Область изобретения
Изобретение относится к пирроло[2,1-f][1,2,4]триазиновым производным, представленным общей формулой I, их изомерам или их фармацевтически приемлемым солям, сложным эфирам или гидратам, и способу их получения и применения. Соединения I, представленные общей формулой I могут ингибировать фосфатидилинозитол 3-киназный (PI3K) сигнальный путь, таким образом применяться для приготовления лекарственных средств для лечения заболеваний, связанных в фосфатидилинозитол 3-киназой, таких как рак.
Предшествующий уровень техники
PI3K представляет собой липидную киназу и может фосфорилировать по 3-позиции инозитольного кольца в фосфатидилинозитоле с образованием фосфатидилинозитол-3-фосфата (PIP), фосфатидилинозитол-3,4-дифосфата (PIP2) и фосфатидилинозитол-3,4,5-трифосфата (PIP3). PIP, PIP2 и PIP3, будучи важными вторичными мессенджерами, связываются и активируют различные белки, содержащие домен РН (плекстрин-гомологичный домен), домен FYVE (названный по первым буквам в названиях белков Fablp, YOTB, Vaclp и ЕЕА1, которые, как исходно обнаружили, содержат домен FYVE, смотри Gaullier, J.М.; Simonsen, A.; D′ Arrigo, A.; Bremnes, В.; Stenmark, Н., Chem. Phys. Lipids, 1999, 98: 87-94.), домен РХ (Phox-гомологичный домен) и другую связывающуюся с фосфолипидом область с образованием комплекса сигнального каскада, и наконец регулируют клеточные функции, такие как пролиферация, дифференциация, выживание и миграция и т.п. (смотри Vanhaesebroeck, В.; Leevers, S.J.; Ahmadi, К.; Timms, J.; Katso, R.; Driscoll, P.C; Woscholski, R.; Parker, P.J.; Waterfield, M.D., Annu. Rev. Biochem., 2001, 70: 535-602).
В зависимости от различий в генной последовательности, субстратной специфичности и функции суперсемейство PI3K группируется в три класса: I, II и III PI3K. Класс I PI3K представляет собой наиболее широко исследованный до настоящего времени класс. Субстраты PI3K представляют собой фосфатидилинозитол (PI), фосфатидилинозитол-4-фосфат (PI(4)P), фосфатидилинозитол 4,5-бисфосфат (PI(4,5)P2). Класс I PI3K представляют собой гетеродимерные молекулы, состоящие из одной каталитической субъединицы и одной регуляторной субъединицы. Класс I PI3K может быть дополнительно разделен на две категории ввиду различий в регуляторной субъединице и в механизме активации: PI3K IA и PI3K IB. Где PI3K IA содержит PI3Kα, PI3Kβ и PI3Kδ и активируется рецепторной тирозинкиназой; хотя PI3K IB состоит только из PI3Ky и активируется рецепторами, связанными с G белком. PI и PI(4)Р представляют собой подклассы класса II PI3K. Класс II PI3K включает PI3KC2α, PI3KC2β и PI3KC2y. Они характеризуются доменом С2 по С-концу, что указывает на то, что их активности регулируются ионом кальция. Субстрат для класса III PI3K представляет собой PI. Механизм его активации до настоящего времени остается невыясненным (смотри Engelman, J.A.; Luo, J.; Cantley, L.С, Nat Rev. Genet, 2006, 7: 606-619).
Гиперактивация PI3K инициирует сигнальный путь через фосфатидилинозитол 3-киназу/белковую киназу В/белок-мишень млекопитающих для рапамицина (PI3K/Akt/mTOR) и способствует клеточному выживанию и пролиферации, которые часто присутствуют приблизительно в 60% человеческих опухолей. PTEN (гомолог фосфата и тензина с делецией на хромосоме 10) действует в качестве агента, подавляющего опухоли, и дефосфорилирует 3-позицию в инозитольном кольце фосфатидилинозитола и оказывают антагонистическую активность в отношении PI3K. Такая функция утрачивается при множестве форм рака. Активная мутация в гене PIK3CA, кодирующем р110α, представлена свыше чем при 30% видов рака. Кроме того, амплификации генов PI3K3CA и протеинкиназы В (Akt) часто обнаруживаются при других формах рака, которые также вносят вклад в экспрессию белка (смотри Engelman, J.A., Nat. Rev. Рак, 2009, 9: 550-562). Эти факты свидетельствуют о том, что PI3K тесно связана с онкогенезом и активацией. Белок-мишень для рапамицина (mTOR) представляет собой один из важных последующих белков каскада протеинкиназы В, представляющей собой сериновую/треониновую киназу. Протеинкиназа В дополнительно активирует белок-мишень рапамицина путем прямого фосфорилирования mTOR; или опосредованно, усиливая активацию mTOR путем инактивации подавляющего опухоль гена TSC2 (белок туберозного склероза 2). Активный mTOR прямо или опосредованно принимает участие в регуляциях различных процессов, связанных с клеточной пролиферацией и ростом, таких как исходная фаза трансляции, транскрипции, разборки микрофиламента, мембранного транспорта, деградации белка, пути протеинкиназы С (РКС), синтеза рибосомального белка и синтеза tRNA и т.п. путем регуляции последующих сигнальных путей, таких как рибосомальная S6 киназа (S6K1 или P70S6K), белок 1 (4Е-ВР1), связывающийся с фактором инициации трансляции эукариотических клеток 4Е (elF-4 е), фактор 3 трансдукции сигнала и активации транскрипции (STAT3) и т.п. Таким образом, mTOR представляет собой центральный регуляторный белок клеточного роста и пролиферации и становится новой мишенью для нового противоопухолевого лекарства.
Ингибиторы PI3K и последующего сигнального белка mTOR представляют собой класс многообещающих противоопухолевых лекарств. В настоящее время, несколько ингибиторов пан-PI3K, таких как GDC-0941, XL-147, РХ-866, и т.п. были вовлечены в клинические исследования. Тем не менее, количество и структурное разнообразие должны быть увеличены для того, чтобы удовлетворять потребностям исследования разработки нового противоракового лекарства. В то же время, существуют дефекты, присутствующие у известных ингибиторов. Например РХ-866, происходящий из вортманнина, сложно синтезировать; и активность GDC-0941 должна быть улучшена. Таким образом, открытие и разработка противоопухолевых лекарств, нацеленных на PI3K с более высокой активностью, большей безопасностью привлекают растущий интерес по всему миру.
Пирроло[2,1-f][1,2,4]триазин представляет собой исключительную структуру в медицинской химии. После того, как об этой исключительной структуре сообщали в качестве пуриновых аналогов (смотри: Hayashi, М.; Araki, A.; Maeba, I., Heterocycles, 1992, 34: 569-574. Patil, S.A.; Otter, В.A.; Klein, R.S., Tempahedron Lett., 1994, 35: 5339-5342), все больше и больше соединений, содержащих такую исключительную структуру, были синтезированы и демонстрировали разнообразие биологических активностей, например действующие в качестве ингибиторов JAK2 (смотри: Weinberg, L.R.; Albom, М.S.; Angeles, Т.S. et al., Bioorg. Med. Chem. Lett. 2001, 21: 7325-7330), ингибиторов киназы пан-аврора (Abraham, S.; Hadd, M.J.; Tran, L. et al., Bioorg. Med. Chem. Lett. 2011, 21: 5296-5300), ингибиторов митогенактивируемой протеинкиназы р38α (р38α МАРК) (Liu, С; Lin, J.; Wrobleski, S.Т. et al., J. Med. Chem., 2010, 53: 6629-6639), ингибиторов киназы лимфомы ALK (Mesaros, E.R; Thieu, Т.V; Wells, G.J. et al., J. Med. Chem., 2012, 55: 115-125), двойных ингибиторов VEGFR-2/FGFR-1 (Cai, Z. - w.; Zhang, Y; Borzilleri, R.M. et al., J. Med. Chem., 2008, 5: 1976-1980), ингибиторов VEGFR-2 (Hunt, J.Т.; Mitt, Т.; Borzilleri, R. et al., J. Med. Chem., 2004, 47: 4054-4059), ингибиторов EGFR1/2 (Gavai, A.V.; Fink, В.E.; Fairfax, D.J. et al., J. Med. Chem., 2009, 52: 6527-6530), ингибиторов IGF-1R (смотри: Wittman, M.D.; Carboni, J.M.; Yang, Z. et al. J. Med. Chem., 2009, 52: 7360-7363), или ингибиторов киназы Met (смотри: Schroeder, G.M.; Chen, X. - T.; Williams, D.K. et al., Bioorg. Med. Chem. Lett, 2007, 18: 1945-1951). Кроме того, соединения, содержащие эту пирроло[2,1-f][1,2,4]триазиновую исключительную структуру, такие как EGFR ингибитор АС-480 (WO-2004054514), антагонист рецептора VEGF-2 BMS-690514 (WO 2005/066176 A1), и антагонист IGF-1R BMS-754807 (US 2008/0009497 А1) и т.п. были включены в клинические исследования. Также сообщали о способах синтеза коровой структуры пирроло[2,1-f][1,2,4]триазина, например Thieu, Т; Sclafani, J.A.; Levy, D.V. et al., Org. Lett, 2011, 13: 4204-4207. Дополнительно к вышеупомянутым литературным источникам существует множество заявок на патенты, связанных с коровой структурой пирроло[2,1-f][1,2,4]триазина, например, действующих в качестве ингибиторов киназы (публикация No.: US 2006/0084650 A1), ингибиторов киназы EGFR (публикация No.: US 2006/0089358 A1, WO 2006/069395), ингибиторов VEGFR-2 и FGFR-1 (публикация No.: WO 2004/009784, WO 2004/043912), и заявок на патенты, связанных со способами синтеза промежуточных соединений (WO 2007/005709, WO 2008/083398), ингибиторов тирозинкиназного рецептора (WO 2007/061882, WO 2008/131050), ингибиторов киназы аврора (публикация №: WO 2009/136966), ингибиторов киназы JAK (публикация No.: WO 2010/002472). Вышеупомянутые пирроло[2,1-f][1,2,4]триазины, о которых сообщалось, не охватывают и не относятся к соединениям по настоящему изобретению и их применению в качестве ингибиторов PI3K.
На основе вышеприведенных соображений авторы изобретения разработали и синтезировали серии ингибиторов PI3K с пиррол[2,1-f][1,2,4]триазином в качестве коровой структуры. Соединения по настоящему изобретению продемонстрировали превосходную биологическую активность in vitro и in vivo, и, как предполагается, будут преобразованы в новое лекарственное средство для лечения рака.
Краткое изложение сущности изобретения
Задача настоящего изобретения заключается в том, чтобы предложить новый тип пирроло[2,1-f][1,2,4]триазиновых производных, представленных общей формулой I.
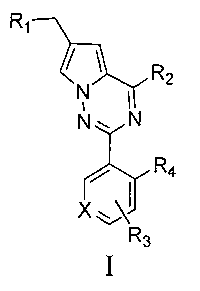
где X представляет собой СН или N;
R1 представляет собой -NR5R6;
R2 представляет собой 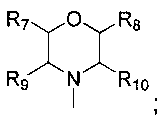
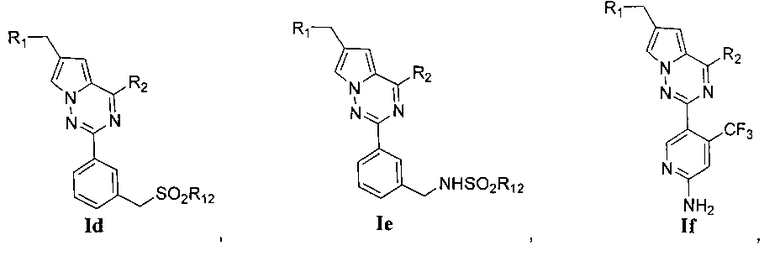
R3 представляет собой -NH2, -NHC(O)NHR11, -NHC(O)OR11, -СН2ОН, -CH2S(O)2R12, -CH2OS(O)2R12 или -CH2NHS(O)2R12;
R4 представляет собой H или CF3;
каждый из R5 и R6 независимо представляет собой C1-C4 ал кил или вместе с атомом азота, к которому они присоединены, образуют незамещенный насыщенный гетероцикл или насыщенный гетероцикл, замещенный заместителем(ями), предпочтительно пирролидил, пиперидинил и пиперазинил, более предпочтительно пиперазинил; где заместитель представляет собой -S(O)2R12;
каждый из R7, R8, R9 и R10 независимо представляет собой Н или C1-C3 алкил; альтернативно R7 и R8 или R9 и R10 комбинированы с атомом углерода, к которому они присоединены, с образованием 5-8-членного насыщенного кольца; предпочтительно R7 и R8 или R9 и R10 с атомами азота, к которым они присоединены в качестве мостиковых атомов углерода, образуют связанный мостиковыми связями бицикло-гетероцикл с морфолиновым кольцом;
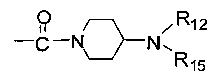
R11 представляет собой C1-C4 алкил, незамещенный C3-C6 циклоалкил или C3-C6 циклоалкил, замещенный одним или более чем одним заместителем, незамещенный бензил или бензил, замещенный одним или более чем одним заместителем, незамещенный фенил или фенил, замещенный одним или более чем одним заместителем, незамещенный изоксазолил или изоксазолил, замещенный одним или более чем одним заместителем, или незамещенный пиридил или пиридил, замещенный одним или более чем одним заместителем, где один или более чем один заместитель выбран из галогена, C1-C3 алкила или C1-C3 алкоксила, -CF3, -C(O)OR12, -C(O)NR12R15, 
 или
или 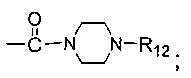
каждый из R12 и R15 независимо представляет собой C1-C3 алкил. Предпочтительно, структура общей формулы I представлена следующим образом:
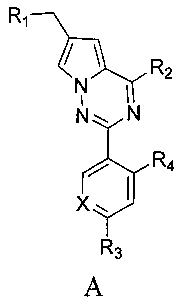 или
или 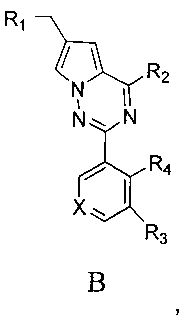
где X, R1, R2, R3 и R4 являются такими как определено выше.
Более предпочтительно, R1 представляет собой диметиламино или 1-метилсульфонилпиперазинил;
R2 представляет собой морфолинил, (S)-3-метилморфолинил или 8-окса-3-азабицикло[3,2,1]октан-3-ил;
R3 представляет собой -NH2, -NHC(O)NHR11, -NHC(O)OR11, -СН2ОН, -CH2S(O)2Me, или -CH2NHS(O)2Me;
R4 представляет собой Н или -CF3;
R11 представляет собой метил, этил, пропил, циклопропил, трет-бутил, изобутил, 4-фторбензил, незамещенный фенил или фенил, замещенный одним или более чем одним заместителем, незамещенный изоксазолил или изоксазолил, замещенный одним или более чем одним заместителем, или незамещенное пиридиновое кольцо или пиридиновое кольцо, замещенное одним или более чем одним заместителем, и заместитель выбран из фтора, хлора, трифторметила, метила, метокси, этоксикарбонила, диметиламинокарбонила, 4-метил-пиперазин-1-карбонила, пиперидин-1-карбонила и 4-диметиламинопиперидин-1-карбонила.
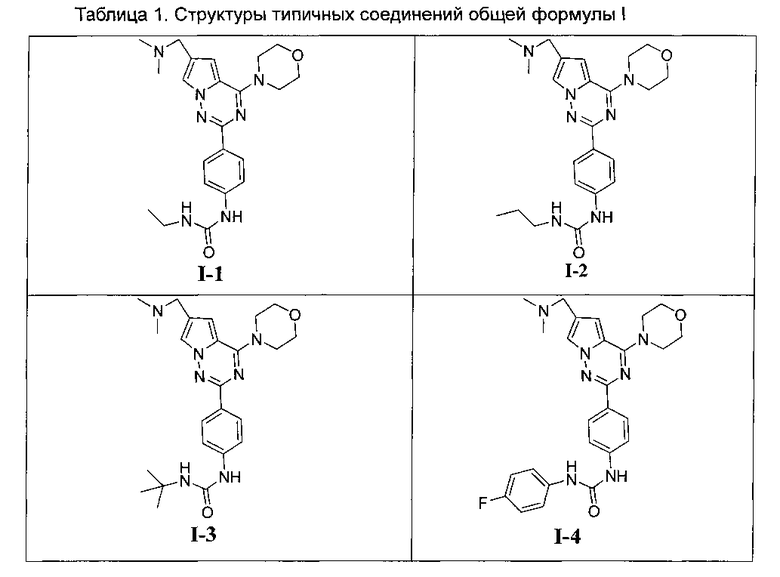
Более предпочтительно, соединения, представленные общей формулой I, имеют следующие структуры:
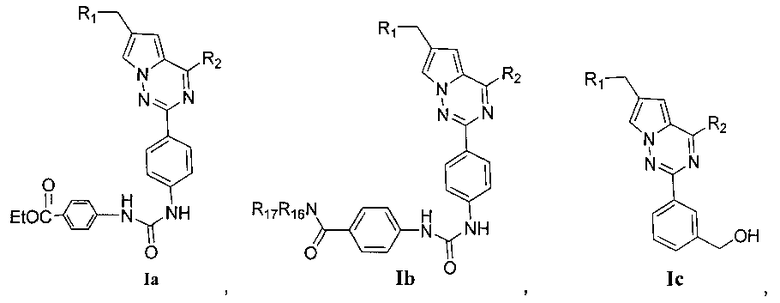
или
где R1, R2, R11 и R12 являются такими как определено выше,
R16 и R17 идентичны или отличаются, и каждый независимо выбран из C1-C4 алкила, или R16 и R17 вместе с атомом азота, к которому они присоединены, образуют 4-метил-пиперазинил, 4-диметиламино-пиперидил или пиперидин-1-ил.
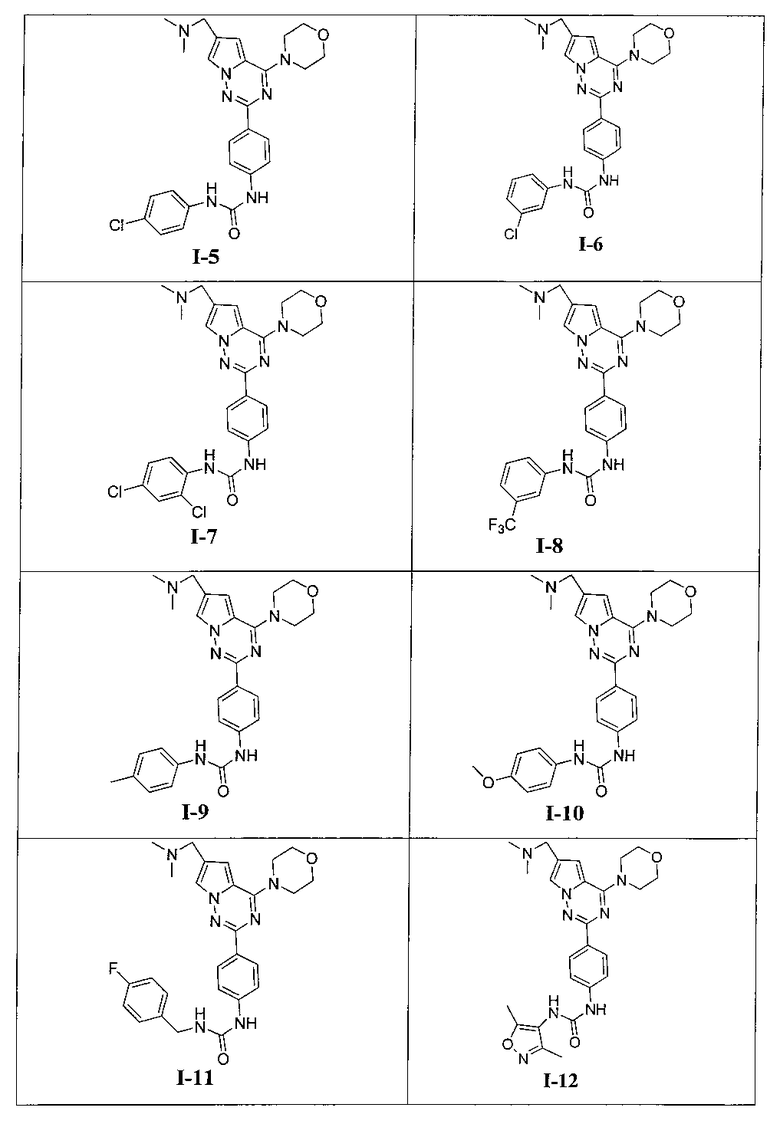
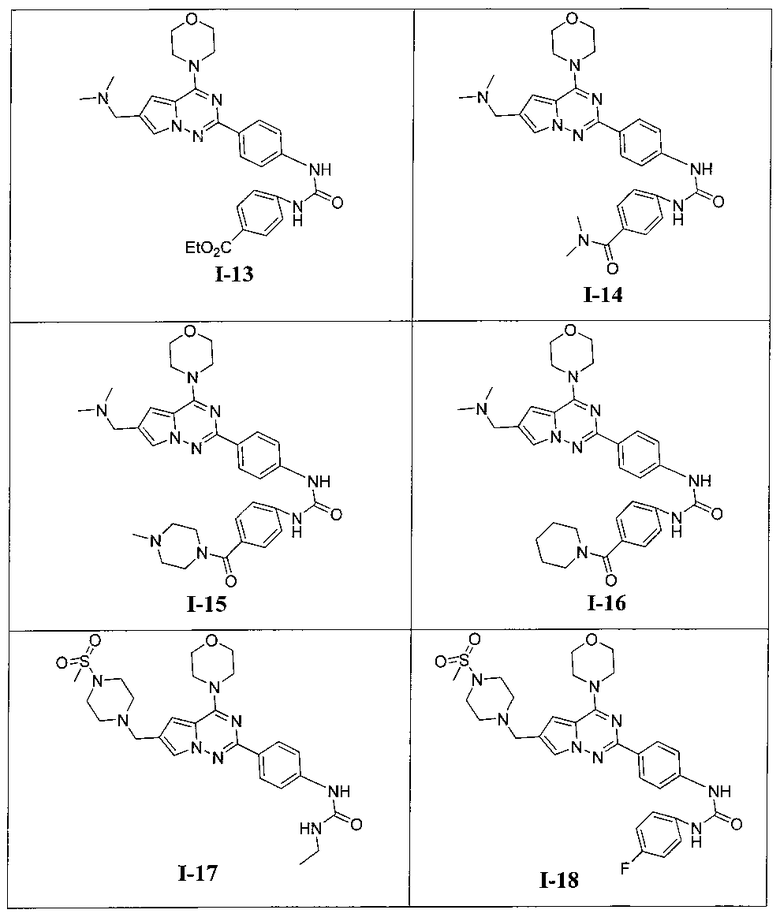
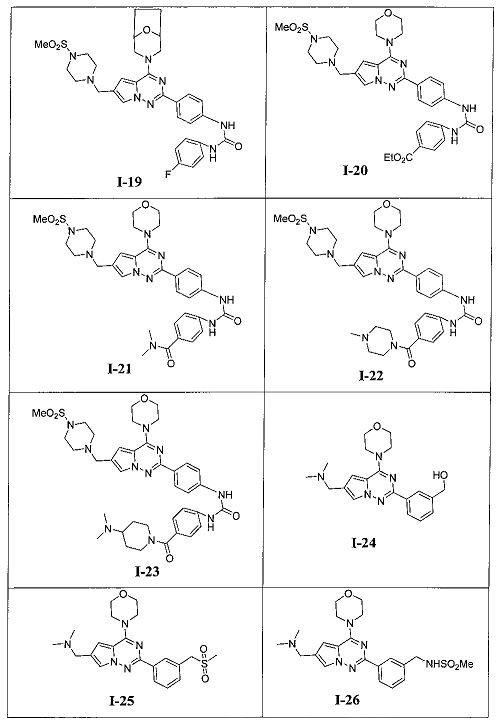
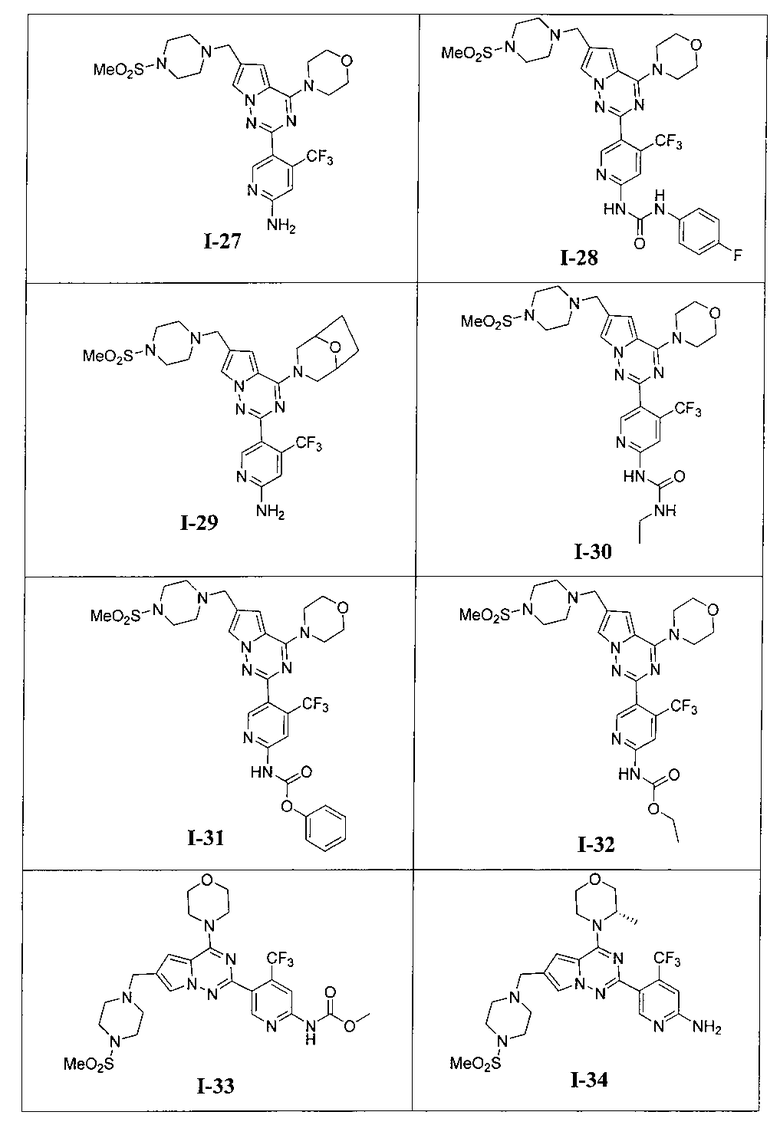
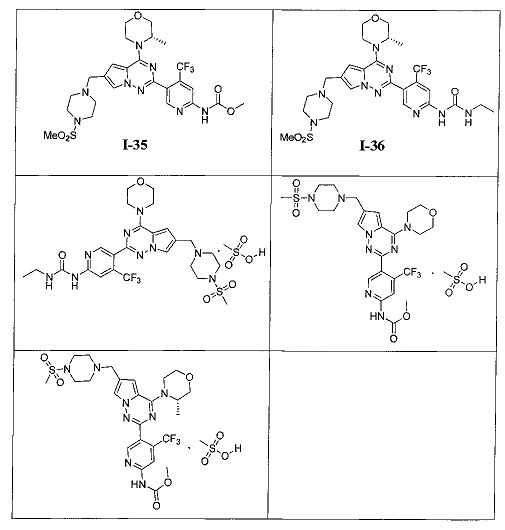
Более предпочтительно, в настоящем изобретении предложены соединения, представленные в таблице 1.
Еще одна задача настоящего изобретения заключается в том, чтобы предложить способ получения соединений, представленных общей формулой I, где в способе получения осуществляют следующие стадии:
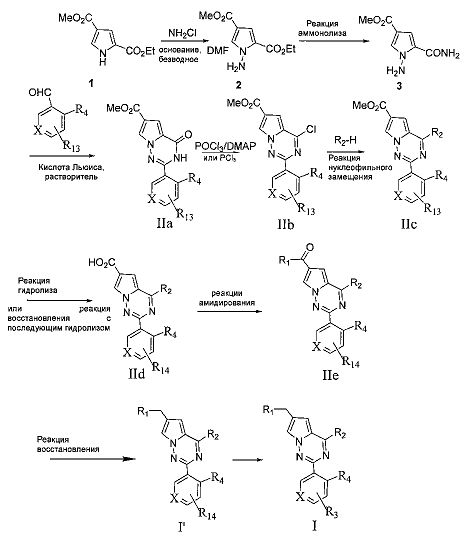
где R13 представляет собой нитро или -СН2ОАс; R14 представляет собой амино или -СН2ОН;
1. Пиррольное производное 1 и хлорамин в безводном N,N-диметилформамиде подвергают реакции N-аминирования в присутствии основания с получением соединения 2;
Основание может представлять собой гидрид натрия, карбонат калия или трет-бутоксид калия;
2. Соединение, представленное формулой 2, не подвергнутое дополнительной очистке, подвергают аммонолизу с получением соединения 3;
3. соединение 3 подвергают взаимодействию с ароматическим альдегидом под действием кислоты Льюиса с металлом с получением соединения IIa, или соединение 3 и альдегид под действием кислоты Льюиса в качестве катализатора, такого как раствор трифторида бора в диэтиловом эфире, подвергают конденсации с получением основания Шиффа, и затем осуществляют окислительную циклизацию с получением соединения IIa;
Кислота Льюиса с металлом может представлять собой реагент одновалентной или двухвалентной меди, такой как бромид одновалентной меди, хлорид одновалентной меди, моногидрат ацетата меди, бромид меди, безводный хлорид меди, дигидрат хлорида меди и т.п. Предпочтительно дигидрат хлорида меди используют ввиду его более высокого выхода и легкости последующей обработки по сравнению с другими медными реагентами. Реакционный растворитель представляет собой диметилсульфоксид или N,N-диметилформамид, N,N-диметилацетамид, и реакционная температура составляет 80-150°C;
4. Соединение IIa подвергают хлорированию с получением соединения IIb; Хлорирующий агент представляет собой оксихлорид фосфора или пентахлорид фосфора, и используемое основание представляет собой N,N-диметиланилин или 4-диметиламинопиридин (DMAP);
5. Соединение IIb и морфолин или морфолиновое производное подвергают реакции нуклеофильного замещения с получением соединения IIc;
6. Сложный эфир соединения Не подвергают реакции гидролиза; или нитросоединения IIc сначала подвергают восстановлению и затем сложноэфирную группу IIc гидролизуют;
7. Соединение IId подвергают взаимодействию с амином или замещенным/незамещенным насыщенным гетероциклом, содержащим один атом азота, с получением соединения IIe;
амин здесь представляет собой диметиламин или метилсульфонил пиперазин;
8. Соединение IIe восстанавливают при помощи восстановителя с получением соединения I′;
восстановитель здесь представляет собой боран-тетрагидрофурановый комплекс или боран-диметилсульфидный комплекс;
9. соединение I′ далее подвергают (реакции присоединения с R11NCO, этерификации или амидирования с R11OC(O)Cl, этерификации или амидирования с R11C(O)OH или R11C(O)Cl, или реакции этерификации или реакции амидирования с R12S(O)2Cl), с образованием соединений, представленных общей формулой I.
В частности, соединения, представленные общей формулой Ia-If, могут быть получены при помощи следующих стадий:
(1) Синтез соединений, представленных общей формулой IIIa-IIIc, и синтез N-(5-формил-4-(трифторметил)пиридин-2-ил)пиваламида (7):
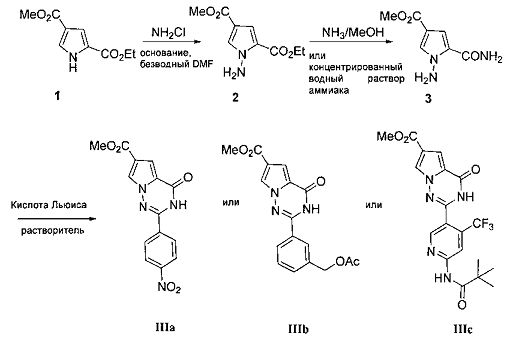
пиррольное производное 1 и хлорамин в безводном N,N-диметилформамиде подвергают реакции N-аминирования в присутствии основания с получением соединения 2, где основание может представлять собой гидрид натрия, карбонат калия или трет-бутоксид калия. Без дополнительной очистки неочищенный продукт 2 непосредственно подвергают реакции аммонолиза в запаянной пробирке путем использования насыщенного раствора аммиака в метаноле или имеющегося в продаже концентрированного водного раствора аммиака с получением соединения 3. Соединение 3 подвергают взаимодействию с соответствующим ароматическим альдегидом (например пара-нитробензальдегид, 3-формил-бензилацетат, N-(5-формил-4-(трифторметил)пиридин-2-ил)пиваламид) в присутствии подходящей кислоты Льюиса с металлом с получением соединения IIIa, IIIb или IIIc. Соединения IIIa, IIIb или IIIc также получают путем конденсации соединения 3 и различных альдегидов в присутствии кислоты Льюиса, такой как раствор трифторида бора в диэтиловом эфире, с получением основания Шиффа, и последующей окислительной циклизации. Где кислота Льюиса с металлом может представлять собой реагент одновалентной или двухвалентной меди, такой как бромид одновалентной меди, хлорид одновалентной меди, моногидрат ацетата меди, бромид меди, безводный хлорид меди, дигидрат хлорида меди и т.п. По сравнению с другим медным реагентом может быть получены более высокие выходы путем использования дигидрата хлорида меди, и облегчается последующая обработка. Реакционный растворитель представляет собой диметилсульфоксид или N,N-диметилформамид, N,N-диметилацетамид, и реакционная температура составляет 80-150°C.
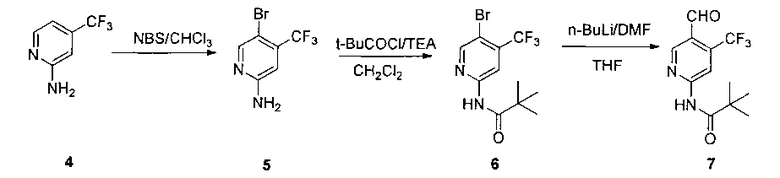
Где альдегид 7 получают при помощи трехстадийного способа 2-амино-4-трифторметил-пиридин (4) бромируют при помощи N-бромсукцинимида в хлороформе с получением соединения 5. Аминогруппу соединения 5 защищают при помощи пивалоильной группы, и затем соединение 7 получают с использованием н-бутиллития и N,N-диметилформамида в безводном тетрагидрофуране.
(2) синтез пирроло[2,1-f][1,2,4]триазиновых производных Ia и Ib, где X представляет собой СН и R3 представляет собой NHC(O)NHR11:
Соединение IIIa хлорируют при помощи оксихлорида фосфора или пентахлорида фосфора с получением продукта 8, который подвергают взаимодействию с морфолином или его аналогом при комнатной температуре в тетрагидрофуране с получением соединения 9. Соединение 9 восстанавливают при помощи 5% или 10% палладия на углероде с получением соединения 10, сложноэфирную группу которого гидролизуют в основных условиях с получением кислоты 11. Соединение 11 и амин, такой как диметиламин или метилсульфонил пиперазин, и т.п, подвергают конденсации с получением соединения 12. Затем 12 восстанавливают при помощи восстановителя с получением соединения 13, где восстановитель может представлять собой боран-тетрагидрофурановый комплекс или боран-диметилсульфидный комплекс. Продукт восстановления 13 подвергают взаимодействию с сериями изоцианатов в безводном дихлорметане при комнатной температуре, или с соответствующим ацилазидом в диоксане при кипячении с обратным холодильником с получением Ia. Когда R11 представляет собой 4-этоксикарбонил-фенил, тогда Ia гидролизуют с получением соединения 14. Соединение 14 и диметиламин, N-метилпиперазин или 4-диметиламинопиперидин подвергают конденсации с получением Ib.
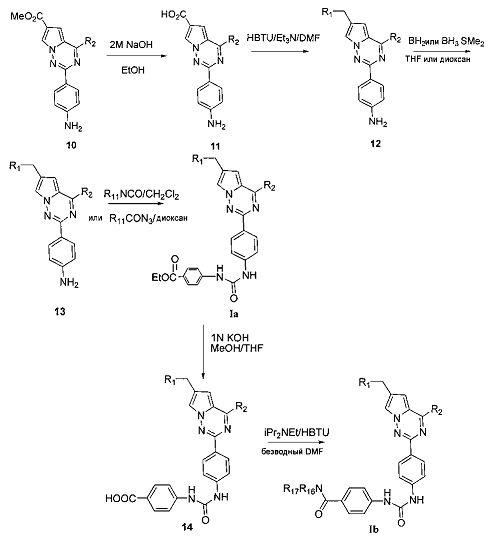
(3) синтез пирроло[2,1-f][1,2,4]триазиновых производных Ic-e, где X представляет собой СН и R3 представляет собой СН2ОН, CH2S(O)2R12 или CH2NHS(O)2R12:
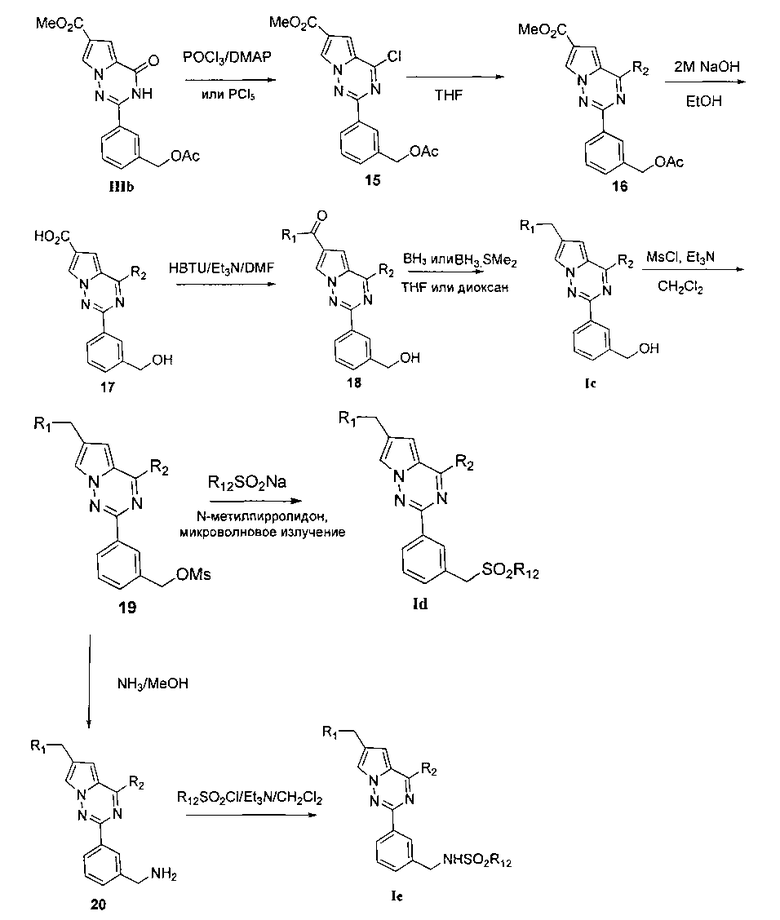
Хлорированное соединение IIIb подвергают взаимодействию с морфолином или его аналогом при комнатной температуре в тетрагидрофуране с получением соединения 16. Сложноэфирную группу соединения 16 гидролизуют с получением соединения 17. 17 и амин, такой как диметиламин или метилсульфонил пиперазин и т.п, подвергают конденсации с получением соединения 18, и затем 18 восстанавливают при помощи боран-тетрагидрофуранового раствора или боран-диметилсульфидного раствора с получением Ic. При использовании метилсульфонил хлорида гидроксил в Ic превращают для хорошего высвобождения метансульфонатной группы, и превращают в соединение 19. Соединение 19 подвергают взаимодействию с алкилсульфонатом натрия при воздействии микроволнового излучения в N-метилпирролидоне при 120°C в течение 30 минут с получением соединения Id. Кроме того, соединение 19 подвергают аммонолизу с получением соединения 20, которое затем подвергают взаимодействию с алкилсульфонилхлоридом с получением соединения Ie.
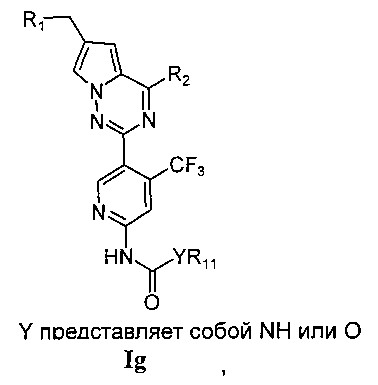
(4) синтез пирроло[2,1-f][1,2,4]триазиновых производных If-Ig, где X представляет собой N, R3 представляет собой NH2, NHC(O)NHR11 или NHC(O)OR11, и R4 представляет собой CF3:
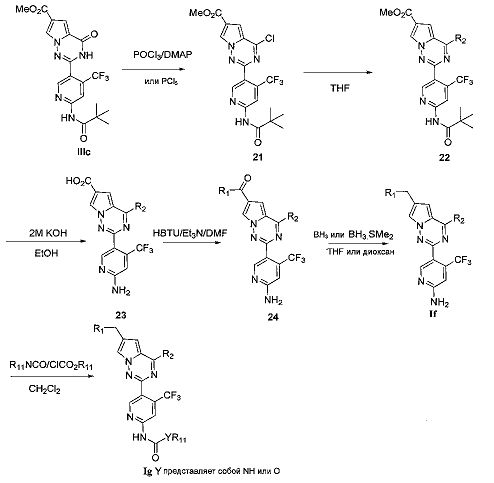
Хлорированное соединение IIIc подвергают взаимодействию с морфолином или его аналогом в тетрагидрофуране при комнатной температуре с получением соединения 22. Сложноэфирную группу соединения 22 гидролизуют с получением продукта 23. 23 и амин или замещенный или незамещенный насыщенный гетероцикл подвергают конденсации с получением соединения 24, которое затем восстанавливают с использованием боран-тетрагидрофуранового раствора или боран-диметилсульфидного раствора с получением If. Соединение If подвергают взаимодействию с изоцианатом или хлороформиатом в безводном дихлорметане при комнатной температуре с получением Ig.
Соединения по настоящему изобретению могут эффективно ингибировать активность киназы PI3K. Таким образом, эти соединения могут быть использованы в лечении заболеваний, ассоциированных с путем PI3K, в частности для лечения опухолей. Таким образом, еще одна задача настоящего изобретения заключается в том, чтобы предложить применение соединений общей формулы I или их фармацевтически приемлемых солей для приготовления лекарственных средств для фосфатидилинозитол 3-киназы и белка-мишени млекопитающих рапамицинового ингибитора, т.е. для приготовления лекарственных средств для лечения заболеваний, связанных с фосфатидилинозитол 3-киназой. Заболевания, связанные с фосфатидилинозитол 3-киназой, включают опухоли. Опухоли включают человеческую рабдомиосаркому, немелкоклеточный рак легкого, человеческую глиому, рак предстательной железы, рак яичников, рак печени, рак толстой кишки, рак молочной железы и т.п.
Кроме того, в настоящем изобретении предложена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения общей формулы I, и фармацевтическая композиция также может включать другие ингредиенты, такие как носитель, эксципиент и т.п.
В настоящем изобретении предложен способ лечения заболеваний, связанных с фосфатидилинозитол 3-киназой, при котором осуществляют введение терапевтически эффективного количества соединения, представленного общей формулой I.
Краткое описание графических материалов
Фиг. 1 демонстрирует действия I-33 на сигнальный путь PI3K в клетках человеческой рабдомиосаркомы Rh30 и клетках человеческой глиомы U87MG.
Фиг. 2 демонстрирует ингибирующее рост действие I-30 и I-33 на подкожно трансплантированные опухоли человеческой глиомы U87MG бестимусным мышам.
Подробное описание изобретения
Настоящее изобретение дополнительно проиллюстрировано при помощи следующих примеров, но эти примеры никоим образом не ограничивают изобретение. Во всех примерах 1H ЯМР (ядерный магнитный резонанс) регистрировали при помощи спектрометров ядерного магнитного резонанса Brucher АМ-400 или GEMINI-300, где химический сдвиг представлен посредством δ (млн-1). Масс-спектр регистрировали при помощи масс-спектрометра МАТ-95. Для разделения использовали силикагели 200-300 меш.
Примеры
1. Получение метил 1-амино-5-карбамоил-1Н-пиррол-3-карбоксилата (3)
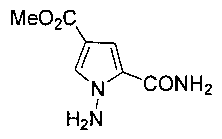
Смесь 9 г хлорида аммония и 330 мл диэтилового эфира охлаждали до -20°C и при помощи пипетки добавляли 15 мл концентрированного водного раствора аммиака. 216 мл 5% (процент по массе) раствора гипохлорита натрия добавляли по каплям через капельную воронку при постоянном давлении. Смесь перемешивали при -10°C в течение 30 минут. После разделения органический слой промывали насыщенным рассолом (хлорамин нестабилен и рассол должен быть предварительно охлажден). Безводный хлорид кальция добавляли в органический слой, и смесь сушили при -40°C в течение 1 часа до применения.
Соединение пиррол-1,3-дикарбоксилат 1 (5 г, 25,4 ммоль, полученный в соответствии с Kamijo, S., Kanazawa, С, and Yamamoto Y.J. AM. CHEM. SOC. 2005, 127, 9260-9266, где исходные вещества метилпропиолят и этилизоцианоацетат приобретены в Darui chemical Co., Ltd) растворяли в 25 мл безводного N,N-диметилформамида и охлаждали в ледяной бане до 0°С. Гидрид натрия (60%, диспенсированный в минеральном масле, 1,22 г, 30,5 ммоль) добавляли порциями. Смесь перемешивали в течение 1 часа при комнатной температуре. Затем одной порцией добавляли 300 мл раствора хлорамина в диэтиловом эфире, приготовленного заранее, и перемешивали в течение ночи при комнатной температуре в атмосфере азота. Реакционную смесь гасили насыщенным раствором тиосульфата натрия и разбавляли водой. Слой диэтилового эфира отделяли, и водный слой однократно экстрагировали этилацетатом. Органические слои комбинировали и трижды промывали водой, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением 6,2 г неочищенного продукта, который непосредственно подвергали аммонолизу без очистки. К каждым 3 г неочищенного продукта добавляли 80 мл насыщенного раствора аммиака в метаноле, и реакцию осуществляли при 80°C в запаянной пробирке в течение 2 суток. Реакционную смесь концентрировали для осаждения твердых веществ, затем оставляли осаждаться в течение приблизительно 1 часа, и фильтровали с получением 3 г продукта в виде белого твердого вещества. Выход после двух стадий составил 64,6%. т.пл. (точка плавления) 222-224°C.
1H ЯМР (300 МГц, DMSO-d6): δ 7,95 (br s, 1Н), 7,38 (s, 1Н), 7,33 (br s, 1Н), 7,15 (s, 1Н), 6,87 (s, 2Н), 3,70 (s, 3Н). MS (EI(ионизация распылением электронов)) m/z (%): 183 (М+, 100).
2. Получение 2-амино-4-трифторметил-5-бромпиридина (5)
2-Амино-4-трифторметилпиридин (5 г, 30,8 ммоль, Langfang Beixin Chemical Co., Hebei) растворяли в 100 мл хлороформа и порциями добавляли N-бромсукцинимид (5,92 г, 33,3 ммоль). Смесь перемешивали в темноте или вдали от света при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали и очищали при помощи колоночной хроматографии с градиентной элюцией (петролейный эфир: этилацетат = 10:1 и дихлорметан) с получением 4,33 г красного твердого вещества. Выход: 58,2%. LC-MS (жидкостная хроматография/масс-спектрометрия): 240 (М+1), 242 (М+2+1).
3. Получение N-(5-бром-4-(трифторметил)пиридин-2-ил)пиваламида (6) 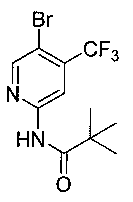
В ледяной бане 29,8 г пивалоилхлорида (226 ммоль) добавляли по каплям к раствору соединения 5 (50,0 г, 207 ммоль) и триэтиламина (37,9 мл) в дихлорметане (300,0 мл) в течение одного часа и затем перемешивали в течение 2 часов до исчезновения исходных веществ. В реакционный раствор добавляли 150 мл воды и перемешивали при комнатной температуре в течение 10 минут. Органический слой отделяли, сушили над безводным сульфатом натрия, концентрировали и разделяли на короткой колонке с этилацетатом с получением белого твердого вещества (57,7 г, 85,6%).
т.пл. 126-128°C. 1Н ЯМР (300 МГц, CDCl3): δ 8,67 (s, 1Н), 8,50 (s, 1Н), 8,14 (brs, 1Н), 1,33 (s, 9Н).
4. Получение N-(5-формил-4-(трифторметил)пиридин-2-ил)пиваламида (7)
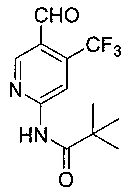
Соединение 6 (15,0 г, 46,2 ммоль) растворяли в 350 мл безводного тетрагидрофурана и охлаждали до -78°C в атмосфере азота. 45 мл 2,5 М раствора н-бутиллития в тетрагидрофуране медленно добавляли в реакционный раствор в течение одного часа. Реакционный раствор перемешивали при -78°C в течение 1 часа, и затем 15 мл безводного N,N-диметилформамида медленно добавляли по каплям и перемешивали в течение еще 2,5 ч при -78°C. К реакционному раствору добавляли 120 мл 1М разбавленной соляной кислоты для гашения реакции. Реакционную смесь экстрагировали этилацетатом (200 мл ×3). Органические слои комбинировали и промывали соответственно водой (200 мл ×3), насыщенным рассолом (200 мл), затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали и очищали при помощи колоночной хроматографии (петролейный эфир: дихлорметан: этилацетат = 60: 10: 1) с получением 7,1 г белого твердого вещества (56,1%). т.пл. 96-98°C. 1H ЯМР (300 МГц, CDCl3): δ 10,32 (brs, 1 Н), 9,01 (s, 1 Н), 8,73 (s, 1 Н), 8,40 (s, 1 Н), 1,38 (s, 9 Н).
5. Общий способ получения соединений IIIa-IIIc
5 мл диметилсульфоксида добавляли к смеси соединения 3 (55 мг, 0,3 ммоль), соответствующего альдегида (0,3 ммоль) и дигидрата хлорида меди (51 мг, 0,3 ммоль), и реакцию осуществляли при 80-150°C. После завершения реакции реакционную смесь охлаждали и выливали в воду, осажденные твердые вещества фильтровали. Если неочищенный продукт обладал плохой растворимостью, то его промывали метанолом. Если он обладал хорошей растворимостью, то его очищали при помощи колоночной хроматографии (дихлорметан: метанол = 50:1).
Получение метил 2-лара-нитрофенил-4-оксо-3,4-дигидропирроло[2,1 -f][1,2,4]триазин-6-формиата (IIIa)
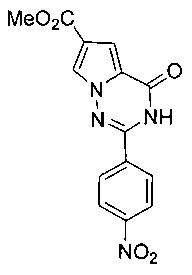
В соответствии с общим способом получения, описанным в примере 5 выше, пара-нитробензальдегид подвергают взаимодействию с соединением 3 с получением соединения IIIa в виде светло-желтого твердого вещества с выходом 72,0%. т.пл. больше 300°C. 1H ЯМР (300 МГц, DMSO (диметилсульфоксид)-d6): δ 12,52 (s, 1Н), 8,38 (d, J=8,5 Гц, 2Н), 8,22 (s, 1Н), 8,21 (d, J=8,5 Гц, 2Н), 7,26 (s, 1Н), 3,80 (s, 3Н). LRMS (масс-спектр низкого разрешения) (EI) m/z (%): 314 (М+, 85), 283 (100). HRMS (масс-спектр высокого разрешения) вычисл. C14H10N4O5: 314,0651; найдено: 314,0659.
Получение метил 2-(3-(ацетоксиметил)фенил)-4-оксо-3,4-дигидропирроло[2,1-f][1,2,4] триазин-6-карбоксилата (IIIb)
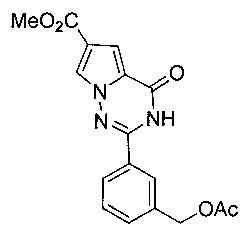
В соответствии с общими способами получения, описанным в примере 5 выше, 3-формилбензилацетат подвергают взаимодействию с соединением 3 с получением соединения IIIb в виде не совсем белого твердого вещества с выходом 39,0%. т.пл. 202-203°C. 1Н ЯМР (300 МГц, DMSO-d6): δ 12,29 (s, 1Н), 8,19 (d, J=1,7 Гц, 1Н), 7,96 (s, 1Н), 7,92 (dt, J=1,7, 7,2 Гц, 1Н), 7,58-7,53 (m, 2Н), 7,24 (d, J=1,7 Гц, 1Н), 5,16 (s, 2Н), 3,81 (s, 3Н), 2,10 (s, 3Н). LC-MS: 342 (М+1).
Получение метил 4-оксо-2-(6-пиваламидо-4-(трифторметил)-пиридин-3-ил)-3,4-дигидропирроло[2,1-f][1,2,4]триазин-6-карбоксилата (IIIc)
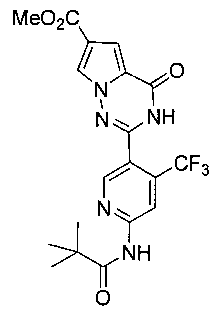
В соответствии с общим способом получения, описанным в примере 5 выше, соединение 7 подвергают взаимодействию с соединением 3 с получением соединения IIIc в виде светло-желтого твердого вещества с выходом 25,9%. т.пл. 244-245°C. 1Н ЯМР (300 МГц, DMSO-d6): δ 12,47 (s, 1Н), 10,68 (s, 1Н), 8,86 (s, 1Н), 8,57 (s, 1Н), 8,20 (d, J=1,7 Гц, 1Н), 7,29 (d, J=1,7 Гц, 1Н), 3,81 (s, 3Н), 1,28 (s, 9Н). LC-MS: 438 (М+1).
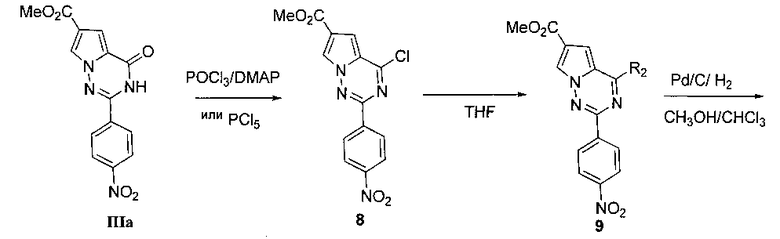
6. Получение метил 2-лара-нитрофенил-4-хлорпирроло[2,1-f][1,2,4]триазин-6-карбоксилата (8)
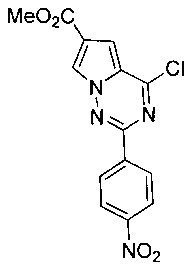
20 мл оксихлорида фосфора добавляли к смеси соединения IIIa (4,74 г, 15,1 ммоль) и 4-диметиламинопиридина (4,34 г, 35,6 ммоль) и кипятили с обратным холодильником в течение 5 часов. После охлаждения реакционной смеси порцию оксихлорид фосфора отгоняли при пониженном давлении. Остаток выливали в измельченный лед, фильтровали и сушили с получением желтого твердого вещества (4,6 г, 91,8%). т.пл. 218-223°C. 1H ЯМР (300 МГц, CDCl3): δ 8,54 (d, J=8,9 Гц, 2Н), 8,36 (s, 1Н), 8,34 (d, J=8,9 Гц, 2Н), 7,46 (d, J=1,3 Гц, 1Н), 3,96 (s, 3Н). MS (EI) m/z (%): 332 (М+ 100), 334 (М+2, 33).
7. Получение соединения 9
Метил 2-(пара-нитрофенил)-4-(морфолинил)-пирроло[2,1-f][1,2,4]триазин-6-карбоксилат (9а)
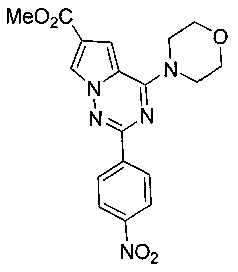
Соединение 8 (4,6 г, 13,8 ммоль) суспендировали в 150 мл тетрагидрофурана, по каплям добавляли 3,6 мл морфолина и подвергали взаимодействию в течение 5 часов при комнатной температуре. 3,9 г твердого вещества получали путем фильтрования, и фильтрат разделяли при помощи колоночной хроматографии с дихлорметаном с получением 1,1 г желтого соединения 9а (94,3%). т.пл. 296-300°C. 1H ЯМР (300 МГц, CDCl3): δ 8,45 (d, J=8,7 Гц, 2Н), 8,29 (d, J=8,7 Гц, 2Н), 8,14 (d, J=1,3 Гц, 1Н), 7,23 (d, J=1,3 Гц, 1Н), 4,17 (t, J=4,8 Гц, 4Н), 3,91 (t, J=4,8 Гц, 7Н). LC-MS: 384 (М+1).
Метил 4-(8-окса-3-азабицикло[3,2,1]октан-3-ил)-2-(пара-нитрофенил)пирроло[2,1-f][1,2,4]триазин-6-карбоксилат (9b)
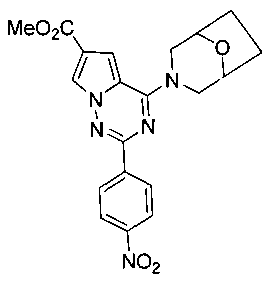
Соединение 8 (100 мг, 0,3 ммоль) суспендировали в 15 мл тетрагидрофурана, и добавляли 8-окса-3-азабицикло[3,2,1]октанхлоргидрат (54 мг, 0,36 ммоль) и одну каплю триэтиламина и подвергали взаимодействию в течение 3-4 ч при комнатной температуре. Растворитель удаляли при пониженном давлении. Остаток промывали водой, сушили и очищали при помощи колоночной хроматографии с дихлорметаном с получением 109 мг желтого твердого вещества 9b (88,6%). т.пл. 278-280°C. 1H ЯМР (300 МГц, CDCl3): δ 8,45 (d, J=9,0 Гц, 2Н), 8,29 (d, J=9,0 Гц, 2Н), 8,13 (d, J=1,3 Гц, 1Н), 7,21 (d, J=1,3 Гц, 1Н), 4,60 (br s, 4Н), 3,91 (s, 3Н), 3,63 (br, s, 2H), 2,08-2,04 (m, 2H), 1,91-1,84 (m, 2H). LC-MS: 410 (М+1).
8. Общий способ получения соединения 10
Смесь растворителей метанола и хлороформа (500 мл, 1: 1) и 10 масс. % палладия на углероде исходного вещества (10% Pd-углерод) добавляли к соединению 9 (13 ммоль) и восстанавливали в течение 24 часов в атмосфере водорода при комнатной температуре. Палладий-углерод отфильтровывали при помощи целита, и фильтрат концентрировали при пониженном давлении для количественного получения Соединения 10.
Метил 2-(пара-аминофенил)-4-(морфолинил)пирроло[2,1-f][1,2,4]триазин-6-карбоксилат (10а)
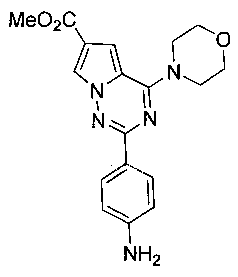
10а получали из 9а в соответствии с общим способом получения соединения 10. Белое твердое вещество, т.пл. 238-240°C. 1H ЯМР (300 МГц, DMSO-d6): δ 8,13 (d, J=1,6 Гц, 1Н), 7,93 (d, J=8,5 Гц, 2Н), 7,31 (d, J=1,6 Гц, 1Н), 6,62 (d, J=8,5 Гц, 2Н), 5,70 (br, s, 2Н), 4,04 (t, J=4,5 Гц, 4Н), 3,80 (s, 3Н), 3,77 (t, J=4,5 Гц, 4Н). MS (EI) m/e (отношение массы иона m к его заряду e) (%): 353 (М+, 100).
Метил 2-(пара-аминофенил)-4-(8-окса-3-азабицикло[3,2,1]октан-3-ил)пирроло[2,1-f][1,2,4] триазин-6-карбоксилат (10b)
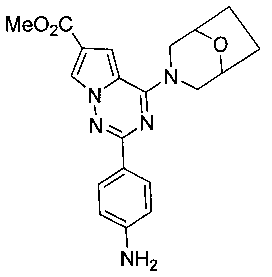
10b получали из 9b в соответствии с общим способом получения соединения 10. Желтое твердое вещество, т.пл. 254-256°C. 1H ЯМР (300 МГц, DMSO-d6): δ 8,21 (s, 1Н), 8,19 (d, J=8,7 Гц, 2Н), 7,34 (s, 1Н), 7,22 (d, J=8,7 Гц, 2Н), 4,53 (br s, 4Н), 3,81 (s, 3Н), 3,49 (br, s, 2Н), 1,89-1,85 (m, 2Н), 1,78-1,75 (m, 2Н). LC-MS: 380 (М+1).
9. Способ получения соединения 11
2-(пара-аминофенил)-4-(морфолинил)пирроло[2,1-f][1,2,4]триазин-6-карбоновая кислота (11а)
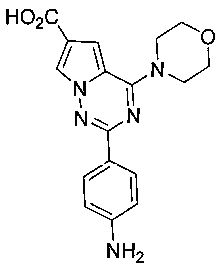
Соединение 10а (14 ммоль) суспендировали в 150 мл этанола и добавляли 30 мл 2М водного раствора гидроксида натрия. Реакционную смесь кипятили с обратным холодильником с получением прозрачного раствора, и реакция по существу завершалась. Добавляли 2 мл уксусной кислоты. Большую часть растворителя отгоняли при пониженном давлении, и осадки фильтровали с получением соединения 11а (3,25 г, 68,5%). т.пл. >300°C. 1H ЯМР (300 МГц, DMSO-d6): δ 7,91 (d, J=8,8 Гц, 2Н), 7,71 (d, J=1,2 Гц, 1Н), 6,99 (d, J=1,2 Гц, 1Н), 6,59 (d, J=8,8 Гц, 2Н), 5,49 (s, 2Н), 4,02 (t, J=4,4 Гц, 4Н), 3,77 (t, J=4,4 Гц, 4Н). MS (EI) m/e (%): 339 (М+, 100).
2-(пара-аминофенил)-4-(8-окса-3-азабицикло[3,2,1]октан-3-ил)пирроло[2,1-f][1,2,4]триазин-6-карбоновая кислота (11b)
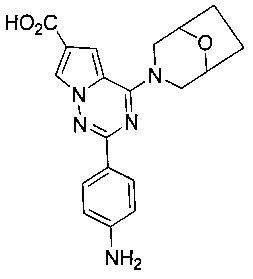
В соответствии с тем же самым способом, как способ получения соединения 11а, 360 мг 10b (0,95 ммоль) в качестве исходного вещества гидролизовали с получением соединения 11b (270 мг, 77,9%). т.пл. 278-280°C. 1H ЯМР (300 МГц, DMSO-d6): δ 12,46 (s, 1Н), 8,04 (d, J=1,7 Гц, 1Н), 7,92 (d, J=8,8 Гц, 2Н), 7,22 (d, J=1,7 Гц, 1Н), 6,60 (d, J=8,8 Гц, 2Н), 5,55 (br s, 2Н), 4,51 (br s, 4Н), 3,48 (br, s, 1H), 3,44(br, s, 1H), 1,88-1,85 (m, 2H), 1,79-1,75 (m, 2H). LC-MS: 366 (M+1).
10. Общий способ получения соединения 12
2-(пара-аминофенил)-N,N-диметил-4-морфолинопирроло[2,1-f][1,2,4]триазин-6-карбоксамид (12а)
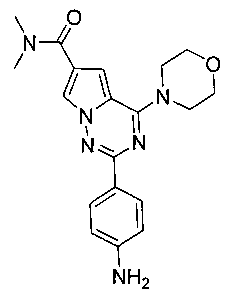
Диметиламин хлоргидрат (686 мг, 8,4 ммоль) добавляли к 30 мл безводного N,N-диметилформамида, и добавляли карбонат калия (3,48 г, 25,2 ммоль) и перемешивали в течение 30 минут при комнатной температуре. Затем добавляли соединение 11а (4,2 ммоль), HBTU (бензотриазол-N,N,N′,N′-тетраметилуроний гексафторфосфат, 4,77 г, 12,6 ммоль), и триэтиламин (2,9 мл, 21 ммоль) и подвергали взаимодействию в течение ночи при комнатной температуре в атмосфере азота. Реакционную смесь выливали в воду и фильтровали. Фильтрат однократно экстрагировали этилацетатом, сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха. Остаток комбинировали с остатком на фильтре. Неочищенный продукт очищали при помощи колоночной хроматографии (дихлорметан:метанол = 100:1) с получением белого соединения 12а (922 мг, 60,0%). т.пл. 238-239°C. 1H ЯМР (300 МГц, CDCl3): δ 8,08 (d, J=8,6 Гц, 2Н), 7,82 (d, J=1,4 Гц, 1Н), 7,02 (d, J=1,4 Гц, 1Н), 6,73 (d, J=8,6 Гц, 2Н), 4,11 (t, J=4,4 Гц, 4H), 3,86 (t, J=4,4 Гц, 4H), 3,20 (br, s, 6H). MS (EI) m/e (%): 366 (M+, 100).
(2-(пара-аминофенил)-4-морфолинопирроло[2,1-f][1,2,4]триазин-6-ил)(4-(метилсульфонил)пиперазин-1-ил)метанон (12b)
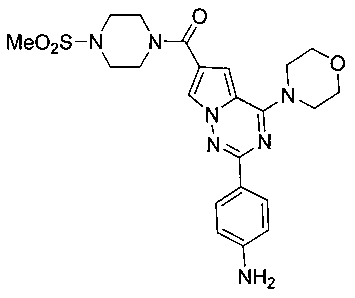
Белое твердое вещество 12b (540 мг, 31,4%) получали в соответствии с тем же самым способом как способ получения соединения 12а, где 1,2 г соединения 11а (3,54 ммоль) использовали в качестве исходного вещества и метилсульфонилпиперазин трифторметансульфонат (1,85 г, 7,1 ммоль) использовали вместо диметиламинхлоргидрата. т.пл. 185-186°C. 1H ЯМР (300 МГц, CDCl3): δ 8,07 (d, J=8,6 Гц, 2Н), 7,75 (d, J=1,5 Гц, 1Н), 6,93 (d, J=1,5 Гц, 1Н), 6,71 (d, J=8,6 Гц, 2Н), 4,09 (t, J=4,8 Гц, 4Н), 3,90 (t, J=4,8 Гц, 4Н), 3,86 (t, J=4,8 Гц, 4Н), 3,27 (t, J=4,8 Гц, 4Н), 2,80 (s, 3Н). LC-MS: 508 (М+23).
(2-(пара-аминофенил)-4-(8-окса-3-азабицикло[3,2,1]октан-3-ил)пирроло[2,1-f][1,2,4]триазин-6-ил)(4-(метилсульфонил)пиперазин-1-ил)метанон (12c)
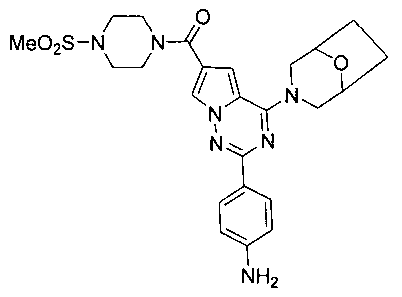
183 мг соединения 11b (0,5 ммоль) использовали вместо соединения 11а, и соединение 12с (120 мг, 46,8%) получали тем же самым образом как получение соединения 12b. т.пл. >300°C. 1H ЯМР (300 МГц, DMSO-d6): δ 7,96 (s, 1Н), 7,91(d, J=8,5 Гц, 2Н), 7,06 (s, 1Н), 6,61 (d, J=8,5 Гц, 2Н), 5,53 (s, 2Н), 4,51 (br, s, 4Н), 3,75 (t, J=4,5 Гц, 4H), 3,48 (br s, 1H), 3,43 (br s, 1H), 3,18 (t, J=4,5 Гц, 4H), 2,91 (s, 3H), 1,90-1,75 (m, 4H). LC-MS: 511 (M+), 512 (M+1).
11. Общий способ получения соединения 13
2-(пара-аминофенил)-6-(диметиламинометил)-4-орфолинопирроло[2,1-f][1,2,4]триазин (13а)
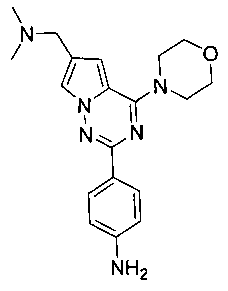
1 г соединения 12а (2,7 ммоль) и 50 мл тетрагидрофурана или диоксана добавляли в двухгорлую колбу объемом 150 мл и кипятили с обратным холодильником в атмосфере азота. 2М раствор боран-диметилсульфида (10,8 ммоль) медленно добавляли по каплям и кипятили с обратным холодильником в течение 2 часов. Реакционную смесь гасили метанолом и очищали при помощи колоночной хроматографии с дихлорметаном с получением белого твердого вещества 13а (930 мг, 96,7%). т.пл. 205°C (декомпозиция). 1H ЯМР (300 МГц, DMSO-d6): δ 7,92 (d, J=8,5 Гц, 2Н), 7,86 (s, 1Н), 7,05 (s, 1Н), 6,60 (d, J=8,5 Гц, 2Н), 5,48 (br s, 2Н), 4,02 (t, J=4,5 Гц, 4Н), 3,92 (s, 2Н), 3,77 (t, J=4,5 Гц, 4Н), 2,42 (s, 6Н). MS (EI) m/e (%): 352 (М+, 24).
2-(пара-аминофенил)-6-[((4-метилсульфонил)пиперазин-1-ил)метил]-4-морфолинопирроло[2,1-f][1,2,4]триазин (13b)
Соединение 12b (540 мг, 1,1 ммоль) использовали вместо соединения 12а в качестве исходного вещества, и соединение 13b (288 мг, 55,0%, белое твердое вещество) получали в соответствии с тем же самым способом получения как способ получения соединения 13а. т.пл. 199-200°C. 1H ЯМР (300 МГц, DMSO-d6): δ 7,92 (d, J=8,6 Гц, 2Н), 7,85 (d, J=1,4 Гц, 1Н), 7,03 (d, J-1,4 Гц, 1Н), 6,60 (d, J=8,6 Гц, 2Н), 5,47 (s, 2Н), 4,08 (s, 2Н), 4,03 (t, J=4,6 Гц, 4Н), 3,78 (t, J=4,6 Гц, 4Н), 3,49-3,35 (m, 4Н), 2,95 (s, 3Н), 2,89 (t, J=5,7 Гц, 4Н). MS (EI) m/e (%): 471 (М+, 12).
4-(4-(8-окса-3-азабицикло[3,2,1]октан-3-ил)-6-((4-(метилсульфонил) пиперазин-1-ил)метил)пирроло[2,1-f][1,2,4]триазин-2-ил)анилин (13с)
Соединение 12с (100 мг, 0,195 ммоль) использовали вместо соединения 12а в виде неочищенного вещества, и соединение 13c получали в соответствии с тем же самым способом получения как способ получения соединения 13а в виде светло-желтого твердого вещества (39 мг, 40,1%). 1H ЯМР (300 МГц, DMSO-d6): δ 7,89(d, J=8,4 Гц, 2Н), 7,61 (s, 1Н), 6,78 (s, 1Н), 6,59 (d, J=8,4 Гц, 2Н), 5,46 (br s, 2Н), 4,50 (br, s, 4H), 3,56 (s, 2H), 3,43 (br s, 1H), 3,39 (br s, 1H), 3,30 (br s, 4H), 3,11 (br s, 4H), 2,86 (s, 3H), 1,88-1,85 (m, 2H), 1,78-1,75 (m, 2H). MS (EI) m/e (%): 497 (M+, 12).
12. Общий способ получения соединения I (1-13, 17-20)
1-этил-3-[4-(6-(диметиламинометил)-4-морфолинопирроло[2,1-f][1,2,4]триазин-2-ил)фенил]мочевина (I-1)
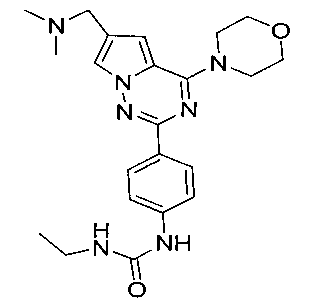
Соединение 13а (0,15 ммоль) растворяли в 10 мл безводного дихлорметана, и добавляли 3 эквив этилизоцианата и перемешивали при комнатной температуре в течение ночи. Желаемое соединение получали путем фильтрования.
Белое твердое вещество (22 мг, 34,6%). т.пл. 222-224°C. 1H ЯМР (300 МГц, DMSO-d6): δ 8,65 (s, 1Н), 8,09 (d, J=8,8 Гц, 2Н), 7,93 (s, 1Н), 7,48 (d, J=8,8 Гц, 2Н), 7,11 (s, 1Н), 6,16 (t, J=5,8 Гц, 1Н), 4,05 (t, J=4,5 Гц, 4Н), 3,94 (s, 2Н), 3,78 (t, J=4,5 Гц, 4Н), 3,12 (квинт., J=5,8, 7,1 Гц, 2Н), 2,43 (s, 6Н), 1,06 (t, J=7,1 Гц, 3Н). ESI-MS (масс-спектрометрия с ионизацией распылением электронов): 424 (М+1).
1-пропил-3-[4-(6-(диметиламинометил)-4-морфолинопирроло[2,1-f][1,2,4]триазин-2-ил)фенил]мочевина (I-2)
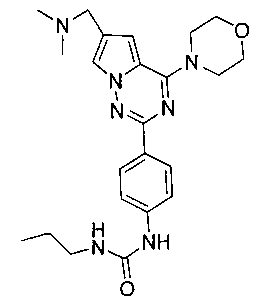
Способ получения был идентичен способу получения I-1 за исключением того, что этилизоцианат заменяли на пропилизоцианат. Белое твердое вещество (23 мг, 35,1%). т.пл. 224-225°C. 1H ЯМР (300 МГц, DMSO-d6): δ 8,64 (s, 1Н), 8,09 (d, J=8,9 Гц, 2Н), 7,93 (s, 1Н), 7,48 (d, J=8,9 Гц, 2Н), 7,11 (s, 1Н), 6,20 (t, J=5,8 Гц, 1Н), 4,05 (t, J=5,1 Гц, 4Н), 3,94 (s, 2Н), 3,78 (t, J=5,1 Гц, 4Н), 3,05 (q, J=5,8, 7,2 Гц, 2Н), 2,43 (s, 6Н), 1,44 (секстет, J=7,2 Гц, 2Н), 0,88 (t, J=7,2 Гц, 3Н). ESI-MS: 438 (М+1).
1-трет-6утил-3-[4-(6-(диметиламинометил)-4-морфолинопирроло[2,1-f][1,2,4]триазин-2-ил)фенил]мочевина (I-3)
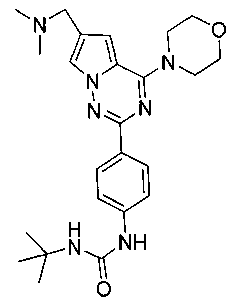
Способ получения был идентичен способу получения I-1 за исключением того, что этилизоцианат заменяли на трет-бутил изоцианат. Белое твердое вещество (11 мг, 16,3%). т.пл. 220-224°C. 1H ЯМР (300 МГц, DMSO-d6): δ 8,49 (s, 1Н), 8,08 (d, J=8,8 Гц, 2Н), 7,93 (d, J=1,0 Гц, 1Н), 7,44 (d, J=3,8 Гц, 2Н), 7,11 (d, J=1,0 Гц, 1Н), 6,07 (s, 1Н), 4,05 (t, J=4,4 Гц, 4Н), 3,94 (s, 2Н), 3,78 (t, J=4,4 Гц, 4Н), 2,42 (s, 6Н), 1,30 (s, 9Н). ESI-MS: 452 (М+1).
1-[4-(6-диметиламинометил-4-морфолинопирроло[2,1-f][1,2,4]триазин-2-ил)фенил]-3-(пара-фторфенил)мочевина (I-4)
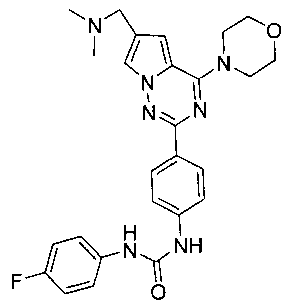
Способ получения был идентичен способу получения I-1 за исключением того, что этилизоцианат заменяли на пара-фторфенилизоцианат. Белое твердое вещество (30 мг, 40,9%). т.пл. 217-219°C. 1H ЯМР (300 МГц, DMSO-d6): δ 8,90 (s, 1Н), 8,75 (s, 1Н), 8,15 (d, J=8,7 Гц, 2Н), 7,95 (s, 1Н), 7,55 (d, J=8,7 Гц, 2Н), 7,47 (dd, J=4,6, 8,8 Гц, 2Н), 7,13 (t, J=8,8 Гц, 2Н), 7,13 (s, 1Н), 4,06 (t, J=4,6 Гц, 4Н), 3,94 (s, 2Н), 3,79 (t, J=4,6 Гц, 4Н), 2,43 (s, 6Н). ESI-MS: 490 (М+1).
1-[4-(6-диметиламинометил-4-морфолинопирроло[2,1-f][1,2,4]триазин-2-ил)фенил]-3-(пара-хлорфенил)мочевина (I-5)
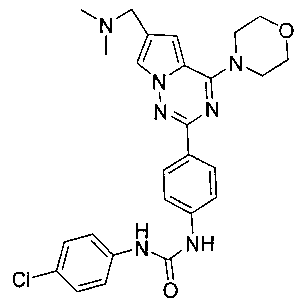
Способ получения был идентичен способу получения I-1 за исключением того, что этилизоцианат заменяли на пара-хлорфенилизоцианат. Белое твердое вещество (43 мг, 56,8%). т.пл. 237°C (декомпозиция). 1H ЯМР (300 МГц, DMSO-d6): δ 8,95 (s, 1Н), 8,87 (s, 1Н), 8,15 (d, J=8,5 Гц, 2Н), 7,95 (s, 1Н), 7,55 (d, J=8,5 Гц, 2Н), 7,50 (d, J=8,7 Гц, 2Н), 7,34 (d, J=8,7 Гц, 2Н), 7,13 (s, 1Н), 4,06 (t, J=4,5 Гц, 4Н), 3,94 (s, 2Н), 3,79 (t, J=4,5 Гц, 4Н), 2,43 (s, 6Н). ESI-MS: 506 (М+1), 508 (М+2+1).
1-(3-хлорфенил)-3-(4-(6-((диметиламино)метил)-4-морфолинопирроло[2,1-f][1,2,4]триазин-2-ил)фенил)мочевина (I-6)
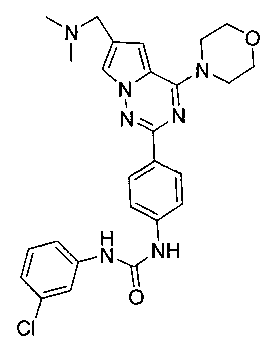
Способ получения был идентичен способу получения I-1 за исключением того, что этилизоцианат заменяли на мета-хлорфенилизоцианат. Белое твердое вещество (25 мг, 33,0%). т.пл. 173-176°C. 1H ЯМР (300 МГц, DMSO-d6): δ 9,00 (s, 1Н), 8,94 (s, 1Н), 8,16 (d, J=8,8 Гц, 2Н), 7,95 (d, J=1,3 Гц, 1Н), 7,73 (t, J=1,9 Гц, 1Н), 7,56 (d, J=8,8 Гц, 2Н), 7,34-7,25 (m, 2Н), 7,13 (d, J=1,3 Гц, 1Н), 7,03 (dt, J=1,9, 7,0 Гц, 1Н), 4,07 (t, J=4,6 Гц, 4Н), 3,94 (s, 2Н), 3,79 (t, J=4,6 Гц, 4Н), 2,43 (s, 6Н). ESI-MS: 506 (М+1), 508 (М+2+1).
1-(2,4-дихлорфенил)-3-(4-(6-((диметиламино)метил)-4-морфолинопирроло[2,1-f][1,2,4]триазин-2-ил)фенил)мочевина (I-7)
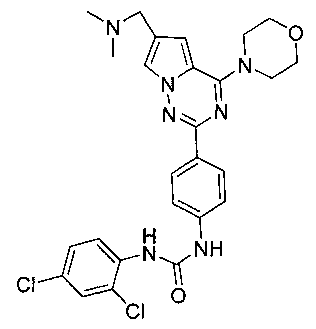
Способ получения был идентичен способу получения I-1 за исключением того, что этилизоцианат заменяли на 2,4-дихлорфенилизоцианат.Белое твердое вещество (29 мг, 35,8%). т.пл. 225°C (декомпозиция). 1H ЯМР (300 МГц, DMSO-d6): δ 9,66 (s, 1Н), 8,45 (s, 1Н), 8,22 (d, J=9,0 Гц, 1Н), 8,18 (d, J=8,8 Гц, 2Н), 7,95 (d, J=1,4 Гц, 1Н), 7,64 (d, J=2,4 Гц, 1Н), 7,57 (d, J=8,8 Гц, 2Н), 7,40 (dd, J=2,4, 9,0 Гц, 1Н), 7,13 (d, J=1,4 Гц, 1Н), 4,07 (t, J=4,7 Гц, 4Н), 3,95 (s, 2Н), 3,79 (t, J=4,7 Гц, 4Н), 2,43 (s, 6Н). ESI-MS: 540 (М+1), 542 (М+2+1).
1-(4-(6-((диметиламино)метил)-4-морфолинопирроло[2,1-f][1,2,4]триазин-2-ил)фенил)3-(3-(трифторметил)фенил)мочевина (I-8)
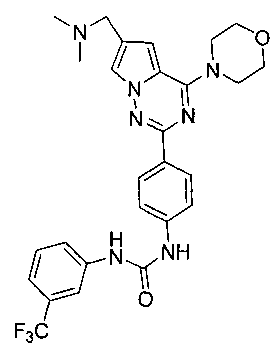
Способ получения был идентичен способу получения I-1 за исключением того, что этилизоцианат заменяли на мета-трифторфенилизоцианат. Белое твердое вещество (81 мг, 100%). т.пл. 160-163°C. 1H ЯМР (300 МГц, DMSO-d6): δ 9,73 (s, 1Н), 9,55 (s, 1Н), 8,16 (d, J=8,8 Гц, 2Н), 8,01 (s, 1Н), 7,96 (d, J=1,1 Гц, 1Н), 7,61 (d, J=8,6 Гц, 1Н), 7,57 (d, J=8,8 Гц, 2Н), 7,52 (t, J=7,5, 8,6 Гц, 1Н), 7,31 (d, J=7,5 Гц, 1Н), 7,12 (d, J=1,1 Гц, 1Н), 4,07 (t, J=4,4 Гц, 4Н), 3,94 (s, 2Н), 3,79 (t, J=4,4 Гц, 4Н), 2,43 (s, 6Н). ESI-MS: 540 (М+1).
1-(4-(6-((диметиламино)метил)-4-морфолинопирроло[2,1-г][1,2,4]триазин-2-ил)фенил)-3-(пара-толил)мочевина (I-9)
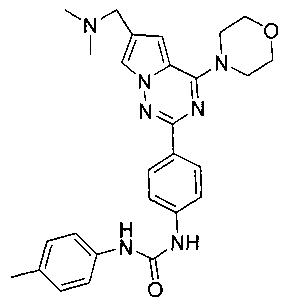
Способ получения был идентичен способу получения I-1 за исключением того, что этилизоцианат заменяли на пара-толилизоцианат. Белое твердое вещество (37 мг, 50,8%). т.пл. 230°C (декомпозиция). 1H ЯМР (300 МГц, DMSO-d6): δ 8,86 (s, 1Н), 8,61 (s, 1Н), 8,15 (d, J=8,8 Гц, 2Н), 7,95 (d, J=1,2 Гц, 1Н), 7,55 (d, J=8,8 Гц, 2Н), 7,35 (d, J=8,6 Гц, 2Н), 7,13 (d, J=1,2 Гц, 1Н), 7,09 (d, J=8,6 Гц, 2Н), 4,06 (t, J=4,6 Гц, 4Н), 3,94 (s, 2Н), 3,79 (t, J=4,6 Гц, 4Н), 2,43 (s, 6Н), 2,24 (s, 3Н). ESI-MS: 486 (М+1).
1-(4-(6-((диметиламино)метил)-4-морфолинопирроло[2,1-f][1,2,4]триазин-2-ил)фенил)-3-(4-метоксифенил)мочевина (I-10)
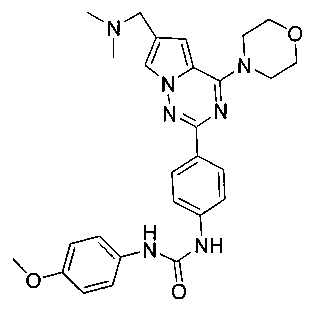
Способ получения был идентичен способу получения I-1 за исключением того, что этилизоцианат заменяли на пара-метоксифенилизоцианат. Белое твердое вещество (49 мг, 65,2%). т.пл. 235°C (декомпозиция). 1H ЯМР (300 МГц, DMSO-d6): δ 8,82 (s, 1Н), 8,52 (s, 1Н), 8,14 (d, J=8,8 Гц, 2Н), 7,94 (s, 1Н), 7,55 (d, J=8,8 Гц, 2Н), 7,37 (d, J=8,8 Гц, 2Н), 7,12 (s, 1Н), 6,87 (d, J=8,8 Гц, 2Н), 4,06 (t, J=4,5 Гц, 4Н), 3,95 (s, 2Н), 3,79 (t, J=4,5 Гц, 4Н), 3,72 (s, 3Н), 2,43 (s, 6Н). ESI-MS: 502 (М+1).
1-(4-(6-((диметиламино)метил)-4-морфолинопирроло[2,1-f][1,2,4]триазин-2-ил)фенил)-3-(4-фторбензил)мочевина (I-11)
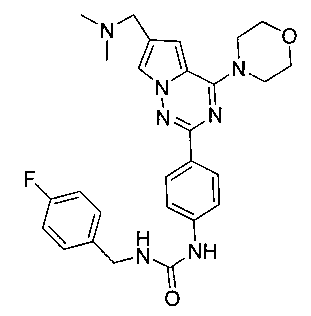
Способ получения был идентичен способу получения I-1 за исключением того, что этилизоцианат заменяли на пара-фторбензилизоцианат. Белое твердое вещество (32 мг, 42,4%). т.пл. 219-223°C. 1H ЯМР (300 МГц, DMSO-d6): δ 8,83 (s, 1Н), 8,10 (d, J=8,8 Гц, 2Н), 7,93 (s, 1Н), 7,50 (d, J=8,8 Гц, 2Н), 7,35 (dd, J=5,7, 8,6 Гц, 2Н), 7,16 (t, J=8,6 Гц, 2Н), 6,70 (t, J=5,5 Гц, 1Н), 7,11 (s, 1Н), 4,29 (d, J=5,5 Гц, 2Н), 4,05 (t, J=4,4 Гц, 4Н), 3,94 (s, 2Н), 3,78 (t, J=4,4 Гц, 4Н), 2,42 (s, 6Н). ESI-MS: 504 (М+1).
1-(4-(6-((диметиламино)метил)-4-морфолинопирроло[2,1-f][1,2,4]триазин-2-ил)фенил)-3-(3,5-диметилизоксазол-4-ил)мочевина (I-12)
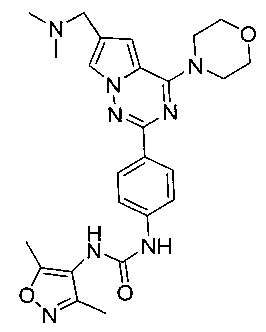
Способ получения был идентичен способу получения I-1 за исключением того, что этилизоцианат заменяли на 3,5-диметилизоксазол-4-изоцианат. Белое твердое вещество (24 мг, 31,7%). т.пл. 236°C (декомпозиция). 1H ЯМР (300 МГц, DMSO-d6): δ 9,05 (s, 1Н), 8,14 (d, J=8,8 Гц, 2Н), 7,94 (d, J=1,6 Гц, 1Н), 7,76 (s, 1Н), 7,55 (d, J=8,8 Гц, 2Н), 7,12 (d, J=1,6 Гц, 1Н), 4,06 (t, J=4,5 Гц, 4Н), 3,94 (s, 2Н), 3,79 (t, J=4,5 Гц, 4Н), 2,43 (s, 6Н), 2,30 (s, 3Н), 2,13 (s, 3Н). ESI-MS: 505 (М+1).
Этил 4-(3-(4-(6-((диметиламино)метил)-4-морфолинопирроло[2,1-f][1,2,4]триазин-2-ил)фенил)-уреидо)бензоат (I-13)
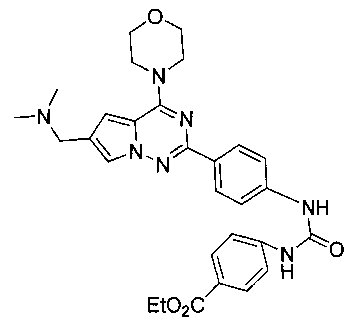
Способ получения был идентичен способу получения I-1 за исключением того, что этилизоцианат заменяли на пара-этоксикарбонилфенилизоцианат. Белое твердое вещество (47 мг, 57,7%). т.пл. 175-176°C. 1H ЯМР (300 МГц, DMSO-d6): δ 8,24 (d, J=8,5 Гц, 2Н), 8,01 (d, J=8,7 Гц, 2Н), 7,68 (s, 1Н), 7,49 (d, J=8,5 Гц, 2Н), 7,42 (d, J=8,7 Гц, 2Н), 6,92 (s, 1Н), 6,78 (s, 1Н), 6,70 (s, 1Н), 4,36 (q, J=7,0 Гц, 2Н), 4,10 (t, J=4,5 Гц, 4Н), 4,03 (s, 2Н), 3,89 (t, J=4,5 Гц, 4Н), 2,58 (s, 6Н), 1,39 (t, J=7,0 Гц, ЗН). ESI-MS: 544 (М+1).
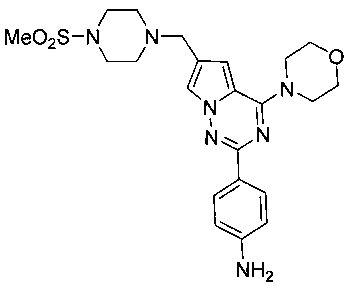
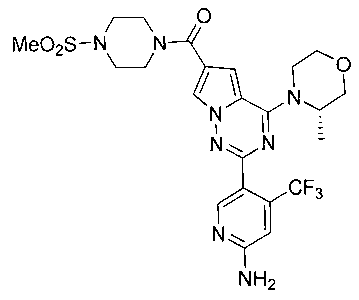
1-Этил-3-(4-(6-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-морфолинопирроло[2,1-f][1,2,4]триазин-2-ил)фенил)мочевина (I-17)
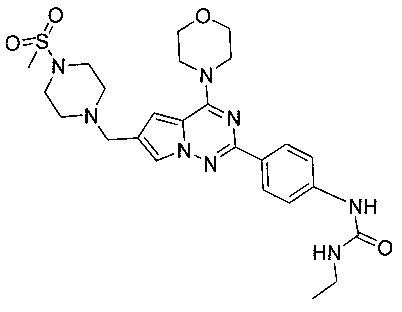
Способ получения был идентичен способу получения I-1 за исключением того, что 13а заменяли на соединение 13b. Белое твердое вещество (34 мг, 41,8%). т.пл. 200°C (декомпозиция). 1H ЯМР (300 МГц, DMSO-d6): δ 8,92 (s, 1Н), 8,08 (d, J=8,8 Гц, 2Н), 7,92 (s, 1Н), 7,49 (d, J=8,8 Гц, 2Н), 7,09 (s, 1Н), 6,36 (t, J=5,6 Гц, 1Н), 4,10 (s, 2Н), 4,05 (t, J=4,5 Гц, 4Н), 3,79 (t, J=4,5 Гц, 4Н), 3,49-3,37 (m, 4Н), 3,11 (квинтет, J=5,6, 7,0 Гц, 2Н), 2,96 (s, 3Н), 2,89 (br, s, 4Н), 1,05 (t, J=7,0 Гц, 3Н). ESI-MS: 543 (М+1)
1-(4-фторфенил)-3-(4-(6-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-морфолинопирроло[2,1-f][1,2,4]триазин-2-ил)фенил)мочевина I-18)
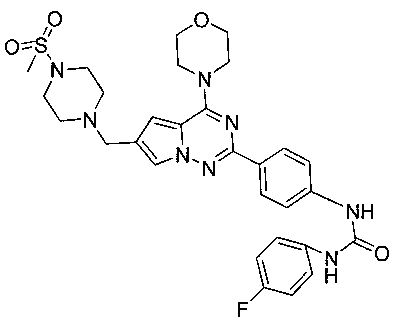
Способ получения был идентичен способу получения I-4 за исключением того, что 13а заменяли на соединение 13b. Белое твердое вещество (45 мг, 49,3%). т.пл. 255-256°C. 1H ЯМР (300 МГц, DMSO-d6): δ 8,89 (s, 1Н), 8,74 (s, 1Н), 8,15 (d, J=8,8 Гц, 2Н), 7,93 (s, 1Н), 7,56 (d, J=8,8 Гц, 2Н), 7,47 (dd, J=4,8, 8,8 Гц, 2Н), 7,13 (t, J=8,8 Гц, 2Н), 6,50 (s, 1Н), 4,10 (s, 2Н), 4,07 (br, s, 4Н), 3,80 (br, s, 4Н), 3,58-3,38 (m, 4Н), 2,96 (s, 3Н), 2,90 (br, s, 4H). ESI-MS: 609 (М+1).
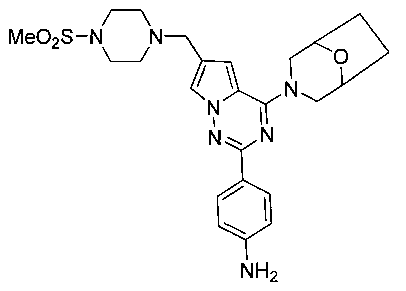
1-(4-(4-(8-окса-3-азабицикло[3,2,1]октан-3-ил)-6-((4-(метилсульфонил)пиперазин-1-ил)метил)-пирроло[2,1-f][1,2,4]триазин-2-ил)фенил)-3-(4-фторфенил)мочевина (I-19)
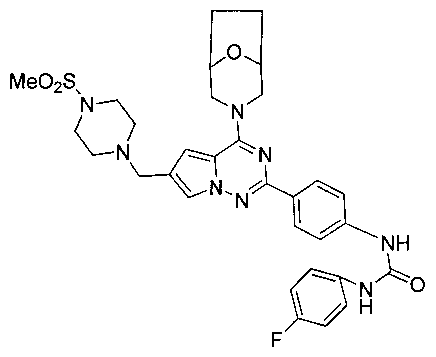
Способ получения был идентичен способу получения I-4 за исключением того, что 13а заменяли на соединение 13с. Белое твердое вещество (31 мг, 32,6%). т.пл. 266-267°C. 1H ЯМР (300 МГц, DMSO-d6): δ 8,89 (s, 1Н), 8,75 (s, 1Н), 8,13 (d, J=8,5 Гц, 2Н), 7,70 (s, 1Н), 7,54 (d, J=8,5 Гц, 2Н), 7,47 (dd, J=5,0, 8,8 Гц, 2Н), 7,13 (t, J=8,8 Гц, 2Н), 6,84 (s, 1Н), 4,51 (br, s, 4Н), 3,57 (s, 2Н), 3,47 (br s, 1Н), 3,43 (br s, 1Н), 3,32 (br s, 4Н), 3,11 (t, J=4,0 Гц, 4H), 2,87 (s, 3Н), 1,89-1,85 (m, 2Н), 1,80-1,76 (m,2H). ESI-MS: 635 (М+1).
Этил 4-(-3-(4-(6-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-морфолинопирроло[2,1-f][1,2,4]триазин-2-ил)фенил)уреидо)бензоат (I-20)
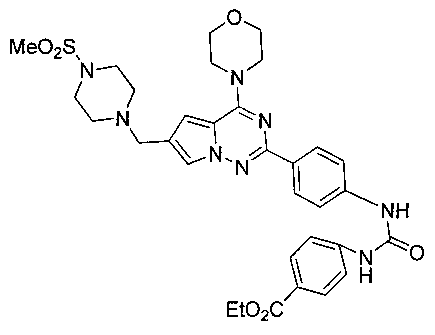
Способ получения был идентичен способу получения I-13 за исключением того, что 13а заменяли на соединение 13b. Белое твердое вещество (40 мг, 40,2%). т.пл. 260-262°C. 1H ЯМР (300 МГц, DMSO-d6): δ 9,15 (s, 1Н), 9,02 (s, 1Н), 8,16 (d, J=8,8 Гц, 2Н), 7,90 (d, J=8,8 Гц, 2Н), 7,72 (s, 1Н), 7,62-7,56 (m, 4Н), 6,90 (s, 1Н), 6,52 (s, 3Н), 4,28 (q, J=7,0 Гц, 2Н), 4,05 (br s, 4Н), 3,79 (br s, 4Н), 3,58 (s, 2H), 3,11 (br s, 4H), 2,87 (br s, 4H), 1,31 (t, J=7,0 Гц, 3Н). ESI-MS: 663 (M+1).
13. Общий способ получения соединений I-14~I-16
Соответствующую карбоновую кислоту (0,5 ммоль) растворяли в безводном N,N-диметилформамиде и триэтиламине (101 мг, 1 ммоль) и добавляли дифенилазидофосфат (165 мг, 0,6 ммоль), и подвергали взаимодействию при комнатной температуре в течение 1 часа. Реакционную смесь выливали в воду и фильтровали, и остаток на фильтре сушили в вакуумной печи при комнатной температуре в течение 24 часов с получением пара-карбамоилбензоилазида. Соединение 13 (0,1 ммоль) и пара-карбамоилбензоилазид (0,2 ммоль) в безводном диоксане кипятили с обратным холодильником в течение 3 часов. Растворитель отгоняли при пониженном давлении. Остаток растворяли в смешанном растворителе дихлорметана и метанола и очищали при помощи препаративной тонкослойной хроматографии (дихлорметан: метанол = 8:1), с получением чистого желаемого продукта.
4-(3-(4-(6-((диметиламино)метил)-4-морфолинопирроло[2,1-f][1,2,4]триазин-2-ил)фенил)уреидо)-N,N-диметилбензамид (I-14)
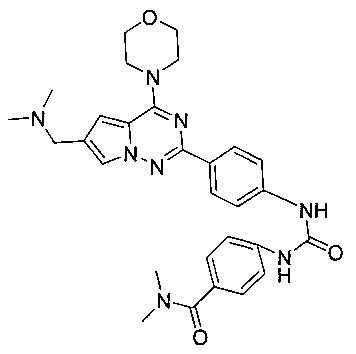
В соответствии с общим способом, описанным в примере 13, 4-(диметилкарбамоил)бензойную кислоту использовали в качестве исходного вещества, и получающийся в результате 4-(диметилкарбамоил)бензоилазид подвергали взаимодействию с 13а с получением светло-желтого твердого вещества (14 мг, 25,8%). т.пл. 240°C. 1H ЯМР (300 МГц, DMSO-d6): δ 9,59 (s, 2Н), 8,16 (d, J=8,5 Гц, 2Н), 7,99 (s, 1Н), 7,58 (d, J=8,5 Гц, 2Н), 7,52 (d, J=8,5 Гц, 2Н), 7,36 (d, J=8,5 Гц, 2Н), 7,19 (s, 1Н), 4,25 (s, 2Н), 4,08 (t, J=4,4 Гц, 4Н), 3,80 (t, J=4,4 Гц, 4Н), 2,96 (s, 6Н), 2,69 (s, 6Н). ESI-MS: 543 (М+1).
1-(4-(6-((диметиламино)метил)-4-морфолинопирроло[2,1-f][1,2,4]триазин-2-ил)фенил)-3-(4-(4-метилпиперазин-1-карбонил)фенил)мочевина (I-15)
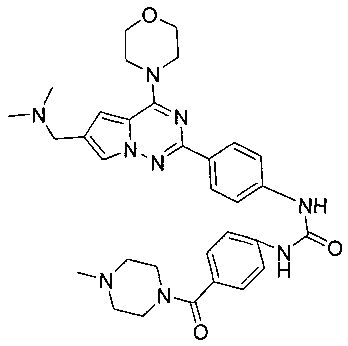
В соответствии с общими способами, описанным в примере 13, 4-(4-метилпиперазин-1-карбонил)бензойную кислоту использовали в качестве исходного вещества, и получающийся в результате 4-(4-метилпиперазин-1-карбонил)бензоилазид подвергали взаимодействию с 13а с получением белого твердого вещества (9 мг, 15,1%). 1H ЯМР (300 МГц, DMSO-d6): δ 9,63 (br, s, 2Н), 8,16 (d, J=8,9 Гц, 2H), 7,92 (s, 1Н), 7,57 (d, J=8,9 Гц, 2H), 7,53 (d, J=8,5 Гц, 2H), 7,34 (d, J=8,5 Гц, 2H), 7,13 (s, 1Н), 4,07 (br, s, 6H), 3,80 (br, s, 4H), 3,51 (br, s, 4H), 3,06 (br s, 2H), 2,55 (s, 6H), 2,38 (br s, 2H), 2,24 (s, 3H). ESI-MS: 598 (M+1).
1-(4-(6-((диметиламино)метил)-4-морфолинопирроло[2,1-г][1,2,4]триазин-2-ил)фенил)-3-(4-(пиперидин-1-карбонил)фенил)мочевина (I-16)
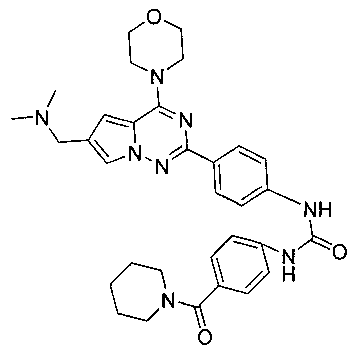
В соответствии с общим способом, описанным в примере 13, 4-(пиперидин-1-карбонил)бензойную кислоту использовали в качестве исходного вещества, и получающийся в результате 4-(пиперидин-1-карбонил)бензоилазид подвергали взаимодействию с 13а с получением желтого твердого вещества (11 мг, 18,9%). т.пл. 184-186°C. 1H ЯМР (300 МГц, DMSO-d6): δ 9,57 (s, 2Н), 8,15 (d, J=8,8 Гц, 2Н), 7,93 (s, 1Н), 7,57 (d, J=8,8 Гц, 2H), 7,52 (d, J=8,6 Гц, 2H), 7,31 (d, J=8,6 Гц, 2H), 7,13 (s, 1Н), 4,07 (br, s, 6H), 3,79 (t, J=4,3 Гц, 4H), 3,38 (br, s, 4H), 2,58 (s, 6H), 1,60 (br, s, 2H), 1,50 (br, s, 4H). ESI-MS: 583 (M+1).
14. Получение 4-(3-(-4(6-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-морфолинопирроло[2,1-f][1,2,4]триазин-2-ил)фенил)уреидо)бензойной кислоты (14)
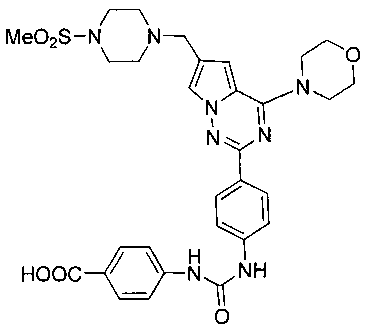
Соединение I-20 (395 мг, 0,6 ммоль) суспендировали в 20 мл тетрагидрофурана и 10 мл метанола, и добавляли 4 мл 1М раствора гидроксида калия и кипятили с обратным холодильником в течение 3 часов. Реакционную смесь охлаждали, и добавляли 2 мл уксусной кислоты для осаждения твердых веществ. После фильтрования получали белые твердые вещества (312 мг, 82,0%). т.пл. 207-209°C. 1H ЯМР (300 МГц, DMSO-d6): δ 9,16 (s, 1Н), 9,08 (s, 1Н), 8,16 (d, J=8,7 Гц, 2Н), 7,88 (d, J=8,7 Гц, 2Н), 7,71 (s, 1Н), 7,58 (d, J=7,9 Гц, 4Н), 6,87 (s, 1Н), 4,04 (br s, 4Н), 3,79 (br s, 4Н), 3,58 (s, 2Н), 3,34 (br, s, 4H), 3,11 (brs, 4H), 2,87 (s, 3Н). LC-MS: 635 (М+1).
15. Общие способы получения соединения I-21~I-23.
Соединение 14 (95 мг, 0,15 ммоль), диизопропилэтиламин (116 мг, 0,9 ммоль), и HBTU (бензотриазол-N,N,N′,N′-тетраметилуроний гексафторфосфат, 284 мг, 0,75 ммоль) растворяли в 5 мл N,N-диметилформамида и перемешивали в течение 1 часа при комнатной температуре. Добавляли каждый соответствующий амин (0,6 ммоль) и перемешивали при комнатной температуре в течение 4-6 часов. Реакционную смесь выливали в 50 мл воды и экстрагировали этилацетатом. Получающуюся в результате смесь разделяли при помощи препаративной пластины (дихлорметан: метанол = 10:1) с получением продукта.
N,N-диметил-4-(3-(4-(6-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-морфолинопирроло[2,1-f][1,2,4]триазин-2-ил)фенил)уреидо)бензамид (I-21)
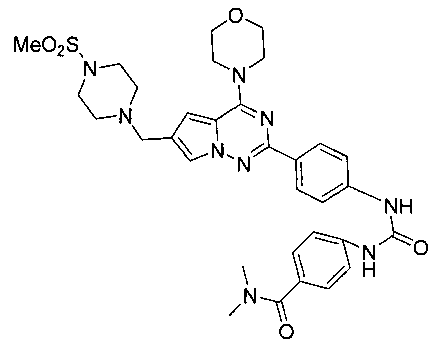
Светло-желтое твердое вещество (38 мг, 38,3%). т.пл. 248-250°C. 1H ЯМР (300 МГц, DMSO-d6): δ 9,40 (s, 2Н), 8,15 (d, J=8,8 Гц, 2Н), 7,72 (s, 1Н), 7,57 (d, J=8,9 Гц, 2Н), 7,52 (d, J=8,9 Гц, 2Н), 7,36 (d, J=8,8 Гц, 2Н), 6,89 (s, 1Н), 4,05 (t, J=4,4 Гц, 4Н), 3,79 (t, J=4,4 Гц, 4Н), 3,59 (s, 2Н), 3,32 (br s, 4Н), 3,12 (br s, 4H), 2,96 (s, 6H), 2,87 (s, 3Н). ESI-MS: 684 (M+23).
1-(4-(4-метилпиперазин-1-карбонил)фенил)-3-(4-(6-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-морфолинопирроло[2,1-f][1,2,4]триазин-2-ил)фенил)мочевина (I-22)
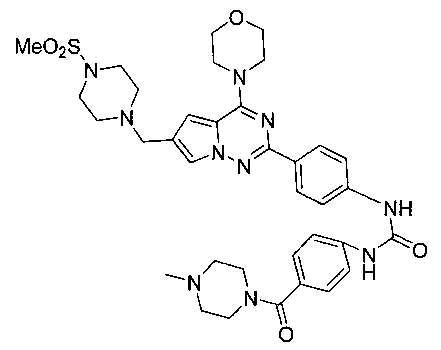
Светло-желтое твердое вещество (40 мг, 37,2%). т.пл. 185-188°C. 1H ЯМР (300 МГц, DMSO-d6): δ 9,03 (s, 1Н), 9,02 (s, 1Н), 8,15 (d, J=8,7 Гц, 2Н), 7,71 (s, 1Н), 7,58-7,52 (m, 4Н), 7,34 (d, J=8,7 Гц, 2Н), 6,89 (s, 1Н), 4,04 (t, J=4,7 Гц, 4Н), 3,79 (t, J=4,7 Гц, 4Н), 3,59 (s, 2Н), 3,50 (br s, 4Н), 3,32 (br s, 4Н), 3,12 (br s, 4H), 2,87 (s, 3Н), 2,38 (br s, 4H), 2,24 (s, 3H). ESI-MS: 717 (M+1).
1-(4-(4-(диметиламино)пиперидин-1-карбонил)фенил)-3-(4-(6-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-морфолинопирроло[2,1-f][1,2,4]триазин-2-ил)фенил)мочевина (I-23)
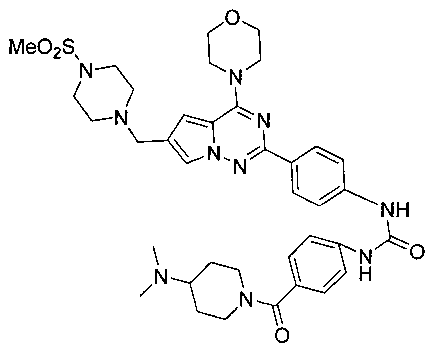
Белое твердое вещество (44 мг, 39,4%). т.пл. 200-202°C. 1H ЯМР (300 МГц, DMSO): δ 9,12 (s, 2Н), 8,15 (d, J=8,6 Гц, 2Н), 7,71 (s, 1Н), 7,58-7,53 (m, 4Н), 7,37 (d, J=8,6 Гц, 2Н), 6,90 (s, 1Н), 4,05 (t, J=4,2 Гц, 4Н), 3,79 (t, J=4,2 Гц, 4Н), 3,59 (s, 2Н), 3,32 (br s, 8Н), 3,12 (br s, 4Н), 2,87 (s, 3Н), 2,94 (s, 1Н), 2,68 (s, 6Н), 2,05-1,91 (m, 2Н), 1,63-1,48 (m, 2Н). ESI-MS: 745 (М+1).
16. Получение метил 2-(3-(ацетоксиметил)фенил)-4-хлорпирроло[2,1-f][1,2,4] триазин-6-карбоксилата (15)
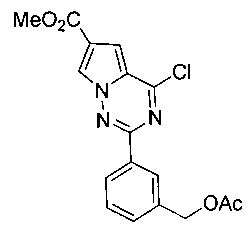
Соединение 15 получали при помощи способа, который был идентичен получению соединения 8, где соединение IIIb (341 мг, 1 ммоль) использовали в качестве исходного вещества. Неочищенный продукт очищали при помощи колоночной хроматографии (петролейный эфир: этилацетат = 5:1) с получением белого твердого вещества (197 мг, 54,8%). т.пл. 154-155°C. 1H ЯМР (300 МГц, CDCl3): δ 8,33-8,29 (m, 3Н), 7,52-7,50 (m, 2Н), 7,41 (d, J=1,3 Гц, 1Н), 5,20 (s, 2Н), 3,94 (s, 3Н), 2,14 (s, 3Н). LC-MS: 360 (М+1), 362 (М+2+1).
17. Получение метил 2-(3-(ацетоксиметил)фенил)-4-морфолинопирроло[2,1-f][1,2,4]триазин-6-карбоксилата (16)
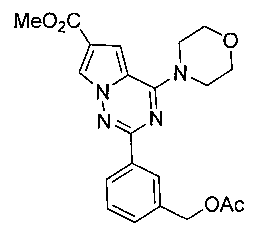
Соединение 16 получали при помощи способа, который был идентичен получению соединения 9, где соединение 15 (180 мг, 0,5 ммоль) использовали в качестве исходного вещества. Белое твердое вещество (177 мг, 86,3%). т.пл. 204-205°C. 1H ЯМР (300 МГц, CDCl3): δ 8,26-8,22 (m, 2Н), 8,13 (s, 1Н), 7,46 (d, J=4,5 Гц, 2Н), 7,19 (s, 1Н), 5,19 (s, 2Н), 4,15 (t, J=4,8 Гц, 4Н), 3,90 (s, 3Н), 3,90 (t, J=4,8 Гц, 4Н), 2,13 (s, 3Н). LC-MS: 411 (М+1).
18. Получение 2-(3-(гидроксиметил)фенил)-4-морфолинопирроло[2,1-f][1,2,4] триазин-6-карбоновой кислоты (17)
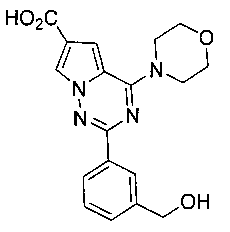
Соединение 17 получали при помощи способа, который был идентичен получению соединения 11, где соединение 16 (150 мг, 0,366 ммоль) использовали в качестве исходного вещества. Белое твердое вещество (111 мг, 85,7%). т.пл. 254°C (декомпозиция). 1H ЯМР (300 МГц, DMSO-d6): δ 12,64 (s, 1Н), 8,21 (s, 1Н), 8,19 (d, J=1,6 Гц, 1Н), 8,13-8,10 (m, 1Н), 7,44-7,42 (m, 2Н), 7,36 (d, J=1,6 Гц, 1Н), 5,30 (s, 1Н), 4,58 (s, 2Н), 4,10 (t, J=4,5 Гц, 4Н), 3,80 (t, J=4,5 Гц, 4Н). LC-MS: 355 (М+1).
19. Получение 2-(3-(гидроксиметил)фенил)-N,N-диметил-4-морфолинопирроло[2,1-f][1,2,4]триазин-6-карбоксамида (18)
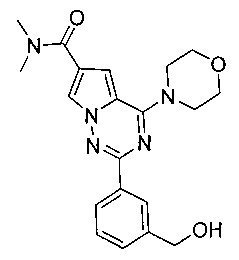
Соединение 18 получали при помощи способа, который был идентичен получению соединения 12а, где соединение 17 (92 мг, 0,26 ммоль) использовали в качестве исходного вещества. Белое твердое вещество (80 мг, 80,8%). т.пл. 170-171°C. 1H ЯМР (300 МГц, CDCl3): δ 8,22 (s, 1Н), 8,20-8,17 (m, 1Н), 7,84 (d, J=1,5 Гц, 1Н), 7,46-7,43 (m, 2Н), 7,04 (d, J=1,5 Гц, 1Н), 4,77 (s, 2Н), 4,11 (t, J=4,7 Гц, 4Н), 3,87 (t, J=4,7 Гц, 4Н), 3,20 (br, s, 6Н). LC-MS: 382 (М+1).
20. Получение (3-(6-((диметиламино)метил)-4-морфол инопирроло[2,1-f][1,2,4]триазин-2-ил)фенил)метанола (I-24)
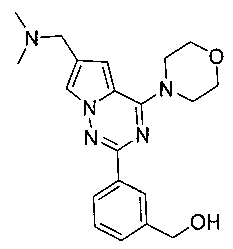
Соединение I-24 получали при помощи способа, который был идентичен получению соединения 13а, где соединение 18 (70 мг, 0,184 ммоль) использовали в качестве исходного вещества. Белое твердое вещество (100%). т.пл. 163-165°C. 1H ЯМР (300 МГц, DMSO-d6): δ 8,20 (s, 1Н), 8,11 (dt, J=2,4, 6,0 Гц, 1Н), 7,99 (d, J=1,3 Гц, 1Н), 7,45-7,41 (m, 2Н), 7,15 (d, J=1,3 Гц, 1Н), 5,27 (t, J=5,8 Гц, 1Н), 4,57 (d, J=5,8 Гц, 2Н), 4,07 (t, J=4,6 Гц, 4Н), 3,95 (s, 2Н), 3,80 (t, J=4,6 Гц, 4Н), 2,44 (s, 6Н). MS (EI) m/e(%): 367 (М+, 16).
21. Получение 3-(6-((диметиламино)метил)-4-морфолинопирроло[2,1-f][1,2,4]триазин-2-ил)бензилметансульфоната (19)
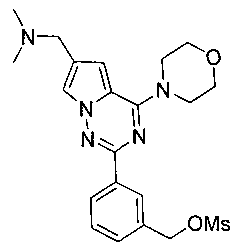
Соединение I-24 (36,7 мг, 0,1 ммоль) растворяли в 5 мл безводного дихлорметана и охлаждали до 0°C. Добавляли метансульфонилхлорид (14 мкл, 0,12 ммоль) и триэтиламин (16 мкл, 0,12 ммоль). Реакцию осуществляли при 0°C в течение 30 минут. Реакционную смесь последовательно промывали насыщенным раствором бикарбоната натрия и водой, и органический слой сушили над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении с получением 45 мг белого твердого вещества (100%). т.пл. 178-179°C. 1H ЯМР (300 МГц, CDCl3): δ 8,33 (s, 1Н), 8,31 (dd, J=1,8, 5,5 Гц, 1Н), 7,70 (d, J=1,5 Гц, 1Н), 7,51 (d, J=5,5 Гц, 2Н), 6,72 (d, J=1,5 Гц, 1Н), 5,34 (s, 2Н), 4,14 (t, J=4,8 Гц, 4Н), 4,04 (s, 2Н), 3,91 (t, J=4,8 Гц, 4Н), 2,95 (s, 3Н), 2,56 (s, 6Н). MS (EI) m/e (%): 350 (M-MeSO3, 30).
22. Получение N,N-диметил-1-(2-(3-((метилсульфонил)метил)фенил)-4-морфолинопирроло[2,1-f][1,2,4]триазин-6-ил)метанамина (I-25)
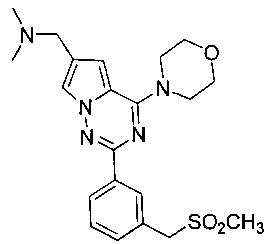
Соединение 19 (45 мг, 0,1 ммоль) и метилсульфинат натрия (41 мг, 0,4 ммоль) растворяли в 2 мл N-метилпирролидона. На смесь воздействовали микроволновым излучением в течение 30 минут при 120°C при мощности 100 ватт. После завершения реакции реакционную смесь выливали в воду, затем трижды экстрагировали этилацетатом и затем трижды экстрагировали дихлорметаном. Органические слои комбинировали и сушили над безводные сульфатом натрия. Растворитель концентрировали при пониженном давлении до небольшого объема. Неочищенный продукт очищали при помощи препаративной пластины (дихлорметан: метанол = 6:1) с получением 12 мг белого твердого вещества (28,0%). т.пл. 143-144°C. 1H ЯМР (300 МГц, CDCl3): δ 8,32 (d, J=5,2 Гц, 1Н), 8,29 (s, 1Н), 7,63 (d, J=1,2 Гц, 1Н), 7,50 (d, J=5,2 Гц, 2Н), 6,78 (d, J=1,2 Гц, 1Н), 4,34 (s, 2Н), 4,11 (t, J=4,4 Гц, 4Н), 3,88 (t, J=4,4 Гц, 4Н), 3,66 (s, 2Н), 2,78 (s, 3Н), 2,36 (s, 6Н). MS (EI) m/e (%): 429 (М+, 12).
23. Получение 1-(2-(3-(аминометил)фенил)-4-морфолинопирроло[2,1-f][1,2,4]триазин-6-ил)-N,N-диметилметанамина (20)
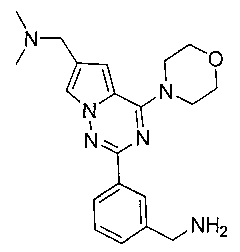
Соединение 19 (80 мг, 0,18 ммоль) добавляли к 20 мл насыщенного аммиачного раствора метанола. Реакцию осуществляли при 80°C в запаянной пробирке в течение 8 ч. После того, как реакционную смесь концентрировали, ее очищали при помощи препаративной пластины (дихлорметан: метанол = 6:1) с получением 37 мг бесцветного масла (56,2%). 1H ЯМР (300 МГц, CDCl3): δ 8,20 (s, 1Н), 8,17-8,13 (m, 1Н), 7,64 (s, 1Н), 7,41-7,39 (m, 2Н), 6,86 (s, 1Н), 4,11 (t, J=4,7 Гц, 4Н), 3,97 (s, 2Н), 3,87 (t, J=4,7 Гц, 4Н), 3,69 (s, 2Н), 2,41 (s, 6Н). LC-MS: 367 (М+1).
24. Получение N-(3-(6-((диметиламино)метил)-4-морфол инопирроло[2,1-f][1,2,4] триазин-2-ил)бензил)метансульфонамида (I-26)
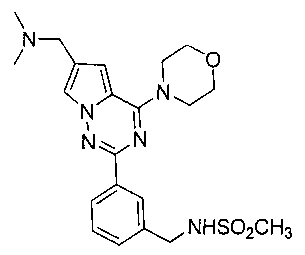
Соединение I-26 получали при помощи способа, который был идентичен получению соединения 19, где соединение 20 (37 мг, 0,1 ммоль) использовали в качестве исходного вещества. Светло-желтое твердое вещество (12 мг, 26,7%) получали при помощи препаративной пластины (дихлорметан: метанол = 8:1). т.пл. 238-240°C. 1H ЯМР (300 МГц, CDCl3): δ 8,20 (s, 1Н), 8,19 (d, J=7,1 Гц, 1Н), 7,60 (s, 1Н), 7,47-7,38 (m, 2 Н), 6,88 (s, 1Н), 4,41 (s, 2Н), 4,02 (br, s, 4Н), 3,81 (t, J=4,4 Гц, 4Н), 2,95 (s, 2Н), 2,94 (s, 3Н), 2,87 (s, 1Н), 2,51 (s, 6Н). MS (EI) m/e (%): 444 (М+, 16).
25. Получение метил 4-хлор-2-(6-пиваламидо-4-(трифторметил)пиридин-3-ил)пирроло [2,1-f][1,2,4]триазин-6-карбоксилата (21)
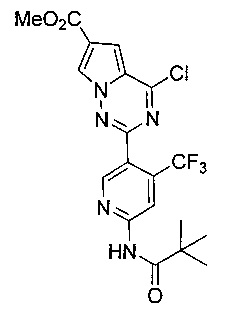
Соединение 21 получали при помощи способа, который был идентичен получению соединения 8, где IIIc (300 мг, 0,686 ммоль) использовали в качестве исходного вещества. Светло-желтое твердое вещество (290 мг, 92,9%). т.пл. 172-173°C. 1H ЯМР (300 МГц, CDCl3): δ 8,79 (s, 1Н), 8,77 (s, 1Н), 8,33 (d, J=1,6 Гц, 1Н), 8,30 (s, 1Н), 7,47 (d, J=1,6 Гц, 1Н), 3,94 (s, 3Н), 1,36 (s, 9Н). LC-MS: 478 (М+23), 480 (М+2+23).
26. Получение соединения 22
Получение соединения 22 было идентично получению соединения 9. Метил 4-морфолино-2-(6-пиваламидо-4-(трифторметил)пиридин-3-ил)пирроло[2,1-f][1,2,4] триазин-6-карбоксилат (22а)
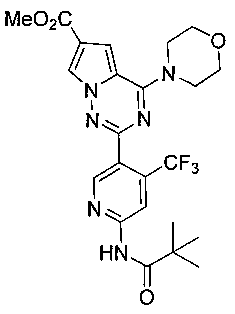
Соединение 22а получали из 100 мг соединения 21 (0,22 ммоль). Белое твердое вещество (73 мг, 65,8%). т.пл. 205°C. 1H ЯМР (300 МГц, DMSO-d6): δ 10,53 (s, 1H), 8,79 (s, 1H), 8,57 (s, 1H), 8,30 (d, J=1,2 Гц, 1H), 7,46 (d, J=1,2 Гц, 1H), 4,03(t, J=4,2 Гц, 4H), 3,83 (s, 3Н), 3,75 (t, J=4,2 Гц, 4H), 1,27 (s, 9H). LC-MS: 507 (М+1).
Метил-4-(8-окса-3-азабицикло[3,2,1]октан-3-ил)-2-(6-пиваламидо-4-(трифторметил)пиридин-3-ил)пирроло[2,1-f][1,2,4]триазин-6-карбоксилат (22b)
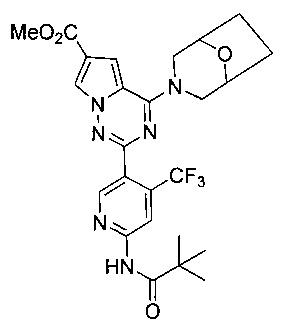
Соединение 22b получали из 276 мг соединения 21 (0,61 ммоль). Светло-желтое твердое вещество (274 мг, 84,8%). т.пл. 175-176°C. 1H ЯМР (300 МГц, CDCl3): δ 8,71 (s, 1Н), 8,70 (s, 1Н), 8,27 (s, 1Н), 8,09 (s, 1Н), 7,21 (s, 1Н), 4,52 (br s, 4Н), 3,90 (s, 3Н), 3,54 (br s, 2Н), 2,04-1,99 (m, 2Н), 1,89-1,78 (m, 2Н), 1,36 (s, 9Н). LC-MS: 533 (М+1).
(S)-метил 4-(3-метилморфолино)-2-(6-пиваламидо-4-(трифторметил)пиридин-3-ил)пирроло[2,1-f][1,2,4]триазин-6-карбоксилат (22с)
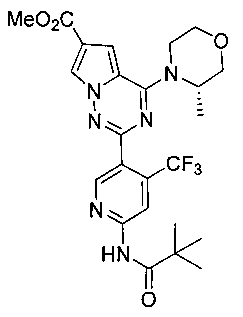
Соединение 22с получали из 210 мг соединения 21 (0,46 ммоль). Светло-желтое твердое вещество (145 мг, 60,4%). т.пл. 225-226°C. 1H ЯМР (300 МГц, DMSO-d6): δ 10,53 (s, 1Н), 8,79 (s, 1Н), 8,57 (s, 1Н), 8,29 (d, J=1,5 Гц, 1Н), 7,42 (s, 1Н), 4,92 (br, 1Н), 4,61 (br, 1Н), 4,03-3,96(m, 1Н), 3,83 (s, 3Н), 3,81-3,64(m, 3Н), 3,53 (t, J=11,4 Гц, 1Н), 1,43-1,31 (m, 3Н), 1,27 (s, 9Н). LC-MS: 521 (М+1).
27. Получение соединения 23
2-(6-амино-4-(трифторметил)пиридин-3-ил)-4-морфолинопирроло[2,1-f][1,2,4]триазин-6-карбоновая кислота (23а)
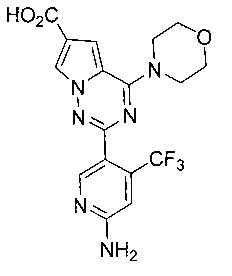
Соединение 22а (300 мг, 0,59 ммоль) суспендировали в 10 мл этанола. Добавляли 1,5 мл 2М раствора гидроксида калия и кипятили с обратным холодильником в течение 1 часа. Реакция по существу завершалась. Добавляли 2 мл уксусной кислоты. Большую часть растворителя отгоняли при пониженном давлении для осаждения твердых веществ. Осажденные твердые вещества фильтровали с получением соединение 23а. Белое твердое вещество (224 мг, 92,6%). т.пл. 244-245°C. 1H ЯМР (300 МГц, DMSO-d6): δ 12,54 (s, 1Н), 8,37 (s, 1Н), 8,11 (s, 1Н), 7,34 (s, 1Н), 6,83 (s, 3Н), 4,00(t, J=4,0 Гц, 4Н), 3,73(t, J=4,0 Гц, 4Н). LC-MS: 409 (М+1).
2-(6-амино-4-(трифторметил)пиридин-3-ил)-4-(8-окса-3-азабицикло[3,2,1]октан-3-ил)пирроло[2,1-f][1,2,4]триазин-6-карбоновая кислота (23b)
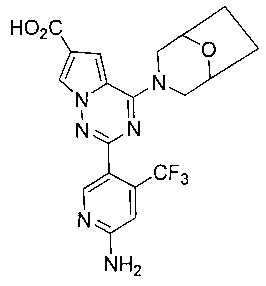
Соединение 23b получали при помощи способа, который был идентичен получению соединения 23а, где соединение 22b (250 мг, 0,45 ммоль) использовали в качестве исходного вещества. Белое твердое вещество (102 мг, 51,9%). т.пл. 284-285°C. 1H ЯМР (300 МГц, DMSO-d6): δ 12,44(br s, 1Н), 8,36 (s, 1Н), 8,11 (s, 1Н), 7,30 (s, 1Н), 6,82 (s, 3Н), 4,47 (br s, 2H), 4,42 (br s, 2H), 3,32 (br s, 2H), 1,87-1,84 (m, 2H), 1,75-1,68 (m, 2H). LC-MS: 435 (M+1).
(S)-2-(6-амино-4-(трифторметил)пиридин-3-ил)-4-(3-метилморфолино)пирроло[2,1-f][1,2,4]триазин-6-карбоновая кислота (23с)
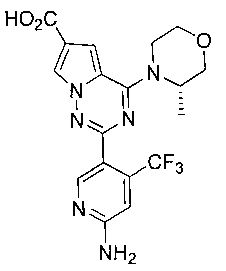
Соединение 23с получали при помощи способа, который был идентичен получению соединения 23а, где соединение 22с (1,8 г, 3,46 ммоль) использовали в качестве исходного вещества. Белое твердое вещество (1,33 г, 91,2%). т.пл. >300°C. 1H ЯМР (400 МГц, DMSO-d6): δ 12,76 (s, 1Н), 8,36 (s, 1Н), 8,11 (d, J=1,3 Гц, 1Н), 7,30 (s, 1Н), 6,85 (s, 2Н), 6,82 (s, 1Н), 4,89 (br, 1Н), 4,52 (br, 1Н), 3,97 (d, J=8,2 Гц, 1Н), 3,76-3. 63(m, 2Н), 3,51 (t, J=10,7 Гц, 2H),1,36(s, 3Н). LC-MS: 423(М+1).
28. Получение соединения 24
Получение соединения 24 было идентично получению соединения 12.
(2-(6-амино-4-(трифторметил)пиридин-3-ил)-4-морфолинопирроло[2,1-f][1,2,4]триазин-6-ил)(4-(метилсульфонил)пиперазин-1-ил)метанон (24а)
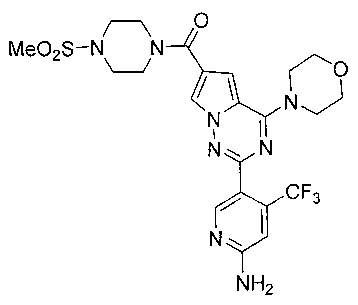
Соединение 24а получали из 220 мг соединения 23а (0,54 ммоль). Белое твердое вещество (253 мг, 84,7%). т.пл. 197-198°C. 1H ЯМР (300 МГц, DMSO-d6): δ 8,36 (s, 1Н), 8,04 (s, 1Н), 7,18 (s, 1Н), 6,83 (s, 3Н), 4,04-3,92 (m, 4Н), 3,73 (br s, 8Н), 3,20-3,11 (m, 4Н), 2,91 (s, 3Н). MS (EI) m/e (%): 554 (68, М+).
(2-(6-амино-4-(трифторметил)пиридин-3-ил)-4-(8-окса-3-азабицикло[3,2,1]октан-3-ил)пирроло[2,1-f][1,2,4]триазин-6-ил)(4-(метилсульфонил)пиперазин-1-ил)метанон (24b)
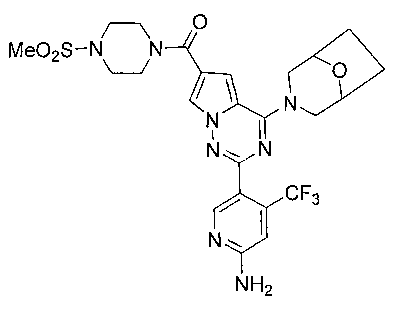
Соединение 24b получали из 90 мг соединения 23b (0,21 ммоль). Белое твердое вещество (113 мг, 93,8%). т.пл. 175°C. 1H ЯМР (300 МГц, CDCl3): δ 8,52 (s, 1Н), 7,77 (d, J=1,7 Гц, 1Н), 6,96 (d, J=1,7 Гц, 1Н), 6,81 (s, 1Н), 4,86 (s, 2Н), 4,49 (br s, 4Н), 3,91 (t, J=4,6 Гц, 4Н), 3,55 (br s, 2Н), 3,28 (t, J=4,6 Гц, 4Н), 2,81 (s, 3Н), 2,04-1,95 (m, 2Н), 1,89-1,79 (m, 2Н). LC-MS: 581 (М+1).
(S)-(2-(6-амино-4-(трифторметил)пиридин-3-ил)-4-(3-метилморфолино)пирроло[2,1-f][1,2,4]триазин-6-ил)(4-(метилсульфонил)пиперазин-1-ил)метанон (24с)
Соединение 24 с получали из 2,42 г соединения 23с (4,86 ммоль) в качестве исходного вещества. Светло-желтое твердое вещество (3 г, 92,2%). т.пл. 200-202°C. 1H ЯМР (300 МГц, CDCl3): δ 8,52 (s, 1Н), 7,78 (d, J=1,7 Гц, 1Н), 6,97 (d, J=1,7 Гц, 1Н), 6,81 (s, 1Н), 4,90 (s, 3Н), 4,81-4,40 (m, 1Н), 4,03 (d, J=7,4 Гц, 1Н), 3,91 (t, J=4,8 Гц, 4Н), 3,83-3,72 (m, 2Н), 3,66-3,56 (m, 2Н), 3,28 (t, J=4,8 Гц, 4Н), 2,81 (s, 3Н), 1,48 (d, J=6,2 Гц, 3Н). LC-MS: 569 (М+1).
29. Получение 5-(6-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-морфолинопирроло [2,1-f][1,2,4]триазин-2-ил)-4-(трифторметил)пиридин-2-амина (I-27)
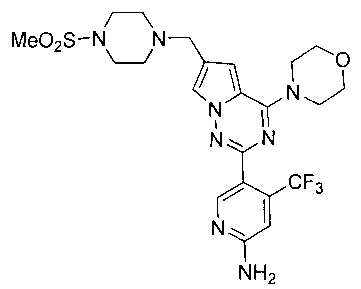
Соединение I-27 получали из 200 мг соединения 24а (0,36 ммоль) в качестве исходного вещества. Способ получения был аналогичен способу получения соединения 13. Препаративную пластину (дихлорметан: метанол = 20:1) использовали для очистки. Светло-желтое твердое вещество (43 мг, 22,0%). т.пл. 122-123°C. 1H ЯМР (300 МГц, CDCl3): δ 8,51 (s, 1Н), 7,59 (s, 1Н), 6,80 (s, 1Н), 6,67 (s, 1Н), 4,89 (s, 2Н), 4,04(t, J=4,4 Гц, 4Н), 3,82(t, J=4,4 Гц, 4Н), 3,64 (s, 2Н), 3,27 (br s, 4Н), 2,78 (s, 3Н), 2,62 (br s, 4Н). MS (EI) m/e (%): 540 (М+, 5).
30. Получение 1-(4-фторфенил)-3-(5-(6-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-морфолинопирроло[2,1-f][1,2,4]триазин-2-ил)-4-(трифторметил)пиридин-2-ил)мочевины (I-28)
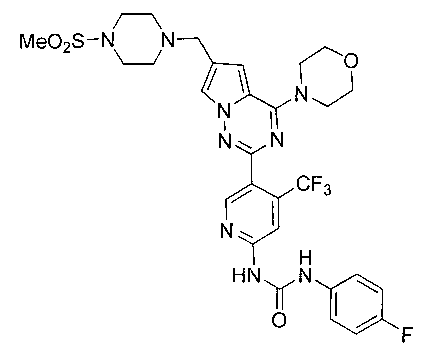
I-28 получали из 30 мг I-27 (0,055 ммоль) в качестве исходного вещества. Способ получения был тем же самым как синтез соединения I-1. Препаративную пластину (дихлорметан: метанол = 10:1) использовали для очистки. Белое твердое вещество (7 мг, 18,6%). т.пл. 204-205°C. 1H ЯМР (300 МГц, CDCl3): δ 11,38 (s, 1Н), 9,37 (s, 1Н), 8,75 (s, 1Н), 7,62 (s, 1Н), 7,57 (dd, J=8,5, 4,4 Гц, 2Н), 7,33 (s, 1Н), 7,06 (t, J=8,5 Гц, 2Н), 6,72 (s, 1Н), 4,10-4,01 (m, 4Н), 3,89-3,81 (m, 4Н), 3,67 (s, 2Н), 3,30 (br s, 4H), 2,79 (s, 3Н), 2,64 (br s, 4H). ESI-MS: 678 (M+1).
31. Получение 5-(4-(8-окса-3-азабицикло[3,2,1]октан-3-ил)-6-((4-(метилсульфонил)пиперазин-1-ил)метил)пирроло[2,1-f][1,2,4]триазин-2-ил)-4-(трифторметил)пиридин-2-амина (I-29)
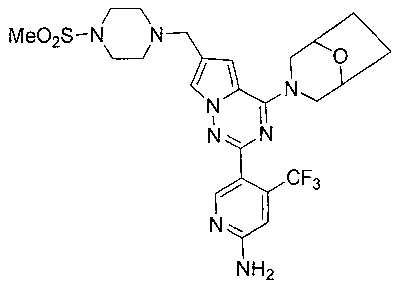
I-29 получали из 100 мг соединения 24b (0,17 ммоль). Способ получения был тем же самым как способ получения соединения 13. Препаративную пластину (дихлорметан: метанол = 20:1) использовали для очистки. Белое твердое вещество (17 мг, 17,4%). т.пл. 138°C. 1H ЯМР (300 МГц, CDCl3): δ 8,50 (s, 1Н), 7,57 (d, J=1,2 Гц, 1Н), 6,79 (s, 1Н), 6,63 (d, J=1,2 Гц, 1Н), 4,92 (s, 2Н), 4,48 (br s, 4Н), 3,61 (s, 2Н), 3,52 (br s, 1Н), 3,47 (br s, 1Н), 3,26 (t, J=4,8 Гц, 4H), 2,77 (s, 3Н), 2,60 (t, J=4,8 Гц, 4H), 2,00-1,96 (m, 2Н), 1,89-1,79 (m, 2Н). MS (EI) m/e (%): 566 (M+, 5).
32. Получение 1-этил-3-(5-(6-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-морфолинопирроло[2,1-f][1,2,4]триазин-2-ил)-4-(трифторметил)пиридин-2-ил)мочевины (1-30)
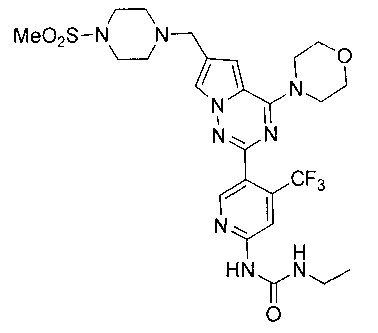
К раствору I-27 (110 мг, 0,2 ммоль) добавляли этилизоцианат (85 мг, 1,2 ммоль) и 1,8-диазацикло[5,4,0]ундец-7-ен (DBU, 183 мг, 1,2 ммоль) в дихлорметане и перемешивали в течение двух суток при комнатной температуре. Для перекристаллизации использовали диэтиловый эфир, и получали 74 мг (60,5%) белого твердого вещества. т.пл. 208°C (декомпозиция). 1Н ЯМР (300 МГц, DMSO-d6): δ 9,99 (br s, 1Н), 8,62 (s, 1Н), 8,18 (s, 1Н), 7,78 (br s, 1Н), 7,73 (s, 1Н), 6,95 (s, 1Н), 3,98 (br s, 4Н), 3,73 (br s, 4Н), 3,59 (s, 2Н), 3,33 (br s, 2Н), 3,16 (q, J=7,5 Гц, 2H), 3,11 (br s, 4H), 2,86 (s, 3Н), 1,62 (br s, 2H), 1,09 (t, J=7,5 Гц, 3Н). LC-MS: 612 (M+1).
33. Получение соединения (I-31 ~ I-33)
Общий способ получения: К раствору I-27 (54 мг, 0,1 ммоль) и триэтиламина (101 мг, 1 ммоль) в хлороформе добавляли соответствующий хлороформиат (0,3 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение четырех суток. Неочищенный продукт очищали при помощи колоночной хроматографии с дихлорметаном/метанолом (10:1).
Фенил (5-(6-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-морфолинопирроло[2,1-f][1,2,4]триазин-2-ил)-4-(трифторметил)пиридин-2-ил)карбамат (I-31)
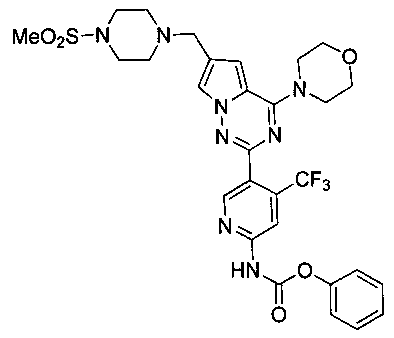
Светло-желтое твердое вещество. Выход 37,1%. т.пл. 108-110°C. 1Н ЯМР (300 МГц, DMSO-d6): δ 11,42 (s, 1Н), 8,79 (s, 1Н), 8,27 (s, 1Н), 7,76 (s, 1Н), 7,49-7,44 (m, 2Н), 7,32-7,27 (m, 3Н), 6,98 (s, 1Н), 3,99 (br s, 4Н), 3,74 (br s, 4H), 3,60 (s, 2H), 3,31 (br s, 4H), 3,12 (br s, 4H), 2,87 (s, 3H). LC-MS: 661 (M+1).
Этил (5-(6-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-морфолинопирроло[2,1-f][1,2,4]триазин-2-ил)-4-(трифторметил)пиридин-2-ил)карбамат (I-32)
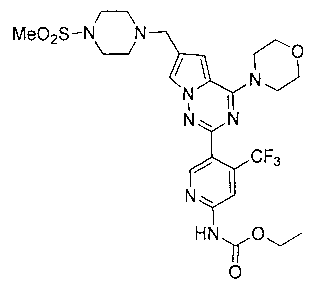
Белое твердое вещество. Выход 50,7%. т.пл. 211-213°C. 1H ЯМР (300 МГц, CDCl3): δ 8,71 (s, 1Н), 8,41 (s, 1Н), 7. 92(s, 1Н), 7,60 (s, 1Н), 6,67 (s, 1Н), 4,30 (q, J=7,2 Гц, 2Н), 4,05 (t, J=4,5 Гц, 4Н), 3,83 (t, J=4,5 Гц, 4Н), 3,63 (s, 2Н), 3,26 (br s, 4Н), 2,78 (s, 3Н), 2,61 (br s, 4Н), 1,36 (t, J=7,2 Гц, 3Н). LC-MS: 613(М+1).
Метил (5-(6-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-морфолинопирроло[2,1-f][1,2,4]триазин-2-ил)-4-(трифторметил)пиридин-2-ил)карбамат (I-33)
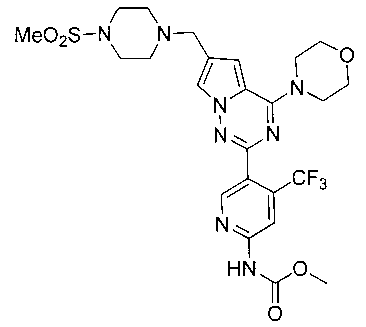
Белое твердое вещество (43,4%). т.пл. 212-214°C. 1H ЯМР (300 МГц, CDCl3): δ 8,70 (s, 1Н), 8,40 (s, 1Н), 7. 60 (d, J=1,5 Гц, 1Н), 6,67 (s, 1Н), 4,05 (t, J=4,5 Гц, 4Н), 3,85 (s, 3Н), 3,83 (t, J=4,5 Гц, 4Н), 3,63 (s, 2Н), 3,27 (br s, 4Н), 2,78 (s, 3Н), 2,61 (br s, 4Н). LC-MS: 599 (М+1).
34. Получение (S)-5-(4-(3-метилморфолино)-6-((4-(метилсульфонил)пиперазин-1-ил)метил)пирроло[2,1-f][1,2,4]триазин-2-ил)-4-(трифторметил)пиридин-2-амина (I-34)
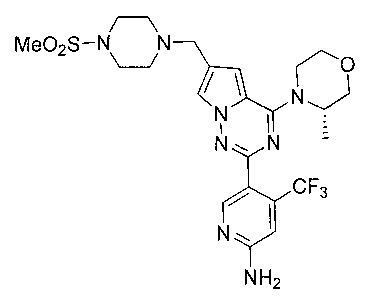
При -40°C 1М боран-THF (80 мл) добавляли по каплям к раствору соединения 24с (1,8 г, 3,17 ммоль) в 50 мл тетрагидрофурана. После прохождения реакции в течение 0,5 часов при этой температуре реакционную систему кипятили с обратным холодильником в течение 2 часов, и затем охлаждали до температуры меньше 0°C. По каплям добавляли 100 мл концентрированной соляной кислоты и после этого кипятили с обратным холодильником в течение 1 часа. Когда большую часть соляной кислоты удаляли при помощи роторного испарителя, рН раствора корректировали до приблизительно 8 путем использования насыщенного раствора карбоната натрия. После трехкратного экстрагирования этилацетатом органическую фазу сушили над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали при помощи колоночной хроматографии с дихлорметаном/метанолом (40:1) с получением белого твердого вещества (56,9%). т.пл. 262°C. 1H ЯМР (300 МГц, DMSO-d6): δ 8,34 (s, 1Н), 7,68 (s, 1Н), 6,88 (s, 1Н), 6,82 (s, 1Н), 6,77 (s, 2Н), 4,87 (br s, 1Н), 4,47 (br s, 1Н), 3,96 (br s, 1H), 3,77-3,41 (m, 8H), 3,11 (s, 6H), 2,87 (s, 3Н), 1,32 (s, 3Н). LC-MS: 555 (М+1).
35. Получение (S)-метил (5-(4-(3-метилморфолино)-6-((4-(метилсульфонил)пиперазин-1-ил)метил)пирроло[2,1-f][1,2,4]триазин-2-ил)-4-(трифторметил)пиридин-2-ил)карбамата (I-35)
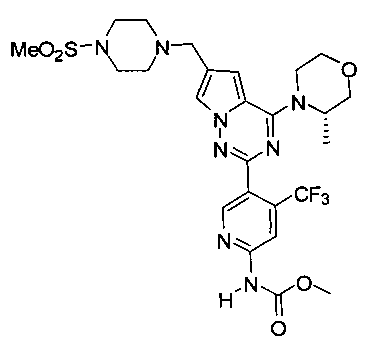
При -40°C метилхлороформиат (28,85 ммоль) добавляли к раствору I-34 (880 мг, 1,44 ммоль) и триэтиламина (1,5 г, 14,4 ммоль) в хлороформе и перемешивали в течение 2 ч. Неочищенный продукт очищали при помощи колоночной хроматографии с дихлорметаном/метанолом (60: 1) с получением белого твердого вещества (44,2%). т.пл. 150-152°C. 1Н ЯМР (400 МГц, CDCl3): δ 8,70 (s, 1Н), 8,41 (s, 1Н), 8,22 (s, 1Н), 7,60 (s, 1Н), 6,67 (s, 1Н), 4,92 (br s, 1Н), 4,56 (br s, 1Н), 4,03 (d, J=7,9 Гц, 1Н), 4,00-3,52 (m, 9Н), 3,27 (s, 4Н), 2,78 (s, 3Н), 2,62 (s, 4Н), 1,47 (d, J=6,6 Гц, 3Н). LC-MS: 613 (М+1).
36. Получение (S)-1-этил-3-(5-(4-(3-метилморфолино)-6-((4-(метилсульфонил)пиперазин-1-ил)метил)пирроло[2,1-f][1,2,4]триазин-2-ил)-4-(трифторметил)пиридин-2-ил)мочевины (I-36)
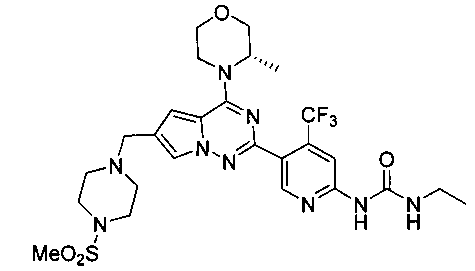
Соединение I-36 получали при помощи того же самого способа как способ получения соединения I-30. Белое твердое вещество, 1Н ЯМР (300 МГц, CDCl3) δ 9,45 (s, 1Н), 9,01 (br s, 1Н), 8,63 (s, 1Н), 7,59 (s, 1Н), 7,30 (s, 1Н), 6,67 (s, 1Н), 4,93 (br s, 1Н), 4,56 (br s, 1Н), 4,03 (d, J=7,5 Гц, 1Н), 3,89-3,69 (m, 2Н), 3,64 (s, 2Н), 3,44 (q, J=6,9 Гц, 2Н), 3,39-3,11 (m, 6Н), 2,78 (s, 3Н), 2,62 (s, 4Н), 1,47 (d, J=6,7 Гц, 3Н), 1,26 (t, J=7,2 Гц, 3Н).
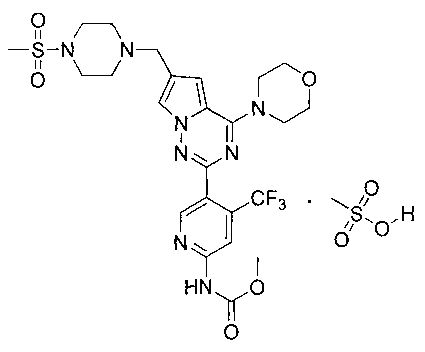
37. 1-Этил-3-(5-(6-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-морфолинопирроло[2,1-f][1,2,4]триазин-2-ил)-4-(трифторметил)пиридин-2-ил)мочевины мезилат
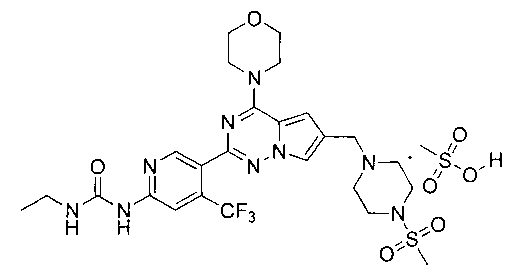
5% раствор метансульфоновой кислоты в тетрагидрофуране (600 мкл, 0,47 ммоль) добавляли к раствору I-30 (220 мг, 0,36 ммоль) в 10 мл хлороформа и перемешивали в течение 1 ч при комнатной температуре. Чистый продукт получали путем фильтрования. Белый порошок (99,0%). т.пл. 220°C (декомпозиция). 1H ЯМР (300 МГц, DMSO-d6): δ 9,83 (br s, 1Н), 9,63 (s, 1Н), 8,63 (s, 1Н), 8,12 (s, 1Н), 7,94 (s, 1Н), 7,37 (s, 1Н), 7,17 (s, 1Н), 4,43 (s, 2Н), 4,01 (s, 4Н), 3,75 (s, 6Н), 3,50 (s, 2Н), 3,28-3,01 (m, 6Н), 3,0 (s, 3Н), 2,32 (s, 3Н), 1,08 (t, J=7,2 Гц, 3Н). 13C ЯМР (126 МГц, DMSO) δ 155,03, 154,57, 153,91, 153,00, 150,65, 136,78 (q, J=32,1 Гц, CF3C), 123,24 (q, J=274,8 Гц, CF3), 123,51, 121,78, 113,99, 113,09, 108,30 (q, J=5,2 Гц, CF3CCH), 107,81, 66,37, 52,23, 50,49, 46,00, 42,93, 35,55, 34,45, 15,67, 15,67.
38. Метил (5-(6-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-морфол инопирроло[2,1-f][1,2,4]триазин-2-ил)-4-(трифторметил)пиридин-2-ил)карбамата мезилат
5% раствор метансульфоновой кислоты в тетрагидрофуране (978 мкл, 0,77 ммоль) добавляли к раствору I-33 (300 мг, 0,50 ммоль) в 15 мл тетрагидрофурана и перемешивали в течение 5 ч при комнатной температуре. Диэтиловый эфир добавляли в систему до осаждения большого количества твердых веществ. Чистый продукт получали путем фильтрования. Белое твердое вещество (100%). т.пл. 240°C (декомпозиция)., 1Н ЯМР (300 МГц, DMSO-d6): δ 10,94 (s, 1 Н), 9,82 (s, 1 Н), 8,74 (s, 1 Н), 8,32 (s, 1 Н), 7,99 (s, 1Н), 7,19 (s, 1 Н), 4,46 (s, 2 Н), 4,03 (t, J=4,8 Гц, 4 Н), 3,78 (t, J=4,8 Гц, 4Н), 3,75 (s, 3 Н), 3,67-3,43 (m, 4Н), 3,27-3,05 (m, 4 Н), 3,02 (s, 3 Н), 2,33 (s, 3 Н). 13C ЯМР (126 МГц, DMSO-d6): δ 154,58, 154,16, 153,88, 152,90, 151,07, 137,029 (q, J=31 Гц, CF3C), 125,19, 123,23 (q, J=274,9 Гц, CF3), 121,84, 113,99, 113,07, 108,60 (q, J=5 Гц, CF3CH), 107,88, 66,35, 52,78, 52,22, 50,49, 46,03, 42,95, 35,57, 25,59.
39. (S)-метил (5-(4-(3-метилморфолино)-6-((4-(метилсульфонил)пиперазин-1-ил)метил)пирроло[2,1-f][1,2,4]триазин-2-ил)-4-(трифторметил)пиридин-2-ил)карбамата мезилат
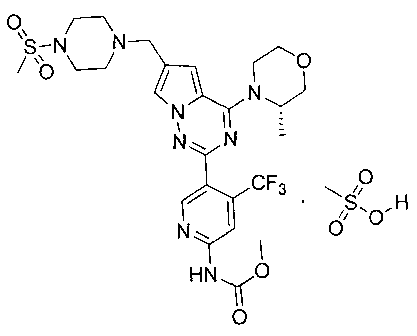
5% раствор метансульфоновой кислоты в тетрагидрофуране (589 мкл, 0,46 ммоль) добавляли к раствору I-35 (231 мг, 0,38 ммоль) в 15 мл тетрагидрофурана и перемешивали в течение 3 ч при комнатной температуре. Тетрагидрофуран удаляли при пониженном давлении с получением геля. Небольшое количество метанола добавляли для растворения геля. А затем диэтиловый эфир добавляли по каплям до осаждения твердого вещества. Чистый продукт получали путем фильтрования. Белый порошок (80,72%). т.пл. 210°C (декомпозиция). 1H ЯМР (300 МГц, DMSO-d6): δ 10,91 (s, 1Н), 9,86 (s, 1Н), 8,73 (s, 1Н), 8,31 (s, 1Н), 7,98 (s, 1Н), 7,17 (s, 1Н), 4,90 (br s, 1Н), 4,45 (br s, 3Н), 4,00 (d, J=7,9 Гц, 1H), 3,88-3,61 (m, 7Н), 3,53 (s, 4Н), 3,15 (br s, 4H), 3,01 (s, 3Н), 2,41-2,26 (m, 3Н), 1,39 (br s, 3Н). 13C ЯМР (126 МГц, DMSO-d6): δ 154,60, 154,15, 153,68, 152,96, 151,09, 136,91 (q, J=32,5 Гц, CF3C), 125,26, 123,24 (q, J=274,9 Гц, CF3) 121,89, 114,05, 113,09, 108,60 (q, J=5,5 Гц, CF3CH), 107,95, 70,51, 66,46, 52,78, 52,20, 50,48, 42,93, 35,58, 31,17, 13,19.
Тестирование биологической активности
Ингибирование активности PI3Kα киназы
Экспериментальный способ
Активность очищенной киназы определяли при помощи люминесцентного анализа Kinase-Glo® Plus путем измерения количества оставшейся киназы в растворе после завершения киназной реакции. Киназную реакцию осуществляли в 384-луночном белом планшете (Greiner), 1 мкл тестируемого соединения или DMSO (диметилсульфоксид) в качестве контроля добавляли в каждую лунку, содержащую 5 мкл реакционного буфера [10 мМ Tris-HCl рН 7,5, 50 мМ NaCl, 3 мМ MgCl2, 1 мМ DTT (дитиотреитол), 0,05% CHAPS (3-[(3-холамидопропил)диметиламмонио]-1-пропансульфонат, 3-(3-холаминопропил)-диметиламино-1-пропансульфоновая кислота), и реакционный буфер дополняли 12 мкМ субстратом, D-мио-фосфатидилинозитол-4,5-бисфосфатом (4,5-фосфатидилинозитолдифосфатом) и 2 мкМ АТР (аденозинтрифосфатом). И затем 4 мкл реакционного буфера, содержащего 62,5 нМ PI3Ka или контроля, не представляющего собой PI3Ka, добавляли для инициации киназной реакции. После осуществления реакции в течение 1 часа при комнатной температуре 10 мкл смеси Kinase Glo-Plus добавляли и инкубировали в течение 1 часа для гашения реакции. Величину хемилюминесценции обнаруживали с использованием многофункционального микропланшетного ридера EnVision 2104 (Perkinelmer).
Результаты экспериментов
Результаты экспериментов. (таблица 2) продемонстрировали, что ингибирующие активности следующих семи соединений по настоящему изобретению в отношении PI3Kα были сравнимы или превосходили активность PI-103. Они представляли собой соответственно I-22 (9,59 нМ), I-27 (8,37 нМ), I-28 (9,30 нМ), I-30 (8,19 нМ), I-32 (7,15 нМ), I-33 (2,84 нМ), и I-35 (5,67nM). 
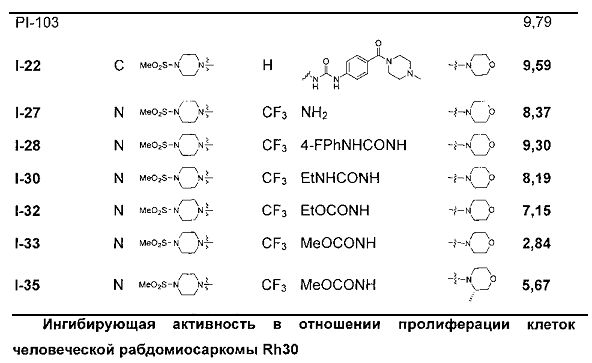
Клетки Rh30 высевали в 96-луночный планшет в количестве 3000 клеток/лунку. После прикрепления клеток добавляли тестируемые соединения в концентрации 10, 3, 1, 0,3, или 0,1 мкМ и инкубировали в течение 72 ч. Культуральные среды удаляли, и клетки фиксировали трихлоруксусной кислотой. После пятикратного промывания дистиллированной водой и сушки добавляли сульфонилродамин В. После пятикратного промывания 1% ледяной уксусной кислотой и сушки добавляли тригидроксиметиламинометановый буфер. Величину OD (оптической плотности) измеряли путем использования микропланшетного ридера при длине волны 560 нм. Рассчитывали скорость ингибирования, и результаты представлены в таблице 3. Вышеприведенные результаты продемонстрировали сильную ингибирующую в отношении пролиферации клеток Rh30, где величины IC50 для I-24, I-25 и I-28 даже достигали десятков наномоль.
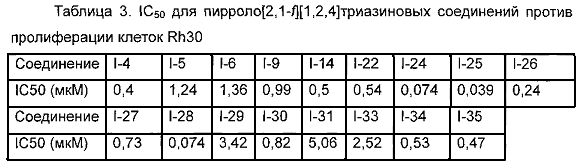
Действия I-33 в отношении сигнальных путей PI3K в клетках человеческой рабдомиосаркомы Rh30 и клетках человеческой глиомы U87MG
Экспериментальный способ
Клетки Rh30 и U87MG засевали в 12-луночные планшеты в количестве 2×105 клеток/лунку. На следующие сутки клетки инкубировали в свежих бессывороточных культуральных средах, подвергали выращиванию на минимальных средах в течение 24 часов. Клетки затем обрабатывали различными концентрациями соединения I-33 в течение 1 ч. После стимулирования IGF-1 в течение 10 минут лизированные клетки собирали, и добавляли 4× загрузочный буфер с SDS (додецилсульфат натрия) [200 мМ Tris. Cl (рН 6,8), 400 мМ DTT, 8% SDS (додецилсульфат натрия), 0,4% бромфеноловый синий, 40% глицерин]. Клеточные лизаты кипятили в течение 10 минут. Аликвоту наносили на полиакриламидный гель и осуществляли электрофорез в Tris-глициновом буфере для электрофореза (25 мМ Tris, 250 мМ глицин, 0,1% SDS) при 80-100В в течение 2-2,5 часов. Белки переносили из геля на нитроцеллюлозную мембрану путем использования способа полусухого переноса. Мембрану блокировали блокирующим раствором, содержащим 5% сухое обезжиренное молоко (5% сухое обезжиренное молоко, 20 мМ Tris-HCl (рН 7,2-7,4), 150 мМ NaCl, 0,1% (об./об.) Tween20) в шейкере при комнатной температуре в течение 2 часов. Затем добавляли специфическое первичное антитело и гибридизовали при 4°C в течение ночи. Мембрану трижды промывали промывающим буфером [20 мМ Tris-HCl (рН 7,2-7,4), 150 мМ NaCl, 0,1% (об./об.) Tween20] при комнатной температуре, по 15 минут каждый раз. Добавляли вторичное антитело, меченое пероксидазой хрена, и систему помещали в шейкер и осторожно встряхивали при комнатной температуре в течение 1 часа. После тройного промывания промывающим буфером мембрану инкубировали с хемилюминесцентным субстратом SuperSignal West Dura (Pierce Inc, Rochford, IL). И затем мембрану проявляли и фиксировали, и получали изображения.
Результаты экспериментов
Результаты экспериментов (фиг. 1) продемонстрировали, что I-33 значительно ингибировал трансдукцию через сигнальные пути PI3K в клетках человеческой рабдомиосаркомы Rh30 и клетках человеческой глиомы U87MG.
Ингибирующая активность I-30 и I-33 на пролиферацию человеческих раковых клеток из различных тканей
I-30 и I-33 выбраны для тестирования их активности на пролиферацию человеческих раковых клеток, происходящих из различных типов тканей. Результаты представлены в таблице 4.
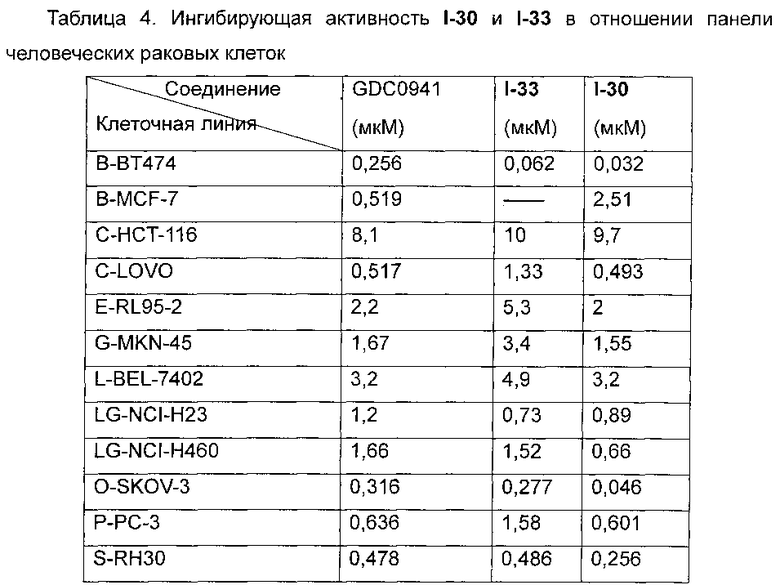
Как представлено в таблице 4, активность соединений I-30 и I-33 сравнима с активностью положительного контроля GDC0941 (приобретенного в Shanghai han-xiang chemical со., LTD., димезилат) в отношении пролиферации различных клеточных линий, таких как клетки рака толстой кишки С-НСТ-116, клетки колоректального рака C-LOVO, клетки рака эндометрия E-RL95-2, клетки рака желудка G-MKN-45, клеток гепатомы L-BEL-7402 и клеток рабдомиосаркомы S-RH30. Тем не менее, активности соединений значительно сильнее положительного контроля в отношении ингибирования пролиферации клеток В-ВТ474.
Ингибирующие действия в отношении роста ксенотрансплантата человеческой нейроглиомы U87-MG, подкожно трансплантированного бестимусным мышам
Экспериментальный способ
Хорошо развивающиеся опухоли у бестимусных мышей нарезали на фрагменты 1,5 мм3 и подкожно трансплантировали в правый бок бестимусных мышей в стерильных условиях. Диаметры опухолей, подкожно трансплантированных бестимусным мышам, измеряли при помощи штангенциркуля. Когда опухоль достигала объема 100-200 мм3, мышей произвольным образом распределяли на несколько групп: группа I-33 мезилат 50 мг/кг и группа 25 мг/кг; группа I-30 мезилат 50 мг/кг; группа GDC0941 димезилат 50 мг/кг и контрольная группа. Контрольным группам давали то же самое количество растворителя в качестве бланка, и обрабатываемые группы получали тестируемые соединения (перорально). Соединения вводили один раз в сутки в течение трех недель. В процессе эксперимента размер опухолей измеряли дважды в неделю и одновременно определяли массы мышей. Объем опухоли (TV) рассчитывали следующим образом: TV=1/2×a×b2, где a и b представляют соответственно длину и ширину. Индивидуальный относительный объем опухоли (RTV) рассчитывали следующим образом: RTV=Vt/V0, где V0 представляет собой объем в начале обработки и Vt представляет объем, измеряемый в конкретный момент времени. Индекс оценки противоопухолевой активности представлял собой относительную скорость пролиферации опухоли Т/С (%), формула для расчета которой была следующей: Т/С(%)=(TRTV/CRTV)×100%, TRTV: RTV обрабатываемой группы; CRTV: RTV отрицательной контрольной группы.
Результаты экспериментов
Результаты экспериментов представлены в таблице 5 и фиг. 2. I-33 мезилат перорально вводили в дозе 50 мг/кг или 20 мг/кг каждые сутки. Через одну неделю рост опухоли в обрабатываемой группе I-33 значительно замедлялся. После последовательного введения в течение трех недель мезилат I-33 значительно ингибировал рост подкожно трансплантированного бестимусным мышам ксенотрансплантата U87mG (фиг. 2). Величина Т/С на 21е сутки составляла 25,65% и 32,61%, соответственно. Мезилат I-33 демонстрировал более сильную активность по сравнению с GDC0941 (50 мг/кг) в отношении ингибирования опухолевого роста. Мезилат I-30 в концентрации 50 мг/кг также демонстрировал значительные ингибирующие действия в отношении опухолевого роста. Величина Т/С для мезилата 1-30 в концентрации 50 мг/кг на 21е сутки составила 37,26%, тогда как аналогичная величина для группы GDC0941 50 мг/кг составила 51,40%.
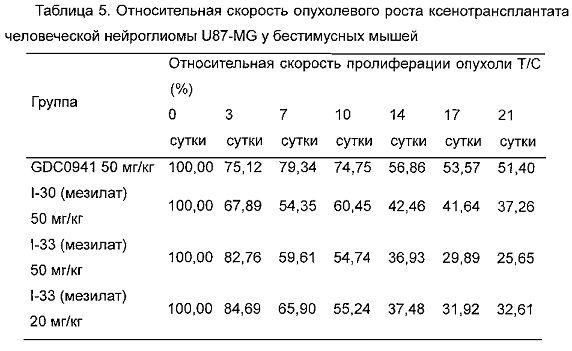
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ 2-АРИЛБЕНЗОФУРАН-7-ФОРМАМИДА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2013 |
|
RU2583900C2 |
| ИНГИБИТОР FGFR И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2018 |
|
RU2771311C2 |
| СОЕДИНЕНИЯ НА ОСНОВЕ 7-ЗАМЕЩЕННОГО ПИРРОЛОТРИАЗИНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2018 |
|
RU2745548C1 |
| ГИДРОКСИПУРИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2685417C1 |
| АГОНИСТ S1P1 И ЕГО ПРИМЕНЕНИЕ | 2017 |
|
RU2754845C2 |
| АНАЛОГ ПИРИДИНО[1,2-А]ПИРИМИДОНА, ИСПОЛЬЗУЕМЫЙ В КАЧЕСТВЕ ИНГИБИТОРА mTOR/PI3K | 2015 |
|
RU2658912C1 |
| СОЕДИНЕНИЕ НАФТИЛАМИДА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2015 |
|
RU2655607C2 |
| СИНТЕЗ И ПРОТИВОРАКОВАЯ АКТИВНОСТЬ ПРОИЗВОДНЫХ АРИЛ И ГЕТЕРОАРИЛХИНОЛИНОВ | 2011 |
|
RU2584688C2 |
| ХИНОЛИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ SMO | 2015 |
|
RU2695815C2 |
| Ингибиторы ErbB/BTK | 2019 |
|
RU2764069C1 |
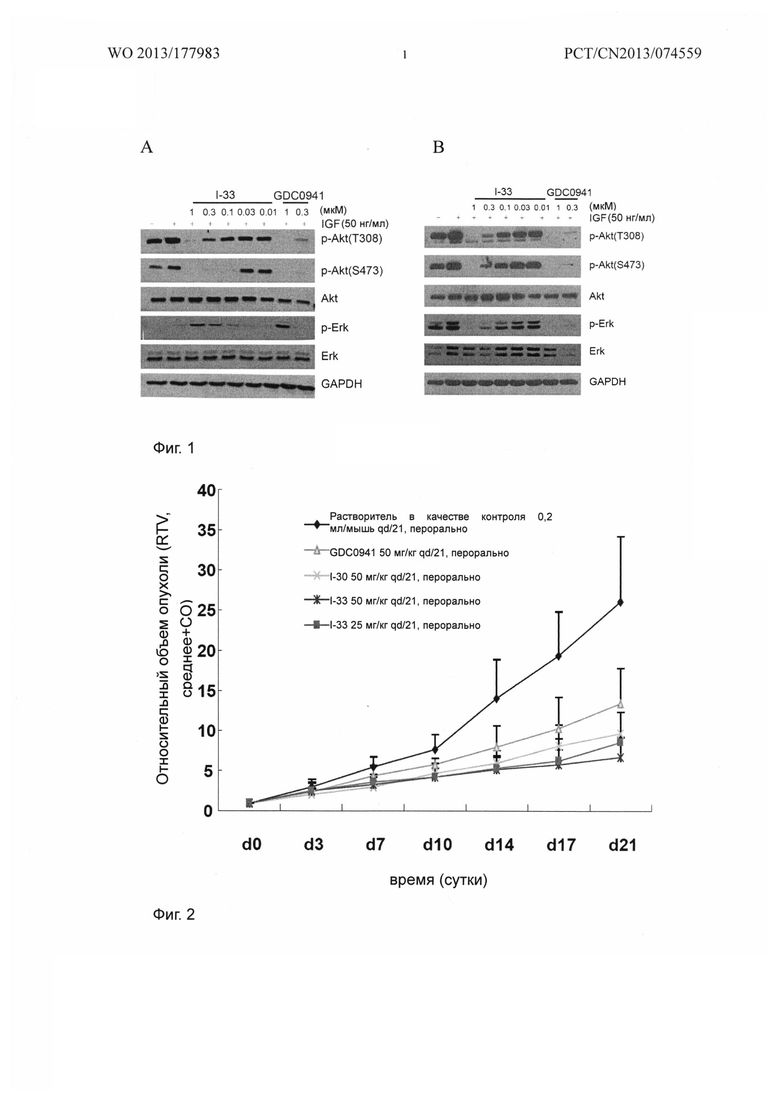
Изобретение относится к пирроло[2,1-f][1,2,4]триазиновому соединению общей формулы I, его изомеру, фармацевтически приемлемой соли, сложному эфиру или гидрату. Соединение общей формулы I обладает свойствами ингибитора активности киназы PI3K. В общей формуле I
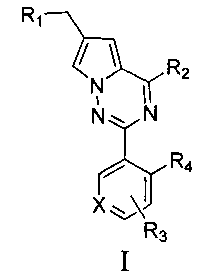
X представляет собой СН или N; R1 представляет собой -NR5R6; R2 представляет собой 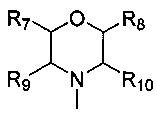 ; R3 представляет собой -NH2, -NHC(O)NHR11, -NHC(O)OR11, -NHC(O)R11, -CH2OH, -CH2S(O)2R12 или -CH2NHS(O)2R12; R4 представляет собой H или CF3. Изобретение также относится к способу получения пирроло[2,1-f][1,2,4]триазиновых соединений общей формулы I и к их применению в получении лекарственных средств для лечения заболевания, связанного с фосфатидилинозитол 3-киназой и ингибитора белка-мишени рапамицина. Технический результат: получены новые пирроло[2,1-f][1,2,4]триазиновые соединения общей формулы I, обладающие свойствами ингибитора активности киназы PI3K. 3 н. и 8 з.п. ф-лы, 2 ил., 5 табл., 39 пр.
; R3 представляет собой -NH2, -NHC(O)NHR11, -NHC(O)OR11, -NHC(O)R11, -CH2OH, -CH2S(O)2R12 или -CH2NHS(O)2R12; R4 представляет собой H или CF3. Изобретение также относится к способу получения пирроло[2,1-f][1,2,4]триазиновых соединений общей формулы I и к их применению в получении лекарственных средств для лечения заболевания, связанного с фосфатидилинозитол 3-киназой и ингибитора белка-мишени рапамицина. Технический результат: получены новые пирроло[2,1-f][1,2,4]триазиновые соединения общей формулы I, обладающие свойствами ингибитора активности киназы PI3K. 3 н. и 8 з.п. ф-лы, 2 ил., 5 табл., 39 пр.
1. Пирроло[2,1-f][1,2,4]триазиновое соединение общей формулы I, его изомер или его фармацевтически приемлемая соль, сложный эфир или гидрат
где
X представляет собой СН или N;
R1 представляет собой -NR5R6;
R2 представляет собой ;
R3 представляет собой -NH2, -NHC(O)NHR11, -NHC(O)OR11, -NHC(O)R11, -CH2OH, -CH2S(O)2R12 или -CH2NHS(O)2R12;
R4 представляет собой H или CF3;
каждый из R5 и R6 независимо представляет собой С1-С4алкил или вместе с атомом азота, к которому они присоединены, образуют пиперазинил, замещенный -S(O)2R12;
каждый из R7, R8, R9 и R10 независимо представляет собой Н или С1-С3алкил; альтернативно R7 и R8 образуют связанный мостиковыми связями бицикло-гетероцикл с морфолиновым кольцом;
R11 представляет собой С1-С4 алкил, бензил или бензил, замещенный одним или более чем одним заместителем, незамещенный фенил или фенил, замещенный одним или более чем одним заместителем, или изоксазолил, замещенный одним или более чем одним заместителем, и указанный один или более чем один заместитель выбран из галогена, С1-С3 алкила или С1-С3 алкоксила, -CF3, -C(O)OR12, -C(O)NR12R15), 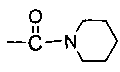 ,
, 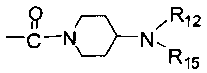 или
или 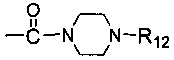 ;
;
каждый из R12 и R15 независимо представляет собой С1-С3алкил.
2. Пирроло[2,1-f][1,2,4]триазиновое соединение, его изомер или его фармацевтически приемлемая соль, сложный эфир или гидрат по п. 1, где соединение имеет следующую структуру, представленную общей формулой А или общей формулой В:
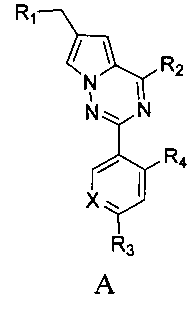 или
или 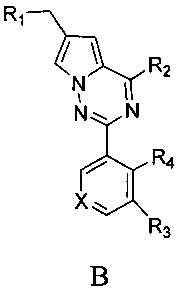
3. Пирроло[2,1-f][1,2,4]триазиновое соединение, его изомер или его фармацевтически приемлемая соль, сложный эфир или гидрат по п. 1 или 2, где
R2 представляет собой 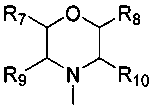 ;
;
R5 и R6 вместе с атомом азота, к которому они присоединены, образуют пиперазинил, замещенный -S(O)2R12.
4. Пирроло[2,1-f][1,2,4]триазиновое соединение, его изомер или его фармацевтически приемлемая соль, сложный эфир или гидрат по п. 3, где
R1 представляет собой диметиламино или 1-метилсульфонил пиперазинил;
R2 представляет собой  ,
, 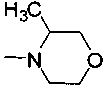 или
или 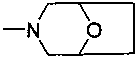 ;
;
R11 представляет собой метил, этил, пропил, трет-бутил, изобутил, 4-фторбензил, незамещенный фенил или фенил, замещенный одним или более чем одним заместителем, изоксазолил, замещенный одним или более чем одним заместителем, и заместитель выбран из фтора, хлора, трифторметила, метила, метокси, этоксикарбонила, диметиламинокарбонила, 4-метил-пиперазин-1-карбонила, пиперидин-1-карбонила и 4-диметиламино-пиперидин-1-карбонила.
5. Пирроло[2,1-f][1,2,4]триазиновое соединение, его изомер или его фармацевтически приемлемая соль, сложный эфир или гидрат по п. 1, где соединение имеет следующую структуру общей формулы:
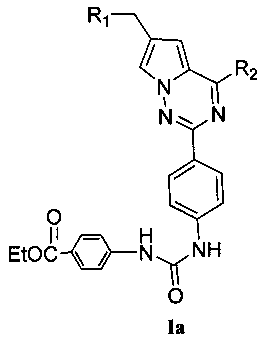 ,
, 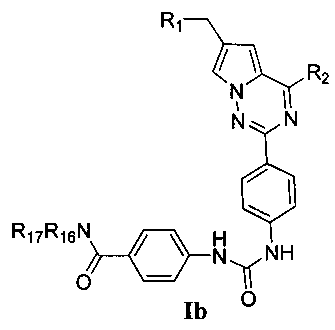 ,
, 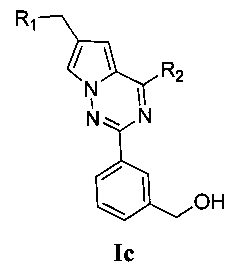 ,
,
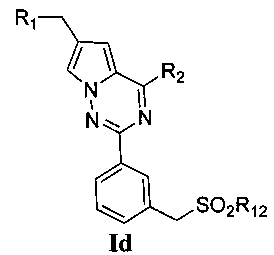 ,
, 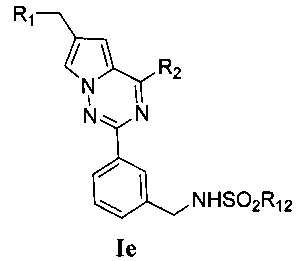 ,
, 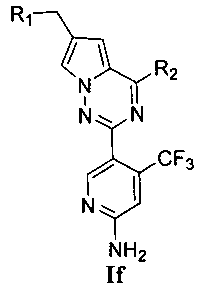 ,
,
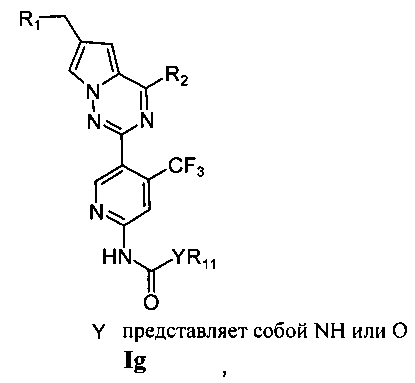
где R1, R2, R11 и R12 определены как в п. 1,
R16 и R17 идентичны или отличаются, и каждый независимо выбран из С1-С4 алкила, или R16 и R17 вместе с атомом азота, к которому они присоединены, образуют 4-метил-пиперазинил, 4-диметиламино-пиперидинил или пиперидин-1-ил.
6. Пирроло[2,1-f][1,2,4]триазиновое соединение, его изомер или его фармацевтически приемлемая соль, сложный эфир или гидрат по п. 1, где соединение имеет следующую структуру общей формулы:
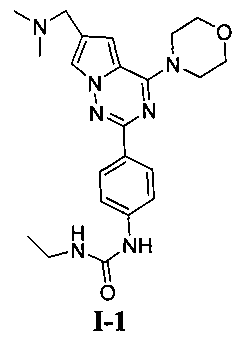
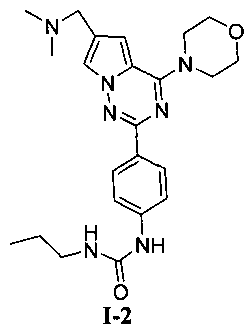
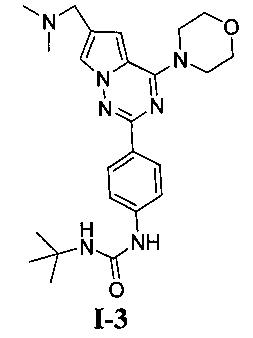
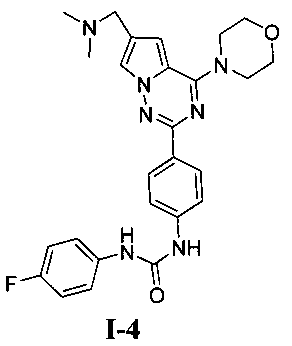
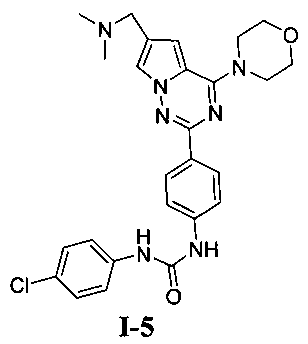
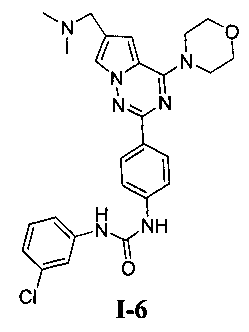
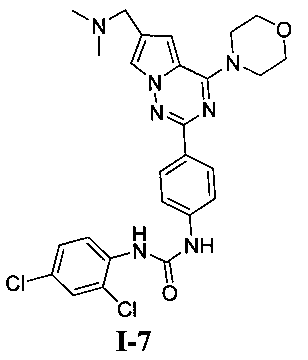
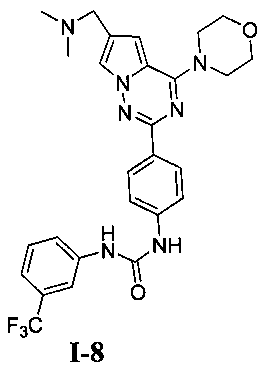
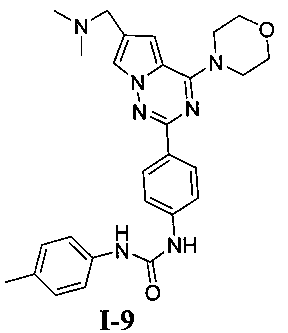
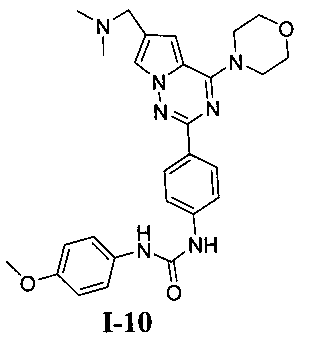
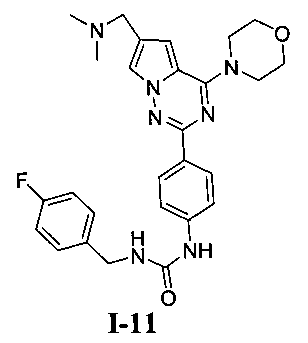
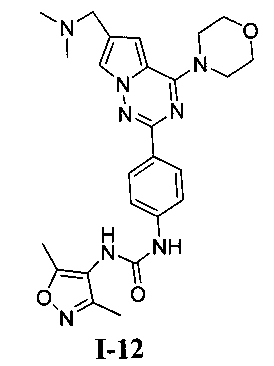
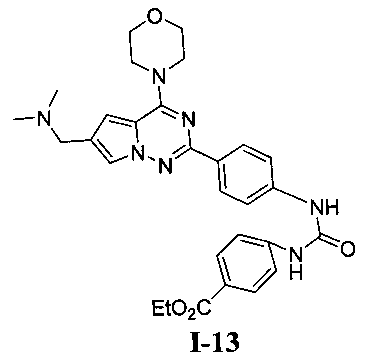
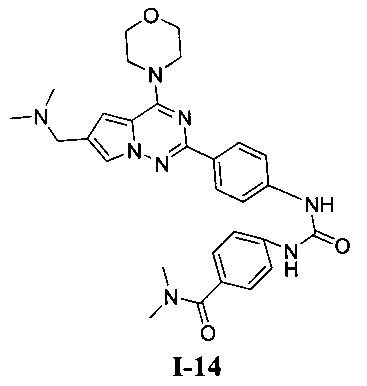
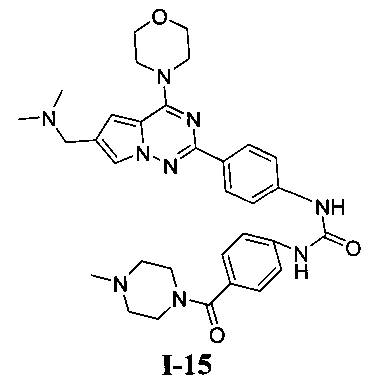
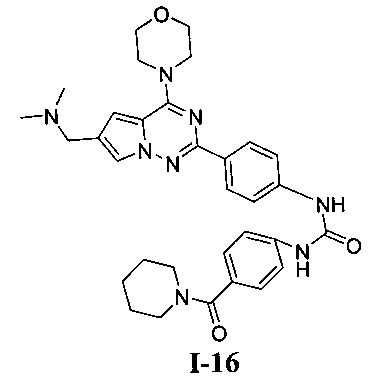
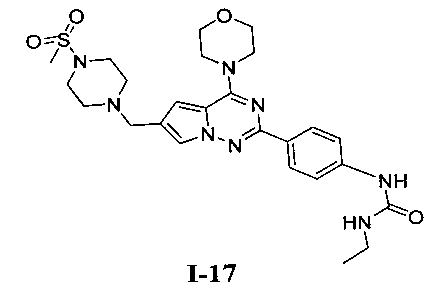
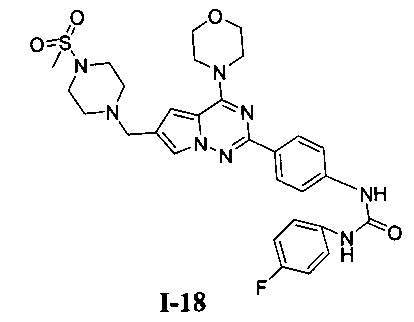
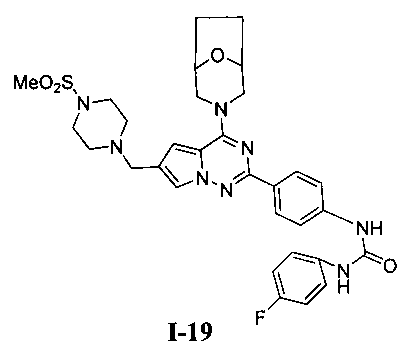
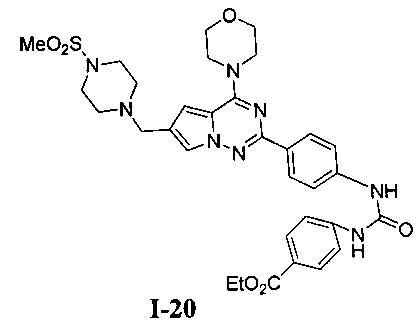
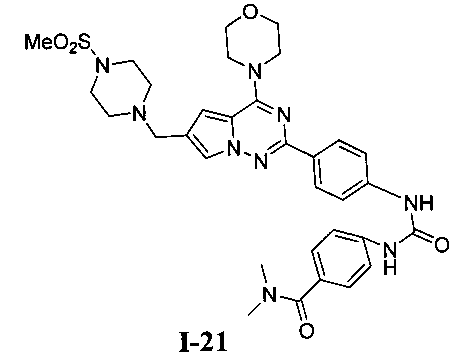
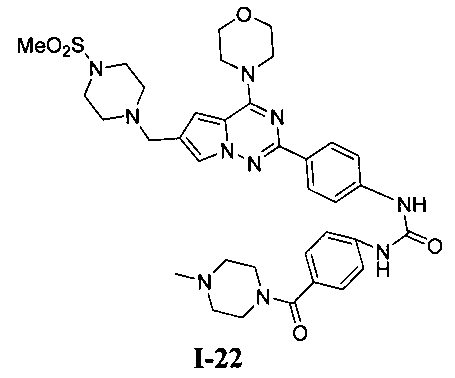
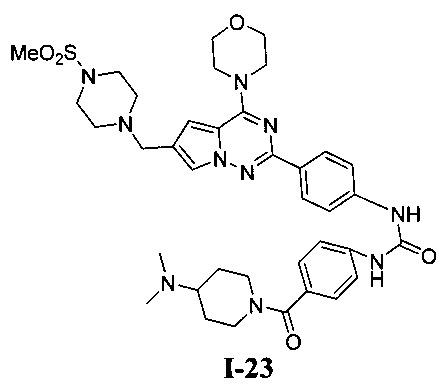
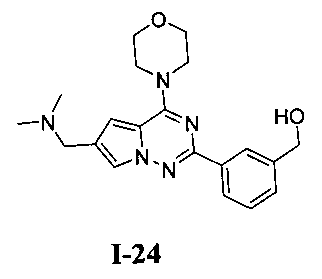
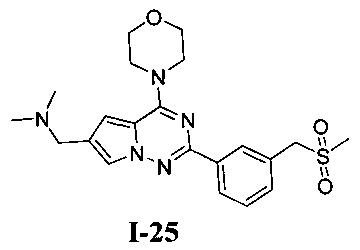
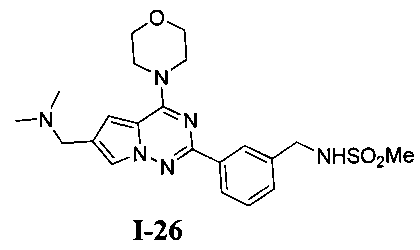
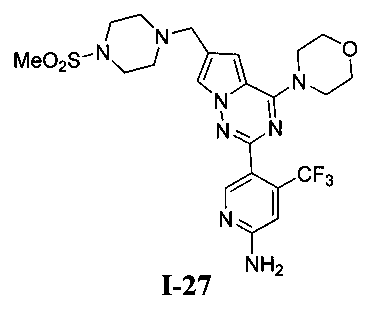
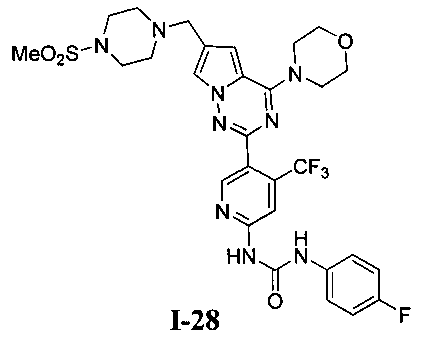
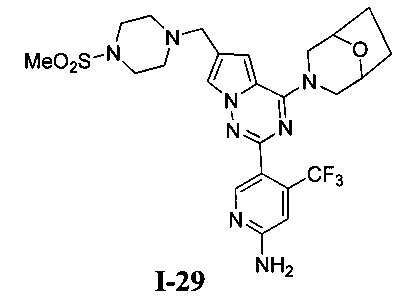
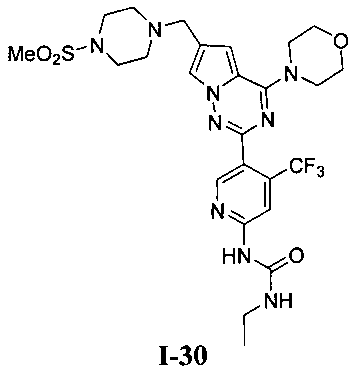
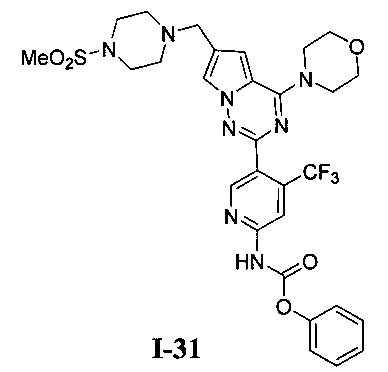
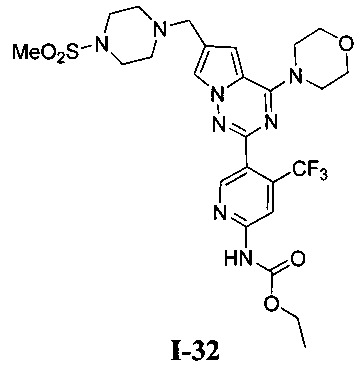
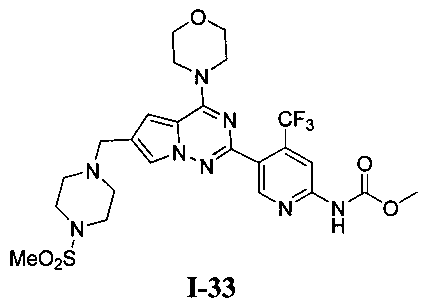
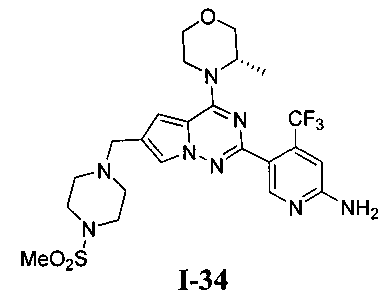
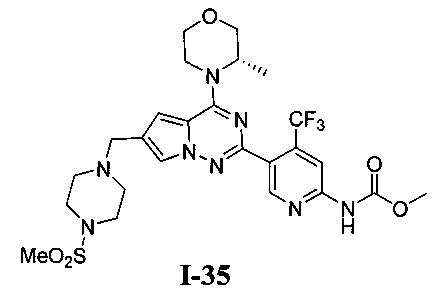
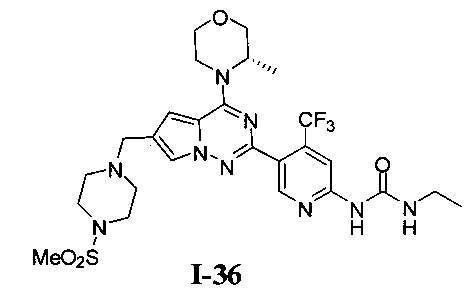
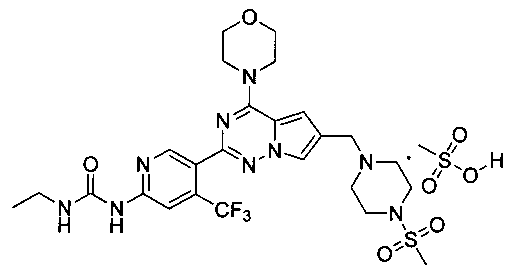
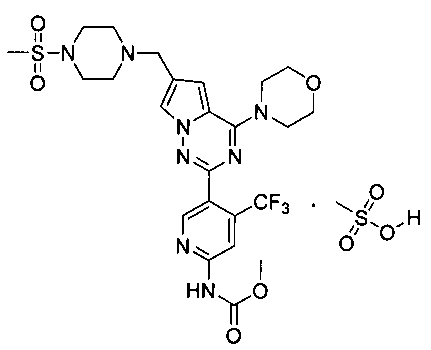
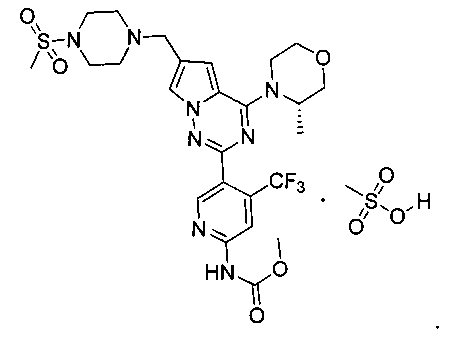
7. Способ получения пирроло[2,1-f][1,2,4]триазиновых соединений по п. 1, включающий следующие стадии:
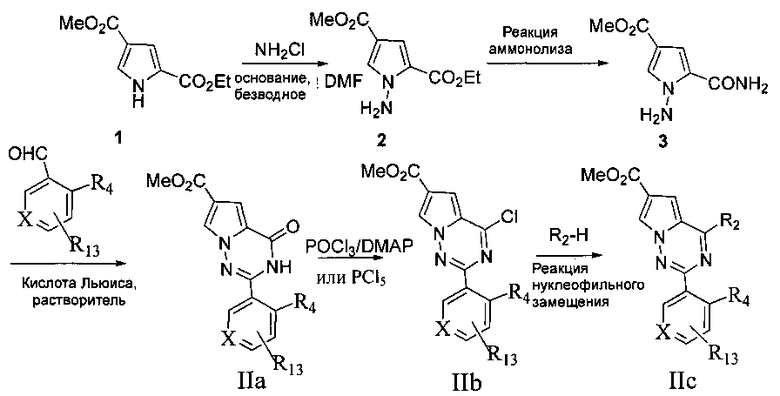
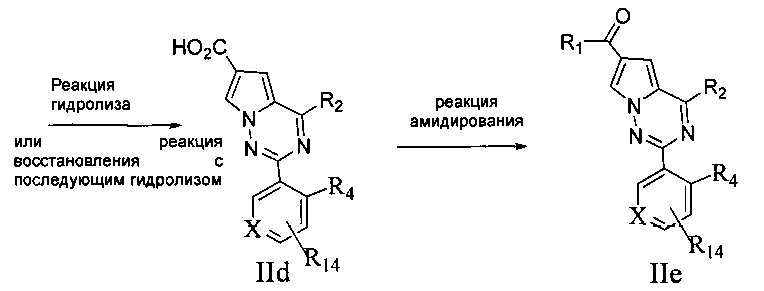
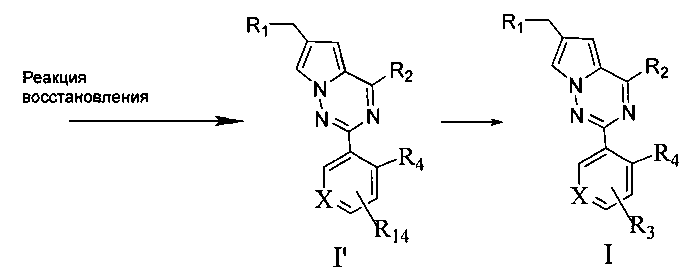
где R13 представляет собой нитро или -СН2ОАс; R14 представляет собой амино или -СН2ОН;
1) пиррольное производное 1 и хлорамин в безводном N,N-диметилформамиде подвергают реакции N-аминирования в присутствии основания с получением соединения 2;
2) соединение, представленное формулой 2, не подвергнутое очистке, подвергают аммонолизу с получением соединения 3;
3) соединение 3 подвергают взаимодействию с ароматическим альдегидом под действием кислоты Льюиса с металлом с получением соединения IIa, или соединение 3 и альдегид под действием кислоты Льюиса в качестве катализатора, такого как раствор трифторида бора в диэтиловом эфире, подвергают конденсации с получением основания Шиффа, и затем осуществляют окислительную циклизацию с получением соединения IIa;
4) соединение IIa подвергают реакции хлорирования в присутствии основания с получением соединения IIb;
5) соединение IIb и R2-H подвергают реакции нуклеофильного замещения с получением соединения IIc;
6) сложноэфирную группу соединения IIc подвергают реакции гидролиза; или нитрогруппу соединения IIc подвергают реакции восстановления, и затем сложноэфирную группу соединения IIc гидролизуют;
7) соединение IId подвергают взаимодействию с R1-H амином или пиперазином, замещенным S(O)2R12, с получением соединения IIe;
8) соединение IIe восстанавливают при помощи восстановителя с получением соединения I′;
9) соединение I′ далее подвергают реакции присоединения с R11NCO, реакции этерификации или реакции амидирования с R11OC(O)Cl, реакции этерификации или реакции амидирования с R11C(O)OH или R11C(O)Cl, или реакции этерификации или реакции амидирования с R12S(O)2Cl, с образованием соединений, представленных общей формулой I.
8. Способ получения по п. 7, где
основание на стадии 1) представляет собой гидрид натрия, карбонат калия или трет-бутоксид калия;
кислота Льюиса с металлом на стадии 3) может представлять собой реагент одновалентной или двухвалентной меди, и реакционная температура составляет 80-150°C;
хлорирующий агент на стадии 4) представляет оксихлорид фосфора или пентахлорид фосфора, и используемое основание представляет собой N,N-диметиланилин или 4-диметиламинопиридин;
амин на стадии 7) представляет собой диметиламин или 1-метилсульфонил пиперазин;
восстановитель на стадии 8) представляет собой боран-тетрагидрофурановый комплекс или боран-диметилсульфидный комплекс.
9. Способ получения по п. 8, где медный реагент представляет собой бромид одновалентной меди, хлорид одновалентной меди, моногидрат ацетата меди, бромид меди, безводный хлорид меди или дигидрат хлорида меди; и реакционный растворитель представляет собой диметилсульфоксид или N,N-диметилформамид, N,N-диметилацетамид.
10. Применение пирроло[2,1-f][1,2,4]триазинового соединения по п. 1 в получении лекарственных средств для лечения заболевания, связанного с фосфатидилинозитол 3-киназой и ингибитора белка-мишени рапамицина.
11. Применение по п. 10, где заболевание, связанное с фосфатидилинозитол 3-киназой, представляет собой человеческую рабдомиосаркому, немелкоклеточный рак легкого, человеческую глиому, рак предстательной железы, рак яичников, рак печени, рак толстой кишки или рак молочной железы.

