Область техники, к которой относится изобретение
[1] Настоящее изобретение относится к ряду хинолиновых производных в качестве ингибиторов пути hedgehog, в особенности в качестве ингибиторов SMO, соединения согласно настоящему изобретению можно применять в лечении заболеваний, связанных с путем hedgehog, в том числе рака.
Предыдущий уровень техники
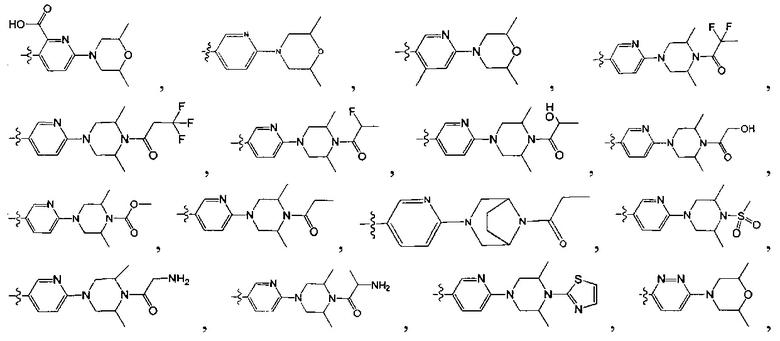
[2] Белки hedgehog являются секретированными сигнальными белками, первоначально обнаруженными у дрозофилы, они являются высокогидрофобными белками и выполняют важную роль в развитии зародыша. У людей уже были идентифицированы три типа гомологичных белков hedgehog, которые включают sonic hedgehog (Shh), Indian hedgehog (Ihh) и Desert hedgehog (Dhh), соответственно. Среди них Shh является необходимым не только для развития зародыша, также существует много доказательств, свидетельствующих о том, что он также выполняет важную роль в механизме канцерогенеза некоторых видов рака, в том числе базально-клеточной карциномы и т.д. (Caro, I. and J.A. Low, Clin Cancer Res, 2010. 16 (13): 3335-9). In vivo Shh синтезирует белок-предшественник с молекулярным весом 45 кДа, продуцирует N-концевой фрагмент с молекулярным весом 20 кДа путем саморазрезания, причем N-концевой фрагмент обеспечивает все его известные биологические активности in vivo, в том числе активируя внутриклеточный сигнальный путь hedgehog, чьи ключевые члены охватывают patched (РТСН), онкоген smoothened (SMO), являющиеся GPCR-подобными рецепторами, и транскрипционный фактор Gli и т.д. (Bale, А.Е. and K.P. Yu, Hum Mol Genet, 2001. 10 (7): 757-62). Результаты анализа изменения сигнального пути hedgehog базально-клеточной карциномы свидетельствуют о том, что большинство изменений происходят в РТСН-1 и SMO. (Von Hoff, D.D., et al., N Engl J Med, 2009. 361 (12): 1164-72). PTCH-1 представляет собой мембранный белок с 12-трансмембранной структурой, который является прямым рецептором Shh. В отсутствие Shh РТСН-1 взаимодействует с SMO, который представляет собой 7-трансмембранный белок, для ингибирования биологической активности SMO. Связывание Shh и РТСН-1 приводит к удалению РТСН-1 из SMO, тем самым освобождая SMO от ингибирования. Транскрипционный фактор Gli управляется SMO, который представлен в качестве переключателя транскрипции гена и чьи ключевые члены включают Gli1, Gli2 и Gli3. Весь путь hedgehog выполняет важную роль в нормальном развитии зародыша. Прерывание данного сигнального пути вызовет серьезный порок развития, природное тератогенное соединение циклопамин является примером, который представляет собой ингибитор hedgehog. Обычно концентрация белка hedgehog в организме взрослого человека является очень низкой. В случае низкой концентрации белка hedgehog РТСН-1 связывается с SMO, тем самым ингибируя биологическую активность SMO, так что весь путь является неактивным или имеет очень низкую активность. Когда клетки секретируют белок hedgehog, связывание hedgehog с рецептором РТСН-1 приводит к его удалению из SMO, тем самым снижая эффект ингибирования, направленный на SMO. SMO дополнительно активирует транскрипционный фактор Gli-1 для регуляции транскрипции гена и роста клеток. Увеличивающееся количество доказательств указывает, что большая часть базально-клеточной карциномы является результатом сверхвысокой активности сигнального пути hedgehog, вызванной мутациями или другими причинами. Таким образом, ингибирование сверхвысокой активности сигнального пути hedgehog может ингибировать рост раковых клеток для достижения лечения базально-клеточной карциномы или других видов рака, вызванных тем же механизмом. Доказательство того, что конститутивная активация SMO приводит к раку (например, ВСС), а также ослабление ингибирования Ptch приводит к тому, что SMO вызывает рак, иллюстрирует применение антагониста SMO в качестве терапевтических средств в лечении заболеваний (Stone, et al, (1996) Nature 384: 129). Ряд научных и клинических испытаний свидетельствует о том, что ингибиторы hedgehog являются эффективными в лечении многих видов рака. Последние данные клинических испытаний свидетельствуют о том, что ингибитор hedgehog GDC-0449 является эффективным в лечении базально-клеточной карциномы и мозговидной клеточной карциномы (LorussoPM. et al. Clin Cancer Res. 2011; 17 (8): 2502-11), на что было выдано разрешение FDA в январе 2012 года, и другие виды рака вызваны тем же механизмом, такие как синдром базально-клеточного невуса (BCNS) (Goldberg LH. et al. Arch Dermatol. 2011 Mar 21.). Биохимические испытания свидетельствуют о том, что положение ингибирования для GDC-0449 размещено на SMO, при этом ингибирование активности SMO означает ингибирование активности всего пути hedgehog для достижения цели противоракового применения. В дополнение к двум типам видов рака, базально-клеточной карциноме и мозговидной клеточной карциноме, существует много других видов рака, связанных со сверхвысокой активностью сигнального пути hedgehog, в том числе рак поджелудочной железы, рак желудка, колоректальный рак, рак яичников и рак предстательной железы, и часть видов лейкоза и т.д. (De Smaele Е. et al. Curr Opin Investig Drugs. 2010; 11 (6): 707-18). Таким образом, существует перспективный потенциал разработки ингибиторов hedgehog в качестве новых противораковых лекарственных средств.
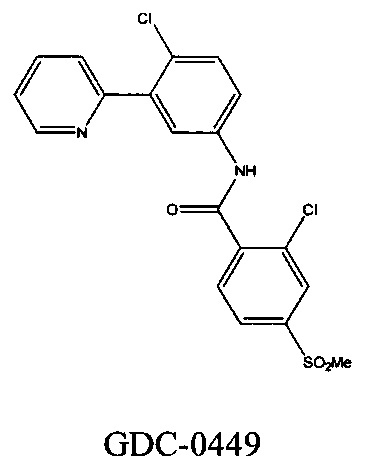
[3] Хотя в предыдущем уровне техники существуют некоторые ингибиторы SMO, все еще существует необходимость в дополнительном улучшении аспектов активности, растворимости, фармакокинетики, применимости в качестве лекарственного средства и т.д.
Содержание описания настоящего изобретения
[4] Целью настоящего изобретения является обеспечения соединения, представленного формулой (I), или его фармацевтически приемлемой соли,
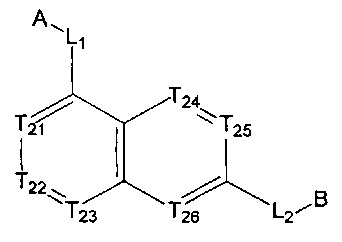
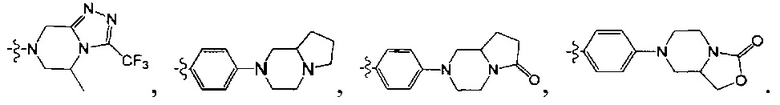
[5] где А выбран из
(R20)3С-Е13-;
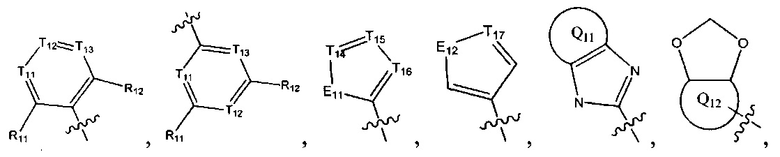
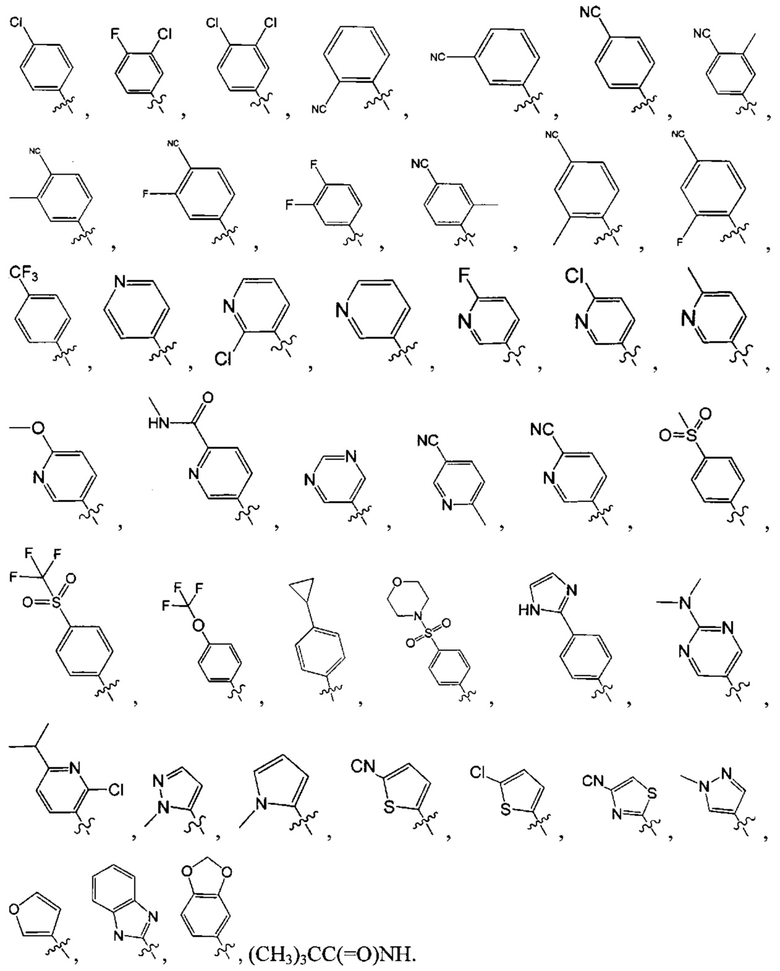
[6] каждый из Т11-17 независимо выбран из N, С(R13);
[7] каждый из Е11-13, L1, L2 независимо выбран из N(R14), С(=O)N(R15), S(=O)2N(R16), C=N(R17), С(R18)(R19), S, С(=O)O, С(=O), С=S, S(=O) или S(=O)2;
[8] каждый из L1, L2 также может быть независимо выбран из одинарной связи;
[9] каждый из R11-13, R18-19 независимо выбран из Н, F, Cl, Br, I, CN, ОН, SH, NH2, необязательно R01-замещенного С1-6алкила, или гетероалкила, или группы алкил-гетероатом, или группы гетероалкил-гетероатом, необязательно R02-замещенной С3-8циклической группы, или гетероциклической группы, или группы цикл-гетероатом, или группы гетероцикл-гетероатом; предпочтительно выбран из метила, трифторметила, трифторметокси, F, Cl, Br, I, CN, метиламинокарбонила, метилсульфонила, трифторметилсульфонила, трифторметокси, циклопропила, морфолинилсульфонила, 2-имидазолила, диметиламино, н- или изо-пропила;
[10] каждый из R14-17 независимо выбран из Н, необязательно Р01-замещенного С1-6алкила или С3-6циклоалкила;
[11] R20 выбран из Н, F, Cl, Br, I, CN, ОН, SH, NH2, метокси, метиламино, диметиламино;
[12] каждый из Q11-12 независимо выбран из фенила, пиридила, тиенила, фурила;
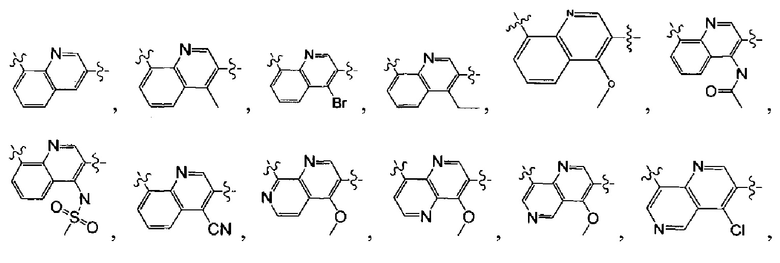
[13] структурная единица 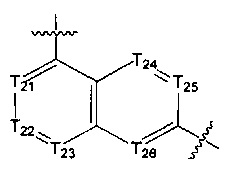 может быть замещена
может быть замещена
 ;
;
[14] каждый из Т21-26 независимо выбран из N, С(R25); Т25 также может быть выбран из N⊕(R25);
[15] каждый из R21-25 независимо выбран из Н, F, Cl, Br, I, CN, ОН, SH, NH2, необязательно R01-замещенного С1-6алкила, или гетероалкила, или группы алкил-гетероатом, или группы гетероалкил-гетероатом, необязательно R02-замещенной С3-8циклической группы, или гетероциклической группы, или группы цикл-гетероатом, или группы гетероцикл-гетероатом; предпочтительно выбран из F, Cl, Br, I, CN, ОН, метила, этила, изопропила, метокси, трифторметила, дифторметокси, н- или изо-пропокси, циклопропила, формамидо, метансульфониламино, диметиламино, диметиламиноэтокси, метилсульфонила, карбометокси, 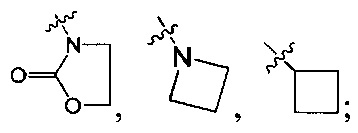
[16] каждый из m21-24 независимо выбран из 0, 1 или 2;
[17] В выбран из 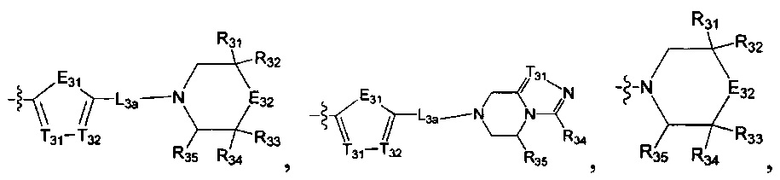
[18] R3 выбран из 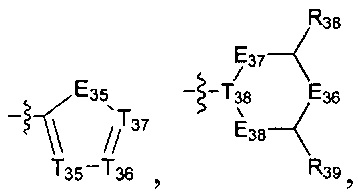 Н, необязательно R01-замещенного С1-6алкила, или гетероалкила, или группы алкил-гетероатом, или группы гетероалкил-гетероатом, необязательно R02-замещенной С3-8циклической группы, или гетероциклической группы, или группы цикл-гетероатом, или группы гетероцикл-гетероатом;
Н, необязательно R01-замещенного С1-6алкила, или гетероалкила, или группы алкил-гетероатом, или группы гетероалкил-гетероатом, необязательно R02-замещенной С3-8циклической группы, или гетероциклической группы, или группы цикл-гетероатом, или группы гетероцикл-гетероатом;
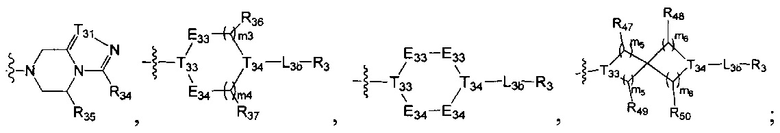
[19] каждый из Е31-38, L3a, L3b независимо выбран из N(R40), N(R40) (СН2)1-3, С(=О)N(R40)(СН2)1-3, С(=O)N(R41), S(=O)2N(R42), С=N(R43), С(R44)(R45), S, С(=O)O, С(=O), C=S, S(=O), S(=O)2, О или С(=O)С(R44)(R45);
[20] каждый из L3a, L3b также может быть независимо выбран из одинарной связи или С1-5алкила;
[21] каждый из Е31, Е35 также может быть независимо выбран из -Т39=Т40-;
[22] каждый из T31-40 независимо выбран из N, C(R46);
[23] каждый из R40-43 независимо выбран из Н, необязательно R01-замещенного С1-6алкила или С3-6циклоалкила, необязательно R01-замещенного С1-6алкиладила или С3-6циклоалкилацила, необязательно R01-замещенного С1-6алкилсульфонила или С3-6циклоалкилсульфонила, необязательно R01-замещенного 5-6-членного арилацила, необязательно R01-замещенного 5-6-членного гетероарилацила, необязательно R01-замещенного 5-6-членного арилсульфонила, необязательно R01-замещенного С1-6алкилоксикарбонила, необязательно R01-замещенного С1-6алкиламинокарбонила;
[24] каждый из R31-49, R44-50 независимо выбран из H, F, Cl, Br, I, CN, ОН, SH, NH2, С(=O)ОН, необязательно R01-замещенного С1-6алкила, или гетероалкила, или группы алкил-гетероатом, или группы гетероалкил-гетероатом, необязательно R02-замещенной С0-3алкил-С3-8циклической группы, или гетероциклической группы, или группы цикл-гетероатом, или группы гетероцикл-гетероатом;
[25] каждый из R01, R02 независимо выбран из F, Cl, Br, I, CN, ОН, SH, NH2, R03;
[26] R03 выбран из С1-6алкиламино, ди(С1-6алкил)амино, С1-6алкокси, С3-8циклоалкиламино, С3-8гетероциклоалкиламино, С3-8циклоалкокси;
[27] гетероатом или группа гетероатома независимо выбраны из C(=O)NR04, N(R05), C=N(R06), О, S, С(=O)O, С(=O), C=S, S(=O), S(=O)2 и/или S(=O)2N(R07);
[28] каждый из R04-07 независимо выбран из H, R08;
[29] R08 выбран из С1-6алкила или С3-8циклоалкила;
[30] R03, R08 необязательно замещены R001, R001 выбран из ОН, N(CH3)2, NH(CH3), NH2, F, Cl, Br, I, CN, метила, метокси;
[31] число R01, R02, R001, гетероатома или группы гетероатома независимо выбрано из 0, 1, 2 или 3;
[32] каждый из m3, m4 независимо выбран из 0 или 1, если m3 или m4 выбраны из 0, то соответствующая структурная единица представляет собой одинарную связь только для соединения;
[33] каждый из m5, m6 независимо выбран из 1 или 2;
[34] необязательно R31 и R32, R31 и R33, R31 и R35, Е33 и Е34 вместе образуют соединяющую связь (CH2)1-6, предпочтительно (СН2)2, (СН2)3, (СН2)4, (СН2)5;
[35] необязательно Е32 и R32 соединены друг с другом с образованием 5-членного карбоциклического кольца или гетероциклического кольца;
[36] необязательно, если Е32 выбран из N(R40) или О, R31 и R34 представляют цис-расположение; если Е32 выбран из C(R43)(R44), R31 и R34 представляют транс-расположение; если Т34 выбран из N, R36 и R37 представляют цис-расположение; если Т34 выбран из С(R45), R36 и R37 представляют транс-расположение;
[37] предпочтительно С3-8циклическая группа, или гетероциклическая группа, или группа цикл-гетероатом, или группа гетероцикл-гетероатом выбраны из фенила, пиридила, тиенила, фурила, имидазолила, оксазолила, тиазолила, изотиазолила.
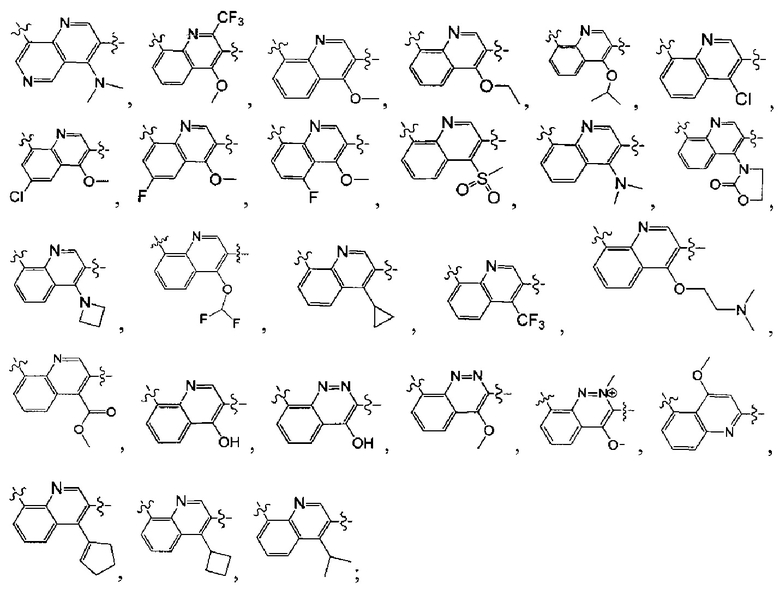
[38] Предпочтительным является соединение или его фармацевтически приемлемая соль, где А выбран из 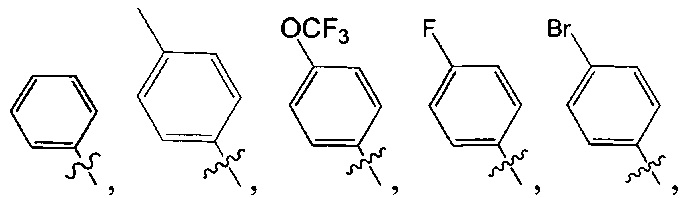
[39] Необязательно соединение или его фармацевтически приемлемая соль, где структурная единица 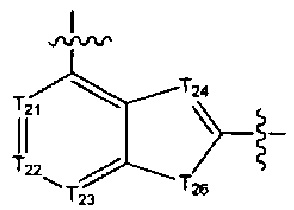 выбрана из
выбрана из  ,
,
 выбран из
выбран из 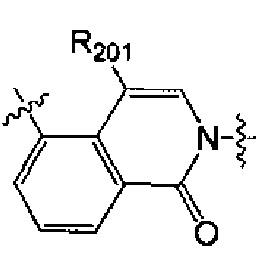 , структурная единица
, структурная единица 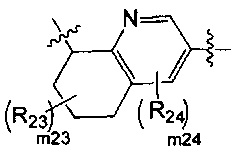
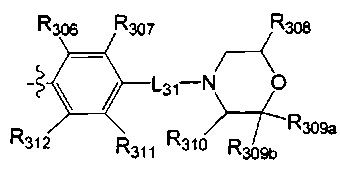
выбрана из 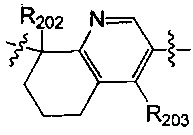 , где
, где
[40] каждый из R201, R202, R203 независимо выбран из Н, F, Cl, Br, I, CN, ОН, SH, NH2, необязательно R01-замещенного С1-6алкила, или гетероалкила, или группы алкил-гетероатом, или группы гетероалкил-гетероатом, необязательно R02-замещенной С3-8циклической группы, или гетероциклической группы, или группы цикл-гетероатом, или группы гетероцикл-гетероатом;
[41] предпочтительно R201 выбран из метила, F, Cl, Br, I; каждый из R202, R203 независимо выбран из С1-6алкокси, более предпочтительно - метокси.
[42] Предпочтительным является соединение или его фармацевтически приемлемая соль, где структурная единица 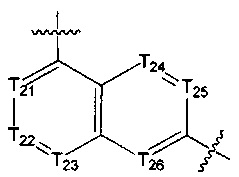 выбрана из:
выбрана из:
[43] структурная единица  выбрана из:
выбрана из: 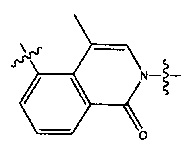 ,
, 
[44] структурная единица 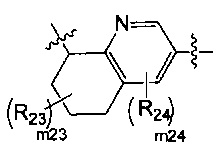 выбрана из:
выбрана из: 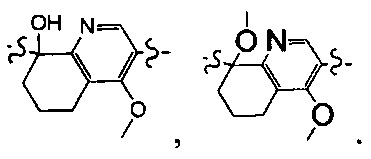
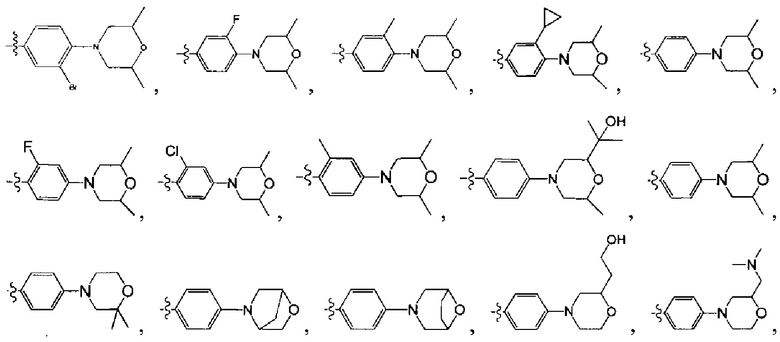
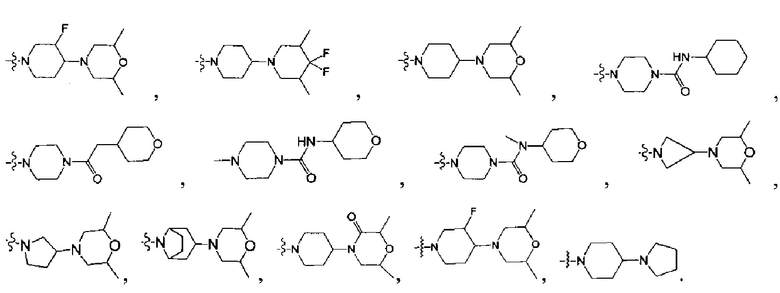
[45] Предпочтительным является соединение или его фармацевтически приемлемая соль, где В выбран из 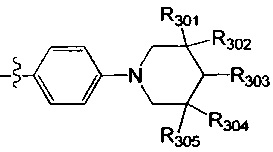 , где
, где
[46] каждый из R301-305 независимо выбран из Н, ОН, NH2, F, Cl, Br, I, CN, необязательно R3001-замещенного С1-3алкила, или алкокси, или алкиламино, или циклопропила;
[47] R3001 выбран из ОН, N(СН3)2, NH(СН3), NH2, F, Cl, Br, I, CN, метила, метокси, число R3001 выбрано из 1, 2 или 3;
[48] необязательно структурная единица  в общей структуре может быть замещена пиридилом, тиенилом, фурилом;
в общей структуре может быть замещена пиридилом, тиенилом, фурилом;
[49] необязательно
[50] каждый из R301-305 независимо выбран из метила, Н, ОН, NH2, F, Cl, Br, I, CN;
[51] кроме того, необязательно
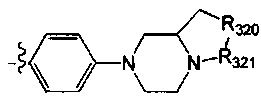
[52] В выбран из 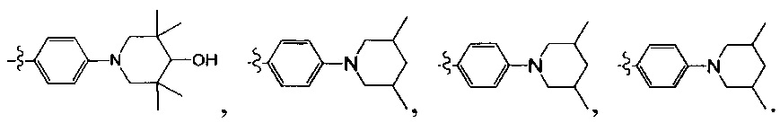
[53] Необязательным является соединение или его фармацевтически приемлемая соль, где В выбран из
 , где
, где
[54] каждый из R306-308, R309a, R309b, R310-312 независимо выбран из Н, ОН, NH2, F, Cl, Br, I, CN, необязательно R3001-замещенного С1-3алкила, или алкокси, или алкиламино, или циклопропила;
[55] R3001 выбран из ОН, N(СН3)2, NH(СН3), NH2, F, Cl, Br, I, CN, метила, метокси, число R3001 выбрано из 1, 2 или 3;
[56] L31 выбран из одинарной связи, R3002N(R3003)R3004, О, S, С(=O)O, С(=O), С=S, S(=O) и/или S(=O)2, R3002 выбран из одинарной связи или С(=O);
[57] R3003 выбран из Н, С1-3алкила или циклопропила;
[58] R3004 выбран из (СН2)0-3;
[59] необязательно R308 и R309a, R308 и R310 вместе образуют соединяющую связь (СН2)1-3;
[60] предпочтительно R308 и R310 вместе образуют соединяющую связь СН2, R308 и R309a вместе образуют соединяющую связь СН2СН2.
[61] Предпочтительно, каждый из R306-308, R309a, R309b, R310-312 независимо выбран из метила, циклопропила, С(СН3)2(ОН), СН2СН2ОН, CH2N(СН3)2, Н, ОН, NH2, F, Cl, Br, I, CN.
[62] Предпочтительно L31 выбран из одинарной связи, NHCH2CH2;
[63] предпочтительно В выбран из 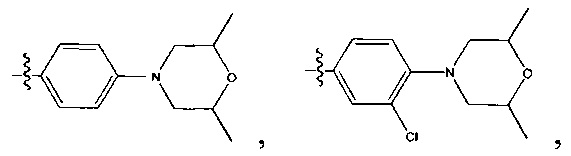
 .
.
[64] Предпочтительным является соединение или его фармацевтически приемлемая соль, где В выбран из 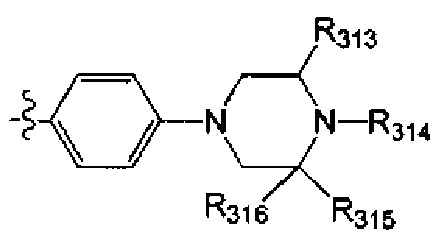 , где
, где
каждый из R313, R315, R316 независимо выбран из Н, ОН, NH2, F, Cl, Br, I, CN, необязательно R3001-замещенного С1-3алкила, или алкокси, или алкиламино, или циклопропила;
[65] R314 выбран из Н, ОН, NH2, F, Cl, Br, I, CN, необязательно R3001-замещенного С1-3алкила, или алкокси, или алкиламино, или циклопропила,
(СН2)0-3R3005,  , где R3005 необязательно замещен R3001;
, где R3005 необязательно замещен R3001;
[66] R3005 выбран из С3-6циклоалкила, фенила, пиридила, тиенила, фурила, имидазолила, оксазолила, тиазолила, изотиазолила, каждый из вышеупомянутой группы необязательно конденсирован бензольным кольцом;
[67] R3001 выбран из ОН, N(СН3)2, NH(СН3), NH, F, Cl, Br, I, CN, метила, метокси, число R3001 выбрано из 1, 2 или 3;
[68] необязательно  в общей структуре может быть замещен пиридилом, тиенилом, фурилом, имидазолилом, оксазолилом, тиазолилом, изотиазолилом.
в общей структуре может быть замещен пиридилом, тиенилом, фурилом, имидазолилом, оксазолилом, тиазолилом, изотиазолилом.
[69] Предпочтительно каждый из R313, R315, R316 независимо выбран из Н, метила;
[70] предпочтительно R314 выбран из Н, метила, этила, фенилметилена, циклопропилметилена, метоксифенила,  .
.
[71] Предпочтительно В выбран из 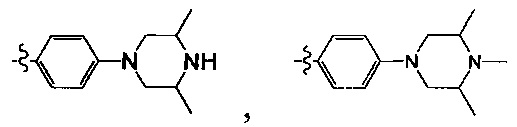 ,
,

[72] Предпочтительным является соединение или его фармацевтически приемлемая соль, где В выбран из 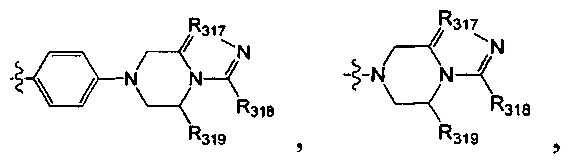
 , где
, где
[73] R317 выбран из CR3006 или N;
[74] каждый из R317, R319, R3006-3008 независимо выбран из Н, ОН, NH2, F, Cl, Br, I, CN, необязательно R3001-замещенного С1-3алкила, или алкокси, или алкиламино, или циклопропила;
[75] каждый из R320-321 независимо выбран из С(R3007)(R3008), О, CON(R3009), N(R3010), С=N(R3011), S, С(=O)O, С(=O), С=S, S(=O) и/или S(=O)2;
[76] каждый из R3009-3011 независимо выбран из Н, необязательно R3001-замещенного С1-3алкила или циклопропила;
[77] R3001 выбран из ОН, N(СН3)2, NH(СН3), NH2, F, Cl, Br, I, CN, метила, метоксила, число R3001 выбрано из 1, 2 или 3;
[78] необязательно  в общей структуре может быть замещен пиридилом, тиенилом, фурилом, имидазолилом, оксазолилом, тиазолилом, изотиазолилом.
в общей структуре может быть замещен пиридилом, тиенилом, фурилом, имидазолилом, оксазолилом, тиазолилом, изотиазолилом.
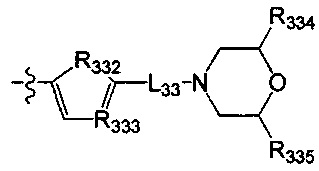
[79] Предпочтительно R317 выбран из CR3006 или N; каждый из R3006, R318, R319 выбран из метила, трифторметила;
[80] каждый из R320-321 независимо выбран из СН2, О, С(=O).
[81] Предпочтительно В выбран из 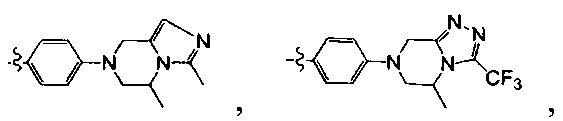
[82] Предпочтительным является соединение или его фармацевтически приемлемая соль, где В выбран из 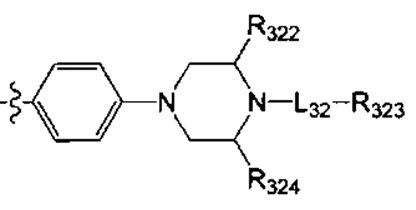 ,
,
[83] где
[84] L32 выбран из С(R3007)(R3008), О, CON(R3009), N(R3010), С=N(R3011), S, С(=O)O, С(=O), С=S, S(=O) и/или S(=O)2;
[85] R323 выбран из С1-6алкила, С3-6циклоалкила, фенила, пиридила, имидазолила, тиенила, фурила, оксазолила, тиазолила, изотиазолила, каждый из вышеупомянутой группы необязательно замещен R3012;
[86] каждый из R322, R324, R3007, R3008, R3012 независимо выбран из Н, ОН, NH2, F, Cl, Br, I, CN, необязательно R3001-замещенного С1-3алкила, или алкокси, или алкиламино, или циклопропила;
[87] каждый из R3009-3011 независимо выбран из Н, необязательно R3001-замещенного С1-3алкила или циклопропила;
[88] R3001 выбран из ОН, N(СН3)2, NH(СН3), NH2, F, Cl, Br, I, CN, метила, метокси;
[89] число каждого из R3001, R3012 выбрано из 1, 2 или 3.
[90] Необязательно  в общей структуре может быть замещен пиридилом, тиенилом, фурилом, имидазолилом, оксазолилом, тиазолилом, изотиазолилом.
в общей структуре может быть замещен пиридилом, тиенилом, фурилом, имидазолилом, оксазолилом, тиазолилом, изотиазолилом.
[91] Предпочтительно каждый из R322, R324 независимо выбран из Н, метила, фенила, С(СН3)2ОН.
[92] Предпочтительно L32 выбран из С(=O), S(=O)2;
[93] предпочтительно R323 выбран из трет-бутокси, метила, метокси, этила, этокси, пропокси, изопропила, н-пропила, изопропокси, циклопропила, метиламино, фенила, пиридила, 3-метилпиридила, имидазолила, С(СН3)2ОН;
[94] предпочтительно В выбран из 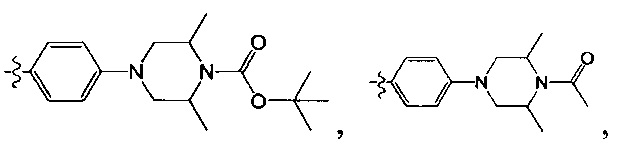
[95] Предпочтительным является соединение или его фармацевтически приемлемая соль, где В выбран из 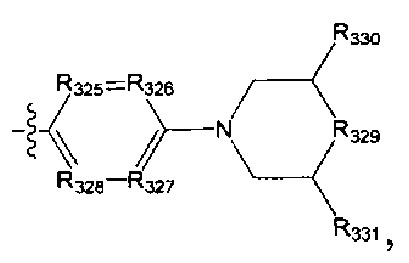
[96] где
[97] один или два из R325-328 выбраны из N, остальные выбраны из С(R3013);
[98] R329 выбран из N(R3014), О, С(R3015)(R3016), CON(R3017), R(R3018), С=N(R3019), S, С(=O)O, С(=O), С=S, S(=O) и/или S(=O)2;
[99] R3014 выбран из С(=O)R3020, S(=О)2R3020, тиазолила, изотиазолила, фенила, пиридила, имидазолила, тиенила, фурила, оксазолила;
[100] R3020 выбран из необязательно R3001-замещенного С1-3алкила или алкоксила;
[101] каждый из R3013, R3015, R3016, R330-331 независимо выбран из Н, ОН, NH2, F, Cl, Br, I, CN, С(=O)ОН, необязательно R3001-замещенного С1-3алкила, или алкокси, или алкиламино, или циклопропила;
[102] каждый из R3017-3019 независимо выбран из Н, необязательно R3001-замещенного С1-3алкила или циклопропила;
[103] R3001 выбран из ОН, N(CH3)2, NH(CH3), NH2, F, Cl, Br, I, CN, метила, метокси, число R3001 выбрано из 1, 2 или 3;
[104] необязательно R330 и R331 вместе образуют соединяющую связь (СН2)1-3;
[105] необязательно, если R329 выбран из N(R3014) или О, R330 и R331 представляют цис-расположение; если R329 выбран из C(R3015)(R3016), R330 и R331 представляют транс-расположение;
[106] предпочтительно один или два из R325-328 выбраны из N, остальные выбраны из СН, СС(=O)ОН или ССН3.
[107] Предпочтительно R329 выбран из N(R3014), О.
[108] Предпочтительно R3014 выбран из C(=O)R3020,  .
.
[109] Предпочтительно R3020 выбран из C(CH3)(F)2, СН3, CF3, СН2СН3, CH2CF3, CH(F)(CH3), СН(ОН)(СН3), СН2(ОН), CH(NH2)(CH3), метокси, этокси, аминометила.
[110] Предпочтительно R330-331 выбран из метила.
[111] Предпочтительно В выбран из 
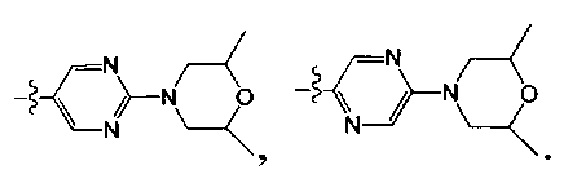
[112] Предпочтительным является соединение или его фармацевтически приемлемая соль, где В выбран из
 , где
, где
[113] R332 выбран из S, N(R3021), О, C(R3022)(R3023), CON(R3024), N(R3025), C=N(R3026), S, C(=O)O, C(=O), C=S, S(=O) и/или S(=O)2;
[114] L33 выбран из одинарной связи, С(=O), S, О, C(R3022)(R3023), CON(R3024), N(R3025), C=N(R3026), S, C(=O)O, C=S, S(=O) и/или S(=O)2;
[115] R333 выбран из N, C(R3027);
[116] каждый из R3027, R334, R335, R3022, R3023 независимо выбран из Н, ОН, NH2, F, Cl, Br, I, CN, необязательно R3001-замещенного С1-3алкила, или алкокси, или алкиламино, или циклопропила;
[117] каждый из R3021, R3024-3026 независимо выбран из Н, необязательно R3001-замещенного С1-3алкила или циклопропила;
[118] R3001 выбран из ОН, N(CH3)2, NH(CH3), NH2, F, Cl, Br, I, CN, метила, метокси, число R3001 выбрано из 1, 2 или 3;
[119] предпочтительно R332 выбран из S, R333 выбран из N или СН, L33 выбран из одинарной связи, С(=O), каждый из R334, R335 выбран из метила.
[120] Предпочтительно В выбран из 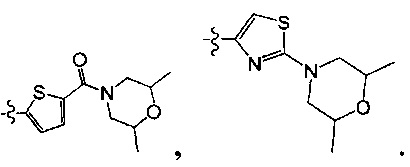
[121] Предпочтительным является соединение или его фармацевтически приемлемая соль, где В выбран из 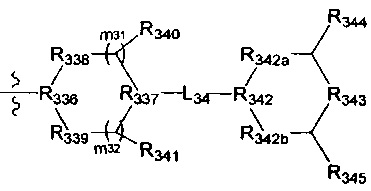 , где
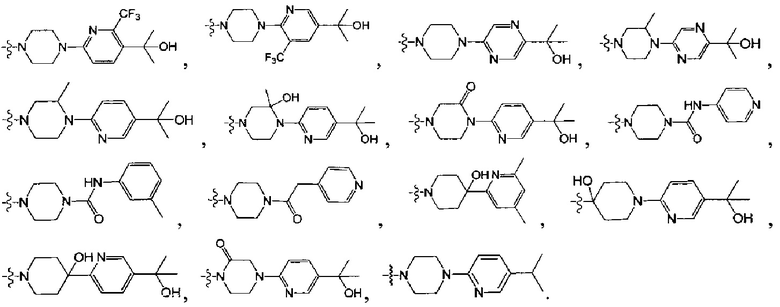
, где
[122] каждый из R336, R337, R342 независимо выбран из N или C(R3028);
[123] каждый из R338, R339 выбран из C(R3029) (R3030), необязательно R338 и R339 вместе соединены с одним и тем же (CH2)1-3 с образованием кольца;
[124] каждый из m31, m32 независимо выбран из 0 или 1;
[125] каждый из R342a, R342b, R343, L34 независимо выбран из одинарной связи, C(=O)N(R3031), C(=O)C(R3032)(R3033), C(R3034)(R3035), CON(R3036), N(R3037), C=N(R3038), O, S, C(=O)O, C(=O), C=S, S(=O) и/или S(=O)2;
[126] L34 также может быть выбран из одинарной связи;
[127] каждый из R340, R341, R344, R345, R3028, R3029, R3030, R3032-3035 независимо выбран из Н, ОН, NH2, F, О, Br, I, CN, необязательно R3001-замещенного С1-3алкила, или алкокси, или алкиламино, или циклопропила;
[128] каждый из R3031, R3036-3038 независимо выбран из Н, необязательно R3001-замещенного С1-3алкила или циклопропила;
[129] R3001 выбран из ОН, N(CH3)2, NH(CH3), NH2, F, Cl, Br, I, CN, метила, метокси, число R3001 выбрано из 1, 2 или 3;
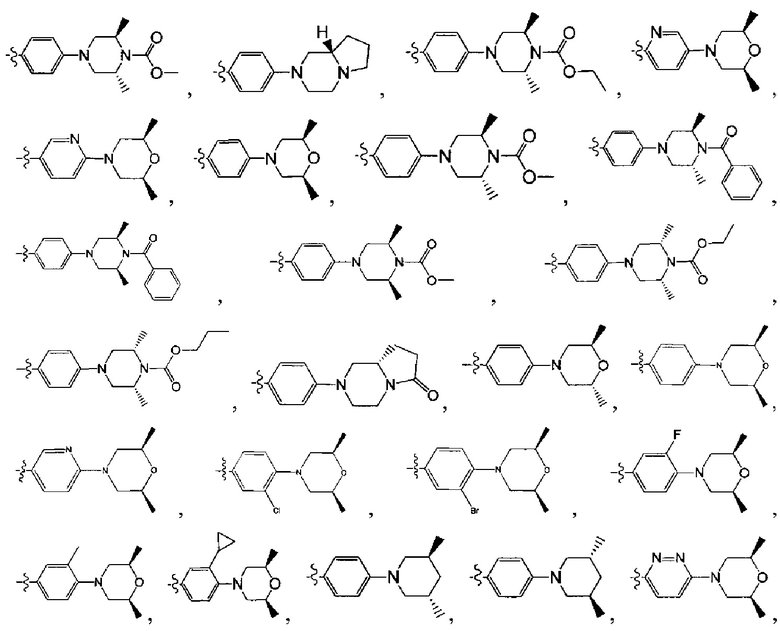
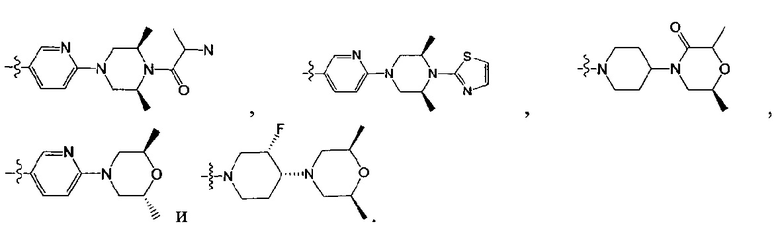
[130] предпочтительно каждый из R336, R337, R342 независимо выбран из N или СН.
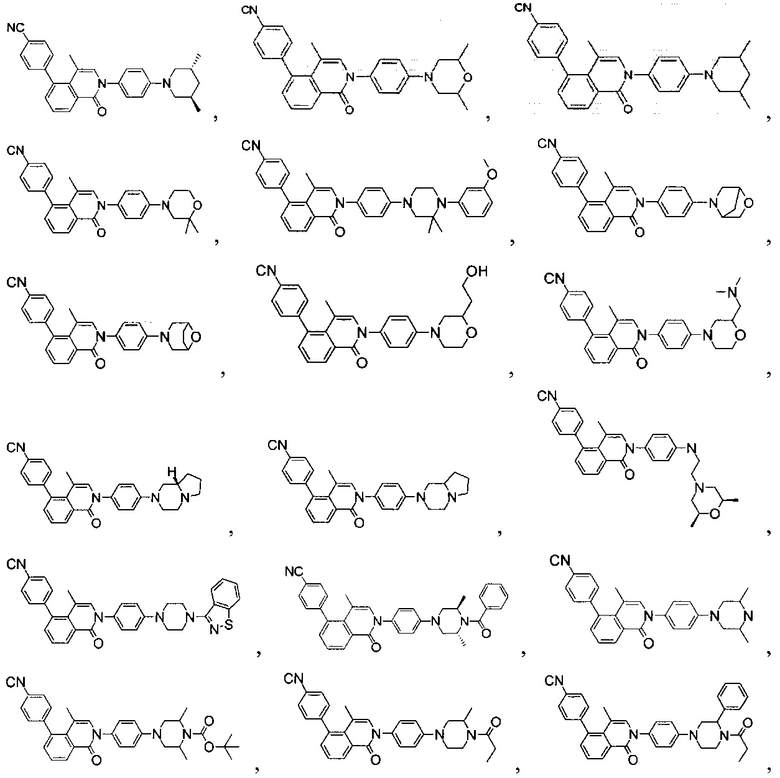
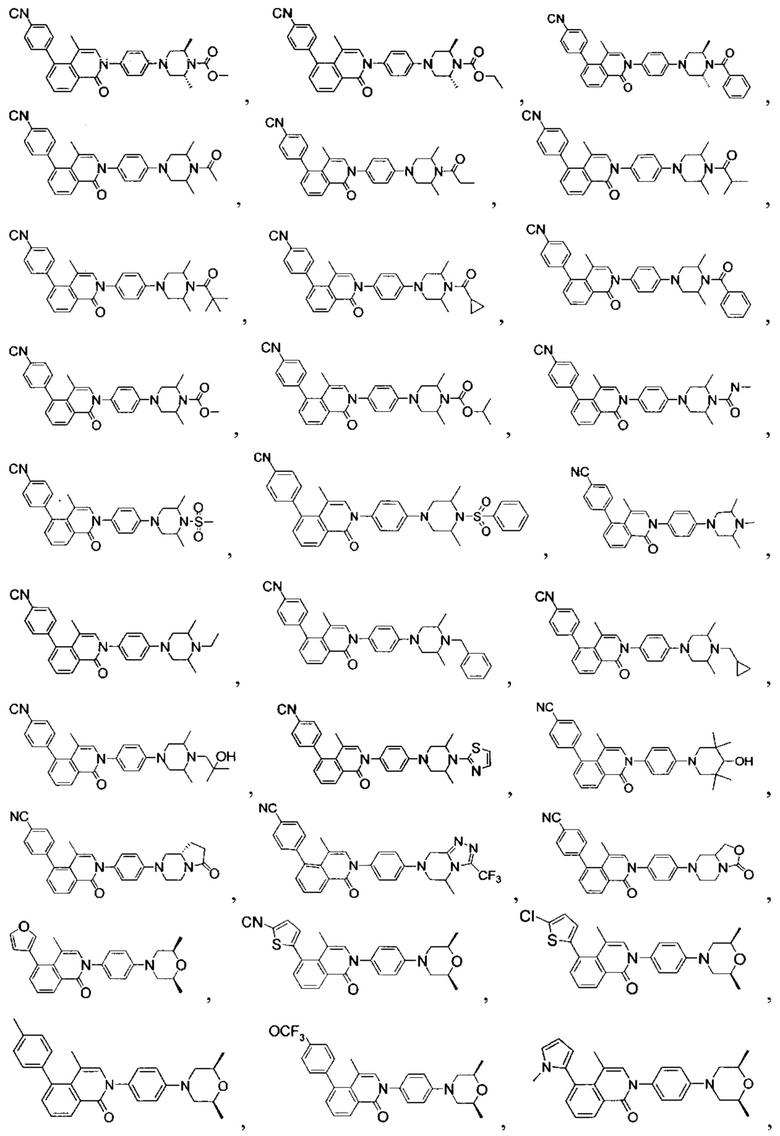
[131] Предпочтительно каждый из R338, R339 независимо выбран из СН2, необязательно R338 и R339 вместе соединены с одним и тем же СН2СН2 с образованием кольца.
[132] Предпочтительно каждый из m31, m32 независимо выбран из 0 или 1.
[133] Предпочтительно L34 независимо выбран из одинарной связи, С(=O)NH, С(=O)N(СН3), С(=O)СН2.
[134] Предпочтительно каждый из R340, R341, R344, R345 независимо выбран из Н или метила.
[135] Предпочтительно каждый из R342a, R342b выбран из С(=O) или СН2.
[136] Предпочтительно В выбран из 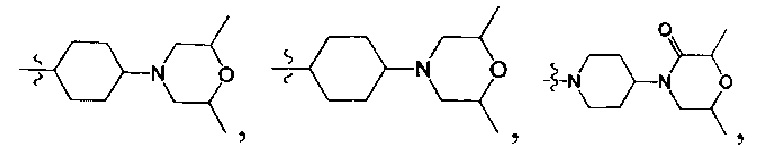
[137] Предпочтительным является соединение или его фармацевтически приемлемая соль, где В выбран из 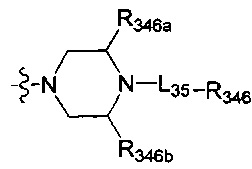 , где
, где
[138] L35 независимо выбран из одинарной связи, С(=O)N(R3031), С(=O)С(R3032)(R3033), С(R3034)(R3035), N(R3037), C=N(R3038), О, S, С(=O)O, С(=O), С=S, S(=O) и/или S(=O)2;
[139] L35 также может быть выбран из одинарной связи;
[140] каждый из R3032-3035 независимо выбран из Н, ОН, NH2, F, Cl, Br, I, CN, необязательно R3001-замещенного С1-3алкила, или алкокси, или алкиламино, или циклопропила;
[141] каждый из R346, R346a, R346b независимо выбран из Н, необязательно R3001-замещенного С1-3алкила, или алкокси, или алкилацила, или алкилсульфонила, или алкиламино, или циклопропила, необязательно R3001-замещенного циклопропилацила или циклопропилсульфонила;
[142] R346 также может быть выбран из фенила, пиридила, имидазолила, тиенила, фурила, оксазолила, тиазолила, изотиазолила, пирролидила, 2-пиридонила;
[143] каждый из R3031, R3037, R3038 независимо выбран из Н, необязательно R3001-замещенного С1-3алкила или циклопропила;
[144] R3001 выбран из ОН, N(СН3)2, NH(СН3), NH2, F, Cl, Br, I, CN, -S(=O)СН3, -S(=O)2СН3, метила, метокси, число R3001 выбрано из 1, 2 или 3;
[145] предпочтительно L35 выбран из одинарной связи, С(=O), СН2;
[146] предпочтительно R346 выбран из Н, метила, фенила, С(СН3)2(ОН), СН2С(СН3)2(ОН), циклопропилацила, изопропилсульфонила, пирролидила, 2-пиридонила, 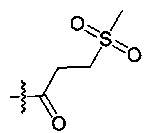 ;
;
[147] предпочтительно В выбран из 
[148] Предпочтительным является соединение или его фармацевтически приемлемая соль, где В выбран из 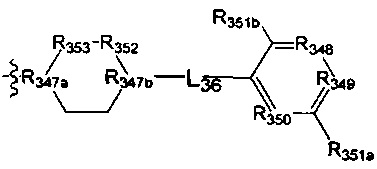 , где
, где
[149] каждый из R347a, R347b независимо выбран из N или C(R3028);
[150] ни один, один или два из R348-350 выбраны из N, остальные выбраны из C(R3039);
[151] каждый из R352, R353, L36 независимо выбран из одинарной связи, -C(=O)N(R3031)-, C(=O)C(R3032)(R3033)-, C(R3034)(R3035), CON(R3036), N(R3037), C=N(R3038), О, S, C(=O)O, C(=O), C=S, S(=O) и/или S(=O)2;
[152] L36 также может быть выбран из одинарной связи;
[153] каждый из R3028, R3032-3035, R3039, R351a, R351b независимо выбран из Н, ОН, NH2, F, Cl, Br, I, CN, необязательно R3001-замещенного С1-3алкила, или алкокси, или алкиламино, или циклопропила;
[154] каждый из R3031, R3036-3038 независимо выбран из Н, необязательно R3001-замещенного С1-3алкила или циклопропила;
[155] R3001 выбран из ОН, N(CH3)2, NH(CH3), NH2, F, Cl, Br, I, CN, метила, метокси, число R3001 выбрано из 1, 2 или 3; предпочтительно L36 выбран из одинарной связи, C(=O)NH, С(=O)СН2.
[156] Предпочтительно R347a, R347b независимо выбраны из N, СН или С(ОН).
[157] Предпочтительно ни один, один или два из R348-350 выбраны из N, остальные выбраны из C(R3039).
[158] Предпочтительно R3039 выбран из Н, метила, трифторметила, С(СН3)2(ОН).
[159] Предпочтительно каждый из R351a, R351b независимо выбран из Н, метила, трифторметила, изопропила.
[160] Предпочтительно каждый из R352, R353 независимо выбран из С(=O), С(СН3)(ОН).
[161] Предпочтительно В выбран из 
[162] Предпочтительным является соединение или его фармацевтически приемлемая соль, где В выбран из 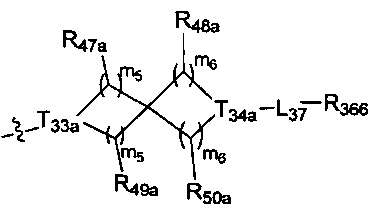 , где
, где
[163] каждый из T33a, Т34а независимо выбран из N, СН или С(R4001);
[164] L37 независимо выбран из одинарной связи, С1-3алкила, О, S, С(=O), С=S, S(=O) и/или S(=O)2;
[165] каждый из R366, R47a, R48a, R49a, R50a независимо выбран из Н, необязательно R4001-замещенного С1-3алкила, или алкокси, или алкилацила, или алкилсульфонила, или алкиламино, или циклопропила, необязательно R4001-замещенного циклопропилацила или циклопропилсульфонила;
[166] R366 также может быть выбран из группы, включающей фенил, пиридил, имидазолил, тиенил, фурил, оксазолил, тиазолил и изотиазолил, которые необязательно замещены R4001;
[167] R4001 выбран из ОН, N(СН3)2, NH(СН3), NH2, С(СН3)2(ОН), F, Cl, Br, I, CN, метила, метокси, число R4001 выбрано из 1, 2 или 3;
[168] предпочтительно L37 выбран из одинарной связи, С(=O), СН2.
[169] Предпочтительно В выбран из  ,
, 
[170] Предпочтительным является соединение или его фармацевтически приемлемая соль, где В выбран из группы, включающей

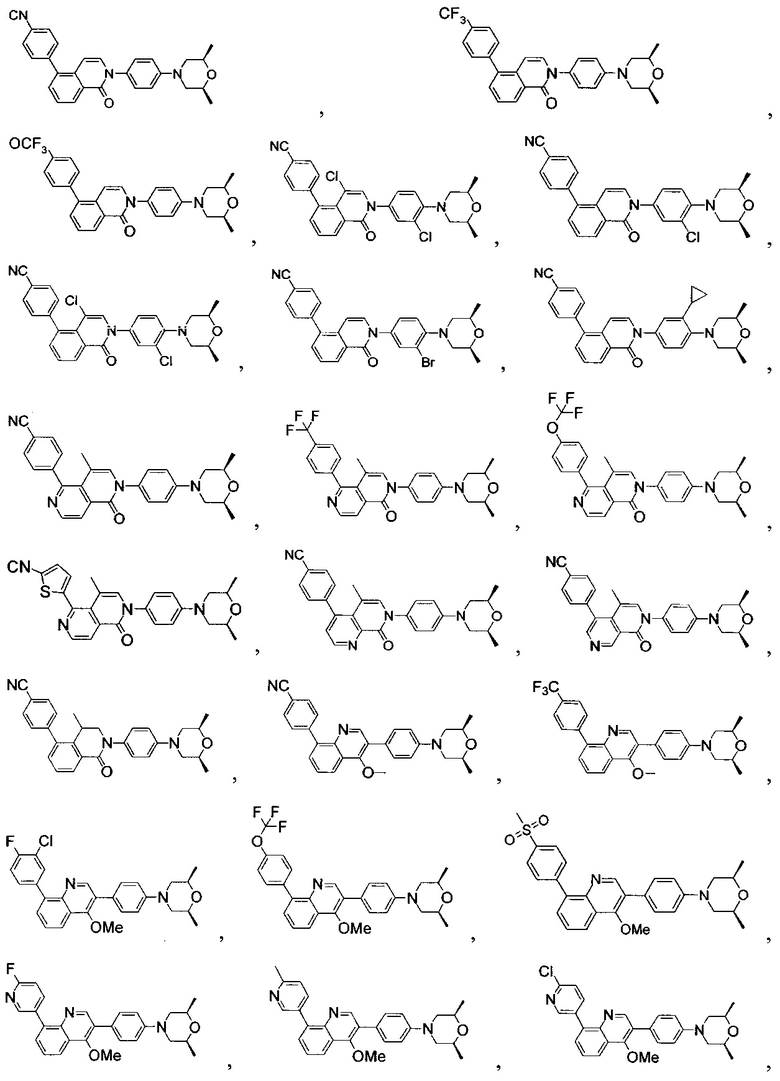
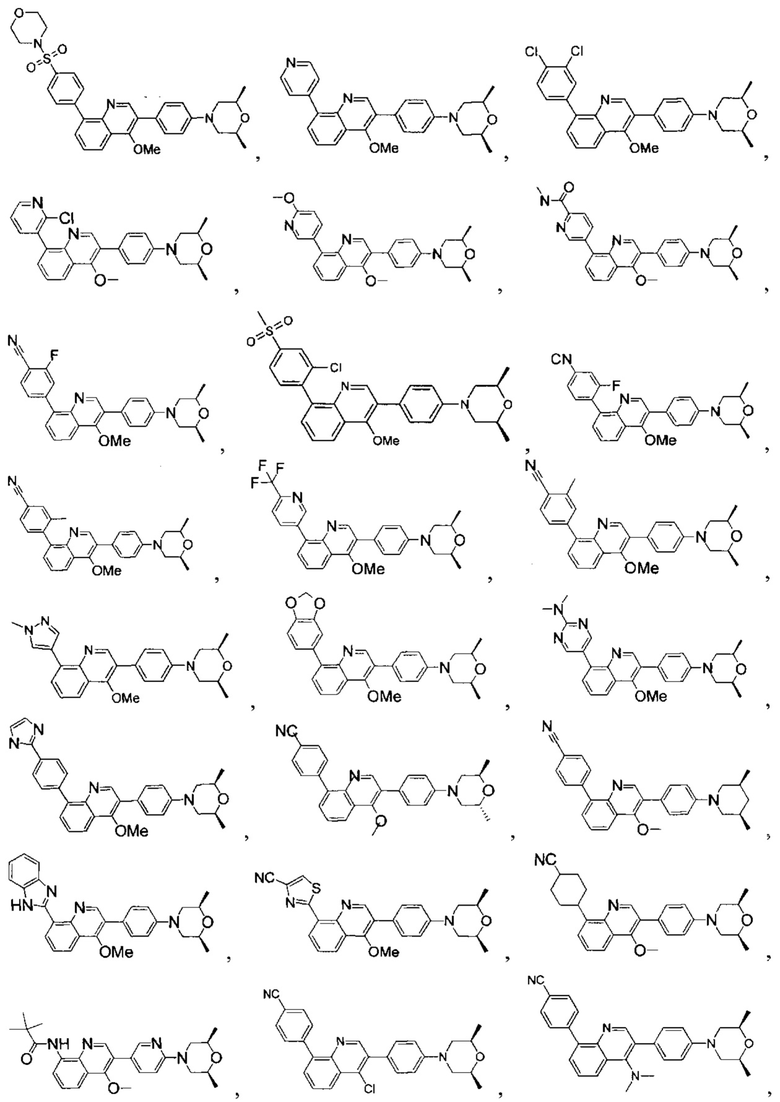
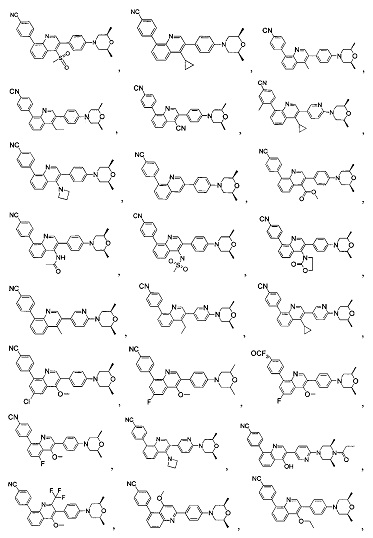
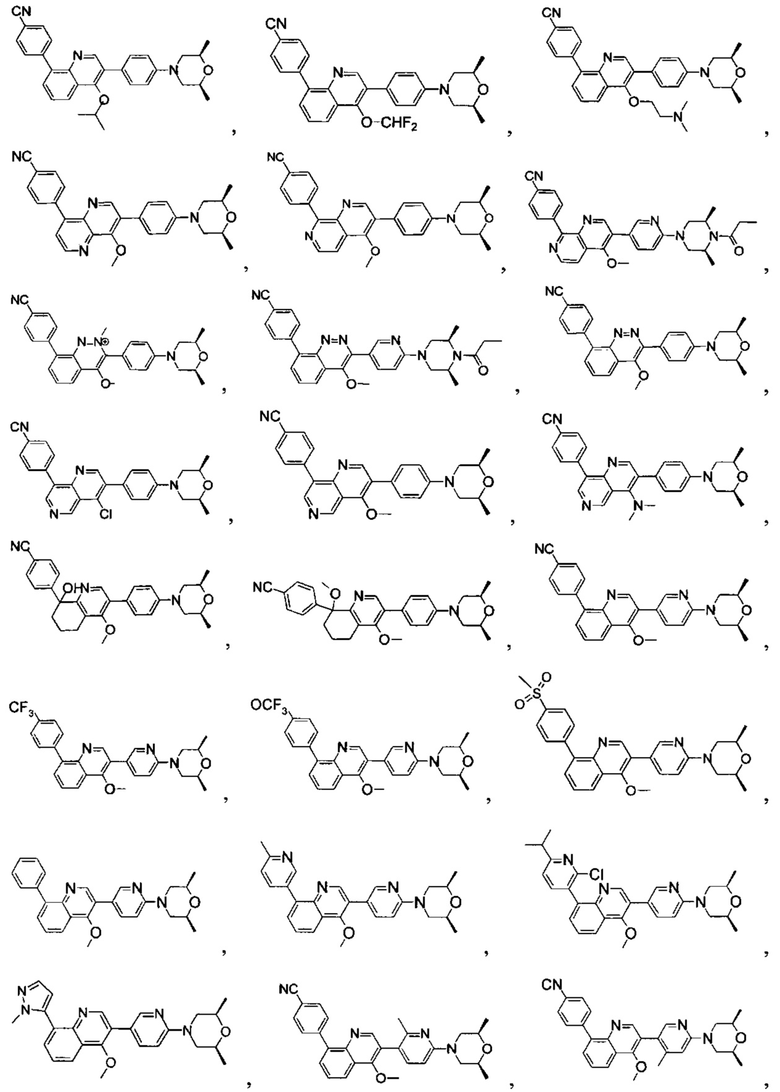
[171] Предпочтительно соединение или его фармацевтически приемлемая соль выбраны из группы, включающей


[172] Термин «фармацевтически приемлемая соль» относится к соли соединения согласно настоящему изобретению, которую получают с помощью соединения с конкретным заместителем, раскрытым в настоящем изобретении, и относительно нетоксичной кислоты или щелочи. Если соединение согласно настоящему изобретению содержит относительно кислотную функциональную группу, соль присоединения щелочи можно получить путем приведения соединения в нейтральной форме в контакт с достаточным количеством щелочи в чистом растворе или подходящем инертном растворе. Фармацевтически приемлемая соль присоединения щелочи включает соль натрия, калия, кальция, аммония, органического аммония или магния или т.п. Если соединение согласно настоящему изобретению содержит относительно щелочную функциональную группу, соль присоединения кислоты можно получить путем приведения соединения в нейтральной форме в контакт с достаточным количеством кислоты в чистом растворе или подходящем инертном растворителе. Примеры фармацевтически приемлемой соли присоединения кислоты включают соль неорганической кислоты, причем неорганическая кислота включает кислоты, такие как хлористоводородная кислота, бромистоводородная кислота, азотная кислота, угольная кислота, бикарбонат, фосфорная кислота, гидрофосфат, дигидрофосфат, серная кислота, гидросульфат, йодистоводородная кислота, фосфористая кислота и т.д; и соль органической кислоты, причем органическая кислота включает кислоты, такие как уксусная кислота, пропионовая кислота, изомасляная кислота, малеиновая кислота, малоновая кислота, бензойная кислота, янтарная кислота, пробковая кислота, фумаровая кислота, молочная кислота, миндальная кислота, фталевая кислота, фенилсульфоновая кислота, n-толуолсульфоновая кислота, лимонная кислота, винная кислота, метилсульфоновая кислота и т.п; и также включают соль аминокислоты (например, аргинина и т.д.), и соль органической кислоты, такой как глюкуроновая кислота и т.п. (см. Berge et al., "Pharmaceutical Salts", Journal of Pharmaceutical Science 66: 1-19 (1977)). Некоторые конкретные соединения согласно настоящему изобретению содержат как щелочную, так и кислотную функциональные группы, с возможностью превращения в любую соль присоединения щелочи или присоединения кислоты.
[173] Предпочтительно нейтральную форму соединения регенерируют путем приведения соли в контакт с основанием или кислотой традиционным способом, а затем отделения исходного соединения. Разница между исходной формой соединения и различными солевыми формами заключена в некоторых физических свойствах, как, например, растворимость в полярном растворителе является отличающейся.
[174] «Фармацевтически приемлемая соль» в настоящем изобретении представляет собой производные соединения согласно настоящему изобретению, где исходное соединение модифицировано путем образования соли с помощью кислоты или щелочи. Примеры фармацевтически приемлемой соли включают, но без ограничения, соль неорганической кислоты или органической кислоты и щелочи, такой как амин, щелочной металл, или органическую соль кислотного радикала, такого как карбоновой кислоты и т.п. Фармацевтически приемлемая соль включает традиционные нетоксичные соли или соли четвертичного аммония исходного соединения, такие как соль, образованная с помощью нетоксичной неорганической кислоты или органической кислоты. Традиционная нетоксичная соль включает, но без ограничения, те соли, полученные из неорганических кислот и органических кислот, причем неорганические кислоты или органические кислоты выбраны из 2-ацетоксибензойной кислоты, 2-изэтионовой кислоты, уксусной кислоты, аскорбиновой кислоты, фенилсульфоновой кислоты, бензойной кислоты, бикарбоната, угольной кислоты, лимонной кислоты, этилендиаминтетрауксусной кислоты, этандисульфоновой кислоты, этансульфоновой кислоты, фумаровой кислоты, глюкогептозы, глюконовой кислоты, глутаминовой кислоты, гликолевой кислоты, бромистоводородной кислоты, хлористоводородной кислоты, гидройодата, гидроксила, гидроксинафтойной кислоты, изэтионовой кислоты, молочной кислоты, лактозы, додекансульфоновой кислоты, малеиновой кислоты, яблочной кислоты, миндальной кислоты, метансульфоновой кислоты, азотной кислоты, щавелевой кислоты, памоевой кислоты, пантотеновой кислоты, фенилуксусной кислоты, фосфорной кислоты, полигалактуронана, пропионовой кислоты, салициловой кислоты, стеариновой кислоты, фолиновой кислоты, янтарной кислоты, аминосульфоновой кислоты, сульфаниловой кислоты, серной кислоты, дубильной кислоты, винной кислоты и n-толуолсульфоновой кислоты.
[175] Фармацевтически приемлемая соль согласно настоящему изобретению может быть получена посредством традиционного способа с помощью исходного соединения, содержащего кислотную или щелочную группу. В общем, способ получения соли включает получение в воде или органическом растворителе или смеси воды и органического растворителя, приведение данных соединений в форме свободных кислот или щелочей в реакцию со стехиометрическим количеством подходящих щелочей или кислот. В общем, предпочтительно выбирать неводные среды, такие как простой эфир, этилацетат, этанол, изопропанол или ацетонитрил и т.п.
[176] За исключением формы соли в настоящем изобретении представлена форма пролекарства для соединения. Пролекарство соединения, описанного в настоящем изобретении, легко превращают в соединение согласно настоящему изобретению посредством химических изменений в физиологических условиях. Кроме того, пролекарство можно превращать в соединение согласно настоящему изобретению посредством химического или биохимического способа в окружающей среде in vivo.
[177] Некоторые соединения согласно настоящему изобретению могут существовать в виде несольватной или сольватной форм, в том числе гидратных форм. В общем, сольватная форма подобна несольватной форме, обе из которых включены в объем настоящего изобретения. Некоторые соединения согласно настоящему изобретению могут существовать в поликристаллической или аморфной форме.
[178] Некоторые соединения согласно настоящему изобретению могут содержать асимметрические атомы углерода (оптический центр) или двойные связи. Рацемические изомеры, диастереомеры, геометрические изомеры и одинарные изомеры включены в объем настоящего изобретения.
[179] Если не указано иное, термин «замещенный» относится к одному или более атомов водорода в конкретном атоме, необязательно замещенном заместителем, в том числе дейтерием и вариантом водорода, при условии, что валентное состояние конкретного атома является нормальным и соединение, полученное после замещения, является стабильным. Если заместитель представляет собой кетонную группу (т.е.=O), это означает, что два атома водорода являются замещенными. Замещение кетонной группы не происходит в ариле. Термин «необязательно замещенный» означает, что он может быть замещенным или не быть замещенным, если не указано иное, тип и число заместителей может быть произвольным при допущении стабильности, доступной в химической структуре.
[180] Если не указано иное, когда любой параметр (например, R) демонстрирует встречаемость более одного раза в композиции или структуре соединения, определение каждой встречаемости является независимым. Следовательно, например, если группа является замещенной 0~2 R, группа может быть необязательно замещенной не более чем двумя R, и R имеет независимый вариант в каждом случае. Кроме того, комбинация заместителей и/или их вариантов возможна, только если такая комбинация приведет к стабильному соединению.
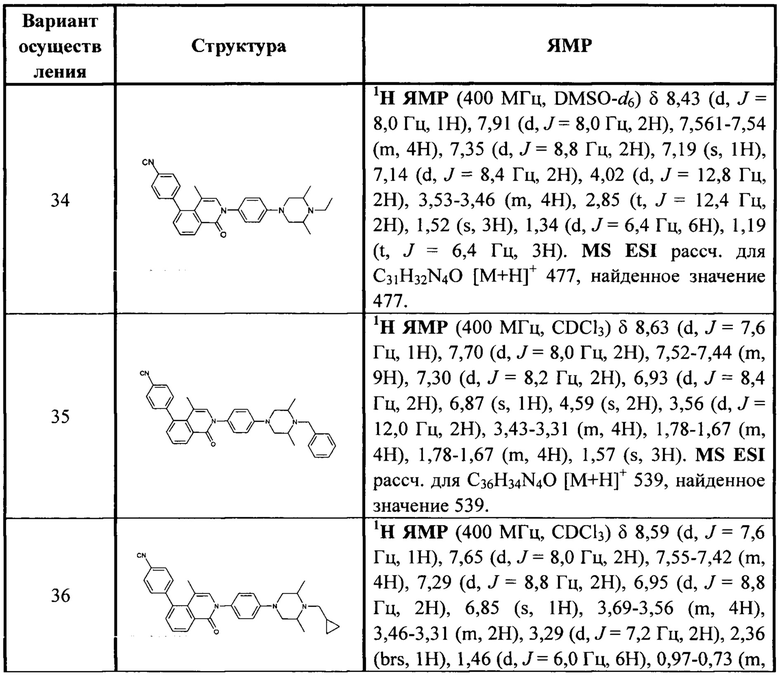
[181] Если один параметр в нем выбран из одинарной связи, это означает, что для двух групп параметр обеспечивает непосредственное соединение друг с другом, например, если L в A-L-Z относится к одинарной связи, это означает, что структура фактически представляет собой A-Z.
[182] Если не указано иное, когда связи заместителя могут быть поперечно соединены с двумя атомами кольца, заместитель может быть связан с произвольными атомами в кольце. Если не указано, посредством какого атома, содержащегося в перечисленном заместителе, он соединен с общей структурной формулой, включая соединение, которое конкретно не упомянуто, заместитель может быть связан посредством любого из его атомов. Комбинация заместителей и/или их вариантов возможна, только если такая комбинация приведет к стабильному соединению.
[183] Если не указано иное, термин «углеводородная группа» или его конкретное представление (такое как алкил, алкенил, алкинил, фенил и т.д.) само по себе или как часть другого заместителя представляет собой линейную, разветвленную или циклическую углеводородную группы или их комбинацию, которая может быть полностью насыщенной, моноциклической или полициклической ненасыщенной, может быть монозамещенной, дизамещенной или полизамещенной, может быть одновалентной (такой как метил), бивалентной (такой как метилен) или многовалентной (такой как метенил), может включать бивалентные или многовалентные атомные группы с конкретным числом атомов углерода (как, например, C1-С10 относится к группе, имеющей 1~10 атомов углерода). Термин «алкил» включает алифатическую углеводородную группу и ароматическую углеводородную группу, причем алифатическая углеводородная группа включает линейные и циклические структуры, конкретно включает, но без ограничения, алкил, алкенил и алкинил, ароматическая углеводородная группа включает, но без ограничения, 6-12-членную ароматическую углеводородную группу, такую как бензол, нафталин и т.п. В некоторых вариантах осуществления термин «алкил» относится к линейной или разветвленной или циклической группам или их комбинации, которые могут быть полностью насыщенными, моноциклическими или полициклическими ненасыщенными, могут включать дивалентные и поливалентные группы. Примеры насыщенных углеводородных групп включают, но без ограничения, гомологи или изомеры метила, этила, н-пропила, изопропила, н-бутила, трет-бутила, изобутила, втор-бутила, изобутила, циклогексила, (циклогексил)метила, циклопропилметила, н-амила, н-гексила, н-гептила, н-октила и т.п. Ненасыщенный алкил имеет одинарную, или двойную, или тройную связь, примеры которого включают, но без ограничения, винил, 2-пропенил, бутенил, кротил, 2-изопентенил, 2-бутадиенил, 2,4-(пентадиенил), 3-(1,4-пентадиенил), ацетенил, 1- и 3-пропинил, 3-бутинил и более сложные гомологи и изомеры.
[184] Если не указано иное, термины «гетероуглеводородная группа», «гетероциклическая группа», «группа углеводород-гетероатом», «группа цикл-гетероатом», «группа гетероуглеводород-гетероатом», «группа гетероцикл-гетероатом» означают, что гетероатом или группа гетероатома содержатся в конкретной группе, гетероатом или группа гетероатома включают, но без ограничения, N, NH, замещенный или защищенный NH, О, S, S(=O), S(=O)2, С(=O), С(=O)O, для кольцевой системы гетероатом или группа гетероатома могут быть размещены во внутреннем положении или внешнем положении кольцевой системы (например, циклопропилсульфонила, циклопропилацила), где «гетероуглеводородная группа», «гетероциклическая группа» присоединены к остальной части молекулы посредством атома углерода, то есть гетероатом может быть размещен в любом положении группы (исключая положение, в котором углеводородная группа прикреплена к остальной части молекулы); «группа углеводород-гетероатом», «группа цикл-гетероатом» присоединены к остальной части молекулы посредством гетероатома, то есть гетероатом размещен в положении, в котором группа прикреплена к остальной части молекулы); «группа гетероуглеводород-гетероатом», «группа гетероцикл-гетероатом» присоединены к остальной части молекулы посредством гетероатома, где гетероатом может быть размещен в любом положении группы (включая положение, в котором углеводородная группа прикреплена к остальной части молекулы).
[185] Если не указано иное, термин «гетероуглеводородная группа» или его конкретные представления (такие как гетероалкил, гетероалкенил, гетероалкинил, гетероарил и т.д.) сам по себе или термин, объединяющийся с другим термином, относится к стабильной, линейной, разветвленной или циклической углеводородной группам или их комбинациям, которые состоят из конкретного числа атомов углерода и по меньшей мере одного гетероатома. В некоторых вариантах осуществления термин «гетероуглеводородная группа» или его конкретные представления (такие как гетероалкил, гетероалкенил, гетероалкинил, гетероарил и т.д.) сам по себе или термин, объединяющийся с другим термином, относится к стабильной, линейной, разветвленной углеводородной группам или их комбинациям, которые состоят из конкретного числа атомов углерода и по меньшей мере одного гетероатома. В типичном варианте осуществления гетероатом выбран из группы, включающей В, О, N и S, в которой атомы азота и серы необязательно окислены, и атом азота необязательно кватернизован. Гетероатомы В, О, N и S могут быть размещены в любом внутреннем положении гетероуглеводородной группы (исключая положение, в котором углеводородная группа присоединена к остальной части молекулы). Примеры включают, но без ограничения, -СН2-СН2-О-СН3, -CH2-CH2-NH-CH3, -CH2-CH2-N(CH3)-CH3, -CH2-S-CH2-CH3, -СН2-СН2, -S(O)-CH3, -CH2-CH2-S(O)2-CH3, -CH=CH-O-CH3, -CH2-CH=N-OCH3 и -CH=CH-N(CH3)-CH3. He более чем два гетероатома являются смежными, как, например, -CH2-NH-OCH3.
[186] Если не указано иное, термины «алкокси», «алкиламино» и «алкилтио» (или «тиоалкокси») являются идиоматическими выражениями, которые относятся к алкильной группе, присоединенной к остальной части молекулы посредством атома кислорода, амино или серы, соответственно.
[187] Если не указано иное, термины «циклоуглеводородная группа», «гетероциклоуглеводородная группа», «группа циклоуглеводород-гетероатом» или их конкретные представления (такие как арил, гетероарил, группа арил-гетероатом, циклоалкил, гетероциклоалкил, группа циклоалкил-гетероатом, циклоалкенил, гетероциклоалкенил, группа циклоалкенил-гетероатом, циклоалкинил, гетероциклоалкинил, группа циклоалкинил-гетероатом и т.д.) сами по себе или термин, объединяющийся с другими терминами, соответственно, относятся к циклической «углеводородной группе», «гетероуглеводородной группе» или «группе углеводород-гетероатом». Примеры циклоалкила включают, но без ограничения, циклопентил, циклогексил, 1-циклогексенил, 3-циклогексенил, циклогептил и т.д. Неограниченные примеры гетероциклической группы включают 1-(1,2,5,6-тетрагидропиридинил), 1-пиперидил, 2-пиперидил, 3-пиперидил, 4-морфолинил, 3-морфолинил, тетрагидрофуран-2-ил, тетрагидрофуранилиндол-3-ил, тетрагидротиофен-2-ил, тетрагидротиофен-3-ил, 1-пиперазинил и 2-пиперазинил.
[188] Если не указано иное, термины «галогенированный» или «галоген» сами по себе или как часть другого заместителя относятся к атому фтора, хлора, брома или йода. Кроме того, термин «галогенированный алкил» предназначен для включения моногалогенированного алкила и полигалогенированного алкила. Например, термин «галогенированный (С1-С4)алкил» предназначен для включения, но без ограничения, трифторметила, 2,2,2-трифторэтила, 4-хлорбутила и 3-бромпропила и т.д.
[189] Если не указано иное, термин «арил» относится к полиненасыщенному ароматическому углеводородному заместителю, который может быть монозамещенным, дизамещенным или многозамещенным, он может быть моноциклическим или полициклическим (предпочтительно от 1 до 3 колец), они конденсированы вместе или соединены посредством ковалентной связи. Термин «гетероарил» относится к арилу (или кольцу), содержащему 1-4 гетероатома. В иллюстративном варианте осуществления гетероатом выбран из группы, включающей В, N, О и S, в которой атомы азота и серы необязательно окислены, и атом азота необязательно кватернизован. Гетероарильная группа может быть присоединена к остальной части молекулы посредством гетероатома. Неограниченные примеры арила или гетероарила включают фенил, 1-нафтил, 2-нафтил, 4-бифенил, 1-пирролил, 2-пирролил, 3-пирролил, 3-пиразолил, 2-имидазолил, 4-имидазолил, пиразинил, 2-оксазолил, 4-оксазолил, 2-фенил-4-оксазолил, 5-оксазолил, 3-изоксазолил, 4-изоксазолил, 5-изоксазолил, 2-тиазолил, 4-тиазолил, 5-тиазолил, 2-фуранил, 3-фуранил, 2-тиенил, 3-тиенил, 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидинил, 4-пиримидинил, 5-бензотиазолил, пуринил, 2-бензоимидазолил, 5-индолил, 1-изохинолил, 5-изохинолил, 2-хиноксалил, 5-хиноксалил, 3-хинолил и 6-хинолил. Любой из заместителей в арильной и гетероарильной кольцевой системе выбран из приемлемых заместителей, описанных ниже.
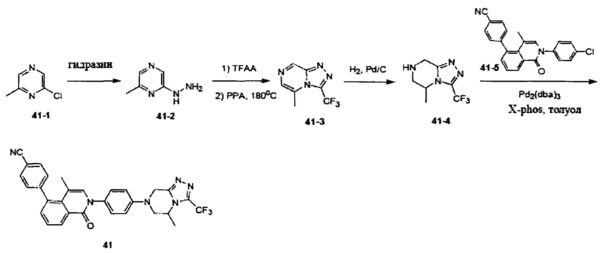
[190] Если не указано иное, в целях краткости при использовании в объединении с. другими терминами (например, арилокси, арилтио, аралкил), арил включает определение арильного и гетероарильного кольца, определенного выше. Следовательно, термин «аралкил» предназначен для включения групп, в которых арил присоединен к алкилу (например, бензил, фенилэтил, пиридилметил), включая те алкилы, где атомы углерода (такие как метилен) замещены такими атомами, как атомы кислорода, такими как феноксиметил, 2-пиридилоксиметил-3-(1-нафтокси)пропил и т.д.
[191] Если не указано иное, «кольцо» относится к замещенному или незамещенному циклоалкилу, замещенному или незамещенному гетероциклоалкилу, замещенному или незамещенному арилу или замещенному или незамещенному гетероарилу. Кольцо включает конденсированное кольцо. Число атомов в кольце обычно определено как член кольца, например, «5-7-членное кольцо» представляет собой кольцо с замкнутой структурой с 5-7 атомами. Если не указано иное, кольцо необязательно содержит 1-3 гетероатома. Следовательно, «5-7-членное кольцо» включает, например, фенилпиридин и пиперидинил; с другой стороны, термин «5-7-членное гетероциклоалкильное кольцо» включает пиридил и пиперидинил, но не включает фенил. Термин «кольцо» также включает кольцевую систему, содержащую по меньшей мере одно кольцо, где каждое кольцо независимо выбрано из вышеуказанного определения.
[192] Если не указано иное, термин «гетероатом» включает атомы, за исключением углерода (С) и водорода (Н), такие как включающие кислород (О), азот (N), серу (S), диоксид кремния (Si), германий (Ge), алюминий (Al), бор (В) и т.д.
[193] Если не указано иное, термин «уходящая группа» относится к функциональной группе или атому, которые могут быть замещены с помощью другой функциональной группы или атома посредством реакции замещения (например, реакции нуклеофильного замещения). Например, иллюстративные уходящие группы включают трифлат; хлор, бром, йод; сульфонат, как, например, мезилат, тозилат, п-бромбензолсульфонат, п-тозилат и т.д.; ацилокси, как, например, ацетокси, трифторацетокси и т.п.
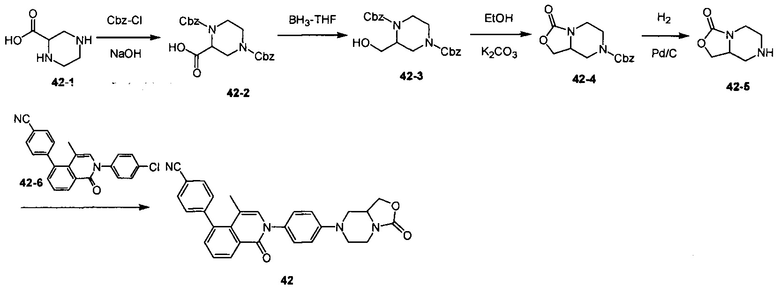
[194] Если не указано иное, термин «защитная группа» включает, но без ограничения, «защитную группу амино», «защитную группу гидроксила» или «защитную группу меркапто». Термин «защитная группа амино» относится к защитной группе, которая является подходящей для предупреждения побочных реакций, которые возникают при атоме азота аминогруппы. Иллюстративная защитная группа амино включает, но без ограничения, формил; ацил, такой как алканоил (такой как ацетил, трихлорацетил или трифторацетил); алкоксикарбонил, такой как трет-бутоксикарбонил (Boc); арилметоксикарбонил, такой как бензилоксикарбонил (Cbz) и 9-фторенилметоксикарбонил (Fmoc); арилметил, такой как бензил (Bn), трифенилметил (Tr), 1,1-бис-(4'-метоксифенил)метил; силил, такой как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS) и т.д. Термин «защитная группа гидроксила» относится к защитной группе, которая является подходящей для предупреждения побочных реакций гидроксильной группы. Иллюстративная защитная группа гидроксила включает, но без ограничения, алкил, такой как метил, этил и трет-бутил; ацил, такой как алканоил (такой как ацетил); арилметил, такой как бензил (Bn), п-метоксибензил (РМВ), 9-фторенилметил (Fm) и дифенилметил (дифенилметил, DPM); силил, такой как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS) и т.д.
[195] Если не указано иное, примеры галогенированного алкила включают, но без ограничения, трифторметил, трихлорметил, пентафторэтил и пентахлорэтил. Алкоксил представляет, что алкильная группа с конкретным числом атомов углерода присоединена с помощью кислородного мостика. С1-6алкоксил включает С1, С2, С3, С4, С5 и С6алкоксил. Примеры алкоксила включают, но без ограничения, метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси, трет-бутокси, н-пентилокси и 5-пентилокси. «Циклоалкил» включает насыщенную циклическую группу, такую как циклопропил, циклобутил или циклопентил. 3-7-членный циклоалкил включает С3, С4, С5, С6 и С7циклоалкил. «Алкенил» включает линейную или разветвленную углеводородную цепь, где любые стабильные участки на цепи характеризуются одной или более С-С двойными связями, такими как винил и пропенил.
[196] Если не указано иное, термин «галоген» или «галогенид» относится к фтору, хлору, брому и йоду.
[197] Если не указано иное, термины «гетероцикл» или «гетероциклическая группа» относятся к стабильному моноциклическому, бициклическому или бициклическому гетеро-кольцу, они могут быть насыщенными, частично ненасыщенными или ненасыщенными (ароматическими), они содержат атомы углерода и 1, 2, 3 или 4 гетероатома в кольце, которые независимо выбраны из группы, включающей N, О и S, где любой из гетероцикла может быть конденсирован до бензольного кольца с образованием бициклического кольца.
[198] Если не указано иное, примеры гетероциклического соединения включают, но без ограничения, акридинил, азоцинил, бензимидазолил, бензофуранил, бензомеркаптофуранил, бензомеркаптофенил, бензоксазолил, бензоксазолинил, бензотиазолил, бензотриазолил, бензотетразолил, бензоизоксазолил, бензоизотиазолил, бензоимидазолинил, карбазолил, 4aH-карбазолил, карболинил, хроманил, хромен, циннолинила декагидрохинолил, 2H,6H-1,5,2-дитиазинил, дигидрофуро[2,3-b]тетрагидрофуранил, фуранил, фуразанил, имидазолидинил, имидазолинил, имидазолил, 1H-индазолил, индоалкенил, индолинил, индолизинил, индолил, 3H-индолил, изатиногруппу, изобензофуранил, пиран, изоиндолил, изоиндолинил, изоиндолил, индолил, изохинолил, изотиазолил, изоксазолил, метилендиоксифенил, морфолинил, нафтиридинил, октагидроизохинолил, оксадиазолил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,5-оксадиазолил, 1,3,4-оксадиазолил, оксазолидинил, оксазолил, изооксазолил, гидроксилиндил, пиримидил, фенантридинил, фенантролинил, феназин, фенотиазин, бензопуринил, феноксазинил, фталазинил, пиперазинил, пиперидил, оксопиперидинил, 4-оксопиперидинил, пиперонил, птеридил, пуринил, пиранил, пиразинил, пиразолидинил, пиразолинил, пиразолил, пиридазинил, оксазолопиридин, пиридиноимидазол, пиридинотиазол, пиридил, пиримидил, пирролидинил, пирролинил, 2H-пирролил, пирролил, пиразолил, хиназолинил, хинолил, 4H-хинолизинил, хиноксалинил, хинуклидинил, тетрагидрофурил, тетрагидроизохинолинил, тетрагидрохинолинил, тетразолил, 6H-1,2,5-тиадиазинил, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, 1,2,5-тиадиазолил, 1,3,4-тиадиазолил, тиантренил, тиазил, изотиазолилтиенил, тиенил, тиофеноксазолил, тиофенотиазолил, тиофеноимидазолил, тиенил, триазинил, 1,2,3-триазолил, 1,2,4-триазолил, 1,2,5-триазолил, 1,3,4-триазолил и ксантенил. Конденсированные кольцевые и спирокольцевые соединения также включены. Если не указано иное, соединение согласно настоящему изобретению можно получить посредством многих способов синтеза, которые хорошо известны специалисту в данной области техники, включая конкретные варианты осуществления, перечисленные далее, и их комбинацию с другими химическими способами синтеза и эквивалентными альтернативными способами, которые известны специалисту в данной области техники, предпочтительные варианты осуществления включают, но без ограничения, варианты осуществления согласно настоящему изобретению.
[199] Если не указано иное, применяемые в настоящем изобретении растворители являются коммерчески доступными.
[200] Если не указано иное, в настоящем изобретении приняты следующие сокращения: водн. относится к воде; HATU представляет собой 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат; EDC представляет собой N-(3-диметиламинопропил)-N'-этилкарбодиимида гидрохлорид; m-СРВА представляет собой м-хлорпербензойную кислоту; экв. представляет собой эквивалентный, эквивалентно-количественный; CDI представляет собой карбонилдиимидазол; DCM представляет собой дихлорметан; РЕ представляет собой петролейный эфир; DIAD представляет собой диизопропилазодикарбоксилат; DMF представляет собой N,N-диметилформамид; DMSO представляет собой диметилсульфоксид; EtOAc представляет собой этилацетат; EtOH представляет собой этанол; МеОН представляет собой метанол; Cbz представляет собой бензилоксикарбонил, защитную группу амино; Boc представляет собой трет-бутоксикарбонил, защитную группу амина; НОАс представляет собой уксусную кислоту; NaCNBH3 представляет собой цианоборогидрид натрия; к.т. представляет собой комнатную температуру; О/N представляет собой в течение ночи; THF представляет собой тетрагидрофуран; Boc2O представляет собой ди-трет-бутилдикарбонат; TFA представляет собой трифторуксусную кислоту; DIPEA представляет собой диизопропилэтиламин; SOCl2 представляет собой тионилхлорид; CS2 представляет собой сероуглерод; TsOH представляет собой п-толуолсульфоновую кислоту; NFSI представляет собой N-фторбензолсульфонимид; NCS представляет собой N-хлорсукцинимид; п-Bu4NF представляет собой фторид тетрабутиламмония; iPrOH представляет собой 2-пропанол; т.пл. представляет собой температуру плавления.
[201] Если не указано иное, соединения названы вручную или с помощью программного обеспечения ChemDraw®, коммерчески доступные соединения названы в соответствии с каталогом поставщиков.
[202] В сравнении с предыдущим уровнем техники соединения согласно настоящему изобретению являются эффективными, имеют низкую токсичность и добиваются огромных и даже невероятных результатов относительно аспектов активности, периода полувыведения, растворимости, фармакокинетики и т.д., которые являются более подходящими для фармацевтической промышленности.
Подробное описание предпочтительного варианта осуществления
[203] Следующие примеры дополнительно иллюстрируют настоящее изобретение, но это не означает какое-либо неблагоприятное ограничение настоящего изобретения. Настоящее изобретение уже было подробно описано в данной заявке, в которой также были раскрыты варианты осуществления, для специалиста в данной области техники очевидна возможность изменения и улучшения вариантов осуществления настоящего изобретения без отступления от сущности и объема настоящего изобретения.
Вариант осуществления 1
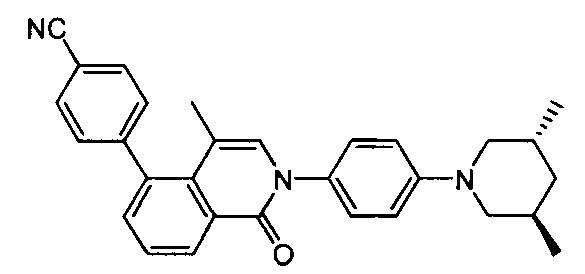
4-(2-(4-((3R,5R)-3,5-диметилпиперидин-1-ил)фенил)-4-метил-1-оксо-1,2-дигидроизохинолин-5-ил)бензонитрил

[204] Стадия 1. Соединение 1-1 (60 г, 0,36 моль) растворяли в растворе ледяной воды (900 мл), ацетона (300 мл) и водного раствора HCl (180 мл, 2,23 моль), затем смесь медленно каплями добавляли в водный раствор (360 мл) нитрита натрия (50 г, 0,72 моль) при поддержании температуры при 0-10°C. После перемешивания в течение 2 ч. непосредственно добавляли твердое вещество йодида калия (120 г, 0,72 моль), и температуру поддерживали при 7-10°С в течение 30 мин. Реакционную смесь нагревали до 80-90°C до исчезновения пурпурного газа, а затем охлаждали до комнатной температуры. Реакционную смесь фильтровали с получением соединения 1-2 (85 г, выход 85%) в виде желтого твердого вещества. MS ESI рассч. для C8H7IO3 [М+Н]+ 279, найденное значение 279.
[205] Стадия 2. Соединение 1-2 (34,5 г, 0,12 моль) растворяли в DCM (300 мл), добавляли DMF (0,1 мл), а затем по каплям добавляли соединение 1-3 (12 мл, 0,135 моль), реакционную смесь перемешивали в течение 1 ч. с получением соединения 1-4 в виде желтого масла. Неочищенный продукт применяли непосредственно на следующей стадии. MS ESI рассч. для C8H6ClIO2 [М+Н]+ 297, найденное значение 297.
[206] Стадия 3. Соединение 1-5 (15 г, 0,12 моль) растворяли в DCM (150 мл), затем добавляли DIPEA (77 г, 0,6 моль), реакционную смесь перемешивали при комнатной температуре в течение 5 мин. Соединение 1-4 (46 г, 0,12 моль) растворяли в DCM (300 мл) и по каплям добавляли в реакционную смесь при 0°C.Через 3 ч. реакцию завершали, как обнаружено с помощью TLC (РЕ : EtOAc=3:1), реакционную смесь выливали в воду и экстрагировали с помощью DCM. Органическую фазу промывали солевыми растворами, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (РЕ : EtOAc=3:1) с получением целевого соединения 1-6 (38 г, выход 75%) в виде желтого твердого вещества. MS ESI рассч. для C13H9ClINO2 [М+Н]+ 374, найденное значение 374.
[207] Стадия 4. NaH (4,8 г, 0,2 моль) добавляли в раствор соединения 1-6 (38 г, 0,1 моль) в DMF (400 мл), в реакционную смесь добавляли соединение 1-7 (24 г, 0,2 моль) и перемешивали при 0°C в течение 2 ч. После завершения реакции, как обнаружено с помощью LC-MS, реакционную смесь медленно выливали в воду и экстрагировали с помощью EtOAc. Органическую фазу промывали солевыми растворами, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (РЕ : EtOAc=5:1) с получением целевого соединения 1-8 (29 г, выход 69%) в виде желтого масла. MS ESI рассч. для C17H15ClINO2 [М+Н]+ 428, найденное значение 428.
[208] Стадия 5. Соединение 1-8 (28 г, 66 ммоль), бромид тетрабутиламмония (53 г, 165 ммоль), KOAc (9,7 г, 0,1 моль) и Pd(OAc)2 (1,48 г, 6,6 ммоль) растворяли в DMF (250 мл), реакционную смесь перемешивали при 100°C в течение 16 ч. Реакцию завершали, как обнаружено с помощью TLC (РЕ : EtOAc=3:1). Реакционную смесь гасили с помощью воды и экстрагировали с помощью EtOAc. Органическую фазу промывали солевыми растворами, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт очищали посредством рекристаллизации с получением соединения 1-9 в виде белого твердого вещества (16 г, выход 81%). MS ESI рассч. для C17H14ClNO2 [М+Н]+ 300, найденное значение 300.
[209] Стадия 6. BBr3 (12,5 мл, 0,134 моль) стекал в раствор соединения 1-9 (16 г, 0,054 моль) в DCM (150 мл) при 0°C, реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакцию обнаруживали с помощью TLC (РЕ : EtOAc=3:1). Реакционную смесь гасили с помощью насыщенного раствора карбоната натрия и фильтровали с получением соединения 1-10 в виде белого твердого вещества (13 г, выход 84%). MS ESI рассч. для C16H12ClNO2 [М+Н]+ 286, найденное значение 286.
[210] Стадия 7. DIPEA (4,5 г, 35 ммоль) и соединение 1-11 (9,4 г, 26 ммоль) добавляли в раствор соединения 1-10 (5 г, 17,5 ммоль) в THF (50 мл), реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакцию обнаруживали с помощью TLC (РЕ : EtOAc=2:1). Реакционную смесь выливали в воду и экстрагировали с помощью EtOAc, органическую фазу промывали солевыми растворами, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (РЕ : EtOAc=5:1) с получением соединения 1-12 в виде желтого масла (3,6 г, выход 50%). MS ESI рассч. для C17H11ClF3NO4S [М+Н]+ 418, найденное значение 418.
[211] Стадия 8. Соединение 1-12 (3,6 г, 8,6 ммоль), соединение 1-13 (1,7 г, 12 ммоль), Pd(dppf)Cl2 (630 мг, 0,86 ммоль) и карбонат натрия (2,1 г, 19,8 ммоль) растворяли в диоксане/H2O (48 мл), реакционную смесь перемешивали при 110°C и нагревали с обратным холодильником в течение 16 ч. Реакцию обнаруживали с помощью TLC (РЕ : EtOAc=3:1). Реакционную смесь выливали в воду и фильтровали с собиранием остатка, неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (РЕ : EtOAc=5:1) с получением соединения 1-14 в виде белого твердого вещества (1,5 г, выход 50%). MS ESI рассч. для C23H15ClN2O [М+Н]+ 371, найденное значение 371.
[212] Стадия 9. В атмосфере газа азота соединение 1-14 (100 мг, 0,27 ммоль), соединение 1-15 (46 мг, 0,40 ммоль), Pd2(dba)3 (25 мг, 0,03 ммоль), Xantphos (26 мг, 0,06 ммоль) и трет-бутоксида калия (61 мг, 0,54 ммоль) растворяли в толуоле (10 мл), реакционную смесь нагревали с обратным холодильником при 120°C и перемешивали в течение 2 ч. Затем реакционную смесь фильтровали с помощью диатомита, концентрировали с помощью роторного испарителя для удаления растворителя, экстрагировали с помощью EtOAc (50 мл) и H2O (20 мл), промывали солевыми растворами и сушили над безводным сульфатом натрия. После концентрирования при пониженном давлении остаток очищали с помощью препаративной HPLC с получением титульного соединения в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,65 (d, J=7,2 Гц, 1Н), 7,69 (d, J=8,0 Гц, 2Н), 7,50-7,35 (m, 4Н), 7,23 (d, J=8,4 Гц, 2Н), 6,93 (t, J=8,8 Гц, 3Н), 3,20 (dd, J=3,2 Гц, J=12,0 Гц, 2Н), 2,85 (dd, J=2,4 Гц, J=11,6 Гц, 2Н), 2,02 (dd, J=5,6 Гц, J=9,2 Гц, 2Н), 1,56 (s, 3Н), 1,45 (t, J=6,0 Гц, 2Н), 1,01 (d, J=6,8 Гц, 6Н). MS ESI рассч. для C30H29N3O [М+Н]+ 448, найденное значение 448.
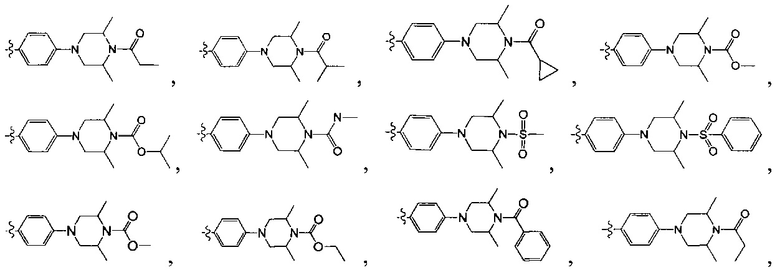
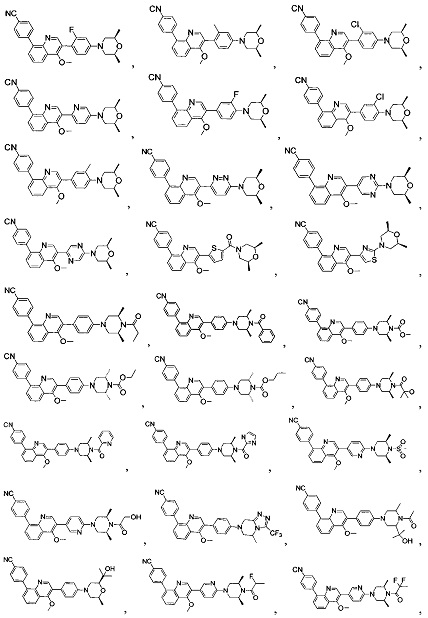
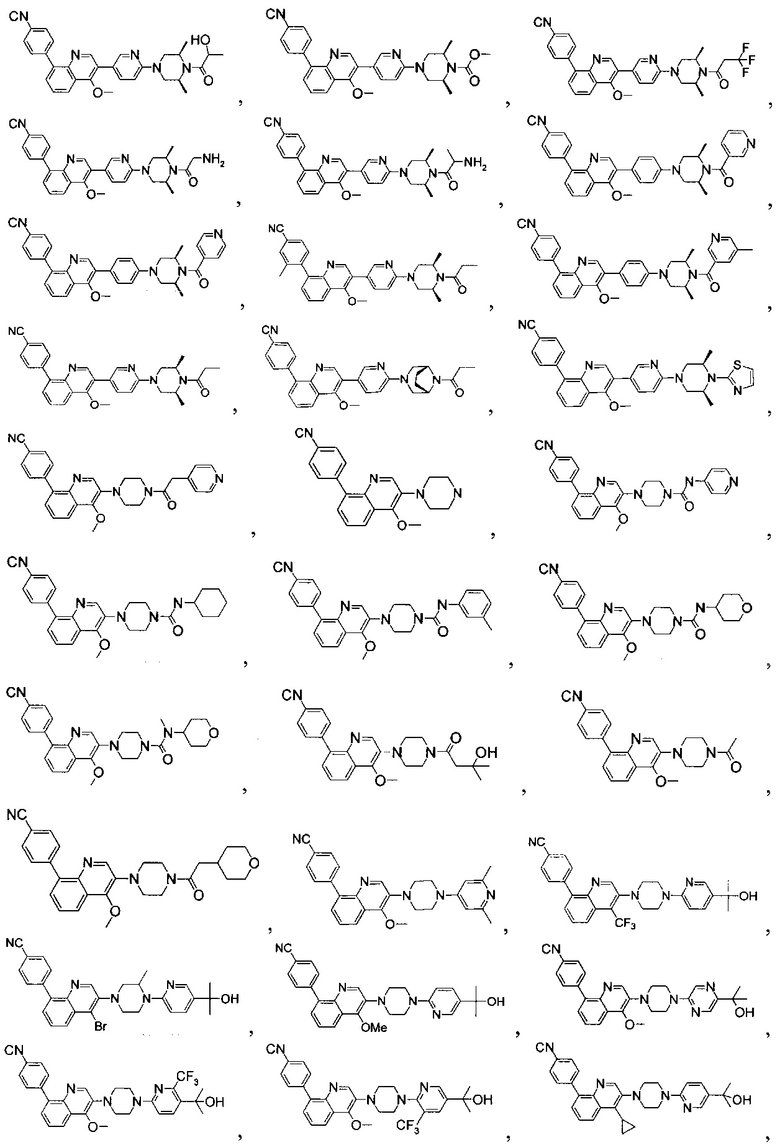
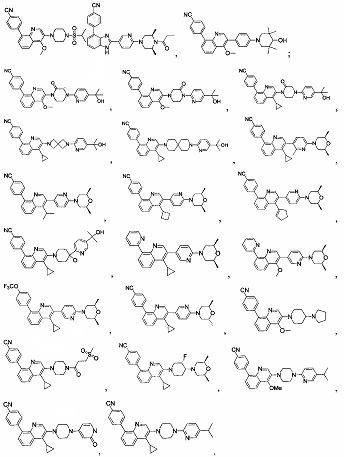
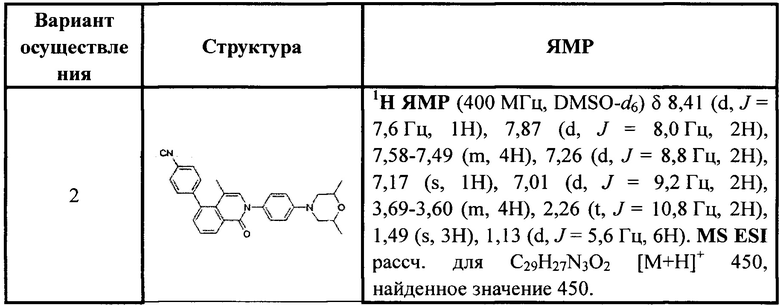
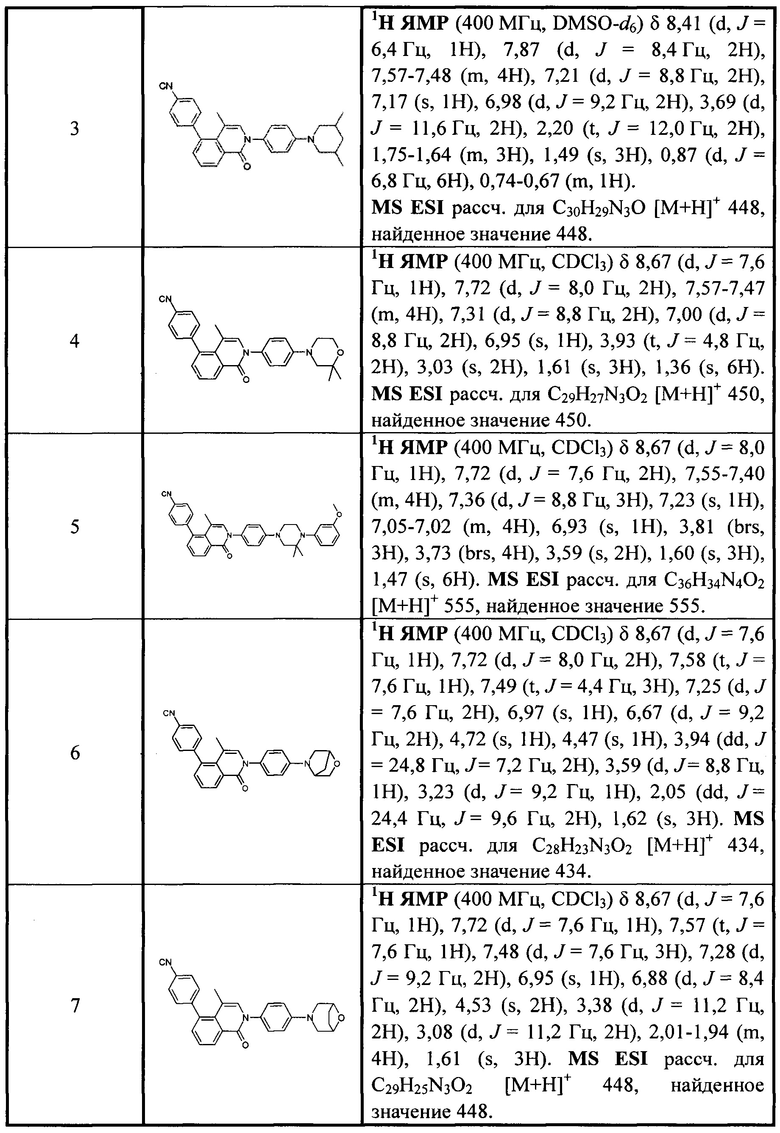
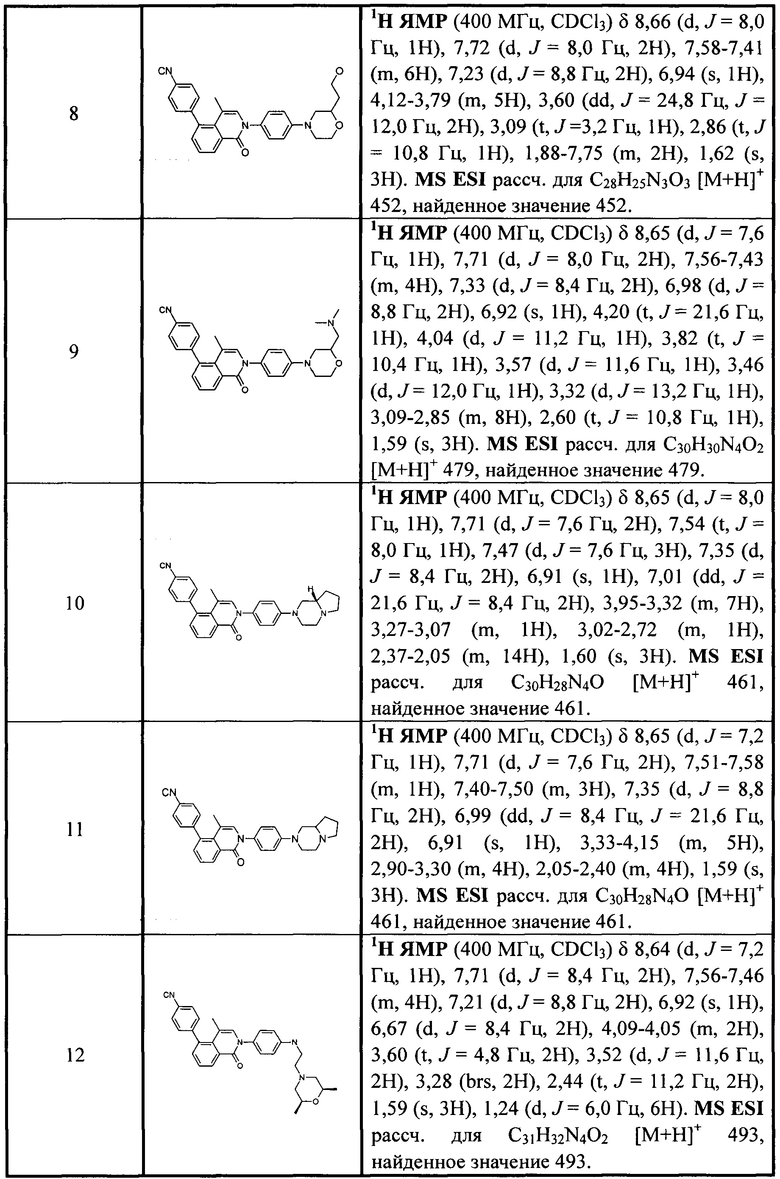
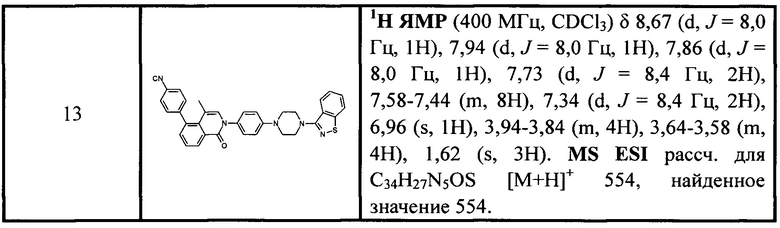
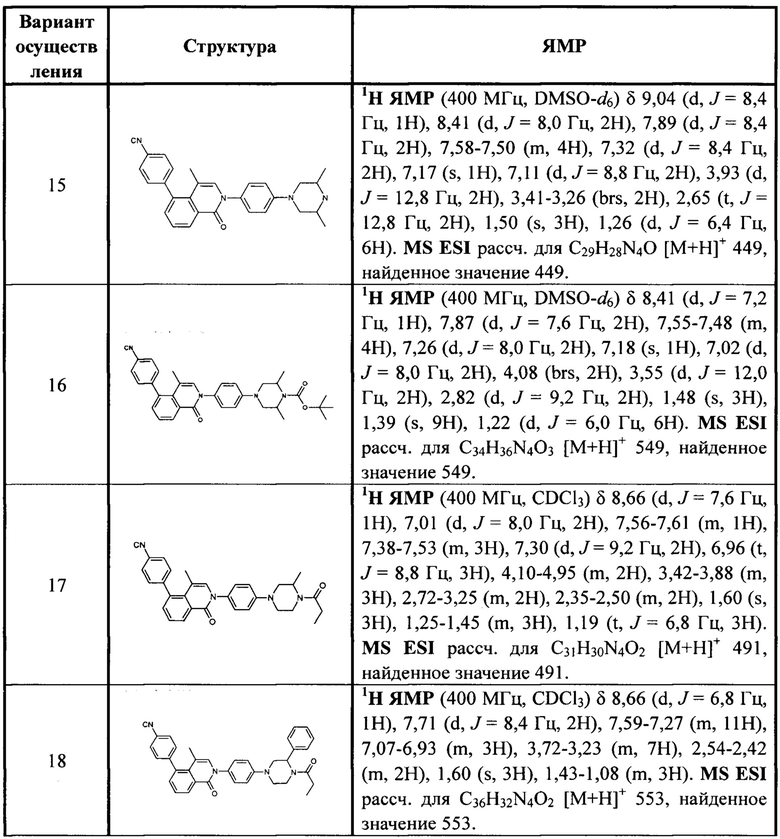
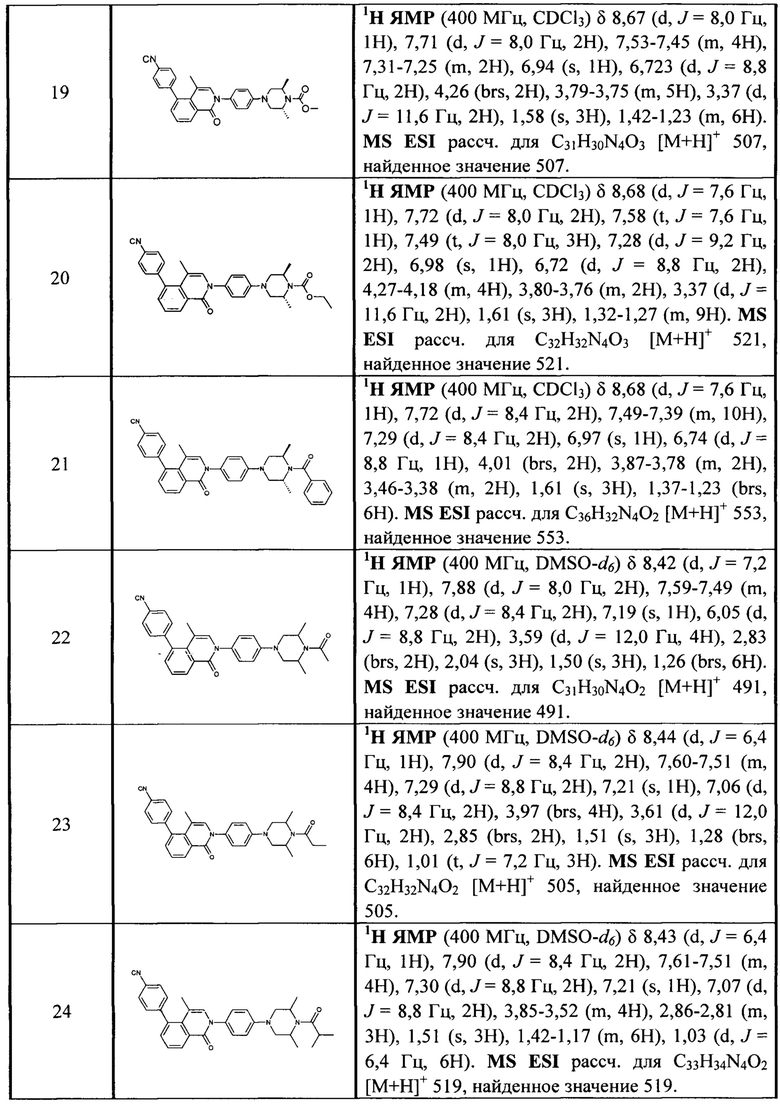
[213] Соединения, перечисленные в таблице 1, можно синтезировать с помощью соединения 1-14 и соответствующих аминов.
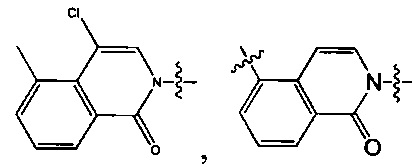
Вариант осуществления 14
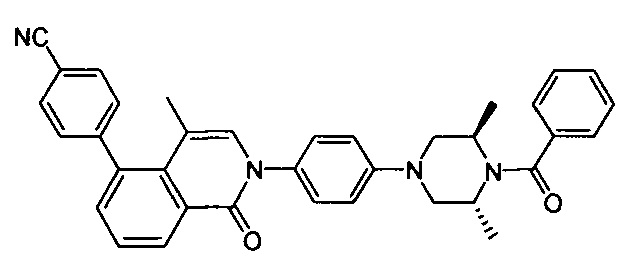
4-(2-(4-((3R,5R)-4-бензоил-3,5-диметилпиперазин-1-ил)фенил)-4-метил-1-оксо-1,2-дигидроизохинолин-5-ил)бензонитрил
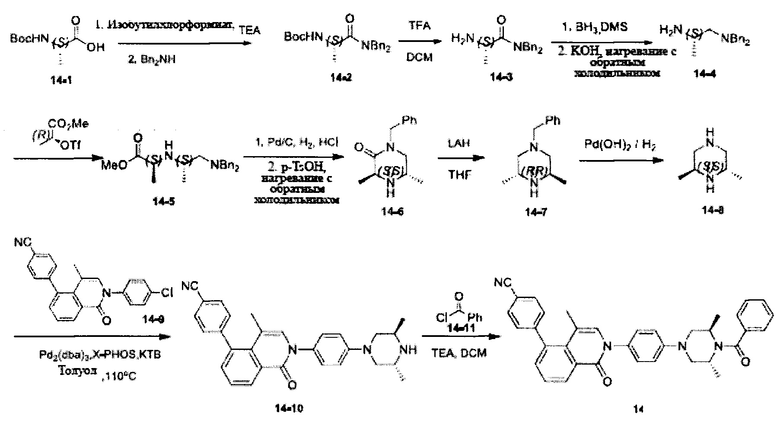
[214] Стадия 1. Соединение 14-1 (75 г, 0,4 моль) и TEA (60 мл, 0,45 моль) растворяли в THF (500 мл) и охлаждали до -30°C, по каплям добавляли раствор изобутилхлорформиата (54 мл, 0,42 моль) в THF (100 мл) и полученную в результате смесь перемешивали при -30°C в течение 0,5 ч., а затем нагревали до комнатной температуры, перемешивали в течение дополнительных 5 ч. Реакционную смесь снова охлаждали до 0°C, Bn2NH (88 мл, 0,43 моль) и TEA (70 мл, 500 ммоль) растворяли в THF (100 мл) и добавляли по каплям, реакционную смесь перемешивали при комнатной температуре в течение 10 ч. Реакцию обнаруживали с помощью LC-MS. Реакционную смесь выливали в воду и экстрагировали с помощью EtOAc. Органическую фазу промывали солевыми растворами, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением соединения 14-2 в виде белого твердого вещества (84 г, выход 60%). MS ESI рассч. для C22H28N2O3 [М+Н]+ 369, найденное значение 369.
[215] Стадия 2. TFA (120 мл) добавляли в раствор соединения 14-2 (53 г, 0,15 моль) в DCM (450 мл), реакционную смесь перемешивали при комнатной температуре в течение 5 ч. Реакцию обнаруживали с помощью TLC (РЕ : EtOAc=5:1). Реакционную смесь выливали в воду, добавляли бикарбонат натрия для регуляции рН до значения более 7 и экстрагировали с помощью DCM. Органическую фазу промывали солевыми растворами, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением соединения 14-3 в виде желтого масла (38 г, выход 98%). MS ESI рассч. для C17H20N2O [М+Н]+ 269, найденное значение 269.
[216] Стадия 3. Боран-метилсульфид (98 мл, 0,98 моль) добавляли в раствор соединения 14-3 (38 г, 0,14 моль) в THF (400 мл), реакционную смесь перемешивали при комнатной температуре в течение 48 ч. Реакцию обнаруживали с помощью LC-MS. Реакционную смесь гасили с помощью хлористоводородной кислоты и регулировали рН до значения более 7 с помощью водного раствора NaOH, а затем добавляли KOH (100 г), смесь нагревали до температуры флегмы в течение 24 ч. Реакционную смесь экстрагировали с помощью EtOAc, органическую фазу промывали солевыми растворами, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (DCM : МеОН=10:1) с получением соединения 14-4 в виде белого твердого вещества (25 г, выход 69%). MS ESI рассч. для C17H22N2 [М+Н]+ 255, найденное значение 255.
[217] Стадия 4. Раствор соединения 14-4 (25 г, 98,4 ммоль) и TEA (15,9 г, 157,44 ммоль) в DCM (150 мл) добавляли в раствор 2-трифторметансульфонилоксиметилпропионата (27,9 г, 118,1 ммоль) в DCM (100 мл) при 0°C, реакционную смесь перемешивали при 0°C в течение 2 ч. и при комнатной температуре в течение 2 ч. Реакцию обнаруживали с помощью LC-MS и выливали в DCM и NaHCO3. Реакционную смесь экстрагировали с помощью DCM, органическую фазу промывали насыщенными солевыми растворами, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (EtOAc) с получением соединения 14-5 в виде желтого масла (13,5 г, выход 40%). MS ESI рассч. для C21H28N2O2 [М+Н]+ 341, найденное значение 341.
[218] Стадия 5. Соединение 14-5 (12 г, 35,2 ммоль), хлористоводородную кислоту (36%) (5 мл) и Pd/C (2,5 г) растворяли в EtOH (100 мл), реакционную смесь подвергали реакции при 40 фунтов/кв. дюйм в течение 2 ч. Реакционную смесь фильтровали, фильтрат концентрировали. Остаток растворяли в EtOH (100 мл), добавляли п-толуолсульфоновую кислоту (2 г), реакционную смесь перемешивали при 90°C и нагревали с обратным холодильником в течение 16 ч. Реакцию обнаруживали с помощью LC-MS, реакционную смесь концентрировали при пониженном давлении, добавляли DCM и NaHCO3. Реакционную смесь экстрагировали с помощью DCM, органическую фазу промывали солевыми растворами, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением соединения 14-6 в виде желтого твердого вещества (6 г, выход 78%). MS ESI рассч. для C13H18N2O [М+Н]+ 219, найденное значение 219.
[219] Стадия 6. Соединение 14-6 (5,5 г, 25,2 ммоль) порциями добавляли в раствор LAH (2,9 г, 75,6 ммоль) в THF (50 мл), реакционную смесь перемешивали при комнатной температуре в течение 0,5 ч., затем нагревали до 70°C и перемешивали в течение 6 ч. Реакцию обнаруживали с помощью LC-MS. Реакционную систему гасили с помощью Н2О (2,9 мл), 15% NaOH (2,9 мл) и Н2О (8,8 мл). Реакционную смесь перемешивали в течение 0,5 ч., твердое вещество фильтровали и промывали с помощью THF. Фильтрат сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением соединения 14-7 в виде желтого твердого вещества (4,9 г, выход 96%). MS ESI рассч. для C13H20N2 [М+Н]+ 205, найденное значение 205.
[220] Стадия 7. Соединение 14-7 (2 г, 9,8 ммоль) и Pd(OH)2/C (0,9 г) растворяли в МеОН (20 мл), реакционную смесь подвергали реакции при 35 фунтов/кв. дюйм в течение 24 ч. Реакционную смесь фильтровали, фильтрат концентрировали при пониженном давлении с получением соединения 14-8 в виде бесцветного масла (1 г, выход 91%). MS ESI рассч. для C6H14N2 [М+Н]+ 115, найденное значение 115.
[221] Стадия 8. Соединение 14-10 получали в соответствии с вышеупомянутым способом в виде желтого твердого вещества (600 мг, выход 84%). MS ESI рассч. для C29H28N4O [М+Н]+ 449, найденное значение 449.
[222] Стадия 9. Соединение 14-11 (93 мг, 0,66 ммоль) добавляли в раствор соединения 14-10 (150 мг, 0,33 ммоль) и TEA (100 мг, 0,99 ммоль) в DCM (5 мл), реакционную смесь перемешивали при комнатной температуре в течение 5 ч. Реакцию обнаруживали с помощью LC-MS. Неочищенный продукт очищали с помощью препаративной HPLC с получением титульного соединения в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,68 (d, J=7,6 Гц, 1Н), 7,72 (d, J=7,2 Гц, 2H), 7,67-7,39 (m, 8H), 7,28 (t, J=8,0 Гц, 3Н), 6,96 (s, 1H), 6,74 (d, J=8,4 Гц, 2H), 4,25 (brs, 2H), 3,85 (d, J=10,4 Гц, 2H), 3,40 (d, J=11,2 Гц, 2H), 1,61 (s, 3H), 1,31 (brs, 6H). MS ESI рассч. для C36H32N4O2 [M+H]+ 553, найденное значение 553.
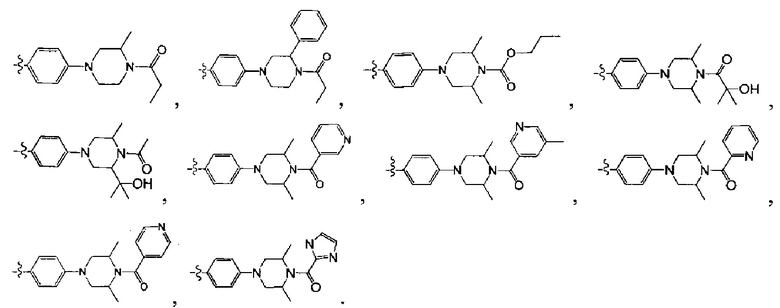
[223] Соединения, перечисленные в таблице 2, можно синтезировать с помощью соединения 14-10 и соответствующих ацилхлоридов и сульфонилхлоридов.
Вариант осуществления 33
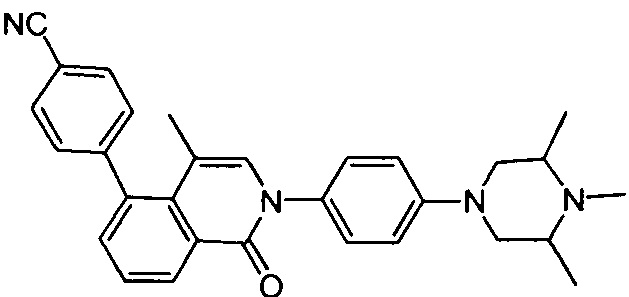
4-(4-метил-1-оксо-2-(4-(3,4,5-триметилпиперазин-1-ил)фенил)-1,2-дигидроизохинолин-5-ил)бензонитрил
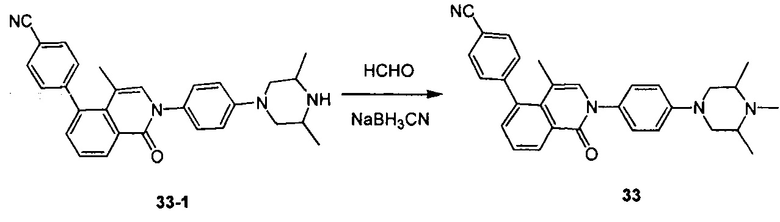
[224] Соединение 33-1 (200 мг, 0,45 ммоль), формальдегид (41 мг, 1,35 ммоль) и NaBH3CN (43 мг, 0,675 ммоль) растворяли в THF (5 мл), и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакцию обнаруживали с помощью LC-MS. Неочищенный продукт очищали с помощью препаративной HPLC с получением титульного соединения в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ 8,43 (d, J=8,0 Гц, 1Н), 7,91 (d, J=8,0 Гц, 2H), 7,61-7,54 (m, 4H), 7,35 (d, J=8,8 Гц, 2H), 7,19 (s, 1H), 7,14 (d, J=8,4 Гц, 2H), 4,01 (d, J=12,8 Гц, 2H), 3,35 (d, J=6,4 Гц, 2H), 2,85 (t, J=12,4 Гц, 2H), 1,52 (s, 3H), 1,34 (d, J=6,4 Гц, 6H), 1,19 (t, J=6,4 Гц, 3H). MS ESI рассч. для C30H30N4O [M+H]+ 463, найденное значение 463.
[225] Соединения, перечисленные в таблице 3, можно синтезировать с помощью соединения 33-1 и соответствующих альдегидов.

Вариант осуществления 39

4-(2-(4-(4-гидрокси-3,3,5,5-тетраметилпиперидин-1-ил)фенил)-4-метил-1-оксо-1,2-дигидроизохинолин-5-ил)бензонитрил
[226] Стадия 1. Соединение 39-1 (4 г, 20 ммоль) растворяли в THF (50 мл), охлаждали до 0°C, добавляли NaH (6,4 г, 160 ммоль), а затем реакционную смесь перемешивали при комнатной температуре в течение 0,5 ч., снова охлаждали до 0°C, по каплям добавляли йодметан (10 мл, 160 ммоль), затем нагревали до комнатной температуры и перемешивали в течение дополнительных 2 ч. Реакцию обнаруживали с помощью TLC, реакционную смесь гасили с помощью воды и экстрагировали с помощью EtOAc, органическую фазу промывали солевыми растворами, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением соединения 39-2 в виде желтого твердого вещества (4,5 г, выход 88%). MS ESI рассч. для C14H25NO3 [М+Н]+ 256, найденное значение 256.
[227] Стадия 2. Соединение 39-2 (410 мг, 1,6 ммоль) растворяли в растворе хлористоводородной кислоты/метанола (10 мл) и перемешивали при комнатной температуре в течение 2 ч. После завершения реакции смесь концентрировали при пониженном давлении с получением соединения 39-3 в виде желтого масла (330 мг, выход 86%). MS ESI рассч. для C9H17NO [М+Н]+ 156, найденное значение 156.
[228] Стадия 3. Соединение 39-5 получали в соответствии с вышеупомянутым способом в виде желтого твердого вещества (300 мг, выход 75%). MS ESI рассч. для C32H31N3O2 [М+Н]+ 490, найденное значение 490.
[229] Стадия 4. Соединение 39-5 (490 мг, 1 ммоль) растворяли в THF (10 мл), добавляли NaBH4 (57 мг, 1,5 ммоль), затем смесь перемешивали при комнатной температуре в течение 0,5 ч. Реакцию обнаруживали с помощью LC-MS, остаток выливали в H2O, экстрагировали с помощью DCM. Органическую фазу промывали солевыми растворами, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением неочищенного соединения. Неочищенный продукт очищали с помощью препаративной HPLC с получением титульного соединения в виде белого твердого вещества (64 мг, выход 13%). 1H ЯМР (400 МГц, CDCl3) δ 8,69 (d, J=7,2 Гц, 1H), 7,74 (d, J=7,6 Гц, 2Н), 7,60-7,45 (m, 4Н), 7,27 (d, J=6,4 Гц, 2Н), 7,00-6,90 (m, 3Н), 3,39 (d, J=12,0 Гц, 2Н), 3,12 (s, 1H), 2,60 (d, J=12,0 Гц, 2Н), 1,62 (s, 3Н), 1,09 (s, 6Н), 1,04 (s, 6Н). MS ESI рассч. для C32H33N3O2 [М+Н]+ 492, найденное значение 492.
Вариант осуществления 40
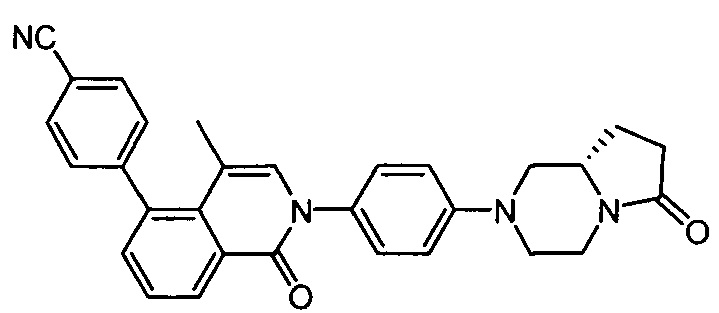
(S)-4-(4-метил-1-оксо-2-(4-(6-оксо-гексагидропирроло[1,2-а]пиразин-2(1Н)-ил)фенил)-1,2-дигидроизохинолин-5-ил)бензонитрил
[230] Стадия 1. Соединение 40-1 (10 г, 4,20 ммоль) и DIEA (10,8 г, 84 ммоль) растворяли в толуоле (200 мл) и охлаждали до 0°C, добавляли этил-2,3-дибромпропионат (13,1 г, 50,4 ммоль), а затем реакционную смесь перемешивали при 100°C в течение ночи. Реакцию обнаруживали с помощью TLC. Реакционную смесь гасили с помощью H2O, экстрагировали с помощью EtOAc. Органическую фазу промывали солевыми растворами, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении, остаток очищали с помощью колоночной хроматографии на силикагеле (РЕ/EtOAc=10/1) с получением соединения 40-2 в виде желтого масла (8,5 г, выход 61%). MS ESI рассч. для C21H26N2O2 [М+Н]+ 339, найденное значение 339.
[231] Стадия 2. В атмосфере газа азота DIBAL-H (45 мл, 45 ммоль) добавляли в раствор соединения 40-2 (8,4 г, 24,85 ммоль) в толуоле (100 мл) при -78°C и перемешивали при -78°C в течение 1 ч. После завершения реакции добавляли 20%-ный раствор гидроксида натрия (30,7 мл). Реакционную смесь нагревали до комнатной температуры и добавляли 20%-ный водный раствор гидроксида натрия (76,8 мл). Реакционный раствор экстрагировали с помощью EtOAc и органическую фазу промывали солевыми растворами, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением соединения 40-3 в виде желтого масла (7,4 г, неочищенное). MS ESI рассч. для C19H22N2O [М+Н]+ 295, найденное значение 295.
[232] Стадия 3. В атмосфере газа азота соединение 40-3 (7,3 г, 24,75 ммоль) растворяли в толуоле (100 мл), добавляли соединение 40-4 (17,2 г, 49,50 ммоль) и смесь перемешивали при 80°C в течение ночи. Реакцию обнаруживали с помощью LC-MS. Остаток выливали в H2O, экстрагировали с помощью EtOAc. Органическую фазу промывали солевыми растворами, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении, остаток очищали с помощью колоночной хроматографии на силикагеле (РЕ/EtOAc= от 10/1 до 4/1) с получением соединения 40-5 в виде желтого масла (5,6 г, выход 62%). MS ESI рассч. для C23H28N2O2 [М+Н]+ 365, найденное значение 365.
[233] Стадия 4. Pd/C (2,8 г, 10%) добавляли в раствор соединения 40-5 (5,6 г, 15,34 ммоль) в этаноле (100 мл), реакционную смесь нагревали до 70°C при 55 фунтов/кв. дюйм и перемешивали в течение ночи. Реакцию обнаруживали с помощью TLC и фильтровали с помощью диатомита, фильтрат концентрировали с получением соединения 40-6 в виде желтого масла (2 г, выход 71%). MS ESI рассч. для C7H12N2O [М+Н]+ 141, найденное значение 141.
[234] Стадия 5. Титульное соединение получали в соответствии с вышеупомянутым способом в виде белого твердого вещества (30 мг, выход 12%). 1H ЯМР (400 МГц, CDCl3) δ 8,66 (d, J=7,6 Гц, 1Н), 7,72 (d, J=8,0 Гц, 2Н), 7,42-7,58 (m, 4Н), 7,34 (d, J=8,8 Гц, 2Н), 7,04 (d, J=8,8 Гц, 2Н), 6,94 (s, 1Н), 4,13-4,18 (m, 1H), 3,76-3,90 (m, 2Н), 2,69 (d, J=12,0 Гц, 1H), 3,05-3,18 (m, 1Н), 2,75-2,85 (m, 1Н), 2,52-2,63 (m, 3Н), 2,25-2,35 (m, 1Н), 1,65-1,78 (m, 1Н), 1,61 (s, 3Н). MS ESI рассч. для C30H26N4O2 [М+Н]+ 475, найденное значение 475.
Вариант осуществления 41
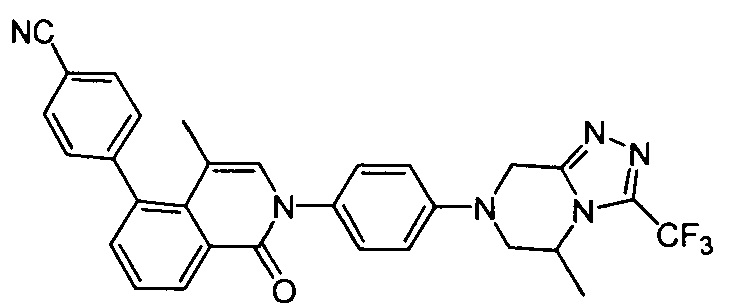
4-(4-метил-2-(4-(5-метил-3-(трифторметил)-5,6-дигидро-[1,2,4]триазоло[4,3-а]пиразин-7(8Н)-ил)фенил)-1-оксо-1,2-дигидроизохинолин-5-ил)бензонитрил
[235] Стадия 1. Соединение 41-1 (2,9 г, 22,75 ммоль) добавляли в гидрат гидразина (15 мл), реакционную смесь помещали в предварительно нагретую масляную баню (50°C), затем нагревали до 100°C свыше 30 мин. После охлаждения до комнатной температуры реакционную смесь охлаждали до 0°C в течение 1 ч. Остаток собирали путем фильтрования и сушили с получением соединения 41-2 в виде белого твердого вещества (1,6 г, выход 58%). MS ESI рассч. для C5H8N4 [М+Н]+ 125, найденное значение 125.
[236] Стадия 2. TFAA (35 мл) по каплям добавляли в соединение 41-2 (1,6 г, 13,2 ммоль) при 0°C, реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении с получением желтого твердого вещества. Затем добавляли РРА (40 мл). Реакционную смесь нагревали до 120°C и перемешивали в течение 18 ч. Горячий раствор РРА выливали в ледяную воду и нейтрализовали с помощью аммиака. Водную фазу экстрагировали с помощью EtOAc, органическую фазу промывали солевыми растворами, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (РЕ : EtOAc=1:1) с получением соединения 41-3 в виде желтого твердого вещества (2 г, выход 77%). MS ESI рассч. для C7H5F3N4 [М+Н]+ 203, найденное значение 203.
[237] Стадия 3. Соединение 41-3 (2 г, 10 ммоль) и Pd/C (1,1 г) растворяли в EtOH (30 мл) и THF (15 мл), в атмосфере газа водорода реакционную смесь перемешивали при комнатной температуре в течение 18 ч. Реакционную смесь фильтровали и фильтрат концентрировали с получением соединения 41-4 в виде желтого масла (1,9 г, выход 90%). MS ESI рассч. для C7H9F3N4 [М+Н]+ 207, найденное значение 207.
[238] Стадия 4. Титульное соединение получали в соответствии с вышеупомянутым способом в виде белого твердого вещества (25 мг, выход 6,8%). 1H ЯМР (400 МГц, CDCl3) δ 8,66 (d, J=7,6 Гц, 1H), 7,71 (d, J=8,4 Гц, 2Н), 7,54 (t, J=8,0 Гц, 1Н), 7,45 (dd, J=23,2 Гц, J=8,0 Гц, 5Н), 7,06 (d, J=8,8 Гц, 2Н), 6,92 (s, 1Н),, 4,91 (d, J=16,0 Гц, 1Н), 4,70 (brs, 1H), 4,44 (d, J=16,0 Гц, 1Н), 3,83 (d, J=13,2 Гц, 1Н), 3,53 (d, J=10,8 Гц, 1Н), 1,68 (d, J=6,0 Гц, 3Н), 1,59 (s, 3Н). MS ESI рассч. для C30H23F3N6O [М+Н]+ 541, найденное значение 541.
Вариант осуществления 42
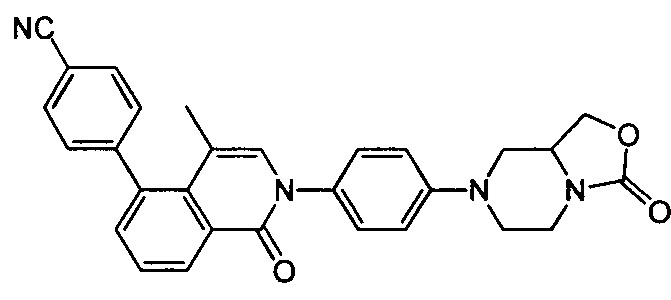
4-(4-метил-1-оксо-2-(4-(3-оксотетрагидро-1Н-оксазоло[3,4-а]пиразин-7(3Н)-ил)фенил)-1,2-дигидроизохинолин-5-ил)бензонитрил
[239] Стадия 1. Соединение 42-1 (17 г, 84 ммоль) растворяли в диоксане (340 мл) и Н2О (210 мл), раствор регулировали до рН>11 с помощью водного раствора гидроксида натрия (50%), добавляли CbzCl (24 мл, 168 ммоль), реакционную смесь перемешивали в течение 2 ч., а затем выливали в 1 л H2O. Водную фазу экстрагировали с помощью DCM, органическую фазу промывали солевыми растворами, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением соединения 42-2 в виде бесцветного клейкого масла (18 г, 51%). MS ESI рассч. для C21H22N2O6 [М+Н]+ 399, найденное значение 399.
[240] Стадия 2. Соединение 42-2 (14 г, 35 ммоль) растворяли в THF (150 мл), затем по каплям добавляли BH3-THF (70 мл, 70 ммоль), реакционную смесь перемешивали при 50°C в течение 3 ч. После завершения реакции, как обнаружено с помощью LC-MS, медленно добавляли МеОН для гашения реакции. В отсутствие высвобождения газа реакционную смесь нагревали до 50°C и перемешивали в течение 1 ч., а затем концентрировали при пониженном давлении с получением соединения 42-3 в виде бесцветного масла (4 г, 30%). MS ESI рассч. для C21H24N2O5 [М+Н]+ 385, найденное значение 385.
[241] Стадия 3. Соединение 42-3 (4 г, 10,4 ммоль) и карбонат калия (1,7 г, 12,5 ммоль) растворяли в EtOH (40 мл), реакционную смесь перемешивали при 70°C в течение ночи. После завершения реакции, как обнаружено с помощью LC-MS, смесь фильтровали с удалением оставшегося карбоната калия. Фильтрат концентрировали при пониженном давлении, очищали с помощью колоночной хроматографии на силикагеле (РЕ : EtOAc=1:1) с получением соединения 42-4 в виде желтого твердого вещества (2,2 г, 78%). MS ESI рассч. для C14H16N2O4 [М+Н]+ 277, найденное значение 277.
[242] Стадия-4. Pd/C (100 мг) добавляли в раствор соединения 42-4 (500 мг, 1,8 ммоль) в МеОН (50 мл), в атмосфере газа водорода (40 фунтов/кв. дюйм) реакционную смесь перемешивали при комнатной температуре в течение 12 ч. Pd/C удаляли путем фильтрования, фильтрат концентрировали при пониженном давлении с получением соединения 42-5 в виде желтого масла (240 мг, 95%). MS ESI рассч. для C6H10N2O2 [М+Н]+ 143, найденное значение 143.
[243] Стадия 5. Титульное соединение получали в соответствии с вышеупомянутым способом в виде белого твердого вещества (55 мг, выход 20%). 1Н ЯМР (400 МГц, CDCl3) δ 8,67 (d, J=6,8 Гц, 1Н), 7,72 (d, J=8,0 Гц, 2Н), 7,55 (t, J=8,0 Гц, 1H), 7,48 (d, J=7,6 Гц, 3Н), 7,35 (d, J=8,4 Гц, 2Н), 7,02 (d, J=8,4 Гц, 2Н), 6,92 (s, 1H), 4,51 (t, J=10,4 Гц, 1Н), 4,04 (d, J=5,6 Гц, 2Н), 3,96 (d, J=2,4 Гц, 1Н), 3,71 (d, J=12,0 Гц, 1Н), 3,61 (d, J=12,0 Гц, 1H), 3,37-3,22 (m, 1H), 2,94-2,78 (m, 1H), 2,76 (t, J=10,04 Гц, 1Н), 1,59 (s, 3Н). MS ESI рассч. для C29H24N4O3 [М+Н]+ 477, найденное значение 477.
Вариант осуществления 43
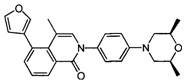
2-(4-((2S,6R)-2,6-диметилморфолино)фенил)-5-(фуран-3-ил)-4-метилизохинолин-1(2Н)-он
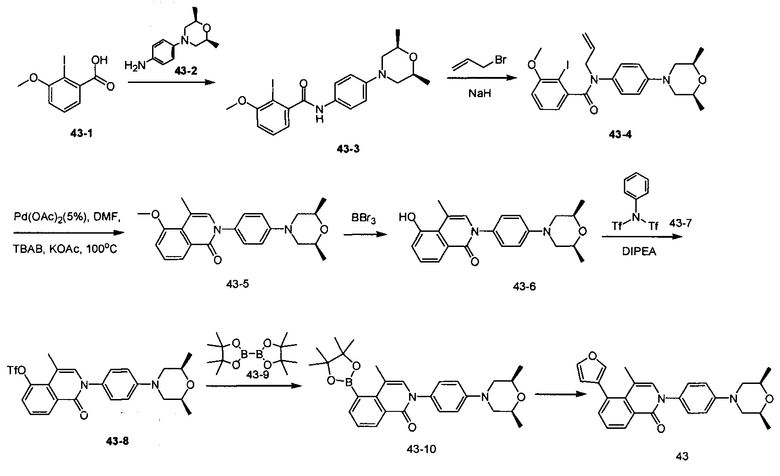
[244] Стадия 1. HATU (5,5 г, 40,86 ммоль) добавляли в раствор соединения 43-1 (9,7 г, 34,9 ммоль) в DMF (100 мл) при 0°C. После перемешивания при комнатной температуре в течение 30 мин. добавляли DIEA (18 г, 69,8 ммоль) и соединение 43-2 (7,2 г, 34,9 ммоль) при 0°C, а затем реакционную смесь подвергали реакции при комнатной температуре в течение ночи. Реакционную смесь выливали в H2O, экстрагировали с помощью EtOAc. Органическую фазу промывали с помощью H2O, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле с получением соединения 43-3 (13 г, выход 80%) в виде белого твердого вещества. MS (ESI) масса/заряд (М+Н)+ 467.
[245] Стадия 2. В атмосфере газа азота NaH (1,34 г, 33,5 ммоль), 3-бром-1-пропилен (6,8 г, 55,8 ммоль) добавляли в раствор соединения 43-3 (13 г, 27,9 ммоль) в DMF (100 мл) при 0°C, а затем реакционную смесь подвергали реакции при комнатной температуре в течение ночи. Реакционную смесь выливали в Н2О, экстрагировали с помощью EtOAc, органическую фазу промывали с помощью H2O, сушили над безводным сульфатом натрия, фильтровали и концентрировали, остаток очищали с помощью колоночной хроматографии на силикагеле с получением соединения 43-4 (12 г, выход 85,7%). MS (ESI) масса/заряд (M+H)+ 507.
[246] Стадия 3. Соединение 43-4 (11,1 г, 21,74 ммоль), ТВАВ (17,50 г, 54,35 ммоль), ацетат калия (3,2 г, 32,61 ммоль), ацетат палладия (487 мг, 2,17 ммоль) добавляли в DMF (700 мл), нагревали до 100°C и перемешивали в течение ночи. H2O добавляли в реакционную смесь, EtOAc использовали для экстракции, органическую фазу промывали с помощью H2O, сушили над безводным сульфатом натрия, фильтровали и концентрировали, остаток очищали с помощью колоночной хроматографии на силикагеле с получением соединения 43-5 (4,5 г, выход 50%). MS (ESI) масса/заряд (М+Н)+ 379.
[247] Стадия 4. Трибромид бора (12,6 г, 4,7 мл) добавляли в раствор соединения 43-5 (3,8 г, 10,05 ммоль) в DCM (40 мл) при 0°C, реакционную смесь перемешивали при комнатной температуре в течение ночи, затем гасили с помощью насыщенного раствора карбоната натрия, смесь фильтровали с получением соединения 43-6 (1,9 г, выход 53%). MS (ESI) масса/заряд (М+Н)+ 365.
[248] Стадия 5. DIPEA (1,1 г, 8,24 ммоль) и соединение 43-7 (2,2 г, 6,18 ммоль) добавляли в раствор соединения 43-6 (1,5 г, 4,12 ммоль) в DMF (60 мл), реакционную смесь подвергали реакции при комнатной температуре в течение ночи, а затем концентрировали при пониженном давлении с удалением растворителя. Остаток очищали с помощью колоночной хроматографии на силикагеле с получением соединения 43-8 (1,5 г, выход 75%). MS (ESI) масса/заряд (М+Н)+ 497.
[249] Стадия 6. Соединение 43-9 (508 мг, 2,0 ммоль), Pd(dppf)Cl2 (73 мг, 0,10 ммоль) и ацетат калия (196 мг, 2,0 ммоль) добавляли в раствор соединения 43-8 (496 мг, 1,0 ммоль) в 1,4-диоксане. В атмосфере газа азота реакционная смесь реагировала при 110°C в течение ночи, а затем растворитель выпаривали. Остаток очищали с помощью колоночной хроматографии на силикагеле с получением соединения 43-10 (270 мг, выход 60%). MS (ESI) масса/заряд (М+Н)+ 475.
[250] Стадия 7. 3-фуранбороновую кислоту (24 мг, 1,5 экв.), фосфат калия (59 мг, 0,28 ммоль) и Pd(dppf)Cl2 (10 мг) добавляли в раствор соединения 43-10 (70 мг, 0,14 ммоль) в DMF и H2O (6 мл, 5:1), затем реакционную смесь нагревали с обратным холодильником в течение ночи, H2O добавляли в реакционную смесь, DCM использовали для экстракции 3 раза, органические фазы объединяли и промывали солевыми растворами, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, остаток очищали с помощью препаративной HPLC с получением титульного соединения в виде светло-желтого твердого вещества. 1Н ЯМР (400 МГц, метанол-d4) δ 8,57-8,59 (d, J=8,0 Гц, 1Н) 7,55-7,60 (m, 4Н) 7,33-7,35 (mz, 2Н) 7,16-7,18 (d, J=8,4 Гц, 2Н) 7,13 (s, 1Н) 6,52 (s, 1Н) 3,79-3,83 (d, J=14,4 Гц 2Н) 3,60-3,63 (t, J=11,2 Гц, 2Н) 2,50-2,56 (t, J=22,4 Гц, 2Н) 1,97 (s, 3Н) 1,24-1,25 (d, J=6,4 Гц, 6Н). MS ESI рассч. для C26H26NO3 [М+Н]+ 416, найденное значение 416.
[251] Соединения, перечисленные в таблице 4, можно синтезировать с помощью соединения 43-10 и соответствующих борных кислот.
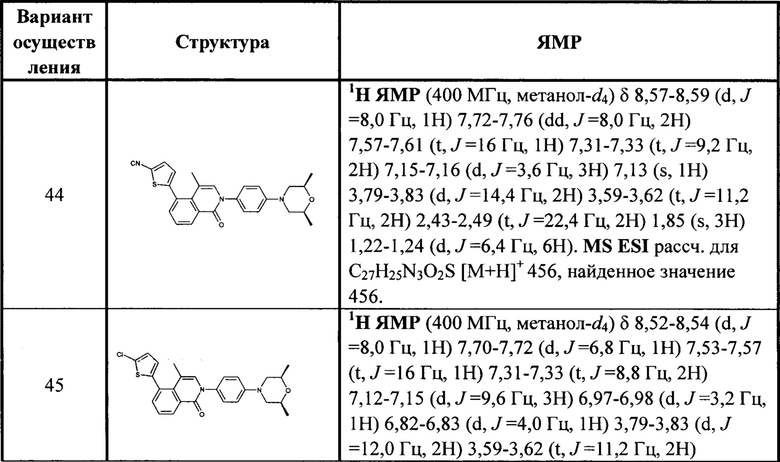
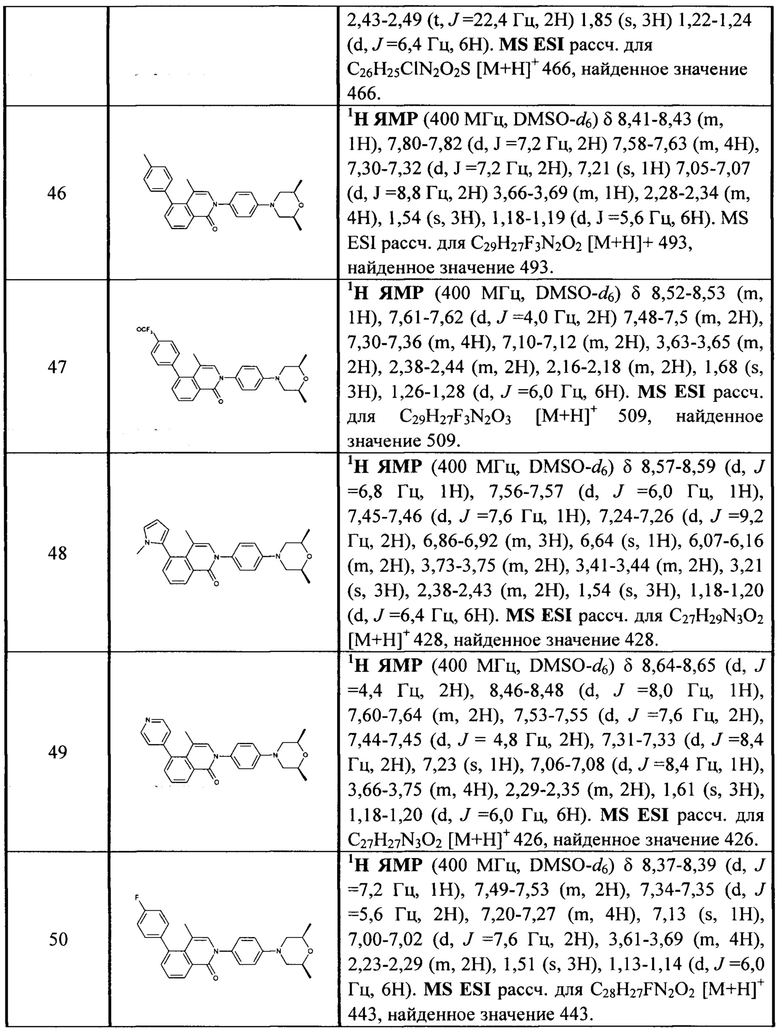

Вариант осуществления 69
5-(4-циклопропилфенил)-2-(4-((2R,6S)-2,6-диметилморфолино)фенил)-4-метилизохинолин-1(2Н)-он
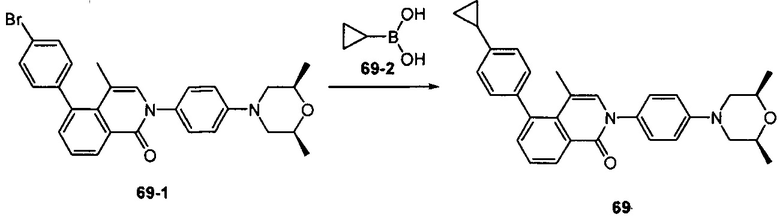
[252] Соединение 69-1 (106 мг, 0,20 ммоль), соединение 69-2 (52 мг, 0,60 ммоль), н-BuPAd2 (6,5 мг, 0,02 ммоль), ацетат палладия (2,7 мг, 0,01 ммоль) и карбонат цезия (197 мг, 0,6 ммоль) добавляли в смесь толуола/H2O (5:1, 3 мл), а затем в атмосфере газа азота реакционную смесь перемешивали при 110°C в течение 2 ч. Реакционную смесь выливали в H2O, экстрагировали с помощью EtOAc (10 мл), жидкость после экстракции промывали солевыми растворами, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали с помощью препаративной HPLC с получением титульного соединения (20 мг, выход 15%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,60-8,62 (t, J=9,6 Гц, 1Н) 7,55-7,56 (d, J=3,6 Гц, 2Н) 7,46-7,48 (d, J=8,4 Гц, 2Н), 7,27-7,32 (m, 2Н) 7,20-7,22 (d, J=8,0 Гц, 2Н), 7,09-7,11 (d, J=8,0 Гц, 2Н), 6,89 (s, 1Н), 4,00-4,04 (t, J=14,4 Гц, 2Н) 3,57-3,60 (t, J=11,2 Гц, 2Н) 2,68-2,73 (t, J=22,4 Гц, 2Н), 1,96-1,98 (m, 1H), 1,66 (s, 3Н), 1,30-1,31 (d, J=6,0 Гц, 6Н), 1,03-1,05 (m, 2Н), 0,77-0,79 (m, 2Н). MS ESI рассч. для C31H32N2O2 [М+Н]+ 465, найденное значение 465.
Вариант осуществления 70
4-(2-(6-(2,6-диметилморфолино-4-ил)пиридин-3-ил)-1-оксо-1,2-дигидроизохинолин-5-ил)бензонитрил
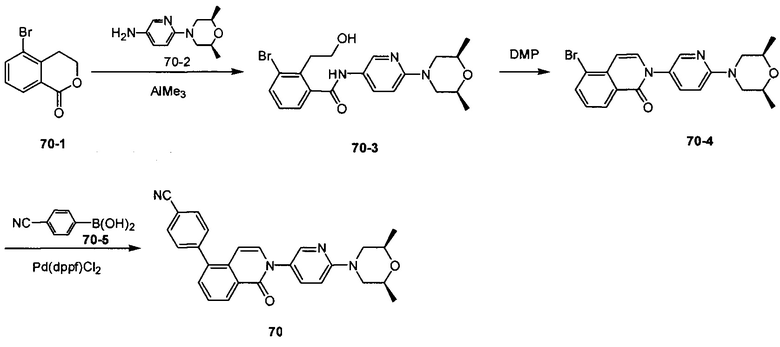
[253] Стадия 1. Раствор триметилалюминия (2 моль/л, 3,85 мл) в толуоле добавляли в раствор соединения 70-2 в сухом DCM (20 мл) при 0°C, реакционную смесь перемешивали при комнатной температуре в течение 15 мин. Затем по каплям добавляли раствор соединения 70-1 (0,7 г, 3,1 ммоль) в DCM (5 мл) и полученный раствор перемешивали при комнатной температуре в течение 3 ч. После завершения реакции, как обнаружено с помощью LCMS, добавляли раствор сегнетовой соли. Полученную смесь экстрагировали с помощью DCM, промывали солевыми растворами, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (РЕ/EtOAc=5:1) с получением соединения 70-3 (1,1 г, 85%) в виде желтого твердого вещества. MS ESI рассч. для C20H24BrN3O3 [М+Н]+ 435, найденное значение 435.
[254] Стадия 2. Периодинан Десс-Мартина (507 мг, 1,2 ммоль) добавляли в раствор соединения 70-3 (434 мг, 1 ммоль) в сухом DCM (20 мл) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем реакционную смесь гасили с помощью насыщенного водного раствора тиосульфата натрия и экстрагировали с помощью DCM, органическую фазу сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (РЕ/EtOAc=5:1) с получением соединения 70-4 (320 мг, 77%) в виде желтого твердого вещества. MS ESI рассч. для C20H20BrN3O2 [М+Н]+ 415, найденное значение 415.
[255] Стадия 3. В атмосфере газа азота соединение 70-5 (53 мг, 0,36 ммоль), карбонат натрия (51 мг, 0,48 ммоль) и Pd(dppf)Cl2 (18 мг, 0,024 ммоль) добавляли в раствор соединения 70-4 (0,10 г, 0,24 ммоль) в 1,4-диоксане (5 мл) и H2O (1 мл). Затем реакционную смесь нагревали до температуры флегмы и перемешивали в течение ночи. После завершения реакции остаток очищали с помощью препаративной HPLC с получением титульного соединения (56 мг, 53%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,43 (d, J=8,0 Гц, 1Н), 8,18 (d, J=2,0 Гц, 1H), 7,90-7,82 (m, 3Н), 7,75-7,61 (m, 4H), 7,32 (d, J=8,0 Гц, 1H), 7,16 (d, J=8,8 Гц, 1H), 6,60 (d, J=7,6 Гц, 1H), 4,13 (d, J=12,8 Гц, 2H), 3,75-3,71 (m, 2H), 2,68 (t, J=12,0 Гц, 2H), 1,24 (d, J=5,6 Гц, 6H). MS ESI рассч. для C27H24N4O2 [M+H]+ 437, найденное значение 437.
[256] Соединения, перечисленные в таблице 5, можно синтезировать с помощью соединения 70-4 и соответствующих борных кислот.
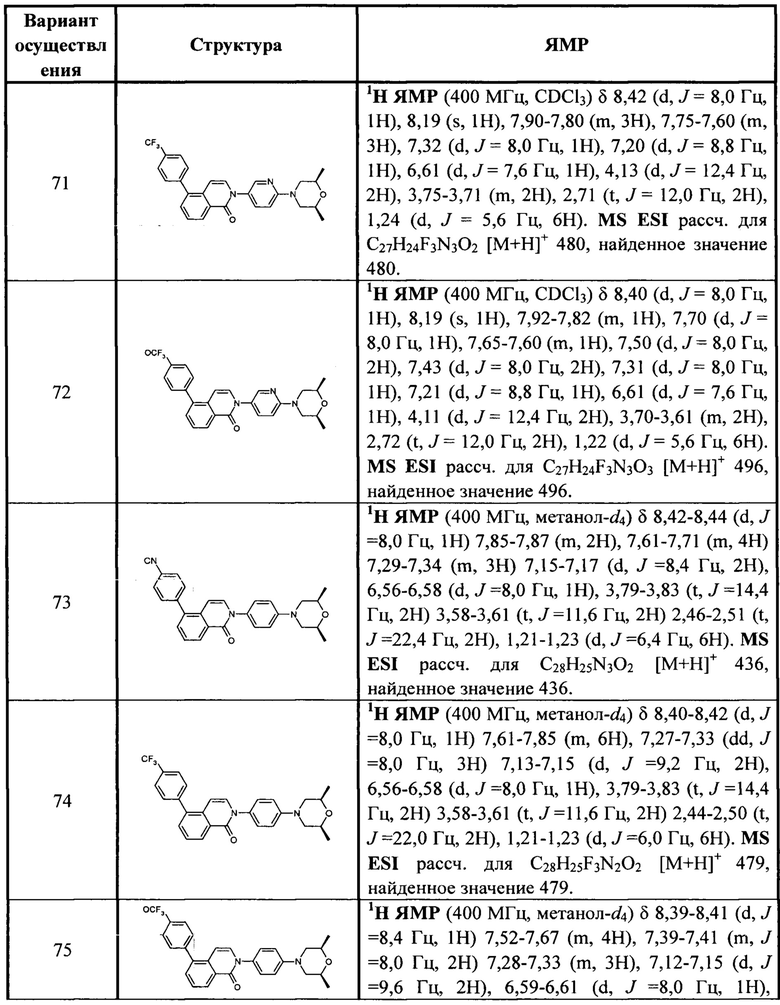

Вариант осуществления 76
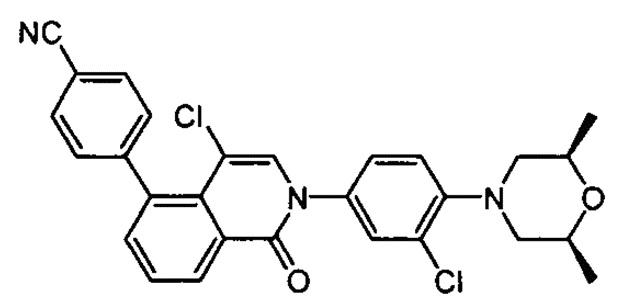
4-(4-хлор-2-(3-хлор-4-((2S,6R)-2,6-диметилморфолино)фенил)-1-оксо-1,2-дигидроизохинолин-5-ил)бензонитрил
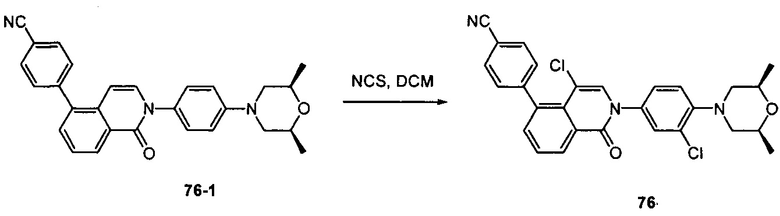
[257] NCS (48 мг, 0,36 ммоль) добавляли в раствор соединения 76-1 (100 мг, 0,24 ммоль) в сухом DCM (10 мл). Затем реакционную смесь перемешивали при комнатной температуре в течение ночи. После завершения реакции остаток очищали с помощью препаративной HPLC с получением титульного соединения в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,65 (d, J=8,0 Гц, 1Н), 7,70 (d, J=8,4 Гц, 2H), 7,50-7,68 (m, 2H), 7,40-7,49 (m, 3Н), 7,27-7,35 (m, 2H), 7,12 (d, J=8,0 Гц, 1H), 3,85-4,00 (m, 2H), 3,20-3,35 (m, 2H), 2,40-2,55 (m, 2H), 1,25 (s, 3Н), 1,24 (s, 3Н). MS ESI рассч. для C28H27N5O [M+H]+ 450, найденное значение 450.
Варианты осуществления 77, 78
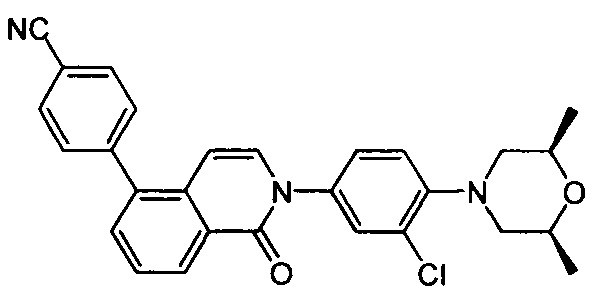
4-(2-(3-хлор-4-((2S,6R)-2,6-диметилморфолино)фенил)-1-оксо-1,2-дигидроизохинолин-5-ил)бензонитрил
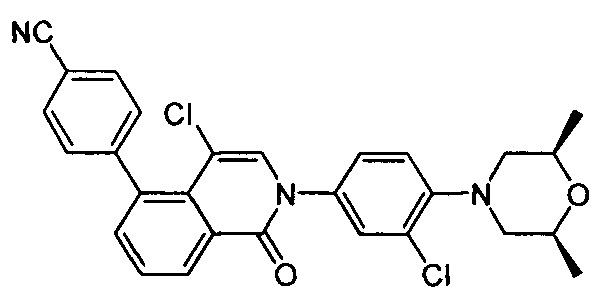
4-(4-хлор-2-(3-хлор-4-((2S,6R)-2,6-диметилморфолино)фенил)-1-оксо-1,2-дигидроизохинолин-5-ил)бензонитрил
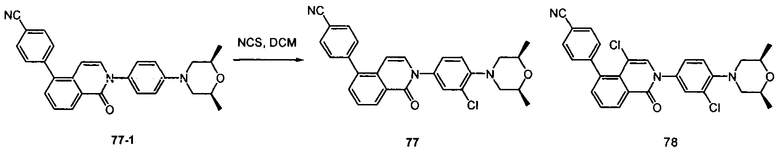
[258] NCS (48 мг, 0,36 ммоль) порциями добавляли в раствор соединения 77-1 (100 мг, 0,24 ммоль) в DCM (10 мл), а затем реакционную смесь перемешивали при комнатной температуре в течение ночи и растворитель выпаривали, остаток очищали с помощью препаративной HPLC с получением титульного соединения варианта осуществления 77 в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,57-8,59 (d, J=8,0 Гц, 1Н) 7,80-7,82 (d, J=8,0 Гц, 2Н) 7,63-7,64 (t J=6,4 Гц, 2Н), 7,56-7,58 (t, J=8,0 Гц, 2Н), 7,46 (s, 1Н) 7,29-7,34 (m, 1Н) 7,12-7,14 (d, J=8,4 Гц, 2Н), 6,52-6,54 (d, J=8,0 Гц, 1Н), 3,93-3,95 (t, J=14,4 Гц, 2Н) 3,27-3,30 (t, J=10,8 Гц, 2Н) 2,47-2,53 (t, J=23,2 Гц, 2Н), 1,24-1,26 (d, J=6,0 Гц, 6Н). MS ESI рассч. для C28H24ClN3O2 [М+Н]+ 471, найденное значение 471. В то же время титульное соединение варианта осуществления 78 выделяли в виде желтого твердого вещества. 1Н ЯМР (400 МГц, метанол-d4) δ 8,64-8,66 (d, J=7,2 Гц, 1Н) 7,57-7,71 (m, 4Н), 7,44-7,47 (dd, J=8,0 Гц, 3Н) 7,31 (s, 1Н), 7,11-7,13 (d, J=8,0 Гц, 1Н), 3,89-3,93 (t, J=14,4 Гц, 2Н), 3,26-3,29 (t, J=11,6 Гц, 2Н) 2,44-2,50 (t, J=21,6 Гц, 2Н), 1,24-1,26 (d, J=6,0 Гц, 6Н). MS ESI рассч. для C28H23Cl2N3O2 [М+Н]+ 505, найденное значение 505.
Вариант осуществления 79
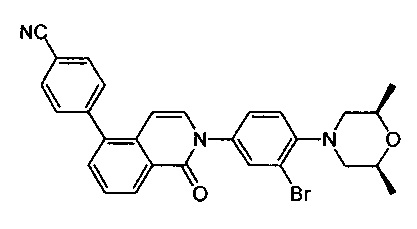
4-(2-(3-бром-4-((2S,6R)-2,6-диметилморфолино)фенил)-1-оксо-1,2-дигидроизохинолин-5-ил)бензонитрил
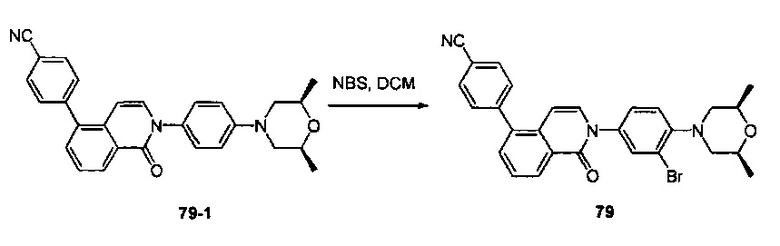
[259] NBS (43 мг, 0,24 ммоль) порциями добавляли в раствор соединения 79-1 (100 мг, 0,24 ммоль) в DCM (10 мл), затем реакционную смесь перемешивали при комнатной температуре в течение ночи и растворитель выпаривали, остаток очищали с помощью препаративной HPLC с получением титульного соединения (20 мг, выход 25%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, метанол-d4) δ 8,43-8,45 (d, J=7,6 Гц, 1Н) 7,87-7,89 (d, J=8,4 Гц, 2Н), 7,64-7,71 (m, 5Н), 7,24-7,40 (m, 3Н), 6,58-6,60 (d, J=7,6 Гц, 2Н), 3,87-3,91 (t, J=14,4 Гц, 2Н), 2,42-2,47 (t, J=22,4 Гц, 2Н), 1,18-1,20 (d, J=6,4 Гц, 6Н). MS ESI рассч. для C28H24BrN3O2 [М+Н]+ 515, найденное значение 515.
Вариант осуществления 80
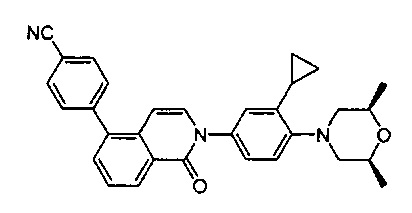
4-(2-(3-циклопропил-4-((2S,6R)-2,6-диметилморфолино)фенил)-1-оксо-1,2-дигидроизохинолин-5-ил)бензонитрил
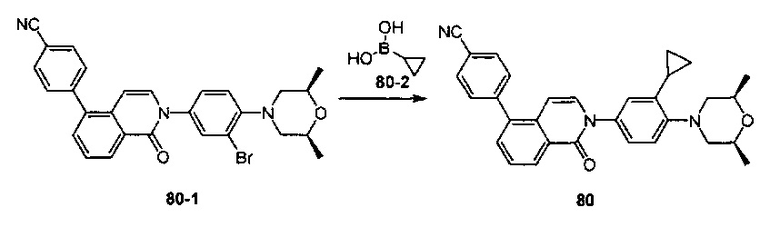
[260] Соединение 80-1 (100 мг, 0,19 ммоль), 80-2 (49 мг, 0,57 ммоль), H-BuPAd2 (7,2 мг, 0,02 ммоль), ацетат палладия (3 мг, 0,01 ммоль) и карбонат цезия (187 мг, 0,57 ммоль) добавляли в раствор толуола/H2O (5:1, 3 мл), затем в атмосфере газа азота реакционную смесь перемешивали при 110°С в течение 2 ч. Реакционную смесь выливали в H2O, экстрагировали с помощью EtOAc (10 мл), жидкость после экстракции промывали солевыми растворами, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали с помощью препаративной HPLC с получением титульного соединения (250 мг, выход 20%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,57-8,59 (t, J=9,2 Гц, 1Н) 7,80-7,82 (d, J=8,0 Гц, 2Н) 7,57-7,63 (m, 4Н), 7,11-7,18 (m, 3Н) 6,83-6,84 (d, J=1,6 Гц, 1Н), 6,50-6,52 (d, J=7,6 Гц, 1Н), 3,94-3,98 (t, J=14,4 Гц, 2Н) 3,27-3,30 (t, J=10,8 Гц, 2Н) 2,55-2,60 (t, J=21,6 Гц, 2Н), 2,29-2,34 (m, 2Н), 1,24-1,26 (d, J=6,4 Гц, 6Н), 1,03-1,05 (m, 2Н), 0,75-0,76 (m, 2Н). MS ESI рассч. для C31H29N3O2 [М+Н]+ 476, найденное значение 476.
Вариант осуществления 81
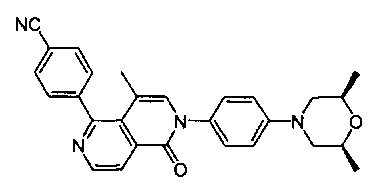
4-(6-(4-((2S,6R)-2,6-диметилморфолино)фенил)-8-метил-5-оксо-5,6-дигидро-2,6-нафтиридин-1-ил)бензонитрил
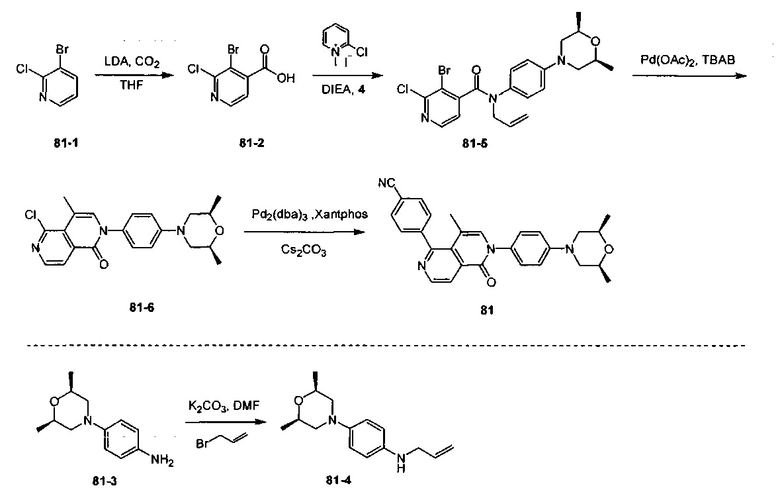
[261] Стадия 1. LDA (60 мл, 0,12 моль) стекал в раствор соединения 81-1 (20 г, 0,1 моль) в THF (200 мл) при -78°С, реакционную смесь перемешивали при -78°С в течение 2 ч. Реакционную смесь быстро выливали в сухой лед. Затем смесь перемешивали при комнатной температуре в течение 30 мин. и в конце гасили с помощью H2O. Водную фазу экстрагировали с помощью EtOAc. Объединенную органическую фазу промывали солевыми растворами, сушили над сульфатом натрия и концентрировали при пониженном давлении с получением соединения 81-2 (10 г, выход 43%) в виде грязно-белого твердого вещества. MS ESI рассч. для C6H3BrClNO2 [М+Н]+ 236, найденное значение 236.
[262] Стадия 2. Смесь соединения 81-3 (5 г, 24,3 ммоль), аллилбромида (2,1 г, 17 ммоль) и K2CO3 (6,7 г, 48,6 ммоль) добавляли в DMF (50 мл), реакционную смесь перемешивали при 80°С в течение 5 ч. Реакционную смесь выливали в H2O. Водную фазу экстрагировали с помощью EtOAc. Объединенную органическую фазу промывали солевыми растворами, сушили над сульфатом натрия и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали с помощью хроматографии на силикагеле (РЕ:EtOAc = 15:1) с получением соединения 81-4 (2,5 г, выход 42%) в виде коричневого твердого вещества. MS ESI рассч. для C15H22N2O [М+Н]+ 247, найденное значение 247.
[263] Стадия 3. CMPI (2,2 г, 8,9 ммоль) добавляли в раствор соединения 81-2 (2,1 г, 8,9 ммоль), соединение 81-4 (2,2 г, 8,9 ммоль) и DIEA (2,3 г, 17,8 ммоль) в ацетонитриле (20 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Неочищенный продукт применяли непосредственно на следующей стадии. MS ESI рассч. для C21H23BrClN3O2 [М+Н]+ 464, найденное значение 464.
[264] Стадия 4. Соединение 81-5 (4,1 г, 8,9 ммоль), тетра-бутиламмония бромид (7,2 г, 22,3 ммоль), ацетат палладия (200 мг, 0,89 ммоль) и TEA (2,7 г, 26,7 ммоль) в ацетонитриле (40 мл) нагревали до температуры флегмы и перемешивали в течение 48 ч. Реакционную смесь выливали в Н2О. Водную фазу экстрагировали с помощью EtOAc. Объединенную органическую фазу промывали солевыми растворами, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле (РЕ:EtOAc = 2:1) с получением соединения 81-6 (1 г, выход 30%) в виде коричневого твердого вещества. MS ESI рассч. для C21H22ClN3O2 [М+Н]+ 384, найденное значение 384.
[265] Стадия 5. Соединение 81-6 (100 мг, 0,26 ммоль), 4-цианофенилбороновую кислоту (45,6 мг, 0,31 ммоль), Pd2(dba)3(24 мг, 0,026 ммоль), Xantphos(25 мг, 0,052 ммоль) и карбонат цезия (171 мг, 0,52 ммоль) добавляли в диоксан (5 мл) и смесь нагревали до температуры флегмы, затем перемешивали в течение 16 ч. Неочищенный продукт очищали с помощью препаративной HPLC с получением титульного соединения в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ 8,79 (d, J=5,6 Гц, 1H), 7,77 (d, J=8,0 Гц, 2Н), 7,67 (d, J=5,2 Гц, 1Н), 7,61 (s, 1H), 7,54 (d, J=8,0 Гц, 2Н), 7,17 (d, J=8,4 Гц, 2Н), 6,97 (d, J=8,8 Гц, 2Н), 3,45-3,52 (m, 4Н), 2,15-2,30 (m, 4Н), 1,13 (s, 3Н), 1,11 (s, 3Н). MS ESI рассч. для C28H26N4O2 [М+Н]+ 451, найденное значение 451.
[266] Соединения, перечисленные в таблице 6, можно синтезировать с помощью соединения 81-6 и соответствующих борных кислот.
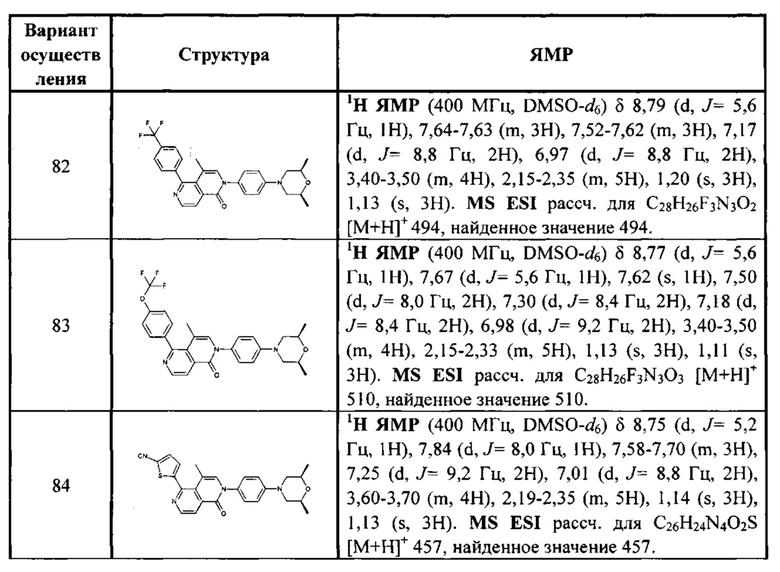
Вариант осуществления 85

4-(7-(4-((2S,6R)-2,6-диметилморфолино)фенил)-5-метил-8-оксо-7,8-дигидро-1,7-нафтиридин-4-ил)бензонитрил
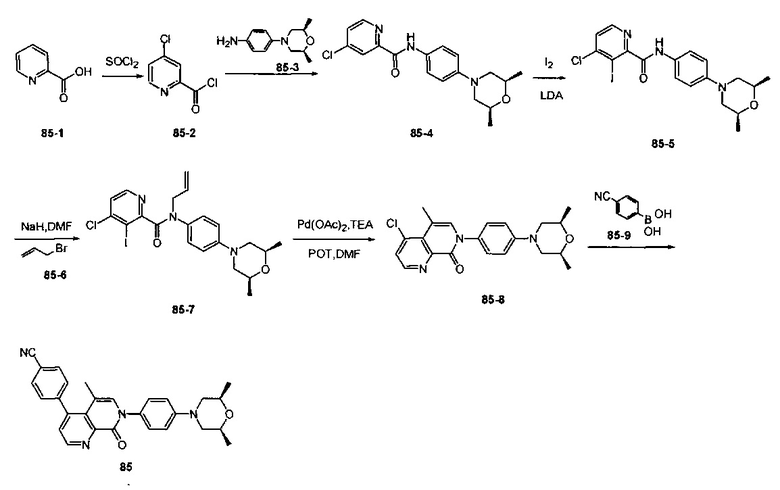
[267] Стадия 1. Соединение 85-1 (4,3 г, 35 ммоль), DMF (0,5 мл) и тионилхлорид (13 мл, 181 ммоль) перемешивали при 80°С в течение 12 ч. Реакционную смесь концентрировали досуха при пониженном давлении с получением соединения 85-2 (6,1 г, выход 100%) в виде коричневого твердого вещества. MS ESI рассч. для C6H3Cl2NO [М+Н]+ 176, найденное значение 176.
[268] Стадия 2. Соединение 85-2 (6,1 г, 35 ммоль) добавляли в раствор соединения 85-3 (6 г, 29,1 ммоль) в DCM (60 мл) при 0°С. После добавления реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь выливали в H2O (200 мл) и осадок собирали путем фильтрования. Отфильтрованный осадок растворяли в EtOAc, сушили над сульфатом натрия, фильтровали и концентрировали досуха. В конце остаток очищали с помощью хроматографии на силикагеле (РЕ:EtOAc = 2:1) с получением соединения 85-4 (5,6 г, выход 56%) в виде белого твердого вещества. MS ESI рассч. для C18H20ClN3O2 [М+Н]+ 346, найденное значение 346.
[269] Стадия 3. В атмосфере газа азота LDA (22 мл, 43,5 ммоль) добавляли в раствор соединения 85-4 (5 г, 14,5 ммоль) в THF (20 мл) при -78°С. Через 30 мин. раствор I2 (5,5 г, 22,7 ммоль) в THF (20 мл) добавляли в раствор. Полученную реакционную смесь перемешивали при -78°С в течение дополнительных 2 ч. Затем реакционную смесь выливали в H2O, экстрагировали с помощью EtOAc, органические фазы объединяли и промывали солевыми растворами, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле (РЕ:EtOAc = 2:1) с получением соединения 85-5 (5 г, выход 74%) в виде желтого твердого вещества. MS ESI рассч. для C18H19ClIN3O2 [М+Н]+ 472, найденное значение 472.
[270] Стадия 4. NaH (848 мг, 21,2 ммоль, содержащий 40% минеральное масло) добавляли в раствор соединения 85-5 (5 г, 10,6 ммоль) в DMF при 0°С. Реакционную смесь перемешивали в атмосфере газа азота в течение 10 мин. Соединение 85-6 (2,56 г, 21,2 ммоль) добавляли в раствор и полученную смесь перемешивали при 0°С в течение дополнительных 2 ч. Затем реакционную смесь выливали в H2O, экстрагировали с помощью EtOAc. Объединенную органическую фазу промывали солевыми растворами, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения 85-7 (5,3 г, выход 99%) в виде белого твердого вещества. MS ESI рассч. для C21H23ClIN3O2 [М+Н]+ 512, найденное значение 512.
[271] Стадия 5. Соединение 85-7 (5,4 г, 10,6 ммоль), ацетат палладия (237 мг, 1,06 ммоль), TEA (2,1 г, 21,2 ммоль) и РОТ (322 мг, 1,06 ммоль) добавляли в DMF (40 мл) и реакционную смесь перемешивали при 120°С в течение 12 ч. Реакционную смесь выливали в Н2О, экстрагировали с помощью EtOAc. Объединенную органическую фазу промывали солевыми растворами, сушили над безводным сульфатом натрия, фильтровали и концентрировали, в конце очищали с помощью хроматографии на силикагеле (РЕ:EtOAc = 1:1) с получением соединения 85-8 (3 г, выход 75%) в виде грязно-белого твердого вещества. MS ESI рассч. для C21H22ClN3O2 [М+Н]+ 384, найденное значение 384.
[272] Стадия 6. Соединение 85-8 (300 мг, 0,78 ммоль), соединение 85-9 (160 мг, 1,09 ммоль), К3РО4 (413 мг, 1,95 ммоль), Pd(dppf)Cl2 (57 мг, 0,078 ммоль) добавляли в DMF (5 мл), а затем в атмосфере газа азота смесь перемешивали при 120°С в течение 12 ч. Реакционную смесь выливали в H2O, экстрагировали с помощью EtOAc. Объединенную органическую фазу промывали солевыми растворами, сушили над безводным сульфатом натрия, фильтровали и концентрировали, очищали с помощью препаративной HPLC с получением титульного соединения (75 мг, выход 21%) в виде белого твердого вещества. MS ESI рассч. для C28H26N4O2 [М+Н]+ 451, найденное значение 451.
Вариант осуществления 86
4-(7-(4-((2S,6R)-2,6-диметилморфолино)фенил)-5-метил-8-оксо-7,8-дигидро-2,7-нафтиридин-4-ил)бензонитрил
[273] Стадия 1. В атмосфере газа азота н-бутиллитий (1,8 мл, 4,4 ммоль) по каплям добавляли в раствор 2,2,6,6-тетраметилпиперидина (0,81 мл, 4,8 ммоль) в THF (10 мл) при -50°С. Через 5 мин. добавляли 5-бромникотиновую кислоту (0,40 г, 2,0 ммоль). Через 30 мин. раствор I2 (0,61 г, 2,4 ммоль) в THF (5 мл) по каплям добавляли при -50°С. Полученную реакционную смесь перемешивали при -50°С в течение 2 ч. Реакционную смесь гасили с помощью H2O (10 мл), водную фазу отделяли и экстрагировали с помощью Et2O (10 мл), затем водную фазу окисляли с помощью 1 М хлористоводородной кислоты до рН=3. Осадок фильтровали и отфильтрованный осадок сушили с получением соединения 86-2 в виде бежевого порошка. MS ESI рассч. для C6H3BrINO2 [М+Н]+ 328, найденное значение 328.
[274] Стадия 2. Соединение 86-2 (650 мг, 25 ммоль), соединение 86-3 (500 мг, 2 ммоль), HATU (770 мг, 2 ммоль) и DIEA (2 мл) добавляли в DMF (10 мл) и смесь перемешивали при 20°С в течение 4 ч. Реакционную смесь выливали в H2O и экстрагировали с помощью EtOAc. Объединенную органическую фазу сушили над сульфатом натрия. После концентрирования остаток очищали с помощью хроматографии на силикагеле с получением соединения 86-4 (выход 70%) в виде грязно-белого твердого вещества. MS ESI рассч. для C21H23BrIN3O2 [М+Н]+ 556, найденное значение 556.
[275] Стадия 3. Соединение 86-5 в виде белого твердого вещества синтезировали в соответствии с предварительно описанным способом. MS ESI рассч. для C21H22BrN3O2 [М+Н]+ 428, найденное значение 428.
[276] Стадия 4. Титульное соединение (выход 48%) в виде белого твердого вещества синтезировали в соответствии с предварительно описанным способом. 1H ЯМР (400 МГц, CDCl3) δ 9,66 (s, 1H), 8,56 (s, 1Н), 7,72 (d, J=8,4 Гц, 2Н), 7,44 (d, J=8,4 Гц, 2Н), 7,23 (d, J=8,4 Гц, 2Н), 6,95 (d, J=8,8 Гц, 2Н), 3,55-3,75 (m, 3Н), 3,40-3,50 (m, 2Н), 2,35-2,50 (m, 2Н), 1,57 (s, 3Н), 1,22 (s, 3Н), 1,21 (s, 3Н). MS ESI рассч. для C28H26N4O2 [М+Н]+ 451, найденное значение 451.
Вариант осуществления 87
4-(2-(4-((2S,6R)-2,6-диметилморфолино)фенил)-4-метил-1-оксо-1,2,3,4-тетрагидроизохинолин-5-ил)бензонитрил
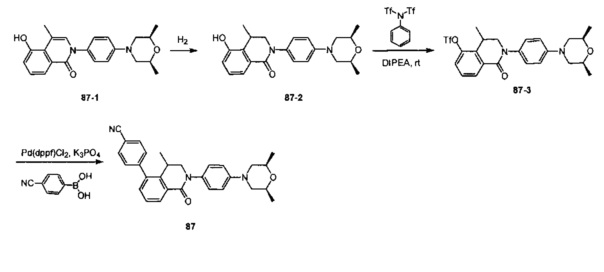
[277] Стадия 1. 10% Pd/C (50 мг, 50% влажность) добавляли в раствор соединения 87-1 (500 мг, 1,37 ммоль) в МеОН (50 мл). Затем при давлении газа водорода 55 фунтов/кв. дюйм реакционную смесь перемешивали при 40°С в течение 48 ч. После завершения реакции реакционную смесь фильтровали с помощью диатомита и фильтрат концентрировали при пониженном давлении с получением соединения 87-2 (400 мг, выход 80%) в виде белого твердого вещества. MS ESI рассч. для C22H26N2O3 [М+Н]+ 367, найденное значение 367.
[278] Стадия 2. Соединение 87-3 (150 мг, выход 30%) в виде грязно-белого твердого вещества синтезировали в соответствии с предварительно описанным способом. MS ESI рассч. для C22H26N2O3 [М+Н]+ 499, найденное значение 499.
[279] Стадия 3., Титульное соединение в виде белого твердого вещества синтезировали в соответствии с предварительно описанным способом. 1Н ЯМР (400 МГц, CDCl3) δ 8,25 (d, J=7,6 Гц, 1H), 7,78 (d, J=8,4 Гц, 2Н), 7,35-7,56 (m, 8Н), 4,33 (dd, J=8,4, 3,6 Гц, 1Н), 4,06-4,16 (m, 2Н), 3,50-3,57 (m, 3Н), 3,07-3,16 (m, 1H), 2,75-2,85 (m, 2Н), 1,31 (s, 3Н), 1,30 (s, 3Н), 1,24 (d, J=6,8 Гц, 1H). MS ESI рассч. для C29H29N3O2 [М+Н]+ 452, найденное значение 452.
Вариант осуществления 88

4-(3-(4-((2S,6R)-2,6-диметилморфолино)фенил)-4-метоксихинолин-8-ил)бензонитрил
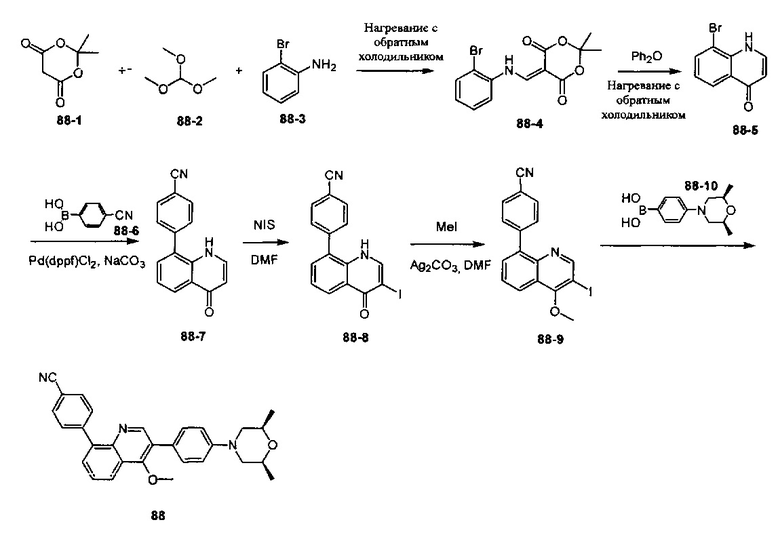
[280] Стадия 1. Соединение 88-1 (50 г, 350 ммоль) добавляли в соединение 88-2 (170 мл, 1 моль), затем реакционную систему нагревали до температуры флегмы в течение 3 ч. После охлаждения реакционной системы до комнатной температуры добавляли соединение 88-3 (48 г, 280 ммоль). Реакционную смесь перемешивали в течение 12 ч., наблюдали с помощью LC-MS до завершения реакции. Затем систему концентрировали, остаток промывали с помощью МеОН, сушили с получением светло-желтого соединения 88-4 (80 г, 90%). MS ESI рассч. для C13H12BrNO4 [М+Н]+ 326, найденное значение 326.
[281] Стадия 2. Соединение 88-4 (80 г, 245 ммоль) растворяли в Ph2O (560 мл), затем смесь нагревали до температуры флегмы в течение 1 ч. После завершения реакции, как наблюдали с помощью LC-MS, 560 мл н-гексана порциями добавляли в раствор с получением продукта. Продукт промывали с помощью н-гексана, сушили при пониженном давлении с получением соединения 88-5 (48 г, 87%) в виде серого твердого вещества. MS ESI рассч. для C9H6BrNO [М+Н]+ 225, найденное значение 225.
[282] Стадия 3. Соединение 88-5 (20 г, 89 ммоль), соединение 88-6 (14,4 г, 98 ммоль), Pd(dppf)Cl2 (6,5 г, 8,9 ммоль) и Na2CO3 (18,8 г, 178 ммоль) растворяли в DMF (200 мл) и 40 мл H2O добавляли в раствор. Реакционная система реагировала при 90°С в течение ночи. После завершения реакции, как наблюдали с помощью LC-MS, реакционную систему выливали в 500 мл H2O, фильтровали с получением коричневого твердого вещества (19 г, 87%). MS ESI рассч. для C16H10N2O [М+Н]+ 247, найденное значение 247.
[283] Стадия 4. NIS (17,4 г, 77,2 ммоль) порциями добавляли в раствор 88-7 (19 г, 77,2 ммоль) в DMF (200 мл). Реакционную систему перемешивали при комнатной температуре в течение 3 ч. Реакцию наблюдали с помощью LC-MS и TLC до ее завершения. Раствор выливали в H2O (500 мл), фильтровали с получением коричневого остатка. Остаток промывали с помощью МеОН и рекристаллизовали с помощью РЕ:EtOAc = 1:1 с получением целевого соединения 88-8 (25 г, 87%) в виде коричневого твердого вещества. MS ESI рассч. для C16H9IN2O [М+Н]+ 373, найденное значение 373.
[284] Стадия 5. Соединение 88-8 (15 г, 40,3 ммоль) и Ag2CO3 (25 г, 80,6 ммоль) растворяли в DMF (150 мл). Затем MeI (3,8 мл, 60,5 ммоль) добавляли в реакционную систему. Реакционную смесь перемешивали при 80°С в течение 3 ч. Реакцию наблюдали с помощью LC-MS до ее завершения, реакционную смесь добавляли в H2O и экстрагировали с помощью EtOAc. Органические фазы объединяли и промывали солевыми растворами, сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха. Неочищенный продукт очищали посредством колоночной хроматографии (РЕ:EtOAc = 10:1) с получением белого соединения 88-9 (3 г, 19%). MS ESI рассч. для C17H11IN2O [М+Н]+ 387, найденное значение 387.
[285] Стадия 5. Соединение 88-9 (200 мг, 0,52 ммоль), соединение 88-13 (246 мг, 1,04 ммоль), Pd(dppf)Cl2 (38 мг, 0,052 ммоль) и Na2CO3 (110 мг, 1,04 ммоль) растворяли в THF (5 мл) и H2O (1 мл). Смесь перемешивали при 60°С в течение ночи. Реакцию наблюдали с помощью LC-MS до ее завершения.
Реакционную систему экстрагировали с помощью EtOAc, органические фазы объединяли и промывали солевыми растворами, сушили над безводным сульфатом натрия, фильтровали, концентрировали, неочищенный продукт очищали с помощью препаративной хроматографии с получением титульного соединения в виде светло-желтого твердого вещества (23 мг, выход 10%). 1Н ЯМР (400 МГц, CDCl3) δ 8,89 (s, 1Н), 8,33-8,31 (m, 1H), 7,84-7,56 (m, 8Н), 7,04 (d, J=8,8 Гц, 2Н), 3,86-3,82 (m, 2Н), 3,70 (s, 3Н), 3,57 (d, J=11,2 Гц, 2Н), 2,51 (t, J=11,2 Гц, 2Н), 1,30 (d, J=6,0 Гц, 6Н). MS ESI рассч. для C29H27N3O2 [М+Н]+ 450, найденное значение 450.
Вариант осуществления 89
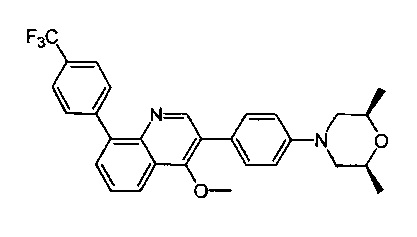
(2R,6S)-4-(4-(4-метокси-8-(4-(трифторметил)фенил)хинолин-3-ил)фенил)-2,6-диметилморфолин
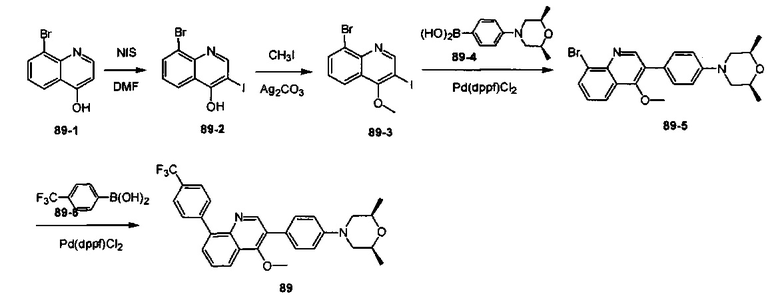
[286] Стадия 1. NIS (3,8 г, 22 ммоль) порциями добавляли в раствор соединения 89-1 (4,9 г, 22 ммоль) в DMF (40 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. После завершения реакции, как обнаружено с помощью LC-MS, реакционную смесь выливали в H2O (500 мл). Продукт 89-2 получали путем фильтрования (5,5 г, 71%) в виде коричневого твердого вещества. MS ESI рассч. для C9H5BrINO [М+Н]+ 350, найденное значение 350.
[287] Стадия 2. CH3I (1,2 г, 8,7 ммоль) добавляли в раствор соединения 89-2 (2,0 г, 5,8 ммоль) и Ag2CO3 (2,4 г, 8,7 ммоль) в DMF (20 мл). Реакционную смесь перемешивали при 60°С в течение 2 ч. После завершения реакции, как обнаружено с помощью LC-MS, реакционную смесь выливали в H2O, затем экстрагировали с помощью EtOAc (80×3 мл), органическую фазу сушили над безводным сульфатом натрия. После концентрирования неочищенный продукт дополнительно очищали с помощью колоночной хроматографии (РЕ:EtOAc = 5:1) с получением соединения 89-3 (464 мг, 22%) в виде белого твердого вещества. MS ESI рассч. для C10H7BrINO [М+Н]+ 364, найденное значение 364.
[288] Стадия 3. Соединение 89-3 (1089 мг, 3 ммоль), соединение 89-4 (1050 мг, 3,3 ммоль), Pd(dppf)Cl2 (110 мг, 0,15 ммоль) и Na2CO3 (636 мг, 6 ммоль) добавляли в THF (20 мл) / H2O (2 мл), в атмосфере газа азота реакционную смесь перемешивали при 60°С в течение ночи. После завершения реакции смесь фильтровали, фильтрат экстрагировали с помощью EtOAc, органическую фазу промывали солевыми растворами, сушили над безводным сульфатом натрия, концентрировали при пониженном давлении с получением неочищенного продукта 4 (140 мг, 11%) в виде белого твердого вещества. MS ESI рассч. для C22H23BrN2O2 [М+Н]+ 427, найденное значение 427.
[289] Стадия 4. Соединение 89-5 (70 мг, 0,16 ммоль), соединение 89-6 (46 мг, 0,24 ммоль) добавляли в раствор Pd(dppf)Cl2 (15 мг, 0,02 ммоль), Na2CO3 (34 мг, 0,32 ммоль) в THF (10 мл) / H2O (1 мл), в атмосфере газа азота реакционную смесь перемешивали при 80°С в течение ночи. После завершения реакции смесь фильтровали, фильтрат экстрагировали с помощью EtOAc, органическую фазу промывали солевыми растворами, сушили над безводным сульфатом натрия, концентрировали при пониженном давлении с получением титульного соединения (35 мг, 45%) в виде светло-желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,91 (s, 1H), 8,32-8,30 (m, 1H), 7,83-7,57 (m, 8Н), 7,04 (d, J=8,8 Гц, 2Н), 3,84-3,83 (m, 2Н), 3,71 (s, 3Н), 3,57 (d, J=11,2 Гц, 2Н), 2,51 (t, J=11,2 Гц, 2Н), 1,30 (d, J=6,0 Гц, 6Н). MS ESI рассч. для C29H27F3N2O2 [М+Н]+ 493, найденное значение 493.
[290] Соединения, перечисленные в таблице 7, синтезировали с помощью соединения 89-5 и соответствующих борных кислот.
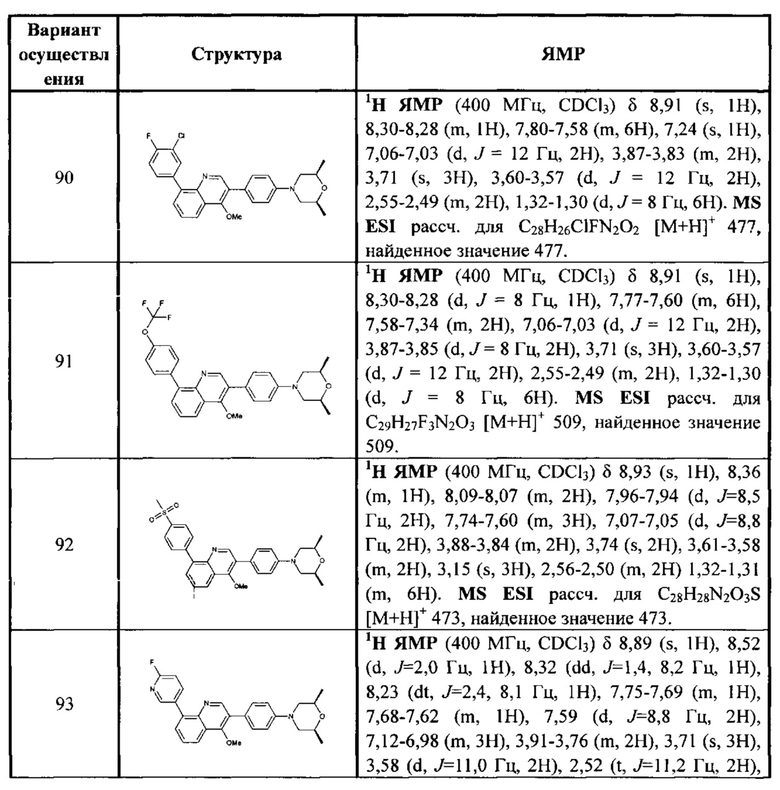
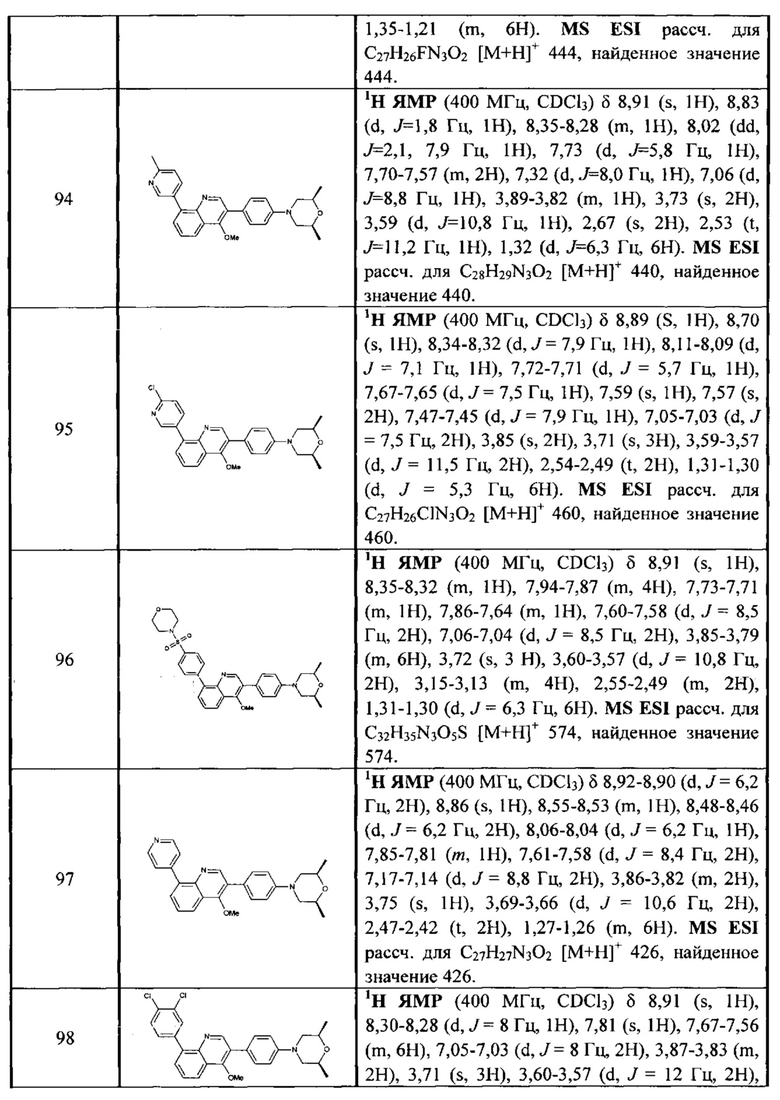
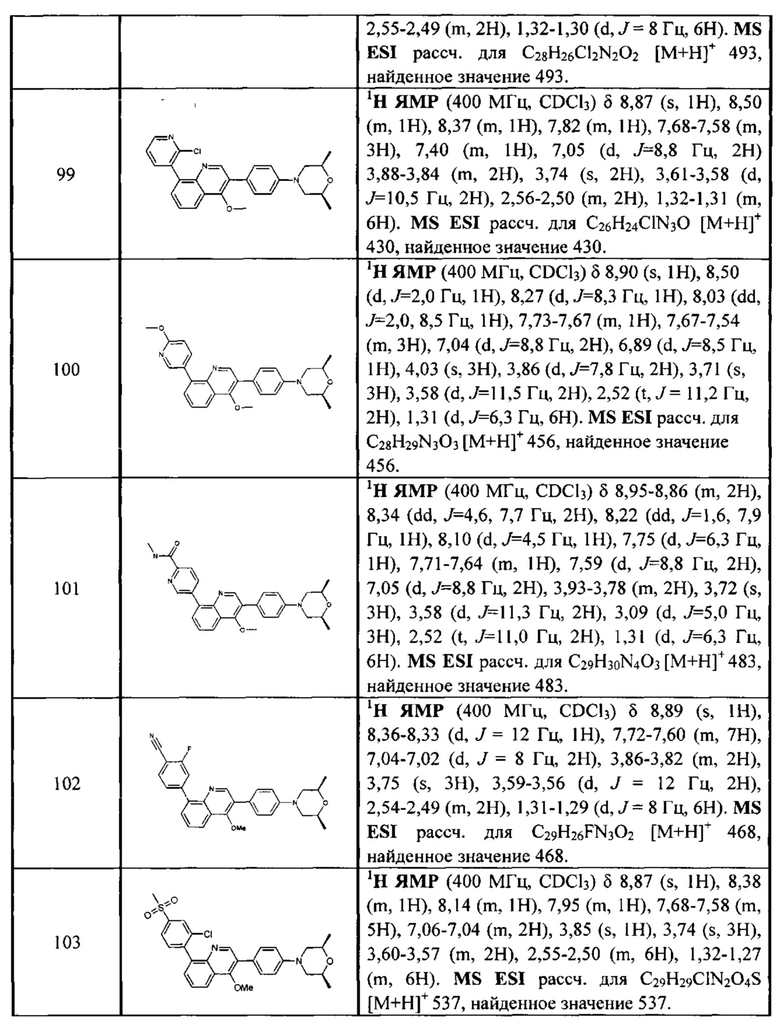
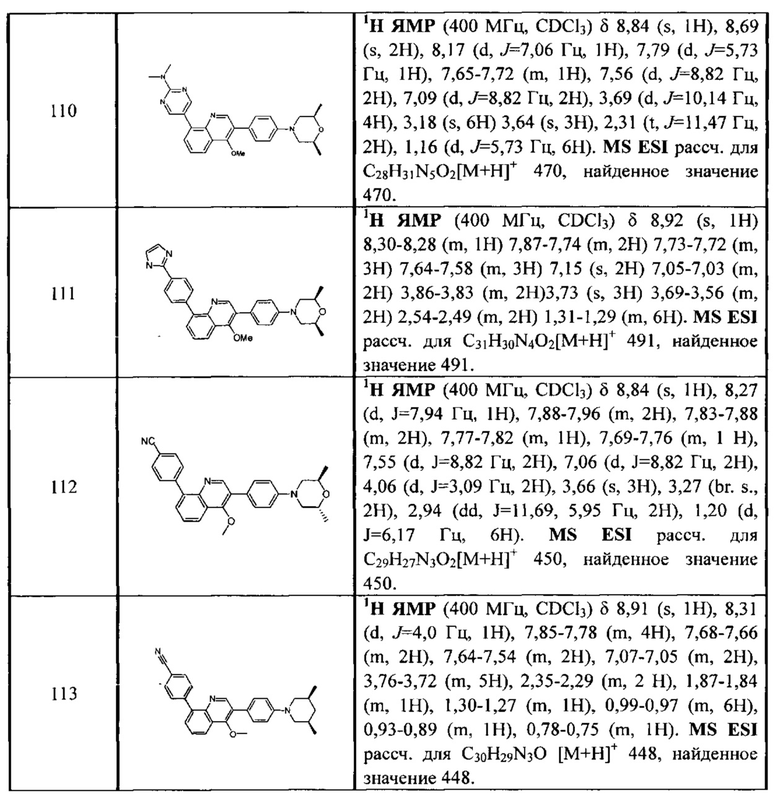
Вариант осуществления 114
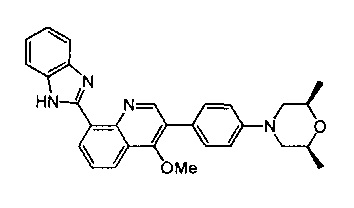
(2S,6R)-4-(4-(8-(1Н-бензо[d]имидазол-2-ил)-4-метоксихинолин-3-ил)фенил)-2,6-диметилморфолин
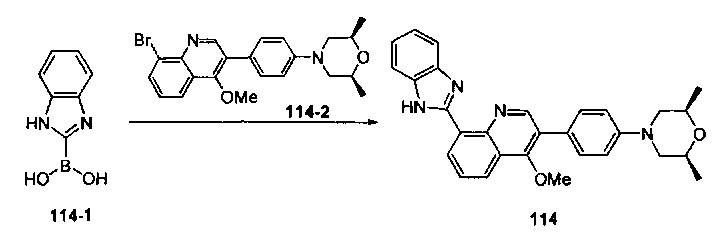
[291] Смесь соединения 114-1 (100 мг, 0,61 ммоль), Pd(dppf)Cl2 (23,1 мг, 0,061 ммоль), соединение 114-2 (263,5 мг, 0,61 ммоль) и Na2CO3 (161 мг, 1,53 ммоль) добавляли в диоксан (2 мл). В атмосфере газа азота полученную реакционную смесь нагревали до 120°С и перемешивали в течение 16 ч. Реакционную смесь выливали в H2O, экстрагировали с помощью EtOAc. Объединенную жидкость после экстракции промывали солевыми растворами, сушили над сульфатом натрия и концентрировали досуха, в конце очищали с помощью колоночной хроматографии (РЕ:EtOAc = 3:1) с получением титульного соединения (15 мг, выход 5,4%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 9,10-9,20 (m, 1H), 9,02 (s, 1Н), 8,37 (d, J=8,0 Гц, 1Н), 7,65-7,80 (m, 1Н), 7,62 (d, J=8,4 Гц, 2Н), 7,20-7,35 (m, 5Н), 7,08 (d, J=8,4 Гц, 1Н), 3,80-3,92 (m, 2Н), 3,74 (s, 3Н), 3,55-3,62 (m, 2Н), 2,50-2,60 (m, 2Н), 1,20-1,40 (m, 6Н). MS ESI рассч. для C29H28N4O2 [М+Н]+ 465, найденное значение 465.
Вариант осуществления 115
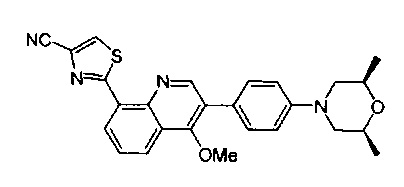
2-(3-(4-((2S,6R)-2,6-диметилморфолино)фенил)-4-метоксихинолин-8-ил)тиазол-4-карбонитрил
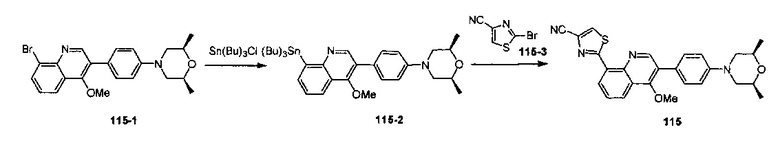
[292] Стадия 1. В атмосфере газа азота н-BuLi (0,22 мл, 0,552 ммоль) по каплям добавляли в раствор соединения 115-1 (200 мг, 0,46 ммоль) в THF (10 мл) при -65°С. После добавления реакционную смесь перемешивали при данной температуре в течение дополнительного 1 ч. Затем SnBu3Cl (150 мг, 0,46 ммоль) добавляли в раствор. Реакционную смесь перемешивали при -65°С в течение дополнительных 4 ч. После завершения реакции реакционную смесь гасили с помощью насыщенного раствора NH4Cl и экстрагировали с помощью EtOAc. Объединенную жидкость после экстракции промывали солевыми растворами, сушили над сульфатом натрия и концентрировали досуха, в конце очищали с помощью колоночной хроматографии (РЕ:EtOAc = 1:1) с получением соединения 115-2 (120 мг, выход 41%) в виде белого твердого вещества. MS ESI рассч. для C34H50N2O2Sn [М+Н]+ 639, найденное значение 639.
[293] Стадия 2. Соединение 115-2 (100 мг, 0,16 ммоль), соединение 115-3 (30 мг, 0,16 ммоль), Na2CO3 (43 мг, 0,4 ммоль) и Pd(dppf)Cl2 (10 мг, 0,016 ммоль) добавляли в диоксан (2 мл). В атмосфере газа азота реакционную смесь нагревали до 120°С и перемешивали в течение 16 ч. Реакционную смесь выливали в H2O, экстрагировали с помощью EtOAc. Объединенную жидкость после экстракции промывали солевыми растворами, сушили над сульфатом натрия и концентрировали досуха, в конце очищали с помощью колоночной хроматографии (РЕ:EtOAc = 1:1) с получением титульного соединения (25 мг, выход 34%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,95 (s, 1Н), 8,85 (d, J=2,0 Гц, 1H), 8,33 (d, J=2,0 Гц, 1H), 8,10 (s, 1H), 7,60-7,70 (m, 1H), 7,56 (d, J=8,4 Гц, 2H), 7,02 (d, J=8,4 Гц, 2H), 3,75-3,85 (m, 2H), 3,57 (s, 3H), 3,52-3,60 (m, 2H), 3,40-3,50 (m, 2H), 1,29 (s, 3H), 1,27 (s, 3H). MS ESI рассч. для C26H24N4O2S [M+H]+ 457, найденное значение 457.
Вариант осуществления 116
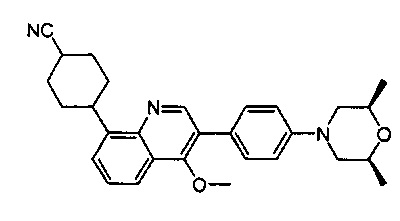
4-(3-(4-((2S,6R)-2,6-диметилморфолино)фенил)-4-метоксихинолин-8-ил)циклогексанкарбонитрил
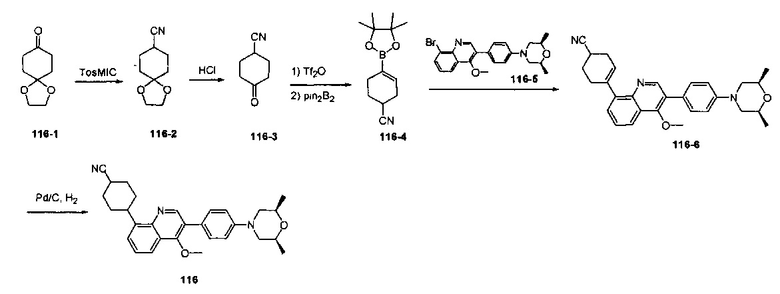
[294] Стадия 1. Трет-BuOK (18,5 г, 165,2 ммоль) добавляли в раствор соединения 116-1 (10 г, 64 ммоль) и TosMIC (17,6 г, 90,27 ммоль) в DME (10 мл) при 0°С. Реакционную смесь перемешивали при 0°С в течение 2 ч. После завершения реакции реакционную смесь гасили с помощью насыщенного раствора NH4Cl и экстрагировали с помощью EtOAc. Объединенную жидкость после экстракции промывали солевыми растворами, сушили над сульфатом натрия и концентрировали досуха, в конце очищали с помощью колоночной хроматографии (РЕ:EtOAc = 1:1) с получением 116-2 (6,7 г, выход 50%) в виде бесцветного масла.
[295] Стадия 2. Смесь соединения 116-2 (3 г, 18 ммоль) в конц. хлористоводородной кислоте / H2O (объем/объем = 1:1, 20 мл) перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь регулировали до рН=8 с помощью NaOH и экстрагировали с помощью EtOAc. Объединенную жидкость после экстракции промывали солевыми растворами, сушили над сульфатом натрия и концентрировали досуха, в конце очищали с помощью колоночной хроматографии (РЕ:EtOAc = 1:1) с получением соединения 116-3 (1,5 г, выход 68%) в виде бесцветного масла.
[296] Стадия 3. LiHMDS (9,5 мл, 9,5 ммоль) по каплям добавляли в раствор соединения 116-3 (1 г, 8 ммоль) в THF (20 мл) при 0°С. После добавления реакционную смесь перемешивали в течение дополнительных 30 мин. Затем PhN(SO2CF3) (3,6 г, 8,6 ммоль) добавляли в раствор. Полученную смесь перемешивали при 25°С в течение 6 ч., в конце гасили с помощью H2O и экстрагировали с помощью EtOAc. Объединенную жидкость после экстракции промывали солевыми растворами, сушили над сульфатом натрия и концентрировали досуха с получением сульфоната в виде желтого масла. Полученный сульфонат, Pd(dppf)Cl2 (80,4 мг, 0,11 ммоль), бис(пинаколато)дибор (0,67 г, 2,6 ммоль) и КОАс (0,65 г, 6,6 ммоль) добавляли в диоксан (15 мл), в атмосфере газа азота реакционную смесь нагревали до 100°С и перемешивали в течение 18 ч. Реакционную смесь выливали в H2O, экстрагировали с помощью EtOAc. Объединенную жидкость после экстракции промывали солевыми растворами, сушили над сульфатом натрия и концентрировали досуха, в конце очищали с помощью колоночной хроматографии (РЕ:EtOAc = 5:1) с получением соединения 116-4 (0,5 г, выход 28%) в виде белого твердого вещества.
[297] Стадия 4. Смесь Pd(dppf)Cl2 (180,8 мг, 0,23 ммоль), соединение 116-4 (500 мг, 2,1 ммоль), соединение 116-5 (1 г, 2,3 ммоль) и Na2CO3 (556 мг, 5,25 ммоль) добавляли в диоксан (20 мл). В атмосфере газа азота полученную суспензию нагревали до 80°С и перемешивали в течение 4 ч. Реакционную смесь выливали в H2O, экстрагировали с помощью EtOAc. Объединенную жидкость после экстракции промывали солевыми растворами, сушили над сульфатом натрия и концентрировали досуха, в конце очищали с помощью колоночной хроматографии (РЕ:EtOAc = 5:1) с получением соединения 116-6 (400 мг, выход 42%) в виде белого твердого вещества. MS ESI рассч. для C29H31N3O2 [М+Н]+ 454, найденное значение 454.
[298] Стадия 5. Pd/C (100 мг, 10%) и соединение 116-6 (100 мг, 0,22 ммоль) добавляли в EtOAc (20 мл). В атмосфере газа водорода, обеспеченной посредством баллона с водородом, реакционную смесь перемешивали при комнатной температуре в течение 50 ч. Реакционную смесь фильтровали, фильтрат концентрировали досуха, в конце очищали с помощью препаративной HPLC с получением титульного соединения (50 мг, выход 46%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,90 (s, 1Н), 8,14 (d, J=8,4 Гц, 1H), 7,50-7,65 (m, 6H), 7,06 (d, J=8,8 Гц, 2H), 4,00-4,15 (m, 1H), 3,80-3,95 (m, 2H), 3,69 (s, 3H), 3,55-3,65 (m, 2H), 3,15-3,20 (m, 1H), 2,45-2,55 (m, 2H), 1,90-2,25 (m, 7H), 1,55-1,65 (m, 1H), 1,33 (s, 3H), 1,31 (s, 3H). MS ESI рассч. для C29H33N3O2 [M+H]+ 456, найденное значение 456.
Вариант осуществления 117
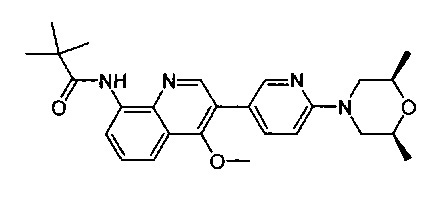
N-(3-(6-((2S,6R)-2,6-диметилморфолино)пиридин-3-ил)-4-метоксихинолин-8-ил)пиваламид
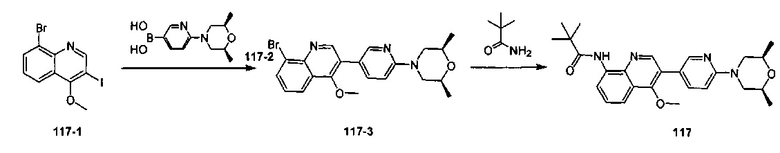
[299] Стадия 1. Смесь Na2CO3 (430 мг, 4 ммоль), соединение 117-1 (730 мг, 2 ммоль), соединение 117-2 (480 мг, 2 ммоль) и Pd(dppf)Cl2 (150 мг, 0,2 ммоль) добавляли в смешанный растворитель THF7DMF/H2O. Затем реакционную смесь нагревали до 80°С и перемешивали в течение 4 ч. Реакционную смесь выливали в H2O, экстрагировали с помощью EtOAc. Объединенную жидкость после экстракции сушили над сульфатом натрия и концентрировали досуха, наконец-то очищали с помощью колоночной хроматографии (РЕ:EtOAc = 5:1) с получением соединения 117-3 (430 мг, выход 50%) в виде коричневого твердого вещества. MS ESI рассч. для C21H22BrN3O2 [М+Н]+ 428, найденное значение 428.
[300] Стадия 2. Смесь соединения 117-3 (130 мг, 0,3 ммоль), триметилацетамида (60 мг, 0,6 ммоль), Cs2CO3 (190 мг, 0,6 ммоль), Pd2(dba)3 (30 мг, 0,03 ммоль) и Xantphos (36 мг, 0,06 ммоль) добавляли в диоксан (5 мл). В атмосфере газа азота реакционную смесь нагревали до 120°С и перемешивали в течение 4 ч. Реакционную смесь выливали в H2O, экстрагировали с помощью EtOAc. Объединенную жидкость после экстракции сушили над сульфатом натрия и концентрировали досуха, в конце очищали с помощью препаративной HPLC с получением титульного соединения (30 мг, выход 21%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 10,23 (brs, 1H), 8,77 (d, J=7,6 Гц, 1H), 8,74 (s, 1H), 8,47 (d, J=2,0 Гц, 1H), 7,90-7,95 (m, 2H), 7,69-7,80 (m, 1H), 6,78 (d, J=8,4 Гц, 1H), 4,05-4,10 (m, 2H), 3,35-3,46 (m, 5H), 2,26-2,85 (m, 2H), 1,42 (s, 9H), 1,31 (s, 9H), 1,29 (s, 3H). MS ESI рассч. для C26H32N4O3 [M+H]+ 449, найденное значение 449.
Вариант осуществления 118
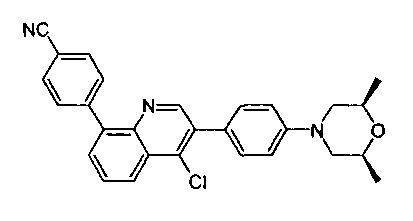
4-(4-хлор-3-(4-((2S,6R)-2,6-диметилморфолино)фенил)хинолин-8-ил)бензонитрил
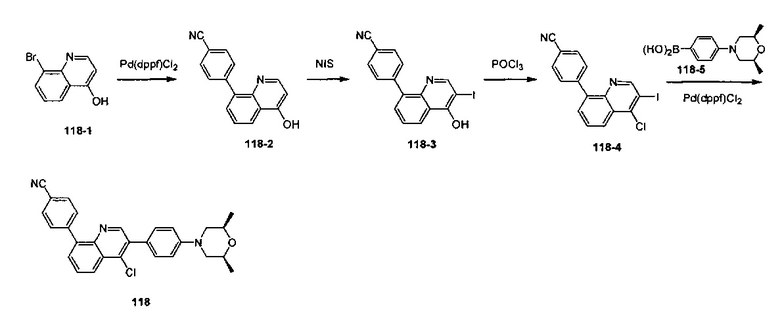
[301] Стадия 1. Pd(dppf)Cl2 (1,6 г, 0,1 ммоль), карбонат натрия (9,5 г, 89,23 ммоль) и DMF / H2O (100:20, 120 мл) добавляли в суспензию соединения 118-1 (10 г, 44,64 ммоль), 4-цианофенилбороновой кислоты (7,2 г, 49,11 ммоль). В атмосфере газа азота реакционную смесь перемешивали при 90°С в течение ночи. После завершения реакции, как обнаружено с помощью LC-MS, реакционную смесь выливали в H2O (500 мл). Продукт 2 получали после фильтрования в виде коричневого твердого вещества (10,8 г, 98%). MS ESI рассч. для C16H10N2O [М+Н]+ 247, найденное значение 247.
[302] Стадия 2. NIS (11,96 г, 53,17 ммоль) порциями добавляли в раствор соединения 118-2 (10,9 г, 44,31 ммоль) в DMF (110 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. После завершения реакции, как обнаружено с помощью LC-MS и TLC, реакционную смесь выливали в H2O (500 мл). Продукт получали после фильтрования в виде коричневого остатка (14 г и 85%). MS ESI рассч. для C16H9IN2O [М+Н]+ 373, найденное значение 373.
[303] Стадия 3. Соединение 118-3 (5 г, 13,4 ммоль) добавляли в оксихлорид фосфора (20 мл). Реакционную смесь перемешивали при температуре флегмы в течение 3 ч. После завершения реакции, как обнаружено с помощью LC-MS, реакционную смесь выливали в H2O (500 мл). Смесь фильтровали и сушили под вакуумом с получением соединения 4 (5,2 г, 99%) в виде коричневого твердого вещества. MS ESI рассч. для C16H8ClIN2 [М+Н]+ 391, найденное значение 391.
[304] Стадия 4. Соединение 118-1 (200 мг, 0,5 ммоль), соединение 118-5 (176 мг, 0,75 ммоль) добавляли в суспензию Pd(dppf)Cl2 (18 мг, 0,03 ммоль), карбоната натрия (106 мг, 1,0 ммоль) в THF/H2O (10:1, 11 мл), затем в атмосфере газа азота реакционную смесь перемешивали при 90°С в течение ночи. Реакцию завершали, как обнаружено с помощью LC-MS. Смесь очищали с помощью препаративной HPLC с получением необходимого продукта в виде коричневого твердого вещества (40 мг, 33%). 1H ЯМР (400 МГц, CDCl3) δ 8,84 (s, 1H), 8,38-8,49 (m, 1H), 7,37-7,62 (m, 6H), 7,54 (d, 2H, J=8,0 Гц), 7,25 (d, 2Н, J=8,4 Гц), 3,97 (t, 2Н, J=7,6 Гц), 3,61 (d, 2Н, J=11,6 Гц), 2,67 (t, 2Н, J=11,6 Гц), 1,28 (d, 6Н, J=6,4 Гц). MS ESI рассч. для C28H24ClN3O [М+Н]+ 454, найденное значение 454.
Вариант осуществления 119
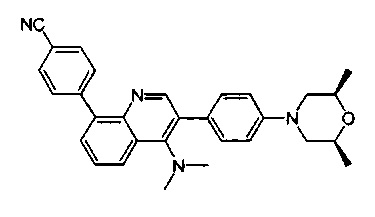
4-(4-(диметиламино)-3-(4-((2S,6R)-2,6-диметилморфолино)фенил)хинолин-8-ил)бензонитрил
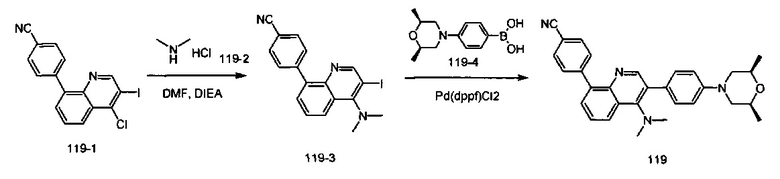
[305] Стадия 1. Соединение 119-2 (1,1 г, 13 ммоль) добавляли в раствор соединения 119-1 (500 мг, 1,3 ммоль) в DMF (5 мл). Добавляли DIEA (5 г, 39 ммоль). Реакционную смесь перемешивали при 120°С в течение 10 ч. Затем реакционную смесь выливали в H2O (50 мл) и экстрагировали с помощью EtOAc. Органическую фазу сушили над сульфатом натрия и концентрировали при пониженном давлении с получением необходимого соединения в виде белого твердого вещества (100 мг, 20%). MS ESI рассч. для C18H14IN3 [М+Н]+ 400, найденное значение 400.
[306] Стадия 2. В атмосфере газа азота смесь соединения 119-3 (18 мг, 0,03 ммоль), карбоната натрия (100 мг, 0,25 ммоль), соединения 119-4 (88 мг, 0,37 ммоль), Pd(dppf)Cl2 (53 мг, 0,5 ммоль) в THF/H2O (11 мл, 10:1) перемешивали при 90°С в течение ночи. После завершения реакции, как обнаружено с помощью LC-MS, реакционную смесь очищали с помощью препаративной HPLC с получением титульного соединения (20 мг, 20%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,37 (s, 1H), 8,26 (t, 1H, J=4,8 Гц), 7,80 (d, 2H, J=6,8 Гц), 7,74 (d, 2H, J=4,4 Гц), 7,59 (d, 2H, J=7,2 Гц), 7,19 (d, 2H, J=7,2 Гц), 7,05 (d, 2H, J=8,0 Гц), 3,85 (t, 2H, J=7,2 Гц), 3,12 (s, 6H), 2,53 (t, 2H, J=11,2 Гц), 1,28 (d, 6H, J=10,0 Гц). MS ESI рассч. для C30H30N4O [M+H]+ 463, найденное значение 463.
Вариант осуществления 120
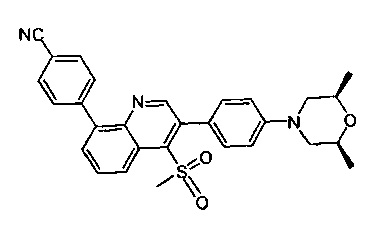
4-(3-(4-((2S,6R)-2,6-диметилморфолино)фенил)-4-(метилсульфонил)хинолин-8-ил)бензонитрил
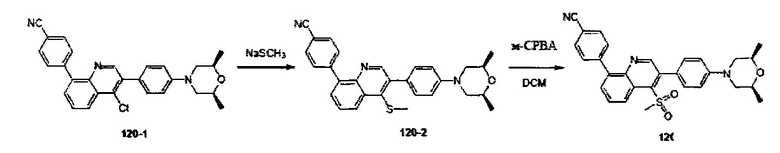
[307] Стадия 1. NaSCH3 (31 мг, 0,44 ммоль) добавляли в раствор соединения 120-1 (200 мг, 0,44 ммоль) в DMF (5 мл). Затем реакционную смесь перемешивали при 60°С в течение 3 ч. После завершения реакции, как обнаружено с помощью LC-MS, реакционную смесь выливали в H2O и экстрагировали с помощью EtOAc. Органическую фазу сушили над сульфатом натрия и концентрировали под вакуумом с получением необходимого соединения (200 мг, 97,6%) в виде белого твердого вещества. MS ESI рассч. для C29H27N3OS [М+Н]+ 466, найденное значение 466.
[308] Стадия 2. м-хлорпербензойную кислоту (186 мг, 1,08 ммоль, 80%) добавляли в раствор соединения 120-2 (200 мг, 0,43 ммоль) в DCM (20 мл). Затем реакционную смесь перемешивали при температуре флегмы в течение ночи. После завершения реакции, как обнаружено с помощью LC-MS, реакционную смесь выливали в H2O и экстрагировали с помощью DCM. Органическую фазу сушили над сульфатом натрия и концентрировали под вакуумом, очищали с помощью препаративной HPLC с получением необходимого продукта (120 мг, 55%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 9,36 (d, 1Н, J=8,0 Гц), 8,77 (s, 1H), 7,99 (d, 2Н, J=8,4 Гц), 7,68-7,88 (m, 6Н), 7,62 (d, 2Н, J=8,4 Гц), 4,67 (s, 2Н), 4,19 (d, 2Н, J=14,0 Гц), 3,48 (dd, 2Н, J=10,0 Гц, 22,0 Гц), 3,06 (s, 3Н), 1,22-1,42 (m, 6Н). MS ESI рассч. для C29H27N3O3S [М+Н]+ 498, найденное значение 498.
Вариант осуществления 121
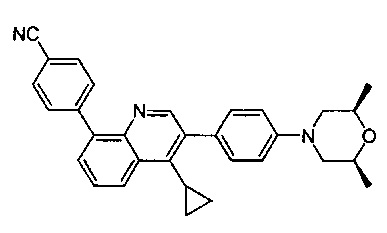
4-(4-циклопропил-3-(4-((2S,6R)-2,6-диметилморфолино)фенил)хинолин-8-ил)бензонитрил
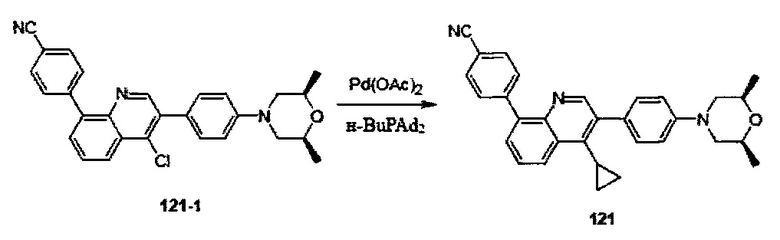
[309] В атмосфере газа азота смесь соединения 121-1 (100 мг, 0,22 ммоль), ацетата палладия (4,5 мг, 0,02 ммоль), H-BuPAd2 (4,7 мг, 0,01 ммоль), карбоната цезия (143 мг, 0,44 ммоль) в толуоле/H2O (10:1, 11 мл) перемешивали при 120°С в течение ночи. После завершения реакции, как обнаружено с помощью LC-MS, реакционную смесь очищали с помощью препаративной HPLC с получением необходимого продукта (50 мг, 42%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,80 (s, 1Н), 8,70 (d, 1Н, J=8,4 Гц), 7,77 (t, 2Н, J=8,4 Гц), 7,65-7,75 (m, 3Н), 7,50 (d, 2Н, J=8,0 Гц), 7,38 (d, 2Н, J=8,4 Гц), 7,04-7,13 (m, 2Н), 3,85 (dd, 2Н, J=6,0 Гц, J=8,0 Гц), 3,50 (d, 2Н, J=11,2 Гц), 2,56 (t, 2Н, J=11,2 Гц), 2,34 (t, 1Н, J=6,4 Гц), 1,18 (d, 6Н, J=6,0 Гц), 1,08 (d, 2Н, J=8,0 Гц), 0,33 (d, 2Н, J=5,6 Гц). MS ESI рассч. для C31H29N3O [М+Н]+ 460, найденное значение 460.
[310] Соединения, перечисленные в таблице 8, синтезировали с помощью соединения 121-1 и соответствующих борных кислот.
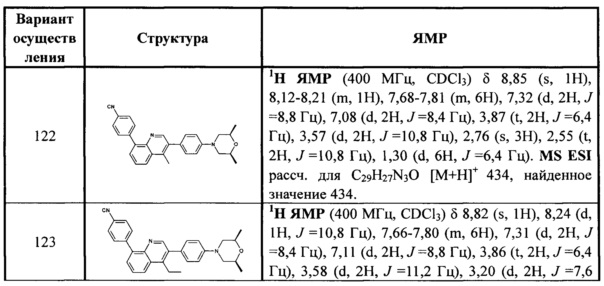

Вариант осуществления 126
4-(4-(азетидин-1-ил)-3-(4-((2S,6R)-2,6-диметилморфолино)фенил)хинолин-8-ил)бензонитрил
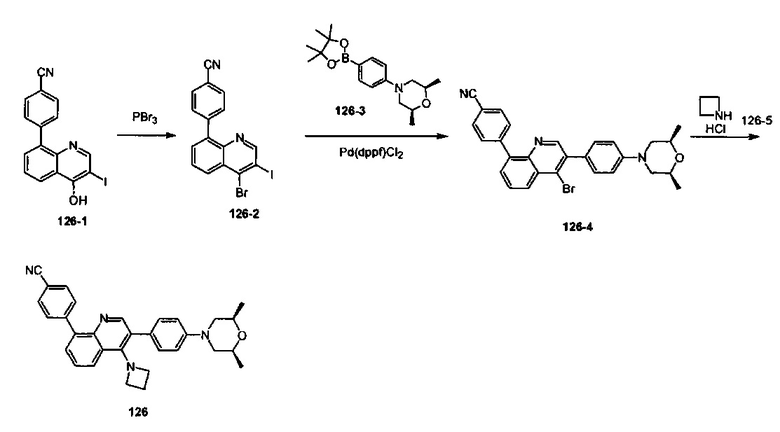
[311] Стадия 1. Раствор PBr3 (26,0 г, 97 ммоль) по каплям добавляли в раствор соединения 126-1 (24,0 г, 65 ммоль) в DMF (320 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч., а затем фильтровали с получением продукта 126-5 (26,0 г, выход 93%) в виде белого твердого вещества. MS ESI рассч. для C16H8BrIN2 [М+Н]+ 434, найденное значение 434.
[312] Стадия 2. К раствору соединения 126-2 (11 г, 25 ммоль), соединения 126-3 (9 г, 28 ммоль) и карбоната натрия (5,3 г, 50 ммоль) в THF/H2O (5:1, 180 мл) добавляли Pd(dppf)Cl2 (1,83 г, 2,5 ммоль). Реакционную смесь перемешивали при 66°С в течение 12 ч. Реакцию завершали, как обнаружено с помощью LCMS. Затем реакционную смесь выливали в H2O, экстрагировали с помощью EtOAc, сушили над безводным сульфатом натрия. Неочищенный продукт 126- очищали с помощью колоночной хроматографии с получением соединения 126-4 (9,3 г, выход 74%) в виде желтого твердого вещества. MS ESI рассч. для C28H24BrN3O [М+Н]+ 498, найденное значение 498.
[313] Стадия 3. К раствору соединения 126-4 (100 мг, 0,2 ммоль), соединения 126-5 (37,6 мг, 0,4 ммоль) и трет-бутоксида калия (112 мг, 1 ммоль) в PhCH3 (5 мл) добавляли Pd2(dba)3 (18,3 мг, 0,02 ммоль) и Xantphos (19,1 мг, 0,04 ммоль). Реакционную смесь нагревали до температуры флегмы в течение 12 ч. Реакцию завершали, как обнаружено с помощью LCMS. Затем реакционную смесь выливали в H2O, экстрагировали с помощью эфира (3×3 мл), органическую фазу сушили над безводным сульфатом натрия. После концентрирования остаток очищали с помощью препаративной HPLC с получением титульного соединения (выход 21%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,30 (brs, 1H), 8,00 (d, 1H, J=8,0 Гц), 7,70-7,80 (m, 3Н), 7,50-7,65 (m, 5H), 6,92 (d, 2H, J=5,6 Гц), 3,79 (t, 3H, J=8,0 Гц), 3,45-3,53 (m, 4H), 2,30-2,50 (m, 5H), 1,27 (d, 6H, J=6,0 Гц). MS ESI рассч. для C31H30N4O [M+H]+ 475, найденное значение 475.
Вариант осуществления 127

4-(3-(4-((2S,6R)-2,6-диметилморфолино)фенил)хинолин-8-ил)бензонитрил

[314] н-BuLi (0,5 ммоль, 0,2 мл) добавляли в раствор соединения 127-1 (150 мг, 0,3 ммоль) в THF при -70°С. Реакционную смесь выливали в H2O. Водную фазу экстрагировали с помощью EtOAc. Объединенную органическую фазу промывали насыщенными солевыми растворами, сушили над безводным сульфатом натрия, концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью препаративной HPLC с получением продукта (50 мг, выход 50%) в виде белого твердого вещества. MS ESI рассч. для C28H25N3O [М+Н]+ 420, найденное значение 420.
Вариант осуществления 128
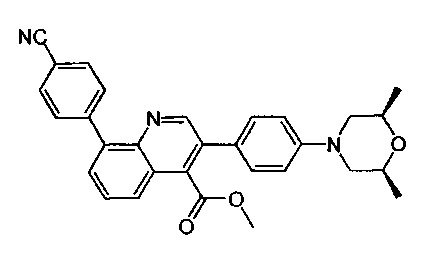
метил-8-(4-цианофенил)-3-(4-((2S,6R)-2,6-диметилморфолино)фенил)хинолин-4-карбоксилат

[315] Pd(PPh3)Cl2 (21 мг, 0,03 ммоль) и TEA (303 мг, 3 ммоль) добавляли в раствор соединения 128-1 (150 мг, 0,3 ммоль) в метаноле/DMF/THF(25/5/5 мл), в атмосфере газа СО с давлением 50 фунтов/кв. дюйм реакционную смесь перемешивали при 70°С в течение 48 ч. После завершения реакции, как обнаружено с помощью LC-MS, неочищенный продукт очищали с помощью препаративной HPLC с получением титульного соединения в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 9,00 (s, 1H), 7,95 (d, 1H, J=7,6 Гц), 7,68-7,85 (m, 6H), 7,50 (d, 2H, J=8,8 Гц), 7,27 (d, 2H, J=8,8 Гц), 3,98 (d, 2H, J=6,4 Гц), 3,86 (s, 3H), 3,62 (d, 2H, J=11,6 Гц), 2,62-2,73 (m, 2H), 1,30 (d, 6H, J=6,0 Гц). MS ESI рассч. для C30H27N3O3 [M+H]+ 478, найденное значение 478.
Вариант осуществления 129
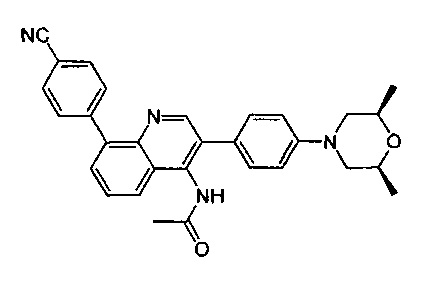
N-(8-(4-цианофенил)-3-(4-((2S,6R)-2,6-диметилморфолино)фенил)хинолин-4-ил)ацетамид

[316] Карбонат цезия (100 мг, 0,3 ммоль), Pd2dba3 (9 мг, 0,01 ммоль) и Xantphos (12 мг, 0,02 ммоль) добавляли в раствор соединения 129-1 (50 мг, 0,1 ммоль) и NH2Ac (30 мг, 0,5 ммоль) в диоксане (5 мл). Реакционную смесь нагревали до температуры флегмы в течение 12 ч. После завершения реакции, как обнаружено с помощью LC-MS, реакционную смесь выливали в H2O, экстрагировали с помощью EtOAc, сушили над безводным сульфатом натрия и растворяли в DMF. Неочищенный продукт очищали с помощью препаративной HPLC с получением титульного соединения (21 мг, выход 44%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,93 (s, 1H), 8,00 (d, 1H, J=8,4 Гц), 7,65-7,80 (m, 6Н), 7,37 (d, 2Н, J=8,8 Гц), 7,10 (d, 2Н, J=8,8 Гц), 3,87 (t, 2Н, J=7,6 Гц), 3,58 (d, 2Н, J=11,6 Гц), 3,56 (t, 2Н, J=11,6 Гц), 2,22 (brs, 2Н), 1,93 (s, 3Н), 1,18 (d, 6Н, J=6,0 Гц). MS ESI рассч. для C30H28N4O2 [М+Н]+ 477 найденное значение 477.
[317] Соединения, перечисленные в таблице 9, синтезировали с помощью соединения 129-1 и соответствующих амидов.
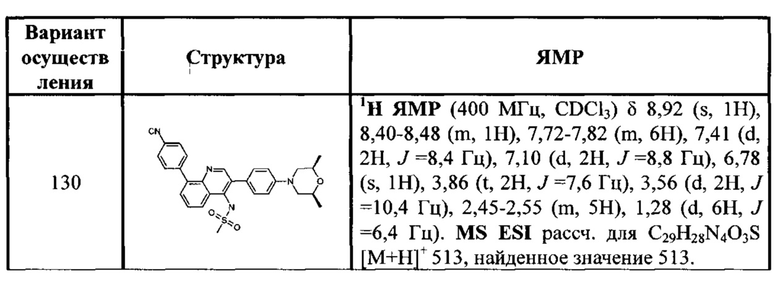
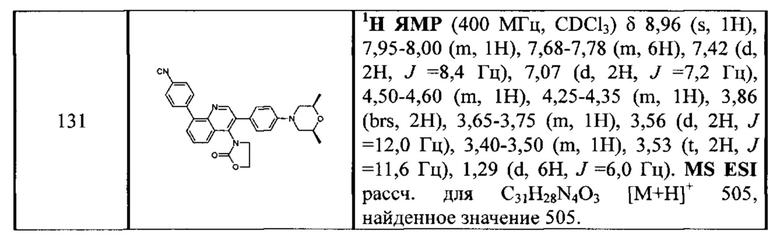
Вариант осуществления 132
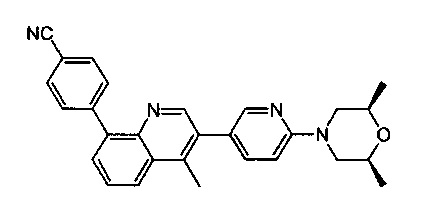
4-(3-(6-((2S,6R)-2,6-диметилморфолино)пиридин-3-ил)-4-метилхинолин-8-ил)бензонитрил
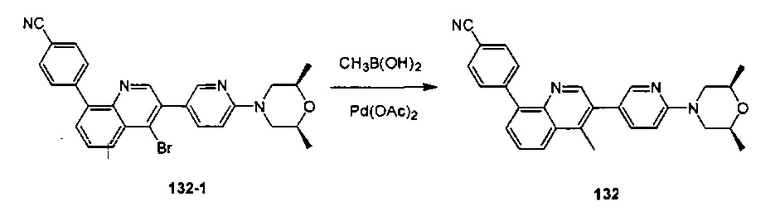
[318] Смесь н-BuPAd2 (20 мг, 0,04 ммоль), ацетата палладия (5 мг, 0,02 ммоль) и карбоната цезия (130 мг, 0,4 ммоль) добавляли в раствор соединения 132-1 (100 мг, 0,2 ммоль) и СН3В(ОН)2 (120 мг, 2 ммоль) в толуоле/H2O (3 мл/1 мл), затем реакционную смесь перемешивали при 110°С в течение ночи. После выливания в H2O смесь экстрагировали с помощью EtOAc, сушили над сульфатом натрия, концентрировали с получением неочищенного продукта, который очищали с помощью препаративной HPLC с получением титульного соединения (30 мг, выход 30%) в виде желтого твердого вещества. MS ESI рассч. для C28H26N4O [М+Н]+ 435 найденное значение 435.
[319] Соединения, перечисленные в таблице 10, синтезировали с помощью соединения 132-1 и соответствующих борных кислот.
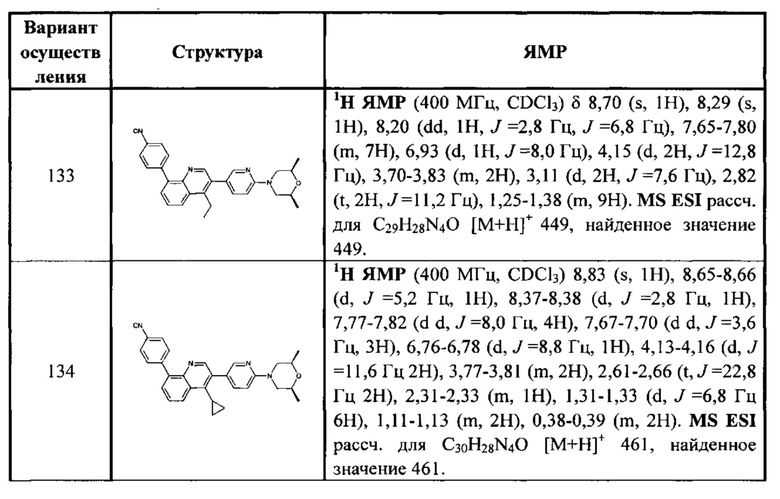
Вариант осуществления 135
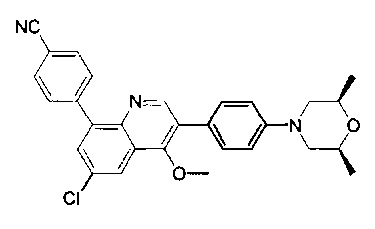
4-(6-хлор-3-(4-((2S,6R)-2,6-диметилморфолино)фенил)-4-метоксихинолин-8-ил)бензонитрил
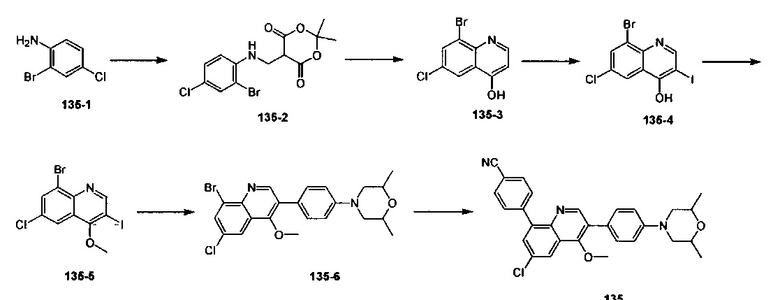
[320] Стадия 1. Смесь 2,2-диметил-1,3-диоксан-4,6-диона (19,2 г, 133 ммоль) и триметоксиметана (70 мл, 680 ммоль) нагревали с обратным холодильником в течение 2,5 ч., затем соединение 135-1 (20 г, 97 ммоль) добавляли в раствор при 100°С и перемешивали в течение ночи, твердое вещество собирали после фильтрования с получением соединения 135-2 (35,6 г, выход 98%) в виде желтого твердого вещества. MS ESI рассч. для C13H13BrClNO4 [М+Н]+ 362, найденное значение 362.
[321] Стадия 2. Смесь соединения 135-2 (35,6 г, 100 ммоль) и соединения 135-Ph2O (170 г, 1 моль) нагревали до 250°С в течение 0,5 ч., затем охлаждали до комнатной температуры, твердое вещество собирали после фильтрования с получением продукта в виде коричневого твердого вещества (12,2 г, выход 48%). MS ESI рассч. для C9H5BrClNO [М+Н]+ 258, найденное значение 258.
[322] Стадия 3. К раствору соединения 135-3 (3,56 г, 14 ммоль) и NIS (3,1 г, 14 ммоль) в DMF (10 мл) смесь перемешивали при комнатной температуре в течение 2 ч., твердое вещество собирали после фильтрования с получением 4,2 г продукта в виде желтого твердого вещества (выход 80%). MS ESI рассч. для C9H4BrClINO [М+Н]+ 384, найденное значение 384.
[323] Стадия 4. MeI (2,0 г, 14 ммоль) и Ag2CO3 (6,6 г, 24 ммоль) добавляли в раствор соединения 135-4 (4,5 г, 12 ммоль) в DMF (20 мл), затем смесь перемешивали при 70°С в течение 2 ч., после фильтрования смесь экстрагировали с помощью EtOAc, сушили над сульфатом натрия, концентрировали с получением 4,8 г неочищенного продукта, который очищали с помощью колоночной хроматографии с получением 2,2 г продукта в виде красного твердого вещества (выход 47%). MS ESI рассч. для C10H6BrClINO [М+Н]+ 397, найденное значение 397.
[324] Стадия 5. Pd(dppf)Cl2 (40 мг, 0,05 ммоль) и карбонат натрия (107 мг, 1,0 ммоль) добавляли в раствор соединения 135-5 (200 мг, 0,5 ммоль) и 2,6-диметил-4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)морфолин а (160 мг, 0,5 ммоль) в THF/H2O (6 мл, 5:1). Реакционную смесь перемешивали при 70°С в течение ночи, затем выливали в H2O, смесь экстрагировали с помощью EtOAc, сушили над сульфатом натрия, концентрировали с получением соединения 135-6 в виде желтого твердого вещества (200 мг, выход 40%). MS ESI рассч. для C22H22BrClN2O2 [М+Н]+ 461, найденное значение 461.
[325] Стадия 6. Pd(dppf)Cl2 (37 мг, 0,05 ммоль) и карбонат натрия (92 мг, 0,86 ммоль) добавляли в раствор соединения 135-6 (200 мг, 0,43 ммоль) и 4-цианофенилбороновой кислоты (63,7 мг, 0,43 ммоль) в THF/H2O (6 мл, 5:1). После перемешивания при 70°С в течение 3 ч. смесь выливали в H2O, экстрагировали с помощью EtOAc, сушили над сульфатом натрия, концентрировали с получением неочищенного продукта, который очищали с помощью препаративной HPLC с получением титульного соединения (55 мг, выход 26%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,84 (s, 1H), 8,27 (d, 1H, J=2,4 Гц), 7,78 (dd, 4H, J=8,4 Гц, J=10,8 Гц), 7,63 (d, 1H, J=2,0 Гц), 7,54 (d, 2H, J=8,8 Гц), 7,02 (d, 2H, J=8,4 Гц), 3,75-3,85 (m, 2H), 3,68 (s, 3H), 3,56 (d, 2H, J=10,8 Гц), 2,50 (t, 2H, J=11,2 Гц), 1,28 (d, 6H, J=6,4 Гц). MS ESI рассч. для C29H26ClN3O2 [M+H]+ 484, найденное значение 484.
Вариант осуществления 136

4-(3-(4-(2,6-диметилморфолино)фенил)-6-фтор-4-метоксихинолин-8-ил)бензонитрил
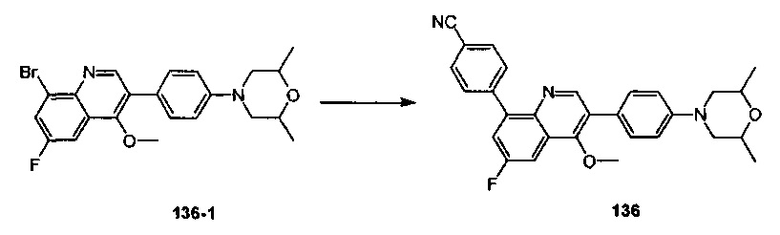
[326] Pd(dppf)Cl2 (33 мг, 0,045 ммоль) и карбонат натрия (96 мг, 0,9 ммоль) добавляли в раствор соединения 136-1 (200 мг, 0,45 ммоль) и 4-цианофенилбороновой кислоты (66 мг, 0,45 ммоль) в THF/H2O (6 мл, 5:1). Реакционную смесь перемешивали при 70°С в течение ночи, затем выливали в H2O, экстрагировали с помощью EtOAc, сушили над сульфатом натрия, концентрировали с получением неочищенного продукта, который очищали с помощью препаративной HPLC с получением титульного соединения (45 мг, выход 21,4%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,82 (s, 1Н), 7,65-7,70 (m, 1H), 7,70-7,80 (m, 4Н), 7,50-7,60 (m, 2Н), 7,47 (dd, 1Н, J=2,8 Гц, J=8,8 Гц), 7,03 (brs, 2Н), 3,85 (brs, 2Н), 3,68 (s, 3Н), 3,56 (d, 2Н, J=10,8 Гц), 2,53 (t, 2Н, J=10,4 Гц), 1,29 (d, 6Н, J=6,4 Гц). MS ESI рассч. для C29H26FN3O2 [М+Н]+ 468 найденное значение 468.
[327] Соединения, перечисленные в таблице 11, синтезировали с помощью соединения 136-1 и соответствующих борных кислот.
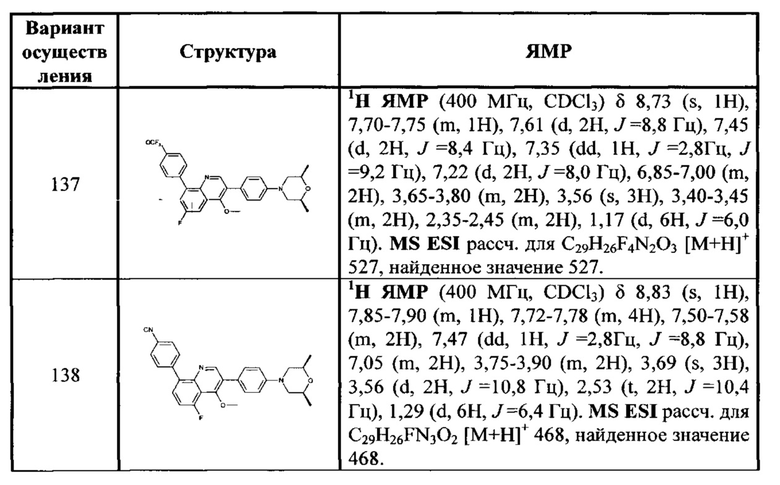
Вариант осуществления 139
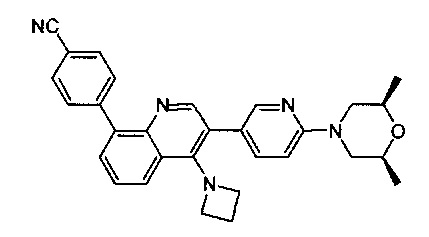
4-(4-(азетидин-1-ил)-3-(6-((2S,6R)-2,6-диметилморфолино)пиридин-3-ил)хинолин-8-ил)бензонитрил
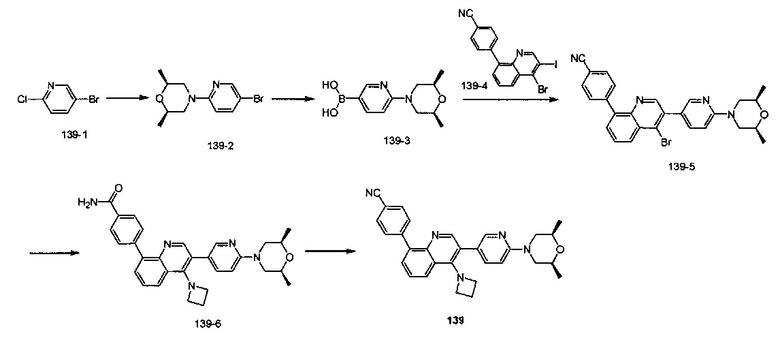
[328] Стадия 1. 2,6-диметилморфолин (18 г, 0,15 моль) и К2СО3 (27,6 г, 0,2 моль) добавляли в раствор соединения 139-1 (19,2 г, 0,1 моль) в DMF (200 мл), реакционную смесь перемешивали при 120°С в течение 4 ч. После выливания в H2O реакционную смесь экстрагировали с помощью EtOAc, сушили над сульфатом натрия, концентрировали с получением неочищенного продукта, который очищали с помощью колоночной хроматографии (EtOAc:РЕ = от 1:5 до 1:1) с получением соединения 139-2 (15,4 г, выход 57%) в виде желтого твердого вещества. MS ESI рассч. для C11H15BrN2O [М+Н]+ 271, найденное значение 271.
[329] Стадия 2. Pd(dppf)Cl2 (732 мг, 1 ммоль) и АсОК (4,0 г, 40 ммоль) добавляли в раствор соединения 139-2 (5,4 г, 20 ммоль) и бис(пинаколато)дибора (7,6 г, 30 ммоль) в 1,4-диоксане (100 мл), смесь перемешивали при 120°С в течение 6 ч. Затем растворитель удаляли, остаток очищали с помощью колоночной хроматографии (EtOAc:РЕ = от 1:10 до 1:2) с получением соединения 139-3 (5,0 г, выход 98%) в виде белого твердого вещества. MS ESI рассч. для C11H17BN2O3 [М+Н]+ 237, найденное значение 237.
[330] Стадия 3. Смесь карбоната натрия (214 мг, 4 ммоль) добавляли в раствор соединения 139-3 (236 мг, 0,7 ммоль) и соединения 139-4 (300 мг, 2 ммоль) в DMF/H2O/THF (14:1:5 мл) и смесь перемешивали при -80°С в течение 2 ч. После выливания в H2O реакционную смесь экстрагировали с помощью EtOAc, сушили над сульфатом натрия, концентрировали с получением неочищенного продукта, который очищали с помощью колоночной хроматографии (РЕ:EtOAc = 5:1) с получением соединения 139-5 (200 мг, выход 57%) в виде белого твердого вещества. MS ESI рассч. для C27H23BrN4O [М+Н]+ 499, найденное значение 499.
[331] Стадия 4. Xantphos (350 мг, 0,6 ммоль), Pd2(dba)3 (275 мг, 0,3 ммоль) и трет-дутоксид калия (1,3 г, 12 ммоль) добавляли в раствор соединения 139-2 (1,5 г, 3 ммоль) и азетидина (560 мг, 6 ммоль) в толуоле (20 мл), реакционную смесь перемешивали при 110°С в течение 3 ч. После выливания в H2O реакционную смесь экстрагировали с помощью EtOAc, сушили над сульфатом натрия, концентрировали с получением неочищенного продукта, который очищали с помощью колоночной хроматографии с получением необходимого продукта, соединения 139-6, в виде желтого твердого вещества (500 мг, выход 30%). MS ESI рассч. для C30H31N5O2 [М+Н]+ 494, найденное значение 494.
[332] Стадия 5. TFAA (4 мл) и DIPEA (4 мл) добавляли в раствор соединения 139-6 (1,5 г, 3 ммоль) в DCM (20 мл), реакционную смесь перемешивали при комнатной температуре в течение 30 мин., затем выливали в H2O, экстрагировали с помощью EtOAc, сушили над сульфатом натрия, концентрировали с получением неочищенного продукта, который перемещали для очистки с помощью препаративной HPLC с получением титульного соединения в виде желтого твердого вещества (500 мг, выход 30%). 1Н ЯМР (400 МГц, CDCl3) δ 8,29 (s, 1Н), 8,19 (s, 1Н), 8,05 (d, 1Н, J=8,0 Гц), 7,93 (brs, 1Н), 7,80 (d, 2Н, J=7,6 Гц), 7,50-7,70 (m, 4Н), 6,98 (brs, 1Н), 4,70 (brs, 3Н), 4,17 (d, 3Н, J=12,4 Гц), 3,65-3,80 (m, 2Н), 2,78 (t, 2Н, J=11,6 Гц), 2,47 (brs, 2Н), 1,30 (d, 6Н, J=6,0 Гц). MS ESI рассч. для C30H29N5O [М+Н]+ 476 найденное значение 476.
Вариант осуществления 140
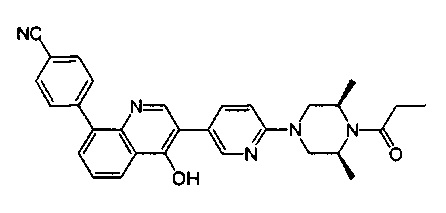
4-(3-(6-((3S,5R)-3,5-диметил-4-пропионилпиперазин-1-ил)пиридин-3-ил)-4-гидроксихинолин-8-ил)бензонитрил
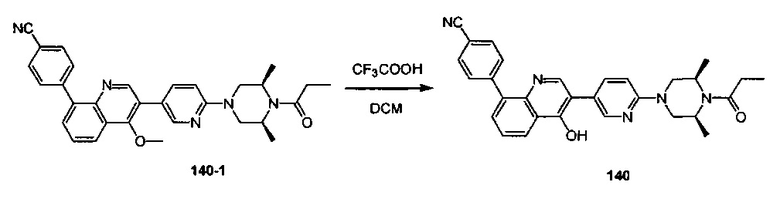
[333] CF3COOH (114 мг, 1 ммоль) добавляли в раствор соединения 140-1 (100 мг, 0,2 ммоль) в DCM (5 мл). Реакционную смесь перемешивали при 50°С в течение 2 ч., реакцию завершали, как обнаружено с помощью LC-MS. Реакционную смесь выливали в H2O, экстрагировали с помощью EtOAc (30×3 мл), органическую фазу сушили над сульфатом натрия. После концентрирования остаток очищали с помощью препаративной HPLC с получением титульного соединения (30 мг) - продукта в виде белого твердого вещества. MS ESI рассч. для C30H29N5O2 [М+Н]+ 492 найденное значение 492.
Вариант осуществления 141
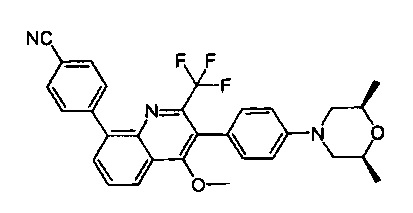
4-(3-(4-((2S,6R)-2,6-диметилморфолино)фенил)-4-метокси-2-(трифторметил)хинолин-8-ил)бензонитрил
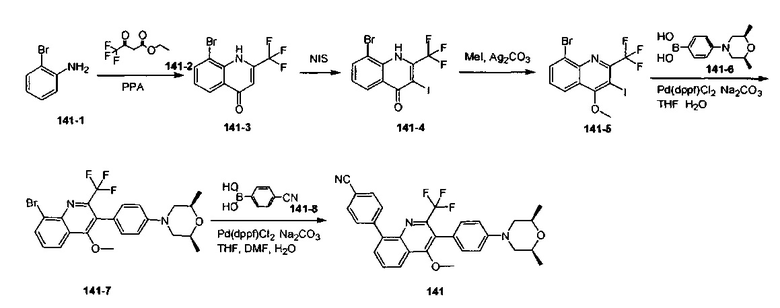
[334] Стадия 1. Соединение 141-1 (10 г, 58,5 ммоль) по каплям добавляли в смесь соединения 140-2 (10,7 г, 58,5 ммоль) и РРА (40 г) при 100°С, период добавления составлял не менее 15 мин. После добавления реакционную смесь нагревали до 150°С и перемешивали в течение 12 ч. После охлаждения реакционную смесь разбавляли с помощью гидроксида натрия (160 мл, 10 вес. %). Полученный осадок фильтровали, фильтрат окисляли с помощью конц. хлористоводородной кислоты. Полученный осадок фильтровали, рекристаллизовали с помощью этанола с получением соединения 141-3 (10,5 г, выход 62%) в виде белого твердого вещества. MS ESI рассч. для C10H5BrF3NO [М+Н]+ 292, найденное значение 292.
[335] Стадия 2. NIS (8,1 г, 36 ммоль) порциями добавляли в раствор соединения 141-3 (10,5 г, 36 ммоль) в DMF (100 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Затем реакционную смесь выливали в H2O (1 л) и фильтровали, отфильтрованный осадок растворяли в EtOAc, сушили над сульфатом натрия, концентрировали с получением соединения 141-4 (14,3 г, выход 95%) в виде желтого твердого вещества. MS ESI рассч. для C10H4BrF3INO [М+Н]+ 418, найденное значение 418.
[336] Стадия 3. MeI (0,22 мл, 3,6 ммоль) по каплям добавляли в суспензию соединения 141-4 (1 г, 2,4 ммоль) и Ag2CO3 (1,5 г, 4,8 ммоль) в толуоле (10 мл). Затем реакционную смесь перемешивали при 70°С в течение 2 ч. Реакционную смесь выливали в H2O, экстрагировали с помощью EtOAc. Объединенную жидкость после экстракции промывали солевыми растворами, сушили над Na2SO4 и концентрировали досуха, в конце очищали с помощью колоночной хроматографии (РЕ:EtOAc = 10:1) с получением соединения 141-5 (550 мг, выход 55%) в виде желто-коричневого твердого вещества. MS ESI рассч. для C11H6BrF3INO [М+Н]+ 432, найденное значение 432.
[337] Стадия 4. Смесь соединения 141-5 (550 мг, 1,27 ммоль), соединения 140-6 (299 мг, 1,27 ммоль), Pd(dppf)Cl2 (93 мг, 0,127 ммоль) и Na2CO3 (269 мг, 2,54 ммоль) добавляли в перемешанный растворитель THF (5 мл) и H2O (1 мл), в атмосфере газа азота реакционную смесь нагревали до 60°C и перемешивали в течение ночи. Реакционную смесь выливали в H2O, экстрагировали с помощью EtOAc. Объединенную жидкость после экстракции промывали солевыми растворами, сушили над Na2SO4 и концентрировали досуха с получением соединения 141-7 (170 мг, выход 27%) в виде желто-коричневого твердого вещества. MS ESI рассч. для C23H22BrF3N2O2 [М+Н]+ 495, найденное значение 495.
[338] Стадия 5. Смесь соединения 141-7 (170 мг, 0,34 ммоль), соединения 140-8 (60,6 мг, 0,41 ммоль), Pd(dppf)Cl2 (25 мг, 0,034 ммоль) и Na2CO3 (72 мг, 0,68 ммоль) добавляли в перемешанный растворитель THF (10 мл), DMF (2 мл) и H2O (2 мл), в атмосфере газа азота реакционную смесь нагревали до 110°C и перемешивали в течение ночи. Органическую фазу отделяли и промывали солевыми растворами, сушили над Na2SO4 и концентрировали досуха, в конце очищали с помощью препаративной HPLC с получением титульного соединения (60 мг, выход 34%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,26 (d, J=8,0 Гц, 1Н), 7,82-7,95 (m, 3Н), 7,70-7,80 (m, 3Н), 7,35-7,50 (m, 4Н), 3,95-4,20 (m, 3Н), 3,65-3,80 (m, 2Н), 3,63 (s, 3Н), 2,76-2,90 (m, 2Н), 1,33 (s, 3Н), 1,32 (s, 3Н). MS ESI рассч. для C30H26F3N3O2 [М+Н]+ 518, найденное значение 518.
Вариант осуществления 142
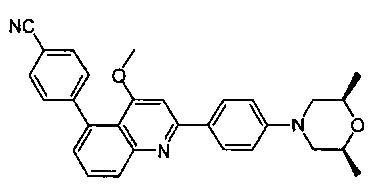
4-(2-(4-((2R,6S)-2,6-диметилморфолино)фенил)-4-метоксихинолин-5-ил)бензонитрил
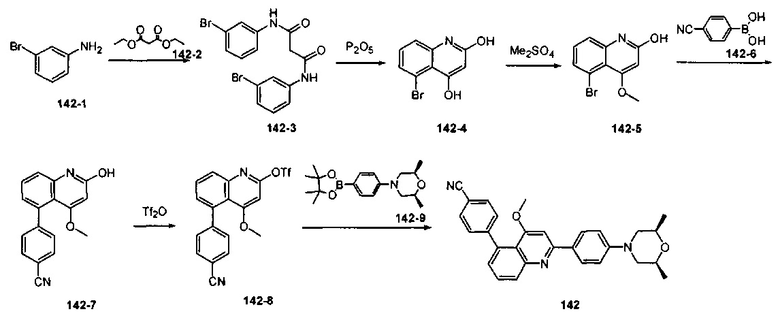
[339] Стадия 1. Соединение 142-1 (3,0 г, 17,4 ммоль) и соединение 142-2 (1,4 г, 8,7 ммоль) нагревали до 180°C под воздействием микроволнового излучения и перемешивали в течение 3 ч. После охлаждения полученное твердое вещество промывали с помощью EtOAc с получением соединения 142-3 (2,4 г, выход 22,2%) в виде желто-коричневого твердого вещества. MS ESI рассч. для C15H12Br2N2O2 [М+Н]+ 411, найденное значение 411.
[340] Стадия 2. Раствор соединения 142-3 (3,5 г, 8,46 ммоль) и P2O5 (2,4 г, 16,9 ммоль) в CH3SO3H (15 мл) нагревали до 160°C и перемешивали в течение 3 ч. Реакционную смесь выливали в лед. Полученное после фильтрования твердое вещество промывали с помощью H2O. Затем твердое вещество растворяли в 1 М растворе NaOH. Нерастворенное вещество фильтровали. Водную фазу регулировали рН до 3 с помощью конц. хлористоводородной кислоты. Осадок фильтровали и промывали с помощью небольших количеств H2O, в конце сушили при пониженном давлении с получением соединения 142-4 (1,85 г, выход 91%) в виде белого твердого вещества. MS ESI рассч. для C9H6BrNO2 [М+Н]+ 240, найденное значение 240.
[341] Стадия 3. Смесь соединения 142-4 (1,84 г, 7,62 ммоль) и K2CO3 (2,11 г, 15,24 ммоль) добавляли в ацетон (300 мл). Диметилсульфат (1,152 г, 9,15 ммоль) по каплям добавляли в смесь при перемешивании при комнатной температуре. После добавления реакционную смесь нагревали до 50°C и перемешивали в течение 2 ч. Реакционную смесь концентрировали досуха, затем регулировали рН до 4 с помощью 1 М водного раствора HCl. Осадок фильтровали и промывали с помощью н-гексана с получением смеси соединения 142-5 (0,84 г, выход 43%). Смесь очищали с помощью препаративной HPLC с получением соединения 142-5 (420 мг) в виде белого твердого вещества. MS ESI рассч. для C10H8BrNO2 [М+Н]+ 254, найденное значение 254.
[342] Стадия 4. Смесь соединения 142-5 (300 мг, 1,18 ммоль), соединение 142-6 (208 мг, 1,41 ммоль), Na2CO3 (313 мг, 2,95 ммоль) и Pd(dppf)Cl2 (86 мг, 0,118 ммоль) добавляли в перемешенный растворитель CH3CN (5 мл) и H2O (1 мл). Затем в атмосфере газа азота реакционную смесь нагревали до 80°C и перемешивали в течение 8 ч. Реагенты фильтровали, фильтрат концентрировали досуха, в конце очищали с помощью колоночной хроматографии с получением соединения 142-7 (223 мг, выход 69%) в виде желто-коричневого твердого вещества. MS ESI рассч. для C17H12N2O2 [М+Н]+ 277, найденное значение 277.
[343] Стадия 5. Трифторметансульфоновый ангидрид (644 мг, 2,28 ммоль) по каплям добавляли в раствор соединения 142-7 (350 мг, 1,27 ммоль) и пиридина (201 мг, 2,54 ммоль) в DCM (15 мл). Затем реакционную смесь перемешивали в течение ночи. Реакционную смесь концентрировали досуха, в конце очищали с помощью колоночной хроматографии с получением соединения 142-8 (254 мг, выход 49%) в виде белого твердого вещества. MS ESI рассч. для C18H11F3N2O4S [М+Н]+ 409, найденное значение 409.
[344] Стадия 6. Смесь соединения 142-8 (100 мг, 0,245 ммоль), соединения 142-9 (116 мг, 0,367 ммоль), Na2CO3 (78 мг, 0,735 ммоль) и Pd(dppf)Cl2 (36 мг, 0,049 ммоль) добавляли в перемешанный растворитель CH3CN (5 мл) и H2O (1 мл). В атмосфере газа азота реакционную смесь нагревали до 80°C и перемешивали в течение 3 ч. Реагенты фильтровали, фильтрат концентрировали досуха, в конце очищали с помощью препаративной HPLC с получением титульного соединения (95 мг, выход 86%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,18 (d, J=7,6 Гц, 1Н), 8,09 (d, J=8,8 Гц, 2Н), 7,62-7,72 (m, 3Н), 7,40-7,50 (m, 2Н), 7,19 (d, J=6,4 Гц, 1Н), 7,09 (s, 1Н), 7,03 (d, J=8,8 Гц, 2Н), 3,75-3,83 (m, 2Н), 3,65 (s, 3Н), 3,55-3,64 (m, 2Н), 2,45-2,57 (m, 2Н), 1,25-1,30 (m, 6Н). MS ESI рассч. для C29H27N3O2 [М+Н]+ 450, найденное значение 450.
Вариант осуществления 143
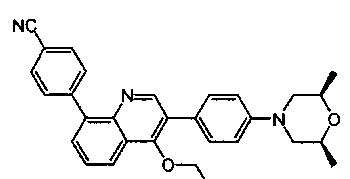
4-(3-(4-((2S,6R)-2,6-диметилморфолино)фенил)-4-этоксихинолин-8-ил)бензонитрил
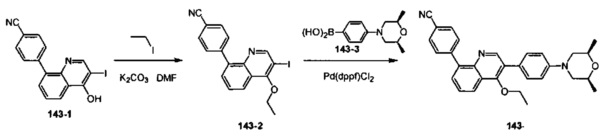
[345] Стадия 1. K2CO3 (745 мг, 5,4 ммоль) и этилйодид добавляли в раствор соединения 143-1 в DMF (20 мл), реакционную смесь перемешивали при 90°C в течение 2 ч. После завершения реакции добавляли H2O, затем реакционную смесь экстрагировали с помощью EtOAc. Объединенную органическую фазу промывали солевыми растворами, сушили над Na2SO4 и концентрировали под вакуумом с получением необходимого соединения 2 (0,9 г, 83%) в виде твердого вещества. MS ESI рассч. для C18H13IN2O [М+Н]+ 401, найденное значение 401.
[346] Стадия 2. Соединение 143-2 (200 мг, 0,5 ммоль), соединение 143-3 (176 мг, 0,74 ммоль), Pd(dppf)Cl2 (36 мг, 0,05 ммоль) и Na2CO3 (106 мг, 1 ммоль) суспендировали в перемешанном растворителе THF/H2O (10:1, 11 мл), затем в атмосфере газа азота реакционную смесь перемешивали при 90°C в течение 12 ч. После охлаждения реакционную смесь разбавляли с помощью EtOAc, затем промывали солевыми растворами, сушили над Na2SO4, концентрировали при пониженном давлении, остаток очищали с помощью RP-HPLC (ацетонитрил/H2O с помощью 0,05% модификатора гидроксида аммония) с получением титульного соединения (150 мг, 70%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,89 (s, 1Н), 8,51-8,49 (m, 1H), 7,82 (s, 4H), 7,64 (d, J=8,0 Гц, 2H), 7,49 (d, J=7,6 Гц, 2H), 7,08 (d, J=7,6 Гц, 2H), 4,08-4,03 (m, 2H), 3,86 (br. s., 2H), 3,58 (d, J=12 Гц, 2H), 2,56 (t, J=11,2 Гц, 2H), 1,37 (t, J=7,2 Гц, 3H), 1,0 (d, J=6,4 Гц, 6H). MS ESI рассч. для C30H29N3O2 [M+H]+ 464, найденное значение 464.
[347] Соединения, перечисленные в таблице 12, синтезировали с помощью соединения 143-1 и соответствующих галогеналканов.
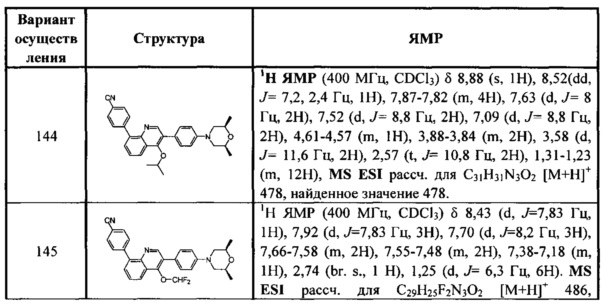
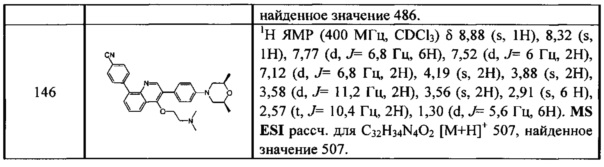
Вариант осуществления 147
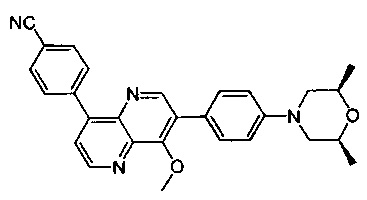
4-(7-(4-((2S,6R)-2,6-диметилморфолино)фенил)-8-метокси-1,5-нафтиридин-4-ил)бензонитрил
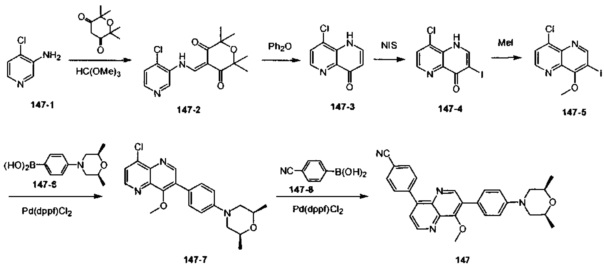
[348] Стадия 1. 2,2,6,6-тетраметил-2H-пиран-3,5(4H,6H)-дион (13,8 г, 96 ммоль) добавляли в НС(ОМе)3 (50 мл, 288 ммоль). После перемешивания смеси при 80°C в течение 3 ч. добавляли соединение 147-1 (10 г, 76,9 ммоль), смесь перемешивали в течение дополнительных 5 ч. После обнаружения с помощью LC-MS смесь концентрировали при пониженном давлении, неочищенный продукт промывали с помощью МеОН с получением соединения 147-2 (15 г, выход 57%). MS ESI рассч. для C15H17ClN2O3 [М+Н]+ 309, найденное значение 309.
[349] Стадия 2. Соединение 147-2 (13 г, 46 ммоль) порциями добавляли в предварительно нагретый Ph2O (250°C) (500 мл). После перемешивания в течение 5 мин. смесь обнаруживали с помощью LC-MS. После охлаждения реакционной смеси до комнатной температуры петролейный эфир добавляли к фильтрату, фильтрат концентрировали при пониженном давлении с получением соединения 147-3 (4 г, выход 48%). MS ESI рассч. для C8H5ClN2O [М+Н]+ 309, найденное значение 309.
[350] Стадия 3. Раствор соединения 147-3 (3 г, 16,7 ммоль) и NIS (4,1 г, 18,3 ммоль) в DMF (20 мл) перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь фильтровали и фильтрат концентрировали при пониженном давлении с получением 147-4 (2,5 г, выход 49%). MS ESI рассч. для C8H4ClIN2O [M+H]+ 307, найденное значение 307.
[351] Стадия 4. Раствор соединения 147-4 (2,5 г, 8,2 ммоль), MeI (1,75 г, 12,3 ммоль) и Ag2CO3 (4,5 г, 16,4 ммоль) в DMF (20 мл) перемешивали при 90°C в течение 3 ч. Реакционную смесь обнаруживали с помощью LC-MS. Смесь выливали в H2O, водную фазу экстрагировали с помощью EtOAc. Объединенную органическую фазу промывали солевыми растворами, сушили над Na2SO4, концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью хроматографии на силикагеле (РЕ:EtOAc = 10:1) с получением соединения 147-5 (300 мг, выход 12%). MS ESI рассч. для C9H6ClIN2O [М+Н]+ 321, найденное значение 321.
[352] Стадия 5. Соединение 147-5 (320 мг,1 ммоль), соединение 147-6 (350 мг, 1,1 ммоль), Pd(dppf)Cl2 (74 мг, 0,1 ммоль) и Na2CO3 (212 мг, 2 ммоль) растворяли в перемешанном растворителе THF/H2O (10:2 мл) и перемешивали при 70°C в течение 16 ч. Реакционную смесь обнаруживали с помощью LC-MS. Смесь выливали в H2O, водную фазу экстрагировали с помощью EtOAc. Объединенную органическую фазу промывали солевыми растворами, сушили над Na2SO4, концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью хроматографии на силикагеле (РЕ:EtOAc = 2:1) с получением соединения 147-7 (200 мг, выход 52%). MS ESI рассч. для C21H22ClN3O2 [М+Н]+ 384, найденное значение 384.
[353] Стадия 6. Соединение 147-7 (200 мг, 0,52 ммоль), соединение 147-8 (84 мг, 0,57 ммоль), Pd(dppf)Cl2 (38 мг, 0,052 ммоль) и Na2CO3 (110 мг, 1,04 ммоль) растворяли в перемешанном растворителе THF/H2O/DMF (10:2:2 мл) и смесь перемешивали при 90°C в течение 4 ч. Реакционную смесь обнаруживали с помощью LC-MS. Смесь выливали в Н2О, водную фазу экстрагировали с помощью EtOAc. Объединенную органическую фазу сушили над Na2SO4, концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью препаративной HPLC с получением титульного соединения (80 мг, выход 34%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 9,05 (d, J=4,8 Гц, 1H), 8,95 (s, 1Н), 7,89-7,81 (m, 4Н), 7,59 (t, J=4,4 Гц, 3Н), 7,04 (d, J=8,8 Гц, 2Н), 4,09 (s, 3Н), 3,85-3,82 (m, 2Н), 3,58 (d, J=11,2 Гц, 2Н), 2,51 (t, J=11,2 Гц, 2Н), 1,30-1,25 (m, 6Н). MS ESI рассч. для C28H26N4O2 [М+Н]+ 451, найденное значение 451.
Вариант осуществления 148
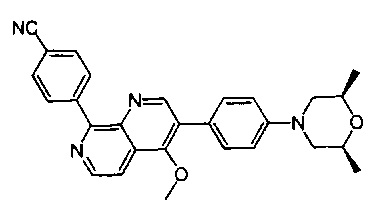
4-(3-(4-((2S,6R)-2,6-диметилморфолино)фенил)-4-метокси-1,7-нафтиридин-8-ил)бензонитрил
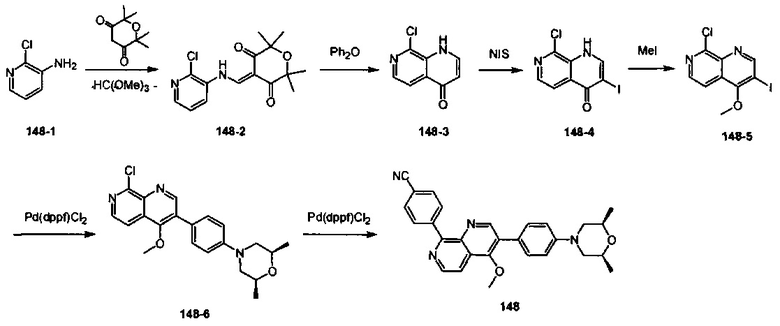
[354] Титульное соединение синтезировали в соответствии с вышеупомянутым способом в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 9,05 (s, 1Н), 8,82 (d, J=5,2 Гц, 1Н), 8,32 (d, J=5,6 Гц, 1Н), 8,14 (d, J=7,6 Гц, 2Н), 7,87 (d, J=8 Гц, 2Н), 7,62 (d, J=8 Гц, 2Н), 7,16 (d, J=8 Гц, 2Н), 3,89 (s, 2Н), 3,77 (s, 3Н), 3,62 (d, J=12 Гц, 2Н), 2,61 (d, J=10,8 Гц, 2Н), 1,31 (d, J=6 Гц, 6Н). MS ESI рассч. для C28H26N4O2 [М+Н]+ 451, найденное значение 451.
Вариант осуществления 149
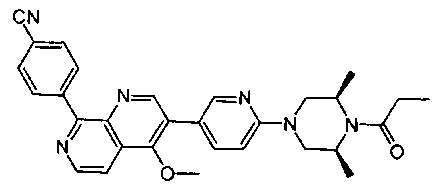
4-(3-(6-((3S,5R)-3,5-диметил-4-пропионилпиперазин-1-ил)пиридин-3-ил)-4-метокси-1,7-нафтиридин-8-ил)бензонитрил
[355] Титульное соединение синтезировали в соответствии с вышеупомянутым способом в виде белого твердого вещества. 1Н ЯМР (400 МГц, метанол-d4) δ 8,96 (s, 1Н), 8,69 (d, J=5,6 Гц, 1Н), 8,46 (d, J=2,4 Гц, 1Н), 8,18 (dd, J=11,6, 8,4 Гц, 3Н), 7,95 (dd, J=8,8, 2,4 Гц, 2Н), 7,87 (d, J=8,4 Гц, 2Н), 7,06 (d, J=8,8 Гц, 1Н), 4,31-4,42 (m, 3Н), 3,81 (s, 3Н), 3,16 (d, J=11,6 Гц, 2Н), 2,50 (m, 2Н), 1,31 (s, 6Н), 1,15 (t, J=7,6 Гц, 3Н). MS ESI рассч. для C30H30N6O2 [М+Н]+ 507, найденное значение 507.
Вариант осуществления 150

8-(4-цианофенил)-3-(4-((2R,6S)-2,6-диметилморфолино)фенил)-2-метилциннолин-2-ия-4-олат

[356] Стадия 1. K2CO3 (1,9 г, 13,7 ммоль) и метилйодид добавляли в раствор соединения 150-1 (2,3 г, 6,84 ммоль) в DMF (20 мл). Реакционную смесь перемешивали при 90°C в течение 4 ч. После завершения реакции, как обнаружено с помощью LC-MS, добавляли H2O, а затем реакционную смесь экстрагировали с помощью EtOAc. Объединенную органическую фазу промывали солевыми растворами, сушили над Na2SO4 и концентрировали под вакуумом. Остаток очищали с помощью хроматографии на силикагеле (РЕ/EtOAc = 3:1) с получением необходимого соединения 2 (1,5 г, 68%) в виде твердого вещества. MS ESI рассч. для C9H6ClIN2O [М+Н]+ 321, найденное значение 321.
[357] Стадия 2. Соединение 150-2 (321 мг, 1 ммоль), соединение 150-3 (282 мг, 1,2 ммоль), Pd(dppf)Cl2 (73,2 мг, 0,1 ммоль) и Na2CO3 (212 мг, 2 ммоль) суспендировали в перемешанном растворителе THF/H2O (10:1 мл), в атмосфере газа азота реакционную смесь перемешивали при 90°C в течение ночи. После завершения реакции, как обнаружено с помощью LC-MS, добавляли H2O, а затем реакционную смесь экстрагировали с помощью EtOAc. Объединенную органическую фазу промывали солевыми растворами, сушили над Na2SO4 и концентрировали под вакуумом с получением неочищенного продукта 4 (320 мг, 83,6%). MS ESI рассч. для C21H22ClN3O2 [М+Н]+ 384, найденное значение 384.
[358] Стадия 3. Соединение 150-4 (320 мг, 0,84 ммоль), соединение 150-5 (184 мг, 1,25 ммоль), Pd2(dba)3 (77 мг, 0,08 ммоль), Xantphos (86 мг, 0,16 ммоль) и Na2CO3 (178 мг, 1,68 ммоль) суспендировали в перемешанном растворителе 1,4-диоксана/H2O (10:1 мл), в атмосфере газа азота реакционную смесь нагревали до 120°C и нагревали с обратным холодильником в течение 3 ч. После завершения реакции смесь фильтровали с помощью диатомита, фильтрат выпаривали, экстрагировали с помощью EtOAc. Объединенную фазу EtOAc промывали солевыми растворами, сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали с помощью препаративной HPLC с получением титульного соединения (80 мг, 20%). 1Н ЯМР (400 МГц, CDCl3) δ 8,48 (d, J=8 Гц, 1Н), 7,80-7,69 (m, 6H), 7,89-7,81 (m, 4H), 7,36 (d, J=8,8 Гц, 2H), 7,15 (d, J=8,4 Гц, 2H), 4,19 (s, 3Н), 3,90-3,86 (m, 2H), 3,60 (d, J=11,6 Гц, 2H), 2,61 (t, J=10,8 Гц, 2H), 1,29 (d, J=6,4 Гц, 6H). MS ESI рассч. для C28H26N4O2 [M+H]+ 451, найденное значение 451.
Вариант осуществления 151
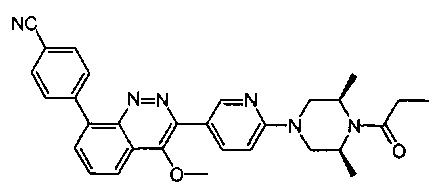
4-(3-(6-(3,5-диметил-4-пропионилпиперазин-1-ил)пиридин-3-ил)-4-метоксициннолин-8-ил)бензонитрил
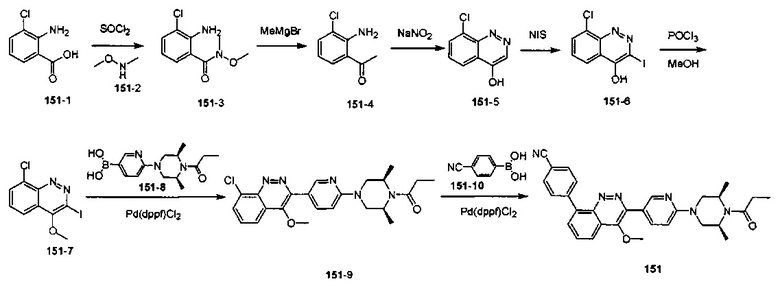
[359] Стадия 1. Соединение 151-1 (51 г, 0,3 моль), тионилхлорид (200 мл) нагревали до температуры флегмы в течение 2 ч., затем тионилхлорид выпаривали, остаток растворяли в DCM, раствор соединения 151-2 (42,3 г, 0,45 моль), DIPEA (116 г, 0,9 моль) добавляли в DCM, реакционная смесь реагировала при комнатной температуре в течение 1 ч., затем ее выливали в Н2О, смесь промывали с помощью Н2О и солевых растворов, соответственно, органическую фазу сушили и концентрировали, остаток очищали с помощью колоночной хроматографии с получением соединения 151-3 (26 г, выход 40%) в виде желтого масла. MS ESI рассч. для C9H11ClN2O2 [М+Н]+ 215, найденное значение 215.
[360] Стадия 2. Соединение 151-3 (26 г, 0,12 моль) растворяли в THF (300 мл), MeMgBr (120 мл, 3 М в Et2O) медленно добавляли в раствор при 0°C. После добавления реакционную смесь поддерживали при 0°C в течение 2 ч., а затем выливали в водный раствор хлорида аммония, экстрагировали с помощью EtOAc, жидкость после экстракции промывали солевыми растворами, сушили над безводным сульфатом натрия и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии с получением соединения 151-4 (10,5 г, выход 51,4%) в виде желтого твердого вещества. MS ESI рассч. для C8H8ClNO [М+Н]+ 169, найденное значение 169.
[361] Стадия 3. Соединение 151-4 (1,69 г, 10 ммоль) добавляли в H2O (10 мл), конц. хлористоводородную кислоту (17 мл) добавляли при 0°C, затем раствор нитрита натрия (2,1 г, 30 ммоль) в H2O (10 мл) по каплям добавляли при температуре в диапазоне от -5 до 0°C. После добавления реакционная смесь реагировала при 0°C в течение 1 ч., затем ее нагревали до 80°C и она реагировала в течение 6 ч., затем ее охлаждали на ледяной бане. Смесь фильтровали, сушили с получением 1,2 г соединения 151-5, выход 67% в виде желтого твердого вещества. MS ESI рассч. для C8H5ClN2O [М+Н]+ 181, найденное значение 181.
[362] Стадия 4. NIS (1,5 г, 0,67 ммоль) добавляли в раствор соединения 151-5 (1,2 г, 0,67 ммоль) в DMF (10 мл), реакционную смесь перемешивали при комнатной температуре в течение 2 ч., раствор выливали в H2O, фильтровали, отфильтрованный осадок сушили с получением 1,5 г соединения 151-6, выход 75% в виде желтого твердого вещества. MS ESI рассч. для C8H4ClIN2O [М+Н]+ 307, найденное значение 307.
[363] Стадия 5. Соединение 151-6 (1,2 г, 0,67 ммоль), оксихлорид фосфора (5 мл) нагревали до 120°C и перемешивали в течение 2 ч., затем реакционную смесь охлаждали и выливали в метанол, H2O и EtOAc добавляли, соответственно, органическую фазу отделяли, сушили и концентрировали с получением 1,0 г соединения 151-7, выход 62,5% в виде желтого твердого вещества. MS ESI рассч. для C9H6ClIN2O [М+Н]+ 321, найденное значение 321.
[364] Стадия 6. Соединение 151-7 (320 мг, 1,0 ммоль), соединение 151-8 (292 мг, 1,0 ммоль), Pd(dppf)Cl2 (73 мг, 0,1 ммоль) и карбонат натрия (212 мг, 2,0 ммоль) добавляли в раствор THF/H2O/DMF (10:1:1, 12 мл), в атмосфере газа азота реакционную смесь перемешивали при 80°C в течение ночи. Смесь фильтровали с помощью диатомита, фильтрат промывали с помощью H2O (10 мл) и экстрагировали с помощью EtOAc (20 мл), жидкость после экстракции промывали солевыми растворами и сушили над безводным сульфатом натрия, концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии с получением соединения 151-9 (200 мг, выход 68%) в виде желтого твердого вещества. MS ESI рассч. для C23H26ClN5O2 [М+Н]+ 441, найденное значение 441.
[365] Стадия 7. Титульное соединение (10 мг, выход 9%) синтезировали в соответствии с вышеупомянутым способом в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 9,04-9,05 (d, J=2,0 Гц, 1Н), 8,42-8,43 (d, J=6,8 Гц, 1Н), 8,28-8,30 (d, J=8,8 Гц, 1H), 7,91-7,93 (d, J=8,4 Гц, 2Н), 7,55-7,85 (m, 4Н) 6,86-6,88 (d, J=8,8 Гц, 1Н) 4,08-4,13 (m, 2Н), 3,90 (s, 3Н) 3,17-3,21 (d d, J=4,0 Гц 2Н), 2,25-2,28 (m, 2Н) 2,03-2,06 (m, 1Н) 1,19-1,28 (m, 9Н) 0,98-0,99 (d, J=6,4 Гц 2Н), MS ESI рассч. для C30H30N6O2 [М+Н]+ 507, найденное значение 507.
Вариант осуществления 152
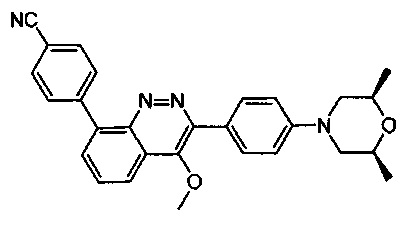
4-(3-(4-((2R,6S)-2,6-диметилморфолино)фенил)-4-метоксициннолин-8-ил)бензонитрил
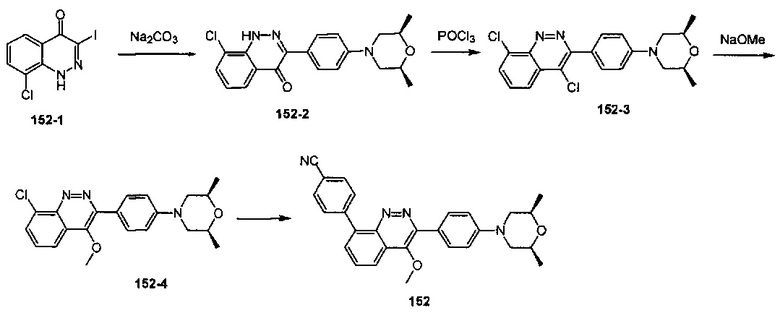
[366] Стадия 1. Соединение 152-1 (1,2 г, 4 ммоль), карбонат натрия (4,3 г, 40,2 ммоль), Pd(dppf)Cl2 (200 мг, 0,2 ммоль) добавляли в раствор 4-((2R,6S)-2,6-диметилморфолинил)бензоборной кислоты (1,15 г, 5 ммоль) в перемешанном растворителе THF/H2O (20:4 мл) и перемешивали при 70°C в течение ночи. После завершения реакции, как обнаружено с помощью LC-MS, добавляли H2O, затем экстрагировали с помощью EtOAc. Объединенную органическую фазу промывали солевыми растворами, сушили над сульфатом натрия, концентрировали под вакуумом. Остаток очищали с помощью хроматографии на силикагеле (РЕ:EtOAc = 5:1-2:1) с получением соединения 152-2 (0,62 г, выход 44%). MS ESI рассч. для C20H20ClN3O2 [М+Н]+ 370, найденное значение 370.
[367] Стадия 2. Раствор соединения 152-2 (3,7 г, 10 ммоль) в POCl3 (35 мл) нагревали при 110°C в течение 6 ч. Растворитель удаляли под вакуумом. Остаток гасили с помощью H2O. Раствор разделяли между EtOAc и H2O, органическую фазу объединяли, промывали солевыми растворами, сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали с помощью хроматографии на силикагеле (РЕ:EtOAc = 2:1) с получением соединения 152-3 (0,78 г, выход 21%). MS ESI рассч. для C20H19Cl2N3O [М+Н]+ 389, найденное значение 389.
[368] Стадия 3. NaOMe (0,055 г, 1 ммоль) порциями добавляли в раствор соединения 152-3 (0,19 г, 0,5 ммоль) в МеОН (10 мл) при -10°C в течение 10 мин. Реакционную смесь перемешивали при 20°C в течение 1 ч., затем перемешивали при 50°C в течение 3 ч. После завершения реакции добавляли H2O (100 мл), затем реакционную смесь экстрагировали с помощью EtOAc. Объединенную органическую фазу промывали солевыми растворами, сушили над сульфатом натрия и концентрировали под вакуумом с получением соединения 152-4 (0,11 г, 58%) в виде коричневого твердого вещества, которое непосредственно применяли на следующей стадии. MS ESI рассч. для C21H22ClN3O2 [М+Н]+ 384, найденное значение 384.
[369] Стадия 4. Соединение 152-4 (40 мг, 0,1 ммоль), 4-цианофенилбороновой кислоты (30 мг, 0,2 ммоль), Pd2(dba)3 (9 мг, 0,01 ммоль), Xantphos (10 мг, 0,02 ммоль) и карбонат натрия (4,3 г, 40,2 ммоль) растворяли в перемешанном растворителе диоксана/H2O (5:1 мл), в атмосфере газа азота реакционную смесь перемешивали при 110°C в течение 6 ч. Смесь разбавляли с помощью H2O (10 мл), экстрагировали с помощью EtOAc. Объединенную органическую фазу промывали солевыми растворами, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали с помощью препаративной HPLC с получением титульного соединения в виде белого твердого вещества (выход 32%). 1H ЯМР (400 МГц, CDCl3) δ 8,23 (d, J=8 Гц, 1Н), 8,06 (d, J=8,4 Гц, 2Н), 7,85 (d, J=8 Гц, 2Н), 7,77-7,69 (m, 4Н), 6,99 (d, J=8,8 Гц, 2Н), 3,74 (s, 5Н), 3,57 (d, J=12 Гц, 2Н), 2,47 (t, J=10,8 Гц, 2Н), 1,23 (d, J=6,4 Гц, 6Н). MS ESI рассч. для C28H26N4O2 [М+Н]+ 451, найденное значение 451.
Вариант осуществления 153

4-(4-хлор-3-(4-((2S,6R)-2,6-диметилморфолино)фенил)-1,6-нафтиридин-8-ил)бензонитрил
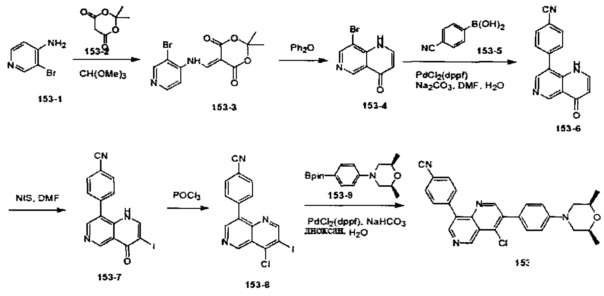
[370] Стадия 1. Раствор соединения 153-2 (21 г, 0,146 моль) в СН(ОМе)3 (50 мл) перемешивали при 60°C в течение 0,5 ч. Реакционную смесь охлаждали до комнатной температуры, добавляли соединение 153-1 (21,05 г, 0,122 моль). Смесь нагревали с обратным холодильником при 100°C в течение 2 ч. TLC (РЕ:EtOAc = 1:1) показала, что исходный материал полностью потребляли. Реакционную смесь охлаждали до 0°C и фильтровали. Отфильтрованный осадок размалывали с МеОН с получением соединения 153-2 (24 г, 62%) в виде темно-желтого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ 11,48 (brs, 1Н), 8,96-8,81 (m, 1H), 8,77 (s, 1H), 8,51 (d, J=5,29 Гц, 1Н), 7,90 (d, J=5,29 Гц, 1H), 7,37 (t, J=7,9 Гц, 1H), 7,15-6,90 (m, 1H), 1,68 (s, 6H). MS ESI рассч. для C12H11BrN2O4 [M+H]+ 326, 328, найденные значения 326, 328.
[371] Стадия 2. Раствор соединения 153-2 (0,4 г, 1,223 моль) в PH2O (3 мл) нагревали с обратным холодильником при 220°C в течение 0,5 ч. TLC показала, что исходный материал полностью потребляли. Реакционную смесь охлаждали до комнатной температуры и добавляли гексан. Смесь фильтровали и отфильтрованный осадок промывали гексаном с получением соединения 153-4 (200 мг, 72%) в виде темно-желтого твердого вещества. MS ESI рассч. для C8H5BrN2O [М+Н]+ 224 и 226, найденные значения 224 и 226.
[372] Стадия 3. Карбонат натрия (780 мг, 7,365 ммоль) добавляли в раствор соединения 153-4 (550 мг, 2,455 ммоль) и соединения 153-5 (397 мг, 2,701 ммоль) в перемешанном растворителе DMF (5 мл) и H2O (1 мл) при комнатной температуре. Реакционную смесь продували с помощью газа азота 3 раза, добавляли PdCl2(dppf) (90 мг, 0,123 ммоль). Затем реакционную смесь снова продували с помощью газа азота 3 раза и перемешивали при температуре флегмы в течение ночи. TLC (EtOAc) показала, что исходный материал полностью потребляли. Реакционную смесь фильтровали с помощью подушки из диатомита. Фильтрат концентрировали досуха с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле (EtOAc:МеОН = 100:4) с получением соединения 153-6 (310 мг, 35%) в виде бежевого твердого вещества. MS ESI рассч. для C15H9N3O [М+Н]+ 248, найденное значение 248.
[373] Стадия 4. NIS (200 мг, 0,887 ммоль) порциями добавляли в раствор соединения 153-6 (210 мг, 0,85 ммоль) в DMF (2 мл) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение ночи. TLC (EtOAc) показала, что большую часть исходного материала полностью потребляли. Реакционную смесь фильтровали, фильтрат концентрировали с получением соединения 153-7 (250 мг, 79%) в виде белого твердого вещества. MS ESI рассч. для C15H8IN3O [М+Н]+ 374, найденное значение 374.
[374] Стадия 5. Раствор соединения 153-7 (310 мг, 0,831 ммоль) в POCl3 (10 мл) перемешивали при температуре флегмы в течение 4 ч. TLC показала, что исходный материал полностью потребляли. Реакционную смесь концентрировали досуха и разбавляли с помощью EtOAc. Полученный раствор подщелачивали с помощью NaHCO3 (водн.) при 0°C. Смесь разделяли между EtOAc и H2O. Органическую фазу концентрировали досуха с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле (РЕ:EtOAc = 1:1) с получением соединения 153-8 (290 мг, 90%) в виде белого твердого вещества. MS ESI рассч. для C15H7ClIN3 [М+Н]+ 392, найденное значение 392.
[375] Стадия 6. Соединение 153-8 (27 мг, 0,691 ммоль) и соединение 153-9 (262 мг, 0,828 ммоль) растворяли в перемешанном растворителе диоксана (3 мл) и H2O (0,6 мл), добавляли бикарбонат натрия (145 мг, 1,727 ммоль). Реакционную смесь продували с помощью газа азота 3 раза, добавляли Pd(dppf)Cl2 (50 мг, 0,069 ммоль), затем реакционную смесь снова продували с помощью газа азота 3 раза и перемешивали при 80°C в течение 2 ч. TLC (РЕ:EtOAc = 3:1) показала, что исходный материал полностью потребляли. Реакционную смесь фильтровали с помощью подушки из диатомита. Фильтрат концентрировали досуха с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле (РЕ:EtOAc = 2:1) с получением титульного соединения (120 мг, 30%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 9,85 (s, 1Н), 9,08 (s, 1Н), 8,87 (s, 1Н), 7,90-7,83 (m, 5Н), 7,52 (d, J=8,53 Гц, 2Н), 7,09 (d, J=7,78 Гц, 2Н), 3,87 (br. s., 2Н), 3,62 (d, J=11,29 Гц, 2Н), 2,57 (t, J=11,17 Гц, 2Н), 1,32 (d, J=6,27 Гц, 6Н). MS ESI рассч. для C27H23ClN4O [М+Н]+ 455, найденное значение 455.
Вариант осуществления 154
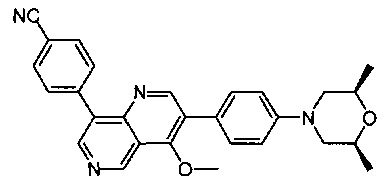
4-(3-(4-((2S,6R)-2,6-диметилморфолино)фенил)-4-метоксихинолин-8-ил)бензонитрил
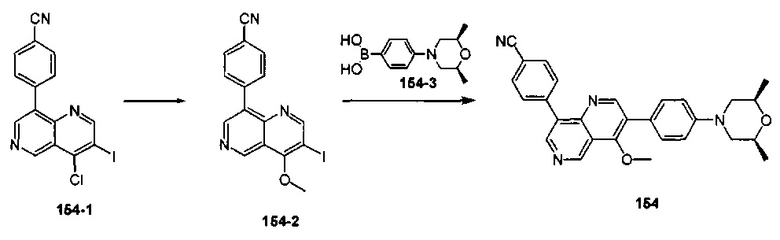
[376] Стадия 1. NaOMe (32 мг, 6 ммоль) добавляли в раствор соединения 154-1 (120 мг, 3 ммоль) в МеОН (15 мл) и смесь перемешивали при 45°C в течение 16 ч. После того, как LC-MS показала, что исходный материал полностью потребляли, раствор концентрировали под вакуумом и разделяли между EtOAc и H2O. Органические фазы объединяли и промывали солевыми растворами, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения 154-2 (0,1 г, 83%) в виде белого твердого вещества. MS ESI рассч. для C16H10IN3O [М+Н]+ 387, найденное значение 387.
[377] Стадия 2. Соединение 154-2 (100 мг, 0,28 ммоль), соединение 154-3 (91 мг, 0,39 ммоль), Pd(dppf)Cl2 (22 мг, 0,032 ммоль) и карбонат натрия (45 мг, 0,52 ммоль) растворяли в перемешанном растворителе THF (20 мл) и H2O (3 мл) и смесь перемешивали при 80°C в течение ночи. После того, как LC-MS показала, что исходный материал полностью потребляли, раствор разделяли между EtOAc и H2O. Органические фазы объединяли и промывали солевыми растворами, сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали с помощью препаративной HPLC с получением титульного соединения (30 мг, 25%). 1Н ЯМР (400 МГц, CDCl3) δ 9,71 (s, 1H), 9,03 (s, 1Н), 8,76 (s, 1Н), 7,87 (d, J=8 Гц, 2Н), 7,81 (d, J=8 Гц, 2Н), 7,53 (d, J=8,4 Гц, 2Н), 7,04 (d, J=8,8 Гц, 2Н), 3,86-3,82 (m, 5Н), 3,59 (d, J=10,8 Гц, 2Н), 2,52 (t, J=11,2 Гц, 2Н), 1,30 (d, J=6,4 Гц, 6Н). MS ESI рассч. для C28H26N4O2 [М+Н]+ 451, найденное значение 451.
Вариант осуществления 155
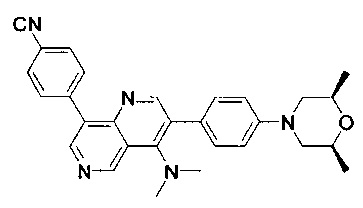
4-(4-(диметиламино)-3-(4-((2S,6R)-2,6-диметилморфолино)фенил)-1,6-нафтиридин-8-ил)бензонитрил

[378] Соединение 155-2 (146 мг, 1,79 ммоль) и DIPEA (282 мг, 2,186 ммоль) добавляли в раствор соединения 155-1 (80 мг, 0,176 ммоль) в DMF (2 мл). Смесь перемешивали при 120°C в течение ночи. TLC (РЕ:EtOAc = 1:1) показала, что исходный материал полностью потребляли. Смесь концентрировали с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле (РЕ:EtOAc = 1:1) с получением титульного соединения (35 мг, 43%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 9,62 (br. s., 1Н), 8,77 (s, 1Н), 8,61 (s, 1Н), 7,91 (d, J=7,06 Гц, 2Н), 7,84-7,69 (m, 5Н), 7,40 (d, J=7,06 Гц, 2Н), 4,58 (br. s., 2Н), 3,51 (d, J=11,47 Гц, 2H), 3,01 (s, 6Н), 2,91 (t, J=11,25 Гц, 2Н), 2,55 (s, 1Н), 1,94 (s, 1Н), 1,25 (d, J=6,62 Гц, 6Н). MS ESI рассч. для C29H29N5O [М+Н]+ 464, найденное значение 464.
Вариант осуществления 156
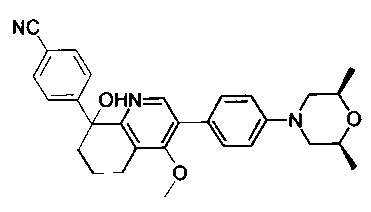
4-(3-(4-((2S,6R)-2,6-диметилморфолино)фенил)-8-гидрокси-4-метокси-5,6,7,8-тетрагидрохинолин-8-ил)бензонитрил
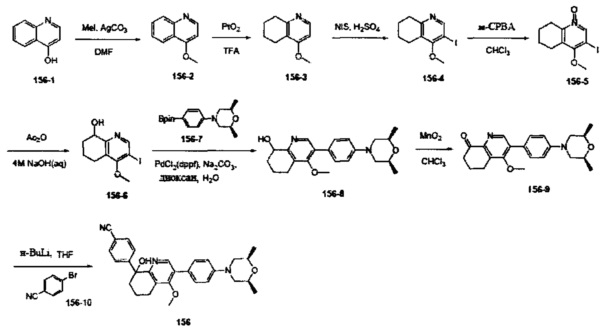
[379] Стадия 1. Ag2CO3 (55,55 г, 0,201 моль) добавляли в раствор соединения 156-1 (14,62 г, 0,101 моль) в DMF (150 мл). Реакционную смесь продували с помощью газа азота и добавляли MeI (12,88 г, 0,091 моль). Затем реакционную смесь перемешивали при 80°C в течение ночи. TLC (EtOAc) показала, что исходный материал полностью потребляли. Смесь фильтровали, фильтрат разделяли между EtOAc и H2O. Органическую фазу концентрировали с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле (РЕ:EtOAc = 2:1) с получением соединения 156-2 (10 г, 56%) в виде бесцветного масла. 1Н ЯМР (400 МГц, CDCl3) δ 8,72 (d, J=3,53 Гц, 1Н), 8,17 (d, J=7,94 Гц, 1Н), 8,02 (d, J=8,38 Гц, 1Н), 7,71-7,62 (m, 1Н), 7,52-7,42 (m, 1Н), 6,75-6,59 (m, 1Н), 3,99 (d, J=1,76 Гц, 4Н). MS ESI рассч. для C10H9NO [М+Н]+ 160, найденное значение 160.
[380] Стадия 2. В атмосфере газа азота PtO2 (1 г) добавляли в раствор соединения 156-2 (5 г, 0,031 моль) в TFA (45 мл). Смесь перемешивали при давлении газа водорода 50 фунтов/кв. дюйм при комнатной температуре. TLC (РЕ:EtOAc = 1:1) показала, что исходный материал полностью потребляли. Смесь фильтровали, фильтрат концентрировали под вакуумом. Остаток растворяли в H2O, подщелачивали до рН=9 с помощью 8 н. NaOH (водн.) при 0°C, водную фазу экстрагировали с помощью DCM, органическую фазу промывали солевыми растворами, сушили над безводным сульфатом натрия и концентрировали с получением соединения 156-3 (4 г, 80%) в виде желтого масла, которое непосредственно применяли на следующей стадии. 1Н ЯМР (400 МГц, CDCl3) δ 8,18 (d, J=5,73 Гц, 1Н), 6,50 (d, J=5,73 Гц, 1Н), 3,75 (s, 3Н), 2,78 (t, J=6,17 Гц, 2Н), 2,53 (t, J=6,17 Гц, 2Н), 1,79-1,62 (m, 5Н). MS ESI рассч. для C10H13NO [М+Н]+ 164, найденное значение 164.
[381] Стадия 3. NIS (9,6 г, 0,043 моль) порциями добавляли в раствор соединения 156-3 (5,8 г, 0,0356 моль) в серной кислоте (30 мл) при 0°C, смесь перемешивали при комнатной температуре в течение 0,5 ч., затем нагревали до 60°C и перемешивали в течение 2 ч. TLC (РЕ:EtOAc = 1:1) показала, что исходный материал полностью потребляли. Смесь выливали в ледяную воду и подщелачивали до рН=9 с помощью 8 н. NaOH (водн.). Затем водную фазу экстрагировали с помощью DCM. Органическую фазу промывали нас. NaHCO3 (водн.) и солевыми растворами, концентрировали с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле (РЕ:EtOAc = 15:1) с получением соединения 156-4 нас. NaHCO3 (водн.) в виде коричневого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,55 (brs, 1Н), 3,80-3,73 (m, 2H), 2,84-2,77 (m, 2H), 2,76-2,70 (m, 2H), 1,85-1,69 (m, 4H), MS ESI рассч. для C10H12INO [M+H]+ 290, найденное значение 290.
[382] Стадия 4. м-СРВА (2,6 г, 0,015 моль) порциями добавляли в раствор соединения 156-4 (1,44 г, 0,005 моль) в CHCl3 (10 мл) при 0°C, смесь перемешивали при 40°C в течение 2 ч. Смесь гасили с помощью H2O, регулировали рН до 14 с помощью 8 н. NaOH (водн.). Затем водную фазу экстрагировали с помощью DCM. Органическую фазу промывали нас. NaHCO3 (водн.) и солевыми растворами, концентрировали с получением соединения 156-5 (1,3 г, 88%) в виде желтого твердого вещества, которое непосредственно применяли для следующей стадии. MS ESI рассч. для C10H12INO2 [М+Н]+ 306, найденное значение 306.
[383] Стадия 5. Раствор соединения 156-5 (1,3 г, 0,0043 ммоль) в Ac2O (6 мл) перемешивали при 90°C в течение 2 ч. Смесь концентрировали и добавляли 4 н. NaOH (водн.) (4,3 мл, 0,017 моль). Смесь перемешивали при 80°C в течение 2 ч. Затем водную фазу экстрагировали с помощью DCM. Органическую фазу концентрировали с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле (РЕ : EtOAc = 3:1) с получением соединения 156-6 (0,82 г, 33%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,61 (s, 1H), 4,59 (brs, 1Н), 3,79 (s, 3Н), 3,67 (br. s., 1Н), 2,77 (br. s., 2H), 2,19 (br. s., 1H), 1,94 (br. s., 1H), 1,71 (br. s., 2H). MS ESI рассч. для C10H12INO2 [M+H]+ 306, найденное значение 306.
[384] Стадия 6. Карбонатнатрия (0,69 г, 0,0065 моль) добавляли в раствор соединения 156-6 (0,8 г, 0,0026 моль) и соединения 156-7 в перемешанном растворителе диоксана (8 мл) и H2O (2 мл). Смесь продували с помощью газа азота и добавляли Pd(dppf)Cl2 (0,19 г, 0,26 ммоль). Затем смесь перемешивали при 100°C и нагревали с обратным холодильником в течение 2 ч. Смесь фильтровали с помощью подушки из диатомита, фильтрат концентрировали с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле (РЕ:EtOAc = 1:2) с получением соединения 156-8 (0,66 г, 68%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,28 (s, 1Н), 7,37 (d, J=8,82 Гц, 2Н), 6,91 (d, J=8,82 Гц, 2Н), 4,65 (t, J=6,84 Гц, 1H), 3,88 (s, 1Н), 3,79-3,68 (m, 2Н), 3,47 (d, J=11,03 Гц, 2Н), 3,39 (s, 3Н), 2,82-2,63 (m, 2Н), 2,41 (t, J=11,03 Гц, 2Н), 2,27-2,14 (m, 1Н), 2,01-1,91 (m, 1Н), 1,73 (t, J=8,16 Гц, 2Н), 1,22 (d, J=6,17 Гц, 7Н). MS ESI рассч. для C22H28N2O3 [М+Н]+ 369, найденное значение 369.
[385] Стадия 7. MnO2 (1,56 г, 0,018 моль) добавляли в раствор соединения 156-8 (0,66 г, 0,0018 моль) в CHCl3 (10 мл). Смесь перемешивали при температуре флегмы в течение ночи. Смесь фильтровали с помощью подушки из диатомита, фильтрат концентрировали с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле (РЕ:EtOAc = 1:2) с получением соединения 156-9 (0,57 г, 86%) в виде желтого твердого вещества. MS ESI рассч. для C22H26N2O3 [М+Н]+ 367, найденное значение 367.
[386] Стадия 8. В атмосфере газа азота н-BuLi (0,3 мл, 0,75 ммоль) по каплям добавляли в раствор соединения 156-10 (124 мг, 0,682 ммоль) в THF (2 мл) при -65°C. Смесь перемешивали при -65°C в течение 0,5 ч., затем по каплям добавляли раствор соединения 156-9 (200 мг, 0,546 ммоль) в THF (1 мл). Смесь перемешивали при -65°C в течение 1 ч. и перемешивали при 0°C в течение еще 1 ч. TLC показала, что исходный материал полностью потребляли. Реакционную смесь гасили с помощью нас. NaHCO3 (водн.) и разбавляли с помощью EtOAc. Органическую фазу концентрировали с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле (РЕ:EtOAc = 3:1) с получением титульного соединения (65 мг, 30%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,28 (s, 1Н), 7,51 (d, J=8,38 Гц, 2Н), 7,39 (d, J=8,82 Гц, 2Н), 7,27 (d, J=8,38 Гц, 2Н), 6,92 (d, J=8,82 Гц, 2Н), 4,08 (s, 1H), 3,76 (ddd, J=10,14, 6,17, 2,21 Гц, 2Н), 3,49 (s, 1Н), 3,46 (s, 4Н), 2,91-2,74 (m, 2Н), 2,42 (t, J=11,25 Гц, 2Н), 2,21-2,11 (m, 2Н), 1,85 (brs, 1H), 1,55 (brs, 1Н), 1,22 (d, J=6,17 Гц, 6Н). MS ESI рассч. для C29H31N3O3 [М+Н]+ 470, найденное значение 470.
Вариант осуществления 157

4-(3-(4-((2S,6R)-2,6-диметилморфолино)фенил)-4,8-диметокси-5,6,7,8-тетрагидрохинолин-8-ил)бензонитрил
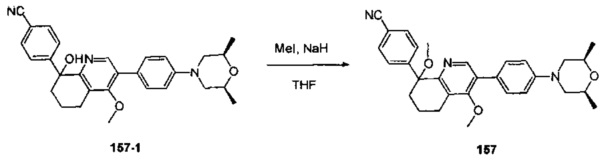
[387] NaH (27,3 мг, 0,682 ммоль) добавляли в раствор соединения 157-1 (80 мг, 0,171 ммоль) в THF (3 мл) при 0°C, суспензию перемешивали при 0°C в течение 10 мин. Добавляли раствор MeI (29 мг, 0,204 ммоль) в THF (1 мл). Смесь перемешивали при комнатной температуре в течение ночи. TLC показала, что соединение 157-1 полностью потребляли. Смесь концентрировали с получением неочищенного продукта, который очищали с помощью препаративной TLC (РЕ : EtOAc = 3:1) с получением титульного соединения (40 мг, 40%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,43 (s, 1Н), 7,63 (d, J=8,28 Гц, 2Н), 7,56-7,49 (m, 2Н), 7,41 (d, J=8,28 Гц, 2Н), 7,02 (d, J=8,78 Гц, 2Н), 3,90-3,79 (m, 2Н), 3,59 (br. s., 1Н), 3,57-3,51 (m, 4Н), 3,27 (s, 3Н), 3,10-3,00 (m, 1Н), 2,87-2,76 (m, 1Н), 2,51 (t, J=11,17 Гц, 2Н), 2,33 (dd, J=13,30, 4,02 Гц, 1Н), 2,14 (br. s., 1Н), 2,00-1,90 (m, 1Н), 1,84 (d, J=6,78 Гц, 1Н), 1,31 (d, J=6,27 Гц, 6Н). MS ESI рассч. для C30H33N3O3 [М+Н]+ 484, найденное значение 484.
Вариант осуществления 158
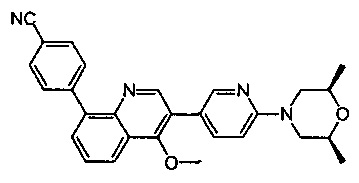
4-(3-(6-((2S,6R)-2,6-диметилморфолино)пиридин-3-ил)-4-метоксихинолин-8-ил)бензонитрил
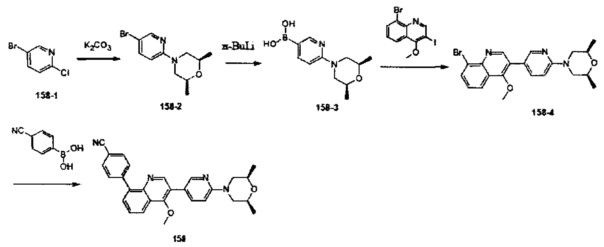
[388] Стадия 1. (2S,6R)-2,6-диметилморфолин (4,0 г, 34,8 ммоль) и карбонат калия (8,0 г, 58 ммоль) добавляли в раствор 5-бром-2-хлорпиридина (5,0 г, 26,2 ммоль) в DMF (18 мл). Реакционную смесь перемешивали при 90°C в течение 6 ч. и фильтровали, отфильтрованный осадок промывали с помощью EtOAc. Фильтрат концентрировали, неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с получением соединения 158-2 (6 г, выход 84,9%) в виде желтого масла. MS ESI рассч. для C11H15BrN2O [М+Н]+ 271, найденное значение 271.
[389] Стадия 2. н-BuLi (8,3 мл, 20,6 ммоль) добавляли в раствор соединения 158-2 (3,7 г, 13,7 ммоль) в THF (30 мл), в атмосфере газа азота реакционную смесь перемешивали при -78°C в течение 30 мин. Триизопропилборат (7,8 г, 41,2 ммоль) добавляли в реакционную смесь, затем перемешивали при -78°C в течение 3 ч. Смесь выливали в H2O, экстрагировали с помощью EtOAc, сушили над сульфатом натрия и фильтровали, концентрировали при пониженном давлении, остаток очищали с помощью колоночной хроматографии на силикагеле с получением соединения 158-3 (1,3 г, выход 40,2%) в виде коричневого твердого вещества. MS ESI рассч. для C11H17BN2O3 [М+Н]+ 237, найденное значение 237.
[390] Стадия 3. Соединение 158-3 (324 мг, 1,37 ммоль), Pd(dppf)Cl2 (100,3 мг, 1,37 ммоль) и карбонат натрия (290 мг, 2,74 ммоль) добавляли в раствор 8-бром-3-йод-4-метоксилхинолина (500 мг, 1,37 ммоль) в перемешанном растворителе THF (4 мл), Н2О (1 мл) и DMF (1 мл), реакционную смесь перемешивали при 60°C в течение 3 ч. Затем реакционную смесь выливали в Н2О, экстрагировали с помощью EtOAc, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта 158-4 (400 мг, выход 68,4%) в виде коричневого твердого вещества. MS ESI рассч. для C21H22BrN3O2 [М+Н]+ 428, найденное значение 428.
[391] Стадия 4. 4-цианофенилбороновую кислоту (206 мг, 1,4 ммоль), Pd(dppf)Cl2 (34 мг, 0,0466 ммоль) и карбонат натрия (100 мг, 0,932 ммоль) добавляли в раствор соединения 158-4 (200 мг, 0,466 ммоль) в перемешанном растворителе THF (3 мл), H2O (0,5 мл) и DMF (0,5 мл). Реакционную смесь перемешивали при 70°C в течение 4 ч. Затем смесь выливали в H2O и экстрагировали с помощью EtOAc, органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, неочищенный продукт очищали с помощью препаративной HPLC с получением титульного соединения (100 мг, выход 47,8%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,89 (s, 1Н), 8,50 (s, 1H), 8,31 (d, J=7,2 Гц, 1Н), 7,78-7,87 (m, 5Н), 7,66-7,71 (m, 2Н), 6,78 (d, J=8,8 Гц, 1H), 4,15 (d, J=12 Гц, 2Н), 3,76 (s, 5Н), 2,63 (t, J=11,6 Гц, 2Н), 1,31 (d, J=6,4 Гц, 7Н). MS ESI рассч. для C28H26N4O2 [М+Н]+ 451, найденное значение 451.
[392] Соединения, перечисленные в таблице 13, синтезировали с помощью соединения 158-4 и соответствующих борных кислот.
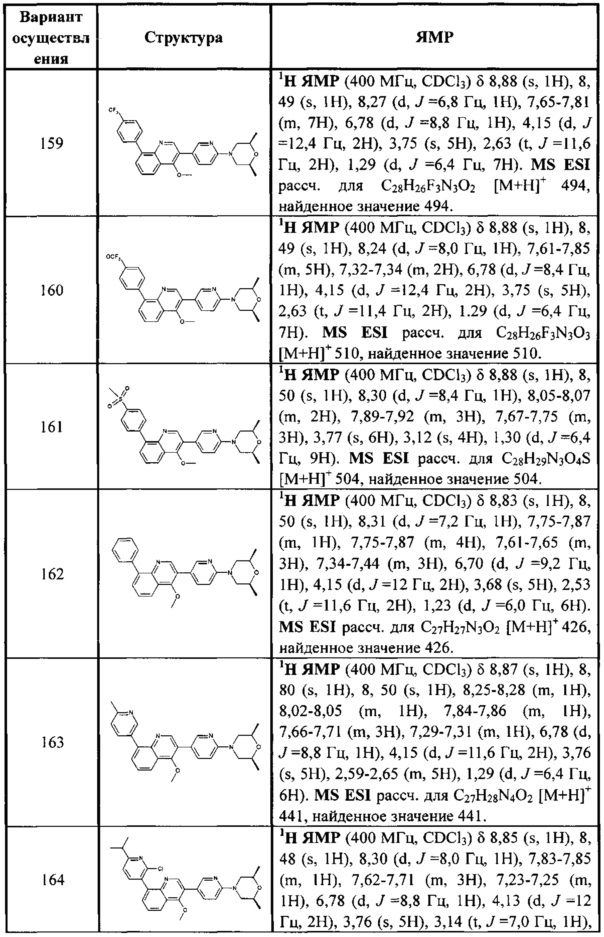
Вариант осуществления 166
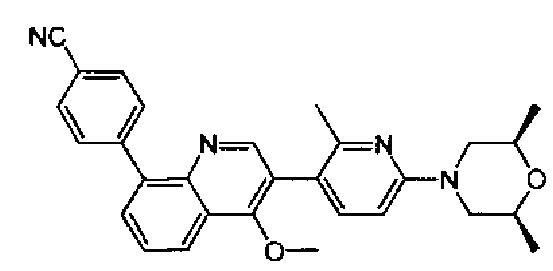
4-(3-(6-((2S,6R)-2,6-диметилморфолино)-2-метилпиридин-3-ил)-4-метоксихинолин-8-ил)бензонитрил
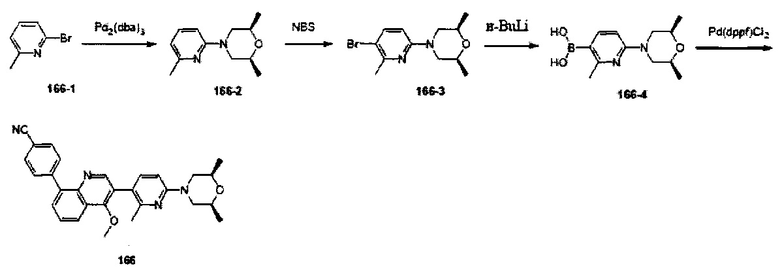
[393] Стадия 1. Xantphos (1,7 г, 3,48 ммоль), (2S,6R)-2,6-диметилморфолин (6 г, 52,2 ммоль), трет-бутоксид калия (3,9 г, 34,8 ммоль), Pd2(dba)3 (1,6 г, 1,74 ммоль) добавляли в раствор 2-бром-6-метилпиридина (3 г, 17,4 ммоль) в толуоле (50 мл), соответственно. Реакционную смесь перемешивали при 110°C в течение 2 ч., затем выливали в H2O, экстрагировали с помощью EtOAc (50 мл×3), органическую фазу промывали солевыми растворами, фильтровали и сушили над сульфатом натрия. После концентрирования остаток очищали с помощью колоночной хроматографии на силикагеле с получением соединения 166-2 (2,4 г, выход 66,7%) в виде белого твердого вещества. MS ESI рассч. для C12H18N2O [М+Н]+ 207, найденное значение 207.
[394] Стадия 2. NBS (1,65 г, 9,3 ммоль) добавляли в раствор соединения 166-2 (2,4 г, 11,6 ммоль) в DMF (30 мл). Реакционную смесь перемешивали при комнатной температуре в течение 6 ч., выливали в H2O, экстрагировали с помощью EtOAc (30×3 мл), органическую фазу сушили над сульфатом натрия, после концентрирования неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с получением соединения 166-3 (0,86 г, выход 25,9%) в виде белого твердого вещества. MS ESI рассч. для C12H17BrN2O [М+Н]+ 285, найденное значение 285.
[395] Стадия 3. Раствор н-BuLi (1,8 мл, 4,5 ммоль) добавляли в раствор соединения 166-3 (0,86 г, 3 ммоль) в THF (20 мл) при -78°C. Реакционную смесь перемешивали при -78°C в течение 30 мин. Затем добавляли триизопропилборат (846 мг, 4,5 ммоль). Затем реакционную смесь выливали в H2O, экстрагировали с помощью EtOAc (50 мл×3), органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта 166-4 (375 мг) в виде белого твердого вещества. MS ESI рассч. для C12H19BN2O3 [М+Н]+ 251, найденное значение 251.
[396] Стадия 4. (6-((2S,6R)-2,6-диметилморфолино)-2-метилпиридин-3-ил)борную кислоту (375 мг, 1,5 ммоль), 4-(3-бром-4-метоксихинолин-8-ил)бензонитрил (340 мг, 1 ммоль) и карбонат натрия (212 мг, 2 ммоль) растворяли в DMF (3 мл), H2O (3 мл) и THF (15 мл), Pd(dppf)Cl2 (73 мг, 0,1 ммоль) добавляли в раствор, затем реакционную смесь перемешивали при 70°C в течение 2 ч. Затем реакционную смесь выливали в Н2О, экстрагировали с помощью EtOAc, органическую фазу промывали солевыми растворами, сушили над сульфатом натрия. Неочищенный продукт очищали с помощью препаративной HPLC с получением титульного соединения (55 мг, выход 8%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,64 (s, 1H), 8,29 (d, J=8,0 Гц, 1H), 7,93-7,95 (m, 2Н), 7,84-7,89 (m, 3Н), 7,52-7,54 (m, 1Н), 7,45-7,50 (m, 1H), 6,82 (d, J=8,4 Гц, 1H), 4,23 (d, J=12 Гц, 2Н), 3,63-3,65 (m, 5Н), 2,62-2,66 (m, 2Н), 2,22 (s, 2Н), 1,19 (d, J=6,0 Гц, 7Н). MS ESI рассч. для C29H28N4O2 [М+Н]+ 465, найденное значение 465.
[397] Соединение, перечисленное в таблице 14, синтезировали с помощью соответствующего арилгалогенида.
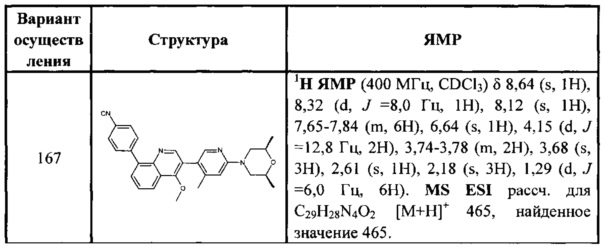
Вариант осуществления 168
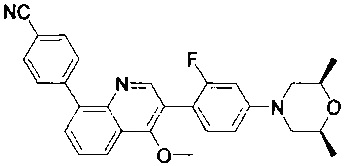
4-(3-(4-((2S,6R)-2,6-диметилморфолино)-2-фторфенил)-4-метоксихинолин-8-ил)бензонитрил
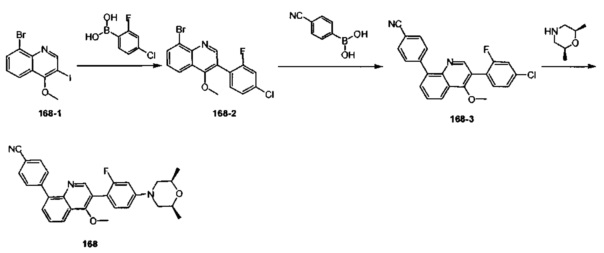
[398] Стадия 1. (4-хлор-2-фторфенил)борную кислоту (0,87 г, 5 ммоль) добавляли в раствор соединения 168-1 (1,8 г, 5 ммоль) в THF/H2O/DMF (15/3/3 мл), Pd(dppf)Cl2 (350 мг, 0,5 ммоль) и карбонат натрия (1,1 г, 10 ммоль) добавляли при комнатной температуре. Реакционную смесь перемешивали при 70°C в течение 4 ч. Реакционную смесь выливали в H2O, экстрагировали с помощью EtOAc, органическую фазу сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле с элюированием PE/EtOAc (15/1) с получением соединения 168-2 (1,158 г, выход 63,3%) в виде коричневого твердого вещества. MS ESI рассч. для C16H10BrClFNO [М+Н]+ 366, найденное значение 366.
[399] Стадия 2. 4-цианофенилборную кислоту (0,2 г, 1,4 ммоль), Pd(dppf)Cl2 (73 мг, 0,1 ммоль) и карбонат натрия (0,4 г) добавляли в раствор соединения 168-2 (0,4 г, 1 ммоль) в THF/H2O/DMF (5/1/1 мл) при комнатной температуре. Реакционную смесь перемешивали при 70°C в течение 5 ч. Реакционную смесь выливали в H2O, экстрагировали с помощью EtOAc, органическую фазу сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле с элюированием РЕ/EtOAc (10/1) с получением соединения 168-3 (158 мг, выход 43,2%) в виде коричневого твердого вещества. MS ESI рассч. для C23H14ClFN2O [М+Н]+ 389, найденное значение 389.
[400] Стадия 3. Титульное соединение (100 мг, выход 55,6%) синтезировали в соответствии с вышеупомянутым способом в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,78 (s, 1H), 8,34 (d, J=6,8 Гц, 1H), 7,85-7,60 (m, 6H), 7,37 (t, J=6,4 Гц, 1H), 6,82-6,71 (m, 2H), 3,90-3,75 (m, 2H), 3,75 (s, 3Н), 2,53 (t, J=11,2 Гц, 2H), 1,31 (d, J=6,8 Гц, 6H). MS ESI рассч. для C29H26FN3O2 [M+H]+ 468, найденное значение 468.
[401] Соединения, перечисленные в таблице 15, синтезировали с помощью соединения 168-1 и соответствующих борных кислот
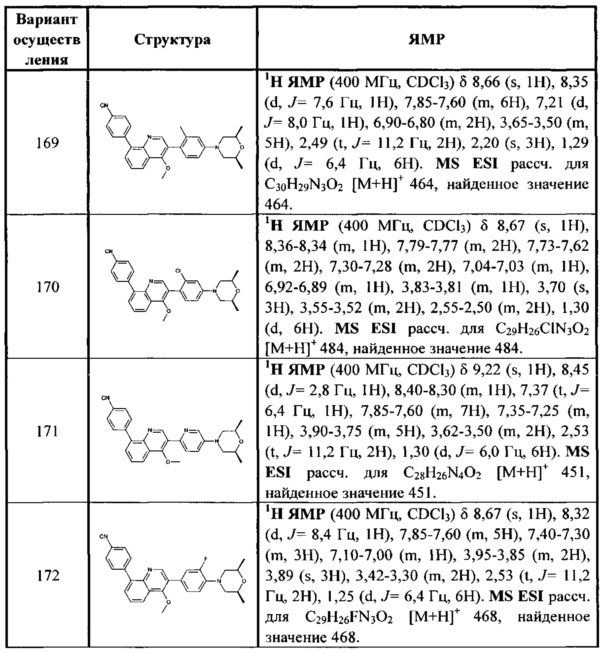
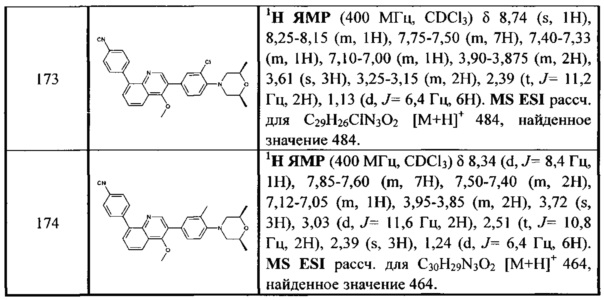
Вариант осуществления 175
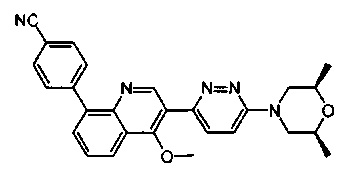
4-(3-(6-((2S,6R)-2,6-диметилморфолино)пиридазин-3-ил)-4-метоксихинолин-8-ил)бензонитрил
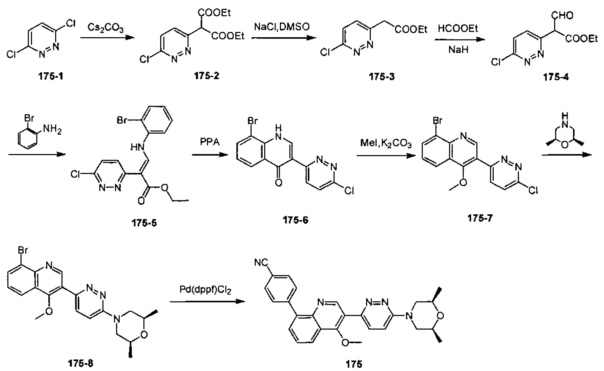
[402] Стадия 1. Карбонат цезия (176 г, 540 ммоль) добавляли в раствор соединения 175-1 (40 г, 270 ммоль) и диэтилмалоната (61 мл, 410 ммоль) в DMSO (80 мл) при комнатной температуре. Реакционную смесь перемешивали при 110°C в течение 1 ч. Реакционную смесь выливали в H2O, экстрагировали с помощью EtOAc. Органическую фазу сушили над сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью хроматографии на силикагеле (PE:EtOAc = 10:1) с получением соединения 175-2 (52 г, выход 71%) в виде бесцветного масла. MS ESI рассч. для C11H13ClN2O4 [М+Н]+ 273, найденное значение 273.
[403] Стадия 2. Соединение 2 (52 г, 190 ммоль), NaCl (45 г, 760 ммоль) и H2O (5 мл) добавляли в DMSO (300 мл), реакционную смесь перемешивали при температуре в диапазоне от 150 до 160°C в течение 2 ч. Реакционную смесь выливали в H2O, экстрагировали с помощью EtOAc. Органическую фазу концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью хроматографии на силикагеле (РЕ:EtOAc = 5:1) с получением соединения 175-3 (30 г, выход 79%) в виде бесцветного масла. MS ESI рассч. для C8H9ClN2O2 [М+Н]+ 201, найденное значение 201.
[404] Стадия 3. В атмосфере газа азота NaH (8,4 г, 350 ммоль) добавляли в раствор соединения 175-3 (20 г, 100 ммоль) в HCO2Et (200 мл) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Затем реакционную смесь выливали в 5%-ный водный раствор HCl, экстрагировали с помощью EtOAc. Органическую фазу промывали с помощью водного раствора Na2CO3 и сушили над сульфатом натрия. Неочищенный продукт очищали с помощью хроматографии на силикагеле (PE:EtOAc = 8:1) с получением соединения 175-4 (6 г, выход 26%) в виде бесцветного масла. ESI рассч. для C9H9ClN2O3 [М+Н]+ 229, найденное значение 229.
[405] Стадия 4. Раствор соединения 175-4 (5 г, 22 ммоль) и 2-бромфениламина (3,8 г, 22 ммоль) в EtOH (100 мл) перемешивали при 70°C в течение 20 ч. Реакционную смесь охлаждали до комнатной температуры. Неочищенный продукт собирали путем фильтрования с получением соединения 175-5 (5 г, выход 57%) в виде желтого твердого вещества. MS ESI рассч. для C15H13BrClN3O2 [М+Н]+ 382, найденное значение 382.
[406] Стадия 5. Соединение 175-5 (4,4 г, 11,5 ммоль) растворяли в РРА (50 мл). Реакционную смесь перемешивали при комнатной температуре в течение 12 ч. Реакционную смесь выливали в ледяную воду. Продукт собирали путем фильтрования с получением соединения 175-6 (3 г, выход 77%) в виде желтого твердого вещества. MS ESI рассч. для C13H7BrClN3O [М+Н]+ 335, найденное значение 335.
[407] Стадия 6. Раствор соединения 175-6 (1,4 г, 4,2 ммоль), CH3I (887 мг, 6,2 ммоль) и Ag2CO3 (2,3 г, 8,4 ммоль) в DMF (20 мл) перемешивали при 70°C в течение 4 ч. Реакционную смесь выливали в H2O, экстрагировали с помощью DCM. Органическую фазу промывали солевыми растворами, сушили над сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью хроматографии на силикагеле (РЕ:EtOAc = 1:1) с получением соединения 175-7 (540 мг, выход 37%) в виде желтого твердого вещества. MS ESI рассч. для C14H9BrClN3O [М+Н]+ 350, рассч. для 350.
[408] Стадия 7. Раствор соединения 175-7 (540 мг, 1,55 ммоль) в (2S,6R)-2,6-диметилморфолине (1 мл) перемешивали при 120°C в течение 2 ч. Реакционную смесь выливали в H2O, экстрагировали с помощью DCM. Объединенную органическую фазу промывали солевыми растворами, сушили над сульфатом натрия и концентрировали при пониженном давлении с получением соединения 175-8 (663 мг, 100%) в виде желтого твердого вещества. Неочищенный продукт непосредственно применяли на следующей стадии без дополнительной очистки. MS ESI рассч. для C20H21BrN4O2 [М+Н]+ 429, найденное значение 429.
[409] Стадия 8. Раствор Pd(dppf)Cl2 (117 мг, 0,16 ммоль) и карбоната натрия (329 мг, 3,1 ммоль) в THF/H2O/DMF (10/2/2) добавляли в раствор соединения 175-8 (663 мг, 1,55 ммоль), 4-цианофенилборной кислоты (251 мг, 1,7 ммоль), перемешивали при 90°C в течение 3 ч. Реакционную смесь фильтровали и разделяли между EtOAc и H2O, органическую фазу сушили и концентрировали. Неочищенный продукт очищали с помощью препаративной HPLC с получением титульного соединения (90 мг, выход 13%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, метанол-d4) δ 8,96 (s, 1Н), 8,65 (d, J=9,6 Гц, 1Н), 8,56 (d, J=10,0 Гц, 1H), 8,06 (d, J=8,4 Гц, 1Н), 7,92 (d, J=8,4 Гц, 1H), 7,81-7,61 (m, 6Н), 4,29 (d, J=12,8 Гц, 2Н), 3,81-3,71 (m, 2Н), 3,30 (s, 3Н), 2,81 (t, J=12,0 Гц, 2Н), 1,28 (d, J=5,6 Гц, 6Н). MS ESI рассч. для C27H25N5O2 [М+Н]+ 452, найденное значение 452.
Вариант осуществления 176
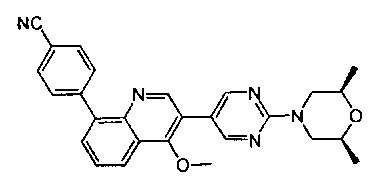
4-(3-(2-((2S,6R)-2,6-диметилморфолино)пиримидин-5-ил)-4-метоксихинолин-8-ил)бензонитрил
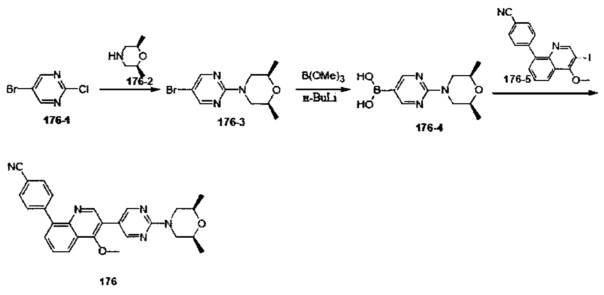
[410] Стадия 1. Соединение 176-1 (3 г, 15,5 ммоль) растворяли в соединении 176-2 (1,8 г, 15,5 ммоль), затем перемешивали в течение 2 ч. при 100°C. Раствор разделяли между EtOAc и H2O, органическую фазу промывали солевыми растворами, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения 176-3 (3,1 г, 74%) в виде белого твердого вещества. MS ESI рассч. для C10H14BrN3O [М+Н]+ 272, найденное значение 272.
[411] Стадия 2. Соединение 176-3 (4,2 г, 15,4 ммоль) растворяли в THF (40 мл), н-BuLi (9,2 мл, 23,1 ммоль) по каплям добавляли в раствор при -78°C. Смесь перемешивали в течение 30 мин., затем по каплям добавляли В(ОМе)3 (4,8 г, 46,2 ммоль). Затем реакционную смесь нагревали до 0°C и перемешивали в течение 3 ч. Реакционную смесь разделяли между EtOAc и H2O, органическую фазу отделяли, объединяли, промывали солевыми растворами, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением соединения 176-4 (2,6 г, 72%) в виде коричневого твердого вещества. MS ESI рассч. для C10H16BN3O3 [М+Н]+ 237, найденное значение 237.
[412] Стадия 3. Титульное соединение синтезировали в соответствии с вышеупомянутым способом (23 мг, 10%) в виде коричневого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,83 (s, 1Н), 8,65 (s, 2Н), 8,27 (d, J=7,2 Гц, 1Н), 7,85-7,60 (m, 6Н), 4,63 (d, J=12,8 Гц, 2Н), 3,80 (s, 3Н), 3,75-3,61 (m, 2Н), 2,68 (t, J=12,0 Гц, 2Н), 1,27 (d, J=6,0 Гц, 6Н), MS ESI рассч. для C27H25N5O2 [М+Н]+ 452, найденное значение 452.
[413] Соединение, перечисленное в таблице 16, синтезировали с помощью соответствующего арилгалогенида.
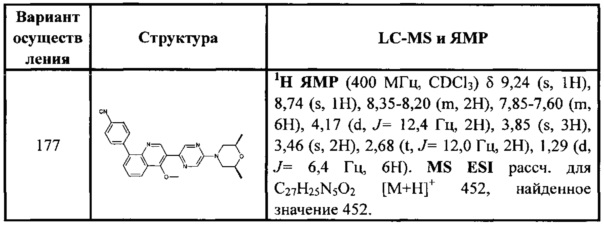
Вариант осуществления 178

4-(3-(5-((2S,6R)-2,6-диметилморфолин-4-карбонил)тиофен-2-ил)-4-метоксихинолин-8-ил)бензонитрил
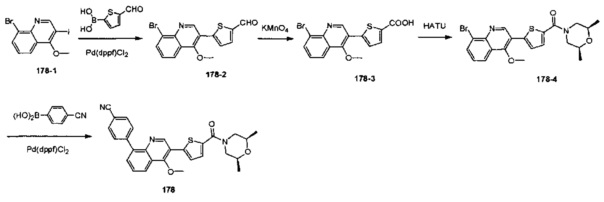
[414] Стадия 1. Pd(dppf)Cl2 (102 мг, 0,14 ммоль) добавляли в раствор соединения 178-1 (500 мг, 1,4 ммоль), (5-формилтиофен-2-ил)борной кислоты (270 мг, 1,7 ммоль) и карбоната натрия (300 мг, 2,8 ммоль) в DMF (2 мл)/H2O (2 мл)/THF (10 мл). Смесь перемешивали при 70°C в течение 2 ч. Смесь выливали в H2O, экстрагировали с помощью EtOAc, органическую фазу промывали солевыми растворами, сушили над сульфатом натрия, концентрировали. Остаток очищали с помощью колоночной хроматографии (РЕ/EtOAc = 5:1) с получением соединения 178-2 (438 мг, 91,6%) в виде коричневого твердого вещества. MS ESI рассч. для C15H10BrNO2S [М+Н]+ 348, найденное значение 348.
[415] Стадия 2. Дигидрофосфат натрия (300 мг, 2,5 ммоль) добавляли в раствор соединения 178-2 (438 мг, 1,25 ммоль) и перманганата калия (200 мг, 1,25 ммоль) в ацетоне (15 мл). Смесь перемешивали при комнатной температуре в течение 4 ч. Коричневый осадок MnO2 фильтровали, ацетон удаляли под вакуумом, смесь окисляли с помощью 1 н. HCl и экстрагировали с помощью EtOAc. Органическую фазу сушили над безводным сульфатом натрия, концентрировали с получением соединения 178-3 (350 мг, 76,4%) в виде коричневого твердого вещества. MS ESI рассч. для C15H10BrNO3S [М+Н]+ 364, найденное значение 364.
[416] Стадия 3. К раствору соединения 178-3 (350 мг, 1 ммоль), HATU (384 мг, 1 ммоль) и (2S,6R)-2,6-диметилморфолина (138 мг, 1,2 ммоль) в DMF (10 мл) добавляли DIPA (387 мг, 3 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь выливали в H2O, экстрагировали с помощью EtOAc, органическую фазу сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью колоночной хроматографии (РЕ/EtOAc = 2:1) с получением соединения 178-4 (265 мг, 59,8%) в виде коричневого твердого вещества. MS ESI рассч. для C21H21BrN2O3S [М+Н]+ 461, найденное значение 461.
[417] Стадия 4. Pd(dppf)Cl2 (45 мг, 0,06 ммоль) добавляли в раствор соединения 178-4 (265 мг, 0,6 ммоль), 4-цианофенилборной кислоты (106 мг, 0,72 ммоль) и карбоната натрия (127 мг, 1,2 ммоль) в DMF (2 мл)/H2O (растворитель 2 мл)/THF (10 мл). Смесь перемешивали при 70°C в течение 2 ч. Реакционную смесь выливали в H2O, экстрагировали с помощью EtOAc, органическую фазу промывали солевыми растворами, сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью препаративной HPLC с получением титульного соединения (32 мг, выход 11%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 9,20 (s, 1Н), 8,26-8,24 (m, 1H), 7,83-7,78 (m, 4H), 7,74-7,68 (m, 2H), 7,56-7,55 (m, 1H), 7,35-7,34 (m, 1H), 4,03 (s, 3H), 3,67 (s, 1H), 1,24 (d, 6H). MS ESI рассч. для C28H25N3O3S [M+H]+ 484, найденное значение 484.
Вариант осуществления 179
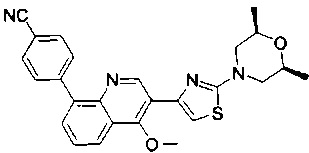
4-(3-(2-(2,6-диметилморфолин-4-ил)тиазол-4-ил)-4-метоксихинолин-8-ил)бензонитрил
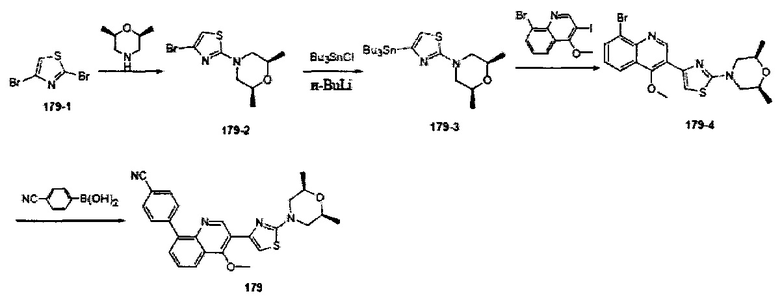
[418] Стадия 1. K2CO3 (5,7 г, 41 ммоль) добавляли в раствор соединения 179-1 (5,0 г, 20,5 ммоль) и (2S,6R)-2,6-диметилморфолина (3,55 г, 30,9 ммоль) в DMF (60 мл). Смесь перемешивали при 70°C в течение ночи. Реакционную смесь охлаждали до комнатной температуры, затем выливали в H2O, экстрагировали с помощью EtOAc, сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью колоночной хроматографии с получением соединения 179-2 (5,56 г, выход 99%) в виде желтого масла. MS ESI рассч. для C9H13BrN2OS [М+Н]+ 277, найденное значение 277.
[419] Стадия 2. н-BuLi (0,8 мл, 2,5 М в растворе THF) добавляли в раствор соединения 179-2 (500 мг, 1,8 ммоль) в THF (10 мл) при -78°C, смесь перемешивали при -78°C в течение 1 ч., затем добавляли SnBu3Cl (650 мг, 2 ммоль) при -78°C и перемешивали при -78°C в течение 1 ч. Реакционную смесь выливали в H2O, экстрагировали с помощью EtOAc, сушили над сульфатом натрия, концентрировали с получением соединения 179-3 (0,7 г, выход 79%) в виде бесцветного масла. MS ESI рассч. для C21H40N2OSSn [М+Н]+ 489, найденное значение 489.
[420] Стадия 3. Pd(PPh3)4 (80 мг, 0,08 ммоль) добавляли в раствор соединения 179-3 (500 мг, 1,2 ммоль) и 8-бром-3-йод-4-метоксихинолина (230 мг, 0,6 ммоль) в толуоле (10 мл). Смесь перемешивали при 110°C в течение ночи. Реакционную смесь разделяли между H2O и EtOAc. Органическую фазу сушили над сульфатом натрия, концентрировали с получением соединения 179-4 (50 мг, выход 12%) в виде желтого твердого вещества. MS ESI рассч. для C19H20BrN3O2S [М+Н]+ 433, найденное значение 433.
[421] Стадия 4. Pd(dppf)Cl2 (25 мг, 0,03 ммоль) и карбонат натрия (50 мг, 0,5 ммоль) добавляли в раствор соединения 179-4 (50 мг, 0,14 ммоль) и 4-цианофенилборной кислоты (48 мг, 0,14 ммоль) в THF/H2O/DMF (42 мл, 61 секунду после пяти). Смесь перемешивали при 70°C в течение ночи. Реакционную смесь выливали в H2O, экстрагировали с помощью EtOAc, органическую фазу сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью препаративной HPLC с получением титульного соединения (6 мг, выход 10%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 9,45 (s, 1Н), 8,15-8,10 (m, 1Н), 7,75-7,50 (m, 6Н), 7,20 (s, 1H), 3,85 (s, 3Н), 3,75-3,60 (m, 4Н), 2,68 (t, J=10,8 Гц, 2Н), 1,15 (d, J=6,4 Гц, 6Н). MS ESI рассч. для C26H24N4O2S [М+Н]+ 457, найденное значение 457.
Вариант осуществления 180
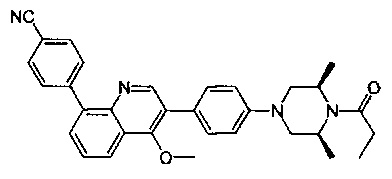
4-(3-(4-((3S,5R)-3,5-диметил-4-пропионилпиперазин-1-ил)фенил)-4-метоксихинолин-8-ил)бензонитрил
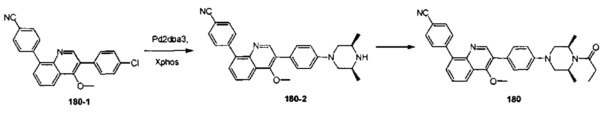
[422] Стадия 1. Pd2(dba)3 (330 мг, 0,3 ммоль), Xantphos (350 мг, 0,7 ммоль) и KOtBu (1,6 г, 15 ммоль) добавляли в раствор соединения 180-1 (2,0 г, 5,4 ммоль) и (2S,6R)-2,6-диметилпиперазина (0,8 г, 7 ммоль) в толуоле (30 мл). В атмосфере газа азота реакционную смесь перемешивали при 120°C в течение 5 ч., затем выливали в H2O. Смесь экстрагировали с помощью эфира (3×30 мл), органическую фазу сушили над сульфатом натрия. Остаток очищали с помощью колоночной хроматографии с получением соединения 180-2 (1,7 г, выход 70%) в виде желтого твердого вещества. MS ESI рассч. для C29H28N4O [М+Н]+ 449, найденное значение 449.
[423] Стадия 2. Пропионилхлорид (130 мг, 0,1 ммоль) добавляли в раствор соединения 180-2 (90 мг, 0,2 ммоль) и TEA (200 мг, 2 ммоль) в DCM (10 мл) при комнатной температуре. Реакционную смесь перемешивали в течение 1 ч., а затем выливали в H2O, экстрагировали с помощью DCM (2×10 мл), органическую фазу сушили над сульфатом натрия. Остаток очищали с помощью препаративной HPLC с получением титульного соединения (50 мг, выход 40%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,89 (s, 1H), 8,47-8,45 (m, 2Н), 7,97-7,81 (m, 4H), 7,62 (d, J=3,6 Гц, 2H), 7,46 (d, J=4,8 Гц, 2H), 7,02 (d, J=8,8 Гц, 2H), 3,89 (s, 3Н), 3,56 (d, J=12,4 Гц, 2H), 3,00 (d, J=9,6 Гц, 2H), 2,51-2,38 (m, 2H), 1,49-1,36 (m, 6H), 1,19 (t, J=7,6 Гц, 3Н). MS ESI рассч. для C32H32N4O2 [M+H]+ 505, найденное значение 505.
[424] Соединения, перечисленные в таблице 17, синтезировали с помощью соединения 180-2 и соответствующих ацилхлоридов.
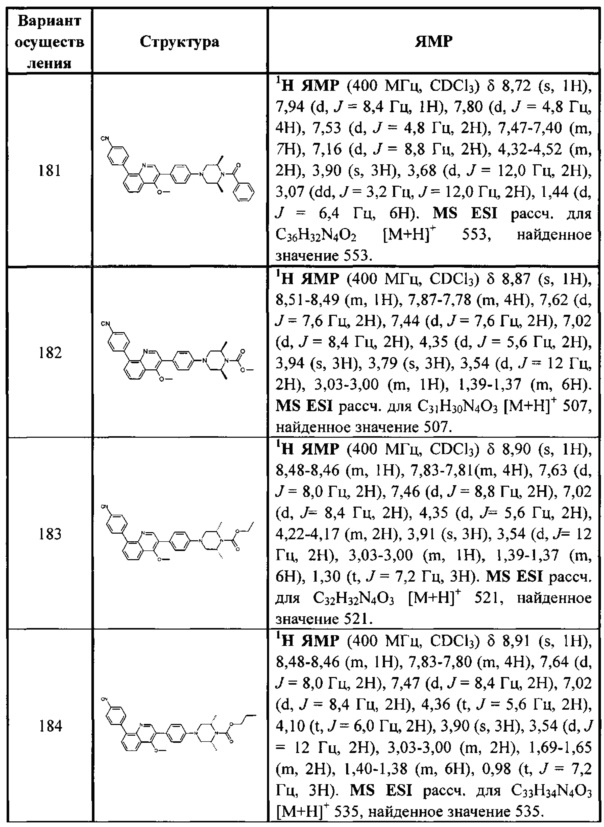
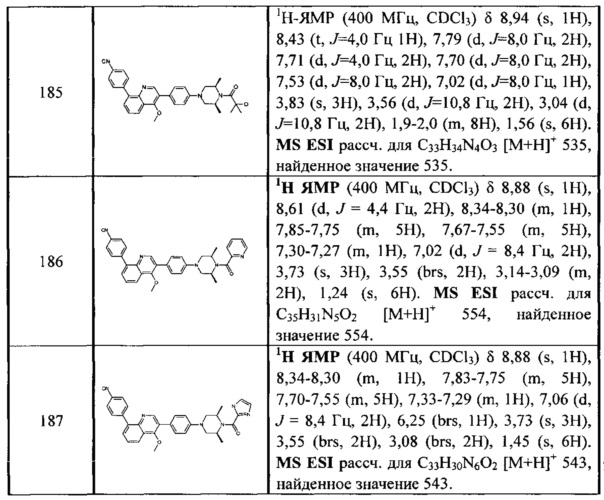
Вариант осуществления 188
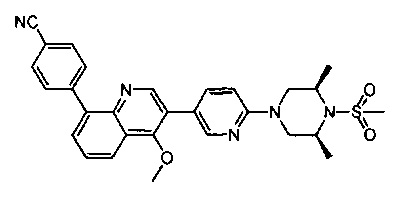
4-(3-(6-(4-метилсульфонил-3,5-диметилпиперазин-1-ил)пиридин-3-ил)-4-метоксихинолин-8-ил)бензонитрил
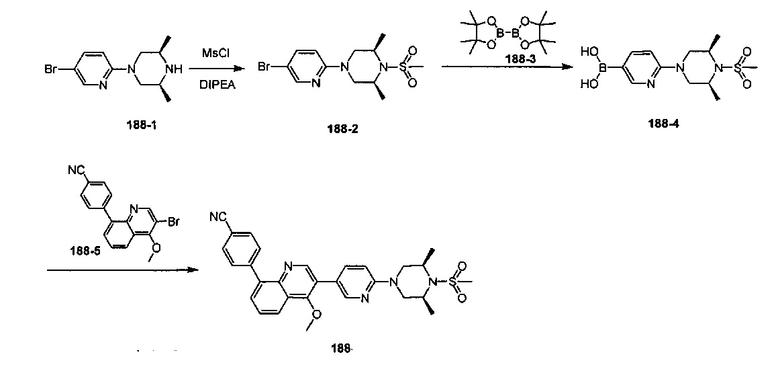
[425] Стадия 1. MsCl (290 мг, 2,4 ммоль) по каплям добавляли в раствор соединения 188-1 (540 мг, 2 ммоль) и DIPEA (774 мг, 6 ммоль) в DCM (10 мл), реакционную смесь перемешивали при комнатной температуре в течение 30 мин. и наблюдали с помощью LCMS до завершения реакции. Смесь выливали в H2O, экстрагировали с помощью EtOAc, органическую фазу сушили над безводным сульфатом натрия, концентрировали с получением неочищенного продукта, который очищали с помощью колоночной хроматографии на силикагеле (МеОН:EtOAc=5:1) с получением соединения 188-2 (500 мг, выход 72%) в виде белого твердого вещества. MS ESI рассч. для C12H18BrN3O2S [М+Н]+ 350, найденное значение 350.
[426] Стадия 2. Pd(dppf)Cl2 (102 мг, 0,14 ммоль) добавляли в раствор соединения 188-2 (500 мг, 1,4 ммоль), 188-3 (432 мг, 1,7 ммоль) и AcOK (300 мг, 2,8 ммоль) в диоксане (10 мл), реакционную смесь перемешивали при 70°C в течение 2 ч. и наблюдали с помощью LCMS до завершения реакции. Затем смесь выливали в H2O, экстрагировали с помощью EtOAc, органическую фазу сушили над безводным сульфатом натрия, концентрировали с получением неочищенного продукта, который очищали с помощью колоночной хроматографии на силикагеле (РЕ:EtOAc=1:1) с получением соединения 188-4 (460 мг, выход 100%) в виде желтого твердого вещества. MS ESI рассч. для C12H20BN3O4S [М+Н]+ 314, найденное значение 314.
[427] Стадия 3. Титульное соединение (120 мг, 23%) синтезировали в соответствии с вышеупомянутым способом в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,90 (s, 1Н), 8,50 (d, J=2,0 Гц, 1H), 8,33 (d, J=6,8 Гц, 1Н), 7,90-7,73 (m, 7Н), 6,82 (d, J=8,8 Гц, 1H), 4,28-4,22 (m, 4Н), 3,80 (s, 3Н), 3,29 (dd, J=4,4, 12,8 Гц, 2Н), 2,96 (s, 3Н), 1,51 (d, J=7,2 Гц, 6Н). MS ESI рассч. для C29H29N5O3S [М+Н]+ 528, найденное значение 528.
Вариант осуществления 189
4-(3-(6-(4-(2-гидроксиацетил)-3,5-диметилпиперазин-1-ил)пиридин-3-ил)-4-метоксихинолин-8-ил)бензонитрил
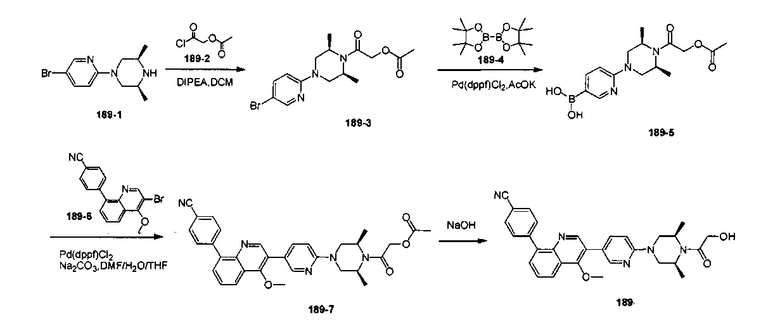
[428] Стадия 1. Соединение 189-2 (227 мг, 1,7 ммоль) добавляли в раствор соединения 189-1 (300 мг, 1,1 ммоль) и DIPEA (430 мг, 3,3 ммоль) в DCM (30 мл), реакционную смесь перемешивали при 0°C в течение 30 мин. и наблюдали с помощью LCMS до завершения реакции. Затем смесь выливали в H2O, экстрагировали с помощью EtOAc, органическую фазу сушили над безводным сульфатом натрия, концентрировали с получением неочищенного продукта, который очищали с помощью колоночной хроматографии на силикагеле (РЕ:EtOAc = 5:1) с получением соединения 189-3 (260 мг, выход 63%) в виде красного масла. MS ESI рассч. для C15H20BrN3O3 [M+H]+ 370, найденное значение 370.
[429] Стадия 2. Pd(dppf)Cl2 (102 мг, 0,14 ммоль) добавляли в раствор соединения 189-3 (260 мг, 0,7 ммоль), 189-4 (213 мг, 0,84 ммоль) и AcOK (205 мг, 2,1 ммоль) в диоксане (6 мл), реакционную смесь перемешивали при 100°C в течение 2 ч. и наблюдали с помощью LCMS до завершения реакции. Затем смесь выливали в H2O, экстрагировали с помощью EtOAc, органическую фазу сушили над безводным сульфатом натрия, концентрировали с получением неочищенного продукта, который очищали с помощью колоночной хроматографии на силикагеле (РЕ:EtOAc = 1:1) с получением соединения 189-5 (170 мг, выход 70%). MS ESI рассч. для C15H22BN3O5 [М+Н]+ 336, найденное значение 336.
[430] Стадия 3. Соединение 189-7 (60 мг, 22%) синтезировали в соответствии с вышеупомянутым способом в виде белого твердого вещества. MS ESI рассч. для C32H31N5O4 [М+Н]+ 550, найденное значение 550.
[431] Стадия 4. NaOH (132 мг, 3,3 ммоль) добавляли в раствор соединения 189-7 (60 мг, 0,11 ммоль) в МеОН (2 мл)/THF (2 мл)/H2O (2 мл), реакционную систему перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь наблюдали с помощью LC-MS до завершения реакции. Смесь выливали в H2O, промывали с помощью EtOAc, водную фазу окисляли до рН=4 с помощью 1 н. HCl, затем экстрагировали с помощью EtOAc, органическую фазу сушили над безводным сульфатом натрия, концентрировали с получением неочищенного продукта, который дополнительно очищали с помощью TLC (DCM:MeOH = 20:1) с получением титульного соединения (10 мг) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,91 (s, 1Н), 8,51 (s, 1H), 8,35-8,33 (m, 1H), 7,86-7,70 (m, 7H), 6,87 (d, J=8,8 Гц, 1H), 3,80 (s, 3H), 1,67 (s, 3H), 1,44 (m, 6Н). MS ESI рассч. для C30H29N5O3 [M+H]+ 508, найденное значение 508.
Вариант осуществления 190
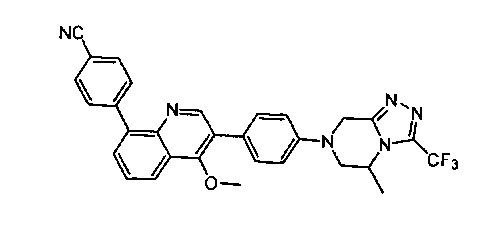
4-(4-метокси-3-(4-(5-метил-3-(трифторметил)-5,6-дигидро-[1,2,4]триазоло[4,3-а]пиразин-7(8Н)-ил)фенил)хинолин-8-ил)бензонитрил
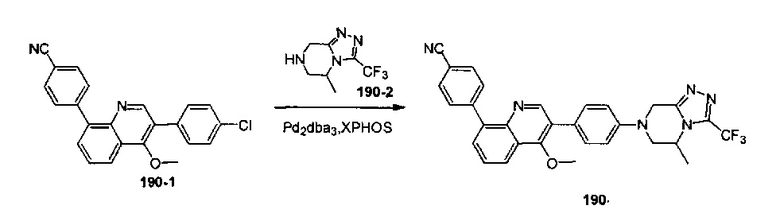
[432] Pd2(dba)3 (45 мг, 0,05 ммоль), Xantphos (50 мг, 0,1 ммоль) и KOtBu (0,23 г, 2 ммоль) добавляли в раствор соединения 190-1 (0,185 г, 0,5 ммоль) и 190-2 (0,12 г, 0,6 ммоль) в толуоле (30 мл), в атмосфере газа азота реакционную смесь перемешивали при 120°C в течение 5 ч., затем выливали в H2O, смесь экстрагировали с помощью эфира (3×30 мл), органическую фазу сушили над сульфатом натрия, остаток очищали с помощью препаративной HPLC с получением титульного соединения (70 мг, выход 30%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,88 (s, 1Н), 8,40 (t, J=4,8 Гц, 1H), 7,75-7,74 (m, 4H), 7,62-7,52 (m, 4H), 7,04 (d, J=8,4 Гц, 2H), 4,93 (d, J=16 Гц, 1H), 4,69 (s, 1H), 4,45 (d, J=12,0 Гц, 1H), 3,88-3,81 (m, 5H), 1,64 (d, J=9,8 Гц, 3H). MS ESI рассч. для C30H23F3N6O [M+H]+ 541, найденное значение 541.
Вариант осуществления 191
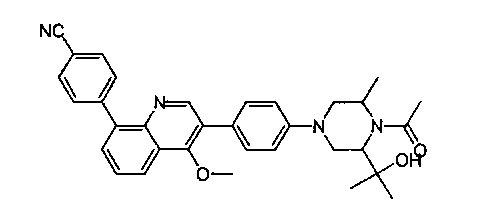
4-(3-(4-(4-ацетил-3-(2-гидроксипропан-2-ил)-5-метилпиперазин-1-ил)фенил)-4-метоксихинолин-8-ил)бензонитрил
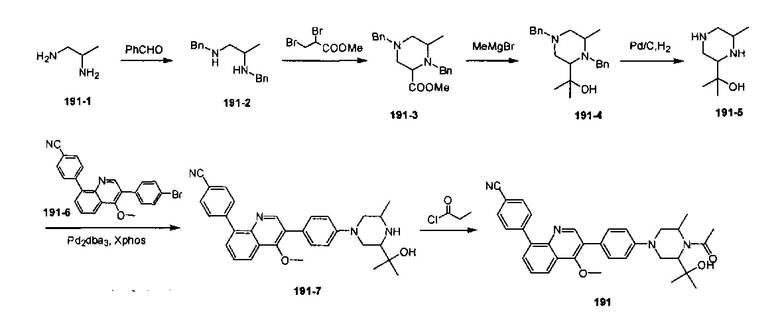
[433] Стадия 1. Бензальдегид (29 г, 270 ммоль) добавляли в раствор соединения 191-1 (10 г, 135 ммоль) в метаноле (100 мл) при 0°C, реакционную смесь перемешивали при комнатной температуре в течение 2 ч., а затем снова охлаждали до 0°C, порциями добавляли боргидрид натрия (10 г, 270 ммоль). Смесь перемешивали при комнатной температуре в течение 16 ч. и концентрировали при пониженном давлении. Остаток разбавляли с помощью H2O и DCM и фильтровали. Органическую фазу собирали и промывали солевыми растворами, сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии с получением соединения 191-2 (14 г, выход 40%) в виде бесцветного масла. MS ESI рассч. для C17H22N2 [М+Н]+ 255, найденное значение 255.
[434] Стадия 2. DIPEA (33 мл, 239 ммоль) добавляли в раствор 2,3-дибромметилпропионата (16,5 мл, 55 ммоль) и соединения 191-2 (14 г, 55 ммоль) в толуоле (50 мл), смесь перемешивали при комнатной температуре в течение ночи, затем разбавляли с помощью EtOAc и промывали солевыми растворами. Органическую фазу собирали и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии с получением соединения 191-3 (12,5 г, выход 66%) в виде желтой жидкости. MS ESI рассч. для C21H26N2O2 [М+Н]+ 339, найденное значение 339.
[435] Стадия 3. Бромид метилмагния (48 мл, 143 ммоль) по каплям добавляли в раствор соединения 191-3 (5,6 г, 15,9 ммоль) в THF (50 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Смесь выливали в насыщенный хлорид аммония, водную фазу экстрагировали с помощью EtOAc. Органическую фазу промывали солевыми растворами, сушили над сульфатом натрия, концентрировали при пониженном давлении с получением соединения 191-4 (3,3 г, выход 61%), которое непосредственно применяли для следующей стадии. MS ESI рассч. для C22H30N2O [М+Н]+ 339, найденное значение 339.
[436] Стадия 4. В атмосфере газа водорода (50 фунтов/кв. дюйм) раствор соединения 191-4 (3,3 г, 9,8 ммоль) и Pd/C (330 мг) в метаноле (50 мл) перемешивали при комнатной температуре в течение 24 ч. Реакционную смесь фильтровали и концентрировали с получением соединения 191-5 (1,4 г, выход 82%) в виде светло-желтого масла. MS ESI рассч. для C8H18N2O [М+Н]+ 159, найденное значение 159.
[437] Стадия 5. Соединение 191-7 (120 мг, выход 25%) синтезировали в соответствии с вышеупомянутым способом в виде светло-желтого масла. MS ESI рассч. для C31H32N4O2 [М+Н]+ 493, найденное значение 493.
[438] Стадия 6. Титульное соединение (13 мг, выход 12%) синтезировали в соответствии с вышеупомянутым способом в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,89 (s, 1H), 8,34-8,31 (m, 1Н), 7,84-7,77 (m, 4Н), 7,69-7,65 (m, 2Н), 7,58 (d, J=8,4 Гц, 2Н), 6,86 (d, J=8,4 Гц, 2Н), 4,41-4,31 (m, 1Н), 4,11-4,01 (m, 1H), 3,91-3,81 (m, 2Н), 3,72 (s, 3Н), 3,71-3,62 (m, 1Н), 3,32-3,21 (m, 1H), 2,23 (s, 3 Н), 1,35 (d, J=7,6 Гц, 3Н), 1,29 (s, 6Н). MS ESI рассч. для C33H34N4O3 [М+Н]+ 535, найденное значение 535.
Вариант осуществления 192
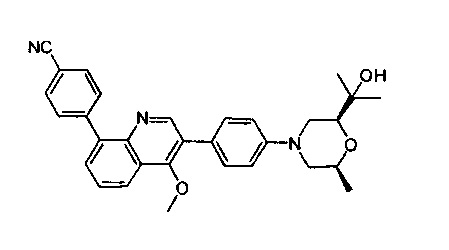
4-(3-(4-((2S,6S)-2-(2-гидроксипропан-2-ил)-6-метилморфолино)фенил)-4-метоксихинолин-8-ил)бензонитрил
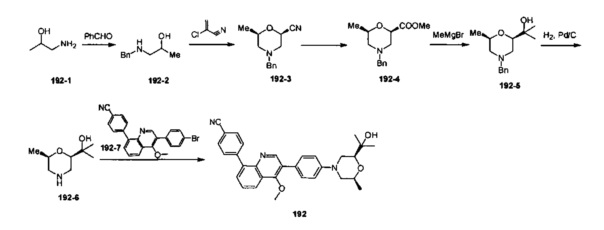
[439] Стадия 1. Раствор соединения 192-1 (2 г, 26,67 ммоль), бензальдегида (2,8 мл, 29 ммоль) и сульфата магния (8 г) в DCM (70 мл) перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь фильтровали и фильтрат концентрировали досуха. Неочищенный продукт растворяли в МеОН (70 мл) и добавляли боргидрид натрия (1 г, 26,6 ммоль), смесь перемешивали при комнатной температуре в течение 5 ч. Смесь концентрировали досуха и экстрагировали с помощью 1 н. разбавленной HCl и EtOAc. Водную фазу подщелачивали до рН=11 с помощью NaOH (2 М) и экстрагировали с помощью DCM. Органическую фазу сушили над безводным сульфатом натрия, концентрировали с получением неочищенного соединения 192-2 (3,17 г, 72%), которое применяли для следующей стадии без дополнительной очистки. MS ESI рассч. для C10H15NO [М+Н]+ 166, найденное значение 166.
[440] Стадия 2. Раствор соединения 192-2 (15 г, 91 ммоль) и 2-хлоракрилонитрила (9,5 г, 109 ммоль) в THF (300 мл) перемешивали при комнатной температуре в течение 36 ч., KOtBu (15,3 г, 136,5 ммоль) добавляли в реакционную систему, смесь нагревали до 70°C и перемешивали в течение дополнительных 2 ч. Реакционную смесь экстрагировали с помощью EtOAc и H2O. Органическую фазу концентрировали, остаток очищали с помощью колоночной хроматографии с получением соединения 192-4 (3,5 г, выход 16%) в виде бесцветного масла. MS ESI рассч. для C13H16N2O [М+Н]+ 217, найденное значение 217.
[441] Стадия 3. Раствор соединения 192-3 (1 г, 4,63 ммоль) и конц. серной кислоты (2,5 мл) в МеОН (15 мл) перемешивали при 100°C в течение ночи. После охлаждения реакционной смеси добавляли насыщенный бикарбонат натрия (20 мл), затем экстрагировали с помощью EtOAc (100 мл). Органическую фазу сушили над безводным сульфатом натрия и концентрировали. Неочищенный продукт очищали с помощью хроматографии с получением соединения 192-4 (460 мг, выход 40%) в виде светло-желтого масла. MS ESI рассч. для C14H19NO3 [М+Н]+ 250, найденное значение 250.
[442] Стадия 4. Бромид метилмагния (1,4 мл, 4,16 ммоль) добавляли в раствор соединения 192-4 (260 мг, 1,04 ммоль) в THF (4 мл) при комнатной температуре и перемешивали в течение 1 ч. Реакционную смесь гасили с помощью насыщенного хлорида аммония (1 мл). Смесь сушили над безводным сульфатом натрия и концентрировали с получением соединения 192-5 (200 мг, выход 77%), которое непосредственно применяли для следующей стадии. MS ESI рассч. для C15H23NO2 [М+Н]+ 250, найденное значение 250.
[443] Стадия 5. Раствор соединения 192-5 (280 мг, 1,12 ммоль) и Pd/C (100 мг) в EtOH (10 мл) нагревали до 75°C при давлении газа водорода 50 фунтов/кв. дюйм и перемешивали в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и фильтровали, фильтрат концентрировали с получением соединения 192-6 (170 мг, выход 95%) в виде бесцветного масла. MS ESI рассч. для C8H17NO2 [М+Н]+ 160, найденное значение 160.
[444] Стадия 6. Титульное соединение (50 мг, выход 28%) синтезировали в соответствии с вышеупомянутым способом в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ ppm 8,90 (s, 1Н) 8,36-8,32 (m, 1H) 7,85-7,78 (m, 4Н) 7,70-7,65 (m, 2Н) 7,60-7,58 (d, J=S, 2Н) 7,07-7,05 (d, J=8, 2Н) 3,87-3,76 (m, 1Н) 3,72 (s, 3Н) 3,642-3,52 (m, 3Н) 2,73-2,68 (m, 1Н) 2,55-2,49 (m, 2Н) 1,32-1,28 (m, 9Н). MS ESI рассч. для C31H31N3O3 [М+Н]+ 494, найденное значение 494.
Вариант осуществления 193

4-(3-(6-((3S,5R)-4-(2-фторпропаноил)-3,5-диметилпиперазин-1-ил)пиридин-3-ил)-4-метоксихинолин-8-ил)бензонитрил

[445] Стадия 1. В атмосфере газа азота Pd(dppf)Cl2 (1,7 г, 2 ммоль) добавляли в раствор соединения 193-1 (8,0 г, 23,6 ммоль), соединения 193-2 (4,5 г, 28,3 ммоль) и карбоната натрия (6,3 г, 59 ммоль) в THF (160 мл) и H2O (32 мл). Реакционную смесь нагревали до 80°C и перемешивали в течение 16 ч. Реакционную смесь охлаждали и экстрагировали с помощью EtOAc, органическую фазу сушили над сульфатом натрия и концентрировали, остаток очищали с помощью колоночной хроматографии с получением очищенного соединения 193-3 (8,51 г, выход 97%) в виде желтого твердого вещества. MS ESI рассч. для C22H14ClN3O [М+Н]+ 372, найденное значение 372.
[446] Стадия 2. В атмосфере газа азота Pd2(dba)3 (2,1 г, 2,3 ммоль) добавляли в раствор соединения 193-3 (8,51 г, 23 ммоль), соединения 193-4 (2,88 г, 25 ммоль), NaOtBu (5,51 г, 57 ммоль) и Xantphos (2,65 г, 4,6 ммоль) в толуоле (100 мл). Реакционную смесь нагревали до 130°C в течение 16 ч. Смесь охлаждали и экстрагировали с помощью EtOAc, органическую фазу сушили над сульфатом натрия и концентрировали, остаток очищали с помощью колоночной хроматографии с получением соединения 193-5 (4 г, выход 39%) в виде желтого твердого вещества. MS ESI рассч. для C28H27N5O [М+Н]+ 450, найденное значение 450.
[447] Стадия 3. Соединение 193-6 (294 мг, 3,34 ммоль) и пиридин (440 мг, 5,57 ммоль) добавляли в раствор соединения 193-5 (1 г, 2,23 ммоль) и HATU (1,35 г, 3,56 ммоль) в DCM (15 мл), реакционную смесь перемешивали при температуре флегмы в течение ночи. Остаток экстрагировали с помощью EtOAc и H2O, органическую фазу сушили над сульфатом натрия и концентрировали, неочищенный продукт очищали с помощью колоночной хроматографии с получением соединения 193-7 (226 мг, выход 20%) в виде светло-желтого твердого вещества. MS ESI рассч. для C31H29N5O3 [М+Н]+ 520, найденное значение 520.
[448] Стадия 4. NaBH4 (33 мг, 0,87 ммоль) добавляли в раствор соединения 193-7 (226 мг, 0,44 ммоль) в THF (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч., гасили с помощью H2O (2 капли). Смесь сушили над сульфатом натрия и концентрировали досуха. Остаток очищали с помощью препаративной HPLC с получением соединения 193-8 (220 мг, выход 97%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,90 (s, 1Н), 8,50 (d, J=4,0 Гц, 1H), 8,31 (s, 1H), 7,90-7,79 (m, 5H), 7,72-7,68 (m, 2H), 6,87-6,85 (d, J=8,0 Гц, 1H), 4,75-4,74 (m, 1H), 4,46-4,43 (m, 2H), 4,22-4,19 (m, 1H), 4,02-4,00 (m, 1H), 3,83-3,81 (m, 1H), 3,79 (s, 3H), 3,20-3,12 (m, 2H), 1,53-1,47 (m, 3H), 1,43-1,40 (m, 6H). MS ESI рассч. для C31H31N5O3 [M+H]+ 522, найденное значение 522.
[449] Стадия 5. DAST (740 мг, 4,6 ммоль) добавляли в раствор соединения 193-8 (120 мг, 0,23 ммоль) в DCM (10 мл) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч., смесь разбавляли с помощью DCM и промывали с помощью насыщенного бикарбоната натрия 3 раза, органическую фазу сушили и концентрировали, остаток очищали с помощью препаративной TLC с получением титульного соединения (41 мг, выход 33%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,94 (s, 1H), 8,55 (s, 1Н), 8,37 (d, J=8,0 Гц, 1Н), 7,94-7,83 (m, 5Н), 7,76-7,72 (m, 2Н), 6,90 (d, J=8,4 Гц, 1H), 5,46-5,35 (m, 1Н), 4,84 (brs, 1H), 4,51-4,21 (m, 3Н), 3,83 (s, 3Н), 3,36-3,19 (m, 2Н), 1,72-1,64 (m, 3Н), 1,53-1,45 (m, 6Н). MS ESI рассч. для C31H30FN5O2 [М+Н]+ 524, найденное значение 524.
[450] Соединения, перечисленные в таблице 18, синтезировали с помощью соединения 193-5 и соответствующих кислот.
Вариант осуществления 205

4-(4-метокси-3-(6-(8-пропионил-3,8-диазабицикло[3.2.1]октан-3-ил)пиридин-3-ил)хинолин-8-ил)бензонитрил
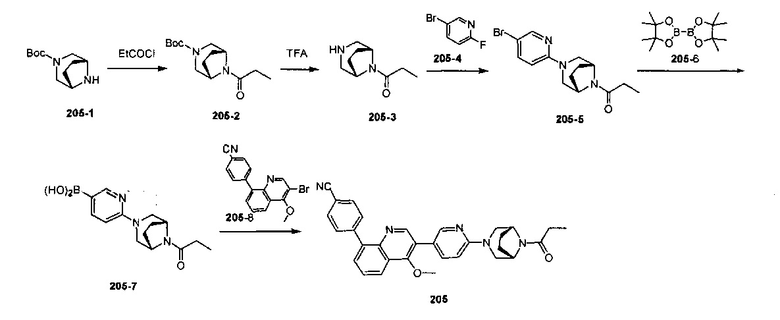
[451] Стадия 1. K2CO3 (27,6 г, 0,2 моль) добавляли в раствор соединения 205-1 (100 мг, 0,47 ммоль), пропионилхлорида (87 мг, 0,94 ммоль) в DCM (3 мл), после перемешивания реакционной смеси при комнатной температуре в течение 1 ч. смесь фильтровали, фильтрат концентрировали с получением 120 мг соединения 205-2 в виде желтого масла (95%).
[452] Стадия 2. CF3COOH (3 мл) по каплям добавляли в раствор соединения 205-2 (120 мг, 0,47 ммоль) в DCM (6 мл), после добавления реакционную смесь перемешивали при комнатной температуре в течение 1 ч., затем растворитель выпаривали с получением 80 мг соединения 205-3 в виде коричневого масла, выход 95%.
[453] Стадия 3. K2CO3 (100 мг, 0,8 ммоль) добавляли в раствор соединения 205-3 (70 мг, 0,4 ммоль) и соединения 205-4 (73 мг, 0,4 ммоль) в DMF (2 мл), затем смесь перемешивали при 120°C в течение 2 ч., после охлаждения реакционную смесь выливали в H2O и экстрагировали с помощью EtOAc (10 мл), жидкость после экстракции промывали солевыми растворами, сушили над безводным сульфатом натрия и концентрировали под вакуумом с получением 40 мг соединения 205-5 в виде белого твердого вещества, выход 31%. MS ESI рассч. для C14H18BrN3O [М+Н]+ 325, найденное значение 325.
[454] Стадия 4. Соединение 205-5 (33 мг, 0,1 ммоль), соединение 205-6 (50 мг, 0,2 ммоль), Pd(dppf)Cl2 (8 мг, 0,01 ммоль) и ацетат калия (20 мг, 0,2 ммоль) добавляли в 1,4-диоксан (2 мл), в атмосфере газа азота реакционную смесь перемешивали при 110°C в течение 2 ч. Смесь фильтровали с помощью диатомита, фильтрат концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии с получением соединения 205-7 (20 мг, выход 95%) в виде белого твердого вещества. MS ESI рассч. для C14H20BN3O3 [М+Н]+ 290, найденное значение 290.
[455] Стадия 5. Титульное соединение (10 мг, выход 20%) синтезировали в соответствии с вышеупомянутым способом в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,89 (s, 1H), 8,49-8,50 (d, J=2,4 Гц, 1H), 8,31-8,33 (m, 1Н) 7,78-7,86 (m, 5Н) 7,68-7,72 (m, 2Н) 6,73-6,75 (d, J=8,8 Гц, 1H) 4,93-4,95 (d, J=5,2 Гц, 2Н), 4,36-4,38 (d, J=6,0 Гц, 2Н), 4,24-4,27 (m, 2Н) 3,92-3,94 (m, 2Н) 3,78 (s, 3Н) 3,12-3,25 (d d, J=10,8 Гц 2Н) 2,38-2,47 (m, 2Н) 1,95-2,03 (m, 4Н) 1,22-1,25 (t, J=14,8 Гц 3Н). MS ESI рассч. для C31H29N5O2 [М+Н]+ 504, найденное значение 504.
Вариант осуществления 206
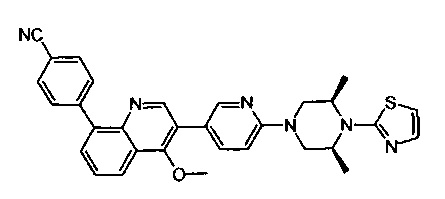
4-(3-(6-((3S,5R)-3,5-диметил-4-(тиазол-2-ил)пиперазин-1-ил)пиридин-3-ил)-4-метоксихинолин-8-ил)бензонитрил

[456] Стадия 1. В атмосфере газа азота Pd(dppf)2Cl2 (50 мг, 0,07 ммоль) и KOAc (150 мг, 1,42 ммоль) добавляли в раствор соединения 206-1 (250 мг, 0,71 ммоль) и соединения 206-2 (216 мг, 0,85 ммоль) в диоксане (5 мл). Реакционную смесь перемешивали при 120°C в течение 4 ч. и непосредственно применяли для следующей стадии.
[457] Стадия 2. Титульное соединение (30 мг, выход 12%) синтезировали в соответствии с вышеупомянутым способом в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,86 (s, 1H), 8,49 (s, 1Н), 8,33 (d, J=8,0 Гц, 1Н), 7,85-7,55 (m, 7Н), 7,23 (d, J=4,0 Гц, 1Н), 6,87 (d, J=8,8 Гц, 1Н), 6,57 (d, J=4,0 Гц, 1 Н), 4,33-4,28 (m, 4Н), 3,78 (s, 3Н), 3,35-3,30 (m, 2Н), 1,43 (d, J=6,8 Гц, 6 Н). MS ESI рассч. для C31H28N6OS [М+Н]+ 533, найденное значение 533.
Вариант осуществления 207
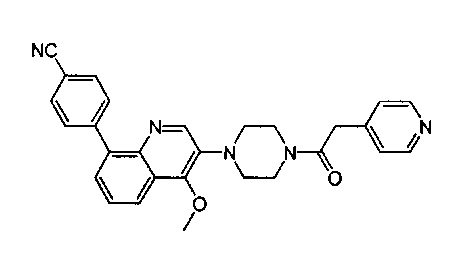
4-(4-метокси-3-(4-(2-(пиридин-4-ил)ацетил)пиперазин-1-ил)хинолин-8-ил)бензонитрил
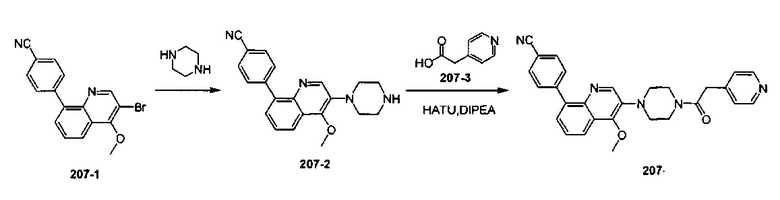
[458] Стадия 1. В атмосфере газа азота трет-бутоксид натрия (900 мг, 9 ммоль), Pd2(dba)3 (270 мг, 0,3 ммоль) и Xantphos (350 мг, 0,6 ммоль) добавляли в раствор соединения 207-1 (1 г, 3 ммоль) и пиперазина (270 мг, 3 ммоль) в толуоле (15 мл). Реакционную смесь перемешивали при 130°C в течение 3 ч. Смесь концентрировали под вакуумом, неочищенный продукт очищали с помощью колоночной хроматографии (EtOAc/метанол = 1:1) с получением соединения 207-2 (420 мг, выход 42%) в виде желтого твердого вещества. MS ESI рассч. для C21H20N4O [М+Н]+ 345, найденное значение 345.
[459] Стадия 2. DIPEA (260 мг, 2 ммоль) и HATU (380 мг, 1 ммоль) добавляли в раствор соединения 207-2 (69 мг, 0,2 ммоль), соединения 207-3 (144 мг, 1 ммоль) в DMF. Смесь перемешивали при комнатной температуре в течение 30 мин., очищали с помощью препаративной HPLC с получением титульного соединения (20 мг, 21%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,68 (s, 1Н), 8,59 (d, J=8,0 Гц, 2Н), 7,75-7,73 (m, 4Н), 7,61-7,59 (m, 2Н), 7,24-7,22 (m, 2Н), 4,09 (s, 3Н), 3,88-3,85 (m, 2 Н), 3,80 (s, 2 Н), 3,68-3,65 (m, 2 Н), 3,28-3,25 (m, 2 Н), 3,18-3,15 (m, 2 Н). MS ESI рассч. для C28H25N5O2 [М+Н]+ 464, найденное значение 464.
[460] Соединения, перечисленные в таблице 19, синтезировали с помощью соединения 207-2 и соответствующих кислот.
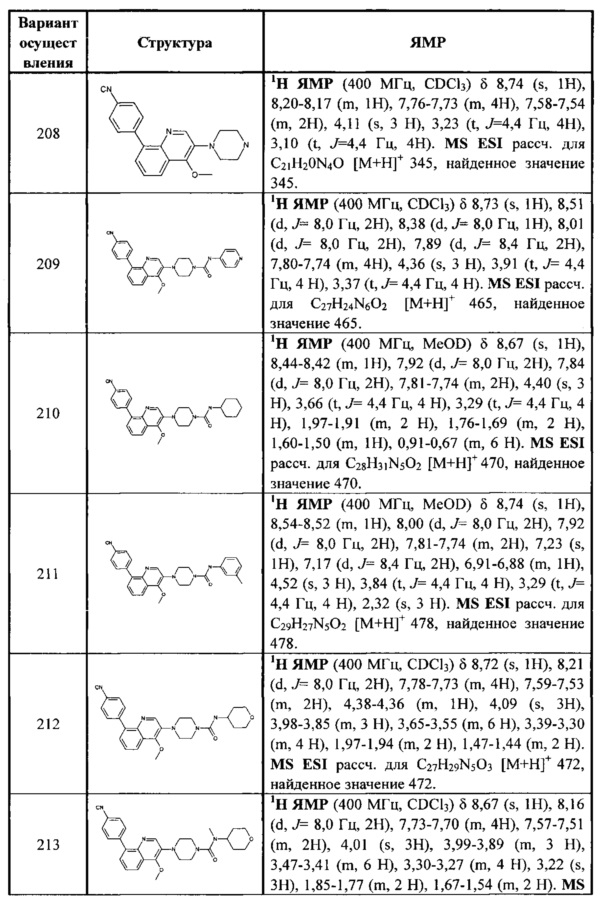
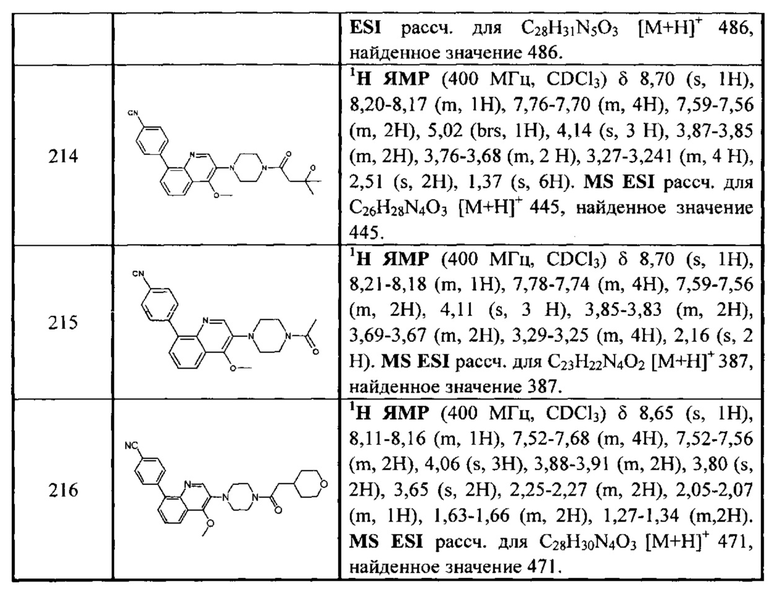
Вариант осуществления 217
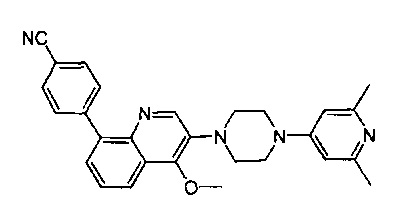
4-(3-(4-(2,6-диметилпиридин-4-ил)пиперазин-1-ил)-4-метоксихинолин-8-ил)бензонитрил
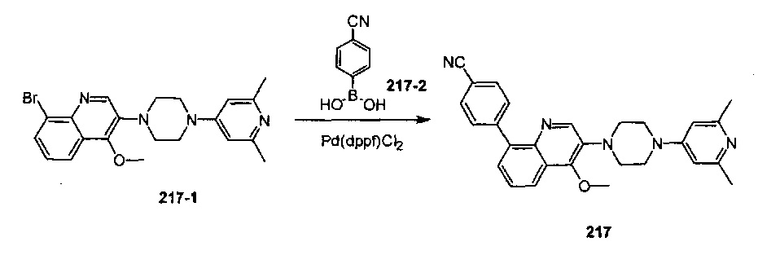
[461] Соединение 217-1 (490 мг, 1,1 ммоль), соединение 217-2 (253 мг, 1,7 ммоль), Pd(dppf)Cl2 (81 мг, 0,11 ммоль) и Na2CO3 (233 мг, 2,2 ммоль) добавляли в THF/H2O (10:1, 11 мл), в атмосфере газа азота смесь перемешивали при 80°C в течение ночи. Смесь фильтровали с помощью диатомита, фильтрат промывали с помощью H2O (50 мл) и экстрагировали с помощью EtOAc (100 мл), жидкость после экстракции промывали солевыми растворами и сушили над безводным сульфатом натрия и концентрировали под вакуумом. Остаток очищали с помощью препаративной HPLC с получением титульного соединения (200 мг, выход 45%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,93 (s, 1H), 8,31 (d, J=8,0 Гц, 1H), 7,76-7,74 (m, 6Н), 7,48-7,38 (m, 1H), 6,60 (s, 2Н), 4,27 (s, 3Н), 3,83 (s, 4Н), 3,41 (s, 4Н), 2,51 (s, 6Н). MS ESI рассч. для C28H27N5O [М+Н]+ 450, найденное значение 450.
Вариант осуществления 218
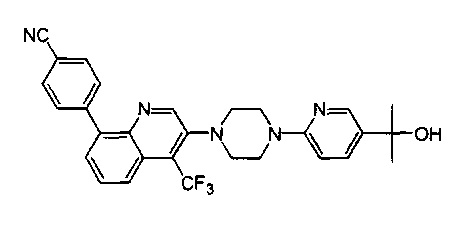
4-(3-(4-(5-(2-гидроксипропан-2-ил)пиридин-2-ил)пиперазин-1-ил)-4-(трифторметил)хинолин-8-ил)бензонитрил
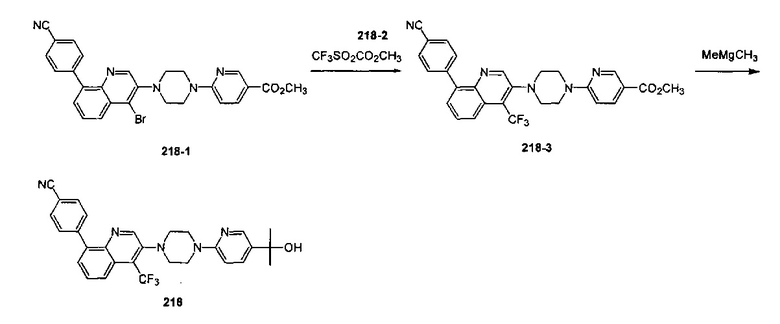
[462] Стадия 1. CuI (155 мг, 0,82 ммоль) и KF (32 мг, 0,55 ммоль) добавляли в раствор соединения 218-1 (300 мг, 0,55 ммоль) и соединения 218-2 (210 мг, 1,1 ммоль) в DMF (10 мл). Суспензию перемешивали при 110°C в течение 3 ч., затем выливали в H2O, экстрагировали с помощью EtOAc, сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии с получением соединения 218-3 в виде желтого твердого вещества (200 мг, выход 70%). MS ESI рассч. для C28H22F3N5O2 [М+Н]+ 518, найденное значение 518.
[463] Стадия 2. Раствор MeMgBr (3 М, 0,13 мл, 0,38 ммоль) добавляли в раствор соединения 218-3 (200 мг, 0,38 ммоль) в THF (20 мл) при 0°C. Смесь перемешивали при 0°C в течение 2 ч., затем выливали в насыщенный хлорид аммония, экстрагировали с помощью EtOAc, сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали с помощью препаративной HPLC с получением титульного соединения (30 мг, выход 15%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,96 (s, 1Н), 8,36 (s, 1H), 8,22 (d, J=8,0 Гц, 1Н), 7,75-7,53 (m, 7Н), 6,75-6,70 (m, 1H), 3,77 (t, J=4,4 Гц, 4 Н), 3,40 (t, J=4,4 Гц, 4 Н), 1,56 (s, 6Н). MS ESI рассч. для C29H26F3N5O [М+Н]+ 518, найденное значение 518.
Вариант осуществления 219
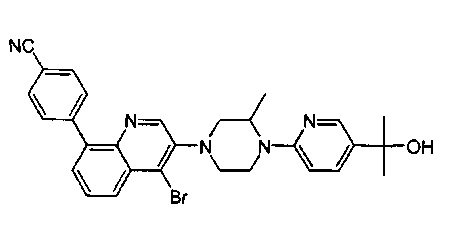
4-(4-бром-3-(4-(5-(2-гидроксипропан-2-ил)пиридин-2-ил)-3-метилпиперазин-1-ил)хинолин-8-ил)бензонитрил
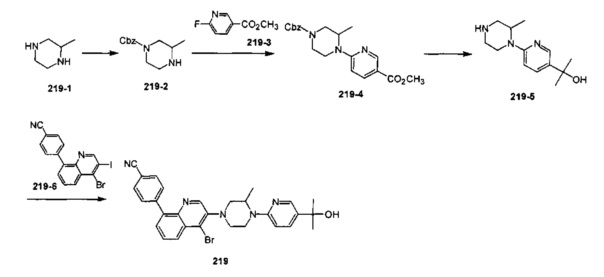
[464] Стадия 1. CbzCl (17 г, 0,1 моль) добавляли в раствор соединения 219-1 (30 г, 0,3 моль) и DIPEA (40 г, 0,3 моль) в DCM (200 мл) при 0°C, затем смесь перемешивали при комнатной температуре в течение 3 ч. и растворитель удаляли, неочищенный продукт очищали с помощью колоночной хроматографии с получением соединения 2 (11 г, выход 47%) в виде желтого масла. MS ESI рассч. для C13H18N2O2 [М+Н]+ 235, найденное значение 235.
[465] Стадия 2. K2CO3 (4 г, 0,029 моль) добавляли в раствор соединения 219-2 (2,25 г, 14,5 ммоль) и соединения 219-3 (3,41 г, 14,5 ммоль) в DMF (10 мл). Смесь перемешивали при 160°C в течение 4 ч., затем выливали в H2O. Смесь экстрагировали с помощью EtOAc, сушили над безводным сульфатом натрия и концентрировали, неочищенный продукт очищали с помощью колоночной хроматографии с получением соединения 219-4 (2,8 г, выход 52%). MS ESI рассч. для C20H23N3O4 [М+Н]+ 370, найденное значение 370.
[466] Стадия 3. Раствор MeMgBr (3,3 мл, 11 ммоль) медленно добавляли в раствор соединения 219-4 (370 мг, 1 ммоль) в THF (10 мл) при 0°C. Смесь перемешивали при 0°C в течение 1 ч., затем нагревали до 80°C и перемешивали в течение 2 ч., затем выливали в насыщенный раствор хлорида аммония, смесь экстрагировали с помощью EtOAc, сушили над безводным сульфатом натрия и концентрировали с получением соединения 219-5 (100 мг, выход 42%) в виде белого твердого вещества. MS ESI рассч. для C13H21N3O [М+Н]+ 236, найденное значение 236.
[467] Стадия 4. Титульное соединение (120 мг, выход 23%) синтезировали в соответствии с вышеупомянутым способом в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,74 (s, 1H), 8,39-8,34 (m, 2Н), 7,76-7,63 (m, 5Н), 6,67-6,61 (m, 1H), 4,65 (brs, 1Н), 4,22-4,19 (m, 1Н), 3,62-3,32 (m, 4 Н), 3,20-3,01 (m, 4 Н), 1,60 (s, 6Н), 1,47 (d, J=6,4 Гц, 3Н). MS ESI рассч. для C29H28BrN5O [М+Н]+ 542, найденное значение 542.
Вариант осуществления 220
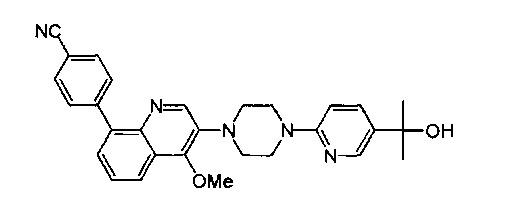
4-(3-(4-(5-(2-гидроксипропан-2-ил)пиридин-2-ил)пиперазин-1-ил)-4-метоксихинолин-8-ил)бензонитрил
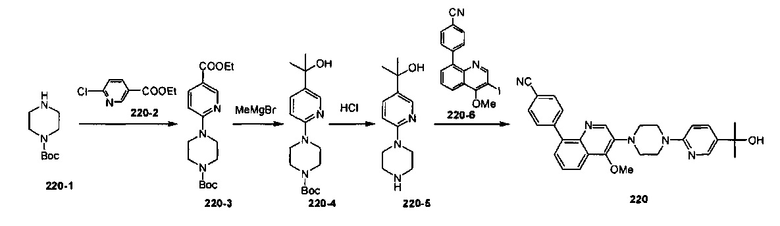
[468] Стадия 1. Раствор соединения 220-1 (5 г, 36,9 ммоль), соединения 220-2 (5,34 г, 28,8 ммоль) и DIPEA (7 мл, 40 ммоль) в DME (30 мл) нагревали до 130°C в течение ночи в автоклаве. Смесь разбавляли с помощью EtOAc (200 мл), дважды промывали 10%-ным водным раствором NaOH, один раз лимонной кислотой и солевыми растворами, соответственно. Органическую фазу сушили над сульфатом натрия и концентрировали, неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с получением соединения 220-3 (7,7 г, выход 85%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,81 (s, 1Н) 8,04 (d, J=12,0 Гц, 1H) 6,59 (d, J=8,0 Гц, 1Н) 4,37-4,31 (m, 2Н) 3,70-3,67 (m, 4Н) 3,56-3,54 (m, 4Н) 1,49 (s, 9Н) 1,39-1,36 (m, 3Н). MS ESI рассч. для C17H25N3O4 [М+Н]+ 336, найденное значение 336.
[469] Стадия 2. Раствор MeMgBr (24 мл, 71,6 ммоль) по каплям добавляли в раствор соединения 220-3 (6 г, 17,9 ммоль) в THF (80 мл) и смесь перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь гасили с помощью насыщенного хлорида аммония (10 мл) и экстрагировали с помощью EtOAc (200 мл). Органическую фазу сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии с получением очищенного соединения 220-4 (4,3 г, выход 74%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,30 (s, 1H) 7,68-7,60 (m, 1H) 6,64 (d, J=8,0 Гц, 1H) 3,54-3,52 (m, 8H) 1,57 (s, 6H) 1,49 (s, 9H). MS ESI рассч. для C17H27N3O3 [M+H]+ 322, найденное значение 322.
[470] Стадия 3. CF3COOH (15 мл) добавляли в раствор соединения 220-4 (3 г, 9,33 ммоль) в DCM (30 мл). Затем смесь перемешивали при комнатной температуре в течение 30 мин. NaOH (6 М) добавляли для подщелачивания смеси до рН=9, затем смесь экстрагировали с помощью CHCl3 3 раза. Объединенную органическую фазу сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии с получением соединения 220-5 (1,6 г, выход 75%) в виде светло-желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,22 (s, 1Н) 7,71-7,68 (m, 1Н) 6,80-6,75 (m, 1Н) 3,47-3,40 (m, 4Н) 2,94-2,88 (m, 4Н), 1,51 (s, 6Н). MS ESI рассч. для C12H19N3O [М+Н]+ 222, найденное значение 222.
[471] Стадия 4. Титульное соединение (21 мг, выход 11%) синтезировали в соответствии с вышеупомянутым способом в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,79 (s, 1H) 8,36-8,35 (m, 1Н) 8,24-8,22 (m, 1Н) 7,81-7,78 (m, 4Н) 7,76-7,70 (m, 1Н) 7,60-7,59 (m, 2Н) 6,75-6,73 (m, 1Н) 4,16 (s, 3Н) 3,78-3,76 (m, 4H) 3,42-3,39 (m, 4H) 1,67 (s, 1H) 1,60 (s, 6H). MS ESI рассч. для C29H29N5O2 [M+H]+ 480, найденное значение 480.
[472] Соединения, перечисленные в таблице 20, синтезировали с помощью соединения 220-6 и соответствующих аминов.
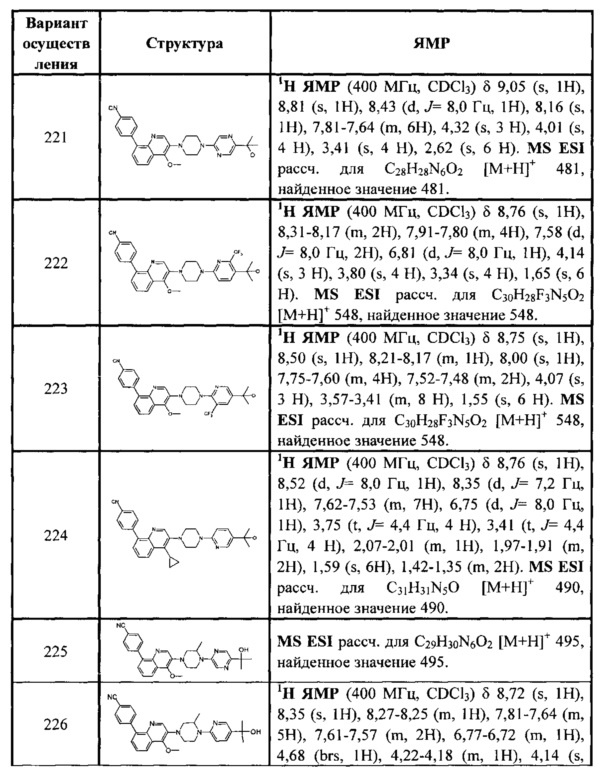

Вариант осуществления 227
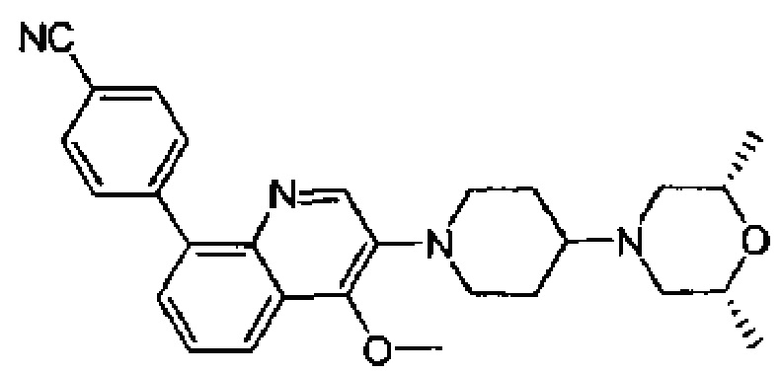
4-(3-(4-(2,6-диметилморфолино)пиперидин-1-ил)-4-метоксихинолин-8-ил)бензонитрил
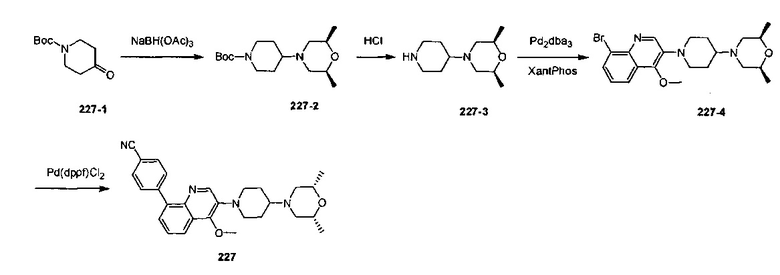
[473] Стадия 1. Раствор соединения 227-1 (2,0 г, 10 ммоль) и цис-2,6-диметилморфолина (1,25 г, 11 ммоль) в DCM (40 мл) перемешивали при 0°C в течение 1 ч., затем триацетоксиборгидрид натрия (5,3 г, 25 ммоль) добавляли при 20°C и смесь перемешивали при 20°C в течение 5 ч. Затем реакционную смесь гасили с помощью насыщенного водного раствора карбоната натрия, экстрагировали с помощью DCM (30×2 мл). Органические фазы объединяли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (РЕ/EtOAc = 5:1-1:1) с получением соединения 227-2 (1,5 г, 50%) в виде белого твердого вещества. MS ESI рассч. для C16H30N2O3 [М+Н]+ 299, найденное значение 299.
[474] Стадия 2. HCl (4 М метанольный раствор, 10 мл) добавляли в раствор соединения-227-2 (3,0 г, 10 ммоль) в метаноле (20 мл). Полученный раствор перемешивали при 20°C в течение 4 ч., затем концентрировали при пониженном давлении. Полученное желтое твердое вещество непосредственно применяли для следующей стадии без дополнительной очистки (1,9 г, 99%). MS ESI рассч. для C11H22N2O [М+Н]+ 199, найденное значение 199.
[475] Стадия 3. В атмосфере газа азота Pd2(dba)3 (180 мг, 0,2 ммоль), Xantphos (290 мг, 0,5 ммоль) и трет-бутоксид натрия (1,0 г, 10 ммоль) добавляли в раствор соединения 227-3 (1,0 г, 5,0 ммоль) и 8-бром-3-йод-4-метоксихинолина (1,4 г, 4 ммоль) в толуоле (30 мл), реакционную смесь перемешивали при 120°C в течение 5 ч., затем выливали в H2O и экстрагировали с помощью эфира (3×3 мл), органические фазы объединяли, сушили над сульфатом натрия и концентрировали при пониженном давлении, остаток очищали с помощью колоночной хроматографии на силикагеле (РЕ/EtOAc = 1:1-1:2) с получением соединения 227-4 (0,36 г, 50%) в виде желтого твердого вещества. MS ESI рассч. для C21H28BrN3O2 [М+Н]+ 435, найденное значение 435.
[476] Стадия 4. Титульное соединение (0,1 г, выход 40%) синтезировали в соответствии с вышеупомянутым способом в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,75 (s, 1H), 8,23-8,15 (m, 1Н), 7,79-7,74 (m, 4Н), 7,58-7,46 (m, 2Н), 4,11 (s, 3Н), 3,72-3,65 (m, 4Н), 2,90-2,84 (m, 4Н), 2,35-2,28 (m, 1Н), 2,20-2,10 (m, 2Н), 1,90-1,80 (m, 2Н), 1,77-1,67 (m, 2Н), 1,21 (d, J=5,6 Гц, 6Н). MS ESI рассч. для C28H3N4O2 [М+Н]+ 457, найденное значение 457.
Вариант осуществления 228
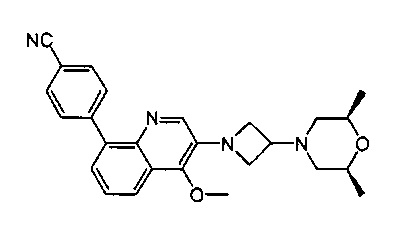
4-(3-(3-(2,6-диметилморфолино-4-ил)азетидин-1-ил)-4-метоксихинолин-8-ил)бензонитрил
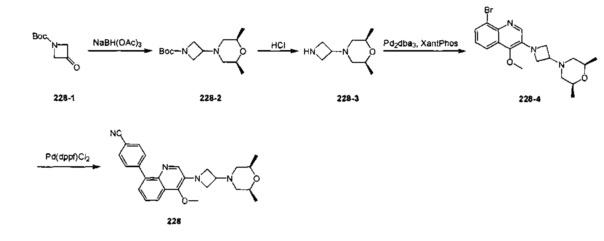
[477] Стадия 1. Раствор соединения 228-1 (1,7 г, 10 ммоль) и цис-2,6-диметилморфолина (1,4 г, 12 ммоль) в DCM (30 мл) перемешивали при 0°C в течение 1 ч., затем триацетоксиборгидрид натрия (6,0 г, 30 ммоль) добавляли при 20°C и смесь перемешивали при 20°C в течение 5 ч. Затем реакционную смесь гасили с помощью насыщенного водного раствора карбоната натрия, экстрагировали с помощью DCM (30×2 мл). Органические фазы объединяли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (РЕ/EtOAc = 5:1-1:1) с получением соединения 228-2 (1,8 г, 67%) в виде белого твердого вещества. MS ESI рассч. для C14H26N2O3 [М+Н]+ 271, найденное значение 271.
[478] Стадия 2. HCl (4 М метанольный раствор, 10 мл) добавляли в раствор соединения 228-2 (2,6 г, 10 ммоль) в метаноле (20 мл). Полученный раствор перемешивали при 20°C в течение 4 ч., затем концентрировали при пониженном давлении. Полученное желтое твердое вещество непосредственно применяли для следующей стадии без дополнительной очистки (1,6 г, 99%). MS ESI рассч. для C9H18N2O3 [М+Н]+ 171, найденное значение 171.
[479] Стадия 3. В атмосфере газа азота Pd2(dba)3 (90 мг, 0,1 ммоль), Xantphos (120 мг, 0,2 ммоль) и трет-бутоксид натрия (0,6 г, 6 ммоль) добавляли в раствор соединения 228-3 (1,1 г, 3 ммоль) и 8-бром-3-йод-4-метоксилхинолина (0,55 г, 3,3 ммоль) в толуоле (10 мл), реакционную смесь перемешивали при 120°C в течение 5 ч., затем выливали в H2O и экстрагировали с помощью эфира (3×3 мл), органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении, остаток очищали с помощью колоночной хроматографии на силикагеле (РЕ/EtOAc = 1:1-1:2) с получением соединения 228-4 (0,57 г, 47%) в виде желтого твердого вещества. MS ESI рассч. для C19H24BrN3O2 [М+Н]+ 407, найденное значение 407.
[480] Стадия 4. Титульное соединение (0,045 г, выход 10%) синтезировали в соответствии с вышеупомянутым способом в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,50 (s, 1H), 8,09 (d, J=8,0 Гц, 1Н), 7,80-7,65 (m, 6Н), 4,56 (brs, 2Н), 4,41 (brs, 2Н), 4,06 (brs, 2Н), 3,94 (s, 3Н), 3,91 (brs, 1H), 3,49 (d, J=7,6 Гц, 2Н), 2,34 (t, J=11,6 Гц, 2Н), 1,26 (d, J=6,0 Гц, 6Н). MS ESI рассч. для C26H28N4O2 [М+Н]+ 429, найденное значение 429.
Вариант осуществления 229
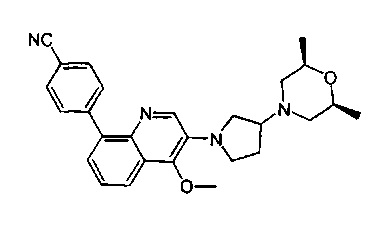
4-(3-(3-(2,6-диметилморфолино)пирролидин-1-ил)-4-метоксихинолин-8-ил)бензонитрил
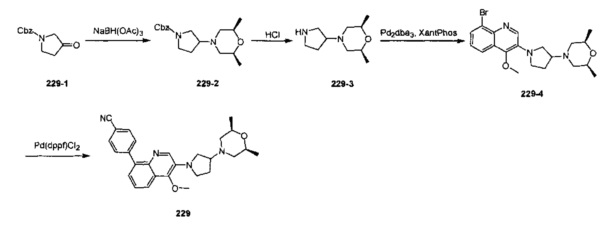
[481] Стадия 1. Раствор соединения 229-1 (2,0 г, 10 ммоль) и цис-2,6-диметилморфолина (1,25 г, 11 ммоль) в DCM (40 мл) перемешивали при 0°C в течение 1 ч., затем триацетоксиборгидрид натрия (6,0 г, 30 ммоль) добавляли при 20°C и смесь перемешивали при 20°C в течение 5 ч. Затем реакционную смесь гасили с помощью насыщенного водного раствора карбоната натрия, экстрагировали с помощью DCM (30×2 мл). Органические фазы объединяли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (РЕ/EtOAc = 5:1-1:1) с получением соединения 229-2 (1,5 г, 60%) в виде белого твердого вещества. MS ESI рассч. для C18H26N2O3 [М+Н]+ 319, найденное значение 319.
[482] Стадия 2. Pd/C (100 мг) добавляли в раствор соединения 229-2 (0,65 г, 2 ммоль) в метаноле (10 мл) и смесь гидрогенизировали при 30 фунтов/кв. дюйм при 20°C в течение 4 ч. Затем смесь фильтровали и концентрировали при пониженном давлении. Полученное желтое твердое вещество непосредственно применяли для следующей стадии без дополнительной очистки (0,37 г, 99%). MS ESI рассч. для C10H20N2O [М+Н]+ 185, найденное значение 185.
[483] Стадия 3. В атмосфере газа азота Pd2(dba)3 (90 мг, 0,1 ммоль), Xantphos (120 мг, 0,2 ммоль) и трет-бутоксид натрия (0,4 г, 3 ммоль) добавляли в раствор соединения 229-3 (0,4 г, 1,1 ммоль) и 8-бром-3-йод-4-метоксихинолина (0,2 г, 1,1 ммоль) в толуоле (10 мл), реакционную смесь перемешивали при 120°C в течение 5 ч., затем выливали в H2O и экстрагировали с помощью эфира (3×3 мл), органические фазы объединяли, сушили над сульфатом натрия и концентрировали при пониженном давлении, остаток очищали с помощью колоночной хроматографии на силикагеле (РЕ/EtOAc = 1:1-1:2) с получением соединения 229-4 (0,36 г, 42%) в виде желтого твердого вещества. MS ESI рассч. для C20H26BrN3O2 [М+Н]+ 421, найденное значение 421.
[484] Стадия. 4. Титульное соединение (0,13 г, 60%) синтезировали в соответствии с вышеупомянутым способом в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,68 (s, 1Н), 8,14-8,12 (m, 1H), 7,72-7,65 (m, 4Н), 7,49-7,47 (m, 2Н), 4,04 (s, 3Н), 3,64-3,60 (m, 4Н), 2,82-2,78 (m, 4Н), 2,24-2,20 (m, 1H), 2,00-1,92 (m, 2Н), 1,86-1,72 (m, 2Н), 1,68-1,56 (m, 2Н), 1,16 (d, J=6,0 Гц, 6Н). MS ESI рассч. для C27H30N4O2 [М+Н]+ 443, найденное значение 443.
Вариант осуществления 230
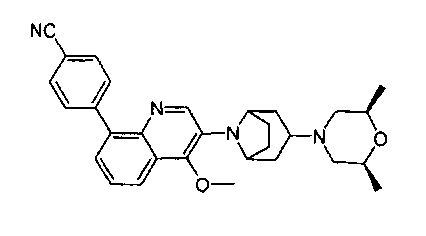
4-(3-(3-(2,6-диметилморфолино-4-ил)-8-азабицикло[3.2.1]октан-8-ил)-4-метоксихинолин-8-ил)бензонитрил
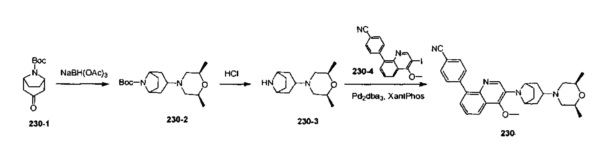
[485] Стадия 1. Триацетоксиборгидрид натрия (6,3 г, 30 ммоль) добавляли в раствор соединения 230-1 (4,5 г, 20 ммоль) и цис-2,6-диметилморфолина (2,5 г, 22 ммоль) в DCM (60 мл) при 0°C и смесь перемешивали при 25°C в течение 12 ч. Затем смесь выливали в H2O и экстрагировали с помощью EtOAc. Органические фазы объединяли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (РЕ/EtOAc = 10:1) с получением соединения 230-2 (1,1 г, 17%) в виде белого твердого вещества. MS ESI рассч. для C18H32N2O3 [М+Н]+ 325, найденное значение 325.
[486] Стадия 2. HCl (4 М метанольный раствор, 10 мл) добавляли в раствор соединения 230-2 (324 мг, 1 ммоль) в метаноле (5 мл). Полученный раствор перемешивали при 25°C в течение 2 ч., затем концентрировали при пониженном давлении. Полученное желтое твердое вещество непосредственно применяли для следующей стадии без дополнительной очистки (224 мг, 100%). MS ESI рассч. для C13H24N2O [М+Н]+ 225, найденное значение 225.
[487] Стадия 3. Титульное соединение (40 мг, 8,3%) синтезировали в соответствии с вышеупомянутым способом в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,71 (s, 1H), 8,24-8,21 (m, 1H), 7,82-7,75 (m, 4Н), 7,59-7,57 (m, 2Н), 4,75-4,60 (m, 2Н), 3,97 (s, 3Н), 3,74-3,68 (m, 3Н), 3,52 (d, J=12,0 Гц, 2Н), 2,62-2,50 (m, 2Н), 2,20-2,18 (m, 4Н), 2,00-1,89 (m, 2Н), 1,21 (d, J=6,4 Гц, 6Н). MS ESI рассч. для C30H34N4O2 [М+Н]+ 483, найденное значение 483.
Вариант осуществления 231
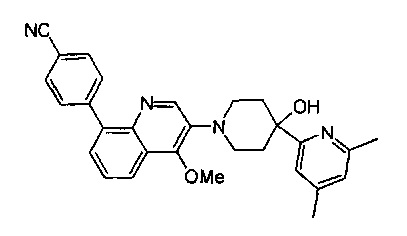
4-(3-(4'-гидрокси-4,6-диметил-3',4',5',6'-тетрагидро-2'Н-[2,4']бипиридил-1'-ил)-4-метоксихинолин-8-ил)бензонитрил
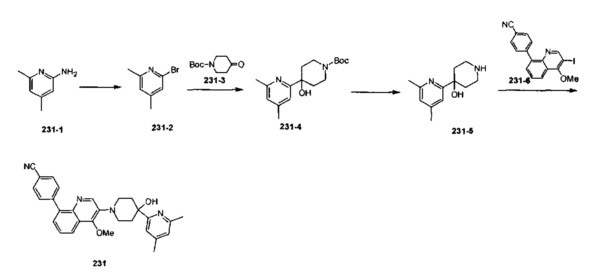
[488] Стадия 1. Br2 (36,8 г, 0,23 ммоль) добавляли в раствор соединения 231-1 (10 г, 82 ммоль) в HBr (41 мл) и смесь перемешивали при -10°C в течение 10 мин. Затем NaNO2 (14,1 г, 210 ммоль) добавляли при поддержании температуры реакции не более 0°C. Реакционную смесь перемешивали в течение 30 мин. Затем добавляли раствор NaOH (35 г, растворенные в 35 мл H2O) и смесь экстрагировали с помощью EtOAc. Органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (РЕ/EtOAc = 10:1) с получением соединения 231-2 (3,36 г, 22%) в виде светло-желтого масла. MS ESI рассч. для C7H8BrN [М+Н]+ 187, найденное значение 187.
[489] Стадия 2. н-BuLi (8 мл, 19,91 ммоль) добавляли в раствор соединения 231-2 (3,36 г, 18,1 ммоль) в THF (70 мл) при -70°C и перемешивали в течение 1 ч. Затем добавляли соединение 231-3 (4,32 г, 21,71 ммоль) при -70°C. Реакционную смесь медленно нагревали до комнатной температуры и перемешивали при комнатной температуре в течение 2 ч. Затем реакционную смесь гасили с помощью насыщенного водного раствора хлорида аммония (2 мл) и экстрагировали с помощью EtOAc (200 мл). Органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (РЕ/EtOAc = 5:1) с получением соединения 231-4 (3,11 г, 56%) в виде светло-желтого масла. MS ESI рассч. для C17H26N2O3 [М+Н]+ 307, найденное значение 307.
[490] Стадия 3. CF3COOH (6 мл) по каплям добавляли в раствор соединения 231-4 (2 г, 6,5 ммоль) в DCM (30 мл). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Затем реакционную смесь подщелачивали с помощью 6 н. NaOH до рН=9, экстрагировали с помощью CHCl3 (70×3 мл). Органические фазы объединяли, сушили над сульфатом натрия и концентрировали при пониженном давлении с получением неочищенного соединения 231-5 (1,197 г, 89%) в виде желтого твердого вещества, которое непосредственно применяли для следующей стадии без дополнительной очистки. MS ESI рассч. для C12H18N2O [М+Н]+ 207, найденное значение 207.
[491] Стадия 4. Титульное соединение (18 мг, 9,5%) синтезировали в соответствии с вышеупомянутым способом в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,89 (s, 1H), 8,24-8,21 (m, 1H), 7,83-7,76 (m, 4Н), 7,60-7,56 (m, 2Н), 7,01 (s, 1H), 6,94 (s, 1H), 5,74 (s, 1H), 4,19 (s, 3Н), 3,63-3,49 (m, 4Н), 2,54 (s, 3Н), 2,37 (s, 3Н), 2,28-2,21 (m, 2Н), 1,79-1,76 (m, 2Н). MS ESI рассч. для C29H28N4O2 [М+Н]+ 465, найденное значение 465.
Вариант осуществления 232
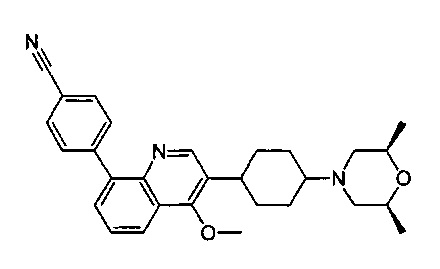
4-(3-(4-(2,6-диметилморфолино-4-ил)циклогексил)-4-метоксихинолин-8-ил)бензонитрил
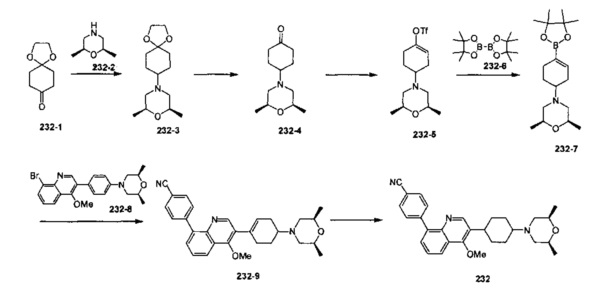
[492] Стадия 1. Триацетоксиборгидрид натрия (54,3 г, 0,26 моль) добавляли в раствор соединения 232-1 (20 г, 0,13 моль), цис-2,6-диметилморфолина (14,74 г, 0,13 моль) и уксусной кислоты (7,69 мг, 0,13 моль) в 1,2-дихлорэтане (400 мл) при 0°C и смесь перемешивали при 25°C в течение 16 ч. Затем 10%-ный водный раствор NaOH (200 мл) по каплям медленно добавляли (через 20 мин.) для гашения реакции, затем смесь экстрагировали с помощью EtOAc. Органические фазы объединяли, сушили над сульфатом натрия и концентрировали при пониженном давлении с получением соединения 232-3 (30 г, 92%) в виде белого твердого вещества. MS ESI рассч. для C14H25NO3 [М+Н]+ 256, найденное значение 256.
[493] Стадия 2. 7 н. водный раствор HCl (40 мл) добавляли в раствор соединения 232-3 (10 г, 39,2 ммоль) в THF (200 мл) и смесь перемешивали при 25°C в течение 16 ч. Затем 2 н. водный раствор NaOH (500 мл) добавляли для гашения реакции, затем смесь экстрагировали с помощью EtOAc (1X) и DCM (3Х). Органические фазы объединяли, сушили над сульфатом натрия и концентрировали при пониженном давлении с получением неочищенного соединения 232-4 (8 г, 96%) в виде белого твердого вещества. MS ESI рассч. для C21H12NO2 [М+Н]+ 212, найденное значение 212.
[494] Стадия 3. LiHMDS (17 мл, 17 ммоль) добавляли в раствор соединения 232-4 (3 г, 14,2 ммоль) в THF (50 мл) при 0°C, затем реакционную смесь перемешивали в течение 30 мин. Затем добавляли N,N-бис(трифторметилсульфонил)анилин (5,58 г, 15,6 ммоль) и реакционную смесь перемешивали при 25°C в течение 6 ч. Реакционную смесь выливали в H2O и экстрагировали с помощью EtOAc. Органические фазы объединяли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (РЕ/EtOAc = 5:1) с получением соединения 232-5 (2,3 г, 47%) в виде белого твердого вещества. MS ESI рассч. для C13H20F3NO4S [М+Н]+ 344, найденное значение 344.
[495] Стадия 4. В атмосфере газа азота Pd(dppf)Cl2 (0,4 мг, 0,11 ммоль) и KOAc (0,65 г, 6,6 ммоль) добавляли в раствор соединения 232-5 (0,76 г, 2,2 ммоль) и соединения 232-6 (0,67 г, 2,6 ммоль) в 1,4-диоксане (15 мл). Реакционную смесь перемешивали при 100°C в течение 15 ч., затем выливали в H2O и экстрагировали с помощью EtOAc (50×2 мл). Органические фазы объединяли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (РЕ/EtOAc = 5:1) с получением соединения 232-7 (0,35 г, 49%) в виде желтого твердого вещества. MS ESI рассч. для C18H32BNO3 [М+Н]+ 322, найденное значение 322.
[496] Стадия 5. В атмосфере газа азота Pd(dppf)Cl2 (113,8 мг, 0,16 ммоль) и Na2CO3 (412 мг, 3,89 ммоль) добавляли в раствор соединения 232-7 (600 мг, 1,87 ммоль) и соединения 232-8 (601 мг, 1,56 ммоль) в THF (10 мл) и H2O (2 мл). Реакционную смесь перемешивали при 80°C в течение 4 ч., затем экстрагировали с помощью EtOAc (50×2 мл). Органические фазы объединяли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (РЕ/EtOAc = 5:1) с получением соединения 232-7 (400 мг, 48%) в виде белого твердого вещества. MS ESI рассч. для C29H31N3O2 [М+Н]+ 454, найденное значение 454.
[497] Стадия 6. Pd/C (100 мг) добавляли в раствор соединения 232-9 (100 мг, 0,22 ммоль) в EtOAc (20 мл) и реакционную смесь гидрогенизировали в баллоне водорода при 20°C в течение 50 ч. Затем смесь фильтровали и концентрировали при пониженном давлении. Полученное желтое твердое вещество очищали с помощью препаративной HPLC с получением титульного соединения (27 мг, 54%) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO) δ 8,99 (s, 1H), 8,17-8,15 (J=8 Гц, d, 1Н), 7,94-7,92 (d, J=12 Гц, 2Н), 7,85-7,83 (d, J=8 Гц, 2Н), 7,80-7,78 (J=8 Гц, d, 1Н), 7,74-7,72 (J=8 Гц, d, 1H), 3,98 (s, 3Н) 3,54 (s, 2Н), 3,18-3,17 (J=4 Гц, d, 1H), 3,54 (s, 1Н), 2,76-2,74 (J=8 Гц, d, 2Н), 1,95-1,71 m, 8Н), 1,49-1,44 (m, 2 Н), 1,07-1,05 (J=8 Гц, d, 6Н). MS ESI рассч. для C29H33N3O2 [М+Н]+ 465, найденное значение 465.
Вариант осуществления 233
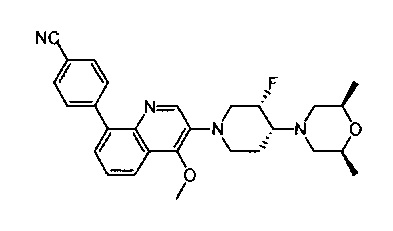
4-(3-(4-(2,6-диметилморфолино-4-ил)-3-фторпиперидин-1-ил)-4-метоксихинолин-8-ил)бензонитрил
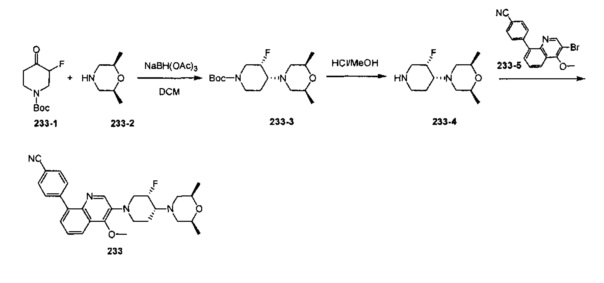
[498] Стадия 1. Раствор соединения 1 (3,0 г, 13,8 ммоль) и цис-2,6-диметилморфолина (1,7 г, 15,2 ммоль) в DCM (15 мл) перемешивали при 20°C в течение 0,5 ч., затем добавляли триацетоксиборгидрид натрия (4,4 г, 20,7 ммоль) при 20°C и реакционную смесь перемешивали при 20°C в течение 15 ч. Затем реакционную смесь гасили с помощью насыщенного водного раствора карбоната натрия (50 мл), экстрагировали с помощью DCM (50 мл). Органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (РЕ/EtOAc = 8:1-1:1) с получением соединения 233-3 (1,2 г, 27%) в виде белого твердого вещества. MS ESI рассч. для C16H29FN2O3 [М+Н]+ 317, найденное значение 317.
[499] Стадия 2. HCl (4 М метанольный раствор, 60 мл) добавляли в раствор соединения 233-3 (1,2 г, 3,8 ммоль) в метаноле (10 мл). Полученный раствор перемешивали при 20°C в течение 0,5 ч., затем концентрировали при пониженном давлении. Полученное желтое твердое вещество непосредственно применяли для следующей стадии без дополнительной очистки (0,6 г, 75%). MS ESI рассч. для C11H21FN2O [М+Н]+ 217, найденное значение 217.
[500] Стадия 3. Титульное соединение (27 мг, 15%) синтезировали в соответствии с вышеупомянутым способом в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,73 (s, 1H), 8,24-8,21 (m, 1H), 7,80-7,75 (m, 4Н), 7,59-7,58 (d, J=4 Гц, 2Н), 4,15 (s, 3Н), 4,03-4,00 (m, 1Н), 3,74-3,71 (m, 3H), 3,07-2,92 (m, 4Н), 2,25-1,94 (m, 6Н), 1,21-1,19 (d, J=8 Гц, 6Н). MS ESI рассч. для C28H31FN4O2 [М+Н]+ 475, найденное значение 475.
[501] Соединение, перечисленное в таблице 21, синтезировали с помощью соединения 233-5 и соответствующего амина.
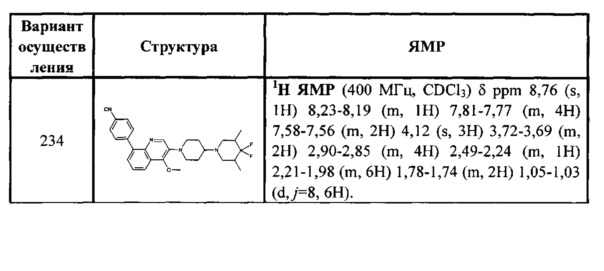
Вариант осуществления 235
4-(3-(4-(2,6-диметил-3-оксо-морфолино-4-ил)пиперидин-1-ил)-4-метоксихинолин-8-ил)бензонитрил
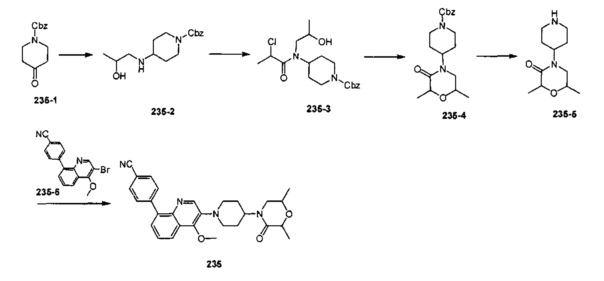
[502] Стадия 1. Раствор соединения 235-1 (15 г, 64 ммоль), соединения 235-А (12 мл, 192 ммоль) и уксусной кислоты (11,04 мл, 192 ммоль) в метаноле (200 мл) перемешивали при 20°C в течение 1 ч., затем добавляли NaBH3CN (12,11 г, 192 ммоль) весь сразу и смесь перемешивали при 25°C в течение ночи. После завершения реакции, как обнаружено с помощью LCMS, реакционную смесь экстрагировали с помощью DCM и промывали с помощью насыщенного водного раствора бикарбоната натрия. Органические фазы объединяли, сушили над сульфатом натрия и концентрировали при пониженном давлении с получением соединения 235-2 (15,6 г, 83%) в виде желтого масла. MS ESI рассч. для C16H24N2O3 [М+Н]+ 293, найденное значение 293.
[503] Стадия 2. TEA (0,7 г, 7 ммоль) и соединение 235-В (0,59 г, 4,65 ммоль) добавляли в раствор соединения 235-2 (1,36 г, 4,65 ммоль) в DCM (20 мл), смесь перемешивали при комнатной температуре в течение 2 ч. После завершения реакции, как обнаружено с помощью LCMS, реакционную смесь экстрагировали с помощью DCM. Органические фазы объединяли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (РЕ/EtOAc = 5:1) с получением соединения 235-3 (1,2 г, 61,4%) в виде желтого масла. MS ESI рассч. для C19H27ClN2O4 [М+Н]+ 383, найденное значение 383.
[504] Стадия 3. Трет-бутоксид калия (225 мг, 2 ммоль) добавляли в раствор соединения 235-3 (760 мг, 2 ммоль) в THF (10 мл), смесь перемешивали при комнатной температуре в течение 2 ч. После завершения реакции, как обнаружено с помощью LCMS, реакционную смесь экстрагировали с помощью EtOAc. Органические фазы объединяли, сушили над сульфатом натрия и концентрировали при пониженном давлении с получением соединения 235-4 (500 мг, 70%) в виде желтого масла. MS ESI рассч. для C19H26N2O4 [М+Н]+ 347, найденное значение 347.
[505] Стадия 4. Pd/C (300 мг) добавляли в раствор соединения 235-4 (3,0 г, 8,6 ммоль) в метаноле (10 мл) и смесь гидрогенизировали при 40 фунтов/кв. дюйм при комнатной температуре в течение ночи. Затем смесь фильтровали и концентрировали при пониженном давлении. Полученное бесцветное масло непосредственно применяли для следующей стадии без дополнительной очистки (1,3 г, 71%). MS ESI рассч. для C11H20N2O2 [М+Н]+ 213, найденное значение 213.
[506] Стадия 5. Титульное соединение (20 мг, 1,8%) синтезировали в соответствии с вышеупомянутым способом в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,93 (s, 1Н), 8,35-8,32 (m, 1H), 7,83-7,72 (m, 4Н), 7,59-7,57 (m, 2Н), 4,55-4,50 (m, 1H), 4,49-4,45 (m, 1Н), 4,32 (s, 3Н), 4,15-4,02 (m, 2Н), 3,75-3,52 (m, 2Н), 3,22-3,02 (m, 4Н), 2,15-2,07 (m, 2Н), 2,05-1,92 (m, 2Н), 1,48 (d, J=7,2 Гц, 3Н). MS ESI рассч. для C28H30N4O3 [М+Н]+ 471, найденное значение 471.
Вариант осуществления 236
4-(4-циклопропил-3-(4-(5-(1-гидрокси-1-метилэтил)пиридин-2-ил)-3-оксопиперазин-1-ил)хинолин-8-ил)бензонитрил
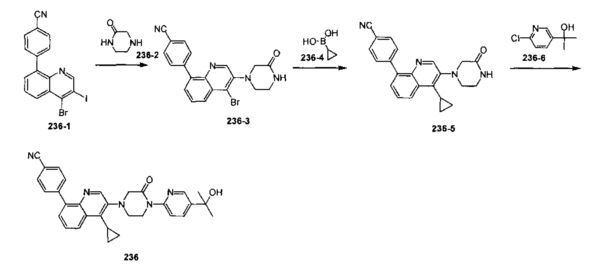
[507] Стадия 1. Соединение 236-1 (4,35 г, 10 ммоль), соединение 236-2 (1,0 г, 10 ммоль), Pd2(dba)3 (920 мг, 1,0 ммоль), Xantphos (1,15 г, 2,0 ммоль) и трет-бутоксид натрия (2,0 г, 20 ммоль) добавляли в толуол (30 мл), в атмосфере газа азота реакционную смесь перемешивали при 120°C в течение 2 ч. Смесь выливали в Н2О, экстрагировали с помощью EtOAc (100 мл), жидкость после экстракции промывали солевыми растворами, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле с получением 1,5 г соединения 236-3, выход 37,5%, в виде желтого твердого вещества. MS ESI рассч. для C20H15BrN4O [М+Н]+ 407, найденное значение 407.
[508] Стадия 2. Соединение 236-3 (407 мг, 1,0 ммоль), соединение 236-4 (870 мг, 10 ммоль), н-BuPAd2 (71 мг, 0,2 ммоль), ацетат палладия (23 мг, 0,1 ммоль) и карбонат цезия (650 мг, 2,0 ммоль) добавляли в толуол/H2O (5:1, 12 мл), в атмосфере газа азота реакционную смесь перемешивали при 110°C в течение 2 ч. Смесь выливали в H2O, экстрагировали с помощью EtOAc (50 мл), жидкость после экстракции промывали солевыми растворами, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле с получением 230 мг соединения 236-5, выход 64%, в виде желтого твердого вещества. MS ESI рассч. для C23H20N4O [М+Н]+ 369, найденное значение 369.
[509] Стадия 3. Соединение 236-5 (185 мг, 0,5 ммоль), соединение 236-6 (86 мг, 0,5 ммоль), Pd2(dba)3 (46 мг, 0,05 ммоль), Xantphos (57,7 мг, 0,1 ммоль) и карбонат цезия (325 мг, 1,0 ммоль) добавляли в толуол (10 мл), в атмосфере газа азота реакционную смесь перемешивали при 120°C в течение 2 ч. Смесь выливали в H2O, экстрагировали с помощью EtOAc (100 мл), жидкость после экстракции промывали солевыми растворами, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали с помощью препаративной HPLC с получением титульного соединения (80 мг, выход 32%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,78 (s, 1Н), 8,61-8,62 (d, J=2,0 Гц, 1H), 8,53-8,55 (d, J=7,6 Гц, 1H), 8,01-8,04 (d, J=8,4 Гц, 1H), 7,78-7,80 (m, 4H) 7,61-7,67 (m, 2H) 4,26-4,28 (t, J=10,4 Гц 2H), 4,23 (s, 2H) 3,74-3,76 (t, J=10,8 Гц 2H), 2,11-2,15 (m, 1H) δ 1,65 (s, 6H), 1,34-1,37 (m, 2H) 0,98-1,00 (d, J=6,4 Гц 2H). MS ESI рассч. для C31H29N5O2 [M+H]+ 504, найденное значение 504.
Вариант осуществления 237
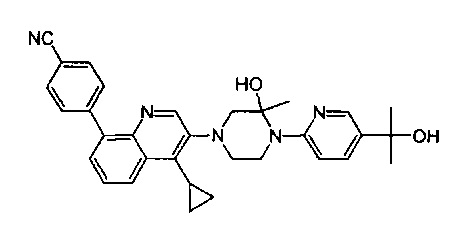
4-(4-циклопропил-3-(3-гидрокси-4-(5-(1-гидрокси-1-метилэтил)пиридин-2-ил)-3-метилпиперазин-1-ил)хинолин-8-ил)бензонитрил
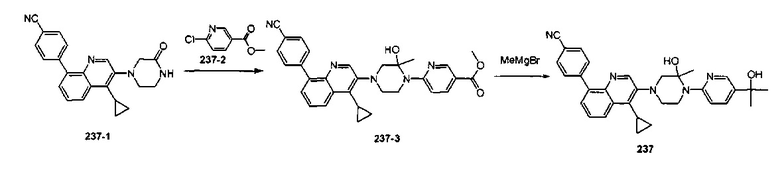
[510] Стадия 1. Соединение 237-1 (150 мг, 0,4 ммоль), соединение 237-2 (70 мг, 0,4 ммоль), Pd2(dba)3 (37 мг, 0,04 ммоль), Xantphos (45 мг, 0,08 ммоль) и карбонат цезия (260 мг, 0,8 ммоль) добавляли в толуол (2 мл), в атмосфере газа азота реакционную смесь перемешивали при 120°C в течение 2 ч. Смесь выливали в H2O, экстрагировали с помощью EtOAc (10 мл), жидкость после экстракции промывали солевыми растворами, сушили над безводным сульфатом натрия и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии на силикагеле с получением 80 мг соединения 237-3, выход 39%, в виде желтого твердого вещества. MS ESI рассч. для C30H25N5O3 [М+Н]+ 504, найденное значение 504.
[511] Стадия 2. Соединение 237-3 (50 мг, 0,1 ммоль) растворяли в THF (5 мл), бромид метилмагния (0,1 мл, 3 М в Et2O) медленно добавляли в раствор при -76°C, перемешивали при данной температуре в течение 1 ч., реакционную смесь выливали в водный раствор хлорида аммония, экстрагировали с помощью EtOAc (10 мл), жидкость после экстракции промывали солевыми растворами, сушили над безводным сульфатом натрия и концентрировали под вакуумом. Остаток очищали с помощью препаративной HPLC с получением титульного соединения (10 мг, выход 20%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,76 (s, 1Н), 8,66 (s, 1H), 8,38-8,39 (d, J=6,4 Гц, 1H), 7,83-7,85 (d d, J=2,0 Гц, 1H), 7,80-7,83 (m, 4H) 7,54-7,57 (m, 2H) 6,25-6,33 (m, 2H) 4,29 (s, 2H), 3,84 (s, 3H) 3,72-3,75 (t, J=11,2 Гц 2H), 3,47-3,49 (d, J=6,8 Гц, 2H), 2,16 (s, 6H) 1,92-1,95 (m, 1H), 1,22-1,26 (m, 2H) 0,75-0,79 (m, 2H). MS ESI рассч. для C32H33N5O2 [M+H]+ 520, найденное значение 520.
Вариант осуществления 238

4-(3-(4-гидрокси-4-(5-(2-гидроксипропан-2-ил)пиридин-2-ил)пиперидин-1-ил)-4-метоксихинолин-8-ил)бензонитрил
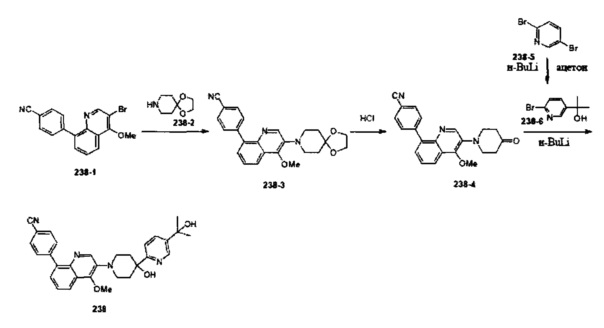
[512] Стадия 1. Соединение 238-2 (930 мг, 6,5 ммоль) и трет-бутоксид натрия (1,9 г, 19,5 ммоль) добавляли в раствор соединения 238-1 (2,64 г, 7,8 ммоль) в толуоле (70 мл), затем в атмосфере газа азота добавляли Pd2(dba)3 (595 мг, 0,65 ммоль) и Xantphos (750 мг, 1,3 ммоль), реакционную смесь нагревали до 100°C и перемешивали в течение 3 ч. Растворитель выпаривали, добавляли H2O (100 мл), смесь экстрагировали с помощью EtOAc (50 мл × 3). Органическую фазу промывали солевыми растворами, сушили над сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (РЕ/EtOAc = 5/1) с получением соединения 238-3 (1 г, неочищенное) в виде желтого твердого вещества. MS ESI рассч. для C24H23N3O3 [М+Н]+ 402, найденное значение 402.
[513] Стадия 2. 2 н. HCl (15 мл) добавляли в раствор соединения 238-3 (1 г, неочищенное, 2,5 ммоль) в THF (15 мл), реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Смесь гасили с помощью 2 н. водного раствора NaOH и регулировали до рН=7-8. Добавляли H2O (20 мл), смесь экстрагировали с помощью EtOAc (20 мл × 2). Органическую фазу промывали солевыми растворами, сушили над сульфатом натрия и концентрировали с получением соединения 238-4 (380 мг, неочищенное) в виде желтого масла. MS ESI рассч. для C22H19N3O2 [М+Н]+ 358, найденное значение 358.
[514] Стадия-3. Соединение 238-5 растворяли в THF (150 мл), охлаждали до -70°C, по каплям добавляли н-BuLi (5 мл, 12,5 ммоль), после проведения реакции в течение 30 мин. по каплям добавляли ацетон (2 мл, 20 ммоль), затем реакционную смесь перемешивали при -70°C в течение 1 ч. Реакционную смесь гасили с помощью насыщенного раствора хлорида аммония, добавляли H2O (30 мл), затем экстрагировали с помощью EtOAc (30 мл × 3). Органическую фазу промывали солевыми растворами, сушили над сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (РЕ/EtOAc = 3/1) с получением соединения 238-6 (462 мг, 17,1%) в виде белого твердого вещества. MS ESI рассч. для C8H10BrNO [М+Н]+ 216, найденное значение 216.
[515] Стадия 4. Соединение 238-6 (30 мг, 0,14 ммоль) растворяли в THF (5 мл), смесь охлаждали до -70°C, по каплям добавляли н-BuLi (0,12 мл, 0,31 ммоль), после проведения реакции в течение 30 мин. по каплям добавляли соединение 238-4 (50 мг, неочищенное, 0,14 ммоль) в THF (2 мл), реакционную смесь перемешивали при -70°C в течение 1 ч. Реакционную смесь гасили с помощью насыщенного раствора хлорида аммония, добавляли Н2О (10 мл), затем экстрагировали с помощью EtOAc (20 мл). Органическую фазу промывали солевыми растворами, сушили над сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной HPLC с получением титульного соединения (3 мг, 4,3%) в виде желтого твердого вещества. MS ESI рассч. для C30H30N4O3 [М+Н]+ 495, найденное значение 495.
Вариант осуществления 239

3-(8-(4-цианофенил)-4-циклопропилхинолин-3-ил)-6-(2,6-диметилморфолино-4-ил)пиридин-2-карбоновая кислота
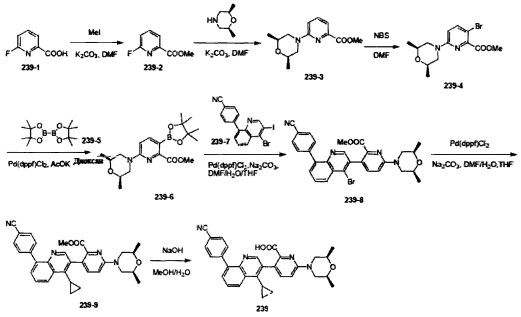
[516] Стадия 1. Соединение 239-1 (5 г, 35 ммоль) и K2CO3 (10 г, 70 ммоль) добавляли в раствор MeI (5 г, 35 ммоль) в DMF (40 мл), реакционную смесь перемешивали при 60°С в течение 2 ч., реакцию наблюдали с помощью LCMS до ее завершения. Затем 2,6-диметилморфолин (6,1 г, 53 ммоль) добавляли в реакционную систему, затем смесь перемешивали при 110°С в течение 2 ч., после завершения реакции, как обнаружено с помощью TLC, смесь выливали в Н2О и экстрагировали с помощью EtOAc, органическую фазу сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (MeOH:EtOAc=5:1) с получением соединения 239-3 (3,5 г, выход 40%) в виде масла. MS ESI рассч. для C13H18N2O3 [М+Н]+ 251, найденное значение 251.
[517] Стадия 2. NBS (1,6 г, 9 ммоль) медленно добавляли в раствор соединения 239-3 (5 г, 35 ммоль) в DMF (15 мл), реакционную смесь перемешивали при комнатной температуре в течение 6 ч. и реакцию наблюдали с помощью LCMS до ее завершения. Затем смесь выливали в H2O и экстрагировали с помощью EtOAc (100×3 мл), органическую фазу сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (РЕ:EtOAc=5:1) с получением соединения 239-4 (3 г, выход 91%) в виде белого твердого вещества. MS ESI рассч. для C13H17BrN2O3 [M+H]+ 329, найденное значение 329.
[518] Стадия 3. Соединение 239-4 (2 г, 6 ммоль), соединение 239-5 (1,82 г, 7,2 ммоль) и КОАс (1,2 г, 12 ммоль) растворяли в диоксане (50 мл), затем в атмосфере газа азота добавляли Pd(dppf)Cl2 (440 мг, 0,6 ммоль), реакционную смесь перемешивали при 120°С в течение 3 ч. После завершения реакции смесь выливали в H2O и экстрагировали с помощью EtOAc. Органическую фазу сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (РЕ:EtOAc=5:1) с получением соединения 239-6 (1,3 г, 60%) в виде желтого твердого вещества. MS ESI рассч. для C19H29BN2O5 [M+H]+ 377, найденное значение 377.
[519] Стадия 4. Соединение 239-7 (1,74 г, 4 ммоль), соединение 239-6 (1,5 г, 4 ммоль) и Na2CO3 (848 мг, 8 ммоль) растворяли в DMF (2 мл)/H2O (2 мл)/THF (10 мл), затем Pd(dppf)Cl2 (293 мг, 0,4 ммоль) добавляли в раствор. Реакционная система реагировала при 70°С в течение ночи. После завершения реакции, как обнаружено с помощью LC-MS, смесь выливали в H2O и экстрагировали с помощью EtOAc. Органическую фазу сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (РЕ:EtOAc=2:1) с получением соединения 239-8 (474 мг, 21%) в виде красного твердого вещества. MS ESI рассч. для C29H25BrN4O3 [М+Н]+ 557, найденное значение 557.
[520] Стадия 5. Соединение 239-8 (474 мг, 0,85 ммоль), циклопропилборную кислоту (731 мг, 8;5 ммоль) и Na2CO3 (180 мг, 1,7 ммоль) растворяли в DMF (2 мл)/H2O (2 мл)/THF (10 мл), затем Pd(dppf)Cl2 (73 мг, 0,1 ммоль) добавляли в раствор. Реакционную систему подвергали реакции при 70°С в течение ночи. После завершения реакции, как обнаружено с помощью LC-MS, смесь выливали в H2O и экстрагировали с помощью EtOAc. Органическую фазу сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (РЕ:EOtAc=2:1) с получением соединения 239-9 (200 мг, 21%) в виде красного твердого вещества. MS ESI рассч. для C32H30N4O3 [М+Н]+ 519, найденное значение 519.
[521] Стадия 6. NaOH (80 мг, 2 ммоль) добавляли в раствор соединения 239-9 (200 мг, 0,4 ммоль) в МеОН (5 мл)/H2O (1 мл), реакционная система реагировала при комнатной температуре в течение ночи. После завершения реакции, как обнаружено с помощью LC-MS, неочищенный продукт очищали с помощью HPLC с получением титульного соединения (30 мг, 15%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,77 (d, J=8,4 Гц, 1H), 7,86-7,82 (m, 5Н), 7,76-7,06 (m, 3Н), 7,07 (d, J=8,4 Гц, 1Н), 4,10 (d, J=12,0 Гц, 2H), 3,81 (m, 2H), 2,76 (m, 2H), 2,32 (m, 2H), 1,37 (d, J=6,0 Гц, 6H), 1,08 (m, 2H), 0,49 (d, J=5,2 Гц, 2H). MS ESI рассч. для C31H28N4O3 [M+H]+ 505, найденное значение 505.
Вариант осуществления 240
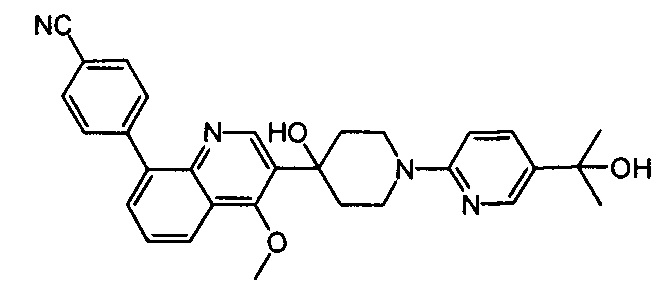
4-(3-(4-гидрокси-5'-(1-гидрокси-1-метилэтил)-3,4,5,6-тетрагидро-2Н-[1,2']бипиридил-4-ил)4-метоксихинолин-8-ил)бензонитрил
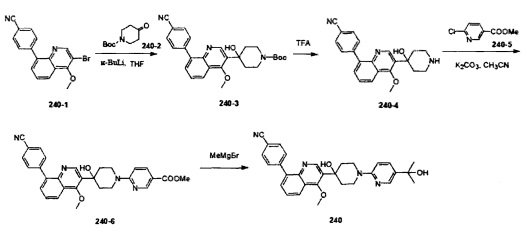
[522] Стадия 1. н-BuLi (2,5 М, 1,25 ммоль) медленно добавляли в раствор соединения 240-1 (170 мг, 0,5 ммоль) в THF (10 мл) при -78°С, реакционную смесь перемешивали при -78°С в течение 0,5 ч., затем 2 (100 мг, 0,5 ммоль) добавляли в. реакционную систему, смесь перемешивали при -78°С в течение 2 ч., после завершения реакции, как обнаружено с помощью TLC, смесь выливали в NH4Cl (водн.), экстрагировали с помощью EtOAc, органическую фазу сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (МеОН:EtOAc=2:1) с получением соединения 240-3 (100 мг, выход 33%) в виде масла. MS ESI рассч. для C27H29N3O4 [M+H]+ 460, найденное значение 460.
[523] Стадия 2. CF3COOH (2 мл) добавляли в раствор соединения 240-3 (400 мг, 0,9 ммоль) в DCM (4 мл), реакционную смесь перемешивали при комнатной температуре в течение 1 ч., реакцию наблюдали с помощью TLC до ее завершения. Затем растворитель выпаривали досуха, неочищенный продукт непосредственно применяли для следующей стадии.
[524] Стадия 3. Соединение 240-4 (250 мг, 0,7 ммоль), K2CO3 (292 мг, 2,1 ммоль) растворяли в ACN (10 мл), затем добавляли 240-5 (120 мг, 0,7 ммоль), реакционную смесь перемешивали при 100°С в течение 8 ч. После завершения реакции, как обнаружено с помощью LCMS, фильтрат концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (РЕ:EtOAc=1:1) с получением соединения 240-6 (200 мг, 58%) в виде белого твердого вещества. MS ESI рассч. для C29H26N4O4 [М+Н]+ 495, найденное значение 495.
[525] Стадия 4. MeMgBr (1 мл, 3,0 ммоль) медленно по каплям добавляли в раствор соединения 240-6 (200 мг, 0,4 ммоль) в THF (10 мл) при комнатной температуре, реакционную смесь перемешивали при комнатной температуре в течение 30 мин. После завершения реакции, как обнаружено с помощью LC-MS, смесь выливали в NH4Cl (водн.) и экстрагировали с помощью EtOAc (100×3 мл). Органическую фазу сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью HPLC с получением титульного соединения (25 мг, 13%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,95 (s, 1Н), 8,35 (d, J=3,2 Гц, 1H), 8,15 (dd, J=1,2, 8,0 Гц, 1Н), 7,79 (s, 4Н), 7,74-7,68 (m, 3Н), 6,76 (d, J=7,2 Гц, 1Н), 4,29-4,26 (m, 2Н), 4,20 (s, 3Н), 3,86 (s, 1H), 3,51 (t, J=12 Гц, 2Н), 2,40-2,33 (m, 2Н), 2,09-2,03 (m, 2Н), 1,61 (s, 6Н). MS ESI рассч. для C30H30N4O3 [М+Н]+ 495, найденное значение 495.
Вариант осуществления 241
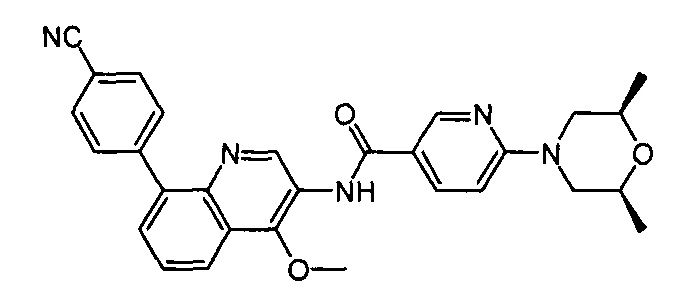
N-(8-(4-цианофенил)-4-метоксихинолин-3-ил)-6-(2,6-диметилморфолино-4-ил)никотинамид
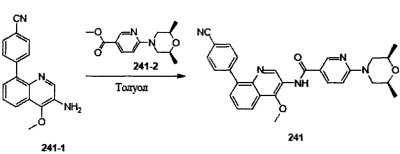
[526] Соединение 241-1 (100 мг, 0,36 ммоль), соединение 241-2 (100 мг, 0,4 ммоль), раствор триметилалюминия (0,9 мл, 0,9 ммоль) растворяли в толуоле (2 мл), реакционную смесь перемешивали при 800°С в течение 12 ч. Реакционную смесь охлаждали до комнатной температуры, добавляли H2O, раствор экстрагировали с помощью EtOAc (50 мл×2), органическую фазу промывали с помощью H2O, солевых растворов, соответственно, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с получением титульного соединения (80 мг, выход 44%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CD3OD) δ ppm 8,89 (s, 1Н), 8,84 (s, 1H), 8,33-8,36 (d, J=8,4 Гц, 1 H), 8,17 (d, J=8,8 Гц, 1H), 7,71-7,85 (m, 5 H), 6,94 (d, J=9,2 Гц, 1 H), 5,51 (s, 1H), 4,35 (d, J=13,2 Гц, 2H), 4,16 (s, 3H), 2,66 (t, J=12,8 Гц, 2H), 1,28 (d, J=6,4 Гц, 6H). MS ESI рассч. для C29H27N5O3 [M+H]+ 494, найденное значение 494.
Вариант осуществления 242
8-(4-цианофенил)-N-(4-((2S,6R)-2,6-диметилморфолино)фенил)-4-метоксихинолин-3-карбоксамид
[527] Стадия 1. (Диэтилэтоксиметилен)малонат (13 г, 60 ммоль), 2-броманилин (10,3 г, 60 ммоль) смешивали вместе и перемешивали при 100°С в течение 2 ч. После охлаждения до комнатной температуры реакционную смесь выливали в метанол, рекристаллизовали с метанолом с получением соединения 242-2 (14,5 г, выход 70,7%) в виде желтого твердого вещества. MS ESI рассч. для C14H16BrNO4 [М+Н]+ 342, найденное значение 342.
[528] Стадия 2. Соединение 242-2 (10 г, 29,2 ммоль) в PH2O (100 мл) перемешивали при температуре флегмы в течение 30 мин., затем охлаждали до комнатной температуры, добавляли гексан, смесь фильтровали с получением соединения 242-3 (2,8 г, выход 32,6%) в виде коричневого твердого вещества. MS ESI рассч. для C12H10BrNO3 [М+Н]+ 295, найденное значение 295.
[529] Стадия 3. Соединение 242-3 (500 мг, 1,7 ммоль) растворяли в POCl3 (4 мл) и смесь перемешивали при 110°С в течение 1 ч. Затем реакционную смесь выливали в метанол и добавляли водный раствор карбоната натрия. Смесь экстрагировали с помощью EtOAc, сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом с получением соединения 242-4 (400 мг, выход 76%) в виде коричневого твердого вещества. MS ESI рассч. для C13H12BrNO3 [М+Н]+ 310, найденное значение 310.
[530] Стадия-4. Соединение 242-4 (527 мг, 1,7 ммоль) растворяли в метаноле (5 мл), H2O (1 мл), добавляли NaOH (340 мг, 8,5 ммоль), реакционную смесь перемешивали при комнатной температуре в течение 5 ч. Затем смесь выливали в H2O, затем раствор HCl добавляли для регулирования до рН=7, смесь экстрагировали с помощью EtOAc, сушили над сульфатом натрия, фильтровали и концентрировали под вакуумом с получением соединения 242-5 (478 мг, выход 99%) в виде желтого твердого вещества. MS ESI рассч. для C11H8BrNO3 [М+Н]+ 282, найденное значение 282.
[531] Стадия 5. Соединение 242-5 (478 мг, 1,7 ммоль), 4-((2S,6R)-2,6-диметилморфолино)анилин 6 (525 мг, 2,55 ммоль), TEA (515 мг, 5,1 ммоль) растворяли в DMF (5 мл), добавляли HATU (980 мг, 2,55 ммоль), реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Затем реакционную смесь выливали в H2O, экстрагировали с помощью EtOAc, органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали под вакуумом, остаток очищали с помощью колоночной хроматографии на силикагеле с получением соединения 242-7 (400 мг, выход 50%) в виде желтого твердого вещества. MS ESI рассч. для C23H24BrN3O3 [М+Н]+ 470, найденное значение 470.
[532] Стадия 6. Титульное соединение (100 мг, выход 32%) синтезировали в соответствии с вышеупомянутым способом в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 9,49 (s, 1H), 9,23 (s, 1H), 8,28 (d, J=8,0 Гц, 1H), 7,74-7,84 (m, 7H), 7,65-7,67 (m, 2H), 7,02 (s, 1H), 4,21 (s, 3H), 3,90 (s, 1H), 3,44-3,49 (m, 3H), 2,48 (s, 2H), 1,28 (d, J=6,0 Гц, 6H). MS ESI рассч. для C30H28N4O3 [M+H]+ 493, найденное значение 493.
Вариант осуществления 243
8-(4-цианофенил)-N-(6-((2S,6R)-2,6-диметилморфолино)пиридин-3-ил)-4-метоксихинолин-3-карбоксамид
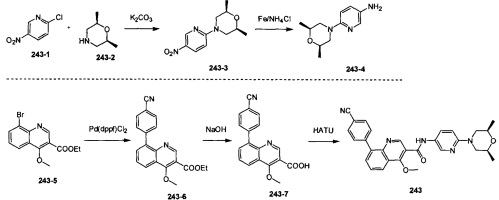
[533] Стадия 1. Соединение 2-хлор-5-нитропиридин (5 г, 32 ммоль) и K2CO3 (8,8 г, 64 ммоль) растворяли в MeCN (40 мл), добавляли (2S,6R)-2,6-диметилморфолин (4,4 г, 38,4 ммоль). Реакционную смесь перемешивали при 110°С в течение 2 ч. Затем реакционную смесь фильтровали, фильтрат концентрировали с получением соединения 243-3 (7,3 г) в виде желтого твердого вещества. MS ESI рассч. для C11H15N3O3 [М+Н]+ 238, найденное значение 238.
[534] Стадия 2. Соединение 243-3 (7,3 г, 31 ммоль) и хлорид аммония (16,7 г, 310 ммоль) растворяли в МеОН (80 мл) и H2O (80 мл), добавляли железный порошок (20,8 г, 372 ммоль). Реакционную смесь перемешивали при 80°С в течение 3 ч. Затем смесь фильтровали, фильтрат концентрировали, остаток растворяли в ацетоне, фильтровали и концентрировали под вакуумом, неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с получением соединения 243-4 (800 мг) в виде коричневого твердого вещества. MS ESI рассч. для C11H17N3O [М+Н]+ 208, найденное значение 208.
[535] Стадия 3. 4-цианофенилборную кислоту (1,8 г, 12 ммоль), соединение 243-4 (3,1 г, 10 ммоль) и карбонат натрия (2,1 г, 20 ммоль) растворяли в DMF (5 мл), H2O (5 мл) и THF (25 мл), добавляли Pd(dppf)Cl2 (732 мг, 1 ммоль). Реакционную смесь перемешивали при 70°С в течение 2 ч. Затем смесь выливали в H2O, экстрагировали с помощью EtOAc, органическую фазу промывали солевыми растворами, сушили над сульфатом натрия, фильтровали и концентрировали, остаток очищали с помощью колоночной хроматографии на силикагеле с получением соединения 243-6 (2,8 г) в виде белого твердого вещества. MS ESI-рассч. для C20H16N2O3 [М+Н]+ 333, найденное значение 333.
[536] Стадия 4. Соединение 243-6 (1,8 г, 5,4 ммоль) растворяли в МеОН (10 мл) и H2O (4 мл), добавляли NaOH (1 г, 27 ммоль), реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Затем смесь выливали в H2O, экстрагировали с помощью EtOAc, водную фазу окисляли до рН=4 с помощью 1 н. хлористоводородной кислоты, а затем экстрагировали с помощью EtOAc, органическую фазу промывали солевыми растворами, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения 243-7 (580 мг) в виде белого твердого вещества. MS ESI рассч. для C18H12N2O3 [М+Н]+ 305, найденное значение 305.
[537] Стадия 5. Соединение 243-7 (200 мг, 0,65 ммоль), HATU (494 мг, 1,3 ммоль) и соединение 243-4 (136 мг, 0,65 ммоль) растворяли в DMF (5 мл), добавляли DIPEA (387 мг, 3 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, реакционную смесь очищали с помощью препаративной HPLC с получением титульного соединения (100 мг) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 9,49 (s, 1Н), 9,23 (s, 1H), 8,27-8,30 (m, 3H), 7,73-7,85 (m, 7H), 6,73-6,75 (m, 1H), 4,21 (s, 3H), 4,05-4,08 (m, 2H), 3,74-3,79 (m, 2H), 2,56-2,61 (m, 2H), 1,29 (d, J=6,0 Гц, 6H). MS ESI рассч. для C29H27N5O3 [M+H]+ 494, найденное значение 494.
Вариант осуществления 244
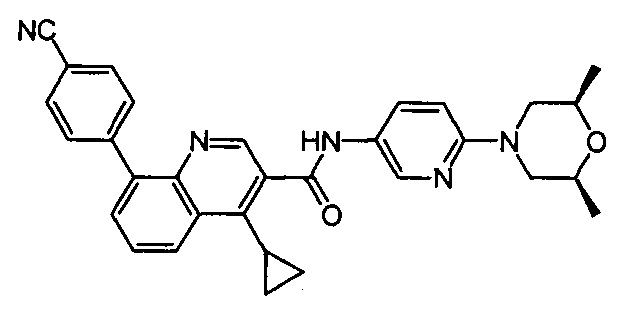
8-(4-цианофенил)-4-циклопропил-N-(6-((2S,6R)-2,6-диметилморфолино)пиридин-3-ил)хинолин-3-карбоксамид
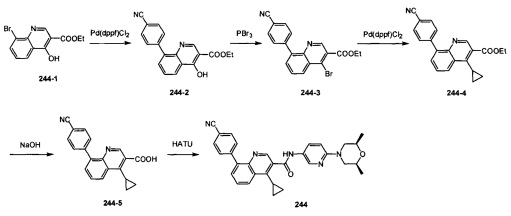
[538] Стадия 1. 4-цианофенилборную кислоту (2 г, 14,4 ммоль), сложный этиловый эфир этил-8-бром-4-гидроксихинолил-3-карбоновой кислоты (3,5 г, 12 ммоль) и карбонат натрия (2,5 г, 24 ммоль) растворяли в DMF (5 мл), H2O (5 мл) и THF (25 мл), добавляли Pd(dppf)Cl2 (878 мг, 1,2 ммоль). Реакционную смесь перемешивали при 70°С в течение 2 ч. Затем смесь выливали в H2O, фильтровали с получением соединения 244-2 (3,2 г) в виде коричневого твердого вещества. MS ESI рассч. для C19H14N2O3 [М+Н]+ 319, найденное значение 319.
[539] Стадия 2. Соединение 244-2 (3,2 г, 10 ммоль) растворяли в DMF (40 мл), добавляли PBr3 (4 мл, 10 М), реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Затем смесь выливали в H2O, твердое вещество фильтровали и промывали с помощью H2O, сушили на воздухе с получением соединения 244-3 (3,0 г) в виде коричневого твердого вещества. MS ESI рассч. для C19H13BrN2O2 [М+Н]+ 381, найденное значение 381.
[540] Стадия 3. Циклопропилборную кислоту (6,7 г, 78 ммоль), соединение 244-3 (3,0 г, 7,8 ммоль) и карбонат натрия (1,7 г, 15,6 ммоль) растворяли в DMF (5 мл), H2O (5 мл) и THF (25 мл), добавляли Pd(dppf)Cl2 (571 мг, 0,78 ммоль). Реакционную смесь перемешивали при 70°С в течение 2 ч. Затем смесь выливали в H2O, экстрагировали с помощью EtOAc, органическую фазу промывали солевыми растворами, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении, неочищенное вещество очищали с помощью колоночной хроматографии на силикагеле с получением соединения 244-4 (1,8 г) в виде желтого твердого вещества. MS ESI рассч. для C22H18N2O2 [М+Н]+ 343, найденное значение 343.
[541] Стадия 4. Соединение 244-4 (1,8 г, 5,4 ммоль) растворяли в МеОН (10 мл) и H2O (4 мл), добавляли NaOH (1 г, 27 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Затем смесь выливали в H2O, экстрагировали с помощью EtOAc, водную фазу окисляли с помощью 1 н. раствора HCl, экстрагировали с помощью EtOAc, органическую фазу промывали солевыми растворами, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения 244-5 (570 мг) в виде желтого твердого вещества. MS ESI рассч. для C20H14N2O2 [М+Н]+ 315, найденное значение 315.
[542] Стадия 5. Соединение 244-5 (200 мг, 0,65 ммоль), HATU (494 мг, 1,3 ммоль) и 6-((2S,6R)-2,6-диметилморфолино)пиридин-3-амин (136 мг, 0,65 ммоль) растворяли в DMF (5 мл), добавляли DIPEA (387 мг, 3 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, очищали с помощью препаративной HPLC (100 мг) с получением титульного соединения в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 10,2 (s, 1H), 8,95 (s, 1H), 8,61-8,74 (m, 3H), 7,75-7,78 (m, 4H), 7,63-7,65 (m, 2H), 7,08 (d, J=9,2 Гц, 1H), 4,04 (d, J=13,2 Гц, 2H), 3,74 (s, 2H), 2,83-2,89 (m, 2H), 2,64 (s, 1H), 1,27 (d, J=6,0 Гц, 8H), 0,72-0,73 (m, 1H). MS ESI рассч. для C31H29N5O2 [M+H]+ 504, найденное значение 504.
Вариант осуществления 245
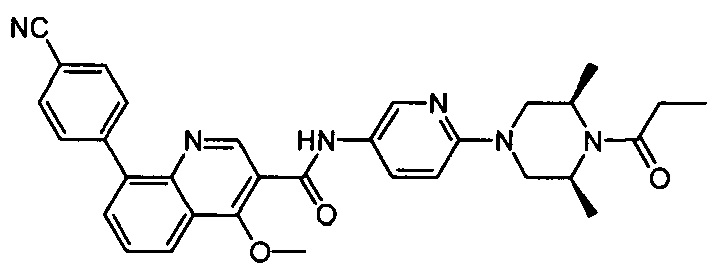
8-(4-цианофенил)-N-(6-((3S,5R)-3,5-диметил-4-пропионилпиперазин-1-ил)пиридин-3-ил)-4-метоксихинолин-3-карбоксамид
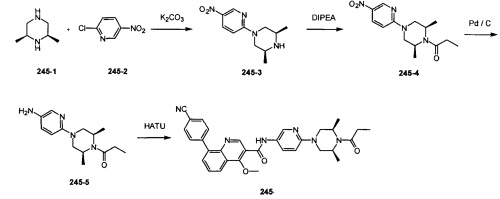
[543] Стадия 1. 2-хлор-5-нитропиридин (5 г, 32 ммоль) и K2CO3 (8,8 г, 64 ммоль) растворяли в ацетонитриле (40 мл), добавляли соединение 245-1 (4,3 г, 38,4 ммоль). Реакционную смесь перемешивали при 80°С в течение 1 ч., затем реакционную смесь фильтровали, концентрировали с получением соединения 245-3 (4,5 г) в виде желтого твердого вещества. MS ESI рассч. для C11H16N4O2 [М+Н]+ 237, найденное значение 237.
[544] Стадия 2. Соединение 245-3 (4,5 г, 19 ммоль) и DIPEA (7,4 г, 57 ммоль) в DCM (40 мл), добавляли пропионилхлорид (2,2 г, 23 ммоль). Реакционную смесь перемешивали при 0°С в течение 30 мин. Затем реакционную смесь выливали в H2O, экстрагировали с помощью EtOAc, органическую фазу промывали солевыми растворами, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения 245-4 (3,2 г) в виде желтого твердого вещества. MS ESI рассч. для C14H20N4O3 [М+Н]+ 293, найденное значение 293.
[545] Стадия 3. Соединение 245-4 (3,2 г, 11 ммоль) и Pd/C (0,32 г) растворяли в метаноле (500 мл), реакционную смесь перемешивали при комнатной температуре. при давлении газа водорода 40 фунтов/кв. дюйм в течение ночи. Затем реакционную смесь фильтровали, растворитель концентрировали при пониженном давлении с получением неочищенного соединения 245-5 (800 мг) в виде красного масла. MS ESI рассч. для C14H22N4O [М+Н]+ 263, найденное значение 263.
[546] Стадия 4. Раствор 8-(4-цианофенил)-4-метоксихинолин-3-карбоновой кислоты (100 мг, 0,33 ммоль), HATU (251 мг, 0,66 ммоль) и соединение 245-5 (86 мг, 0,33 ммоль) растворяли в DMF (5 мл), добавляли DIPEA (86 мг, 0,33 ммоль). После перемешивания при комнатной температуре в течение ночи реакционную смесь очищали с помощью HPLC с получением титульного соединения (80 мг) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 9,47 (s, 1Н), 9,24 (s, 1H), 8,26-8,28 (m, 3H), 7,72-7,84 (m, 6H), 6,77 (d, J=9,2 Гц, 1H), 4,21 (s, 3H), 3,05-3,10 (m, 2H), 1,34 (s, 6H), 1,19 (t, J=7,4 Гц, 1H). MS ESI рассч. для C32H32N6O3 [M+H]+ 548, найденное значение 548.
[547] Соединение, перечисленное в таблице 22, синтезировали с помощью соединения 245-5 и соответствующей кислоты.
Вариант осуществления 247
4-(4-метокси-3-(3-трифторметил-5,6-дигидро-8Н-[1,2,4]триазоло[4,3-а]пиразин-7-ил)хинолин-8-ил)бензонитрил
[548] Соединение 247-1 (339 мг, 1 ммоль), Xantphos (115 мг, 0,2 ммоль), 247-2 (192 мг, 1 ммоль) и трет-BuONa (200 мг, 2 ммоль) растворяли в толуоле (10 мл), затем в атмосфере газа азота добавляли Pd2(dba)3 (92 мг, 0,1 ммоль), реакционную смесь перемешивали при 110°С в течение 2 ч. После завершения реакции смесь выливали в H2O, экстрагировали с помощью EtOAc. Органическую фазу сушили над безводным сульфатом натрия, концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью HPLC с получением титульного соединения (30 мг, 7%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 9,48 (s, 1Н), 8,45 (d, J=8,0 Гц, 1H), 7,87 (m, 4H), 7,66 (m, 2H), 4,98 (s, 1H), 4,45-4,39 (m, 5H), 4,02 (s, 2H). MS ESI рассч. для C23H17F3N6O [M+H]+ 451, найденное значение 451.
Вариант осуществления 248
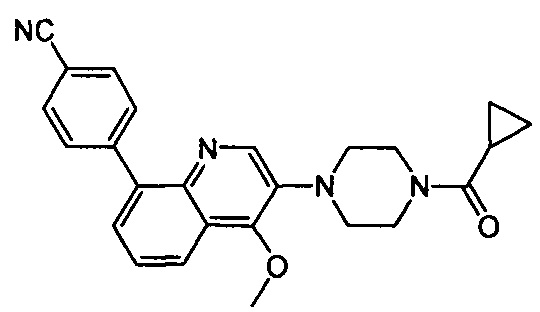
4-(3-(4-циклопропанкарбонилпиперазин-1-ил)-4-метоксихинолин-8-ил)бензонитрил
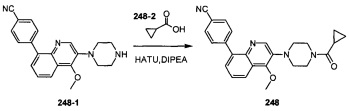
[549] Соединение 248-1 (172 мг, 0,5 ммоль), HATU (380 мг, 1 ммоль) и 248-2 (86 мг, 1 ммоль) растворяли в DMF (4 мл), затем добавляли DIPEA (194 мг, 1,5 ммоль), реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь наблюдали с помощью LCMS до ее завершения. Реакционную систему очищали с помощью HPLC с получением титульного соединения (40 мг, 20%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,76 (s, 1H), 8,4 (t, J=4,8 Гц, 1H), 7,82-7,79 (m, 4Н), 7,63-7,59 (m, 2Н), 4,16 (s, 3Н), 3,92 (m, 4Н), 3,33-3,30 (m, 4Н), 1,85-1,80 (m, 1H), 1,08-1,05 (m, 2Н), 0,86-0,83 (m, 2Н). MS ESI рассч. для C25H24N4O2 [M+H]+ 413, найденное значение 413.
Вариант осуществления 249
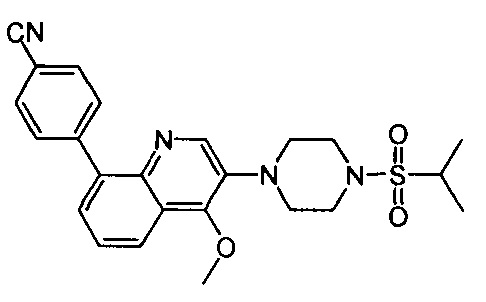
4-(4-метокси-3-(4-(пропан-2-сульфонил)пиперазин-1-ил)-хинолин-8-ил)бензонитрил
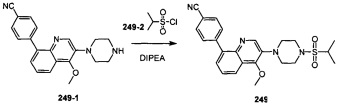
[550] Соединение 249-1 (172 мг, 0,5 ммоль) и DIPEA (194 мг, 1,5 ммоль) растворяли в DCM (5 мл), затем добавляли 2 (142 мг, 1 ммоль), реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Реакционную смесь наблюдали с помощью LCMS до ее завершения. Реакционную смесь выливали в H2O, экстрагировали с помощью EtOAc. Органическую фазу сушили над безводным сульфатом натрия, концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью HPLC с получением титульного соединения (20 мг, 10%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,75 (s, 1H), 8,22 (dd, J=4,4, 5,6 Гц, 1H), 7,82-7,77 (m, 4H), 7,63-7,61 (m, 2H), 4,13 (s, 3Н), 3,62-3,60 (m, 4H), 3,33-3,30 (m, 4H), 3,31-3,26 (m, 1H), 1,43 (d, J=7,2 Гц, 6H). MS ESI рассч. для C24H26N4O3S [M+H]+ 451, найденное значение 451.
Вариант осуществления 250
4-(2-(6-(3,5-диметил-4-пропионилпиперазин-1-ил)пиридин-3-ил)-1H-бензо[d]имидазол-4-ил)бензонитрил
[551] Стадия 1. Соединение 250-2 (1,14 г, 10 ммоль) добавляли в раствор соединения 250-1 (1,71 г, 10 ммоль) и K2CO3 (2,78 г, 20 ммоль) в DMF (20 мл), реакционную смесь перемешивали при 110°С в течение 2 ч., наблюдали с помощью LCMS до ее завершения. Затем смесь выливали в H2O, экстрагировали с помощью EtOAc, органическую фазу сушили над безводным сульфатом натрия, концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (РЕ:EtOAc=1:1) с получением соединения 250-3 (2,3 г, выход 92%) в виде масла. MS ESI рассч. для C13H19N3O2 [M+H]+ 250, найденное значение 250.
[552] Стадия 2. 250-4 (1,0 г, 10,8 ммоль) медленно добавляли в раствор соединения 250-3 (2,3 г, 9 ммоль) и DIPEA (2,3 г, 18 ммоль) в DCM (30 мл), реакционную смесь перемешивали при 0°С в течение 30 мин., наблюдали с помощью LCMS до ее завершения. Затем смесь выливали в H2O, экстрагировали с помощью EtOAc (100×3 мл), органическую фазу сушили над безводным сульфатом натрия, концентрировали с получением неочищенного продукта 250-5 (2,5 г, выход 91%). MS ESI рассч. для C16H23N3O3 [М+Н]+306, найденное значение 306.
[553] Стадия 3. NaOH (1,28 г, 32 ммоль) добавляли в раствор соединения 250-5 (2,5 г, 8 ммоль) в МеОН (10 мл)/H2O (2 мл), реакционную смесь перемешивали при 40°С в течение 1 ч. После завершения реакции смесь выливали в H2O, промывали с помощью EtOAc, водную фазу окисляли до рН=4 с помощью 1 н. раствора HCl, затем экстрагировали с помощью EtOAc, органическую фазу сушили над безводным сульфатом натрия, концентрировали с получением неочищенного продукта 250-6 (1 г, 43%). Неочищенный продукт применяли непосредственно на следующей стадии.
[554] Стадия 4. Соединение 250-10 (374 мг, 2 ммоль), соединение 250-11 (294 мг, 2 ммоль) и Na2CO3 (424 мг, 4 ммоль) растворяли в DMF (2 мл)/H2O (2 мл)/THF (10 мл), затем Pd(dppf)Cl2 (146 мг, 0,2 ммоль) добавляли в раствор. Реакционную систему перемешивали при 70°С в течение ночи, наблюдали с помощью LC-MS до ее завершения. Смесь выливали в H2O, экстрагировали с помощью EtOAc. Органическую фазу сушили над безводным сульфатом натрия, концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (РЕ:EtOAc=1:1) с получением соединения 250-7 (200 мг, 50%) в виде красного твердого вещества. MS ESI рассч. для C13H11N3 [M+H]+ 210, найденное значение 210.
[555] Стадия 5. Соединение 250-7 (200 мг, 1 ммоль), HATU (760 мг, 2 ммоль) и 250-6 (291 мг, 1 ммоль) растворяли в DMF (10 мл), затем DIPEA (387 мг, 3 ммоль) добавляли в раствор. Реакционную систему перемешивали при комнатной температуре в течение 1 ч., наблюдали с помощью LC-MS до ее завершения. Смесь выливали в H2O, экстрагировали с помощью EtOAc. Органическую фазу сушили над безводным сульфатом натрия, концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (РЕ:EtOAc=1:1) с получением соединения 250-5 (200 мг, 42%) в виде коричневого твердого вещества. MS ESI рассч. для C28H30N6O2 [M+H]+ 483, найденное значение 483.
[556] Стадия 6. Соединение 250-5 (80 мг, 0,16 ммоль) растворяли в АсОН (2 мл), реакционная система реагировала под воздействием микроволнового излучения при 180°С в течение 40 мин. Реакционную смесь наблюдали с помощью LC-MS до ее завершения. Неочищенный продукт очищали с помощью HPLC с получением титульного соединения (10 мг, 14%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,83-8,82 (m, 1H), 8,30-8,26 (m, 3Н), 7,78-7,62 (m, 3Н), 7,52-7,32 (m, 2Н), 6,79-6,75 (m, 1H), 3,23-3,18 (m, 2Н), 1,67 (s, 3Н), 1,34 (s, 6Н), 1,22 (t, J=7,6 Гц, 3Н). MS ESI рассч. для C28H28N6O [М+Н]+ 465, найденное значение 465.
Вариант осуществления 251
4-(2-(6-(3,5-диметил-4-пропионилпиперазидин-1-ил)пиридин-3-ил)-1Н-бензимидазол-4-ил)бензонитрил
[557] Стадия 1. На ледяной бане NaH (6,4 г, 160 ммоль) добавляли в раствор соединения 251-1 (4 г, 20 ммоль) в THF (50 мл). Реакционную смесь перемешивали при комнатной температуре в течение 0,5 ч., затем снова охлаждали до 0°С, по каплям добавляли MeI (10 мл, 160 ммоль). Затем реакционную смесь нагревали до комнатной температуры и перемешивали в течение еще 2 ч. Реакцию обнаруживали с помощью TLC. Смесь гасили с помощью H2O. Водную фазу экстрагировали с помощью EtOAc. Объединенную органическую фазу промывали с помощью насыщенного раствора хлорида натрия, сушили над сульфатом натрия, концентрировали при пониженном давлении с получением необходимого продукта 251-2 (4,5 г, 88%). MS ESI рассч. для C14H25NO3 [М+Н]+ 256, найденное значение 256.
[558] Стадия 2. Соединение 251-2 (410 мг, 1,6 ммоль) растворяли в HCl/метаноле (4 М, 10 мл) и перемешивали при комнатной температуре в течение 2 ч. После завершения реакции реакционную смесь концентрировали при пониженном давлении с получением необходимого продукта 251-3 (330 мг, 86%). MS ESI рассч. для C9H17NO[М+Н]+ 156, найденное значение 156.
[559] Стадия 3. Соединение 251-5 (300 мг, выход 75%) синтезировали как упомянуто выше. MS ESI рассч. для C32H31N3O2 [М+Н]+ 490, найденное значение 490.
[560] Стадия 4. NaBH4 (57 мг, 1,5 ммоль) добавляли в раствор соединения 251-5 (490 мг, 1 ммоль) в THF (10 мл), затем перемешивали при комнатной температуре в течение 0,5 ч. После завершения реакции, как обнаружено с помощью LC-MS, остаток выливали в H2O. Водную фазу экстрагировали с помощью DCM. Объединенную органическую фазу промывали с помощью насыщенного раствора хлорида натрия, сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали с помощью препаративной HPLC с получением титульного соединения (64 мг, выход 13%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,93 (s, 1H), 8,47-8,44 (m, 1H), 7,83-7,78 (m, 4Н), 7,66 (d, J=7,6 Гц, 2Н), 7,43 (d, J=8,4 Гц, 2Н), 7,00 (d, J=8,8 Гц, 2Н), 3,88 (s, 3Н), 3,47 (d, J=12,4 Гц, 2Н), 3,12 (s, 1Н), 2,64 (s, 2Н), 1,08 (s, 6Н), 1,05 (s, 6Н).
Вариант осуществления 252
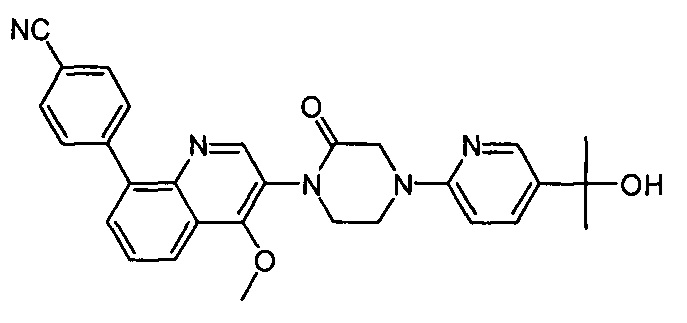
4-(3-(4-(5-(2-гидроксипропан-2-ил)пиридин-2-ил)-2-оксопиперазин-1-ил-4-метоксихинолин-8-ил))бензонитрил
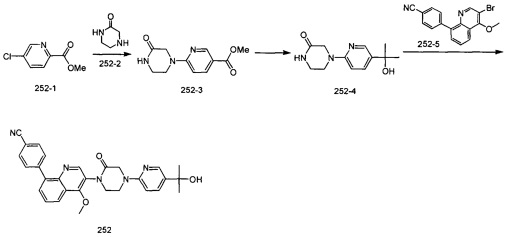
[561] Стадия 1. K2CO3 (13,8 г, 0,1 моль) добавляли в раствор соединения 252-1 (8,6 г, 50 ммоль) и 252-2 (5 г, 50 ммоль) в DMF (50 мл), затем смесь перемешивали при 120°С в течение ночи. Смесь охлаждали и выливали в H2O. Осадок собирали после фильтрования, промывали с помощью H2O и метанола с получением сложного метилового эфира 6-(3-оксопиперазин-1-ил)никотиновой кислоты (8,5 г, выход 72,6%) в виде желтого твердого вещества. MS ESI рассч. для C11H13N3O3 [М+Н]+ 236, найденное значение 236.
[562] Стадия 2. При -78°С MeMgBr (3 мл, 3 М в THF, 9 ммоль) добавляли в раствор 252-3 (708 мг, 3 ммоль) в THF (50 мл). Смесь перемешивали при комнатной температуре в течение 1 ч. и выливали в насыщенный раствор NH4Cl. Смесь экстрагировали с помощью EtOAc, сушили над сульфатом натрия, концентрировали с получением 252-4 (260 мг, выход 37%) в виде желтого твердого вещества. MS ESI рассч. для C12H17N3O2 [М+Н]+ 236, найденное значение 236.
[563] Стадия 3. В атмосфере газа азота N1,N1,N3,N3-тетраметилпропан-1,3-диамин (3мг, 0,03 ммоль), CuI (4 мг, 0,02 ммоль) и K2CO3 (70 мг, 0,6 ммоль) добавляли в раствор соединения 252-4 (100 мг, 0,3 ммоль) и 252-5 (72 мг, 0,3 ммоль) в толуоле (5 мл). Реакционную смесь перемешивали при 120°С в течение ночи. После выливания в H2O смесь дважды экстрагировали с помощью EtOAc. Органическую фазу сушили над сульфатом натрия и концентрировали. Концентрированное вещество очищали с помощью препаративной HPLC с получением титульного соединения (10 мг, выход 7%) в виде желтого твердого вещества. MS ESI рассч. для C29H27N5O3 [М+Н]+ 494, найденное значение 494. 1НЯМР (400 МГц, CDCl3) δ ppm 8,69 (s, 1Н), 8,39-8,40 (d, J=2,0 Гц, 1H), 8,27 (m, 1Н), 7,67-7,80 (m, 7Н), 6,69-6,71 (d, J=8,8 Гц, 1H), 4,39 (s, 2H), 4,15-4,18 (m, 2H), 4,11 (s, 3H), 3,88 (s, 2H), 1,62 (m, 6H).
Вариант осуществления 253
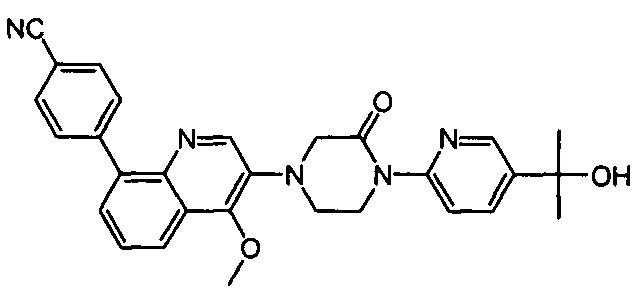
4-(3-(4-(5-(2-гидроксипропан-2-ил)пиридин-2-ил)-3-оксопиперазин-1-ил-4-метоксихинолин-8-ил))бензонитрил

[564] Стадия 1. В атмосфере газа азота Pd2(dba)3 (184 мг, 0,2 ммоль), Xantphos (240 мг, 0,4 ммоль) и карбонат цезия (1,3 г, 4 ммоль) добавляли в раствор соединения 253-1 (768 мг, 2 ммоль) и 253-2 (200 мг, 2 ммоль) в толуоле (10 мл). Смесь перемешивали при 120°С в течение ночи. После выливания в H2O смесь дважды экстрагировали с помощью EtOAc. Органическую фазу сушили над сульфатом натрия и концентрировали. Концентрированное вещество очищали с помощью колоночной хроматографии на силикагеле (РЕ/EtOAc 1:1) с получением 253-3 (300 мг, выход 44%) в виде желтого твердого вещества. MS ESI рассч. для C21H18N4O2 [М+Н]+ 359, найденное значение 359.
[565] Стадия 2. 253-3 (180 мг, 0,5 ммоль) и 253-4 (86 мг, 0,5 ммоль) добавляли в толуол (5 мл), Pd2(dba)3 (46 мг, 0,05 ммоль), Xantphos (57,7 мг, 0,1 ммоль) и карбонат цезия (325 мг, 1 ммоль) добавляли в полученный раствор в атмосфере газа азота. Смесь перемешивали при 120°С в течение ночи. После выливания в H2O смесь дважды экстрагировали с помощью EtOAc. Органическую фазу сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью препаративной HPLC с получением целевого продукта (80 мг, выход 12%) в виде желтого твердого вещества. MS ESI рассч. для C29H27N5O3 [М+Н]+ 494, найденное значение 494. 1Н ЯМР (400 МГц, CDCl3) δ ppm 8,69 (s, 1Н), 8,39-8,40 (d, J=2,0 Гц, 1H), 8,27 (m, 1Н), 7,67-7,80 (m, 7Н), 6,69-6,71 (d, J=8,8 Гц, 1H), 4,39 (s, 2Н), 4,15-4,18 (m, 2Н), 4,11 (s, 3Н), 3,88 (s, 2Н), 1,64 (m, 6Н).
Вариант осуществления 254
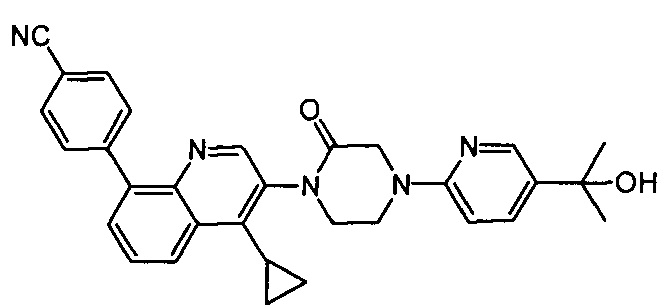
4-(4-циклопропил-3-(4-(5-(2-гидроксипропан-2-ил)пиридин-2-ил)-2-оксопиперазин-1-ил)хинолин-8-ил)бензонитрил
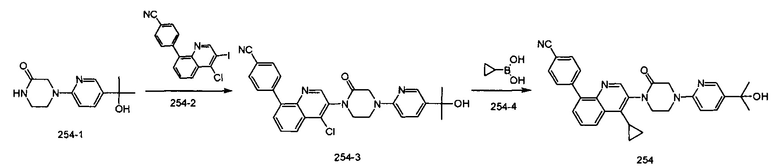
[566] Стадия 1. 254-1 (472 мг, 2 ммоль) и 254-2 (780 мг, 2 ммоль), Pd2(dba)3 (180 мг, 0,2 ммоль), Xantphos (240 мг, 0,4 ммоль) и карбонат цезия (1,3 мг, 4 ммоль) добавляли в толуол (20 мл). Смесь перемешивали при 120°С в течение ночи в атмосфере газа азота. После выливания в H2O смесь дважды экстрагировали с помощью EtOAc. Органическую фазу сушили над сульфатом натрия и концентрировали. Концентрированное вещество очищали с помощью колоночной хроматографии на силикагеле (РЕ:EtOAc=1:1) с получением соединения 254-3 (520 мг, выход 52%) в виде желтого твердого вещества. MS ESI рассч. для C28H24ClN5O2 [М+Н]+ 498, найденное значение 498.
[567] Стадия 2. 254-3 (150 мг, 0,3 ммоль) и циклопропилборную кислоту (262 мг, 3 ммоль), н-BuPAd (22 мг, 0,06 ммоль), Pd(OAc)2 (7 мг, 0,03 ммоль) и карбонат цезия (195 мг, 0,6 ммоль) добавляли в толуол/H2O (5 мл/1 мл). Смесь перемешивали при 120°С в течение ночи в атмосфере газа азота. После выливания в H2O смесь дважды экстрагировали с помощью EtOAc. Органическую фазу сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью препаративной HPLC с получением целевого соединения (50 мг, выход 33%) в виде желтого твердого вещества. MS ESI рассч. для C31H29N5O2 [М+Н]+ 504, найденное значение 504. 1Н NMR(400 МГц, CDCL3) δ ppm 8,71 (s, 1Н), 8,59-8,61 (d, J=8,0 Гц, 1Н), 8,39 (dd, J=2,0 Гц, 1Н), 7,68-7,78 (m, 7Н), 6,69-6,71 (d, J=8,8 Гц, 1Н), 4,31-4,65 (m, 2Н), 4,12-4,23 (m, 2Н), 3,91-3,96 (m, 2Н), 2,13 (m, 1Н), 1,62 (s, 6Н), 1,18-1,24 (m, 2Н), 0,84 (m, 1H), 0,64 (m, 1Н).
Вариант осуществления 255
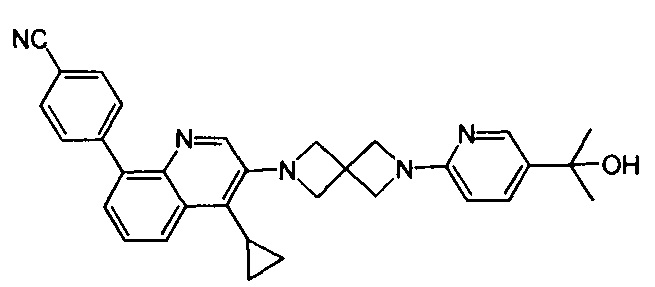
4-(4-циклопропил-3-(6-(5-(2-гидроксипропан-2-ил)пиридин-2-ил)-2,6-диазаспиро[3.3]гептан-2-ил)хинолин-8-ил)бензонитрил

[568] Стадия 1. Pd2(dba)3 (92 мг, 0,1 ммоль), Xantphos (120 мг, 0,2 ммоль) и карбонат цезия (650 мг, 2 ммоль) добавляли в раствор 255-1 (390 мг, 1 ммоль) и 255-2 (299 мг, 1 ммоль) в толуоле (10 мл). В атмосфере газа азота смесь перемешивали при 120°С в течение ночи. После выливания в H2O смесь дважды экстрагировали с помощью EtOAc. Органическую фазу сушили над безводным сульфатом натрия и концентрировали. Концентрированное вещество очищали с помощью колоночной хроматографии на силикагеле (РЕ/EtOAc=1:1) с получением 255-3 (400 мг, выход 87%) в виде желтого твердого вещества. MS ESI рассч. для C26H25ClN4O2 [М+Н]+ 461, найденное значение 461.
[569] Стадия 2. 255-3 (400 мг, 0,87 ммоль) и циклопропилборную кислоту (378 мг, 4,35 ммоль), н-BuPAd (62 мг, 0,174 ммоль), Pd(OAc)2 (20 мг, 0,09 ммоль) и карбонат цезия (565 мг, 1,74 ммоль) добавляли в толуол/H2O (10 мл/1 мл). В атмосфере газа азота смесь перемешивали при 120°С в течение ночи. После выливания в H2O смесь дважды экстрагировали с помощью EtOAc. Органическую фазу сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле (РЕ/ EtOAc=3:1) с получением 255-5 (400 мг, выход 98%) в виде желтого твердого вещества. MS ESI рассч. для C29H30N4O2 [М+Н]+ 467, найденное значение 467.
[570] Стадия 3. 255-5 (400 мг, 0,87 ммоль) растворяли в DCM (10 мл) и добавляли TFA (5 мл), смесь перемешивали при комнатной температуре в течение 2 ч. Смесь концентрировали и сушили с получением соединения 255-6 (300 мг, выход 94%) в виде желтого масла. MS ESI рассч. для C24H22N4 [М+Н]+ 367, найденное значение 367.
[571] Стадия 4. 255-6 (184 мг, 0,5 ммоль) и 255-7 (86 мг, 0,5 ммоль), Pd2(dba)3 (46 мг, 0,05 ммоль), Xantphos (57,7 мг, 0,1 ммоль) и карбонат цезия (325 мг, 1 ммоль) добавляли в толуол (5 мл). В атмосфере газа азота смесь перемешивали при 120°С в течение ночи. После выливания в H2O смесь дважды экстрагировали с помощью EtOAc. Органическую фазу сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью препаративной HPLC с получением целевого соединения (30 мг, выход 12%) в виде желтого твердого вещества. MS ESI рассч. для C32H31N5O [М+Н]+ 502, найденное значение 502. 1Н NMR (400 МГц, CDCL3) δ ppm 8,30-8,38 (m, 3Н), 7,70-7,77 (m, 4H), 7,68 (d, J=2,0 Гц, 1H), 7,56-7,58 (d, J=8,4 Гц, 1H), 6,35-6,37 (d, J=8,8 Гц, 1H), 4,37-4,40 (m, 4H), 4,11-4,13 (m, 4H), 2,05-2,07 (m, 1H), 1,59 (s, 6H), 1,23-1,27 (m, 2H), 0,78-0,79 (m, 2H).
Вариант осуществления 256
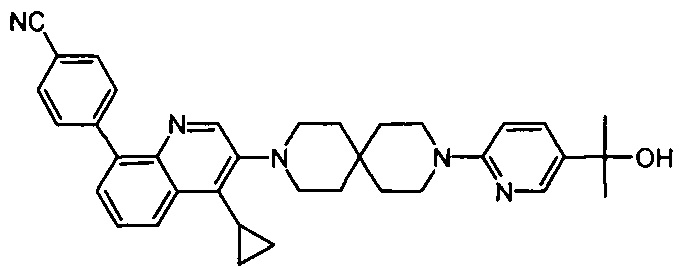
4-(4-циклопропил-3-(9-(5-(2-гидроксипропан-2-ил)пиридин-2-ил)-3,9-диазаспиро[5.5]ундекан-3-ил)хинолин-8-ил)бензонитрил
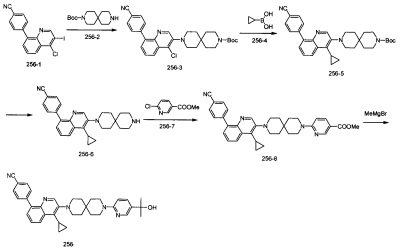
[572] Стадия 1. 256-1 (780,00 мг, 2,0 ммоль) и 256-2 (507,96 мг, 2,0 ммоль) растворяли в толуоле (20 мл), в атмосфере газа азота добавляли Pd2(dba)3 (115,00 мг, 200,00 мкмоль). Смесь перемешивали при комнатной температуре в течение 10 мин., затем нагревали до 110°С и подвергали реакции в течение 3 ч. После охлаждения до комнатной температуры смесь выливали в H2O (200 мл) и перемешивали в течение 20 мин. Затем смесь экстрагировали с помощью EtOAc (400 мл*3). Объединенную органическую фазу промывали с помощью H2O и солевых растворов (200 мл*2), соответственно, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью тонкослойной хроматографии (РЕ/EtOAc экстрагированная часть =5:1) с получением 256-3 (100 мг, выход 9,67%) в виде желтого твердого вещества. MS ESI рассч. для C30H33ClN4O2 [М+Н]+ 517, найденное значение 517.
[573] Стадия 2. 256-3 (100 мг, 193,4 мкмоль), циклопропилборную кислоту (83,07 мг, 967 мкмоль) растворяли в толуоле (10 мл), в атмосфере газа азота добавляли Pd(OAc)2 (0,20 мг, 38,68 мкмоль). Смесь перемешивали при комнатной температуре в течение 10 мин., затем нагревали до 110°С и подвергали реакции в течение 1 ч. После охлаждения до комнатной температуры смесь выливали в H2O (50 мл) и перемешивали в течение 20 мин. Затем смесь экстрагировали с помощью EtOAc (100 мл*3). Объединенную органическую фазу промывали с помощью H2O и солевых растворов (50 мл*2), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (РЕ/ EtOAc =5:1) с получением 256-5 (80,00 мг, выход 79,14%) в виде желтого твердого вещества. MS ESI рассч. для C33H38N4O2 [М+Н]+ 523, найденное значение 523.
[574] Стадия 3. 256-5 (80 мг, 153,06 мкмоль) растворяли в DCM (5 мл) и добавляли HCl/EtOAc (4 М, 2 мл). Затем смесь перемешивали при комнатной температуре в течение 2 ч. Неочищенный продукт 256-6, полученный после концентрации, непосредственно применяли для следующей стадии. MS ESI рассч. для C28H30N4 [М+Н]+ 423, найденное значение 423.
[575] Стадия 4. 256-6 (50 мг, 118,33 мкмоль) и 256-7 (24,36 мг, 142 мкмоль) растворяли в ацетонитриле (5 мл) и добавляли K2CO3 (163,54 мг, 1,18 ммоль). Смесь перемешивали при комнатной температуре в течение 10 мин., затем нагревали до 80°С в течение 12 ч. После охлаждения до комнатной температуры смесь выливали в H2O (50 мл) и перемешивали в течение 20 мин., полученную смесь экстрагировали с помощью EtOAc (50 мл*3). Органические фазы объединяли и промывали солевыми растворами (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали с помощью тонкослойной хроматографии (РЕ/EtOAc =5:1) с получением 256-8 (30 мг, выход 45,46%) в виде желтого твердого вещества. MS ESI рассч. для C35H35N5O2 [М+Н]+ 558, найденное значение 558.
[576] Стадия 5. 256-8 (30 мг, 53,79 мкмоль) растворяли в THF (5 мл), бромид метилмагния (32,07 мг, 268,95 мкмоль) по каплям добавляли при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 1 ч., затем выливали в водный раствор хлорида аммония (50 мл), перемешивали в течение 20 мин., полученную смесь экстрагировали с помощью EtOAc (50 мл×3). Органические фазы объединяли и промывали солевыми растворами (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали с помощью препаративной HPLC с получением целевого соединения (2 мг, выход 6,67%) в виде желтого твердого вещества. MS ESI рассч. для C36H39N5O [М+Н]+ 558, найденное значение 558. 1H ЯМР (400 МГц, метанол-d4) δ ppm 8,76 (s, 1H), 8,49 (d, J=8 Гц, 1Н), 8,31 (d, J=2,0 Гц, 1Н), 7,76 (s, 4Н), 7,66 (t, J=6,4 Гц, 1H), 7,51-7,58 (m, 2Н), 3,50-3,58 (m, 4Н), 3,25-3,29 (m, 4Н), 2,01-2,05 (m, 2Н), 1,63 (s, 6Н), 1,23-1,26 (m, 2Н).
Вариант осуществления 257
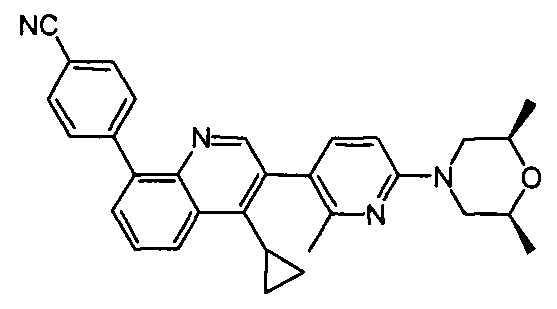
4-(4-циклопропил-3-(6-((2S,6R)-2,6-диметилморфолино)-2-метилпиридин-3-ил)хинолин-8-ил)бензонитрил
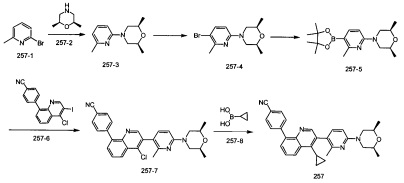
[577] Стадия 1. 257-1 (10 г, 58,13 ммоль), 257-2 (8,03 г, 69,76 ммоль), Pd2(dba)3 (53,23 г, 58,13 ммоль), Xantphos (33,64 г, 58,13 ммоль) и трет-бутоксид калия (6,52 г, 58,13-ммоль) перемешивали в толуоле (40 мл), дегазировали и продували с помощью газа азота 3 раза. В атмосфере газа азота смесь перемешивали при 110°С в течение 12 ч. Реакционную смесь разбавляли с помощью H2O (100 мл) и экстрагировали с помощью EtOAc (100 мл*3). Органические фазы объединяли и промывали солевыми растворами (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле (РЕ/EtOAc =10:1) с получением соединения 257-3 (8 г, выход 66,72%) в виде желтого масла. MS ESI рассч. для C12H18N2O [М+Н]+ 207, найденное значение 207.
[578] Стадия 2. 257-3 (11,40 г, 55,26 ммоль) растворяли в DMF (20 мл), добавляли NBS (9,34 г, 52,50 ммоль). Смесь перемешивали при 25°С в течение 0,5 ч., разбавляли с помощью H2O (100 мл), затем экстрагировали с помощью EtOAc (100 мл × 3). Объединенную органическую фазу промывали солевыми растворами (100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле (РЕ/EtOAc =10:1) с получением соединения 257-4 (12 г, выход 76,15%) в виде белого твердого вещества. MS ESI рассч. для C12H17BrN2O [М+Н]+ 285, найденное значение 285.
[579] Стадия 3. 257-4 (16 г, 56,10 ммоль), бис(пинаколато)дибор (21,37 г, 84,15 ммоль), Pd(dppf)Cl2 (2,05 г, 2,81 ммоль) и KOAc (11,01 г, 112,20 ммоль) перемешивали в диоксане (50 мл). В атмосфере газа азота смесь перемешивали при 100°С в течение 2 ч. Реакционную смесь разбавляли с помощью H2O (100 мл) и экстрагировали с помощью EtOAc (100 мл*3). Органическую фазу объединяли и промывали солевыми растворами (20 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле (РЕ/ EtOAc=10:1) с получением соединения 257-5 (10 г, выход 53,65%) в виде белого твердого вещества. MS ESI рассч. для C18H29BN2O3 [М+Н]+ 333, найденное значение 333.
[580] Стадия 4. Pd(dppf)Cl2 (561,95 мг, 768 мкмоль) и Na2CO3 (3,26 г, 30,72 ммоль) добавляли в раствор 257-6 (6 г, 15,36 ммоль) и 257-5 (7,65 г, 23,04 ммоль) в. DMF (5 мл)/H2O (5 мл)/THF (25 мл). Смесь перемешивали при 70°С в течение 2 ч. Реакционную смесь разбавляли с помощью H2O (100 мл) и экстрагировали с помощью EtOAc (100 мл*3). Объединенную органическую фазу промывали солевыми растворами (20 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии (РЕ/EtOAc =5:1) с получением 257-7 (3 г, выход 41,65%) в виде желтого твердого вещества. MS ESI рассч. для C28H25ClN4O [М+Н]+ 469, найденное значение 469.
[581] Стадия 5. Бис(адамантил)бутилфосфин (229,47 мг, 640 мкмоль), Cs2CO3 (2,09 г, 6,40 ммоль) и Pd(OAc)2 (71,84 мг, 320 мкмоль) добавляли в раствор 257-7 (1,50 г, 3,20 ммоль) и циклопропилборной кислоты (1,37 г, 16 ммоль) в толуоле (15 мл). Смесь перемешивали при 110°С в течение 2 ч. Реакционную смесь разбавляли с помощью H2O (100 мл) и экстрагировали с помощью EtOAc (100 мл*3). Объединенную органическую фазу промывали солевыми растворами (20 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии (РЕ/EtOAc=10:1) с получением целевого продукта (1 г, выход 65,85%) в виде светло-красного твердого вещества. MS ESI рассч. для C31H30N4O [М+Н]+ 475, найденное значение 475. 1НЯМР (400 МГц, CDCl3) δ ppm 8,69 (s, 1H), 8,64 (dd, J=3,0, 6,5 Гц, 1H), 7,74-7,90 (m, 4Н), 7,67-7,72 (m, 2Н), 7,35 (d, J=8,5 Гц, 1Н), 6,58 (d, J=8,5 Гц, 1Н), 4,06-4,26 (m, 2Н), 3,69-3,79 (m, 2Н), 2,58 (t, J=11,5 Гц, 2Н), 2,29 (s, 3Н), 2,09-2,21 (m, 1Н), 1,31 (d, J=6,5 Гц, 6Н), 0,99 (q, J=5,3, 8,6 Гц, 2Н), 0,27-0,54 (m, 2Н).
Вариант осуществления 258
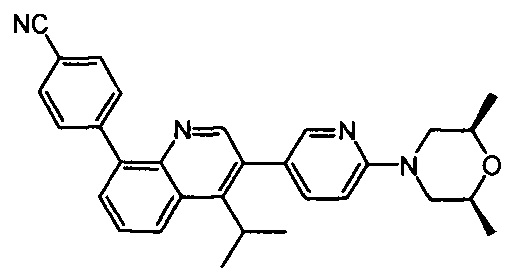
4-(3-(6-((2S,6R)-2,6-диметилморфолино)пиридин-3-ил)-4-изопропилхинолин-8-ил)бензонитрил
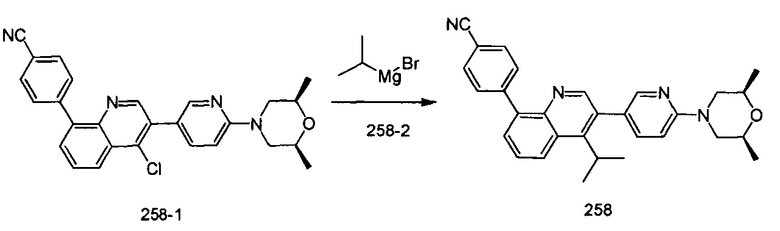
[582] 258-1 (227,48 мг, 500 мкмоль) растворяли в THF (4 мл) и NMP (1 мл), затем при -40°С по каплям добавляли изопропилмагния бромид (73,65 мг, 500 мкмоль). Реакционную смесь нагревали до 0°С и перемешивали в течение 10 мин., затем при комнатной температуре в течение 2 ч. Реакционную смесь медленно гасили с помощью льда, затем экстрагировали с помощью EtOAc (5 мл × 3). Органические фазы объединяли и промывали солевыми растворами (100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью HPLC с получением целевого соединения (20 мг, выход 8,79%) в виде белого твердого вещества. MS ESI рассч. для C30H30N4O [М+Н]+ 463, найденное значение 463. 1H ЯМР (400 МГц, хлороформ-d) δ ppm 8,68 (s, 1Н), 8,46-8,48 (t, J=5,02 Гц, 1Н), 8,22 (s, 1Н), 7,73-7,82 (m, 7Н), 7,06-7,08 (d, J=9,2 Гц, 1Н), 4,13-4,16 (m, 2Н), 3,81-3,84 (m, 2Н), 3,65 (m, 1Н), 2,91-2,97 (m, 2Н), 1,60-1,62 (d, J=7,2 Гц, 6Н), 1,33-1,35 (d, J=6,0 Гц, 6Н).
Вариант осуществления 259

4-(4-циклобутил-3-(6-((2S,6R)-2,6-диметилморфолино)пиридин-3-ил)хинолин-8-ил)бензонитрил
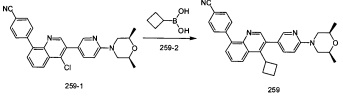
[583] Pd(dppf)Cl2 (36,59 мг, 50 мкмоль) и Na2CO3 (105,99 мг, 1 ммоль) добавляли в раствор 259-1 (227,48 мг, 500 мкмоль), циклобутилборной кислоты (99,92 мг, 1 ммоль) в THF/H2O (2 мл). В атмосфере газа азота смесь нагревали до 80°С и подвергали реакции в течение 2 ч., затем выливали в H2O (10 мл). Реакционную смесь экстрагировали с помощью EtOAc (10 мл*3). Органическую фазу промывали солевыми растворами (30 мл), сушили над безводным MgSO4, концентрировали под вакуумом. Остаток очищали с помощью препаративной HPLC с получением целевого продукта (50 мг, выход 21,07%). MS ESI рассч. для C31H30N4O [М+Н]+ 475, найденное значение 475. 1Н ЯМР (400 МГц, хлороформ-d) δ ppm 8,70 (s, 1Н), 8,09-8,23 (m, 2Н), 7,79 (d, J=3,01 Гц, 4Н), 7,58-7,71 (m, 2Н), 7,46-7,57 (m, 1Н), 6,63-6,84 (m, 1Н), 4,26-4,50 (m, 1Н), 4,12 (d, J=12,55 Гц, 2Н), 3,76 (d, J=6,53 Гц, 2Н), 2,54-2,69 (m, 2Н), 2,37 (d, J=8,53 Гц, 2Н), 1,84-2,15 (m, 4Н), 1,65-1,78 (m, 1Н), 1,17-1,38 (m, 8Н).
Вариант осуществления 260
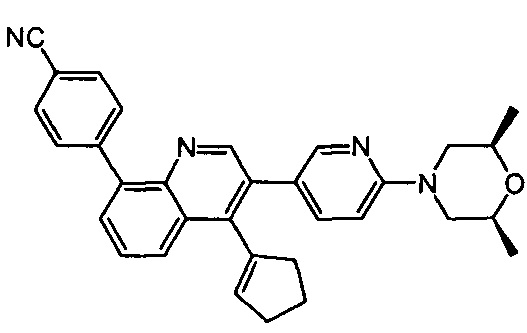
4-(4-(1-циклопентен-1-ил)-6-(3-(-(2S,6R)-2,6-диметилморфолино)пиридин-3-ил)хинолин-8-ил)бензонитрил
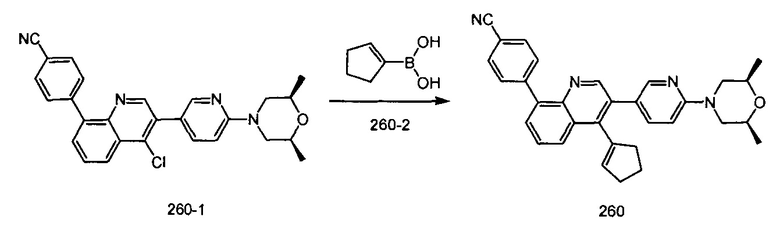
[584] Pd(dppf)Cl2 (80,49 мг, 110 мкмоль) и Na2CO3 (233,18 мг, 2,20 ммоль) добавляли в раствор 260-1 (500 мг, 1,10 ммоль), 260-2 (256,19 мг, 1,32 ммоль) в THF (10 мл). В атмосфере газа азота смесь нагревали до 80°С и подвергали реакции в течение 2 ч. Реакционную смесь выливали в H2O (10 мл) и экстрагировали с помощью EtOAc (10 мл*3). Органическую фазу промывали солевыми растворами (10 мл), сушили над безводным сульфатом магния и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии с получением целевого соединения (450 мг, выход 84,07%) в виде желтого твердого вещества. MS ESI рассч. для C32H30N4O [М+Н]+ 487, найденное значение 487. 1H ЯМР (400 МГц, хлороформ-d) δ ppm 8,78-8,91 (m, 1Н), 8,31 (d, J=2,26 Гц, 1Н), 8,13 (dd, J=8,28, 1,25 Гц, 1Н), 7,74-7,88 (m, 5Н), 7,60-7,73 (m, 2Н), 6,72 (d, J=8,78 Гц, 1Н), 5,96 (s, 1Н), 4,05-4,29 (m, 2Н), 3,72-3,90 (m, 2Н), 2,64 (d, J=10,54 Гц, 4Н), 2,32-2,47 (m, 1Н), 2,01 (t, J=7,28 Гц, 2Н), 1,32 (d, J=6,27 Гц, 6Н).
Вариант осуществления 261
4-(4-((4-циклопропил-35-изопропилпиридин-2-ил)-3-)-хинолин-8-ил)бензонитрил
[585] Стадия 1. В атмосфере газа азота Pd(dppf)Cl2 (1,14 г, 1,56 ммоль, 0,10 экв.) добавляли в смесь 261-1 (3 г, 15,59 ммоль, 1 экв.), карбонат натрия (3,30 г, 31,18 ммоль, 2 экв.) и 261-2 (2,77 г, 18,71 ммоль, 1,20 экв.) в DMF (2 мл)/H2O (2 мл)/ТНР (10 мл). Смесь перемешивали при 70°С в течение 12 ч., выливали в H2O (150 мл). Полученную смесь экстрагировали с помощью EtOAc (100×3). Объединенную органическую фазу промывали солевыми растворами (150 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии (РЕ/EtOAc=20:1) с получением 261-3 (1,50 г, выход 62,60%) в виде бесцветного масла. MS ESI рассч. для C8H8ClN [М+Н]+ 154, найденное значение 154.
[586] Стадия 2. 261-3 (1,50 г, 9,76 ммоль, 1 экв.) растворяли в этаноле (30 мл), в атмосфере газа азота добавляли PtO2 (2,22 г, 9,76 ммоль, 1 экв.) (10%, 0,15 г). Суспензию дегазировали под вакуумом и продували с помощью газа водорода 3 раза. Смесь перемешивали при 25°С в течение 4 ч. в атмосфере Н2 (30 фунтов/кв. дюйм). Реакционную смесь фильтровали, фильтрат концентрировали. Неочищенный продукт применяли непосредственно на следующей стадии. MS ESI рассч. для C8H10ClN [М+Н]+ 156, найденное значение 156.
[587] Стадия 3. Смесь 261-5 (100 мг, 282,13 мкмоль, 1 экв.), карбонат цезия (183,85 мг, 564,26 мкмоль, 2 экв.), Xantphos (32,65 мг, 56,43 мкмоль, 0,20 экв.) и 261-4 (65,86 мг, 423,20 мкмоль, 1,50 экв.) растворяли в толуоле (10 мл), в атмосфере газа азота добавляли Pd2(dba)3 (25,84 мг, 28,21 ммоль, 0,10 экв.). Смесь перемешивали при 110°С в течение 2 ч., выливали в H2O (100 мл). Полученную смесь экстрагировали с помощью EtOAc (100 мл × 3). Органические фазы объединяли и промывали солевыми растворами (100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали (кислотой) с помощью HPLC с получением целевого соединения (20 мг, выход 14,97%) в виде желтого твердого вещества. MS ESI рассч. для C31H31N5 [М+Н]+ 474, найденное значение 474. 1Н ЯМР (400 МГц, метанол-d4) δ ppm 8,86 (d, J=8,3 Гц, 1Н), 8,73 (s, 1H), 8,14 (dd, J=2,0, 9,3 Гц, 1H), 7,95-8,03 (m, 3Н), 7,75-7,91 (m, 4Н), 7,52 (d, J=9,5 Гц, 1H), 4,04 (brs, 4Н), 3,64 (brs, 4Н), 1,59 (d, J=7,3 Гц, 2Н), 1,32 (d, J=7,0 Гц, 7Н), 1,10 (d, J=4,8 Гц, 2Н).
4-(4-циклопропил-3-(4-гидрокси-(5-(2-гидроксипропан-2-ил)пиридин-2-ил)пиперидин-1-ил)хинолин-8-илбензонитрил
[588] Стадия 1. В атмосфере газа азота Pd2(dba)3 (234,42 мг, 256 мкмоль, 0,10 экв.) добавляли в смесь 262-1 (1 г, 2,56 ммоль, 1 экв.), Xantphos (296,25 мг, 512 мкмоль, 0,20 экв.), карбоната цезия (1,67 г, 5,12 ммоль, 2 экв.) и 262-2 (439,85 мг, 3,07 ммоль, 1,20 экв.) в толуоле (20 мл). Смесь перемешивали при 110°С в течение 2 ч., выливали в H2O (150 мл). Полученную смесь экстрагировали с помощью EtOAc (400 мл × 3). Органические фазы объединяли и промывали солевыми растворами (200 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии (РЕ/EtOAc =3:1) с получением соединения 262-3 (800 мг, выход 76,99%) в виде желтого твердого вещества. MS ESI рассч. для C23H20ClN3O2 [М+Н]+ 406, найденное значение 406.
[589] Стадия 2. 262-3 (400 мг, 985,51 мкмоль, 1 экв.) растворяли в THF (10 мл) и добавляли раствор HCl (4 М EtOAc, 2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 12 ч., затем нагревали до 45°С и перемешивали в течение еще 1 ч. Смесь выливали в H2O (150 мл) и экстрагировали с помощью EtOAc (400 мл × 3). Органические фазы объединяли и промывали солевыми растворами (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии (РЕ/EtOAc =2:1) с получением 262-4 (320 мг, выход 89,-74%) в виде белого твердого вещества. MS ESI рассч. для C22H19N3O2 [М+Н]+ 358, найденное значение 358.
[590] Стадия 3. В атмосфере газа азота бис(адамантил)бутилфосфин (79,27 мг, 221,10 мкмоль) добавляли в смесь 262-4 (200 мг, 552,76 мкмоль, 1 экв.) и 262-5 (237,41 мг, 2,76 ммоль, 5 экв.) в толуоле (10 мл). Смесь перемешивали при 110°С в течение 2 ч., выливали в H2O (150 мл). Полученную смесь экстрагировали с помощью EtOA (400 мл × 3). Органические фазы объединяли и промывали солевыми растворами (200 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии (РЕ/EtOAc =2:1) с получением 262-6 (150 мг, выход 73,85%) в виде белого твердого вещества. MS ESI рассч. для C24H21N3O [М+Н]+ 368, найденное значение 368.
[591] Стадия 4. В атмосфере газа азота н-BuLi (52,30 мг, 816,45 мкмоль, 3 экв.) по каплям добавляли в раствор 262-7 (70,56 мг, 326,58 мкмоль, 1,20 экв.), растворенный в THF (5 мл) при -78°С. Через 1 ч. 262-6 (100 мг, 272,15 мкмоль, 1 экв.) добавляли в смесь и смесь перемешивали при -78°С в течение дополнительного 1 ч. Смесь нагревали до 25°С, выливали в насыщенный раствор NH4Cl (50 мл), затем перемешивали в течение 20 мин., полученную смесь экстрагировали с помощью EtOAc (100 мл × 3). Органические фазы объединяли и промывали солевыми растворами (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали с помощью HPLC с получением целевого соединения (3 мг, выход 2,18%) в виде желтого твердого вещества. MS ESI рассч. для C32H32N4O2 [М+Н]+ 505, найденное значение 505. 1Н ЯМР (400 МГц, CDCl3) δ ppm 8,56-9,23 (m, 2Н), 7,53-7,95 (m, 10Н), 3,68 (brs., 4Н), 2,21 (brs, 1Н), 1,58 (brs, 11Н), 1,06 (brs., 2H).
Вариант осуществления 263
(2S,6R)-4-(4-(4-циклопропил-8-(пиридин-2-ил)хинолин-3-ил)фенил)-2,6-диметилморфолин
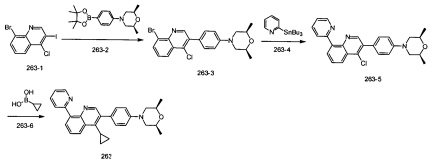
[592] Стадия 1. 263-1 (266,27 мг, 722,77 мкмоль, 1 экв.) и 263-2 (230 мг, 722,77 мкмоль, 1 экв.) растворяли в DMF (2 мл)/THF (2 мл). В атмосфере газа азота Pd(dppf)Cl2 (52,89 мг, 72,28 мкмоль, 0,10 экв.) добавляли в реакционную систему. Смесь перемешивали при 70°С в течение 2 ч., выливали в H2O (100 мл). Смесь экстрагировали с помощью EtOAc (200 мл × 3). Органические фазы объединяли и промывали солевыми растворами (100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии (РЕ/EtOAc=5:1) с получением 263-3 (270 мг, выход 86,33%) в виде белого твердого вещества. MS ESI рассч. для C21H20BrClN2O [М+Н]+ 431, найденное значение 431.
[593] Стадия 2. В атмосфере газа азота Pd(PPh3)4 (72,10 мг, 62,39 мкмоль, 0,10 экв.) добавляли в раствор 263-3 (270 мг, 623,93 мкмоль, 1 экв.) и 263-4 (229,69 мг, 623,93 мкмоль, 1 экв.) в толуоле (10 мл). Смесь перемешивали при 110°С в течение 2 ч., выливали в H2O (50 мл). Смесь экстрагировали с помощью EtOAc (100 мл × 3). Органические фазы объединяли и промывали солевыми растворами (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии (РЕ/EtOAc=2:1) с получением 263-5 (120 мг, выход 44,63%) в виде белого твердого вещества. MS ESI рассч. для C26H24ClN3O [М+Н]+ 430, найденное значение 430.
[594] Стадия. 3. В атмосфере газа азота Pd(OAc)2 и бис(адамантил)бутилфосфин (66,56 мг, 185,65 мкмоль, 1 экв.) добавляли в раствор 263-5 (80 мг, 185,65 мкмоль, 1 экв.) и циклопропилборной кислоты (159,47 мг, 1,86 ммоль, 10 экв.) в толуоле (5 мл). Смесь перемешивали при 110°С в течение 2 ч., затем выливали в H2O (50 мл). Смесь экстрагировали с помощью EtOAc (100мл × 3). Органические фазы объединяли и промывали солевыми растворами (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали с помощью HPLC с получением целевого соединения (15 мг, выход 18,51%) в виде белого твердого вещества. MS ESI рассч. для C29H29N3O [М+Н]+ 436, найденное значение 436. 1H ЯМР (400 МГц, метанол-d4) δ ppm 9,14 (s, 1Н), 9,03-9,13 (m, 2Н), 8,75-8,81 (m, 1Н), 8,65-8,72 (m, 2Н), 8,41-8,51 (m, 1Н), 8,35 (s, 1Н), 8,03-8,11 (m, 2Н), 7,68 (d, J=9,5 Гц, 1Н), 4,19-4,29 (m, 2Н), 3,89 (dd, J=6,3, 8,5 Гц, 2Н), 3,04 (dd, J=11,2, 12,4 Гц, 2Н), 2,57-2,75 (m, 1H), 1,28-1,37 (m, 8Н), 0,52 (q, J=5,5 Гц, 2Н).
Вариант осуществления 264
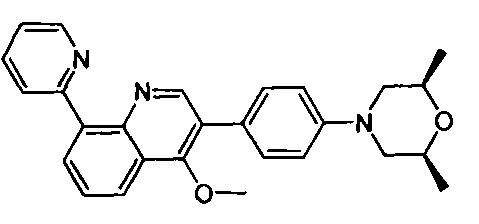
(2S,6R)-4-(4-(4-метокси-8-(пиридин-2-ил)хинолин-3-ил)фенил)-2,6-диметилморфолин

[595] 264-1 (40 мг, 92,82 мкмоль) растворяли в метаноле (5 мл), затем добавляли NaOMe (50,14 мг, 928,20 мкмоль). Смесь перемешивали при 25°С в течение 48 ч., гасили с помощью H2O. После концентрирования остаток очищали с помощью препаративной HPLC с получением целевого соединения (3 мг, выход 7,58%) в виде белого твердого вещества. MS ESI рассч. для C27H27N3O2 [М+Н]+ 426, найденное значение 426. 1Н ЯМР (400 МГц, CDCl3) δ ppm 8,91 (s, 1H), 8,79 (d, J=4,5 Гц, 1H), 8,51 (d, J=2,0 Гц, 1H), 8,31 (d, J=8,5 Гц, 1H), 8,08 (dd, J=7,3, 13,8 Гц, 2H), 7,79-7,90 (m, 2H), 7,70 (t, J=7,8 Гц, 1H), 7,28-7,36 (m, 1H), 6,79 (d, J=9,0 Гц, 1H), 4,16 (d, J=12,0 Гц, 2H), 3,68-3,84 (m, 5H), 2,63 (t, J=11,8 Гц, 2H), 1,31 (d, J=6,0 Гц, 6H).
Вариант осуществления 265
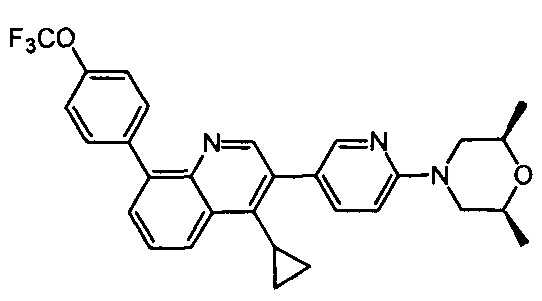
(2S,6R)-4-(5-(4-циклопропил-8-(4-(трифторметокси)фенил)хинолин-3-ил)пиридин-2-ил)-2,6-диметилморфолин
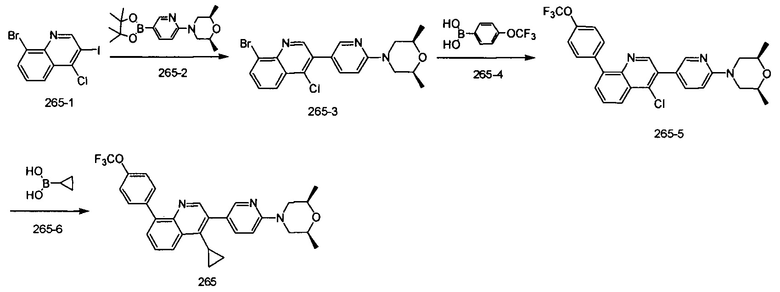
[596] Стадия 1. В атмосфере газа азота смесь 265-1 (500 мг, 1,36 ммоль, 1 экв.), 265-2 (431,89 мг, 1,36 ммоль, 1 экв.), Pd(dppf)Cl2 (99,51 мг, 136 мкмоль, 0,10 экв.) и Na2CO3 (288,29 мг, 2,72 ммоль, 2 экв.) в THF (10 мл) нагревали до 80°С и подвергали реакции в течение 2 ч. Реакционную смесь выливали в H2O (10 мл). Смесь дважды экстрагировали с помощью EtOAc. Органическую фазу промывали солевыми растворами (10 мл), сушили над безводным MgSO4, концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии (РЕ/EtOAc=3:1) с получением 265-3 (500 мг, выход 84,96%) в виде желтого твердого вещества. MS ESI рассч. для C20H19BrClN3O [M+H]+ 432, найденное значение 432.
[597] Стадия 2. В атмосфере газа азота смесь 265-3 (500 мг, 1,16 ммоль, 1 экв.), 265-4 (237,94 мг, 1,16 ммоль, 1 экв.), Pd(dppf)Cl2 (84,88 мг, 116 мкмоль, 0,10 экв.) и Na2CO3 (245,90 мг, 2,32 ммоль, 2 экв.) в THF (10 мл) и H2O (2 мл) нагревали до 80°С и подвергали реакции в течение 2 ч. Реакционную смесь выливали в H2O (10 мл), экстрагировали с помощью EtOAc (10 мл × 3). Органическую фазу промывали солевыми растворами (10 мл), сушили над безводным MgSO4 и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии (РЕ/EtOAc=3:1) с получением 265-5 (560 мг, 1,09 ммоль, выход 93,93%) в виде желтого твердого вещества. MS ESI рассч. для C27H23ClF3N3O2 [М+Н]+ 514, найденное значение 514.
[598] Стадия 3. В атмосфере газа азота смесь 265-5 (100 мг, 194,58 мкмоль, 1 экв.), циклопропилборной кислоты (83,57 мг, 972,90 мкмоль, 5 экв.), Pd(OAc)2 (4,37 мг, 19,46 мкмоль, 0,10 экв.) и бис(адамантил)бутилфосфина (13,95 мг, 38,92 мкмоль, 0,20 экв.) в толуоле (5 мл) нагревали до 120°С и подвергали реакции в течение 2 ч. Реакционную смесь выливали в H2O (10 мл), экстрагировали с помощью EtOAc (10 мл × 3). Органическую фазу промывали солевыми растворами (10 мл), сушили над безводным Na2SO4 и концентрировали под вакуумом. Остаток очищали с помощью препаративной HPLC (TFA) с получением целевого соединения (60 мг, выход 59,35%) в виде белого твердого вещества. MS ESI рассч. для C30H28F3N3O2 [М+Н]+ 520, найденное значение 520. 1Н ЯМР (400 МГц, хлороформ-d) δ ppm 8,87 (s, 1Н), 8,63 (dd, J=7,28, 2,51 Гц, 1Н), 8,40 (d, J=2,26 Гц, 1Н), 7,63-7,80 (m, 5Н), 7,36 (d, J=8,28 Гц, 2Н), 6,79 (d, J=8,78 Гц, 1Н), 4,17 (d, J=11,29 Гц, 2Н), 3,81 (dd, J=4,02, 2,26 Гц, 2Н), 2,65 (dd, J=12,42, 10,92 Гц, 2Н), 2,34 (s, 1Н), 1,33 (d, J=6,27 Гц, 6Н), 1,13 (d, J=7,53 Гц, 2Н), 0,41 (d, J=5,02 Гц, 2Н).
Вариант осуществления 266

4-(4-циклопропил-3-(6-((2S,6S)-2,6-диметилморфолино)пиридин-3-ил)хинолин-8-ил)бензонитрил
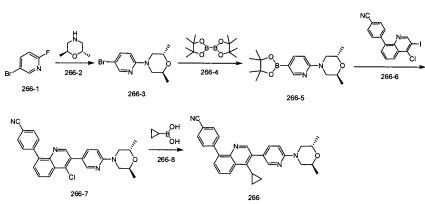
[599] Стадия 1. 266-1 (600 мг, 3,41 ммоль, 1 экв.) и 266-2 (471,28 мг, 4,09 ммоль, 1,20 экв.) растворяли в ацетонитриле (10 мл), затем добавляли K2CO3 (942,59 мг, 6,82 ммоль, 2 экв.). Смесь перемешивали при 80°С в течение 24 ч. Смесь охлаждали, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии (РЕ/EtOAc=10:1) с получением 266-3 (450 мг, выход 48,67%) в виде белого твердого вещества. MS ESI рассч. для C11H15BrN2O [М+Н]+ 271, найденное значение 271.
[600] Стадия 2. В атмосфере газа азота Pd(dppf)Cl2 (121,46 мг, 166 мкмоль, 0,10 экв.) добавляли в раствор 266-3 (450 мг, 1,66 ммоль, 1 экв.), KOAc (325,82 мг, 3,32 ммоль, 2 экв.) и 266-4 (843,08 мг, 3,32 ммоль, 2 экв.) в диоксане (10 мл). Смесь перемешивали при 80°С в течение 2 ч., выливали в H2O (100 мл). Смесь экстрагировали с помощью EtOAc (100×3). Органические фазы объединяли и промывали солевыми растворами (100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии (РЕ/EtOAc=5:1) с получением 266-5 (200 мг, выход 37,86%) в виде желтого твердого вещества. MS ESI рассч. для C17H27BN2O3 [М+Н]+ 319, найденное значение 319.
[601] Стадия 3. 266-6 (200 мг, 512,03 мкмоль, 1 экв.) и 266-5 (195,53 мг, 614,44 мкмоль, 1,20 экв.) растворяли в DMF (2 мл)/H2O (2 мл)/THF (10 мл), в атмосфере газа азота добавляли Pd(dppf)Cl2 (37,47 мг, 51,20 мкмоль, 0,10 экв.). Смесь перемешивали при 70°С в течение 2 ч., выливали в H2O (150 мл). Смесь экстрагировали с помощью EtOAc (150 мл × 3). Органические фазы объединяли и промывали солевыми растворами (100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии (РЕ/EtOAc=5:1) с получением 266-7 (150 мг, выход 64,39%) в виде желтого твердого вещества. MS ESI рассч. для C27H23ClN4O [М+Н]+ 455, найденное значение 455.
[602] Стадия 4. 266-7 (150 мг, 329,71 мкмоль, 1 экв.), карбонат цезия (214,85 мг, 659,42 мкмоль, 2 экв.), бис(адамантил)бутилфосфин (118,21 мг, 329,71 мкмоль, 1 экв.) и циклопропилборную кислоту (141,61 мг, 1,65 ммоль, 5 экв.) растворяли в толуоле (10 мл), в атмосфере газа азота добавляли Pd(OAc)2 (37,01 мг, 164,86 мкмоль, 0,50 экв.). Смесь перемешивали при 110°С в течение 2 ч., выливали в H2O (100 мл). Полученную смесь экстрагировали с помощью EtOAc (100 мл × 3). Органические фазы объединяли и промывали солевыми растворами (100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали с помощью HPLC (кислоты) с получением 266-9 (50 мг, выход 32,93%) в виде желтого твердого вещества. MS ESI рассч. для C30H28N4O [М+Н]+ 461, найденное значение 461. 1H ЯМР (CDCl3, 400 МГц): δ ppm 8,83 (s, 1H), 8,65 (t, J=4,8 Гц, 1H), 8,36 (s, 1H), 7,74-7,85 (m, 4Н), 7,64-7,73 (m, 3Н), 6,74 (d, J=9,0 Гц, 1Н), 4,20 (td, J=6,3, 3,5 Гц, 2Н), 3,78 (d, J=3,0 Гц, 1Н), 3,69-3,74 (m, 1Н), 3,33 (dd, J=12,5, 6,5 Гц, 2Н), 2,28-2,41 (m, 1H), 1,33 (d, J=6,5 Гц, 6Н), 1,13 (d, J=8,0 Гц, 2Н), 0,39 (q, J=5,5 Гц, 2Н).
Вариант осуществления 267

4-(4-циклопропил-3-(4-(3-(метилсульфонил)-3-пропаноил))хинолин-8-ил)бензонитрил
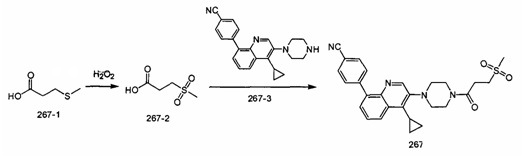
[603] Стадия 1. 267-1 (3 г, 24,96 ммоль, 1 экв.) растворяли в уксусном ангидриде (20 мл)/уксусной кислоте (20 мл), добавляли Н2О2 (40%, 2 мл). Смесь перемешивали при 25°С в течение 12 ч. После фильтрования фильтрат концентрировали с получением 267-2 (3 г, выход 78,99%) в виде белого твердого вещества. MS ESI рассч. для C25H25ClN4O2 [M+H]+ 449, найденное значение 153.
[604] Стадия-2. 267-3 (1,08 г, 3,05 ммоль, 1 экв.) и 267-2 (556,94 мг, 3,66 ммоль, 1,20 экв.) растворяли в DMF (10 мл), добавляли HATU (2,32 г, 6,10 ммоль, 2 экв.) и DIEA (1,18 г, 9,15 ммоль, 3 экв.). Смесь перемешивали при 25°С в течение 0,5 ч., затем смесь разделяли между Н2О (100 мл) и EtOAc (100 мл). Фазу EtOAc промывали солевыми растворами (30×3), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии (РЕ/EtOAc=3:1) с получением целевого соединения (800 мг, выход 53,68%) в виде желтого твердого вещества. MS ESI рассч. для C27H28N4O3S [М+Н]+ 489, найденное значение 489. 1Н ЯМР (400 МГц, CDCl3) δ ppm 8,69 (s, 1H), 8,51 (d, J=8,5 Гц, 1H), 7,76 (s, 4H), 7,56-7,63 (m, 2H), 3,88 (brs, 2H), 3,74 (brs, 2H), 3,48 (t, J=7,0 Гц, 2H), 3,18-3,36 (m, 4H), 3,01 (s, 3Н), 2,99 (d, J=7,5 Гц, 2H), 2,00-2,16 (m, 1H), 1,32 (d, J=8,0 Гц, 2H), 0,98 (d, J=4,5 Гц, 2Н).
Вариант осуществления 268
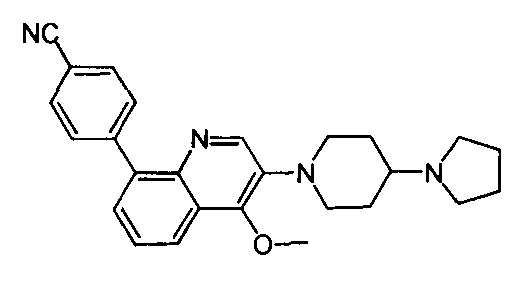
4-(4-метокси-3-(4-(пирролидин-1-ил)пиперидин-1-ил)хинолин-8-ил)бензонитрил

[605] Стадия 1. 268-1 (10 г, 50,19 ммоль, 1 экв.) и пирролидин (3,57 г, 50,19 ммоль, 1 экв.) растворяли в DCM (100 мл) и добавляли NaBH(ОАс)3 (15,96 г, 75,29 ммоль, 1,50 экв.) и уксусную кислоту (1,05 г, 17,49 ммоль). Смесь перемешивали при 25°С в течение 16 ч., выливали в смесь льда-воды (вес/вес=1/1) (100 мл) и перемешивали в течение 10 мин. Смесь экстрагировали с помощью DCM (100 мл × 3). Органические фазы объединяли и промывали солевыми растворами (100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии на силикагеле (РЕ/EtOAc = от 5:1 до 1:1) с получением 268-3 (8,50 г, выход 66,58%) в виде желтого твердого вещества. MS ESI рассч. для C14H26N2O2 [M+H]+ 255, найденное значение 255.
[606] Стадия 2. 268-3 (8,50 г, 33,42 ммоль, 1 экв.) растворяли в DCM (50 мл) и добавляли TFA (50 мл). Полученное соединение перемешивали при 25°С в течение 5 ч., концентрировали и сушили с получением неочищенного продукта 268-4 в виде желтого масла, которое непосредственно применяли для следующей стадии без дополнительной очистки. MS ESI рассч. для C9H18N2 [M+H]+ 155, найденное значение 155.
[607] Стадия 3. 268-5 (500 мг, 1,15 ммоль, 1 экв.) и 268-4 (308,31 мг, 1,15 ммоль, 1 экв.) растворяли в толуоле (10 мл), добавляли Xantphos (133 мг, 229,85 мкмоль, 0,20 экв.), Pd2(dba)3 (105,24 мг, 114,93 мкмоль, 0,10 экв.) и карбонат цезия (1,50 г, 4,60 ммоль, 4 экв.). Смесь перемешивали при 120°С в течение 2 ч., охлаждали до 60°С и концентрировали. Концентрат очищали с помощью колоночной хроматографии на силикагеле (РЕ/EtOAc = от 10:1 до 1:1) с получением 268-6 (220 мг, выход 41,46%) в виде желтого твердого вещества. MS ESI рассч. для C25H25BrN4 [М+H]+ 461, найденное значение 461.
[608] Стадия 4. KOMe (7,60 мг, 108,37 мкмоль, 1 экв.) порциями добавляли в раствор 268-6 (50 мг, 108,37 мкмоль, 1 экв.) в DMF (2 мл). Соединение перемешивали при 25°С в течение 2 ч. и концентрировали под вакуумом. Остаток очищали с помощью HPLC (TFA) с получением целевого соединения (10 мг, выход 22,37%) в виде желтого твердого вещества. MS ESI рассч. для C26H28N4O [М+Н]+ 413, найденное значение 413. 1H ЯМР (400 МГц, хлороформ-d) δ ppm 9,07 (brs, 1H), 2,13 Гц, 1H), 8,36 (d, J=7,65, 7,82 (d, J=8,03 Гц, 2Н), 7,71-7,78 (m, 2Н), 7,65 (d, J=7,78 Гц, 2Н), 4,34 (s, 3Н), 3,86-4,05 (m, 3Н), 3,57-3,72 (m, 2Н), 2,95-3,16 (m, 5Н), 2,05-2,43 (m, 9Н).
Вариант осуществления 269
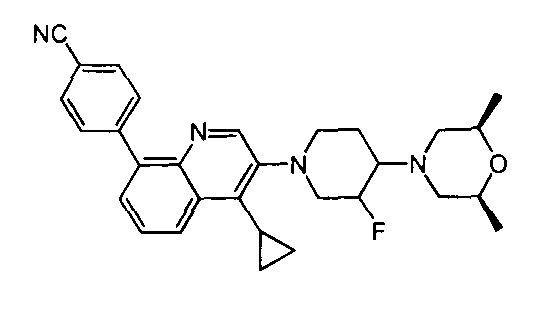
4-(4-циклопропил-3-((3S,4R)-4-((2S,6R)-2,6-диметилморфолино)-3-фторпиперидин-1-ил)хинолин-8-ил)бензонитрил
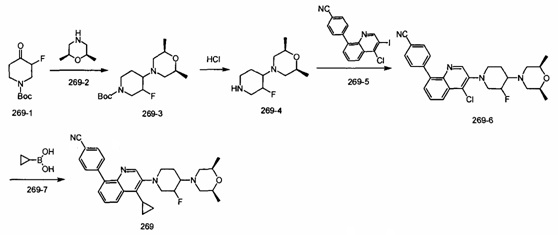
[609] Стадия 1. 269-1 (3 г, 1,00 ммоль) и 269-2 (1,91 г, 1,20 ммоль) растворяли в DCM (15 мл) и перемешивали при комнатной температуре в течение 0,5 ч., затем добавляли NaBH(OAc)3 (4,39 г, 16,57 ммоль), смесь перемешивали в течение 17 ч. Смесь выливали в Н2О (100 мл), экстрагировали с помощью DCM (150 мл*3). Органическую фазу промывали солевыми растворами (150 мл), сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии на силикагеле (РЕ/EtOAc = от 10:1 до 2:1) с получением 269-3 (2 г, выход 45,77%) в виде бесцветного масла. MS ESI рассч. для C16H29FN2O3 [М+Н]+ 317, найденное значение 317.
[610] Стадия 2. 269-3 (720 мг, 2,28 ммоль) растворяли в DCM (10 мл) и добавляли HCl/диоксан (4 М, 2 мл). Смесь перемешивали при комнатной температуре в течение 30 мин. После концентрации получали 269-4, которое непосредственно применяли для следующей стадии. MS ESI рассч. для C11H21FN2O [М+Н]+ 217, найденное значение 217.
[611] Стадия 3. 269-5 (700 мг, 1,79 ммоль), Xantphos (207,15 мг, 358,00 мкмоль), Cs2CO3 (2,33 г, 7,16 ммоль) и 269-4 (464,5 мг, 2,15 ммоль) растворяли в толуоле (15 мл) и добавляли Pd2(dba)3 (163,91 мг, 179 мкмоль). Смесь перемешивали при 110°С в течение 2 ч., затем выливали в Н2О (150 мл). Смесь экстрагировали с помощью EtOAc (200 мл*3). Органическую фазу промывали солевыми растворами (200 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии (РЕ/EtOAc = от 8:1 до 1:1) с получением 269-3 (200 мг, выход 23,33%) в виде белого твердого вещества. MS ESI рассч. для C27H28ClFN4O [М+Н]+ 479, найденное значение 479.
[612] Стадия 4. 269-6 (50 мг, 104,39 мкмоль), карбонат цезия (68,02 мг, 208,78 мкмоль), бис(адамантил)бутилфосфин (37,43 мг, 104,39 мкмоль) и 269-7 (44,84 мг, 521,955 мкмоль) растворяли в толуоле (3 мл), добавляли Pd(OAc)2 (11,72 мг, 52,20 мкмоль). Смесь перемешивали при 110°С в течение 2 ч., затем выливали в Н2О (50 мл). Смесь экстрагировали с помощью EtOAc (20 мл*3). Органическую фазу промывали солевыми растворами (50 мл), сушили над безводным сульфатом натрия и концентрировали под вакуумом. Остаток очищали с помощью HPLC с получением соединения (2 мг, выход 3,95%) в виде желтого твердого вещества. MS ESI рассч. для C30H33FN4O [М+Н]+ 485, найденное значение 485. 1H ЯМР (400 МГц, CDCl3) δ ppm 8,72 (s, 1H), 8,51 (d, J=7,8 Гц, 1H), 7,77 (s, 5H), 7,56-7,62 (m, 2H), 3,73-3,91 (m, 4H), 3,17-3,23 (m, 1H), 2,95-3,12 (m, 3Н), 2,28-2,48 (m, 2H), 1,98-2,17 (m, 4H), 1,91 (d, J=9,8 Гц, 1H), 1,22 (d, J=6,0 Гц, 7Н), 0,90 (t, J=6,7 Гц, 3Н).
Вариант осуществления 270
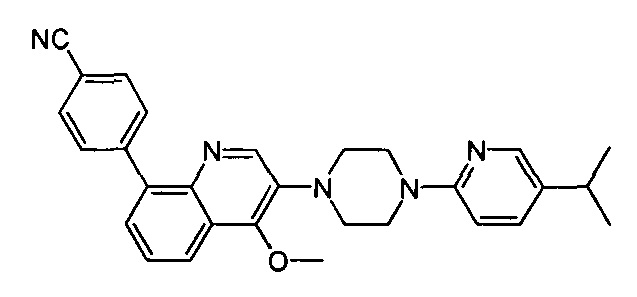
4-(3-(4-(5-изопропилпиридин-2-ил)-3-)-4-метоксихинолин-8-ил)бензонитрил
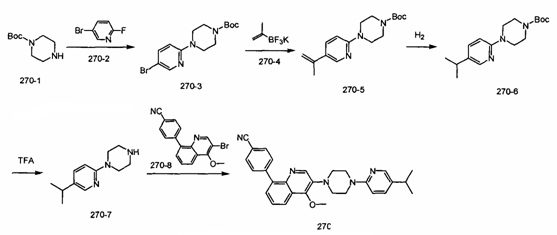
[613] Стадия 1. K2CO3 (2,76 г, 20 ммоль) добавляли в смесь 270-2 (1,76 г, 10 ммоль) и 270-1 (2,24 г, 12 ммоль) в DMF (20 мл). Смесь перемешивали при 110°С в течение 12 ч., выливали в Н2О (100 мл). Полученную смесь экстрагировали с помощью EtOAc (150 мл × 3). Органические фазы объединяли и промывали солевыми растворами (150 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии (РЕ/EtOAc=7:1) с получением 270-3 (3 г, выход 87,66%) в виде бесцветного масла. MS ESI рассч. для C14H2OBrN3O2 [М+Н]+ 342, найденное значение 342.
[614] Стадия 2. 270-3 (1,70 г, 4,97 ммоль), Na2CO3 (1,05 г, 9,94 ммоль) и изопропенилтрифторборат калия (1,49 г, 9,94 ммоль) растворяли в DMF (2 мл), в атмосфере газа азота добавляли Pd(dppf)Cl2 (363,65 мг, 497 мкмоль). Смесь перемешивали при 70°С в течение 2 ч., затем выливали в Н2О (150 мл). Полученную смесь экстрагировали с помощью EtOAc (150 мл × 3). Органические фазы объединяли и промывали солевыми растворами (150 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии (РЕ/EtOAc=10:1) с получением 270-5 (1 г, выход 66,32%) в виде бесцветного масла. MS ESI рассч. для C17H25N3O2 [M+H]+ 304, найденное значение 304.
[615] Стадия 3. 270-5 (1 г, 3,30 ммоль) растворяли в метаноле (30 мл)/EtOAc (30 мл) и добавляли Pd(OH)2/C (10%, 0,1 г). Суспензию дегазировали и продували с помощью газа водорода 3 раза. Смесь перемешивали при 25°С в течение 2 ч. в атмосфере Н2 (40 фунтов/кв. дюйм). TLC показала, что исходный материал полностью потребляли. Смесь фильтровали, фильтрат концентрировали. Неочищенный продукт применяли непосредственно на следующей стадии. MS ESI рассч. для C17H27N3O2 [М+Н]+ 306, найденное значение 306.
[616] Стадия 4. 270-6 (500 мг, 1,64 ммоль) растворяли в DCM (5 мл) и порциями добавляли TFA (2,43 г, 21,32 ммоль). Смесь перемешивали в течение 30 мин. и концентрировали. Остаток выливали в насыщенный раствор Na2CO3 (100 мл). Полученную смесь экстрагировали с помощью EtOAc (100 мл × 3). Органические фазы объединяли и промывали солевыми растворами (100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Неочищенный продукт применяли непосредственно на следующей стадии. MS ESI рассч. для C12H19N3 [М+Н]+ 206, найденное значение 206.
[617] Стадия 5. В атмосфере газа азота Pd2(dba)3 (40,50 мг, 44,22 мкмоль, 0,10 экв.) добавляли в раствор 270-8 (150 мг, 442,23 мкмоль, 1 экв.), карбоната цезия (288,17 мг, 884,46 мкмоль, 2 экв.), Xantphos (51,18 мг, 88,45 мкмоль, 0,20 экв.) и 270-7 (108,95 мг, 530,68 мкмоль, 1,20 экв.) в толуоле (10 мл). Смесь перемешивали при 110°С в течение 2 ч., выливали в Н2О (100 мл). Полученную смесь экстрагировали с помощью EtOAc (150 мл × 3). Органические фазы объединяли и промывали солевыми растворами (100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали с помощью HPLC (кислоты) с получением целевого продукта (50 мг, выход 24,39%) в виде желтого твердого вещества. MS ESI рассч. для C29H29N5O [М+H]+ 464, найденное значение 464. 1H ЯМР (400 МГц, CDCl36) δ ppm 8,79 (s, 1H), 8,16-8,27 (m, 1H), 8,05-8,14 (m, 1H), 7,74-7,82 (m, 4H), 7,53-7,64 (m, 2H), 7,35-7,45 (m, 1H), 6,72 (dd, J=6,3, 8,3 Гц, 1H), 4,15 (s, 3Н), 3,72 (d, J=4,0 Гц, 4H), 3,34-3,44 (m, 4H), 2,49 (t, J=7,5 Гц, 1H), 1,21-1,31 (m, 5H).
Вариант осуществления 271
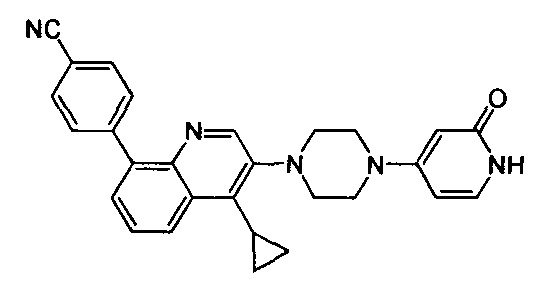
4-(4-циклопропил-3-(4-(2-оксо-1,2-дигидропиридин-4-ил)пиперазин-1-ил)хинолин-8-ил)бензонитрил
[618] Стадия 1. В атмосфере газа азота Pd2(dba)3 (38,75 мг, 42,32 мкмоль) добавляли в смесь 271-1 (150 мг, 423,19 мкмоль), трет-бутоксида натрия (81,34 мг, 846,38 мкмоль), Xantphos (48,97 мг, 84,64 мкмоль) и 271-2 (134,13 мг, 507,83 мкмоль) в толуоле (10 мл). Смесь перемешивали при 110°С в течение 2 ч., выливали в H2O (150 мл). Полученную смесь экстрагировали с помощью EtOAc (100×3). Органические фазы объединяли и промывали солевыми растворами (100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии (РЕ/EtOAc=3:1) с получением 271-3 (150 мг, выход 65,93%) в виде желтого твердого вещества. MS ESI рассч. для C35H31N5O [М+Н]+ 538, найденное значение 538.
[619] Стадия 2. FeCl3 (452,52 мг, 2,79 ммоль) порциями добавляли в смесь 271-3 (150 мг, 278,99 мкмоль) в DCM (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 60 мин., затем фильтровали, экстрагировали с помощью EtOAc (100 мл × 3). Органические фазы объединяли и промывали солевыми растворами (100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали с помощью препаративной HPLC (кислоты) с получением целевого соединения (50 мг, выход 40,05%) в виде желтого твердого вещества. MS ESI рассч. для C28H25N5O [M+H]+ 448, найденное значение 448. 1Н ЯМР (400 МГц, метанол-d4) δ ppm 8,77 (d, J=7,8 Гц, 1H), 8,67 (s, 1H), 7,93 (d, J=8,3 Гц, 2Н), 7,82-7,89 (m, 1H), 7,76-7,83 (m, 3Н), 7,72 (d, J=7,5 Гц, 1H), 6,86 (d, J=5,5 Гц, 1H), 6,25 (d, J=2,3 Гц, 1H), 3,91 (brs., 4H), 3,55 (d, J=4,8 Гц, 4Н), 1,52 (brs, 1H), 0,99-1,09 (m, 2H), 0,13 (brs, 2H).
[620] Эксперимент 1. Анализ in vitro
[621] Цель эксперимента
[622] Репортерный ген люциферазы (Gli-Luc) с чувствительным элементом Gli трансфицировали в клетки С3Н10Т1/2 в условиях стимуляции Shh-N, определяли ингибиторы сигнального пути hedgehog посредством выявления экспрессии генов в отчете клеточной активности. В данном исследовании оценивали ингибирующие эффекты соединений на сигнальном пути hedgehog в величине IC50 соединения в качестве индекса.
[623] Экспериментальные материалы
Клеточные линии: стабильная клеточная линия C3H10T1/2/Gli-Luc
среда для клеточных культур C3H10T1/2/Gli-Luc (DMEM/HyClone с высоким уровнем глюкозы, № SH30022.01B; 10% сыворотка Hyclone, № SV30087; 0,4% гигромицин В Roche, №13398200)
0,25% трипсин-EDTA (Gibco, №25200)
PBS (KH2PO4 0,24 г, Na2HPO4 1,44 г, NaCl 8,0 г, KCl 0,2 г, H2O, добавленный до 1 л и отрегулированный до рН 7,4)
Shh-N, стабильная надосадочная жидкость культуры клеток HEK293/SHH-N
лизаты (Promega, № Е1531)
реакционный раствор (Promega, № Е1501)
384-луночный планшет, Greiner №781074
96-луночный культуральный планшет, Greiner №655180
96-луночный планшет с отверстиями малого диаметра, Bi Yuntian № FPT019
инкубатор CO2, Thermo №3423
бокс, AIRTECH № А10051560
инвертированный микроскоп, Nikon № TS100
центрифуга, Xiangyi № L530
сверхбыстрый мультифункциональный считывающий прибор Therom VarioSkan
[624] Процедура и способ эксперимента
Инокулировали логарифмическую фазу роста клеток C3H10T1/2/Gli-Luc в 96-луночный культуральный планшет с 20000 клеток на лунку, клетки культивировали при 37°С, 5% инкубатора СО2 в течение ночи. На следующий день соединения разбавляли следующими способами: положительное соединение GDC0449 (1 мМ) и соединения (1 мМ), подлежащие измерению, серийно разбавляли до 7 концентраций с помощью DMSO согласно 1:3 и 1:10, соответственно, восьмая - для контроля DMSO, а затем разбавляли с помощью свежей среды в 100 раз. Среду культивированных в течение ночи клеток удаляли, на лунку добавляли 80 мкл свежей культуральной среды, затем добавляли 20 мкл градиентно разбавленного положительного соединения и соединений, подлежащих измерению, и 100 мкл 30 нМ содержащей Shh-N кондиционированной среды, каждую концентрацию повторяли в 2 лунках в одно и тоже время, были установлены положительные и отрицательные контрольные лунки (те. контр. Shh: 80 мкл свежей среды + 20 мкл 1% содержащей DMSO среды + 100 мкл 30 нМ содержащей Shh-N кондиционированной среды; нестимулированный контр.: 180 мкл свежей среды + 20 мкл 1% содержащей DMSO среды), клетки культивировали в инкубаторе в течение дополнительных 24 ч.
Выявление внутриклеточной активности люциферазы: вытаскивали клетки из 96-луночного планшета, сливали культуральную среду, клетки дважды промывали с помощью PBS, затем в каждую лунку добавляли 20 мкл лизата (Promega E1531), осуществляли пиролиз при комнатной температуре в течение 30 мин. 5 мкл клеточных лизатов трансфицировали в 384-луночный планшет (Greiner 781074), затем в каждую лунку добавляли 25 мкл реакционной жидкости люциферазы (Promega E1501), после быстрого перемешивания их немедленно помещали в сверхбыстрый мультифункциональный считывающий прибор VarioSkan для считывания величины относительных световых единиц (RLU).
Данные анализа. Применение Prism's GraphPad 5 для анализа данных. Считывание RLU к логарифмическому составлению концентрации соединения в зависимости от активности люциферазы, а затем подбор кривой с использованием следующего уравнения с получением величины IC50, Y (RLU) = минимум + (максимум - минимум)/(1+10^(log концентрации соединения -LogIC50)).







Примечание: А≤50 нМ; 50 нМ<В≤100 нМ; 100 нМ<С≤500 нМ; 500 нМ<D≤1000 нМ; 1000 нМ<Е≤5000 нМ.
Заключение. Соединения согласно настоящему изобретению имеют значительный эффект ингибирования на пути hedgehog.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГИДРОКСИПУРИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2685417C1 |
| ПРОИЗВОДНОЕ НА ОСНОВЕ ДИГИДРОПИРИМИДО-КОЛЬЦА В КАЧЕСТВЕ ИНГИБИТОРА HBV | 2015 |
|
RU2693897C2 |
| МОДУЛЯТОРЫ АКТИВНОСТИ НЕС1 И СПОСОБЫ ДЛЯ НИХ | 2011 |
|
RU2576036C2 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ ПИРИМИДИН-4(3H)-ОНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2021 |
|
RU2839367C1 |
| ЗАМЕЩЁННЫЕ ИНДОЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ПОНИЖАЮЩИХ РЕГУЛЯТОРОВ РЕЦЕПТОРОВ ЭСТРОГЕНА | 2017 |
|
RU2722441C2 |
| Имидазопиридиновые соединения в качестве ингибиторов PAD | 2018 |
|
RU2782743C2 |
| МАКРОЦИКЛИЧЕСКИЕ АНТИБИОТИКИ ШИРОКОГО СПЕКТРА ДЕЙСТВИЯ | 2016 |
|
RU2766543C2 |
| ГИДРОКСИПУРИНОВЫЕ СОЕДИНЕНИЯ И ПУТИ ИХ ПРИМЕНЕНИЯ | 2015 |
|
RU2673458C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ ДЛЯ ОПОСРЕДОВАНИЯ АКТИВНОСТИ ТИРОЗИНКИНАЗЫ 2 | 2020 |
|
RU2826012C2 |
| АГОНИСТ S1P1 И ЕГО ПРИМЕНЕНИЕ | 2017 |
|
RU2754845C2 |
Изобретение относится к области органической химии, а именно к гетероциклическому соединению формулы (I) или к его фармацевтически приемлемой соли, где A выбран из
 ,
, 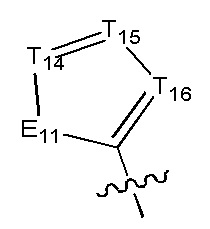 ,
, 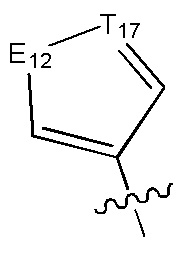 ,
, 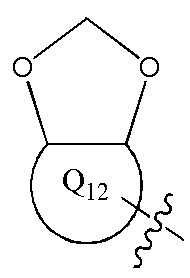 ,
,  ;
;
каждый из T11-13 независимо выбран из N, C(R13); где T11-13 представляют собой C(R13) или один или два из T11-13 представляют собой N, а остальные из T11-13 представляют собой C(R13); T14 и T17 представляют собой N; каждый из T15 и T16 представляют собой СН; каждый из E11-12 независимо выбран из N(СН3); L1 представляет собой одинарную связь; L2 выбран из одинарной связи или C(=O)NН; R11 выбран из H или метила; R12 выбран из Н, метила, F, Cl; R13 выбран из Н, метила, трифторметила, трифторметокси, F, Cl, CN, метиламинокарбонила, метилсульфонила, морфолинилсульфонила, 2-имидазолила или диметиламино; Q12 выбран из фенила; где структурная единица
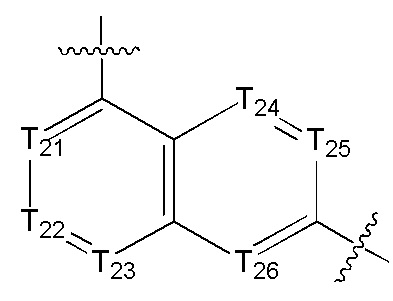
и B выбраны из структур, указанных в п.1 формулы. Также изобретение относится к конкретным соединениям формулы (I). Технический результат: получены новые гетероциклические соединения, полезные в качестве ингибиторов пути hedgehog, в особенности в качестве ингибиторов SMO и, соответственно, полезны при лечении рака. 2 н. и 12 з.п. ф-лы, 22 табл.
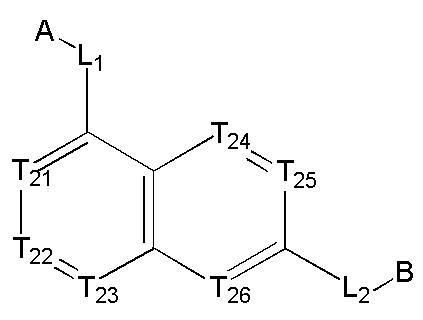
(I)
1. Соединение, представленное формулой (I), или его фармацевтически приемлемая соль,
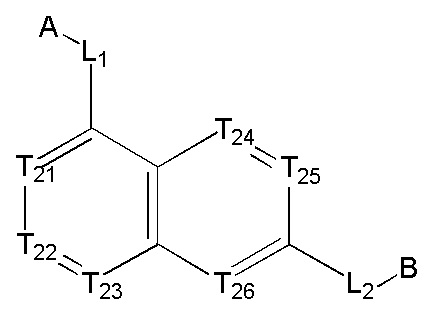 , (I)
, (I)
где A выбран из
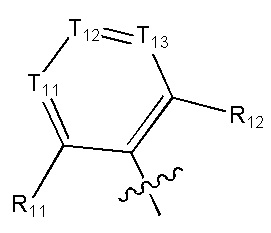 ,
,  ,
, 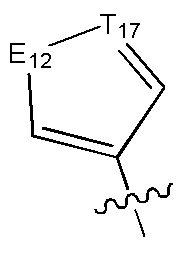 ,
, 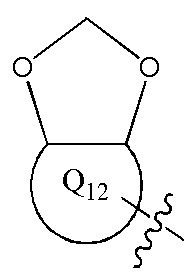 ,
,  ;
;
каждый из T11-13 независимо выбран из N, C(R13); где T11-13 представляют собой C(R13) или один или два из T11-13 представляют собой N, а остальные из T11-13 представляют собой C(R13);
T14 и T17 представляют собой N;
каждый из T15 и T16 представляют собой СН;
каждый из E11-12 независимо выбран из N(СН3);
L1 представляет собой одинарную связь;
L2 выбран из одинарной связи или C(=O)NН;
R11 выбран из H или метила;
R12 выбран из Н, метила, F, Cl;
R13 выбран из Н, метила, трифторметила, трифторметокси, F, Cl, CN, метиламинокарбонила, метилсульфонила, морфолинилсульфонила, 2-имидазолила или диметиламино;
Q12 выбран из фенила;
где структурная единица
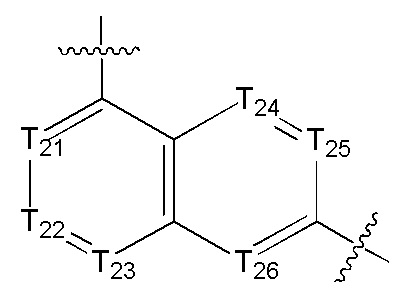
выбрана из
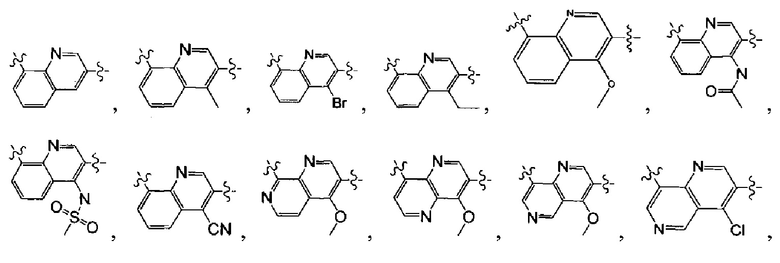
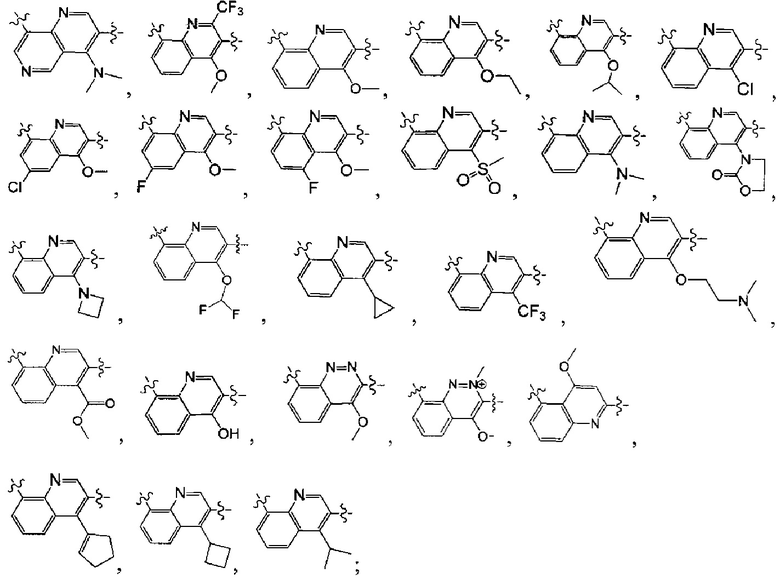
B выбран из
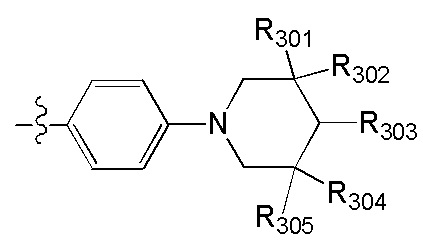 ,
,
где каждый из R301 и R305 независимо выбран из H или С1-3алкила;
каждый из R302 и R304 независимо выбран из С1-3алкила;
R303 представляет собой Н или OH;
или B выбран из
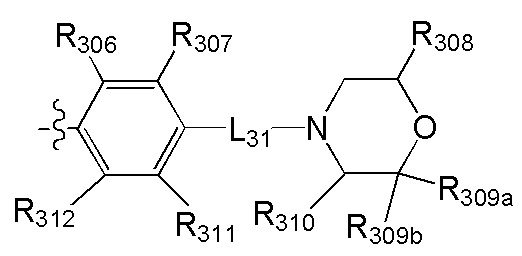 ,
,
где каждый из R306 и R307 независимо выбран из H, F, Cl или C1-3алкила;
каждый из R308 и R309a независимо выбран из необязательно R3001-замещенного C1-3алкила;
каждый из R309b, R310-312 независимо представляет собой H или C1-3алкил;
R3001 выбран из OH, при этом число R3001 представляет собой 1;
L31 представляет собой одинарную связь;
или B выбран из
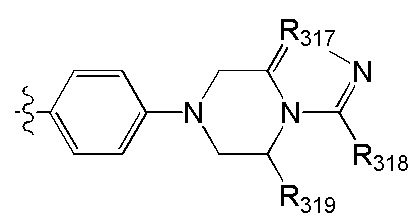 ,
,
где R317 представляет собой N; каждый из R318, R319 независимо выбран из Н или необязательно R3001-замещенного C1-3алкила; R3001 выбран из F, Cl, Br или I, число R3001 выбрано из 1, 2 или 3;
или B выбран из
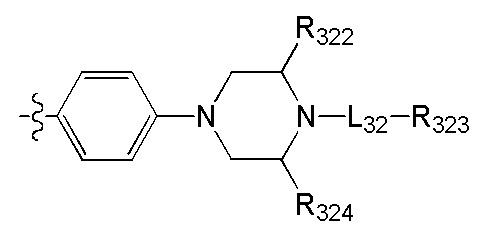 ,
,
где L32 выбран из C(=O)O или C(=O);
R323 выбран из C1-6алкила, фенила, пиридила, имидазолила, при этом указанный алкил и пиридил необязательно замещен R3012;
R3012 выбран из OH или C1-3алкила; каждый из R322 и R324 независимо выбран из необязательно R3001-замещенного C1-3алкила;
R3001 выбран из OH;
при этом число каждого из R3001, R3012 выбрано из 1, 2 или 3;
или B выбран из
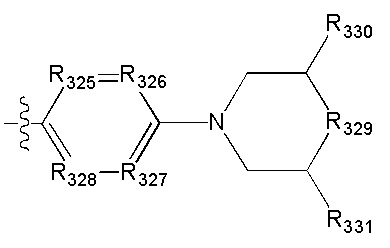 ,
,
где один или два из R325-328 выбраны из N, остальные выбраны из C(R3013);
R329 выбран из N(R3014), O;
R3014 выбран из C(=O)R3020, S(=O)2R3020, тиазолила;
R3020 выбран из необязательно R3001-замещенного C1-3алкила или алкоксила;
R3013 выбран из H, C(=O)OH или C1-3алкила;
каждый из R330 и R331 выбран из C1-3алкила; или R330 и R331 вместе образуют соединяющую связь (CH2)1-3;
R3001 выбран из OH, N(CH3)2, NH(CH3), NH2, F, Cl, Br, I, метокси, при этом число R3001 выбрано из 1, 2 или 3;
или B выбран из
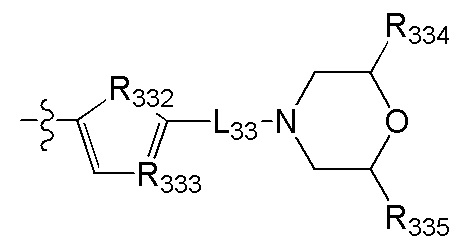 ,
,
где R332 представляет собой S;
L33 представляет собой одинарную связь или C(=O);
R333 представляет собой N, CН;
каждый из R334, R335 независимо выбран из C1-3алкила;
или B выбран из
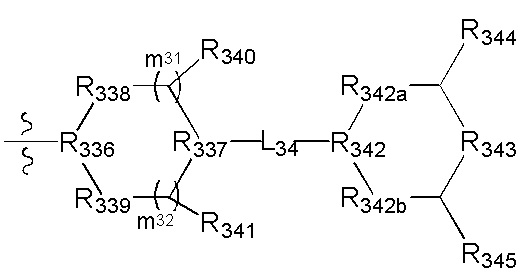 ,
,
где каждый из R336, R337, R342 независимо выбран из N или CН;
каждый из R338, R339 и R342b выбран из CН2 или R338 и R339 вместе соединены с одним и тем же (CH2)1-3 с образованием кольца;
каждый из m31, m32 независимо выбран из 0 или 1;
R342a выбран из одинарной связи, CH2 или С(=О); R343 выбран из C(R3034)(R3035) или О; при этом каждый из R3034 и R3035 независимо выбран из H, F, Cl, Br или I;
L34 представляет собой одинарную связь, C(=O)NH, C(=O)CH2 или C(=O)NCH3;
каждый из R340, R344, R345 независимо выбран из H, F, Cl, Br, I или C1-3алкила;
R341 представляет собой Н;
или B выбран из
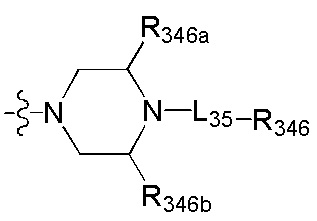 ,
,
где L35 выбран из одинарной связи, C(=O) или S(=O)2;
каждый из R346a, R346b представляет собой H;
R346 выбран из Н, необязательно R3001-замещенного C1-3алкила или циклопропила;
R3001 выбран из OH или -S(=O)2CH3, при этом число R3001 представляет собой 1;
или B выбран из
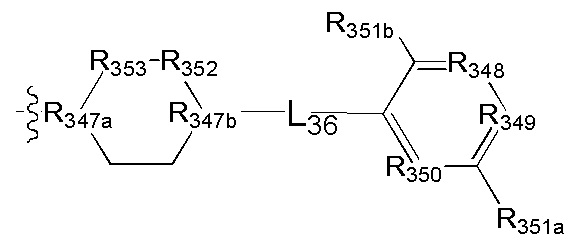 ,
,
где каждый из R347a, R347b независимо выбран из N или C(R3028);
ни один, один или два из R348-350 выбраны из N, остальные выбраны из C(R3039);
каждый из R351а и R351b независимо выбран из Н, необязательно R3001-замещенного C1-3алкила;
L36 выбран из одинарной связи, -C(=O)NН, -C(=O)СН2;
R352 выбран из СН2, C(=O), СН(СН3) или C(OН)(СН3),
R353 выбран из СН2 или C(=O);
R3028 выбран из Н или ОН;
R3039 выбран из H или необязательно R3001-замещенного C1-3алкила;
R3001 выбран из OH, F, Cl, Br или I, при этом число R3001 выбрано из 1, 2 или 3;
или B выбран из
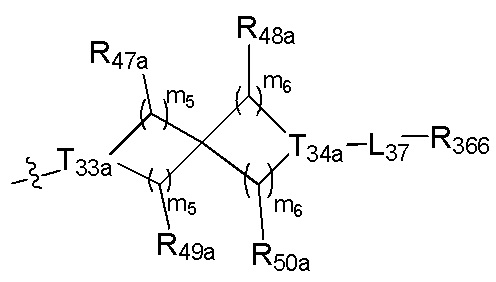 ,
,
где каждый из T33a, T34a представляет собой N;
каждый из m5 и m6 независимо выбран из 1 или 2;
L37 представляет собой одинарную связь;
каждый из R47a, R48a, R49a, R50a представляет собой H,
R366 представляет собой пиридил, замещенный R4001;
R4001 представляет собой C(CH3)2(OH).
2. Соединение, представленное формулой (I), или его фармацевтически приемлемая соль по п.1, причем A выбран из
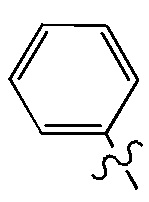 ,
,  ,
, 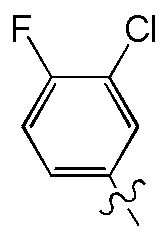 ,
, 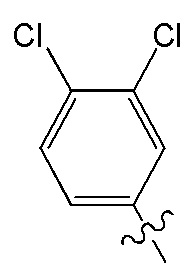 ,
,  ,
, 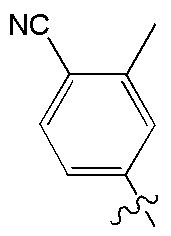 ,
, 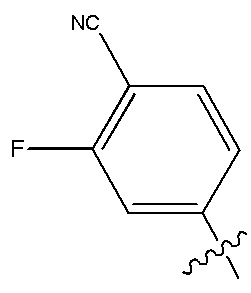 ,
, 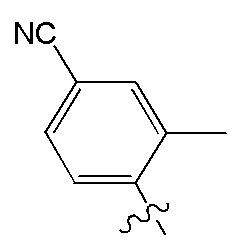 ,
, 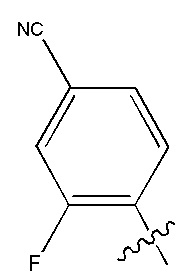 ,
, 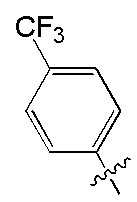 ,
, 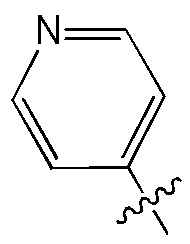 ,
, 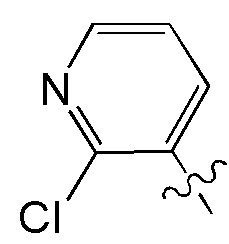 ,
, 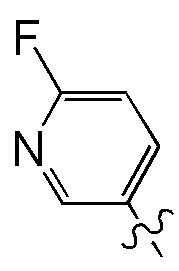 ,
, 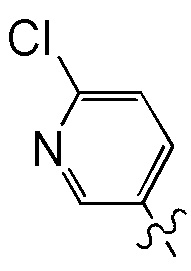 ,
, 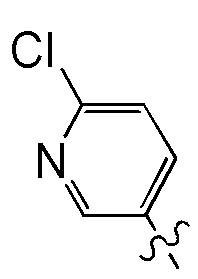 ,
,  ,
, 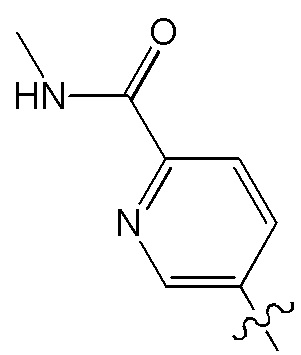 ,
, 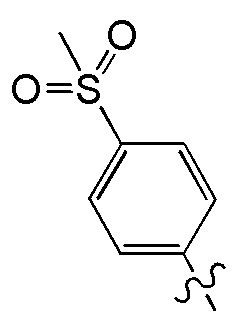 ,
, 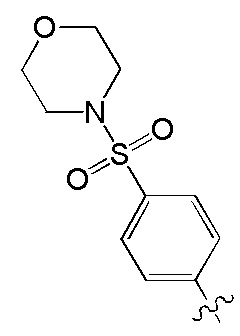 ,
, 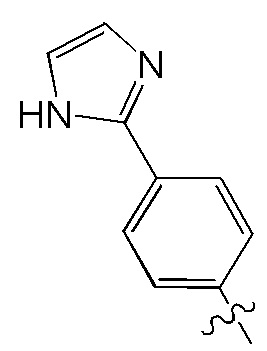 ,
, 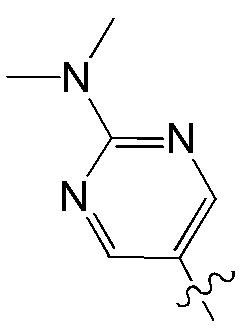 ,
, 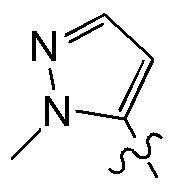 ,
,  ,
, 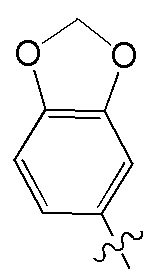 .
.
3. Соединение, представленное формулой (I), или его фармацевтически приемлемая соль по п.1, где B выбран из
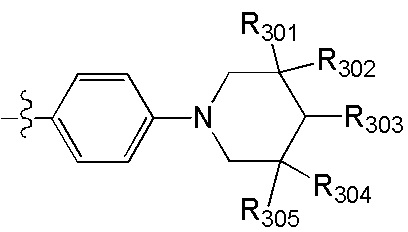 ,
,
где каждый из R301 и R305 независимо выбран из H или метила;
каждый из R302 и R304 независимо представляет собой метил;
или B выбран из
 ,
,  .
.
4. Соединение, представленное формулой (I), или его фармацевтически приемлемая соль по п.1, где B выбран из
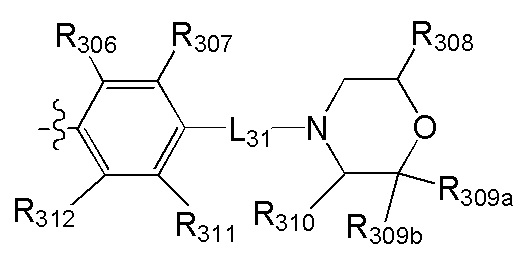 ,
,
где каждый из R306 и R307 независимо выбран из Н, F, Cl или метила;
каждый из R308 и R309а независимо выбран из Н, метила, C(CH3)2(OH);
каждый из R309b, R310-312 независимо выбран из Н;
L31 представляет собой одинарную связь;
или B выбран из
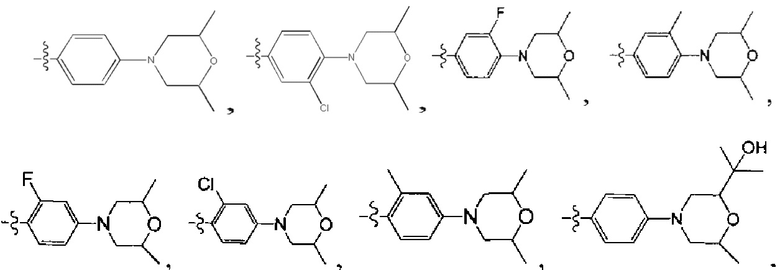
5. Соединение, представленное формулой (I), или его фармацевтически приемлемая соль по п.1, где B выбран из
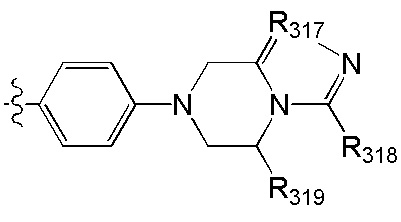 ,
,
где R317 представляет собой N; каждый из R318, R319 выбран из метила, трифторметила;
или B выбран из
 .
.
6. Соединение, представленное формулой (I), или его фармацевтически приемлемая соль по п.1, где B выбран из
 ,
,
где каждый из R322, R324 независимо выбран из метила, C(CH3)2OH;
L32 выбран из C(=O)О, C(=O);
R323 выбран из метила, этила, н-пропила, фенила, пиридила, 3-метилпиридила, имидазолила, C(CH3)2OH;
или B выбран из
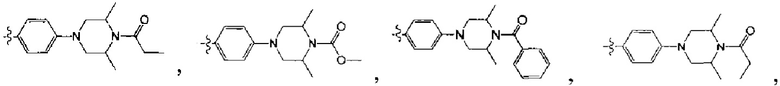
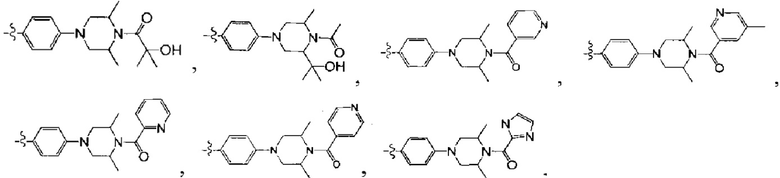
7. Соединение, представленное формулой (I), или его фармацевтически приемлемая соль по п.1, где B выбран из
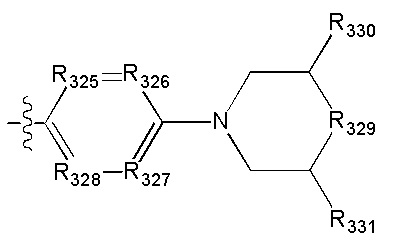 ,
,
где если R329 выбран из N(R3014) или O, то R330 и R331 находятся в цис-положении; или
один или два из R325-328 выбраны из N, остальные выбраны из CH, CC(=O)OH или CCH3;
R329 выбран из N(R3014), O;
R3014 выбран из C(=O)R3020, S(=O)2R3020,  ;
;
R3020 выбран из C(CH3)(F)2, CH3, CF3, CH2CH3, CH2CF3, CH(F)(CH3), CH(OH)(CH3), CH2(OH), CH(NH2)(CH3), CH2NH2, метокси;
каждый из R330-331 независимо выбран из метила; или R330 и R331 вместе образуют соединяющую связь (CH2)2;
или B выбран из
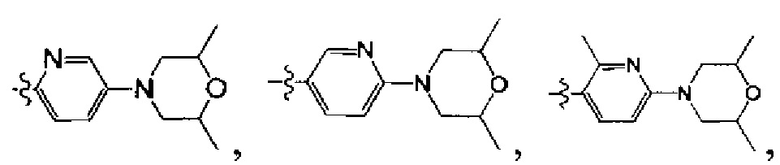


8. Соединение, представленное формулой (I), или его фармацевтически приемлемая соль по п.1, где B выбран из
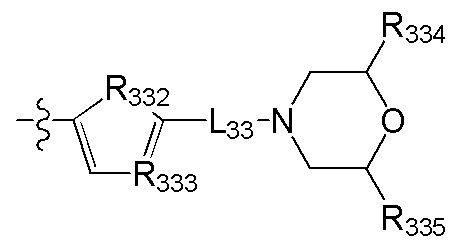 ,
,
где R332 представляет собой S,
R333 выбран из N или CH,
L33 выбран из одинарной связи, C(=O),
каждый из R334, R335 независимо выбран из метила;
или B выбран из
 ,
,  .
.
9. Соединение, представленное формулой (I), или его фармацевтически приемлемая соль по п.1, где B выбран из
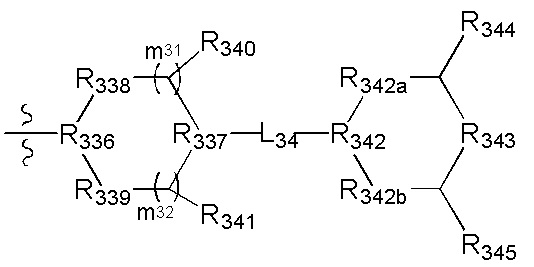 ,
,
где каждый из R336, R337, R342 независимо выбран из N или CH;
каждый из R338, R339 или R342b независимо выбран из CH2 или R338 и R339 вместе соединены с одним и тем же CH2CH2 с образованием кольца;
каждый из m31, m32 независимо выбран из 0 или 1;
L34 выбран из одинарной связи, C(=O)NH, C(=O)N(CH3), C(=O)CH2;
каждый из R340, R344, R345 независимо выбран из H, F или метила;
R342a выбран из одинарной связи, C(=O) или CH2;
R341 представляет собой Н;
или B выбран из
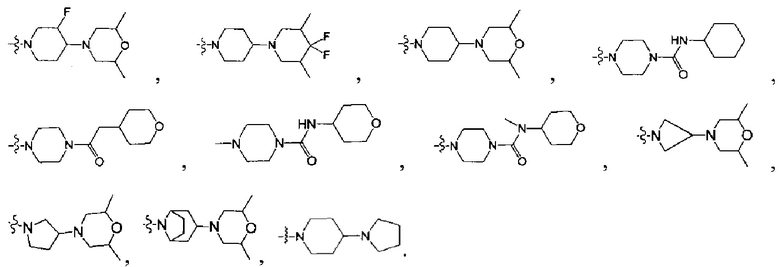
10. Соединение, представленное формулой (I), или его фармацевтически приемлемая соль по п.1, где B выбран из
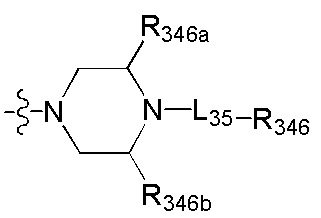 ,
,
где L35 выбран из одинарной связи или C(=O);
или B выбран из

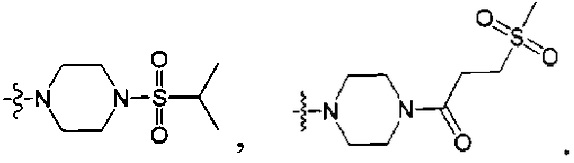
11. Соединение, представленное формулой (I), или его фармацевтически приемлемая соль по п.1, где B выбран из
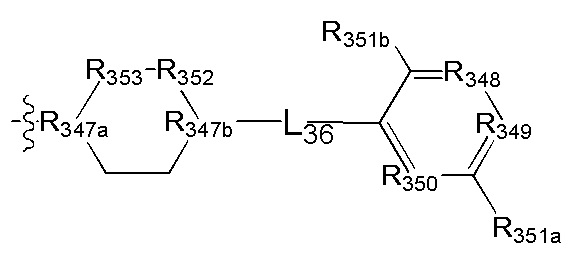 ,
,
где L36 выбран из одинарной связи, C(=O)NH, C(=O)CH2;
каждый из R347a, R347b независимо выбран из N, CH или C(OH);
ни один, один или два из R348-350 выбраны из N, остальные выбраны из C(R3039);
R3039 выбран из H, метила, трифторметила, C(CH3)2(OH);
каждый из R351a, R351b независимо выбран из H, метила, трифторметила;
R352 выбран из CH2, C(=O), CН(CH3) или С(OH)(CH3),
R353 выбран из CH2 или C(=O);
или B выбран из
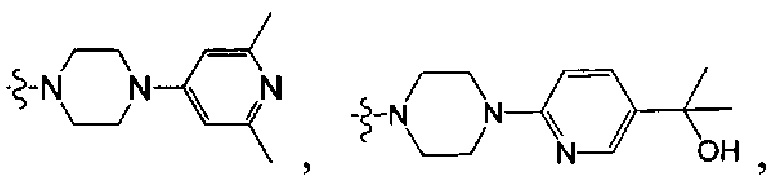
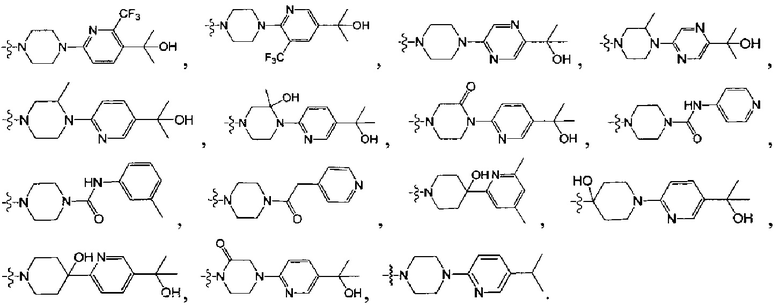
12. Соединение, представленное формулой (I), или его фармацевтически приемлемая соль по п.1, где B выбран из
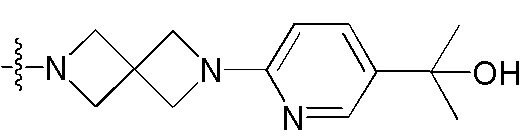 ,
, 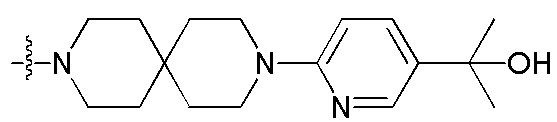 .
.
13. Соединение, представленное формулой (I), или его фармацевтически приемлемая соль по п. 1, где B выбран из группы, состоящей из
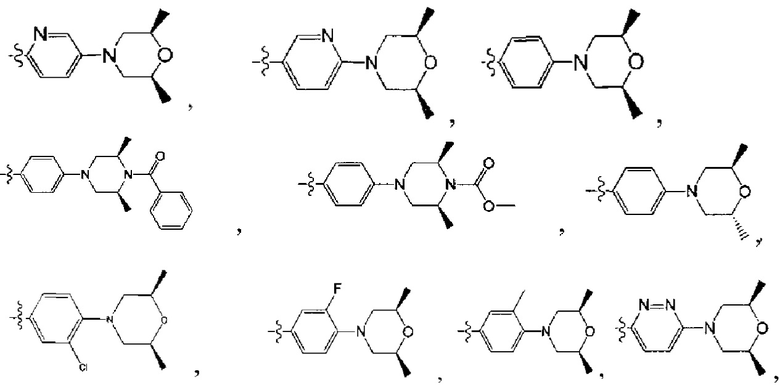
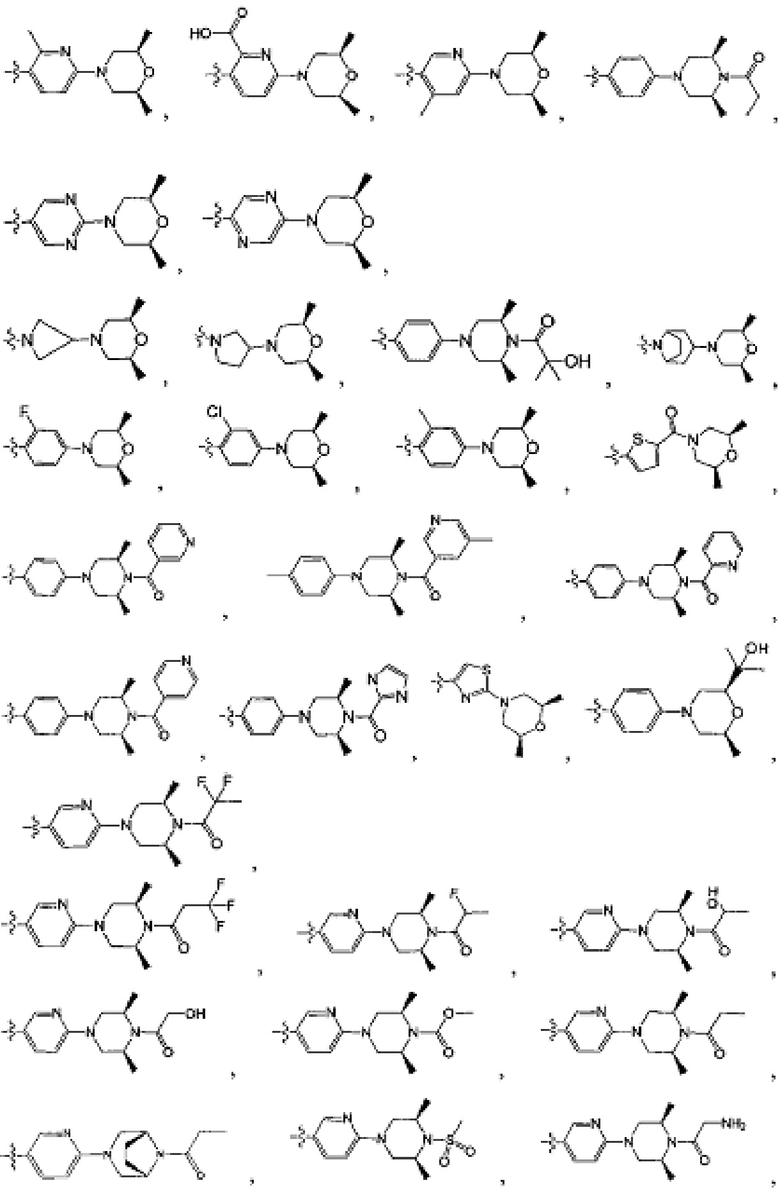
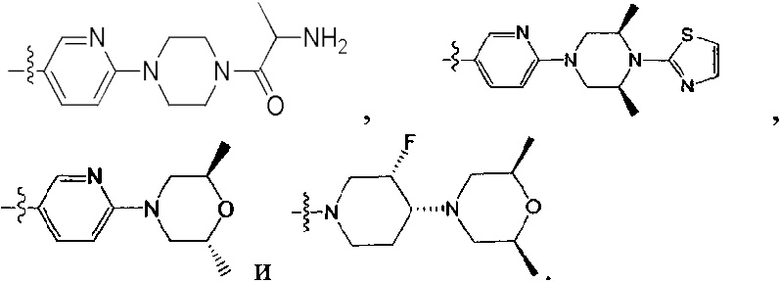
14. Соединение или его фармацевтически приемлемая соль, которое выбрано из группы, состоящей из
 ,
, 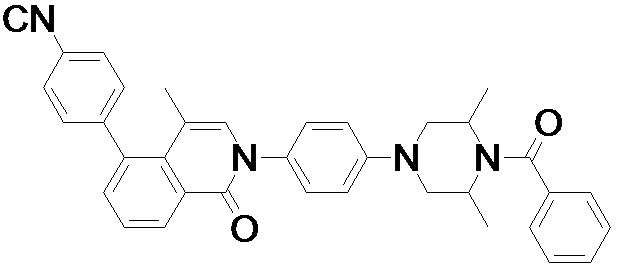 ,
,
 ,
, 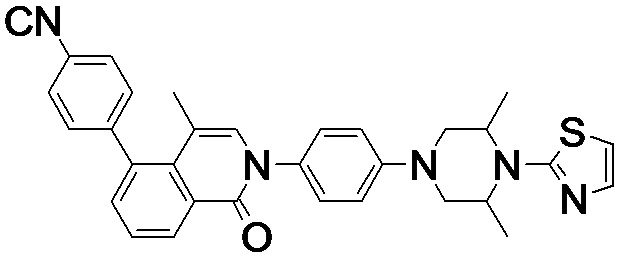 ,
,
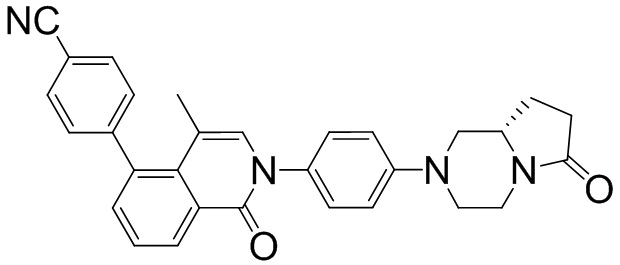 ,
, 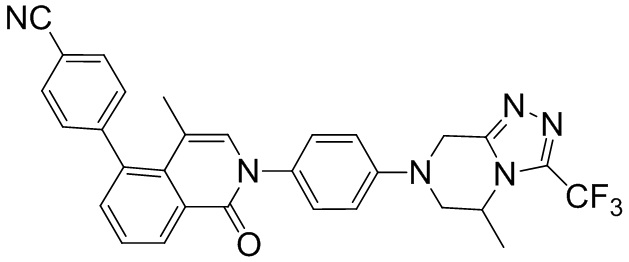 ,
,
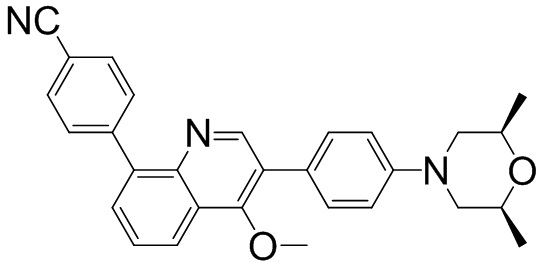 ,
, 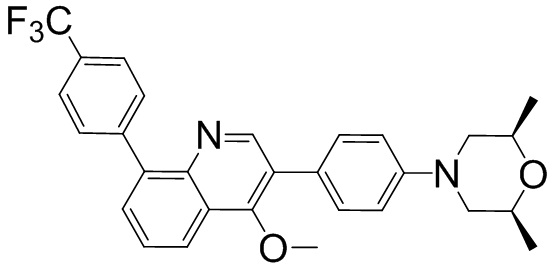 ,
, 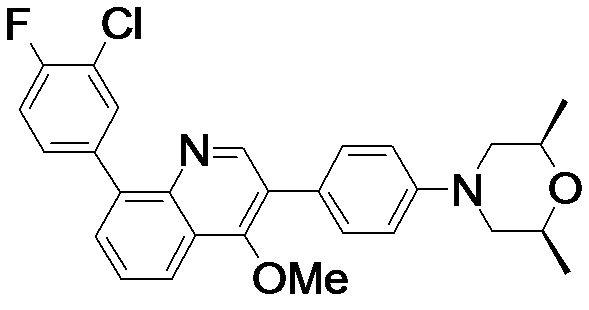 ,
,
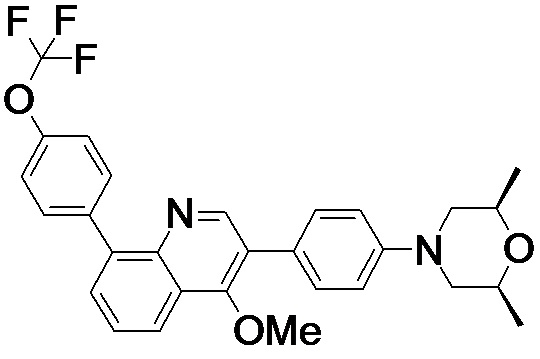 ,
, 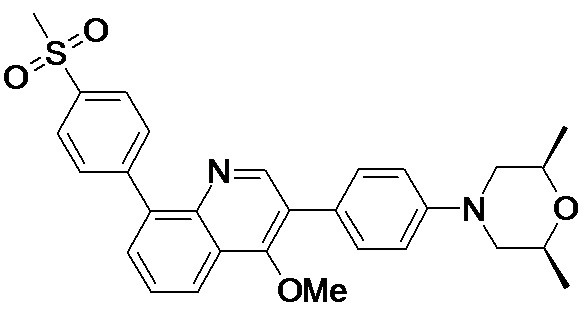 ,
, 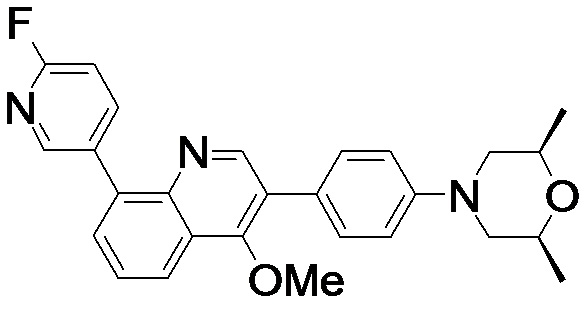 ,
,
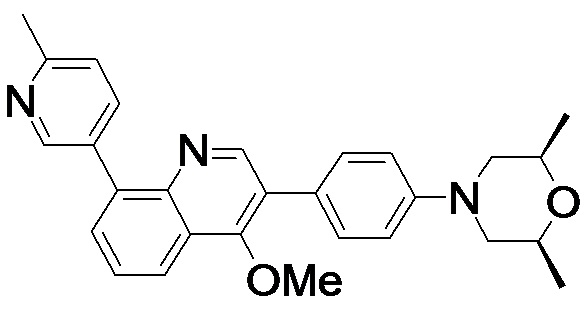 ,
,  ,
, 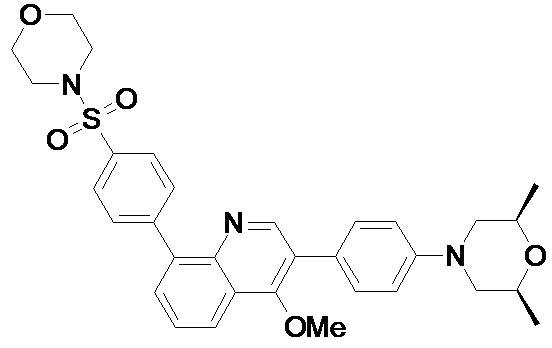 ,
,
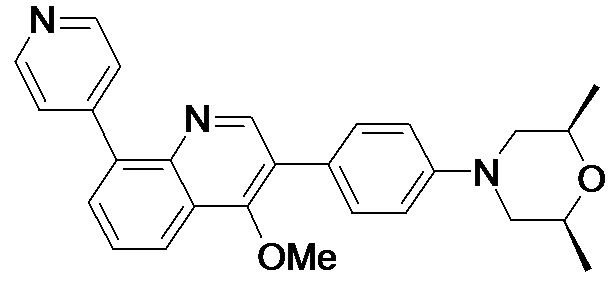 ,
, 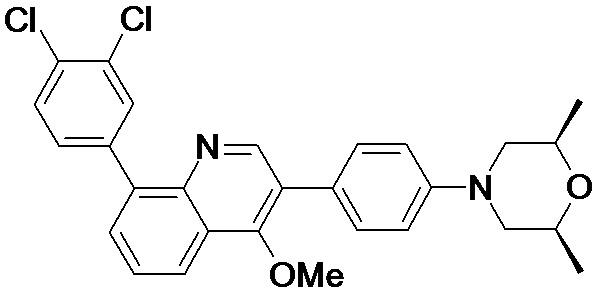 ,
, 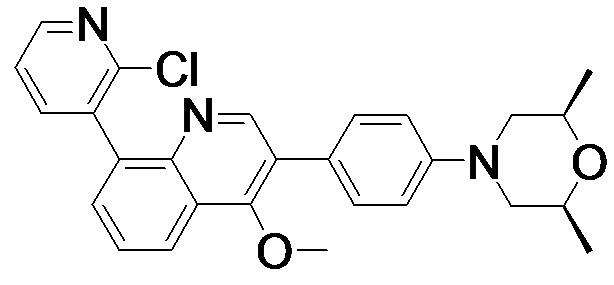 ,
,
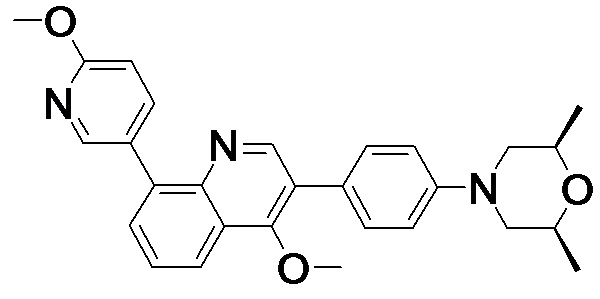 ,
, 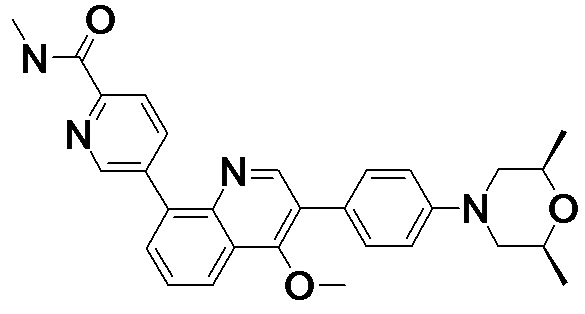 ,
, 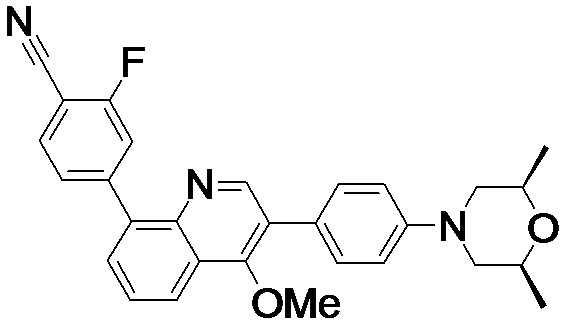 ,
,
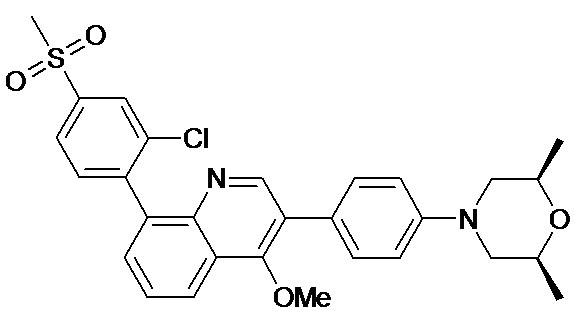 ,
, 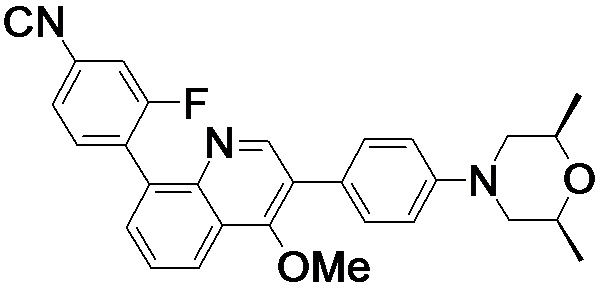 ,
, 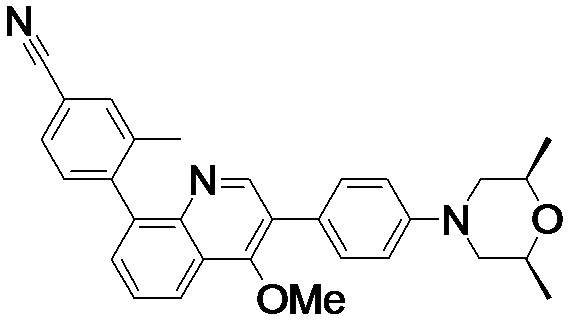 ,
,
 ,
,  ,
, 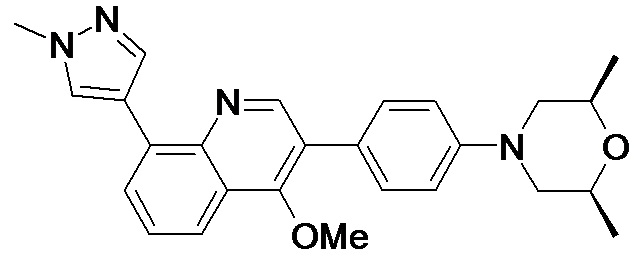 ,
,
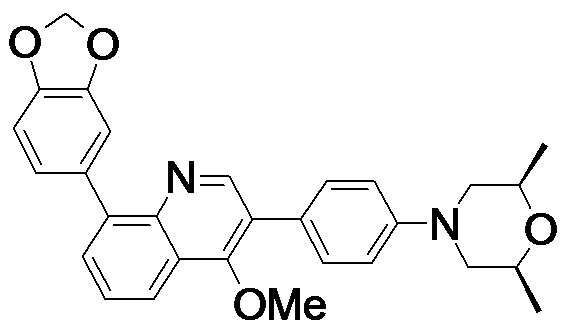 ,
, 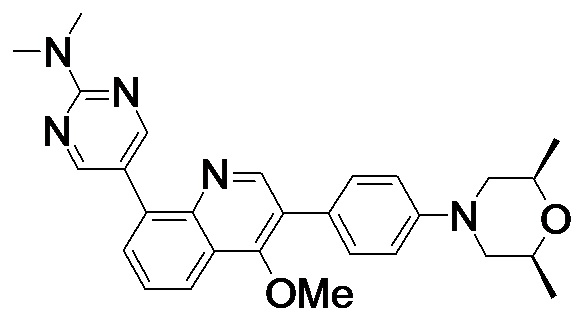 ,
, 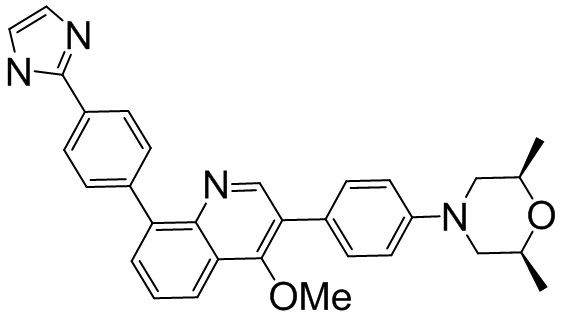 ,
,
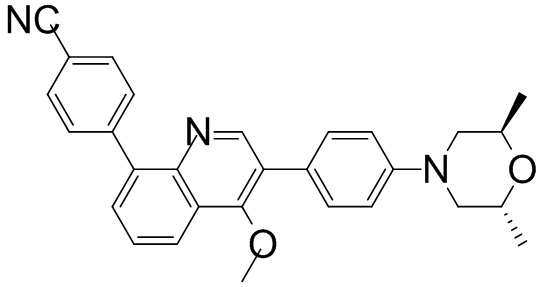 ,
, 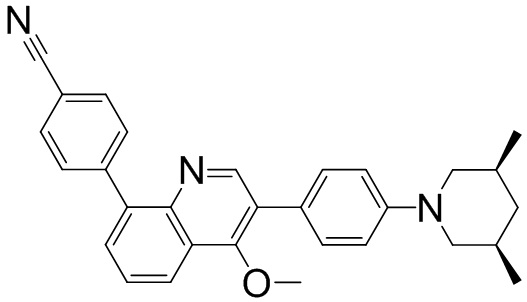 ,
,
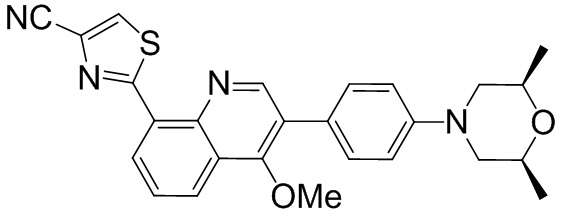 ,
, 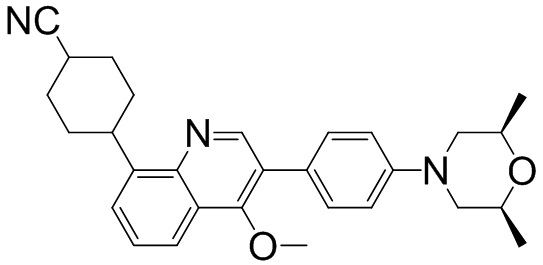 ,
,
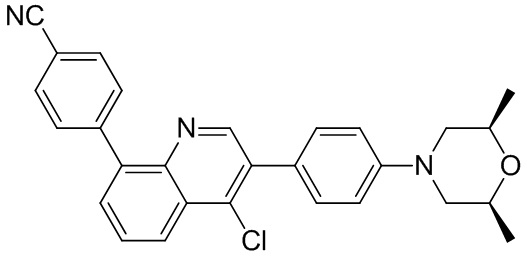 ,
, 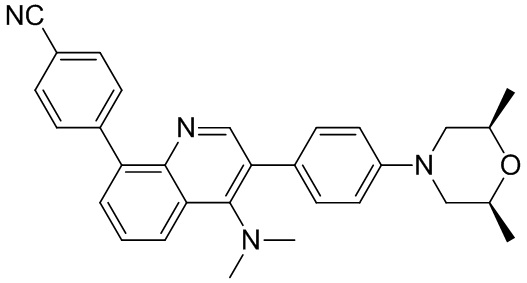 ,
,
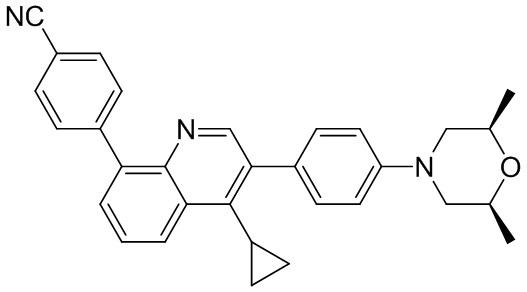 ,
, 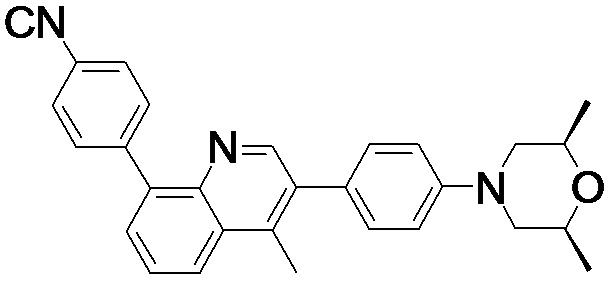 ,
, 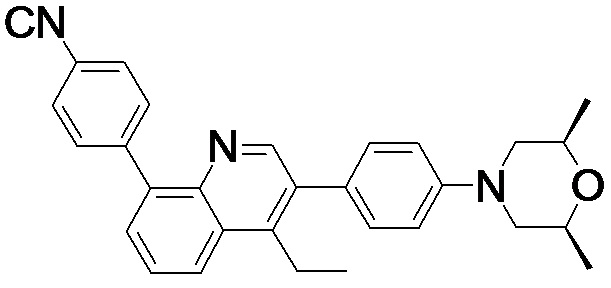 ,
,
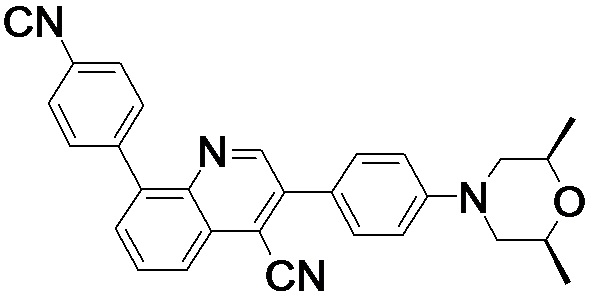 ,
, 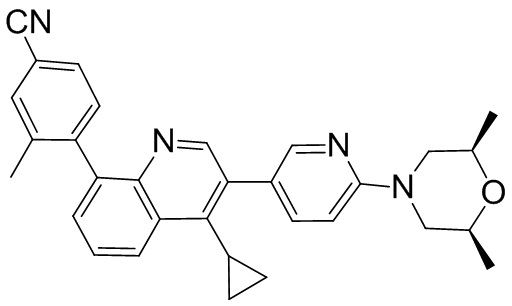 ,
,  ,
,
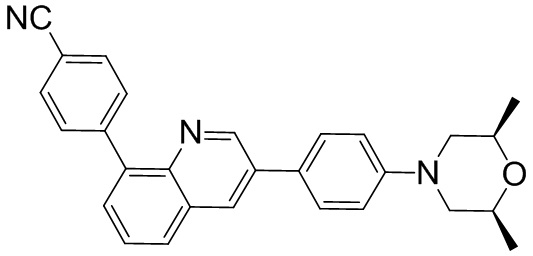 ,
, 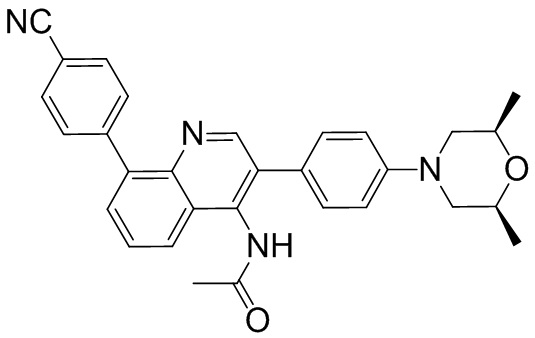 ,
,
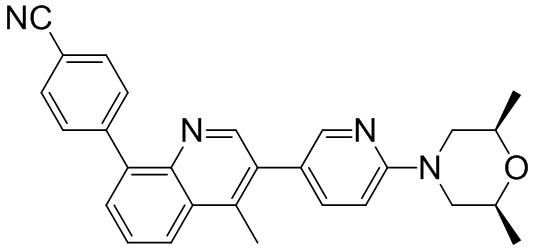 ,
,
 ,
, 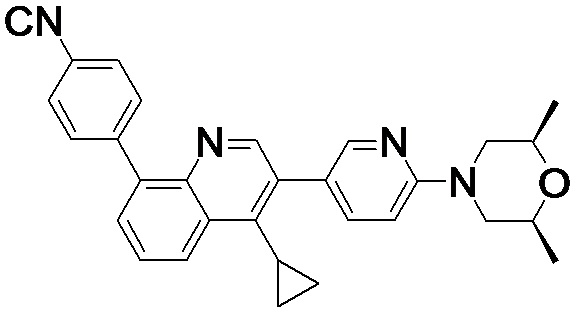 ,
, 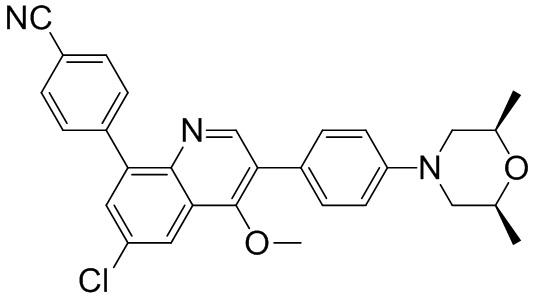 ,
,
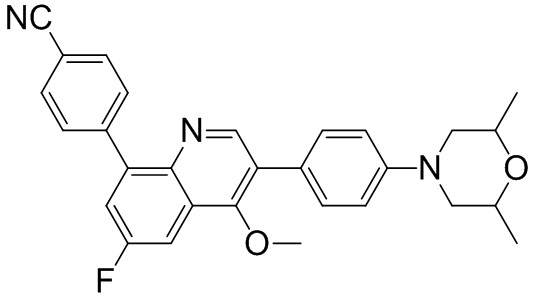 ,
, 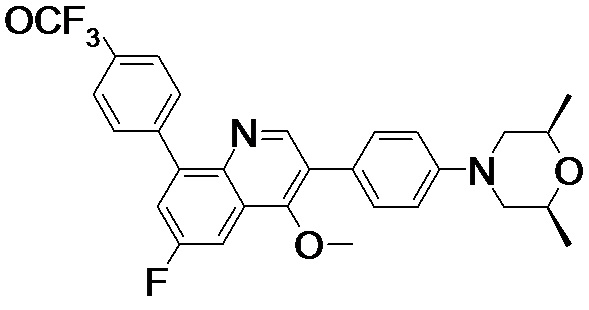 ,
, 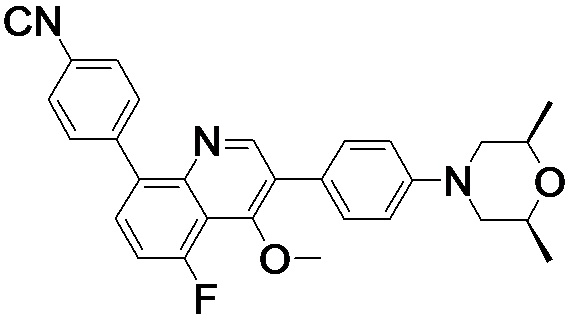 ,
,
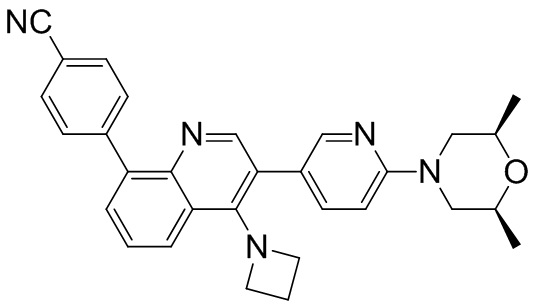 ,
, 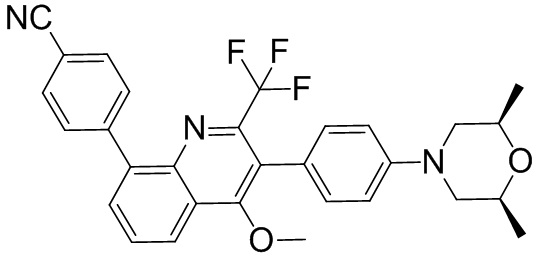 ,
,
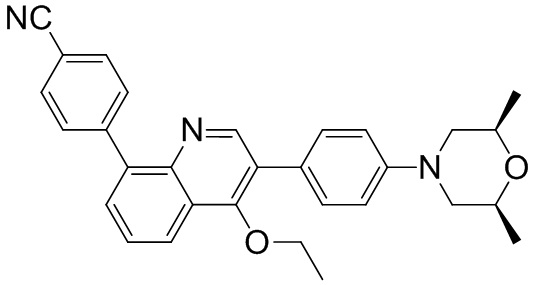 ,
, 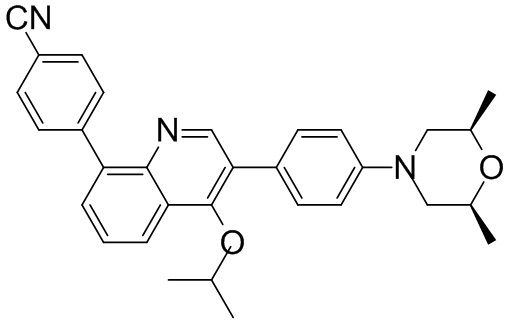 ,
,
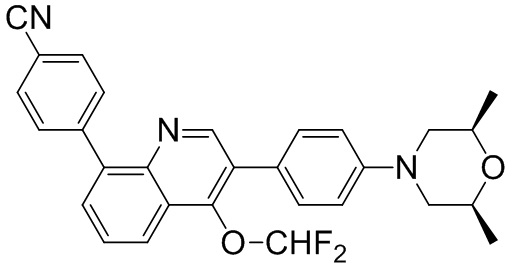 ,
,  ,
,
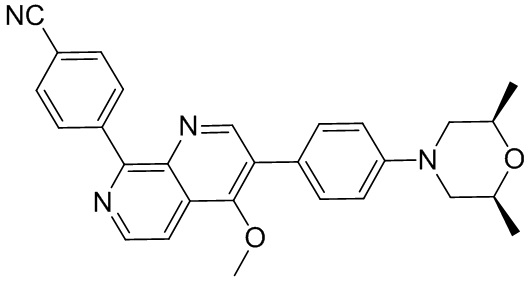 ,
, 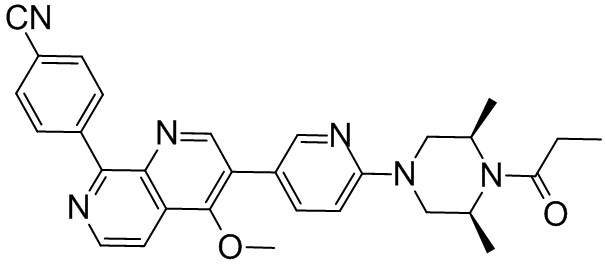 ,
,
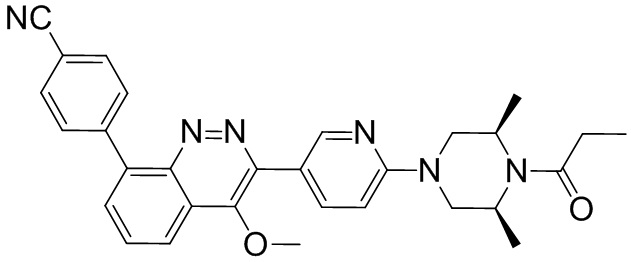 ,
, 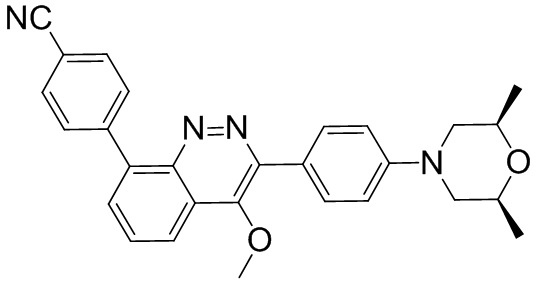 ,
, 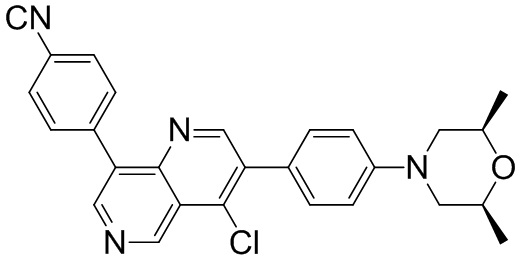 ,
,
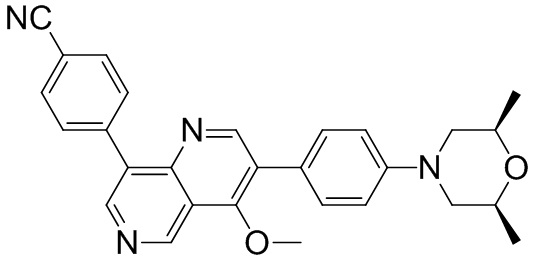 ,
, 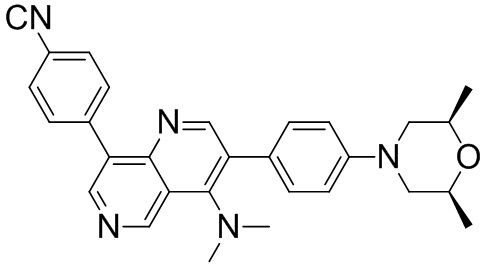 ,
, 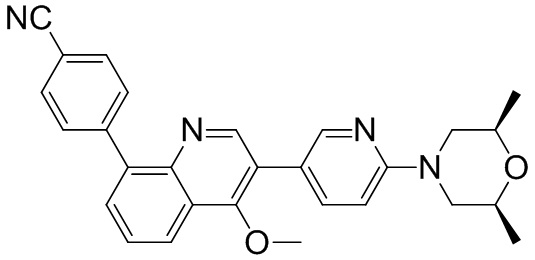 ,
, 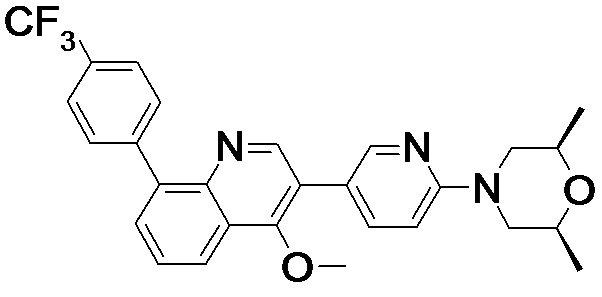 ,
,
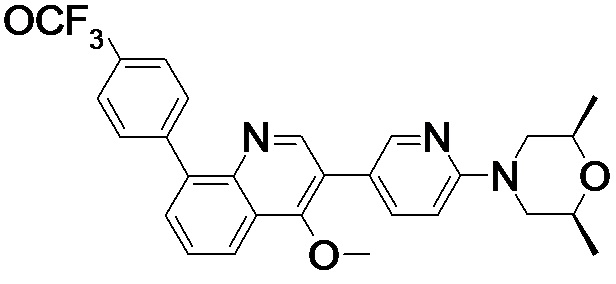 ,
,  ,
, 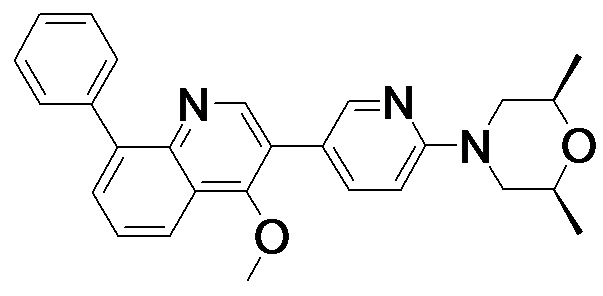 ,
,
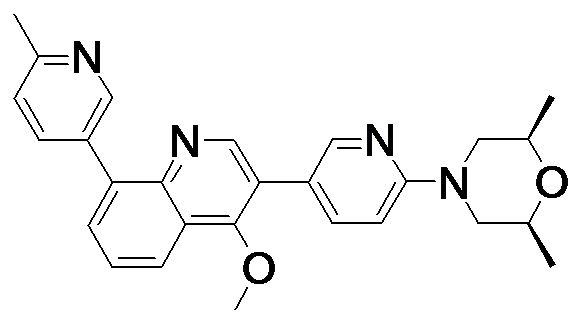 ,
,  ,
,  ,
,
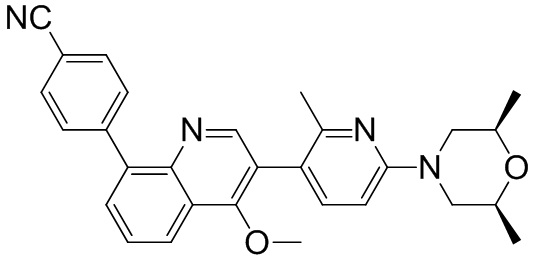 ,
, 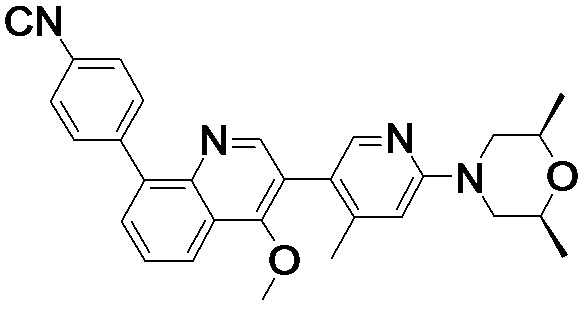 ,
, 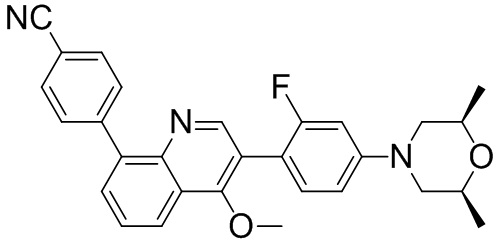 ,
,
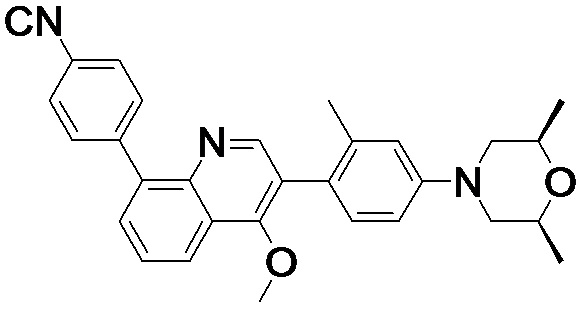 ,
, 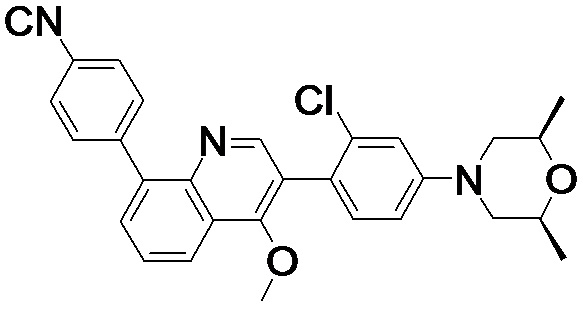 ,
, 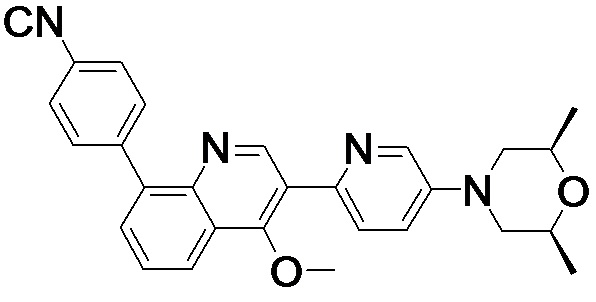 ,
,
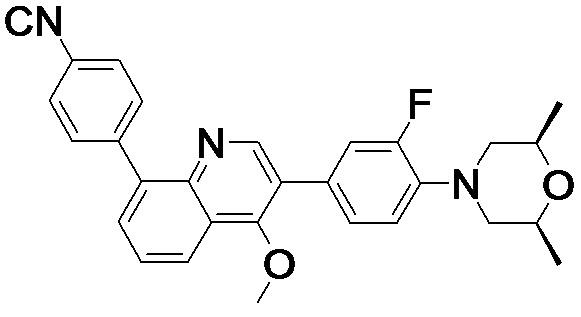 ,
, 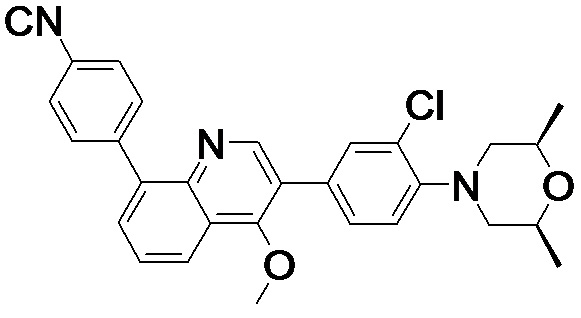 ,
, 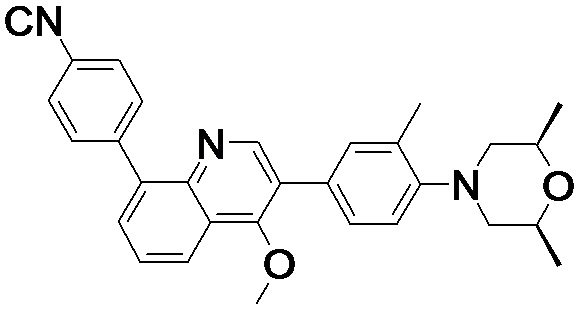 ,
,
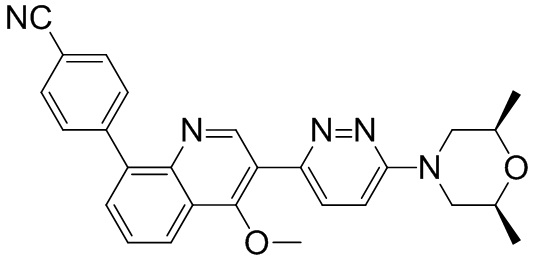 ,
, 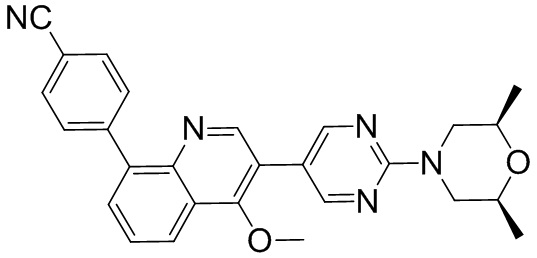 ,
, 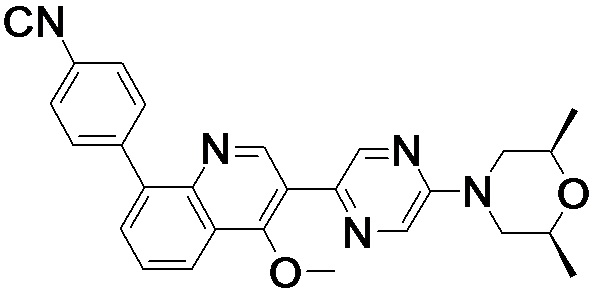 ,
,
 ,
, 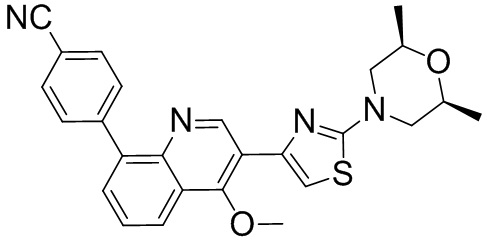 ,
, 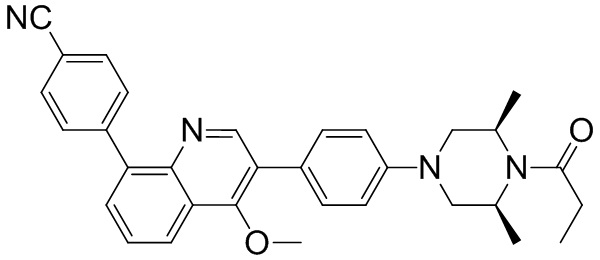 ,
,
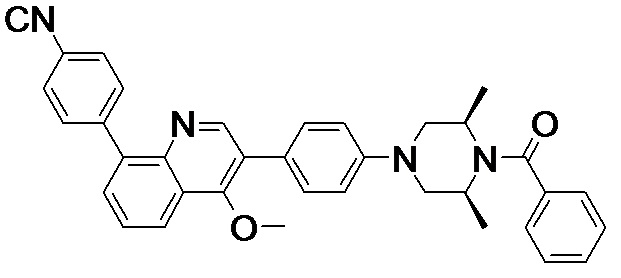 ,
, 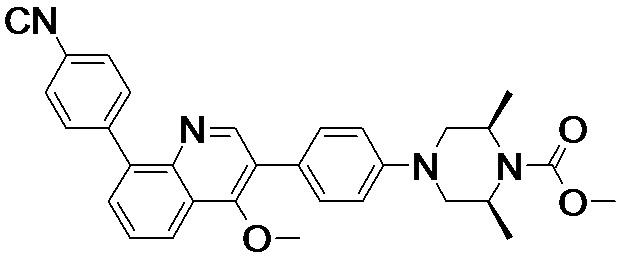 ,
,  ,
,  ,
,
 ,
, 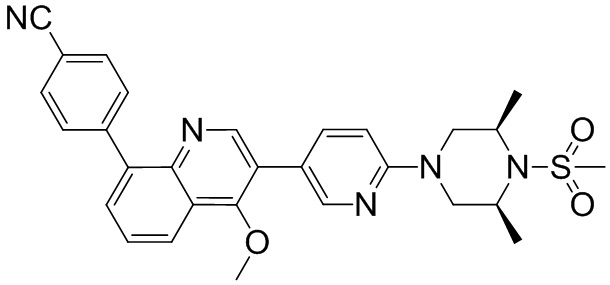 ,
, 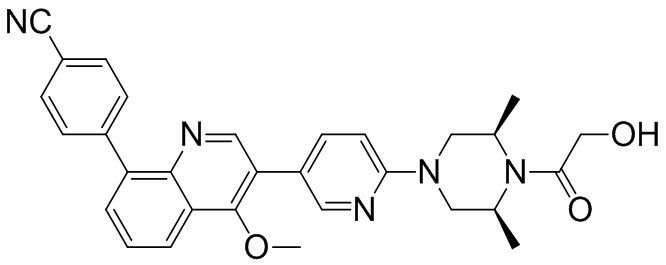 ,
,
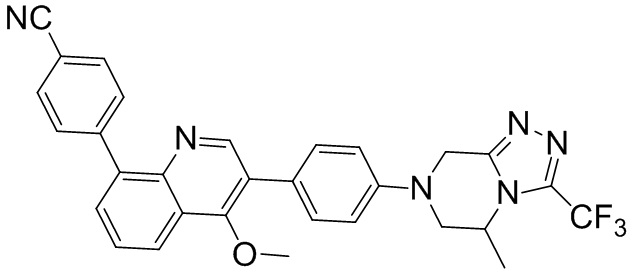 ,
,  ,
, 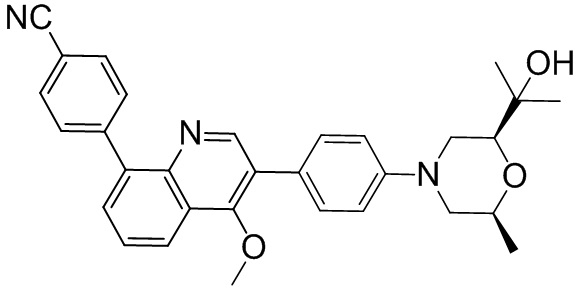 ,
,
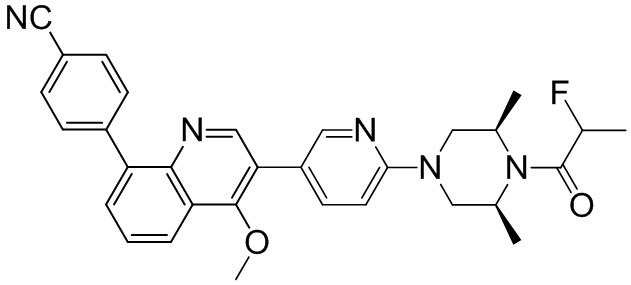 ,
, 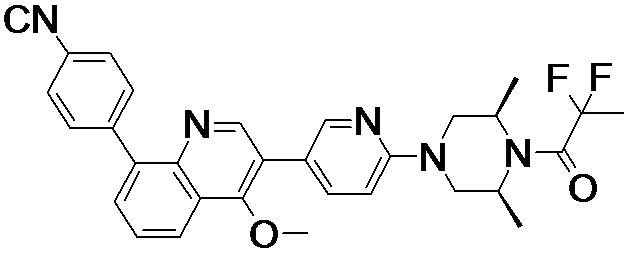 ,
, 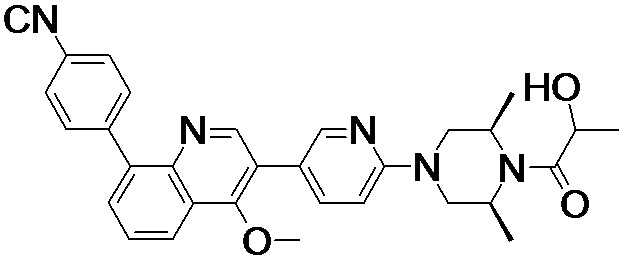 ,
,
 ,
, 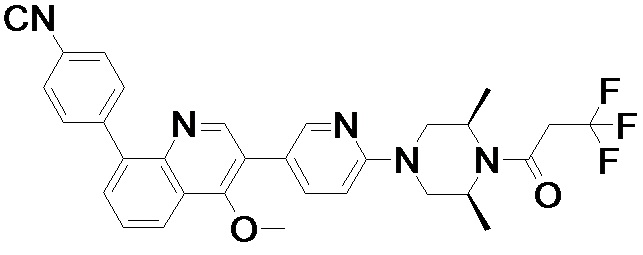 ,
, 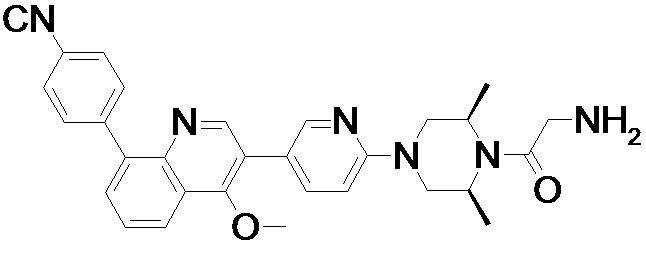 ,
,
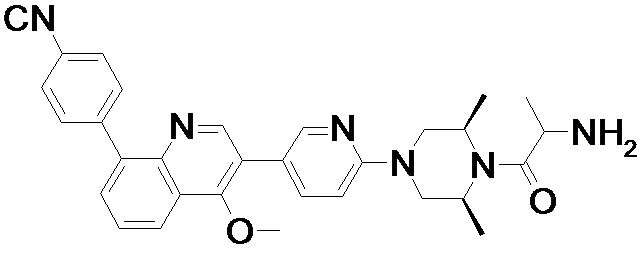 ,
, 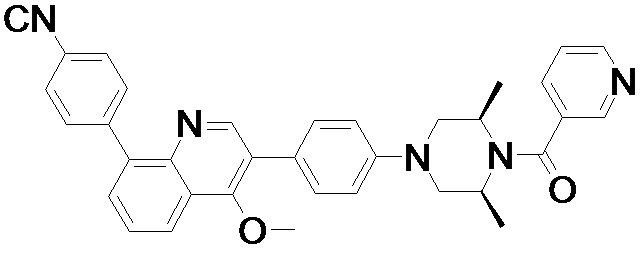 ,
, 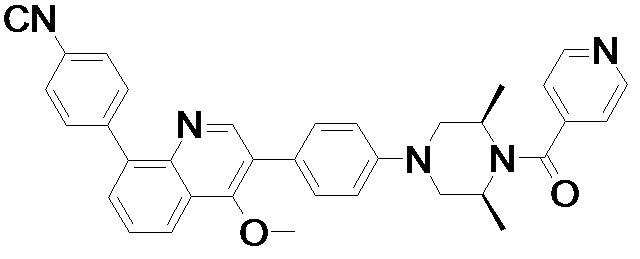 ,
,
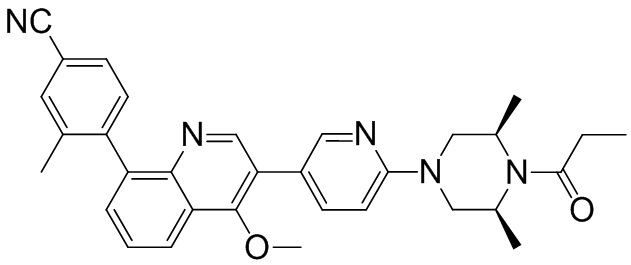 ,
, 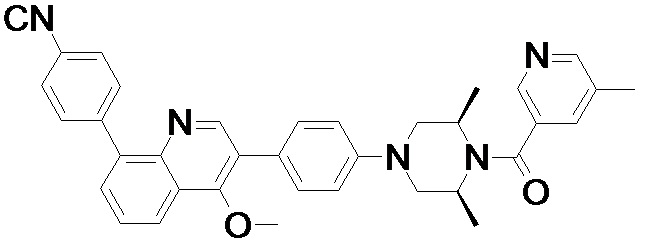 ,
, 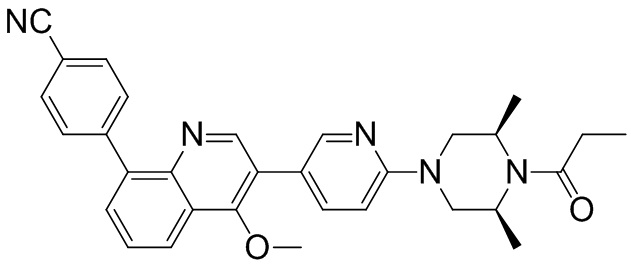 ,
,
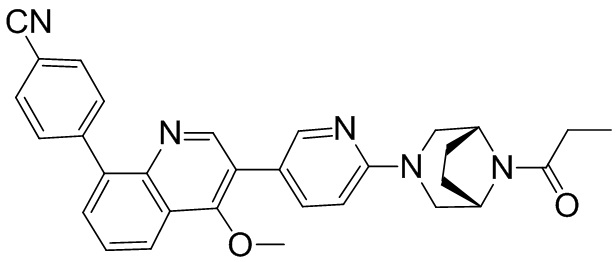 ,
,  ,
,  ,
,
 ,
,  ,
,  ,
,
 ,
,  ,
,  ,
,
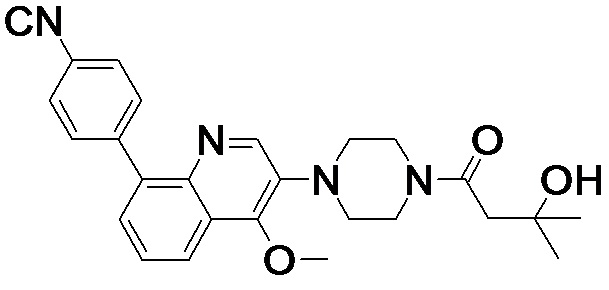 ,
, 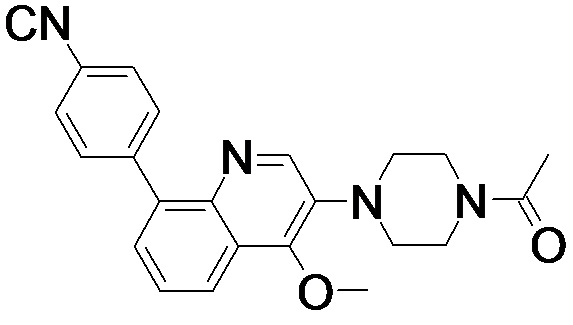 ,
, 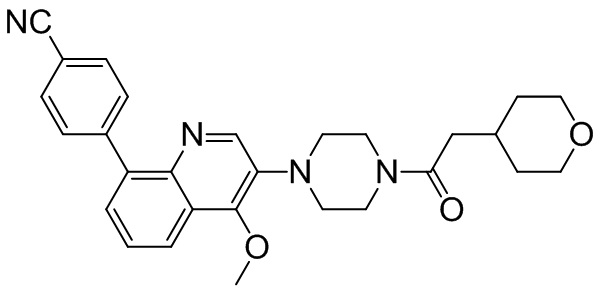 ,
,
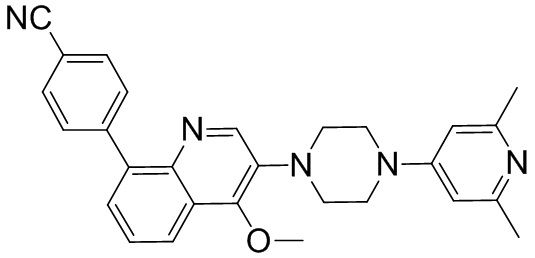 ,
, 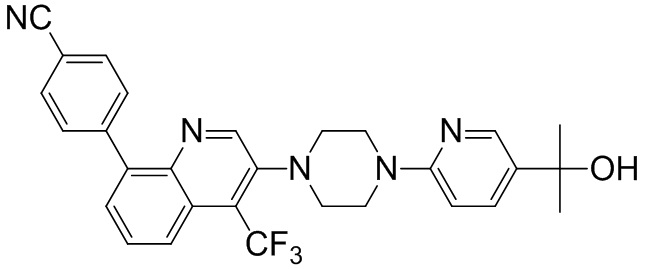 ,
, 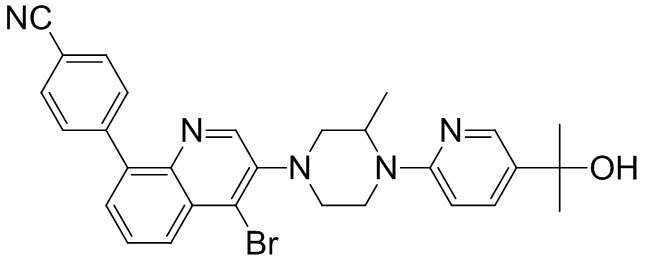 ,
,
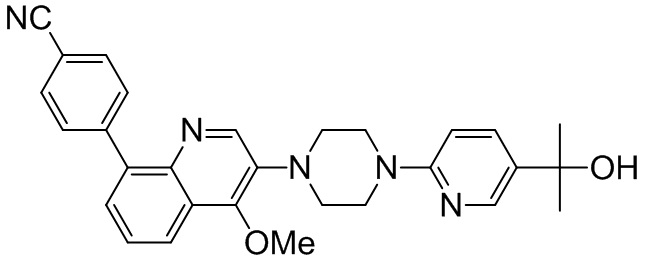 ,
, 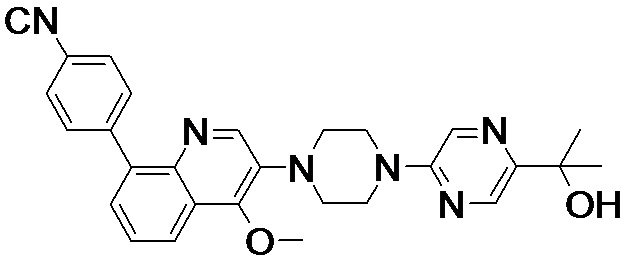 ,
, 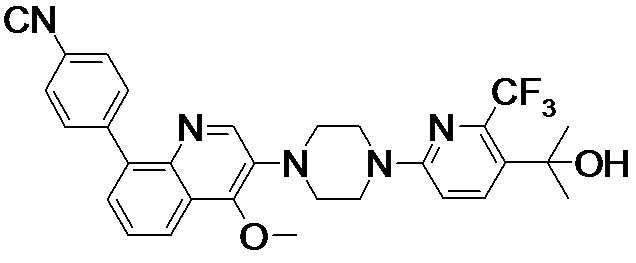 ,
,
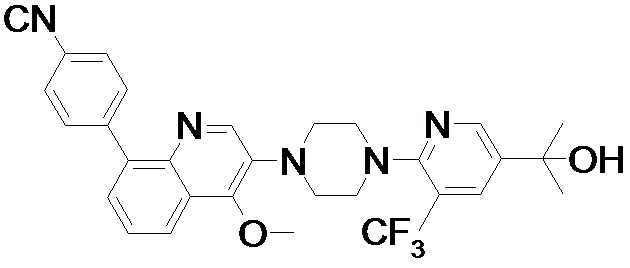 ,
, 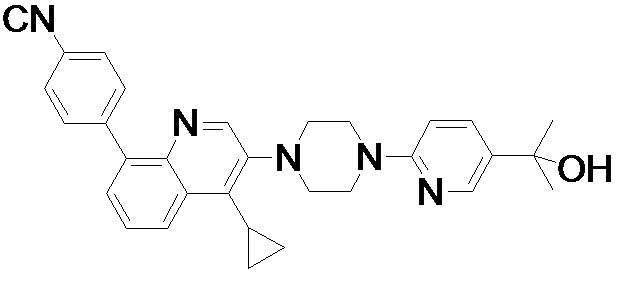 ,
, 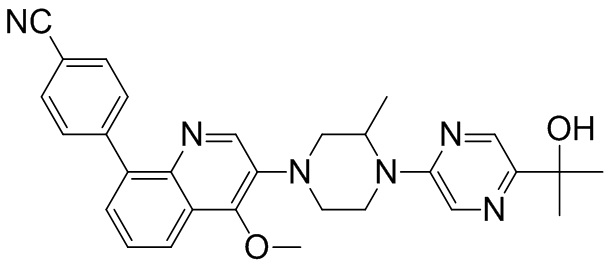 ,
,
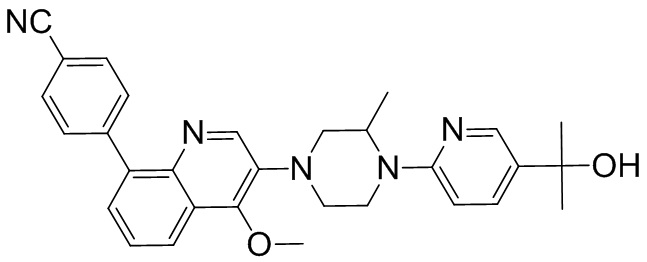 ,
,  ,
, 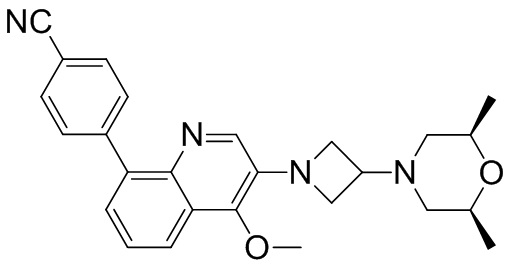 ,
,
 ,
, 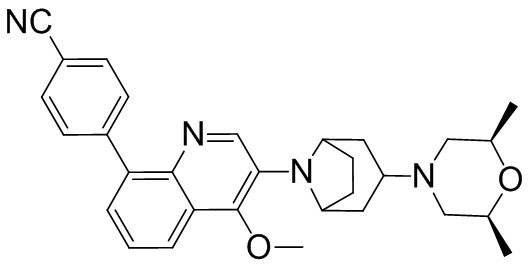 ,
, 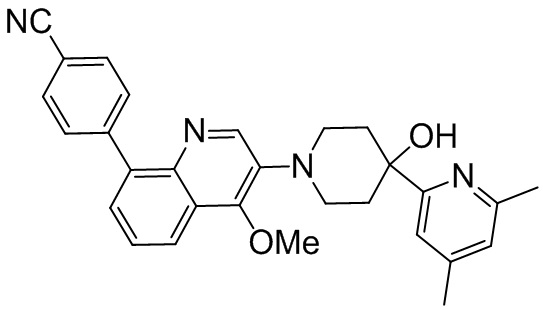 ,
,
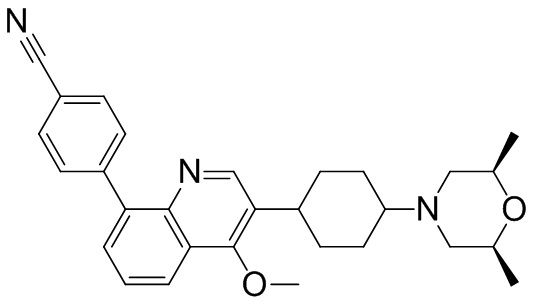 ,
, 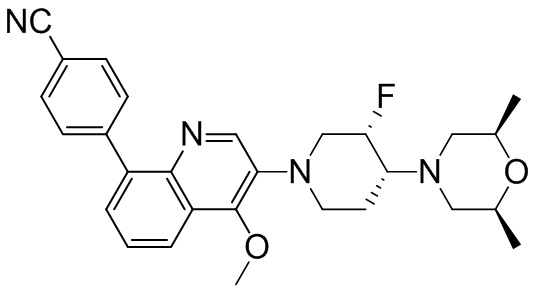 ,
, 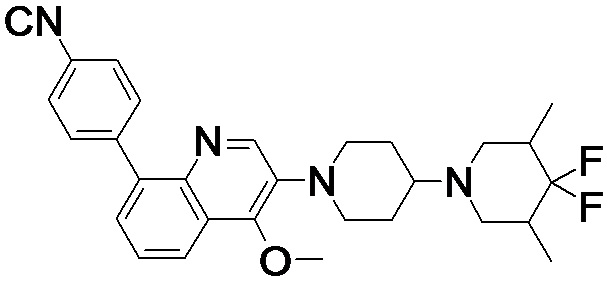 ,
,
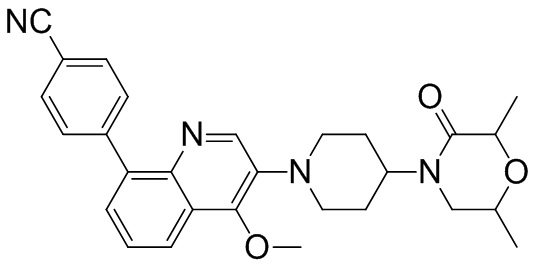 ,
, 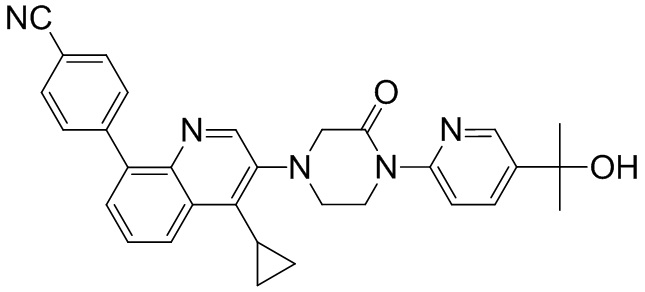 ,
,  ,
,
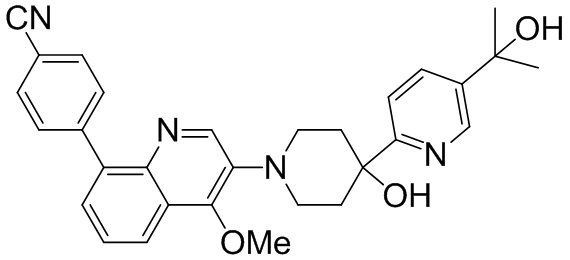 ,
, 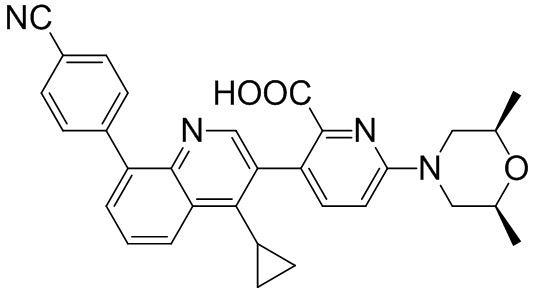 ,
,  ,
,
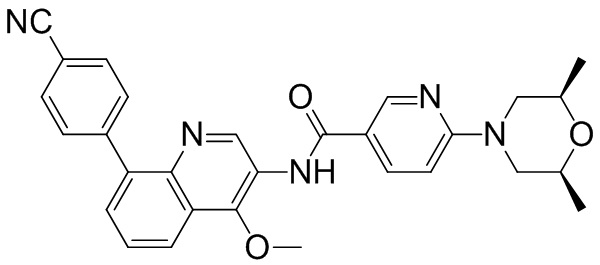 ,
, 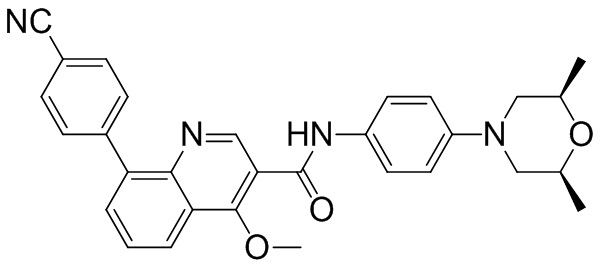 ,
, 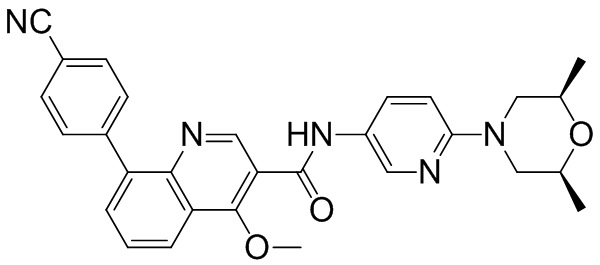 ,
,
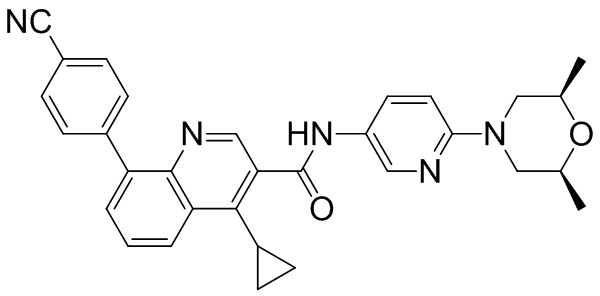 ,
, 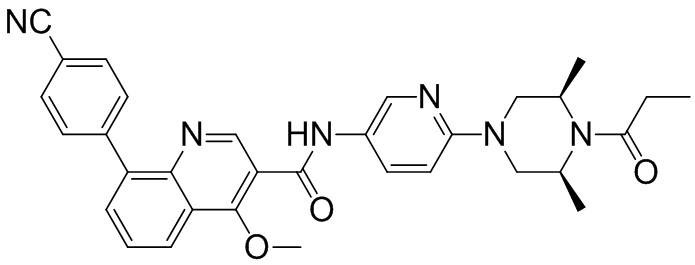 ,
, 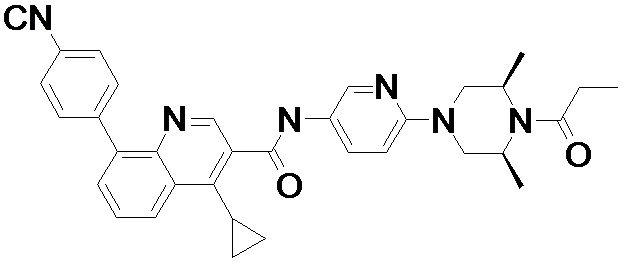 ,
,
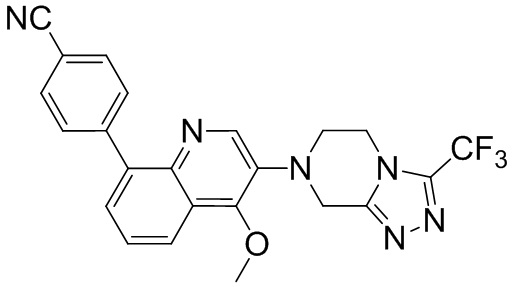 ,
, 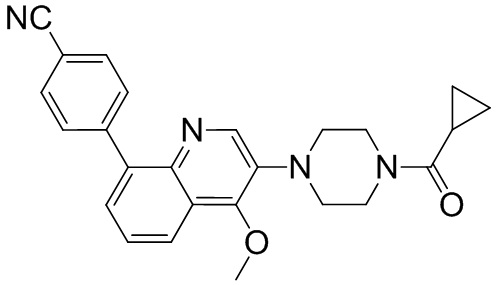 ,
, 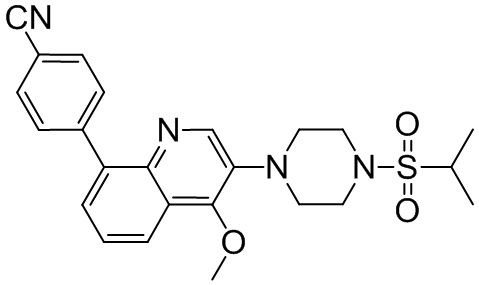 ,
,
 ;
;
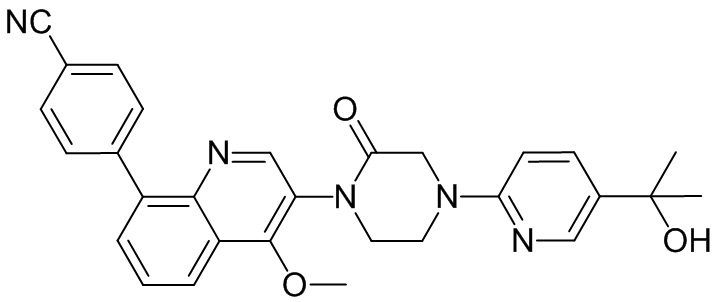 ,
, 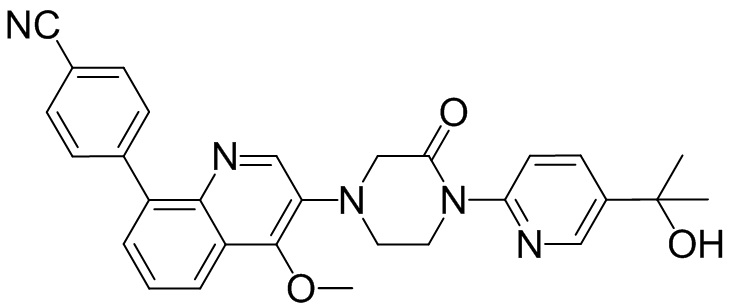 ,
, 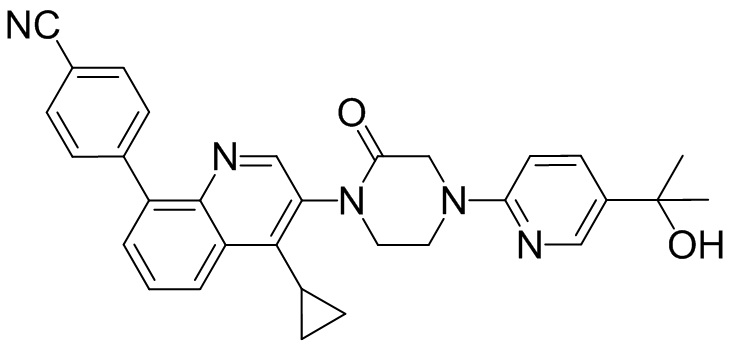 ,
,
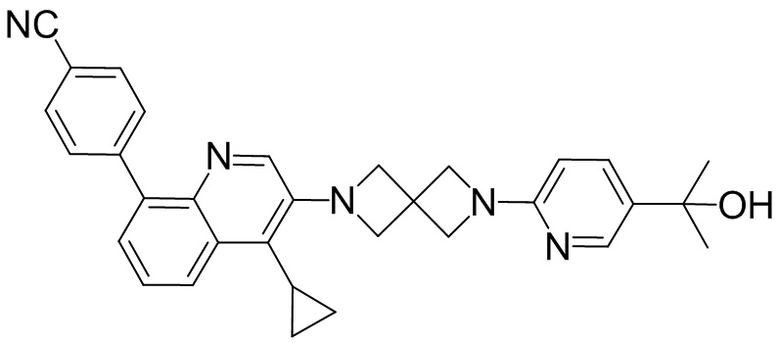 ,
, 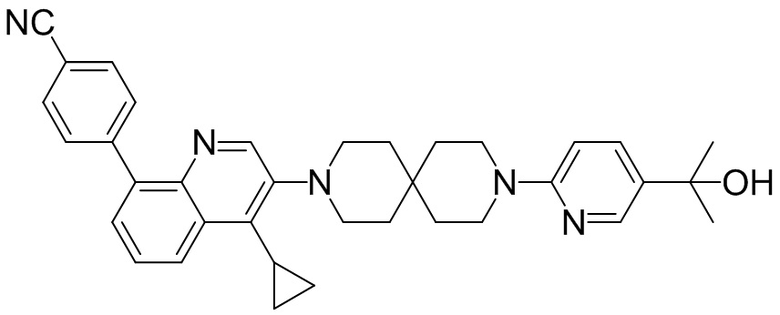 ,
, 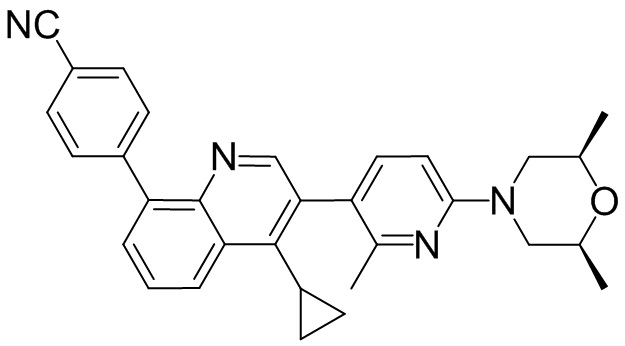 ,
,
 ,
,  ,
, 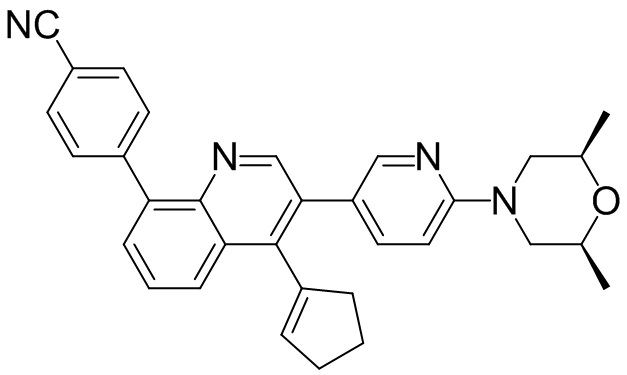 ,
,
 ,
,
 ,
, 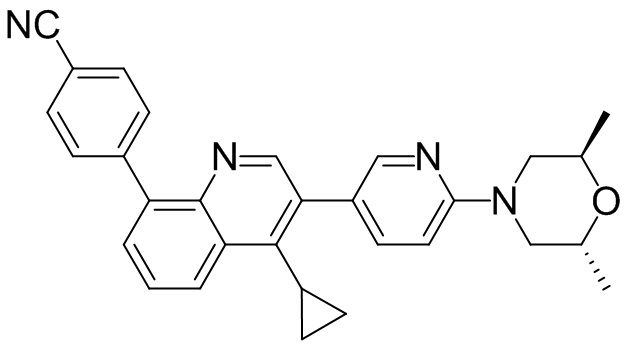 ,
, 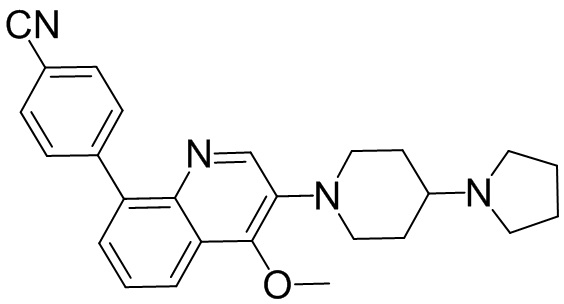 ,
,
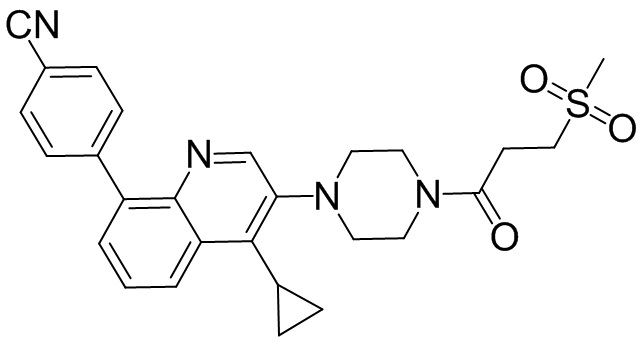 ,
, 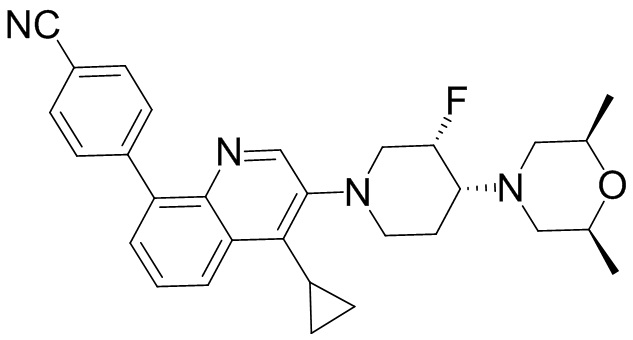 ,
, 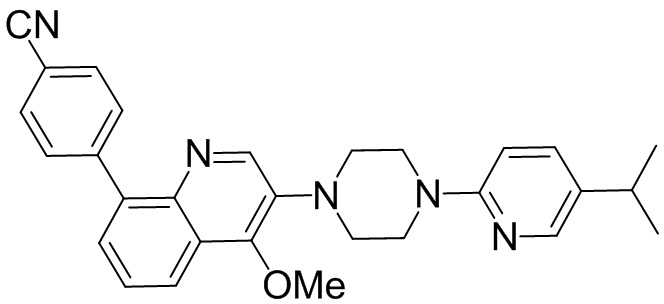 ,
,
 и
и  .
.
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Паровой двигатель | 1928 |
|
SU17262A1 |

