Область техники
Настоящее изобретение относится к новому производному гиалуроновой кислоты, которое подходит для получения гидрогелей, и к способу его получения. Кроме того, настоящее изобретение относится к гидрогелям на основе указанного производного, к их свойствам, применению и способу их получения.
Предшествующий уровень техники
Гиалуроновая кислота представляет собой полисахарид, содержащий дисахаридные единицы, состоящие из D-глюкуроновой кислоты и D-N-ацетилглюкозамина, которые связаны переменными β-1,4- и β-1,3-гликозидными связями. Среднемассовая молекулярная масса (если далее будет упоминаться молекулярная масса, это всегда будет среднемассовая молекулярная масса) in vivo находится в пределах диапазона 3 кДа - 20 МДа. Это полисахарид, который легко растворяется в водной среде, в которой он образует высоковязкие растворы в зависимости от своей молекулярной массы и концентрации.
Гидрогели представляют собой материалы, которые образуются в воде при помощи нерастворимой сетки, по меньшей мере, частично гидрофильных полимеров1. Существует несколько способов получения нерастворимой решетки из первоначально гидрофильного полимера. Это гидрофобизация полимера2 или применение производного растворимого в воде полимера, несущего реакционноспособные функциональные группы, которые могут участвовать в дальнейших химических реакциях, которые приводят к образованию 3-мерной полимерной сетки3-5.
Получение растворимых производных гиалуроновой кислоты и их последующее сшивание было описано множеством авторов3-6. Также ранее было описано применение фенольных производных гиалуроновой кислоты для реакций сшивания и получения гидрогелей. Calabro et. al.4,7,8 раскрыли способ получения фенольных производных гиалуроновой кислоты путем осуществления взаимодействия карбоксилов, которые представлены в пределах структуры D-глюкуроновой кислоты гиалуроновой кислоты, с аминоалкильными производными фенола. При этом взаимодействии получали амиды гиалуроновой кислоты. Определяющим элементом для осуществления указанного синтеза является активация карбоксила гиалуроновой кислоты, для которой применяли реакцию с дегидратирующими агентами карбодиимидного типа (такими как EDC). Наиболее часто используемым аминоалкильным фенолом является тирамин6.
В общем случае, сшивание фенольных производных гиалуроновой кислоты инициировано добавлением пероксидазы (такой как пероксидаза хрена - HRP) и разбавленного раствора пероксида водорода. Пероксидаза хрена (пероксидаза хрена, HRP, Е.С.1.11.1.7) в настоящее время широко применяется как катализатор органических и биотрансформационных реакций". Она характеризуется очень широкой специфичностью субстрата и, таким образом, она способна окислять ряд органических и неорганических соединений13-15.
Она представляет собой фермент, который содержит железо в гемовой форме как простетическую группу. Железо обладает степенью окисления (III) в неактивном состоянии фермента. Взаимодействие с пероксидами приводит к образованию промежуточного соединения, которое называется HRP-I. Входящий в состав гем-группы атом железа Fe(III) окисляется до оксиферрильной группы (Fe(IV)=O) при одновременном образовании катионного π-радикала на порфириновом цикле. Такой активированный фермент способен образовывать комплексы с молекулами субстрата, которые в течение этого взаимодействия подвергались оксилению14,16-18.
Превращение окисленного фермента обратно в свою начальную форму проходит в две стадии. На первой стадии происходит взаимодействие между молекулой субстрата (S) и HRP-I, что приводит к образованию радикала субстрата (R·) и частично редуцированной формы фермента HRP-II. HRP-II все еще удерживает оксиферрильную группу (Fe(IV)=O), но он больше не содержит порфирин π-радикал. Во время перехода электрона к порфириновому радикалу один Н+ принимается белком. HRP-II снова подвергается взаимодействию с субстратом, что приводит к образованию R·. В течение этой реакции оксиферрильная группа (Fe(IV)=O) снова восстанавливается до Fe(III). Этот процесс связан с переносом 2Н+ к кислороду оксиферрильной группы. Один протон происходит из субстрата (или растворителя), а другой из белка. Это приводит к образованию молекулы воды (уравнение I и схема I).
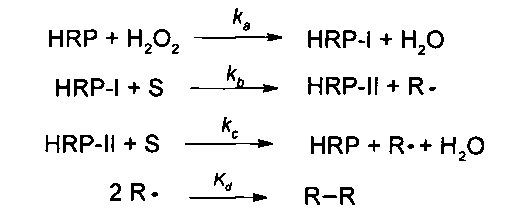
Уравнение I: Основное описание механизма катализа окисления субстрата при помощи HRP
Полученные радикалы субстрата в ряде случаев способны взаимодействовать вместе с образованием димеров R-R. Этот процесс больше не находится под действием ферментов и связан со стабильностью и реакционной способностью полученных радикалов 14,16-26.
Таким образом, в случае ферментативной реакции сшивания фенольного производного полисахарида, субстрат (фенольный реакционноспособный лиганд, связанный с полимером) при помощи фермента превращался в реакционноспособный радикал. Затем этот радикал может взаимодействовать с другим фенольным радикалом с образованием дитирамина. Допуская свободную подвижность молекул субстрата (лиганда) ферментативной реакции и то, что осуществление взаимодействия точно копирует уравнение I, фермент должен постепенно превращать (при условии использования достаточного количества пероксида) все молекулы субстрата в реакционноспособные радикалы, и они все должны постепенно подвергаться димеризации (или олигомеризации), если обеспечено достаточно длительное время реакции. В случае соединения субстрата (лиганда) с полимером степень сшивания полимера должна всегда достигать того же значения, даже если время достижения этого значения будет отличаться в зависимости от количества используемого фермента. Впрочем, на практике она отличается. В литературе подробно описана взаимосвязь между предполагаемым соотношением внутримолекулярного и межмолекулярного сшивания и молекулярной массы сегментов полимерной цепи между местами сшивания (плотность поперечных связей, расстояние между узлами сети), в то время как приводящие к сшиванию внутримолекулярные взаимодействия определены как эластично не эффективные по сравнению с межмолекулярным сшиванием.
Кроме того, из литературы известно, что в случае применения фенольных производных НА количество фермента воздействует не только на скорость реакции сшивания, а также в значительной степени воздействует на полученные в результате механические свойства гидрогелей4,6,7,28. В литературе указано, что путем реологических измерений было обнаружено, что модули упругости при сдвиге (G') были выше при использовании фермента в более высокой концентрации. Согласно утверждениям авторов, причиной этого явления является более высокая плотность поперечных связей гидрогелей. Если необходимо получить максимально твердый гидрогель, реакция сшивания должна быть осуществлена при относительно высокой концентрации пероксидазы и, таким образом, также быстрее. Тем не менее, слишком быстрое проведение реакции может затем привести к образованию негомогенного сшитого гидрогеля. Тогда в образцах могут появляться места, которые совсем не сшиты. Более того, слишком быстрое проведение реакции также может вызывать проблемы при помещении геля на место его конечного применения и т.п.
Причиной этого является небольшое расстояние от реакционноспособного центра до основной полимерной цепи. Низкая подвижность лиганда снижает вероятность эффективного столкновения радикалов лиганда для образования дитирамина. Таким образом, при низкой концентрации фермента в системе за единицу времени может быть образовано небольшое количество форм реакционноспособного лиганда. Таким образом, реакция сшивания проходит медленно и является малоэффективной.
Park et. al. 29 старались увеличить реакционную способность лигандов, связанных с полимером, вставкой подходящего спейсера между реакционноспособным лигандом и полимерной цепью. Данный документ описывает вставку гидрофильной цепи между полисахаридной цепью и фенольным или анилиновым кольцом для усиления реакционной способности этих заместителей. Основной причиной для введения гидрофильной цепи в структуру полимера было улучшение его растворимости и улучшение доступности реакционноспособных центров (фенольное или анилиновое кольцо). Более легкая пространственная доступность реакционноспособных центров увеличивает вероятность взаимодействия между лигандами. Наиболее часто, поддерживая ту же ферментативную активность, эта стадия приводит к более высокой степени замещения, более высокой концентрации и лучшей гомогенности сшивания гидрогеля. Более того, как указывают авторы, благодаря введению этой гидрофильной цепи в структуру гидрогеля, биостойкость и механические свойства гидрогеля были усилены. Тем не менее, Park et al. в качестве «спейсера» использовали гидрофильный полимер PEG с молекулярной массой 3500 Да, и, таким образом, в конце он скорее представляет собой сополимер. Тем не менее, такое вмешательство в структуру гидрогеля, даже при низкой степени замещения, приводит к существенным изменениям физических свойств основного полимера. Более того, в случае гиалуроновой кислоты, более высокие концентрации сшивания приводят к увеличенной твердости гидрогеля, но в то же время они также приводят к его увеличенной хрупкости, что является нежелательным для предполагаемого использования при технологиях культивирования тканей. При рассмотрении материала, предназначенного для клеточных каркасов, например, но не только для клеточных каркасов для суставного хряща, делали акцент на его достаточную прочность и стойкость, в то время как материал, который является хрупким, необратимо деформировался при более высокой нагрузке и в случае гидрогелей даже возникает его полное разрушение.
Раскрытие настоящего изобретения
Таким образом, целью настоящего изобретения являлось выявление материала, который будет достаточно крепким и в то же время упругим и который не будет проявлять никаких существенных изменений биологических и физических свойств по сравнению с основным полимером. Прочность гидрогеля на основе гиалуроновой кислоты в целом может быть усилена увеличением концентрации сшивания, например увеличением концентрации полимера в растворе, из которого был образован гидрогель, или увеличением степени замещения полимера. Тем не менее, в существующем уровне техники в случае гиалуроновой кислоты оба эти способа приводили также к повышенной хрупкости полученного гидрогеля, что существенно ограничивает его возможные применения.
Проблема, которая решается настоящим изобретением, состоит в том, чтобы найти такие производные, которые приведут к повышенной реакционной способности лигандов и усиленной прочности гидрогеля, при этом сохраняя физические и биологические свойства основного полимера. Неожиданным образом было обнаружено, что введение относительно короткого спейсера (с молекулярной массой приблизительно 130 Да) согласно настоящему изобретению между реакционноспособным лигандом и НА приводит к значительному усилению упругости готовых гидрогелей уже при очень низкой степени замещения.
Таким образом, в одном аспекте настоящее изобретение относится к производному НА, несущему реакционноспособные лиганды, связанные через гидрофобные спейсеры с целью увеличения подвижности лиганд и, таким образом, увеличения вероятности их эффективного столкновения, даже в случае их низкой концентрации (низкой степени замещения и низкой ферментативной активности). Было обнаружено, что, несмотря на распространенность очень небольшой массы спейсера в пределах гидрогеля, составляющей, например, только от 0,01 до 0,02%, достигалось значительное увеличение упругости и прочности гидрогеля по сравнению с гидрогелем на основе аналогичного производного НА без вставленного спейсера (т.е. идентичной концентрации, молярной массы и степени замещения/сшивания). Таким образом, настоящее изобретение относится к этому новому производному гиалуроновой кислоты, подходящему для получения гидрогелей, и к способу их получения. Кроме того, оно относится к гидрогелям на основе этого производного, к их применению и к способу их получения.
Гидрогель получали способом с применением сшивания цепей модифицированной гиалуроновой кислоты при помощи реакции, которая катализируется пероксидазой хрена или ее аналогом. Подходящие производные гиалуроновой кислоты содержат в своей структуре фенольные или гетероарильные фенольные кольца, ковалентно связанные с основной полисахаридной цепью. Процедура сшивания может быть описана в виде каскада последовательных химических реакций, который начинается с образования в системе реакционноспособных форм кислорода (ROS). Их добавляли к смеси или их образование обеспечивали присутствием химических соединений, которые служат как их «генератор». ROS активизирует фермент пероксидазу или его аналоги, который впоследствии катализирует димеризацию (или олигомеризацию) ароматических или гетероароматических колец, присутствующих в структуре производного гиалуроновой кислоты. Это приводит к образованию трехмерной полимерной сети.
Согласно настоящему изобретению гиалуроновую кислоту, модифицированную связыванием лиганда, содержащего аминоалкилфенол или аминоалкилгетероарилфенол (например, тирамин, 5-гидрокситрипрофан, серотонин), использовали для получения этого гидрогеля. Описанные в настоящем изобретении производные гиалуроновой кислоты содержат лиганд, который связан с полисахаридом при помощи спейсера. Присутствие этого спейсера в структуре производного НА вследствие его гибкости приводит к увеличению эластичности и свободе возможностей конформационного расположения причастных сегментов полимерной цепи, и, таким образом, также к возможности рассеивания энергии деформации. Введение спейсера также увеличивает расстояние реакционноспособного ароматического центра (фенола, гетероарилфенола) от основной полимерной цепи, улучшает его доступность для взаимодействия с ферментом и значительно влияет на ход реакции сшивания и свойства полученного гидрогеля.
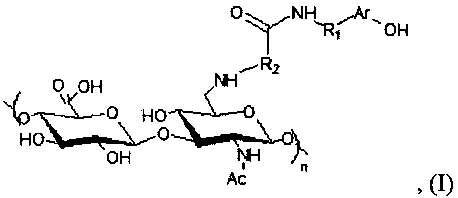
В своем первом аспекте настоящее изобретение относится к производному на основе гиалуроновой кислоты общей формулы (I)

где Ar представляет собой фенил и R1 представляет собой этилен, или Ar представляет собой индол и R1 представляет собой этилен, или Ar представляет собой индол и R1 представляет собой карбоксиэтилен, и где R2 представляет собой алкил, содержащий от 3 до 7 атомов углерода, и где n находится в диапазоне от 1 до 7500.
В другом аспекте настоящее изобретение относится к способу получения производного общей формулой (I), при котором сначала получали альдегидное производное гиалуроновой кислоты формулы (II)

причем альдегидное производное получали с применением окислительной системы 4-ацетамидо-ТЕМРО/NaClO в протонной среде, и оно характеризовалось степенью замещения 5-15% и молекулярной массой в диапазоне от 10000 г/моль до 2000000 г/моль,
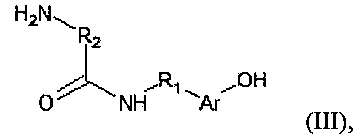
затем отдельно получали соединение общей формулы (III)

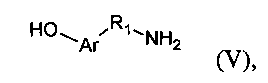
где Ar представляет собой фенил и R1 представляет собой этилен, или Ar представляет собой индол и R1 представляет собой этилен, или Ar представляет собой индол и R1 представляет собой карбоксиэтилен, и где R2 представляет собой алкил, содержащий от 3 до 7 атомов углерода, и где n находится в диапазоне от 1 до 7500, причем соединение общей формулы (III) получали путем осуществления взаимодействия промежуточного прекурсора формулы (IV)

где Ζ представляет собой защитную группу, обычно используемую для защиты первичной аминогруппы,
с лигандом формулы (V)
в апротонной среде при температуре в диапазоне от 40°С до 150°С в течение от 1 до 24 часов в присутствии агента, активирующего карбоксильные функциональные группы,
с получением соединения общей формулы (VI)
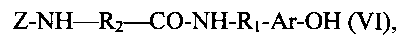
из которого получали соединение общей формулы (III) путем удаления защитной группы Ζ,
и затем осуществляли взаимодействие альдегидного производного гиалуроновой кислоты формулы (II) с соединением общей формулы (III) при значении рН в диапазоне от 3 до 8 при комнатной температуре в течение от 1 до 72 часов в присутствии пиколин-боранового комплекса с получением производного формулы (I).
Таким образом, производное по настоящему изобретению содержит лиганд, способный подвергаться олигомеризации посредством обработки подходящим агентом, и гибкий спейсер, который вставлен между цепью гиалуроновой кислоты и лигандом. Лиганд общей формулы (V) по настоящему изобретению предпочтительно выбран из группы, включающей в себя тирамин, серотонин и 5-гидрокситриптофан. Соединение общей формулы (IV), т.е. промежуточный прекурсор, предпочтительно выбрано из группы аминокислот, включающей в себя производные ω-[(трет-бутоксикарбонил)амино]карбоновых кислот, где R2 представляет собой алкил, содержащий от 3 до 7 атомов углерода.
Согласно другому предпочтительному варианту осуществления способа по настоящему изобретению взаимодействие промежуточного прекурсора с лигандом происходит в среде THF или DMF при температуре 50°С в течение от 2 до 6 часов в присутствии 1,1'-карбодиимидазола.
Кроме того, является предпочтительным, чтобы удаление защитной группы Ζ выполняли с помощью трифторуксусной кислоты или хлорводородной кислоты.
Для настоящего изобретения промежуточный спейсер-лиганд представлен соединениями общей формулы:
HO-Ar-R1-NH-CO-R2-NH2
Предпочтительно, соединение общей формулы:
-CO-R2-NH2,
где R2 представляет собой алкил, содержащий от 3 до 7 атомов углерода, используют в качестве спейсера.
Способ получения производного по настоящему изобретению может быть описан на схеме 1:

Схема 1: Пример возможного способа получения производного НА-спейсер-лиганд согласно настоящему изобретению
Кроме того, настоящее изобретение относится к гидрогелю, образованному сшиванием производного общей формулы (I), и способу его получения. Этот способ получения гидрогеля предусматривает обработку производного общей формулы (I) генератором реакционноспособных феноксильных радикалов, предпочтительно, системой пероксидазы хрена и источником гидроксильных радикалов, который может быть раствором пероксида водорода в воде, или системой оксидаза-кислород-субстрат, например, галактозоксидаза-галактоза или глюкозоксидаза-глюкоза, при значении рН в диапазоне от 4 до 10.
Таким образом, для олигомеризации реакционноспособных лиганд использовали агенты, которые способны вызвать образование феноксирадикалов из ароматических колец лиганд. Согласно настоящему изобретению предпочтительно использовали систему пероксид/пероксидаза хрена. Пероксид может быть введен в систему в форме разбавленного раствора или быть образован химической реакцией in situ. Пероксид водорода может быть образован в смеси при помощи различных видов ферментов (оксидаз) из кислорода как акцептор электронов и соответствующий донор электронов в окислительно-восстановительной реакции. Предпочтительно, может быть использована комбинация галактозоксидазы или глюкозоксидазы и их субстратов: галактозы и глюкозы. Другие агенты, которые способны обуславливать образование феноксирадикалов в присутствии молекулярного кислорода, представляют собой ферменты тирозиназы, лактазы и т.д.
Является общеизвестным, что на свойства этих гидрогелей влияет химическая структура полимера и его концентрация, а также выбранные типы сшивающих агентов и их используемое количество. Физико-химические свойства полимера (производного НА), прежде всего, затронуты структурой мономера, конформацией сегментов полимерной цепи, степенью сшивания и молекулярной массой. Механические свойства полимера также находились под влиянием этого. При воздействии механического напряжения на полимер происходит его деформация, причем часть абсорбированной энергии деформации рассеивается - потребляется для изменения конформации узлов сети и сегментов полимерной цепи, а часть энергии безвозвратно превращается в теплоту. Количество рассеянной энергии, и, таким образом, также возможность принимать различные конформационные расположения в пределах структуры полимера, связаны с густотой макромолекулярных цепей и отражают степень упругого сопротивления материала деформации. Материалы полимера, состоящие из жестких негибких цепей и их сегментов, затем могут проявлять низкую степень упругого сопротивления деформации и хрупкость.
Увеличение эластичности этих полимеров проводили способом по настоящему изобретению, при котором гибкие сегменты вводили в структуру полимера. Указанные сегменты характеризуются повышенной свободой отдельных молекул вокруг своих связей, тем самым достигая увеличения возможностей своего конформационного расположения при подвергании воздействия энергии деформации, и возможностей рассеивания указанной энергии. Следовательно, введение подходящего гибкого спейсера между лигандом и основной цепью гиалуроновой кислоты приводит к достижению более высокой эластичности, упругости и прочности готового материала, который являлся очень полезным для, например, гидрогелей, предназначенных для клеточных каркасов (scaffolds) для лечения дефектов определенных тканей, которые подвержены более сильным нагрузкам, таких как суставной хрящ или кости. Как описано выше, введение гибкого спейсера между лигандом и основной цепью гиалуроновой кислоты предпочтительно может использоваться также в случае, если механические свойства гидрогелей зависят от концентрации фермента, используемого как катализатор реакции сшивания. Введение гибкого спейсера между лигандом и основной цепью гиалуроновой кислоты обеспечивает достаточную стерическую доступность реакционноспособных групп производного для взаимной димеризации даже после частичного сшивания полимера.
Это решение приводит к более эффективной реакции сшивания, которая обусловливает более высокую гомогенность полученных гидрогелей и, таким образом, приводит к преодолению проблем технологии, связанных со сшиванием гиалуроновой кислоты, модифицированной гидроксифенилом или гетероарилфенолом (тирамином, серотонином и т.д.), в случае если агентами сшивания являются пероксидаза хрена и пероксид водорода (или другой тип генератора феноксирадикалов).
Тем не менее, неожиданным образом далее было обнаружено, что введение выбранных изобретателями спейсеров между лигандом и основной цепью гиалуроновой кислоты приводит даже при очень низкой степени замещения к значительному увеличению степени эластичности, упругости и прочности готового гидрогеля на основе производного НА.
Кроме того, настоящее изобретение относится к применению гидрогелей на основе производных согласно настоящему изобретению, особенно в области технологии культивирования тканей, для косметических средств, в медицине или регенеративной медицине. Применение описанных в настоящей заявке гидрогелей главным образом нацелено на основной материал для образования клеточных каркасов при технологии культивирования тканей, в основном в области лечения суставных дефектов и дефектов кости, такие как покрытия для заживления ран, например, препятствующие образованию послеоперационных срастаний биоразлагаемые барьеры, препараты для аугментации мягких тканей и заполнители дефектов тканей и т.п. При использовании гидрогеля в качестве материала для клеточных каркасов клеточные каркасы могут быть или высеянными, или не высеянными. Если они являются высеянными клеточными каркасами, тип клеток, который включен в клеточный каркас, выбирали в зависимости от предназначенного места применения.
Краткое раскрытие графического материала
На фиг. 1 представлены деформационные свойства (кривые «деформация-напряжение»), полученные в течение измерения деформации гидрогелей на основе производных, полученных согласно примерам VIII, IX, XI и XII, при сжатии.
Предпочтительные варианты осуществления настоящего изобретения
1. Пример синтеза производных
Синтез производных гиалуроновой кислоты проводили в несколько стадий (см. Схему 1). Первой стадией является получение альдегидного производного гиалуроновой кислоты (Пример 1.7). Другой стадией является синтез различных промежуточных соединений спейсер-лигандов (Примеры 1.1-1.6), которые затем связывали с гиалуроновой кислотой при помощи процесса восстановительного аминирования (Примеры 1.9-1.14).
Примеры также включают в себя синтез производных гиалуроновой кислоты, в которых лиганд (тирамин, гидрокситриптофан) связан непосредственно с полисахаридом без применения любого спейсера (пример VIII). Эти производные и полученные из них гидрогели служили для сравнения их свойств со свойствами описанных в настоящей заявке производных (производные НА-спейсер-лиганд-производные IX-XIV).
Пример 1.1: Синтез 6-амино-N-[2-(4-гидроксифенил)этил]гексанамида (промежуточный спейсер-лиганд (I))
6-[(трет-бутоксикарбонил)амино]гексановую кислоту (1,00 г, 4,3 ммоль) растворяли в 50 мл тетрагидрофурана (THF). К этому раствору кислоты добавляли 1,1'-карбодиимидазол (0,70 г, 4,3 ммоль). Смесь нагревали до 50°С в течение 60 минут. Затем реакционный сосуд промывали инертным газом. К реакционной смеси добавляли тирамин (0,59 г, 4,3 ммоль). Смесь дополнительно нагревали еще в течение 2 часов. Затем THF удаляли при помощи дистилляции при пониженном давлении. Осадок после выпаривания растворяли в 50 мл этилацетата. Раствор промывали 150 мл очищенной воды (разделенными на три части). Органический слой сушили над молекулярным ситом. Этилацетат удаляли дистилляцией при пониженном давлении. Осадок после выпаривания растворяли в 50 мл МеОН и добавляли 2 мл трифторуксусной кислоты (TFA). Раствор нагревали в течение 6 часов с обратным холодильником. Растворитель удаляли дистилляцией под сниженным давлением. Осадок после выпаривания растворяли в 50 мл этилацетата. Полученный раствор промывали 150 мл очищенной воды (разделенными на три части). Органический слой сушили над молекулярным ситом. Этилацетат удаляли дистилляцией под сниженным давлением.
m=0,75 г (70% теоретически)
1Н ЯМР (D2O, ppm) δ: 1,17 (m, 2H, γ-СН2-гексановая кислота); 1,48 (m, 2Н, β-СН2-гексановая кислота); 1,58 (т, 2Н, δ-СН2-гексановая кислота); 2,17 (t, 2 Η, -СН2-СО-); 2,73 (m, 2Н, -CH2-Ph); 2,91 (m, 2H, -CH2-NH2); 3,42 (m, 2H, -CH2-NH-CO-); 6,83 (d, 2H, ароматическое соединение); 7.13 (d, 2 Η, ароматическое соединение).
13С ЯМР (D2O, ppm) δ: 24 (γ-С-гексановая кислота); 26 (δ-С-гексановая кислота); 33 (β-С-гексановая кислота); 35 (-С-СО-); 39 (-C-NH2); 40 (C-Ph); 63 (-C-NH-CO-); 115 (С3 ароматическое соединение); 126 (С1 ароматическое соединение); 130 (С2 ароматическое соединение); 153 (С4 ароматическое соединение); 176 (-СО-).
Пример 1.2: Синтез 4-амино-N-[2-(4-гидроксифенил)этил]бутанамида (промежуточный спейсер-лиганд (II))
4-[(трет-бутоксикарбонил)амино]бутановую кислоту (0,50 г, 2,5 ммоль) растворяли в 25 мл тетрагидрофурана (THF). К раствору кислоты добавляли 1,1'-карбодиимидазол (0,40 г, 25 ммоль). Смесь нагревали до 50°С в течение шестидесяти минут. Затем реакционный сосуд промывали инертным газом. К реакционной смеси добавляли тирамин (0,34 г, 25 ммоль). Смесь дополнительно нагревали еще в течение 2 часов. Затем THF удаляли дистилляцией под сниженным давлением. Осадок после выпаривания растворяли в 50 мл этилацетата. Раствор промывали 150 мл очищенной воды (разделенными на три части). Органический слой сушили над молекулярным ситом. Этилацетат удаляли дистилляцией при пониженном давлении. Осадок после выпаривания растворяли в 50 мл МеОН и добавляли 2 мл трифторуксусной кислоты. Раствор нагревали в течение 6 часов с обратным холодильником. Растворитель удаляли дистилляцией под сниженным давлением. Осадок после выпаривания растворяли в 50 мл этилацетата. Раствор промывали 150 мл очищенной воды (разделенными на три части). Органический слой сушили над молекулярным ситом. Этилацетат удаляли дистилляцией при пониженном давлении.
m=0,44 г (80% теоретически)
1Н ЯМР (D2O, ppm) δ: 1,75 (m, 2H, β-СН2-бутановая кислота); 2,16 (t, 2Н, -СН2-СО-); 2,59 (m, 2Н, -СН2-In); 2,78 (m, 2Н, -CH2-NH2); 3,20 (m, 2Н, -CH2-NH-CO-); 6,69 (d, 2Н, ароматическое соединение); 6,99 (d, 2Н, ароматическое соединение).
13С ЯМР (D2O, ppm) δ: 23 (β-С-бутановая кислота); 25 (t, 2 Η, -C-CO-); 32 (-C-NH2); 45 (CH2-Ar); 60 (-C-NH-CO-); 115 (С3 ароматическое соединение); 117 (С1 ароматическое соединение); 129 (С2 ароматическое соединение); 155 (С4 ароматическое соединение); 171 (-СО-).
Пример 1.3: Синтез 8-амино-N-[2-(4-гидроксифенил)этил]октанамида (промежуточный спейсер-лиганд (III))
8-[(трет-бутоксикарбонил)амино]октановую кислоту (0,50 г, 1,9 ммоль) растворяли в 25 мл тетрагидрофурана (THF). К раствору кислоты добавляли 1,1'-карбодиимидазол (0,31 г, 1,9 ммоль). Смесь нагревали до 50°С в течение шестидесяти минут. Затем реакционный сосуд промывали инертным газом. К реакционной смеси добавляли тирамин (0,26 г, 1,9 ммоль). Смесь дополнительно нагревали еще в течение 2 часов. Затем THF удаляли дистилляцией под сниженным давлением. Осадок после выпаривания растворяли в 50 мл этилацетата. Раствор промывали 150 мл очищенной воды (разделенными на три части). Органический слой сушили над молекулярным ситом. Этилацетат удаляли дистилляцией при пониженном давлении. Осадок после выпаривания растворяли в 50 мл МеОН и добавляли 2 мл трифторуксусной кислоты. Раствор нагревали в течение 6 часов с обратным холодильником. Растворитель удаляли дистилляцией под сниженным давлением. Осадок после выпаривания растворяли в 50 мл этилацетата. Раствор промывали 150 мл очищенной воды (разделенными на три части). Органический слой сушили над молекулярным ситом. Этилацетат удаляли дистилляцией при пониженном давлении.
m=0,40 г (75% теоретически)
1H ЯМР (CDCl3, ppm) δ: 1,16-1,34 (m, 6H, С4-С6-СН2-октановая кислота); 1,56-1,44 (m, 4Н, С3-С7-октановая кислота); 2,58 (m, 2Н, -СН2-Ar); 2,78 (m, 2Н, -CH2-NH2); 3,19 (m, 2H, -СН2-NH-CO-); 6,68 (d, 2 Η, ароматическое соединение); 6,98 (d, 2Н, ароматическое соединение).
13С ЯМР (CDCl3, ppm) δ: 21 (С7 октановая кислота); 24 (С4 октановая кислота); 26 (С6-октановая кислота); 28 (С5-октановая кислота); 33 (С3-октановая кислота); 35 (-С-СО-); 39 (-C-NH2); 40 (C-Ph); 63 (-C-NH-CO-); 115 (С3 ароматическое соединение); 126 (С1 ароматическое соединение); 130 (С2 ароматическое соединение); 153 (С4 ароматическое соединение); 176 (-СО-).
Пример 1.4: Синтез 4-амино-N-[2-(5-гидрокси-1Н-индол-3-ил)этил]бутанамида (промежуточный спейсер-лиганд (IV))
4-[(трет-бутоксикарбонил)амино]бутановую кислоту (0,50 г, 2,5 ммоль) растворяли в 25 мл N,N-диметил формамида (DMF). К раствору кислоты добавляли 1,1'-карбодиимидазол (0,40 г, 2,5 ммоль). Смесь нагревали до 50°С в течение шестидесяти минут. Затем реакционный сосуд промывали инертным газом. К реакционной смеси добавляли раствор 5-гидрокситриптамина гидрохлорида (0,52 г, 2,5 ммоль) и триэтиламина (0,68 мл; 4,9 ммоль) в 25 мл DMF. Смесь дополнительно нагревали еще в течение 2 часов. Смесь разбавляли добавлением этилацетата (100 мл). Полученный раствор промывали 300 мл очищенной воды (разделенными на три части). Органический слой сушили над молекулярным ситом. Этилацетат удаляли дистилляцией под сниженным давлением. Осадок после выпаривания растворяли в 50 мл МеОН и добавляли 2 мл трифторуксусной кислоты. Раствор нагревали в течение 6 часов с обратным холодильником. Растворитель удаляли дистилляцией под сниженным давлением. Осадок после выпаривания растворяли в 50 мл этилацетата. Раствор промывали 150 мл очищенной воды (разделенными на три части). Органический слой сушили над молекулярным ситом. Этилацетат удаляли дистилляцией при пониженном давлении.
m=0,43 г (65% теоретически)
1Н ЯМР: (DMSO, ppm) δ: 1,77 (m, 2Н, β-СН2-бутановая кислота); 2,20 (t, 2Н, -СН2-СО-); 2,73 (m, 2Н, -СН2-In); 2,81 (m, 2Н, -CH2-NH2); 3,30 (m, 2Н, -CH2-NH-CO-); 6,60 (d, 1Н, С6-ароматическое соединение); 6,82 (s, 1Н, С4-ароматическое соединение); 7,03 (s, 1Н, С2-ароматическое соединение); 7,13 (d, 1Н, С7-ароматическое соединение).
13С ЯМР (DMSO, ppm) δ: 23 (β-С-бутановая кислота); 25 (t, 2 Η, -С-СО-); 32 (-С-NH2); 39 (CH2-In); 60 (-C-NH-CO-); 102 (C4 ароматическое соединение); 110 (C6 ароматическое соединение); 111 (С7 ароматическое соединение); 111 (С3 ароматическое соединение); 123 (С2 ароматическое соединение); 127 (С7 -С-NH-ароматическое соединение); 131 (С4-С-С3-ароматическое соединение); 150 (С5-ароматическое соединение); 171 (-СО-).
Пример 1.5: Синтез 6-амино-N-[2-(5-гидрокси-1Н-индол-3-ил)этил]гексанамида (промежуточный спейсер-лиганд (V))
6-[(трет-бутоксикарбонил)амино]гексановую кислоту (1,00 г, 4,3 ммоль) растворяли в 50 мл N,N-диметилформамида (DMF). К раствору кислоты добавляли 1,1'-карбодиимидазол (0,70 г, 4,3 ммоль). Смесь нагревали до 50°С в течение шестидесяти минут. Затем реакционный сосуд промывали инертным газом. К реакционной смеси добавляли раствор 5-гидрокситриптамина гидрохлорида (0,91 г, 4,3 ммоль) и триэтиламина (0,68 мл, 49 ммоль) в 25 мл DMF. Смесь дополнительно нагревали еще в течение 2 часов. Смесь разбавляли добавлением этилацетата (100 мл). Полученный раствор промывали 300 мл очищенной воды (разделенными на три части). Органический слой сушили над молекулярным ситом. Этилацетат удаляли дистилляцией при пониженном давлении. Осадок после выпаривания растворяли в 50 мл МеОН и добавляли 2 мл трифторуксусной кислоты. Раствор нагревали в течение 6 часов с обратным холодильником. Растворитель удаляли дистилляцией под сниженным давлением. Осадок после выпаривания растворяли в 50 мл этилацетата. Раствор промывали 150 мл очищенной воды (разделенными на три части). Органический слой сушили над молекулярным ситом. Этилацетат удаляли дистилляцией при пониженном давлении.
m=0,75 г (60% теоретически)
1H ЯМР: (DMSO, ppm) δ: 1,17 (m, 2H, γ-СН2-гексановая кислота); 1,48 (m, 2Н, β-СН2-гексановая кислота); 1,58 (m, 2Н, δ-СН2-гексановая кислота); 2,17 (t, 2Н, -СН2-СО-); 2,73 (m, 2Н, -СН2-In); 2,91 (m, 2Н, -CH2-NH2); 3,42 (m, 2Н, -CH2-NH-CO-); 6,60 (d, 1H, С6-ароматическое соединение); 6,82 (s, 1Н, С4-ароматическое соединение); 7,03 (s, 1Н, С2-ароматическое соединение); 7,13 (d, 1Н, С7-ароматическое соединение).
13С ЯМР (DMSO, ppm) δ: 24 (γ-С-гексановая кислота); 26 (δ-С-гексановая кислота); 33 (β-С-гексановая кислота); 35 (-С-СО-); 39 (-C-NH2); 40 (C-In); 63 (-C-NH-СО-); 102 (С4 ароматическое соединение); 110 (С6 ароматическое соединение); 111 (С7 ароматическое соединение); 111 (С3 ароматическое соединение); 123 (С2 ароматическое соединение); 127 (С7 -С-NH- ароматическое соединение); 131 (С4-С-С3-ароматическое соединение); 150 (С5 ароматическое соединение); 171 (-СО-).
Пример 1.6: Получение 2-[(6-аминогексаноил)амино]-3-(5-гидрокси-1H-индол-3-ил)-пропановой кислоты (промежуточный спейсер-лиганд VI)
6-[(трет-бутоксикарбонил)амино]гексановую кислоту (0,50 г, 2,2 ммоль) растворяли в 50 мл тетрагидрофурана (THF). К раствору кислоты добавляли 1,1'-карбодиимидазол (0,35 г, 2,2 ммоль). Смесь нагревали до 50°С в течение шестидесяти минут. Затем реакционный сосуд промывали инертным газом. К реакционной смеси добавляли 5-гидрокситриптофан (0,48 г, 2,2 ммоль). Смесь дополнительно нагревали еще в течение 2 часов. Смесь разбавляли добавлением этилацетата (100 мл). Полученный раствор промывали 300 мл очищенной воды (разделенными на три части). Органический слой сушили над молекулярным ситом. Этилацетат удаляли дистилляцией под сниженным давлением. Осадок после выпаривания растворяли в 50 мл МеОН и добавляли 2 мл трифторуксусной кислоты. Раствор нагревали в течение 6 часов с обратным холодильником. Растворитель удаляли дистилляцией под сниженным давлением. Осадок после выпаривания растворяли в 50 мл этилацетата. Раствор промывали 150 мл очищенной воды (разделенными на три части). Органический слой сушили над молекулярным ситом. Этилацетат удаляли дистилляцией при пониженном давлении.
m=0,62 г (85% теоретически)
1Н ЯМР: (DMSO, ppm) δ: 1,17 (m, 2H, γ-СН2-гексановая кислота); 1,48 (m, 2Н, β-СН2-гексановая кислота); 1,58 (m, 2Н, δ-СН2-гексановая кислота); 2,19 (t, 2Н, -СН2-СО-); 2,51 (m, 2Н, -СН2-In); 2,90 (m, 2Н, -CH2-NH2); 3,81 (m, 2Н, -CH2-NH-CO-); (m, 2Н, -CH2-NH-CO-); 6,61 (d, 1Н, С6-ароматическое соединение); 6,95 (s, 1Н, С4-ароматическое соединение); 7,02 (s, 1Н, С2-ароматическое соединение); 7,13 (d, 1Н, С7-ароматическое соединение).
13С ЯМР (DMSO, ppm) δ: 24 (γ-С-гексановая кислота); 26 (δ-С-гексановая кислота); 33 (β-С-гексановая кислота); 35 (-С-СО-); 39 (-C-NH2); 40 (C-Ph); 55 (-C-NH-СО-); 102 (С4 ароматическое соединение); 110 (С6 ароматическое соединение); 111 (С7 ароматическое соединение); 111 (С3 ароматическое соединение); 123 (С2 ароматическое соединение); 127 (С7 - С-NH- ароматическое соединение); 131 (С4-С-С3- ароматическое соединение); 150 (С5 ароматическое соединение); 171 (-СО-).
Пример 1.7: Получение альдегидного производного (НА-СНО) - общий способ (VII)
Гиалуроновую кислоту (10,00 г, Mw=2 МДа) растворяли в 750 мл 2,5% (масса/масса) раствора Na2HPO4·12H2O. Раствор охлаждали до 5°С. К полученному раствору добавляли 2,60 г NaBr и 0,05 г 4-ацетамидо-2,2,6,6-тетраметилпипперидин-1-оксила. После тщательной гомогенизации раствора к реакционной смеси добавляли 3 мл раствора NaClO (10-15% доступного Cl2). Реакцию продолжали при непрерывном перемешивании в течение 15 мин. Реакцию гасили добавлением 100 мл 40% раствора пропан-2-ола. Продукт очищали ультрафильтрацией и выделяли осаждением при помощи пропан-2-ола.
ИК (KBr): 3417, 2886, 2152, 1659, 1620, 1550, 1412, 1378, 1323, 1236, 1204, 1154, 1078, 1038, 945, 893 см-1.
1Н ЯМР (D2O) δ: 2,01 (s, 3 Η, CH3-), 3,37-3,93 (m, основа из гиалуроновой кислоты), 4,46 (s, 1Н, аномер), 4,54 (s, 1Н, аномер, -О-СН(ОН)-), 5,27 (геминальный гликоль -СН-(ОН)2).
Пример 1.8: Синтез тираминового производного (VIII)
Альдегидное производное НА (VII) (5,00 г) растворяли в 500 мл деминерализованной воды. Значение рН раствора при помощи уксусной кислоты доводили до 3. Затем к реакционной смеси добавляли тирамин (1,70 г) в форме раствора в 100 мл 40% пропан-2-ола. Смесь дополнительно перемешивали в течение 1 часа при комнатной температуре. Затем к смеси добавляли раствор пиколин-боранового комплекса (0,50 г) в 50 мл 40% пропан-2-ола. Реакционную смесь дополнительно перемешивали в течение 12 часов при комнатной температуре. Низкомолекулярные балластные вещества удаляли из продукта при помощи ультрафильтрации. Продукт получали осаждением при помощи пропан-2-ола. Осадок лишали влаги и остаточного пропан-2-ола сушкой в сушилке с обогревом горячим воздухом (40°С, 3 дня).
ИК (KBr): 3400, 2893, 2148, 1660, 1620, 1549, 1412, 1378, 1323, 1236, 1204, 1154, 1078, 1038, 945, 893 см-1.
1Н ЯМР (D2O) δ: 2,01 (s, 3Н, СН3-), 2,66-2,77 (m, 4Н, -CH2-CH2-NH-), 3,00 (s, 1Н, H-CH-NH-), 3,37-3,93 (m, основа из гиалуроновой кислоты), 4,46 (s, 1Н, аномер), 4,54 (s, 1Н, аномер, -O-CH(OH)-), 6,59 (d, 2Н, ароматическое соединение), 7,04 (d, 2Н, ароматическое соединение).
Пример 1.9: Получение тираминового производного НА со спейсером С6 (IX)
Альдегидное производное НА (VII) (5,00 г) растворяли в 500 мл деминерализованной воды. Значение рН раствора при помощи уксусной кислоты доводили до 3. Затем к раствору НА-СНО добавляли 6-амино-N-[2-(4-гидроксифенил)этил]гексанамид (промежуточное соединение (I)) (0,625 г, 2,5 ммоль). Смесь перемешивали в течение 2 часов при комнатной температуре. Затем к реакционной смеси добавляли пиколин-борановый комплекс (0,270 г, 2,5 ммоль). Смесь дополнительно перемешивали в течение 12 часов при комнатной температуре. Продукт очищали ультрафильтрацией и выделяли из ретентата осаждением при помощи пропан-2-ола. Осадок лишали влаги и остаточного пропан-2-ола сушкой в сушилке с обогревом горячим воздухом (40°С, 3 дня).
ИК (KBr): 3425, 2893, 2148, 1660, 1620, 1549, 1412, 1378, 1323, 1236, 1204, 1154, 1078, 1038, 945, 893 см-1.
1Н ЯМР (D2O) δ: 1,25 (t, 2Н, γ-СН2-аминогексановая кислота), 1,48 (m, 2Н, δ-СН2-аминогексановая кислота) 1,51 (m, 2Н, β-СН2-аминогексановая кислота), 2,01 (s, 3Н, СН3-), 2,65 (m, 2Н, Ph-CH2-), 2,73 (m, 2Н, ε-СН2-аминогексановая кислота), 3,37-3,93 (т, основа из гиалуроновой кислоты), 4,46 (s, 1Н, аномер), 4,54 (s, 1Н, аномер, -О-СН(ОН)-), 6,59 (d, 2Н, ароматическое соединение), 7,01 (d, 2Н, ароматическое соединение).
Пример 1.10: Получение производного НА со спейсером С4 и 5-гидрокситриптамином (X)
Альдегидное производное НА (VII) (3,00 г), Na2HPO4·12H2O (7,50 г) растворяли в 300 мл деминерализованной воды. Затем к раствору НА-СНО добавляли 4-амино-N-[2-(5-гидрокси-1H-индол-3-ил)этил]бутанамид (0,40 г, 1,5 ммоль) - (промежуточное соединение (IV)). Смесь перемешивали в течение 2 часов при комнатной температуре. Затем к реакционной смеси добавляли пиколин-борановый комплекс (0,16 г, 1,5 ммоль). Смесь дополнительно перемешивали в течение 12 часов при комнатной температуре. Продукт очищали ультрафильтрацией и выделяли из ретентата осаждением при помощи пропан-2-ола. Осадок лишали влаги и остаточного пропан-2-ола сушкой в сушилке с обогревом горячим воздухом (40°С, 3 дня).
ИК (KBr): 3400, 2893, 2148, 1660, 1620, 1549, 1412, 1378, 1323, 1236, 1204, 1154, 1078, 1038, 945, 893 см-1.
1Н ЯМР (D2O) δ: 1,73 (m, 2Н, β-СН2-аминобутановая кислота), 2,01 (s, 3Н, СН3), 2,60 (m, 2Н, γ-СН2-аминобутановая кислота), 2,93 (m, 2Н, Ind-CH2-), 3,37-3,93 (m, основа из гиалуроновой кислоты), 4,46 (s, 1Н, аномер), 4,54 (s, 1Н, аномер, -О-СН(ОН)-), 6,85 (d, 1Н, ароматическое соединение), 7,09 (s, 1Н, ароматическое соединение), 7,21 (s, 1Н, ароматическое соединение), 7,40 (s, 1Н, ароматическое соединение).
Пример 1.11: Получение тираминированного производного НА со спейсером С4 (XI)
Альдегидное производное НА (VII) (3,50 г) растворяли в 350 мл деминерализованной воды. Значение рН раствора при помощи уксусной кислоты доводили до 3. Затем к раствору НА-СНО добавляли 4-амино-N-[2-(4-гидроксифенил)этил]бутанамид (0,40 г, 1,8 ммоль) - (промежуточное соединение (II)). Смесь перемешивали в течение 2 часов при комнатной температуре. Затем к реакционной смеси добавляли пиколин-борановый комплекс (0,19 г, 1,8 ммоль). Смесь дополнительно перемешивали в течение 12 часов при комнатной температуре. Продукт очищали ультрафильтрацией и выделяли из ретентата осаждением при помощи пропан-2-ола. Осадок лишали влаги и остаточного пропан-2-ола сушкой в сушилке с обогревом горячим воздухом (40°С, 3 дня).
ИК (KBr): 3425, 2893, 2148, 1660, 1620, 1549, 1412, 1378, 1323, 1236, 1204, 1154, 1078, 1038, 945, 893 см-1.
1Н ЯМР (D2O) δ: 1,25 (t, 2Н, γ-СН2-аминогексановая кислота), 1,48 (m, 2Н, δ-СН2-аминогексановая кислота) 1,51 (m, 2Н, β-СН2-аминогексановая кислота), 2,01 (s, 3Н, СН3-), 2,65 (m, 2Н, Ph-CH2-), 2,73 (т, 2Н, ε-СН2-аминогексановая кислота), 3,37-3,93 (m, основа из гиалуроновой кислоты), 4,46 (s, 1Н, аномер), 4,54 (s, 1Н, аномер, -О-СН(ОН)-), 6,59 (d, 2Н, ароматическое соединение), 7,01 (d, 2Н, ароматическое соединение).
Пример 1.12: Получение тираминированного производного НА со спейсером C8 (XII)
Альдегидное производное НА (VII) (2,90 г) растворяли в 300 мл деминерализованной воды. Значение рН раствора при помощи уксусной кислоты доводили до 3. Затем к раствору НА-СНО добавляли 8-амино-N-[2-(4-гидроксифенил)этил]октанамид (0,40 г, 1,4 ммоль) - (промежуточное соединение (III)). Смесь перемешивали в течение 2 часов при комнатной температуре. Затем к реакционной смеси добавляли пиколин-борановый комплекс (0,15 г, 1,4 ммоль). Смесь дополнительно перемешивали в течение 12 часов при комнатной температуре. Продукт очищали ультрафильтрацией и выделяли из ретентата осаждением при помощи пропан-2-ола. Осадок лишали влаги и остаточного пропан-2-ола сушкой в сушилке с обогревом горячим воздухом (40°С, 3 дня).
ИК(KBr): 3425, 2893, 2148, 1660, 1620, 1549, 1412, 1378, 1323, 1236, 1204, 1154, 1078, 1038, 945, 893 см-1.
1H ЯМР (D2O) δ: 1,16-1,34 (m, 6Н, С4-С6 -СН2-октановая кислота); 1,56-1,44 (m, 4Н, С3-С7 октановая кислота); 2,01 (s, 3Н, СН3-), 2,58 (m, 2Н, -СН2-Ar); 2,78 (m, 2Н, -CH2-NH-), 3,37-3,93 (m, основа из гиалуроновой кислоты), 4,46 (s, 1Н, аномер), 4,54 (s, 1H, аномер, -О-СН(ОН)-), 6,59 (d, 2Н, ароматическое соединение), 7,01 (d, 2Н, ароматическое соединение).
Пример 1.13: Получение производного НА со спейсером С6 и 5-гидрокситрипамином (XIII)
Альдегидное производное НА (VII) (5,00 г) и Na2HPO4·12H2O (12,5 г) растворяли в 500 мл деминерализованной воды. Затем к раствору НА-СНО добавляли 6-амино-N-[2-(5-гидрокси-1H-индол-3-ил)этил]гексанамид (0,73 г, 2,5 ммоль) - (промежуточное соединение (V)). Смесь перемешивали в течение 2 часов при комнатной температуре. Затем к реакционной смеси добавляли пиколин-борановый комплекс (0,27 г, 2,5 ммоль). Смесь дополнительно перемешивали в течение 12 часов при комнатной температуре. Продукт очищали ультрафильтрацией и выделяли из ретентата осаждением при помощи пропан-2-ола. Осадок лишали влаги и остаточного пропан-2-ола сушкой в сушилке с обогревом горячим воздухом (40°С, 3 дня).
ИК (KBr): 3400, 2893, 2148, 1660, 1620, 1549, 1412, 1378, 1323, 1236, 1204, 1154, 1078, 1038, 945, 893 см-1.
1Н ЯМР (D2O) δ: 1,25 (t, 2Н, γ-СН2-аминогексановая кислота), 1,48 (m, 2Н, δ-СН2-аминогексановая кислота) 1,51 (m, 2Н, β-СН2-аминогексановая кислота), 2,01 (s, 3Н, СН3-), 2,65 (m, 2Н, Ph-CH2-), 2,73 (m, 2Н, 8-СН2-аминогексановая кислота), 3,37-3,93 (m, основа из гиалуроновой кислоты), 4,46 (s, 1Н, аномер), 4,54 (s, 1Н, аномер, -О-СН(ОН)-), 6,85 (d, 1Н, ароматическое соединение), 7,09 (s, 1Н, ароматическое соединение), 7,21 (s, 1Н, ароматическое соединение), 7,40 (s, 1Н, ароматическое соединение).
Пример 1.14: Получение производного НА со спейсером С6 и 5-гидрокситриптофаном (XIV)
Альдегидное производное НА (VII) (3,50 г) и Na2HPO4·12H2O (8,75 г) растворяли в 350 мл деминерализованной воды. Затем к раствору НА-СНО добавляли 2-[(6-аминогексаноил)амино]-3-(5-гидрокси-1Н-индоле-3 -ил)пропановую кислоту (0,60 г, 1,8 ммоль) - (промежуточное соединение (VI)). Смесь перемешивали в течение 2 часов при комнатной температуре. Затем к реакционной смеси добавляли пиколин-борановый комплекс (0,19 г, 1,8 ммоль). Смесь дополнительно перемешивали в течение 12 часов при комнатной температуре. Продукт очищали ультрафильтрацией и выделяли из ретентата осаждением при помощи пропан-2-ола. Осадок лишали влаги и остаточного пропан-2-ола сушкой в сушилке с обогревом горячим воздухом (40 С°, 3 дня).
ИК (KBr): 3400, 2893, 2148, 1660, 1620, 1549, 1412, 1378, 1323, 1236, 1204, 1154, 1078, 1038, 945, 893 см-1.
1Н ЯМР (D2O) δ: 1,25 (t, 2Н, γ-СН2-аминогексановая кислота), 1,48 (m, 2Η, δ-СН2-аминогексановая кислота) 1,51 (m, 2Н, β-СН2-аминогексановая кислота), 2,01 (s, 3Н, СН3-), 2,65 (m, 2Н, Ph-CH2-), 2,73 (m, 2Н, ε-СН2-аминогексановая кислота), 3,37-3,93 (m, основа из гиалуроновой кислоты), 4,46 (s, 1H, аномер), 4,54 (s, 1Н, аномер, -О-СН(ОН)-), 6,85 (d, 1Н, ароматическое соединение), 7,09 (s, 1Н, ароматическое соединение), 7,21 (s, 1Н, ароматическое соединение), 7,40 (s, 1Н, ароматическое соединение).
Пример 1.15: Общий способ получения гидрогеля на основе производного НА со спейсером и 5-гидрокситриптофаном и на основе тираминового производного
Выбранное производное НА растворяли в 0,1 Μ PBS при рН 7,4. Количество производного выбирали согласно желаемой концентрации. К раствору производного добавляли желаемое количество фермента. После тщательной гомогенизации добавляли разбавленный раствор пероксида водорода. Смесь снова гомогенизировали и образовывали прозрачный гель.
Пример 1.16: Получение гидрогеля на основе тираминового производного
40-60 мг (согласно желаемой концентрации раствора полимера) производного НА, полученного согласно примеру 1.8 (VIII), растворяли в 2 мл 0,1 Μ PBS со значением рН 7,4. Затем к раствору производного добавляли 20 мкл раствора фермента HRP (24 мг фермента HRP, растворенного в 1 мл 0,1 Μ PBS со значением рН 7,4). После тщательной гомогенизации добавляли 100 мкл раствора H2O2 (33 мкл 30% H2O2, растворенного в 10 мл 0,1 Μ PBS со значением рН 7,4). Смесь гомогенизировали и образовывали прозрачный гель.
Пример 1.17: Получение гидрогеля на основе тираминового производного НА со спейсером
40-60 мг (согласно желаемой концентрации раствора полимера) производного НА, полученного согласно примеру 1.9 (IX), 1.11 (XI) или 1.12 (XII), растворяли в 2 мл 0,1 Μ PBS со значением рН 7,4. К раствору производного добавляли 10 мкл раствора фермента HRP (2,4 мг фермента HRP, растворенного в 1 мл 0,1 Μ PBS со значением рН 7,4). После тщательной гомогенизации добавляли 100 мкл раствора H2O2 (33 мкл 30% H2O2, растворенного в 10 мл 0,1 Μ PBS со значением рН 7,4). Смесь гомогенизировали и образовывали прозрачный гель.
2. Различия в свойствах гидрогеля
Пример 2.1: Различие в механических свойствах гидрогелей в зависимости от типа используемого производного НА и количества добавленного фермента
Образцы гидрогелей из производных VIII (тирамин, без спейсера), IX, XI и XII (со спейсером) получали согласно примерам 1.16 или 1.17 в зависимости от типа используемого производного. После тщательной гомогенизации образцы выдерживали в течение 120 минут при комнатной температуре. Аналоги производных, используемые для получения сравниваемых гидрогелей, всегда обладали сопоставимой молекулярной массой и степенью замещения. Все образцы обладали одинаковыми размерами и исследовались при постоянных лабораторных условиях (температура, давление, влажность).
Модули Юнга эластичности при сжатии, упругости, прочности при сжатии и соответствующую деформацию образца измеряли для каждого образца; и для определения вязкоэластичных свойств образцов измеряли модули сдвига и тангенс угла потерь.
Полученные данные четко показывают, что введение гибкого спейсера между лигандом и основной цепью гиалуроновой кислоты приводит к более высокой эластичности, стойкости и прочности гидрогелей на основе указанных производных по сравнению с гидрогелями на основе аналогичных производных гиалуроновой кислоты без какого-либо спейсера.
В таблице 1 показано сравнение механических свойств гидрогеля в зависимости от типа производного, используемого для его получения. Концентрация означает (%) концентрацию полимера в растворе, из которого получали гидрогель, степень замещения (%) означает степень замещения реакционноспособным/сшивающим лигандом, т.е. число связанных лиганд на 100 структурных единиц полимера, где в случае НА структурная единица полимера представляет собой дисахарид (или димер) глюкозамина + глюкуроновая кислота.

Источники информации
1. Slaughter, В.V.; Khurshid, S.S.; Fisher, Ο.Z.; Khademhosseini, Α.; Peppas, Ν.Α., Hydrogels in Regenerative Medicine. Advanced Materials 2009, 21 (32-33), 3307-3329.
2. Benedetti, L.; Cortivo, R.; Berti, Т.; Berti, Α.; Pea, F.; Mazzo, M.; Moras, M.; Abatangelo, G., Biocompatibility and biodegradation of different hyaluronan derivatives (Hyaff) implanted in rats. Biomaterials 1993, 14 (15), 1154-1160.
3. Calabro, Α.; Gross, R.Α.; Darr, A.B. Hydroxyphenyl cross-linked macromolecular network and applications thereof. 2004.
4. Calabro, Α.; Akst, L.; Alam, D.; Chan, J.; Darr, А.В.; Fukamachi, K.; Gross, R.Α.; Haynes, D.; Kamohara, K.; Knott, D.P.; Lewis, H.; Melamud, Α.; Miniaci, Α.; Strome, M. Hydroxyphenyl cross-linked macromolecular network and applications thereof. 2008 (WO 2006/010066).
5. Tan, H.; Chu, C.R.; Payne, Κ.Α.; Marra, K.G., Injectable in situ forming biodegradable chitosan-hyaluronic acid based hydrogels for cartilage tissue engineering. Biomaterials 2009, 30 (13), 2499-2506.
6. Darr, Α.; Calabro, Α., Synthesis and characterization of tyramine-based hyaluronan hydrogels. Journal of Materials Science: Materials in Medicine 2009, 20 (1), 33-44.
7. Kurisawa, M.; Lee, F.; Chung, J.E. Formation of Hydrogel in the Presence of Peroxidase and Low Concentration of Hydrogen Peroxide 2009 (WO 2009/148405).
8. Lee, F.; Chung, J.E.; Kurisawa, M., An injectable enzymatically crosslinked hyaluronic acid-tyramine hydrogel system with independent tuning of mechanical strength and gelation rate. Soft Matter 2008, 4, 880-887.
9. Akkara, J.Α.; Senecal, K.J.; Kaplan, D.L., Synthesis and characterization of polymers produced by horseradish peroxidase in dioxane. Journal of Polymer Science Part A: Polymer Chemistry 1991, 29 (11), 1561-1574.
10. Shutava, Т.; Zheng, Z.; John, V.; Lvov, Y., Microcapsule modification with peroxidase-catalyzed phenol polymerization. Biomacromolecules 2004, 5 (3), 914-21.
11. Ghan, R.; Shutava, Т.; Patel, Α.; John, V.Т.; Lvov, Y., Enzyme-Catalyzed Polymerization of Phenols within Polyelectrolyte Microcapsules. Macromolecules 2004, 37 (12), 4519-4524.
12. Higashimura, H.; Kobayashi, S., Oxidative Polymerization. John Wiley & Sons, Inc.: 2002.
13. Veitch, N.C, Horseradish peroxidase: a modern view of a classic enzyme. Phytochemistry 2004, 65 (3), 249-259.
14. Gilabert, Μ.Α.; Phenoll, L.G.; Garcia-Molina, F.; Garcia-Ruiz, P.Α.; Tudela, J.; Garcia-Canovas, F.; Rodriguez-Lopez, J.N., Stereospecificity of horseradish peroxidase. Biol Chem 2004, 385 (12), 1177-84.
15. Uyama, H.; Kobayashi, S., Enzymatic Synthesis of Polyphenols. Current Organic Chemistry 2003, 7, 1387.
16. Gilabert, Μ.Α.; Phenoll, L.G.; Garcia-Molina, F.; Tudela, J.; Garcia-Canovas, F.; Rodriguez-Lopez, J.N., Kinetic characterization of phenol and aniline derivates as substrates of peroxidase. Biol Chem 2004,385 (9), 795-800.
17. Gilabert, Μ.Α.; Hiner, A.N.; Garcia-Ruiz, P.Α.; Tudela, J.; Garcia-Molina, F.; Acosta, M.; Garcia-Canovas, F.; Rodriguez-Lopez, J.N., Differential substrate behaviour of phenol and aniline derivatives during oxidation by horseradish peroxidase: kinetic evidence for a two-step mechanism. Biochim Biophys Acta 2004, 1699 (1-2), 235-43.
18. Hewson, W.D.; Dunford, H.В., Oxidation of p-cresol by horseradish peroxidase compound I. J Biol Chem 1976,251 (19), 6036-42.
19. Burner, U.; Obinger, С, Transient-state and steady-state kinetics of the oxidation of aliphatic and aromatic thiols by horseradish peroxidase. FEBS Letters 1997, 411 (2-3), 269-274.
20. Patel, P.K.; Mondal, M.S.; Modi, S.; Behere, D.V., Kinetic studies on the oxidation of phenols by the horseradish peroxidase compound II. Biochim Biophys Acta 1997, 1339 (1), 79-87.
21. Hewson, W.D.; Dunford, H.В., Stoichiometry of the reaction between horseradish peroxidase and p-cresol. J Biol Chem 1976, 251 (19), 6043-52.
22. Job, D.; Dunrord, H.В., Substituent effect on the oxidation of phenols and aromatic amines by horseradish peroxidase compound I. Eur J Biochem 1976, 66 (3), 607-14.
23. Dunrord, H.В.; Cotton, M.L., Kinetics of the oxidation of p-aminobenzoic acid catalyzed by horseradish peroxidase compounds I and II. J Biol Chem 1975, 250 (8), 2920-32.
24. Kalyanaraman, В.; Felix, С.C; Sealy, R.C, Peroxidatic oxidation of catecholamines. A kinetic electron spin resonance investigation using the spin stabilization approach. Journal of Biological Chemistry 1984, 259 (12), 7584-7589.
25. Won, K.; Kim, Υ.H.; An, E.S.; Lee, Y.S.; Song, В.K., Horseradish Peroxidase-Catalyzed Polymerization of Cardanol in the Presence of Redox Mediators. Biomacromolecules 2003, 5 (1), 1-4.
26. Xu, Y. - P.; Huang, G. - L.; Yu, Y. - T., Kinetics of phenolic polymerization catalyzed by peroxidase in organic media. Biotechnology and Bioengineering 1995, 47 (1), 117-119.
27. Tonelli, A.E., Effects of crosslink density and length on the number of intramolecular crosslinks (defects) introduced into a rubbery network. Polymer 1974, 15 (4), 194-196.
28. Jin, R.; Hiemstra, C; Zhong, Z.; Feijen, J., Enzyme-mediated fast in situ formation of hydrogels from dextran-tyramine conjugates. Biomaterials 2007, 28 (18), 2791-2800.
29. Park, K. - D.; Joung, Y. - K.; Park, K. - M. In situ Forming Hydrogel and Biomedical Use Thereof 2011 (WO 2011/028031).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ КОРИЧНОЙ КИСЛОТЫ, ПРОИЗВОДНОЕ КОРИЧНОЙ КИСЛОТЫ И ПОЛИСАХАРИДА, СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО КОРИЧНОЙ КИСЛОТЫ И ГЛИКОЗАМИНОГЛИКАНА, СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО КОРИЧНОЙ КИСЛОТЫ И ПОЛИАМИНОСАХАРИДА, СПОСОБ ПОЛУЧЕНИЯ СШИТОГО ПРОИЗВОДНОГО КОРИЧНОЙ КИСЛОТЫ И ПОЛИСАХАРИДА | 1995 |
|
RU2169136C2 |
| Способ получения производных N-гидроксибутанамида | 2020 |
|
RU2769320C1 |
| СРЕДСТВО ПЕПТИДНОЙ ПРИРОДЫ, ВКЛЮЧАЮЩЕЕ ПСМА-СВЯЗЫВАЮЩИЙ ЛИГАНД НА ОСНОВЕ ПРОИЗВОДНОГО МОЧЕВИНЫ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ ДЛЯ ПОЛУЧЕНИЯ КОНЪЮГАТА С ЛЕКАРСТВЕННЫМ И ДИАГНОСТИЧЕСКИМ АГЕНТОМ | 2018 |
|
RU2697519C1 |
| СПОСОБ ПОЛУЧЕНИЯ ФУНКЦИОНАЛИЗОВАННЫХ ПРОИЗВОДНЫХ ГИАЛУРОНОВОЙ КИСЛОТЫ И ОБРАЗОВАНИЯ ИХ ГИДРОГЕЛЕЙ | 2009 |
|
RU2523182C2 |
| С-С-АЦИЛИРОВАННОЕ ПРОИЗВОДНОЕ ГИАЛУРОНОВОЙ КИСЛОТЫ, СПОСОБ ЕГО ПОЛУЧЕНИЯ, НАНОМИЦЕЛЛЯРНАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ СТАБИЛИЗИРОВАННОЙ НАНОМИЦЕЛЛЯРНОЙ КОМПОЗИЦИИ И ЕЕ ПРИМЕНЕНИЕ | 2013 |
|
RU2640287C2 |
| ПРОИЗВОДНОЕ БИСАМИДОВ ДИКАРБОНОВЫХ КИСЛОТ, ЕГО ПРИМЕНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ, СПОСОБЫ ЕГО ПОЛУЧЕНИЯ | 2018 |
|
RU2725881C2 |
| ПРОИЗВОДНОЕ БИСАМИДОВ ДИКАРБОНОВЫХ КИСЛОТ, ЕГО ПРИМЕНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ, СПОСОБЫ ЕГО ПОЛУЧЕНИЯ | 2018 |
|
RU2721421C2 |
| Способ получения гидроксамовых кислот, производных 2-арил-2,3-дигидрохиназолин-4(1Н)-онов | 2020 |
|
RU2744750C1 |
| ПРОИЗВОДНОЕ ГИАЛУРОНОВОЙ КИСЛОТЫ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ ЕГО МОДИФИКАЦИИ | 2010 |
|
RU2550602C2 |
| СОЕДИНЕНИЕ ПЕПТИДНОЙ ПРИРОДЫ, ОБЛАДАЮЩЕЕ СПОСОБНОСТЬЮ СВЯЗЫВАТЬСЯ С ПСМА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2021 |
|
RU2823164C2 |

Изобретение относится к новому производному гиалуроновой кислоты общей формулы (I), способу его получения, к гидрогелю на его основе, к способу получения гидрогеля и к применению гидрогеля для получения препаратов для косметических средств, медицины или регенеративной медицины. Причем Ar представляет собой фенил и R1 представляет собой этилен, или Ar представляет собой индол и R1 представляет собой этилен, или Ar представляет собой индол и R1 представляет собой карбоксиэтилен. R2 представляет собой алкил с 3-7 атомами углерода, а n находится в диапазоне от 1 до 7500. Изобретение позволяет получить материал, достаточно крепкий и одновременно упругий, не проявляющий существенных изменений биологических и физических свойств и образующий прочный гидрогель. 5 н. и 7 з.п. ф-лы, 1 ил., 1 табл., 18 пр.

1. Производное на основе гиалуроновой кислоты общей формулы (I)
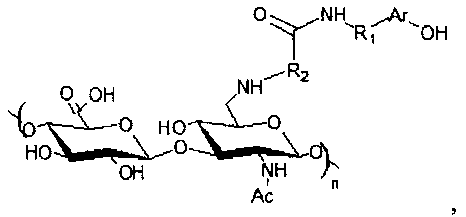 (I)
(I)
где Ar представляет собой фенил и R1 представляет собой этилен, или Ar представляет собой индол и R1 представляет собой этилен, или Ar представляет собой индол и R1 представляет собой карбоксиэтилен, и где R2 представляет собой алкил, содержащий от 3 до 7 атомов углерода, и где n находится в диапазоне от 1 до 7500.
2. Способ получения производного, обозначенного общей формулой (I), который предусматривает сначала получение альдегидного производного гиалуроновой кислоты формулы (II)

причем альдегидное производное получают с применением окислительной системы 4-ацетамидо-ТЕМРО/NaClO в протонной среде, и характеризующегося степенью замещения 5-15% и молекулярной массой в диапазоне от 10000 г/моль до 2000000 г/моль, затем отдельное получение соединения общей формулы (III)
где Ar представляет собой фенил и R1 представляет собой этилен, или Ar представляет собой индол и R1 представляет собой этилен, или Ar представляет собой индол и R1 представляет собой карбоксиэтилен, и где R2 представляет собой алкил, содержащий от 3 до 7 атомов углерода, и где n находится в диапазоне от 1 до 7500, причем соединение общей формулы (III) получают путем осуществления взаимодействия промежуточного прекурсора формулы (IV)

где Ζ представляет собой защитную группу, обычно используемую для защиты первичной аминогруппы,
с лигандом формулы (V)
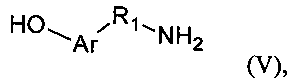
в апротонной среде при температуре в диапазоне от 40°C до 150°C в течение от 1 до 24 часов в присутствии агента, активирующего карбоксильные функциональные группы,
с получением соединения общей формулы (VI)

из которого получают соединение общей формулы (III) путем удаления защитной группы Ζ,
и затем осуществляют взаимодействие альдегидного производного гиалуроновой кислоты формулы (II) с соединением общей формулы (III) при значении pH в диапазоне от 3 до 8 при комнатной температуре в течение от 1 до 72 часов в присутствии пиколин-боранового комплекса с получением производного формулы (I).
3. Способ получения по п. 2, характеризующийся тем, что лиганд общей формулы (V) выбран из группы, включающей в себя тирамин, серотонин и 5-гидрокситриптофан.
4. Способ получения по любому из пп. 2 и 3, характеризующийся тем, что соединение общей формулы (IV), представляющее собой промежуточный прекурсор, выбрано из группы аминокислот, включающей в себя производные ω-[(трет-бутоксикарбонил)амино]карбоновых кислот, где R2 представляет собой алкил, содержащий от 3 до 7 атомов углерода.
5. Способ получения по любому из пп. 2 и 3, характеризующийся тем, что взаимодействие промежуточного прекурсора с лигандом происходит в среде THF или DMF при температуре 50°C в течение от 2 до 6 часов в присутствии 1,1′-карбодиимидазола.
6. Способ получения по любому из пп. 2 и 3, характеризующийся тем, что удаление защитной группы Ζ выполняют с помощью трифторуксусной кислоты или хлорводородной кислоты.
7. Гидрогель на основе сшитого производного общей формулы (I)
где Ar представляет собой фенил и R1 представляет собой этилен, или Ar представляет собой индол и R1 представляет собой этилен, или Ar представляет собой индол и R1 представляет собой карбоксиэтилен, и где R2 представляет собой алкил, содержащий от 3 до 7 атомов углерода, и где n находится в диапазоне от 1 до 7500.
8. Способ получения гидрогеля по п. 7, характеризующийся тем, что производное общей формулы (I) обрабатывают генератором реакционноспособных феноксильных радикалов при значении pH в диапазоне от 4 до 10.
9. Способ получения по п. 8, характеризующийся тем, что генератор реакционноспособных феноксильных радикалов выбран из группы, включающей в себя систему пероксидазы хрена и источник гидроксильных радикалов, причем источником гидроксильных радикалов может быть раствор пероксида водорода в воде, или систему оксидаза-кислород-субстрат, например галактозоксидаза-галактоза или глюкозоксидаза-глюкоза.
10. Применение гидрогеля по п. 7 для получения препаратов для косметических средств, в медицине или регенеративной медицине.
11. Применение по п. 10, при котором препараты включают в себя покрытия для заживления ран, препятствующие образованию послеоперационных срастаний биоразлагаемые барьеры, препараты для аугментации мягких тканей, заполнители дефектов тканей, клеточные каркасы для технологий культивирования тканей.
12. Применение по п. 10, при котором препараты включают в себя засеянные или не засеянные клеточные каркасы для лечения дефектов суставного хряща и дефектов костей.
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| ПОПЕРЕЧНОСШИТЫЕ ГИАЛУРОНОВЫЕ КИСЛОТЫ И ИХ ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 1999 |
|
RU2230752C2 |

