н), 5,33 (с, 2Н), 7,21 (д, J=8,8 Гц, 2Н), 7,68-7,74 (м, 2Н), 7,75-7,81 (м, 2Н), 7,93 (д, J=8,8 Гц, 2Н), 10,50-12,32 (уш.с, 1Н); 13C ЯМР (101 МГц, ДМСО-d6), δ ч/млн: 16,35 (кв., 1C), 19,25 (т, 1C), 35,14 (д, 1C), 49,04 (уш.т, 1C), 49,76 (уш.т, 1C), 57,23 (т, 1C), 68,66 (т, 1C), 115,60 (д, 2C), 119,65 (с, 1C), 124,21 (с, 1JCF=272,5 Гц, 1C), 125,34 (д, 3JCF=3,6 Гц, 2C), 127,63 (д, 2C), 128,05 (д, 2C), 128,51 (с, 2JCF=31,7 Гц, 1C), 128,63 (с, 1C), 141,51 (с, 1C), 142,86 (с, 1C), 160,11 (с, 1C), 160,76 (с, 1C), 174,95 (с, 1C); LC-MS (способ A): Rt 1,82, [M+H] 461.
3-{2-[4-(4-Хлорбензилокси)фенил]-6,7-дигидро-H-оксазоло[4,5-c]пиридин-5-ил}-2-метилпропионовая кислота (157). Соединение 157 получали, следуя пути C. 1H ЯМР (400 МГц, ДМСО-d6), δ ч/млн: 1,19 (д, J=7,4 Гц, 3Н), 3,00-3,12 (м, 3Н), 3,18 (дд, J=13,2, 5,3 Гц, 1Н), 3,52 (дд, J=13,2, 7,1 Гц, 1Н), 3,56-3,66 (уш.с, 2Н), 4,24 (уш.с, 2Н), 5,11 (с, 2Н), 7,08 (д, J=9,0 Гц, 2Н), 7,35-7,43 (м, 4Н), 7,81 (д, J=9,0 Гц, 2Н), 10,57-11,92 (уш.с, 1Н); 13С ЯМР (101 МГц, ДМСО-d6), δ ч/млн: 16,44 (кв., 1С), 19,19 (т, 1С), 35,09 (д, 1С), 49,1 (уш.т, 1С), 49,6 (уш.т, 1С), 57,17 (т, 1С), 68,72 (т, 1С), 115,59 (д, 2С), 119,50 (с, 1С), 127,60 (д, 2С), 128,45 (д, 2С), 128,52 (с, 1С), 129,51 (д, 2С), 132,58 (с, 1С), 135,69 (с, 1С), 142,79 (с, 1С), 160,22 (с, 1С), 160,80 (с, 1С), 174,92 (с, 1С); LC-MS (способ А): Rt 1,72, [М+Н] 427.
3-{2-[4-(2,3-Дифторбензилокси)фенил]-6,7-дигидро-H-оксазоло[4,5-с]пиридин-5-ил}масляная кислота (175). Соединение 175 получали, следуя пути С. 1Н ЯМР (400 МГц, ДМСО-d6), δ ч/млн: 1,27 (д, J=6,6 Гц, 3Н), 2,5-2,6 (м, 1Н), 2,8-3,1 (м, 3Н), 3,1-3,5 (уш.с, 2Н), 3,6-3,7 (уш.с, 1Н), 3,9-4,1 (уш.с, 2Н), 5,28 (с, 2Н), 7,0-7,2 (м, 2Н), 7,2-7,3 (м, 1Н), 7,4-7,5 (м, 2Н), 7,9-8,0 (м, 2Н), 12,16 (уш.с, 1Н); LC-MS (способ А): 1,3, [М+Н] 429.
3-{2-[4-(4-Трифторметилбензилокси)-2-фторфенил]-6,7-дигидро-H-оксазоло[4,5-с]пиридин-5-ил}-2-метилпропионовая кислота (271). Соединение 271 получали, следуя пути С. 1Н ЯМР (400 МГц, ДМСО-d6), δ ч/млн: 1,29 (д, J=7,2 Гц, 3Н), 3,06-3,36 (м, 4Н), 3,51-3,68 (м, 2Н), 3,70-3,90 (уш.с, 1Н), 4,18-4,58 (уш.с, 2Н), 5,33 (с, 2Н), 7,03 (дд, J=8,7, 2,2 Гц, 1Н), 7,10 (дд, J=12,9, 2,2 Гц, 1Н), 7,65-7,71 (м, 2Н), 7,73-7,77 (м, 2Н), 7,92 (т, J=8,7 Гц, 1Н), 11,01 (уш.с, 1Н); LC-МБ (способ А): 1,56, [М+Н] 479.
Трет-бутиловый эфир 2-(4-бромфенил)-6,7-дигидро-H-оксазоло[4,5-с]пиридин-5-карбоновой кислоты (28, Rb=Н). К
суспензии соединения 20 (Rb=H, 14,3 г, 48,2 ммоль) в DCM (300 мл) добавляли DIPEA (1 экв.) и ди-трет-бутилдикарбонат (11,59 г, 53 ммоль, 1,1 экв.). Смесь перемешивали при комнатной температуре в течение 16 ч, после чего анализ TLC выявил завершение реакции. Смесь концентрировали в вакууме с получением неочищенного соединения 28 (Rb=H), которое использовали на следующей стадии без дальнейшей очистки.
Трет-бутиловый эфир 2-(4-гидроксифенил)-6,7-дигидро-H-оксазоло[4,5-c]пиридин-5-карбоновой кислоты (29, Rb=H). Соединение 28 (Rb=H, 9 г, 20,4 ммоль) растворяли в 1,4-диоксане (90 мл) и к раствору добавляли гидроксид калия (4,58 г, 81 ммоль, в 90 мл воды), и раствор дегазировали. К раствору добавляли 2-ди-трет-бутилфосфино-2',4',6'-триизопропилбифенил (346 мг, 0,82 ммоль, 0,04 экв.) и трис-(дибензилиденацетон)дипалладий(0) (373,5 мг, 0,41 ммоль, 0,02 экв.). Смесь перемешивали при 80°C в течение 16 ч, после чего анализ LC-MS показал, что преобразование завершилось. Смесь охлаждали до комнатной температуры, разбавляли EtOAc, подкисляли до pH 6 с помощью 0,1 Н HCl и экстрагировали EtOAc. Органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме. Осадок очищали колоночной хроматографией на силикагеле (DCM/MeOH, 97/3, об./об.) с получением чистого соединения 29 (Rb=H, 6 г, 83%).
Трет-бутиловый эфир 2-[4-(4-трифторметилбензилокси)фенил)-6,7-дигидро-H-оксазоло[4,5-c]пиридин-5-карбоновой кислоты (30, Ra=4CF3, Rb=H). Соединение 29 (Rb=H, 6 г, 17 ммоль) растворяли в смеси DCM/вода, 2/1, об./об. (78 мл/ммоль) и к этому раствору добавляли гидроксид натрия (27 мл, 2 Н, 3 экв.). К этой смеси добавляли бромид тетрабутиламмония (549 мг, 0,1 экв.) и 4-трифторметилбензилбромид (4,48 г, 18,75 ммоль, 1,1 экв.). Смесь перемешивали в течение 16 ч при комнатной температуре, после чего анализ LC-MS показал завершение реакции. Смесь разбавляли DCM (200 мл), слои разделяли, и водный слой экстрагировали DCM. Органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме. Осадок очищали колоночной хроматографией на силикагеле (элюент: EtOAc/петролейный эфир, 1/3) с получением чистого соединения 30 (Rb=H, 9,0 г, 96%) в качестве бесцветной пены.
2-[4-(4-Трифторметилбензилокси)фенил)-6,7-дигидро-H-оксазоло[4,5-c]пиридин (30, Ra=4CF3, Rb=H). Соединение 30 (9,6 г, 18,5 ммоль) растворяли в DCM (150 мл) и добавляли трифторуксусную кислоту (8,5 мл, 111 ммоль, 6 экв.). Раствор кипятили с обратным холодильником в течение 16 ч, после чего анализ TLC показал завершение реакции. Смесь нейтрализовывали 5% водн. NaHCO3. Смесь экстрагировали DCM (3×), и объединенные органические слои промывали рассолом, сушили над Na2SO4 и концентрировали в вакууме с получением соединения 27 (Ra=4CF3, Rb=H), которое использовали на следующей стадии без дальнейшей очистки.
3-{2-[4-(4-Трифторметоксибензилокси)фенил]-6,7-дигидро-H-оксазоло[4,5-c]пиридин-5-ил}циклобутанкарбоновая кислота (306). Соединение 27 (Ra=4CF3, Rb=H, 0,71 г, 1,8 ммоль) суспендировали в 1,2-дихлорэтане (40 мл). К этой суспензии добавляли 3-оксоциклобутанкарбоновую кислоту (0,27 г, 2,34 ммоль, 1,3 экв.) и триацетоксиборгидрид натрия (0,61 г, 2,88 ммоль, 1,6 экв.). Смесь перемешивали при комнатной температуре в течение 16 ч, после чего анализ TLC выявил завершение реакции. Раствор разбавляли 5% NaHCO3 (15 мл/ммоль) и смесь экстрагировали DCM. Объединенные органические слои сушили над Na2SO4, концентрировали в вакууме, и осадок очищали колоночной хроматографией на силикагеле с получением смеси цис- и транс-стереоизомеров в соотношении 2 к 1. Вторая очистка колоночной хроматографией на силикагеле (DCM/MeOH, 9/1, об./об.) привела к выделению двух обогащенных стереоизомерных фракций. Соединение 307-цис (Rf 0,2, 0,46 г, 51%, цис/транс=95/5) и 306-транс (Rf 0,25, 0,21 г, 25%, цис/транс=5/95). 307-цис: 1H ЯМР (400 МГц, ДМСО-d6): δ ч/млн 1,92-2,05 (м, 2Н); 2,26-2,37 (м, 2Н); 2,68 (т, J=4,5 Гц, 2Н); 2,70-2,79 (м, 3Н); 2,89-3,00 (м, 1Н); 3,30-3,41 (уш.с, 2Н); 5,21 (с, 2Н); 7,14 (д, J=8,8 Гц, 2Н); 7,41 (д, J=8,5 Гц, 2Н); 7,61 (д, J=8,5 Гц, 2Н); 7,87 (д, J=8,8 Гц, 2Н); 12,15 (уш.с, 1Н); 306-транс: 1H ЯМР (400 МГц, ДМСО-d6) δ ч/млн: 2,10-2,21 (м, 2Н); 2,22-2,34 (м, 2Н); 2,67 (т, J=4,8 Гц, 2Н); 2,71-2,78 (м, 2Н); 2,84-2,95 (м, J=9,6 Гц, 1Н); 3,17 (квинт., J=7,4 Гц, 1Н); 3,31-3,40 (уш.с, 2Н); 5,21 (с, 2Н); 7,14 (д, J=8,8 Гц, 2Н); 7,41 (д, J=8,5 Гц, 2Н); 7,61 (д, J=8,5 Гц, 2Н); 7,87 (д, J=8,8 Гц, 2Н); 12,20 (уш.с, 1Н). LC-MS Rt 1,41, [M+H]=489.
3-{2-[4-(3,4-Дифторбензилокси)фенил]-6,7-дигидро-H-оксазоло[4,5-c]пиридин-5-ил}циклобутанкарбоновая кислота (277). Соединения 278-цис и 277-транс получали, как описано для 306. 278-цис: 1H ЯМР (400 МГц, ДМСО-d6): δ ч/млн 1,93-2,04 (м, 2Н), 2,26-2,36 (м, 2Н), 2,68 (т, J=4,8 Гц, 2Н), 2,70-2,80 (м, 3Н), 2,88-3,00 (м, 1Н), 3,33-3,40 (уш.с, 2Н), 5,16 (с, 2Н), 7,14 (д, J=8,8 Гц, 2Н), 7,29-7,38 (м, 1Н), 7,47 (дт, J=10,7, 8,4 Гц, 1Н), 7,56 (ддд, J=11,5, 8,0, 2,0 Гц, 1Н), 7,87 (д, J=8,8 Гц, 2Н), 12,15 (уш.с, 1Н); LC-MS (способ A): Rt 1,33, [M+H]=441; 277-транс: 1H ЯМР (400 МГц, ДМСО-d6), δ ч/млн: 2,09-2,20 (м, 2Н), 2,23-2,31 (м, 2Н), 2,67 (т, J=4,5 Гц, 2Н), 2,70-2,77 (м, 2Н), 2,84-2,93 (м, 1Н), 3,12-3,22 (м, 1Н), 3,32-3,39 (уш.с, 2Н), 5,16 (с, 2Н), 7,14 (д, J=8,8 Гц, 2Н), 7,30-7,37 (м, 1Н), 7,47 (дт, J=10,8, 8,4 Гц, 1Н), 7,56 (ддд, J=11,4, 8,0, 1,9 Гц, 1Н), 7,87 (д, J=8,8 Гц, 2Н), 12,20 (уш.с, 1Н); LC-MS (способ A): Rt 1,31, [M+H]=441.
3-Бром-N-бензилоксикарбонил-4-пиперидон (502). К охлажденному (0°C) раствору N-бензилоксикарбонил-4-пиперидона (501, 27,3 г, 117 ммоль) в DCM (3 мл/ммоль) добавляли DIPEA (25,5 мл, 146,2 ммоль, 1,25 г. э.) и триметилсилилтрифторметансульфонат (25,4 мл, 141 ммоль, 1,2 г. э.). Смесь перемешивали в течение 30 минут при 0°C. Далее добавляли N-бромсукцинимид (21,2 г, 119,3 мл, 1,02 г. э.) и перемешивание продолжали в течение 16 ч при комнатной температуре. Реакционную смесь промывали 5% NaHCO3, и органический слой сушили (MgSO4), а затем концентрировали при пониженном давлении. Полученное масло очищали хроматографией на силикагеле (диэтиловый эфир/петролейный эфир, от 1/1 до 1/0, об./об., Rf 0,1) с получением 502 с выходом 90% в виде желтого масла.
Бензиловый эфир 2-(4-метоксифенил)-6,7-дигидро-4H-тиазоло[5,4-c]пиридин-5-карбоновой кислоты (504). Соединение 502 (2,0 г, 6,5 ммоль) растворяли в этаноле (20 мл). К раствору добавляли 4-метокситиобензамид (503, 1,09 г, 6,5 ммоль, 1 экв.) и желтую смесь кипятили с обратным холодильником в течение 17 ч, после чего анализ TLC показал полное преобразование 502. Реакционную смесь концентрировали в вакууме, и осадок растворяли в EtOAc, промывали 5% Na2SO4, и органический слой сушили (MgSO4), а затем концентрировали при пониженном давлении. Полученное масло очищали хроматографией на силикагеле (диэтиловый эфир, Rf 0,3) с получением соединения 504 с выходом 49% в виде бесцветного масла. Соединения, имеющие заместитель Rb, можно получать, выбирая надлежащим образом замещенный (Rb) - 4-метокситиобензамид (503).
4-(4,5,6,7-Тетрагидротиазоло[5,4-c]пиридин-2-ил)фенол 505. Соединение 504 (18,0 г, 47,3 ммоль) растворяли в этаноле (300 мл). К охлажденному раствору (-78°C) капельно добавляли трибромид бора (5 г. э., 236 ммоль). Через один час охлаждение прекращали, и смесь перемешивали в течение 16 ч при комнатной температуре. Реакционную смесь гасили MeOH, смесь концентрировали в вакууме с получением соединения 505 в качестве масла, которое использовали на следующей стадии без дальнейшей очистки.
Общая методика синтеза соединений 508. Соединения 508 получают, начиная с соединений 505, аналогично тому, как описано для синтеза соединений 7 и 21 (см. схемы 1-4). В качестве типичного примера авторы настоящего изобретения описывают синтез соединения 508 (Ra=F, Rb=H, Rc=C2).
Трет-бутиловый эфир 3-[2-(4-(гидроксифенил)-6,7-дигидро-4H-тиазоло[5,4-c]пиридин-5-ил]пропионовой кислоты (506, Rb=H, Rc=C2). К суспензии 505 (Rb=H, 5,8 г, 25,1 ммоль) в MeOH (100 мл) и DIPEA (5,1 мл, 30,1 ммоль, 1,2 экв.) добавляли трет-бутилакрилат (4,4 мл, 30,1 ммоль, 1,2 экв.) и смесь кипятили с обратным холодильником в течение 16 ч, после чего анализ TLC (диэтиловый эфир, Rf 0,2) показал полное преобразование 505. Реакционную смесь концентрировали в вакууме, и осадок растворяли в EtOAc, промывали 5% NaHCO3, и органический слой сушили (Na2SO4), а затем концентрировали при пониженном давлении. Полученное масло очищали хроматографией на силикагеле (диэтиловый эфир) с получением соединения 506 (Rb=H, Rc=C2) с выходом 79% в виде белого твердого материала.
Трет-бутиловый эфир 3-[2-(4-(2-фторбензилокси)фенил)-6,7-дигидро-4H-тиазоло[5,4-c]пиридин-5-ил]пропионовой кислоты (507, Ra=2F, Rb=H, Rc=C2). К суспензии 506 (Rb=H, Rc=C2, 0,5 г, 1,4 ммоль) в DMA (4 мл) добавляли трифенилфосфин (0,45 г, 1,7 ммоль, 1,25 г. э.), диизопропилазодикарбоксилат (0,33 мл, 1,7 ммоль, 1,25 экв.) и 2-фторбензиловый спирт (0,17 мл, 1,56 ммоль, 1,15 г. э.) и смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь промывали диэтиловым эфиром (150 мл) и 3×50 мл воды. Объединенные органические слои сушили (Na2SO4), а затем концентрировали при пониженном давлении. Полученное масло очищали хроматографией на силикагеле (диэтиловый эфир/петролейный эфир, 2/1, об./об.) с получением соединения 507 (Ra=2F, Rb=H, Rc=C2) с выходом 67% в виде белого твердого материала.
3-[2-(4-(2-Фторбензилокси)фенил)-6,7-дигидро-4H-тиазоло[5,4-c]пиридин-5-ил]пропионовая кислота (38, Ra=2F, Rb=H, Rc=C2). Соединение 507, Ra=2F, Rb=H, Rc=C2, 0,43 г, 0,9 ммоль) растворяли в 4 Н HCl в диоксане (10 мл). Смесь перемешивали в течение 16 ч при комнатной температуре. Смесь концентрировали в вакууме, и полученное твердое вещество промывали диизопропиловым эфиром с получением 38 в виде белого твердого вещества (Ra=2F, Rb=H, Rc=C2, 0,42 г, 97%). Соединение 38: 1H ЯМР (400 МГц, ДМСО-d6), δ ч/млн: 2,88-3,03 (м, 2H), 3,06-3,14 (м, 1Н), 3,20 (м, 1Н), 3,48 (ушир. сигнал, 3Н), 3,73-3,87 (м, 1Н), 4,44 (уш.д, J=15,9, 6,0 Гц, 1Н), 4,72 (уш., J=15,9 Гц, 1Н), 5,22 (с, 2Н), 7,16 (д, J=8,6 Гц, 2Н), 7,23-7,32 (м, 2Н), 7,41-7,49 (м, 1Н), 7,59 (дт, J=7,6, 1,5 Гц, 1Н), 7,87 (д, J=8,6 Гц, 2Н), 11,63 (уш.с, 1Н); LC-MS (способ A): Rt 1,3, [M+H] 473.
4-Бром-N-(4-хлорпиридин-3-ил)бензамид (510). Смесь 4-амино-3-хлорпиридина (509, 5,9 г, 46,2 ммоль), 4-бромбензоилхлорида (15, 11,15 г, 50,8 ммоль) и карбоната калия (22,4 г, 161,7 ммоль) в ацетонитриле (150 мл) кипятили с обратным холодильником в течение 24 ч. Реакционную смесь концентрировали в вакууме, перерастворяли в DCM, и раствор промывали водой. Органический слой сушили (MgSO4), концентрировали, и полученное масло очищали колоночной хроматографией на силикагеле (DCM/MeOH, 99/1, об./об., Rf 0,2) с получением чистого 510 (10,4 г, 72%) в виде масла. Когда используют замещенное соединение 15, в соединение 510 вводится Rb.
2-(4-Бромфенил)тиазоло[4,5c]пиридин (511). Соединение 510 (10,4 г, 33,4 ммоль) суспендировали в толуоле (400 мл) и добавляли реагент Лоуссона (9,4 г, 23,4 ммоль, 0,7 экв.) и смесь кипятили с обратным холодильником в течение 24 ч, после чего анализ TLC (DCM/MeOH, 97/3, об./об., Rf 0,7) показал завершение реакции. Смесь концентрировали в вакууме, и к маслу добавляли NaHCO3 (5% раствор, 200 мл), и суспензию экстрагировали DCM (3×200 мл). Объединенные органические слои сушили над MgSO4, концентрировали, и масло очищали колоночной хроматографией на силикагеле (DCM/MeOH, от 99/1 до 97/3, об./об.) с получением чистого 511 (8,7 г, 89%) в качестве масла.
2-(4-Бензилоксифенил)-4,5,6,7-тетрагидротиазоло-[4,5c]-пиридин (518). Соединения 518 получали, начиная с 511, аналогично тому, как описано для синтеза соединений 27 на схемах 4 и 7. Соответствующие концевые части Rc можно было присоединять к 518 аналогично тому, как описано для соединений 27.
4-{2-[4-(3,5-Дифторбензилокси)фенил]-6,7-дигидро-4H-тиазоло-[4,5c]-пиридин-5-ил}масляная кислота (446, Ra=3,5-F, Rb=H, Rc=C3). Соединение 519 (Ra=3,5-F, Rb=H, Rc=C3, 0,79 г, 1,58 ммоль) растворяли в 4 Н HCl в диоксане (25 мл) и перемешивали в течение 18 ч при 50°C и в течение 70 ч при комнатной температуре. Смесь концентрировали в вакууме, и полученное масло перемешивали в диэтиловом эфире с получением 446 в виде белого твердого вещества (0,72 г, 94%). 1H ЯМР (400 МГц, ДМСО-d6), δ ч/млн: 2,03-2,15 (м, 2Н), 2,40 (т, J=7,3 Гц, 2Н), 3,12-3,22 (м, 1Н), 3,32 (ушир. сигнал, 3Н), 3,40-3,51 (м, 1Н), 3,75-3,90 (м, 1Н), 4,36 (дд, J=15,2, 6,8 Гц, 1Н), 4,59 (д, J=15,2 Гц, 1Н), 5,21 (с, 2Н), 7,05 (тт, J=9,0, 2,3 Гц, 1Н), 7,09-7,18 (м, 4Н), 7,84 (д, J=8,9 Гц, 2Н), 11,17 (уш.с, 1Н); Rt 1,33, [M+H] 502.
2-{2-[4-(3,5-Дифторбензилокси)фенил]-6,7-дигидро-H-оксазоло[4,5-c]пиридин-5-ил}уксусная кислота (463). 2-[4-(3,5-Дифторбензилокси)фенил]-4,5,6,7-тетрагидрооксазоло[4,5-c]пиридин (27, Ra=3,5-ди-F, Rb=H) синтезировали, следуя пути C, как описано выше. Соединение 27 (Ra=3,5-ди-F, Rb=H, 0,4 г, 1,17 ммоль) растворяли в ацетонитриле (20 мл). К раствору добавляли DIPEA (0,51 мл, 2,9 ммоль, 2,5 экв.) и трет-бутилбромацетат (0,19 мл, 1,29 ммоль, 1,1 экв.). Смесь кипятили с обратным холодильником в течение 6 ч, после чего анализ LCMS показал завершение реакции. Смесь концентрировали в вакууме, добавляли воду, и суспензию экстрагировали DCM (3×200 мл). Объединенные органические слои сушили над MgSO4, концентрировали, и масло очищали колоночной хроматографией на силикагеле (диэтиловый эфир/петролейный эфир, 3/1, об./об.). Полученное масло суспендировали в 4 Н HCl в диоксане (20 мл). Смесь перемешивали при 45°C в течение 5 ч и при комнатной температуре в течение 16 ч. Смесь концентрировали в вакууме с получением белого твердого вещества. Твердый материал промывали 3 раза диэтиловым эфиром с получением 463 (0,39 г, 91%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6): δ ч/млн: 3,10-3,20 (м, 2Н), 3,70-3,80 (м, 2Н), 4,31 (с, 2Н), 4,44 (с, 2Н), 5,22 (с, 2Н), 7,02-7,10 (м, 1Н), 7,13-7,20 (м, 4Н), 7,92 (д, J=8,8 Гц, 2Н), 9,53-12,44 (уш.с, 1Н); Rt 1,51; [M+H] 401.
2-{2-[4-(3,5-Дифторбензилокси)фенил]-6,7-дигидро-H-оксазоло[4,5-c]пиридин-5-ил}пропионовая кислота (464). Соединение 27 (Ra=3,5-ди-F, Rb=H, 0,4 г, 1,17 ммоль) растворяли в ацетонитриле (20 мл). К раствору добавляли DIPEA (0,51 мл, 2,9 ммоль, 2,5 экв.) и трет-бутиловый эфир 2-бромпропионовой кислоты (0,27 мл, 1,29 ммоль, 1,1 экв.). Смесь кипятили с обратным холодильником в течение 24 ч, после чего анализ LCMS показал завершение реакции. Смесь концентрировали в вакууме, добавляли воду, и суспензию экстрагировали DCM (3×200 мл). Объединенные органические слои сушили над MgSO4, концентрировали, и масло очищали колоночной хроматографией на силикагеле (диэтиловый эфир/петролейный эфир, 3/2, об./об., Rf 0,3). Полученное масло суспендировали в 4 Н HCl в диоксане (20 мл). Смесь перемешивали при 45°C в течение 5 ч и при комнатной температуре в течение 16 ч. Смесь концентрировали в вакууме с получением белого твердого вещества. Твердый материал промывали 3 раза диэтиловым эфиром с получением 464 (0,41 г, 89%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6), δ ч/млн: 1,66 (д, J=7,1 Гц, 3Н), 3,17 (уш.с, 2Н), 3,74 (уш.с, 2Н), 4,33-4,52 (м, 4Н), 5,22 (с, 2Н), 7,02-7,11 (м, 1Н), 7,13-7,25 (м, 4Н), 7,92 (д, J=8,8 Гц, 2Н); Rt 1,53, [M+H] 415.
3-{2-[4-(3,5-Дифторбензилокси)фенил]-6,7-дигидро-4H-оксазоло[4,5-c]-пиридин-5-ил}циклопентанкарбоновая кислота (465, смесь цис-транс 1/1). Соединение 27 (Ra=3,5-ди-F, Rb=H, 0,1 г, 0,29 ммоль) растворяли в 1,2-дихлорэтане (5 мл). К раствору добавляли 3-оксоциклопентанкарбоновую кислоту (0,05 г, 0,41 ммоль, 1,4 экв.), триацетоксиборгидрид натрия (0,11 г, 0,53 ммоль, 1,8 экв.) и AcOH (0,03 мл, 0,58 ммоль, 2 экв.). Смесь перемешивали в течение 16 ч, после чего анализ LCMS показал завершение реакции. К смеси добавляли насыщенный раствор NH4Cl и смесь экстрагировали DCM. Объединенные органические слои концентрировали в вакууме, и масло очищали колоночной хроматографией на силикагеле (DCM/MeOH, 9/1, об./об.) с получением соединения 465 в качестве смеси цис/транс в соотношении 1/1.
Rt 1,27, [M+H] 455.
4-{2-[4-(2,3-Дифторбензилокси)-2-метилфенил]-6,7-дигидро-4H-оксазоло-[4,5c]-пиридин-5-ил}пентановая кислота (393). Соединение 22 (Rb=2Me, Rc=H) получали, перемешивая 29 (Rb=2Me) в смеси TFA и DCM в течение 16 ч. Растворители выпаривали с получением неочищенного 22 (Rb=2Me, Rc=H) в виде масла, которое использовали на следующей стадии без дальнейшей очистки. Для получения свободного основания соединения 22 использовали колонку SCX-2 (Rb=2Me, Rc=H, 1 г, 4,3 ммоль), и его растворяли в MeOH. К этому раствору добавляли этиллевулинат (2,46 мл, 17,4 ммоль, 4 экв.), AcOH (0,5 мл, 8,7 ммоль, 2 экв.) и гидроксид палладия на угле. Реакционную смесь встряхивали при давлении водорода 4 атм. Через 16 ч ELSD показала, что реакция еще не завершилась. Добавляли другую порцию этиллевулината (2,46 мл, 17,4 ммоль, 4 экв.) и гидроксид палладия на угле и гидрогенизацию при 4 атм. продолжали в течение 72 ч. Смесь дегазировали, фильтровали через Hyflo, и осадок промывали MeOH. Фильтрат концентрировали в вакууме, и полученное масло очищали колоночной хроматографией на силикагеле с получением 522 (Rb=2Me, Rc=xCH2(CH3)CH2CH2C(=O)OEt) с выходом 41%. Последующее бензилирование 522 2,3-дифторбензилбромидом проводили аналогично тому, как описано для соединений согласно схемам 1-5, с получением соединения 523. Наконец, соединение 523 растворяли в растворе гидроксида натрия в этаноле (18 мл, 0,3M), и раствор перемешивали при 50°C в течение 16 ч. Смесь концентрировали в вакууме, и осадок перемешивали в диэтиловом эфире с получением 393 в виде белого твердого вещества (Ra=2,3-ди-F, Rb=2Me, Rc=CH2(CH3)CH2CH2C(=O)OH). 1H ЯМР (400 МГц, ДМСО-d6), δ ч/млн: 1,40 (д, J=5,9 Гц, 3Н), 1,74-1,93 (м, 1Н), 2,15-2,30 (м, 1Н), 2,31-2,50 (м, 3Н), 2,63 (с, 3Н), 3,04-3,15 (м, 1Н), 3,22-3,35 (м, 1Н), 3,45-3,69 (м, 2Н), 3,72-3,85 (м, 1Н), 4,22-4,46 (м, 2Н), 5,25 (с, 2Н), 6,98-7,07 (м, 2Н), 7,20-7,29 (м, 1Н), 7,34-7,45 (м, 2Н), 7,87 (д, J=8,6 Гц, 1Н), 10,49-10,69 (ушир. сигнал, 1Н), 11,47-13,29 (ушир. сигнал, 1Н). Rt 1,37, [M+H] 457.
4-{2-[4-(2,3-дифторбензилокси)-2-метилфенил]-6,7-дигидро-4H-оксазоло[4,5-c]-пиридин-5-ил}-2-метилмасляная кислота (394). Соединение 22 (Rb=2Me, 1 г, 3,8 ммоль) растворяли в ацетонитриле (10 мл) и к раствору добавляли DIPEA (1,93 мл, 11,25 ммоль), йодид натрия (0,56 г, 3,75 ммоль) и метиловый эфир 4-хлор-2-метилмасляной кислоты (0,85 г, 5,6 ммоль). Смесь перемешивали при 60°C в течение 16 ч, после чего вновь добавляли DIPEA (1,93 мл, 11,25 ммоль), йодид натрия (0,56 г, 3,75 ммоль) и добавляли метиловый сложный эфир 4-хлор-2-метилмасляной кислоты (0,85 г, 5,6 ммоль), и перемешивание продолжали в течение 30 ч при 60°C. Смесь концентрировали в вакууме и очищали колоночной хроматографией на силикагеле (DCM/MeOH, от 95/5 до 9/1, об./об.) с получением 525 (Rb=2Me; Rd=Me) в виде масла. Соединение 525 (Rb=2Me; Rd=Me) бензилировали в условиях Мицунобу аналогично тому, как описано выше для синтеза 23. Полученные соединения 526 (0,5 ммоль) растворяли в этаноле (20 мл) и добавляли 2 Н раствор NaOH (4 мл) и смесь перемешивали в течение 2 ч при 50°C. Раствор нейтрализовывали 1 M HCl, экстрагировали DCM (3×20 мл), и органические слои сушили над MgSO4. Выпаривание растворителя приводило к выделению чистого соединения 394. 1H ЯМР (400 МГц, ДМСО-d6), δ ч/млн: 1,09 (д, J=7,1 Гц, 3Н), 1,51-1,61 (м, 1Н), 1,80-1,92 (м, 1Н), 2,36-2,46 (м, 1Н), 2,56 (т, J=7,1 Гц, 2Н), 2,60 (с, 3Н), 2,72-2,78 (м, 2Н), 2,79-2,85 (м, 2Н), 3,39-3,47 (м, 2Н), 5,24 (с, 2Н), 6,95-7,04 (м, 2Н), 7,20-7,29 (м, 1Н), 7,35-7,47 (м, 2Н), 7,82 (д, J=8,6 Гц, 1Н), 10,85-13,06 (ушир. сигнал, 1Н). LC-MS: Rt 1,38, [M+H] 457.
4-{2-[4-(2,3-Дифторбензилокси)-2-метилфенил]-6,7-дигидро-4H-оксазоло-[4,5c]-пиридин-5-ил}-3-метилмасляная кислота (437). Синтез метилового эфира 4-хлор-3-метилмасляной кислоты.
4-Метилдигидрофуран-2-он (4 г, 39,95 ммоль) растворяли в MeOH (10 мл) и раствор охлаждали до -10°C. Капельно добавляли тионилхлорид (3,6 мл, 49,9 ммоль). Смесь перемешивали при -10°C в течение 2 ч, а затем в течение 16 ч при комнатной температуре. Смесь концентрировали в вакууме с получением неочищенного метилового эфира 4-хлор-3-метилмасляной кислоты (4 г, 87%), который использовали на следующей стадии без дальнейшей очистки.
Соединение 22 (Rb=2Me, Rd=Me) селективно алкилировали в положении азота метиловым эфиром 4-хлор-3-метилмасляной кислоты с получением 528. Последующее бензилирование 528 в ранее описанных условиях Мицунобу давало 529. Соединение 529 деметилировали аналогично тому, как описано выше для синтеза соединения 394, с получением соединения 437 (Ra=2,3-ди-F, Rb=2Me, Rd=H) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6), δ ч/: 1,12 (д, J=6,6 Гц, 3Н), 2,26 (дд, J=16,3, 7,3 Гц, 1Н), 2,44-2,57 (м, 2Н), 2,63 (с, 3Н), 2,99-3,96 (м, 6Н), 4,10-4,65 (ушир. сигнал, 2Н), 5,24 (с, 2Н), 6,96-7,08 (м, 2Н), 7,18-7,27 (м, 1Н), 7,31-7,43 (м, 2Н), 7,88 (д, J=8,6 Гц, 1Н), 10,17-11,21 (ушир. сигнал, 1Н), 11,46-13,30 (ушир. сигнал, 1Н). LC-MS: Rt 1,44, [M+H] 457.
3-{2-[4-(2-Фенилциклопропил)-3-фторфенил]-6,7-дигидро-4H-оксазоло[4,5-c]пиридин-5-ил}циклобутанкарбоновая кислота (459 транс).
2-Фенилциклопропилтрифторборан. 4,4,5,5-Тетраметил-2-(2-фенилциклопропил)-[1,3,2]диоксаборолан (3,1 г, 12,7 ммоль) в MeOH (48 мл) и воде (12 мл) охлаждали до 0°C. Добавляли бифторид калия (6,96 г, 88,9 ммоль, 7 экв.) и смесь перемешивали в течение 16 ч при комнатной температуре. Смесь концентрировали в вакууме, и осадок совместно упаривали с ацетонитрилом (3×40 мл). Осадок промывали теплым ацетонитрилом (3×40 мл), и объединенные смывы ацетонитрила концентрировали с получением 2-фенилциклопропилтрифторборана (2 г, 71%). Это производное трифторборана (1,8 г, 8,2 ммоль, 1,3 ммоль) растворяли в дегазированной смеси толуол/вода (137,5 мл, 10/1, об./об.) и добавляли трехосновный фосфат калия (5,2 г, 24,7 ммоль, 3,9 экв.). Смесь перемешивали в течение 15 минут и к этому раствору добавляли соединение 28 (Rb=3F, 2,5 г, 6,3 ммоль), ацетат палладия(II) (71,2 мг, 0,3 ммоль, 0,05 экв.) и 2-дициклогексилфосфино-2,6-диизопропокси-1,1-бифенил (296 мг, 0,6 ммоль, 0,1 экв., RuPhos). Смесь перемешивали при 115°C в течение 3 ч, после чего анализ TLC (Et2O/PA, 3/7, об./об.) показал завершение реакции. Смеси позволяли достигнуть комнатной температуры и разбавляли EtOAc (300 мл), и раствор промывали водой. Органический слой сушили над MgSO4 и концентрировали в вакууме с получением масла, которое очищали колоночной хроматографией на силикагеле (Et2O/PA, 3/7, об./об.) с получением чистого 530 (2,54 г, 92%) в виде белого твердого вещества. BOC в 530 удаляли в кислотных условиях с получением 531, которое преобразовывали в 532 в условиях введения концевой части Rc, как описано выше. Соединение 532 (чистый транс, 0,45 г, 0,94 ммоль) растворяли в 20 мл 4 M HCl в диоксане. Раствор перемешивали в течение 16 ч при комнатной температуре. Смесь концентрировали в вакууме и перемешивали в диэтиловом эфире. Полученное твердое вещество отфильтровывали с получением соединения 459 (транс, 0,36 г, 83%). 1H ЯМР (400 МГц, ДМСО-d6), δ ч/млн: 1,27 (д, J=7,3 Гц, 3Н), 1,51-1,70 (м, 2Н), 2,26-2,40 (м, 2Н), 3,05-3,23 (м, 3Н), 3,27 (дд, J=13,1, 4,8 Гц, 2Н), 3,55-3,84 (м, 3Н), 4,37 (уш.с, 2Н), 7,14-7,23 (м, 3Н), 7,26-7,35 (м, 3Н), 7,64 (д, J=11,1, Гц, 1Н), 7,74 (д, J=8,3 Гц, 1Н), 10,00-11,79 (ушир. сигнал, 1Н), 11,93-13,53 (ушир. сигнал, 1Н). LC-MS: Rt 1,36, [M+H] 433.
4-(2-{4-[2-(3,5-Дифторфенил)винил]фенил}-6,7-дигидро-4H-оксазоло-[4,5c]-пиридин-5-ил)масляная кислота (451). Раствор соединения 28 (Rb=H, 4,3 г, 11,1 ммоль), транс-2-(3,5-дифторфенил)винилбороновой кислоты пинаколового эфира (4,09 мл, 16,7 ммоль, 1,5 экв.) в толуоле (100 мл) дегазировали. К этому раствору добавляли аддукт хлор-(2-дициклогексилфосфино-2,4,6-триизопропил-1,1-бифенил)[2-(2-аминоэтил)пентил]палладия(II) с метил-трет-бутиловым эфиром (183,8 мг, 0,22 ммоль, 0,02 экв.) и трехосновный фосфат калия (7,08 г, 33,3 ммоль, 3,0 экв.). Смесь перемешивали в течение 24 ч при 115°C, после чего анализ LC-MS показал завершение реакции. Смеси позволяли достигнуть комнатной температуры и разбавляли EtOAc (300 мл), и раствор промывали 5% раствором NaHCO3. Органический слой сушили над MgSO4 и концентрировали в вакууме с получение масла, которое очищали колоночной хроматографией на силикагеле (DCM/MeOH, 99,5/0,5, об./об.) с получением чистого 533 (3,4 г, 69%) в виде белого твердого вещества. BOC в 533 удаляли в кислотных условиях с получением 534, которое преобразовывали в 535 в условиях введения концевой части Rc, как описано выше. Соединение 535 (0,45 г, 0,94 ммоль) растворяли в 20 мл 4 M HCl в диоксане. Раствор перемешивали в течение 16 ч при 55°C. Смесь концентрировали в вакууме и перемешивали в диэтиловом эфире. Полученное твердое вещество отфильтровывали с получением соединения 451 (0,426 г, 95%).
1H ЯМР (400 МГц, ДМСО-d6), δ ч/млн: 2,02-2,14 (м, 2Н), 2,40 (т, J=7,1 Гц, 2Н), 3,07-3,18 (м, 1Н), 3,23-3,38 (м, 3Н), 3,65-3,72 (м, 1H), 3,78-3,89 (м, 1Н), 4,23-4,35 (м, 1Н), 4,41-4,54 (м, 1Н), 6,96-7,05 (м, 1Н), 7,28-7,37 (м, 3Н), 7,41 (д, J=16,4 Гц, 1Н), 7,74 (д, J=8,3 Гц, 2Н), 7,98 (д, J=8,3 Гц, 2Н), 11,25 (уш.с, 1Н). LC-MS: Rt 1,39, [M+H] 425.
4-(2-{4-[2-(3,5-Дифторфенил)этил]фенил}-6,7-дигидро-4H-оксазоло[4,5c]пиридин-5-ил)масляная кислота (452). Соединение 533 (1,5 г, 3,4 ммоль) растворяли в MeOH (250 мл). К суспензии добавляли гидроксид палладия на угле (20%, 0,2 г, 1,42 ммоль, 0,42 экв.). Смесь помещали под слой водорода при комнатной температуре и 1 атм. Через 24 ч анализ LC-MS показал завершение реакции (анализ TLC: DCM/MeOH, 99/1, об./об.). Смесь фильтровали над Hyflo, и фильтрат концентрировали в вакууме с получением 536 (1,43 г, 95%) в качестве масла. Соединение 536 конвертировали в соответствующее производное с концевой частью 538, как описано выше. Защитные группы tBu или метиловых эфиров удаляли аналогично тому, как описано выше. Соединение 452: 1H ЯМР (400 МГц, ДМСО-d6), δ ч/млн: 1,99-2,13 (м, 2Н), 2,39 (т, J=7,2 Гц, 2Н), 2,96 (с, 4Н), 3,18 (ушир. сигнал, 2Н), 3,25-3,33 (м, 2Н), 3,51-3,95 (ушир. сигнал, 2Н), 4,34 (уш.с, 2Н), 6,85-6,98 (м, 3Н), 7,37 (д, J=8,3 Гц, 2Н), 7,88 (д, J=8,3 Гц, 2Н), 10,67-11,61 (ушир. сигнал, 1Н), 11,86-12,71 (ушир. сигнал, 1Н). LC-MS: Rt 1,36, [M+H] 427.
Получение чистых энантиомеров хиральных соединений 1 и 2. Соединения, имеющие, например, замещение метилом в концевой части Rc, дают смеси энантиомеров в качестве исследуемого соединения. Разделение этой смеси на чистые энантиомеры можно проводить способами хиральной ВЭЖХ, начиная с того момента, как хиральную концевую часть вводят в ядро, т.е., например, с соединений 1, 2, 7, 8, 9, 21, 22, 23. Для специалистов в данной области очевидно, что небольшие изменения в ядре могут привести к отличающемуся поведению в процессе хирального разделения. Таким образом, каждое соединение подвергали скринингу с использованием широкого набора условий, таких как материал хиральной колонки и элюенты. Таким образом, для каждого соединения определяли, на какой стадии и в каких условиях хирального разделения разделение является наиболее успешным. Разделенные конечные продукты были названы Rel1 и Rel2, поскольку абсолютная конфигурация еще не определена. Этот процесс проиллюстрирован с помощью следующих типичных примеров.
(+) и (-) энантиомеры трет-бутилового эфира 3-[2-(4-гидроксифенил)-4,5,6,7-тетрагидрооксазоло[4,5-c]пиридин]-2-метилпропионовой кислоты 22b (Rb=H). Соединение 22b получали в качестве энантиомерной смеси, как описано на схеме 5. Энантиомерную смесь разделяли с помощью хиральной ВЭЖХ с использованием следующей системы хиральной ВЭЖХ. Неподвижная фаза: Chiralcel OD-H (5 микрон); подвижная фаза: н-гептан/2-пропанол (90/10, об./об.)+0,1% TFA; скорость потока 1 мл/мин; детекция с помощью УФ-излучения при 280 нм. Соединение 22b-rel1: [α]25=+37,1; 1H ЯМР (400 МГц, Хлороформ-d), δ ч/млн: 1,14 (д, J=6,8 Гц, 3Н), 1,43 (с, 9Н), 2,54 (дд, J=12,1, 6,2 Гц, 1Н), 2,60-2,70 (м, 1Н), 2,73-2,77 (м, 2Н), 2,80-2,97 (м, 3Н), 3,49 (д, J=14,1 Гц, 1Н), 3,60 (д, J=14,1 Гц, 1Н), 6,71-6,96 (ушир. сигнал, 1Н), 6,84 (д, J=8,6 Гц, 2Н), 7,82 (д, J=8,6 Гц, 2Н). Соединение 22-rel2: [α]25=-35,9; 1H ЯМР (400 МГц, Хлороформ-d), δ ч/млн: 1,14 (д, J=6,8 Гц, 3Н), 1,42 (с, 9Н), 2,53 (дд, J=12,4, 6,2 Гц, 1Н), 2,60-2,70 (м, 1Н), 2,72-2,79 (м, 2Н), 2,80-2,96 (м, 3Н), 3,49 (д, J=14,1 Гц, 1Н), 3,59 (д, J=14,1 Гц, 1Н), 6,83 (д, J=8,6 Гц, 2Н), 7,45-7,75 (ушир. сигнал, 1Н), 7,79 (д, J=8,6 Гц, 2Н).
(+) и (-) энантиомеры трет-бутилового эфира 3-[2-(4-гидрокси-3-F-фенил)-4,5,6,7-тетрагидрооксазоло[4,5-c]пиридин]-2-метилпропионовой кислоты 22b (Rb=3F). Соединение 22b (Rb=H) получали в качестве энантиомерной смеси, как описано на схеме 5. Энантиомерную смесь разделяли с помощью хиральной ВЭЖХ с использованием следующей системы хиральной ВЭЖХ. Неподвижная фаза: Chiralcel OD-H (5 микрон); подвижная фаза: н-гептан/2-пропанол (90/10, об./об.) + 0,1% TFA; скорость потока 1 мл/мин; детекция с помощью УФ-излучения при 280 нм. Соединение 22b-rel1: [α]25=-36,4 (МеОН). Соединение 22-rel2: [α]25=+34,8; 1Н ЯМР (400 МГц, Хлороформ-d), δ ч/млн: 1,15 (д, J=6,8 Гц, 3Н), 1,43 (с, 9Н), 2,54 (дд, J=11,9, 6,2 Гц, 1Н), 2,60-2,71 (м, 1Н), 2,73-2,79 (м, 2Н), 2,81-2,96 (м, 3Н), 3,49 (д, J=14,1 Гц, 1Н), 3,59 (д, J=14,1 Гц, 1Н), 4,88-6,89 (ушир. сигнал, 1Н), 7,02 (т, J=8,5 Гц, 1Н), 7,61-7,73 (м, 2Н).
3-{2-[4-(2,3-Ди-F-бензилокси)фенил]-6,7-дигидро-H-оксазоло[4,5-с]пиридин-5-ил}масляная кислота, rel 1 и rel 2 соединения 175. Трет-бутил-защищенное производное 175 разделяли в его чистом виде с помощью хиральной ВЭЖХ. Неподвижная фаза: Chiralpak 1С (5 мкм); код колонки no.: WJH022830; размеры: 250×4,6 мм; подвижная фаза: н-гептан/DCM/этанол (50/50/1)+0,1% DEA; скорость потока: 1 мл/мин; инжекция: 5 мкл; детекция: УФ (290 нм). Удаление защитной группы из полученных таким образом энантиомеров привело к выделению исследуемых соединений 426 и 425. Соединение 426, rell: [α]25=-22 (МеОН), Rt 1,3, [М+Н] 429. Соединение 425 rel2: [α]25=+19,9 (МеОН); Rt 1,3, [М+Н] 429.
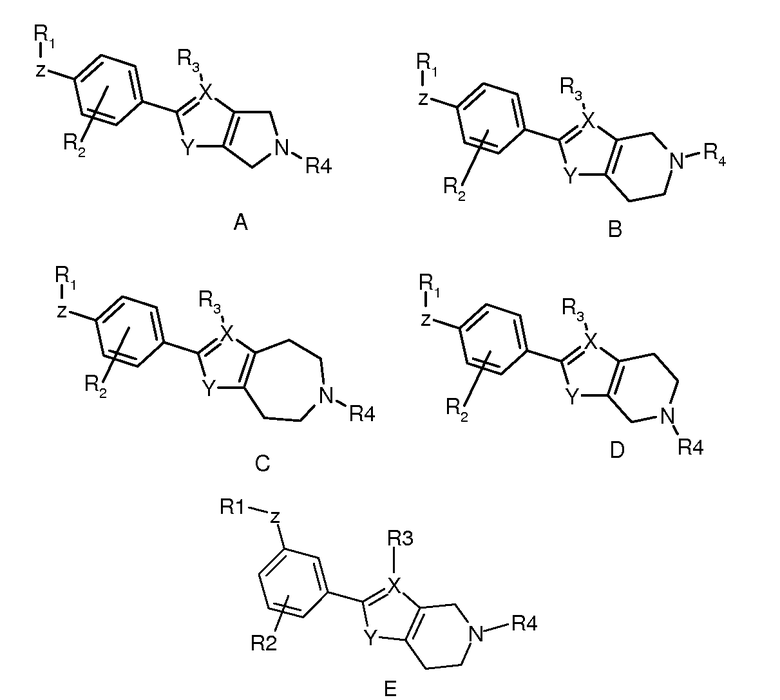
Cl-фенил

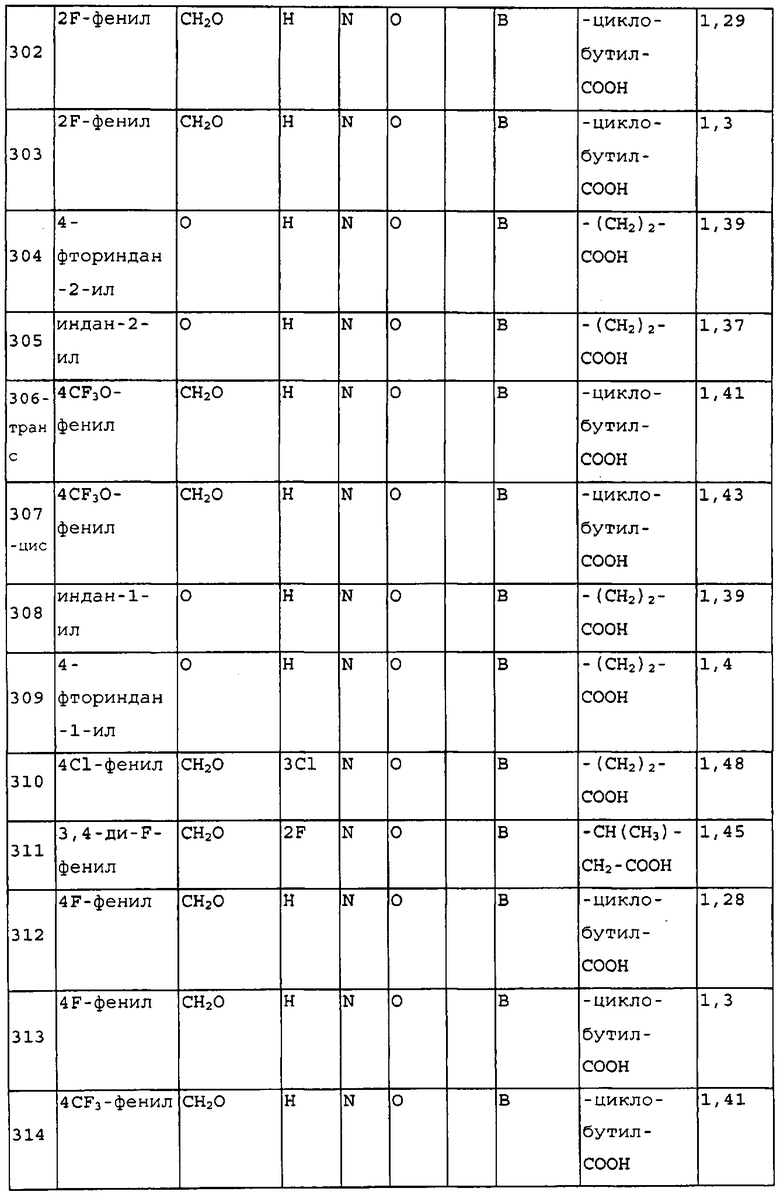
хиральный 2
хиральный 1
#=ЯМР согласуется с предполагаемой структурой
§6. ФАРМАКОЛОГИЧЕСКИЕ ИСПЫТАНИЯ И ДАННЫЕ
Функциональная активность (агонизм) in vitro на рецепторах S1P5 человека
Систему для анализа CHO-S1P5 человека-экворин приобретали от Euroscreen, Brussels (Euroscreen, технический справочник, рецептор лизофосфолипида S1P5 (Edg8) человека, клон ДНК и рекомбинантная клеточная линия CHO AequoScreenTM, каталожный номер №: ES-593-A, сентябрь 2006 года). Клетки S1P5 человека-экворин экспрессируют нацеленный на митохондрии апоэкворин. Для восстановления активного экворина клетки должны быть нагружены коэлантеразином. После связывания агонистов рецептора S1P5 человека внутриклеточная концентрация кальция возрастает и связывание кальция с комплексом апоэкворин/коэлентеразин приводит к реакции окисления коэлентеразина, которая приводит к образованию апоэкворина, коэлентерамида, CO2 и света (max 469 нм). Этот люминесцентный ответ зависит от концентрации агониста. Люминесценцию измеряют с использованием MicroBeta Jet (Perkin Elmer). Агонистические эффекты соединений выражают в качестве pEC50. Соединения исследовали в полулогарифмическом диапазоне концентраций из 10 точек, и для получения данных для отдельных точек проводили 3 независимых эксперимента.
Функциональная активность (агонизм) in vitro на рецепторах S1P3 человека
Анализ CHO-human-S1P3-Aeqorin (CHO/Gα16/AEQ/h-S1P3) был разработан в Solvay Pharmaceuticals. Плазмидную ДНК, кодирующую рецептор S1P3 (номер доступа в GenBank NM_005226), приобретали от UMR cDNA resource Centre (Rolla, MO). Конструкцию pcDNA3.1/hS1P3, несущую нацеленный на митохондрии апоэкворин и белок Gα16, трансфицировали в клеточную линию CHO K1. Клетки S1P3 человека-экворин экспрессируют нацеленный на митохондрии апоэкворин. Для восстановления активного экворина клетки должны быть нагружены коэлантеразином. После связывания агонистов рецептора S1P3 человека внутриклеточная концентрация кальция возрастает и связывание кальция с комплексом апоэкворин/коэлентеразин приводит к реакции окисления коэлентеразина, которая приводит к образованию апоэкворина, коэлентерамида, CO2 и света (max 469 нм). Этот люминесцентный ответ зависит от концентрации агониста. Люминесценцию измеряют с использованием MicroBeta Jet (Perkin Elmer). Агонистические эффекты соединений выражают в качестве pEC50. Соединения исследовали в полулогарифмическом диапазоне концентраций из 10 точек, и для получения данных для отдельных точек проводили 3 независимых эксперимента.
Функциональная активность (агонизм) in vitro на рецепторах S1P1 человека (способ A)
Систему для анализа CHO-S1P1 человека-экворин приобретали от Euroscreen, Brussels (Euroscreen, технический справочник, рецептор лизофосфолипида S1P1 (Edg8) человека, клон ДНК и рекомбинантная клеточная линия CHO AequoScreenTM, каталожный номер №: FAST-0197L, февраль 2010 года). Клетки S1P1 человека-экворин экспрессируют нацеленный на митохондрии апоэкворин. Для восстановления активного экворина клетки должны быть нагружены коэлантеразином. После связывания агонистов рецептора S1P1 человека внутриклеточная концентрация кальция возрастает, и связывание кальция с комплексом апоэкворин/коэлентеразин приводит к реакции окисления коэлентеразина, которая приводит к образованию апоэкворина, коэлентерамида, CO2 и света (max 469 нм). Этот люминесцентный ответ зависит от концентрации агониста. Люминесценцию измеряют с использованием MicroBeta Jet (Perkin Elmer). Агонистические эффекты соединений выражают в качестве pEC50. Соединения исследовали в полулогарифмическом диапазоне концентраций из 10 точек, и для получения данных для отдельных точек проводили 2 независимых эксперимента.
Функциональная активность (агонизм) in vitro на рецепторах S1P1 человека (способ B)
Анализ CHO-K1-S1P1 человека-c-AMP проводили в Euroscreenfast, Brussels (Euroscreen, рецептор S1P1, сопряженный с Gi/0, (Edg1), каталожный номер №: FAST-0197C, декабрь 2009 года).
Рекомбинантные клетки CHO-K1, экспрессирующие S1P1, выращенные до середины log-фазы в культуральной среде без антибиотиков, отделяли, центрифугировали и ресуспендировали. Для исследования агонистов клетки смешивают с соединением и Forskolin и инкубируют при комнатной температуре. Клетки лизируют и оценивают концентрацию cAMP согласно описанию производителя с помощью набора HTRF от CIS-BIO International (каталожный номер № 62AM2PEB).
Агонистические эффекты соединений выражают в качестве процента активности эталонного соединения в его концентрации EC100, EC50 вычисляют, и результаты приводят в качестве pEC50. Соединения исследовали в полулогарифмическом диапазоне концентраций из 10 точек, дублированных в одном 1 эксперименте.
Фармакологические данные (агонизм рецепторов) для отдельных соединений:
S1P1B: определяли с использованием способа B
Модель лечения in vivo; T-лабиринт
Связанные со старением расстройства памяти встречаются у человека и грызунов. Спонтанное чередование является врожденной тенденцией у грызунов к чередованию свободного выбора в T-лабиринте на протяжении серии последовательных прохождений. Эта последовательная процедура основана на кратковременной памяти и является чувствительной к различным фармакологическим манипуляциям, влияющим на процессы памяти (Aging and the physiology of spatial memory. Barnes C.A. Neurobiol. Aging 1988:563-8; Dember W.N., Fowler H. Spontaneous alternation behavior. Psychol. Bull. 1958, 55(6):412-427; Gerlai R. A new continuous alternation task in T-maze detects hippocampal dysfunction in mice. A strain comparison and lesion study. Behav Brain Res 1998 95(1):91-101).
Для этого исследования использовали самцов мышей C57BL/6J в возрасте 2 месяца или 12 месяцев в исследовании спонтанного чередования в T-лабиринте. В кратком изложении, мышей подвергали 1 сеансу, включающему 15 испытаний, состоящих из 1 испытания “принудительного выбора”, с последующими 14 испытаниями “свободного выбора”. Животное считали вошедшим в один из рукавов лабиринта, когда все четыре лапы находились в этом рукаве. Сеанс завершали, и животное удаляли из лабиринта, как только было проведено 14 испытаний свободного выбора или проходило 15 мин, в зависимости от того, что происходило раньше. Для каждой мыши определяли процент чередования на протяжении 14 испытаний свободного выбора и использовали в качестве индекса эффективности кратковременной памяти. Соединение по изобретению вводили п/о в течение 21 суток до анализа в T-лабиринте и в день анализа в T-лабиринте в момент времени t=-30 мин. Было выявлено, что соединения по изобретению в дозах в диапазоне 0,01-15 мг/кг/сутки обращают вспять связанное со старением снижение когнитивной способности у мышей в возрасте 12-месяцев C57BL6J вплоть до 100%. Таким образом, мыши в возрасте 12 месяцев, которым проводили введение соединений, были идентичны по их характеристикам мышам в возрасте 2 месяцев, которым вводили носитель. (См. фиг.1).
Заключение: соединения по настоящему изобретению обладают положительным эффектом на связанное со старением снижение когнитивной способности.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПИРОЦИКЛИЧЕСКИЕ АМИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ S1P | 2011 |
|
RU2602800C2 |
| АНТИГИПЕРТЕНЗИВНЫЕ СОЕДИНЕНИЯ ДВОЙНОГО ДЕЙСТВИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 2008 |
|
RU2476427C2 |
| ЗАМЕЩЕННЫЕ АМИНОМАСЛЯНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ НЕПРИЛИЗИНА | 2012 |
|
RU2604522C2 |
| НОВЫЕ ПОВЕРХНОСТНО-АКТИВНЫЕ ВЕЩЕСТВА И ИХ ПРИМЕНЕНИЕ | 2004 |
|
RU2395493C2 |
| ТЕТРАЦИКЛИЧЕСКИЕ ГЕТЕРОСОЕДИНЕНИЯ В КАЧЕСТВЕ МОДУЛЯТОРОВ ЭСТРОГЕННЫХ РЕЦЕПТОРОВ | 2002 |
|
RU2305099C2 |
| ИНГИБИТОРЫ НЕПРИЛИЗИНА | 2011 |
|
RU2622288C2 |
| КОНЪЮГАТЫ: ТЕРАПЕВТИЧЕСКОЕ СОЕДИНЕНИЕ - ЖИРНАЯ КИСЛОТА | 1994 |
|
RU2137755C1 |
| ИНГИБИТОРЫ НЕПРИЛИЗИНА | 2012 |
|
RU2629930C2 |
| СРЕДСТВО ПЕПТИДНОЙ ПРИРОДЫ, ВКЛЮЧАЮЩЕЕ ПСМА-СВЯЗЫВАЮЩИЙ ЛИГАНД НА ОСНОВЕ ПРОИЗВОДНОГО МОЧЕВИНЫ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ ДЛЯ ПОЛУЧЕНИЯ КОНЪЮГАТА С ЛЕКАРСТВЕННЫМ И ДИАГНОСТИЧЕСКИМ АГЕНТОМ | 2018 |
|
RU2697519C1 |
| МОНОМЕРНЫЕ ПРОИЗВОДНЫЕ ГЛИКОПЕПТИДНОГО АНТИБИОТИКА | 2005 |
|
RU2424248C2 |
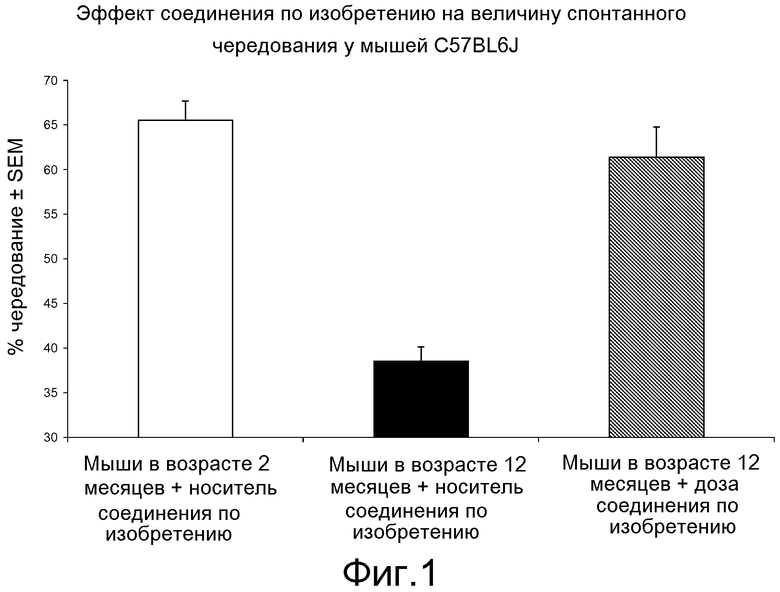
Изобретение относится к конденсированному гетероциклическому производному формулы (I), выбранной из структур А, В, С, D и Е, где R1 выбран из (1-4С)алкила; (3-6С)циклоалкила; инданила, необязательно замещенного галогеном; фенила, необязательно замещенного одним - тремя заместителями, независимо выбранными из галогена, циано, (1-4С)алкила, необязательно замещенного одним или несколькими атомами фтора, (1-4С)алкокси, необязательно замещенного одним или несколькими атомами фтора; пиридила, необязательно замещенного одним атомом галогена; Z представляет собой линкерную группу -W-(Cn-алкилен)-Т-, где W связан с R1 и выбран из связи, -О-, -S-, -SO2-, -NH-, -СН=СН-, -С≡С- и транс-циклопропилена; n представляет собой целое число от 0 до 2; Т связан с фрагментом фенилена и выбран из связи, -О-, -S-, -SO2-, -NH-, -СО-, -С=С-, -С≡С- и транс-циклопропилена; R2 представляет собой Н или один или два заместителя, независимо выбранных из галогена, (1-4С)алкила, необязательно замещенного одним - тремя атомами галогена, или (1-4С)алкокси; X выбран из С или N; если X представляет собой С, R3 выбран из Н и (1-4С)алкила, в ином случае R3 отсутствует; Y выбран из NH, О и S; R4 представляет собой (1-4С)алкилен-R5, где один или несколько атомов углерода в алкиленовой группе может быть независимо замещен одним или двумя атомами галогена или (СН2)2, образуя циклопропильную часть, R5 представляет собой -СООН. Также изобретение относится к фармацевтически приемлемым солям соединений формулы (I), фармацевтической композиции, содержащей соединения формулы (I), и применению соединений формулы (I) для изготовления лекарственного средства. Технический результат - соединения формулы (I), проявляющие свойства агонистов рецептора S1P (рецептор сфингозин-1-фосфата). 10 н. и 14 з.п. ф-лы, 1 ил., 1 табл.
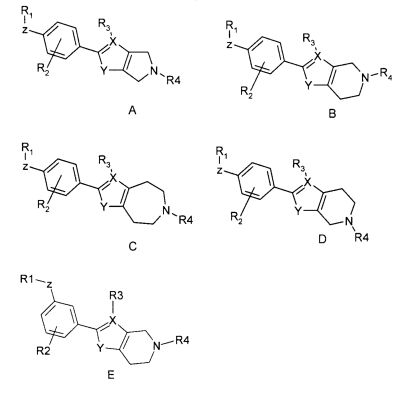
1. Конденсированное гетероциклическое производное формулы (I):

где формулу (I) выбирают из формул А, В, С, D и Е:
и
где
R1 выбран из
(1-4С)алкила,
(3-6С)циклоалкила,
инданила, необязательно замещенного галогеном,
фенила, необязательно замещенного одним - тремя заместителями, независимо выбранными из галогена, циано, (1-4С)алкила, необязательно замещенного одним или несколькими атомами фтора, (1-4С)алкокси, необязательно замещенного одним или несколькими атомами фтора, и
пиридила, необязательно замещенного одним атомом галогена,
Z представляет собой линкерную группу -W-(Cn-алкилен)-Т-,
где
W связан с R1 и выбран из связи, -О-, -S-, -SO2-, -NH-, -СН=СН-, -С≡С- и транс-циклопропилена;
n представляет собой целое число от 0 до 2; и
Т связан с фрагментом фенилена и выбран из связи, -О-, -S-, -SO2-, -NH-, -СО-, -С=С-, -С≡С- и транс-циклопропилена;
R2 представляет собой Н или один или два заместителя, независимо выбранных из галогена, (1-4С)алкила, необязательно замещенного одним - тремя атомами галогена, или (1-4С)алкокси;
X выбран из С или N; если X представляет собой С, R3 выбран из Н и (1-4С)алкила, в ином случае R3 отсутствует;
Y выбран из NH, О и S;
и
R4 представляет собой (1-4С)алкилен-R5, где один или несколько атомов углерода в алкиленовой группе может быть независимо замещен одним или двумя атомами галогена или (СН2)2, образуя циклопропильную часть, где R5 представляет собой -СООН;
или его фармацевтически приемлемая соль.
2. Соединение по п. 1, где
R1 выбран из
(3-6С)циклоалкила, инданила, необязательно замещенного галогеном, (1-4С)алкила, и
фенила, необязательно замещенного одним - тремя заместителями, независимо выбранными из галогена, циано, (1-4С)алкила, (1-4С)алкокси, трифторметила и трифторметокси;
и
R2 представляет собой Н или один или два заместителя, независимо выбранных из галогена, (1-4С)алкила, необязательно замещенного одним - тремя атомами фтора или (1-4С)алкокси.
3. Соединение по п. 1, где соединение имеет структуру (Ib)
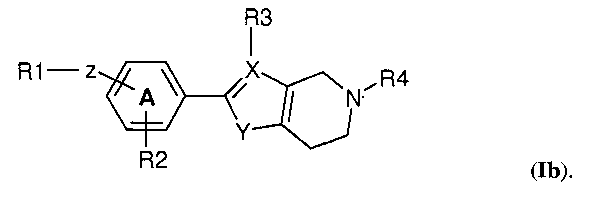
4. Соединение по п. 1, где Y представляет собой О.
5. Соединение по п. 1, где R4 выбран из -(СН2)2-СООН, -(СН2)3-СООН, -СН2-СНСН3-СООН, -СН2-С(СН3)2-СООН, -СНСН3-СН2-СООН, -СН2-CF2-COOH и 1,3-циклобутилен-СООН.
6. Соединение по п. 1, где R1 представляет собой необязательно замещенный фенил.
7. Соединение по п. 6, где необязательные заместители представляют собой один - три заместителя, независимо выбранных из галогена, циано, (1-4С)алкила, (1-4С)алкокси, трифторметила и трифторметокси.
8. Соединение по п. 1, где Z представляет собой -CH2O-.
9. Соединение по п. 1, где X представляет собой N.
10. Соединение по п. 1, выбранное из:
3-{2-[4-(2-фторбензилокси)фенил]-6,7-дигидро-4Н-фуро[3,2-с]пиридин-5-ил}пропионовая кислота,
3-{2-[4-(4-хлорбензилокси)фенил]-6,7-дигидро-4Н-фуро[3,2-с]пиридин-5-ил}-2-метилпропионовая кислота,
3-{2-[4-бензилоксифенил]-6,7-дигидро-4Н-фуро[3,2-с]пиридин-5-ил}масляная кислота,
4-{2-[4-(2-фторбензилокси)фенил]-6,7-дигидро-4Н-фуро[3,2-с]пиридин-5-ил}масляная кислота,
4-{2-[4-(2-фторбензилокси)-2-фторфенил]-6,7-дигидро-4Н-фуро[3,2-с]пиридин-5-ил}масляная кислота,
3-{2-[4-(бензилокси)фенил)]-6,7-дигидро-Н-оксазоло[4,5-с]пиридин-5-ил}пропионовая кислота,
4-{2-[4-(2,3-дифторбензилокси)фенил)]-6,7-дигидро-Н-оксазоло[4,5-с]пиридин-5-ил}масляная кислота,
4-{2-[4-бензилоксифенил]-6,7-дигидро-Н-оксазоло[4,5-с]пиридин-5-ил}масляная кислота,
4-{2-[4-(4-трифторметилбензилокси)-2-фторфенил]-6,7-дигидро-Н-оксазоло[4,5-с]пиридин-5-ил}масляная кислота,
4-{2-[4-(2-фторбензилокси)-2-метилфенил]-6,7-дигидро-Н-оксазоло[4,5-с]пиридин-5-ил}масляная кислота,
4-{2-[4-(3,4-дихлорбензилокси)-2-фторфенил]-6,7-дигидро-Н-оксазоло[4,5-с]пиридин-5-ил}масляная кислота,
4-{2-[4-(2-фторбензилокси)-3-хлорфенил]-6,7-дигидро-Н-оксазоло[4,5-с]пиридин-5-ил}масляная кислота,
4-{2-[4-(4-хлорбензилокси)-3-фторфенил]-6,7-дигидро-Н-оксазоло[4,5-с]пиридин-5-ил}масляная кислота,
3-[2-(4-бензилоксифенил)-6,7-дигидро-Н-оксазоло[4,5-с]пиридин-5-ил]-2-метилпропионовая кислота,
3-{2-[4-(4-трифторметилбензилокси)фенил]-6,7-дигидро-Н-оксазоло[4,5-с]пиридин-5-ил}-2-метилпропионовая кислота,
3-{2-[4-(4-хлорбензилокси)фенил]-6,7-дигидро-Н-оксазоло[4,5-с]пиридин-5-ил}-2-метилпропионовая кислота,
3-{2-[4-(2,3-дифторбензилокси)фенил]-6,7-дигидро-Н-оксазоло[4,5-с]пиридин-5-ил}масляная кислота,
3-{2-[4-(4-трифторметилбензилокси)-2-фторфенил]-6,7-дигидро-Н-оксазоло[4,5-с]пиридин-5-ил}-2-метилпропионовая кислота,
цис- и транс-3-{2-[4-(4-трифторметоксибензилокси)фенил]-6,7-дигидро-Н-оксазоло[4,5-с]пиридин-5-ил}циклобутанкарбоновая кислота,
цис- и транс-3-{2-[4-(3,4-дифторбензилокси)фенил]-6,7-дигидро-Н-оксазоло[4,5-с]пиридин-5-ил}циклобутанкарбоновая кислота,
3-[2-(4-(2-фторбензилокси)фенил)-6,7-дигидро-4Н-тиазоло[5,4-с]пиридин-5-ил]пропионовая кислота,
4-{2-[4-(3,5-дифторбензилокси)фенил]-6,7-дигидро-4Н-тиазоло[4,5-с]пиридин-5-ил}масляная кислота,
2-{2-[4-(3,5-дифторбензилокси)фенил]-6,7-дигидро-Н-оксазоло[4,5-с]пиридин-5-ил}уксусная кислота,
2-{2-[4-(3,5-дифторбензилокси)фенил]-6,7-дигидро-Н-оксазоло[4,5-с]пиридин-5-ил}пропионовая кислота,
3-{2-[4-(3,5-дифторбензилокси)фенил]-6,7-дигидро-4Н-оксазоло[4,5-с]пиридин-5-ил}циклопентанкарбоновая кислота,
4-{2-[4-(2,3-дифторбензилокси)-2-метилфенил]-6,7-дигидро-4Н-оксазоло[4,5-с]пиридин-5-ил}пентановая кислота,
4-{2-[4-(2,3-дифторбензилокси)-2-метилфенил]-6,7-дигидро-4Н-оксазоло[4,5-с]пиридин-5-ил}-2-метилмасляная кислота,
4-{2-[4-(2,3-дифторбензилокси)-2-метилфенил]-6,7-дигидро-4Н-оксазоло[4,5-с]пиридин-5-ил}-3-метилмасляная кислота,
3-{2-[4-(2-фенилциклопропил)-3-фторфенил]-6,7-дигидро-4Н-оксазоло[4,5-с]пиридин-5-ил}циклобутанкарбоновая кислота,
4-(2-{4-[2-(3,5-дифторфенил)винил]фенил}-6,7-дигидро-4Н-оксазоло[4,5-с]пиридин-5-ил)масляная кислота,
4-(2-{4-[2-(3,5-дифторфенил)этил]фенил}-6,7-дигидро-4Н-оксазоло[4,5-с]пиридин-5-ил)масляная кислота, или их фармацевтически приемлемая соль.
11. Соединение формулы

или его фармацевтически приемлемая соль.
12. Соединение по п. 1 формулы
или его фармацевтически приемлемая соль.
13. Соединение формулы

или его фармацевтически приемлемая соль.
14. Соединение формулы

или его фармацевтически приемлемая соль.
15. Соединение формулы
или его фармацевтически приемлемая соль.
16. Соединение формулы
или его фармацевтически приемлемая соль.
17. Соединение, которое представляет собой (+) энантиомер формулы

или его фармацевтически приемлемая соль.
18. Соединение, которое представляет собой (-) энантиомер формулы

или его фармацевтически приемлемая соль.
19. Соединение по любому из пп. 1-18 для получения лекарственного средства для лечения, смягчения или профилактики заболеваний и состояний, в которые вовлечен любой из рецепторов S1P или в которые вовлечено модулирование эндогенной системы передачи сигнала S1P через любой из рецепторов S1P.
20. Соединение по любому из пп. 1-18 для применения в лечении, смягчении или профилактике заболеваний и состояний, в которые вовлечен любой из рецепторов S1P или в которые вовлечено модулирование эндогенной системы передачи сигнала S1P через любой из рецепторов S1P.
21. Соединение по п. 20, где заболевание представляет собой нарушение ЦНС, такое как нейродегенеративное нарушение, в частности, выбранное из когнитивного нарушения, болезни Альцгеймера, (сосудистой) деменции, болезни Ниманна-Пика и когнитивных расстройств при шизофрении, обсессивно-компульсивного поведения, большой депрессии, аутизма, рассеянного склероза и боли, и, в частности, когнитивного нарушения, такого как связанное со старением снижение когнитивной способности.
22. Фармацевтическая композиция, обладающая агонистической активностью в отношении рецепторов S1P, содержащая в качестве активного ингредиента соединение по любому из пп. 1-18 и по меньшей мере один фармацевтически приемлемый эксципиент, носитель, адъювант или наполнитель.
23. Применение соединения по любому из пп. 1-18 для изготовления лекарственного средства для лечения, смягчения или профилактики заболеваний и состояний, в которые вовлечен любой из рецепторов S1P или в которые вовлечено модулирование эндогенной системы передачи сигнала S1P через любой из рецепторов S1P.
24. Применение по п. 23, где заболевание представляет собой нарушение ЦНС, такое как нейродегенеративное нарушение, в частности, выбранное из когнитивного нарушения, болезни Альцгеймера, (сосудистой) деменции, болезни Ниманна-Пика и когнитивных расстройств при шизофрении, обсессивно-компульсивного поведения, большой депрессии, аутизма, рассеянного склероза и боли, и, в частности, когнитивного нарушения, такого как связанное со старением снижение когнитивной способности.
| Колосоуборка | 1923 |
|
SU2009A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Дигидрохлорид 5-/ @ -( @ -анизил)- @ -оксиэтил/ 2-(3,4-диметоксифенил)-1-метил-4,5,6,7-тетрагидроимидазо(4,5- @ )пиридина,обладающий спазмолитическим действием | 1982 |
|
SU1069387A1 |
| ЮТИЛОВ Ю.М | |||
| и др., Синтез и противовирусная активность производных спинацеамина, ХИМИКО-ФАРМАЦЕВТИЧЕСКИЙ ЖУРНАЛ, 1989, том23, N1, с.56-59 | |||
| ЮТИЛОВ Ю.М | |||
| и др., Синтез и биологическая активность N(5)-бетта-оксиэтилспинацеаминов, ХИМИКО-ФАРМАЦЕВТИЧЕСКИЙ ЖУРНАЛ, 1989, том 23, N2, с.160-163 | |||
| RU 2008135125 А, 10.03.2010. | |||

