Область техники, к которой относится изобретение
Изобретение раскрывает промежуточные соединения и способы получения некоторых C5aR антагонистов.
Предшествующий уровень техники
Система комплемента играет центральную роль в клиренсе иммунных комплексов и иммунных ответах на возбудителей инфекций, чужеродные антигены, зараженные вирусом клетки и опухолевые клетки. Ненадлежащая или избыточная активация системы комплемента может привести к вредным и даже потенциально угрожающим жизни последствиям из-за сильного воспаления и вызываемого им разрушения ткани. Эти последствия клинически проявляются в различных нарушениях, включая септический шок; ишемии миокарда и кишечника/реперфузионном повреждении; отторжении трансплантата; отказе органов; нефрите; патологическом воспалении и аутоиммунных заболеваниях.
Активация системы комплемента генерирует биологически активные фрагменты белков комплемента, например С3а, С4а и С5а анафилотоксинов и C5b-9 мембраноатакующих комплексов (МАК), которые все вызывают воспалительный ответ путем воздействия на хемотаксис лейкоцитов; активации макрофагов, нейтрофилов, тромбоцитов, тучных клеток и клеток эндотелия; и усиления сосудистой проницаемости, цитолиза и поражения ткани.
Комплемент C5a представляет собой один из наиболее мощных провоспалительных медиаторов в системе комплемента. (Анафилактический C5a пептид в 100 раз активнее, в расчете на мольные количества, в создании воспалительного ответа, чем C3a.) C5a представляет собой активированную форму C5 (молекулярный вес 190 кДа). C5a присутствует в плазме крови человека в количестве примерно 80 мкг/мл (Kohler, P. F. et al., J. Immunol. 99: 1211-1216 (1967)). Он состоит из двух полипептидных цепочек, α и β, с приблизительными молекулярными весами 115 кДа и 75 кДа, соответственно (Tack, B. F. et al., Biochemistry 18: 1490-1497 (1979)). Биосинтезируясь в виде одноцепочечной промолекулы, C5 ферментативно расщепляется на двухцепочечную структуру во время процессинга и секреции. После расщепления, две полученные цепочки удерживаются вместе благодаря по меньшей мере одной дисульфидной связи, а также нековалентным взаимодействиям (Ooi, Y. M. et al., J. Immunol. 124: 2494-2498(1980)).
C5 расщепляется на фрагменты C5a и C5b во время активации комплементной реакции. Ферменты конвертазы, отвечающие за активацию C5, представляют собой много-субъединичные комплексы из C4b, C2a и C3b для классического пути, и из (C3b)2, Bb и P для альтернативного пути (Goldlust, M. B. et al., J. Immunol. 113: 998-1007 (1974); Schreiber, R. D. et al, Proc. Natl. Acad. Sci. 75: 3948-3952 (1978)). C5 активируется расщеплением по положению 74-75 (Arg-Leu) в α-цепи. После активации высвобождается 74-аминокислотный пептид C5a весом 11.2 кДа из амино-терминального участка α-цепи. C5a и C3a оба являются сильными стимуляторами нейтрофилов и моноцитов (Schindler, R. et al., Blood 76: 1631-1638 (1990); Haeffner-Cavaillon, N. et al., J. Immunol. 138: 794-700 (1987); Cavaillon, J. M. et al., Eur. J. Immunol. 20: 253-257 (1990)).
В дополнение к своим анафилотоксическим свойствам, C5a вызывает хемотаксичную миграцию нейтрофилов (Ward, P. A. et al., J. Immunol. 102: 93-99 (1969)), эозинофилов (Kay, A. B. et al., Immunol. 24: 969-976 (1973)), базофилов (Lett-Brown, M. A. et al., J. Immunol. 117: 246-252 1976)) и моноцитов (Snyderman, R. et al., Proc. Soc. Exp. Biol. Med. 138: 387-390 1971)).
Считается, что анафилактические и хемотактические эффекты C5a работают через его взаимодействие с C5a рецептором. Человеческий C5a рецептор (C5aR) представляет собой 52 кДа мембраносвязанный рецептор, связанный с G-белком, который экспрессирован на нейтрофилах, моноцитах, базофилах, эозинофилах, гепатоцитах, гладких мышцах легких и эндотелиальных клетках, а также в тканях почечных клубочков (Van-Epps, D. E. et al., J. Immunol. 132: 2862-2867 (1984); Haviland, D. L. et al., J. Immunol. 154:1861-1869 (1995); Wetsel, R. A., Immunol. Leff. 44: 183-187 (1995); Buchner, R. R. et al., J. Immunol. 155: 308-315 (1995); Chenoweth, D. E. et al., Proc. Natl. Acad. Sci. 75: 3943-3947 (1978); Zwirner, J. et al., Мол. Immunol. 36:877-884 (1999)). Связывающийся с лигандом сайт C5aR сложный и состоит из по меньшей мере двух физически разделимых связывающихся доменов. Один связывается с амино-концом C5a (аминокислоты 1-20) и дисульфидно-связанным ядром (аминокислоты 21-61), а второй связывается с карбоксильным концом C5a (аминокислоты 62-74) (Wetsel, R. A., Curr. Opin. Immunol. 7: 48-53 (1995)).
Только недавно в литературе были описаны непептидные антагонисты C5a рецептора (например, Sumichika, H., et al., J. Biol. Chem. (2002), 277, 49403-49407). Сообщалось, что непептидный антагонист C5a рецептора эффективен для лечения эндотоксинового шока у крыс (Stracham, A.J., et al., J. of Immunol. (2000), 164(12): 6560-6565); и для лечения воспалительной болезни кишечника в крысиной модели (Woodruff, T.M., et al., J of Immunol., 2003, 171: 5514-5520). Непептидные модуляторы C5a рецептора также были описаны в патентной литературе компанией Neurogen Corporation (например, WO2004/043925, WO2004/018460, WO2005/007087, WO03/082826, WO03/08828, WO02/49993, WO03/084524); Dompe S.P.A. (WO02/029187); и Университетом Квинланда (WO2004/100975).
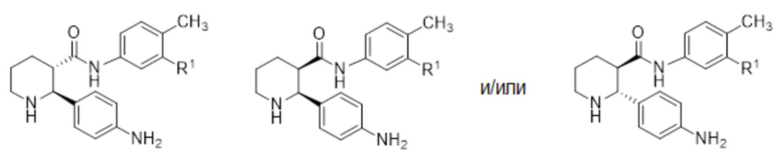
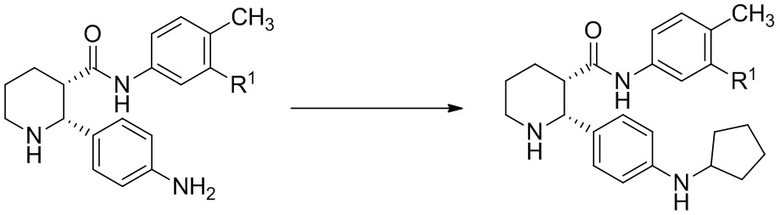
Соединения, проявляющие активность как C5aR антагонисты, были позднее обнаружены и описаны в Патенте США № 8,445,515 B2. В целом, эти соединения представлены формулой A, но некоторые варианты осуществления описаны как имеющие формулу B:
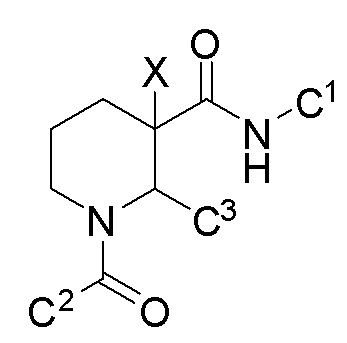
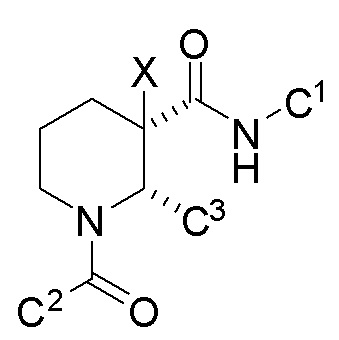
Некоторые описанные в указанном документе соединения особенно активны после расщепления до их (2R,3S) изомеров и описаны как:
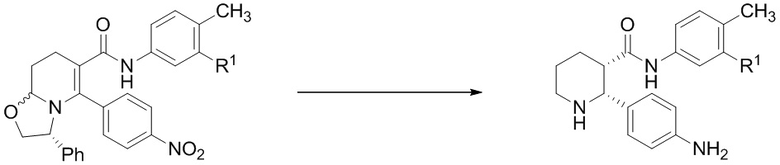
Получение соединения IA было осуществлено, как описано на Фиг. 4, и включает продолжительный синтез с привлечением классического расщепления изомеров (смотри, например, превращение 6 в 7).
В данной области существует потребность в более эффективных способах получения соединений IA, IB и IC. В настоящем изобретении описаны такие способы, а также промежуточные соединения в этих методах синтеза.
Краткое описание изобретения
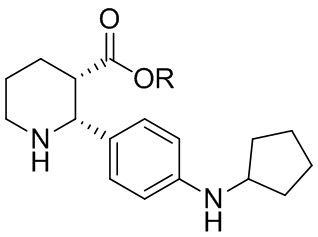
В одном аспекте, в настоящем изобретении описано соединение, которое может применяться в получении нескольких C5aR антагонистов, имеющее формулу (i-3):
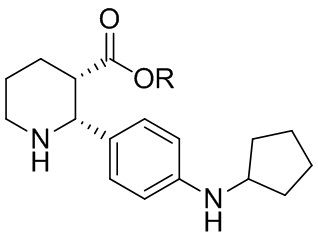 (i-3)
(i-3)
(2R,3S)
где R выбран из H, C1-8 алкила, арила и арил-C1-4 алкила, или его соль, которое практически не содержит энантиомерных или диастереомерных примесей ((2R,3R), (2S,3R) и (2S,3S) изомеров).
В другом аспекте, в настоящем изобретении описано соединение, которое может применяться в получении нескольких C5aR антагонистов, имеющее формулу (ii-4):
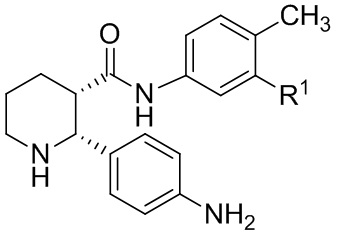 (ii-4)
(ii-4)
(2R,3S)
или его соль, где R1 представляет собой Cl или CF3, и где указанное соединение практически не содержит энантиомерных или диастереомерных примесей (соответствующих (2R,3R), (2S,3R) и (2S,3S) изомеров).
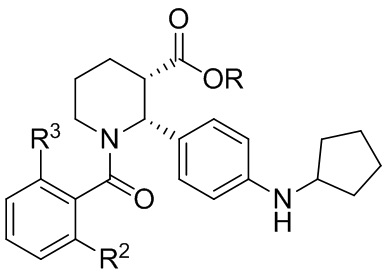
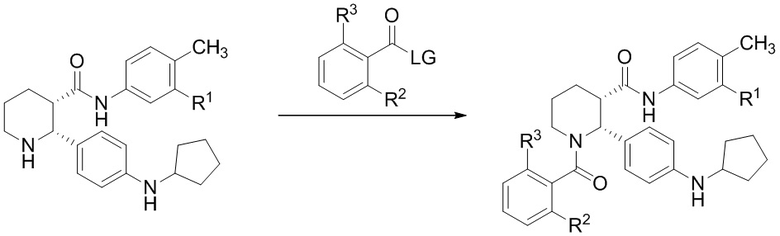
В другом аспекте, в настоящем изобретении описан способ получения соединения, имеющего формулу (I):
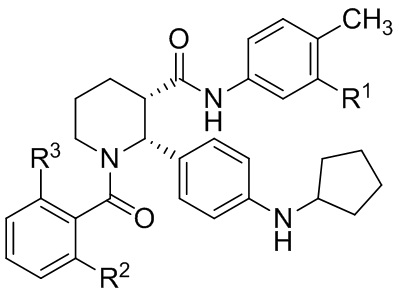 (I)
(I)
или его соли, где R1 представляет собой Cl или CF3; R2 представляет собой F или Cl; и R3 представляет собой H или CH3; и где указанное соединение формулы (I) практически не содержит энантиомерных или диастереомерных примесей, включающий:
(a) взаимодействие соединения, имеющего формулу (i-3):
(i-3)
где R выбран из C1-8 алкила, арила и арил-C1-4 алкила, которое практически не содержит энантиомерных или диастереомерных примесей, или его соли, с соединением, имеющим формулу:
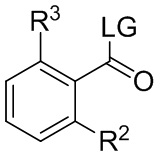
где LG представляет собой уходящую группу; в условиях, достаточных для образования соединения формулы (i-4):
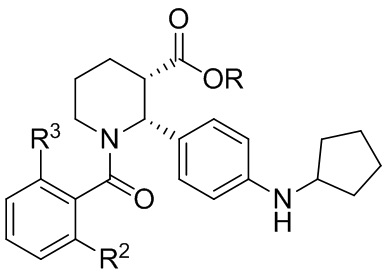 (i-4); и
(i-4); и
(b) превращение соединения формулы (i-4) в указанное соединение формулы (I), где указанное соединение формулы (I) практически не содержит энантиомерных или диастереомерных примесей.
В другом аспекте, в настоящем изобретении описан другой способ получения соединения, имеющего формулу (I):
(I)
или его соли, где R1 представляет собой Cl или CF3; R2 представляет собой F или Cl; и R3 представляет собой H или CH3; и где соединение формулы (I) практически не содержит энантиомерных или диастереомерных примесей, включающий:
(a) взаимодействие соединения, имеющего формулу (ii-4):
(ii-4)
или его соли, где указанное соединение практически не содержит энантиомерных или диастереомерных примесей, с циклопентаноном и восстановителем в условиях, достаточных для образования соединения, имеющего формулу (ii-5):
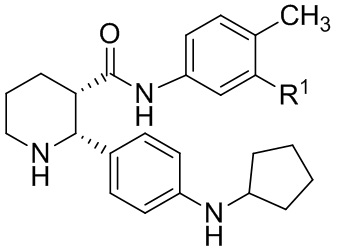 (ii-5); и
(ii-5); и
(b) взаимодействие указанного соединения формулы (ii-5) с соединением, имеющим формулу:
где LG представляет собой уходящую группу; в условиях, достаточных для образования соединения формулы (I), которое практически не содержит энантиомерных или диастереомерных примесей.
Описаны также представленные ниже другие способы, включающие два, три или четыре или более синтетических превращения, приводящие к получению соединений IA, IB и/или IC, или их фармацевтически приемлемых солей.
Краткое описание чертежей
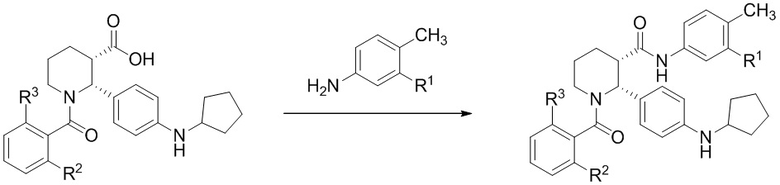
На Фиг. 1 изображена схема, иллюстрирующая стадии получения соединений IA, IB и IC, в которой применяется стадия гидрирования для достижения (2R,3S) стереохимии, за которой следует восстановительное аминирование циклопентанона, бензоилирование пиперидинового атома азота и присоединение анилина с образованием С3 амида.
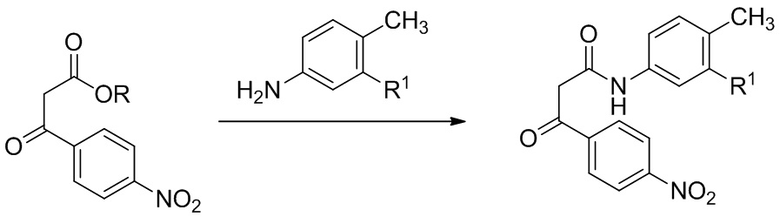
На Фиг. 2 приведена схема, в которой образование С3 амида (финальная стадия на Схеме 1) проводится через превращение C3 эфира в C3 карбоновую кислоту, которая после обработки соответствующим анилином дает такие соединения как IA, IB и IC.
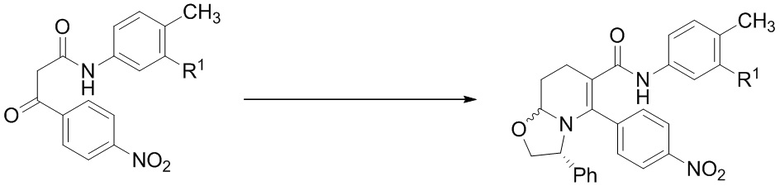
На Фиг. 3 приведена схема, в которой С3 амид конструируют на более ранней стадии синтеза, после чего следуют стадии, в которых применяется гидрирование для достижения (2R,3S) стереохимии, за которым следует восстановительное аминирование циклопентанона, и в заключение бензоилирование пиперидинового атома азота, с получением таких соединений как IA, IB и IC.
На Фиг. 4 приведена схема получения IA, описанная в Патенте США № 8,445,515 B2, в которой применяется классическая стадия расщепления для получения соединения 7, имеющего (2R,3S) стереохимию.
Подробное описание изобретения
Определения
Термин "алкил", сам по себе и как часть другого заместителя, означает, если не указано иное, линейный или разветвленный углеводородный радикал, имеющий обозначенное число атомов углерода (например, C1-8 означает 1-8 атомов углерода). Примеры алкильных групп включают метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил, н-пентил, н-гексил, н-гептил, н-октил и т.п. Термин "алкенил" означает ненасыщенную алкильную группу, содержащую одну или больше двойных связей. Аналогично, термин «алкинил» означает ненасыщенную алкильную группу, содержащую одну или больше тройных связей. Примеры таких ненасыщенных алкильных групп включают винил, 2-пропенил, кротил, 2-изопентенил, 2-(бутадиенил), 2,4-пентадиенил, 3-(1,4-пентадиенил), этинил, 1- и 3-пропинил, 3-бутинил и их высшие гомологи и изомеры.
Термин "арил" означает, если не указано иное, полиненасыщенную, в типичном случае ароматическую углеводородную группу, которая может представлять собой один цикл или несколько циклов (до трех циклов), сопряженные или связанные ковалентно. Неограничивающие примеры арильных групп включают фенил, нафтил и бифенил. Заместители в перечисленных выше арильных циклических систем выбраны из группы приемлемых заместителей, описанных выше.
Термин "арилалкил" или "арил-C1-4 алкил” охватывает радикалы, в которых арильная группа присоединена к алкильной группе (например, бензил, фенетил и т.п.).
Указанные выше термины (например, "алкил" и "арил") в некоторых вариантах осуществления включают как замещенные, так и незамещенные формы указанного радикала. Предпочтительные заместители для каждого типа радикала перечислены ниже.
Заместителями в алкильных радикалах (включая группы, которые часто именуются алкилен, алкенил, алкинил и циклоалкил) могут быть различные группы, выбранные из:
-галоген, -OR’, -NR’R”, -SR’, -SiR’R”R”’, -OC(O)R’, -C(O)R’, -CO2R’, -CONR’R”,
-OC(O)NR’R”, -NR”C(O)R’, -NR’-C(O)NR”R”’, -NR”C(O)2R’, -NH-C(NH2)=NH,
-NR’C(NH2)=NH, -NH-C(NH2)=NR’, -S(O)R’, -S(O)2R’, -S(O)2NR’R”, -NR’S(O)2R”, -CN и -NO2 в количестве от нуля до (2 m’+1), где m’ это общее число атомов углерода в таком радикале. R’, R” и R”’ каждый независимо означают атом водорода, незамещенный C1-8 алкил, незамещенный арил, арил, замещенный 1-3 галогенами, незамещенный C1-8 алкил, C1-8 алкокси или C1-8 тиоалкокси группу, или незамещенные арил-C1-4 алкильные группы. Когда R’ и R” присоединены к одному и тому же атому азота, они могут объединяться с атомом азота с образованием 3-, 4-, 5-, 6- или 7-членного цикла. Например, -NR’R” включает 1-пирролидинил и 4-морфолинил.
Аналогично, заместители в арильных группах варьируются и обычно выбраны из:
-галоген, -OR’, -OC(O)R’, -NR’R”, -SR’, -R’, -CN, -NO2, -CO2R’, -CONR’R”, -C(O)R’, -OC(O)NR’R”, -NR”C(O)R’, -NR”C(O)2R’, ,-NR’-C(O)NR”R”’, -NH-C(NH2)=NH, -NR’C(NH2)=NH, -NH-C(NH2)=NR’, -S(O)R’, -S(O)2R’, -S(O)2NR’R”, -NR’S(O)2R”, -N3, перфтор(C1-C4)алкокси, и перфтор(C1-C4)алкил, в количестве от нуля до общего числа незанятых валентностей в ароматической циклической системе; и где R’, R” и R”’ независимо выбраны из атома водорода, C1-8 алкила, C1-8 галогеналкила, C3-6 циклоалкила, C2-8 алкенила и C2-8 алкинила. Другие подходящие заместители включают каждый из перечисленных выше заместителей для арила, присоединенных к атому в цикле алкиленовым мостиком из 1-4 атомов углерода.
Два из заместителей у соседних атомов арильного или гетероарильного кольца могут опционально быть замещены заместителем формулы -T-C(O)-(CH2)q-U-, где T и U независимо представляют собой -NH-, -O-, -CH2- или одинарную связь, и q представляет собой целое число от 0 до 2. Альтернативно, два из заместителей у соседних атомов арильного или гетероарильного кольца могут опционально быть замещены заместителем формулы -A-(CH2)r-B-, где A и B независимо представляют собой -CH2-, -O-, -NH-, -S-, -S(O)-, -S(O)2-, -S(O)2NR’- или одинарную связь, и r представляет собой целое число от 1 до 3. Одна из простых связей в новом цикле, образующемся таким образом, может опционально быть заменена на двойную связь. Альтернативно, два из заместителей у соседних атомов арильного или гетероарильного кольца могут опционально быть замещены заместителем формулы -(CH2)s-X-(CH2)t-, где s и t независимо представляют собой целые числа от 0 до 3, и X представляет собой -O-, -NR’-, -S-, -S(O)-, -S(O)2- или -S(O)2NR’-. Заместитель R’ в -NR’- и -S(O)2NR’- выбран из атома водорода или незамещенного C1-6 алкила.
При использовании в настоящем тексте, волнистая линия " ", пересекающая простую, двойную или тройную связь в любой изображенной в настоящем тексте химической структуре, означает точку присоединения простой, двойной или тройной связи к остальной части молекулы.
", пересекающая простую, двойную или тройную связь в любой изображенной в настоящем тексте химической структуре, означает точку присоединения простой, двойной или тройной связи к остальной части молекулы.
Термин "галоген" сам по себе или как часть другого заместителя означает, если не указано иное, атом фтора, хлора, брома или иода. Кроме того, такие термины как "галогеналкил," включают моногалогеналкил и полигалогеналкил. Например, термин "C1-4 галогеналкил" включает трифторметил, 2,2,2-трифторэтил, 4-хлорбутил, 3-бромпропил и т.п.
Термин "взаимодействие" относится к процессу введения в контакт по меньшей мере двух разных веществ таким образом, что они могут вступать в реакцию. Однако следует понимать, что получаемый продукт может образовываться напрямую в реакции между добавляемыми реагентами, или из промежуточного продукта, который может образовываться в реакционной смеси из одного или более добавляемых реагентов.
Термин "в условиях, достаточных для" относится к подбору условий реакции (включая растворители или смеси растворителей, выбор температуры, включая изменение температуры по мере протекания реакции, концентрацию реагентов, порядок добавления реагентов в реакционную смесь, время прохождения реакции и т.д.), которые могут обеспечить протекание желаемой реакции или превращение одной молекулы в другую.
Термин "превращение" относится к проведению трансформации соединения с целью превращения соединения в другое соединение, например путем модификации одной функциональной группы с получением другой функциональной группы, объединения двух молекул с образованием новой молекулы, или, в некоторых случаях, образования соли. Однако "превращение" может также включать более одной трансформации.
Термин "растворитель" относится к веществу, такому как жидкость, способному растворять растворенный компонент. Растворители могут полярными или неполярными, протонными или апротонными. Полярные растворители обычно имеют диэлектрическую константу выше примерно 5 или дипольный момент больше примерно 1.0, и неполярные растворители обычно имеют диэлектрическую константу ниже примерно 5 или дипольный момент меньше примерно 1.0. Протонные растворители отличаются наличием протона, доступного для удаления из молекулы, например наличием гидрокси- или карбокси-группы. В апротонных растворителях нет таких групп. Репрезентативные полярные протонные растворители включают спирты (метанол, этанол, пропанол, изопропанол и т.д.), кислоты (муравьиная кислота, уксусная кислота и т.д.) и воду. Репрезентативные полярные апротонные растворители включают дихлорметан, хлороформ, тетрагидрофуран, диэтиловый эфир, ацетон, этилацетат, диметилформамид, ацетонитрил и диметилсульфоксид. Репрезентативные неполярные растворители включают алканы (пентаны, гексаны и т.д.), циклоалканы (циклопентан, циклогексан и т.д.), бензол, толуол и 1,4-диоксан.
Термин "восстановитель" относится к агенту, способному восстанавливать атом из более высокой степени окисления в более низкую степень окисления. Восстановители могут включать (но не ограничены только ими) цинк, железо, никель Ренея, платину, иридий, родий, палладий, сульфид натрия, дитионит натрия, сульфид аммония и доноры водорода, такие как литийалюминий гидрид, боргидрид натрия и триацетоксиборгидрид натрия.
Термин "уходящая группа" относится к группам, сохраняющим связывающую электронную пару при гетеролитическом разрыве связи. Например, уходящая группа легко замещается во время реакции нуклеофильного замещения. Подходящие уходящие группы включают (но не ограничиваются только ими) хлор, бром, иод, гидроксил, метансульфонат (или мезилат), трифторметансульфонат (трифлат), бензосульфонат, 4-метилбензолсульфонат (тозилат), 4-нитробензолсульфонат, 4-хлорбензолсульфонат, и карбоксилатный компонент в смешанном или симметричном ангидриде. Квалифицированному специалисту в данной области известны другие уходящие группы, которые можно применять по настоящему изобретению.
Термин "практически не содержащий энантиомерных или диастереомерных примесей" относится к соединению, содержащему по меньшей мере один хиральный центр, которое существует в виде одного энантиомера или диастереомера в количестве по меньшей мере 80% относительно других энантиомеров или диастереомеров данного соединения. В некоторых вариантах осуществления, данный термин относится также к соединению, которое существует в виде одного энантиомера или диастереомера в количестве по меньшей мере 90%, 95%, 96%, 97%, 98%, 99% или 99.5% относительно других энантиомеров или диастереомеров данного соединения.
Термин "нитрующее средство" относится к реагенту, способному добавлять нитрогруппу -NO2 к соединению. Репрезентативные нитрующие средства включают (но не ограничиваются только ей) азотную кислоту.
Термин "хлорирующее средство" относится к реагенту, способному добавлять хлоро-группу -Cl к соединению. Репрезентативные хлорирующие средства включают (но не ограничиваются только ими) оксихлорид фосфора, тионилхлорид, оксалилхлорид и сульфурилхлорид.
Термин "фармацевтически приемлемые соли" или "соли" включает соли веществ, полученные с относительно нетоксичными кислотами или основаниями, в зависимости от конкретных заместителей в описанных в настоящем тексте соединениях. Когда соединения содержат относительно кислые функциональные группы, можно получить основно-аддитивные соли путем взаимодействия нейтральной формы таких соединений с достаточным количество желаемого основания, даже без растворителя или в подходящем инертном растворителе. Примеры солей, являющихся производными фармацевтически приемлемых неорганических оснований, включают соли алюминия, аммония, кальция, меди, железа(II), железа (III), лития, магния, марганца, калия, натрия, цинка и т.д. Соли, являющиеся производными фармацевтически приемлемых органических оснований, включают соли первичных, вторичных и третичных аминов, включая замещенные амины, циклические амины, природные амины и т.д., такие как аргинин, бетаин, кофеин, холин, N,N’-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и т.п. Когда соединения по настоящему изобретению содержат относительно основные функциональные группы, можно получить кислотно-аддитивные соли путем взаимодействия нейтральной формы таких соединений с достаточным количеством желаемой кислоты, без растворителя или в подходящем инертном растворителе. Примеры фармацевтически приемлемых кислотно-аддитивных солей включают соли с неорганическими кислотами, такими как хлористоводородная, бромистоводородная, азотная, угольная, моногидроугольная, фосфорная, моногидрофосфорная, дигидрофосфорная, серная, моногидросерная, иодистоводородная или фосфористая кислота и т.п., а также соли с относительно нетоксичными органическими кислотами, такими как уксусная, пропионовая, изомасляная, малоновая, бензойная, янтарная, субериновая, фумаровая, миндальная, фталевая, бензолсульфоновая, пара-толуолсульфоновая, лимонная, винная, метансульфоновая и т.п. Также охватываются соли с аминокислотами, такие как аргинаты и т.п., и соли таких органических кислот, как глюкуроновая или галактуроновая кислоты и т.п. (см, например, Berge, S.M., et al, “Pharmaceutical Salts”, Journal of Pharmaceutical Science, 1977, 66, 1-19). Некоторые частные соединения по настоящему изобретению содержат и основные, и кислотные функциональные группы, что позволяет таким соединениям образовывать как основно-аддитивные, так и кислотно-аддитивные соли.
Нейтральные формы соединений можно регенерировать путем взаимодействия соли с основанием или кислотой и выделения материнского соединения обычным способом. Материнская форма соединения отличается от различных солевых форм определенными физическими характеристиками, такими как растворимость в полярных растворителях, но во всем остальном соли эквивалентны материнским соединениям, в терминах настоящего изобретения.
Помимо солевых форм, в настоящем изобретении описаны соединения, представляющие собой со-кристаллические формы. Со-кристаллы представляют собой комплексы соединений, описанных в настоящем тексте, в которых соединение кристаллизовано в присутствии второго соединения, такого как аминокислота, гликоль или низший спирт.
Некоторые соединения по настоящему изобретению могут существовать в несольватированных формах, а также в сольватированных формах, включая гидратированные формы. В целом, сольватированные формы эквивалентны несольватированным формам, и все они охватываются настоящим изобретением. Некоторые соединения по настоящему изобретению могут существовать в нескольких кристаллических или аморфных формах. В целом, все физические формы эквивалентны для областей применения, охватываемых настоящим изобретением, и входят в объем настоящего изобретения.
Некоторые соединения по настоящему изобретению имеют асимметрические атомы углерода (оптические центры) или двойные связи; все рацематы, диастереомеры, геометрические изомеры, региоизомеры и индивидуальные изомеры (например, отдельные энантиомеры) входят в объем настоящего изобретения, если не указано иное. Соединения по настоящему изобретению могут также иметь неприродные соотношения изотопов по одному или больше атомов, составляющих эти соединения. Неприродные соотношения изотопов можно определить как находящиеся в диапазоне от природного количества до количества рассматриваемого атома равного 100%. Например, соединения могут быть радиоактивно мечены радиоактивными изотопами, такими как, например, тритий (3H), иод-125 (125I) или углерод-14 (14C), или нерадиоактивными изотопами, такими как дейтерий (2H) или углерод-13 (13C). Такие вариации изотопов могут открыть дополнительные области применения к описанным в других разделах настоящего описания. Например, изотопные модификации соединений по настоящему изобретению могут найти дополнительное применение, включая (но не ограничиваясь только ими) применение в качестве диагностических и/или визуализирующих реагентов, или в качестве цитотоксических/радиотоксических терапевтических средств. Кроме того, изотопные варианты соединений по настоящему изобретению могут иметь измененные фармакокинетические и фармакодинамические характеристики, которые могут вносить свой вклад в улучшение характеристик безопасности, переносимости или эффективности при лечении. Все изотопные вариации соединений по настоящему изобретению, радиоактивные и нерадиоактивные, входят в объем настоящего изобретения.
Термин “килограммовые масштабы” относится к реакции, в которой по меньшей мере один из реагентов применяется в количестве по меньшей мере 1 килограмм.
Общее
Как указано выше, в настоящем изобретении описаны промежуточные соединения и способы, которые могут использоваться для получения C5aR антагонистов, которые могут применяться в лечении заболеваний или нарушений, в целом характеризующихся как воспалительные заболевания или нарушения, сердечно-сосудистые заболевания или нарушения, и аутоиммунные заболевания или нарушения.
Конкретные промежуточные соединения, имеющие (2R, 3S) конфигурацию, можно получить согласно описанным в настоящем тексте способам и затем превратить в C5aR антагонисты.
Варианты осуществления настоящего изобретения
Промежуточные соединения в синтезе C5aR антагонистов
В одном аспекте, в настоящем изобретении описано соединение, которое может применяться в получении нескольких C5aR антагонистов, имеющее формулу (i-3):
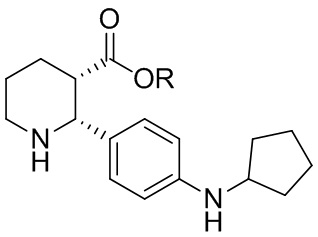 (i-3)
(i-3)
(2R,3S)
где R выбран из H, C1-8 алкила, арила и арил-C1-4 алкила, или его соль. В одной группе вариантов осуществления, данное соединение практически не содержит энантиомерных или диастереомерных примесей. В другой группе вариантов осуществления, соединение (i-3) получают в виде L-DTTA соли (соли (-)-O,O’-ди-п-толуоил-L-винной кислоты); и в некоторых вариантах осуществления соединение (i-3) получают в виде бис L-DTTA соли.
Соединение формулы (i-3), которое практически не содержит энантиомерных или диастереомерных примесей, означает соединение, которое практически не содержит одного или больше из следующих изомеров (или любых их солевых форм):
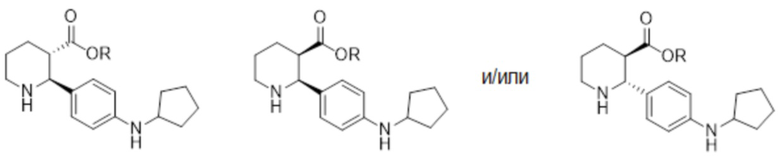 .
.
(2S,3S)
(2S,3R)
(2R,3R)
В настоящем изобретении, общее количество любого из (i-a), (i-b) или (i-c) предпочтительно ниже примерно 5% по весу относительно суммы весов (i-3), (i-a), (i-b) и (i-c). Более предпочтительно, количество любой комбинации (i-a), (i-b) и/или (i-c) ниже примерно 5%, ниже примерно 4%, ниже примерно 3%, и в некоторых вариантах осуществления ниже примерно 2.5, 2.0, 1.5 или 1.0 % по весу относительно (i-3).
В другом аспекте, в настоящем изобретении описано соединение, которое может применяться в получении нескольких C5aR антагонистов, имеющее формулу (ii-4):
(ii-4)
(2R,3S)
или его соль, где R1 представляет собой Cl или CF3. В одной группе вариантов осуществления, данное соединение практически не содержит энантиомерных или диастереомерных примесей. В другой группе вариантов осуществления, соединение (ii-4) получают в форме L-DTTA соли (соли (-)-O,O’-ди-п-толуоил-L-винной кислоты); и в некоторых вариантах осуществления соединение (ii-4) получают в форме бис L-DTTA соли.
Соединение формулы (ii-4), которое практически не содержит энантиомерных или диастереомерных примесей, означает соединение, которое практически не содержит одного или больше из следующих изомеров (или любых их солевых форм):
(2S,3S)
(2S,3R)
(2R,3R)
В настоящем изобретении, общее количество любого из (ii-a), (ii-b) или (ii-c) предпочтительно ниже примерно 5% по весу относительно суммы весов (ii-4), (ii-a), (ii-b) и (ii-c). Более предпочтительно, количество любой комбинации (ii-a), (ii-b) и/или (ii-c) ниже примерно 5%, ниже примерно 4%, ниже примерно 3%, и в некоторых вариантах осуществления ниже примерно 2.5, 2.0, 1.5 или 1.0% по весу относительно (ii-4).
Способы получения C5aR антагонистов
В другом аспекте, в настоящем изобретении описан способ получения соединения, имеющего формулу (I):
(I)
или его соли, где R1 представляет собой Cl или CF3; R2 представляет собой F или Cl; и R3 представляет собой H или CH3; и где указанное соединение формулы (I) практически не содержит энантиомерных или диастереомерных примесей, включающий:
(a) взаимодействие соединения, имеющего формулу (i-3):
 (i-3)
(i-3)
где R выбран из C1-8 алкила, арила и арил-C1-4 алкила, или его соли, которое практически не содержит энантиомерных или диастереомерных примесей, с соединением, имеющим формулу:
где LG представляет собой уходящую группу; в условиях, достаточных для образования соединения формулы (i-4):
 (i-4); и
(i-4); и
(b) превращение соединения формулы (i-4) в указанное соединение формулы (I), где указанное соединение формулы (I) практически не содержит энантиомерных или диастереомерных примесей.
Рассматривая сначала стадию (a), в одной группе вариантов осуществления, получают соединение формулы (i-3), которое практически не содержит изомеров (i-a), (i-b) и (i-c). В некоторых предпочтительных вариантах осуществления, получают соединение (i-3) по меньшей мере 95%-ной чистоты, более предпочтительно по меньшей мере 96%, 97% или по меньшей мере 98%-ной чистоты, относительно других изомеров. В еще более предпочтительных вариантах осуществления, получают соединение (i-3) по меньшей мере 99%-ной или 99.5%-ной чистоты, относительно других изомеров.
На стадии (a), соединение (i-3) взаимодействует с соединением, имеющим формулу:
, где LG представляет собой уходящую группу. Квалифицированному специалисту в данной области будет понятно, что подходящая уходящая группа представляет собой такую, которая облегчает участие соединения в целевом образовании амидной связи. Более конкретно, LG представляет собой уходящую группу, которая облегчает реакцию по карбонильному центру, несущему LG. В одной группе вариантов осуществления, LG представляет собой галоген. В другой группе вариантов осуществления, LG представляет собой Cl. В другой группе вариантов осуществления, -LG выбрана из -OH, -OAc, -O-S(O)2-(4-толил) и -O-S(O)2метил. В другой группе вариантов осуществления, -LG представляет собой -OC(O)Ph(R2)(R3), формируя симметричный ангидрид с остальной частью молекулы. В некоторых вариантах осуществления, взаимодействие проводят в органическом растворителе или смеси растворителей, или в смеси воды и растворителя – например, в смеси воды и простого эфира, такого как метил-трет-бутиловый эфир (MTBE). В других вариантах осуществления, смесь растворителей представляет собой смесь воды и ТГФ, диоксана или ацетонитрила. В других вариантах осуществления, взаимодействие проводят в присутствии основания. Подходящие основания включают триэтиламин, N,N-диизопропилэтиламин, DBU и N-метилморфолин, а также карбонат калия (K2CO3), бикарбонат калия (KHCO3), карбонат натрия (Na2CO3) или бикарбонат натрия (NaHCO3). В одной группе вариантов осуществления, взаимодействие проводят при температурах от -20°C до примерно 50°C. В другой группе вариантов осуществления, взаимодействие проводят при комнатной температуре (около 25°C ± 5°C). После смешивания реакционную смесь можно отслеживать вплоть до завершения реакции, что, в зависимости от конкретных применяемых условий (и растворителей) может занимать период времени от примерно 20 минут до примерно 3 дней. Обычно получение соединения (i-4) завершается примерно за 1-2 часа. В некоторых вариантах осуществления, соединение (i-4) выделяют по стандартным методикам, таким как описанные ниже в Примерах.
Соединение (i-4) можно затем превратить в соединение формулы (I) либо непосредственным амидированием сложного эфира (фрагмент присутствует в (i-4)), либо сначала превращая сложный эфир в карбоновую кислоту и затем формируя амид с применением подходящего анилина. В настоящем изобретении, подходящий анилин выбран из группы, состоящей из 4-метил-3-(трифторметил)анилина и 3-хлор-4-метиланилина.
Для непосредственного амидирования, анилин обычно объединяют с соединением (i-4) в присутствии металлорганического реагента, такого как алюминийорганические реагенты или соединения алюминия (соли), алкиллитиевые соединения, реагенты Гриньяра, цинкорганические реагенты или соединения цинка (соли), гидрид натрия или натриевые, калиевые или литиевые соли HMDS. В некоторых вариантах осуществления, металлорганический реагент представляет собой алюминийорганический реагент, такой как Al(Me)3 или DABAL-Me3 (комплекс триметилалюминия с DABCO). В некоторых частных вариантах осуществления, металлорганический реагент представляет собой Al(Me)3.
Для тех вариантов осуществления, в которых сложноэфирная форма соединения (i-4) превращается в карбоновую кислоту, применяют гидролиз с использованием водного раствора кислоты, такой как серная кислота. В некоторых вариантах осуществления, можно применять температуры выше комнатной, например до 100°C. Сочетание карбоксильной формы (i-4) с анилином, например с 4-метил-3-(трифторметил)анилином или 3-хлор-4-метиланилином, можно проводить либо методом активированного сложного эфира (применяя метансульфонил хлорид с основанием, таким как N,N-диизопропилэтиламин), либо с использованием другого каплинг-реагента, такого как HATU, с основанием, таким как N-метилморфолин.
В другом аспекте, в настоящем изобретении описан другой способ получения соединения, имеющего формулу (I):
(I)
или его соли, где R1 представляет собой Cl или CF3; R2 представляет собой F или Cl; и R3 представляет собой H или CH3; и где соединение формулы (I) практически не содержит энантиомерных или диастереомерных примесей, включающий:
(a) взаимодействие соединения, имеющего формулу (ii-4):
(ii-4)
или его соли, где указанное соединение практически не содержит энантиомерных или диастереомерных примесей, с циклопентаноном и восстановителем в условиях, достаточных для образования соединения, имеющего формулу (ii-5):
(ii-5); и
(b) взаимодействие указанного соединения формулы (ii-5) с соединением, имеющим формулу:
где LG представляет собой уходящую группу; в условиях, достаточных для образования соединения формулы (I), которое практически не содержит энантиомерных или диастереомерных примесей.
В одной группе вариантов осуществления, R1 представляет собой CF3; R2 представляет собой F; и R3 представляет собой CH3. В другой группе вариантов осуществления, R1 представляет собой CF3; R2 представляет собой Cl; и R3 представляет собой H. В другой группе вариантов осуществления, R1 представляет собой Cl; R2 представляет собой F; и R3 представляет собой CH3.
Рассматривая сначала стадию (a), в одной группе вариантов осуществления, получают соединение формулы (ii-4), которое практически не содержит изомеров (ii-a), (ii-b) и (ii-c). В некоторых предпочтительных вариантах осуществления, получают соединение (ii-4) по меньшей мере 95%-ной чистоты, более предпочтительно по меньшей мере 96%, 97% или по меньшей мере 98%-ной чистоты, относительно других изомеров. В еще более предпочтительных вариантах осуществления, получают соединение (ii-4) по меньшей мере 99%-ной или 99.5%-ной чистоты, относительно других изомеров.
Соединение (ii-4) обычно сначала вводят в реакцию с циклопентаноном и кислотой для образования промежуточного имина, который затем восстанавливают до соответствующего амина с применением подходящего восстановителя. Примеры подходящих восстановителей включают газообразный водород (с палладием или другим металлическим катализатором), боргидридные реагенты и алюминийгидридные реагенты. В одной группе вариантов осуществления, восстановители представляют собой боргидридные реагенты, такие как боргидрид натрия или лития, цианоборгидрид натрия, или триацетоксиборгидрид натрия. Условия как для формирования имина, так и для последующего восстановления можно варьировать согласно общеизвестным методам. Например, формирование имина можно проводить в одном растворителе или в смеси растворителей (таких как дихлорметан и п-диоксан). Аналогично, температуры реакций подбирают таким образом, чтобы уменьшить количество побочных продуктов и получить хороший выход. Обычно такие реакции проводят при комнатной температуре в течение периода времени от 1-2 часов до 18 часов или больше.
Соединение формулы (ii-5) можно выделить с применением стандартной обработки для реакции восстановительного аминирования. Такая обработка может включать, например, нейтрализацию кислоты в реакционной (или взаимодействующей) смеси и отделение соединения (ii-5) путем экстракции смеси органическим растворителем и последующего удаления растворителя из органического слоя. Обычно дальнейшей очистки соединения (ii-5) не требуется перед стадией (b).
На стадии (b), продукт со стадии (a) взаимодействует с соединением, имеющим формулу:
, где LG представляет собой уходящую группу. Как описано для представленного выше способа, квалифицированному специалисту в данной области будет понятно, что подходящая уходящая группа представляет собой такую, которая облегчает участие соединения в целевом образовании амидной связи. Более конкретно, LG представляет собой уходящую группу, которая облегчает реакцию по карбонильному центру, несущему LG. В одной группе вариантов осуществления, LG представляет собой галоген. В другой группе вариантов осуществления, LG представляет собой Cl. В другой группе вариантов осуществления, -LG выбрана из -OH, -OAc, -O-S(O)2-(4-толил) и -O-S(O)2метил. В другой группе вариантов осуществления, -LG представляет собой -OC(O)Ph(R2)(R3) -, формируя симметричный ангидрид с остальной частью молекулы. В некоторых вариантах осуществления, взаимодействие проводят в органическом растворителе или смеси растворителей, или в смеси воды и растворителя – например, в смеси воды и простого эфира, такого как метил-трет-бутиловый эфир (MTBE). В некоторых вариантах осуществления, растворитель представляет собой органический растворитель, такой как ТГФ или другой простоэфирный растворитель. В других вариантах осуществления, взаимодействие проводят в присутствии основания. Подходящие основания включают триэтиламин, диизопропилэтиламин, DBU и N-метил морфолин, а также карбонат калия (K2CO3), бикарбонат калия (KHCO3), карбонат натрия (Na2CO3) или бикарбонат натрия (NaHCO3). В одной группе вариантов осуществления, взаимодействие проводят при температурах от -20°C до примерно 50°C. В другой группе вариантов осуществления, взаимодействие проводят при комнатной температуре (около 25°C ± 5°C). После смешивания реакционную смесь можно отслеживать вплоть до завершения реакции, что, в зависимости от конкретных применяемых условий (и растворителей) может занимать период времени от примерно 20 минут до примерно 3 дней. Обычно, получение соединения (I) завершается примерно за 1-2 часа. В некоторых вариантах осуществления, соединение формулы (I) выделяют по стандартным методикам, таким как описанные ниже в Примерах.
В другом аспекте, в настоящем изобретении описаны способы получения соединений формулы (I), или их фармацевтически приемлемых солей, сольватов, гидратов или ротамеров, включающие любые две, три или четыре из стадий (a), (b), (c), (d), (d1) и (d2), которые могут следовать или не следовать одна за другой:
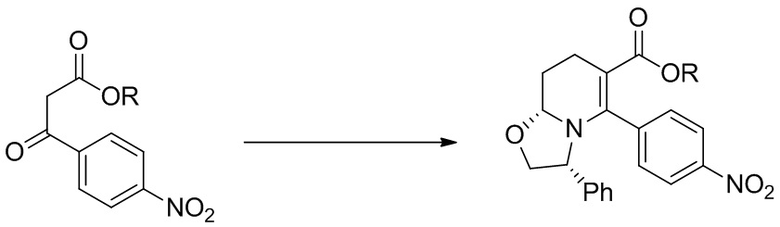
(a) реакция сложного эфира (R представляет собой алкил, предпочтительно C1-8 алкил, или арил или арил-C1-4 алкил) 3-(4-нитрофенил)-3-оксо-пропаноата (i-1), (R)-(−)-2-фенилглицинола и диэтилацеталя акролеина или его эквивалента, с получением соединения (i-2);
(b) гидрирование или восстановление (i-2) с получением промежуточного амина и превращение полученного промежуточного соединения в (i-3) реакцией с циклопентаноном и восстановителем;
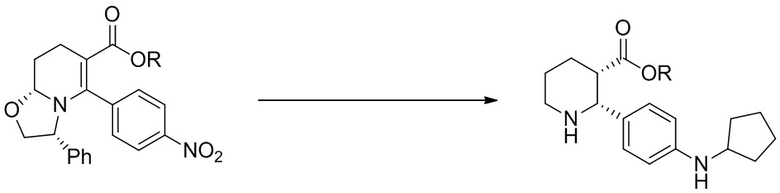
(c) взаимодействие (i-3) с 2-фтор-6-метилбензоил хлоридом или 2-хлорбензоил хлоридом, с получением (i-4);
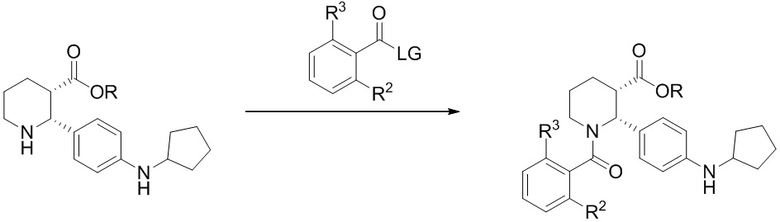
(d) взаимодействие (i-4) с 3-хлор-4-метиланилином или 3-трифторметил-4-метиланилином в условиях, достаточных для получения соединения формулы (I).
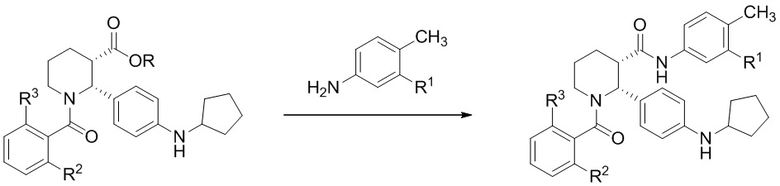
Опционально, в некоторых вариантах осуществления, превращение, осуществляемое на стадии (d), можно проводить в виде двухстадийного процесса, включающего:
(d)(1) превращение сложного эфира (i-4) в карбоновую кислоту (i-5):
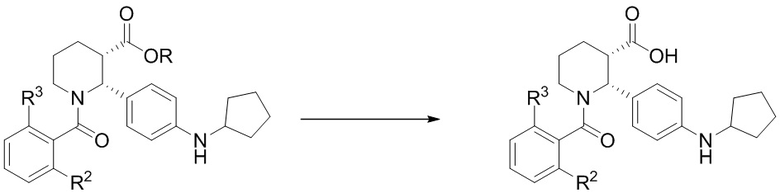
(d)(2) взаимодействие (i-5) с 3-хлор-4-метиланилином или 3-трифторметил-4-метиланилином в условиях, достаточных для получения соединения формулы (I).
В некоторых вариантах осуществления, способ получения соединений формулы (I) включает стадии (a) и (b). В других вариантах осуществления, способ получения соединений формулы (I) включает стадии (b) и (c). В других вариантах осуществления, способ получения соединений формулы (I) включает стадии (c) и (d). В других вариантах осуществления, способ получения соединений формулы (I) включает стадии (c) и (d)(1). В других вариантах осуществления, способ получения соединений формулы (I) включает стадии (c) и (d)(2). В других вариантах осуществления, способ получения соединений формулы (I) включает стадии (b) и (d). В других вариантах осуществления, способ получения соединений формулы (I) включает стадии (b) и (d1). В других вариантах осуществления, способ получения соединений формулы (I) включает стадии (b) и (d2).
В некоторых вариантах осуществления, способ получения соединений формулы (I) включает стадии (a) и (c). В других вариантах осуществления, способ получения соединений формулы (I) включает стадии (a) и (d). В других вариантах осуществления, способ получения соединений формулы (I) включает стадии (a) и (d1). В других вариантах осуществления, способ получения соединений формулы (I) включает стадии (a) и (d)(2).
В других вариантах осуществления, способ получения соединений формулы (I) включает по меньшей мере три из стадий (a), (b), (c) и (d), или, необязательно, (d)(1) и (d)(2). В других вариантах осуществления, способ получения соединений формулы (I) включает стадии (a), (b) и (c). В других вариантах осуществления, способ получения соединений формулы (I) включает стадии (a), (b) и (d). В других вариантах осуществления, способ получения соединений формулы (I) включает стадии (a), (b) и (d1). В других вариантах осуществления, способ получения соединений формулы (I) включает стадии (a), (b) и (d2). В другой группе вариантов осуществления, способ получения соединений формулы (I) включает стадии (b), (c) и (d). В других вариантах осуществления, способ получения соединений формулы (I) включает стадии (b), (c) и (d1). В других вариантах осуществления, способ получения соединений формулы (I) включает стадии (b), (c) и (d2).
В другом родственном аспекте, в настоящем изобретении описаны способы получения соединений формулы (I) или их фармацевтически приемлемых солей, сольватов, гидратов или ротамеров, включающие любые две, три или четыре из стадий (a’), (b’), (c’), (d’) и (e’), которые могут следовать или не следовать одна за другой в общем пути синтеза:
(a’) реакция сложного эфира (R представляет собой алкил, предпочтительно C1-8 алкил, арил или арил- C1-4 алкил) 3-(4-нитрофенил)-3-оксо-пропаноата (i-1, или ii-1, где R представляет собой алкил) с 3-хлор-4-метиланилином или 3-трифторметил-4-метиланилином в условиях, достаточных для получения соединения формулы (ii-2);
(b’) реакция (ii-2), (R)-(−)-2-фенилглицинола и диэтилацеталя акролеина или его эквивалента, с получением соединения (ii-3);
(c’) восстановление (ii-3) в условиях, достаточных для получения промежуточного диамина (ii-4), который практически не содержит энантиомерных или диастереомерных примесей;
(d’) превращение (ii-4) в (ii-5) реакцией с циклопентаноном и восстановителем; и
(e’) взаимодействие (ii-5) с 2-фтор-6-метилбензоил хлоридом или 2-хлорбензоил хлоридом, с получением (I);
В описанных выше способах с использованием любых из стадий (a), (b), (c), (d), (d1), (d2), (a’), (b’), (c’), (d’) или (e’), квалифицированному специалисту в данной области будет понятно, что можно применять указанные соединения, в некоторых случаях в виде солей, гидратов или сольватных форм; и что можно подбирать такие условия, которые способствуют указанным реакциям и повышают выход продукта на конкретной стадии и/или чистоту продукта, образующегося на конкретной стадии.
В некоторых вариантах осуществления, способ получения соединений формулы (I) включает стадии (a’) и (b’). В других вариантах осуществления, способ получения соединений формулы (I) включает стадии (b’) и (c’). В других вариантах осуществления, способ получения соединений формулы (I) включает стадии (c’) и (d’). В других вариантах осуществления, способ получения соединений формулы (I) включает стадии (d’) и (e’).
В некоторых вариантах осуществления, способ получения соединений формулы (I) включает стадии (a’) и (c’). В других вариантах осуществления, способ получения соединений формулы (I) включает стадии (a’) и (d’). В других вариантах осуществления, способ получения соединений формулы (I) включает стадии (a’) и (e’).
В других вариантах осуществления, способ получения соединений формулы (I) включает стадии (b’) и (d’). В других вариантах осуществления, способ получения соединений формулы (I) включает стадии (b’) и (e’). В других вариантах осуществления, способ получения соединений формулы (I) включает стадии (c’) и (e’).
В некоторых вариантах осуществления, способ получения соединений формулы (I) включает стадии (a’), (b’) и (c’). В некоторых вариантах осуществления, способ получения соединений формулы (I) включает стадии (a’), (b’) и (d’). В некоторых вариантах осуществления, способ получения соединений формулы (I) включает стадии (a’), (b’) и (e’). В некоторых вариантах осуществления, способ получения соединений формулы (I) включает стадии (a’), (c’) и (d’). В некоторых вариантах осуществления, способ получения соединений формулы (I) включает стадии (a’), (c’) и (e’). В других вариантах осуществления, способ получения соединений формулы (I) включает стадии (a’), (d’) и (e’). В других вариантах осуществления, способ получения соединений формулы (I) включает стадии (b’), (c’) и (d’). В других вариантах осуществления, способ получения соединений формулы (I) включает стадии (b’), (c’) и (e’). В других вариантах осуществления, способ получения соединений формулы (I) включает стадии (c’), (d’) и (e’).
В других вариантах осуществления, способ получения соединений формулы (I) включает по меньшей мере четыре из стадий (a’), (b’), (c’), (d’) и (e’). В некоторых вариантах осуществления, способ получения соединений формулы (I) включает стадии (a’), (b’), (c’) и (d’). В других вариантах осуществления, способ получения соединений формулы (I) включает стадии (a’), (b’), (c’) и (e’). В других вариантах осуществления, способ получения соединений формулы (I) включает стадии (a’), (b’), (d’) и (e’). В других вариантах осуществления, способ получения соединений формулы (I) включает стадии (a’), (c’), (d’) и (e’). В другой группе вариантов осуществления, способ получения соединений формулы (I) включает стадии (b’), (c’), (d’) и (e’). В других вариантах осуществления, способ получения соединений формулы (I) включает стадии (a’), (b’), (c’), (d’) и (e’).
Описанные выше и в настоящем тексте способы делают возможными экономически эффективные, безопасные, эффективные и/или легко масштабируемые процессы, которые можно применять в крупномасштабном или промышленном производстве каждого из IA, IB и IC, или их фармацевтически приемлемой соли, сольвата, гидрата или ротамера.
В одном варианте осуществления, в настоящем изобретении описаны способы получения соединений формулы (I) или их фармацевтически приемлемой соли, сольвата, гидрата или ротамера, которые являются практически чистыми. В одном варианте осуществления, в настоящем изобретении описаны способы получения соединений формулы (I) или их фармацевтически приемлемой соли, сольвата, гидрата или ротамера, которые являются практически химически чистыми. В одном варианте осуществления, в настоящем изобретении описаны способы получения соединений формулы (I) или их фармацевтически приемлемой соли, сольвата, гидрата или ротамера, которые могут применяться на людях, например для лечения, профилактики и/или смягчения степени тяжести заболеваний или патологических состояний, включая (но не ограничиваясь только ими), заболевания, опосредуемые антагонистами C5a рецептора.
В одном варианте осуществления, в настоящем изобретении описаны способы получения соединений формулы (I) или их фармацевтически приемлемой соли, сольвата, гидрата или ротамера, в масштабе более 1 грамма, более 10 грамм, более 100 грамм, более 1000 грамм, более 10000 грамм или более 100000 грамм.
В одном варианте осуществления, в настоящем изобретении описаны способы получения соединений формулы (I) или их фармацевтически приемлемой соли, сольвата, гидрата или ротамера, с общим выходом выше примерно 10%, выше примерно 15%, выше примерно 20%, выше примерно 25%, выше примерно 30%, выше примерно 40%, выше примерно 50%, выше примерно 60%, выше примерно 70%, выше примерно 80%, выше примерно 90% или выше примерно 95%, где выход вычисляют по лимитирующему исходному соединению.
В одном варианте осуществления, в настоящем изобретении описаны способы получения соединений формулы (I) или их фармацевтически приемлемой соли, сольвата, гидрата или ротамера, которые являются практически чистыми. В одном варианте осуществления, чистота соединений формулы IA, IB и IC, или их фармацевтически приемлемой соли, сольвата, гидрата или ротамера, выше примерно 95% вес/вес, выше примерно 96% вес/вес, выше примерно 97% вес/вес, выше примерно 98% вес/вес, выше примерно 99% вес/вес, выше примерно 99.5% вес/вес, выше примерно 99.8% вес/вес, выше примерно 99.9% вес/вес, выше примерно 99.95% вес/вес, выше примерно 99.98% вес/вес, или выше примерно 99.99% вес/вес из расчета на общий вес продукта.
В одном варианте осуществления, общее количество примесей в соединениях формулы IA, IB и/или IC, или в их фармацевтически приемлемой соли, сольвате, гидрате или ротамере, полученных описанным в настоящем тексте способом, ниже примерно 5% вес/вес, ниже примерно 4% вес/вес, ниже примерно 3% вес/вес, ниже примерно 2% вес/вес, ниже примерно 1% вес/вес, ниже примерно 0.5% вес/вес, ниже примерно 0.2% вес/вес, ниже примерно 0.1% вес/вес, ниже примерно 0.05% вес/вес, ниже примерно 0.02% вес/вес, ниже примерно 0.01% вес/вес, ниже примерно 0.005% вес/вес, или ниже примерно 0.001% вес/вес из расчета на общий вес продукта.
В одном варианте осуществления, количество индивидуальной примеси в соединениях формулы (I), или в их фармацевтически приемлемой соли, сольвате, гидрате или ротамере, полученных описанным в настоящем тексте способом, ниже примерно 5% вес/вес, ниже примерно 2% вес/вес, ниже примерно 1% вес/вес, ниже примерно 0.9% вес/вес, ниже примерно 0.8% вес/вес, ниже примерно 0.7% вес/вес, ниже примерно 0.6% вес/вес, ниже примерно 0.5% вес/вес, ниже примерно 0.4% вес/вес, ниже примерно 0.3% вес/вес, ниже примерно 0.2% вес/вес, ниже примерно 0.1% вес/вес, ниже примерно 0.05% вес/вес, ниже примерно 0.01% вес/вес, ниже примерно 0.005% вес/вес, ниже примерно 0.001% вес/вес, ниже примерно 0.0005% вес/вес, или ниже примерно 0.0001% вес/вес из расчета на общий вес продукта.
В одном варианте осуществления, описанные в настоящем тексте способы обеспечивают получение соединений формулы (I) или их фармацевтически приемлемой соли, сольвата, гидрата или ротамера, которые практически химически чистые. В одном варианте осуществления, химическая чистота соединений формулы IA, IB или IC, или их фармацевтически приемлемой соли, сольвата, гидрата или ротамера, выше примерно 95% вес/вес, выше примерно 96% вес/вес, выше примерно 97% вес/вес, выше примерно 98% вес/вес, выше примерно 99% вес/вес, выше примерно 99.5% вес/вес, выше примерно 99.8% вес/вес, выше примерно 99.9% вес/вес, выше примерно 99.95% вес/вес, выше примерно 99.98% вес/вес, или выше примерно 99.99% вес/вес из расчета на общий вес продукта.
В одном варианте осуществления, описанный профиль чистоты реакционной смеси или выделенного продукта в способах по настоящему изобретению проанализирован одним или более аналитическими методами, такими как, например, ВЭЖХ (высокоэффективная жидкостная хроматография), ГХ (газовая хроматография) и ТСХ (тонкослойная хроматография). В одном варианте осуществления, примесь детектируется аналитическим методом, таким как, например, ВЭЖХ, ГХ или ТСХ. В одном варианте осуществления, примесь или предполагаемая примесь в реакционной среде или выделенном продукте, полученном описанными в настоящем тексте способами, включает (но не ограничивается только им) исходное вещество, применяемое в реакции, или любое исходное вещество, применяемое на предыдущих стадиях.
В некоторых случаях, примесь в выделенном продукте, полученном описанными в настоящем тексте способами, может представлять собой летучее органическое соединение, такое как, например, метанол, диметилформамид, дихлорметан, толуол, ацетон, метил-трет-бутиловый эфир, этанол или тетрагидрофуран.
В некоторых случаях, потеря веса при высушивании (LOD) для соединения формулы IA, IB или IC, или его фармацевтически приемлемой соли, сольвата, гидрата или ротамера, полученных описанным в настоящем тексте способом, ниже примерно 5% вес/вес, ниже примерно 2% вес/вес, ниже примерно 1% вес/вес, ниже примерно 0.9% вес/вес, ниже примерно 0.8% вес/вес, ниже примерно 0.7% вес/вес, ниже примерно 0.6% вес/вес, ниже примерно 0.5% вес/вес, ниже примерно 0.4% вес/вес, ниже примерно 0.3% вес/вес, ниже примерно 0.2% вес/вес, ниже примерно 0.1% вес/вес, ниже примерно 0.05% вес/вес, или ниже примерно 0.01% вес/вес из расчета на общий вес продукта.
В одном варианте осуществления, остаток при сжигании для соединения формулы IA, IB или IC, или его фармацевтически приемлемой соли, сольвата, гидрата или ротамера, полученных описанным в настоящем тексте способом, ниже примерно 1% вес/вес, ниже примерно 0.9% вес/вес, ниже примерно 0.8% вес/вес, ниже примерно 0.7% вес/вес, ниже примерно 0.6% вес/вес, ниже примерно 0.5% вес/вес, ниже примерно 0.4% вес/вес, ниже примерно 0.3% вес/вес, ниже примерно 0.2% вес/вес, ниже примерно 0.1% вес/вес, ниже примерно 0.05% вес/вес, или ниже примерно 0.01% вес/вес из расчета на общий вес продукта.
В одном варианте осуществления, общее количество примесей тяжелых металлов в соединении формулы IA, IB или IC, или его фармацевтически приемлемой соли, сольвате, гидрате или ротамере, полученных описанным в настоящем тексте способом, ниже примерно 500 ч/млн (частей на миллион) вес/вес, ниже примерно 200 ч/млн вес/вес, ниже примерно 100 ч/млн вес/вес, ниже примерно 50 ч/млн вес/вес, ниже примерно 20 ч/млн вес/вес, ниже примерно 10 ч/млн вес/вес, ниже примерно 5 ч/млн вес/вес, ниже примерно 2 ч/млн вес/вес, ниже примерно 1 ч/млн вес/вес, ниже примерно 0.5 ч/млн вес/вес, ниже примерно 0.2 ч/млн вес/вес, или ниже примерно 0.1 ч/млн вес/вес из расчета на общий вес продукта.
В одном варианте осуществления, в настоящем изобретении описаны способы получения соединения формулы IA, IB или IC, или их фармацевтически приемлемой соли, сольвата, гидрата или ротамера, которое практически не содержит одного или больше остаточных растворителей, включая (но не ограничиваясь только ими) метанол, этанол, диметилформамид, толуол, дихлорметан, ацетон, метил-трет-бутиловый эфир и тетрагидрофуран. В одном варианте осуществления, содержание остаточного растворителя или предполагаемого остаточного растворителя ниже примерно 5000 ч/млн вес/вес, ниже примерно 2000 ч/млн вес/вес, ниже примерно 1000 ч/млн вес/вес, ниже примерно 500 ч/млн вес/вес, ниже примерно 200 ч/млн вес/вес, ниже примерно 100 ч/млн вес/вес, ниже примерно 50 ч/млн вес/вес, ниже примерно 20 ч/млн вес/вес, ниже примерно 10 ч/млн вес/вес, ниже примерно 5 ч/млн вес/вес, ниже примерно 2 ч/млн вес/вес, ниже примерно 1 ч/млн вес/вес, ниже примерно 0.5 ч/млн вес/вес, ниже примерно 0.2 ч/млн вес/вес, или ниже примерно 0.1 ч/млн вес/вес из расчета на общий вес продукта. В одном варианте осуществления, предполагаемый остаточный растворитель, такой как, например, метанол, этанол, диметилформамид, толуол, дихлорметан, ацетон, метил-трет-бутиловый эфир и тетрагидрофуран, невозможно детектировать.
В одном варианте осуществления, в настоящем изобретении описаны способы получения соединений формулы IA, IB или IC, или их фармацевтически приемлемой соли, сольвата, гидрата или ротамера, в которых содержание воды ниже примерно 5% вес/вес, ниже примерно 4% вес/вес, ниже примерно 3% вес/вес, ниже примерно 2% вес/вес, ниже примерно 1% вес/вес, ниже примерно 0.9% вес/вес, ниже примерно 0.8% вес/вес, ниже примерно 0.7% вес/вес, ниже примерно 0.6% вес/вес, ниже примерно 0.5% вес/вес, ниже примерно 0.4% вес/вес, ниже примерно 0.3% вес/вес, ниже примерно 0.2% вес/вес, или ниже примерно 0.1% вес/вес из расчета на общий вес продукта.
В одном варианте осуществления, в настоящем изобретении описаны способы получения соединений формулы IA, IB или IC, или их фармацевтически приемлемой соли, сольвата, гидрата или ротамера, которые по внешнему виду представляют собой белые или не совсем белые твердые вещества.
В одном варианте осуществления, одна или больше стадий в способах по настоящему изобретению проводятся в соответствии с условиями GMP (Good Manufacturing Process). В одном варианте осуществления, одна или больше стадий в способах по настоящему изобретению проводятся не в соответствии с условиями GMP.
Примеры
Применяющиеся в описанных ниже примерах сокращения имеют следующие значения:
водн: водный; BBr3: трибромид бора; CH2Cl2 или ДХМ: дихлорметан; CH3CN: ацетонитрил; CH3OH или MeOH: метанол; DIEA: N,N-диизопропилэтиламин; ДМФА: диметилформамид; ДМСО: диметилсульфоксид; экв.: эквиваленты; Et3N: триэтиламин; Et2O: диэтиловый эфир; EtOH: этанол; ч: час(ы); HATU, O-(7-Азабензотриазол-1-ил)-N,N,N’,N’-тетраметилурония гексафторфосфат; HCl: хлороводород; H2O: вода; K2CO3: карбонат калия; KHSO4: бисульфат калия; MgSO4: сульфат магния; мл: миллилитр; NaCl: хлорид натрия; NaH: гидрид натрия; NaHCO3: бикарбонат натрия; NaOEt: этоксид натрия; NaOH: гидроксид натрия; NaOMe: метоксид натрия; Na2SO4: сульфат натрия; NH4Cl: хлорид аммония; NMP: N-метил-пирролидинон; pH: -log [H+]; POCl3: фосфорил трихлорид; PPTS: пиридиния п-толуолсульфонат; ОФ-ВЭЖХ: обращенно-фазная высокоэффективная жидкостная хроматография; RT: комнатная температура; ТФУК: трифторуксусная кислота; ТГФ: тетрагидрофуран; ТСХ: тонкослойная хроматография.
Пример 1
Данный пример иллюстрирует получение (2R,3S)-2-[4-(циклопентиламино)фенил]-1-(2-фтор-6-метил-бензоил)-N-[4-метил-3-(трифторметил)фенил]пиперидин-3-карбоксамида способом, более подробно описанным на Фиг. 1 (Схема 1) с применением описанных ниже реагентов:
Путь 1:
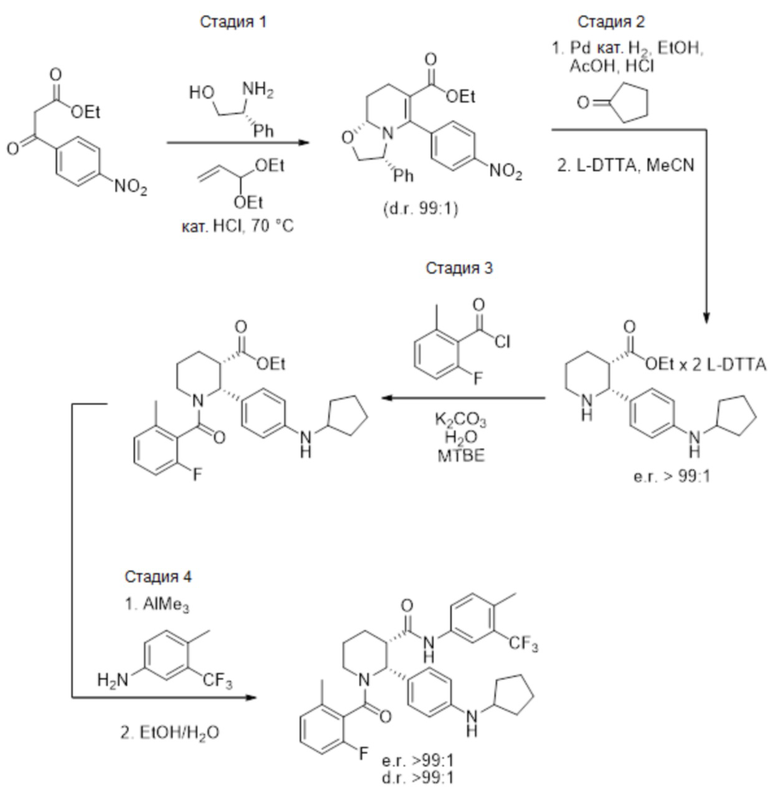
Стадия 1: В высушенную в термошкафу 12-литровую 3-горлую колбу, оснащенную механической мешалкой, обратным холодильником и термометром, загружали диэтилацеталь акролеина (1127 г, 8.666 моль, 1.05 экв.) и нагревали до 40°C. Смесь твердого этил 3-(4-нитрофенил)-3-оксо-пропаноата (1956 г, 8.253 моль) и (R)-(−)-2-фенилглицинола (>99.5% e.e., 1187 г, 8.666 моль, 1.05 экв.) добавляли порциями в течение 40 минут, поддерживая в перемешиваемой смеси внутреннюю температуру примерно 40°C. После добавления всех твердых веществ, смесь перемешивали при 40°C 10 минут. Затем добавляли 4M раствор HCl в диоксане (206.2 мл, 0.825 моль, 10 мол.%) через обратный холодильник в течение 2 минут, и внутреннюю температуру повышали до 70°C. Реакционную смесь перемешивали 22 часа, после чего метод LC-MS показал полное исчезновение исходных соединений и енаминового промежуточного соединения. Нагрев отключали и добавляли этанол (6.6 л). В полученный раствор затем добавляли в качестве затравки 4 г этил (3R,8aR)-5-(4-нитрофенил)-3-фенил-3,7,8,8a-тетрагидро-2H-оксазоло[3,2-a]пиридин-6-карбоксилата и перемешивали при комнатной температуре 18 часов. Затем твердый осадок отфильтровывали и использовали 0.1 л этанола для промывки колбы и оборудования на фильтре. Выделенное твердое вещество затем промывали три раза на фильтре этанолом (по 250 мл) и сушили в вакууме, получая 1253 г этил (3R,8aR)-5-(4-нитрофенил)-3-фенил-3,7,8,8a-тетрагидро-2H-оксазоло[3,2-a]пиридин-6-карбоксилата в виде ярко-желтого твердого вещества (38% выход, 98.5% ВЭЖХ вес/вес чистота, 0.15 вес.% EtOH).
Стадия 2: 260 г этил (3R,8aR)-5-(4-нитрофенил)-3-фенил-3,7,8,8a-тетрагидро-2H-оксазоло[3,2-a]пиридин-6-карбоксилата (0.659 моль), 0.66 л этанола и 56 г палладиевого катализатора (10% Pd/C, Degussa type E101 NE/W, 50% воды, 21.5 вес.% порошка, 4.0 мол.% Pd) помещали в аппарат Парра (2.2 л) и продували азотом. Реактор помещали в шейкер Парра и добавляли водород с такой скоростью, чтобы внешняя температура реактора не превышала 30°C. Через 4 часа потребление водорода замедлилось. Реактор встряхивали под давлением водорода 50 фунт/кв.дюйм 2 часа. Затем в реактор добавляли 94 мл ледяной уксусной кислоты (1.65 моль, 2.5 экв.) и продували реактор три раза водородом под давлением 50 фунт/кв.дюйм. Затем реактор встряхивали под давлением водорода 35 – 55 фунт/кв.дюйм 48 часов, поддерживая температуру ниже 30°C. Реактор вынимали из аппарата и добавляли 55 мл 12M водного раствора HCl (0.659 моль, 1 экв.) затем добавляли 87 мл циклопентанона (0.989 моль, 1.5 экв.). Реактор три раза продували водородом под давлением 50 фунт/кв.дюйм и затем встряхивали под давлением водорода 50 фунт/кв.дюйм 16 – 20 часов. Реакционную смесь извлекали из аппарата и фильтровали через стеклянный фильтр с целитом (80 г), и затем промывали три раза по 0.125 л этанола. Добавляли 54.1 г безводного ацетата натрия (0.659 моль, 1 экв.), и смесь упаривали в вакууме при 40 – 55°C, удаляя 0.9 л летучих компонентов. Добавляли 2.0 л ацетонитрила, и 2.0 л летучих компонентов упаривали в вакууме. Полученное сырое вещество разбавляли добавлением 1.0 л ацетонитрила и механически перемешивали при комнатной температуре 30 минут. Реакционную смесь фильтровали через целит (40 г) и осадок на фильтре промывали 0.28 л ацетонитрила. Объединенные фильтраты дали раствор сырого ацетата амина (Раствор A, e.e. = 78%). Растворы A из двух независимых опытов объединяли для дальнейшей работы.
В 12-литровой 3-горлой колбе, оснащенной механической мешалкой, внутренним термометром и обратным холодильником, растворяли (−)-O,O′-ди-п-толуоил-L-винную кислоту (1.019 кг, 2.64 моль, 2 экв.) в 5.8 л ацетонитрила. Реакционную смесь нагревали при 60°C при перемешивании, затем быстро добавляли 1 л Раствора A. В полученный раствор вносили в качестве затравки 4 г кристаллической соли этил (2R,3S)-2-[4-(циклопентиламино)фенил]пиперидин-3-карбоксилата с (−)-O,O′-ди-п-толуоил-L-винной кислотой (1:2) и перемешивали при 60°C 15 минут. После 15 минут при 60°C образовывался осадок. Остальное количество Раствора A добавляли в течение 2.5 часов, поддерживая внутреннюю температуру 60°C. По окончании добавления, источник нагрева отключали и смесь перемешивали 17 часов, достигая финальной температуры 22.5°C. Полученную суспензию фильтровали и осадок промывали 0.50 л ацетонитрила для промывки оборудования и переносили весь твердый осадок на фильтр. Полученный влажный осадок промывали на фильтре 3.0 л ацетонитрила и сушили в вакуумном термошкафу при 45°C 48 часов с получением 1.005 кг соли этил (2R,3S)-2-[4-(циклопентиламино)фенил]пиперидин-3-карбоксилата с (−)-O,O′-ди-п-толуоил-L-винной кислотой (1:2) в виде не совсем белого твердого вещества (70% выход, содержит 1 вес.% ацетонитрила). Энантиомерное соотношение в продукте составляло 99.4:0.6.
Стадия 3: В 5-литровой 3-горлой колбе, оснащенной механической мешалкой и капельной воронкой, твердый безводный карбонат калия (K2CO3, 226 г, 1.64 моль, 4.1 экв.) растворяли в H2O (0.82 л) и охлаждали до комнатной температуры. Добавляли MTBE (0.82 л), затем твердую соль этил (2R,3S)-2-[4-(циклопентиламино)фенил]пиперидин-3-карбоксилата с (−)-O,O′-ди-п-толуоил-L-винной кислотой (1:2) (436 г, 0.400 моль). Реакционную смесь интенсивно перемешивали при комнатной температуре 1 час, затем добавляли 2-фтор-6-метилбензоил хлорид (72.5 г, 0.420 ммоль, 1.05 экв.) в MTBE (0.14 л) по каплям в течение 1 часа. Продукт начинал выпадать в осадок из реакционной смеси до окончания добавления хлорангидрида. Реакционную смесь интенсивно перемешивали при комнатной температуре 30 минут и контролировали методом LC-MS исчезновение исходного соединения. Реакционную смесь затем переносили в 5-литровую колбу для упаривания с помощью 0.3 л MTBE для промывки оборудования и удаления всего осадка. Реакционную смесь упаривали в вакууме для удаления MTBE, затем добавляли 0.3 л гептана, и смесь снова упаривали, оставляя только продукт, суспендированный в водном растворе. Колбу вынимали из роторного испарителя и добавляли воду (0.82 л) и гептан (0.82 л). Полученную суспензию интенсивно перемешивали 16 часов механической мешалкой. Затем смесь фильтровали, и осадок промывали водой (2 x 0.42 л) и гептаном (0.42 л). Твердый продукт сушили в вакуумном термошкафу при 45°C с получением 172 г этил (2R,3S)-2-[4-(циклопентиламино)фенил]-1-(2-фтор-6-метил-бензоил)пиперидин-3-карбоксилата в виде не совсем белого порошка (95% выход).
Стадия 4: 0.5-литровую 3-горлую круглодонную колбу сушили в течение ночи в термошкафу при 200°C и затем охлаждали в токе азота. Колбу оснащали магнитной мешалкой, вводом азота и термометром. В колбу загружали 30.2 г этил (2R,3S)-2-[4-(циклопентиламино)фенил]-1-(2-фтор-6-метил-бензоил)пиперидин-3-карбоксилата (66.7 ммоль), 11.5 мл 4-метил-5-трифторметиланилина (80 ммоль, 1.2 экв.) и 141 мл сухого толуола в атмосфере азота. Азот пропускали через полученный раствор в течение 10 минут, и затем раствор нагревали до 30°C. Масляную баню удаляли, и через канюлю добавляли в реакционную смесь 100 мл 2 M раствора AlMe3 в толуоле (Aldrich, 200 ммоль, 3 экв.) с такой скоростью, чтобы поддерживалась температура реакции 35 – 40°C, данный процесс занимал примерно 45 минут. Температуру реакционной смеси затем повышали до 55°C в течение 1 часа, реакционную смесь перемешивали при 55°C 8 часов, за это время расходовался весь исходный сложный эфир (контролировали методом LC-MS). Реакционную смесь затем охлаждали в течение ночи до комнатной температуры, и полученный раствор затем переносили через канюлю в механически перемешиваемую 1-литровую колбу, содержащую раствор 67.8 г тартрата натрия-калия тетрагидрата (240 ммоль, 3.6 экв.) в 237 мл воды, охлажденный до 10°C на ледяной бане. Процесс добавления занимал примерно 30 минут, во время него реакционная смесь самопроизвольно разогревалась до 57°C. Пустую реакционную колбу затем промывали 20мл сухого толуола, и полученный раствор объединяли с остальной смесью. Реакционную смесь затем охлаждали до комнатной температуры при перемешивании, добавляли 91 мл этилацетата, и смесь перемешивали еще 15 минут. Реакционную смесь затем фильтровали через слой целита, и оставляли фильтрат разделяться на два слоя. Органический слой затем отделяли и промывали раствором 5.7 г тартрата натрия-калия тетрагидрата (20 ммоль) в 120 мл воды и затем двумя порциями по 120 мл воды. Влажный органический раствор упаривали в вакууме до веса ~150 г, и осуществляли замену растворителя на этанол, сохраняя общий объем 0.2 – 0.3 л, до тех пор пока не достигали содержания < 1 мол.% толуола относительно этанола, по данным 1H ЯМР. Полученный раствор затем упаривали при повышенной температуре до веса 223 г и нагревали до кипения. Начинали механическое перемешивание и добавляли 41 мл воды. В полученный раствор вносили в качестве затравки кристаллы (2R,3S)-2-[4-(циклопентиламино)фенил]-1-(2-фтор-6-метил-бензоил)-N-[4-метил-3-(трифторметил)фенил]пиперидин-3-карбоксамида при 60°C и затем медленно охлаждали до комнатной температуры в течение 2 часов. Суспензию перемешивали 18 часов и отфильтровывали твердый осадок. Осадок затем промывали двумя порциями по 30 мл смеси 7:3 этанол/вода и сушили в вакуумном термошкафу 24 часа при 50°C, получая 31.0 г (2R,3S)-2-[4-(циклопентиламино)фенил]-1-(2-фтор-6-метил-бензоил)-N-[4-метил-3-(трифторметил)фенил]пиперидин-3-карбоксамид в виде не совсем белых кристаллов (80% выход). Данные анализа: ВЭЖХ чистота: 99.59%; >99.8% d.e.и e.e. по ВЭЖХ; ICP-OES Pd: <1 ч/млн; Al: 6 ч/млн; остаточный толуол по данным GC-MS: 15 ч/млн; зола <0.1%; K-F 0.1%. 1H ЯМР (400 МГц, ТФУК-d) δ 7.91 (д, J = 8.6 Гц, 1 H), 7.84 (д, J = 8.6 Гц, 1 H), 7.58-6.82 (м, 8 H), 6.75 (т, J = 8.6 Гц, 1 H), 4.10-4.00 (м, 1H), 3.60-3.47 (м, 1H), 3.45-3.41 (м, 1H), 3.33-3.25 (м, 1H), 2.44-2.22 (м, 7H), 2.04-1.92 (м, 4H), 1.82-1.69 (м, 7H), MS: (ES) m/z 582 (M+H+).
Пример 2
Данный пример иллюстрирует получение (2R,3S)-2-[4-(циклопентиламино) фенил]-1-(2-фтор-6-метил-бензоил)-N-[4-метил-3-(трифторметил)фенил]пиперидин-3-карбоксамида по общей методике, представленной на Фиг. 2 (Схема 2), применяя реагенты, показанные в Пути 2:
Путь 2:
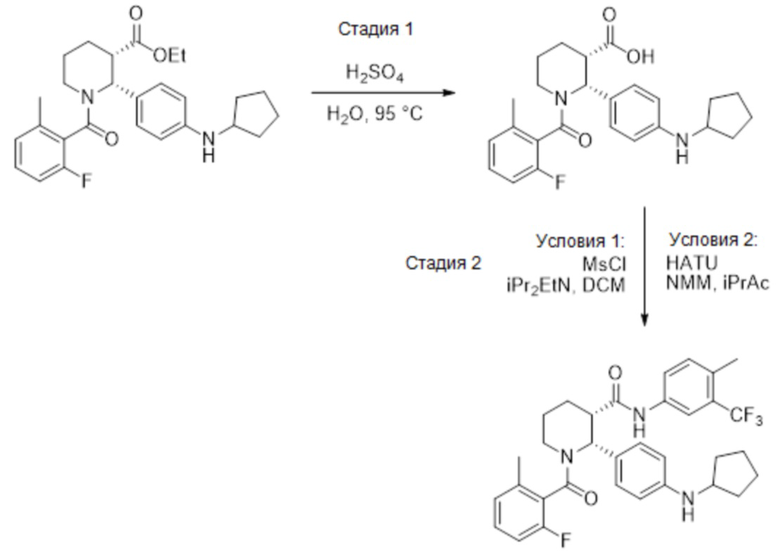
Стадия 1: Твердый этил (2R,3S)-2-[4-(циклопентиламино)фенил]-1-(2-фтор-6-метил-бензоил)пиперидин-3-карбоксилат (316 г, 0.698 моль) добавляли порциями в 12-литровую колбу, содержащую механически перемешиваемый 0.44 M раствор H2SO4 в воде (2.80 л), при нагревании до 70°C. Использовали дополнительно 0.36 л 0.44 M раствора H2SO4 для смывания твердого вещества с воронки. Полученную суспензию доводили до 95°C и перемешивали при этой температуре 21 час, при этом наблюдалось полное растворение и оставалось не более 4% исходного вещества. Реакционную смесь охлаждали до комнатной температуры. В полученную смесь добавляли 2.80 л 1 M раствора NaOH в воде в течение 30 минут, поддерживая температуру около 20°C, затем 1.58 л MTBE и снова 1.40 л 1M раствора NaOH. Реакционную смесь интенсивно перемешивали 1 час до полного растворения всех твердых веществ (финальный pH 13.1). Слои разделяли, и органический слой отбрасывали. Водный слой снова экстрагировали с помощью 1.58 л MTBE. Водный слой упаривали в вакууме для удаления избытка MTBE. Раствор переносили обратно в механически перемешиваемую колбу, и раствор подкисляли добавлением 1 M раствора H2SO4 в течение 25 минут до значения pH 4.8 (примерно 0.71 л 1M раствора H2SO4), и полученную смесь перемешивали при комнатной температуре 1 час. Полученную суспензию фильтровали, и осадок промывали двумя 2-литровыми порциями воды, затем 1.0 л гептана. Твердый осадок сушили в вакуумном термошкафу при 45°C, получая 279 г (2R,3S)-2-[4-(циклопентиламино)фенил]-1-(2-фтор-6-метил-бензоил)пиперидин-3-карбоновую кислоту в виде белого твердого вещества (94% выход).
Стадия 2, условия 1: В 12-литровую колбу, содержащую (2R,3S)-2-[4-(циклопентиламино)фенил]-1-(2-фтор-6-метил-бензоил)пиперидин-3-карбоновую кислоту (277 г, 0.652 моль) в 1.66 л дихлорметана, добавляли 4-метил-3-(трифторметил)анилин (112 мл, 137 г, 0.782 моль, 1.2 экв.), затем N,N-диизопропилэтиламин (204 мл, 152 г, 1.17 моль, 1.8 экв.). Раствор охлаждали до 0°C и добавляли по каплям метансульфонил хлорид (65.6 мл, 97.1 г, 0.848 моль, 1.3 экв.). После перемешивания в течение 18 часов при комнатной температуре, добавляли N,N-диизопропилэтиламин (114 мл, 84.2 г, 0.652 моль, 1.0 экв.). Реакционную смесь перемешивали при комнатной температуре 15 минут и добавляли в колбу 2.22 л изопропилацетата и 0.55 л ДХМ. Раствор промывали 2.22 л воды и затем снова 1.11 л воды. Органический слой дважды промывали порциями по 2.22 л 0.1M раствора гидроксида натрия в воде. Добавляли в органический слой 139 г безводного сульфата натрия и перемешивали 15 минут. В полученную суспензию добавляли 544 г силикагеля (230-400 меш), и смесь перемешивали 30 минут. Полученную суспензию фильтровали с помощью высокого стеклянного фильтра, и слой силикагеля промывали на фильтре, используя 2.50 л смеси изопропилацетат/ДХМ (1:1). Объединенный раствор упаривали в вакууме при 40°C до примерного веса 670 г. Колбу оснащали механической мешалкой, и в этот момент времени происходила спонтанная кристаллизация. После 15 минут перемешивания, в течение 20 минут добавляли 1.14 л гептана в полученную суспензию. После 16 часов перемешивания при комнатной температуре получали бесцветные кристаллы, которые отфильтровывали и последовательно промывали на фильтре двумя порциями гептана (0.76 л и 0.38 л). Твердый продукт сушили в вакуумном термошкафу 16 часов при 45°C, получая 289 г (2R,3S)-2-[4-(циклопентиламино)фенил]-1-(2-фтор-6-метил-бензоил)-N-[4-метил-3-(трифторметил)фенил]пиперидин-3-карбоксамида в виде бесцветных кристаллов (76% выход, e.e. 98.6%, 1.4 вес.% изопропилацетата (по 1H ЯМР); 98.1% чистота по ВЭЖХ при 220 нм). 1H ЯМР (400 МГц, ТФУК-d) δ 7.91 (д, J = 8.6 Гц, 1 H), 7.84 (д, J = 8.6 Гц, 1 H), 7.58-6.82 (м, 8 H), 6.75 (т, J = 8.6 Гц, 1 H), 4.10-4.00 (м, 1H), 3.60-3.47 (м, 1H), 3.45-3.41 (м, 1H), 3.33-3.25 (м, 1H), 2.44-2.22 (м, 7H), 2.04-1.92 (м, 4H), 1.82-1.69 (м, 7H), MS: (ES) m/z 582 (M+H+).
Стадия 2, условия 2: В 250-миллилитровую колбу, содержащую (2R,3S)-2-[4-(циклопентиламино)фенил]-1-(2-фтор-6-метил-бензоил)пиперидин-3-карбоновую кислоту (8.01 г, 18.8 ммоль) в 40 мл изопропилацетата, добавляли 4-метил-3-(трифторметил)анилин (2.97 мл, 3.62 г, 20.7 ммоль, 1.1 экв.), затем N-метилморфолин (3.10 мл, 2.85 г, 28.2 ммоль, 1.5 экв.) и HATU (9.29 г, 24.4 моль, 1.3 экв.). После 44 часов перемешивания при комнатной температуре, реакционную смесь разбавляли добавлением 100 мл изопропилацетата и 60 мл воды, и перемешивали 15 минут. Нерастворившийся твердый осадок отфильтровывали, и водный слой отбрасывали. Органическую фазу дважды промывали по 60 мл воды и затем упаривали в вакууме до веса 58 г. Затем растворитель заменяли на этанол посредством совместного упаривания, и раствор упаривали в вакууме до веса 74 г (оставалось 0.6 вес.% изопропилацетата). Реакционную смесь нагревали до кипения и добавляли 14 мл воды. В полученный раствор добавляли в качестве затравки кристаллы (2R,3S)-2-[4-(циклопентиламино)фенил]-1-(2-фтор-6-метил-бензоил)-N-[4-метил-3-(трифторметил)фенил]пиперидин-3-карбоксамида при 60°C и затем медленно охлаждали до комнатной температуры в течение 2 часов. Полученную суспензию затем перемешивали 18 часов и осадок отфильтровывали. Осадок промывали двумя порциями (по 8 мл) смеси 7:3 этанол/вода и сушили в вакуумном термошкафу 24 часа при 50°C, получая 7.91 г (2R,3S)-2-[4-(циклопентиламино)фенил]-1-(2-фтор-6-метил-бензоил)-N-[4-метил-3-(трифторметил)фенил]пиперидин-3-карбоксамида в виде бесцветных кристаллов (72% выход). Данные анализа: ВЭЖХ чистота: 99.26%; >99.8% d.e.и e.e. по ВЭЖХ. 1H ЯМР (400 МГц, ТФУК-d) δ 7.91 (д, J = 8.6 Гц, 1 H), 7.84 (д, J = 8.6 Гц, 1 H), 7.58-6.82 (м, 8 H), 6.75 (т, J = 8.6 Гц, 1 H), 4.10-4.00 (м, 1H), 3.60-3.47 (м, 1H), 3.45-3.41 (м, 1H), 3.33-3.25 (м, 1H), 2.44-2.22 (м, 7H), 2.04-1.92 (м, 4H), 1.82-1.69 (м, 7H), MS: (ES) m/z 582 (M+H+).
Пример 3
Данный пример иллюстрирует получение (2R,3S)-2-[4-(циклопентиламино) фенил]-1-(2-фтор-6-метил-бензоил)-N-[4-метил-3-(трифторметил)фенил]пиперидин-3-карбоксамида по общей методике, представленной на Фиг.3 (Схема 3), применяя реагенты, показанные в Пути 3:
Путь 3:

Стадия 1: В 500-миллилитровую 3-горлую круглодонную колбу, оснащенную термометром, добавляли этил 3-(4-нитрофенил)-3-оксо-пропаноат (50 г, 211 ммоль), o-ксилол (100 мл), затем 4-метил-3-(трифторметил)анилин (33.25 мл, 232 ммоль), и реакционную смесь перемешивали при 130°C в течение 6 часов (этанол, выделяющийся в ходе реакции, удаляли дистилляцией по мере его образования). Реакционную смесь охлаждали до комнатной температуры и оставляли на ночь. Выпавшие кристаллы отделяли фильтрованием, промывали диэтиловым эфиром (500 мл), сушили в высоком вакууме, получая N-[4-метил-3-(трифторметил)фенил]-3-(4-нитрофенил)-3-оксо-пропанамид (74.4 г) с выходом 96% в виде ярко-желтого кристаллического вещества. 1H ЯМР показал присутствие ~2:1 смеси кето-енольных таутомеров. 1H ЯМР (400 МГц, ДМСО-d6) δ 10.61 (ушир.с, 1H), 10.48 (с, 1H), 8.37-8.31 (м, 2 H), 8.21 (д, J = 9 Гц, 1H), 8.0-7.95 (м, 2H), 7.65 (дд, J = 21.2, 8.2 Гц, 1 H), 7.37 (дд, J = 13.3, 8.2 Гц, 1 H), 6.06 (с, 1H), 4.25 (с, 2 H), 2.37, 2.36 (2с, 3 H); MS: (ES) m/z 367 (M+H+).
Стадия 2: Смесь (R)-(−)-2-фенилглицинола (3.02 г, 22 ммоль), N-[4-метил-3-(трифторметил)фенил]-3-(4-нитрофенил)-3-оксо-пропанамида (7.32 г, 20 ммоль), диэтилацеталя акролеина (4 мл, 28.6 ммоль) и муравьиной кислоты (0.8 мл, 20 ммоль) в п-диоксане (10 мл) перемешивали при 90°C в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли дихлорметаном (20 мл), адсорбировали на силикагеле и очищали методом колоночной хроматографии (продукт элюировали 30%-ным этилацетатом в гексане), получая (3R)-N-[4-метил-3-(трифторметил)фенил]-5-(4-нитрофенил)-3-фенил-3,7,8,8a-тетрагидро-2H-оксазоло[3,2-a]пиридин-6-карбоксамид (8.4 г) с выходом 80% в виде желтой пены с соотношением диастереомеров ~3:2. 1H ЯМР (400 МГц, CDCl3) δ 7.96-786 (ушир.с, 1H), 7.20-7.15 (м, 3H), 7.15-7.0 (м, 6H), 6.9 (дд, J = 7.81, 1.57 Гц, 1H), 6.7 (д, J = 8.6 Гц, 1H), 6.44 (д, J = 29.7 Гц, 1H), 5.26 (дд, J = 8.6, 3.5 Гц, 0.6 H), 5.06 (дд, J = 9.77, 2.73 Гц, 0.4 H), 4.48 (д, J = 6.25 Гц, 0.5H), 4.36-4.28 (м, 1H), 4.22-4.17 (м, 0.5H), 4.02 (дд, J = 8.99, 1.56 Гц, 0.5H), 3.8 (дд, J = 8.6, 5.08 Гц, 0.5H), 3.2-2.8 (м, 1H), 2.7-2.4 (м, 2H), 2.14 (с, 3H), 1.95-1.85 (м, 1H); MS: (ES) m/z 524 (M+H+).
Стадия 3: (3R)-N-[4-метил-3-(трифторметил)фенил]-5-(4-нитрофенил)-3-фенил-3,7,8,8a-тетрагидро-2H-оксазоло[3,2-a]пиридин-6-карбоксамид (2.1 г, 4 ммоль), ДМФА (12 мл), палладиевый катализатор (10% Pd/C, Degussa type E101 NE/W, 50% воды, 800 мг, 45 вес.% порошка, 0.36 ммоль) и уксусную кислоту (0.6 мл, 10 ммоль) помещали в реактор Парра и перемешивали в атмосфере водорода (60 фунт/кв.дюйм) 20 часов при комнатной температуре. Реакционную смесь пропускали через стеклянный фильтр для удаления палладиевого катализатора, промывали метанолом (2 × 20 мл) и упаривали досуха на роторном испарителе в вакууме, получая сырой продукт. В полученный сырой продукт добавляли этилацетат (30 мл), дихлорметан (60 мл), (−)-O,O′-ди-п-толуоил-L-винную кислоту (L-DTTA, 1.55 г, 4 ммоль), и полученную смесь выдерживали при комнатной температуре в течение ночи. Полученные кристаллы отделяли фильтрованием, промывали холодным этилацетатом (2 × 10 мл) и сушили в высоком вакууме, получая (2R,3S)-2-(4-аминофенил)-N-[4-метил-3-(трифторметил)фенил]пиперидин-3-карбоксамид в виде 1:1 L-DTTA соли (1.32 г) с выходом 43% и соотношением энантиомеров 98:2 (хиральная колонка: Regis Cell, ВЭЖХ система: Agilent 1200 Series Model G1312A, растворитель: 0.1% диэтиламин в MeOH, изократический режим, скорость потока: 1 мл/мин, комнатная температура, время удерживания основного изомера: 6.86 мин). 1H ЯМР (400 МГц, CD3OD) δ 8.01 (д, J = 6.6 Гц, 4H), 7.88 (д, J = 2.35 Гц, 1H), 7.5 (дд, J = 8.4, 2.34 Гц, 1H), 7.28 (д, J = 7.8 Гц, 2H), 7.26 (д, J = 8.99 Гц, 2H), 7.14 (д, J = 8.6 Гц, 2H), 6.68 (д, J = 8.6 Гц, 2H), 5.87 (с, 2H), 4.35 (д, J = 3.12 Гц, 1H), 3.52-3.58 (м, 1H), 3.06-3.23 (м, 2H), 2.40 (с, 9H), 2.18-2.12 (м, 2H), 1.84 (д, J = 14.46 Гц, 1H); MS: (ES) m/z 378 (M+H+).
Стадия 4: К соли (2R,3S)-2-(4-аминофенил)-N-[4-метил-3-(трифторметил)фенил] пиперидин-3-карбоксамида с (−)-O,O′-ди-п-толуоил-L-винной кислотой (1:1) (15.17 г, 19.85 ммоль) в дихлорметане (100 мл) добавляли циклопентанон (1.93 мл, 21.84 ммоль), 4н. раствор HCl в п-диоксане (6.31 мл, 25.24 ммоль) и уксусную кислоту (3.57 мл, 59.55 ммоль), затем триацетоксиборгидрид натрия (6.31 г, 29.78 ммоль) при комнатной температуре, и полученную смесь перемешивали при комнатной температуре в течение ночи. Медленно добавляли насыщенный водный раствор бикарбоната натрия (100 мл) и отделяли органический слой. Водный слой экстрагировали дихлорметаном (2 × 100 мл), и объединенные органические слои сушили над безводным сульфатом натрия и упаривали в вакууме, получая сырой (2R,3S)-2-[4-(циклопентиламино)фенил]-N-[4-метил-3-(трифторметил)фенил]пиперидин-3-карбоксамид (9.5 г), который использовали в следующей стадии без дополнительной очистки. 1H ЯМР (400 МГц, CD3OD) δ 7.71 (д, J = 1.96 Гц, 1H), 7.43 (дд, J = 8.2, 1.95 Гц, 1H), 7.22 (д, J = 8.6 Гц, 1H), 7.08 (д, J = 8.6 Гц, 2H), 6.58 (д, J = 8.6 Гц, 2H), 3.88 (д, J = 3.52 Гц, 1H), 3.69 (кв, J = 12.1, 6.3 Гц, 1H), 3.33-3.35 (м, 1H), 2.88-2.78 (м, 2H), 2.39 (с, 3H), 2.16-1.85 (м, 5H), 1.75-1.5 (м, 5H), 1.45-1.35 (м, 2H); MS: (ES) m/z 446 (M+H+).
Стадия 5: В колбу, содержащую раствор бикарбоната натрия (1.9 г, 22.62 ммоль) в 45 мл воды, добавляли раствор сырого (2R,3S)-2-[4-(циклопентиламино)фенил]-N-[4-метил-3-(трифторметил)фенил]пиперидин-3-карбоксамида (5.0 г, 11.23 ммоль) в 90 мл тетрагидрофурана в течение 10 минут. Полученную смесь перемешивали при комнатной температуре 1 час. По каплям добавляли 2-фтор-6-метилбензоил хлорид (1.73 г, 8.98 ммоль) в 5 мл тетрагидрофурана в течение 10 минут. После окончания реакции отфильтровывали нерастворившийся осадок и упаривали фильтрат в вакууме. Гептан (50 мл) добавляли к оставшемуся водному слою, и смесь интенсивно перемешивали 16 часов при комнатной температуре. Смесь фильтровали и промывали осадок водой (2 x 30 мл), затем гептаном (30 мл). Осадок сушили в высоком вакууме, получая сырой продукт (3.68 г), который растворяли в этаноле (22 мл) при аккуратном нагреве и затем добавляли воду (4 мл). Полученный коричневый прозрачный раствор охлаждали до комнатной температуры и перемешивали в течение ночи. Отфильтровывали выпавшие кристаллы, промывали холодным этанолом (5 мл), сушили в высоком вакууме, получая (2R,3S)-2-[4-(циклопентиламино)фенил]-1-(2-фтор-6-метил-бензоил)-N-[4-метил-3-(трифторметил) фенил]пиперидин-3-карбоксамид (1.66 г) с выходом 28% за две стадии, с соотношением энантиомеров 98:2 (хиральная колонка: Pirkle Covalent, (S,S) Whelk-O1, 5/100, 25 см x 4.6 мм Kromasil, S/N 50404, ВЭЖХ система: Agilent 1200 Series Model G1312A, растворитель: 15% гексана в изопропаноле, изократический режим, скорость потока: 1 мл/мин, температура колонки: 75°C, время удерживания основного изомера: 9.9 мин). 1H ЯМР (400 МГц, ТФУК-d) δ 7.91 (д, J = 8.6 Гц, 1 H), 7.84 (д, J = 8.6 Гц, 1 H), 7.58-6.82 (м, 8 H), 6.75 (т, J = 8.6 Гц, 1 H), 4.10-4.00 (м, 1H), 3.60-3.47 (м, 1H), 3.45-3.41 (м, 1H), 3.33-3.25 (м, 1H), 2.44-2.22 (м, 7H), 2.04-1.92 (м, 4H), 1.82-1.69 (м, 7H), MS: (ES) m/z 582 (M+H+).
Пример 4
Данный пример иллюстрирует синтез (2R,3S)-2-[4-(циклопентиламино)фенил]-1-(2-фтор-6-метил-бензоил)-N-[4-метил-3-хлорфенил]пиперидин-3-карбоксамида:
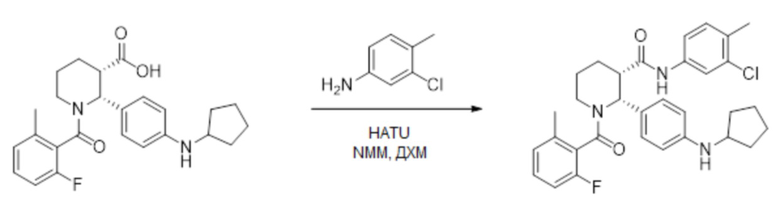
В 100-миллилитровую колбу, содержащую (2R,3S)-2-[4-(циклопентиламино) фенил]-1-(2-фтор-6-метил-бензоил)пиперидин-3-карбоновую кислоту (2.71 г, 6.38 ммоль) в 20 мл дихлорметана, добавляли 3-хлор-4-метиланилин (0.85 мл, 7.01 ммоль, 1.1 экв.), затем N-метилморфолин (1.05 мл, 968 мг, 9.57 ммоль, 1.5 экв.) и HATU (2.91 г, 7.66 моль, 1.2 экв.). После 24 часов перемешивания при комнатной температуре, реакционную смесь упаривали в вакууме, разбавляли добавлением 50 мл изопропилацетата и 20 мл воды, и перемешивали 15 минут. Нерастворившийся осадок отфильтровывали, и водный слой отбрасывали. Органическую фазу дважды промывали по 20 мл воды и затем упаривали в вакууме досуха. Твердый остаток дважды упаривали с 30 мл этанола. Полученный остаток растворяли в 22 мл кипящего этанола и добавляли 4 мл воды. Полученный раствор затем кипятили 15 минут (до получения затравочного количества кристаллов) и затем медленно охлаждали до комнатной температуры. Полученную суспензию затем перемешивали 3 часа и отфильтровывали осадок. Твердый осадок затем промывали 10 мл смеси 7:3 этанол/вода и сушили в вакуумном термошкафу 24 часа при 50°C, получая 2.95 г (2R,3S)-2-[4-(циклопентиламино)фенил]-1-(2-фтор-6-метил-бензоил)-N-[4-метил-3-хлорфенил]пиперидин-3-карбоксамида в виде бесцветных кристаллов (84% выход).
Следует понимать, что описанные в настоящем тексте примеры и варианты осуществления служат исключительно иллюстративным целям, и что в их свете квалифицированный специалист в данной области может предложить различные модификации или видоизменения, которые охватываются сутью настоящего изобретения и входят в объем притязаний.
Все публикации, патенты и заявки на патенты, процитированные в настоящем тексте, включены в настоящий текст во всем своем объеме посредством ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| РАСТВОРИМЫЕ С5аR АНТАГОНИСТЫ | 2017 |
|
RU2748260C2 |
| ПРОИЗВОДНЫЕ МАННОЗЫ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2013 |
|
RU2667060C2 |
| [9,10-ДИМЕТОКСИ-3-(2-МЕТИЛПРОПИЛ)-1H,2H,3H,4H,6H,7H,11BH- ПИРИДО-[2,1-A]ИЗОХИНОЛИН-2-ИЛ]МЕТАНОЛ И СВЯЗАННЫЕ С НИМ СОЕДИНЕНИЯ, КОМПОЗИЦИИ И СПОСОБЫ | 2016 |
|
RU2736509C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ИММУНОЛОГИЧЕСКИХ И ОНКОЛОГИЧЕСКИХ СОСТОЯНИЙ | 2009 |
|
RU2545023C9 |
| МОДУЛЯТОРЫ SHIP1 И ОТНОСЯЩИЕСЯ К НИМ СПОСОБЫ | 2014 |
|
RU2679805C2 |
| 4'-ЗАМЕЩЕННЫЕ НУКЛЕОЗИДНЫЕ ИНГИБИТОРЫ ОБРАТНОЙ ТРАНСКРИПТАЗЫ И ИХ ПОЛУЧЕНИЕ | 2016 |
|
RU2720811C2 |
| БИАРИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ БЕЛОК-БЕЛКОВОГО ВЗАИМОДЕЙСТВИЯ YAP/TAZ-TEAD | 2021 |
|
RU2830596C1 |
| ПРОИЗВОДНЫЕ МАННОЗЫ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2014 |
|
RU2678327C2 |
| ИНГИБИТОРЫ КАСПАЗ И ИХ ПРИМЕНЕНИЕ | 2005 |
|
RU2382780C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ИНГИБИРОВАНИЯ ВИРУСНОЙ ПОЛИМЕРАЗЫ | 2013 |
|
RU2654482C2 |
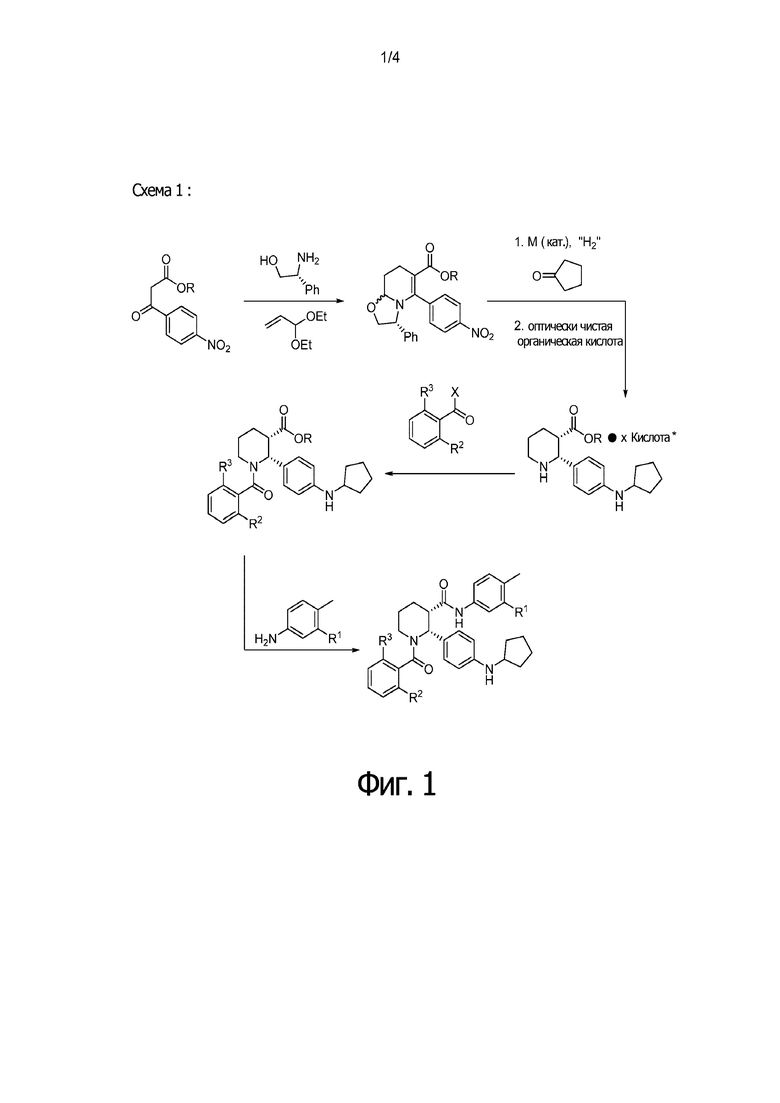
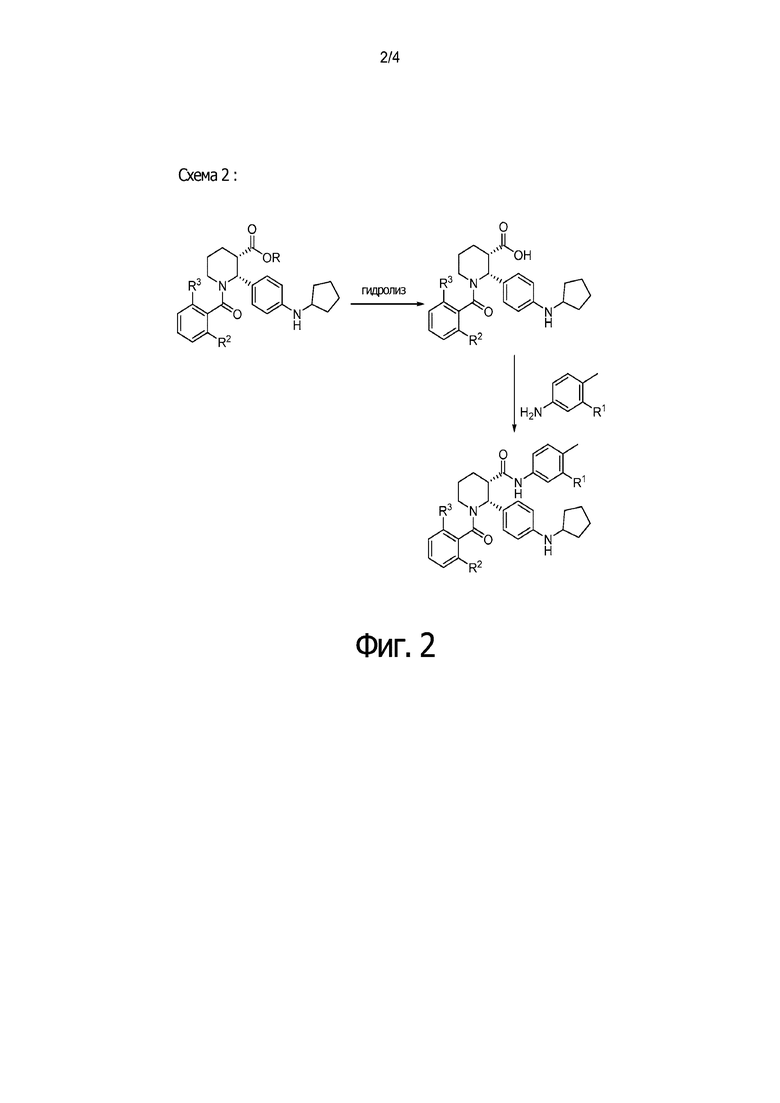
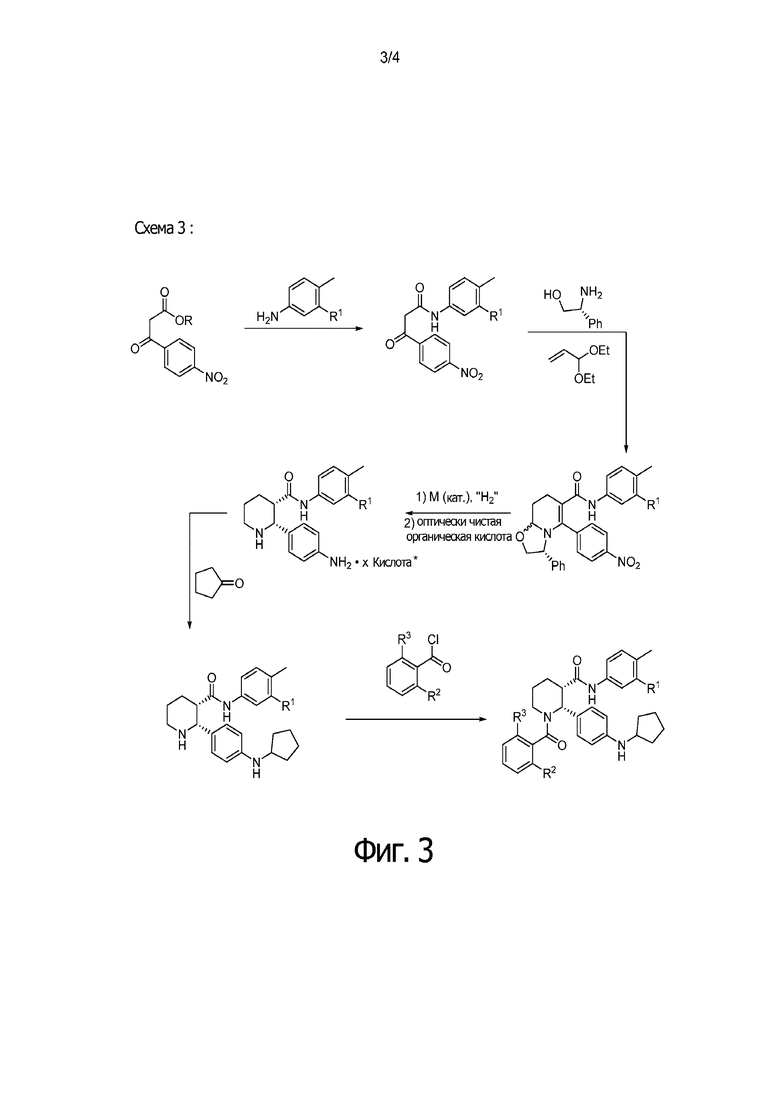
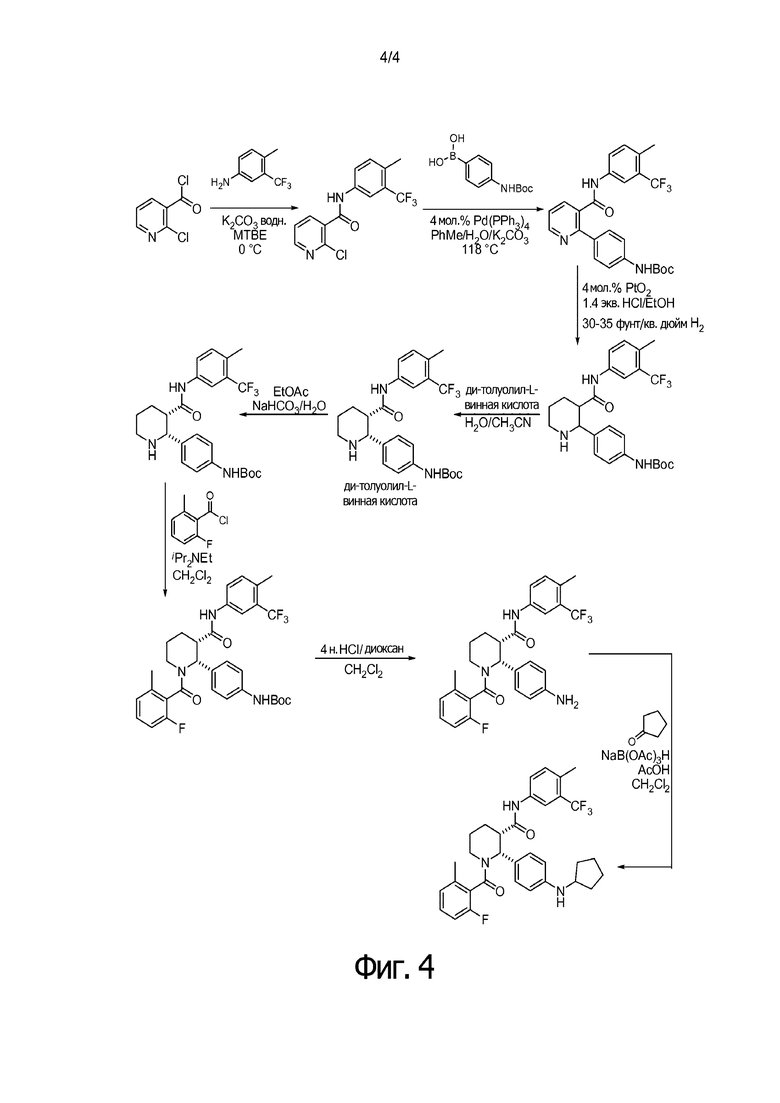
Изобретение относится к промежуточным соединениям формулы (i-3) и (ii-4), в которых R выбран из группы, состоящей из H, C1-8 алкила, арила и арил-C1-4 алкила, R1 представляет собой Cl или CF3, или к их соли. Также изобретение относится к способу получения соединения формулы (I), где R1 представляет собой Cl или CF3, R2 представляет собой F или Cl и R3 представляет собой H или CH3 или к его соли. Технический результат – получены новые соединения, которые могут быть использованы в получении конечного соединения формулы (I), которое может найти применение в медицине в качестве C5aR антагонистов. 5 н. и 30 з.п. ф-лы, 4 ил., 4 пр.
 (i-3)
(i-3)  (ii-4)
(ii-4)
 (I)
(I)
1. Соединение, имеющее формулу (i-3):
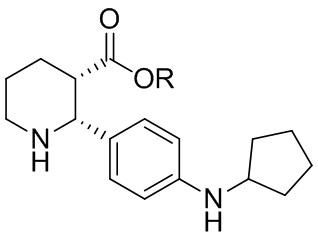 (i-3)
(i-3)
где R выбран из группы, состоящей из H, C1-8 алкила, арила и арил-C1-4 алкила, или его соль.
2. Соединение по п. 1, в форме бис L-DTTA соли.
3. Способ получения соединения, имеющего формулу (I):
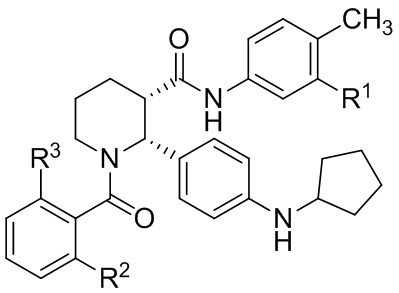 (I)
(I)
или его соли, где
R1 представляет собой Cl или CF3;
R2 представляет собой F или Cl; и
R3 представляет собой H или CH3;
и где указанное соединение формулы (I) практически не содержит энантиомерных или диастереомерных примесей, включающий:
(a) взаимодействие соединения, имеющего формулу (i-3):
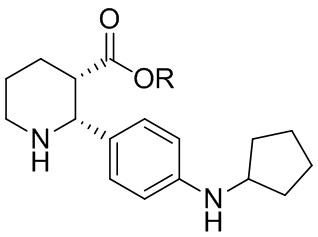 (i-3)
(i-3)
где R выбран из группы, состоящей из H, C1-8 алкила, арила и арил-C1-4 алкила, которое практически не содержит энантиомерных или диастереомерных примесей, или его соли, с соединением, имеющим формулу:
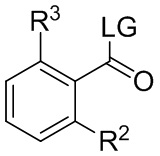
где LG представляет собой уходящую группу; R2 представляет собой F или Cl; и R3 представляет собой H или CH3; в присутствии основания с образованием соединения формулы (i-4):
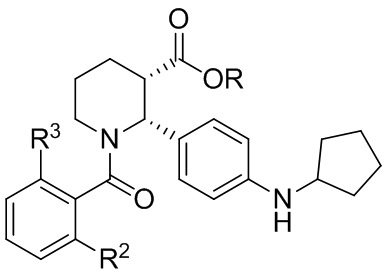 (i-4); и
(i-4); и
(b) взаимодействие соединения формулы (i-4) с анилином, имеющим формулу:
где R1 представляет собой Cl или CF3; в присутствии металлсодержащего реагента с образованием соединения формулы (I), где указанное соединение формулы (I) практически не содержит энантиомерных или диастереомерных примесей.
4. Способ по п. 3, где стадия (b) включает
(b)(1) гидролиз указанного соединения формулы (i-4), в которой R представляет собой C1-8 алкил, арил и арил-C1-4 алкил, с образованием соединения формулы (i-4), в которой R представляет собой Н; и
(b)(2) взаимодействие соединения формулы (i-4), в которой R представляет собой Н, с анилином, имеющим формулу:
где R1 представляет собой Cl или CF3; в присутствии реагента сочетания и второго основания с получением указанного соединения формулы (I).
5. Способ по любому из пп. 3, 4, где R1 представляет собой CF3, R2 представляет собой F, и R3 представляет собой CH3.
6. Способ по любому из пп. 3, 4, где R1 представляет собой CF3, R2 представляет собой Cl, и R3 представляет собой H.
7. Способ по любому из пп. 3, 4, где R1 представляет собой Cl, R2 представляет собой F, и R3 представляет собой CH3.
8. Способ получения соединения формулы (I), или его фармацевтически приемлемых солей, включающий:
(a) реакцию сложного эфира 3-(4-нитрофенил)-3-оксо-пропаноата (i-1), где R выбран из группы, состоящей из C1-8 алкила, арила и арил-C1-4 алкила, с (R)-(−)-2-фенилглицинолом и диэтилацеталем акролеина с получением соединения (i-2);
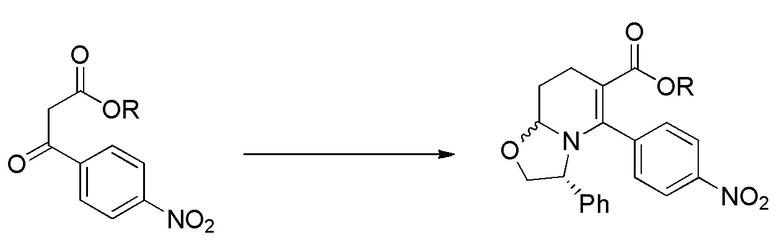
(b) гидрирование или восстановление (i-2) с получением промежуточного амина и превращение полученного промежуточного амина в (i-3) реакцией с циклопентаноном и восстановителем;
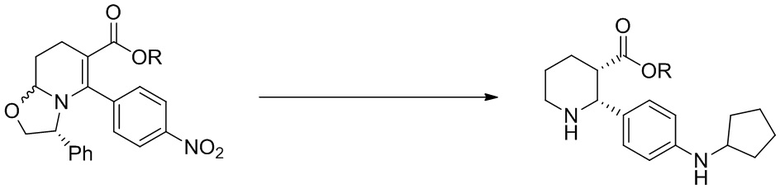
(c) взаимодействие (i-3) или с 2-фтор-6-метилбензоил хлоридом или 2-хлорбензоил хлоридом в присутствии первого основания с получением (i-4);
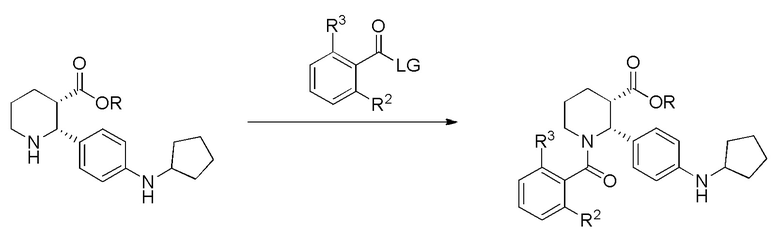
где R2 представляет собой F, R3 представляет собой метил, и LG представляет собой Cl; или R2 представляет собой Cl, R3 представляет собой водород, и LG представляет собой Cl;
(d) взаимодействие (i-4) с 3-хлор-4-метиланилином или 3-трифторметил-4-метиланилином в присутствии металлсодержащего реагента с получением соединения формулы (I);
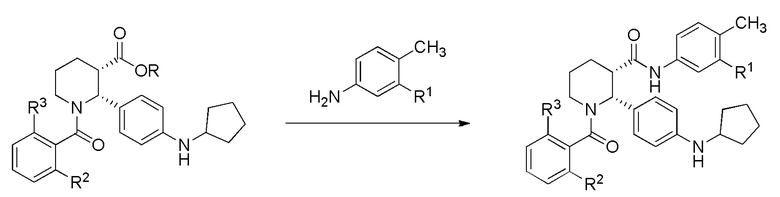
где R1 представляет собой Cl или СF3; или
(d)(1) преобразование сложного эфира (i-4) в карбоновую кислоту (i-5):
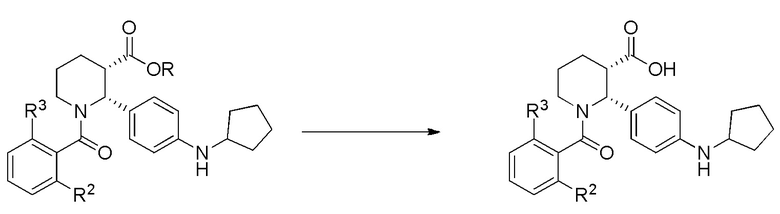
(d)(2) взаимодействие (i-5) с 3-хлор-4-метиланилином или 3-трифторметил-4-метиланилином в присутствии реагента сочетания и второго основания с получением соединения формулы (I)
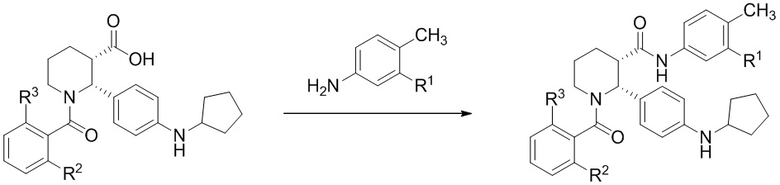
где R1 представляет собой Cl или СF3; и R2 представляет собой Cl, R3 представляет собой водород, или R2 представляет собой F, R3 представляет собой метил.
9. Способ по п. 3, где LG выбран из группы, включающей галоген, гидроксил, метансульфонат (или мезилат), трифторметансульфонат (трифлат), бензосульфонат, 4-метилбензолсульфонат (тозилат), 4-нитробензолсульфонат, 4-хлорбензолсульфонат, и карбоксилатный компонент в смешанном или симметричном ангидриде.
10. Способ по п. 3 или 9, где LG представляет собой галоген.
11. Способ по п. 3 или 8, где металлсодержащий реагент выбран из группы, включающей алюминийорганические реагенты, алкиллитиевые соединения, реагенты Гриньяра, цинкорганические реагенты, гидрид натрия и натриевые, калиевые или литиевые соли HMDS.
12. Способ по п. 3 или 8, где металлсодержащий реагент представляет собой алюминийорганический реагент.
13. Способ по п. 3 или 8, где металлсодержащий реагент представляет собой Al(Me)3 или комплекс триметилалюминия с DABCO (DABAL-Me3).
14. Способ по п. 8, где соединение формулы (I) получают с чистотой выше чем примерно 95% вес/вес.
15. Способ по п. 8, где соединение формулы (I) получают с чистотой выше чем 99% вес/вес.
16. Способ по п. 8, где соединение формулы (I) представлено соединением формулы IA
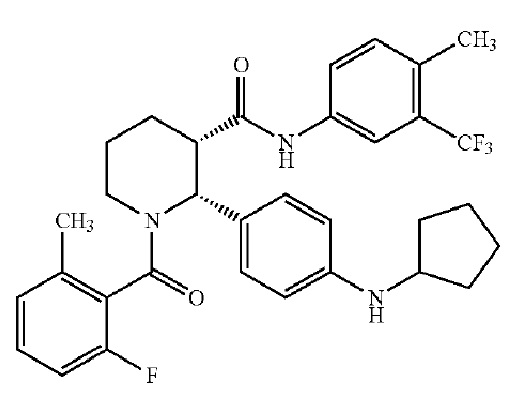 (IA).
(IA).
17. Способ по п. 8, где соединение формулы (I) представлено соединением формулы IB
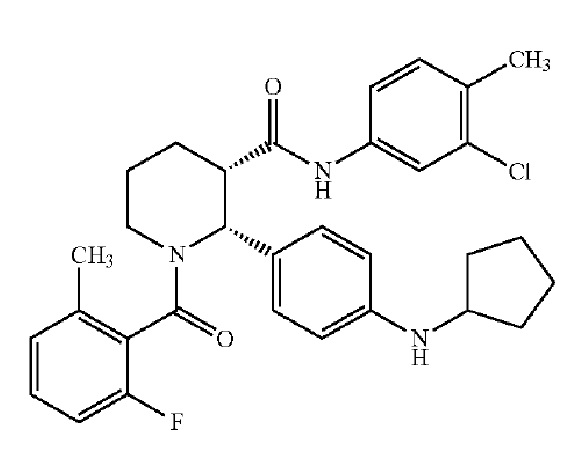 (IB).
(IB).
18. Способ по п. 8, где соединение формулы (I) представлено соединением формулы IC
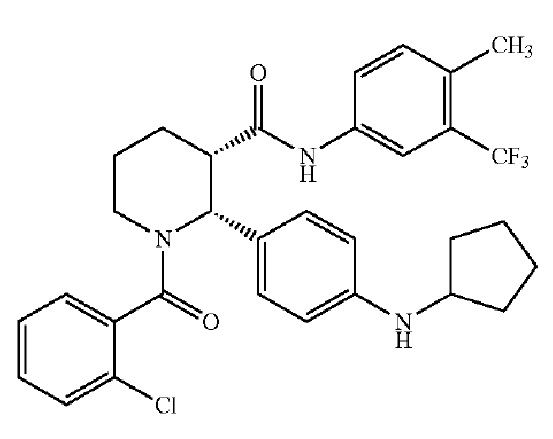 (IC).
(IC).
19. Способ по п. 3, где основание выбрано из группы, включающей триэтиламин, N,N-диизопропилэтиламин, DBU, N-метилморфолин, карбонат калия (K2CO3), бикарбонат калия (KHCO3), карбонат натрия (Na2CO3) и бикарбонат натрия (NaHCO3).
20. Способ по п. 4 или 8, где реагент сочетания представляет собой O-(7-Азабензотриазол-1-ил)-N,N,N’,N’-тетраметилурония гексафторфосфат (HATU) или метансульфонил хлорид (MsCl).
21. Способ по п. 4 или 8, где второе основание представляет собой N-метилморфолин или N,N-диизопропилэтиламин.
22. Способ по п. 8, где первое основание выбрано из группы, включающей триэтиламин, N,N-диизопропилэтиламин, DBU, N-метилморфолин, карбонат калия (K2CO3), бикарбонат калия (KHCO3), карбонат натрия (Na2CO3) и бикарбонат натрия (NaHCO3).
23. Способ по п. 8, где восстановитель выбран из группы, включающей газообразный водород с металлическим катализатором, сульфид натрия, дитионит натрия, сульфид аммония, литийалюминий гидрид, литийборгидрид, боргидрид натрия, цианоборгидрид натрия и триацетоксиборгидрид натрия.
24. Соединение, имеющее формулу (ii-4):
 (ii-4)
(ii-4)
где R1 представляет собой Cl или CF3, или его соль.
25. Соединение по п. 24, в форме соли с L-DTTA.
26. Соединение по п. 24, где R1 представляет собой CF3.
27. Соединение по п. 24, где R1 представляет собой Cl.
28. Способ получения соединения, имеющего формулу (I):
(I)
или его соли, где
R1 представляет собой Cl или CF3;
R2 представляет собой F или Cl; и
R3 представляет собой H или CH3;
и где указанное соединение формулы (I) практически не содержит энантиомерных или диастереомерных примесей, включающий:
(a) взаимодействие соединения, имеющего формулу (ii-4):
(ii-4)
где R1 представляет собой Cl или CF3; или его соли, где указанное соединение практически не содержит энантиомерных или диастереомерных примесей, с циклопентаноном и восстановителем с образованием соединения, имеющего формулу (ii-5):
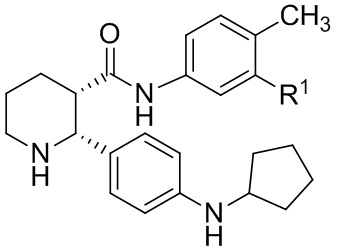 (ii-5); и
(ii-5); и
(b) взаимодействие указанного соединения формулы (ii-5) с соединением, имеющим формулу:
где LG представляет собой уходящую группу, R2 представляет собой F или Cl; и R3 представляет собой H или CH3; в присутствии основания с образованием соединения формулы (I), которое практически не содержит энантиомерных или диастереомерных примесей.
29. Способ по п. 28, где R1 представляет собой CF3, R2 представляет собой F, и R3 представляет собой CH3.
30. Способ по п. 28, где R1 представляет собой CF3, R2 представляет собой Cl, и R3 представляет собой H.
31. Способ по п. 28, где R1 представляет собой Cl, R2 представляет собой F, и R3 представляет собой CH3.
32. Способ по п. 28, где восстановитель выбран из группы, включающей газообразный водород с металлическим катализатором, сульфид натрия, дитионит натрия, сульфид аммония, литийалюминий гидрид, литийборгидрид, боргидрид натрия, цианоборгидрид натрия и триацетоксиборгидрид натрия.
33. Способ по п. 28, где LG выбран из группы, включающей галоген, гидроксил, метансульфонат (или мезилат), трифторметансульфонат (трифлат), бензосульфонат, 4-метилбензолсульфонат (тозилат), 4-нитробензолсульфонат, 4-хлорбензолсульфонат, и карбоксилатный компонент в смешанном или симметричном ангидриде.
34. Способ по п. 28, где LG представляет собой галоген.
35. Способ по п. 28, где основание выбрано из группы, включающей триэтиламин, N,N-диизопропилэтиламин, DBU, N-метилморфолин, карбонат калия (K2CO3), бикарбонат калия (KHCO3), карбонат натрия (Na2CO3) и бикарбонат натрия (NaHCO3).
| US 20110275639 A1, 10.11.2011; | |||
| ROMAIN NOЁL ET AL, European journal of Organic Chemistry, vol | |||
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Электрический аппарат для охраны касс, основанный на действии катодного реле | 1922 |
|
SU476A1 |
| WO 2010075257 A1, 01.07.2010 | |||
| Предохранитель к замку бомбодержателя | 1927 |
|
SU11088A1 |

