Область техники
Настоящее изобретение относится к способу получения гидрофобизированной гиалуроновой кислоты и ее применению для переноса биологически активных гидрофобных веществ, причем биологически активное вещество инкапсулировано в наномицеллы гидрофобизированного гиалуронана. Гидрофобизацию гиалуронана проводили с помощью реакции эстерификации гиалуронана с длинноцепочечными карбоновыми кислотами, последние были активированы галогенидным производным 2,4,6-трихлорбензойной кислоты или другом органическим хлоридом R3-CO-Cl. В подходящей водной среде растворимые в воде гидрофобизированные производные могут образовывать наномицеллы, в которых неполярные вещества могут быть связаны посредством нековалентных физических взаимодействий. Ядро наномицеллы образовано гидрофобными ацильными функциональными группами, в то же время оболочка наномицеллы образована гиалуроновой кислотой. Инкапсуляция веществ в наномицеллы может быть выполнена при помощи способа замены растворителя или при помощи обработки ультразвуком. Гиалуроновые наномицеллы способствуют проникновению связывающих веществ при местных применениях и позволяют связывающим веществам переноситься в отдельные клетки. Настоящее изобретение дополнительно относится к способу получения стабилизированных наномицеллярных композиций. Наномицеллы, полученные из производных гидрофобизированного гиалуронана, применимы при косметических и фармацевтических применениях.
Уровень техники
Гиалуроновая кислота является важным полисахаридом, включающим в себя две повторяющиеся единицы β-(1,3)-D-глюкуроновой кислоты и β-(1,4)-N-ацетил-D-глюкозамина. Она характеризуется высокой молекулярной массой в диапазоне от 5,104 до 5,106 г. моль-1, которая зависит от способа выделения и от используемого начального вещества. Гиалуроновая кислота, а в частности ее натриевая соль, известна как гиалуронан, и является основным составляющим соединительных тканей и синовиальной жидкости. Более того, она играет значительную роль в многочисленных биологических процессах, таких как гидратация, группировка протеогликанов, клеточная дифференцировка, пролиферация и ангиогенез. Такой полисахарид, который является сильно гидрофильным, растворим в воде в форме солей в пределах всего масштаба значений рН.
Системы носителя на основе гиалуроновой кислоты
Вследствие гидрофильной природы своей нативной формы гиалуроновая кислота не может служить эффективным носителем для гидрофобных веществ. По этой причине гидрофобные функциональные группы должны быть связаны с полимерной цепью гиалуроновой кислоты. В случае если такие гидрофобные функциональные группы обладали достаточными количествами и длиной, может быть начат процесс автоагрегации, включающий в себя то же самое, что приводило к образованию гидрофобных доменов в структуре гиалуронана. После этого небольшие молекулы нерастворимых в воде веществ могут быть связаны с такими доменами при помощи нековалентных связей. Полученная структура в литературе часто была обозначена как полимерная наномицелла, при этом ядро мицеллы было гидрофобным, тем самым позволяя прохождение растворения небольших неполярных молекул, в то время как ее оболочка была гидрофильной, тем самым позволяя самой полимерной мицелле растворяться в водной среде. Полимерная мицелла, которая в размере не превышает 200 нм (диаметр), может называться как наномицелла.
Системы носителя, образованные полимерными мицеллами на основе модифицированной гиалуроновой кислоты, известны из конъюгатов гиалуронана с алкиламинами (Liu et al., 2011) и фолиевой кислотой. При этом полагали, что присутствие высокотоксичного и тератогенного формамида было существенным для получения вышеуказанного гиалуронанового производного. Подобный способ получения полимерных мицелл использовали для получения редокс-чувствительных мицелл (Li et al., 2011). Является очевидным, что такие мицеллярные системы не применимы при биологических применениях из-за присутствия высокотоксичных реагентов.
Конъюгирование гиалуроновой кислоты с другими полимерами (например, молочных и гликолевых кислот) посредством связи, опосредованной карбоксильной группой D-глюкуроновой кислоты и, возможно, посредством включения низкомолекулярных веществ, заявлено в патенте США №7767806, причем авторы упоминают биологическую совместимость полимера, что, тем не менее, не было ни описано, ни доказано испытаниями. В другом случае, низкомолекулярный гиалуронан (9-45 кДа) был ковалентно модифицирован (в положении карбоксильной группы глюкуроновой кислоты) гидрофобными аминами с различной длиной цепи и положительно заряженным спермином, используемым в качестве катионного сегмента (Shen, Li, Tu & Zhu, 2009). Целью последнего было получения носителя для генов. Тем не менее применение спермина для вышеуказанной цели ограничено вследствие его острой и подострой токсичности (Til, Falke, Prinsen & Willems, 1997). Определяли критическую мицеллярную концентрацию образованных полимерных мицелл с диаметром 125-555, которая была выше 0,04 мг.мл-1. В дополнение к тому факту, что значение критической мицеллярной концентрации является слишком высоким и, таким образом, не позволяет никакого чрезмерного разбавления полученной мицеллярной системы (например, в кровообращении), мицеллы с вышеуказанным размером не рассматривались как подходящие кандидаты для пассивного распределения медикаментов в человеческом организме, поскольку размер полимерной мицеллы является ограничивающим ее способность достигать местоположения опухоли или инфарктного поражения через разорванную венозную стенку. В таких случаях полимерные мицеллы с диаметром 20-100 нм являются предпочтительными (Wang, Mongayt & Torchilin, 2005).
Модификацию карбоксильной группы глюкуроновой кислоты использовали для получения полимерных гиалуронановых мицелл на основе электростатического взаимодействия между отрицательно заряженной гиалуроновой кислоты и положительно заряженного стирилпиридиния (Tao, Xu, Chen, Bai & Liu, 2012). Тем не менее, применение таких полимерных мицеллы in vivo ограничено вследствие того факта, что взаимодействия стирилпиридиния с нервными окончаниями и мускариновыми рецепторами до настоящего времени не были в достаточной степени пояснены, хотя такие взаимодействия играют важную роль в связи с регулированием высвобождения нейротрансмиттеров из нейронов (Mazzone, Mori, Burman, Palovich, Belmonte & Canning, 2006).
В патентных документах США №7993678 и WO 2007/033677 заявлен способ получения алкил/арилянтарных производных гиалуронана, такие производные также были применимы для инкапсуляции активных неполярных веществ. В последнем случае модификация включает в себя первичные гидроксильные группы гиалуронана, в то время как карбоксильная группа остается неизменной. Недостатком вышеуказанной реакции модификации является щелочной диапазон значений рН (рН 8,5-9,0), в котором проходит реакция циклических ангидридов с гиалуронаном. Фактически, такие щелочные значения рН могут вызывать гидролиз ангидридов и, таким образом, быть причиной уменьшения эффективности процесса модификации. Это будет особенно существенным в промышленном масштабе. В алкил/арилянтарных производных гиалуронана, которые получали вышеуказанным способом, и они обладали степенью замещения 44%, способность образовывать мицеллы (для агрегирования) в водной среде была доказана, если соответствующая концентрация была выше чем 0,003-0,004 мг.мл-1. Наблюдаемый размер полимерных мицелл находился в диапазоне от 50 до 200 нм. Тем не менее, недостаток таких производных заключается в увеличении общего отрицательного заряда гиалуронана, вызванного присутствием дополнительной СОО- группы при модификации алкил/арилянтарной функциональной группы. Отрицательный заряд молекулы может обладать значительным неблагоприятным влиянием на взаимодействие между клетками и соответствующей системой носителя (Wang, Mongayt & Torchilin, 2005). Один из ограничивающих факторов для инъекционного применения производных, полученных вышеуказанным способом, заключается в их низкой растворимости (Eenschooten, Guillaumie, Kontogeorgis, Stenby & Schwach-Abdellaoui, 2010). Другой недостаток таких производных заключается в нестабильности сложноэфирных связей в течение термических процессов стерилизации, таких как автоклавирование. В патентных документах США №7993678 и WO 2007/033677 описан только способ прямого растворения неполярных веществ в растворах алкил/арилянтарных производных гиалуронана, включая в себя образование стабильной эмульсии. Главный недостаток прямого способа инкапсулирования неполярных веществ заключается в низкой связующей способности полимерных мицелл (Kedar, Phutane, Shidhaye & Kadam, 2010). Хотя в вышеуказанных патентах заявлено использование структуры модифицированного гиалуронана для систем носителя, в них не обеспечено никакого дополнительного возможного способа связывания гидрофобного вещества до представленной структуры гиалуронана в дополнение к эмульсионной системе. По этой причине, обеспечение обеих полимерных систем носителя с достаточной связующей способностью полностью отсутствует, что будет одной из основных характеристик, требуемых для реальной оценки применимости. Более того, не были упомянуты детали в отношении цитотоксичности и клеточных взаимодействий, и, таким образом, невозможно прийти к заключению о том, будет ли заявленная структура в действительности применимой для активного переноса гидрофобных веществ в клетки, а также заключением является необходимым в фармацевтических применениях.
В дальнейшей публикации (Šmejkalová, Hermannová, Šuláková, Průšová, Kučerík & Velebný, 2012) описаны гидрофобные домены гиалуронана, которые происходят из агрегирования С6-ацильных цепей, связанных с гиалуронаном ковалентными связями. Тем не менее, описанные в более поздней публикации производные не полностью свободны от остаточных растворителей. Кроме того, ни образование, ни характеристика полимерных мицелл не обсуждались в вышеуказанной публикации. Более того, упомянутые в вышеуказанной публикации симметричные ангидриды не являются применимыми для образования связей между длинными алкильными цепями. Карбоновые кислоты с длинными алифатическими цепями являются очень дорогостоящими и, вдобавок ко всему, по меньшей мере один моль кислоты теряется в течение получения одного моля конечного реагента. Также в публикации не обсуждается цитотоксичность полученных производных.
Получение масляных сложных эфиров полисахаридов, включая соответствующую фармацевтическую композицию, было заявлено в патенте ЕР 0941253. Тем не менее заявленный способ получения позволяет достижение только очень низких степеней замещения (макс. 3%). Количество гидрофобного вещества, которое связано с полученными производными посредством нековалентной связи, находится под неблагоприятным влиянием такой низкой степени замещения. Масляные сложные эфиры гиалуронана дополнительно получали согласно патентному документу WO 2005/092929, в котором, тем не менее, не водные условия. Следовательно, превращение гиалуронана в четвертичную аммонийную соль может сопровождаться распадом гиалуронана. Достигнутая степень замещения была ниже 0,1% и, таким образом, такие сложноэфирные производные не подходят для получения систем носителя. Подобные результаты получали при проведении одновременной эстерификации гиалуронана с ангидридом масляной кислоты и хлоридом ретиноевой кислоты (WO 2004/056877).
Композиция полимерных мицелл на основе модифицированного гиалуронана (HA)-[O(C=O)NH-M]p, где M представляет собой модифицированную единицу, включающую в себя алкильную функциональную группу С2-16, и p представляет собой величину, равную 3-4, и фармацевтически активные молекулы заявлены в патентных документах США 2010/0316682 и ЕР 1538166 А1. Основной недостаток таких производных заключается в том, что лаурат дибутилолова, который является известным как вещество с иммунотоксическим и тератогенным потенциалом, использовали для осуществления модификации гиалуронана. Последнее вещество по большей части использовали в способах модификации в отношении получения адгезивов, и оно внесено в список Европейского агентства по вопросам окружающей среды в результате своей острой токсичности (Boyer, 1989). Другой недостаток заявленных производных заключается в их конъюгации с полимерами, например, с полиэтиленгликолем, которые являются инородными по отношению к человеческому организму, и могут вызывать воспалительные реакции или обусловливать цитотоксические продукты распада при использовании для внутривенных или местных применений. Более того, многократное применение полимерных мицелл, где полиэтиленгликоль образовывал гидрофильный сегмент, приводило к ускоренному устранению таких мицелл из кровообращения вследствие образования анти-PEG IgM антител (Gong, Chen, Zheng, Wang & Wang, 2012).
Паклитаксел был успешным образом включен в мицеллы модифицированного гиалуронана посредством сополимера молочной и гликолевой кислот (PLGA) (Kim, Lee, Jang & Park, 2009). Включение проводили с применением диалитического способа, при котором и полимер, и соответствующее связанное вещество растворяли в DMSO, и полученный раствор диализировали по отношению к Н2О. В последнем случае получали связующую способность полученных полимерных мицелл 4,5% по массе. Основной недостаток таких систем носителя заключается в присутствии PLGA полимеров, которые являются инородными по отношению к человеческому организму и могут не представлять собой полностью биоразлагаемую систему. Другой недостаток заключается в присутствии остаточного DMSO в конечных продуктах.
Модификация гиалуронана с длинноцепочечными карбоновыми кислотами
Для модификации полисахаридов с карбоновыми кислотами по большей части требуется коммерчески доступный ангидрид данной кислоты (WO 1996/035720, WO 2007/033677 (Šmejkalová, Hermannová, Šuláková, Průšová, Kučerík & Velebný, 2012), EP 0893451). Основные недостатки таких коммерчески доступных ангидридов заключаются в их чувствительности к гидролизу и в возможном присутствии примесей. Более того, ангидриды некоторых кислот (например, ундекан-карбоновые кислоты) не являются коммерчески доступными. Некоторые доступные ангидриды являются очень дорогостоящими (например, ангидриды олеиновой, линолевой или линоленовой кислот). Таким образом, недоступность, высокая цена и нестабильность таких ангидридов делает крупномасштабное получение модифицированных полисахаридов сложным.
Кислотные ангидриды могут быть замещены другими кислотными производными, которые применимы для эстерификации гиалуронана. В патентном документе WO 2010/105582 заявлен способ активации карбоновых кислот при помощи этилхлорформиата при неводных условиях, где образованы О-ацил-О'-алкилкарбонаты, которые впоследствии применяли для эстерификации гиалуронана. Недостаток такой активации заключается в образовании токсичных и теоретически взрывоопасных газов. Подобный способ активации этилхлорформиатом раскрыт в патентах США №3720662 и CZ 20060605.
Другой известный способ основан на эстерификации полисахаридов с карбоновыми кислотами в присутствии имидазола (США 2012/0172587). Тем не менее, заявленный способ получения требует высоких температур реакции (90-200°С), которые не применимы по отношению к гиалуронану по причине его распада при повышенных температурах.
В европейском патенте ЕР 0893451 заявлена эстерификация полисахаридов с ангидридами карбоновых кислот при помощи способа сверхкритической экстракции. Недостатки последней процедуры эстерификации заключаются в необходимости высокого давления и в высокой стоимости оборудования.
По вышеуказанным причинам является очень важным найти альтернативный способ активации длинноцепочечных карбоновых кислот, при этом способ будет применим in-situ. Одно из возможных технических решений основано на активации карбоновых кислот производным 2,4,6-трихлорбензойной кислоты, сопровождаемой образованием ангидрида. В первый раз ангидрид 2,4,6-трихлорбензойной кислоты использовали в комбинации с DMAP катализатором для быстрой эстерификации макроциклических веществ при умеренных условиях реакции (Inanaga, Hirata, Saeki, Katsuki & Yamaguchi, 1979). Тем не менее такой способ эстерификации еще не использовали для модификации полисахаридов, в частности, гиалуроновой кислоты, поскольку возможна экзотермическая реакция, сопровождаемая распадом соответствующего полисахарида.
Сущность изобретения
Объектом настоящего изобретения является синтез гидрофобизированного производного гиалуроновой кислоты и применение указанного производного в форме полимерного носителя для биологически активных гидрофобных веществ в водной среде, причем природный характер и биологическая активность связанных веществ должны оставаться неизменными.
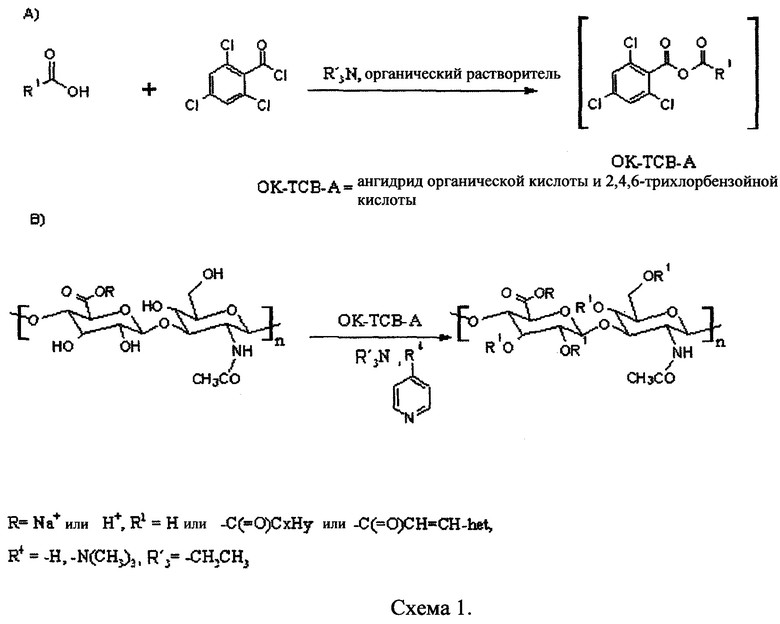
Гидрофобизация гиалуронана с длинноцепочечными алифатическими сложными эфирами (С6-С18) основана на прямой активации длинноцепочечных карбоновых кислот, которая сопровождается образованием ангидрида 2,4,6-трихлорбензойной кислоты (схема 1). На следующей стадии полученный ангидрид взаимодействует с гиалуроновой кислотой с образованием ацилированного производного гиалуроновой кислоты. Основное преимущество указанной активации заключается в возможности прямого применения алифатических карбоновых кислот. В отличие от подобных гидрофобизирующих реакций гиалуронана, которые составляют известный уровень техники, представленная модификация не требует использования никакого коммерчески доступного ангидрида. Другое преимущество представленной активации заключается в том факте, что соответствующая реакция может проходить при умеренных условиях (от комнатной температуры до 50°С) и быть короткой. Более того, настоящая активация не требует, в отличие от раскрытого в патентной заявке WO 2010/105582 способа, ни избыточного количества алифатических кислот, ни особых безводных условий. Активирующий реагент является стабильным и, в отличие от этилхлорформиата, не вызывает образование токсичных или взрывоопасных газов при использовании в реакции. Другое преимущество представленного активирующего реагента заключается в том, что реагент не образует никаких побочных продуктов реакции и не вызывает реакции сшивания.
Активация карбоновых кислот производным 2,4,6-трихлорбензойной кислоты с последующим применением полученного активированного продукта в эстерификации гиалуроновой кислоты представлена на схеме 1 ниже:
Более того, настоящее изобретение относится к применению гидрофобизированной гиалуроновой кислоты для получения наномицеллярных систем и к применению веществ, связанных в наномицеллах гиалуроновой кислоты для лечения кожи, волос и слизистых оболочек, другие местные применения также были возможны. Инкапсулированные биологически активные вещества связаны в наномицеллах гиалуронана нековалентными физическими взаимодействиями с гидрофобными функциональными группами полимерного носителя. При инкапсуляции вышеуказанным способом такие вещества эффективно проникают в поверхностный слой кожи (эпидермис), в полную структуру волос и в эпителий слизистых оболочек. Одно из преимуществ заключается в том, что глубина проникновения активных веществ, связанных в наномицеллах, больше глубины проникновения тех же веществ, когда они непосредственно растворены в неполярных растворителях.
По сравнению с подобными полимерными системами (такими, которые раскрыты в патентных документах США №7993678 и WO 2007/033677), наномицеллярные конъюгаты гиалуронана неожиданно проявляли от 3-кратной до 10-кратной низшей критической мицеллярной концентрации и, одновременно, относительно высокую стабильность в соленой и водной средах. Такое очень низкое значение критической мицеллярной концентрации в особенности является предпочтительным по отношению к возможным внутривенным применениям гиалуронановых мицелл. В отличие от подобных систем на основе большинства из известных полимеров, как описано со ссылкой на известный уровень техники, высокая стабильность гиалуронановых мицелл может быть предпочтительной по отношению к лиофилизации различных веществ, поскольку желаемая инкапсуляция может быть сохранена без добавления любого лиопротектора к раствору перед застыванием.
Другое преимущество заявленных гидрофобизированных производных гиалуронана относится к отсутствию синтетических полимеров (PLGA, PEG и т.п.) и сополимеров, которые иным образом часто необходимы для образования наномицелл и которые могут инициировать образование антител при неоднократном введении (Gong, Chen, Wang & Wang).
В отличие от подобных полимерных мицеллярных систем, образование наномицелл не основано на модифицированном гиалуронане с повышенным общим отрицательным зарядом, и, таким образом, такой заряд не оказывает неблагоприятного влияния на взаимодействия между системой носителя и клетками.
Некоторые из наномицеллярных структур, в особенности те, которые содержат более длинные ацильные цепи, связанные с гиалуронаном, могут образовывать гелевую фазу в водной среде. Такая гелевая фаза может быть преимущественно используемой при определенных требованиях применения и повышенной вязкости соответствующей системы носителя.
Размеры полученных наномицеллярных систем по большей части находятся в диапазоне от 20 до 100 нм, которые являются оптимальными размерами для фармацевтических применений, в которых необходимо преимущество усиленной проницаемости и эффект удерживания (эффект EPR). Такой размер не всегда может быть достижим в полимерных мицеллах, описанных со ссылкой на известный уровень техники.
Другое преимущество гиалуронановых мицелл относится к переносу связанных веществ из гидрофобных доменов наномицеллы в клетку.
Новый способ инкапсулирования основан на получении смеси гиалуронана, растворенного в воде, и биологически активного вещества, растворенного в органическом растворителе, с последующим энергичным разрушением гидратирующей оболочки гиалуронана и последующим удалением растворителя из раствора. В отличие от общих способов инкапсулирования, вышеуказанный способ не основан на растворимости полимеров в органическом растворителе и, таким образом, не требуется, чтобы природное поведение гиалуроновой кислоты было подавлено, и полимерная цепь была в значительной мере модифицирована, в частности, в местоположении важных карбоксильных групп, что позволяет гиалуронану распознаваться клеточными рецепторами. Предпочтительными органическими растворителями являются такие, у которых более низкая точка кипения, чем у воды. Связующая способность гиалуронановых наномицелл была заметно усилена полным выпариванием остаточной водной фазы с последующей регидратацией наномицелл. Несвязанное биологически активное вещество может быть удалено в процессе фильтрации, и полученные наномицеллы могут быть непосредственно лиофилизированы и храниться в сухом состоянии до следующей регидратации.
Главным образом, настоящее изобретение относится к С6-С18-ацилированному производному гиалуроновой кислоты общей формулы (I)
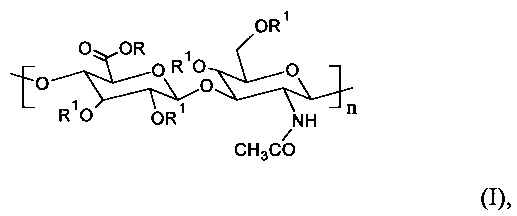
где R представляет собой Н+ или Na+ и R1 представляет собой Н или -С(=O)СхНy или -C(=O)CH=CH-het, где х представляет собой целое число в диапазоне от 5 и 17, и y представляет собой целое число в диапазоне от 11 и 35, и CxHy представляет собой неразветвленную или разветвленную, насыщенную или ненасыщенную С5-С17 цепь, и het представляет собой гетероциклическую или гетероароматическую группу с произвольным содержанием атомов N, S или О, по меньшей мере с одной повторяющейся единицей, содержащей одну или несколько R1 -С(=O)СхНу или -C(=O)CH=CH-het групп, и где n находится в диапазоне от 12 и 4000. Согласно предпочтительному варианту осуществления настоящего изобретения С6-С18-ацилированное производное представляет собой олеиловое производное, это означает, что R представляет собой Н+ или Na+, и R1 представляет собой -С(=O)(СН2)7-СН=СН-(СН2)7CH3 в формуле (I).
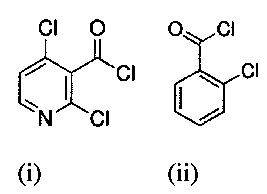
Кроме того, настоящее изобретение относится к способу получения вышеуказанного производного гиалуроновой кислоты, при котором осуществляют взаимодействие гиалуроновой кислоты с С6-С18-карбоновой кислотой, активированной хлоридом 2,4,6-трихлорбензойной кислоты или активированной органическим хлоридом R3-CO-Cl, в присутствии основания и катализатора в смеси воды и смешивающегося с водой апротонного растворителя, где R3 представляет собой алифатический или разветвленный C1-С30-алкил, иногда содержащий гетероароматические или ароматические функциональные группы. Типичной гетероароматической функциональной группой может быть пиридин вместе со своими производными (например, формулы (i)), типичной ароматической функциональной группой может быть бензол вместе со своими галогеновыми производными (например, формулы (ii)).
Гиалуроновая кислота может быть в свободной кислотной форме или в форме фармацевтически приемлемой соли, такой как Na, K, Са, Mg, Zn или Li соль, и быть с молекулярной массой в диапазоне предпочтительно от 5×103 г/моль до 1,6×106 г/моль, более предпочтительно, от 15×103 г/моль до 250×103 г/моль, и наиболее предпочтительно, от 15×103 до 50×103 г/моль. Способ получения по настоящему изобретению заключается в том, что гиалуроновую кислоту растворяли в смеси воды и смешивающегося с водой апротонного растворителя, последнее представляет собой полярный органический растворитель, содержание воды находилось в диапазоне от 10 до 99% по объему, предпочтительно, 50% по объему. Смешивающимся с водой апротонным растворителем может быть, например, диметилсульфоксид (DMSO), тетрагидрофуран (THF), ацетон, ацетонитрил или изопропанол (IPA). Реакционная смесь содержит R'3N основание, где R' представляет собой неразветвленную или разветвленную CnHm углеводородную цепь, где n представляет собой целое число в диапазоне от 1 и 4 и m представляет собой целое число в диапазоне от 3 и 9, например, триэтиламин, в количестве от 0,01 до 20 эквивалентов, предпочтительно, 6 эквивалентов, по отношению к димеру гиалуроновой кислоты, и катализатор выбран из группы, включающей в себя замещенные пиридинины, такие как диметиламинопиридин, в количестве от 0,01 до 1 эквивалента, предпочтительно, 0,05 эквивалента, по отношению к димеру гиалуроновой кислоты. При способе получения по настоящему изобретению сначала активацию С6-С18-карбоновой кислоты проводили в полярном органическом растворителе в присутствии основания и 2,4,6-трихлорбензойной кислоты или ее производных или в присутствии основания и органического хлорида, и впоследствии смесь, содержащую активированную С6-С18-карбоновую кислоту, добавляли к гиалуроновой кислоте, которая была растворена в смеси воды, органического растворителя, основания и катализатора, продуктом в результате реакции является производное общей формулы (I). Указанная С6-С18-карбоновая кислота выбрана из группы, содержащей капроновые, энантовые, каприловые, каприновые, пальмитиновые, стеариновые, олеиновые, линолевые и линоленовые кислоты. Количество активированной С6-С18-карбоновой кислоты находится в диапазоне от 0,01 до 5 эквивалентов, предпочтительно, от 0,5 до 2 эквивалентов, по отношению к димеру гиалуроновой кислоты. Активация С6-С18-карбоновой кислоты происходит в течение от 5 до 120 минут, предпочтительно, в течение 30 минут, при температуре от 20 до 60°С, предпочтительно, при температуре 25°С. Взаимодействие гиалуроновой кислоты с активированной С6-С18-карбоновой кислотой происходили в течение от 1 до 24 часов, предпочтительно, в течение от 2 до 3 часов, при температуре от 20 до 60°С, предпочтительно, при температуре 25°С. С6-С18-ацилированное производное гиалуроновой кислоты впоследствии может быть отделено от реакционной смеси, промыто, высушено и лиофилизировано. Производное может быть отделено от реакционной смеси в процессе осаждения с применением NaCl и спирта. Впоследствии производное может быть промыто спиртом, в частности, изопропанолом или этанолом.
Согласно дополнительному аспекту настоящее изобретение относится к наномицеллярной композиции на основе С6-С18-ацилированного производного гиалуроновой кислоты общей формулы (I), такая композиция содержит наномицеллы, которые включают в себя гидрофобное ядро, образованное С6-С18-ацильными группами, связанными с гиалуроновой кислотой, и гидрофильную оболочку, образованную гидрофильными функциональными группами гиалуроновой кислоты, одно или несколько биологически активных веществ были физически связаны в наномицелле. Композиция дополнительно содержит воду, и также может содержать соли (например, 0,9% NaCl). Согласно предпочтительному варианту осуществления наномицеллярная композиция содержит от 0,3 до 50% по массе биологически активного вещества по отношению к массовому содержанию С6-С18-ацилированного производного гиалуроновой кислоты, биологически активное вещество было выбрано из группы, включающей в себя фармацевтически и косметически активные вещества, в частности, витамины, медикаменты, цитостатические средства, фитоэкстракты, фитокомплексы или фитоактивные вещества, минеральные или растительные масла, или их смесь. Примеры применимых биологически активных веществ включают в себя, например, токоферол, паклитаксел, фосфатидилхолин или кофермент Q10. Согласно предпочтительному варианту осуществления композиция содержит С6-С18-ацилированное производное гиалуроновой кислоты в концентрации, которая выше его критической концентрации агрегации. Концентрация С6-С18-ацилированного производного гиалуроновой кислоты находится в диапазоне от 0,0001 мг.мл-1 до 30 мг.мл-1, предпочтительно, от 1 до 20 мг.мл-1, если композиция представляет собой водный раствор. Согласно другому предпочтительному варианту осуществления биологически активное вещество представляет собой минеральное или растительное масло, содержащееся в количестве от 0,05 до 40% по массе, предпочтительно, от 1 до 20% по массе, по отношению к массовому содержанию С6-С18-ацилированного производного гиалуроновой кислоты. Согласно еще одному предпочтительному варианту осуществления композиция содержит биологически активное вещество, которое является жидким и нерастворимым в воде, указанное вещество содержит дополнительное биологически активное вещество, растворенное в нем. Таким биологически активным веществом, которое является жидким и нерастворимым в воде, может быть, например, минеральное или растительное масло, и дополнительное биологически активное вещество может относиться, например, к фармацевтически или косметически активным веществам, в частности, витаминам, медикаментам, цитостатическим средствам, фитоэкстрактам, фитокомплексам или фитоактивным веществам, или к их смесям. Наномицеллярная композиция согласно настоящему изобретению может находиться в форме раствора, наноэмульсии, микроэмульсии, коацервата или геля.
Настоящее изобретение дополнительно относится к способу получения наномицеллярной композиции, как определено выше, при котором С6-С18-ацилированное производное гиалуроновой кислоты общей формулы (I) растворяли в воде, биологически активное вещество растворяли в органическом растворителе, полученные растворы смешивали вместе, и после этого органический растворитель удаляли. Органическим растворителем может быть летучий хлорированный растворитель, такой как трихлорметан, или спирт, такой как этанол или изопропанол, и его удаление может проходить при помощи выпаривания под вакуумом. Впоследствии водную фазу сушили и регидратировали, и полученные наномицеллярные структуры фильтровали и в конечном итоге лиофилизировали. Альтернативно, органический растворитель может быть удален при помощи диализа. И вновь, полученные наномицеллярные структуры последовательно фильтровали и в конечном итоге лиофилизировали. Согласно предпочтительному варианту осуществления С6-С18-ацилированное производное гиалуроновой кислоты общей формулы (I) растворяли в воде, и впоследствии смешивали вместе с биологически активным веществом, которое является жидким и нерастворимым в воде, после чего полученную смесь гомогенизировали при помощи обработки ультразвуком с образованием микроэмульсии или наноэмульсии. Согласно другому предпочтительному варианту осуществления С6-С18-ацилированное производное гиалуроновой кислоты общей формулы (I) растворяли в воде, и впоследствии смешивали вместе с биологически активным веществом, которое является жидким и нерастворимым в воде, и в котором растворено дополнительное биологически активное вещество, после чего полученную смесь гомогенизировали при помощи обработки ультразвуком с образованием микроэмульсии или наноэмульсии.
Согласно еще одному аспекту настоящее изобретение относится к применению наномицеллярной композиции при фармацевтических или косметических применениях, предпочтительно, к местному применению.
Более того, может быть получена стабилизированная наномицеллярная композиция. Способ получения такой стабилизированной наномицеллярной композиции заключается в том, что получали С6-С18-ацилированный гиалуронан общей формулы (II)

где R представляет собой Н+ или Na+, один или несколько R1 членов представляют собой неразветвленную С6-С18-цепь по меньшей мере в одной повторяющейся единице, при этом неразветвленная цепь может содержать ненасыщенные связи, и 3-(2-тиенил)акриловую кислоту или 3-(2-фурил)акриловую кислоту или производные указанных кислот в другой по меньшей мере одной повторяющейся единице, при этом наномицеллярную композицию получали из С6-С18-ацилированного гиалуронана общей формулы (II), такую композицию затем стабилизировали при помощи реакции сшивания.
В частности, стабилизацию проводили следующим способом: сначала получали производное общей формулы (III):
I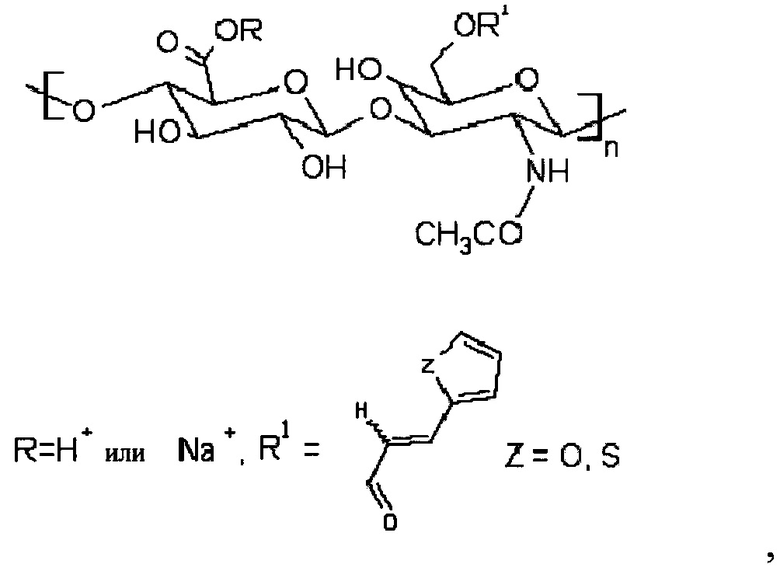
при этом гиалуроновую кислоту растворяли в воде, и после этого основание (такое как TEA) и катализатор (DMAP); отдельной процедурой получали активированную 3-(2-тиенил)акриловую кислоту или 3-(2-фурил)акриловую кислоту или производное любой кислоты, активацию проводили в смеси органического растворителя (например, THF) и основания (например, TEA) с добавлением хлорида 2,4,6-трихлорбензойной кислоты, и в конечном итоге обе смеси смешивали вместе с образованием акрилированного гиалуронана формулы (III). Впоследствии активированную С6-С18-карбоновую кислоту получали для ацилирования указанного акрилированного гиалуронана формулы (III) способом, который подобный описанному выше со ссылкой на способ получения С6-С18-ацилированного гиалуронана общей формулы (I), с образованием ацилированного гиалуронана формулы (II). В конечном итоге наномицеллы могут быть аналогично получены из ацилированного гиалуронана, который был получен вышеуказанным способом. Такие наномицеллы могут быть впоследствии поперечно сшитыми при реакциях радикального типа, например, с применением пероксодисульфата аммония. Такой поперечно сшитый гиалуронан не растворим в воде.
Список литературы
Boyer, I.J. (1989). Toxicity of dibutyltin, tributyltin and other organotin compounds to humans and to experimental animals. Toxicology, 55(3), 253-298.
Eenschooten, C., Guillaumie, F., Kontogeorgis, G.M., Stenby, E.H., & Schwach-Abdellaoui, K. (2010). Preparation and structural characterisation of novel and versatile amphiphilic octenyl succinic anhydride-modified hyaluronic acid derivatives. Carbohydrate Polymers, 79(3), 597-605.
Gong, J., Chen, M., Zheng, Y., Wang, S., & Wang, Y. (2012). Polymeric micelles drug delivery system in oncology. Journal of Controlled Release, 159(3), 312-323.
Inanaga, J., Hirata, K., Saeki, H., Katsuki, T., & Yamaguchi, M. (1979). A Rapid Esterification by Means of Mixed Anhydride and Its Application to Large-ring Lactonization. Bulletin of the Chemical Society of Japan, 52(7), 1989-1993.
Kedar, U., Phutane, P., Shidhaye, S., & Kadam, V. (2010). Advances in polymeric micelles for drug delivery and tumor targeting. Nanomedicine: Nanotechnology, Biology and Medicine, 6(6), 714-729.
Kim, T.G., Lee, H., Jang, Y., & Park, T.G. (2009). Controlled Release of Paclitaxel from Heparinized Metal Stent Fabricated by Layer-by-Layer Assembly of Polylysine and Hyaluronic Acid-g-Poly(lactic-co-glycolic acid) Micelles Encapsulating Paclitaxel. Biomacromolecules, 10(6), 1532-1539.
Li, J., Huo, M., Wang, J., Zhou, J., Mohammad, J.M., Zhang, Y., Zhu, Q., Waddad, A.Y., & Zhang, Q. (2012). Redox-sensitive micelles self-assembled from amphiphilic hyaluronic acid-deoxycholic acid conjugates for targeted intracellular delivery of paclitaxel. Biomaterials, 33(7), 2310-2320.
Liu, Y., Sun, J., Cao, W., Yang, J., Lian, H., Li, X., Sun, Y., Wang, Y., Wang, S., & He, Z. (2011) Dual targeting folate-conjugated hyaluronic acid polymeric micelles for paclitaxel delivery. International Journal of Pharmaceutics, 421(1), 160-169.
Mazzone, S.В., Mori, N., Burman, M., Palovich, M., Belmonte, K.E., & Canning, B.J. (2006). Fluorescent styryl dyes FM1-43 and FM2-10 are muscarinic receptor antagonists: intravital visualization of receptor occupancy. The Journal of Physiology, 575(1), 23-35.
Shen, Y., Li, Q., Tu, J., & Zhu, J. (2009). Synthesis and characterization of low molecular weight hyaluronic acid-based cationic micelles for efficient siRNA delivery. Carbohydrate Polymers, 77(1), 95-104.
Šmejkalová, D., Hermannová, M., Šuláková, R., Průšová, Α., Kučerik, J., & Velebný, V. (2012). Structural and conformational differences of acylated hyaluronan modified in protic and aprotic solvent system. Carbohydrate Polymers, 87(2), 1460-1466.
Tao, Y., Xu, J., Chen, M., Bai, H., & Liu, X. Core cross-linked hyaluronan-styrylpyridinium micelles as a novel carrier for paclitaxel. (2012). Carbohydrate Polymers, 88(1), 118-124.
Til, H.P., Falke, H.E., Prinsen, M.K., & Willems, M. I. (1997). Acute and subacute toxicity of tyramine, spermidine, spermine, putrescine and cadaverine in rats. Food and Chemical Toxicology, 35(3-4), 337-348.
Wang, J., Mongayt, D., & Torchilin, V.P. (2005). Polymeric micelles for delivery of poorly soluble drugs: Preparation and anticancer activity in vitro of paclitaxel incorporated into mixed micelles based on poly(ethylene glycol)-lipid conjugate and positively charged lipids. Journal of Drug Targeting, 13(1), 73-80.
Краткое описание графического материала
Фигура 1. Определение критической мицеллярной (агрегационной) концентрации (CMC) с применением флюоресцентного метода по отношению к производному ацилированного гиалуронана (С6) и (С16) с инкапсулированным Нильским красным в воде.
Фигура 2. Определение критической мицеллярной (агрегационной) концентрации (CMC) с применением метода статического светорассеивания по отношению к производному ацилированного гиалуронана (С6) и (С16) с инкапсулированным масляным красным в воде.
Фигура 3. Cry-SEM изображения гиалуронановых наномицелл с инкапсулированным витамином Ε (верхнее изображение) и с инкапсулированным паклитакселом (нижнее изображение).
Фигура 4. Цитотоксичность паклитаксела и паклитаксела, инкапсулированного в НАС6 и НАС16, в зависимости от концентрации и от времени клеточного взаимодействия.
Фигура 5. Перенос доксорубицина в НСТ 116 и MCF-7 клетки после 1 часа воздействия.
Фигура 6. Клеточная интернализация с 7AAD в живых клетках с применением НА(С6) носителей с инкапсулированным 7AAD.
Фигура 7. Профиль высвобождения инкапсулированного масляного красного из НАС6 носителя в PBS и с PBS, содержащий 1% добавленного TWEEN 80. Концентрация НАС6 + масляный красный: 1 или 10 мг.мл-1.
Фигура 8. Профиль высвобождения инкапсулированного паклитаксела из НАС6 и НАС16 (с=10 мг.мл-1) носителей в PBS.
Фигура 9. Проникновение инкапсулированного Нильского красного (NR) из гиалуронановых наномицелл НА(С6) и НА (С10) по сравнению с проникновением Нильского красного, растворенного в масле и диспергированного в воде, в кожу, любой образец Нильского красного обладал одинаковой концентрацией.
Фигура 10. Проникновение инкапсулированного Нильского красного (NR) из гиалуронановых наномицелл НА(С6) и НА (С10) по сравнению с проникновением Нильского красного, растворенного в масле и диспергированного в воде, в волосы, любой образец Нильского красного обладал одинаковой концентрацией (на микроскопическом изображении был показан поперечный разрез волоса).
Фигура 11. Проникновение инкапсулированного Нильского красного (NR) из полимерных гиалуронановых наномицелл НА(С6) и НА (С10) по сравнению с проникновением Нильского красного, растворенного в масле и диспергированного в воде, в трансбуккальную слизистую оболочку, любой образец Нильского красного обладал одинаковой концентрацией.
Фигура 12. Проникновение инкапсулированного Нильского красного (NR) из полимерных гиалуронановых наномицелл НА(С6) и НА (С10) по сравнению с проникновением Нильского красного, растворенного в масле и диспергированного в воде, в вагинальную слизистую оболочку, любой образец Нильского красного обладал одинаковой концентрацией.
Примеры
DS = степень замещения = 100% * молярное количество связанного заместителя/ молярное количество все полисахаридных димеров
Если не отмечено иное, используемое в настоящем описании выражение «эквивалент» (экв.) относится к димеру гиалуроновой кислоты. Если не отмечено иное, процентное отношение рассчитывали на основе масса/масса.
Молекулярную массу первичной гиалуроновой кислоты (источник: Contipro Biotech spol. s r.о., Dolní Dobrouč, Czech Republic) определяли при помощи способа SEC-MALLS.
Пример 1. Получение капронилового (С6) производного гиалуроновой кислоты посредством смешанного ангидрида 2,4,6-трихлорбензойной кислоты и капроновой кислоты
1 г гиалуроната натрия (2,5 ммоль, 15 кДа) растворяли в 10 мл деминерализованной воды. После этого постепенно добавляли 5 мл DMSO. Затем к раствору добавляли TEA (1,05 мл, 3 экв.) и DMAP (8,0 мг, 0,05 экв.). Одновременно гексановую кислоту (0,63 мл, 2 экв.) растворяли в 5 мл DMSO, и к раствору добавляли TEA (1,05 мл, 3 экв.), а затем 2,4,6-трихлорбензоилхлорид (1,6 мл, 4 экв.). После активации кислоты осадок фильтровали в полученный раствор НА. Реакцию проводили при комнатной температуре в течение 3 часов. После этого реакционную смесь разбавляли 5 мл деминерализованной воды, содержащей добавку 0,25 г NaCl. Ацилированное производное выделяли из реакционной смеси последующим способом осаждения с использованием 4-кратного абсолютного изопропанола. После подвергания декантированию осадок неоднократно промывали сначала водным раствором изопропанола (85% по объему) для удаления DMSO и DMAP из производного, а затем абсолютным изопропанолом для удаления воды из производного. После этого осадок сушили при температуре 40°С в течение 48 часов, а затем его лиофилизировали для удаления остаточных растворителей.
DS 60% (определено при помощи ЯМР)
1Н ЯМР (D2O) сигналы ацила: δ 2.4 ppm (m, 2Н, α СН2), δ 1.6 ppm (m, 2H, β СН2), δ 1.3 ppm (m, 4H, γ, δ СН2), δ 0.8 (m, 3Η, CH3).
Пример 2. Получение капронилового (С6) производного гиалуроновой кислоты посредством смешанного ангидрида 2,4,6-трихлорбензойной кислоты и капроновой кислоты
1 г гиалуроната натрия (2,5 ммоль, 38 кДа) растворяли в 10 мл деминерализованной воды. После этого постепенно добавляли 5 мл изопропанола. Затем к раствору добавляли TEA (1,05 мл, 3 экв.) и пиридин (0,4 мл, 2,0 экв.). Одновременно гексановую кислоту (0,32 мл, 1 экв.) растворяли в 5 мл изопропанола, а затем к раствору добавляли TEA (1,05 мл, 3 экв.) и 2,4,6-трихлорбензоилхлорид (0,391 мл, 1 экв.). После активации кислоты осадок фильтровали в полученный раствор НА. Реакцию проводили при комнатной температуре в течение 3 часов. После этого реакционную смесь разбавляли 5 мл деминерализованной воды, содержащей добавку 0,50 г NaCl. Ацилированное производное выделяли из реакционной смеси последующим способом осаждения с использованием 4-кратного абсолютного изопропанола. После подвергания декантированию осадок неоднократно промывали сначала водным раствором изопропанола (85% по объему) для удаления пиридина из производного, а затем абсолютным изопропанолом для удаления воды из производного. После этого осадок сушили при температуре 40°С в течение 48 часов, а затем его лиофилизировали для удаления остаточных растворителей.
DS 15% (определено при помощи ЯМР)
1Н ЯМР (D2O) сигналы ацила: δ 2.4 ppm (m, 2Н, α СН2), δ 1.6 ppm (m, 2Н, β СН2), δ 1.3 ppm (m, 4Н, γ, δ СН2), δ 0.8 (m, 3Н, CH3).
Пример 3. Получение энантилового (С7) производного гиалуроновой кислоты посредством смешанного ангидрида 2,4,6-трихлорбензойной кислоты и энантовой кислоты
1 г гиалуроната натрия (2,5 ммоль, 15 кДа) растворяли в 10 мл деминерализованной воды. После этого постепенно добавляли 5 мл ацетонитрила. Затем к раствору добавляли TEA (0,70 мл, 2 экв.) и DMAP (15,0 мг, 0.05 экв.). Одновременно энантовую кислоту (0,35 мл, 1 экв.) растворяли в 5 мл ацетонитрила, а затем к раствору добавляли TEA (0,70 мл, 2 экв.) и 2,4,6-трихлорбензоилхлорид (0,39 мл, 1 экв.). После активации кислоты осадок фильтровали в полученный раствор НА. Реакцию проводили при комнатной температуре в течение 3 часов. После этого реакционную смесь разбавляли 5 мл деминерализованной воды, содержащей добавку 0,75 г NaCl. Ацилированное производное выделяли из реакционной смеси последующим способом осаждения с использованием 4-кратного абсолютного изопропанола. После подвергания декантированию осадок неоднократно промывали сначала водным раствором изопропанола (85% по объему) для удаления ацетонитрила и DMAP из производного, а затем абсолютным изопропанолом для удаления воды из производного. После этого осадок сушили при температуре 40°С в течение 48 часов, а затем его лиофилизировали для удаления остаточных растворителей.
DS 12% (определено при помощи ЯМР)
1Н ЯМР (D2O) сигналы ацила: δ 2.4 ppm (m, 2Н, α СН2), δ 1.6 ppm (m, 2Н, β СН2), δ 1.3 ppm (m, 6Н, γ, δ, ε (СН2)3), δ 0.8 (m, 3Н, CH3).
Пример 4. Получение каприлового (С8) производного гиалуроновой кислоты посредством смешанного ангидрида 2,4,6-трихлорбензойной кислоты и каприловой кислоты
1 г гиалуроната натрия (2,5 ммоль, 15 кДа) растворяли в 10 мл деминерализованной воды. После этого постепенно добавляли 5 мл ацетонитрила. Затем к раствору добавляли TEA (1,05 мл, 3 экв.) и DMAP (8,0 мг, 0,05 экв.). Одновременно октановую кислоту (0,63 г, 4 экв.) растворяли в 5 мл ацетонитрила, а затем к раствору добавляли TEA (1,05 мл, 3 экв.) и 2,4,6-трихлорбензоилхлорид (0,8 мл, 4 экв.). После активации кислоты осадок фильтровали в полученный раствор НА. Реакцию проводили при комнатной температуре в течение 3 часов. После этого реакционную смесь разбавляли 5 мл деминерализованной воды, содержащей добавку 0,50 г NaCl. Ацилированное производное выделяли из реакционной смеси последующим способом осаждения с использованием 4-кратного абсолютного изопропанола. После подвергания декантированию осадок неоднократно промывали сначала водным раствором изопропанола (85% по объему) для удаления ацетонитрила и DMAP из производного, а затем абсолютным изопропанолом для удаления воды из производного. После этого осадок сушили при температуре 40°С в течение 48 часов, а затем его лиофилизировали для удаления остаточных растворителей.
DS 40% (определено при помощи ЯМР)
1Н ЯМР (D2O) сигналы ацила: δ 2.4 ppm (m, 2Н, α СН2), δ 1.6 ppm (m, 2H, β СН2), δ 1.3 ppm (m, 8H, (СН2)4), δ 0.8 (m, 3Н, CH3).
Пример 5. Получение капринилового (С10) производного гиалуроновой кислоты посредством смешанного ангидрида 2,4,6-трихлорбензойной кислоты и каприновой кислоты
1 г гиалуроната натрия (2,5 ммоль, 15 кДа) растворяли в 10 мл деминерализованной воды. После этого постепенно добавляли 5 мл THF. Затем к раствору добавляли TEA (1,05 мл, 3 экв.) и DMAP (8,0 мг, 0,025 экв.). Одновременно декановую кислоту (0,8 г, 2 экв.) растворяли в 5 мл THF, а затем к раствору добавляли TEA (1,05 мл, 3 экв.) и 2,4,6-трихлорбензоилхлорид (0,8 мл, 2 экв.). После активации кислоты осадок фильтровали в полученный раствор НА. Реакцию проводили при комнатной температуре в течение 3 часов. После этого реакционную смесь разбавляли 5 мл деминерализованной воды, содержащей добавку 0,25 г NaCl. Ацилированное производное выделяли из реакционной смеси последующим способом осаждения с использованием 4-кратного абсолютного изопропанола. После подвергания декантированию осадок неоднократно промывали сначала водным раствором изопропанола (85% по объему) для удаления DMAP из производного, а затем абсолютным изопропанолом для удаления воды из производного. После этого осадок сушили при температуре 40°С в течение 48 часов, а затем его лиофилизировали для удаления остаточных растворителей.
DS 15% (определено при помощи ЯМР)
1Н ЯМР (D2O) сигналы ацила: δ 2.4 ppm (m, 2H, α СН2), δ 1.6 ppm (m, 2H, β СН2), δ 1.3 ppm (m, 12H, γ, δ, ε, ζ, η, θ СН2), δ 0.8 (m, 3Н, CH3).
Пример 6. Получение капринилового (С10) производного гиалуроновой кислоты посредством смешанного ангидрида 2,4,6-трихлорбензойной кислоты и каприновой кислоты
1 г гиалуроната натрия (2,5 ммоль, 15 кДа) растворяли в 10 мл деминерализованной воды. После этого постепенно добавляли 5 мл THF. Затем к раствору добавляли TEA (1,05 мл, 3 экв.) и DMAP (8,0 мг, 0,025 экв.). Одновременно декановую кислоту (0,8 г, 4 экв.) растворяли в 5 мл THF, а затем к раствору добавляли TEA (1,05 мл, 3 экв.) и 2,4,6-трихлорбензоилхлорид (0,8 мл, 4 экв.). После активации кислоты осадок фильтровали в полученный раствор НА. Реакцию проводили при комнатной температуре в течение 3 часов. После этого реакционную смесь разбавляли 5 мл деминерализованной воды, содержащей добавку 0,25 г NaCl. Ацилированное производное выделяли из реакционной смеси последующим способом осаждения с использованием 4-кратного абсолютного изопропанола. После подвергания декантированию осадок неоднократно промывали сначала водным раствором изопропанола (85% по объему) для удаления THF и DMAP из производного, а затем абсолютным изопропанолом для удаления воды из производного. После этого осадок сушили при температуре 40°С в течение 48 часов, а затем его лиофилизировали для удаления остаточных растворителей.
DS 40% (определено при помощи ЯМР)
1Н ЯМР (D2O) сигналы ацила: δ 2.4 ppm (m, 2Н, α СН2), δ 1.6 ppm (m, 2H, β СН2), δ 1.3 ppm (m, 12H, γ, δ, ε, ζ, η, θ СН2), δ 0.8 (m, 3Н, CH3).
Пример 7. Получение пальмитоилового (С16) производного гиалуроновой кислоты посредством смешанного ангидрида 2,4,6-трихлорбензойной кислоты и пальмитиновой кислоты
0,5 г гиалуроната натрия (1,25 ммоль, 38 кДа) растворяли в 20 мл деминерализованной воды. После этого постепенно добавляли 10 мл THF. Затем к раствору добавляли TEA (0,52 мл, 3 экв.) и DMAP (8,0 мг, 0,05 экв.). Одновременно пальмитиновую кислоту (0,16 г, 0,5 экв.) растворяли в 10 мл THF, а затем к раствору добавляли TEA (0,52 мл, 3 экв.) и 2,4,6-трихлорбензоилхлорид (0,098 мл, 0,5 экв.). После активации кислоты осадок фильтровали в полученный раствор НА. Реакцию проводили при комнатной температуре в течение 3 часов. После этого реакционную смесь разбавляли 5 мл деминерализованной воды, содержащей добавку 0,25 г NaCl. Ацилированное производное выделяли из реакционной смеси последующим способом осаждения с использованием 4-кратного абсолютного изопропанола. После подвергания декантированию осадок неоднократно промывали сначала водным раствором изопропанола (85% по объему) для удаления DMAP из производного, а затем абсолютным изопропанолом для удаления воды из производного. После этого осадок сушили при температуре 40°С в течение 48 часов, а затем его лиофилизировали для удаления остаточных растворителей.
1Н ЯМР (D2O) сигналы ацила: δ 2.4 ppm (m, 2Н, α СН2), δ 1.6 ppm (m, 2H, β СН2), δ 1.3 ppm (m, 24H, (СН2)12), δ 0.8 (m, 3H, CH3). DS 14% (определено при помощи ЯМР)
Пример 8. Получение стеарилового (С18) производного гиалуроновой кислоты посредством смешанного ангидрида 2,4,6-трихлорбензойной кислоты и стеариновой кислоты
0,5 г гиалуроната натрия (1,25 ммоль, 15 кДа) растворяли в 10 мл деминерализованной воды. После этого постепенно добавляли 5 мл THF. Затем к раствору добавляли TEA (0,52 мл, 3 экв.) и DMAP (8,0 мг, 0,05 экв.). Одновременно стеариновую кислоту (0,711 г, 2 экв.) растворяли в 5 мл THF, а затем к раствору добавляли TEA (0,52 мл, 3 экв.) и 2,4,6-трихлорбензоилхлорид (0,391 мл, 2 экв.). После активации кислоты осадок фильтровали в полученный раствор НА. Реакцию проводили при комнатной температуре в течение в течение 3 часов, и впоследствии реакционную смесь нагревали до 50°С в течение 1 часа. После этого реакционную смесь разбавляли 5 мл деминерализованной воды, содержащей добавку 0,25 г NaCl. Ацилированное производное выделяли из реакционной смеси последующим способом осаждения с использованием 4-кратного абсолютного изопропанола. После подвергания декантированию осадок неоднократно промывали сначала водным раствором изопропанола (85% по объему) для удаления DMAP из производного, а затем абсолютным изопропанолом для удаления воды из производного. После этого осадок сушили при температуре 40°С в течение 48 часов, а затем его лиофилизировали для удаления остаточных растворителей.
DS 7% (определено при помощи ЯМР)
1Н ЯМР (D2O) сигналы ацила: δ 2.4 ppm (m, 2Н, α СН2), δ 1.6 ppm (m, 2H, β СН2), δ 1.3 ppm (m, 28H, (СН2)14), δ 0.8 (m, 3Н, CH3).
Пример 9. Получение олеилового (С18:1) производного гиалуроновой кислоты посредством смешанного ангидрида 2,4,6-трихлорбензойной кислоты и олеиновой кислоты
0,5 г гиалуроната натрия (1,25 ммоль, 15 кДа) растворяли в 10 мл деминерализованной воды. После этого постепенно добавляли 5 мл THF. Затем к раствору добавляли TEA (0,52 мл, 3 экв.) и DMAP (15,0 мг, 0,1 экв.). Одновременно олеиновую кислоту (0,18 г, 0,5 экв.) растворяли в 5 мл THF, а затем к раствору добавляли TEA (0,52 мл, 3 экв.) и 2,4,6-трихлорбензоилхлорид (0,098 мл, 0,5 экв.). После активации кислоты осадок фильтровали в полученный раствор НА. Реакцию проводили при комнатной температуре в течение 3 часов. После этого реакционную смесь разбавляли 5 мл деминерализованной воды, содержащей добавку 0,25 г NaCl. Ацилированное производное выделяли из реакционной смеси последующим способом осаждения с использованием 4-кратного абсолютного изопропанола. После подвергания декантированию осадок неоднократно промывали сначала водным раствором изопропанола (85% по объему) для удаления DMAP из производного, а затем абсолютным изопропанолом для удаления воды из производного. После этого осадок сушили при температуре 40°С в течение 48 часов, а затем его лиофилизировали для удаления остаточных растворителей.
DS 10% (определено при помощи ЯМР)
1Н ЯМР (D2O): δ 0.88 (t, 3Н, -СН2-CH3), δ 1.22-1.35 (m, 20Н, (-СН2-)10, δ 1.60 (m, 2Η, -СН2-СН2-СО-), δ 2,0 ppm (m, 4H, (СН2)2), δ 2.41 (t, 2H, -СН2-CO-), δ 5.41 (d, 2H, CH=CH)
Пример 10. Получение олеилового (C18:1) производного гиалуроновой кислоты посредством смешанного ангидрида 2,4,6-трихлорбензойной кислоты и олеиновой кислоты
0,5 г гиалуроната натрия (1,25 ммоль, 130 кДа) растворяли в 5 мл деминерализованной воды. После этого постепенно добавляли 3 мл изопропанола. Затем к раствору добавляли TEA (0,52 мл, 3 экв.) и DMAP (15,0 мг, 0,1 экв.). Одновременно олеиновую кислоту (0,4 мл, 1 экв.) растворяли в 5 мл изопропанола, а затем к раствору добавляли TEA (0,52 мл, 3 экв.) и 2,4,6-трихлорбензоилхлорид (0,195 мл, 1 экв.). После активации кислоты осадок фильтровали в полученный раствор НА. Реакцию проводили при комнатной температуре в течение 3 часов. После этого реакционную смесь разбавляли 5 мл деминерализованной воды, содержащей добавку 0,25 г NaCl. Ацилированное производное выделяли из реакционной смеси последующим способом осаждения с использованием 4-кратного абсолютного изопропанола. После подвергания декантированию осадок неоднократно промывали сначала водным раствором изопропанола (85% по объему) для удаления DMAP из производного, а затем абсолютным изопропанолом для удаления воды из производного. После этого осадок сушили при температуре 40°С в течение 48 часов, а затем его лиофилизировали для удаления остаточных растворителей.
DS 12% (определено при помощи ЯМР)
1Н ЯМР (D2O): δ 0.88 (t, 3H, -СН2-CH3), δ 1.22-1.35 (m, 20Н, (-СН2-)10, δ 1.60 (m, 2Η, -CH2-СН2-СО-), δ 2,0 ppm (m, 4Н, (СН2)2), δ 2.41 (t, 2Н, -СН2-СО-), δ 5.41 (d, 2Н, СН=СН)
Пример 11. Получение олеилового (C18:1) производного гиалуроновой кислоты посредством смешанного ангидрида изобутирилхлорида и олеиновой кислоты
1,0 г гиалуроната натрия (2,5 ммоль, 15 кДа) растворяли в 10 мл деминерализованной воды. После этого постепенно добавляли 5 мл THF. Затем к раствору добавляли TEA (1,05 мл, 3 экв.) и DMAP (15,0 мг, 0,05 экв.). Одновременно олеиновую кислоту (0,787 мл, 1 экв.) растворяли в 5 мл THF, а затем к раствору добавляли TEA (1,05 мл, 3 экв.) и изобутирилхлорид (0,26 мл, 1 экв.). После активации кислоты осадок фильтровали в полученный раствор НА. Реакцию проводили при комнатной температуре в течение 3 часов. После этого реакционную смесь разбавляли 5 мл деминерализованной воды, содержащей добавку 0,50 г NaCl. Ацилированное производное выделяли из реакционной смеси последующим способом осаждения с использованием 4-кратного абсолютного изопропанола. После подвергания декантированию осадок неоднократно промывали сначала водным раствором изопропанола (85% по объему) для удаления DMAP из производного, а затем абсолютным изопропанолом для удаления воды из производного. После этого осадок сушили при температуре 40°С в течение 48 часов, а затем его лиофилизировали для удаления остаточных растворителей.
DS 11% (определено при помощи ЯМР)
1H ЯМР (D2O): δ 0.88 (t, 3H, -СН2-CH3), δ 1.22-1.35 (m, 20Н, (-СН2-)10, δ 1.60 (m, 2H, -CH2-СН2-СО-), δ 2,0 ppm (m, 4H, (СН2)2), δ 2.41 (t, 2H, -СН2-CO-), δ 5.41 (d, 2H, CH=CH)
Пример 12. Получение олеилового (С18:1) производного гиалуроновой кислоты посредством смешанного ангидрида 2,4,6-трихлорбензойной кислоты и олеиновой кислоты 0,5 г гиалуроната натрия (1,25 ммоль, 130 кДа) растворяли в 5 мл деминерализованной воды. После этого постепенно добавляли 3 мл THF. Затем к раствору добавляли TEA (1,2 мл, 3 экв.) и DMAP (15,0 мг, 0,1 экв.). Одновременно олеиновую кислоту (0,787 мл, 2 экв.) растворяли в 10 мл THF, а затем к раствору добавляли TEA (0,52 мл, 3 экв.) и 2,4,6-трихлорбензоилхлорид (0,391 мл, 2 экв.). После активации кислоты осадок фильтровали в полученный раствор НА. Реакцию проводили при комнатной температуре в течение 3 часов. После этого реакционную смесь разбавляли 5 мл деминерализованной воды, содержащей добавку 0,25 г NaCl. Ацилированное производное выделяли из реакционной смеси последующим способом осаждения с использованием 4-кратного абсолютного изопропанола. После подвергания декантированию осадок неоднократно промывали сначала водным раствором изопропанола (85% по объему) для удаления DMAP из производного, а затем абсолютным изопропанолом для удаления воды из производного. После этого осадок сушили при температуре 40°С в течение 48 часов, а затем его лиофилизировали для удаления остаточных растворителей.
DS 18% (определено при помощи ЯМР)
1Н ЯМР (D2O): δ 0.88 (t, 3Н, -СН2-CH3), δ 1.22-1.35 (m, 20Н, (-СН2-)10, δ 1.60 (m, 2Η, -CH2-СН2-СО-), δ 2,0 ppm (m, 4Н, (СН2)2), δ 2.41 (t, 2Н, -СН2-СО-), δ 5.41 (d, 2Н, СН=СН)
Пример 13. Получение линолеилового (С18:2) производного гиалуроновой кислоты посредством смешанного ангидрида 2,4,6-трихлорбензойной кислоты и линолевой кислоты
0,5 г гиалуроната натрия (1,25 ммоль, 15 кДа) растворяли в 10 мл деминерализованной воды. Затем к раствору добавляли TEA (0,52 мл, 3 экв.) и DMAP (8 мг, 0,05 экв.). Одновременно линолевую кислоту (0,77 мл, 2 экв.) растворяли в 3 мл THF, а затем к раствору добавляли TEA (1,2 мл, 7 экв.) и 2,4,6-трихлорбензоилхлорид (0,391 мл, 2 экв.). После активации кислоты осадок фильтровали в полученный раствор НА. Реакцию проводили при комнатной температуре в течение 3 часов. После этого реакционную смесь разбавляли 5 мл деминерализованной воды, содержащей добавку 0,5 г NaCl. Ацилированное производное выделяли из реакционной смеси последующим способом осаждения с использованием 4-кратного абсолютного изопропанола. После подвергания декантированию осадок неоднократно промывали сначала водным раствором изопропанола (85% по объему) для удаления DMAP из производного, а затем абсолютным изопропанолом для удаления воды из производного. После этого осадок сушили при температуре 40°С в течение 48 часов, а затем его лиофилизировали для удаления остаточных растворителей.
DS 16% (определено при помощи ЯМР)
1Н ЯМР (D2O): δ 0.88 (t, 3Н, -СН2-CH3), δ 1.22-1.35 (m, 14Н, (-СН2-)7), δ 1.63 (m, 2Η, -CH2-СН2-СО-), δ 2,0 ppm (m, 4Н, (СН2)2), δ 2.44 (t, 2Н, -СН2-СО-), δ 2.83 (m, 2Н, =СН-СН2-СН=), δ 5.45 (m, 4Н, СН=СН)
Пример 14. Получение линолеилового (С18:2) производного гиалуроновой кислоты посредством смешанного ангидрида 2,4,6-трихлорбензойной кислоты и линолевой кислоты
0,5 г гиалуроната натрия (1,25 ммоль, 15 кДа) растворяли в 10 мл деминерализованной воды. Затем к раствору добавляли TEA (0,52 мл, 3 экв.) и DMAP (8 мг, 0,05 экв.). Одновременно линолевую кислоту (0,77 мл, 2 экв.) растворяли в 3 мл THF, а затем к раствору добавляли TEA (1,2 мл, 7 экв.) и 2,4,6-трихлорбензоилхлорид (0,391 мл, 2 экв.). После активации кислоты осадок фильтровали в полученный раствор НА. Реакцию проводили при комнатной температуре в течение 3 часов, и впоследствии реакционную смесь нагревали до 50°С в течение 1 часа. После этого реакционную смесь разбавляли 5 мл деминерализованной воды, содержащей добавку 0,75 г NaCl. Ацилированное производное выделяли из реакционной смеси последующим способом осаждения с использованием 4-кратного абсолютного изопропанола. После подвергания декантированию осадок неоднократно промывали сначала водным раствором изопропанола (85% по объему) для удаления DMAP из производного, а затем абсолютным изопропанолом для удаления воды из производного. После этого осадок сушили при температуре 40°С в течение 48 часов, а затем его лиофилизировали для удаления остаточных растворителей.
DS 20% (определено при помощи ЯМР)
1Н ЯМР (D2O): δ 0.88 (t, 3Н, -СН2-CH3), δ 1.22-1.35 (m, 14Η, (-СН2-)7), δ 1.63 (m, 2Η, -СН2-СН2-СО-), δ 2,0 ppm (m, 4Н, (СН2)2), δ 2.44 (t, 2Н, -СН2-СО-), δ 2.83 (m, 2Н, =СН-СН2-СН=), δ 5.45 (m, 4Н, СН=СН)
Пример 15. Получение линоленилового (С18:3) производного гиалуроновой кислоты посредством смешанного ангидрида 2,4,6-трихлорбензойной кислоты и линоленовой кислоты
0,5 г гиалуроната натрия (1,25 ммоль, 15 кДа) растворяли в 10 мл деминерализованной воды. Затем к раствору добавляли TEA (0,52 мл, 3 экв.) и DMAP (8 мг, 0,05 экв.). Одновременно линоленовую кислоту (0,765 мл, 2,0 экв.) растворяли в 5 мл THF, а затем к раствору добавляли TEA (0,52 мл, 3 экв.) и 2,4,6-трихлорбензоилхлорид (0,391 мл, 2,0 экв.). После активации кислоты осадок фильтровали в полученный раствор НА. Реакцию проводили при комнатной температуре в течение 3 часов. После этого реакционную смесь разбавляли 5 мл деминерализованной воды, содержащей добавку 0,75 г NaCl. Ацилированное производное выделяли из реакционной смеси последующим способом осаждения с использованием 4-кратного абсолютного изопропанола. После подвергания декантированию осадок неоднократно промывали сначала водным раствором изопропанола (85% по объему) для удаления DMAP из производного, а затем абсолютным изопропанолом для удаления воды из производного. После этого осадок сушили при температуре 40°С в течение 48 часов, а затем его лиофилизировали для удаления остаточных растворителей.
DS 15% (определено при помощи ЯМР)
1Н ЯМР (D2O): δ 0.88 (t, 3H, -СН2-CH3), δ 1.22-1.35 (m, 8Н, (-СН2-)4), δ 1.61 (m, 2Н, -СН2-СН2-СО-), δ 2,0 ppm (m, 4Н, (СН2)2), δ 2.43 (t, 2Н, -СН2-СО-), δ 2.83 (m, 4Н,=СН-СН2-СН=), δ 5.45 (m, 6Н, СН=СН)
Пример 16. Получение линоленилового (С18:3) производного гиалуроновой кислоты посредством смешанного ангидрида 2,4,6-трихлорбензойной кислоты и линоленовой кислоты
0,5 г гиалуроната натрия (1,25 ммоль, 15 кДа) растворяли в 10 мл деминерализованной воды. Затем к раствору добавляли TEA (0,52 мл, 3 экв.) и DMAP (8 мг, 0,05 экв.). Одновременно линоленовую кислоту (0,382 мл, 1 экв.) растворяли в 5 мл THF, а затем к раствору добавляли TEA (0,52 мл, 3 экв.) и 2,4,6-трихлорбензоилхлорид (0,195 мл, 1 экв.). После активации кислоты осадок фильтровали в полученный раствор НА. Реакцию проводили при комнатной температуре в течение 3 часов, и впоследствии реакционную смесь нагревали до 50°С в течение 1 часа. После этого реакционную смесь разбавляли 5 мл деминерализованной воды, содержащей добавку 0,25 г NaCl. Ацилированное производное выделяли из реакционной смеси последующим способом осаждения с использованием 4-кратного абсолютного изопропанола. После подвергания декантированию осадок неоднократно промывали сначала водным раствором изопропанола (85% по объему) для удаления DMAP из производного, а затем абсолютным изопропанолом для удаления воды из производного. После этого осадок сушили при температуре 40°С в течение 48 часов, а затем его лиофилизировали для удаления остаточных растворителей.
DS 10% (определено при помощи ЯМР)
1Н ЯМР (D2O): δ 0.88 (t, 3Н, -СН2-CH3), δ 1.22-1.35 (m, 8Н, (-СН2-)4), δ 1.61 (m, 2Н, -СН2-СН2-СО-), δ 2,0 ppm (m, 4Н, (СН2)2), δ 2.43 (t, 2Н, -СН2-СО-), δ 2.83 (m, 4Н, =СН-СН2-СН=), δ 5.45 (m, 6Н, СН=СН)
Пример 17. Инкапсулирование токоферола (витамин Е) в капронильное (С6) производное гиалуроновой кислоты
100 мг ацилированного производного гиалуронана, полученного согласно примеру 1, растворяли в 5 мл воды при непрерывном перемешивании в течение 3 часов. Полученный раствор постепенно дополняли раствором токоферола (10 мг в 3 мл CHCl3) при непрерывном перемешивании и при температуре в диапазоне от 25 до 40°С, и после этого постепенно добавляли еще 3 мл CHCl3. Впоследствии CHCl3 удаляли из раствора при непрерывном процессе выпаривания. После удаления CHCl3 водную фазу полностью сушили, регидратировали над водной баней и фильтровали через 1 мкм стеклянный фильтр. Фильтрат лиофилизировали.
Количество связанного токоферола (определено при помощи ВЭЖХ способа) составляло: 2,3% (масса/масса)
Пример 18. Инкапсулирование Нильского красного в капронильное (С6) производное гиалуроновой кислоты
100 мг ацилированного производного гиалуронана, полученного согласно примеру 1, растворяли в 5 мл воды при непрерывном перемешивании в течение 3 часов. Полученный раствор постепенно дополняли раствором Нильского красного (10 мг в 3 мл CHCl3) при непрерывном перемешивании и при температуре в диапазоне от 25 до 40°С, и после этого постепенно добавляли еще 3 мл CHCl3. Впоследствии CHCl3 удаляли из раствора при непрерывном процессе выпаривания. После удаления CHCl3 водную фазу полностью сушили, регидратировали над водной баней и фильтровали через 1 мкм стеклянный фильтр. Фильтрат лиофилизировали.
Количество связанного Нильского красного (определено при помощи способа в УФ/видимом диапазоне) составляло: 0,4% (масса/масса)
Пример 19. Инкапсулирование паклитаксела в капронильное (С6) производное гиалуроновой кислоты
100 мг ацилированного производного гиалуронана, полученного согласно примеру 1, растворяли в 5 мл воды при непрерывном перемешивании в течение 3 часов. Полученный раствор постепенно дополняли раствором паклитаксела (10 мг в 3 мл CHCl3) при непрерывном перемешивании и при температуре в диапазоне от 25 до 40°С, и после этого постепенно добавляли еще 3 мл CHCl3. Впоследствии CHCl3 удаляли из раствора при непрерывном процессе выпаривания. После удаления CHCl3 водную фазу полностью сушили, регидратировали над водной баней и фильтровали через 1 мкм стеклянный фильтр. Фильтрат лиофилизировали.
Количество связанного паклитаксела (определено при помощи ВЭЖХ способа): 5% (масса/масса)
Пример 20. Инкапсулирование фосфатидилхолина в капронильное (С6) производное гиалуроновой кислоты
100 мг ацилированного производного гиалуронана, полученного согласно примеру 1, растворяли в 5 мл воды при непрерывном перемешивании в течение 3 часов. Полученный раствор постепенно (по каплям) дополняли раствором фосфатидилхолина (10 мг в 5 мл EtOH) при непрерывном перемешивании. EtOH удаляли из раствора при непрерывном процессе выпаривания. Впоследствии остаточную водную фазу полностью сушили, регидратировали над водной баней и фильтровали через 1 мкм стеклянный фильтр. Фильтрат лиофилизировали.
Количество связанного фосфатидилхолина (определено при помощи ВЭЖХ способа): 3,0% (масса/масса).
Пример 21. Инкапсулирование кофермента Q10 в пальмитоиловое (С16) производное гиалуроновой кислоты
100 мг ацилированного производного гиалуронана, полученного согласно примеру 7, растворяли в 10 мл воды при условиях перемешивания всю ночь. Полученный раствор постепенно дополняли раствором кофермента Q10 (20 мг в 5 мл CHCl3) при непрерывном перемешивании и при температуре в диапазоне от 30 до 40°С, и после этого постепенно добавляли еще 3 мл CHCl3. Впоследствии CHCl3 удаляли из раствора при непрерывном процессе выпаривания. После удаления CHCl3 водную фазу полностью сушили, регидратировали над водной баней и фильтровали через 1 мкм стеклянный фильтр. Фильтрат лиофилизировали.
Количество связанного кофермента Q10 (определено при помощи способа в УФ/видимом диапазоне) составляло: 12% (масса/масса)
Если продукт растворяли в 0,9% растворе NaCl, образовывался коацерват или гелеобразный раствор в зависимости от концентрации растворенного продукта.
Пример 22. Инкапсулирование токоферола (витамин Е) в стеариловое (С18) производное гиалуроновой кислоты
100 мг ацилированного производного гиалуронана, полученного согласно примеру 8, растворяли в 10 мл воды при условиях перемешивания всю ночь. Полученный раствор постепенно дополняли раствором токоферола (приблизительно 50 мг в 5 мл этанола) при непрерывном перемешивании и при температуре в диапазоне от 25 до 40°С. Впоследствии этанол удаляли из раствора при непрерывном процессе выпаривания. После удаления EtOH водную фазу полностью сушили, регидратировали над водной баней и фильтровали через 1 мкм стеклянный фильтр. Фильтрат лиофилизировали.
Количество связанного токоферола (определено при помощи способа в УФ/видимом диапазоне) составляло: 30% (масса/масса)
Пример 23. Инкапсулирование токоферола (витамин Е) в олеильное (С18:1) производное гиалуроновой кислоты
100 мг ацилированного производного гиалуронана, полученного согласно примеру 9, растворяли в 10 мл воды при условиях перемешивания всю ночь. Полученный раствор постепенно дополняли раствором токоферола (приблизительно 50 мг в 5 мл изопропанола) при непрерывном перемешивании и при температуре в диапазоне от 25 до 40°С. Впоследствии изопропанол удаляли из раствора при непрерывном процессе выпаривания. После удаления изопропанола водную фазу полностью сушили, регидратировали над водной баней и фильтровали через 1 мкм стеклянный фильтр. Фильтрат лиофилизировали.
Количество связанного токоферола (определено при помощи способа в УФ/видимом диапазоне) составляло: 40% (масса/масса)
Пример 24. Инкапсулирование кофермента Q10 в пальмитоиловое (С16) производное гиалуроновой кислоты
100 мг ацилированного производного гиалуронана, полученного согласно примеру 7, растворяли в 10 мл воды при условиях перемешивания всю ночь. Полученный раствор постепенно дополняли раствором кофермента Q10 (приблизительно 30 мг в 2 мл EtOH) при непрерывном перемешивании. После перемешивания в течение 3 часов полученную смесь подвергали обработке ультразвуком (100 Вт) в течение 30 минут. Впоследствии смесь подвергали интенсивному диализу (в течение 2 дней) по отношению к дистиллированной воде, и ее фильтровали через 1 мкм стеклянный фильтр и лиофилизировали.
Количество связанного кофермента Q10 (определено при помощи способа в УФ/видимом диапазоне) составляло: 4,6% (масса/масса)
Если продукт растворяли в 0,9% растворе NaCl, образовывался коацерват или гелеобразный раствор в зависимости от концентрации растворенного продукта.
Пример 25. Инкапсулирование паклитаксела в пальмитоиловое (С16) производное гиалуроновой кислоты
100 мг ацилированного производного гиалуронана, полученного согласно примеру 7, растворяли в 10 мл воды при условиях перемешивания всю ночь. Полученный раствор постепенно дополняли раствором паклитаксела Q10 (приблизительно 40 мг в 2 мл EtOH) при непрерывном перемешивании. После перемешивания в течение 3 часов полученную смесь подвергали обработке ультразвуком (100 Вт) в течение 30 минут. Впоследствии смесь подвергали интенсивному диализу (3,5 кДа, отключали) по отношению к дистиллированной воде, и ее фильтровали через S4 фарфоровую фритту и лиофилизировали.
Количество связанного паклитаксела (определено при помощи ВЭЖХ способа): 25% (масса/масса)
Пример 26. Инкапсулирование экстракта хмеля в олеильное (C18:1) производное гиалуроновой кислоты
100 мг ацилированного производного гиалуронана, полученного согласно примеру 9, растворяли в 10 мл воды при условиях перемешивания всю ночь. Полученный раствор постепенно дополняли раствором смеси экстракта хмеля (приблизительно 50 мг в 5 мл изопропанола) при непрерывном перемешивании. Впоследствии изопропанол удаляли из раствора при непрерывном процессе выпаривания. После удаления изопропанола водную фазу полностью сушили, регидратировали над водной баней и фильтровали через 1 мкм стеклянный фильтр. Фильтрат лиофилизировали.
Количество связанного токоферола (определено при помощи способа в УФ/видимом диапазоне) составляло: 40% (масса/масса)
Пример 27. Определение критических мицеллярных (агрегационных) концентраций ацилированных производных гиалуроновой кислоты
(a) Флюоресцентный метод
Критическую мицеллярную (агрегационную) концентрацию определяли в зависимости от интенсивности флуоресценции по отношению к концентрациям раствора (фигура 1). Спектры испускания (580-700 нм) водных растворов ацилированных производных НАС6 (DS=60%) и НАС16 (DS=14%) со связанным Нильским красным, который получали в концентрации в диапазоне от 0,00002 до 1,5 мг.мл-1 согласно способу, описанному в примере 18, измеряли при помощи флуориметрического аппарата RF-5301 (Shimadzu), работающим с длиной волны возбуждения 543 нм.
Определяли следующие критические мицеллярные (агрегационные) концентрации: для НА С6: 0,001-0,003 мг.мл-1, для НА С16: 0,00006-0,0002 мг.мл-1 (фигура 1). Тот же результат измерения получали для PBS с 0,9% раствором NaCl.
(b) Метод статического светорассеивания
Критическую мицеллярную (агрегационную) концентрацию определяли в зависимости от интенсивностей рассеянного света (I90) по отношению к концентрациям раствора (фигура 2). Интенсивность рассеянного света в водных растворах ацилированных производных НАС6 (DS=60%) и НАС16 (DS=14%) со связанным Нильским красным, который получали в концентрации в диапазоне от 0,00002 до 0,06 мг.мл-1 согласно способу, описанному в примере 18, измеряли под углом 90° при помощи фотометрического аппарата DAWN EOS (Wyatt Technology Corporation), работающим с длиной волны 632 нм.
Определяли следующие критические мицеллярные (агрегационные) концентрации: для НА С6: 0,002-0,004 мг.мл-1, для НА С16: 0,00006-0,0001 мг.мл-1 (фигура 2). Тот же результат измерения получали для PBS с 0,9% раствором NaCl.
Пример 28. Определение зета-потенциала гиалуронановых наномицелл
Зета-потенциал определяли при помощи аппарата Zetasizer Nano-ZS (Malvern Instruments), оборудованного лазером He-Ne (633 нм). Независимо от инкапсулированного вещества зета-потенциал, проявляемый наномицеллами в водных растворах, составлял ~ -50 мВ при 5 мг.мл-1 и от ~ -60 до -70 мВ после 10-кратного разбавления. В 0,9% растворе NaCl диапазон зета-потенциала был понижен (от -30 до -23 мВ). Таким образом, абсолютное значение зета-потенциала означает высокую стабильность полученных наномицелл в водных растворах и их относительно высокую стабильность в солевых растворах.
Пример 29. Морфологический анализ гиалуронановых наномицелл
Микроскопическое исследование проводили при -135°С при помощи сканирующего микроскопа JEOL 7401F, работающего с ускоряющим напряжением луча 2 кВ (т.е., в режиме узкого пучка). Для вышеуказанного анализа 2-3 мкл концентрированного образца (приблизительно 20 мг/0,4 мл) капали на Al пластину и погружали в жидкий азот, заполненный в криокамеру Alta 2500 (Gatan). Впоследствии это покрывали смесью Pt/Pd в течение 2 минут.
Размер наномицелл гиалуронана С6 с инкапсулированным витамином Ε (пример 17) и гиалуронана С16 с инкапсулированным паклитакселом (пример 25) находился в диапазоне: 20-50 нм (фигура 3).
Пример 30. Распределение ацилированных цепей и неполярных веществ в гиалуронановых наномицеллах
Разделение наномицелл гидрофобизированного гиалуронана с инкапсулированным витамином Е проводили способом проточного фракционирования в переменном поле (FlFFF) с применением канала разделения с фриттой на входе. В целях анализа 10 мг лиофилизированного ацилированного гиалуронана со связанным витамином Е (полученным из перечисленных в таблице 1 производных способом, описанным в примере 23) растворяли в 1 мл подвижной фазы (50 мМ NaNO3 с 0,02% NaN3) и фильтровали через стеклянный шприцевой фильтр с размером пор 1 мкм. Впоследствии 100 мкл вводили в аппарат FlFFF.
Разделение проводили при помощи градиента поперечного потока от 2 мл/мин до 0,1 мл/мин в течение 5-минутного интервала. Скорость потока подвижной фазы, которую подавали в детектор, сохраняли постоянной, рабочая точка была 1 мл/мин.
Разделение проходило при лабораторной температуре. Элюат наблюдали при помощи детектора рассеивания света DAWN EOS, дифференциального рефрактометра Optilab rEX (оба изготовлены Wyatt Technology Corporation) и УФ-детектора, работающего с длиной волны 292 ни (Shimadzu).
При применении вышеуказанного способа является возможным определение как процентного отношения связанного вещества и гидрофобизированного гиалуронана, прочно содержащегося внутри наномицелл, так и их процентного отношения снаружи агрегатных структур (см. таблицу 1).
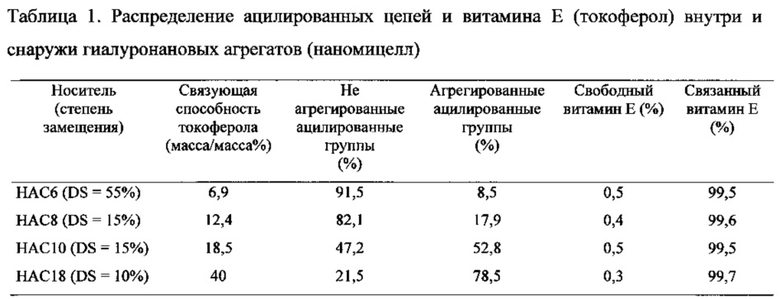
Результаты, представленные в таблице 1, четко показывают, что на распределение ацилированных цепей в гиалуронановых наномицеллах в первую очередь влияет длина ацильной цепи. Степень агрегации ацильных цепей возрастает с увеличением их длины. Включение неполярного вещества в наномицеллу происходит независимо от длины соответствующей ацильной цепи. В этом конкретном случае всегда полностью преобладает распределение неполярного вещества в мицелле (>99,5%).
Пример 31. Цитотоксичность наномицелл, несущих цитостатическое лекарственное средство на основе паклитаксела
Паклитаксел, связанный с ацилированными производными гиалуронана С6 и С16, который получали способами, описанными в примерах 19 и 25, соответственно, растворяли в среде для культивирования (содержащей 10% FBS), конечная концентрация составляла 100 мкг/мл. Клетки фибробласта кожи человека (NHDF), клеточную линию карциномы молочной железы человека (MCF-7) и клеточную линию карциномы толстой кишки человека (НСТ 116) использовали для исследования концентраций паклитаксела 0,001, 0,01, 0,1, 1,0, 10,0 и 100,0 мкг/мл, которое проводили при помощи производных ацилированного гиалуронана С6 и С16, исследование основывалось на определении жизнеспособности клетки. Влияние паклитаксела, переносимого производными ацилированного гиалуронана С6 и С16, сравнивали с влиянием паклитаксела самого по себе (фигура 4). Определение жизнеспособности клетки было основано на выявлении активности фермента дегидрогеназы, который активен в живых клетках и переносит желтый субстрат в фиолетовый раствор. Поглощение последнего, которое было определено при 540 нм, является пропорциональным процентному отношению живых клеток.
Увеличивающая концентрация носителя вызывала, особенно при использовании НАС16, небольшое снижение цитостатической эффективности паклитаксела (фигура 4).
Ацилированные производные сами по себе не проявляли никаких цитостатических эффектов.
Пример 32. Перенос инкапсулированных веществ в клетки
Вещества доксорубицин и 7-аминоактиномицин D (7-AAD, которое представляет собой вещество, что проникает только в фиксированные и пермеабилизированные клетки) инкапсулировали в ацилированное производное гиалуронана С6 способом, описанным в примере 18 (где 7-AAD использовали вместо Нильского красного). Клетки фибробласта кожи человека (NHDF), клеточную линию карциномы молочной железы человека (MCF-7) и клеточную линию карциномы толстой кишки человека (НСТ 116) использовали для исследования существования различия между проникновением вещества в клетку (если вещество применяют в растворе, в котором оно находится само по себе) или в ацилированное производное гиалуронана С6. Исследования проводили при помощи способа люминесцентной микроскопии (инвертационный микроскоп Nikon Eclipse Ti). Доксорубицин исследовали в концентрации 5,0 мкг/мл (фигура 5) и 7-AAD исследовали в концентрации 15 мкг/мл (фигура 6).
Перенос веществ из носителя в клетки был эффективным.
Пример 33. Высвобождение инкапсулированного масляного красного из наномицелл в раствор
Высвобождение масляного красного (Масляный Красный О, сольвент красный 27), инкапсулированного в НАС6 способом, описанным в примере 18 (где Масляный Красный О использовали вместо Нильского красного), в растворы изучали in vitro. Заданными используемыми растворами были PBS и PBS с добавлением 1% TWEEN 80. Водные растворы ацилированных производных со связанным масляным красным (в диапазоне концентраций от 1 до 10 мг.мл-1) растворяли в PBS или в PBS с добавлением 1% TWEEN 80, количественно переносили в трубки для диализа (MWCO 12-14 кДа, Spectrum Laboratories) и диализировали при температуре 37°С относительно PBS или относительно PBS с добавлением 1% TWEEN 80. В предпочтительных промежутках времени отбирали 4 мл диализата и заменяли свежей средой. Высвобожденное количество масляного красного было определено при помощи способа в УФ/видимом диапазоне (фигура 7).
Медленное высвобождение связанного вещества указывало на высокую стабильность систем носителя в PBS.
Пример 34. Высвобождение инкапсулированного паклитаксела из наномицелл в раствор
Высвобождение паклитаксела, инкапсулированного в НАС6 и НАС16 способом, описанным в примерах 19 и 25, соответственно, в раствор изучали in vitro при температуре 37°С. Водные растворы ацилированных производных, содержащих паклитаксел в общей концентрации 0,2 мг, растворяли в PBS, количественно переносили в трубки для диализа (MWCO 12-14 кДа, Spectrum Laboratories) и диализировали при температуре 37°С относительно 50 мл PBS. В предпочтительных промежутках времени диализат заменяли свежей средой. Высвобожденное количество паклитаксела определяли после его подвергания экстракции в хлороформ, выпариванию и последующему растворению в ацетонитриле при помощи ВЭЖХ (фигура 8).
Высвобождение, регистрируемое для НАС16, было более медленным по сравнению с НАС6. Медленное высвобождение связанного вещества указывало на высокую стабильность систем носителя в PBS.
Пример 35. Получение наноэмульсий и микроэмульсий из олеилового производного (С18:1) гиалуроновой и олеиновой кислот
80 мг ацилированного производного гиалуронана, полученного согласно примеру 11, растворяли в 4 мл воды при условиях перемешивания всю ночь. Полученный раствор постепенно дополняли 8 мг олеиновой кислоты при непрерывном перемешивании. После перемешивания полученную смесь подвергали двухстадийной обработке ультразвуком (ультразвуковая установка, UPS 200S, мощность 200 Вт). За первой стадией, которая была непрерывной (амплитуда 50%), следовала вторая стадия, которая была импульсной (частота импульсов 0,8 с, амплитуда 70%), каждая стадия длилась 25 минут. В течение процесса обработки ультразвуком емкость, содержащую обработанную смесь, погружали в ледяную баню для защиты от перегрева.
Размер частиц (определен при помощи Zetasizer): 200-300 нм. Размер зависит от количества масляной фазы в смеси.
Пример 36. Получение наноэмульсий и микроэмульсий из олеилового производного (С18:1) гиалуроновой и олеиновой кислот с растворенным коферментом Q10
80 мг ацилированного производного гиалуронана, полученного согласно примеру 11, растворяли в 4 мл воды при условиях перемешивания всю ночь. Полученный раствор постепенно дополняли 8 мг олеиновой кислоты, содержащей уже растворенный в ней кофермент Q10, (приблизительно 0,5 мг), при непрерывном перемешивании. После перемешивания полученную смесь подвергали двухстадийной обработке ультразвуком (ультразвуковая установка, UPS 200S, мощность 200 Вт). За первой стадией, которая была непрерывной (амплитуда 50%), следовала вторая стадия, которая была импульсной (частота импульсов 0,8 с, амплитуда 70%), каждая стадия длилась 25 минут. В течение процесса обработки ультразвуком емкость, содержащую обработанную смесь, погружали в ледяную баню для защиты от перегрева.
Размер частиц (определен при помощи Zetasizer): 200-300 нм. Размер зависит от количества масляной фазы в смеси.
Пример 37. Получение стабилизированных наномицелл при помощи ковалентных поперечных связей
10 г гиалуроната натрия (25 ммоль, 38 кДа) растворяли в 200 мл деминерализованной воды. Затем к раствору добавляли TEA (6,97 мл, 2 экв. по отношению к димеру НА) и DMAP (153 мг, 0,05 экв.). Активация: 3,5 г 3-(2-фурил)акриловой кислоты (25 ммоль) растворяли в 50 мл тетрагидрофурана и 19,2 мл TEA (2 экв.). Впоследствии полученный раствор охлаждали в ледяной бане и дополняли трихлорбензоилхлоридом (1,2 мл). Реакция проходила в течение 15 минут. После этого активированную кислоту добавляли к гиалуронановому раствору, и реакция проходила при температуре 25°С в течение 24 часов. Полученную реакционную смесь разбавляли эфиром. Акрилированное производное выделяли из реакционной смеси последующим способом осаждения с использованием 4-кратного абсолютного изопропанола. После подвергания декантированию осадок неоднократно промывали водным раствором изопропанола (85% по объему). После этого осадок сушили при температуре 40°С в течение 48 часов. Полученное акрилированное производное было ацилировано (см. пример 1), и впоследствии растворено в воде и лиофилизировано для удаления остаточных растворителей.
Систему носителя, которая была получена из масляного красного (Масляный Красный О) и из акрилированного производного согласно примеру 18 (где масляный красный использовали вместо Нильского красного), растворяли с образованием 1% водного раствора. В полученном растворе система носителя была поперечно связана посредством пероксодисульфата аммония (10 экв.), используемого как инициатор.
DS 20% активной группы для фотосшивания (определено при помощи ЯМР)
1Н ЯМР (D2O): δ 7.83, 6.87 (d, J=3,5), 6.87, 6.61 (bs), 7.83, 7.59 (Jтранс=16.01), 6.39 (Jтранс=15.85).
Пример 38. Местное применение гиалуронановых наномицелл - проникновение в кожу, волосы и слизистые оболочки
Свиная кожа, вагинальная слизистая оболочка крупного рогатого скота и трансбуккальная слизистая оболочка крупного рогатого скота были получены от компании MasoEko, s.r.o., которая находится в Kunčice 243, Letohrad. Сразу же после взятия образцы подвергали пассивному действию раствора ацилированного гиалуронана С6 и С10 (10 мг.мл-1) с инкапсулированным Нильским красным (полученным способом, описанным в примере 18), и проникновение в образцы сравнивали при помощи измерения значений флуоресценции в инвертационном микроскопе Nikon Eclipse Ti (оборудованном объективами Plan Fluor 4х). Последующая инкубация при температуре 37°С: образцы кожи в течение 20 часов (фигура 9), образцы волос в течение 15 минут (фигура 10) и образцы слизистых оболочек в течение 4 часов (фигуры 11 и 12).
Носители способствовали достижению более эффективного проникновения в кожу, волосы и слизистые оболочки по сравнению с масляными и водными растворителями.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОТИВООПУХОЛЕВАЯ КОМПОЗИЦИЯ НА ОСНОВЕ ГИАЛУРОНОВОЙ КИСЛОТЫ И НЕОРГАНИЧЕСКИХ НАНОЧАСТИЦ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И ЕЕ ПРИМЕНЕНИЕ | 2015 |
|
RU2686679C2 |
| САМОПОДДЕРЖИВАЮЩАЯ БИОРАЗЛАГАЕМАЯ ПЛЕНКА НА ОСНОВЕ ГИДРОФОБИЗОВАННОЙ ГИАЛУРОНОВОЙ КИСЛОТЫ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2016 |
|
RU2712174C2 |
| ПРОИЗВОДНОЕ ГИАЛУРОНОВОЙ КИСЛОТЫ, СПОСОБ ЕГО ПОЛУЧЕНИЯ, СПОСОБ ЕГО МОДИФИКАЦИИ И ЕГО ПРИМЕНЕНИЕ | 2013 |
|
RU2647859C2 |
| ПРОИЗВОДНОЕ ГИАЛУРОНОВОЙ КИСЛОТЫ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ ЕГО МОДИФИКАЦИИ | 2010 |
|
RU2550602C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОКИСЛЕННОГО ПРОИЗВОДНОГО ГИАЛУРОНОВОЙ КИСЛОТЫ И СПОСОБ ЕГО МОДИФИКАЦИИ | 2010 |
|
RU2559447C2 |
| ПРОИЗВОДНЫЕ НА ОСНОВЕ ГИАЛУРОНОВОЙ КИСЛОТЫ, СПОСОБНЫЕ ОБРАЗОВЫВАТЬ ГИДРОГЕЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ГИДРОГЕЛИ НА ОСНОВЕ УКАЗАННЫХ ПРОИЗВОДНЫХ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2013 |
|
RU2586931C2 |
| НЕНАСЫЩЕННЫЕ ПРОИЗВОДНЫЕ ПОЛИСАХАРИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2017 |
|
RU2725500C1 |
| ДИГИДРОБЕНЗОДИАЗЕПИНЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2000 |
|
RU2247115C2 |
| ОКСАЗОЛИДИНОНОВЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2006 |
|
RU2417223C2 |
| НОВЫЕ БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2016 |
|
RU2760554C1 |
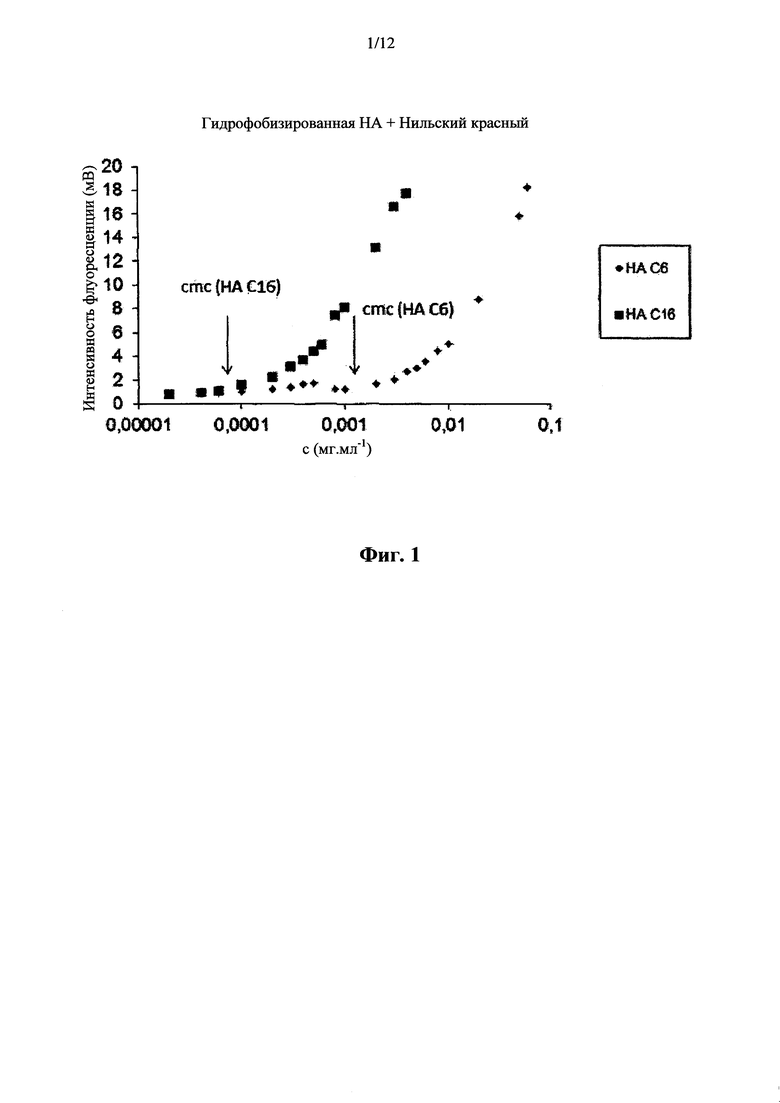
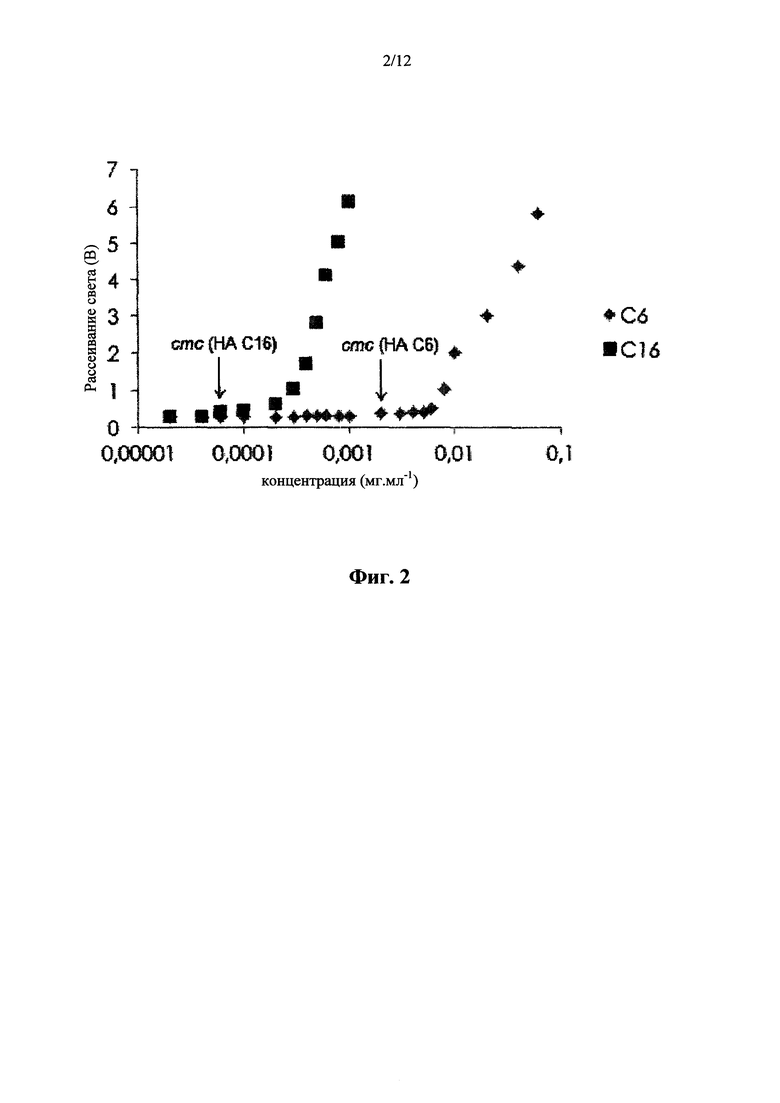




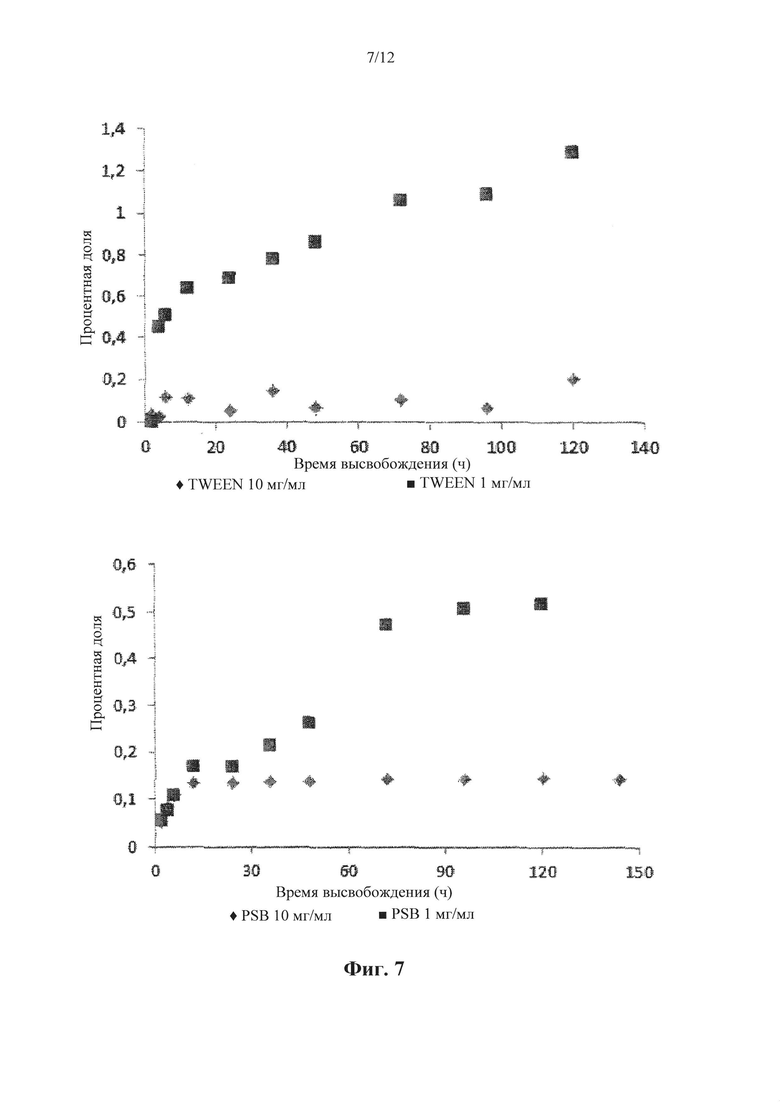



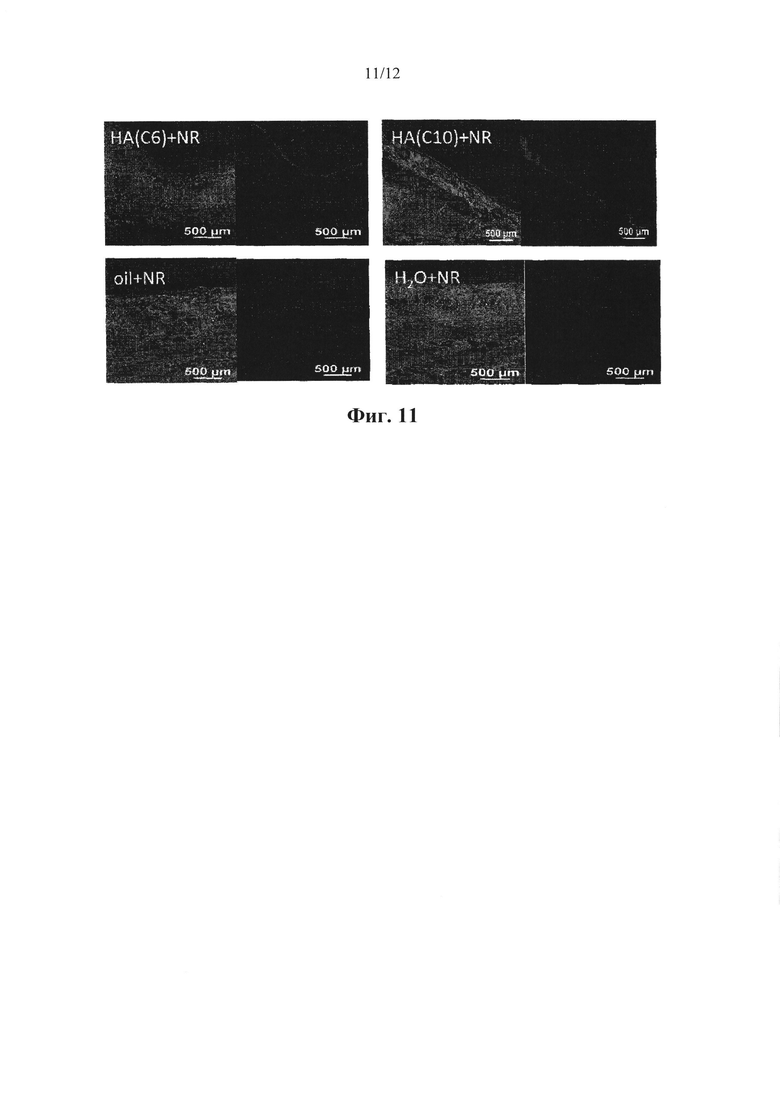

Настоящее изобретение относится к способу получения гидрофобизированной гиалуроновой кислоты (формула I). Причем R представляет собой H+ или Na+ и R1 представляет собой H или -С(=O)CxHy или -C(=O)CH=CH-het, где x представляет собой целое число в диапазоне от 5 до 17, и y представляет собой целое число в диапазоне от 11 до 35, и CxHy представляет собой неразветвленную или разветвленную, насыщенную или ненасыщенную C5-C17 цепь, и het представляет собой гетероциклическую или гетероароматическую группу с произвольным содержанием атомов N, S или О, по меньшей мере с одной повторяющейся единицей, содержащей одну или несколько R1 -С(=O)CxHy или -C(=O)CH=CH-het групп, и где n находится в диапазоне от 12 до 4000. Также предложены способ получения этого прозводного гиалуроновой кислоты, наномицеллярная композиция на ее основе, способ получения наномицеллярной композиции, ее применение при фармацевтических и косметических применениях, а также способ получения ее стабилизированной формы. Изобретение позволяет получить гидрофобный носитель на основе гиалуроновой кислоты для биологически активных веществ и обеспечивает перенос связанных веществ из гидрофобных доменов наномицеллы в клетку. 6 н. и 29 з.п. ф-лы, 12 ил., 1 табл., 38 пр.
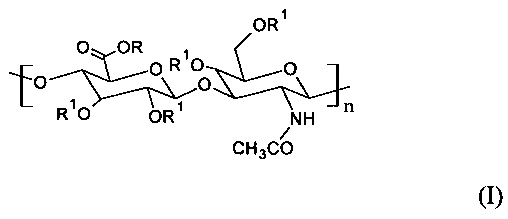
1. С6-С18-ацилированное производное гиалуроновой кислоты общей формулы (I)
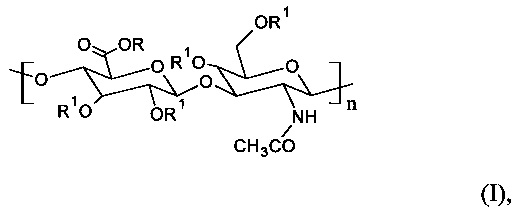
где R представляет собой H+ или Na+ и R1 представляет собой H или -С(=O)CxHy или -C(=O)CH=CH-het, где x представляет собой целое число в диапазоне от 5 до 17, и y представляет собой целое число в диапазоне от 11 до 35, и CxHy представляет собой неразветвленную или разветвленную, насыщенную или ненасыщенную C5-C17 цепь, и het представляет собой гетероциклическую или гетероароматическую группу с произвольным содержанием атомов N, S или О, по меньшей мере с одной повторяющейся единицей, содержащей одну или несколько R1 -С(=O)CxHy или -C(=O)CH=CH-het групп, и где n находится в диапазоне от 12 до 4000.
2. С6-С18-ацилированное производное гиалуроновой кислоты по п. 1, характеризующееся тем, что R представляет собой Н+ или Na+, и R1 представляет собой -С(=O)(СН2)7-СН=СН-(СН2)7-СН3.
3. Способ получения производного гиалуроновой кислоты общей формулы (I), отличающийся тем, что осуществляют взаимодействие гиалуроновой кислоты с С6-C18-карбоновой кислотой, активированной хлоридом 2,4,6-трихлорбензойной кислоты или активированной органическим хлоридом R3-CO-Cl, где R3 представляет собой алифатический или разветвленный C1-С30-алкил, гетероароматическую функциональную группу или ароматическую функциональную группу, в присутствии основания и катализатора в смеси воды и смешивающегося с водой апротонного растворителя.
4. Способ получения по п. 3, отличающийся тем, что гиалуроновая кислота находится в свободной кислотной форме или в форме фармацевтически приемлемой соли и обладает молекулярной массой в предпочтительном диапазоне от 5×103 г/моль до 1,6×106 г/моль, более предпочтительно, от 15×103 г/моль до 250×103 г/моль, и наиболее предпочтительно, от 15×103 до 50×103 г/моль.
5. Способ получения по п. 3 или 4, отличающийся тем, что гиалуроновую кислоту растворяют в смеси воды и смешивающегося с водой апротонного растворителя, последнее представляет собой полярный органический растворитель, содержание воды находилось в диапазоне от 10 до 99% по объему, предпочтительно, 50% по объему.
6. Способ получения по п. 3 или 4, отличающийся тем, что смешивающийся с водой апротонный растворитель представляет собой диметилсульфоксид, тетрагидрофуран, ацетон, ацетонитрил или изопропанол.
7. Способ получения по п. 3 или 4, отличающийся тем, что реакционная смесь содержит R'3N основание, где R' представляет собой неразветвленную или разветвленную CnHm-углеводородную цепь, где n представляет собой целое число в диапазоне от 1 до 4 и m представляет собой целое число в диапазоне от 3 до 9, например триэтиламин, в количестве от 0,01 до 20 эквивалентов, предпочтительно, 6 эквивалентов, по отношению к димеру гиалуроновой кислоты, и катализатор выбран из группы, включающей в себя замещенные пиридинины, такие как диметиламинопиридин, в количестве от 0,01 до 1 эквивалента, предпочтительно, 0,05 эквивалента, по отношению к димеру гиалуроновой кислоты.
8. Способ получения по п. 3 или 4, отличающийся тем, что сначала активацию C6-C18-карбоновой кислоты проводят в полярном органическом растворителе в присутствии основания и 2,4,6-трихлорбензойной кислоты или ее производных, или в присутствии основания и органического хлорида, и впоследствии смесь, содержащую активированную С6-С18-карбоновую кислоту, добавляют к гиалуроновой кислоте, которая была растворена в смеси воды, органического растворителя, основания и катализатора, продуктом в результате реакции является производное общей формулы (I).
9. Способ получения по п. 3 или 4, отличающийся тем, что указанная С6-С18-карбоновая кислота выбрана из группы, содержащей капроновые, энантовые, каприловые, каприновые, пальмитиновые, стеариновые, олеиновые, линолевые и линоленовые кислоты.
10. Способ получения по п. 3 или 4, отличающийся тем, что количество активированной С6-С18-карбоновой кислоты находится в диапазоне от 0,01 до 5 эквивалентов, предпочтительно, от 0,5 до 2 эквивалентов, по отношению к димеру гиалуроновой кислоты.
11. Способ получения по п. 3 или 4, отличающийся тем, что активация С6-С18-карбоновой кислоты происходит в течение от 5 до 120 минут, предпочтительно, в течение 30 минут, при температуре от 20 до 60°С, предпочтительно, при температуре 25°С.
12. Способ получения по п. 3 или 4, отличающийся тем, что взаимодействие гиалуроновой кислоты с активированной С6-С18-карбоновой кислотой происходит в течение от 1 до 24 часов, предпочтительно, в течение от 2 до 3 часов, при температуре от 20 до 60°C, предпочтительно, при температуре 25°С.
13. Способ получения по п. 3 или 4, отличающийся тем, что С6-С18-ацилированное производное гиалуроновой кислоты впоследствии отделяют от реакционной смеси, промывают, сушат и лиофилизируют.
14. Способ получения по п. 13, отличающийся тем, что С6-С18-ацилированное производное гиалуроновой кислоты отделяют от реакционной смеси в процессе осаждения с применением NaCl и спирта.
15. Способ получения по п. 13, характеризующийся тем, что С6-С18-ацилированное производное гиалуроновой кислоты промывают спиртом, в частности изопропанолом или этанолом.
16. Наномицеллярная композиция на основе С6-С18-ацилированного производного гиалуроновой кислоты общей формулы (I), отличающаяся тем, что композиция содержит наномицеллы, которые включают в себя гидрофобное ядро, образованное С6-C18-ацильными группами, связанными с гиалуроновой кислотой, и гидрофильную оболочку, образованную гидрофильными функциональными группами гиалуроновой кислоты, и одно или несколько биологически активных веществ, физически связанных в наномицелле.
17. Наномицеллярная композиция по п. 16, отличающаяся тем, что она содержит от 0,3 до 50% по массе биологически активного вещества по отношению к массовому содержанию С6-С18-ацилированного производного гиалуроновой кислоты, биологически активное вещество было выбрано из группы, включающей в себя фармацевтически и косметически активные вещества, в частности витамины, медикаменты, цитостатические средства, фитоэкстракты, фитокомплексы или фитоактивные вещества, минеральные или растительные масла, или их смесь.
18. Наномицеллярная композиция по п. 17, отличающаяся тем, что биологически активное вещество представляет собой токоферол, паклитаксел, фосфатидилхолин или кофермент Q10.
19. Наномицеллярная композиция по п. 16, отличающаяся тем, что она содержит С6-C18-ацилированное производное гиалуроновой кислоты в концентрации, которая выше его критической концентрации агрегации.
20. Наномицеллярная композиция по п. 16, отличающаяся тем, что концентрация С6-С18-ацилированного производного гиалуроновой кислоты находится в диапазоне от 0,0001 мг⋅мл-1 до 30 мг⋅мл-1, предпочтительно, от 1 до 20 мг⋅мл-1, если композиция представляет собой водный раствор.
21. Наномицеллярная композиция по п. 16, отличающаяся тем, что биологически активное вещество представляет собой минеральное или растительное масло в количестве от 0,05 до 40% по массе, предпочтительно, от 1 до 20% по массе, по отношению к массовому содержанию С6-С18-ацилированного производного гиалуроновой кислоты.
22. Наномицеллярная композиция по любому из пп. 16-21, отличающаяся тем, что она содержит биологически активное вещество, которое является жидким и нерастворимым в воде, указанное вещество содержит дополнительное биологически активное вещество, растворенное в нем.
23. Наномицеллярная композиция по п. 22, отличающаяся тем, что биологически активное вещество, которое является жидким и нерастворимым в воде, представляет собой минеральное или растительное масло, и дополнительное биологически активное вещество относится к фармацевтически или косметически активным веществам, в частности витаминам, медикаментам, цитостатическим средствам, фитоэкстрактам, фитокомплексам или фитоактивным веществам, или к их смесям.
24. Наномицеллярная композиция по любому из пп. 16-21 или п. 23, отличающаяся тем, что она находится в форме раствора, наноэмульсии, микроэмульсии, коацервата или геля.
25. Способ получения наномицеллярной композиции по любому из пп. 16-24, отличающийся тем, что С6-С18-ацилированное производное гиалуроновой кислоты общей формулы (I) растворяют в воде, биологически активное вещество растворяют в органическом растворителе, полученные растворы смешивают вместе, и впоследствии органический растворитель удаляют.
26. Способ получения по п. 25, отличающийся тем, что органический растворитель удаляют при помощи выпаривания под вакуумом, впоследствии водную фазу сушат и регидратируют, и полученные наномицеллярные структуры фильтруют и в конечном итоге лиофилизируют.
27. Способ получения по п. 24, отличающийся тем, что органический растворитель удаляют при помощи диализа, впоследствии полученные наномицеллярные структуры фильтруют и в конечном итоге лиофилизируют.
28. Способ получения по любому из пп. 25-27, отличающийся тем, что органический растворитель представляет собой летучий хлорированный растворитель, такой как трихлорметан, или спирт, такой как этанол или изопропанол.
29. Способ получения наномицеллярной композиции по п. 16, отличающийся тем, что С6-С18-ацилированное производное гиалуроновой кислоты общей формулы (I) растворяют в воде, и впоследствии смешивают вместе с биологически активным веществом, которое является жидким и нерастворимым в воде, после чего полученную смесь гомогенизируют при помощи обработки ультразвуком с образованием микроэмульсии или наноэмульсии.
30. Способ получения наномицеллярной композиции по п. 22 или 23, отличающийся тем, что С6-С18-ацилированное производное гиалуроновой кислоты общей формулы (I) растворяют в воде, и впоследствии смешивают вместе с биологически активным веществом, которое является жидким и нерастворимым в воде, и в котором растворено дополнительное биологически активное вещество, после чего полученную смесь гомогенизируют при помощи обработки ультразвуком с образованием микроэмульсии или наноэмульсии.
31. Применение наномицеллярной композиции по любому из пп. 16-24 при фармацевтических или косметических применениях, предпочтительно, при местных применениях.
32. Способ получения стабилизированной наномицеллярной композиции, отличающийся тем, что получают С6-С18-ацилированный гиалуронан общей формулы (II)
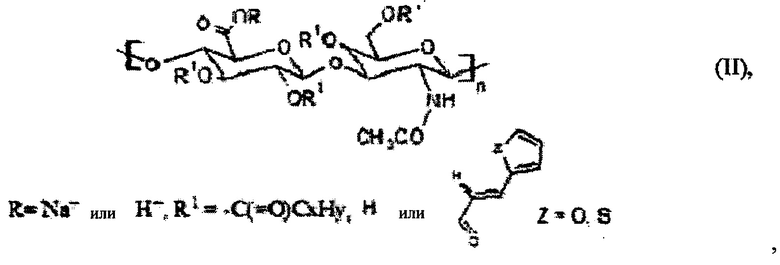
где R представляет собой H+ или Na+, один или несколько R1 членов представляют собой неразветвленную С6-С18-цепь по меньшей мере в одной повторяющейся единице, при этом неразветвленная цепь может содержать ненасыщенные связи, и 3-(2-тиенил)акриловую кислоту или 3-(2-фурил)акриловую кислоту или производные указанных кислот в другой по меньшей мере одной повторяющейся единице, при этом наномицеллярную композицию получают из С6-С18-ацилированного гиалуронана общей формулы (II), такую композицию затем стабилизируют при помощи реакции сшивания.
33. Способ получения по п. 32, отличающийся тем, что сначала осуществляют взаимодействие гиалуроновой кислоты с 3-(2-тиенил)акриловой кислотой или 3-(2-фурил)акриловой кислотой или с производным любой кислоты в присутствии основания и катализатора в смеси воды и смешивающегося с водой апротонного растворителя, указанные кислоты были активированы хлоридом 2,4,6-трихлорбензойной кислоты или активированы органическим хлоридом R3-CO-Cl, где R3 представляет собой алифатический или разветвленный C1-С30-алкил, возможно содержащий гетероароматическую или ароматическую функциональную группу, с образованием акрилированного гиалуронана формулы (III)
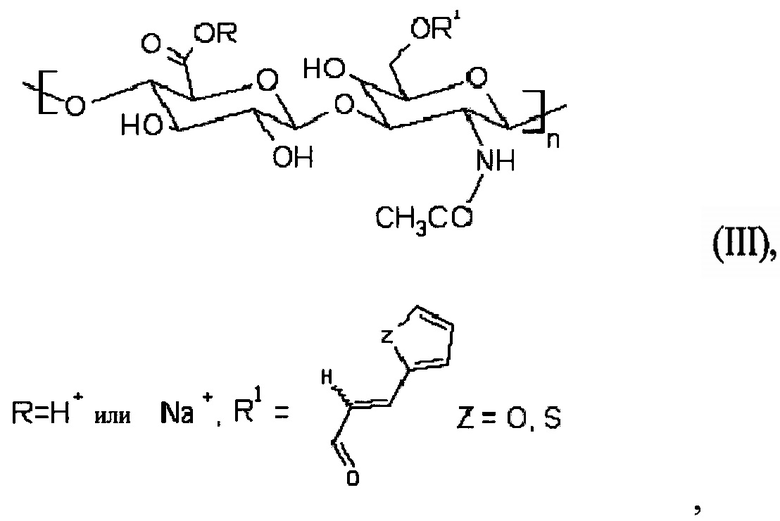
после этого осуществляют взаимодействие акрилированного гиалуронана формулы (III) с С6-С18-карбоновой кислотой, активированной хлоридом 2,4,6-трихлорбензойной кислотой или активированной органическим хлоридом R3-CO-Cl, где R3 представляет собой алифатический или разветвленный C1-С30-алкил, по мере необходимости содержащий гетероароматическую или ароматическую функциональную группу, в присутствии основания и катализатора в смеси воды и смешивающегося с водой апротонного растворителя с образованием С6-С18-ацилированного гиалуронана формулы (II), впоследствии наномицеллярную композицию получают из указанного гиалуронана формулы (II) с применением способа по любому из пп. 25-30, при этом композицию затем подвергают реакции сшивания при помощи реакций радикального типа.
34. Способ получения по любому из пп. 32 и 33, отличающийся тем, что реакцию радикального типа катализируют при помощи сшивающего агента.
35. Способ получения по п. 34, отличающийся тем, что сшивающий агент представляет собой пероксодисульфат аммония.
| C | |||
| EENSCHOOTEN; F | |||
| GUILLAUMIEA; G | |||
| M.KONTOGEORGIS; E | |||
| H.STENBY; K | |||
| SCHWACH-ABDELLAOUI "Preparation and structural characterisation of novel and versatile amphiphilic octenyl succinic anhydride-modified hyaluronic acid derivatives", CARBOHYDRATE POLYMERS; vol | |||
| Цилиндрический сушильный шкаф с двойными стенками | 0 |
|
SU79A1 |
| WO 2007033677 A1, 29.03.2007 | |||
| Линейный интерполятор | 1988 |
|
SU1538166A2 |
| CN 101897976 A, 01.12.2010 | |||
| EP 1994062 B1, 15.07.2009. | |||

