Область техники
В данной заявке заявлен приоритет заявки на патент Кореи № 10-2010-0041436, поданной 3 мая 2010, в Korean Intellectual Property Office, описание которой включено в настоящее описание в качестве ссылки.
Данное изобретение относится к фармацевтической композиции для ингибирования апоптоза нейронов или нейродегенерации.
Уровень техники
Апоптоз нейронов может быть вызван при обычных физиологических функциях, таких как развитие нервной системы, или при патологических процессах, таких как заболевания. Во время процесса развития нейронов избыток нейронов удаляют посредством апоптоза для оптимальной точной связи между пресинапсом и постсинапсом (Neuron, 40:401-413(2003); Neuron, 20:633-647(1998)). Широкий спектр апоптоза нейронов наблюдают при нейродегенеративных заболеваниях, таких как амиотрофический латеральный склероз, болезнь Альцгеймера и болезнь Паркинсона, удар и внешние повреждения. Прямые причины этих заболеваний еще не найдены, однако они связаны с апоптозом, и на апоптоз влияют различные факторы, такие как окислительный стресс, дисрегулирование гомеостаза кальция, дисфункция митохондрий, повышение образования реакционно-способных видов кислорода, эксайтотоксичность, активация каспазы и трофические поражения (Nature Reviews Molecular Cell Biology, 1:120-130(2000), Neurotoxicology and Teratology, 24:675-682(2002)).
При болезни Паркинсона описано, что дисфункция митохондрии повышает секрецию кальция и дегенерацию реакционно-способных видов кислорода, тем самым вызывая окислительный стресс для снижения активности антиокислительных систем. Кроме того, имеется доклад, касающийся связи между эксайтотоксичностью глутаматом и болезнью Паркинсона (Neurotoxicology and Teratology, 24:675-682(2002)).
При болезни Альцгеймера описано, что апоптоз нейронов связан с окислительным стрессом, дисрегулированием гомеостаза ионов, депривацией фактора роста, аккумуляцией амилоида Aβ, ухудшением метаболизма, дисфункцией митохондрии и агрегацией белка (Nat. Rev. Neurosci., 7:278-294(2006); Cerebellum, 2:270-278(2003)).
В настоящее время предложены различные типы нейропротекторных агентов, применяемых для защиты нейронов от апоптоза, вызванного различными механизмами (Neurotoxicology and Teratology, 24:675-682(2002)). Примеры нейропротекторных агентов включают антиоксиданты, хелатирующие ион агенты, ловушки свободных радикалов, нейротрофические факторы, возбуждающие антагонисты аминокислоты, биоэнергетические добавки, иммунодепрессанты и композиции, предотвращающие агрегацию или аккумуляцию белка. Однако лекарственные средства, которые эффективно ингибируют апоптоз нейронов или нейродегенерацию, все еще не являются коммерчески доступными, и таким образом, все еще сохраняется необходимость в разработке фармацевтической композиции для ингибирования апоптоза нейронов или нейродегенерации.
Описание изобретения
Техническая проблема
В данном изобретении представлена фармацевтическая композиция для ингибирования апоптоза нейронов или нейродегенерации.
В данном изобретении также представлена фармацевтическая композиция для нейропротекции или восстановления функции нервной ткани.
Дополнительные аспекты представлены частично в описании, представленном ниже, и частично будут очевидны из описания или могут быть изучены при практическом осуществлении представленных вариантов.
Решение проблемы
Далее данное изобретение подробно описано со ссылкой на приложенные чертежи.
В данном изобретении представлена фармацевтическая композиция для ингибирования апоптоза нейронов или нейродегенерации, включающая терапевтически эффективное количество соединения, выбранного из группы, состоящей из замещенного производного азола, представленного формулой 1 ниже, его фармацевтически приемлемых солей, изомеров замещенного производного азола, сольватов замещенного производного азола, и их сочетания; и фармацевтически приемлемый носитель.
В данном изобретении также представлена фармацевтическая композиция для нейропротекции, включающая терапевтически эффективное количество соединения, выбранного из группы, состоящей из замещенного производного азола, представленного формулой 1 ниже, его фармацевтически приемлемых солей, изомеров замещенного производного азола, сольватов замещенного производного азола, и их сочетания; и фармацевтически приемлемый носитель.
В данном изобретении также представлена фармацевтическая композиция для восстановления функций нервной ткани, включающая терапевтически эффективное количество соединения, выбранного из группы, состоящей из замещенного производного азола, представленного формулой 1 ниже, его фармацевтически приемлемых солей, изомеров замещенного производного азола, сольватов замещенного производного азола, и их сочетания; и фармацевтически приемлемый носитель.
В данном изобретении также представлена фармацевтическая композиция для предупреждения или лечения нейродегенеративных заболеваний или заболеваний, связанных с ишемией или реперфузией, где композиция включает терапевтически эффективное количество соединения, выбранного из группы, состоящей из замещенного производного азола, представленного формулой 1 ниже, его фармацевтически приемлемых солей, изомеров замещенного производного азола, сольватов замещенного производного азола, и их сочетания; и фармацевтически приемлемый носитель.
В данном изобретении также представлена фармацевтическая композиция для предупреждения или лечения заболеваний, выбранных из группы, состоящей из удара, болезни Альцгеймера, болезни Хантингтона, болезни Паркинсона, болезни Пика, болезни Крейтцфельда-Якоба, комплекса Паркинсон-БАС-слабоумие, болезни Уилсона, рассеянного склероза, прогрессирующего надъядерного паралича, биполярных расстройств, связанных с невропатической болью, кортико-базальной дегенерации, шизофрении, синдрома гиперактивности с дефицитом внимания (ADHD), слабоумия, амиотрофического латерального склероза, болезни сетчатки, эпилепсии, апоплексии, преходящего нарушения мозгового кровообращения, ишемии миокарда, мышечной ишемии, ишемии, вызванной хирургическим вмешательством, требующим длительного прекращения подачи крови к мозгу, повреждения головы, повреждения спинного мозга, гипоксии и депрессии, где композиция включает терапевтически эффективное количество соединения, выбранного из группы, состоящей из замещенного производного азола, представленного формулой 1 ниже, его фармацевтически приемлемых солей, изомеров замещенного производного азола, сольватов замещенного производного азола, и их сочетания; и фармацевтически приемлемый носитель.
Формула I
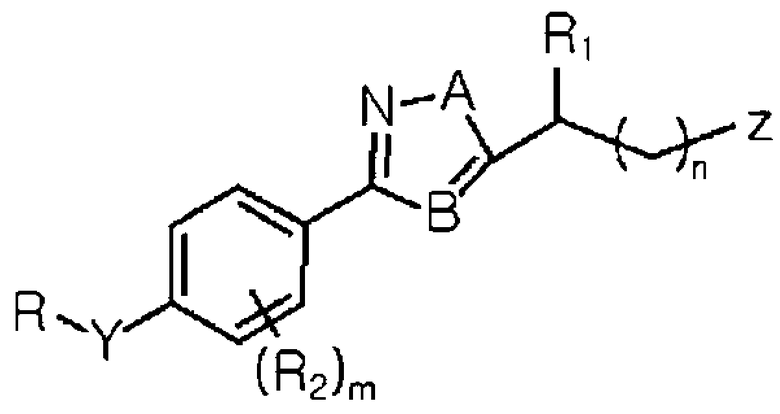
где R выбран из группы, состоящей из замещенной или незамещенной C1-C15 арилалкильной группу, замещенной или незамещенной C1-C10 гетероарилалкильной группу и замещенной или незамещенной, линейной, разветвленной или циклической C1-C10 алкильной группы;
Y выбран из группы, состоящей из O и -N-R1;
R1 является, по меньшей мере, одним, выбранным из группы, состоящей из H и линейной или разветвленной C1-C3 алкильной группы;
R2 выбран из группы, состоящей из H и галогена;
A выбран из группы, состоящей из N, O и S;
B является C или N;
Z выбран из группы, состоящей из замещенной или незамещенной гетероциклической группы, карбамата, -OC(=O)NR3R4, NH2, NR5R6, NC(=NH)NH2 и -NC(=O)NH2;
каждый из R3 и R4 независимо выбран из группы, состоящей из H; C1-C5 алкила, незамещенного или замещенного, по меньшей мере, одним заместителем, выбранным из группы, состоящей из NH2 и NR7R8; гетероциклического кольца, незамещенного или замещенного C1-C3 алкилом; или R3 и R4 вместе могут образовывать 5- или 7-членное гетероциклическое кольцо, незамещенное или замещенное C1-C3 алкилом;
каждый из R5 и R6 независимо выбран из группы, состоящей из H; C2-C3 алкена; C2-C3 алкина; и линейного или разветвленного C1-C7 алкила, незамещенного или замещенного, по меньшей мере, одним заместителем, выбранным из группы, состоящей из -OH, -C(O)NH2, C1-C3 алкокси и карбамата, или R5 и R6 вместе могут образовывать замещенный или незамещенный алифатический циклический амин или ароматический циклический амин;
каждый из R7 и R8 независимо является, по меньшей мере, одним, выбранным из группы, состоящей из H и линейной или разветвленной C1-C3 алкильной группы;
m является целым числом в интервале от 0 до 4; и
n является целым числом в интервале от 0 до 5.
Фармацевтическая композиция может включать терапевтически эффективное количество соединения, выбранного из группы, состоящей из замещенного производного азола, представленного формулой 1 ниже, его фармацевтически приемлемых солей, изомеров замещенного производного азола, сольватов замещенного производного азола, и их сочетания.
Термин "лечение" в данном описании включает, у животных, у которых никогда не диагностировали заболевания, расстройства или состояния, вызванные апоптозом нейронов или нейродегенерацией, но у которых имеется риск развития таких заболеваний, расстройств или состояний, предупреждение развития таких болезней, расстройств или состояний, ингибирование развития заболеваний, расстройств или состояний, то есть ингибирование развития заболеваний, расстройств или состояний, и облегчение заболеваний, расстройств или состояний, то есть дегенерацию заболеваний, расстройств или состояний. Поэтому термин "терапевтически эффективное количество" в данном описании относится к достаточному количеству, применяемому для достижения фармакологического действия, описанного выше.
Замещенное производное азола формулы I может быть получено с применением известных соединений или соединений, которые могут быть легко получены из них специалистами в области синтеза соединения в области, к которой относится данное изобретение. Поэтому способ получения замещенного производного азола формулы I, который описан ниже, является примерным вариантом, данным в иллюстративных целях, и порядок стадий в нем может быть селективно изменен, при необходимости, не ограничивая объем данного изобретения.
Схема 1: Синтез азола
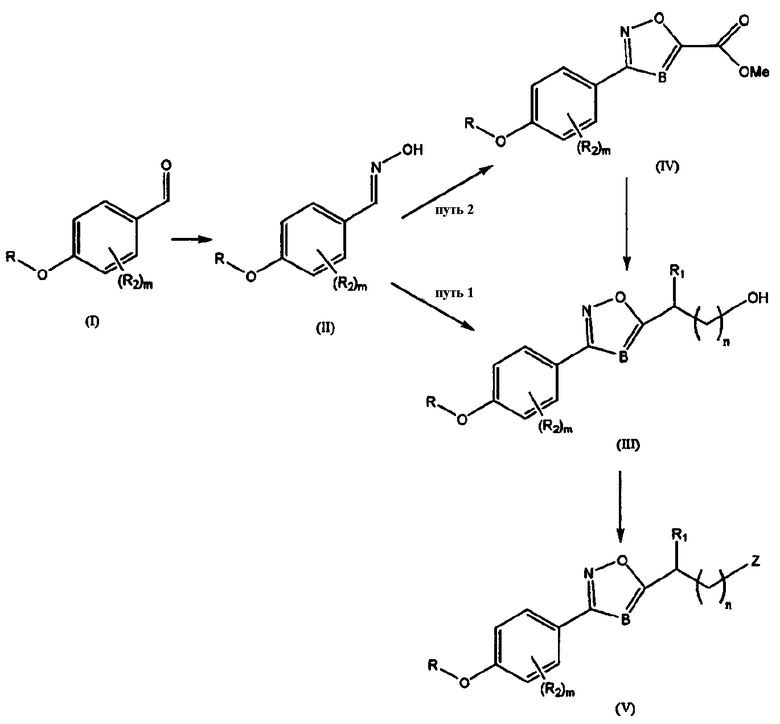
R может быть бензильной группой, и R2, Z, B, m и n такие, как определены выше. Общий способ синтеза азола может проводиться следующим образом: оксим (II) получают из альдегида (I), применяемого в качестве исходного материала, полученный оксим подвергают [3+2] циклоприсоединению с алкинами или нитрилами в присутствии NaOCl с получением соединения азола (III или IV), и затем желаемые функциональные группы вводят в соединение азола с получением конечного соединения (V).
Схема 2: Синтез тиазола
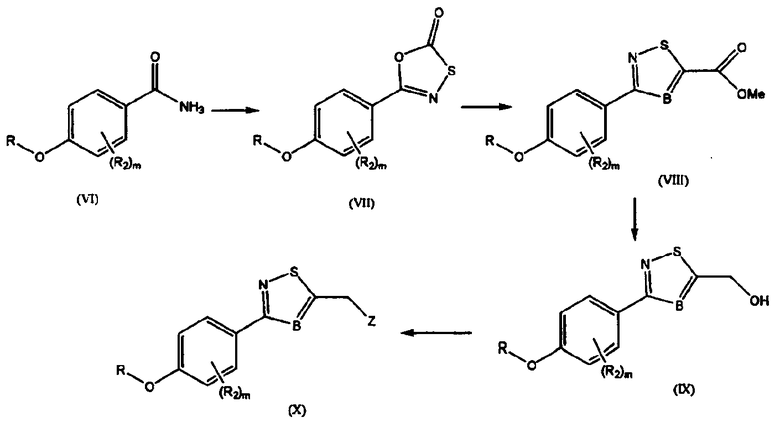
R может быть бензильной группой и R2, Z, B и m такие, как определены выше. Общий способ синтеза тиазола может проводиться следующим образом: оксатиазолон (VII) получают из амида (VI), применяемого в качестве исходного материала, полученное соединение подвергают [3+2] циклоприсоединению с алкинами или нитрилами в присутствии NaOCl с получением соединения тиазола (VIII), и соединение тиазола восстанавливают (IX), и желаемые функциональные группы вводят туда с получением конечного соединения (X).
Схема 3:
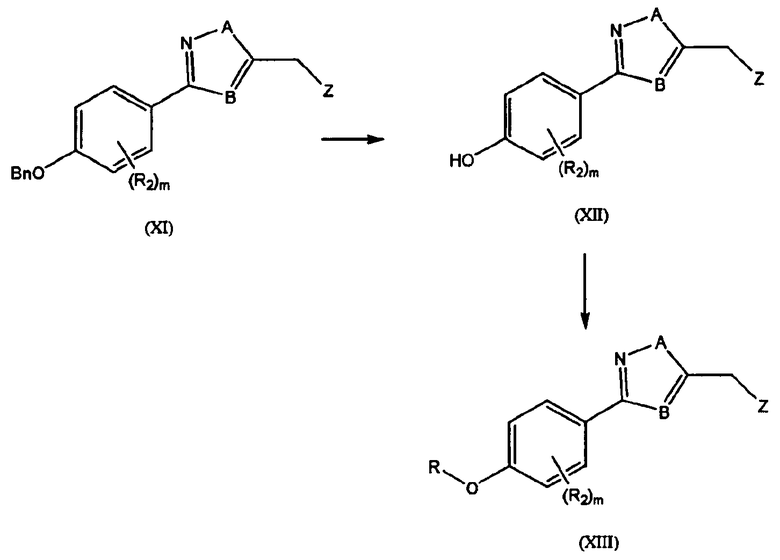
R, R2, Z, A, m и B такие, как определены выше. Общий способ синтеза конечного соединения (XIII) может проводиться следующим образом: производное гидроксифенила (XII) получают дебензилированием соединения (XI), применяемого в качестве исходного материала, и желаемые функциональные группы вводят туда с получением конечного соединения (XIII).
Схема 4:
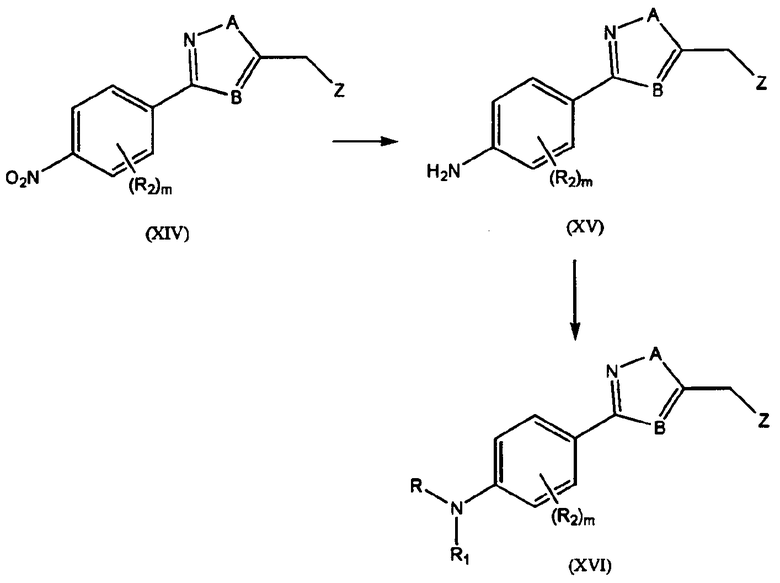
R, R1, R2, Z, A, m и B такие, как определены выше. Общий способ синтеза конечного соединения (XVI) может проводиться следующим образом: производное аминофенила (XV) синтезируют восстановлением производного нитрофенила (XIV), применяемого в качестве исходного материала, и синтезированное соединение затем подвергают восстановительному аминированию с желаемым альдегидом с получением конечного соединения (XVI).
Производное азола включает, в дополнение к производному азота формулы 1, его фармацевтически приемлемые соли, то есть аддитивные соли кислоты или основания, и их стереохимические изомеры, и солями могут быть любые соли, которые сохраняют активность исходного соединения у пациента, которому их вводят, без нежелательных эффектов. Такими солями могут быть неорганические или органические соли. Примеры солей включают соли уксусной кислоты, азотной кислоты, аспартиновой кислоты, сульфоновой кислоты, серной кислоты, малеиновой кислоты, глутаминовой кислоты, муравьиной кислоты, янтарной кислоты, фосфорной кислоты, фталевой кислоты, дубильной кислоты, винной кислоты, бромистоводородной кислоты, пропионовой кислоты, бензолсульфоновой кислоты, бензойной кислоты, стеариновой кислоты, эзилат, молочной кислоты, бикарбоновой кислоты, бисерной кислоты, бивинной кислоты, щавелевой кислоты, масляной кислоты, эдетат кальция, камзиловой кислоты, карбоновой кислоты, хлорбензойной кислоты, лимонной кислоты, эдетиновой кислоты, толуолсульфоновой кислоты, эдизиловой кислоты, эзиловой кислоты, фумаровой кислоты, глуцептиновой кислоты, памоат, глюконовой кислоты, гликоллиларсанилиновой кислоты, метилазотной кислоты, полигалактуроновой кислоты, гексилрезорциновой кислоты, малоновой кислоты, гидрабаминовой кислоты, хлористоводородной кислоты, йодистоводородной кислоты, гидроксинафтокислоты, изетионовой кислоты, лактобионовой кислоты, миндальной кислоты, эстолевой кислоты, слизевой кислоты, напсиловой кислоты, муконовой кислоты, п-нитрометансульфоновой кислоты, гексамовой кислоты, пантотеновой кислоты, моногидрофосфорной кислоты, дигидрофосфорной кислоты, салициловой кислоты, сульфаминовой кислоты, сульфанилиновой кислоты, метансульфоновой кислоты и теоклиновой кислоты. Также форма щелочной соли включает, например, аммониевую соль, соли щелочных металлов и соли щелочноземельных металлов, такие как соли лития, натрия, калия, магния и кальция, соли органических оснований, такие как соли бензатина, N-метил-D-глюкамина и гидрабамина, и соли, содержащие аминокислоты, такие как аргинин и лизин. Также формы солей могут быть превращены в свободные формы обработкой подходящими основаниями или кислотами. Термин "аддитивная соль" в данном описании означает соли, которые включают сольваты, которые могут образовывать замещенные производные азота формулы 1 или их соли. Сольваты могут быть гидратами или алкоголятами.
В данном описании термин "стереохимические изомеры замещенного производного азола формулы I" относится ко всем возможным формам, которые может иметь замещенное производное азола формулы I. Если не определено или указано иное, химические наименования замещенного производного азола формулы I указывают на смеси всех возможных стереохимических изомеров, включая все диастереомеры и энантиомеры основных молекулярных структур. В частности, каждый хиральный центр может иметь либо R-, либо S-конфигурацию, и заместители на двухвалентных циклических (частично) насыщенных радикалах может иметь цис- или транс-конфигурацию. Соединения, имеющие двойные связи, могут иметь E- или Z-стереохимию. Все стереохимические изомеры замещенного производного азола формулы I включены в объем данного изобретения.
Согласно определению формулы I выше, примеры замещенных производных азола могут включать 3-(4-бензилоксифенил)изоксазол-5-илметиловый эфир карбаминовой кислоты, 3-(4-бензилоксифенил)-[1,2,4]оксадиазол-5-илметиловый эфир карбаминовой кислоты, 3-(4-бензилоксифенил)изотиазол-5-илметиловый эфир карбаминовой кислоты, 3-(4-бензилоксифенил)-[1,2,4]тиадиазол-5-илметиловый эфир карбаминовой кислоты, 3-(4-бензилокси-2-хлорфенил)изоксазол-5-илметиловый эфир карбаминовой кислоты, 3-(4-бензилокси-3-хлорфенил)изоксазол-5-илметиловый эфир карбаминовой кислоты, 3-(4-бензилокси-3-бромфенил)изоксазол-5-илметиловый эфир карбаминовой кислоты, 3-(4-бензилокси-3-фторфенил)изоксазол-5-илметиловый эфир карбаминовой кислоты, 3-(4-бензилокси-3,5-диметилфенил)изоксазол-5-илметиловый эфир карбаминовой кислоты, 3-[4-(1-фенилэтокси)фенил]изоксазол-5-илметиловый эфир карбаминовой кислоты, 3-[4-(2-фторбензилокси)фенил]изоксазол-5-илметиловый эфир карбаминовой кислоты, 3-[4-(3-фторбензилокси)фенил]изоксазол-5-илметиловый эфир карбаминовой кислоты, 3-[4-(4-фторбензилокси)фенил]изоксазол-5-илметиловый эфир карбаминовой кислоты, 3-[4-(2,6-дифторбензилокси)фенил]изоксазол-5-илметиловый эфир карбаминовой кислоты, 3-[4-(2,3-дифторбензилокси)фенил]изоксазол-5-илметиловый эфир карбаминовой кислоты, 3-[4-(3,5-дифторбензилокси)фенил]изоксазол-5-илметиловый эфир карбаминовой кислоты, 3-[4-(3,4-дифторбензилокси)фенил]изоксазол-5-илметиловый эфир карбаминовой кислоты, 3-[4-(2,4,6-трифторбензилокси)фенил]изоксазол-5-илметиловый эфир карбаминовой кислоты, 3-[4-(3-трифторметилбензилокси)фенил]изоксазол-5-илметиловый эфир карбаминовой кислоты, 3-[4-(3-хлорбензилокси)фенил]изоксазол-5-илметиловый эфир карбаминовой кислоты, 3-[4-(2-хлорбензилокси)фенил]изоксазол-5-илметиловый эфир карбаминовой кислоты, 3-[4-(4-хлорбензилокси)фенил]изоксазол-5-илметиловый эфир карбаминовой кислоты, 3-[4-(2,6-дихлорбензилокси)фенил]изоксазол-5-илметиловый эфир карбаминовой кислоты, 3-[4-(2,5-дихлорбензилокси)фенил]изоксазол-5-илметиловый эфир карбаминовой кислоты, 3-[4-(2-хлор-5-фторбензилокси)фенил]изоксазол-5-илметиловый эфир карбаминовой кислоты, 3-[4-(3-нитробензилокси)фенил]изоксазол-5-илметиловый эфир карбаминовой кислоты, метиловый эфир 4-[4-(5-карбамоилоксиметилизоксазол-3-ил)феноксиметил]бензойной кислоты, 3-[4-(4-метилбензилокси)фенил]изоксазол-5-илметиловый эфир карбаминовой кислоты, 3-[4-(2-метилбензилокси)фенил]изоксазол-5-илметиловый эфир карбаминовой кислоты, 3-[4-(3-метоксибензилокси)фенил]изоксазол-5-ил метиловый эфир карбаминовой кислоты, 3-[4-(3-трифторметилбензилокси)фенил]изоксазол-5-илметиловый эфир, 3-[4-(4-изопропилбензилокси)фенил]изоксазол-5-илметиловый эфир карбаминовой кислоты и 3-[4-(4-трет-бутилбензилокси)фенил]изоксазол-5-илметиловый эфир карбаминовой кислоты. Способы получения этих производных азола описаны в заявке на патент Кореи № 2009-15856, поданной авторами данного изобретения, описание которой включено в настоящее описание в качестве ссылки полностью.
Согласно одному из вариантов данного изобретения, замещенным производным азота формулы I может быть 3-(4-бензилоксифенил)изоксазол-5-илметиловый эфир карбаминовой кислоты (CBI), представленный формулой II ниже:
Формула II

Фармацевтическая композиция в соответствии с вариантом данного изобретения может включать фармацевтически приемлемый носитель.
Фармацевтически приемлемый носитель в фармацевтической композиции, который обычно используют в композиции, может включать лактозу, декстрозу, сахарозу, сорбит, маннит, крахмал, аравийскую камедь, фосфат кальция, альгинаты, желатин, силикат кальция, микрокристаллическую целлюлозу, поливинилпирролидон, целлюлозу, воду, сироп, метилцеллюлозу, метилгидроксибензоат, пропилгидроксибензоат, тальк, стеарат магния и минеральное масло, но не ограничены ими. Фармацевтическая композиция также может включать смазывающий агент, смачивающий агент, подсластитель, усилитель вкуса, эмульгирующий агент, суспендирующий агент и консервант. Подходящие фармацевтически приемлемые носители и композиции описаны в Remington's Pharmaceutical Sciences (19th ed., 1995).
Фармацевтическая композиция в соответствии с одним из вариантов данного изобретения может вводиться перорально или парентерально. Парентеральное введение может включать внутривенные инъекции, подкожные инъекции, внутримышечные инъекции, внутрибрюшинные инъекции, эндотелиальное введение, местное введение, интраназальное введение, внутрилегочное введение и ректальное введение. Для перорального введения активное лекарственное средство, сформированное из фармацевтической композиции, может быть покрыто оболочкой, или фармацевтическая композиция может быть составлена для предотвращения переваривания. Кроме того, фармацевтическая композиция может вводиться устройством, способным переносить активное вещество в целевую клетку.
Подходящая доза фармацевтической композиции в соответствии с одним вариантом данного изобретения может зависеть от множества факторов, таких как способы составления композиции, способы введения, возраст пациента, масса тела, пол, патологическое состояние, режим питания, время введения, способ введения, скорость выведения и чувствительность, и доза фармацевтической композиции, которая является эффективной для желаемого лечения или предупреждения, может быть легко определена и прописана обычным лечащим врачом.
Фармацевтическая композиция может быть составлена с применением фармацевтически приемлемого носителя и/или добавки хорошо известным в данной области техники способом, и получена в стандартной лекарственной форме или в многоразовом контейнере. В связи с этим композиция может быть раствором в масле или водной среде, суспензией, эмульгирующим раствором, экстрактом, порошком, гранулами, таблеткой или капсулой, и может также включать диспергирующий или стабилизирующий агент. Кроме того, фармацевтическая композиция может вводиться в виде отдельного лекарственного средства или вместе с другими лекарственными средствами, или может вводиться последовательно или одновременно с ранее существующими лекарственными средствами.
Фармацевтическую композицию применяют для ингибирования смерти нейронов или нейродегенерации.
Термин "нейрон" в данном описании относится к животной клетке, состоящей из клеточного тела, одного из ответвлений, которые выходят из клеточного тела, т.е. аксона или нейрита, и нескольких дендритов, и примеры нейрона могут включать сенсорные нейроны, мотонейроны и интерейроны. Кроме того, нейрон может включать нейроны, составляющие центральную нервную систему, мозг, ствол мозга, спинной мозг и синаптические области центральной нервной системы и периферийной нервной системы, нейроподдерживающие клетки, нейроглию и шванновские клетки.
Термин "смерть нейрона" в данном описании включает смерть нейронов посредством апоптоза. Кроме того, термин "нейродегенерация" в данном описании означает частичную дегенерацию структуры или функций нейронов, включая смерть нейронов.
Тот факт, что апоптоз нейронов или нейродегенерация вызывает различные заболевания мозга, такие как боковой амиотрофический склероз, болезнь Альцгеймера и болезнь Паркинсона, хорошо известен в данной области техники, и проводятся исследования, касающиеся механизма апоптоза нейронов для предупреждения или лечения этих заболеваний. В Nature Reviews Molecular Cell Biology 1:120-130 (2000) и Journal of Cellular and Molecular Medicine,12:2263-2280(2008) описано, что апоптоз нейронов является причиной различных заболеваний, таких как болезнь Альцгеймера, болезнь Паркинсона, болезнь Хантингтона, ишемия, удар и склероз, и через исследования, касающиеся механизма апоптоза нейронов, вызывающего окислительный стресс и дисфункцию митохондрий, был обнаружен способ предупреждения или лечения нейродегенеративных заболеваний. Таким образом, специалисту в области медицины понятно, что фармацевтическая композиция, включающая соединение, обладающее ингибирующим апоптоз нейронов или нейродегенерацию действием, может применяться для предупреждения или лечения описанных выше заболеваний.
Фармацевтическая композиция согласно одному из вариантов данного изобретения может применяться для нейропротекции.
Термин "нейропротекция" в данном описании означает механизмы в нервной системе, которые защищают нейроны от апоптоза или дегенерации, и в частности, означает снижение, ингибирование или облегчение нервных повреждений, а также означает защиту, восстановление или регенерацию нейронов в нервной ткани, поврежденной нервными повреждениями. Кроме того, термин "нейропротекция" является стандартным термином, который обычно применяется специалистами в области техники, к которой относится данное изобретение (Neuro Report, 9:3955-3959(1998); Chen, J-F., J. Neurosci., 21:RC143(2001)). Термин "защита нервной клетки" в данном описании означает механизмы снижения или облегчения нервного поражения, или механизмы защиты или восстановления нейронов, поврежденных нервным поражением. Кроме того, термин "нервное повреждение" в данном описании означает повреждение нейронов или нервной ткани, вызванное различными факторами (например, метаболическим фактором, токсическим фактором, нейротоксическим фактором и химическим фактором). Примеры нервного повреждения могут включать окислительный стресс, дисрегулирование гомеостаза кальция, дисфункцию митохондрии, эксайтотоксичность, активацию каспазы и алиментарную депривацию (Nature Reviews Molecular Cell Biology 1:120-130(2000), Neurotoxicology and Teratology 24:675-682(2002)). Фармацевтическая композиция обладает действием ингибирования апоптоза нейронов или нейродегенерации такими различными нервными повреждениями, или действием защиты нейронов от нервных повреждений. Например, среди нервных повреждений, описанных выше, окислительный стресс представляет собой заболевание, связанное с апоптозом или дегенерацией нейронов, и может вызывать различные заболевания, такие как болезнь Альцгеймера, боковой амиотрофический склероз, демиелинизирующие заболевания, диабетическая полиневропатия, синдром Дауна, ВИЧ невропатия, болезнь Хантингтона, множественная системная атрофия, болезнь Паркинсона, удар и ишемически-реперфузионное повреждение, таупатия и травматическое повреждение мозга. Также известно, что повышение активности антиокислительного фермента против реакционно-способных видов кислорода является одним из механизмов нейропротекции (Free radical Biology & Medicine, 33(2):182-191(2002)). Поэтому фармацевтическая композиция ингибирует окислительный стресс, вызывая снижение реакционно-способных видов кислорода, тем самым предотвращая апоптоз нейрона, и поэтому может применяться для предупреждения или лечения различных описанных здесь заболеваний.
Следовательно, фармацевтическая композиция может применяться в качестве нейропротекторных терапевтических средств, которые являются лекарственными или химическими средствами, предназначенными для защиты мозга или спинного мозга от повреждения вследствие ишемии, эпилептического припадка, конвульсий или травматических повреждений.
Фармацевтическая композиция в соответствии с данным изобретением может применяться для восстановления функций нервной ткани.
Термин "восстановление функций нервной ткани" в данном описании относится к восстановлению поврежденной нервной системы усилением образования новых синаптических соединений от нейронов. Восстановление функций нервной системы может означать восстановление дисфункции, вызванной поврежденными нейронами. Например, восстановление функций нервной системы может означать образование и рост нейритов из нервной клетки, которые необходимы для коммуникации с окружающими клетками, или повышение количества спинов.
Тот факт, что различные заболевания нервной системы могут быть предотвращены или лечиться восстановлением функций нервной ткани, хорошо известен в данной области. В Neurotoxicity Research, 2:71-84(2000) описана возможность предупреждения или лечения конкретных заболеваний, таких как болезнь Хантингтона, болезнь Паркинсона, боковой амиотрофический склероз и болезнь Альцгеймера, лекарственными средствами, применяемыми для восстановления функций нервной системы, и в WO 07/022182 описано, что заболевания, такие как болезнь Хантингтона и подобные, могут лечиться восстановлением функций центральной нервной системы.
Как описано выше, апоптоз нейронов или нейродегенерация вызывается различными нервными повреждениями и связан с различными нейродегенеративными заболеваниями, и таким образом, фармацевтическая композиция в соответствии с данным изобретением может предотвращать или лечить нейродегенеративные заболевания посредством ингибирования различных нервных повреждений.
Фармацевтическая композиция в соответствии с данным изобретением может применяться для предупреждения или лечения нейродегенеративных заболеваний или заболеваний, связанных с ишемией или реперфузией.
Примеры нейродегенеративных заболеваний, которые могут лечиться фармацевтической композицией, могут включать слабоумие, Болезнь Хантингтона, Болезнь Паркинсона и боковой амиотрофический склероз, но не ограничены ими. Кроме того, примеры заболеваний, связанных с ишемией или реперфузией, которые могут лечиться фармацевтической композицией, могут включать ишемический удар, преходящее нарушение мозгового кровообращения, ишемию миокарда, мышечную ишемию и ишемию, вызванную хирургическими вмешательствами, требующими длительного перекрытия потока крови к мозгу, но не ограничены ими.
Фармацевтическая композиция в соответствии с данным изобретением может применяться для предупреждения или лечения заболеваний, выбранных из группы, состоящей из удара, болезни Альцгеймера, болезни Хантингтона, болезни Паркинсона, болезни Пика, болезни Крейтцфельда-Якоба, комплекса Паркинсон-БАС-слабоумие, болезни Уилсона, рассеянного склероза, прогрессирующего надъядерного паралича, биполярных расстройств, связанных с невропатической болью, кортико-базальной дегенерации, шизофрении, синдрома гиперактивности с дефицитом внимания (ADHD), слабоумия, бокового амиотрофического склероза, болезни сетчатки, эпилепсии, апоплексии, преходящего нарушения мозгового кровообращения, ишемии миокарда, мышечной ишемии, ишемии, вызванной хирургическими вмешательствами, требующими длительного перекрытия потока крови к мозгу, повреждения головы, повреждения спинного мозга, гипоксии и депрессии.
Согласно одному из вариантов данного изобретения, представлен способ лечения заболеваний, связанных с апоптозом нейронов или нейродегенерацией, где способ включает введение субъекту фармацевтической композиции. Способ может включать способ ингибирования апоптоза нейронов или нейродегенерации, включающий введение субъекту фармацевтической композиции. Заболевания могут быть выбраны из группы, состоящей из удара, болезни Альцгеймера, болезни Хантингтона, болезни Паркинсона, болезни Пика, болезни Крейтцфельда-Якоба, комплекса Паркинсон-БАС-слабоумие, болезни Уилсона, рассеянного склероза, прогрессирующего надъядерного паралича, биполярных расстройств, связанных с невропатической болью, кортико-базальной дегенерации, шизофрении, синдрома гиперактивности с дефицитом внимания (ADHD), слабоумия, бокового амиотрофического склероза, болезни сетчатки, эпилепсии, апоплексии, преходящего нарушения мозгового кровообращения, ишемии миокарда, мышечной ишемии, ишемии, вызванной хирургическими вмешательствами, требующими длительного перекрытия потока крови к мозгу, повреждения головы, повреждения спинного мозга, гипоксии и депрессии.
Контактирование может осуществляться in vitro или in vivo, и при осуществлении контактирования in vivo способ может включать введение фармацевтической композиции субъекту.
Субъектом может быть клетка, ткань, орган или индивидуум. Кроме того, введение может проводиться растворением фармацевтической композиции в подходящем буфере с последующим прямым контактированием клетки, ткани или органа с полученным раствором, или парентеральным введением индивидууму. Подробное описание фармацевтической композиции и способа ее введения, применяемых в способе лечения, описанном выше, также представлены выше, и поэтому не представлены здесь во избежании усложнения описания.
Субъекты, которым вводят фармацевтическую композицию, могут включать всех животных. Например, животными могут быть человек, собака, кошка или мышь.
Один или более вариантов данного изобретения более подробно описаны в представленных ниже примерах. Эти примеры представлены только в иллюстративных целях и не ограничивают объем одного или более вариантов данного изобретения.
Краткое описание чертежей
Представленные выше и другие признаки и преимущества данного изобретения станут более очевидными при подробном описании примерных вариантов осуществления со ссылкой на прилагаемые чертежи, в которых:
На ФИГ.1 представлен график, показывающий степень болезни Паркинсона у индуцированных MPTP обезьян, которым вводят CBI, согласно одному из вариантов данного изобретения;
На ФИГ.2 представлены микроскопические изображения, показывающие, существует или нет допаминовый транспортер в центральном полосатом теле и хвостовом полосатом теле у индуцированных MPTP обезьян, которым вводят CBI, согласно одному из вариантов данного изобретения;
На ФИГ.3 представлен график, показывающий результаты теста подвешивания за хвост у индуцированных MPTP обезьян, которым вводят CBI, согласно одному из вариантов данного изобретения;
На ФИГ.4 представлен график, показывающий концентрацию допамина в полосатом теле у индуцированных MPTP мышей, которым вводят CBI, согласно одному из вариантов данного изобретения;
На ФИГ.5 представлен график, показывающий степень снижения нейронов в черной субстанции у индуцированных MPTP мышей, которым вводят CBI, согласно одному из вариантов данного изобретения;
На ФИГ.6 представлен график, показывающий степень снижения нейронов в компактной черной субстанции у индуцированных 6-OHDA крыс, которым вводят CBI, полученный иммуностереохимическим окрашиванием с применением тирозингидроксилазы в качестве антитела, и окрашиванием крезил-виолетом, согласно одному из вариантов данного изобретения;
На ФИГ.7 представлены изображения, показывающие, восстанавливается или нет после повреждения полосатое тело у мышей, индуцированных малонатом, которым вводят CBI, согласно одному из вариантов данного изобретения;
На ФИГ.8 представлен график, показывающий степень восстановления после повреждения поврежденного полосатого тела у мышей, индуцированных малонатом, которым вводят CBI, согласно одному из вариантов данного изобретения;
На ФИГ.9 представлен график, показывающий степень апоптоза в SH-SY5Y клетках с дефицитом MAO-B, обработанных CBI, согласно одному из вариантов данного изобретения;
На ФИГ.10 представлен график, показывающий результаты измерения количества мРНК Bcl-2 в SH-SY5Y клетках с дефицитом MAO-B, обработанных CBI, согласно одному из вариантов данного изобретения;
На ФИГ.11 представлены изображения, показывающие результаты измерения количества белков Bcl-2 и Bcl-xL в SH-SY5Y клетках с дефицитом MAO-B, обработанных CBI, согласно одному из вариантов данного изобретения;
На ФИГ.12 представлен график, показывающий результаты измерения количества мРНК BDNF в SH-SY5Y клетках с дефицитом MAO-B, обработанных CBI, согласно одному из вариантов данного изобретения;
На ФИГ.13 представлен график, показывающий результаты измерения количества мРНК GDNF в SH-SY5Y клетках с дефицитом MAO-B, обработанных CBI, согласно одному из вариантов данного изобретения;
На ФИГ.14 представлен график, показывающий результаты измерения количества мРНК NGF в SH-SY5Y клетках с дефицитом MAO-B, обработанных CBI, согласно одному из вариантов данного изобретения;
На ФИГ.15 представлен график, показывающий результаты измерения количества мРНК NGF у мышей, которым вводят CBI, согласно одному из вариантов данного изобретения;
На ФИГ.16 представлен график, показывающий потенциал мембраны митохондрии в SH-SY5Y клетках с дефицитом MAO-B, обработанных CBI и MPP+, согласно одному из вариантов данного изобретения;
На ФИГ.17 представлены изображения, показывающие результаты измерения количества цитоплазмического цитохрома c в SH-SY5Y клетках с дефицитом MAO-B, обработанных CBI и MPP+, согласно одному из вариантов данного изобретения;
На ФИГ.18 представлен график, показывающий результаты измерения активности каспазы-3 в SH-SY5Y клетках с дефицитом MAO-B, обработанных CBI и MPP+, согласно одному из вариантов данного изобретения;
На ФИГ.19 представлены микроскопические изображения реакционно-способных видов кислорода в SH-SY5Y клетках с дефицитом MAO-B, обработанных CBI и MPP+, согласно одному из вариантов данного изобретения;
На ФИГ.20 представлен график, показывающий, что реакционно-способные виды кислорода существуют в SH-SY5Y клетках с дефицитом MAO-B, обработанных CBI и MPP+, согласно одному из вариантов данного изобретения;
На ФИГ.21 представлены графики, показывающие результаты измерения активности каталазы, супероксиддисмутазы (SOD) и глутатион-пероксидазы (GPx) в SH-SY5Y клетках с дефицитом MAO-B, обработанных CBI и MPP+, согласно одному из вариантов данного изобретения;
На ФИГ.22 представлены графики, показывающие результаты измерения активности каталазы, SOD и GPx в полосатом теле и черной субстанции у мышей, которым вводят CBI, согласно одному из вариантов данного изобретения;
На ФИГ.23 представлены микроскопические изображения и аналитический график, показывающий, восстановлен ли нейрит у мышей, индуцированных MPTP, которым вводят CBI, согласно одному из вариантов данного изобретения; и
На ФИГ.24 представлены микроскопические изображения и аналитический график, показывающий, восстановлен ли спин у мышей, индуцированных MPTP, которым вводят CBI, согласно одному из вариантов данного изобретения;
Способ осуществления данного изобретения
Далее более подробно представлены варианты, примеры которых проиллюстрированы на прилагаемых чертежах, где ссылочные числовые обозначения совпадают с обозначением этих же элементов в описании. В связи с этим представленные варианты могут иметь различные формы и не должны рассматриваться как ограничивающие представленным описанием. Следовательно, варианты представлены здесь, со ссылками на фигуры, только для объяснения некоторых аспектов данного изобретения.
Пример 1: Получение 3-(4-бензилоксифенил)изоксазол-5-илметилового эфира карбаминовой кислоты (Формула II)
Формула II
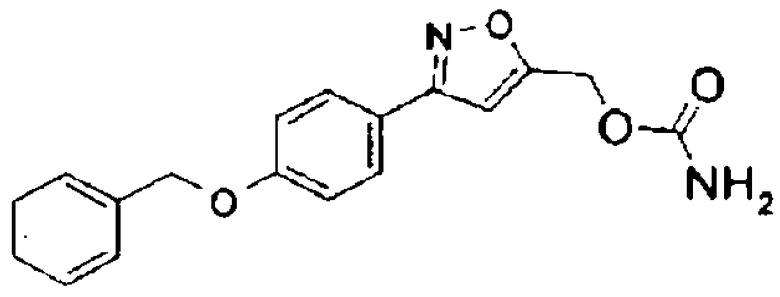
1.1. Синтез оксима 4-бензилоксибензальдегида
4,24 г 4-бензилоксибензальдегида (20 ммоль) растворяют в 0,2 M смешанном растворе этанола и воды (3:1, 100 мл), затем перемешивают. Добавляют 2,78 г NH2OH-HCl (40 ммоль) и 2,46 г ацетата натрия (30 ммоль) и перемешивают при комнатной температуре в течение примерно 30 минут. Затем завершение реакции подтверждают жидкостной хроматографией, и воду и этанол удаляют дистилляцией при пониженном давлении с получением бледно-желтого твердого соединения. Бледно-желтое твердое соединение три раза экстрагируют водой и этилацетатом, органический слой растворителя сушат при пониженном давлении с получением неочищенного продукта, и неочищенный продукт затем очищают раствором гексана/этилацетата (10:1) с получением белого твердого соединения. Полученное твердое соединение применяют в последующей реакции без дополнительной очистки.
1.2. Синтез [3-(4-бензилоксифенил)изоксазол-5-ил]метанола
2,27 г оксима 4-бензилоксибензальдегида (10 ммоль; 92% чистота) растворяют в 40 мл метиленхлорида (0,25 M), и затем к полученному раствору добавляют 1,77 мл пропаргилового спирта (30 ммоль). Затем 13,7 мл 10% NaOCl (20 ммоль) по каплям добавляют очень медленно к полученному раствору с применением капельной воронки при 0°C. После завершения добавления NaOCl полученную смесь перемешивают в течение примерно 5 часов при медленном повышении температуры до комнатной температуры. Затем завершение реакции подтверждают жидкостной хроматографией, полученный продукт дистиллируют при пониженном давлении для выпаривания метиленхлорида, к остатку добавляют 200 мл воды, и полученное твердое вещество затем фильтруют. Отфильтрованное соединение промывают большим количеством воды и затем промывают диэтиловым эфиром с получением твердого соединения. Полученное твердое соединение очищают раствором этилацетата/гексана (1:2) с получением белого твердого вещества [3-(4-бензилоксифенил)изоксазол-5-ил]метанола (выход: 2,5 г).
1H-ЯМР (CDCl3, 200 МГц) δ 7,7 (д, 2H), 7,4 (м, 4H), 7,1 (д,2H), 6,5 (с, 1H), 5,1 (с, 2H), 4,8 (с, 2H).
1.3. Синтез 3-(4-бензилоксифенил)изоксазол-5-илметилового эфира карбаминовой кислоты
1,04 мл (12 ммоль) хлорсульфонилизоцианата медленно добавляют при -78°C к раствору ТГФ (50 мл, 0,2 M) и [3-(4-бензилоксифенил)изоксазол-5-ил]метанола (2,813 г, 10 ммоль), помещенному в 250 мл колбу. Затем полное удаление исходного материала подтверждают жидкостной хроматографией, затем к полученному реакционному раствору добавляют воду. Через один час полученный раствор дистиллируют при пониженном давлении для выпаривания ТГФ, к полученному раствору добавляют 100 мл воды и полученное твердое вещество фильтруют. Отфильтрованное твердое вещество промывают 100 мл воды и раствор этилацетата/гексана (1:2), соответственно, и сушат с получением 3,4 г неочищенного продукта (чистота: 95,9%). Неочищенный продукт очищают раствором этилацетат/гексан/метиленхлорид (1:4:1), содержащим 1% метанол с получением 2,743 г 3-(4-бензилоксифенил)изоксазол-5-илметилового эфира карбаминовой кислоты (CBI) с чистотой 99%.
1H-ЯМР (CDCl3, 200 МГц) δ 7,7 (д, 2H), 7,4 (м, 4H), 7,1 (д, 2H), 6,6 (с, 1H), 5,2 (с, 2H), 5,1 (с, 2H), 4,8 (шир.с, 2H)
Пример 2: Подтверждение нейропротекторного действия CBI у обезьян, индуцированных MPTP
Описано, что 1-метил-4-фенил-1,2,3,6-тетрагидропиридин (MPTP) вызывает клинические, биохимические и патологические реакции, похожие на реакции у пациентов с болезнью Паркинсона, и он известен как нейротоксин, который широко применяют для получения животной модели болезни Паркинсона у грызунов и приматов (J. Neural Transm., 103:987-1041(1996); Neurotoxicol. Teratol. 24:607-620(2002)). MPTP превращают в 1-метил-4-фенилпиридиний (MPP+) моноаминоксидазой (MAO)-B, и MPP+ имеет высокое сродство с допаминовым транспортером (DAT) и вызывает дисфункцию митохондрии и окислительный стресс, вызывая апоптоз допаминергических нейронов, который вызывает образование допамина (J.N eurochem., 61:1191-1206(1993); J. Neural Transm., 103:987-1041(1996); Mov. Disord., 13:35-38(1998); Restor. Neurol. Neurosci., 16:135-142(2000)).
Макак (n=35, возраст 3-4 года) используют в качестве экспериментальной модели. Макак делят на три группы и 0,2 мг/кг MPTP вводят каждой группе (один раз в сутки каждый день до тех пор, пока значение болезни Паркинсона не достигнет 8 или в течение 14 дней) внутривенной инъекцией. На следующий день после 14-дневого введения наполнитель (контроль), 1 мг/кг CBI и 1 мг/кг разагилина (полученного с применением способа получения R(+)-N-пропаргил-1-аминоинданразагилина, описанного в заявке на патент США № 5457133) соответственно вводят перорально трем группам в течение 4 недель, и измеряют изменения значения болезни Паркинсона. Кроме того, для подтверждения действия CBI в качестве агониста допамина, транспортеры допамина, существующие в медиальном полосатом теле и хвостовом полосатом теле, взятые из модели обезьян, индуцированных MPTP, подвергают анализу связывания транспортера допамина.
Пример 2-1: Измерение степени болезни Паркинсона
Степень болезни Паркинсона измеряют, анализируя записанное на видео поведение каждой группы обезьян, основываясь на четырех стандартах: a) интервал движения, b) гипокинезия, c) степень аномальности позы, и d) дрожание. Степень болезни Паркинсона оценивают по сумме (4-интервал движения) + гипокинезия + степень аномальности позы + дрожание. Следовательно, максимальное значение степени болезни Паркинсона составляет 10. Степень болезни Паркинсона измеряют в течение 10 минут каждые 30 минут в течение 2 часов. То есть, максимальное значение наивысшей степени болезни Паркинсона составляет 40. Способ измерения, основанный на четырех стандартах, описан ниже, и указанные ниже результаты оценки представляют типовое поведение, отмеченное в период наблюдения:
a) Степень интервала движения: 0 = нет движения; 1 = только движение головы; 2 = движение головы, конечностей и/или тела без упражнений не менее 30% времени наблюдения; 3 = ходьба/ходьба или лазание по стене клетки не более 30% времени наблюдения; 4 = ходьба/ходьба или лазание по стене клетки не менее 30% времени наблюдения.
b) Степень гипокинезии: 0 = нормальная скорость движения и нормальное начало движения; 1 = незначительное замедление движения; 2 = среднее или медленное движение, затрудненное начало и поддержание движения, видимая скованность тела; 3 = невозможность упражнений, непрерывная скованность тела, не дающая возможность двигаться.
c) Степень аномальности позы: 0 = нормальные хорошие позы, возможность поднимания головы, нормальное равновесие; 1 = согнутое тело, возможность поднимать голову; 2 = согнутое тело, невозможность поднимать шею и голову, потеря равновесия.
d) Степень дрожания: 0 = отсутствует; 1 = существует.
В результате измерения степени болезни Паркинсона было подтверждено, что группа обезьян, которым вводили CBI, демонстрировала значительно пониженную степень болезни Паркинсона по сравнению с группой обезьян, которым вводили разагилин, известный как лекарственное средство, применяемое в лечении болезни Паркинсона (ссылка на ФИГ.1).
Пример 2-2: Анализ связывания транспортера допамина
У каждой группы обезьян берут мозг, оттуда отделяют ствол мозга, затем полушарие головного мозга делят на два по средней линии. Ткань погружают в изопентан при -45°C, немедленно замораживают и затем хранят при -80°C. Коронарный срез полушария головного мозга получают до толщины 20 в криостате при -17°C, затем размораживают. Размороженный препарат устанавливают на предметное стекло, покрытое желатином, сушат на нем и затем хранят при -80°C.
Для связывания транспортера допамина проводят мечение радиоактивным элементом [125I]-(E)-N-(3-йодпро-2-фенил)-2βкарбоксиметил-3β(4'-метилфенил)нортропаном (PE2I) с применением станнилового предшественника в соответствии с обычным методом, применяемым для подтверждения допаминового окончания нерва (D. Guilloteau et al.,1998). Препарат очищают с получением формы без добавления носителя [125I]PE2I, имеющей активность 2000 Ки/ммоль. Коронарный срез полушария головного мозга инкубируют со 100 пМ [125I]PE2I в pH 7,4 фосфатном буфере (NaH2PO4 10,14 мМ, NaCl 137 мМ, KCl 2,7 мМ, KH2PO4 1,76 мМ) при 25°C в течение 90 минут с применением способа, описанного в известном уровне техники (S. Chalon et al., 1999; E. Bezard et al., 2001). Соседний срез инкубируют в присутствии 100 кокаина (Sigma, StLouis, MO), подтверждая неспецифическое связывание. После инкубирования срез дважды промывают фосфатным буфером при 4°C в течение 20 минут, и промывают дистиллированной водой при 4°C в течение одной секунды. Полученный срез сушат при комнатной температуре и затем проявляют на чувствительной к β излучению пленке (Hyperfilm β max, Amersham, Buckingamshire, UK) вместе с калиброванными [125I]-микрообъектами (Amersham) в рентгеновских кассетах для пленок в течение 3 дней, измеряя таким образом связывание радиоактивности с желаемыми областями.
Как показано на ФИГ.2, группа обезьян, которым вводили CBI, демонстрирует большее количество транспортеров допамина в срединном полосатом теле и хвостовом полосатом теле, и демонстрируют значительно большее количество транспортеров допамина, чем то, которое демонстрирует группа обезьян, которым вводили такое же количество разагилина, как и CBI. Эти результаты показывают, что CBI эффективно ингибирует апоптоз допаминергических нейронов, и поэтому потеря транспортеров допамина из-за MPTP относительно ниже, по сравнению с контролем или группой обезьян, которым вводили разагилин. Результаты подтверждают, что CBI способен осуществлять нейропротекцию, включая ингибирование развития болезни Паркинсона.
Пример 3: Нейропротекторное действие CBI на модели мышей, индуцированных MPTP
C57BL/6 мышей (n=94, возраст 8 недель; самцы) используют в экспериментальное модели (субштамм: C57BL/6NCrljBgi, ORIENT BIO INC.). 30 мг/кг MPTP вводят мышам внутрибрюшинной инъекцией один раз в сутки в течение 4 дней. Через 4 дня после последнего дня введения мышей делят на три группы, и наполнитель (контроль), 1 мг/кг CBI и 1 мг/кг разагилина соответственно вводят перорально трем группам один раз в сутки в течение 10 дней. На следующий день после последнего дня введения проводят тест подвешивания за хвост (TST) для каждой группы, и мозговую ткань из полосатого тела и черной субстанции берут у каждой группы мышей для измерения концентраций доапмина и продукта его метаболизма в полосатом теле, и степени снижения нейронов в черной субстанции.
Пример 3-1: Анализ поведения мышей в тесте подвешивания за хвост
Тест подвешивания за хвост проводят для измерения степени причинной зависимости поведенческого нарушения от введения MPTP и лекарственных средств. TST проводят следующим образом: через 7 дней после соответствующего введения описанных выше соединений трем группам, круглый стержень из нержавеющей стали шириной 1 см закрепляют в клетке шириной 16 см и высотой 40 см на высоте 35 см, и ее левую и правую сторону покрывают черным деревом. Время движения мыши измеряют в течение 6 минут во второй единице, и оценивают эффективность соединений.
Результаты анализа TST подтверждают, что хотя группа мышей, которым давали MPTP, демонстрирует значительное нарушение поведения, группа мышей, индуцированных MPTP, которым вводили CBI демонстрируют такую же степень нарушения поведения, как и нормальные мыши, и демонстрируют превосходную способность к восстанавливанию поведения, по сравнению с группой мышей, индуцированных MPTP, которым вводили разагилин (ссылка на ФИГ.3).
Пример 3-2: Измерение количеств допамина и продукта его метаболизма в полосатом теле
Изменение количества допамина и продукта его метаболизма в полосатом теле вследствие введения MPTP и описанных выше соединений измеряют высокоэффективной жидкостной хроматографией (ВЭЖХ). Через 7 дней после соответствующего введения соединений, описанных выше, трем группам, мышей в каждой группе умерщвляют смещением шейных позвонков и сразу же берут мозговую ткань. Полосатое тело собирают с мозговой ткани, 0,5 мл разбавителя для анализа ВЭЖХ (0,1 M HClO4, 0,1 мМ EDTA) добавляют к полосатому телу, и гомогенат ткани готовят с применением ультразвукового процессора. Гомогенат центрифугируют при 12000 об/мин в течение 15 минут, и надосадочную жидкость фильтруют через нитроцеллюлозный фильтр (0,2 мкм, Millipore). Для ВЭЖХ анализа применяют колонку HR-80 (80 мм × 4,6 мм, размер частиц: 3 мкм, ESA, USA), скорость потока подвижной фазы (0,07 M одноосновный фосфат натрия, 1 мМ октасульфоновой кислоты натрия, 0,1 мкМ EDTA, 5% ацетонтрила, pH 3,2) выдерживают на уровне 0,7 мл/мин, и электродный потенциал электрохимического детектора (Coulochem III, ESA, USA) составляет E1 = -100 мВ, E2 = 350 мВ.
В результате анализа концентрации допамина в полосатом теле в течение эксперимента было подтверждено, что группа мышей, которой вводили разагилин, демонстрируют восстановление концентрации допамина на примерно 40% по сравнению с нормальными мышами, в то время как группа мышей, индуцированных MPTP, которым вводят CBI, демонстрируют восстановление концентрации допамина на примерно 70% по сравнению с нормальными мышами (ссылка на ФИГ.4).
Пример 3-3: Иммунохимическое окрашивание с применением антитела для тирозингидроксилазы
Изменение экспрессии антитела против тирозингидроксилазы в полосатом теле и черной субстанции в результате введения описанных выше соединений измеряют иммунохимическим окрашиванием. Каждую группу мышей анестезируют фенобарбиталом натрия (50 мг/кг), вскрывают грудную клетку и 200 мл 0,1 M PBS (pH 7,4) вливают в сердце, удаляя кровь в кровяных сосудах. После полного удаления крови 250-300 мл 4% фиксирующего раствора параформальдегида/PBS вливают в сердце, берут мозг, и мозг подвергают вторичной фиксации фиксирующим раствором параформальдегида/PBS в замороженном состоянии в течение 24 часов. Затем ткань мозга тщательно промывают PBS для удаления фиксирующего раствора и для предотвращения образования ледяных кристаллов во время замораживания, полученную мозговую ткань помещают в 30% раствор сахарозы и выдерживают в нем до погружения. Полученную ткань заливают заливкой для замораживания (OCT соединение) и замораживают при -40°C, затем готовят коронарный срез средней области мозга, содержащей полосатое тело и черную субстанцию, толщиной 40 мкм с применением Криостата (Reichert Frigocut model 2000). Коронарный срез выдерживают в 3% H2O2/PBS в течение 30 минут, затем выдерживают в 0,1 M PBS, содержащем 0,3% Triton X-100 и 3% альбумина бычьей сыворотки в течение 30 минут. Для селективного окрашивания клеток, содержащих допамин, срез подвергают взаимодействию с анти-мышиным моноклональным TH (Chemicon International, Temecula, CA; 1:500) в качестве первичного антитела при комнатной температуре в течение ночи, и биотинилированный козий анти-мышиный IgG (Vector Lab, Burlingame, CA, 1:200) используют в качестве вторичного антитела. Затем авидин-биотиновое связывание индуцируют с помощью набора Vectastain elite ABC (Vector Lab, Burlingame, CA), и цвет проявляют на ткани с применением 3,4-диаминобензидина (ДАБ). Полученную ткань помещают в PBS, помещают на предметное стекло, и препарат сушат и покрывают покровным стеклом. Черную субстанцию средней области мозга в полученном наблюдают с применением микроскопа, оборудованного цифровой камерой (Olympus BX-60, Olympus Optical, Tokyo, Japan) с увеличением 200×, клетки, которые демонстрируют положительную реакцию на антитело против тирозингидроксилазы, наблюдают и записывают, и статистический анализ (One-way ANOVA) проводят с применением программы Graph pad Prism 4.
В результате анализа степени снижения нейронов в черной субстанции иммунохимическим окрашиванием с применением антитела против тирозингидроксилазы, как описано выше, группа мышей, индуцированных MPTP, которым вводили CBI, демонстрирует такую же степень снижения нейронов в черной субстанции, как и группа мышей, индуцированных MPTP, которым вводили разагилин (ссылка на ФИГ.5). Результаты подтверждают, что CBI действует в качестве агониста допамина, и тем самым способно осуществлять нейропротекцию, и поэтому обладает действием, превосходящим действие разагилина, который широко известен как агонист допамина.
Пример 4: Подтверждение нейропротекторного действия CBI с применением модели крыс, индуцированных 6-OHDA
6-Гидроксидопамин (6-OHDA) известен как нейротоксин, который повышает образование гидроксильных радикалов, тем самым вызывая дегенерацию нейронов в черной субстанции и полосатом теле. Гидроксильные радикалы быстро разрушают концевую область нейрона (J. Neural. Transm., 103:987-1041(1996); J. Neurosci., 19:1284-1293(1999)), тем самым вызывая постепенную потерю клеток в компактной части черной субстанции (SNpc), и известно. что такая потеря похожа на постепенную дегенерацию черной субстанции и полосатого тела, наблюдаемую у пациентов с болезнью Паркинсона на ранних стадиях (Brain Res., 26:301-307(1991); Neurosci., 59:401-415(1994); Neurosci., 67:631-647(1995); Neurosci., 72:641-653(1996)).
Крыс Wistar от ORIENT BIO INC. (наполнитель и CBI n=7, разагилин n=6; возраст 6 недель; 20 самцов крыс) используют в качестве экспериментальной модели. Одностороннюю инъекцию 3 мкл раствора, содержащего 20 мкг/мкл 6-OHDA, делают на полосатое тело каждой крысе (положение: спереди -1,0 мм, сзади -3,0 мм, от задней части живота -5,0 мм), тем самым вызывая дегенерацию нейронов в полосатом теле. Крыс делят на три группы, и наполнитель (контроль), 1 мг/кг CBI и 1 мг/кг разагилина, соответствено, вводят перорально трем группам за 1 час до введения 6-OHDA и один раз через день в течение 6 недель. Через 4, 5 и 6 недель после последнего дня введения для каждой группы проводят тест на вызванное апоморфином вращение. Тест на вызванное апоморфином вращение проводят следующим образом: 0,5 мг/кг апоморфина вводят каждой группе крыс внутрибрюшинной инъекцией, каждую группу крыс помещают в роторную камеру, и роторное движение записывают в течение 45 минут, таким образом измеряют количество оборотов в минуту для каждой группы крыс для определения среднего значения. Кроме того, после теста на вызванное апоморфином вращение, проводимого через 6 недель после последнего дня введения, каждую группу крыс умерщвляют, таким образом подтверждая степень снижения нейронов в компактной части черной субстанции, используя иммуногистохимическое окрашивание с применением антитела против тирозингидроксилазы и окрашивания крезил-виолетом. Как описано в Примере 3-3 выше, получают коронарный срез средней области мозга, содержащей черную субстанцию, срез помещают в PBS, полученную часть закрепляют на покрытом силаном предметном стекле и сушат, и предметное стекло помещают в ксилол, 100% спирт, 95% спирт, 70% спирт и дистиллированную воду на 5 минут, 2 минуты, 1 минуту, 1 минуту и 2 минуты, соответственно. Затем полученное предметное стекло погружают в 1% раствор крезил-виолета на 5 минут и промывают дистиллированной водой, 70% спиртом, 95% спиртом, 100% спиртом и ксилолом в течение 2 минут, 1 минуту, 1 минуту, 2 минуты и 5 минут, соответственно, и предметное стекло накрывают покровным стеклом и наблюдают в микроскопе, оборудованном цифровой камерой (Olympus BX-60, Olympus Optical, Tokyo, Japan). Черную субстанцию средней области мозга наблюдают с увеличением 200×, клетки, которые демонстрируют положительную реакцию на крезил-виолет, наблюдают и записывают, и затем проводят статистический анализ (One-way ANOVA) с использованием программы Graph pad Prism 4.
Как показано на ФИГ.6, подтверждено, что группа крыс, индуцированных 6-OHDA, которым вводят CBI, демонстрирует значительно меньшую степень снижения нейронов, и превосходит по действию группу крыс, индуцированных 6-OHDA, которым вводили разагилин.
Пример 5: Подтверждение нейропротекторного действия CBI с применением модели мышей, индуцированных малонатом
Малонат является обратимым ингибитором сукцинатдегидрогеназы, которая является ферментом митохондрии, и известно, что он ингибирует систему транспорта электрона в митохондрии, вызывая дегенерацию эксайтотоксичных нейронов, или повышает высвобождение допамина из полосатого тела, вызывая потерю полосатого тела. Дефицит биоэнергии в митохондрии связан с патологическим явлением различных нейродегенеративных заболеваний, таких как болезнь Паркинсона, болезнь Хантингтона, болезнь Альцгеймера и боковой амиотрофический склероз (Ann. Neurol., 58:495-505(2005); Nat. Rev. Neurosci., 7:278-294(2006)), и инъекция малоната в полосатое тело животных вызывает потерю, такую же, которая наблюдается при очаговой ишемии или болезни Хантингтона (Experimental Neurology, 178:301-305(2002)). Это вызывает метаболический стресс в некоторых группах нейронов, что вызывает снижение количества допамина и в теле клетки допамина черной субстанции, и в полосатом теле (J. Neurochem., 61:1147-1150(1993); Brain Res., 773:223-226(1997); Neuroscience, 96:309-316(2000)).
Мышей ICR (n=34, возраст 10 недель; самцы) используют в качестве экспериментальной модели. 5 мл/кг эквитезина вводят каждой мыши через внутрибрюшинную инъекцию для анестезии, два выравнивателя стереотактического инструмента устанавливают на 0 мм от обоих наружных слуховых проходов, и череп мышей перфорируют в стереотактическом инструменте. 0,2 мг/мл аскорбиновой кислоты применяют в качестве контроля, и 2,4 мкмоль/2 малоната вводят поврежденной группе и группе, обработанной соединением, 0,5 мм спереди (AP) и 1,2 мм вбок от полосатого тела с правой стороны (bregma) и 3,1 мм вниз от твердого вещества с применением шприца Гамильтона (10 мкл, 26G игла) со скоростью 1 мкл/мин. Группы, которым вводят малонат, делят на две группы, и вводят 0,5 мл/кг наполнителя (n=12) и 5 мг/кг CBI (n=14), соответственно, двум группам внутрибрюшинной инъекцией за 2 часа до операции, и снова вводят через 1 час, 1 день, 2 дня и 3 дня после операции, т.е. всего пять раз. Малонат вводят в полосатое тело мыши, мышей умерщвляют через 3 дня, и мозг вынимают из мыши для получения среза. Затем срез окрашивают хлоридом 2,3,5-трифенилтетразолия (TTC).
Как показано на ФИГ.7 и 8, было подтверждено, что CBI ингибирует апоптоз нейронов в полосатом теле, вызванный малонатом, тем самым значительно снижая поврежденные области полосатого тела. Таким образом, результаты показывают, что CBI облегчает апоптоз нейронов, вызванный повреждением митохондрии.
Пример 6: Подтверждение нейропротекторного действия CBI анти-апоптозом нейронов
Многие нейродегенеративные заболевания, такие как удар, повреждение мозга, повреждения спинного мозга, боковой амиотрофический склероз, болезнь Хантингтона, болезнь Альцгеймера и болезнь Паркинсона характеризуются апоптозом нейронов (The New England Journal of Medicine, 348:1365(2003)), и известно, что хронические нейродегенеративные заболевания вызываются индукцией путей апоптоза несколькими внутренними или внешними факторами. Для объяснения биохимических и молекулярных биологических изменений, возникающих при апоптозе нейронов, было предпринято несколько подходов к поиску продуктов, демонстрирующих разнонаправленные механизмы в нескольких стадиях апоптоза нейронов или лечения нейропротекторными лекарственными средствами (CNS drugs, 19:723(2005); Nat. Rev. Neurosci., 7:295(2006)).
Согласно ФИГ.6-10, подтверждается, что CBI ингибирует апоптоз нейронов, вызванный несколькими внутренними или внешними факторами, тем самым оказывая терапевтическое действие при нейродегенеративных заболеваниях.
В этом варианте используют MAO-B-дефицитные SH-SY5Y клетки нейробластомы человека (Korean Cell Line Bank). Апоптоз клеток нейробластомы человека вызывают сывороточным голоданием. MAO-B-дефицитные SH-SY5Y клетки нейробластомы человека, культивированные в нормальной среде, распределяют на 6-луночный планшет в концентрации 1,8×105 клеток/лунку и инкубируют в течение 1 дня, среду меняют на не содержащую сыворотку среду, содержащую CBI (0,1, 1 и 10), не содержащую сыворотку среду, содержащую разагилин (0,1, 1 и 10), или не содержащую сыворотку среду, не содержащую ни CBI, ни разагилин, и клетки далее инкубируют в 5% CO2 при 37°C в течение 48 часов. Затем количество мертвых клеток представляют как процент по сравнению с контролем, в котором не вызывали апоптоз нейронов.
Как показано на ФИГ.9, подтверждают, что когда в нейрон с вызванным апоптозом вводят CBI, степень апоптоза нейронов снижается. В частности, подтверждается, что когда в нейрон с вызванным апоптозом вводят 10 CBI, он демонстрирует более высокую степень снижения апоптоза нейронов, т.е. примерно 33%, по сравнению с вариантом, в котором в нейрон с вызванным апоптозом вводят 10 разагилина.
Множество белков трансдукции сигнала вовлечено в процесс медиирования или ингибирования апоптоза, и их типовые примеры включают белки семейства генов Bcl-2 (Journal of Bioenergetics and Biomembranes, 37:179-190(2005); J. Cell Mol. Med., 7:249-257(2003); Genes and Development, 13:1899-1911(1999)). Таким образом, 0,1, 1 и 10 CBI или 0,1, 1 и 10 разагилина, соответственно, добавляют в клетки с вызванным апоптозом, как описано выше, полученные клетки далее инкубируют в 5% CO2 при 37°C в течение 24 часов, мРНК экстрагируют из культивированных клеток или из них получают экстракт клеток, измеряют количество Bcl-2 мРНК и количество белков Bcl-2 и Bcl-xL. Количество Bcl-2 мРНК измеряют ОТ-ПЦР в режиме реального времени, и количества белков Bcl-2 и Bcl-xL измеряют вестерн-блоттингом.
Общее РНК клеток SH-SY5Y экстрагируют с применением RNeasy MiniKit (Qiagen) после обработки клеток SH-SY5Y CBI или разагилином в не содержащей сыворотку среде в течение 24 часов. 2 мкг общей РНК подвергают обратной транскрипции с применением набора High Capacity cDNA Reverse Transcription Kit (Applied Biosystems), и проводят ПЦР в режиме реального времени (Applied Biosystems, 7500 Real Time PCR SYSTEM), используя пробы TaqMan (Applied Biosystems, USA) для Bcl-2. В качестве внутреннего контроля амплифицируют мРНК для 18SRNA. Относительный количественный анализ уровней мРНК целевых генов определяют методом ddCt (Takekawa, 1998).
Для вестерн-блоттинга клетки SH-SY5Y лизируют буфером RIPA (50 мМ Трис-Cl pH 7,4, 1% NP-40, 0,25% деоксихолата натрия, 0,1% SDS, 150 мМ NaCl, 1 мМ EDTA) и затем центрифугируют с получением экстракта клеток. Затем экстракт клеток количественно анализируют, такое же количество экстракта клеток загружают в гель SDS-PAGE с последующим электрофорезом, гель переносят на нитроцеллюлозную мембрану, мембрану подвергают блоттингу с применением антитела против Bcl-2 (Cat#: 2872, Cell Signaling, USA) и антитела против Bcl-xL (Cat#: 2762, Cell Signaling, USA), затем соответственно разводят до 1:5000 с применением хорошо известного в данной области техники метода, и количество экспрессии белков Bcl-2 и Bcl-xL подтверждают с применением набора ECL (Amersham Pharmacia). Антитело против β-актина (Cat#: A2228, Sigma, USA) применяют в качестве контроля, и подтверждают количество экспрессии β-актинового белка.
Как показано на ФИГ.10 и 11, было подтверждено, что в случае введения CBI в нейроны с вызванным апоптозом, количество мРНК белка Bcl-2, имеющего анти-апоптозное действие, в 1,5-2 раза больше, чем в контроле, и количество белка Bcl-2 также в 1,5-2 раза больше, чем в контроле. Кроме того, было подтверждено, что количество Bcl-xL, который является другим анти-апоптозным белком, также больше, чем в контроле. Результаты подтверждают, что CBI обладает действием на ингибирование апоптоза нейронов и, таким образом, обладает нейропротекторным действием.
Пример 7: Подтверждение нейропротекторного действия CBI, вызывая экспрессию нейротрофического фактора
Нейротрофический фактор является белком, который играет критическую роль в развитии, регенерации и восстановлении нейронов, и примеры нейротрофического фактора включают нейротрофический фактор головного мозга (BDNF), нейротрофический фактор глиальной клеточной линии (GDNF) и фактор роста нервов (NGF). Индукция нейротрофического фактора позволяет ингибировать апоптоз нейронов (Nature medicine, 15:331-337(2009); Brain Research Bulletin, 57:817-822(2002); The Journal of Neuroscience, 21:8108-8118(2001); The Journal of Pharmacology and Experimental Therapeutics, 322:59-69(2007); TRENDS in Pharmacological Sciences, 27:619-625(2006)).
Применяя способ из Примера 6, 0,1, 1 и 10 CBI или 0,1, 1 и 10 разагилина, соответственно, добавляют к клеткам с индуцированным апоптозом, полученные клетки далее инкубируют в 5% CO2 при 37°C в течение 24 часов, мРНК экстрагируют из культивированных клеток и измеряют количества мРНК BDNF, GDNF и NGF с помощью ОТ-ПЦР в режиме реального времени.
Как показано на ФИГ.12-14, было подтверждено, что при обработке нейрона с индуцированным апоптозом CBI, количества мРНК BDNF, GDNF и NGF, соответственно, в 1,5-2 раза, 4-8 раз и 2-2,5 раза больше, чем в контроле.
Кроме того, для подтверждения in vivo того, индуцируется или нет нейротрофический фактор обработкой CBI, используют мышей C57BL/6 (n=12, возраст 8 недель, самцы, ORIENT BIO INC.). Мышей делят на три группы (n=4 для каждой группы), и наполнитель (контроль), 1 мг/кг CBI и 1 мг/кг разагилина соответствующе вводят перорально трем группам без обработки нейротоксином один раз в сутки в течение 8 дней. На следующий день после последнего дня введения ткани полосатого тела и черной субстанции берут у каждой группы мышей. мРНК экстрагируют из клетки взятой ткани и измеряют количество мРНК NGF ОТ-ПЦР в режиме реального времени.
Как показано на ФИГ.15, подтверждается, что для группы мышей, обработанных CBI, количество мРНК NGF в полосатом теле в примерно 1,7-2,5 раз больше, чем в другой группе мышей. В частности, подтверждено, что в группе мышей, обработанных CBI, экспрессия NGF в полосатом теле значительно выше, чем в группе мышей, обработанных разагилином. Результаты показывают, что CBI вызывает экспрессию нейротрофических факторов нейронов, тем самым ингибирует апоптоз нейронов.
Пример 8: Подтверждение нейропротекторного действия CBI улучшением функций митохондрии в нейронах
В этом варианте применяют SH-SY5Y клетки человеческой нейробластомы с дефицитом MAO-B (Korean Cell Line Bank). Клетки инкубируют в среде DMEM в 5% CO2 при 37°С в течение 24 часов. Культивированные клетки делят на пять групп, к каждой группе добавляют 2 мМ 1-метил-4-фенилпиридиния (MPP+), три из пяти групп соответственно обрабатывают 1 нМ, 10 нМ и 50 нМ CBI, одну из оставшихся двух групп обрабатывают 10 нМ разагилина, и обработанные CBI или разагилином четыре группы далее инкубируют в 5% CO2 при 37°C в течение 24 часов. Группу с добавлением только MPP+ инкубируют в тех же условиях, как описано выше. Далее, согласно протоколу производителя, трансмембранный потенциал митохондрии определяют с применением набора MitoPTtm (Immunochemistry Technology). Трансмембранный потенциал митохондрии подтверждают с применением флуоресцентного планшетного ридера (Tecan, Austria). Если трансмембранный потенциал митохондрии является низким, имеется зеленая флуоресценция, с другой стороны, если он является высоким, имеется красная флуоресценция. Таким образом, значения RFU (значение красной флуоресценции/значение зеленой флуоресценции) определяют из результатов, и результаты показаны на ФИГ.16.
MPP+ снижает трансмембранный потенциал митохондрии, тем самым вызывая нестабильность мембраны митохондрии. Проницаемость мембраны митохондрии является жизненно важным процессом в апоптозе, таким образом стабильность мембраны митохондрии может стать механизмом анти-апоптоза (Brain Res. Rev., 29:1-25(1999)). Как показано на ФИГ.16, было подтверждено, что в случае добавления MPP+ к нейронам, трансмембранный потенциал митохондрии стабилизируется обработкой CBI зависимо от концентрации, и эффект стабильности трансмембранного потенциала митохондрии не примерно менее чем в 2 раза выше, чем в группе нейронов, обработанных той же концентрацией разагилина, как и CBI.
Как описано выше, многие нейродегенеративные заболевания, такие как удар, повреждения мозга, повреждения спинного мозга, боковой амиотрофический склероз, болезнь Хантингтона, болезнь Альцгеймера и болезнь Паркинсона, характеризуются апоптозом нейронов (The New England Journal of Medicine, 348:1365(2003)), и апоптоз нейронов вызывается индукцией путей апоптоза несколькими внутренними или внешними факторами. Кроме того, стабильность мембраны митохондрии является механизмом анти-апоптоза, и известно, что апоптоз нейронов ингибируется белком Bcl-2 или Bcl-xL, и мембрана митохондрии стабилизируется белком (Biochem Biophys Res Commun., 304(3):433-435(2003); The New England Journal of Medicine, 348(14):1365-1375(2003); Brain Res Rev., 29(1):1-25(1999); Journal of Neurological Sciences, 283:240-320(2009)). Таким образом, результаты показывают, что CBI стабилизирует трансмембранный потенциал митохондрии, тем самым предотвращая или вылечивая нейродегенеративные заболевания, описанные выше.
Кроме того известно, что апоптоз вызывается механизмами выделения цитохрома c из митохондрии и активацией каспазы 3 (The New England journal of Medicine, 348:1365-1375(2003)). Для подтверждения того, связана ли стабильность трансмембранного потенциала митохондрии, вызванная обработкой CBI, с анти-апоптозом нейронов, из групп клеток получают экстракты клеток, и измеряют количество цитохрома c и активность каспазы 3 в цитоплазме клетки.
Пример 8-1: Измерение выделения цитохрома c
Клетки SH-SY5Y инкубируют в тех же условиях, которые описаны в Примере 7, и затем промывают PBS. Коктейль ингибитора протеазы (Roche) и коктейль ингибитора фосфатазы (Roche) добавляют к гипертоническому буферу (20 мМ HEPES, 10 мМ KCl, 2 мМ MgCl2, 1 мМ EDTA), и клетки SH-SY5Y обрабатывают 100 мкл полученного раствора и затем однородно суспендируют. Полученное выдерживают на льду в течение 30 минут и затем центрифугируют при 12000 об/мин в течение 20 минут. Затем то же количество надосадочной жидкости загружают в гель SDS-PAGE для последующего электрофореза, гель переносят в нитроцеллюлозную мембрану, мембрану подвергают блоттингу с применением цитохрома c (Santacruz, sc13156), разведенного до 1:2000 с применением способа, хорошо известного в данной области техники, и экспрессию цитохрома c оценивают с применением набора ECL (Amersham Pharmacia).
Как показано на ФИГ.17, подтверждено, что для нейронов, к которым добавлен MPP+, количество цитохрома c в цитоплазме снижается при обработке CBI, и количество цитохрома c, выделяемого из митохондрии, меньше, чем для нейронов, обработанных MPP+, обработанных тем же количеством разагилина, что и CBI.
Пример 8-2: Измерение активности каспазы 3/7
Клетки SH-SY5Y инкубируют в 96-луночном планшете в концентрации 5×105 клеток/лунку в тех же условиях, которые описаны выше, клетки обрабатывают 2 мМ MPP+ и CBI (1, 5, 10 и 50 нМ) или разагилином (50 нМ), и полученные клетки инкубируют в течение 24 часов. Затем туда добавляют 100 мкл реагента Apo-ONE каспазы 3/7 (Promega, G7790) и перемешивают, полученную смесь далее инкубируют в течение 4 часов. После окончания инкубирования измеряют флуоресценцию при длине волны возбуждения 495 нм и длине волны испускания 521 нм с применением флуоресцентного планшетного ридера (GeminiXPS, Molecular Devices). Как показано на ФИГ.18, подтверждено, что активность каспазы 3 снижается при обработке CBI, так же как и при обработке клеток разагилином.
Результаты Примера 8 подтверждают, что CBI стабилизирует мембрану митохондрии в нейронах, тем самым предотвращая высвобождение цитохрома c из митохондрии, и снижает активность каспазы 3, ингибируя апоптоз нейронов.
Как описано выше, многие нейродегенеративные заболевания, такие как удар, повреждения мозга, повреждения спинного мозга, боковой амиотрофический склероз, болезнь Хантингтона, болезнь Альцгеймера и болезнь Паркинсона, характеризуются апоптозом нейронов, и результаты показывают, что CBI ингибирует апоптоз нейронов, и таким образом способен предотвращать и лечить нейродегенеративные заболевания.
Пример 9: Подтверждение нейропротекторного действия CBI ингибированием реакционно-способных видов кислорода в нейронах
В этом варианте применяют SH-SY5Y клетки человеческой нейробластомы с дефицитом MAO-B (Korean Cell Line Bank). Клетки культивируют в среде DMEM в 5% CO2 при 37°С в течение 24 часов. Культивированные клетки делят на три группы, к каждой группе добавляют 2 мМ MPP+, одну из трех групп обрабатывают 50 нМ CBI, одну из оставшихся двух групп обрабатывают 50 нМ разагилина и три группы далее инкубируют в 5% CO2 при 37°C в течение 24 часов. Группу с добавлением только MPP+ инкубируют в тех же условиях, как описано выше. Затем клетки окрашивают диацетатом 2,7-дихлорфлуоресцеина (DCF-DA), который представляет собой флуоресцентный краситель, способный определять реакционно-способные виды кислорода, и затем наблюдают с применением конфокального микроскопа (Nikon Co., Japan).
Как показано на ФИГ.19 и 20, при добавлении к нейронам MPP+, реакционно-способные виды кислорода значительно снижаются путем обработки CBI, что также происходит при обработке нейронов, к которым добавлен MPP+, той же концентрацией разагилина, что и CBI.
Известно, что образование и повышение реакционно-способных видов кислорода в клетках вызывает апоптоз, и таким образом, полученные выше результаты подтверждают, что CBI вызывает снижение реакционно-способных видов кислорода, тем самым ингибируя апоптоз. Известно, что такой окислительный стресс вызывает различные заболевания, связанные с апоптозом нейронов или нейродегенерацией, например, болезнь Альцгеймера, боковой амиотрофический склероз, демиелинирующие заболевания, диабетическую полиневропатию, синдром Дауна, ВИЧ невропатию, болезнь Хантингтона, множественную системную атрофию, болезнь Паркинсона, удар и ишемическое реперфузионное повреждение, таупатию и травматические повреждения мозга (Free radical Biology & Medicine, 33(2):182-191(2002)), и эти результаты показывают, что CBI вызывает снижение реакционно-способных видов кислорода, тем самым ингибируя окислительный стресс, и, таким образом, используется для предупреждения или лечения различных нейродегенеративных заболеваний.
Пример 10: Подтверждение нейропротекторного действия CBI повышением активности антиокислительного фермента
В этом варианте применяют клетки SH-SY5Y человеческой нейробластомы с дефицитом MAO-B (Korean Cell Line Bank). Клетки распределяют в 6-луночный планшет в концентрации 1,8×105 клеток/лунку и затем инкубируют в 5% CO2 при 37°С в течение 24 часов. Полученные клетки делят на три группы, к каждой группе добавляют 2 мМ MPP+, в две из трех групп соответственно добавляют 0,1, и полученные клетки далее инкубируют в 5% CO2 при 37°C в течение 24 часов. Контрольную группу, которую не обрабатывали MPP+, инкубируют в тех же условиях, которые описаны выше. Затем из клеток получают клеточные экстракты и измеряют активность антиокислительных ферментов, т.е. каталазы, супероксиддисмутазы (SOD) и глутатионпероксидазы (GPx).
Как показано на ФИГ.21, нейроны в добавлением MPP+ демонстрируют тенденцию к снижению активности каталазы и GPx по сравнению с группой клеток, которую не обрабатывали MPP+, и нейроны с добавлением MPP+, обработанные CBI, демонстрируют повышение пониженной активности антиокислительных ферментов. В частности подтверждено, что активность GPx в этой группе в 2,5-4 раза выше, чем в контрольной группе.
Кроме того, для подтверждения in vivo повышается ли активность антиокислительных ферментов при обработке CBI, используют мышей C57BL/6 ((n=12), возраст 8-9 недель, самцы) (ORIENT BIO INC.). Мышей C57BL/6 делят на три группы (n=4 для каждой группы) и наполнитель (контроль), 1 мг/кг CBI и 1 мг/кг разагилина соответственно вводят перорально трем группам без обработки нейротоксином в течение 8 дней. На следующий день после последнего дня введения у каждой группы мышей берут полосатое тело и черную субстанцию. Экстракты клеток получают из клеток тканей и измеряют активность каталазы, SOD и GPx.
Как показано на ФИГ.22, в группе мышей, которым вводили CBI, отсутствует значительное изменение в активности SOD, однако активность каталазы повышается на примерно 13% в полосатом теле, и активность GPx повышается на примерно 28% в черной субстанции.
Известно, что образование и повышение реакционно-способных видов кислорода в клетках вызывает апоптоз, и известно, что антиокислительные ферменты разрушают реакционно-способные виды кислорода, таким образом результаты подтверждают, что CBI повышает активность антиокислительных ферментов в нейронах, в частности, активность GPx в черной субстанции, тем самым вызывая снижение реакционно-способных видов кислород, и, следовательно, способен ингибировать апоптоз. Кроме того, результаты показывают, что CBI вызывает снижение реакционно-способных видов кислорода, тем самым ингибируя окислительный стресс для предупреждения апоптоза нейронов, и поэтому может применяться для предупреждения или лечения различных описанных выше заболеваний.
Пример 11: Подтверждение восстановления функций нервной системы с помощью CBI
C57BL/6 мышей (n=8/группу (всего 4 группы), возраст 8 недель; самцы) используют в качестве экспериментальной модели (субштамм: C57BL/6NCrljBgi, от ORIENT BIO INC.). 30 мг/кг MPTP вводят мышам внутрибрюшинной инъекцией один раз в сутки в течение 4 дней. Через 4 дня после последнего дня введения мышей делят на три группы (n=4/группу), и наполнитель (контроль), 1 мг/кг CBI и 1 мг/кг разагилина соответственно вводят перорально один раз в сутки в течение 10 дней. На следующий день после последнего дня введения у каждой группы мышей берут ткани полосатого тела и черной субстанции. Для определения того, увеличилось или нет количество невритов и спинов на нейрон в тканях, получают коронарный срез толщиной 200 мкм, содержащий полосатое тело или черную субстанцию, с применением вибратома. Каждую группу мышей умерщвляют, и мозг берут у каждой группы и затем помещают в холодный, высоко концентрированный сахарозный диссекционный буфер, барботируемый карбогеном (5% CO2 и 95% O2) (87 мМ NaCl, 2,5 мМ KCl, 1,25 мМ NaH2PO4, 25 мМ NaHCO3, 1 мМ CaCl2, 3 мМ MgCl2, 10 мМ декстрозы и 75 мМ сахарозы). При проведении вибратома камеру для разделения наполняют однородно карбогенированным сахарозным диссекционным буфером. После завершения разделения все срезы промывают PBS и фиксируют 4% параформальдегидом в растворе PBS. Фиксированные срезы мозга помещают на блок 1,5% агарозы (в PBS). Их изображения получают с применением конфокального микроскопа (ZEISS LSM 510 META) с применением программного обеспечения Zeiss LSM (Carl Zeiss Microimaging, Germany, версия 4.0 SP2).
Как показано на ФИГ.23 и 24, в группе индуцированных MPTP мышей, которым вводили CBI, количество невритов повышается, и количество спинов также восстанавливается до уровня нормальных клеток. В частности, в группе индуцированных MPTP мышей, которым вводили разагилин, наблюдается повышение количества спинов, однако повышение количества невритов не является статистически значимым. Результаты подтверждают, что CBI ингибирует апоптоз, а также улучшает невральную пластичность.
Пример 12: Введение CBI и получение таблеток, содержащих CBI (прогнозирование)
Фармацевтическую композицию в соответствии с настоящим изобретением применяют для ингибирования апоптоза нейронов или нейродегенерации, или для защиты или восстановления функций нервной системы. Клинически подходящая доза (пероральное введение) фармацевтической композиции составляет 25 мг - 100 мг для взрослого человека.
На основании дозы таблетку, содержащую компоненты, показанные в Таблице 1, получают с использованием обычного способа. В качестве наполнителя используют Avicel 102 (микрокристаллическую целлюлозу).
Подходящая доза компонентов составляет 1 или 2 таблетки, содержащих компоненты, в сутки для взрослого человека с массой тела 60 кг.
Согласно одному или более вариантам настоящего изобретения, фармацевтическая композиция может эффективно предотвращать или лечить заболевания, связанные с апоптозом нейронов или нейродегенерацией.
Хотя данное изобретение было показано и описано со ссылкой на его типовые варианты, специалисту в данной области техники должно быть понятно, что различные изменения в форме и деталях могут быть проведены, не выходя за рамки сущности и объема настоящего изобретения, определенные в формуле изобретения.
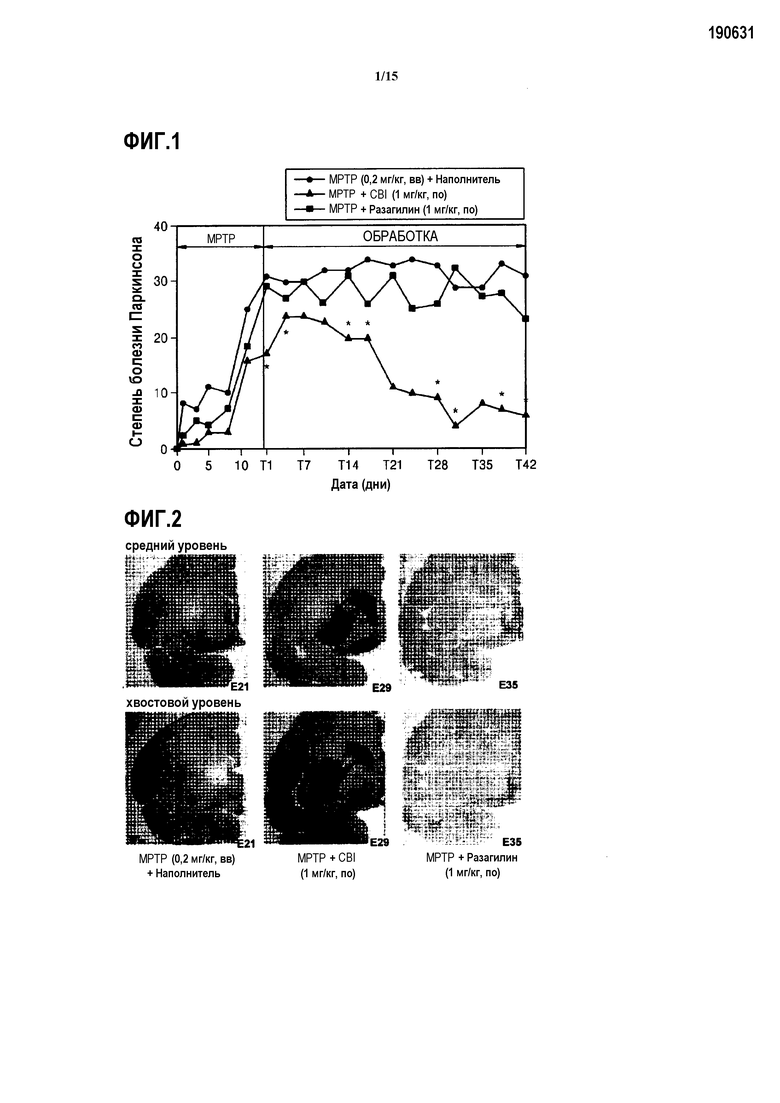
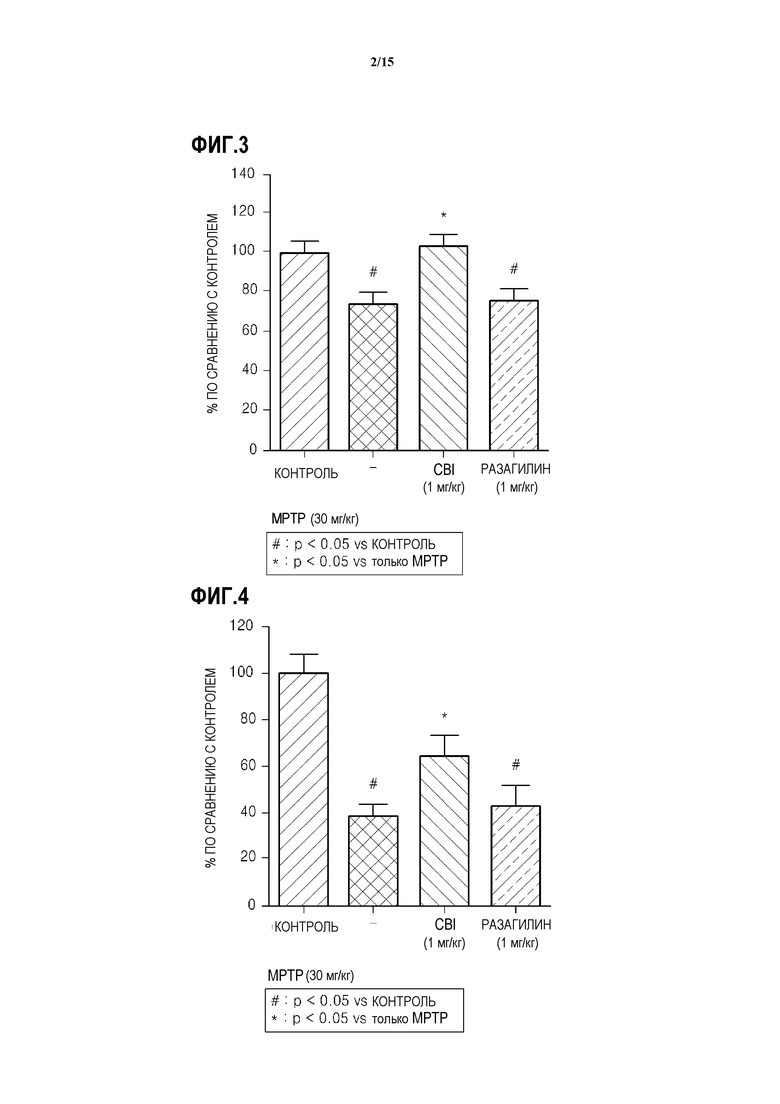
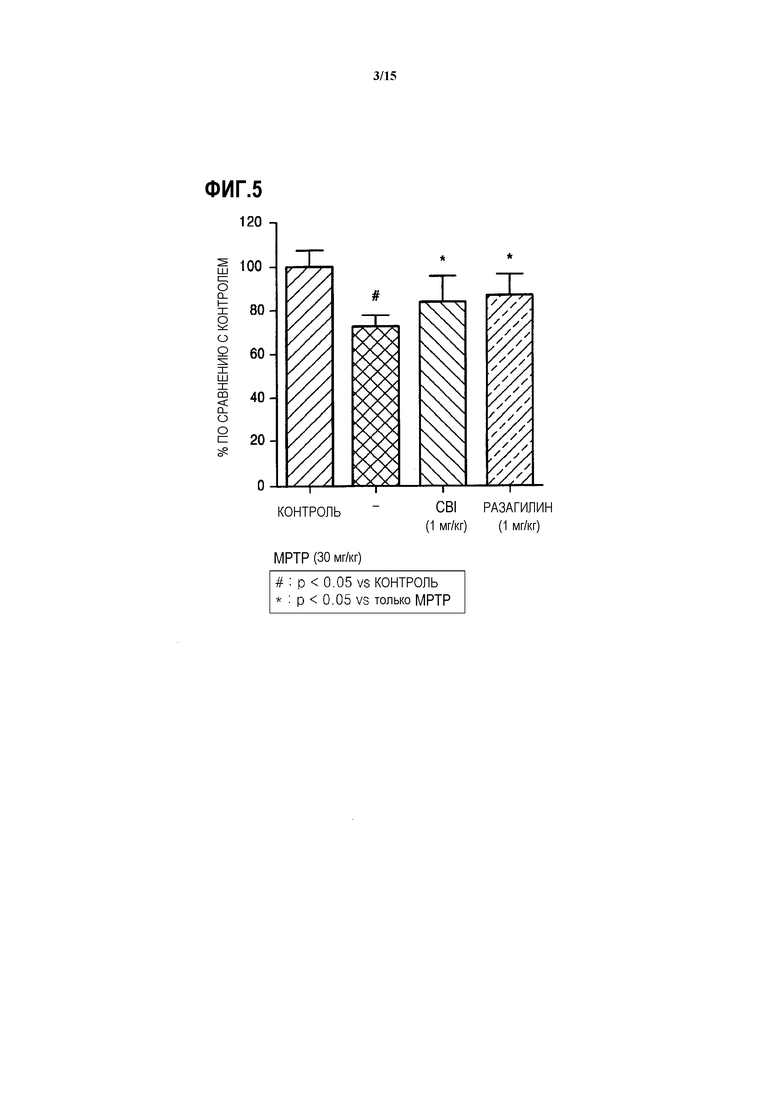
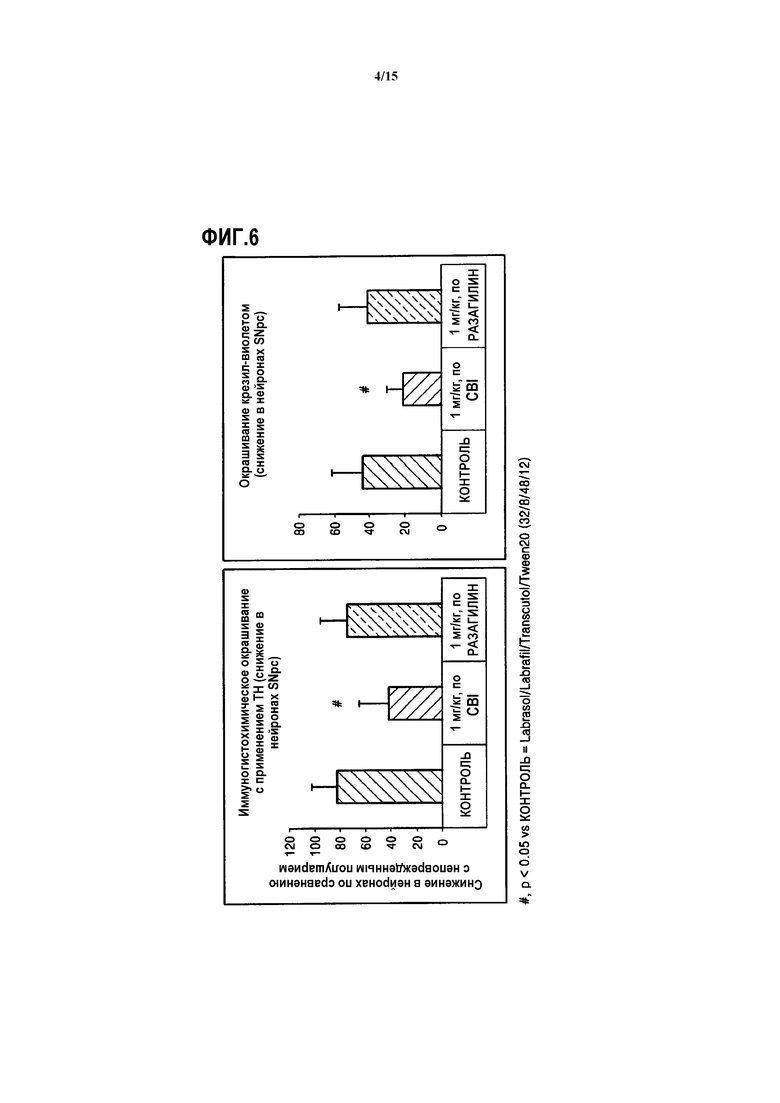
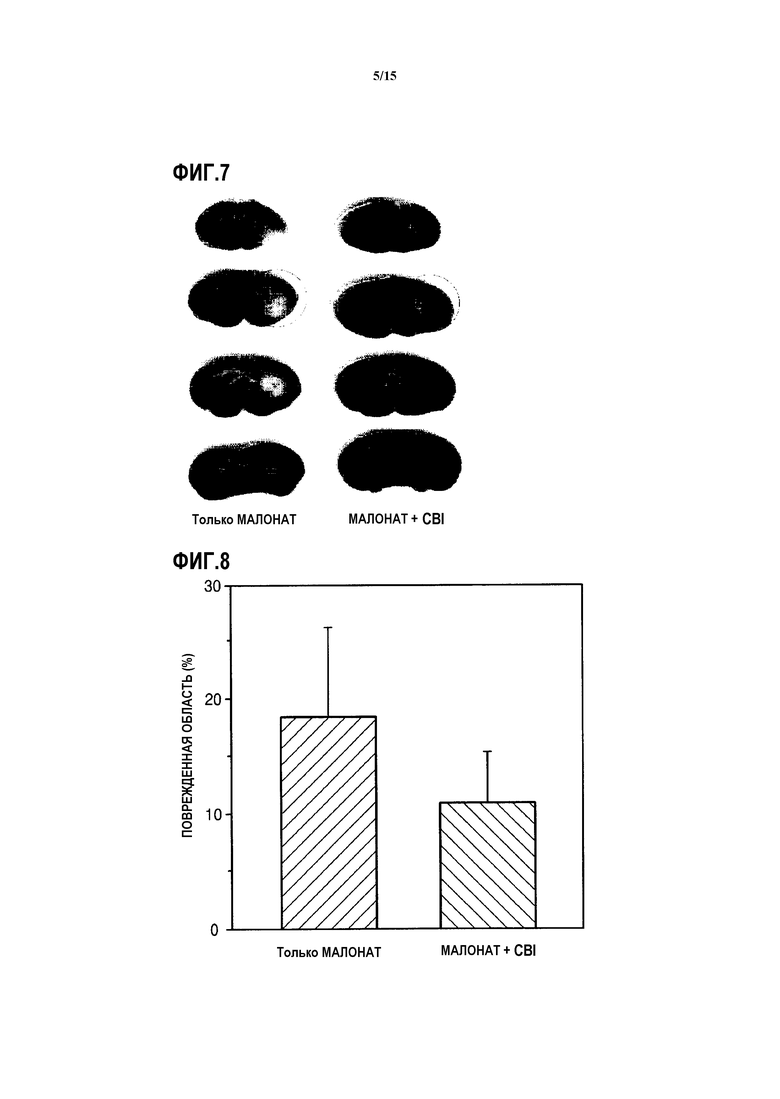
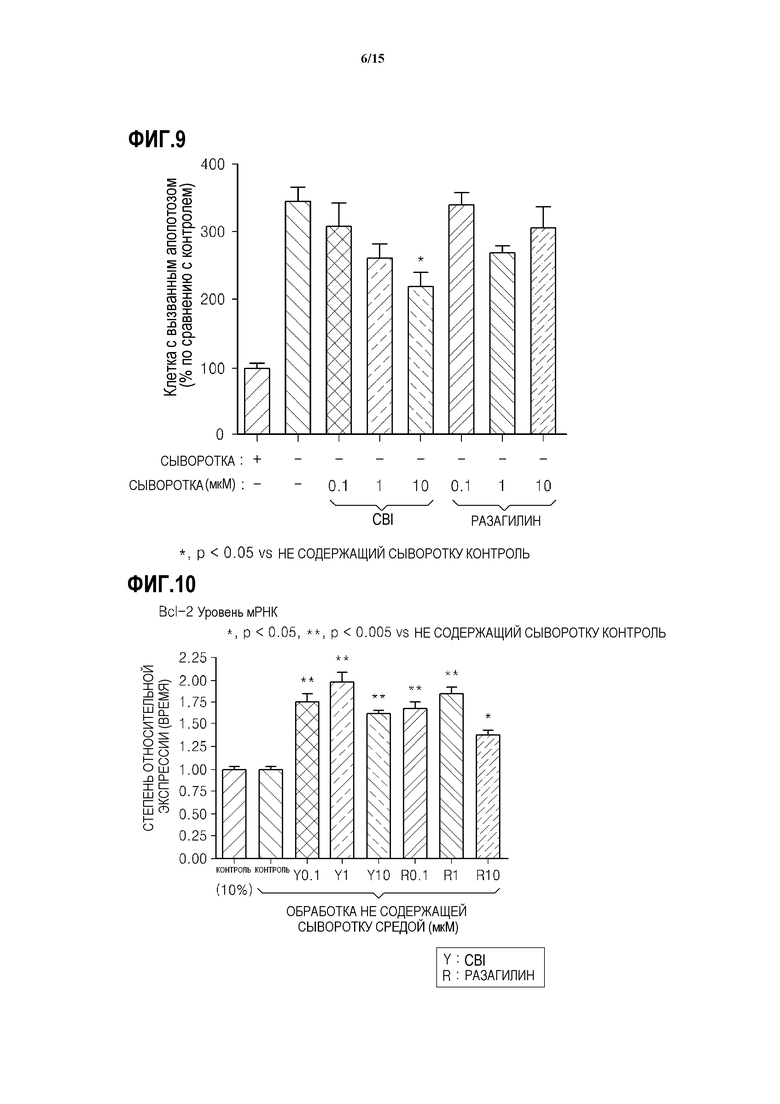
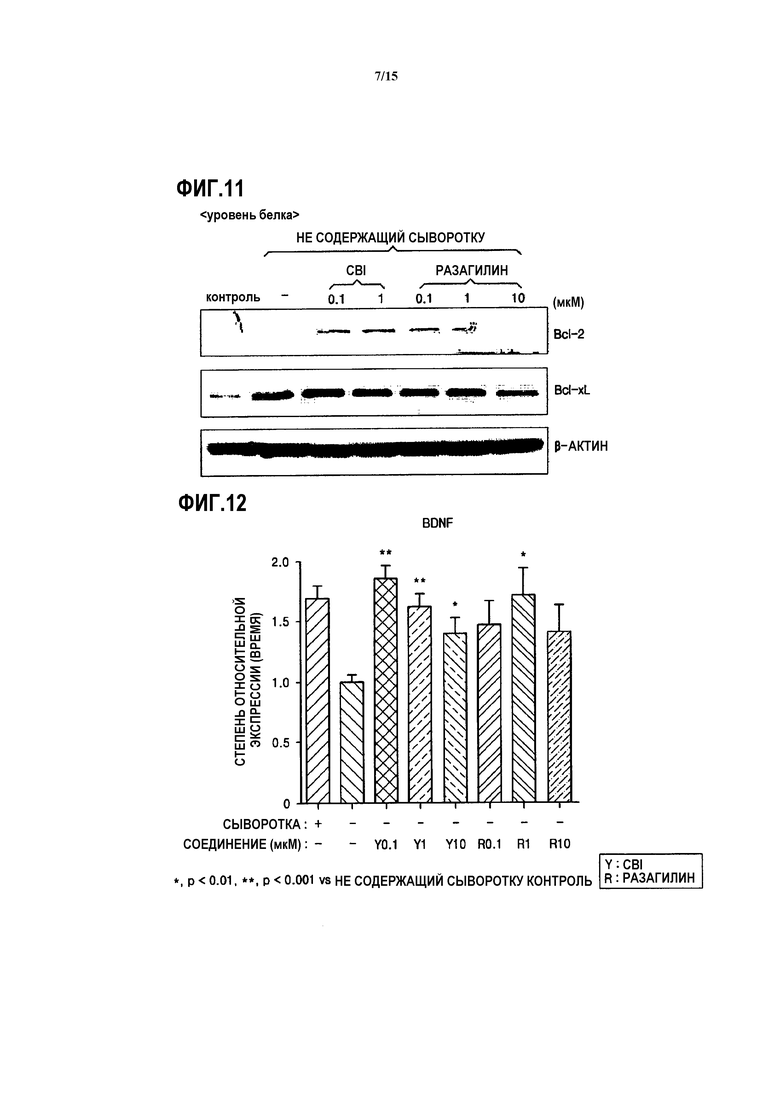
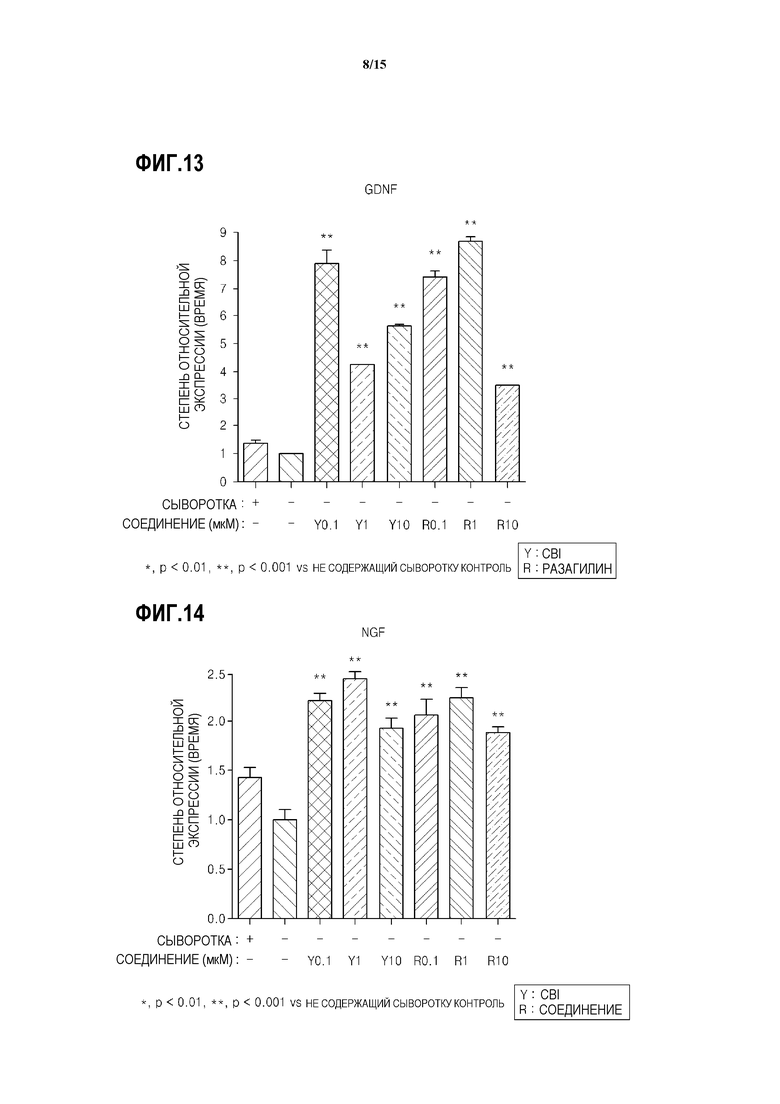
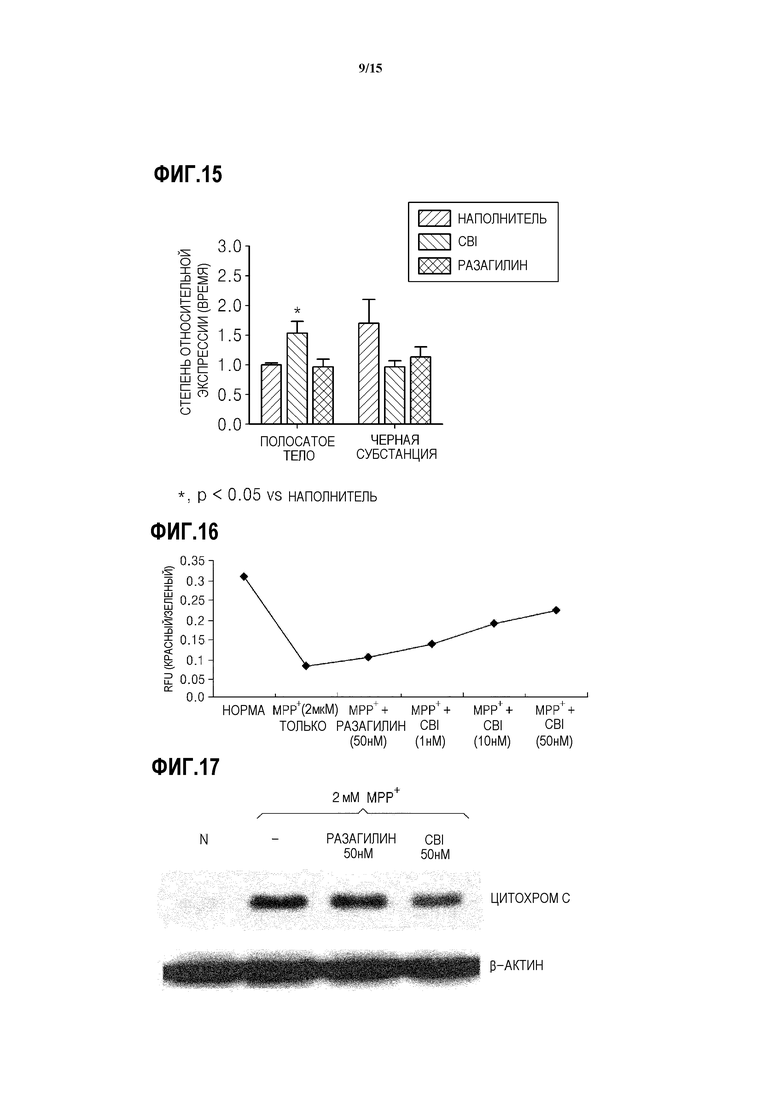
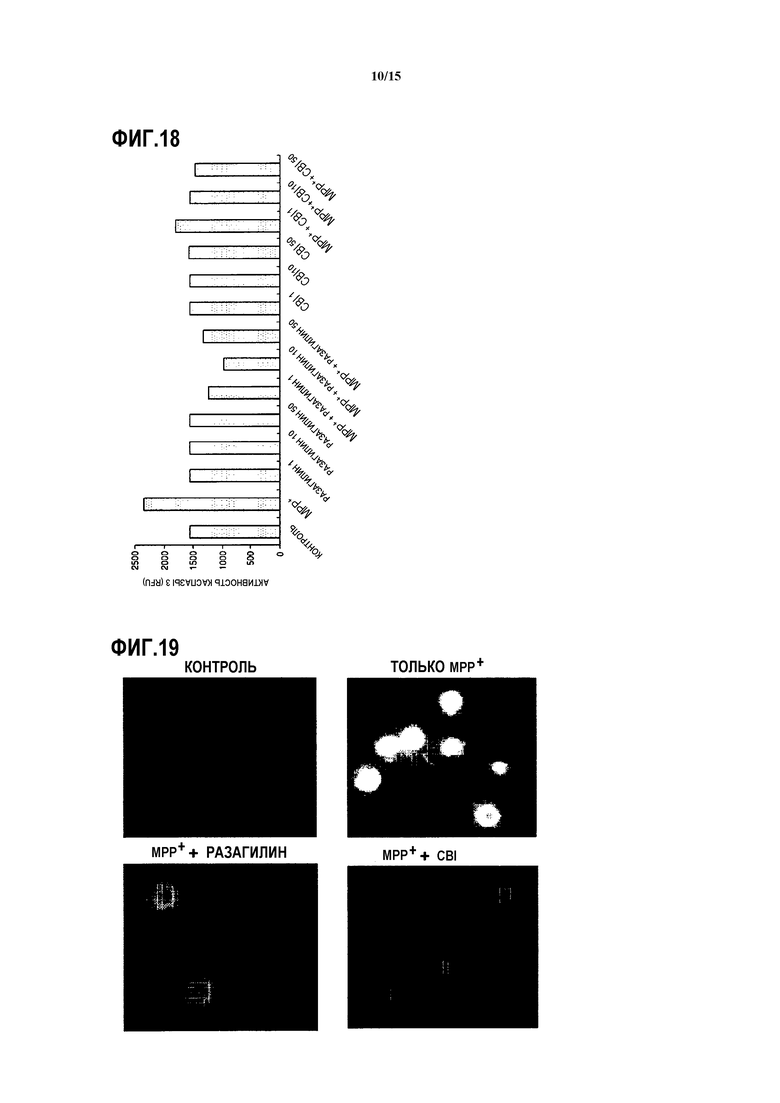
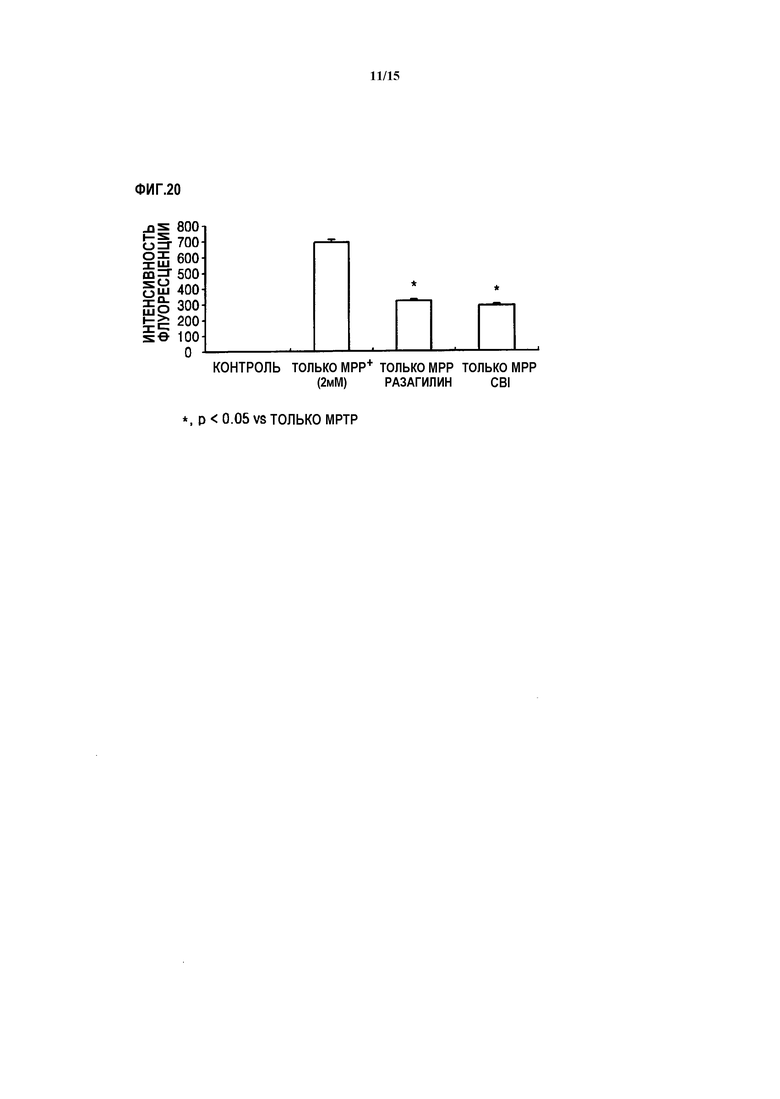
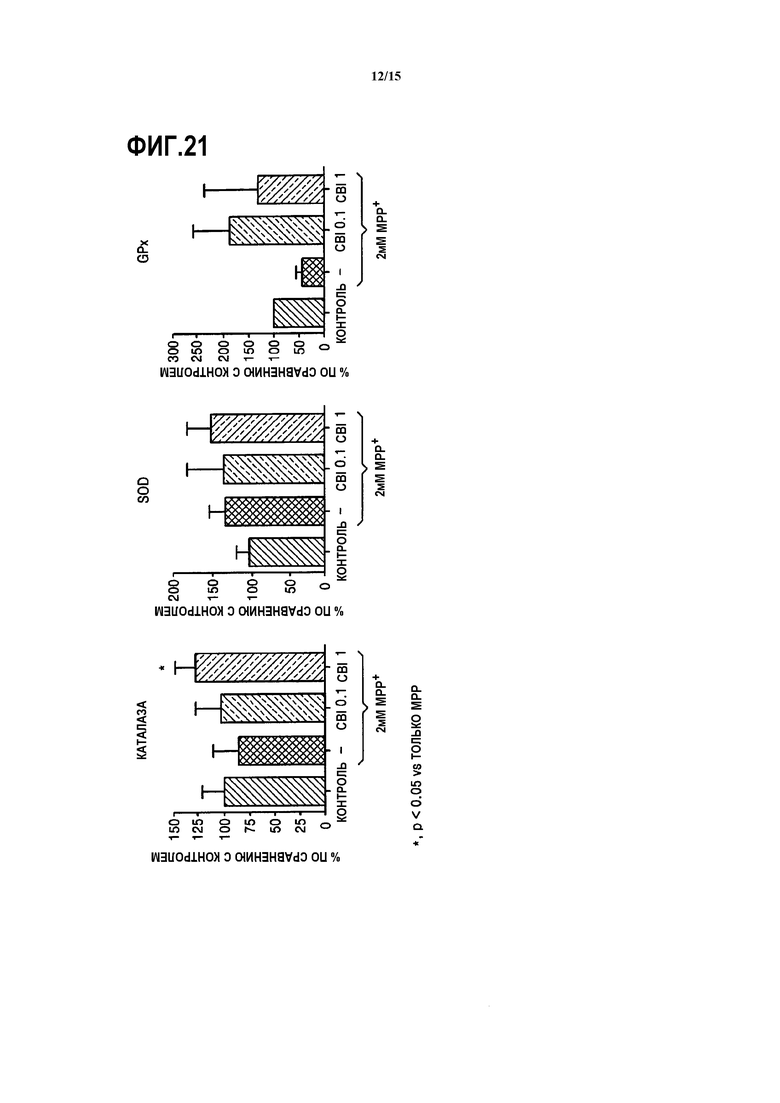
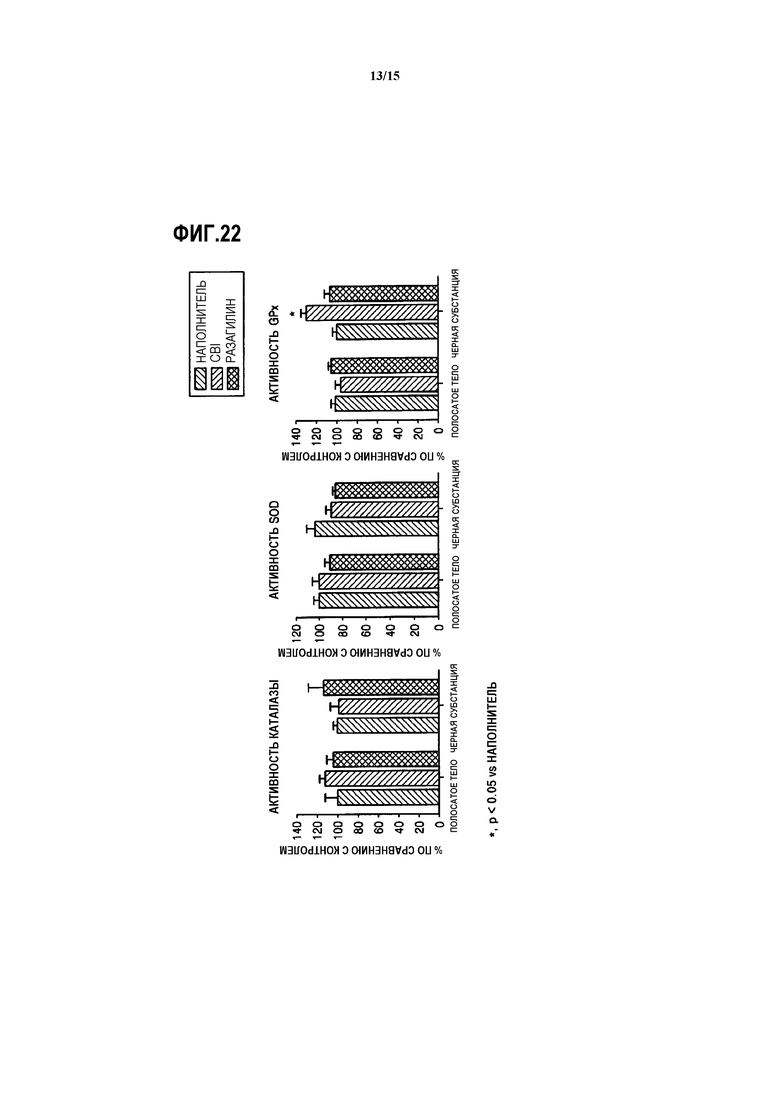
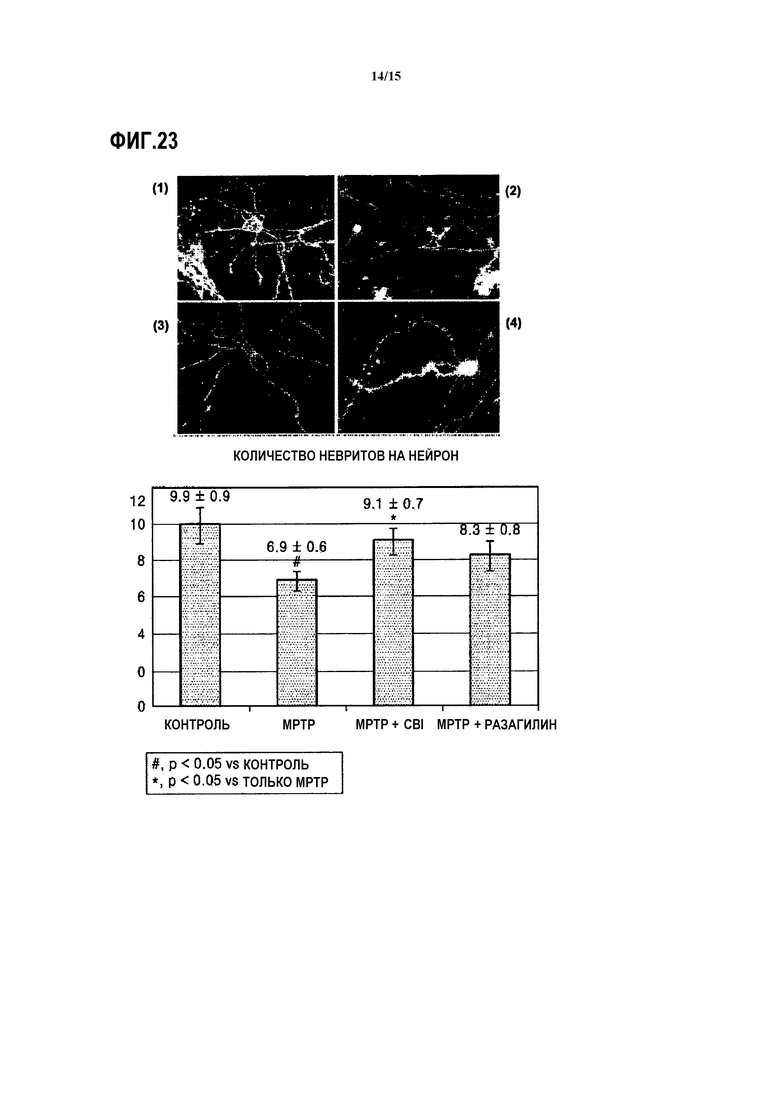
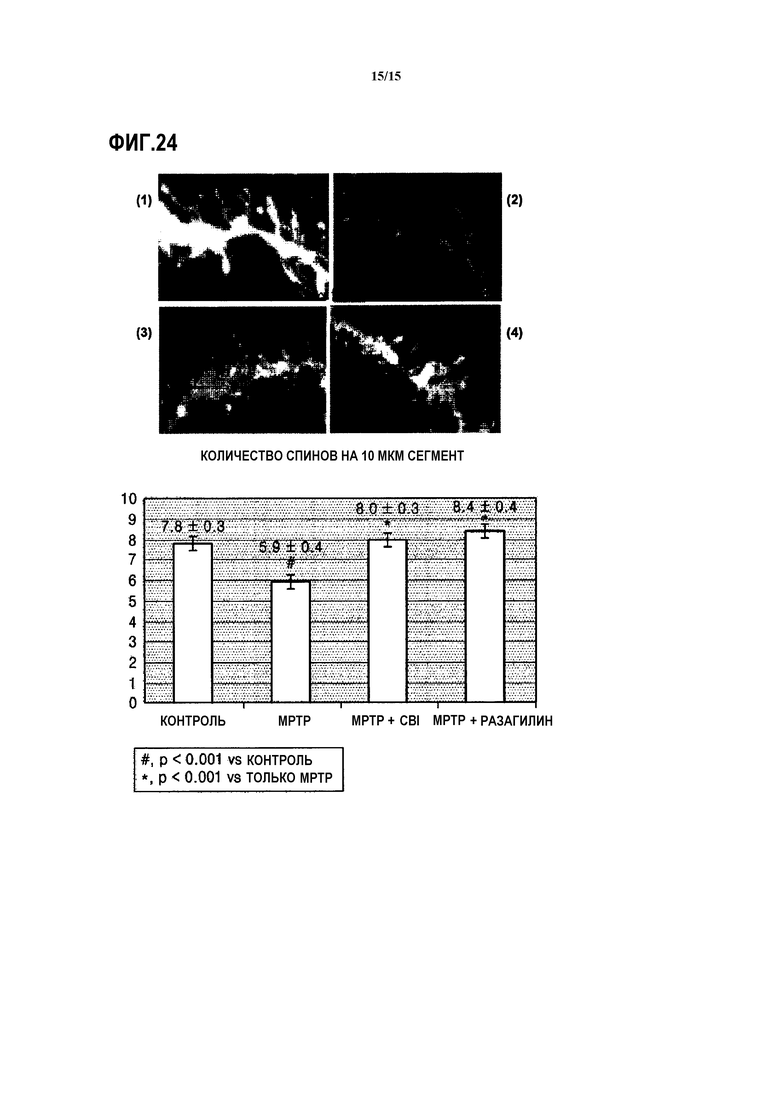
Изобретение относится к применению соединения формулы I
Формула I
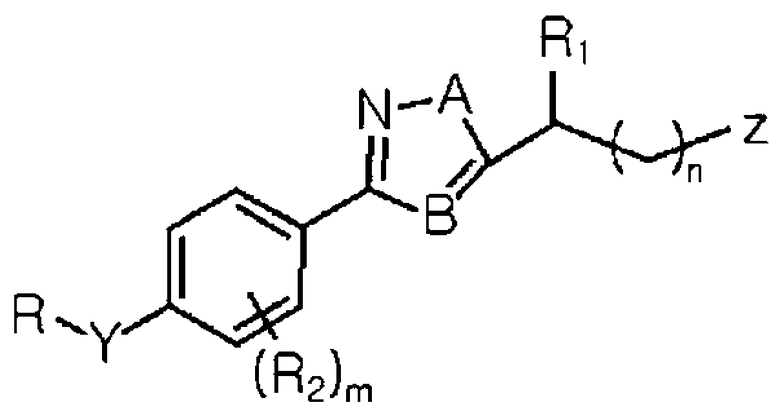
где R представляет собой С7-С15 фенилалкильную группу; Y является О; R1 и R2 являются Н; А является О; В является С; Z является -OC(=O)NR3R4; каждый из R3 и R4 является Н; m и n являются 0, его фармацевтически приемлемой соли, изомера, сольвата или гидрата или любой их комбинации для изготовления лекарственного средства для предупреждения или лечения заболеваний, выбранных из группы, состоящей из удара, болезни Альцгеймера, болезни Хантингтона, болезни Пика, болезни Крейтцфельда-Якоба, комплекса Паркинсон-БАС-слабоумие, болезни Уилсона, рассеянного склероза, прогрессирующего надъядерного паралича, биполярных расстройств, связанных с невропатической болью, кортико-базальной дегенерации, шизофрении, синдрома гиперактивности с дефицитом внимания (ADHD), слабоумия, амиотрофического латерального склероза, болезни сетчатки, эпилепсии, апоплексии, преходящего нарушения мозгового кровообращения, ишемии миокарда, мышечной ишемии, ишемии, вызванной хирургическим вмешательством, требующим длительного прекращения подачи крови к мозгу, повреждения головы, повреждения спинного мозга, гипоксии и депрессии. 2 н. и 1 з.п. ф-лы, 12 пр., 1 табл., 24 ил.
1. Применение соединения формулы I
Формула I
где R представляет собой С7-С15 фенилалкильную группу;
Y является О;
R1 является Н;
R2 является Н;
А является О;
В является С;
Z является -OC(=O)NR3R4;
каждый из R3 и R4 является Н;
m является 0; и
n является 0,
его фармацевтически приемлемой соли, изомера, сольвата или гидрата или любой их комбинации для изготовления лекарственного средства для предупреждения или лечения заболеваний, выбранных из группы, состоящей из удара, болезни Альцгеймера, болезни Хантингтона, болезни Пика, болезни Крейтцфельда-Якоба, комплекса Паркинсон-БАС-слабоумие, болезни Уилсона, рассеянного склероза, прогрессирующего надъядерного паралича, биполярных расстройств, связанных с невропатической болью, кортико-базальной дегенерации, шизофрении, синдрома гиперактивности с дефицитом внимания (ADHD), слабоумия, амиотрофического латерального склероза, болезни сетчатки, эпилепсии, апоплексии, преходящего нарушения мозгового кровообращения, ишемии миокарда, мышечной ишемии, ишемии, вызванной хирургическим вмешательством, требующим длительного прекращения подачи крови к мозгу, повреждения головы, повреждения спинного мозга, гипоксии и депрессии.
2. Применение по п. 1, где соединением формулы I является соединение, представленное формулой II:
Формула II
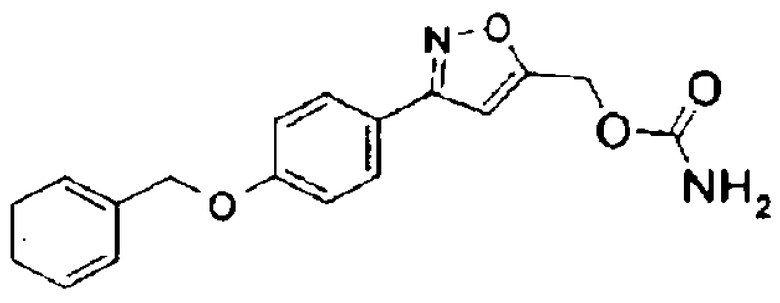
3. Способ лечения заболеваний, выбранных из группы, состоящей из удара, болезни Альцгеймера, болезни Хантингтона, болезни Пика, болезни Крейтцфельда-Якоба, комплекса Паркинсон-БАС-слабоумие, болезни Уилсона, рассеянного склероза, прогрессирующего надъядерного паралича, биполярных расстройств, связанных с невропатической болью, кортико-базальной дегенерации, шизофрении, синдрома гиперактивности с дефицитом внимания (ADHD), слабоумия, амиотрофического латерального склероза, болезни сетчатки, эпилепсии, апоплексии, преходящего нарушения мозгового кровообращения, ишемии миокарда, мышечной ишемии, ишемии, вызванной хирургическим вмешательством, требующим длительного прекращения подачи крови к мозгу, повреждения головы, повреждения спинного мозга, гипоксии и депрессии, включающий введение субъекту фармацевтической композиции, которая включает соединение формулы I, его фармацевтически приемлемую соль, изомер, сольват или гидрат или любую их комбинацию и фармацевтически приемлемый носитель, субъекту, нуждающемуся в этом, где соединением формулы I является соединение

где R представляет собой C7-C15 фенилалкильную группу;
Y является О;
R1 является Н;
R2 является Н;
А является О;
В является С;
Z является -ОС(=O)NR3R4; каждый из R3 и R4 является Н;
m является 0; и
n является 0.
| WO 2010039947 A1, 08.04.2010 | |||
| WO 2007150025 A2, 27.12.2007 | |||
| WO 2010001365 A1, 07.01.2010 | |||
| WO 2008074384 A1, 26.06.2008 | |||
| WO 2006042245 A1, 20.04.2006 | |||
| ПРОИЗВОДНЫЕ ГЕТЕРОАРИЛАЛКИЛКАРБАМАТОВ, ИХ ПОЛУЧЕНИЕ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ФЕРМЕНТА FAAH | 2005 |
|
RU2364586C2 |

