ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННУЮ ЗАЯВКУ
По данной патентной заявке испрашивается приоритет по временной патентной заявке № 61/333540, поданной 11 мая 2010 года, которая включена в настоящее описание в качестве ссылки в полном объеме.
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Изобретение относится к технической области молекулярной генетики, связанной с нарушением экспрессии генов. Более конкретно, было открыто, что мутация в гене spnK преобразует продуцирующий спиносад штамм в штамм, продуцирующий предшественник спинеторама.
УРОВЕНЬ ТЕХНИКИ
Как описано в патенте США № 5362634, продукт ферментации A83543 представляет собой семейство родственных соединений, продуцируемых Saccharopolyspora spinosa. Известные представители этого семейства называют факторами или компонентами, и каждому из них присвоено идентификационное буквенное обозначение. Эти соединения далее в настоящем описании называют спинозином A, B и т.д. Соединения спинозина пригодны для борьбы с паукообразными, круглыми червями и насекомыми, в частности видами Lepidoptera и Diptera, и они являются довольно благоприятными для окружающей среды и обладают привлекательным токсикологическим профилем.
Продуцируемые в природе соединения спинозина состоят из 5,6,5-трициклической кольцевой системы, конденсированной с 12-членным макроциклическим лактоном, нейтрального сахара (рамноза) и аминосахара (форозамин) (см. Kirst et al. (1991). Если аминосахар не присутствует, соединения называют псевдоагликоном A, D и т.д., и, если нейтральный сахар не присутствует, тогда соединения называют обратным псевдоагликоном A, D и т.д. Более предпочтительной номенклатурой является называние псевдоагликонов спинозином A 17-Psa, спинозином D 17-Psa и т.д. и называние обратных псевдоагликонов - спинозином A 9-Psa, спинозином D 9-Psa и т.д.
Природные соединения спинозинов можно получать ферментацией культур NRRL 18395, 18537, 18538, 18539, 18719, 18720, 18743 и 18823. Эти культуры были депонированы и являются частью коллекции исходных культур Midwest Area Northern Regional Research Center, Agricultural Research Service, United States Department of Agriculture, 1815 North University Street, Peoria, Ill., 61604.
Патент США № 5362634 и соответствующая патентная заявка Европы № 375316 A1 относятся к спинозинам A, B, C, D, E, F, G, H и J. Описано, что эти соединения получают культивированием штамма нового микроорганизма Saccharopolyspora spinosa, выбранного из NRRL 18395, NRRL 18537, NRRL 18538 и NRRL 18539.
WO 93/09126 относится к спинозинам L, M, N, Q, R, S и T. Также в ней обсуждаются штаммы, продуцирующие спинозин J: NRRL 18719 и NRRL 18720, и штамм, который продуцирует спинозины Q, R, S и T: NRRL 18823.
WO 94/20518 и патент США № 56704486 относятся к спинозинам K, O, P, U, V, W и Y и их производным. Также обсуждается продуцирующий спинозин K штамм NRRL 18743.
Проблема при продуцировании соединений спинозина возникает из того факта, что требуется очень большой объем ферментации для получения очень небольшого количества спинозинов. Высоко желательным является увеличение эффективности продуцирования спинозина и, тем самым, увеличение доступности спинозинов при одновременном снижении их стоимости.
Было бы преимущественным предоставление клонированных генов биосинтеза, которые обеспечивают способ продуцирования новых производных спинозинов, которые могут обладать отличающимся спектром инсектицидной активности. Новые производные являются желательными, поскольку, хотя известные спинозины ингибируют широкий спектр насекомых, они не осуществляют борьбу со всеми вредителями. Различные профили контроля могут быть предоставлены с помощью промежуточных соединений биосинтеза спинозинов или с помощью их производных, продуцированных in vivo, или с помощью производных, полученных их химической модификацией in vitro.
Также было бы преимущественным предоставление новых промежуточных соединений, синтезируемых мутантными штаммами S. spinosa, в которых части определенных генов, кодирующих ферменты для биосинтеза спинозина, заменены частями тех же генов, которые были подвергнуты определенным мутациям in vitro, или соответствующими частями генов из других организмов.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способам преобразования штамма, продуцирующего спиносад, такой как спинозин A и D, в штамм, продуцирующий спинеторам, такой как спинозин J и L. Такой способ может включать обеспечение модификации в гене spnK для устранения активности 3'-O-метилтрансферазы. Модификацию можно проводить с помощью делеций в рамке считывания, мутаций, замен, делеций, инсерций и т.п. Делеции в рамке считывания можно проводить по всему гену, включая делеции на 5'-конце, 3'-конце или в кодирующей области spnK. Одна такая делеция в рамке считывания может включать SEQ ID NO:9. Точковые мутации могут включать, но не ограничиваться ими, мутации в положениях пар оснований 528, 589, 602, 668, 721, 794, 862, 895, 908, 937 и 1131. Эти мутации могут приводить к изменению трансляции гена spnK. Такие изменения могут представлять собой изменения аминокислот, замены или создание стоп-кодонов. Такие модификации приводят к продуцированию соединения спинозина J и L относительно спинозина A и D.
Конкретные способы по настоящему изобретению включают преобразование продуцирующего спиносад штамма в штамм, продуцирующий предшественник спинеторама, путем выключения гена spnK при одновременном сохранении продуцирования спинозина J и L. Выключение или нарушение нормальной активности белка spnK может происходить с помощью делеций в рамке считывания, мутаций, замен, делеций, инсерций и т.п. Также оно может быть вызвано манипуляциями с последовательностями промотора или участка связывания рибосом.
Кроме того, изобретение относится к генетически модифицированной клетке-хозяину, которая продуцирует предшественник спинеторама. Этого генетически модифицированного хозяина можно получать путем модификации гена spnK для устранения активности 3'-O-метилтрансферазы. Модификация может быть осуществлена с помощью делеций в рамке считывания, мутаций, замен, делеций, инсерций и т.п. Делеции в рамке считывания могут включать делеции на 5'-конце, 3'-конце или в кодирующей области spnK.
Также изобретение относится к способам преобразования продуцирующих спиносад штаммов в штаммы, продуцирующие предшественник спинеторама, путем модификации гена spnK для устранения активности 3'-O-метилтрансферазы. Этот процесс может включать делеции в рамке считывания, точковые мутации, делеции и инсерции. Такие делеции в рамке считывания могут включать делеции в рамке считывания на 5'-конце, делеции в рамке считывания на 3'-конце и делеции в рамке считывания в кодирующей области spnK. Делеции могут включать делеции одного или нескольких нуклеотидных оснований, которые нарушают нормальную рамку считывания гена spnK. Инсерции могут включать инсерции одного или нескольких нуклеотидных оснований, которые нарушают нормальную рамку считывания гена spnK. Точковые мутации могут находиться в положениях пар оснований 528, 589, 602, 668, 721, 794, 862, 895, 908, 937 и 1131. Эти точковые мутации могут приводить к аминокислотным заменам в активном центре или участке связывания субстрата гена spnK.
Также изобретение включает генетически модифицированные клетки-хозяева, которые продуцируют предшественник спинеторама вследствие обеспечения модификации в гене spnK для устранения активности 3'-O-метилтрансферазы, где генетически модифицированная клетка-хозяин представляет собой прокариотическую клетку-хозяина, которая в норме не продуцирует значительные количества предшественника спинеторама. Другие варианты осуществления включают способы преобразования продуцирующего спиносад штамма в штамм, продуцирующий предшественник спинеторама, путем выключения гена spnK при одновременном сохранении продуцирования спинозина J и L. Такие способы могут включать делеции в рамке считывания, точковые мутации, делеции и инсерции. Такие делеции в рамке считывания могут включать делеции в рамке считывания на 5'-конце, делеции в рамке считывания на 3'-конце и делеции в рамке считывания в кодирующей области spnK. Делеции могут включать делеции одного или нескольких нуклеотидных оснований, которые нарушают нормальную рамку считывания гена spnK. Инсерции могут включать инсерции одного или нескольких нуклеотидных оснований, которые нарушают нормальную рамку считывания гена spnK. Точковые мутации могут находиться в положениях пар оснований 528, 589, 602, 668, 721, 794, 862, 895, 908, 937 и 1131. Эти точковые мутации могут приводить к заменам аминокислот в активном центре или участке связывания субстрата гена spnK. Другие способы выключения spnK могут осуществляться путем манипулирования с участком связывания рибосом или путем манипулирования с промотором гена spnK.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фиг.1 представлено расположение точковых мутаций spnK. Мутации выделены в последовательности spnK дикого типа (SEQ ID NO:17).
На фиг.2 представлена физическая карта spnJ, spnK, spnL и spnM. Продукты ПЦР, которые были получены, указаны линиями ниже карты хромосомы.
На фиг.3 представлено встраивание конструкции с делецией spnK в рамке считывания в область spnLM с помощью гомологичной рекомбинации с одним кроссинговером согласно варианту осуществления настоящего изобретения. (Звездочка указывает на неполную кодирующую последовательность spnJ и spnM).
На фиг.4 представлены мутанты с двойным кроссинговером, которые приводили к делеции гена spnK согласно одному варианту осуществления настоящего изобретения. Размер и последовательность ДНК фрагмента ПЦР указывает на делецию гена spnK в рамке считывания.
На фиг.5 представлена диаграмма встраивания кассеты, содержащей кассету гена устойчивости к апрамицину (aac(3)IV) в рамке считывания, в spnK в соответствии с одним вариантом осуществления настоящего изобретения.
На фиг.6 представлен участок связывания рибосом (обозначенный как последовательность Шайна-Дальгарно), который расположен выше кодирующей последовательности spnK согласно одному варианту осуществления настоящего изобретения (SEQ ID NO:16). Эта последовательность выделена на фиг.6.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Существует множество применений для клонированной ДНК Saccharopolyspora spinosa. Клонированные гены можно использовать для увеличения выхода спинозинов и получения новых спинозинов. Увеличения выхода можно достигать путем встраивания в геном конкретного штамма удвоенной копии гена для фермента, который является скоростьограничивающим в этом штамме. В случаях, где биосинтетический каскад блокирован в конкретном мутантном штамме вследствие нехватки требуемого фермента, продуцирование желаемых спинозинов может быть восстановлено путем встраивания копии требуемого гена. Когда нарушен биосинтетический путь, можно создавать другой штамм-предшественник. Более конкретно, нарушение гена spnK может приводить к продуцированию спинозина J и L относительно продуцирования спинозина A и D.
Новые спинозины можно получать, используя фрагменты клонированной ДНК для нарушения стадий в биосинтезе спинозинов. Такое нарушение может приводить к накоплению предшественников или “шунтовых” продуктов (природным образом процессируемые производные предшественников). Фрагменты, пригодные для осуществления нарушения, являются внутренними фрагментами гена с отсутствующими основаниями на 5'- и 3'-концах гена, а также по всему гену. События гомологичной рекомбинации, в которых используются такие фрагменты, приводят к двум неполным копиям гена: в одной из них отсутствуют основания на 5'-конце и в другой из них отсутствуют основания на 3'-конце. Количество оснований, пропущенных на каждом конце фрагмента, должно быть достаточным, чтобы ни одна из неполных копий гена не сохраняла активность.
Следующие определения используют в настоящем описании, и на них следует ссылаться для интерпретации формулы изобретения и описания. Если нет иных указаний, все патенты США и патентные заявки США, упоминаемые в настоящем описании, включены в качестве ссылок в полном объеме.
Как используют в рамках изобретения, подразумевают, что форма единственного числа у элемента или компонента по изобретению является неограничивающей в отношении количества случаев (т.е. встречаемости) элемента или компонента. Таким образом, форму единственного числа следует истолковывать как включающую один или по меньшей мере один, и словоформа элемента или компонента в единственном числе также включает множественное число, если явно не предполагается, что число является единственным.
Как используют в рамках изобретения, термины “содержащий” и “включающий” означают присутствие указываемых признаков, чисел, стадий или компонентов, упоминаемых в формуле изобретения, но они не исключают присутствия или добавления одного или нескольких других признаков, чисел, стадий, компонентов или их групп. Это означает, что композиция, смесь, процесс, способ, изделие или устройство, которые “содержат” или “включают” перечень элементов, не ограничиваются только этими элементами, а могут включать другие, явно не перечисленные или не присущие ему. Как используют в рамках изобретения, “или” относится к включающему и исключающему “или”. Например, условие A или B удовлетворяется одним из следующих: A справедливо (или присутствует) и B ошибочно (или отсутствует), A ошибочно (или отсутствует) и B справедливо (или присутствует), и как A, так и B являются справедливыми (или присутствуют).
Как используют в рамках изобретения, термин “приблизительно” относится к модификации количества ингредиента или реагента по изобретению или используемого ингредиента или реагента и относится к варьированию числового количества, которое может возникать, например, при типических процессах измерения или обработки жидкостей, используемых для получения концентратов или растворов для применения: в реальном мире; вследствие неизбежных ошибок в этих процессах; вследствие различий в производстве, источнике и чистоте ингредиентов, используемых для получения композиций или осуществления способов и т.п. Термин “приблизительно” также охватывает количества, которые отличаются благодаря различным равновесным условиям для композиции, получаемой из конкретной первоначальной смеси. Независимо от модификации термином “приблизительно” формула изобретения включает эквиваленты количествам.
Как используют в рамках изобретения, термин “изобретение” или “настоящее изобретение” является неограничивающим термином, и подразумевают, что он охватывает все возможные варианты, как описано в описании и перечислено в формуле изобретения.
Как используют в рамках изобретения, термины “полипептид” и “пептид” используют взаимозаменяемо для обозначения полимера из двух или более аминокислот, связанных друг с другом пептидной связью. В одном аспекте этот термин также включает модификации после экспрессии полипептида, например гликозилирование, ацетилирование, фосфорилирование и т.п. В это определение включены, например, пептиды, содержащие один или несколько аналогов аминокислот или меченые аминокислоты и пептидомиметики. Пептиды могут содержать L-аминокислоты.
Как используют в рамках изобретения, термины “представляющий интерес пептид”, “POI”, “продукт гена”, “целевой продукт гена” и “целевой генный продукт кодирующей области” относятся к желаемому гетерологичному пептидному/белковому продукту, кодируемому рекомбинантно экспрессируемым чужеродным геном. Представляющий интерес пептид может включать любой пептидный/белковый продукт, включающий, но не ограничивающийся ими, белки, слитые белки, ферменты, пептиды, полипептиды и олигопептиды. Размер представляющего интерес пептида находится в диапазоне от 2 до 398 аминокислоты в длину.
Как используют в рамках изобретения, термин “генетическая конструкция” относится к серии последовательно расположенных нуклеиновых кислот, пригодных для модулирования генотипа или фенотипа организма. Неограничивающие примеры генетических конструкций включают, но не ограничиваются ими, молекулу нуклеиновой кислоты и открытую рамку считывания, ген, экспрессирующую кассету, вектор, плазмиду и т.п.
Как используют в рамках изобретения, термин “эндогенный ген” относится к нативному гену в его природном положении в геноме организма.
Как используют в рамках изобретения, “чужеродный ген” относится к гену, который в норме не встречается в организме-хозяине, но введен в организм-хозяин путем переноса генов. Чужеродные гены могут включать нативные гены, встроенные в ненативный организм, или химерные гены.
Как используют в рамках изобретения, термин “гетерологичный” в отношении последовательности в конкретном организме/геноме указывает на то, что последовательность происходит из чужеродных видов, или, если она из того же вида, существенно модифицирована относительно ее нативной формы с точки зрения состава и/или геномного локуса путем преднамеренного вмешательства человека. Таким образом, например, экспрессия гетерологичного гена относится к процессу экспрессии гена из одного организма/генома путем помещения его в геном другого организма/генома.
Как используют в рамках изобретения, термин “рекомбинантный” относится к искусственному комбинированию двух в ином случае разделенных сегментов последовательности, например, путем химического синтеза или путем манипулирования с выделенными сегментами нуклеиновых кислот способами генетической инженерии. “Рекомбинантный” также включает указание на клетку или вектор, которые были модифицированы путем встраивания гетерологичной нуклеиновой кислоты, или клетку, происходящую из клетки, модифицированной таким путем, но не охватывает изменение клетки или вектора вследствие природных событий (например, спонтанная мутация, природная трансформация, природная трансдукция, природная транспозиция), таких как события, происходящие без преднамеренного вмешательства человека.
Термин “полученный способами генетической инженерии” или “генетически измененный” означает научное изменение структуры генетического материала в живом организме. Он вовлекает получение и применение рекомбинантной ДНК. Более конкретно, его используют для отграничения полученного способами генетической инженерии или модифицированного организма от встречающегося в природе организма. Получение способами генетической инженерии можно проводить с помощью ряда способов, известных в данной области, например таких как замена генов, амплификация генов, нарушение генов, трансфекция, трансформация с использованием плазмид, вирусов или других векторов. Генетически модифицированный организм, например генетически модифицированный микроорганизм, часто называют рекомбинантным организмом, например рекомбинантным микроорганизмом.
Как используют в рамках изобретения, термин “нарушенный” или “нарушение” относится к гену, подвергнутому манипулированию или модифицированному способами генетической инженерии или вследствие природных причин, которые изменяют активность гена. Такая активность гена может быть увеличенной или сниженной. Кроме того, такое нарушение может устранять функцию белка. Для способствования такому снижению можно снижать число копий гена, например, путем недостаточной экспрессии или нарушения гена. Ген называют геном с “недостаточной экспрессией”, если уровень транскрипции указанного гена снижен по сравнению с геном дикого типа. Это можно измерять, например, с помощью нозерн-блот-анализа, количественно определяющего количество мРНК в качестве признака экспрессии гена. Как используют в рамках изобретения, экспрессия гена является недостаточной, если количество образовавшейся мРНК снижено по меньшей мере на 1%, 2%, 5% 10%, 25%, 50%, 75%, 100%, 200% или даже более чем на 500% по сравнению с количеством мРНК, образовавшейся из гена дикого типа. Альтернативно, для регуляции экспрессии полинуклеотида можно использовать слабый промотор. В другом варианте осуществления для достижения сниженной экспрессии можно изменять промотор, регуляторную область и/или участок связывания рибосом выше гена. Экспрессию также можно снижать путем снижения относительного времени полужизни матричной РНК. В другом варианте осуществления можно снижать активность самого полипептида с использованием одной или нескольких мутаций в аминокислотной последовательности полипептида, которые снижают активность. Например, к сниженной активности может приводить изменение аффинности полипептида в отношении его соответствующего субстрата. Аналогично, может быть снижено относительное время полужизни полипептида. В любом сценарии, где снижают экспрессию гена или снижают активность, снижения можно достигать путем изменения состава клеточной культуральной среды и/или способов, используемых для культивирования. “Сниженная экспрессия” или “сниженная активность”, как используют в рамках изобретения, означает снижение по меньшей мере на 5%, 10%, 25%, 50%, 75%, 100%, 200% или даже более чем на 500% по сравнению с белком, полинуклеотидом, геном дикого типа; или активностью и/или концентрацией белка, присутствующего до снижения содержания полинуклеотидов или полипептидов. Активность белка SpnK также можно снижать путем контактирования белка со специфическим или общим ингибитором его активности. Термины “сниженная активность”, “сниженная или устраненная активность” используют в настоящем описании взаимозаменяемо.
Выражение “последовательности контроля” относится в совокупности к промоторным последовательностям, участкам связывания рибосом, последовательностям терминации транскрипции, вышерасположенным регуляторным доменам, энхансерам и т.п., которые в совокупности обеспечивают транскрипцию и трансляцию кодирующей последовательности в клетке-хозяине. Не все из этих последовательностей контроля всегда должны присутствовать в рекомбинантном векторе при условии, что желаемый ген способен быть транскрибированным и транслированным.
“Рекомбинация” относится к перераспределению фрагментов последовательностей ДНК или РНК между двумя молекулами ДНК или РНК. “Гомологичная рекомбинация” происходит между двумя молекулами ДНК, которые гибридизуются благодаря гомологичным или комплементарным нуклеотидным последовательностям, присутствующим в каждой молекуле ДНК.
Термины “жесткие условия” или “гибридизация в жестких условиях” относятся к условиям, в которых зонд гибридизуется предпочтительно с его подпоследовательностью-мишенью и в меньшей степени гибридизуется или вообще не гибридизуется с другими последовательностями. “Жесткая гибридизация” и “жесткие условия промывания при гибридизации” в контексте экспериментов по гибридизации нуклеиновых кислот, таких как саузерн-гибридизация и нозерн-гибридизация, являются зависимыми от последовательности и различаются при различных параметрах окружающей среды. Обширное руководство по гибридизации нуклеиновых кислот представлено в Tijssen (1993) Laboratory Techniques in Biochemistry and Molecular Biology-Hybrydisation with Nucleic Acid Probes part I chapter 2 Overview of principles of hybridization and the strategy of Nucleic acid probe assays, Elsevier, New York. Как правило, в высокой степени жесткие условия гибридизации и промывания выбирают так, чтобы температура была приблизительно на 5°C ниже, чем температура плавления (Tm) для конкретной последовательности при заданной ионной силе и pH. Tm представляет собой температуру (при определенной ионной силе и pH), при которой 50% последовательности-мишени гибридизуется с абсолютно совпадающим зондом. В высокой степени жесткие условия выбирают так, чтобы температура была равна Tm для конкретного зонда.
Примером жестких условий гибридизации для гибридизации комплементарных нуклеиновых кислот, которые имеют более 100 комплементарных остатков, на фильтре при саузерн-блоттинге или нозерн-блоттинге, является 50% формамид с 1 мг гепарина при 42°C, причем гибридизацию проводят в течение ночи. Примером в высокой степени жестких условий промывания является 0,15M NaCl при 72°C в течение приблизительно 15 минут. Примером жестких условий промывания является промывание 0,2×SSC при 65°C в течение 15 минут (см., Sambrook et al. (1989) Molecular Cloning-A Laboratory Manual (2nd ed.) Vol. 1-3, Cold Spring Harbor Laboratory, Cold Spring Harbor Press, NY, для описания буфера SSC). Часто промыванию высокой жесткости предшествует промывание низкой жесткости для устранения фонового сигнала зонда. Примером промывания средней жесткости для дуплекса, например, более чем из 100 нуклеотидов является 1×SSC при 45°C в течение 15 минут. Примером промывания низкой жесткости для дуплекса, например, более чем из 100 нуклеотидов является 4-6xSSC при 40°C в течение 15 минут. Как правило, отношение сигнала к шуму 2× (или более) относительно того, что наблюдают для неродственного зонда в конкретном анализе гибридизации, указывает на детекцию специфической гибридизации. Нуклеиновые кислоты, которые не гибридизуются друг с другом в жестких условиях, все еще являются идентичными, если полипептиды, которые они кодируют, являются по существу идентичными. Это происходит, например, когда создают копию нуклеиновой кислоты с использованием максимальной вырожденности кодонов, допускаемой генетическим кодом.
Также изобретение относится к выделенному полинуклеотиду, гибридизующемуся в жестких условиях, предпочтительно в высокой степени жестких условиях, с полинуклеотидом по настоящему изобретению.
Как используют в рамках изобретения, термин “гибридизующийся” предназначен для описания условий гибридизации и промывания, в которых нуклеотидные последовательности, по меньшей мере приблизительно на 50%, по меньшей мере приблизительно на 60%, по меньшей мере приблизительно на 70%, более предпочтительно по меньшей мере приблизительно на 80%, еще более предпочтительно по меньшей мере приблизительно на 85%-90%, наиболее предпочтительно по меньшей мере на 95% гомологичные друг другу, как правило, остаются гибридизованными друг с другом.
В одном варианте осуществления нуклеиновая кислота по изобретению по меньшей мере на 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% или более гомологична последовательности нуклеиновой кислоты, представленной в данной заявке, или комплементарной ей последовательности.
Другим неограничивающим примером жестких условий гибридизации является гибридизация в 6x хлориде натрия/цитрате натрия (SSC) при приблизительно 45°C, с последующим одним или несколькими промываниями в 1×SSC, 0,1% SDS при 50°C, предпочтительно при 55°C, более предпочтительно при 60°C и еще более предпочтительно при 65°C.
В высокой степени жесткие условия могут включать инкубацию при 42°C в течение нескольких суток, например 2-4 суток, с использованием меченого ДНК-зонда, такого как меченный дигоксигенином (DIG) ДНК-зонд, с последующими одним или несколькими промываниями в 2×SSC, 0,1% SDS при комнатной температуре и одним или несколькими промываниями в 0,5×SSC, 0,1% SDS или 0,1×SSC, 0,1% SDS при 65-68°C. В частности, в высокой степени жесткие условия включают, например, инкубацию в течение от 2 ч до 4 суток при 42°C с использованием меченого DIG ДНК-зонда (полученного, например, с использованием системы для мечения DIG; Roche Diagnostics GmbH, 68298 Mannheim, Германия) в растворе, таком как раствор DigEasyHyb (Roche Diagnostics GmbH) с 100 мкг/мл ДНК спермы лосося или без нее, или в растворе, содержащем 50% формамид, 5xSSC (150 мМ NaCl, 15 мМ трицитрат натрия), 0,02% додецилсульфат натрия, 0,1% N-лауроилсаркозин и 2% блокирующий реагент (Roche Diagnostics GmbH), с последующим промыванием фильтров два раза в течение от 5 до 15 минут в 2×SSC и 0,1% SDS при комнатной температуре, а затем промыванием два раза в течение 15-30 минут в 0,5xSSC и 0,1% SDS или 0,1×SSC и 0,1% SDS при 65-68°C.
В некоторых вариантах осуществления выделенная молекула нуклеиновой кислоты по изобретению, которая гибридизуется в высокой степени жестких условиях с нуклеотидной последовательностью по изобретению, может соответствовать встречающейся в природе молекуле нуклеиновой кислоты. Как используют в рамках изобретения, “встречающаяся в природе” молекула нуклеиновой кислоты относится к молекуле РНК или ДНК, имеющей нуклеотидную последовательность, которая встречается в природе (например, кодирует природный белок).
Квалифицированному специалисту известно, какие условия применять для жестких или в высокой степени жестких условий гибридизации. Дополнительное руководство по таким условиям легко доступно в данной области, например в Sambrook et al., 1989, Molecular Cloning, A Laboratory Manual, Cold Spring Harbor Press, N.Y.; и Ausubel et al. (eds.), 1995, Current Protocols in Molecular Biology (John Wiley & Sons, N.Y.).
Клонированный фрагмент ДНК, содержащий гены ферментов биосинтеза спинозина, может обеспечить дупликацию генов, кодирующих скоростьограничивающие ферменты при продуцировании спинозинов. Это можно использовать для увеличения выхода в любых обстоятельствах, когда одно из кодируемых активных веществ ограничивает синтез желаемого спинозина. Увеличение выхода этого типа достигали при ферментации Streptomyces fradiae путем удвоения гена, кодирующего скоростьограничивающую метилтрансферазу, которая конвертирует макроцин в тилозин (Baltz et al., 1997).
Конкретные промежуточные соединения (или их природные производные) могут синтезироваться мутантными штаммами S. spinosa, в которых нарушены определенные гены, кодирующие ферменты для биосинтеза спинозина. Такие штаммы можно получать встраиванием путем гомологичной рекомбинации мутагенной плазмиды, содержащей внутренний фрагмент гена-мишени. При встраивании плазмиды образуются две неполных копии гена биосинтеза, тем самым устраняя ферментативную функцию, которую он кодирует. При ферментации мутантного штамма должен накапливаться субстрат для этого фермента или его природное производное. Такую стратегию эффективно использовали для получения штамма Saccharopolyspora erythraea, продуцирующего новые производные 6-дезоксиэритромицина (Weber & McAlpine, 1992).
Такие штаммы можно получать путем замены заданной области посредством гомологичной рекомбинации с двойным кроссинговером на мутагенную плазмиду, содержащую новый фрагмент между немутантными последовательностями, которые фланкируют заданную область. Гибридный ген будет продуцировать белок с измененными функциями либо лишенный активности, либо выполняющий новое ферментативное преобразование. При ферментации такого мутантного штамма будет накапливаться новое производное. Такую стратегию использовали для получения штамма Saccharopolyspora erythraea, продуцирующего новое производное ангидроэритромицина (Donadio et al., 1993).
Гены биосинтеза спинозина и родственные ORF были клонированы, и была определена последовательность ДНК каждого из них. Клонированные гены и ORF обозначаются далее как spnA, spnB, spnC, spnD, spnE, spnF, spnG, spnH, spnI, spnJ, spnK, spnL, spnM, spnN, spnO, spnP, spnQ, spnR, spnS, ORFL15, ORFL16, ORFR1, ORFR2, S. spinosa gtt, S. spinosa gdh, S. spinosa epi и S. spinosa kre.
Saccharapolyspora spinosa продуцируют смесь из девяти близкородственных соединений в совокупности называемых “спинозинами”. В смеси спинозин A и D, известные как спиносад, являются основными компонентами и обладают наиболее высокой активностью против ключевых насекомых-мишеней. Спинозин J и L, два из второстепенных компонентов в смеси спинозина, являются предшественниками спинеторама, инсектицида на основе спинозина второго поколения. Варианты осуществления изобретения касаются прямого преобразования продуцирующего спиносад штамма в штамм, продуцирующий предшественник спинеторама, с помощью манипуляций с spnK, который кодирует 3'-O-метилтрансферазу.
Спиносад представляет собой инсектицид, производимый Dow AgroSciences (Indianapolis, Ind.), который содержит, главным образом, приблизительно 85% спинозина A и приблизительно 15% спинозина D. Спинозин A и D являются природными продуктами, продуцируемыми ферментацией Saccharopolyspora spinosa, как описано в патенте США № 5362634. Спиносад является активным ингредиентом нескольких инсектицидных составов, коммерчески доступных от Dow AgroSciences, включая продукты для борьбы с насекомыми TRACERTM, SUCCESSTM, SPINTORTM и CONSERVETM. Например, продукт TRACER содержит от приблизительно 44% до приблизительно 48% спиносада (масс./об.), или приблизительно 4 фунтов спиносада на галлон (0,5 кг на 1 л). Соединения спинозина в гранулярных и жидких составах имеют общепризнанную применимость для борьбы с паукообразными, круглыми червями и насекомыми, в частности видами Lepidoptera, Thysanoptera и Diptera. Спинозин A и D также называют в настоящем описании спинозином A/D.
Спинеторам представляет собой смесь 5,6-дигидро-3'-этоксиспинозина J (основной компонент) и 3'-этоксиспинозина L, производимую Dow AgroSciences. Смесь можно получать путем этоксилирования смеси спинозина J и спинозина L с последующей гидрогенизацией. 5,6-двойная связь спинозина J и его 3'-этоксипроизводного гидрогенизируется значительно более легко, чем у спинозина L и его 3'-этоксипроизводного вследствие пространственного препятствия в виде метильной группы на C-5 в спинозине L и его 3'-этоксипроизводном. См. патент США № 6001981. Спинозин J и L также называют в настоящем описании спинозином J/L.
Недавно было продемонстрировано, что spnK кодирует 3'-O-метилтрансферазу. См., Kim et al., JACS, 132(9): 2901-3 (2010). Заявители открыли, что spnK может быть удален из кластера генов биосинтеза спинозина путем гомологичной рекомбинации с двойным кроссинговером в рамке считывания без полярного эффекта на транскрипцию нижележащих генов spnL и spnM. Это позволяет получение способами инженерии продуцирующего спиносад штамма для получения штамма, продуцирующего предшественник спинеторама. Это также указывает на то, что штамм с нокаутом spnK утратил активность 3'-O-метилтрансферазы.
Варианты осуществления настоящего изобретения могут включать манипуляции в гене spnK, которые приводят к делеции в рамке считывания гена spnK путем удаления одного или нескольких кодонов в продуцирующем спиносад штамме. Делеция гена spnK в рамке считывания может включать любое укорочение любой части гена spnK. Делеции в рамке считывания в соответствии с настоящим изобретением включают делеции, которые удаляют сегмент кодирующей белок последовательности, но сохраняют надлежащую рамку считывания после делеции. Некоторые варианты осуществления настоящего изобретения могут включать делеции, которые представляют собой “чистые делеции”, т.е. они не содержат экзогенных последовательностей ДНК, встроенных в ген. Делеция гена spnK в рамке считывания может включать удаления из любого положения от 1 до 397 аминокислот. Она может включать удаление инициирующего кодона. Кроме того, она может включать удаление любого консервативного домена или любой области инициации транскрипции.
Для описания полинуклеотидных последовательностей в настоящем описании используются общепринятые обозначения: левый конец одноцепочечной полинуклеотидной последовательности представляет собой 5'-конец; направление двухцепочечной полинуклеотидной последовательности влево называют 5'-направлением. Направление добавления нуклеотидов от 5'- к 3'- к образующимся РНК-транскриптам называют направлением транскрипции. Цепь ДНК, имеющая ту же последовательность, что и мРНК, называют “кодирующей цепью”; последовательности на ДНК-цепи, имеющие ту же последовательность, что и мРНК, транскрибированная с этой ДНК и расположенные с 5'-стороны от 5'-конца РНК-транскрипта, называют “вышележащими последовательностями”; последовательности на цепи ДНК, имеющие ту же последовательность, что и РНК, и располагающиеся с 3'-сторны от 3'-конца кодирующего РНК-транскрипта называют “нижележащими последовательностями”.
Варианты осуществления настоящего изобретения могут включать манипуляции в гене spnK, которые приводят к делеции в рамке считывания 5'-конца гена spnK путем удаления одного или нескольких кодонов в продуцирующем спиносад штамме. Эти кодоны могут включать первый, второй или третий встречающийся кодон ATG.
Дополнительные варианты осуществления настоящего изобретения могут включать манипуляции в гене spnK, которые приводят к делеции в рамке считывания 3'-конца гена spnK путем удаления одного или нескольких кодонов в продуцирующем спиносад штамме.
Другие варианты осуществления настоящего изобретения могут включать манипуляции в гене spnK с делецией в рамке считывания в кодирующей области spnK либо одного кодона, либо нескольких кодонов, при сохранении как 5'-конца, так и 3'-конца гена, неизмененными.
Дополнительные варианты осуществления настоящего изобретения могут включать манипуляции в гене spnK, которые включают единичные или множественные точковые мутации, которые приводит к преждевременной терминации транскрипции или замене(ам) аминокислот во множестве участков, включая, но не ограничиваясь ими, активный центр и/или участок связывания субстрата. Такие единичные или множественные точковые мутации могут встречаться в SAM-связывающем мотиве, что приводит к ранней терминации в активном центре или участке связывания субстрата. Такие единичные или множественные точковые мутации также могут располагаться в области, которая влияет на общую структуру SpnK или влияет на надлежащее сворачивание, которое может устранить функцию SpnK. Такие единичные или множественные точковые мутации можно создавать путем детекции функциональных полиморфизмов или путем мутагенеза.
“Функциональный полиморфизм”, как используют в рамках изобретения, относится к изменению последовательности пар оснований гена, которое вызывает качественное или количественное изменение активности белка, кодируемого этим геном (например, изменение специфичности активности; изменение уровня активности). Термин “функциональный полиморфизм” включает мутации, делеции и инсерции.
Как правило, стадию детекции полиморфизма можно проводить путем взятия биологического образца, содержащего ДНК, из источника, а затем определения присутствия или отсутствия ДНК, содержащей представляющий интерес полиморфизм, в биологическом образце.
Определение присутствия или отсутствия ДНК, кодирующей конкретную мутацию, можно проводить с помощью олигонуклеотидного зонда, меченного пригодной поддающейся детекции группой, и/или с помощью реакции амплификации, такой как полимеразная цепная реакция или лигазная цепная реакция (продукт этой реакции амплификации затем можно выявлять с помощью меченого олигонуклеотидного зонда или ряда других способов). Кроме того, стадия детекции может включать стадию детекции того, является ли индивидуум гетерозиготным или гомозиготным по конкретной мутации. Известны многочисленные различные форматы анализа с олигонуклеотидными зондами, которые можно использовать для осуществления настоящего изобретения. См., например, патент США № 4302204, выданный Wahl et al.; патент США № 4358535, выданный Falkow et al.; патент США № 4563419, выданный Ranki et al.; и патент США № 4994373, выданный Stavrianopoulos et al.
Амплификацию выбранной или целевой последовательности нуклеиновой кислоты можно проводить любым пригодным способом. См., главным образом, Kwoh et al., Am. Biotechnol. Lab. 8, 14-25 (1990). Примеры пригодных способов амплификации включают, но не ограничиваются ими, полимеразную цепную реакцию, лигазную цепную реакцию, амплификацию с вытеснением нити (см., главным образом G. Walker et al., Proc. Natl. Acad. Sci. USA 89, 392-396 (1992); G. Walker et al., Nucleic Aids Res. 20, 1691-1696 (1992)), амплификацию на основе транскрипции (см. D. Kwoh et al., Proc. Natl. Acad Sci. USA 86, 1173-1177 (1989)), самоподдерживающуюся репликацию последовательности (или “3SR”) (см. J. Guatelli et al., Proc. Natl. Acad. Sci. USA 87, 1874-1878 (1990)), систему Qβ-репликазы (см. P. Lizardi et al., BioTechnology 6, 1197-1202 (1988)), амплификацию на основе последовательности нуклеиновой кислоты (или “NASBA”) (см. R. Lewis, Genetic Engineering News 12 (9), 1 (1992)), реакцию с репарацией цепей (или “RCR”) (см. R. Lewis, выше) и амплификацию ДНК по принципу бумеранга (или “BDA”) (см. R. Lewis, выше). Главным образом, предпочтительной является полимеразная цепная реакция.
Полимеразную цепную реакцию (ПЦР) можно проводить в соответствии с известными способами. См., например, патенты США № 4683195; 4683202; 4800159 и 4965188. Как правило, ПЦР вовлекает сначала обработку образца нуклеиновой кислоты (например, в присутствии термостабильной ДНК-полимеразы) олигонуклеотидным праймером для каждой нити конкретной последовательности, подлежащей детекции, в условиях гибридизации, так чтобы синтезировался продукт удлинения каждого праймера, который комплементарен каждой нити нуклеиновой кислоты, с праймерами, достаточно комплементарными каждой нити конкретной последовательности, чтобы гибридизоваться с ней, так чтобы продукт удлинения, синтезированный с каждого праймера, когда он отделен от его комплементарной последовательности, мог служить в качестве матрицы для синтеза продукта удлинения другого праймера, а затем обработку образца в денатурирующих условиях для отделения продуктов удлинения праймеров от их матриц, если последовательность или последовательности, подлежащие детекции, присутствуют. Эти стадии повторяют циклически до тех пор, пока не достигают желаемой степени амплификации. Детекцию амплифицированной последовательности можно проводить добавлением к продукту реакции олигонуклеотидного зонда, способного гибридизоваться с продуктом реакции (например, олигонуклеотидным зондом по настоящему изобретению), где зонд содержит поддающуюся детекции метку, а затем детекцией метки в соответствии с известными способами или путем прямой визуализации на геле. Такие зонды могут иметь длину от 5 до 500 нуклеотидов, предпочтительно от 5 до 250, более предпочтительно от 5 до 100 или от 5 до 50 нуклеотидов. Когда условия ПЦР позволяют амплификацию всех аллельных типов, типы можно различить гибридизацией с аллелеспецифическим зондом путем расщепления эндонуклеазой рестрикции, путем электрофореза на денатурирующих градиентных гелях или другими способами.
Лигазную цепную реакцию (LCR) также проводят в соответствии с известными способами. См., например, R. Weiss, Science 254, 1292 (1991). Как правило, реакцию проводят с двумя парами олигонуклеотидных зондов: одна пара связывается с одной нитью последовательности, подлежащей детекции; а другая пара связывается с другой нитью последовательности, подлежащей детекции. Каждая пара вместе полностью перекрывает нить, которой она соответствует. Реакцию проводят сначала путем денатурации (например, разделения) нитей последовательности, подлежащих детекции, а затем реакции нитей с двумя парами олигонуклеотидных зондов в присутствии термостабильной лигазы, так чтобы каждая пара олигонуклеотидных зондов лигировалась вместе, а затем разделением продукта реакции, и затем циклическим повторением процесса до тех пор, пока последовательность не амплифицируется до желаемой степени. Затем можно проводить детекцию подобно тому, как описано выше в отношении ПЦР.
Способы амплификации ДНК, такие как указанные выше способы, могут вовлекать применение зонда, пары зондов или двух пар зондов, которые специфично связываются с ДНК, содержащей функциональный полиморфизм, но не связываются с ДНК, которая не содержит функциональный полиморфизм. Альтернативно зонд или пара зондов могут связываться с ДНК которая как содержит, так и не содержит функциональный полиморфизм, но продуцирует или амплифицирует продукт (например, продукт элонгации), в котором могут быть выявлены поддающиеся детекции различия (например, более короткий продукт, где функциональным полиморфизмом является мутация с делецией). Такие зонды можно получать стандартными способами из известных последовательностей ДНК в гене, связанном с spnK, или последовательностей, ассоциированных с геном, связанным с spnK, или из последовательностей, которые можно получать из таких генов в соответствии со стандартными способами.
Понятно, что стадии детекции, описанные в настоящем описании, можно проводить прямо или непрямо. Другие способы непрямого определения аллельного типа включают измерение полиморфных маркеров, которые связаны с конкретным функциональным полиморфизмом, как было продемонстрировано для VNTR (вариабельные по числу тандемные повторы).
Молекулярная биология включает широкое множество способов анализа последовательностей нуклеиновых кислот и белков. Многие из этих способов и методик формируют основу диагностических анализов и тестов. Эти способы включают анализ гибридизации нуклеиновых кислот, анализ с ферментом рестрикции, анализ генетической последовательности и разделение и очистку нуклеиновых кислот и белков (см., например, J. Sambrook, E. F. Fritsch, and T. Maniatis, Molecular Cloning: A Laboratory Manual, 2 Ed., Cold spring Harbor Laboratory Press, Cold Spring Harbor, N.Y., 1989).
Большинство из этих способов вовлекают проведение многочисленных действий (например, пипетирование, центрифугирование и электрофорез) на большом количестве образцов. Они часто являются сложными и требующими много времени и обычно требуют высокой степени точности. Многие способы ограничены в их применении вследствие нехватки чувствительности, специфичности или воспроизводимости.
Анализ гибридизации нуклеиновых кислот, как правило, вовлекает детекцию очень небольшого количества специфических нуклеиновых кислот-мишеней (ДНК или РНК) с помощью избытка зонда ДНК среди относительно большого количества комплексных не являющихся мишенями нуклеиновых кислот. Снижение комплексности нуклеиновой кислоты в образце способствует детекции низких количеств копий (т.е. от 10000 до 100000) нуклеиновых кислот-мишеней. Снижения комплексности ДНК достигают до некоторой степени путем амплификации последовательностей нуклеиновых кислот-мишеней (см. M.A. Innis et al., ПЦР Protocols: A Guide to Methods and Applications, Academic Press, 1990, Spargo et al., 1996, Molecular & Cellular Probes, что касается амплификации SDA). Это является следствием того, что амплификация нуклеиновых кислот-мишеней приводит к огромному количеству последовательностей нуклеиновых кислот-мишеней относительно не являющихся мишенями последовательностей, тем самым улучшая последующую стадию гибридизации мишени.
Стадия гибридизации вовлекает приведение полученного образца ДНК в контакт со специфическим репортерным зондом в наборе оптимальных условий для осуществления гибридизации между последовательностью ДНК-мишени и зондом. Гибридизацию можно проводить в любом из ряда форматов. Например, анализ гибридизации нуклеиновых кислот с множеством образцов проводили с различными форматами фильтров и твердых подложек (см. Beltz et al., Methods in Enzymology, Vol. 100, Part et al., Eds., Academic Press, New York, Chapter 19, pp. 266-308, 1985). Один формат, так называемый “дот-блот” гибридизация, вовлекает нековалентное присоединения ДНК-мишеней к фильтру с последующей гибридизацией с меченным радиоизотопом зондом(ами). “Дот-блот” гибридизация достигла широкого применения в течение последних двух десятилетий, на протяжении которых было разработано много версий (см. Anderson and Young, Nucleic Acid Hybridisation-A Practical Approach, Hames and Higgins, Eds., IRL Press, Washington, D.C. Chapter 4, pp. 73-111, 1985). Например, дот-блот способ был разработан для множественных анализов геномных мутаций (EPA 0228075, выданный Nanibhushan et al.) и для детекции перекрывающихся клонов и конструирования геномных карт (патент США № 5219726, выданный Evans).
Дополнительные способы для осуществления анализа гибридизации нуклеиновых кислот с множеством образцов включают мультиплексные или матричные устройства в микроформате (например, ДНК-чипы) (см. M. Barinaga, 253 Science, pp. 1489, 1991; W. Bains, 10 Bio/Technology, pp. 757-758, 1992). В этих способах обычно специфические последовательности ДНК присоединяют к очень небольшим конкретным областям твердой подложки, таким как микролунки ДНК-чипа. Эти форматы гибридизации представляют собой микроверсии общепринятых “дот-блот”- и “сэндвич”-систем гибридизации.
Гибридизацию в микроформате можно использовать для осуществления “секвенирования гибридизацией” (SBH) (см. M. Barinaga, 253 Science, pp. 1489, 1991; W. Bains, 10 Bio/Technology, pp. 757-758, 1992). В SBH используются все возможные олигомеры из n нуклеотидов (n-меры) для идентификации n-меров в неизвестном образце ДНК, которые затем выравнивают с помощью алгоритмического анализа с получением последовательности ДНК (см. Drmanac, патент США № 5202231).
Существуют два формата для проведения SBH. Первый формат вовлекает создание набора всех возможных n-меров на подложке, которые затем гибридизуют с последовательностью-мишенью. Второй формат вовлекает присоединение последовательности-мишени к подложке, которую затем исследуют с помощью всех возможных n-меров. Southern (патентная заявка Великобритании GB 8810400, 1988; E.M. Southern et al., 13 Genomics 1008, 1992), предложил использовать первый формат для анализа или секвенирования ДНК. Southern идентифицировал известную единичную точковую мутацию с использованием ПЦР амплифицированной геномной ДНК. Southern также описал способ синтеза набора олигонуклеотидов на твердой подложке для SBH. Drmanac et al., (260 Science 1649-1652, 1993), использовал второй формат для секвенирования нескольких коротких (116 п.н.) последовательностей ДНК. ДНК-мишени связывали с мембранными подложками (“дот-блот”-формат). Каждый фильтр последовательно гибридизовали с 272 мечеными 10-мерными и 1-мерными олигонуклеотидами. Использовали широкие диапазоны условий жесткости для достижения специфической гибридизации для каждого n-мерного зонда. Время промывания варьировало от 5 минут до промывания в течение ночи при использовании температур от 0°C до 16°C. Для большинства зондов требовалось промывание в течение 3 часов при 16°C. Фильтры необходимо было проявлять в течение от 2 до 18 часов для детекции сигналов гибридизации.
Главным образом, доступно множество способов для детекции и анализа событий гибридизации. В зависимости от репортерной группы (флуорофор, фермент, радиоактивный изотоп и т.д.), используемой для мечения ДНК-зонда, детекию и анализ проводят флуориметрически, калориметрически и радиоавтографически. Путем наблюдения и измерения испускаемого излучения, такого как флуоресцентное излучение или испускание частиц, может быть получена информация о событиях гибридизации. Даже когда способы детекции имеют очень высокую присущую им чувствительность, детекция событий гибридизации является трудной вследствие присутствия фона от неспецифически связанных материалов. Таким образом, детекция событий гибридизации зависит от того, как можно проводить специфическую и чувствительную гибридизацию. Что касается генетического анализа, также было разработано несколько способов, в которых предпринимали попытку увеличить специфичность и чувствительность.
Одной из форм генетического анализа является анализ, сосредоточенный на выявлении однонуклеотидных полиморфизмов или (“SNP”). Факторами, способствующими использованию SNP, являются их широкая распространенность в геноме человека (особенно по сравнению с короткими тандемными повторами (STR)), их частое расположение в кодирующих или регуляторных областях генов (что может влиять на структуру белка или уровни экспрессии) и их стабильность, когда они проходят от одного поколения к следующему (Landegren et al., Genome Research, Vol. 8, pp. 769-776, 1998).
SNP определяют как любое положение в геноме, которое существует в двух вариантах, и наиболее распространенный вариант встречается менее чем в 99% случаев. Для использования SNP в качестве широко распространенных генетических маркеров важно иметь возможность генотипировать их легко, быстро, точно и эффективно по стоимости. В настоящее время доступны многочисленные способы для типирования SNP (для обзора см. Landegren et al., Genome Research, Vol. 8, pp. 769-776, (1998), все из которых требуют амплификации мишени. Они включают прямое секвенирование (Carothers et al., BioTechniques, Vol. 7, pp. 494-499, 1989), однонитевой конформационный полиморфизм (Orita et al., Proc. Natl. Acad. Sci. USA, Vol. 86, pp. 2766-2770, 1989), аллелеспецифическую амплификацию (Newton et al., Nucleic Acids Research, Vol. 17, pp. 2503-2516, (1989), рестрикционное расщепление (Day and Humphries, Analytical Biochemistry, Vol. 222, pp. 389-395, 1994) и анализы гибридизации. В их наиболее основной форме анализы гибридизации функционируют путем различения с помощью коротких олигонуклеотидных репортеров совпадающих и несовпадающих мишеней. Были разработаны многие адаптации основного протокола. Они включают лигазную цепную реакцию (Wu and Wallace, Gene, Vol. 76, pp. 245-254, 1989) и минисеквенирование (Syvanen et al., Genomics, Vol. 8, pp. 684-692, 1990). Другие усовершенствования включают использование 5'-нуклеазной активности ДНК-полимеразы Taq (Holland et al., Proc. Natl. Acad. Sci. USA, Vol. 88, pp. 7276-7280, 1991), молекулярных маяков (Tyagi and Kramer, Nature Biotechnology, Vol. 14, pp. 303-308, 1996), кривых термической денатурации (Howell et al., Nature Biotechnology, Vol. 17, pp. 87-88, 1999) и ДНК-“чипов” (Wang et al., Science, Vol. 280, pp. 1077-1082, 1998).
Дополнительным явлением, которое можно использовать для различения SNP, являются энергии взаимодействия нуклеиновых кислот или энергии стэкинга оснований, генерируемые при гибридизации множества специфичных к мишени зондов с одной мишенью (см. R. Ornstein et al., “An Optimized Potential Функция for the Calculation of Nucleic Acid Interaction Energies,” Biopolymers, Vol.17, 2341-2360 (1978); J. Norberg and L. Nilsson, Biophysical Journal, Vol. 74, pp. 394-402, (1998); и J. Pieters et al., Nucleic Acids Research, Vol. 17, no. 12, pp. 4551-4565 (1989)). Это явление стэкинга оснований используют в уникальном формате в рамках настоящего изобретения для обеспечения высокочувствительных перепадов Tm, позволяющих детекцию SNP в образце нуклеиновых кислот.
Дополнительные способы используют для различения последовательностей нуклеиновых кислот в родственных организмах или для секвенирования ДНК. Например, в патенте США № 5030557, выданном Hogan et al., описано, что на вторичную и третичную структуру одноцепочечной нуклеиновой кислоты-мишени можно влиять путем связывания олигонуклеотидов-“помощников” в дополнение к олигонуклеотидам-“зондам”, вызывающих возникновение более высокой Tm между зондом и нуклеиновой кислотой-мишенью. Однако настоящая заявка была ограничена в этом подходе использованием энергий гибридизации только для изменения вторичной и третичной структуры подвергающихся самоотжигу цепей РНК, которые, если остаются неизмененными, будут иметь тенденцию к предотвращению гибридизации зонда с его мишенью.
Что касается секвенирования ДНК, K. Khrapko et al., Federation of European Biochemical Societies Letters, Vol. 256, no. 1,2, pp. 118-122 (1989), например, описали, что непрерывная стэкинг-гибридизация приводила к стабилизации дуплекса. Кроме того, J. Kieleczawa et al., Science, Vol. 258, pp. 1787-1791 (1992) описали применение последовательно расположенных нитей гексамеров для затравки синтеза ДНК, где оказалось, что последовательно расположенные нити стабилизируют процесс затравки. Аналогично, L. Kotler et al., Proc. Natl. Acad. Sci. USA, Vol. 90, pp. 4241-4245, (1993) описали специфичность последовательностей в процессе затравки в реакциях секвенирования ДНК с использованием гексамерных и пентамерных олигонуклеотидных модулей. Further, S. Parinov et al., Nucleic Acids Research, Vol. 24, no. 15, pp. 2998-3004, (1996) описали использование олигомеров со стэкингом оснований для секвенирования ДНК совместно с микрочипами для пассивного секвенирования ДНК. Более того, G. Yershov et al., Proc. Natl. Acad. Sci. USA, Vol. 93, pp. 4913-4918 (1996), описали применение энергий стэкинга оснований в SBH на пассивном микрочипе. В примере Yershov 10-мерные ДНК-зонды заякоривали на поверхности микрочипа и гибридизовали с последовательностями-мишенями совместно с дополнительными короткими зондами, комбинация которых, как оказалось, стабилизирует связывание зондов. В этом формате могут быть установлены короткие сегменты последовательности нуклеиновой кислоты для секвенирования ДНК. Далее Yershov отметил, что в их системе дестабилизирующий эффект несоответствующих оснований увеличивался при использовании более коротких зондов (например 5-меров). Использование таких коротких зондов при секвенирвоании ДНК обеспечило способность отличить присутствие несоответствующих оснований вдоль последовательности, подвергаемой исследованию с помощью зонда, вместо только одного несовпадения в конкретной области гибридизационного комплекса зонд/мишень. Использование более длинных зондов (например, 8-мерные, 10-мерные и 13-мерные олигонуклеотиды) было менее функциональным для таких целей.
Дополнительный пример технологий, в которых используется стэкинг оснований при анализе нуклеиновых кислот, включает патент США № 5770365, выданный Lane et al., где описан способ улавливания нуклеиновых кислот-мишеней с использованием одномолекулярного зонда для улавливания, имеющего одноцепочечную петлю и двухцепочечную область, которая действует вместе со связываемой мишенью, стабилизируя образование дуплекса с помощью энергий стэкинга.
Нуклеотидную последовательность можно удобным образом модифицировать сайт-направленным мутагенезом в соответствии с общепринятыми способами. Альтернативно нуклеотидную последовательность можно получать с помощью химического синтеза, включая, но не ограничиваясь ими, использование устройства для синтеза олигонуклеотидов, где олигонуклеотиды сконструированы на основе аминокислотной последовательности желаемого полипептида, и предпочтительно селекцию тех кодонов, которые являются благоприятными в клетке-хозяине, в которой рекомбинантный полипептид будет продуцироваться.
Новые спинозины также можно получать путем мутагенеза клонированных генов и замены мутантными генами их немутантных аналогов в продуцирующем спинозин организме. Мутагенез может вовлекать, например: 1) делецию или инактивацию KR-, DH- или ER-домена, так чтобы одна или несколько из этих функций блокировались и штамм продуцировал спинозин, имеющий лактонное ядро с двойной связью, гидроксильную группу или кетогруппу, которая не присутствует в ядре спинозина A (см. Donadio et al., 1993); 2) замену AT-домена так, чтобы в лактонное ядро встраивалась другая карбоновая кислота (см. Ruan et al., 1997); 3) вставку KR-, DH- или ER-домена в существующий модуль PKS, так чтобы штамм продуцировал спинозин, имеющий лактонное ядро с насыщенной связью, гидроксильной группой или двойной связью, которая не присутствует в ядре спинозина A; или 4) вставку или удаление полного модуля PKS так, чтобы циклическое лактонное ядро имело большее или меньшее число атомов углерода. Гибридный PKS можно создавать путем замены нагрузочного домена PKS спинозина гетерологичным нагрузочным доменом PKS. См., например, патент США № 7626010. Кроме того, отмечается, что спинозины, посредством модификации сахаров, которые связаны с лактонным остовом спинозина, могут включать модификации части в виде рамнозы и/или форозамина или присоединение других дезоксисахаров. Группа Salas в Испании продемонстрировала, что можно получать новые поликетидные соединения путем замены существующей молекулы сахара отличающимися молекулами сахаров. Rodriguez et al. J. Mol. Microbiol Biotechnol. 2000 Jul; 2(3): 271-6. Примеры, которые представлены на протяжении заявки, помогают проиллюстрировать использование мутагенеза для получения спинозина с модифицированной функциональностью.
ДНК из кластерной области генов спинозина можно использовать в качестве гибридизационного зонда для идентификации гомологичных последовательностей. Таким образом, ДНК, клонированные в рамках настоящего изобретения, можно использовать для определения дополнительных плазмид из библиотек генов Saccharopolyspora spinosa, которые охватывают область, описанную в настоящем описании, но также содержат ранее неклонированную ДНК из соседних областей в геноме Saccharopolyspora spinosa. Кроме того, ДНК из области, клонированной в рамках настоящего изобретения, можно использовать для идентификации неидентичных, но сходных последовательностей в других организмах. Гибридизационные зонды обычно имеют длину по меньшей мере приблизительно 20 оснований и являются мечеными, чтобы позволить детекцию.
Различные типы мутагенеза можно использовать в рамках настоящего изобретения для различных целей. Они включают, но не ограничиваются ими, сайт-направленный мутагенез, случайный точковый мутагенез, гомологичную рекомбинацию, шаффлинг ДНК или другие способы возвратного мутагенеза, конструирование химерных конструкций, мутагенез с использованием урацилсодержащих матриц, олигонуклеотиднаправленный мутагенез, фосфоротиоатмодифицированный мутагенез ДНК, мутагенез с использованием дуплексной ДНК с пропусками или сходные с ними или любые их комбинации. Дополнительные пригодные способы включают репарацию точечных несовпадений оснований, мутагенез с использованием штаммов-хозяев с дефектом репарации, рестрикцию-селекцию и рестрикцию-очистку, мутагенез с делецией, мутагенез путем тотального синтеза гена, репарацию двуцепочечного разрыва и т.п. Мутагенез, включая, но не ограничиваясь ими, вовлечение химерных конструкций, также включен в настоящее изобретение. В одном варианте осуществления мутагенез может определяться известной информацией о встречающейся в природе молекуле или измененной или мутантной встречающейся в природе молекуле, включая, но не ограничиваясь ими, последовательность, сравнение последовательностей, физические свойства, кристаллическую структуру или сходные с ними.
В тексте и примерах в настоящем описании описаны эти методики. Дополнительная информация представлена в следующих цитированных публикациях и ссылках: Ling et al., Approaches to DNA mutagenesis: an overview, Anal Biochem. 254(2): 157-178 (1997); Dale et al., Oligonucleotide-directed random mutagenesis using the phosphorothioate method, Methods Mol. Biol. 57:369-374 (1996); Smith, In vitro mutagenesis, Ann. Rev. Genet. 19:423-462(1985); Botstein & Shortle, Strategies and applications of in vitro mutagenesis, Science 229:1193-1201(1985); Carter, Site-directed mutagenesis, Biochem. J. 237:1-7 (1986); Kunkel, The efficiency of oligonucleotide directed mutagenesis, in Nucleic Acids & Molecular Biology (Eckstein, F. and Lilley, D.M.J. eds., Springer Verlag, Berlin) (1987); Kunkel, Rapid and efficient site-specific mutagenesis without phenotypic selection, Proc. Natl. Acad. Sci. USA 82:488-492 (1985); Kunkel et al., Rapid and efficient site-specific mutagenesis without phenotypic selection, Methods in Enzymol. 154, 367-382 (1987); Bass et al., Mutant Trp repressors with new DNA-binding specificities, Science 242: 240-245 (1988); Methods in Enzymol. 100: 468-500 (1983); Methods in Enzymol. 154: 329-350 (1987); Zoller & Smith, Oligonucleotide-directed mutagenesis using M13-derived vectors: an efficient and general procedure for the production of point mutations in any DNA fragment, Nucleic Acids Res. 10: 6487-6500 (1982); Zoller & Smith, Oligonucleotide-directed mutagenesis of DNA fragments cloned into M13 vectors, Methods in Enzymol. 100: 468-500 (1983); Zoller & Smith, Oligonucleotide-directed mutagenesis: a simple method using two oligonucleotide primers and a single-stranded DNA template, Methods in Enzymol. 154: 329-350 (1987); Taylor et al., The use of phosphorothioate-modified DNA in restriction enzyme reactions to prepare nicked DNA, Nucl. Acids Res. 13: 8749-8764 (1985); Taylor et al., The rapid generation of oligonucleotide-directed mutations at high frequency using phosphorothioate-modified DNA, Nucl. Acids Res. 13: 8765-8787 (1985); Nakamaye & Eckstein, Inhibition of restriction endonuclease Nci I cleavage by phosphorothioate groups and its application to oligonucleotide-directed mutagenesis, Nucl. Acids Res. 14: 9679-9698 (1986); Sayers et al., Y-T Exonucleases in phosphorothioate-based oligonucleotide-directed mutagenesis, Nucl. Acids Res. 16: 791-802 (1988); Sayers et al., Strand specific cleavage of phosphorothioate-containing DNA by reaction with restriction endonucleases in the presence of ethidium bromide, (1988) Nucl. Acids Res. 16: 803-814; Kramer et al., The gapped duplex DNA approach to oligonucleotide-directed mutation construction, Nucl. Acids Res. 12: 9441-9456 (1984); Kramer & Fritz Oligonucleotide-directed construction of mutations via gapped duplex DNA, Methods in Enzymol. 154: 350-367 (1987); Kramer et al., Improved enzymatic in vitro reactions in the gapped duplex DNA approach to oligonucleotide-directed construction of mutations, Nucl. Acids Res. 16: 7207 (1988); Fritz et al., Oligonucleotide-directed construction of mutations: a gapped duplex DNA procedure without enzymatic reactions in vitro, Nucl. Acids Res. 16: 6987-6999 (1988); Kramer et al., Point Mismatch Repair, Cell 38: 879-887 (1984); Carter et al., Improved oligonucleotide site-directed mutagenesis using M13 vectors, Nucl. Acids Res. 13: 4431-4443 (1985); Carter, Improved oligonucleotide-directed mutagenesis using M13 vectors, Methods in Enzymol. 154: 382-403 (1987); Eghtedarzadeh & Henikoff, Use of oligonucleotides to generate large deletions, Nucl. Acids Res. 14: 5115 (1986); Wells et al., Importance of hydrogen-bond formation in stabilizing the transition state of subtilisin, Phil. Trans. R. Soc. Lond. A 317: 415-423 (1986); Nambiar et al., Total synthesis and cloning of a gene coding for the ribonuclease S protein, Science 223: 1299-1301 (1984); Sakamar and Khorana, Total synthesis and expression of a gene for the a-subunit of bovine rod outer segment guanine nucleotide-binding protein (transducin), Nucl. Acids Res. 14: 6361-6372 (1988); Wells et al., Cassette mutagenesis: an efficient method for generation of multiple mutations at defined sites, Gene 34:315-323 (1985); Grundstrom et al., Oligonucleotide-directed mutagenesis by microscale “shot-gun” gene synthesis, Nucl. Acids Res. 13: 3305-3316 (1985); Mandecki, Oligonucleotide-directed double-strand break repair in plasmids of Escherichia coli: a method for site-specific mutagenesis, Proc. Natl. Acad. Sci. USA, 83: 7177-7181 (1986); Arnold, Protein engineering for unusual environments, Current Opinion in Biotechnology 4:450-455 (1993); Sieber, et al., Nature Biotechnology, 19:456-460 (2001). W.P.C. Stemmer, Nature 370, 389-91 (1994); и I.A. Lorimer, I. Pastan, Nucleic Acids Res. 23, 3067-8 (1995). Дополнительные детали о многих из описанных выше способах могут быть найдены в Methods in Enzymology Volume 154, в которой описаны пригодные пути решения проблем, связанных с ошибками, при различных способах мутагенеза.
Термины “гомология” или “процентная идентичность” используют в настоящем описании взаимозаменяемо. Для целей этого изобретения в настоящем описании определено, что для определения процентной идентичности двух аминокислотных последовательностей или двух последовательностей нуклеиновых кислот последовательности выравнивают для целей оптимального сравнения (например, пропуски можно вносить в последовательность первой аминокислотной последовательности или последовательности нуклеиновой кислоты для оптимального выравнивания со второй аминокислотной последовательностью или последовательностью нуклеиновой кислоты). Затем сравнивают аминокислотные остатки или нуклеотиды в соответствующих положениях аминокислот или положениях нуклеотидов. Когда положение в первой последовательности занимает тот же аминокислотный остаток или нуклеотид, что и в соответствующем положении во второй последовательности, тогда молекулы являются идентичными в этом положении. Процентная идентичность между двумя последовательностями является функцией количества идентичных положений, общих для последовательностей (т.е. % идентичность=количество идентичных положений/общее количество положений (т.е. перекрывающиеся положения ×100). Предпочтительно, эти две последовательности имеют одну длину.
Квалифицированному специалисту известно, что для определения гомологии между двумя последовательностями доступно несколько различных компьютерных программ. Например, сравнение последовательностей и определение процентной идентичности между двумя последовательностями можно проводить с использованием математического алгоритма. В предпочтительном варианте осуществления процентную идентичность между двумя аминокислотными последовательностями определяют с использованием алгоритма Needleman и Wunsch (J. Mol. Biol. (48): 444-453 (1970)), который включен в программу GAP в пакете программ GCG (доступном через web-сайт accelrys, более конкретно на http://www.accelrys.com), с использованием либо матрицы Blossom 62, либо матрицы PAM250, и веса пропусков 16, 14, 12, 10, 8, 6 или 4, и веса длины 1, 2, 3, 4, 5 или 6. Квалифицированному специалисту понятно, что все эти различные параметры приводят к немного отличающимся результатам, но что общая процентная идентичность двух последовательностей значительно не изменяется при использовании различных алгоритмов.
В другом варианте осуществления процентную идентичность между двумя нуклеотидными последовательностями определяют с использованием программы GAP в пакете программ GCG (доступном через интернет на web-сайте accelrys, более конкретно на http://www.accelrys.com), с использованием a матрицы NWSgapDNA.CMP и веса пропуска 40, 50, 60, 70 или 80 и веса длины 1, 2, 3, 4, 5 или 6. В другом варианте осуществления процентную идентичность двух аминокислотных или нуклеотидных последовательностей определяют с использованием алгоритма E. Meyers и W. Miller (CABIOS, 4: 11-17 (1989), который включен в программу ALIGN (версии 2.0) (доступную через интернет на web-сайте vega, более конкретно ALIGN - IGH Montpellier, или более конкретно на http://vega.igh.cnrs.fr/bin/align-guess.cgi) с использованием таблицы веса остатков PAM120, штрафа за продолжение пропуска 12 и штрафа за делецию 4.
Последовательности нуклеиновых кислот и белков по настоящему изобретению можно далее использовать в качестве “последовательности запроса” для проведения поиска против общественных баз данных, например, для идентификации других представителей семейств или родственных последовательностей. Такой поиск можно проводить с использованием программ BLASTN и BLASTX (версия 2.0) Altschul, et al. (1990) J. Mol. Biol. 215: 403-10. Поиск нуклеотидов BLAST можно проводить с помощью программы BLASTN, сумма баллов=100, длина слова=12 для получения нуклеотидных последовательностей, гомологичных молекулам нуклеиновых кислот по настоящему изобретению. Поиск белков BLAST можно проводить с помощью программы BLASTX, сумма баллов=50, длина слова=3 для получения аминокислотных последовательностей, гомологичных молекулам белка по настоящему изобретению. Для проведения выравнивания с пропусками для целей сравнения можно использовать Gapped BLAST, как описано в Altschul et al., (1997) Nucleic Acids Res. 25 (17): 3389-3402. При использовании программ BLAST и Gapped BLAST можно использовать параметры по умолчанию соответствующих программ (например, BLASTX и BLASTN). (Доступны на web-сайте ncbi, более конкретно на http://www.ncbi.nlm.nih.gov).
Другие варианты осуществления настоящего изобретения могут включать манипуляции в гене spnK, которые могут включать делецию(и) одного или нескольких нуклеотидов, которая может нарушать нормальную рамку считывания spnK. Такие делеции могут включать делеции в любом положении нуклеотидов с 1 по 1194. Такая делеция влияет на нормальную рамку считывания spnK, что приводит к получению штамма, продуцирующего предшественник спинеторама.
Другой вариант осуществления настоящего изобретения может включать манипуляции в гене spnK, которые могут включать инсерцию(и) одного или нескольких нуклеотидов в кодирующей области spnK, которая нарушает рамку считывания spnK. Такая инсерция влияет на нормальную рамку считывания spnK, что приводит к получению штамма, продуцирующего предшественник спинеторама.
Дополнительные варианты осуществления настоящего изобретения могут включать манипуляции в гене spnK, которые включают использование антисмысловой или смысловой технологии для устранения или значительного препятствования продуцированию белка SpnK. Специалисту в данной области известно, как достигнуть антисмыслового эффекта и эффекта косупрессии. Например, способ ингибирования путем косупрессии описан в Jorgensen (Trends Biotechnol. 8 (1990), 340-344), Niebel et al., (Curr. Top. Microbiol. Immunol. 197 (1995), 91-103), Flavell et al. (Curr. Top. Microbiol. Immunol. 197 (1995), 43-46), Palaqui и Vaucheret (Plant. Mol. Biol. 29 (1995), 149-159), Vaucheret et al., (Mol. Gen. Genet. 248 (1995), 311-317), de Borne et al. (Mol. Gen. Genet. 243 (1994), 613-621).
Таким образом, настоящее изобретение дополнительно относится к способам подавления генов путем экспрессии в организме, таком как S. spinosa, нуклеиновой кислоты, имеющей инвертированный повтор, с 5'- или 3'-стороны относительно смысловой или антисмысловой нацеливающей последовательности, где смысловая или антисмысловая нацеливающая последовательность обладает значительной идентичностью последовательности с геном-мишенью, подлежащим подавлению, однако инвертирвоанный повтор не является родственным по последовательности гену-мишени. В другом варианте осуществления гетерологичный инвертированный повтор фланкируется 5'- и 3'-нацеливающей последовательностью.
Конструкцию для подавления генов можно экспрессировать в выбираемом организме, например бактериальной клетке, клетке грибов, эукариотической клетке, например растительной клетке или клетке млекопитающих.
Пригодные экспрессирующие векторы для применения в настоящем изобретении включают прокариотические и эукариотические векторы (например, плазмида, фагминда или бактериофаг), векторы млекопитающих и векторы растений. Пригодные прокариотические векторы включают плазмиды, такие как, но не ограничиваясь ими, плазмиды, обычно используемые для манипулирования с ДНК в Actinomyces, (например, pSET152, pOJ260, pIJ101, pJV1, pSG5, pHJL302, pSAM2, pKC1250. Такие плазмиды описаны Kieser et al. (“Practical Streptomyces Genetics,” 2000). Другие пригодные векторы могут включать плазмиды, такие как плазмиды, способные к репликации в E. coli (например, pBR322, ColE1, pSC101, PACYC 184, itVX, pRSET, pBAD (Invitrogen, Carlsbad, Calif.) и т.п.). Такие плазмиды описаны Sambrook (cf. “Molecular Cloning: A Laboratory Manual”, второе издание под редакцией Sambrook, Fritsch, & Maniatis, Cold Spring Harbor Laboratory, (1989)), и многие такие векторы являются коммерчески доступными. Плазмиды Bacillus включают pC194, pC221, pT127 и т.п. и описаны Gryczan (The Molecular Biology of the Bacilli, Academic Press, NY (1982), pp. 307-329). Пригодные плазмиды Streptomyces включают pli101 (Kendall et al., J. Bacteriol. 169: 4177-4183, 1987) и бактериофаги Streptomyces включают, но не ограничиваясь ими, такие как ψC31 (Chatter et al., Sixth International Symposium on Actinomycetales Biology, Akademiai Kaido, Budapest, Hungary (1986), pp. 45-54). Плазмиды Pseudomonas рассмотрены John et al. (Rev. Infect. Dis. 8:693-704, 1986) и Izaki (Jpn. J. Bacteriol. 33: 729-742, 1978).
Подавление экспрессии конкретных генов является важным инструментом как для исследования, так и для разработки сконструированных способами генетической инженерии организмов, более подходящих для конкретной цели. Подавление гена можно осуществлять путем введения трансгена, соответствующего представляющему интерес гену, в антисмысловой ориентации относительно его промотора (см., например, Sheehy et al., Proc. Nat'l Acad. Sci. USA 85:8805 8808 (1988); Smith et al., Nature 334: 724 726 (1988)) или в смысловой ориентации относительно его промотора (Napoli et al., Plant Cell 2:279 289 (1990); van der Krol et al., Plant Cell 2:291 299 (1990); патент США № 5034323; патент США № 5231020; и патент США № 5283184), причем оба из этих вариантов приводят к сниженной экспрессии трансгена, а также эндогенного гена.
Было описано, что посттрансляционное подавление гена сопровождается накоплением небольших (от 20 до 25 нуклеотидов) фрагментов антисмысловой РНК, которые можно синтезировать с РНК-матрицы, и они являются детерминантами специфичности и подвижности в процессе (Hamilton & Baulcombe, Science 286:950 952 (1999)). Стало очевидным, что в ряде организмов введение дцРНК (dsRNA (двухцепочечная РНК)) является важным компонентом, ведущим к подавлению генов (Fire et al., Nature 391:806 811 (1998); Timmons & Fire, Nature 395:854 (1998); WO99/32619; Kennerdell & Carthew, Cell 95:1017 1026 (1998); Ngo et al., Proc. Nat'l Acad. Sci. USA 95:14687 14692 (1998); Waterhouse et al., Proc. Nat'l Acad. Sci. USA 95:13959 13964 (1998); WO99/53050; Cogoni & Macino, Nature 399:166 169 (1999); Lohmann et al., Dev. Biol. 214:211 214 (1999); Sanchez-Alvarado & Newmark, Proc. Nat'l Acad. Sci. USA 96:5049 5054 (1999)). Не требуется, чтобы в бактериях подавляемый ген представлял собой эндогенный бактериальный ген, поскольку как репортерные трансгены, так и вирусные гены, подвергаются посттранскрипционному подавлению генов с помощью введенных трансгенов (English et al., Plant Cell 8: 179-188 (1996); Waterhouse et al., выше). Однако во всех из описанных выше способах некоторое сходство последовательностей может быть предпочтительным между введенным трансгеном и геном, подвергаемым подавлению.
Ранее введение смыслового трансгена, состоящего из 5'-UTR (“нетранслируемая область”), кодирующей области и 3'-UTR гена оксидазы ACC под контролем промотора CaMV 35S, приводило к активности фермента оксидазы ACC в 15% популяции растений томата (Hamilton et al., Plant J. 15: 737-746 (1998); WO98/53083). Однако если в эту конструкцию включали инвертированные и смысловые повторы части 5'-UTR этой оксидазы ACC, подавление наблюдали в 96% растений (Hamilton et al., выше). Кроме того, происходило подавление другого гена оксидазы ACC, сходного по последовательности с кодирующей областью трансгена, но не с 5'-UTR трансгена, демонстрируя, что двухцепочечная РНК любой части транскрипта нацеливает весь РНК-транскрипт на деградацию. Кроме того, высокая частота и высокий уровень посттранскрипционного подавления генов были выявлены при введении либо конструкций, содержащих инвертированные повторы кодирующих областей вируса, либо репортерных генов, или при скрещивании растений, экспрессирующих смысловые и антисмысловые транскрипты кодирующей области гена-мишени (Waterhouse et al., Proc. Nat'l Acad. Sci. USA 95: 13959-13964 (1998)). Сходные результаты были получены путем экспрессии смысловых и антисмысловых трансгенов под контролем различных промоторов в одном и том же растении (Chuang & Meyerowitz, Proc. Nat'l Acad. Sci. USA 97: 4985-4990 (2000)).
Другие варианты осуществления настоящего изобретения могут включать манипуляции в гене spnK, которые включают подавление гена. Выражение “подавление гена” относится к процессу, посредством которого экспрессия конкретного продукта гена снижается или ослабевает. Подавление гена может происходить различными путями. Если нет иных указаний, как используют в рамках изобретения, подавление генов относится к снижению экспрессии продуктов генов в результате РНК-интерференции (РНК-i) - определенного, хотя и частично охарактеризованного, каскада, посредством которого малая ингибиторная РНК (миРНК (siRNA)) действует согласованно с белками хозяина (например, индуцируемый РНК комплекс подавления, RISC) для деградации матричной РНК (мРНК) зависимым от последовательности образом. Уровень подавления генов можно измерять различными способами, включая, но не ограничиваясь ими, измерение уровней транскрипта нозерн-блот-анализом, способы B-ДНК, чувствительные к транскрипции репортерные конструкции, определение профиля экспрессии (например ДНК-чипы) и сходные технологии. Альтернативно уровень подавления можно измерять путем оценки уровня белка, кодируемого конкретным геном. Это можно осуществлять путем проведения ряда исследований, включающих вестерн-анализ, измерение уровней экспрессии репортерного белка, который обладает, например, флуоресцентными свойствами (например GFP) или ферментативной активностью (например щелочные фосфатазы), или несколько других методик.
Дополнительные варианты осуществления включают замену(ы) одной или нескольких аминокислот в активном центре или участке связывания субстрата гена spnK, которые нарушают ген spnK и приводят к продуцированию спинозина J/L. Как правило, специалистам в данной области будет понятно, что незначительные делеции или замены можно вносить в аминокислотные последовательности пептидов по настоящему изобретению без чрезмерного неблагоприятного влияния на их активность. Таким образом, белки и пептиды, содержащие такие делеции или замены, являются следующим аспектом настоящего изобретения. В пептидах, содержащих замены или замещения аминокислот, одна или несколько аминокислот в последовательности пептида могут быть заменены одной или несколькими другими аминокислотами, где такая замена не влияет на функцию этой последовательности. Такие изменения могут определяться известным сходством между аминокислотами по физическим признакам, таким как плотность заряда, гидрофобность/гидрофильность, размер и конфигурация, так что аминокислоты заменяют другими аминокислотами, обладающими по существу теми же функциональными свойствами. Например: Ala может быть заменен на Val или Ser; Val может быть заменен Ala, Leu, Met или Ile, предпочтительно Ala или Leu; Leu может быть заменен Ala, Val или Ile, предпочтительно Val или Ile; Gly может быть заменен Pro или Cys, предпочтительно Pro; Pro может быть заменен Gly, Cys, Ser или Met, предпочтительно Gly, Cys или Ser; Cys может быть заменен Gly, Pro, Ser или Met, предпочтительно Pro или Met; Met может быть заменен Pro или Cys, предпочтительно Cys; His может быть заменен Phe или Gln, предпочтительно Phe; Phe может быть заменен His, Tyr или Trp, предпочтительно His или Tyr; Tyr may be replaced with His, Phe или Trp, предпочтительно Phe или Trp; Trp может быть заменен Phe или Tyr, предпочтительно Tyr; Asn может быть заменен Gln или Ser, предпочтительно Gln; KGln может быть заменен His, Lys, Glu, Asn или Ser, предпочтительно Asn или Ser; Ser может быть заменен Gln, Thr, Pro, Cys или Ala; Thr может быть заменен Gln или Ser, предпочтительно Ser; Lys может быть заменен Gln или Arg; Arg может быть заменен Lys, Asp или Glu, предпочтительно Lys или Asp; Asp может быть заменен Lys, Arg или Glu, предпочтительно Arg или Glu; и Glu может быть заменен Arg или Asp, предпочтительно Asp. После внесения изменения подвергают скринингу общепринятыми способами для определения их эффектов на функцию.
Другие варианты осуществления настоящего изобретения могут включать манипуляции в гене spnK, которые включают манипуляцию с участками связывания рибосом (RBS). Можно проводить такие манипуляции с участком связывания рибосом (обозначаемый как последовательность Шайна-Дальгарно), который расположен выше кодирующей последовательности spnK, чтобы ген spnK нарушался, что приводит к получению штамма, продуцирующего предшественник спинеторама.
Дополнительные варианты осуществления настоящего изобретения могут включать ферментативное ингибирование, влияющее на множество каскадов передачи сигнала для гена spnK, которое может приводить к получению штамма, продуцирующего спинеторам. Способы детекции ферментативной активности, ассоциированной с мишенью, могут включать использование ферментных анализов.
Другой вариант осуществления настоящего изобретения включает прерывание промоторной последовательности, которая кодирует ген spnK. Такое прерывание может быть осуществлено путем любого типа манипулирования, включая, но не ограничиваясь ими, укорочения, делеции, точковые мутации и инсерции. Такие манипуляции могут быть в рамке считывания или вне рамки считывания. Такие манипуляции приводят к получению штамма, продуцирующего спинеторам.
Настоящее изобретение более подробно объяснено в следующих неограничивающих примерах.
Пример 1: Внесение точковых мутаций в spnK
Точковые мутации вносили в ген spnK случайным мутагенезом штамма Saccharopolyspora spinosa, продуцирующего A и D (Kieser et al., 2000). Далее мутантные штаммы, продуцирующие спинозин J и L вместо спинозин A и D, охарактеризовывали амплификацией способом ПЦР гена spnK с последующим секвенированием ДНК. Ген spnK амплифицировали способом ПЦР с spnKF (SEQ ID NO:1; GGGAATTCCATATGTCCACAACGCACGAGATCGA) и spnKR (SEQ ID NO:2; GCCGCTCGAGCTCGTCCTCCGCGCTGTTCACGTCS) с использованием системы для ПЦР FailSafe PCR System (Epicentre Biotechnologies; Madison, WI). Полученный продукт ПЦР очищали с использованием набора для очистки MoBio Ultraclean PCR Clean-up DNA Purification Kit (MoBio Laboratories; Solana Beach, CA) и клонировали в клонирующий вектор TA с использованием ДНК-лигазы T4 (Invitrogen Life Technologies; Carlsbad, CA). Бактериальные колонии, которые предположительно содержали продукт ПЦР, выделяли и подтверждали расщеплением ферментом рестрикции. Секвенирование ДНК положительных клонов плазмиды проводили, как описано в протоколе изготовителя с использованием набора CEQTM DTCS-Quick Start Kit (Beckman-Coulter; Palo Alto, CA). Реакционные смеси очищали с использованием кассет Performa DTR Gel Filtration Cartridges (Edge BioSystems; Gaithersburg, MD), как описано в протоколах изготовителя. Реакционные смеси для секвенирования анализировали на системе Beckman-Coulter CEQTM 2000 XL DNA Analysis System и охарактеризацию нуклеотидов проводили с использованием SEQUENCHER™ (Gene Codes Corporation; Ann Arbor, MI). Результаты секвенирования подтверждали расположение точковых мутаций в последовательности гена spnK. Полученные точковые мутации приведены в таблице 1 и затемнены на фиг.1.
Ферментацию мутантных по spnK штаммов Saccharopolyspora spinosa можно проводить в условиях, описанных Burns et al. (WO 2003070908). Анализ ферментационного бульона в отношении присутствия спинозиновых факторов можно проводить в условиях, описанных Baltz et al. (патент США № 6143526). Для подтверждения присутствия спинозиновых факторов в супернатанте экстракты ферментационного бульона высушивали в SpeedVac в течение ночи, а затем осадок распределяли между водой и простым эфиром. Слой простого эфира сушили упариванием в потоке N2. Затем образец растворяли в ацетоне-d6 и переносили в пробирку для ЯМР для получения данных 1D протонной ЯМР. Профили ЯМР сравнивали с профилями стандартов спинозина. Результаты ЯМР показали присутствие избытка J/L относительно A/D. Ферментация штаммов, которые содержат точковую мутацию, приводила к смеси спинозинов, содержащей спинозин J и L, по сравнению с контрольным Saccharopolyspora spinosa, который продуцировал смесь спинозинов, содержащую спинозин A и D.
Пример 2: Внесение делеционной мутации spnK
Конструирование вектора для делеции spnK в рамке считывания
Фрагмент ДНК размером 1595 п.н. амплифицировали способом ПЦР с использованием геномной ДНК штамма, продуцирующего спинозин A и D (Hopwood et al., 1985). Этот фрагмент охватывал инициирующий кодон spnK и содержал кодирующую область spnJ без 5'-конца spnJ (фиг.2). Реакцию ПЦР проводили с использованием набора FailSafe PCR kit (Epicentre Biotechnologies; Madison, WI) и Forward Primer #1 (SEQ ID NO:3; CGGTGCCCGAATTCCATGACCCG) и Reverse Primer #1 (SEQ ID NO:4; GTGCGTTCTAGACATATGAGCTCCTCATGGCTG).
Проводили вторую реакцию ПЦР, в которой получали фрагмент ДНК размером 1951 п.н.; этот фрагмент содержал 3'-конец spnK, целый spnL и 5'-конец spnM (фиг.2). Реакцию ПЦР проводили с использованием набора FailSafe PCR kit и Forward Primer #2 (SEQ ID NO:5; GTGCCATCTAGACTGGACGACATATTGCACCTG) и Reverse Primer #2 (SEQ ID NO:6; GAATGCGAAGCT TACGATCTCGTCGTCCGTG). Продукты ПЦР очищали с использованием набора для очистки QIAquick PCR Purification Kit (Qiagen; Valencia, CA) согласно инструкциям изготовителя.
Фрагмент ПЦР размером 1595 п.н. расщепляли с помощью EcoRI и XbaI. Фрагмент ПЦР размером 1951 п.н. расщепляли XbaI и HindIII. После завершения расщепления ферментом рестрикции фрагменты очищали с использованием набора для очистки QIAquick PCR Purification Kit. Расщепленные фрагменты лигировали с соответствующими участками рестрикции EcoRI и HindIII плазмиды pOJ260 с использованием набора для лигирования FastLink DNA Ligation Kit (Epicentre; Madison, WI) и трансформировали в компетентные клетки E. coli TOP10 (Invitrogen; Carlsbad, CA). Колонии отбирали и подвергали скринингу в отношении желаемого продукта лигирования посредством расщепления ферментом рестрикции и анализа последовательности ДНК. Положительные клоны идентифицировали, и отобранный клон использовали для последующей делеции spnK в рамке считывания в Saccharopolyspora spinosa. Полученная последовательность удаленного фрагмента гена spnK в плазмиде pOJ260 представлена в таблице 2.



Таким образом, одна делеция spnK включает последовательность
ATGTCTAGACTGGACGACATATTGCACCTGGCCGACGTGAACAGCGCGGAGGACGAGTGA (SEQ ID NO:9).
Конъюгация вектора с делецией spnK в Saccharopolyspora spinosa
Конструкцию spnK с делецией в рамке считывания трансформировали в штамм E. coli, являющийся донором для конъюгации, ET12567/pUZ8002. Положительно трансформированный штамм идентифицировали и использовали для инокуляции колбы со средой с бульоном Луриа (содержавшей соответствующие антибиотики) для выращивания в течение ночи при 37°C при встряхивании при 225 об/мин. Подтверждение отличительных признаков плазмиды проводили путем выделения плазмидной ДНК и проведения расщепления ферментом рестрикции из донорного штамма E. coli. После подтверждения того, что этот клон является правильным, оставшуюся культуру хранили в 20% глицерине при -80°C для дальнейшего применения.
Конъюгацию клеток E. coli, имеющих конструкцию с делецией spnK в рамке считывания, с Saccharopolyspora spinosa проводили в соответствии со способом, описанным в Matsushima et al., (1994). Отбирали предполагаемые трансконъюгаты, устойчивые к апрамицину, вследствие присутствия маркера в виде гена устойчивости к апрамицину в остове вектора конструкции с делецией в spnK в рамке считывания.
Подтверждение трансконъюгатов и амплификация области spnK для определения участка встраивания
Единый первичный трансконъюгат, выращенный в среде R6, переносили в чашки с агаром с сердечно-мозговой вытяжкой (BHI), дополненным 50 мкг/мл апрамицина и 25 мкг/мл налидиксовой кислоты для подтверждения фенотипа устойчивости. Мицелии трансконъюгатов инокулировали из чашки BHI в среду с триптическим соевым бульоном (TSB), дополненную 50 мкг/мл апрамицина. Культуру инкубировали при 29°C при встряхивании при 250 об/мин в течение 72 часов. Мицелии собирали после инкубации в течение 72 часов и геномную ДНК выделяли с использованием набора для выделения ДНК Edge BioSystem's Genomic DNA Isolation Kit в соответствии с инструкциями изготовителя (Edge Biosystems; Gaithersburgh, MD). ПЦР проводили с использованием геномной ДНК, выделенной из трансконъюгата, в качестве матрицы с праймерами SpnK Del Validation 1Forward (SEQ ID NO:7; GTTCACGGTGATTCCGGTGACTCG) и SpnK Del Validation 1Reverse (SEQ ID NO:8; ACCTGCACTGCTTCCTGGAGCTTC). Кроме того, геномную ДНК, выделенную из исходного контрольного штамма Saccharopolyspora spinosa, и плазмидную ДНК конструкции с делецией spnK в рамке считывания использовали в качестве матриц для контрольной реакции ПЦР. Продукты амплификации способом ПЦР секвенировали. Данные секвенирования показали, что конструкция с делецией spnK в рамке считывания встраивалась в область spnLM путем гомологичной рекомбинации с однократным кроссинговером (фиг.3). Встраивание в область spnLM в хромосоме приводило к неизмененной копии spnJ, spnK, spnL и укороченному spnM выше остова вектора pOJ260 и укороченному spnJ, и неизмененному spnL и spnM ниже остова вектора.
Выделение мутанта с делецией spnK в рамке считывания с двойным кроссинговером
Мутант с однократным кроссинговером, устойчивый к апрамицину, инокулировали на чашки с агаром с BHI в отсутствие апрамицина и инкубировали при 29°C в течение 14 суток. Споры собирали с планшетов в соответствии с Hopwood et al. (1985) и хранили в 20% глицерине при -80°C. Споры инокулировали в десять новых планшетов с агаром с BHI без апрамицина и планшеты инкубировали при 29°C в течение 14 суток. Эту стадию повторяли три раза. Препарат спор разбавляли до 10-6 с использованием 20% глицерина и разбавленные споры высевали на десять чашек с агаром BHI. Планшеты инкубировали при 29°C в течение 10 суток для формирования единичных колоний. Отдельные колонии наносили пятнами на новые чашки с агаром с BHI с апрамицином и без него. Все чашки инкубировали при 29°C в течение 10 суток для развития мицелия. Колонии, которые не росли на чашках с агаром с BHI, содержащим 50 мкг/мл апрамицина, идентифицировали в качестве кандидатов в мутанты с двойным кроссинговером и отбирали для подтверждения с использованием ПЦР.
Идентификация и подтверждение мутантов с двойным кроссинговером
Мутанты с двойным кроссинговером подтверждали с помощью ПЦР. Праймеры: SpnK Del Validation 1Forward (SEQ ID NO:7) и SpnK Del Validation 1Reverse (SEQ ID NO:8), сконструированные так, чтобы они связывались в генах spnL и spnJ, использовали для амплификации способом ПЦР с использованием системы для ПЦР FailSafe PCR System. Размеры продуктов ПЦР определяли с помощью агарозного гель-электрофореза. Мутанты с двойным кроссинговером, которые приводили к делеции гена spnK (фиг.4), идентифицировали и отбирали на основании размера продукта ПЦР. Размер и последовательность ДНК фрагмента ПЦР указывает на делецию гена spnK в рамке считывания.
Продуцирование спинозина мутантами с двойным кроссинговером путем ферментации во вращающихся колбах
Ферментацию мутанта с двойным кроссинговером можно проводить в условиях, описанных Burns et al. (WO 2003070908). Анализ ферментационного бульона в отношении присутствия спинозиновых факторов можно проводить в условиях, описанных Baltz et al. (патент США № 6143526). Для подтверждения присутствия спинозиновых факторов в супернатанте экстракты ферментационного бульона высушивали в SpeedVac в течение ночи, а затем осадок распределяли между водой и простым эфиром. Слой простого эфира сушили упариванием в потоке N2. Затем образец растворяли в ацетоне-d6 и переносили в пробирку для ЯМР для получения данных 1D протонной ЯМР. Профили ЯМР сравнивали с профилями стандартов спинозина. Ферментация мутанта с двойным кроссинговером приводила к продуцированию спинозина J и L. Результаты ЯМР указали на присутствие избытка J/L относительно A/D.
Пример 3: Получение мутации с инсерцией spnK
Мутанты Saccharopolyspora spinosa получают путем мутации с инсерцией в гене spnK. Конструируют фрагмент ДНК, содержащий кассету с геном устойчивости к апрамицину (aac(3)IV) в рамке считывания в гене spnK и непрерывающиеся вышележащие и нижележащие фланкирующие последовательности генов spnJ и spnL (фиг.5). Фрагмент гена aac(3)IV клонируют в плазмиду и трансформируют в штамм E. coli, являющийся донором для конъюгации, ET12567/pUZ8002. Положительно трансформированный штамм идентифицируют и используют для инокуляции колбы со средой с бульоном Луриа (содержащей соответствующие антибиотики) для выращивания в течение ночи при 37°C при встряхивании при 225 об/мин. Подтверждение отличительных признаков плазмиды проводят путем выделения плазмидной ДНК и проведения расщепления ферментом рестрикции. После подтверждения того, что плазмида, содержащая кассету для встраивания гена устойчивости к апрамицину, является правильной, оставшуюся культуру хранят в 20% глицерине при -80°C.
Конъюгацию донорных клеток E. coli с Saccharopolyspora spinosa проводят в соответствии со способом, описанным в Matsushima et al., (1994). Перенос кассеты с геном устойчивости к апрамицину из E. coli и последующее встраивание этой плазмиды в геном Saccharopolyspora spinosa определяют с использованием устойчивости к апрамицину.
Единый первичный трансконъюгат выращивают в среде R6 и переносят в чашки с агаром с сердечно-мозговой вытяжкой (BHI), дополненным 50 мкг/мл апрамицина и 25 мкг/мл налидиксовой кислоты для подтверждения фенотипа устойчивости. Мицелии трансконъюгатов инокулируют из чашки BHI в среду с триптическим соевым бульоном (TSB), дополненную 50 мкг/мл апрамицина. Культуру инкубируют при 29°C при встряхивании при 250 об/мин в течение 72 часов. Мицелии собирают после инкубации в течение 72 часов и геномную ДНК выделяют с использованием набора для выделения ДНК Edge BioSystem's Genomic DNA Isolation Kit в соответствии с инструкциями изготовителя (Edge Biosystems; Gaithersburgh, MD). ПЦР проводят с использованием геномной ДНК, выделенной из трансконъюгата, в качестве матрицы. Желаемый продукт ПЦР клонируют в плазмиду с использованием технологии клонировании TOPO® Cloning Technology (Invitrogen; Carlsbad CA). Бактериальные колонии, которые предположительно содержат продукт ПЦР, клонированный в вектор TOPO®, выделяют и подтверждают расщеплением ферментом рестрикции. Проводят секвенирование ДНК положительных клонов плазмид. Результаты секвенирования показывают, что кассета устойчивости к апрамицину встраивается в ген spnK Saccharopolyspora spinosa путем гомологичной рекомбинации с двойным кроссинговером. Полученное встраивание путем гомологичной рекомбинации нарушает транскрипцию spnK, тем самым устраняя функцию гена spnK.
Ферментацию мутантных по spnK штаммов Saccharopolyspora spinosa можно проводить в условиях, описанных Burns et al. (WO 2003070908). Анализ ферментационного бульона в отношении присутствия спинозиновых факторов можно проводить в условиях, описанных Baltz et al. (патент США № 6143526). Для подтверждения присутствия спинозиновых факторов в супернатанте экстракты ферментационного бульона высушивают в SpeedVac в течение ночи, а затем осадок распределяют между водой и простым эфиром. Слой простого эфира сушат упариванием в потоке N2. Затем образец растворяют в ацетоне-d6 и переносят в пробирку для ЯМР для получения данных 1D протонной ЯМР. Профили ЯМР сравнивают с профилями стандартов спинозина. Результаты ЯМР показали присутствие избытка J/L относительно A/D. Ферментация штаммов, которые содержат мутацию с инсерцией, приводит к смеси спинозинов, содержащей спинозин J и L, по сравнению с контрольными Saccharopolyspora spinosa, которые продуцируют смесь спинозинов, содержащую спинозин A и D.
Пример 4: Нарушение последовательности Шайна-Дальгарно в spnK
Нарушают последовательность Шайна-Дальгарно, расположенную выше spnK (фиг.6), посредством чего получают снижение трансляции мРНК spnK. Мутантный штамм Saccharopolyspora spinosa, содержащий последовательность spnK с делецией последовательности Шайна-Дальгарено, получают с использованием протокола, сходного с протоколом, описанным в примере 2. Два фрагмента размером по меньшей мере 1500 п.н., расположенные выше и ниже последовательности Шайна-Дальгарно в spnK, амплифицируют способом ПЦР. Эти фрагменты не содержат следующую последовательность 5'-AGGAGCTC-3'. Эти два фрагменты лигируют вместе в плазмиде, такой как pOJ260, которую можно использовать для конъюгации с Saccharopolyspora spinosa.
Желаемую плазмиду трансформируют в штамм E. coli, являющийся донором для конъюгации, ET12567/pUZ8002. Положительно трансформированный штамм подтверждают расщеплением ферментом рестрикции. После подтверждения того, что штамм E. coli, содержащий плазмиду, является правильным, проводят конъюгацию клеток E. coli с Saccharopolyspora spinosa в соответствии со способом, описанным в Matsushima et al., (1994). Перенос плазмиды из донорных клеток E. coli и последующее встраивание этой плазмиды в геном Saccharopolyspora spinosa определяют с использованием устойчивости к антибиотику.
Встраивание плазмиды в хромосому Saccharopolyspora spinosa молекулярно охарактеризовывают амплификацией способом ПЦР определенной области геномной ДНК. В кратком изложении, геномную ДНК выделяют и инсерцию, содержащую последовательность Шайна-Дальгарно spnK, амплифицируют способом ПЦР, клонируют и секвенируют. Данные секвенирования указывают на то, что конструкция с делецией последовательности Шайна-Дальгарно в spnK встраивается в область spnJK путем гомологичной рекомбинацией с однократным кроссинговером.
Получают мутанты с двойным кроссинговером, которые содержат нарушенную последовательность Шайна-Дальгарно в spnK, с использованием протокола, описанного в примере 2. Колонии, которые не способны расти на чашках с агаром с BHI, содержащих антибиотики, которые отбирают благодаря маркеру, присутствующему на остове вектора, идентифицируют в качестве кандидатов в мутанты с двойным кроссинговером и отбирают для подтверждения с использованием ПЦР. Используют праймеры, сконструированные для связывания в гене spnK и spnJ. Полученный продукт ПЦР субклонируют в плазмиду с использованием технологии клонирования TOPO® Cloning Technology (Invitrogen; Carlsbad CA). Бактериальные колонии, которые предположительно содержат продукт ПЦР, клонированный в вектор TOPO®, выделяют и подтверждают расщеплением ферментом рестрикции. Проводят секвенирование ДНК положительных клонов плазмид. Результаты секвенирвоания указывают на то, что нуклеотидная последовательность Шайна-Дальгарно в spnK нарушена в геноме Saccharopolyspora spinosa. Ферментацию штаммов Saccharopolyspora spinosa с мутацией последовательности Штамма-Дальгарно в spnK можно проводить в условиях, описанных Burns et al. (WO 2003070908). Анализ ферментационного бульона в отношении присутствия спинозиновых факторов можно проводить в условиях, описанных Baltz et al. (патент США № 6143526). Для подтверждения присутствия спинозиновых факторов в супернатанте экстракты ферментационного бульона высушивают в SpeedVac в течение ночи, а затем осадок распределяют между водой и простым эфиром. Слой простого эфира сушат упариванием в потоке N2. Затем образец растворяют в ацетоне-d6 и переносят в пробирку для ЯМР для получения данных 1D протонной ЯМР. Результаты ЯМР показали присутствие избытка J/L относительно A/D. Профили ЯМР сравнивают с профилями стандартов спинозина. Ферментация штаммов, которые содержат мутацию в виде делеции последовательности Шайна-Дальгарно в spnK, приводит к смеси спинозинов, содержащей спинозин J и L, по сравнению с контрольными Saccharopolyspora spinosa, которые продуцируют смесь спинозинов, содержащую спинозин A и D.
Пример 5: Снижение экспрессии 3'-O-метилтрансферазы путем подавления spnK с помощью антисмысловой РНК
Конструируют плазмиду, продуцирующую асРНК (антисмысловая РНК), комплементарную кодирующей последовательности spnK. Полученное подавление экспрессии гена spnK приводит к снижению активности spnK.
Кодирующую последовательность для spnK амплифицируют способом ПЦР и клонируют в плазмиду, такую как pOJ260, для встраивания в хромосому Saccharopolyspora spinosa. Альтернативно кодирующую последовательность spnK можно клонировать в плазмиду, которая стабильно поддерживается и реплицируется в цитозоле Saccharopolyspora spinosa. Полученную плазмиду конструируют так, чтобы она продуцировала асРНК spnK путем экспрессии антисмысловой нити spnK с использованием сильного конститутивного бактериального промотора. Эту плазмиду с асРНК spnK трансформируют в штамм E. coli, являющийся донором для конъюгации, ET12567/pUZ8002. Положительно трансформированный штамм подтверждают расщеплением ферментом рестрикции. После подтверждения того, что штамм E. coli, содержащий плазмиду, является правильным, проводят конъюгацию клеток E. coli с Saccharopolyspora spinosa в соответствии со способом, описанным в Matsushima et al., (1994). Перенос плазмиды с асРНК spnK из донорных клеток E. coli Saccharopolyspora spinosa определяют с использованием устойчивости к антибиотику; которая кодируется плазмидой с асРНК spnK. Геномную ДНК выделяют из трансконъюгатов и используют в качестве матрицы для амплификации способом ПЦР в целях подтверждения существования плазмиды.
Ферментацию штаммов Saccharopolyspora spinosa, содержащих плазмиду с асРНК spnK, можно проводить в условиях, описанных Burns et al. (WO 2003070908). Анализ ферментационного бульона в отношении присутствия спинозиновых факторов можно проводить в условиях, описанных Baltz et al. (патент США № 6143526). Для подтверждения присутствия спинозиновых факторов в супернатанте, экстракты ферментационного бульона высушивают в SpeedVac в течение ночи, а затем осадок распределяют между водой и простым эфиром. Слой простого эфира сушат упариванием в потоке N2. Затем образец растворяют в ацетоне-d6 и переносят в пробирку для ЯМР для получения данных 1D протонной ЯМР. Результаты ЯМР показали присутствие избытка J/L относительно A/D. Профили ЯМР сравнивают с профилями стандартов спинозина. Ферментация штаммов, которые содержат плазмиду с асРНК spnK, приводит к смеси спинозинов, содержащей спинозин J и L, по сравнению с контрольными Saccharopolyspora spinosa, которые продуцируют смесь спинозинов, содержащую спинозин A и D.
Пример 6: Получение дополнительных делеционных мутаций в spnK
Пример 6.1 Конструирование вектора с делецией 5'-конца spnK
Геномную ДНК штамма, продуцирующего спинозин A и D (Hopwood et al., 1985), амплифицируют способом ПЦР с получением двух фрагментов ДНК. Первый амплифицированный фрагмент имеет длину приблизительно 1500 п.н. и расположен непосредственно выше инициирующего кодона ATG. Второй амплифицированный фрагмент имеет длину приблизительно 1500 п.н. и расположен непосредственно ниже 61 пары оснований spnK. Амплификации способом ПЦР проводят с использованием способов, известных специалистам в данной области. Олигонуклеотидные праймеры синтезируют так, чтобы они включали последовательности связывания ферментов рестрикции. Полученные продукты ПЦР расщепляют ферментами рестрикции, которые расщепляют последовательности связывания, включаемые с помощью праймеров. Фрагменты лигируют вместе, а затем лигируют в соответствующие участки рестрикции плазмиды pOJ260. Полученный продукт лигирования клонируют в компетентные клетки E. coli. Колонии отбирают и подвергают скринингу в отношении желаемого продукта лигирования путем расщепления ферментом рестрикции и анализа последовательности ДНК. Положительные клоны идентифицируют и отобранный клон используют для последующей делеции 5'-конца spnK в Saccharopolyspora spinosa. Полученная последовательность делетированного фрагмента гена spnK в плазмиде pOJ260 представлена в таблице 3. Таким образом, делеция инициирующего кодона spnK включает последовательность (SEQ ID NO:10).

Конъюгация вектора для spnK с делецией в Saccharopolyspora spinosa
Конъюгацию клеток E. coli, имеющих конструкцию с делецией 5'-конца spnK, с Saccharopolyspora spinosa проводят в соответствии со способом, описанным в Matsushima et al., (1994) и проиллюстрированным в примере 2. Отбирают предполагаемые трансконъюгаты, устойчивые к апрамицину, вследствие присутствия маркера в виде гена устойчивости к апрамицину в остове вектора конструкции с делецией 5'-конца spnK.
Подтверждение трансконъюгатов и амплификация области spnK для определения участка встраивания
Единый первичный трансконъюгат, выращенный в среде R6, переносят в чашки с агаром с сердечно-мозговой вытяжкой (BHI), дополненным 50 мкг/мл апрамицина и 25 мкг/мл налидиксовой кислоты для подтверждения фенотипа устойчивости. Мицелии трансконъюгатов инокулируют из чашки BHI в среду с триптическим соевым бульоном (TSB), дополненную 50 мкг/мл апрамицина. Культуру инкубируют при 29°C при встряхивании при 250 об/мин в течение 72 часов. Мицелии собирают после инкубации в течение 72 часов и геномную ДНК выделяют. ПЦР проводят с использованием геномной ДНК, выделенной из трансконъюгата, в качестве матрицы с праймерами, сконструированными для детекции мутанта с однократным кроссинговером. Полученные продукты амплификации способом ПЦР секвенируют. Данные секвенирования указывают на то, что конструкция с делецией 5'-конца spnK встраивается в область spnJK путем гомологичной рекомбинации с однократным кроссинговером.
Выделение spnK с двойным кроссинговером в мутанте с делецией 5'-конца spnK
Мутант с однократным кроссинговером, устойчивый к апрамицину, инокулируют в чашки с агаром с BHI в отсутствие апрамицина и инкубируют при 29°C в течение 14 суток. Споры собирают с планшетов в соответствии с Hopwood et al., (1985) и хранят в 20% глицерине при -80°C. Споры инокулируют в новые чашки с агаром с BHI без апрамицина и чашки инкубируют при 29°C в течение 14 суток. Эту стадию повторяют несколько раз. Препарат спор разбавляют с использованием 20% глицерина и разбавленные споры высевают на десять чашек с агаром BHI. Планшеты инкубируют при 29°C в течение 10 суток для формирования единичных колоний. Отдельные колонии наносят пятнами на новые чашки с агаром с BHI с апрамицином и без него. Все чашки инкубируют при 29°C в течение 10 суток для развития мицелия. Колонии, которые не росли на чашках с агаром с BHI, содержащих 50 мкг/мл апрамицина, идентифицируют в качестве кандидатов в мутанты с двойным кроссинговером и отбирают для подтверждения с использованием ПЦР.
Идентификация и подтверждение мутантов с двойным кроссинговером
Мутанты с двойным кроссинговером подтверждают с помощью ПЦР. Для амплификации используют праймеры, которые сконструированы так, чтобы они связывались в генах spnJ и spnK. Размеры продуктов ПЦР определяют с помощью агарозного гель-электрофореза. Мутанты с двойным кроссинговером, которые приводят к делеции 5'-конца гена spnK, идентифицируют и отбирают на основании размера продукта ПЦР. Размер и последовательность ДНК фрагмента ПЦР указывает на делецию стартового кодона ATG и 5'-конца гена spnK.
Продуцирование спинозина путем ферментации во вращающихся колбах
Ферментацию мутанта с двойным кроссинговером можно проводить в условиях, описанных Burns et al. (WO 2003070908). Анализ ферментационного бульона в отношении присутствия спинозиновых факторов можно проводить в условиях, описанных Baltz et al. (патент США № 6143526). Ферментация мутанта с двойным кроссинговером приводит к продуцированию спинозина J и L.
Пример 6.2 Конструирование вектора с делецией в spnK в рамке считывания
Два фрагмента ДНК амплифицируют способом ПЦР с использованием геномной ДНК штамма, продуцирующего спинозин A и D (Hopwood et al., 1985). Первый амплифицированный фрагмент имеет длину приблизительно 1500 п.н. и расположен непосредственно выше первого предполагаемого домена S-аденозилметионин-зависимой метилтрансферазы. Второй амплифицированный фрагмент имеет длину приблизительно 1500 п.н. и расположен непосредственно ниже первого предполагаемого домена S-аденозилметионин-зависимой метилтрансферазы. Амплификации способом ПЦР проводят с использованием способов, известных специалистам в данной области. Олигонуклеотидные праймеры синтезируют так, чтобы они включали последовательности связывания ферментов рестрикции. Полученные продукты ПЦР расщепляют ферментами рестрикции, которые расщепляют последовательности связывания, включаемые с помощью праймеров. Фрагменты лигируют вместе, а затем лигируют в соответствующие участки рестрикции плазмиды pOJ260. Полученный продукт лигирования клонируют в компетентные клетки E. coli. Колонии отбирают и подвергают скринингу в отношении желаемого продукта лигирования путем расщепления ферментом рестрикции и анализа последовательности ДНК. Положительные клоны идентифицируют и отобранный клон используют для последующей делеции в рамке считывания первого предполагаемого домена S-аденозилметионин-зависимой метилтрансферазы в spnK в Saccharopolyspora spinosa. Полученная последовательность делетированного фрагмента гена spnK в плазмиде pOJ260 представлена в таблице 4. Таким образом, делеция в spnK включает последовательность SEQ ID NO:11.




Конъюгация вектора для spnK с делецией в Saccharopolyspora spinosa
Конъюгацию клеток E. coli, имеющих конструкцию с делецией в spnK в рамке считывания, с Saccharopolyspora spinosa проводят в соответствии со способом, описанным в Matsushima et al., (1994) и проиллюстрированным в примере 2. Отбирают предполагаемые трансконъюгаты, устойчивые к апрамицину, вследствие присутствия маркера в виде гена устойчивости к апрамицину в остове вектора конструкции с делецией в spnK в рамке считывания.
Подтверждение трансконъюгатов и амплификация области spnK для определения участка встраивания
Единый первичный трансконъюгат, выращенный в среде R6, переносят в чашки с агаром с сердечно-мозговой вытяжкой (BHI), дополненным 50 мкг/мл апрамицина и 25 мкг/мл налидиксовой кислоты для подтверждения фенотипа устойчивости. Мицелии трансконъюгатов инокулируют из чашки BHI в среду с триптическим соевым бульоном (TSB), дополненную 50 мкг/мл апрамицина. Культуру инкубируют при 29°C при встряхивании при 250 об/мин в течение 72 часов. Мицелии собирают после инкубации в течение 72 часов и геномную ДНК выделяют. ПЦР проводят с использованием геномной ДНК, выделенной из трансконъюгата, в качестве матрицы с праймерами, сконструированными для детекции мутанта с однократным кроссинговером. Полученные продукты амплификации способом ПЦР секвенируют. Данные секвенирования показывают, что конструкция с делецией в spnK в рамке считывания встраивается в область spnK путем гомологичной рекомбинации с однократным кроссинговером.
Выделение spnK с двойным кроссинговером в мутанте с делецией в spnK в рамке считывания
Мутант с однократным кроссинговером, устойчивый к апрамицину, инокулируют в чашки с агаром с BHI в отсутствие апрамицина и инкубируют при 29°C в течение 14 суток. Споры собирают с планшетов в соответствии с Hopwood et al., (1985) и хранят в 20% глицерине при -80°C. Споры инокулируют в новые чашки с агаром с BHI без апрамицина и чашки инкубируют при 29oC в течение 14 суток. Эту стадию повторяют несколько раз. Препарат спор разбавляют с использованием 20% глицерина и разбавленные споры высевают на десять чашек с агаром BHI. Планшеты инкубируют при 29°C в течение 10 суток для формирования единичных колоний. Отдельные колонии наносят пятнами на новые чашки с агаром с BHI с апрамицином и без него. Все чашки инкубируют при 29°C в течение 10 суток для развития мицелия. Колонии, которые не растут на чашках с агаром с BHI, содержащих 50 мкг/мл апрамицина, идентифицируют в качестве кандидатов в мутанты с двойным кроссинговером и отбирают для подтверждения с использованием ПЦР.
Идентификация и подтверждение мутантов с двойным кроссинговером
Мутанты с двойным кроссинговером подтверждают с помощью ПЦР. Для амплификации используют праймеры, которые сконструированы так, чтобы они связывались в гене spnK. Размеры продуктов ПЦР определяют с помощью агарозного гель-электрофореза. Мутанты с двойным кроссинговером, которые приводят к делеции первого предполагаемого домена S-аденозилметионин-зависимой метилтрансферазы в гене spnK, идентифицируют и отбирают на основании размера продукта ПЦР. Размер и последовательность ДНК фрагмента ПЦР указывает на делецию первого предполагаемого домена S-аденозилметионин-зависимой метилтрансферазы в гене spnK.
Продуцирование спинозина путем ферментации во вращающихся колбах
Ферментацию мутанта с двойным кроссинговером можно проводить в условиях, описанных Burns et al. (WO 2003070908). Анализ ферментационного бульона в отношении присутствия спинозиновых факторов можно проводить в условиях, описанных Baltz et al. (патент США № 6143526). Ферментация мутанта с двойным кроссинговером приводит к продуцированию спинозина J и L.
Пример 6.3 Конструирование вектора с делецией в spnK в рамке считывания
Два фрагмента ДНК амплифицируют способом ПЦР с использованием геномной ДНК штамма, продуцирующего спинозин A и D (Hopwood et al., 1985). Первый амплифицированный фрагмент имеет длину приблизительно 1500 п.н. и расположен непосредственно выше второго предполагаемого домена S-аденозилметионин-зависимой метилтрансферазы. Второй амплифицированный фрагмент имеет длину приблизительно 1500 п.н. и расположен непосредственно ниже второго предполагаемого домена S-аденозилметионин-зависимой метилтрансферазы. Амплификации способом ПЦР проводят с использованием способов, известных специалистам в данной области. Олигонуклеотидные праймеры синтезируют так, чтобы они включали последовательности связывания ферментов рестрикции. Полученные продукты ПЦР расщепляют ферментами рестрикции, которые расщепляют последовательности связывания, включаемые с помощью праймеров. Фрагменты лигируют вместе, а затем лигируют в соответствующие участки рестрикции плазмиды pOJ260. Полученный продукт лигирования клонируют в компетентные клетки E. coli. Колонии отбирают и подвергают скринингу в отношении желаемого продукта лигирования путем расщепления ферментом рестрикции и анализа последовательности ДНК. Положительные клоны идентифицируют и отобранный клон используют для последующей делеции в рамке считывания второго предполагаемого домена S-аденозилметионин-зависимой метилтрансферазы в spnK в Saccharopolyspora spinosa. Полученная последовательность делетированного фрагмента гена spnK в плазмиде pOJ260 представлена в таблице 5. Таким образом, делеция в spnK включает последовательность SEQ ID NO:12.




Конъюгация вектора для spnK с делецией в Saccharopolyspora spinosa
Конъюгацию клеток E. coli, имеющих конструкцию с делецией в spnK в рамке считывания, с Saccharopolyspora spinosa проводят в соответствии со способом, описанным в Matsushima et al., (1994) и проиллюстрированным в примере 2. Отбирают предполагаемые трансконъюгаты, устойчивые к апрамицину, вследствие присутствия маркера в виде гена устойчивости к апрамицину в остове вектора конструкции с делецией в spnK в рамке считывания.
Подтверждение трансконъюгатов и амплификация области spnK для определения участка встраивания
Единый первичный трансконъюгат, выращенный в среде R6, переносят в чашки с агаром с сердечно-мозговой вытяжкой (BHI), дополненным 50 мкг/мл апрамицина и 25 мкг/мл налидиксовой кислоты для подтверждения фенотипа устойчивости. Мицелии трансконъюгатов инокулируют из чашки BHI в среду с триптическим соевым бульоном (TSB), дополненную 50 мкг/мл апрамицина. Культуру инкубируют при 29°C при встряхивании при 250 об/мин в течение 72 часов. Мицелии собирают после инкубации в течение 72 часов и геномную ДНК выделяют. ПЦР проводят с использованием геномной ДНК, выделенной из трансконъюгата, в качестве матрицы с праймерами, сконструированными для детекции мутанта с однократным кроссинговером. Полученные продукты амплификации способом ПЦР секвенируют. Данные секвенирования показывают, что конструкция с делецией в spnK в рамке считывания встраивается в область spnK путем гомологичной рекомбинации с однократным кроссинговером.
Выделение spnK с двойным кроссинговером в мутанте с делецией в spnK в рамке считывания
Мутант с однократным кроссинговером, устойчивый к апрамицину, инокулируют в чашки с агаром с BHI в отсутствие апрамицина и инкубируют при 29°C в течение 14 суток. Споры собирают с планшетов в соответствии с Hopwood et al., (1985) и хранят в 20% глицерине при -80°C. Споры инокулируют в новые чашки с агаром с BHI без апрамицина и чашки инкубируют при 29°C в течение 14 суток. Эту стадию повторяют несколько раз. Препарат спор разбавляют с использованием 20% глицерина и разбавленные споры высевают на десять чашек с агаром BHI. Планшеты инкубируют при 29°C в течение 10 суток для формирования единичных колоний. Отдельные колонии наносят пятнами на новые чашки с агаром с BHI с апрамицином и без него. Все чашки инкубируют при 29°C в течение 10 суток для развития мицелия. Колонии, которые не растут на чашках с агаром с BHI, содержащих 50 мкг/мл апрамицина, идентифицируют в качестве кандидатов в мутанты с двойным кроссинговером и отбирают для подтверждения с использованием ПЦР.
Идентификация и подтверждение мутантов с двойным кроссинговером
Мутанты с двойным кроссинговером подтверждают с помощью ПЦР. Для амплификации используют праймеры, которые сконструированы так, чтобы они связывались в гене spnK. Размеры продуктов ПЦР определяют с помощью агарозного гель-электрофореза. Мутанты с двойным кроссинговером, которые приводят к делеции второго предполагаемого домена S-аденозилметионин-зависимой метилтрансферазы в гене spnK, идентифицируют и отбирают на основании размера продукта ПЦР. Размер и последовательность ДНК фрагмента ПЦР указывает на делецию второго предполагаемого домена S-аденозилметионин-зависимой метилтрансферазы в гене spnK.
Продуцирование спинозина путем ферментации во вращающихся колбах
Ферментацию мутанта с двойным кроссинговером можно проводить в условиях, описанных Burns et al., (WO 2003070908). Анализ ферментационного бульона в отношении присутствия спинозиновых факторов можно проводить в условиях, описанных Baltz et al. (патент США № 6143526). Ферментация мутанта с двойным кроссинговером приводит к продуцированию спинозина J и L.
Пример 6.3 Конструирование вектора с делецией 3'-конца spnK
Два фрагмента ДНК амплифицируют способом ПЦР с использованием геномной ДНК штамма, продуцирующего спинозин A и D (Hopwood et al., 1985). Первый амплифицированный фрагмент имеет длину приблизительно 1500 п.н. и расположен непосредственно выше 1141 пары оснований spnK. Второй амплифицированный фрагмент имеет длину приблизительно 1500 п.н. и расположен непосредственно ниже кодона терминации spnK и включает часть spnL. Амплификации способом ПЦР проводят с использованием способов, известных специалистам в данной области. Олигонуклеотидные праймеры синтезируют так, чтобы они включали последовательности связывания ферментов рестрикции. Полученные продукты ПЦР расщепляют ферментами рестрикции, которые расщепляют последовательности связывания, включаемые с помощью праймеров. Фрагменты лигируют вместе, а затем лигируют в соответствующие участки рестрикции плазмиды pOJ260. Полученный продукт лигирования клонируют в компетентные клетки E. coli. Колонии отбирают и подвергают скринингу в отношении желаемого продукта лигирования путем расщепления ферментом рестрикции и анализа последовательности ДНК. Положительные клоны идентифицируют и отобранный клон используют для последующей делеции 3'-конца spnK в Saccharopolyspora spinosa. Полученная последовательность делетированного фрагмента гена spnK в плазмиде pOJ260 представлена в таблице 6. Таким образом, делеция в spnK включает последовательность SEQ ID NO:13.

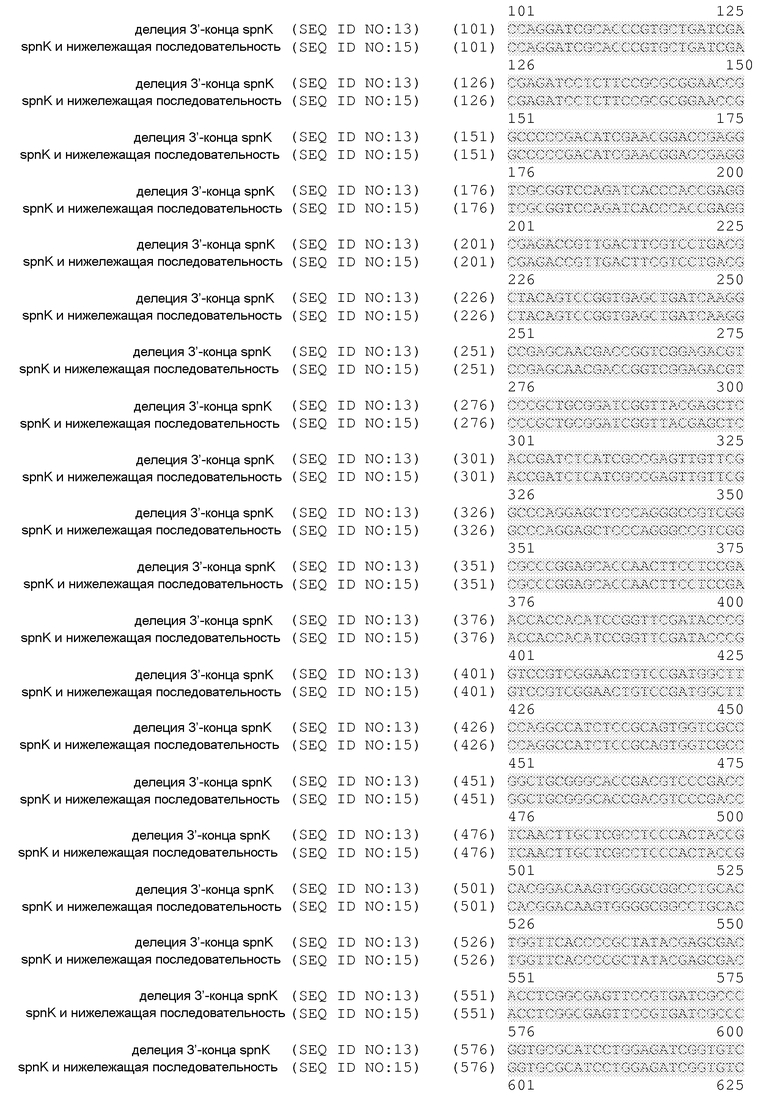

Конъюгация вектора для spnK с делецией в Saccharopolyspora spinosa
Конъюгацию клеток E. coli, имеющих конструкцию с делецией 5'-конца spnK, с Saccharopolyspora spinosa проводят в соответствии со способом, описанным в Matsushima et al., (1994) и проиллюстрированным в примере 2. Отбирают предполагаемые трансконъюгаты, устойчивые к апрамицину, вследствие присутствия маркера в виде гена устойчивости к апрамицину в остове вектора конструкции с делецией 3'-конца spnK.
Подтверждение трансконъюгатов и амплификация области spnK для определения участка встраивания
Единый первичный трансконъюгат, выращенный в среде R6, переносят в чашки с агаром с сердечно-мозговой вытяжкой (BHI), дополненным 50 мкг/мл апрамицина и 25 мкг/мл налидиксовой кислоты для подтверждения фенотипа устойчивости. Мицелии трансконъюгатов инокулируют из чашки BHI в среду с триптическим соевым бульоном (TSB), дополненную 50 мкг/мл апрамицина. Культуру инкубируют при 29°C при встряхивании при 250 об/мин в течение 72 часов. Мицелии собирают после инкубации в течение 72 часов и геномную ДНК выделяют. ПЦР проводят с использованием геномной ДНК, выделенной из трансконъюгата, в качестве матрицы с праймерами, сконструированными для детекции мутанта с однократным кроссинговером. Полученные продукты амплификации способом ПЦР секвенируют. Данные секвенирования показали, что конструкция с делецией 3'-конца spnK встраивается в область spnKL путем гомологичной рекомбинации с однократным кроссинговером.
Выделение spnK с двойным кроссинговером в мутанте с делецией 3'-конца spnK
Мутант с однократным кроссинговером, устойчивый к апрамицину, инокулируют в чашки с агаром с BHI в отсутствие апрамицина и инкубируют при 29°C в течение 14 суток. Споры собирают с планшетов в соответствии с Hopwood et al., (1985) и хранят в 20% глицерине при -80°C. Споры инокулируют в новые чашки с агаром с BHI без апрамицина и чашки инкубируют при 29°C в течение 14 суток. Эту стадию повторяют несколько раз. Препарат спор разбавляют с использованием 20% глицерина и разбавленные споры высевают на десять чашек с агаром BHI. Планшеты инкубируют при 29°C в течение 10 суток для формирования единичных колоний. Отдельные колонии наносят пятнами на новые чашки с агаром с BHI с апрамицином и без него. Все чашки инкубируют при 29°C в течение 10 суток для развития мицелия. Колонии, которые не росли на чашках с агаром с BHI, содержащих 50 мкг/мл апрамицина, идентифицируют в качестве кандидатов в мутанты с двойным кроссинговером и отбирают для подтверждения с использованием ПЦР.
Идентификация и подтверждение мутантов с двойным кроссинговером
Мутанты с двойным кроссинговером подтверждают с помощью ПЦР. Для амплификации используют праймеры, которые сконструированы так, чтобы они связывались в генах spnK и spnL. Размеры продуктов ПЦР определяли с помощью агарозного гель-электрофореза. Мутанты с двойным кроссинговером, которые приводят к делеции 3'-конца гена spnK, идентифицируют и отбирают на основании размера продукта ПЦР. Размер и последовательность ДНК фрагмента ПЦР указывает на делецию и 3'-конца гена spnK.
Продуцирование спинозина путем ферментации во вращающихся колбах
Ферментацию мутанта с двойным кроссинговером можно проводить в условиях, описанных Burns et al. (WO 2003070908). Анализ ферментационного бульона в отношении присутствия спинозиновых факторов можно проводить в условиях, описанных Baltz et al. (патент США № 6143526). Ферментация мутанта с двойным кроссинговером приводит к продуцированию спинозина J и L.
Все упоминаемые патенты и публикации включены в настоящее описание в качестве ссылок в полном объеме. Вышеуказанное иллюстрирует настоящее изобретение, и его не следует истолковывать как его ограничение. Изобретение определяется следующей формулой изобретения, и она включает эквиваленты пунктов формулы изобретения.








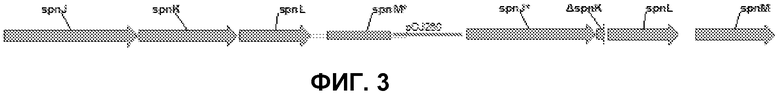
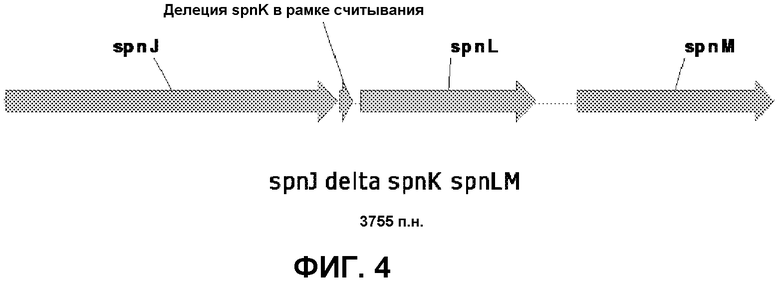
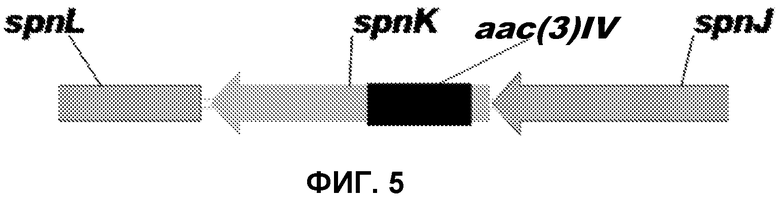
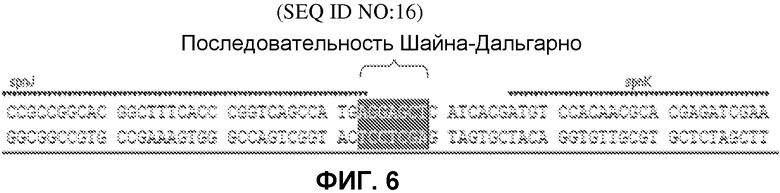
Изобретения относятся к области молекулярной генетики и касаются способов преобразования продуцирующего спиносад штамма Saccharopolyspora spinosa в штамм, продуцирующий предшественника спинеторама (варианты), и генетически модифицированной клетки-хозяина Saccharopolyspora spinosa. Охарактеризованные способы включают внесение модификации в ген spnK или выключение гена spnK при сохранении продуцирования спинозина J и L для устранения активности 3′-O-метилтрансферазы с получением генетически модифицированной клетки-хозяина Saccharopolyspora spinosa, продуцирующей предшественник спинеторама. Причем указанная модификация является точковой мутацией, находящейся в положении пары оснований, выбранном из группы, состоящей из положений 528, 589, 602, 668, 721, 794, 862, 895, 908, 937 и 1131. Изобретения позволяют продуцировать спинозин с большей эффективностью. 3 н. и 2 з.п.ф-лы, 6 ил., 6 табл., 6 пр.
1. Способ преобразования продуцирующего спиносад штамма Saccharopolyspora spinosa в штамм, продуцирующий предшественника спинеторама, включающий внесение модификации в ген spnK для устранения активности 3′-O-метилтрансферазы, где указанная модификация является точковой мутацией, находящейся в положении пары оснований, выбранном из группы, состоящей из положений 528, 589, 602, 668, 721, 794, 862, 895, 908, 937 и 1131.
2. Способ по п. 1, где точковая мутация получена в результате химического мутагенеза.
3. Способ по п. 1, где ген spnK выключен с использованием антисмысловой технологии.
4. Способ преобразования продуцирующего спиносад штамма Saccharopolyspora spinosa в штамм, продуцирующий предшественник спинеторама, включающий выключение гена spnK при сохранении продуцирования спинозина J и L, где выключение гена spnK осуществляют точковой мутацией, находящейся в положении пары оснований, выбранном из группы, состоящей из положений 528, 589, 602, 668, 721, 794, 862, 895, 908, 937 и 1131.
5. Генетически модифицированная клетка-хозяин Saccharopolyspora spinosa, которая продуцирует предшественник спинеторама, где указанная клетка получена способом по п. 1 или 4 и имеет модификацию в гене spnK для устранения активности 3′-О-метилтрансферазы, где указанная модификация является точковой мутацией, находящейся в положении пары оснований, выбранном из группы, состоящей из положений 528, 589, 602, 668, 721, 794, 862, 895, 908, 937 и 1131.
| HUANG KX et al., Recent advances in the biochemistry of spinosyns, Appl Microbiol Biotechnol., 2009 Feb, Vol.82, No.1, pp.13-23 | |||
| HAK JOONG KIM et al., The Biosynthesis of Spinosyn in Saccharopolyspora spinosa: Synthesis of Permethylated Rhamnose and Characterization of the Functions of SpnH, SpnI, and SpnK, J Am Chem Soc., 2010 March 10, Vol.132, No.9, pp.2901-2903; | |||
| CN 1986768 A, 27.06.2007 | |||
| СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЯ ДЛЯ УНИЧТОЖЕНИЯ НАСЕКОМЫХ ИЛИ КЛЕЩЕЙ (ВАРИАНТЫ), ИНСЕКТИЦИДНАЯ И ПРОТИВОКЛЕЩЕВАЯ КОМПОЗИЦИЯ, СПОСОБ УНИЧТОЖЕНИЯ НАСЕКОМЫХ ИЛИ КЛЕЩЕЙ, ШТАММ SACCHAROPOLYSPORA SPINOSA, ИСПОЛЬЗУЕМЫЙ ДЛЯ ПОЛУЧЕНИЯ СОЕДИНЕНИЯ ДЛЯ УНИЧТОЖЕНИЯ НАСЕКОМЫХ ИЛИ КЛЕЩЕЙ | 1992 |
|
RU2165704C2 |

