Данное изобретение относится к новому способу получения пан-ЦЗК (циклин зависимой киназы)-ингибиторов формулы (I), а также к промежуточным соединениям для получения.
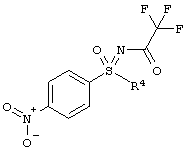
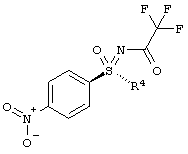
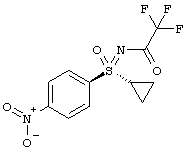
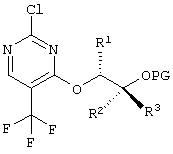
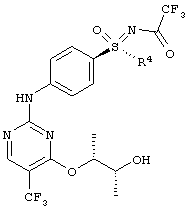
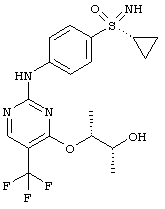
Новый способ относится к соединениям формулы (I), в частности, к соединению (2R,3R)-3-{[2-{[4-(S-циклопропилсульфонимидоил)-фенил]амино}-5-(трифторметил)пиримидин-4-ил]окси}бутан-2-олу (соединение А), которое проявляет свою противоопухолевую активность через клеточнотоксичный механизм. Был открыт способ получения соединения общей формулы (I)
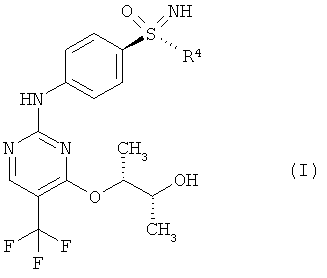 ,
,
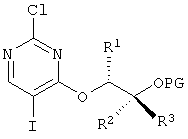
в которой
R4 означает (C1-С6)-алкильную группу или (C3-C7)-циклоалкильное кольцо,
который подходит для крупного масштабного производства и в котором преодолены недостатки способа уровня техники для получения соединений этого класса веществ.
Этот способ получения особенно подходит для получения соединения А
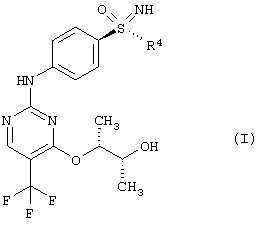
В данном изобретении использованы следующие обозначения:
(С1-С6)-алкил
Под (C1-С6)-алкильной группой в каждом случае понимают линейный или разветвленный алкильный радикал, такой как, например, метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, изопентил или гексил.
(С3-С7)-циклоалкил
Под (С3-С7)-циклоалкильным кольцом понимают циклопропильное, циклобутильное, циклопентильное, циклогексильное или циклогептильное кольцо.
Соединения общей формулы (I), в частности, также соединение А и способ их получения опубликованы в WO 2010/046035 A1, эта публикация соответствует современному уровню техники.
Способ согласно WO 2010/046035 A1 представляет собой 10-стадийный конвергентный способ с общим выходом для самой длинной последовательности около 7%.
Способ согласно WO 2010/046035 A1 включает, как минимум, одну из следующих стадий:
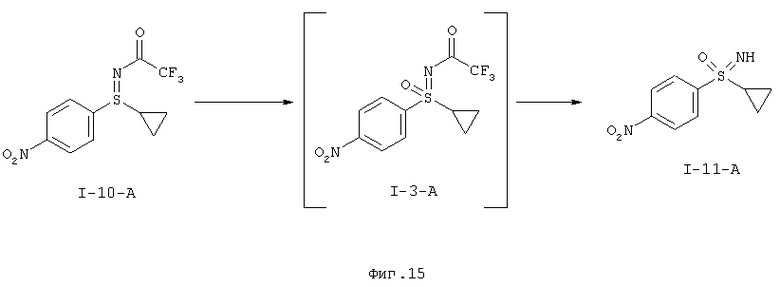
а) Окисление нитрофенилсульфида формулы (I-1) в нитрофенил-сульфоксид формулы (I-2).
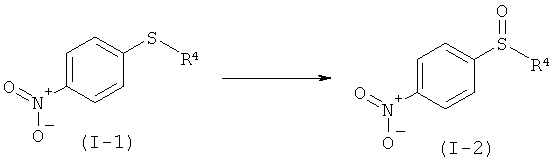
b1) Прямое иминирование нитрофенилсульфоксида формулы (I-2) в защищенный трифторацетатом нитрофенилсульфоксимин формулы (I-3).
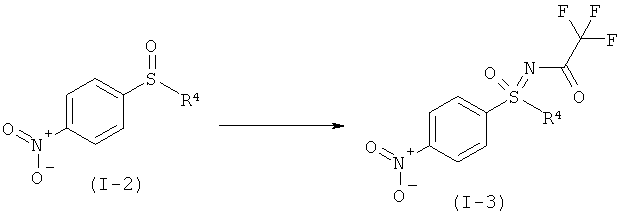
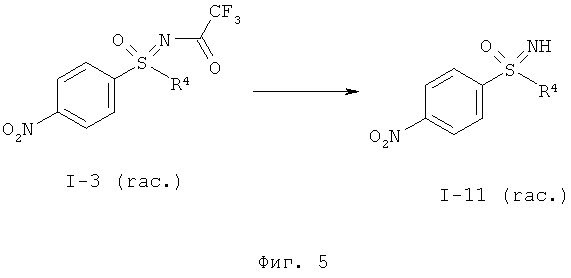
b2) Иминирование нитрофенилсульфоксида формулы (I-2) в нитрофенилсульфоксимин формулы (I-11) и последующее введение защитной группы с образованием защищенного трифторацетатом нитрофенилсульфоксимина формулы (I-3).
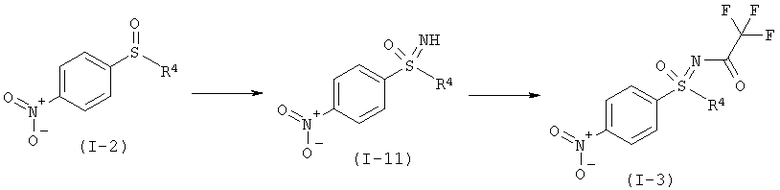
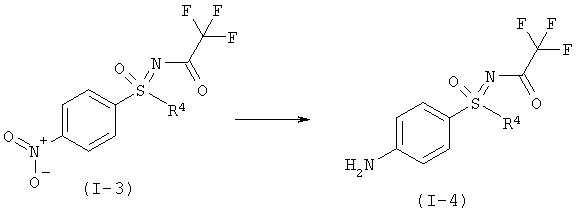
с) Восстановление соединения формулы (I-3) в соединение формулы (I-4).
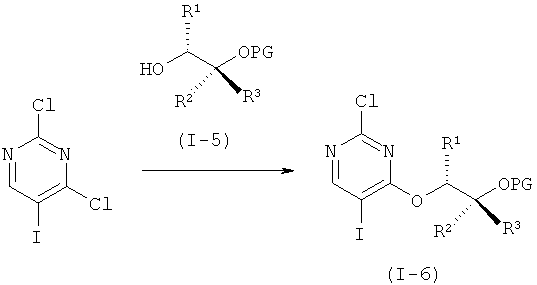
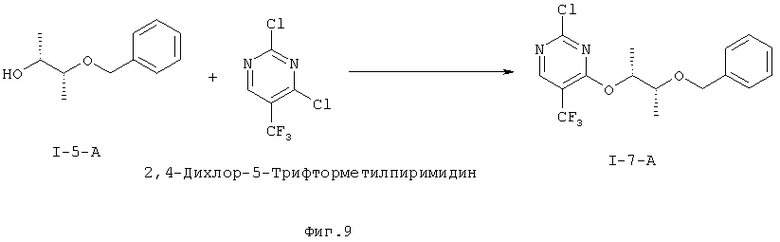
d) Функционализирование 4-положения 2,4-дихлор-5-йодпиримидина путем взаимодействия с монозащищенным диолом формулы (I-5) с образованием защищенного гидрокси-алкокси-пиримидина формулы (I-6).
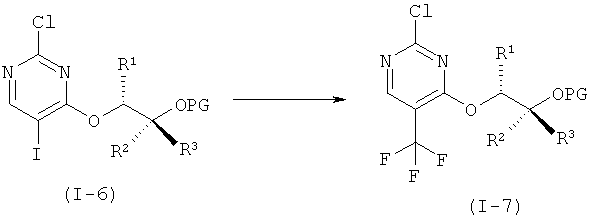
e) Получение защищенного 5-CF3 промежуточного соединения (I-7).
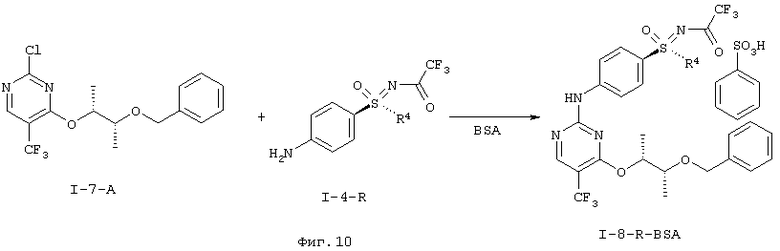
f) Связывание соединений формулы (I-7) и (I-4) с образованием двукратно защищенного анилино-пиримидина формулы (I-8).
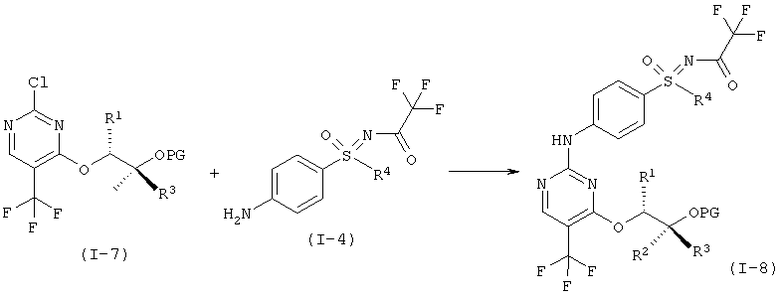
g) Отщепление защитной группы (PG) с образованием однократно защищенного анилино-пиримидина (I-9).
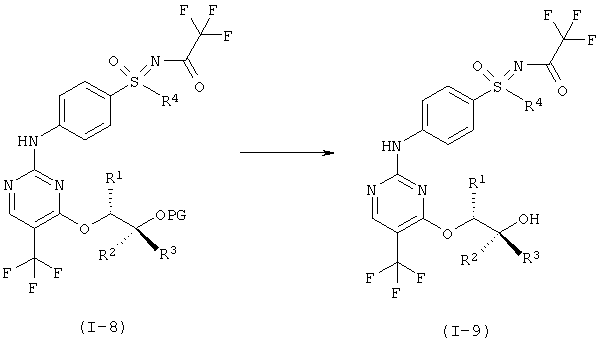
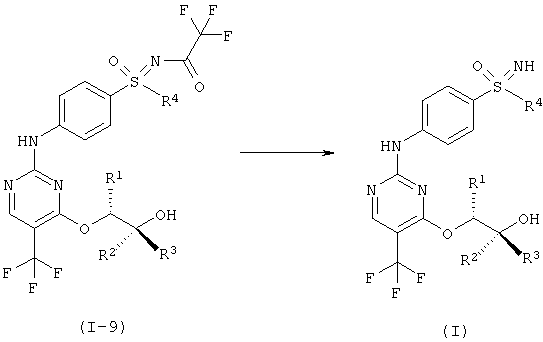
h) Отщепление защитной группы у сульфоксимина с образованием соединений формулы (I).
Причем, в WO 2010/046035A1
R1 означает метил, этил, пропил или изопропил, и
R2 и R3 независимо один от другого означают водород, метил или этил, и
R4 означает (C1-С6)-алкил или (С3-С7)-циклоалкильное кольцо.
Диастереомеры формулы I разделяют с помощью препаративной хроматографии. Подробности эксперимента приведены в WO 2010/046035 A1.
Для соединения А в WO 2010/046035 A1 опубликованы следующие условия для отдельных стадий синтеза.
Получение промежуточных продуктов
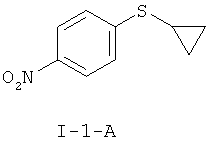
1-Циклопропилсульфанил-4-нитробензол (I-1-А)
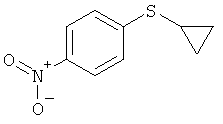
К циклопропантиолу в тетрагидрофуране (ТГФ) / диэтиловом эфире добавляют по порциям гидрид натрия и перемешивают при комнатной температуре. Затем добавляют по порциям 1-фтор-4-нитробензол. Реакционную смесь перемешивают в течение 2 часов при 40°С. После охлаждения реакционную смесь подают в воду и проводят трехкратную экстракцию бензолом. Отгоняют растворитель из объединенных органических фаз и остаток чистят хроматографически (гексан / уксусный эфир 95:5). (Выход: 61%).
(RS)-1-Циклопропансульфинил-4-нитробензол (I-2-A)
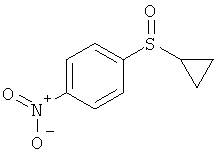
К 1-циклопропилсульфанил-4-нитробензолу в ацетонитриле добавляют хлорид железа(III) и перемешивают при комнатной температуре. Затем добавляют по порциям перйодную кислоту. Реакционную смесь перемешивают в течение 30 минут и затем выливают при перемешивании в охлажденный насыщенный раствор тиосульфата натрия. Проводят двукратную экстракцию эфиром уксусной кислоты. Объединенные органические фазы сушат над Na2SO4, фильтруют и отгоняют растворитель. Полученный остаток чистят на хроматографе (гексан / уксусный эфир 1:1). (Выход: 76%).
(RS)-S-Циклопропил-S-(4-нитрофенил)-N-(трифторацетил)-сульфоксимид (I-3-А)
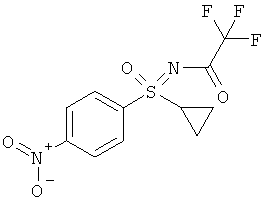
К суспензии (RS)-1-циклопропансульфинил-4-нитробензола, трифторацетамида, йодбензолдиацетата и оксида магния в дихлорметане (ДХМ) добавляют в атмосфере аргона димер ацетата родия(II) и перемешивают в течение ночи при комнатной температуре. Реакционную смесь фильтруют через целит и отгоняют растворитель. Полученный остаток чистят хроматографически (гексан / эфир уксусной кислоты 2:1) (выход: 78%).
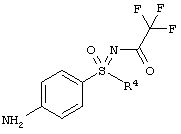
(RS)-S-(4-аминофенил)-S-циклопропил-N-(трифторацетил)-сульфоксимид (I-4-А)
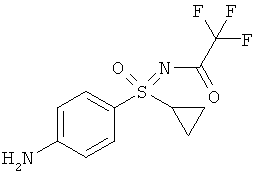
К раствору (RS)-S-циклопропил-S-(4-нитрофенил)-N-(трифторацетил)сульфоксимида в этаноле и тетрагидрофуране добавляют палладий на угле и гидрируют в течение 1 часа при нормальном давлении и температуре 25°С. Добавляют еще палладия на угле и продолжают гидрирование еще 4,5 часа при нормальном давлении. Реакционную смесь фильтруют, к фильтрату снова добавляют палладий на угле и гидрируют еще 45 минут. Реакционную смесь фильтруют и отгоняют растворитель (выход: 93%).
(2R,3R)-3-бензилоксибутан-2-ол (I-5-A)
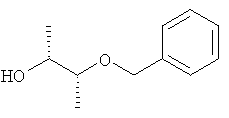
К раствору (2R,3R)-бутан-2,3-диола в ТГФ при комнатной температуре добавляют трет-бутилат калия и реакционную смесь нагревают в течение 15 минут в условиях рефлюкса. Реакционную смесь охлаждают до температуры около 50°С и добавляют бензилбромид. Нагревают в условиях рефлюкса в течение 3 часов, после этого перемешивают в течение ночи при комнатной температуре. Реакционную смесь разбавляют эфиром уксусной кислоты и раствором хлористого натрия, а затем промывают однократно 1 Н раствором хлористого водорода и двукратно раствором хлористого натрия. Органическую фазу сушат над Na2SO4, фильтруют и отгоняют растворитель. Полученный остаток чистят хроматографически (гексан / эфир уксусной кислоты 1:1) (выход: 43%).
4-((1R,2R)-2-бензилокси-1-метилпропокси)-2-хлор-5-йодпиримидин (I-6-A)
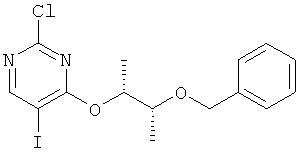
К (2R,3R)-3-бензилоксибутан-2-олу в диэтиловом эфире добавляют при температуре 0°С при перемешивании по порциям гидрид натрия. Через 10 минут убирают ледяную баню и перемешивают еще 3 минуты при комнатной температуре. Образовавшуюся суспензию добавляют при температуре 0°С к раствору 2,4-дихлор-5-йодпиримидина. Реакционную смесь перемешивают в течение 4 часов при температуре 40°С и затем добавляют разбавленный раствор хлористого натрия. Экстрагируют двукратно эфиром уксусной кислоты. Объединенные органические фазы сушат над Na2SO4, фильтруют и отгоняют растворитель. Полученный остаток чистят хроматографически (гексан / эфир уксусной кислоты 4:1) (выход: 41%).
4-((1R,2R)-2-бензилокси-1-метилпропокси)-2-хлор-5-трифторметил-пиримидин (I-7-А)
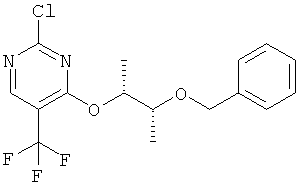
К раствору 4-((1R,2R)-2-бензилокси-1-метилпропокси)-2-хлор-5-йодпиримидина в N-метилпирролидиноне (NMП) и ТГФ добавляют при комнатной температуре при перемешивании йодид меди(1), фторид калия и (трифторметил)триметилсилан. Реакционную смесь перемешивают в течение 5,5 часов при температуре 80°С. После охлаждения реакционную смесь подают в разбавленный раствор хлористого натрия и 2 раза экстрагируют эфиром уксусной кислоты. Объединенные органические фазы сушат над Na2SO4, фильтруют и отгоняют растворитель. Полученный остаток чистят хроматографически (гексан / эфир уксусной кислоты 4:1) (выход: 54%).
(RS)-S-(4-{[4-{[(1R,2R)-2-(бензилокси)-1-метилпропил]окси}-5-(трифторметил)пиримидин-2-ил]амино)фенил)-S-циклопропил-N-(трифторацетил)сульфоксимид (I-8-А)
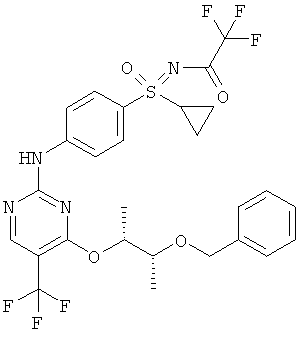
К 4-((1R,2R)-2-бензилокси-1-метилпропокси)-2-хлор-5-трифтор-метилпиримидину и (RS)-S-(4-аминофенил)-S-циклопропил-N-(трифторацетил)сульфоксимиду в ацетонитриле добавляют 4Н раствор хлористого водорода в диоксане и перемешивают в течение 5 часов при температуре 80°С. После охлаждения реакционную смесь разбавляют эфиром уксусной кислоты и промывают насыщенным раствором гидрокарбоната натрия и насыщенным раствором хлористого натрия, сушат над Na2SO4, фильтруют и отгоняют растворитель. Полученный остаток чистят хроматографически (гексан / эфир уксусной кислоты 4:1) (выход: 56%).
(RS)-S-циклопропил-S-(4-{[4-{[(1R,2R)-2-гидрокси-1-метилпропил]-окси}-5-(трифторметил)пиримидин-2-ил]амино)фенил)-N-(трифторацетил)сульфоксимид (I-9-А)
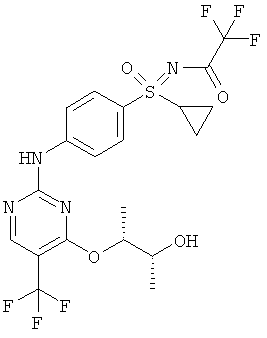
К раствору (RS)-S-(4-{[4-{[(1R,2R)-2-(бензилокси)-1-метил-пропил]окси}-5-(трифторметил)пиримидин-2-ил]амино}фенил)-S-циклопропил-N-(трифторацетил)сульфоксимида в этаноле добавляют палладий на угле (10-процентный) и гидрируют при нормальном давлении при комнатной температуре. Реакционную смесь фильтруют и отгоняют растворитель (выход: 79%).
Получение соединения А
К (RS)-S-циклопропил-S-(4-{[4-{[(1R,2R)-2-гидрокси-1-метил-пропил]окси}-5-(трифторметил)пиримидин-2-ил]амино}фенил)-N-(трифторацетил)сульфоксимиду в 35 мл метанола добавляют карбонат калия и перемешивают в течение 1,5 часов при комнатной температуре. Разбавляют насыщенным раствором хлористого натрия и экстрагируют 3 раза эфиром уксусной кислоты. Объединенные органические фазы сушат над Na2SO4, фильтруют и отгоняют растворитель.
Смесь диастереомеров разделяют препаративной жидкостной хроматографией высокого разрешения (ЖХВР) на чистые стереоизомеры:
колонка: Chiralpak IA 5 мкм 250×30 мм,
элюент: гексан / этанол 8:2,
поток: 40,0 мл/мин,
детектор: УФ 254 нм,
температура: комнатная температура,
время удерживания: 10,8-13,4 мин; стереоизомер 1, 13,6-18,5 мин; стереоизомер 2 (соединение А).
Это получение соединения формулы (I) согласно WO 2010/046035А1 не подходит для способа производства.
К критическим пунктам относятся:
- На большей части промежуточных стадий проводится хроматографическая очистка. Это является дорогостоящим и затратным при больших масштабах.
- Исходный материал (I-1) получают из циклопропилсульфида, который отсутствует в продаже в больших количествах.
- Для получения (I-2) применяется рацемический способ окисления. Поэтому стереоизомеры по окончании синтеза должны быть разделены хроматографически. В связи с тем, что разделение проводят только после окончания синтеза, общий выход для последовательного синтеза сильно уменьшается.
- При получении (I-3) применяют большие количества димера ацетата родия (II). Это дорого и родий необходимо удалить, для того чтобы не загрязнять биологически активное вещество. Йодбензолдиацетат не подходит для крупного масштабного производства, так как в больших количествах его трудно получить и он потенциально является взрывчатым веществом.
- Альтернативное получение (I-3) через (I-2) и (I-2/3) непросто осуществить, исходя из оснований безопасности, так как применяются токсичные и взрывчатые вещества, такие как азид натрия или о-мезитиленсульфонилгидроксиламин (МСГ).
- Синтез (I-5-А) не является селективным, так как также происходит двойное алкилирование. Выход составляет поэтому только 43%.
- Последовательность (I-6)-(I-7) не конвергентна, так как трифторметильная группа не переносится вместе с пиримидиновой основой. Выходы обеих стадий плохие. В связи с тем, что превращения связаны с образованием многих побочных компонентов, приходится проводить затратную хроматографию.
- Промежуточное соединение (I-8) образуется в виде масла, которое чистят только хроматографически. Масло трудно получить в промышленном масштабе, и оно отличается плохой стабильностью при хранении по сравнению с твердым веществом.
- На стадии (I-9) проводят препаративное разделение диастереомеров. Это очень затратно и дорого. Кроме того, теряется много от общего выхода, так как разделение предпринимают на последней стадии синтеза.
Эти аспекты следовало бы оптимизировать при расширении синтеза в многограммовом, соответственно, килограммовом масштабе.
В связи с этим задача данного изобретения состояла в том, чтобы представить в распоряжение способ получения пан-ЦЗК-ингибиторов общей формулы (I), в частности, соединения А, у которого отсутствуют приведенные выше недостатки.
I. Стадии способа согласно данному изобретению для получения соединения общей формулы (I)
Способ получения согласно данному изобретению отличается различными предпочтительными стадиями получения, а также промежуточными веществами.
Способ получения согласно данному изобретению соединений общей формулы (I) характеризуется, как минимум, одной из следующих стадий:
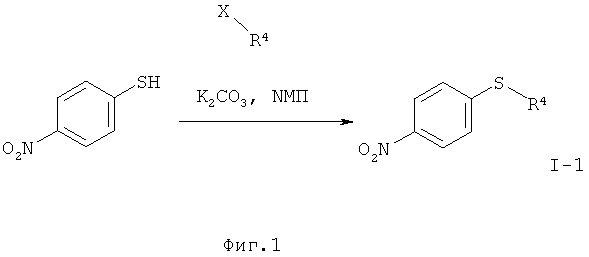
I.а). Алкилирование 4-нитротиофенола в присутствии карбоната калия в N-метилпирролидиноне (NMП) с получением нитрофенил-сульфида формулы (I-1)
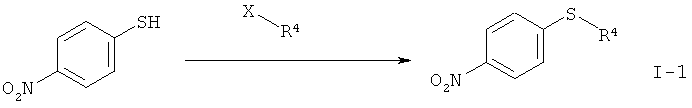 ,
,
причем, X означает Br, Cl, I, О-SO2-СН3 или O-SO2-(4-метилфенил).
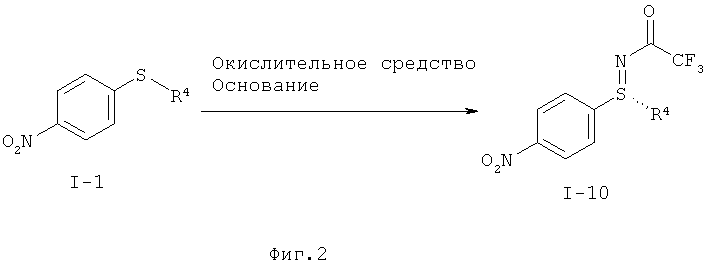
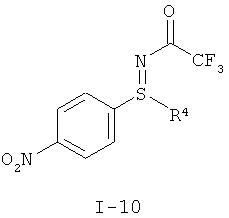
I.b). Окислительное аминирование нитрофенилсульфида формулы (I-1) в защищенный трифторацетатом нитрофенилсульфилимин формулы (I-10)
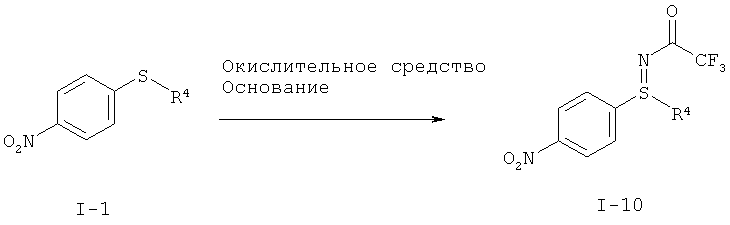 ,
,
I.с) Окисление защищенного трифторацетатом нитрофенилсульфилимина формулы (I-10) в защищенный трифторацетатом нитрофенилсульфоксимин формулы (I-3) и последующее снятие защиты с образованием нитрофенилсульфоксимина формулы (I-11)
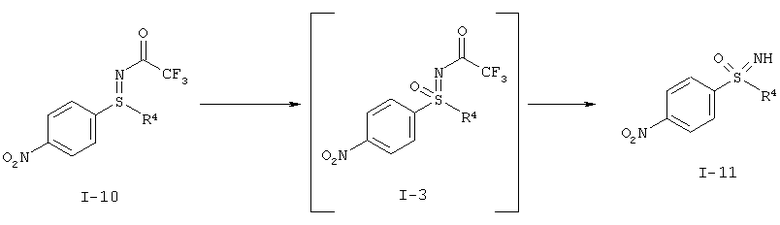 .
.
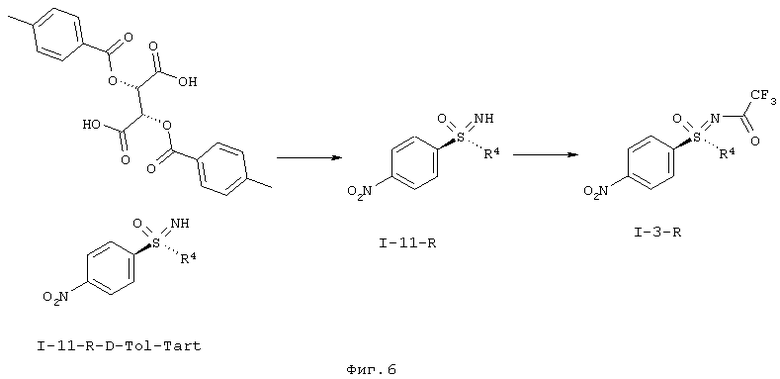
I.d) Расщепление рацематов (гас.) нитрофенилсульфоксимина формулы (I-11) с помощью (+)-ди-О-п-толуол-D-винной кислоты (D-Tol-Tart)
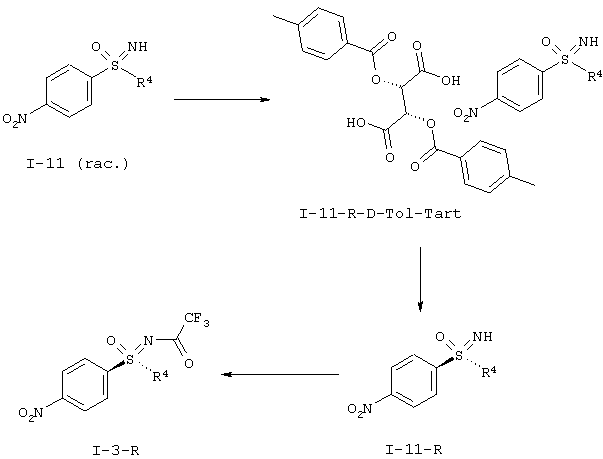 ,
,
причем, R-энантиомер нитрофенилсульфоксимина формулы (I-11-R) высвобождают исключительно из солей и вновь вводят трифторацетатную защитную группу с образованием R-энантиомера защищенного трифторацетатом нитрофенилсульфоксимина формулы (I-3-R).
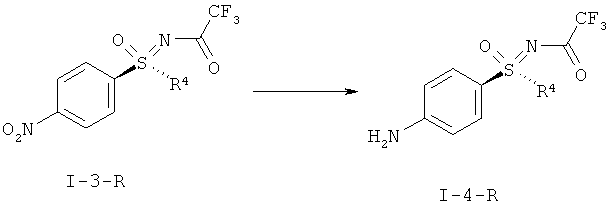
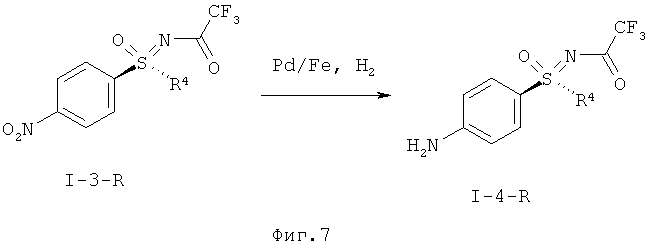
I.e) Гидрирование защищенного трифторацетатом нитрофенилсульфоксимина формулы (I-3-R) с образованием защищенного трифторацетатом анилиносульфоксимина формулы (I-4-R) в присутствии легированного железом палладиевого катализатора
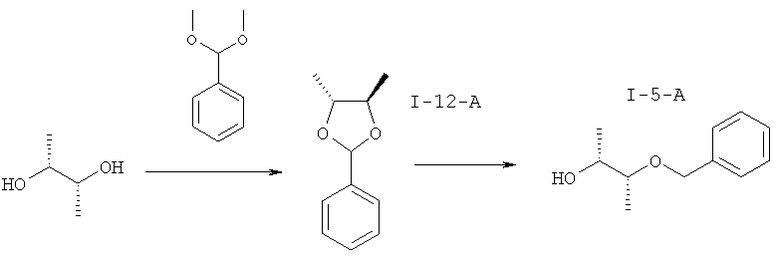
I.f). Получение (2R,3R)-3-(бензилокси)бутан-2-ола (I-5-A) в двухстадийном процессе через (4R,5R)-4,5-диметил-2-фенил-1,3-диоксолан (I-12-A), причем, первую стадию с пиридиний-п-толуолсульфонатом проводят в толуоле и затем проводят восстановление диизобутилалюминийгидридом в толуоле
 .
.
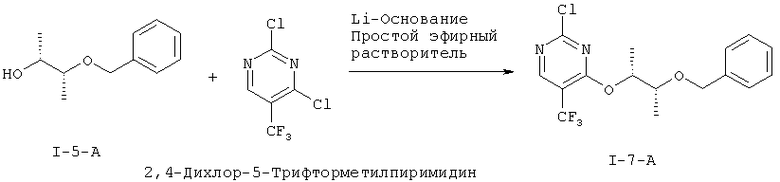
I.g). Присоединение (I-5-А) к 2,4-дихлор-5-трифторметил-пиримидину происходит с образованием 4-{[(2R,3R)-3-(бензилокси)бутан-2-ил]окси}-2-хлор-5-(трифторметил)-пиримидина (I-7-A) в присутствии Li-оснований в простых эфирных растворителях
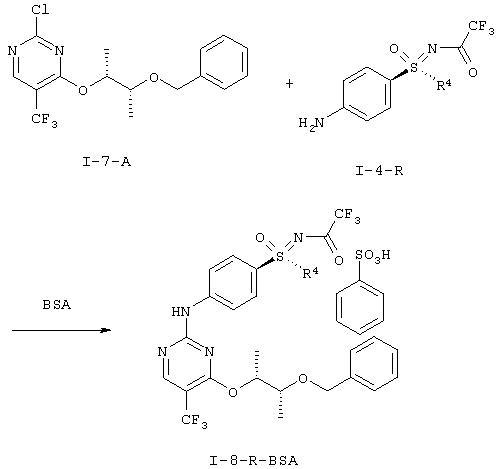
I.h) Получение солей бензолсульфоновой кислоты (BSA) двукратно защищенных анилинопиримидинов формулы (I-8-R-BSA) путем катализируемого бензолсульфоновой кислотой присоединения (I-7-A) к (I-4-R)
I.i) Отщепление защитных групп в солях бензолсульфоновой кислоты (BSA) двукратно защищенных анилинопиримидинов формулы (I-8-R-BSA) путем гидрирования водородом в присутствии паладия на активном угле в метаноле, а также обработки карбонатом калия в метаноле с получением соединений формулы (I)
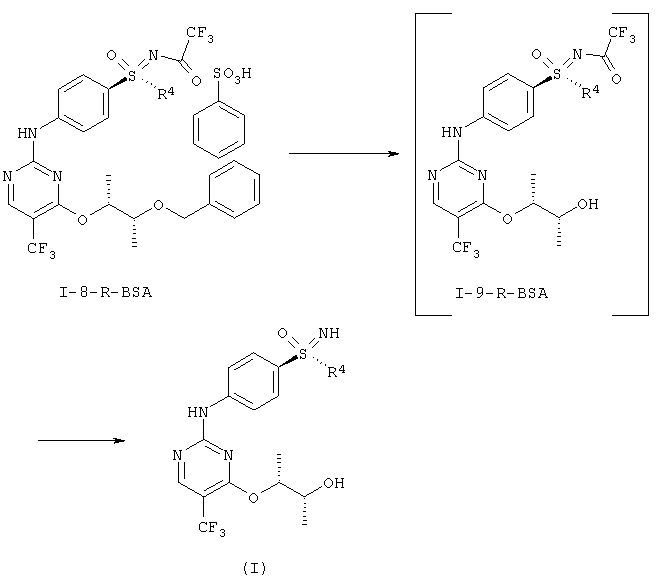
Стадии получения "северной половины" соединений формулы (I)
I.a) Получение нитрофенилсульфидов формулы (I-1)
Одним из объектов данного изобретения относится к стадии алкилирования 4-нитрофенола.
Согласно WO 2010/046035 А1 исходный материал (I-1) получен из циклопропилсульфида. Последний имеется в продаже только в небольших количествах. В связи с этим перестроились на алкилирование имеющегося в продаже 4-нитротиофенола с применением алкилирующих агентов (X-R4) в присутствии вспомогательного основания, причем, Х означает Br, Cl, I, O-SO2-СН3 или O-SO2-(4-метилфенил). В качестве оснований подходят карбонат натрия, карбонат калия или карбонат цезия, более предпочтителен карбонат калия. В качестве растворителя подходят N,N-диметилформамид. N-метилпирролидинон, диметилсульфоксид, N,N-диметилацетамид, более предпочтителен N-метилпирролидинон (NMП).
Другие объекты данного изобретения относятся к окислительному аминированию нитрофенилсульфидов формулы (I-1) с образованием защищенных трифторацетатом нитрофенилсульфилиминов формулы (I-10) (фиг.2) и заключительное окисление в нитрофенил-сульфоксимины формулы (I-11) (фиг.3).
I.b) Получение защищенных трифторацетатом нитрофенилсульфилиминов формулы (I-10)
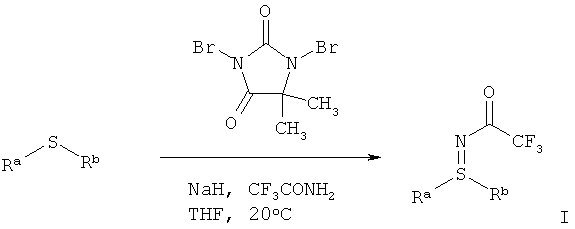
Современный уровень техники для получения сульфилиминов
Цель состояла в прямом аминировании сульфидов для препаративно хорошо применяемых защищенных трифторацетатом сульфилиминов с применением простых исходных материалов, таких как, например, 2,2,2-трифторацетамид (CF3CONH2). Carreira и др. (Orgr. Lett. 1999, 1, 149-151) описывают Cu-катализируемое прямое аминирование в защищенные трифторацетатом сульфилимины с помощью литиированного гидроксиламина трифторуксусной кислоты (ТФУК), который однако до этого надо получить в две стадии и который отсутствует в продаже. Энантиоселективно удается осуществить это превращение со стехиометрическим количеством нитридо-Mn комплекса (Helv. Chim. Acta. 2002, 3773-3783).
Bolm и др. сообщают (Tetrahedron Letters 2005), что прямое без участия металлов иминирование сульфидов возможно. Предлагаются п-нитрофенилсульфонамид (носиламид, Nos-NH2) и (диацетоксийод)бензол (PhI(OAc)2) и получают носил-защищенные сульфинимины после 16 часов нагревания в условиях рефлюкса. Для реакции в крупных промышленных масштабах эти условия однако мало подходят, так как п-нитрофенилсульфонамидная защитная группа с трудом удаляется и (диацетоксийод)бензол имеется в продаже в небольших количествах.
В качестве окислительного средства в превращении согласно данному изобретению согласно фиг.2 применяют среди других N-бромсукцинимид, йод, гипобромид натрия, 1,3-дибром-5,5-диметилгидантоин, N-хлорсукцинимид и трихлорциануровую кислоту в присутствии таких оснований, как карбонат цезия, трет-бутилат калия, трет-бутилат натрия, водный натронный щелок, метанолат натрия, этанолат натрия, гидрид натрия (NaH) в таких растворителях, как метанол, дихлорметан, тетрагидрофуран-вода, ацетонитрил, ацетонитрил-вода, тетрагидрофуран (ТГФ), пропионитрил, метил-трет-бутиловый эфир, 1,4-диоксан, хлорбензол.
Предпочтительным окислительным средством является 1,3-дибром-5,5-диметилгидантоин.
В качестве комбинации растворитель-основание предпочтительны комбинации ацетонитрил-карбонат цезия, 1,4-диоксан-гидрид натрия, дихлорметан-трет-бутилат калия, ацетонитрил-гидрид натрия, тетрагидрофуран-гидрид натрия или метил-трет-бутиловый эфир-гидрид натрия.
Желательная реакция протекает полностью уже при температуре около 20°С в течение нескольких часов без добавления катализатора.
По сравнению с известными из литературы способами новый способ окислительного аминирования, показанный на фиг.2, обладает следующими преимуществами:
- можно отказаться от применения дорогого и потенциально взрывоопасного (диацетоксийод)бензола, а также от добавок солей металлов;
- образуется только небольшое количество сульфоксида и реакция протекает при мягких условиях уже при температуре около 20°С с использованием коммерчески доступных базовых веществ и реагентов;
- трифторацетатная группа легко гидролизуется (например, карбонатом калия в метаноле) и в связи с этим представляет собой большую препаративную ценность.
Не только нитрофенилсульфиды формулы (1-1) удается окислительно аминировать согласно стадии I.b).
Этим способом можно также получить другие защищенные трифторацетатом сульфилимины.
В таблице 1 показаны другие, получаемые с помощью этой стадии доступные сульфилимины.
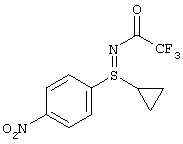
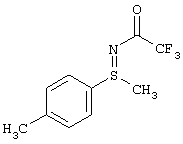
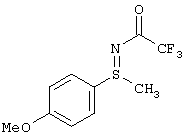
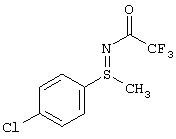
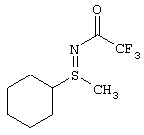
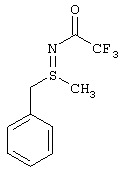
I.с) Получение нитрофенилсульфоксиминов формулы (I-11)
Окисление защищенного трифторацетатом нитрофенилсульфилимина (I-10) в нитрофенилсульфоксимин (I-11) происходит предпочтительно при применении пероксомоносульфата калия (оксон®) в качестве окислительного средства.
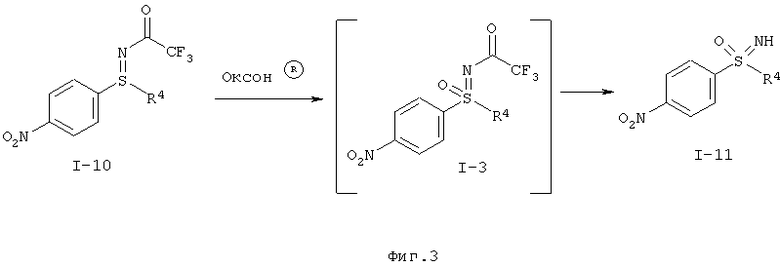
Желательное окисление происходит особенно быстро в основной области рН-значений. В этих условиях одновременно отщепляется трифторацетатная группа, так что при необходимости примыкающая стадия снятия защиты может быть проведена как реакция, происходящая в одном реакторе.
Реакцию предпочтительно проводят в смеси метанол-вода с добавлением тетраметиленсульфона (сульфолан), в качестве вещества, способствующего растворению. Пероксомоносульфат калия (оксон®) добавляют по порциям и рН-значение после каждого дозирочного шага устанавливают равным рН 10.
I.d) Расщепление рацематов нитрофенилсульфоксиминов формулы (I-11).
Другой объект данного изобретения относится к расщеплению рацематов нитрофенилсульфоксиминов формулы (I-11).
Расщепление рацематов опирается на следующую стадию:
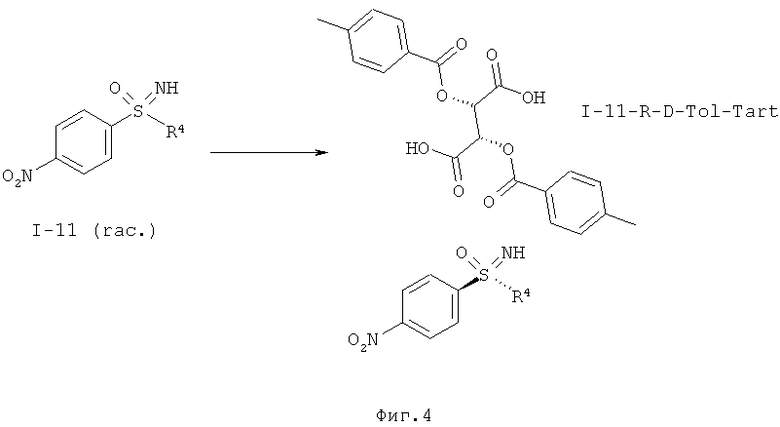
Неожиданно, например, для нитрофенилсульфоксимина формулы (I-11-А) было обнаружено, что с помощью (+)-ди-О-п-толуоил-D-винной кислоты (D-Tol-Tart) удается получить соотношение энантиомеров 95:5 в кристаллизате. В качестве растворителя можно применять ацетонитрил, пропионитрил или толуол. Более предпочтительны ацетонитрил или пропионитрил.
Интегрирование процесса кристаллизации в способ получения может происходить в результате того, что защитную трифторацетатную группу в (I-3) отщепляют карбонатом калия в метаноле и неочищенный нитрофенилсульфоксимин (I-11) подвергают взаимодействию с (+)-ди-О-п-толуоил-D-винной кислотой с образованием (I-11-R-D-Tol-Tart).
Толуоил-О-винную кислоту удаляют основной экстракцией из соли и оптически активный нитрофенилсульфоксимин формулы (I-11-R) можно с помощью способа, проводимого в одном реакторе, защитить ангидридом трифторуксусной кислоты в присутствии триэтиламина с образованием (I-3-R).
I.e) Гидрирование защищенного трифторацетатом нитрофенил-сульфоксимина формулы (I-3-R) в защищенный трифторацетатом анилиносульфоксимин формулы (I-4-R)
Другим объектом данного изобретения является гидрирование защищенного трифторацетатом нитрофенилсульфоксимина формулы (I-3-R) с получением защищенного трифторацетатом анилино-сульфоксимина формулы (I-4-R) в присутствии легированного железом палладиевого катализатора.
Восстановление нитрогруппы в соединении (I-3-R) в соответствующую анилиновую группу (I-4-R) удается эффективно провести путем гидрирования с помощью иммобилизированного палладиевого катализатора. При этом предпочтительны легированные железом палладиевые катализаторы на угле. В качестве растворителей могут применяться метанол, этанол, изо-пропанол, тетрагидрофуран или уксусная кислота. Более предпочтителен метанол.
Стадии получения "южной половины" соединении формулы (I)
I.f) Получение (R,R)-диметилдиоксолана (I-12-A) и (R,R)-бензилбурандиола (I-5-А)
Другой объект данного изобретения относится к получению (4R,5R)-4,5-диметил-2-фенил-1,3-диоксолана (I-12-A) и (2R,3R)-3-(бензилокси)бутан-2-ола (I-5-A) для "южной половины" соединений формулы (I).
Согласно WO 2010/046035A1 подвергают взаимодействию имеющийся в продаже (R,R)-бутан-2,3-диол с бензилхлоридом в одну стадию с получением монобензилированного (I-5-A). В связи с тем, что взаимодействие протекает не селективно с получением моносоединения, реакционную смесь необходимо чистить хроматографически и в связи с этим выходы составляют около менее 50%.
В качестве альтернативы вводится двухстадийный процесс (Bloorg. Med. Cheia. Lett. 2006, 16, 186-190).
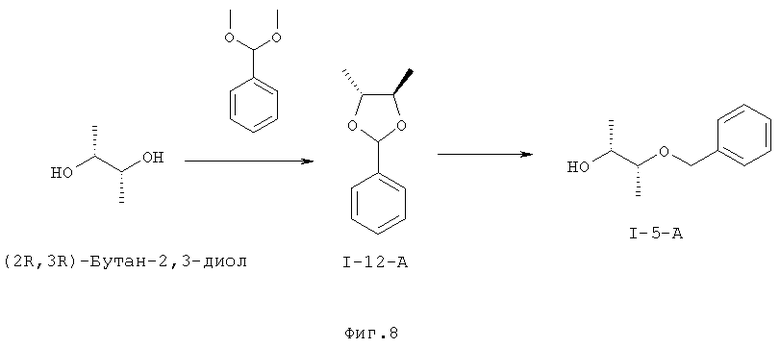
Объектом изобретения является выяснение экспериментальных условий для полного превращения, а также простое выделение и очистка, которые подходят для промышленных масштабов производства.
Промежуточное соединение (4R,5R)-4,5-диметил-2-фенил-1,3-диоксолан (I-12-A) получают подходящим образом путем превращения бензальдегиддиметилацеталя и избытка (2R,3R)-бутан-2,3-диола в присутствии пиридиний-п-толуолсульфоната в толуоле в качестве растворителя. Реакция протекает при температуре около 50°С полностью в течение 3 часов, причем, метанол следует постоянно отгонять при пониженном давлении.
В рамках водной переработки избыток диола удаляют путем экстракции. Остающуюся толуольную фазу можно напрямую применять на следующей стадии.
Для последующего восстановления с помощью диизобутилалюминийгидрида (ДИБАЛ) добавляют 1,5 М раствор диизобутилалюминийгидрида в толуоле при температуре 55-60°С. Для переработки дозируют декагидрат сульфата натрия и растворитель отгоняют после фильтрования. Получают соединение (I-5-A) с хорошей чистотой и выходом. Продукт без дальнейшей очистки можно применять на следующей стадии.
I.g) Получение 4-{[(2R,3R)-3-(бензилокси)бутан-2-ил]окси)-2-хлор-5-(трифторметил)пиримидина (I-7-А)
Нуклеофильное монозамещение одного атома хлора в коммерчески доступном 2,4-дихлор-5-трифторметилпиримидине происходит предпочтительно во 2-положении
Неожиданно было открыто, что вариацией условий замещения можно направить замещение в желательное 4-положение. Удалось показать, что Li-основания в растворах простых эфиров при температуре -30°С дает хорошие выходы и в лучшем случае соотношение 4-изомера / 2-изомеру, равное 1,2:1. В качестве растворителя можно применять, например, тетрагидрофуран, 1,2-диметоксиэтан, 1,4-диоксан, метил-трет-бутиловый эфир, диизопропиловый эфир, ди-н-бутиловый эфир, 2-метилтетрагидрофуран или циклопентилметиловый эфир. Более предпочтителен тетрагидрофуран. В качестве оснований можно, например, применять гексаметилдисилазид лития, н-бутиллитий, диизопропиламид лития или литий-2,2,6,6-тетраметилпиперидин. Более предпочтителен гексаметилдисилазид лития. Работают в температурном интервале от -78°С до +20°С. Из этого выводится стадия способа согласно данному изобретению, которая проводится с гексаметилдисилазидом лития в тетрагидрофуране при температуре -30°С с получением после хроматографирования желательного изомера (I-7-A) с выходом до 46% и чистотой более 95 F1% (процентная доля площади).
Соединение вместе северной и южной половины и получение соединений формулы (I)
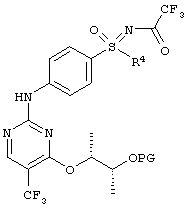
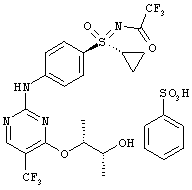
I.h) Получение анилинопиримидинов формулы (I-8-R-BSA)
Оба структурных элемента (I-7-A) и (I-4-R) соединяют в (I-8-R). Это взаимодействие проводят в присутствии кислоты. К подходящим кислотам относятся, например, хлористоводородная кислота, п-толуолсульфоновая кислота, бензолсульфоновая кислота, метансульфоновая кислота. Более предпочтительна бензолсульфоновая кислота.
Свободные основания (I-8-R) обычно являются маслами, что затрудняет их очистку, а также хранение. Неожиданно было открыто, что при применении бензолсульфоновой кислоты (BSA) из реакционной смеси кристаллизуются образовавшиеся соли бензолсульфоновой кислоты (I-8-R-BSA). Соли (I-8-R-BSA) можно чистить кристаллизацией и стабильны при хранении.
Альтернативно можно применять толуолсульфоновую кислоту или метансульфоновую кислоту.
I.i) Получение соединений формулы (I)
На последних двух стадиях отщепляют защитные группы (фиг.11).
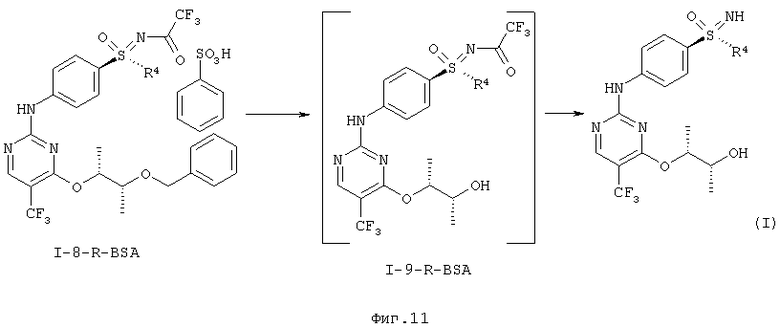
Гидрирование при нормальном давлении проводят с палладием / углеродом и водородом в метаноле в течение нескольких часов с образованием промежуточного соединения формулы (I-9-R-BSA).
Промежуточное соединение формулы (I-9-R-BSA) можно далее напрямую превратить в конечную стадию. Отщепление группы можно полностью завершить с помощью карбоната калия, и кристаллизация конечной стадии проводится из этилового эфира уксусной кислоты/н-гептана.
II. Промежуточные соединения
Далее предметом данного изобретения являются следующие промежуточные соединения
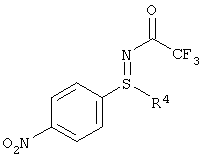
II.а) Защищенные трифторацетатом нитрофенилсульфинилимины формулы (I-10), в частности (I-10-A)
 ,
, 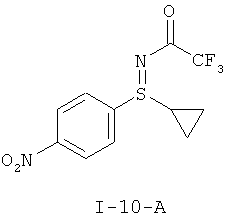 .
.
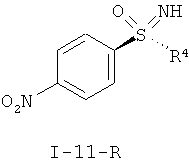
II.b) Нитрофенилсульфоксимины формулы (I-11-R), в частности (I-11-A) и (I-11-A-R)
 ,
, 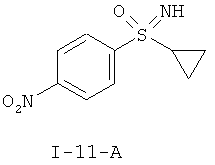 ,
, 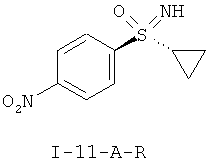 .
.
II.с) (R)-энантиомеры защищенных трифторацетатом нитрофенил-сульфоксиминов формулы (I-3-R), в частности (I-3-A-R)
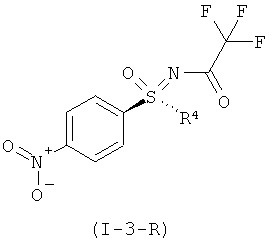 ,
, 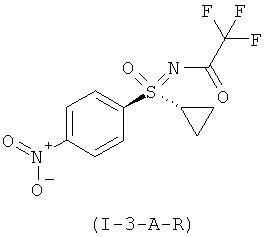 .
.
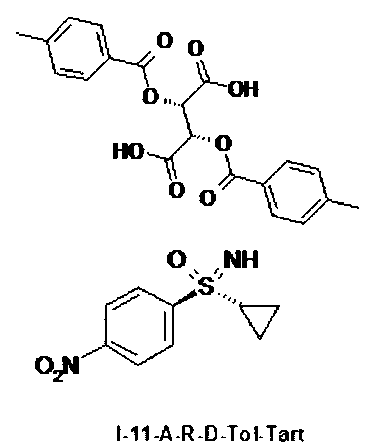
II.d) Соли (I-11-R-D-Tol-Tart) нитрофенилсульфоксиминов формулы (I-11-R) с (+)-ди-О-п-толуоил-D-винной кислоты (D-Tol-Tart), в частности (I-11-A-R-D-Tol-Tart)
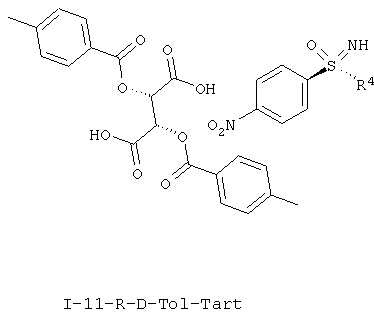 ,
, 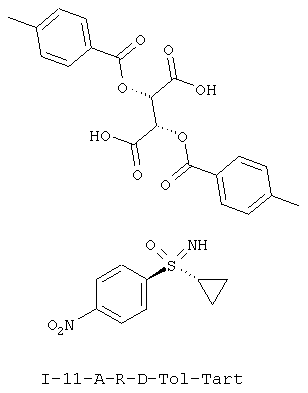 .
.
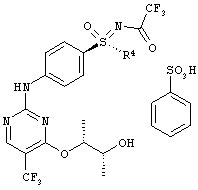
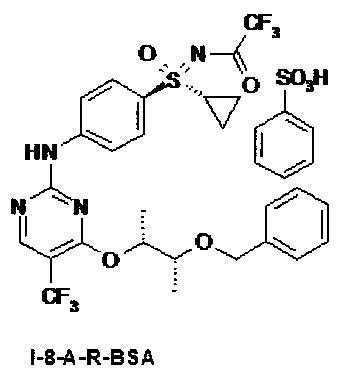
I.e) Анилинопиримидины формулы (I-8-R-BSA), в частности (I-8-A-R-BSA)
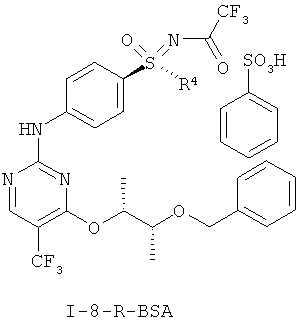 ,
,  .
.
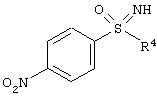
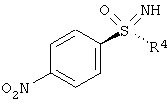
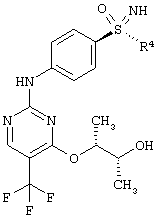
причем, R4 в каждом случае означает (C1-C6)-алкильную группу или (С3-С7)-циклоалкильное кольцо.
1-А. Получение соединения А
Получение "северной половины" соединения А
1-А.а) Получение циклопропилнитрофенилсульфида (I-1-A)
На первой стадии последовательности реакций 4-нитротиофенол алкилируют бромциклопропаном в присутствии карбоната калия. Желательное превращение проводят в N-метилпирролидиноне (NMП) в течение 8-10 часов при предпочтительной температуре 135°С.
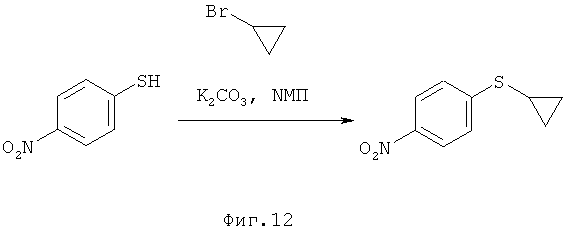
Выделение (I-1-A) происходит путем дозирования реакционной смеси в ледяную воду и выделения сырого кристаллизата с хорошей чистотой типично 89-93 F1% с выходом 82-87%.
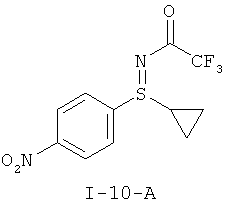
I-A.b) Получение защищенного трифторацетатом циклопропил-нитрофенилсульфилимина (I-10-А)

На второй стадии проводят окислительное аминирование с образованием защищенного трифторацетатом циклопропилнитрофенил-сульфилимина (I-10-A).
При широком скрининге было исследовано превращение циклопропил-нитрофенилсульфида (I-1-A) в защищенный трифторацетатом циклопропилнитрофенилсульфилимин (I-10-A).
В качестве окислительного средства при превращении согласно данному изобретению по фиг.13 можно применять N-бромсукцинимид, 1,3-дибром-5,5-диметилгидантион в присутствии таких оснований, как калий-трет-бутилат, гидрид натрия в таких растворителях, как дихлорметан, тетрагидрофуран или ацетонитрил. Желательная реакция протекает в температурном интервале 0-50°С, причем, температура 20°С более предпочтительна.
В качестве окислительного средства тестировали 1,3-дибром-5,5-диметилгидантоин, N-хлорсукцинимид и трихлорциануровую кислоту в присутствии оснований трет-бутилата калия, трет-бутилата натрия, водного натронного щелока, гидроксида натрия, метанолата натрия, гидрида натрия в растворителях метаноле, дихлорметане, тетрагидрофуране-воде, ацетонитриле, ацетонитриле-воде, тетрагидрофуране, пропионитриле, метил-трет-бутиловом эфире, диоксане, хлорбензоле.

В ходе превращения вначале в реакционный сосуд помещают гидрид натрия в тетрагидрофуране, а затем по каплям добавляют циклопропилнитрофенилсульфид (I-1-A) с трифторацетамидом. При охлаждении дозируют раствор 1,3-дибром-5,5-диметилгидантоина в тетрагидрофуране и перемешивают при комнатной температуре. Переработку проводят восстановительно (сульфит натрия) и кристаллизуют из диизопропилэтилового эфира / н-гептана. Получают продукт (I-10-A) с хорошими выходами и чистотой.
I-А.с) Получение 1-(циклопропилсульфонимидоил)-4-нитробензол (I-II-А)
Окисление защищенных трифторацетатом нитрофенилсульфилиминов ((I-10-A)) с получением 1-(циклопропилсульфонимидоил)-4-нитробензола (I-11-A) проводят предпочтительно с помощью пероксомоносульфата калия (оксон®) в качестве окислительного средства.
Реакцию проводят в смеси метанола-воды и добавляют тетраметиленсульфон (сульфолан) в качестве вспомогательного средства для растворения. Пероксомоносульфат калия (оксон®) добавляют по порциям и рН-значение после каждой стадии дозировки устанавливают равным рН 10.
Через 5 часов наблюдают 99% превращение в желательный рацемический 1-(циклопропилсульфонимидоил)-4-нитробензол (I-11-А). Перерабатывают водой (сульфит натрия) и продукт кристаллизуют из органической фазы (метиленхлорид) после сушки над сульфатом магния из н-гептана.
I-A.d) Расщепление рацематов 1-(циклопропилсульфонимидоил)-4-нитробензола (I-11-A)
Расщепление рацематов опирается на следующую стадию:
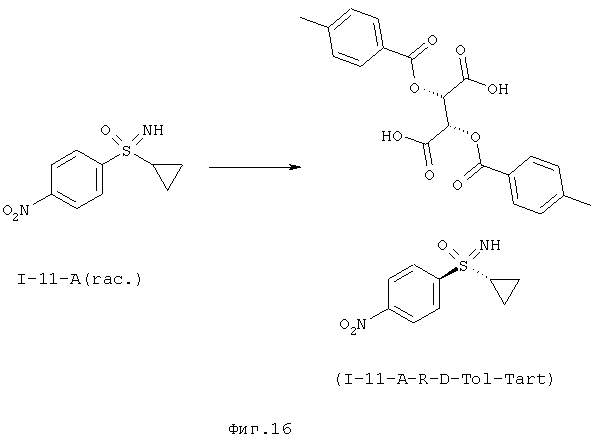
Неожиданно было обнаружено, что с помощью (+)-ди-О-п-толуоил-D-винной кислоты в ацетонитриле получают соотношение энантиомеров как минимум 95:5 в кристаллизате. Выходы составляют около 40-45%. Альтернативно ацетонитрилу можно также применять пропионитрил. Путем перекристаллизации из ацетонитрила или пропионитрила можно еще более улучшить оптическую чистоту.
Интегрирование процесса кристаллизации в способ получения может происходить, если защитную трифторацетатную группу в (I-3-A) отщепляют с помощью карбоната калия в метаноле и взаимодействия неочищенного нитрофенилсульфоксимина (I-11-A) с (+)-ди-O-п-толуоил-D-винной кислотой в ацетонитриле с образованием (I-11-A-D-Tol-Tart).
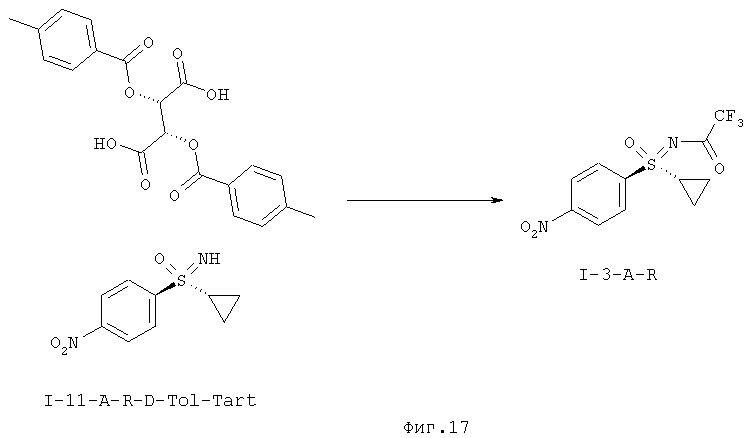
Оптически активный нитрофенилсульфоксимин выделяют основной экстракцией и затем в ходе реакции проводимой в одном сосуде защищают ангидридом трифторуксусной кислоты в присутствии триэтиламина с образованием (I-3-A-R).
Описанный в WO 2010/046035A1 путь получения защищенного трифторацетатом нитрофенилсульфоксимина (I-3-A) дает северную структурную группу в виде рацемата.
1-А.е) Гидрирование защищенного трифторацетатом анилино-сульфоксимина (I-4-A-R)
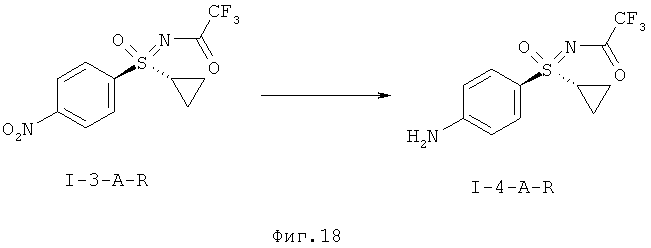
Превращение нитро-группы в соединении (I-3-A-R) в соответствующую анилиновую группу (I-4-A-R) проводят путем гидрирования в присутствии иммобилизированных палладиевых катализаторов. Особенно чистый продукт получают при применении легированного железом палладиевого катализатора на угле. В качестве растворителя предпочтителен метанол. Защищенный трифторацетатом анилинсульфоксимин (I-4-A-R) можно выделить путем кристаллизации с выходом, как минимум, 88%.
Получение "южной половины" соединения А
Создание структуры южной половины соединения А происходит в соответствии с изобретением согласно I.g) и I.f).
Соединение в одно целое северной и южной половины
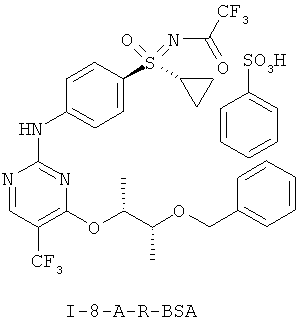
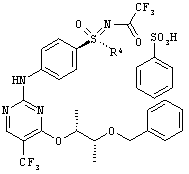
I-A.h) Получение соли N-[(4-{[4-{[(2R,3R)-3-(бенаилокси)бутан-2-ил]окси}-5-(трифторметил)пиримидин-2-ил]амино)фенил)-(циклопропил)оксидо-ламбда6-сульфанилиден]-2,2,2-трифторацетамида и бензолсульфоновой кислоты (I-8-A-R-BSA)
На первой стадии проводят соединение в одно целое обеих структурных групп (I-7-A) и (I-4-A-R) с образованием (I-8-A-R). Это взаимодействие проводят в присутствии кислоты. Свободное основание (I-8-A-R) представляет собой масло. Неожиданно было открыто, что в случае применения 1,4-диоксана в качестве растворителя, образующаяся соль бензолсульфоновой кислоты (I-8-A-R-BSA) кристаллизуется из реакционной смеси.
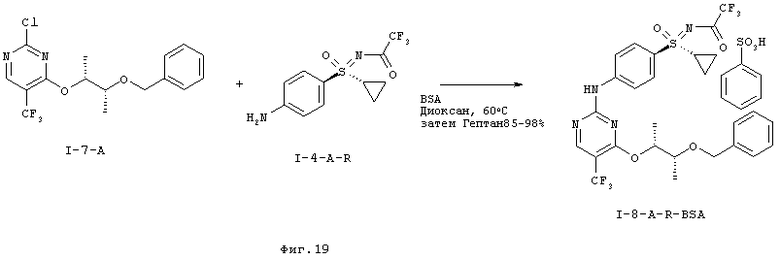
Кристаллизацию можно с помощью н-гептана полностью завершить, и получить желательную соль N-[(4-{[4-{[(2R,3R)-3-(бензилокси)бутан-2-ил]окси}-5-(трифторметил)пиримидин-2-ил]амино}фенил)-(циклопропил)оксидо-ламбда6-сульфанилиден]-2,2,2-трифторацетамида с бензолсульфоновой кислотой с хорошим выходом.
Соль (I-8-A-R-BSA) является кристаллической и хорошо хранится и ее выделяют с типичной чистотой около 90 Fl%.
I-A.i) Получение соединения А
На двух последних стадиях отщепляют защитные группы (фиг.20).
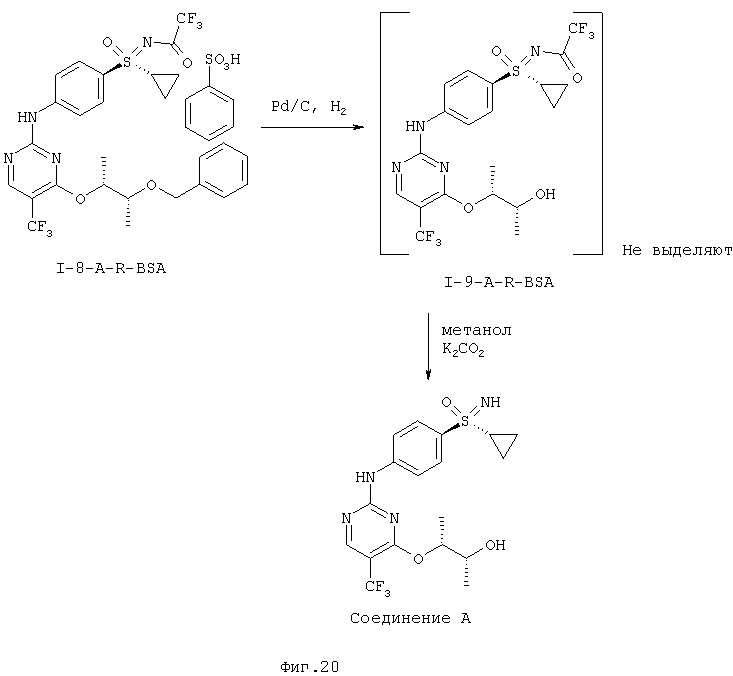
Промежуточное соединение I-9-A-R-BSA не выделяют, а напрямую подвергают превращению на конечной стадии. Отщепление трифторацетатной группы проводят с помощью карбоната калия в метаноле и кристаллизацию последней стадии проводят из смеси этилового эфира уксусной кислоты /гептана.
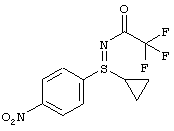
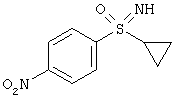
Пример
Получение 1-(циклопропилсульфанил)-4-нитробензола (1-1-А)
Раствор 80 г (0,51 моля) 4-нитротиофенола (80-процентного) в 400 мл N-метилпирролидинона (NMn) добавляют в течении 30 минут к суспензии 92,6 г (0,67 моля) карбоната калия в 400 мл NMП. Температура возрастает при этом на 30°С.
К реакционной смеси добавляют 93,6 г (0,77 моля) циклопропилбромида и перемешивают в течение 8 часов при температуре 135-140°С. Реакционную смесь охлаждают до температуры 20°С и добавляют 4,0 г активного угля. Нагревают до температуры 65°С, перемешивают в течение одного часа, фильтруют и еще раз промывают 80 мл NMП. Смесь охлаждают до температуры 20°С и в течение 1 часа добавляют по дозам в 3 л воды. Фильтруют и фильтровальный остаток промывают три раза по 400 мл воды. В заключение перемешивают с 800 мл 1 М водного раствора соляной кислоты, снова фильтруют и три раза промывают водой по 400 мл. После этого сушат при температуре 40°С в вакууме и получают 86,6 г (86%) титульного соединения (I-1-A) с чистотой 91,2 Fl.%. Сырой материал можно подвергнуть дальнейшей очистке. Для этого растворяют 90 г сырого материала в 700 мл н-гептана, нагревают до температуры 65°С, отгоняют около 400 мл н-гептана и добавляют кристаллы для затравки во время охлаждения до температуры 20°С.Перемешивают при температуре 0-5°С в течение одного часа, фильтруют и остаток промывают 100 мл холодного н-гептана. После сушки получают 81 г титульного соединения (I-1-A) с чистотой 100 F1.%.
Анализ с помощью ЯМР-спектрометрии и масс-спектрометрии: Luecking, Ulrich; Krueger, Martin; Jautelat, Rolf; Siemeister, Gerhard. Получение пиримидиниламиноарилсульфоксиминов в качестве циклин зависимой киназы (ЦЗК) и/или васкуляр эндотелиального ростового фактора (ВЭРФ) ингибиторов: WO 2005/037800, р. 105.
ЖХВР (жидкостной хроматографии высокого разрешения) - способ А: колонка Zorbax SB-Aq 150×3 мм, 3,5 мкм; градиент: 0-20 мин. от 95% водного фосфатного буфера рН 2,4 / 5% ацетонитрила до 20% водного фосфатного буфера рН 2,4 / 80% ацетонитрила; поток: 0,5 мл /мин; детектирование при 210 нм; Т=45°С;
Время удерживания для (I-1-A) при способе А: 16,7 мин.
Получение N-[циклопропил(4-нитрофенил)-ламбда4-сульфанилиден]-2,2,2-трифторацетамида (I-10-А)
Раствор 11,5 г (58,9 ммоля) 1-(циклопропилсульфанил)-4-нитробензола (I-1-А) и 10,0 г (88,4 ммоля) 2,2,2-трифторацетамида в 46 мл тетрагидрофурана (ТГФ) добавляют по порциям при температуре 0-5°С в течение 30 минут к суспензии 2,1 г (53 ммоля) гидрида натрия (60-процентного в минеральном масле) в 50 мл тетрагидрофурана.
К реакционной смеси добавляют раствор 25,3 г (88,4 ммоля) 1,3-дибром-5,5-диметилгидантиона в 86 мл тетрагидрофурана при температуре 20-25°С в течение 15 минут и перемешивают в течение 14 часов.
Для переработки добавляют 70 мл 25-процентного раствора сульфита натрия и 140 мл толуола. Органическую фазу промывают три раза по 140 мл водой, концентрируют в вакууме до около 80 г, добавляют 40 мл н-гептана и перемешивают еще 1,5 часа при температуре 20°С. Фильтруют, промывают два раза по 25 мл н-гептаном и сушат при температуре 40°С в вакууме. Получают 14,5 г титульного соединения (I-10-A) с чистотой 100 F1%. Это соответствует выходу 80,8%.
Больший масштаб:
Раствор 80,0 г (409,8 ммоля) 1-(циклопропилсульфанил)-4-нитробензола (I-1-А) и 69,5 г (614,6 ммоля) 2,2,2-трифторацетамида в 320 мл ТГФ добавляют по порциям при температуре 0-5°С в течение 45 минут к суспензии 14,8 г (368,8 ммоля) гидрида натрия (60-процентного в минеральном масле) в 360 мл ТГФ.
Затем добавляют раствор 175,7 г (614,6 ммоля) 1,3-дибром-5,5-диметилгидантоина в 550 мл ТГФ при температуре 0-5°С в течение 45 минут. Нагревают до температуры 20°С и оставляют стоять в течение 14 часов.
Переработку проводят в 560 мл 10-процентного раствора лимонной кислоты и добавляют 1,1 л толуола. Органическую фазу промывают 560 мл 25-процентного раствора сульфита натрия и трехкратно водой по 640 мл. Органическую фазу концентрируют в вакууме до около 750 г и добавляют 525 г н-гептана. Перемешивают в течение 1 часа при комнатной температуре, отсасывают и промывают 50 мл смеси 1:1 толуола / гептана. После сушки в вакууме получают 90,7 г (выход 72%) титульного соединения (I-10-A) с чистотой 99,1 F1%.
ЖХВР-способ А: Время удерживания для (I-10-A): 14,8 мин.
МС (ХИ): [М+Н]+=307, [М+NH4]+=324.
1H ЯМР (400 МГц, ДМСО-d6) δ млн. долей 1,09-1,19 (m, 1H) 1,21-1,38 (m, 3H) 3,03-3,15 (m, 1H) 8,13-8,23 (m, 2H) 8,42-8,54 (m, 2H).
Получение 1-(S-циклопропилсульфонимидоил)-4-нитробензол (I-11-А)
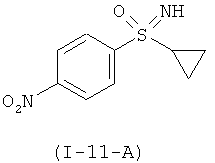
К раствору 100,0 г (326,5 ммоля) (I-10-A) в 850 мл метанола, 130 мл тетраметиленсульфона (сульфолана) и 590 мл воды добавляют 341,2 г (555,1 ммоля) пероксомоносульфата калия (оксон®), разделенного на восемь порций, при температуре 25°С. После каждого добавления рН-значение устанавливают с помощью 47-процентного водного раствора карбоната калия на значение рН 10. В сумме израсходовано около 350 мл раствора карбоната калия. Превращение при температуре около 25°С в течение одного часа было полным.
Добавляют 960 мл дихлорметана и перемешивают в течение 1 часа при температуре 20°С. Отсасывают и остаток промывают два раза по 400 мл дихлорметана. Объединенные органические фазы промывают 400 мл 10-процентного раствора, водного сульфита натрия и четыре раза по 1 л водой. После разделения фаз сушат над сульфатом магния и концентрируют до 450 г.Добавляют 100 мл н-гептана, концентрируют в вакууме до 400 мл и перемешивают в течение часа при температуре 0-5°С.Отсасывают и остаток промывают два раза 100 мл холодного н-гептана. После этого сушат в вакууме при температуре 40°С и получают 68,5 г (92,8%) титульного соединения (I-11-A) с чистотой 100 F1%.
ЖХВР-способ А: время удерживания для (I-11-A): 9,5 мин.
МС (ХИ): [М+Н]+=227 (МС=масс-спектрометрия, ХИ=химическая ионизация).
1H ЯМР (400 МГц, ДМСО-d6) δ млн. долей 0,86-1,06 (m, 3H) 1,15 (dt, J=9,96, 4,19 Гц, 1H) 2,69-2,88 (m, 1H) 4,65 (s, широкая, 1H) 8,15 (d, J=8,80 Гц, 2H) 8,41 (d, J=8,56 Гц, 2H).
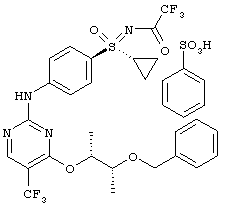
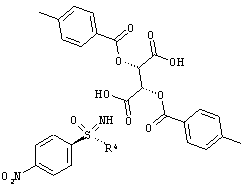
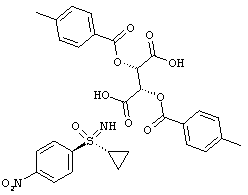
Получение (2S,3S)-2,3-бис[(4-метилбензоил)окси]бутандикислота-N-[(R)-циклопропил(4-нитрофенил) оксидо-ламбда6-сульфанилиден]-2,2,2-трифторацетамид (1:1) (I-11-A-R-D-Tol-Tart)
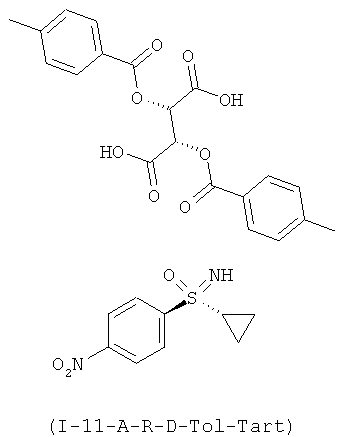
К суспензии 64,9 г (286,8 ммоля) (I-11-A) в 1,3 л ацетонитрила добавляют при температуре 20°С 116,4 г (301,2 ммоля) ди-п-толуоил-О-винной кислоты (D-Tol-Tart) и перемешивают в течение 16 часов при температуре 20°С. Отсасывают и промывают два раза по 90 мл ацетонитрилом. Сушат в вакууме при температуре 40°С до сухости и получают 71,1 г (I-11-A-R-D-Tol-Tart) Это соответствует выходу 40,4%.
ЖХВР-способ А: время удерживания для (I-11-A-D-Tol-Tart): 9,6 мин. (36,2%) и 14,7 мин. (63,8%).
ЖХВР-способ В (L159-16EE): Chiralpak 1C (фирмы DAICEL), длина: 250 мм, внутренний диаметр: 4,6 мм, размер зерен: 5 мкм, градиент: 1:1 н-гептан/ изо-пропанол изократически; поток: 1,0 мл /мин., детектирование при 252 нм; Т=35°С.
Время удерживания для R-энантиомера: 9,3 мин; время удерживания для 3-энантиомера: 8,4 мин; избыток энантиомеров (ее): 99,6%.
МС (ИЭУ+): [М+Н]+=227; МС (ИЭУ-): [М-Н]-=385.
1H ЯМР (400 МГц, ДМСО-d6) δ млн. долей 0,91-1,06 (m, 3H) 1,10-1,20 (m, 1H) 2,41 (s, 6H) 2,72-2,84 (m, 1H) 4,64 (s, широкий, 1H) 5,82 (s, 2H) 7,40 (d, J=8,07 Гц, 4H) 7,90 (d, J=8,07 Гц, 4H) 8,09-8,20 (m, 2H) 8,33-8,52 (m, 2H) 13,85 (s, широкий, 2Н).
Получение N-[(R)-циклопропил(4-нитрофенил)оксидо-ламбда6-сульфанилиден]-2,2,2-триф-горацетамида (I-3-A-R)
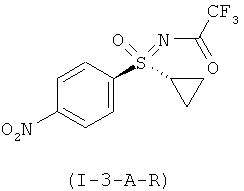
Раствор 72,1 г (117,7 ммоля) (I-11-A-R-D-Tol-Tart.) в 720 мл дихлорметана перемешивают в течение 60 минут с раствором 24,4 г карбоната калия в 350 мл воды при температуре 20°С. Водную фазу экстрагируют 360 мл дихлорметана и объединенные органические фазы промывают 720 мл воды и сушат над сульфатом магния. Фильтруют и добавляют к фильтрату 49 мл (353,1 ммоля) триэтиламина и затем в течение 40 минут добавляют 49,9 мл (353,1 ммоля) ангидрида трифторуксусной кислоты при температуре 20-25°С. Перемешивают еще 10 минут и затем добавляют по дозам в 1,1 л насыщенного раствора гидрокарбоната натрия. После разделения фаз промывают 1,0 л воды, сушат над сульфатом магния и отгоняют растворитель в вакууме. Остаток помещают в 120 мл изо-пропанола и полученную суспензию перемешивают в течение 1 часа при температуре 0-5°С. Фильтруют и остаток промывают два раза по 30 мл холодным изо-пропанолом. Сушат при температуре 40°С в вакууме и получают 27,3 г (75%) (I-3-A-R).
ЖХВР-способ А: время удерживания для (I-3-A-R): 16,4 мин (99,6%).
ЖХВР-способ С (L159-10EE): Chiralpak 1C (фирмы DAICEL), длина: 250 мм, внутренний диаметр: 4,6 мм, размер зерен: 5 мкм, градиент: 1:1 н-гептан/ этанол изократически; поток: 1,0 мл /мин, детектирование при 240 нм, Т=35°С.
Время удерживания для R-энантиомера (I-3-A-R): 4,5 мин; время удерживания для S-энантиомера: 3,7 мин; избыток энантиомера (ее): 100%.
МС (ОХИ): [M+H]+=323, [M+NH4]+=340.
1H ЯМР (400 МГц, хлороформ-d) δ млн. долей 1,11-1,29 (m, 1H) 1,36-1,52 (m, 2H) 1,74-1,88 (m, 1H) 2,69-2,89 (m, 1H) 8,14 (d, J=8,80 Гц, 2H) 8,47 (d, J=8,80 Гц, 2H).
Получение N-[(R)-(4-аминофенил)(циклопропил)оксидо-ламбда6-сульфанилиден]-2,2,2-трифторацетамида (I-4-A-R):
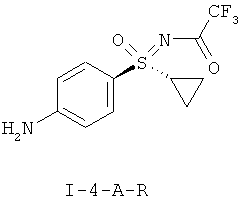
Суспензию из 40,0 г (124,1 ммоля) (I-3-A-R) и 10,0 г палладия на угле (Pd/C: 5% Pd, 1% Fe, 55% воды) в 800 мл метанола гидрируют семь часов при давлении 2,5 бара. Фильтруют через кизельгур и два раза промывают по 200 мл метанолом. Фильтрат концентрируют в вакууме, а после этого добавляют 800 мл воды. Перемешивают в течение одного часа, фильтруют и промывают два раза по 400 мл водой. Кристаллы сушат при 40°С в вакууме. Получают 33,4 г (292,3 ммоля) желательного анилина (I-4-A-R). Это соответствует выходу 92%.
ЖХВР-способ А: Время удерживания для (I-4-A-R): 14,4 минут (96,1%).
ЖХВР-способ С: Время удерживания для R-энантиомера (I-4-A-R): 13,7 мин;
Время удерживания для S-энантиомера: 12,2 мин; избыток энантиомера (ее): 100%.
МС (ОХИ): [M+H]+=293, [M+NH4]+=310.
1Н ЯМР (400 МГц, хлороформ-d) δ млн. долей 1,00-1,16 (m, 1H) 1,17-1,40 (m, 2H) 1,56-1,71 (m, 1H) 2,74 (tt, J=7,79, 4,92 Гц, 1H) 4,33 (br. s., 2H) 6,67-6,79 (m, 2H) 7,58-7,74 (m, 2H).
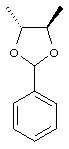
Получение (4R,5R)-4,5-диметил-2-фенил-1,3-диоксолана (I-12-А)
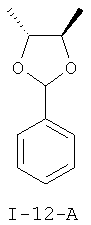
Раствор 300 г (3,33 моля) 2R,3R-бутандиола, 422 г (2,78 моля) бензальдегиддиметилацеталя и 7,0 г (27,7 ммоля) пиридиний-п-толуолсульфоната в 1,2 л толуола нагревают при температуре 50°С. При давлении 600-800 мбар в течение 3 часов отгоняют около 400 мл дистиллята. Охлаждают и реакционную смесь дозируют до 500 мл с помощью 1М натронного щелока. Проводят разделение фаз и органическую фазу промывают два раза по 500 мл водой. Органическую фазу сушат азеотропно, причем отгоняют около 250 мл толуола.
Полученный таким образом продукт в толуоле (1061 г) применяют напрямую на следующей стадии. Для проведения анализа брали часть вещества и упаривали до сухости.
Получение соединения (I-12-A) можно среди прочего провести согласно литературным данным (Chemistry Letters (1995), 4, 263-4; Journal of Organic Chemistry (2003), 68(9), 3413-3415, Tetrahedron (1989), 45(2), 507-16; Journal of Organic Chemistry (2005), 70(20), 8009-8016; Bioorganic & Medicinal Chemistry Letters (2006), 16(1), 186-190; Journal of Organic Chemistry (1999), 64 (20), 7594-7600).
ГХ (газовой хроматографии)-способ А: Колонка RTX-50 (Fused Silca, 100% метилфенилполисилоксан), длина: 30 м, внутренний диаметр: 0,32 мм, толщина пленки: 1,0 мкм; поток: 3 мл/мин; газ-носитель водород; детектор FID 320°С, температура инжектора 280°С; программа анализа: Т=80°С, время выдерживания 2 мин, скорость повышения температуры 10°С/мин до Т=300°С, время выдерживания 6 мин.
Время удерживания (I-12-A): 12,1 мин (98%), толуола 3,3 мин.
МС (ОХИ): [М+Н]+=179.
1H ЯМР (400 МГц, хлороформ-d) δ млн. долей 1,30-1,35 (m, 3H) 1,36-1,41 (m, 3H) 3,74-3,87 (m, 2H) 5,94 (s, 1H) 7,32-7,41 (m, 3H) 7,49 (dd, J=7,70, 1,83 Гц, 2H).
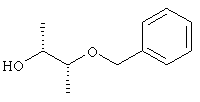
Получение (2R,3R)-3-(фенилметокси)-2-бутанол (I-5-А)
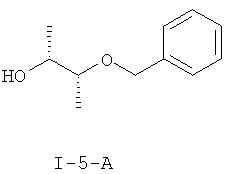
Полученный на предыдущей стадии толуольный раствор (I-12-A) разделяют и в ходе двух подходов подвергают дальнейшему превращению:
Берут 530 г раствора (I-12-A) и разбавляют 500 мл толуола, нагревают до температуры 55°С и в течение одного часа добавляют 2,2 л 1,5 М раствора диизобутилалюминия в толуоле. Перемешивают в течение двух часов при температуре 50-60°С. Реакционную смесь добавляют по дозам в течение одного часа при температуре 20-25°С в суспензию 500 г натрий сульфат-декагидрата в 500 мл толуола. Фильтруют, промывают десять раз по 500 мл толуола. Объединенные органические фазы фильтруют через кизельгур и отгоняют растворитель в вакууме. Получают 230 г (1,27 моля) (I-5-А) в одном подходе. Это соответствует выходу около 76% на двух стадиях.
Получение соединения (I-5-А) можно среди прочего также провести согласно литературным данным: Bioorganic & Medicinal Chemistry Letters (2006), 16(1), 186-190; EP 1291336A2; Journal of the American Chemical Society (1997), 119(19), 4541-4542; Journal of Organic Chemistry (1990), 55(10), 3129-37.
ЖХВР-способ А: Время удерживания (I-5-А): 12,5 минут (97,4%). МС (ЭУ+): [M+H]+=181.
1H ЯМР (400 МГц, хлороформ-d) δ млн. долей 1,16 (t, J=5,99 Гц, 6Н) 2,77 (d, J=2,69 Гц, 1H) 3,25-3,36 (m, 1H) 3,61 (quind, J=6,54, 6,54, 6,54, 6,54, 2,69 Гц, 1Н) 4,43 (d, J=11.49 Гц, 1Н) 4,66 (d, J=11,49 Гц, 1Н) 7,26-7,38 (m, 5Н).
Получение 4-{[(2R,3R)-3-(бензилокси)бутан-2-ил]окси}-2-хлор-5-(трифторметил)пиримидина (I-7-А)
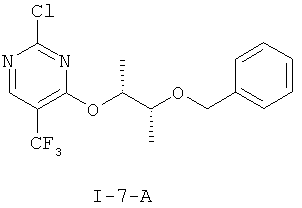
К раствору 140,4 г (647,3 ммоля) 2,4-дихлор-5-(трифторметил)пиримидина и 122,5 г (679,6 ммоля) (I-5-A) в 1,0 л ТГФ добавляют по порциям при температуре -35°С в течение 30 минут 680 мл 1 М раствора гексаметилдисилазида лития. Перемешивают в течение трех часов при температуре -30°С. Нагревают до температуры 0°С и в течение 15 минут добавляют 1,0 л воды. Добавляют 1,0 л эфира уксусной кислоты, разделяют фазы и водную фазу экстрагируют 300 мл эфира уксусной кислоты. Отгоняют растворитель из объединенных органических фаз в вакууме до объема около 1,5 л и промывают 1,0 л воды. Сушат над сульфатом натрия, отсасывают через кизельгур и упаривают в вакууме. Получают 238,4 г сырого продукта в виде коричневого масла. Вторая порция в тех же масштабах дала следующие 236 г сырого продукта.
Обе порции объединяют и растворяют в 470 мл гептана / эфира уксусной кислоты 1:1. Отсасывают через силикагель 60 и два раза промывают по 1,0 л уксусной кислоты. Объединенные фильтраты фильтруют через силикагель, сушат над сульфатом магния и отгоняют растворители в вакууме. Остаток хроматографируют через 15 кг силикагеля 60 с н-гептаном/эфиром уксусной кислоты 15:1. Получают 171 г (I-7-А) (35%), а также 63 г смешанной фракции, которая содержала еще 58 Fl% (I-7-А).
ЖХВР-способ А: Время удерживания (I-7-A) (4-изомер) 21,6 минут (95%); время удерживания 2-изомера: 21,0 минуты.
МС (ES-API): [М+Н]+=361.
1H ЯМР (400 МГц, ДМСО-d6) δ млн. долей 1,16 (d, J=6,36 Гц, 3Н) 1,32 (d, J=6,36 Гц, 3Н) 3,74 (quin, J=6,17 Гц, 1Н) 4,46 (d, J=11,98 Гц, 1Н) 4,61 (d, J=11,98 Гц, 1Н) 5,44 (quin, J=6,24 Гц, 1Н) 7,11-7,46 (m, 5Н) 8,85 (s, 1Н).
Получение соли N-[(4-{[4-{[(2R,3R)-3-(бензилокси)буран-2-ил]окси}-5-(трифторметил)пиримидин-2-ил]амино)фенил)-(циклопропил)оксидо-ламбда6-сульфанилиден]-2,2,2-трифторацетамида с бенэолсульфоновой кислотой (1:1) (I-8-A-R-BSA)
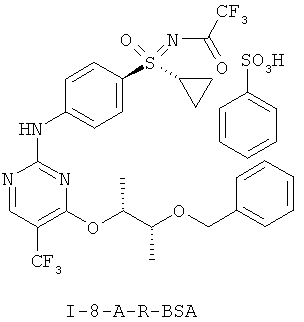
Суспензию 52,0 г (144 ммоля) (I-7-A), 42,1 г (144 ммоля) (I-4-A-R) и 22,8 г (144 ммоля) бензолсульфоновой кислоты в 842 мл диоксана перемешивают в течение 16 часов при температуре 20°С. После этого нагревают в течение 19 часов при температуре 55-60°С. Для выделения добавляют при температуре 20°С кристаллы для инициирования кристаллизации и разбавляют 1,68 л н-гептана. Перемешивают в течение одного часа, отсасывают, промывают 240 мл диоксана/н-гептана (1:1), а также 240 мл н-гептана. Сушат до постоянства массы в вакууме при температуре 40°С. Получают 113,7 г (100%) желательного вещества в виде бежевых кристаллов.
ЖХВР-способ А: Время удерживания (I-8-A-R-BSA) 22,3 минут (94%); время удерживания для продукта распада с частично снятой трифторацетатной защитой 19,1 минут (2,4%).
МС (ЭУ+): [М+Н]+=617.
1Н ЯМР (500 МГц, ДМСО-d6) δ млн. долей 1,10 (m, 2H) 1,19 (m, 3H) 1,34 (m, 3H), 1,45 (m, 1H) 3,38 (m, 1H) 3,77 (m, 1H) 4,49 (d, 1H) 4,60 (d, 1H) 5,52 (m, 1H) 7,30 (m, 7H) 7,60 (m, 2H) 7,92 (m, 2H) 8,09 (m, 2H) 8,63 (m, 1H) 10,71 (m, 1H).
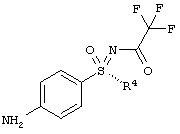
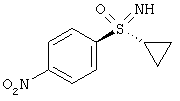
Получение (2R,3R)-3-{[2-{[4-(S-циклопропилсульфонимидоил)фенил]-амино}-5-(трифторметил)пиримидин-4-ил]окси}бутан-2-ола (соединение А)
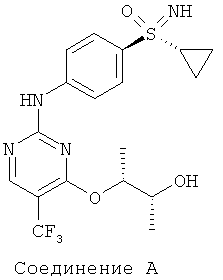
Через суспензию 120 г (I-8-A-R-BSSA) и 60 г 10% Pd/C в 640 г метанола пробулькивают в течение 6 часов водород при нормальном давлении. Фильтруют через кизельгур и два раза промывают по 100 г метанола. К фильтрату добавляют 53 г карбоната калия и перемешивают в течение 1 часа при температуре 20°С. Для переработки добавляют 900 г воды и 700 г дихлорметана. Водную фазу экстрагируют 700 г дихлорметана и объединенные органические фазы промывают 900 г воды и сушат над сульфатом натрия. Фильтруют и дополнительно промывают 400 г дихлорметана. Отгоняют растворитель в вакууме. Остаток растворяют в 10 г эфира уксусной кислоты и добавляют 125 г н-гептана. Перемешивают в течение 10 минут при температуре 20°С и снова добавляют 125 г н-гептана. Перемешивают в течение двух часов при температуре 20°С, фильтруют, промывают смесью н-гептана (70 г)/эфира уксусной кислоты (45 г) и 100 г н-гептана. Сушат в вакууме до постоянства массы при температуре 30°С. Получают 47 г (69%) целевого соединения.
ЖХВР-способ А: Время удерживания соединения А 14,24 мин. (100%).
МС (ИЭП+): [М+Н]+=431.
1H ЯМР (500 МГц, ДМСО-d6) δ млн. долей 0,92 (m, 3Н) 1,10 (m, 4H) 1,30 (d, 3Н) 2,62 (m, 1H) 3,85 (m, 1H) 4,08 (s, 1H) 4,91 (d, 1H) 5,31 (m, 1H) 7,83 (d, 2H) 7,94 (d, 2H) 8,59 (s, 1H) 10,50 (m, 1H).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКИХ АМИНОВ В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРА ЕР4 | 2011 |
|
RU2565596C2 |
| ЗАМЕЩЕННЫЕ N-ФЕНИЛБИПИРРОЛИДИНКАРБОКСАМИДЫ И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕБНЫХ ЦЕЛЯХ | 2008 |
|
RU2477720C2 |
| СОЕДИНЕНИЯ ПИРИМИДИНА И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ РАКА | 2019 |
|
RU2807277C2 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛОВ В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2689777C1 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ, ПРИМЕНЯЕМЫЕ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ, СВЯЗАННЫХ С NTRK | 2016 |
|
RU2744974C2 |
| ЗАМЕЩЕННЫЕ ДИАМИНОКАРБОКСАМИДНЫЕ И ДИАМИНОКАРБОНИТРИЛЬНЫЕ ПРОИЗВОДНЫЕ ПИРИМИДИНОВ, ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ИХ ПОМОЩЬЮ | 2012 |
|
RU2697712C2 |
| Макрогетероциклические нуклеозидные производные и их аналоги, получение и применение | 2017 |
|
RU2731385C1 |
| 2-ОКСА-5-АЗАБИЦИКЛО[2.2.1]ГЕПТАН-3-ИЛЬНЫЕ ПРОИЗВОДНЫЕ | 2015 |
|
RU2697651C2 |
| АНТИПРОЛИФЕРАТИВНЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ PAH | 2020 |
|
RU2786588C1 |
| СПОСОБЫ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ В ПОЛУЧЕНИИ C5aR АНТАГОНИСТОВ | 2015 |
|
RU2712233C2 |
Изобретение относится к новому способу получения ингибиторов пан-ЦЗК (циклинзависимой киназы) общей формулы (I). Соединения общей формулы (I)
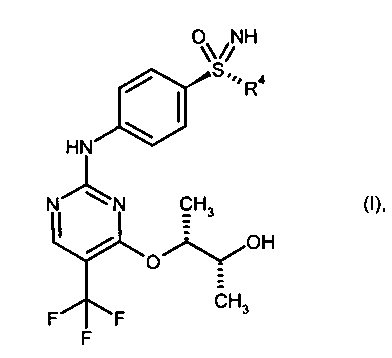
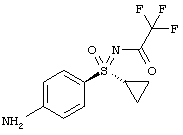
в которой R4 означает (С1-С6)-алкильную группу или (С3-С7)-циклоалкильное кольцо, получают путем катализируемого бензолсульфоновой кислотой присоединения предварительно полученных соединений (I-7-A) к (I-4-R) с получением солей бензолсульфоновой кислоты (BSA) защищенных анилинопиримидинов формулы (I-8-R-BSA)
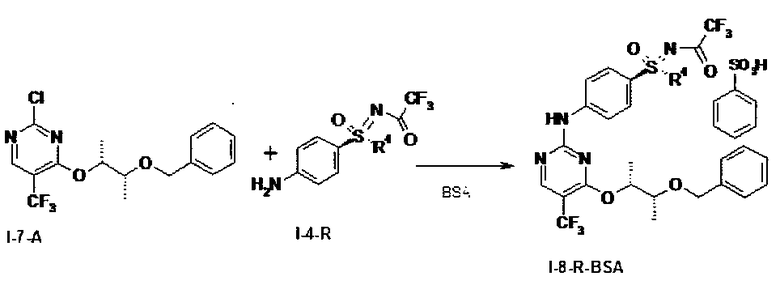 ,
,
с отщеплением защитных групп в солях формулы (I-8-R-BSA) гидрированием водородом в присутствии палладия на активном угле в метаноле и обработкой карбонатом калия в метаноле с получением соединений формулы (I).
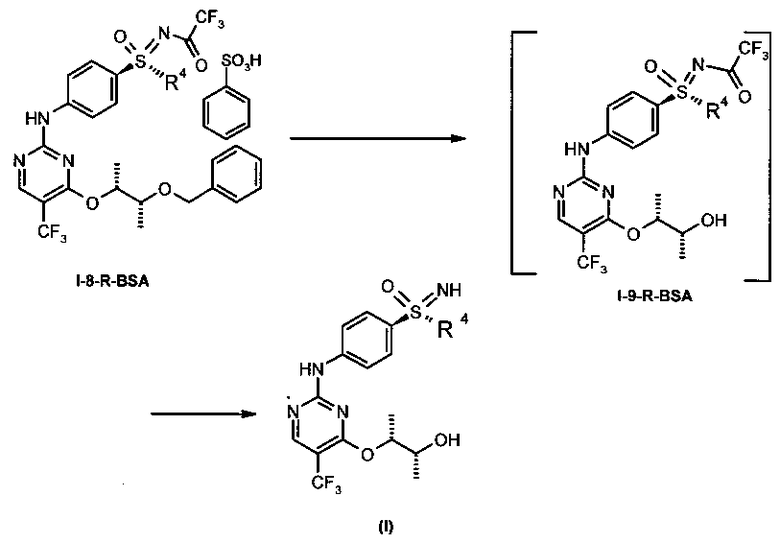
Изобретение также относится к новым промежуточным соединениям структурных формул (I-8-А-R-BSA) и (I-11-A-R-D-Tol-Tart).
 ,
, 
Способ упрощает условия получения промежуточных и целевых продуктов, что позволяет его использовать в промышленном масштабе. 3 н. и 4 з.п. ф-лы, 2 табл.
1. Способ получения соединений общей формулы (I)
в которой
R4 означает (С1-С6)алкильную группу или (С3-С7)циклоалкильное кольцо,
путем катализируемого бензолсульфоновой кислотой присоединения (I-7-A) к (I-4-R) с получением солей бензолсульфоновой кислоты (BSA) двукратно защищенных анилинопиримидинов формулы (I-8-R-BSA)
и отщепления защитных групп в солях бензолсульфоновой кислоты двукратно защищенных анилинопиримидинов формулы (I-8-R-BSA) путем гидрирования водородом в присутствии палладия на активном угле в метаноле, а также обработки карбонатом калия в метаноле с получением соединений формулы (I),
при этом используют соединение (I-4-R), полученное
I.a) алкилированием 4-нитротиофенола в присутствии карбоната калия в N-метилпирролидиноне (NMП) с получением нитрофенилсульфида формулы (I-1)
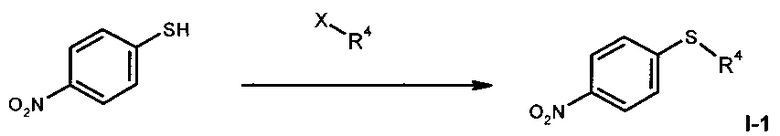 ,
,
причем X означает Br, Cl, I, O-SO2-CH3 или O-SO2-(4-метилфенил),
I.b) окислительным аминированием нитрофенилсульфида формулы (I-1) в защищенный трифторацетатом нитрофенилсульфилимин формулы (I-10)
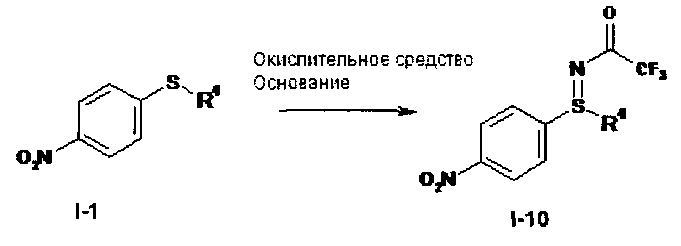
I.c) окислением защищенного трифторацетатом нитрофенилсульфилимина формулы (I-10) в защищенный трифторацетатом нитрофенилсульфоксимин формулы (I-3) и последующим снятием защиты с образованием нитрофенилсульфоксимина формулы (I-11)
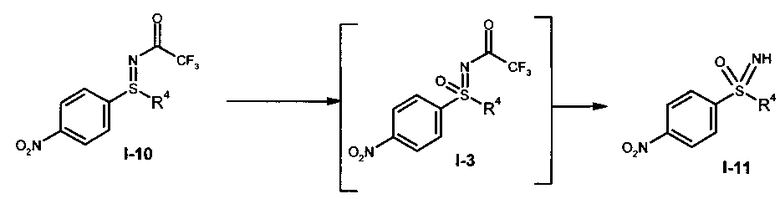 ,
,
I.d) расщеплением рацематов (rac.) нитрофенилсульфоксимина
формулы (I-11) с помощью (+)-ди-O-п-толуол-D-винной кислоты (D-Tol-Tart)
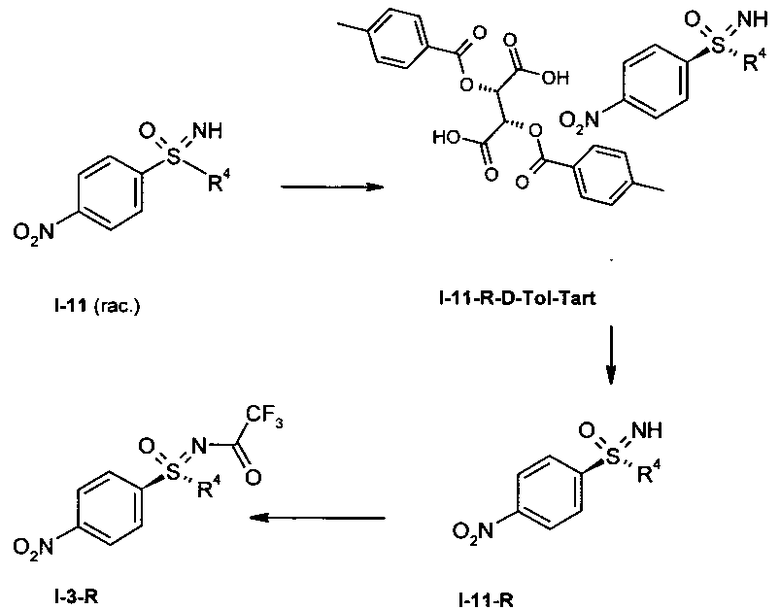 ,
,
причем R-энантиомер нитрофенилсульфоксимина формулы (I-11-R) высвобождают затем из солей и вновь вводят трифторацетатную защитную группу с образованием R-энантиомера защищенного трифторацетатом нитрофенилсульфоксимина формулы (I-3-R),
I.e) гидрированием защищенного трифторацетатом нитрофенилсульфоксимина формулы (I-3-R) с образованием защищенного трифторацетатом анилиносульфоксимина формулы (I-4-R) в присутствии легированного железом палладиевого катализатора

и соединение (I-7-А), полученное
I.f) получением (2R,3R)-3-(бензилокси)бутан-2-ола (I-5-А) в двухстадийном процессе через (4R,5R)-4,5-диметил-2-фенил-1,3-диоксолан (I-12-А), причем первую стадию проводят с пиридиний-п-толуолсульфонатом в толуоле и затем проводят восстановление диизобутилалюминийгидридом в толуоле
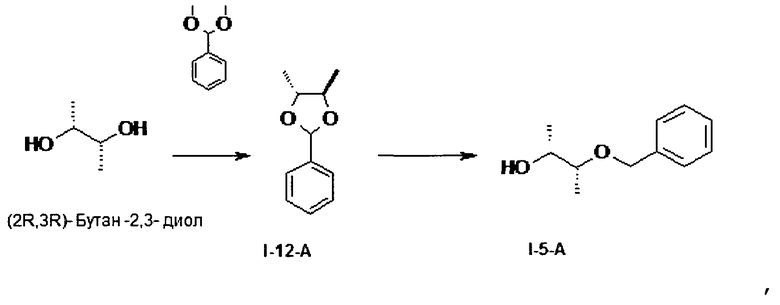
I.g) присоединением (I-5-A) к 2,4-дихлор-5-трифторметилпиримидину с образованием 4-{[(2R,3R)-3-(бензилокси)бутан-2-ил]окси}-2-хлор-5-(трифторметил)пиримидина (I-7-A) в присутствии Li-оснований в простых эфирных растворителях
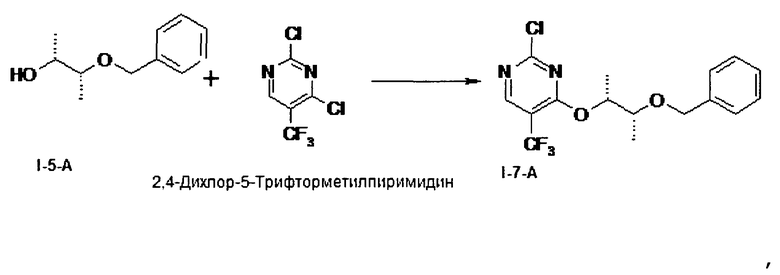
2. Способ по п. 1, в котором на стадии I.b) используют 1,3-дибром-5,5-диметилгидантоин в качестве окислительного средства и трифторацетамид в качестве реагента.
3. Способ по п. 1, в котором на стадии I.c) окисление проводят пероксомоносульфатом калия (оксон®).
4. Способ по п. 1, в котором на стадии I.d) нитрофенил-сульфоксимин формулы (I-11) с (+)-ди-O-п-толуоил-D-винной кислотой кристаллизуют в ацетонитриле или пропионитриле.
5. Способ по п. 1, в котором на стадии I.g) в качестве литиевого основания используют гексаметилдисилазид лития, а в качестве простого эфирного растворителя - тетрагидрофуран.
6. Соль формулы (I-11-A-R-D-Tol-Tart)
.
7. Промежуточное соединение формулы (I-8-A-R-BSA)
.
| WO 2010046035 A1, 29.04.2010 &αµπ; EA019230 &αµπ; EP 2179991 A1 |

