ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям, которые ингибируют ErbB (рецептор эпидермального фактора роста) (например, EGFR или Her2), в частности мутантные формы ErbB, и/или ингибируют тирозинкиназу Брутона (BTK). Настоящее изобретение также относится к фармацевтической композиции, содержащей одно или более соединений в качестве активного ингредиента, и к применению соединений в изготовлении лекарственных средств для лечения расстройств, ассоциированных с мутантными формами ErbB (например, EGFR или Her2) и/или с BTK.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Рецепторные тирозинкиназы семейства ErbB действуют, осуществляя передачу сигналов извне клетки внутрь клетки путем активации вторичных эффекторов передачи через событие фосфорилирования по их тирозиновым остаткам фосфорилирования. Эти сигналы модулируют различные клеточные процессы, в том числе пролиферацию, утилизацию углеводов, синтез белков, ангиогенез, рост клеток и выживание клеток. Дерегуляция передачи сигналов рецепторами семейства ErbB модулирует пролиферацию, инвазию, метастазирование, ангиогенез и выживание опухолевых клеток и может быть связана со многими видами рака человека, включая рак легкого, рак в области головы и шеи и рак молочной железы. Подробные обзоры по передаче сигналов рецепторами ErbB и ее улучшению при опухолегенезе представлены в New England Journal of Medicine, 2008, Vol. 358: 1160-74, и в Biochemical and Biophysical Research Communications, 2004, Vol. 319: 1-11.
Было обнаружено, что EGFR сверхэкспрессируется и/или мутирует при многих видах рака, таких как глиомы и не мелкоклеточная карцинома легкого. Теперь клинически доступны противораковые лекарственные средства, направленно воздействующие на EGFR, в том числе, например, гефитиниб (IRESSAT®), эрлотиниб (TARCEVA®), лапатиниб (TYKERB®, TYVERB®), панитумумаб (VECTIBIX), цетуксимаб (ERBITUX), осимертиниб (TAGRISSO, AZD9291) и афатиниб (GIOTRIF). У большинства рецидивирующих пациентов с приобретенной устойчивостью к лекарственным средствам мутация EGFR по остатку Т790М была обнаружена по меньшей мере у половины таких клинически устойчивых пациентов. Более того, также может быть, что мутация Т790М уже существовала ранее. Например, есть пациенты с мутацией L858R/T790M, которые никогда не получали лечения гефитинибом, а мутации EGFR Т790М зародышевого типа связаны с некоторыми семейными видами рака легкого, и это дает основания предполагать, что мутация Т790М может играть независимую онкогенную роль. Находящиеся в настоящее время в разработке лекарственные средства, включающие ковалентные ингибиторы второго поколения, такие как BIBW2992, HKI-272 и PF-0299804, эффективны против мутации Т790М, устойчивой к существующим лекарственным средствам, но проявляют доза-ограничивающую токсичность вследствие параллельного ингибирования EGFR дикого типа (WT). Сообщалось также о неблагоприятных эффектах, таких как кожная сыпь и диарея, которые, как считается, связаны с ингибированием путей передачи сигналов WT EGFR (EGFR дикого типа) в нормальных клетках кожи и кишечника у более чем 60% пациентов с NSCLC (немелкоклеточный рак легкого), которых лечили гефитинибом или эрлотинибом (Zhou СС et al. Journal of Clinical Oncology, 2011, Vol. 12: 735-42; Mok TS et al. New England Journal of Medicine, 2009, Vol. 361: 947-57).
Примерно в 4-9,2% случаев рака легкого, обусловленного мутантным EGFR, были обнаружены вставки в экзоне 20 EGFR (Mitsudomin and Yatabe FEBS J., 2010; 277 (2): 301-8), большая часть из которых встречается в области, кодирующей аминокислоты 767-774 в экзоне 20, в петле, которая следует за С-спиралью киназного домена EGFR (Yasuda et al. Lancet Oncol., 2012; 13(1): e23-31). Сообщалось, что пациенты с раком легкого, экспрессирующим типичные инсерционные мутации в экзоне 20 EGFR, не реагировали на гефитиниб, или эрлотиниб, или афатиниб (Yasuda et al. Lancet Oncol., 2012, 13(1): e23-31; Yasuda et al. Sci Transl Med., 2013, 5(216): 216ra177).
Сверхэкспрессия Her2 может иметь место в раковых опухолях молочной железы, яичника, мочевого пузыря, в немелкоклеточной карциноме легкого, а также в других типах опухолей. Клинически доступные противораковые лекарственные средства, направленно воздействующие на Her2, включают трастузумаб (также известный как герцептин). Хотя две трети пациентов с раком молочной железы хорошо реагировали на герцептин, некоторые пациенты с Her2-положительным раком молочной железы не реагировали на это лекарственное средство. Возможно, что нереагирующая группа пациентов может иметь устойчивую к лекарственным средствам мутацию в Her2. Мутация, представляющая собой вставку четырех аминокислот YVMA в кодоне 775 в экзоне 20 Her2, также была обнаружена у примерно 2-4% некурящих пациентов с немелкоклеточным раком легкого, и у пациентов, имеющих такую Her2 YVMA-мутацию, была обнаружена сильная устойчивость к известным ингибиторам EGFR (Arcila et al. Clin Cancer Res., 2012, 18: 4910-4918).
Тирозинкиназа Брутона (BTK) является членом src-родственного Тес-семейства цитоплазматических тирозинкиназ, которые преимущественно экспрессируются в В-клетках и распределяются в лимфатической системе, гематопоэтической и гематологической системах. BTK играет ключевую роль в сигнальном пути В-клеточного рецептора В-клеток, который требуется для развития, активации и выживания В-клеток. Поэтому ингибиторы BTK разрабатывались с целью лечения В-клеточных злокачественных заболеваний, которые зависят от сигнального пути BCR (В-клеточный рецептор), таких как хронический лимфоцитарный лейкоз (CLL) и неходжкинская лимфома (NHL), мантийноклеточная лимфома (MCL) и диффузная большая В-клеточная лимфома (DLBCL). BTK также участвует в промотировании сигнального пути Toll-подобного рецептора, который регулирует активацию макрофагов и продуцирование провоспалительных цитокинов. Несколько исследований продемонстрировали перекрестные взаимодействия между сигнальными путями BTK и TLR (Toll-подобный рецептор), опосредующими трансактивацию нижележащих каскадов. Кроме того, установлено, что BTK играет решающую роль в регуляции иммунитета. BTK стала привлекательной мишенью для лечения не только В-клеточных злокачественных заболеваний, но и для лечения аутоиммунных заболеваний (смотри Ping et al., Oncotarget, 2017, 8(24): 39218-39229). Теперь клинически доступны противораковые лекарственные средства, направленно воздействующие на BTK, включающие ибрутиниб, который является необратимым ингибитором BTK, имеющим небольшую молекулу, и который разрешен для лечения CLL, мантийноклеточной лимфомы (MCL) и макроглобулинемии Вальденстрема (WM).
Соответственно, остается потребность в разработке новых ингибиторов ErbB (в частности EGFR или Her2), которые имеют лучшую селективность в отношении мутантного EGFR по сравнению с WT EGFR, или имеют лучшую селективность в отношении мутантного Her2 по сравнению с WT Her2. Также остается потребность в разработке новых ингибиторов BTK.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В одном аспекте настоящего изобретения предложено соединение, представленное формулой (I):
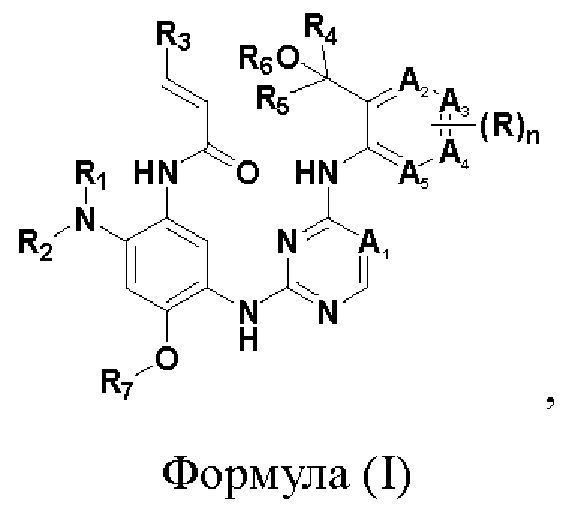
или его фармацевтически приемлемая соль, сложный эфир, гидрат, сольват или стереоизомер.
В другом аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая одно или более соединений формулы (I), их фармацевтически приемлемых солей, сложных эфиров, гидратов, сольватов или стереоизомеров.
В другом аспекте настоящего изобретения также предложено соединение формулы (I) или его фармацевтически приемлемая соль, сложный эфир, гидрат, сольват или стереоизомер, или фармацевтическая композиция одного или более из вышеуказанных для применения в качестве лекарственного средства для ингибирования ErbB, предпочтительно EGFR или HER2, более предпочтительно для ингибирования одной или более мутантных форм EGFR или HER2.
В еще одном другом аспекте настоящего изобретения предложено применение соединения формулы (I), его фармацевтически приемлемых солей, сложных эфиров, гидратов, сольватов или стереоизомеров, или фармацевтической композиции одного или более из вышеуказанных в изготовлении лекарственного средства для ингибирования ErbB, предпочтительно EGFR или HER2, более предпочтительно одной или более мутантных форм EGFR или HER2 у субъекта.
В другом аспекте настоящего изобретения предложен способ ингибирования ErbB, предпочтительно EGFR или HER2, более предпочтительно одной или более мутантных форм EGFR или HER2, путем использования одного или более соединений формулы (I), их фармацевтически приемлемых солей, сложных эфиров, гидратов, сольватов или стероизомеров, или фармацевтической композиции одного или более из вышеуказанных.
В другом аспекте настоящего изобретения предложен способ лечения ассоциированного с ErbB расстройства (например, рака), путем использования соединений формулы (I), их фармацевтически приемлемых солей, сложных эфиров, гидратов, сольватов или стереоизомеров, или фармацевтической композиции одного или более из вышеуказанных.
В еще одном аспекте настоящего изобретения предложено соединение формулы (I), его фармацевтически приемлемая соль, сложный эфир, гидрат, сольват или стереоизомер в комбинации со вторым терапевтическим агентом, предпочтительно противоопухолевым агентом.
В другом аспекте настоящего изобретения предложено комбинированное применение соединения формулы (I), его фармацевтически приемлемой соли, сложного эфира, гидрата, сольвата или стереоизомера и второго терапевтического агента, предпочтительно противоопухолевого агента.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Соединения
В одном аспекте настоящего изобретения предложено соединение формулы (I):
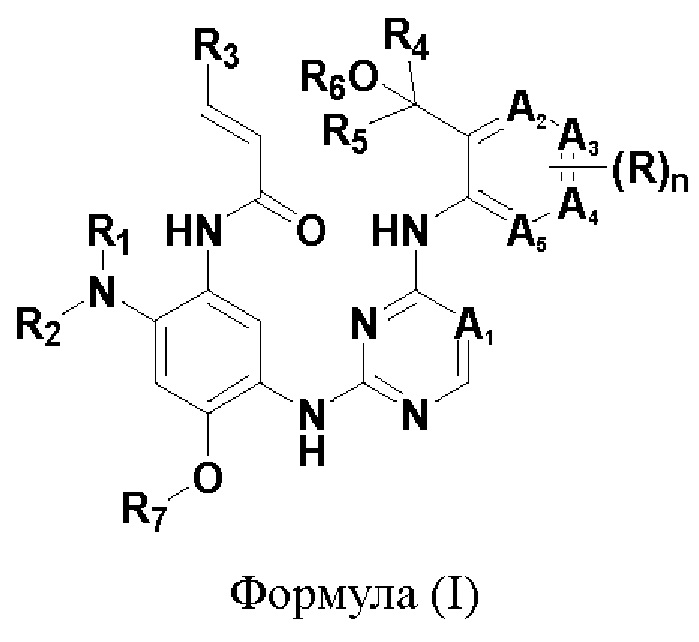
или его фармацевтически приемлемая соль, сложный эфир, гидрат, сольват или стереоизомер, где
A1 представляет собой N или CR8;
каждый из А2, A3, А4 и А5 независимо представляет собой N или CR9, причем не более чем один из А2, А3, А4 и А5 представляет собой N;
каждый из R1 и R2 независимо представляет собой водород или С1-12алкил, возможно монозамещенный или независимо полизамещенный одним или более чем одним галогеном, гидроксилом, -NRaRb, С1-12алкилом, С1-12алкокси, 3-10-членным насыщенным или ненасыщенным карбоциклилом, 3-10-членным насыщенным или ненасыщенным гетероциклилом, где каждый из С1-12алкила, С1-12алкокси, 3-10-членного насыщенного или ненасыщенного карбоциклила, 3-10-членного насыщенного или ненасыщенного гетероциклила может быть не замещен или моно- или полизамещен С1-12алкилом,
где каждый из Ra и Rb независимо выбран из водорода или С1-12алкила, который может быть возможно монозамещен или независимо полизамещен дейтерием, тритием, галогеном, гидроксилом или С1-12алкокси,
или Ra и Rb, взятые вместе с атомом азота, с которым они связаны, образуют 3-10-членный насыщенный или ненасыщенный гетероциклил, возможно моно- или полизамещенный галогеном, гидроксилом или С1-12алкилом;
или R1 и R2, взятые вместе с атомом азота, с которым они связаны, образуют 3-12-членное моноциклическое или полициклическое кольцо, возможно содержащее один или более дополнительных гетероатомов, выбранных из N, О и S, которое может быть возможно монозамещено или независимо полизамещено галогеном, гидроксилом, С1-12алкилом, С1-12алкокси, -NRaRb или группой -С1-12алкил-NRaRb;
R3 представляет собой Н, С1-12алкил или -С1-12алкил-NRaRb;
каждый из R4 и R5 независимо представляет собой С1-6алкил, возможно монозамещенный или независимо полизамещенный одним или более чем одним дейтерием, тритием, галогеном, гидроксилом, С1-12алкилом или С1-12алкокси,
или R4 и R5, взятые вместе с атомом углерода, с которым они связаны, образуют 3-10-членное моноциклическое или полициклическое кольцо, возможно содержащее один или более гетероатомов, выбранных из N, О и S, которое может быть возможно монозамещено или независимо полизамещено одним или более чем одним дейтерием, тритием, галогеном, гидроксилом, С1-12алкилом или С1-12алкокси,
R6 представляет собой водород или С1-12алкил, который может быть возможно монозамещен или независимо полизамещен дейтерием, тритием, галогеном, гидроксилом, С1-12алкилом или С1-12алкокси,
R7 представляет собой водород или С1-12алкил, который может быть возможно моно- или полизамещен дейтерием, тритием, галогеном или гидроксилом,
R8 представляет собой водород, дейтерий, тритий, галоген, циано, гидроксил, С1-12алкил, С1-12алкоксил, который может быть возможно монозамещен или независимо полизамещен одним или более чем одним дейтерием, тритием, галогеном или С1-12алкилом;
R9 отсутствует или представляет собой водород, дейтерий, тритий, галоген, циано, гидроксил, С1-12алкил или С1-12алкоксил, который может быть возможно монозамещен или независимо полизамещен одним или более чем одним дейтерием, тритием, галогеном или С1-12алкилом;
n равно 0, 1, 2, 3 или 4;
каждый R независимо представляет собой водород, дейтерий, тритий, галоген, циано, гидроксил, С1-12алкил, С1-12алкокси, 3-10-членный насыщенный или ненасыщенный карбоциклил или 3-10-членный насыщенный или ненасыщенный гетероциклил, конденсированный с кольцом, с которым он связан, которые могут быть возможно монозамещены или независимо полизамещены одним или более чем одним дейтерием, тритием, галогеном или С1-12алкилом.
В некоторых воплощениях A1 в формуле (I) представляет собой N. В некоторых воплощениях A1 в формуле (I) представляет собой CR8, где R8 представляет собой водород, дейтерий, тритий, галоген, циано, гидроксил, С1-12алкил, С1-12алкоксил, который может быть возможно монозамещен или независимо полизамещен одним или более чем одним дейтерием, тритием, галогеном или С1-12алкилом. В некоторых воплощениях A1 в формуле (I) представляет собой СН.
В некоторых воплощениях соединения по настоящему изобретению представлены формулой (Ia):

или его фармацевтически приемлемая соль, сложный эфир, гидрат, сольват или стереоизомер, где
каждый из А2, A3, А4 и А5 независимо представляет собой N или CR9, причем не более чем один из А2, A3, А4 и А5 представляет собой N;
каждый из R1 и R2 независимо представляет собой водород или С1-12алкил, возможно монозамещенный или независимо полизамещенный одним или более чем одним галогеном, гидроксилом, -NRaRb, С1-12алкилом, С1-12алкокси, 3-10-членным насыщенным или ненасыщенным карбоциклилом, 3-10-членным насыщенным или ненасыщенным гетероциклилом, где каждый из С1-12алкила, С1-12алкокси, 3-10-членного насыщенного или ненасыщенного карбоциклила, 3-10-членного насыщенного или ненасыщенного гетероциклила может быть не замещен или моно- или полизамещен С1-12алкилом,
где каждый из Ra и Rb независимо выбран из водорода или С1-12алкила, который может быть возможно монозамещен или независимо полизамещен дейтерием, тритием, галогеном, гидроксилом или С1-12алкокси,
или Ra и Rb, взятые вместе с атомом азота, с которым они связаны, образуют 3-10-членный насыщенный или ненасыщенный гетероциклил, возможно моно- или полизамещенный галогеном, гидроксилом или С1-12алкилом;
или R1 и R2, взятые вместе с атомом азота, с которым они связаны, образуют 3-12-членное моноциклическое или полициклическое кольцо, возможно содержащее один или более дополнительных гетероатомов, выбранных из N, О и S, которое может быть возможно монозамещено или независимо полизамещено галогеном, гидроксилом, С1-12алкилом, С1-12алкокси, -NRaRb или группой -С1-12алкил-NRaRb;
R3 представляет собой Н, С1-12алкил или -С1-12алкил-NRaRb;
каждый из R4 и R5 независимо представляет собой С1-6алкил, возможно монозамещенный или независимо полизамещенный одним или более чем одним дейтерием, тритием, галогеном, гидроксилом, С1-12алкилом или С1-12алкокси,
или R4 и R5, взятые вместе с атомом углерода, с которым они связаны, образуют 3-10-членное моноциклическое или полициклическое кольцо, возможно содержащее один или более гетероатомов, выбранных из N, О и S, которое может быть возможно монозамещено или независимо полизамещено одним или более чем одним дейтерием, тритием, галогеном, гидроксилом, С1-12алкилом или С1-12алкокси,
R6 представляет собой водород или С1-12алкил, который может быть возможно монозамещен или независимо полизамещен дейтерием, тритием, галогеном, гидроксилом, С1-12алкилом или С1-12алкокси,
R7 представляет собой водород или С1-12алкил, который может быть возможно моно- или полизамещен дейтерием, тритием, галогеном или гидроксилом,
R9 отсутствует или представляет собой водород, дейтерий, тритий, галоген, циано, гидроксил, С1-12алкил или С1-12алкоксил, который может быть возможно монозамещен или независимо полизамещен одним или более чем одним дейтерием, тритием, галогеном или С1-12алкилом;
n равно 0, 1, 2, 3 или 4;
каждый R независимо представляет собой водород, дейтерий, тритий, галоген, циано, гидроксил, С1-12алкил, С1-12алкокси, 3-10-членный насыщенный или ненасыщенный карбоциклил или 3-10-членный насыщенный или ненасыщенный гетероциклил, конденсированный с кольцом, с которым он связан, которые могут быть возможно монозамещены или независимо полизамещены одним или более чем одним дейтерием, тритием, галогеном или С1-12алкилом.
В некоторых воплощениях соединения по настоящему изобретению представлены формулой (Ib):

или его фармацевтически приемлемая соль, сложный эфир, гидрат, сольват или стереоизомер, где
каждый из А2, A3, А4 и А5 независимо представляет собой N или CR9, причем не более чем один из А2, A3, А4 и А5 представляет собой N;
каждый из R1 и R2 независимо представляет собой водород или С1-12алкил, возможно монозамещенный или независимо полизамещенный одним или более чем одним галогеном, гидроксилом, -NRaRb, С1-12алкилом, С1-12алкокси, 3-10-членным насыщенным или ненасыщенным карбоциклилом, 3-10-членным насыщенным или ненасыщенным гетероциклилом, где каждый из С1-12алкила, С1-12алкокси, 3-10-членного насыщенного или ненасыщенного карбоциклила, 3-10-членного насыщенного или ненасыщенного гетероциклила может быть не замещен или моно- или полизамещен С1-12алкилом,
где каждый из Ra и Rb независимо выбран из водорода или С1-12алкила, который может быть возможно монозамещен или независимо полизамещен дейтерием, тритием, галогеном, гидроксилом или С1-12алкокси,
или Ra и Rb, взятые вместе с атомом азота, с которым они связаны, образуют 3-10-членный насыщенный или ненасыщенный гетероциклил, возможно моно- или полизамещенный галогеном, гидроксилом или С1-12алкилом;
или R1 и R2, взятые вместе с атомом азота, с которым они связаны, образуют 3-12-членное моноциклическое или полициклическое кольцо, возможно содержащее один или более дополнительных гетероатомов, выбранных из N, О и S, которое может быть возможно монозамещено или независимо полизамещено галогеном, гидроксилом, С1-12алкилом, С1-12алкокси, -NRaRb или группой -С1-12алкил-NRaRb;
R3 представляет собой Н, С1-12алкил или -С1-12алкил-NRaRb;
каждый из R4 и R5 независимо представляет собой С1-6алкил, возможно монозамещенный или независимо полизамещенный одним или более чем одним дейтерием, тритием, галогеном, гидроксилом, С1-12алкилом или С1-12алкокси,
или R4 и R5, взятые вместе с атомом углерода, с которым они связаны, образуют 3-10-членное моноциклическое или полициклическое кольцо, возможно содержащее один или более гетероатомов, выбранных из N, О и S, которое может быть возможно монозамещено или независимо полизамещено одним или более чем одним дейтерием, тритием, галогеном, гидроксилом, С1-12алкилом или С1-12алкокси,
R6 представляет собой водород или С1-12алкил, который может быть возможно монозамещен или независимо полизамещен дейтерием, тритием, галогеном, гидроксилом, С1-12алкилом или С1-12алкокси,
R7 представляет собой водород или С1-12алкил, который может быть возможно моно- или полизамещен дейтерием, тритием, галогеном или гидроксилом,
R8 представляет собой водород, дейтерий, тритий, галоген, циано, гидроксил, С1-12алкил, С1-12алкоксил, который может быть возможно монозамещен или независимо полизамещен одним или более чем одним дейтерием, тритием, галогеном или С1-12алкилом;
R9 отсутствует или представляет собой водород, дейтерий, тритий, галоген, циано, гидроксил, С1-12алкил или С1-12алкоксил, который может быть возможно монозамещен или независимо полизамещен одним или более чем одним дейтерием, тритием, галогеном или С1-12алкилом;
n равно 0, 1, 2, 3 или 4;
каждый R независимо представляет собой водород, дейтерий, тритий, галоген, циано, гидроксил, С1-12алкил, С1-12алкокси, 3-10-членный насыщенный или ненасыщенный карбоциклил или 3-10-членный насыщенный или ненасыщенный гетероциклил, конденсированный с кольцом, с которым он связан, которые могут быть возможно монозамещены или независимо полизамещены одним или более чем одним дейтерием, тритием, галогеном или С1-12алкилом.
В некоторых воплощениях один из А2, А3, А4 и А5 в формуле (I), формуле (Ia) или формуле (Ib) представляет собой N, а остальные, каждый независимо, представляют собой CR9. В некоторых воплощениях А2, A3, А4 и А5 в формуле (I), формуле (Ia) или формуле (Ib), каждый независимо, представляют собой CR9. В некоторых воплощениях, когда А2, A3, А4 или А5 в формуле (I), формуле (Ia) или формуле (Ib) представляют собой CR9, который дополнительно замещен R, тогда R9 отсутствует.
В некоторых воплощениях n равно 2, А2 и А5 представляют собой СН, каждый из A3 и А4 независимо представляет собой СН, который дополнительно замещен R, R независимо представляет собой галоген.
В некоторых воплощениях каждый из R1 и R2 в формуле (I), формуле (Ia) или формуле (Ib) независимо замещен или не замещен С1-12алкилом, который может быть возможно монозамещен или независимо полизамещен одним или более чем одним галогеном, гидроксилом, -NRaRb, С1-12алкилом, С1-12алкокси, 3-10-членным насыщенным или ненасыщенный карбоциклилом, 3-10-членным насыщенным или ненасыщенным гетероциклилом, где каждый из С1-12алкила, С1-12алкокси, 3-10-членного насыщенного или ненасыщенного карбоциклила, 3-10-членного насыщенного или ненасыщенного гетероциклила может быть не замещен или моно- или полизамещен С1-12алкилом,
где каждый из Ra и Rb независимо выбран из водорода или С1-12алкила, который может быть возможно монозамещен или независимо полизамещен дейтерием, тритием, галогеном, гидроксилом или С1-12алкокси,
или Ra и Rb, взятые вместе с атомом азота, с которым они связаны, образуют 3-10-членный насыщенный или ненасыщенный гетероциклил, возможно моно- или полизамещенный галогеном, гидроксилом или С1-12алкилом.
В некоторых воплощениях R1 и R2 в формуле (I), формуле (Ia) или формуле (Ib), взятые вместе с атомом азота, с которым они связаны, образуют 3-12-членное моноциклическое или полициклическое кольцо, возможно содержащее один или более дополнительных гетероатомов, выбранных из N, О и S, которое может быть возможно монозамещено или независимо полизамещено галогеном, гидроксилом, С1-12алкилом, С1-12алкокси, -NRaRb или группой -С1-12алкил-NRaRb,
где каждый из Ra и Rb независимо выбран из водорода или С1-12алкила, который может быть возможно монозамещен или независимо полизамещен дейтерием, тритием, галогеном, гидроксилом или С1-12алкокси,
или Ra и Rb, взятые вместе с атомом азота, с которым они связаны, образуют 3-10-членный насыщенный или ненасыщенный гетероциклил, возможно моно- или полизамещенный галогеном, гидроксилом или С1-12алкилом.
В некоторых воплощениях каждый из R1 и R2 в формуле (I), формуле (Ia), формуле (Ib) независимо выбран из:
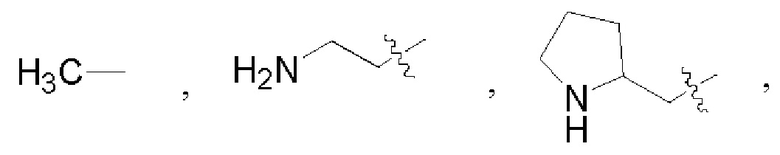
которые могут быть возможно монозамещены или независимо полизамещены дейтерием, тритием, галогеном, гидроксилом, С1-12алкокси или С1-12алкилом, где С1-12алкил может быть возможно монозамещен или независимо полизамещен дейтерием, тритием, галогеном или гидроксилом.
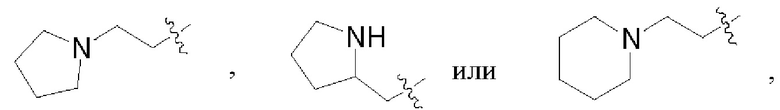
В некоторых воплощениях каждый из R1 и R2 в формуле (I), формуле (Ia), формуле (Ib) независимо выбран из:


В некоторых воплощениях R1 и R2 в формуле (I), формуле (Ia), формуле (Ib), взятые вместе с атомом азота, с которым они связаны, образуют 3-12-членное моноциклическое или полициклическое кольцо, выбранное из:


которые могут быть возможно монозамещены или независимо полизамещены галогеном, гидроксилом, С1-12алкилом, С1-12алкокси, -NRaRb или -С1-12алкил-NRaRb.
В некоторых воплощениях R1 и R2 в формуле (I), формуле (Ia), формуле (Ib), взятые вместе с атомом азота, с которым они связаны, образуют:
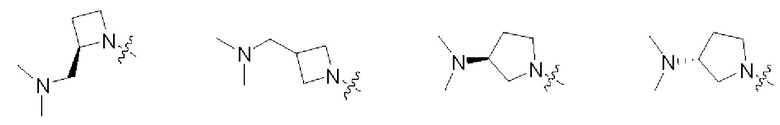
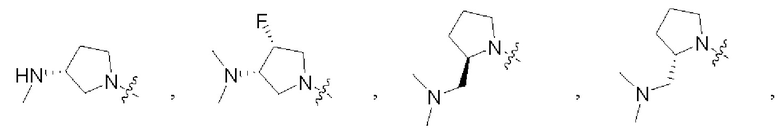
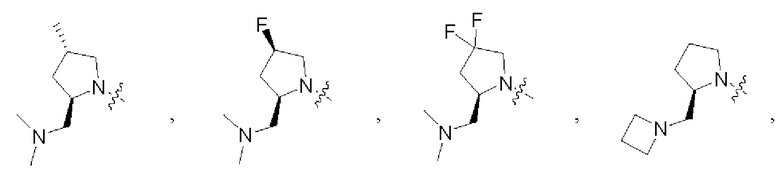



В некоторых воплощениях каждый из R4 и R5 в формуле (I), формуле (Ia) или формуле (Ib) независимо замещен или не замещен С1-6алкилом, который может быть возможно монозамещен или независимо полизамещен одним или более чем одним дейтерием, тритием, галогеном, гидроксилом, С1-12алкилом или С1-12алкокси. В некоторых воплощениях каждый из R4 и R5 в формуле (I), формуле (Ia) или формуле (Ib) независимо представляет собой незамещенный С1-6алкил.
В некоторых воплощениях R4 и R5 в формуле (I), формуле (Ia) или формуле (Ib), взятые вместе с атомом углерода, с которым они связаны, образуют 3-10-членное моноциклическое или полициклическое кольцо, возможно содержащее один или более гетероатомов, выбранных из N, О и S, которое может быть возможно монозамещено или независимо полизамещено одном или более чем одним дейтерием, тритием, галогеном, гидроксилом, С1-12алкилом или С1-12алкокси.
В некоторых воплощениях R6 в формуле (I), формуле (Ia) или формуле (Ib) представляет собой водород, дейтерий или тритий.
В некоторых воплощениях R7 в формуле (I), формуле (Ia) или формуле (Ib) представляет собой метил, дифторметил или трифторметил.
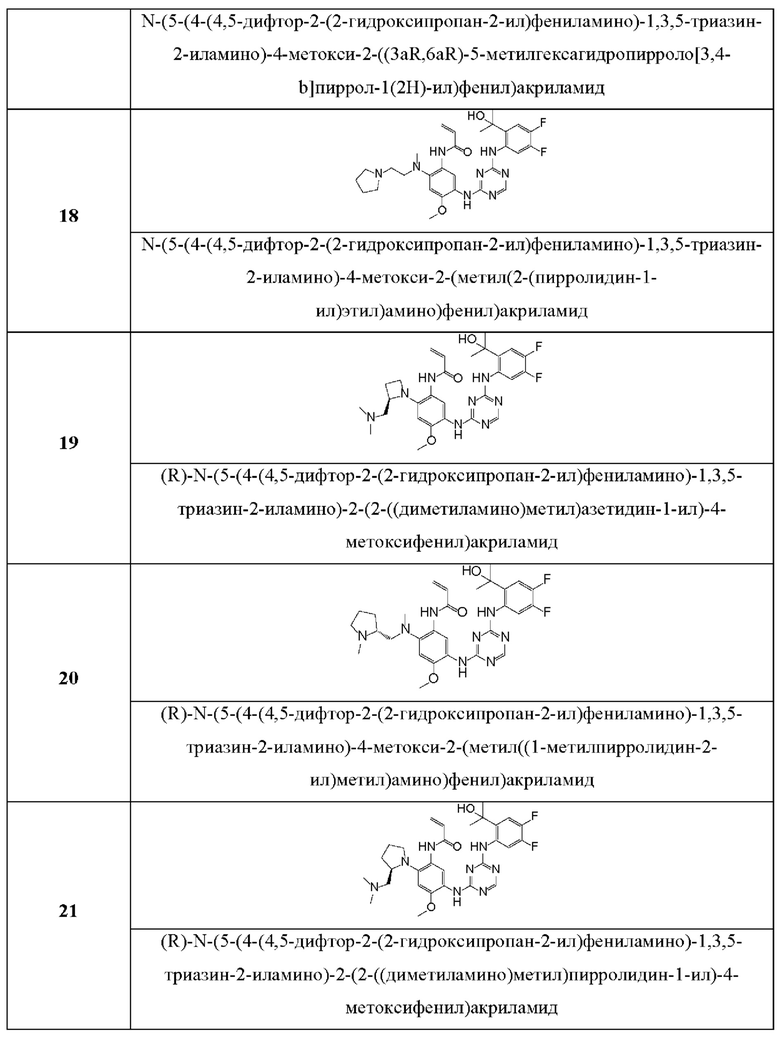
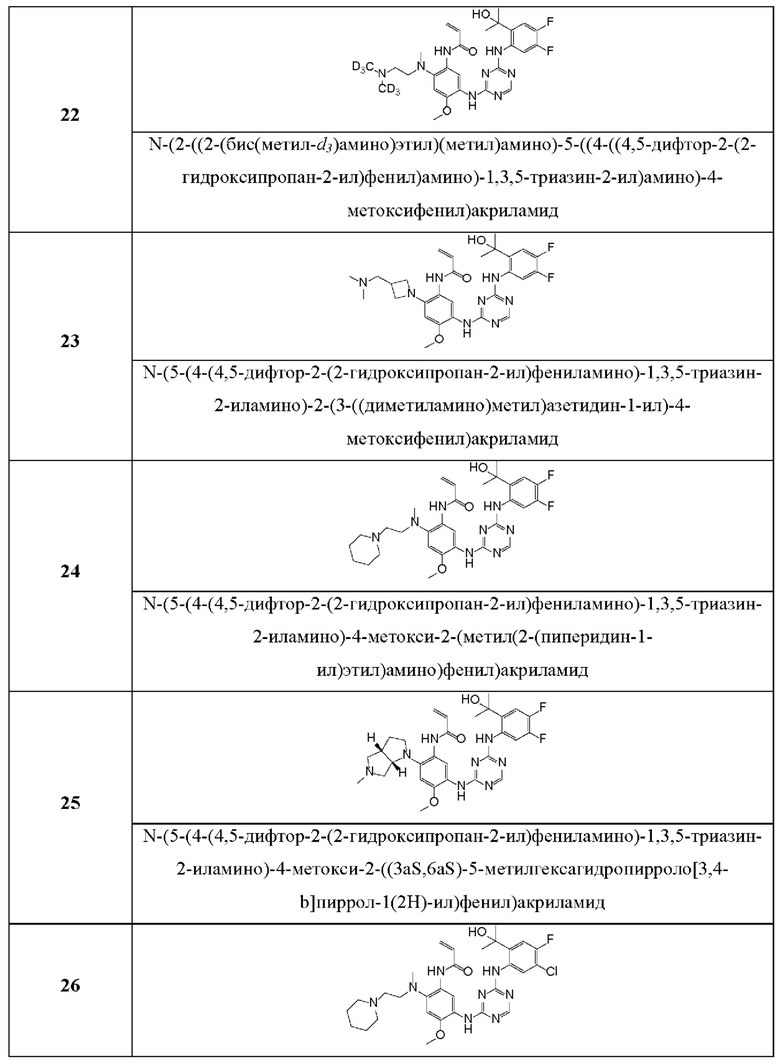
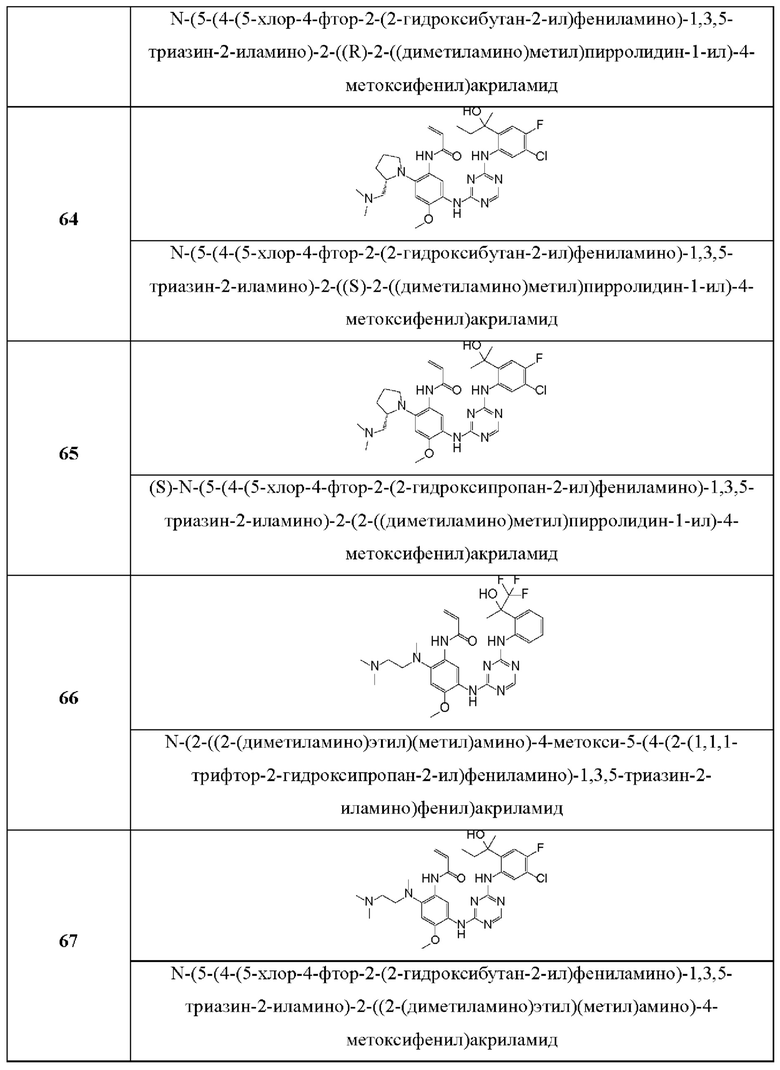
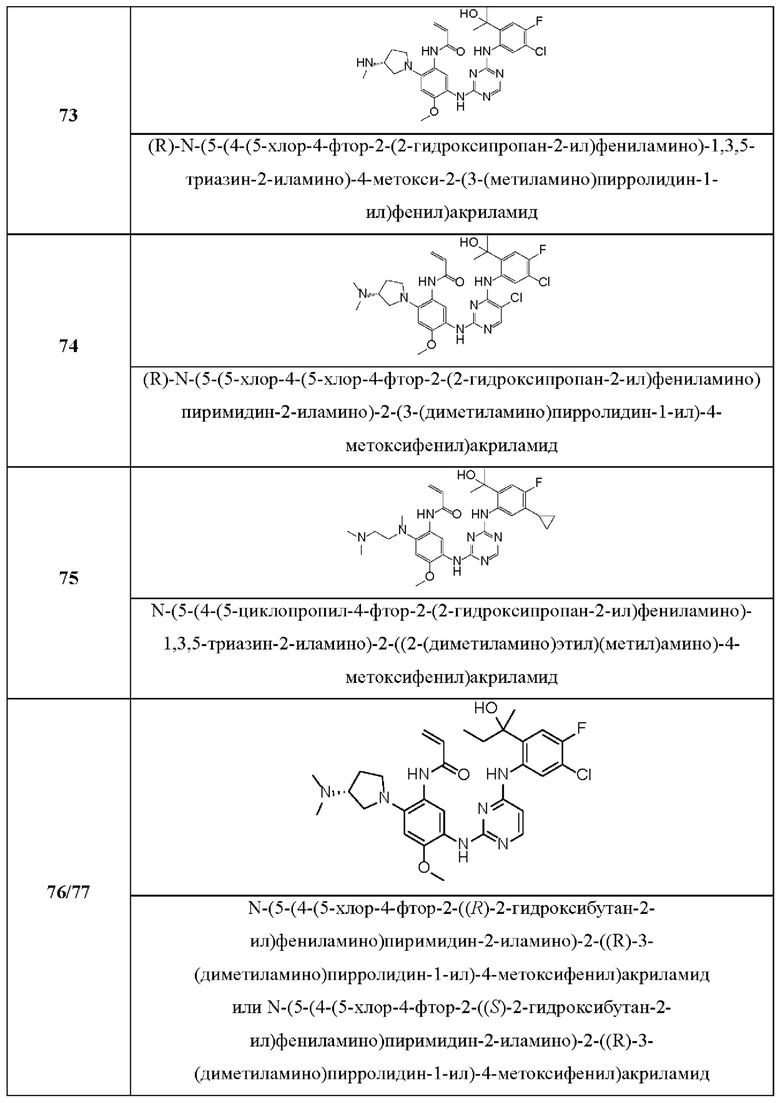
Иллюстративные соединения 1-98 формулы (I) представлены в Таблице 1 ниже.


















Следует иметь в виду, что некоторые признаки настоящего изобретения, которые для ясности описаны в контексте отдельных воплощений, могут быть предусмотрены также в комбинации в единственном воплощении. И наоборот, различные признаки настоящего изобретения, которые для краткости описаны в контексте единственного воплощения, могут быть предусмотрены также отдельно или в любой подходящей подкомбинации.
В разных местах настоящего описания изобретения описаны линкерные заместители. В тех случаях, когда структура четко требует наличия линкерной группы, должно быть понятно, что переменные Маркуша, перечисленные для этой группы, представляют собой линкерные группы. Например, если структура требует наличия линкерной группы, и в определении группы Маркуша для этой переменной указан "алкил", то понятно, что этот "алкил" представляет собой линкерную алкиленовую группу.
Использованный в данном документе термин "замещенный", когда он относится к химической группе, означает, что химическая группа имеет один или более атомов водорода, которые удалены и замещены заместителями. Использованный в данном документе термин "заместитель" имеет обычное значение, известное в данной области, и относится к химической группировке, которая ковалентно присоединена к родительской группе или, в соответствующем случае, конденсирована с родительской группой. Использованный в данном документе термин "возможно замещенный" или "возможно…замещенный" означает, что химическая группа может не иметь заместителей (т.е. является незамещенной) или может иметь один или более заместителей (т.е. является замещенной). Следует понимать, что замещение по данному атому ограничено валентностью.
Использованный в данном документе термин "Ci-j" показывает диапазон количества атомов углерода, где i и j означают числа, и диапазон количества атомов углерода включает в себя конечные точки (т.е. i и j) и каждое целое число между ними, и где i ∈{1, 2, 3, 4, 5, 6, 7, 8, 9 или 10}, j больше, чем i, j ∈ {2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39 или 40}. Например, C1-6 показывает диапазон от одного до шести атомов углерода, включая один атом углерода атом, два атома углерода, три атома углерода атомы, четыре атома углерода, пять атомов углерода и шесть атомов углерода.
Использованный в данном документе термин "алкил", как часть другого термина или использованный независимо, относится к насыщенной или ненасыщенной углеводородной цепи, при этом последняя может быть дополнительно подразделена на углеводородную цепь, имеющую по меньшей мере одну двойную или тройную связь (алкенил или алкинил). Углеводородная цепь, упомянутая выше, может представлять собой прямую цепь или разветвленную цепь. Термин "Ci-jалкил" относится к алкилу, имеющему от i до j атомов углерода. В некоторых воплощениях алкильная группа содержит 1-12, 1-8, 1-6, 1-4, 1-3 или 1-2 атомов углерода. Примеры насыщенной алкильной группы включают, без ограничения, метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил; высшие гомологи, такие как 2-метил-1-бутил, н-пентил, 3-пентил, н-гексил, 1,2,2-триметилпропил и т.п. Примеры ненасыщенных алкильных групп включают, без ограничения, этенил, н-пропенил, изопропенил, н-бутенил, втор-бутенил, этинил, пропин-1-ил, пропин-2-ил и т.п.
Использованные в данном документе термины "галоген" и "галогено" относятся к атому, выбранному из атомов фтора, хлора, брома и йода.
Использованный в данном документе термин "циано" относится к группе формулы -CN.
Использованный в данном документе термин "гидроксил" относится к группе формулы -ОН.
Использованный в данном документе термин "алкокси", как часть другого термина или использованный независимо, относится к группе формулы -О-алкил. Термин "Qi-jaлкoкcи" означает, что алкильная группировка группы алкокси имеет от i до j атомов углерода. В некоторых воплощениях алкильная группировка имеет 1-12, 1-10, 1-8, 1-6, 1-5, 1-4, 1-3 или 1-2 атома углерода. Примеры групп алкокси включают, без ограничения, метокси, этокси, пропокси (например, н-пропокси и изопропокси), трет-бутокси и т.п.
Использованный в данном документе термин "карбоциклил", как часть другого термина или использованный независимо, относится к любому кольцу, в котором все кольцевые атомы представляют собой атомы углерода, и которое содержит по меньшей мере три образующих кольцо атома углерода. В некоторых воплощениях карбоциклил может содержать 3-12 образующих кольцо атомов углерода, 3-10 образующих кольцо атомов углерода, 3-8 образующих кольцо атомов углерода или 4-8 образующих кольцо атомов углерода. Карбоциклильные группы могут быть насыщенными или частично ненасыщенными. В некоторых воплощениях карбоциклильная группа может представлять собой насыщенную циклическую алкильную группу. В некоторых воплощениях карбоциклильная группа может представлять собой ненасыщенную циклическую алкильную группу, которая содержит по меньшей мере одну двойную связь в ее кольцевой системе. В некоторых воплощениях ненасыщенная карбоциклильная группа может содержать одно или более ароматических колец.
Карбоциклильные группы могут включать в себя моно- или полициклическое(ие) кольцо(а) (например, имеющее 2, 3 или 4 конденсированных, мостиковых или спироколец). Примеры моноциклических карбоциклильных групп включают, без ограничения, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклопентенил, циклогексенил, циклогексадиенил, циклогептатриенил и т.п. Использованный в данном документе термин "спирокольца" относится к кольцевым системам, имеющим два кольца, соединенные через один единственный общий атом; термин "конденсированные кольца" относится к кольцевым системам, имеющим два кольца, имеющие два общих соседних атома; и термин "мостиковые кольца" относится к кольцевым системам с двумя кольцами, имеющими три или более общих атомов. Примеры спирокарбоциклила включают, без ограничения, спиро[5.5]ундекан, спиропентадиен, спиро[3.6]декан и т.п. Примеры конденсированного карбоциклила включают, без ограничения, нафталин, бензопирен, антрацен, аценафтен, флуорен, нен и т.п. Примеры мостикового карбоциклила включают, без ограничения, бицикло[2,2,1]гептенил, бицикло [2.2.1] гептан, бицикло[2.2.2]октан, бицикло [3.3.1]нонан, бицикло[3.3.3]ундекан и т.п.
Использованный в данном документе термин "гетероциклил" относится к карбоциклильной группе, где один или более (например, 1, 2 или 3) кольцевых атомов заменены гетероатомами, которые включают, без ограничения, атомы кислорода, серы, азота, фосфора и т.п. В некоторых воплощениях гетероциклил представляет собой насыщенный гетероциклил. В некоторых воплощениях гетероциклил представляет собой ненасыщенный гетероциклил, имеющий одну или более двойных связей в его кольцевой системе. В некоторых воплощениях ненасыщенная гетероциклильная группа может содержать одно или более ароматических колец.
Гетероциклильные группы могут включать в себя моно- или полициклическое(ие) кольцо(а) (например, имеющие 2, 3 или 4 конденсированных, мостиковых или спироколец). Иллюстративные моноциклические гетероциклильные группы включают, без ограничения, пирролидил, тетрагидрофуран, пиперидил, пиперазинил, морфолинил и т.п. Примеры спирогетероциклила включают, без ограничения, спиропираны, спирооксазины и т.п. Примеры конденсированного гетероциклила включают, без ограничения, группы хинолина, изохинолина, хинолизина, хиназолина, птеридина, хромена, изохромена, индола, изоиндола, индолизина, индазола, пурина, бензофурана, изобензофурана, бензимидазола, бензотиенила, карбазола, феназина, фенотиазина, фенантридина и т.п.
Примеры мостикового гетероциклила включают, без ограничения, морфан, гексаметилентетрамин, 1,4-диазабицикло[2.2.2]октан (DABCO) и т.п.
Использованный в данном документе термин "i-j-членный" относится к карбоциклильным или гетероциклильным группам, имеющим от i до j образующих кольцо атомов. Например, "3-8-членный карбоциклил" относится к карбоциклильным группам, имеющим от 3 до 10 (например, 3, 4, 5, 6, 7, 8, 9 или 10) образующих кольцо членов; "3-10-членный гетероциклил" относится к гетероциклилу, имеющему от 3 до 10 (например, 3, 4, 5, 6, 7, 8, 9 или 10) образующих кольцо членов. В некоторых воплощениях карбоциклильные или гетероциклильные группы являются 3-10-членными, 3-8-членными, 3-6-членными или 4-6-членными. Например, пиперидинил является примером 6-членного гетероциклила, пиразолил является примером 5-членного гетероциклила, пиридил является примером 6-членного гетероциклила, и 1,2,3,4-тетрагидро-нафталин является примером 10-членного карбоциклила.
Использованный в данном документе термин "ароматическая группа" или "ароматическое кольцо" относится к моно- или полициклической карбоциклильной или гетероциклильной группировке, имеющей перемежающиеся двойные и одинарные связи между образующими кольцо атомами по меньшей мере в одном кольце. В некоторых воплощениях ароматические кольца имеют от 5 до 12, от 5 до 10, от 5 до 8, от 6 до 12, от 6 до 10 или от 6 до 8 образующих кольцо атомов (т.е. 5-12, 5-10, 5-8, 6-12, 6-10 или 6-8-членные). Примеры карбоциклических ароматических групп включают, без ограничения, фенил, нафтил, тетрагидронафтил, инданил и инденил и т.п. В некоторых воплощениях гетероциклическая ароматическая группа является 5-членной или 6-членной. Иллюстративными 5-членными гетероциклическими ароматическими группами являются тиенил, фурил, пирролил, имидазолил, тиазолил, оксазолил, пиразолил, изотиазолил, изоксазолил, 1,2,3-триазолил, 1,2,4-триазолил, 1,3,4-триазолил, тетразолил, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, 1,3,4-тиадиазолил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,3,4-оксадиазолил и т.п. Иллюстративными 6-членными гетероциклическим ароматическими группами являются пиридил, пиразинил, пиримидинил, триазинил и пиридазинил.
"Соединение" по настоящему изобретению охватывает все стереоизомеры, геометрические изомеры и таутомеры изображенных структур, если конкретно не указано иное.
Термин "стереоизомер" относится к любым различным стереоизомерным конфигурациям (например, энантиомерам, диастереомерам и рацематам) асимметрических соединений (например, соединений, имеющих один или более асимметрически замещенных атомов углерода, т.е. "асимметрических центров"). Соединения по настоящему изобретению, которые содержат асимметрические центры, могут быть выделены в оптически активной (энантиомеры или диастереомеры) или оптически неактивной (рацемической) форме. Термин "энантиомер" охватывает пары стереоизомеров, зеркальные отображения которых не накладываются друг на друга. Смесь 1:1 пары энантиомеров представляет собой "рацемическую смесь". Термины "диастереомеры" или "диастереоизомеры" охватывают стереоизомеры, которые имеют по меньшей мере два асимметрических атома, но которые не являются зеркальными отображениями друг друга. Некоторые соединения, содержащие один или более асимметрических центров, могут приводить к энантиомерам, диастереомерам или другим стереоизомерным формам, которые могут быть определены в терминах абсолютной конфигурации как (R)- или (S)- по каждому асимметрическому центру согласно R-S системе Кана-Ингольда-Прелога. Разделенные соединения, чья абсолютная конфигурация неизвестна, могут быть обозначены с использованием термина "или" по асимметрическому центру. В данной области известны способы получения оптически активных форм из рацемических смесей, такие как разделение методом ВЭЖХ или стереоселективный синтез.
"Геометрические изомеры" или "цис и транс изомеры" относятся к соединениям с одинаковой формулой, но их функциональные группы повернуты в разном направлении в трехмерном пространстве. Термин "таутомеры" охватывает прототропные таутомеры, которые представляют собой изомерные состояния протонирования соединений, имеющих одинаковую формулу и суммарный заряд. Примеры прототропных таутомеров включают, без ограничения, кетон-енольные пары, амид-имидные кислотные пары, пары лактам-лактим, пары енамин-имин и кольцеобразные формы, где протон может занимать два или более положений гетероциклической системы, например 1Н- и 3Н-имидазол, 1Н-, 2Н- и 4Н-1,2,4-триазол, 1Н- и 2Н-изоиндол и 1Н- и 2Н-пиразол. Таутомеры могут находиться в равновесии или могут быть стерически заблокированы в одну форму в результате соответствующего замещения. Соединения по настоящему изобретению, идентифицированные по названию или структуре как одна конкретная таутомерная форма, включают в себя другие таутомерные формы, если конкретно не указано иное.
"Соединение" по настоящему изобретению также охватывает все изотопы атомов в соединениях. Изотопы атома включают атомы, имеющие тот же атомный номер, но другие массовые числа. Например, подразумевается, что водород, углерод, азот, кислород, фосфор, сера, фтор, хлор, бром или йод в "соединении" по настоящему изобретению также включают их изотопы, такие как, но без ограничения, 1Н, 2Н, 3Н, 11С, 12С, 13С, 14С, 14N, 15N, 16О, 17O, 18O, 31Р, 32Р, 32S, 33S, 34S, 36S, 17F, 19F, 35Cl, 37Cl, 79Br, 81Br, 127I и 131I. В некоторых воплощениях водород включает протий, дейтерий и тритий. В некоторых воплощениях углерод включает 12С и 13С.
Должно быть понятно также, что "соединение" по настоящему изобретению может существовать в сольватированных, а также в несольванированных формах, таких как, например, гидратированные формы, твердотельные формы, и настоящее изобретение охватывает все такие сольватированные и несольватированные формы.
Должно быть также понятно, что "соединение" по настоящему изобретению может существовать в формах фармацевтически приемлемых солей или сложных эфиров.
Использованный в данном документе термин "фармацевтически приемлемый" относится к тем соединениям, веществам, композициям и/или лекарственным формам, которые в рамках обоснованного медицинского суждения являются подходящими для применения в контакте с тканями человеческих существ и животных без излишних токсичности, раздражения, аллергической реакции или другой проблемы или осложнения, соизмеримых с разумным соотношением польза/риск. В некоторых воплощениях соединения, вещества, композиции и/или лекарственные формы, которые являются фармацевтически приемлемыми, относятся к тем из них, которые разрешены для применения регулирующим органом (таким как Управление по контролю качества пищевых продуктов и лекарственных средств США (U.S. Food и Drag Administration), Управление по контролю качества пищевых продуктов и лекарственных средств Китая (China Food и Drug Administration) или Европейское агентство по лекарственным средствам (European Medicines Agency)) или перечислены в общепризнанных фармакопеях (таких как Фармакопея США, Фармакопея Китая или Европейская Фармакопея) для применения у животных и, более конкретно, у людей.
В данном документе "фармацевтически приемлемые соли" относятся к производным соединений по настоящему изобретению, где родительское соединение модифицировано путем превращения существующей кислотной группировки (например, карбоксильной и т.п.) или основной группировки (например, амин, щелочь и т.п.) в ее солевую форму. Во многих случаях соединения по настоящему изобретению способны образовывать соли с кислотами и/или основаниями благодаря присутствию амино и/или карбоксильных групп, или групп, подобных им. И "фармацевтически приемлемая соль" включает соли присоединения кислоты или соли присоединения основания, которые сохраняют биологическую эффективность и свойства родительского соединения, и которые обычно не являются биологически или иным образом нежелательными.
В данном документе "фармацевтически приемлемые сложные эфиры" относятся к сложным эфирам, которые гидролизуются in vivo и включают те из них, которые легко разлагаются в организме человека с образованием родительского соединения или его соли. Такие сложные эфиры могут действовать как пролекарства, как определено в данном документе. Сложные эфиры могут быть образованы с аминной, гидрокси или карбоксильной боковой цепью на соединениях, описанных в данном документе. Например, если раскрытое соединение содержит спиртовую функциональную группу, то сложный эфир может быть образован в результате замещения атома водорода спиртовой группы кислотной группой, такой как, без ограничения, группы карбоновых кислот, фосфорных кислот, фосфоновых кислот, сульфиновых кислот, сульфоновых кислот и бороновых кислот. Методики и конкретные группы для получения таких сложных эфиров известны специалистам в данной области и легко могут быть найдены в источниках информации, таких как Greene and Wuts, Protective Groups in Organic Synthesis, 3rd Ed., John Wiley & Sons, New York, N.Y., 1999, который во всей его полноте включен в данное описание изобретения посредством ссылки.
Подходящие фармацевтически приемлемые соли соединения по настоящему изобретению включают, например, соль присоединения кислоты, которая может быть производной от, например, неорганической кислоты (например, соляной, бромистоводородной, серной, азотной, фосфорной кислоты и т.п.) или органической кислоты (например, муравьиной, уксусной, пропионовой, гликолевой, оксалиновой, малеиновой, яблочной, янтарной, фумаровой, винной, тримезиновой, лимонной, молочной, фенилуксусной, бензойной, миндальной, метансульфоновой, нападисиловой (нафталин-1,5-дисульфоновой), этансульфоновой, толуолсульфоновой, трифторуксусной, салициловой, сульфосалициловой кислот и т.п.). В некоторых воплощениях фармацевтически приемлемая соль соединения по настоящему изобретению представляет собой соль муравьиной кислоты. В некоторых воплощениях фармацевтически приемлемая соль соединения по настоящему изобретению представляет собой TFA соль. Подходящие фармацевтически приемлемые соли соединения по настоящему изобретению также включают, например, соль присоединения основания, которая может быть производной, например, от неорганических оснований (например, натриевая, калиевая, аммониевая соли и соли гидроксид, карбонат, бикарбонат металлов из групп I-XII Периодической таблицы, таких как кальций, магний, железо, серебро, цинк, медь и т.п.) или органических оснований (например, первичных, вторичных и третичных аминов, замещенных аминов, в том числе встречающихся в природе замещенных аминов, циклических аминов, основных ионообменных смол и т.п.). Некоторые органические амины включают, без ограничения, изопропиламин, бензатин, холинат, диэтаноламин, диэтиламин, лизин, меглумин, пиперазин и трометамин. Специалист поймет, что дополнительные кислоты или основания для образования солей присоединения кислоты/основания, иные, чем те, которые представлены в примерах, также возможны. Списки дополнительных подходящих солей можно найти, например, в "Remington's Pharmaceutical Sciences," 20th ed., Mack Publishing Company, Easton, Pa., (1985); и в "Handbook of Pharmaceutical Salts: Properties, Selection, and Use" by Stahl and Wermuth (Wiley-VCH, Weinheim, Germany, 2002).
Настоящее изобретение также охватывает активные промежуточные соединения, активные метаболиты и пролекарства соединений по настоящему изобретению. В данном документе "активное промежуточное соединение" относится к промежуточному в процессе синтеза соединению, которое проявляет такую же или по существу такую же биологическую активность, как и конечное синтезированное соединение.
В данном документе "активный метаболит" относится к разложившемуся или конечному продукту соединения по настоящему изобретению или его соли или пролекарства, образовавшемуся в результате метаболизма или биотрансформации в организме животного или человека, который проявляет такую же или по существу такую же биологическую активность, как и конкретно указанное соединение. Такие метаболиты могут образовываться в результате, например, окисления, восстановления, гидролиза, амидирования, деамидирования, этерификации, деэтерификации, ферментативного расщепления и т.п. введенного соединения или соли или пролекарства.
В данном документе "пролекарства" относятся к любым соединениям или конъюгатам, которые высвобождают активное родительское лекарственное средство после введения субъекту-животному или человеку. Пролекарства могут быть получены путем модифицирования функциональных групп, присутствующих в соединениях, таким образом, чтобы модификации расщеплялись либо при рутинной манипуляции, либо in vivo до родительских соединений. Пролекарства включают соединения, где гидроксильная, амино, сульфгидрильная или карбоксильная группа связана с любой группой, которая при введении субъекту-млекопитающему отщепляется с образованием свободной гидроксильной, амино, сульфгидрильной или карбоксильной группы соответственно. Примеры пролекарств включают, без ограничения, ацетатные, формиатные и бензоатные производные функциональных спиртовых и аминогрупп в соединениях по настоящему изобретению. Получение и применение пролекарств рассматривается в Т. Higuchi and V. Stella, "Pro-drags as Novel Delivery Systems," Vol. 14 of A.C.S. Symposium Series, и в Bioreversible Carriers in Drag Design, ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987, которые оба во всей их полноте включены в данное описание посредством ссылки.
Если конкретно не указано иное, "ErbB" или "ErbB дикого типа" относится к нормальным членам семейства ErbB. В одном аспекте настоящего изобретения предложены соединения-ингибиторы киназы семейства ErbB (например, EGFR, Her2, Her3 и/или Her4). В некоторых воплощениях соединения по настоящему изобретению могут ингибировать дикий тип (WT) и мутантные формы киназы семейства ErbB. В некоторых воплощениях соединения по настоящему изобретению являются селективными ингибиторами по меньшей мере одной мутации киназы семейства ErbB по сравнению с соответствующей киназой семейства ErbB дикого типа. Использованный в данном документе термин "мутации" относится к любым мутациям белка ErbB; "мутант" или "мутантная форма" относится к белку, который содержит указанную мутацию. Иллюстративные мутации ErbB включают, без ограничения, L858R, Т790М, G719S, G719X, delE746-A750, А763_Y764insFQEA, V769_D770insASV, Н773_V774insNPH и т.п. в EGFR, и Exon 20 insYVMA в Her2. В некоторых воплощениях соединения по настоящему изобретению являются селективными ингибиторами по меньшей мере одной мутации EGFR по сравнению с EGFR дикого типа. В некоторых воплощениях соединения по настоящему изобретению являются селективными ингибиторами по меньшей мере одной мутации Her2 по сравнению с WT Her2 (Her2 дикого типа). В некоторых воплощениях по меньшей мере одна мутация EGFR представляет собой точковую мутацию (например, L858R, Т790М). В некоторых воплощениях по меньшей мере одна мутация EGFR представляет собой делеционную мутацию (например, delE746-A750). В некоторых воплощениях по меньшей мере одна мутация EGFR представляет собой инсерционную мутацию (например, V769_D770insASV в экзоне 20 EGFR, Н773_V774insNPH в экзоне 20). В некоторых воплощениях по меньшей мере одна мутация EGFR представляет собой активирующую мутацию (например, L858R, G719S или delE746-A750). В некоторых воплощениях по меньшей мере одна мутация EGFR представляет собой мутацию, обуславливающую лекарственную устойчивость (например, Т790М в экзоне 20). В некоторых воплощениях по меньшей мере одна мутация EGFR представляет собой Т790М. В некоторых воплощениях предложенное соединение селективно ингибирует сомутацию T790M/L858R и является щадящим в том, что касается ингибирования WT EGFR (EGFR дикого типа).
Использованный в данном документе термин "селективно ингибирует" при использовании в сравнении с ингибированием WT EGFR/Her2 означает, что предложенное соединение является более сильнодействующим в качестве ингибитора по меньшей мере одной мутации EGFR/Her2 (т.е. по меньшей мере одной точковой мутации, по меньшей мере одной делеционной мутации, по меньшей мере одной инсерционной мутации, по меньшей мере одной активирующей мутации, по меньшей мере одной мутации, обуславливающей устойчивость, или комбинации по меньшей мере одной делеционной мутации и по меньшей мере одной точковой мутации) по меньшей мере в одном анализе, описанном в данном документе (например, биохимическом или клеточном). В некоторых воплощениях термин "селективно ингибирует" при использовании в сравнении с ингибированием WT EGFR означает, что предложенное соединение по меньшей мере в 100 раз более сильнодействующее, по меньшей мере в 50 раз, по меньшей мере в 45 раз, по меньшей мере в 40 раз, по меньшей мере в 35 раз, по меньшей мере в 30 раз, по меньшей мере в 25 раз, по меньшей мере в 20 раз, по меньшей мере в 15 раз, по меньшей мере в 10 раз, по меньшей мере в 5 раз, по меньшей мере в 4 раза, по меньшей мере в 3 раза, по меньшей мере в 2 раза, по меньшей мере в 1,5 раза или по меньшей мере в 1,25 раза более сильнодействующее в качестве ингибитора по меньшей мере одной мутации EGFR, как определено и описано в данном документе, по сравнению с WT EGFR. В некоторых воплощениях термин "селективно ингибирует" при использовании в сравнении с ингибированием WT EGFR означает, что предложенное соединение вплоть до 1500 раз более сильнодействующее, вплоть до 1200 раз, вплоть до 1000 раз, вплоть до 800 раз, вплоть до 600 раз, вплоть до 400 раз, вплоть до 200 раз, вплоть до 100 раз, вплоть до 50 раз, вплоть до 10 раз более сильнодействующее в качестве ингибитора по меньшей мере одной мутации EGFR, как определено и описано в данном документе, по сравнению с WT EGFR. Использованный в данном документе термин "щадящий в том, что касается WT EGFR" означает, что указанный селективный ингибитор по меньшей мере одной мутации EGFR, как определено и описано выше в данном документе, не может ингибировать WT EGFR на верхнем пределе детектирования по меньшей мере одного анализа, как описано в данном документе (например, биохимического или клеточного, как описано подробно в Примерах). В некоторых воплощениях термин "щадящий в том, что касается WT EGFR" означает, что предложенное соединение ингибирует WT EGFR при IC50 (концентрация полумаксимального ингибирования) по меньшей мере 10 мкМ, по меньшей мере 9 мкМ, по меньшей мере 8 мкМ, по меньшей мере 7 мкМ, по меньшей мере 6 мкМ, по меньшей мере 5 мкМ, по меньшей мере 3 мкМ, по меньшей мере 2 мкМ или по меньшей мере 1 мкМ.
В некоторых воплощениях соединения по настоящему изобретению ингибируют фосфорилирование WT EGFR и/или мутантного EGFR при значении IC50 0,1-1000 нМ, предпочтительно 0,1-600 нМ, 1-600 нМ, 0,1-500 нМ, 1-500 нМ, 0,1-400 нМ, 1-400 нМ, 0,1-300 нМ, 1-300 нМ, 0,1-200 нМ, 1-200 нМ, 0,1-100 нМ, 1-100 нМ, 0,1-80 нМ, 0,1-50 нМ, 0,1-40 нМ, 0,1-30 нМ, 0,1-20 нМ, 0,1-10 нМ или 0,1-5 нМ, более предпочтительно 0,1-20 нМ, 0,1-10 нМ или 0,1-5 нМ.
В некоторых воплощениях соединения по настоящему изобретению ингибируют фосфорилирование WT Her2 и/или мутантного Her2 при значении IC50 0,1-1000 нМ, предпочтительно 0,1-600 нМ, 1-600 нМ, 0,1-500 нМ, 1-500 нМ, 0,1-400 нМ, 1-400 нМ, 0,1-300 нМ, 1-300 нМ, 0,1-200 нМ, 1-200 нМ, 0,1-100 нМ, 1-100 нМ, 0,1-80 нМ, 0,1-50 нМ, 0,1-40 нМ, 0,1-30 нМ, 0,1-20 нМ, 0,1-10 нМ или 0,1-5 нМ, более предпочтительно 0,1-20 нМ, 0,1-10 нМ или 0,1-5 нМ.
В некоторых воплощениях соединения по настоящему изобретению ингибируют пролиферацию клеток, несущих WT EGFR и/или мутантный EGFR, при значении GI50 (концентрация полумаксимального ингибирования пролиферации клеток) 1-1000 нМ, предпочтительно 1-800 нМ, 1-600 нМ, 1-500 нМ, 1-400 нМ, 1-300 нМ, 1-300 нМ, 1-200 нМ, 1-100 нМ, 1-80 нМ, 1-60 нМ, 1-40 нМ, 1-20 нМ или 1-10 нМ, более предпочтительно 1-300 нМ, 1-200 нМ, 1-100 нМ, 1-80 нМ, 1-60 нМ, 1-40 нМ, 1-20 нМ или 1-10 нМ.
В некоторых воплощениях соединения по настоящему изобретению ингибируют пролиферацию клеток, несущих WT Her2 и/или мутантный Her2, при значении GI50 1-1000 нМ, предпочтительно 1-800 нМ, 1-600 нМ, 1-500 нМ, 1-400 нМ, 1-300 нМ, 1-300 нМ, 1-200 нМ, 1-100 нМ, 1-80 нМ, 1-60 нМ, 1-40 нМ, 1-20 нМ или 1-10 нМ, более предпочтительно 1-300 нМ, 1-200 нМ, 1-100 нМ, 1-80 нМ, 1-60 нМ, 1-40 нМ, 1-20 нМ или 1-10 нМ.
В некоторых воплощениях соединения по настоящему изобретению ингибируют пролиферацию клеток, несущих BTK, при значении GI5 1-1000 нМ, более чем 1000 нМ, более чем 2000 нМ или более чем 3000 нМ, предпочтительно 1-800 нМ, 1-600 нМ, 1-500 нМ, 1-400 нМ, 1-300 нМ, 1-300 нМ, 1-200 нМ, 1-100 нМ, 1-80 нМ, 1-60 нМ, 1-40 нМ, 1-20 нМ или 1-10 нМ, более предпочтительно 1-300 нМ, 1-200 нМ, 1-100 нМ, 1-80 нМ, 1-60 нМ, 1-40 нМ, 1-20 нМ или 1-10 нМ.
В некоторых воплощениях IC50 и/или GI50 соединений для мутанта EGFR по меньшей мере в 2 раза, 3 раза, 4 раза, 5 раз, предпочтительно в 10 раз, 20 раз, 30 раз, 50 раз или 100 раз выше, чем IC50 и/или GI50 соединений для EGFR дикого типа.
Способ синтеза
Синтез соединений, предложенных в данном документе, включая их соли, сложные эфиры, гидраты или сольваты или стереоизомеры, иллюстрируется на схемах синтеза в примерах. Соединения, предложенные в данном документе, могут быть получены с использованием известных методов органического синтеза и могут быть синтезированы в соответствии с любыми многочисленными известными путями синтеза, и поэтому эти схемы являются только иллюстративными и не исключают другие возможные способы, которые могут быть использованы для получения соединений, предложенных в данном документе. Дополнительно, стадии на Схемах предназначены для лучшего иллюстрирования и могут быть изменены, если это целесообразно. Воплощения соединений в Примерах были синтезированы в Китае в целях исследования и потенциального представления регулирующим органам.
Реакции для получения соединений по изобретению могут быть проведены в подходящих растворителях, которые легко может выбрать специалист в области органического синтеза. Подходящие растворители могут быть по существу нереакционноспособными с исходными веществами (реагентами), промежуточными соединениями или продуктами при температурах, при которых проводятся реакции, например при температурах в диапазоне от температуры замерзания растворителя до температуры кипения растворителя. Данная реакция может быть проведена в одном растворителе или в смеси растворителей больше одного. В зависимости от конкретной реакционной стадии подходящие растворители для конкретной реакционной стадии могут быть выбраны специалистом.
Получение соединений по изобретению может включать в себя защиту и снятие защиты с различных химических групп. Необходимость в защите и снятии защиты и выбор соответствующих защитных групп легко может определить специалист в данной области. Химию защитных групп можно найти, например, в Т. W. Greene and P. G. M. Wilts, Protective Groups in Organic Synthesis, 3rd Ed., Wiley & Sons, Inc., New York (1999), который во всей полноте включен в данное описание посредством ссылки.
Реакции можно контролировать любыми методами, известными в данной области. Например, образование продукта можно контролировать спектроскопическими методами, такими как спектроскопия ядерного магнитного резонанса (например, 1Н или 13С), инфракрасная спектроскопия, спектрофотометрия (например, в УФ-видимом диапазоне), масс-спектрометрия, или хроматографическими методами, такими как высокоэффективная жидкостная хроматография (ВЭЖХ), жидкостная хроматография/масс-спектроскопия (ЖХ/МС) или тонкослойная хроматография (ТСХ). Соединения могут быть очищены специалистами в данной области различными методами, в том числе методом высокоэффективной жидкостной хроматографии (ВЭЖХ) ("Preparative LC-MS Purification: Improved Compound Specific Method Optimization" Karl F. Blom, Brian Glass, Richard Sparks, Andrew P. Combs J. Combi. Chem. 2004, 6(6), 874-883, который во всей его полноте включен в данное описание посредством ссылки) и нормально-фазной хроматографии на диоксиде кремния.
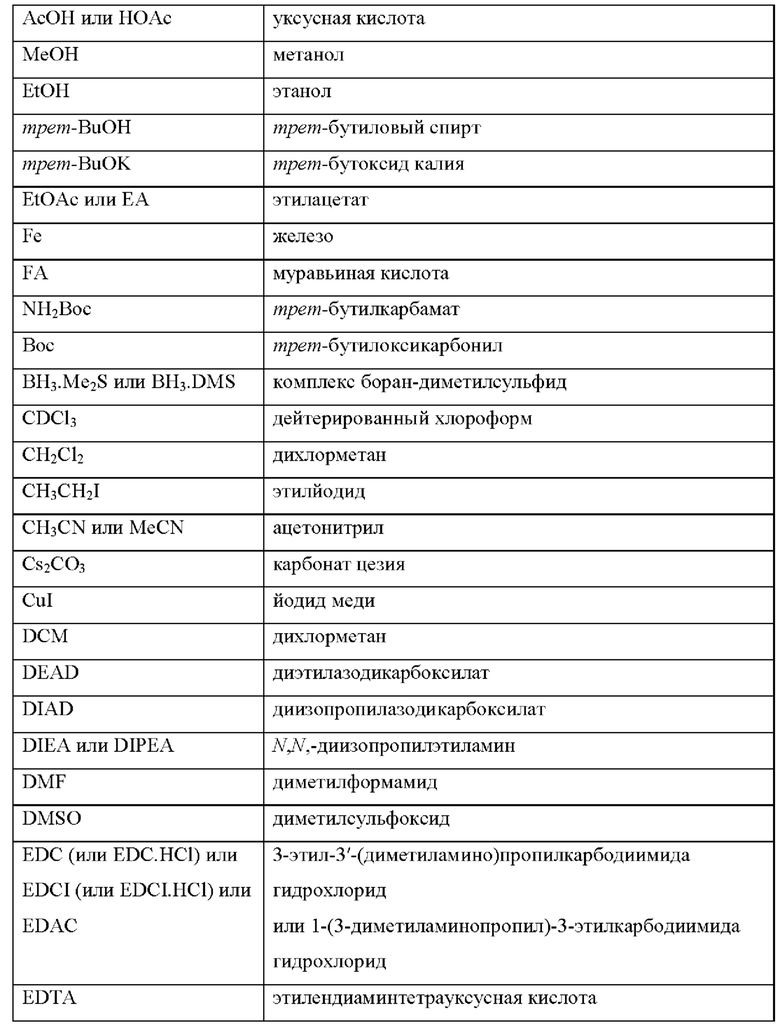
Сокращения, использованные в данном документе, определены следующим образом: "1 х" или "х 1" означают один раз; "2 х" или "х 2" означают дважды; "3 х" или "х 3" означают трижды; "4 х" или "х 4" означают четыре раза; "5 х" или "х 5" означают пять раз; "°С" означает градусы Цельсия; "эк." или "экв." означают эквивалент или эквиваленты; "г" означает грамм или граммы; "мг" означает миллиграмм или миллиграммы; "л" означает литр или литры; "мл" или "мл" означает миллилитр или миллилитры; "мкл" означает микролитр или микролитры; "н." означает нормальный; "М" означает молярный; "ммоль" означает миллимоль или миллимоли; "мин" означает минута или минуты; "ч" означает час или часы; "к.т." или "кт" означают комнатная температура; "атм" означает атмосфера; "фунт/кв.дюйм" означает фунты на квадратный дюйм; "конц." означает концентрировать; "насыщ." означает насыщенный; "МС" или "масс-спек." означает масс-спектрометрия; "ЭРИ" означает масс-спектроскопия с электрораспылительной ионизацией; "ЖХ/МС" означает жидкостная хроматография-масс-спектрометрия; "ЖХВД" означает жидкостная хроматография высокого давления; "ОФ" означает обращенно-фазная; "ТСХ" или "тех" означает тонкослойная хроматография; "SM" означает исходное вещество; "ЯМР" означает спектроскопия ядерного магнитного резонанса; "1Н" означает протон; "δ" означает дельта; "s" означает синглет; "d" означает дублет; "t" означает триплет; "q" означает квартет; "m" означает мультиплет; "br" означает уширенный; и "Гц" означает герцы, "α", "β", "R", "S", "Е" и "Z" являются стереохимическими обозначениями, известными специалисту в данной области.

Фармацевтическая композиция
Согласно настоящему изобретению предложена фармацевтическая композиция, содержащая по меньшей мере одно соединение по настоящему изобретению. В некоторых воплощениях фармацевтическая композиция содержит более чем одно соединение по настоящему изобретению. В некоторых воплощениях фармацевтическая композиция содержит одно или более соединений по настоящему изобретению и фармацевтически приемлемый носитель.
Фармацевтически приемлемые носители представляют собой традиционные в данной области медицинские носители, которые могут быть получены способом, общеизвестным в фармацевтической области. В некоторых воплощениях соединения по настоящему изобретению могут быть смешаны с фармацевтически приемлемым носителем для приготовления фармацевтической композиции.
Термин "фармацевтически приемлемый носитель" в данном документе относится к фармацевтически приемлемому веществу, композиции или несущей среде, таким как жидкий или твердый наполнитель, разбавитель, эксципиент, растворитель или инкапсулирующий материал, задействованный в переносе или транспортировке соединения, предложенного в данном документе, из одного местоположения, жидкости организма, ткани, органа (внутреннего или внешнего) или участка организма в другое местоположение, жидкость организма, ткань, орган или участок организма. Фармацевтически приемлемыми носителями могут быть несущие среды, разбавители, эксципиенты или другие вещества, которые можно применять в контакте с тканями животного без излишней токсичности или неблагоприятных эффектов. Иллюстративные фармацевтически приемлемые носители включают сахара, крахмал, целлюлозы, солод, трагакант, желатин, раствор Рингера, альгиновую кислоту, изотонический физиологический раствор, буферные агенты и т.п. Фармацевтически приемлемый носитель, который может быть использован в настоящем изобретении, включает общеизвестные в данной области носители, такие как те, которые описаны в "Remington Pharmaceutical Sciences" Mack Pub. Co., New Jersey (1991), который включен в данное описание посредством ссылки.
Некоторые примеры веществ, которые могут служить фармацевтически приемлемыми носителями, включают: (1) сахара, такие как лактоза, глюкоза и сахароза; (2) крахмалы, такие как кукурузный крахмал и картофельный крахмал; (3) целлюлозу и ее производные, такие как натрий-карбоксиметилцеллюлоза, этилцеллюлоза и ацетат целлюлозы; (4) порошкообразный трагакант; (5) солод; (6) желатин; (7) тальк; (8) эксципиенты, такие как масло какао и суппозиторные воски; (9) масла, такие как арахисовое масло, хлопковое масло, подсолнечное масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; (10) гликоли, такие как пропиленгликоль; (11) пол иолы, такие как глицерин, сорбит, маннит и полиэтиленгликоль; (12) сложные эфиры, такие как этилолеат и этиллаурат; (13) агар; (14) буферные агенты, такие как гидроксид магния и гидроксид алюминия; (15) альгиновую кислоту; (16) апирогенную воду; (17) изотонический физиологический раствор; (18) раствор Рингера; (19) спирт, такой как этиловый спирт и пропиловый спирт; (20) фосфатные буферные растворы; и (21) другие нетоксичные совместимые вещества, используемые в фармацевтических препаратах, такие как ацетон.
Фармацевтические композиции могут содержать фармацевтически приемлемые вспомогательные вещества, если это необходимо для приближения к физиологическим условиям, такие как регулирующие рН и буферные агенты, агенты, регулирующие токсичность, и т.п., например ацетат натрия, хлорид натрия, хлорид калия, хлорид кальция, лактат натрия и т.п.
Форма фармацевтической композиции зависит от ряда критериев, включающих, без ограничения, путь введения, степень заболевания или дозу, которую нужно вводить.
Фармацевтические композиции могут быть приготовлены для перорального, назального, ректального, подкожного, внутривенного или внутримышечного введения. В соответствии с желаемым путем введения фармацевтические композиции могут быть приготовлены в форме таблеток, капсул, пилюль, драже, порошка, гранул, саше, облаток, лепешек, суспензий, эмульсий, растворов, сиропов, аэрозолей (в виде твердого вещества или жидкой среды), спрея, мази, пасты, крема, лосьона, геля, пластыря, ингалятора или суппозитория.
Фармацевтические композиции, обеспечивающие быстрое, длительное или отсроченное высвобождение активного ингредиента после введения пациенту, могут быть приготовлены с использованием методик, известных в данной области. В некоторых воплощениях фармацевтическая композиция приготовлена в форме длительного высвобождения. Использованный в данном документе термин "форма длительного высвобождения" относится к высвобождению активного агента из фармацевтической композиции таким образом, чтобы он был доступным для биологического всасывания у субъекта, в основном в желудочно-кишечном тракте субъекта, в течение пролонгированного периода времени (замедленное высвобождение) или в определенном местоположении (контролируемое высвобождение). В некоторых воплощениях пролонгированный период времени может составлять примерно от 1 часа до 24 часов, от 2 часов до 12 часов, от 3 часов до 8 часов, от 4 часов до 6 часов, от 1 до 2 суток или больше. В некоторых воплощениях пролонгированный период времени составляет по меньшей мере примерно 4 часа, по меньшей мере примерно 8 часов, по меньшей мере примерно 12 часов или по меньшей мере примерно 24 часа. Фармацевтическая композиция может быть приготовлена в форме таблетки. Например, скорость высвобождения активного агента может контролироваться не только растворением активного агента в желудочно-кишечном тракте и последующей диффузией из таблетки или пилюли независимо от рН, но может также зависеть от физических процессов разрыхления и эрозии таблетки. В некоторых воплощениях для длительного высвобождения могут быть использованы полимерные материалы, которые описаны в "Medical Applications of Controlled Release," Langer and Wise (eds.), CRC Pres., Boca Raton, Florida (1974); "Controlled Drug Bioavailability," Drug Product Design and Performance, Smolen and Ball (eds.), Wiley, New York (1984); Ranger and Peppas, 1983, J Macromol. Sci. Rev. Macromol Chem. 23:61; смотри также Levy et al., 1985, Science 228:190; During et al., 1989, Ann. Neurol. 25:351; Howard et al., 1989, J. Neurosurg. 71:105. Вышеуказанные источники информации во всей их полноте включены в данное описание посредством ссылки.
В некоторых воплощениях фармацевтические композиции содержат от примерно 0,0001 мг до примерно 5000 мг соединения по настоящему изобретению (например, от примерно 0,0001 мг до примерно 10 мг, от примерно 0,001 мг до примерно 10 мг, от примерно 0,01 мг до примерно 10 мг, от примерно 0,1 мг до примерно 10 мг, от примерно 1 мг до примерно 10 мг, от примерно 5 мг до примерно 10 мг, от примерно 5 мг до примерно 20 мг, от примерно 5 мг до примерно 30 мг, от примерно 5 мг до примерно 40 мг, от примерно 5 мг до примерно 50 мг, от примерно 10 мг до примерно 100 мг, от примерно 20 мг до примерно 100 мг, от примерно 30 мг до примерно 100 мг, от примерно 40 мг до примерно 100 мг, от примерно 50 мг до примерно 100 мг, от примерно 50 мг до примерно 200 мг, от примерно 50 мг до примерно 300 мг, от примерно 50 мг до примерно 400 мг, от примерно 50 мг до примерно 500 мг, от примерно 100 мг до примерно 200 мг, от примерно 100 мг до примерно 300 мг, от примерно 100 мг до примерно 400 мг, от примерно 100 мг до примерно 500 мг, от примерно 200 мг до примерно 500 мг, от примерно 300 мг до примерно 500 мг, от примерно 400 мг до примерно 500 мг, от примерно 500 мг до примерно 1000 мг, от примерно 600 мг до примерно 1000 мг, от примерно 700 мг до примерно 1000 мг, от примерно 800 мг до примерно 1000 мг, от примерно 900 мг до примерно 1000 мг, от примерно 1000 мг до примерно 2000 мг, от примерно 2000 мг до примерно 3000 мг, от примерно 3000 мг до примерно 4000 мг или от примерно 4000 мг до примерно 5000 мг). Подходящие дозировки на субъекта в сутки могут составлять от примерно 5 мг до примерно 500 мг, предпочтительно от примерно 5 мг до примерно 50 мг, от примерно 50 мг до примерно 100 мг или от примерно 50 мг до примерно 500 мг.
В некоторых воплощениях фармацевтические композиции могут быть приготовлены в стандартных лекарственных формах, каждая из которых содержит от примерно 0,0001 мг до примерно 10 мг, от примерно 0.001 мг до примерно 10 мг, от примерно 0,01 мг до примерно 10 мг, от примерно 0,1 мг до примерно 10 мг, от примерно 1 мг до примерно 10 мг, от примерно 5 мг до примерно 10 мг, от примерно 5 мг до примерно 20 мг, от примерно 5 мг до примерно 30 мг, от примерно 5 мг до примерно 40 мг, от примерно 5 мг до примерно 50 мг, от примерно 10 мг до примерно 100 мг, от примерно 20 мг до примерно 100 мг, от примерно 30 мг до примерно 100 мг, от примерно 40 мг до примерно 100 мг, от примерно 50 мг до примерно 100 мг, от примерно 50 мг до примерно 200 мг, от примерно 50 мг до примерно 300 мг, от примерно 50 мг до примерно 400 мг, от примерно 50 мг до примерно 500 мг, от примерно 100 мг до примерно 200 мг, от примерно 100 мг до примерно 300 мг, от примерно 100 мг до примерно 400 мг, от примерно 100 мг до примерно 500 мг, от примерно 200 мг до примерно 500 мг, от примерно 300 мг до примерно 500 мг, от примерно 400 мг до примерно 500 мг, от примерно 500 мг до примерно 1000 мг, от примерно 600 мг до примерно 1000 мг, от примерно 700 мг до примерно 1000 мг, от примерно 800 мг до примерно 1000 мг, от примерно 900 мг до примерно 1000 мг, от примерно 1000 мг до примерно 2000 мг, от примерно 2000 мг до примерно 3000 мг, от примерно 3000 мг до примерно 4000 мг или от примерно 4000 мг до примерно 5000 мг соединения по настоящему изобретению. Термин "стандартные лекарственные формы" относится к физически дискретным единицам, подходящим в качестве однократных дозировок для субъектов-людей и других млекопитающих, причем каждая единица содержит предопределенное количество активного вещества, рассчитанного на получение желаемого терапевтического эффекта, совместно с подходящим фармацевтическим носителем. В некоторых воплощениях фармацевтические композиции содержат одно или более соединений по настоящему изобретению в качестве первого активного ингредиента и дополнительно содержат второй активный ингредиент. Вторым активным ингредиентом может быть любой противораковый агент, известный в данной области, например ингибиторы трансдукции клеточных сигналов, алкилирующие агенты, ингибиторы топоизомеразы, иммунотерапевтические агенты, ингибиторы митоза, антигормональные агенты, химиотерапевтические лекарственные средства, ингибиторы EGFR, ингибиторы BTK, ингибиторы CTLA-4, ингибиторы MEK, ингибиторы PD-L1; агонисты ОХ40 и т.п. Репрезентативные примеры противораковых агентов для лечения рака и опухолей могут включать, без ограничения, сорафениб, сунитиниб, дазатиниб, вориностат, темсиролимус, эверолимус, пазопаниб, трастузумаб, адо-трастузумаб эмтанзин, пертузумаб, бевацизумаб, цетуксимаб, ранибизумаб, пегаптаниб, панитумумаб, тремелимумаб, пембролизумаб, ниволумаб, ипилимумаб, атезолизумаб, авелумаб, дуравалумаб, кризотиниб, руксолитиниб, паклитаксел, винкристин, винбластин, цисплатин, карбоплатин, гемцитабин, тамоксифен, ралоксифен, циклофосфамид, хлорамбуцил, кармустин, метотрексат, фторурацил, актиномицин, доксорубицин, эпирубицин, антрациклин, блеомицин, митомицин-С, иринотекан, топотекан, тенипозид, интерлейкин, интерферон и т.п. В некоторых воплощениях второй активный агент представляет собой один или более из бевацизумаба, пембролизумаба, ниволумаба, ипилимумаба, атезолизумаба, авелумаба, дурвалумаба, кризотиниба.
Способ лечения
Согласно настоящему изобретению предложен способ лечения заболевания, ассоциированного с ErbB (включая, например, EGFR или Her2), в частности с мутацией ErbB, включающий введение субъекту эффективного количества одного или более соединений, их фармацевтически приемлемых солей, сложных эфиров, гидратов, сольватов или стереоизомеров или фармацевтической композиции по настоящему изобретению.
Согласно настоящему изобретению предложен также способ лечения заболевания, ассоциированного с BTK. В некоторых воплощениях этот способ включает введение субъекту эффективного количества одного или более соединений, их фармацевтически приемлемых солей, сложных эфиров, гидратов, сольватов или стереоизомеров или фармацевтической композиции по настоящему изобретению.
Использованные в данном документе термины "заболевания, ассоциированные с ErbB" или "ассоциированные с ErbB заболевания" относятся к заболеваниям, чье начало или развитие или и то, и другое связано с экспрессией или активностью ErbB. Примеры включают, без ограничения, ассоциированные с иммунитетом заболевания, пролиферативные расстройства, рак и другие заболевания.
Использованные в данном документе термины "заболевания, ассоциированные с EGFR", или "ассоциированные с EGFR заболевания", или "заболевания, ассоциированные с Her2", или "ассоциированные с Her2 заболевания" относятся к заболеваниям, чье начало или развитие или и то, и другое связано с геномными изменениями, экспрессией или активностью EGFR или Her2, в зависимости от обстоятельств. Примеры включают, без ограничения, ассоциированные с иммунитетом заболевания, пролиферативные расстройства, рак и другие заболевания.
Использованные в данном документе термины "заболевания, ассоциированные с BTK" или "ассоциированные с BTK заболевания" относятся к заболеваниям, чье начало или развитие или и то, и другое связано с геномными изменениями, экспрессией или активностью BTK, в зависимости от обстоятельств. В некоторых воплощениях ассоциированные с BTK заболевания включают онкологические заболевания и аутоиммунные заболевания. Онкологические заболевания включают, без ограничения, лимфому и лейкоз. Аутоиммунные заболевания включают, без ограничения, ревматоидный артрит, системную красную волчанку и синдром Шегрена.
Использованные в данном документе термины "лечение", "лечить" и "проведение лечения" относятся к реверсированию, ослаблению, задержке начала или торможению прогрессирования заболевания или расстройства или одного или более его симптомов, как описано в данном документе. В некоторых воплощениях лечение может быть назначено после проявления одного или более симптомов. В других воплощениях лечение может быть назначено в отсутствии симптомов. Например, лечение может быть назначено восприимчивой личности до появления симптомов (например, в свете истории симптомов и/или в свете генетических или других факторов восприимчивости). Лечение также может быть продолжено после устранения симптомов, например с целью предупреждения или задержки их рецидива.
В некоторых воплощениях предложенные в данном документе одно или более соединений, их фармацевтически приемлемых солей, сложных эфиров, гидратов, сольватов или стереоизомеров или фармацевтическую композицию вводят парентеральным путем или не парентеральным путем. В некоторых воплощениях одно или более соединений, их фармацевтически приемлемых солей, сложных эфиров, гидратов, сольватов или стереоизомеров или фармацевтическую композицию вводят перорально, энтерально, трансбуккально, назально, интраназально, трансмукозально, эпидермально, трансдермально, дермально, офтальмически, пульмонарно, сублингвально, ректально, вагинально, местно, подкожно, внутривенно, внутримышечно, внутриартериально, интратекально, интракапсулярно, интраорбитально, интракардиально, интрадермально, интаперитонеально, транстрахеально, субкутикулярно, интраартикулярно, субкапсулярно, субарахноидально, интраспинально или интрастернально.
Соединения, предложенные в данном документе, можно вводить в чистой форме, в комбинации с другими активными ингредиентами или в форме фармацевтической композиции по настоящему изобретению. В некоторых воплощениях соединения, предложенные в данном документе, можно вводить нуждающемуся субъекту одновременно или последовательно в комбинации с одним или более противораковым(и) агентом(ами), известным(и) в данной области. В некоторых воплощениях введение проводят один раз в сутки, дважды в сутки, три раза в сутки или один раз каждые двое суток, один раз каждые трое суток, один раз каждые четверо суток, один раз каждые пять суток, один раз каждые шесть суток, один раз в неделю.
В некоторых воплощениях предложенные в данном документе одно или более соединений, их фармацевтически приемлемых солей, сложных эфиров, гидратов, сольватов или стереоизомеров или фармацевтическую композицию вводят перорально. Для перорального введения подходит любая доза, которая обеспечивает достижение желаемой цели. В некоторых воплощениях подходящие суточные дозировки составляют от примерно 0,001-5000 мг, предпочтительно от 0,1 мг до 5 г, более предпочтительно от 5 мг до 1 г, более предпочтительно от 10 мг до 500 мг, и введение проводят один раз в сутки, дважды в сутки, три раза в сутки, каждый день или 3-5 суток в неделю. В некоторых воплощениях доза предложенных в данном документе одного или более соединений, их фармацевтически приемлемых солей, сложных эфиров, гидратов, сольватов или стереоизомеров или фармацевтической композиции находится в диапазоне от примерно 0,0001 мг, предпочтительно 0,001 мг, 0,01 мг, 0,1 мг, 1 мг, 10 мг, 50 мг, 100 мг, 200 мг, 250 мг, 500 мг, 750 мг, 1000 мг, 2000 мг, 3000 мг, 4000 мг или вплоть до примерно 5000 мг в сутки.
Применение соединений
В некоторых воплощениях настоящего изобретения предложено применение соединений, их фармацевтически приемлемых солей, сложных эфиров, гидратов, сольватов или стереоизомеров или фармацевтической композиции по настоящему изобретению в изготовлении лекарственных средств для лечения заболеваний, ассоциированных с ErbB (например, EGFR, Her2, Her3 или Her4). В некоторых воплощениях настоящего изобретения предложено применение соединений, их фармацевтически приемлемых солей, сложных эфиров, гидратов, сольватов или стереоизомеров или фармацевтической композиции по настоящему изобретению в изготовлении лекарственных средств для лечения заболеваний, ассоциированных с мутантным ErbB. В некоторых воплощениях мутантный ErbB представляет собой мутантный EGFR. В некоторых воплощениях мутантный ErbB представляет собой мутантный Her2. В некоторых воплощениях заболевания, ассоциированные с ErbB, представляют собой заболевания, ассоциированные с мутантным ErbB, включая раковые заболевания.
В некоторых воплощениях настоящего изобретения предложено применение соединений, их фармацевтически приемлемых солей, сложных эфиров, гидратов, сольватов или стереоизомеров или фармацевтической композиции по настоящему изобретению в изготовлении лекарственных средств для лечения заболеваний, ассоциированных с BTK. В некоторых воплощениях настоящего изобретения предложено применение соединений, их фармацевтически приемлемых солей, сложных эфиров, гидратов, сольватов или стереоизомеров или фармацевтической композиции по настоящему изобретению в изготовлении лекарственных средств для лечения заболеваний, ассоциированных с BTK. В некоторых воплощениях заболевания, ассоциированные с BTK, включают раковые заболевания.
В частности, раковые заболевания включают, без ограничения, лейкоз, глиобластому, меланому, хондросаркому, холангиокарциному остеосаркому лимфому, рак легкого, аденому, миелому печеночноклеточную карциному, адренокортикальную карциному, рак поджелудочной железы, рак молочной железы, рак мочевого пузыря, рак предстательной железы, рак печени, рак желудка, рак ободочной кишки, колоректальный рак, рак яичника, рак шейки матки, рак головного мозга, рак пищевода, рак кости, рак яичка, рак кожи, рак почки, мезотелиому нейробластому, рак щитовидной железы, рак в области головы и шеи, рак пищевода, рак глаза, рак предстательной железы, назофарингеальный рак или рак ротовой полости. В некоторых воплощениях раковые заболевания представляют собой рак легкого, рак молочной железы, рак яичника, рак мочевого пузыря или глиобластому. В некоторых воплощениях рак представляет собой рак легкого (например, не мелко клеточный рак легкого, мелкоклеточный рак легкого, аденокарциному, плоскоклеточный рак легкого и крупноклеточный рак легкого). В некоторых воплощениях рак представляет собой метастатический рак легкого. В некоторых воплощениях рак представляет собой рак с одной или более мутациями ErbB (например, точковыми мутациями, делеционными мутациями, инсерционными мутациями, активирующими мутациями или обуславливающими лекарственную устойчивость мутациями EGFR или Her2).
Соединения и их фармацевтические композиции по настоящему изобретению можно применять в предупреждении или лечении начала или развития любого из заболеваний или состояний, ассоциированных с ErbB/BTK (экспрессией или активностью), у млекопитающих, в частности у людей. В некоторых воплощениях соединения и их фармацевтические композиции по настоящему изобретению можно применять в предупреждении или лечении начала или развития любого из заболеваний или состояний, ассоциированных с мутантным ErbB, у млекопитающих, в частности у людей.
В такой ситуации согласно настоящему изобретению предложен также способ скрининга пациентов, подходящих для лечения соединениями или фармацевтической композицией по настоящему изобретению, одними или совместно с другими ингредиентами (например, со вторым активным ингредиентом, например противораковым агентом). Способ включает секвенирование образцов опухолей, взятых у пациентов, и детектирование накопления ErbB (например, EGFR или Her2) или BTK у пациента или детектирование мутационного статуса ErbB (например, EGFR или Her2) или BTK у пациента.
ПРИМЕРЫ
Нижеследующие примеры дополнительно объясняют общие способы по настоящему изобретению. Соединения по настоящему изобретению могут быть получены способами, известными в данной области. Нижеследующие примеры подробно иллюстрирует способы получения предпочтительных соединений по настоящему изобретению. Однако они никоим образом не ограничивают способы получения соединений по настоящему изобретению.
ПРИМЕРЫ СИНТЕЗА
Структуры соединений в нижеследующих примерах были охарактеризованы спектроскопией ядерного магнитного резонанса (ЯМР) и/или масс-спектрометрией (МС). ЯМР сдвиг (δ) приведен в единицах 10-6 (м.д. (миллионные доли)). 1Н-ЯМР спектры регистрировали в диметилсульфоксиде-d6 (DMSO-d6), или CDCl3, или CD3OD, или D2O (от Aldrich или Cambridge Isotope Lab., Inc.) на спектрометрах Bruker AVANCE NMR (400 МГц) с использованием ICON-NMR (управляемых программой TopSpin), или на спектрометрах Varian 400 MR NMR или Varian VNMR400 NMR (400 МГц) (управляемых программой VnmrJ) с тетраметилсиланом в качестве внутреннего стандарта.
МС измерение проводили с использованием масс-спектрометра Shimadzu 2010, или Agilent 6110А MSD, или 1969А TOF с использованием методов электрораспылительной ионизации, химической ионизации и ионизации электронным ударом исходя из предела измерения приборов.
Измерения методом высокоэффективной жидкостной хроматографии (ВЭЖХ) проводили на системах серии Shimadzu LC-20A или Shimadzu LC-2010HT, или серии Agilent 1200 LC или Agilent 1100 с использованием колонки Ultimate ХВ-С18 (3,0×50 мм, 3 мкм, или 3,0×150 мм, 3 мкм), или колонки Xbridge shieldRPIS (5 мкм, 50 мм × 2,1 мм), или колонки Xtimate С18 (3 мкм, 2,1×30 мм), или колонки MERCK RP18 2,5-2 мм, или колонки Agilent Zorbax Eclipse Plus CI8 (4,6 мм × 150 мм, 5 мкм) и т.д.
Тонкослойную хроматографию проводили с использованием силикагелевых пластин Yantai Huanghai HSGF254 или Anhui Liang Chen Gui Yuans. Силикагелевые пластины, использованные для тонкослойной хроматографии (ТСХ), были 0,15 мм-0,2 мм. Силикагелевые пластины, использованные для разделения и очистки продуктов методом ТСХ, были 0,4 мм-0,5 мм.
Для очистки использовали колонку с силикагелем в качестве носителя (100-200, 200-300 или 300-400 меш, компании Yantai Huanghai со. или Anhui Liang Chen Gui Yuan со., etc.), или флэш-колонку (флэш-колонка с диоксидом кремния-CS 40-60 мкм, или обращенно-фазную С18 колонку 20-35 мкм компании Agela Technologies, etc.), или флэш-колонку с диоксидом кремния-CS (40-60 мкм), или С18 колонку (20-40 мкм) компании Agela Technologies в комби-флэш-системе Teledyne ISCO или флэш-системе Biotage. Размеры колонок подбирали исходя из количества соединений.
Известные исходные вещества по настоящему изобретению могут быть синтезированы с использованием способов, известных в данной области или согласно им, или они могут быть приобретены у Alfa Aesar, Langcaster, TCI, Aldrich, Bepharm и Scochem (или PharmaBlock, Bide, Amatek, Stru Chem, Firster Pharmaceutical, Titan (Adamas) и т.д.).
Если конкретно не указано иное, реакции в примерах все проводили в атмосфере аргона или азота. В атмосфере аргона или азота означает, что реакционная колба соединена с аргоновым или азотным баллоном объемом примерно 1 л. Гидрирование обычно проводили под давлением. Если конкретно не указано иное, реакционной температурой в примерах была температура окружающей среды, которая составляла 20°С-30°С.
Протекание реакций, описанных в примерах, отслеживали методом ТСХ. Использованные для реакций системы элюентов включают систему дихлорметан-метанол и систему петролейный эфир-этилацетат. Объемные соотношения растворителей подбирали исходя из различной полярности соединений.
Система элюирования для колоночной хроматографии, использованной для очистки соединений, и система элюентов для ТСХ включают систему дихлорметан-метанол и систему петролейный эфир-этилацетат. Объемные соотношения растворителей подбирали исходя из различной полярности соединений. Небольшое количество щелочного или кислотного агента (0,1%-1%), такого как муравьиная кислота, или уксусная кислота, или TFA, или аммиак, может быть добавлено для корректировки.
Пример 1
(R)-N-(2-(3-(диметиламино)пиперидин-1-ил)-5-(4-(2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-4-метоксифенил)акриламид

Методика получения соединения 1b:
В раствор соединения 1а (25 г, 1,0 экв., 133,59 ммоль) в THF (250 мл) добавляли CH3MgBr (222,65 мл, 5,0 экв., 667,95 ммоль) при 0-5°С в бане лед-вода. Полученную черную смесь перемешивали при 26-36°С в течение 2 ч до тех пор, пока ТСХ (петролейный эфир/EtOAc = 5/1 (об./об.)) не показала, что исходное вещество (Rf = 0,70) израсходовалось. Реакционную смесь разбавляли насыщенным водным NH4Cl (500 мл) и экстрагировали EtOAc (2×300 мл). Объединенные органические слои концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле (петролейный эфир/EtOAc = 10/1 (об./об.)) с получением соединения 1b (22 г, 88%-ный выход) в виде желтого масла.
ЖХ/МС: Rt = 0,678 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z = 169,9 [М+Н-18]+.
1Н ЯМР (400 МГц, MeOH-d4) δ 6.97 (dd, J=8.9, 12,7 Гц, 1Н), 6.52 (dd, J=7.3, 12,8 Гц, 1Н), 1.57 (s, 6Н).
Методика получения соединения 1с:
В раствор соединения 1b (1 г, 5,3 ммоль) и DIEA (1 г, 7,9 ммоль) в CH2Cl2 (10 мл) добавляли 2,4-дихлор-1,3,5-триазин (0,96 г, 6,4 ммоль). Смесь перемешивали при 26-34°С в течение 12 часов. Реакционную смесь напрямую очищали колоночной хроматографией на силикагеле (20% EtOAc в петролейном эфире (об./об.)) с получением указанного в заголовке продукта 1с в виде белого твердого вещества (900 мг, 56%-ный выход).
ЖХ/МС: Rt = 0,803 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z = 300,9 [М+Н]+.
1Н ЯМР (400 МГц, DMSO-d6) δ 10.84 (s, 1Н), 8.68 (s, 1Н), 8.10 (s, 1Н), 7.46 (dd, J=8.8, 12,4 Гц, 1Н), 6.35 (br s, 1H), 1.51 (s, 6H).
Методика получения соединения 1d:
В раствор соединения 1 с (300 мг, 1,0 ммоль) и 4-фтор-2-метокси-5-нитроанилина (185 мг, 1,0 ммоль) в n-BuOH (10 мл) добавляли TFA (0,1 мл). Полученную смесь перемешивали при 26-33°С в течение 2 часов. Выпавшее в осадок твердое вещество собирали фильтрованием и затем сушили в глубоком вакууме с получением указанного в заголовке продукта 1d в виде желтого твердого вещества (400 мг, чистота 99%, 88%-ный выход).
ЖХ/МС: Rt = 0,828 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z = 451,1 [М+Н]+.
Методика получения соединения 1е:
Раствор соединения 1d (1,144 г, 2,76 ммоль), (R)-N,N-диметилпиперидин-3-амина гидрохлорида (500 мг, 3,04 ммоль) и K2CO3 (764 мг, 5,52 ммоль) в DMSO (30 мл) перемешивали при 100°С в течение 12 часов. Реакционную смесь добавляли в холодную воду (200 мл). Желтое твердое вещество выпало в осадок, затем остаток очищали колоночной хроматографией на силикагеле (градиентное элюирование: CH2Cl2/МеОН от 100/0 до 90/10) с получением указанного в заголовке продукта 1е в виде желтого твердого вещества (298 мг, чистота 84,0%, 20%-ный выход).
ЖХ/МС: Rt = 0,717 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z = 523,5 [М+Н]+
Методика получения соединения 1f:
Раствор соединения 1е (298 мг, 0,57 ммоль) и Pd/C (30 мг, 0,1 экв.) в МеОН (10 мл) перемешивали под давлением Н2 1 атм при 25°С в течение 1 часа. Реакционную массу фильтровали через целит и промывали метанолом (5 мл × 3). Фильтраты объединяли и выпаривали под вакуумом с получением указанного в заголовке продукта в виде коричневого масла (206 мг, 73%-ный выход).
ЖХ/МС: Rt = 0,630 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z = 493,3 [М+Н]+
Методика получения соединения Примера 1:
В раствор 1f (194 мг, 0,39 ммоль) и DIEA (78 мг, 1,5 экв., 0,59 ммоль) в DMF (5 мл) по каплям добавляли акрилоилхлорид (39 мг, 0,43 ммоль) при 0°С. Смесь перемешивали при 25°С в течение 2 часов. Реакционную смесь очищали препаративной ВЭЖХ (колонка: Phenomenex Gemini С18 150×25 мм, 5 мкм; условия: 30-40% В (А: 0,05% аммиака в воде; В: CH3CN); скорость потока: 25 мл/мин). Фракции, содержащие целевое соединение лиофилизировали с получением соединения Примера 1 в виде белого твердого вещества (47,2 мг, 20%-ный выход).
ЖХ/МС: Rt = 2,100 мин при 0-60АВ_220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z = 547,1 [М+Н]+.
ВЭЖХ: Rt = 3,88 мин при 0-60_АВ_1,2 мл. мет хроматографии (Ultimate С18 3,0 мкм, 3,0×50 мм); СФХ (сверхкритическая флюидная хроматография): Rt = 3,763 мин, чистота 96,5%.
1Н ЯМР (400 МГц, CDCl3) δ 10.67 (br s, 1Н), 9.91 (br s, 1Н), 8.85 (s, 1Н), 8.41 (s, 1Н), 8.30 (d, J=8,0 Гц, 1Н), 7.61 (br s, 1Н), 7.35-7.30 (m, 2Н), 7.04 (t, J=7,5 Гц, 1Н), 6.72 (s, 1Н), 6.50-6.27 (m, 2H), 5.82 (br d, J=11,0 Гц, 1H), 5.60 (s, 1H), 4.06-3.95 (m, 1H), 3.07 (br s, 1H), 2.89 (br s, 1H), 2.67 (br s, 2H), 2.45-2.38 (m, 1H), 2.34 (s, 6H), 1.98 (br s, 2H), 1.80 (s, 6H), 1.72-1.68 (m, 2H), 1.68-1.64 (m, 2H).
Пример 2
N-(2-((2-(диметиламино)этил)(метил)амино)-5-(4-(2-(1-гидроксициклопропил)фениламино)-1,3,5-триазин-2-иламино)-4-метоксифенил)акриламид
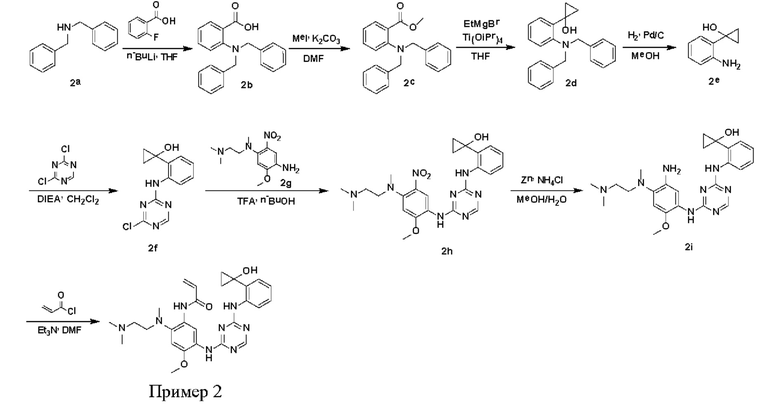
Методика получения соединения 2b:
В смесь соединения 2а (50 г, 253,5 ммоль) в THF (500 мл) по каплям добавляли n-BuLi (140 мл, 2,5 М, 354,8 ммоль) при -60°С в течение 30 мин. Смесь перемешивали при -60°С в течение 1 ч. Затем добавляли 2-фторбензойную кислоту (32 г, 228,1 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 20 ч. Реакционную смесь гасили водным NH4Cl (1000 мл) при 0°С. Полученную смесь экстрагировали EtOAc (300 мл × 3). Объединенные органические слои сушили над Mg2SO4 и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир : EtOAc = 3:1) с получением соединения 2b (25 г, 31%-ный выход) в виде бесцветного масла.
ЖХ/МС: Rt = 0,753 мин (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 318,0 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 8.17 (dd, J=8.0, 1,6 Гц, 1Н), 7.64-7.56 (m, 1Н), 7.49 (d, J=7,6 Гц, 1Н), 7.40-7.35 (m, 2Н), 7.31-7.25 (m, 5Н), 7.24-7.16 (m, 4Н), 4.18 (s, 4Н).
Методика получения соединения 2с:
В смесь соединения 2b (25 г, 70,89 ммоль) и K2CO3 (29,4 г, 212,67 ммоль) в DMF (500 мл) добавляли MeI (20,1 г, 141,78 ммоль). Затем реакционную смесь перемешивали при 25°С в течение 1 ч. Реакционную смесь вливали в воду (1200 мл) и экстрагировали EtOAc (400 мл × 3). Объединенные органические слои промывали рассолом (300 мл × 3), сушили над сульфатом натрия, фильтровали, и фильтрат концентрировали с получением соединения 2 с (27 г, неочищенное) в виде желтого масла.
ЖХ/МС: Rt = 0,822 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 332,4 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 7.72 (dd, J=1,6 Гц, 8,0 Гц, 1Н), 7.34-7.23 (m, 11Н), 7.00-6.95 (m, 2Н), 4.27 (s, 4Н), 3.91 (s, 3Н).
Методика получения соединения 2d:
В смесь соединения 2с (27 г, 81,47 ммоль) в THF (250 мл) добавляли по каплям Ti(OiPr)4 (6,9 мг, 24,44 ммоль) при 80°С в атмосфере N2. Затем добавляли EtMgBr (109 мл, 325,88 ммоль), и реакционную смесь перемешивали при 80°С в течение 1 ч. Реакционную смесь вливали в насыщенный раствор NH4Cl (200 мл) и экстрагировали EtOAc (80 мл × 3). Объединенные органические слои промывали рассолом (60 мл), сушили над сульфатом натрия, фильтровали, и фильтрат концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (10% EtOAc в петролейном эфире) с получением соединения 2d (17 г, 63%-ный выход) в виде желтого масла.
ЖХ/МС: Rt = 0,760 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 330,1 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 7.87 (brs, 1Н), 7.25-7.15 (m, 6Н), 7.14-7.08 (m, 5Н), 7.05-6.97 (m, 2H), 6.92 (dd, J=1,2 Гц, 8,0 Гц, 1Н), 4.11 (s, 4Н), 1.09-1.03 (m, 2Н), 0.93-0.87 (m, 2Н).
Методика получения соединения 2е:
В смесь соединения 2d (0,5 г, 1,52 ммоль) в МеОН (8 мл) добавляли Pd/C (влажный) (100 мг) и АсОН (0,1 мл) в атмосфере Н2. Реакционную смесь перемешивали при 20-25°С в течение 40 ч. Реакционную смесь фильтровали, и фильтрат концентрировали и очищали колоночной хроматографией на силикагеле (петролейный эфир EtOAc = 3:1) с получением соединения 2е (200 мг, 29%-ный выход) в виде бесцветного масла.
1Н ЯМР (400 МГц, CDCl3) δ 7.19-7.12 (m, 2Н), 6.75-6.70 (m, 2Н), 4.30 (brs, 2Н), 1.19-1.15 (m, 2Н), 0.10-0.95 (m, 2Н).
Методика получения соединения 2f:
В смесь соединения 2е (200 мг, 1,34 ммоль) и DIEA (346 мг, 2,68 ммоль) в CH2Cl2 (8 мл) добавляли 2,4-дихлор-1,3,5-триазин (241 мг, 1,61 ммоль). Затем реакционную смесь перемешивали при 25°С в течение 12 ч. Реакционную смесь концентрировали и очищали колоночной хроматографией на силикагеле (30% EtOAc в петролейном эфире) с получением 1-(2-((4-хлор-1,3,5-триазин-2-ил)амино)фенил)циклопропанола 2f (220 мг, 62%-ный выход) в виде желтого масла.
ЖХ/МС: Rt = 0,718 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 262,9 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 8.79 (brs, 1Н), 8.55 (s, 1Н), 8.19 (s, 1Н), 7.41-7.33 (m, 1Н), 7.33-7.30 (m, 1Н), 7.18-7.13 (m, 1H), 1.28-1.24 (m, 2H), 1.03-0.99 (m, 2Н).
Методика получения соединения 2g:

В раствор 4-фтор-2-метокси-5-нитроанилина (20 г, 0,11 моль) и K2CO3 (29 г, 0,22 моль) в DMF (200 мл) добавляли N1,N1,N2-триметилэтан-1,2-диамин (13 г, 0,13 моль).
Смесь перемешивали при 25°С в течение 48 часов. Реакционную смесь обрабатывали 1 л воды и экстрагировали EtOAc (300 мл × 3). Объединенные органические слои промывали рассолом (500 мл × 3). Органический слой сушили над сульфатом натрия, фильтровали, и фильтрат концентрировали в вакууме с получением соединения 2g (30 г, 100%-ный выход) в виде красного масла.
ЖХ/МС: Rt=0,125 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z=269,0 [М+Н]+.
1Н ЯМР: (400 МГц, CDCl3) δ 7.28 (s, 1H), 6.62 (s, 1H), 3.92 (s, 3Н), 3.17 (t, J=7,2 Гц, 2Н), 2.50 (t, J=7,2 Гц, 2Н), 2.25 (s, 6Н).
Методика получения соединения 2h:
В смесь соединения 2f (140 мг, 0,53 ммоль) и соединения 2g (56 мг, 0,58 ммоль) в n-BuOH (5 мл) добавляли TFA (0,5 мл). Затем реакционную смесь перемешивали при 22-28°С в течение 12 ч. Реакционную смесь концентрировали и очищали колоночной хроматографией на силикагеле (5% МеОН в CH2Cl2) с получением соединения 2h (160 мг, 61%-ный выход) в виде коричневого масла.
ЖХ/МС: Rt=0,682 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z=495,3 [М+Н]+.
Методика получения соединения 2i:
В смесь соединения 2h (110 мг, 0,22 ммоль) и NH4Cl (59 мг, 1,10 ммоль) в МеОН (5 мл) и воде (0,5 мл) добавляли Zn (72 мг, 1,10 ммоль). Затем реакционную смесь перемешивали при 90°С в течение 1 ч. Реакционную смесь фильтровали, и фильтрат концентрировали с получением соединения 2i (140 мг, неочищенное) в виде коричневого масла.
ЖХ/МС: Rt=0,636 мин при 5-95АВ_1,5 мин 220&254 хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z=465,2 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 8.24 (brs, 1 Н), 7.83 (brs, 1 Н), 7.60 (brs, 1 Н), 7.71-7.40 (m, 2 Н), 7.20-6.93 (m, 1 Н), 6.61 (s, 1 Н), 3.80 (s, 3 Н), 3.20-2.85 (m, 2 Н).2.70-2.25 (m, 11 Н),1.16-1.08 (m, 2 Н), 0.91-0.82 (m, 2 Н).
Методика получения соединения Примера 2:
В смесь соединения 2i (140 мг, 0,21 ммоль) и DIEA (54 мг, 0,42 ммоль) в DMF (2 мл) добавляли акрилоилхлорид (21 мг, 0,23 ммоль) при 0°С. Реакционную смесь перемешивали при 24-27°С в течение 0,5 ч. Реакционную смесь очищали препаративной ВЭЖХ: [колонка: Phenomenex Gemini C18 250 × 50 мм, 10 мкм; условия: 35-65% В (А: 0,05%-ный аммиак; В: CH3CN); скорость потока: 30 мл/мин]. Фракции, содержащие целевое соединение, лиофилизировали с получением соединения Примера 2 (32,0 мг, 6%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt=1,999 мин при 10-80CD_3 мин 220&254 хроматографии (В: XBrige Shield RP18 2,1 × 50 мм), МС (ЭРИ) m/z=519,3 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 10.34 (brs, 1 Н), 10.05 (brs, 1 Н), 8.78 (brs, 1 Н), 8.37-8.31 (m, 2 Н), 7.60 (brs, 1 Н), 7.30-7.24 (m, 2 Н), 6.95 (t, J=7,6 Гц, 1 H), 6.71 (s, 1 Н), 6.23-6.21 (m, 2 Н), 5.64 (t, J=6,0 Гц, 1 H), 3.81 (s, 3 Н), 2.81-2.78 (m, 2 Н), 2.63 (s, 3 Н), 2.23-2.18 (m, 9 Н), 1.31-1.22 (m, 2 Н), 0.97-0.94 (m, 2 Н).
ВЭЖХ: Rt=3,56 мин при 10-80CD_1,2 мл хроматографии (XBridge Shield RP 18 2,1 × 50 мм, 5 мкм).
Пример 3
N-(2-((2-(диметиламино)этил)(метил)амино)-5-(4-(6-фтор-4-(2-гидроксипропан-2-ил)пиридин-3-иламино)-1,3,5-триазин-2-иламино)-4-метоксифенил)акриламид
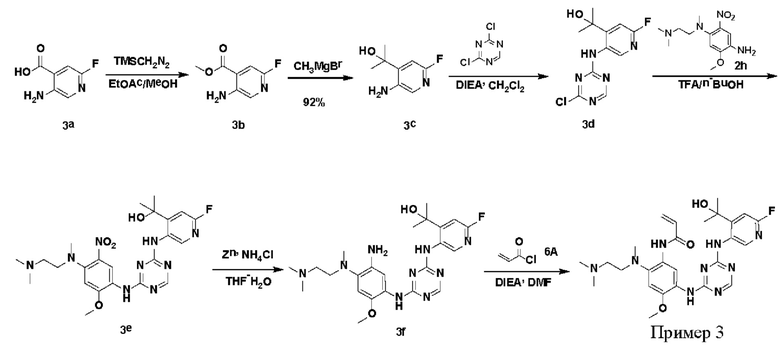
Методика получения соединения 3b:
В раствор соединения 3а (700 мг, 4,48 ммоль) в EtOAc (20 мл) и МеОН (20 мл) добавляли TMSCH2N2 (4,48 мл, 8,97 ммоль, 2М в гексане). Смесь перемешивали при 27-34°С (комнатная температура) в течение 1,5 ч. Реакционную смесь концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле (петролейный эфир/EtOAc=5/1) с получением соединения 3b (650 мг,: 85%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt=0,576 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z=170,8 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 7.78 (s, 1Н), 7.30 (d, J=2,6 Гц, 1H), 5.47 (br s, 2Н), 3.94 (s, 3Н).
Методика получения соединения 3с:
В раствор соединения 3b (650 мг, 3,82 ммоль) в THF (40 мл) добавляли CH3MgBr (5,1 мл, 3 М в диэтиловом эфире) при 0-5°С. Смесь перемешивали при 26-33°С в течение 1,5 ч. Реакционную смесь гасили добавлением водного NH4Cl (20 мл), затем экстрагировали EtOAc (3 × 40 мл). Органические слои промывали рассолом, концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле (петролейный эфир/EtOAc=3/1) с получением соединения 3с (600 мг, 92%-ный выход) в виде светло-желтого твердого вещества.
ЖХ/МС: Rt=1,572 мин при 10-80_7_мин 220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z=283,9 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 7.47 (d, J=1,4 Гц, 1H), 6.58 (d, J=1,6 Гц, 1H), 1.62-1.58 (m, 6Н).
Методика получения соединения 3d:
В раствор соединения 3с (600 мг, 3,52 ммоль) и DIEA (683 мг, 5,95 ммоль) в CH2Cl2 (40 мл) добавляли 2,4-дихлор-1,3,5-триазин (581 мг, 3,87 ммоль). Полученную смесь перемешивали при 26-33°С (комнатная температура) в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле (петролейный эфир/EtOAc=1/1) с получением соединения 3d (650 мг, 65%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt=1,299 мин при 10-80CD_3 мин_220&254.lcm хроматографии (XBrige Shield RP18 2,1 × 50 мм), МС (ЭРИ) m/z=283,9 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 9.85-9.51 (m, 1Н), 8.97 (br s, 1Н), 8.54 (br s, 1H), 6.85 (d, J=1,6 Гц, 1H), 2.97-2.37 (m, 1H), 1.70 (s, 6Н).
Методика получения соединения 3е:
В раствор соединения 3d (600 мг, 2,11 ммоль) и соединения 2g (624 мг, 2,32 ммоль) в n-BuOH (10 мл) добавляли TFA (0,1 мл). Полученную смесь перемешивали при 26-32°С в течение 18 ч. Реакционную смесь гасили добавлением водного NH4Cl (20 мл), затем экстрагировали EtOAc (3 × 40 мл). Органические слои промывали рассолом (40 мл), сушили и концентрировали при пониженном давлении с получением неочищенного остатка, который очищали колоночной хроматографией на силикагеле (СН2 Cl2/МеОН=10/1) с получением соединения 3е (80 мг, 7,3%-ный выход) в виде красного твердого вещества.
ЖХ/МС: Rt=0,662 мин при 5-95АВ_220&254.lcm хроматографии (XBrige Shield RP18 2,1 × 50 мм), МС (ЭРИ) m/z=516,2 [М+Н]+.
Методика получения соединения 3f:
В раствор соединения 3е (80 мг, ОД 6 ммоль) в THF (1 мл) и Н2 О (1 мл) добавляли Zn (30 мг, 0,47 ммоль) и NH4Cl (25 мг, 0,47 ммоль). Смесь перемешивали при 50°С в течение 1,5 ч в атмосфере N2. Реакционную смесь гасили добавлением водного NH4Cl (20 мл), затем экстрагировали EtOAc (3 × 40 мл). Органические слои промывали рассолом, сушили и концентрировали при пониженном давлении с получением соединения 3f (35 мг) в виде коричневого твердого вещества.
ЖХ/МС: Rt=0,596 мин при 5-95АВ_220&254.lcm хроматографии (XBrige Shield RP18 2,1 × 50 мм), МС (ЭРИ) m/z=486,2 [М+Н]+.
Методика получения соединения Примера 3:
В раствор соединения 3f (35 мг, 0,07 ммоль) и DIEA (14 мг, 0,11 ммоль) в DMF (1 мл) добавляли акрилоилхлорид (6,5 мг, 0,07 ммоль) в DMF (1 мл). Полученную смесь перемешивали при 0°С в течение 30 мин. Реакционную смесь очищали препаративной ВЭЖХ [колонка: Phenomenex Gemini С18 250 × 50 мм, 10 мкм; условия: 43-53% В (А: 0,05%-ный аммиак; В: CH3CN); скорость потока: 30 мл/мин]. Фракции, содержащие целевое соединение, лиофилизировали с получением соединения Примера 3 (4,2 мг, 10,8%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt=3,921 мин при 10-80CD_7 мин_220&254.lcm хроматографии (XBrige Shield RP18 2,1 × 50 мм), МС (ЭРИ) m/z=540,3 [М+Н]+.
ВЭЖХ: Rt=3,26 мин при 10-80_cd_1,2 мл. мет. хроматографии (XBridge Shield RP 18 2,1 × 50 мм, 5 мкм).
1Н ЯМР (400 МГц, CDCl3) δ 10.40 (br s, 1H), 10.11 (br s, 1H), 9.88 (br s, 1H), 9.01 (s, 1H), 8.34 (s, 1H), 7.62 (br s, 1H), 6.78 (s, 1H), 6.71 (s, 1H), 6.30 (br s, 3H), 5.75-5.63 (m, 1H), 3.81 (s, 3H), 2.81 (br s, 2H), 2.63 (s, 3H), 2.21 (br s, 8H), 1.71 (s, 6H).
Пример 4
N-(5-(4-(4,5-дифтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид

Методика получения соединения 4а:
Раствор соединения 1d (400 мг, 0,89 ммоль), N1,N1,N2-триметилэтан-1,2-диамина (91 мг, 0,89 ммоль) и К2СО3 (184 мг, 1,45 ммоль) в DMF (5 мл) перемешивали при 27-34°С в атмосфере N2 в течение 2 часов. Смесь по каплям добавляли в воду (50 мл), и полученную смесь перемешивали при комнатной температуре в течение 15 мин. Выпавшее в осадок твердое вещество собирали фильтрованием и затем сушили в глубоком вакууме с получением указанного в заголовке продукта 4а в виде оранжевого твердого вещества (270 мг, 57%-ный выход).
ЖХ/МС: Rt=0,707 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z=533,1 [М+Н]+.
Методика получения соединения 4b:
Раствор соединения 4а (270 мг, 0,50 ммоль) и Pd/C (20 мг) в МеОН (10 мл) продували и дегазировали Н2 3 раза, затем перемешивали в атмосфере Н2 (15 фунт/кв.дюйм) при 27-34°С в течение 2 часов. Реакционную массу фильтровали через целит и промывали метанолом (3×5 мл). Фильтраты объединяли и выпаривали под вакуумом с получением указанного в заголовке продукта 4b в виде черного масла (200 мг, 93%-ный выход).
ЖХ/МС: Rt=1,224 мин при 10-80АВ_220&254.lcm хроматографии (MERCK RP-18е 25-2 мм), МС (ЭРИ) m/z=503,3 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 10.02 (s, 1Н), 8.17-8.12 (m, 2Н), 7.66 (s, 1H), 6.98 (dd, J=11.2, 8,8 Гц, 2Н), 6.58 (s, 1H), 3.73 (s, 3H), 2.87-2.84 (m, 2Н), 2.55 (s, 3H), 2.32 (t, J=6,8 Гц, 2Н), 2.15 (s, 6Н), 1.55 (s, 6Н).
Методика получения соединения Примера 4:
В раствор соединения 4b (100 мг, 0,20 ммоль) и DIEA (38 мг, 0,30 ммоль) в DMF (3 мл) по каплям добавляли акрилоилхлорид (18 мг, 0,20 ммоль) при 0°С. Смесь перемешивали при 26-33°С в течение 2 часов. Реакционную смесь очищали препаративной ВЭЖХ (колонка: Phenomenex Gemini C18 150 × 25 мм, 5 мкм; 45-75% В (А: 0,05% аммиака в воде; В: CH3CN); скорость потока: 30 мл/мин). Фракции, содержащие целевое соединение, лиофилизировали с получением соединения Примера 4 в виде белого твердого вещества (14,5 мг, чистота 95,7%, 13%-ный выход).
ЖХ/МС: Rt=2,053 мин при 10-80_CD 3 мин_220&254.lcm хроматографии (Xtimate С18, 2,1 × 30 мм, 3 мкм), МС (ЭРИ) m/z=557,3 [М+Н]+.
ВЭЖХ: Rt=4,88 мин при 10-80_CD_1,2 мл.мет.хроматографии (Ultimate С18 3,0 мкм, 3,0 × 50 мм).
1Н ЯМР (400 МГц, CDCl3) δ 10.69 (s, 1H), 10.45 (s, 1H), 9.99 (s, 1H), 8.43 (s, 1H), 8.34 (dd, J=8.0, 12,8 Гц, 1Н), 7.70 (s, 1H), 7.13 (dd, J=8.4, 12,0 Гц, 1H), 6.80 (s, 1H), 6.39 (s, 2Н), 5.78 (d, J=11,2 Гц, 1H), 3.90 (s, 3H), 2.90 (s, 2Н), 2.72 (s, 3H), 2.29 (s, 8Н), 1.79 (s, 6Н).
Пример 5
(E)-N-(5-(4-(4,5-дифтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)-4-(диметиламино)бут-2-енамид

Методика получения соединения Примера 5:
Смесь соединения 4b (80 мг, 0,16 ммоль), соединения 5а (31 мг, 0,19 ммоль), HATU (91 мг, 0,24 ммоль) и DIEA (62 мг, 0,48 ммоль) в CH2Cl2 (4 мл) перемешивали при 28-35°С в течение 2 часов. Реакционную смесь очищали препаративной ВЭЖХ (колонка: Phenomenex Gemini C18 150 × 25 мм, 5 мкм; 55-85% В (А: 0,05% аммиака в воде; В: CH3CN); скорость потока: 30 мл/мин). Фракции, содержащие целевое соединение, лиофилизировали с получением соединения Примера 5 в виде белого твердого вещества (17,8 мг, 18%-ный выход).
ЖХ/МС: Rt=1,755 мин при 0-60 АВ 220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z=614.3 [М+Н]+.
ВЭЖХ: Rt=2,15 мин при 0-60CD_1,2 мл.мет. хроматографии (Ultimate С18 3,0 мкм, 3,0 × 50 мм).
1Н ЯМР (400 МГц, CDCl3) δ 10.69 (s, 1H), 10.29 (s, 1Н), 9.98 (s, 1Н), 8.42 (s, 1Н), 8.33 (dd, J=13.2, 8,0 Гц, 1H), 7.69 (s, 1H), 7.12 (dd, J=12.0, 8,8 Гц, 1H), 6.92 (td, J=15.2, 6,0 Гц, 1H), 6.79 (s, 1H), 6.25 (s, 1H), 3.89 (s, 3H), 3.73 (s, 1H), 3.18 (d, J=5,6 Гц, 2Н), 2.89 (t, J=5,2 Гц, 2Н), 2.71 (s, 3H), 2.33 (s, 8Н), 2.30 (s, 6Н), 1.79 (s, 6Н).
Пример 6
N-(2-((2-(диметиламино)этил)(метил)амино)-5-(4-(4-(2-гидроксипропан-2-ил)-6-метилпиридин-3-иламино)-1,3,5-триазин-2-иламино)-4-метоксифенил)акриламид

Методика получения соединения 6b:
В раствор соединения 6а (4,0 г, 23,2 ммоль) в смеси EtOAc/МеОН=1:1 (60 мл) добавляли TMSCH2N2 (23,2 мл, 46,4 ммоль, 2 M в гексане). Полученную смесь перемешивали при 24-30°С в течение 30 мин. Реакционную смесь вливали в Н2О (30 мл) и экстрагировали EtOAc (50 мл × 3). Объединенные органические слои промывали водой (20 мл × 3) и рассолом (20 мл), сушили над Na2SO4 и концентрировали в вакууме с получением указанного в заголовке соединение 6b (3,1 г, 67,7%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=0,672 мин при 5-95 АВ_220&254.lcm хроматографии (ACSSH-LCMS-АВ MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z=186,9 [М+Н]+.
1Н ЯМР (400 МГц, DMSO-d6) δ 8.06 (s, 1H), 7.50 (s, 1Н), 6.79 (br s, 2H), 3.84 (s, 3H).
Методика получения соединения 6c:
В раствор соединения 6b (3,1 г, 16,6 ммоль) в THF (40 мл) добавляли по каплям CH3MgBr (22,2 мл, 66,5 ммоль, 3 М в диэтиловом эфире) при 0°С. Полученную смесь перемешивали при 26-34°С в течение 1,5 ч в атмосфере N2. Реакционную смесь вливали в насыщенный NH4Cl (100 мл) и затем экстрагировали EtOAc (100 мл × 3). Объединенные органические слои промывали водой (30 мл × 3) и рассолом (30 мл), сушили над Na2SO4 и концентрировали в вакууме с получением указанного в заголовке соединения 6с (2,79 г, 85,6%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=0,471 мин при 5-95АВ_220&254.lcm хроматографии (ACSSH-LCMS-Q MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z=187,0 [М+Н]+.
1Н ЯМР (400 МГц, DMSO-d6) δ 7.71 (s, 1H), 6.98 (s, 1H), 5.63 (s, 2Н), 5.52 (s, 1H), 1.46 (s, 6Н).
Методика получения соединения 6d:
В раствор соединения 6с (1,0 г, 5,36 ммоль), метилбороновой кислоты (1,28 г, 21,44 ммоль) и К2СО3 (1,48 г, 10,72 ммоль) в смеси Н2О/диоксан = 1:5 (20 мл) добавляли Pd(PPh3)4 (929 мг, 0,15 экв., 0,80 ммоль). Реакционную смесь нагревали при 100°С в течение 24 ч в атмосфере N2. Реакционную смесь фильтровали, и фильтрат концентрировали в вакууме с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (4% МеОН в CH2Cl2) с получением соединения 6d (235 мг, 26,1%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=0,142 мин при 5-95АВ_220&254.lcm хроматографии (ACSSH-LCMS-АВ MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z=167,0 [М+Н]+.
1Н ЯМР (400 МГц, DMSO-d6) δ 7.79 (s, 1H), 6.81 (s, 1H), 5.34 (s, 1Н), 5.25 (br s, 2H), 2.26 (s, 3H), 1.46 (s, 6H).
Методика получения соединения 6е:

В перемешиваемый раствор соединения 6е1 (100 г, 537,2 ммоль) и DIEA (138,9 г, 1074,4 ммоль) в CH2Cl2 (1500 мл) добавляли соединение 6е2 (96,7 г, 644,6 ммоль) при 10°С, затем перемешивали при 25°С в течение 1 ч. Реакционную смесь концентрировали в вакууме напрямую с получением неочищенного продукта, который растирали с CH2Cl2 (800 мл) в течение 30 мин, фильтровали, и твердый остаток на фильтре промывали CH2Cl2 (100 мл × 2). Остаток на фильтре сушили в вакууме с получением указанного в заголовке соединение 6е (118 г, 73%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=0,760 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z=299,9 [М+Н]+.
1Н ЯМР: (400 МГц, DMSO-d6) δ 10.27 (s, 1 Н), 8.60 (s, 1 Н), 8.37 (d, J=8,0 Гц, 1 Н), 7.41 (d, J=13,6 Гц, 1 Н), 3.95 (s, 3 Н).
Методика получения соединения 6f:
Смесь соединения 6d (200 мг, 1,20 ммоль) и соединения 6е (433 мг, 1,44 ммоль) в пиридине (6 мл) нагревали при 50°С в течение 12 ч. Реакционную смесь концентрировали в вакууме с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (5% МеОН в CH2Cl2) с получением соединения 6f (240 мг, 16%-ный выход) в виде красновато-коричневого твердого вещества.
ЖХ/МС: Rt=0,688 мин при 5-95АВ_220&254.lcm хроматографии (ACSSH-LCMS-АВ MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z=430,1 [М+Н]+.
Методика получения соединения 6g:
В раствор соединения 6f (240 мг, 0,191 ммоль) и соединения N1,N1,N2-триметилэтан-1,2-диамин (29 мг, 0,286 ммоль) в DMF (5 мл) добавляли К2СО3 (53 мг, 0,382 ммоль). Реакционную смесь перемешивали при 27-34°С в течение 12 ч. Реакционную смесь вливали в Н2O (30 мл) и экстрагировали EtOAc (30 мл × 3). Объединенные органические слои промывали водой (20 мл × 3) и рассолом (20 мл), сушили над Na2SO4 и концентрировали в вакууме с получением соединения 6g (120 мг, 91,8%-ный выход) в виде оранжевого масла.
ЖХ/МС: Rt=0,633 мин при 5-95 АВ_220&254.lcm хроматографии (ACSSH-LCMS-АВ MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z=512,2 [М+Н]+.
Методика получения соединения 6h:
В раствор соединения 6g (90 мг, 0,131 ммоль) в смеси МеОН/Н2О=2/1 (9 мл) добавляли Zn (26 мг, 0,394 ммоль) и NH4Cl (21 мг, 0,394 ммоль). Полученную смесь нагревали при 90°С в течение 3 ч до тех пор, пока ТСХ (CH2Cl2/МеОН=5/1(об./об.)) не показала одно главное пятно (Rf=0,25) и полное расходование исходного вещества (Rf=0,5). Реакционную смесь экстрагировали смесью CHCl3/изопропанол = 3/1 (20 мл × 3). Объединенные органические слои сушили над Na2SO4, затем концентрировали в вакууме с получением соединения 6h (85 мг, 94%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt=0,571 мин при 5-95АВ 220&254.lcm хроматографии (ACSSH-LCMS-АВ MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z=482,3 [М+Н]+.
Методика получения соединения Примера 6:
В раствор соединения 6h (85 мг, 0,123 ммоль) и DIE А (24 мг, 0,185 ммоль) в DMF (2 мл) добавляли акрилоилхлорид (11 мг, 0,123 ммоль) при 0°С. Полученную смесь перемешивали при 0°С в течение 1 ч. Реакционную смесь фильтровали, и фильтрат очищали препаративной ВЭЖХ (колонка: Phenomenex Gemini C18 250 × 50 мм, 10 мкм; условия: 25-55% В (А: 0,05%-ный аммиак, В: CH3CN); скорость потока: 30 мл/мин) и лиофилизировали с получением продукта с примесями в виде белого твердого вещества, который дополнительно очищали препаративной ТСХ (CH2Cl2:МеОН=7:1 (об./об.)) с получением соединения Примера 6 (18,4 мг, 27,9%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt=1,969 мин при 10-80CD_3 мин_220&254 хроматографии (ACSSH-LCMS-AS A: Xtimate С18, 2,1 × 30 мм, 3 мкм; В: XBrige Shield RP18 2,1 × 50 мм), МС (ЭРИ) m/z=537,3 [М+Н]+.
1Н ЯМР: (400 МГц, CDCl3) δ 10.40 (br s, 1H), 10.20 (br s, 1H), 9.93 (br s, 1H), 9.32 (s, 1H), 8.39 (s, 1H), 7.67 (br s, 1H), 7.04 (s, 1H), 6.77 (s, 1H), 6.47-6.27 (m, 2H), 6.14 (br s, 1H), 5.80-5.69 (m, 1H), 3.87 (s, 3H), 2.90-2.84 (m, 2H), 2.69 (s, 3H), 2.53 (s, 3H), 2.31-2.25 (m, 8H), 1.75 (s, 6H).
Пример 7
N-(2-((2-(диметиламино)этил)(метил)амино)-5-(4-(2-(2-гидроксипропан-2-ил)-6-метилпиридин-3-иламино)-1,315-триазин-2-иламино)-4-метоксифенил)акриламид

Методика получения соединения 7b:
В смесь соединения 7а (5 г, 26,73 ммоль) в безводном THF (70 мл) добавляли DMAP (6,35 г, 53,46 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 10 минут, добавляли (Вос)2O (17,50 г, 80,19 ммоль), затем перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали в вакууме с получением неочищенного остатка, который очищали колоночной хроматографией на силикагеле (5-10% EtOAc в петролейном эфире) с получением целевого продукта 7b (10 г, 96,6%-ный выход) в виде светло-желтого твердого вещества.
ЖХ/МС: Rt=0,875 мин при 5-95 АВ_220&254.lcm хроматографии (ACSSH-LCMS-Q MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z=388,9 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 7.40 (d, J=7,6 Гц, 1H), 7.14 (d, J=7,6 Гц, 1H), 2.57 (s, 3H), 1.41 (s, 18Н)
Методика получения соединения 7с:
В смесь соединения 7b (3,5 г, 9,04 ммоль) в безводном МеОН (50 мл) и безводном DMF (150 мл) добавляли Pd(OAc)2 (150 мг, 0,1 экв., 0,904 ммоль) в атмосфере азота, затем добавляли DPPF (501 мг, 0,904 ммоль) и Et3N (1,37 г, 13,56 ммоль). Полученную смесь перемешивали при 80°С в течение 16 часов в атмосфере СО (50 фунт/кв.дюйм). Реакционную смесь фильтровали, и фильтрат вливали в воду (10 мл), перемешивали в течение дополнительных 10 минут, экстрагировали EtOAc (20 мл × 3). Объединенные органические слои промывали рассолом (30 мл), сушили над безводным Na2SO4 и концентрировали в вакууме с получением неочищенного остатка, который очищали колоночной хроматографией на силикагеле (5-8% EtOAc в петролейном эфире) с получением соединения 7 с (2,0 г, 60,4%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt=0,812 мин при 5-95 АВ_220&254.lcm хроматографии (ACSSH-LCMS-Q MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z=367,1 [М+Н]+.
Методика получения соединения 7d:
В смесь соединения 7с (2,0 г, 5,46 ммоль) в безводном THF (20 мл) добавляли MeMgBr (9,1 мл, 27,3 ммоль, 3 М в диэтиловом эфире). Полученную смесь перемешивали при 0°С в течение 2,5 часов в атмосфере N2. Реакционную смесь вливали в водный NH4Cl (25 мл), перемешивали в течение 10 минут, затем экстрагировали EtOAc (30 мл × 3). Объединенные органические слои промывали рассолом (30 мл), сушили над безводным Na2SO4 и концентрировали под вакуумом с получением указанного в заголовке продукта 7d (1,4 г, 49,8%-ный выход) в виде желтого масла, которое напрямую использовали на следующей стадии без дополнительной очистки.
ЖХ/МС: Rt=0,616 мин при 5-95АВ_220&254.lcm хроматографии (ACSSH-LCMS-Q MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z=267,1 [М+Н]+.
Методика получения соединения 7е:
В смесь соединения 7d (1,4 г, 1,13 ммоль) в безводном CH2Cl2 (9 мл) добавляли TFA (3 мл). Полученную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь вливали в водный NaHCO3 (25 мл) до установления рН=8,0, перемешивали в течение 10 минут, затем экстрагировали EtOAc (25 мл × 3). Объединенные органические слои промывали рассолом (30 мл), сушили над безводным Na2SO4 и концентрировали в вакууме с получением неочищенного остатка, который очищали колоночной хроматографией на силикагеле (градиентное элюирование: петролейный эфир: этил ацетат = 20/1 (об./об.)) с получением соединения 7е (230 мг, 26,3%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt=0,129 мин при 5-95АВ_220&254.lcm хроматографии (ACSSH-LCMS-Q MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z=167,0 [М+Н]+.
1H ЯМР (400 МГц, Метанол-d4) δ 6.97 (d, J=8,4 Гц, 1H), 6.86 (d, J=8,0 Гц, 1H), 2.35 (s, 3H), 1.58 (s, 6Н).
Методика получения соединения 7f:
В смесь соединения 7е (311 мг, 2,076 ммоль) в безводном CH2Cl2 (5 мл) добавляли DIEA (358 мг, 2,768 ммоль), затем добавляли соединение 2,4-дихлор-1,3,5-триазин (230 мг, 1,0 экв., 1,384 ммоль). Полученную смесь перемешивали при 25°С в течение 1,5 часов. Реакционную смесь вливали в воду (20 мл) и EtOAc (20 мл), перемешивали в течение 5 минуты и затем экстрагировали EtOAc (30 мл × 3). Объединенные органические слои промывали рассолом (40 мл), сушили над безводным Na2SO4 и концентрировали в вакууме с получением неочищенного остатка, который очищали колоночной флэш-хроматографией на силикагеле (15-30% EtOAc в петролейном эфире) с получением соединения 7f (290 мг, 74,9%-ный выход) в виде не совсем белого твердого вещества.
ЖХ/МС: Rt=0,352 мин при 5-95АВ_220&254.lcm хроматографии (ACSSH-LCMS-Q MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z=279,9 [М+Н]+.
1H ЯМР (400 МГц, CDCl3) δ 9.93-9.54 (m, 1H), 8.50 (s, 1Н), 8.30 (br d, J=14,4 Гц, 1H), 7.12 (brd, J=8,4 Гц, 1H), 3.07-2.87 (m, 1H), 2.51 (s, 3H), 1.69 (s, 6Н).
Методика получения соединения 7g:
В смесь соединения 7f (290 мг, 1,038 ммоль) в n-BuOH (10 мл) добавляли соединение 2g (289 мг, 1,038 ммоль) и TFA (0,1 мл). Полученную смесь перемешивали при комнатной температуре в течение 16 часов с получением темно-красного мутного раствора. Смесь концентрировали в вакууме напрямую с получением неочищенного остатка, который очищали колоночной флэш-хроматографией на силикагеле (от 0 до 15% МеОН в CH2Cl2 с 1% аммиака) с получением соединения 7g (110 мг, 20,7%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=0,604 мин при 5-95АВ_220&254.lcm хроматографии (ACSSH-LCMS-Q MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z=512,2 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 8.91 (br s, 1Н), 8.36 (br s, 1H), 8.24 (br s, 1H), 7.47 (br s, 1H), 7.15 (br d, J=8,4 Гц, 1H), 6.66 (s, 1H), 3.96 (s, 3H), 3.29 (t, J=7,2 Гц, 2H), 2.89 (s, 3H), 2.54 (s, 3H), 2.28 (s, 6H), 1.69 (s, 8H).
Методика получения соединения 7h:
В раствор соединения 7g (110 мг, 0,215 ммоль) в смеси МеОН/Н2О=5/1 (6 мл) добавляли Zn (70 мг, 1,075 ммоль) и NH4Cl (58 мг, 5,0 экв., 1,075 ммоль). Полученную смесь нагревали при 90°С в течение 3 ч. Реакционную смесь фильтровали, и фильтрат экстрагировали CH2Cl2 (20 мл × 3). Объединенные органические слои промывали водой (10 мл × 3) и рассолом (10 мл), сушили над Na2SO4 и концентрировали в вакууме с получением соединения 7h (93 мг, 79,6%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt=0,554 мин при 5-95 АВ_220&254.lcm хроматографии (ACSSH-LCMS-АВ MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z=482,1 [М+Н]+. Методика получения соединения Примера 7:
В раствор соединения 7h (93 мг, 0,171 ммоль) и DIEA (33 мг, 1,5 экв., 0,256 ммоль) в DMF (2 мл) добавляли акрилоилхлорид (15 мг, 1,0 экв., 0,171 ммоль) при 0°С. Полученную смесь перемешивали при 0°С в течение 1 ч. Реакционную смесь концентрировали в вакууме с получением неочищенного продукта, который очищали препаративной ТСХ (CH2Cl2:МеОН=7:1 (об./об.)) с получением продукта с примесями в виде желтого твердого вещества. Этот неочищенный продукт дополнительно очищали препаративной ВЭЖХ (колонка: Waters Xbridge 150 × 25, 5 мкм; условия: 30-55% В (А: 0,05%-ный аммиак, В: CH3CN); скорость потока: 25 мл/мин) и лиофилизировали с получением соединения Примера 7 (20,3 мг, 22,2%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=2,002 мин при 10-80CD_3 мин_220&254 хроматографии (ACSSH-LCMS-AS A: Xtimate C18 2,1 × 30 мм, 3 мкм; В: XBrige Shield RP18 2,1 × 50 мм), МС (ЭРИ) m/z=536,3 [M+H]+.
1H ЯМР (400 МГц, CDCl3) δ 10.34 (br s, 2H), 9.95 (br s, 1H), 8.43 (d, J=8,4 Гц, 1H), 8.37 (br s, 1H), 7.64 (br s, 1H), 7.07 (d, J=8,4 Гц, 1H), 6.77 (s, 1H), 6.52-6.31 (m, 2H), 5.77 (br d, J=10,4 Гц, 1H), 3.87 (s, 3H), 2.88 (t, J=4,8 Гц, 2H), 2.70 (s, 3H), 2.49 (s, 3H), 2.28 (s, 8H), 1.78 (s, 6H).
Пример 8
N-(2-((2-(диметиламино)этил)(метил)амино)-5-(4-(2-(2-гидроксипропан-2-ил)пиридин-3-иламино)-1,3,5-триазин-2-иламино)-4-метоксифенил)акриламид

Методика получения соединения 8b:
В раствор соединения 8а (1,0 г, 6,57 ммоль) в CH2Cl2 (20 мл) добавляли (Вос)2О (3,6 г, 16,42 ммоль) и DMAP (1,2 г, 1,5 экв., 9,85 ммоль). Полученную смесь перемешивали при 28-38°С в течение 4 ч. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного остатка, который очищали колоночной хроматографией на силикагеле (петролейный эфир/EtOAc=50/1 (об./об.)) с получением соединения 8b (2,5 г, 99%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt=0,784 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z=353,0 [М+Н]+.
1Н ЯМР (400 МГц, Метанол-d4) δ 8.63 (dd, J=1.4, 4,6 Гц, 1Н), 7.85 (dd, J=1.5, 8,0 Гц, 1Н), 7.69 (dd, J=4.8, 8,0 Гц, 1Н), 3.94 (s, 3H), 1.36 (s, 18Н).
Методика получения соединения 8с:
Раствор соединения 8b (2,3 г, 6,52 ммоль) в THF (30 мл) продували и дегазировали N2 3 раза, и в него по каплям добавляли MeMgBr (10,87 мл) при 0°С. Полученную смесь перемешивали при 29-34°С в течение 1 ч. Реакционную смесь разбавляли насыщенным водным NH4Cl (10 мл) и экстрагировали EtOAc (2 × 20 мл). Объединенные органические слои концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле (петролейный эфир/EtOAc=10/1) с получением соединения 8с (1 г, 61%-ный выход) в виде белого твердого вещества
ЖХ/МС: Rt=0,697 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z=253,0[М+Н]+.
Методика получения соединения 8d:
В раствор соединения 8с (1 г, 1,0 экв., 3,70 ммоль) в CH2Cl2 (6 мл) добавляли TFA (2 мл). Полученную смесь перемешивали при 29-36°С в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении с получением соединения 8d (1,2 г, 94%-ный выход) в виде желтого масла (TFA соль).
1Н ЯМР (400 МГц, Метанол-d4) δ 7.85 (dd, J=1.0, 5,3 Гц, 1H), 7.72 (dd, J=1.3, 8,5 Гц, 1H), 7.61 (dd, J=5.5, 8,5 Гц, 1H), 1.70 (s, 6Н).
Методика получения соединения 8е:
В раствор соединения 8d (200 мг, 1,0 экв., 0,67 ммоль) в пиридине (5 мл) добавляли соединение 6е (256 мг, 1,1 экв., 0,74 ммоль). Полученную смесь перемешивали при 28-36°С в течение 6 ч. Реакционную смесь концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле (CH2Cl2/МеОН=10/1) с получением соединения 8е (240 мг, 58%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=0,687 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z=438,1 [М+Н]+.
Методика получения соединения 8f:
В раствор соединения 8е (240 мг, 1,0 экв., 0,58 ммоль) в DMF (10 мл) добавляли К2СО3 (160 мг, 2,0 экв., 1,16 ммоль) и соединение N1,N1,N2-триметилэтан-1,2-диамин (118 мг, 2,0 экв., 1,16 ммоль). Полученную смесь перемешивали при 28-36°С в течение 2 ч. Реакционную смесь разбавляли H2O (200 мл) и экстрагировали EtOAc (2 × 100 мл). Объединенные органические слои промывали рассолом (100 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением неочищенного остатка, который очищали колоночной хроматографией на силикагеле (CH2Cl2/МеОН=10/1) с получением соединения 8f (170 мг, 59%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=0,643 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z=498,2[М+Н]+.
Методика получения соединения 8g:
В раствор соединения 8f (170 мг, 1,0 экв., 0,34 ммоль) в МеОН (10 мл) и Н2 0 (5 мл) добавляли Zn (111 мг, 5,0 экв., 1,70 ммоль) и NH4Cl (182 мг, 10,0 экв., 3,40 ммоль). Полученную смесь перемешивали при 80°С в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении и экстрагировали EtOAc (2 × 10 мл). Объединенные органические слои сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением соединения 8g (140 мг, 88%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt=0,554 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z=468,2 [М+Н]+.
Методика получения соединения Примера 8:
В раствор соединения 8g (140 мг, 1,0 экв., 0,30 ммоль) и DIEA (58 мг, 1,5 экв., 0,45 ммоль) в DMF (3 мл) добавляли акрилоилхлорид (27 мг, 1,0 экв., 0,30 ммоль). Полученную смесь перемешивали при 0°С в течение 30 мин. Реакционную смесь очищали препаративной ВЭЖХ [колонка: Waters Xbridge 150 × 25, 5 мкм; условия: 28-58% В (А: 0,05% NH3H2O; В: CH3CN); скорость потока: 25 мл/мин]. Фракции, содержащие целевое соединение, лиофилизировали с получением соединения Примера 8 (58,7 мг, 37%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt=1,844 мин при 10-80CD_3 мин_220&254 хроматографии (XBrige Shield RP18 2,1 × 50 мм), МС (ЭРИ) m/z=522,3 [М+Н]+.
ВЭЖХ: Rt=3,65 мин при 10-80_CD_1,2 мл хроматографии (XBridge Shield RP 18 2,1 × 50 мм, 5 мкм).
1Н ЯМР (400 МГц, CDCl3) δ 10.85 (br s, 1H), 10.41 (br s, 1H), 9.99 (br s, 1H), 8.69 (d, J=7,0 Гц, 1H), 8.41 (s, 1H), 8.23 (dd, J=1.5, 4,5 Гц, 1H), 7.68 (br s, 1H), 7.23 (dd, J=4.9, 8,2 Гц, 1H), 6.79 (s, 1Н), 6.48-6.41 (m, 1H), 6.37 (br d, J=10,0 Гц, 1H), 5.78 (br d, J=11,0 Гц, 1H), 3.88 (s, 3H), 2.88 (br s, 2H), 2.71 (s, 3H), 2.28 (s, 8H), 1.83 (s, 6H).
Пример 9
N-(5-(4-(3,4-дифтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид

Методика получения соединения 9b:
В раствор соединения 9а (340 мг, 1,0 экв., 1,18 ммоль) в CH2Cl2 (6 мл) добавляли TFA (2 мл). Полученную смесь перемешивали при 26-32°С в течение 2 ч. Полученную смесь концентрировали при пониженном давлении с получением соединения 9b в виде TFA соли (400 мг, 94%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=0,698 мин при 5-95АВ_220&254.lcm хроматографии (Agilent Pursit 5 C18 20 × 2,0 мм), МС (ЭРИ) m/z=188,1 [М+Н]+.
Методика получения соединения 9с:
В раствор соединения 9b (286 мг, 1,0 экв., 0,95 ммоль) в пиридине (10 мл) добавляли соединение 6е (400 мг, 1,1 экв., 1,05 ммоль). Полученную смесь перемешивали при 50°С в течение 6 ч. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного остатка, который очищали колоночной хроматографией на силикагеле (петролейный эфир/EtOAc=1/1 (об./об.)) с получением соединения 9 с (200 мг, 42%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=0,946 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18 25-2 мм), МС (ЭРИ) m/z=451,1 [М+Н]+.
Методика получения соединения 9d:
В раствор соединения 9 с (200 мг, 0,44 ммоль) в DMF (5 мл) добавляли К2СО3 (122 мг, 0,88 ммоль) и соединение N1,N1,N2-триметилэтан-1,2-диамин (54 мг, 0,53 ммоль). Полученную смесь перемешивали при 26-33°С в течение 2 ч. Реакционную смесь вливали в Н2О (50 мл) и фильтровали. Остаток на фильтре концентрировали при пониженном давлении с получением соединения 9d (200 мг, 85%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=0,807 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18 25-2 мм), МС (ЭРИ) m/z=533,2 [М+Н]+.
Методика получения соединения 9е:
В раствор соединения 9d (200 мг, 0,38 ммоль) в МеОН (5 мл) добавляли Pd/C (20 мг). Полученную смесь продували и дегазировали Н2 3 раза, затем перемешивали при 26-34°С (комнатная температура) в атмосфере Н2 (водородный баллон, 30 фунт/кв.дюйм) в течение 2 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением соединения 9е (150 мг, 78%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt=0,752 мин при 5-95АВ_1,5 мин_220&254 хроматографии (Xtimate С18 2,1 × 30 мм), МС (ЭРИ) m/z=503,2 [М+Н]+.
Методика получения соединения Примера 9:
В раствор соединения 9е (150 мг, 0,30 ммоль) и DIEA (58 мг, 0,45 ммоль) в DMF (3 мл) добавляли соединение акрилоилхлорид (27 мг, 0,30 ммоль). Полученную смесь перемешивали при 0°С в течение 30 мин. Реакционную смесь очищали препаративной ВЭЖХ [колонка: Waters Xbridge 150 × 25, 5 мкм; условия: 42-72% В (А: 0,05% NH3H2O; В: CH3CN); скорость потока: 25 мл/мин]. Фракции, содержащие целевое соединение, лиофилизировали с получением соединения Примера 9 (39,4 мг, 23%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt=1,784 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С18 2,1 × 30 мм), МС (ЭРИ) m/z=557,0 [М+Н]+.
ВЭЖХ: Rt=3,12 мин в 10-80_ab_1,2 мл хроматографии (Ultimate С18 3 × 50 мм, 3 мкм).
1Н ЯМР (400 МГц, CDCl3) δ 11.29 (br s, 1H), 10.45 (br s, 1H), 9.89 (br s, 1H), 8.38 (s, 1H), 8.14 (br dd, J=5.2, 7,2 Гц, 1H), 7.65 (br s, 1H), 7.09 (q, J=9,2 Гц, 1H), 6.78 (s, 1H), 6.44-6.29 (m, 2H), 6.08 (br s, 1H), 5.81-5.74 (m, 1H), 3.88 (s, 3H), 2.92-2.84 (m, 2H), 2.70 (s, 3H), 2.35-2.20 (m, 8H), 1.88 (d,J=3,6 Гц, 6H).
Пример 10
N-(2-((2-(диметиламино)этил)(метил)амино)-5-(4-(2-(3-гидроксиоксетан-3-ил)фениламино)-1,3,5-триазин-2-иламино)-4-метоксифенил)акриламид

Методика получения соединения 10а:
В смесь анилина (5 г, 53,69 ммоль) в CHCl3 (80 мл) добавляли Boc2O (23,4 г, 107,38 ммоль). Реакционную смесь перемешивали при 70°С в течение 12 ч. Реакционную смесь напрямую концентрировали в вакууме с получением неочищенного остатка, который очищали колоночной хроматографией на силикагеле (2% EtOAc в петролейном эфире) с получением указанного в заголовке соединение 10а (8 г, 100%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt=0,793 мин при 5-95АВ_220&254 хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z=137,9 [М+55]+.
1Н ЯМР (400 МГц, CDCl3) δ 7.38 (d, J=8,0 Гц, 2 Н), 7.33-7.28 (m, 2 H), 7.08-7.03 (m, 1 H), 6.50 (brs, 1 H), 1.54 (s, 9 H).
Методика получения соединения 10b:
В раствор соединения 10а (3,0 г, 1,55 ммоль) в THF (50 мл) добавляли по каплям mpem-BuLi (30 мл, 38,81 ммоль) при -78°С в течение 1 ч. Реакционную смесь нагревали до 0°С в течение 15 минут и затем охлаждали -78°С. Добавляли по каплям оксетан-3-он (3,36 г, 46,61 ммоль) при -78°С в течение 15 минут. Полученную смесь перемешивали при -78°С в течение 1 ч. Реакционную смесь гасили добавлением 100 мл воды и затем экстрагировали EtOAc (3 × 100 мл). Органические слои промывали рассолом (3 × 100 мл), сушили и концентрировали в вакууме с получением неочищенного остатка, который очищали колоночной хроматографией на силикагеле (петролейный эфир:EtOAc=3/1 (об./об.)) с получением соединения 10b (580 мг, 14,1%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt=0,722 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z=209,9 [М-55]+.
1H ЯМР (400 МГц, DMSO-d6) δ 8.09 (s, 1H), 7.65 (d, J=7,8 Гц, 1H), 7.40 (dd, J=1.4, 7,8 Гц, 1H), 7.34-7.29 (m, 1H), 7.16-7.11 (m, 1H), 4.88 (d, J=7,0 Гц, 2H), 4.71 (d, J=7,0 Гц, 2H), 1.45 (s, 9H).
Методика получения соединения 10c:
В раствор соединения 10b (500 мг, 1,89 ммоль) в МеОН (20 мл) и Н2О (10 мл) добавляли К2СО3 (780 мг, 5,66 ммоль). Полученную смесь перемешивали при 70°С в течение 2 ч. Реакционную смесь экстрагировали EtOAc (3 × 50 мл). Объединенные органические слои промывали рассолом, сушили и концентрировали в вакууме с получением неочищенного остатка, который очищали колоночной хроматографией на силикагеле (петролейный эфир/EtOAc=3/1 (об./об.)) с получением соединения 10 с (240 мг, 77,1%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt=0,553 мин при 10-80CD_3 мин_220&254; XBrige Shield RP18 2,1 × 50 мм, МС (ЭРИ) m/z=166,1 [М+Н]+.
1Н ЯМР (400 МГц, DMSO-d6) δ 7.14 (d, J=7,8 Гц, 1Н), 7.05-6.99 (m, 1H), 6.70 (d, J=7,8 Гц, 1H), 6.59 (dt, J=1.0, 7,4 Гц, 1Н), 6.18 (brs, 1Н), 4.89 (d, J=6,8 Гц, 2Н), 4.79 (s, 2Н), 4.71 (d, J=6,8 Гц, 2Н).
Методика получения соединения 10d:
В раствор соединения 10с (500 мг, 1,89 ммоль) в МеОН (20 мл) и Н2О (10 мл) добавляли К2СО3 (780 мг, 5,66 ммоль). Полученную смесь перемешивали при 70°С в течение 2 ч. Реакционную смесь экстрагировали EtOAc (3 × 50 мл). Объединенные органические слои промывали рассолом, сушили и концентрировали в вакууме с получением неочищенного остатка, который очищали колоночной хроматографией на силикагеле (петролейный эфир/EtOAc=3/1 (об./об.)) с получением соединения 10d (240 мг, 77,1%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt=0,553 мин при 10-80CD_3 мин_220&254; XBrige Shield RP18 2,1 × 50 мм, МС (ЭРИ) m/z=166,1 [М+Н]+.
1Н ЯМР (400 МГц, DMSO-d6) δ 7.14 (d, J=7,8 Гц, 1Н), 7.05-6.99 (m, 1H), 6.70 (d, J=7,8 Гц, 1H), 6.59 (dt, J=1.0, 7,4 Гц, 1Н), 6.18 (brs, 1Н), 4.89 (d, J=6,8 Гц, 2Н), 4.79 (s, 2Н), 4.71 (d, J=6,8 Гц, 2Н).
Методика получения соединения 10е:
В раствор соединения 10d (220 мг, 1,33 ммоль) в пиридине (10 мл) добавляли соединение 6е (439 мг, 1,47 ммоль). Полученную смесь перемешивали при 50°С в течение 18 ч. Реакционную смесь концентрировали при пониженном давлении и очищали препаративной ТСХ (CH2Cl2/МеОН=15/1 (об./об.)) на силикагеле с получением соединения 10е (150 мг, 26,3%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt=0,723 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z=429,1 [М+Н]+.
Методика получения соединения 10f:
В раствор соединения 10е (190 мг, 0,44 ммоль) и К2СО3 (122 мг, 0,89 ммоль) в DMF (2 мл) добавляли соединение N1,N1,N2-триметилэтан-1,2-диамин (54 мг, 0,53 ммоль). Смесь перемешивали при 25-33°С в течение 2 часов. В реакционную смесь добавляли 10 мл воды, и смесь экстрагировали EtOAc (10 мл × 3). Объединенные органические слои промывали рассолом (10 мл × 3), сушили и концентрировали в вакууме с получением неочищенного остатка, который очищали препаративной ТСХ (CH2Cl2/МеОН=15/1(об./об.)) на силикагеле с получением соединения 10f (140 мг, 62%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt=0,714 мин при 5-95АВ_1,5_мин 220&254 хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z=511,3 [М-ОН]+.
Методика получения соединения 10g:
В раствор соединения 10f (130 мг, 0,25 ммоль) в МеОН (3 мл) добавляли Pd/C (13 мг) в атмосфере N2. Смесь перемешивали при 26-31°С в атмосфере H2 (15 фунт/кв.дюйм) в течение 1 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением соединения 10g (95 мг, 78%-ный выход) в виде коричневого масла.
ЖХ/МС: Rt=0,669 мин при 5-95АВ_1,5 мин_220&254 хроматографии (5-95АВ_1,5 мин_1500), МС (ЭРИ) m/z=481,3 [М+Н]+.
Методика получения соединения Примера 10:
В раствор соединения 10h (85 мг, 0,18 ммоль) и DIEA (34 мг, 0,26 ммоль) в DMF (1 мл) добавляли акрилоилхлорид (16 мг, 0,18 ммоль) в DMF (1 мл). Полученную смесь перемешивали при 0°С в течение 30 мин. Реакционную смесь очищали препаративной ВЭЖХ [колонка: Waters Xbridge 150 × 25 5 мкм; условия: 25-50% В (А: 0,05%-ный аммиак; В: CH3CN); скорость потока: 25 мл/мин]. Фракции, содержащие целевое соединение, лиофилизировали с получением соединения Примера 10 (25,2 мг, 26,7%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt=3,452 мин при 10-80_CD_7 мин_220&254 хроматографии (XBrige Shield RP18 2,1 × 50 мм), МС (ЭРИ) m/z=535,3 [М+Н]+.
ВЭЖХ: Rt=2,91 мин при 10-80_CD_1,2 мл хроматографии (XBridge Shield RP 18 2,1 × 50 мм, 5 мкм).
1Н ЯМР (400 МГц, DMSO-d6) δ 10.08 (s, 1H), 8.85 (br s, 1H), 8.47-8.33 (m, 2H), 8.23 (s, 1H), 7.92 (br s, 1H), 7.38 (brd, J=6,6 Гц, 1H), 7.08 (br s, 1H), 6.98 (s, 1H), 6.83 (br s, 1H), 6.47-6.33 (m, 1H), 6.27-6.17 (m, 1H), 5.75 (br d, J=l 1,4 Гц, 1H), 4.86 (brd, J=6,8 Гц, 2H), 4.70 (brd, J=7,0 Гц, 2H), 3.76 (s, 3H), 2.85 (br t, J=5,6 Гц, 2H), 2.71 (s, 3H), 2.34-2.28 (m, 2H), 2.20 (s, 6H).
Пример 11
N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид
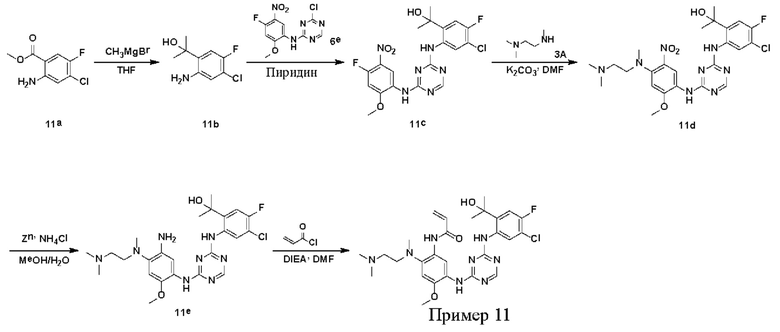
Методика получения соединения 11b:
В раствор соединения 11а (2,0 г, 9,83 ммоль) в THF (20 мл) добавляли по каплям CH3MgBr (16,4 мл, 49,2 ммоль, 3 М в диэтиловом эфире) при 0°С. Полученную смесь перемешивали при 27-34°С в течение 2 ч в атмосфере N2. Реакционную смесь вливали в насыщенный NH4Cl (100 мл) и экстрагировали EtOAc (30 мл × 3). Объединенные органические слои промывали водой (20 мл × 3) и рассолом (20 мл), сушили над Na2SO4 и концентрировали в вакууме с получением соединения 11b (2,0 г, 95%-ный выход) в виде коричневого масла.
ЖХ/МС: Rt=0,558 мин при 5-95АВ_220&254.lcm хроматографии (ACSSH-LCMS-АВ MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z=185,9 [М-ОН]+.
1Н ЯМР (400 МГц, CDCl3) δ 6.91 (d, J=10,8 Гц, 1H), 6.63 (d, J=6,4 Гц, 1H), 1.64 (s, 6Н).
Методика получения соединения 11с:
Смесь соединения 11b (1,9 г, 9,3 ммоль) и соединения 6е (2,8 г, 9,3 ммоль) в пиридине (20 мл) нагревали при 50°С в течение 12 ч. Реакционную смесь концентрировали в вакууме с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (50% EtOAc в петролейном эфире) с получением соединения 11с (2,6 г, 60%-ный выход) в виде светло-желтого твердого вещества.
ЖХ/МС: Rt=0,821 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z=467,1 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 9.73 (br s, 1H), 9.42 (br s, 1H), 8.49 (s, 1H), 8.33 (br s, 1Н), 7.54 (s, 1H), 7.09 (d, J=10,4 Гц, 1H), 6.78 (d, J=12,0 Гц, 1H), 4.03 (s, 3H), 2.51 (br s, 1Н), 1.71 (s, 6Н).
Методика получения соединения 11d:
В раствор соединения 11d (740 мг, 1,59 ммоль) и К2СО3 (439 мг, 3,18 ммоль) в DMF (10 мл) добавляли N1,N1,N2-триметилэтан-1,2-диамин (244 мг, 2,39 ммоль). Реакционную смесь перемешивали при 26-31°С в течение 4 ч. Реакционную смесь добавляли по каплям в Н2О (100 мл) в бане лед-вода, затем фильтровали, остаток на фильтре промывали Н2О (15 мл × 3) и сушили в глубоком вакууме с получением соединения 11е (800 мг, 91,65%-ный выход) в виде оранжевого твердого вещества.
ЖХ/МС: Rt=0,717 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z=549,1 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 9.70 (br s, 1Н), 9.00 (br s, 1H), 8.39 (br s, 2H), 7.42 (br s, 1H), 7.08 (d, J=10,5 Гц, 1H), 6.64 (s, 1H), 3.95 (s, 3H), 3.28 (t, J=7,2 Гц, 2H), 2.88 (s, 3H), 2.57 (t, J=7,2 Гц, 2H), 2.25 (s, 6H), 1.69 (s, 6H).
Методика получения соединения 11е:
В раствор соединения 11d (200 мг, 0,364 ммоль) в 8 мл смеси МеОН/H2O=5/1 (об./об.) добавляли Zn (119 мг, 1,82 ммоль) и NH4Cl (97 мг, 1,82 ммоль). Полученную смесь нагревали при 90°С в течение 1 ч. Реакционную смесь фильтровали, и фильтрат концентрировали в вакууме с получением остатка, который растворяли в CH2Cl2 (30 мл), промывали водой (20 мл × 3) и рассолом (20 мл), сушили над Na2SO4 и концентрировали в вакууме с получением соединения 11е (180 мг, 95,3%-ный выход) в виде коричневого масла.
ЖХ/МС: Rt=0,679 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z=519,1 [М+Н]+.
Методика получения соединения Примера 11:
В раствор соединения 11е (180 мг, 0,35 ммоль) и DIEA (68 мг, 0,53 ммоль) в DMF (3 мл) добавляли акрилоилхлорид (32 мг, 0,35 ммоль) при 0°С. Полученную смесь перемешивали при 0°С в течение 1 ч. Реакционную смесь гасили Н2O (0,1 мл) и затем фильтровали. Фильтрат очищали препаративной ВЭЖХ (колонка: Waters Xbridge 150 × 25, 5 мкм; условия: 52-82% В (А: 0,05%-ный аммиак, В: CH3CN); скорость потока: 25 мл/мин) и лиофилизировали с получением соединения Примера 11 (52,9 мг, 26,4%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt=2,124 мин при 10-80CD_3 мин_220&254 хроматографии (XBrige Shield RP18 2,1 × 50 мм), МС (ЭРИ) m/z=573,3 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 10.60 (br s, 1H), 10.44 (br s, 1H), 9.96 (br s, 1H), 8.47 (d, J=7,6 Гц, 1H), 8.42 (s, 1H), 7.69 (br s, 1H), 7.10 (d, J=10,8 Гц, 1H), 6.78 (s, 1H), 6.49-6.28 (m, 2H), 6.18 (br s, 1H), 5.91-5.65 (m, 1H), 3.88 (s, 3H), 2.88 (t, J=5,6 Гц, 2H), 2.70 (s, 3H), 2.27 (s, 8H), 1.77 (s, 6H).
Пример 12
N-(5-(4-(4-хлор-5-фтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид

Методика получения соединения 12b:
В раствор соединения 12а (2 г, 13,74 ммоль) в CH2Cl2 (20 мл) добавляли IC1 (3,3 г, 20,61 ммоль) при 0°С. Полученную смесь перемешивали при 27-34°С в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле (петролейный эфир/EtOAc=100/1) с получением соединения 12b (2,5 г, 67%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt=0,811 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP-18 25-2 мм), МС (ЭРИ) m/z=271,8 [М+Н]+.
1H ЯМР (400 МГц, Метанол-d4) δ 7.59 (d, J=8,0 Гц, 1Н), 6.62 (d, J=11,6 Гц, 1H).
Методика получения соединения 12с:
В раствор соединения 12b (450 мг, 1,66 ммоль) в МеОН (10 мл) и DMF (20 мл) добавляли Pd(OAc)2 (38 мг, 0,17 ммоль), DPPF (94 мг, 0,17 ммоль) и Et3N (504 мг, 4,98 ммоль). Полученную смесь продували и дегазировали СО 3 раза, затем перемешивали при 80°С в атмосфере СО (50 фунт/кв.дюйм) в течение 24 ч. Реакционную смесь фильтровали и разбавляли EtOAc (50 мл). Органический слой промывали рассолом (50 мл), сушили и концентрировали при пониженном давлении с получением неочищенного остатка, который очищали колоночной хроматографией на силикагеле (петролейный эфир/EtOAc=50/1(об./об.)) с получением соединения 12с (320 мг, 95%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt=0,941 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18 25-2 мм), МС (ЭРИ) m/z=203,9[М+Н]+.
1Н ЯМР (400 МГц, МеОН-d4) δ 7.83 (d, J=8,4 Гц, 1H), 6.59 (d, J=11,6 Гц, 1H), 3.85 (s, 3H).
Методика получения соединения 12d:
В раствор соединения 12с (320 мг, 1,57 ммоль) в THF (10 мл) добавляли CH3MgBr (2,62 мл, 7,85 ммоль) при 0°С. Полученную смесь перемешивали при 26-31°С в течение 2 ч. Реакционную смесь разбавляли насыщ. водн. NH4Cl (10 мл) и экстрагировали EtOAc (2 × 20 мл). Объединенные органические слои концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле (петролейный эфир/EtOAc=10/1(об./об.)) с получением соединения 12d (250 мг, 78%-ный выход) в виде бесцветного масла.
ЖХ/МС: Rt=0,826 мин при 5-95АВ 1,5 мин 220&254 хроматографии (MERCK RP18 25-2 мм), МС (ЭРИ) m/z=185,9 [М+Н-18]+.
1H ЯМР (400 МГц, MeOH-d4) δ 7.08 (d, J=8,4 Гц, 1H), 6.49 (d, J=11,6 Гц, 1H), 1.57 (s, 6Н).
Методика получения соединения 12е:
В раствор соединения 12d (200 мг, 0,98 ммоль) в CH2Cl2 (10 мл) добавляли DIEA (253 мг, 1,96 ммоль) и 2,4-дихлор-1,3,5-триазин (162 мг, 1,08 ммоль). Полученную смесь перемешивали при 26-32°С в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле (петролейный эфир/EtOAc=10/1(об./об.)) с получением соединения 12е (210 мг, 67%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt=0,971 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18 25-2 мм), МС (ЭРИ) m/z=316,9 [М+Н]+.
Методика получения соединения 12f:
В раствор соединения 12е (210 мг, 0,66 ммоль) и соединения 2g (177 мг, 0,66 ммоль) в n-BuOH (5 мл) добавляли TFA (0,05 мл). Полученную смесь перемешивали при 26-33°С в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле (CH2Cl2/МеОН=10/1 (об./об.)) с получением соединения 12f (150 мг, 41%-ный выход) в виде коричневого масла.
ЖХ/МС: Rt=0,821 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18 25-2 мм), МС (ЭРИ) m/z=549,1[М+Н]+.
Методика получения соединения 12g:
В раствор соединения 12f (150 мг, 0,27 ммоль) в МеОН (5 мл) добавляли 10% Pd/C (15 мг). Полученную смесь продували и дегазировали Н2 3 раза, затем перемешивали при 26-34°С (комнатная температура) в атмосфере Н2 (30 фунт/кв.дюйм) в течение 2 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением соединения 12g (110 мг, 78%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt=0,771 мин при 5-95АВ_1,5 мин_220&254 хроматографии (Xtimate С18 2,1 × 30 мм), МС (ЭРИ) m/z=519,1 [М+Н]+.
Методика получения соединения Примера 12:
В раствор соединения 12g (110 мг, 0,21 ммоль) и DIE А (41 мг, 0,32 ммоль) в DMF (3 мл) добавляли акрилоилхлорид (19 мг, 0,21 ммоль). Полученную смесь перемешивали при 0°С в течение 30 мин. Реакционную смесь очищали препаративной ВЭЖХ [колонка: Waters Xbridge 150 × 25, 5 мкм; условия: 50-80% В (А: 0,05% NH3H2O; В: CH3CN); скорость потока: 25 мл/мин]. Фракции, содержащие целевое соединение, лиофилизировали с получением соединения Примера 12 (13,5 мг, 11%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt=2,167 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С18 2,1 × 30 мм), МС (ЭРИ) m/z=573,0 [М+Н]+.
ВЭЖХ: Rt=3,38 мин при 10-80_ab_1,2 мл хроматографии (Ultimate С18 3 × 50 мм, 3 мкм).
1Н ЯМР (400 МГц, CDCl3) δ 10.85 (br s, 1H), 10.45 (br s, 1H), 9.96 (br s, 1H), 8.43 (s, 1H), 8.39 (d, J=11,8 Гц, 1H), 7.72 (br s, 1H), 7.29 (d, J=8,4 Гц, 1H), 6.78 (s, 1H), 6.43-6.32 (m, 2H), 6.10 (br s, 1H), 5.81-5.73 (m, 1H), 3.88 (s, 3H), 2.91-2.85 (m, 2H), 2.70 (s, 3H), 2.35-2.20 (m, 8H), 1.78 (s, 6H).
Пример 13
N-(2-((2-(диметиламино)этил)(метил)амино)-5-(4-(2-(2-гидроксипропан-2-ил)-4-(трифторметил)фениламино)-1,3,5-триазин-2-иламино)-4-метоксифенил)акриламид
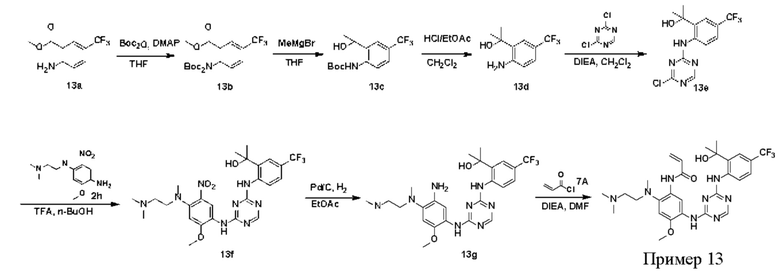
Методика получения соединения 13b:
В раствор соединения 13а (0,92 г, 4,2 ммоль) и Boc2O (3,17 г, 14,7 ммоль) в THF (15 мл) добавляли DMAP (256 мг, 2,1 ммоль) при 26-32°С. Реакционную смесь перемешивали при 26-32°С в течение 4 ч. Реакционный раствор концентрировали в вакууме с получением неочищенного остатка, который очищали колоночной хроматографией на силикагеле (0-10% EtOAc в петролейном эфире) с получением целевого продукта - соединения 13b (1,43 г, 81,2%-ный выход) в виде не совсем белого твердого вещества.
ЖХ/МС: Rt=1,078 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z=442,1 [М+23]+.
1Н ЯМР (400 МГц, CDCl3) δ 8.28 (d, J=1,6 Гц, 1 H), 7.95 (dd, J=2.0, 8,4 Гц, 1 Н), 7.53 (d, J=8,4 Гц, 1 H), 3.93 (s, 3 H), 1.36 (s, 18 Н).
Методика получения соединения 13с:
В раствор соединения 13b (1,43 г, 3,41 ммоль) в THF (25 мл) добавляли по каплям MeMgBr (4,54 мл, 13,63 ммоль, 3 М в диэтиловом эфире) в бане лед-вода. Смесь перемешивали в течение 3 ч при 26-32°С. В реакционную смесь добавляли насыщенный водный NH4Cl (30 мл) и экстрагировали EtOAc (50 мл × 3). Объединенный органический слой промывали рассолом (30 мл) и сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного остатка, который очищали колоночной хроматографией на силикагеле (3-30% EtOAc в петролейном эфире) с получением целевого продукта - соединения 13 с (0,84 г, 77,8%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=0,889 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z=246,0 [М+Н]+.
1Н ЯМР (400 МГц, Метанол-d4): δ 8.22 (d, J=8,8 Гц, 1 Н), 7.54-7.51 (m, 2 Н), 1.64 (s, 6 H), 1.54 (s, 9 H).
Методика получения соединения 13d:
В раствор соединения 13с (640 мг, 2,0 ммоль) в CH2Cl2 (10 мл) добавляли по каплям HCl/EtOAc (20 мл, 4 М) в бане лед-вода. Смесь перемешивали в течение 6 ч при 26-34°С. Реакционную смесь подщелачивали насыщенным NaHCO3 до рН=8 и экстрагировали EtOAc (70 мл × 3). Объединенный органический слой промывали рассолом (50 мл), сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта, который очищали колоночной флэш-хроматографией (3-30% EtOAc в петролейном эфире) с получением целевого продукта - соединения 13d (260 мг, 45,1%-ный выход) в виде желтого масла.
ЖХ/МС: Rt=0,726 мин при 5-95 АВ_220&254. lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z=202,0 [М-17]+.
1H ЯМР (400 МГц, Метанол-d4): δ 7.33 (s, 1H), 7.23 (d, J=8,8 Гц, 1 Н), 6.74 (d, J=8,4 Гц, 1 H), 1.63 (s, 6 H).
Методика получения соединения 13е:
В раствор соединения 13d (260 мг, 1,18 ммоль) и DIEA (304 мг, 2,56 ммоль) добавляли 2,4-дихлор-1,3,5-триазин (267 мг, 1,78 ммоль) при 0°С. После перемешивания реакционной смеси при 28-34°С в течение 4 ч реакционную смесь гасили насыщенным водным NH4Cl (5 мл) и экстрагировали EtOAc (50 мл × 3). Объединенный органический слой промывали рассолом (50 мл) и сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта (320 мг), который очищали колоночной флэш-хроматографией (3-30% EtOAc в петролейном эфире) с получением соединения 13е (200 мг, 50,8%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt=0,832 мин при 5-95 АВ_220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z=332,9 [М+Н]+.
Методика получения соединения 13f:
В раствор соединения 13е (200 мг, 0,61 ммоль) и соединения 2g (183 мг, 0,68 ммоль) в n-BuOH (10 мл) добавляли по каплям TFA (0,8 мл) в бане лед-вода. Смесь перемешивали в течение 2 ч при 28-34°С. Реакционный раствор напрямую концентрировали в вакууме с получением неочищенного остатка, который очищали колоночной флэш-хроматографией на силикагеле (элюируя 0-15% МеОН в дихлорметане) с получением загрязненного примесями продукта (350 мг), который дополнительно очищали препаративной ТСХ (МеОН/CH2Cl2=10/1) с получением соединения 13f (80 мг, 50,8%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt=0,755 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z=565,1 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3): δ м.д. 8.48-8.43 (m, 2 Н), 7.63 (d, J=8,4 Гц, 1 Н), 7.52-7.48 (m, 2 Н), 6.96 (s, 1 Н), 4.00 (s, 3 Н), 3.40 (t, J=6,4 Гц, 2 Н), 2.90 (s, 3 Н), 2.65 (t, J=6,4 Гц, 2 Н), 2.30 (s, 6 Н), 1.75 (s, 6 Н).
Методика получения соединения 13g:
Смесь соединения 13f (80 мг, 0,14 ммоль) и Pd/C (70 мг, 10%) в EtOAc (30 мл) перемешивали в атмосфере Н2 (15 фунт/кв.дюйм) в течение 30 мин. Реакционную смесь фильтровали, и остаток на фильтре промывали EtOAc (50 мл). Фильтрат концентрировали в вакууме с получением соединения 13g (60 мг, 80,0%-ный выход) в виде серого твердого вещества.
ЖХ/МС: Rt=0,712 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z=535,2 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 9.83-9.80 (m, 1 Н), 8.39 (d, J=8,8 Гц, 1 H), 8.27-8.25 (m, 1 H), 7.77 (s, 1 H), 7.68-7.49 (m, 2 Н), 7.41 (s, 1 Н), 6.59 (s, 1 H), 3.95(s, 3 H), 2.88 (t, J=6,8 Гц, 2 Н), 2.59 (s, 3 Н), 2.37 (t, J=6,8 Гц, 2 Н), 2.35-2.34 (m, 2 Н), 2.25 (s, 6 Н), 2.21-2.20 (m, 1 Н), 1.67 (s, 6 Н).
Методика получения соединения Примера 13:
В смесь соединения 12g (60 мг, 0,112 ммоль) и DIEA (29 мг, 0,224 ммоль) в DMF (1 мл) добавляли по каплям соединение акрилоилхлорид (16,5 мг в 1 мл DMF) на протяжении 30 мин в бане лед-вода. После перемешивания реакционной смеси в течение 30 мин при 0-5°С реакционная смесь превращалась в коричневый раствор. Реакционную смесь гасили Н2О (45 мг) и затем напрямую очищали препаративной ТСХ (CH2Cl2/МеОН=10/1) с получением соединения Примера 13 (18,2 мг, 27,5%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt=2,698 мин при 0-60АВ_4,0 мин_220&254 хроматографии (Xtimate С18 2,1 × 30 мм), МС (ЭРИ) m/z=589,0 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3): δ 10.88 (br s, 1Н), 10.37 (br s, 1H), 9.89 (br s, 1H), 8.46 (d, J=8,4 Гц, 1H), 8.36 (s, 1H), 7.64 (br s, 1H), 7.54-7.43 (m, 2H), 6.71 (s, 1H), 6.39-6.22 (m, 2H), 5.71 (br d, J=11,2 Гц, 1H), 3.81 (s, 3H), 2.81 (m, 2H), 2.63 (s, 3H), 2.21 (m, 8H), 1.75 (s, 6H).
Пример 14
N-(5-(4-(4-хлор-3-фтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид

Методика получения соединения 14b:
Раствор соединения 14а (5 г, 34,35 ммоль) и Boc2O (15 г, 68,70 ммоль) в THF (80 мл) перемешивали при 60°С в течение 12 ч. Реакционную смесь концентрировали в вакууме напрямую с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (2% EtOAc в петролейном эфире) с получением соединения 14b (8,2 г, 69%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=0,866 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18 2,5-2 мм).
1Н ЯМР (400 МГц, CDCl3) δ 7.36 (d, J=9,6 Гц, 1 И), 7.19 (t, J=6,8 Гц, 1 Н), 6.87 (d, J=7,6 Гц, 1 Н), 6.45 (s, 1 H), 1.45 (s, 9 H).
Методика получения соединения 14с:
В раствор соединения 14b (8 г, 32,56 ммоль) в THF (120 мл) добавляли трет-BuLi (1,3 М) (50 мл, 65,12 ммоль) при -78°С в атмосфере N2. После перемешивания в течение 1 ч добавляли DMF (3,6 г, 48,84 ммоль), и реакционную смесь перемешивали при -78-33°С в течение 16 ч. Реакционную смесь гасили насыщенным раствором NH4Cl (300 мл), затем экстрагировали EtOAc (200 мл × 3). Объединенные органические слои промывали рассолом (100 мл), сушили над сульфатом натрия и концентрировали в вакууме с получением соединения 14с (8,2 г, 92%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=0,929 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм).
1H ЯМР (400 МГц, Метанол-d4) δ 10.49 (brs, 1 Н), 10.28 (s, 1 Н), 8.18 (dd, J=0,8 Гц, 9,2 Гц, 1 Н), 7.48(t, J=8,8 Гц, 1 Н), 1.46 (s, 9 Н).
Методика получения соединения 14d:
В раствор соединения 14с (4 г, 14,62 ммоль) в THF (40 мл) добавляли MeMgBr (3,0 М) (19,5 мл, 58,48 ммоль) при 0°С в атмосфере N2. Реакционную смесь перемешивали при 0°С в течение 2 ч. Реакционную смесь гасили насыщенным раствором NH4Cl (80 мл) и экстрагировали EtOAc (30 мл × 3). Объединенные органические слои промывали рассолом (30 мл), сушили над сульфатом натрия и концентрировали в вакууме с получением соединения 14d (4 г, 94%-ный выход) в виде желтого масла, которое использовали на следующей стадии напрямую без дополнительной очистки.
ЖХ/МС: Rt=0,871 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм).
Методика получения соединения 14е:
В раствор соединения 14d (4 г, 14,62 ммоль) в CH2Cl2 (40 мл) добавляли DMP (9,3 г, 21,93 ммоль) при 0°С в атмосфере N2. Реакционную смесь перемешивали при 0°С в течение 2 ч. Добавляли насыщенный раствор NaHCO3 (40 мл) и насыщенный раствор Na2SO3 (30 мл), и смесь перемешивали при 25-33°С в течение 30 мин. Органический слой отделяли и экстрагировали CH2Cl2 (30 мл). Объединенные органические слои промывали рассолом (20 мл), сушили над сульфатом натрия и концентрировали в вакууме с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (1% EtOAc в петролейном эфире) с получением соединения 14е (2,5 г, 59%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=0,924 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z=187,9 [М-101+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 10.20 (brs, 1 Н), 8.19 (d, J=9,6 Гц, 1 H), 7.47 (t, J=9,2 Гц, 1 H), 2.67 (d, J=8,0 Гц, 1 H), 1.52 (s, 9 Н).
Методика получения соединения 14f:
В раствор соединения 14е (2,5 г, 8,69 ммоль) в THF (40 мл) добавляли MeMgBr (3,0 М) (10 мл, 30,42 ммоль) при 0°С в атмосфере N2. Реакционную смесь перемешивали при 0°С в течение 2 ч. Реакционную смесь гасили насыщенным раствором NH4Cl (80 мл) и экстрагировали EtOAc (30 мл × 3). Объединенные органические слои промывали рассолом (30 мл), сушили над сульфатом натрия и концентрировали в вакууме с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (0,5% EtOAc в петролейном эфире) с выделением соединения 14е (1,2 г) с последующим элюированием 2%-ным EtOAc в петролейном эфире с получением указанного в заголовке продукта 14f (1,5 г, 57%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=1,066 мин при 5-95 АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z=270,1 [M-55+Na]+.
1Н ЯМР (400 МГц, CDCl3) δ 9.67 (brs, 1 Н), 7.94 (d, J=9,2 Гц, 1 Н), 7.23 (t, J=8,4 Гц, 1 H), 1.75 (d, J=3,6 Гц, 6 Н), 1.51 (s, 9 Н).
Методика получения соединения 14g:
В раствор соединения 14f (1,5 г, 4,94 ммоль) в CH2Cl2 (15 мл) добавляли TFA (5 мл) при 0°С в атмосфере N2. Реакционную смесь перемешивали при 36-34°С в течение 1 ч. Реакционную смесь концентрировали в вакууме с получением соединения 14g (700 мг, 94%-ный выход) в виде желтого масла.
ЖХ/МС: Rt=0,652 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z=185,9 [М -ОН]+.
1Н ЯМР (400 МГц, CDCl3) δ 6.92 (t, J=8,0 Гц, 1 И), 6.24 (dd, J=1,2 Гц, 8,4 Гц, 1 H), 1.65 (d, J=3,6 Гц, 6 Н).
Методика получения соединения 14h:
Раствор соединения 14g (200 мг, 0,98 ммоль) и соединения 6е (294 мг, 0,98 ммоль) в пиридине (1,5 мл) перемешивали при 50°С в течение 4 ч. Реакционную смесь напрямую концентрировали в вакууме с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (EtOAc:петролейный эфир = 7:3 (об./об.)) с получением указанного в заголовке продукта 14h (150 мг, 21%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt=0,864 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z=467,0 [М+Н]+.
Методика получения соединения 14i:
В раствор соединения 14h (150 мг, 0,32 ммоль) и K2CO3 (89 мг, 0,64 ммоль) в DMF (3 мл) добавляли N1,N1,N2-триметилэтан-1,2-диамин (39 мг, 0,39 ммоль). Реакционную смесь перемешивали при 28-33°С в течение 2 ч. Реакционную смесь вливали в воду (10 мл) и перемешивали в течение 30 мин. Смесь фильтровали, и остаток на фильтре промывали водой (10 мл × 3). Остаток на фильтре сушили в глубоком вакууме с получением соединения 14i (240 мг, неочищенное) в виде желтого твердого вещества.
ЖХ/МС: Rt=0,738 мин при 5-95АВ_220&254 хроматографии (В: XBrige Shield RP18 2,1 × 50 мм), МС (ЭРИ) m/z=549,1 [М+Н]+.
Методика получения соединения 14j:
В раствор соединения 14i (200 мг, 0,35 ммоль) и NH4Cl (95 мг, 1,75 ммоль) в МеОН (4 мл) и воде (0,5 мл) добавляли Zn (115 мг, 1,75 ммоль). Реакционную смесь перемешивали при 90°С в течение 2 ч. Реакционную смесь фильтровали, и фильтрат вливали в воду (50 мл) и экстрагировали EtOAc (25 мл × 3). Объединенные органические слои промывали рассолом (20 мл), сушили над сульфатом натрия, фильтровали, и фильтрат концентрировали в вакууме с получением соединения 14j (180 мг, неочищенное) в виде черного твердого вещества.
ЖХ/МС: Rt=0,699 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z=519,1 [М+Н]+.
Методика получения соединения Примера 14:
В смесь соединения 14j (180 мг, неочищенное, 0,35 ммоль) и DIEA (90 мг, 0,70 ммоль) в DMF (3 мл) добавляли акрилоилхлорид (32 мг, 0,35 ммоль) при 0°С. Реакционную смесь перемешивали при 26-32°С в течение 0,5 ч. Реакционную смесь очищали препаративной ВЭЖХ: [колонка: Waters Xbridge 150 × 25, 5 мкм; условия: 65-95% В (А: 0,05%-ный аммиак; В: CH3CN); скорость потока: 30 мл/мин]. Фракции, содержащие целевое соединение, лиофилизировали с получением соединения Примера 14 (24,5 мг, 12%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt=2,204 мин при 10-80CD_3 мин_220&254 хроматографии (В: XBrige Shield RP18 2,1 × 50 мм), МС (ЭРИ) m/z=573,2 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 11.61 (brs, 1 Н), 10.30 (brs, 1 Н), 9.86 (s, 1 Н), 8.40 (s, 1 Н), 8.27 (d, J=8,8 Гц, 1 Н), 7.66 (brs, 1 Н), 7.35-7.26 (m, 1 Н), 6.75 (brs, 1 Н), 6.40 (d,.J=16,0 Гц, 1 Н), 6.01 (brs, 1 Н), 5.78 (d, J=11,2 Гц, 1 Н), 3.88 (s, 3 Н), 3.07-2.87 (m, 2 Н), 2.71 (s, 3 Н), 2.58-2.13 (m, 8Н), 1.87 (d, J-3,6 Гц, 6Н).
ВЭЖХ: Rt = 3,86 мин при 10-80CD_1,2 мл хроматографии (XBridge Shield RP 18 2,1×50 мм, 5 мкм).
Пример 15
N-(5-(4-(4,5-дихлор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид
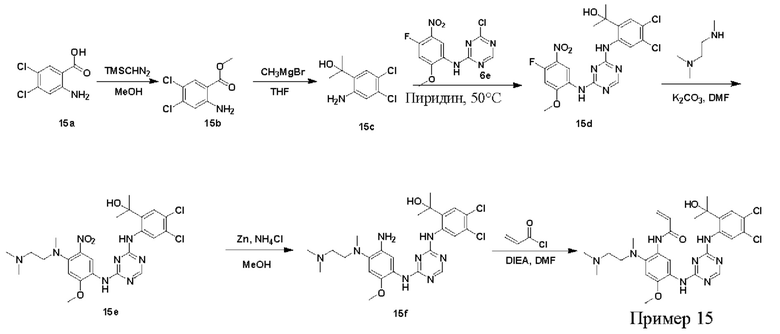
Методика получения соединения 15b:
В раствор соединения 15а (2,0 г, 9,71 ммоль) в EtOAc (40 мл) и МеОН (40 мл) добавляли TMSCHN2 (9,7 мл, 25,78 ммоль, 2М в гексане). Смесь перемешивали при 26-33°С в течение 1,5 ч. Реакционную смесь концентрировали при пониженном давлении с получением соединения 15b (2,0 г, 94,1%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 0,988 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 187,8 [М-31]+.
1H ЯМР (400 МГц, CDCl3) δ 7.85 (s, 1Н), 6.72 (s, 1H), 5.71 (br s, 2H), 3.80 (s, 3H).
Методика получения соединения 15с:
В раствор соединения 15b (1,0 г, 4,54 ммоль) в THF (30 мл) добавляли CH3MgBr (6 мл, 3 М в диэтиловом эфире) при 0-5°С. Смесь перемешивали при 26-34°С в течение 1,5 ч (желтый раствор). Реакционную смесь гасили добавлением водного NH4Cl (20 мл), затем экстрагировали EtOAc (3×100 мл). Органические слои промывали рассолом (3 х 100 мл) и концентрировали при пониженном давлении с получением соединения 15 с (950 мг, 95%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 0,867 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 201,8 [M-OH]+.
1H ЯМР (400 МГц, CDCl3) δ 7.13 (s, 1H), 6.70 (s, 1H), 1.64 (s, 6H).
Методика получения соединения 15d:
В раствор соединения 15 с (500 мг, 2,27 ммоль) в пиридине (5 мл) добавляли соединение 6е (749 мг, 2,50 ммоль). Полученную смесь перемешивали при 50°С в течение 18 ч. Смесь реакционную смесь концентрировали при пониженном давлении и очищали препаративной ТСХ на силикагеле (CH2Cl2/MeOH = 15/1 (об./об.)) с получением соединения 15d (300 мг; 29,2%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,991 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 483,1 [M+H]+.
Методика получения соединения 15е:
В раствор соединения 15d (300 мг, 0,62 ммоль) и K2CO3 (171 мг, 1,24 ммоль) в DMF (4 мл) добавляли N1,N1,N2-триметилэтан-1,2-диамин (76 мг, 0,74 ммоль). Смесь перемешивали при 28-33°С в течение 2 часов. В реакционную смесь добавляли 10 мл воды и экстрагировали EtOAc (10 мл × 3). Объединенные органические слои промывали рассолом (10 мл × 3), сушили над сульфатом натрия и концентрировали в вакууме с получением неочищенного остатка, который очищали препаративной ТСХ на силикагеле (CH2Cl2/МеОН = 15/1(об./об.)) с получением соединения 15е (250 мг, 71%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,745 мин при 5-95АВ_1,5 мин 220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 565,1 [М+Н]+.
Методика получения соединения 15f:
В раствор соединения 15е (220 мг, 0,39 ммоль) в МеОН (5 мл) и H2O (1 мл) добавляли Zn (127 мг, 1,95 ммоль) и NH4Cl (208 мг, 3,89 ммоль). Смесь перемешивали при 70°С в течение 1,5 ч в атмосфере N2 (превращалась в коричневую смесь). Реакционную смесь гасили добавлением водного NH4Cl (20 мл), затем экстрагировали EtOAc (3×10 мл). Органический слой промывали рассолом (3×10 мл), сушили и концентрировали в вакууме с получением соединения 15f (190 мг, 91,4%-ный выход).
ЖХ/МС: Rt = 0,799 мин при 5-95АВ_1,5 мин 220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 535,1 [М+Н]+.
Методика получения соединения Примера 15:
В раствор соединения 15f (190 мг, 0,35 ммоль) и DIEA (69 мг, 0,53 ммоль) в DMF (1 мл) добавляли акрилоилхлорид (32 мг, 0,35 ммоль) в DMF (1 мл). Полученную смесь перемешивали при 0°С в течение 30 мин. Реакционную смесь очищали препаративной ВЭЖХ [колонка: Waters Xbridge 150×25, 5 мкм; условия: 25-55% В (А: 0,05%-ный аммиак; В: CH3CN); скорость потока: 25 мл/мин]. Фракции, содержащие целевое соединение, лиофилизировали с получением соединения Примера 15 (37,8 мг, 12,2%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 5,084 мин при 10-80CD_7 мин_220&254 хроматографии (XBrige Shield RP18 2,1×50 мм), МС (ЭРИ) m/z = 589,3 [M+H]+.
ВЭЖХ: Rt = 4.32 мин при 10-80_CD_1,2 мл хроматография (XBridge Shield RP 18 2,1×50 мм, 5 мкм).
1H ЯМР (400 МГц, DMSO-d6) δ 10.32 (br s, 1H), 10.03 (s, 1H), 9.15 (br s, 1H), 8.42 (br s, 1H), 8.29 (br d, J=14,8 Гц, 2Н), 7.38 (br s, 1H), 7.03 (br s, 1H), 6.43-6.34 (m, 1H), 6.33 (s, 1H), 6.25-6.13 (m, 1H), 5.73 (br d, J=11,6 Гц, 1H), 3.78 (s, 3H), 2.86 (br t, J=5,6 Гц, 2Н), 2.72 (s, 3H), 2.37-2.30 (m, 2Н), 2.21 (s, 6H), 1.52 (s, 6H).
Пример 16
N-(5-(4-(4-хлор-3,5-дифтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид
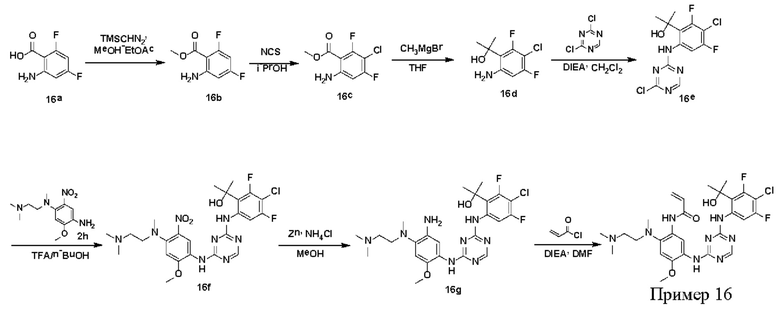
Методика получения соединения 16b:
В раствор соединения 16а (5,0 г, 28,89 ммоль) в EtOAc (50 мл) и МеОН (50 мл) добавляли TMSCHN2 (29 мл, 57,76 ммоль, 2М в гексане) при 0-5°С. Смесь перемешивали при 28-36°С (комнатная температура) в течение 1,5 ч. Реакционную смесь концентрировали при пониженном давлении с получением соединения 16b (4,8 г, 82,5%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,882 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 187,8 [M+H]+.
1H ЯМР: (400 МГц, CDCl3) δ 6.05-5.94 (m, 2Н), 5.93-5.72 (m, 2Н), 3.76 (s, 3H).
Методика получения соединения 16с:
Перемешиваемый раствор соединения 16b (2,70 г, 14,42 ммоль) в i-PrOH (50 мл) охлаждали до 0°С и порциями добавляли NCS (2,02 г, 15,15 ммоль), полученную желтую суспензию перемешивали при 0°С-30°С в течение 12 ч, одновременно отслеживая реакцию методом ЖХ/МС (постепенно превращалась в прозрачную). Реакционную смесь напрямую концентрировали в вакууме с получением неочищенного продукта, который очищали колоночной флэш-хроматографией на силикагеле (условия: 65-75% В (А: 0,05% TFA в воде; В: МеОН); скорость потока: 40 мл/мин)) с получением соединения 16 с (1000 мг чистота 87,20%, и 500 мг исходного вещества) в виде белого твердого вещества.
ЖХ/МС: Rt = 0,905 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 189,9 [М-32]+.
1H ЯМР (400 МГц, DMSO-d6) δ 7.04 (br s, 2H), 6.61 (dd, J-1.8, 11,8 Гц, 1Н), 3.82 (s, 3Н).
Методика получения соединения 16d:
В раствор соединения 16с (500 мг, 2,26 ммоль) в THF (10 мл) добавляли CH3MgBr (3 мл, 3 М в диэтиловом эфире) при 0-5°С. Желтый раствор перемешивали при 24-29°С в течение 1,5 ч (желтый раствор). Реакционную смесь гасили добавлением водного NH4Cl (20 мл), затем экстрагировали EtOAc (3×10 мл). Органические слои промывали рассолом (3×10 мл) и напрямую концентрировали в вакууме с получением соединения 16d (450 мг, 74%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 0,900 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 203,9 [M-OH]+.
Методика получения соединения 16е:
В раствор соединения 16d (450 мг, 2,03 ммоль) и DIEA (394 мг, 3,05 ммоль) в CH2Cl2 (10 мл) добавляли 2,4-дихлор-1,3,5-триазин (335 мг, 2,23 ммоль). Полученную белую смесь перемешивали при 24-29°С (комнатная температура) в течение 2 ч. Реакционную смесь гасили добавлением водного NH4Cl (20 мл), затем экстрагировали EtOAc (3×10 мл). Органические слои промывали рассолом (3×10 мл) и напрямую концентрировали в вакууме с получением соединения 16е (500 мг, 30%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 0,927 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 334,9 [М+Н]+.
Методика получения соединения 16f:
В три отдельных раствора соединения 16е (100 мг × 3, 0,90 ммоль) и соединения 2g (80 мг, 0,30 ммоль) в n-BuOH (2 мл) добавляли TFA (0,02 мл). Полученную смесь перемешивали при 25-33°С в течение 3 ч (красная смесь). Реакционную смесь напрямую концентрировали в вакууме с получением неочищенного продукта, который очищали колоночной флэш-хроматографией на силикагеле (условия: 87-89% В (А: 0,05% TFA в воде; В: МеОН); скорость потока: 40 мл/мин)) с получением соединения 16f (150 мг, чистота 97,18%, 30%-ный выход) в виде красного твердого вещества.
ЖХ/МС: Rt = 0,792 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 567,3 [М+H]+.
Методика получения соединения 16g:
В раствор соединения 16f (150 мг, 0,26 ммоль) в МеОН (5 мл) и Н2О (1 мл) добавляли Zn (87 мг, 1,32 ммоль) и NH4Cl (142 мг, 2,65 ммоль). Смесь перемешивали при 70°С в течение 1,5 ч в атмосфере N2 (черная смесь). Реакционную смесь гасили добавлением водного NH4Cl (20 мл), затем экстрагировали EtOAc (3×10 мл). Органические слои промывали рассолом (3×10 мл), сушили и напрямую концентрировали в вакууме с получением соединения 16g (120 мг, 84,6%-ный выход) в виде коричневого масла.
ЖХ/МС: Rt = 0,802 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 537,1 [М+Н]+.
Методика получения соединения Примера 16:
В раствор соединения 16g (120 мг, 0,22 ммоль) и DIEA (43 мг, 0,33 ммоль) в DMF (1 мл) добавляли акрилоилхлорид (20 мг, 0,22 ммоль) в DMF (1 мл). Полученную коричневую смесь перемешивали при 0°С в течение 30 мин. Реакционную смесь очищали препаративной ВЭЖХ [колонка: Waters Xbridge 150×25, 5 мкм; условия: 62-92% В (А: 0,05%-ный аммиак; В: CH3CN); скорость потока: 25 мл/мин]. Фракции, содержащие целевое соединение, лиофилизировали с получением соединения Примера 16 (27,6 мг, 21%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 2,310 мин при 10-80CD_3 мин_220&254 хроматографии (XBrige Shield RP18 2,1×50 мм), МС (ЭРИ) m/z = 591,2 [M+H]+.
ВЭЖХ: Rt = 4,78 мин в 10-80_CD_1,2 мл хроматография (XBridge Shield RP 18 2,1×50 мм).
1H ЯМР (400 МГц, DMSO-d6) δ 11.23 (br s, 1H), 10.05 (br s, 1H), 9.24 (brs, 1H), 8.35-8.27 (m, 2H), 7.01 (s, 1H), 6.82 (br s, 1H), 6.42 (br s, 1H), 6.18 (br d, J=16,8 Гц, 1H), 5.72 (br d, J=10,8 Гц, 1H), 3.77 (s, 3Н), 2.90 (br s, 2H), 2.77-2.68 (m, 3Н), 2.45-2.31 (m, 2H), 2.24 (br s, 6Н), 1.61 (brs, 6H).
Пример 17
N-(5-(4-(4,5-дифтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-4-метокси-2-((3aR,6aR)-5-метилгексагидропирроло[3,4-b]пиррол-1(2Н)-ил)фенил)акриламид

Методика получения соединения 17b:
В раствор соединения 1d (200 мг, 0,444 ммоль) и K2CO3 (123 мг, 0,888 ммоль) в DMSO (6 мл) добавляли соединение 17а (67 мг, 0,533 ммоль). Реакционную смесь перемешивали при 85°С в течение 1 ч (цвет менялся с бледно-желтого на оранжевый). Реакционную смесь добавляли по каплям в Н2О (80 мл) в бане лед-вода, и выпавшее в осадок твердое вещество собирали фильтрованием, остаток на фильтре промывали H2O (15 мл × 3) и затем сушили в глубоком вакууме с получением соединения 17b (200 мг, 81%-ный выход) в виде оранжевого твердого вещества.
ЖХ/МС: Rt = 0,717 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 557,1 [М+Н]+.
1H ЯМР (400 МГц, Метанол-d4) δ 8.24 (br s, 2H), 8.03 (br s, 1H), 7.21 (dd, J=8.4, 12,0 Гц, 1H), 6.58 (s, 1H), 4.56-4.39 (m, 1H), 3.96 (s, 3Н), 3.61 (dt, J=6.8, 10,4 Гц, 1H), 3.18 (t, J=8,8 Гц, 1H), 3.07 (quin, J=7,6 Гц, 1H), 2.83 (t, J=8,8 Гц, 1H), 2.79-2.69 (m, 1H), 2.39 (dd, J=7.2, 9,6 Гц, 1H), 2.26 (s, 3Н), 2.20 (dd, J=3.2, 10,0 Гц, 1H), 2.14-2.02 (m, 1H), 1.92 (dd, J=6.4, 12,4 Гц, 1H), 1.60 (s, 6H).
Методика получения соединения 17с:
В раствор соединения 17b (200 мг, 0,36 ммоль) в МеОН (15 мл) добавляли Pd/C (10%, 20 мг). Реакционную смесь перемешивали при 25-31°С в течение 3 ч в атмосфере Н2 (15 фунт/кв.дюйм). Реакционную смесь фильтровали, и фильтрат концентрировали в вакууме с получением соединения 17с (168 мг, 88,6%-ный выход) в виде серовато-зеленого твердого вещества.
ЖХ/МС: Rt = 0,675 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 527,3 [M+H]+.
1H ЯМР (400 МГц, Метанол-d4) δ 8.21 (br s, 2H), 7.47 (br s, 1H), 7.22 (dd, J=9.0, 12,4 Гц, 1H), 6.86 (s, 1H), 4.15-4.03 (m, 1H), 3.79 (s, 3Н), 3.48-3.38 (m, 1H), 3.00-2.88 (m, 1H), 2.74-2.65 (m, 2H), 2.62 (d, J-10,0 Гц, 1H), 2.45 (dd, J=7.8, 9,2 Гц, 1H), 2.33 (s, 3Н), 2.19-2.09 (m, 2H), 1.85-1.71 (m, 1H), 1.60 (s, 6H).
Методика получения соединения Примера 17:
В раствор соединения 17с (155 мг, 0,294 ммоль) и DIEA (57 мг, 0,441 ммоль) в DMF (2 мл) добавляли акрилоилхлорид (27 мг, 0,294 ммоль) в бане лед-вода. Полученную смесь перемешивали при 5-10°С в течение 0,5 ч. Реакционную смесь гасили H2O (0,1 мл) и затем фильтровали. Фильтрат очищали препаративной ВЭЖХ (колонка: Waters Xbridge 150×25 5 мкм; условия: 30-60% В (А: 0,05%-ный аммиак, В: CH3CN); скорость потока: 25 мл/мин) и лиофилизировали с получением соединения Примера 17 (39,1 мг, 22,90%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 1,990 мин при 10-80CD_3 мин_220&254. lcm хроматографии (XBrige Shield RP18 2,1×50 мм), МС (ЭРИ) m/z = 581,3 [M+H]+.
1H ЯМР (400 МГц, CDCl3) δ 10.65 (br s, 1H), 9.90 (br s, 1H), 9.59 (br s, 1H), 8.40 (s, 1H), 8.32 (dd, J=8.0, 12,8 Гц, 1H), 7.67 (br s, 1H), 7.10 (dd, J=8.8, 12,0 Гц, 1H), 6.78 (s, 1H), 6.56-6.44 (m, 1H), 6.44-6.33 (m, 1H), 6.00 (br s, 1H), 5.76 (d, J=10,0 Гц, 1H), 3.87 (s, 3H), 3.68 (dd, J=4.4, 7,8 Гц, 1H), 3.22 (t, J=7,2 Гц, 1H), 2.93-2.78 (m, 3H), 2.72 (d, J-10,0 Гц, 1H), 2.29 (s, 5H), 1.90 (dd, J=4.4, 10,4 Гц, 1H), 1.87-1.80 (m, 1H), 1.76 (s, 6H).
Пример 18
N-(5-(4-(4,5-дифтор-2-(2-г1адроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-4-метокси-2-(метил(2-(пирролидин-1-ил)этил)амино)фенил)акриламид
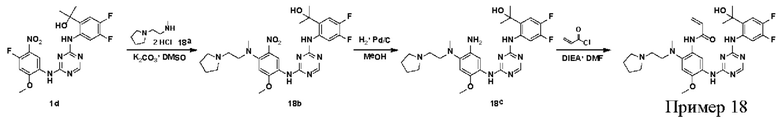
Методика получения соединения 18b:
В раствор соединения 1d (200 мг, 1,0 экв., 0,44 ммоль) и K2CO3 (243 мг, 4,0 экв., 1,76 ммоль) в DMSO (5 мл) добавляли соединение 18а (133 мг, 1,5 экв., 0,66 ммоль). Полученную смесь перемешивали при 85°С в течение 4 ч, при этом цвет менялся с бледно-желтого на насыщенный желтый. Реакционную смесь вливали в ледяную воду (50 мл), и желтое твердое вещество осаждалось. Выпавшее в осадок желтое твердое вещество собирали фильтрованием и затем растворяли в CH2Cl г (30 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением соединения 18b (230 мг, 93%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 0,754 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z = 559,3 [М+Н]+.
Методика получения соединения 18с:
В раствор соединения 18b (230 мг, 1,0 экв., 0,41 ммоль) в МеОН (10 мл) добавляли Pd/C (23 мг). Полученную смесь продували и дегазировали Н2 3 раза, затем перемешивали при 25-28°С (комнатная температура) в атмосфере водорода (15 фунт/кв.дюйм) в течение 1 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением соединения 18с (200 мг, 92%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,678 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 529,1 [М+Н]+.
Методика получения соединения Примера 18:
В раствор соединения 18с (200 мг, 1,0 экв., 0,38 ммоль) и DIEA (98 мг, 2,0 экв., 0,76 ммоль) в DMF (2,5 мл) добавляли акрилоилхлорид (34 мг, 1,0 экв., 0,38 ммоль) в DMF (0,5 мл). Полученную смесь перемешивали при 0°С в бане лед-вода в течение 30 мин. Реакционную смесь очищали методом ОФ-ВЭЖХ (обращенно-фазная ВЭЖХ) [колонка: обращенно-фазная колонка; условия: 50-70% В (А: 0,25% NH3HCO3; В: МеОН); скорость потока: 40 мл/мин]. Фракции концентрировали при пониженном давлении и лиофилизировали с получением соединения Примера 18 (45,7 мг, 20%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 1,716 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 583,2 [М+Н]+.
ВЭЖХ: Rt = 3,24 мин при 10-80_ab_1,2 мл хроматографии (Ultimate C18 3×50 мм, 3 мкм).
1H ЯМР (400 МГц, COCl3) δ 10.65 (br s, 1H), 9.89 (br s, 2H), 8.40 (s, 1H), 8.32 (dd, J=8.0, 12,8 Гц, 1H), 7.67 (br s, 1H), 7.10 (dd, J=8.4, 12,0 Гц, 1H), 6.74 (s, 2H), 6.38 (d, J=16,8 Гц, 1H), 5.77 (br d, J=10,8 Гц, 1H), 3.88 (s, 3H), 3.06 (br s, 2H), 2.91-2.74 (m, 4H), 2.70 (s, 5H), 1.94 (br s, 4H), 1.76 (s, 6H).
Пример 19
(R)-N-(5-(4-(4,5-дифтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-(2-((диметиламино)метил)азетидин-1-ил)-4-метоксифенил)акриламид
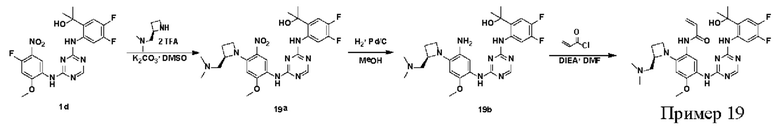
Методика получения соединения 19а:
В раствор соединения 1d (300 мг, 1,0 экв., 0,67 ммоль) и K2CO3 (926 мг, 10,0 экв., 6,70 ммоль) в DMSO (5 мл) добавляли (R)-1-(азетидин-2-ил)-N,N-диметилметанамина TFA соль (2,0 г, 10,0 экв., 6,70 ммоль). Полученную смесь перемешивали при 85°С в течение 4 ч, при этом цвет менялся с бледно-желтого на насыщенный желтый. Реакционную смесь вливали в воду (50 мл), и желтое твердое вещество осаждалось. Выпавшее в осадок твердое вещество собирали фильтрованием и растворяли в CH2Cl2 (20 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением соединения 19а (300 мг) в виде желтого твердого вещества.
ЖХ/МС: Rt = 0,734 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 567,2 [M+Na]+.
Методика получения соединения 19b:
В раствор соединения 19а (300 мг, 1,0 экв., 0,55 ммоль) в МеОН (10 мл) добавляли Pd/C (30 мг). Полученную смесь продували и дегазировали Н2 3 раза, затем перемешивали при 24-29°С в атмосфере Н2 (водородный баллон, 15 фунт/кв.дюйм) в течение 2 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением соединения 19b (270 мг) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,679 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 515,3[М+Н]+.
Методика получения соединения Примера 19:
В раствор соединения 19b (270 мг, 1,0 экв., 0,52 ммоль) и DIEA (134 мг, 2,0 экв., 1,04 ммоль) в DMF (2,5 мл) добавляли акрилоилхлорид (47 мг, 1,0 экв., 0,52 ммоль) в DMF (0,5 мл). Полученную смесь перемешивали при 0°С в бане лед-вода в течение 30 мин. Реакционную смесь очищали препаративной ВЭЖХ [колонка: Waters Xbridge 150×25, 5 мкм; условия: 20-50% В (А: 0,05% NH3H2O; В: CH3CN); скорость потока: 25 мл/мин]. Фракции, содержащие целевое соединение, лиофилизировали с получением соединения Примера 19 (77,5 мг) в виде белого твердого вещества.
ЖХ/МС: Rt = 1,658 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate C18 2,1×30 мм), МС (ЭРИ) m/z = 568,8 [M+H]+.
ВЭЖХ: Rt = 2,89 мин при 10-80_ab_1,2 мл хроматографии (Ultimate C18 3×50 мм 3 мкм).
1H ЯМР (400 МГц, CDCl3) δ 10.49 (br s, 1H), 9.40 (br s, 1H), 8.94 (br s, 1H), 8.38 (s, 1H), 8.30 (dd, J=7.9, 12,9 Гц, 1H), 7.67-7.44 (m, 1H), 7.09 (dd, J=8.7, 12,2 Гц, 1H), 6.63 (s, 1H), 6.43-6.29 (m, 2H), 5.80 (br d, J=10,3 Гц, 1H), 4.30-4.17 (m, 1H), 3.90 (s, 3H), 3.86 (br s, 1H), 3.58 (q, J=8,0 Гц, 1H), 2.66 (dd, J=5.9, 12,9 Гц, 1H), 2.49-2.37 (m, 2H), 2.26 (s, 6H), 2.15-2.05 (m, 1H), 1.74 (s, 6H).
Пример 20
(R)-N-(5-(4-(4,5-дифтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-4-метокси-2-(метил((1-метилпирролидин-2-ил)метил)амино)фенил)акриламид
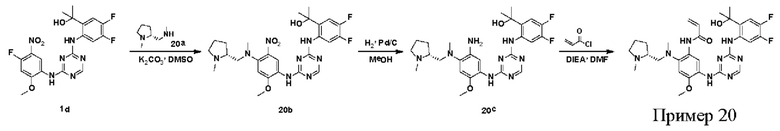
Методика получения соединения 20b:
В раствор соединения 1d (300 мг, 0,666 ммоль) и K2CO3 (184 мг, 1,332 ммоль) в DMSO (6 мл) добавляли соединение 20а (160 мг, 0,793 ммоль). Реакционную смесь перемешивали при 85°С в течение 6 ч, при этом цвет менялся с бледно-желтого на оранжевый. Реакционную смесь по каплям добавляли в Н2О (80 мл) в бане лед-вода, и твердое вещество выпадало в осадок, его собирали фильтрованием, и остаток на фильтре промывали H2O (15 мл × 3) и сушили под вакуумом с получением соединения 20b (300 мг, 81%-ный выход) в виде оранжевого твердого вещества.
ЖХ/МС: Rt = 0,731 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 559,1 [М+Н]+.
1H ЯМР (400 МГц, Метанол-d4) δ 8.37 (br s, 1H), 8.27 (br s, 1H), 8.05 (br s, 1H), 7.22 (dd, J=8.8, 12,4 Гц, 1H), 6.83 (s, 1H), 3.97 (s, 3H), 3.54 (dd, J=5.2, 13,6 Гц, 1H), 3.23-3.00 (m, 2H), 2.90 (s, 3H), 2.67-2.54 (m, 1H), 2.44 (s, 3H), 2.29 (q, J=9,2 Гц, 1H), 2.12-1.97 (m, 1H), 1.81-1.68 (m, 2H), 1.60 (s, 6H), 1.57-1.47 (m, 1H).
Методика получения соединения 20с:
В раствор соединения 20b (300 мг, 0,537 ммоль) в МеОН (15 мл) добавляли Pd/C (10%, 30 мг). Реакционную смесь перемешивали при 25-31°С в течение 3 ч в атмосфере Н2 (15 фунт/кв.дюйм). Реакционную смесь фильтровали, и фильтрат концентрировали в вакууме с получением соединения 20с (260 мг, 79,4%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,686 мин при 5-95AB_220&254.lcm хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 529,2 [M+H]+.
Методика получения соединения Примера 20:
В раствор соединения 20с (260 мг, 0,427 ммоль) и DIEA (83 мг, 0,64 ммоль) в DMF (3 мл) добавляли акрилоилхлорид (39 мг, 0,427 ммоль) в бане лед-вода. Полученную смесь перемешивали при 5-10°С в течение 0,5 ч. Реакционную смесь гасили H2O (0,1 мл) и затем фильтровали. Фильтрат очищали препаративной ВЭЖХ (колонка: Waters Xbridge 150×25, 5 мкм; условия: 50-80% В (А: 0,05%-ный аммиак. В: CH3CN); скорость потока: 25 мл/мин) и лиофилизировали с получением соединения Примера 20 (44,4 мг, 17,9%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 2,208 мин при 10-80CD_3 мин_220&254.lcm хроматографии (XBrige Shield RP18 2,1×50 мм), МС (ЭРИ) m/z = 583,3 [M+H]+.
ВЭЖХ: Rt = 4,39 мин при 10-80_CD_1,2 мл хроматографии (XBridge Shield RP 18 2,1×50 мм, 5 мкм).
1H ЯМР (400 МГц, CDCl3) δ 10.58 (br s, 1H), 10.08 (br s, 1H), 10.01 (br s, 1H), 8.40 (s, 1H), 8.31 (dd, J=8.0, 13,2 Гц, 1H), 7.65 (br s, 1H), 7.10 (dd, J=8.8, 12,0 Гц, 1H), 6.71 (s, 1H), 6.39 (br s, 1H), 6.38 (br s, 1H), 6.04 (br s, 1H), 5.77 (t, J=5,6 Гц, 1H), 3.88 (s, 3H), 3.18-3.03 (m, 1H), 2.87-2.80 (m, 1H), 2.74 (s, 3H), 2.71-2.64 (m, 2H), 2.56 (s, 3H), 2.41-2.33 (m, 1H), 1.97 (qd, J=8.8, 12,4 Гц, 1H), 1.79-1.72 (m, 8H), 1.43-1.35 (m, 1H).
Пример 21
(R)-N-(5-(4-(4,5-дифтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-(2-((диметиламино)метил)пирролидин-1-ил)-4-метоксифенил)акриламид
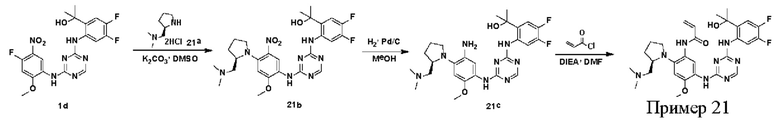
Методика получения соединения 21b:
Раствор соединения 1d (150 мг, 0,33 ммоль), соединения 21а (101 мг, 0,50 ммоль) и K2CO3 (91 мг, 0,66 ммоль) в DMSO (3 мл) перемешивали при 90°С в течение 3 ч. Реакционную смесь вливали в ледяную воду (15 мл), затем перемешивали в течение 30 мин. Выпавшее в осадок твердое вещество собирали, и остаток на фильтре растворяли в CH2Cl2 (35 мл), сушили над сульфатом натрия, фильтровали, и фильтрат концентрировали в вакууме с получением соединения 21b (220 мг, неочищенное) в виде желтого твердого вещества.
1H ЯМР (400 МГц, COCl3) δ 9.75 (brs, 1H), 8.88 (brs, 1H), 8.30 (s, 1H), 8.17 (s, 1H), 7.26 (s, 1H), 7.01 (dd, J-8,4 Гц, 12,0 Гц, 1H), 6.63 (s, 1H), 3.88 (s, 3H), 3.58-3.50 (m, 1H), 2.61-2.56 (m, 2H), 2.36-2.28 (m, 1H), 2.26-2.19 (m, 7H), 1.96-1.91 (m, 1H), 1.83-1.65 (m, 3H), 1.62 (s, 6H).
Методика получения соединения 21с:
Раствор соединения 21b (200 мг неочищенного, 0,33 ммоль) и Pd/C (20 мг) в МеОН (3 мл) перемешивали в атмосфере Н2 при 25-31°С в течение 1 ч (темная смесь). Реакционную смесь фильтровали, и фильтрат концентрировали в вакууме с получением соединения 21с (180 мг, неочищенное) в виде зеленого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9.75 (brs, 1H), 8.29-8.15 (m, 2H), 7.80-7.50 (m, 2H), 7.01 (dd, J=9,2 Гц, 12,4 Гц, 1H), 6.64 (s, 1H), 3.74 (s, 3H), 3.36-3.31 (m, 2H), 2.60-2.53 (m, 1H), 2.25-2.15 (m, 2H), 2.13 (s, 6H), 1.87-1.82 (m, 2H), 1.68-1.63 (m, 1H), 1.61-1.56 (m, 7H).
Методика получения соединения Примера 21:
В раствор соединения 21с (180 мг, неочищенное, 0,33 ммоль) и DIEA (85 мг, 0,66 ммоль) в DMF (3 мл) по каплям добавляли акрилоилхлорид (30 мг, 0,33 ммоль) при 0°С. Черную смесь перемешивали при 0°С в течение 1 ч. Реакционную смесь очищали препаративной ВЭЖХ: [колонка: Waters Xbridge 150×25, 5 мкм; условия: 40-70% В (А: 0,05%-ный аммиак; В: CH3CN); скорость потока: 25 мл/мин]. Фракции, содержащие целевое соединение, лиофилизировали с получением соединения Примера 21 (40,6 мг, 21% за 3 стадии) в виде белого твердого вещества.
ЖХ/МС: Rt = 1,856 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate C18 2,1×30 мм), МС (ЭРИ) m/z = 583,0 [M+H]+.
1H ЯМР (400 МГц, CDCl3) δ 11.65 (brs, 1H), 10.06 (brs, 1H), 9.88 (s, 1H), 8.40 (s, 1H), 8.32 (dd, J=8,0 Гц, 12,8 Гц, 1H), 7.68 (s, 1H), 7.10 (dd, J=8,8 Гц, 12,4 Гц, 1H), 6.71 (s, 1H), 6.41-6.30 (m, 2H), 6.05 (brs, 1H), 5.78 (dd, J=3,2 Гц, 8,0 Гц, 1H), 3.86 (s, 3H), 3.33-3.28 (m, 2H), 3.02-2.93 (m, 1H), 2.29 (dd, J=8,0 Гц, 12,4 Гц, 1H), 2.17-2.07 (m, 7H), 2.01-1.84 (m, 3H), 1.76 (s, 6H), 1.72-1.64 (m, 1H).
ВЭЖХ: Rt = 3,26 мин при 10-80АВ_1,2 мл хроматографии (Ultimate C18 3×50 мм, 3 мкм).
Пример 22
N-(2-((2-(бис(метил-d3)амино)этил)(метил)амино)-5-((4-((4,5-дифтор-2-(2-гидроксипропан-2-ил)фенил)амино)-1,3,5-триазин-2-ил)амино)-4-метоксифенил)акриламид

Методика получения соединения 22b:
В раствор соединения 22а (500 мг, 2,87 ммоль) и K2CO3 (793 мг, 5,74 ммоль) в THF (20 мл) добавляли CD3I (624 мг, 4,30 ммоль). Смесь перемешивали при 26-33°С в течение 1 ч, при этом белое твердое вещество выпадало в осадок. Затем в смесь добавляли CD3I (208 мг, 0,5 экв., 1,43 ммоль) и перемешивали при 26-33°С в течение еще 1 ч. Реакционную смесь фильтровали, и органический слой концентрировали при пониженном давлении с получением соединения 22b (300 мг, 50%-ный выход) в виде бесцветного твердого вещества.
ЖХ/МС: Rt = 0,743 мин при 0-60АВ_2 мин_Е.М хроматографии (Xtimate C18, 2,1×30 мм, 3 мкм), МС (ЭРИ) m/z = 209,2 [M+H]+.
1H ЯМР (400 МГц, DMSO-d6) δ 3.24-3.18 (m, 1H), 3.12 (t, J=6,8 Гц, 1Н), 2.78 (br s, 3H), 2.62 (brt, J=6,7 Гц, 1H), 2.34-2.26 (m, 1H), 1.38 (s, 9H).
Методика получения соединения 22с:
В раствор соединения 22b (300 мг, 1,0 экв., 1,44 ммоль) в CH2Cl2 (5 мл) добавляли HCl-диоксан (5 мл, 4 M). Полученную смесь перемешивали при 22-27°С в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении с получением соединения 22с (250 мг, 95%-ный выход) в виде желтого масла.
ЖХ/МС: Rt = 0,098 мин при 0-60АВ_2 мин_Е.М хроматографии (Xtimate C18, 2,1×30 мм, 3 мкм), МС (ЭРИ) m/z = 109,2 [М+Н]+.
1H ЯМР (400 МГц, DMSO-d6) δ 3.43-3.37 (m, 1H), 3.36-3.22 (m, 1H), 3.16 (br s, 2H), 2.56 (br s, 3H).
Методика получения соединения 22d:
В раствор соединения 22с (250 мг, 0,55 ммоль) и K2CO3 (304 мг, 2,20 ммоль) в DMSO (5 мл) добавляли соединение 6d (250 мг, 1,38 ммоль). Полученную смесь перемешивали при 22-27°С в течение 12 ч, при этом цвет менялся с бледно-коричневого на темно желтый. Реакционную смесь разбавляли EtOAc (20 мл) и промывали рассолом (2×30 мл). Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением неочищенного остатка, который очищали колоночной хроматограф ией на силикагеле (CH2Cl2/МеОН = 10/1) с получением соединения 22d (90 мг, 30%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 2,414 мин при 10-80АВ_7 мин_220&254.lcm хроматографии (Xtimate С 18 2,1×30 мм), МС (ЭРИ) m/z = 539,0 [М+Н]+.
Методика получения соединения 22е:
В раствор соединения 22d (90 мг, 1,0 экв., 0,17 ммоль) в МеОН (10 мл) добавляли Pd/C (9 мг). Полученную смесь продували и дегазировали Н2 3 раза, затем перемешивали при 23-28°С в атмосфере водорода (15 фунт/кв.дюйм) в течение 2 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением соединения 22е (70 мг, 81%-ный выход) в виде бесцветного масла. Структура и чистота были подтверждены методом ЖХ/МС (Rt = 0,698 мин, 509,4 [M+H]+).
ЖХ/МС: Rt = 0,698 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 509,4 [М+Н]+.
Методика получения соединения Примера 22:
В раствор соединения 22е (70 мг, 0,14 ммоль) и DIEA (36 мг, 0,28 ммоль) в DMF (2,5 мл) по каплям добавляли раствор соединения акрилоилхлорид (13 мг, 0,14 ммоль) в DMF (0,5 мл) при 0°С. Полученную смесь перемешивали при 0°С в бане лед-вода в течение 30 мин. Реакционную смесь очищали препаративной ВЭЖХ [колонка: Waters Xbridge 150×25, 5 мкм; условия: 38-68% В (А: 0,05% NH3H2O; В: CH3CN); скорость потока: 25 мл/мин]. Фракции, содержащие целевое соединение, лиофилизировали с получением соединения Примера 22 (21,9 мг, 28%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 1,790 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 563,0 [М+Н]+.
ВЭЖХ: Rt = 3,06 мин при 10-80_ab_1,2 мл хроматографии (Ultimate C18 3×50 мм, 3 мкм).
1H ЯМР (400 МГц, CDCl3) δ 10.68 (br s, 1Н), 10.46 (br s, 1H), 9.97 (br s, 1H), 8.41 (s, 1H), 8.32 (dd, J=8.0, 12,8 Гц, 1H), 7.69 (br s, 1H), 7.11 (dd, J=8.8, 12,2 Гц, 1H), 6.78 (s, 1H), 6.44-6.30 (m, 2H), 6.11 (br s, 1H), 5.80-5.73 (m, 1H), 3.88 (s, 3H), 2.88 (br s, 2H), 2.71 (s, 3H), 2.29 (br s, 2H), 1.77 (s, 6H).
Пример 23
N-(5-(4-(4,5-дифтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-(3-((диметиламино)метил)азетидин-1-ил)-4-метоксифенил)акриламид
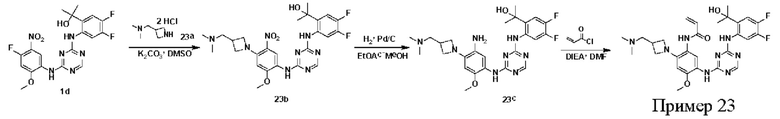
Методика получения соединения 23b:
Смесь соединения 1d (150 мг, 0,318 ммоль), соединения 23а (60 мг, 0,318 ммоль) и K2CO3 (175 мг, 1,27 ммоль) в DMSO (2 мл) перемешивали при 90°С в течение 3 ч (коричневая суспензия). Реакционную смесь гасили ледяной водой (8 мл), затем фильтровали, и остаток на фильтре промывали H2O (5 мл) и сушили в глубоком вакууме с получением соединения 23b (180 мг, 74,3%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 0,710 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z = 545,2 [М+Н]+.
1H ЯМР (400 МГц, CDCl3) δ 10.05 (brs, 1H), 8.98 (brs, 1H), 8.37 (s, 1H), 8.24 (s, 1H), 7.28 (m, 1H), 7.11-7.05 (m, 1H), 5.97 (s, 1H), 4.17 (d, J=8,0 Гц, 2H), 3.93 (s, 3H), 3.67-3.63 (m, 2H), 2.95-2.88 (m, 1H), 2.57 (d, J=7,6 Гц, 2H), 2.43 (s, 6H), 1.69 (s, 6H).
Методика получения соединения 23с:
Смесь соединения 23b (180 мг, 0,33 ммоль) и Pd/C (180 мг, 10%) в EtOAc (20 мл) и МеОН (5 мл) в атмосфере Н2 перемешивали в течение 3 ч при 23-28°С (превращалась в черную смесь). Реакционную смесь фильтровали, остаток на фильтре промывали EtOAc (50 мл), и фильтрат концентрировали в вакууме с получением соединения 23 с (140 мг, 82,3%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 0,660 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z = 515,2 [М+Н]+.
1H ЯМР (400 МГц, CDCl3): δ м.д. 9.63 (brs, 1H), 8.23-8.17 (m, 2H), 7.59 (s, 1H), 7.49 (s, 1H), 7.04-6.98 (m, 1H), 6.17 (m, 1H), 3.94 (t, J=7,2 Гц, 2H), 3.50 (s, 3H), 3.41 (t, J-7,6 Гц, 2H), 2.85-2.77 (m, 1H), 2.55-2.48 (m, 2H), 2.17 (s, 6H), 1.59 (s, 6H).
Методика получения соединения Примера 23:
В смесь соединения 23с (140 мг, 0,27 ммоль) и DIEA (105 мг, 0,81 ммоль) в DMF (3 мл) по каплям добавляли раствор акрилоилхлорида (29,9 мг, 0,33 ммоль) в DMF (1 мл) в бане лед-вода в течение 1 ч. После перемешивания реакционной смеси в течение 30 мин при 0-5°С (коричневый раствор) реакционную смесь гасили H2O (0,1 мл), и полученный раствор напрямую очищали препаративной ВЭЖХ [Waters Xbridge 150×25,5 мкм; условия: 37-57% В (А: 0,05%-ный аммиак; В: CH3CN); скорость потока: 25 мл/мин]. Фракции, содержащие целевое соединение, лиофилизировали с получением соединения Примера 23 (32,4 мг, 20,9%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 2,172 мин при 0-60АВ_4,0 мин_220&254 хроматографии (Xtimate С 18 2,1×30 мм), МС (ЭРИ) m/z = 569,0 [М+Н]+.
1H ЯМР (400 МГц, CDCl3) δ 8.35-8.27 (m, 2H), 7.50 (s, 1H), 7.30-7.27 (m, 1H), 7.10-7.25 (m, 1H), 6.41-6.27 (m, 3Н), 5.80 (d, J=10,0 Гц, 2H), 3.99 (t, J=7,6 Гц, 2H), 3.87 (s, 3H), 3.50 (t, J=7,2 Гц, 2H), 2.94-2.87 (m, 1H), 2.54 (d, J=8,0 Гц, 2H), 2.23 (s, 6H), 1.71 (s, 6H).
ВЭЖХ: Rt = 2,24 мин. ВЭЖХ-Р Venusil XBP C18 3×50 мм, метод\0-60АВ_1,2 мл. мет
Пример 24
N-(5-(4-(4,5-дифтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-4-метокси-2-(метил(2-(пиперидин-1-ил)этил)амино)фенил)акриламид

Методика получения соединения 24b:
В раствор соединения 24а (1,0 г, 6,77 ммоль) и метанамина (1,4 г, 20,31 ммоль) в МеОН (10 мл) добавляли KOH (760 мг, 13,55 ммоль) в 5 мл воды с каталитическим количеством KI (225 мг, 1,35 ммоль). Полученную смесь нагревали при 50°С-80°С в течение 18 ч. Реакционную смесь обрабатывали HCl (1М) до установления рН = 7, затем экстрагировали EtOAc (3×10 мл). Органические слои промывали рассолом (3×10 мл), сушили и концентрировали при пониженном давлении с получением неочищенного остатка, который очищали колоночной хроматографией на силикагеле (CH2Cl2/МеОН = 10/1 (об./об.)) с получением соединения 24b (120 мг, 12,5 выход) в виде бесцветного масла.
ЖХ/МС: Rt = 1,688 мин (MSD TIC) при 10-80CD_4 мин (XBrige Shield RP18 2,1×50 мм), МС (ЭРИ) m/z = 143,2 [M+H]+.
1H ЯМР (400 МГц, CDCl3) δ 2.57-2.47 (m, 2H), 2.46-2.30 (m, 8H), 2.28-2.15 (m, 2H), 1.59-1.54 (m, 5H), 1.42 (br d, J=5,2 Гц, 2H).
Методика получения соединения 24с:
В раствор соединения 1d (190 мг, 0,42 ммоль) и K2CO3 (116 мг, 0,84 ммоль) в DMSO (2 мл) добавляли соединение 24b (60 мг, 0,42 ммоль). Смесь перемешивали при 28-33°С в течение 2 часов. Реакционную смесь очищали колоночной флэш-хроматографией на силикагеле (условия: 65-75% В (А: 0,05% TFA в воде; В: МеОН); скорость потока: 40 мл/мин)) с получением соединения 24с (90 мг, 37,3%-ный выход) в виде красного твердого вещества.
ЖХ/МС: Rt = 0,805 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 573,2 [М+Н]+.
Методика получения соединения 24d:
В раствор соединения 24с (90 мг, 0,16 ммоль) в МеОН (3 мл) добавляли Pd/C (10 мг) в защитной атмосфере N2. Черную смесь перемешивали при 26-33°С в атмосфере Н2 (15 фунт/кв.дюйм) в течение 1 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением соединения 24d (70 мг, 82,1%-ный выход) в виде коричневого масла.
ЖХ/МС: Rt = 0,764 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 543,1 [M-OH]+.
Методика получения соединения Примера 24:
В раствор соединения 24d (70 мг, 0,13 ммоль) и DIEA (25 мг, 0,19 ммоль) в DMF (1 мл) добавляли раствор акрилоилхлорида (12 мг, 0,13 ммоль) в DMF (1 мл). Полученную коричневую смесь перемешивали при 0°С в течение 30 мин. Реакционную смесь очищали препаративной ВЭЖХ [колонка: Waters Xbridge 150×25, 5 мкм; условия: 40-70% В (А: 0,05%-ный аммиак; В: CH3CN); скорость потока: 25 мл/мин]. Фракции, содержащие целевое соединение, лиофилизировали с получением соединения Примера 24 (12,9 мг, 16,8%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 5,224 мин при 10-80CD_7 мин_220&254 хроматографии (XBrige Shield RP18 2,1×50 мм), МС (ЭРИ) m/z = 597,3 [М+Н]+.
ВЭЖХ, Rt = 4,81 чистота 96,87% (220 нм); 10-80_CD_1,2 мл.мет (XBridge Shield RP 18 2,1×50 мм, 5 мкм).
1H ЯМР (400 МГц, DMSO-d6) δ 10.26 (br s, 1H), 9.33 (s, 1H), 9.14 (br s, 1H), 8.25 (s, 1H), 8.14 (s, 1H), 7.26 (br s, 1H), 6.96 (br s, 1H), 6.59 (dd, J=10.4, 16,8 Гц, 1H), 6.31-6.12 (m, 2H), 5.72 (br d, J=10,4 Гц, 1H), 3.77 (s, 3H), 2.98 (br t, J=6,4 Гц, 2H), 2.70 (s, 3H), 2.40-2.22 (m, 6H), 1.57-1.44 (m, 10Н), 1.37 (br d, J=4,0 Гц, 2H), 1.24 (br s, 1H).
Пример 25
N-(5-(4-(4,5-дифтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-4-метокси-2-((3aS,6aS)-5-метилгексагидропирроло[3,4-b]пиррол-1(2Н)-ил)фенил)акриламид
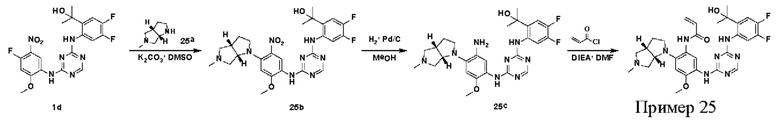
Методика получения соединения 25b:
В раствор соединения 1d (200 мг, 0,444 ммоль) и K2CO3 (123 мг, 0,888 ммоль) в DMSO (4 мл) добавляли соединение 25а (196 мг, 1,554 ммоль). Бледно-желтую реакционную смесь перемешивали при 85°С в течение 3 ч (цвет менялся с коричневого на оранжевый). Реакционную смесь по каплям добавляли в Н2О (40 мл) в бане лед-вода, при этом твердое вещество выпадало в осадок. Выпавшее в осадок твердое вещество собирали фильтрованием и промывали H2O (15 мл × 3), затем сушили в глубоком вакууме с получением соединения 25b (230 мг, 89,8%-ный выход) в виде оранжевого твердого вещества.
ЖХ/МС: Rt = 0,709 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 557,2 [М+Н]+.
1H ЯМР (400 МГц, CDCl3) δ 10.04 (br s, 1H), 8.30 (br s, 1H), 8.19 (s, 1H), 7.48 (br s, 1H), 7.06 (dd, J=8.4, 12,0 Гц, 1H), 6.37 (s, 1H), 4.45-4.33 (m, 1H), 3.92 (s, 3H), 3.53 (dt, J=6.4, 10,4 Гц, 1Н), 3.15 (t, J=8,4 Гц, 1H), 3.01 (quin, J=7,2 Гц, 1H), 2.67 (t, J=8,8 Гц, 1H), 2.61-2.51 (m, 1H), 2.39 (br dd, J=6.4, 9,2 Гц, 1H), 2.23 (d, J=9,2 Гц, 1H), 2.19 (s, 3H), 2.12-1.99 (m, 1H), 1.86 (dd, J=6.0, 12,4 Гц, 1H), 1.65 (s, 6H).
Методика получения соединения 25с:
В раствор соединения 25b (230 мг, 0,338 ммоль) в МеОН (15 мл) добавляли Pd/C (10%, 25 мг). Реакционную смесь перемешивали при 22-28°С в течение 3 ч в атмосфере Н2 (15 фунт/кв.дюйм). Реакционную смесь фильтровали, и фильтрат концентрировали в вакууме с получением соединения 25с (180 мг, 82%-ный выход) в виде серовато-зеленого масла.
ЖХ/МС: Rt = 0,667 мин при 5-95АВ_220&254.lcm хроматографии (ACSSH-LCMS-АВ MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 527,2 [M+H]+.
Методика получения соединения Примера 25:
В раствор соединения 25с (170 мг, 0,33 ммоль) и DIEA (64 мг, 0,50 ммоль) в DMF (2 мл) добавляли акрилоилхлорид (29 мг, 0,33 ммоль) в бане лед-вода. Полученную смесь перемешивали при 5-10°С в течение 0,5 ч. Реакционную смесь гасили H2O (0,1 мл) и затем фильтровали, фильтрат очищали препаративной ВЭЖХ (колонка: Waters Xbridge 150×25, 5 мкм; условия: 30-60% В (А: 0,05%-ный аммиак. В: CH3CN); скорость потока: 25 мл/мин) и лиофилизировали с получением соединения Примера 25 (48,0 мг, 25,1%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 1,982 мин при 10-80CD_3 мин_220&254 (XBrige Shield RP18 2,1×50 мм), МС (ЭРИ) m/z = 581,3 [M+H]+.
ВЭЖХ: Rt = 3,94 чистота 96,04% (220 нм); 10-80_CD_1,2 мл.мет (XBridge Shield RP 18 2,1×50 мм, 5 мкм).
1H ЯМР (400 МГц, CDCl3) δ 10.66 (br s, 1Н), 9.92 (br s, 1H), 9.59 (br s, 1H), 8.40 (s, 1H), 8.33 (dd, J-8.0, 12,8 Гц, 1H), 7.68 (br s, 1H), 7.10 (dd, J=8.8, 12,0 Гц, 1H), 6.78 (s, 1H), 6.53-6.34 (m, 2H), 5.99 (br s, 1H), 5.76 (d, J=9,6 Гц, 1H), 3.87 (s, 3Н), 3.72-3.60 (m, 1H), 3.21 (t, J=7,2 Гц, 1H), 2.92-2.78 (m, 3Н), 2.71 (d, J=10,0 Гц, 1H), 2.33-2.31 (m, 1H), 2.29 (s, 3Н), 2.23-2.17 (m, 1H), 1.90 (dd, J=4.0, 10,0 Гц, 1H), 1.87-1.80 (m, 1H), 1.76 (br s, 6H).
Пример 26
N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-4-метокси-2-(метил(2-(пиперидин-1-ил)этил)амино)фенил)акриламид
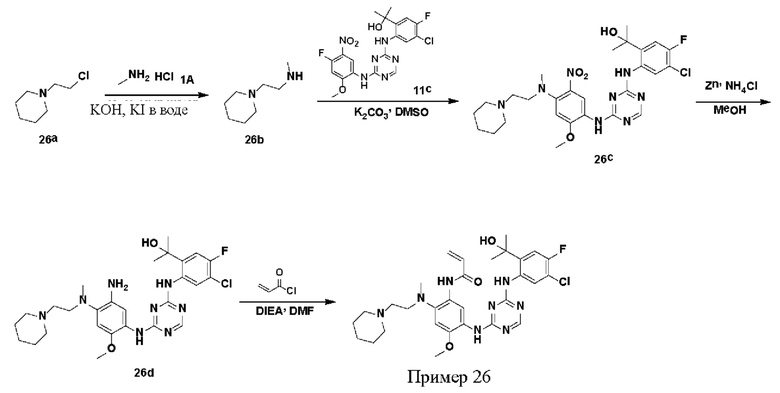
Методика получения соединения 26b:
В раствор соединения 26а (1,0 г, 6,77 ммоль) в МеОН (10 мл) добавляли DIEA (1,8 г, 13,55 ммоль) и метанамина гидрохлорид (10 мл, 20,31 ммоль, 2М в THF). Полученную смесь перемешивали в течение 18 ч. Реакционную смесь обрабатывали HCl (1М) до установления рН = 7, затем экстрагировали EtOAc (3×10 мл). Органические слои промывали рассолом (3×10 мл), сушили и концентрировали при пониженном давлении с получением неочищенного остатка, который очищали колоночной хроматографией на силикагеле (CH2Cl/МеОН = 10/1 (об./об.)) с получением соединения 26b (120 мг, 12,5%-ный выход) в виде бесцветного масла.
ЖХ/МС: Rt = 1,688 мин при 10-80CD_4 мин (XBrige Shield RP18 2,1×50 мм), МС (ЭРИ) m/z = 143,2 [M+H]+.
1H ЯМР (400 МГц, COCl3) δ 2.57-2.47 (m, 2H), 2.46-2.30 (m, 8H), 2.28-2.15 (m, 2H), 1.59-1.54 (m, 5H), 1.42 (br d, J=5,0 Гц, 2H).
Методика получения соединения 26с:
В раствор соединения 26b (197 мг, 0,42 ммоль) и K2CO3 (116 мг, 0,84 ммоль) в DMSO (2 мл) добавляли соединение 11с (60 мг, 0,42 ммоль). Смесь перемешивали при 23-28°С в течение 2 часов. Смесь очищали обращенно-фазной С-18 колоночной хроматографией на флэш-системе Biotage, элюируя смесью МеОН/Н2О (МеОН в воде от 55% до 60%) с получением соединения 26с (150 мг, 60,4%-ный выход) в виде красного твердого вещества.
ЖХ/МС: Rt = 0,826 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 589,1 [M+H]+.
Методика получения соединения 26d:
В раствор соединения 26с (150 мг, 0,26 ммоль) в МеОН (5 мл) и H2O (1 мл) добавляли Zn (87 мг, 1,32 ммоль) и NH4Cl (142 мг, 2,65 ммоль). Смесь перемешивали при 70°С в течение 1,5 ч в атмосфере N2 (черная смесь). Реакционную смесь гасили добавлением водного NH4Cl (20 мл), затем экстрагировали EtOAc (3×10 мл). Органические слои промывали рассолом (3×10 мл), сушили и напрямую концентрировали в вакууме с получением соединения 26d (120 мг, 84,4%-ный выход) в виде коричневого масла.
ЖХ/МС: Rt = 0,783 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 559,1 [М-ОН]+.
Методика получения соединения Примера 26:
В раствор соединения 26d (120 мг, 0,21 ммоль) и DIEA (42 мг, 0,32 ммоль) в DMF (1 мл) по каплям добавляли раствор акрилоилхлорида (19 мг, 0,21 ммоль) в DMF (1 мл) при 0°С. Полученную коричневую смесь перемешивали при 0°С в течение 30 мин. Реакционную смесь очищали обращенно-фазной С-18 колоночной хроматографией на флэш-системе Biotage [условие: 75-80% В (А: 0,05% водный NH4HCO3; В: CH3CN); скорость потока: 40 мл/мин]. Фракции, содержащие целевое соединение, лиофилизировали с получением соединения Примера 26 (47,2 мг, 35,9%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 5,365 мин при 10-80CD_7 мин_220&254 хроматографии (XBrige Shield RP18 2,1×50 мм), МС (ЭРИ) m/z = 613,3 [M+H]+.
ВЭЖХ: Rt = 4,83 чистота 97,09% (220 нм), 10-80_CD_1,2 мл. мет (XBridge Shield RP 18 2,1×50 мм, 5 мкм).
1H ЯМР (400 МГц, DMSO-d6) δ 10.19 (br s, 1H), 9.34 (br s, 1H), 9.08 (br s, 1H), 8.25 (s, 1H), 8.12 (s, 1H), 7.26 (br s, 1H), 6.97 (br s, 1H), 6.60 (br s, 1H), 6.30 (s, 1H), 6.19 (br d, J=17,1 Гц, 1H), 5.72 (br d, J=10,3 Гц, 1H), 3.79 (s, 3Н), 2.98 (br s, 2H), 2.69 (s, 3H), 2.34 (br s, 6H), 1.57-1.31 (m, 13H).
Пример 27
(R)-N-(5-(4-(4,5-дифтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-(3-(диметиламино)пирролидин-1-ил)-4-метоксифенил)акриламид

Методика получения соединения 27а:
В раствор соединения 1d (180 мг, 0.40 ммоль), K2CO3 (110,6 мг, 0,80 ммоль) в DMSO (4 мл) добавляли (R)-N,N-диметилпирролидин-3-амин (54,8 мг, 0,48 ммоль). Полученную смесь перемешивали при 24-27°С в течение 2 ч. Реакционную смесь объединяли с партией 1359-035 и осторожно вливали в воду (50 мл) при перемешивании; желтое твердое вещество выпадало в осадок. Выпавшее в осадок твердое вещество собирали фильтрованием и растворяли в CH2Cl2 (20 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением соединения 27а (210 мг, 87%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 0,705 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 545,1 [М+Н]+.
Методика получения соединения 27b:
В раствор соединения 27а (210 мг, 0,39 ммоль) в МеОН (5 мл) добавляли Pd/C (35 мг). Полученную смесь продували и дегазировали Н2 3 раза, затем перемешивали при 23-29°С в атмосфере Н2 (15 фунт/кв.дюйм) в течение 2 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением соединения 27b (170 мг, 84,7%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,667 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 515,2 [M+H]+.
Методика получения соединения Примера 27:
В раствор соединения 27b (100 мг, 0,19 ммоль) и DIEA (50 мг, 0,38 ммоль) в DMF (2 мл) добавляли акрилоилхлорид (17 мг, 0,19 ммоль) в DMF (1 мл). Полученную смесь перемешивали при 0°С в бане лед-вода в течение 20 мин. Реакционную смесь гасили тремя каплями воды и очищали препаративной ТСХ (CH2Cl2/MeOH = 7/1 (об./об.)) с получением загря3Ненного примесями продукта (100 мг) в виде белого твердого вещества, которое дополнительно очищали препаративной ВЭЖХ (колонка: Waters Xbridge 150x25, 5 мкм; 30-60% В (А: вода (0,05% гидроксида аммония об./об.), В: CH3CN), скорость потока: 25 мл/мин) с получением соединения Примера 27 (15 мг, 13,9%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 1,651 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С 18 2,1×30 мм), МС (ЭРИ) m/z = 568,9 [М+Н]+.
ВЭЖХ: Rt = 3,17 мин в 10-80_CD_1,2 мл хроматографии (Ultimate C18 3×50 мм, 3 мкм).
1H ЯМР (400 МГц, CDCl3) δ 10.59 (br s, 1H), 9.80 (br s, 1H), 8.49 (br s, 1H), 8.40 (s, 1H), 8.33 (dd, J=8.0, 13,2 Гц, 1H), 7.64 (br s, 1H), 7.10 (dd, J=8.4, 12,2 Гц, 1H), 6.76 (s, 1H), 6.40-6.36 (m, 2H), 5.84-5.78 (m, 1H), 3.88 (s, 3H), 3.15-3.07 (m, 4H), 2.90 (br t, J=7,2 Гц, 1Н), 2.31 (s, 6H), 2.19 (dt, J=7.2, 12,8 Гц, 1H), 2.00-1.90 (m, 1H), 1.76 (s, 6H), 3.87 (s, 3H), 2.40 (s, 6H).
Пример 28
N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-4-метокси-2-((3aR,6aR)-5-метилгексагидропирроло[3,4-b]пиррол-1(2Н)-ил)фенил)акриламид

Методика получения соединения 28b:
В раствор соединения 11с (300 мг, 0,64 ммоль) в DMSO (5 мл) добавляли карбонат калия (450 мг, 3,25 ммоль) и соединение 28а (120 мг, 0,95 ммоль). Смесь перемешивали при 85°С в течение 3 ч. Смесь вливали в ледяную воду (30 мл), и твердое вещество выпадало в осадок. Твердое вещество отделяли фильтрованием под разряжением и сушили в вакууме с получением соединения 28b (320 мг, 87%-ный выход) в виде оранжевого твердого вещества.
ЖХ/МС: Rt = 1,868 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С 18, 2,1×30 мм, 3 мкм), МС (ЭРИ) m/z = 573,2 [M+H]+.
1H ЯМР: (400 МГц, CDCl3) δ 9.60 (s, 1H), 8.85 (s, 1H), 8.30 (s, 1H), 7.32 (s, 1H), 7.00 (d, J=10,4 Гц, 1H), 6.32 (s, 1H), 4.40-4.29 (m, 1H), 3.88 (s, 3H), 3.53-3.43 (m, 1H), 3.18-3.08 (m, 1H), 3.01-2.90 (m, 1H), 2.61-2.55 (m, 1H), 2.47-2.40 (m, 1H), 2.39-2.33 (m, 1H), 2.24-2.17 (m, 1H), 2.13 (s, 3H), 2.05-1.95 (m, 1H), 1.86-1.75 (m, 1H), 1.62 (s, 6H).
Методика получения соединения 28с:
В раствор соединения 28b (200 мг, 0,35 ммоль) в смеси метанол/вода (5 мл/1 мл) добавляли NH4Cl (112 мг, 2,09 ммоль) и Zn (114 мг, 1,75 ммоль). Полученную смесь перемешивали при 90°С в течение 1 ч в атмосфере азота. Смесь вливали в воду (20 мл) и экстрагировали дихлорметаном (15 мл × 4). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением коричневой смолы, которую очищали колоночной флэш-хроматографией на силикагеле (от 0 до 10% метанола в CH2Cl2) с получением соединения 28с (110 мг, 57,8%-ный выход) в виде зеленого твердого вещества.
ЖХ/МС: Rt = 0,716 мин при 5-95АВ_220&254 хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 543,0 [M+H]+.
1H ЯМР: (400 МГц, DMSO-d6) δ 10.15 (s, 1H), 8.82 (br. s, 1H), 8.42 (s, 1H), 8.24 (s, 1H), 7.27 (d, J=10,4 Гц, 1H), 6.80 (br. s, 3H), 6.29 (s, 1H), 4.28-4.20 (m, 1H), 3.69 (s, 3H), 3.07-2.95 (m, 2H), 2.70-2.59 (m, 2H), 2.53 (s, 3H), 2.49-2.36 (m, 2H), 2.32-2.18 (m, 1H), 2.15-2.02 (m, 1H), 1.85-1.72 (m, 1H), 1.52 (s, 6H).
Методика получения соединения Примера 28:
В раствор соединения 28с (90 мг, 0,17 ммоль) в DMF (3 мл) добавляли DIEA (43 мг, 0,33 ммоль), затем добавляли акрилоилхлорид (15 мг, 0,17 ммоль) за три раза при 0°С. Смесь гасили водой и объединяли с предыдущей партией для дополнительной очистки препаративной ВЭЖХ (колонка: Waters Xbridge 150×25, 5 мкм, условие: 46%-66% В (А: вода/10 мМ NH4HCO3, В: CH3CN), скорость потока: 25 мл/мин) с получением соединения Примера 28 (29,4 мг, 23,8%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 1,870 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С 18 2,1×30 мм), МС (ЭРИ) m/z = 597,1 [M+H]+.
ВЭЖХ: Rt = 3,84 мин при 10-80_CD_1,2 мл хроматографии (XBridge Shield RP 18 2,1×50 мм, 5 мкм).
1H ЯМР (400 МГц, CDCl3) δ 10.53 (s, 1H), 9.85 (s, 1H), 9.52 (s, 1H), 8.40 (d, J=7,2 Гц, 1H), 8.34 (s, 1H), 7.60 (s, 1H), 7.02 (d, J=10,8 Гц, 1H), 6.70 (s, 1H), 6.42 (br. s, 1H), 6.35-6.26 (m, 1H), 5.98 (br. s, 1H), 5.69 (d, J=10,8 Гц, 1H), 3.79 (s, 3H), 3.65-3.53 (m, 1H), 3.19-3.09 (m, 1H), 2.85-2.73 (m, 3H), 2.72-2.56 (m, 1H), 2.31-2.08 (m, 5H), 1.84-1.75 (m, 2H), 1.69 (s, 6H).
Пример 29
N-(5-(4-(5-хлор-4-фтор-2-(2 -гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-4-метокси-2-(метил(2-(пирролидин-1-ил)этил)амино)фенил)акриламид

Методика получения соединения 29b:
В раствор соединения 11с (200 мг, 0,43 ммоль) и K2CO3 (119 мг, 0,86 ммоль) в DMSO (5 мл) добавляли соединение 29а (105 мг, 0,52 ммоль). Полученную смесь перемешивали при 85°С в течение 4 ч, при этом цвет менялся с бледно-желтого на насыщенный желтый. Реакционную смесь вливали в воду (50 мл), и желтое твердое вещество выпадало в осадок. Выпавшее в осадок твердое вещество собирали фильтрованием и растворяли в CH2Cl2 (30 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением соединения 29b (230 мг, 93%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 0,814 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z = 575,1 [M+H]+.
Методика получения соединения 29с:
В раствор соединения 29b (230 мг, 0,40 ммоль) в МеОН (10 мл) и Н2О (2 мл) добавляли Zn (130 мг, 2,00 ммоль) и NH4Cl (214 мг, 4,00 ммоль). Полученную смесь перемешивали при 80°С в течение 2 ч. Реакционную смесь фильтровали, и фильтрат концентрировали при пониженном давлении с получением остатка, который распределяли между EtOAc (2×10 мл) и водой (10 мл). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением соединения 29с (180 мг, 82%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 0,766 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 545,1 [М+H]+.
Методика получения соединения Примера 29:
В раствор соединения 29с (180 мг, 0,33 ммоль) и DIEA (85 мг, 0,66 ммоль) в DMF (2,5 мл) добавляли раствор акрилоилхлорида (30 мг, 0,33 ммоль) в DMF (0,5 мл). Полученную смесь перемешивали при 0°С в бане лед-вода в течение 30 мин. Реакционную смесь очищали методом ОФ-ВЭЖХ (обращенно-фазная ВЭЖХ) [колонка: обращенно-фазная колонка; условия: 42-72% В (А: 0,25% NH3HCO3; В: МеОН); скорость потока: 40 мл/мин]. Фракции концентрировали при пониженном давлении и лиофилизировали с получением соединения Примера 29 (47,4 мг, 20%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 1,912 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С 18 2,1×30 мм), МС (ЭРИ) m/z = 599,0 [M+H]+.
ВЭЖХ: Rt = 3,41 мин при 10-80_ab_1,2 мл хроматографии (Ultimate C18 3×50 мм, 3 мкм).
1H ЯМР (400 МГц, CDCl3) δ 10.61 (br s, 1H), 10.06 (br s, 1H), 9.96 (br s, 1H), 8.48 (d, J=7,6 Гц, 1H), 8.43 (s, 1H), 7.69 (br s, 1H), 7.10 (d, J=10,8 Гц, 1H), 6.78 (s, 1H), 6.38 (br d, J=4,8 Гц, 2Н), 6.13 (br s, 1H), 5.80-5.72 (m, 1H), 3.88 (s, 3Н), 2.95 (br s, 2H), 2.69 (s, 3H), 2.55 (br s, 4H), 2.42 (br s, 2H), 1.83 (br s, 4H), 1.78 (s, 6H).
Пример 30
(R)-N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-4-метокси-2-(метил((1-метилпирролидин-2-ил)метил)амино)фенил)акриламид
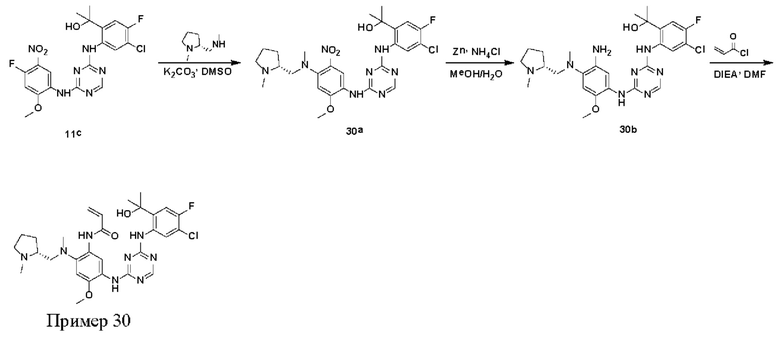
Методика получения соединения 30а:
В раствор соединения 11с (180 мг, 0,386 ммоль) и K2CO3 (107 мг, 0,772 ммоль) в DMSO (3 мл) добавляли (R)-N-метил-1-(1-метилпирролидин-2-ил)метанамин (93 мг, 0,463 ммоль). Реакционную смесь перемешивали при 85°С в течение 1 ч (цвет менялся с желтого на темно-оранжевый). Реакционную смесь по каплям добавляли в H2O (40 мл) в бане лед-вода, и твердое вещество выпадало в осадок. Твердое вещество собирали фильтрованием и промывали H2O (15 мл × 3), затем сушили в вакууме с получением соединения 30а (227 мг, 82%-ный выход) в виде оранжевого твердого вещества.
ЖХ/МС: Rt = 0,743 мин при 5-95AB_220&254.lcm хроматографии (ACSSH-LCMS-АВ MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 575,1 [М+Н]+.
1H ЯМР (400 МГц, CDCl3) δ 9.96 (br s, 1H), 8.88 (br s, 1H), 8.29 (br s, 2H), 7.51 (br s, 1H), 7.02 (d, J=10,4 Гц, 1H), 6.62 (s, 1H), 3.89 (s, 3H), 3.48-3.39 (m, 1H), 3.07 (dd, J=7.6, 13,2 Гц, 1H), 3.01-2.95 (m, 1H), 2.80 (s, 3H), 2.54-2.43 (m, 1H), 2.34 (s, 3H), 2.22-2.15 (m, 1H), 2.02-1.91 (m, 1H), 1.75-1.65 (m, 2H), 1.63 (s, 6H), 1.54-1.46 (m, 1H).
Методика получения соединения 30b:
В раствор соединения 30а (227 мг, 0,316 ммоль) в МеОН/Н2О = 5/1 (5 мл) добавляли Zn (124 мг, 6,0 экв., 1,896 ммоль) и NH4Cl (101 мг, 1,896 ммоль). Полученную смесь нагревали при 90°С в течение 1 ч (цвет менялся с оранжевого на коричневый). Реакционную смесь фильтровали, и фильтрат концентрировали в вакууме с получением неочищенного остатка, который растворяли в CH2Cl2 (20 мл), промывали водой (15 мл × 3), затем сушили над Na2SO4 и концентрировали в вакууме с получением соединения 30b (170 мг, 98%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,700 мин при 5-95АВ_220&254.lcm хроматографии (ACSSH-LCMS-АВ MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 545,2 [М+Н]+.
Методика получения соединения Примера 30:
В раствор соединения 30b (170 мг, 0,312 ммоль) и DIEA (60 мг, 0,468 ммоль) в DMF (2 мл) добавляли акрилоилхлорид (28 мг, 0,312 ммоль) в бане лед-вода. Полученную смесь перемешивали при 5-10°С в течение 10 мин. Реакционную смесь гасили Н2О (0,1 мл) и затем фильтровали. Фильтрат очищали препаративной ВЭЖХ (колонка: Waters Xbridge 150×25, 5 мкм; условия: 50-80% В (А: 0,05%-ный аммиак. В: CH3CN); скорость потока: 25 мл/мин) и затем лиофилизировали с получением соединения Примера 30 (78,1 мг, 41,8%-ный выход).
ЖХ/МС: Rt = 1,983 мин при 10-80АВ_4 мин_220&254. lcm хроматографии (ACSSH-LCMS-D Xtimate С 18 2,1×30 мм), МС (ЭРИ) m/z = 599,0 [М+H]+.
1H ЯМР (400 МГц, CDCl3) δ 10.52 (br s, 1H), 10.09 (br s, 1H), 10.02 (br s, 1H), 8.46 (d, J=7,2 Гц, 1H), 8.41 (s, 1H), 7.65 (br s, 1H), 7.09 (d, J=10,8 Гц, 1H), 6.71 (s, 1H), 6.39 (br s, 1H), 6.38 (br s, 1H), 6.11 (br s, 1H), 5.77 (t, J=5,6 Гц, 1H), 3.88 (s, 3H), 3.20-3.02 (m, 1H), 2.89-2.78 (m, 1H), 2.74 (s, 3H), 2.71-2.63 (m, 2H), 2.56 (s, 3H), 2.45-2.30 (m, 1H), 2.06-1.88 (m, 1H), 1.77 (br s, 6H), 1.73-1.69 (m, 2H), 1.45-1.32 (m, 1H).
Пример 31
(R)-N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-(2-((диметиламино)метил)пирролидин-1-ил)-4-метоксифенил)акриламид

Методика получения соединения 31а:
Раствор соединения 11с (180 мг, 0,38 ммоль), (Р)-N,N-диметил-1-(пирролидин-2-ил)метанамина дигидрохлорида (74 мг, 0,58 ммоль) и K2CO3 (106,6 мг, 0,77 ммоль) в DMSO (2 мл) перемешивали при 85°С в течение 1 ч. Реакционную смесь добавляли в H2O (10 мл) в бане лед-вода при перемешивании. Выпавшее в осадок твердое вещество собирали фильтрованием и затем растворяли в DCM (30 мл), сушили над Na2SO4 и концентрировали в вакууме с получением продукта 31а (310 мг, 88%-ный выход) в виде оранжевого масла.
ЖХ/МС: Rt = 0,758 мин при 5-95АВ_220&254.lcm хроматографии (ACSSH-LCMS-АВ MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 575,4 [M+H]+.
1H ЯМР (400 МГц, CDCl3) δ 9.79 (br s, 1H), 8.86 (br s, 1H), 8.34 (br s, 2H), 7.33 (br s, 1H), 7.05 (d, J=10,4 Гц, 1H), 6.68 (s, 1H), 4.13-3.98 (m, 1H), 3.93 (s, 4H), 3.70-3.58 (m, 1H), 3.58 (dt, J=6.0, 10,4 Гц, 2H), 2.46-2.31 (m, 2H), 2.05-1.93 (m, 1H), 1.92-1.79 (m, 9H), 1.73-1.62 (m, 6H).
Методика получения соединения 31b:
В раствор соединения 31а (300 мг, 0,52 ммоль) в МеОН/Н2О = 5/1 (9 мл) добавляли Zn (170,55 мг, 2,61 ммоль) и NH4Cl (139,5 мг, 2,61 ммоль). Полученную смесь нагревали при 90°С в течение 1 ч. Реакционную смесь фильтровали, и фильтрат концентрировали в вакууме с получением неочищенного остатка, который растворяли в CH2Cl2 (30 мл), промывали водой (30 мл × 2) и рассолом (30 мл), затем сушили над Na2SO4 и концентрировали в вакууме с получением продукта 31b (160 мг, 56%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,693 мин при 5-95АВ_220&254.lcm хроматографии (ACSSH-LCMS-АВ MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 545,1 [М+Н]+.
1H ЯМР (400 МГц, CDCl3) β 9.58 (br s, 1H), 8.35 (br d, J=7.2 Гц, 1H), 7.81 (br s, 1H), 7.71-7.48 (m, 1H), 7.07 (br d, J=10,4 Гц, 1H), 6.71 (br s, 1H), 5.30 (s, 1H), 4.05-3.85 (m, 2H), 3.81 (br s, 3H), 3.52-3.33 (m, 2H), 2.69-2.59 (m, 2H), 2.29-2.15 (m, 8H), 1.68 (br d, J=4,4 Гц, 6H).
Методика получения соединения Примера 31:
В раствор соединения 31b (90 мг, 0,165 ммоль) и DIEA (31,9 мг, 0,53 ммоль) в DMF (2 мл) по каплям добавляли акрилоилхлорид (14,9 мг, 0,165 ммоль) при 0°С. Полученную смесь перемешивали при 0°С в течение 1 ч. Реакционную смесь гасили Н2О (0,1 мл) и затем фильтровали. Фильтрат объединяли и дополнительно очищали препаративной ВЭЖХ (колонка: Gemini 150×25, 5 мкм; подвижная фаза А: вода с 0,05% гидроксида аммония об./об.; подвижная фаза В: DMF; скорость потока: 25 мл/мин; время градиента: 10 мин; описание профиля: 35%-65%) с получением соединения Примера 31 (20,1 мг, 11,4%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 1,826 мин при 10-80АВ_4 мин 220&254 хроматографии (ACSSH-LCMS-AS A: Xtimate С 18, 2,1×30 мм, 3 мкм; В: XBrige Shield RP18 2,1×50 мм), МС (ЭРИ) m/z = 599,3 [M+H]+.
ВЭЖХ: Rt = 3,98 10-80_CD_1,2 мл.мет XBridge Shield RP 18 2,1×50 мм, 5 мкм.
1H ЯМР (400 МГц, CDCl3) δ 10.59 (br s, 1H), 10.05 (br s, 1H), 9.89 (br s, 1H), 8.56-8.32 (m, 2H), 7.67 (br s, 1H), 7.09 (d, J=10,8 Гц, 1H), 6.69 (s, 1H), 6.37 (br s, 2H), 6.08 (br s, 1H), 5.86-5.72 (m, 1H), 3.86 (s, 3H), 3.31 (br s, 2H), 3.08-2.86 (m, 1H), 2.39 (br s, 1H), 2.29-2.13 (m, 6H), 2.09-1.91 (m, 3H), 1.76 (s, 8H).
Пример 32
N-(2-((2-(диметиламино)этил)(метил)амино)-5-(4-(6-(2-гидроксипропан-2-ил)-1-метил-1Н-индол-5-иламино)-1,3,5-триазин-2-иламино)-4-метоксифенил)акриламид
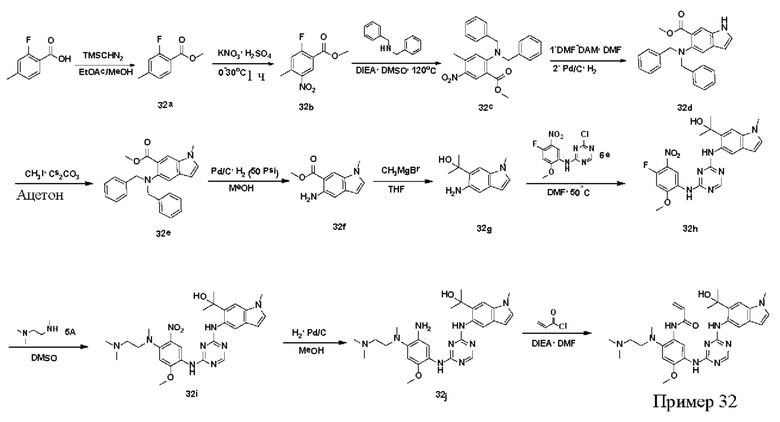
Методика получения соединения 32а:
В раствор 2-фтор-4-метилбензойной кислоты (10,0 г, 64,87 ммоль) в EtOAc (100 мл) и МеОН (100 мл) добавляли TMSCHN2 (64,87 мл, 129,75 ммоль, 2М в гексане). Смесь перемешивали при 28-36°С (комнатная температура) в течение 1,5 ч. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного остатка, который очищали колоночной хроматографией на силикагеле (петролейный эфир/EtOAc = 10/1(об./об.)) с получением соединения 32а (9 г, 82,5%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 0,780 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 169,0 [M+H]+.
1H ЯМР (400 МГц, CDCl3) δ 7.76 (t, J=7,8 Гц, 1Н), 6.93 (d, J=7,4 Гц, 1Н), 6.88 (d, J=12,0 Гц, 1Н), 3.84 (s, 3H), 2.32 (s, 3H).
Методика получения соединения 32b:
В раствор соединения 32а (9,0 г, 53,52 ммоль) в H2SO4 (100 мл) затем добавляли KMO3 (10,8 г, 107,04 ммоль) несколькими порциями при 0-5°С. Полученную смесь перемешивали при 28-36°С в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного остатка, который очищали колоночной хроматографией на силикагеле (петролейный эфир/EtOAc = 10/1) с получением соединения 32b (10 г, 87%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,781 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z NA (нет данных).
1H ЯМР (400 МГц, CDCl3) δ 8.66 (d, J=6,6 Гц, 1H), 7.16 (d, J=10,6 Гц, 1H), 3.98 (s, 3Н), 2.69 (s, 3H).
Методика получения соединения 32с:
В раствор соединения 32b (6,0 г, 28,15 ммоль) в DMSO (100 мл) добавляли соединение дибензиламин (8,3 г, 42,22 ммоль) и DIEA (7,3 г, 56,29 ммоль). Смесь перемешивали при 120°С (комнатная температура) в течение 1,5 ч. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного остатка, который очищали колоночной хроматографией на силикагеле (петролейный эфир/EtOAc = 10/1(об./об.)) с получением соединения 32с (4,8 г, 44%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,947 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 391,1 [M+H]+.
1H ЯМР (400 МГц, Метанол-d4) δ 8.46 (s, 1H), 7.33-7.22 (m, 10Н), 6.96 (s, 1H), 4.44 (s, 4H), 3.91 (s, 3H), 2.54 (s, 3H).
Методика получения соединения 32d:
В раствор соединения 32с (4,8 г, 12,30 ммоль) в DMF (20 мл) добавляли DMF-DMA (4,4 г, 36,88 ммоль,), смесь перемешивали при 150°С в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении, затем добавляли CH2Cl2 (20 мл) и Pd/C (480 мг), и смесь перемешивали в атмосфере водорода (15 фунт/кв.дюйм) в течение 2 ч. Реакционную смесь фильтровали, и в фильтрат добавляли CH2Cl2 (100 мл) и H2O (300 мл). Органическую фазу отделяли и промывали рассолом (3×100 мл), сушили и концентрировали в вакууме с получением неочищенного остатка, который очищали колоночной хроматографией на силикагеле (петролейный эфир/EtOAc = 5/1 (об./об.)) с получением соединения 32d (3,0 г, 66,7%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 1,395 мин при 10-80АВ_3 мин_220&254 хроматографии (XBrige Shield RP18 2,1×50 мм), МС (ЭРИ) m/z = 371,2 [M+H]+.
1H ЯМР (400 МГц, DMSO-d6) δ 11.18 (br s, 1H), 7.98-7.93 (m, 1H), 7.63 (s, 1H), 7.42 (t, J=2,8 Гц, 1H), 7.35 (s, 2H), 7.34 (s, 2H), 7.31 (s, 1H), 7.27 (s, 1H), 7.25 (s, 2H), 7.23 (s, 1H), 7.18 (s, 1H), 6.31 (brs, 1H), 4.12 (s, 4H), 3.86 (s, 3H).
Методика получения соединения 32е:
В раствор соединения 32d (2,7 г, 7,29 ммоль) и Cs2CO3 (7,1 г, 21.87 ммоль) в ацетоне (50 мл) добавляли CH3I (1,6 г, 10,93 ммоль). Полученную суспензию перемешивали при 18-25°С в течение 18 ч. Реакционную смесь фильтровали, и фильтрат обрабатывали водным NH4Cl (20 мл), затем экстрагировали CH2Cl2 (25 мл × 2). Объединенный органический слой сушили и концентрировали при пониженном давлении с получением соединения 32е (2,6 г, 92,9%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,849 мин при 5-95АВ_1,5 мин_220&254.lcm; хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 385,2 [М+H]+.
Методика получения соединения 32f:
В раствор соединения 32е (3,5 г, 9,10 ммоль) в МеОН (50 мл) добавляли Pd/C (10%, 300 мг). Полученную смесь продували и дегазировали Н2 три раза, затем перемешивали при 29-40°С в атмосфере Н2 (15 фунт/кв.дюйм) в течение 18 ч. Реакционную смесь фильтровали, и фильтрат концентрировали при пониженном давлении с получением соединения 32f (1,6 г, 86,5%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,536 мин при 5-95АВ_1,5 мин_220&254.lcm хроматографии (Xtimate С 18 2,1×30 мм), МС (ЭРИ) m/z = 205,0 [М+H]+.
1H ЯМР (400 МГц, CDCl3) δ 7.91 (s, 1Н), 7.11 (d, J=3,2 Гц, 1H), 6.87 (s, 1H), 6.24 (d, J=2,4 Гц, 1H), 5.31 (br s, 2H), 3.93 (s, 3H), 3.76 (s, 3H).
Методика получения соединения 32g:
В раствор соединения 32f (1,0 г, 4,90 ммоль) в THF (50 мл) по каплям добавляли CH3MgBr (8,2 мл, 3 М в диэтиловом эфире) при 0-5°С. Смесь перемешивали при 17-21°С в течение 1,5 ч. Реакционную смесь гасили добавлением водного NH4Cl (100 мл), затем экстрагировали EtOAc (3×100 мл). Объединенные органические слои промывали рассолом (3×100 мл), сушили и концентрировали при пониженном давлении с получением неочищенного остатка, который очищали колоночной хроматографией на силикагеле (петролейный эфир/EtOAc = 20/1 (об./об.)) с получением соединения 32g (700 мг, 70%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,865 мин при 10-80АВ_7 мин_220&254.lcm хроматографии (Xtimate С 18 2,1×30 мм), МС (ЭРИ) m/z = 186,9 [М-17]+.
1H ЯМР (400 МГц, CDCl3) δ 7.14 (s, 1H), 6.98-6.96 (m, 2H), 6.27 (d, J=2,4 Гц, 1H), 4.11-3.80 (m, 2H), 3.74 (s, 3H), 1.77 (s, 6H).
Методика получения соединения 32h:
В раствор соединения 32g (200 мг, 0,98 ммоль) в DMF (3 мл) добавляли соединение 6е (323 мг, 1,08 ммоль). Полученную смесь перемешивали при 50°С в течение 2 ч. Реакционную смесь вливали в воду (50 мл) при перемешивании, и желтое твердое вещество выпадало в осадок. Твердое вещество собирали фильтрованием и растворяли в CH2Cl2 (20 мл), затем сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением соединения 32h (380 мг, 83,1%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 0,796 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 490,1 [M+Na]+.
Методика получения соединения 32i:
В раствор соединения 32h (300 мг, 0,64 ммоль) в DMSO (4 мл) добавляли N1,N1,N2-триметилэтан-1,2-диамин (131 мг, 1,28 ммоль), затем перемешивали при 14-19°С в течение 2 часов. Реакционную смесь вливали в ледяную воду (50 мл) при перемешивании, и желтое твердое вещество выпадало в осадок. Твердое вещество собирали фильтрованием и растворяли в CH2Cl2 (60 мл), затем сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением соединения 32i (280 мг, 80%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 0,703 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 550,2 [М+Н]+.
Методика получения соединения 32j:
В раствор соединения 32i (250 мг, 0,45 ммоль) в МеОН (5 мл) добавляли Pd/C (25 мг, 10%) в атмосфере N2. Полученную черную смесь перемешивали при 15-21°С в атмосфере Н2 (15 фунт/кв.дюйм) в течение 1 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением соединения 32j (200 мг, 84,7%-ный выход) в виде коричневого масла.
ЖХ/МС: Rt = 0,653 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 520,1 [M+H]+.
1H ЯМР (400 МГц, CDCl3) δ 9.47 (br s, 1H), 8.24 (s, 2H), 7.96 (br s, 1H), 7.73 (br s, 1H), 7.07 (d, J=2,4 Гц, 1H), 6.67-6.64 (m, 1H), 6.50 (d, J=2,4 Гц, 1H), 3.81-3.79 (m, 6H), 3.49 (s, 3H), 2.91 (br t, J=6,8 Гц, 2H), 2.62 (s, 4H), 2.37 (br t, J=6,8 Гц, 2H), 2.26-2.22 (m, 6H), 1.77 (s, 6H).
Методика получения соединения Примера 32:
В раствор соединения 32j (200 мг, 0,38 ммоль) и DIEA (75 мг, 0,58 ммоль) в DMF (1 мл) по каплям добавляли акрилоилхлорид (35 мг, 0,38 ммоль) в DMF (1 мл) при 0°С. Полученную коричневую смесь перемешивали при 0°С в течение 30 мин. Реакционную смесь очищали препаративной ВЭЖХ [колонка: Waters Xbridge 150 × 25 5 мкм; условия: 30-50% В (А: 0,05%-ный аммиак; В: CH3CN); скорость потока: 25 мл/мин] и затем лиофилизировали с получением соединения Примера 32 (57,8 мг, 26,2%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 4,478 мин при 10-80CD_7 мин_220&254 хроматографии (XBrige Shield RP18 2,1×50 мм), МС (ЭРИ) m/z = 574,3 [М+Н]+.
ВЭЖХ: Rt = 3,90 мин при 10-80_CD_1,2 мл. мет (XBridge Shield RP 18 2,1×50 мм 5 мкм).
1Н ЯМР (400 МГц, CDCl3) δ 10.61 (br s, 1H), 10.42 (br s, 1H), 10.03 (br s, 1H), 8.41-8.39 (m, 2Н), 7.59 (br s, 1H), 7.02 (d, J=2,8 Гц, 1H), 6.78 (s, 1H), 6.48-6.44 (m, 2H), 6.37-6.30 (m, 1H), 6.00 (br s, 1H), 5.78 (br d,.7=10,8 Гц, 1H), 3.88 (s, 3H), 3.79 (s, 3H), 2.88 (br t, J=5,2 Гц, 2H), 2.71 (s, 3H), 2.27 (s, 8H), 1.90 (s, 6H).
Пример 33
(R)-N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-(3-(диметиламино)пирролидин-1-ил)-4-метоксифенил)акриламид

Методика получения соединения 33а:
В раствор соединения 11с (200 мг, 0,43 ммоль) и К2СО3 (119 мг, 0,86 ммоль) в DMSO (4 мл) добавляли соединение (R)-N,N-диметилпирролидин-3-амин (59 мг, 0,51 ммоль). Полученную смесь перемешивали при 24-27°С в течение 1 ч. Реакционную смесь вливали в воду (50 мл), и желтое твердое вещество выпадало в осадок. Желтое твердое вещество собирали фильтрованием и растворяли в CH2Cl2 (20 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением указанного в заголовке продукта 33а (220 мг, 91%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 0,789 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 583,1 [M+Na]+.
1Н ЯМР (400 МГц, CDCl3) δ 9.69 (br s, 1H), 8.91 (br s, 1H), 8.38 (br s, 2Н), 7.35 (br s, 1Н), 7.08 (d, J=10,4 Гц, 1H), 6.28 (s, 1Н), 3.92 (s, 3Н), 3.59-3.51 (m, 1H), 3.37-3.31 (m, 1Н), 3.23-3.18 (m, 1Н), 3.15-3.07 (m, 1H), 2.86-2.75 (m, 1Н), 2.29 (s, 6Н), 2.25-2.15 (m, 1H), 1.99-1.88 (m, 1H), 1.69 (s, 6Н).
Методика получения соединения 33b:
В раствор соединения 33а (220 мг, 0,39 ммоль), Zn (224 мг, 4,19 ммоль) в МеОН/Н2О (6 мл, 5/1) добавляли NH4Cl (236 мг, 3,6 ммоль). Полученную смесь перемешивали при 90°С в течение 2 ч. Реакционную смесь фильтровали и концентрировали под вакуумом с получением остатка, который растворяли в CH2Cl2 (20 мл), промывали водой (15 мл × 3), сушили над Na2SO4 и концентрировали при пониженном давлении с получением указанного в заголовке продукта 33b (180 мг, 87%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,732 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 531,2 [М+Н]+.
Методика получения соединения Примера 33:
В раствор соединения ЗЗЬ (180 мг, 0,34 ммоль) и DIEA (66 мг, 0,51 ммоль) в DMF (2 мл) по каплям добавляли раствор акрилоилхлорида (31 мг, 0,34 ммоль) в DMF (0,5 мл). Полученную смесь перемешивали при 0°С в бане лед-вода в течение 20 мин. Реакционную смесь гасили тремя каплями воды и очищали препаративной ВЭЖХ (колонка: Waters Xbridge 150×25, 5 мкм: 30-60% В (А: вода (0,05% гидроксида аммония об./об.), В: CH3CN), скорость потока: 25 мл/мин) с получением указанного в заголовке продукта - соединения Примера 33 (53,6 мг, 27%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 1,726 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 584,9 [М+Н]+.
ВЭЖХ: Rt = 2,85 мин в 10-80_CD_1,2 мл хроматографии (Ultimate С18 3×50 мм 3 мкм).
1Н ЯМР (400 МГц, CDCl3) δ 10.53 (br s, 1H), 9.79 (br s, 1H), 8.54-8.45 (m, 2H), 8.41 (s, 1H), 7.64 (br s, 1H), 7.09 (d, J=10,8 Гц, 1H), 6.75 (s, 1H), 6.40-6.35 (m, 2H), 5.84-5.79 (m, 1H), 3.87 (s, 3H), 3.14-3.06 (m, 4H), 2.93-2.84 (m, 1H), 2.30 (s, 6H), 2.23-2.13 (m, 1H), 2.00-1.90 (m, 1H), 1.76 (s, 6H).
Пример 34
N-(5-(4-(4-циклопропил-5-фтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид

Методика получения соединения 34b:
В раствор соединения 34а (1,0 г, 4,91 ммоль) в диоксан (20 мл) и Н2О (4 мл) добавляли циклопропилбороновую кислоту (1,1 г, 12,28 ммоль), затем добавляли Pd(OAc)2 (772 мг, 3,44 ммоль), РСу3 (1,9 г, 6,87 ммоль) и Cs2CO3 (4,8 г, 3,0 экв., 14,73 ммоль) в атмосфере N2. Полученную смесь дегазировали N2 в течение 1 мин и перемешивали при 100°С под воздействием микроволнового излучения в течение 1 ч. Реакционную смесь фильтровали и разбавляли EtOAc (50 мл), промывали рассолом (50 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением остатка, который очищали колоночной хроматографией на силикагеле (петролейный эфир/EtOAc = 100/1 (об./об.)) с получением соединения 34b (700 мг, 68%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,797 мин при 5-95 АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 209,9 [М+Н]+.
1Н ЯМР (400 МГц, Метанол-d4) δ 7.45 (d, J=8,8 Гц, 1H), 6.41 (d, J=12,4 Гц, 1H), 3.82 (s, 3Н), 1.88-1.81 (m, 1H), 0.89-0.81 (m, 2Н), 0.60-0.50 (m, 2Н).
Методика получения соединения 34 с:
В раствор соединения 34b (700 мг, 3,35 ммоль) в THF (10 мл) добавляли CH3MgBr (5,58 мл, 16,75 ммоль) при 0°С. Полученную смесь перемешивали при 24-26°С в течение 2 ч. Реакционную смесь разбавляли насыщенным водным NH4Cl (10 мл) и экстрагировали EtOAc (2×20 мл), объединенные органические слои концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле (петролейный эфир/EtOAc = 10/1) с получением соединения 34 с (450 мг, 64%-ный выход) в виде бесцветного масла.
ЖХ/МС: Rt = 0,672 мин при 5-95 АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 191,9 [М+Н-18]+.
1Н ЯМР (400 МГц, DMSO-d6) δ 6.65-6.56 (m, 1H), 6.34 (d, J=12,8 Гц, 1H), 1.84-1.72 (m, 1H), 1.44 (s, 6Н), 0.82-0.75 (m, 2Н), 0.55-0.48 (m, 2Н).
Методика получения соединения 34d:
В раствор соединения 34 с (400 мг, 1,91 ммоль) в CH2Cl2 (10 мл) добавляли DIEA (494 мг, 3,82 ммоль) и 2,4-дихлор-1,3,5-триазин (315 мг, 2,10 ммоль). Полученную смесь перемешивали при 24-27°С в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле (петролейный эфир/EtOAc = 10/1 (об./об.)) с получением соединения 34d (450 мг, 73%-ный выход) в виде бесцветного масла.
ЖХ/МС: Rt = 0,999 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 323,0 [М+Н]+.
1Н ЯМР (400 МГц, Метанол-d4) δ 8.69-8.44 (m, 1H), 7.99 (br d, J=14,0 Гц, 1H), 6.93 (d, J=8,4 Гц, 1H), 2.08-2.02 (m, 1Н), 1.59 (s, 6Н), 1.02-0.93 (m, 2Н), 0.77-0.68 (m, 2Н).
Методика получения соединения 34е:
В раствор соединения 34d (450 мг, 1,39 ммоль) и В (259 г, 1,39 ммоль) в n-BuOH (5 мл) добавляли TFA (0,05 мл). Полученную смесь перемешивали при 25-30°С в течение 3 ч, при этом серое твердое вещество выпадало в осадок. Реакционную смесь фильтровали, и остаток на фильтре промывали 30 мл петролейного эфира и сушили при пониженном давлении с получением соединения 34е (450 мг, 68%-ный выход) в виде серого твердого вещества.
ЖХ/МС: Rt = 0,890 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 473,2 [М+Н]+.
1Н ЯМР (400 МГц, Метанол-d4) δ 8.80 (s, 1H), 8.31 (br s, 1H), 8.16-7.58 (m, 1H), 7.15 (d, J=12,8 Гц, 1H), 6.91 (d, J=8,4 Гц, 1H), 4.02 (s, 3H), 1.99 (br s, 1H), 1.59 (s, 6H), 0.95 (br d,.7=7,2 Гц, 2H), 0.74-0.67 (m, 2H).
Методика получения соединения 34f:
В раствор соединения 34е (150 мг, 0,32 ммоль) и К2СО3 (88 мг, 0,64 ммоль) в DMSO (5 мл) добавляли N1,N1,N2-триметилэтан-1,2-диамин (49 мг, 0,48 ммоль). Полученную смесь перемешивали при 22-32°С в течение 12 ч, при этом цвет менялся с бледно-коричневого на насыщенный желтый. Реакционную смесь вливали в воду (50 мл), и желтое твердое вещество выпадало в осадок. Выпавшее в осадок твердое вещество отфильтровывали и растворяли в CH2Cl2 (30 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением соединения 34f (150 мг, 84%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 0,831 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 555,2 [М+Н]+.
Методика получения соединения 34g:
В раствор соединения 34f (150 мг, 0,27 ммоль) в МеОН (10 мл) добавляли Pd/C (15 мг). Полученную смесь продували и дегазировали Н2 3 раза, затем перемешивали при 23-29°С в атмосфере Н2 (15 фунт/кв. дюйм) в течение 2 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением соединения 34g (130 мг, 92%-ный выход) в виде коричневого масла.
ЖХ/МС: Rt = 0,765 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 525,1 [М+Н]+.
Методика получения соединения Примера 34:
В раствор соединения 34g (130 мг, 0,25 ммоль) и DIE А (85 мг, 0,66 ммоль) в DMF (2,5 мл) добавляли акрилоилхлорид (30 мг, 0,33 ммоль) в DMF (0,5 мл). Полученную смесь перемешивали при 0°С в бане лед-вода в течение 30 мин. Реакционную смесь очищали ОФ-ВЭЖХ (обращенно-фазная ВЭЖХ) [колонка: обращенно-фазная колонка; условия: 0-30% В (А: 0,25% NH3HCO3; В: МеОН); скорость потока: 40 мл/мин] и затем лиофилизировали с получением соединения Примера 34 (24,4 мг, 17%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 1,994 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 601,3 [M+Na]+.
ВЭЖХ: Rt = 3,41 мин при 10-80_ab_1,2 мл хроматографии (Ultimate С18 3×50 мм 3 мкм).
1Н ЯМР (400 МГц, CDCl3) δ 10.72 (br s, 1H), 10.39 (br s, 1H), 9.97 (br s, 1H), 8.41 (s, 1H), 8.11 (d, J=12,4 Гц, 1H), 7.66 (br s, 1H), 6.86 (d, J=8,4 Гц, 1H), 6.78 (s, 1H), 6.45-6.30 (m, 2H), 5.93 (br s, 1H), 5.79-5.72 (m, 1H), 3.88 (s, 3H), 2.92-2.83 (m, 2H), 2.70 (s, 3H), 2.33-2.23 (m, 8H), 2.06-1.97 (m, 1H), 1.76 (s, 6H), 0.97-0.86 (m, 2H), 0.73-0.65 (m, 2H).
Пример 35
(S)-N-(5-(4-(4,5-дифтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-(3-((диметиламино)метил)морфолино)-4-метоксифенил)акриламид
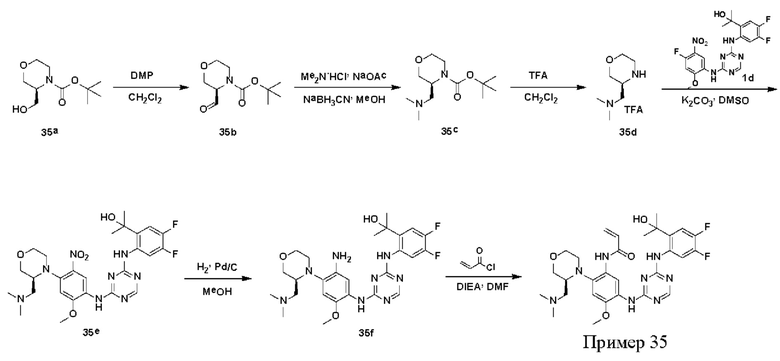
Методика получения соединения 35b:
В раствор соединения 35а (1,4 г, 6,44 ммоль) в CH2Cl2 (40 мл) порциями добавляли DMP (4,1 г, 9,67 ммоль) при 0-5°С. Полученную белую смесь перемешивали при 0-5°С в течение 1 ч. Реакционную смесь обрабатывали водным NaHCO3 (30 мл) и экстрагировали EtOAc (3×20 мл). Объединенные органические слои промывали рассолом (3×20 мл), сушили и концентрировали при пониженном давлении с получением неочищенного остатка, который очищали колоночной хроматографией на силикагеле (петролейный эфир/EtOAc = 5/1 (об./об.)) с получением соединения 35b (1,1 г, 79,7%-ный выход) в виде светло-желтого масла.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.60 (br d,.7=10,0 Гц, 1H), 4.52-4.36 (m, 2Н), 3.94-3.67 (m, 2Н), 3.60 (dd, 7=4.4, 12,2 Гц, 2Н), 3.05-2.79 (m, 1H), 1.43-1.36 (m, 9Н).
Методика получения соединения 35с:
В раствор соединения 35b (1,0 г, 4,65 ммоль) в МеОН (10 мл) добавляли Me2N.HCl (1,1 г, 13,94 ммоль) и NaOAc (572 мг, 6,97 ммоль). Эту белую смесь перемешивали при 24-26°С в течение 2 ч, затем добавляли NaBH3CN (584 мг, 9,29 ммоль). Полученную смесь перемешивали при 22-27°С в течение 18 ч. Реакционную смесь гасили добавлением водного NH4Cl (20 мл) и экстрагировали EtOAc (3×10 мл). Объединенные органические слои промывали рассолом (3×10 мл), сушили и концентрировали в вакууме с получением соединения 35с (800 мг, 70,5%-ный выход) в виде бесцветного масла.
ЖХ/МС: Rt = 2,519 мин при 10-80CD_4 мин_Е.М, XBrige Shield RP18 2,1×50 мм, МС (ЭРИ) m/z = 245,1 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 3.99-3.63 (m, 2Н), 3.46-3.33 (m, 1H), 2.98 (br s, 1H), 2.71 (br t, J=10,8 Гц, 1H), 2.31-2.18 (m, 6Н), 1.41 (s, 9Н).
Методика получения соединения 35d:
В раствор соединения 35с (500 мг, 2,30 ммоль) в CH2Cl2 (6 мл) добавляли TFA (2 мл). Полученный бесцветный раствор перемешивали при 22-32°С в течение 3 ч. Реакционный раствор концентрировали в вакууме напрямую с получением соединения 35d в виде TFA соли (300 мг, 61,1%-ный выход) в виде бесцветного масла.
ЖХ/МС: Rt = 0,306 мин при 10-80CD_4 мин_Е.М; XBrige Shield RP18 2,1×50 мм, МС (ЭРИ) m/z = 145,2 [М-ОН]+.
Методика получения соединения 35е:
В раствор соединения 35d (187 мг, 0,42 ммоль) и K2CO3 (116 мг, 0,84 ммоль) в DMSO (2 мл) добавляли соединение 1d (60 мг, 0,42 ммоль). Смесь перемешивали при 28-33°С в течение 2 часов. Смесь напрямую очищали обращенно-фазной С-18 колоночной хроматографией на флэш-системе Biotage, элюируя смесью МеОН/Н2О (МеОН в воде от 56% до 60%) с получением целевого продукта 35е (50 мг, 20,9%-ный выход) в виде красного твердого вещества, которое использовали на следующей стадии напрямую.
Методика получения соединения 35f:
В раствор соединения 35е (90 мг, 0,16 ммоль) в МеОН (3 мл) добавляли Pd/C (10 мг) в защитной атмосфере N2. Черную смесь перемешивали при 26-33°С в атмосфере водорода (15 фунт/кв. дюйм) в течение 1 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением соединения 35f (70 мг, 82,1%-ный выход) в виде коричневого масла.
ЖХ/МС: Rt = 1,791 мин при 10-80CD_3 мин_220&254; XBrige Shield RP18 2,1×50 мм; МС (ЭРИ) m/z = 545,3 [М+Н]+.
Методика получения соединения Примера 35:
В раствор соединения 35f (70 мг, 0,13 ммоль) и DIEA (25 мг, 0,19 ммоль) в DMF (1 мл) по каплям добавляли раствор акрилоилхлорида (12 мг, 0,13 ммоль) в DMF (1 мл). Полученную коричневую смесь перемешивали при 0°С в течение 30 мин. Реакционную смесь очищали препаративной ВЭЖХ [колонка: Waters Xbridge 150×25, 5 мкм; условия: 35-65% В (А: 0,05%-ный аммиак; В: CH3CN); скорость потока: 25 мл/мин]. Фракции, содержащие целевое соединение, лиофилизировали с получением соединения Примера 35 (11,9 мг, 16,8%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 1,816 мин при 10-80CD_3 мин_220&254; XBrige Shield RP18 2,1×50 мм, МС (ЭРИ) m/z = 599,3 [М+Н]+.
ВЭЖХ, Rt = 3,45 чистота 92,56% (220 нм), 10-80_CD_1,2 мл. мет (XBridge Shield RP 18 2,1×50 мм, 5 мкм).
1Н ЯМР (400 МГц, DMSO-d6) δ 10.29 (br s, 1H), 9.14 (s, 2H), 8.31 (s, 1H), 8.27 (s, 1H), 7.26 (br s, 1H), 7.11 (s, 1H), 6.64 (br dd, J=10.4, 17,1 Гц, 1H), 6.27 (s, 1H), 6.18 (br d, J=16,8 Гц, 1H), 5.73 (br d, J=10,4 Гц, 1H), 4.02 (br d, J=9,2 Гц, 1H), 3.78 (s, 5H), 3.55-3.48 (m, 1H), 3.28 (br s, 1H), 2.85 (br s, 2H), 2.68 (br s, 1H), 2.34 (s, 1H), 2.25 (br t, J=11,2 Гц, 1H), 2.01 (s, 6H), 1.92 (br d, J=9,2 Гц, 1H), 1.52 (br s, 6H).
Пример 36
(N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)пиримидин-2-иламино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид)
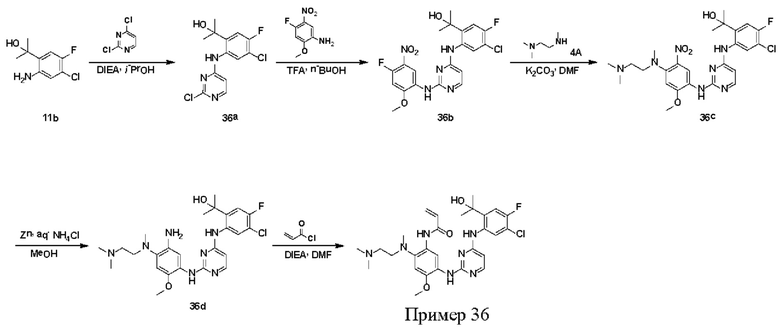
Методика получения соединения 36а:
В раствор соединения 11b (500 мг, 2,46 ммоль) и DIEA (634 мг, 4,92 ммоль) в изопропаноле (10 мл) добавляли 2,4-дихлорпиримидин (442 мг, 2,95 ммоль). Полученную смесь нагревали при 90°С в течение 27 ч. Реакционную смесь концентрировали в вакууме с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (25-40% EtOAc в петролейном эфире) с получением соединения 36а (390 мг, 50,14%-ный выход) в виде не совсем белого твердого вещества.
ЖХ/МС: Rt = 0,796 мин при 5-95АВ_220&254.lcm хроматографии (ACSSH-LCMS-АВ MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 315,9 [М+Н]+.
1Н ЯМР (400 МГц, Метанол-d4) δ 8.12 (d, J=6,0 Гц, 1H), 8.08 (br d, J=6,8 Гц, 1H), 7.31 (d, J=10,8 Гц, 1H), 6.70 (d, J=6,0 Гц, 1Н), 1.57 (s, 6Н).
Методика получения соединения 36b:
Раствор соединения 36а (760 мг, 2,4 ммоль) и 4-фтор-2-метокси-5-нитроанилина (448 мг, 2,4 ммоль) в смеси TFA/n-BuOH = 1/10 (11 мл) нагревали при 50°С в течение 4 ч, дополнительно добавляли 30 мг соединения 4-фтор-2-метокси-5-нитроанилин, и продлевали время реакции при 50°С еще на 22 ч. Реакционную смесь фильтровали, и твердый остаток на фильтре промывали петролейным эфиром (20 мл × 3), затем сушили в глубоком вакууме с получением соединения 36b (960 мг, 86%-ный выход) в виде не совсем белого твердого вещества.
ЖХ/МС: Rt = 0,753 мин при 5-95АВ_220&254.lcm хроматографии (ACSSH-LCMS-АВ MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 466,1 [М+Н]+.
1Н ЯМР (400 МГц, Метанол-d4) δ 8.52 (d, J=8,0 Гц, 1H), 7.96 (d, J=6,8 Гц, 1H), 7.84 (d, J=7,2 Гц, 1H), 7.33 (d, J=10,8 Гц, 1H), 7.20 (d, J=12,8 Гц, 1H), 6.48 (d, J=7,2 Гц, 1H), 4.00 (s, 3Н), 1.59 (s, 6Н).
Методика получения соединения 36с:
В раствор соединения 36b (200 мг, 0,43 ммоль) и К2СО3 (119 мг, 0,86 ммоль) в DMF (3 мл) добавляли N1,N1,N2-триметилэтан-1,2-диамин (66 мг, 0,644 ммоль). Реакционную смесь перемешивали при 22-32°С в течение 15 ч (цвет менялся с коричневого на насыщенный оранжевый). Реакционную смесь добавляли по каплям в H2O (40 мл) в бане лед-вода при перемешивании, и твердое вещество выпадало в осадок. Твердое вещество собирали фильтрованием и промывали H2O (15 мл × 3), затем сушили в глубоком вакууме с получением соединения 36с (210 мг, 89%-ный выход) в виде оранжевого твердого вещества.
ЖХ/МС: Rt = 0,669 мин при 5-95АВ_220&254.lcm хроматографии (ACSSH-LCMS-АВ MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 548,1 [М+Н]+.
1Н ЯМР (400 МГц, Метанол-d4) δ 8.60 (s, 1H), 7.99 (d, J=6,0 Гц, 1H), 7.95 (d, J=7,6 Гц, 1H), 7.26 (d, J=10,8 Гц, 1H), 6.82 (s, 1H), 6.20 (d, J=6,0 Гц, 1H), 3.99 (s, 3Н), 3.24 (t, J=7,4 Гц, 2Н), 2.85 (s, 3Н), 2.59 (t, J=7,2 Гц, 2Н), 2.28 (s, 6Н), 1.59 (s, 6Н).
Методика получения соединения 36d:
В раствор соединения 36с (200 мг, 0,365 ммоль) в МеОН/Н2О = 5/1 (5 мл) добавляли Zn (143 мг, 2,190 ммоль) и NH4Cl (117 мг, 2,190 ммоль). Полученную смесь нагревали при 90°С в течение 2 ч (цвет менялся с оранжевого на коричневый). Реакционную смесь фильтровали, и фильтрат концентрировали в вакууме с получением неочищенного остатка, который растворяли в CH2Cl2 (20 мл) и промывали водой (15 мл × 3), сушили над Na2SO4 и концентрировали в вакууме с получением соединения 36d (170 мг, 89,9%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,646 мин при 5-95АВ_220&254.lcm хроматографии (ACSSH-LCMS-АВ MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 518,2 [М+Н]+.
Методика получения соединения Примера 36:
В раствор соединения 36d (170 мг, 0,329 ммоль) и DIEA (64 мг, 0,494 ммоль) в DMF (2 мл) добавляли акрилоилхлорид (30 мг, 1,0 экв., 0,329 ммоль) в бане лед-вода. Полученную смесь перемешивали при 5-10°С в течение 10 мин. Реакционную смесь гасили Н2О (0,1 мл) и затем фильтровали. Фильтрат очищали препаративной ВЭЖХ (колонка: Xtimate С18 150×25 мм, 5 мкм; условия: 53-83% В (A: 0,04%NH3⋅H2O+10 мМ NH4HCO3, В: CH3CN); скорость потока: 25 мл/мин) и затем лиофилизировали с получением соединения Примера 36 (17,0 мг, 8,1%-ный выход) в виде не совсем белого твердого вещества.
ЖХ/МС: Rt = 2,356 мин при 10-80CD_3 мин_220&254 хроматографии (ACSSH-LCMS-AS A: Xtimate С18, 2,1×30 мм, 3 мкм; В: XBrige Shield RP18 2,1×50 мм), МС (ЭРИ) m/z = 572,2 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 10.34 (br s, 1H), 9.72 (s, 1H), 9.43 (s, 1H), 8.02 (d, J=6,0 Гц, 1H), 7.46 (s, 1H), 7.41 (d, 7=7,2 Гц, 1H), 7.08 (d, J=10,8 Гц, 1H), 6.69 (s, 1H), 6.36-6.21 (m, 3H), 6.02 (br s, 1H), 5.70-5.63 (m, 1H), 3.79 (s, 3H), 2.85-2.78 (m, 2H), 2.61 (s, 3H), 2.20 (s, 8H), 1.66 (s, 6H).
Пример 37
N-(2-((2-(бис(метил-d3)амино)этил)(метил)амино)-5-((4-((4-хлор-5-фтор-2-(2-гидроксипропан-2-ил)фенил)амино)-1,3,5-триазин-2-ил)амино)-4-метоксифенил)акриламид

Методика получения соединения 37b:
В три отдельных раствора соединения 37а (500 мг, 2,87 ммоль) и K2CO3 (793 мг, 5,74 ммоль) в THF (10 мл) добавляли CD3I (624 мг, 4,30 ммоль). Смесь перемешивали при 24-26°С в течение 1 ч, при этом белое твердое вещество выпадало в осадок. Три реакционных смеси фильтровали, и органический слой концентрировали при пониженном давлении с получением неочищенного остатка, который разбавляли EtOAc (20 мл) и промывали Н2О (20 мл), затем сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением соединения 37b (350 мг, 19%-ный выход) в виде бесцветного масла.
ЖХ/МС: Rt = 0,744 мин при 0-60АВ_2 мин_Е.М хроматографии (Xtimate С18, 2,1×30 мм, 3 мкм), МС (ЭРИ) m/z = 209,2 [М+Н]+.
1Н ЯМР (400 МГц, DMSO-d6) δ 3.19-3.13 (m, 1Н), 3.13-3.10 (m, 1H), 2.68 (br d, J=6,4 Гц, 3H), 2.56 (t, J=6,8 Гц, 1H), 2.32-2.24 (m, 1H), 1.30-1.23 (m, 9H).
Методика получения соединения 37c:
В раствор соединения 37b (300 мг, 1,44 ммоль) в CH2Cl2 (5 мл) добавляли HCl/диоксан (5 мл, 4 М) при 0°С. Полученную смесь перемешивали при 22-32°С в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении с получением соединения 37 с (260 мг, неочищенное) в виде белого твердого вещества.
ЖХ/МС: Rt = 0,097 мин при 0-60 АВ_2 мин_Е.М хроматографии (Xtimate С18, 2,1×30 мм, 3 мкм), МС (ЭРИ) m/z = 109,1 [М+Н]+.
1Н ЯМР (400 МГц, DMSO-d6) δ 3.38 (s, 1H), 3.35 (br d, J=4,0 Гц, 1H), 3.27-3.11 (m, 2Н), 2.58 (br d, J=4,4 Гц, 3Н).
Методика получения соединения 37d:
В раствор соединения 37с (250 мг, 0,54 ммоль) и К2СО3 (149 мг, 1,08 ммоль) в DMSO (5 мл) добавляли соединение 11d (230 мг, 1,27 ммоль). Полученную смесь перемешивали при 22-29°С в течение 12 ч, при этом цвет менялся с бледно-коричневого на насыщенный желтый. Реакционную смесь разбавляли EtOAc (20 мл) и промывали рассолом (2×30 мл). Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением неочищенного остатка, который очищали колоночной хроматографией на силикагеле (CH2Cl2/МеОН = 10/1) с получением соединения 37d (90 мг, 30%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 0,820 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 555,2 [М+Н]+.
Методика получения соединения 37е:
В раствор соединения 37d (90 мг, 0,16 ммоль) в МеОН (4 мл) и Н2О (2 мл) добавляли Zn (52 мг, 0,80 ммоль) и NH4Cl (85 мг, 1,60 ммоль). Полученную смесь продували и дегазировали N2 3 раза, затем перемешивали при 80°С в течение 1 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении, остаток растворяли в EtOAc (20 мл) и промывали H2O (10 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением соединения 37е (80 мг, 95%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,709 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 525,0 [М+Н]+.
Методика получения соединения Примера 37:
В раствор соединения 37е (80 мг, 0,15 ммоль) и DIE А (39 мг, 0,30 ммоль) в DMF (2,5 мл) по каплям добавляли раствор акрилоилхлорида (14 мг, 0,15 ммоль) в DMF (0,5 мл). Полученную смесь перемешивали при 0°С в бане лед-вода в течение 30 мин. Реакционную смесь очищали препаративной ВЭЖХ [колонка: Waters Xbridge 150×25, 5 мкм; условия: 43-73% В (А: 0,05% NH3H2O; В: CH3CN); скорость потока: 25 мл/мин]. Фракции, содержащие целевое соединение, лиофилизировали с получением соединения Примера 37 (35,0 мг, 40%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 1,882 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 579,3 [М+Н]+.
ВЭЖХ: Rt = 3,23 мин при 10-80_ab_1,2 мл хроматографии (Ultimate С18 3×50 мм 3 мкм).
1H ЯМР (400 МГц, CDCl3) δ 10.60 (br s, 1H), 10.46 (br s, 1H), 9.96 (br s, 1H), 8.47 (d, J=7,6 Гц, 1H), 8.42 (s, 1H), 7.69 (br s, 1H), 7.10 (d, J=10,8 Гц, 1H), 6.78 (s, 1H), 6.45-6.30 (m, 2H), 6.18 (br s, 1H), 5.81-5.70 (m, 1H), 3.88 (s, 3H), 2.87 (br d, J=4,8 Гц, 2H), 2.70 (s, 3H), 2.29 (brs, 2H), 1.78 (s, 6H).
Пример 38
(R)-N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-(2-((диметиламино)метил)азетидин-1-ил)-4-метоксифенил)акриламид
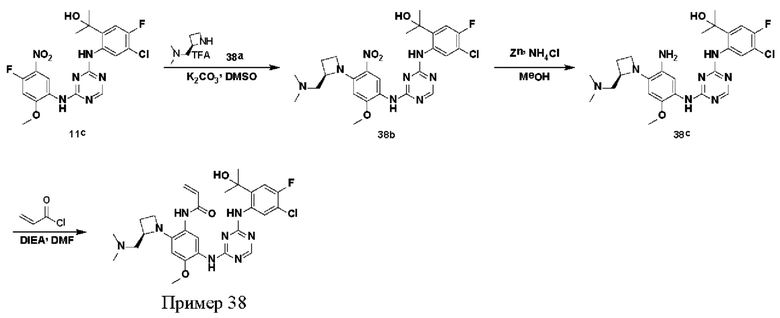
Методика получения соединения 38b:
Раствор соединения 11 с (180 мг, 0,385 ммоль), соединения 38а (440,3 мг, 3,86 ммоль) и К2СО3 (532,9 мг, 3,86 ммоль) в DMSO (2 мл) перемешивали при 85°С в течение 2 ч. Реакционную смесь добавляли в Н2О (10 мл) в бане лед-вода при перемешивании. Выпавшее в осадок твердое вещество отфильтровывали, и остаток на фильтре растворяли в DCM (30 мл), затем сушили и концентрировали в вакууме с получением продукта 38b (230 мг, 95,7%-ный выход) в виде оранжевого твердого вещества.
ЖХ/МС: Rt = 0,718 мин при 5-95АВ_220&254.lcm хроматографии (ACSSH-LCMS-АВ MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 560,1, 583,1 [М+Н, M+Na]+.
1Н ЯМР (400 МГц, CDCl3) δ 9.71 (br s, 1H), 8.98 (br s, 1H), 8.37 (br s, 2H), 7.35 (br s, 1H), 7.06 (d, J=10,4 Гц, 1H), 6.83 (s, 1H), 4.45-4.35 (m, 1H), 4.30 (dt,.7=5.1, 9,2 Гц, 1H), 3.93 (s, 3H), 3.33-3.24 (m, 1H), 2.80 (m, 1H), 2.49 (dd, J=5.6, 13,2 Гц, 1H), 2.46-2.36 (m, 1H), 2.30 (s, 6H), 2.20-2.08 (m, 1H), 1.69 (s, 6H).
Методика получения соединения 38c:
В раствор соединения 38b (230 мг, 0,410 ммоль) в 6 мл смеси МеОН/Н2О=5/1 (об./об.) добавляли Zn (134,02 мг, 2,05 ммоль) и NH4Cl (109,65 мг, 2,05 ммоль). Полученную смесь нагревали при 90°С в течение 1 ч. Реакционную смесь фильтровали, и фильтрат концентрировали в вакууме с получением неочищенного остатка, который растворяли в CH2Cl2 (30 мл), промывали водой (20 мл × 2) и рассолом (20 мл), затем сушили над Na2SO4 и концентрировали в вакууме с получением продукт 38с (197 мг, 90,5%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,657 мин при 5-95АВ_220&254.lcm хроматографии (ACSSH-LCMS-АВ MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 531,3 [М+Н]+.
Методика получения соединения Примера 38:
В раствор соединения 38с (197 мг, 0,351 ммоль) и DIEA (67,9 мг, 0,527 ммоль) в DMF (3 мл) добавляли акрилоилхлорид (31,7 мг, 0,351 ммоль) при 0°С. Полученную смесь перемешивали при 0°С в течение 1 ч. Реакционную смесь гасили H2O (0,1 мл) и очищали препаративной ВЭЖХ (прибор: колонка DB: Gemini 150×25, 5 мкм; подвижная фаза А: вода с 0,05% гидроксида аммония об./об.; подвижная фаза В: DMF; скорость потока: 25 мл/мин; время градиента: 10 мин; описание профиля: 30%-60%) с получением соединения Примера 38 (36,6 мг, 18%-ный выход) в виде светло-желтого твердого вещества.
ЖХ/МС: Rt = 1,295 мин при 10-80АВ_4 мин_220&254 хроматографии (ACSSH-LCMS-AS A: Xtimate С18, 2,1×30 мм, 3 мкм; В: XBrige Shield RP18 2,1×50 мм), МС (ЭРИ) m/z = 585,3 [М+Н]+.
ВЭЖХ: Rt = 3,37 мин при 10-80_CD_1,2 мл. MeTXBridge Shield RP 18 2,1×50 мм, 5 мкм.
1Н ЯМР (400 МГц, CDCl3) δ 10.41 (br s, 1H), 9.38 (br s, 1H), 8.95 (br s, 1H), 8.38 (m, 1H), 7.56 (m, 1H), 7.08 (m, 1H), 6.62 (m, 1H), 6.37 (m, 1H), 6.82-6.21 (m, 2H), 5.80 (br s, 1H), 4.23 (m, 1H), 3.89 (m, 5H), 3.56 (m, 1H), 2.64 (m, 1H), 2.39 (m, 1H), 2.25 (m, 7H), 1.90-1.73 (s, 6H).
Пример 39
N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-(3-((диметиламино)метил)азетидин-1-ил)-4-метоксифенил)акриламид

Методика получения соединения 39b:
Смесь соединения 11с (100 мг, 0,22 ммоль), соединения 39а (40 мг, 0,22 ммоль) и K2CO3 (ПО мг, 0,85 ммоль) в DMSO (2 мл) перемешивали при 90°С в течение 3 ч (желтая суспензия). После завершения в смесь добавляли ледяную воду (10 мл). Выпавшее в осадок твердое вещество отфильтровывали, и остаток на фильтре промывали Н2О (10 мл) и сушили в глубоком вакууме получением соединения 39b (150 мг, 83,4%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 0,710 мин при 5-95 АВ 220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z = 583,1 [M+Na]+.
1Н ЯМР: (400 МГц, DMSO-d6) δ м.д. 10.23 (br s, 1H), 9.27-8.91 (m, 1Н), 8.27 (s, 1H), 7.90 (s, 1H), 7.29 (s, 1H), 6.30 (br s, 1H), 6.21 (s, 1H), 4.09 (t, J=8,4 Гц, 2H), 3.86 (s, 3H), 3.62 (t, J=8,4 Гц, 2H), 2.91-2.82 (m, 1H), 2.55 (s, 2H), 2.15 (s, 6H), 1.51 (s, 6H).
Методика получения соединения 39c:
В смесь соединения 39b (150 мг, 0,267 ммоль) в МеОН/H2O (50/10 мл) добавляли Zn (87 мг, 1,34 ммоль) и NH4Cl (71 мг, 1,34 ммоль). Полученную суспензию перемешивали при 85°С в течение 3 ч. Реакционную смесь фильтровали, и фильтрат концентрировали в вакууме с получением неочищенного остатка, который обрабатывали водой (20 мл) и экстрагировали CH2Cl2 (30 мл × 3). Объединенные органические слои промывали рассолом (20 мл), сушили над Na2SO4 и концентрировали в вакууме с получением продукта 39 с (120 мг, 85,1%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 0,665 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 529,1 [М+Н-2]+.
1Н ЯМР: (400 МГц, DMSO-d6) δ 10.11 (br s, 1H), 8.84 (br s, 1H), 8.33 (s, 1H), 8.19 (s, 1H), 7.26 (s, 1H), 6.53 (br s, 1H), 6.27-6.21 (m, 2H), 4.05-3.91 (m, 4H), 3.67 (s, 3H), 3.46-3.39 (m, 2H), 2.76-2.67 (m, 1H), 2.16 (s, 6H), 1.51 (s, 6H).
Методика получения соединения Примера 39:
В смесь соединения 39с (120 мг, 0,225 ммоль) и DIEA (87 мг, 0,675 ммоль) в DMF (3 мл) по каплям добавляли акрилоилхлорид (26,6 мг, 0,293 ммоль) в DMF (1 мл) в бане лед-вода в течение 1 ч. Полученную смесь перемешивали в течение 30 мин при 0-5°С (коричневый раствор), затем гасили Н20 (0,05 мл) и напрямую очищали препаративной ВЭЖХ [Waters Xbridge 150×25.5 мкм; условия: 28-58% В (А: 0,05%-ный аммиак; В: CH3CN); скорость потока: 25 мл/мин]. Фракции, содержащие целевое соединение, лиофилизировали с получением соединения Примера 39 (48,0 мг, 20,9%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 2,325 мин при 0-60АВ_4,0 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 585,1 [М+Н]+.
ВЭЖХ: Rt = 4,92 мин при 0-60_CD_1,2 мл хроматографии (XBridge Shield RP 18 2,1×50 мм, 5 мкм).
1Н ЯМР: (400 МГц, CDCl3) δ м.д. 10.23 (brs, 1H), 9.07 (brs, 1H), 8.36-8.28 (m, 2Н), 743-7.32 (m, 2Н), 6.99 (d, J=10,8 Гц, 1H), 6.34-6.19 (m, 3Н), 5.72 (d, J=9,2 Гц, 1H), 3.90 (t, J=7,2 Гц, 2Н), 3.80 (s, 3Н), 3.42 (t, 7=6,4 Гц, 2Н), 2.86-2.79 (m, 1H), 2.45 (d, 7=7,2 Гц, 2Н), 2.16 (s, 6Н), 1.64 (s, 6Н).
Пример 40
(S)-N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-(3-((диметиламино)метил)морфолино)-4-метоксифенил)акриламид

Методика получения соединения 40b:
В раствор соединения 40а (1,4 г, 6,44 ммоль) в CH2Cl2 (40 мл) порциями добавляли DMP (4,1 г, 9,67 ммоль) при 0-5°С. Полученную белую смесь перемешивали при 0-5°С в течение 1 ч. Реакционную смесь обрабатывали водным NaHCO3 (30 мл) и экстрагировали EtOAc (3×20 мл). Объединенные органические слои промывали рассолом (3×20 мл), сушили и концентрировали при пониженном давлении с получением неочищенного остатка, который очищали колоночной хроматографией на силикагеле (петролейный эфир/EtOAc = 5/1(об./об.)) с получением соединения 40b (1,1 г, 79,7%-ный выход) в виде светло-желтого масла.
ЖХ/МС: Rt = 1,300-1,400 мин при 10-80CD_3 мин_220&254 хроматографии (XBrige Shield RP18 2,1×50 мм).
1Н ЯМР: (400 МГц, DMSO-d6) δ 9.60 (br d, J=10,0 Гц, 1Н), 4.52-4.36 (m, 2H), 3.94-3.67 (m, 2H), 3.63-3.52 (m, 2H), 3.08-2.79 (m, 1H), 1.43-1.36 (m, 9H).
Методика получения соединения 40c:
В раствор соединения 40b (1,0 г, 4,65 ммоль) в МеОН (10 мл) добавляли Me2N.HCl (1,1 г, 13,94 ммоль) и NaOAc (572 мг, 6,97 ммоль). Эту белую смесь перемешивали при 24-26°С в течение 2 ч, затем добавляли NaBH3CN (584 мг, 9,29 ммоль). Полученную смесь перемешивали при 22-27°С в течение 18 ч. Реакционную смесь гасили добавлением водного NH4Cl (20 мл) и экстрагировали EtOAc (3×10 мл). Объединенные органические слои промывали рассолом (3×10 мл), сушили и концентрировали в вакууме с получением соединения 40 с (800 мг, 70,5%-ный выход) в виде бесцветного масла.
ЖХ/МС: Rt = 2,475 мин при 10-80CD_7 мин_220&254, хроматографии (XBrige Shield RP18 2,1×50 мм), МС (ЭРИ) m/z = 245,1 [М+Н]+.
1Н ЯМР: (400 МГц, CDCl3) δ 4.15-3.40 (m, 5Н), 3.53-3.44 (m, 2Н), 3.06 (br s, 1H), 2.79 (br t, J=10,8 Гц, 1H), 2.32 (br s, 6H), 1.48 (s, 9H).
Методика получения соединения 40d:
В раствор соединения 40с (500 мг, 2,30 ммоль) в CH2Cl2 (6 мл) добавляли TFA (2 мл). Полученный бесцветный раствор перемешивали при 22-32°С в течение 3 ч. Реакционный раствор концентрировали в вакууме напрямую с получением соединения 40d в виде TFA соли (300 мг, 61,1%-ный выход) в виде бесцветного масла.
ЖХ/МС: Rt = 0,561 мин при 10-80CD_4 мин_Е, хроматографии (XBrige Shield RP18 2,1×50 мм), МС (ЭРИ) m/z = 145,2 [М+Н]+.
Методика получения соединения 40е:
В раствор соединения 40d (375 мг, 0,42 ммоль) и K2CO3 (230 мг, 1,67 ммоль) в DMSO (2 мл) добавляли соединение 11с (120 мг, 0,83 ммоль). Смесь перемешивали при 28-33°С в течение 8 часов. Смесь очищали обращенно-фазной С-18 колоночной хроматографией на флэш-системе Biotage, элюируя смесью МеОН/Н2О (МеОН в воде от 56% до 60%) с получением соединения 40е (120 мг, 20,9%-ный выход) в виде красного твердого вещества.
ЖХ/МС: Rt = 0,809 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18 2,5-2 мм). МС (ЭРИ) m/z = 591,3 [М+Н]+.
Методика получения соединения 40f:
В раствор соединения 40е (120 мг, 0,20 ммоль) в МеОН (5 мл) и Н2О (1 мл) добавляли Zn (67 мг, 1,02 ммоль) и NH4Cl (109 мг, 2,04 ммоль). Черную суспензию перемешивали при 70°С в течение 1,5 ч в атмосфере N2. Реакционную смесь гасили добавлением водного NH4Cl (20 мл), экстрагировали EtOAc (10 мл × 3). Органические слои промывали рассолом (10 мл × 3), сушили и напрямую концентрировали в вакууме с получением соединения 40f (70 мг, 82,1%-ный выход) в виде коричневого масла.
ЖХ/МС: Rt = 0,760 мин при 5-95АВ_1,5 мин_220&254.lcm хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 561,2 [М+Н]+.
Методика получения соединения Примера 40:
В раствор соединения 40f (70 мг, 0,12 ммоль) и DIEA (24 мг, 0,19 ммоль) в DMF (1 мл) по каплям добавляли раствор акрилоилхлорида (11 мг, 0,12 ммоль) в DMF (1 мл). Полученную коричневую смесь перемешивали при 0°С в течение 30 мин. Реакционную смесь очищали препаративной ВЭЖХ [колонка: Waters Xbridge 150×25, 5 мкм; условия: 35-65% В (А: 0,05%-ный аммиак; В: CH3CN); скорость потока: 25 мл/мин]. Фракции, содержащие целевое соединение, лиофилизировали с получением соединения Примера 40 (10,2 мг, 15,5%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 4,015 мин при 10-80CD_7 мин_220&254 хроматографии (XBrige Shield RP18 2,1×50 мм), МС (ЭРИ) m/z = 615,3 [М+Н]+.
ВЭЖХ: Rt = 3,76 мин, 10-80_CD_1,2 мл. мет (XBridge Shield RP 18 2,1×50 мм, 5 мкм).
1Н ЯМР: (400 МГц, DMSO-d6) δ 10.20 (br s, 1H), 9.15-9.00 (m, 2Н), 8.29 (br d,.7=15,6 Гц, 2H), 7.26 (br s, 1H), 7.12 (br s, 1H), 6.63 (dd, J=10.0, 16,8 Гц, 1H), 6.30 (s, 1H), 6.17 (br d, J=16,8 Гц, 1H), 5.73 (br d, J=11,0 Гц, 1H), 4.01 (br d, J=9,4 Гц, 1H), 3.82-3.77 (m, 5H), 3.55-3.49 (m, 1H), 3.32-3.23 (m, 1H), 2.84 (br s, 2H), 2.29-2.18 (m, 1H), 2.02 (s, 6H), 1.91 (br d, J=9,4 Гц, 1H), 1.51 (d, J=7,2 Гц, 6H).
Пример 41
N-(5-(4-(4-хлор-5-фтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-4-метокси-2-((3aR,6aR)-5-метилгексагидропирроло[3,4-b]пиррол-1(2Н)-ил)фенил)акриламид

Методика получения соединения 41а:
В раствор 4-хлор-3-фтор анилина (15 г, 103,05 ммоль) в дихлорметане (150 мл) по каплям добавляли ICl (25 г, 154,57 ммоль). Полученную черную смесь перемешивали при 24-29°С в течение 2 ч. Смесь разбавляли 100 мл дихлорметана и промывали насыщенным раствором бикарбоната натрия (200 мл). Органический слой концентрировали с получением неочищенного остатка, который очищали колоночной флэш-хроматографией на силикагеле (от 0 до 0,5% этилацетата в петролейном эфире) с получением соединения 41а (8 г коричневого твердого вещества и 12 г черного твердого вещества, 71,5%-ный выход в сумме).
ЖХ/МС: Rt = 0,974 мин при 5-95АВ_220&254 хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 271,8 [М+Н]+.
1H ЯМР: (400 МГц, CDCl3) δ 7.53 (d, J=8,0 Гц, 1H), 6.46 (d, J=10,4 Гц, 1Н), 4.12 (s, 2H).
Методика получения соединения 41b:
В раствор соединения 41а (20 г, 73,67 ммоль) в DMF (60 мл) и метаноле (120 мл) добавляли DPPF (4,08 г, 7,37 ммоль), Et3N (31 мл, 221,03 ммоль) и Pd(OAc)2 (1,65 г, 7,37 ммоль) в атмосфере азота. Реакционную смесь продували и дегазировали СО три раза и перемешивали при 80°С в атмосфере СО (50 фунт/кв. дюйм) в течение 24 ч. Смесь фильтровали, и метанол удаляли в вакууме с получением остатка, который вливали в рассол (300 мл), и черное твердое вещество выпадало в осадок. Твердое вещество собирали фильтрованием и дополнительно очищали колоночной флэш-хроматографией на силикагеле (от 0 до 1% этилацетата в петролейном эфире) с получением соединения 41b (11,8 г, 78,6%-ный выход) в виде розового твердого вещества.
1Н ЯМР: (400 МГц, CDCl3) δ 7.83 (d, J=8,4 Гц, 1H), 6.36 (d, J=10,8 Гц, 1H), 5.80 (s, 2Н), 3.79 (s, 3Н).
Методика получения соединения 41с:
В раствор соединения 41b (6 г, 29,47 ммоль) в THF (100 мл) по каплям добавляли CH3MgBr (49,1 мл, 147,35 ммоль) при 0°С в атмосфере азота. Полученную коричневую смесь перемешивали при 24-31°С в течение 2 ч. Смесь гасили насыщенным раствором NH4Cl (300 мл) и экстрагировали этилацетатом (200 мл × 3). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали колоночной флэш-хроматографией на силикагеле (от 0 до 15% этилацетата в петролейном эфире) с получением соединения 41с (5,17 г, 84%-ный выход) в виде желтого масла.
ЖХ/МС: Rt = 0,821 мин при 5-95АВ_220&254 хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 185,9 [М+Н]+.
1Н ЯМР: (400 МГц, CDCl3) δ 7.09 (d, J=8,4 Гц, 1H), 6.42 (d, J=11,2 Гц, 1H), 4.84 (s, 2Н), 1.66 (s, 6Н).
Методика получения соединения 41d:
В раствор соединения 41с (5.17 г, 25,39 ммоль) в дихлорметане (100 мл) добавляли DIEA (6,56 г, 50,78 ммоль) и 2,4-дихлор-1,3,5-триазин (4,19 г, 27,93 ммоль). Полученный раствор перемешивали при 23-29°С в течение 3 ч. Раствор концентрировали в вакууме с получением неочищенного продукта, который очищали колоночной флэш-хроматографией на силикагеле (от 0 до 10% этилацетата в петролейном эфире) с получением соединения 41d (5,74 г, 71,3%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 2,614 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 316,7 [М+Н]+.
1Н ЯМР: (400 МГц, CDCl3) δ 10.20 (s, 1H), 8.49 (s, 1H), 8.16 (d, J=10,8 Гц, 1H), 7.22 (d, J=7,6 Гц, 1H), 2.24 (s, 1H), 1.62 (s, 6Н).
Методика получения соединения 41е:
В желтый раствор соединения 41d (4,74 г, 14,95 ммоль) в смеси n-BuOH/TFA (50 мл/0,5 мл) добавляли 4-фтор-2-метокси-5-нитроанилин (2,8 г, 15,04 ммоль). Полученную смесь перемешивали при 21-24°С в течение 1 ч, при этом цвет менялся с коричневого на желтый, и твердое вещество выпадало в осадок. Твердое вещество собирали фильтрованием и промывали петролейным эфиром (20 мл). Твердое вещество сушили в вакууме с получением соединения 41е (5,42 г, 77,7%-ный выход).
ЖХ/МС: Rt = 0,971 мин при 5-95АВ_220&254 хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 467,1 [М+Н]+.
1Н ЯМР: (400 МГц, DMSO-d6) δ 10.56 (s, 1H), 9.49 (s, 1H), 8.45 (s, 1H), 8.39 (s, 1H), 7.45-7.34 (m, 1H), 3.95 (s, 3Н), 1.53 (s, 6H).
Методика получения соединения 41f:
В раствор соединения 41е (200 мг, 0,43 ммоль) и К2СО3 (119 мг, 0,86 ммоль) в DMSO (5 мл) добавляли (3aR,6aR)-5-метилоктагидропирроло[3,4-b]пиррол (65 мг, 0,52 ммоль). Полученную смесь перемешивали при 85°С в течение 4 ч, при этом цвет менялся с бледно-желтого на насыщенный желтый. Реакционную смесь вливали в ледяную воду (50 мл), и желтое твердое вещество выпадало в осадок. Желтый осадок фильтровали и растворяли в CH2Cl2 (20 мл), затем сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением соединения 41f (230 мг, 93%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 0,755 мин при 5-95АВ_220&254.lcm хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 572,9 [М+Н]+.
Методика получения соединения 41g:
В раствор соединения 41f (230 мг, 0,40 ммоль) в МеОН (10 мл) и H2O (5 мл) добавляли Zn (131 мг, 2,00 ммоль) и NH4Cl (214 мг, 4,00 ммоль). Полученную смесь продували и дегазировали N2 3 раза, затем перемешивали при 80°С в течение 1 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением остатка, который растворяли в EtOAc (20 мл) и промывали H2O (10 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением соединения 41g (190 мг, 87%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,701 мин при 5-95АВ_1,5 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 542,9 [М+Н]+.
Методика получения соединения Примера 41:
В раствор соединения 41g (190 мг, 0,35 ммоль) и DIEA (90 мг, 0,70 ммоль) в DMF (2,5 мл) по каплям добавляли акрилоилхлорид (32 мг, 0,35 ммоль) в DMF (0,5 мл). Полученную смесь перемешивали при 0°С в бане лед-вода в течение 30 мин. Реакционную смесь очищали препаративной ВЭЖХ [колонка: Waters Xbridge 150×25, 5 мкм; условия: 40-70% В (А: 0,05% NH3H2O; В: CH3CN); скорость потока: 25 мл/мин]. Фракции, содержащие целевое соединение, лиофилизировали с получением соединения Примера 41 (74,4 мг, 36%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 1,924 мин при 10-80АВ_4 мин 220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 597,1 [М+Н]+.
ВЭЖХ: Rt = 3,30 мин при 10-80_ab_1,2 мл хроматографии (Ultimate С18 3×50 мм, 3 мкм).
1Н ЯМР: (400 МГц, CDCl3) δ 10.83 (br s, 1H), 9.91 (br s, 1H), 9.60 (br s, 1H), 8.42 (s, 1H), 8.40 (d, J=11,6 Гц, 1H), 7.70 (br s, 1H), 7.29 (s, 1H), 6.78 (s, 1H), 6.56-6.33 (m, 2H), 5.96 (br s, 1H), 5.77 (br d, J=10,8 Гц, 1H), 3.87 (s, 3H), 3.73-3.64 (m, 1H), 3.22 (br t, J=7,6 Гц, 1H), 2.93-2.79 (m, 3H), 2.72 (br d, J=10,4 Гц, 1H), 2.33 (br s, 1H), 2.30 (s, 3H), 2.25-2.16 (m, 1H), 1.95-1.86 (m, 2H), 1.78 (s, 6H).
Пример 42
(S)-N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-4-метокси-2-(метил(2-(2-метилпирр олидин-1-ил)этил)амино)фенил)акриламид

Методика получения соединения 42b:
В раствор соединения 42а (300 мг, 2,47 ммоль) в МеОН (8 мл) добавляли NaOAc (404,74 мг, 4,93 ммоль). Смесь перемешивали при 23-28°С в течение 10 мин и добавляли трет-бутилметил(2-оксоэтил)карбамат (512,76 мг, 2,96 ммоль). Полученную смесь перемешивали при 23-28°С в течение 1 ч и затем в вышеуказанную смесь добавляли NaBH3CN (310,05 мг, 4,93 ммоль). Смесь перемешивали при 23-28°С в течение 15 ч. Смесь концентрировали в вакууме с получением остатка, который растворяли в CH2Cl2 (15 мл) и промывали водой (6 мл), рассолом (6 мл), сушили над Na2SO4 и концентрировали в вакууме с получением неочищенного продукта. Его очищали колоночной хроматографией на силикагеле (0,5% МеОН в CH2Cl2) с получением соединения 42b (350 мг, 58,5%-ный выход) в виде желтого масла.
1H ЯМР: (400 МГц, CDCl3) δ 3.65-2.93 (m, 5Н), 2.89 (s, 3Н), 2.75-2.32 (m, 2Н), 2.20 (br s, 1H), 2.06-1.65 (m, 3Н), 1.46 (s, 9Н), 1.26-1.06 (m, 3Н).
Методика получения соединения 42с:
В раствор соединения 42b (350 мг, 1,44 ммоль) в CH2Cl2 (3 мл) добавляли HCl-EtOAc (4 мл). Полученную смесь перемешивали при 22-32°С в течение 12 ч. Реакционную смесь концентрировали в вакууме с получением соединения 42с (200 мг, 64%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 0,101 мин при 0-60 АВ_2 мин_50_Е.lcm хроматографии (Xtimate С18, 2,1×30 мм, 3 мкм), МС (ЭРИ) m/z = 143,1 [М+Н]+.
1H ЯМР: (400 МГц, D2O) δ 3.81-3.61 (m, 2Н), 3.60-3.33 (m, 4Н), 3.24-3.12 (m, 1Н), 2.77 (s, 3Н), 2.39-2.23 (m, 1H), 2.17-2.04 (m, 1H), 2.03-1.94 (m, 1H), 1.79-1.63 (m, 1H), 1.41 (d, J=6,4 Гц, 3Н).
Методика получения соединения 42d:
В раствор соединения 11с (200 мг, 0,428 ммоль) и K2CO3 (118 мг, 0,856 ммоль) в DMSO (3 мл) добавляли соединение 42с (111 мг, 0,514 ммоль). Реакционную смесь нагревали при 50°С в течение 3 ч, при этом цвет менялся с коричневого на оранжевый. Реакционную смесь по каплям добавляли в H2O (40 мл) в бане лед-вода при перемешивании, при этом твердое вещество выпадало в осадок, затем фильтровали. Остаток на фильтре промывали H2O (15 мл × 3), сушили в глубоком вакууме с получением соединения 42d (210 мг, 83%-ный выход) в виде оранжевого твердого вещества.
ЖХ/МС: Rt = 0,748 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 589,1 [М+Н]+.
1Н ЯМР: (400 МГц, CD3OD) δ 8.53 (br s, 1H), 8.26 (br s, 1H), 8.18 (br s, 1H), 7.21 (d, J=10,8 Гц, 1H), 6.81 (s, 1H), 3.96 (s, 3H), 3.42-3.33 (m, 2H), 3.19-3.08 (m, 2H), 2.90 (s, 3H), 2.51-2.34 (m, 2H), 2.24 (q, J=9,2 Гц, 1H), 2.04-1.91 (m, 1H), 1.81-1.70 (m, 2H), 1.60 (s, 6H), 1.40 (qd, J=8.4, 12,4 Гц, 1H), 1.12 (d, J=6,0 Гц, 3Н).
Методика получения соединения 42е:
В раствор соединения 42d (200 мг, 0,34 ммоль) в МеОН/H2O (5 мл, 5/1) добавляли Zn (133 мг, 2,04 ммоль) и NH4Cl (109 мг, 2,04 ммоль). Полученную смесь нагревали при 90°С в течение 3 ч, при этом цвет менялся с оранжевого на коричневый. Реакционную смесь фильтровали, и затем фильтрат концентрировали в вакууме с получением неочищенного остатка, который растворяли в CH2Cl2 (20 мл) и промывали водой (15 мл × 3), затем сушили над Na2SO4 и концентрировали в вакууме с получением соединения 42е (118 мг, 62%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,703 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 559,1 [М+Н]+.
Методика получения соединения Примера 42:
В раствор соединения 42е (115 мг, 0,206 ммоль) и DIEA (40 мг, 0,309 ммоль) в DMF (2 мл) по каплям добавляли акрилоилхлорид (19 мг, 0,206 ммоль) в бане лед-вода. Полученную смесь перемешивали при 5-10°С в течение 15 мин. Реакционную смесь гасили H2O (0,1 мл) и затем фильтровали, фильтрат очищали препаративной ВЭЖХ (колонка: Waters Xbridge 150×25, 5 мкм; условия: 42-72% В (А: 0,05%-ный аммиак, В: CH3CN); скорость потока: 25 мл/мин) и лиофилизировали с получением соединения Примера 42 (23,7 мг, 16,8%-ный выход) в виде не совсем белого твердого вещества.
ЖХ/МС: Rt = 4,638 мин при 30-90CD_7 мин_220&254.lcm хроматографии (Xtimate 2,1×30 мм, 3 мкм), МС (ЭРИ) m/z = 613,3 [М+Н]+.
ВЭЖХ: Rt = 4,56 мин при 10-80CD_1,2 мл хроматографии (XBridge Shield RP 18 2,1×50 мм 5 мкм).
1Н ЯМР: (400 МГц, CDCl3) δ 10.60 (br s, 1Н), 9.92 (br s, 1H), 9.73 (br s, 1H), 8.48 (d, J=7,2 Гц, 1H), 8.42 (s, 1H), 7.69 (br s, 1H), 7.10 (d, J=10,8 Гц, 1H), 6.78 (s, 1H), 6.53-6.30 (m, 2H), 6.10 (br s, 1H), 5.84-5.70 (m, 1H), 3.88 (s, 3H), 3.22-3.18 (m, 1H), 3.02-2.90 (m, 2H), 2.88-2.77 (m, 1H), 2.67 (s, 3H), 2.37-2.28 (m, 1H), 2.16-1.91 (m, 3H), 1.77 (s, 6H), 1.66 (br s, 1H), 1.49-1.40 (m, 1H), 1.36-1.20 (m, 1H), 1.04 (d, J=5,6 Гц, 3H).
Пример 43
(R)-N-(5-(4-(4-хлор-5-фтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-(2-((диметиламино)метил)пирролидин-1-ил)-4-метоксифенил)акриламид

Методика получения соединения 43а:
В раствор соединения 41е (200 мг, 0,428 ммоль) и К2СО3 (118 мг, 0,856 ммоль) в DMSO (3 мл) добавляли (R)-N,N-диметил-1-(пирролидин-2-ил)метанамин (103 мг, 0,514 ммоль). Реакционную смесь нагревали при 85°С в течение 2,5 ч, при этом цвет менялся с коричневого на оранжевый. Реакционную смесь по каплям добавляли в H2O (40 мл) в бане лед-вода при перемешивании, при этом твердое вещество выпадало в осадок, затем фильтровали. Остаток на фильтре промывали H2O (15 мл × 3), затем сушили в глубоком вакууме с получением соединения 43а (200 мг, 73,7%-ный выход) в виде оранжевого твердого вещества.
ЖХ/МС: Rt = 0,745 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 575,2 [М+Н]+.
Методика получения соединения 43b:
В раствор соединения 43а (200 мг, 0,348 ммоль) в смеси МеОН/H2O = 5/1 (5 мл) добавляли Zn (136 мг, 2,087 ммоль) и NH4Cl (112 мг, 2,087 ммоль). Полученную смесь нагревали при 90°С в течение 1,5 ч, при этом цвет менялся с оранжевого на коричневый. Реакционную смесь фильтровали, фильтрат концентрировали в вакууме с получением остатка, который растворяли в CH2Cl2 (20 мл) и промывали водой (15 мл × 3), затем сушили над Na2SO4 и концентрировали в вакууме с получением соединения 43b (175 мг, 92,3%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,725 мин при 5-95АВ_220&254.lcm хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 545,0 [М+Н]+.
Методика получения соединения Примера 43:
В раствор соединения 43b (175 мг, 0,321 ммоль) и DIEA (62 мг, 0,482 ммоль) в DMF (2 мл) добавляли акрилоилхлорид (29 мг, 0,321 ммоль) в бане лед-вода. Полученную смесь перемешивали при 5-10°С в течение 15 мин. Реакционную смесь гасили H2O (0,1 мл) и затем фильтровали, фильтрат очищали препаративной ВЭЖХ (колонка: Waters Xbridge 150×25, 5 мкм; условия: 42-72% В (А: 0,05%-ный аммиак, В: CH3CN); скорость потока: 25 мл/мин) и затем лиофилизировали с получением соединения Примера 43 (46,0 мг, 21,44%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 2,066 мин при 10-80АВ_4 мин_220&254.lcm хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 599,2 [М+Н]+.
ВЭЖХ: Rt = 4,12 мин при 10-80CD_1,2 мл хроматография (XBridge Shield RP 18 2,1×50 мм, 5 мкм).
1Н ЯМР: (400 МГц, CDCl3) δ 10.82 (br s, 1H), 10.07 (br s, 1H), 9.88 (br s, 1H), 8.41 (s, 1H), 8.39 (d, J=12,0 Гц, 1H), 7.70 (br s, 1H), 7.32-7.25 (m, 1H), 6.71 (s, 1H), 6.44-6.28 (m, 2H), 6.03 (br s, 1H), 5.84-5.71 (m, 1H), 3.86 (s, 3H), 3.39-3.25 (m, 2H), 3.03-2.92 (m, 1H), 2.37 (dd, J=7.8, 12,0 Гц, 1H), 2.33-2.21 (m, 1H), 2.19 (s, 6H), 2.06 (dd, J=5.4, 12,0 Гц, 1H), 2.02-1.92 (m, 2H), 1.78 (s, 6H), 1.70-1.65 (m, 1H).
Пример 44
(S)-N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-4-метокси-2-(метил((1-метилпирролидин-2-ил)метил)амино)фенил)акриламид

Методика получения соединения 44b:
В раствор соединения 36b (180 мг, 0,39 ммоль), K2CO3 (216 мг, 1,56 ммоль) в DMSO (10 мл) добавляли соединение 44а (117 мг, 0,58 ммоль). Полученную смесь перемешивали при 50°С в течение 12 ч. Реакционную смесь объединяли с предыдущей партией и по каплям добавляли в Н2О (100 мл) в бане лед-вода при перемешивании. Выпавшее в осадок твердое вещество отфильтровывали, и остаток на фильтре растворяли в CH2Cl2 (15 мл × 3), затем сушили и концентрировали в вакууме с получением целевого соединения 44b (220 мг, средний выход 89,0%) в виде оранжевого твердого вещества.
ЖХ/МС: Rt = 0,738 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 575,1 [M+Na]+.
1Н ЯМР (400 МГц, CDCl3) δ 9.64 (s, 1H), 9.04 (s, 1H), 8.40 (br s, 1H), 8.38-8.30 (m, 1Н), 7.42 (br s, 1H), 7.08 (d, J=10,4 Гц, 1H), 6.68 (s, 1H), 3.95 (s, 3H), 3.50 (dd, J=4.8, 13,6 Гц, 1H), 3.17-3.05 (m, 2H), 2.86 (s, 3H), 2.63 (s, 2H), 2.59-2.52 (m, 1H), 2.42 (s, 3H), 2.30-2.21 (m, 1H), 2.07-1.96 (m, 1H), 1.80-1.74 (m, 1H), 1.70 (s, 6H).
Методика получения соединения 44c:
В раствор соединения 44b (220 мг, 0,38 ммоль), Zn (125 мг, 1,9 ммоль) в 6 мл смеси метанол/вода = 5:1 (об./об.) добавляли NH4Cl (102 мг, 1,9 ммоль). Полученную смесь перемешивали при 75°С в течение 2 ч. Реакционную смесь фильтровали и концентрировали под вакуумом с получением остатка, который обрабатывали водой (10 мл) и экстрагировали CH2Cl2 (15 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением соединения 44с (90 мг, 43,4%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,701 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 545,1 [М+Н]+.
Методика получения соединения Примера 44:
В раствор соединения 44с (90 мг, 0,17 ммоль) и DIEA (33 мг, 0,26 ммоль) в DMF (2,5 мл) по каплям добавляли акрилоилхлорид (31 мг, 0,34 ммоль) в DMF (0,5 мл). Полученную смесь перемешивали при 0°С (баня лед-вода) в течение 20 мин. Реакционную смесь гасили тремя каплями воды и затем напрямую очищали препаративной ВЭЖХ (колонка: Waters Xbridge 150×25, 5 мкм: 53-83% В (А: вода (0,05%-ный аммиак гидроксид об./об.), В: CH3CN), скорость потока: 25 мл/мин) с получением соединения Примера 44 (11,1 мг, 10,9%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 1,994 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм) МС (ЭРИ) m/z = 599,1 [М+Н]+.
ВЭЖХ: Rt = 4,42 мин при 10-80_CD_1,2 мл хроматографии (XBridge Shield RP 18 2,1×50 мм, 5 мкм).
1H ЯМР (400 МГц, CDCl3) δ 10.53 (br s, 1H), 10.10 (br s, 1H), 10.03 (br s, 1H), 8.47 (d, J=7,2 Гц, 1H), 8.41 (s, 1H), 7.64 (br s, 1H), 7.10 (d, J=10,8 Гц, 1H), 6.71 (s, 1H), 6.43-6.32 (m, 2H), 6.12 (br s, 1H), 5.81-5.74 (m, 1H), 3.88 (s, 3H), 3.14-3.08 (m, 1H), 2.88-2.78 (m, 1H), 2.74 (s, 3H), 2.71-2.63 (m, 2H), 2.56 (s, 3H), 2.41-2.32(m, 1H), 2.06-1.90 (m, 1H), 1.81-1.72 (m, 7H), 1.45-1.23 (m, 2H).
Пример 45
N-(5-(4-(4-циклопропил-5-фтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-4-метокси-2-((3aR,6aR)-5-метилгексагидропирроло[3,4-b]пиррол-1(2Н)-ил)фенил)акриламид

Методика получения соединения 45b:
В раствор соединения 34е (150 мг, 0,32 ммоль) и К2СО3 (88 мг, 0,64 ммоль) в DMSO (5 мл) добавляли соединение 45а (48 мг, 0,38 ммоль). Полученную смесь перемешивали при 85°С в течение 4 ч, при этом цвет менялся с бледно-желтого на насыщенный желтый. Реакционную смесь вливали в воду (50 мл), и желтое твердое вещество выпадало в осадок. Твердое вещество отфильтровывали, и остаток на фильтре растворяли в CH2Cl2 (20 мл), затем сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением соединения 45b (150 мг, 82%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 0,834 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z = 579,3 [М+Н]+.
Методика получения соединения 45с:
В раствор соединения 45b (150 мг, 0,26 ммоль) в МеОН (10 мл) и Н20 (5 мл) добавляли Zn (85 мг, 1,30 ммоль) и NH4Cl (139 мг, 2,60 ммоль). Полученную смесь продували и дегазировали N2 3 раза, затем перемешивали при 80°С в течение 2 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении, затем экстрагировали EtOAc (10 мл × 2). Объединенные органические слои промывали Н2О (10 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением соединения 45с (130 мг, 91%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,715 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 549,1 [М+Н]+.
Методика получения соединения Примера 45:
В раствор соединения 45с (130 мг, 0,24 ммоль) и DIEA (62 мг, 0,48 ммоль) в DMF (2,5 мл) по каплям добавляли акрилоилхлорид (22 мг, 0,24 ммоль) в DMF (0,5 мл). Полученную смесь перемешивали при 0°С в бане лед-вода в течение 30 мин. Реакционную смесь очищали препаративной ВЭЖХ [колонка: Waters Xbridge 150×25 5 мкм; условия: 35-65% В (А: 0,05% NH3H2O; В: CH3CN); скорость потока: 25 мл/мин]. Фракции, содержащие целевое соединение, лиофилизировали с получением соединения Примера 45 (28,1 мг, 19%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 1,778 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 603,3 [М+Н]+.
ВЭЖХ: Rt = 2,79 мин при 10-80_ab_1,2 мл хроматографии (Ultimate С18 3×50 мм 3 мкм).
1Н ЯМР: (400 МГц, CDCl3) δ 10.72 (br s, 1H), 9.93 (br s, 1H), 9.56 (br s, 1H), 8.41 (s, 1H), 8.12 (d, J=12,4 Гц, 1H), 7.64 (br s, 1H), 6.86 (d, J=8,4 Гц, 1H), 6.78 (s, 1H), 6.55-6.45 (m, 1H), 6.44-6.35 (m, 1H), 5.76 (br d, J=11,2 Гц, 1H), 3.87 (s, 3H), 3.67 (br s, 1H), 3.22 (br t, J=7,6 Гц, 1H), 2.96-2.79 (m, 3H), 2.73 (br d, 7=10,0 Гц, 1H), 2.30 (br s, 4H), 2.25-2.17 (m, 1H), 2.08-1.94 (m, 2H), 1.89 (br s, 1H), 1.76 (s, 6H), 1.05-0.88 (m, 2H), 0.75-0.55 (m, 2H).
Пример 46
(R)-N-(5-(4-(4-циклопропил-5-фтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-(2-((диметиламино)метил)пирролидин-1-ил)-4-метоксифенил)акриламид

Методика получения соединения 46b:
В раствор соединения 34е (150 мг, 0,32 ммоль) и К2СО3 (88 мг, 0,64 ммоль) в DMSO (5 мл) добавляли соединение 46а (96 мг, 0,48 ммоль). Полученную смесь перемешивали при 85°С в течение 4 ч, при этом цвет менялся с бледно-желтого на насыщенный желтый. Реакционную смесь вливали в ледяную воду (50 мл), и желтое твердое вещество выпадало в осадок. Желтое твердое вещество отфильтровывали, и остаток на фильтре разбавляли CH2Cl2 (20 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением соединения 46b (140 мг, 75%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 0,766 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z = 581,1 [М+Н]+.
Методика получения соединения 46с:
В раствор соединения 46b (140 мг, 0,24 ммоль) в МеОН (5 мл) добавляли Pd/C (15 мг). Полученную смесь продували и дегазировали Н2 3 раза, затем перемешивали при 21-24°С в атмосфере Н2 (15 фунт/кв. дюйм) в течение 2 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением соединения 46с (130 мг, 98%-ный выход) в виде коричневого масла.
ЖХ/МС: Rt = 0,738 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 551,1 [М+Н]+.
Методика получения соединения Примера 46:
В раствор соединения 46с (130 мг, 0,24 ммоль) и DIEA (62 мг, 0,48 ммоль) в DMF (2,5 мл) по каплям добавляли акрилоилхлорид (22 мг, 0,24 ммоль) в DMF (0,5 мл). Полученную смесь перемешивали при 0°С в бане лед-вода в течение 30 мин.
Реакционную смесь очищали препаративной ВЭЖХ [колонка: Waters Xbridge 150×25, 5 мкм; условия: 42-72% В (А: 0,05% NH3H2O; В: CH3CN); скорость потока: 25 мл/мин]. Фракции, содержащие целевое соединение, лиофилизировали с получением соединения Примера 46 (32,5 мг, 22%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 1,883 мин при 10-80АВ4 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 605,3 [М+Н]+.
ВЭЖХ: Rt = 3,00 мин при 10-80_ab_1,2 мл хроматографии (Ultimate С18 3×50 мм, 3 мкм).
1Н ЯМР: (400 МГц, CDCl3) δ 10.71 (br s, 1H), 10.00 (br s, 1H), 9.91 (br s, 1H), 8.41 (s, 1H), 8.11 (d, J=12,4 Гц, 1H), 7.65 (br s, 1H), 6.86 (d, J=8,4 Гц, 1H), 6.70 (s, 1H), 6.44-6.30 (m, 2H), 5.77 (br d, J=11,2 Гц, 1H), 3.86 (s, 3H), 3.39-3.24 (m, 2H), 3.03-2.90 (m, 1H), 2.41-2.31 (m, 1H), 2.25-2.15 (m, 7H), 2.10-1.96 (m, 4H), 1.76 (s, 6H), 1.69 (br d, J=4,4 Гц, 1H), 0.98-0.88 (m, 2H), 0.73-0.66 (m, 2H).
Пример 47
N-(5-(4-(4,5-дифтор-2-(2-гидроксибутан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-4-метокси-2-((3aR,6aR)-5-метилгексагидропирроло[3,4-b]пиррол-1(2Н)-ил)фенил)акриламид

Методика получения соединения 47с:
В смесь соединения 47а (10 г, 48,08 ммоль) и соединения 47b (20,84 г, 57,69 ммоль) в толуоле (200 мл) добавляли Pd(PPh3)4 (2,78 г, 2,4 ммоль) одной порцией в атмосфере N2. Полученную черную смесь перемешивали при 100°С в течение 12 ч в атмосфере N2. Реакционную смесь охлаждали до 25°С и затем обрабатывали 6 н. HCl (10 мл) при перемешивании при 25°С в течение 1 ч. Смесь разбавляли водой (400 мл) и экстрагировали EtOAc (120 мл × 3). Объединенные органические слои промывали последовательно 20%-ным раствором KF (200 мл) и рассолом (100 мл), сушили над сульфатом натрия и концентрировали в вакууме с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (5% EtOAc в петролейном эфире) с получением соединения 47с (4,2 г, 51%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 0,709 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 171,9 [М+Н]+.
1Н ЯМР: (400 МГц, CDCl3) δ 7.50 (dd, J=8,8 Гц, 11,2 Гц, 1H), 6.41 (dd, J=6,8 Гц, 12,0 Гц, 1H), 6.37-6.17 (m, 2Н), 2.52 (s, 3Н).
Методика получения соединения 47d:
В раствор соединения 47с (2 г, 11,68 ммоль) в THF (100 мл) добавляли EtMgBr (3,0 М) (16 мл, 46,72 ммоль) при 0°С в атмосфере N2. Реакционную смесь перемешивали при 23-27°С в течение 2 ч. Реакционную смесь гасили насыщенным раствором NH4Cl (50 мл) и экстрагировали EtOAc (30 мл × 3). Объединенные органические слои сушили над Na2SO4, фильтровали, и фильтрат концентрировали при пониженном давлении с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (0-30% EtOAc в петролейном эфире (об./об.)) с получением соединения 47d (1,7 г, 72%-ный выход) в виде желтого масла.
ЖХ/МС: Rt = 0,559 мин при 5-95АВ_220&254.lcm хроматографии MERCK RP18 2,5-2 мм, МС (ЭРИ) m/z = 184,0 [М+Н-18]+.
1Н ЯМР: (400 МГц, MeOD-d4) δ 6.91 (dd, J=8,8 Гц, 12,8 Гц, 1H), 6.51 (dd, J=7,6 Гц, 12,8 Гц, 1H), 2.07-2.01 (m, 1H), 1.86-1.76 (m, 1Н), 1.53 (s, 3Н), 0.80 (t, J=7,6 Гц, 3Н).
Методика получения соединения 47е:
В раствор соединения 47d (1,68 г, 8,4 ммоль) в CH2Cl2 (15 мл) добавляли DIEA (1,6 г, 12,6 ммоль) и 2,4-дихлорпиримидин (1,5 г, 10,0 ммоль). Полученную смесь перемешивали при 22-30°С в течение 0,5 ч. Реакционную смесь концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле (0-20% EtOAc в петролейном эфире) с получением соединения 47е (1,58 г, 60%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 0,825 мин при 5-95AB_220&254.lcm хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 314,9 [М+Н]+.
Методика получения соединения 47f:
В раствор соединения 47е (1,58 г, 5,0 ммоль) и 4-фтор-2-метокси-5-нитроанилина (935 мг, 5,0 ммоль) в n-BuOH (10 мл) добавляли TFA (0,1 мл). Полученную смесь перемешивали при 18-25°С в течение 2 ч, при этом серое твердое вещество выпадало в осадок. Реакционную смесь фильтровали, и остаток на фильтре собирали и сушили при пониженном давлении с получением соединения 47f (1,5 г, 97%-ный выход) в виде палевого твердого вещества.
ЖХ/МС: Rt = 0,911 мин при 5-95АВ_220&254.lcm хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 464,9 [М+Н]+.
1Н ЯМР: (400 МГц, DMSO-d6) δ 10.55 (br s, 1H), 9.46 (br s, 1H), 8.46 (br s, 1H), 8.37 (s, 1H), 7.39 (d, J=13,6 Гц, 1H), 7.33-7.29 (m, 1H), 7.18 (s, 1H), 7.05 (s, 1H), 3.94 (s, 3H), 1.82-1.70 (m, 2H), 1.51 (s, 3H), 0.71 (brt, J=7,6 Гц, 3Н).
Методика получения соединения 47h:
В раствор соединения 47f (150 мг, 0,32 ммоль) и К2СО3 (88 мг, 0,64 ммоль) в DMSO (3 мл) добавляли соединение 47g (48 мг, 0,38 ммоль). Полученную смесь перемешивали при 85°С в течение 2 ч, при этом цвет менялся с бледно-желтого на насыщенный желтый. Реакционную смесь вливали в воду (50 мл), и выпавшее в осадок желтое твердое вещество отфильтровывали, остаток на фильтре растворяли в CH2Cl2 (20 мл), затем сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением соединения 47h (160 мг, 88%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 0,766 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z = 571,0 [М+Н]+.
Методика получения соединения 47i:
В раствор соединения 47h (160 мг, 0,28 ммоль) в МеОН (5 мл) добавляли Pd/C (16 мг). Полученную смесь продували и дегазировали Н2 3 раза, затем перемешивали при 22-30°С в атмосфере Н2 (водородный баллон, 15 фунт/кв. дюйм) в течение 1 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединение 47i (140 мг, 92%-ный выход) в виде бесцветного твердого вещества.
ЖХ/МС: Rt = 0,732 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 541,0 [М+Н]+.
Методика получения соединения Примера 47:
В раствор соединения 47i (140 мг, 0,26 ммоль) и DIEA (67 мг, 0,52 ммоль) в DMF (2,5 мл) по каплям добавляли акрилоилхлорид (24 мг, 0,26 ммоль) в DMF (0,5 мл). Полученную смесь перемешивали при 0°С в бане лед-вода в течение 30 мин. Реакционную смесь очищали препаративной ВЭЖХ [колонка: Waters Xbridge 150×25, 5 мкм; условия: 45-75% В (А: 0,05% NH3H2O; В: CH3CN); скорость потока: 25 мл/мин]. Фракции, содержащие целевое соединение, лиофилизировали с получением соединения Примера 47 (50,6 мг, 33%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 1,855 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 595,1 [М+Н]+.
ВЭЖХ: Rt = 2,69 мин при 10-80_ab_1,2 мл хроматографии (Ultimate С18 3×50 мм 3 мкм).
1Н ЯМР: (400 МГц, CDCl3) δ 10.91 (br s, 1Н), 9.94 (br s, 1H), 9.57 (br s, 1H), 8.45-8.35 (m, 2H), 7.69 (br s, 1H), 7.03 (dd, J=8.8, 12,4 Гц, 1H), 6.79 (s, 1H), 6.49-6.33 (m, 2H), 5.78-5.73 (m, 1H), 3.96-3.81 (m, 3H), 3.62 (br dd, J=4.4, 7,6 Гц, 1H), 3.19 (brt, J=7,6 Гц, 1H), 2.98-2.76 (m, 3H), 2.70 (br d, J=10,4 Гц, 1H), 2.36-2.26 (m, 4H), 2.26-2.10 (m, 2H), 2.09-2.00 (m, 1H), 1.89 (br dd, J=4.0, 10,2 Гц, 1H), 1.86-1.78 (m, 1H), 1.70 (s, 3H), 0.89 (td, J=7.2, 11,2 Гц, 3H).
Пример 48
N-(5-(4-(4,5-дифтор-2-(2-гидроксибутан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-((R)-3-(диметиламино)пирролидин-1-ил)-4-метоксифенил)акриламид

Пример 48 Методика получения соединения 48а:
В раствор соединения 48а (200 мг, 0,43 ммоль) и K2CO3 (119 мг, 0,86 ммоль) в DMSO (5 мл) добавляли соединение (R)-N,N-диметилпирролидин-3-амин (60 мг, 0,52 ммоль). Полученную смесь перемешивали при 23-29°С в течение 0,5 ч. Реакционную смесь по каплям добавляли в Н2О (100 мл) в бане лед-вода при перемешивании. Выпавшее в осадок твердое вещество отфильтровывали, и остаток на фильтре растворяли в CH2Cl2 (45 мл), затем сушили и концентрировали в вакууме с получением соединения 48b (230 мг, 96%-ный выход) в виде оранжевого твердого вещества.
ЖХ/МС: Rt = 0,722 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 559,2 [М+Н]+.
Методика получения соединения 48b:
К раствору 48а (230 мг, 0,41 ммоль) в МеОН (10 мл) добавляли Pd/C (35 мг). Полученную смесь продували и дегазировали Н2 3 раза, затем перемешивали при 23-30°С в атмосфере Н2 (15 фунт/кв.дюйм) в течение 0,5 ч. Реакционную смесь фильтровали, и фильтрат концентрировали при пониженном давлении с получением соединения 48b (205 мг, 94%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,680 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 529,2 [М+Н]+.
Методика получения соединения Примера 48:
В раствор соединения 48b (205 мг, 0,4 ммоль) и DIEA (78 мг, 0,4 ммоль) в DMF (2,5 мл) по каплям добавляли акрилоилхлорид (37 мг, 0,4 ммоль) в DMF (0,5 мл). Полученную смесь перемешивали при 0°С в бане лед-вода в течение 20 мин. Реакционную смесь гасили тремя каплями воды и затем напрямую очищали препаративной ВЭЖХ (колонка: Waters Xbridge 150×25, 5 мкм, 40-70% В (А: вода (0,05%-ный аммиак гидроксид об./об.), В: CH3CN), скорость потока: 25 мл/мин) с получением соединения Примера 48 (65,1 мг, 28%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 1,795 мин при 10-80CD_4 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм, МС (ЭРИ) m/z = 583,1 [М+Н]+.
ВЭЖХ: Rt = 3,45 мин при 10-80_CD_1,2 мл хроматографии (XBridge Shield RP 18 2,1×50 мм, 5 мкм).
1Н ЯМР: (400 МГц, CDCl3) δ 10.84 (br s, 1Н), 9.78 (br s, 1Н), 8.43-8.35 (m, 2Н), 7.64 (br s, 1Н), 7.02 (dd, J=8,4 Гц, 12,0 Гц, 1Н), 6.76 (s, 1Н), 6.37-6.32 (m, 2Н), 5.82-5.77 (m, 1Н), 3.87 (s, 3Н), 3.16-3.04 (m, 4Н), 2.94-2.85 (m, 1Н), 2.30 (s, 6Н), 2.24-2.08 (m, 2Н), 2.05-1.90 (m, 2Н), 1.69 (s, 3Н), 0.92-0.86 (m, 3Н).
Пример 49
N-(5-(4-(4,5-дифтор-2-(2-гидроксибутан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид

Пример 49 Методика получения соединения 49а:
В раствор соединения 47f (200 мг, 0,43 ммоль) и K2CO3 (119 мг, 0,86 ммоль) е DMSO (5 мл) добавляли соединение N1,N1,N2-триметилэтан-1,2-диамин (53 мг, 0,52 ммоль). Полученную смесь перемешивали при 18-25°С в течение 0,5 ч. Реакционную смесь по каплям добавляли в Н2О (50 мл) в бане лед-вода при перемешивании. Выпавшее в осадок твердое вещество отфильтровывали, и остаток на фильтре растворяли в CH2Cl2 (50 мл), сушили и концентрировали при пониженном давлении с получением соединения 49а (230 мг, 97%-ный выход) в виде оранжевого твердого вещества.
ЖХ/МС: Rt = 0,778 мин при 5-95АВ_220&254.lcm хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 547,0 [М+Н]+.
Методика получения соединения 49b:
В раствор соединения 49а (230 мг, 0,42 ммоль) в МеОН (10 мл) добавляли Pd/C (35 мг). Полученную смесь продували и дегазировали Н2 3 раза, затем перемешивали при 18-20°С в атмосфере Н2 (15 фунт/кв.дюйм) в течение 0,5 ч. Реакционную смесь фильтровали, и фильтрат концентрировали при пониженном давлении с получением соединения 49b [150 мг, 69%-ный выход) в виде бордового твердого вещества.
ЖХ/МС: Rt = 0,691 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP18 2,5-2 и), МС (ЭРИ) m/z = 517,2 [М+Н]+.
1Н ЯМР: (400 МГц, MeOH-d4) δ 8.20 (br s, 2Н), 7.16 (dd, J=8,8 Гц, 12,0 Гц, 1Н), 6.83 (s, 1H), 3.80 (s, 3H), 3.07-3.01 (m, 2H), 2.65 (s, 3H), 2.48 (t, J=7,2 Гц, 2H), 2.27 (s, 6H), 1.90-1.83 (m, 2H), 1.58 (s, 3H), 0.81 (t, J=7,2 Гц, 3H).
Методика получения соединения Примера 49:
В раствор соединения 49b (150 мг, 0,29 ммоль) и DIEA (56 мг, 0,44 ммоль) в DMF (2,5 мл) по каплям добавляли акрилоилхлорид (26 мг, 029 ммоль) в DMF (0,5 мл). Полученную смесь перемешивали при 0°С в бане лед-вода в течение 20 мин. Реакционную смесь гасили тремя каплями воды и напрямую очищали препаративной ВЭЖХ (колонка: Xbridge ВЕН С18, 250×50 мм, 10 мкм; условия: 44-84% В (А: вода (0,04% NH3H2O +10 мМ NH4HCO3)), В: CH3CN), скорость потока: 25 мл/мин) с получением соединения Примера 49 (31,3 мг, 20%-ный выход) в виде бледно-желтого твердого вещества.
ЖХ/МС: Rt = 1,877 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 571,1 [М+Н]+.
ВЭЖХ: Rt = 2,72 мин при 10-80_CD_1,2 мл хроматографии (Ultimate С18 3×50 мм 3 мкм).
1Н ЯМР: (400 МГц, CDCl3) δ 10.91 (br s, 1Н), 10.42 (br s, 1Н), 9.94 (br s, 1Н), 8.40 (s, 1Н), 8.39-8.33 (m, 1Н), 7.70 (br s, 1Н), 7.02 (dd, J=8,4 Гц, 12,0 Гц, 1Н), 6.78 (s, 1Н), 6.44-6.26 (m, 2Н), 5.93 (br s, 1Н), 5.78-5.72 (m, 1Н), 3.88 (s, 3Н), 2.87 (br t, J=4,8 Гц, 2H), 2.70 (s, 3Н), 2.35-2.20 (m, 8Н), 2.19-2.09 (m, 1Н), 2.07-1.99 (m, 1Н), 1.70 (s, 3Н), 0.88 (t, J=7,6 Гц, 3Н).
Пример 50
N-(5-(4-(4,5-дифтор-2-(2-гидроксибутан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-4-метокси-2-((3aS,6aS)-5-метилгексагидропирроло[3,4-b]пиррол-1(2Н)-ил)фенил)акриламид
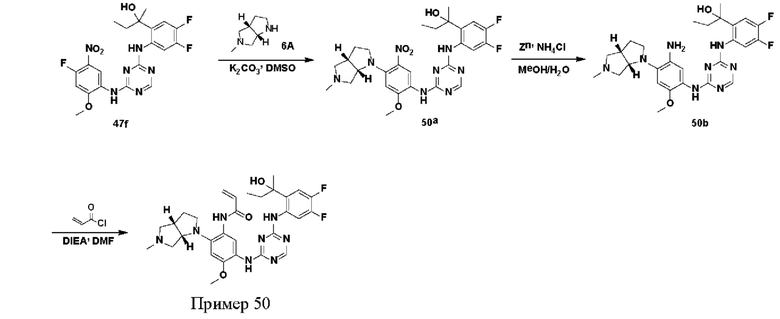
Пример 50 Методика получения соединения 50а:
В смесь соединения 47f (150 мг, 0,32 ммоль) в DMSO (3 мл) добавляли K2CO3 (134 мг, 0,97 ммоль) и (3aS,6aS)-5-метилоктагидропирроло[3,4-b]пиррол (60 мг, 0,48 ммоль). Полученную смесь перемешивали при 50°С в течение 12 ч, затем при 80°С в течение 2 ч. Реакционную смесь вливали в воду (15 мл), при этом оранжевое твердое вещество выпадало в осадок. Твердое вещество собирали фильтрованием под разряжением и сушили в вакууме с получением соединения 50а (190 мг, 91,7%-ный выход).
ЖХ/МС: Rt = 2,019 мин при 10-80АВ_4 мин 220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 571,1 [М+Н]+.
1Н ЯМР: (400 МГц, CDCl3) δ 9.83 (br. s, 1Н), 8.45-8.05 (m, 2H), 7.29 (br. s, 1H), 7.02-6.88 (m, 1H), 6.33 (s, 1H), 4.39-4.31 (m, 1H), 3.89 (s, 3H), 3.54-3.41 (m, 1H), 3.19-3.10 (m, 1H), 3.02-2.92 (m, 1H), 2.63-2.55 (m, 1H), 2.48-2.42 (m, 1H), 2.40-2.34 (m, 1H), 2.24-2.18 (m, 1H), 2.15 (d, J=2,4 Гц, 3Н), 2.07-1.99 (m, 1H), 1.90-1.87 (m, 1H), 1.82-1.76 (m, 2H), 1.59 (s, 3H), 0.81 (t, J=7,2 Гц, 3Н).
Методика получения соединения 50b:
В желтый раствор соединения 50а (190 мг, 0,33 ммоль) в смеси метанол/вода (5 мл/1 мл) добавляли NH4Cl (125 мг, 2,33 ммоль) и Zn (109 мг, 1,66 ммоль). Полученную смесь перемешивали при 90°С в течение 1 ч. Реакционную смесь вливали в воду (15 мл) и экстрагировали смесью дихлорметан/метанол (3/1, 10 мл × 4). Объединенные органические слои сушили и концентрировали в вакууме с получением соединения 50b (170 мг, 95,2%-ный выход) в виде черного твердого вещества.
ЖХ/МС: Rt = 1,526 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 541,2 [М+Н]+.
1Н ЯМР: (400 МГц, CDCl3) δ 9.86 (br. s, 1Н), 8.29-8.15 (m, 2Н), 7.78-7.42 (m, 2Н), 7.01-6.88 (m, 1Н), 6.62 (s, 1Н), 4.06-3.96 (m, 1Н), 3.74 (s, 3Н), 3.42-3.32 (m, 1Н), 2.88-2.79 (m, 1Н), 2.77-2.68 (m, 1Н), 2.63-2.55 (m, 1Н), 2.54-2.48 (m, 1Н), 2.47-2.38 (m, 1Н), 2.22 (s, 3Н), 2.15-2.07 (m, 1H), 2.06-1.97 (m, 1H), 1.85 (q, J=7,2 Гц, 2H), 1.77-1.73 (m, 1H), 1.57 (s, 3H), 0.79 (t, J=7,2 Гц, 3Н).
Методика получения соединения Примера 50:
В раствор соединения 50b (170 мг, 0,31 ммоль) в DMF (3 мл) добавляли DIEA (81 мг, 0,63 ммоль), затем добавляли акрилоилхлорид (28 мг, 0,31 ммоль) при 0°С за три раза и затем перемешивали в течение 2 ч. Смесь гасили 3 каплями воды и очищали препаративной ВЭЖХ (колонка: Waters Xbridge 150×25, 5 мкм, условия: 45%-75% В (А: вода/10 мМ NH4HCO3, В: CH3CN), скорость потока: 25 мл/мин) с получением соединения Примера 50 (39,8 мг, 21,6%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 1,697 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 595,1 [М+Н]+.
ВЭЖХ: Rt = 3,94 мин при 10-80_CD_1,2 мл хроматографии (XBridge Shield RP 18 2,1×50 мм, 5 мкм).
1Н ЯМР (400 МГц, CDCl3) δ 10.84 (br. s, 1Н), 9.87 (s, 1Н), 9.49 (s, 1Н), 8.37-8.25 (m, 2H), 7.60 (br. s, 1H), 6.95 (dd, J=8.8, 12,4 Гц, 1H), 6.71 (s, 1H), 6.46-6.22 (m, 1H), 5.68 (d, J=11,6 Гц, 1H), 3.79 (s, 3H), 3.60-3.53 (m, 1H), 3.17-3.08 (m, 1H), 2.88-2.78 (m, 2H), 2.77-2.72 (m, 1H), 2.68-2.60 (m, 1H), 2.28-2.24 (m, 1H), 2.22 (s, 3H), 2.18-2.03 (m, 2H), 2.00-1.89 (m, 1H), 1.85-1.72 (m, 2H), 1.65 (s, 3H), 0.86-0.77 (m, 3H).
Пример 51
(R)-N-(5-(4-(4-хлор-5-фтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-(3-(диметиламино)пирролидин-1-ил)-4-метоксифенил)акриламид

Методика получения соединения 51а:
В смесь соединения 41е (200 мг, 0,43 ммоль) в DMSO (5 мл) добавляли K2CO3 (178 мг, 1,29 ммоль) и (R)-N,N-диметилпирролидин-3-амин (59 мг, 0,51 ммоль). Полученную оранжевую смесь перемешивали при 23-29°С в течение 4 ч. Смесь вливали в воду (25 мл), и выпавшее в осадок оранжевое твердое вещество собирали фильтрованием под разряжением. Твердое вещество сушили в вакууме с получением соединения 51а (230 мг, 95%-ный выход).
ЖХ/МС: Rt = 0,778 мин при 5-95АВ_220&254 хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 583,0 [M+Na]+.
1Н ЯМР: (400 МГц, CDCl3) δ 10.06 (br. s, 1H), 8.92 (br. s, 1H), 8.46-8.25 (m, 2H), 7.35 (br. s, 1H), 7.26 (s, 1H), 6.31 (s, 1H), 3.95 (s, 3H), 3.61-3.52 (m, 1H), 3.39-3.32 (m, 1H), 3.25-3.12 (m, 2H), 2.89-2.78 (m, 1H), 2.31 (s, 6H), 2.02-1.81 (m, 2H), 1.71 (d, J=5,2 Гц, 6H).
Методика получения соединения 51b:
В желтый раствор соединения 51а (230 мг, 0,41 ммоль) в смеси метанол/вода (5 мл/1 мл) добавляли NH4Cl (154 мг, 2,87 ммоль) и Zn (134 мг, 2,05 ммоль). Полученную смесь перемешивали при 90°С в течение 1 ч. Смесь вливали в воду (20 мл) и экстрагировали дихлорметаном (15 мл × 5). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением соединения 51b (180 мг, 82,7%-ный выход) в виде зеленого твердого вещества.
ЖХ/МС: Rt = 0,713 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 531,0 [М+Н]+.
1Н ЯМР: (400 МГц, CDCl3) δ 9.86 (br. s, 1Н), 8.30-8.16 (m, 2Н), 7.79-7.39 (m, 2Н), 7.18 (s, 1Н), 6.59 (s, 1Н), 3.75 (s, 3Н), 3.16-2.98 (m, 4H), 2.95-2.84 (m, 1H), 2.29 (s, 6H), 1.88-1.79 (m, 2H), 1.61 (s, 6H).
Методика получения соединения Примера 51:
В раствор соединения 51b (160 мг, 0,3 ммоль) в DMF (2 мл) добавляли DIEA (78 мг, 0,6 ммоль) и перемешивали при 0°С, затем добавляли акрилоилхлорид (27 мг, 0,3 ммоль) при 0°С за три раза и перемешивали в течение 0,5 ч. Реакционную смесь объединяли с реакционной смесью предыдущей партии и очищали препаративной ВЭЖХ (колонка: Waters Xbridge 150×25, 5 мкм, условие: 42%-72% В (А: вода/10 мМ NH4HCO3, В: CH3CN), скорость потока: 25 мл/мин) с получением соединения Примера 51 (91,4 мг, 46%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 1,660 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 585,2 [М+Н]+.
ВЭЖХ: Rt = 3,46 мин при 10-80_CD_1,2 мл хроматографии (XBridge Shield RP 18 2,1×50 мм, 5 мкм).
1Н ЯМР (400 МГц, CDCl3) δ 10.67 (br. s, 1Н), 9.63 (s, 1Н), 8.53 (s, 1Н), 8.38-8.26 (m, 2H), 7.57 (s, 1H), 7.22-7.16 (m, 1H), 6.63 (s, 1H), 6.51-6.38 (m, 1H), 6.35-6.25 (m, 1H), 5.73 (d, J=10,0 Гц, 1H), 3.79 (s, 3H), 3.15-3.06 (m, 2H), 3.04-2.96 (m, 2H), 2.94-2.85 (m, 1H), 2.30 (s, 6H), 2.15-2.10 (m, 1H), 1.99-1.94 (m, 1H), 1.68 (s, 6H).
Пример 52
(R)-N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)пиримидин-2-иламино)-2-(3-(диметиламино)пирролидин-1-ил)-4-метоксифенил)акриламид

Методика получения соединения 52a:
В раствор соединения 36b (200 мг, 0,429 ммоль) и K2CO3 (119 мг, 0,858 ммоль) в DMSO (3 мл) добавляли (R)-N,N-диметилпирролидин-3-амин (59 мг, 0,515 ммоль). Реакционную смесь перемешивали при 22-30°С в течение 4 ч, затем при 50°С в течение 1 ч, при этом цвет менялся с коричневого на насыщенный зеленый. Реакционную смесь по каплям добавляли в Н2О (40 мл) в бане лед-вода. Выпавшее в осадок твердое вещество собирали фильтрованием и промывали Н2О (15 мл × 3). Остаток на фильтре растворяли в CH2Cl2 (20 мл), сушили над Na2SO4 и концентрировали в вакууме с получением соединения 52а (210 мг, 87,4%-ный выход) в виде оранжевого твердого вещества.
ЖХ/МС: Rt = 0,677 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 559,9 [М+Н]+.
1Н ЯМР (400 МГц, Метанол-d4) δ 8.42 (s, 1Н), 7.99 (d, J=7,6 Гц, 1Н), 7.96 (d, J=5,6 Гц, 1Н), 7.24 (d, J=10,8 Гц, 1Н), 6.50 (s, 1Н), 6.15 (d, J=5,6 Гц, 1Н), 3.96 (s, 3Н), 3.54 (dt, J=6.4, 10,4 Гц, 1Н), 3.36 (t, J=9,2 Гц, 1Н), 3.28-3.24 (m, 1Н), 3.09 (dd, J=6.8, 10,0 Гц, 1Н), 2.93-2.81 (m, 1Н), 2.33 (s, 6Н), 2.30-2.25 (m, 1Н), 1.97-1.82 (m, 1Н), 1.59 (4.7=3,6 Гц, 6Н).
Методика получения соединения 52b:
В раствор соединения 52а (210 мг, 0,375 ммоль) в 5 мл смеси МеОН/Н2О = 5/1 (об./об.) добавляли Zn (147 мг, 2,25 ммоль) и NH4Cl (120 мг, 2,25 ммоль). Полученную смесь нагревали при 90°С в течение 2 ч, при этом цвет менялся с оранжевого на коричневый. Реакционную смесь фильтровали, и фильтрат концентрировали в вакууме с получением неочищенного остатка, который растворяли в CH2Cl2 (20 мл), промывали водой (15 мл ×3), затем сушили над Na2SO4 и концентрировали в вакууме с получением соединения 52b (125 мг, 63%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,629 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 530,1 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 8.85 (s, 1Н), 8.15 (d, J=7,2 Гц, 1Н), 8.03 (d, J=6,0 Гц, 1Н), 7.87 (s, 1Н), 7.43 (s, 1Н), 7.08 (d, J=10,4 Гц, 1Н), 6.66 (s, 1Н), 6.05 (d, J=5,6 Гц, 1Н), 3.81 (s, 3Н), 3.20-3.11 (m, 2Н), 3.06-2.97 (m, 2Н), 2.91-2.83 (m, 1Н), 2.28 (s, 6Н), 2.17-2.09 (m, 1Н), 1.92-1.81 (m, 1Н), 1.65 (s, 6Н).
Методика получения соединения Примера 52:
В раствор соединения 52b (125 мг, 0,236 ммоль) и DIEA (46 мг, 0,354 ммоль) в DMF (1,5 мл) добавляли акрилоилхлорид (21 мг, 0,236 ммоль) в бане лед-вода. Полученную смесь перемешивали при 5-10°С в течение 15 мин. Реакционную смесь гасили Н2О (0,1 мл) и затем фильтровали. Фильтрат напрямую очищали препаративной ВЭЖХ (колонка: Xtimate С18 150×25 мм, 5 мкм; условия: 35-65% В (А: 0,04% NH3⋅H2O + 10 мМ NH4HCO3, В: CH3CN); скорость потока: 30 мл/мин) и затем лиофилизировали с получением соединения Примера 52 (14,5 мг, 10,5%-ный выход) в виде не совсем белого твердого вещества.
ЖХ/МС: Rt = 2,028 мин при 10-80CD_3 мин_220&254. lcm хроматографии (XBrige Shield RP18 2,1×50 мм, 5 мкм), МС (ЭРИ) m/z = 584,3 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 9.62 (s, 1Н), 9.43 (br s, 1Н), 8.59 (br s, 1Н), 8.08 (d, J=5,2 Гц, 1Н), 7.53 (d, J=6,8 Гц, 1Н), 7.47 (br s, 1Н), 7.14 (d, J=10,8 Гц, 1Н), 6.75 (s, 1H), 6.41-6.31 (m, 3H), 5.77 (t, J=5,4 Гц, 1H), 3.86 (s, 3H), 3.15-3.02 (m, 4H), 2.97-2.84 (m, 1H), 2.30 (s, 6H), 2.21-2.14 (m, 1H), 1.99-1.94 (m, 1H), 1.72 (s, 6H).
Пример 53
(R)-N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)пиримидин-2-иламино)-2-(2-((диметиламино)метил)азетидин-1-ил)-4-метоксифенил)акриламид

Пример 53 Методика получения соединения 53а:
В раствор соединения 36b (150 мг, 0,32 ммоль) и K2CO3 (442 мг, 3,20 ммоль) в DMSO (5 мл) добавляли (R)-1-(азетидин-2-ил)-N,N-диметилметанамина TFA соль (986 мг, 3,20 ммоль). Полученную смесь перемешивали при 85°С в течение 4 ч, при этом цвет менялся с бледно-желтого на насыщенный желтый. Реакционную смесь вливали в воду (50 мл), и желтое твердое вещество выпадало в осадок. Твердое вещество отфильтровывали и растворяли в CH2Cl2 (20 мл), затем сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением соединения 53а (150 мг, 84%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 0,692 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z = 559,9 [М+Н]+.
Методика получения соединения 53b:
В раствор соединения 53а (150 мг, 0,27 ммоль) в МеОН (5 мл) добавляли Pd/C (15 мг). Полученную смесь продували и дегазировали Н2 3 раза, затем перемешивали при 22-30°С в атмосфере водорода (15 фунт/кв.дюйм) в течение 1 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением соединения 53b (100 мг, 70%-ный выход) в виде черного твердого вещества.
ЖХ/МС: Rt = 0,663 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 530,0 [М+Н]+.
Методика получения соединения Примера 53:
В раствор соединения 53b (100 мг, 0,19 ммоль) и DIEA (49 мг, 2,0 экв., 0,38 ммоль) в DMF (2,5 мл) добавляли акрилоилхлорид (17 мг, 0,19 ммоль) в DMF (0,5 мл). Полученную смесь перемешивали при 0°С в бане лед-вода в течение 30 мин. Реакционную смесь очищали препаративной ВЭЖХ [колонка: Waters Xbridge 150×25, 5 мкм; условия: 36-66% В (А: 0,05% NH3H2O; В: CH3CN); скорость потока: 25 мл/мин]. Фракции, содержащие целевое соединение, лиофилизировали с получением соединения Примера 53 (12,2 мг, 11%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 1,378 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 584,1 [М+Н]+.
ВЭЖХ: Rt = 2,74 мин при 10-80_ab_1,2 мл хроматографии (Ultimate С18 3×50 мм, 3 мкм).
1Н ЯМР: (400 МГц, CDCl3) δ 9.41-9.33 (m, 2Н), 8.95 (br s, 1Н), 8.08 (d, J=6,0 Гц, 1Н), 7.52 (br d, J=6,8 Гц, 1H), 7.44 (s, 1H), 7.15 (d, J=10,4 Гц, 1H), 6.68 (s, 1H), 6.38 (d, J=2,8 Гц, 1H), 6.36-6.30 (m, 2H), 5.81-5.75 (m, 1H), 5.62-5.36 (m, 1H), 4.19 (br d, J=6,8 Гц, 1H), 3.90 (s, 3H), 3.83-3.78 (m, 1H), 3.56 (q, J=8,4 Гц, 1H), 2.65 (br dd, J=5.6, 12,4 Гц, 1H), 2.45 (br dd, J=6.8, 12,8 Гц, 1H), 2.37 (br d, J=7,6 Гц, 1H), 2.26 (s, 6H), 2.16-2.08 (m, 1H), 1.71 (d, J=2,8 Гц, 6H).
Пример 54
(R)-N-(5-(4-(4,5-дихлор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-(2-((диметиламино)метил)азетидин-1-ил)-4-метоксифенил)акриламид
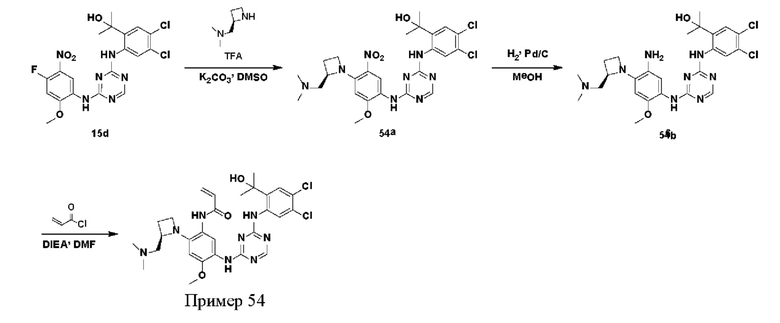
Синтез осуществляли в соответствии с экспериментальной методикой, описанной в Примере 19, с получением соединения Примера 54 в виде бледно-белого твердого вещества.
ЖХ/МС: Rt = 4,386 мин при 10-80CD_7 мин_220&254.lcm; XBrige Shield RP18 2,1×50 мм, МС (ЭРИ) m/z = 601,3 [М+Н]+.
ВЭЖХ: Rt = 3,72 мин при 10-80_ab_1,2 мл хроматографии (Ultimate С18 3×50 мм 3, мкм).
1Н ЯМР (400 МГц, DMSO-d6) δ 10.54 (br s, 1Н), 9.32 (br s, 1Н), 8.84 (br s, 1H), 8.51 (s, 1H), 8.33 (s, 1H), 7.52 (br s, 1H), 7.25 (s, 1H), 6.59-6.52 (m, 1H), 6.34-6.26 (m, 2H), 5.76-5.68 (m, 1H), 4.22-4.12 (m, 1H), 3.82 (s, 3H), 3.78 (br s, 1H), 3.48 (q, J=8,0 Гц, 1H), 2.60 (br dd, J=5.6, 12,8 Гц, 1H), 2.45-2.26 (m, 2H), 2.20 (s, 6H), 2.09-1.90 (m, 2H), 1.67 (br s, 6H).
Пример 55
N-(5-(4-(4,5-дифтор-2-(2-гидроксибутан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-((R)-2-((диметиламино)метил)азетидин-1-ил)-4-метоксифенил)акриламид

Синтез осуществляли в соответствии с экспериментальной методикой, описанной в Примере 47, с получением соединения Примера 55 в виде белого твердого вещества. ЖХ/МС: Rt = 1,741 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 583,1 [М+Н]+.
ВЭЖХ: Rt = 3,49 мин в 10-80_CD_1,2 мл хроматографии (XBridge Shield RP 18 2,1×50 мм, 5 мкм).
1Н ЯМР: (400 МГц, CDCl3) δ 10.70 (br s, 1Н), 9.43 (br s, 1Н), 8.87 (br s, 1H), 8.39-8.26 (m, 2H), 7.62-7.48 (m, 1H), 6.99 (dd, J=8,8 Гц, 12,4 Гц, 1Н), 6.61 (br d, J=6,0 Гц, 1H), 6.40-6.26 (m, 2H), 5.80-5.74 (m, 1H), 4.27-4.16 (m, 1H), 3.88 (s, 3H), 3.84-3.79 (m, 1H), 3.60-3.51 (m, 1H), 2.65 (br dd, J=5,6 Гц, 13,6 Гц, 1Н), 2.49-2.32 (m, 2H), 2.24 (s, 6H), 2.16-1.91 (m, 3Н), 1.66 (s, 3Н), 0.86 (q, J=7,6 Гц, 3Н).
Пример 56
N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксибутан-2-ил)фениламино)-1,315-триазин-2-иламино)-2-((R)-2-((диметиламино)метил)азетидин-1-ил)-4-метоксифенил)акриламид

Методика получения соединения 56b:
В раствор соединения 56а (5 г, 22,28 ммоль) в DMF (50 мл) добавляли Pd(PPh3)2Cl2 (1,0 г, 1,42 ммоль) и соединение 47b (8,85 г, 24,5 ммоль) в атмосфере азота. Смесь перемешивали при 100°С в атмосфере азота в течение 12 ч, затем охлаждали до комнатной температуры, добавляли водный HCl (6 М, 8 мл), и смесь перемешивали при 25°С в течение еще 3 ч. Смесь разбавляли водой (250 мл) и экстрагировали этилацетатом (200 мл × 3). Объединенные органические слои промывали водой (300 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (от 0 до 10% EtOAc в РЕ (об./об.)) с получением соединения 56b (1,89 г, 45%-ный выход).
ЖХ/МС: Rt = 2,071 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 187,9 [М+Н]+.
1H ЯМР (400 МГц, CDCl3) δ 7.39 (d, J=10,0 Гц, 1Н), 6.65 (d, J=4,0 Гц, 1Н), 2.47 (s, 3Н).
Методика получения соединения 56с:
В желтый раствор соединения 56b (1,69 г, 9,01 ммоль) в THF (20 мл) по каплям добавляли EtMgBr (12 мл, 36,03 ммоль) в атмосфере азота при 0°С. Полученную смесь перемешивали при 19-24°С в течение 3 ч. Смесь гасили насыщенным раствором NH4Cl (100 мл) и экстрагировали EtOAc (30 мл × 3). Объединенные органические слои объединяли с предыдущей партией и концентрировали в вакууме с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (от 0 до 10% EtOAc в РЕ) с получением соединения 56 с (1,1 г, 50%-ный выход) в виде желтого масла.
ЖХ/МС: Rt = 1,429 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С18 2.1×30 мм), МС (ЭРИ) m/z = 199,9 [М-H2O + Н]+.
1Н ЯМР (400 МГц, DMSO-d6) δ 6.94 (d, J=11,6 Гц, 1Н), 6.71 (d, J=6,8 Гц, 1Н), 5.50 (s, 2Н), 1.88-1.72 (m, 2Н), 1.43 (s, 3Н), 0.72 (t, J=7,2 Гц, 3Н).
Методика получения соединения 56d:
В желтый раствор соединения 56с (1,34 г, 6,16 ммоль) в дихлорметане (20 мл) последовательно добавляли DIEA (1,59 г, 12,31 ммоль) и 2,4-дихлор-1,3,5-триазин (1,02 г, 6,77 ммоль). Полученный желтый раствор перемешивали при 21-29°С в течение 2 ч. Смесь концентрировали в вакууме с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (от 0 до 10% EtOAc в РЕ) с получением соединения 56d (1,28 г, 58%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 0,922 мин при 5-95АВ_220&254 хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 330,7 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 10.35-10.03 (m, 1Н), 8.62-8.44 (m, 1Н), 8.35 (d, J=7,2 Гц, 1Н), 7.02 (d, J=10,4 Гц, 1Н), 2.44 (br. s, 1Н), 1.95-1.88 (m, 2Н), 1.66 (s, ЗН), 0.87 (t, J=7.2 Гц, 3Н).
Методика получения соединения 56е:
В смесь соединения 56d (1,28 г, 3,87 ммоль) в n-BuOH/TFA (10 мл/0,1 мл) добавляли 4-фтор-2-метокси-5-нитроанилин (720 мг, 3,87 ммоль). Смесь перемешивали при 23-29°С в течение 4 ч, при этом серое твердое вещество выпадало в осадок. Твердое вещество собирали фильтрованием под разряжением и сушили в глубоком вакууме с получением соединения 56е (1,07 г, 57,5%-ный выход).
ЖХ/МС: Rt = 0,984 мин при 5-95АВ_220&254 хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 481,1 [М+Н]+.
1Н ЯМР: (400 МГц, DMSO-d6) δ 10.46 (br s, 1Н), 8.36 (s, 1Н), 7.40 (d, J=13,6 Гц, 1H), 7.30 (s, 1H), 7.26 (br d, J=10,8 Гц, 1H), 7.17 (s, 1H), 7.05 (s, 1H), 3.93 (s, 3H), 1.85-1.68 (m, 2H), 1.51 (s, 3H), 0.72 (t, J=7,2 Гц, 3Н).
Методика получения соединения 56f:
В смесь соединения 56е (150 мг, 0,31 ммоль) в DMSO (3 мл) добавляли K2CO3 (474 мг, 3,43 ммоль) и (R)-1-(азетидин-2-ил)-N,N-диметилметанамина TFA соль (1,07 мг, 3,12 ммоль). Полученную смесь перемешивали при 85°С в течение 1 ч. Реакционную смесь объединяли с предыдущей партией и вливали в ледяную воду (20 мл). Выпавшее в осадок оранжевое твердое вещество собирали фильтрованием под разряжением и промывали водой (10 мл), затем сушили в вакууме с получением соединения 7 (160 мг, 79%-ный выход).
ЖХ/МС: Rt = 2,003 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С18 2.1×30 мм), МС (ЭРИ) m/z = 575,1 [М+Н]+.
1Н ЯМР: (400 МГц, CDCl3) δ 9.66 (s, 1Н), 8.90 (s, 1Н), 8.45-8.18 (m, 2Н), 7.28 (br. s, 1Н), 6.93 (d, J=10,4 Гц, 1Н), 6.75 (br. s, 1Н), 4.35 (br. s, 1H), 4.29-4.18 (m, 1H), 3.87 (s, 3H), 3.28-3.15 (m, 1H), 2.82-2.74 (m, 1H), 2.68-2.60 (m, 1H), 2.50-2.23 (m, 2H), 2.26 (s, 3H), 2.14-2.02 (m, 1H), 1.92-1.85 (m, 2H),1.54 (s, 6H), 0.81 (t, J=7,2 Гц, 3Н).
Методика получения соединения 56g:
В желтый раствор соединения 56f (160 мг, 0,28 ммоль) в смеси метанол/вода (5 мл/1 мл) добавляли NH4Cl (89 мг, 1,67 ммоль) и Zn (90 мг, 1,39 ммоль). Полученную смесь перемешивали при 90°С в течение 1 ч. Реакционную смесь вливали в воду (20 мл) и экстрагировали смесью дихлорметан/метанол = 3:1 (об./об.) (10 мл × 3). Объединенные органические слои сушили и концентрировали в вакууме с получением соединения 56g (150 мг, 98%-ный выход) в виде черного твердого вещества.
ЖХ/МС: Rt = 2,144 мин при 0-60АВ_4 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 545,2 [М+Н]+.
1Н ЯМР: (400 МГц, CDCl3) δ 9.66 (br. s, 1Н), 8.30 (br. s, 1Н), 8.21 (br. s, 1H), 7.78-7.37 (m, 2H), 6.93 (d, J=10,4 Гц, 1H), 6.45 (s, 1H), 4.21-4.09 (m, 1H), 4.00-3.88 (m, 1H), 3.76 (s, 3H), 3.34-3.20 (m, 1H), 2.43-2.30 (m, 1H), 2.22-2.17 (m, 5H), 2.14-2.00 (m, 2H), 1.89-1.83 (m, 2H), 1.61-1.51 (m, 6H), 0.82-0.77 (m, 3H).
Методика получения соединения Примера 56:
В черный раствор соединения 56g (150 мг, 0,27 ммоль) в DMF (2 мл) добавляли DIEA (71 мг, 0,55 ммоль), затем добавляли акрилоилхлорид (25 мг, 0,27 ммоль) при 0°С за три раза и перемешивали при 0°С в течение 1 ч. Смесь гасили 3 каплями воды и очищали препаративной ВЭЖХ напрямую (колонка: Waters Xbridge 150×25 5 мкм, условие: 41%-61% В (А: вода/10 мМ NH4HCO3, В: CH3CN), скорость потока: 25 мл/мин) с получением соединения Примера 56 (31,9 мг, 19,7%-ный выход) в виде серого твердого вещества. 11,6 мг было предоставлено, а остальную часть партии 20,3 мг дополнительно очищали методом СФХ.
ЖХ/МС: Rt = 1,709 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 621,1 [M+Na]+.
ВЭЖХ: Rt = 3,65 мин при 10-80_CD_1,2 мл хроматографии (XBridge Shield RP 18 2,1×50 мм, 5 мкм).
1Н ЯМР (400 МГц, CDCl3) δ 10.68 (br s, 1Н), 9.41 (br s, 1Н), 8.90 (br s, 1H), 8.61-8.25 (m, 2H), 7.59 (br s, 1H), 7.02 (d, J=9,6 Гц, 1H), 6.64 (br s, 1H), 6.39 (br s, 2H), 5.80 (br s, 1H), 4.41-4.16 (m, 1H), 4.02-3.78 (m, 4H), 3.66-3.45 (m, 1H), 2.83-2.62 (m, 1H), 2.58-2.46 (m, 1H), 2.44-2.37 (m, 1H), 2.30 (s, 6H), 2.21-2.05 (m, 3H), 1.69 (s, 3H), 0.94-0.83 (m, 3H).
Пример 57
N-(5-(4-(4,5-дифтор-2-(2-гидроксибутан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-((R)-2-((диметиламино)метил)пирролидин-1-ил)-4-метоксифенил)акриламид

Синтез осуществляли в соответствии с экспериментальной методикой, описанной в Примере 47, с получением соединения Примера 57 в виде бледно-желтого твердого вещества.
ЖХ/МС: Rt = 1,928 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 597,1 [М+Н]+.
ВЭЖХ: Rt = 4,09 мин при 10-80_CD_1,2 мл хроматографии (XBridge Shield RP 18 2,1×50 мм, 5 мкм).
1Н ЯМР: (400 МГц, CDCl3) δ 10.92 (br s, 1H), 10.14-9.97 (m, 1H), 9.95-9.80 (m, 1H), 8.40 (s, 1H), 8.39-8.34 (m, 1H), 7.68 (br d, J=9,6 Гц, 1H), 7.03 (dd, J=8,4 Гц, 12,4 Гц, 1Н), 6.71 (s, 1H), 6.36 (br d, J=5,2 Гц, 2H), 6.04-5.63 (m, 2H), 3.86 (s, 3H), 3.39-3.26 (m, 2H), 3.01-2.92 (m, 1H), 2.42-2.32 (m, 1H), 2.19 (s, 6H), 2.13-1.91 (m, 5H), 1.70 (s, 3H), 1.68-1.63 (m, 2H), 0.94-0.84 (m Гц, 3Н).
Пример 58
N-(5-(4-(4,5-дифтор-2-(2-гидроксибутан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-((S)-2-((диметиламино)метил)пирролидин-1-ил)-4-метоксифенил)акриламид
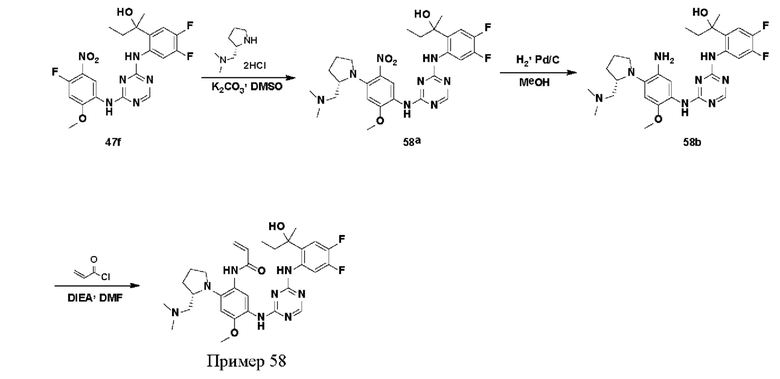
Синтез осуществляли в соответствии с экспериментальной методикой, описанной в Примере 47, с получением соединения Примера 58 в виде белого твердого вещества.
ЖХ/МС: Rt = 1,959 мин при 10-80АВ_4м мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 619,1 [M+Na]+.
ВЭЖХ: Rt = 2,74 мин при 10-80_ab_1,2 мл хроматографии (Ultimate С18 3×50 мм, 3 мкм).
1Н ЯМР: (400 МГц, CDCl3) δ 10.91 (br s, 1Н), 10.24-9.80 (m, 2H), 8.48-8.33 (m, 2H), 7.68 (br d, J=10,4 Гц, 1H), 7.03 (dd, J=8.8, 12,4 Гц, 1H), 6.71 (s, 1H), 6.36 (br d, J=4,4 Гц, 2H), 5.79-5.75 (m, 1H), 3.87 (s, 3H), 3.32 (br d, J=10,8 Гц, 2H), 3.02-2.91 (m, 1H), 2.44-2.32 (m, 1H), 2.23-2.16 (m, 7H), 2.14-1.86 (m, 6H), 1.70 (s, 3H), 0.89 (td, J=7.6, 12,0 Гц, 3Н).
Пример 59
N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксибутан-2-ил)фениламино)-1,315-триазин-2-иламино)-2-((R)-3-(диметиламино)пирролидин-1-ил)-4-метоксифенил)акриламид
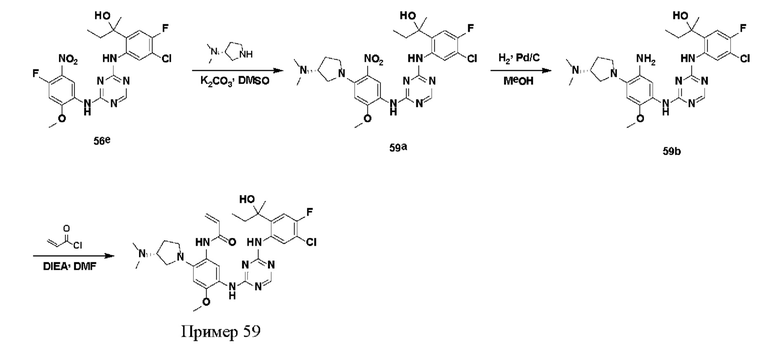
Синтез осуществляли в соответствии с экспериментальной методикой, описанной в Примере 56, с получением соединения Примера 59 в виде белого твердого вещества.
ЖХ/МС: Rt = 1,713 мин при 10-80АВ_4 мин_220&254.1cm хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 599,1 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 10.78 (br s, 1Н), 9.77 (br s, 1Н), 8.54 (br s, 1Н), 8.53 (br s, 1H), 8.41 (s, 1H), 7.64 (br s, 1H), 7.01 (d, J=10,8 Гц, 1H), 6.74 (s, 1H), 6.48-6.29 (m, 2H), 5.86-5.76 (m, 1H), 5.62 (br s, 1H), 3.87 (s, 3H), 3.17-3.03 (m, 4H), 2.98-2.86 (m, 1H), 2.33 (s, 6H), 2.24-2.15 (m, 1H), 2.14-2.07 (m, 1H), 2.05-1.95 (m, 2H), 1.69 (s, 3H), 0.89 (dt, J=3.2, 7,6 Гц, 3Н).
Пример 60
(S)-N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-(3-(диметиламино)пирролидин-1-ил)-4-метоксифенил)акриламид
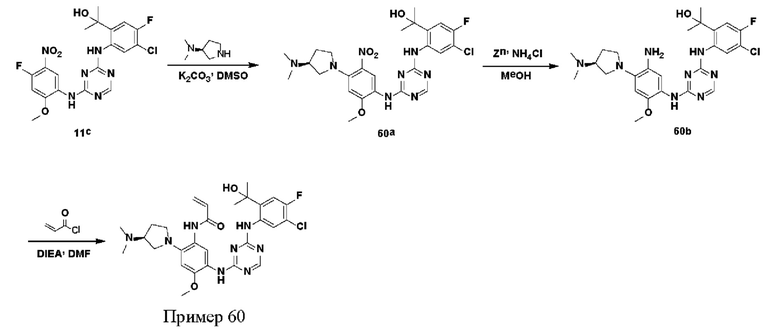
Синтез осуществляли в соответствии с экспериментальной методикой, описанной в Примере 33, с получением соединения Примера 60 в виде белого твердого вещества.
ЖХ/МС: Rt = 1,568 мин при 10-80АВ_4 мин_220&254. lcm хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 585,2 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 10.51 (br s, 1Н), 9.72 (br s, 1Н), 8.56 (br s, 1H), 8.47 (d, J=7,2 Гц, 1H), 8.40 (s, 1H), 7.63 (br s, 1H), 7.08 (d, J=10,8 Гц, 1H), 6.71 (s, 1H), 6.55-6.26 (m, 2H), 5.88 (br s, 1H), 5.79 (d, J=9,2 Гц, 1H), 3.86 (s, 3H), 3.20-2.99 (m, 4H), 2.90 (quin, J=6,8 Гц, 1H), 2.31 (s, 6H), 2.23-2.12 (m, 1H), 2.03-1.90 (m, 1H), 1.75 (br s, 6H).
Пример 61
(R)-N-(5-(4-(4,5-дихлор-2-(2-гидроксипропан-2-ил)фениламино)-1,315-триазин-2-иламино)-2-(3-(диметиламино)пирролидин-1-ил)-4-метоксифенил)акриламид

Синтез осуществляли в соответствии с экспериментальной методикой, описанной в Примере 33, с получением соединения Примера 61 в виде белого твердого вещества.
ЖХ/МС: Rt = 2,535 мин при 0-60АВ_4,0 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 601,1 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 10.71 (br s, 1Н), 9.80 (br s, 1H), 8.63 (s, 1H), 8.52 (br s, 1H), 8.44 (s, 1H), 7.67 (br s, 1H), 7.35 (s, 1H), 6.76 (s, 1H), 6.39-6.38 (m, 2H), 5.95-5.70 (m, 2H), 3.89 (s, 3H), 3.15-3.08 (m, 4H), 2.94-2.87 (m, 1H), 2.32 (s, 6H), 2.24-2.13 (m, 1H), 1.96-1.93 (m, 1H), 1.78 (s, 6H).
Пример 62
(R)-N-(5-(4-(4-хлор-5-фтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-(2-((диметиламино)метил)азетидин-1-ил)-4-метоксифенил)акриламид

Синтез осуществляли в соответствии с экспериментальной методикой, описанной в Примере 41, с получением соединения Примера 62 в виде белого твердого вещества.
ЖХ/МС: Rt = 1,493 мин при 10-80АВ_3 мин_220&254 хроматографии (Xtimate С18, 2,1×30 мм, 3 мкм), МС (ЭРИ) m/z = 585,3 [М+Н]+.
ВЭЖХ: Rt = 3,52 мин при 10-80_CD_1,2 мл. мет XBridge Shield RP 18 2,1×50 мм, 5 мкм.
1Н ЯМР: (400 МГц, CDCl3) δ 10.66 (br s, 1Н), 9.50-9.19 (m, 1Н), 8.93 (br s, 1H), 8.50-8.28 (m, 2H), 7.58 (br s, 1H), 7.26 (br s, 1H), 6.62 (s, 1H), 6.44-6.28 (m, 2H), 5.80 (br d, J=9,6 Гц, 1H), 4.29-4.17 (m, 1H), 3.90 (s, 3H), 3.87-3.79 (m, 1H), 3.58 (q, J=8,4 Гц, 1H), 2.73-2.62 (m, 1H), 2.56-2.40 (m, 2H), 2.26 (s, 6H), 2.18-2.00 (m, 1H), 1.75 (s, 6H).
Пример 63
N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксибутан-2-ил)фениламино)-1,315-триазин-2-иламино)-2-((R)-2-((диметиламино)метил)пирролидин-1-ил)-4-метоксифенил)акриламид
Синтез осуществляли в соответствии с экспериментальной методикой, описанной в Примере 56, с получением соединения Примера 63 в виде белого твердого вещества.
ЖХ/МС: Rt = 1,870 мин при 10-80 АВ_4 мин_220&254. 1cm хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 613,1 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 10.86 (br s, 1Н), 10.04 (d, J=27,6 Гц, 1Н), 9.89 (br d, J=20,0 Гц, 1H), 8.53 (dd, J=2.8, 7,2 Гц, 1H), 8.41 (s, 1H), 7.69 (d, J=8,8 Гц, 1H), 7.02 (d, J=10,8 Гц, 1H), 6.70 (s, 1H), 6.36 (s, 1H), 6.35 (br s, 1H), 5.92 (br s, 1H), 5.77 (t, J=5,8 Гц, 1H), 3.86 (s, 3H), 3.39-3.24 (m, 2H), 3.02-2.92 (m, 1H), 2.37 (dd, J=7.8, 12,0 Гц, 1H), 2.23-2.16 (m, 7H), 2.13-1.88 (m, 5H), 1.70 (s, 3H), 1.69-1.64 (m, 1H), 0.89 (td, J=7.4, 20,0 Гц, 3Н).
Пример 64
N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксибутан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-((S)-2-((диметиламино)метил)пирролидин-1-ил)-4-метоксифенил)акриламид
Синтез осуществляли в соответствии с экспериментальной методикой, описанной в Примере 56, с получением соединения Примера 64 в виде светло-желтого твердого вещества.
ЖХ/МС: Rt = 1,473 мин при 10-80АВ_3 мин_220&254 хроматографии (Xtimate С18, 2,1×30 мм, 3 мкм), МС (ЭРИ) m/z = 613,3 [М+Н]+.
ВЭЖХ: Rt = 4,27 мин, 10-80_CD_1,2 мл. мет XBridge Shield RP 18 2,1×50 мм, 5 мкм.
*Н ЯМР: (400 МГц, CDCl3) δ 10.86 (br s, 1Н), 10.15-9.97 (m, 1Н), 9.93-9.79 (m, 1H), 8.53 (br d, J=7,4 Гц, 1H), 8.41 (s, 1H), 7.73-7.63 (m, 1H), 7.02 (d, J=10,8 Гц, 1H), 6.71 (s, 1H), 6.36 (br d, J=5,2 Гц, 2H), 5.91 (br s, 1H), 5.82-5.73 (m, 1H), 3.87 (s, 3H), 3.40-3.22 (m, 2H), 2.97 (q, J=7,2 Гц, 1H), 2.46-2.30 (m, 2H), 2.22-2.16 (m, 7H), 2.13-1.89 (m, 6H), 1.71 (s, 3H), 0.94-0.85 (m, 3H).
Пример 65
(S)-N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-(2-((диметиламино)метил)пирролидин-1-ил)-4-метоксифенил)акриламид

Синтез осуществляли в соответствии с экспериментальной методикой, описанной в Примере 31, с получением соединения Примера 65 в виде белого твердого вещества.
ЖХ/МС: Rt = 2,553 мин при 0-60АВ_4,0 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 621,0 [M+Na]+.
1Н ЯМР (CDCl3 400 МГц) δ 10.60 (br s, 1Н), 10.09 (br s, 1H), 9.91 (br s, 1H), 8.48 (d, J=7,2 Гц, 1H), 8.42 (s, 1H), 7.68 (br s, 1H), 7.10 (d, J=10,8 Гц, 1H), 6.70 (s, 1H), 6.45-6.29 (m, 2H), 6.11 (br s, 1H), 5.85-5.73 (m, 1H), 3.87 (s, 3H), 3.38-3.25 (m, 2H), 3.02-2.93 (m, 1H), 2.42-2.34 (m, 1H), 2.24-2.16 (m, 7H), 2.16-1.91 (m, 4H), 1.77 (s, 6H).
Пример 66
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-(4-(2-(1,1,1-трифтор-2-гидрооксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)фенил)акриламид
Методика получения соединения 66b:
В раствор соединения 66а (5,0 г, 29 ммоль) и триэтиламина (3,82 г, 38 ммоль) в DCM (125 мл) добавляли пивалоилхлорид (3,855 г, 32 ммоль) при 0°С. Полученную смесь перемешивали при 20°С в течение 16 ч. Реакционную смесь разбавляли водой (100 мл), и органическую фазу отделяли. Водную фазу экстрагировали DCM (50 мл × 3), и объединенные органические слои промывали рассолом (200 мл × 1), сушили над безводным Na2SO4, фильтровали и выпаривали при пониженном давлении. Остаток очищали флэш-хроматографией на диоксиде кремния с градиентным элюированием от 10% до 30% EtOAc в петролейном эфире (об./об.). Чистые фракции упаривали досуха с получением соединения 66b (7,271 г, 98%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 1,44 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3), Waters Acquity UPLC ВЕН CI8 1,7 мкм, 2,1×50 мм, 40°С), МС (ЭРИ) m/z = 257,2 [М+Н]+.
Методика получения соединения 66с:
В раствор соединения 66b (2,0 г, 7,8 ммоль) в THF (46 мл) добавляли n-BuLi (12 мл, 1,6 М в гексане) при -30°С. Полученную смесь перемешивали при -30°С в течение 1 ч. Добавляли TFAA (2,5 г, 12 ммоль) при -30°С, затем смесь перемешивали при 20°С в течение еще 16 ч. Смесь гасили 1 М раствором HCl (32 мл) и экстрагировали ЕА (20 мл × 5). Объединенные органические фазы сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток обрабатывали конц. HCl (20 мл) и THF (20 мл), затем полученную смесь нагревали при 80°С в течение 16 ч. Реакционную смесь разбавляли насыщенным раствором NaHCO3 (100 мл) и экстрагировали ЕА (50 мл × 5). Объединенные органические фазы промывали рассолом (200 мл×1) и выпаривали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле с градиентом элюирования от 0% до 30% EtOAc в петролейном эфире. Чистые фракции упаривали досуха с получением соединения 66 с (863 мг, 58%-ный выход) в виде коричневого масла.
1Н ЯМР (500 МГц, CDCl3) δ м.д. 6.4 (br s, 2Н) 6.7-6.8 (m, 2Н) 7.3-7.4 (m, 1Н) 7.7-7.8 (m, 1Н).
Методика получения соединения 66d:
В раствор соединения 66 с (850 мг, 4,5 ммоль) в THF (17 мл) добавляли метилмагния бромид (7,48 мл, 3М в THF) при 0°С. Полученную реакционную смесь перемешивали при 20°С в течение 1 ч. Реакционную смесь гасили насыщенным раствором NH4Cl (50 мл) и экстрагировали ЕА (50 мл × 3). Объединенные органические фазы промывали рассолом, сушили над безводным сульфатом натрия, фильтровали и выпаривали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле с градиентом элюирования от 10% до 50% EtOAc в петролейном эфире. Чистые фракции упаривали досуха с получением соединения 66d (342 мг, 21%-ный выход) в виде коричневого масла.
ЖХ/МС: Rt = 1,16 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3), Waters Acquity UPLC ВЕН C18 1,7 мкм, 2,1×50 мм, 40°С), МС (ЭРИ) m/z = 206,1 [М+Н]+.
1Н ЯМР (500 МГц, DMSO-d6) δ 1.7 (s, 3Н) δ.5 (s, 2Н) 6.5-6.5 (m, 1Н) 6.6 (dd, J=7.9, 1,3 Гц, 1Н) 6.9 (s, 1Н) 7.0-7.0 (m, 1Н) 7.1 (d, J=8,2 Гц, 1Н).
Методика получения соединения 66е:
Раствор соединения 6е (174 мг, 0,58 ммоль) и соединения 66d (120 мг, 0,58 ммоль) в NMP (1 мл) герметично закрывали и нагревали при 80°С в течение 40 мин. Реакционную смесь разбавляли водой (50 мл) и экстрагировали ЕА (20 мл × 5). Объединенные органические слои промывали рассолом, сушили над безводным сульфатом натрия, фильтровали и выпаривали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле с градиентом элюирования от 30% до 100% EtOAc в петролейном эфире. Чистые фракции упаривали досуха с получением соединения 66е (235 мг, 85%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 1,90 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1×50 мм, 40°С) МС (ЭРИ) m/z = 469,3 [М+Н]+.
Методика получения соединения 66f:
Раствор соединения 66е (235 мг, 0,50 ммоль), N1,N1,N2-триметилэтан-1,2-диамина (72 мг, 0,70 ммоль) и DIE А (194 мг, 1,5 ммоль) в NMP (3,0 мл) нагревали при 50°С в течение 80 мин. Реакционную смесь разбавляли водой (100 мл) и экстрагировали ЕА (20 мл × 5). Объединенные органические фазы промывали рассолом, сушили над безводным сульфатом натрия, фильтровали и выпаривали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле с градиентом элюирования от 0% до 10% МеОН в DCM. Чистые фракции упаривали досуха с получением соединения 66f (191 мг, 69%-ный выход) в виде светло-коричневого твердого вещества.
ЖХ/МС: Rt = 1,28 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3), Waters Acquity UPLC ВЕН C18 1,7 мкм, 2,1×50 мм, 40°C), МС (ЭРИ) m/z = 551,2 [M+H]+.
Методика получения соединения 66g:
Палладий на углероде (37 мг) добавляли в раствор соединения 66f (95 мг, 0,17 ммоль) в МеОН (10 мл). Полученную реакционную смесь перемешивали при 20°С в атмосфере водорода в течение 30 мин. Смесь фильтровали, и фильтрат выпаривали при пониженном давлении с получением соединения 66 (90 мг, неочищенное) в виде светло-коричневого твердого вещества.
ЖХ/МС: Rt = 1,19 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3), Waters Acquity UPLC ВЕН C18 1,7 мкм, 2,1×50 мм, 40°С), МС (ЭРИ) m/z = 521,3 [М+Н]+.
Методика получения соединения Примера 66:
В раствор соединения 66g (90 мг, 0,17 ммоль) и DIEA (67 мг, 0,52 ммоль) в NMP (3 мл) добавляли раствор акрилоилхлорида (21 мг, 0,23 ммоль) в NMP (0,5 мл) при 0°С. Реакционную смесь перемешивали при 0°С в течение 30 мин. Затем раствор очищали С18-флэш-хроматографией с градиентом элюирования от 0% до 60% MeCN в воде (0,02% аммиака). Чистые фракции лиофилизировали досуха с получением соединения Примера 66 (23 мг, 23%-ный выход) в виде светло-желтого твердого вещества.
ЖХ/МС: Rt = 1,24 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3), Waters Acquity UPLC ВЕН C18 1,7 мкм, 2,1×50 мм, 40°C), МС (ЭРИ) m/z = 575,3 [М+Н]+.
1Н ЯМР: (500 МГц, DMSO-d6) δ 1.8 (br s, 3H) 2.2 (s, 6H) 2.3 (br t, J=5,7 Гц, 2H) 2.7 (s, 3H) 2.9 (br d, J=5,0 Гц, 2H) 3.8 (s, 3H) 5.7 (br d, J=10,7 Гц, 1H) 6.2 (br d, J=17,3 Гц, 1H) 6.4 (br dd, J=16.7, 10,1 Гц, 1H) 7.0 (s, 2H) 7.0-7.3 (m, 1H) 7.3-7.4 (m, 1H) 8.4 (s, 1H) 8.2 (s, 1H) 8.2-8.4 (m, 1H) 8.9 (br s, 1H) 10.1 (br s, 1H).
Пример 67
N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксибутан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид

Пример 67
Синтез осуществляли в соответствии с экспериментальной методикой, описанной в Примере 56, с получением соединения Примера 67 в виде белого твердого вещества.
ЖХ/МС: Rt = 1,585 мин при 10-80АВ_3 мин_220&254 хроматографии (Xtimate С18, 2,1×30 мм), МС (ЭРИ) m/z = 609,1 [M+Na]+.
ВЭЖХ: Rt = 3,45 10-80АВ_1,2 мл. мет (Ultimate С18 3×50 мм, 3 мкм).
1Н ЯМР: (400 МГц, CDCl3) δ 10.84 (br s, 1Н), 10.42 (br s, 1Н), 9.94 (br s, 1H), 8.52 (br d, J=7,4 Гц, 1H), 8.41 (s, 1H), 7.70 (br s, 1H), 7.02 (d, J=10,8 Гц, 1H), 6.78 (s, 1H), 6.42-6.29 (m, 2H), 5.97 (br s, 1H), 5.82-5.72 (m, 1H), 3.88 (s, 3H), 2.92-2.82 (m, 2H), 2.70 (s, 3H), 2.42-2.31 (m, 8H), 2.18-2.02 (m, 2H), 1.70 (s, 3H), 0.88 (br t, J=7,6 Гц, 3Н).
Пример 68
(R)-N-(5-(4-(4,5-дихлор-2-(2-гидроксипропан-2-ил)фениламино)пиримидин-2-иламино)-2-(3-(диметиламино)пирролидин-1-ил)-4-метоксифенил)акриламид
Методика получения соединения 68а:
В раствор соединения 15с (900 мг, 4,09 ммоль) и DIEA (1,1 г, 8,18 ммоль) в изопропаноле (2 мл) добавляли 2,4-дихлорпиримидин (730 мг, 4,90 ммоль). Полученную смесь нагревали при 90°С в течение 48 ч. Реакционную смесь концентрировали в вакууме с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (25-35% EtOAc в петролейном эфире) с получением соединения 68а (1,1 г, 81%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 0,883 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z = 331,8/333,8 [М+Н]+.
1H ЯМР (400 МГц, Метанол-d4) δ 8.27 (s, 1Н), 8.14 (d, J=6,0 Гц, 1Н), 7.50 (s, 1Н), 6.74 (d, J=6,0 Гц, 1Н), 1.58 (s, 6Н).
Методика получения соединения 68b:
В раствор соединения 68а (1,1 г, 3,31 ммоль) и 4-фтор-2-метокси-5-нитроанилина (616 мг, 3,31 ммоль) в n-BuOH (10 мл) добавляли TFA (0,1 мл). Полученную смесь перемешивали при 50°С в течение 12 ч. Реакционную смесь фильтровали, и остаток на фильтре сушили при пониженном давлении с получением соединения 68b (1,3 г, 81%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 0,807 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 482,0 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 8.53 (br d, J=8,8 Гц, 1Н), 8.02-7.92 (m, 2Н), 7.53 (s, 1Н), 7.22 (d, J=12,8 Гц, 1Н), 6.49 (d, J=7,3 Гц, 1H), 4.00 (s, 3H), 1.59 (s, 6H).
Методика получения соединения 68c:
В раствор соединения 68b (200 мг, 0,41 ммоль) и K2CO3 (113 мг, 0,82 ммоль) в DMSO (5 мл) добавляли (R)-N,N-диметилпирролидин-3-амин (56 мг, 0,49 ммоль). Полученную смесь перемешивали при 16-21°С в течение 12 ч, при этом цвет менялся с бледно-желтого на насыщенный желтый. Реакционную смесь вливали в воду (50 мл), и желтое твердое вещество выпадало в осадок. Твердое вещество собирали фильтрованием и растворяли в CH2Cl2 (20 мл), затем сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением соединения 68 с (200 мг, 85%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 0,867 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 576,0 [М+Н]+.
Методика получения соединения 68d:
В раствор соединения 68с (200 мг, 0,35 ммоль) в EtOAc (5 мл) добавляли Pd/C (40 мг). Полученную смесь продували и дегазировали Н2 3 раза, затем перемешивали при 12-17°С в атмосфере Н2 (15 фунт/кв.дюйм) в течение 1 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением соединения 68d (150 мг, 78%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 0,670 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 546,1 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 8.96 (s, 1Н), 8.30 (s, 1Н), 8.07 (d, J=5,6 Гц, 1Н), 7.87 (s, 1Н), 7.45 (s, 1Н), 7.33 (s, 1Н), 6.68 (s, 1Н), 6.09 (d, J=5,6 Гц, 1Н), 3.82 (s, 3Н), 3.15 (br d, J=6,8 Гц, 2Н), 3.05 (br d, J=6,4 Гц, 2H), 2.89 (s, 1Н), 2.31 (s, 6Н), 2.14 (br s, 1Н), 1.90 (br s, 1H), 1.68 (s, 6H).
Методика получения соединения 68e:
В раствор соединения 68d (150 мг, 0,27 ммоль) в CH2Cl2 (3 мл) добавляли 3-хлорпропаноилхлорид (35 мг, 0,27 ммоль) в бане лед-вода. Полученную смесь перемешивали при 5-10°С в течение 5 мин, при этом цвет менялся с коричневого на желтый. Реакционную смесь вливали в насыщенный NaHCO3 (5 мл) и экстрагировали CH2Cl2 (15 мл × 2). Объединенные органические слои сушили над Na2SO4 и концентрировали при пониженном давлении с получением неочищенного остатка, который очищали колоночной хроматографией на силикагеле (10% МеОН в CH2Cl2) с получением соединения 68е (90 мг, 52%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 0,709 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 636,1/638.1 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 9.60-9.43 (m, 2Н), 8.63-8.45 (m, 1Н), 8.11 (d, J=5,6 Гц, 1Н), 7.67 (s, 1Н), 7.49 (s, 1Н), 7.40 (s, 1Н), 6.76 (s, 1Н), 6.37 (s, 1Н), 3.91 (br d, J=6,4 Гц, 2Н), 3.86 (s, 3Н), 3.10 (br s, 4Н), 2.91 (br s, 2H), 2.33 (br s, 8H), 2.06 (s, 1H), 1.77-1.70 (m, 6H).
Методика получения соединения Примера 68:
В раствор соединения 68е (90 мг, 0,14 ммоль) в CH3CN (3 мл) добавляли TEA (42 мг, 0,42 ммоль). Полученную смесь перемешивали при 80°С в течение 18 ч. Реакционную смесь концентрировали при пониженном давлении и очищали колоночной флэш-хроматографией на силикагеле (5% МеОН в CH2Cl2). Элюенты концентрировали при пониженном давлении и затем лиофилизировали с получением соединения Примера 68 (33,1 мг, 31%-ный выход) в виде светло-желтого твердого вещества.
ЖХ/МС: Rt = 1,589 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 600,0 [М+Н]+.
ВЭЖХ: Rt = 2,42 мин при 10-80_ab_1,2 мл хроматографии (Ultimate С18 3×50 мм 3 мкм).
1Н ЯМР (400 МГц, CDCl3) δ 9.57 (br d, J=11,8 Гц, 2Н), 8.04 (d, J=5,8 Гц, 1Н), 7.52 (s, 1Н), 7.41 (s, 1Н), 7.32 (s, 1Н), 6.65 (br s, 1Н), 6.37-6.27 (m, 2H), 5.71 (br d, J=11,2 Гц, 1H), 3.79 (s, 3H), 3.12 (br d, J=12,8 Гц, 2H), 3.07-2.98 (m, 2H), 2.98-2.92 (m, 1H), 2.39 (br s, 6H), 2.15 (brs, 2H), 1.66 (s, 6H).
Пример 69/Пример 70
N-(5-(4-(5-хлор-4-фтор-2-((R)-2-гидроксибутан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-((R)-3-(диметиламино)пирролидин-1-ил)-4-метоксифенил)акриламид и
N-(5-(4-(5-хлор-4-фтор-2-((S)-2-гидроксибутан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-((R)-3-(диметиламино)пирролидин-1-ил)-4-метоксифенил)акриламид
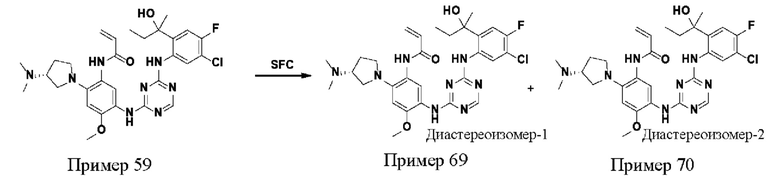
Соединение Примера 59 (50 мг, 0,0835 ммоль) разделяли методом СФХ с получением соединения Примера 69 (21,0 мг, 42%-ный выход) в виде белого твердого вещества и соединения Примера 70 (24,6 мг, 49,2%-ный выход) в виде белого твердого вещества.
Пример 69:
ЖХ/МС: Rt = 1,840 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 621,0 [M+Na]+.
ВЭЖХ: Rt = 3,88 мин при 10-80_CD_1,2 мл хроматографии (XBridge Shield RP 18 2,1×50 мм, 5 мкм).
1H ЯМР (400 МГц, CDCl3) δ 10.79 (br s, 1Н), 9.69 (br s, 1Н), 8.79 (br s, 1H), 8.53 (d, J=7,2 Гц, 1H), 8.40 (s, 1H), 7.61 (br s, 1H), 7.01 (d, J=10,8 Гц, 1H), 6.73 (br s, 1H), 6.67 (s, 1H), 6.37 (d, J=16,4 Гц, 1H), 5.79 (d, J=10,8 Гц, 1H), 5.70 (br s, 1H), 3.87 (s, 3H), 3.41-3.24 (m, 2H), 3.18 (br s, 1H), 3.07 (dd, J=6.4, 10,0 Гц, 1H), 2.98 (q, J=8,0 Гц, 1H), 2.55 (s, 6H), 2.30-2.20 (m, 2H), 2.14-2.08 (m, 1H), 2.04-1.99 (m, 1H), 1.69 (s, 3H), 0.88 (t, J=7,6 Гц, 3Н).
СФХ: Rt = 6,855 мин при AD-3_IPA(DEA)_5_40_2,5 мл.
Пример 70:
ЖХ/МС: Rt = 1,850 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 599,0 [М+Н]+.
ВЭЖХ: Rt = 3,85 мин при 10-80_CD_1,2 мл хроматографии (XBridge Shield RP 18 2,1×50 мм, 5 мкм).
1Н ЯМР (400 МГц, CDCl3) δ 10.79 (br s, 1Н), 9.68 (br s, 1Н), 8.86 (br s, 1H), 8.52 (d, J=7,2 Гц, 1H), 8.41 (s, 1H), 7.61 (br s, 1H), 7.01 (d, J=10,8 Гц, 1H), 6.84 (br s, 1H), 6.66 (s, 1H), 6.37 (d, J=16,4 Гц, 1H), 5.80 (d, J=11,2 Гц, 1H), 5.71 (br s, 1H), 3.87 (s, 3H), 3.45-3.31 (m, 2H), 3.25 (br s, 1H), 3.06 (dd, J=6.6, 10,4 Гц, 1H), 2.96 (q, J=8,0 Гц, 1H), 2.60 (br s, 6H), 2.35-2.19 (m, 2H), 2.15-2.09 (m, 1H), 2.04-2.00 (m, 1H), 1.69 (s, 3H), 0.87 (t, J=7,6 Гц, 3Н).
СФХ: Rt = 7,292 мин при AD-3_IPA(DEA)_5_40_2,5 мл.
Пример 71
(R)-N-(5-(4-(4,5-дихлор-2-(2-гидроксипропан-2-ил)фениламино)пиримидин-2-иламино)-2-(2-((диметиламино)метил)азетидин-1-ил)-4-метоксифенил)акриламид
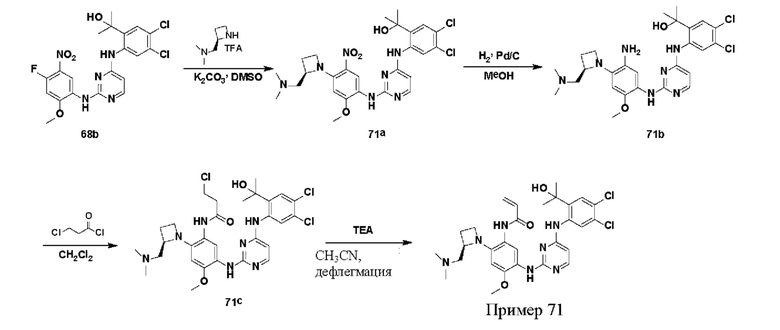
Методика получения соединения 71а:
В раствор соединения 68b (200 мг, 0,42 ммоль) и K2CO3 (1,2 г, 8,4 ммоль) в DMSO (5 мл) добавляли (R)-1-(азетидин-2-ил)-N,N-диметилметанамина TFA соль (887 мг, 4,2 ммоль). Полученную смесь перемешивали при 50°С в течение 1 ч. Реакционную смесь добавляли по каплям в H2O (100 мл) в бане лед-вода при перемешивании. Выпавшее в осадок твердое вещество отфильтровывали, и остаток на фильтре растворяли в CH2Cl2 (45 мл), затем сушили и концентрировали в вакууме с получением неочищенного продукта, который дополнительно очищали обращенно-фазной С-18 колоночной флэш-хроматографией с использованием MeOH/TFA/H2O (МеОН в воде от 10% до 100%) с получением соединения 71а (180 мг, 74%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 0,710 мин при 5-95АВ_1,5 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 576,1 [М+Н]+.
Методика получения соединения 71b:
В раствор 71а (180 мг, 0.31 ммоль) в МеОН (10 мл) добавляли Pd/C (40 мг). Полученную смесь продували и дегазировали Н2 3 раза, затем перемешивали при 15-21°С в атмосфере Н2 (15 фунт/кв.дюйм) в течение 1 ч. Реакционную смесь фильтровали, и фильтрат концентрировали при пониженном давлении с получением соединения 71b (140 мг, 73%-ный выход) в виде черного твердого вещества.
ЖХ/МС: Rt = 0,689 мин при 5-95АВ_1,5 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 546,1 [М+Н]+.
Методика получения соединения 71с:
В раствор соединения 71b (110 мг, 0,2 ммоль) в CH2Cl2 (3 мл) добавляли 3-хлорпропаноилхлорид (26 мг, 0,2 ммоль) в бане лед-вода. Полученную смесь перемешивали при 5-10°С в течение 45 мин, при этом цвет менялся с черного на коричневый. Реакционную смесь вливали в насыщенный NaHCO3 (5 мл) и экстрагировали CH2Cl2 (15 мл × 2). Объединенные органические слои последовательно промывали водой (10 мл × 2) и рассолом (10 мл), сушили и концентрировали в вакууме с получением неочищенного остатка, который очищали колоночной хроматографией на силикагеле (0-5% МеОН в CH2Cl2) с получением соединения 71с (50 мг, 39%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,883 мин при 10-80АВ_2 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 636,1 [М+Н]+.
Методика получения соединения Примера 71:
В раствор соединения 71с (50 мг, 0,078 ммоль) в CH3CN (5 мл) добавляли Et3N (32 мг, 0,31 ммоль). Полученную смесь перемешивали при 80°С в течение 12 ч. Реакционную смесь концентрировали в вакууме с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (0-5% МеОН в CH2Cl2) и затем сушили и лиофилизировали с получением соединения Примера 71 (24,1 мг, 51%-ный выход) в виде не совсем белого твердого вещества.
ЖХ/МС: Rt = 1,648 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 600,0 [М+Н]+.
ВЭЖХ: Rt = 2,42 мин при 10-80АВ_1,2 мл хроматографии (Ultimate С18 3×50 мм, 3 мкм).
1Н ЯМР (400 МГц, CDCl3) δ 9.76 (br s, 1Н), 9.40 (br s, 1H), 8.76 (br s, 1H), 8.06 (d, J=5,6 Гц, 1H), 7.86-7.55 (m, 2H), 7.39 (s, 1H), 6.86 (br s, 1H), 6.67 (s, 1H), 6.43-6.35 (m, 2H), 5.80 (d, J=11,6 Гц, 1H), 5.70-5.43 (m, 1H), 4.69 (br s, 1H), 4.09 (br s, 1H), 3.95 (s, 3H), 3.45-3.33 (m, 1H), 3.16-3.08 (m, 2H), 2.75 (br s, 6H), 2.56-2.45 (m, 1H), 2.43-2.34 (m, 1H), 1.71 (s, 6H).
Пример 72
N-(5-(4-(5-хлор-4-фтор-2-(1-гидроксициклобутил)фениламино)-1,3,5-триазин-2-иламино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида муравьинокислая соль

Методика получения соединения 72b:
В смесь соединения 72а (2,0 г, 9,0 ммоль) в толуоле (15 мл) добавляли муравьиную кислоту (1,0 г, 22 ммоль). Полученную смесь кипятили с обратным холодильником в течение 8 часов. Смесь концентрировали в вакууме, и неочищенный продукт промывали метанолом (10 мл) и затем фильтровали. Твердое вещество сушили в вакууме с получением соединения 72b (1,6 г, 71%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 1,12 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (0,02% FA), Waters Acquity UPLC ВЕН C18 1,7 мкм, 2,1×50 мм, 40°С), МС (ЭРИ) m/z = 252,0, 254,0 [М+Н]+.
Методика получения соединения 72с:
В смесь соединения 72b (1,6 г, 6,3 ммоль) добавляли n-BuLi (10 мл, 1,6 М в гексане) при -78°С в атмосфере азота за 3 мин, затем добавляли циклобутанон (1 г, 14 ммоль). Полученную смесь перемешивали при -78°С в течение 10 мин в атмосфере азота. Смесь гасили насыщенным водным раствором NHCl (50 мл) и экстрагировали EtOAc (50 мл). Органический слой концентрировали в вакууме с получением соединения 2 с (500 мг, 32%-ный выход) в виде желтого масла.
ЖХ/МС: Rt = 1,06 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (0,02% FA), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1×50 мм, 40°С), МС (ЭРИ) m/z = 226,0 [М+Н]+.
Методика получения соединения 72d:
В смесь соединения 72с (500 мг, 2,1 ммоль) в этаноле (3 мл) добавляли гидроксид калия (200 мг, 3,6 ммоль) и воду (2 мл). Полученную смесь нагревали при 70°С в течение 15 мин в атмосфере азота. Смесь разбавляли насыщенным водным раствором NH4Cl (50 мл) и экстрагировали EtOAc (50 мл). Органический слой концентрировали в вакууме, остаток очищали С18-флэш-хроматографией с градиентом элюирования от 5% до 60% MeCN в воде (0,02% FA). Чистые фракции упаривали досуха с получением соединения 72d (320 мг, 72%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 1,12 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (0,02% FA), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1×50 мм, 40°С), МС (ЭРИ) m/z = 198,2 [M+H]+.
Методика получения соединения 72е:
В смесь соединения 72d (302 мг, 1,4 ммоль) в DCM (5 мл) добавляли 2,4-дихлор-1,3,5-триазин (300 мг, 2,0 ммоль) и DIEA (300 мг, 2,3 ммоль). Полученную смесь перемешивали при 20°С в течение 16 часов. Смесь затем очищали флэш-хроматографией на диоксиде кремния с градиентом элюирования от 5% до 50% EtOAc в петролейном эфире. Чистые фракции упаривали досуха с получением соединения 72е (350 мг, 77%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 1,39 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (0,02% FA), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1×50 мм, 40°С), МС (ЭРИ) m/z = 329,1 [M+H]+.
Методика получения соединения 72f:
В смесь соединения 72е (160 мг, 0,49 ммоль) в 1-бутаноле (8 мл) добавляли 4-фтор-2-метокси-5-нитроанилин (80 мг, 0,54 ммоль) и TEA (20 мг, 0,21 ммоль). Полученную смесь перемешивали при 55°С в течение 2 часов. Смесь затем очищали С18-флэш-хроматографией с градиентом элюирования от 5% до 70% MeCN в воде (0,02% FA). Чистые фракции упаривали досуха с получением соединения 72f (150 мг, 64%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 1,40 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (0,02% FA), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1×50 мм, 40°С), МС (ЭРИ) m/z = 479,1 [M+H]+.
Методика получения соединения 72g:
В смесь соединения 72f (80 мг, 0,17 ммоль) в NMP (5 мл) добавляли N,N,N'-триметилэтилендиамин (30 мг, 0,29 ммоль) и DIEA (40 мг, 0,31 ммоль). Полученную смесь перемешивали при 60°С в течение 1 часа. Смесь затем очищали С18-флэш-хроматографией с градиентом элюирования от 5% до 60% MeCN в воде (0,02% FA). Чистые фракции упаривали досуха с получением соединения 72g (70 мг, 75%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 1,00 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (0,02% FA), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1×50 мм, 40°С), МС (ЭРИ) m/z = 560,2 [M+H]+.
Методика получения соединения 72h:
В смесь соединения 72g (70 мг, 0,13 ммоль) в метаноле (5 мл) добавляли палладий на углероде (20 мг). Полученную смесь перемешивали при 20°С в течение 1 часа в атмосфере водорода. Смесь затем фильтровали, и фильтрат концентрировали в вакууме с получением соединения 72h (50 мг, 76%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 0,92 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (0,02% FA), Waters Acquity UPLC ВЕН С 18 1,7 мкм, 2,1×50 мм, 40°С), МС (ЭРИ) m/z = 531,3 [М+Н]+.
Методика получения соединения Примера 72:
В смесь соединения 72h (50 мг, 0,094 ммоль) в NMP (2 мл) добавляли акрилоилхлорид (10 мг, 0,11 ммоль) и DIEA (30 мг, 0,23 ммоль) при 0°С. Полученный раствор перемешивали при 0°С в течение 10 мин. Смесь очищали С18-флэш-хроматографией с градиентом элюирования от 5% до 60% MeCN в воде (0,02% FA). Чистые фракции упаривали досуха с получением соединения Примера 72 в форме муравьинокислой соли (28 мг, 51%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 0,97 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (0,02% FA), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1×50 мм, 40°С), МС (ЭРИ) m/z = 585,3 [M+H]+.
1H ЯМР: (500 МГц, DMSO-d6) δ 1.46 (br d, J=7.57 Гц, 1H) 1.83 (br s, 1H) 2.23-2.32 (m, 2H) 2.35-2.44 (m, 2H) 2.51-2.57 (m, 2H) 2.59 (s, 3H) 2.79 (s, 3H) 2.80 (s, 3H) 3.26-3.27 (m, 2H) 3.81 (s, 3H) 5.76 (br d, J=10.40 Гц, 1H) 6.25 (br d, J=17.34 Гц, 1H) 6.52 (br s, 1H) 6.64 (br dd, J=17.18, 10.25 Гц, 1H) 7.00 (br s, 1H) 7.40 (br s, 1H) 8.04 (br s, 1H) 8.26 (s, 1H) 8.81-9.22 (m, 2H) 9.24-9.51 (m, 2H).
Пример 73
(R)-N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-4-метокси-2-(3-(метиламино)пирролидин-1-ил)фенил)акриламид
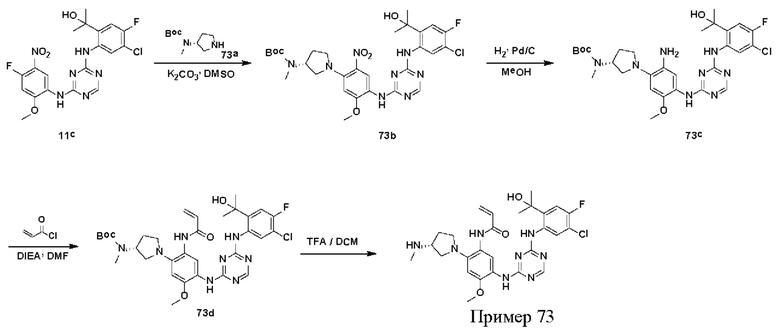
Методика получения соединения 73b:
Раствор соединения 11 с (200 мг, 0,428 ммоль), соединения 73а (128,71 мг, 0,642 ммоль) и K2CO3 (118,4 мг, 0,856 ммоль) в DMSO (2 мл) перемешивали при 85°С в течение 2 ч. Реакционную смесь объединяли с предыдущей партией и добавляли в Н2О (20 мл) в бане лед-вода при перемешивании. Выпавшее в осадок твердое вещество отфильтровывали, и остаток на фильтре растворяли в CH2Cl (50 мл), сушили и концентрировали в вакууме с получением соединения 73b (430 мг, 88,6%-ный выход) в виде оранжевого твердого вещества.
ЖХ/МС: Rt = 0,873 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 647,1 [M+H]+.
1H ЯМР (400 МГц, CDCl3) δ 9.71 (br s, 1H), 8.96 (br s, 1H), 8.38 (m, 2H), 7.36 (s, 1H), 7.08 (d, J=10,4 Гц, 1H), 6.32 (s, 1H), 4.82 (m, 1H), 3.96 (s, 3Н), 3.43-3.28 (m, 3H), 3.14-3.04 (m, 1H), 2.86 (s, 3Н), 2.22-2.10 (m, 2H), 1.70 (s, 6H), 1.47 (s, 9H).
Методика получения соединения 73с:
В раствор соединения 73b (380 мг, 0,587 ммоль) в МеОН (5 мл) добавляли Pd/C (200 мг). Полученную смесь перемешивали при 16-21°С в течение 3 ч в атмосфере H2 (15 фунт/кв.дюйм). Реакционную смесь фильтровали, фильтрат объединяли с предыдущей партией и концентрировали в вакууме с получением соединения 73 с (380 мг, 92,7%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,823 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 617,2 [M+H]+.
1H ЯМР: (400 МГц, CDCl3) δ 9.61 (br s, 1H), 8.33 (m, 2H), 7.87-7.33 (m, 2H), 7.09 (d, J=10,4 Гц, 1H), 6.63 (s, 1H), 4.91 (br s, 1H), 3.83 (s, 3Н), 3.80-3.55 (m, 2H), 3.20 (m, 1H), 3.12-3.06 (m, 2H), 2.92 (s, 3Н), 2.32-2.21 (m, 1H), 1.98-1.87 (m, 1H), 1.68 (s, 6H), 1.48 (s, 9H).
Методика получения соединения 73d:
В раствор соединения 73с (280,0 мг, 0,453 ммоль) и DIEA (87,65 мг, 0,679 ммоль) в DMF (2 мл) по каплям добавляли акрилоилхлорид (41.07 мг, 0,453 ммоль) при 0°С. Полученную смесь перемешивали при 0°С в течение 1 ч. Реакционную смесь гасили Н2О (0,2 мл), объединяли с предыдущей партией и очищали обращенно-фазной С18 колоночной флэш-хроматографией (МеОН и вода) с получением соединения 73d (114 мг, 27,6%-ный выход).
ЖХ/МС: Rt = 0,868 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 671,3 [M+H]+.
1H ЯМР (400 МГц, CDCl3) δ 10.55 (br s, 1H), 9.87 (br s, 1H), 8.72-8.51 (m, 1H), 8.49 (br d, J=7,6 Гц, 1H), 8.42 (s, 1H), 7.66 (br s, 1H), 7.10 (d, J=10,8 Гц, 1H), 6.72 (s, 1H), 6.39 (br s, 2H), 6.02-5.63 (m, 2H), 4.71 (br s, 1H), 3.89 (s, 3Н), 3.19-3.11 (m, 2H), 3.05-2.99 (m, 2H), 2.95 (s, 3Н), 2.38-2.25 (m, 1H), 2.10-2.00 (m, 1H), 1.77 (s, 6H), 1.50 (s, 9H).
Методика получения соединения Примера 73:
В раствор соединения 73d (94 мг, 0,146 ммоль) в CH2Cl2 (3 мл) добавляли TEA (1 мл) при 15-21°С. Полученную смесь затем перемешивали при этой температуре в течение 12 ч. Реакционную смесь объединяли с предыдущей партией и очищали препаративной ВЭЖХ (прибор: ВН колонка: Gemini 150×25, 5 мкм; подвижная фаза А: вода с 0,05% гидроксида аммония об./об.; подвижная фаза В: CH3CN; скорость потока: 25 мл/мин; время градиента: 7 мин; описание профиля: 42%-72%) с получением соединения Примера 73 (27,0 мг, 27,8%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 1,317 мин при 10-80АВ_4 мин 220&254 хроматографии (A: Xtimate С18, 2,1×30 мм, 3 мкм; В: XBrige Shield RP18 2,1×50 мм), МС (ЭРИ) m/z = 571,2 [M+H]+.
ВЭЖХ: Rt = 3,02 мин при 10-80_CD_1,2 мл. мет, XBridge Shield RP 18 2,1×50 мм, 5 мкм.
1H ЯМР: (400 МГц, CDCl3) δ 10.54 (br s, 1H), 9.77 (br s, 1H), 8.72 (br s, 1H), 8.48 (d, J=7,2 Гц, 1H), 8.41 (s, 1H), 7.63 (br s, 1H), 7.09 (d, J-10,4 Гц, 1H), 6.72 (s, 1H), 6.51-6.30 (m, 2H), 5.80 (br d, J=11,4 Гц, 1H), 6.04-5.68 (m, 1H), 3.87 (s, 3H), 3.73 (m, 1H), 3.72-3.66 (m, 1H), 3.49 (s, 1H), 2.98 (m, 3H), 2.48 (s, 3H), 2.32-2.24 (m, 1H), 1.85-1.79 (m, 1H), 1.76 (s, 6H).
Пример 74
(R)-N-(5-(5-хлор-4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)пиримидин-2-иламино)-2-(3-(диметиламино)пирролидин-1-ил)-4-метоксифенил)акриламид
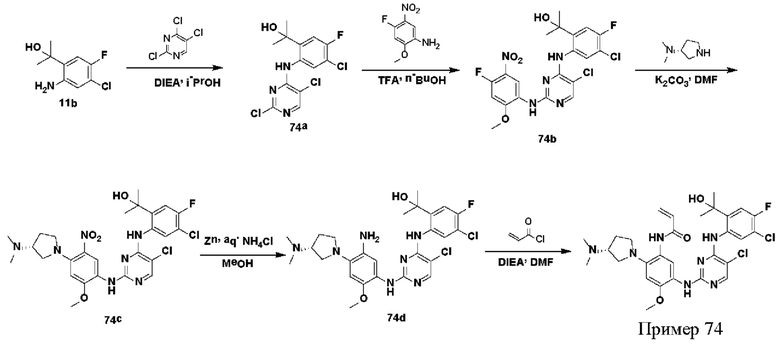
Методика получения соединения 74а:
Смесь соединения 11b (350 мг, 1,72 ммоль), 2,4,5-трихлорпиримидина (314,5 мг, 1,72 ммоль) и DIEA (444 мг, 3,44 ммоль) в i-PrOH (5 мл) перемешивали при 80°С в течение 5 ч. Реакционную смесь объединяли с предыдущей партией и концентрировали в вакууме с получением неочищенного остатка, который очищали колоночной флэш-хроматографией на силикагеле дважды (0-10% EtOAc в петролейном эфире) с получением соединения 74а (580 мг, 84,6%-ный выход) в виде не совсем белого твердого вещества.
ЖХ/МС: Rt = 0,870 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 351,8 [М+Н+2]+.
1H ЯМР: (400 МГц, CDCl3) δ 8.45 (d, J=7,2 Гц, 1H), 8.17 (s, 1H), 7.09 (d, J=10,4 Гц, 1H), 2.77 (br s, 1H), 1.68 (s, 6H).
Методика получения соединения 74b:
Смесь соединения 74а (500 мг, 1.43 ммоль), 4-фтор-2-метокси-5-нитроанилина (345 мг, 1,85 ммоль) и TFA (350 мкл) в n-BuOH (3,5 мл) перемешивали при 100°С в течение 5 ч. Реакционную смесь объединяли с предыдущей партией и затем фильтровали, остаток на фильтре промывали n-BuOH (2 мл) и сушили в вакууме с получением соединения 74b (560 мг, 71,4%-ный выход) в виде серого твердого вещества.
ЖХ/МС: Rt = 0,836 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z = 500,1 [М+Н]+.
Методика получения соединения 74с:
Смесь соединения 74b (150 мг, 0,3 ммоль), (R)-N,N-диметилпирролидин-3-амина (41,1 мг, 0,36 ммоль) и K2CO3 (83,0 мг, 0,6 ммоль) в DMSO (2 мл) перемешивали при 80°С в течение 5 ч (коричневая суспензия). Реакционную смесь объединяли с предыдущей партией и добавляли в Н2О (10 мл), при этом твердое вещество выпадало в осадок. Твердое вещество собирали фильтрованием, промывали Н2О (5 мл) и сушили в глубоком вакууме с получением соединения 74с (220 мг, 92.8%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,709 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z = 594.1 [М+Н]+.
Методика получения соединения 74d:
Смесь соединения 74с (180 мг, 0,3 ммоль), Zn (98,4 мг, 1,5 ммоль) и NH4Cl (81 мг, 1,5 ммоль) в смеси МеОН/Н2О (5,0/2,0 мл) перемешивали при 80°С в течение 3 ч (черная суспензия). Реакционную смесь фильтровали, и остаток на фильтре последовательно промывали CH2Cl2 (20 мл) и Н2О (5 мл), фильтрат экстрагировали CH2Cl2 (30 мл × 3). Объединенный органический слой промывали рассолом (20 мл), сушили над Na2SO4 и концентрировали в вакууме с получением неочищенного остатка, который очищали колоночной флэш-хроматографией на силикагеле (элюируя смесью МеОН/CH2Cl2 = 10/1(об./об.)) с получением соединения 74d (130 мг, 76,0%-ный выход) в виде серого твердого вещества.
ЖХ/МС: Rt = 0,708 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z = 564,1 [М+Н]+.
Методика получения соединения Примера 74:
В смесь соединения 74d (130 мг, 0,23 ммоль) и DIEA (89,1 мг, 0,69 ммоль) в DMF (3 мл) по каплям добавляли акрилоилхлорид (31,0 мг, 0,34 ммоль) в бане лед-вода. После перемешивания реакционной смеси при 0-5°С в течение 4 ч коричневый раствор гасили МеОН (0,05 мл) и напрямую очищали препаративной ВЭЖХ [Xtimate С 18 150×25 мм, 5 мкм; условия: 44-74% В (А: 0,04% аммиак + 10 мМ NH4HCO3 В: CH3CN); скорость потока: 30 мл/мин]. Фракции, содержащие целевое соединение, лиофилизировали с получением соединения Примера 74 (32,4 мг, 22,8%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 2,266 мин при 0-60АВ_4,0 мин_220&254 хроматографии (Xtimate С 18 2,1×30 мм), МС (ЭРИ) m/z = 618,0 [M+H]+.
ВЭЖХ: Rt = 5,68 мин при 0-60_CD_1,2 мл хроматографии (XBridge Shield RP 18 2,1×50 мм, 5 мкм).
1H ЯМР: (400 МГц, CDCl3) δ 9.76 (br s, 1H), 8.93 (br s, 1H), 8.46 (d, J=7,2 Гц, 1Н), 8.20 (br s, 1H), 8.11 (s, 1H), 7.18 (s, 1H), 6.87 (d, J=10,4 Гц, 1H), 6.68 (s, 1H), 6.40-6.20 (m, 2H), 5.72 (d, J=10,8 Гц, 1H), 3.85 (s, 3Н), 3.70-3.47 (m, 1H), 3.20-3.04 (m, 4H), 2.95-2.84 (m, 1H), 2.34 (s, 6H), 2.22-2.10 (m, 1H), 1.98-1.86 (m, 1H), 1.61 (d, J=2,0 Гц, 6Н).
Пример 75
N-(5-(4-(5-циклопропил-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид
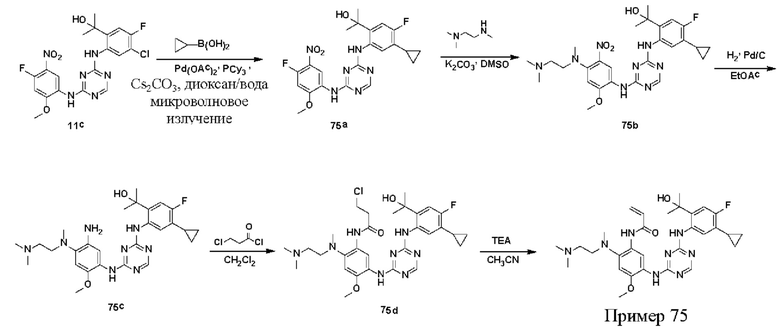
Методика получения соединения 75а:
В раствор соединения 11с (500 мг, 1,07 ммоль) в диоксане (10 мл) и Н2О (2 мл) добавляли циклопропилбороновую кислоту (229 мг, 2,67 ммоль), Pd(OAc)2 (168 мг, 0,75 ммоль), РСу3 (420 мг, 1,50 ммоль) и Cs2CO3 (1,1 г, 3,21 ммоль). Полученную смесь дегазировали N2 в течение 1 мин и перемешивали при 130°С под воздействием микроволнового излучения в течение 1 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (петролейный эфир/EtOAc = 2/1(об./об.)) с получением соединения 75а (300 мг, 59%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,878 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z = 473,1 [M+H]+.
1H ЯМР: (400 МГц, CDCl3) δ 9.32 (s, 1H), 8.55-8.28 (m, 1H), 7.72 (s, 1H), 7.51 (br d, J=13,6 Гц, 1H), 7.26-7.23 (m, 1H), 6.97 (br d, J=11,2 Гц, 1H), 6.76 (d, J=12,0 Гц, 1H), 4.02 (s, 3Н), 2.16 (br d, J=7,2 Гц, 1H), 1.69-1.64 (m, 6H), 0.96 (br s, 2H), 0.73 (br s, 2H).
Методика получения соединения 75b:
В раствор соединения 75а (300 мг, 0,64 ммоль) и K2CO3 (177 мг, 1,28 ммоль) в DMSO (5 мл) добавляли N1,N1,N2-триметилэтан-1,2-диамин (79 мг, 0,77 ммоль). Полученную смесь перемешивали при 14-23°С в течение 4 ч, при этом цвет менялся с коричневого на насыщенный желтый. Реакционную смесь разбавляли EtOAc (20 мл) и промывали водой (30 мл). Органический слой сушили и концентрировали при пониженном давлении с получением неочищенного остатка, который очищали колоночной хроматографией на силикагеле (5% МеОН в CH2Cl2) с получением соединения 75b (200 мг, 56%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 0,760 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 555,1 [M+H]+.
1H ЯМР: (400 МГц, CDCl3) δ 9.47-8.93 (m, 1H), 8.38 (br s, 1H), 7.86-7.51 (m, 1H), 7.40 (br s, 1H), 7.21 (s, 1H), 7.04-6.88 (m, 1H), 6.65 (s, 1H), 3.96 (s, 3Н), 3.28 (bR t, J=7,2 Гц, 2H), 2.87 (s, 3Н), 2.58 (bR t, J=7,2 Гц, 2H), 2.27 (s, 7H), 1.73-1.65 (m, 6H), 1.00-0.90 (m, 4H).
Методика получения соединения 75с:
В раствор соединения 75b (200 мг, 0,36 ммоль) в EtOAc (10 мл) добавляли Pd/C (40 мг). Полученную смесь продували и дегазировали Н; 3 раза, затем перемешивали при 12-21°С в атмосфере Н2 (15 фунт/кв.дюйм) в течение 12 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением соединения 75с (160 мг, 85%-ный выход) в виде светло-желтого твердого вещества.
ЖХ/МС: Rt = 1,799 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С 18 2,1×30 мм), МС (ЭРИ) m/z = 525,2 [M+H]+.
1H ЯМР: (400 МГц, CDCl3) δ 9.12 (s, 1H), 8.34 (br s, 1H), 7.85 (s, 1H), 7.62 (s, 1H), 7.26-7.23 (m, 1H), 7.06-6.90 (m, 1H), 6.67 (s, 1H), 3.82 (s, 3Н), 3.00-2.93 (m, 2H), 2.69-2.64 (m, 3H), 2.43 (br s, 2H), 2.29 (br s, 6H), 2.10 (br s, 1H), 1.67 (s, 6H), 0.96 (br s, 2H), 0.77 (br s, 2H).
Методика получения соединения 75d:
В раствор соединения 75с (160 мг, 0,30 ммоль) в CH2Cl2 (5 мл) добавляли соединение 3-хлорпропаноилхлорид (42 мг, 0,33 ммоль) в бане лед-вода. Полученную смесь перемешивали при 0-5°С в течение 30 мин, при этом немного нерастворенного масла выпало в осадок. Реакционную смесь вливали в насыщенный NaHCO3 (10 мл) и перемешивали при 15-22°С в течение 30 мин, затем экстрагировали CH2Cl2 (15 мл × 2). Объединенные органические слои сушили над Na2SO4 и концентрировали при пониженном давлении с получением неочищенного остатка, который очищали колоночной хроматографией на силикагеле (5% МеОН в CH2Cl2) с получением соединения 75d (150 мг, 81%-ный выход) в виде светло-желтого твердого вещества.
ЖХ/МС: Rt = 0,751 мин при 5-95АВ_1,5 мин_220&254 хроматографии (Xtimate С 18 2,1×30 мм), МС (ЭРИ) m/z = 615,2 [М+Н]+.
1H ЯМР: (400 МГц, CDCl3) δ 10.31 (br s, 1Н), 10.08 (s, 1H), 9.86 (s, 1H), 8.38 (s, 1H), 7.80 (br d, J=8,0 Гц, 1H), 7.65 (br s, 1H), 7.32 (s, 1H), 7.02-6.90 (m, 1H), 6.74 (s, 1H), 3.92 (t, J=6,4 Гц, 2H), 3.87 (s, 3H), 3.80 (t, J=6,8 Гц, 1H), 2.98 (br s, 3H), 2.80 (t, J=6,8 Гц, 1Н), 2.68 (s, 3H), 2.41 (br s, 8H), 1.74 (s, 6H), 1.03-0.94 (m, 2H), 0.82 (br d, J=6,0 Гц, 2Н).
Методика получения соединения Примера 75:
В раствор соединения 75d (150 мг, 0,24 ммоль) в CH3CN (5 мл) добавляли TEA (97 мг, 4,0 экв., 0,96 ммоль). Полученную смесь перемешивали при 80°С в течение 12 ч. Реакционную смесь очищали препаративной ВЭЖХ [колонка: Waters Xbridge 150×25, 5 мкм; условия: 55-85% В (А: 0,05% NH3H2O; В: CH3CN); скорость потока: 25 мл/мин]. Фракции, содержащие целевое соединение, лиофилизировали с получением соединения Примера 75 (51,6 мг, 43%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 1,867 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С 18 2,1×30 мм), МС (ЭРИ) m/z = 579,0 [М+Н]+.
ВЭЖХ: Rt = 3,29 мин при 10-80_ab_1,2 мл хроматографии (Ultimate C18 3 × 50 мм, 3 мкм).
1H ЯМР: (400 МГц, CDCl3) δ 10.43 (br s, 2H), 9.98 (br s, 1H), 8.38 (s, 1H), 7.83 (br d, J=7,6 Гц, 1H), 7.63 (br s, 1H), 6.96 (d, J=11,6 Гц, 1H), 6.78 (s, 1H), 6.44-6.28 (m, 2H), 6.05 (br s, 1H), 5.80-5.73 (m, 1H), 3.87 (s, 3H), 2.91-2.84 (m, 2H), 2.70 (s, 3H), 2.27 (s, 7H), 2.14-2.04 (m, 1H), 1.76 (s, 6H), 1.04-0.94 (m, 2H), 0.86-0.73 (m, 2H).
Пример 76/Пример 77
N-(5-(4-(5-хлор-4-фтор-2-((R)-2-гидроксибутан-2-ил)фениламино)пиримидин-2-иламино)-2-((R)-3-(диметиламино)пирролидин-1-ил)-4-метоксифенил)акриламид и N-(5-(4-(5-хлор-4-фтор-2-((S)-2-гидроксибутан-2-ил)фениламино)пиримидин-2-иламино)-2-((R)-3-(диметиламино)пирролидин-1-ил)-4-метоксифенил)акриламид
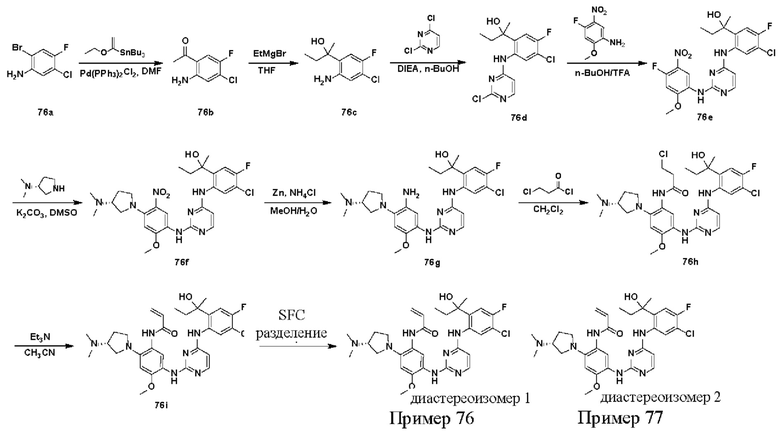
Методика получения соединения 76b:
В раствор соединения 76а (5,0 г, 22,28 ммоль) в DMF (50 мл) добавляли бис(трифенилфосфин)палладий(II) (1,0 г, 1,42 ммоль) и трибутил(1-этоксивинил)олово (8,85 г, 24,51 ммоль) в атмосфере азота. Полученную смесь перемешивали при 110°С в атмосфере азота в течение 12 ч. После охлаждения до комнатной температуры реакционную смесь обрабатывали водным HCl (6 М, 10 мл) и перемешивали при 25°С в течение еще 4 ч. Смесь вливали в воду (300 мл) и экстрагировали этилацетатом (100 мл × 3). Объединенные органические слои последовательно промывали раствором KF (20%, 100 мл × 2) и рассолом (100 мл), сушили над сульфатом натрия и концентрировали с получением черного неочищенного продукта, который очищали колоночной хроматографией на силикагеле (от 0 до 5% этилацетата в РЕ) с получением соединения 76b (1,7 г, 40,6%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 2,030 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 187,9 [М+Н]+.
1H ЯМР (400 МГц, DMSO-d6) δ 7.74 (d, J=10,4 Гц, 1Н), 7.20 (s, 2H), 6.92 (d, J=6,4 Гц, 1Н), 2.48 (s, 3H).
Методика получения соединения 76с:
В желтый раствор соединения 76b (1,7 г, 9,06 ммоль) в THF (20 мл) по каплям добавляли EtMgBr (12 мл, 36,25 ммоль) в атмосфере азота при 0°С. Полученную смесь перемешивали при 19-21°С в течение 2 ч. Смесь гасили насыщенным раствором NH4Cl (100 мл) и экстрагировали EtOAc (50 мл × 3). Объединенные органические слои сушили и концентрировали в вакууме с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (от 0 до 10% EtOAc в РЕ (об./об.)) с получением соединения 76 с (1,2 г, 60,8%-ный выход) в виде желтого масла.
ЖХ/МС: Rt = 0,758 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate C18 2,1×30 мм), МС (ЭРИ) m/z = 199,8 [M-H2O+H]+.
1H ЯМР (400 МГц, CDCl3) δ 6.76 (d, J = 10,8 Гц, 1Н), 6.55 (d, J=6,4 Гц, 1Н), 4.53 (brs, 2H), 1.95-1.80 (m, 2H), 1.51 (s, 3H), 0.79 (t, J=7,2 Гц, 3H).
Методика получения соединения 76d:
В светло-желтый раствор соединения 76с (1,0 г, 4,59 ммоль) в n-BuOH (15 мл) добавляли DIEA (1.19 г, 9,19 ммоль) и 2,4-дихлорпиримидин (753 мг, 5,05 ммоль). Полученную смесь перемешивали при 120°С в течение 10 ч. Смесь концентрировали в вакууме и очищали колоночной хроматографией на силикагеле (от 0 до 20% EtOAc в РЕ) с получением соединения 76d (780 мг, 51,5%-ный выход) в виде светло-желтого твердого вещества.
ЖХ/МС: Rt = 0,872 мин при 5-95АВ_220&254 хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 330,0 [M+H]+.
1H ЯМР (400 МГц, CDCl3) δ 9.18 (s, 1Н), 8.05 (d, J=6,0 Гц, 1Н), 7.89 (d, J=6,8 Гц, 1Н), 6.98 (d, J=10,4 Гц, 1Н), 6.47 (d, J=6,0 Гц, 1Н), 2.36 (s, 1Н), 1.81 (q, J=7,6 Гц, 2H), 1.56 (s, 3H), 0.76 (t, J=7,6 Гц, 3H).
Методика получения соединения 76е:
В смесь соединения 76d (700 мг, 2,12 ммоль) в смеси n-BuOH/TFA (5 мл/0,05 мл) добавляли 4-фтор-2-метокси-5-нитроанилин (415 мг, 2,23 ммоль). Полученную смесь перемешивали при 50°С в течение 3 ч и при 80°С в течение 3 ч. Смесь охлаждали до 25°С, при этом серое твердое вещество выпадало в осадок. Твердое вещество собирали фильтрованием под разряжением и затем сушили в вакууме с получением соединения 76е (680 мг, 62,9%-ный выход) в виде серого твердого вещества.
ЖХ/МС: Rt = 0,755 мин при 5-95АВ_220&254 хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 480,1 [M+H]+.
1H ЯМР: (400 МГц, DMSO-d6) δ 10.28 (br s, 1H), 8.60-8.47 (m, 1H), 8.08 (d, J=6,4 Гц, 1H), 7.85-7.76 (m, 1H), 7.45-7.34 (m, 2H), 6.47 (br. s, 1H), 5.89 (s, 1H), 3.97 (s, 3H), 1.84-1.65 (m, 2H), 1.49 (s, 3H), 0.67 (t, J=7,2 Гц, 3Н).
Методика получения соединения 76f:
В оранжевый раствор соединения 76е (160 мг, 0,33 ммоль) в DMSO (3 мл) добавляли K2CO3 (92 мг, 0,67 ммоль) и (R)-N,N-диметилпирролидин-3-амин (46 мг, 0,40 ммоль). Полученную смесь перемешивали при 50°С в течение 3 ч. Смесь вливали в ледяную воду (30 мл), и оранжевое твердое вещество выпадало в осадок. Его отделяли фильтрованием под разряжением и сушили в вакууме с получением соединения 76f (130 мг, 68,6%-ный выход).
ЖХ/МС: Rt = 0,674 мин при 5-95АВ_220&254 хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 574,1 [M+H]+.
1H ЯМР: (400 МГц, CDCl3) δ 8.98 (d, J=4,8 Гц, 1H), 8.88 (d, J=10,8 Гц, 1H), 7.98 (dd, J=6.0, 1,6 Гц, 1H), 7.90-7.82 (m, 1H), 7.11 (d, J=7,6 Гц, 1H), 6.96 (dd, J=10.8, 2,0 Гц, 1H), 6.22 (s, 1H), 6.06 (dd, J=6.0, 3,6 Гц, 1H), 3.85 (s, 3H), 3.52-3.42 (m, 1H), 3.30-3.23 (m, 1H), 3.12-3.02 (m, 2H), 2.78-2.69 (m, 1H), 2.22 (d, J=3,2 Гц, 6Н), 2.16-2.07 (m, 1H), 1.90-1.75 (m, 3H),1.55 (s, 3H), 0.78 (t, J- 7,2 Гц, 3Н).
Методика получения соединения 76g:
В смесь соединения 76f (200 мг, 0,36 ммоль) в смеси метанол/вода (5 мл/1 мл) добавляли Zn (114 мг, 1,74 ммоль) и NH4Cl (186 мг, 3,48 ммоль). Полученную смесь перемешивали при 90°С в течение 1 ч. Смесь вливали в воду (30 мл) и экстрагировали смесью дихлорметан/метанол (3/1, 20 мл × 4). Объединенные органические слои сушили и концентрировали с получением соединения 76g (220 мг, 95%-ный выход) в виде черного твердого вещества.
ЖХ/МС: Rt = 1,345 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С 18 2,1×30 мм), МС (ЭРИ) m/z = 544,1 [M+H]+.
1H ЯМР: (400 МГц, CDCl3) δ 8.87 (s, 1H), 8.06 (d, J=7,2 Гц, 1H), 7.93 (d, J=6,0 Гц, 1H), 7.77 (s, 1H), 7.34 (s, 1H), 6.94 (d, J=10,8 Гц, 1H), 6.58 (s, 1H), 5.95 (d, J=5,2 Гц, 1H), 3.73 (s, 3Н), 3.15-3.01 (m, 2H), 2.99-2.87 (m, 2H), 2.84-2.74 (m, 1H), 2.20 (s, 6Н), 2.11-1.99 (m, 1H), 1.88-1.77 (m, 3Н), 1.54 (s, 3Н), 0.78 (t, J=7,6 Гц, 3Н).
Методика получения соединения 76h:
В коричневый раствор соединения 76g (200 мг, 0,31 ммоль) в дихлорметане (5 мл) добавляли 3-хлорпропионилхлорид (40 мг, 0,31 ммоль). Полученную смесь перемешивали при 5-10°С в ледяной бане в течение 1 ч. Смесь гасили насыщенным раствором бикарбоната натрия (30 мл) и экстрагировали дихлорметаном (10 мл × 3). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением соединения 76h (200 мг, 86%-ный выход) в виде коричневой смолы.
ЖХ/МС: Rt = 0,715 мин при 5-95АВ_220&254 хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 634,5 [M+H]+.
1H ЯМР: (400 МГц, CDCl3) δ 9.48 (d, J=5,2 Гц, 1Н), 9.31 (br. s, 1H), 8.49 (br. s, 1H), 8.00 (d, J=6,0 Гц, 1H), 7.52-7.46 (m, 1H), 7.42-7.38 (m, 1H), 7.34 (s, 1H), 7.01 (d, J=10,8 Гц, 1H), 6.67 (s, 1H), 6.24 (d, J=6,4 Гц, 1H), 3.84-3.75 (m, 5H), 3.12-2.92 (m, 4H), 2.88-2.76 (m, 3Н), 2.26 (s, 6H), 2.16-2.06 (m, 2H), 1.93-1.85 (m, 2H), 1.59 (s, 3H), 1.19-1.17 (m, 3H).
Методика получения соединения 76i:
В коричневую смесь соединения 76h (200 мг, 0,27 ммоль) в CH3CN (5 мл) добавляли Et3N (81 мг, 0,80 ммоль). Полученную смесь перемешивали при 80°С в течение 2 ч. Реакционную смесь очищали препаративной ВЭЖХ (колонка: Waters Xbridge 150×25, 5 мкм, условия: 42%-62% В (А: вода/10 мМ NH4HCO3, В: CH3CN), скорость потока: 25 мл/мин) и затем лиофилизировали с получением соединения 76i (73,2 мг, 44,7%-ный выход) в виде желтого твердого вещества. Его дополнительно очищали методом СФХ.
ЖХ/МС: Rt = 1,595 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С 18 2,1×30 мм), МС (ЭРИ) m/z = 597,9 [М+Н]1.
ВЭЖХ: Rt = 4.12 мин при 10-80_CD_1,2 мл хроматографии (XBridge Shield RP 18 2,1×50 мм, 5 мкм).
1H ЯМР (400 МГц, CDCl3) δ 9.53 (s, 1H), 9.46 (s, 1H), 8.69 (s, 1H), 8.00 (d, J=5,6 Гц, 1H), 7.47-7.35 (m, 2H), 7.01 (d, J=10,8 Гц, 1H), 6.69-6.48 (m, 2H), 6.35-6.34 (m, 2H), 5.70 (d, J=10,8 Гц, 1H), 5.42 (br s, 1H), 3.79 (s, 3H), 3.28-2.84 (m, 5H), 2.44 (s, 6H), 2.23-2.09 (m, 2H), 2.06-1.99 (m, 1H), 1.95-1.88 (m, 1H), 1.58 (s, 3Н), 0.77 (t, J=7,2 Гц, 3Н).
Хиральная СФХ: Rt = 6,233 мин и 6,903 мин при IC-3_MeOH(DEA)_40_2,5 мл.
Методика получения соединения Примера 76 и соединения Примера 77:
Соединение 76i (73,2 мг) разделяли методом СФХ (колонка: DAICEL CHIRALPAK IC 250 мм × 30 мм, 5 мкм; условия: 40% В (А: СО2, В: 0,1% аммиака/метанол); скорость потока: 60 мл/мин) с получением соединения Примера 76 (13,5 мг, 18,4%-ный выход) и соединения Примера 77 (18,2 мг, 24,9%-ный выход) в виде белого твердого вещества.
Пример 76:
ЖХ/МС: Rt = 1,588 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С 18 2,1×30 мм), МС (ЭРИ) m/z = 598,0 [M+H]+.
ВЭЖХ: Rt = 4,15 мин при 10-80_CD_1,2 мл хроматографии (XBridge Shield RP 18 2,1×50 мм, 5 мкм).
1H ЯМР (400 МГц, CDCl3) δ 9.55 (s, 1H), 9.43 (s, 1H), 8.53 (s, 1H), 8.00 (d, J=6,0 Гц, 1H), 7.44 (d, J=6,8 Гц, 1H), 7.39 (s, 1H), 7.01 (d, J=10,8 Гц, 1H), 6.66 (s, 1H), 6.42-6.32 (m, 1H), 6.30 (s, 1H), 6.26 (d, J=5,2 Гц, 1H), 5.69 (d, J=10,8 Гц, 1H), 5.35 (br s, 1H), 3.79 (s, 3Н), 3.13-2.95 (m, 4H), 2.93-2.82 (m, 1H), 2.29 (s, 6H), 2.18-2.09 (m, 1H), 2.06-1.99 (m, 1H), 1.98-1.88 (m, 2H), 1.58 (s, 3Н), 0.78 (t, J=7,2 Гц, 3Н).
Хиральная СФХ: Rt = 6,292 мин при IC-3_MeOH(DEA)_40 2,5 мл.
Пример 77:
ЖХ/МС: Rt = 1,599 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 597,9 [М+H]+.
ВЭЖХ: Rt = 4,14 мин при 10-80_CD_1,2 мл хроматографии (XBridge Shield RP 18 2,1×50 мм, 5 мкм).
1H ЯМР (400 МГц, CDCl3) δ 9.56 (s, 1H), 9.44 (s, 1H), 8.56 (s, 1H), 8.00 (d, J=6,0 Гц, 1H), 7.43 (d, J=7,2 Гц, 1H), 7.40 (s, 1H), 7.01 (d, J=10,8 Гц, 1H), 6.65 (s, 1H), 6.48-6.35 (m, 1H), 6.30 (s, 1H), 6.26 (d, J=5,6 Гц, 1H), 5.69 (d, J=11,2 Гц, 1H), 5.38 (br s, 1H), 3.78 (s, 3Н), 3.14-3.04 (m, 2H), 3.03-2.96 (m, 2H), 2.95-2.88 (m, 1H), 2.32 (s, 6H), 2.19-2.10 (m, 1H), 2.07-2.00 (m, 1H), 1.98-1.88 (m, 2H), 1.58 (s, 3Н), 0.77 (t, J=7,2 Гц, 3Н).
Хиральная СФХ: Rt = 6,980 мин при IC-3_MeOH(DEA)_40_2,5 мл.
Пример 78
N-(5-(5-хлор-4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)пиримидин-2-иламино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид

Синтез осуществляли в соответствии с экспериментальной методикой, описанной в Примере 74, с получением соединения Примера 78 в виде белого твердого вещества.
ЖХ/МС: Rt = 1,336 мин при 10-80АВ_3 мин_220&254 хроматографии (Xtimate С18, 2,1×30 мм, 3 мкм), МС (ЭРИ) m/z = 606,2 [M+H]+.
ВЭЖХ: Rt = 3,13 мин в 10-80_АВ_1,2 мл. мет Ultimate С 18 3×50 мм, 3 мкм.
1H ЯМР: (400 МГц, CDCl3) δ 10.15 (br s, 1Н), 9.66 (br s, 1H), 9.16 (s, 1H), 8.44 (br d, J=7,4 Гц, 1H), 8.15 (s, 1H), 7.24 (s, 1H), 6.87 (d, J=10,8 Гц, 1H), 6.72 (s, 1H), 6.43-6.17 (m, 2H), 5.69 (d, J=11,2 Гц, 1H), 3.86 (s, 3Н), 3.78-3.66 (m, 1H), 2.88 (br s, 2H), 2.70 (s, 3H), 2.33-2.21 (m, 8H), 1.60 (s, 6H).
Пример 79
N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)пиримидин-2-иламино)-2-((3S,4R)-3-(диметиламино)-4-фторпирролидин-1-ил)-4-метоксифенил)акриламид

Методика получения соединения 79а:

В смесь соединения 79а1 (120 мг, 0,59 ммоль) в метаноле (3 мл) добавляли палладий на углероде (40 мг) и формальдегид (0,3 мл, 37%-ный водный раствор). Полученную смесь перемешивали при 20°С в течение 16 часов в атмосфере водорода. Смесь фильтровали, фильтрат концентрировали в вакууме с получением соединения 79а2 (120 мг, 88%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 1,10 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (0,02% FA), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1×50 мм, 40°С), МС (ЭРИ) m/z = 233,2 [M+H]+.
В смесь соединения 79а2 (120 мг, 0,52 ммоль) в DCM (1 мл) добавляли TFA (1 мл). Полученную смесь перемешивали при 20°С в течение 30 мин. Смесь затем концентрировали в вакууме и разбавляли водой (2 мл). Смесь лиофилизировали с получением соединения 79а (110 мг, 86%-ный выход) в виде белого твердого вещества (TFA соль).
ЖХ/МС: Rt = 0,27 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (0,02% FA), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1×50 мм, 40°С), МС (ЭРИ) m/z = 133,2 [М+Н]+.
Методика получения соединения 79b:
В смесь соединения 79а (40 мг, 0,30 ммоль) в DMF (3 мл) добавляли соединение 36b (100 мг, 0,21 ммоль) и карбонат калия (90 мг, 0,65 ммоль). Полученную смесь перемешивали при 100°С в течение 2 часов. Смесь затем очищали С18-флэш-хроматографией с градиентом элюирования от 5 до 50% CH3CN в воде (0,02% FA). Чистые фракции упаривали досуха с получением соединения 79b (95 мг, 77%-ный выход) в виде оранжевого твердого вещества.
ЖХ/МС: Rt = 0,64 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (0,02% FA), Waters Acquity UPLC ВЕН С 18 1,7 мкм, 2,1×50 мм, 40°С), МС (ЭРИ) m/z = 578,1 [М+Н]+.
Методика получения соединения 79с:
В смесь соединения 79b (95 мг, 0,16 ммоль) в метаноле (5 мл) добавляли палладий на углероде (30 мг). Полученную смесь перемешивали при 20°С в течение 3 часов в атмосфере водорода. Смесь затем фильтровали, и фильтрат концентрировали в вакууме с получением соединения 79 с (75 мг, 83%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 0,99 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (0,02% FA), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1×50 мм, 40°С), МС (ЭРИ) m/z = 548,2 [M+H]+.
Методика получения соединения Примера 79:
В смесь соединения 79с (70 мг, 0,13 ммоль) в NMP (2 мл) добавляли акрилоилхлорид (12 мг, 0,13 ммоль) и DIEA (40 мг, 0,31 ммоль) при 0°С. Полученный раствор перемешивали при 0°С в течение 1 часа. Смесь очищали С18-флэш-хроматографией с градиентом элюирования от 5% до 60% CH3CN в воде (6 ммоль/л NH4HCO3). Чистые фракции упаривали досуха с получением соединения Примера 79 (22 мг, 29%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 1,31 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1×50 мм, 40°С), МС (ЭРИ) m/z = 602,3 [М+Н]+.
1H ЯМР: (500 МГц, DMSO-d6) δ 1.49 (s, 6H) 2.24 (s, 6H) 2.53-2.65 (m, 1H) 3.21-3.33 (m, 2H) 3.41-3.45 (m, 1H) 3.64-3.78 (m, 1H) 3.80 (s, 3H) 5.17-5.35 (m, 1H) 5.68 (dd, J=10.25, 1.73 Гц, 1H) 6.05 (d, J=5.67 Гц, 1H) 6.12-6.23 (m, 2H) 6.39-6.55 (m, 2H) 7.29 (d, J=11.03 Гц, 1H) 7.47 (br s, 1H) 7.82 (br s, 1H) 7.95 (d, J=5.67 Гц, 1H) 8.14 (br d, J=7.25 Гц, 1H) 9.34 (s, 1H) 9.57 (s, 1H).
Пример 80
(R)-N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)пиримидин-2-иламино)-4-метокси-2-(3-(метиламино)пирролидин-1-ил)фенил)акриламида TFA

Методика получения соединения 80а:
В раствор соединения 36b (300 мг, 0,644 ммоль) и К2СО3 (178 мг, 1,29 ммоль) в DMSO (3 мл) добавляли соединение 73а (155 мг, 0,644 ммоль). Полученную смесь перемешивали при 85°С в течение 2 ч, при этом цвет менялся с бледно-желтого на насыщенный желтый. Реакционную смесь вливали в ледяную воду (20 мл) при перемешивании, и желтое твердое вещество выпадало в осадок. Выпавшее в осадок твердое вещество собирали фильтрованием и затем растворяли в CH2Cl2 (50 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением соединения 80а (380 мг, 71%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 0,800 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z = 646,1 [М+Н]+.
Методика получения соединения 80b:
В раствор соединения 80а (350 мг, 0,542 ммоль) в EtOAc (5 мл) добавляли Pd/C (200 мг, 10%, влажный). Полученную смесь продували и дегазировали Н2 3 раза, затем перемешивали при 13-20°С в атмосфере Н2 (15 фунт/кв.дюйм) в течение 2 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением соединения 80b (300 мг, 70%-ный выход) в виде светло-желтого твердого вещества.
ЖХ/МС: Rt = 0,758 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 616,2 [М+Н]+.
Методика получения соединения 80с:
В раствор соединения 80b (300 мг, 0,377 ммоль) в CH2Cl2 (5 мл) по каплям добавляли акрилоилхлорид (47,9 мг, 0,377 ммоль) в бане лед-вода. Полученную смесь перемешивали при 0-5°С в течение 30 мин. Реакционную смесь вливали в насыщенный NaHCO3 (5 мл) и перемешивали при 12-17°С в течение 2 ч, затем экстрагировали CH2Cl2 (5 мл × 2). Объединенные органические слои сушили над Na2SO4 и концентрировали при пониженном давлении с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (8% МеОН в CH2Cl2) с получением соединения 80с (200 мг, 60%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 0,825 мин при 5-95АВ_1,5 мин_220&254 хроматографии (Agilent Pursit 5 С18 20×2,0 мм), МС (ЭРИ) m/z = 670,5 [M+H]+.
1H ЯМР: (400 МГц, CDCl3) δ 9.68-9.56 (m, 1Н), 9.37 (s, 1H), 8.49 (br s, 1H), 8.01 (d, J=6,0 Гц, 1H), 7.43 (s, 1H), 7.37-7.27 (m, 1H), 7.08 (d, J=10,8 Гц, 1H), 6.64 (s, 1H), 6.32-6.26 (m, 2H), 5.74-5.65 (m, 1H), 4.68 (br s, 1H), 3.80 (s, 3H), 3.20-3.12 (m, 2H), 2.97-2.90 (m, 2H), 2.87 (s, 3H), 2.27-2.18 (m, 1H), 1.97-1.89 (m, 1H), 1.63 (s, 8H), 1.40 (s, 9H).
Методика получения соединения Примера 80:
В раствор соединения 80с (200 мг, 2,12 ммоль) в CH2Cl2 (10 мл) добавляли TFA (1,57 г, 21,2 ммоль) при 0°С. Смесь перемешивали при 18-22°С в течение 6 ч. Смесь концентрировали в вакууме с получением неочищенного продукта в виде коричневого масла, которое очищали препаративной ВЭЖХ (прибор: ВН колонка: Gemini 150×25, 5 мкм; подвижная фаза А: вода с 0,05% гидроксида аммония об./об.; подвижная фаза В: DMF; скорость потока: 25 мл/мин; время градиента: 7 мин; описание профиля: 42%-72%) с получением соединения Примера 80 в форме TFA соли (90 мг, 78%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 1,455 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate C18 2,1×30 мм), МС (ЭРИ) m/z = 570,1 [М+Н]+.
ВЭЖХ: Rt = 2,15 мин при 10-80_ab_1,2 мл хроматографии (Ultimate C18 3×50 мм 3 мкм).
1H ЯМР: (400 МГц, DMSO-d6) δ 10.53 (br s, 1H), 9.86 (br s, 1H), 9.14 (br s, 1H), 8.90 (br s, 2H), 7.90 (br s, 2H), 7.68 (br s, 1H), 7.40 (br s, 1H), 6.71 (s, 1H), 6.55 (dd, J=10,4 Гц, 17,2 Гц, 1H), 6.43 (br s, 1H), 6.28-6.01 (m, 2H), 5.76-5.70 (m, 1H), 3.82-3.77 (m, 4H), 3.39-3.25 (m, 3H), 3.10-3.01 (m, 1H), 2.68-2.62 (m, 3H), 2.37-2.28 (m, 1H), 2.09-1.98 (m, 1H), 1.47 (s, 6H).
Пример 81
N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)-5-метоксипиримидин-2-иламино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид

Методика получения соединения 81b:
Раствор соединения 81а (200 мг, 1,1 ммоль), соединения 11b (240 мг, 1,2 ммоль) и DIEA (434 мг, 3,4 ммоль) в NMP (6 мл) герметично закрывали и нагревали при 120°С в течение 2 ч. Реакционную смесь разбавляли водой (100 мл) и экстрагировали ЕА (20 мл × 5). Объединенные органические фазы промывали рассолом, сушили над безводным сульфатом натрия, фильтровали, и фильтрат выпаривали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле с градиентом элюирования от 30% до 100% EtOAc в петролейном эфире. Чистые фракции упаривали досуха с получением соединения 81b (50 мг, 13%-ный выход) в виде светло-желтого твердого вещества.
ЖХ/МС: Rt = 1,53 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1×50 мм, 40°С) МС (ЭРИ) m/z = 346,1 [М+Н]+.
1H ЯМР: (500 МГц, DMSO-d6) δ 1.5 (s, 6H) 3.9 (s, 3H) 6.4 (s, 1H) 7.3 (d, J=11,0 Гц, 1H) 8.0 (s, 1H) 8.5 (d, J=7,3 Гц, 1H) 10.8 (s, 1H).
Методика получения соединения 81с:
Раствор соединения 81b (47 мг, 0,14 ммоль), 4-фтор-2-метокси-5-нитроанилина (28 мг, 0,15 ммоль) и TEA (15 мг, 0,136 ммоль) в пропан-2-оле (2,0 мл) герметично закрывали и нагревали при 100°С в течение 48 ч. Реакционную смесь выпаривали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле с градиентом элюирования от 0% до 10% МеОН в DCM. Чистые фракции упаривали досуха с получением соединения 81 с (67 мг, 79%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 1,20 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1×50 мм, 40°С), МС (ЭРИ) m/z = 496,1 [М+Н]+.
Методика получения соединения 81d:
Раствор соединения 81 с (67 мг, 0,14 ммоль), N1,N1,N2-триметилэтан-1,2-диамина (28 мг, 0,27 ммоль) и DIEA (52 мг, 0,41 ммоль) в NMP (0,6 мл) нагревали при 80°С в течение 2 ч. Реакционную смесь разбавляли водой (50 мл) и экстрагировали ЕА (10 мл × 5). Объединенные органические фазы промывали рассолом, сушили над безводным сульфатом натрия, фильтровали, и фильтрат выпаривали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле с градиентом элюирования от 0% до 10% МеОН в DCM. Чистые фракции упаривали досуха с получением соединения 81d (30 мг, 38%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 1,46 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1×50 мм, 40°С), МС (ЭРИ) m/z = 578,1 [М+Н]+.
Методика получения соединения 81е:
Палладий на углероде (11 мг) добавляли в раствор соединения 81d (30 мг, 0,050 ммоль) в МеОН (5 мл). Реакционную смесь перемешивали при 20°С в течение 30 мин в атмосфере водорода. Полученную смесь фильтровали, и фильтрат выпаривали при пониженном давлении с получением соединения 81е (22 мг, неочищенный) в виде светло-коричневого твердого вещества.
ЖХ/МС: Rt = 1,22 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1×50 мм, 40°С), МС (ЭРИ) m/z = 548,2 [М+Н]+.
Методика получения соединения Примера 81:
В раствор соединения 81е (22 мг, 0,04 ммоль) и DIEA (16 мг, 0,12 ммоль) в NMP (0,8 мл) добавляли раствор акрилоилхлорида (21 мг, 0,23 ммоль) в NMP (0,2 мл) при 0°С. Реакционную смесь перемешивали при 0°С в течение 30 мин. Раствор очищали С18-флэш-хроматографией с градиентом элюирования от 0% до 80% MeCN в воде (0,02% аммиака). Чистые фракции лиофилизировали досуха с получением соединения Примера 81 (3,8 мг, 16%-ный выход) в виде не совсем белого твердого вещества.
ЖХ/МС: Rt = 1,34 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3), Waters Acquity UPLC ВЕН С 18 1,7 мкм, 2,1×50 мм, 40°С), МС (ЭРИ) m/z = 602,1 [М+Н]+.
1H ЯМР: (500 МГц, DMSO-d6) δ 1.5 (s, 6H) 2.2 (s, 6H) 2.3 (bR t, J=5,7 Гц, 2Н) 2.7 (s, 3H) 2.8 (bR t, J=5,5 Гц, 2H) 3.8 (s, 3H) 3.8 (s, 3H) 5.7-5.7 (m, 1H) 6.1 (dd, J=16.9, 1,7 Гц, 1Н) 6.2 (s, 1H) 6.3 (dd, J=16.9, 10,2 Гц, 1Н) 7.0 (s, 1H) 7.2 (d, J=11,0 Гц, 1Н) 7.6 (s, 1H) 7.8 (s, 1H) 8.6 (d, J=7,6 Гц, 1Н) 8.7 (s, 1H) 10.0 (s, 1H) 10.4 (s, 1H).
Пример 82
(R)-N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)пиримидин-2-иламино)-2-(2-((диметиламино)метил)-4,4-дифторпирролидин-1-ил)-4-метоксифенил)акриламид
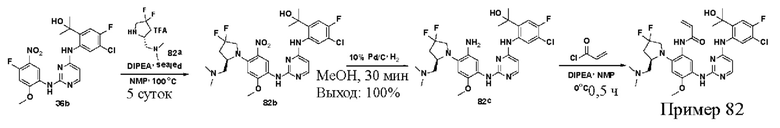
Методика получения соединения 82а:

К перемешиваемому раствору соединения 82а1 (250 мг, 1,054 ммоль) и TEA (213 мг, 2,108 ммоль) в DCM (5 мл) при 0°С в бане лед/вода по каплям добавляли MsCl (127 мг, 1,106 ммоль, 1,05 экв.) на протяжении 2 мин. Затем реакционную смесь перемешивали при этой температуре в течение 1 ч. Затем реакционную смесь разбавляли 50 мл насыщенного раствора NaHCO3 и экстрагировали DCM (20 мл x 3). Объединенный органический слой промывали рассолом (60 мл × 1), сушили над безводным сульфатом натрия, фильтровали и выпаривали при пониженном давлении с получением соединения 82а2 (330 мг, неочищенное) в виде коричневого масла.
ЖХ/МС: Rt = 1,21 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3, Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1×50 мм, 40°С), МС (ЭРИ) m/z=216,1 [M-Boc]+.
В раствор соединения 82а2 (330 мг, 1,05 ммоль) в THE добавляли 40% масс. раствора диметиламина в воде (3 мл). Полученную смесь герметично закрывали и нагревали при 80°С в течение 16 ч. Реакционную смесь разбавляли насыщенным раствором NaHCO3 (50 мл), экстрагировали ЕА (20 мл × 3), промывали рассолом (50 мл × 1), сушили над безводным сульфатом натрия, фильтровали и выпаривали при пониженном давлении с получением соединения 82а3 (275 мг, неочищенное) в виде коричневого масла.
ЖХ/МС: Rt = 0,64 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (0,02% FA, Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1×50 мм, 40°С), МС (ЭРИ) m/z = 208,7 [М-55]+.
Перемешиваемый раствор соединения 82а3 (275 мг, 1,04 ммоль) в DCM (3 мл) охлаждали до 0°С в бане лед/вода. Затем в реакционную смесь по каплям добавляли TFA. Полученную смесь перемешивали и нагревали медленно до комнатной температуры в течение 2 ч. После завершения реакционный раствор концентрировали при пониженном давлении с получением соединения 82а (270 мг, неочищенное) в виде коричневого масла.
ЖХ/МС: Rt = 0,25 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (0,02% FA, Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1×50 мм, 40°С), МС (ЭРИ) m/z = 165,1 [M+1]+.
Методика получения соединения 82b:
Раствор соединения 36b (152 мг, 0,270 ммоль), соединения 82а (106 мг, 0,404 ммоль) и DIPEA (105 мг, 0,810 ммоль) в NMP (2,5 мл) герметично закрывали и нагревали при 100°С в течение 5 суток. Реакционную смесь разбавляли 100 мл воды и экстрагировали ЕА (20 мл × 5). Объединенный органический слой промывали рассолом (100 мл × 1), сушили над безводным сульфатом натрия, фильтровали и выпаривали при пониженном давлении. Остаток очищали на силикагелевой флэш-колонке (MeOH/DCM=0%-10%) с получением соединения 82b (84 мг, 51%-ный выход) в виде светло-коричневого твердого вещества.
ЖХ/МС: Rt = 1,63 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3, Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1×50 мм, 40°С), МС (ЭРИ) m/z = 610,1 [М+Н]+.
Методика получения соединения 82с:
10% Pd/C (29 мг, 0,028 ммоль) добавляли в раствор соединения 82b (84 мг, 0,138 ммоль) в 7 мл МеОН в атмосфере азота. Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода в течение 30 мин. После завершения реакции полученную смесь фильтровали через целит и промывали метанолом. Фильтрат выпаривали при пониженном давлении с получением соединения 82 с (80 мг, неочищенное) в виде светло-коричневого твердого вещества.
ЖХ/МС: Rt = 1,51 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3, Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1×50 мм, 40°С), МС (ЭРИ) m/z = 580,2 [М+Н]+.
Методика получения соединения Примера 82:
Раствор соединения 82с (54 мг, 0,093 ммоль) и DIPEA (36 мг, 0,279 ммоль) в NMP (2 мл) охлаждали и перемешивали при 0°С в бане лед/вода, затем по каплям добавляли раствор акрилоилхлорида (8,4 мг, 0,093, 1,00 экв.) в NMP (0,2 мл). Реакционную смесь перемешивали при 0°С в течение 30 мин. Затем раствор очищали С18/40 G (MeCN/вода = 0%-80%, 0,02% раствор гидроксида аммония в воде) и лиофилизировали с получением соединения Примера 82 (8,50 мг, 14%-ный выход) в виде не совсем белого твердого вещества.
ЖХ/МС: Rt = 1,56 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3, Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1×50 мм, 40°С), МС (ЭРИ) m/z = 634,2 [М+Н]+.
ВЭЖХ: Rt = 10,203 мин (15 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3, Agilent Eclipse Plus С18, 5 мкм, 4,6×150 мм, 30°С).
1H ЯМР (500 МГц, DMSO-d6) δ 1.4 (s, 6H) 2.0 (s, 6H) 2.1-2.4 (m, 4H) 2.5-2.7 (m, 1H) 3.6-3.7 (m, 1H) 3.7 (s, 3H) 3.8 (br d, J=6,3 Гц, 1H) 5.6 (br d, J=10,1 Гц, 1H) 6.0-6.1 (m, 3H) 6.5 (br dd, J=16.7, 10,4 Гц, 1H) 6.8 (s, 1H) 7.2 (br d, J=11,0 Гц, 1H) 7.8 (br s, 1H) 7.9 (br d, J=5,7 Гц, 1H) 8.1 (br d, J=6,9 Гц, 1H) 8.2 (s, 1H) 9.2 (s, 1H) 9.5 (br s, 1H).
Пример 83
(R)-N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)пиримидин-2-иламино)-2-(7-(диметиламино)-5-азаспиро[2.4]гептан-5-ил)-4-метоксифенил)акриламида муравьинокислая соль

Методика получения соединения 83b:
В смесь соединения 36b (120 мг, 0,26 ммоль) в NMP (3 мл) добавляли соединение 83а (55 мг, 0,26 ммоль) и DIEA (74 мг, 0,57 ммоль). Полученную смесь нагревали при 100°С в течение 2 часов. Смесь очищали С18-флэш-хроматографией с градиентом элюирования от 5% до 70% CH3CN в воде (0,02% FA). Чистые фракции упаривали досуха с получением соединения 83b (120 мг, 70%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 1,19 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (0,02% FA), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1×50 мм, 40°С), МС (ЭРИ) m/z = 658,1 [М+Н]+.
Методика получения соединения 83с:
В смесь соединения 83b (120 мг, 0,18 ммоль) в DCM (1 мл) добавляли TFA (1 мл). Полученную смесь перемешивали при 20°С в течение 30 мин. Смесь разбавляли насыщенным водным раствором карбоната натрия (50 мл) и EtOAc (50 мл). Органический слой промывали рассолом, сушили над сульфатом натрия, концентрировали в вакууме с получением соединения 83с (96 мг, 94%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 1,34 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1×50 мм, 40°С), МС (ЭРИ) m/z = 558,1 [М+Н]+.
Методика получения соединения 83d:
В смесь соединения 83с (95 мг, 0,17 ммоль) в метаноле (3 мл) добавляли формальдегид (1 мл) и NaBH3CN (54 мг, 0,85 ммоль). Полученную смесь перемешивали при 20°С в течение 1 часа. Смесь очищали С18-флэш-хроматографией с градиентом элюирования от 5% до 70% CH3CN в воде (0,02% FA). Чистые фракции упаривали досуха с получением соединения 83d (82 мг, 82%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 1,55 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1×50 мм, 40°С), МС (ЭРИ) m/z = 586,0 [М+Н]+.
Методика получения соединения 83е:
В смесь соединения 83d (81 мг, 0,15 ммоль) в метаноле (3 мл) добавляли палладий на углероде (20 мг). Полученную смесь перемешивали при 20°С в течение 1 часа в атмосфере водорода. Смесь затем фильтровали, и фильтрат концентрировали в вакууме с получением соединения 83е (67 мг, 87%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 1,44 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1×50 мм, 40°С), МС (ЭРИ) m/z=556,1 [M+H]+.
Методика получения соединения Примера 83:
В смесь соединения 83е (67 мг, 0,12 ммоль) в NMP (2 мл) добавляли 3-хлорпропионилхлорид (17 мг, 0,13 ммоль) и DIEA (19 мг, 0,15 ммоль) при 0°С. Полученный раствор перемешивали при 0°С в течение 1 часа. Добавляли MeCN (2 мл) и TEA (0,5 мл). Смесь нагревали при 80°С в течение 16 часов. Смесь разбавляли водой (30 мл) и ЕА (30 мл). Органический слой концентрировали, остаток очищали С18-флэш-хроматографией с градиентом элюирования от 5% до 60% CH3CN в воде (0,02% FA). Чистые фракции упаривали досуха с получением соединения Примера 83 в форме муравьинокислой соли (38 мг, 52%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 0,74 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (0,02% FA), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1×50 мм, 40°С), МС (ЭРИ) m/z = 610,2 [M+H]+.
1H ЯМР (500 МГц, DMSO-d6) δ 0.53-0.59 (m, 2Н) 0.66-0.72 (m, 1H) 0.91-0.96 (m, 1H) 1.49 (s, 6H) 2.23 (s, 6H) 2.77 (br dd, J=5.36, 2.84 Гц, 1Н) 2.80 (d, J=8.83 Гц, 1Н) 3.35-3.37 (m, 3H) 3.79 (s, 3H) 5.63-5.73 (m, 1H) 6.06 (d, J=5.67 Гц, 1H) 6.16 (dd, J=17.18, 2.05 Гц, 1H) 6.48 (dd, J=17.02, 10.40 Гц, 1H) 6.56 (s, 1H) 7.29 (d, J=11.03 Гц, 1H) 7.65 (s, 1H) 7.84 (brs, 1H) 7.96 (d, J=5.67 Гц, 1H) 8.14 (d, J=7.25 Гц, 1H) 8.21 (s, 1H) 9.21 (s, 1H) 9.58 (s, 1H).
Пример 84
(R)-N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)-5-(трифторметил)пиримидин-2-иламино)-2-(3-(диметиламино)пирролидин-1-ил)-4-метоксифенил)акриламид

Методика получения соединения 84b:
В раствор соединения 11b (1000 мг, 4,91 ммоль) и DIEA (1269 мг, 9,82 ммоль) в i-PrOH (20 мл) добавляли соединение 84а (1065 мг, 4,91 ммоль). Полученную смесь перемешивали при 50°С в течение 2 ч. Реакционную смесь очищали обращенно-фазовой С-18 колоночной флэш-хроматографией с использованием смеси МеОН/Н2О (МеОН в воде от 10% до 100%) с получением соединения 84b (300 мг, 16%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,944 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 366,1 [M-OH]+.
Методика получения соединения 84с:
В раствор соединения 84b (300 мг, 0,78 ммоль) в n-BuOH (5 мл) с TFA (0,05 мл) добавляли 4-фтор-2-метокси-5-нитроанилин (160 мг, 0,86 ммоль). Полученную смесь перемешивали при 50°С в течение 18 ч. Реакционную смесь вливали в воду (50 мл), и желтое твердое вещество выпадало в осадок. Твердое вещество отфильтровывали и растворяли в CH2Cl2 (60 мл), затем сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением соединения 84с (320 мг, 77%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 0,931 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z = 534,1 [M+H]+.
Методика получения соединения 84d:
В раствор соединения 84с (300 мг, 0,56 ммоль) в DMSO (4 мл) добавляли (R)-N,N-диметилпирролидин-3-амин (64 мг, 0,56 ммоль) и К2СО3 (233 мг, 1,69 ммоль). Смесь перемешивали при 80°С в течение 18 часов. Реакционную смесь вливали в воду (50 мл), и желтое твердое вещество выпадало в осадок. Желтое твердое вещество отфильтровывали и растворяли в CH2Cl2 (60 мл), затем сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением соединения 84d (280 мг, 80%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 0,773 мин при 5-95АВ_1,5 мин_220&254 хроматографии (XBrige Shield RP18 2,1×50 мм), МС (ЭРИ) m/z = 628,4 [М+Н]+.
Методика получения соединения 84е:
В раствор соединения 84d (280 мг, 0,45 ммоль) в МеОН (5 мл) добавляли Pd/C (28 мг) в атмосфере N2. Черную смесь перемешивали при 6-13°С в атмосфере Н2 (15 фунт/кв.дюйм) в течение 1 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением соединения 84е (250 мг, 95,4%-ный выход) в виде коричневого масла.
ЖХ/МС: Rt = 0,720 мин при 10-80CD_3 мин_220&254 хроматографии (XBrige Shield RP18 2,1×50 мм), МС (ЭРИ) m/z = 598,1 [М+Н]+.
Методика получения соединения 84f:
В раствор соединения 84е (250 мг, 1,0 экв., 0,42 ммоль) в CH2Cl2 (10 мл) добавляли 3-хлорпропаноилхлорид (53 мг, 0.42 ммоль) в бане лед-вода. Полученную смесь перемешивали при 0-5°С в течение 45 мин, при этом цвет менялся с черного на коричневый. Реакционную смесь вливали в насыщенный NaHCO3 (5 мл) и экстрагировали CH2Cl2 (15 мл × 2). Объединенные органические слои последовательно промывали водой (10 мл × 2) и рассолом (10 мл), сушили и концентрировали в вакууме с получением соединения 84f (200 мг, 69%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,736 мин при 5-95АВ_1,5 мин_220&254.lcm хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z = 688,1 [М+H]+.
Методика получения соединения Примера 84:
В раствор соединения 84f (200 мг, 0,29 ммоль) в CH3CN (10 мл) добавляли EtsN (118 мг, 1,16 ммоль). Полученную смесь перемешивали при 80°С в течение 12 ч. Реакционную смесь очищали препаративной ВЭЖХ [колонка: Waters Xbridge 150×25, 5 мкм; условия: 42-72% В (А: 0,05%-ный аммиак; В: CH3CN); скорость потока: 25 мл/мин]. Фракции, содержащие целевое соединение, лиофилизировали с получением соединения Примера 84 (85,7 мг, 45,3%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt = 2,312 мин при 0-60АВ_4 мин_220&254.lcm хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 652,0 [M+H]+.
ВЭЖХ: Rt = 3,32 мин при 10-80АВ_1,2 мл.мет хроматографии (Ultimate C18 3×50 мм, 3 мкм).
1H ЯМР: (400 МГц, CDCl3) δ 9.38 (br s, 1H), 8.75 (br s, 1H), 8.36 (br s, 1H), 8.15 (br s, 1H), 7.96 (br s, 1H), 7.43 (s, 1H), 7.04 (d, J=10,6 Гц, 1H), 6.70 (s, 1H), 6.33-6.20 (m, 2H), 5.75-5.68 (m, 1H), 3.87 (s, 3H), 3.21-3.06 (m, 4H), 2.93-2.82 (m, 1H), 2.72 (br s, 1H), 2.30 (s, 6H), 2.23-2.12 (m, 1H), 1.98-1.87 (m, 1H), 1.68 (br d, J=4,0 Гц, 6Н)
Пример 85
(R)-N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)-5-цианопиримидин-2-иламино)-2-(3-(диметиламино)пирролидин-1-ил)-4-метоксифенил)акриламид
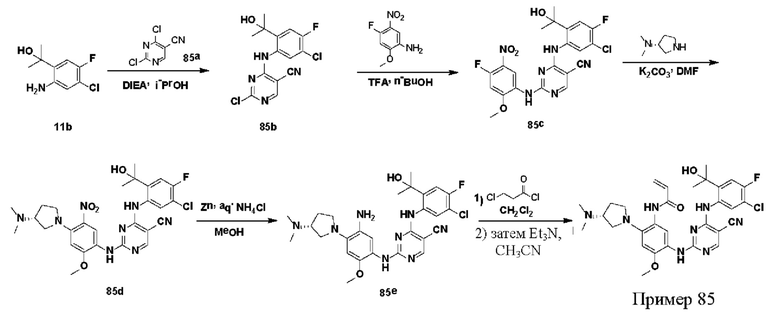
Методика получения соединения 85b:
В раствор соединения 11b (500 мг, 2,46 ммоль) в i-PrOH (5 мл) добавляли DIEA (635 мг, 4,91 ммоль) и соединение 85а (470 мг, 2,70 ммоль). Полученную смесь перемешивали при 80°С в течение 1 ч. Смесь вливали в воду (20 мл) и экстрагировали EtOAc (100 мл × 3). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта, который очищали препаративной ВЭЖХ [колонка Boston Green ODS 150 × 30, 5 мкм, условия: 65% В (А, вода/0,1%TF А, В: CH3CN); скорость потока: 25 мл/мин]. РН фракции доводили до 7-8 насыщенным NaHCO3 и экстрагировали EtOAc (20 мл × 3). Объединенные органические слои промывали рассолом (50 мл), сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением целевого соединения 85b (280 мг, 33%-ный выход) и его региоизомера.
ЖХ/МС: Rt = 3,661 при 10-80АВ_7,0 мин_220&254 хроматографии (Xtimate C18 2,1×30 мм), МС (ЭРИ) m/z = 322,8 [M+H-18]+.
1H ЯМР (400 МГц, DMSO-d6) δ 11.22 (br s, 1H), 8.82 (s, 1H), 8.23 (d, J=7,2 Гц, 1Н), 7.44 (d, J=11,2 Гц, 1H), 6.63 (br s, 1H), 1.53 (s, 6H).
Методика получения соединения 85с:
Смесь соединения 85b (280 мг, 0,82 ммоль), 4-фтор-2-метокси-5-нитроанилина (168,0 мг, 0,90 ммоль) и TFA (300 мкл) в n-BuOH (3,0 мл) перемешивали при 35°С в течение 15 ч. Реакционную смесь фильтровали, и остаток на фильтре промывали n-BuOH (3 мл), затем сушили в вакууме с получением целевого продукта 85с (270 мг, 67,2%-ный выход) в виде серого твердого вещества. Точная структура была подтверждена на этой стадии методом 20-ЯМР (НМВС (гетероядерная многосвязевая корреляционная спектроскопия) и т.д.).
ЖХ/МС: Rt = 0,899 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z = 491,1 [M+H]+.
1H ЯМР: (400 МГц, DMSO-d6) δ 10.81 (s, 1Н), 9.47 (br s, 1H), 8.57 (s, 1H), 8.31 (br d, J=6,4 Гц, 1H), 8.02 (br s, 1H), 7.39 (d, J=13,6 Гц, 1H), 7.31 (br d, J=11,2 Гц, 1H), 7.28-7.14 (m, 1H), 6.56 (br s, 1H), 3.89 (s, 3H), 1.52 (s, 6H).
Методика получения соединения 85d:
Смесь соединения 85с (240 мг, 0,489 ммоль), (R)-N,N-диметилпирролидин-3-амина (66,3 мг, 0,586 ммоль) и К2СО3 (135,0 мг, 0,978 ммоль) в DMSO (3 мл) перемешивали при 80°С в течение 20 ч (коричневая суспензия). Реакционную смесь гасили Н2О (10 мл), разбавляли EtOAc (10 мл), перемешивали в течение 3 ч и затем фильтровали. Остаток на фильтре промывали H2O (5 мл) и затем сушили в глубоком вакууме с получением указанного в заголовке соединения 85d (190 мг, 66,4%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt = 0,760 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z = 607,2 [M+Na]+.
Методика получения соединения 85е:
Смесь соединения 85d (140 мг, 0,239 ммоль), Zn (77,8 мг, 1,196 ммоль) и NH4Cl (64,5 мг, 1,196 ммоль) в МеОН/Н2О (5,0 мл/2,0 мл) перемешивали при 80°С в течение 2,0 ч (белая суспензия). Реакционную смесь фильтровали, остаток на фильтре промывали CH2Cl2 (10 мл) и H2O (10 мл), фильтрат экстрагировали CH2Cl2 (20 мл × 3), объединенные органические слои промывали рассолом (20 мл), сушили над Na2SO4 и концентрировали в вакууме с получением соединения 85е (120 мг, 90,9%-ный выход) в виде серого твердого вещества.
ЖХ/МС: Rt = 0,712 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP-18e 25-2 мм), МС (ЭРИ) m/z = 555,1 [M+H]+.
Методика получения соединения Примера 85:
Смесь соединения 85е (120 мг, 0,217 ммоль) и 3-хлорпропаноилхлорида (34,1 мг, 0,238 ммоль) в CH2Cl2 (3 мл) перемешивали при 0°С в течение 1 ч (коричневая суспензия). Реакционную смесь разбавляли CH3CN (5 мл). Полученную смесь концентрировали в вакууме до объема 4 мл, к остатку добавляли TEA (218 мг, 2,16 ммоль), и полученную смесь перемешивали при 100°С в течение 4 ч (черная суспензия). Реакционную смесь напрямую очищали препаративной ВЭЖХ [Waters Xbridge 150×25, 5 мкм; условия: 42-72% В (А: 0,05%-ный гидроксид аммония; В: CH3CN); скорость потока: 25 мл/мин]. Фракции, содержащие целевое соединение, лиофилизировали с получением соединения Примера 85 (59,0 мг, 32,6%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 2,482 мин при 0-60АВ_4,0 мин_220&254 хроматографии (Xtimate С18 2,1×30 мм), МС (ЭРИ) m/z = 609,1 [M+H]+.
ВЭЖХ: Rt = 5,13 мин при 0-60_CD_1,2 мл хроматографии (XBridge Shield RP 18 2,1×50 мм, 5 мкм).
1H ЯМР: (400 МГц, CDCl3) δ 10.08 (br s, 1H), 9.30-8.03 (m, 2H), 8.01-7.71 (m, 1H), 7.48 (br s, 1H), 6.96 (d, J=10,4 Гц, 1H), 6.64 (s, 1H), 6.21 (br s, 2H), 5.67 (br d, J=6,0 Гц, 1Н), 3.82 (s, 3H), 3.70-3.36 (m, 1H), 3.25-2.98 (m, 4H), 2.82 (q, J=7,2 Гц, 1H), 2.24 (s, 6H), 2.17-2.06 (m, 1H), 1.88-1.82 (m, 1H), 1.62 (br s, 6H).
Пример 86
(S)-N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)пиримидин-2-иламино)-2-(3-((диметиламино)метил)пирролидин-1-ил)-4-метоксифенил)акриламид
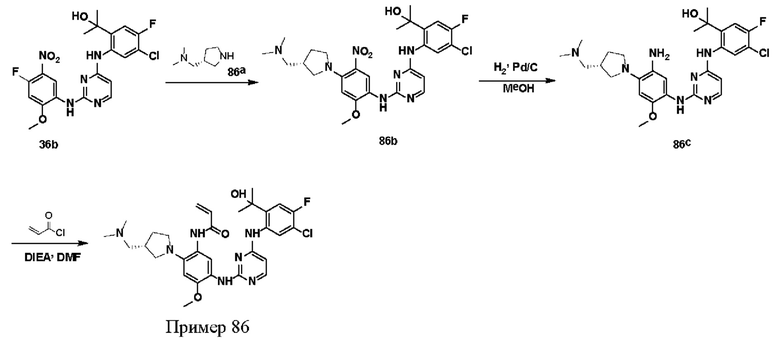
Методика получения соединения 86а:

В раствор соединения 86а1 (1 г, 4,98 ммоль) и TEA (1,0 г, 9,96 ммоль) в CH2Cl2 (10 мл) добавляли MsCl (800 мг, 6,98 ммоль) в бане лед-вода. Полученную смесь перемешивали при 0-5°С в течение 30 мин. Реакционный раствор вливали в рассол (5 мл) и экстрагировали CH2Cl2 (10 мл × 2). Объединенные органические слои последовательно промывали водой (10 мл × 2) и рассолом (10 мл), затем сушили над Na2SO4 и концентрировали в вакууме с получением соединения 86а2 (1,2 г, 86%-ный выход) в виде желтого масла, которое напрямую использовали на следующей стадии без дополнительной очистки.
Смесь соединения 86а2 (1,2 г, 4,30 ммоль) и Me2NH.HCl (1,75 г, 21,5 ммоль) и К2СО3 (4,16 г, 30,1 ммоль) в DMF (10 мл) перемешивали при 100°С в течение 5 ч. Реакционную смесь разбавляли EtOAc (40 мл) и перемешивали в течение еще 30 мин, фильтровали, и остаток на фильтре промывали EtOAc (20 мл). Фильтрат концентрировали в вакууме с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (0-10% МеОН в CH2Cl2) с получением соединения 86а3 (800 мг, 66,7%-ный выход) в виде бесцветного масла.
1H ЯМР: (400 МГц, CDCl3) δ 3.52-3.42 (m, 1Н), 3.41-3.31 (m, 1H), 3.28-3.16 (m, 1H), 2.94-2.85 (m, 1H), 2.34-2.13 (m, 9H), 1.95-1.85 (m, 1H), 1.59-1.44 (m, 1H), 1.38 (s, 9H).
В раствор соединения 86а3 (800 мг, 3,51 ммоль) в CH2Cl2 (10 мл) добавляли TFA (2,6 г, 35,1 ммоль) при 0°С. Смесь перемешивали при 6-13°С в течение 6 ч. Реакционную смесь концентрировали в вакууме с получением указанного в заголовке соединение 86а в форме TFA соли (400 мг, 80,2%-ный выход) в виде бесцветного масла.
1H ЯМР: (400 МГц, CDCl3) δ 3.70 (br s, 1H), 3.51 (br s, 1H), 3.34-3.26 (m, 1H), 3.26-3.22 (m, 1H), 3.00-2.92 (m, 7H), 2.83-2.72 (m, 1H), 2.66-2.54 (m, 1H), 2.16 (br s, 1H), 1.73 (br s, 1H).
Методика получения соединения 86b:
В раствор соединения 36b (200 мг, 0,429 ммоль) и К2СО3 (118 мг, 0,858 ммоль) в DMSO (3 мл) добавляли соединение 86а (65,9 мг, 0,515 ммоль). Полученную смесь перемешивали при 85°С в течение 2 ч, при этом цвет менялся с бледно-желтого на насыщенный желтый. Реакционную смесь вливали в ледяную воду (20 мл) при перемешивании, и желтое твердое вещество выпадало в осадок. Выпавшее в осадок твердое вещество собирали фильтрованием и затем растворяли в CH2Cl2 (50 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением соединения 86b (200 мг, 70,6%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt = 0,691 мин при 5-95АВ_220&254.lcm хроматографии (Agilent Pursit 5 С18 20×2,0 мм), МС (ЭРИ) m/z = 574,5 [M+H]+.
1H ЯМР: (400 МГц, CDCl3) δ 8.97 (s, 1H), 8.90 (s, 1H), 8.09 (d, J=5,6 Гц, 1H), 7.92 (d, J=7,2 Гц, 1H), 7.18 (s, 1H), 7.10 (d, J=10,4 Гц, 1H), 6.33 (s, 1H), 6.17 (d, J=5,6 Гц, 1H), 3.94 (s, 3H), 3.48-3.41 (m, 1H), 3.32-3.27 (m, 1H), 3.14-3.07 (m, 2H), 2.53-2.43 (m, 1H), 2.38-2.33 (m, 2H), 2.27-2.23 (m, 7H), 2.16-2.10 (m, 1H), 1.67 (s, 6H).
Методика получения соединения 86c:
В раствор соединения 86b (200 мг, 0,348 ммоль) в EtOAc (5 мл) добавляли Pd/C (50 мг, 10%, влажный). Полученную смесь продували и дегазировали Н2 3 раза, затем перемешивали при 13-20°С в атмосфере Н2 (15 фунт/кв.дюйм) в течение 2 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением соединения 86 с (150 мг, 72,8%-ный выход) в виде светло-желтого твердого вещества.
ЖХ/МС: Rt=0,667 мин при 5-95АВ_1,5 мин_220&254 хроматографии (Agilent Pursit 5 С18 20 × 2,0 мм), МС (ЭРИ) m/z=544,5 [М+Н]+.
1Н ЯМР: (400 МГц, CDCl3) δ 8.08 (d, J=6,8 Гц, 1H), 7.95 (d, J=5,6 Гц, 1Н), 7.77 (s, 1H), 7.39 (s, 1H), 7.00 (d, J=10,4 Гц, 1H), 6.56 (s, 1H), 5.97 (d, J=5,6 Гц, 1H), 3.74 (s, 3H), 3.52-3.43 (m, 1H), 3.40-3.31 (m, 1H), 3.26-3.16 (m, 1H), 3.08-3.01 (m, 2H), 2.89 (m, 1H), 2.75 (m, 1H), 2.43 (s, 1H), 2.31-2.26 (s, 5H), 2.18-2.13 (s, 3H), 1.95-1.92 (m, 1H), 1.58 (s, 6H).
Методика получения соединения Примера 86:
В раствор соединения 86с (150 мг, 0,276 ммоль) в CH2Cl2 (150 мл) добавляли акрилоилхлорид (150 мг, 0,276 ммоль) в бане лед-вода. Полученную смесь перемешивали при 0-5°С в течение 30 мин. Реакционную смесь вливали в насыщенный NaHCO3 (5 мл) и перемешивали при 12-17°С в течение 2 ч, затем экстрагировали CH2Cl2 (15 мл × 2). Объединенные органические слои сушили над Na2SO4 и концентрировали при пониженном давлении с получением неочищенного остатка, который очищали колоночной хроматографией на силикагеле (8% МеОН в CH2Cl2) с получением соединения Примера 86 (24,5 мг, 15,7%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=1,547 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С18 2,1 × 30 мм), МС (ЭРИ) m/z=598,1 [М+Н]+.
ВЭЖХ: Rt=2,26 мин при 10-80_ab_1,2 мл хроматографии (Ultimate С18 3 × 50 мм 3 мкм).
1Н ЯМР: (400 МГц, CDCl3) δ 9.52-9.33 (m, 2Н), 8.39 (s, 1H), 7.99 (d, J=6,0 Гц, 1H), 7.52-7.41 (m, 2Н), 7.07 (d, J=10,8 Гц, 1H), 6.63 (s, 1H), 6.37-6.24 (m, 3H), 5.75-5.67 (m, 1H), 3.79 (s, 3H), 3.18-3.11 (m, 1Н), 3.05-2.94 (m, 2Н), 2.91-2.85 (m, 1H), 2.81-2.59 (m, 3H), 2.51 (s, 6Н), 2.22-2.14 (m, 1H), 1.82-1.74 (m, 1H), 1.64 (s, 6Н).
Пример 87
(S)-N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)пиримидин-2-иламино)-2-(3-(диметиламино)пирролидин-1-ил)-4-метоксифенил)акриламид
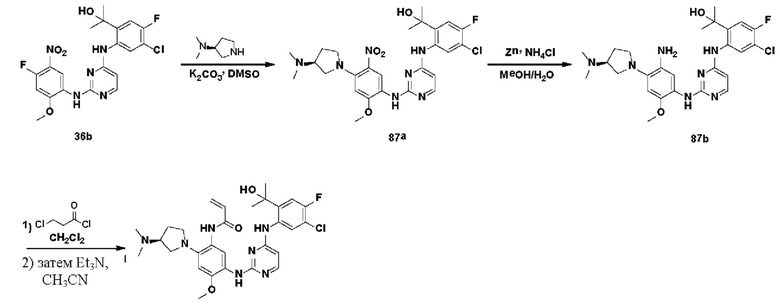
Синтез осуществляли в соответствии с экспериментальной методикой, описанной в Примере 76, с получением соединения Примера 87 в виде белого твердого вещества.
ЖХ/МС: Rt=1,446 мин при 10-80АВ_4 мин_220&254 хроматографии (Xtimate С18 2,1 × 30 мм), МС (ЭРИ) m/z=584,1 [М+Н]+.
ВЭЖХ: Rt=3,74 мин при 10-80_CD_1,2 мл хроматографии (XBridge Shield RP 18 2,1 × 50 мм, 5 мкм).
1Н ЯМР: (400 МГц, CDCl3) δ 9.66 (s, 1H), 9.43 (s, 1H), 8.56 (s, 1Н), 8.09 (d, J=5,6 Гц, 1H), 7.52 (d, J=7,2 Гц, 1H), 7.47 (s, 1H), 7.15 (d, J=10,4 Гц, 1H), 6.76 (s, 1H), 6.41-6.29 (m, 3H), 5.81-5.75 (m, 1H), 5.64 (br s, 1H), 3.86 (s, 3H), 3.16-3.01 (m, 4H), 2.89 (q, J=6,8 Гц, 1H), 2.30 (s, 6H), 2.23-2.12 (m, 1H), 2.00-1.87 (m, 1H), 1.73 (s, 6H).
Пример 88
N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)пиримидин-2-иламино)-2-((2R,4R)-2-((диметиламино)метил)-4-фторпирролидин-1-ил)-4-метоксифенил)акриламида муравьинокислая соль

В перемешиваемый раствор соединения 88а1 (440 мг, 2,0 ммоль) и TEA (404 мг, 4,0 ммоль) в DCM (5 мл) при 0°С в бане лед/вода по каплям добавляли MsCl (276 мг, 2,4 ммоль). Затем реакционную смесь перемешивали при 0°С в течение 1 ч. Затем реакционную смесь разбавляли 50 мл насыщенного раствора NaHCO3 и экстрагировали DCM (30 мл × 3). Объединенный органический слой промывали рассолом (60 мл × 2), сушили над безводным сульфатом натрия, фильтровали и выпаривали при пониженном давлении с получением соединения 88а2 (500 мг, 84%-ный выход) в виде коричневого масла.
ЖХ/МС: Rt=1,32 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3, Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=198,1 [М-Вос]+.
В раствор соединения 88а2 (500 мг, 1,7 ммоль) в THF добавляли водный раствор диметиламина (3 мл, 40% масс). Полученную смесь герметично закрывали и нагревали при 90°С в течение 16 ч. Реакционную смесь разбавляли насыщенным раствором NaHCO3 (50 мл), экстрагировали ЕА (20 мл × 3), промывали рассолом (50 мл × 1), сушили над безводным сульфатом натрия, фильтровали и выпаривали при пониженном давлении с получением соединения 88а3 (320 мг, 77%-ный выход) в виде бесцветного масла.
ЖХ/МС: Rt=1,18 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3, Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=247,2 [М+Н]+.
В перемешиваемый раствор соединения 88а3 (320 мг, 1,3 ммоль) в DCM (5 мл) по каплям добавляли TFA (2 мл) при 0°С. Полученную смесь перемешивали при 20°С в течение 2 ч, затем реакционный раствор упаривали при пониженном давлении с получением соединения 88а (180 мг, 95%-ный выход) в виде бесцветного масла.
ЖХ/МС: Rt=0,25 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3, Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=147,2 [М+Н]+.
Методика получения соединения 88b:
Смесь соединения 36b (225 мг, 0,48 ммоль), соединения 88а (106 мг, 0,73 ммоль) и К2СО3 (200 мг, 1,4 ммоль) в DMSO (3,0 мл) герметично закрывали и нагревали при 85°С в течение 24 ч. Реакционную смесь разбавляли 100 мл воды и экстрагировали ЕА (20 мл × 5). Объединенный органический слой промывали рассолом (100 мл × 1), сушили над безводным сульфатом натрия, фильтровали и выпаривали при пониженном давлении. Остаток очищали флэш-хроматографией на диоксиде кремния с градиентом элюирования от 0% до 10% МеОН в DCM. Чистые фракции упаривали досуха с получением соединения 88b (240 мг, 85%-ный выход) в виде светло-коричневого твердого вещества.
ЖХ/МС: Rt=1,48 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3, Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=592,2 [М+Н]+.
Методика получения соединения 88с:
Палладий на углероде (25 мг, 10% масс.) добавляли в раствор соединения 88b (70 мг, 0,12 ммоль) в МеОН (7 мл) в атмосфере азота. Полученную реакционную смесь перемешивали при 20°С в течение 30 мин в атмосфере водорода. Смесь фильтровали через целит, и фильтрат выпаривали при пониженном давлении с получением соединения 88с (62 мг, 94%-ный выход) в виде светло-коричневого твердого вещества.
ЖХ/МС: Rt=1,34 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3, Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=562,2 [М+Н]+.
Методика получения соединения 88d:
В раствор соединения 88 с (62 мг, 0,11 ммоль) и DIPEA (14 мг, 0,11 ммоль) в NMP (5 мл) добавляли 3-хлорпропионилхлорид (14 мг, 0,11 ммоль) при 0°С. Полученную смесь перемешивали при 0°С в течение 10 мин. Реакционную смесь разбавляли водой (50 мл) и экстрагировали DCM (10 мл × 3). Органический слой промывали рассолом (30 мл × 1), сушили над безводным сульфатом натрия, фильтровали и выпаривали при пониженном давлении с получением соединения 88d (70 мг, 91%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt=1,37 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3, Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=652, 2 [М+Н]+.
Методика получения соединения Примера 88:
Раствор соединения 88d (70 мг, 0,11 ммоль) и TEA (110 мг, 1,1 ммоль) в CH3CN (5 мл) нагревали при 80°С в течение 12 ч. Затем раствор очищали С18-флэш-хроматографией с градиентом элюирования от 0% до 80% CH3CN в воде (0,02% FA). Чистые фракции упаривали досуха с получением соединения Примера 88 в форме муравьинокислой соли (38 мг, 57%-ный выход) в виде не совсем белого твердого вещества.
ЖХ/МС: Rt=1,33 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3, Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=616,2 М+Н]+.
1Н ЯМР (500 МГц, DMSO-d6) δ 1.50 (d, J=2.21 Гц, 6 Н) 1.89-1.99 (m, 1 Н) 2.12 (s, 6 Н)2.18 (dd, J=12.30, 7.57 Гц, 1 Н) 2.34 (br dd, J=12.14, 5.83 Гц, 1 Н) 3.17-3.31 (m, 2 Н) 3.42-3.51 (m, 1 Н) 3.55-3.62 (m, 1 Н) 3.79 (s, 3 Н) 5.33-5.49 (m, 1 Н) 5.72 (br d, J=11.66 Гц, 1 Н) 6.10-6.13 (m, 1 Н)6.13-6.20 (m, 2 Н) 6.38 (dd, J=17.02, 10.40 Гц, 1 Н) 6.86 (s, 1 Н) 7.29 (d, J=11.03 Гц, 1 Н) 7.85 (s, 1 Н) 7.97-8.01 (m, 1 Н) 8.11-8.17 (m, 1 Н) 8.36 -8.45 (m, 1 Н) 9.53-9.56 (m, 1 Н) 9.57-9.61 (m, 1 Н).
Пример 89
N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)пиримидин-2-иламино)-4-метокси-2-((1R,5R)-6-метил-3,6-диазабицикло[3.2.0]гептан-3-ил)фенил)акриламида муравьинокислая соль
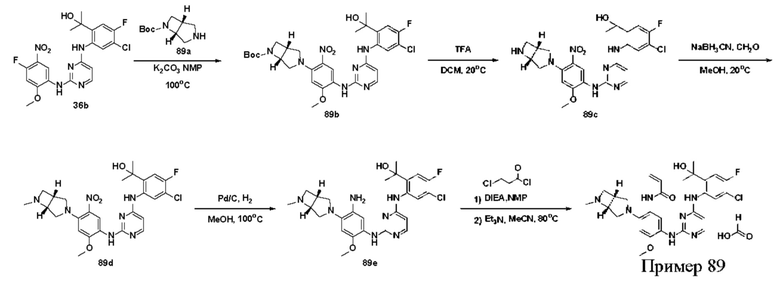
Методика получения соединения 89b:
В смесь соединения 36b (80 мг, 0,17 ммоль) в NMP (3 мл) добавляли соединение 89а (34 мг, 0,17 ммоль) и карбонат калия (60 мг, 0.43 ммоль). Полученную смесь нагревали при 100°С в течение 3 часов. Смесь очищали С18-флэш-хроматографией с градиентом элюирования от 5% до 70% CH3CN в воде (0,02% FA). Чистые фракции упаривали досуха с получением соединения 89b (75 мг, 68%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=1,15 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (0,02% FA), Waters Acquity UPLC ВЕН C18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=644,1 [М+Н]+.
Методика получения соединения 89с:
В смесь соединения 5с (75 мг, 0,11 ммоль) в DCM (1 мл) добавляли TFA (1 мл), Полученную смесь перемешивали при 20°С в течение 30 мин. Смесь разбавляли насыщенным водным раствором карбоната натрия (50 мл) и EtOAc (50 мл). Органический слой промывали рассолом, сушили над сульфатом натрия и концентрировали в вакууме с получением соединения 89с (59 мг, 93%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=1,21 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3), Waters Acquity UPLC ВЕН C18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=544,1 [М+Н]+.
Методика получения соединения 89d:
В смесь соединения 89с (59 мг, 0,11 ммоль) в метаноле (3 мл) добавляли формальдегид (0,5 мл) и NaBH3CN (34 мг, 0,54 ммоль), Полученную смесь перемешивали при 20°С в течение 1 часа. Смесь очищали С18-флэш-хроматографией с градиентом элюирования от 5% до 70% CH3CN в воде (0,02% FA). Чистые фракции упаривали досуха с получением соединения 89d (44 мг, 73%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=1,27 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3), Waters Acquity UPLC ВЕН C18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=558,2 [М+Н]+.
Методика получения соединения 89е:
В смесь соединения 89d (59 мг, 0,11 ммоль) в метаноле (3 мл) добавляли палладий на углероде (20 мг). Полученную смесь перемешивали при 20°С в течение 1 часа в атмосфере водорода. Смесь затем фильтровали, и фильтрат концентрировали в вакууме с получением соединения 89е (42 мг, 75%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=1,17 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3), Waters Acquity UPLC ВЕН C18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=528,1 [М+Н]+.
Методика получения соединения Примера 89:
В смесь соединения 5f (42 мг, 0,080 ммоль) в NMP (2 мл) добавляли акрилоилхлорид (8 мг, 0,089 ммоль) и DIEA (12 мг, 0,093 ммоль) при 0°С. Полученный раствор перемешивали при 0°С в течение 1 часа. Смесь очищали С18-флэш-хроматографией с градиентом элюирования от 5% до 50% CH3CN в воде (0,02% FA). Чистые фракции упаривали досуха с получением соединения Примера 89 в форме муравьинокислой соли (14 мг, 30%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt=0,78 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (0,02% FA), Waters Acquity UPLC ВЕН C18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=582,0 [М+Н]+.
1Н ЯМР (500 МГц, DMSO-d6) δ 0.99 (t, J=7.09 Гц, 1 Н) 1.43 (s, 6 Н) 2.63-2.83 (m, 2 Н) 3.07 (br s, 1 Н) 3.15 (br d, J=9.46 Гц, 1 H) 3.32-3.54 (m, 5 H) 3.66-3.80 (m, 4 H) 5.65 (br d, J=11.03 Гц, 1 H) 6.04 (d, J=5.67 Гц, 1 H) 6.11 (s, 1 H) 6.14 (br d, J=17.02 Гц, 1 H) 6.34-6.47 (m, 1 H) 6.79 (s, 1 H) 7.22 (d, J=11.03 Гц, 1 H) 7.84 (s, 1 H) 7.92 (d, J=5.67 Гц, 1 H) 8.04-8.21 (m, 2 H) 9.27 (s, 1 H) 9.54 (s, 1 H).
Пример 90
N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)пиримидин-2-иламино)-2-((((2R,4S)-1,4-диметилпирролидин-2-ил)метил)(метил)амино)-4-метоксифенил) акриламида муравьинокислая соль
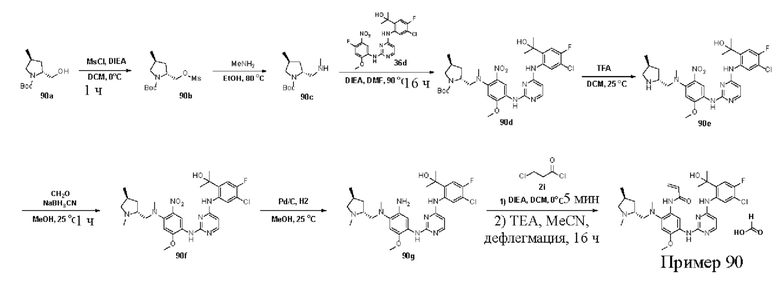
Методика получения соединения 90b:
В перемешиваемый раствор соединения 90а (215 мг, 1,0 ммоль) и DIEA (258 мг, 2,0 ммоль) в DCM (2 мл) добавляли метансульфонилхлорид (127 мг, 1,1 ммоль) при 0°С. Полученный раствор перемешивали при 0°С в течение 1 часа. Затем реакционную смесь гасили водой (10 мл) и экстрагировали DCM (10 мл) дважды. Органический слой затем сушили над сульфатом натрия и концентрировали с получением соединения 90b (300 мг, неочищенный) в виде бесцветного масла, которое может быть использовано на следующей стадии без дополнительной очистки.
ЖХ/МС: Rt=1,39 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л бикарбоната аммония), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=294,2 [М+Н]+.
Методика получения соединения 90с:
В смесь соединения 90b (150 мг, неочищенное, 0,50 ммоль) в этаноле (3 мл) добавляли 25%-ный водный раствор диметиламина (1 мл). Полученную смесь перемешивали при 80°С в течение 16 часов в атмосфере азота. Смесь затем концентрировали, и остаток вливали в воду (10 мл) и экстрагировали этилацетатом (10 мл) дважды. Органический слой затем промывали рассолом, сушили над сульфатом натрия и концентрировали с получением соединения 90с (100 мг, неочищенное) в виде бесцветного масла, которое может быть напрямую использовано без дополнительной очистки.
ЖХ/МС: Rt=1,16 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л бикарбоната аммония), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=229,3 [М+Н]+.
Методика получения соединения 90d:
В смесь соединения 36d (120 мг, 0,26 ммоль) в DMF (3 мл) добавляли соединение 90с (60 мг, 0,26 ммоль) и DIEA (74 мг, 0,57 ммоль). Полученную смесь нагревали при 90°С в течение 16 часов. Смесь очищали С18-флэш-хроматографией с градиентом элюирования от 20% до 100% MeCN в воде (6 ммоль/л бикарбоната аммония). Чистые фракции упаривали досуха с получением соединения 90d (100 мг, 58%-ный выход) в виде оранжевого твердого вещества.
ЖХ/МС: Rt=1,96 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л бикарбоната аммония), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=674,1 [М+Н]+.
Методика получения соединения 90е:
В смесь соединения 90d (100 мг, 0,15 ммоль) в DCM (1 мл) добавляли TFA (1 мл). Полученную смесь перемешивали при 20°С в течение 90 мин. Раствор затем концентрировали, и остаток разбавляли насыщенным водным раствором карбоната натрия (20 мл) и этилацетатом (20 мл). Органический слой промывали рассолом, сушили над сульфатом натрия, концентрировали в вакууме с получением соединения 90е (80 мг, 93%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=1,28 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л бикарбоната аммония), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=574,1 [М+Н]+.
Методика получения соединения 90f:
В смесь соединения 90е (80 мг, 0,14 ммоль) в метаноле (3 мл) добавляли 37%-ный формалин (1 мл) и NaBH3CN (54 мг, 0,85 ммоль). Полученную смесь затем концентрировали, и остаток разбавляли насыщенным водным раствором карбоната натрия (20 мл) и ЕА (20 мл). Органический слой промывали рассолом, сушили над сульфатом натрия и концентрировали в вакууме с получением соединения 90f (80 мг, 98%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=1,53 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л бикарбоната аммония), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=588,1 [М+Н]+.
Методика получения соединения 90g:
В смесь соединения 90f (80 мг, 0,14 ммоль) в метаноле (3 мл) добавляли палладий на углероде (20 мг). Полученную смесь перемешивали при 20°С в течение 1 часа в атмосфере водорода. Смесь затем фильтровали, и фильтрат концентрировали в вакууме с получением соединения 90g (70 мг, 92%-ный выход) в виде желтоватого твердого вещества.
ЖХ/МС: Rt=0,85 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л бикарбоната аммония), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=558,1 [М+Н]+.
Методика получения соединения Примера 90:
В охлажденный перемешиваемый раствор соединения 90g (70 мг, 0,13 ммоль) и DIEA (17 мг, 0,13 ммоль) в DCM (2 мл) добавляли 3-хлорпропионилхлорид (17 мг, 0,13 ммоль) при 0°С. Полученный раствор перемешивали при 0°С в течение 1 часа. Добавляли MeCN (2 мл) и TEA (0,5 мл). Смесь нагревали при 80°С в течение 16 часов. Смесь концентрировали, и остаток очищали С18-флэш-хроматографией с градиентом элюирования от 5% до 50% MeCN в воде (0,05% FA). Чистые фракции упаривали досуха с получением соединения Примера 90 в форме муравьинокислой соли (36,3 мг, 52%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt=1,68 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль бикарбоната аммония), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=612,0 [М+Н]+.
1Н ЯМР (500 МГц, DMSO-d6) δ 9.93 (s, 1H), 9.66 (s, 1H), 8.71 (s, 1Н), 8.20 (s, 1Н), 8.17 (d, J=7,4 Гц, 1H), 7.97 (d, J=5,7 Гц, 1H), 7.91 (s, 1H), 7.24 (d, J=11,0 Гц, 1H), 6.86 (s, 1H), 6.34 (dd, J=16.9, 10,1 Гц, 1H), 6.16 (dd, J=16.9, 2,1 Гц, 1H), 6.08 (d, J=5,7 Гц, 1H), 5.72 (dd, J=10.1, 2,1 Гц, 1H), 3.78 (s, 3H), 3.13-3.10 (m, 1H), 2.88-2.82 (m, 1H), 2.81-2.75 (m, 1H), 2.72 (s, 3H), 2.63 (dd, J=12.9, 4,4 Гц, 1Н), 2.11-1.97 (m, 2Н), 1.61-1.47 (m, 8Н), 0.96 (d, J=6,2 Гц, 3H).
Пример 91
N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)пиримидин-2-иламино)-2-((2R,4S)-2-((диметиламино)метил)-4-метилпирролидин-1-ил)-4-метоксифенил)акриламид

Методика получения соединения 91b:
В перемешиваемый раствор соединения 91а (215 мг, 1,0 ммоль) и DIEA (258 мг, 2,0 ммоль) в DCM (2 мл) добавляли метансульфонилхлорид (127 мг, 1,1 ммоль) при 0°С.Полученный раствор перемешивали при 0°С в течение 1 часа. Затем реакционную смесь гасили водой (10 мл) и экстрагировали DCM (10 мл) дважды. Органический слой затем сушили над сульфатом натрия и концентрировали с получением соединения 91b (300 мг, неочищенное) в виде бесцветного масла, которое может быть использовано на следующей стадии без дополнительной очистки.
ЖХ/МС: Rt=1,39 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л бикарбоната аммония), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=294,2 [М+Н]+.
Методика получения соединения 91с:
В смесь соединения 91b (150 мг неочищенного, 0,50 ммоль) в этаноле (3 мл) добавляли 25%-ный водный раствор диметиламина (1 мл). Полученную смесь перемешивали при 80°С в течение 16 часов в атмосфере азота. Смесь затем концентрировали, и остаток вливали в воду (10 мл) и экстрагировали этилацетатом (10 мл) дважды. Органический слой промывали рассолом и сушили над сульфатом натрия. Раствор концентрировали с получением соединения 91 с (120 мг, неочищенное) в виде бесцветного масла, которое может быть использовано напрямую без дополнительной очистки.
ЖХ/МС: Rt=1,29 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л бикарбоната аммония), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=243,3 [М+Н]+.
Методика получения соединения 91d:
В смесь соединения 1 с (120 мг, 0,50 ммоль) в DCM (1 мл) добавляли TFA (1 мл). Полученную смесь перемешивали при 25°С в течение 3 часов. Смесь затем концентрировали в вакууме и разбавляли водой (2 мл). Смесь лиофилизировали с получением соединения 91d (180 мг, 97%) в виде белого твердого вещества (TFA соль).
ЖХ/МС: Rt=0,27 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (0,02% FA), Waters Acquity UPLC ВЕН C18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=143,2 [М+Н]+.
Методика получения соединения 91е:
В смесь соединения 91d (110 мг, 0,30 ммоль) в DMF (3 мл) добавляли соединение 36d (100 мг, 0,21 ммоль) и DIEA (129 мг, 1,0 ммоль). Полученную смесь перемешивали при 90°С в течение 16 часов. Смесь затем очищали С18-флэш-хроматографией с градиентом элюирования от 20% до 90% MeCN в воде (6 ммоль/л бикарбоната аммония). Чистые фракции упаривали досуха с получением соединения 91е (80 мг, 63%-ный выход) в виде оранжевого твердого вещества.
ЖХ/МС: Rt=1,63 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л бикарбоната аммония), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=588,1 [М+Н]+.
Методика получения соединения 91f:
В смесь соединения 91е (80 мг, 0,14 ммоль) в метаноле (6 мл) добавляли палладий на углероде (20 мг). Полученную смесь перемешивали при 20°С в течение 1 часа в атмосфере водорода. Смесь затем фильтровали, и фильтрат концентрировали в вакууме с получением соединения 91f (76 мг, неочищенное) в виде желтоватого твердого вещества, которое может быть использовано напрямую без дополнительной очистки.
ЖХ/МС: Rt=0,99 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (0,02% FA), Waters Acquity UPLC ВЕН C18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=558,2 [М+Н]+.
Методика получения соединения Примера 91:
В охлажденный перемешиваемый раствор соединения 91f (76 мг, 0,14 ммоль) и DIEA (17,6 мг, 0,14 ммоль) в DCM (2 мл) добавляли 3-хлорпропионилхлорид (17 мг, 0,14 ммоль) и при 0°С. Полученный раствор перемешивали при 0°С в течение 1 часа. Добавляли MeCN (2 мл) и TEA (0,5 мл). Смесь нагревали при 80°С в течение 16 часов. Смесь концентрировали, и остаток очищали С18-флэш-хроматографией с градиентом элюирования от 10% до 70% MeCN в воде (6 ммоль/л бикарбоната аммония). Чистые фракции упаривали досуха с получением соединения Примера 91 (17,6 мг, 21%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt=1,68 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л бикарбоната аммония), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=612,0 [М+Н]+.
1Н ЯМР (500 МГц, DMSO-d6) δ 9.61 (s, 1Н), 9.37 (s, 1H), 8.22-8.15 (m, 2H), 8.00 (d, J=5,7 Гц, 1Н), 7.86 (s, 1H), 7.31 (d, J=11,0 Гц, 1H), 6.86 (s, 1H), 6.53 (dd, J=17.0, 10,2 Гц, 1H), 6.19 (s, 1H), 6.13 (dd, J=16.5, 3,9 Гц, 1H), 5.70 (dd, J=10.1, 2,0 Гц, 1H), 3.81 (s, 3H), 3.70 (dq, J=13.6, 7.0, 5,6 Гц, 1H), 3.45 (dd, J=8.9, 6,5 Гц, 1H), 2.38 (h, J=6,8 Гц, 1H), 2.25 (dd, J=11.9, 4,8 Гц, 1H), 2.12(s, 1H), 1.93-1.75 (m, 2H), 1.52 (s, 6H), 1.05 (d, J=6,7 Гц, 3H).
Пример 92
N-(5-(4-(5-хлор-4-фтор-2-(1,1,1-трифтор-2-гидроксипропан-2-ил)фениламино)пиримидин-2-иламино)-2-((R)-3-(диметиламино)пирролидин-1-ил)-4-метоксифенил)акриламид

Методика получения соединения 92а:
В раствор 2-бром-5-хлор-4-фторанилина (2,0 г, 8,9 ммоль) и триэтиламина (1,2 г, 12 ммоль) в DCM (40 мл) добавляли пивалоилхлорид (1,2 г, 9,8 ммоль) при 0°С. Полученную смесь перемешивали при 20°С в течение 16 ч. Смесь гасили водой (100 мл) и экстрагировали DCM (50 мл × 3). Объединенные органические фазы промывали рассолом, сушили над безводным Na2SO4, фильтровали, и фильтрат выпаривали в вакууме. Остаток очищали флэш-хроматографией на силикагеле с градиентом элюирования от 10% до 20% EtOAc в петролейном эфире. Чистые фракции упаривали досуха с получением соединения 92а (2,7 г, 97%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt=1,51 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3), Waters Acquity UPLC ВЕН C18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=309,0 [М+Н]+.
Методика получения соединения 92b:
В раствор соединения 92а (2,7 г, 8,6 ммоль) в THF (53 мл) добавляли n-BuLi (13 мл, 1,6 М в гексане) при -30°С. Полученную смесь перемешивали при -30°С в течение 1 ч. Добавляли TFAA (2,7 г, 13 ммоль) при -30°С, затем смесь перемешивали при 20°С в течение еще 16 ч. Смесь гасили 1 М раствором HCl (35 мл) и экстрагировали ЕА (30 мл × 5). Объединенные органические фазы сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток обрабатывали конц. HCl (60 мл) и THF (30 мл). Затем полученную смесь нагревали при 100°С в течение 2 ч. Реакционную смесь разбавляли насыщенным раствором NaHCO3 и экстрагировали EtOAc (50 мл × 5). Объединенные органические фазы промывали рассолом и выпаривали в вакууме. Остаток очищали флэш-хроматографией на силикагеле с градиентом элюирования от 0% до 30% EtOAc в петролейном эфире. Чистые фракции упаривали досуха с получением соединения 92b (576 мг, 28%-ный выход) в виде коричневого масла.
1Н ЯМР (500 МГц, DMSO-d6) δ 7.2 (d, J=6,3 Гц, 1 Н) 7.4 (dd, J=10.6, 1,7 Гц, 1 Н) 7.8 (br s, 1 Н).
Методика получения соединения 92с:
В раствор соединения 92b (576 мг, 2,4 ммоль) в THF (12 мл) добавляли метилмагния бромид (4,0 мл, 3М в THF) при 0°С. Полученную реакционную смесь перемешивали при 20°С в течение 1 ч. Реакционную смесь гасили насыщенным раствором NH4Cl (50 мл) и экстрагировали ЕА (50 мл × 3). Объединенные органические фазы промывали рассолом, сушили над безводным сульфатом натрия, фильтровали и выпаривали в вакууме. Остаток очищали флэш-хроматографией на силикагеле с градиентом элюирования от 10% до 50% EtOAc в петролейном эфире. Чистые фракции упаривали досуха с получением соединения 92с (401 мг, 65%-ный выход) в виде коричневого масла.
ЖХ/МС: Rt=1,22 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3), Waters Acquity UPLC ВЕН C18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=258,1 [М+Н]+.
Методика получения соединения 92d:
В раствор соединения 92 с (401 мг, 1,6 ммоль) в NMP (7 мл) добавляли NaH (120 мг, 0,58 ммоль, 60%-ная дисперсия в минеральном масле) при 0°С. Реакционную смесь перемешивали при 0°С в течение 15 мин, затем добавляли 2,4-дихлорпиримидин (232 мг, 1,6 ммоль). Полученную смесь перемешивали при 20°С в течение 1 ч. Реакционную смесь гасили водой (100 мл) при 0°С и экстрагировали ЕА (20 мл × 5). Объединенные органические фазы промывали рассолом, сушили над безводным сульфатом натрия, фильтровали, и фильтрат выпаривали в вакууме. Остаток очищали флэш-хроматографией на силикагеле с градиентом элюирования от 30% до 100% EtOAc в петролейном эфире. Чистые фракции упаривали досуха с получением соединения 92d (374 мг, 65%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt=1,36 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1 × 50 мм, 40°С) МС (ЭРИ) m/z=370,0 [М+Н]+.
Методика получения соединения 92е:
Раствор соединения 92d (374 мг, 1,0 ммоль), 4-фтор-2-метокси-5-нитроанилина (207 мг, 1,1 ммоль) и TFA (576 мг, 5,1 ммоль) в пропан-2-оле (5,7 мл) герметично закрывали и нагревали при 50°С в течение 16 ч. Реакционную смесь упаривали в вакууме. Остаток очищали флэш-хроматографией на силикагеле с градиентом элюирования от 0% до 10% МеОН в DCM. Чистые фракции упаривали досуха с получением соединения 92е (420 мг, 80%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt=1,21 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3), Waters Acquity UPLC ВЕН C18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=520,1 [М+Н]+.
Методика получения соединения 92f:
В раствор соединения 92е (420 мг, 0,81 ммоль) и К2СО3 (447 мг, 3,2 ммоль) в DMSO (8 мл) добавляли (R)-N,N-диметилпирролидин-3-амин (129 мг, 1,1 ммоль). Полученную смесь перемешивали при 50°С в течение 16 ч. Реакционную смесь разбавляли ледяной водой (100 мл) и экстрагировали ЕА (20 мл × 5). Объединенные органические фазы промывали рассолом, сушили над безводным сульфатом натрия, фильтровали, и фильтрат выпаривали в вакууме с получением соединения 92f (500 мг, неочищенный) в виде желтого твердого вещества.
ЖХ/МС: Rt=1,51 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3), Waters Acquity UPLC ВЕН C18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=613,9 [М+Н]+.
Методика получения соединения 92g:
Палладий на углероде (88 мг) добавляли в раствор соединения 92f (253 мг, 0,41 ммоль) в МеОН (8 мл). Полученную смесь перемешивали при 20°С в течение 30 мин в атмосфере водорода. Смесь затем фильтровали, и фильтрат выпаривали в вакууме с получением неочищенного соединения 92g (241 мг, неочищенное) в виде светло коричневого твердого вещества.
ЖХ/МС: Rt=1,10 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (0,02% FA), Waters Acquity UPLC ВЕН C18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=583,9 [М+Н]+.
Методика получения соединения 92h:
В раствор соединения 92g (241 мг, 0,41 ммоль) в DCM (4 мл) по каплям добавляли 3-хлорпропаноилхлорид (55 мг, 0.43 ммоль) при 0°С. Реакционную смесь перемешивали при 0°С в течение 0,5 ч. Смесь затем разбавляли насыщенным водным раствором NaHCO3 (10 мл), и полученную смесь перемешивали при 12-17°С в течение 2 ч. Смесь экстрагировали DCM (20 мл × 3). Объединенные органические фазы промывали рассолом, сушили над безводным сульфатом натрия, фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле с градиентом элюирования от 0% до 3% МеОН в DCM. Чистые фракции упаривали досуха с получением соединения 92h (270 мг, 97%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=1,36 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3), Waters Acquity UPLC ВЕН C18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=673,9 [М+Н]+.
Методика получения соединения Примера 92:
В раствор соединения 92h (270 мг, 0,40 ммоль) в MeCN (4 мл) добавляли TEA (162 мг, 1,60 ммоль). Реакционную смесь нагревали при 80°С в течение 16 ч. Реакционный раствор упаривали в вакууме. Остаток очищали С18-флэш-хроматографией с градиентом элюирования от 0% до 60% MeCN в воде (0,02% аммиака). Чистые фракции лиофилизировали досуха с получением соединения Примера 92 (120 мг, 47%-ный выход) в виде светло-коричневого твердого вещества.
ЖХ/МС: Rt=1,33 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3), Waters Acquity UPLC ВЕН C18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=638,0 [М+Н]+.
1Н ЯМР (500 МГц, DMSO-d6) δ м.д. 1.7-1.8 (m, 1 Н) 1.8 (s, 3 Н) 2.0-2.1 (m, 1 Н) 2.2 (s, 6 Н) 2.7 (quin, J=7,6 Гц, 1 Н) 3.1-3.2 (m, 3 Н) 3.3-3.3 (m, 1 Н) 3.3 (br s, 1 Н) 3.8 (d, J=6,9 Гц, 3 Н) 5.7 (br d, J=10,4 Гц, 1 H) 6.1 (dd, J=5.5, 3,0 Гц, 1 H) 6.2 (dd, J=17.0, 1,6 Гц, 1 H) 6.4-6.5 (m, 2 Н) 7.5 (br dd, J=11.2, 2,0 Гц, 1 Н) 7.5 (br d, J=7,6 Гц, 1 H) 7.9 (br d, J=6,3 Гц, 1 H) 7.9 (d, J=5,7 Гц, 2 H) 8.3 (dd, J=12.1, 7,4 Гц, 1 H) 9.3 (s, 1 H) 9.4 (br s, 1 H).
Пример 93
N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)-1,3,5-триазин-2-иламино)-2-((3S,4R)-3-(диметиламино)-4-фторпирролидин-1-ил)-4-метоксифенил)акриламид

Методика получения соединения 93а:
В смесь соединения 1d (100 мг, 0,21 ммоль) в NMP (3 мл) добавляли соединение 79а (28 мг, 0,21 ммоль) и карбонат калия (73 мг, 0,53 ммоль). Полученную смесь нагревали при 100°С в течение 2 часов. Смесь очищали С18-флэш-хроматографией с градиентом элюирования от 5% до 60% CH3CN в воде (0,02% FA). Чистые фракции упаривали досуха с получением соединения 93а (91 мг, 74%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=0,99 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (0,02% FA), Waters Acquity UPLC ВЕН C18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=579,0 [М+Н]+.
Методика получения соединения 93b:
В смесь соединения 93а (90 мг, 0,16 ммоль) в метаноле (5 мл) добавляли палладий на углероде (30 мг). Полученную смесь перемешивали при 20°С в течение 1 часа в атмосфере водорода. Смесь затем фильтровали, и фильтрат концентрировали в вакууме с получением соединения 93b (78 мг, 91%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=1,24 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3), Waters Acquity UPLC ВЕН C18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=549,1 [М+Н]+.
Методика получения соединения Примера 93:
В смесь соединения 93b (78 мг, 0,14 ммоль) в NMP (2 мл) добавляли акрилоилхлорид (13 мг, 0,14 ммоль) и DIEA (20 мг, 0,16 ммоль) при 0°С. Полученный раствор перемешивали при 0°С в течение 1 часа. Смесь очищали С18-флэш-хроматографией с градиентом элюирования от 5% до 50% CH3CN в воде (0,02% FA). Чистые фракции упаривали досуха с получением соединения Примера 93 (45 мг, 52%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt=0,92 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (0,02% FA), Waters Acquity UPLC ВЕН C18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=603,1 [М+Н]+.
1Н ЯМР (500 МГц, DMSO-d6) δ 1.50 (br s, 6 Н) 2.25 (s, 6 Н) 2.54-2.66 (m, 1 Н) 3.38-3.48 (m, 3 Н) 3.78 (s, 3 Н) 3.79-3.87 (m, 1 Н) 5.17-5.34 (m, 1 Н) 5.21 (br s, 1 Н) 5.32 (br s, 1 Н) 5.68 (br d, J=11.03 Гц, 1 H) 6.18 (br d, J=16.71 Гц, 1 H) 6.27 (br s, 1 H) 6.46 (br dd, J=17.02, 10.09 Гц, 2H) 6.95-7.40 (m, 2 H) 8.21 (brs, 2 H) 8.52-9.11 (m, 1 H)9.37(brs, 1 H) 10.12 (br s, 1H).
Пример 94
N-(2-((2-(диметиламино)этил)(метил)амино)-5-(4-(4-(2-гидроксипропан-2-ил)-1H-индол-5-иламино)-1,3,5-триазин-2-иламино)-4-метоксифенил)акриламид

Методика получения соединения 94b:
В смесь соединения 94а (3,0 г, 12,9 ммоль) в DMF (10 мл) добавляли NBS (2,3 г, 12,9 ммоль). Полученную смесь перемешивали при 20°С в течение 30 мин. Смесь разбавляли водой (50 мл) и экстрагировали EtOAc (50 мл). Органический слой концентрировали в вакууме, остаток очищали флэш-хроматографией на диоксиде кремния с градиентом элюирования от 5% до 50% EtOAc в петролейном эфире. Чистые фракции упаривали досуха с получением соединения 94b (2,0 г, 50%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=1,54 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (0,02% FA), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=313,2 [М+Н]+.
Методика получения соединения 94с:
В смесь соединения 94b (1,6 г, 5,1 ммоль) и комплекса [1,1'-бис(дифенил-фосфино)ферроцен]дихлорпалладия(II) с дихлорметаном (200 мг, 0,25 ммоль) в метаноле (15 мл) добавляли TEA (1 г, 9,9 ммоль). Полученную смесь нагревали при 80°С в течение 16 часов в атмосфере монооксида углерода. Смесь концентрировали в вакууме, и остаток очищали С18-флэш-хроматографией с градиентом элюирования от 5% до 80% MeCN в воде (0,02% FA). Чистые фракции упаривали досуха с получением соединения 94 с (350 мг, 24%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=1,49 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (0,02% FA), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=291,3 [М+Н]+.
Методика получения соединения 94d:
В смесь соединения 1с (300 мг, 1,0 ммоль) в THF (10 мл) добавляли MeMgBr (10 мл, 3М в THF). Полученную смесь перемешивали при 20°С в течение 1 часа в атмосфере азота. Смесь затем разбавляли водой (50 мл) и экстрагировали EtOAc (50 мл). Органический слой концентрировали в вакууме, и остаток очищали С18-флэш-хроматографией с градиентом элюирования от 5% до 70% MeCN в воде (0,02% FA). Чистые фракции упаривали досуха с получением соединения 1d (120 мг, 40%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=1,39 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (0,02% FA), Waters Acquity UPLC ВЕН C18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=291,3 [М+Н]+.
Методика получения соединения 94е:
В смесь соединения 94d (120 мг, 0,41 ммоль) в NMP (5 мл) добавляли соединение 6е (130 мг, 0,43 ммоль) и DIEA (150 мг, 1,2 ммоль). Полученную смесь перемешивали при 100°С в течение 30 мин. Смесь затем очищали С18-флэш-хроматографией с градиентом элюирования от 5% до 70% MeCN в воде (0,02% FA). Чистые фракции упаривали досуха с получением соединения 94е (110 мг, 48%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=1,52 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (0,02% FA), Waters Acquity UPLC ВЕН C18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=554,2 [М+Н]+.
Методика получения соединения 94f:
В смесь соединения 94е (110 мг, 0,20 ммоль) в NMP (5 мл) добавляли N,N,N'-триметилэтилендиамин (30 мг, 0,29 ммоль) и DIEA (40 мг, 0,31 ммоль). Полученную смесь перемешивали при 60°С в течение 2 часов. Смесь затем очищали С18-флэш-хроматографией с градиентом элюирования от 5% до 60% MeCN в воде (0,02% FA). Чистые фракции упаривали досуха с получением соединения 94f (120 мг, 95%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=1,04 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (0,02% FA), Waters Acquity UPLC ВЕН C18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=636,2 [М+Н]+.
Методика получения соединения 94g:
В смесь соединения 94f (110 мг, 0,17 ммоль) в метаноле (5 мл) добавляли метоксид натрия (500 мг, 4,0 ммоль). Полученную смесь перемешивали при 20°С в течение 6 часов. Смесь затем разбавляли водой (50 мл) и экстрагировали EtOAc (50 мл). Органический слой концентрировали в вакууме с получением соединения 94g (75 мг, 81%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=0,78 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (0,02% FA), Waters Acquity UPLC ВЕН C18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=536,2 [М+Н]+.
Методика получения соединения 94h:
В смесь соединения 94g (40 мг, 0,075 ммоль) в метаноле (5 мл) добавляли палладий на углероде (20 мг). Полученную смесь перемешивали при 20°С в течение 2 часов в атмосфере водорода. Смесь затем фильтровали, и фильтрат концентрировали в вакууме с получением соединения 94h (32 мг, 85%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt=0,67 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (0,02% FA), Waters Acquity UPLC ВЕН C18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=506,3 [М+Н]+.
Методика получения соединения Примера 94:
В смесь соединения 94h (32 мг, 0,063 ммоль) в NMP (2 мл) добавляли акрилоилхлорид (7 мг, 0,077 ммоль) и DIEA (20 мг, 0,15 ммоль) при 0°С. Полученный раствор перемешивали при 0°С в течение 10 мин. Смесь очищали С18-флэш-хроматографией с градиентом элюирования от 5% до 60% MeCN в воде (0,02% FA). Чистые фракции упаривали досуха с получением соединения Примера 94 (14 мг, 39%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt=0,70 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (0,02% FA), Waters Acquity UPLC ВЕН C18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=560,3 [М+Н]+.
1Н ЯМР (500 МГц, DMSO-d6) δ 1.69 (s, 7 Н) 2.22 (s, 7 Н) 2.34 (br t, J=5.83 Гц, 2 H) 2.70 (s, 3 Н) 2.87 (br t, J=5.67 Гц, 2 H) 3.77 (br s, 3 H) 5.69-5.78 (m, 1 H) 5.92 (br s, 1 H) 6.23 (dd, J=17.02, 1.89 Гц, 1 H) 6.30-6.47 (m, 1 H) 6.56-6.71 (m, 1 H) 6.96 (s, 1 H) 7.06-7.29 (m, 2 H) 7.65 (br s, 1 H) 8.09-8.27 (m, 1 H) 8.12-8.57 (m, 1 H) 8.46 (br s, 1 H) 10.09 (br s, 2 H) 10.86-11.07 (m, 1 H).
Пример 95
N-(2-((2-(диметиламино)этил)(метил)амино)-5-(4-(6-(2-гидроксипропан-2-ил)-1H-индол-5-иламино)-1,3,5-триазин-2-иламино)-4-метоксифенил)акриламид

Методика получения соединения 95а:
В раствор соединения 32d (3,0 г, 8,10 ммоль) в МеОН (50 мл) добавляли Pd/C (300 мг). Полученную смесь продували и дегазировали Н2 3 раза, затем перемешивали при 29-40°С в атмосфере Н2 (водородный баллон, 15 фунт/кв.дюйм) в течение 1 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением соединения 95а (1,48 г, 95,5%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt=1,440 мин при 10-80CD_3 мин_220&254 хроматографии (XBrige Shield RP18 2,1 × 50 мм), МС (ЭРИ) m/z=191,1 [М+Н]+.
1Н ЯМР (400 МГц, DMSO-d6) δ 10.82 (br s, 1Н), 7.84 (s, 1H), 7.42 (t, J=2,8 Гц, 1H), 6.80 (s, 1H), 6.16 (br s, 1H), 5.91 (br s, 2H), 3.80 (s, 3H).
Методика получения соединения 95b:
В раствор соединения 95а (1,3 г, 6,84 ммоль) в THF (100 мл) добавляли CH3MgBr (9,1 мл, 3 М в диэтиловом эфире) при 0-5°С. Смесь перемешивали при 29-40°С в течение 1,5 ч. Реакционную смесь гасили добавлением водного NH4Cl (20 мл), затем экстрагировали EtOAc (3 × 100 мл). Органические слои промывали рассолом (3 × 100 мл), сушили и концентрировали в вакууме с получением неочищенного остатка, который очищали колоночной хроматографией на силикагеле (петролейный эфир/EtOAc=20/1) с получением соединения 95b (1,1 г, 84,6%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt=0,880 мин при 10-80CD_7 мин_220&254 хроматографии (XBrige Shield RP18 2,1 × 50 мм), МС (ЭРИ) m/z=191,1 [М+Н]+.
1Н ЯМР (400 МГц, DMSO-d6) δ 10.47 (br s, 1H), 7.09 (s, 2H), 6.78-6.62 (m, 1H), 6.07 (s, 1H), 5.18 (br s, 1H), 4.92 (br s, 2H), 1.56 (s, 6H).
Методика получения соединения 95c:
В раствор соединения 95b (200 мг, 1,05 ммоль) и DIEA (204 мг, 1,58 ммоль) в CH2Cl2 (5 мл) добавляли 2,4-дихлор-1,3,5-триазин (174 мг, 1,16 ммоль). Полученную смесь перемешивали при 25-33°С (комнатная температура) в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного остатка, который очищали колоночной хроматографией на силикагеле (петролейный эфир/EtOAc=5/1) с получением соединения 95 с (200 мг, 62.7%-ный выход) в виде коричневого твердого вещества.
ЖХ/МС: Rt=0,717 мин при 5-95АВ 1,5 мин 220&254 хроматографии (Xtimate С18 2,1 × 30 мм), МС (ЭРИ) m/z=285,9 [М-18]+.
1Н ЯМР (400 МГц, CDCl3) δ 10.13-9.72 (m, 1Н), 8.56-8.41 (m, 1H), 8.35-8.09 (m, 2Н), 7.40 (s, 1H), 7.26 (t, J=2,6 Гц, 1H), 6.65-6.51 (m, 1Н), 1.76 (s, 6Н).
Методика получения соединения 95d:
В раствор соединения 95с (190 мг, 0,63 ммоль) и соединения 2g (168 мг, 0,63 ммоль) в n-BuOH (5 мл) добавляли TFA (0,05 мл). Полученную смесь перемешивали при 25-33°С в течение 3ч. В реакционную смесь добавляли 10 мл воды, и смесь экстрагировали EtOAc (10 мл × 3). Объединенные органические слои промывали рассолом (10 мл × 3), сушили и концентрировали в вакууме с получением неочищенного остатка, который очищали препаративной ТСХ (CH2Cl2/МеОН=15/1(об./об.)) на силикагеле с получением соединения 95d (250 мг, 73%-ный выход) в виде красного твердого вещества.
ЖХ/МС: Rt=0,728 мин при 5-95АВ_1,5 мин_220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z=518,3 [М-18]+.
Методика получения соединения 95е:
В раствор соединения 95d (190 мг, 0,35 ммоль) в МеОН (3 мл) добавляли Pd/C (20 мг) в защитной атмосфере N2. Смесь перемешивали при 26-33°С в атмосфере Н2 (водородный баллон, 15 фунт/кв.дюйм) в течение 1 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением соединения 95е (100 мг, 75%-ный выход) в виде коричневого масла.
ЖХ/МС: Rt=0,703 мин при 5-95АВ 1,5 мин 220&254 хроматографии (MERCK RP18e 25-2 мм), МС (ЭРИ) m/z=506,4 [М+Н]+.
Методика получения соединения Примера 95:
В раствор соединения 95d (100 мг, 0,20 ммоль) и DIEA (38 мг, 0,30 ммоль) в DMF (1 мл) добавляли акрилоилхлорид (18 мг, 0,20 ммоль) в DMF (1 мл). Полученную смесь перемешивали при 0°С в течение 30 мин. Реакционную смесь очищали препаративной ВЭЖХ [колонка: Waters Xbridge 150 × 25, 5 мкм; условия: 25-55% В (А: 0,05%-ный аммиак; В: CH3CN); скорость потока: 25 мл/мин]. Фракции, содержащие целевое соединение, лиофилизировали с получением соединения Примера 95 (13,6 мг, 12,2%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt=1,908 мин при 10-80_CD_3 мин 220&254 хроматографии (XBrige Shield RP18 2,1 × 50 мм), МС (ЭРИ) m/z=560,3 [М+Н]+.
ВЭЖХ: Rt=3,44 минут при 10-80_CD_1,2 мл.мет (XBridge Shield RP 18 2,1 × 50 мм 5 мкм).
1Н ЯМР (400 МГц, DMSO-d6) δ 10.87 (br s, 1H), 10.04 (br s, 2Н), 8.43 (br s, 1H), 8.19 (br s, 2Н), 7.28 (br s, 2Н), 7.02 (br s, 1H), 6.41 (br d, J=9,4 Гц, 1H), 6.22 (br d, J=16,4 Гц, 2H), 5.91 (br s, 1H), 5.74 (br d, J=10,8 Гц, 1H), 3.80 (s, 3H), 2.90 (br s, 2H), 2.71 (s, 3H), 2.42-2.12 (m, 8H), 1.57 (s, 6H).
Пример 96
N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)пиримидин-2-иламино)-4-метокси-2-((1S,5R)-3-метил-3,6-диазабицикло[3.2.0]гептан-6-ил)фенил)акриламид
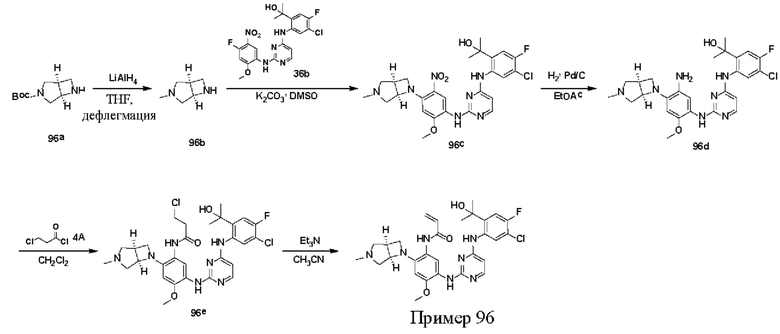
Методика получения соединения 96b:
В раствор соединения LiAlH4 (230 мг, 6,05 ммоль) в THF (8 мл) добавляли соединение 96а (300 мг, 1,51 ммоль). Полученную серую смесь нагревали при 75°С в течение 14 ч. После охлаждения до комнатной температуры реакционную смесь разбавляли THF (15 мл) и последовательно обрабатывали H2O (0,2 мл), 15%-ным водным NaOH (0,2 мл) и H2O (0,6 мл), затем перемешивали в течение еще 30 мин и добавляли Na2SO4. Смесь фильтровали, и фильтрат концентрировали в вакууме с получением соединения 96b (130 мг, 77%-ный выход) в виде желтого масла.
ЖХ/МС: Rt=0,079 мин при 0-60 АВ_2 мин_50_Е хроматографии (Xtimate С18, 2,1 × 30 мм, 3 мкм), МС m/z=113,1 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 4.24 (dd, J=4.4, 6,4 Гц, 1H), 3.68-3.66 (m, 1H), 3.38 (dd, J=4.8, 8,8 Гц, 1H), 3.08-3.03 (m, 1Н), 2.98-2.93 (m, 2Н), 2.45 (s, 3H), 2.05-2.01 (m, 2Н).
Методика получения соединения 96с:
В раствор соединения 36b (200 мг, 0,43 ммоль) и K2CO3 (119 мг, 0,86 ммоль) в DMSO (2 мл) добавляли соединение 96b (58 мг, 0,86 ммоль). Реакционную смесь перемешивали при 50°С в течение 14 ч, при этом цвет менялся с желтого на оранжевый. Реакционную смесь по каплям добавляли в H2O (30 мл) при перемешивании, и выпавшее в осадок твердое вещество собирали фильтрованием. Твердое вещество растворяли в CH2Cl2 (20 мл), сушили над Na2SO4 и концентрировали в вакууме с получением соединения 96 с (220 мг, 92%-ный выход) в виде оранжевого твердого вещества.
ЖХ/МС: Rt=0,667 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z=558,1 [M+H]+.
1Н ЯМР (400 МГц, CDCl3) δ 9.09 (s, 1H), 8.92 (s, 1Н), 8.09 (d, J=6,0 Гц, 1H), 7.95 (d, J=6,8 Гц, 1H), 7.19 (s, 1H), 7.09 (d, J=10,4 Гц, 1H), 6.16 (d, J=6,0 Гц, 1H), 5.94 (s, 1H), 5.02 (dd, J=4.4, 6,4 Гц, 1H), 4.07 (t, J=8,0 Гц, 1H), 3.91 (s, 3H), 3.89-3.83 (m, 1H), 3.15-3.06 (m, 1Н), 2.96 (d, J=10,0 Гц, 1H), 2.89 (d, J=11,2 Гц, 1H), 2.29 (s, 3H), 2.07 (dd, J=5.2, 10,0 Гц, 1Н), 1.85 (dd, J=4.4, 11,2 Гц, 1H), 1.67 (br s, 3H), 1.66 (s, 3H).
Методика получения соединения 96d:
В раствор соединения 96 с (220 мг, 0,39 ммоль) в EtOAc (10 мл) добавляли Pd/C (10%, 30 мг). Реакционную смесь перемешивали в атмосфере Н2 (15 фунт/кв.дюйм) при 8-13°С в течение 15 ч, затем при 35°С в течение 15 ч. Реакционную смесь фильтровали, и фильтрат концентрировали в вакууме с получением соединения 96d (180 мг, 87%-ный выход) в виде черного твердого вещества.
ЖХ/МС: Rt=0,640 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z=528,1 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 8.82 (s, 1Н), 8.15 (d, J=7,2 Гц, 1H), 8.01 (d, J=5,6 Гц, 1Н), 7.75 (s, 1H), 7.33 (s, 1H), 7.07 (d, J=10,8 Гц, 1Н), 6.34 (s, 1H), 6.01 (d, J=5,6 Гц, 1H), 4.65 (dd, J=4.8, 6,4 Гц, 1H), 3.91 (dd, J=4.8, 7,2 Гц, 1H), 3.87-3.79 (m, 4Н), 3.13-3.05 (m, 1H), 3.01 (br d, J=10,4 Гц, 2Н), 2.35 (s, 3H), 2.10 (br dd, J=6.0, 10,0 Гц, 1H), 1.96 (dd, J=4.4, 10,8 Гц, 1Н), 1.65 (s, 3H), 1.65 (s, 3H).
Методика получения соединения 96е:
В раствор соединения 96d (176 мг, 0,33 ммоль) в CH2Cl2 (3 мл) по каплям добавляли соединение 3-хлорпропаноилхлорид (42 мг, 0,33 ммоль) в бане лед-вода. Полученную черную смесь перемешивали при 5-10°С в течение 1,5 ч, и твердое вещество выпадало в осадок. Насыщенный водный NaHCO3 (10 мл) добавляли в реакционную смесь и перемешивали в течение еще 2 ч, затем выдерживали при комнатной температуре в течение 12 ч. Водную фазу отделяли и экстрагировали CH2Cl2 (15 мл × 3). Объединенные органические слои последовательно промывали водой (10 мл) и рассолом (10 мл), сушили над Na2SO4 и концентрировали в вакууме с получением указанного в заголовке соединение 96е (164 мг, 59%-ный выход) в виде темно-зеленого твердого вещества.
ЖХ/МС: Rt=0,683 мин при 5-95АВ_220&254.lcm хроматографии (MERCK RP18 2,5-2 мм), МС (ЭРИ) m/z=618,1 [М+Н]+.
Методика получения соединения Примера 96:
В раствор соединения 96е (164 мг, 0,19 ммоль) в CH3CN (5 мл) добавляли Et3N (79 мг, 0,78 ммоль). Полученную черную смесь перемешивали при 80°С в течение 14 ч. Растворитель удаляли при пониженном давлении с получением неочищенного остатка, который растворяли в МеОН (2 мл) и сначала очищали препаративной ТСХ (CH2Cl2: МеОН=5:1(об./об.)) с получением загрязненного примесями продукта в виде темно-красного твердого вещества. Его дополнительно очищали препаративной ВЭЖХ (колонка: Xbridge 150 × 30 мм, 10 мкм; условия: 37-67% В (А: 0,05%-ный гидроксид аммония, В: CH3CN); скорость потока: 25 мл/мин) и затем лиофилизировали с получением соединения Примера 96 (31,3 мг, 28%-ный выход) в виде светло-желтого твердого вещества.
ЖХ/МС: Rt=1,225 мин при 10-80АВ_4 мин_220&254.lcm хроматографии (Xtimate С18 2,1 × 30 мм), МС (ЭРИ) m/z=582,1[М+Н]+.
ВЭЖХ: Rt=2,10 мин при 10-80АВ_1,2 мл.мет (Ultimate С18 3,0 мкм, 3,0 × 50 мм).
1Н ЯМР (400 МГц, CDCl3) δ 9.35 (br s, 1H), 9.08 (br s, 1H), 8.16 (br s, 1H), 8.05 (d, J=6,0 Гц, 1H), 7.60 (d, J=6,0 Гц, 1H), 7.35 (s, 1H), 7.12 (d, J=10,8 Гц, 1H), 6.46-6.31 (m, 3H), 6.25 (d, J=5,2 Гц, 1H), 5.75 (dd, J=3.6, 7,6 Гц, 1H), 5.40 (br s, 1H), 4.61 (dd, J=4.0, 6,4 Гц, 1H), 3.92-3.84 (m, 4H), 3.83-3.76 (m, 1H), 3.17-3.01 (m, 3H), 2.43 (s, 3H), 2.18 (dd, J=6.4, 10,0 Гц, 1H), 2.04-1.94 (m, 1H), 1.75-1.63 (m, 6H).
Пример 97
N-(5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)пиримидин-2-иламино)-4-метокси-2-((1S,5S)-6-метил-3,6-диазабицшгло[3.2.0]гептан-3-ил)фенил)акриламида муравьинокислая соль

Синтез осуществляли в соответствии с экспериментальной методикой, описанной в Примере 89, с получением соединения Примера 97 в форме муравьинокислой соли в виде бледно-желтого твердого вещества.
ЖХ/МС: Rt=0,75 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (0,02% FA), Waters Acquity UPLC ВЕН C18 1,7 мкм, 2,1 × 50 мм, 40°C), МС (ЭРИ) m/z=582,0 [M+H]+.
1H ЯМР (500 МГц, DMSO-d6) δ 9.61 (s, 1H), 9.55 (s, 1H), 8.31 (s, 1H), 8.20 (s, 1H), 8.15 (d, J=7,4 Гц, 1H), 7.99 (d, J=5,7 Гц, 1H), 7.91 (s, 1H), 7.28 (d, J=11,0 Гц, 1H), 6.84 (s, 1H), 6.55 (dd, J=17.1, 10,3 Гц, 1H), 6.20 (dd, J=17.1, 2,0 Гц, 1H), 6.10 (d, J=5,7 Гц, 1H), 5.70 (dd, J=10.2, 2,0 Гц, 1H), 4.25-4.16 (m, 1H), 3.79 (s, 3H), 3.58-3.53 (m, 4H), 3.21 (d, J=9,8 Гц, 2H), 3.17-3.11 (m, 2H), 2.88-2.82 (m, 1H), 2.77-2.72 (m, 1H), 1.50 (s, 6H).
Пример 98
(R)-N-(2-(2-(азетидин-1-илметил)пирролидин-1-ил)-5-(4-(5-хлор-4-фтор-2-(2-гидроксипропан-2-ил)фениламино)пиримидин-2-иламино)-4-метоксифенил)акриламида муравьинокислая соль
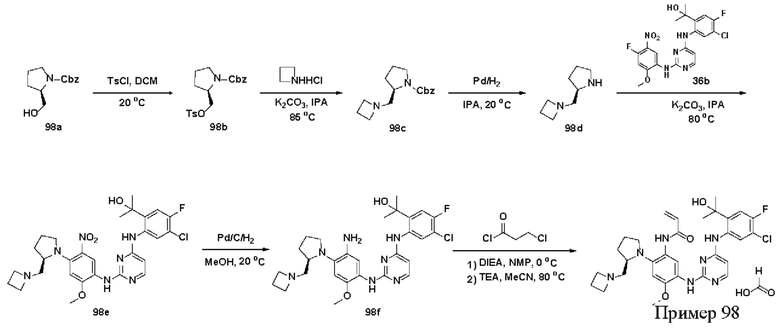
Методика получения соединения 98b:
В раствор соединения 98а (0,70 г, 3,0 ммоль) в DCM (5 мл) добавляли тозилхлорид (684 мг, 3,6 ммоль) и пиридин (1,79 мл, 22 ммоль) при 0°С. Полученную смесь перемешивали при 20°С в течение 16 ч. Смесь разбавляли 1 н. HCl (10 мл) и экстрагировали DCM (20 мл × 3). Объединенные органические фазы сушили над безводным сульфатом натрия, фильтровали, и фильтрат выпаривали при пониженном давлении с получением неочищенного продукта, который очищали флэш-хроматографией на диоксиде кремния с градиентом элюирования от 0% до 10% EtOAc в петролейном эфире. Чистые фракции упаривали досуха с получением соединения 98b (1,1 г, 75%-ный выход) в виде бесцветного масла.
ЖХ/МС: Rt=1,55 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3), Waters Acquity UPLC ВЕН C18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=390 [M+H]+.
Методика получения соединения 98с:
В раствор соединения 98b (0,47 г, 1,2 ммоль) в NMP (10 мл) добавляли азетидина гидрохлорид (673 мг, 7,2 ммоль) и карбонат калия (995 мг, 7,2 ммоль). Смесь перемешивали при 85°С в течение 16 ч. Смесь разбавляли водой (20 мл) и экстрагировали EtOAc (20 мл × 3). Объединенные органические фазы сушили над безводным сульфатом натрия, фильтровали, и фильтрат выпаривали при пониженном давлении с получением неочищенного продукта, который очищали С18-флэш-хроматографией с градиентом элюирования от 0 до 50% CH3CN в воде (0,02% FA). Чистые фракции упаривали досуха с получением соединения 98 с (0.20 г, 60%-ный выход) в виде бесцветного масла.
ЖХ/МС: Rt=0,94 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3), Waters Acquity UPLC ВЕН C18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=275 [М+Н]+.
Методика получения соединения 98d:
В раствор соединения 98с (90 мг, 0,30 ммоль) в IPA (5 мл) добавляли палладий на углероде (10 мг). Полученную смесь перемешивали при 20°С в атмосфере водорода в течение 16 ч. Смесь фильтровали, и фильтрат концентрировали в вакууме с получением неочищенного соединения 98d (70 мг, неочищенное), которое напрямую использовали на следующей стадии.
ЖХ/МС: Rt=0,24 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3), Waters Acquity UPLC ВЕН C18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=141 [М+Н]+.
Методика получения соединения 98е:
В раствор соединения 98d (70 мг, 0,50 ммоль) в IPA (10 мл) добавляли карбонат калия (90 мг, 0,65 ммоль) и соединение 36b (150 мг, 0,32 ммоль). Полученную смесь нагревали при 80°С в атмосфере азота в течение 16 ч. Затем смесь фильтровали, и фильтрат удаляли при пониженном давлении. Остаток разбавляли водой (20 мл) и экстрагировали EtOAc (20 мл × 3). Объединенные органические фазы сушили над безводным сульфатом натрия, фильтровали, и фильтрат выпаривали при пониженном давлении с получением соединения 98е (150 мг, 80%-ный выход) в виде красного твердого вещества.
ЖХ/МС: Rt=1,52 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3), Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1 × 50 мм, 40°С) МС (ЭРИ) m/z=586 [M+H]+.
Методика получения соединения 98f:
В раствор соединения 98е (70 мг, 0,12 ммоль) в МеОН (15 мл) добавляли палладий на углероде (20 мг). Полученную смесь перемешивали при 20°С в атмосфере водорода в течение 2 ч. Смесь фильтровали, и фильтрат концентрировали в вакууме с получением соединения 98f (50 мг, 60%-ный выход) в виде желтого твердого вещества.
ЖХ/МС: Rt=1,16 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3, Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=556 [М+Н]+.
Методика получения соединения Примера 98:
В раствор соединения 98f (40 мг, 0,070 ммоль) и DIEA (10 мг, 0,080 ммоль) в NMP (3 мл) добавляли 3-хлорпропионилхлорид (10 мг, 0,080 ммоль) при 0°С. Затем добавляли MeCN (3 мл) и TEA (1 мл), и полученную смесь нагревали при 80°С в течение 16 ч. Смесь концентрировали в вакууме, и остаток очищали С18-флэш-хроматографией с градиентом элюирования от 0 до 30% CH3CN в воде (0,02% FA). Чистые фракции упаривали досуха с получением соединения Примера 98 в форме муравьинокислой соли (3,9 мг, 8,9%-ный выход) в виде белого твердого вещества.
ЖХ/МС: Rt=1,16 мин при 3 мин хроматографии (3 мин-5-95% MeCN в воде (6 ммоль/л NH4HCO3, Waters Acquity UPLC ВЕН С18 1,7 мкм, 2,1 × 50 мм, 40°С), МС (ЭРИ) m/z=610,1 [М+Н]+.
1Н ЯМР (500 МГц, DMSO-d6) δ м.д. 9.61 (d, J=8,1 Гц, 1H), 8.28 (s, 1H), 8.16 (d, J=7,4 Гц, 1H), 7.98 (d, J=5,7 Гц, 1H), 7.86 (s, 1H), 7.28 (d, J=11,1 Гц, 1H), 6.78 (s, 1H), 6.57 (dd, J=17.0, 10,2 Гц, 1H), 6.35-6.03 (m, 3H), 5.70 (dd, J=10.4, 1,9 Гц, 1H), 3.97-3.68 (m, 4Н), 3.11 (dq, J=22.2, 6,8 Гц, 4Н), 2.96-2.85 (m, 1H), 2.48-2.34 (m, 2Н), 2.27-2.15 (m, 1H), 2.11-2.00 (m, 1H), 1.98-1.71 (m, 4Н), 1.69-1.59 (m, 1H), 1.55-1.40 (m, 6Н).
БИОЛОГИЧЕСКИЕ ПРИМЕРЫ:
Ниже описаны анализы, использованные для измерения биологической активности предложенных соединений в качестве селективных ингибиторов мутантного EGFR по сравнению с WT EGFR (и другими протеинкиназами).
ПРИМЕР 99: ЛИНИИ КЛЕТОК С МУТАЦИЯМИ EGFR ИЛИ HER2
В целях начального анализа активности in vitro может быть использована лентивирусная система для создания линий клеток с разными мутациями EGFR или Her2.
Конкретно, линию клеток с мутацией человеческого EGFR, представляющей собой инсерцию ASV в экзоне 20, создают путем субклонирования человеческого EGFR с V769_D770insASV в экзоне 20 в лентивирусный вектор переноса рМТ143 (Sunbio Co., Ltd.) и последующего трансфицирования указанного лентивирусного вектора переноса и лентивирусных упаковочных плазмид в клетки 293Т/17 (Американская коллекция типовых культур (АТСС), CRL-11268) для создания рекомбинантного лентивируса, кодирующего EGFR V769_D770insASV человека. Ввиду IL-3-зависимой пролиферации родительские клетки Ba/F3 (DSMZ, номер по каталогу АСС-300) культивировали в среде RPMI1640 (Life Technology, номер по каталогу 11835-055), дополненной 10% FBS (фетальная коровья сыворотка) и 10% об. кондиционированной средой из клеток линии WEHI-3B (DSMZ, номер по каталогу АСС-26) в качестве источника мышиного IL-3. Для создания линии клеток Ba/F3, трансдуцированных EGFR с V769_D770insASV в экзоне 20, родительские клетки Ba/F3 инфицировали рекомбинантным лентивирусом, несущим EGFR V769 D770insASV, и затем следовал отбор с использованием 1 мкг/мл пуромицина и истощение IL-3. Полученная линия клеток экспрессирует конститутивно фосфорилированный белок EGFR и пролиферирует в отсутствие IL-3, что может быть использовано для анализов PD (фармакодинамика) и антипролиферации in vitro.
Линию клеток с мутацией человеческого Her2, представляющей собой инсерцию YVMA в экзоне 20, создают путем субклонирования мутации человеческого Her2, представляющей собой YVMA в экзоне 20, в лентивирусный вектор переноса рМТ143 (Sunbio Co., Ltd.) с последующим трансфицированием указанного лентивирусного вектора переноса и лентивирусных упаковочных плазмид в клетки 293Т/17 (Американская коллекция типовых культур (АТСС), CRL-11268) для создания рекомбинантного лентивируса, кодирующего мутацию человеческого Her2, представляющую собой инсерцию YVMA в экзоне 20. Родительские клетки NIH-3T3 культивировали в среде DMEM (Life Technology, номер по каталогу 11835-055), дополненной 10% NBCS (сыворотка новорожденных телят). Для создания линии клеток NIH-3T3, транс дуциро ванных Her2 с инсерцией YVMA в экзоне 20, родительские клетки инфицировали рекомбинантным лентивирусом, несущим Her2 с инсерцией YVMA в экзоне 20, и затем следовал отбор с использованием 1,0 мкг/мл пуромицина. Полученная линия клеток экспрессировала фосфорилированный белок Her2, и его пролиферация зависела от гена с мутацией, что может быть использовано для анализов PD и антипролиферации in vitro.
Рекомбинантная линия клеток Ba/F3, имеющих EGFR H773_V774insNPH в экзоне 20 человека, была приобретена у Crown Bioscience, Inc., China (номер по каталогу С2058). Подобно рекомбинантной линии клеток Ba/F3, имеющей EGFR H769_V770insASV в экзоне 20 человека, рекомбинантная линия клеток Ba/F3, имеющая Н773_V774insNPH EGFR в экзоне 20 человека, также экспрессирует конститутивно фосфорилированный белок EGFR и пролиферирует в отсутствие IL-3, что может быть использовано для анализов PD и антипролиферации in vitro.
Линия клеток NCI-H1975 (CRL5908™), содержащая обе точковые мутации EGFR L858R и Т790М, была получена из Американской коллекции типовых культур (АТСС).
ПРИМЕР 100: ОЦЕНКА АКТИВНОСТИ В ОТНОШЕНИИ EGFR (WT) И МУТАНТОВ EGFR
Ингибирующую активность и селективность соединений против мутантного EGFR и WT EGFR оценивали с использованием несущей WT EGFR линии клеток А-431 (Американская коллекция типовых культур (АТСС), CRL-1555™) и мутантных линий клеток, которые описаны в Примере 99.
За сутки до тестирования клетки WT или мутантные клетки высевали в 96-луночные планшеты в целесообразных концентрациях с соответствующей ростовой средой, дополненной 1% FBS, и инкубировали в течение ночи. На следующий день тестируемые соединения в последовательных концентрациях добавляли в каждую индивидуальную лунку планшетов, и планшеты инкубировали в течение 4 ч при 37°С с 5% CO2. Что касается линии клеток, несущих WT EGFR, после инкубирования с тестируемыми соединениями и перед анализом с использованием наборов MSD следует выполнить дополнительное стимулирование взятым в количестве 100 нг/мл рекомбинантным hEGF (RD, номер по каталогу 236-EG) в течение 10 мин.
Уровень фосфорилирования (активность) EGFR (Y1068) клеток в каждой лунке затем измеряли с использованием набора MSD, именуемого "MULTI-SPOT®96 4-Spot НВ Prototype EGFR Triplex ANALYTES: PEGFR(Tyrl068), pEGFR(Tyr1173), Total EGFR", согласно инструкции производителя (MESO SCALE DISCOVERY, номер по каталогу N45ZB-1). Анализ представляет собой электрохемилюминесцентный метод определения как фосфорилированного, так и общего EGFR клеток с использованием визуализатора MSD SECTOR® Imager, и соотношение p-EGFR/общий EGFR может быть определено автоматически. Уровень фосфорилирования (активность) Her2 (Y1248) клеток в каждой лунке измеряли с использованием набора MSD, именуемого "Phospho-ErbB2 (Tyr1248) Assay Whole Cell Lysate Kit", согласно инструкции производителя (MESO SCALE DISCOVERY, номер по каталогу K151CLD-3). Анализ представляет собой электрохемилюминесцентный метод (MESO SCALE DISCOVERY) определения как фосфорилированного, так и общего Her2 клеток с использованием виазулизатора MSD SECTOR® Imager, и затем соотношение р-Her2/общий Her2 может быть определено автоматически. Процент ингибирования вычисляли по формуле: % ингибирования = 100 × [1-(соотношение для лунки с образцом - соотношение Мин для лунки с контролем)/(соотношение Макс - соотношение Мин для лунки с контролем)]. Значения IC50 вычисляли как концентрацию соединения, необходимую для 50%-ного ингибирования, на наилучшим образом выровненных кривых с использованием программного обеспечения Prism GraphPad 7.0 или Microsoft Xlfit.

В Таблице 3 показана активность иллюстративных соединений по настоящему изобретению и других ингибиторов EGFR в качестве положительного контроля в анализе ингибирования EGFR, описанном выше. Селективность протестированных соединений по отношению к мутантному EGFR и WT EGFR может быть оценена исходя из данных для каждого протестированного соединения. Номера соединений соответствуют номерам соединений в Таблице 1.
и WT) для иллюстративных соединений

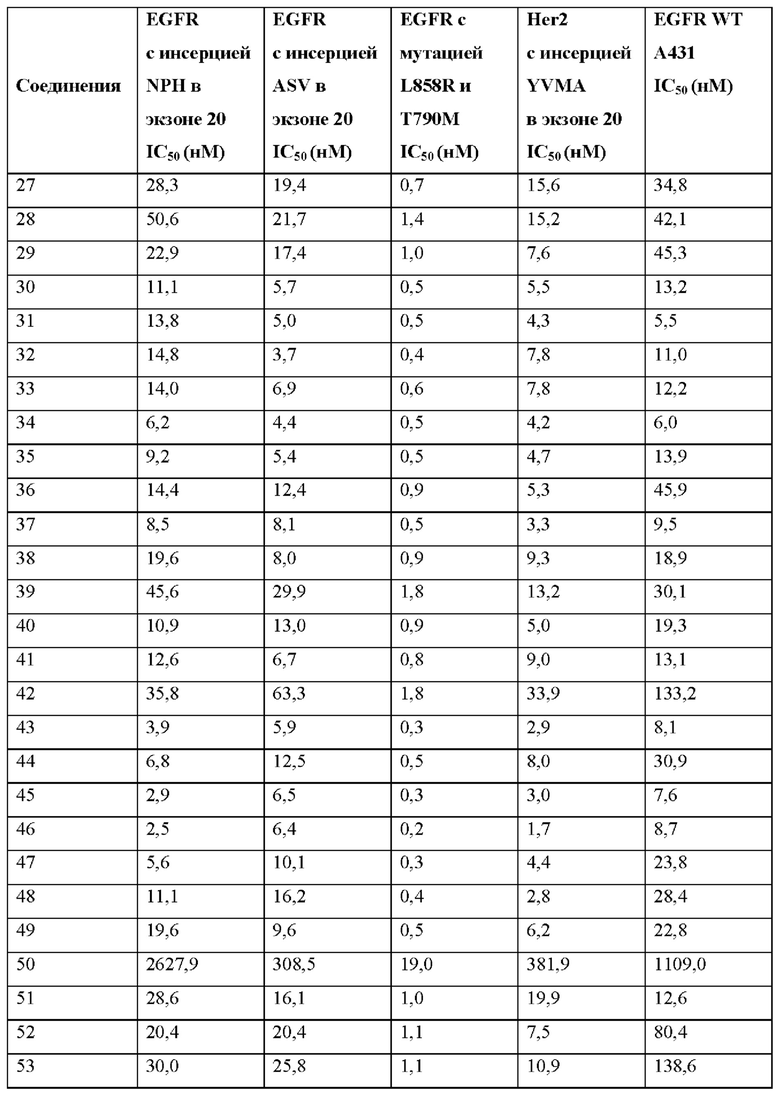
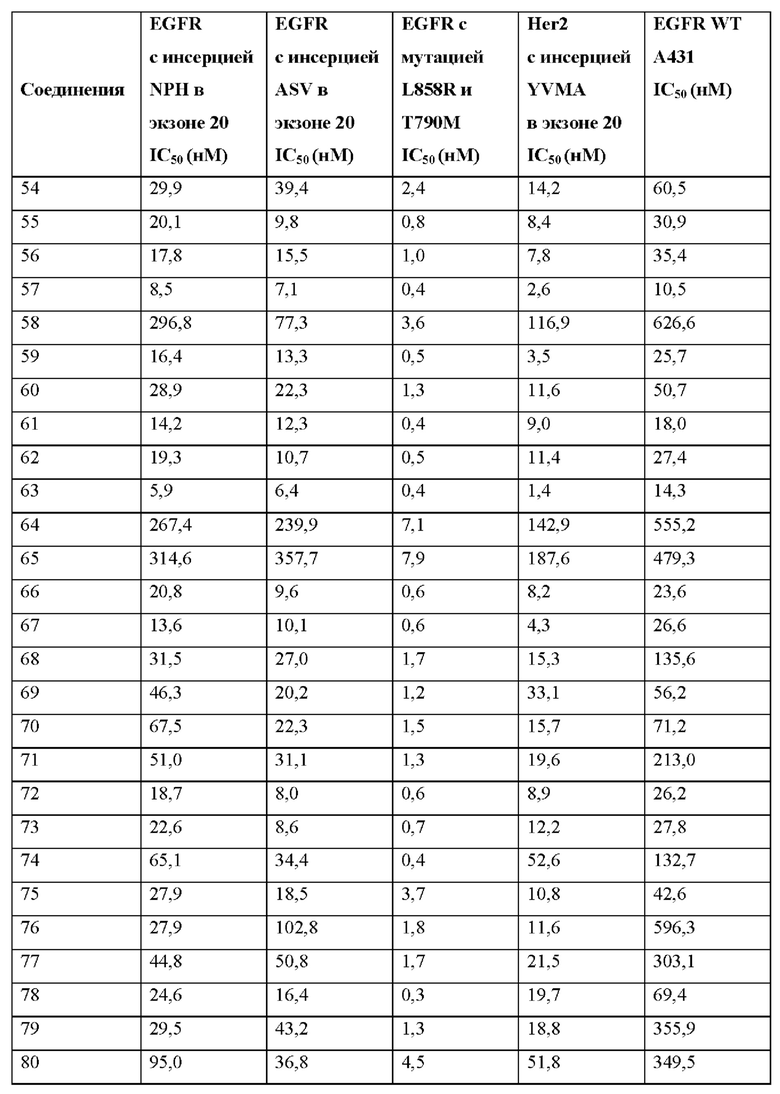
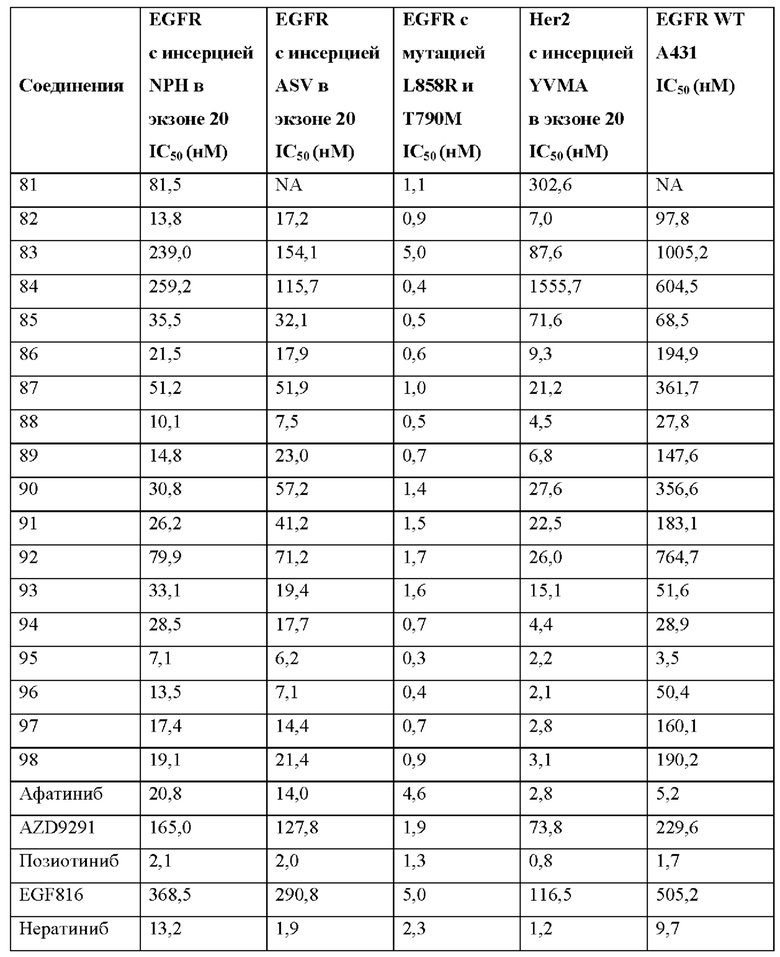
IC50 соединений по отношению к клеткам, имеющим двойную мутацию EGFR L858R и Т790М, может вплоть до 1500 раз более действенной, чем IC50 соединений по отношению к EGFR дикого типа. Большинство соединений по настоящему изобретению демонстрируют более высокую ингибирующую активность по отношению к EGFR с инсерциями NPH и ASV в экзоне 20 и к Her2 с инсерцией YVMA в экзоне 20 по сравнению с AZD9291.
ПРИМЕР 101: ПРОЛИФЕРАЦИЯ КЛЕТОК С EGFR (WT) И МУТАНТАМИ EGFR
Клетки WT или мутантные клетки высевали в 384-луночные планшеты в целесообразных концентрациях с соответствующими ростовыми средами, дополненными 10% FBS. Для каждого эксперимента подготавливали по два идентичных планшета-дубликата, и эти планшеты инкубировали в течение ночи. На следующий день в один из каждых двух планшетов-дубликатов добавляли тестируемые соединения в последовательных концентрациях, а другой идентичный планшет анализировали для получения значения G0. Планшеты с соединениями инкубировали в течение еще 72 ч при 37°C с 5% СО2, и количество жизнеспособных клеток в каждой лунке G0 планшетов или планшетов с соединениями измеряли люминесцентным методом анализа жизнеспособности клеток CellTiter-Glo® Luminescent Cell Viability Assay (Promega). Этот анализ представляет собой люминесцентный метод определения количества жизнеспособных клеток путем измерения клеточной концентрации АТР (аденозинтрифосфат) посредством детектирования активности люциферазы. Детекционный реагент (15 мкл) добавляли в каждую лунку, и планшеты инкубировали в течение 30 мин при комнатной температуре. Затем люминесценцию в каждой лунке измеряли с использованием планшет-ридера Envision (PerkinElmer). Процент пролиферации вычисляли по формуле: % пролиферации = 100 × (значение G3 для лунки с образцом значение Go)/(значение G3 для DMSO-контроля значение Go). Значения GI50 далее вычисляли как концентрацию соединения, необходимую для 50%-ной пролиферации, на кривых наилучшего соответствия с использованием программного обеспечения Genedata Screener®.

В Таблице 5 показано ингибирование пролиферации иллюстративными соединениями по настоящему изобретению и другими ингибиторами EGFR в качестве положительного контроля в анализе ингибирования пролиферации, описанном выше.




GI50 соединений по отношению к клеткам, имеющим двойную мутацию EGFR L858R и Т790М, могут быть вплоть до 25 раз более действенными, чем GI50 соединений по отношению к EGFR дикого типа. Большинство соединений по настоящему изобретению демонстрируют более высокую активность по отношению к EGFR с инсерциями NPH и ASV в экзоне 20 и Her2 с инсерцией YVMA в экзоне 20 по сравнению с AZD9291,
ПРИМЕР 102: ПРОЛИФЕРАЦИЯ КЛЕТОК С ВТК (WT)
OCI-LY-10 при 3750 клеток на лунку, TMD-8 и Ri-1 при 2000 клеток на лунку и DB при 1250 клеток на лунку соответственно сортировали в 384-луночные планшеты в среде RPMI1640 с 10% FBS. После инкубирования в течение ночи все клетки инкубировали с соединением в последовательных концентрациях. Вместе с тем, подготавливали планшет-дубликат для каждой линии клеток для измерения значения G0. Планшеты с соединениями дополнительно инкубировали в течение 72 часов, и количество жизнеспособных клеток измеряли путем проведения анализа CellTiter-Glo® Luminescent Cell Viability Assay (Promega), представляющего собой люминесцентный метод определения количества жизнеспособных клеток посредством измерения клеточной концентрации АТР с использованием планшет-ридера Envision (PerkinElmer). Процент пролиферации вычисляли следующим образом: % пролиферации = 100 × (значение G3 для лунки с образцом - значение G0)/(значение G3 для DMSO-контроля - значение G0). Значения GI50 далее вычисляли как концентрацию соединения, необходимую для 50%-ного ингибирования пролиферации, на наилучшим образом выровненных кривых с использованием программного обеспечения XLFit.
В Таблице 6 представлено ингибирование пролиферации иллюстративными соединениями по настоящему изобретению и другими ингибиторами ВТК в качестве положительного контроля в анализе ингибирования пролиферации, описанном выше.


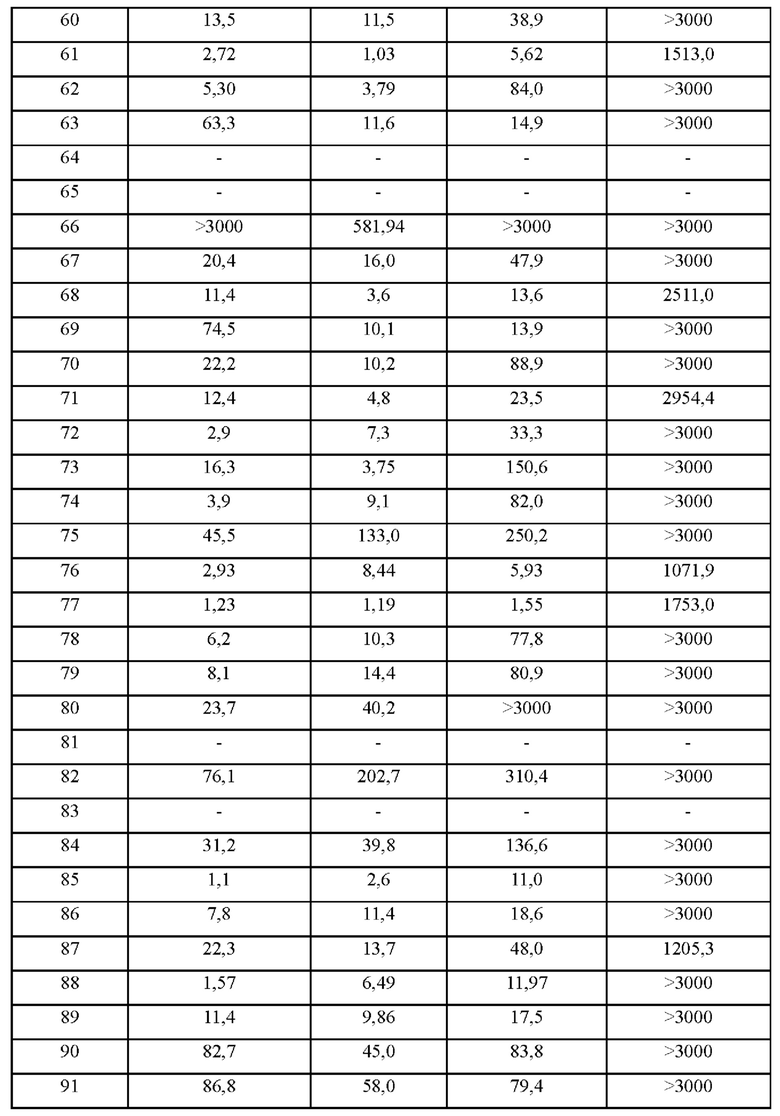

Как показано в Таблице 7 ниже, иллюстративные соединения, раскрытые в данном изобретении, продемонстрировали лучшую активность и более высокую занятость рецептора при более низких концентрациях лекарственных средств по сравнению с АСР-196 и ибрутинибом.
Анализ р-ВТК
Суспензии клеток Рамоса в RPMI1640 (Gibco) с 1,5% FBS (Invitrogen) равномерно распределяли в 384-луночный белый планшет небольшого объема. Соединения разбавляли с помощью Platemate plus (Thermo) и добавляли в планшеты для анализа с использованием жидкостного манипулятора ECHO 555 (Labcyte) с получением конечной концентрации DMSO 0,1%. Планшеты для анализа затем инкубировали в течение 30 минут при 37°C, 5% CO2. 300 мкМ перванадата (3Х) добавляли в каждую лунку, и планшеты для анализа снова инкубировали в течение 60 минут при 37°C, 5% СО2. Phospho-BTK (фосфорилированная форма ВТК) (Tyr223) детектировали с использованием реагентов HTRF® (Cisbio) согласно протоколу однопланшетного анализа производителя.
Твердофазный иммуноферментный анализ занятости мишени Btk
Метод твердофазного иммуноферментного анализа (ELISA) для детектирования свободной, не ингибированной Btk в мышиных и человеческих лизатах. Цельную кровь человека или мыши инкубировали с соединением в течение 2 часов при 37°C. Цельную кровь подвергали лизису в охлажденном во льду буфере для лизиса, содержащем 50 мМ трис-HCl, рН 7,5, 150 мМ NaCl, 1 мМ EDTA, 1% Triton Х-100. Клеточные лизаты затем инкубировали с ибрутиниб-биотином в конечной концентрации 1 мкМ. Образцы переносили в покрытый стрептавидином 96-луночный планшет для ELISA и смешивали, встряхивая в течение 1 часа при комнатной температуре. Затем в лунку добавляли анти-Btk антитело (1:500; номер клона D3H5, Cell signaling Technology, Danvers, MA, USA) и инкубировали в течение 1 часа при комнатной температуре. После промывки в каждую лунку добавляли конъюгат козьего антикроличьего антитела с HRP (пероксидаза хрена) (1:5000 разведение в PBS + 0,05% Tween-20 + 1% BSA) и инкубировали в течение 1 ч при комнатной температуре. ELIS А проводили с добавлением тетраметилбензидина (ТМВ) и с последующим добавлением стоп-раствора и считыванием при OD 450 нм.

ПРИМЕР 103: АНАЛИЗ НА КЛИРЕНС ГЕПАТОЦИТОВ КРЫСЫ/ЧЕЛОВЕКА IN VITRO
Протокол анализа на метаболическую стабильность гепатоцитов крысы/человека использован для определения клиренса соединений по настоящему изобретению in vitro.
Гепатоциты крысы мужского пола и гепатоциты человека смешанного пола были получены от коммерческих поставщиков (например, BioreclamationIVT) и до использования хранились при -150°C. Подготавливали 10 мМ исходные растворы тестируемых соединений в DMSO. Перед использованием размораживающую среду и дополнительную инкубационную среду (не содержащую сыворотку) помещали в водяную баню при 37°C по меньшей мере на 15 минут. Исходные растворы разводили до 100 мкМ объединением 198 мкл ацетонитрила и 2 мкл 10 мМ исходного раствора.
Виалы с криоконсервированными гепатоцитами извлекали из хранилища и убеждались, что виалы оставались при криогенных температурах. Виалы размораживали в водяной бане при 37°C, слегка встряхивая. Виалы держали в водяной бане до тех пор, пока все ледяные кристаллы не растворились и стали невидимыми. Виалы опрыскивали 70%-ным этанолом, после чего переносили в бокс биологической безопасности. Затем содержимое вливали в 50 мл размораживающей среды в конической пробирке. Виалы центрифугировали при 100 g в течение 10 минут при комнатной температуре. Размораживающую среду аспирировали, и гепатоциты ресуспендировали в не содержащей сыворотку инкубационной среде с получением примерно 1,5 × 106 клеток/мл.
Жизнеспособные клетки и плотность подсчитывали, используя исключение триптанового синего, и затем клетки разводили в не содержащей сыворотку инкубационной среде до рабочей плотности 1 × 106 живых клеток/мл. Порцию гепатоцитов при 1 × 106 живых клеток/мл кипятили в течение 10 мин, затем добавляли в планшет в качестве отрицательного контроля для устранения ферментативной активности, чтобы практически никакого метаболизма субстрата не наблюдалось. Инактивированные гепатоциты использовали для приготовления отрицательных образцов, которые использовали, чтобы исключить вводящий в заблуждение фактор, возникающий в результате нестабильности самого химического вещества.
Аликвоты по 247,5 мкл гепатоцитов вносили в каждую лунку 96-луночного непокрытого планшета. Планшет помещали в инкубатор на орбитальном шейкере приблизительно на 10 минут. Аликвоты по 2,5 мкл 100 мкМ тестируемых соединений добавляли в соответствующие лунки непокрытого 96-луночного планшета, чтобы начать реакцию. Этот анализ осуществляли в двух повторах. Планшет инкубировали на орбитальном шейкере до намеченых точек времени. 20 мкл содержимого переносили и смешивали с 6 объемами (120 мкл) холодного ацетонитрила с внутренним стандартом для остановки реакции в моменты времени 5, 15, 30, 45, 60, 80 и 100 минут. Образцы центрифугировали в течение 20 минут при 4000 g, и аликвоты по 100 мкл надосадочных жидкостей использовали для ЖХ/МС/МС анализа для измерения тестируемых соединений.
Клиренс гепатоцитов in vitro оценивали, основываясь на определении периода полувыведения (T1/2) соединений, исчезновения из их первоначальных концентраций. Вычисляли отношения площадей пиков каждого соединения (тестируемого или контрольного) к IS. Строили кривую зависимости Ln (% контроля) от времени инкубирования (мин) и вычисляли тангенс угла наклона (угловой коэффициент) линии линейного выравнивания. Константу скорости выведения лекарственного средства k (мин-1), Т1/2 (мин) и собственный клиренс in vitro CLint (мкл/мин/Е6) вычисляли согласно следующим уравнениям:
k=- угловой коэффициент
Т1/2=0,693/k
CLint=k/Chep
где Chep (клетки × мкл-1) означает концентрацию клеток в инкубационной системе.
Иллюстративные данные представлены в приведенной ниже Таблице 8.

Поскольку гепатоциты содержат ферменты фазы I и II метаболизма, анализ клиренса гепатоцитов крысы/человека может отражать собственный клиренс в печени. Первичный скрининговый анализ был использован для выбора соединений для следующего раунда фармакокинетических исследований на крысах in vivo и прогнозирования печеночного клиренса у человека при условии, что экстраполяция in vitro-in vivo у крысы установлена.
ПРИМЕР 104: ФАРМАКОКИНЕТИЧЕСКИЕ ИССЛЕДОВАНИЯ IN VIVO
Для фармакокинетических исследований шести самцам крыс Han Wistar, приобретенных у Beijing Vital River, перорально вводили тестируемое соединение в дозе 10 мг/кг в 0,5% Tween-80 и 0,5% гидроксипропилметилцеллюлозы (об./масс). Через 0,5, 1, 2, 4, 7 и 16 часов после введения образцы крови (>100 мкл/момент времени) собирали из воротной вены и посредством пункции сердца в отдельные K2EDTA коагулированные пробирки и затем сразу центрифугировали при 1500 g в течение 5 мин при 4°C для отделения плазмы. Образцы плазмы депротеинизировали 4-кратным ацетонитрилом (включающим внутренний стандарт), вортексировали в течение 10 мин и центрифугировали в течение 10 минут при 4000 g при 4°C. Надосадочные жидкости анализировали методом LC/MC/MC (API 5500, Applied Biosystems, Foster City). Два комплекта стандартных кривых снимали в начале и в конце каждого периода анализа образцов плазмы.
Всасывание соединения оценивали по показателям AUC воротной вены (AUCHPV) и системной AUC (AUCSYS) и максимальной наблюдаемой концентрации (Cmax) у крыс после перорального введения. Доза представляет собой фактическую дозу, использованную в SOA исследовании. Найдите ниже в Таблице 9 данные для протестированных иллюстративных соединений.

Исследование направлено на оценку системного воздействия тестируемых соединений в показателях Cmax и AUC, чему способствуют пероральное всасывание, кишечный метаболизм и печеночная экстракция. Оно будет использовано для выбора соединений для фармакокинетического исследования in vivo.
Хотя настоящее изобретение было конкретно продемонстрировано и описано со ссылкой на конкретные воплощения (некоторые из которых являются предпочтительными воплощениями), специалистам в данной области будет понятно, что различные изменения в форме и деталях могут быть сделаны без отклонения от замысла и объема настоящего изобретения, раскрытого в данном документе.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ РЕЦЕПТОРОВ ERBB | 2019 |
|
RU2810215C2 |
| 3,6-ДИАМИНОПИРИДАЗИН-3-ИЛЬНЫЕ ПРОИЗВОДНЫЕ, СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ПРОАПОПТОТИЧЕСКИХ СРЕДСТВ | 2020 |
|
RU2830186C2 |
| 6,7-ДИГИДРО-5H-ПИРИДО[2,3-C]ПИРИДАЗИНОВЫЕ ПРОИЗВОДНЫЕ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ БЕЛКОВ BCLXL И ПРОАПОПТОТИЧЕСКИХ СРЕДСТВ ДЛЯ ЛЕЧЕНИЯ ЗЛОКАЧЕСТВЕННЫХ НОВООБРАЗОВАНИЙ | 2020 |
|
RU2832191C2 |
| 1-(2-ИЗОПРОПОКСИЭТИЛ)-2-ТИОКСО-1,2,3,5-ТЕТРАГИДРО-ПИРРОЛО[3,2-d]ПИРИМИДИН-4-ОН И ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 2005 |
|
RU2409578C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ПИРРОЛО[3,2-d]ПИРИМИДИН-4-ОНА И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2005 |
|
RU2577858C2 |
| ПРОИЗВОДНЫЕ ТИОФЕНА | 2003 |
|
RU2296758C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛХИНАЗОЛИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2011 |
|
RU2652638C2 |
| Соединение диарилтиогидантоина в качестве антагониста андрогенового рецептора | 2018 |
|
RU2804108C9 |
| Производные тетрагидротриазолопиримидина в качестве ингибиторов нейтрофильной эластазы человека | 2012 |
|
RU2622643C2 |
| C-3 и C-17 модифицированные тритерпеноиды в качестве ингибиторов ВИЧ-1 | 2017 |
|
RU2716502C2 |
Изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли, сложному эфиру, гидрату, сольвату или стереоизомеру. В формуле (I): A1 представляет N или CR8, каждый из А2, А3, А4 и А5 независимо представляет N или CR9, причем не более чем один из A2, А3, А4 и A5 представляет N, каждый из R1 и R2 независимо представляет водород или С1-12алкил, возможно монозамещенный -NRaRb, С1-12алкилом, 3-10-членным насыщенным карбоциклилом или 3-6-членным насыщенным гетероциклилом, содержащим один гетероатом, выбранный из N и О, где каждый из 3-10-членного насыщенного карбоциклила и 3-6-членного насыщенного гетероциклила может быть не замещен или моно- или полизамещен С1-12алкилом, где каждый из Ra и Rb независимо выбран из водорода или С1-12алкила, который может быть возможно монозамещен или независимо полизамещен дейтерием или тритием, или Ra и Rb, взятые вместе с атомом азота, с которым они связаны, образуют 3-10-членный насыщенный гетероциклил, возможно монозамещенный С1-12алкилом, или R1 и R2, взятые вместе с атомом азота, с которым они связаны, образуют 3-12-членное моноциклическое или полициклическое кольцо, возможно содержащее один дополнительный гетероатом, выбранный из N и О, которое может быть возможно монозамещено или независимо полизамещено галогеном, С1-12алкилом, -NRaRb или группой -С1-12алкил-NRaRb, R3 представляет Н, С1-12алкил или -C1-12алкил-NRaRb, каждый из R4 и R5 независимо представляет C1-6алкил, возможно монозамещенный или независимо полизамещенный одним или более чем одним дейтерием, тритием или галогеном, или R4 и R5, взятые вместе с атомом углерода, с которым они связаны, образуют 3-10-членное моноциклическое кольцо, возможно содержащее один гетероатом О, которое может быть возможно монозамещено или независимо полизамещено одним или более чем одним дейтерием или тритием, R6 представляет водород, R7 представляет С1-12алкил, который может быть возможно моно- или полизамещен дейтерием или тритием, R8 представляет водород, дейтерий, тритий, галоген, циано, С1-12алкил, C1-12алкоксил, который может быть возможно монозамещен или независимо полизамещен одним или более чем одним дейтерием, тритием или галогеном, R9 отсутствует или представляет водород, дейтерий или тритий, n равно 0, 1, 2, 3 или 4, каждый R независимо представляет галоген, С1-12алкил, 3-10-членный насыщенный карбоциклил или 3-6-членный насыщенный или ненасыщенный гетероциклил, содержащий один гетероатом N, при этом указанный гетероциклил конденсирован с кольцом, с которым он связан, которые могут быть возможно монозамещены или независимо полизамещены одним или более чем одним дейтерием, тритием, галогеном или С1-12алкилом. Также предложены фармацевтическая композиция и способ лечения ассоциированного с тирозинкиназой заболевания. Предложенные соединения, ингибирующие ErbB и ВТК, могут эффективно лечить связанные с ErbB (в частности мутантными формами ErbB) или ВТК заболевания, включая рак. 5 н. и 20 з.п. ф-лы, 9 табл., 104 пр.

1. Соединение формулы (I):

или его фармацевтически приемлемая соль, сложный эфир, гидрат, сольват или стереоизомер,
где
A1 представляет собой N или CR8;
каждый из А2, А3, А4 и А5 независимо представляет собой N или CR9, причем не более чем один из A2, А3, А4 и A5 представляет собой N;
каждый из R1 и R2 независимо представляет собой водород или С1-12алкил, возможно монозамещенный -NRaRb, С1-12алкилом, 3-10-членным насыщенным карбоциклилом или 3-6-членным насыщенным гетероциклилом, содержащим один гетероатом, выбранный из N и О, где каждый из 3-10-членного насыщенного карбоциклила и 3-6-членного насыщенного гетероциклила может быть не замещен или моно- или полизамещен С1-12алкилом,
где каждый из Ra и Rb независимо выбран из водорода или С1-12алкила, который может быть возможно монозамещен или независимо полизамещен дейтерием или тритием,
или Ra и Rb, взятые вместе с атомом азота, с которым они связаны, образуют 3-10-членный насыщенный гетероциклил, возможно монозамещенный С1-12алкилом;
или R1 и R2, взятые вместе с атомом азота, с которым они связаны, образуют 3-12-членное моноциклическое или полициклическое кольцо, возможно содержащее один дополнительный гетероатом, выбранный из N и О, которое может быть возможно монозамещено или независимо полизамещено галогеном, С1-12алкилом, -NRaRb или группой -С1-12алкил-NRaRb;
R3 представляет собой Н, С1-12алкил или -C1-12алкил-NRaRb;
каждый из R4 и R5 независимо представляет собой C1-6алкил, возможно монозамещенный или независимо полизамещенный одним или более чем одним дейтерием, тритием или галогеном,
или R4 и R5, взятые вместе с атомом углерода, с которым они связаны, образуют 3-10-членное моноциклическое кольцо, возможно содержащее один гетероатом О, которое может быть возможно монозамещено или независимо полизамещено одним или более чем одним дейтерием или тритием,
R6 представляет собой водород,
R7 представляет собой С1-12алкил, который может быть возможно моно- или полизамещен дейтерием или тритием,
R8 представляет собой водород, дейтерий, тритий, галоген, циано, С1-12алкил, C1-12алкоксил, который может быть возможно монозамещен или независимо полизамещен одним или более чем одним дейтерием, тритием или галогеном;
R9 отсутствует или представляет собой водород, дейтерий или тритий;
n равно 0, 1, 2, 3 или 4;
каждый R независимо представляет собой галоген, С1-12алкил, 3-10-членный насыщенный карбоциклил или 3-6-членный насыщенный или ненасыщенный гетероциклил, содержащий один гетероатом N, при этом указанный гетероциклил конденсирован с кольцом, с которым он связан, которые могут быть возможно монозамещены или независимо полизамещены одним или более чем одним дейтерием, тритием, галогеном или С1-12алкилом.
2. Соединение по п. 1, где A1 представляет собой N.
3. Соединение по п. 1, где A1 представляет собой СН.
4. Соединение по п. 1, где каждый из А2, А3, А4 и A5 независимо представляет собой CR9.
5. Соединение по п. 1, где каждый из R1 и R2 независимо выбран из:
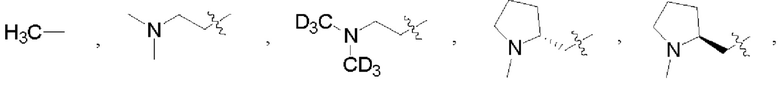
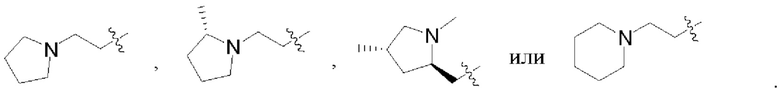
6. Соединение по п. 1, где R1 и R2, взятые вместе с атомом азота, с которым они связаны, образуют 3-12-членное моноциклическое или полициклическое кольцо, выбранное из:
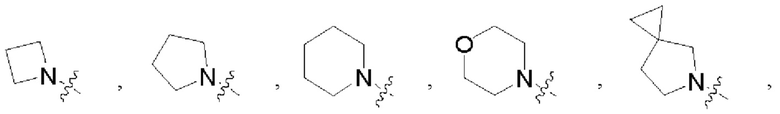

которое возможно монозамещено или независимо полизамещено галогеном, C1-12алкилом, -NRaRb или группой -C1-12алкил-NRaRb.
7. Соединение по п. 1, где R1 и R2, взятые вместе с атомом азота, с которым они связаны, образуют:
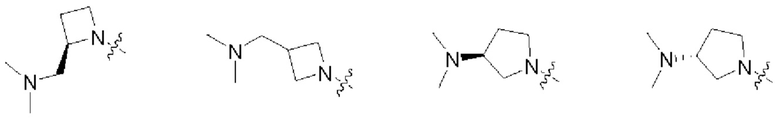

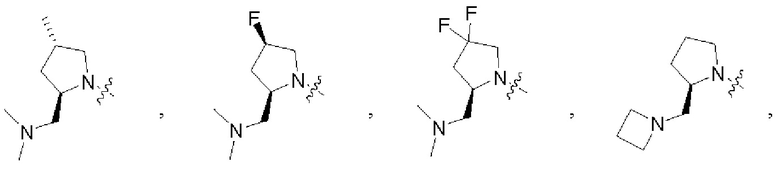
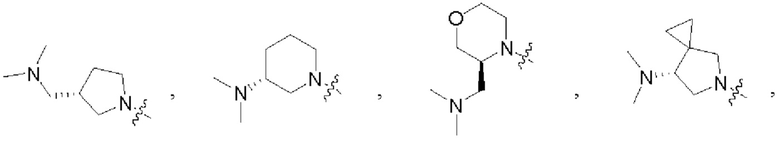


8. Соединение по п. 1, где n равно 2, А2 и А5 представляют собой СН, каждый из А3 и А4 независимо представляет собой СН, замещенный R, каждый R независимо представляет собой галоген и каждый из R4 и R5 независимо представляет собой незамещенный C1-6алкил.
9. Соединение по п. 1, выбранное из группы, состоящей из
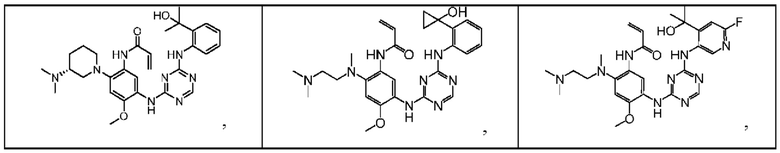


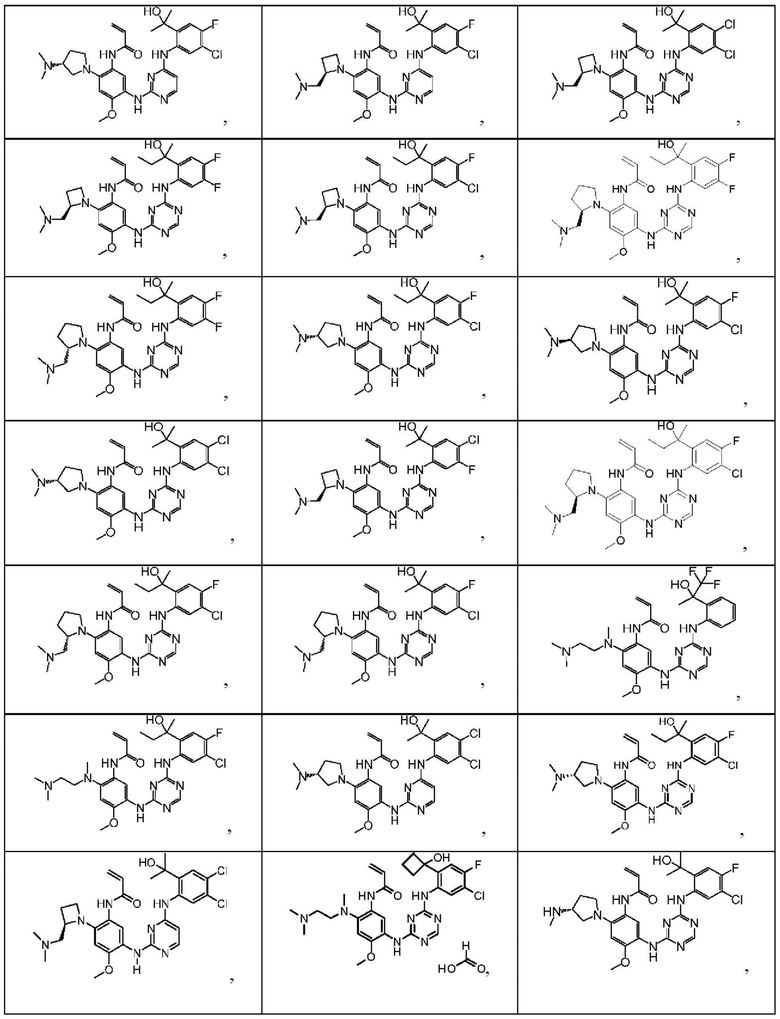
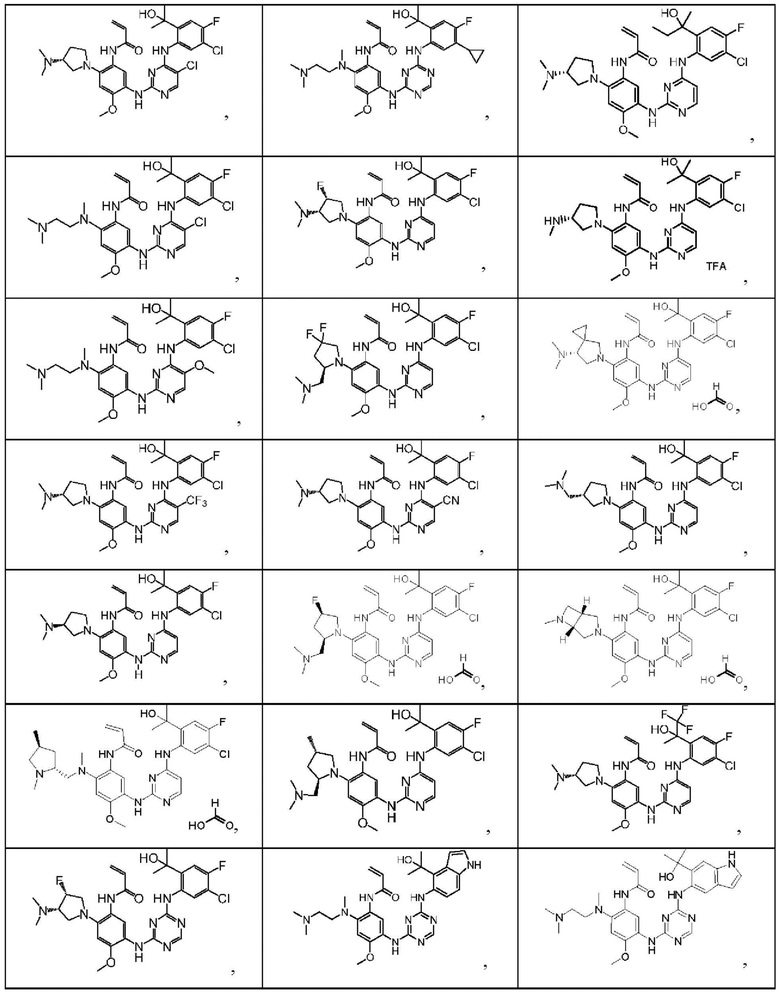
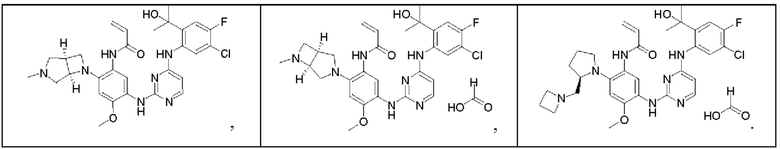
10. Соединение формулы (I) или его фармацевтически приемлемая соль, сложный эфир, гидрат, сольват или стереоизомер по любому из пп. 1-9 в кристаллической форме.
11. Соединение формулы (I) или его фармацевтически приемлемая соль, сложный эфир, гидрат, сольват или стереоизомер по любому из пп. 1-10 для применения в качестве лекарственного средства для ингибирования тирозинкиназы, где тирозинкиназа выбрана из ErbB (рецептор эпидермального фактора роста) и BTK (тирозинкиназа Брутона).
12. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении тирозинкиназы, где тирозинкиназа выбрана из ErbB и BTK и композиция содержит одно или более соединений формулы (I), его фармацевтически приемлемых солей, сложных эфиров, гидратов, сольватов или стереоизомеров по любому из пп. 1-10 в качестве первого активного ингредиента и фармацевтически приемлемый разбавитель, эксципиент или носитель.
13. Фармацевтическая композиция по п. 12 для применения в качестве лекарственного средства для ингибирования ErbB.
14. Фармацевтическая композиция по п. 13, где ErbB представляет собой EGFR или Her2, предпочтительно мутантный EGFR или мутантный Her2.
15. Способ ингибирования тирозинкиназы, где тирозинкиназа выбрана из ErbB и BTK путем использования одного или более соединений, его фармацевтически приемлемых солей, сложных эфиров, гидратов, сольватов или стереоизомеров по любому из пп. 1-10 или фармацевтической композиции по п. 12.
16. Способ лечения ассоциированного с тирозинкиназой заболевания, где ассоциированное с тирозинкиназой заболевание выбрано из ассоциированного с ErbB заболевания и ассоциированных с BTK заболеваний у субъекта, включающий введение этому субъекту эффективного количества одного или более соединений, их фармацевтически приемлемых солей, сложных эфиров, гидратов, сольватов или стереоизомеров по любому из пп. 1-10 или фармацевтической композиции по п. 12.
17. Способ по п. 16, где субъект представляет собой теплокровное животное, такое как человек.
18. Способ по п. 16, где ассоциированное с ErbB заболевание представляет собой рак.
19. Способ по п. 16, где ассоциированное с BTK заболевание представляет собой онкологическое заболевание или аутоиммунное заболевание.
20. Способ по п. 19, где онкологическое заболевание представляет собой лимфому или лейкоз.
21. Способ по п. 19, где аутоиммунное заболевание представляет собой ревматоидный артрит, системную красную волчанку или синдром Шегрена.
22. Способ по любому из пп. 15-18, где ErbB представляет собой EGFR или Her2, предпочтительно мутантный EGFR или мутантный Her2.
23. Способ по п. 22, где мутантный EGFR выбран из группы, состоящей из EGFR D761_E762insEAFQ, EGFR А763_Y764insHH, EGFR M766_A767instAI, EGFR A767_V769dupASV, EGFR A767_S768insTLA, EGFR S768_D770dupSVD, EGFR S768_V769insVAS, EGFR S768_V769insAWT, EGFR V769_D770insASV, EGFR V769_D770insGV, EGFR V769_D770insCV, EGFR V769_D770insDNV, EGFR V769_D770insGSV, EGFR V769_D770insGVV, EGFR V769_D770insMASVD, EGFR D770_N771insSVD, EGFR D770_N771insNPG, EGFR D770_N771insAPW, EGFR D770_N771insD, EGFR D770_N771insDG, EGFR D770_N771insG, EGFR D770_N771insGL, EGFR D770_N771insN, EGFR D770_N771insNPH, EGFR D770_N771insSVP, EGFR D770_N771insSVQ, EGFR D770_N771insMATP, EGFR delD770insGY, EGFR N771_P772insH, EGFR N771_P772insN, EGFR N771_H773dupNPH, EGFR delN771insGY, EGFR delN771insGF, EGFR P772_H773insPR, EGFR P772_H773insYNP, EGFR P772_H773insX, EGFR P772_H773insDPH, EGFR P772_H773insDNP, EGFR P772_H773insQV, EGFR P772_H773insTPH, EGFR P772_H773insN, EGFR P772_H773insV, EGFR H773_V774insNPH, EGFR H773_V774insH, EGFR H773_V774insPH, EGFR H773_V774insGNPH, EGFR H773_V774dupHV, EGFR H773_V774insG, EGFR H773_V774insGH, EGFR V774_C775insHV, EGFR с делецией в экзоне 19, EGFR L858R, EGFR T790M, EGFR L858R/T790M, EGFR с делецией в экзоне 19/Т790М, EGFR S768I, EGFR G719S, EGFR G719A, EGFR G719C, EGFR E709A/G719S, EGFR E709A/G719A, EGFR E709A/G719C и EGFR L861Q.
24. Способ по п. 22, где мутантный Her2 выбран из группы, состоящей из Her2 A775_G776insYVMA, Her2 delG776insVC, Her2 V777_G778insCG и Her2 P780_Y781insGSP.
25. Фармацевтическая комбинация, обладающая ингибирующей активностью в отношении тирозинкиназы, где тирозинкиназа выбрана из ErbB и BTK, которая содержит соединение формулы (I) или его фармацевтически приемлемую соль, сложный эфир, гидраты, сольваты или стереоизомеры по любому из пп. 1-10 или фармацевтическую композицию по п. 12 и второй терапевтический агент, представляющий собой противораковый агент.
| WO 2017219500 A1, 28.12.2017 | |||
| CN 105384694 A, 09.03.2016 | |||
| WO 2017101803 A1, 22.06.2017 | |||
| CN 106905245 A, 30.06.2017 | |||
| НОВЫЕ ПИРИДИНОВОЕ ПРОИЗВОДНОЕ И ПИРИМИДИНОВОЕ ПРОИЗВОДНОЕ (1) | 2005 |
|
RU2330021C2 |

