РОДСТВЕННЫЕ ЗАЯВКИ
Данной заявкой является не предварительная заявка, поданная по 37 CFR 1,53(b)(1), испрашивающая приоритет по 35 USC 119(e) для предварительной заявки номер 61/316326, поданной 22 марта 2010 года, содержимое которой включено в данную заявку посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Изобретение относится к использованию соединений без поверхностно-активных веществ, включая, например, полиоксиэтилен (ПОЭ) сорбитаны и полиэтиленгликоли (ПЭГ), для стабилизации белоксодержащих готовых форм и для предотвращения агрегации белков в подобных готовых формах.
УРОВЕНЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
Когда с целью защиты белка от денатурации при встряхивании, взбалтывании, расщеплении и замерзании-оттаивании, или в состоянии покоя на границе раздела для белкового препарата необходим стабилизатор, часто используют неионный детергент (т.е., поверхностно-активное вещество) (см., например, патент США № 5183746). Примером этого является использование полисорбатов во многих белоксодержащих продуктах. Например, полисорбаты 20 и 80 (Tween® 20 и Tween® 80) используют в составе биотерапевтических продуктов как для предотвращения поверхностной адсорбции, так и в качестве стабилизаторов против агрегации белка (Kerwin, J. Pharm. Sci. 97(8):2924-2936 (2008)). Полисорбаты представляют собой амфипатические, неионные поверхностно-активные вещества, состоящие из сложных эфиров жирных кислот полиоксиэтилен (ПОЭ) сорбитана, представляющего собой полиоксиэтиленсорбитана монолаурат для полисорбата 20 и полиоксиэтиленсорбитана моноолеат для полисорбата 80.
К сожалению, однако, полисорбаты могут подвергаться распаду либо посредством окисления, либо гидролиза. Когда молекула полисорбата распадается, она образует различные побочные продукты распада, содержащие, например, жирные кислоты, ПОЭ сорбитан, ПЭГ, ПЭГ сложные эфиры и алкиловые кислоты. Некоторые из этих побочных продуктов распада полисорбата, включая свободные жирные кислоты, могут являться причиной повышения мутности и агрегации белка в белоксодержащих готовых формах. Вследствие этого, несмотря на то, что полисорбаты широко используются в качестве стабилизаторов белка, жирные кислоты и другие побочные продукты распада, высвобождающиеся со временем при распаде полисорбата, могут неблагоприятно влиять на защитное действие, которое полисорбаты демонстрируют в белоксодержащих готовых формах.
В связи с этим существует потребность в дополнительных композициях, используемых для предотвращения агрегации белков в белоксодержащих водных готовых формах.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Представленное изобретение основано на новом открытии, что полиоксиэтилен (ПОЭ) сорбитаны и полиэтиленгликоли (ПЭГ), присутствующие в определенных концентрациях в водной готовой форме, применимы для стабилизации белок- и пептидсодержащих готовых форм и для предотвращения агрегации белков в подобных готовых формах.
Соответственно, в одном аспекте, изобретение относится к композиции, содержащей антитело или другой белок или пептид и ПОЭ сорбитан. В одном варианте осуществления, полиоксиэтиленсорбитан присутствует в концентрации, составляющей от приблизительно 20 м.д. до приблизительно 100000 м.д. В еще одном варианте осуществления, композиция, содержащая антитело или другой белок или пептид и ПОЭ сорбитан, не содержит неионогенные поверхностно-активные вещества, например, не содержит полисорбат. Необязательно, композиция также может не содержать лиопротектанты, такие как, например, сахара, свободные аминокислоты и их варианты и т.д. В некоторых вариантах осуществления, композиция может быть в водной или твердой форме.
В еще одном аспекте, изобретение относится к композиции, содержащей антитело, или другой белок или пептид, и полиэтиленгликоль. В некоторых вариантах осуществления, полиэтиленгликоль присутствует в указанной композиции в концентрации, ниже чем приблизительно 10000 м.д., предпочтительно между приблизительно 20 м.д. и приблизительно 10000 м.д. В еще одном варианте осуществления, композиция необязательно содержит по меньшей мере один ПОЭ сорбитан, который необязательно может присутствовать в указанной композиции в концентрации, составляющей от приблизительно 20 м.д. до приблизительно 100000 м.д. Необязательно, композиция может также не содержать неионогенные поверхностно-активные вещества, например, не содержать полисорбат. Необязательно, композиция также может не содержать лиопротектанты, такие как, например, сахара, свободные аминокислоты и их варианты и т.д. В некоторых вариантах осуществления, композиция может быть в водной или твердой форме.
В еще одном аспекте, изобретение относится к изделию, содержащему контейнер, вмещающий любую из описанных в данном случае композиций.
В еще одном аспекте, предоставлен способ получения стабилизированного антитела (или другого белка или пептида), включающий композицию посредством смешивания антитела или другого белка вместе с ПОЭ сорбитаном или ПЭГ. В еще одном аспекте, представленное изобретение направлено на способ повышения стабильности антитела или другого белка или пептида в водной готовой форме, включающий смешивание антитела или другого белка или пептида, со стабилизирующим количеством ПОЭ сорбитана, при этом ПОЭ сорбитан повышает стабильность антитела или другого белка или пептида. В одном варианте осуществления, ПОЭ сорбитан присутствует в концентрации, составляющей от приблизительно 20 м.д. до приблизительно 100000 м.д., при этом необязательно может не содержать неионогенные поверхностно-активные вещества, например, не содержит полисорбат, или лиопротектанты, такие как, например, сахара, свободные аминокислоты и их варианты и т.д.
В еще одном аспекте, представленное изобретение направлено на способ повышения стабильности антитела в водной готовой форме, или другого белка или пептида, включающий смешивание антитела, или другого белка или пептида, со стабилизирующим количеством полиэтиленгликоля, при этом полиэтиленгликоль повышает стабильность антитела или другого белка или пептида. В одном варианте осуществления, полиэтиленгликоль присутствует в концентрации, составляющей ниже чем приблизительно 10000 м.д., предпочтительно между приблизительно 20 м.д. и приблизительно 10000 м.д. В еще одном варианте осуществления, композиция необязательно содержит по меньшей мере один ПОЭ сорбитан, который может необязательно присутствовать в концентрации, составляющей от приблизительно 20 м.д. до приблизительно 100000 м.д., и необязательно может не содержать неионогенные поверхностно-активные вещества, например, не содержит полисорбат, или лиопротектанты, такие как, например, сахара, свободные аминокислоты и их варианты и т.д.
В еще одном аспекте, представленное изобретение направлено на способ предотвращения или уменьшения агрегации антитела, или другого белка или пептида, в водной готовой форме, включающий смешивание антитела, или другого белка или пептида, с ПОЭ сорбитаном. В одном варианте осуществления, ПОЭ сорбитан присутствует в концентрации, составляющей от приблизительно 20 м.д. до приблизительно 100000 м.д. Водная готовая форма необязательно не содержит неионогенные поверхностно-активные вещества, например, не содержит полисорбат, или лиопротектанты, такие как, например, сахара, свободные аминокислоты и их варианты и т.д. В еще одном варианте осуществления, агрегацию антитела или другого белка индуцируют взбалтыванием водного раствора.
В еще одном аспекте, представленное изобретение направлено на способ предотвращения или уменьшения агрегации антитела, или другого белка или пептида, в водной готовой форме, включающий смешивание антитела, или другого белка или пептида, с полиэтиленгликолем. В одном варианте осуществления, полиэтиленгликоль присутствует в концентрации, ниже чем приблизительно 10000 м.д., предпочтительно между приблизительно 20 м.д. и приблизительно 10000 м.д. В еще одном варианте осуществления, водная готовая форма необязательно содержит по меньшей мере один ПОЭ сорбитан, который может необязательно присутствовать в концентрации, составляющей от приблизительно 20 м.д. до приблизительно 100000 м.д., и необязательно может не содержать неионогенные поверхностно-активные вещества, например, не содержит полисорбат, или лиопротектанты, такие как, например, сахара, свободные аминокислоты и их варианты и т.д. В еще одном варианте осуществления, агрегацию антитела или другого белка индуцируют взбалтыванием водного раствора.
В способе очистки белка, пептида или антитела от рекомбинантных клеточных белков или других загрязняющих белков, вариант осуществления изобретения относится к усовершенствованию, включающему добавление ПОЭ сорбитана или полиэтиленгликоля к белку, пептиду или антителу во время процесса очистки.
Еще одним аспектом изобретения является способ изготовления неагрегированных водных растворов аутоагрегирующего иным образом антитела или другого белка или пептида посредством смешивания по меньшей мере одного ПОЭ сорбитана или полиэтиленгликоля в водном растворе, содержащем аутоагрегирующее антитело или другой белок или пептид, а затем концентрирования водного раствора.
Другие признаки и преимущества изобретения будут очевидны из следующего подробного описания и примеров, которые не следует трактовать, как ограничивающие. Содержание всех ссылок, патентов и опубликованных патентных заявок, цитируемых на протяжении данной заявки, в прямой форме включено в данную заявку посредством ссылки.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фигура 1 показывает результаты, полученные из анализа мутности антитела анти-IL13 в комбинации с различными добавками, включая полисорбат 20 (PS), лауриновую кислоту (LA) или ПОЭ сорбитан 20 (ПОЭ).
Фигура 2 показывает результаты, полученные из анализа концентрации белка антитела анти-IL13 в комбинации с различными добавками, включая ПОЭ сорбитан 20, ПЭГ 1000 или ПЭГ 6000, все с различными концентрациями.
Фигура 3 показывает результаты, полученные из анализа концентрации белка антитела анти-IgE в комбинации с различными добавками, включая ПОЭ сорбитан 20, ПЭГ 1000 или ПЭГ 6000, все с различными концентрациями.
Фигура 4 показывает результаты, полученные из анализа мутности антитела анти-IL13 в комбинации с различными добавками, включая ПОЭ сорбитан 20, ПЭГ 1000 или ПЭГ 6000, все с различными концентрациями.
Фигура 5 показывает результаты, полученные из анализа мутности антитела анти-IgE в комбинации с различными добавками, включая ПОЭ сорбитан 20, ПЭГ 1000 или ПЭГ 6000, все с различными концентрациями.
Фигура 6 показывает результаты, полученные из анализа размера частиц (частиц больше, чем 2 мкм) антитела анти-IL13 в комбинации с различными добавками, включая ПОЭ сорбитан 20, ПЭГ 1000 или ПЭГ 6000, все с различными концентрациями.
Фигура 7 показывает результаты, полученные из анализа размера частиц (частиц больше, чем 2 мкм) антитела анти-IgE в комбинации с различными добавками, включая ПОЭ сорбитан 20, ПЭГ 1000 или ПЭГ 6000, все с различными концентрациями.
Фигура 8 показывает результаты, полученные из анализа размера частиц (частиц больше, чем 10 мкм) антитела анти-IL13 в комбинации с различными добавками, включая ПОЭ сорбитан 20, ПЭГ 1000 или ПЭГ 6000, все с различными концентрациями.
Фигура 9 показывает результаты, полученные из анализа размера частиц (частиц больше, чем 10 мкм) антитела анти-IgE в комбинации с различными добавками, включая ПОЭ сорбитан 20, ПЭГ 1000 или ПЭГ 6000, все с различными концентрациями.
Фигура 10 показывает результаты, полученные из анализа размера частиц (частиц больше, чем 50 мкм) антитела анти-IL13 в комбинации с различными добавками, включая ПОЭ сорбитан 20, ПЭГ 1000 или ПЭГ 6000, все с различными концентрациями.
Фигура 11 показывает результаты, полученные из анализа размера частиц (частиц больше, чем 50 мкм) антитела анти-IgE в комбинации с различными добавками, включая ПОЭ сорбитан 20, ПЭГ 1000 или ПЭГ 6000, все с различными концентрациями.
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНОГО ВАРИАНТА ОСУЩЕСТВЛЕНИЯ
Представленное изобретение может быть понято более легко с помощью ссылки на следующее подробное описание специфических вариантов осуществления и содержащихся в них примеров.
Если не определено иное, все технические и научные термины, использованные в данном случае, имеют такое же значение, которое широко понятно рядовым специалистам в области, к которой относится данное изобретение. Хотя при практическом использовании или испытании изобретения могут быть использованы любые способы и материалы, аналогичные или эквивалентные способам и материалам, описанным в данном случае, далее описаны предпочтительные способы и материалы. Все публикации, упомянутые в данном случае, включены в данную заявку посредством ссылки во всей полноте.
Агрегация антител и других белков вызывается главным образом гидрофобными взаимодействиями, которые в конечном счете приводят к денатурации. Когда гидрофобная область частично или полностью развернутого белка подвергается воздействию воды, это создает термодинамически неблагоприятную ситуацию, вследствие того, что обычно скрытая гидрофобная внутренняя часть теперь подвергается воздействию гидрофильной водной среды. Следовательно, уменьшение энтропии в результате структурирования молекул воды вокруг гидрофобной области вызывает агрегирование денатурированного белка, главным образом через подвергнутые воздействию гидрофобные области. Таким образом, также может быть подвергнута риску растворимость белка. В некоторых случаях, самоассоциация белковых субъединиц, либо нативных, либо неправильно свернутых, может происходить в определенных условиях, и это может приводить к осаждению и потере активности.
Факторы, которые влияют на агрегацию белка в растворе, обычно включают концентрацию белка, pH, температуру, другие эксципиенты и механические напряжения. Некоторые факторы (например, температуру) можно более легко контролировать в процессе очистки, составления смеси, изготовления, хранения и применения, чем другие (например, механические напряжения). Исследования технологии приготовления лекарственных средств будут предписывать правильный выбор (выборы) pH и эксципиентов, которые не будут вызывать агрегацию и/или, в действительности, будут содействовать предотвращению агрегации. Концентрация белка предписывается необходимой терапевтической дозой и, в зависимости от того, какой является данная концентрация, будет определять, существует ли возможность повышенных объединенных состояний (димеры, тетрамеры и т.д.), которые затем могут приводить к агрегации в растворе. в процессе разработки технологии приготовления лекарственного средства должны быть проведены тщательные исследования с целью определения, какие факторы оказывают влияние на агрегацию белка, а затем, как эти факторы можно устранить или контролировать.
Необходимость идентифицирования препаратов антитела или другого белка со стабильным раствором для использования при парентеральном или ином введении может приводить к разработке методики испытаний для оценки влияния различных добавок на физическую стабильность. Основываясь на известных факторах, влияющих на агрегацию белка, и требованиях к подобному применению, физическую стабильность можно оценивать, используя механические процедуры, включающие взбалтывание или вращение белковых растворов. Методология для испытания с физическим напряжением с целью идентификации способности различных добавок предотвращать агрегацию может включать подвергание встряхиванию или помешиванию в горизонтальной плоскости или вращению x см от оси колеса, вращающегося при n об/мин в вертикальной плоскости. мутность, являющаяся результатом агрегации, обычно определяют, как функцию времени при визуальном осмотре или посредством анализа рассеяния света. В качестве альтернативы, уменьшение содержания растворимого белка вследствие осаждения может быть количественно оценено посредством анализа ЖХВР в качестве функции времени.
Представленное изобретение основано на новом открытии, что полиоксиэтилен (ПОЭ) сорбитаны и полиэтиленгликоли (ПЭГ), присутствующие в определенных концентрациях в жидкой готовой форме, применимы для стабилизации белоксодержащих готовых форм и для предотвращения агрегации белков в подобных готовых формах.
Соответственно, в одном аспекте, представленное изобретение описывает композиции, содержащие антитело или другой белок, либо в высокой, либо в низкой концентрации, и ПОЭ сорбитан. Как используется в данном описании, «полиоксиэтиленсорбитан» или «ПОЭ сорбитан» относится к соединению не являющемуся поверхностно-активным веществом, имеющего следующую химическую структуру:
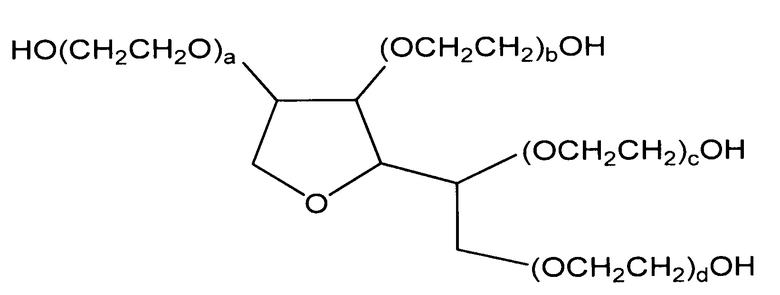 ,
,
где a+b+c+d предпочтительно составляет от приблизительно 6 до приблизительно 80, более предпочтительно от приблизительно 8 до приблизительно 60, еще более предпочтительно от приблизительно 10 до приблизительно 40, и еще более предпочтительно от приблизительно 10 до приблизительно 20. В отношении сказанного выше, в данной области понятно, что результатом химического синтеза соединений, таких как ПОЭ сорбитаны, описанные в данном документе, является весьма гетерогенная смесь соединений, а не полностью гомогенный препарат. В связи с этим, когда в данном случае описано, что a+b+c+d, например, предпочтительно составляет приблизительно от 6 до 80, необходимо учитывать что данное определение относится к большинству составных элементов гетерогенной смеси, которые являются результатом их химического синтеза.
ПОЭ сорбитаны могут быть использованы раздельно в качестве агента, стабилизирующего антитело или другой белок, или могут быть использованы в комбинации с другими ПОЭ сорбитанами для стабилизации антитела или другого белка в водном растворе. ПОЭ сорбитаны находят применение в качестве агентов, стабилизирующих антитело или другой белок (или антиагрегационных) в широком диапазоне концентраций в водном растворе. В некоторых вариантах осуществления представленного изобретения, ПОЭ сорбитан (если используется в качестве единственного стабилизирующего агента) или ПОЭ сорбитаны (если используются в комбинации) может присутствовать в водной содержащей антитело или другой белок готовой форме в концентрации, составляющей от приблизительно 20 м.д. до приблизительно 100000 м.д., более предпочтительно от 100 м.д. до приблизительно 50000 м.д., еще более предпочтительно от 150 м.д. до приблизительно 10000 м.д., еще более предпочтительно от 200 м.д. до приблизительно 5000 м.д., еще более предпочтительно от 200 м.д. до приблизительно 1000 м.д.
В еще одном аспекте, представленное изобретение описывает композиции, содержащие антитело или другой белок либо в высокой, либо в низкой концентрации, и полиэтиленгликоль.
Как используется в данном описании, «полиэтиленгликоль», «ПЭГ» и аналогичные термины предназначены охватывать полиэтиленгликоль и различные его производные, такие как метокси-ПЭГ-амин, диамин-ПЭГ и тому подобное. Более конкретно и в некоторых вариантах осуществления представленного изобретения, термин «полиэтиленгликоль» или «ПЭГ» относится к соединению, не являющемуся поверхностно-активным веществом, имеющему следующую химическую структуру:
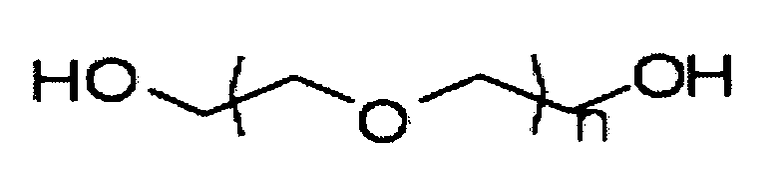 ,
,
где n составляет от приблизительно 5 до приблизительно 240 и может необязательно включать некоторую степень ненасыщенности. ПЭГ, предназначенные для использования в представленном изобретении, могут быть разветвленными или линейными, предпочтительно линейными. В отношении сказанного выше, в данной области понятно, что результатом химического синтеза соединений, таких как ПЭГ, описанных в данном случае, является весьма гетерогенная смесь соединений, а не полностью гомогенный препарат. В связи с этим, когда в данном случае описано, что n предпочтительно составляет от приблизительно 5 до 240, должно быть понятно, что данное определение относится к большинству составных элементов гетерогенной смеси, которые являются результатом их химического синтеза.
ПЭГ могут быть использованы раздельно в качестве агента, стабилизирующего антитело или другой белок, или могут быть использованы в комбинации с другими ПЭГ для стабилизации антитела или другого белка в водном растворе. ПЭГ находят применение в качестве агентов, стабилизирующих антитело или другой белок (или антиагрегационных) в широком диапазоне концентраций в водном растворе. В некоторых вариантах осуществления представленного изобретения, ПЭГ (если используется в качестве единственного стабилизирующего агента) или ПЭГ (если используются в комбинации) может присутствовать в водной готовой форме, содержащей антитело или другой белок, в концентрации, равной менее чем приблизительно 10000 м.д., предпочтительно от приблизительно 20 м.д. до приблизительно 10000 м.д., более предпочтительно от приблизительно 200 м.д. до приблизительно 10000 м.д., более предпочтительно от приблизительно 200 м.д. до приблизительно 5000 м.д., более предпочтительно от приблизительно 200 м.д. до приблизительно 1000 м.д., более предпочтительно от приблизительно 200 м.д. до приблизительно 500 м.д.
Предпочтительные ПЭГ включают полимеры молекулярной массы, составляющей приблизительно 200-12000, но полимеры с более высокой молекулярной массой также находятся в пределах объема правовых притязаний изобретения. ПЭГ включает линейные и разветвленные полимеры, молекулы в виде звезд и ПЭГ блок-сополимеры, образованные посредством соединения по меньшей мере двух различных ПЭГ полимеров с образованием полимера с более высокой молекулярной массой, все из которых хорошо известны в данной области.
Под термином «полипептид» или «белок» подразумевается последовательность аминокислот, для которой длина цепи является достаточной для получения более высоких уровней третичной и/или четвертичной структуры. Таким образом, белки отличаются от «пептидов», которые также представляют собой основанные на аминокислотах молекулы, которые не имеют подобной структуры. Как правило, белок для использования в данном случае будет иметь молекулярную массу, равную по меньшей мере приблизительно 5-20 кД, в качестве альтернативы по меньшей мере приблизительно 15-20 кД, предпочтительно по меньшей мере приблизительно 20 кД. «Пептид» подразумевает последовательность аминокислот, которая в общем не демонстрирует более высокий уровень третичной и/или четвертичной структуры. В общем, пептиды имеют молекулярную массу, равную менее чем приблизительно 5 кД.
Примеры полипептидов, попадающих в пределы определения в данном случае, включают белки млекопитающих, такие как, например, ренин; гормон роста, включая человеческий гормон роста и бычий гормон роста; фактор, стимулирующий выделение гормона роста; паратиреоидный гормон; тиреостимулирующий гормон; липопротеины; альфа-1-антитрипсин; A-цепь инсулина; B-цепь инсулина; проинсулин; фолликулостимулирующий гормон; кальцитонин; лютеинизирующий гормон; глюкагон; факторы свертывающей системы крови, такие как фактор VIIIC, фактор IX, тканевой фактор и фактор фон Виллебранда; факторы, препятствующие свертыванию крови, такие как C-белок; предсердный натрийуретический фактор; легочный сурфактант; активатор плазминогена, такой как активатор плазминогена урокиназного типа человеческой мочи или тканевого типа (t-PA); бомбезин; тромбин; гемопоэтический фактор роста; фактор некроза опухоли альфа-и-бета; энкефалиназа; RANTES (хемокин, выделяемый T-клетками при активации); человеческий макрофагальный белок воспаления (MIP-1-альфа); сывороточный альбумин, такой как человеческий сывороточный альбумин; мюллерова ингибирующая субстанция; A-цепь релаксина; B-цепь релаксина; прорелаксин; мышиный гонадотропин-связанный пептид; белок микроорганизмов, такой как бета-лактамаза; ДНКаза; IgE; антиген цитотоксических T-лимфоцитов (CTLA), такой как CTLA-4; ингибин; активин; фактор роста эндотелия сосудов (VEGF); рецепторы для гормонов или факторов роста; белок А или D; ревматоидные факторы; нейротрофический фактор, такой как костный нейротрофический фактор (BDNF), нейротрофин-3,-4,-5, или-6 (NT-3, NT-4, NT-5, или NT-6), или фактор роста нервной ткани, такой как NGF-β; тромбоцитарный фактор роста (PDGF); фибробластный фактор роста, такой как aFGF и bFGF; эпидермальный фактор роста (EGF); трансформирующий фактор роста (TGF), такой как TGF-альфа и TGF-бета, включая TGF-β1, TGF-β2, TGF-β3, TGF-β4 или TGF-β5; инсулиноподобный фактор роста-I и-II (IGF-I и IGF-II); des(l-3)-IGF-I (IGF-I мозга), белки, связывающие инсулиноподобный фактор роста (IGFBP); CD-белки, такие как CD3, CD4, CD8, CD19 и CD20; эритропоэтин; остеоиндуктивные факторы; иммунотоксины; костный морфогенетический белок (BMP); интерферон, такой как интерферон-альфа, -бета и -гамма; колониестимулирующие факторы (CSF), например, M-CSF, GM-CSF, и G-CSF; интерлейкины (ILs), например, от IL-1 до IL-10; супероксиддисмутаза; рецепторы T-клеток; белки поверхности мембран; фактор распада; вирусный антиген, такой как, например, участок оболочки вируса СПИД; транспортные белки; «хоминг»-рецепторы; адрессины; регуляторные белки; интегрины, такие как CD11a, CD11b, CD11c, CD18, ICAM, VLA-4 и VCAM; опухолевый специфический антиген, такой как CA125 (антиген рака яичников) или рецептор HER2, HER3 или HER4; иммуноадгезины; и фрагменты и/или варианты любого из перечисленных выше белков, а также антител, включая фрагменты антител, связанные с любым из перечисленных выше белков.
Белком, который содержится в готовой форме, предпочтительно является по существу чистый и желательно по существу гомогенный (т.е., не содержащий загрязняющих белков). «По существу чистый» белок означает композицию, содержащую по меньшей мере приблизительно 90% белка по массе, в расчете на общую массу композиции, предпочтительно по меньшей мере приблизительно 95% по массе. «По существу гомогенный» белок означает композицию, содержащую по меньшей мере приблизительно 99% белка по массе, в расчете на общую массу композиции.
В некоторых вариантах осуществления, белком является антитело. Антитело в данном случае направлено против представляющего интерес «антигена». Предпочтительно, антиген представляет собой биологически важный белок, а результатом введения антитела млекопитающему, страдающему от заболевания или расстройства, может быть терапевтическая польза для данного млекопитающего. Однако также предполагаются антитела, направленные против небелковых антигенов (таких как опухолеспецифические гликолипидные антигены; см. Патент США 5091178). Когда антигеном является белок, это может быть трансмембранная молекула (например, рецептор) или лиганд, такой как фактор роста. Приведенные для примера антигены включают те белки, которые обсуждались выше. Предпочтительные молекулярные мишени для антител, охватываемых представленным изобретением, включают полипептиды CD, такие как CD3, CD4, CD8, CD19, CD20 и CD34; члены семейства рецепторов HER, такие как рецептор EGF (HER1), рецептор HER2, HER3 или HER4; молекулы клеточной адгезии, такие как LFA-1, Macl, pl50,95, VLA-4, ICAM-1, VCAM и интегрин av/b3, включая его субъединицы либо а, либо b (например, антитела анти-CD11a, анти-CD18 или анти-CDllb); факторы роста, такие как VEGF; IgE; антигены групп крови; рецептор flk2/flt3; рецептор ожирения (OB); mpl-рецептор; CTLA-4; полипептид C и т.д. Для получения антител в качестве иммуногенов могут быть использованы растворимые антигены или их фрагменты, необязательно конъюгированные с другими молекулами. Для трансмембранных молекул, таких как рецепторы, в качестве иммуногена могут быть использованы их фрагменты (например, внеклеточный домен рецептора). В виде альтернативы, в качестве иммуногена могут быть использованы клетки, экспрессирующие трансмембранную молекулу. Подобные клетки могут быть получены из естественного источника (например, линий раковых клеток) или могут представлять собой клетки, которые были трансформированы с помощью рекомбинантных технологий для экспрессирования трансмембранной молекулы.
Примеры антител, подлежащих очистке, в данном случае включают, но без ограничения: антитела против HER2, включая трастузумаб (HERCEPTIN®) (Carter et al, Proc. Natl. Acad. Sci. USA, 89:4285-4289 (1992), патент США № 5725856) и пертузумаб (OMNITARG™) (WO 01/00245); антитела против CD20 (см. ниже); антитела против IL-8 (St John et al., Chest, 103:932 (1993), и международная публикация № WO 95/23865); антитела против VEGF или против рецепторов VEGF, включая гуманизированные и/или аффинно развитые антитела против VEGF, такие как гуманизированное антитело против VEGF huA4.6.1 бевацизумаб (AVASTIN®) и ранибизумаб (LUCENTIS®) (Kim et al., Growth Factores, 7:53-64 (1992), международная публикация № WO 96/30046, и WO 98/45331, опубликованная 15 октября 1998 года); антитела против PSCA (WOO 1/40309); антитела против Cdlla, включая эфализумаб (RAPTIVA®) (Патент США № 6037454, Патент США № 5622700, WO 98/23761, Stoppa et al., Transplant Intl. 4:3-7 (1991), и Hourmant et al., Transplantation 58:377-380 (1994)); антитела, которые связывают IgE, включая омализумаб (XOLAIR®) (Presta et al., J. Immunol. 151:2623-2632 (1993), и международная публикация № WO 95/19181; Патент США № 5714338, выданный 3 февраля 1998 года или Патент США № 5091313, выданный 25 февраля 1992 года, WO 93/04173 опубликованной 4 марта 1993 года, или Международная заявка № PCT/US98/13410, поданная 30 июня 1998 года, Патент США № 5714338); антитела против CD18 (Патент США № 5622700, выданный 22 апреля 1997 года, или как в WO 97/26912, опубликованной 31 июля 1997 года); антитела против рецептора Apo-2 (WO 98/51793, опубликованный 19 ноября 1998 года); антитела против Тканевого Фактора (TF) (Европейский Патент № 0420937 B1, выданный 9 Ноября 1994 года); антитела против α4-α7 интегрина (WO 98/06248, опубликованной 19 Февраля 1998 года); антитела против EGFR (например, химеризованное или гуманизированное антитело 225, цетуксимаб, ERBUTIX® как в WO 96/40210 опубликованной 19 Декабря 1996 года); антитела против CD3, такие как OKT3 (Патент США № 4515893, выданный 7 мая 1985 года); антитела против CD25 или Tac, такие как CHI-621 (SIMULECT®) и ZENAPAX® (см. Патент США № 5693762, выданный 2 декабря 1997 года); антитела против CD4, такие как антитело cM-7412 (Choy et al., Arthritis Rheum 39(l):52-56 (1996)); антитела против CD52, такие как CAMPATH-1H (ILEX/Berlex) (Riechmann et al, Nature 332:323-337 (1988)); антитела против рецептора Fc, такие как антитело M22, направленное против Fc(RI, как у Graziano et al., J. Immunol. 155(10):4996-5002 (1995)); антитела против карциноэмбрионного антигена (CEA), такие как hMN-14 (Sharkey et al., Cancer Res. 55(23Suppl): 5935s-5945s (1995)); антитела, направленные против эпителиальных клеток молочной железы, включая huBrE-3, hu-Mc 3 и CHL6 (Ceriani et al, Cancer Res. 55(23): 5852s-5856s (1995); и Richman et al, Cancer Res. 55(23 Supp): 5916s-5920s (1995)); антитела, которые связываются с клетками карциномы толстой кишки, такими как C242 (Litton et al, Eur J. Immunol. 26(1):1-9 (1996)); антитела против CD38, например, АТ13/5 (Ellis et al., J.Immunol. 155(2): 925-937 (1995)); антитела против CD33, такие как Hu M195 (Jurcic et al., Cancer Res. 55 (23Suppl): 5908s-5910s (1995) и СМА-676 или CDP771; антитела против EpCAM, такие как 17-1А (PANOREX®); антитела против GpIIb/IIIa, такие как абциксимаб или с7Е3 Fab (REOPRO®); антитела против RSV, такие как MEDI-493 (SYNAGIS®); антитела против CMV, такие как PROTOVIR®; антитела против ВИЧ, такие как PRO542; антитела против гепатита, такие как антитело против Нер В OSTAVIR®; антитело против CA125, включая анти-MUC16 (WO2007/001851; Yin, BWT и Lloyd, KO, J. Biol. Chem. 276:27371-27375 (2001)) и OvaRex; антитело ВЕС2 против идиотипического эпитопа CD3; антитело против ανβ3 (например, VITAXIN®; Medimmune); антитело против карциномы почечных клеток человека, такое как ch-G250; ING-1; антитело против 17-1А человека (3622W94); антитело против колоректальной опухоли человека (A33); антитело против меланомы человека R24, направленное против ганглиозида GD3; антитело против карциномы сквамозных клеток человека (SF-25); и антитела против лейкоцитарного антигена человека (HLA), такие как Smart ID10, и антитело Oncolym (Lym-1) против HLA DR; антитело против CD37, такое как TRU 016 (Trubion); антитело против IL-21 (Zymogenetics/Novo Nordisk); антитело против B клеток (Impheron); выделяющее В-клетки MAb (Immunogen/Aventis); 1D09C3 (Morphosys/GPC); LymphoRad 131 (HGS); антитело Lym-1, такое как Lym-1Y-90 (USC) или анти-Lym-1 Oncolym (USC/Peregrine); LIF 226 (Enhanced Lifesci.); антитело против BAFF (например, WO 03/33658); антитело против рецептора BAFF (см. например, WO 02/24909); антитело BR3; антитело Blys, такое как белимумаб; LYMPHOSTAT-B™; ISF 154 (UCSD/Roche/Tragen); гомиликсима (Idec 152; Biogen Idee); антитело против рецептора IL-6, такое как атлизумаб (ACTEMRA™; Chugai/Roche); антитело против IL-15, такое как HuMax-II-15 (Genmab/Amgen); антитело против хемокинового рецептора, такое как антитело CCR2 (например, MLN1202; Millieneum); антикомплементное антитело, такое как антитело C5 (например, экулизумаб, 5G1.1; Alexion); пероральная готовая форма человеческого иммуноглобулина (например, IgPO; Protein Therapeutics); антитело против IL-12, такое как ABT-874 (CAT/Abbott); Teneliximab (BMS-224818; BMS); антитела против CD40, включая S2C6 и их гуманизированные варианты (WO 00/75348) и TNX 100 (Chiron/Tanox); антитела против TNF-α, включая cA2 или инфликсимаб (REMICADE®), CDP571, MAK-195, адалимумаб (HUMIRA™), пегилированный фрагмент антитела против TNF-α, такой как CDP-870 (Celltech), D2E7 (Knoll), поликлональное антитело против TNF-α (например, PassTNF; Verigen); антитела против CD22, такие как LL2 или эпратузумаб (LYMPHOCIDE®; Immunomedics), включая эпратузумаб Y-90 и эпратузумаб I-131, антитело Abiogen против CD22 (Abiogen, Italy), CMC 544 (Wyeth/Celltech), комботокс (UT Soutwestern), BL22 (NIH), и LympoScan Tc99 (Immunomedics).
Примеры антител против CD20 включают: «C2B8», называемые в настоящее время «ритуксимаб» («RITUXAN®») (Патент США № 5736137); иттрий-[90]-меченое 2В8 мышиное антитело, обозначенное «Y2D8» или «Ибритумомаб Тиуксетан» (ZEVALIN®), коммерчески доступный от IDEC Pharmaceuticals, Inc. (Патент США № 5736137; 2B8 депонируемый в ATCC под № HB11388 поступления от 22 июня 1993 года); мышиный IgG2a «B1», также называемый «Тозитумомаб», необязательно меченый 131I для образования 131I-B1 антитела или иод I131 тозитумомаб, (BEXXARTM), коммерчески доступный от Corixa (см., также, Патент США № 5595721); мышиное моноклональное антитело «1F5» (Press et al, Blood 69(2):584-591 (1987)) и его варианты, включая «имеющее «очажки» каркасной области» или гуманизированное 1F5 (WO03/002607, Leung, S.; депозит ATCC НВ-96450); мышиное антитело 2Н7 и химерное антитело 2Н7 (Патент США 5677180); гуманизированное антитело 2Н7; (WO 2004/056312, Lowman et al.); 2F2 (HuMax-CD20), полностью человеческое, высокоаффинное антитело, направленное на молекулу CD20 в клеточной мембране B-клеток (Genmab, Denmark; см., например, Glennie and van de Winkel, Drug Discovery Today 8:503-510 (2003) и Cragg et al, Blood 101: 1045-1052 (2003); WO 2004/035607; US2004/0167319); человеческие моноклональные антитела, изложенные в WO 2004/035607 и US2004/0167319 (Teeling et al.); антитела, имеющие комплекс сцепленных с N-гликозидом сахарных цепей, связанных с Fc-областью, описанной в US 2004/0093621 (Shitara et al.); моноклональные антитела и антигенсвязывающие фрагменты, связанные с CD20 (WO 2005/000901, Tedder et al.), такие как HB20-3, HB20-4, HB20-25 и MB20-11; CD20-связывающие молекулы, такие как серия антител AME, например, антитела AME 33, как изложено в WO 2004/103404 и US2005/0025764 (Watkins et al, Eli Lilly/Applied Molecular Evolution, AME); CD20-связывающие молекулы, такие как молекулы, описанные в US 2005/0025764 (Watkins et al.); антитело A20 или его варианты, такие как химерное или гуманизированное антитело A20 (cA20, hA20, соответственно) или IMMU-106 (US 2003/0219433, Immunomedics); CD20-связывающие антитела, включая эпитоп-истощенные Leu-16, 1H4 или 2B8, необязательно конъюгированные с IL-2, как в US 2005/0069545A1 и WO 2005/16969 (Carr et al.); биспецифическое антитело, которое связывает CD22 и CD20, например, hLL2xhA20 (WO2005/14618, Chang et al.); моноклональные антитела L27, G28-2, 93-1B3, B-Cl или NU-B2, доступные от International Leukocyte Typing Workshop (Valentine et al, In: Leukocyte Typing III (McMichael, Ed., p. 440, Oxford University Press (1987)); 1H4 (Haisma et al, Blood 92: 184 (1998)); конъюгат анти-CD20 ауристан E (Seattle Genetics); анти-CD20-IL2 (EMD/Biovation/City of Hope); анти-CD20 MAb терапевтик (EpiCyte); анти-CD20 антитело TRU 015 (Trubion).
Термин «антитело», в том смысле, в котором он используется в данном описании, включает моноклональные антитела (включая антитела с полной длиной, которые имеют Fc-область иммуноглобулина), композиции антител с полиэпитопной специфичностью, мультиспецифические антитела (например, биспецифические антитела, диатела и молекулы с единственной цепью, а также фрагменты антител (например, Fab, F(ab')2 и Fv). Термин «иммуноглобулин» (Ig) используется в данном описании взаимозаменяемо с «антителом».
Основная 4-цепочечная единица антитела представляет собой гетеротетрамерный гликопротеин, состоящий из двух идентичных легких (L) цепей и двух идентичных тяжелых (H) цепей. Антитело IgM состоит из 5 основных гетеротетрамерных единиц наряду с дополнительным полипептидом, называющимся цепь J, и содержит 10 антиген связывающих сайтов, тогда как антитела IgA содержат от 2 до 5 основных 4-цепочечных единиц, которые могут полимеризоваться с образованием поливалентных комплексов в комбинации с цепью J. в случае IgGs, 4-цепочечная единица составляет в общем приблизительно 150000 дальтон. Каждая L-цепь соединена с H-цепью одной ковалентной дисульфидной связью, тогда как две H-цепи сцеплены друг с другом одной или более дисульфидными связями в зависимости от изотипа H-цепи. Каждая H-и L-цепь также имеет расположенные с равными интервалами внутрицепочечные дисульфидные мостики. Каждая H-цепь на N-конце имеет вариабельный домен (VH), за которым следуют три константных домена (CH) для каждой из α и γ цепей и четыре CH домена для μ и ε изотипов. Каждая L-цепь на N-конце имеет вариабельный домен (VL), за которым следуют константный домен на другом ее конце. VL выровнен с VH, а CL выровнен с первым константным доменом тяжелой цепи (CH1). Полагают, что отдельные аминокислотные остатки образуют область контакта между вариабельными доменами легкой цепи и тяжелой цепи. Объединение VH и VL вместе образует единый антигенсвязывающий сайт. О структуре и свойствах различных классов антител, см. например, Basic and Clinical Immunology, 8th Edition, Daniel P. Sties, Abba I. Terr and Tristram G. Parsolw (eds), Appleton & Lange, Norwalk, Conn., 1994, page 71 and Chapter 6.
L-цепь любого вида позвоночных может быть отнесена к одному из двух четко отличающихся типов, называющихся kappa и lambda, на основании аминокислотных последовательностей их константных доменов. В зависимости от аминокислотной последовательности константного домена их тяжелых цепей (CH), иммуноглобулины могут быть отнесены к различным классам или изотипам. Существует пять классов иммуноглобулинов: IgA, IgD, IgE, IgG и IgM, имеющих тяжелые цепи, обозначенные α, δ, ε, γ и μ, соответственно. Классы γ и α дополнительно подразделяют на подклассы на основании относительно небольших различий в последовательности и функционировании CH, например, у людей экспрессируются следующие подклассы: IgGl, IgG2, IgG3, IgG4, IgAl и IgA2.
Термин «вариабельный» относится к факту, что определенные участки вариабельных доменов в значительной степени различаются по последовательности среди антител. V-домен опосредует связывание антигена и определяет специфичность конкретного антитела к его конкретному антигену. Однако вариабельность распределена неравномерно по всему диапазону вариабельных доменов. Вместо этого, V-области состоят из относительно неизменных участков, называемых каркасные области (FR), из приблизительно 15-30 аминокислотных остатков, разделенных более короткими областями с выраженной вариабельностью, называемыми «гипервариабельные области» или иногда «определяющие комплементарность области» (CDRs), каждая из которых составляет приблизительно 9-12 аминокислотных остатков в длину. Каждый из вариабельных доменов природных тяжелых и легких цепей содержит четыре FR, пронимающие в значительной степени β-листовую конфигурацию, соединенные тремя гипервариабельными областями, которые образуют петли, соединяющие, а в некоторых случаях образующие часть, β-листовой структуры. Гипервариабельные области в каждой цепи удерживаются вместе в непосредственной близости за счет FR, а с гипервариабельными областями из другой цепи принимают участие в образовании антигенсвязывающего сайта антител (см. Kabat et al, Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, Md. (1991). Константные домены непосредственно не вовлечены в связывание антитела с антигеном, но демонстрируют различные эффекторные функции, такие как участие антитела в антителозависимой клеточной цитотоксичности (ADCC).
Термин «гипервариабельная область» (также известный, как «определяющие комплементарность области» или CDR), при использовании в данном случае относится к аминокислотным остаткам антитела, которые находятся (обычно три или четыре короткие области с чрезвычайной вариабельностью последовательность) в пределах V-области домена иммуноглобулина, которые образуют антигенсвязывающий сайт и являются основными детерминанты специфичности антигенов. Существует по меньшей мере два способа идентификации CDR остатков: (1) подход на основании вариабельности последовательностей при перекрестных скрещиваниях (т.е., Kabat et al, Sequences of Proteins of Immunological Interest (National Institute of Health, Bethesda, M S 1991); и (2) подход на основании кристаллографических исследований комплексов антиген-антитело (Chothia, C. et al, J. Mol. Biol. 196: 901-917 (1987)). Однако, в случае, если две методики идентификации остатков определяют области перекрывающихся, но не идентичных областей, они могут быть объединены для определения гибридной CDR.
Термин «моноклональное антитело», в том смысле, в котором он используется в данном описании, относится к антителу, полученному из популяции по существу гомогенных антител, т.е. отдельные антитела, составляющие популяцию, являются идентичными за исключением возможных естественных мутаций и/или посттрансляционных модификаций (например, изомеризаций, амидирований), которые могут присутствовать в небольших количествах. Моноклональные антитела являются высоко специфичными, будучи направленными против одного антигенного сайта. Кроме того, в противоположность обычным (поликлональным) препаратам антител, которые, как правило, содержат различные антитела, направленные против различных детерминант (эпитопов), каждое моноклональное антитело направлено против одной детерминанты в антигене. В дополнение к их специфичности, моноклональные антитела обладают тем преимуществом, что они синтезируются гибридомной культурой, незагрязненной другими иммуноглобулинами. Определение «моноклональное» обозначает характер антитела, полученного по существу из гомогенной популяции антител, и не ограничивается необходимостью получения антитела каким-либо определенным способом. Например, моноклональные антитела для применения в соответствии с представленным изобретением могут быть получены гибридомным способом, впервые описанным Kohler et ah, Nature, 256:495 (1975), или могут быть получены методами рекомбинантной ДНК (см., например, Патент США № 4816567). «Моноклональные антитела» также можно выделить из фаговых библиотек антител, с использованием методик, описанных, например, у Clackson et al, Nature, 352:624-628 (1991) и Marks et al., J. Mol. Biol, 222:581-597 (1991).
В данном случае моноклональные антитела особо включают «химерные» антитела (иммуноглобулины), в которых участок тяжелой и/или легкой цепи является идентичным или гомологичным соответствующим последовательностям в антителах, полученных от определенных видов или относящихся к конкретному классу или подклассу антител, в то время как оставшаяся часть цепи (цепей) является (являются) идентичным или гомологичным соответствующим последовательностям в антителах, полученных от других видов или относящихся к другому классу или подклассу антител, а также фрагментов подобных антител, при условии, что они демонстрируют необходимую биологическую активность (Патент США № 4816567; Morrison et al, Proc. Natl. Acad. Sci. USA, 81:6851-6855 (1984)). В данном случае интересующие химерные антитела включают «приматизированные» антитела, содержащие последовательности антигенсвязывающего вариабельного домена, полученные от приматов, не относящихся к человеку (например, низших узконосых обезьян, Ape и т.д.) и последовательности человеческих константных областей.
«Интактное» антитело представляет антитело, которое содержит антигенсвязывающий сайт, а также CL и по меньшей мере домены CH1, CH2 и CH3 тяжелой цепи. Константные домены могут представлять константные домены с природной последовательностью (например, человеческие константные домены с природной последовательностью) или их варианты аминокислотной последовательности. Предпочтительно, интактное антитело имеет одну или более эффекторные функции.
«Фрагмент антитела» содержит участок интактного антитела, предпочтительно антигенсвязывающую и/или вариабельную область интактного антитела. Примеры фрагментов антител включают Fab, Fab', F(ab')2 и Fv фрагменты; диатела; линейные антитела (см. Патент США № 5641870, Пример 2; Zapata et al, Protein Eng. 8(10): 1057-1062 [1995]); молекулы одноцепочечных антител и мультиспецифические антитела, образованные из фрагментов антител.
В результате расщепления антител папаином получается два одинаковых антигенсвязывающих фрагмента, называемых «Fab»-фрагментами, и остаточный «Fc»-фрагмент, название которого отражает его способность к легкой кристаллизации. Fab-фрагмент состоит из всей L-цепи наряду с доменом вариабельной области H-цепи (VH) и первым константным доменом одной тяжелой цепи (CH1). Каждый Fab-фрагмент является моновалентным по отношению к связыванию антигена, т.е. он имеет единственный антигенсвязывающий сайт. Обработка антитела пепсином дает один большой F(ab')2-фрагмент, который приблизительно соответствует двум Fab-фрагментам с дисульфидным сцеплением, имеющим различную антигенсвязывающую активность и по-прежнему способным перекрестно связываться с антигеном. Fab'-фрагменты отличаются от Fab-фрагментов за счет наличия нескольких дополнительных остатков на карбоксильном конце домена CH1, включая один или более цистеинов из шарнирной области антитела. В данном случае Fab'-SH является обозначением Fab', в котором цистеиновый остаток (остатки) константных доменов имеет свободную тиоловую группу. F(ab')2-фрагменты антитела первоначально были получены в виде пары Fab'-фрагментов, которые имеют между ними шарнирные цистеины. Известны также другие химические связи фрагментов антител.
Fc-фрагмент содержит карбоксил-концевые участки обеих H-цепей, удерживаемых вместе дисульфидными связями. Эффекторные функции антител определяются последовательностями в Fc-области, области, которая также распознается Fc-рецепторами (FcR), обнаруженными в некоторых типах клеток.
«Fv» представляет минимальный фрагмент антитела, который содержит полный антигенраспознающий и антигенсвязывающий сайт. данный фрагмент состоит из димера домена вариабельной области одной тяжелой и одной легкой цепи в близкой нековалентной связи. из складывания двух данных доменов происходит шесть гипервариабельных петель (каждые 3 петли из H и L-цепей), аминокислотные остатки которых принимают участие в связывании антигена и которые придают антителу антигенсвязывающую специфичность. Однако даже единственный вариабельный домен (или половина Fv, содержащая только три CDRs, специфичных для антигена) обладает способностью распознавать и связывать антиген, хотя и с меньшей аффинностью, чем связывающий сайт целиком.
«Одноцепочечные Fv», также обозначаемые «sFv» или «scFv», представляют фрагменты антитела, которые содержат домены VH и VL антитела, соединенные в одну полипептидную цепь. Предпочтительно, чтобы полипептид sFv дополнительно содержал полипептидный линкер между доменами VH и VL, который обеспечивает возможность sFv образовывать необходимую структуру для связывания антигена. Обзор по sFv, см. у Pluckthun в Pharmacology of Monoclonal Antibodies, vol. 113, Rosenburg and Moore eds., Springer-Verlag, New York, pp. 269-315 (1994).
Термин «диатела» относится к небольшим фрагментам антитела, полученным посредством конструирования sFv фрагментов (см. предшествующий абзац) с помощью коротких линкеров (приблизительно 5-10) остатков) между доменами VH и VL таким образом, чтобы достигалось межцепочечное, но не внутрицепочечное спаривание V-доменов, результатом чего является бивалентный фрагмент, т.е., фрагмент, имеющий два антигенсвязывающих сайта. Биспецифические диатела представляют гетеродимеры двух «перекрещивающихся» sFv фрагментов, в которых в различных полипептидных цепях присутствуют домены VH и VL двух антител. более подробно диатела описаны, например, в EP 404097; WO 93/11161; Hollinger et al, Proc. Natl. Acad. Sci. USA 90: 6444-6448 (1993).
Антитело, которое «специфически связывается» или является «специфичным для» конкретного полипептида или эпитопа на конкретном полипептиде представляет антитело, которое связывается с данным конкретным полипептидом или эпитопом на конкретном полипептиде по существу без связывания с каким-либо другим полипептидом или эпитопом полипептида.
Термин «твердая фаза» описывает безводную матрицу, к которой может прилипать антитело представленного изобретения. Примеры твердых фаз, охватываемых в данном описании, включают твердые фазы, которые частично или полностью образованы из стекла (например, стекла с контролируемым размером пор), полисахаридов (например, агарозы), полиакриламидов, полистирола, поливинилового спирта и силиконов. В некоторых вариантах осуществления, в зависимости от контекста, твердая фаза может включать лунку аналитического планшета; в других она представляет собой колонку для очистки (например, аффинную хроматографическую колонку). Данный термин также включает дисперсную твердую фазу из дискретных частиц, такую как твердые фазы, описанные в Патенте США № 4275149.
«Гуманизированные» формы отличных от человеческих (например, мышиных) антител представляют собой химерные иммуноглобулины, цепи иммуноглобулинов или их фрагменты (такие как Fv, Fab, Fab', F(ab')2 или другие антигенсвязывающие подпоследовательности антител) по большей части человеческих последовательностей, которые включают минимальную последовательность, происходящую из не принадлежащего человеку иммуноглобулина. Главным образом, гуманизированные антитела являются человеческими иммуноглобулинами (реципиентное антитело), в которых остатки из гипервариабельной области (также CDR) реципиента заменены остатками из гипервариабельной области не относящихся к человеку (донорного антитела) видов, таких как мышь, крыса или кролик, обладающих необходимой специфичностью, аффинностью и емкостью. В некоторых случаях, остатки Fv каркасной области (FR) человеческого иммуноглобулина заменены соответствующими не принадлежащими человеку остатками. Кроме того, «гуманизированные антитела», в том смысле, в котором используется в данном описании, также могут содержать остатки, которые не обнаружены ни в реципиентном антителе, ни в донорском антителе. Эти модификации вводят для дополнительного улучшения и оптимизации эффективности антитела. Оптимально, гуманизированные антитело также будет содержать по меньшей мере участок константной области (Fc) иммуноглобулина, как правило, участок константной области человеческого иммуноглобулина. Для дополнительных подробностей см. Jones et al, Nature, 321:522-525 (1986); Reichmann et al, Nature, 332:323-329 (1988); и Presta, Curr. Op. Struct. Biol, 2:593-596 (1992).
«Видозависимое антитело», например, антитело против человеческого IgE млекопитающих, представляет собой антитело, которое имеет более сильную аффинность связывания для антигена из первого вида млекопитающих, чем оно имеет для гомолога данного антигена из второго вида млекопитающих. Обычно, видозависимое антитело «специфично связывается» с человеческим антигеном (т.е., имеет значение аффинности связывания (Kd) не более, чем приблизительно 1×10-7M, в качестве альтернативы не более, чем приблизительно 1×10-8M, в качестве альтернативы не более, чем приблизительно l×l0-9M), но обладает аффинностью связывания для гомолога антигена из второго не относящегося к человеку вида млекопитающих, которая по меньшей мере приблизительно в 50 раз, по меньшей мере приблизительно в 500 раз или по меньшей мере приблизительно в 1000 раз слабее, чем его аффинность связывания для не принадлежащего человеку антигена. Видозависимым антителом может быть любое из различных типов антител, которые определены выше, но предпочтительно гуманизированное или человеческое антитело.
«Эффекторные функции» антител относятся к таким видам биологической активности, которые могут быть приписаны Fc-области (Fc-области, имеющей нативную последовательность или Fc-области, имеющей вариант аминокислотной последовательности) антитела, и варьируют с изотипом антитела. Примеры эффекторных функций антитела включают: связывание с C1q и комплементзависимая цитотоксичность; связывание с Fc-рецептором; антителозависимая клеточно-опосредуемая цитотоксичность (ADCC); фагоцитоз; негативная регуляция рецепторов клеточной поверхности (например, В-клеточных рецепторов); и активация В-клеток.
«Антителозависимая клеточно-опосредуемая цитотоксичность» или ADCC относится к форме цитотоксичности, при которой секретированный Ig, связанный с Fc-рецепторами (FcRs), присутствующими на некоторых цитотоксических клетках (например, природных клетках-киллерах (NK), нейтрофилах и макрофагах) сообщает этим цитотоксическим эффекторным клеткам способность специфически связываться с антиген-содержащими клетками-мишенями, а впоследствии уничтожать клетки-мишени с цитотоксинами. Антитела «нейтрализуют» цитотоксические клетки и являются необходимыми для уничтожения клеток-мишеней посредством данного механизма. Первичные клетки, опосредующие ADCC, NK-клетки экспрессируют только FcyRIII, тогда как моноциты экспрессируют FcyRI, FcyRII и FcyRIII. Экспрессия Fc на гемапоэтических клетках систематизирована в Таблице 3 на странице 464 Ravetch and Kinet, Annu. Rev. Immunol 9: 457-92 (1991). Для оценки ADCC-активности представляющей интерес молекулы, может быть проведен анализ на ACDD in vitro, такой как анализ, описанный в Патентах США № 5500362 или 5821337. Эффекторными клетками, подходящими для таких анализов, являются мононуклеарные клетки периферической крови (МКПК) и природные клетки-киллеры (NK). В качестве альтернативы или дополнительно, ADCC-активность представляющей интерес молекулы может быть оценена in vivo, например, у животного-модели, такого как, животное, раскрытое в публикации Clynes et al, PNAS USA 95:652-656 (1998).
«Fc-рецептор» или «FcR» описывает рецептор, который связывается с Fc-областью антитела. Предпочтительным FcR является FcR человека с нативной последовательностью. более того, предпочтительным FcR является рецептор, который связывается с антителом IgG (гамма-рецептор) и включает рецепторы подклассов FcyRI, FcyRII, и FcyRIII, включая аллельные варианты и альтернативно сплайсированные формы этих рецепторов. Рецепторы FcyRII включают FcyRIIA («активирующий рецептор») и FcyRIIB («ингибирующий рецептор»), которые имеют аналогичные аминокислотные последовательности, которые отличаются главным образом в их цитоплазматических доменах. Активирующий рецептор FcyRIIA содержит в своем цитоплазматическом домене иммунорецепторный активирующий мотив на основе тирозина (ITAM). Ингибирующий рецептор FcyRIIB содержит в своем цитоплазматическом домене иммунорецепторный ингибирующий мотив на основе тирозина (ITIM), (см. M. Daeron, Annu. Rev. Immunol. 15:203-234 (1997). FcRs описаны в публикациях Ravetch and Kinet, Annu. Rev. Immunol 9: 457-92 (1991); Capel et al, Immunmethods 4: 25-34 (1994); и de Haas et al, J. Lab. Clin. Med. 126: 330-41 (1995). Используемый в данном описании термин «FcR» также охватывает и другие FcR, включая FcR, которые будут идентифицированы в будущем. Термин также включает неонатальный рецептор, FcRn, ответственный за передачу материнских IgG плоду. Guyer et al, J. Immunol. 117: 587 (1976) и Kim et al, J. Immunol. 24: 249 (1994).
«Эффекторными клетками человека» являются лейкоциты, которые экспрессируют один или более FcR и обладают эффекторными функциями. Предпочтительно, клетки экспрессируют по меньшей мере FcyRIII и обладают эффекторной ADCC-функцией. Примеры лейкоцитов человека, которые опосредуют ADCC, включают мононуклеарные клетки периферической крови (МКПК), природные клетки-киллеры (NK) клетки, моноциты, цитотоксические T-клетки и нейтрофилы, при этом предпочтительными являются МКПК и MNK-клетки. Эффекторные клетки могут быть выделены из природного источника, например, крови.
«Комплементзависимая цитотоксичность» или «CDC» относится к лизису клеток-мишеней в присутствии комплемента. Активация классического пути комплемента инициируется связыванием первого компонента системы комплемента (C1q) с антителами (соответствующего подкласса), которые связываются с их когнатным антигеном. Для оценки активации комплемента может быть проведен анализ на CDC, например, который описан в публикации Gazzano-Santoro et al, J. Immunol. Methods 202:163 (1996).
«Выделенный» при использовании для описания различных полипептидов и антител, раскрытых в данном документе, означает полипептид или антитело, которое было идентифицировано, отделено и/или извлечено из компонента его продуктивной среды. Предпочтительно, выделенный полипептид не содержит связи со всеми другими составными элементами из его продуктивной среды. Загрязняющие компоненты его продуктивной среды, например компоненты, происходящих из рекомбинантных трансфицированных клеток, представляют собой материалы, которые как правило, будут мешать диагностическим или терапевтическим способам применения полипептида и могут включать в себя ферменты, гормоны и другие белковые или небелковые растворы. В предпочтительных вариантах осуществления полипептид будет очищен (1) до степени, достаточной для получения, по меньшей мере, 15 остатков N-концевой или внутренней аминокислотной последовательности при использовании секвенатора с вращающимся стаканом, или (2) до гомогенности в SDS-PAGE в невосстанавливающих или восстанавливающих условиях с использованием окраски Кумасси синим или предпочтительно серебром. Обычно, однако, выделенный полипептид будут получать посредством, по меньшей мере, одной стадии очистки.
«Выделенная» молекула нуклеиновой кислоты, кодирующая полипептиды и антитела, в данном случае представляет собой молекулу нуклеиновой кислоты, которую идентифицируют и отделяют, по меньшей мере, от одной загрязняющей молекулы нуклеиновой кислоты, с которой она обычно связана в среде, в которой она была получена. Предпочтительно, выделенная нуклеиновая кислота не содержит связи со всеми компонентами, связанными с продуктивной средой. Выделенные молекулы нуклеиновой кислоты, кодирующей полипептиды и антитела в данном случае являются отличными от формы или окружения, в которых их обнаружили в природе. Вследствие этого, выделенные молекулы нуклеиновой кислоты отличаются от нуклеиновой кислоты, кодирующей полипептиды и антитела данного описания, существующие в естественных условиях в клетках.
Термин «контрольные последовательности» относится к последовательностям ДНК, необходимым для экспрессии функционально связанной кодирующей последовательности в конкретном организме-хозяине. Контрольные последовательности, подходящие для прокариот, например, включают в себя последовательность промотора, необязательно, оператора и участка связывания рибосомы. Известно, что эукариотические клетки используют промоторы, сигналы полиаденилирования, и энхансеры.
Нуклеиновая кислота является «функционально связанной», когда ее помещают в функциональное взаимодействие с другой последовательностью нуклеиновой кислоты. Например, ДНК для препоследовательности или секреторный лидер является функционально связанной с ДНК для полипептида, если она экспрессируется как пребелок, который участвует в секреции полипептида; промотор или энхансер является функционально связанным с кодирующей последовательностью, если он влияет на транскрипцию последовательности; или участок связывания рибосомы является функционально связанным с кодирующей последовательностью, если он расположен так, чтобы облегчать трансляцию. Как правило, «функционально связанные» означает последовательности ДНК, связанные по соседству, и в случае секреторного лидера - по соседству и в фазе считывания. Однако энхансеры не должны находиться по соседству. Связывание осуществляют посредством лигирования в подходящие участки рестрикции. Если таких участков не существует, используют синтетические олигонуклеотидные адапторы или линкеры в соответствии с общепринятой практикой.
Термин «меченный эпитопом» при использовании в данном описании здесь относится к химерному полипептиду, содержащему полипептид или антитело, описанные в данном документе, слитые с «полипептидом-меткой». Полипептид-метка имеет достаточно остатков, чтобы обеспечивать эпитоп, против которого можно получить антитело, однако является достаточно коротким, чтобы не мешать активности полипептида, с которым он слит. Полипептид-метка предпочтительно также является достаточно уникальным, так что антитело по существу не реагирует перекрестно с другими эпитопами. Подходящие полипептиды-метки, как правило, содержат, по меньшей мере, шесть аминокислотных остатков, а обычно между приблизительно 8 и 50 аминокислотными остатками (предпочтительно между приблизительно 10 и 20 аминокислотными остатками).
Как используется в данном описании, термин «иммуноадгезин» обозначает антитело-подобные молекулы, сочетающие связывающую специфичность гетерологичного белка («адгезин») с эффекторными функциями константных доменов иммуноглобулина. Структурно иммуноадгезины содержат слитые аминокислотную последовательность с желаемой связывающей специфичностью, отличную от участка узнавания и связывания антигена из антитела (т.е. являются «гетерологичными»), и последовательность константного домена иммуноглобулина. Часть адгезина молекулы иммуноадгезина, как правило, представляет собой непрерывную аминокислотную последовательность, содержащую, по меньшей мере, участок связывания рецептора или лиганда. Последовательность константного домена иммуноглобулина в иммуноадгезине можно получить из любого иммуноглобулина, такого как иммуноглобулин подтипов IgG-1, IgG-2, IgG-3, или IgG-4, IgA (включая IgA-I и IgA-2), IgE, IgD или IgM. Слитые белки Ig предпочтительно содержат замену домена полипептида или антитела, описанных в данном документе, вместо по меньшей мере одной вариабельной области в молекуле Ig. В особенно предпочтительном варианте осуществления, слитый с иммуноглобулином белок содержит шарнирную CH2 и CH3 или шарнирную CH1, CH2 и CH3 области молекулы IgG1. Получение слитных с иммуноглобулином белков см. также в патенте США No. 5428130, поданном 27 июня 1995 г.
Термин «фармацевтическая готовая форма» относится к препарату, который находится в такой форме, которая обеспечивает, чтобы биологическая активность активного ингредиента была эффективной, и которая не содержит дополнительные компоненты, которые являются неприемлемо токсичными для пациента, которому должна быть введена готовая форма.
Антитело обладает «биологической активностью» в фармацевтической готовой форме, если биологическая активность антитела в данное время находится в пределах приблизительно 10% (в пределах ошибки анализа) биологической активности, проявленной во время, когда была получена фармацевтическая готовая форма, которая определялась способностью антитела in vitro или in vivo связываться с антигеном и приводить к измеримому биологическому ответу.
«Стабильной» или «стабилизированной» готовой формой является готовая форма, в которой белок по существу сохраняет свою физическую и/или химическую стабильность при хранении. Стабильность может быть измерена при выбранной температуре в течение выбранного временного периода. Предпочтительно, готовая форма является стабильной при комнатной температуре (~30°C) или при 40°C в течение по меньшей мере 1 месяца и/или стабильной приблизительно при 2-8°C в течение по меньшей мере 1 года и предпочтительно в течение по меньшей мере 2 лет. Например, в качестве индикатора стабильности белка может быть использована степень агрегации во время хранение. Таким образом, «стабильной» готовой формой может быть готовая форма, в которой менее чем приблизительно 10%, а предпочтительно менее чем приблизительно 5% белка присутствует в готовой форме в виде агрегата. В данной области доступны различные аналитические методики для измерения стабильности белка, обзор которых произведен, например, в Peptide and Protein Drug Delivery, 247-301, Vincent Lee Ed., Marcel Dekker, Inc., New York, N.Y., Pubs. (1991) и Jones, A. Adv. Drug Delivery Rev. 10: 29-90 (1993).
Повышение «стабильности» белоксодержащей готовой формы относится к уменьшению (по сравнению с необработанной белоксодержащей готовой формой) или предотвращению образования белковых агрегатов в данной готовой форме.
Термин «водный раствор» относится к раствору, в котором растворяющей средой и растворителем является вода. Когда вещество растворяется в жидкости, смесь называется раствор. Разложенное вещество является растворенным веществом, а жидкость, которая производит растворение (в данном случае вода) является растворителем.
Термин, «стабилизирующий агент» или «стабилизатор», в том смысле, в котором он используется в данном описании, представляет химическое вещество или соединение, которое добавляют в раствор или смесь или суспензию или композицию или терапевтическую композицию для сохранения ее в стабильном или неизменном состоянии; или соединение, которое используется, потому что производит реакцию, включающую изменения в атомах или молекулах, приводящую к более стабильному или неизменному состоянию.
Термин «агрегат» или «агрегация», в том смысле, в котором он используется в данном описании, предусматривает схождение вместе или скапливание в массив или целое, например, как в агрегации пептидов, полипептидов, антител или их вариантов. Агрегаты могут быть аутоагрегирующимися или агрегирующимися вследствие других факторов, например, вызывающих агрегацию агентов, осаждающих агентов, взбалтывания или других средств и способов, являющихся причиной, по которой пептиды, полипептиды, антитела или их варианты сходятся вместе.
Индуцируемая взбалтыванием агрегация представляет собой образование агрегатов в белоксодержащем растворе, вызванное взбалтыванием, при этом взбалтывание запускается за счет встряхивания или помешивания.
Антитело, которое «подвержено агрегации» представляет собой антитело, которое, как замечено агрегирует с молекулой (молекулами) другого антитела, особенно при взбалтывании.
Под «ингибированием» индуцируемой взбалтыванием агрегации предполагается предотвращение, уменьшение или сокращение величины индуцируемой взбалтыванием агрегации, измеренной посредством сравнения величины агрегата, присутствующего в белоксодержащем растворе, который содержит по меньшей мере один ингибитор индуцируемой взбалтыванием агрегации, с количеством агрегата, присутствующего в белоксодержащем растворе, который не содержит по меньшей мере один ингибитор индуцируемой взбалтыванием агрегации.
Ингибирующая величина индуцируемой взбалтыванием агрегации представляет собой величину, которая ингибирует индуцируемую взбалтыванием агрегацию.
Методы, которые могут найти применение в представленном изобретении для измерения индуцируемой взбалтыванием агрегации, включают гель-электрофорез, изоэлектрическое фокусирование, капиллярный электрофорез, хроматографию, например эксклюзионную хроматографию, ионообменную хроматографию и высокоэффективную жидкостную хроматографию с обращенными фазами, пептидное картирование, олигосахаридное картирование, масс-спектрометрию, спектроскопию поглощения в ультрафиолетовой области, флуоресцентную спектроскопию, спектроскопиию кругового дихроизма, изотермическую титрационную калориметрию, дифференциальную сканирующую калориметрию, аналитическое ультрацентрифугирование, динамическое рассеяние света, протеолиз и образование поперечных межмолекулярных связей, измерение мутности, анализы замедления фильтрования, иммунологические анализы, анализы связывания флуоресцентных красителей, анализы окрашивания белка, микроскопия и обнаружение агрегатов посредством ELISA или другого анализа связывания.
«Изотоническая» готовая форма представляет собой готовую форму, которая имеет по существу такое же осмотическое давление, как человеческая кровь. Изотонические готовые формы будут в общем иметь осмотическое давление, составляющее от приблизительно 250 до 350 мОсмоль. Термин «гипотонический» описывает готовую форму с осмотическим давлением ниже давления человеческой крови. Соответственно, термин «гипертоническая» используется для описания готовой формы с осмотическим давлением выше давления человеческой крови. Изотоничность может быть измерена, используя, например, тип осмометра давления пара или криоскопический. Готовые формы представленного изобретения являются гипертоническими в результате добавления соли и/или буфера.
«Восстановленная» готовая форма представляет готовую форму, которая была получена посредством растворения лиофилизированной готовой формы, содержащей белок или антитело, в разбавителе таким образом, чтобы белок в восстановленной готовой форме был диспергирован. Восстановленная готовая форма подходит для введения (например, парентерального введения) пациенту, подлежащего лечению представляющим интерес белком, а в некоторых вариантах осуществления изобретения может представлять собой готовую форму, которая подходит для подкожного введения.
«Поверхностно-активные вещества» представляют собой поверхностно-активные агенты, которые могут оказывать свое действие на поверхностях твердое вещество-твердое вещество, твердое вещество-жидкость, жидкость-жидкость и жидкость-воздух, вследствие их химического состава, содержащего как гидрофильную, так и гидрофобную группы. Данные материалы уменьшают концентрацию белков в разбавленных растворах на поверхностях раздела воздух-вода и/или вода-твердое вещество, при этом белки могут быть адсорбированы и потенциально агрегированы. В содержащих белок готовых формах поверхностно-активные вещества могут связываться с гидрофобными поверхностями раздела. Белки на поверхности воды будут агрегировать, особенно при агрегации вследствие разворачивания и последующей агрегации белкового монослоя.
«Поверхностно-активные вещества» могут денатурировать белки, но могут также стабилизировать их против поверхностной денатурации. В общем, ионные поверхностно-активные вещества могут денатурировать белки. Однако неионные поверхностно-активные вещества обычно не денатурируют белки даже при относительно высоких концентрациях (1% м/о). Наиболее парентерально приемлемые неионные поверхностно-активные вещества происходят либо от полисорбата, либо от сложных полиэфирных групп. В имеющихся на рынке содержащих белок готовых формах современными стабилизаторами - поверхностно-активными веществами являются полисорбат 20 и 80. Однако другие поверхностно-активные вещества, используемые в содержащих белок готовых формах, включают Pluronic F-68 и мембраны класса «Brij». Неионогенные поверхностно-активные вещества могут иметь в основе сахар. Основанными на сахаре поверхностно-активными веществами могут быть алкиловые гликозиды. Общая структура алкилового гликозида представляет собой R1-0-(CH2)x-R, где R независимо представляет собой CH3 или циклогексил (C6H11), а R1 независимо представляет собой глюкозу или мальтозу. Приведенные для примера алкиловые гликозиды включают гликозиды, в которых R1 представляет собой глюкозу, R представляет собой CH3, а x представляет собой 5 (n-гексил-β-D-глюкопиранозид), x представляет собой 6 (n-гептил-β-D-глюкопиранозид), x представляет собой 7 (n-октил-β-D-глюкопиранозид), x представляет собой 8 (n-нонил-β-D-глюкопиранозид), x представляет собой 9 (n-децил-β-D-глюкопиранозид), а x представляет собой 11 (n-додецил-β-D-глюкопиранозид). Иногда глюкопиранозиды называют глюкозиды. Приведенные для примера алкиловые гликозиды дополнительно включают гликозиды, у которых R1 представляет собой мальтозу, R представляет собой CH3, а x представляет собой 5 (n-гексил-β-D-мальтопиранозид), x представляет собой 7 (n-октил-β-D-мальтопиранозид), x представляет собой 8 (n-нонил-β-D-мальтопиранозид), x представляет собой 9 (n-децил-β-D-мальтопиранозид), x представляет собой 10 (n-ундецил-β-D-мальтопиранозид), x представляет собой 11 (n-додецил-β-D-мальтопиранозид), x представляет собой 12 (n-тридецил-β-D-мальтопиранозид), x представляет собой 13 (n-тетрадецил-β-D-мальтопиранозид), и x представляет собой 15 (n-гексадецил-β-D-мальтопиранозид). Иногда мальтопиранозиды называют мальтозиды. Приведенные для примера алкиловые гликозиды дополнительно содержат гликозиды, в которых R1 представляет собой глюкозу, x представляет собой 3, а R представляет собой циклогексил (3-циклогексил-1-пропил-β-D-глюкозид); и в которых R1 представляет собой мальтозу, x представляет собой 4, а R представляет собой циклогексил (4-циклогексил-l-бутил-β-D-мальтозид).
«Фармацевтически приемлемая кислота» включает неорганические и органические кислоты, которые являются нетоксичными в концентрации и при способе, с помощью которого их вводят в готовую форму. Например, подходящие неорганические кислоты включают соляную, хлорную, бромистоводородную, иодистоводородную, азотную, серную, сульфоновую, серную, сульфаниловую, фосфорную, углекислую и т.д. Подходящие органические кислоты включают алкиловые, ароматические, циклические, циклоалифатические, арилалифатические, гетероциклические, насыщенные, ненасыщенные, моно, ди- и три-карбоновые с прямыми и разветвленными цепями, включая, например, муравьиную, уксусную, 2-гидроксиуксусную, трифторуксусную, фенилуксусную, триметилуксусную, t-бутилуксусную, антраниловую, пропановую, 2-гидроксипропановую, 2-оксопропановую, пропандиовую, циклопентанпропионовую, циклопентан пропионовую, 3-фенилпропионовую, бутановую, бутандиовую, бензойную, 3-(4-гидроксибензоил)бензойную, 2-ацетоксибензойную, аскорбиновую, коричную, лауриловую, серную, стеариновую, муконовую, миндальную, янтарную, эмбоновую, фумаровую, яблочную, малеиновую, гидроксималеиновую, малоновую, молочную, лимонную, винную, гликолевую, гликоновую, глюконовую, пировиноградную, глиоксиловую, щавелевую, мезиловую, янтарную, салициловую, фталевую, пальмоновую, пальмеиновую, тиоциановую, метансульфокислоту, этансульфокислоту, 1,2-этандисульфоновую, 2-гидроксиэтансульфоновую, бензолсульфокислоту, 4-хлорбензолсульфоновую, нафтален-2-сульфокислоту, p-толуенсульфокислоту, камфорсульфокислоту, 4-метилбицикло[2,2,2]-окт-2-ен-1-карбоновую, глюкогептоновую, 4,4'-метиленбис-3-(гидрокси-2-ен-l-карбоновую кислоту), гидроксинафтойную.
«Фармацевтически приемлемые основания» включают неорганические и органические основания, которые являются нетоксичными в концентрации и при способе, с помощью которого их вводят в готовую форму. Например, подходящие основания включают основания, образованные из образующих неорганические основания металлов, таких как литий, натрий, калий, магний, кальций, аммоний, железо, цинк, медь, магний, алюминий, N-метилглюкамин, морфолин, пиперидин и органические нетоксичные основания, включая первичный, вторичный и третичный амин, замещенные амины, циклические амины и катионообменные смолы, [например, N(R')4 + (где R' независимо представляет собой H или C1-4 алкил, например, аммоний, трис)], например, изопропиламин, триметиламин, диэтиламин, триэтиламин, трипропиламин, этаноламин, 2-диэтиламинэтан, триметамин, дициклогексиламин, лизин, аргинин, гистидин, каффеин, прокаин, гидрабамин, холин, бетаин, этилендиамин, глюкозамин, метилглюкамин, теобромин, пурин, пиперазин, пиперидин, N-этилпиперидин, полиаминовые смолы и тому подобное. Особенно предпочтительными органическими нетоксичными основаниями являются изопропиламин, диэтиламин, этаноламин, триметамин, дициклогексиламин, холин, и каффеин.
Дополнительные фармацевтически приемлемые кислоты и основания, применимые с представленным изобретением, включают кислоты и основания, которые получают из аминокислот, например, гистидин, глицин, фенилаланин, аспарогиновая кислота, глютаминовая кислота, лизин и аспарагин.
«Фармацевтически приемлемые» буферы и соли включают буферы и соли, полученные из солей присоединения как кислот, так и оснований указанных выше кислот и оснований. Конкретные буферы и/или соли включают гистидин, сукцинат и ацетат.
«Лиопротектант» представляет собой молекулу, которая при соединении с представляющим интерес белком, существенно предотвращает или уменьшает физикохимическую нестабильность белка при лиофилизации и последующем хранении. Приведенные для примера лиопротектанты включают сахара и их соответствующие сахарные спирты; аминокислоту, такую как мононатрия глютамат или гистидин; метиламин, такой как бетаин; лиотропную соль, такую как магния сульфат; полиол, такой как трехатомные сахарные спирты или сахарные спирты с более высокой молекулярной массой, например, глицерин, декстран, эритритол, глицерол, арабитол, ксилитол, сорбитол и маннитол; пропиленгликоль; полиэтиленгликоль; Pluronics®; и их комбинации. Дополнительные приведенные для примера лиопротектанты включают глицерин и желатин, и сахара мелибиозу, мелезитозу, раффинозу, маннотриозу и стахиозу. Примеры восстанавливающих сахаров включают глюкозу, мальтозу, лактозу, мальтулозу, изомальтулозу и лактулозу. Примеры невосстанавливающих сахаров включают невосстанавливающие гликозиды полигидрокси соединений, выбранных из сахарных спиртов и других полиспиртов с прямой цепью. Предпочтительными сахарными спиртами являются моногликозиды, особенно те соединения, которые получены путем восстановления из дисахаридов, таких как лактоза, мальтоза, лактулоза и мальтулоза. Гликозидная боковая группа может быть либо глюкозидной, либо галактозидной. Дополнительными примерами сахарных спиртов являются глюцитол, мальтитол, лактитол и изомальтулоза. Предпочтительным лиопротектантом являются невосстанавливающие сахара трегалоза или сахароза.
Лиопротектант добавляют к предварительно лиофилизированной готовой форме в «лиопротективном количестве», что означает, что вслед за лиофилизацией белка в присутствии лиопротективного количества лиопротектанта, белок по существу сохраняет свою физикохимическую стабильность при лиофилизации и хранении.
«Фармацевтически приемлемым сахаром» является молекула, которая, при соединении с представляющим интерес белком, в значительной степени предотвращает или понижает физико-химическую нестабильность белка при хранении. Когда предполагается лиофилизация, а затем восстановление готовой формы, «фармацевтически приемлемые сахара» также могут быть известны, как «лиопротектант». Приведенные для примера сахара и их соответствующие сахарные спирты включают: аминокислоту, такую как мононатрия глютамат или гистидпин; метиламин, такой как бетаин; лиотропную соль, такую как магния сульфат; полиол, такой как трехатомные сахарные спирты или сахарные спирты с более высокой молекулярной массой, например, глицерин, декстран, эритритол, глицерол, арабитол, ксилитол, сорбитол и маннитол; пропиленгликоль; полиэтиленгликоль; Pluronics® и их комбинации. Дополнительные приведенные для примера лиопротектанты включают глицерин и желатин, и сахара мелибиозу, мелезитозу, раффинозу, маннотриозу и стахиозу. Примеры восстанавливающих сахаров включают глюкозу, мальтозу, лактозу, мальтулозу, изомальтулозу и лактулозу. Примеры невосстанавливающих сахаров включают невосстанавливающие гликозиды полигидроксисоединений, выбранных из сахарных спиртов и других полиспиртов с прямой цепью. Предпочтительными сахарными спиртами являются моногликозиды, особенно соединения, которые получены посредством восстановления дисахаридов, таких как лактоза, мальтоза, лактулоза и мальтулоза. Гликозидная боковая группа может быть либо глюкозидной, либо галактозидной. Дополнительными примерами сахарных спиртов являются глюцитол, мальтитол, лактитол и изомальтулоза. Предпочтительными фармацевтически приемлемыми сахарами являются невосстанавливающие сахара трегалоза или сахароза.
Фармацевтически приемлемые сахара добавляют в готовую форму в «защищающем количестве» (например, предварительная лиофилизация), что означает, что белок по существу сохраняет свою физико-химическую стабильность во время хранения (например, после восстановления и хранения).
Представляющим интерес «разбавителем» в данном случае является разбавитель, который является фармацевтически приемлемым (безопасным и нетоксичным для введения человеку) и может быть использован для приготовления жидкой готовой формы, такой как готовая форма, восстановленная после лиофилизации. Приведенные для примера разбавители включают стерильную воду, бактериостатическую воду для инъекций (BWFI), кислотно-основной буферный раствор (например, фосфат-буферный раствор натрия хлорида), стерильный раствор натрия хлорида, раствор Рингера или раствор декстрозы. В альтернативном варианте осуществления, разбавители могут включать водные растворы солей и/или буферы.
«Консервантом» является соединение, которое может быть добавлено в готовые формы в данном случае для уменьшения бактериальной активности. Добавление консерванта, например, может облегчить получение готовой формы многоразового использования (содержащей множество доз). Примеры возможных консервантов включают октадецилдиметилбензилхлорид аммония, гексаметония хлорид, бензалкония хлорид (смесь алкилбензилдиметилхлоридов аммония, в которых алкиловыми группами являются длинноцепочечные соединения) и бензетония хлорид. Другие виды консервантов включают ароматические спирты, такие как фенолспирт, бутиловый и бензиловый спирт, алкиловые парабены, такие как метилпарабен или пропилпарабен, катехол, резорцинол, циклогексанол, 3-пентанол и m-крезол. Наиболее предпочтительным консервантом в данном случае является бензиловый спирт.
«Лечение» относится как к терапевтическому лечению, так и к профилактическим или превентивным мерам. Пациенты, нуждающиеся в лечении включают пациентов, которые уже имеют расстройство, а также пациентов, у которых расстройство необходимо предотвратить.
«Млекопитающее» для целей лечения относится к любому животному, классифицируемому как млекопитающее, включая людей, домашних и сельскохозяйственных животных, и животных, содержащихся в зоопарках, связанных со спортом или прирученных животных, таких как собаки, лошади, кролики, домашний скот, свиньи, хомяки, песчанки, мыши, хорьки, крысы, кошки и т.д. Предпочтительно, млекопитающим является человек.
«Расстройством» является любое состояние, на которое будет оказано благоприятное воздействие при лечении белком. Оно включает хронические и острые расстройства или заболевания, включая такие патологические состояния, которые провоцируют расстройство млекопитающего, о котором идет речь. Неограничивающие примеры расстройств, подлежащих лечению, в данном случае включают злокачественные новообразования и воспаления.
«Терапевтически эффективное количество» представляет собой по меньшей мере минимальную концентрацию, необходимую для оказания измеримого улучшения или предотвращения конкретного расстройства. Терапевтически эффективные количества известных белков хорошо известны в данной области, при этом эффективные количества белков раскрываемых далее в данном случае могут быть определены посредством стандартных методик, которые вполне находятся в пределах компетентности квалифицированного специалиста, такого как рядовой врач.
Способы получения антител (включая антитела, которые конъюгируют с токсинами) и других белков, которые могут быть введены в готовую форму, как описано в данном случае, хорошо известны в данной области и подробно описаны, например, в WO2007/001851.
В соответствии с представленным изобретением, антитела и другие белки могут быть введены в готовую форму либо в водной, либо в лиофилизированной форме, причем последнюю можно восстановить в водную форму.
Готовые формы, описанные в данном документе, можно приготовить в виде восстановленных лиофилизированных готовых форм. Для получения жидких готовых форм изобретения, белки или антитела, описанные в данном документе, лиофилизируют, а затем восстанавливают. В данном конкретном варианте осуществления, после получения представляющего интерес белка, как описано выше, получают «предварительно лиофилизированную готовую форму». Количество белка, присутствующего в предварительно лиофилизированной готовой форме, определяют, принимая в расчет необходимые объемы дозы, режим (режимы) введения и т.д. например, начальная концентрация исходного антитела может составлять от приблизительно 2 мг/мл до приблизительно 50 мг/мл, предпочтительно от приблизительно 5 мг/мл до приблизительно 40 мг/мл и наиболее предпочтительно от приблизительно 20 до 30 мг/мл.
В общем, белок, подлежащий введению в готовую форму, находится в растворе. Например, в жидких готовых формах изобретения, белок может присутствовать в кислотно-основном буферном растворе при pH от приблизительно 4 до 8, и предпочтительно от приблизительно 5 до 7. Концентрация буфера может составлять от приблизительно 1 мМ до приблизительно 20 мМ, в качестве альтернативы от приблизительно 3 мМ до приблизительно 15 мМ, в зависимости, например, от буфера и необходимой тоничности готовой формы (например, восстановленной готовой формы). Приведенные для примера буферы и/или соли представляют собой буферы и/или соли, которые являются фармацевтически приемлемыми и могут быть созданы из подходящих кислот, оснований и их солей, например, таких, которые определены под термином «фармацевтически приемлемые» кислоты, основания или буферы.
В одном варианте осуществления, к предварительно лиофилизированной готовой форме добавляют лиопротектант. Количество лиопротектанта в предварительно лиофилизированной готовой форме в общем такое, что при восстановлении, полученная в результате готовая форма будет изотонической. Однако подходящими также могут быть гипертонические восстановленные готовые формы. В дополнение, количество лиопротектанта не должно быть слишком низким, чтобы при лиофилизации не произошел распад/агрегация белка в неприемлемом объеме. Однако приведенные для примера концентрации лиопротектанта в предварительно лиофилизированной готовой форме составляют от приблизительно 10 мМ до приблизительно 400 мМ, в качестве альтернативы от приблизительно 30 мМ до приблизительно 300 мМ, в качестве альтернативы от приблизительно 50 мМ до приблизительно 100 мМ. Приведенные для примера лиопротектанты включают сахара и сахарные спирты, такие как сахароза, манноза, трегалоза, глюкоза, сорбитол, маннитол. Однако в конкретных обстоятельствах, некоторые лиопротектанты также могут принимать участие в повышении вязкости готовой формы. В связи с этим, необходимо соблюдать осторожность чтобы выбрать конкретные лиопротектанты, которые минимизируют или нейтрализуют данное действие. Дополнительные лиопротектанты описаны выше под определением «лиопротектантов», также упоминаемых в данном случае, как «фармацевтически приемлемые сахара».
Для каждой конкретной комбинации белка или антитела и лиопротектанта соотношение белка и лиопротектанта может варьировать. В случае выбора антитела в качестве белка и сахара (например, сахарозы или трегалозы) в качестве лиопротектанта для получения изотонической восстановленной готовой формы с высокой концентрацией белка, молярное соотношение лиопротектанта и антитела может составлять от приблизительно 100 до приблизительно 1500 молей лиопротектанта на 1 моль антитела, и предпочтительно от приблизительно 200 до приблизительно 1000 молей лиопротектанта на 1 моль антитела, например от приблизительно 200 до приблизительно 600 молей лиопротектанта на 1 моль антитела.
При приготовлении предварительно лиофилизированной готовой формы может быть использована смесь лиопротектанта (такого как сахароза или трегалоза) и объемообразующего агента (например, маннитола или глицина). Объемообразующий агент может обеспечить возможность получения однородной лиофилизированной таблетки без излишних полостей в них и т.д. Другие фармацевтически приемлемые носители, эксципиенты или стабилизаторы, например, те, которые описаны в Remington's Pharmaceutical Sciences 16th edition, Osol, A. Ed. (1980) могут быть включены в предварительно лиофилизированную готовую форму (и/или лиофилизированную готовую форму и/или восстановленную готовую форму) при условии, что они не оказывают вредного воздействия на необходимые характеристики готовой формы. Приемлемые носители, эксципиенты или стабилизаторы являются нетоксичными к реципиентам в используемых дозировках и концентрациях и включают: дополнительные буферные агенты; консерванты; вспомогательные растворители; антиоксиданты, включая аскорбиновую кислоту и метионин; комплексообразующие агенты, такие как EDTA; металлокомплексы (например, комплексы Zn-белок); биоразлагаемые полимеры, такие как сложные полиэфиры и/или солеобразующие противоионы, такие как натрий.
Готовая форма в данном случае может также включать более чем один белок в случае необходимости для конкретного показания, подлежащего лечению, предпочтительно белки с взаимодополняющими функциями, которые не оказывают вредного воздействия на другой белок. Например, в одной готовой форме может быть необходимо предоставить два или более антител, которые связываются с требуемой мишенью (например, рецептором или антигеном). Подобные белки присутствуют соответствующим образом в комбинации в количествах, которые являются эффективными для предполагаемой цели.
Готовые формы, подлежащие использованию для введения in vivo, должны быть стерильными. Это легко осуществить посредством фильтрования через стерильные фильтровальные мембраны, перед или после лиофилизации и восстановления. В качестве альтернативы, стерильность всей смеси может быть достигнута посредством автоклавирования ингредиентов, за исключением белка, приблизительно при 120°C в течение, например, приблизительно 30 минут.
После того, как белок, необязательный лиопротектант и другие необязательные составные элементы смешивают вместе, готовую форму лиофилизируют. Для данной цели доступно множество различных лиофилизаторов, таких как лиофилизаторы Hull50™ (Hull, USA) или GT20™ (Leybold-Heraeus, Germany). Лиофилизацию осуществляют посредством замораживания готовой формы, а затем сублимации льда из замороженного содержимого при температуре, подходящей для первичной сушки. В данном состоянии, температура продукта ниже эвтектической точки или температуры разрушения готовой формы. Как правило, температура на полках для первичной сушки будет колебаться от приблизительно -30 до 25°C (при условии, что продукт остается замороженным в процессе первичной сушки) при подходящем давлении, варьирующем, как правило, от приблизительно 50 до 250 мТорр. готовая форма, размер и тип контейнера, вмещающего образец (например, стеклянный флакон) и объем жидкости будут главным образом предписывать время, необходимое для сушки, которое может колебаться от нескольких часов до нескольких дней (например, 40-60 часов). Необязательно, также может быть выполнена стадия дополнительной сушки в зависимости от необходимого уровня остаточной влажности продукта. Температура, при которой проводят дополнительную сушку, варьирует от приблизительно 0 до 40°C, главным образом в зависимости от типа и размера контейнера и типа используемого белка. Например, температура на полке на протяжении всей фазы удаления воды лиофилизации может составлять от приблизительно 15 до 30°C (например, приблизительно 20°C). Время и давление, необходимые для дополнительной сушки, будут такими, чтобы обеспечить подходящую лиофилизированную таблетку, и зависят, например, от температуры и других параметров. Время дополнительной сушки предписывается требуемым уровнем остаточной влажности продукта и, как правило, занимает по меньшей мере приблизительно 5 часов (например, 10-15 часов). Давление может быть таким же, как давление, используемое во время стадии первичной сушки. Условия лиофилизации могут колебаться в зависимости от готовой формы и размера флакона.
Перед введением пациенту, лиофилизированную готовую форму восстанавливают с помощью фармацевтически приемлемого разбавителя таким образом, чтобы концентрация белка в восстановленной готовой форме составляла по меньшей мере приблизительно 50 мг/мл, например, от приблизительно 50 мг/мл до приблизительно 400 мг/мл, в качестве альтернативы от приблизительно 80 мг/мл до приблизительно 300 мг/мл, в качестве альтернативы от приблизительно 90 мг/мл до приблизительно 150 мг/мл. Считается, что подобные высокие концентрации белка в восстановленной готовой форме особенно подходят, когда предполагается подкожная доставка восстановленной готовой формы. Однако, для других способов введения, таких как внутривенное введение, могут быть необходимы более низкие концентрации белка в восстановленной готовой форме (например, от приблизительно 5 до 50 мг/мл, или от приблизительно 10 до 40 мг/мл белка в восстановленной готовой форме). В некоторых вариантах осуществления, концентрация белка в восстановленной готовой форме значительно выше, чем концентрация в предварительно лиофилизированной готовой форме. Например, концентрация белка в восстановленной готовой форме может быть приблизительно в 2-40 раз, в качестве альтернативы в 3-10 раз, в качестве альтернативы в 3-6 раз (например, по меньшей мере трехкратно или по меньшей мере четырехкратно), выше концентрации предварительно лиофилизированной готовой формы.
В общем, для обеспечения полной гидратации, восстановление происходит при температуре, равной приблизительно 25°C, хотя при необходимости могут быть использованы другие температуры. Время, необходимое для восстановления, будет зависеть, например, от типа разбавителя, количества эксципиента (эксципиентов) и белка. Приведенные для примера разбавители включают стерильную воду, бактериостатическую воду для инъекции (BWF), кислотно-основной буферный раствор (например, забуференный фосфатом физиологический раствор), стерильный раствор натрия хлорида, раствор Рингера или раствор декстрозы. Необязательно разбавитель содержит консервант. Приведенные для примера консерванты были описаны выше, при этом предпочтительными консервантами являются ароматические спирты, такие как бензиловый или фенолспирт. Количество используемого консерванта определяют с помощью оценки различных концентраций консервантов на совместимость с белком и испытания эффективности консервации. Например, если консервантом является ароматический спирт (такой как бензиловый спирт), он может присутствовать в количестве от приблизительно 0,1 до 2,0% и предпочтительно от приблизительно 0,5 до 1,5%, но наиболее предпочтительно приблизительно 1,0-1,2%.
Предпочтительно, восстановленная готовая форма имеет менее чем 6000 частиц на флакон, размер которых составляет >10 мкм.
Терапевтические готовые формы готовят к хранению посредством смешивания активного ингредиента, имеющего необходимую степень чистоты, с необязательными фармацевтически приемлемыми носителями, эксципиентами или стабилизаторами (Remington's Pharmaceutical Sciences 18th edition, Mack Publishing Co., Easton, Pa. 18042 [1990]). Приемлемые носители, эксципиенты или стабилизаторы нетоксичны к реципиентам в используемых дозировках и концентрациях и включают буферы, антиоксиданты, включая аскорбиновую кислоту, метионин, витамин E, натрия метабисульфит, консерванты, изотонирующие добавки, стабилизаторы, металлокомплексы (например, комплексы Zn с белком), и/или комплексообразующие агенты, такие как EDTA.
Когда терапевтическим агентом является фрагмент антитела, предпочтительным является наименьший фрагмент, который специфически связывается с доменом связывания белка-мишени. Например, на основании последовательностей вариабельной области антитела, могут быть сконструированы фрагменты антител или даже пептидные молекулы, которые сохраняют способность связывать последовательность белка-мишени. Подобные пептиды могут быть синтезированы химически и/или получены посредством методики рекомбинантных ДНК (см., например, Marasco et al, Proc. Natl. Acad. Sci. USA 90: 7889-7893 [1993]).
Для регулирования pH в диапазоне, который оптимизирует терапевтическую эффективность, используют буферы, особенно если стабильность является pH зависимой. Буферы предпочтительно присутствуют в концентрациях, варьирующих от приблизительно 50 мМ до приблизительно 250 мМ. Подходящие буферные агенты для использования с представленным изобретением включают как органические, так и неорганические кислоты и их соли. Например, цитрат, фосфат, сукцинат, тартрат, фумарат, глюконат, оксалат, лактат, ацетат. Кроме того, буферы могут состоять из гистидина и солей триметиламина, таких как трис.
Для замедления роста микроорганизмов добавляют консерванты, которые, как правило, присутствуют в диапазоне от 0,2% до 1,0% (м/о), подходящие консерванты для использования с представленным изобретением включают октадецилдиметилбензил хлорид аммония; гексаметония хлорид; бензалкония галиды (например, хлорид, бромид, иодид), бензетония хлорид; тимерозал, фенол, бутиловый или бензиловый спирт; алкилпарабены, такие как метилпарабен или пропилпарабен; катехол; резорцинол; циклогексанол, 3-пентанол и m-крезол.
Регулирующие тоничность агенты, иногда называемые «стабилизаторы», присутствуют для регулировки или сохранения тоничности жидкой композиции. При использовании с большими, заряженными биомолекулами, такими как белки и антитела, их часто называют «стабилизаторы», потому что они могут взаимодействовать с заряженными группами боковых цепей аминокислот, уменьшая посредством этого возможность межмолекулярных и внутримолекулярных взаимодействий. Регулирующие тоничность агенты могут присутствовать в любом количестве между 0,1% и 25% по массе, предпочтительно между 1 и 5%, принимая в расчет относительные количества других ингредиентов. Предпочтительные регулирующие тоничность агенты включают многоатомные сахарные спирты, предпочтительно трехатомные или высшие сахарные спирты, такие как глицерин, эритритол, арабитол, ксилитол, сорбитол и маннитол.
Дополнительные эксципиенты включают агенты, которые могут служить в качестве одого или более из следующего: (1) объемообразующих агентов, (2) усилителей растворимости, (3) стабилизаторов и (4) и агентов, предотвращающих денатурацию или прилипание к стенке контейнера. Подобные эксципиенты включают: многоатомные сахарные спирты (перечисленные выше); аминокислоты, такие как аланин, глицин, глютамин, аспарагин, гистидин, аргинин, лизин, орнитин, лейцин, 2-фенилаланин, глютаминовая кислота, треонин и т.д.; органические сахара или сахарные спирты, такие как сахароза, лактоза, лактитол, трегалоза, стахиоза, манноза, сорбоза, ксилоза, рибоза, рибитол, миоинозитоза, миоинозитол, галактоза, галактитол, глицерол, циклитолы (например, инозитол), полиэтиленгликоль; серосодержащие восстанавливающие агенты, такие как мочевина, глютатион, тиоктовая кислота, натрия тиогликолат, тиоглицерол, a-монотиоглицерол и натрия тиосульфат; низкомолекулярные белки, такие как человеческий сывороточный альбумин, бычий сывороточный альбумин, желатин или другие иммуноглобулины; гидрофильные полимеры, такие как поливинилпирролидон; моносахариды (например, ксилоза, манноза, фруктоза, глюкоза; дисахариды (например, лактоза, мальтоза, сахароза); трисахариды, такие как раффиноза; и полисахариды, такие как декстрин или декстран.
Для того, чтобы готовые формы можно было использовать для введения in vivo, они должны быть стерильными. Готовую форму можно привести в стерильное состояние посредством фильтрования через стерильные фильтрующие мембраны. В общем в данном случае терапевтические композиции помещают в контейнер, имеющий стерильное впускное отверстие, например, пакет или флакон с внутривенным раствором, имеющий пробку, поддающуюся прокалыванию иглой для подкожных инъекций.
Способ введения находится в соответствии с известными и признанными способами, например посредством единственного или множества болюсов или инфузии в течение продолжительного периода времени подходящим образом, например, инъекция или инфузия посредством подкожного, внутривенного, внутрибрюшинного, внутримышечного, внутриартериального, внутриочагового или внутрисуставного способов применения, местного введения, ингаляции или с помощью средства замедленного высвобождения или пролонгированного высвобождения.
Готовая форма в данном случае в случае необходимости также может содержать более чем одно активное соединение для конкретного показания, подлежащего лечению, предпочтительно активные соединения с взаимодополняющими функциями, которые не оказывают вредного воздействия друг на друга. В качестве альтернативы, или в дополнение, композиция может содержать цитотоксических агент, цитокин или ингибиторующий рост агент. Подобные молекулы присутствуют соответствующим образом в комбинации в количествах, которые являются эффективными для предполагаемой цели.
Активные ингредиенты также могут быть заключены в микрокапсулы, полученные, например, посредством методик коацервации или посредством межфазной полимеризации, например, гидроксиметилцеллюлозные или желатиновые микрокапсулы и поли-(метилметакрилатные) микрокапсулы, соответственно, в коллоидных системах доставки лекарственных средств (например, липосомах, альбуминовы микросферах, микроэмульсиях, наночастицах и нанокапсулах) или в макроэмульсиях. подобные методики раскрыты в Remington's Pharmaceutical Sciences 18th edition, supra.
Можно изготовить препараты с замедленным высвобождением. Подходящие примеры препаратов с замедленным высвобождением включают полупроницаемые матрицы твердых гидрофобных полимеров, содержащих антитело, причем данные матрицы находятся в виде имеющих определенную форму изделий, например, пленок или микрокапсул. Примеры матриц с замедленным высвобождением включают сложные полиэфиры, гидрогели (например, поли(2-гидроксиэтилметакрилат), или поли(винилспирт)), полилактиды (Патент США № 3773919), сополимеры L-глютаминовой кислоты и γ-этил-L-глютамат, неразлагаемый этиленвинилацетат, разлагаемые сополимеры молочной кислоты-гликолевой кислоты, такие как LUPRON DEPOT™ (инъецируемые микросферы, состоящие из сополимера молочной кислоты-гликолевой кислоты и лейпролид ацетата), и поли-D-(-)-3-гидроксимасляная кислота. Микрокапсулирование рекомбинантных белков для замедленного высвобождения было успешно выполнено с человеческим гормоном роста (rhGH), интерфероном-(rhIFN-), интерлейкином-2, и MN rpg 120. Johnson et al, Nat. Med. 2: 795-799 (1996); Yasuda et al, Biomed. Ther. 27: 1221-1223 (1993); Hora et al, Bio/Technology 8: 755-758 (1990); Cleland, «Design and Production of Single Immunization Vaccines, Using Polylactide Polyglicolide Microsphere Systems,» in Vaccine Design: The Subunit and Adjuvant Approach, Powell and Newman, eds., (Plenum Press: New York, 1995), pp. 439-462; WO 97/03692; WO 96/40072; WO 96/07399; и Патент США № 5654010.
Готовые формы с замедленным высвобождением данных белков могут быть разработаны с использованием полимера полимолочной-когликолевой кислоты (PLGA) вследствие его биосовместимости и широкого диапазона свойств биологического разложения. Продукты распада PLGA, молочную и гликолевую кислоты, можно быстро очистить внутри человеческого организма. Более того, возможность разложения данного полимера можно отрегулировать от месяцев до лет в зависимости от его молекулярной массы и композиции. Lewis, «Controlled release of bioactive agents from lactide/glycolide polymer», в Biodegradable Polymers as Drug Delivery Systems (Marcel Dekker; New York, 1990), M. Chasin and R. Langer (Eds.) pp. 1-41.
Тогда как полимеры, такие как этиленвинилацетат и молочная кислота-гликолевая кислота, обеспечивают возможность высвобождения молекул на протяжении свыше 100 дней, некоторые гидрогели высвобождают белки на протяжении более коротких периодов времени. Когда инкапсулированные антитела остаются в организме в течение долгого времени, они могут денатурировать или агрегировать в результате подверганию действию влаги при 37°C, приводя к потере биологической активности и возможным изменениям иммуногенности. Для стабилизации в зависимости от вовлеченного механизма могут быть разработаны рациональные стратегии. Например, если выявлено, что механизмом агрегации является образование межмолекулярных S--S связей через тиодисульфидный обмен, стабилизация может быть достигнута за счет модифицирования сульфгидрильных остатков, лиофилизации из кислотных растворов, регулирования содержания влаги, использования подходящих добавок и разработки специфичных полимерных матричных композиций.
Для приготовления готовых форм с белками или антителами, раскрытыми в данном случае, также могут быть использованы липосомальные или протеиноидные композиции. См. Патенты США №№ 4925673 и 5013556.
Стабильность белков и антител, описанных в данном случае, может быть повышена через использование нетоксичных «водорастворимых солей поливалентных металлов». Примеры включают Ca2+, Mg2+, Zn2+, Fe2+, Fe3+, Cu2+, Sn2+, Sn3+, Al2+ и Al3+. Примеры анионов, которые могут образовывать водорастворимые соли с приведенными выше катионами поливалентных металлов, включают анионы, образованные из неорганических кислот и/или органических кислот. Подобные водорастворимые соли обладают растворимостью в воде (при 20°C), равной по меньшей мере приблизительно 20 мг/мл, в качестве альтернативы, по меньшей мере приблизительно 100 мг/мл, как вариант по меньшей мере приблизительно 200 мг/мл.
Подходящие неорганические кислоты, которые могут быть использованы для образования «водорастворимых солей поливалентных металлов», включают соляную, уксусную, серную, азотную, тиоциановую и фосфорную кислоту. Подходящие оганические кислоты, которые могут быть использованы, включают алифатическую карбоновую кислоту и ароматические кислоты. Алифатические кислоты в пределах данного определения могут быть определены, как насыщенные или ненасыщенные C2-9 карбоновые кислоты (например, алифатические моно-, ди- и трикарбоновые кислоты). Например, приведенные для примера монокарбоновые кислоты в пределах данного определения включают насыщенные C2-9 монокарбоновые кислоты: уксусную, пропионовую, масляную, валериановую, капроновую, энантовую, каприловую, пеларгоновую и каприоновую, и ненасыщенные C2-9 монокарбоновые кислоты: акриловую, пропиоловую, метакриловую, кротоновую и изокротоновую кислоты. Приведенные для примера дикарбоновые кислоты включают насыщенные C2-9 дикарбоновые кислоты: малоновую, янтарную, глутаровую, адипиновую и пимелиновую, тогда как ненасыщенные C2-9 дикарбоновые кислоты включают малеиновую, фумаровую, цитраконовую и мезаконовую кислоты. Приведенные для примера трикарбоновые кислоты включают насыщенные C2-9 трикарбоновые кислоты трикарбаллиловую и 1,2,3-бутантрикарбоновую кислоту. Кроме того, карбоновые кислоты данного определения также могут включать одну или две гидроксильные группы с образованием гидроксикарбоновых кислот. Приведенные для примера гидроксикарбоновые кислоты включают гликолевую, молочную, глицериновую, тартроновую, яблочную, винную и лимонную кислоту. Ароматические кислоты в пределах данного определения включают бензойную и салициловую кислоту.
Широко используемые водорастворимые соли поливалентных металлов, которые могут быть использованы для помощи в стабилизации инкапсулированных полипептидов данного изобретения, включают, например: (1) соли металлов неорганических кислот галидов (например, хлорид цинка, хлорид кальция), сульфаты, нитраты, фосфаты и тиоцианаты; (2) соли металлов алифатической карбоновой кислоты (например, ацетат кальция, ацетат цинка, пропионат кальция, гликолат цинка, лактат кальция, лактат цинка и тартрат цинка); и (3) соли металлов ароматической карбоновой кислоты бензоатов (например, бензоат цинка) и салицилаты.
Для предотвращения или лечения заболевания, правильная дозировка активного агента будет зависеть от типа заболевания, подлежащего лечению, как определено выше, серьезности и течения заболевания, вводится ли агент с профилактической или терапевтической целями, предшествующего лечения, истории болезни пациента и реакции на агент и усмотрения лечащего врача. Агент соответствующим образом вводят пациенту за один раз или на протяжении серии обработок.
Способ изобретения может быть объединен с известными способами лечения расстройства, либо в виде объединенных или дополнительных стадий лечения, либо в виде дополнительных составных элементов терапевтической готовой формы.
Дозировки и необходимая концентрация лекарственного средства фармацевтических композиций представленного изобретения могут варьировать в зависимости от конкретного предполагаемого использования. Определение правильной дозировки или способа введения вполне попадает в пределы компетентности рядового специалиста. Эксперименты на животных предоставляют надежное руководство для определения эффективных доз для лечения людей. Межвидовую корректировку эффективных доз можно выполнить, следуя принципам, заложенным Mordenti, J. и Chappell, W. «The Use of Interspecies Scaling in Toxicokinetics», в Toxicokinetics and New Drug Development, Yacobi et al, Eds, Pergamon Press, New York 1989, pp. 42-46.
Когда используют введение in vivo полипептидов или антител, описанных в данном случае, нормальные величины дозировки могут варьировать от приблизительно 10 нг/кг до приблизительно 100 мг/кг массы тела млекопитающего или более в день, предпочтительно приблизительно от 1 мг/кг/день до 10 мг/кг/день, в зависимости от способа введения. Руководство, касающееся конкретных дозировок и способов доставки, предоставлено в литературе; см., например, Патент США № 4657760; 5206344; или 5225212. В пределах объема правовых притязаний изобретения находится то, что различные готовые формы будут эффективными для различных способов лечения и различных расстройств, и, что введение, предназначенное для лечения конкретного органа или ткани, может делать необходимой доставку посредством способа, отличающегося от способа для другого органа или ткани. Более того, дозы можно вводить посредством одного или более отдельных введений или посредством непрерывной инфузии. Для многократных введений на протяжении нескольких дней или дольше, в зависимости от состояния, лечение продолжают до тех пор, пока не произойдет требуемое подавление симптомов заболевания. Однако могут быть использованы другие схемы приема. Ход данного лечения легко отслеживать с помощью общепризнанных методик и анализов.
Готовые формы представленного изобретения, включая, но без ограничения, восстановленные готовые формы, вводят млекопитающему, нуждающемуся в лечении, с белком, предпочтительно человеческим, в соответствии с известными способами, такими как внутривенное введение в виде ударной дозы вещества или посредством непрерывной инфузии в течение некоторого периода времени, посредством внутримышечного, внутрибрюшинного, интраспинального, подкожного, интроартикулярного, внутрисуставного, интратекального, перорального, местного или ингаляционного способов применения.
В предпочтительных вариантах осуществления, готовые формы вводят млекопитающему посредством подкожного (т.е., под кожу) введения. Для этих целей, готовая форма может быть инъецирована при использовании шприца. Однако, для введения готовой формы доступны другие устройства, такие как инъекционные устройства (например, устройства Inject-ease™ и Genject™); шприц-ручки (такие как GenPen™); автоинжекторные устройства, безыгольные устройства (например, MediJector™ и BioJector™); и подкожные системы доставки с помощью пластыря.
В специальном варианте осуществления, представленное изобретение направлено на наборы для блока введения однократной дозы. Подобные наборы содержат контейнер с водной готовой формой терапевтического белка или антитела, содержащий как однокамерные, так и многокамерные предварительно заполненные шприцы. Приведенные для примера предварительно заполненные шприцы доступны от Vetter GmbH, Ravensburg, Germany.
Вводится ли белок для профилактической или терапевтической целей, правильная дозировка («терапевтически эффективное количество») белка будет зависеть, например, от состояния, подлежащего лечению, серьезности и течения заболевания, предшествующего лечения, истории болезни пациента и реакции на белок, типа используемого белка и усмотрения лечащего врача. Белок вводят соответствующим образом пациенту за один раз или на протяжении серии обработок и его можно вводить пациенту в любое время с момента диагностирования. Белок может вводиться в виде единственного лечения или в сочетании с другими лекарственными средствами или способами лечения, используемыми для лечения состояния, о котором идет речь.
Когда белком выбора является антитело, первоначальная возможная дозировка для введения пациенту составляет приблизительно от 0,1-20 мг/кг, посредством, например, одного ли или более отдельных введений. Однако могут быть использованы другие схемы приема. Ход данного лечения легко отслеживать с помощью общепризнанных методик.
В еще одном варианте осуществления изобретения, предоставлено изделие, которое содержит готовую форму и предпочтительно предоставляет инструкции для ее использования. Изделие содержит контейнер. Подходящие контейнеры включают, например, бутылки, флаконы (например, двухкамерные флаконы), шприцы (такие как шприцы с одной или двумя камерами) и пробирки. Контейнер может быть образован из множества материалов, таких как стекло или пластмасса. Этикетка, которая находится на контейнере, вмещающем готовую форму, или соединена с ним, может содержать инструкции для восстановления и/или применения. Этикетка может дополнительно сообщать, что готовая форма может быть использована или предназначена для подкожного введения. Контейнер, вмещающий готовую форму, может представлять собой флакон многоразового использования, который предоставляет возможность повторных введений (например, от 2 до 6 введений) восстановленной готовой формы. Изделие может дополнительно содержать второй контейнер, содержащий подходящий разбавитель (например, BWFI). При смешивании разбавителя и лиофилизированной готовой формы, итоговая концентрация белка в восстановленной готовой форме в общем будет составлять по меньшей мере 50 мг/мл. Изделие может дополнительно содержать другие материалы, желательные с коммерческой и потребительской точки зрения, включая другие буферы, разбавители, фильтры, иглы, шприцы и листовки-вкладыши с инструкциями для применения.
Изобретение будет более полно понятно с помощью ссылки на следующие примеры. Однако их не следует трактовать как ограничение объема правовых притязаний изобретения. Все цитаты на протяжении всего раскрытия в прямой форме включены в данную заявку посредством ссылки.
Пример 1-Исследование действия жирных кислот, полисорбатов и ПОЭ-сорбитана на агрегацию белка
Данный пример иллюстрирует, как полисорбаты, жирные кислоты и ПОЭ сорбитан влияют на агрегацию белка в водном растворе.
Защитное действие ПОЭ сорбитана против индуцируемой взбалтыванием агрегации анти-IL13 моноклональных антител в растворе оценивали, используя анализ индуцируемой взбалтыванием агрегации белка. Конкретно, в данном исследовании, буферные растворы, содержащие 1 мг/мл анти-IL13 моноклональных антител (20 мм His-OAc, pH 5,7), готовили в комбинации со следующими потенциально стабилизирующими добавками:
(i) без добавки, контрольные;
(ii) лауриновая кислота (29 м.д.);
(iii) лауриновая кислота (29 м.д.) и полисорбат 20 (24 м.д.);
(iv) ПОЭ сорбитан 20 «(a+b+c+d=20)» (150 м.д.);
(v) ПОЭ сорбитан 20 «(a+b+c+d=20)» (150 м.д.) и полисорбат 20 (24 м.д.);
(vi) ПОЭ сорбитан 20 «(a+b+c+d=20)» (150 м.д.) и лауриновая кислота (29 м.д.);
(vii) ПОЭ сорбитан 20 «(a+b+c+d=20)» (150 м.д.), лауриновая кислота (29 м.д.) и полисорбат 20 (24 м.д.);
(viii) полисорбат 20 (24 м.д.).
Девять мл содержащей моноклональные антитела готовой формы помещали в отдельные 15 мл флаконы Forma Vitrium (в трех повторениях), флаконы герметично закрывали и затем обеспечивали возможность их взбалтывания на настольном шейкере (при 70 об/мин) при комнатной температуре в течение 0 часов, 4 часов или 24 часов. При завершении, содержимое каждой флаконы немедленно подвергали анализу методом УФ-спектрометрии (340-360 нм) для измерения мутности раствора. Мутность получали при 25°C, используя ячейку с толщиной слоя 1 см и спектрометр Agilent 8453 UV. Усредняли значения поглощающей способности при 340, 345, 350, 355 и 360 нм, где ни одна из хромофор содержащих белок готовых форм не поглощает, и нельзя было определить эффекты рассеивания нерастворимых белковых агрегатов. Усредненные значения поглощающей способности при приведенных выше длины волн отображают мутность образца. В этом отношении в данной области хорошо известно, что мутность белоксодержащего раствора прямо и в количественном отношении коррелирует с количеством белка агрегация в растворе (например, см. Dani et al, J. Pharm. Sci., 96(6): 1504-1517 (2007)). Результаты из этих анализов показаны на фигуре 1.
Данные, показанные на фигуре 1, демонстрируют, что некоторые продукты распада полисорбата (включая жирную кислоту, лауриновую кислоту) оказывают неблагоприятное воздействие на стабильность белков в водном растворе и вызывают агрегацию белка при взбалтывании. В отличие от этого, добавление ПОЭ сорбитана в водные содержащие антитела растворы предотвращало образование белковых агрегатов в растворе при взбалтывании. Вследствие этого, эти данные демонстрируют, что ПОЭ сорбитан обладает эффектом предотвращения агрегация белков при взбалтывании и, следовательно, повышения стабильности терапевтических белков в растворе.
Пример 2 - исследование действия ПОЭ-сорбитана и ПЭГ на агрегацию белка
Данный пример иллюстрирует использование ПОЭ сорбитанов и ПЭГ в качестве стабилизаторов для предотвращения или уменьшения агрегации белков.
Защитное действие различных ПОЭ сорбитанов и ПЭГ против индуцируемой взбалтыванием агрегации двух моноклональных антител, анти-IL13 и анти-IgE, в растворе оценивали, используя анализ индуцируемой взбалтыванием агрегации белка.
В данной группе исследований, буферные растворы, содержащие 1 мг/мл либо антитела анти-IL13 (20 мм His-OAc, pH 5,7), либо антитела анти-IgE (His-HisCl, pH 6,0), готовили со следующими добавками:
(i) без добавки, контрольные;
(ii) ПОЭ сорбитан 20 «(a+b+c+d=20)» в концентрации, равной 200 м.д.;
(iii) ПОЭ сорбитан 20 «(a+b+c+d=20)» в концентрации, равной 1000 м.д.;
(iv) ПОЭ сорбитан 20 «(a+b+c+d=20)» в концентрации, равной 5000 м.д.;
(v) ПЭГ 1000 в концентрации, равной 200 м.д.;
(vi) ПЭГ 1000 в концентрации, равной 1000 м.д.;
(vii) ПЭГ 1000 в концентрации, равной 5000 м.д.;
(viii) ПЭГ 6000 в концентрации, равной 200 м.д.;
(ix) ПЭГ 6000 в концентрации, равной 1000 м.д.;
(x) ПЭГ 6000 в концентрации, равной 5000 м.д.
Девять мл каждой содержащей антитела готовой формы помещали в отдельные 15 мл флаконы Forma Vitrium (в трех повторениях), флаконы герметичо закрывали, а затем обеспечивали возможность их взбалтывания на настольном шейкере (при 70 об/мин) при комнатной температуре в течении 0 часов, 4 часов или 24 часов. При завершении, содержимое каждой флаконы немедленно подвергали каждому из следующих анализов, (a) анализ методом УФ-спектрометрии для измерения концентрации белка после фильтрования (b) анализ методом УФ-спектрометрии (340-360 нм) для измерения мутности раствора, и (c) анализ методом светотени для определения размера и распределения белковых частиц.
A. УФ спектрометрия для измерения концентрации белка
Немедленно после подвергания встряхиванию, как описано выше, белоксодержащие растворы фильтровали для удаления белковых агрегатов, а затем определяли концентрацию белка в фильтрате посредством УФ-спектрометрии. Данные по концентрации белка получали при 25°C, используя ячейку с толщиной слоя 0,5 или 1 см и спектрометр Agilent 8453 UV. Коэффициент ослабления E, равный 1,45 и 1,60 мл мг-1 см-1 при 278 нм, использовали с целью определения концентраций антител после фильтрования через 0,2 мкм шприцевой фильтр. Значения поглощающей способности при 320 нм вычитали из значения поглощающей способности при 278 нм для расчета эффектов рассеивания. В этом отношении, в данной области хорошо известно, что концентрация белка в белоксодержащем растворе может быть количественно измерена, с использованием анализа поглощения в УФ области (например, см. Liu et al, J. Pharm. Sci., 94(9): 1928-1940 (2005)). Результаты данных, полученных для антитела анти-IL13 и антитела анти-IgE, показаны на фигурах 2 и 3, соответственно.
Данные на фигурах 2 и 3 демонстрируют, что взбалтывание необработанных контрольных антительных готовых форм индуцировало измеримую и значительную агрегацию и потерю белка при фильтровании. В отличие от этого, добавление ПОЭ сорбитана или ПЭГ во все различные испытываемые концентрации предотвращало образование индуцируемых взбалтыванием белковых агрегатов и, следовательно, потерю белка при фильтровании. Эти данные демонстрируют, что как ПОЭ сорбитан, так и полиэтиленгликоль функционируют, как эффективные стабилизаторы белков в водных растворах за счет предотвращения или уменьшения образования в них белковых агрегатов.
B. УФ-спектрометрия для измерения мутности раствора
Как описано выше, УФ-спектрометрия при 340-360 нм предоставляет эффективное средство для количественного определения количества белкового агрегата, присутствующего в растворе, в котором мутность прямо коррелирует с количеством присутствующего агрегированного белка. Результаты, полученные из анализов мутности для антитела анти-IL13 и антитела анти-IgE, показаны на фигурах 4 и 5, соответственно.
Данные на фигурах 4 и 5 демонстрируют, что взбалтывание необработанных контрольных антительных готовых форм индуцировало измеримую и значительную агрегацию антител в них. В отличие от этого, добавление ПОЭ сорбитана или ПЭГ во все различные испытываемые концентрации предотвращало и/или уменьшало образование индуцируемых взбалтыванием белковых агрегатов. Эти данные демонстрируют, что как ПОЭ сорбитан, так и полиэтиленгликоль функционируют, как эффективные стабилизаторы белков в водных растворах за счет предотвращения или уменьшения образования в них белковых агрегатов.
C. Определение распределения размеров частиц
Описанные выше содержащие антитела водные растворы также анализировали с целью определения распределения размеров частиц содержащихся в них белков. Конкретно, количество и размер нерастворимых частиц между 2 и 50 мкм измеряли при комнатной температуре, используя счетчик частиц в жидкости HIAC/Royco 9703, прикрепленный к HIAC/Royco 3000A шприц для взятия проб жидкости, датчик HRLD-150 и анализировали, используя программное обеспечение PacificSpec Version 2,0. Верхний предел измерения составляет ~18000 частиц/мл, и образцы, которые превышают данное пороговое значение, для измерения надлежащим образом разбавляли. Каждый образец измеряли четыре раза с объемом, равным 1,0 мл на инъекцию. Первую инъекцию отбрасывали, и среднее значение получали из последних трех инъекций. Между каждым анализом образца, систему промывали водой для инъекций до точки, при которой количества 2 мкм частица устройства составляли <10, невидимые частицы ≥2, 5, 10, 15, 25, 35, и 50 мкм представлены в виде кумулятивной встречаемости на мл.
Результаты, полученные из этих анализов, показаны на фигурах 6-11. Данные на фигурах 6-11 демонстрируют, что взбалтывание необработанных контрольных антительных готовых форм индуцировало измеримую и значительную агрегацию антител в них. В отличие от этого, добавление ПОЭ сорбитана или ПЭГ во все различные испытываемые концентрации предотвращало и/или уменьшало образование индуцируемых взбалтыванием белковых агрегатов. Эти данные демонстрируют, что как ПОЭ сорбитан, так и полиэтиленгликоль функционируют как эффективные стабилизаторы белков в водных растворах за счет предотвращения или уменьшения образования в них белковых агрегатов.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПОЗИЦИИ И СПОСОБЫ, ВКЛЮЧАЮЩИЕ АЛКИЛГЛИКОЗИДЫ, ДЛЯ СТАБИЛИЗАЦИИ СОДЕРЖАЩИХ БЕЛКИ СОСТАВОВ | 2011 |
|
RU2575830C2 |
| ВЫСОКОКОНЦЕНТРИРОВАННЫЕ КОМПОЗИЦИИ АНТИТЕЛ И БЕЛКОВ | 2004 |
|
RU2332986C2 |
| Водная фармацевтическая композиция рекомбинантного моноклонального антитела к ФНОα | 2016 |
|
RU2665966C2 |
| Водная фармацевтическая композиция рекомбинантного моноклонального антитела к ФНОа | 2017 |
|
RU2764521C2 |
| ОДНОЦЕПОЧЕЧНЫЕ БЕЛКИ-АГОНИСТЫ РЕЦЕПТОРА CD40 | 2016 |
|
RU2745801C2 |
| СТАБИЛЬНЫЕ БЕЛКОВЫЕ ПРЕПАРАТЫ, СОДЕРЖАЩИЕ МОЛЯРНЫЙ ИЗБЫТОК СОРБИТОЛА | 2015 |
|
RU2689145C2 |
| СПОСОБ ОЧИСТКИ АРО-2 LIGAND/TRAIL ПОСРЕДСТВОМ КРИСТАЛЛИЗАЦИИ НА ХОЛОДЕ | 2006 |
|
RU2435781C2 |
| СОСТАВЫ АНТИТЕЛ | 2014 |
|
RU2694055C2 |
| КОМПОЗИЦИИ АНТИТЕЛ | 2005 |
|
RU2426554C2 |
| ГУМАНИЗИРОВАННЫЕ АНТИТЕЛА С ПОВЫШЕННОЙ СТАБИЛЬНОСТЬЮ | 2015 |
|
RU2684703C2 |
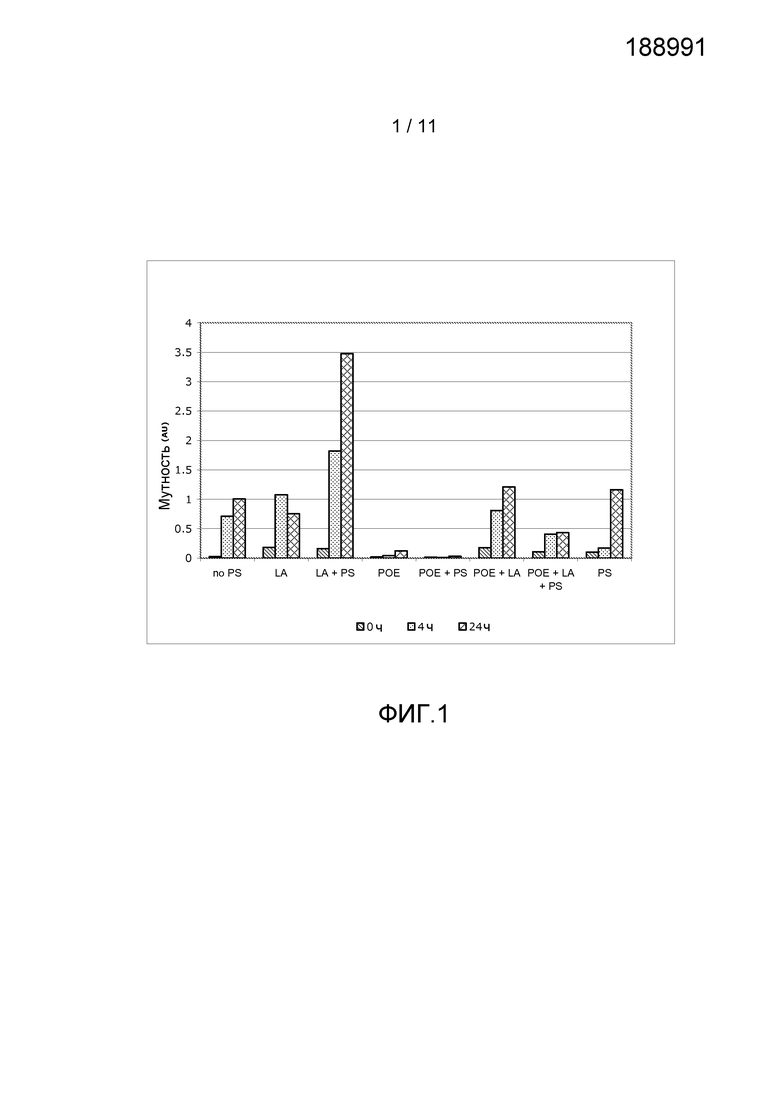
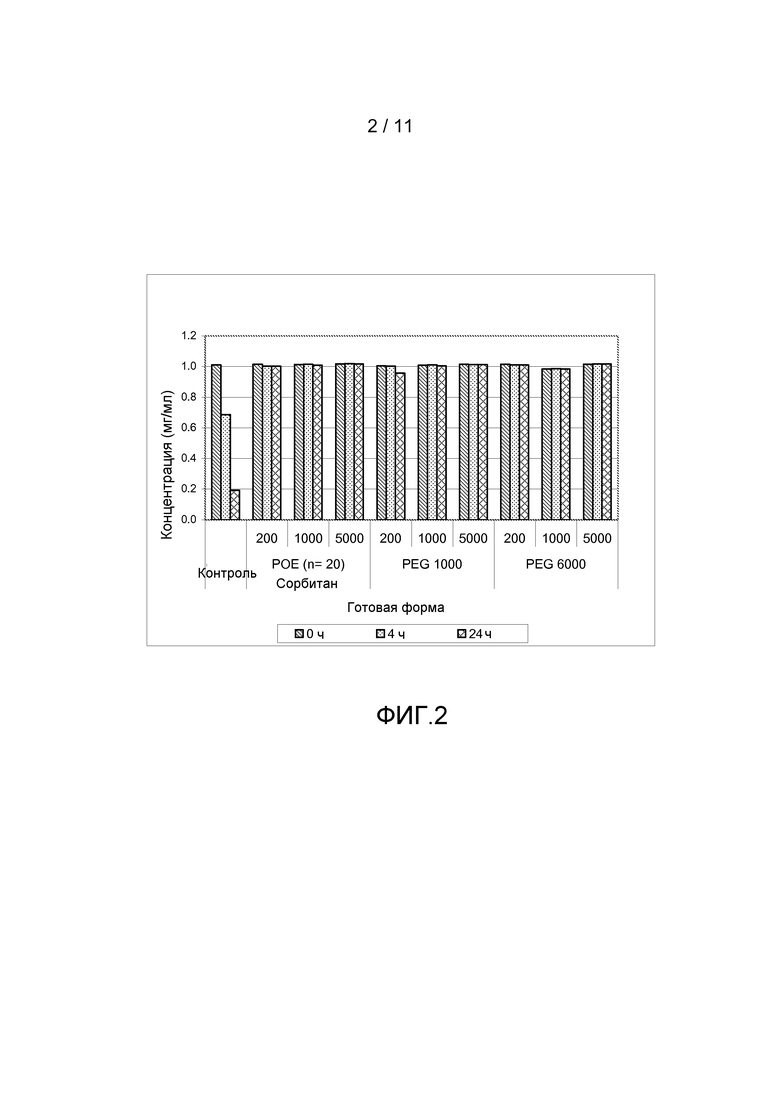
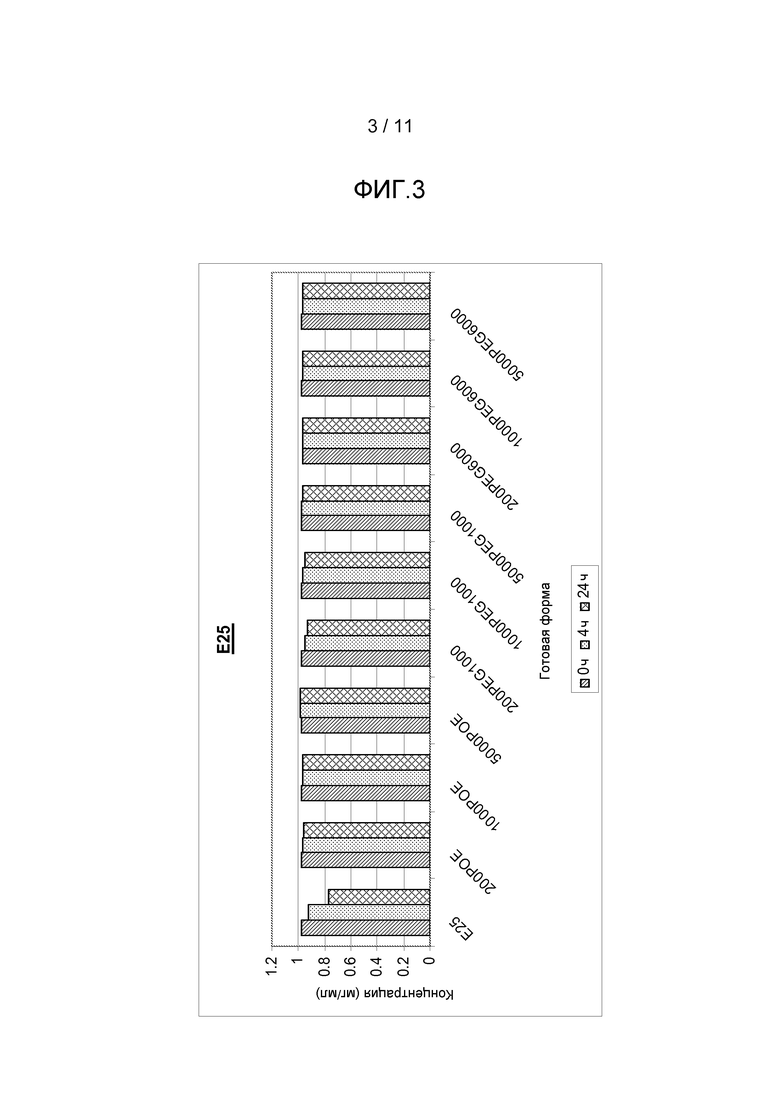
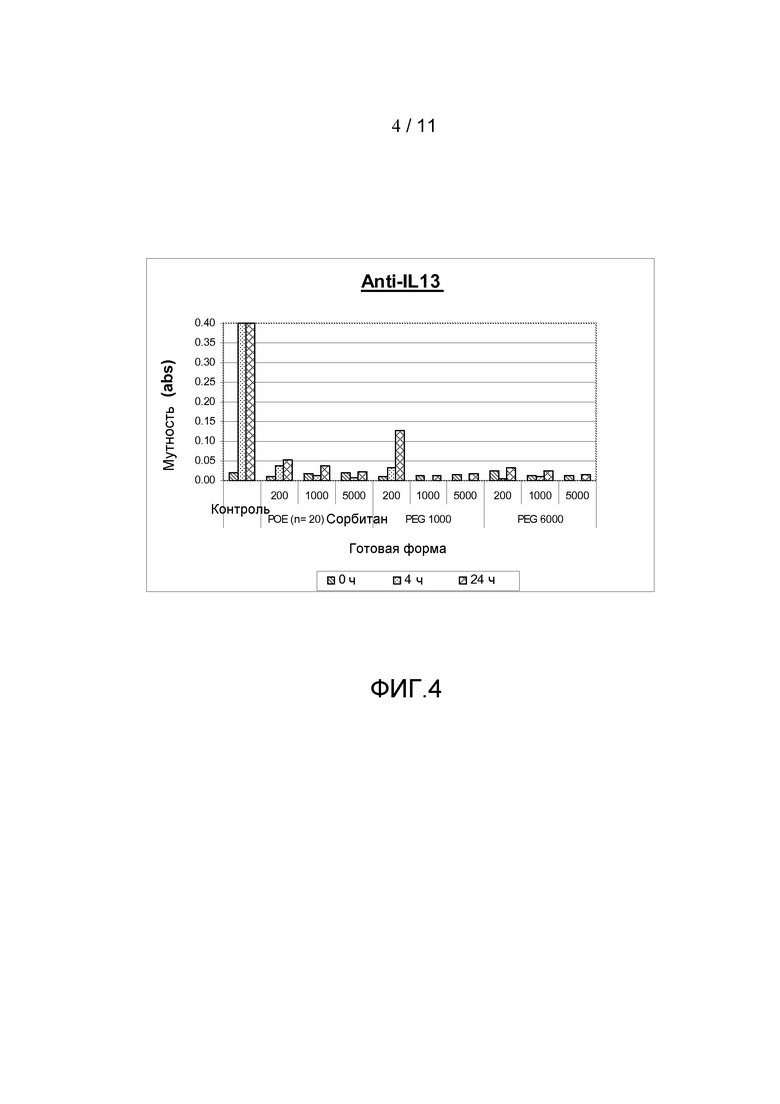
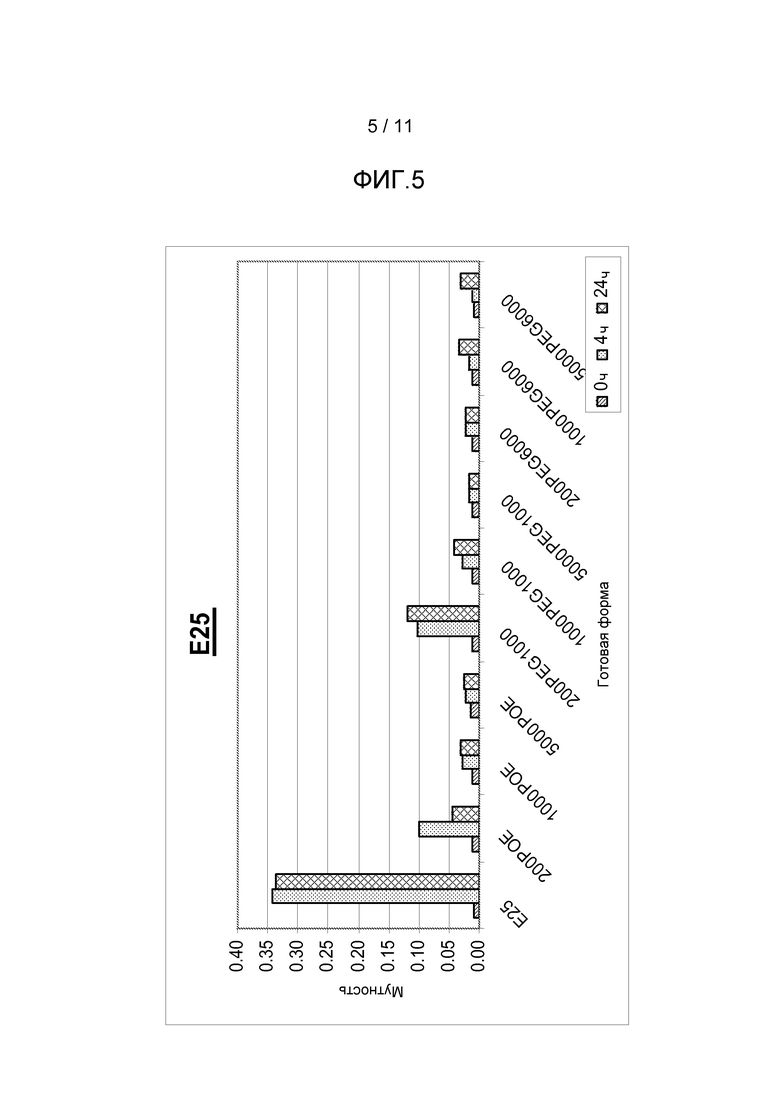
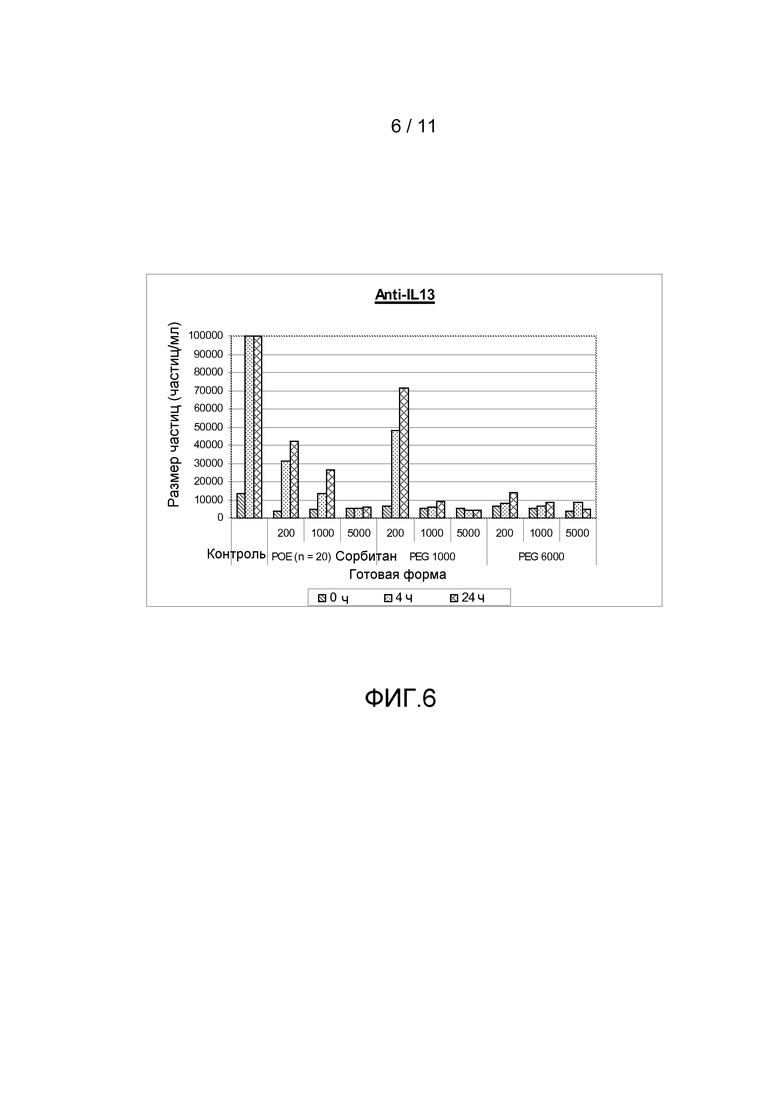
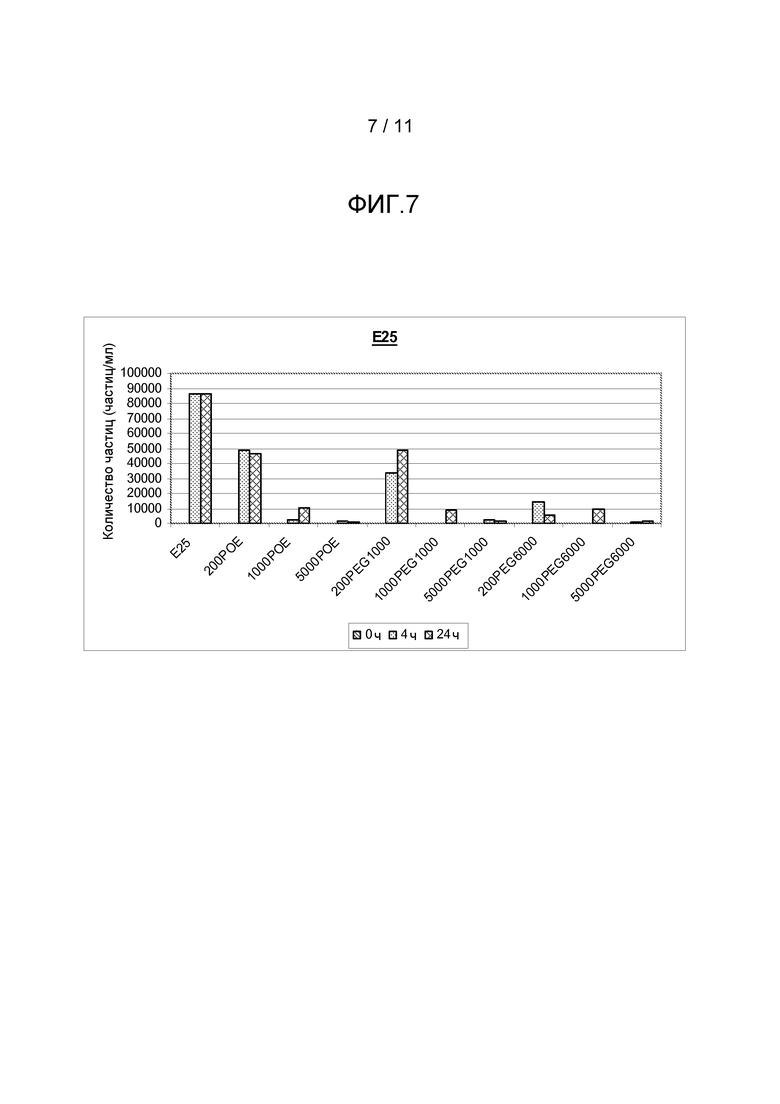
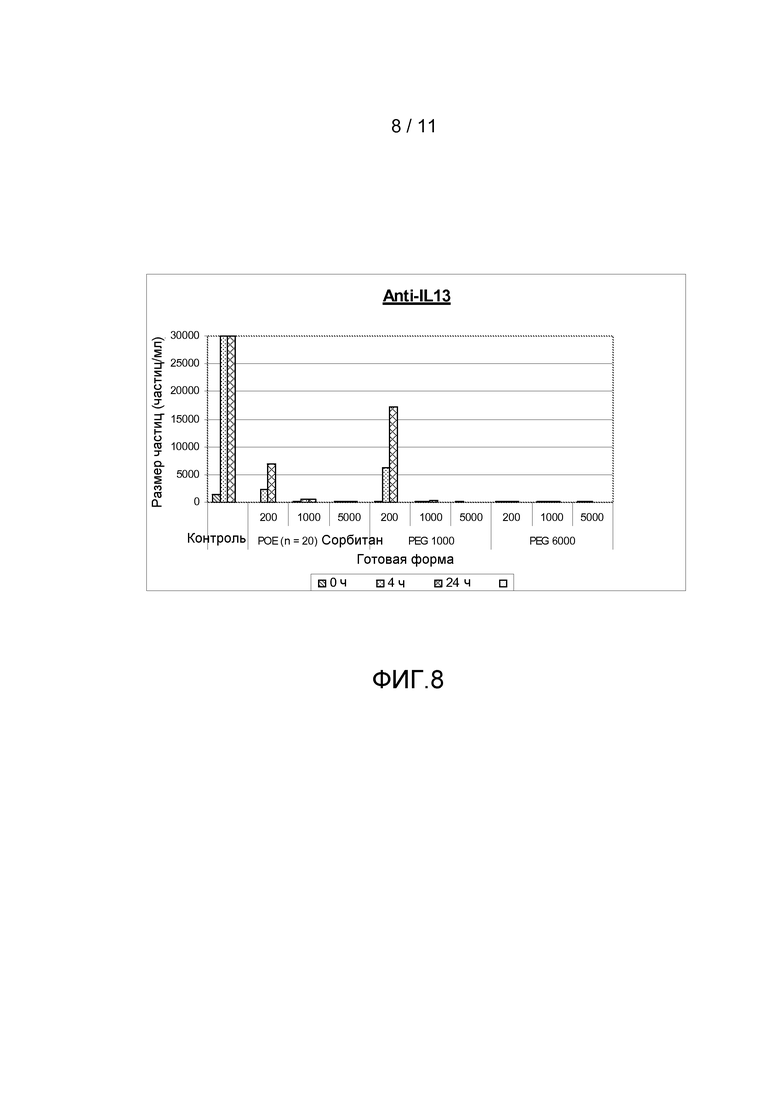

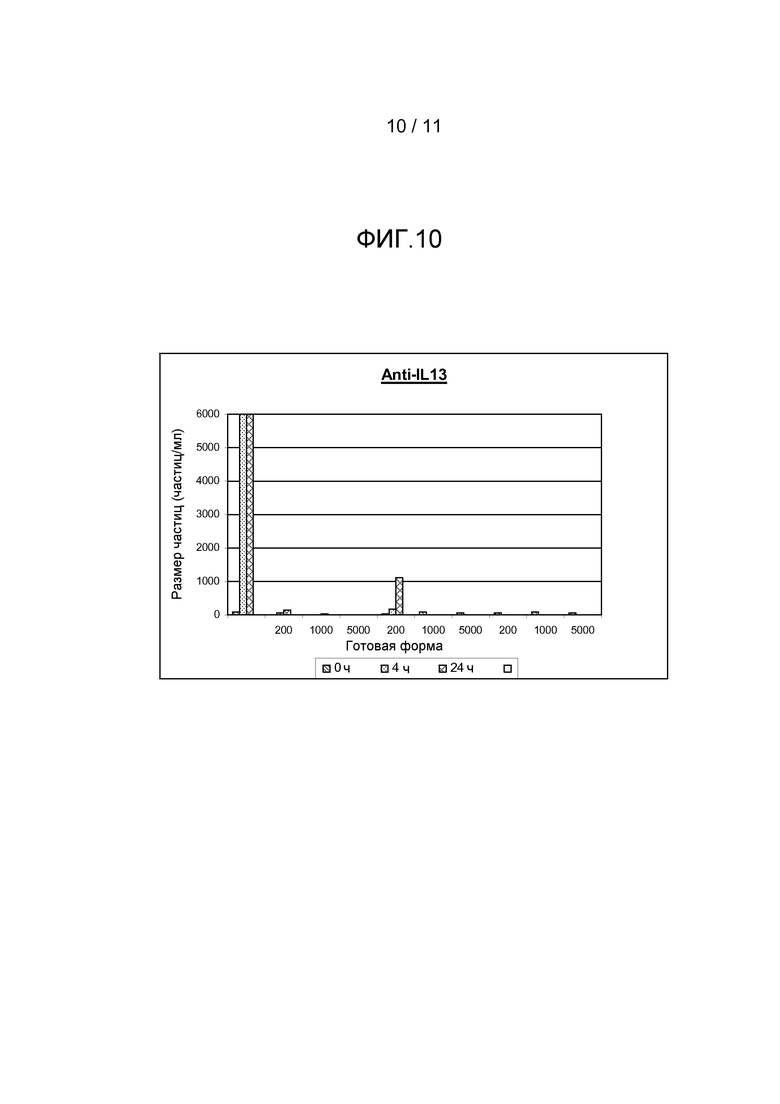

Группа изобретений относится к области фармацевтики и касается фармацевтической композиции, содержащей антитело и полиоксиэтилен (ПОЭ) сорбитан, без полисорбата, а также способа приготовления композиции и способа стабилизации белоксодержащих готовых форм. Группа изобретений обеспечивает улучшенную стабильность путем предотвращения агрегации белков в водных растворах. 5 н. и 9 з.п. ф-лы, 11 ил.
1. Водная композиция, содержащая антитело и полиоксиэтилен (ПОЭ) сорбитан, для пониженной агрегации белков, где композиция не содержит полисорбата.
2. Композиция по п. 1, в которой ПОЭ сорбитан присутствует в концентрации, составляющей от приблизительно 20 частей на миллион (м.д.) до приблизительно 100000 м.д.
3. Композиция по п. 1, в которой ПОЭ сорбитан выбирают из группы, состоящей из ПОЭ сорбитана 10, ПОЭ сорбитана 20, ПОЭ сорбитана 40 и ПОЭ сорбитана 80.
4. Композиция по п. 1, которая не содержит поверхностно-активное вещество.
5. Композиция по п. 1, которая находится в лиофилизированной форме, полученной путем лиофилизации композиции по любому из пп. 1-4.
6. Композиция по п. 1, которая дополнительно содержит полиэтиленгликоль.
7. Набор для пониженной агрегации белков, содержащий водную композицию по п. 1 и инструкции по применению.
8. Способ приготовления водной стабилизированной белоксодержащей готовой формы, не содержащей полисорбата, при этом способ включает смешивание антитела с ПОЭ сорбитаном, предоставляя посредством этого стабилизированную белоксодержащую готовую форму.
9. Способ по п. 8, в котором ПОЭ сорбитан выбирают из группы, состоящей из ПОЭ сорбитана 10, ПОЭ сорбитана 20, ПОЭ сорбитана 40 и ПОЭ сорбитана 80.
10. Способ по п. 8, дополнительно включающий стадию лиофилизации стабилизированной белоксодержащей готовой формы.
11. Способ повышения стабильности белка в водном растворе, не содержащем полисорбата, при этом способ включает смешивание антитела с ПОЭ сорбитаном, причем ПОЭ сорбитан повышает стабильность белка в водном растворе.
12. Способ по п. 11, в котором ПОЭ сорбитан выбирают из группы, состоящей из ПОЭ сорбитана 10, ПОЭ сорбитана 20, ПОЭ сорбитана 40 и ПОЭ сорбитана 80.
13. Способ предотвращения или уменьшения агрегации белка в водном растворе, не содержащем полисорбата, при этом способ включает смешивание антитела с ПОЭ сорбитаном, причем ПОЭ сорбитан предотвращает или уменьшает агрегацию белка в водном растворе.
14. Способ по п. 13, в котором ПОЭ сорбитан выбирают из группы, состоящей из ПОЭ сорбитана 10, ПОЭ сорбитана 20, ПОЭ сорбитана 40 и ПОЭ сорбитана 80.
| Формирователь импульсов | 1979 |
|
SU845274A1 |
| СПОСОБ СВАРКИ ПЛАВЛЕНИЕМ ПО ЩЕЛЕВОЙ РАЗДЕЛКЕ | 2000 |
|
RU2175906C1 |
| US6399385 B1,04.06.2002 | |||
| LEE RC et al "Surfactant copolymer prevent aggregation of heat denatured lysozymе", Ann | |||
| Biomed | |||
| Eng | |||
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |

