ОБЛАСТЬ ИЗОБРЕТЕНИЯ
В настоящем документе представлены соединения 1,6-нафтиридинила и пиридо[2,3-d]пиримидинила, полезные для лечения легочной гипертензии и родственных заболеваний и полезные для лечения аномального роста клеток, такого как рак; способы получения таких соединений; и способы применения таких соединений для лечения легочной гипертензии и родственных заболеваний, таких как легочная артериальная гипертензия, и для лечения аномального роста клеток, такого как рак.
Предпосылки создания изобретения
Легочная гипертензия (PH) ранее классифицировалась как первичная (идиопатическая) или вторичная. Недавно Всемирная организация здравоохранения (ВОЗ) классифицировала легочную гипертензию на пять групп: Группа 1: легочная артериальная гипертензия (PAH); Группа 2: PH с поражением левых отделов сердца; Группа 3: PH с заболеванием легких и/или гипоксемией; Группа 4: PH вследствие хронического тромботического и/или эмболического заболевания; и Группа 5: смешанные состояния (например, саркоидоз, гистиоцитоз X, лимфангиоматоз и компрессия легочных сосудов).
Легочная артериальная гипертензия представляет собой серьезное, сложное, прогрессирующее и опасное для жизни заболевание легочной сосудистой системы, характеризующееся сильным сужением сосудов и аномальным разрастанием гладкомышечных клеток в стенках легочных артерий. Сильное сужение кровеносных сосудов в легких приводит к очень высокому давлению в легочной артерии. Это высокое давление мешает сердцу перекачивать кровь через легкие для насыщения кислородом. Пациенты с PAH страдают от сильной одышки, поскольку сердце изо всех сил пытается справиться с этим высоким давлением. У пациентов с PAH обычно развивается значительное повышение резистентности легочных сосудов (PVR) и устойчивое повышение легочного артериального давления (PAP), что в конечном итоге приводит к правожелудочковой недостаточности и смерти. Пациенты с диагнозом PAH имеют плохой прогноз и в равной степени ухудшенное качество жизни, при отсутствии лечения средняя ожидаемая продолжительность жизни составляет от 2 до 5 лет с момента постановки диагноза.
В настоящее время существует 10 одобренных методов лечения PAH в Соединенных Штатах Америки (США) и Европе. Все эти методы лечения относятся к одному из 3 классов вазодилататоров, включая аналоги простациклина (эпопростенол, трепростинил, илопрост и селексипаг), антагонисты рецепторов эндотелина (ERA) (бозентан, амбрисентан и мацитентан) и средства, влияющие на сигнальный путь оксида азота, включая активатор растворимой гуанилатциклазы риоцигуат и ингибиторы фосфодиэстеразы 5 типа силденафил и тадалафил. Эти лечения направлены преимущественно на эндотелиальную и сосудистую гемодинамическую дисфункцию, связанную с этим заболеванием, и, хотя было продемонстрировано, что эти методы лечения замедляют прогрессирование заболевания, заболеваемость и смертность в этой популяции пациентов остаются высокими. Несмотря на доступность этих методов лечения, пятилетняя выживаемость пациентов с PAH остается на уровне 57%. McGoon MD, Miller DP. REVEAL: A contemporary US pulmonary arterial hypertension registry. Eur Respir Rev. 2012; 21:8-18.
Патофизиология, приводящая к ремоделированию сосудов, характерному для PAH, является многофакторной и включает аномальную пролиферацию васкулярных клеток, снижение апоптоза, воспаление, тромбоз in situ и вазоконстрикцию. Признано, что PAH имеет много общих характеристик злокачественного фенотипа, включая аномальную пролиферацию, образование плексиформных поражений вблизи легочной сосудистой системы, измененный митохондриальный метаболизм и переключение на анаэробные метаболические пути, несмотря на адекватное снабжение кислородом. Двумя ключевыми типами клеток, участвующих в прогрессировании в сторону PAH, являются гладкомышечные клетки легочной артерии (PASMC) и адвентициальные фибробласты легочной артерии (PAAF). В WO2018/073687, ингибиторы циклин-зависимых киназ (CDK), такие как палбоциклиб, идентифицированы как полезные для лечения PAH, путем оценки воздействия на PASMC человека и крысы и PAAF человека. Однако сохраняется потребность в лечении PAH.
Более того, CDK и родственные серин/треониновые протеинкиназы являются важными клеточными ферментами, которые выполняют основные функции в регуляции деления и пролиферации клеток. Каталитические единицы CDK активируются посредством регуляторных субъединиц, известных как циклины. По меньшей мере, идентифицированы шестнадцать циклинов млекопитающих (Johnson DG, Walker CL. Cyclins and Cell Cycle Checkpoints. Annu. Rev. Pharmacol. Toxicol. (1999) 39:295 312). Дополнительные функции гетеродимеров циклин/CDK включают регуляцию транскрипции, репарацию ДНК, дифференцировку и апоптоз (Morgan DO. Cyclin dependent kinases: engines, clocks, and microprocessors. Annu. Rev. Cell. Dev. Biol. (1997) 13:261 291).
Было показано, что ингибиторы CDK полезны при лечении рака. Было показано, что увеличенная активность или временно аномальная стимуляция CDK приводит к развитию опухолей у человека, и развитие опухоли у человека обычно ассоциируется с изменениями или в самих CDK-белках или в их регуляторах (Cordon Cardo C. Mutations of cell cycle regulators: biological and clinical implications for human neoplasia. Am. J. Pathol. (1995) 147:545 560; Karp JE, Broder S. Molecular foundations of cancer: new targets for intervention. Nat. Med. (1995) 1:309 320; Hall M, Peters G. Genetic alterations of cyclins, cyclin dependent kinases, and CDK inhibitors in human cancer. Adv. Cancer Res. (1996) 68:67 108).
CDK4 и CDK6 являются важными регуляторами прогрессии клеточного цикла в контрольной точке G1-S, которые контролируются циклинами D-типа и эндогенными ингибиторами CDK INK4, такими как p16INK4a (CDKN2A). Сообщалось, что нарушение регуляции пути циклин D-CDK4/6-INK4-ретинобластома (Rb) связано с развитием резистентности к эндокринной терапии.
Мутации CDK4 и CDK6 были описаны в подгруппах меланомы и других опухолей (Zuo L, et al., Germline mutations in the p16INK4a binding domain of CDK4 in familial melanoma. Nature Genet. (1996) 12, 97-99; Ortega S, et al. Cyclin D dependent kinases, INK4 inhibitors and cancer. Biochim. Biophys. Acta (2002) 1602:73 87; Smalley KSM et al. Identification of a novel subgroup of melanomas with KIT/cyclin dependent kinase 4 в течение expression. Cancer Res (2008) 68: 5743 52). Амплификации регуляторных субъединиц CDK и циклинов, и мутации, делеции генов или транскрипционный сайленсинг эндогенных ингибиторов CDK INK4 также описаны в качестве механизма, с помощью которого этот путь может быть активирован (Smalley KSM (2008)).
Разработка ингибиторов CDK была рассмотрена в литературе. Например, см. Sánchez-Martínez et al. Cyclin dependent kinase (CDK) inhibitors as anticancer drugs, Bioorg. Med. Chem. Lett. (2015) 25: 3420-3435 (и ссылки, приведенные в нем). Использование ингибиторов CDK4/6 в сочетании с эндокринной терапией продемонстрировало значительную эффективность в лечении гормон-рецептор(HR)-положительного рака, отрицательного по рецептору эпидермального фактора роста человека (HER2) распространенного или метастатического рака молочной железы, а также ингибиторов CDK4/6, включая палбоциклиб, рибоциклиб и абемациклиб, были одобрены в комбинации с эндокринной терапией в условиях первой или второй линии терапии.
Тем не менее, из-за возможности развития приобретенной резистентности к и других механизмов обхода ингибиторов CDK4/6 остается необходимость в идентификации новых ингибиторов CDK4/6, имеющих соответствующий фармакологический профиль, например, с точки зрения эффективности, селективности, фармакокинетики и продолжительность действия.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В одном аспекте изобретение относится к соединению формулы I:

или его фармацевтически приемлемой соли, где:
A представляет собой CH или N;
R1 представляет собой C1-C2 алкил или C1-C2 фторалкил;
R2 представляет собой C1-C4 алкил, необязательно замещенный 1, 2 или 3 заместителями, каждый независимо выбран из группы, состоящей из -OH, C1-C2 алкокси и F;
R3 представляет собой 4-8-членный гетероциклил, C3-C8 циклоалкил, (4-8-членный гетероциклил)-C1-C4 алкил- или (C3-C8 циклоалкил)-C1-C4 алкил-,
где каждый из 4-8-членного гетероциклила и 4-8-членного гетероциклильного фрагмента в (4-8-членный гетероциклил)-C1-C4 алкил- необязательно замещен 1 или 2 R5,
где каждый из C3-C8 циклоалкила и C3-C8 циклоалкильного фрагмента в (C3-C8 циклоалкил)-C1-C4 алкил- необязательно замещен 1 или 2 R5 и дополнительно необязательно замещен 1 -N(R6)2, и
где каждый из C1-C4 алкильных фрагментов в (4-8-членный гетероциклил)-C1-C4 алкил- и (C3-C8 циклоалкил)-C1-C4 алкил- необязательно замещен 1, 2 или 3 R5;
R4 представляет собой фрагмент, имеющий структуру
 ;
;
каждый R5 независимо выбран из группы, состоящей из -F, -OH, -CN, C1-C4 алкила, C1-C4 фторалкила, C1-C4 алкокси, C1-C4 фторалкокси, C1-C4 алкокси-C1-C4 алкил-, C1-C4 фторалкокси-C1-C4 алкил-, C1-C4 алкокси-C1-C4 фторалкила и C1-C4 фторалкокси-C1-C4 фторалкил-;
каждый R6 независимо выбран из группы, состоящей из H и C1-C2 алкила;
каждый R7 независимо представляет собой H или C1-C2 алкила; и
каждый R8 независимо представляет собой H, F или C1-C2 алкил.
Настоящее изобретение также предоставляет фармацевтическую композицию, которая включает соединение формулы I или его фармацевтически приемлемую соль и фармацевтически приемлемый эксципиент или носитель.
Настоящее изобретение также предоставляет способ лечения заболевания или расстройства у субъекта, который включает введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли, где заболевание или расстройство выбрано из группы, состоящей из легочной гипертензии, легочной артериальной гипертензии, легочной гипертензии с поражением левых отделов сердца, легочной гипертензии с заболеванием легких и/или гипоксемией, легочной гипертензии вследствие хронического тромботического и/или эмболического заболевания, и заболевания, связанного с легочной гипертензией, включая саркоидоз, гистиоцитоз X, лимфангиоматоз и компрессию легочных сосудов.
Настоящее изобретение также предоставляет способ лечения аномального роста клеток (например, рака) у субъекта, который включает введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли.
Настоящее изобретение также предоставляет применение соединения формулы I или его фармацевтически приемлемой соли для лечения заболевания или расстройства, или для получения лекарственного средства для лечения заболевания или расстройства, где заболевание или расстройство выбрано из группы, состоящей из легочной гипертензии, легочной артериальной гипертензии, легочной гипертензии с поражением левых отделов сердца, легочной гипертензии с заболеванием легких и/или гипоксемией, легочной гипертензии вследствие хронического тромботического и/или эмболического заболевания, и заболевания, связанного с легочной гипертензией, включая саркоидоз, гистиоцитоз X, лимфангиоматоз и компрессию легочных сосудов.
Настоящее изобретение также предоставляет применение соединения формулы I или его фармацевтически приемлемой соли для лечения аномального роста клеток (такого как рак) или для получения лекарственного средства для лечения аномального роста клеток (например, рака).
Настоящее изобретение также предоставляет способ ингибирования CDK (например, CDK4 и/или CDK6), который включает контактирование CDK с соединением формулы I или его фармацевтически приемлемой солью.
Следует понимать, что как предшествующее общее описание, так и последующее подробное описание являются лишь иллюстративными и пояснительными и не ограничивают заявленное в формуле изобретения.
КРАТКОЕ ОПИСАНИЕ ФИГУР
Фиг. 1 представляет собой наблюдаемую порошковую рентгеновскую дифрактограмму для безводной (ангидрат) кристаллической формы (обозначенной как форма 1) соединения 5.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение может быть более понятно со ссылкой на последующее подробное описание и примеры и схемы, представленные в настоящем документе. Следует понимать, что используемая в настоящем документе терминология предназначена только для описания конкретных вариантов осуществления и не предназначена для ограничения.
Следует понимать, что настоящее изобретение не ограничивается конкретными методами синтеза получения, которые, конечно, могут изменяться. Также следует понимать, что терминология, используемая в настоящем документе, предназначена только для описания конкретных вариантов осуществления и не предназначена для ограничения. В настоящем описании и в следующей формуле изобретения будет сделана ссылка на ряд терминов, которые должны быть определены, как имеющие следующие значения:
Как используется в настоящем документе в описании, форма единственного числа может означать один или несколько. Используемые в настоящем документе в формуле изобретения, при использовании в сочетании со словом «включающий» слова в единственном числе могут означать один или несколько. Используемый в настоящем документе термин «другой» может означать, по меньшей мере, второй или более.
Термин «примерно» относится к относительному термину, обозначающему приближение плюс или минус 10% от номинального значения, которое относится, в одном варианте осуществления к плюс или минус 5%, в другом варианте осуществления к плюс или минус 2%. Для области настоящего раскрытия этот уровень приближения является подходящим, если только значение специально не указано, что требует более узкого диапазона.
Используемый в настоящем описании термин «алкил» включает любые ациклические, насыщенные алифатические углеводороды, включающие прямые и разветвленные цепи. В некоторых вариантах осуществления алкильная группа от 1 до 4 атомов углерода, от 1 до 3 атомов углерода, от 1 до 2 атомов углерода или только один атом углерода. Например, термин "C1-6 алкил” относится к линейным или разветвленным алифатическим углеводородным цепям с 1-6 атомами углерода, включительно (например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил или н-гексил); термин "C1-4 алкил” относится к линейным или разветвленным алифатическим углеводородным цепям с 1-4 атомами углерода, включительно (например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил или трет-бутил); термин "C1-2 алкил” относится к алифатическим углеводородным цепям, содержащим от 1 до 2 атомов углерода, включительно; и термин "C1 алкил” относится к метилу и термин "C2 алкил” относится к этилу. Алкильная группа необязательно может быть замещена одним или несколькими (например, 1-5) подходящими заместителями, если и когда это указано.
Используемый в настоящем описании термин «алкокси» или «алкилокси» относится к -O-алкильной группе. Например, термин “C1-C4 алкокси” или “C1-C4 алкилокси” относится к -O-(C1-C4 алкил) группе; и термин “C1-C2 алкокси” или “C1-C2 алкилокси” относится к -O-(C1-C2 алкил) группе. Примеры алкокси включают метокси, этокси, пропокси (например, н-пропокси и изопропокси), трет-бутокси и тому подобное. C1-C2 алкокси включают метокси и этокси. Алкокси или алкилокси группа необязательно может быть замещена 1 или несколькими (например, 1-5) подходящими заместителями, если и когда это указано.
Используемый в настоящем описании термин "циклоалкил” относится к насыщенным или ненасыщенным, неароматическим, моноциклическим или полициклическим (таким как бициклические) углеводородным кольцам [например, моноциклическим, таким как циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклогептатриенил или бициклическим, включая спиро, конденсированные или мостиковые системы (такие как бицикло[1.1.1]пентанил, бицикло[2.2.1]гептанил, бицикло[3.1.0]гексанил или бицикло[3.2.1]октанил и т.п.)]. В некоторых вариантах осуществления циклоалкил может необязательно содержать одну, две или несколько неароматических двойных или тройных связей в кольцевой структуре и/или необязательно может быть замещен одной или несколькими (например, одной-тремя) оксогруппами. В некоторых вариантах осуществления бициклоалкильная группа имеет от 3 до 8 атомов углерода. Например, термин "C3-8 циклоалкил” относится к насыщенным или ненасыщенным, неароматическим, моноциклическим или полициклическим (например, бициклическим) углеводородным кольцам из 3-8 образующих кольцо атомов углерода (например, циклопропил, циклобутил, циклопентил, циклопентенил. циклогексил, бицикло[3.1.0]гексанил, бицикло[1.1.1]пентанил или бицикло[3.2.1]октанил); и термин "C3-6 циклоалкил” относится к насыщенным или ненасыщенным, неароматическим, моноциклическим или полициклическим (например, бициклическим) углеводородным кольцам из 3-6 образующих кольцо атомов углерода (например, циклопропил, циклобутил, циклопентил, циклогексил, бицикло[3.1.0]гексанил, бицикло[1.1.1]пентан-1-ил или бицикло[1.1.1]пентан-2-ил). В качестве еще одного примера термин "C3-4 циклоалкил” относится к насыщенным или ненасыщенным, неароматическим, моноциклическим углеводородным кольцам из 3-4 образующих кольцо атомов углерода (например, циклопропил или циклобутил). В некоторых вариантах осуществления C3-8 циклоалкильная группа включает насыщенные моноциклические или полициклические (например, бициклические) углеводородные кольца из 3-8 образующих кольцо атомов углерода и ненасыщенные моноциклические или полициклические (например, бициклические) углеводородные кольца из 3-8 образующих кольцо атомов углерода с кольцом с одной двойной связью. В некоторых вариантах осуществления C3-8 циклоалкильная группа включает насыщенные моноциклические или полициклические (например, бициклические) углеводородные кольца из 3-8 образующих кольцо атомов углерода. Циклоалкильная группа необязательно может быть замещена 1 или несколькими (например, 1-5) подходящими заместителями, если и когда это указано.
Термин «гетероциклил» означает насыщенную или частично ненасыщенную, неароматическую кольцевую систему, содержащую один или несколько (например, один, два, три или четыре) атомов углерода, образующих кольцо, и один или несколько (например, один, два, три или четыре) гетероатомов, образующих кольцо, каждый независимо выбран из группы, состоящей из кислорода, азота и серы. Кольцевая система гетероциклила может быть моноциклической или полициклической (включая 2 или более колец, например, бициклическая кольцевуая система), включая спиро, конденсированные и/или мостиковые системы, где каждое отдельное кольцо в кольцевой системе содержит 1 или несколько атомов углерода, образующих кольцо, и 0, 1 или несколько (например, 0, 1, 2 или 3) гетероатомов, образующих кольцо (каждый из которых независимо выбран из кислорода, азота и серы). В некоторых вариантах осуществления гетероциклильная группа включает насыщенную кольцевую систему, содержащую один или несколько (например, один, два, три или четыре) атомов углерода, образующих кольцо, и один или несколько (например, один, два, три или четыре) гетероатомов, образующих кольцо, каждый независимо выбран из группы, состоящей из кислорода, азота и серы. Некоторые примеры «гетероциклила» включают лактоны, лактамы, циклические простые эфиры и циклические амины. Некоторые неограничивающие примеры «гетероциклила» включают пирролидинонил, 2,5-дигидро-1H-пирролил, пиперидинонил, морфолинонил, пиперазинонил, оксазолидинонил, имидазолидинонил, 1,3-оксазинан-2-онил, тетрагидропиримидин-2(1Н)-онил, эпоксидил, тетрагидрофуранил, тетрагидропиранил, диоксанил, азиридинил, азетидинил, оксетанил, пирролидинил, оксазолидинил, тиазолидинил, пиперидинил, морфолинил, пиперазинил, тиоморфолинил, 1,3-оксазинанил, 1,3-тиазинанил, азаспиро[3.3]гептанил, 2-азабицикло[2.1.1]гексанил, 5-азабицикло[2.1.1]гексанил, 6-азабицикло[3.1.1]гептанил, 2-азабицикло[2.2.1]гептанил, 3-азабицикло[3.1.1]гептанил, 2-азабицикло[3.1.1]гептанил, 3-азабицикло[3.1.0]гексанил, 2-азабицикло[3.1.0]гексанил, 3-азабицикло[3.2.1]октанил, 8-азабицикло[3.2.1]октанил, 3-окса-7-азабицикло[3.3.1]нонанил, 3-окса-9-азабицикло[3.3.1]нонанил, 2-окса-5-азабицикло[2.2.1]гептанил, 6-окса-3-азабицикло[3.1.1]гептанил, 2-азаспиро[3.3]гептанил и 2-окса-6-азаспиро[3.3]гептанил. В некоторых вариантах осуществления образующий кольцо атом S в гетероциклиле может быть необязательно замещен одной или двумя оксогруппами, если и когда это указано, и/или образующий кольцо атом углерода в гетероциклиле может быть необязательно замещен одной оксогруппой, если и когда это указано. Гетероциклильная группа необязательно может быть замещена 1 или несколькими (например, от 1 до 5) подходящими заместителями, если и когда это указано.
Используемый в настоящем описании термин «галоген» или «галогеновая» группа включает фтор (F), хлор (Cl), бром (Br) или йод (I).
Используемый в настоящем описании термин “фторалкил” относится к алкильной группе, имеющей один или несколько галогеновых заместителей (вплоть до перфторалкила, т.е. каждый атом водорода алкильной группы заменен атомом фтора, например, от 1 до 5 атомов водорода алкильной группы заменены атомами фтора). Например, используемый в настоящем описании термин "C1-C4 фторалкил" означает C1-C4 алкил, как определено в настоящем описании, имеющий один или несколько фтор (F)-заместителей (вплоть до перфторалкила, т.е. каждый атом водорода C1-C4 алкильной группы заменен атомом фтора, например, от 1 до 5 атомов водорода алкильной группы заменены атомами фтора; и термин "C1-C2 фторалкил" означает C1-C2 алкил, как определено в настоящем описании, имеющий один или несколько фтор (F)-заместителей (вплоть до перфторалкила, т.е. каждый атом водорода C1-C2 алкильной группы заменен атомом фтора). В другом примере термин “C1 фторалкил” означает метил, имеющий один или несколько фтор (F)-заместителей (вплоть до перфторалкила, т.е. каждый атом водорода метильной группы заменен атомом фтора). Примеры C1-C4 фторалкила включают CF3, CHF2, CH2F, CH2CF3, CH2CHF2, C2F5, CH2C2F5, CH2CH2CH2CF3, и тому подобное; C1-C2 фторалкила включают CF3, CHF2, CH2F, CH2CF3, CH2CHF2, CH2CH2F; CHFCF3, CHFCHF2, CHFCH2F, CHFCH3, CF2CF3, CF2CHF2, CF2CH2F и CF2CH3; и C1 фторалкила включают CF3, CHF2 и CH2F.
Используемый в настоящем описании термин “фторалкокси” или “фторалкилокси” относится к -O-фторалкильной группе. Например, термин “C1-C6 фторалкокси” или “C1-C6 фторалкилокси” относится к -O-(C1-C6 фторалкил) группе; термин “C1-C4 фторалкокси” или “C1-C4 фторалкилокси” относится к -O-(C1-C4 фторалкил) группе; и термин “C1-C2 фторалкокси” или “C1-C2 фторалкилокси” относится к -O-(C1-C2 фторалкил) группе. Примеры фторалкокси включают монофторметокси (-OCH2F), дифторметокси (-OCHF2), трифторметокси (-OCF3), -OCH2CF3, -OC2F5, и тому подобное.
«Замещенный» атом или фрагмент указывает, что любой водород на указанном атоме или фрагменте может быть заменен выбранным из указанной группы заместителей (вплоть до того, что каждый атом водорода на указанном атоме или фрагменте заменен выбранным из указанной группы заместителей), при условии, что нормальная валентность указанного атома или фрагмента не превышена, и что замещение приводит к стабильному соединению. Например, если метильная группа (т.е. CH3) замещена, тогда вплоть до 3 атомов водорода на атоме углерода могут быть заменены группами заместителей. В качестве другого примера, если пипердин-4-ил замещен группой заместителей, то любой атом водорода на образующем кольцо атоме углерода или образующем кольцо атоме азота может быть замещен группой заместителей. Некоторые примеры групп заместителей для алкила включают галоген (например, F), OH и -O-C1-C4 алкил (т.е. C1-C4 алкокси). Некоторые примеры групп заместителей для циклоалкила или гетероциклила включают галоген, OH, -NH2, -NH(C1-C4 алкил), N(C1-C4 алкил)2, C1-C4 алкил (который дополнительно может быть замещен 0, 1 или более заместителями, каждый независимо выбран из галогена, OH, C1-C4 алкокси, и C1-C4 галогеналкокси) и C1-C4 алкокси (который дополнительно может быть замещен 0, 1 или более заместителями, каждый независимо выбран из галогена, OH, C1-C4 алкокси и C1-C4 фторалкокси).
Используемый в настоящем описании термин «необязательно замещенный» означает, что замещение является необязательным и, поэтому, включает как незамещенные, так и замещенные атомы и фрагменты.
В настоящем описании, если не указано иное, место присоединения заместителя может быть из любого подходящего положения заместителя. Например, пиперидинил может представлять собой пиперидин-1-ил (присоединенный через атом N пиперидинила), пиперидин-2-ил (присоединенный через атом С в положении 2 пиперидинила), пиперидин-3-ил (присоединенный через атом С в положении 3 пиперидинила) или пиперидин-4-ил (присоединенный через атом С в положении 4 пиперидинила). В качестве другого примера, пиридинил (или пиридил) может представлять собой 2-пиридинил (или пиридин-2-ил), 3-пиридинил (или пиридин-3-ил) или 4-пиридинил (или пиридин-4-ил).
Используемый в настоящем описании термин «n-членный», где n представляет собой целое число, обычно описывает число образующих кольцо атомов в кольцевом фрагменте, где число образующих кольцо атомов равно n. Например, пиперидинил представляет собой пример 6-членного гетероциклильного кольца и тетрагидропиранил представляет собой пример 5-членной гетероциклильной группы. Соответственно, термин «n-m-членный» гетероциклил (где целое число «n» описывает нижнее число образующих кольцо атомов в гетероциклиле, а целое число «m» описывает верхнее число образующих кольцо атомов) специально предназначены для включения любой из n-членной, (n+1)-членной, … . и до m-членной гетероциклильной группы; например, термин «4-8-членный гетероциклил» специально предназначен для включения любой 4-, 5-, 6-, 7- или 8-членной гетероциклильной группы включительно.
В различных местах настоящего описания заместители соединений по изобретению описаны в группах или в диапазонах. В частности, предполагается, что изобретение включает каждую индивидуальную подкомбинацию членов таких групп и диапазонов (включая конечные точки). Например, содержание атомов углерода в алкиле и различных других углеводородсодержащих фрагментах может быть указано с помощью префикса, обозначающего количество атомов углерода в фрагменте, то есть префикс Ci указывает на фрагмент, состоящий из целого числа атомов углерода «i». Так, например, C3 алкил означает любой алкил с 3 атомами углерода, включая пропан-1-ил и пропан-2-ил. В качестве другого примера, содержание атомов углерода в алкиле и различных других углеводородсодержащих фрагментах указано префиксом, обозначающим нижнее и верхнее число атомов углерода в фрагменте, то есть префикс Ci-j указывает фрагмент целого числа "i" до целого числа "j" атомов углерода включительно. Так, например, термин “C1-C6 алкил” специально предназначен для включения C1 алкила (метил), C2 алкила (этил), C3 алкила (т.е., пропан-1-ил или пропан-2-ил), C4 алкила (например, н-бутил, изобутил или трет-бутил), C5 алкила (например, пентан-1-ил или 3-метилбутан-1-ил) и C6 алкила (например, гексан-1-ил или 3-метилпентан- 1-ил). В другом примере, термин "C3-C6 циклоалкил” специально предназначен для включения любого C3 циклоалкила (циклопропил), C4 циклоалкила (например, циклобутил), C5 циклоалкила (например, циклокпентил) или C6 циклоалкила (например, циклогексил).
В различных местах настоящего описания химические термины, такие как алкил, алкокси, фторалкокси (например, C1-C2 алкил, C1-C4 алкил, C1-C4 фторалкил, C1-C2 алкокси, C1-C2 фторалкокси) могут использоваться в сочетании с одним или несколькими другими химическими терминами, например, “C1-C2 алкокси-C1-C2 фторалкил-”, “-C1-C4 алкил-(4-8-членный гетероциклил)”, или “-C1-C4 алкил-(C3-C8 циклоалкил)”. В таком случае специалисты в данной области легко распознают указанную точку присоединения для комбинации [например, указанная точка присоединения для “-C1-C4 алкил-(C3-C8 циклоалкил)” проходит через “C1-C4 алкильный” фрагмент] и что каждый фрагмент комбинации удовлетворяет правилу нормальной валентности [например, “C1-C4 алкильный” фрагмент “-C1-C4 алкил-(C3-C8 циклоалкил)” является двухвалентным, т.е. присоединяется к “C3-C8 циклоалкильному” фрагменту и присоединяется к фрагменту основной молекулы через указанную точку присоединения; и для другого примера, “C1-C2 фторалкильный” фрагмент “C1-C2 алкокси-C1-C2 фторалкил-“ является двухвалентным, связывается с фрагментом “C1-C2 алкокси” и присоединяется к фрагменту основной молекулы через указанную точку присоединения].
Вариант осуществления изобретения представляет собой соединение формулы I или его фармацевтически приемлемую соль, где R2 представляет собой C1-C4 алкил, необязательно замещенный 1, 2 или 3 F, и дополнительно необязательно замещенный 1 OH или C1-C2 алкокси. В другом варианте осуществления R2 представляет собой C1-C4 алкил, необязательно замещенный 1, 2 или 3 F, и дополнительно необязательно замещенный 1 C1-C2 алкокси. В еще одном варианте осуществления R2 представляет собой C1-C2 алкил, необязательно замещенный 1, 2 или 3 F, и дополнительно необязательно замещенный 1 C1-C2 алкокси.
Вариант осуществления изобретения представляет собой соединение формулы I или его фармацевтически приемлемую соль, где R2 представляет собой C1-C2 алкил, необязательно замещенный 1, 2 или 3 F. В другом варианте осуществления R2 представляет собой C1-C2 алкил. В еще одном варианте осуществления R2 представляет собой метил.
Вариант осуществления изобретения представляет собой соединение формулы I или его фармацевтически приемлемую соль, где R3 представляет собой 4-8-членный гетероциклил или C3-C8 циклоалкил, где 4-8-членный гетероциклил необязательно замещен 1 или 2 R5, и где C3-C8 циклоалкил необязательно замещен 1 или 2 R5 и дополнительно необязательно замещен 1 -N(R6)2. В другом варианте осуществления, R3 представляет собой 5-7-членный гетероциклил или C3-C6 циклоалкил, где 5-7-членный гетероциклил необязательно замещен 1 или 2 R5, и где C3-C6 циклоалкил необязательно замещен 1 или 2 R5 и дополнительно необязательно замещен 1 -N(R6)2.
Вариант осуществления изобретения представляет собой соединение формулы I или его фармацевтически приемлемую соль, где R3 представляет собой 4-8-членный гетероциклил, необязательно замещенный 1 или 2 R5.
Вариант осуществления изобретения представляет собой соединение формулы I или его фармацевтически приемлемую соль, где R3 представляет собой C3-C8 циклоалкил, необязательно замещенный 1 или 2 R5, и дополнительно необязательно замещенный 1 -N(R6)2. В другом варианте осуществления R3 представляет собой C3-C8 циклоалкил. В еще одном варианте осуществления R3 представляет собой циклопропил, циклобутил, циклопентил, циклогексил или бицикло[3.1.0]гексанил.
Вариант осуществления изобретения представляет собой соединение формулы I или его фармацевтически приемлемую соль, где:
A представляет собой CH или N;
R1 представляет собой CH3 или C1 фторалкил;
R2 представляет собой CH3;
R3 представляет собой 5-7-членный гетероциклил, C3-C6 циклоалкил, (5-7-членный гетероциклил)-C1-C4 алкил- или (C3-C6 циклоалкил)-C1-C4 алкил-,
где каждый из 5-7-членного гетероциклила и 5-7-членного гетероциклильного фрагмента в (5-7-членный гетероциклил)-C1-C4 алкил- необязательно замещен 1 R5,
где каждый из C3-C6 циклоалкила и C3-C6 циклоалкильного фрагмента в (C3-C6 циклоалкил)-C1-C4 алкил- необязательно замещен 1 или 2 R5 и дополнительно необязательно замещен 1 -N(R6)2, и
где каждый из C1-C4 алкильных фрагментов в (5-7-членный гетероциклил)-C1-C4 алкил- и (C3-C6 циклоалкил)-C1-C4 алкил- необязательно замещен 1, 2 или 3 R5;
R4 представляет собой фрагмент, имеющий структуру

каждый R5 независимо выбран из группы, состоящей из -F, -OH, -CN, C1-C2 алкила, C1-C2 фторалкила, C1-C2 алкокси, C1-C2 фторалкокси, C1-C2 алкокси-C1-C2 алкил-, C1-C2 фторалкокси-C1-C2 алкил-, C1-C2 алкокси-C1-C2 фторалкил- и C1-C2 фторалкокси-C1-C2 фторалкил-;
каждый R6 независимо выбран из группы, состоящей из H и C1-C2 алкила; и
каждый R7 независимо представляет собой H или CH3, при условии, что не более двух R7 представляют собой CH3.
Вариант осуществления соединения формулы I по настоящему изобретению или его фармацевтически приемлемой соли представляет собой соединение формулы II:

или его фармацевтически приемлемую соль (где A и R3 имеют значения, как определено в любом из вариантов осуществления, описанных в настоящем документе). В другом варианте осуществления два заместителя в тетрагидропирановом кольце формулы II находятся в транс-положении по отношению друг к другу (т.е. два заместителя в тетрагидропирановом кольце находятся на противоположных сторонах тетрагидропиранового кольца).
Вариант осуществления соединения формулы I или II или его фармацевтически приемлемой соли представляет собой соединение формулы III:

или его фармацевтически приемлемую соль (где A и R3 имеют значения, как определено в любом из вариантов осуществления, описанных в настоящем документе).
Вариант осуществления соединения формулы I или его фармацевтически приемлемой соли по настоящему изобретению представляет собой соединение формулы II:

или его фармацевтически приемлемую соль, где:
A представляет собой CH или N;
R3 представляет собой 5-7-членный гетероциклил, необязательно замещенный 1 R5; и
R5 выбран из группы, состоящей из F, OH, -CN, C1-C2 алкила, C1-C2 фторалкила, C1-C2 алкокси, C1-C2 фторалкокси, C1-C2 алкокси-C1-C2 алкил-, C1-C2 фторалкокси-C1-C2 алкил-, C1-C2 алкокси-C1-C2 фторалкил- и C1-C2 фторалкокси-C1-C2 фторалкил-.
В другом варианте осуществления соединения формулы II, R3 представляет собой 5-7-членный гетероциклил, необязательно замещенный 1 R5 (и R5 имеет значения, как определено в любом из вариантов осуществления, описанных в настоящем документе), и два заместителя в тетрагидропирановом кольце формулы II находятся в транс-положении по отношению друг к другу (т.е. два заместителя в тетрагидропирановом кольце находятся на противоположных сторонах тетрагидропиранового кольца).
Вариант осуществления соединения формулы I или II или его фармацевтически приемлемой соли по настоящему изобретению представляет собой соединение формулы III:

или его фармацевтически приемлемую соль, где R3 представляет собой 5-7-членный гетероциклил, необязательно замещенный 1 R5 (и R5 имеет значение, как определено в любом из вариантов осуществления, описанных в настоящем документе).
Другой вариант осуществления изобретения включает соединение любой из формул I, II и III или его фармацевтически приемлемую соль, где 4-8-членный гетероциклил или 5-7-членный гетероциклил R3 включает один образующий кольцо гетероатом, выбранный из азота или кислорода, и где гетероциклил необязательно замещен 1 R5, и где R5 выбран из группы, состоящей из F, OH, -CN, C1-C2 алкила, C1-C2 фторалкила, C1-C2 алкокси, C1-C2 фторалкокси, C1-C2 алкокси-C1-C2 алкил-, C1-C2 фторалкокси-C1-C2 алкил-, C1-C2 алкокси-C1-C2 фторалкил- и C1-C2 фторалкокси-C1-C2 фторалкил-.
Другой вариант осуществления изобретения включает соединение любой из формул I, II и III, или его фармацевтически приемлемую соль, где 4-8-членный гетероциклил или 5-7-членный гетероциклил R3 выбран из группы, состоящей из азаспиро[3.3]гептанила, тетрагидрофуранила, пирролидинила и пиперидинила, где каждый из выбранных гетероциклилов необязательно замещен 1 R5, и где R5 выбран из группы, состоящей из F, OH, -CN, C1-C2 алкила, C1-C2 фторалкила, C1-C2 алкокси, C1-C2 фторалкокси, C1-C2 алкокси-C1-C2 алкил-, C1-C2 фторалкокси-C1-C2 алкил-, C1-C2 алкокси-C1-C2 фторалкил- и C1-C2 фторалкокси-C1-C2 фторалкил-. Специалистам в данной области должно быть понятно, что 4-8-членный гетероциклил или 5-7-членный гетероциклил R3 может быть присоединен к основному молекулярному фрагменту через образующий кольцо углерод или образующий кольцо атом азота гетероциклила. Кроме того, специалистам в данной области должно быть понятно, что заместитель R5 может присоединяться к образующему кольцо атому углерода или образующему кольцо атому азота гетероциклила.
Другой вариант осуществления изобретения включает соединение любой из формулы I, II или III, или его фармацевтически приемлемую соль, где 4-8-членный гетероциклил или 5-7-членный гетероциклил R3 выбран из группы, состоящей из 2-азаспиро[3.3]гептан-6-ила, пирролидин-3-ила, пиперидин-4-ила и тетрагидрофуран-3-ила, каждый необязательно замещен 1 R5, и R5 выбран из группы, состоящей из F, OH, -CN, C1-C2 алкила, C1-C2 фторалкила, C1-C2 алкокси, C1-C2 фторалкокси, C1-C2 алкокси-C1-C2 алкил-, C1-C2 фторалкокси-C1-C2 алкил-, C1-C2 алкокси-C1-C2 фторалкил- и C1-C2 фторалкокси-C1-C2 фторалкил-. В еще одном варианте осуществления 4-8-членный гетероциклил или 5-7-членный гетероциклил R3 выбран из группы, состоящей из 2-азаспиро[3.3]гептан-6-ила, пирролидин-3-ила, 1-метилпирролидин-3-ила, 2-(2-метоксиэтил)-2-азаспиро[3.3]гептан-6-ила, 2-(2,2-дифторэтил)-2-азаспиро[3.3]гептан-6-ила и пиперидин-4-ила.
Другой вариант осуществления изобретения включает соединение любой из формулы I, II или III, или его фармацевтически приемлемую соль, где R3 представляет собой 2-азаспиро[3.3]гептан-6-ил, необязательно замещенный 1 R5, который выбран из группы, состоящей из C1-C2 алкила, C1-C2 фторалкила, C1-C2 алкокси-C1-C2 алкил- и C1-C2 фторалкокси-C1-C2 алкил-.
Вариант осуществления изобретения представляет собой соединение формулы I или его фармацевтически приемлемую соль, где A представляет собой N, и R3 представляет собой 4-8-членный гетероциклил, необязательно замещенный 1 R5 (и R5 имеет значение, как определено в любом из вариантов осуществления, описанных в настоящем документе). В другом варианте осуществления соединение или его фармацевтически приемлемая соль представляет собой соединение формулы II или III, где R3 представляет собой 5-7-членный гетероциклил, необязательно замещенный 1 R5.
Вариант осуществления изобретения представляет собой соединение формулы I или его фармацевтически приемлемую соль, где A представляет собой CH, и R3 представляет собой 4-8-членный гетероциклил, необязательно замещенный 1 R5 (и R5 имеет значение, как определено в любом из вариантов осуществления, описанных в настоящем документе). В другом варианте осуществления соединение или его фармацевтически приемлемая соль представляет собой соединение формулы II или III, где R3 представляет собой 5-7-членный гетероциклил, необязательно замещенный 1 R5 (и R5 имеет значение, как определено в любом из вариантов осуществления, описанных в настоящем документе).
Вариант осуществления изобретения представляет собой соединение формулы I или его фармацевтически приемлемую соль, где R3 представляет собой C3-C6 циклоалкил, необязательно замещенный 1 или 2 R5, и дополнительно необязательно замещенный 1 -N(R6)2, где каждый из R5 и R6 имеет значение, как определено в любом из вариантов осуществления, описанных в настоящем документе. В другом варианте осуществления, R3 выбран из группы, состоящей из циклопропила, циклобутила, циклопентила, циклопентенила, циклогексанила и бицикло[3.1.0]гексанила, где каждый из выбранных циклоалкилов необязательно замещен 1 или 2 R5, и дополнительно необязательно замещен 1 -N(R6)2; каждый R5 независимо выбран из группы, состоящей из F, OH, -CN, C1-C2 алкила, C1-C2 фторалкила, C1-C2 алкокси, C1-C2 фторалкокси, C1-C2 алкокси-C1-C2 алкил-, C1-C2 фторалкокси-C1-C2 алкил-, C1-C2 алкокси-C1-C2 фторалкила и C1-C2 фторалкокси-C1-C2 фторалкил-; и каждый R6 независимо выбран из группы, состоящей из H и C1-C2 алкила.
Вариант осуществления изобретения представляет собой соединение формулы I или его фармацевтически приемлемую соль, где R3 представляет собой циклобутил или циклопентил, каждый из которых необязательно замещен 1 или 2 R5; и каждый R5 независимо выбран из группы, состоящей из F, OH, C1-C2 алкила, C1-C2 фторалкила, C1-C2 алкокси, C1-C2 фторалкокси, C1-C2 алкокси-C1-C2 алкил-, C1-C2 фторалкокси-C1-C2 алкил-, C1-C2 алкокси-C1-C2 фторалкила и C1-C2 фторалкокси-C1-C2 фторалкил-.
Вариант осуществления соединения формулы I или его фармацевтически приемлемой соли по настоящему изобретению представляет собой соединение формулы II:

или его фармацевтически приемлемую соль, где:
A представляет собой CH или N;
R3 представляет собой C3-C6 циклоалкил, необязательно замещенный 1 или 2 R5, и дополнительно необязательно замещенный 1 -N(R6)2;
каждый R5 независимо выбран из группы, состоящей из F, OH, -CN, C1-C2 алкила, C1-C2 фторалкила, C1-C2 алкокси, C1-C2 фторалкокси, C1-C2 алкокси-C1-C2 алкил-, C1-C2 фторалкокси-C1-C2 алкил-, C1-C2 алкокси-C1-C2 фторалкила и C1-C2 фторалкокси-C1-C2 фторалкил-; и
каждый R6 независимо выбран из группы, состоящей из H и C1-C2 алкила.
В другом варианте осуществления соединения формулы II, где R3 представляет собой C3-C6 циклоалкил, необязательно замещенный 1 или 2 R5, и дополнительно необязательно замещенный 1 -N(R6)2 (где каждый из R5 и R6 имеет значение, как определено в любом из вариантов осуществления, описанных в настоящем документе), и два заместителя в тетрагидропирановом кольце формулы II находятся в транс-положении по отношению друг к другу (т.е. два заместителя в тетрагидропирановом кольце находятся на противоположных сторонах тетрагидропиранового кольца).
Вариант осуществления соединения формулы I или II или его фармацевтически приемлемой соли по настоящему изобретению представляет собой соединение формулы III:
или его фармацевтически приемлемую соль, где R3 представляет собой C3-C6 циклоалкил, необязательно замещенный 1 или 2 R5, и дополнительно необязательно замещенный 1 -N(R6)2 (где каждый из R5 и R6 имеет значение, как определено в любом из вариантов осуществления, описанных в настоящем документе).
Вариант осуществления изобретения включает соединение любой из формул I, II или III, где C3-C8 циклоалкил или C3-C6 циклоалкил R3 выбран из группы, состоящей из циклопропила, циклобутила, циклопентила, циклопентенила, циклогексанила и бицикло[3.1.0]гексанила, где каждый из выбранных циклоалкилов необязательно замещен 1 или 2 R5, и дополнительно необязательно замещен 1 -N(R6)2; каждый R5 независимо выбран из группы, состоящей из F, OH, -CN, C1-C2 алкила, C1-C2 фторалкила, C1-C2 алкокси, C1-C2 фторалкокси, C1-C2 алкокси-C1-C2 алкил-, C1-C2 фторалкокси-C1-C2 алкил-, C1-C2 алкокси-C1-C2 фторалкила и C1-C2 фторалкокси-C1-C2 фторалкил-; и каждый R6 независимо выбран из группы, состоящей из H и C1-C2 алкила.
Вариант осуществления изобретения включает соединение любой из формул I, II и III, где C3-C8 циклоалкил или C3-C6 циклоалкил R3 выбран из группы, состоящей из циклопропила, циклобутила, циклопентила, циклогексила и бицикло[3.1.0]гексанила, где каждый из выбранных циклоалкилов необязательно замещен 1 или 2 R5 и дополнительно необязательно замещен 1 -N(R6)2; каждый R5 независимо выбран из группы, состоящей из F, OH, -CN, C1-C2 алкила, C1-C2 фторалкила, C1-C2 алкокси, C1-C2 фторалкокси, C1-C2 алкокси-C1-C2 алкил-, C1-C2 фторалкокси-C1-C2 алкил-, C1-C2 алкокси-C1-C2 фторалкила и C1-C2 фторалкокси-C1-C2 фторалкил-; и каждый R6 независимо выбран из группы, состоящей из H и C1-C2 алкила. В другом варианте осуществления, C3-C8 циклоалкил или C3-C6 циклоалкил R3 выбран из группы, состоящей из циклопропила, циклобутила, циклопентила, циклогексила и бицикло[3.1.0]гексан-2-ила, где каждый из выбранных циклоалкилов необязательно замещен 1 или 2 R5 и дополнительно необязательно замещен 1 -N(R6)2. В еще одном варианте осуществления, каждый R5 независимо выбран из группы, состоящей из F, OH, C1-C2 алкила, C1-C2 фторалкила, C1-C2 алкокси, C1-C2 фторалкокси, C1-C2 алкокси-C1-C2 алкил-, C1-C2 фторалкокси-C1-C2 алкил-, C1-C2 алкокси-C1-C2 фторалкила и C1-C2 фторалкокси-C1-C2 фторалкил-. В еще одном варианте осуществления каждый R5 независимо выбран из группы, состоящей из F, OH, C1-C2 алкила, C1-C2 фторалкила и C1-C2 алкокси-C1-C2 алкил-. В еще одном варианте осуществления, каждый R5 независимо выбран из группы, состоящей из F, OH, CH3 и CH2CH3; и каждый R6 независимо выбран из группы, состоящей из H и CH3.
Вариант осуществления изобретения включает соединение любой из формул I, II или III, или его фармацевтически приемлемую соль, где R3 представляет собой бицикло[3.1.0]гексан-2-ил, необязательно замещенный 1 или 2 R5; и каждый R5 независимо выбран из группы, состоящей из F, OH, C1-C2 алкила, C1-C2 фторалкила, C1-C2 алкокси, C1-C2 фторалкокси, C1-C2 алкокси-C1-C2 алкил-, C1-C2 фторалкокси-C1-C2 алкил-, C1-C2 алкокси-C1-C2 фторалкила и C1-C2 фторалкокси-C1-C2 фторалкил-.
Вариант осуществления изобретения включает соединение любой из формул I, II или III, или его фармацевтически приемлемую соль, где C3-C8 циклоалкил или C3-C6 циклоалкил R3 представляет собой циклогексил (также известный как циклогексанил), необязательно замещенный 1 или 2 R5, и дополнительно необязательно замещенный 1 -N(R6)2 (где каждый из R5 и R6 имеет значение, как определено в любом из вариантов осуществления, описанных в настоящем документе). В другом варианте осуществления R3 представляет собой циклогексил, замещенный 1 -N(R6)2 (где каждый из R6 имеет значение, как определено в любом из вариантов осуществления, описанных в настоящем документе). В еще одном варианте осуществления, каждый R6 независимо выбран из группы, состоящей из H и CH3.
Вариант осуществления изобретения включает соединение любой из формул I, II или III, или его фармацевтически приемлемую соль, где C3-C8 циклоалкил или C3-C6 циклоалкил R3 представляет собой циклобутил, необязательно замещенный 1 или 2 R5 (где каждый из R5 имеет значение, как определено в любом из вариантов осуществления, описанных в настоящем документе). В другом варианте осуществления, каждый R5 независимо выбран из группы, состоящей из F, OH, C1-C2 алкила, C1-C2 фторалкила, C1-C2 алкокси, C1-C2 фторалкокси, C1-C2 алкокси-C1-C2 алкил-, C1-C2 фторалкокси-C1-C2 алкил-, C1-C2 алкокси-C1-C2 фторалкила и C1-C2 фторалкокси-C1-C2 фторалкил-.
Вариант осуществления изобретения включает соединение любой из формул I, II или III, или его фармацевтически приемлемую соль, где C3-C8 циклоалкил или C3-C6 циклоалкил R3 представляет собой циклопентил, необязательно замещенный 1 или 2 R5, и дополнительно необязательно замещенный 1 -N(R6)2 (где каждый из R5 и R6 имеет значение, как определено в любом из вариантов осуществления, описанных в настоящем документе). В другом варианте осуществления R3 представляет собой циклопентил, необязательно замещенный 1 или 2 R5 (где каждый из R5 имеет значение, как определено в любом из вариантов осуществления, описанных в настоящем документе). В еще одном варианте осуществления R3 представляет собой циклопентил, необязательно замещенный 1 или 2 R5; и каждый R5 независимо выбран из группы, состоящей из F, OH, C1-C2 алкила, C1-C2 фторалкила, C1-C2 алкокси, C1-C2 фторалкокси, C1-C2 алкокси-C1-C2 алкил-, C1-C2 фторалкокси-C1-C2 алкил-, C1-C2 алкокси-C1-C2 фторалкила и C1-C2 фторалкокси-C1-C2 фторалкил-. В еще одном варианте осуществления R3 представляет собой циклопентил.
Вариант осуществления изобретения включает соединение любой из формул I, II или III, или его фармацевтически приемлемую соль, где A представляет собой CH (и другие переменные, такие как R1, R2, R3, R4, если они присутствуют, имеют значения, указанные в любом из вариантов осуществления, описанных в настоящем документе).
Вариант осуществления изобретения включает соединение любой из формул I, II или III, или его фармацевтически приемлемую соль, где A представляет собой N (и другие переменные, такие как R1, R2, R3, R4, если они присутствуют, имеют значения, указанные в любом из вариантов осуществления, описанных в настоящем документе).
Вариант осуществления изобретения представляет собой соединение формулы I, II или III, где R3 представляет собой (5-7-членный гетероциклил)-C1-C4 алкил- или (C3-C6 циклоалкил)-C1-C4 алкил-, где каждый из 5-7-членного гетероциклильного фрагмента в (5-7-членный гетероциклил)-C1-C4 алкил- необязательно замещен 1 или 2 R5, и где C3-C6 циклоалкильный фрагмент в (C3-C6 циклоалкил)-C1-C4 алкил- необязательно замещен 1 или 2 R5 и дополнительно необязательно замещен 1 -N(R6)2 (где каждый из R5 и R6 имеет значение, как определено в любом из вариантов осуществления, описанных в настоящем документе). Некоторые примеры 5-7-членного гетероциклильного фрагмента включают азаспиро[3.3]гептанил, тетрагидрофуранил, пирролидинил или пиперидинил, каждый из которых необязательно замещен 1 R5, и R5 выбран из группы, состоящей из F, OH, -CN, C1-C2 алкила, C1-C2 фторалкила, C1-C2 алкокси, C1-C2 фторалкокси, C1-C2 алкокси-C1-C2 алкил-, C1-C2 фторалкокси-C1-C2 алкил-, C1-C2 алкокси-C1-C2 фторалкил- и C1-C2 фторалкокси-C1-C2 фторалкил-. Некоторые примеры C3-C6 циклоалкильного фрагмента включают циклопропил, циклобутил, циклопентил, циклогексанил или бицикло[3.1.0]гексанил, каждый из которых необязательно замещен 1 или 2 R5 и дополнительно необязательно замещен 1 -N(R6)2; где каждый R5 независимо выбран из группы, состоящей из F, OH, CH3 и CH2CH3; и каждый R6 независимо выбран из H и CH3.
Вариант осуществления изобретения включает соединение формулы II или III или его фармацевтически приемлемую соль, где A представляет собой N; R3 представляет собой циклобутил, циклопентил или бицикло[3.1.0]гексанил, каждый необязательно замещен 1 или 2 R5; и каждый R5 независимо выбран из группы, состоящей из F, OH, CH3 или CH2CH3. В другом варианте осуществления, R3 представляет собой циклобутил, циклопентил или бицикло[3.1.0]гексан-2-ил, каждый необязательно замещен 1 или 2 R5; и каждый R5 независимо выбран из группы, состоящей из F и CH3. В еще одном варианте осуществления соединение или его фармацевтически приемлемая соль представляет собой соединение формулы III или его фармацевтически приемлемую соль.
Вариант осуществления изобретения включает соединение формулы II или III или его фармацевтически приемлемую соль, где A представляет собой CH; R3 представляет собой циклобутил, циклопентил или бицикло[3.1.0]гексанил, каждый необязательно замещен 1 или 2 R5; и каждый R5 независимо выбран из группы, состоящей из F, OH, CH3 или CH2CH3. В другом варианте осуществления, R3 представляет собой циклобутил, циклопентил или бицикло[3.1.0]гексан-2-ил, каждый необязательно замещен 1 или 2 R5; и каждый R5 независимо выбран из группы, состоящей из F и CH3. В еще одном варианте осуществления соединение или его фармацевтически приемлемая соль представляет собой соединение формулы III или его фармацевтически приемлемую соль.
Вариант осуществления изобретения включает соединение формулы II или III или его фармацевтически приемлемую соль, где A представляет собой N; R3 представляет собой циклопентил, необязательно замещенный 1 или 2 R5; и каждый R5 независимо выбран из группы, состоящей из F, OH, CH3 или CH2CH3. В другом варианте осуществления R3 представляет собой циклопентил, необязательно замещенный 1 или 2 R5; и каждый R5 независимо выбран из группы, состоящей из F и CH3. В еще одном варианте осуществления соединение или его фармацевтически приемлемая соль представляет собой соединение формулы III или его фармацевтически приемлемую соль.
Вариант осуществления изобретения включает соединение формулы II или III или его фармацевтически приемлемую соль, где A представляет собой CH; R3 представляет собой циклопентил, необязательно замещенный 1 или 2 R5; и каждый R5 независимо выбран из группы, состоящей из F, OH, CH3 или CH2CH3. В другом варианте осуществления R3 представляет собой циклопентил, необязательно замещенный 1 или 2 R5; и каждый R5 независимо выбран из группы, состоящей из F и CH3. В еще одном варианте осуществления соединение или его фармацевтически приемлемая соль представляет собой соединение формулы III или его фармацевтически приемлемую соль.
Вариант осуществления изобретения включает соединение формулы II или III или его фармацевтически приемлемую соль, где A представляет собой N; R3 представляет собой пирролидинил, пиперидинил или азаспиро[3.3]гептанил, каждый необязательно замещен 1 R5, и где R5 выбран из группы, состоящей из C1-C2 алкила, C1-C2 фторалкила, C1-C2 алкокси, C1-C2 фторалкокси, C1-C2 алкокси-C1-C2 алкил-, C1-C2 фторалкокси-C1-C2 алкил-, C1-C2 алкокси-C1-C2 фторалкил- и C1-C2 фторалкокси-C1-C2 фторалкил-. В другом варианте осуществления R3 представляет собой пирролидин-3-ил, пиперидин-4-ил или 2-азаспиро[3.3]гептан-6-ил, каждый необязательно замещен 1 R5, и где R5 выбран из группы, состоящей из C1-C2 алкила, C1-C2 фторалкила, C1-C2 алкокси-C1-C2 алкил- и C1-C2 фторалкокси-C1-C2 алкил-. В еще одном варианте осуществления R3 представляет собой 2-азаспиро[3.3]гептан-6-ил, необязательно замещенный 1 R5, который выбран из группы, состоящей из C1-C2 алкила, C1-C2 фторалкила, C1-C2 алкокси-C1-C2 алкил- и C1-C2 фторалкокси-C1-C2 алкил-. В еще одном варианте осуществления соединение или его фармацевтически приемлемая соль представляет собой соединение формулы III или его фармацевтически приемлемую соль.
Вариант осуществления изобретения включает соединение формулы II или III или его фармацевтически приемлемую соль, где A представляет собой CH; R3 представляет собой пирролидинил, пиперидинил или азаспиро[3.3]гептанил, каждый необязательно замещен 1 R5, и где R5 выбран из группы, состоящей из C1-C2 алкила, C1-C2 фторалкила, C1-C2 алкокси, C1-C2 фторалкокси, C1-C2 алкокси-C1-C2 алкил-, C1-C2 фторалкокси-C1-C2 алкил-, C1-C2 алкокси-C1-C2 фторалкил- и C1-C2 фторалкокси-C1-C2 фторалкил-. В другом варианте осуществления R3 представляет собой пирролидин-3-ил, пиперидин-4-ил или 2-азаспиро[3.3]гептан-6-ил, каждый необязательно замещен 1 R5, и где R5 выбран из группы, состоящей из C1-C2 алкила, C1-C2 фторалкила, C1-C2 алкокси-C1-C2 алкил- и C1-C2 фторалкокси-C1-C2 алкил-. В еще одном варианте осуществления R3 представляет собой 2-азаспиро[3.3]гептан-6-ил, необязательно замещенный 1 R5, который выбран из группы, состоящей из C1-C2 алкила, C1-C2 фторалкила, C1-C2 алкокси-C1-C2 алкил- и C1-C2 фторалкокси-C1-C2 алкил-. В еще одном варианте осуществления соединение или его фармацевтически приемлемая соль представляет собой соединение формулы III или его фармацевтически приемлемую соль.
В одном варианте осуществления настоящее изобретение предоставляет соединение, выбранное из примеров 1-34 в разделе «ПРИМЕРЫ», или его фармацевтически приемлемую соль (или его исходное/основное соединение, где иллюстративное соединение, например, представляет собой соль), приведенную ниже.
Один вариант осуществления включает соединение, выбранное из:
6-ацетил-8-циклобутил-2-{[3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она;
6-ацетил-8-циклопентил-2-{[3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она;
6-ацетил-8-(2-азаспиро[3.3]гептан-6-ил)-2-{[3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она;
6-ацетил-2-{[3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метил-8-[2-метилциклопентил]пиридо[2,3-d]пиримидин-7(8H)-она;
3-ацетил-1-циклопентил-7-{[3-гидрокситетрагидро-2H-пиран-4-ил]амино}-4-метил-1,6-нафтиридин-2(1H)-она;
6-ацетил-8-[бицикло[3.1.0]гексан-2-ил]-2-{[3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она;
6-ацетил-8-[2-фтор-2-метилциклопентил]-2-{[3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она; и
6-ацетил-8-[2-(2,2-дифторэтил)-2-азаспиро[3.3]гептан-6-ил]-2-{[3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она,
или его фармацевтически приемлемую соль.
Один вариант осуществления включает соединение, выбранное из:
6-ацетил-8-циклобутил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она;
6-ацетил-8-циклопентил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она;
6-ацетил-8-(2-азаспиро[3.3]гептан-6-ил)-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она;
6-ацетил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метил-8-[(1R,2S)-2-метилциклопентил]пиридо[2,3-d]пиримидин-7(8H)-она;
3-ацетил-1-циклопентил-7-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-4-метил-1,6-нафтиридин-2(1H)-она;
6-ацетил-8-[(1S,2R,5R)-бицикло[3.1.0]гексан-2-ил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она;
6-ацетил-8-[(1R,2S,5S)-бицикло[3.1.0]гексан-2-ил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она;
6-ацетил-8-[(1R,2S)-2-фтор-2-метилциклопентил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она;
6-ацетил-8-[(1S,2R)-2-фтор-2-метилциклопентил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она; и
6-ацетил-8-[2-(2,2-дифторэтил)-2-азаспиро[3.3]гептан-6-ил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она,
или его фармацевтически приемлемую соль.
Один вариант осуществления включает соединение, выбранное из:
6-ацетил-8-циклобутил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она;
6-ацетил-8-циклопентил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она;
6-ацетил-8-(2-азаспиро[3.3]гептан-6-ил)-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она;
6-ацетил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метил-8-[(1R,2S)-2-метилциклопентил]пиридо[2,3-d]пиримидин-7(8H)-она;
3-ацетил-1-циклопентил-7-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-4-метил-1,6-нафтиридин-2(1H)-она;
6-ацетил-8-[(1S,2R,5R)-бицикло[3.1.0]гексан-2-ил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она, или 6-ацетил-8-[(1R,2S,5S)-бицикло[3.1.0]гексан-2-ил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она, или их смеси;
6-ацетил-8-[(1R,2S)-2-фтор-2-метилциклопентил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она, или 6-ацетил-8-[(1S,2R)-2-фтор-2-метилциклопентил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она, или их смеси; и
6-ацетил-8-[2-(2,2-дифторэтил)-2-азаспиро[3.3]гептан-6-ил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она,
или его фармацевтически приемлемую соль.
Один вариант осуществления включает соединение, выбранное из:
6-ацетил-8-циклобутил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она;
6-ацетил-8-циклопентил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она;
6-ацетил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метил-8-[(1R,2S)-2-метилциклопентил]пиридо[2,3-d]пиримидин-7(8H)-она;
6-ацетил-8-[(1S,2S)-2-гидрокси-2-метилциклопентил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она или 6-ацетил-8-[(1R,2R)-2-гидрокси-2-метилциклопентил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она, или их смеси;
6-ацетил-8-[(1R,2S)-2-этилциклопентил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она;
6-ацетил-8-[(1S,2R,5R)-бицикло[3.1.0]гексан-2-ил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она или 6-ацетил-8-[(1R,2S,5S)-бицикло[3.1.0]гексан-2-ил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она, или их смеси;
6-ацетил-8-[(1R,2S)-2-фтор-2-метилциклопентил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она или 6-ацетил-8-[(1S,2R)-2-фтор-2-метилциклопентил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она, или их смеси;
6-ацетил-8-(3,3-диметилциклобутил)-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она;
3-ацетил-1-(3-гидроксициклопентил)-7-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-4-метил-1,6-нафтиридин-2(1H)-она, включая разделенные диастереомеры или в виде смеси;
6-ацетил-8-(цис-4-гидроксициклогексил)-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она; и
6-ацетил-8-(транс-3-гидроксициклобутил)-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она,
или его фармацевтически приемлемую соль.
В некоторых вариантах осуществления настоящее изобретение предоставляет кристаллическую форму 3-ацетил-1-циклопентил-7-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-4-метил-1,6-нафтиридин-2(1H)-она (например, кристаллическую форму как в Примере 5). В некоторых дополнительных вариантах осуществления кристаллическая форма 3-ацетил-1-циклопентил-7-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-4-метил-1,6-нафтиридин-2(1H)-она является безводной. В некоторых дополнительных вариантах осуществления кристаллическая форма безводного (ангидрата) 3-ацетил-1-циклопентил-7-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-4-метил-1,6-нафтиридин-2(1H)-она обозначена как «форма I», которая характеризуется своими уникальными характеристиками твердого состояния в соответствии с, например, порошковой рентгеновской дифракцией (PXRD), описанной в настоящем документе (например, по существу, как показано на фиг. 1). Список дифракционных пиков, выраженных в виде градусов 2θ и относительных интенсивностей с относительной интенсивностью ≥ 3,0% (как показано на Фиг. 1), представлен в Таблице X1 в настоящем документе. Как хорошо известно в области порошковой дифракции, относительные интенсивности пиков (отражений) могут варьироваться в зависимости от методики получения образца, процедуры установки образца и конкретного используемого инструмента. Кроме того, вариации инструмента и другие факторы могут повлиять на значения 2-тета. Следовательно, значения пиков XRPD могут варьироваться на плюс или минус примерно 0,2°.
В некоторых вариантах осуществления форма I демонстрирует порошковую рентгеновскую дифрактограмму, содержащую по меньшей мере два характеристических пика в единицах 2θ, выбранных из 8,0±0,2°; 18,6±0,2°; 19,1±0,2°; и 21,3±0,2°. В некоторых вариантах осуществления форма I демонстрирует порошковую рентгеновскую дифрактограмму, содержащую по меньшей мере три характеристических пика в единицах 2θ, выбранных из 8,0±0,2°; 18,6±0,2°; 19,1±0,2°; и 21,3±0,2°. В некоторых вариантах осуществления форма I демонстрирует порошковую рентгеновскую дифрактограмму, содержащую все четыре характеристических пика в единицах 2θ, выбранных из 8,0±0,2°; 18,6±0,2°; 19,1±0,2°; и 21,3±0,2°.
В некоторых вариантах осуществления форма I демонстрирует порошковую рентгеновскую дифрактограмму, содержащую характеристические пики в единицах 2θ, при 8,0±0,2° и 18,6±0,2°.
В некоторых вариантах осуществления форма I демонстрирует порошковую рентгеновскую дифрактограмму, содержащую характеристические пики в единицах 2θ, при 8,0±0,2° и 19,1±0,2°.
В некоторых вариантах осуществления форма I демонстрирует порошковую рентгеновскую дифрактограмму, содержащую характеристические пики в единицах 2θ, при 18,6±0,2° и 19,1±0,2°.
В некоторых вариантах осуществления форма I демонстрирует порошковую рентгеновскую дифрактограмму, содержащую все три характеристических пика в единицах 2θ, выбранных из 8,0±0,2°; 18,6±0,2°; и 19,1±0,2°.
В некоторых вариантах осуществления, форма I демонстрирует порошковую рентгеновскую дифрактограмму, содержащую по меньшей мере два характеристических пика в единицах 2θ, которые перечислены в Таблице X1.
В некоторых вариантах осуществления форма I демонстрирует порошковую рентгеновскую дифрактограмму, содержащую по меньшей мере три характеристических пика в единицах 2θ, которые перечислены в Таблице X1.
В некоторых вариантах осуществления форма I демонстрирует порошковую рентгеновскую дифрактограмму, содержащую по меньшей мере четыре (например, 4, 5, 6, 7, 8, 9 или 10) характеристических пика в единицах 2θ, которые перечислены в Таблице X1.
В некоторых вариантах осуществления, форма I демонстрирует порошковую рентгеновскую дифрактограмму, содержащую по меньшей мере два характеристических пика в единицах 2θ, выбранных из 8,0±0,2°; 18,6±0,2°; 19,1±0,2°; и 21,3±0,2°; и по меньшей мере один дополнительный характеристический пик в единицах 2θ, которые перечислены в Таблице X1 (например, пик при 23,3).
В некоторых вариантах осуществления форма I демонстрирует порошковую рентгеновскую дифрактограмму по существу такую, как показано на фиг. 1.
Настоящее изобретение включает любое подмножество любого варианта осуществления, описанного в настоящем документе.
Настоящее изобретение включает комбинации двух или более вариантов осуществления, описанных выше, или любое их подмножество.
В другом варианте осуществления изобретение предоставляет фармацевтическую композицию, включающую соединение по изобретению, описанное в настоящем документе, и по меньшей мере один фармацевтически приемлемый эксципиент или носитель.
Настоящее изобретение также относится к применению соединения по изобретению, описанного в настоящем документе, для лечения заболевания или расстройства, описанного в настоящем документе (т.е. способ лечения).
Соединение по настоящему изобретению представляет собой ингибитор CDK (например, ингибитор CDK4 и/или CDK6). Таким образом, настоящее изобретение дополнительно предоставляет способ ингибирования CDK (т.е. активности CDK либо in vitro, либо in vivo), включающий контактирование (включая инкубацию) CDK с соединением изобретения, описанным в настоящем документе.
Как используется в настоящем документе, соединения изобретения в фармацевтических композициях (включая лекарственные формы), применениях, способах и/или наборах по настоящему изобретению, описанных в настоящем документе, включают соединения формулы I или их фармацевтически приемлемые соли (включая соединение формулы II или III или его фармацевтически приемлемую соль, и включая все варианты осуществления и комбинации двух или более вариантов осуществления, описанных в настоящем документе, или любую их подкомбинацию, например, любое из соединений, выбранных из примеров 1-34, или его фармацевтически приемлемую соль).
Используемый в настоящем описании термин «контактирование» относится к объединению указанных фрагментов в системе in vitro или системе in vivo. Например, «контактирование» CDK с соединением по изобретению включает введение соединения по настоящему изобретению индивиду или пациенту, такому как человек, имеющему CDK, а также, например, введение соединения по изобретению в образец, содержащий клеточный или очищенный препарат, содержащий CDK.
Настоящее изобретение дополнительно предоставляет способ лечения заболевания или расстройства у пациента, который включает введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения или фармацевтически приемлемой соли по изобретению, где заболевание или расстройство выбрано из группы, состоящей из легочной гипертензии, легочной артериальной гипертензии, легочной гипертензии с поражением левых отделов сердца, легочной гипертензии с заболеванием легких и/или гипоксемией, легочной гипертензии вследствие хронического тромботического и/или эмболического заболевания, и заболевания, связанного с легочной гипертензией, включая саркоидоз, гистиоцитоз X, лимфангиоматоз и компрессию легочных сосудов.
Настоящее изобретение дополнительно предоставляет способ лечения легочной гипертензии у пациента, который включает введение пациенту, нуждающемуся в этом, эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли. В некоторых вариантах осуществления вводимое соединение находится в форме фармацевтически приемлемой соли. В других вариантах осуществления вводимое соединение не находится в форме фармацевтически приемлемой соли (например, вводят основное/исходное соединение).
При обсуждении лечения легочной гипертензии любое одно или несколько из этих родственных заболеваний включены в это лечение, поскольку они подпадают под категорию легочной гипертензии ВОЗ: группа 1: легочная артериальная гипертензия (PAH); группа 2: PH с поражением левых отделов сердца; группа 3: PH с заболеванием легких и/или гипоксемией; группа 4: PH вследствие хронического тромботического и/или эмболического заболевания; и группа 5: смешанные состояния (например, саркоидоз, гистиоцитоз X, лимфангиоматоз и компрессия легочных сосудов).
Другой вариант осуществления изобретения включает способ лечения легочной артериальной гипертензии у пациента, который включает введение патенту, нуждающемуся в этом, эффективного количества соединения или фармацевтически приемлемой соли изобретения.
В одном из вариантов осуществления настоящее изобретение дополнительно предлагает способ лечения аномального роста клеток у субъекта, который включает введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения или фармацевтически приемлемой соли по настоящему изобретению. В другом варианте осуществления, аномальный рост клеток представляет собой рак. Соединения или соли по изобретению можно вводить в виде одного средства или можно вводить в комбинации с одним или несколькими другими противораковыми терапевтическими средствами, в частности, средствами для стандартного лечения, подходящими для конкретного типа рака. В еще одном варианте осуществления, рак выбран из рака молочной железы, рака яичников, рака мочевого пузыря, рака матки, рака предстательной железы, рака легкого [включая немелкоклеточный рак легкого (NSCLC), мелкоклеточный рак легкого (SCLC), плоскоклеточный рак или аденокарциному], рака пищевода, рака головы и шеи, колоректального рака, рака почки (включая почечно-клеточную карциному или RCC), рака печени (включая гепатоцеллюлярную карциному или HCC), рака поджелудочной железы, рака желудка, и рака щитовидной железы. В дополнительных вариантах осуществления способов, представленных в настоящем документе, рак представляет собой рак молочной железы, рак яичников, рак мочевого пузыря, рак матки, рак предстательной железы, рак легкого, рак пищевода, рак печени, рак поджелудочной железы или рак желудка.
В одном из вариантов осуществления настоящее изобретение дополнительно предоставляет способ лечения рака у субъекта, который включает введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения или фармацевтически приемлемой соли по настоящему изобретению. В другом варианте осуществления, рак представляет собой рак молочной железы, включая, например, ER-положительный/HR-положительный, HER2-негативный рак молочной железы; ER-положительный/HR-положительный, HER2-положительный рак молочной железы; трижды негативный рак молочной железы (TNBC); или воспалительный рак молочной железы. В другом варианте осуществления рак молочной железы представляет собой эндокринно-устойчивый рак молочной железы, устойчивый к трастузумабу рак молочной железы или рак молочной железы, демонстрирующий первичную или приобретенную резистентность к ингибированию CDK4/CDK6. В еще одном варианте осуществления рак молочной железы представляет собой распространенный или метастатический рак молочной железы.
В дополнительном аспекте изобретение предоставляет способ лечения аномального роста клеток, в частности рака, у субъекта, нуждающегося в этом, который включает введение субъекту количества соединения по изобретению в комбинации с количеством дополнительного противоракового терапевтического средства, которое в совокупности эффективно при лечении указанного аномального роста клеток.
Настоящее изобретение, кроме того, предоставляет применение соединения по настоящему изобретению для лечения заболевания или расстройства, описанного в настоящем документе (например, тех, которые описаны в способе лечения в настоящем документе).
Настоящее изобретение, кроме того, предоставляет применение соединения настоящего изобретения в получении лекарственного средства для лечения заболевания или расстройства, описанного в настоящем документе (например, тех, которые описаны в способе лечения в настоящем документе).
Изобретение также включает следующие варианты осуществления:
соединения по настоящему изобретению, включая любой из вариантов осуществления, описанных в настоящем документе, для применения в качестве лекарственного средства;
соединения по настоящему изобретению, включая любой из вариантов осуществления, описанных в настоящем документе, для применения в получении лекарственного средства;
применения соединения по настоящему изобретению, включая любой из вариантов осуществления, описанных в настоящем документе, для лечения аномального роста клеток, такого как рак;
применения соединения по настоящему изобретению, включая любой из вариантов осуществления, описанных в настоящем документе, в получении лекарственного средства для лечения аномального роста клеток, такого как рак;
применения соединения по настоящему изобретению, включая любой из вариантов осуществления, описанных в настоящем документе, в получении лекарственного средства для лечения легочной артериальной гипертензии;
применения соединения по настоящему изобретению, включая любой из вариантов осуществления, описанных в настоящем документе, в получении лекарственного средства для лечения легочной гипертензии (PH) или любого из заболеваний, входящих в категорию PH по классификации ВОЗ;
соединения по настоящему изобретению, включая любой из вариантов осуществления, описанных в настоящем документе, для применения при лечении аномального роста клеток, такого как рак;
соединения по настоящему изобретению, включая любой из вариантов осуществления, описанных в настоящем документе, для применения при лечении легочной артериальной гипертензии; и
соединения по настоящему изобретению, включая любой из вариантов осуществления, описанных в настоящем документе, для применения при лечении легочной гипертензии (PH) или любого из заболеваний, входящих в категорию PH по классификации ВОЗ.
Каждый пример или его фармацевтически приемлемая соль могут быть заявлены индивидуально или сгруппированы вместе в любой комбинации с любым количеством каждого и каждого варианта осуществления, описанного в настоящем документе.
Изобретение также относится к фармацевтической композиции, включающей соединение по настоящему изобретению, включая любой из вариантов осуществления, описанных в настоящем документе, для применения при лечении заболевания или расстройства, описанного в настоящем документе, например, выбранного из: легочной гипертензии, легочной артериальной гипертензии, легочной гипертензии с поражением левых отделов сердца, легочной гипертензии с заболеванием легких и/или гипоксемией, легочной гипертензии вследствие хронического тромботического и/или эмболического заболевания, и заболевания, связанного с легочной гипертензией, включая саркоидоз, гистиоцитоз X, лимфангиоматоз и компрессию легочных сосудов.
В другом варианте осуществления изобретения соединение по настоящему изобретению вводят субъекту с диагнозом или имеющему риск развития легочной гипертензии или PAH, включая, но не ограничиваясь ими, идиопатическую PAH, наследственную или семейную PAH и вторичную легочную гипертензию (например, гипертензия в результате тромбоэмболии легочной артерии, эмфиземы, фиброза легких и врожденного порока сердца). В одном варианте осуществления у субъекта диагностирована идиопатическая PAH или наследственная PAH. В некоторых вариантах осуществления субъект с риском развития PAH имеет мутацию в гене, кодирующем рецептор костного морфогенетического белка типа 2.
Настоящее изобретение также включает набор для лечения или профилактики легочной артериальной гипертензии. В одном варианте осуществления набор включает соединение по настоящему изобретению и устройство для доставки. Устройство для доставки может быть разработано для доставки в легкие, например ингаляторы, небулайзеры, инсуффляторы, капельницы и устройства для образования аэрозоля. В других вариантах осуществления устройство для доставки может быть разработано для внутривенной или внутриартериальной доставки, например катетер. Соединение или фармацевтически приемлемая соль по настоящему изобретению может быть необязательно сформулирована для хранения в устройстве для доставки. Набор может дополнительно включать инструкции по введению соединения или фармацевтически приемлемой соли по настоящему изобретению субъекту (например, человеку) для лечения или профилактики PAH или других заболеваний, описанных в настоящем документе. В одном варианте осуществления в инструкциях подробно описывается и уточняется способ введения, например, путем указания дней введения соединения или фармацевтически приемлемой соли по настоящему изобретению в течение 28-дневного цикла.
Изобретение включает способ лечения расстройства, описанного в настоящем документе (например, PAH или рака), где способ включает применение соединения по настоящему изобретению либо в виде монотерапии, либо в комбинированной терапии, при которой субъекту или пациенту, нуждающемуся в лечении, вводят соединение или соль по настоящему изобретению в комбинации с одним или несколькими лекарственными средствами (также называемыми активным средством), одобренными для лечения заболевания или расстройства, описанного в настоящем документе (например, PAH или рака). Например, дополнительное активное средство может включать, но не ограничивается этим, простагландин (например, эпопростенол, трепростинил, илопрост, селексипаг), антагонист рецепторов эндотелина (например, бозентан, амбрисентан, мацитентан), ингибитор гуанилатциклазы (например, риоцигуат), вазодилататоры (например, простациклин и силденафил), блокаторы кальциевых каналов (например, амлодипин, дилтиазем и нифедипин); антикоагулянты (например, варфарин) и диуретики. Ссылка на лекарственное средство, например силденафил, включает силденафил и все фармацевтически приемлемые соли, например, силденафила цитрат. В другом примере дополнительное активное средство может включать противораковое средство, такое как одно из средств для стандартного лечения, подходящих для лечения рака.
В другом аспекте соединение по настоящему изобретению применяют в получении лекарственного средства для лечения или профилактики PAH. Еще в другом аспекте соединение по настоящему изобретению применяют в получении лекарственного средства для лечения или профилактики связанных заболеваний, как обсуждается в настоящем документе. В различных вариантах осуществления лекарственное средство получено для перорального введения, включая лекарственные формы как с немедленным высвобождением, так и с замедленным (модифицированным) высвобождением. В других вариантах осуществления лекарственное средство сформулировано для введения путем ингаляции. Во всех этих вариантах осуществления изобретение предоставляет формы стандартных доз лекарственного средства.
Изобретение также предоставляет комбинацию, которая включает соединение по настоящему изобретению и другое терапевтическое средство. Такую комбинацию можно использовать в применениях и способах изобретения, описанных в настоящем документе.
Настоящее изобретение также включает изотопно-меченные соединения, которые идентичны соединению по настоящему изобретению (например, соединение формулы I, II или III, или его фармацевтически приемлемая соль), за исключением того факта, что один или несколько атомов замещены атомами, имеющими тот же самый атомный номер, но атомную массу или массовое число, отличающиеся от атомной массы или массового числа, обычно встречающегося в природе. Примеры изотопов, которые могут быть включены в соединения по настоящему изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора и хлора, такие как 2H, 3H, 13C, 11C, 14C, 15N, 18O, 17O, 31P, 32P, 35S, 18F и 36Cl, соответственно. Соединения по настоящему изобретению и фармацевтически приемлемые соли указанных соединений, которые содержат вышеупомянутые изотопы и/или другие изотопы других атомов, входят в объем настоящего изобретения. Некоторые изотопно-меченные соединения по настоящему изобретению, например, соединения, которые включают радиоактивные изотопы, такие как 3H и 14C, полезны в анализах распределения лекарственных средств и/или субстратов в тканях. Изотопы, меченные тритием, т.е. 3H, и углерод-14, т.е. 14C, особенно предпочтительны из-за простоты их получения и обнаружения. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий, т.е. 2H, может дать определенные терапевтические преимущества, обусловленные большей метаболической стабильностью, например, увеличенного периода полувыведения in vivo или снижение требуемых дозировок, и, следовательно, может быть предпочтительным в некоторых обстоятельствах. Изотопно-меченные соединения формулы I, II или III, или их фармацевтически приемлемые соли, обычно можно получить путем проведения процедур, описанных в схемах и/или в примерах и получениях, ниже, путем замены немеченного изотопами реагента легко доступным изотопно-меченным реагентом.
Соединения по настоящему изобретению могут демонстрировать явления таутомерии и структурной изомерии. Например, соединения по изобретению могут существовать в нескольких таутомерных формах. Все такие таутомерные формы включены в объем соединений изобретения. Таутомеры могут существовать в виде смесей таутомеров в растворе. В твердой форме обычно преобладает один таутомер. Несмотря на то, что может быть описан один таутомер, настоящее изобретение включает все таутомеры соединений по изобретению.
Изобретение также относится к пролекарствам соединений настоящего изобретения. Таким образом, некоторые производные соединений по настоящему изобретению, которые сами по себе могут иметь небольшую фармакологическую активность или не иметь фармакологическую активность, при введении пациенту могут быть превращены в соединения по настоящему изобретению, например, путем гидролитического расщепления. Такие производные называются «пролекарствами». Дополнительную информацию о применении пролекарств можно найти в ‘Pro-drugs as Novel Delivery Systems, Vol. 14, ACS Symposium Series (T Higuchi and W Stella) и ‘Bioreversible Carriers in Drug Design’, Pergamon Press, 1987 (ed. E B Roche, American Pharmaceutical Association), описания которых полностью включены в настоящий документ посредством ссылки.
Пролекарства в соответствии с изобретением могут, например, быть получены путем замены соответствующих функциональных групп, присутствующих в соединениях по изобретению, определенными фрагментами, известными специалистам в данной области как «профрагменты», как описано, например, в "Design of Prodrugs" by H Bundgaard (Elsevier, 1985), описание которого полностью включено в настоящий документ посредством ссылки.
Некоторые неограничивающие примеры пролекарств в соответствии с изобретением включают:
(i) где соединение содержит функциональную группу спирта (-OH), его простой эфир, например, замещение водорода (C1-C6)алканоилоксиметилом или группой простого фосфатного эфира; и
(ii) где соединение содержит первичную или вторичную аминофункциональную группу (-NH2 или -NHR, где R ≠ H), ее амид, например, защение одного или обоих атомов водорода метаболически лабильной группой, такой как амид, карбамат, мочевина, фосфонат, сульфонат и т.п.
Термин «субъект» или «пациент», используемый в настоящем описании, относится к млекопитающему, такому как человек, примат, корова, овца или домашнее животное (включая кошку или собаку).
Термин «лечение», используемый в настоящем описании, если не указано иное, означает реверсирование, облегчение, ингибирование прогрессирования или предупреждение расстройства или состояния, к которому применяется такой термин, или одного или нескольких симптомов такого расстройства или состояния. Термин «лечение», используемый в настоящем описании, если не указано иное, относится к действию лечения, как «лечение» определено в настоящем документе. Термин «лечение» также включает адъювантное и неоадъювантное лечение субъекта.
Для целей настоящего изобретения полезные или желаемые клинические результаты при лечении рака включают, но не ограничиваются ими, одно или несколько из следующего: уменьшение пролиферации (или разрушение) неопластических или раковых клеток; ингибирование метастазов или неопластических клеток; сокращение или уменьшение размера опухоли; ремиссия рака; уменьшение симптомов, вызванных раком; повышение качества жизни больных раком; уменьшение дозы других лекарств, необходимых для лечения рака; задержка прогрессирования рака; излечение рака; преодоление одного или нескольких механизмов резистентности рака; и/или продление выживаемости пациентов с раком. Положительный терапевтический эффект при раке можно измерить несколькими способами (см., например, W. A. Weber, Assessing tumor response to therapy, J. Nucl. Med. 50 Suppl. 1:1S-10S (2009). Например, что касается ингибирования роста опухоли (T/C), согласно стандартам National Cancer Institute (NCI), T/C, меньшее или равное 42%, является минимальным уровнем противоопухолевой активности. T/C <10% считается высоким уровнем противоопухолевой активности, где T/C (%)=средний объем получившей лечение опухоли/средний объем контрольной опухоли x 100.
Фраза «терапевтически эффективное количество» означает количество соединения по изобретению, которое (i) лечит или предотвращает конкретное заболевание, (ii) ослабляет, облегчает или устраняет один или несколько симптомов конкретного заболевания или (iii) предотвращает или задерживает начало одного или нескольких симптомов конкретного заболевания, описанного в настоящем документе.
Фраза «фармацевтически приемлемый» указывает на то, что вещество или композиция должны быть химически и токсикологически совместимыми с другими ингредиентами, входящими в лекарственную форму, и с млекопитающим, которого лечат ими.
Подходящие соли присоединения кислоты образуются из кислот, которые образуют нетоксичные соли. Примеры включают соли ацетат, адипат, аспартат, бензоат, безилат, бикарбонат/карбонат, бисульфат/сульфат, борат, камсилат, цитрат, цикламат, эдизилат, эзилат, формиат, фумарат, глюцепат, глюконат, глюкуронат, гексафторфосфат, гибензат, гидрохлорид/хлорид, гидробромид/бромид, гидроиодид/иодид, изетионат, лактат, малат, малеат, малонат, мезилат, метилсульфат, нафтилат, 2-напсилат, никотинат, нитрат, оротат, оксалат, пальмитат, памоат, фосфат/гидрофосфат/дигидрофосфат, пироглутамат, сахарат, стеарат, сукцинат, таннат, тартрат, тозилат, трифторацетат, 1,5-нафаталендисульфоновая кислота и ксинафоат.
Подходящие основные соли образуются из оснований, которые образуют нетоксичные соли. Примеры включают соли алюминия, аргинина, бензатина, кальция, холина, диэтиламина, бис(2-гидроксиэтил)амина (диоламин), глицина, лизина, магния, меглумина, 2-аминоэтанола (оламин), калия, натрия, 2-амино-2-(гидроксиметил)пропан-1,3-диола (трис или трометамин) и цинка.
Гемисоли кислот и оснований могут быть также получены, например гемисульфат и гемикальциевые соли. Обзор подходящих солей см. Handbook of Pharmaceutical Salts: Properties, Selection, and Use by Stahl and Wermuth (Wiley-VCH, 2002).
«Аномальный рост клеток», используемый в настоящем описании, если не указано иное, относится к росту клеток, который не зависит от нормальных регуляторных механизмов (например, потеря контактного ингибирования). Аномальный рост клеток может быть доброкачественным (не злокачественным) или злокачественным (раковидным).
Аномальный рост клеток включает аномальный рост: (1) опухолевых клеток (опухолей), которые демонстрируют повышенную экспрессию CDK4 и/или CDK6; (2) опухолей, которые пролиферируют за счет аберрантной активации CDK4 и/или CDK6; и (3) опухолей, которые резистентны к эндокринной терапии, антагонистам HER2 или ингибированию CDK4/6.
Термин «дополнительное противораковое терапевтическое средство», используемый в настоящем описании, означает любое одно или несколько терапевтических средств, кроме соединения по настоящему изобретению, которые используются или могут быть использованы для лечения рака. В некоторых вариантах осуществления, такие дополнительные противораковые терапевтические средства включают соединения, происходящие из следующих классов: ингибиторы митоза, алкилирующие средства, антиметаболиты, противоопухолевые антибиотики, антиангиогенные средства, ингибиторы топоизомеразы I и II, растительный алкалоид, гормональные средства и антагонисты, ингибиторы факторов роста, облучение, ингибиторы передачи сигнала, такие как ингибиторы протеинтирозинкиназ и/или серин/треонинкиназ, ингибиторы клеточного цикла, модификаторы биологического ответа, ингибиторы ферментов, антисмысловые олигонуклеотиды или производные олигонуклеотидов, цитотоксические средства, иммуноонкологические средства и тому подобное.
В некоторых вариантах осуществления дополнительное противораковое средство представляет собой эндокринное средство, такое как ингибитор ароматазы, SERD или SERM.
В других вариантах осуществления соединение по изобретению можно вводить в сочетании со средством стандартного лечения, таким как тамоксифен, доцетаксел, паклитаксел, цисплатин, капецитабин, гемцитабин, винорелбин, эксеместан, летрозол, фулвестрант, анастрозол или трастузумаб.
В некоторых вариантах осуществления дополнительное противораковое средство представляет собой антиангиогенное средство, включая, например, ингибиторы VEGF, ингибиторы VEGFR, ингибиторы TIE-2, ингибиторы PDGFR, ингибиторы ангиопоэтина, ингибиторы PKCβ, ингибиторы COX-2 (циклооксигеназы II), интегрины (альфа-v/бета-3), ингибиторы MMP-2 (матриксная металлопротеиназа 2) и ингибиторы MMP-9 (матриксная металлопротеиназа 9). Предпочтительные антиангиогенные средства включают сунитиниб (сутент™), бевацизумаб (авастин™), акситиниб (AG 13736), SU 14813 (Pfizer) и AG 13958 (Pfizer). Дополнительные антиангиогенные средства включают ваталаниб (CGP 79787), сорафениб (нексавар™), пегаптаниб октасодиум (макуген™), вандетаниб (зактима™), PF-0337210 (Pfizer), SU 14843 (Pfizer), AZD 2171 (AstraZeneca), ранибизумаб (люцентис™), Neovastat™ (AE 941), тетратиомолибдата (Coprexa™), AMG 706 (Amgen), VEGF Trap (AVE 0005), CEP 7055 (Sanofi-Aventis), XL 880 (Exelixis), телатиниб (BAY 57-9352) и CP-868,596 (Pfizer). Другие антиангиогенные средства включают энзастаурин (LY 317615), мидостаурин (CGP 41251), перифозин (KRX 0401), тепренон (Selbex™) и UCN 01 (Kyowa Hakko). Другие примеры антиангиогенных средств включают целекоксиб (целебрекс™), парекоксиб (династат™), деракоксиб (SC 59046), люмиракоксиб (Preige™), валдекоксиб (Bextra™), рофекоксиб (Vioxx™), игуратимод (Careram™), IP 751 (Invedus), SC-58125 (Pharmacia) и эторикоксиб (Аркоксия™). Еще дополнительные антиангиогенные средства включают эксисулинд (Aptosyn™), салсалат (Amigesic™), дифлунизал (Dolobid™), ибупрофен (мотрин™), кетопрофен (орудис™), набуметон (релафен™), пироксикам (фельден™), напроксен (Aleve™, напросин™), диклофенак (вольтарен™), индометацин (индоцин™), сулиндак (клинорил™), толметин (толектин™), этодолак (Lodine™), кеторолак (торадол™) и оксапрозин (Daypro™). Еще дополнительные антиангиогенные средства включают ABT 510 (Abbott), апратастат (TMI 005), AZD 8955 (AstraZeneca), инциклинид (Metastat™) и PCK 3145 (Procyon). Еще дополнительные антиангиогенные средства включают ацитретин (неотигазон™), плитидепсин (аплидин™), циленгтид (EMD 121974), комбретастатин A4 (CA4P), фенретинид (4 HPR), галофугинон (темпостатин™), Panzem™ (2-метоксиэстрадиол), PF-03446962 (Pfizer), ребимастат (BMS 275291), катумаксомаб (ремоваб™), леналидомид (ревлимид™), скваламин (EVIZON™), талидомид (таломид™), Ukrain™ (NSC 631570), витаксин™ (MEDI 522) и золедроновая кислота (Зомета™).
В других вариантах осуществления, дополнительное противораковое средство представляет собой так называемый ингибитор передачи сигнала (например, ингибирующий средства, с помощью которых регуляторные молекулы, которые регулируют фундаментальные процессы роста, дифференцировки и выживания клеток, передаются внутри клетки). Ингибиторы передачи сигнала включают малые молекулы, антитела и антисмысловые молекулы. Ингибиторы передачи сигнала включают, например, ингибиторы киназ (например, ингибиторы тирозинкиназы или ингибиторы серин/треонинкиназы) и ингибиторы клеточного цикла. Более конкретно, ингибиторы передачи сигнала включают, например, ингибиторы фарнезилпротеинтрансферазы, ингибитор EGF, ErbB-1 (EGFR), ErbB-2, pan erb, ингибиторы IGF1R, MEK, ингибиторы c-Kit, ингибиторы FLT-3, ингибиторы K-Ras, ингибиторы киназы PI3, ингибиторы JAK, ингибиторы STAT, ингибиторы Raf-киназы, ингибиторы Akt, ингибитор mTOR, ингибиторы P70S6-киназы, ингибиторы пути WNT и так называемые многоцелевые ингибиторы киназ. Дополнительные примеры ингибиторов передачи сигнала, которые можно использовать в сочетании с соединением по изобретению и фармацевтическими композициями, описанными в настоящем документе, включают BMS 214662 (Bristol-Myers Squibb), лонафарниб (Sarasar™), пелитрексол (AG 2037), матузумаб (EMD 7200), нимотузумаб (TheraCIM h-R3™), панитумумаб (Vectibix™), вандетаниб (зактима™), пазопаниб (SB 786034), ALT 110 (Alteris Therapeutics), BIBW 2992 (Boehringer Ingelheim) и Cervene™ (TP 38). Другие примеры ингибиторов передачи сигнала включают гефитиниб (иресса™), цетуксимаб (эрбитукс™), эрлотиниб (тарцева™), трастузумаб (герцептин™), сунитиниб (сутент™), иматиниб (гливек™), кризотиниб (Pfizer), дакомитиниб (Pfizer), босутиниб (Pfizer), канертиниб (CI 1033), пертузумаб (Omnitarg™), лапатиниб (Tycerb™), пелитиниб (EKB 569), милтефозин (Miltefosin™), BMS 599626 (Bristol-Myers Squibb), Lapuleucel-T (Neuvenge™), NeuVax™ (противораковая вакцина E75), Osidem™ (IDM 1), мубритиниб (TAK-165), CP-724,714 (Pfizer), панитумумаб (вектибикс™), ARRY 142886 (Array Biopharm), эверолимус (сертикан™), зотаролимус (Endeavor™), темсиролимус (торизел™), AP 23573 (ARIAD) и VX 680 (Vertex), XL 647 (Exelixis), сорафениб (нексавар™), LE-AON (Georgetown University) и GI-4000 (Globelmmune). Другие ингибиторы передачи сигнала включают ABT 751 (Abbott), альвоцидиб (флавопиридол), BMS 387032 (Bristol Myers), EM 1421 (Erimos), индисулам (E 7070), селициклиб (CYC 200), BIO 112 (Onc Bio), BMS 387032 (Bristol-Myers Squibb), палбоциклиб (Pfizer) и AG 024322 (Pfizer).
В других вариантах осуществления дополнительное противораковое средство представляет собой так называемое классическое противоопухолевое средство. Классические противоопухолевые агенты включают, но не ограничиваются ими, гормональные модуляторы, такие как гормональные, антигормональные, агонисты андрогенов, антагонисты андрогенов и антиэстрогенные терапевтические средства, ингибиторы гистондеацетилазы (HDAC), ингибиторы ДНК-метилтрансферазы, средства подавления или средства, активирующие гены, рибонуклеазы, протеосомика, ингибиторы топоизомеразы I, производные камптотецина, ингибиторы топоизомеразы II, алкилирующие средства, антиметаболиты, ингибитор поли(АДФ-рибоза) полимеразы-1 (PARP-1) (например, талазопариб, олапарив, рукапариб, нирапариб, инициариб, велипариб), ингибиторы микротубулина, антибиотики, ингибиторы веретена растительного происхождения, платина-координированные соединения, средства генной терапии, антисмысловые олигонуклеотиды, средства нацеливания на сосуды (VTA) и статины. Примеры классических противоопухолевых средств, используемых в комбинированной терапии с соединением по изобретению, необязательно с одним или несколькими другими средствами, включают, но не ограничиваются ими, глюкокортикоиды, такие как дексаметазон, преднизон, преднизолон, метилпреднизолон, гидрокортизон, и прогестины, такие как медроксипрогестерон, мегестрола ацетат (Megace), мифепристон (RU-486), селективные модуляторы эстрогеновых рецепторов (SERM; такие как тамоксифен, ралоксифен, лазофоксифен, афимоксифен, арзоксифен, базедоксифен, фиспемифен, ормелоксифен, оспемифен, тесмилифен, торемифен, трилостан и CHF 4227 (Cheisi), селективные ингибиторы эстрогеновых рецепторов (SERD; такие как фулвестрант), эксеместан (аромазин), анастрозол (аримидекс), атаместан, фадрозол, летрозол (фемара), форместан; агонисты гонадотропин-рилизинг-гормона (GnRH; гормон, стимулирующий высвобождение лютеинизирующего гормона [LHRH]), такие как бусерелин (супрефакт), гозерелин (золадекс), лейпрорелин (люпрон) и трипторелин (трелстар), абареликс (пленаксис), ципротерон, флутамид (Eulexin), мегестрол, нилутамид (ниландрон) и осатерон, дутастерид, эпристерид, финастерид, Serenoa repens, PHL 00801, абареликс, гозерелин, лейпрорелин, трипторелин, бикалутамид; антиандрогенные средства, такие как энзалутамид, абиратерона ацетат, бикалутамид (касодекс); и их комбинации. Другие примеры классических противоопухолевых средств, используемых в комбинации с соединением по изобретению, включают, но не ограничиваются ими, ингибиторы PARP, такие как талазопариб, олапарив, рукапариб, нирапариб, инипариб, велипариб; субероланилид гидроксамовой кислоты (SAHA, Merck Inc./Aton Pharmaceuticals), депсипептид (FR901228 или FK228), G2M-777, MS-275, пивалоилоксиметилбутират и PXD-101; онконаз (ранпирназа), PS-341 (MLN-341), велкейд (бортезомиб), 9-аминокамптотецин, белотекан, BN-80915 (Roche), камптотецин, дифломотекан, эдотекарин, экзатекан (Daiichi), гиматекан, 10-гидроксикамптотецин, иринотекан HCl (камптозар), луртотекан, оратецин (рубитекан, Supergen), SN-38, топотекан, камптотецин, 10-гидроксикамптотецин, 9-аминокамптотецин, иринотекан, SN-38, эдотекарин, топотекан, акларубицин, адриамицин, амонафид, амрубицин, аннамицин, даунорубицин, доксорубицин, элзамитруцин, эпирубицин, этопозид, идарубицин, галарубицин, гидроксикарбамид, неморубицин, новантрон (митоксантрон), пирарубицин, пиксантрон, прокарбазин, ребеккамицин, собузоксан, тафлупозид, валрубицин, зинкард (дексразоксан), азотистый иприт N-оксид, циклофосфамид, AMD-473, альтретамин, AP-5280, апазиквон, бросталицин, бендамустин, бусульфан, карбоквон, кармустин, хлорамбуцил, дакарбазин, эстрамустин, фотемустин, глюфосфамид, ифосфамид, KW-2170, ломустин, мафосфамид, мехлорэтамин, мелфалан, митобронитол, митолактол, митомицин C, митоксатрон, нимустин, ранимустин, темозоломид, тиотепа и платина-координированные алкилирующие соединения, такие как цисплатин, параплатин (карбоплатин), эптаплатин, лобаплатин, недаплатин, элоксатин (оксалиплатин, Sanofi), стрептозоцин, сатрплатин и их комбинации.
В других вариантах осуществления дополнительное противораковое средство представляет собой так называемые ингибиторы дигидрофолатредуктазы (такие как метотрексат и NeuTrexin (триметрезат глюкуронат)), антагонисты пурина (такие как 6-меркаптопурин рибозид, меркаптопурин, 6-тиогуанин, кладрибин, клофарабин (Clolar), флударабин, неларабин и ралтитрексед), антагонисты пиримидина (такие как 5-фторурацил (5-FU), Алимта (преметрекседа динатрий, LY231514, MTA), капецитабин (кселода™), цитозин арабинозид, гемзар™ (гемцитабин, Eli Lilly), тегафур (UFT Orzel или Uforal и включая TS-1 комбинацию тегафура, гиместата и отостата), доксифлуридин, кармофур, цитарабин (включая окфосфат, стеарат фосфата, пролонгированного действия и липосомальные формы), эноцитабин, 5-азацитидин (видаза), децитабин и этинилцитидин) и другие антиметаболиты, такие как эфлорнитин, гидроксимочевина, лейковорин, нолатрексед (тимитак), триапин, триметрексат, N-(5-[N-(3,4-дигидро-2-метил-4-оксохиназолин-6-илметил)-N-метиламино]-2-теноил)-L-глутаминовая кислота, AG-014699 (Pfizer Inc.), ABT-472 (Abbott Laboratories), INO-1001 (Inotek Pharmaceuticals), KU-0687 (KuDOS Pharmaceuticals) и GPI 18180 (Guilford Pharm Inc) и их комбинации.
Другие примеры классических противоопухолевых цитотоксических средств включают, но не ограничиваются ими, абраксан (Abraxis BioScience, Inc.), батабулин (Amgen), EPO 906 (Novartis), винфлунин (Bristol- Myers Squibb Company), актиномицин D, блеомицин, митомицин C, неокарзиностатин (зиностатин), винбластин, винкристин, виндезин, винорелбин (навельбин), доцетаксел (таксотер), ортатаксел, паклитаксел (в том числе таксопрексин конъюгат DHA/пакилтаксел), цисплатин, карбоплатин, недаплатин, оксалиплатин (элоксатин), сатраплатин, камптозар, капецитабин (кселода), оксалиплатин (элоксатин), таксотер алитретиноин, канфосфамид (Telcyta™), DMXAA (антисома), ибандроновая кислота, L-аспарагиназа, пэгаспаргаза (онкаспар™), эфапроксирал (Efaproxyn™ - радиационная терапия), бексаротен (таргретин™), тесмилифен (DPPE - повышает эффективность цитотоксических средств), тератоп™ (Biomira), третиноин (весаноид™), тирапазамин (Trizaone™), мотексафин гадолиний (Xcytrin™) Cotara™ (mAb), и NBI-3001 (Protox Therapeutics), полиглутамат-паклитаксел (Xyotax™) и их комбинации. Дополнительные примеры классических противоопухолевых средств включают, но не ограничиваются ими, как адвексин (ING 201), TNFerade (GeneVec, соединение, которое экспрессирует TNF-альфа в ответ на лучевую терапию), RB94 (Baylor College of Medicine), Genasense (облимерсен, Genta), комбретастатин A4P (CA4P), Oxi-4503, AVE-8062, ZD-6126, TZT-1027, Аторвастатин (липитор, Pfizer Inc.), провастатин (правахол, Bristol-Myers Squibb), ловастатин (мевакор, Merck Inc.), симвастатин (зокор, Merck Inc.), флувастатин (лескол, Novartis), церивастатин (байкол, Bayer), розувастатин (крестор, AstraZeneca), ловастатин, ниацин (Advicor, Kos Pharmaceuticals), кадуэт, липитор, торцетрапиб и их комбинации.
В других вариантах осуществления дополнительное противораковое средство представляет собой эпигенетический модулятор, например ингибитор или EZH2, SMARCA4, PBRM1, ARID1A, ARID2, ARID1B, DNMT3A, TET2, мл L1/2/3, NSD1/2, SETD2, BRD4, DOT1L, HKMTsanti, PRMT1-9, LSD1, UTX, IDH1/2 или BCL6.
В дополнительных вариантах осуществления дополнительное противораковое средство представляет собой иммуномодулирующее средство, такое как ингибитор CTLA-4, PD-1 или PD-L1 (например, пембролизумаб, ниволумаб или авелумаб), LAG-3, TIM-3, TIGIT, 4-1BB, OX40, GITR, CD40 или CAR-T-клеточная терапия.
Используемый в настоящем описании «рак» относится к любому злокачественному и/или инвазивному росту или опухоли, вызванному аномальным ростом клеток. Рак включает солидные опухоли, названные по типу клеток, которые их образуют, рак крови, костного мозга или лимфатической системы. Примеры солидных опухолей включают саркомы и карциномы. Рак крови включает, но не ограничивается ими, лейкоз, лимфому и миелому. Рак также включает первичный рак, который возникает в определенном месте организма, метастатический рак, который распространился из места, в котором он возник, в другие части организма, рецидив исходного первичного рака после ремиссии и второй первичный рак, который является новым первичным раком у человека с историей предыдущего рака другого типа, чем у последнего.
В некоторых вариантах осуществления способов, представленных в настоящем документе, рак выбран из группы, состоящей из рака молочной железы, рака яичников, рака мочевого пузыря, рака матки, рака предстательной железы, рака легкого (включая NSCLC), рака пищевода, рака печени, рака поджелудочной железы и рака желудка.
Настоящее изобретение также включает фармацевтическую композицию, включающую терапевтически эффективное количество соединения по изобретению вместе с фармацевтически приемлемым носителем. Для получения фармацевтических композиций с соединениями по настоящему изобретению фармацевтически приемлемые носители могут быть твердыми или жидкими. Препараты в твердой форме включают порошки, таблетки, пилюли, капсулы, облатки, суппозитории и расходные гранулы. Твердый носитель может представлять собой одно или несколько веществ, которые также могут действовать как разбавители, ароматизаторы, связующие, консерванты, вещество для улучшения распадаемости таблеток или материал для инкапсулирования. Для таблетированных лекарственных форм, в зависимости от дозы, соединение или фармацевтически приемлемая соль по изобретению может составлять от 1% масс. до 80% масс. лекарственной формы, обычно от 5% масс. до 60% масс., более типично от примерно 10% масс. до примерно 35% масс. или даже более типично от примерно 15% масс. до примерно 25% масс. лекарственной формы. В конкретных вариантах осуществления соединение или фармацевтически приемлемая соль изобретения составляет примерно 20% масс. лекарственной формы по массе.
В твердых лекарственных формах по изобретению носитель может включать множество фармацевтически приемлемых эксципиентов, включая, например, разбавители, разрыхлители, связующие, смазывающие вещества, скользящие вещества и поверхностно-активные вещества. Композиции могут также включать эксципиенты, такие как консерванты, антиоксиданты, ароматизаторы и красители, а также другие эксципиенты, известные в данной области.
Твердые лекарственные формы, такие как таблетки, обычно содержат разбавители, например, лактозу (моногидрат, высушенный распылением моногидрат, безводный и т.п.), маннит, ксилит, декстрозу, сахарозу, сорбит, прессованный сахар, микрокристаллическую целлюлозу, порошкообразную целлюлозу, крахмал, прежелатинизированный крахмал, декстраты, декстран, декстрин, декстроза, мальтодекстрин, карбонат кальция, двухосновный фосфат кальция, трехосновный фосфат кальция, сульфат кальция, карбонат магния, оксид магния, полоксамеры, полиэтиленоксид, гидроксипропилметилцеллюлоза и их смеси. Различные типы микрокристаллической целлюлозы могут подходить для использования в композициях, описанных в настоящем документе. Примеры микрокристаллической целлюлозы включают типы Avicel®: PH101, PH102, PH103, PH105, PH 112, PH113, PH200, PH301 и другие типы микрокристаллической целлюлозы, например силикатизированную микрокристаллическую целлюлозу (SMCC). В некоторых вариантах осуществления, разбавитель выбран из группы, состоящей из микрокристаллическуой целлюлозы, моногидрата лактозы, маннита, сорбита, ксилита, карбоната магния, двухосновного фосфата кальция, трехосновного фосфата кальция, или их смеси. В некоторых вариантах осуществления разбавитель включает микрокристаллическую целлюлозу. В некоторых вариантах осуществления разбавитель включает один или несколько типов микрокристаллической целлюлозы, например Avicel® PH105, Avicel® PH200 или их смеси. В некоторых таких вариантах осуществления разбавитель исключает моногидрат лактозы. В других таких вариантах осуществления разбавитель включает микрокристаллическую целлюлозу и дополнительно включает моногидрат лактозы. Разбавители часто составляют от примерно 25% масс. до примерно 75% масс. твердой лекарственной формы и предпочтительно от примерно 50% масс. до примерно 75% масс. лекарственной формы.
Твердые лекарственные формы часто содержат разрыхлители. Примеры разрыхлителей включают натрия крахмал гликолят, карбоксиметилцеллюлозу натрия, карбоксиметилцеллюлозу кальция, кроскармеллозу натрия, кросповидон, поливинилпирролидон, метилцеллюлозу, микрокристаллическую целлюлозу, низшую алкил-замещенную гидроксипропилцеллюлозу, крахмал, прежелатинизированный крахмал и альгинат натрия. В некоторых вариантах осуществления разрыхлитель представляет собой кросповидон. Можно использовать любую марку кросповидона; например марки CL, CL-SF и XL кросповидона подходят для использования в композициях, описанных в настоящем документе. Конкретные примеры включают Kollidon, Kollidon CL®, Kollidon CL-M®, Polyplasdone XL®, Polyplasdone XL-10®, и Polyplasdone INF-10®. В некоторых вариантах осуществления носитель включает по меньшей мере один разрыхлитель, выбранный из группы, состоящей из кросповидона, кроскармеллозы натрия и натрия крахмал гликолята. В конкретных вариантах осуществления разрыхлитель представляет собой кросповидон. Разрыхлители часто составляют от примерно 1% масс. до примерно 25% масс., предпочтительно от примерно 5% масс. до примерно 20% масс., более предпочтительно от примерно 5% масс. до примерно 10% масс. лекарственной формы.
Связующие вещества могут использоваться для придания композиции таблеток когезионных свойств. Подходящие связующие включают микрокристаллическую целлюлозу, желатин, сахара, полиэтиленгликоль, натуральные и синтетические камеди, поливинилпирролидон, прежелатинизированный крахмал, гидроксипропилцеллюлозу, и гидроксипропилметилцеллюлозу. В некоторых вариантах осуществления, связующее выбрано из группы, состоящей из микрокристаллической целлюлозы, гидроксипропилцеллюлозы и гидроксипропилметилцеллюлозы. В конкретных вариантах осуществления связующее представляет собой микрокристаллическую целлюлозу, например, Avicel® PH105. Когда они присутствуют, связующие могут составлять от примерно 0% масс. до примерно 15% масс., или от примерно 0,2% масс. до примерно 10% масс. лекарственной формы. В некоторых вариантах осуществления связующее составляет от примерно 5% масс. до примерно 10% масс. лекарственной формы. В конкретных вариантах осуществления связующее составляет примерно 10% масс. лекарственной формы.
Твердые лекарственные формы часто включают одно или несколько смазывающих веществ. Примеры смазывающих веществ включают стеарат магния, стеарат кальция, стеарат цинка, натрия стеарилфумарат, смеси стеарата магния с лаурилсульфатом натрия или смеси двух или более из них. В некоторых вариантах осуществления смазывающее вещество представляет собой стеарат магния и/или натрия стеарилфумарат. В некоторых вариантах осуществления смазывающее вещество представляет собой стеарат магния. В некоторых таких вариантах осуществления твердая лекарственная форма представляет собой таблетку, содержащую внутригранулярный и внегранулярный стеарат магния. В других вариантах осуществления твердая лекарственная форма представляет собой таблетку, содержащую внутригранулярный стеарат магния и внегранулярный натрия стеарилфумарат. Когда присутствуют, смазывающие вещества часто составляют от примерно 0,25% масс. до примерно 10% масс., предпочтительно от примерно 0,5% масс. до примерно 6% масс. лекарственной формы.
Таблетки также могут включать скользящие вещества, например диоксид кремния, коллоидный диоксид кремния, силикат магния, трисиликат магния, тальк и другие формы диоксида кремния, такие как агрегированные силикаты и гидратированный диоксид кремния. В некоторых вариантах осуществления скользящее вещество представляет собой диоксид кремния. Когда присутствуют, скользящие вещества могут составлять от примерно 0% масс. до примерно 10% масс., предпочтительно от примерно 0,2% масс. до примерно 5% масс., или от примерно 0,5% масс. до примерно 2% масс. таблетки.
Таблетки могут необязательно включать поверхностно-активные вещества, такие как лаурилсульфат натрия и полисорбат 80. Когда присутствуют, поверхностно-активные агенты могут составлять от 0% масс. до 10% масс., или предпочтительно от 0,2% масс. до 5% масс. таблетки.
Как правило, твердые лекарственные формы по изобретению получают способами, обычными в фармацевтической химии. Выбранный носитель (или эксципиенты) может быть включен вместе с активным фармацевтическим ингредиентом в один или оба внегранулярных или внутригранулярных компартментов.
Терапевтически эффективная доза соединения или фармацевтически приемлемой соли по изобретению будет варьироваться от приблизительно 0,01 мг/кг до приблизительно 100 мг/кг массы тела в день. Типичные дозы для взрослых составляют от примерно 0,1 мг до примерно 3000 мг в день. Более типично терапевтически эффективная доза для взрослых соединения или фармацевтически приемлемой соли по изобретению составляет 50 мг, 75 мг, 100 мг, 125 мг, 175 мг или 200 мг в день. Количество активного компонента в стандартной дозы препарата может варьироваться или регулироваться от приблизительно 0,1 мг до приблизительно 500 мг, предпочтительно от приблизительно 25 мг до приблизительно 200 мг, в зависимости от конкретного применения. При желании композиция может также содержать другие совместимые терапевтические средства. Субъекту, нуждающемуся в лечении соединением или фармацевтически приемлемой солью по изобретению, вводят дозу от примерно 0,6 до примерно 500 мг в день, и предпочтительно от примерно 25 мг до примерно 200 мг в день, либо однократно, либо в нескольких дозах в течение 24 часов. Такое лечение можно повторять с последовательными интервалами столько, сколько необходимо.
Соединения по настоящему изобретению (включая соединения формулы I, II или III или их соли) могут включать асимметричные или хиральные центры и, следовательно, существовать в различных стереоизомерных формах. Если не указано иное, предполагается, что все стереоизомерные формы соединений по настоящему изобретению, а также их смеси, включая рацемические смеси, составляют часть настоящего изобретения. Кроме того, настоящее изобретение охватывает все геометрические и позиционные изомеры. Например, если соединение по настоящему изобретению включает двойную связь или конденсированное кольцо, цис- и транс-формы, а также смеси, входят в объем изобретения.
Хиральные соединения по изобретению (и их хиральные предшественники) могут быть получены в энантиомерно-обогащенной форме с использованием хроматографии, обычно высокоэффективная жидкостная хроматография (ВЭЖХ) или сверхкритической флюидной хроматографии (SFC), на смоле с асимметричной неподвижной фазой и с подвижной фаза, состоящая из углеводорода, обычно гептана или гексана, содержащего от 0 до 50% изопропанола, обычно от 2 до 20%, и от 0 до 5% алкиламина, обычно 0,1% диэтиламина (DEA) или изопропиламина. Концентрирование элюента дает обогащенную смесь.
Смеси диастереомеров можно разделить на их индивидуальные диастереоизомеры на основе их физико-химических различий способами, хорошо известными специалистам в данной области, такими как хроматография и/или фракционная кристаллизация. Энантиомеры можно разделить путем превращения энантиомерной смеси в диастереомерную смесь реакцией с подходящим оптически активным соединением (например, хиральным вспомогательным веществом, таким как хиральный спирт или хлорангидрид кислоты Мошера), разделением диастереоизомеров и превращением (например, гидролизом) индивидуальных диастереоизомеров в соответствующие чистые энантиомеры. Энантиомеры также можно разделить с помощью хиральной ВЭЖХ колонки. Альтернативно, конкретные стереоизомеры могут быть синтезированы с использованием оптически активного исходного вещества, путем асимметричного синтеза с использованием оптически активных реагентов, субстратов, катализаторов или растворителей или путем преобразования одного стереоизомера в другой путем асимметричного превращения.
Примерные варианты осуществления включают следующие группы диастереомеров или энантиомеров, где диастереомеры или энантиомеры в каждой группе могут быть разделены или получены, как описано в настоящем документе, и с использованием методов, хорошо известных в данной области:
(1) 6-ацетил-8-циклобутил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он, 6-ацетил-8-циклобутил-2-{[(3S,4S)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он, 6-ацетил-8-циклобутил-2-{[(3R,4S)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он, и 6-ацетил-8-циклобутил-2-{[(3R,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он;
(2) 6-ацетил-8-циклопентил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он, 6-ацетил-8-циклопентил-2-{[(3S,4S)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он, 6-ацетил-8-циклопентил-2-{[(3R,4S)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он и 6-ацетил-8-циклопентил-2-{[(3R,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он;
(3) 6-ацетил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метил-8-[(1R,2S)-2-метилциклопентил]пиридо[2,3-d]пиримидин-7(8H)-он, 6-ацетил-2-{[(3S,4S)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метил-8-[(1R,2S)-2-метилциклопентил]пиридо[2,3-d]пиримидин-7(8H)-он, 6-ацетил-2-{[(3R,4S)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метил-8-[(1R,2S)-2-метилциклопентил]пиридо[2,3-d]пиримидин-7(8H)-он, 6-ацетил-2-{[(3R,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метил-8-[(1R,2S)-2-метилциклопентил]пиридо[2,3-d]пиримидин-7(8H)-он, 6-ацетил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метил-8-[(1S,2S)-2-метилциклопентил]пиридо[2,3-d]пиримидин-7(8H)-он, 6-ацетил-2-{[(3S,4S)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метил-8-[(1S,2S)-2-метилциклопентил]пиридо[2,3-d]пиримидин-7(8H)-он, 6-ацетил-2-{[(3R,4S)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метил-8-[(1S,2S)-2-метилциклопентил]пиридо[2,3-d]пиримидин-7(8H)-он, 6-ацетил-2-{[(3R,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метил-8-[(1S,2S)-2-метилциклопентил]пиридо[2,3-d]пиримидин-7(8H)-он, 6-ацетил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метил-8-[(1R,2R)-2-метилциклопентил]пиридо[2,3-d]пиримидин-7(8H)-он, 6-ацетил-2-{[(3S,4S)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метил-8-[(1R,2R)-2-метилциклопентил]пиридо[2,3-d]пиримидин-7(8H)-он, 6-ацетил-2-{[(3R,4S)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метил-8-[(1R,2R)-2-метилциклопентил]пиридо[2,3-d]пиримидин-7(8H)-он, 6-ацетил-2-{[(3R,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метил-8-[(1R,2R)-2-метилциклопентил]пиридо[2,3-d]пиримидин-7(8H)-он, 6-ацетил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метил-8-[(1S,2R)-2-метилциклопентил]пиридо[2,3-d]пиримидин-7(8H)-он, 6-ацетил-2-{[(3S,4S)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метил-8-[(1S,2R)-2-метилциклопентил]пиридо[2,3-d]пиримидин-7(8H)-он, 6-ацетил-2-{[(3R,4S)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метил-8-[(1S,2R)-2-метилциклопентил]пиридо[2,3-d]пиримидин-7(8H)-она, 6-ацетил-2-{[(3R,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метил-8-[(1S,2R)-2-метилциклопентил]пиридо[2,3-d]пиримидин-7(8H)-он;
(4) 3-ацетил-1-циклопентил-7-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-4-метил-1,6-нафтиридин-2(1H)-он; 3-ацетил-1-циклопентил-7-{[(3S,4S)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-4-метил-1,6-нафтиридин-2(1H)-он; 3-ацетил-1-циклопентил-7-{[(3R,4S)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-4-метил-1,6-нафтиридин-2(1H)-он и 3-ацетил-1-циклопентил-7-{[(3R,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-4-метил-1,6-нафтиридин-2(1H)-он;
(5) 6-ацетил-8-[2-(2,2-дифторэтил)-2-азаспиро[3.3]гептан-6-ил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он, 6-ацетил-8-[2-(2,2-дифторэтил)-2-азаспиро[3.3]гептан-6-ил]-2-{[(3S,4S)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он, 6-ацетил-8-[2-(2,2-дифторэтил)-2-азаспиро[3.3]гептан-6-ил]-2-{[(3R,4S)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он, и 6-ацетил-8-[2-(2,2-дифторэтил)-2-азаспиро[3.3]гептан-6-ил]-2-{[(3R,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он.
Согласно способам изобретения соединение по настоящему изобретению или комбинацию предпочтительно вводят в форме фармацевтической композиции. Соответственно, соединение по настоящему изобретению или их комбинацию можно вводить пациенту отдельно или вместе в любой обычной лекарственной форме для перорального, ректального, трансдермального, парентерального (например, внутривенного, внутримышечного или подкожного), интрацистернального, интравагинального, внутрибрюшинного, местного (например, порошок, мазь, крем, спрей или лосьон), трансбуккального или назального (например, спрей, капли или средство для ингаляции) введения.
Соединения по изобретению или их комбинации можно вводить отдельно, но обычно их вводят в смеси с одним или несколькими подходящими фармацевтическими эксципиентами, адъювантами, разбавителями или носителями, известными в данной области и выбранными с учетом предполагаемого пути введения и общепринятой фармацевтической практики. Соединение по изобретению или комбинация могут быть сформулированы с получением лекарственных форм с немедленным, отсроченным, модифицированным, замедленным, импульсным или контролируемым высвобождением в зависимости от желаемого пути введения и специфичности профиля высвобождения, соизмеримого с терапевтические потребности.
Фармацевтическая композиция содержит соединение по изобретению или их комбинацию в количестве, как правило, в диапазоне от примерно 1% до примерно 75%, 80%, 85%, 90% или даже 95% (по массе) композиции, обычно в диапазоне от примерно 1%, 2% или 3% до примерно 50%, 60% или 70%, чаще в диапазоне от примерно 1%, 2% или 3% до менее чем 50%, например примерно 25%, 30% или 35%.
Способы получения различных фармацевтических композиций с определенным количеством активного соединения известны специалистам в данной области. Например, см. Remington: The Practice of Pharmacy, Lippincott Williams and Wilkins, Baltimore Md. 20.sup.th ed. 2000.
Композиции, подходящие для парентерального введения, обычно включают фармацевтически приемлемые стерильные водные или неводные растворы, дисперсии, суспензии или эмульсии и стерильные порошки для восстановления в стерильные растворы для инъекций или дисперсии. Примеры подходящих водных и неводных носителей или разбавителей (включая растворители и носители) включают воду, этанол, полиолы (пропиленгликоль, полиэтиленгликоль, глицерин и тому подобное), их подходящие смеси, триглицериды, включая растительные масла, такие как оливковое масло, и инъекционные органические сложные эфиры, такие как этилолеат. Предпочтительным носителем является Miglyol.RTM. марка эфира каприловой/каприновой кислоты с глицерином или пропиленгликолем (например, Miglyol.RTM. 812, Miglyol.RTM. 829, Miglyol.RTM. 840), доступный от Condea Vista Co., Cranford, N.J. Подходящую текучесть можно поддерживать, например, путем использования покрытия, такого как лецитин, путем поддержания требуемого размера частиц в случае дисперсий и за счет использования поверхностно-активных веществ.
Эти композиции для парентерального введения могут также включать эксципиенты, такие как консерванты, смачивающие, эмульгирующие и диспергирующие агенты. Предотвращение загрязнения композиций микроорганизмами может быть достигнуто с помощью различных антибактериальных и противогрибковых агентов, например парабенов, хлорбутанола, фенола, сорбиновой кислоты и тому подобное. Также может быть желательно включение изотонических агентов, например сахара, хлорида натрия и тому подобное. Пролонгированная абсорбция фармацевтических композиций для инъекций может быть достигнута за счет использования агентов, способных задерживать всасывание, например, моностеарата алюминия и желатина.
Твердые лекарственные формы для перорального введения включают капсулы, таблетки, жевательные таблетки, лепешки, пилюли, порошки и препараты из множества частиц (гранулы). В таких твердых лекарственных формах соединение по настоящему изобретению или его комбинацию смешивают по меньшей мере с одним инертным наполнителем, разбавителем или носителем. Подходящие эксципиенты, разбавители или носители включают вещества, такие как цитрат натрия или дикальцийфосфат, и/или (а) один или несколько наполнителей или разбавителей (например, микрокристаллическая целлюлоза (доступная как Avicel.TM. от FMC Corp.) крахмалы, лактоза, сахароза, маннит, кремниевая кислота, ксилит, сорбит, декстроза, гидрофосфат кальция, декстрин, альфа-циклодекстрин, бета-циклодекстрин, полиэтиленгликоль, жирные кислоты со средней длиной цепи, оксид титана, оксид магния, оксид алюминия и тому подобное); (b) одно или несколько связующих (например, карбоксиметилцеллюлоза, метилцеллюлоза, гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза, желатин, гуммиарабик, этилцеллюлоза, поливиниловый спирт, пуллулан, прежелатинизированный крахмал, агар, трагакант, альгинаты, желатин, поливинилпирролидон, сахароза, аравийская камедь и тому подобное); (c) одно или несколько увлажнителей (например, глицерин и тому подобное); (d) один или несколько разрыхлителей (например, агар-агар, карбонат кальция, картофельный или тапиоковый крахмал, альгиновая кислота, некоторые сложные силикаты, карбонат натрия, лаурилсульфат натрия, натрия крахмал гликолят (доступный как Explotab.TM.от Edward Mendell Co.), сшитый поливинилпирролидон, кроскармеллоза натрия А-типа (доступная как Ac-di-sol.TM.), полиакрилин калия (ионообменная смола) и тому подобное); (e) один или несколько замедлителей растворения (например, парафин и тому подобное); (f) один или несколько ускорителей поглощения (например, соединения четвертичного аммония и тому подобное); (g) один или несколько смачивающих агентов (например, цетиловый спирт, глицерилмоностеарат и тому подобное); (h) один или несколько адсорбентов (например, каолин, бентонит и тому подобное); и/или (i) один или несколько скользящих веществ (например, тальк, стеарат кальция, стеарат магния, стеариновая кислота, полиоксилстеарат, цетанол, тальк, гидрогенизированное касторовое масло, сложные эфиры сахарозы и жирной кислоты, диметилполисилоксан, микрокристаллический воск, желтый пчелиный воск, белый пчелиный воск, твердые полиэтиленгликоли, лаурилсульфат натрия и тому подобное). В случае капсул и таблеток лекарственные формы могут также включать буферные агенты.
Твердые композиции подобного типа также могут использоваться в качестве наполнителей в мягких или твердых заполненных желатиновых капсулах с использованием таких вспомогательных веществ, как лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли, и тому подобное.
Твердые лекарственные формы, такие как таблетки, драже, капсулы и гранулы, могут быть получены с покрытиями и оболочками, такими как энтеросолюбильные покрытия и другие, хорошо известные в данной области. Они также могут содержать средства, придающее непрозрачность, и также могут иметь такую композицию, которая высвобождает соединение по настоящему изобретению и/или дополнительное фармацевтическое средство замедленным образом. Примерами композиций для заливки, которые можно использовать, являются полимерные вещества и воски. Лекарственное средство также может быть в микрокапсулированной форме, при необходимости, с одним или несколькими из вышеупомянутых эксципиентов.
Для таблеток активное средство обычно будет составлять менее 50% (по массе) композиции, например, менее примерно 10%, например 5% или 2,5% масс. Преобладающая часть композиции включает наполнители, разбавители, разрыхлители, скользящие вещества и, необязательно, ароматизаторы. Композиция этих эксципиентов хорошо известна в данной области. Часто наполнители/разбавители будут включать смеси двух или более следующих компонентов: микрокристаллическая целлюлоза, маннит, лактоза (все типы), крахмал и дикальцийфосфат. Смеси наполнитель/разбавитель обычно составляют менее 98% композиции и предпочтительно менее 95%, например 93,5%. Предпочтительные разрыхлители включают Ac-di-sol.TM., Explotab.TM., крахмал и лаурилсульфат натрия. Когда присутствует, разрыхлитель обычно составляет менее 10% композиции или менее 5%, например, примерно 3%. Предпочтительным скользящим веществом является стеарат магния. Когда присутствует, скользящее вещество обычно будет составлять менее 5% композиции или менее 3%, например, примерно 1%.
Таблетки могут быть получены стандартными процессами таблетирования, например, прямым прессованием или влажным, сухим гранулированием или гранулированием из расплава, процессом застывания расплава и экструзией. Таблетки-ядра могут быть однослойными или многослойными и могут быть покрыты соответствующими слоями, известными в данной области.
Жидкие лекарственные формы для перорального введения включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры. В дополнение к соединению по настоящему изобретению или комбинации жидкая лекарственная форма может содержать инертные разбавители, обычно используемые в данной области, такие как вода или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как, например, этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (например, хлопковое масло, арахисовое масло, масло зародышей кукурузы, оливковое масло, касторовое масло, кунжутное масло и тому подобное), Miglyole.RTM. (доступны от CONDEA Vista Co., Cran-ford, N.J.), глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот и сорбитана или смеси этих веществ и тому подобное.
Помимо таких инертных разбавителей, композиция может также включать эксципиенты, такие как смачивающие агенты, эмульгирующие и суспендирующие агенты, подсластители, ароматизаторы и отдушки.
Жидкие формы для перорального введения соединений по изобретению или комбинаций включают растворы, в которых активное соединение полностью растворено. Примеры растворителей включают все фармацевтически определенные растворители, подходящие для перорального введения, особенно те, в которых соединения по изобретению проявляют хорошую растворимость, например полиэтиленгликоль, полипропиленгликоль, пищевые масла и системы на основе глицерина и глицеридов. Системы на основе глицерина и глицеридов могут включать, например, следующие фирменные продукты (и соответствующие типовые продукты): Captex.TM. 355 EP (глицерилтрикаприлат/капрат, от Abitec, Columbus Ohio), Crodamol.TM. GTC/C (триглицерид со средней длиной цепи, от Croda, Cowick Hall, UK) или Labrafac.TM. CC (триглиды со средней длиной цепи, от Gattefosse), Captex.TM. 500P (триацетат глицерина, т.е. триацетин, от Abitec), Cap-mul.TM. MCM (моно- и диглицериды со средней длиной цепи, от Abitec), Migyol.TM. 812 (триглицерид каприловой/каприновой кислоты, от Condea, Cranford N.J.), Migyol.TM. 829 (каприловый/каприновый/янтарный триглицерид, от Condea), Migyol.TM. 840 (пропиленгликоль дикаприлат/дикапрат, от Condea), Labrafil.TM. M1944CS (олеоилмакрогол-6 глицериды, от Gattefosse), Peceol.TM. (глицерилмоноолеат, от Gattefosse) и Maisine.TM. 35-1 (глицерилмоноолеат, от Gattefosse). Особый интерес представляют триглицеридные масла со средней длиной цепи (примерно C8-C10). Эти растворители часто составляют преобладающую часть композиции, т.е. более примерно 50%, обычно более примерно 80%, например, примерно 95% или 99%. Адъюванты и добавки также могут быть включены в растворители, главным образом в качестве агентов, маскирующих вкус, агентов для приятного вкуса и ароматизаторов, антиоксидантов, стабилизаторов, модификаторов текстуры и вязкости и солюбилизаторов.
Суспензии, в дополнение к соединению по настоящему изобретению или комбинации, могут дополнительно содержать носители, такие как суспендирующие агенты, например этоксилированные изостеариловые спирты, полиоксиэтиленсорбитол и сложные эфиры сорбитана, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант или смеси этих веществ и тому подобное.
Композиции для ректального или вагинального введения предпочтительно включают суппозитории, которые могут быть получены путем смешивания соединения по настоящему изобретению или комбинации с подходящими нераздражающими эксципиентами или носителями, такими как масло какао, полиэтиленгликоль или воск для суппозиториев, которые являются твердыми при обычной комнатной температуре, но жидкими при температуре тела и, следовательно, плавятся в прямой кишке или вагинальной полости, высвобождая активный компонент(ы).
Лекарственные формы для местного введения соединений по настоящему изобретению или их комбинаций включают мази, кремы, лосьоны, порошки и спреи. Лекарственные средства смешаны с фармацевтически приемлемым эксципиентом, разбавителем или носителем, и с любыми консервантами, буферами или пропеллентами, которые могут потребоваться.
Многие из настоящих соединений плохо растворимы в воде, например, менее чем примерно 1 мкг/мл. Следовательно, жидкие композиции в солюбилизирующих неводных растворителях, таких как триглицеридные масла со средней длиной цепи, обсуждаемые выше, являются предпочтительной лекарственной формой для этих соединений.
Аморфные твердые дисперсии, включая дисперсии, образованные методом сушки распылением, также являются предпочтительной лекарственной формой для малорастворимых соединений по изобретению. Под «аморфной твердой дисперсией» подразумевается твердое вещество, в котором по меньшей мере часть плохо растворимого соединения находится в аморфной форме и диспергирована в водорастворимом полимере. Под «аморфным» подразумевается, что плохо растворимое соединение не является кристаллическим. Под «кристаллическим» подразумевается, что соединение демонстрирует дальний порядок в трех измерениях, по меньшей мере, 100 повторяющихся единиц в каждом измерении. Таким образом, термин аморфный предназначен для включения не только вещества, которое по существу не имеет порядка, но также вещества, которое может иметь некоторую небольшую степень порядка, но порядок имеет менее трех измерений и/или только на короткие расстояния. Аморфное вещество может быть охарактеризовано методами, известными в данной области техники, такими как кристаллография с помощью порошковой рентгеновской дифракции (PXRD), ЯМР твердого тела или термическими методами, такими как дифференциальная сканирующая калориметрия (DSC).
В некоторых вариантах осуществления, по меньшей мере, основная часть (т.е. по меньшей мере примерно 60% масс.) плохо растворимого соединения в аморфной твердой дисперсии является аморфным. Соединение может существовать в аморфной твердой дисперсии в относительно чистых аморфных доменах или областях, в виде твердого раствора соединения, гомогенно распределенного в полимере, или любой комбинации этих состояний или тех состояний, которые находятся между ними. Предпочтительно, чтобы аморфная твердая дисперсия была по существу гомогенной, чтобы аморфное соединение было диспергировано как можно более однородно по всему полимеру. Используемый в настоящем описании, «по существу гомогенный» означает, что доля соединения, которая присутствует в относительно чистых аморфных доменах или областях внутри аморфной твердой дисперсии, относительно мала, порядка менее 20% масс., и предпочтительно менее 10% масс. от общего количества лекарственного средства.
Водорастворимые полимеры, подходящие для использования в аморфных твердых дисперсиях, должны быть инертными в том смысле, что они не вступают в химическую реакцию с плохо растворимым соединением неблагоприятным образом, являются фармацевтически приемлемыми и имеют по меньшей мере некоторую растворимость в водном растворе при физиологически соответствующих значениях pH (например, 1-8). Полимер может быть нейтральным или ионизируемым и должен иметь растворимость в воде по меньшей мере 0,1 мг/мл в части диапазона pH 1-8.
Водорастворимые полимеры, подходящие для использования в настоящем изобретении, могут быть целлюлозными или нецеллюлозными. Полимеры могут быть нейтральными или ионизируемыми в водном растворе. Из них предпочтительными являются ионизируемые и целлюлозные полимеры, более предпочтительными являются ионизируемые целлюлозные полимеры.
Примеры водорастворимых полимеров включают гидроксипропилметилцеллюлозу ацетат сукцинат (HPMCAS), гидроксипропилметилцеллюлозу (HPMC), фталат гидроксипропилметилцеллюлозы (HPMCP), карбоксиметилэтилцеллюлозу (CMEC), ацетатфталат целлюлозы (CAP), ацетат-тримеллитат целлюлозы (CAT), поливинилпирролидон (PVP), гидроксипропилцеллюлоза (HPC), метилцеллюлозу (MC), блок-сополимеры этиленоксида и пропиленоксида (PEO/PPO, также известные как полоксамеры) и их смеси. Особенно предпочтительные полимеры включают HPMCAS, HPMC, HPMCP, CMEC, CAP, CAT, PVP, полоксамеры, и их смеси. Наиболее предпочтительным является HPMCAS. См. Публикацию европейской патентной заявки No. 0 901 786 A2, описание которой включено в настоящий документ посредством ссылки.
Аморфные твердые дисперсии могут быть получены в соответствии с любым способом образования аморфных твердых дисперсий, в результате которого по меньшей мере большая часть (по меньшей мере 60%) плохо растворимого соединения находится в аморфном состоянии. Такие процессы включают механические, термические процессы и процессы с растворителем. Примеры механических процессов включают измельчение и экструзию; процессы плавления, включая высокотемпературную плавку, плавление с модифицированным растворителем и процессы плавления-застывания; и способы с использованием растворителя, включая осаждение без использования растворителя, нанесение покрытия распылением и сушку распылением. См., например, следующие патенты США, соответствующие описания которых включены в настоящий документ посредством ссылки: No. 5456923 и 5939099, которые описывают образование дисперсий с помощью процессов экструзии; No. 5340591 и 4673564, которые описывают формирование дисперсий с помощью процессов измельчения; и No. 5707646 и 4894235, которые описывают образование дисперсий с помощью процессов затвердевания расплава. В предпочтительном способе аморфную твердую дисперсию получают сушкой распылением, как раскрыто в публикации Европейской патентной заявки No. 0 901 786 A2. В этом способе соединение и полимер растворяют в растворителе, таком как ацетон или метанол, и затем растворитель быстро удаляют из раствора с помощью сушки распылением с образованием аморфной твердой дисперсии. Твердые аморфные дисперсии могут быть получены таким образом, чтобы они содержали до примерно 99% масс. соединения, например, 1% масс., 5% масс., 10% масс., 25% масс., 50% масс., 75% масс., 95% масс. или 98% масс. по желанию.
Твердая дисперсия может использоваться в качестве самой лекарственной формы или она может служить в качестве продукта для производства (MUP) при получении других лекарственных форм, таких как капсулы, таблетки, растворы или суспензии. Примером водной суспензии является водная суспензия 1:1 (масс./масс.) соединение/HPMCAS-HF высушенная распылением дисперсия, содержащая 2,5 мг/мл соединения в 2% полисорбате-80. Твердые дисперсии для использования в таблетках или капсулах обычно смешивают с другими эксципиентами или адъювантами, обычно присутствующими в таких лекарственных формах. Например, типичный наполнитель для капсул содержит 2:1 (масс./масс.) соединение/HPMCAS-MF высушенная распылением дисперсия (60%), лактозу (быстро растекающуюся) (15%), микрокристаллическую целлюлозу (например, Avicel.sup.(R0-102) (15,8%), крахмал натрия (7%), лаурилсульфат натрия (2%) и стеарат магния (1%).
Полимеры HPMCAS доступны в низком, среднем и высоком качестве как Aqoa.sup.(R)-LF, Aqoat.sup.(R)-MF и Aqoat.sup.(R)-HF, соответственно от Shin-Etsu Chemical Co., LTD, Tokyo, Japan. Обычно предпочтительны более высокие марки MF и HF.
Следующие параграфы описывают примерные композиции, дозировки и т.п., применимые для пригодные для животных, не являющихся человеком. Введение соединений по настоящему изобретению и комбинаций соединений по настоящему изобретению со средствами против ожирения может осуществляться перорально или не перорально.
Количество соединения по настоящему изобретению или комбинации соединения по настоящему изобретению с другим средством против ожирения вводят таким образом, чтобы была получена эффективная доза. Обычно суточная доза, которую вводят животному перорально, составляет от примерно 0,01 до примерно 1000 мг/кг массу тела, например, от примерно 0,01 до примерно 300 мг/кг, или от примерно 0,01 до примерно 100 мг/кг, или от примерно 0,01 до примерно 50 мг/кг массы тела, или от примерно 0,01 до примерно 25 мг/кг, или от примерно 0,01 до примерно 10 мг/кг, или от примерно 0,01 до примерно 5 мг/кг.
В целях удобства, соединение по настоящему изобретению (или комбинацию) можно вводить в питьевую воду так, чтобы терапевтическая доза соединения принималась с ежедневным потреблением воды. Соединение можно непосредственно дозировать в питьевую воду, предпочтительно в форме жидкого водорастворимого концентрата (такого как водный раствор водорастворимой соли).
В целях удобства, соединение по настоящему изобретению (или ег комбинацию) также можно добавлять непосредственно в корм, как таковое, или в форме добавки к корму для животных, также называемой премиксом или концентратом. Премикс или концентрат соединения в эксципиенте, разбавителе или носителе чаще используется для включения средства в корм. Подходящие эксципиенты, разбавители или носители являются жидкими или твердыми, по желанию, такими как вода, различные виды муки, такие как мука из люцерны, соевая мука, мука из хлопкового масла, мука из жмыха льняного семени, мука из кукурузных початков и кукурузная мука, патока, мочевина, костная мука и минеральные смеси, которые обычно используются в кормах для домашней птицы. Особенно эффективным эксципинтом, разбавителем или носителем является сам соответствующий корм для животных; то есть небольшая порцию такого корма. Носитель способствует равномерному распределению соединения в готовом корме, с которым смешивают премикс. Предпочтительно, чтобы соединение было тщательно смешано с премиксом, а затем и с кормом. В этом отношении соединение может быть диспергировано или растворено в подходящем масляном носителе, таком как соевое масло, кукурузное масло, хлопковое масло и т.п., или в летучем органическом растворителе, а затем смешано с носителем. Следует понимать, что пропорции соединения в концентрате могут варьироваться в широких пределах, поскольку количество соединения в готовом корме можно регулировать путем смешивания соответствующей пропорции премикса с кормом для получения желаемого уровня соединения.
Концентраты с высокой активностью могут быть смешаны производителем кормов с белковым носителем, таким как соевый шрот и другие виды муки, как описано выше, для получения концентрированных добавок, которые подходят для непосредственного кормления животных. В таких случаях животным разрешается употреблять обычный рацион. В качестве альтернативы, такие концентрированные добавки могут быть добавлены непосредственно в корм для получения сбалансированного по питательным веществам готового корма, содержащего терапевтически эффективный уровень соединения настоящего изобретения. Смеси тщательно перемешивают с помощью стандартных процедур, например, в V-образном смесителе, для обеспечения однородности.
Если добавка используется в качестве верхнего слоя для корма, она также помогает обеспечить равномерное распределение соединения по верхней части подготовленного корма.
Питьевую воду и корм, эффективные для увеличения отложений постного мяса и для улучшения соотношения постного мяса и жира, обычно получают путем смешивания соединения по настоящему изобретению с достаточным количеством корма для животных, чтобы обеспечить от примерно 10-3 до примерно 500 частей на миллион соединения в корме или воде.
Предпочтительный лечебный корм для свиней, крупного рогатого скота, овец и коз обычно содержит от примерно 1 до примерно 400 граммов соединения по настоящему изобретению (или комбинации) на тонну корма, оптимальное количество для этих животных обычно составляет примерно от 50 до примерно 300 граммов на тонну корма.
Предпочтительные корма для домашней птицы и домашних животных обычно содержат от примерно 1 до примерно 400 граммов и предпочтительно от примерно 10 до примерно 400 граммов соединения по настоящему изобретению (или их комбинации) на тонну корма.
Для парентерального введения животным соединения настоящего изобретения (или их комбинации) могут быть получены в форме пасты или гранул и введены в виде имплантата, обычно под кожу головы или уха животного, у которого требуется увеличение отложения постного мяса и улучшение соотношения постного мяса и жира.
Составы паст могут быть получены путем диспергирования лекарственного средства в фармацевтически приемлемом масле, таком как арахисовое масло, кунжутное масло, кукурузное масло и т.п.
Гранулы, содержащие эффективное количество соединения по настоящему изобретению, фармацевтической композиции или комбинации, могут быть получены путем смешивания соединения по настоящему изобретению или комбинации с разбавителем, таким как карбовакс, карнубский воск и т.п., и скользящим веществом, таким как, магний или стеарат кальция, могут быть добавлены для улучшения процесса гранулирования.
Конечно, понятно, что животному можно вводить более одной гранулы для достижения желаемого уровня дозы, который обеспечит увеличение отложения постного мяса и желаемое улучшение соотношения постного мяса и жира. Кроме того, имплантаты также могут периодически устанавливаться в течение периода лечения животных, чтобы поддерживать надлежащий уровень лекарственного средства в организме животного.
Настоящее изобретение имеет несколько полезных ветеринарных особенностей. Для владельца домашнего животного или ветеринара, который желает увеличить худобу и/или убрать нежелательный жир у домашних животных, настоящее изобретение предоставляет средства, с помощью которых это может быть достигнуто. Для производителей птицы, говядины и свиней использование способа по настоящему изобретению дает нежирных животных, что приносит более высокие продажные цены в мясной промышленности.
Получение
Соединения по настоящему изобретению могут быть синтезированы способами синтеза, которые включают процессы, аналогичные тем, которые хорошо известны в области химии, особенно в свете описания, содержащегося в настящем документе. Исходные вещества обычно доступны из коммерческих источников, таких как Aldrich Chemicals (Milwaukee, WI) или могут быть легко получены с использованием способов, хорошо известных специалистам в данной области (например, получены способами, в основном описанными в Louis F. Fieser and Mary Fieser, Reagents for Organic Syn-thesis, v. 1-19, Wiley, New York (1967-1999 ed.), или Beilsteins Handbuch der organischen Chemie, 4, Aufl. ed. Springer-Verlag, Berlin, включая приложения (также доступны через онлайн базу данных Beilstein)). Многие из используемых в настоящем документе соединений связаны с соединениями, в отношении которых существует большой научный интерес и коммерческая потребность, или получены из них, и, соответственно, многие такие соединения коммерчески доступны или описаны в литературе, или их легко получить из других общедоступных веществ способами, описанными в литературе.
В иллюстративных целях схемы реакций, изображенные ниже, предоставляют потенциальные пути синтеза соединений по настоящему изобретению, а также ключевых промежуточных соединений. Более подробное описание отдельных стадий реакций см. в разделе «Примеры» ниже. Специалистам в данной области очевидно, что для синтеза соединений по изобретению можно использовать другие пути синтеза. Хотя конкретные исходные вещества и реагенты обсуждаются ниже, их можно легко заменить другими исходными веществами или реагентами для получения различных производных и/или условий реакции. Кроме того, многие соединения, полученные способами, описанными ниже, могут быть дополнительно модифицированы в свете данного описания, с использованием традиционной химии, хорошо известной специалистам в данной области.
Если не указано иное, переменные в схемах A-L имеют те же значения, что определены в настоящем документе. Амины, упомянутые в настоящем документе, могут представлять собой защищенные амины, защита которых снимается в стандартных условиях, известных специалистам в данной области. Необходимость в такой защите/снятии защиты будет варьироваться в зависимости от природы удаленной функциональной группы и условий способов получения. Необходимость в такой защите легко определяется специалистом в данной области техники. Использование таких методов защиты/снятия защиты также находится в компетенции специалистов в данной области. Общее описание защитных групп и их использование см. T.W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991.
Например, некоторые соединения содержат первичные амины или функциональные группы карбоновых кислот, которые могут мешать реакциям на других участках молекулы, если они остаются незащищенными. Соответственно, такие функциональные группы могут быть защищены соответствующей защитной группой, которая может быть удалена на последующей стадии. Подходящие защитные группы для защиты аминов и карбоновых кислот включают такие защитные группы, которые обычно используются в синтезе пептидов (такие как N-трет-бутоксикарбонил (Boc), бензилоксикарбонил (Cbz), и 9-флуоренилметиленоксикарбонил (Fmoc) для аминов и сложный эфир низших алкилов или бензиловые эфиры для карбоновых кислот), которые обычно не являются химически реактивными в описанных условиях реакции и обычно могут быть удалены без химического изменения других функциональных групп в соединениях по настоящему изобретению.
Схемы реакций, описанные ниже, предназначены для предоставления общего описания методологии, применяемой при получении соединений по настоящему изобретению. Некоторые из соединений по настоящему изобретению содержат один хиральный центр. На следующих схемах общие способы получения соединений показаны либо в рацемической, либо в энантиообогащенной форме. Специалисту в данной области очевидно, что все синтетические трансформации могут быть проведены аналогичным образом, независимо от того, являются вещества энантиомерно обогащенными или рацемическими. Кроме того отделение целевого оптически активного вещества может происходить в любой желательной точке последовательности с использованием хорошо известных способов, таких как описанные в данном описании изобретения и в химической литературе.
В следующих схемах реакций переменные R1, R2, R3 и R4 имеют значения, как описано в разделе Сущность изобретения, если не указано иное.
Схема A

Как показано на схеме A, окисление A-1 с получением смеси A-2 [сульфоксид (n=1) и/или сульфон (n=2)] можно осуществлять в соответствующих условиях, таких как пероксимоносульфат калия (Oxone ®) в тетрагидрофуране (THF) и воде в качестве растворителя (это окисление можно использовать, например, в схеме D). Последующее нуклеофильное ароматическое замещение (реакция SNAr) дает 2-замещенные пиридо[2,3-d]пиримидин-7(8H)-оны A-3. Реакции SNAr обычно осуществляют в присутствии подходящего основания, такого как N, N-диизопропилэтиламин (DIPEA) в подходящем растворителе, таком как диметилсульфоксид (DMSO) или тетрагидрофуран (THF).
Схема B

Как показано на схеме B, обработка B-1, такого как 6-бром-2-хлор-8-циклопентил-5-метилпиридо[2,3-d]пиримидин-7(8H)-он (приобретенный или синтезированный способом, аналогичным описанному Duan, S. et al., Org. Process Res. Dev. 2016, 20, 1191-1202) предшественником винилового эфира, таким как трибутил(1-этоксиэтенил)станнан или 1-(этенилокси)бутан, в стандартных условиях перекрестного сочетания, катализируемого палладием, дает виниловый эфир B-2, где R представляет собой H. Альтернативно, реагенты замещенный винилстаннан или алкоксивиниловый эфир могут быть использованы для получения промежуточных соединений B-2, где R является отличным от H, например, где R представляет собой метил, необязательно замещенный одним или несколькими атомами фтора. Реакция SNAr B-2 с различными замещенными аминами в присутствии основания, такого как DIPEA, в подходящем растворителе, таком как DMSO дает B-3. Гидролиз винилового эфира в кислых условиях, таких как водная HCl в THF, дает 2-замещенные пиридо[2,3-d]пиримидин-7(8H)-оны B-4 (где R2 представляет собой, например, CH2R, и R представляет собой H или метил, необязательно замещенный одним или несколькими атомами фтора).
Схема C

Альтернативный путь синтеза B-4 изображен на схеме C. Гидролиз винилового эфира B-2 в присутствии кислоты, такой как водная HCl в THF, дает C-1. Реакция SNAr C-1 с различными замещенными аминами в присутствии основания, такого как DIPEA, в подходящем растворителе, таком как DMSO, дает B-4.
Схема D

На схеме D показана альтернативная реакция SNAr B-1 с различными замещенными аминами с получением пиримидина D-1. Функционализация D-1 в положении C(6) достигается обработкой подходящим замещенным винилстаннаном или алкоксивиниловым эфиром в стандартных условиях перекрестного сочетания, известных специалистам в данной области, с последующим кислотным гидролизом полученного винилового эфира, с получением промежуточного кетона B-4. Альтернативно, реакция SNAr B-1 с метантиолатом натрия в подходящем растворителе, таком как THF или DMSO, дает метилтиопиримидин D-2, за которой следуют стандартные условия перекрестного сочетания, и кислотный гидролиз с получением кетона D-3. Следуя процедуре, описанной в схеме A, можно получить B-4.
Схема E

Как показано на схеме E, реакция SNAr E-1 с различными замещенными аминами в щелочных условиях, например с триэтиламином в ацетонитриле в качестве растворителя, дает E-2. Реакция Хека E-2 с соответствующим образом замещенными еновыми кислотами и сопутствующая циклизация с использованием уксусного ангидрида дает пиридо[2,3-d]пиримидин-7(8H)-оны E-3. Бромирование в положении C(6) с использованием стандартных реагентов, таких как бром, в подходящем растворителе, таком как дихлорметан, дает бромид E-4. Реакция SNAr с различными замещенными аминами, как описано ранее, дает E-5. Установка кетонового фрагмента в положение C(6) достигается обработкой его винилстаннаном или алкоксивиниловым эфиром в стандартных условиях перекрестного сочетания, с последующим кислотным гидролизом соответствующего винилового эфира с получением E-6.
Схема F

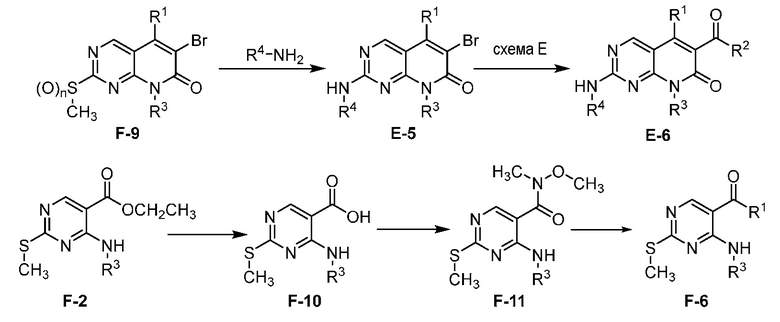
Как показано на схеме F, реакция SNAr этил 4-хлор-2-(метилтио)пиримидин-5-карбоксилата (F-1; приобретенный или синтезированный способом, аналогичным описанному Federico, S. et al., J. Med. Chem. 2014, 57, 6210-6225) с различными замещенными аминами в щелочных условиях дает F-2. Превращение сложноэфирной функциональной группы в F-2 в соответствующий кетон проводят с использованием стандартных методов, известных в данной области, для получения F-6. Например, при восстановлении F-2 алюмогидридом лития (LiAlH4) образуется спирт F-3, за которым следует окисление оксидом марганца(IV) с получением альдегида F-4. Добавление подходящего реагента Гриньяра, такого как метилмагнийхлорид (где R1=Me), к F-4 дает спирт F-5, который затем превращается в кетон F-6 путем окисления оксидом марганца(IV). Пиридо[2,3-d]пиримидин-7(8H)-он F-7 получают циклоконденсацией с использованием либо стандартных условий реакции Хорнера-Вадсворта-Эммонса, либо этилацетата и подходящего основания, такого как натрий бис(триметилсилил)амид. Бромирование в положении C(6), которое дает F-8, сопровождается окислением тиометильной группы по C(2) в стандартных условиях, описанных ранее (схема A), с получением F-9. Реакция SNAr с различными замещенными аминами, как описано ранее, дает E-5. E-6 затем получают из E-5, как описано на схеме E. Альтернативно, введение кетонового фрагмента в F-8 в условиях, аналогичных описанным на схеме D, дает A-1, который затем используют в соответствии с процедурой, описанной на схеме A, для получения A-3. Альтернативный подход к превращению F-2 в F-6 представляет собой гидролиз сложного эфира F-2 с образованием кислоты F-10, которая впоследствии превращается в амид Вайнреба F-11. Добавление подходящего реактива Гриньяра, такого как метилмагнийхлорид (в случае, когда R1=Me) к F-11 в THF дает F-6.
Схема G

Как показано на схеме г, алкилирование 5-метил-2-(метилтио)пиридо[2,3-d]пиримидин-7(8H)-она G-1 (приобретенный или синтезированный способом, аналогичным описанному VanderWel, S. N. et al., J. Med. Chem. 2005, 48, 2371-2387) соединением R3-X, таким как алкилгалогенид или мезилат (т.е. метансульфонат), в присутствии основания, такого как 2-трет-бутил-1,1,3,3-тетраметилгуанидин, дает смесь позиционных изомеров 7-O- и 8-N-алкил пиридо[2,3-d]пиримидинов G-2 и F-7, соответственно, которые разделяются с получением F-7, который можно использовать, как показано на схеме F, для получения E-6.
Схема H

Как показано на схеме H, амидирование/циклизация 1-[4-амино-2-(метилтио)пиримидин-5-ил]этан-1-она H-1 с предшественником дикетена (т.е., 2,2,6-триметил-4H-1,3-диоксин-4-он) с растворителями или без них, такими как толуол, дает метилтиопиримидин H-2. Окисление тиометильной группы H-2 в стандартных условиях (см. выше) с последующей реакцией SNAr с различными замещенными аминами R4-NH2 в щелочных условиях, дает H-4. Стандартная реакция SN2 между H-4 и R3-X (например, алкилгалогенидами) в щелочных условиях, с использованием основания, такого как NaH, дает смесь 7-O- и 8-N-алкил пиридо[2,3-d]пиримидинов H-5 и E-6 (соединение A-3, где R2 представляет собой CH3), соответственно. Разделение этих позиционных изомеров дает E-6, который можно использовать, как обсуждается в настоящем документе.
Схема I

Как показано на схеме I, для улучшения растворимости и обеспечения возможности последующих превращений, H-3 подвергали реакции SNAr с различными алкилтиолами R’-SH, где R’ представляет собой алкил, имеющий по крайней мере 4 атома углерода (например, децилтиол), в щелочных условиях, такие как триэтиламин и THF в качестве растворителя, для образования I-1. Фотокатализируемое региоселективное N-алкилирование I-1 с использованием различных карбоновых кислот R3-CO2H, дает пиридопиримидин I-2. Эта реакция может быть проведена с использованием бис(ацетилокси)(2,4,6-триметилфенил)-λ3-иодана в качестве окислителя и (2,2'-бипиридин-κ2N1,N1'){бис[3,5-дифтор-2-(5-фторпиридин-2-ил)фенил]}иридий гексафторфосфат в качестве катализатора в дихлорметане. Окисление тиометильной группы по C(2) в стандартных условиях, описанных выше, дает I-3. Реакция SNAr I-3 с различными замещенными аминами R4-NH2 в стандартных условиях дает E-6.
Схема J

Как показано на схеме J, обработка коммерчески доступного 4-хлор-2-(метилтио)пиримидина J-1 основанием, таким как диизопропиламид лития (LDA) в подходящем растворителе, таком как THF, с последующим добавлением подходящего замещенного альдегида R1CHO, дает спирт J-2. Окисление спирта J-2 в стандартных условиях, например, оксид марганца(IV) в хлороформе, дает кетон J-3. Реакция SNAr J-3 с различными замещенными аминами R3-NH2 в щелочных условиях дает F-6, который может давать, например, E-6, как показано на схеме F.
Схема K

Как показано на схеме K, реакция SNAr 1-(4,6-дихлорпиридин-3-ил)этан-1-она K-1 (приобретенный или синтезированный способом, аналогичным описанному McCoull, W. et al., J. Med. Chem. 2017, 60, 3187-3197) в положении C-4 с различными замещенными аминами R3-NH2 в щелочных условиях дает K-2. Амидирование/циклизация K-2 с предшественником дикетена (например, 2,2,6-триметил-4H-1,3-диоксин-4-он) дает метилкетон K-3. Стандартная реакция C-N сочетания, катализируемая переходными металлами, K-3 с различными аминами R4-NH2 в подходящем растворителе, таком как толуол или THF, дает 1,6-нафтиридин-2(1H)-он K-4.
Как очевидно для специалиста в данной области, способы, описанные на схемах с E по K для введения R3-заместителя, позволяют вводить широкий спектр R3-заместителей, включая (но не ограничиваясь ими) алкильные, циклоалкильные и гетероциклоалкильные группы. Это может быть осуществлено просто путем использования соответственно замещенного аминного реагента (схемы E, F, J), галогенидного реагента (схемы г, H) или реагента карбоновой кислоты (схема I).
Схема L

Как показано на схеме L, реакция SNAr коммерчески доступного метил 4,6-дихлорникотината L-1 с различными замещенными аминами R3-NH2 в щелочных условиях дает L-2. Превращение сложноэфирной функциональной группы L-2 в соответствующую кетонную группу осуществляли с использованием стандартных способов, известных в данной области (например, описанных на схеме F), с получением кетона L-3. Циклоконденсация L-3 с использованием либо стандартных условий Хорнера-Вадсворта-Эммонса, либо этилацетата и подходящего основания, такого как бис(триметилсилил)амид натрия, дает нафтиридин-2(1H)-он L-4. Стандартная реакция C-N сочетания, катализируемая переходными металлами, L-4 с различными замещенными аминами R4-NH2 в щелочных условиях дает L-5. После бромирования L-5 в положении C(6) кетонный фрагмент вводят обработкой соответствующим образом замещенным винилстаннаном или алкоксивиниловым эфиром в стандартных условиях реакции перекрестного сочетания с последующим кислотным гидролизом с получением аминопиридина L-7.
Примеры
Экспериментальные процедуры
Следующее иллюстрирует синтез различных соединений по настоящему изобретению. Дополнительные соединения в рамках настоящего изобретения могут быть получены с использованием способов, проиллюстрированных в этих примерах, либо по отдельности, либо в сочетании с методами, обычно известными в данной области. Эксперименты обычно осуществляли в инертной атмосфере (азот или аргон), особенно в тех случаях, когда использовали реагенты или промежуточные соединения, чувствительные к кислороду или влаге. Коммерческие растворители и реагенты обычно использовали без дополнительной очистки. При необходимости использовали безводные растворители, как правило, продукты AcroSeal® от Acros Organics, Aldrich® Sure/Seal™ от Sigma-Aldrich, или продукты DriSolv® от EMD Chemicals. В других случаях коммерческие растворители пропускали через колонки, заполненные молекулярными ситами 4Å, до тех пор, пока не были достигнуты следующие стандарты контроля качества для воды: a) <100 частей на миллион для дихлорметана, толуола, N, N-диметилформамида и тетрагидрофурана; b) <180 частей на миллион для метанола, этанола, 1,4-диоксана и диизопропиламина. Для очень чувствительных реакций растворители дополнительно обрабатывали металлическим натрием, гидридом кальция или молекулярными ситами и перегоняли непосредственно перед использованием. Как правило, продукты сушили в вакууме перед тем, как использовать их в дальнейших реакциях или отправить на биологические исследования. Данные масс-спектрометрии представлены с помощью жидкостной хроматографии-масс-спектрометрии (LCMS), химической ионизации при атмосферном давлении (APCI) или газовой хроматографии-масс-спектрометрии (GCMS). Химические сдвиги для данных ядерного магнитного резонанса (ЯМР) выражены в частях на миллион (ppm, δ) относительно остаточных пиков от используемых дейтерированных растворителей (хлороформ, 7,26 ppm; CD2HOD, 3,31 ppm; ацетонитрил-d2, 1,94 ppm; диметилсульфоксид-d5, 2,50 ppm; DHO, 4,79 ppm). В некоторых примерах было выполнено хиральное разделение для разделения энантиомеров определенных соединений по изобретению (в некоторых примерах разделенные энантиомеры обозначены как ENT-1 и ENT-2, в соответствии с их порядком элюирования). В некоторых примерах оптическое вращение энантиомера измеряли с помощью поляриметра. Согласно его наблюдаемым данным вращения (или его данным о удельном вращении), энантиомер с вращением по часовой стрелке был обозначен как (+)-энантиомер, а энантиомер с вращением против часовой стрелки был обозначен как (-)-энантиомер. Аналогичным образом, в некоторых примерах было проведено разделение для разделения диастереомеров определенных соединений изобретения; в некоторых примерах разделенные диастереомеры обозначены как DIAST-1 и DIAST-2 в соответствии с их порядком элюирования. На рацемические соединения указывает либо отсутствие нарисованной или описанной стереохимии, либо присутствие (+/-), примыкающего к структуре; в этом последнем случае указанная стереохимия представляет собой только один из двух энантиомеров, которые составляют рацемическую смесь.
Реакции, протекающие через детектируемые промежуточные соединения, как правило, сопровождались LCMS и продолжались до полного превращения перед добавлением последующих реагентов. Для способов синтеза, относящихся к другим примерам или способам, условия реакции (время реакции и температура) могут изменяться. Как правило, за ходом реакций наблюдали с помощью тонкослойной хроматографии или масс-спектрометрии и реакционные смеси подвергали соответствующей обработке, когда это было необходимо. . Процедуры очистки могут варьировать между экспериментами: в общем, растворители и соотношения растворителей, используемые для элюентов/градиентов, выбирали так, чтобы обеспечить соответствующие значения Rf или времени удерживания. Все исходные вещества в этих разделах Получения и Примеры либо коммерчески доступны, либо могут быть получены способами, известными в данной области техники, или описанными в настоящем документе.
Соединения и промежуточные соединения, описанные ниже, были названы с использованием соглашения о наименованиях, предоставленного ACD/ChemSketch 2017.2.1, File Version N40E41, Build 96719 (Advanced Chemistry Development, Inc., Toronto, Ontario, Canada). Соглашение о наименованиях, предоставляемое ACD/ChemSketch 2017.2.1, хорошо известно специалистам в данной области, и считается, что соглашение о наименованиях, предоставляемое ACD/ChemSketch 2017.2.1, в целом соответствует рекомендациям IUPAC (International Union for Pure and Applied Chemistry) по Nomenclature of Organic Chemistry and the CAS Index rules.
Если не указано иное, исходные вещества обычно доступны из коммерческих источников, таких как Aldrich Chemicals Co. (Milwaukee, WI), Lancaster Synthesis, Inc. (Windham, NH), Acros Organics (Fairlawn, NJ), Maybridge Chemical Company, Ltd. (Cornwall, England) и Tyger Scientific (Princeton, NJ). Были использованы некоторые общепринятые сокращения и аббревиатуры, которые могут включать: AcOH (уксусная кислота), DBU (1,8-диазабицикло[5.4.0]ундец-7-ен), CDI (1,1'-карбонилдиимидазол), DCM (дихлорметан), DEA (диэтиламин), DIPEA (N, N-диизопропилэтиламин), DMAP (4-диметиламинопиридин), DMF (N, N’-диметилформамид), DMSO (диметилсульфоксид), EDCI (N-(3-диметиламинопропил)-N’-этилкарбодиимид), Et2O (диэтиловый эфир), EtOAc (этилацетат), EtOH (этанол), HATU (2-(1H-7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфат метанаминия), HBTU (O-бензотриазол-1-ил-N, N,N’,N’-тетраметилурония гексафторфосфат), HOBT (1-гидроксибензотриазол), IPA (изопропиловый спирт), KHMDS (гексаметилдисилазан калия), MeOH (метанол), MTBE (трет-бутил-метиловый эфир), NaBH(OAc)3 (триацетоксиборгидрид натрия), NaHMDS (натрия гексаметилдисилазан), NMP (N-метилпирролидон), SEM ([2-(Триметилсилил)этокси]метил), TEA (триэтиламин), TFA (трифторуксусная кислота), THF (тетрагидрофуран), 5-FAM-Dyrktide (пептид RRRFRPASPLRGPPK, который мечен трифторацетатной солью на 5’-конце пептида).и T3P (ангидрид пропанфосфоновой кислоты).
Термины «концентрированный», «выпаренный» и «концентрированный в вакууме» относятся к удалению растворителя при пониженном давлении на роторном испарителе с температурой бани менее 60°C. Аббревиатуры «мин» и «ч» обозначают «минуты» и «часы», соответственно. Термин «TLC» относится к тонкослойной хроматографии, «комнатная температура или температура окружающей среды» означает температуру от 18°C до 25°C, “LCMS” относится к жидкостной хроматографии-масс-спектрометрии, “UPLC” относится к сверхэффективной жидкостной хроматографии и “ВЭЖХ” относится к высокоэффективной жидкостной хроматографии (ВЭЖХ).
Гидрирование можно проводить в шейкере Парра под давлением газообразного водорода или в проточном реакторе для гидрирования Thales-nano H-Cube при полном водороде и скорости потока 1-2 мл/мин при заданной температуре.
Время удерживания ВЭЖХ, UPLC, LCMS и SFC измеряли с использованием методов, указанных в способах.
Получение Р1
6-бром-2-хлор-8-циклобутил-5-метилпиридо[2,3-d]пиримидин-7(8H)-он (P1)

Стадия 1. Синтез 5-бром-2-хлор-N-циклобутилпиримидин-4-амина (C1).
Циклобутанамин (1,56 г, 21,9 ммоль) добавляли к суспензии 0°C 5-бром-2,4-дихлорпиримидина (5,00 г, 21,9 ммоль) и триэтиламина (9,51 мл, 68,2 ммоль) в ацетонитриле, и реакционной смеси давали нагреться до комнатной температуры при перемешивании в течение ночи. Смесь затем концентрировали в вакууме и остаток очищали с помощью хроматографии на силикагеле (градиент: 0% - 30% этилацетата в гептане) с получением C1 в виде твердого вещества. Выход: 4,67 г, 17,8 ммоль, 81%. LCMS m/z 262,1 (наблюдали изотопную картину брома-хлора) [M+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 8,10 (с, 1H), 5,61 (шир, с, 1H), 4,67-4,52 (м, 1H), 2,52-2,41 (м, 2H), 2,03-1,89 (м, 2H), 1,86-1,74 (м, 2H).
Стадия 2. Синтез 2-хлор-8-циклобутил-5-метилпиридо[2,3-d]пиримидин-7(8H)-она (C2).
Триэтиламин (16,6 мл, 119 ммоль) и 1-метилпирролидин-2-он (70 мл) добавляли к смеси C1 (4,67 г, 17,8 ммоль), (2E)-бут-2-еновой кислоты (6,13 г, 71,2 ммоль) и ацетата палладия(II) (399 мг, 1,78 ммоль), и полученную смесь продували азотом в течение 20 минут. Реакционную смесь нагревали в течение ночи при 65°C, после чего ее обрабатывали триэтиламином (1,24 мл, 8,90 ммоль) и уксусным ангидридом (3,36 мл, 35,5 ммоль), и нагревали при 65°C в течение дополнительных 2,5 часов. После охлаждения до комнатной температуры реакционную смесь выливали в воду (500 мл) и экстрагировали этилацетатом (4×120 мл). LCMS анализ объединенных органических слоев в этот момент показал преобразование в продукт C2: LCMS m/z 250,1 (наблюдали изотопную картину хлора) [M+H]+. Объединенные органические слои последовательно промывали водой и насыщенным водным раствором хлорида натрия, концентрировали при пониженном давлении, и очищали с помощью хроматографии на силикагеле (градиент: 0% - 45% этилацетата в гептане), с получением C2 в виде твердого вещества. Выход: 3,22 г, 12,9 ммоль, 72%. 1H ЯМР (400 МГц, хлороформ-d) δ 8,73 (с, 1H), 6,51 (кв, J=1,3 Гц, 1H), 5,83-5,71 (м, 1H), 3,15-3,02 (м, 2H), 2,43 (д, J=1,3 Гц, 3H), 2,42-2,32 (м, 2H), 2,12-1,99 (м, 1H), 1,94-1,79 (м, 1H).
Стадия 3. Синтез 6-бром-2-хлор-8-циклобутил-5-метилпиридо[2,3-d]пиримидин-7(8H)-она (P1).
Смесь С2 (2,00 г, 8,01 ммоль), N-бромсукцинимида (2,49 г, 14,0 ммоль) и щавелевой кислоты (147 мг, 1,63 ммоль) в ацетонитриле (40 мл) перемешивали при 55°C в течение 5 часов, после чего снова добавляли щавелевую кислоту (50 мг, 0,55 ммоль) и N-бромсукцинимид (800 мг, 4,49 ммоль). После нагревания в течение дополнительных 3,5 часов, реакционную смесь охлаждали до комнатной температуры, обрабатывали раствором бисульфита натрия (1,68 г, 16,1 ммоль) в воде (приблизительно 10 мл), и перемешивали в течение 10 минут при комнатной температуре. Затем полученную смесь разбавляли водой (150 мл); фильтрация дала P1 в виде твердого вещества. Выход: 2,13 г, 6,48 ммоль, 81%. LCMS m/z 328,0 (наблюдали изотопную картину брома-хлора) [M+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 8,84 (с, 1H), 5,90-5,77 (м, 1H), 3,13-2,99 (м, 2H), 2,66 (с, 3H), 2,48-2,37 (м, 2H), 2,14-2,01 (м, 1H), 1,95-1,81 (м, 1H).
Получение Р2
трет-бутил 6-[6-ацетил-5-метил-2-(метилсульфинил)-7-оксопиридо[2,3-d]пиримидин-8(7H)-ил]-2-азаспиро[3.3]гептан-2-карбоксилат (P2)


Стадия 1. Синтез трет-бутил 6-{[5-ацетил-2-(метилтио)пиримидин-4-ил]амино}-2-азаспиро[3.3]гептан-2-карбоксилата (C3).
1-[4-Хлор-2-(метилтио)пиримидин-5-ил]этанон (2,15 г, 10,6 ммоль), трет-бутил 6-амино-2-азаспиро[3.3]гептан-2-карбоксилат (2,36 г, 11,1 ммоль) и N, N-диизопропилэтиламин (5,54 мл, 31,8 ммоль) растворяли в смеси тетрагидрофурана (20 мл) и ацетонитрила (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов, после чего ее концентрировали досуха, и остаток подвергали хроматографии на силикагеле (градиент: 0% - 20% этилацетата в дихлорметане), с получением C3 (4,18 г) в виде светло-желтой смолы. По данным анализа 1H ЯМР, это вещество содержало некоторое количество N, N-диизопропилэтиламин гидрохлорида. Скорректированный выход с учетом N, N-диизопропилэтиламин гидрохлорида: 3,90 г, 10,3 ммоль, 97%. LCMS m/z 379,0 [M+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 9,26 (ушир.д, J=6,3 Гц, 1H), 8,56 (с, 1H), 4,55-4,42 (м, 1H), 4,00 (с, 2H), 3,88 (с, 2H), 2,73-2,63 (м, 2H), 2,52 (с, 3H), 2,50 (с, 3H), 2,23-2,13 (м, 2H), 1,43 (с, 9H).
Стадия 2. Синтез трет-бутил 6-[5-метил-2-(метилтио)-7-оксопиридо[2,3-d]пиримидин-8(7H)-ил]-2-азаспиро[3.3]гептан-2-карбоксилата (C4).
К охлажденному льдом раствору C3 (1,70 г, 4,49 ммоль) и этилацетата (1,39 г, 15,8 ммоль) в тетрагидрофуране (20 мл) добавляли литий бис(триметилсилил)амид (1 M раствор; 13,5 мл, 13,5 ммоль) частями, со скоростью 0,5 мл каждые 10 секунд. Реакционную смесь перемешивали при охлаждении льдом в течение 30 минут, и затем при 40°C в течение приблизительно 1 часа, после чего ее разбавляли водным раствором хлорида аммония (5 мл), и и объединяли из аналогичной реакции, проводимой с использованием C3 (100 мг, 0,264 ммоль). После разделения полученной смеси между этилацетатом (30 мл) и насыщенным водным раствором хлорида натрия (20 мл), водный слой экстрагировали этилацетатом (2×10 мл), и объединенные органические слои сушили над сульфатом натрия, фильтровали, и концентрировали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (градиент: 0% - 30% этилацетата в дихлорметане) с получением C4 в виде светло-желтого твердого вещества. Общий выход: 1,65 г, 4,10 ммоль, 86%. LCMS m/z 402,9 [M+H]+.
Стадия 3. Синтез трет-бутил 6-[6-бром-5-метил-2-(метилтио)-7-оксопиридо[2,3-d]пиримидин-8(7H)-ил]-2-азаспиро[3.3]гептан-2-карбоксилата (C5).
Щавелевая кислота (36,9 мг, 0,410 ммоль) и N-бромсукцинимид (802 мг, 4,51 ммоль) добавляли порциями к охлажденному льдом раствору C4 (1,65 г, 4,10 ммоль) в смеси ацетонитрила (20 мл) и дихлорметана (10 мл). После перемешивания реакционной смеси в течение 45 минут при 0°C, ее разбавляли дихлорметаном (50 мл) и насыщенным водным раствором хлорида натрия (10 мл), и затем обрабатывали водным раствором сульфита натрия (5 мл). Полученный водный слой экстрагировали дихлорметаном (2×10 мл), и объединенные органические слои сушили над сульфатом натрия, фильтровали, и концентрировали в вакууме. Хроматография на силикагеле (градиент: 0% - 30% этилацетата в дихлорметане) давала C5 в виде светло-желтого твердого вещества. Выход: 1,45 г, 3,01 ммоль, 73%. LCMS m/z 481,1 (наблюдали изотопную картину брома) [M+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 8,79 (с, 1H), 6,03-5,84 (ушир.м, 1H), 4,08 (с, 2H), 4,05 (с, 2H), 3,35-3,22 (м, 2H), 2,67-2,56 (м, 2H), 2,63 (с, 3H), 2,62 (с, 3H), 1,44 (с, 9H).
Стадия 4. Синтез трет-бутил 6-[6-ацетил-5-метил-2-(метилтио)-7-оксопиридо[2,3-d]пиримидин-8(7H)-ил]-2-азаспиро[3.3]гептан-2-карбоксилата (C7).
Азот барботировали через суспензию C5 (2,00 г, 4,15 ммоль) в смеси толуола (30 мл) и 1,4-диоксана (15 мл), и затем добавляли последовательно трибутил(1-этоксиэтенил)станнан (7,50 г, 20,8 ммоль) и тетракис(трифенилфосфин)палладий(0) (480 мг, 0,415 ммоль). Реакционный сосуд закрывали крышкой, и реакционную смесь нагревали при 110°C в течение 6 часов. Затем смесь фильтровали; после концентрирования фильтрата в вакууме остаток растворяли в этилацетате (50 мл), обрабатывали хлористоводродной кислотой (1 M; 10 мл), и перемешивали в течение приблизительно 1 часа, пока анализ тонкослойной хроматографией (элюент: 1:2 этилацетат/петролейный эфир) не укажет на полное превращение промежуточного соединения C6 {трет-бутил 6-[6-(1-этоксиэтенил)-5-метил-2-(метилтио)-7-оксопиридо[2,3-d]пиримидин-8(7H)-ил]-2-азаспиро[3.3]гептан-2-карбоксилат} в C7. Органический слой сушили над сульфатом натрия, фильтровали, и концентрировали при пониженном давлении. Хроматография на силикагеле (градиент: 0% - 70% этилацетата в петролейном эфире) давала C7 в виде светло-желтого твердого вещества. Выход: 1,30 г, 2,92 ммоль, 70%. LCMS m/z 389,1 [(M - 2-метилпроп-1-ен)+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 8,79 (с, 1H), 5,87-5,71 (м, 1H), 4,05 (с, 2H), 4,05 (с, 2H), 3,36-3,23 (м, 2H), 2,66-2,57 (м, 2H), 2,62 (с, 3H), 2,53 (с, 3H), 2,36 (с, 3H), 1,44 (с, 9H).
Стадия 5. Синтез трет-бутил 6-[6-ацетил-5-метил-2-(метилсульфинил)-7-оксопиридо[2,3-d]пиримидин-8(7H)-ил]-2-азаспиро[3.3]гептан-2-карбоксилата (P2).
К 0°C раствору C7 (100 мг, 0,225 ммоль) в тетрагидрофуране (8 мл) добавляли пероксимоносульфат калия (Oxone®; 207 мг, 0,337 ммоль), затем воду (4 мл). Реакционную смесь перемешивали при комнатной температуре (10°C) в течение 1 часа, после чего ее распределяли между этилацетатом (15 мл) и насыщенным водным раствором хлорида натрия (5 мл). Водный слой экстрагировали этилацетатом (2×5 мл), и объединенные органические слои сушили над сульфатом натрия, фильтровали, и концентрировали в вакууме, получая P2 (120 мг) в виде светло-желтого твердого вещества. Это вещество использовали непосредственно в последующей химии.
Получение Р3
6-бром-2-хлор-5-метил-8-[(1R,2S)-2-метилциклопентил]пиридо[2,3-d]пиримидин-7(8H)-она (P3)
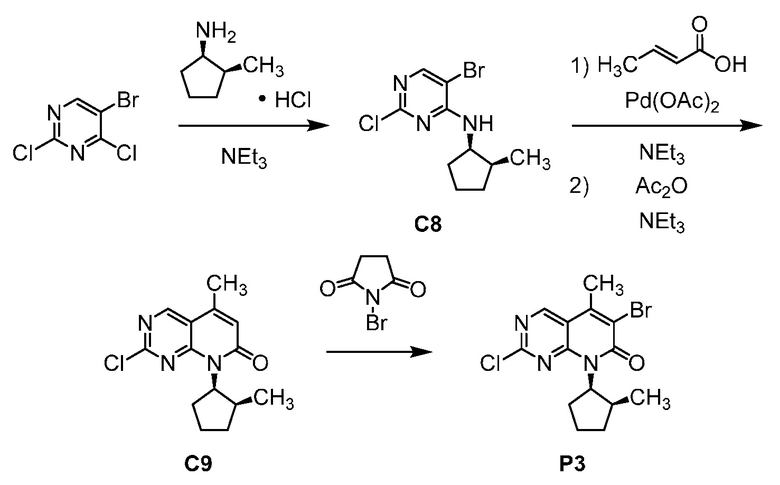
Стадия 1. Синтез 5-бром-2-хлор-N-[(1R,2S)-2-метилциклопентил]пиримидин-4-амина (C8).
Смесь 5-бром-2,4-дихлорпиримидина (21,5 г, 94,4 ммоль) и (1R,2S)-2-метилциклопентанамин гидрохлорида (это вещество получали с использованием способа, описанного W. Wiehl and A. W. Frahm, Chemische Berichte 1986, 119, 2668-2677; его абсолютная стереохимия была дополнительно установлена путем определения рентгеновской кристаллической структуры на производном P3, см. ниже) (12,8 г, 94,4 ммоль) дегазировали азотом в течение 3 минут, после чего добавляли ацетонитрил (500 мл), и смесь охлаждали до 0°C. Добавляли по каплям триэтиламин (38,2 г, 377 ммоль), и реакционную смесь перемешивали при 0°C в течение 1 часа, и затем перемешивали при 15°C в течение 12 часов. Затем реакционную смесь распределяли между насыщенным водным раствором бикарбоната натрия (400 мл) и дихлорметаном (400 мл). Водный слой экстрагировали дихлорметаном (4×200 мл), и объединенные органические слои сушили над сульфатом натрия, фильтровали, и концентрировали в вакууме. Очистку проводили с помощью хроматографии на силикагеле (градиент: 0% - 4% этилацетата в петролейном эфире), с получением C8 в виде масла. Выход: 25,5 г, 87,8 ммоль, 93%. 1H ЯМР (400 МГц, хлороформ-d) δ 8,09 (с, 1H), 5,44 (ушир.д, J=7,3 Гц, 1H), 4,55-4,43 (м, 1H), 2,35-2,23 (м, 1H), 2,16-2,03 (м, 1H), 1,98-1,87 (м, 1H), 1,84-1,71 (м, 1H), 1,70-1,51 (м, 2H), 1,42-1,31 (м, 1H), 0,90 (д, J=7,0 Гц, 3H).
Стадия 2. Синтез 2-хлор-5-метил-8-[(1R,2S)-2-метилциклопентил]пиридо[2,3-d]пиримидин-7(8H)-она (C9).
Эту реакцию проводили в трех одинаковых партиях. Триэтиламин (14,8 г, 146 ммоль) и 1-метилпирролидин-2-он (100 мл) добавляли к смеси C8 (8,50 г, 29,2 ммоль), (2E)-бут-2-еновой кислоты (7,56 г, 87,8 ммоль), и ацетата палладия(II) (656 мг, 2,92 ммоль). Реакционную смесь дегазировали азотом в течение 15 минут, и затем перемешивали в течение 2 часов при 65°C. После охлаждения до комнатной температуры (15°C), реакционную смесь обрабатывали триэтиламином (1,48 г, 14,6 ммоль) и уксусным ангидридом (5,97 г, 58,5 ммоль), и нагревали при 65°C в течение 1 часа. На этом этапе три партии объединяли, выливали в ледяную воду (1,2 л), и экстрагировали этилацетатом (4×300 мл). Объединенные органические слои последовательно промывали насыщенным водным раствором фторида калия (300 мл), водой (300 мл), и насыщенным водным раствором хлорида натрия (300 мл), сушили над сульфатом натрия, фильтровали, и концентрировали в вакууме. Хроматография на силикагеле (элюент: 15% этилацетата в петролейном эфире) давала желтое твердое вещество (17 г), которое нагревали в смеси этилацетата и петролейного эфира 1:10, охлаждали и фильтровали с получением C9 (11,9 г) в виде светло-желтого твердого вещества. Маточный раствор концентрировали при пониженном давлении и очищали с помощью хроматографии на силикагеле, получая дополнительный C9 (4,4 г) в виде светло-желтого твердого вещества. Общий выход: 16,3 г, 58,7 ммоль, 67%. LCMS m/z 277,9 (наблюдали изотопную картину хлора) [M+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 8,73 (с, 1H), 6,56-6,51 (м, 1H), 5,94 (ддд, J=9,9, 9,8, 6,7 Гц, 1H), 2,62-2,35 (м, 2H), 2,45 (д, J=1,3 Гц, 3H), 2,17-2,05 (м, 1H), 2,05-1,93 (м, 2H), 1,91-1,81 (м, 1H), 1,65-1,50 (м, 1H), 0,74 (д, J=7,1 Гц, 3H).
Стадия 3. Синтез 6-бром-2-хлор-5-метил-8-[(1R,2S)-2-метилциклопентил]пиридо[2,3-d]пиримидин-7(8H)-она (P3).
Раствор C9 (11,9 г, 42,8 ммоль) в N, N-диметилформамиде (120 мл) делили на 6 реакционных сосудов, которые индивидуально нагревали до 80°C. N-Бромсукцинимид (3,84 г, 21,6 ммоль) добавляли к каждой реакционной смеси 8 порциями; каждую порцию добавляли в течение 2 минут. Полученные реакционные смеси перемешивали при 80°C в течение ночи, после чего выливали в ледяную воду (1 л) и оставляли на 12 часов. Полученное твердое вещество собирали фильтрацией и очищали с помощью хроматографии на силикагеле (градиент: 10% - 20% этилацетата в петролейном эфире) с получением P3 в виде светло-желтого твердого вещества. Выход: 10,74 г, 30,1 ммоль, 70%. LCMS m/z 356,1 (наблюдали изотопную картину брома) [M+H]+. 1H ЯМР (400 МГц, DMSO-d6) δ 9,21 (с, 1H), 5,91 (ддд, J=9,8, 9,7, 6,6 Гц, 1H), 2,66 (с, 3H), 2,45-2,31 (м, 2H), 2,06-1,86 (м, 3H), 1,86-1,77 (м, 1H), 1,65-1,49 (м, 1H), 0,67 (д, J=7,0 Гц, 3H).
Образец P3 (30 мг) растворяли в диэтиловом эфире (2,0 мл) и обрабатывали гептаном (2,0 мл); смеси давали отстояться в течение выходных в незапечатанном сосуде. Один из полученных кристаллов использовали для рентгеноструктурного анализа (см. ниже).
Монокристальный рентгеноструктурный анализ P3
Рентгеноструктурные анализы монокристалла
Сбор данных проводили на дифрактометре Bruker D8 Quest при комнатной температуре. Сбор данных состоял из омега- и фи- сканирований.
Структуру определяли путем внутреннего фазирования с использованием программного пакета SHELX в моноклинной пространственной группе P21. В дальнейшем структуру уточняли полноматричным методом наименьших квадратов. Все неводородные атомы находили и уточняли с использованием параметров анизотропного смещения.
Атомы водорода помещали в расчетных положениях и позволяли им перемещаться на своих атомах-носителях. Конечное уточнение включало параметры изотропного смещения для всех атомов водорода.
Анализ абсолютной структуры с использованием методов правдоподобия (Hooft, 2008) проводили с использованием PLATON (Spek). Результаты показывают, что абсолютная структура была правильно определена. Метод вычисляет, что вероятность того, что структура является правильной, равна 100%. Параметр Хуфта определяется как 0,018 с esd (8) и параметр Парсона определяется как 0,024 с esd (9). Асимметричная единица содержит две молекулы P3, и абсолютная конфигурация подтверждена для обеих из них.
Конечный R-фактор составлял 2,8%. Конечная разностная карта Фурье не показала отсутствующей или неверно установленной электронной плотности.
Соответствующая информация о кристалле, сборе данных и уточнении представлена в таблице А. Атомные координаты, длины связей, углы связей и параметры смещения приведены в таблицах B - D.
Программное обеспечение и ссылки на источники
SHELXTL, Version 5,1, Bruker AXS, 1997.
PLATON, A. L. Spek, J. Appl. Cryst. 2003, 36, 7-13.
MERCURY, C. F. Macrae, P. R. Edington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler, and J. van de Streek, J. Appl. Cryst. 2006, 39, 453-457.
OLEX2, O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard, and H. Puschmann, J. Appl. Cryst. 2009, 42, 339-341.
R. W. W. Hooft, L. H. Straver, and A. L. Spek, J. Appl. Cryst. 2008, 41, 96-103.
H. D. Flack, Acta Cryst. 1983, A39, 867-881.

Таблица B. Атомные координаты (x 104) и эквивалентные параметры изотропного смещения (Å2×103) для P3. U(eq) представляет собой 1/3 следа ортогонализированного тензора Uij.


Таблица C. Длины [Å] и углы [°] связей для P3.







Таблица D. Параметры анизотропного смещения (Å2×103) для P3. Показатель коэффициента анизотропного смещения принимает вид: -2π2[h2 a*2U11 + ... + 2 h k a* b* U12 ].


Получение Р4
3-ацетил-7-хлор-1-циклопентил-4-метил-1,6-нафтиридин-2(1H)-он (P4)

Стадия 1. Синтез 1-[6-хлор-4-(циклопентиламино)пиридин-3-ил]этанона (C10).
Смесь 1-(4,6-дихлорпиридин-3-ил)этанона (176 г, 926 ммоль), циклопентанамина (197 г, 2,31 моль), и карбоната калия (448 г, 3,24 моль) в ацетонитриле (1,76 л, 10 объемов; для единиц, представленных в этом эксперименте как «объемы», 1 объем=0,176 л) перемешивали при 15°C - 30°C в течение 30 часов, после чего реакционную смесь фильтровали, и фильтровальную подушку промывали ацетонитрилом (3×5 объемов). Объединенные фильтраты концентрировали в вакууме примерно до 2-3 объемов. Затем добавляли трет-бутилметиловый эфир (5 объемов) и концентрировали до 2-3 объемов до тех пор, пока остаток ацетонитрила, по оценке ГХ-анализа, не составлял ≤0,3%. Снова добавляли трет-бутилметиловый эфир (5 объемов), и полученный раствор промывали очищенной водой (2 × 5 объемов) и концентрировали в вакууме примерно до 2-3 объемов. После добавления гептана (5 объемов) смесь концентрировали в вакууме примерно до 2-3 объемов; это добавление/концентрирование повторяли до тех пор, пока процент оставшегося трет-бутилметилового эфира не составлял ≤0,1% по данным ГХ-анализа. Полученную смесь перемешивали при -20°C до -5°C в течение 1-2 часов, после чего твердое вещество собирали фильтрацией с получением C10 в виде твердого вещества. Выход: 204 г, 855 ммоль, 92%. Типичный 1H ЯМР (500 МГц, хлороформ-d) δ 9,20 (шир, с, 1H), 8,56 (с, 1H), 6,58 (с, 1H), 3,87-3,77 (м, 1H), 2,55 (с, 3H), 2,10-1,98 (м, 2H), 1,83-1,72 (м, 2H), 1,72-1,61 (м, 2H), 1,61-1,52 (м, 2H).
Стадия 2. Синтез 3-ацетил-7-хлор-1-циклопентил-4-метил-1,6-нафтиридин-2(1H)-она (P4).
Смесь С10 (177 г, 741 ммоль) и 2,2,6-триметил-4H-1,3-диоксин-4-она (316 г, 2,22 моль) перемешивали при 75°C - 85°C в течение 72 часов, после чего добавляли толуол (354 г, 2 объема), и полученную смесь охлаждали до 15°C - 30°C. Добавляли дихлорметан (2 объемов) и смесь перемешивали при 15°C - 30°C в течение 30 to 40 минут, после чего ее обрабатывали силикагелем (354 г) и перемешивание продолжали при 15°C - 30°C в течение 20 -30 минут. После того как эту смесь фильтровали через подушку из силикагеля (177 г), фильтровальную подушку дополнительно элюировали смесью пропан-2-илацетата и гептана в соотношении 1:1 (приблизительно 45 объемов), и элюент контролировали с помощью тонкослойной хроматографии (элюент: 3:1 петролейный эфир/гептан) до тех пор, пока не перестанет обнаруживаться продукт P4. Соответствующие элюаты объединяли и концентрировали в вакууме до 2-3 объемов. Затем добавляли 2-пропанол (5 объемов) и смесь концентрировали до 2-3 объемов; этот процесс повторяли до тех пор, пока содержание остаточного пропан-2-ил ацетата не составляло ≤0,2% по данным ГХ-анализа. Полученную смесь охлаждали до -5°C - 5°C и перемешивали в течение 1-2 часов, после чего фильтровали с получением P4 в виде твердого вещества. Выход: 69,0 г, 226 ммоль, 30%. Типичный 1H ЯМР (500 МГц, DMSO-d6) δ 8,86 (с, 1H), 7,84 (с, 1H), 5,31-5,20 (м, 1H), 2,43 (с, 3H), 2,36 (с, 3H), 2,16-2,04 (м, 2H), 2,00-1,84 (м, 4H), 1,70-1,58 (м, 2H).
Получение Р5
rac-(1S,2S)-2-[6-бром-5-метил-2-(метилтио)-7-оксопиридо[2,3-d]пиримидин-8(7H)-ил]-1-метилциклопентил ацетат (P5)

Стадия 1. Синтез rac-1-[4-{[(1S,2S)-2-гидрокси-2-метилциклопентил]амино}-2-(метилтио)пиримидин-5-ил]этанона (C11).
Смесь 1-[4-хлор-2-(метилтио)пиримидин-5-ил]этанона (1,24 г, 6,12 ммоль), триэтиламина (2,56 мл, 18,4 ммоль), и транс-2-амино-1-метилциклопентанола (это вещество может быть получено с использованием метода A. Saeed et al., J. Med. Chem. 2016, 59, 750-755) (775 мг, 6,73 ммоль) в этаноле (12 мл) перемешивали при комнатной температуре в течение 2 часов. После концентрирования реакционной смеси в вакууме остаток разбавляли водой и трижды экстрагировали этилацетатом. Объединенные органические слои сушили над сульфатом магния, фильтровали, концентрировали при пониженном давлении, и очищали с помощью хроматографии на силикагеле (градиент: 20% - 60% этилацетата в гептане), с получением C11 в виде бледно-желтого твердого вещества. Выход: 1,52 г, 5,40 ммоль, 88%. LCMS m/z 282,0 [M+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 9,29 (шир, с, 1H), 8,61 (с, 1H), 4,40-4,33 (м, 1H), 4,33 (с, 1H), 2,54 (с, 3H), 2,53 (с, 3H), 2,30-2,19 (м, 1H), 2,03-1,90 (м, 1H), 1,90-1,57 (м, 4H), 1,14 (с, 3H).
Стадия 2. Синтез rac-8-[(1S,2S)-2-гидрокси-2-метилциклопентил]-5-метил-2-(метилтио)пиридо[2,3-d]пиримидин-7(8H)-она (C12).
Этил (диэтоксифосфорил)ацетат (8,99 мл, 45,3 ммоль) медленно добавляли к 0°C смеси гидрида натрия (60% в минеральном масле; 1,73 г, 43,2 ммоль) в тетрагидрофуране (60 мл), после чего охлаждающую баню удаляли и добавляли раствор C11 (2,54 г, 9,03 ммоль) в тетрагидрофуране (30 мл). Реакционную смесь нагревали до 65°C в течение 18 часов, затем охлаждали до комнатной температуры, разбавляли водой, и экстрагировали дважды этилацетатом. Объединенные органические слои сушили над сульфатом магния, фильтровали, и концентрировали в вакууме; хроматография на силикагеле (градиент: 0% - 60% этилацетата в гептане) давала C12 в виде бледно-желтого твердого вещества (2,52 г). По данным анализа 1H ЯМР, это вещество содержало примеси; часть этого вещества переносили непосредственно на следующую стадию. LCMS m/z 306,1 [M+H]+. 1H ЯМР (400 МГц, хлороформ-d), характеристические пики: δ 8,71 (с, 1H), 6,42 (ушир.кв, J=1,2 Гц, 1H), 5,81 (дд, J=9,0, 8,2 Гц, 1H), 2,88-2,75 (м, 1H), 2,64 (с, 3H), 2,43 (д, J=1,2 Гц, 3H), 2,30-2,19 (м, 1H), 1,14 (с, 3H).
Стадия 3. Синтез rac-(1S,2S)-1-метил-2-[5-метил-2-(метилтио)-7-оксопиридо[2,3-d]пиримидин-8(7H)-ил]циклопентил ацетата (C13).
4-(Пирролидин-1-ил)пиридин (124 мг, 0,837 ммоль) добавляли к смеси С12 (из предыдущей стадии; 1,26 г, ≤4,13 ммоль), триэтиламина (1,73 мл, 12,4 ммоль) и уксусного ангидрида (0,780 мл, 8,25 ммоль) в хлороформе (20 мл), и реакционную смесь нагревали до 70°C в течение 16 часов. Смесь гасили водой и экстрагировали дважды этилацетатом. Объединенные органические слои сушили над сульфатом магния, фильтровали, концентрировали в вакууме, и очищали с помощью хроматографии на силикагеле (градиент: 0% - 50% этилацетата в гептане) с получением C13 в виде белого твердого вещества. Выход: 793 мг, 2,28 ммоль, 51% за 2 стадии. 1H ЯМР (400 МГц, DMSO-d6), характеристические пики: δ 8,91 (с, 1H), 6,45-6,41 (м, 1H), 6,28-6,20 (м, 1H), 2,62 (с, 3H), 2,45 (д, J=1,2 Гц, 3H), 2,25 (ддд, J=13,3, 11,1, 7,3 Гц, 1H), 1,95 (с, 3H), 1,35 (с, 3H).
Стадия 4. Синтез rac-(1S,2S)-2-[6-бром-5-метил-2-(метилтио)-7-оксопиридо[2,3-d]пиримидин-8(7H)-ил]-1-метилциклопентил ацетата (P5).
Раствор брома (0,16 мл, 3,1 ммоль) в дихлорметане (10 мл) добавляли к раствору C13 (715 мг, 2,06 ммоль) в дихлорметане (10 мл), и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. LCMS анализ на этой стадии показал присутствие продукта P5: LCMS m/z 426,0 (наблюдали изотопную картину брома) [M+H]+. Реакционную смесь гасили добавлением 10% водного раствора тиосульфата натрия и затем экстрагировали этилацетатом. Органический слой сушили над сульфатом магния, фильтровали, и концентрировали в вакууме. После очистки с помощью хроматографии на силикагеле (градиент: 0% - 35% этилацетата в гептане), P5 выделяли в виде белой пены. Выход: 475 мг, 1,11 ммоль, 54%. 1H ЯМР (400 МГц, хлороформ-d), характеристические пики: δ 8,81 (с, 1H), 6,52-6,34 (ушир.м, 1H), 2,65 (с, 3H), 2,63 (с, 3H), 2,11-2,01 (м, 2H), 2,00 (с, 3H), 1,87-1,69 (м, 2H, предполагаемый; частично скрытый пиком воды), 1,43 (с, 3H).
Получение Р6
6-ацетил-2-хлор-8-циклопентил-5-метилпиридо[2,3-d]пиримидин-7(8H)-он (P6)

Стадия 1. Синтез 2-хлор-8-циклопентил-6-(1-этоксиэтенил)-5-метилпиридо[2,3-d]пиримидин-7(8H)-она (C14).
Бис(три-трет-бутилфосфин)палладий(0) (1,96 г, 3,84 ммоль) и трибутил(1-этоксиэтенил)станнан (36,0 г, 99,7 ммоль) добавляли к раствору 6-бром-2-хлор-8-циклопентил-5-метилпиридо[2,3-d]пиримидин-7(8H)-она (26,3 г, 76,8 ммоль) в 1-метилпирролидин-2-оне (400 мл). После перемешивания реакционной смеси при 45°C в течение 48 часов, ее выливали в смесь насыщенного водного раствора хлорида натрия (1 л) и льда (400 г), и затем разбавляли этилацетатом (1 л). Водный слой экстрагировали этилацетатом (2×1 л), и объединенные органические слои промывали насыщенным водным раствором хлорида натрия (1 л), сушили над сульфатом магния (100 г), фильтровали, и концентрировали в вакууме. Хроматография на силикагеле (градиент: 5% - 9% этилацетата в петролейном эфире) давала C14 в виде светло-желтой смолы. Выход: 23,0 г, 68,9 ммоль, 90%. LCMS m/z 333,9 (наблюдали изотопную картину хлора) [M+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 8,78 (с, 1H), 5,93-5,81 (м, 1H), 4,54 (д, J=2,6 Гц, 1H), 4,18 (д, J=2,6 Гц, 1H), 3,93 (кв, J=7,0 Гц, 2H), 2,46 (с, 3H), 2,27-2,15 (м, 2H), 2,15-2,04 (м, 2H), 1,94-1,83 (м, 2H), 1,70-1,58 (м, 2H), 1,35 (т, J=7,0 Гц, 3H).
Стадия 2. Синтез 6-ацетил-2-хлор-8-циклопентил-5-метилпиридо[2,3-d]пиримидин-7(8H)-она (P6).
К раствору C14 (14,3 г, 42,8 ммоль) в тетрагидрофуране (200 мл) добавляли хлористоводродную кислоту (1 M; 156 мл, 156 ммоль). После перемешивания реакционной смеси при 25°C в течение 30 минут, ее разделяли между насыщенным водным раствором бикарбоната натрия (200 мл) и этилацетатом (400 мл). После прекращении выделения углекислого газа, водный слой экстрагировали этилацетатом (2×200 мл), и объединенные органические слои сушили над сульфатом магния (30 г), фильтровали, концентрировали в вакууме, и очищали хроматографией на силикагеле (градиент: 9% - 25% этилацетата в петролейном эфире), с получением P6 в виде светло-желтого твердого вещества. Выход: 9,1 г, 30 ммоль, 70%. LCMS m/z 305,9 (наблюдали изотопную картину хлора) [M+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 8,85 (с, 1H), 5,91-5,78 (м, 1H), 2,53 (с, 3H), 2,39 (с, 3H), 2,27-2,16 (м, 2H), 2,16-2,04 (м, 2H), 1,97-1,86 (м, 2H), 1,74-1,61 (м, 2H).
Получение Р7
6-бром-2-хлор-8-[(1R,2S)-2-этилциклопентил]-5-метилпиридо[2,3-d]пиримидин-7(8H)-он (P7)

Стадия 1. Синтез 5-бром-2-хлор-N-[(1R,2S)-2-этилциклопентил]пиримидин-4-амина (C15).
Раствор (1R,2S)-2-этилциклопентанамина гидрохлорида (полученный с использованием способа, описанного W. Wiehl and A. W. Frahm, Chemische Berichte 1986, 119, 2668-2677) (250 мг, 1,67 ммоль), 5-бром-2,4-дихлорпиримидина (381 мг, 1,67 ммоль) и N, N-диизопропилэтиламина (864 мг, 6,68 ммоль) в диметилсульфоксиде (40 мл) перемешивали при комнатной температуре в течение 20 часов. Реакционную смесь затем разбавляли водой (20 мл) и экстрагировали этилацетатом (3×50 мл); объединенные органические слои промывали водой (3×50 мл) и насыщенным водным раствором хлорида натрия (30 мл), сушили над сульфатом натрия, фильтровали, и концентрировали в вакууме. Хроматография на силикагеле (градиент: 0% - 20% этилацетата в гептане) давала C15 в виде бесцветного масла. Выход: 162 мг, 0,532 ммоль, 32%. LCMS m/z 303,8 (наблюдали изотопную картину брома-хлора) [M+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 8,09 (с, 1H), 5,43 (ушир.д, J=8,2 Гц, 1H), 4,63-4,51 (м, 1H), 2,12-1,87 (м, 3H), 1,82-1,56 (м, 3H), 1,45-1,29 (м, 2H), 1,28-1,15 (м, 1H), 0,91 (т, J=7,3 Гц, 3H). Отмечается, что абсолютная стереохимия C15 не подтверждена. См. Получение Р3 для определения абсолютной стереохимии P3, полученного из соответствующего конгенера метила (1R,2S)-2-метилциклопентанамина. (1R,2S)-2-Метилциклопентанамин и (1R,2S)-2-этилциклопентанамин синтезировали тем же способом Wiehl и Frahm, и предполагалось, что (1R,2S)-2-этилциклопентанамин, следовательно, будет демонстрировать ту же стереохимию вокруг циклопентана.
Стадия 2. Синтез 2-хлор-8-[(1R,2S)-2-этилциклопентил]-5-метилпиридо[2,3-d]пиримидин-7(8H)-она (C16).
Смесь C15 (162 мг, 0,532 ммоль), (2E)-бут-2-еновой кислоты (137 мг, 1,59 ммоль), ацетата палладия(II) (11,9 мг, 53,0 мкмоль) и триэтиламина (0,371 мл, 2,66 ммоль) в 1-метилпирролидин-2-оне (20 мл) дегазировали азотом в течение 10 минут. Затем реакционный сосуд закрывали крышкой и нагревали до 65°C в течение 20 часов. Добавляли снова (2E)-бут-2-еновую кислоту (137 мг, 1,59 ммоль), ацетат палладия(II) (11,9 мг, 53,0 мкмоль) и триэтиламин (0,371 мл, 2,66 ммоль), с последующей аналогичной дегазацией азотом, после чего реакционную смесь нагревали до 65°C в течение 16 часов. Еще раз добавляли ацетат палладия(II) (11,9 мг, 53,0 мкмоль), продувку азотом повторяли и нагревание при 65°C продолжали в течение 3 часов. Затем добавляли уксусный ангидрид (0,100 мл, 1,06 ммоль) и реакционную смесь нагревали до 65°C в течение 16 часов, после чего ее выливали в воду и нейтрализовали добавлением насыщенного водного раствора бикарбоната натрия. Полученную смесь экстрагировали этилацетатом (3×10 мл) и объединенные органические слои промывали насыщенным водным раствором хлорида натрия (3×10 мл), сушили над сульфатом натрия, фильтровали, и концентрировали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (градиент: 0% - 30% этилацетата в гептане) с получением смеси С16 и (2E)-бут-2-еновой кислоты в виде желтого масла (115 мг). LCMS m/z 291,8 (наблюдали изотопную картину хлора) [M+H]+. 1H ЯМР (400 МГц, хлороформ-d), характеристические пики для C16: δ 8,72 (с, 1H), 6,51 (шир, с, 1H), 6,02-5,91 (м, 1H), 2,41 (д, J=1,3 Гц, 3H), 0,74 (т, J=7,4 Гц, 3H).
Стадия 3. Синтез 6-бром-2-хлор-8-[(1R,2S)-2-этилциклопентил]-5-метилпиридо[2,3-d]пиримидин-7(8H)-она (P7).
N-Бромсукцинимид (105 мг, 0,590 ммоль) и щавелевую кислоту (9,9 мг, 0,11 ммоль) добавляли у раствору C16 (из предыдущей стадии; 115,0 мг, <0,394 ммоль) в ацетонитриле, и реакционную смесь нагревали до 60°C в течение 16 часов. Смесь затем концентрировали в вакууме и остаток очищали с помощью хроматографии на силикагеле (градиент: 0% - 50% этилацетата в гептане) с получением P7 в виде прозрачного желтого масла. По данным анализа 1H ЯМР, это вещество не было полностью чистым. Выход: 80 мг, 0,22 ммоль, 41% за 2 стадии. LCMS m/z 369,7 (наблюдали изотопную картину брома-хлора) [M+H]+. 1H ЯМР (400 МГц, DMSO-d6), только пики продукта: δ 9,20 (с, 1H), 6,04-5,91 (м, 1H), 2,66 (с, 3H), 2,43-2,29 (м, 1H), 2,22-2,08 (м, 1H), 2,07-1,84 (м, 4H), 1,63-1,46 (м, 1H), 1,07-0,90 (м, 2H), 0,74 (т, J=7,3 Гц, 3H).
Получение Р8
7-хлор-4-метил-1-[(3S)-тетрагидрофуран-3-ил]-1,6-нафтиридин-2(1H)-он (P8)
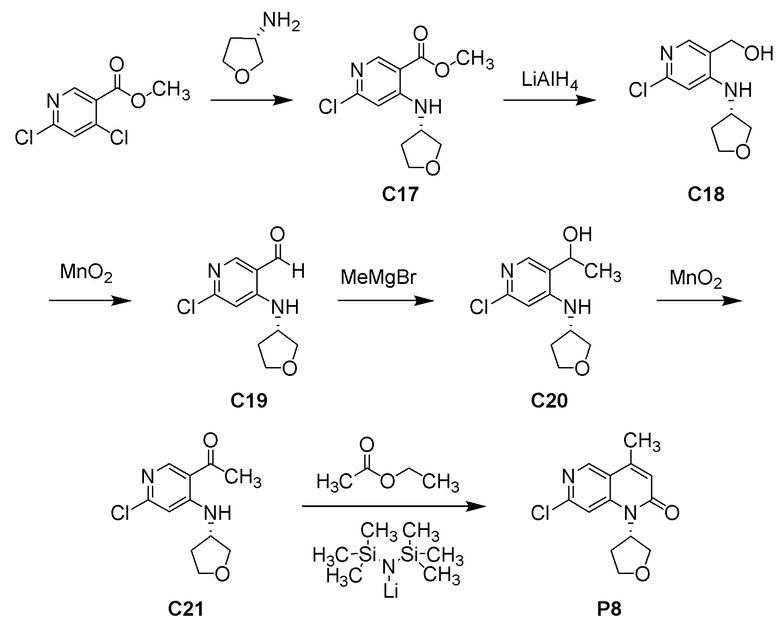
Стадия 1. Синтез метил 6-хлор-4-[(3S)-тетрагидрофуран-3-иламино]пиридин-3-карбоксилата (C17).
(3S)-Тетрагидрофуран-3-амин (1,69 г, 19,4 ммоль) добавляли частями к 0°C раствору метил 4,6-дихлорпиридин-3-карбоксилата (2,0 г, 9,7 ммоль) в ацетонитриле (50 мл). После перемешивания реакционной смеси при 40°C в течение 44 часов, ее разбавляли этилацетатом (50 мл), промывали последовательно насыщенным водным раствором хлорида аммония (20 мл) и насыщенным водным раствором хлорида натрия (20 мл), сушили над сульфатом натрия, фильтровали, и концентрировали в вакууме. Очистка хроматографией на силикагеле (Градиент: 0% - 30% этилацетата в петролейном эфире) давала C17 в виде белого твердого вещества. Выход: 1,70 г, 6,62 ммоль, 68%. LCMS m/z 256,7 (наблюдали изотопную картину хлора) [M+H]+. 1H ЯМР (400 МГц, метанол-d4) δ 8,58 (с, 1H), 8,45 (ушир.д, J=7 Гц, 1H), 6,84 (с, 1H), 4,33-4,25 (м, 1H), 4,01-3,93 (м, 2H), 3,92-3,84 (м, 1H), 3,89 (с, 3H), 3,74 (дд, J=9,4, 2,7 Гц, 1H), 2,44-2,33 (м, 1H), 1,96-1,87 (м, 1H).
Стадия 2. Синтез {6-хлор-4-[(3S)-тетрагидрофуран-3-иламино]пиридин-3-ил}метанола (C18).
К 0°C раствору C17 (1,90 г, 7,40 ммоль) в тетрагидрофуране (30 мл) добавляли алюмогидрид лития (843 мг, 22,2 ммоль) частями. После перемешивания реакционной смеси при комнатной температуре (15°C) в течение 2 часов, смесь гасили декагидратом сульфата натрия и фильтровали. Фильтрпрессную лепешку промывали этилацетатом (5×50 мл), и объединенные фильтраты концентрировали в вакууме с получением C18 (1,96 г) в виде бледно-желтой смолы. По данным анализа 1H ЯМР, это вещество не было чистым. LCMS m/z 270,8 (наблюдали изотопную картину хлора) [M+H]+ для ацетилированного производного. 1H ЯМР (400 МГц, хлороформ-d), только пики продукта: δ 7,76 (с, 1H), 6,46 (с, 1H), 5,82-5,70 (м, 1H), 4,62 (с, 2H), 4,13-4,03 (м, 1H), 4,02-3,94 (м, 2H), 3,93-3,85 (м, 1H), 3,74 (дд, J=9,3, 2,8 Гц, 1H), 2,38-2,27 (м, 1H), 1,96-1,86 (м, 1H).
Стадия 3. Синтез 6-хлор-4-[(3S)-тетрагидрофуран-3-иламино]пиридин-3-карбальдегида (C19).
К раствору C18 (из предыдущей стадии; 1,96 г, ≤7,40 ммоль) в хлороформе (50 мл) добавляли оксид марганца(IV) (6,44 г, 74,1 ммоль), и реакционную смесь перемешивали при 50°C в течение 6 часов. Затем смесь фильтровали, и фильтрат концентрировали в вакууме; очистка остатка с помощью хроматографии на силикагеле (градиент: 10% - 30% этилацетата в петролейном эфире) давала C19 в виде желтого твердого вещества. Выход: 1,17 г, 5,16 ммоль, 70% за 2 стадии. LCMS m/z 227,0 (наблюдали изотопную картину хлора) [M+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 9,84 (д, J=0,7 Гц, 1H), 8,76 (шир, с, 1H), 8,33 (с, 1H), 6,57 (с, 1H), 4,19-4,10 (м, 1H), 4,06-3,97 (м, 2H), 3,91 (ддд, J=8,6, 8,5, 5,3 Гц, 1H), 3,77 (дд, J=9,4, 3,2 Гц, 1H), 2,46-2,30 (м, 1H), 2,01-1,90 (м, 1H).
Стадия 4. Синтез 1-{6-хлор-4-[(3S)-тетрагидрофуран-3-иламино]пиридин-3-ил}этанола (C20).
К 0°C раствору C19 (1,17 г, 5,16 ммоль) в тетрагидрофуране (30 мл) добавляли метилмагний бромид (3 M раствор в диэтиловом эфире; 5,16 мл, 15,5 ммоль). Реакционную смесь перемешивали при 15°C в течение 16 часов, после чего ее обрабатывали водой (15 мл) и экстрагировали этилацетатом (4×50 мл). Объединенные органические слои фильтровали, и фильтрат концентрировали в вакууме с получением C20 в виде белого твердого вещества. Выход: 1,19 г, 4,90 ммоль, 95%. LCMS m/z 243,0 (наблюдали изотопную картину хлора) [M+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 7,68 (с, 1H), 6,42 (с, 1H), 6,25 (ушир.д, J=6,4 Гц, 1H), 4,88 (кв, J=6,7 Гц, 1H), 4,09-4,01 (м, 1H), 4,01-3,85 (м, 3H), 3,76-3,68 (м, 1H), 2,86-2,69 (м, 1H), 2,37-2,24 (м, 1H), 1,97-1,82 (м, 1H), 1,55 (д, J=6,6 Гц, 3H).
Стадия 5. Синтез 1-{6-хлор-4-[(3S)-тетрагидрофуран-3-иламино]пиридин-3-ил}этанона (C21).
Оксид марганца(IV) (4,26 г, 49,0 ммоль) добавляли к раствору C20 (1,19 г, 4,90 ммоль) в хлороформе (70 мл), и реакционную смесь перемешивали при 50°C в течение 16 часов. Затем смесь фильтровали; концентрация фильтрата в вакууме давала C21 в виде белого твердого вещества. Выход: 1,10 г, 4,57 ммоль, 93%. 1H ЯМР (400 МГц, хлороформ-d) δ 9,36 (шир, с, 1H), 8,61 (с, 1H), 6,55 (с, 1H), 4,15-4,07 (м, 1H), 4,06-3,95 (м, 2H), 3,90 (ддд, J=8,6, 8,4, 5,2 Гц, 1H), 3,75 (дд, J=9,3, 3,2 Гц, 1H), 2,58 (с, 3H), 2,41-2,29 (м, 1H), 1,98-1,89 (м, 1H).
Стадия 6. Синтез 7-хлор-4-метил-1-[(3S)-тетрагидрофуран-3-ил]-1,6-нафтиридин-2(1H)-она (P8).
Этилацетат (1,13 мл, 11,6 ммоль) медленно добавляли по каплям к раствору -78°C литий бис(триметилсилил)амида (1 M раствор в тетрагидрофуране; 11,6 мл, 11,6 ммоль) в тетрагидрофуране (10 мл). Перемешивание продолжали при -78°C в течение 30 минут, после чего одной порцией добавляли раствор C21 (929 мг, 3,86 ммоль) в тетрагидрофуране (8 мл). Затем реакционную смесь удаляли из охлаждающей бани и давали ей нагреться до комнатной температуры (15°C); перемешивание продолжали при комнатной температуре в течение дополнительных 16 часов. После гашения реакционной смеси водой (30 мл), ее разбавляли этилацетатом (30 мл), и экстрагировали этилацетатом (3×30 мл). Объединенные органические слои последовательно промывали насыщенным водным раствором хлорида аммония (20 мл) и насыщенным водным раствором хлорида натрия (25 мл), сушили над сульфатом магния, фильтровали, и концентрировали в вакууме. Остаток объединяли с неочищенным продуктом аналогичной реакции, проведенной с использованием C21 (250 мг, 1,04 ммоль) и очищали с помощью хроматографии на силикагеле (градиент: 0% - 50% этилацетата в петролейном эфире). Это давало P8 в виде белого твердого вещества. Общий выход: 1,20 г, 4,53 ммоль, 92%. LCMS m/z 264,9 [M+H]+. 1H ЯМР (400 МГц, метанол-d4) δ 8,78 (с, 1H), 8,01 (с, 1H), 6,57 (кв, J=1,3 Гц, 1H), 6,21-6,08 (м, 1H), 4,44 (ддд, J=8,6, 8,5, 3,1 Гц, 1H), 4,10 (дд, J=10,1, 4,4 Гц, 1H), 3,98 (дд, J=9,7, 9,6 Гц, 1H), 3,82 (ддд, J=9,3, 9,3, 6,8 Гц, 1H), 2,53 (д, J=1,2 Гц, 3H), 2,40-2,21 (м, 2H).
Получение Р9
rac-8-[(1S,2R,5R)-Бицикло[3.1.0]гексан-2-ил]-2-хлор-5-метилпиридо[2,3-d]пиримидин-7(8H)-он (P9)

Стадия 1. Синтез rac-(1S,2R,5R)-N-бензилбицикло[3.1.0]гексан-2-амина (C22).
Смесь бицикло[3.1.0]гексан-2-она (385 мг, 4,00 ммоль), бензиламина (429 мг, 4,00 ммоль), боргидрида натрия (227 мг, 6,00 ммоль), и уксусной кислоты (240 мг, 4,0 ммоль) в смеси дихлорметана (5 мл) и метанола (5 мл) перемешивали при 18°C в течение 10 часов. Концентрирование в вакууме давало C22 в виде смолы желтого цвета. По данным анализа 1H ЯМР, это вещество не было чистым, и содержало остаточный бензиламин. Выход: 700 мг, <3,7 ммоль, <92%. 1H ЯМР (400 МГц, хлороформ-d), характеристические/предполагаемые пики для C22: δ 7,40-7,21 (м, 5H), 3,93-3,78 (м, 2H), 3,48-3,38 (м, 1H), 0,40-0,34 (м, 1H), 0,33-0,25 (м, 1H).
Стадия 2. Синтез rac-(1S,2R,5R)-бицикло[3.1.0]гексан-2-амина, хлористоводородная соль (C23).
Смесь С22, хлористоводородная соль (800 мг, 3,58 ммоль), гидроксида палладия на угле (800 мг) и хлорида водорода (4 M раствор в этилацетате; 1,8 мл, 7,2 ммоль) в метаноле (15 мл) дегазировали 3 раза с водородом. Затем реакционную смесь перемешивали в атмосфере водорода (1 атмосфера) в течение 5 часов при 50°C, после чего смесь фильтровали; фильтрат концентрировали в вакууме с получением C23 в виде белого твердого вещества. Выход: 452 мг, 3,38 ммоль, 94%.
Стадия 3. Синтез rac-N-[(1S,2R,5R)-бицикло[3.1.0]гексан-2-ил]-5-бром-2-хлорпиримидин-4-амина (C24).
Триэтиламин (1,51 г, 14,9 ммоль) добавляли по каплям к раствору 0°C 5-бром-2,4-дихлорпиримидина (850 мг, 3,73 ммоль) и C23 (452 мг, 3,38 ммоль) в ацетонитриле (50 мл). Реакционной смеси давали нагреться до комнатной температуры, и перемешивали при 20°C в течение 18 часов, после чего ее обрабатывали водным раствором хлорида аммония (30 мл) и экстрагировали этилацетатом (2×50 мл). Объединенные органические слои сушили, фильтровали, концентрировали в вакууме, и подвергали хроматографии на силикагеле (градиент: 1% - 4% этилацетата в петролейном эфире), получая C24 в виде белого твердого вещества. Выход: 790 мг, 2,74 ммоль, 81%. LCMS m/z 289,6 (наблюдали изотопную картину брома-хлора) [M+H]+.
Стадия 4. Синтез rac-8-[(1S,2R,5R)-бицикло[3.1.0]гексан-2-ил]-2-хлор-5-метилпиридо[2,3-d]пиримидин-7(8H)-она (P9).
Аргон барботировали через раствор C24 (395 мг, 1,37 ммоль), (2E)-бут-2-еновой кислоты (354 мг, 4,11 ммоль), и N, N-диизопропилэтиламина (1,77 г, 13,7 ммоль) в тетрагидрофуране (20 мл), и затем добавляли бис(бензонитрил)палладия(II) хлорид (52,5 мг, 0,137 ммоль) и три-о-толилфосфин (41,7 мг, 0,137 ммоль). Реакционный сосуд герметично закрывали и нагревали при 70°C в течение 18 часов, после чего добавляли уксусный ангидрид (279 мг, 2,73 ммоль), и нагревание продолжали в течение 3 часов при 70°C. После удаления летучих веществ при пониженном давлении, остаток очищали хроматографией на силикагеле (градиент: 1% - 30% этилацетата в петролейном эфире), с получением P9 в виде светло-желтого твердого вещества. Выход: 290 мг, 1,05 ммоль, 77%. LCMS m/z 275,8 (наблюдали изотопную картину хлора) [M+H]+.
Получение Р10
rac-6-бром-8-[(1R,2S)-2-фтор-2-метилциклопентил]-5-метил-2-(метилтио)пиридо[2,3-d]пиримидин-7(8H)-он (P10)

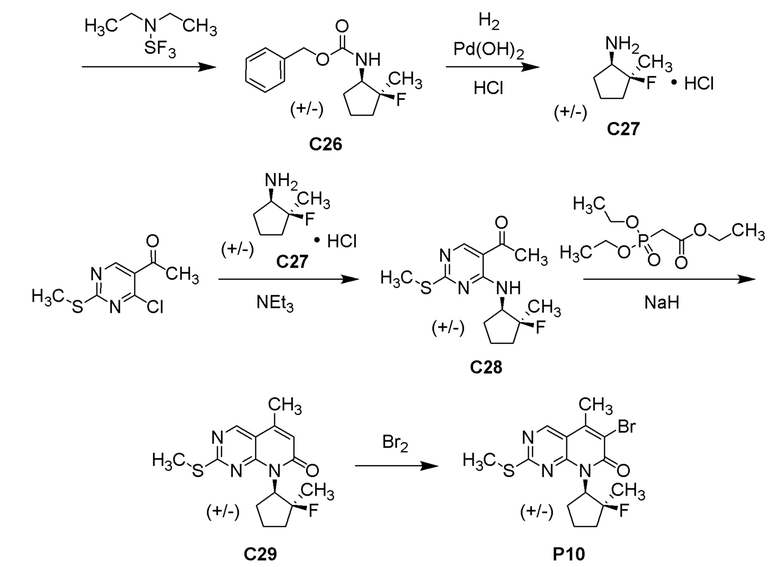
Стадия 1. Синтез rac-бензил [(1R,2R)-2-гидрокси-2-метилциклопентил]карбамата (C25).
Метилмагний бромид (3,0 M; 11,4 мл, 34,2 ммоль) медленно добавляли к -78°C раствору бензил (2-охоциклопентил)карбамата (2,67 г, 11,4 ммоль) в тетрагидрофуране (24 мл), и реакционную смесь перемешивали при -78°C в течение 4 часов; смеси затем давали нагреться до комнатной температуры. К реакционной смеси осторожно добавляли воду, затем хлористоводродную кислоту (1 M; 2 мл), и водный слой экстрагировали дважды этилацетатом. После того, как объединенные органические слои были высушены над сульфатом натрия, отфильтрованы и сконцентрированы в вакууме, остаток очищали с помощью хроматографии на силикагеле (градиент: 0% - 100% этилацетата в гептане) с получением C25 в виде бледно-желтого твердого вещества. Исходное вещество бензил (2-оксоциклопентил)карбамат (1,36 г, 5,83 ммоль) также выделяли после хроматографической очистки. Выход C25: 878 мг, 3,52 ммоль, 31%; в расчете на восстановленное исходное вещество выход C25 составил 63%. 1H ЯМР (400 МГц, хлороформ-d) δ 7,41-7,29 (м, 5H), 5,12 (с, 2H), 4,77 (шир, с, 1H), 3,87 (ддд, J=9,9, 8,2, 6,0 Гц, 1H), 2,19-2,08 (м, 1H), 1,94-1,82 (м, 1H), 1,81-1,68 (м, 2H), 1,68-1,57 (м, 1H), 1,40-1,28 (м, 1H), 1,16 (с, 3H).
Стадия 2. Синтез rac-бензил [(1R,2S)-2-фтор-2-метилциклопентил]карбамата (C26).
(Диэтиламино)серы трифторид (0,40 мл, 3,0 ммоль) добавляли к -78°C раствору C25 (349 мг, 1,40 ммоль) в дихлорметане (8 мл). Реакционную смесь перемешивали при -78°C в течение 4 часов, после чего ее разбавляли 2-пропанолом при -78°C, и затем нагревали до комнатной температуры. После концентрирования реакционной смеси в вакууме, остаток очищали с помощью хроматографии на силикагеле (градиент: 0% - 30% этилацетата в гептане), с получением C26 в виде белого твердого вещества. Указанная относительная стереохимия C26 была установлена на основе монокристального рентгеноструктурного анализа (см. ниже). Выход: 281 мг, 1,12 ммоль, 80%. 1H ЯМР (400 МГц, хлороформ-d) δ 7,41-7,29 (м, 5H), 5,12 (с, 2H), 5,03-4,92 (м, 1H), 3,92-3,75 (м, 1H), 2,18-1,94 (м, 2H), 1,89-1,68 (м, 2H), 1,68-1,54 (м, 2H), 1,40 (д, JHF=21,7 Гц, 3H).
Диффузия паров пентана в дихлорэтановый раствор C26 давала кристаллы для рентгеноструктурного анализа.
Монокристальный рентгеноструктурный анализ C26
Рентгеноструктурные анализы монокристалла
Сбор данных проводили на дифрактометре Kappa APEX-II CCD, оснащенном излучением Mo Kα (λ=0,71073 Å). Кусок бесцветного блока размером 0,313×0,155×0,117 мм устанавливали на CryoLoop с маслом Paratone. Данные собирали в потоке газообразного азота при 100(2) K, используя Φ и ω сканирование. Расстояние от кристалла до детектора составляло 40 мм, а время экспозиции составляло 30 секунд на кадр при ширине сканирования 1,0°. Сбор данных завершали на 99,9% до 25,00° в θ. Всего было собрано 9647 отражений, охватывающих индексы, -21<=h<=25, -5<=k<=5, -15<=l<=13. Было обнаружено, что 2400 отражений не зависят от симметрии с Rint 0,0561. Индексация и уточнение элементарных ячеек указывали на примитивную моноклинную решетку. Было установлено, что пространственная группа равна P21/c. Данные были интегрированы с помощью программного обеспечения Bruker SAINT и масштабированы с помощью программного обеспечения SADABS. Решение прямыми методами (SHELXT) привело к созданию полной модели фазирования, соответствующей предложенной структуре.
Все неводородные атомы уточняли анизотропно методикой наименьших квадратов в полноматричном приближении (SHELXL-2014; G. M. Sheldrick, SHELXTL Version 2014/7. http://shelx.uni-ac.gwdg.de/SHELX/index.php). Все атомы водорода размещали с использованием модели “наездника”. Их положения были ограничены относительно их родительского атома с помощью соответствующей команды HFIX в SHELXL-2014. Кристаллографические данные приведены в таблице E. Атомные координаты, длины связей, углы связей и параметры смещения приведены в таблицах F - K.

Таблица F. Атомные координаты (x 104) и эквивалентные параметры изотропного смещения (Å2×103) для C26. U(eq) представляет собой 1/3 следа ортогонализированного тензора Uij.


Таблица G. Длины [Å] и углы [°] связей для C26.





Таблица H. Параметры анизотропного смещения (Å2×103) для C26. Показатель коэффициента анизотропного смещения принимает вид: -2π2[h2 a*2U11 + ... + 2 h k a* b* U12 ].


Таблица J. Координаты водорода (x 104) и параметры изотропного смещения (Å2×103) для C26.


Таблица K. Водородные связи для C26 [Å и °].

Преобразования симметрии использовали для создания эквивалентных атомов:
#1 x, y+1,z
Стадия 3. Синтез rac-(1R,2S)-2-фтор-2-метилциклопентанамин, хлористоводородная соль (C27).
Смесь С26 (281 мг, 1,12 ммоль), 20% гидроксида палладия на угле (50 мг), и хлорида водорода (2 M раствор в метаноле; 0,671 мл, 1,34 ммоль) перемешивали в атмосфере водорода из баллона в течение ночи, после чего смесь фильтровали через диатомовую землю. Фильтрат повторно подвергали реакционным условиям в течение ночи, и снова фильтровали через диатомитовую землю. Концентрация фильтрата давала C27 в виде бледно-желтого твердого вещества (189 мг). По данным анализа 1H ЯМР, это вещество не было полностью чистым; часть его перенесли непосредственно на следующую стадию. 1H ЯМР (400 МГц, DMSO-d6), характеристические пики: δ 2,12-1,55 (м, 6H), 1,50 (д, JHF=22,0 Гц, 3H).
Стадия 4. Синтез rac-1-[4-{[(1R,2S)-2-фтор-2-метилциклопентил]амино}-2-(метилтио)пиримидин-5-ил]этанона (C28).
Смесь C27 (из предыдущей стадии; 122 мг, ≤0,723 ммоль), 1-[4-хлор-2-(метилтио)пиримидин-5-ил]этанона (175 мг, 0,864 ммоль) и триэтиламина (0,401 мл, 2,88 ммоль) в этаноле (12 мл) перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь затем разбавляли водой и экстрагировали этилацетатом. Объединенные органические слои сушили над сульфатом натрия, фильтровали, и концентрировали в вакууме; хроматография на силикагеле (градиент: 0% - 30% этилацетата в гептане) давала C28 в виде белого твердого вещества. Выход: 180 мг, 0,635 ммоль, 57% за 2 стадии. LCMS m/z 283,9 [M+H]+. 1H ЯМР (400 МГц, хлороформ-d), характеристические пики: δ 9,55 (шир, с, 1H), 8,59 (с, 1H), 4,56-4,40 (м, 1H), 2,54 (с, 3H), 2,52 (с, 3H), 1,41 (д, JHF=21,5 Гц, 3H).
Стадия 5. Синтез rac-8-[(1R,2S)-2-фтор-2-метилциклопентил]-5-метил-2-(метилтио)пиридо[2,3-d]пиримидин-7(8H)-она (C29).
Этил (диэтоксифосфорил)ацетат (0,633 мл, 3,19 ммоль) медленно добавляли к 0°C суспензии гидрида натрия (60% суспензия в минеральном масле; 122 мг, 3,05 ммоль) в тетрагидрофуране (4 мл); охлаждающую баню убирали и добавляли раствор C28 (180 мг, 0,635 ммоль) в тетрагидрофуране (4 мл). Реакционную смесь нагревали при 60°C в течение 18 часов, после чего смесь охлаждали до комнатной температуры, гасили добавлением воды, и экстрагировали дважды этилацетатом. Объединенные органические слои сушили над сульфатом магния, фильтровали, концентрировали в вакууме, и очищали с помощью хроматографии на силикагеле (градиент: 0% - 60% этилацетата в гептане), с получением C29 в виде бледно-желтого твердого вещества. Выход: 190 мг, 0,618 ммоль, 97%. LCMS m/z 307,9 [M+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 8,68 (с, 1H), 6,44 (шир, с, 1H), 5,92-5,72 (м, 1H), 3,29-3,01 (м, 1H), 2,62 (с, 3H), 2,48-2,31 (м, 1H), 2,41 (д, J=1,2 Гц, 3H), 2,31-2,19 (м, 1H), 2,00-1,81 (м, 2H), 1,73-1,6 (м, 1H, предполагаемый; частично скрытый пиком воды), 1,46 (д, JHF=21,8 Гц, 3H).
Стадия 6. Синтез rac-6-бром-8-[(1R,2S)-2-фтор-2-метилциклопентил]-5-метил-2-(метилтио)пиридо[2,3-d]пиримидин-7(8H)-она (P10).
Бром (32 мкл, 0,62 ммоль) добавляли к раствору C29 (128 мг, 0,416 ммоль) в дихлорметане (6,0 мл), и смесь перемешивали при комнатной температуре в течение 15 минут, после чего добавляли 10% водный раствор тиосульфата натрия. Полученную смесь экстрагировали этилацетатом, и объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (градиент: 0% - 100% этилацетата в гептане) давала P10 в виде белого твердого вещества. Выход: 109 мг, 0,282 ммоль, 68%. 1H ЯМР (400 МГц, хлороформ-d) δ 8,79 (с, 1H), 6,04-5,84 (м, 1H), 3,15-2,95 (м, 1H), 2,63 (с, 3H), 2,63 (с, 3H), 2,54-2,37 (м, 1H), 2,35-2,22 (м, 1H), 2,02-1,82 (м, 2H), 1,72-1,6 (м, 1H, предполагаемый; частично скрытый пиком воды), 1,49 (д, JHF=21,9 Гц, 3H).
Получение Р11
трет-бутил (3S)-3-[6-бром-5-метил-2-(метилтио)-7-оксопиридо[2,3-d]пиримидин-8(7H)-ил]пирролидин-1-карбоксилата (P11)

Стадия 1. Синтез трет-бутил (3S)-3-{[5-ацетил-2-(метилтио)пиримидин-4-ил]амино}пирролидин-1-карбоксилата (C30).
1-[4-Хлор-2-(метилтио)пиримидин-5-ил]этанон (4,50 г, 22,2 ммоль) добавляли к раствору трет-бутил (3S)-3-аминопирролидин-1-карбоксилата (4,55 г, 24,4 ммоль) и N, N-диизопропилэтиламина (7,74 мл, 44,4 ммоль) в ацетонитриле (110 мл). Реакционную смесь перемешивали при комнатной температуре (приблизительно 15°C) в течение 2,5 дней, после чего смесь концентрировали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (градиент: 0% - 40% этилацетата в петролейном эфире) с получением C30 в виде бесцветной смолы. Выход: 7,50 г, 21,3 ммоль, 96%. LCMS m/z 353,1 [M+H]+. 1H ЯМР (400 МГц, метанол-d4) δ 8,66 (с, 1H), 4,80-4,71 (м, 1H), 3,81-3,71 (м, 1H), 3,56-3,43 (м, 2H), 3,37-3,31 (м, 1H, предполагаемый; частично скрытый пиком растворителя), 2,56 (с, 3H), 2,53 (с, 3H), 2,37-2,25 (м, 1H), 2,10-1,97 (м, 1H), 1,47 (с, 9H).
Стадия 2. Синтез трет-бутил (3S)-3-[5-метил-2-(метилтио)-7-оксопиридо[2,3-d]пиримидин-8(7H)-ил]пирролидин-1-карбоксилата (C31).
Добавляли литий бис(триметилсилил)амид (1 M раствор в тетрагидрофуране; 63,8 мл, 63,8 ммоль) добавляли по каплям к раствору 0°C C30 (7,50 г, 21,3 ммоль) и этилацетатом (6,56 г, 74,5 ммоль) в тетрагидрофуране (200 мл). Реакционную смесь перемешивали на бане со льдом в течение 30 минут, после чего смесь нагревали до 40°C в течение 1 часа. Добавляли водный раствор хлорида аммония (100 мл), затем воду (100 мл), и полученную смесь экстрагировали этилацетатом (3×150 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия (150 мл), концентрировали в вакууме, и подвергали хроматографии на силикагеле (градиент: 10% - 50% этилацетата в дихлорметане), получая C31 в виде желтоватого твердого вещества. Выход: 6,27 г, 16,7 ммоль, 78%. 1H ЯМР (400 МГц, хлороформ-d) δ 8,71 (с, 1H), 6,47-6,42 (м, 1H), 6,26-6,14 (м, 1H), 3,99 (дд, J=10,2, 8,4 Гц, 1H), 3,88-3,75 (м, 1H), 3,72-3,57 (м, 1H), 3,52-3,40 (м, 1H), 2,97-2,81 (м, 1H), 2,58 (с, 3H), 2,43 (д, J=1,3 Гц, 3H), 2,18-2,06 (м, 1H), 1,52-1,43 (м, 9H).
Стадия 3. Синтез трет-бутил (3S)-3-[6-бром-5-метил-2-(метилтио)-7-оксопиридо[2,3-d]пиримидин-8(7H)-ил]пирролидин-1-карбоксилата (P11).
N-Бромсукцинимид (3,26 г, 18,3 ммоль) добавляли порциями к смеси 0°C C31 (6,27 г, 16,7 ммоль) и щавелевой кислоты (150 мг, 1,67 ммоль) в смеси ацетонитрила (100 мл) и дихлорметана (50 мл). После перемешивания реакционной смеси в течение 45 минут при 0°C, ее обрабатывали насыщенным водным раствором сульфита натрия (45 мл), разбавляли водой (80 мл), и экстрагировали дихлорметан (2×100 мл). Объединенные органические слои концентрировали в вакууме, и остаток очищали с помощью хроматографии на силикагеле (градиент: 10% - 60% этилацетата в петролейном эфире), с получением P11 в виде белого твердого вещества. Выход: 5,30 г, 11,6 ммоль, 69%. 1H ЯМР (400 МГц, хлороформ-d) δ 8,82 (с, 1H), 6,37-6,26 (м, 1H), 3,99-3,78 (м, 2H), 3,78-3,61 (м, 1H), 3,56-3,45 (м, 1H), 2,90-2,71 (м, 1H), 2,64 (с, 3H), 2,60 (с, 3H), 2,24-2,08 (м, 1H), 1,52-1,43 (м, 9H).
Получение Р12
6-ацетил-8-циклопентил-5-метил-2-(метилсульфонил)пиридо[2,3-d]пиримидин-7(8H)-он (P12)

Стадия 1. Синтез 6-бром-8-циклопентил-5-метил-2-(метилтио)пиридо[2,3-d]пиримидин-7(8H)-она (C32).
К 0°C суспензии 6-бром-2-хлор-8-циклопентил-5-метилпиридо[2,3-d]пиримидин-7(8H)-она (9,70 г, 28,3 ммоль) в смеси тетрагидрофурана (95 мл) и воды (50 мл) добавляли метантиолат натрия (3,97 г, 56,6 ммоль). Реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 18 часов, после чего твердое вещество собирали фильтрованием и промывали водой с получением C32 (4,30 г) в виде белого твердого вещества. Фильтрат экстрагировали этилацетатом (3×150 мл), и объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали, и концентрировали в вакууме с получением дополнительного количества C32 (5,60 г) в виде белого твердого вещества. Общий выход: 9,90 г, 27,9 ммоль, 99%. LCMS m/z 353,8 (наблюдали изотопную картину брома) [M+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 8,79 (с, 1H), 6,12-5,99 (м, 1H), 2,63 (с, 6H), 2,36-2,22 (м, 2H), 2,17-2,04 (м, 2H), 1,95-1,83 (м, 2H), 1,74-1,62 (м, 2H).
Стадия 2. Синтез 6-ацетил-8-циклопентил-5-метил-2-(метилтио)пиридо[2,3-d]пиримидин-7(8H)-она (C33).
Суспензию C32 (18,0 г, 50,8 ммоль), трибутил(1-этоксиэтенил)станнана (83,07 г, 230,0 ммоль), и тетракис(трифенилфосфин)палладий(0) (2,94 г, 2,54 ммоль) в толуоле (250 мл) перемешивали при 110°C в течение 16 часов, после чего смесь охлаждали и обрабатывали хлористоводродной кислотой (6 M; 20 мл). После перемешивания полученной смеси в течение 20 минут, смесь фильтровали; pH фильтрата доводили до 7 добавлением 2 М водного раствора гидроксида натрия, а затем экстрагировали этилацетатом, и затем экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия (2×50 мл), сушили над сульфатом натрия, фильтровали, и концентрировали в вакууме. Очистка хроматографией на силикагеле (градиент: 0% - 20% этилацетата в петролейном эфире) давала C33 в виде желтого твердого вещества. Выход: 14,0 г, 44,1 ммоль, 87%. LCMS m/z 317,9 [M+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 8,80 (с, 1H), 5,98-5,88 (м, 1H), 2,63 (с, 3H), 2,55 (с, 3H), 2,38 (с, 3H), 2,37-2,28 (м, 2H), 2,13-2,01 (м, 2H), 1,94-1,84 (м, 2H), 1,75-1,64 (м, 2H).
Стадия 3. Синтез 6-ацетил-8-циклопентил-5-метил-2-(метилсульфонил)пиридо[2,3-d]пиримидин-7(8H)-она (P12).
Раствор 3-хлорпероксибензойной кислоты (16,7 г, 96,8 ммоль) в дихлорметане (300 мл) добавляли в течение 30 минут по каплям к -5°C раствору C33 (14,0 г, 44,1 ммоль) в дихлорметане (300 мл). После перемешивания реакционной смеси при 15°C в течение 16 часов, смесь разбавляли насыщенным водным раствором сульфита натрия (6 мл), затем насыщенным водным раствором карбоната натрия (35 мл). Полученную смесь экстрагировали дихлорметан (2×50 мл), и объединенные органические слои сушили над сульфатом натрия, фильтровали, и концентрировали в вакууме. По данным анализа 1H ЯМР, реакция была неполной, поэтому это вещество растворяли в дихлорметане (150 мл), охлаждали до -5°C, и снова обрабатывали по каплям раствором 3-хлорпероксибензойной кислоты (3,0 г, 17 ммоль) в дихлорметане (10 мл). Эту реакционную смесь перемешивали при 15°C в течение 3 часов, после чего ее обрабатывали насыщенным водным раствором сульфита натрия (2 мл), разбавляли насыщенным водным раствором карбоната натрия (20 мл), и экстрагировали дихлорметаном (2×30 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали, и концентрировали в вакууме с получением P12 в виде желтого твердого вещества (15 г). Это вещество объединяли с продуктом аналогичной реакции, проведенной с использованием C33 (1,47 г, 4,63 ммоль), смешивали с дихлорметаном (60 мл) и петролейным эфиром (400 мл), и перемешивали в течение 40 минут. Фильтрация давала P12 в виде желтого твердого вещества. Общий выход: 14,3 г, 40,9 ммоль, 84%. LCMS m/z 350,3 [M+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 9,10 (с, 1H), 5,96-5,85 (м, 1H), 3,39 (с, 3H), 2,57 (с, 3H), 2,46 (с, 3H), 2,33-2,21 (м, 2H), 2,21-2,10 (м, 2H), 2,01-1,90 (м, 2H), 1,77-1,65 (м, 2H).
Альтернативное получение P12
6-Ацетил-8-циклопентил-5-метил-2-(метилсульфонил)пиридо[2,3-d]пиримидин-7(8H)-он (P12)

Стадия 1. Синтез 6-ацетил-8-циклопентил-5-метил-2-(метилсульфанил)пиридо[2,3-d]пиримидин-7(8H)-она, хлористоводородной соли (C33, хлористоводородная соль).
Смесь С32 (172,7 г, 487,5 ммоль), бутилэтенилового эфира (146,0 г, 1,458 моль), N, N-диизопропилэтиламина (121 мл, 695 ммоль), и {бис[2-(дифенилфосфино)фенил]эфир}палладий(II) дихлорида [Pd(DPEPhos)Cl2; 6,98 г, 9,75 ммоль] в н-бутаноле (1 л) дегазировали в течение 15 минут азотом, и затем нагревали до внутренней температуры 89°C. Реакционную смесь перемешивали в течение ночи с использованием механической мешалки, после чего добавляли водный раствор карбоната натрия (5%; 1 л). Органический слой концентрировали при пониженном давлении, используя водяную баню при 50°C, для удаления н-бутанола, и полученное твердое вещество растворяли в этилацетате (1 л). Добавляли активированный уголь (60 г), и смесь перемешивали в течение 10 минут перед фильтрованием через слой диатомовой земли. Фильтровальную подушку промывали этилацетатом, и объединенные фильтраты обрабатывали акцептором тиолового металла SiliCycle SiliaMetS® (45,0 г), перемешивали при комнатной температуре в течение 30 минут при комнатной температуре и фильтровали. Фильтровальную подушку промывали этилацетатом, и объединенные фильтраты снова обрабатывали акцептором тиолового металла SiliCycle SiliaMetS® (22,5 г), перемешивали при комнатной температуре в течение 30 минут, и фильтровали. Фильтровальную подушку промывали этилацетатом, и объединенные фильтраты концентрировали в вакууме с получением твердого вещества, которое растворяли в этилацетате (1 л) и обрабатывали хлористоводродной кислотой (6 M; 90 мл). После перемешивания этой реакционной смеси при комнатной температуре в течение 2 часов, ее фильтровали, и собранные твердые вещества промывали смесью трет-бутилметилового эфира и этилацетата 1:1 с получением C33, хлористоводородной соли, в виде твердого вещества (132 г). Фильтрат концентрировали досуха при пониженном давлении; остаток растворяли в смеси трет-бутилметилового эфира и этилацетата (1:1, 300 мл), и перемешивали в течение 30 минут. Эту смесь затем фильтровали, и собранные твердые вещества промывали смесью трет-бутилметилового эфира и этилацетата 1:1, получая дополнительно количество C33, хлористоводородной соли (31 г) в виде не совсем белого твердого вещества. Общий выход: 163 г, 460 ммоль, 94%. LCMS m/z 318,2 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ 8,95 (с, 1H), 8,33 (шир, с, 1H), 5,94-5,83 (м, 1H), 2,79 (с, 3H), 2,55 (с, 3H), 2,41 (с, 3H), 2,34-2,23 (м, 2H), 2,14-2,02 (м, 2H), 1,99-1,88 (м, 2H), 1,78-1,66 (м, 2H).
Стадия 2. Синтез 6-ацетил-8-циклопентил-5-метил-2-(метилсульфонил)пиридо[2,3-d]пиримидин-7(8H)-она (P12).
Раствор C33, хлористоводородной соли (154 г, 435 ммоль) в тетрагидрофуране (1,8 л) разбавляли водой (500 мл), обрабатывали фосфатом калия (40,2 г, 189 ммоль), и охлаждали до 15°C при механическом перемешивании. Затем в перемешиваемый раствор в течение нескольких минут медленно добавляли пероксимоносульфат калия (Oxone®; 402,0 г, 654 ммоль). После этого периодически применяли баню со льдом для поддержания внутренней температуры ниже 25°C, и перемешивание продолжали в течение 2 дополнительных часов. LCMS анализ на этой стадиие показал преобразование в продукт: LCMS m/z 350,1 [M+H]+. Реакционную смесь затем фильтровали, и собранное твердое вещество промывали этилацетатом. Водный слой объединенных фильтратов экстрагировали этилацетатом, и объединенные органические слои промывали дважды насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали, и концентрировали в вакууме. Остаток суспендировали в смеси этилацетата в гептане (5%; 1 л) в течение 1 часа; твердое вещество собирали фильтрацией и промывали 5% этилацетатом в гептане, получая P12 (135 г) в виде бледно-желтого твердого вещества. Концентрация фильтрата при пониженном давлении давала остаток, который суспендировали в течение 4 часов в смеси этилацетата в гептане (5%; 250 мл), после чего твердое вещество собирали фильтрованием и промывали 5% этилацетатом в гептане с получением дополнительно P12 (4,0 г) в виде белого твердого вещества. Общий выход: 139 г, 398 ммоль, 91%. 1H ЯМР (400 МГц, CDCl3) δ 9,11 (с, 1H), 5,96-5,85 (м, 1H), 3,39 (с, 3H), 2,57 (с, 3H), 2,46 (с, 3H), 2,33-2,22 (м, 2H), 2,22-2,10 (м, 2H), 2,01-1,91 (м, 2H), 1,78-1,65 (м, 2H).
Получение Р13
6-Ацетил-2-(децилтио)-5-метилпиридо[2,3-d]пиримидин-7(8H)-он (P13)

Стадия 1. Синтез 6-ацетил-5-метил-2-(метилтио)пиридо[2,3-d]пиримидин-7(8H)-она (C34).
Смесь 1-[4-амино-2-(метилтио)пиримидин-5-ил]этанона (17,2 г, 93,9 ммоль) и 2,2,6-триметил-4H-1,3-диоксин-4-она (53,4 г, 376 ммоль) в толуоле (375 мл) нагревали при 100°C в течение ночи, и затем оставляли при комнатной температуре (15°C - 25°C) в течение 2 дней. Реакционную смесь затем разбавляли этилацетатом (300 мл); твердые вещества собирали фильтрованием и промывали этилацетатом (200 мл), с получением C34 в виде коричневатого твердого вещества. Выход: 19,2 г, 77,0 ммоль, 82%. 1H ЯМР (400 МГц, DMSO-d6) δ 12,59 (шир, с, 1H), 9,01 (с, 1H), 2,57 (с, 3H), 2,44 (с, 3H), 2,34 (с, 3H).
Стадия 2. Синтез 6-ацетил-5-метил-2-(метилсульфонил)пиридо[2,3-d]пиримидин-7(8H)-она (C35).
Пероксимоносульфат калия (Oxone®; 128 г, 208 ммоль) добавляли к суспензии C34 (17,2 г, 69,0 ммоль) в смеси тетрагидрофурана (800 мл) и воды (160 мл) при комнатной температуре (20°C). Реакционную смесь перемешивали при 25°C в течение 27 часов, после чего смесь разбавляли водой (800 мл), перемешивали при комнатной температуре (15°C) в течение 2 часов, и фильтровали. Собранное твердое вещество последовательно промывали водой (300 мл) и трет-бутилметиловым эфиром (100 мл), затем смешивали с водой (300 мл), перемешивали при комнатной температуре (20°C) в течение 1 часа, и фильтровали. Фильтрпрессную лепешку промывали водой (100 мл) и объединяли с продуктом аналогичной реакции, проведенной с использованием C34 (2,10 г, 8,42 ммоль). Это вещество снова смешивали с водой (150 мл), перемешивали при комнатной температуре (20°C) в течение 1 часа, и фильтровали. Фильтрпрессную лепешку промывали водой (80 мл) с получением C35 в виде белого твердого вещества. Общий выход: 9,0 г, 32,0 ммоль, 41%. LCMS m/z 282,1 [M+H]+. 1H ЯМР (400 МГц, DMSO-d6) δ 13,31 (шир, с, 1H), 9,39 (с, 1H), 3,43 (с, 3H), 2,46 (с, 3H), 2,41 (с, 3H).
Стадия 3. Синтез 6-ацетил-2-(децилтио)-5-метилпиридо[2,3-d]пиримидин-7(8H)-она (P13)
К раствору C35 (1,12 г, 3,98 ммоль) в тетрагидрофуране (19 мл) добавляли триэтиламин (1,66 мл, 11,9 ммоль), затем декан-1-тиол (96%, 1,08 г, 5,95 ммоль), и реакционную смесь помещали в нагревательный блок при 62°C. Через 3 часа LCMS анализ показал превращение в продукт P13: LCMS m/z 376,3 [M+H]+; реакционной смеси давали остыть до комнатной температуры и перемешивали в течение 15 часов. Смесь затем концентрировали в вакууме и остаток очищали с помощью хроматографии на силикагеле (градиент: 0% - 100% этилацетата в гептане), получая P13 в виде белого твердого вещества. (Было отмечено, что при хроматографической очистке продукт выпадал в осадок.) Выход: 1,37 г, 3,65 ммоль, 92%. 1H ЯМР (400 МГц, хлороформ-d) δ 9,95 (шир, с, 1H), 8,82 (с, 1H), 3,18 (т, J=7,3 Гц, 2H), 2,59 (с, 3H), 2,44 (с, 3H), 1,74 (пентет, J=7,4 Гц, 2H), 1,50-1,39 (м, 2H), 1,37-1,19 (м, 12H), 0,87 (т, J=6,7 Гц, 3H).
Пример 1
6-Ацетил-8-циклобутил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он (1)


Стадия 1. Синтез 6-бром-8-циклобутил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она (C36).
Раствор P1 (150 мг, 0,456 ммоль), (3S,4R)-4-аминотетрагидро-2H-пиран-3-ол гидрохлорида (77,1 мг, 0,502 ммоль) и N, N-диизопропилэтиламина (0,40 мл, 2,3 ммоль) в диметилсульфоксиде (1,8 мл) нагревали при 60°C в течение 5 часов. Реакционную смесь затем охлаждали до комнатной температуры и разбавляли водой (15 мл); полученное твердое вещество собирали фильтрованием с получением C36 в виде светло-красного твердого вещества. Выход: 168 мг, 0,410 ммоль, 90%. LCMS m/z 411,1 (наблюдали изотопную картину брома) [M+H]+. 1H ЯМР (400 МГц, хлороформ-d), характеристические пики: δ 8,58 (с, 1H), 5,89-5,67 (м, 1H), 5,54-5,24 (шир, с, 1H), 4,09 (дд, J=11,4, 4,9 Гц, 1H), 4,04-3,97 (м, 1H), 3,65 (ддд, J=9,3, 9,1, 4,8 Гц, 1H), 3,50 (ушир. дд, J=11,8, 11,5 Гц, 1H), 3,32-3,18 (м, 1H), 2,54 (с, 3H), 2,41-2,28 (м, 2H), 2,16-1,96 (м, 2H), 1,91-1,78 (м, 1H), 1,78-1,66 (м, 2H, предполагаемый; частично скрытый пиком воды).
Стадия 2. Синтез 6-ацетил-8-циклобутил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она (1).
Смесь С36 (165 мг, 0,403 ммоль), тетракис(трифенилфосфин)палладия(0) (23,5 мг, 20,3 мкмоль) и трибутил(1-этоксиэтенил)станнана (204 мг, 0,565 ммоль) в толуоле (4 мл) продували азотом в течение 5 минут и затем нагревали при 110°C в течение 12 часов. Затем реакционную смесь концентрировали в вакууме, и остаток растворяли в 2-метилтетрагидрофуране (8 мл). Добавляли воду (150 мкл) и хлорид водорода (4 M раствор в 1,4-диоксане; 0,20 мл, 0,80 ммоль) и эту реакционную смесь перемешивали при комнатной температуре. Через 2 часа LCMS анализ показал преобразование в 1: LCMS m/z 373,2 [M+H]+. Через 5 часов, реакционную смесь концентрировали при пониженном давлении, и остаток разбавляли насыщенным водным раствором бикарбоната натрия, и трижды экстрагировали этилацетатом. Объединенные органические слои концентрировали в вакууме, и очищали с помощью сверхкритической флюидной хроматографии (Колонка: Nacalai USA Cosmosil 3-гидроксифенил; Подвижная фаза А: диоксид углерода; Подвижная фаза B: метанол; Градиент: 12% B в течение 0,2 минуты, затем увеличение до 28% B при 4% в минуту; Обратное давление: 100 бар) с получением 1. Выход: 83,5 мг, 0,224 ммоль, 56%. 1H ЯМР (400 МГц, 80°C, DMSO-d6) δ 8,75 (с, 1H), 7,49 (шир, с, 1H), 5,80-5,65 (м, 1H), 4,70 (шир, с, 1H), 4,05-3,93 (м, 1H), 3,93-3,81 (м, 2H), 3,65-3,53 (м, 1H), 3,38 (дд, J=11,8, 11,0 Гц, 1H), 3,27-3,05 (м, 3H), 2,41 (с, 3H), 2,36-2,21 (м, 2H), 2,28 (с, 3H), 2,07-1,91 (м, 2H), 1,91-1,75 (м, 1H), 1,69-1,53 (м, 1H).
Пример 2
6-Ацетил-8-циклопентил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он (2)

Стадия 1. Синтез 6-бром-8-циклопентил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она (C37).
К суспензии 6-бром-2-хлор-8-циклопентил-5-метилпиридо[2,3-d]пиримидин-7(8H)-она (12,0 г, 35,0 ммоль) и (3S,4R)-4-аминотетрагидро-2H-пиран-3-ол гидрохлорида (5,92 г, 38,5 ммоль) в диметилсульфоксиде (100 мл) добавляли N, N-диизопропилэтиламин (23,3 мл, 134 ммоль). Реакционную смесь нагревали при 60°C в течение 21 часов, после чего смесь охлаждали до температуры окружающей среды и выливали в воду со льдом (1,5 л). Полученное твердое вещество собирали фильтрованием с получением C37 в виде розового твердого вещества. 1H ЯМР анализ показал, что это соединение существует в виде смеси ротамеров. Выход: 13,1 г, 30,9 ммоль, 88%. LCMS m/z 423,1 (наблюдали изотопную картину брома) [M+H]+. 1H ЯМР (400 МГц, DMSO-d6) δ 8,79 (шир, с, 1H), [7,91-7,77 (м) и 7,74-7,61 (м), общий 1Н], [6,10-5,91 (м) и 5,90-5,74 (м), общий 1Н], 4,95 (д, J=5,2 Гц, 1H), 4,07-3,74 (м, 3H), 3,53 (шир, с, 1H), 3,40-3,24 (м, 1H, предполагаемый; частично скрытый пиком воды), 3,03 (дд, J=11,1, 9,7 Гц, 1H), 2,52 (с, 3H), 2,43-2,24 (м, 1H), 2,23-2,07 (м, 1H), 2,06-1,83 (м, 3H), 1,83-1,69 (м, 2H), 1,69-1,46 (м, 3H).
Стадия 2. Синтез 6-ацетил-8-циклопентил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она (2).
Раствор C37 (450 мг, 1,06 ммоль), тетракис(трифенилфосфин)палладия(0) (62,0 мг, 53,6 мкмоль), и трибутил(1-этоксиэтенил)станнана (537 мг, 1,49 ммоль) в толуоле (11 мл) продували азотом в течение 5 минут и затем нагревали при 110°C в течение 18 часов. Летучие вещества удаляли в вакууме и остаток растворяли в 2-метилтетрагидрофуране (10 мл), обрабатывали водой (0,15 мл) и хлоридом водорода (4 M раствор в 1,4-диоксане; 0,30 мл, 1,2 ммоль), и перемешивали при комнатной температуре в течение 6 часов. Реакционную смесь затем концентрировали, и остаток очищали с помощью хроматографии на силикагеле [градиент: 15% - 100% (9:1 этилацетат/этанол) в гептане]. Очищенное вещество (примерно 500 мг) смешивали с этилацетатом (8 мл) и гептаном (15 мл) и нагревали при 75°C. Добавлен дополнительный гептан (6 мл), и смеси давали остыть до комнатной температуры. Полученное вещество собирали фильтрованием с получением 2 в виде твердого вещества. Выход: 310 мг, 0,802 ммоль, 76%. LCMS m/z 387,2 [M+H]+. 1H ЯМР (400 МГц, 80°C, DMSO-d6) 8,76 (с, 1H), 7,44 (шир, с, 1H), 5,81 (квинтет, J=9,0 Гц, 1H), 4,68 (шир, с, 1H), 3,99-3,80 (м, 3H), 3,64-3,53 (м, 1H), 3,36 (ддд, J=11,7, 11,6, 2,1 Гц, 1H), 3,08 (дд, J=11,2, 9,5 Гц, 1H), 2,45-2,29 (м, 2H), 2,41 (с, 3H), 2,28 (с, 3H), 2,08-1,92 (м, 3H), 1,86-1,71 (м, 2H), 1,71-1,52 (м, 3H).
Пример 3
6-Ацетил-8-(2-азаспиро[3.3]гептан-6-ил)-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он (3)


Стадия 1. Синтез трет-бутил 6-[6-ацетил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метил-7-оксопиридо[2,3-d]пиримидин-8(7H)-ил]-2-азаспиро[3.3]гептан-2-карбоксилата (C38).
Смесь P2 (из Получения Р2, стадия 5; 120 мг, ≤0,225 ммоль), (3S,4R)-4-аминотетрагидро-2H-пиран-3-ола (36,6 мг, 0,312 ммоль), и карбоната натрия (55,2 мг, 0,521 ммоль) в тетрагидрофуране (8 мл) перемешивали при 60°C в течение 18 часов. Реакционную смесь очищали с помощью хроматографии на силикагеле (градиент: 0% - 25% метанола в дихлорметане) с получением C38 в виде светло-желтого стекла. Выход: 100 мг, 0,195 ммоль, 87% за 2 стадии. LCMS m/z 535,9 [M+Na+].
Стадия 2. Синтез 6-ацетил-8-(2-азаспиро[3.3]гептан-6-ил)-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она (3).
Раствор C38 (100 мг, 0,195 ммоль) в смеси трифторуксусной кислоты (2 мл) и дихлорметана (5 мл) перемешивали при комнатной температуре (10°C) в течение приблизительно 1 часа. Затем реакционную смесь концентрировали в вакууме и очищали с помощью обращенно-фазовой ВЭЖХ (Колонка: YMC-Triart C18, 7 мкм; Подвижная фаза А: вода, содержащая 0,05% гидроксида аммония; Подвижная фаза B: ацетонитрил; Градиент: 9% - 49% B) с получением 3 в виде белого твердого вещества. Выход: 49,1 мг, 0,119 ммоль, 61%. LCMS m/z 414,3 [M+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 8,59 (с, 1H), 6,06-5,30 (м, 2H), 4,06 (дд, J=11,4, 4,8 Гц, 1H), 4,02-3,87 (м, 2H), 3,79 (шир, с, 2H), 3,75 (шир, с, 2H), 3,69-3,58 (м, 1H), 3,56-3,43 (м, 1H), 3,31-3,07 (м, 4H), 2,66-2,54 (м, 2H), 2,50 (с, 3H), 2,29 (с, 3H), 2,20-2,05 (м, 1H), 1,76-1,61 (м, 1H).
Пример 4
6-Ацетил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метил-8-[(1R,2S)-2-метилциклопентил]пиридо[2,3-d]пиримидин-7(8H)-он (4)

Стадия 1. Синтез 6-бром-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метил-8-[(1R,2S)-2-метилциклопентил]пиридо[2,3-d]пиримидин-7(8H)-она (C39).
Раствор P3 (150 мг, 0,421 ммоль) (3S,4R)-4-аминотетрагидро-2H-пиран-3-ола (73,9 мг, 0,631 ммоль), и N, N- диизопропилэтиламина (272 мг, 0,366 мл, 2,10 ммоль) в диметилсульфоксиде (2 мл) нагревали при 70°C в течение 18 часов. Реакционную смесь затем разбавляли дихлорметаном (30 мл), промывали последовательно водой (3×10 мл) и насыщенным водным раствором хлорида натрия (10 мл), сушили над сульфатом натрия, фильтровали, и концентрировали в вакууме; хроматография на силикагеле (градиент: 0% - 100% этилацетата в дихлорметане) давала C39 в виде белого стекла. Выход: 130 мг, 0,297 ммоль, 71%. LCMS m/z 437,1 (наблюдали изотопную картину брома) [M+H]+.
Стадия 2. Синтез 6-ацетил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метил-8-[(1R,2S)-2-метилциклопентил]пиридо[2,3-d]пиримидин-7(8H)-она (4).
Азот барботировали через раствор C39 (130 мг, 0,297 ммоль) в толуоле (10 мл), и затем дбавляли трибутил(1-этоксиэтенил)станнан (429 мг, 1,19 ммоль) и тетракис(трифенилфосфин)палладий(0) (34,3 мг, 29,7 мкмоль). Реакционный сосуд герметично закрывали и нагревали при 110°C в течение 18 часов, после чего реакционную смесь охлаждали до комнатной температуры и фильтровали. После концентрирования фильтрата в вакууме остаток растворяли в этилацетате (20 мл), подкисляли до pH приблизительно 5 добавлением 6 М хлористоводродной кислоты, и перемешивали в течение примерно 10 минут. Полученную двухфазную смесь подщелачивали до значения pH примерно 8 путем добавления водного раствора бикарбоната натрия, и водный слой экстрагировали этилацетатом (2×10 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали, и концентрировали при пониженном давлении; остаток очищали с помощью хроматографии на силикагеле (градиент: 0% - 100% этилацетата в дихлорметане) с получением 4 в виде белого твердого вещества. Выход: 58,8 мг, 0,147 ммоль, 49%. LCMS m/z 401,0 [M+H]+. 1H ЯМР (400 МГц, метанол-d4) δ 8,76 (с, 1H), 6,22-6,05 (м, 1H), 4,14-3,89 (м, 3H), 3,73-3,57 (ушир.м, 1H), 3,54-3,45 (м, 1H), 3,22 (ушир. дд, J=10,4, 10,1 Гц, 1H), 2,82-2,56 (ушир.м, 1H), 2,46 (с, 3H), 2,43-2,31 (м, 1H), 2,34 (с, 3H), 2,16-1,98 (м, 2H), 1,98-1,80 (м, 3H), 1,72-1,52 (м, 2H), 0,79 (д, J=7,1 Гц, 3H).
Пример 5
3-Ацетил-1-циклопентил-7-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-4-метил-1,6-нафтиридин-2(1H)-он (5)

Свежеоткрытый флакон с N, N-диметилформамидом продували азотом в течение приблизительно 15 минут. Смесь P4 (63,0 г, 207 ммоль), (3S,4R)-4-аминотетрагидро-2H-пиран-3-ола (29,1 г, 248 ммоль) и трет-бутоксида лития (19,9 г, 249 ммоль) растворяли в продутом N, N-диметилформамиде (300 мл), и колбу продували под легким давлением азота в течение приблизительно 15 минут.
В отдельной колбе смесь ди-трет-бутил[3,6-диметокси-2',4',6'-три(пропан-2-ил)[бифенил]-2-ил] фосфана (tBuBrettPhos; 2,00 г, 4,13 ммоль) и трис(дибензилиденацетон)дипалладий(0) (1,89 г, 2,06 ммоль) продували при слабом давлении азота в течение приблизительно 15 минут. Добавляли N, N-диметилформамид (100 мл; предварительно продували азотом) и полученный раствор нагревали на масляной бане при 70°C в течение приблизительно 7 минут, после чего смесь охлаждали до комнатной температуры. Затем эту каталитическую смесь выливали в колбу, содержащую P4, и реакционную смесь перемешивали при 40°C в течение 1 часа. После охлаждения до комнатной температуры реакционную смесь выливали в воду (700 мл), и доводили до значения pH 5-6 добавлением 1 M хлористоводродной кислоты. Затем добавляли этилацетат (500 мл), и полученную смесь перемешивали для облегчения растворения смолистого твердого вещества. Водный слой экстрагировали этилацетатом (4×400 мл), и объединенные органические слои последовательно промывали водным раствором хлорида лития (7% масс.; 2×400 мл) и насыщенным водным раствором хлорида натрия (250 мл), сушили над сульфатом магния, и фильтровали, используя фриттованную воронку (шириной примерно 12,7 см), которая была покрыта слоем диатомовой земли (около 3,8 см), силикагелем (около 3,8 см) и песком. Фильтровальную подушку промывали этилацетатом (3,5 л), и объединенные фильтраты концентрировали в вакууме; остаток перемешивали с гептаном и концентрировали при пониженном давлении, с получением светло-оранжевого твердого вещества (79,0 г). После того, как это вещество объединяли с продуктами ряда реакций, проведенных аналогичным образом с использованием P4 (всего 160 г, 525 ммоль), смесь очищали с помощью сверхкритической флюидной хроматографии (Колонка: Chiral Technologies DCpak P4VP, 5 мкм; Подвижная фаза: 4:1 диоксид углерода/метанол; Обратное давление: 100 бар). Общий выход: 193,7 г, 502,5 ммоль, 69%. LCMS m/z 386,1 [M+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 8,49 (с, 1H), 6,32 (с, 1H), 5,66 (шир, с, 1H), 5,34 (пентет, J=9,0 Гц, 1H), 4,64 (д, J=5,5 Гц, 1H), 4,10 (дд, J=11,4, 4,9 Гц, 1H), 4,01 (ушир. дд, J=11,7, 4,5 Гц, 1H), 3,88-3,77 (м, 1H), 3,57 (ддд, J=9,4, 9,2, 4,9 Гц, 1H), 3,47 (ддд, J=11,9, 11,8, 2,1 Гц, 1H), 3,20 (дд, J=11,4, 9,9 Гц, 1H), 2,53 (с, 3H), 2,34 (с, 3H), 2,29-2,16 (м, 2H), 2,12-1,99 (м, 3H), 1,99-1,87 (м, 2H), 1,81-1,66 (м, 3H).
Дальнейшую очистку проводили следующим образом. Раствор 5 (185 г, 480 ммоль) в дихлорметане (приблизительно 750 мл) получали путем приложения минимального тепла до тех пор, пока 5 не находилось в растворе. Добавляли SiliaMetS Thiol (SH) (Silicycle; 18,3 г) и полученную смесь перемешивали при слабом кипячении с обратным холодильником в течение 30 минут, после чего ее охлаждали на бане со льдом в течение 5 минут, и фильтровали через слой диатомитовой земли толщиной 1 см, с последующей промывкой фильтровальной подушки дихлорметаном (приблизительно 250 мл). Объединенные фильтраты перегоняли при атмосферном давлении для удаления дихлорметана (приблизительно 250 мл). Добавляли трет-бутилметиловый эфир (500 мл) и затравочные кристаллы 5 (приблизительно 100 мг; см. происхождение этих кристаллов ниже) и дополнительный растворитель (приблизительно 250 мл, температура кипения приблизительно 45°C) удаляли перегонкой. Снова добавляли трет-бутилметиловый эфир (500 мл), и смесь перемешивали, что приводило к осаждению твердого вещества; перегонку продолжали до тех пор, пока не было собрано в общей сложности примерно 1,3 л, а температура кипения дистиллята не достигла 50,6°C. В этот момент смеси давали остыть до комнатной температуры при перемешивании, после чего твердое вещество собирали фильтрованием и промывали трет-бутилметиловым эфиром (2 х 100 мл), получая 5 в виде желтого твердого вещества (163 г, 423 ммоль, 88% для очистки).
Альтернативные условия реакции для получения 5 из P4 и (3S,4R)-4-аминотетрагидро-2H-пиран-3-ола включают использование Pd(OAc)2 из (AllylPdCl)2 и t-BuBrettPhos в качестве катализатора/лиганда; с использованием растворителей, включая изопропилацетат, метилэтилкетон, толуол, THF и 2-метилтетрагидрофуран, который предпочтительно содержит THF [приблизительно до 20% THF]; и с использованием другого основания, включая NaOt-Bu, KOt-Bu, KOt-Amyl, K3PO4 и Cs2CO3. Альтернативные условия обработки включают гашение реакции водным хлоридом аммония и промывание водного слоя органическим растворителем, совместимым с реакционным растворителем. Очистка 5 может быть выполнена кислотно-щелочной обработкой, затем с помощью тиол-силикагелевой подушки. К объединенным органическим слоям добавляют воду и доводят pH примерно до 6 добавлением 1 М хлористоводродной кислоты. трет-Бутилметиловый эфир используется в качестве промывки. Значение pH может быть доведено до примерно 5 с помощью NaOH и K3PO4. Пример 5 экстрагируют органическим растворителем, таким как дихлорметан, и очищают аналогично тому, как описано выше. Спирт с короткой цепью, например, C1-3 спирт, может быть включен в органический слой для элюирования 5 из тиол-силикагелевой подушки. Любой присутствующий цвет можно удалить стандартными процедурами перед кристаллизацией из спирта с короткой цепью.
Получение затравочных кристаллов 5
Суспензию 5 (40 г) в этаноле (235 мл) нагревали с обратным холодильником до получения раствора. К горячему раствору по каплям в течение 35 минут добавляли воду (600 мл), и собирали растворитель (азеотроп этанол/вода), используя насадку для короткой перегонки, при поддержании вакуума приблизительно 450 мбар. После завершения добавления воды нагревание продолжали в течение 45 минут; за это время собирали приблизительно 125 мл растворителя. Затем нагрев отключали, и смеси давали возможность перемешаться и охладиться до комнатной температуры в течение ночи. Фильтрация с последующей промывкой осадка на фильтре водой дала 5 (37,5 г) в виде светло-бежевого твердого вещества, которое, как было показано с помощью рентгено-структурного анализа, является кристаллическим; часть этого материала была использована в качестве затравочных кристаллов, использованных выше.
Получение кристалла 5 для рентгеноструктурного анализа
Образец 5 (150 мг, 0,389 ммоль) растворяли в метаноле (2 мл) и нагревали до кипения с обратным холодильником, получая раствор. Добавляли воду (4 мл) и продолжали нагревание для удаления метанола. Полученное твердое вещество собирали фильтрованием и получали кристалл 5, пригодный для рентгеноструктурного анализа (см. ниже). Кристаллический 5 является безводным из-за отсутствия растворителя, в том числе воды, в кристаллической решетке. Выход: 139 мг, 0,361 ммоль, 93%. 1H ЯМР (400 МГц, DMSO-d6) δ 8,57 (с, 1H), 7,13 (д, J=7,4 Гц, 1H), 6,44 (с, 1H), 5,34 (пентет, J=9,0 Гц, 1H), 5,12 (д, J=5,2 Гц, 1H), 3,91-3,75 (м, 3H), 3,47-3,3 (м, 2H, предполагаемый; частично скрытый пиком воды), 3,07 (дд, J=11,1, 9,5 Гц, 1H), 2,39 (с, 3H), 2,26 (с, 3H), 2,21-2,08 (м, 2H), 2,08-1,94 (м, 3H), 1,91-1,77 (м, 2H), 1,73-1,59 (м, 2H), 1,51-1,38 (м, 1H).
Монокристальный рентгеноструктурный анализ 5
Рентгеноструктурные анализы монокристалла
Сбор данных проводили на дифрактометре D8 Venture при комнатной температуре. Сбор данных состоял из омега- и фи- сканирований.
Структуру определяли путем внутреннего фазирования с использованием программного пакета SHELX в моноклинной пространственной группе P21. В дальнейшем структуру уточняли полноматричным методом наименьших квадратов. Все неводородные атомы находили и уточняли с использованием параметров анизотропного смещения.
Неупорядоченные положения в C17-C15 были отмечены, но не обработаны моделью нарушения.
Атом водорода, расположенный у азота и кислорода, был найден по разностной карте Фурье и уточнен с ограничением расстояний. Оставшиеся атомы водорода помещали в расчетные положения и позволяли им перемещаться на своих атомах-носителях. Конечное уточнение включало параметры изотропного смещения для всех атомов водорода.
Анализ абсолютной структуры с использованием методов правдоподобия (Hooft, 2008) проводили с использованием PLATON (Spek). Была сделана попытка определить абсолютную конфигурацию непосредственно из данных рентгеновской дифракции методом Хоофта, а также методом Парсонса. Окончательный уточненный параметр Хоофта и параметр Парсонса были даны как 0,12 с esd 0,06. Это значение указывает на то, что стереохимия 5, изображенная на схеме выше, представляет собой абсолютную конфигурацию.
Конечный R-фактор составлял 4,2%. Конечная разностная карта Фурье не показала отсутствующей или неверно установленной электронной плотности.
Соответствующая информация о кристалле, сборе данных и уточнении представлена в таблице L. Атомные координаты, длины связей, углы связей и параметры смещения приведены в таблицах M-P.
Программное обеспечение и ссылки на источники
SHELXTL, Version 5.1, Bruker AXS, 1997.
PLATON, A. L. Spek, J. Appl. Cryst. 2003, 36, 7-13.
MERCURY, C. F. Macrae, P. R. Edington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler, and J. van de Streek, J. Appl. Cryst. 2006, 39, 453-457.
OLEX2, O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard, and H. Puschmann, J. Appl. Cryst. 2009, 42, 339-341.
R. W. W. Hooft, L. H. Straver, and A. L. Spek, J. Appl. Cryst. 2008, 41, 96-103.
H. D. Flack, Acta Cryst. 1983, A39, 867-881.

Таблица М. Атомные координаты (x 104) и эквивалентные параметры изотропного смещения (Å2×103) для 5. U(eq) представляет собой 1/3 следа ортогонализированного тензора Uij.

Таблица N. Длины [Å] и углы [°] связей для 5.






Преобразования симметрии использовали для создания эквивалентных атомов.
Таблица P. Параметры анизотропного смещения (Å2×103) для 5. Показатель коэффициента анизотропного смещения принимает вид: -2π2[h2 a*2U11 + ... + 2 h k a* b* U12 ].


Таким образом, один вариант осуществления изобретения предлагает 3-ацетил-1-циклопентил-7-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-4-метил-1,6-нафтиридин-2(1H)-он в виде твердого кристаллического вещества. В другом дополнительном варианте осуществления кристаллическое твердое вещество является безводным.
Получение данных порошковой рентгеновской дифракции (PXRD) для кристалла 5
Образец кристалла 5 (безводный) был представлен на PXRD-анализа, и было обнаружено, что он представляет собой кристаллическое вещество (обозначенное как Форма I). Анализ порошковой рентгеновской дифракции проводили с использованием дифрактометра Bruker D8 Advance, снабженного источником излучения Cu. Щель расходимости устанавливали на 10 мм непрерывного освещения. Дифрагированное излучение регистрировали с помощью детектора LYNXEYE_EX с моторизованными щелями. Напряжение и сила тока рентгеновской трубки были установлены на 40 кВ и 40 мА, соответственно. Данные собирали при длине волны Cu (CuKᾱ=1,5418 λ) в гониометре Theta-Theta от 3,0 до 40,0 градусов 2-тета с использованием шага 0,01 градуса и времени шага 1,0, второй с экраном антирассеивания на месте. Образцы вращались во время сбора данных. Образцы получали, помещая их в кремниевый низкофоновый держатель образцов и вращая во время сбора. Данные собирали с использованием программного обеспечения Bruker DIFFRAC Plus, и анализ выполняли с помощью программного обеспечения EVA diffract plus. Файл данных PXRD не обрабатывался до поиска пика. Используя алгоритм поиска пиков в программном обеспечении EVA, пики, выбранные с пороговым значением 1, использовались для предварительного присвоения пиков. Для обеспечения достоверности корректировки вносились вручную; результаты автоматических назначений проверяли визуально, и положения пиков доводили до максимума. Обычно выбирали пики с относительной интенсивностью ≥ 3%. Обычно пики, которые не были разрешены или соответствовали шуму, не выбирали. Типичная ошибка, связанная с положением пика PXRD, указанным в USP, до +/- 0,2° 2-тета (USP-941). Постоянно наблюдалась одна дифрактограмма, представленная на фиг. 1. Список дифракционных пиков, выраженных в градусах 2θ и относительных интенсивностей с относительной интенсивностью ≥ 3,0% PXRD от образца кристалла 5 (полученного способами, представленными в данном документе) показаны в Таблице X1 ниже.
Примеры 6 и 7
6-ацетил-8-[(1S,2S)-2-гидрокси-2-метилциклопентил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он и 6-ацетил-8-[(1R,2R)-2-гидрокси-2-метилциклопентил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он [6 (DIAST-1) и 7 (DIAST-2)]

Стадия 1. Синтез rac-(1S,2S)-2-[6-(1-этоксиэтенил)-5-метил-2-(метилтио)-7-оксопиридо[2,3-d]пиримидин-8(7H)-ил]-1-метилциклопентил ацетата (C40).
Смесь P5 (447 мг, 1,05 ммоль), трибутил(1-этоксиэтенил)станнана (568 мг, 1,57 ммоль), и тетракис(трифенилфосфин)палладия(0) (72,7 мг, 62,9 мкмоль) в толуоле (10 мл) нагревали с обратным холодильником в течение 5 часов. LCMS-анализ реакционной смеси в этот момент показал присутствие продукта C40: LCMS m/z 418,1 [M+H]+. После фильтрования реакционной смеси через диатомовую землю, фильтрат концентрировали в вакууме и остаток очищали с помощью хроматографии на силикагеле (градиент: 20% - 60% этилацетата в гептане) с получением C40 в виде бледно-желтой пены. Выход: 380 мг, 0,91 ммоль, 87%. 1H ЯМР (400 МГц, хлороформ-d), характеристические пики: δ 8,76 (с, 1H), 6,48-6,23 (м, 1H), 4,54 (д, J=2,5 Гц, 1H), 4,17 (д, J=2,5 Гц, 1H), 3,93 (кв, J=7,0 Гц, 2H), 2,62 (с, 3H), 2,62-2,52 (м, 1H), 2,45 (с, 3H), 1,99 (с, 3H), 1,85-1,71 (м, 1H), 1,44 (шир, с, 3H), 1,35 (т, J=7,0 Гц, 3H).
Стадия 2. Синтез rac-(1S,2S)-2-[6-ацетил-5-метил-2-(метилтио)-7-оксопиридо[2,3-d]пиримидин-8(7H)-ил]-1-метилциклопентил ацетата (C41).
Хлористоводродную кислоту (1,0 M; 3,46 мл, 3,46 ммоль) добавляли к раствору C40 (361 мг, 0,865 ммоль) в тетрагидрофуране (27 мл), и реакционную смесь перемешивали при комнатной температуре в течение 40 минут. LCMS-анализ реакционной смеси в этот момент показал присутствие продукта C41: LCMS m/z 390,1 [M+H]+. Добавляли насыщенный водный раствор бикарбоната натрия и полученную смесь экстрагировали этилацетатом. Объединенные органические слои сушили над сульфатом магния, фильтровали, концентрировали в вакууме, и подвергали хроматографии на силикагеле, получая C41 в виде твердого вещества. Выход: 315 мг, 0,809 ммоль, 94%. 1H ЯМР (400 МГц, хлороформ-d), характеристические пики: δ 8,80 (с, 1H), 6,48-6,24 (шир, с, 1H), 2,61 (с, 3H), 2,51 (с, 3H), 2,38 (с, 3H), 2,36-2,24 (м, 1H), 2,10-1,97 (м, 2H), 1,99 (с, 3H), 1,87-1,73 (м, 1H), 1,45 (шир. с, 3H).
Стадия 3. Синтез rac-(1S,2S)-2-[6-ацетил-5-метил-2-(метилсульфинил)-7-оксопиридо[2,3-d]пиримидин-8(7H)-ил]-1-метилциклопентил ацетата (C42).
Раствор пероксимоносульфата калия (Oxone®; 1,05 г, 1,71 ммоль) в воде (5 мл) добавляли к раствору C41 (315 мг, 0,809 ммоль) в тетрагидрофуране (20 мл), и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. LCMS-анализ реакционной смеси в этот момент показал, что образовался C42: LCMS m/z 346,0 [(M - уксусная кислота) + H]+. Добавляли воду и полученную смесь экстрагировали дважды этилацетатом. Объединенные органические слои сушили над сульфатом натрия, фильтровали, и концентрировали в вакууме с получением C42 в виде бледно-желтого твердого вещества. Выход: 299 мг, 0,737 ммоль, 91%. 1H ЯМР (400 МГц, хлороформ-d), характеристические пики: δ 9,13 (с, 1H), 6,46-6,02 (шир, с, 1H), 3,36 (с, 3H), 2,53 (с, 3H), 2,46 (с, 3H), 2,36-2,20 (м, 2H), 2,16-2,02 (м, 2H), 1,98 (с, 3H), 1,44 (шир. с, 3H).
Стадия 4. Синтез (1S,2S)-2-[6-ацетил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метил-7-оксопиридо[2,3-d]пиримидин-8(7H)-ил]-1-метилциклопентил ацетата и (1R,2R)-2-[6-ацетил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метил-7-оксопиридо[2,3-d]пиримидин-8(7H)-ил]-1-метилциклопентил ацетата (C43).
Смесь С42 (170 мг, 0,419 ммоль), (3S,4R)-4-аминотетрагидро-2H-пиран-3-ол гидрохлорида (129 мг, 0,840 ммоль) и N, N-диизопропилэтиламина (0,348 мл, 2,00 ммоль) в диметилсульфоксиде (3 мл) перемешивали при 50°C в течение 1 часа. LCMS-анализ реакционной смеси в этот момент показал присутствие C43 в виде двух пиков равного размера для двухкомпонентных диастереомеров: LCMS m/z 399,2 [(M - уксусная кислота) + H]+. После разбавления реакционной смеси водой и экстрагирования дихлорметаном объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (градиент: 0% - 10% этилацетата в гептане) давала C43 в виде не совсем белого твердого вещества. Спектр 1H ЯМР C43 соответствовал присутствию 2 диастереомеров. Выход: 170 мг, 0,371 ммоль, 88%. 1H ЯМР (400 МГц, хлороформ-d), характеристические пики: δ [8,63 (с) и 8,62 (с), общий 1Н], 6,35-6,07 (м, 1H), 4,13-4,04 (м, 1H), 4,04-3,91 (м, 2H), 3,71-3,42 (м, 2H), 3,21 (ддд, J=11,3, 9,6, 6,2 Гц, 1H), [2,49 (с) и 2,49 (и), общий 3H], [2,34 (с) и 2,33 (с), общий 3H], [2,04 (с) и 2,02 (с), общий 3H], 1,47 (шир. с, 3H).
Стадия 5. Синтез 6-ацетил-8-[(1S,2S)-2-гидрокси-2-метилциклопентил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она и 6-ацетил-8-[(1R,2R)-2-гидрокси-2-метилциклопентил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она [6 (DIAST-1) и 7 (DIAST-2)].
Карбонат калия (512 мг, 3,70 ммоль) добавляли к раствору C43 (170 мг, 0,371 ммоль) в метаноле (12 мл). Реакционную смесь перемешивали при комнатной температуре в течение 24 часов, после чего смесь разбавляли водой и экстрагировали дихлорметан. Объединенные органические слои сушили над сульфатом натрия, фильтровали, концентрировали в вакууме, и очищали с помощью хроматографии на силикагеле (Градиент: 0% - 10% этилацетата в гептане) с получением смеси 6 и 7 в виде бледно-желтого твердого вещества. Выход: 115 мг, 0,276 ммоль, 74%. Разделение двух диастереомеров проводили с использованием сверхкритической флюидной хроматографии (Колонка: Chiral Technologies Chiralcel OJ-H, 5 мкм; Подвижная фаза: 4:1 диоксид углерода/метанол; Обратное давление: 100 бар). Диастереомер, элюирующийся первым, обозначен как 6, а диастереомер, элюирующийся вторым, как 7; оба выделены в виде твердых веществ.
6 (DIAST-1) - Выход: 23,1 мг, 55,5 мкмоль, 20% для разделения. LCMS m/z 416,9 [M+H]+. 1H ЯМР (400 МГц, 80°C, DMSO-d6), характеристические пики : δ 8,77 (с, 1H), 7,45 (шир. с, 1H), 5,91-5,82 (м, 1H), 4,11 (с, 1H), 4,03-3,93 (м, 1H), 3,90-3,81 (м, 2H), 3,58-3,47 (м, 1H), 3,38 (ддд, J=11,7, 11,6, 2,3 Гц, 1H), 3,08 (дд, J=11,2, 9,6 Гц, 1H), 2,66-2,55 (м, 1H), 2,39 (с, 3H), 2,29 (с, 3H), 2,20-2,10 (м, 1H), 2,05-1,81 (м, 4H), 1,78-1,69 (м, 1H), 1,69-1,56 (м, 1H), 1,03 (с, 3H). Время удерживания: 0,54 минут (Колонка: Chiral Technologies Chiralcel OJ-3, 4,6×100 мм, 3 мкм; Подвижная фаза: 4:1 диоксид углерода/метанол; Скорость потока: 4 мл/минута; Обратное давление: 120 бар).
7 (DIAST-2) - Выход: 23,4 мг, 56,2 мкмоль, 20% для разделения. LCMS m/z 416,9 [M+H]+. 1H ЯМР (400 МГц, 80°C, DMSO-d6), характеристические пики : δ 8,77 (с, 1H), 7,41 (ушир.д, J=7,6 Гц, 1H), 5,88 (дд, J=9,4, 7,5 Гц, 1H), 3,99 (с, 1H), 3,97-3,87 (м, 1H), 3,87 (дд, J=11,1, 4,8, 1H), 3,84-3,77 (м, 1H), 3,64-3,54 (м, 1H), 3,37 (ддд, J=11,8, 11,5, 2,4 Гц, 1H), 3,09 (дд, J=11,0, 9,6 Гц, 1H), 2,39 (с, 3H), 2,29 (с, 3H), 2,27-2,17 (м, 1H), 2,15-2,06 (м, 1H), 2,04-1,93 (м, 1H), 1,93-1,82 (м, 2H), 1,76-1,67 (м, 1H), 1,58-1,44 (м, 1H), 1,04 (с, 3H). Время удерживания: 0,79 минут (Аналитические условия идентичны тем, которые использовали для 6).
Примеры 8 и 9
6-Ацетил-8-циклопентил-2-{[(3S,4R)-3-гидрокси-2,2-диметилтетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он и 6-ацетил-8-циклопентил-2-{[(3R,4S)-3-гидрокси-2,2-диметилтетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он
[8 (ENT-1) и 9 (ENT-2)]
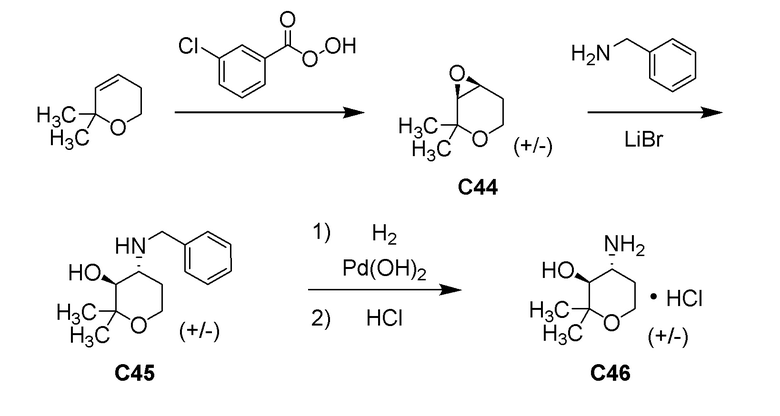

Стадия 1. Синтез rac-(1S,6S)-2,2-диметил-3,7-диоксабицикло[4.1.0]гептана (C44).
3-Хлорпероксибензойную кислоту (70%, 11,2 г, 45,4 ммоль) добавляли к раствору 6,6-диметил-3,6-дигидро-2H-пирана (см. L-I. Olsson and A. Claesson, Synthesis 1979, 743-745) (3,9 г, 35 ммоль) в хлороформе (35 мл), который перемешивали на водяной бане для поддержания реакционной смеси при комнатной температуре. Через 16 часов реакционную смесь, все еще находящуюся на водяной бане, осторожно обрабатывали раствором сульфита натрия (4,4 г, 35 ммоль) в воде (20 мл). Добавляли насыщенный водный раствор бикарбоната натрия (20 мл) и водный слой экстрагировали дихлорметаном (2×80 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали, и осторожно упаривали (10 мм рт. ст., <20°C) с получением C44 (4,49 г), который использовали непосредственно на следующей стадии. По данным анализа 1H ЯМР, это вещество содержало примеси, производные 3-хлорбензойной кислоты. 1H ЯМР (400 МГц, хлороформ-d), только пики C44: δ 3,58 (ддд, J=11,8, 8,6, 4,7 Гц, 1H), 3,44 (ддд, J=11,8, 5,2, 4,2 Гц, 1H), 3,40-3,36 (м, 1H), 2,91 (д, J=4,3 Гц, 1H), 2,00-1,86 (м, 2H), 1,32 (с, 3H), 1,29 (с, 3H).
Стадия 2. Синтез rac-(3S,4R)-4-(бензиламино)-2,2-диметилтетрагидро-2H-пиран-3-ола (C45).
Смесь C44 (из предыдущей стадии; 4,49 г, ≤35 ммоль), бензиламина (11,5 мл, 105 ммоль), и бромид лития (30,4 г, 350 ммоль) в толуоле (35 мл) нагревали при 60°C в течение 16 часов. Затем реакционную смесь концентрировали в вакууме, смешивали с метанолом, и снова концентрировали, после чего остаток распределяли между насыщенным водным раствором бикарбоната натрия (100 мл) и этилацетатом (500 мл). Добавляли воду (400 мл) для облегчения солюбилизации и водный слой экстрагировали этилацетатом (2×400 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением C45 (14,2 г). По данным анализа 1H ЯМР, этот образец содержал значительное количество бензиламина. Выход с поправкой на бензиламин: 7,5 г, 32 ммоль, 91% за 2 стадии. LCMS m/z 236,3 [M+H]+. 1H ЯМР (400 МГц, хлороформ-d), характеристические пики для C45: δ 3,95 (д, J=12,9 Гц, 1H), 3,75-3,68 (м, 2H), 3,62 (ддд, J=12,4, 12,3, 2,4 Гц, 1H), 3,11 (д, J=9,9 Гц, 1H), 2,69 (ддд, J=11,6, 9,9, 4,3 Гц, 1H), 2,08-2,00 (м, 1H), 1,47-1,35 (м, 1H), 1,30 (с, 3H), 1,17 (с, 3H).
Стадия 3. Синтез rac-(3S,4R)-4-амино-2,2-диметилтетрагидро-2H-пиран-3-ол, хлористоводородной соли (C46).
Раствор C45 (2,0 г, 8,5 ммоль) в метаноле (85 мл) дегазировали азотом и затем обработали 20% гидроксидом палладия на угле (400 мг). Эту смесь перемешивали в атмосфере водорода из баллона в течение 16 часов, после чего снова добавляли 20% гидроксид палладия на угле (400 мг) и гидрирование продолжали в течение 3 дней. После удаления катализатора фильтрацией, фильтрпрессную лепешку промывали метанолом (30 мл), и объединенные фильтраты обрабатывали хлористоводродной кислотой (4 M; 10,7 мл, 43 ммоль). Полученную смесь концентрировали в вакууме (10 мм рт. ст., 40°C), получая светло-коричневое твердое вещество. Его смешивали с диэтиловым эфиром (30 мл) и обрабатывали ультразвуком; прозрачный эфирный слой декантировали, а оставшееся смолистое твердое вещество сушили с получением C46 в виде светло-коричневого твердого вещества. Выход: 820 мг, 4,5 ммоль, 53%. LCMS m/z 146,3 [M+H]+. 1H ЯМР (400 МГц, DMSO-d6) δ 8,22 (шир, с, 3H), 3,58-3,44 (м, 2H), 3,23 (д, J=10,2 Гц, 1H), 3,15-3,02 (м, 1H), 1,98-1,89 (м, 1H), 1,73-1,59 (м, 1H), 1,14 (с, 3H), 1,06 (с, 3H).
Стадия 6. Синтез 6-ацетил-8-циклопентил-2-{[(3S,4R)-3-гидрокси-2,2-диметилтетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она и 6-ацетил-8-циклопентил-2-{[(3R,4S)-3-гидрокси-2,2-диметилтетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она [8 (ENT-1) и 9 (ENT-2)].
Флакон, содержащий раствор P6 (317 мг, 1,04 ммоль), C46 (290 мг, 1,6 ммоль) и N, N-диизопропилэтиламин (0,90 мл, 5,2 ммоль) в диметилсульфоксиде (4 мл) закрывали крышкой и нагревали до 80°C (температура блока) при перемешивании в течение 16 часов. Затем реакционную смесь распределяли между дихлорметаном (100 мл) и насыщенным водным раствором бикарбоната натрия (20 мл), и водный слой экстрагировали дихлорметаном (30 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали, и концентрировали в вакууме. Разделение энантиомеров компонентов проводили с использованием сверхкритической флюидной хроматографии (Колонка: Chiral Technologies Chiralpak AD-H, 5 мкм; Подвижная фаза: 3:2 диоксид углерода/метанол; Обратное давление: 100 бар); энантиомер, элюирующийся первым, который проявлял отрицательное (-) вращение, был обозначен как 8, и энантиомер, элюирующийся вторым, который проявлял положительное (+) вращение, был обозначен как 9. При исследовании спектров 1H ЯМР предполагалось, что оба эти вещества существуют в виде смеси ротамеров.
8 (ENT-1) - Выход: 162 мг, 0,391 ммоль, 38%. LCMS m/z 415,0 [M+H]+. 1H ЯМР (700 МГц, DMSO-d6) δ [8,78 (с) и 8,73 (с), общий 1Н], [7,72-7,65 (м) и 7,56-7,50 (м), общий 1Н], [5,91-5,81 (м) и 5,74-5,62 (м), общий 1Н], 5,08-4,99 (м, 1H), [4,14 (ушир.с) и 4,04 (ушир.с), общий 1Н], [3,6-3,49 (м) и 3,31-3,19 (м), общий 1Н, предполагаемый; слабопольный сигнал, частично скрытый пиком воды], 2,38 (с, 3H), 2,36-2,28 (м, 1H), 2,23 (с, 3H), 2,16 (шир, с, 1H), 2,01-1,49 (м, 10H), 1,16 (с, 3H), 1,12 (с, 3H). Время удерживания: 1,27 минут (Колонка: Chiral Technologies Chiralpak AD-3, 4,6×100 мм, 3 мкм; Подвижная фаза: 3:2 диоксид углерода/метанол; Скорость потока: 4 мл/минута; Обратное давление: 120 бар).
9 (ENT-2) - Выход: 151 мг, 0,364 ммоль, 35%. LCMS m/z 415,0 [M+H]+, 1H ЯМР (700 МГц, DMSO-d6) δ [8,78 (с) и 8,73 (с), общий 1Н], [7,69 (ушир.с) и 7,53 (ушир.с), общий 1Н], [5,86 (ушир.с) и 5,68 (ушир.с), общий 1Н], 5,08-4,98 (м, 1H), [4,14 (ушир.с) и 4,04 (ушир.с), общий 1Н], [3,6-3,49 (м) и 3,30-3,18 (м), общий 1Н, предполагаемый; слабопольный сигнал, частично скрытый пиком воды], 2,38 (с, 3H), 2,36-2,29 (м, 1H), 2,23 (с, 3H), 2,20-2,12 (м, 1H), 2,01-1,49 (м, 10H), 1,16 (с, 3H), 1,12 (с, 3H). Время удерживания: 2,24 минут (Аналитические условия идентичны тем, которые использовали для 8).
Пример 10
6-Ацетил-8-[(1R,2S)-2-этилциклопентил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она (10)

Стадия 1. Синтез 6-бром-8-[(1R,2S)-2-этилциклопентил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она (C47).
Раствор P7 (80 мг, 0,22 ммоль), (3S,4R)-4-аминотетрагидро-2H-пиран-3-ола (75,8 мг, 0,647 ммоль) и N, N-диизопропилэтиламина (0,239 мл, 1,37 ммоль) в диметилсульфоксиде (1,5 мл) перемешивали при 60°C в течение 16 часов. Снова добавляли (3S,4R)-4-аминотетрагидро-2H-пиран-3-ол (75,8 мг, 0,647 ммоль) и N, N-диизопропилэтиламин (0,239 мл, 1,37 ммоль) и реакционную смесь перемешивали при 65°C в течение 3 часов, после чего смесь разбавляли водой. Полученные твердые вещества собирали фильтрованием и промывали водой, затем растворяли в этилацетате и сушили над сульфатом натрия, фильтровали, и концентрировали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (градиент: 0% - 100% этилацетата в гептане); полученное вещество растворяли в смеси воды и ацетонитрила, а затем лиофилизировали, с получением C47 в виде светло-желтого твердого вещества. Выход: 60 мг, 0,13 ммоль, 59%. LCMS m/z 452,8 (наблюдали изотопную картину брома) [M+H]+. 1H ЯМР (400 МГц, DMSO-d6) δ 8,76 (с, 1H), 7,39 (шир, с, 1H), 6,15-6,05 (м, 1H), 4,71-4,65 (м, 1H), 3,95-3,79 (м, 3H), 3,64-3,54 (м, 1H), 3,37 (ддд, J=11,7, 11,5, 2,2 Гц, 1H), 3,10 (дд, J=11,1, 9,5 Гц, 1H), 2,54 (с, 3H), 2,14-2,04 (м, 1H), 2,04-1,94 (м, 2H), 1,94-1,82 (м, 3H), 1,62-1,42 (м, 3H), 1,15-1,03 (м, 2H), 0,74 (т, J=7,4 Гц, 3H).
Стадия 2. Синтез 6-ацетил-8-[(1R,2S)-2-этилциклопентил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она (10).
Раствор C47 (60 мг, 0,13 ммоль), трибутил(1-этоксиэтенил)станнана (84,0 мг, 0,233 ммоль) и тетракис(трифенилфосфин)палладий(0) (7,8 мг, 6,8 мкмоль) в толуоле (8 мл) продували азотом и затем нагревали до 110°C в течение 20 часов. После фильтрования реакционной смеси через шприцевой фильтр, фильтрат концентрировали в вакууме, и остаток разбавляли тетрагидрофураном. Добавляли хлорид водорода (4 M раствор в 1,4-диоксане; 0,332 мл, 1,33 ммоль) и воду (0,5 мл), и реакционную смесь перемешивали в течение 1,5 часов при комнатной температуре, после чего смесь концентрировали при пониженном давлении. Очистка с помощью сверхкритической флюидной хроматографии (Колонка: Nacalai Cosmosil 3-гидроксифенил; Подвижная фаза: 88:12 диоксид углерода/метанол) давала 10. На основании анализа 1H ЯМР, предполагалось, что это вещество существует в виде смеси ротамеров. Выход: 30 мг, 72 мкмоль, 55%. LCMS m/z 415,2 [M+H]+. 1H ЯМР (700 МГц, DMSO-d6), характеристические пики : δ [8,80 (с) и 8,77 (с), общий 1Н], [7,91 (ушир.с) и 7,75 (ушир.с), общий 1Н], [6,12 (ушир.с) и 6,00 (ушир.с), общий 1Н], [5,00 (с) и 4,98 (с), общий 1Н], 4,03-3,75 (м, 3H), 3,62-3,48 (м, 1H), 3,11-2,99 (м, 1H), 2,38 (с, 3H), 2,26 (с, 3H), 2,07-1,78 (м, 5H), 1,57-1,40 (м, 2H), 1,15-0,99 (м, 2H), [0,75 (ушир.с) и 0,71 (ушир.с), общий 3H].
Пример 11
3-Ацетил-7-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-4-метил-1-[(3S)-тетрагидрофуран-3-ил]-1,6-нафтиридин-2(1H)-он (11)

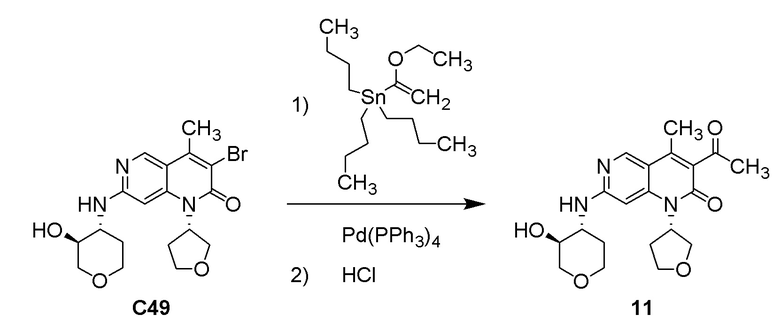
Стадия 1. Синтез 7-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-4-метил-1-[(3S)-тетрагидрофуран-3-ил]-1,6-нафтиридин-2(1H)-она (C48).
К смеси P8 (150 мг, 0,567 ммоль), (3S,4R)-4-аминотетрагидро-2H-пиран-3-ола (79,7 мг, 0,680 ммоль), карбоната цезия (554 мг, 1,70 ммоль), тетракис(трифенилфосфин)палладий(0) (65,5 мг, 56,7 мкмоль), и 4,5-бис(дифенилфосфино)-9,9-диметилксантена (Xantphos; 65,6 мг, 0,113 ммоль) добавляли толуол (6 мл). Через полученную суспензию барботировали азот, которую затем перемешивали при 110°C в течение 16 часов. После еще одного добавления 4,5-бис(дифенилфосфино)-9,9-диметилксантена (32,8 мг, 56,7 мкмоль) и тетракис(трифенилфосфин)палладия(0) (32,8 мг, 28,4 мкмоль), через реакционную смесь снова барботировали азот, и затем ее нагревали при 110°C в течение 4 часов, затем фильтровали. Фильтрат концентрировали в вакууме; анализ LCMS в этот момент показал присутствие C48: LCMS m/z 346,0 [M+H]+. После препаративной тонкослойной хроматографии на силикагеле (элюент: 10:1 дихлорметан/метанол), C48 выделяли в виде желтого масла. Выход: 80 мг, 0,23 ммоль, 41%. 1H ЯМР (400 МГц, хлороформ-d) δ 8,39 (с, 1H), 6,80 (с, 1H), 6,45-6,34 (м, 1H), 6,24 (д, J=1,3 Гц, 1H), 4,76 (ушир.д, J=5,7 Гц, 1H), 4,50-4,42 (м, 1H), 4,14 (дд, J=10,3, 4,0 Гц, 1H), 4,09 (дд, J=11,4, 4,9 Гц, 1H), 4,03-3,97 (м, 1H), 3,92 (дд, J=10,1, 10,0 Гц, 1H), 3,82-3,69 (м, 2H), 3,56 (ддд, J=9,4, 9,2, 4,8 Гц, 1H), 3,47 (ддд, J=11,8, 11,7, 2,1 Гц, 1H), 3,22 (дд, J=11,3, 9,7 Гц, 1H), 2,37 (шир, с, 3H), 2,33-2,23 (м, 2H), 2,08-1,98 (м, 1H), 1,78-1,66 (м, 1H).
Стадия 2. Синтез 3-бром-7-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-4-метил-1-[(3S)-тетрагидрофуран-3-ил]-1,6-нафтиридин-2(1H)-она (C49).
К 0°C раствору C48 (80 мг, 0,23 ммоль) в смеси ацетонитрила (5 мл) и дихлорметана (2,5 мл) добавляли раствор N-бромсукцинимида (45,3 мг, 0,255 ммоль) в ацетонитриле (5 мл). Реакционную смесь перемешивали при 0°C в течение 10 минут; LCMS-анализ реакционной смеси в этот момент показал преобразование в C49: LCMS m/z 425,9 (наблюдали изотопную картину брома) [M+H]+. После разбавления реакционной смеси дихлорметаном (50 мл), смесь промывали последовательно насыщенным водным раствором сульфита натрия (10 мл) и насыщенным водным раствором хлорида натрия (30 мл), сушили над сульфатом натрия, фильтровали, и концентрировали в вакууме. Препаративная тонкослойная хроматография на силикагеле (элюент: 15:1 дихлорметан/метанол) давала C49 в виде белого твердого вещества. Выход: 60 мг, 0,14 ммоль, 61%. 1H ЯМР (400 МГц, хлороформ-d) δ 8,51 (с, 1H), 6,81 (с, 1H), 6,45-6,31 (м, 1H), 4,86-4,76 (м, 1H), 4,52-4,41 (м, 1H), 4,21-4,04 (м, 2H), 4,04-3,89 (м, 2H), 3,86-3,70 (м, 2H), 3,62-3,53 (м, 1H), 3,53-3,43 (м, 1H), 3,27-3,18 (м, 1H), 2,60 (с, 3H), 2,35-2,24 (м, 2H), 2,09-2,00 (м, 1H), 1,78-1,64 (м, 1H).
Стадия 3. Синтез 3-ацетил-7-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-4-метил-1-[(3S)-тетрагидрофуран-3-ил]-1,6-нафтиридин-2(1H)-она (11).
К суспензии C49 (60 мг, 0,14 ммоль) в толуоле (10 мл) добавляли трибутил(1-этоксиэтенил)станнан (255 мг, 0,706 ммоль) и тетракис(трифенилфосфин)палладий(0) (16,3 мг, 14,1 мкмоль). После трехкратной дегазации реакционной смеси азотом, смесь перемешивали при 110°C в течение 16 часов, и охлаждали до комнатной температуры. Добавляли концентрированную хлористоводродную кислоту (0,333 мл) и реакционную смесь перемешивали при 10°C в течение 15 минут, после чего ее разбавляли дихлорметаном (30 мл) и промывали последовательно водой (15 мл), водным раствором гидроксида натрия (2 M; 10 мл), и насыщенным водным раствором хлорида натрия (2×15 мл). Органический слой затем сушили над сульфатом натрия, фильтровали, и концентрировали в вакууме. После препаративной тонкослойной хроматографии на силикагеле (Элюент: 10:1 дихлорметан/метанол), продукт объединяли с продуктом аналогичной реакции, проведенной с использованием C49 (23 мг, 54 мкмоль) и очищали обращенно-фазовой ВЭЖХ (Колонка: Agela Durashell C-18, 5 мкм; Подвижная фаза А: 0,05% гидроксид аммония в воде; Подвижная фаза B: ацетонитрил; Градиент: 14% - 34% B) с получением 11 в виде твердого вещества. Общий выход: 28,5 мг, 73,6 мкмоль, 38%. LCMS m/z 388,3 [M+H]+. 1H ЯМР (400 МГц, метанол-d4) δ 8,57 (с, 1H), 6,89 (с, 1H), 6,27-6,16 (м, 1H), 4,54-4,46 (м, 1H), 4,13 (дд, J=10,1, 4,2 Гц, 1H), 4,01-3,89 (м, 3H), 3,84-3,69 (м, 2H), 3,57 (ддд, J=9,5, 9,3, 4,8 Гц, 1H), 3,50 (ддд, J=11,8, 11,8, 2,2 Гц, 1H), 3,22 (дд, J=11,1, 9,7 Гц, 1H), 2,47 (с, 3H), 2,43-2,33 (м, 1H), 2,35 (с, 3H), 2,32-2,22 (м, 1H), 2,12-2,05 (м, 1H), 1,66-1,55 (м, 1H).
Примеры 12 и 13
6-Ацетил-8-[(1S,2R,5R)-бицикло[3.1.0]гексан-2-ил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он и 6-ацетил-8-[(1R,2S,5S)-бицикло[3.1.0]гексан-2-ил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он [12 (DIAST-1) и 13 (DIAST-2)]

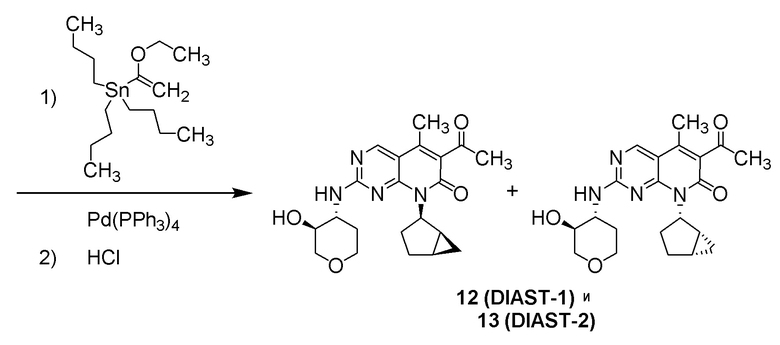
Стадия 1. Синтез 8-[(1S,2R,5R)-бицикло[3.1.0]гексан-2-ил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она и 8-[(1R,2S,5S)-бицикло[3.1.0]гексан-2-ил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она (C50).
Смесь P9 (260 мг, 0,897 ммоль), (3S,4R)-4-аминотетрагидро-2H-пиран-3-ола (158 мг, 1,35 ммоль) и N, N-диизопропилэтиламина (348 мг, 2,69 ммоль) в диметилсульфоксиде (3 мл) перемешивали при 70°C в течение трех часов, после чего реакционную смесь выливали в воду (50 мл) и экстрагировали этилацетатом (2×50 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали, и концентрировали в вакууме. После объединения остатка с продуктом аналогичной реакции, проведенной с использованием P9 (30 мг, 0,10 ммоль), смесь очищали с помощью хроматографии на силикагеле (градиент: 0% - 5% метанола в дихлорметане) с получением C50, которое состояло из указанной смеси 2 диастереомеров, в виде белого твердого вещества. Общий выход: 150 мг, 0,421 ммоль, 42%.
Стадия 2. Синтез 8-[(1S,2R,5R)-бицикло[3.1.0]гексан-2-ил]-6-бром-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она и 8-[(1R,2S,5S)-бицикло[3.1.0]гексан-2-ил]-6-бром-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она (C51).
Раствор C50 (120 мг, 0,337 ммоль), N-бромсукцинимида (65,9 мг, 0,370 ммоль) и щавелевой кислоты (3,0 мг, 33 мкмоль) в ацетонитриле (10 мл) перемешивали в запаянной пробирке в течение 5 часов при 25°C. Добавляли водный раствор бисульфита натрия (10 мл); после перемешивания полученной смеси при комнатной температуре в течение нескольких минут ее разделяли между этилацетатом (50 мл) и водой (50 мл), и водный слой экстрагировали этилацетатом (2×50 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали, и концентрировали в вакууме, получая C51 (150 мг) в виде смолы. Это вещество переносили на следующую стадию.
Стадия 3. Синтез 6-ацетил-8-[(1S,2R,5R)-бицикло[3.1.0]гексан-2-ил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она и 6-ацетил-8-[(1R,2S,5S)-бицикло[3.1.0]гексан-2-ил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она [12 (DIAST-1) и 13 (DIAST-2)].
Аргон барботировали через раствор C51 (из предыдущей стадии; ≤0,337 ммоль) и трибутил(1-этоксиэтенил)станнан (531 мг, 1,47 ммоль) в толуоле (10 мл) в течение нескольких минут, после чего добавляли тетракис(трифенилфосфин)палладий(0) (42,5 мг, 36,8 мкмоль). Реакционную смесь перемешивали в запаянной пробирке при 110°C в течение 18 часов, охлаждали до комнатной температуры, и обрабатывали хлористоводродной кислотой (1,0 M, 5 мл, 5 ммоль). Данной реакционной смеси давали возможность перемешиваться при 25°C в течение 4 часов, и затем подщелачивали до pH 8-9, добавляя водный раствор карбоната натрия. Полученную смесь распределяли между этилацетатом (30 мл) и водой (30 мл), и водный слой экстрагировали этилацетатом (2×30 мл). После того как объединенные органические слои были высушены, отфильтрованы и сконцентрированы в вакууме, остаток очищали обращенно-фазовой ВЭЖХ с получением неочищенного продукта (140 мг) в виде масла; компоненты диастереомеры этой смеси затем разделяли с помощью сверхкритической флюидной хроматографии [Колонка: Regis Technologies, (с,S)-Whelk-01, 5 мкм; Подвижная фаза: 3:2 диоксид углерода / (этанол, содержащий 0,05% диэтиламин)]. Диастереомер, элюирующийся первым, обозначали как 12, а диастереомер, элюирующийся вторым, как 13; оба соединения были получены в виде твердых веществ.
12 (DIAST-1) - Выход: 22 мг, 55 мкмоль, 16% за 2 стадии. LCMS m/z 398,9 [M+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 8,59 (с, 1H), 6,00 (шир, с, 1H), 5,52-5,23 (ушир.м, 1H), 4,08 (дд, J=11,4, 4,8 Гц, 1H), 4,05-3,90 (м, 2H), 3,71-3,59 (м, 1H), 3,55-3,44 (м, 1H), 3,28-3,17 (м, 1H), 2,52 (с, 3H), 2,50-2,37 (м, 1H), 2,30 (с, 3H), 2,15-2,04 (м, 1H), 1,97-1,83 (м, 2H), 1,81-1,68 (м, 1H, предполагаемый; частично скрытый пиком воды), 1,53-1,43 (м, 1H), 1,42-1,27 (м, 3H), 0,65-0,56 (м, 1H). Время удерживания: 4,36 минут [Колонка: Regis Technologies, (с,S)-Whelk-01, 4,6×250 мм; 5 мкм; Подвижная фаза: 3:2 диоксид углерода / (этанол, содержащий 0,05% диэтиламин); Скорость потока: 2,5 мл/минута]. Это соединение показало отрицательное (-) вращение.
13 (DIAST-2) - Выход: 18 мг, 45 мкмоль, 13% за 2 стадии. LCMS m/z 398,9 [M+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 8,59 (с, 1H), 6,01 (шир, с, 1H), 5,51-5,22 (ушир.м, 1H), 4,09 (дд, J=11,4, 5,0 Гц, 1H), 4,05-3,90 (м, 2H), 3,70-3,60 (м, 1H), 3,55-3,44 (м, 1H), 3,28-3,17 (м, 1H), 2,52 (с, 3H), 2,48-2,35 (м, 1H), 2,31 (с, 3H), 2,15-2,03 (м, 1H), 1,96-1,84 (м, 2H), 1,80-1,67 (м, 1H), 1,54-1,44 (м, 1H), 1,41-1,24 (м, 3H), 0,66-0,57 (м, 1H). Время удерживания: 4,90 минут (Аналитические условия идентичны тем, которые использовали для 12). Это соединение показало положительное (+) вращение.
Примеры 14 и 15
3-Ацетил-1-(3-гидроксициклопентил)-7-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-4-метил-1,6-нафтиридин-2(1H)-он, DIAST-1 [14 (DIAST-1)] и 3-ацетил-1-(3-гидроксициклопентил)-7-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-4-метил-1,6-нафтиридин-2(1H)-он, DIAST-2 [15 (DIAST-2)]

В колбу Эрленмейера объемом 500 мл загружали деионизированную воду (27,0 мл), в которую добавляли буферный раствор фосфата калия (pH 7,5, 1,0 M; 4,0 мл), водный раствор хлорида магния (0,165 M; 0,8 мл, 132 мкмоль) и раствор 5 в ацетонитриле (0,005 M; 0,16 мл, 0,8 мкмоль). Смесь обрабатывали микросомами печени самок мышей (4,0 мл, Corning Gentest 452702), с последующим добавлением свежеприготовленного водного раствора NADPH (дигидроникотинамида адениндинуклеотид фосфата; 0,013 M; 4,0 мл, 52 мкмоль). Колбу Эрленмейера без крышки встряхивали с помощью шейкера Thermo Scientific Precision с ходом 2,54 см при 37°C в течение 1 часа. Реакционную смесь делили на равные части (по 20 мл каждая) и выливали в две конические центрифужные пробирки Falcon на 50 мл. Растворы гасили, добавляя ацетонитрил (20 мл) в каждую пробирку Falcon. Пробирки Falcon встряхивали и центрифугировали при 3000 об/мин в течение 5 минут. Супернатант декантировали и переносили равными порциями (по 20 мл каждая) в две конические центрифужные пробирки Falcon объемом 50 мл, а растворитель выпаривали, используя EZ-2 Plus Genevac (настройка ВЭЖХ 1 час, 34°C/238 мбар до 41°C/7 мбар). Оставшиеся водные растворы объединяли (~20 мл) в коническую центрифужную пробирку Falcon объемом 50 мл и обрабатывали ацетонитрилом (0,5 мл) и чистой муравьиной кислотой (0,5 мл), затем загружали деионизированной водой до конечного объема 50 мл. Раствор делили на равные части (по 25 мл каждая), выливали в две высокоскоростные центрифужные пробирки и центрифугировали при 40000 g в течение 30 минут. Супернатант сливали в стеклянную коническую пробирку на 50 мл и раствор адсорбировали на колонке для ВЭЖХ C18 (Zorbax Polaris C18-A, 250×4,6 мм, 5 мкм), с использованием насоса для ВЭЖХ JASCO PU-1580 при скорости потока 0,8 мл/мин в течение ~60 минут. Колонку для ВЭЖХ переносили в масс-спектрометр Thermo LTQ Velos вместе с прибором Waters Acquity UHPLC, состоящим из четвертичного насоса, автодозатора и УФ/видимого детектора с матрицей фотодиодов. Градиент (0,1% муравьиной кислоты в воде (A) и ацетонитриле (B)) применяли для разделения представляющих интерес продуктов. После прохождения через матричный детектор фотодиодов элюент разделяли в соотношении приблизительно 15:1, причем большая часть направлялась в коллектор фракций, а меньшая часть в масс-спектрометр (фракции собирали каждые 20 секунд). Фракции, содержащие интересующие пики, анализировали с помощью UHPLC-UV-HRMS с использованием масс-спектрометра AB Sciex TripleTOF 5600-1 в соответствии с Waters Acquity UPLC System-1. Образцы вводили (2 мкл) в колонку C18 UHPLC (Phenomenex Kinetex C18, 2,1×50 мм, 1,7 мкм) и применяли градиент 0,1% муравьиной кислоты в воде (A) и ацетонитриле (B) при скорости потока 0,4 мл/мин при 40°C. После анализа UHPLC-UV-HRMS, фракции объединяли и растворитель удаляли с использованием EZ-2 Plus Genevac (установка 3 часовой ВЭЖХ, 34 °C/238 мбар до 41 °C/7 мбар). Высушенные образцы анализировали с помощью ЯМР-спектроскопии и количественно определяли с помощью внешней калибровки по спектру 1H ЯМР стандартного раствора 5,0 мМ бензойной кислоты в DMSO-d6 с использованием функции ERETIC2 в Topspin V3.2. Собранный диастереомер, элюирующийся первым, обозначали как 14 (DIAST-1). Выход: 18 мкг, 45 нмоль. 1H ЯМР (600 МГц, DMSO-d6) δ 8,57 (с, 1H), 7,26 (д, J=7,5 Гц, 1H), 6,44 (с, 1H), 5,58-5,44 (м, 1H), 4,49-4,41 (м, 1H), 3,90-3,76 (м, 3H), 3,45-3,33 (м, 2H), 3,08 (т, J=10,3 Гц, 1H), 2,39 (с, 3H), 2,38-2,32 (м, 1H), 2,30-2,23 (м, 4H), 2,12-1,97 (м, 3H), 1,74 (т, J=10,8 Гц, 1H), 1,70-1,63 (м, 1H), 1,50-1,38 (м, 1H). HRMS (TOF, m/z): [M+H]+ рассчитано для C21H28N3O5, 402,2029; найдено, 402,2031 (0,5 ppm). UHPLC Время удерживания 2,605 минут; колонка Phenomenex Kinetex C18 (2,1×50 мм, 1,7 мкм); температура колонки 40°C; скорость потока 0,1 мл/минута; дальность обнаружения УФ 220-400 нм; Подвижная фаза: растворитель A=муравьиная кислота (0,1%), растворитель B=ацетонитрил (100%); градиентное элюирование: 0 - 0,5 минут растворитель A (95%) и растворитель B (5%), 0,5 - 6,5 минут растворитель A (50%) и растворитель B (50%), 6,5 - 7,9 минут растворитель A (20%) и растворитель B (80%), 7,9 - 8,0 минут растворитель A (5%) и растворитель B (95%), 8,0 - 9,1 минут растворитель A (95%) и растворитель B (5%), 9,1 - 10,0 минут растворитель A (100%) и растворитель B (0%); общее время работы 10,0 минут.
Собранный диастереомер, элюирующийся вторым, обозначали как 15. (DIAST-2). Выход: 3 мкг, 7,5 нмоль. 1H ЯМР (600 МГц, DMSO-d6) δ 8,58 (с, 1H), 7,00 (д, J=7,8 Гц, 1H), 6,76 (с, 1H), 5,57-5,35 (м, 1H), 4,27-4,20 (м, 1H), 3,85-3,77 (м, 3H), 3,47-3,35 (м, 2H), 3,08 (т, J=10,4 Гц, 1H), 2,40 (с, 3H), 2,27 (с, 3H), 2,18-2,08 (м, 2H), 2,02-1,96 (м, 1H), 1,96-1,90 (м, 1H), 1,82-1,74 (м, 2H), 1,51-1,38 (м, 2H). HRMS (TOF, m/z): [M+H]+ рассчитано для C21H28N3O5, 402,2029; найдено, 402,2028 (-0,2 ppm). UHPLC Время удерживания 2,796 минут; колонка Phenomenex Kinetex C18, условия идентичны тем, которые использовались для 14 (DIAST-1), выше.
Относительная и абсолютная стереохимия 14 и 15 вокруг циклопентана не определялась; эти соединения представляют собой 2 из 4 возможных диастереомеров. 4 диастереомера, относящиеся к 14 и 15, могут быть индивидуально синтезированы специалистом в данной области из коммерчески доступных исходных материалов (1S,3R)-3-аминоциклопентанол (A1), (1R,3R)-3-аминоциклопентанол (A2), (1S,3S)-3-аминоциклопентанол (A3) и (1R,3S)-3-аминоциклопентанол (A4). Используя A1 в качестве примера, взаимодействие с 1-(4,6-дихлорпиридин-3-ил)этаноном, с использованием способа, описанного в Получении P4, может сопровождаться защитой гидроксигруппы с использованием трет-бутил(диметил)силилхлорида и 1H-имидазола с получением 1-(4-{[(1R,3S)-3-{[трет-бутил(диметил)силил]окси}циклопентил]амино}-6-хлорпиридин-3-ил)этанона. Взаимодействие с 2,2,6-триметил-4H-1,3-диоксин-4-оном, снова с использованием способа, описанного в Получении Р4, дает 3-ацетил-1-[(1R,3S)-3-{[трет-бутил(диметил)силил]окси}циклопентил]-7-хлор-4-метил-1,6-нафтиридин-2(1H)-он (B1). Взаимодействие с B1 с (3S,4R)-4-аминотетрагидро-2H-пиран-3-олом, с использованием способа Примера 5, с последующим удалением силильной группы кислотой, такой как уксусная кислота или трифторуксусная кислота, или, альтернативно, источником фторид-иона, таким как фторид тетрабутиламмония, затем даст 3-ацетил-1-[(1R,3S)-3-гидроксициклопентил]-7-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-4-метил-1,6-нафтиридин-2(1H)-он (D1), один из диастереомеров, представленных структурами 14 и 15.
Аналогичным образом исходные вещества A2, A3 и A4 могут быть соответственно превращены в диастереомеры 3-ацетил-1-[(1R,3R)-3-гидроксициклопентил]-7-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-4-метил-1,6-нафтиридин-2(1H)-он (D2), 3-ацетил-1-[(1S,3S)-3-гидроксициклопентил]-7-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-4-метил-1,6-нафтиридин-2(1H)-он (D3), и 3-ацетил-1-[(1S,3R)-3-гидроксициклопентил]-7-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-4-метил-1,6-нафтиридин-2(1H)-он (D4).


Примеры 16 и 17
6-Ацетил-8-[(1R,2S)-2-фтор-2-метилциклопентил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он и 6-ацетил-8-[(1S,2R)-2-фтор-2-метилциклопентил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он [16 (DIAST-1) и 17 (DIAST-2)]


Стадия 1. Синтез rac-6-(1-этоксиэтенил)-8-[(1R,2S)-2-фтор-2-метилциклопентил]-5-метил-2-(метилтио)пиридо[2,3-d]пиримидин-7(8H)-она (C52).
Смесь P10 (108 мг, 0,280 ммоль), трибутил(1-этоксиэтенил)станнана (151 мг, 0,418 ммоль), и тетракис(трифенилфосфин)палладий(0) (19,4 мг, 16,8 мкмоль) в толуоле (3,0 мл) нагревали при 110°C в течение 2 часов. LCMS-анализ реакционной смеси в этот момент показал присутствие продукта C52: LCMS m/z 377,9 [M+H]+. Реакционную смесь фильтровали через диатомовую землю и фильтрат концентрировали в вакууме. Очистка хроматографией на силикагеле (Градиент: 20% - 60% этилацетата в гептане) давала C52 в виде бледно-желтой пены. Выход: 91 мг, 0,24 ммоль, 86%. 1H ЯМР (400 МГц, хлороформ-d) δ 8,74 (с, 1H), 5,94-5,72 (м, 1H), 4,54 (д, J=2,5 Гц, 1H), 4,20 (д, J=2,5 Гц, 1H), 3,94 (кв, J=7,0 Гц, 2H), 2,62 (с, 3H), 2,51-2,33 (м, 1H), 2,44 (с, 3H), 2,31-2,19 (м, 1H), 1,98-1,81 (м, 3H), 1,71-1,57 (м, 1H), 1,48 (д, JHF=21,8 Гц, 3H), 1,36 (т, J=7,0 Гц, 3H).
Стадия 2. Синтез rac-6-ацетил-8-[(1R,2S)-2-фтор-2-метилциклопентил]-5-метил-2-(метилтио)пиридо[2,3-d]пиримидин-7(8H)-она (C53).
Добавляли хлорид водорода (4,0 M раствор в 1,4-диоксане; 0,181 мл, 0,724 ммоль) к раствору C52 (91 мг, 0,24 ммоль) в дихлорметане (2 мл), и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. LCMS-анализ реакционной смеси в этот момент показал присутствие C53: LCMS m/z 350,2 [M+H]+. Концентрирование в вакууме давало C53 в виде твердого вещества, которое использовали непосредственно на следующей стадии.
Стадия 3. Синтез rac-6-ацетил-8-[(1R,2S)-2-фтор-2-метилциклопентил]-5-метил-2-(метилсульфинил)пиридо[2,3-d]пиримидин-7(8H)-она (C54).
Раствор пероксимоносульфата калия (Oxone®; 326 мг, 0,530 ммоль) в воде (1 мл) добавляли к раствору C53 (из предыдущей стадии; 88 мг, ≤0,24 ммоль) в тетрагидрофуране (4 мл), и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. LCMS-анализ реакционной смеси в этот момент показал присутствие C54: LCMS m/z 345,9 [(M - HF)+H]+. Добавляли воду и полученную смесь экстрагировали дважды этилацетатом. Объединенные органические слои сушили над сульфатом натрия, фильтровали, и концентрировали в вакууме с получением C54 в виде бледно-желтого твердого вещества. Выход: 89,0 мг, 0,24 ммоль, количественный за 2 стадии.
Стадия 4. Синтез 6-ацетил-8-[(1R,2S)-2-фтор-2-метилциклопентил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она и 6-ацетил-8-[(1S,2R)-2-фтор-2-метилциклопентил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она [16 (DIAST-1) и 17 (DIAST-2)].
Смесь C54 (43 мг, 0,12 ммоль), (3S,4R)-4-аминотетрагидро-2H-пиран-3-ола (24,1 мг, 0,206 ммоль) и N, N-диизопропилэтиламина (86 мкл, 0,49 ммоль) в диметилсульфоксиде (2 мл) перемешивали при комнатной температуре в течение 3 часов. Разделение компонентов диастереомеров проводили с использованием сверхкритической флюидной хроматографии [Колонка: Chiral Technologies Chiralpak IC, 5 мкм; Подвижная фаза: 3:2 диоксид углерода / (2-пропанол, содержащий 10 мМ гидроксида аммония)]. Диастереомер, элюирующийся первым, обозначен как 16, и диастереомер, элюирующийся вторым, как 17; оба были получены в виде твердых веществ.
16 (DIAST-1) - Выход: 17 мг, 41 мкмоль, 34%. LCMS m/z 418,9 [M+H]+. 1H ЯМР (400 МГц, 80°C, DMSO-d6), характеристические пики: δ 8,75 (с, 1H), 7,50-7,37 (м, 1H), 5,87-5,73 (м, 1H), 4,72-4,59 (м, 1H), 3,99-3,80 (м, 3H), 3,63-3,54 (м, 1H), 3,35 (ддд, J=11,7, 11,6, 2,3 Гц, 1H), 3,10 (дд, J=11,1, 9,5 Гц, 1H), 2,39 (с, 3H), 2,29 (с, 3H), 2,18-2,06 (м, 1H), 2,00-1,84 (м, 3H), 1,69-1,54 (м, 2H), 1,44 (д, JHF=21,9 Гц, 3H). Время удерживания: 1,22 минут [Колонка: Chiral Technologies Chiralpak IC-3, 4,6×100 мм, 3 мкм; Подвижная фаза: 3:2 диоксид углерода / (2-пропанол, содержащий 10 мМ гидроксида аммония); Скорость потока: 4 мл/минута; Обратное давление: 120 бар]. Это соединение показало отрицательное (-) вращение.
17 (DIAST-2) - Выход: 14 мг, 33 мкмоль, 28%. LCMS m/z 419,0 [M+H]+. 1H ЯМР (400 МГц, 80°C, DMSO-d6), характеристические пики: δ 8,75 (с, 1H), 7,50-7,38 (м, 1H), 5,87-5,73 (м, 1H), 4,66 (шир, с, 1H), 4,00-3,79 (м, 3H), 3,64-3,54 (м, 1H), 3,36 (ддд, J=11,7, 11,5, 2,4 Гц, 1H), 3,10 (дд, J=11,1, 9,5 Гц, 1H), 2,39 (с, 3H), 2,29 (с, 3H), 2,19-2,06 (м, 1H), 2,01-1,81 (м, 3H), 1,70-1,51 (м, 2H), 1,44 (д, JHF=21,9 Гц, 3H). Время удерживания: 1,51 минут (Аналитические условия идентичны тем, которые использовали для 16). Это соединение показало положительное (+) вращение.
Пример 18
6-Ацетил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метил-8-[(3S)-пирролидин-3-ил]пиридо[2,3-d]пиримидин-7(8H)-он (18)

Стадия 1. Синтез трет-бутил (3S)-3-[6-(1-этоксиэтенил)-5-метил-2-(метилтио)-7-оксопиридо[2,3-d]пиримидин-8(7H)-ил]пирролидин-1-карбоксилата (C55).
Трибутил(1-этоксиэтенил)станнан (10,1 г, 28,0 ммоль) и тетракис(трифенилфосфин)палладий(0) (1,34 г, 1,16 ммоль) добавляли к раствору P11 (5,30 г, 11,6 ммоль) в смеси толуола (80 мл) и 1,4-диоксана (40 мл). Реакционную смесь перемешивали при 110°C в течение 5 часов, после чего смесь концентрировали в вакууме и очищали с помощью хроматографии на силикагеле (Градиент: 10% - 60% этилацетата в петролейном эфире), с получением C55 в виде желтого масла. Выход: 4,48 г, 10,0 ммоль, 86%. LCMS m/z 447,2 [M+H]+.
Стадия 2. Синтез трет-бутил (3S)-3-[6-ацетил-5-метил-2-(метилтио)-7-оксопиридо[2,3-d]пиримидин-8(7H)-ил]пирролидин-1-карбоксилата (C56).
Хлористоводродную кислоту (1 M; 40,1 мл, 40,1 ммоль) добавляли к 20°C раствору C55 (4,48 г, 10,0 ммоль) в тетрагидрофуране (150 мл), и реакционную смесь перемешивали в течение 2 часов. После подщелачивания путем добавления насыщенного водного раствора бикарбоната натрия, реакционную смесь экстрагировали этилацетатом (2×100 мл); объединенные органические слои сушили над сульфатом натрия, фильтровали, и концентрировали в вакууме. Хроматография на силикагеле (градиент: 10% - 60% этилацетата в петролейном эфире) давала C56 в виде белой смолы. Выход: 2,64 г, 6,31 ммоль, 63%. LCMS m/z 441,1 [M+Na+]. 1H ЯМР (400 МГц, хлороформ-d) δ 8,81 (с, 1H), 6,27-6,12 (м, 1H), 3,97 (дд, J=10,2, 8,3 Гц, 1H), 3,88-3,74 (м, 1H), 3,74-3,57 (м, 1H), 3,52-3,40 (м, 1H), 2,94-2,75 (м, 1H), 2,58 (с, 3H), 2,53 (с, 3H), 2,38 (с, 3H), 2,20-2,05 (м, 1H), 1,52-1,41 (м, 9H).
Стадия 3. Синтез трет-бутил (3S)-3-[6-ацетил-5-метил-2-(метилсульфонил)-7-оксопиридо[2,3-d]пиримидин-8(7H)-ил]пирролидин-1-карбоксилата (C57).
К раствору C56 (1,00 г, 2,39 ммоль) в смеси тетрагидрофурана (40 мл) и воды (20 мл) добавляли пероксимоносульфат калия (Oxone®; 2,64 г, 4,29 ммоль). Реакционную смесь перемешивали при комнатной температуре (17°C) в течение 3 часов, после чего смесь разбавляли водой (40 мл) и экстрагировали этилацетатом (2×40 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия (50 мл), сушили над сульфатом натрия, фильтровали, и концентрировали в вакууме с получением C57 в виде смолы желтого цвета, которую использовали без дополнительной очистки. Выход: 1,08 г, количественный. LCMS m/z 473,1 [M+Na+].
Стадия 4. Синтез трет-бутил (3S)-3-[6-ацетил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метил-7-оксопиридо[2,3-d]пиримидин-8(7H)-ил]пирролидин-1-карбоксилата (C58).
Суспензию (3S,4R)-4-аминотетрагидро-2H-пиран-3-ола (226 мг, 1,93 ммоль), C57 (700 мг, 1,55 ммоль) и карбоната натрия (341 мг, 3,22 ммоль) в тетрагидрофуране (15 мл) перемешивали при комнатной температуре (20°C) в течение приблизительно 20 часов. Затем реакционную смесь распределяли между водой (35 мл) и этилацетатом (35 мл), и органический слой промывали насыщенным водным раствором хлорида натрия (20 мл), сушили над сульфатом натрия, фильтровали, и концентрировали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (градиент: 0% - 10% метанола в дихлорметане) с получением C58 в виде смолы желтого цвета. Выход: 700 мг, 1,44 ммоль, 93%. LCMS m/z 510,2 [M+Na+].
Стадия 5. Синтез 6-ацетил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метил-8-[(3S)-пирролидин-3-ил]пиридо[2,3-d]пиримидин-7(8H)-она (18).
Раствор C58 (700 мг, 1,44 ммоль) в метаноле (10 мл) обрабатывали хлоридом водорода (4 M раствор в метаноле; 8 мл) и перемешивали при комнатной температуре (20°C) в течение приблизительно 20 часов. Реакционную смесь затем концентрировали при пониженном давлении и очищали с помощью нормально-фазовой ВЭЖХ (Колонка: Agela Durashell NH2, 5 мкм; Подвижная фаза А: 4:1 петролейный эфир/дихлорметан; Подвижная фаза B: метанол; градиент: 5% - 95% B) с получением 18 в виде белого твердого вещества. Выход: 350 мг, 0,903 ммоль, 63%. LCMS m/z 388,2 [M+H]+. 1H ЯМР (400 МГц, хлороформ-d), характеристические пики : δ 8,64 (с, 1H), 6,18-5,97 (м, 1H), 4,15-3,85 (м, 3H), 3,77-3,16 (м, 5H), 3,15 (дд, J=11,0, 10,5 Гц, 1H), 2,92 (ддд, J=10,8, 10,6, 6,6 Гц, 1H), 2,53 (с, 3H), 2,33 (с, 3H), 1,81-1,61 (м, 1H).
Пример 19
6-Ацетил-8-(3,3-диметилциклобутил)-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он (19)




Стадия 1. Синтез ди-μ-хлор{тетракис[3,5-дифтор-2-(5-фторпиридин-2-ил-κN)фенил-κC1]}дииридия (C59).
Смесь 2-(2,4-дифторфенил)-5-фторпиридина (2,46 г, 11,8 ммоль) и хлорида иридия(III) (1,56 г, 5,23 ммоль) в 2-этоксиэтаноле (24 мл) и воде (10 мл) дегазировали под вакуумом и затем в реакционный сосуд загружали азот. Этот цикл вакуумирования повторяли дважды, после чего реакционную смесь нагревали до 120°C в течение 18 часов. Смесь затем охлаждали до комнатной температуры (28°C) и твердые вещества собирали фильтрацией; фильтрпрессную лепешку промывали водой (150 мл) с получением C59 в виде желтого твердого вещества. Выход: 3,11 г, 2,41 ммоль, 92%.
Стадия 2. Синтез (2,2'-бипиридин-κ2N1,N1'){бис[3,5-дифтор-2-(5-фторпиридин-2-ил)фенил]}иридия гексафторфосфата (C60).
Смесь С59 (3,11 г, 2,41 ммоль) и 2,2'-бипиридина (901 мг, 5,77 ммоль) в этан-1,2-диоле (120 мл) дегазировали под вакуумом и продували азотом; этот цикл вакуумирования повторяли и затем реакционную смесь перемешивали в течение 19 часов при 145°C. После охлаждения до комнатной температуры (20°C), реакционную смесь выливали в деионизированную воду (900 мл), и полученную смесь экстрагировали н-гексаном (6×300 мл). Водный слой концентрировали в вакууме для удаления остаточного н-гексана. К этому раствору добавляли водный раствор гексафторфосфата натрия (0,1 г/мл в деионизированной воде; 460 мл); полученное твердое вещество собирали фильтрованием и промывали водой (50 мл) затем н-гексаном (50 мл). Это твердое вещество (3,7 г) растворяли в ацетоне (30 мл) и кипятили с обратным холодильником в течение 20 минут, после чего добавляли н-гептан (20 мл). Полученную смесь фильтровали; фильтрпрессную лепешку промывали н-гексаном (50 мл) и затем очищали хроматографией на силикагеле (градиент: 0% - 10% ацетона в дихлорметане). Это вещество (1,5 г) перекристаллизовывали из ацетона с получением C60 в виде ярко-желтого твердого вещества. Выход: 1,0 г, 1,1 ммоль, 46%. LCMS m/z 765,3 [M+]. 1H ЯМР (400 МГц, DMSO-d6) δ 8,88 (д, J=8,1 Гц, 2H), 8,37-8,27 (м, 4H), 8,11-8,02 (м, 2H), 7,88 (ушир.д, J=5,8 Гц, 2H), 7,74-7,67 (м, 2H), 7,65 (ушир. дд, J=2,7, 2,6 Гц, 2H), 7,00 (ддд, J=12,2, 9,4, 2,4 Гц, 2H), 5,73 (дд, J=8,4, 2,4 Гц, 2H).
Стадия 3. Синтез бис{[(3,3-диметилциклобутил)карбонил]окси}(2,4,6-триметилфенил)-λ3-иодана (C61).
3,3-Диметилциклобутанкарбоновую кислоту (38,4 мг, 0,300 ммоль) обрабатывали раствором бис(ацетилокси)(2,4,6-триметилфенил)-λ3-иодана (56 мг, 0,15 ммоль) в дихлорметане (1 мл). Добавляли толуол (2 мл), реакционный сосуд закрывали крышкой, и реакционную смесь нагревали до 55°C в течение ночи, после чего смесь концентрировали с использованием испарителя Genevac. Остаток смешивали с толуолом (1 мл) и перемешивали в течение 30 минут при 55°C. После повторного выпаривания смеси остаток (C61) хранили в холодильнике для использования на следующей стадии. 1H ЯМР (400 МГц, хлороформ-d) δ 7,06 (с, 2H), 2,93 (пентет, J=8,8 Гц, 2H), 2,69 (с, 6H), 2,34 (с, 3H), 1,99-1,80 (м, 8H), 1,07 (с, 6H), 1,01 (с, 6H).
Стадия 4. Синтез 6-ацетил-2-(децилтио)-8-(3,3-диметилциклобутил)-5-метилпиридо[2,3-d]пиримидин-7(8H)-она (C62).
Реакционный сосуд, содержащий смесь С60 (0,7 мг, 0,8 мкмоль) и меди(I) тиофен-2-карбоксилат (3 мг, 16 мкмоль), продували азотом. Добавляли раствор 4,7-диметокси-1,10-фенантролина (5,5 мг, 22 мкмоль) и 2-трет-бутил-1,1,3,3-тетраметилгуанидина (26 мг, 0,15 ммоль) в 1,4-диоксане (0,25 мл), и сосуд встряхивали и снова продували азотом. Добавляли раствор P13 (28 мг, 75 мкмоль) в 1,4-диоксане (0,5 мл), затем раствор C61 (из предыдущей стадии; ≤0,15 ммоль) в 1,4-диоксане (1 мл); контейнер C61 промывали дополнительным 1,4-диоксаном (0,5 мл), который добавляли к реакционной смеси. После запечатывания реакционного сосуда, его облучали светодиодной лампой Kessil при 440 нм в течение 3 часов. Затем реакционную смесь распределяли между 10% водным раствором лимонной кислоты и этилацетатом (2 мл) при встряхивании. Органический слой отделяли, и экстракцию повторяли; объединенные органические слои элюировали через картридж для твердофазной экстракции из силикагеля SiliCycle этилацетатом. Растворитель удаляли в вакууме и очистку проводили с помощью обращенно-фазовой ВЭЖХ (колонка: Waters XBridge C18, 5 мкм; Подвижная фаза А: вода, содержащая 0,03% гидроксида аммония; Подвижная фаза B: ацетонитрил, содержащий 0,03% гидроксида аммония; Градиент: 80% - 95% B) с получением C62. Выход: 6,1 мг, 13 мкмоль, 17%. LCMS m/z 458,6 [M+H]+. 1H ЯМР (400 МГц, метанол-d4) δ 8,92 (с, 1H), 5,93 (пентет, J=9,2 Гц, 1H), 3,27-3,20 (м, 2H), 3,01-2,92 (м, 2H), 2,49 (с, 3H), 2,38 (с, 3H), 2,16-2,09 (м, 2H), 1,86-1,75 (м, 2H), 1,57-1,47 (м, 2H), 1,44-1,21 (м, 18H), 0,88 (т, J=7 Гц, 3H).
Стадия 5. Синтез 6-ацетил-8-(3,3-диметилциклобутил)-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она (19).
Смесь С62 (6,1 мг, 13 мкмоль) и пероксимоносульфат калия (Oxone®; 15 мг, 24 мкмоль) в смеси тетрагидрофурана и воды (4:1, 0,75 мл) перемешивали при комнатной температуре в течение 66 часов. Затем реакционную смесь распределяли между водой (0,75 мл) и этилацетатом (2 мл), и органический слой элюировали через картридж для твердофазной экстракции, загруженный сульфатом натрия; этот процесс экстракции повторяли и растворитель удаляли в вакууме с получением промежуточного соединения C63 [6-ацетил-2-(децилсульфонил)-8-(3,3-диметилциклобутил)-5-метилпиридо[2,3-d]пиримидин-7(8H)-она]. Это вещество растворяли в метаноле (1 мл), и половину его концентрировали при пониженном давлении и затем обработали раствором (3S,4R)-4-аминотетрагидро-2H-пиран-3-ола (1,2 мг, 10 мкмоль) в тетрагидрофуране (0,25 мл). Реакционный сосуд встряхивали при 65°C в течение ночи, после чего реакционную смесь распределяли между этилацетатом (1,2 мл) и водой (0,60 мл) при встряхивании. Органический слой элюировали через картридж для твердофазной экстракции, загруженный сульфатом натрия; процесс экстракции повторяли, используя этилацетат (0,60 мл), и растворитель удаляли из объединенных органических слоев в вакууме. Очистку проводили с помощью ВЭЖХ с обращенной фазой (Колонка: Waters Sunfire C18, 5 мкм; Подвижная фаза А: 0,05% трифторуксусной кислоты в воде; Подвижная фаза B: 0,05% трифторуксусной кислоты в ацетонитриле; Градиент: 5% - 60% B) с получением 19. Выход: 1,6 мг, 4,0 мкмоль 62%. LCMS m/z 401,4 [M+H]+. Время удерживания: 0,922 минут (Колонка: Phenomenex Kinetex C18, 2,1×30 мм, 2,6 мкм; Подвижная фаза А: 0,05% трифторуксусной кислоты в воде; Подвижная фаза B: 0,05% трифторуксусной кислоты в ацетонитриле; Градиент: 5% - 95% B в течение 2,0 минут, затем удерживание 95% B в течение 0,7 минут; Скорость потока: 1,0 мл/минут).









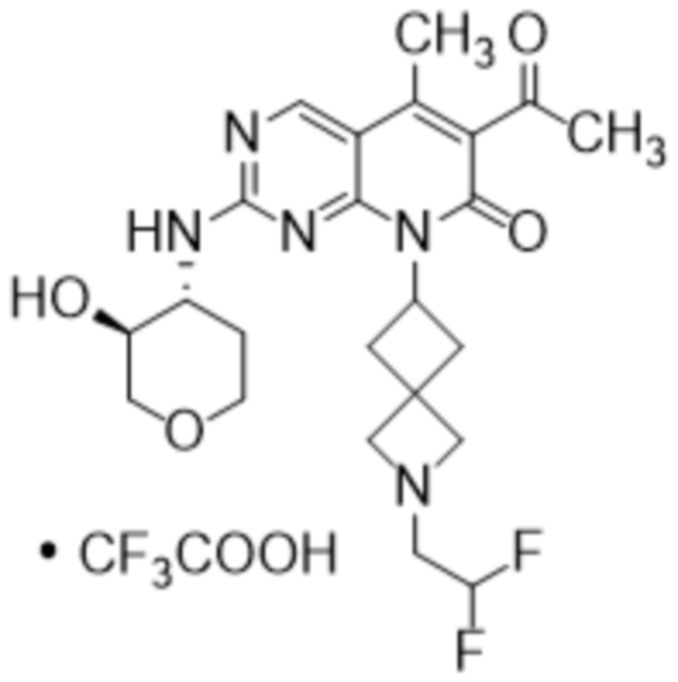


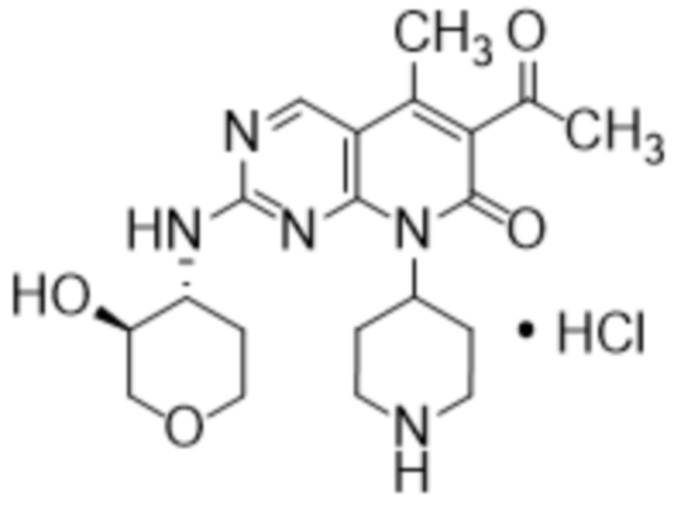
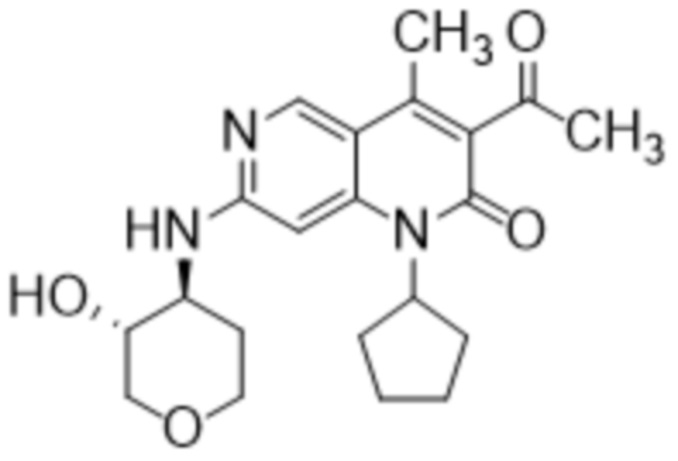

1. Необходимый 6-бром-2-хлор-8-(цис-4-гидроксициклогексил)-5-метилпиридо[2,3-d]пиримидин-7(8H)-он синтезировали с использованием способа, описанного в Получении Р1, с использованием цис-4-аминоциклогексанола вместо циклобутанамина. Гидроксигруппу промежуточного цис-4-[(5-бром-2-хлорпиримидин-4-ил)амино]циклогексанола была защищена в виде его трет-бутилдиметилсилилового эфира; эта защитная группа отделилась на стадии бромирования (в данном случае, проводимого с бромом в дихлорметане)).
2. Взаимодействие 1-[4-хлор-2-(метилтио)пиримидин-5-ил]этанона с транс-3-аминоциклобутанолом и триэтиламином, с последующей гидроксизащитой трет-бутил(дифенил)силилхлоридом и 1H-имидазолом дает 1-{4-[(транс-3-{[трет-бутил(дифенил)силил]окси}циклобутил)амино]-2-(метилтио)пиримидин-5-ил}этанон. Это вещество превращали в необходимый 6-бром-8-(транс-3-{[трет-бутил(дифенил)силил]окси}циклобутил)-5-метил-2-(метилтио)пиридо[2,3-d]пиримидин-7(8H)-он, с использованием химии, описанной для синтеза P10 из C28 в Получении Р10. Силильную защитную группу удаляли из промежуточного соединения 8-(транс-3-{[трет-бутил(дифенил)силил]окси}циклобутил)-6-(1-этоксиэтенил)-5-метил-2-(метилтио)пиридо[2,3-d]пиримидин-7(8H)-она во время его кислотного превращения в 6-ацетил-8-(транс-3-гидроксициклобутил)-5-метил-2-(метилтио)пиридо[2,3-d]пиримидин-7(8H)-он.
3. Взаимодействие бромида метимагния с 2-(дибензиламино)циклопентаноном давало rac-(1S,2R)-2-(дибензиламино)-1-метилциклопентанол, который гидрировали над гидроксидом палладия с получением rac-(1S,2R)-2-амино-1-метилциклопентанола. Защита аминогруппы бензилхлорформиатом давала rac-бензил [(1R,2S)-2-гидрокси-2-метилциклопентил]карбамат, который превращали в rac-6-бром-8-[(1R,2R)-2-фтор-2-метилциклопентил]-5-метил-2-(метилтио)пиридо[2,3-d]пиримидин-7(8H)-он, с использованием химии, описанной в Получении Р10. Подтверждение, что соединения в этой серии являются диастереомерными по сравнению с соединениями из Получения P10, получали путем сравнения физико-химических данных для промежуточного соединения rac-8-[(1R,2R)-2-фтор-2-метилциклопентил]-5-метил-2-(метилтио)пиридо[2,3-d]пиримидин-7(8H)-она {1H ЯМР (400 МГц, хлороформ-d) δ 8,72 (с, 1H), 6,45-6,18 (м, 2H), 2,65 (с, 3H), 2,43 (д, J=1,3 Гц, 3H), [2,60-2,28 (м) и 2,14-1,83 (м), общий 6H], 1,28 (д, JHF=22,9 Гц, 3H). LCMS m/z 308,1 [M+H]+} с таковым rac-(1R,2S) изомера C29 в Получении Р10.
4. Разделение диастереомерных соединений Примеров 22 и 23 осуществляли с использованием сверхкритической флюидной хроматографии (Колонка: Phenomenex Lux Cellulose-4, 3 мкм; Подвижная фаза: 18% метанола в диоксиде углерода; Обратное давление: 100 бар). Элюированный первым диастереомер обозначали как Пример 22, и элюированный вторым диастереомер как Пример 23. Время удерживания для Примера 22: 2,56 минут (Колонка: Chiral Technologies Chiralpak IC, 4,6×100 мм, 5 мкм; Подвижная фаза А: диоксид углерода; Подвижная фаза B: метанол; Градиент: 12% - 70% B в течение 10 минут; Скорость потока: 4,0 мл/минута; Обратное давление: 100 бар). Время удерживания для примера 23: 1,83 минуты (Аналитические условия идентичны тем, которые использовали для примера 22).
5. Взаимодействие 1-[4-хлор-2-(метилтио)пиримидин-5-ил]этанона с трет-бутил (транс-4-аминоциклогексил)карбаматом и N, N-диизопропилэтиламином давало трет-бутил (транс-4-{[5-ацетил-2-(метилтио)пиримидин-4-ил]амино}циклогексил)карбамат, который превращался в трет-бутил {транс-4-[6-бром-5-метил-2-(метилтио)-7-оксопиридо[2,3-d]пиримидин-8(7H)-ил]циклогексил}карбамат в соответствии со способом, описанным в Получении Р10 для синтеза P10 из C28 (за исключением того, что вместо брома использовали N-бромсукцинимид и каталитическую щавелевую кислоту). На последней стадии удаляли трет-бутоксикарбонильную группу с использованием хлорида водорода в метаноле, с получением примера 24.
6. Взаимодействие хлористоводородной соли примера 24 с формальдегидом и триацетоксиборгидридом натрия давало пример 25.
7. Взаимодействие примера 18 с формальдегидом, N, N-диизопропилэтиламином и триацетоксиборгидридом натрия давало пример 26.
8. Необходимый трет-бутил (3R)-3-[6-бром-5-метил-2-(метилтио)-7-оксопиридо[2,3-d]пиримидин-8(7H)-ил]пирролидин-1-карбоксилат синтезировали с использованием способа, описанного в Получении P10, но с использованием трет-бутил (3R)-3-аминопирролидин-1-карбоксилата вместо C27. Реакцию бромирования осуществляли с использованием N-бромсукцинимида и каталитической щавелевой кислоты, а не брома.
9. Алкилирование примера 3 1-бром-2-метоксиэтаном и N, N-диизопропилэтиламином давало пример 28.
10. Условия аналитической ВЭЖХ. Колонка: Waters Atlantis dC18, 4,6×50 мм, 5 мкм; Подвижная фаза А: 0,05% трифторуксусной кислоты в воде (об./об.); Подвижная фаза B: 0,05% трифторуксусной кислоты в ацетонитриле (об./об.); Градиент: 5,0% - 95% B, линейный в течение 4,0 минут; Скорость потока: 2 мл/минута.
11. Алкилирование примера 3 с 2,2-дифторэтил трифторметансульфонатом и N, N-диизопропилэтиламином давало пример 29.
12. Образец транс-4-аминотетрагидро-2H-пиран-3-ола, в основном состоящий из (3S,4R)-4-аминотетрагидро-2H-пиран-3-ола, но содержащий небольшое количество (3R,4S)-4-аминотетрагидро-2H-пиран-3-ола, взаимодействовал с P12 и N, N-диизопропилэтиламином. Разделение полученных энантиомеров осуществляли с помощью сверхкритической флюидной хроматографии (Колонка: Chiral Technologies Chiralpak AS-H, 5 мкм; Подвижная фаза: 4:1 диоксид углерода/метанол). Энантиомер, элюирующийся первым, который демонстрировал положительное (+) вращение, был второстепенным компонентом и был обозначен как Пример 30. Энантиомер, элюирующийся вторым, был 2, который демонстрировал отрицательное (-) вращение. Время удерживания для Примера 30: 0,86 минут (Колонка: Chiral Technologies Chiralpak AS-3, 4,6×100 мм, 3 мкм; Подвижная фаза: 4:1 диоксид углерода/метанол; Скорость потока: 4 мл/минута; Обратное давление: 120 бар). Время удерживания для 2: 1,03 минут (Аналитические условия идентичны тем, которые использовали для Примера 30).
13. В этом случае промежуточное соединение 6-бром-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метил-8-[(3S)-тетрагидрофуран-3-ил]пиридо[2,3-d]пиримидин-7(8H)-он превращали в пример 31 взаимодействием с 1-(этенилокси)бутаном и N, N-диизопропилэтиламином в присутствии дихлор[бис(2-(дифенилфосфино)фенил)эфир]палладия(II) [см. M. T. Maloney et al., Organic Process Research & Development 2016, 20, 1203-1216], с последующим расщеплением енольного эфира хлористоводродной кислотой.
14. Синтез необходимого трет-бутил 4-[6-бром-5-метил-2-(метилтио)-7-оксопиридо[2,3-d]пиримидин-8(7H)-ил]пиперидин-1-карбоксилата осуществляли с использованием способа, описанного Получении Р2, с использованием трет-бутил 4-аминопиперидин-1-карбоксилата вместо трет-бутил 6-амино-2-азаспиро[3.3]гептан-2-карбоксилата.
15. Взаимодействие P4 с (3R,4S)-4-аминотетрагидро-2H-пиран-3-олом, в присутствии [(2-дициклогексилфосфино-3,6-диметокси-2',4',6'-триизопропил-1,1'-бифенил)-2-(2'-амино-1,1'-бифенил)]палладия(II) метансульфонат метансульфоната (BrettPhos Pd G3) и карбонат цезия, при 100°C в толуоле, давало Пример 33.
16. Условия аналитической ВЭЖХ. Колонка: Phenomenex Kinetex C18, 2,1×30 мм, 2,6 мкм; Подвижная фаза А: 0,05% трифторуксусной кислоты в воде; Подвижная фаза B: 0,05% трифторуксусной кислоты в ацетонитриле; Градиент: 5% - 50% B в течение 2,0 минут, затем удерживание 95% B в течение 0,7 минут; Скорость потока: 1,0 мл/минута.
Анализ очищенных ферментов in vitro для определения активности в отношении CDK
Анализ CDK4/циклин D1 и CDK6/циклин D3 CHEF
Усиленная образованием хелатов флуоресценция (CHEF) отслеживает состояние фосфорилирования в режиме реального времени, где уровень флуоресценции прямо пропорционален количеству фосфорилированного субстрата. CHEF использует синтетическую α-аминокислоту с боковой цепью, несущей производное 8-гидроксихинолина (сульфонамидо-оксин, Sox), которое после координации с Mg(II) передает информацию о состоянии фосфорилирования проксимального остатка серина, треонина или тирозина в субстратах киназы на основе пептидов. Фосфорилирование определенного пептида (Catalog # AQT0258) от AssayQuant Technologies приводит к увеличению флуоресценции при длинах волн возбуждения и испускания 360 нм Ex/485 нм Em.
Примеры вместе с DMSO (отрицательный) и палбоциклиб (положительный) контролями добавляли в 384-луночные планшеты в 100-кратной их конечнтеой концентрации с последующим добавлением 10 нМ CDK4/циклина D1 (LJIC-2007F1) или 10 нМ CDK6/циклина D3 (LJIC-2009H2) для 20-минутной предварительной инкубации в буфере для анализа, содержащем 40 мМ HEPES, 1 мМ дитиотрейто (DTT, 10 мМ MgCl2, 1% глицерин, 0,1% BSA. Ферментативные реакции инициировали добавлением пептида AssayQuant Technologies и субстратов АТФ (10 мкМ пептида CHEF (Catalog #AQT0258), 2 мМ АТФ) и давали протекать реакции в течение 2 часов с последующим считыванием флуоресценции реакции.
Определения Ki выполняли по графику относительной скорости как функции концентрации ингибитора, соответствующей уравнению Моррисона с концентрацией фермента в качестве переменной.
Анализ сдвига подвижности (MSA) CDK4/циклин D1
Целью анализа CDK4/циклин D1 является оценка ингибирования (% ингибирования, значения Kiapp и Ki) в присутствии низкомолекулярных ингибиторов с использованием анализа сдвига микрофлюидной подвижности на основе флуоресценции. CDK4/циклин D1 катализирует продуцирование АДФ из АТФ, что сопровождает перенос фосфорила на субстратный пептид 5-FAM-Dyrktide. Анализ сдвига подвижности электрофоретически разделяет флуоресцентно меченые пептиды (субстрат и фосфорилированный продукт) после киназной реакции. И субстрат, и продукт измеряют, и соотношение этих значений используют для определения % превращения субстрата в продукт с помощью LabChip EZ Reader. Типичные реакционные растворы содержали 2% DMSO (±ингибитор), 10 мМ MgCl2, 1 мМ DTT, 3,5 мМ ATP, 0,005% Tween-20, 3 мкМ 5-FAM-Dyrktide, 3 нМ активированного CDK4/циклина D1 в 40 мМ буфере HEPES при pH 7,5.
Определение Ki ингибитора для активированного CDK4/циклина D1 (2007 E1/2008 +PO4) было начато с добавления АТФ (конечный реакционный объем 50 мкл) после восемнадцатиминутной предварительной инкубации фермента и ингибитора при 22°C в реакционной смеси. Реакцию останавливали через 195 минут добавлением 50 мкл 30 мМ EDTA. Определения Ki выполняли по графику относительной скорости как функции концентрации ингибитора, соответствующей уравнению Моррисона с концентрацией фермента в качестве переменной.
Анализ сдвига подвижности CDK6/циклин D3
Целью анализа CDK6/циклин D3 является оценка ингибирования (% ингибирования, значения Kiapp и Ki) в присутствии низкомолекулярных ингибиторов с использованием анализа сдвига микрофлюидной подвижности на основе флуоресценции. CDK6/циклин D3 катализирует продуцирование АДФ из АТФ, что сопровождает перенос фосфорила на субстратный пептид 5-FAM-Dyrktide Анализ сдвига подвижности электрофоретически разделяет флуоресцентно меченые пептиды (субстрат и фосфорилированный продукт) после киназной реакции. И субстрат, и продукт измеряют, и соотношение этих значений используют для определения % превращения субстрата в продукт с помощью LabChip EZ Reader. Типичные реакционные растворы содержали 2% DMSO (± ингибитор), 2% glycerol, 10 мМ MgCl2, 1 мМ DTT, 3,5 мМ АТФ, 0,005% Tween 20 (TW-20), 3 мкМ 5-FAM-Dyrktide, 4 нМ активированный CDK6/циклин D3 в 40 мМ буфере HEPES при pH 7,5.
Определение Ki ингибитора для активированного CDK6/циклина D3 (LJIC-2009G1/2010 +PO4) было начато с добавления АТФ (конечный реакционный объем 50 мкл) после восемнадцатиминутной предварительной инкубации фермента и ингибитора при 22°C в реакционной смеси. Реакцию останавливали через 95 минут добавлением 50 мкл 30 мМ EDTA. Определения Ki выполняли по графику относительной скорости как функции концентрации ингибитора, соответствующей уравнению Моррисона с концентрацией фермента в качестве переменной.
Анализ сдвига подвижности (MSA) CDK6/циклин D1
Целью анализа CDK6/циклин D1 является оценка ингибирования (% ингибирования, значения Kiapp и Ki) в присутствии низкомолекулярных ингибиторов с использованием анализа сдвига микрофлюидной подвижности на основе флуоресценции. CDK6/циклин D1 катализирует продуцирование АДФ из АТФ, что сопровождает перенос фосфорила на субстратный пептид 5-FAM-Dyrktide Анализ сдвига подвижности электрофоретически разделяет флуоресцентно меченые пептиды (субстрат и фосфорилированный продукт) после киназной реакции. И субстрат, и продукт измеряют, и соотношение этих значений используют для определения % превращения субстрата в продукт с помощью LabChip EZ Reader. Типичные реакционные растворы содержали 2% DMSO (± ингибитор), 2% glycerol, 10 мМ MgCl2, 1 мМ DTT, 3,5 мМ АТФ, 0,005% Tween 20 (TW-20), 3 мкМ 5-FAM-Dyrktide, 4 нМ активированный CDK6/циклин D1 в 40 мМ буфере HEPES при pH 7,5.
Определение Ki ингибитора для активированного CDK6/циклина D1 (LJIC-2003 A2/1865) было начато с добавления АТФ (конечный реакционный объем 50 мкл) после пятнадцатиминутной предварительной инкубации фермента и ингибитора при 22°C в реакционной смеси. Реакцию останавливали через 35 минут добавлением 50 мкл 30 мМ EDTA. Определения Ki выполняли по графику относительной скорости как функции концентрации ингибитора, соответствующей уравнению Моррисона с концентрацией фермента в качестве переменной.
Расчет Ki
Для всех анализов CDK4 и CDK6 константу Ki для соединений рассчитывали для каждого фермента с использованием уравнения Моррисона. Фракционную активность измеряли при различных концентрациях соединения [I], и данные соответствовали уравнению Моррисона, где [E] представляет собой концентрацию фермента, [S] представляет собой концентрацию АТФ, и KmApp представляет собой кажущееся Km для АТФ для каждого фермента в каждом формате анализа. С расчетами Ki, зависящими от условий анализа для каждого формата анализа, результирующий Ki становится независимым от анализа с сильной корреляцией один к одному для значений Ki для соединений, исследуемых в обоих форматах анализа.

Для расчетов Ki см. также Morrison, J. F. (1969) Kinetics of the reversible inhibition of enzyme-catalysed reactions by tight-binding inhibitors, Biochimica et biophysica acta 185, 269-286; and Murphy, D. J. (2004) Determination of accurate KI values for tight-binding enzyme inhibitors: an in silico study of experimental error and assay design, Analytical biochemistry 327, 61-67.
В таблице 2 данные анализа представлены двумя (2) значащими цифрами в виде среднего геометрического (Ki), основанного на количестве перечисленных повторов (количество). Ячейка с NA означает, что в указанном анализе не было данных для этого примера.
(CI)
GMean Ki (нМ)
(CI)
(0,8,2,9)
(1,1,2,9)
(0,6,1,0)
(0,9,1,4)
(0,8,1,5)
(1,3,2,0)
(0,6,2,6)
(1,3,3,0)
(0,2,0,5)
(1,1,1,4)
(51,175)
(116,263)
6-ацетил-8-[(1S,2S)-2-гидрокси-2-метилциклопентил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он или
или
6-ацетил-8-[(1R,2R)-2-гидрокси-2-метилциклопентил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он
(4,5,10,0)
(5,2,10,2)
6-ацетил-8-[(1S,2S)-2-гидрокси-2-метилциклопентил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он
или
6-ацетил-8-[(1R,2R)-2-гидрокси-2-метилциклопентил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он
(NA)
(NA)
6-ацетил-8-циклопентил-2-{[(3S,4R)-3-гидрокси-2,2-диметилтетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он
или
6-ацетил-8-циклопентил-2-{[(3R,4S)-3-гидрокси-2,2-диметилтетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он
(1,6,3,9)
(2,8,10,7)
6-ацетил-8-циклопентил-2-{[(3S,4R)-3-гидрокси-2,2-диметилтетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он
или
6-ацетил-8-циклопентил-2-{[(3R,4S)-3-гидрокси-2,2-диметилтетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он
(1,1,4,3)
(2,8,7,3)
(NA)
(NA)
(4,5,12,6)
(6,2,13,1)
6-ацетил-8-[(1S,2R,5R)-бицикло[3.1.0]гексан-2-ил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он
или
6-ацетил-8-[(1R,2S,5S)-бицикло[3.1.0]гексан-2-ил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он
(1,1,3,0)
(2,8,3,8)
6-ацетил-8-[(1S,2R,5R)-бицикло[3.1.0]гексан-2-ил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он
или
6-ацетил-8-[(1R,2S,5S)-бицикло[3.1.0]гексан-2-ил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он
(0,5,1,2)
(0, 5,5)
(3,1,4,5)
(NA)
(1,1,4,1)
(4,5,5,5)
6-ацетил-8-[(1R,2S)-2-фтор-2-метилциклопентил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он
или
6-ацетил-8-[(1S,2R)-2-фтор-2-метилциклопентил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он
(9,5,11,5)
6-ацетил-8-[(1R,2S)-2-фтор-2-метилциклопентил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он
или
6-ацетил-8-[(1S,2R)-2-фтор-2-метилциклопентил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он
(7,7,11,5)
(19,49)
(1,0,8,0)
(0, 9,9)
(18,90)
(40,75)
(7,20)
(11,16)
(2,2,3,4)
или
6-ацетил-8-[(1R,2R)-2-фтор-2-метилциклопентил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он
(N/A)
(N/A)
или
6-ацетил-8-[(1R,2R)-2-фтор-2-метилциклопентил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он
(11,14)
(8,39)
(5,67)
(8,216)
(11,15)
(16,63)
(6,14)
(4,46)
(7,14)
(29,44)
(1,6,2,1)
(3,0,6,9)
(320,540)
(N/A)
(N/A)
(N/A)
(11,21)
(21,33)
(39,85)
(60,200)
(44,59)
(56,115)
aИсследованный в анализе CHEF
bИсследованный в CHEF и MSA
cИсследованный в анализе Ki CDK6/D1 с использованием MSA
CI - 95% доверительный интервал
В настоящей заявке приводятся ссылки на различные публикации. Раскрытие этих публикаций во всей их полноте включены в настоящую заявку посредством ссылки для всех целей.
Специалистам в данной области будет очевидно, что в настоящее изобретение могут быть внесены различные модификации и вариации, без отклонения от сущности или объема изобретения. Другие варианты осуществления изобретения будут очевидны специалистам в данной области в результате рассмотрения спецификации и практики изобретения, раскрытого в настоящем документе. Предполагается, что описание и примеры должны рассматриваться только как иллюстративные, при этом истинный объем и сущность изобретения, определяется указанными нижеследующей формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИРИДОПИРИМИДИНОВЫЕ ИНГИБИТОРЫ CDK2/4/6 | 2017 |
|
RU2726115C1 |
| ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ МЕНИН-MLL | 2017 |
|
RU2799820C2 |
| ЗАМЕЩЕННЫЕ ДИАМИНОКАРБОКСАМИДНЫЕ И ДИАМИНОКАРБОНИТРИЛЬНЫЕ ПРОИЗВОДНЫЕ ПИРИМИДИНОВ, ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ИХ ПОМОЩЬЮ | 2012 |
|
RU2697712C2 |
| 3-КАРБОНИЛАМИНО-5-ЦИКЛОПЕНТИЛ-1H-ПИРАЗОЛЬНЫЕ СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ ИНГИБИТОРОВ CDK2 | 2020 |
|
RU2797889C2 |
| ГЕТЕРОАРИЛ-БИФЕНИЛ АМИНЫ ДЛЯ ЛЕЧЕНИЯ PD-L1 ЗАБОЛЕВАНИЙ | 2020 |
|
RU2837843C1 |
| ИНГИБИТОРЫ РЕЦЕПТОРА ФАКТОРА РОСТА ФИБРОБЛАСТОВ | 2014 |
|
RU2704112C2 |
| ПИРАЗОЛОПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРА КИНАЗЫ | 2017 |
|
RU2714206C1 |
| АГОНИСТЫ РЕЦЕПТОРА МЕЛАНОКОРТИНА | 2007 |
|
RU2411240C2 |
| ЗАМЕЩЕННЫЕ ПУРИНОВЫЕ И 7-ДЕАЗАПУРИНОВЫЕ СОЕДИНЕНИЯ | 2011 |
|
RU2606514C2 |
| РЕАКЦИИ МАКРОЦИКЛИЗАЦИИ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И ДРУГИЕ ФРАГМЕНТЫ, ПРИГОДНЫЕ В ПОЛУЧЕНИИ АНАЛОГОВ ХАЛИХОНДРИНА B | 2014 |
|
RU2710545C2 |
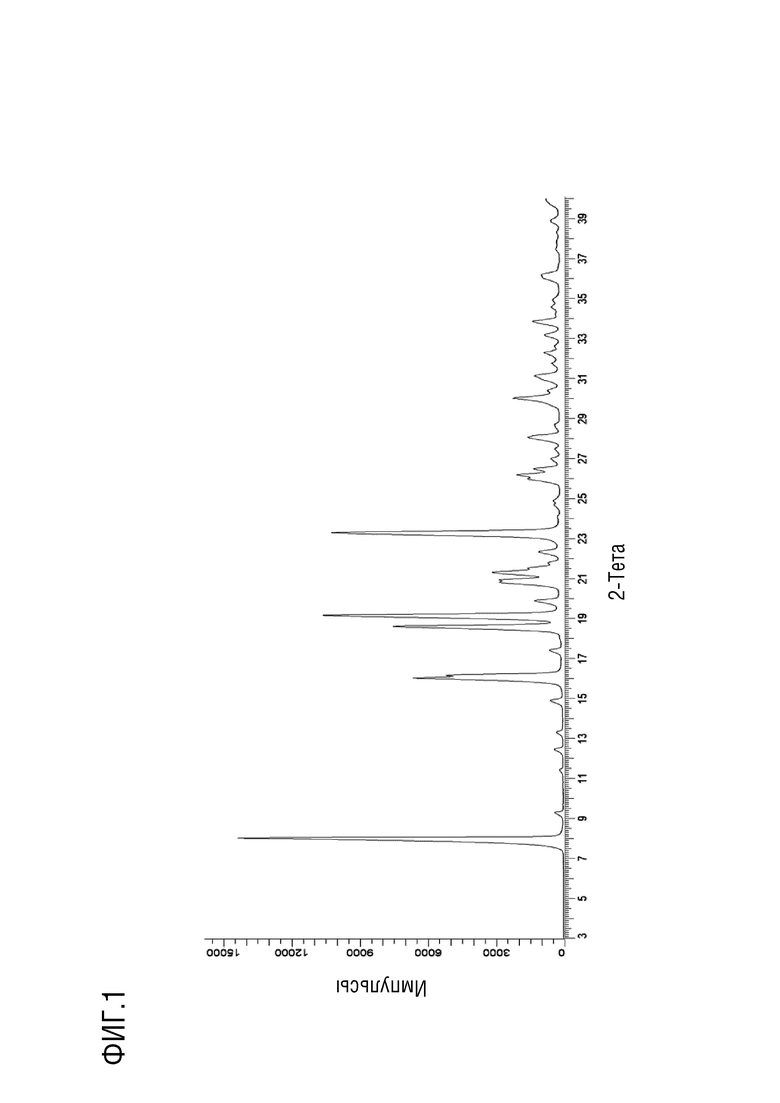
Изобретение относится к органической химии, а именно к соединению формулы I или его фармацевтически приемлемой соли, где A представляет собой CH или N; R1 представляет собой C1-C2 алкил; R2 представляет собой C1-C4 алкил; R3 представляет собой 5-7-членный гетероциклил, содержащий 1 гетероатом, выбранный из азота или кислорода, C3-C8 циклоалкил, где каждый из 5-7-членного гетероциклила необязательно замещен 1 R5, где каждый из C3-C8 циклоалкила необязательно замещен 1 R5 и дополнительно необязательно замещен 1 -N(R6)2; R4 представляет собой фрагмент, имеющий структуру

каждый R5 независимо выбран из группы, состоящей из -F, -OH, C1-C4 алкила, C1-C4 фторалкила, C1-C4 алкокси-C1-C4 алкил-; каждый R6 независимо выбран из группы, состоящей из H и C1-C2 алкила; каждый R7 независимо представляет собой H или C1-C2 алкил; и каждый R8 независимо представляет собой H. Также изобретение относится к конкретным соединениям, кристаллической форме 3-ацетил-1-циклопентил-7-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-4-метил-1,6-нафтиридин-2(1H)-она, фармацевтической композиции на основе соединения формулы I или конкретных указанных соединений, способу лечения заболевания или расстройства, при котором полезно ингибирование CDK4 и/или CDK6 или аномального роста клеток, применению соединения формулы I или конкретных указанных соединений, способу ингибирования CDK. Технический результат: ингибирование CDK4 и/или CDK6 соединением формулы I, полезное при лечении таких заболеваний, как легочная гипертензия, легочная артериальная гипертензия, легочная гипертензия с поражением левых отделов сердца, легочная гипертензия с заболеванием легких и/или гипоксемией, легочная гипертензия вследствие хронического тромботического и/или эмболического заболевания, и заболевания, связанного с легочной гипертензией, включая саркоидоз, гистиоцитоз X, лимфангиоматоз и компрессию легочных сосудов или аномального роста клеток. 13 н. и 31 з.п. ф-лы, 1 ил., 17 табл., 34 пр.
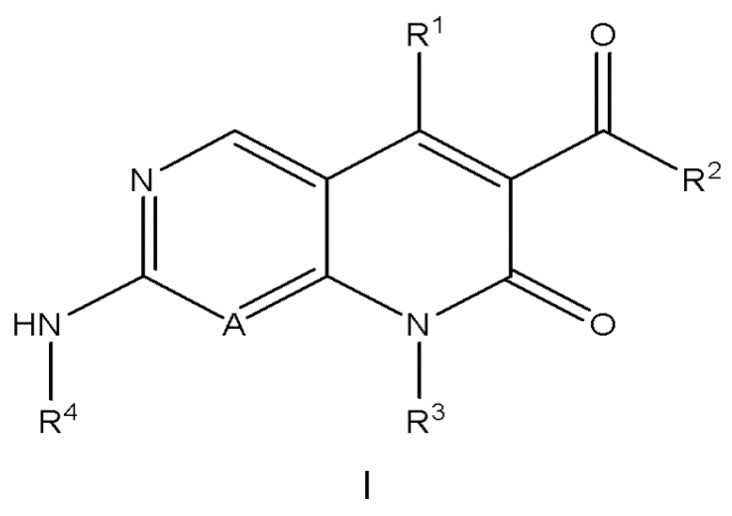
1. Соединение формулы I
или его фармацевтически приемлемая соль, где:
A представляет собой CH или N;
R1 представляет собой C1-C2 алкил;
R2 представляет собой C1-C4 алкил;
R3 представляет собой 5-7-членный гетероциклил, содержащий 1 гетероатом, выбранный из азота или кислорода, C3-C8 циклоалкил,
где каждый из 5-7-членного гетероциклила необязательно замещен 1 R5,
где каждый из C3-C8 циклоалкила необязательно замещен 1 R5 и дополнительно необязательно замещен 1 -N(R6)2;
R4 представляет собой фрагмент, имеющий структуру

каждый R5 независимо выбран из группы, состоящей из -F, -OH, C1-C4 алкила, C1-C4 фторалкила, C1-C4 алкокси-C1-C4 алкил-;
каждый R6 независимо выбран из группы, состоящей из H и C1-C2 алкила;
каждый R7 независимо представляет собой H или C1-C2 алкил; и
каждый R8 независимо представляет собой H.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, где:
A представляет собой CH или N;
R1 представляет собой CH3;
R2 представляет собой CH3;
R3 представляет собой 5-7-членный гетероциклил, содержащий 1 гетероатом, выбранный из азота или кислорода, C3-C6 циклоалкил,
где каждый из 5-7-членного гетероциклила необязательно замещен 1 R5,
где каждый из C3-C6 циклоалкила необязательно замещен 1 R5 и дополнительно необязательно замещен 1 -N(R6)2;
R4 представляет собой фрагмент, имеющий структуру

каждый R5 независимо выбран из группы, состоящей из -F, -OH, C1-C2 алкила, C1-C2 фторалкила, C1-C2 алкокси-C1-C2 алкил;
каждый R6 независимо выбран из группы, состоящей из H и C1-C2 алкила; и
каждый R7 независимо представляет собой H или CH3 при условии, что не более двух R7 представляют собой CH3.
3. Соединение по п. 1 или 2, где соединение представляет собой соединение формулы II
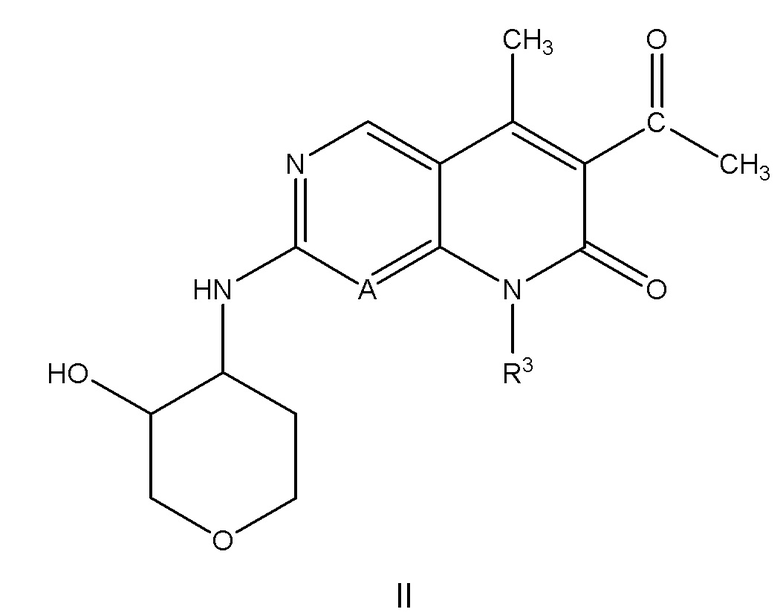
или его фармацевтически приемлемую соль.
4. Соединение по п. 3 или его фармацевтически приемлемая соль, где два заместителя в тетрагидропирановом кольце формулы II находятся в транс-положении по отношению друг к другу.
5. Соединение по любому из пп. 1-4 или его фармацевтически приемлемая соль, где соединение представляет собой соединение формулы III
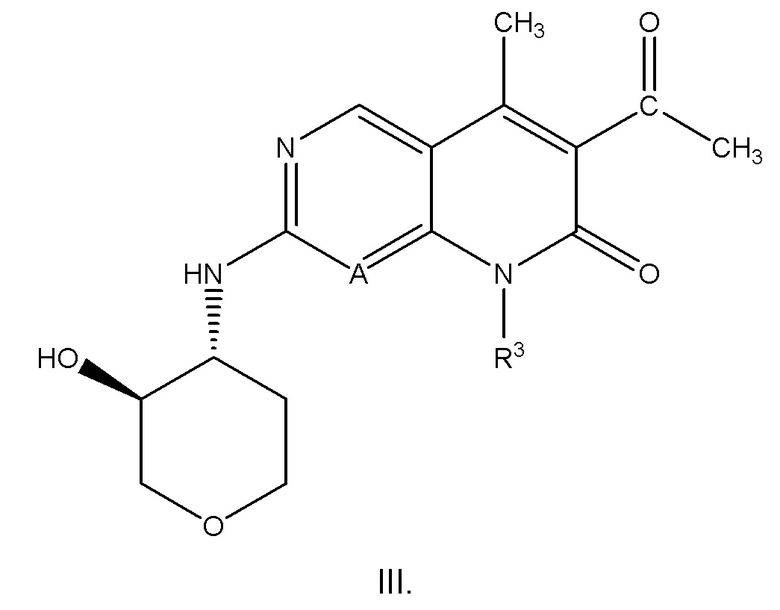
6. Соединение по п. 3 или 4 или его фармацевтически приемлемая соль, где:
A представляет собой CH или N;
R3 представляет собой 5-7-членный гетероциклил, необязательно замещенный 1 R5; и
R5 выбран из группы, состоящей из F, OH, C1-C2 алкила, C1-C2 фторалкила, C1-C2 алкокси-C1-C2 алкил-.
7. Соединение по п. 6 или его фармацевтически приемлемая соль, где соединение представляет собой соединение формулы III
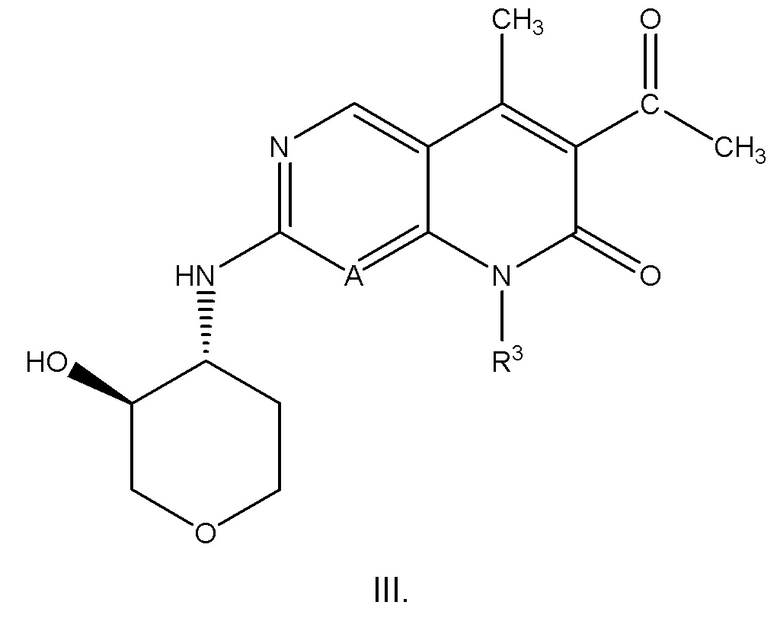
8. Соединение по любому из пп. 1-7 или его фармацевтически приемлемая соль, где R3 выбран из группы, состоящей из азаспиро[3.3]гептанила, тетрагидрофуранила, пирролидинила и пиперидинила, где каждый из выбранных гетероциклилов необязательно замещен 1 R5; и R5 выбран из группы, состоящей из F, OH, C1-C2 алкила, C1-C2 фторалкила, C1-C2 алкокси-C1-C2 алкил.
9. Соединение по любому из пп. 1-8 или его фармацевтически приемлемая соль, где R3 представляет собой 2-азаспиро[3.3]гептан-6-ил, необязательно замещенный 1 R5, который выбран из группы, состоящей из C1-C2 алкила, C1-C2 фторалкила, C1-C2 алкокси-C1-C2 алкил-.
10. Соединение по п. 3 или 4 или его фармацевтически приемлемая соль, где:
A представляет собой CH или N;
R3 представляет собой C3-C6 циклоалкил, необязательно замещенный 1 R5 и дополнительно необязательно замещенный 1 -N(R6)2;
каждый R5 независимо выбран из группы, состоящей из F, OH, C1-C2 алкила, C1-C2 фторалкила, C1-C2 алкокси-C1-C2 алкил-; и
каждый R6 независимо выбран из группы, состоящей из H и C1-C2 алкила.
11. Соединение по п. 10 или его фармацевтически приемлемая соль, где соединение представляет собой соединение формулы III

12. Соединение по любому из пп. 1-5, 10 и 11 или его фармацевтически приемлемая соль, где:
A представляет собой CH или N;
R3 выбран из группы, состоящей из циклопропила, циклобутила, циклопентила, циклогексила и бицикло[3.1.0]гексанила, где каждый из выбранных вариантов необязательно замещен 1 R5 и дополнительно необязательно замещен 1 -N(R6)2;
каждый R5 независимо выбран из группы, состоящей из F, OH, C1-C2 алкила, C1-C2 фторалкила, C1-C2 алкокси-C1-C2 алкил-; и
каждый R6 независимо выбран из группы, состоящей из H и C1-C2 алкила.
13. Соединение по п. 12 или его фармацевтически приемлемая соль, где R3 выбран из группы, состоящей из циклопропила, циклобутила, циклопентила, циклогексила и бицикло[3.1.0]гексанила, где каждый из выбранных вариантов необязательно замещен 1 R5 и дополнительно необязательно замещен 1 -N(R6)2.
14. Соединение по п. 12 или его фармацевтически приемлемая соль, где R3 выбран из группы, состоящей из циклопропила, циклобутила, циклопентила, циклогексила и бицикло[3.1.0]гексан-2-ила, где каждый из выбранных циклоалкилов необязательно замещен 1 R5 и дополнительно необязательно замещен 1 -N(R6)2.
15. Соединение по любому из пп. 12, 13 и 14 или его фармацевтически приемлемая соль, где каждый R5 независимо выбран из группы, состоящей из F, OH, C1-C2 алкила, C1-C2 фторалкила и C1-C2 алкокси-C1-C2 алкил-.
16. Соединение по любому из пп. 12, 13 и 14 или его фармацевтически приемлемая соль, где каждый R5 независимо выбран из группы, состоящей из F, OH, CH3 и CH2CH3, и каждый R6 независимо выбран из группы, состоящей из H и CH3.
17. Соединение по любому из пп. 12-16 или его фармацевтически приемлемая соль, где R3 представляет собой циклобутил, необязательно замещенный 1 R5.
18. Соединение по любому из пп. 10-14 или его фармацевтически приемлемая соль, где R3 представляет собой циклобутил, необязательно замещенный 1 R5, и каждый R5 независимо выбран из группы, состоящей из F, OH, C1-C2 алкила, C1-C2 фторалкила, C1-C2 алкокси-C1-C2 алкил-.
19. Соединение по любому из пп. 10-16 или его фармацевтически приемлемая соль, где R3 представляет собой циклопентил, необязательно замещенный 1 R5.
20. Соединение по любому из пп. 10-14 или его фармацевтически приемлемая соль, где R3 представляет собой циклопентил, необязательно замещенный 1 R5, и каждый R5 независимо выбран из группы, состоящей из F, OH, C1-C2 алкила, C1-C2 фторалкила, C1-C2 алкокси-C1-C2 алкил-.
21. Соединение по любому из пп. 10-16 или его фармацевтически приемлемая соль, где R3 представляет собой циклопентил.
22. Соединение по любому из пп. 1-21 или его фармацевтически приемлемая соль, где A представляет собой CH.
23. Соединение по любому из пп. 1-21 или его фармацевтически приемлемая соль, где A представляет собой N.
24. Соединение по п. 1, выбранное из:
6-ацетил-8-циклобутил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она;
6-ацетил-8-циклопентил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она;
6-ацетил-8-(2-азаспиро[3.3]гептан-6-ил)-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она;
6-ацетил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метил-8-[(1R,2S)-2-метилциклопентил]пиридо[2,3-d]пиримидин-7(8H)-она;
3-ацетил-1-циклопентил-7-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-4-метил-1,6-нафтиридин-2(1H)-она;
6-ацетил-8-[(1S,2R,5R)-бицикло[3.1.0]гексан-2-ил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она;
6-ацетил-8-[(1R,2S,5S)-бицикло[3.1.0]гексан-2-ил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она;
6-ацетил-8-[(1R,2S)-2-фтор-2-метилциклопентил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она;
6-ацетил-8-[(1S,2R)-2-фтор-2-метилциклопентил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она; и
6-ацетил-8-[2-(2,2-дифторэтил)-2-азаспиро[3.3]гептан-6-ил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-она,
или его фармацевтически приемлемая соль.
25. Соединение, которое представляет собой 6-ацетил-8-циклобутил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он.
26. Соединение, которое представляет собой 6-ацетил-8-циклопентил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он.
27. Соединение, которое представляет собой 6-ацетил-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метил-8-[(1R,2S)-2-метилциклопентил]пиридо[2,3-d]пиримидин-7(8H)-он.
28. Соединение, которое представляет собой 3-ацетил-1-циклопентил-7-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-4-метил-1,6-нафтиридин-2(1H)-он.
29. Соединение, которое представляет собой 6-ацетил-8-[2-(2,2-дифторэтил)-2-азаспиро[3.3]гептан-6-ил]-2-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-5-метилпиридо[2,3-d]пиримидин-7(8H)-он.
30. Кристаллическая форма 3-ацетил-1-циклопентил-7-{[(3S,4R)-3-гидрокситетрагидро-2H-пиран-4-ил]амино}-4-метил-1,6-нафтиридин-2(1H)-она, где кристаллическая форма (обозначенная как форма I) имеет порошковую рентгеновскую дифрактограмму (излучение CuKα), содержащую по меньшей мере два характеристических пика в единицах 2θ, выбранных из 8,0±0,2°; 18,6±0,2°; 19,1±0,2°; и 21,3±0,2°.
31. Кристаллическая форма по п. 30, где кристаллическая форма является безводной.
32. Кристаллическая форма по п. 30, имеющая порошковую рентгеновскую дифрактограмму (излучение CuKα), содержащую по меньшей мере три характеристических пика в единицах 2θ, выбранных из 8,0±0,2°; 18,6±0,2°; 19,1±0,2°; и 21,3±0,2°.
33. Кристаллическая форма по п. 30 или 32, имеющая порошковую рентгеновскую дифрактограмму (излучение CuKα), содержащую четыре характеристических пика в единицах 2θ, выбранных из 8,0±0,2°; 18,6±0,2°; 19,1±0,2°; и 21,3±0,2°.
34. Кристаллическая форма по любому из пп. 30-33, имеющая порошковую рентгеновскую дифрактограмму по существу такую, как показано на фиг. 1.
35. Фармацевтическая композиция, обладающая CDK4/6 ингибирующей активностью, содержащая эффективное количество соединения по любому из пп. 1-29 или его фармацевтически приемлемой соли или кристаллическую форму по любому из пп. 30-34 и фармацевтически приемлемый носитель.
36. Способ лечения заболевания или расстройства, при котором полезно ингибирование CDK4 и/или CDK6, у субъекта, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп. 1-29 или его фармацевтически приемлемой соли или кристаллической формы по любому из пп.30-34, где заболевание или расстройство выбрано из группы, состоящей из легочной гипертензии, легочной артериальной гипертензии, легочной гипертензии с поражением левых отделов сердца, легочной гипертензии с заболеванием легких и/или гипоксемией, легочной гипертензии вследствие хронического тромботического и/или эмболического заболевания и заболевания, связанного с легочной гипертензией, включая саркоидоз, гистиоцитоз X, лимфангиоматоз и компрессию легочных сосудов.
37. Способ по п. 36, где заболевание или расстройство представляет собой легочную артериальную гипертензию.
38. Применение соединения по любому из пп. 1-29 или его фармацевтически приемлемой соли или кристаллической формы по любому из пп. 30-34 для лечения заболевания или расстройства, при котором полезно ингибирование CDK4 и/или CDK6, где заболевание или расстройство выбрано из группы, состоящей из легочной гипертензии, легочной артериальной гипертензии, легочной гипертензии с поражением левых отделов сердца, легочной гипертензии с заболеванием легких и/или гипоксемией, легочной гипертензии вследствие хронического тромботического и/или эмболического заболевания и заболевания, связанного с легочной гипертензией, включая саркоидоз, гистиоцитоз X, лимфангиоматоз и компрессию легочных сосудов.
39. Применение по п. 38, где заболевание или расстройство представляет собой легочную артериальную гипертензию.
40. Способ лечения аномального роста клеток у субъекта, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп. 1-29 или его фармацевтически приемлемой соли или кристаллической формы по любому из пп. 30-34.
41. Способ по п. 40, где аномальный рост клеток представляет собой рак.
42. Способ по п. 41, где рак выбран из группы, состоящей из рака молочной железы, рака яичников, рака мочевого пузыря, рака матки, рака предстательной железы, рака легкого, рака пищевода, рака головы и шеи, колоректального рака, рака почки, рака печени, рака поджелудочной железы, рака желудка и рака щитовидной железы.
43. Применение соединения по любому из пп. 1-29 или его фармацевтически приемлемой соли или кристаллической формы по любому из пп. 30-34 для лечения аномального роста клеток.
44. Способ ингибирования CDK, включающий контактирование CDK с соединением по любому из пп. 1-29 или его фармацевтически приемлемой солью или кристаллической формой по любому из пп. 30-34.
| WO 2018033815 A1, 22.02.2018 | |||
| SANCHEZ-MARTINEZ CONCEPCION и др | |||
| Cyclin dependent kinase (CDK) inhibitors as anticancer drugs | |||
| BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, 06.06.2015, 25(17), с.3420-3435 | |||
| WO 2015120049 A1, 13.08.2015 | |||
| ПРОИЗВОДНЫЕ ПИРИДОПИРИМИДИНОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2002 |
|
RU2269527C2 |
| ПИРАЗОЛПИРИМИДИНЫ | 2005 |
|
RU2412186C2 |
| Пустотное устройство для генерирования токов высокой частоты | 1927 |
|
SU7395A1 |

