Перекрестная ссылка на связанные заявки
Данная заявка является национальной стадией международной заявки PCT/CN2018/092419, поданной 22 июня 2018 г., которая испрашивает преимущество приоритета заявки на патент Китая № 201710493411.2, названной “7-SUBSTITUTED PYRROLOTRIAZINE COMPOUNDS OR PHARMACEUTICALLY ACCEPTABLE SALTS THEREOF, AND PREPARATION METHODS AND USES THEREOF” и поданной 23 июня 2017 г. Обе заявки включены в данный документ во всей своей полноте для всех целей.
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к области медицинской химии и, в частности, относится к соединениям на основе 7-замещенного пирролотриазина или их фармацевтически приемлемым солям и к способам их получения и применения.
УРОВЕНЬ ТЕХНИКИ
Сигнальный путь PI3K-Akt-mTOR является одним из основных путей, который передает сигнал от рецепторной тирозинкиназы и рецептора, связанного с G-белком, и играет важную роль в различных функциях клетки. Недавние исследования показали, что несколько ключевых узловых белков в сигнальном пути PI3K-Akt-mTOR избыточно активированы вследствие наличия мутации или амплификации кодирующих генов в различных опухолях человека [Vivancod et al., Nat Rev Cancer 2 (2002), стр. 489-501]. Среди них PI3K, выполняющая функцию мостиковой молекулы, связывающей внеклеточные сигналы и эффекты клеточного ответа, является ключевым фактором, который контролирует рост, метаболизм и выживаемость клеток, избыточная активация которого тесно связана с возникновением множественных опухолей у людей [Sabbah et al., Curr Med Chem 18 (2011), стр. 5528-5544].
PI3K относится к внутриклеточной фосфатидилинозитолкиназе с активностью фосфатидилинозитолкиназы и серин/треонинкиназы. В соответствии с гомологией различных групп генов, субстратной специфичностью и функциями, суперсемейство PI3K в основном делится на три типа: тип I, тип II и тип III [Engelman et al., Nat Rev Genet 7 (2006), стр. 606-619]. PI3K I типа является, безусловно, наиболее широко изученным типом, этот тип PI3K можно дополнительно разделить на четыре подтипа: PI3Kα, PI3Kβ, PI3Kδ и PI3Kγ. Тип I PI3K представляет собой гетеродимерный фермент, состоящий из каталитической субъединицы 110 кДа и регуляторной субъединицы 85 кДа. Из-за отличий регуляторных субъединиц и механизмов активации эти четыре подтипа можно также разделить на две категории: класс IA, класс IB. При этом PI3Kα, PI3Kβ и PI3Kδ относятся к классу IA и активируются рецепторной тирозинкиназой (RTK); тогда как PI3Kγ принадлежит к классу IB и активируется рецептором, связанным с G-белком. 3-положение фосфатидилинозитол-4,5-фосфата (PIP2) может быть фосфорилировано с помощью PI3K I типа с образованием фосфатидилинозитол-3,4,5-фосфата (PIP3). В качестве важного вторичного мессенджера PIP3 может связывать и активировать различные внутриклеточные белки-мишени (такие как AKT и PDK1) с образованием комплекса сигнального каскада и, в конечном итоге, обеспечением контроля пролиферации, дифференциации, выживания и миграции клетки. Киназный домен PI3K II типа или III типа имеет высокую гомологию относительно такового у PI3K I типа, и PI3K II типа и III типа имеют аналогичные функции фосфорилирования, что и PI3K I типа. Однако физиологические функции PI3K II типа или III типа недостаточно изучены. PI3K II типа функционирует как мономер без регуляторной субъединицы и делится на три подтипа: PI3KC2α, PI3KC2β и PI3KC2γ, которые в основном участвуют в транспортировке внутриклеточных материалов, выживании клеток и в процессе интернализации мембранных рецепторных белков. PI3K III типа, которая состоит только из одного подтипа Vps34 (сортировка дефектных белков в вакуоли 34), является общеизвестной за свои функции эндоцитоза и транспорта в аппарате Гольджи [Backer, Biochem J 410 (2008), стр. 1-17]. Недавние исследования показали, что Vps34 играет важную роль в процессе аутофагии клеток [Ma et al., Cell Res 24 (2014), стр. 912-924].
В настоящее время в клинические испытания включают множество ингибиторов PI3K, которые можно условно разделить на три категории: неселективные ингибиторы PI3K, селективные ингибиторы PI3K I типа и селективные по подтипу ингибиторы PI3K I типа. Исходя из анализа существующих клинических данных, хотя неселективные ингибиторы PI3K были самыми первыми кандидатами, которые входили в клиническое испытание, они действовали медленно из-за токсичности, ограничивающей дозу. При этом селективные по подтипу ингибиторы PI3K действовали быстро в клинических испытаниях, особенно ингибиторы PI3Kδ. Первый ингибитор PI3K на рынке, иделалисиб, представляет собой селективный ингибитор PI3Kδ, а другой селективный ингибитор γ/δ, дувелисиб, уже находится на фазе III клинических испытаний [Winkler et al., Chem Biol 20 (2013), стр. 1362-1372]. Среди каталитических субъединиц класса IA каталитическая субъединица p110δ PI3Kδ, отличающаяся от P110α и P110β, которые широко экспрессируются в различных тканях по всему организму, в основном селективно высоко экспрессируется в иммунной системе, такой как B-клетки, T-клетки и т. д., что тесно связано с заболеваниями гемобластоза и воспаления, иммунитета и т. п. [Fruman et al., N Engl J Med 370 (2014), стр. 1061-1062]. Иделалисиб был одобрен FDA США в качестве лекарственного средства для лечения трех видов гемобластоза рецидивирующего хронического лимфоцитарного лейкоза (CLL), рецидивирующей фолликулярной B-клеточной неходжкинской лимфомы (FL) и рецидивирующей мелкоклеточной лимфоцитарной лимфомы. Это подчеркивает важность ингибиторов PI3Kδ в лечении видов гемобластоза.
В настоящее время структуры большинства ингибиторов против PI3Kδ имеют общий основной остов иделалисиба, например, US 20140121224 A1. Для сравнения, токсичность соединений с триазином в качестве основного остова была в значительной степени более низкой, чем таковая у иделалисиба, на основе доступных данных. Другим типом основного остова, который был более изучен, является тиенопиримидин, иллюстративным соединением которого является пан-ингибитор PI3K GDC-0941 [Folkes et al., J Med Chem 51 (2008), стр. 5522-5532]. Хотя соединения на основе тиенопиримидина были описаны как селективные ингибиторы PI3Kδ [Murray et al., J Med Chem 55 (2012), стр. 7866-7695], их основные остовы значительно отличаются от таковых пирролотриазина по свойствам. В настоящее время сообщали только о нескольких случаях получения ингибиторов PI3K на основе пирролотриазина в качестве исходного ядра, таких как случаи, раскрытые в CN 102675323 A, и до настоящего времени не было сообщений об ингибирующей активности в отношении PI3Kδ.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
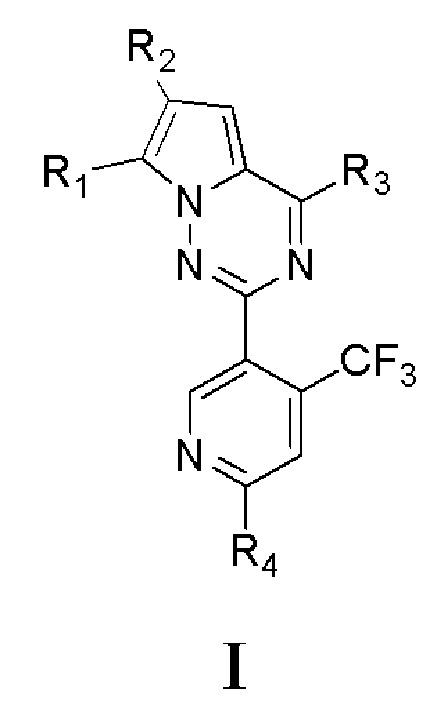
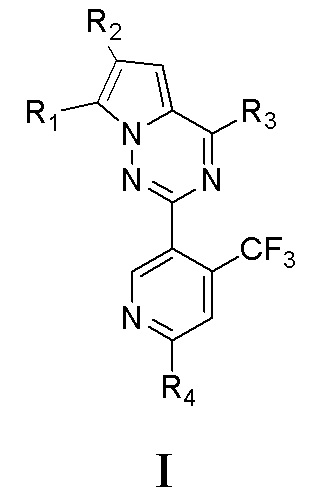
Исходя из этого, целью настоящего изобретения является обеспечение соединений на основе 7-замещенного пирроло[2,1-f][1,2,4]триазина, представленных общей формулой I, или их фармацевтически приемлемых солей:
 ,
,
где
R1 представляет собой галоген, или C1-6алкил, или C3-8циклоалкил, который является незамещенным или замещенным по меньшей мере одним заместителем, при этом заместитель представляет собой галоген;
R2 представляет собой: -C(OH)R5R6; -COC1-6алкил; -CN или незамещенный либо замещенный по меньшей мере одним заместителем C1-6алкил, -CH2NH-C1-6алкил, -CH2N(C1-6алкил)-(C1-6алкил), -CH2-(насыщенный гетероциклил, содержащий 1-2 гетероатома и 3-6 атомов углерода), -CH2-(насыщенный гетероциклил, содержащий 1-2 гетероатома и 3-6 атомов углерода)-(насыщенный гетероциклил, содержащий 1-2 гетероатома и 3-6 атомов углерода), -CH2-(насыщенная спироциклическая группа, содержащая 1-2 гетероатома и 4-12 атомов углерода), -CH2-(насыщенная кольцевая группа с мостиковой связью, содержащая 1-2 гетероатома и 3-12 атомов углерода)-(насыщенный гетероциклил, содержащий 1-2 гетероатома и 3-6 атомов углерода) или -CH2-(насыщенная кольцевая группа с мостиковой связью, содержащая 1-2 гетероатома и 3-12 атомов углерода), где заместитель представляет собой галоген, -N(C1-4алкил)-(C1-4алкил), -O-C1-4алкил, -CN, -COOH, -CHO, -NHS(O)2-C1-4алкил, -N(C1-4алкил)-C(C1-4алкил)-(C1-4алкил)-CONH2, =O, -OH, -S(O)2N(C1-4алкил)-(C1-4 алкил), -S-C1-4алкил, -S(O)2-C1-4алкил, -CO-C3-6циклоалкил, оксетанил, морфолинил, C3-6циклоалкил, -C1-4алкил-N(C1-4алкил)-(C1-4алкил), C1-4алкил-O-C1-4алкил, который является незамещенным или замещен по меньшей мере одним метилом, -CONH2, который является незамещенным или замещен по меньшей мере одним метилом, C1-4алкил-CONH2, который является незамещенным или замещен по меньшей мере одним метилом, -COO-C1-4алкил, который является незамещенным или замещен по меньшей мере одним метилом, -NH2, который является незамещенным или замещен по меньшей мере одним метилом, -NHCO-C1-4алкил, который является незамещенным или замещен по меньшей мере одним метилом, -CO-C1-4алкил, который является незамещенным или замещен по меньшей мере одним заместителем A, где заместитель A представляет собой гидроксил или метил, или C1-4алкил, который является незамещенным или замещен по меньшей мере одним заместителем B, где заместитель B представляет собой -NH2, -OCH3, -CONH2, -OH или -CF3.
В R2 гетероатом выбран из по меньшей мере одного из N, O и S,
каждый из R5 и R6 независимо представляет собой водород или C1-6алкил;
R3 представляет собой 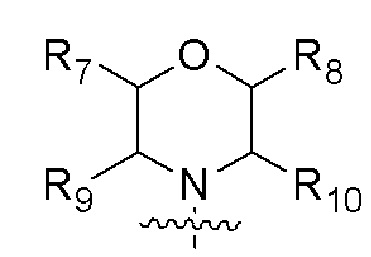 , где каждый из R7, R8, R9 и R10 независимо представляет собой водород или C1-6алкил, который является незамещенным или замещен по меньшей мере одним заместителем, при этом заместитель представляет собой галоген или гидроксил;
, где каждый из R7, R8, R9 и R10 независимо представляет собой водород или C1-6алкил, который является незамещенным или замещен по меньшей мере одним заместителем, при этом заместитель представляет собой галоген или гидроксил;
R4 представляет собой -NH2, -NHCONHR11 или -NHCO2R12, где каждый из R11 и R12 независимо представляет собой C1-6алкил, C3-8циклоалкил или фенил, который является незамещенным или замещен по меньшей мере одним заместителем, выбранным из по меньшей мере одного из галогена и -C(O)OR13, где R13 представляет собой C1-6алкил, который является незамещенным или замещен по меньшей мере одним заместителем, при этом заместитель в R13 представляет собой галоген.
В одном варианте осуществления R1 представляет собой галоген, или C1-4алкил, или C3-6циклоалкил, который является незамещенным или замещенным по меньшей мере одним заместителем.
В одном варианте осуществления R1 представляет собой -Cl, -F, метил, трифторметил или дифторметил.
В одном варианте осуществления R2 представляет собой: -C(OH)R5R6; -COC1-4алкил; -CN или незамещенный либо замещенный по меньшей мере одним заместителем C1-4алкил, -CH2NH-C1-4алкил, -CH2N-(C1-4алкил)-(C1-4алкил), -X-(насыщенный четырех-шести-членный гетероциклил, содержащий 1-2 гетероатома), -X-(насыщенный четырех-шести-членный гетероциклил, содержащий 1-2 гетероатома)-(насыщенный четырех-шести-членный гетероциклил, содержащий 1-2 гетероатома), -X-(насыщенная спиро-бициклическая группа, содержащая 1-2 гетероатома и 4-8 атомов углерода), -X-(насыщенная бициклическая кольцевая группа с мостиковой связью, содержащая 1-2 гетероатома и 3-8 атомов углерода)-(насыщенный четырех-шести-членный гетероциклил, содержащий 1-2 гетероатома) или -X-(насыщенная бициклическая кольцевая группа с мостиковой связью, содержащая 1-2 гетероатома и 3-8 атомов углерода), где X представляет собой CH2, и где гетероциклил, спироциклическая группа и кольцевая группа с мостиковой связью присоединены к X посредством атома N, каждый из R5 и R6 независимо представляет собой водород или C1-4алкил.
В одном варианте осуществления каждый из R5 и R6 представляет собой водород, метил или этил.
В одном варианте осуществления заместитель в R2 представляет собой -F, -Cl, -Br, -I, -CH3, -CH2CH3, -CH2CH(CH3)2, -CH2NH2, -CH2N(CH3)2, -CH2CH2NH2, -CH2CH2N(CH3)2, -CH2OH, -CH2OCH3, -CH2CH2OH, -C(CH3)2OH, -C(CH3)(CF3)OH, -C(CF3)2OH, -C(CH3)2OCH3, -C(CH3)2NH2, -CH2C(CH3)2OH, -CH(OH)CH(CH3)2, -C(CH3)2CH2OH, -C(CH3)2CH2OCH3, -CN, -CF3, -CO2H, -CHO, -COCH3, -CO2CH3, -CO2C(CH3)3, -COCH2OH, -COC(OH)(CH3)2, -COCH(OH)CH3, -CONH2, -CONHCH3, -CON(CH3)2, -CH2CONH2, -CH2CON(CH3)2, -C(CH3)2CONH2, -NH2, -NHCH3, -N(CH3)2, -NHCOCH3, -N(CH3)COCH3, -NHS(O)2CH3, -N(CH3)C(CH3)2CONH2, =O, -OH, -OCH3, -S(O)2N(CH3)2, -SCH3, -S(O)2CH3, -C(O)-циклопропил, циклопропил, циклобутил, оксетанил или морфолинил.
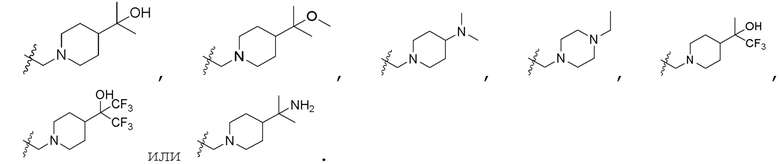
В одном варианте осуществления R2 представляет собой 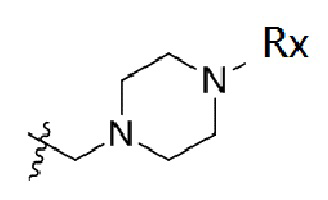 или
или 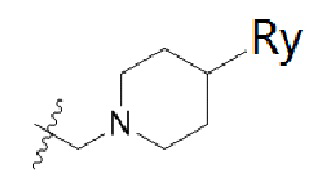 , каждый из Rx и Ry представляет собой -N(CH3)2, -S(O)2CH3 или C1-4алкил, где C1-4алкил является незамещенным или замещен по меньшей мере одним заместителем, при этом заместитель представляет собой галоген, гидроксил, -CONH2, -CF3, амино или -OCH3.
, каждый из Rx и Ry представляет собой -N(CH3)2, -S(O)2CH3 или C1-4алкил, где C1-4алкил является незамещенным или замещен по меньшей мере одним заместителем, при этом заместитель представляет собой галоген, гидроксил, -CONH2, -CF3, амино или -OCH3.
В одном варианте осуществления R2 представляет собой 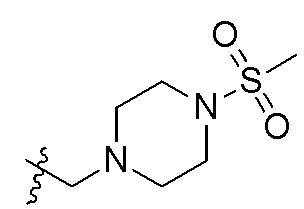 ,
, 
В одном варианте осуществления R3 представляет собой , где каждый из R7, R8, R9, R10 независимо представляет собой водород или C1-4алкил.
В одном варианте осуществления каждый из R7, R8, R9 и R10 независимо представляет собой H или метил.
В одном варианте осуществления R3 представляет собой морфолинил или (S)-3-метилморфолинил.
В одном варианте осуществления R4 представляет собой -NH2, -NHCONHR11 или -NHCO2R12, где каждый из R11 и R12 независимо представляет собой C1-4алкил, C3-6циклоалкил или фенил, который является незамещенным или замещен по меньшей мере одним заместителем, выбранным из по меньшей мере одного из фтора, хлора, брома и -C(O)OR13, где R13 представляет собой C1-4алкил.
В одном варианте осуществления каждый из R11 и R12 независимо представляет собой метил, этил, изопропил, циклопропил, фенил, -Ph-CO2Et-p или 4-фторфенил.
Соединение на основе 7-замещенного пирроло[2,1-f][1,2,4]триазина, представленное общей формулой I, или его фармацевтически приемлемая соль, например, характеризуется структурой, представленной одной из следующих общих формул:
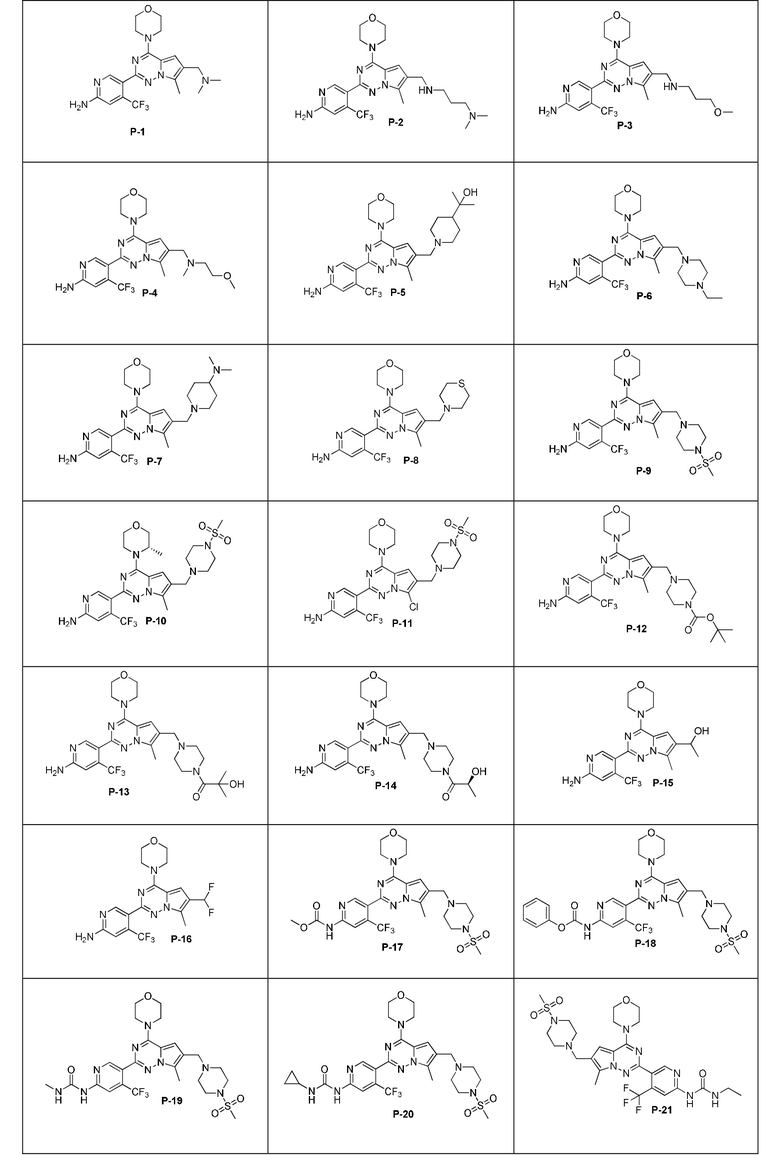
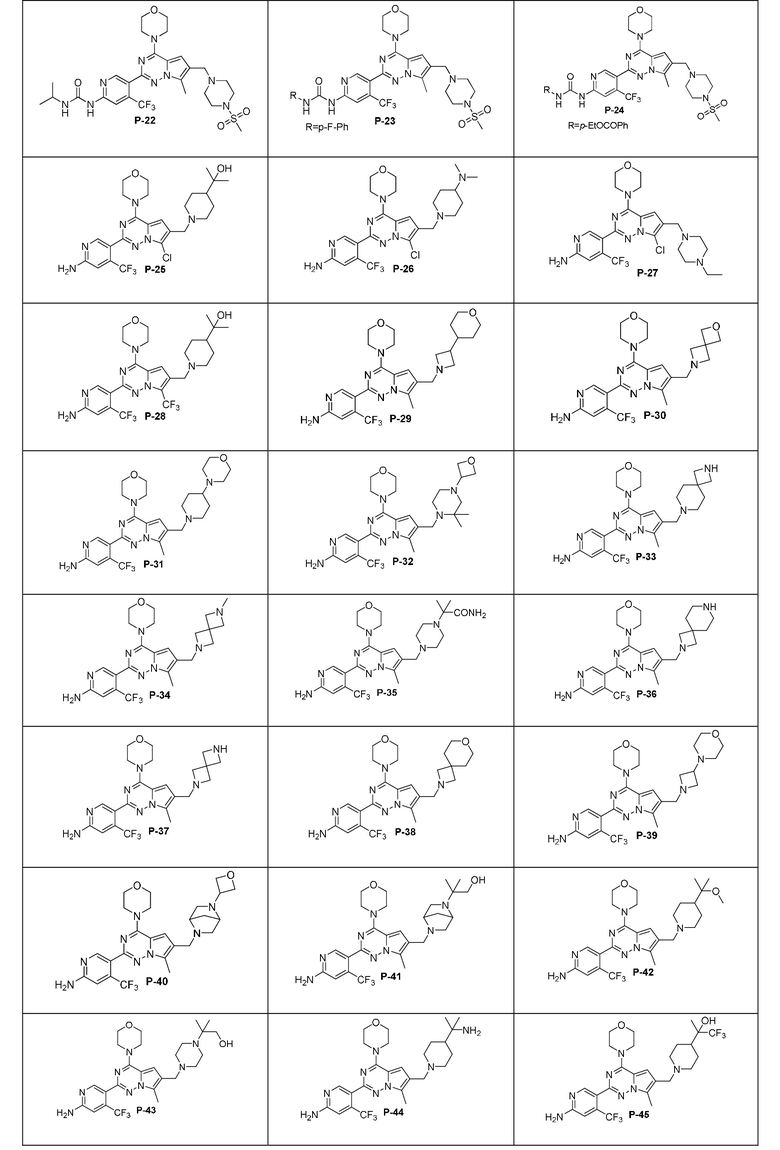

Фармацевтически приемлемые соли включают соли органической кислоты и соли неорганической кислоты, в том числе без ограничения малеат, сукцинат, цитрат, тартрат, фумарат, ацетат, мезилат, гидрохлорид, фосфат, нитрат или сульфат.
Другой целью настоящего изобретения является обеспечение способа получения соединения на основе 7-замещенного пирроло[2,1-f][1,2,4]триазина, представленного общей формулой I, которое получено в соответствии со следующим способом:
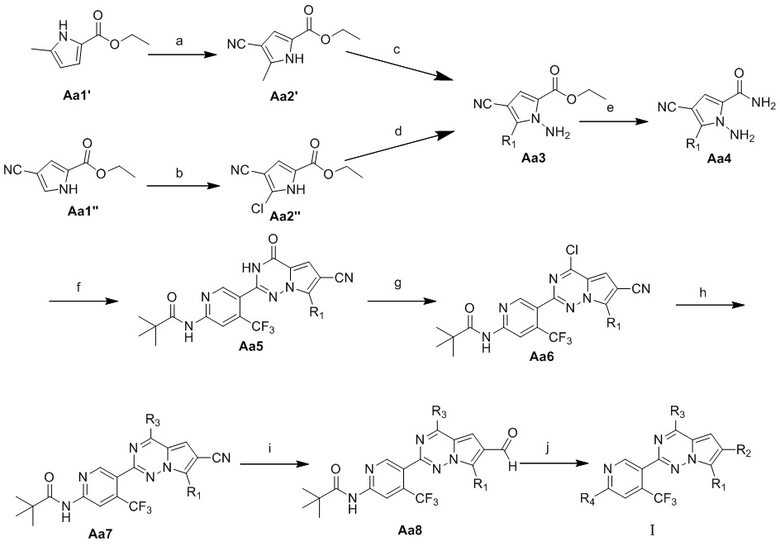
Кроме того, реагенты и условия, необходимые для каждой стадии, включают:
стадия a: добавление хлорсульфонил-изоцианата по каплям в раствор соединения Aa1' и диметилформамида (DMF) в безводном ацетонитриле и осуществление реакции с получением соединения Aa2'; или стадия b: растворение соединения Aa1'' в хлороформе и добавление N-хлорсукцинимида для осуществления реакции с получением соединения Aa2'';
стадия c: добавление 5 вес. % раствора гипохлорита натрия в раствор соединения Aa2', K2CO3, NH4Cl, концентрированного водного раствора аммиака и хлорида метилтриоктиламмония в метил-трет-бутиловом простом эфире и осуществление реакции с получением соединения Aa3; или стадия d: растворение соединения Aa2'' в безводном диметилформамиде, добавление NaH, перемешивание и затем добавление O-(2,4-динитрофенил)гидроксиламина, осуществление реакции с получением соединения Aa3;
стадия e: растворение соединения Aa3 в насыщенном растворе аммиака в метаноле или в растворе аммиака в метаноле и осуществление реакции с получением соединения Aa4;
стадия f: смешивание соединения Aa4 с альдегидом, безводным хлоридом меди и диметилсульфоксидом и осуществление реакции с получением соединения Aa5;
стадия g: добавление оксихлорида фосфора в смесь соединения Aa5 и N, N-диметиламинопиридина или диметиламинопиридина и осуществление реакции с получением соединения Aa6;
стадия h: добавление безводного тетрагидрофурана и морфолина к соединению Aa6 и осуществление реакции с получением соединения Aa7;
стадия i: добавление никеля Ренея в суспензию соединения Aa7 и моногидрата гипофосфита натрия в уксусной кислоте/воде/пиридине и осуществление реакции с получением соединения Aa8; и
стадия j: суспендирование соединения Aa8, цианоборогидрида натрия и амина в метаноле, добавление уксусной кислоты и осуществление реакции с получением продукта; или стадия j: добавление бромида метилмагния в раствор соединения Aa8 в безводном тетрагидрофуране и осуществление реакции с получением продукта; или стадия j: добавление трифторида диэтиламиносеры (DAST) в раствор соединения Aa8 в безводном дихлорметане и осуществление реакции с получением продукта.
В одном варианте осуществления соединение P-28 получали в соответствии со следующим способом:
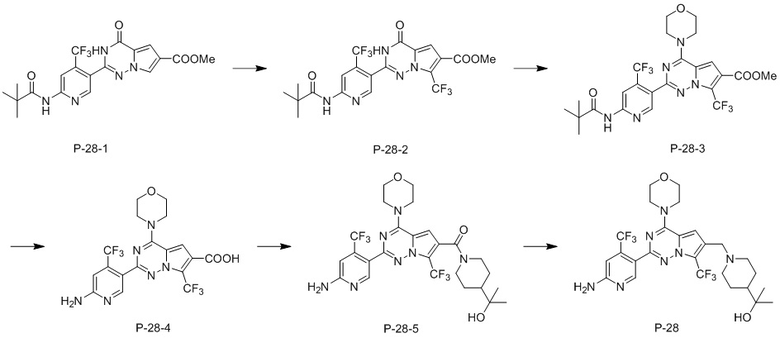
конкретно содержащему следующие стадии:
стадия 1: растворение соединения P-28-1 в смеси дихлорметана и воды, затем добавление трифторметилсульфината натрия, охлаждение вышеуказанной системы и медленное добавление по каплям 70% водного раствора трет-бутилгидропероксида, затем добавление диметилсульфоксида и нагревание реакционной смеси с получением соединения P-28-2;
стадия 2: добавление соединения P-28-2 и оксихлорида фосфора в толуол, затем добавление N, N-диметиланилина, осуществление реакции при нагревании с обратным холодильником с получением неочищенного хлорированного соединения, растворение неочищенного хлорированного соединения в безводном тетрагидрофуране и добавление морфолина при 0°C, перемешивание при комнатной температуре до завершения реакции исходных материалов, затем концентрирование с получением неочищенного продукта, который применяют непосредственно в следующей реакции;
стадия 3: растворение неочищенного продукта, полученного на стадии 2, в метаноле, затем добавление воды и гидроксида натрия, осуществление реакции при нагревании с обратным холодильником до полного израсходования исходных материалов, концентрирование, добавление воды и регулирование значения pH до осаждения твердого вещества, высушивание и затем его применение непосредственно в следующей реакции;
стадия 4: добавление неочищенного продукта со стадии 3, бензотриазол-N,N,N’,N’-тетраметилурония гексафторфосфата (HBTU), триэтиламина и 2-(4-пиперидил)-2-пропанола в N, N-диметилформамид (DMF) и осуществление реакции с получением соединения P-28-5; и
стадия 5: растворение P-28-5 в безводном тетрагидрофуране, затем медленное добавление по каплям борана/тетрагидрофурана (BH3/THF). После завершения добавления по каплям реакционную систему нагревали для осуществления реакции, затем реакционную систему охлаждали и медленно добавляли по каплям концентрированную хлористоводородную кислоту, а после завершения добавления по каплям реакционную систему нагревали с получением соединения P-28.
Другой целью настоящего изобретения является обеспечение применения соединения на основе 7-замещенного пирроло[2,1-f][1,2,4]триазина, представленного общей формулой I, или его фармацевтически приемлемой соли в получении активного ингибитора PI3K.
В одном варианте осуществления ингибитор PI3K характеризуется селективным ингибирующим эффектом в отношении PI3Kδ.
В одном варианте осуществления ингибитор PI3K применяется в лекарственном препарате для лечения заболеваний, связанных с сигнальным путем PI3K.
Предпочтительно заболевания, связанные с сигнальным путем PI3K, включают опухоли, виды лейкоза и аутоиммунные заболевания.
Кроме того, в настоящем изобретении обеспечивается фармацевтическая композиция, содержащая терапевтически эффективное количество соединения на основе 7-замещенного пирроло[2,1-f][1,2,4]триазина, представленного общей формулой I, или его фармацевтически приемлемой соли, при этом фармацевтическую композицию применяют для лечения заболеваний, связанных с сигнальным путем PI3K.
Авторы настоящего изобретения получили новый тип соединений на основе 7-замещенного пирроло[2,1-f][1,2,4]триазина в результате целесообразной разработки и целостного рассмотрения фармакокинетических факторов соединений. Такие соединения проявляют более высокую ингибирующую активность в отношении PI3K и эффективно ингибируют активность PI3K-киназы. Они характеризуются значительно улучшенными фармакокинетическими свойствами, такими как биодоступность, вследствие введения группы в 7-положение; кроме того, соединения по настоящему изобретению проявляют непредсказуемую высокую селективность и высокую ингибирующую активность в отношении PI3Kδ, и, таким образом, такие соединения можно применять для лечения заболеваний, связанных с сигнальным путем PI3K, особенно для борьбы с раком или для лечения опухолей, видов лейкоза и аутоиммунных заболеваний. Ожидается, что после дополнительной оптимизации и скрининга эти соединения превратят в новый тип противоопухолевых лекарственных средств.
ПОДРОБНОЕ ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
Термин “алкил”, описанный в данном документе, означает прямой алкил или разветвленный алкил.
Термин “заместитель” или т. п., описанный в данном документе, означает замещение одного или более атомов водорода. Галоген в данном документе может быть выбран из по меньшей мере одного из F, Cl, Br и I, предпочтительно по меньшей мере одного из F и Cl.
Настоящее изобретение будет дополнительно описано ниже со ссылкой на варианты осуществления, но эти варианты осуществления никоим образом не ограничивают настоящее изобретение. Во всех примерах 1H ЯМР регистрировали с помощью ядерно-магнитного резонансного спектрометра Brucher AM-400 и GEMINI-300, а химические сдвиги выражали в δ (ppm); масс-спектры регистрировали с помощью масс-спектрометра MAT-95; силикагель для разделения составлял 200-300 меш.
Стадия a. Получение этил-4-циано-5-метил-1H-пиррол-2-карбоксилата (Aa2')
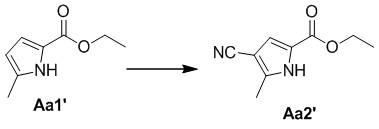
В раствор соединения Aa1' (1,0 г, 6,5 ммоль) и DMF (1,3 мл) в безводном ацетонитриле (20 мл) на ледяной бане добавляли по каплям хлорсульфонил-изоцианат (0,7 мл, 8,0 ммоль), затем смесь помещали в условия комнатной температуры и осуществляли реакцию в течение ночи. Реакционную смесь гасили насыщенным раствором карбоната натрия (20 мл), разбавляли водой, затем дважды экстрагировали этилацетатом (50 мл), органические слои объединяли, один раз промывали насыщенным солевым раствором (100 мл) и высушивали над безводным сульфатом натрия, а затем концентрировали при пониженном давлении с получением неочищенного продукта, который затем очищали с помощью хроматографической колонки (петролейный эфир/этилацетат: 3/1) с получением белого соединения (980 мг, 84%).
1H ЯМР (300 МГц, CDCl3) δ 10,07 (br s 1H), 7,03 (d, J = 2,53 Гц, 1H), 4,34 (q, J = 7,13 Гц, 2H), 2,47 (s, 3H), 1,37 (t, J = 7,13 Гц, 3H).
Стадия c. Получение этил-1-амино-4-циано-5-метил-1H-пиррол-2-карбоксилата (Aa3')
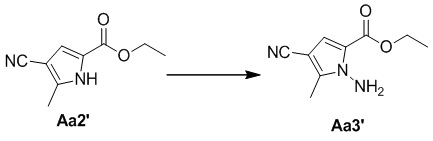
В раствор соединения Aa2' (200 мг, 1,1 ммоль), K2CO3 (840 мг, 6,0 ммоль), NH4Cl (385 мг, 7,2 ммоль), концентрированного водного раствора аммиака (1,2 мл) и хлорида метилтриоктиламмония (0,010 мл) в метил-трет-бутиловом простом эфире (50 мл) в ледяной солевой бане добавляли по каплям 5% (доля в процентах по массе) раствора гипохлорита натрия (12 мл) через капельную воронку с постоянным давлением, затем смесь помещали в условия комнатной температуры и осуществляли реакцию в течение 4 часов. Реакционную смесь гасили насыщенным раствором тиосульфата натрия, слой на основе метил-трет-бутилового простого эфира отделяли, а водный слой один раз экстрагировали этилацетатом (50 мл), затем органические слои объединяли, три раза промывали водой и высушивали над безводным сульфатом натрия, а затем концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали с помощью хроматографической колонки (петролейный эфир/этилацетат: 4/1) с получением бесцветной жидкости (183 мг, 86%), которая затем затвердевала после отстаивания.
1H ЯМР (300 МГц, CDCl3) δ 7,01 (s, 1H), 5,46 (s, 2H), 4,30 (q, J = 7,13 Гц, 2H), 2,42 (s, 3H), 1,35 (t, J = 7,13 Гц, 3H).
Стадия e. Получение 1-амино-4-циано-5-метил-1H-пиррол-2-карбоксамида (Aa4')
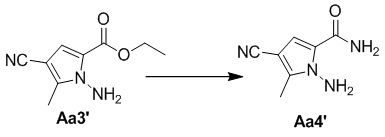
Соединение Aa3' (800 мг, 4,1 ммоль) растворяли в 100 мл насыщенного раствора аммиака в метаноле или раствора аммиака в метаноле, смесь подвергали реакции при 80°C в течение 2 дней в закрытой пробирке, затем охлаждали до комнатной температуры и концентрировали с получением желтого твердого вещества (680 мг, 100%).
1H ЯМР (300 МГц, DMSO-d6) δ 8,01 (br s, 1H), 7,39 br (s, 1H), 7,02 (s, 1H), 6,70 (br s, 2H), 2,27 (s, 3H).
Стадия b. Получение этил-4-циано-5-хлор-1H-пиррол-2-карбоксилата (Aa2'')
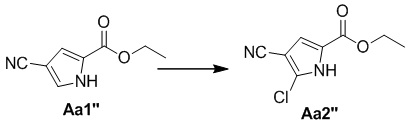
Соединение Aa1'' (2,0 г, 12,2 ммоль) растворяли в хлороформе и добавляли N-хлорсукцинимид (2,0 г, 15,0 ммоль), затем смесь перемешивали в течение ночи при комнатной температуре; смесь разбавляли водой (100 мл), органический слой отделяли и водный слой два раза экстрагировали хлороформом (100 мл), затем органические фазы объединяли, один раз промывали насыщенным солевым раствором (100 мл), высушивали над безводным сульфатом натрия и затем концентрировали при пониженном давлении с получением неочищенного продукта, который затем очищали с помощью хроматографической колонки (петролейный эфир/этилацетат: 20/1) с получением белого твердого вещества (510 мг, 22%).
1H ЯМР (300 МГц, CDCl3) δ 10,68 (s, 1H), 7,09 (d, J = 2,8 Гц, 1H), 4,39 (q, J = 7,1 Гц, 2H), 1,39 (t, J = 7,2 Гц, 3H).
Стадия d. Получение этил-1-амино-4-циано-5-хлор-1H-пиррол-2-карбоксилата (Aa3'')
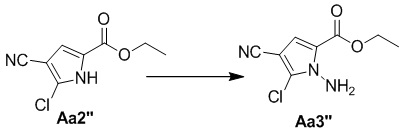
Соединение Aa2'' (300 мг, 1,5 ммоль) растворяли в безводном DMF (10 мл), затем добавляли NaH (60%, диспергированный в минеральном масле, 75 мг, 1,9 ммоль) на ледяной бане после продолжительного перемешивания смеси в течение 30 минут, добавляли O-(2,4-динитрофенил)гидроксиламин (360 мг, 1,8 ммоль) и смесь перемешивали при комнатной температуре в течение 6 часов. Затем реакционный раствор выливали в воду (50 мл), два раза экстрагировали этилацетатом (50 мл), органические слои объединяли, один раз промывали насыщенным солевым раствором (100 мл), высушивали над безводным сульфатом натрия, концентрировали и затем подвергали колоночной хроматографии (петролейный эфир/этилацетат: 10/1) с получением белого твердого вещества (305 мг, 95%).
1H ЯМР (300 МГц, CDCl3) δ 7,09 (s, 1H), 5,80 (s, 2H), 4,34 (q, J = 7,1 Гц, 2H), 1,37 (t, J = 7,1 Гц, 3H).
Стадия e. Получение 1-амино-4-циано-5-хлор-1H-пиррол-2-карбоксамида (Aa4'')
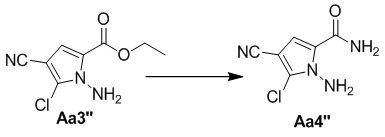
Применяя соединение Aa3'' (350 мг, 1,6 ммоль) в качестве исходного материала в соответствии со способом получения соединения Aa4', получали желтое твердое вещество (290 мг, 100%).
1H ЯМР (300 МГц, DMSO-d6) δ 8,12 (br s, 1H), 7,63 (br s, 1H), 7,19 (s, 1H), 6,86 (br s, 2H).
Стадия f. синтез соединения Aa5
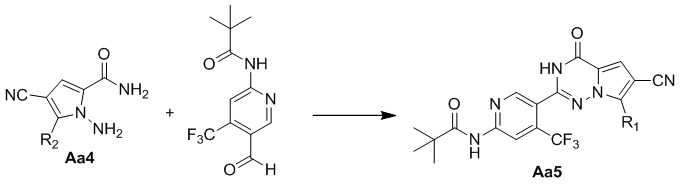
Раствор соединения Aa4 (1,0 экв.), N-(5-формил-4-(трифторметил)пиридил-2)-пиваламида (1,2 экв., полученный исследовательской группой) и безводного хлорида меди (1,0 экв.) в диметилсульфоксиде нагревали до 100°C для проведения реакции. После завершения реакции реакционную смесь охлаждали, выливали в воду и три раза экстрагировали этилацетатом. Органические слои объединяли, один раз промывали насыщенным солевым раствором и высушивали над безводным сульфатом натрия, концентрировали и затем очищали с помощью колоночной хроматографии (дихлорметан/метанол: 50/1) с получением желтого твердого вещества.
Соединение Aa4' (600 мг, 3,7 ммоль), соответствующий альдегид (1,2 г, 4,4 ммоль), безводный хлорид меди (490 мг, 3,7 ммоль), диметилсульфоксид (25 мл); с получением желтого твердого вещества (1,3 г, 85%).
1H ЯМР (300 МГц, CDCl3) δ 8,82 (s, 1H), 8,62 (s, 1H), 8,43 (br s, 1H), 7,27 (s, 1H), 2,60 (s, 3H), 1,38 (s, 9H).
Соединение Aa4'' (200 мг, 1,1 ммоль), соответствующий альдегид (362 мг, 1,3 ммоль), безводный хлорид меди (150 мг, 1,1 ммоль), диметилсульфоксид (15 мл); с получением желтого твердого вещества (408 мг, 85%).
1H ЯМР (300 МГц, CDCl3) δ 8,80 (s, 1H), 8,62 (s, 1H), 8,41 (s, 1H), 7,36 (s, 1H), 1,38 (s, 9H).
Стадии g и h: синтез соединения Aa7
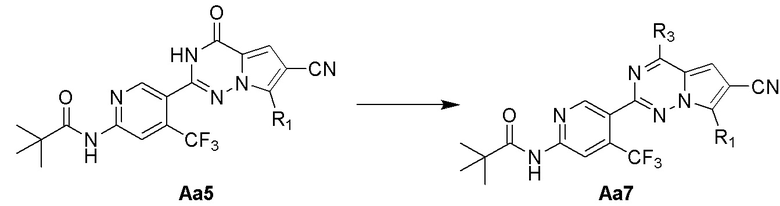
В смесь соединения Aa5 (1,0 экв.) и 4-диметиламинопиридина (2,0 экв.) добавляли оксихлорид фосфора и ее нагревали с обратным холодильником в течение 10 часов. После охлаждения смеси удаляли оксихлорид фосфора при пониженном давлении, затем последовательно добавляли безводный тетрагидрофуран и морфолин, а затем смесь нагревали с обратным холодильником в течение 2 часов. Тетрагидрофуран отгоняли при пониженном давлении, добавляли воду, затем смесь экстрагировали этилацетатом и один раз промывали по очереди насыщенным солевым раствором и водой, высушивали над безводным сульфатом натрия, концентрировали и затем очищали с помощью колоночной хроматографии (петролейный эфир/этилацетат: 4/1) с получением белого твердого вещества.
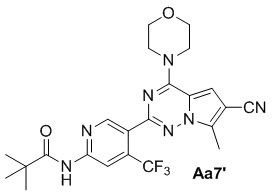
Соединение Aa5' (1,0 г, 2,4 ммоль), 4-диметиламинопиридин (583 мг, 4,8 ммоль), оксихлорид фосфора (20 мл), тетрагидрофуран (100 мл), морфолин (5 мл); с получением белого твердого вещества (980 мг, 84%).
1H ЯМР (300 МГц, CDCl3) δ 8,76 (s, 1H), 8,72 (s, 1H), 8,24 (br s, 1H), 6,99 (s, 1H), 4,06 (t, J = 4,77 Гц, 4H), 3,84 (t, J = 4,73 Гц, 4H), 2,65 (s, 3H), 1,36 (s, 9H).
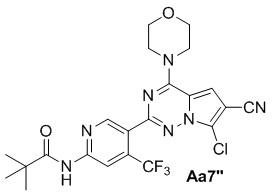
Соединение Aa5'' (360 мг, 0,8 ммоль), 4-диметиламинопиридин (200 мг, 1,6 ммоль), оксихлорид фосфора (5 мл), тетрагидрофуран (50 мл), морфолин (2 мл); с получением белого твердого вещества (370 мг, 88%).
1H ЯМР (300 МГц, CDCl3) δ 8,75 (s, 1H), 8,72 (s, 1H), 8,24 (s, 1H), 7,09 (s, 1H), 4,07 (t, J = 4,8 Гц, 4H), 3,86 (t, J = 4,8 Гц, 4H), 1,36 (s, 9H).
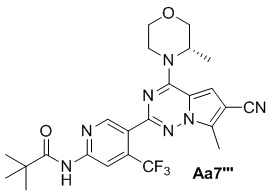
Соединение Aa5' (800 мг, 1,9 ммоль), 4-диметиламинопиридин (466 мг, 3,8 ммоль), оксихлорид фосфора (15 мг), тетрагидрофуран (100 мл), (S)-3-метилморфолин (5 мл); с получением желтого твердого вещества (720 мг, 76%).
Стадия i: синтез соединения Aa8
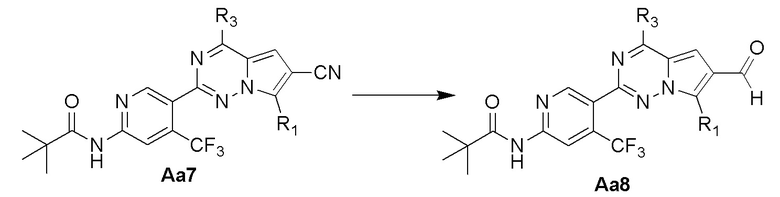
В суспензию соединения Aa7 (1,0 экв.) и моногидрата гипофосфита натрия (6,7 экв.) в уксусной кислоте/воде/пиридине (об./об./об.: 1/1/2) добавляли никель Ренея и смесь нагревали до 60°C для проведения реакции. Реакционную смесь охлаждали до комнатной температуры и фильтровали, экстрагировали этилацетатом, три раза промывали 3 н. хлористоводородной кислотой, концентрировали и затем очищали с помощью колоночной хроматографии (дихлорметан/метанол: 50/1) с получением желтого твердого вещества.
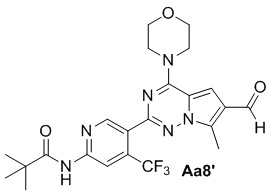
Соединение Aa7' (1,0 г, 1,0 ммоль), моногидрат гипофосфита натрия (710 мг, 6,7 ммоль), уксусная кислота/вода/пиридин (об./об./об.: 1/1/2) (50 мл), никель Ренея (750 мг), с получением желтого твердого вещества (232 мг, 47%).
1H ЯМР (300 МГц, CDCl3) δ 10,15 (s, 1H), 8,78 (br s, 1H), 8,72 (s, 1H), 8,25 (s, 1H), 7,17 (s, 1H), 4,10 (t, J = 4,8 Гц, 8H), 3,84 (t, J = 4,8 Гц, 8H), 2,79 (s, 3H), 1,36 (s, 9H).
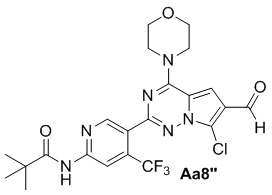
Соединение Aa7'' (300 мг, 0,6 ммоль), моногидрат гипофосфит натрия (426 мг, 4,0 ммоль), уксусная кислота/вода/пиридин (об./об./об.: 1/1/2) (25 мл), никель Ренея (450 мг), с получением желтого твердого вещества (130 мг, 42%).
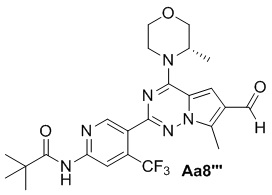
Соединение Aa7''' (600 мг, 1,2 ммоль), моногидрат гипофосфита натрия (852 мг, 8,0 ммоль), уксусная кислота/вода/пиридин (об./об./об.: 1/1/2) (50 мл), никель Ренея (900 мг), с получением желтого твердого вещества (420 мг, 83%).
Стадия j: синтез соединений P-(1-9), 12, (29-32), (34-35), (38-43), (45-46)
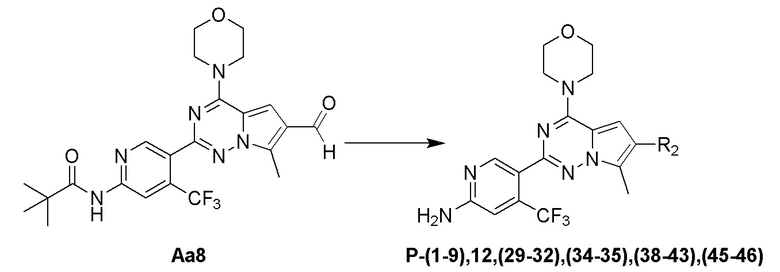
Суспендировали соединение Aa8 (0,2 ммоль), цианоборогидрид натрия (0,4 ммоль) и соответствующий амин (0,24 ммоль) в метаноле (20 мл), добавляли уксусную кислоту (0,05 мл) и затем смесь перемешивали при комнатной температуре. После завершения реакции смесь разбавляли водой (50 мл) и два раза экстрагировали этилацетатом (100 мл). Органические слои объединяли, один раз промывали насыщенным солевым раствором (100 мл) и высушивали над безводным сульфатом натрия, концентрировали и затем подвергали колоночной хроматографии (дихлорметан/метанол: 40/1) с получением продукта.
Полученный продукт растворяли в метаноле (15 мл), добавляли 10 эквивалентов 1 М раствора гидроксида калия и нагревали с обратным холодильником. После завершения реакции реакционную смесь концентрировали и очищали с помощью колоночной хроматографии (дихлорметан/метанол: 40/1).
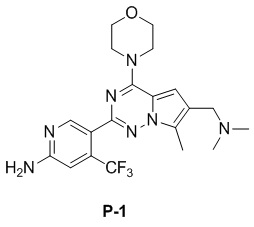
Желтое твердое вещество (38 мг, общий выход в две стадии: 44%). 1H ЯМР (300 МГц, CDCl3) δ 8,62 (s, 1H), 6,80 (s, 1H), 6,73 (s, 1H), 4,90 (br s, 2H), 4,03 (t, J = 4,5 Гц, 4H), 3,81 (t, J = 4,5 Гц, 4H), 3,50 (s, 2H), 2,48 (s, 3H), 2,28 (s, 6H).
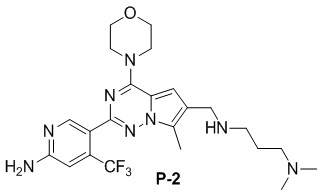
Желтое твердое вещество (44 мг, общий выход в две стадии: 45%). 1H ЯМР (300 МГц, CDCl3) δ 8,63 (s, 1H), 6,80 (s, 1H), 6,77 (s, 1H), 4,89 (br s, 2H), 4,04 (br s, 4H), 3,87 (s, 2H), 3,82 (br s, 4H), 2,77 (t, J = 3,3 Гц, 2H), 2,49 (s, 3H), 2,35 (t, J = 6,3 Гц, 2H), 2,23 (s, 6H), 1,80-1,64 (m, 2H).
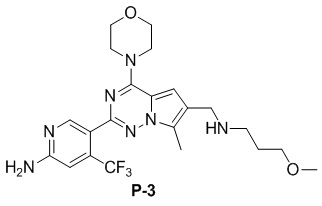
Желтое твердое вещество (49 мг, общий выход в две стадии: 52%). 1H ЯМР (300 МГц, CDCl3) δ 8,62 (s, 1H), 6,79 (s, 1H), 6,72 (s, 1H), 4,88 (br s, 2H), 4,04 (t, J = 3,9 Гц, 4H), 3,91-3,73 (m, 6H), 3,46 (t, J = 6,7 Гц, 2H), 3,32 (s, 3H), 2,77 (t, J = 6,8 Гц, 2H), 2,49 (s, 3H), 1,89-1,72 (m, 2H). 13C NMR (126 МГц, CDCl3) δ 158,83, 153,94, 151,95, 151,83, 138,11 (q, J = 32,4 Гц), 126,24, 122,95 (q, J = 274,3 Гц), 122,44, 121,65, 111,79, 105,21 (q, J = 5,5 Гц), 103,35, 71,40, 66,81, 58,71, 47,06, 45,94, 45,66, 29,90, 9,24.
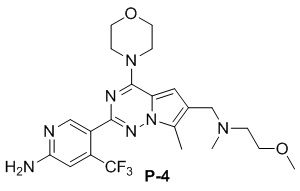
Желтое твердое вещество (38 мг, общий выход в две стадии: 38%). 1H ЯМР (400 МГц, CDCl3) δ 8,63 (s, 1H), 6,80 (s, 1H), 6,76 (s, 1H), 4,84 (br s, 2H), 4,03 (t, J = 4,9 Гц, 4H), 3,82 (t, J = 4,8 Гц, 4H), 3,63 (s, 2H), 3,53 (t, J = 5,6 Гц, 2H), 3,35 (s, 3H), 2,63 (t, J = 5,6 Гц, 2H), 2,48 (s, 3H), 2,33 (s, 3H). 13C ЯМР (101 МГц, CDCl3) δ 158,77, 153,94, 151,95, 151,77, 138,13 (q, J = 32,8 Гц), 127,07, 122,28 (q, J = 275,7 Гц), 121,72, 120,25, 111,93, 105,22 (q, J = 5,6 Гц), 104,45, 70,60, 66,81, 58,92, 56,06, 54,13, 45,91, 42,68, 9,36.
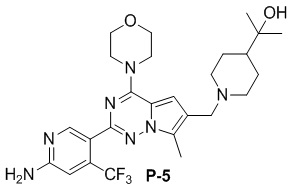
Желтое твердое вещество (68 мг, общий выход в две стадии: 64%). 1H ЯМР (500 МГц, CDCl3) δ 8,65 (s, 1H), 6,82 (s, 1H), 6,74 (s, 1H), 4,94-4,88 (m, 2H), 4,06 (t, J = 4,6 Гц, 4H), 3,84 (t, J = 4,8 Гц, 4H), 3,59 (s, 2H), 3,06 (d, J = 10,7 Гц, 2H), 2,51 (s, 3H), 1,97 (t, J = 11,3 Гц, 2H), 1,81-1,67 (m, 2H), 1,35-1,25 (m, 3H), 1,19 (s, 6H). 13C ЯМР (126 МГц, CDCl3) δ 158,79, 153,93, 151,95, 151,77, 138,12 (q, J = 32,3 Гц), 127,33, 122,95 (q, J = 274,5 Гц), 121,68, 120,06, 111,82, 105,23 (q, J = 6,2 Гц), 104,54, 72,47, 66,81, 54,58, 54,06, 47,40, 45,93, 27,00, 26,84, 9,48.
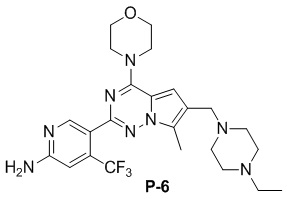
Желтое твердое вещество (58 мг, общий выход в две стадии: 58%). 1H ЯМР (300 МГц, CDCl3) δ 8,62 (s, 1H), 6,80 (s, 1H), 6,69 (s, 1H), 4,84 (br s, 2H), 4,03 (t, J = 4,8 Гц, 4H), 3,82 (t, J = 4,8 Гц, 4H), 3,57 (s, 2H), 2,68-2,45 (m, 8H), 2,41 (q, J = 7,2 Гц, 2H), 1,07 (t, J = 7,2 Гц, 3H). 13C ЯМР (126 МГц, CDCl3) δ 158,78, 153,94, 151,93, 151,79, 138,15 (q, J = 32,5 Гц, CF3C), 127,42, 122,94 (q, J = 274,4 Гц, CF3), 121,71, 119,89, 111,77, 105,23 (q, J = 5,5 Гц, CF3CCH), 104,57, 66,81, 54,42, 52,97, 52,83, 52,28, 45,94, 12,01, 9,45.
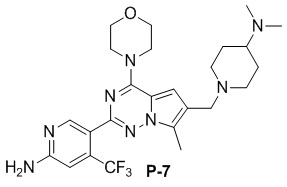
Желтое твердое вещество (55 мг, общий выход в две стадии: 53%). 1H ЯМР (500 МГц, CDCl3) δ 8,64 (s, 1H), 6,80 (s, 1H), 6,71 (s, 1H), 4,99 (br s, 2H), 4,04 (t, J = 4,8 Гц, 4H), 3,83 (t, J = 4,8 Гц, 4H), 3,56 (s, 2H), 3,09-2,86 (m, 2H), 2,50 (s, 3H), 2,29 (s, 6H), 2,21-2,07 (m, 1H), 2,05-1,93 (m, 2H), 1,87-1,75 (m, 2H), 1,65-1,44 (m, 2H). 13C ЯМР (126 МГц, CDCl3) δ 158,88, 153,92, 151,93, 151,77, 138,07 (q, J = 32,5 Гц), 127,26, 122,95 (q, J = 274,6 Гц), 121,58, 120,29, 111,78, 105,21 (q, J = 5,7 Гц), 104,45, 66,80, 62,32, 54,34, 52,93, 45,92, 41,72, 28,29, 9,45.
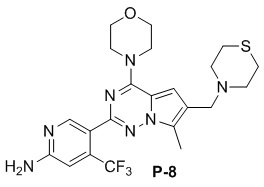
Желтое твердое вещество (46 мг, общий выход в две стадии: 47%). 1H ЯМР (400 МГц, CDCl3) δ 8,62 (s, 1H), 6,81 (s, 2H), 4,87 (br s, 2H), 4,05 (t, J = 4,8 Гц, 4H), 3,83 (t, J = 4,8 Гц, 4H), 3,70 (s, 2H), 2,85 (s, 4H), 2,78 (s, 4H), 2,49 (s, 3H). 13C ЯМР (126 МГц, CDCl3) δ 158,82, 153,96, 152,08, 151,89, 138,16 (q, J = 32,3 Гц), 127,58, 122,93 (q, J = 274,6 Гц), 121,55, 112,14, 105,29 (q, J = 5,4 Гц), 105,03, 66,80, 54,99, 54,49, 45,96, 27,36, 9,47.
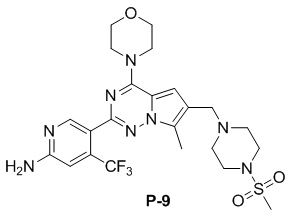
Желтое твердое вещество (79 мг, общий выход в две стадии: 72%). 1H ЯМР (300 МГц, CDCl3) δ 8,62 (s, 1H), 6,81 (s, 1H), 6,64 (s, 1H), 4,84 (br s, 2H), 4,03 (t, J = 4,5 Гц, 4H), 3,82 (t, J = 4,9 Гц, 4H), 3,60 (s, 2H), 3,24 (t, J = 5,0 Гц, 4H), 2,77 (s, 3H), 2,58 (t, J = 4,9 Гц, 4H), 2,49 (s, 3H). 13C ЯМР (126 МГц, CDCl3) δ 158,83, 153,95, 151,99, 151,95, 138,12 (q, J = 32,4 Гц, CF3C), 127,42, 122,95 (q, J = 274,4 Гц, CF3), 121,57, 119,23, 111,90, 105,25 (q, J = 5,5 Гц, CF3CCH), 104,48, 66,79, 54,26, 52,19, 45,95, 45,84, 34,28, 9,44.
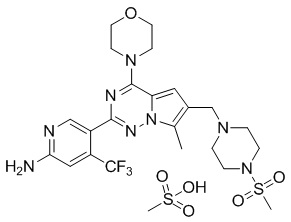
В раствор соединения P-9 (160 мг, 0,3 ммоль) в хлороформе (10 мл) добавляли метансульфоновую кислоту (370 мкл, 0,3 ммоль). После 2 часов прохождения реакции при комнатной температуре реакционную смесь разбавляли безводным простым эфиром (50 мл), фильтровали и высушивали с получением метансульфоната P-9 (145 мг, 77%) в виде желтого соединения.
1H ЯМР (500 МГц, DMSO-d6) δ 9,89 (br s, 1H), 8,51 (s, 1H), 7,25 (s, 1H), 7,23 (s, 1H), 4,47 (br s, 2H), 4,02 (t, J = 5,0 Гц, 4H), 3,76 (t, J = 4,9 Гц, 4H), 3,73 (s, 2H), 3,57-3,47 (m, 2H), 3,30-3,09 (m, 4H), 3,01 (s, 3H), 2,52 (br s, 2H), 2,43 (br s, 6H). 13C ЯМР (126 МГц, DMSO-d6) δ 157,47, 153,93, 151,45, 145,46, 138,57 (q, J = 33,3 Гц), 129,45, 122,77 (q, J = 275,06 Гц), 118,65, 112,36, 111,92, 110,13, 107,64, 66,42, 51,32, 50,27, 46,01, 42,96, 40,23, 35,51, 9,65.
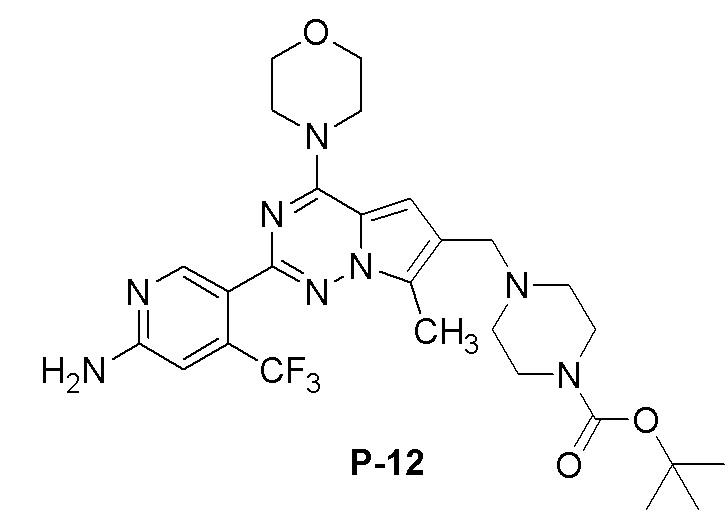
Желтое твердое вещество (63 мг, общий выход в две стадии: 53%). 1H ЯМР (400 МГц, CDCl3) δ 8,63 (s, 1H), 6,83 (s, 1H), 6,70 (s, 1H), 4,99 (br s, 2H), 4,24-3,98 (m, 4H), 3,96-3,78 (m, 4H), 3,60 (s, 2H), 3,46 (br s, 4H), 2,51 (s, 3H), 2,44 (br s, 4H), 1,47 (s, 9H). 13C ЯМР (126 МГц, CDCl3) δ 158,91, 154,77, 153,94, 151,85, 151,72, 138,18 (q, J = 32,6 Гц), 127,40, 122,93 (q, J = 274,1 Гц), 121,50, 119,51, 111,85, 105,39 (q, J = 5,6 Гц), 104,52, 79,65, 66,79, 54,45, 52,74, 45,93, 28,43, 9,45.
Желтое твердое вещество (45 мг, общий выход в две стадии: 43%). 1H ЯМР (500 МГц, хлороформ-d) δ 8,62 (s, 1H), 6,80 (s, 1H), 6,70 (s, 1H), 4,84 (d, J = 7,5 Гц, 2H), 4,06-4,00 (m, 4H), 3,95 (d, J = 11,4 Гц, 2H), 3,83-3,80 (m, 4H), 3,65 (s, 2H), 3,47 (t, J = 7,5 Гц, 2H), 3,36 (t, J = 11,7 Гц, 2H), 2,88 (t, J = 7,3 Гц, 2H), 2,32-2,16 (m, 2H), 1,71-1,57 (m, 2H), 1,52 (ddd, J = 13,2, 3,8, 1,9 Гц, 2H).
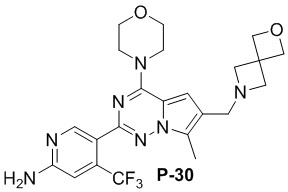
Желтое твердое вещество (40 мг, общий выход в две стадии: 41%). 1H ЯМР (500 МГц, хлороформ-d) δ 8,62 (s, 1H), 6,81 (s, 1H), 6,65 (s, 1H), 4,85 (d, J = 5,3 Гц, 2H), 4,75 (s, 4H), 4,05-4,00 (m, 4H), 3,82 (t, J = 4,9 Гц, 4H), 3,61 (s, 2H), 3,42 (s, 3H), 2,48 (s, 4H).
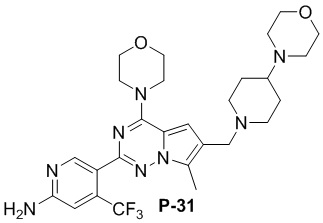
Желтое твердое вещество (36 мг, общий выход в две стадии: 32%). 1H ЯМР (300 МГц, хлороформ-d) δ 8,61 (s, 1H), 6,79 (s, 1H), 6,72 (s, 1H), 4,90 (d, J = 5,8 Гц, 2H), 4,02 (t, J = 4,7 Гц, 4H), 3,81 (t, J = 4,8 Гц, 5H), 3,73-3,68 (m, 4H), 3,57 (s, 2H), 3,00 (d, J = 11,1 Гц, 2H), 2,58-2,46 (m, 8H), 2,18 (dq, J = 11,2, 5,9, 4,2 Гц, 1H), 2,01 (t, J = 11,1 Гц, 2H), 1,82 (d, J = 13,4 Гц, 2H), 1,58 (td, J = 12,1, 3,6 Гц, 2H).
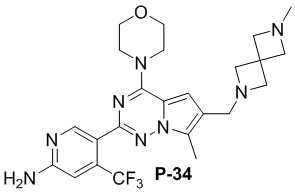
Соответствующий амин синтезировали с использованием ссылки на WO 2010138589 (A1). Желтое твердое вещество (20 мг, общий выход в две стадии: 18%). 1H ЯМР (400 МГц, хлороформ-d) δ 8,61 (s, 1H), 7,74-7,50 (m, 1H), 6,81 (s, 1H), 4,84 (s, 2H), 4,67-4,60 (m, 2H), 4,59-4,51 (m, 2H), 4,32-4,13 (m, 2H), 4,10-3,98 (m, 4H), 3,83 (t, J = 4,9 Гц, 4H), 2,92-2,74 (m, 1H), 2,50 (s, 3H), 2,21 (d, J = 7,7 Гц, 2H), 2,06-1,97 (m, 4H), 1,00-0,84 (m, 6H).
Желтое твердое вещество (52 мг, общий выход в две стадии: 52%). 1H ЯМР (300 МГц, хлороформ-d) δ 8,62 (s, 1H), 6,80 (s, 1H), 6,67 (d, J = 1,3 Гц, 1H), 4,84 (d, J = 4,0 Гц, 2H), 4,08-3,98 (m, 4H), 3,81 (t, J = 4,8 Гц, 4H), 3,62 (s, 2H), 3,37 (dd, J = 4,3, 1,5 Гц, 8H), 2,47 (d, J = 1,5 Гц, 3H), 2,32 (d, J = 1,5 Гц, 3H).
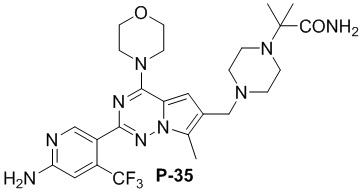
Соответствующий амин синтезировали с использованием ссылки на WO 2010138589 (A1). Желтое твердое вещество (75 мг, общий выход в две стадии: 67%). 1H ЯМР (400 МГц, хлороформ-d) δ 8,61 (s, 1H), 7,14 (d, J = 5,3 Гц, 1H), 6,80 (s, 1H), 6,68 (s, 1H), 5,54 (d, J = 5,3 Гц, 1H), 4,93 (s, 2H), 4,03 (dd, J = 5,7, 4,0 Гц, 4H), 3,82 (dd, J = 5,7, 4,0 Гц, 4H), 3,58 (s, 2H), 2,59-2,52 (m, 6H), 2,49 (s, 3H), 1,20 (s, 8H).
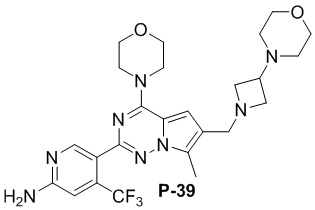
Желтое твердое вещество (49 мг, общий выход в две стадии: 47%). 1H ЯМР (400 МГц, хлороформ-d) δ 8,61 (s, 1H), 6,85 (s, 1H), 6,81 (s, 1H), 4,86 (s, 2H), 4,04 (t, J = 4,9 Гц, 4H), 3,82 (q, J = 4,8, 3,6 Гц, 8H), 3,58 (t, J = 5,3 Гц, 4H), 3,28-3,25 (m, 2H), 2,51 (s, 3H), 1,81 (t, J = 5,6 Гц, 4H).
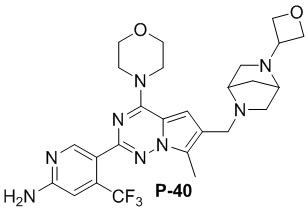
Соответствующий амин синтезировали с использованием ссылки на WO 2010138589 (A1). Желтое твердое вещество (47 мг, общий выход в две стадии: 44%). 1H ЯМР (400 МГц, хлороформ-d) δ 8,61 (s, 1H), 6,80 (s, 1H), 6,77 (s, 1H), 4,88 (s, 2H), 4,03 (dd, J = 5,7, 4,1 Гц, 4H), 3,82 (dd, J = 5,6, 4,0 Гц, 4H), 3,75 (d, J = 1,5 Гц, 2H), 3,47 (s, 2H), 3,42 (d, J = 12,1 Гц, 2H), 3,32 (d, J = 10,1 Гц, 1H), 3,20 (d, J = 10,2 Гц, 1H), 3,01 (s, 1H), 2,78 (dd, J = 9,2, 2,4 Гц, 1H), 2,66 (d, J = 2,4 Гц, 2H), 2,50 (s, 3H), 2,24-2,14 (m, 1H), 1,75 (d, J = 9,7 Гц, 1H), 1,51 (d, J = 9,7 Гц, 1H).
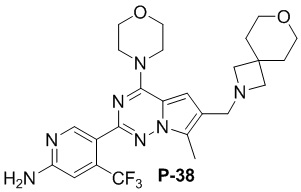
Желтое твердое вещество (56 мг, общий выход в две стадии: 51%). 1H ЯМР (400 МГц, хлороформ-d) δ 8,59 (s, 1H), 7,62 (s, 1H), 6,82 (s, 1H), 4,97 (s, 2H), 4,74 (t, J = 6,3 Гц, 1H), 4,67 (d, J = 6,4 Гц, 1H), 4,55-4,44 (m, 2H), 4,26-4,14 (m, 2H), 4,07 (dd, J = 5,7, 4,0 Гц, 4H), 4,03-3,87 (m, 2H), 3,86-3,80 (m, 4H), 3,49 (s, 1H), 3,35 (d, J = 11,7 Гц, 1H), 2,82-2,62 (m, 1H), 2,51 (s, 3H), 2,21 (t, J = 7,6 Гц, 1H), 2,04-1,97 (m, 2H), 0,90-0,82 (m, 1H).
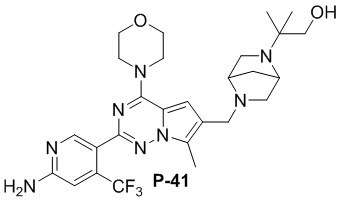
Соответствующий амин синтезировали с использованием ссылки на WO 2010138589 (A1). Желтое твердое вещество (53 мг, общий выход в две стадии: 47%). 1H ЯМР (400 МГц, хлороформ-d) δ 8,62 (s, 1H), 6,81 (s, 1H), 6,77 (s, 1H), 4,81 (s, 2H), 4,03 (dd, J = 5,7, 4,1 Гц, 4H), 3,83-3,81 (m, 4H), 3,72 (d, J = 4,7 Гц, 4H), 3,60 (s, 2H), 3,04 (s, 3H), 2,50 (s, 3H), 2,32 (d, J = 5,3 Гц, 4H), 1,70 (s, 6H).
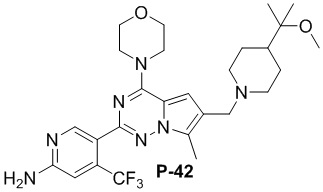
Соответствующий амин синтезировали с использованием ссылки на WO 2010138589 (A1). Желтое твердое вещество (52 мг, общий выход в две стадии: 48%). 1H ЯМР (400 МГц, хлороформ-d) δ 8,61 (d, J = 3,7 Гц, 1H), 6,81 (s, 1H), 6,71 (s, 1H), 4,88 (s, 2H), 4,03 (dq, J = 6,3, 2,8, 2,3 Гц, 4H), 3,85-3,79 (m, 4H), 3,59 (s, 2H), 3,34 (s, 2H), 2,67-2,42 (m, 15H), 1,04 (s, 6H).
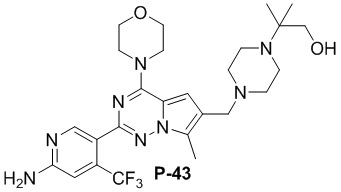
Соответствующий амин синтезировали с использованием ссылки на WO 2010138589 (A1). Желтое твердое вещество (52 мг, общий выход в две стадии: 48%). 1H ЯМР (400 МГц, хлороформ-d) δ 8,62 (s, 1H), 6,80 (s, 1H), 4,86 (s, 2H), 4,04 (dd, J = 5,7, 4,1 Гц, 4H), 3,84-3,79 (m, 4H), 3,62 (s, 2H), 3,16 (d, J = 1,4 Гц, 2H), 3,09-3,04 (m, 2H), 2,48 (d, J = 1,5 Гц, 3H), 2,02 (d, J = 13,2 Гц, 2H), 1,67 (d, J = 8,8 Гц, 2H), 1,45 (s, 2H), 1,08 (s, 6H).
Соответствующий амин синтезировали с использованием ссылки на WO 2013134226. Желтое твердое вещество (50 мг, общий выход в две стадии: 43%). LC-MS масса/заряд: [M+H]+ = 588,0.
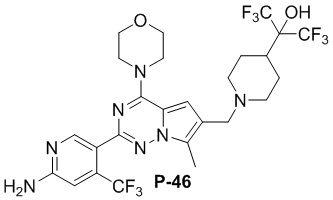
Соответствующий амин синтезировали с использованием ссылки на WO 2013134226. Желтое твердое вещество (61 мг, общий выход в две стадии: 48%). LC-MS масса/заряд: [M+H]+ = 641,9.
Стадия j: синтез соединений P-33, 36, 37
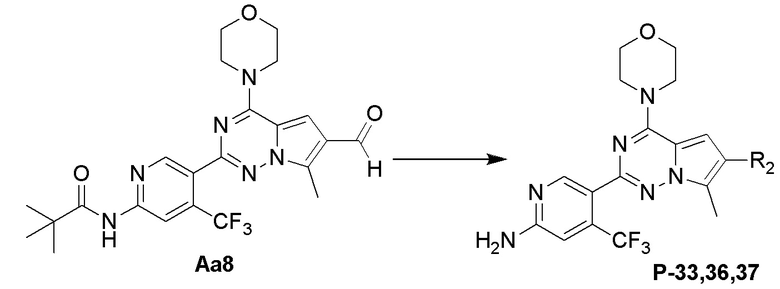
Суспендировали соединение Aa8 (0,2 ммоль), цианоборогидрид натрия (0,4 ммоль) и соответствующий Boc-защищенный амин (0,24 ммоль) в метаноле (20 мл), добавляли уксусную кислоту (0,05 мл) и затем смесь перемешивали при комнатной температуре. После завершения реакции реакционную смесь разбавляли водой (50 мл) и два раза экстрагировали этилацетатом (100 мл). Органические слои объединяли, один раз промывали насыщенным солевым раствором (100 мл) и высушивали над безводным сульфатом натрия, концентрировали и затем подвергали колоночной хроматографии (дихлорметан/метанол: 40/1) с получением продукта.
Продукт растворяли в дихлорметане (10 мл) и добавляли 10-кратный эквивалент трифторуксусной кислоты, затем смесь перемешивали при комнатной температуре в течение 2 дней и затем выпаривали непосредственно после завершения реакции.
Полученный продукт растворяли в метаноле (15 мл) и добавляли 10 эквивалентов 1 М раствора гидроксида калия, затем смесь нагревали с обратным холодильником. После завершения реакции реакционную смесь концентрировали и очищали с помощью колоночной хроматографии (дихлорметан/метанол: 40/1).
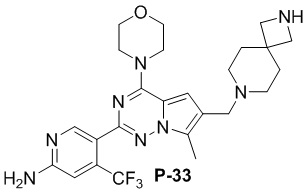
Желтое твердое вещество (52 мг, общий выход в две стадии: 50%). 1H ЯМР (400 МГц, хлороформ-d) δ 8,62 (s, 1H), 6,81 (s, 1H), 6,68 (d, J = 3,4 Гц, 1H), 4,83 (s, 2H), 4,03 (t, J = 4,8 Гц, 4H), 3,82 (t, J = 5,0 Гц, 4H), 3,49 (d, J = 3,3 Гц, 2H), 3,36 (s, 2H), 2,47 (d, J = 3,4 Гц, 3H), 2,34 (s, 2H), 1,93 (s, 4H), 1,79 (t, J = 5,3 Гц, 4H).
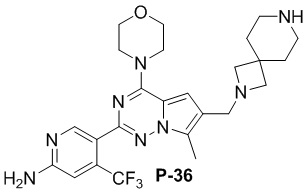
Желтое твердое вещество (50 мг, общий выход в две стадии: 48%). 1H ЯМР (400 МГц, хлороформ-d) δ 8,60 (s, 1H), 6,79 (s, 1H), 6,67 (s, 1H), 4,90 (s, 2H), 4,02 (dd, J = 5,7, 4,1 Гц, 4H), 3,84-3,77 (m, 4H), 3,67 (s, 2H), 3,05 (s, 4H), 2,77 (dd, J = 6,6, 4,0 Гц, 4H), 2,49 (s, 3H), 1,76-1,69 (m, 4H).
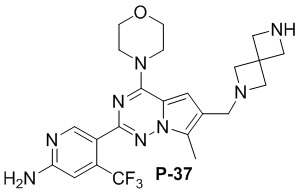
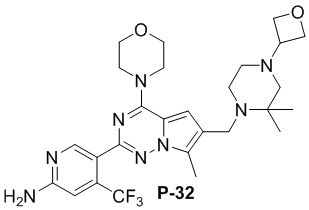
Желтое твердое вещество (45 мг, общий выход в две стадии: 46%). 1H ЯМР (400 МГц, хлороформ-d) δ 8,62 (d, J = 2,0 Гц, 1H), 6,81 (s, 1H), 6,65 (d, J = 3,8 Гц, 1H), 4,80 (s, 2H), 4,03 (q, J = 4,1, 3,2 Гц, 4H), 3,82 (t, J = 4,8 Гц, 4H), 3,74 (s, 2H), 3,60 (d, J = 2,5 Гц, 2H), 3,35 (s, 3H), 3,30 (d, J = 8,3 Гц, 2H), 2,51-2,46 (m, 4H).
Стадия j: синтез соединения P-44
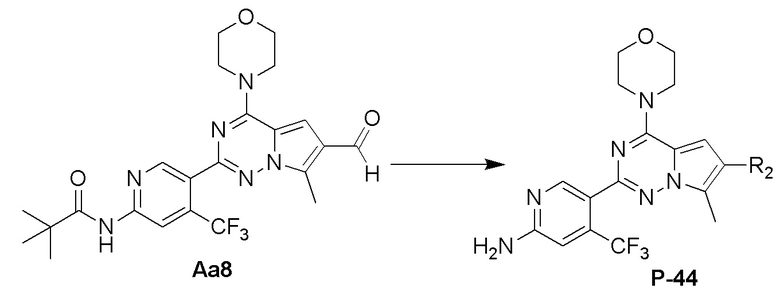
Суспендировали соединение Aa8 (0,2 ммоль), цианоборогидрид натрия (0,4 ммоль) и N-ацетил-2-(4-пиперидил)-2-пропиламин (0,24 ммоль) в метаноле (20 мл) и добавляли уксусную кислоту (0,05 мл), затем смесь перемешивали при комнатной температуре. После завершения реакции реакционную смесь разбавляли водой (50 мл) и два раза экстрагировали этилацетатом (100 мл). Органические слои объединяли, один раз промывали насыщенным солевым раствором (100 мл) и высушивали над безводным сульфатом натрия, концентрировали и затем подвергали колоночной хроматографии (дихлорметан/метанол: 40/1) с получением продукта.
Полученный продукт растворяли в концентрированной хлористоводородной кислоте (10 мл) и нагревали с обратным холодильником в течение 7 дней. После завершения реакции реакционную смесь нейтрализовали и очищали с помощью колоночной хроматографии (дихлорметан/метанол: 10/1).
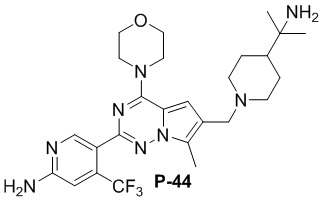
N-ацетил-2-(4-пиперидил)-2-пропиламин синтезировали с использованием ссылки на WO 2014043068. Желтое твердое вещество (12 мг, общий выход в две стадии: 11%). LC-MS масса/заряд: [M+H]+ = 533,0.
Стадия j: синтез соединения P-10
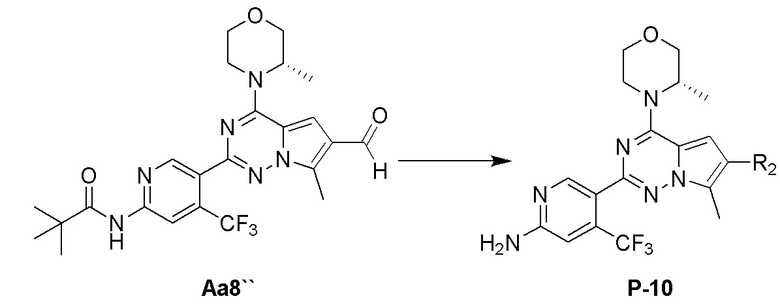
Суспендировали соединение Aa8`` (0,2 ммоль), цианоборогидрид натрия (0,4 ммоль) и соответствующий амин (0,24 ммоль) в метаноле (20 мл) и добавляли уксусную кислоту (0,05 мл), затем смесь перемешивали при комнатной температуре. После завершения реакции реакционную смесь разбавляли водой (50 мл) и два раза экстрагировали этилацетатом (100 мл). Органические слои объединяли, один раз промывали насыщенным солевым раствором (100 мл) и высушивали над безводным сульфатом натрия, концентрировали и затем подвергали колоночной хроматографии (дихлорметан/метанол: 40/1) с получением продукта.
Полученный продукт растворяли в метаноле (15 мл) и добавляли 10 эквивалентов 1 М раствора гидроксида калия, затем смесь нагревали с обратным холодильником. После завершения реакции реакционную смесь концентрировали и очищали с помощью колоночной хроматографии (дихлорметан/метанол: 40/1).
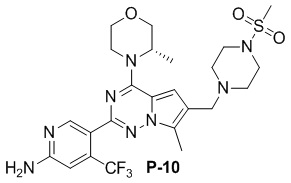
Желтое твердое вещество (55 мг, общий выход в две стадии: 48%). 1H ЯМР (300 МГц, хлороформ-d) δ 8,63 (d, J = 3,1 Гц, 1H), 6,82 (d, J = 3,1 Гц, 1H), 6,65 (s, 1H), 4,93 (br s, 1H), 4,83 (s, 2H), 4,56 (d, J = 11,3 Гц, 1H), 4,09-3,97 (m, 1H), 3,89-3,74 (m, 2H), 3,72-3,53 (m, 4H), 3,26 (s, 4H), 2,78 (s, 3H), 2,60 (s, 4H), 2,49 (s, 3H), 1,45 (d, J = 6,8 Гц, 3H). 13C ЯМР (126 МГц, хлороформ-d) δ 158,81, 153,66, 152,06, 151,91, 138,12 (q, J = 32,3 Гц), 127,41, 122,94 (d, J = 274,5 Гц), 121,65, 111,92, 105,26 (d, J = 6,7 Гц), 104,58, 71,03, 66,98, 54,26, 52,16, 45,83, 34,28, 15,27, 9,49.
Стадия j: синтез соединений P-11, (25-27)
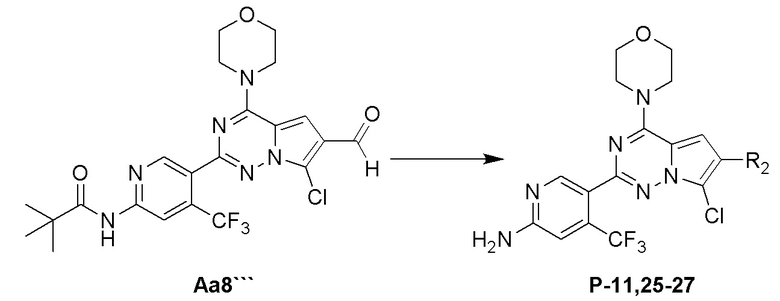
Суспендировали соединение Aa8``` (0,2 ммоль), цианоборогидрид натрия (0,4 ммоль) и соответствующий амин (0,24 ммоль) в метаноле (20 мл) и добавляли уксусную кислоту (0,05 мл), затем смесь перемешивали при комнатной температуре. После завершения реакции реакционную смесь разбавляли водой (50 мл) и два раза экстрагировали этилацетатом (100 мл). Органические слои объединяли, один раз промывали насыщенным солевым раствором (100 мл) и высушивали над безводным сульфатом натрия, концентрировали и затем подвергали колоночной хроматографии (дихлорметан/метанол: 40/1) с получением продукта.
Полученный продукт растворяли в метаноле (15 мл) и добавляли 10 эквивалентов 1 М раствора гидроксида калия, затем смесь нагревали с обратным холодильником. После завершения реакции реакционную смесь концентрировали и очищали с помощью колоночной хроматографии (дихлорметан/метанол: 40/1).
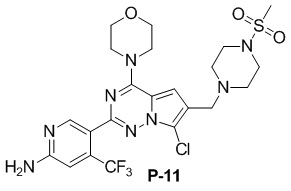
Желтое твердое вещество (34 мг, общий выход в две стадии: 30%). 1H ЯМР (300 МГц, CDCl3) δ 8,59 (s, 1H), 6,80 (s, 1H), 6,74 (s, 1H), 4,87 (br s, 2H), 4,04 (t, J = 4,6 Гц, 4H), 3,83 (t, J = 4,7 Гц, 4H), 3,67 (s, 2H), 3,27 (t, J = 4,9 Гц, 4H), 2,78 (s, 2H), 2,64 (t, J = 5,1 Гц, 4H). 13C ЯМР (126 МГц, CDCl3) δ 159,02, 153,50, 153,30, 151,82, 138,25 (q, J = 32,8 Гц), 122,85 (q, J = 274,3 Гц), 120,96, 118,53, 115,86, 112,74, 105,24 (q, J = 6,1 Гц), 104,61, 66,69, 53,04, 52,06, 46,00, 45,79, 34,38.
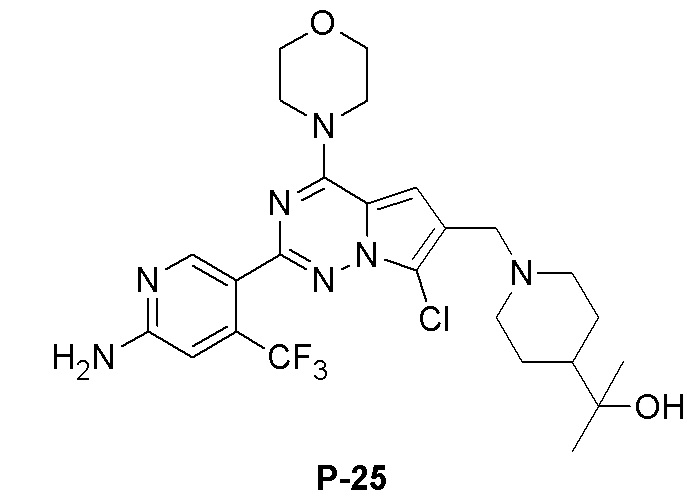
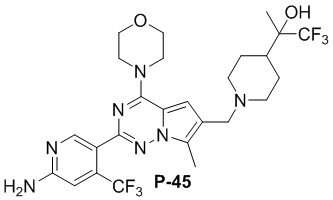
Белое твердое вещество (35 мг, выход: 45%). 1H ЯМР (400 МГц, CD3OD) δ 8,31 (s, 1H), 7,00 (s, 1H), 6,82 (s, 1H), 3,97 (m, 4H), 3,72 (m, 4H), 3,68 (s, 2H), 3,23 (s, 2H), 3,06 (d, 2H), 2,16 (t, 2H), 1,72 (d, 2H), 1,33 (m, 4H), 1,05 (s, 6H).
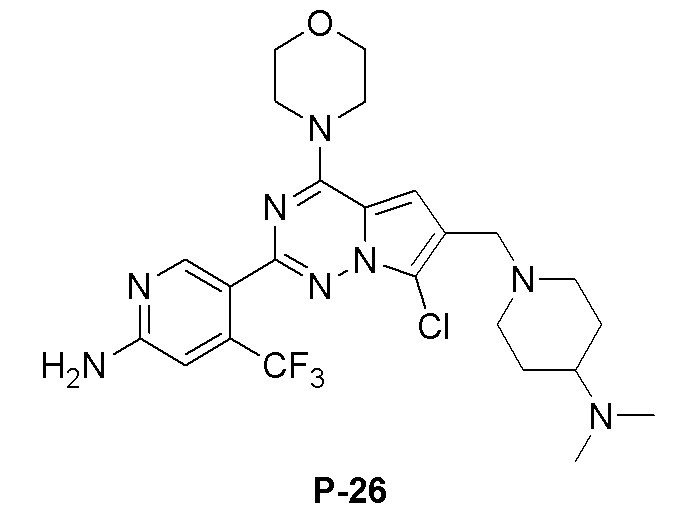
Белое твердое вещество (30 мг, выход: 40%). 1H ЯМР (400 МГц, CD3OD) δ 8,32 (s, 1H), 6,94 (s, 1H), 6,83 (s, 1H), 3,97 (m, 4H), 3,72 (m, 4H), 3,55 (br, s, 1H), 3,22 (d, 2H), 2,92 (d, 2H), 2,23 (s, 6H), 2,03 (d, 2H), 1,78 (d, 2H), 1,47 (d, 2H), 1,18 (d, 2H).
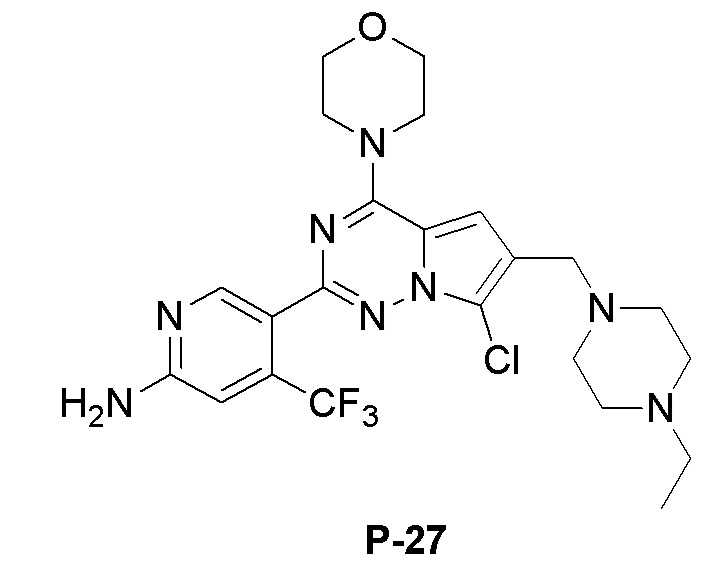
Белое твердое вещество (38 мг, выход: 42%). 1H ЯМР (400 МГц, CD3OD) δ 8,30 (s, 1H), 6,97 (s, 1H), 6,83 (s, 1H), 4,00 (m, 4H), 3,73 (m, 4H), 3,65 (s, 2H), 3,23 (d, 2H), 2,66 (m, 4H), 1,16 (t, 3H).
Синтез соединений P-13, 14
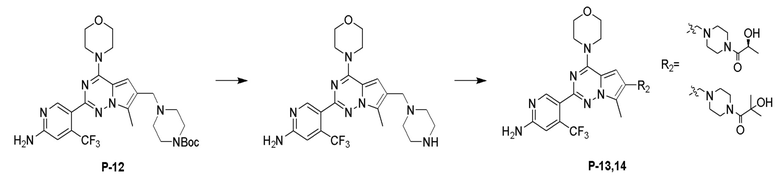
Соединение P-12 (500 мг) растворяли в 40 мл дихлорметана, добавляли 2,5 мл трифторуксусной кислоты и затем перемешивали при комнатной температуре в течение 2 часов. После завершения реакции растворитель удаляли при пониженном давлении с получением масла. Масло растворяли путем добавления 10 мл метанола и добавляли диэтиловый эфир (80 мл) для осаждения твердого вещества, которое фильтровали с получением 420 мг трифторацетатной соли Boc-незащищенного продукта.
Суспендировали полученную выше соль (подсчитанную как содержащую одну молекулу трифторуксусной кислоты, 114 мг, 0,2 ммоль), соответствующую кислоту (1,5 экв.), 1-этил-(3-диметиламинопропил)карбодиимида гидрохлорид (EDC·HCl) (1,5 экв.), 1-гидроксибензотриазол (HOBT) (1,5 экв.) и N,N-диизопропилэтиламин (DIPEA) (3 экв.) в DMF, затем проводили реакцию при комнатной температуре. После завершения реакции реакционную смесь разбавляли водой и три раза экстрагировали этилацетатом. Органические слои объединяли, высушивали над безводным сульфатом натрия, концентрировали и затем очищали с помощью колоночной хроматографии (дихлорметан/метанол: 20/1) с получением продукта.
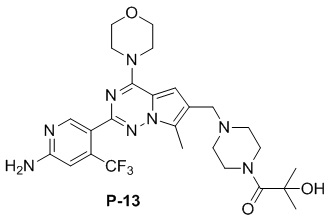
Желтое твердое вещество (67 мг, 60%). 1H ЯМР (500 МГц, CDCl3) δ 8,65 (s, 1H), 6,84 (s, 1H), 6,68 (s, 1H), 4,91 (s, 2H), 4,53 (br s, 1H), 4,06 (t, J = 4,8 Гц, 4H), 3,85 (t, J = 4,8 Гц, 4H), 3,79-3,65 (m, 4H), 3,60 (s, 2H), 2,51 (s, 7H), 1,50 (s, 6H). 13C ЯМР (126 МГц, CDCl3) δ 174,93, 158,85, 153,94, 151,94, 151,92, 138,12 (q, J = 32,6 Гц), 127,36, 122,90 (q, J = 274,06 Гц), 121,55, 119,38, 111,87, 105,27 (q, J = 6,3 Гц), 104,42, 71,53, 66,79, 54,40, 52,96, 45,94, 27,83, 9,44.
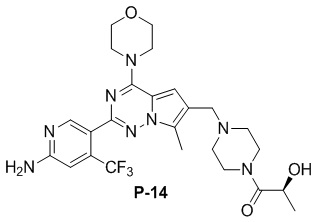
Желтое твердое вещество (55 мг, 50%). 1H ЯМР (500 МГц, CDCl3) δ 8,66 (s, 1H), 6,84 (s, 1H), 6,68 (s, 1H), 4,87 (s, 2H), 4,47 (t, J = 6,7 Гц, 1H), 4,07 (t, J = 4,8 Гц, 4H), 3,90 (d, J = 7,3 Гц, 1H), 3,86 (t, J = 4,8 Гц, 4H), 3,82-3,71 (m, 1H), 3,69-3,62 (m, 1H), 3,61 (s, 1H), 3,43 (d, J = 5,2 Гц, 2H), 2,58-2,35 (m, 7H), 1,34 (d, J = 6,6 Гц, 3H). 13C ЯМР (126 МГц, CDCl3) δ 173,50, 158,83, 153,95, 151,97, 138,26, 138,13 (d, J = 32,8 Гц), 127,37, 122,95 (q, J = 274,6 Гц), 121,59, 119,28, 111,89, 105,27, 104,41, 66,79, 64,02, 54,42, 52,70, 52,56, 45,94, 44,87, 42,53, 21,50, 9,44.
Синтез соединения P-15
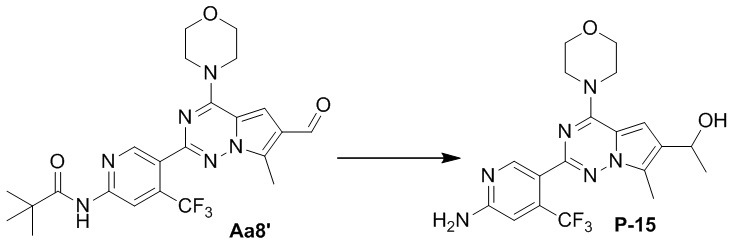
В раствор соединения Aa8' (600 мг, 1,2 ммоль) в безводном тетрагидрофуране (40 мл) добавляли 1 М бромид метилмагния (4,8 мл, 4,8 ммоль) на ледяной бане в защитной атмосфере азота и смесь помещали в условия комнатной температуры для проведения реакции. После завершения реакции реакционную смесь разбавляли с помощью 50 мл воды и два раза экстрагировали этилацетатом (100 мл). Органические слои объединяли, один раз промывали насыщенным солевым раствором (100 мл) и высушивали над безводным сульфатом натрия, концентрировали и затем подвергали колоночной хроматографии (дихлорметан/метанол: 40/1) с получением желтого соединения (520 мг, 86%).
Полученный выше продукт (400 мг, 0,8 ммоль) растворяли в 15 мл метанола и добавляли 10 эквивалентов 1 М раствора гидроксида калия (8 мл), затем смесь вводили в реакцию при нагревании с обратным холодильником. После завершения реакции реакционную смесь концентрировали и очищали с помощью колоночной хроматографии (дихлорметан/метанол=40/1) с получением желтого соединения (270 мг, 80%).
1H ЯМР (300 МГц, CDCl3). δ 8,59 (s, 1H), 6,80 (s, 1H), 6,75 (s, 1H), 5,14 (q, J = 4,8 Гц, 1H), 4,82 (s, 2H), 4,04 (t, J = 4,8 Гц, 4H), 3,82 (t, J = 4,8 Гц, 4H), 2,53 (s, 3H), 1,99 (br s, 1H), 1,58 (d, J = 6,4 Гц, 3H). 13C ЯМР (126 МГц, CDCl3) δ 158,78, 154,14, 152,05, 151,93, 128,13, 125,24, 111,85, 105,25 (q, J = 5,5 Гц), 100,27, 66,80, 63,82, 45,97, 24,52, 9,38.
Синтез соединения P-16
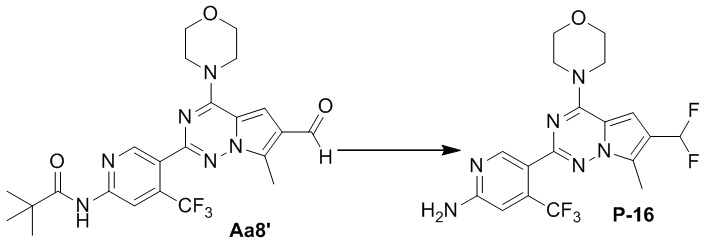
В раствор соединения Aa8' (100 мг, 0,2 ммоль) в безводном дихлорметане (20 мл) добавляли 1 мл трифторида диэтиламиносеры (DAST) на ледяной бане в защитной атмосфере азота и смесь помещали в условия комнатной температуры для проведения реакции. После завершения реакции медленно добавляли 10 мл ледяной воды для гашения, затем реакционную смесь разбавляли водой (50 мл) и два раза экстрагировали дихлорметаном (50 мл). Органические слои объединяли, один раз промывали насыщенным солевым раствором (100 мл) и высушивали над безводным сульфатом натрия, концентрировали и затем применяли непосредственно в следующей реакции.
Полученный выше продукт растворяли в 15 мл метанола и добавляли 10 эквивалентов 1 М раствора гидроксида калия (2 мл), затем смесь нагревали с обратным холодильником. После завершения реакции реакционную смесь концентрировали и очищали с помощью колоночной хроматографии (дихлорметан/метанол=40/1) с получением желтого соединения (38 мг, общий выход в две стадии: 44%).
1H ЯМР (300 МГц, CDCl3). δ 8,63 (s, 1H), 6,86 (t, J = 56,4 Гц, 1H), 6,85 (s, 1H), 6,82 (s, 1H), 4,85 (br s, 2H), 4,06 (t, J = 4,9 Гц, 4H), 3,84 (t, J = 4,8 Гц, 4H), 2,58 (s, 3H). 13C ЯМР (101 МГц, CDCl3) δ 158,98, 154,52, 152,94, 151,96, 138,17 (q, J = 32,6 Гц), 127,09 (t, J = 6,3 Гц), 122,88 (q, J = 274,3 Гц), 121,13, 117,39 (t, J = 25,8 Гц), 112,40 (t, J = 233,7 Гц), 112,24, 105,29 (q, J = 6,1 Гц), 101,21 (t, J = 5,0 Гц), 66,71, 45,96, 9,46.
Синтез соединений P-17, 18
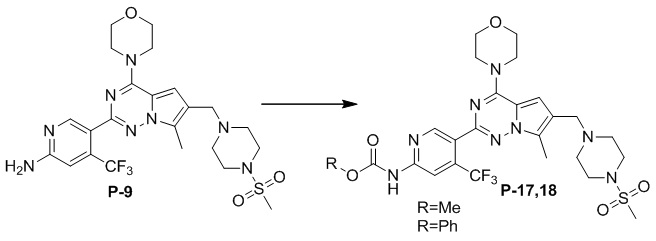
В раствор соединения P-9 (110 мг, 0,2 ммоль) в безводном хлороформе (10 мл) добавляли безводный триэтиламин (30 мкл, 0,25 ммоль) при −30°C и добавляли по каплям раствор соответствующего хлорформиата (0,6 ммоль) в хлороформе (5 мл), затем реакцию поддерживали при данной температуре. После завершения реакции медленно добавляли 10 мл ледяной воды для гашения и добавляли дихлорметан (40 мл) для разбавления. Органический слой отделяли, один раз промывали насыщенным солевым раствором (50 мл) и высушивали над безводным сульфатом натрия, концентрировали и очищали с помощью колоночной хроматографии (дихлорметан/метанол: 50/1) с получением желтого твердого вещества.
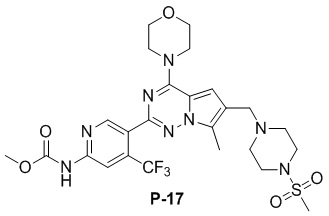
Желтое твердое вещество (55 мг, 45%). 1H ЯМР (400 МГц, CDCl3) δ 9,18 (br s, 1H), 8,84 (s, 1H), 8,45 (s, 1H), 6,68 (s, 1H), 4,05 (t, J = 4,5 Гц, 4H), 3,86 (s, 3H), 3,84-3,78 (m, 4H), 3,62 (s, 2H), 3,25 (s, 4H), 2,78 (s, 3H), 2,59 (s, 4H), 2,50 (s, 3H). 13C ЯМР (101 МГц, CDCl3) δ 153,87, 153,54, 152,67, 151,39, 150,80, 138,69 (q, J = 32,9 Гц, CF3C), 127,50, 126,60, 122,72 (q, J = 275,83 Гц, CF3), 119,55, 111,79, 109,21 (q, J = 5,2 Гц, CF3CCH), 104,69, 66,70, 54,19, 52,72, 52,18, 45,89, 45,84, 34,22, 9,40.
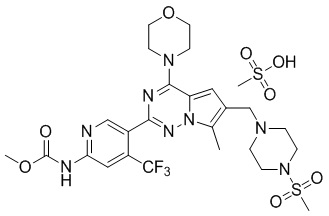
Сделана ссылка на способ получения метансульфоната соединения P-9. С применением соединения P-17 (200 мг, 0,3 ммоль) в качестве исходного материала получали метансульфонат соединения P-17 (190 мг, 81%) в виде желтого соединения.
1H ЯМР (400 МГц, DMSO-d6) δ 10,91 (s, 1H), 9,83 (br s, 1H), 8,79 (s, 1H), 8,31 (s, 1H), 7,23 (s, 1H), 4,46 (s, 2H), 4,01 (t, J = 4,8 Гц, 4H), 3,82-3,68 (m, 7H), 3,62-3,41 (m, 4H), 3,28-3,04 (m, 4H), 3,00 (s, 3H), 2,50 (s, 3H), 2,38 (s, 3H). 13C ЯМР (126 МГц, DMSO-d6) δ 154,58, 154,11, 153,99, 152,04, 151,38, 136,87 (q, J = 31,6 Гц, CF3C), 129,53, 125,47, 123,08 (q, J = 274,9 Гц, CF3), 112,40, 112,00, 108,63 (q, J = 6,9 Гц, CF3CCH), 107,63, 66,41, 52,76, 51,36, 50,31, 46,02, 42,98, 40,23, 35,54, 9,69.
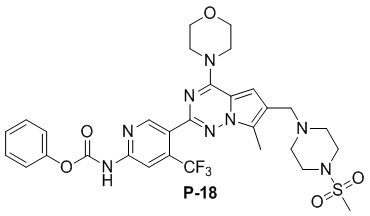
Желтое твердое вещество (55 мг, 74%). 1H ЯМР (300 МГц, CDCl3) δ 9,69 (s, 1H), 8,96 (s, 1H), 8,51 (s, 1H), 7,41 (t, J = 7,2 Гц, 2H), 7,36-7,19 (m, 3H), 6,67 (s, 1H), 4,13-3,91 (m, 4H), 3,79 (s, 4H), 3,61 (s, 2H), 3,25 (s, 4H), 2,78 (s, 3H), 2,58 (s, 4H), 2,41 (s, 3H).
Синтез соединения P-19
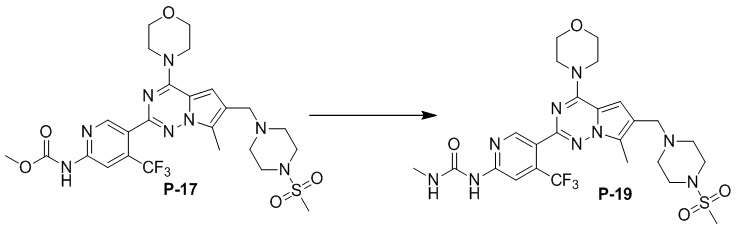
В пробирку для микроволновой обработки объемом 10 мл добавляли соединение P-17 (122 мг, 0,2 ммоль), безводный хлорид кальция (10 мг, 0,09 ммоль) и раствор метиламина в метаноле (2 мл), проводили реакцию при 100°C в течение 30 минут при микроволновом излучении, концентрировали и очищали с помощью тонкослойной хроматографии (дихлорметан/метанол: 20/1) с получением белого твердого вещества (30 мг, 33%).
1H ЯМР (400 МГц, CDCl3). δ 9,37 (s, 1H), 9,06 (br s, 1H), 8,74 (s, 1H), 7,28 (s, 1H), 6,68 (s, 1H), 4,05 (t, J = 4,6 Гц, 4H), 3,84 (t, J = 4,9 Гц, 4H), 3,61 (s, 2H), 3,25 (br s, 4H), 3,01 (d, J = 4,6 Гц, 3H), 2,78 (s, 3H), 2,59 (br s, 4H), 2,50 (s, 3H). 13C ЯМР (126 МГц, CDCl3) δ 156,42, 153,99 (q, J = 13,5 Гц), 151,34, 149,71, 138,66, 138,39, 127,59, 124,36, 122,56 (q, J = 274,93 Гц), 119,56, 111,84, 109,33 (q, J = 5,4 Гц), 104,70, 66,78, 54,26, 52,23, 45,95, 45,90, 34,27, 26,54, 9,45.
Синтез соединения P-20
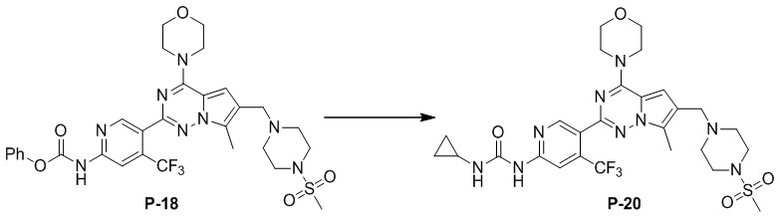
В раствору соединения P-18 (75 мг, 0,1 ммоль) в DMF (3 мл) добавляли циклопропиламин (1 мл) и смесь нагревали до 60°C. После завершения реакции реакционную смесь разбавляли водой (30 мл) и два раза экстрагировали этилацетатом (50 мл). Органические слои объединяли, один раз промывали насыщенным солевым раствором (100 мл) и высушивали над безводным сульфатом натрия, концентрировали и затем подвергали тонкослойной хроматографии (дихлорметан/метанол: 40/1) с получением желтого соединения (26 мг, 40%).
1H ЯМР (400 МГц, CDCl3). δ 9,47 (br s, 1H), 9,09 (br s, 1H), 8,73 (s, 1H), 7,40 (s, 1H), 6,67 (s, 1H), 4,05 (t, J = 4,7 Гц, 4H), 3,84 (t, J = 4,8 Гц, 4H), 3,61 (s, 2H), 3,25 (t, J = 4,7 Гц, 4H), 2,78 (br s, 4H), 2,59 (s, 4H), 2,49 (s, 3H), 0,95-0,78 (m, 2H), 0,71-0,61 (m, 2H). 13C ЯМР (126 МГц, CDCl3) δ 156,70, 153,93, 153,85, 151,32, 149,81, 138,53 (q, J = 33,2 Гц), 127,59, 124,65, 122,57 (q, J = 274,7 Гц), 119,57, 111,83, 109,48 (q, J = 6,6 Гц), 104,69, 66,77, 54,26, 52,23, 45,95, 45,90, 34,27, 22,58, 9,45, 6,80.
Синтез соединений P-(21-24)
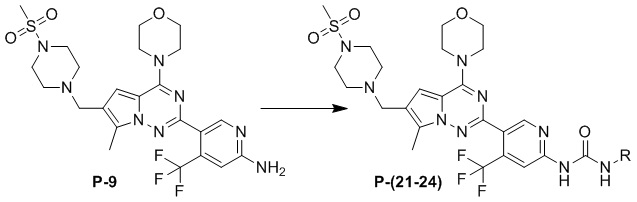
В раствор соединения P-9 (1 экв.) и 1,8-диазацикло[5.4.0]ундец-7-ена (DBU, 6 экв.) в дихлорметане добавляли соответствующий изоцианат (6 экв.) и проводили реакцию при комнатной температуре. После завершения реакции реакционную смесь разбавляли водой, один раз экстрагировали дихлорметаном, один раз промывали насыщенным солевым раствором и высушивали над безводным сульфатом натрия, концентрировали и затем очищали с помощью колоночной хроматографии (дихлорметан/метанол: 50/1) с получением желтого твердого вещества.
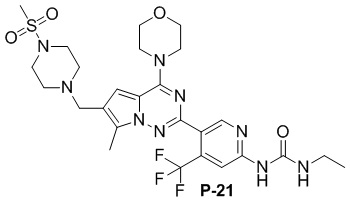
С применением соединения P-9 (160 мг, 0,3 ммоль) в качестве исходного материала получали желтое твердое вещество (54 мг, 29%). 1H ЯМР (300 МГц, DMSO-d6) δ 9,62 (s, 1H), 8,71 (s, 1H), 8,08 (s, 1H), 7,45 (d, J = 5,7 Гц, 1H), 6,92 (s, 1H), 3,96 (t, J = 4,8 Гц, 4H), 3,73 (t, J = 4,8 Гц, 4H), 3,56 (s, 2H), 3,18 (t, J = 6,8 Гц, 2H), 3,08 (s, 4H), 2,85 (s, 3H), 2,48 (s, 4H), 2,42 (s, 3H), 1,09 (t, J = 7,1 Гц, 3H). 13C ЯМР (126 МГц, DMSO-d6) δ 154,81, 154,58, 153,78, 151,31, 150,88, 136,71 (q, J = 31,8 Гц), 127,20, 124,08, 123,23 (q, J = 274,81 Гц), 120,27, 111,34, 108,29 (q, J = 6,2 Гц), 106,21, 66,46, 53,80, 52,14, 45,94, 34,44, 34,06, 15,69, 9,67.
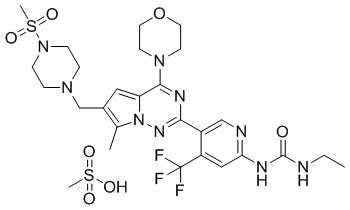
Сделана ссылка на способ получения метансульфоната соединения P-9. С применением соединения P-21 (75 мг, 0,31 ммоль) в качестве исходного материала получали метансульфонат соединения P-21 (70 мг, 81%) в качестве желтого соединения.
1H ЯМР (300 МГц, DMSO-d6) δ 9,80 (br s, 1H), 9,65 (s, 1H), 8,72 (s, 1H), 8,11 (s, 1H), 7,44 (br s, 1H), 7,20 (s, 1H), 4,45 (s, 2H), 4,13 (s, 2H), 4,01 (t, J = 4,9 Гц, 4H), 3,75 (s, 6H), 3,49 (s, 2H), 3,27-3,03 (m, 7H), 3,00 (s, 3H), 2,37 (s, 3H), 1,09 (t, J = 7,0 Гц, 3H). 13C ЯМР (126 МГц, DMSO-d6) δ 155,00, 154,56, 153,98, 152,13, 150,91, 136,74 (q, J = 31,4 Гц), 129,46, 123,69, 123,29 (q, J = 274,3 Гц), 112,39, 111,93, 108,36 (q, J = 6,4 Гц), 107,53, 66,41, 51,36, 40,24, 50,30, 46,00, 42,99, 35,51, 34,45, 15,68, 9,69.
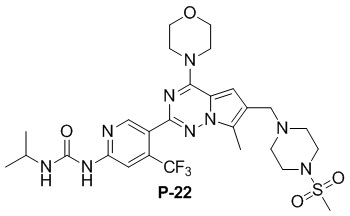
С применением соединения P-9 (160 мг, 0,3 ммоль) в качестве исходного материала получали желтое твердое вещество (128 мг, 67%). 1H ЯМР (300 МГц, CDCl3) δ 9,45 (s, 1H), 8,93 (br s, 1H), 8,74 (s, 1H), 7,30 (s, 1H), 6,67 (s, 1H), 4,10-4,16 (m, 1H), 4,05 (t, J = 4,4 Гц, 4H), 3,84 (t, J = 4,3 Гц, 4H), 3,61 (s, 2H), 3,25 (t, J = 4,5 Гц, 4H), 2,78 (s, 3H), 2,59 (t, J = 4,7 Гц, 4H), 2,50 (s, 3H), 1,30 (d, J = 6,4 Гц, 6H). 13C ЯМР (126 МГц, CDCl3) δ 155,16, 154,33, 153,93, 151,41, 149,67, 138,39 (q, J = 32,9 Гц), 127,55, 124,19, 122,61 (q, J = 274,4 Гц), 119,53, 111,83, 109,53 (q, J = 5,6 Гц), 104,66, 66,78, 54,26, 52,23, 45,95, 45,91, 42,21, 34,25, 23,08, 9,45.
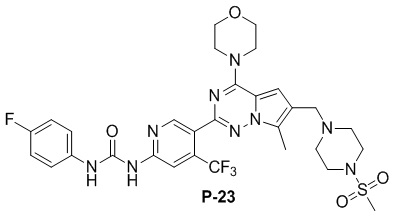
С применением соединения P-9 (110 мг, 0,2 ммоль) в качестве исходного материала получали желтое твердое вещество (40 мг, 29%). 1H ЯМР (300 МГц, CDCl3) δ 11,45 (s, 1H), 9,44 (s, 1H), 8,85 (s, 1H), 7,60 (dd, J = 8,9, 4,7 Гц, 2H), 7,35 (s, 1H), 7,07 (t, J = 8,2 Гц, 2H), 6,69 (s, 1H), 4,07 (br s, 4H), 3,86 (t, J = 5,1 Гц, 4H), 3,63 (s, 2H), 3,26 (s, 4H), 2,79 (s, 3H), 2,60 (s, 4H), 2,52 (s, 3H). 13C ЯМР (126 МГц, CDCl3) δ 159,29 (d, J = 242,9 Гц), 153,74 (d, J = 54,9 Гц), 153,50, 151,09, 149,67, 138,93 (q, J = 32,9 Гц), 133,94 (q, J = 2,9 Гц), 127,67, 125,13, 122,05 (q, J = 7,8 Гц), 119,69, 115,66 (q, J = 22,6 Гц), 111,82, 109,78, 104,83, 66,78, 54,27, 52,25, 45,96, 45,91, 34,28, 9,47.
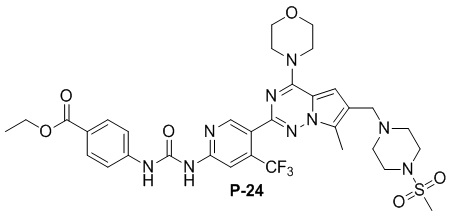
С применением соединения P-9 (110 мг, 0,2 ммоль) в качестве исходного материала получали желтое твердое вещество (46 мг, 31%). 1H ЯМР (300 МГц, DMSO-d6) δ 10,04 (s, 1H), 9,92 (s, 1H), 8,82 (s, 1H), 8,22 (s, 1H), 7,91 (d, J = 8,7 Гц, 2H), 7,65 (d, J = 8,8 Гц, 2H), 6,94 (s, 1H), 4,27 (q, J = 6,9 Гц, 2H), 3,97 (t, J = 4,9 Гц, 4H), 3,82-3,66 (m, 4H), 3,57 (s, 2H), 3,08 (s, 4H), 2,85 (s, 3H), 2,48-2,43 (m, 7H), 1,30 (t, J = 7,0 Гц, 3H). 13C ЯМР (126 МГц, DMSO-d6) δ 165,79, 153,90, 153,79, 152,07, 151,17, 143,65, 136,95 (q, J = 32,2 Гц), 130,89, 127,27, 125,30, 123,23 (q, J = 274,93 Гц), 124,16, 120,35, 118,45, 111,35, 108,74, 106,31, 66,47, 60,89, 53,80, 52,14, 45,94, 34,07, 14,71, 9,68.
Синтез соединения P-28
Стадия 1. Растворяли P-28-1 (437,0 мг, 1,0 ммоль способ его получения см. в CN 201210177980.3) в смеси дихлорметан:вода (10:1, 20 мл) и затем добавляли трифторметилсульфинат натрия (936,0 мг, 6 ммоль), вышеуказанную систему охлаждали до 0°C и медленно добавляли по каплям 70% водный раствор трет-бутилгидропероксида (1,4 мл, 10,0 ммоль). После завершения добавления по каплям продолжали перемешивание в течение 30 мин.; затем добавляли диметилсульфоксид (4,0 мл) и нагревали до 40°C с проведением реакции в течение ночи. Определяли завершение реакции и анализировали с помощью LC-MS. Добавляли дихлорметан и воду и перемешивали, обеспечивали отстаивание и отделяли органическую фазу, высушивали и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью хроматографии с элюированием смесью петролейный эфир:этилацетат (от 10:1 до 4:1) с получением P-28-2 (300 мг). LC-MS: 506,2 (M+1).
Стадия 2. Добавляли P-28-2 (300 мг, 0,6 ммоль) и оксихлорид фосфора (1,85 г, 11,9 ммоль) в толуол (20 мл), затем добавляли N,N-диметиланилин (215,6 мг, 18,0 ммоль) и нагревали с обратным холодильником в течение ночи. После завершения реакции, что определяли посредством TCL-анализа, реакционную систему концентрировали. Затем добавляли этилацетат и ледяную воду и перемешивали и затем получали фазу на основе этилацетата в результате отделения. Фазу на основе этилацетата высушивали и концентрировали с получением неочищенного хлорированного соединения. Неочищенное хлорированное соединение растворяли в безводном тетрагидрофуране (10 мл), добавляли морфолин (155,0 мг, 1,8 ммоль) при 0°C и смесь перемешивали при комнатной температуре до полного израсходования исходных материалов, затем концентрировали с получением неочищенного продукта, который применяли непосредственно на следующей стадии.
Стадия 3. Растворяли неочищенный продукт, полученный на стадии 2, в метаноле (20 мл), затем добавляли воду (1 мл) и гидроксид натрия (237,6 мг) и нагревали с обратным холодильником до полного исчезновения исходных материалов, концентрировали, добавляли воду, регулировали значение pH до 5-6 для осаждения твердого вещества, высушивали и применяли непосредственно на следующей стадии. LC-MS: 477,2 (M+H)+.
Стадия 4. Добавляли неочищенный продукт со стадии 3, HBTU (455,0 мг, 1,2 ммоль), триэтиламин (121 мг, 1,2 ммоль) и 2-(4-пиперидил)-2-пропанол (Aa9`, 93,2 мг, 0,65 ммоль) в DMF (10 мл) и проводили реакцию при комнатной температуре в течение ночи. Завершение реакции контролировали с помощью LC-MS. Реакционную систему выливали в ледяную воду и экстрагировали этилацетатом. Органическую фазу высушивали, концентрировали и пропускали через колонку (DCM: MeOH = от 100:1 до 40:1) с получением P-28-5 (140 мг) в виде светлоокрашенного твердого вещества.
1H ЯМР (300 МГц, CD3OD) δ 8,48 (s, 1H), 7,79 (d, 1H), 7,65 (d, 1H), 7,34-7,42 (brs, 2H), 6,81 (s, 1H), 6,70 (s, 1H), 5,26 (s, 1H), 4,76 (d, 1H), 4,02 (s, 4H), 3,80 (s, 4H), 1,82-1,95 (m, 2H), 1,47-1,69 (m, 2H), 1,24 (s, 6H). LC-MS: 602,3 (M+1).
Стадия 5. В защитной атмосфере N2 растворяли P-28-5 (100 мг, 0,17 ммоль) в безводном тетрагидрофуране (10 мл) и охлаждали до −20°C, затем медленно добавляли по каплям BH3/THF (1 н., 1,7 мл, 1,7 ммоль). После завершения добавления по каплям реакционную систему нагревали до 60°C и проводили реакцию в течение 1 ч., а затем реакционную систему охлаждали до −20°C и медленно добавляли по каплям концентрированную хлористоводородную кислоту (0,8 мл) , а затем нагревали до 60°C с проведением реакции после добавления. С помощью LC-MS определяли, что все промежуточные соединения превращались в целевые соединения. Смесь охлаждали и затем добавляли этилацетат и воду для разделения жидкости с получением водной фазы. Регулировали pH водной фазы до приблизительно 10 с помощью гидроксида натрия, экстрагировали с помощью DCM, высушивали и концентрировали и затем быстро очищали с помощью колонки (элюирование DCM: MeOH = от 100:1 до 40:1) с получением P-28 (20 мг).
1H ЯМР (300 МГц, CD3OD) δ: 8,36 (s, 1H), 7,09 (s, 1H), 6,84 (s, 1H), 5,42 (s, 1H), 4,05 (t, 4H), 3,99 (s, 2H), 3,77 (t, 4H), 3,50 (brs, 1H), 2,39 (t, 2H), 1,80 (t, 2H), 1,36-1,52 (m, 4H), 1,08 (s, 6H), LC-MS: 588,2 (M+1).
Эффект соединений в отношении киназной активности PI3K
Установка концентрации для предварительного скрининга. Соединения, подлежащие тестированию, растворяли в DMSO (диметилсульфоксид) до 100 мкМ или 10 мкМ в качестве растворов для хранения; для каждого отбирали 2 мкл для добавления к 48 мкл 1× реакционного буфера с получением 1 мкМ или 100 нМ раствора соединения, содержащего 4% DMSO. После тщательного смешивания смесь разбавляли с помощью 4% DMSO в 1× реакционном буфере с получением 4 мкМ или 400 нМ раствора соединения. Отбирали 5 мкл каждого разбавленного раствора для добавления в 384-луночный планшет, так что конечная концентрация соединения в конечных 20 мкл киназной реакционной системы составляла 1 мкМ и 100 нМ или 100 нМ и 20 нМ соответственно и содержала 1% DMSO.
Установка концентрации для определения IC50. Для различных ферментов выбирали 10 мкМ и 100 мкМ в качестве исходных концентраций. Соединения, подлежащие испытанию, растворяли в DMSO до 1 мМ или 10 мМ в качестве рабочих растворов, отбирали 1 мкл каждого рабочего раствора для добавления к 24 мкл 1× киназного буфера A с получением 40 мкМ или 400 мкМ раствора соединения, содержащего 4% DMSO. Раствор соединения разбавляли с применением 4x градиента в последовательности, 10 концентраций разбавления. Отбирали 2,5 мкл каждого разбавленного раствора для добавления в 384-луночный планшет, так что конечная концентрация соединения в конечных 10 мкл киназной реакционной системы составляла последовательно 10 мкМ, 2,5 мкМ и 0,625 мкМ и содержала 1% DMSO.
Стадии эксперимента. При комнатной температуре PI3K-киназу подвергали реакции в течение 30 минут и затем ее прекращали, добавляли растворы для обнаружения в каждую лунку, тщательно перемешивали и инкубировали в течение 2 часов при комнатной температуре, затем исследовали с помощью мультифункционального микропланшет-ридера (ридер для считывания нескольких меток 2104 EnVision® (№ по кат.: 2104-0010, PerkinElmer)) и записывали результаты теста.
Результаты эксперимента:
Расчет соотношения значений интенсивности излучения для каждой лунки
Соотношения значений интенсивности излучения (ER) = сигнал излучения при 665 нм/сигнал излучения при 620 нм
Среднее соотношение значений интенсивности излучения для контроля с 100% ингибирования записывали как: ER100%.
Среднее соотношение значений интенсивности излучения для контроля с 0% ингибирования записывали как: ER0%.
Расчет степени ингибирования
Степень ингибирования рассчитывали с помощью следующей формулы:
Степень ингибирования = (ERобразца − ER0%) / (ER100% − ER0%) × 100%
Как показано в таблице 1 и таблице 2, все соединения в таблицах проявляли высокие ингибирующие эффекты в отношении PI3K-киназы, особенно соединения P-5/6/7, которые проявляли селективный ингибирующий эффект в отношении PI3Kδ.
Таблица 1. Ингибирующий эффект некоторых соединений в отношении различных изоформ PI3K-киназы
при указанных концентрациях

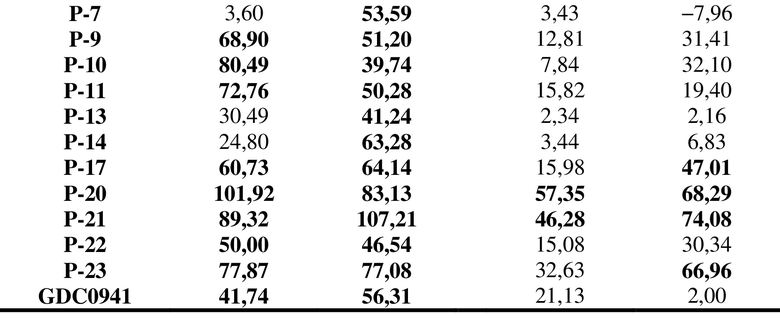
Таблица 2. Ингибирующие эффекты некоторых соединений в отношении различных изоформ PI3K-киназы
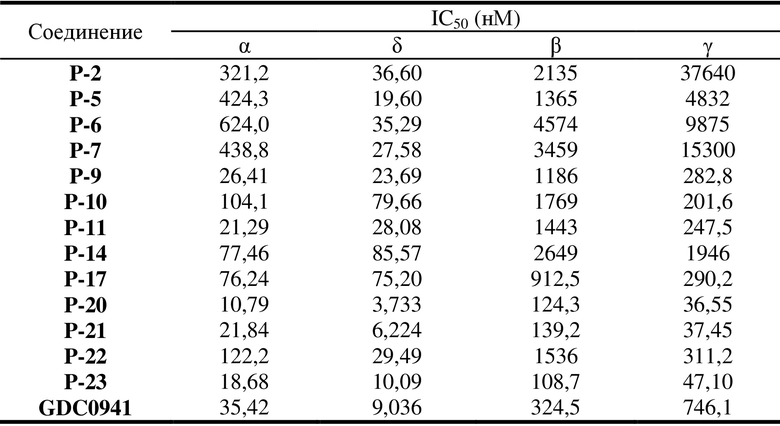
Из таблиц можно видеть, что соединения P-5, P-6, P-7 и т. д. характеризуются надлежащими уровнями селективности в отношении PI3Kδ. GDC0941 представляет собой положительное соединение с химическим названием 2-(1H-индазол-4-ил)-6-[[4-(метилсульфонил)-1-пиперазинил]метил]-4-(4-морфолинил)тиено[3,2-D]пиримидин(4-[2-(1H-индазол-4-ил)-6-[(4-метилсульфонилпиперазин-1-ил)метил]тиено[3,2-d]пиримидин-4-ил]морфолин).
Предварительный фармакокинетический тест
1. 14 здоровых крыс SD, самцов, весом 200-220 г, произвольным образом делили на 4 группы, по 3-4 в каждой группе, и вводили тестируемое соединение P-17 и соединение CYH33 (по сравнению с соединением P-17, у которого отсутствует метиловая группа в 7-положении) путем внутрижелудочного введения (10 мг/кг) и внутривенной инъекции (5 мг/кг) соответственно, конкретные схемы показаны в следующей таблице 3.

Крыс подвергали голоданию в течение 12 ч. перед экспериментом, воду позволяли употреблять свободно. Крыс кормили через 2 ч. после введения дозы.
2. Моменты времени отбора образцов крови и обработки образца
Введение позы путем внутрижелудочного введения: 0,25 ч., 0,5 ч., 1,0 ч., 2,0 ч., 3,0 ч., 4,0 ч., 6,0 ч., 8,0 ч. и 24 ч. после введения дозы.
Введение дозы внутривенно: 5 мин., 0,25 ч., 0,5 ч., 1,0 ч., 2,0 ч., 4,0 ч., 6,0 ч., 8,0 ч. и 24 ч. после введения дозы.
В вышеуказанные моменты времени 0,3 мл венозной крови отбирали из ретроорбитального венозного сплетения крысы и помещали в гепаринизированную пробирку, центрифугировали при 11000 об./мин. в течение 5 мин., затем плазму отделяли и замораживали в холодильнике при −20°C.
3. Тестирование образца и анализ данных
Способ LC/MS/MS применяли для определения концентрации соединений в плазме крыс.
Некомпартментную модель с применением программного обеспечения Phoenix 1.3 (Pharsight Corporation, США) использовали для подсчета фармакокинетических параметров после введения дозы.
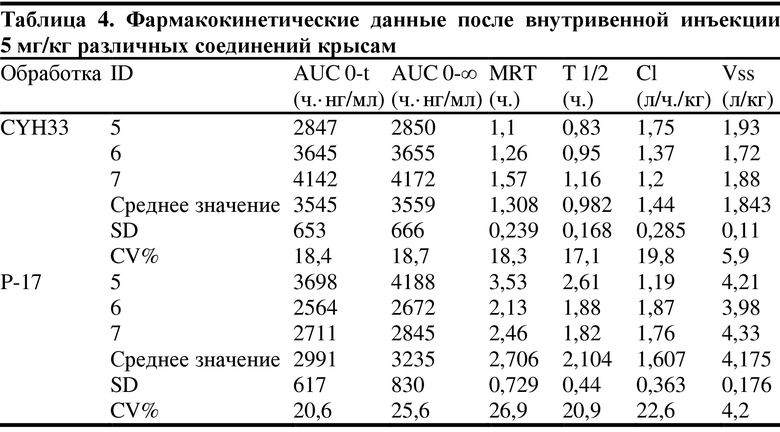
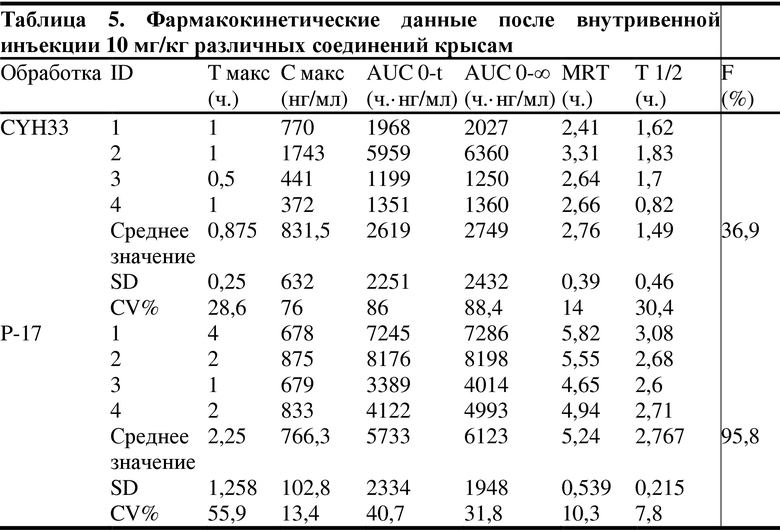
(ID представляет собой количество животных, T макс. представляет собой пиковое время, C макс. представляет собой пик концентрации лекарственного средства в крови, AUC представляет собой площадь под кривой во время медикаментозного лечения, MRT представляет собой среднее время удержания, Cl представляет собой скорость выведения лекарственного средства, Vss представляет собой объем распределения при устойчивом уровне, F представляет собой абсолютную биодоступность)
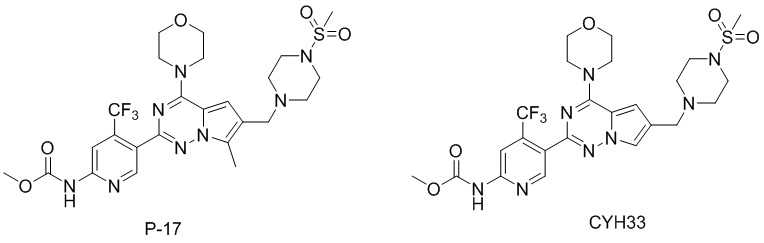
Как можно видеть из таблицы выше, хотя P-17 отличается от CYH33 одним метилом, объем распределения при устойчивом уровне Vss, среднее время удержания MRT, период полувыведения T 1/2 и абсолютная биодоступность F P-17 – все были улучшены, особенно абсолютная биодоступность была значительно увеличена (тестируемые соединения представляли собой мономезилаты CYH33 и P17, структуры соединений были следующими).
Вышеуказанные примеры всего лишь иллюстрируют несколько вариантов осуществления настоящего изобретения, которые описаны конкретно и подробно, но их не следует считать ограничивающими объем настоящего изобретения. Следует отметить, что специалистами в данной области техники несколько вариаций и улучшений могут быть выполнены без отступления от идеи настоящего изобретения, и все они находятся в пределах объема правовой охраны настоящего изобретения. Следовательно, объем правовой охраны настоящего изобретения будет определяться прилагаемой формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИРРОЛО[2,1-f][1,2,4]ТРИАЗИНОВОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2013 |
|
RU2589053C1 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ, АКТИВИРУЮЩИЕ ФЕРМЕНТЫ | 2013 |
|
RU2652989C2 |
| ПРОИЗВОДНОЕ НА ОСНОВЕ ДИГИДРОПИРИМИДО-КОЛЬЦА В КАЧЕСТВЕ ИНГИБИТОРА HBV | 2015 |
|
RU2693897C2 |
| Ингибиторы ErbB/BTK | 2019 |
|
RU2764069C1 |
| БЕНЗОКСАЗИН-ОКСАЗОЛИДИНОНОВОЕ СОЕДИНЕНИЕ, ЗАМЕЩЕННОЕ АЗОТСОДЕРЖАЩИМ ГЕТЕРОЦИКЛОМ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2018 |
|
RU2744784C1 |
| ТЕРАПЕВТИЧЕСКИЕ СРЕДСТВА, НАЦЕЛЕННЫЕ НА УКОРОЧЕННЫЕ БЕЛКИ АДЕНОМАТОЗНОГО ПОЛИПОЗА ТОЛСТОГО КИШЕЧНИКА (АРС) | 2014 |
|
RU2727516C2 |
| СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ ПО ОТНОШЕНИЮ К PDE9A, И ИХ ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2019 |
|
RU2788147C2 |
| ПРОЛЕКАРСТВА ИНГИБИТОРОВ ТРОМБИНА | 1996 |
|
RU2176644C2 |
| ЛЕЧЕНИЕ ИНФЕКЦИЙ H. PYLORI С ПРИМЕНЕНИЕМ ИНГИБИТОРОВ MTAN | 2015 |
|
RU2663803C2 |
| НОВЫЕ СПИРОБИЦИКЛИЧЕСКИЕ АНАЛОГИ | 2018 |
|
RU2789377C2 |
Изобретение относится к области органической химии, а именно к соединению на основе 7-замещенного пирроло[2,1-f][1,2,4]триазина общей формулой I или к его фармацевтически приемлемой соли, где R1 представляет собой галоген, или C1-6алкил, который является незамещенным или замещенным по меньшей мере одним заместителем, при этом заместитель представляет собой галоген; R2 представляет собой: незамещенный либо замещенный одним или двумя заместителями -CH2NH-C1-6алкил, -CH2N(C1-6алкил)-(C1-6алкил), -CH2-(насыщенный гетероциклил, содержащий 1-2 гетероатома и 4-5 атомов углерода), -CH2-(насыщенный гетероциклил, содержащий 1-2 гетероатома и 3-5 атомов углерода)-(насыщенный гетероциклил, содержащий 1-2 гетероатома и 3-5 атомов углерода), -CH2-(насыщенная спироциклическая группа, содержащая 2 гетероатома и 5-7 атомов углерода), -CH2-(насыщенная кольцевая группа с мостиковой связью, содержащая 2 гетероатома и 5 атомов углерода)-(насыщенный гетероциклил, содержащий 1 гетероатом и 3 атома углерода), или -CH2-(насыщенная кольцевая группа с мостиковой связью, содержащая 2 гетероатома и 5 атомов углерода), где заместитель представляет собой: -N(C1-4алкил)-(C1-4алкил), -S(O)2-C1-4алкил, -C1-4алкил-N(C1-4алкил)-(C1-4алкил), C1-4алкил-O-C1-4алкил, который замещен двумя метилами, -C1-4алкил-CONH2, который замещен двумя метилами, -COO-C1-4алкил, -CO-C1-4алкил, который замещен одним заместителем A, где заместитель A представляет собой гидроксил, или C1-4алкил, который является незамещенным или замещен одним-тремя заместителями B, где заместитель B представляет собой -NH2, -OCH3, -CONH2, -OH или -CF3; в R2 гетероатом выбран из группы, состоящей из: (i) N, и (ii) комбинации N и одного из O и S; R3 представляет собой 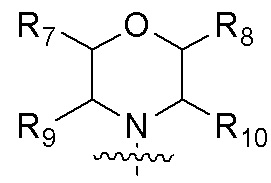 , где каждый из R7, R8, R9 и R10 независимо представляет собой водород или C1-6алкил, который является незамещенным; R4 представляет собой -NH2, -NHCONHR11 или -NHCO2R12, где каждый из R11 и R12 независимо представляет собой C1-6алкил, C3-8циклоалкил или фенил, который является незамещенным или замещен одним заместителем, выбранным из галогена и -C(O)OR13, где R13 представляет собой C1-6алкил, который является незамещенным. Также изобретение относится к применению соединения формулы I и к фармацевтической композиции на его основе. Технический результат: получены новые гетероциклические соединения, эффективно ингибирующие активность PI3K-киназы и полезные для лечения заболеваний, связанных с путем PI3K, особенно для борьбы с раком или для лечения опухолей, в том числе и лейкоза, и аутоиммунных заболеваний. 3 н. и 14 з.п. ф-лы, 5 табл.
, где каждый из R7, R8, R9 и R10 независимо представляет собой водород или C1-6алкил, который является незамещенным; R4 представляет собой -NH2, -NHCONHR11 или -NHCO2R12, где каждый из R11 и R12 независимо представляет собой C1-6алкил, C3-8циклоалкил или фенил, который является незамещенным или замещен одним заместителем, выбранным из галогена и -C(O)OR13, где R13 представляет собой C1-6алкил, который является незамещенным. Также изобретение относится к применению соединения формулы I и к фармацевтической композиции на его основе. Технический результат: получены новые гетероциклические соединения, эффективно ингибирующие активность PI3K-киназы и полезные для лечения заболеваний, связанных с путем PI3K, особенно для борьбы с раком или для лечения опухолей, в том числе и лейкоза, и аутоиммунных заболеваний. 3 н. и 14 з.п. ф-лы, 5 табл.
1. Соединение на основе 7-замещенного пирроло[2,1-f][1,2,4]триазина, представленное общей формулой I, или его фармацевтически приемлемая соль:
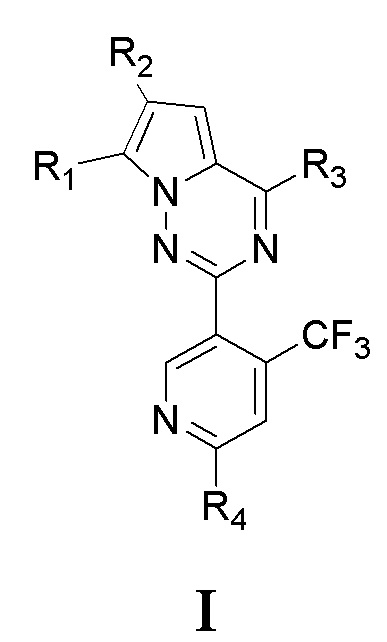 ,
,
где
R1 представляет собой галоген, или C1-6алкил, который является незамещенным или замещенным по меньшей мере одним заместителем, при этом заместитель представляет собой галоген;
R2 представляет собой: незамещенный либо замещенный одним или двумя заместителями -CH2NH-C1-6алкил, -CH2N(C1-6алкил)-(C1-6алкил), -CH2-(насыщенный гетероциклил, содержащий 1-2 гетероатома и 4-5 атомов углерода), -CH2-(насыщенный гетероциклил, содержащий 1-2 гетероатома и 3-5 атомов углерода)-(насыщенный гетероциклил, содержащий 1-2 гетероатома и 3-5 атомов углерода), -CH2-(насыщенная спироциклическая группа, содержащая 2 гетероатома и 5-7 атомов углерода), -CH2-(насыщенная кольцевая группа с мостиковой связью, содержащая 2 гетероатома и 5 атомов углерода)-(насыщенный гетероциклил, содержащий 1 гетероатом и 3 атома углерода), или -CH2-(насыщенная кольцевая группа с мостиковой связью, содержащая 2 гетероатома и 5 атомов углерода), где заместитель представляет собой: -N(C1-4алкил)-(C1-4алкил), -S(O)2-C1-4алкил, -C1-4алкил-N(C1-4алкил)-(C1-4алкил), C1-4алкил-O-C1-4алкил, который замещен двумя метилами, -C1-4алкил-CONH2, который замещен двумя метилами, -COO-C1-4алкил, -CO-C1-4алкил, который замещен одним заместителем A, где заместитель A представляет собой гидроксил, или C1-4алкил, который является незамещенным или замещен одним-тремя заместителями B, где заместитель B представляет собой -NH2, -OCH3, -CONH2, -OH или -CF3;
в R2 гетероатом выбран из группы, состоящей из: (i) N, и (ii) комбинации N и одного из O и S,
R3 представляет собой 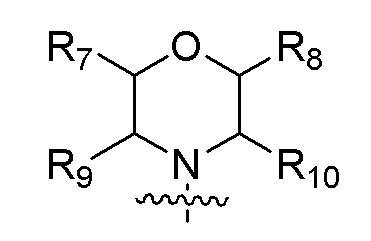 , где каждый из R7, R8, R9 и R10 независимо представляет собой водород или C1-6алкил, который является незамещенным;
, где каждый из R7, R8, R9 и R10 независимо представляет собой водород или C1-6алкил, который является незамещенным;
R4 представляет собой -NH2, -NHCONHR11 или -NHCO2R12, где каждый из R11 и R12 независимо представляет собой C1-6алкил, C3-8циклоалкил или фенил, который является незамещенным или замещен одним заместителем, выбранным из галогена и -C(O)OR13, где R13 представляет собой C1-6алкил, который является незамещенным.
2. Соединение на основе 7-замещенного пирроло[2,1-f][1,2,4]триазина, представленное общей формулой I, по п. 1 или его фармацевтически приемлемая соль, где R1 представляет собой галоген, или C1-4алкил, который является незамещенным или замещен по меньшей мере одним заместителем, где заместитель представляет собой галоген.
3. Соединение на основе 7-замещенного пирроло[2,1-f][1,2,4]триазина, представленное общей формулой I, по п. 2 или его фармацевтически приемлемая соль, где R1 представляет собой -Cl, -F, метил, трифторметил или дифторметил.
4. Соединение на основе 7-замещенного пирроло[2,1-f][1,2,4]триазина, представленное общей формулой I, по п. 1 или его фармацевтически приемлемая соль, где R2 представляет собой: незамещенный либо замещенный одним или двумя заместителями -CH2NH-C1-4алкил, -CH2N-(C1-4алкил)-(C1-4алкил), -X-(насыщенный четырех-шестичленный гетероциклил, содержащий 1-2 гетероатома), -X-(насыщенный четырех-шестичленный гетероциклил, содержащий 1-2 гетероатома)-(насыщенный четырех-шестичленный гетероциклил, содержащий 1-2 гетероатома), -X-(насыщенная бициклическая спироциклическая группа, содержащая 2 гетероатома и 5-7 атомов углерода), -X-(насыщенная бициклическая кольцевая группа с мостиковой связью, содержащая 2 гетероатома и 5 атомов углерода)-(насыщенный четырехчленный гетероциклил, содержащий 1 гетероатом) или -X-(насыщенная бициклическая кольцевая группа с мостиковой связью, содержащая 2 гетероатома и 5 атомов углерода), где X представляет собой CH2, и где гетероциклил, спироциклическая группа и кольцевая группа с мостиковой связью присоединены к X посредством атома N.
5. Соединение на основе 7-замещенного пирроло[2,1-f][1,2,4]триазина, представленное общей формулой I, по п. 4 или его фармацевтически приемлемая соль, где заместитель в R2 представляет собой -CH3, -CH2CH3, -CH2CH(CH3)2, -CH2NH2, -CH2N(CH3)2, -CH2CH2NH2, -CH2CH2N(CH3)2, -CH2OH, -CH2OCH3, -CH2CH2OH, -C(CH3)2OH, -C(CH3)(CF3)OH, -C(CF3)2OH, -C(CH3)2OCH3, -C(CH3)2NH2, -CH2C(CH3)2OH, -CH(OH)CH(CH3)2, -C(CH3)2CH2OH, -C(CH3)2CH2OCH3, -COCH3, -CO2CH3, -CO2C(CH3)3, -COCH2OH, -COC(OH)(CH3)2, -COCH(OH)CH3, -CH2CONH2, -C(CH3)2CONH2, -N(CH3)2, или -S(O)2CH3.
6. Соединение на основе 7-замещенного пирроло[2,1-f][1,2,4]триазина, представленное общей формулой I, по п. 1 или его фармацевтически приемлемая соль, где R2 представляет собой 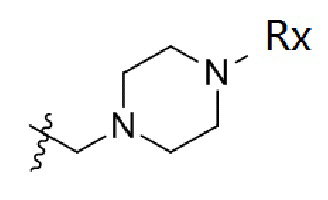 или
или 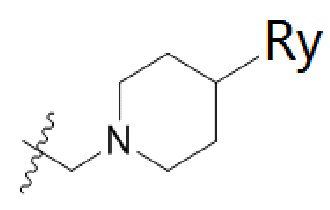 , каждый из Rx и Ry представляет собой -N(CH3)2, -S(O)2CH3 или C1-4алкил, где C1-4алкил является незамещенным или замещен одним-тремя заместителями, при этом заместитель представляет собой гидроксил, -CONH2, -CF3, амино или -OCH3.
, каждый из Rx и Ry представляет собой -N(CH3)2, -S(O)2CH3 или C1-4алкил, где C1-4алкил является незамещенным или замещен одним-тремя заместителями, при этом заместитель представляет собой гидроксил, -CONH2, -CF3, амино или -OCH3.
7. Соединение на основе 7-замещенного пирроло[2,1-f][1,2,4]триазина, представленное общей формулой I, по п. 1 или его фармацевтически приемлемая соль, где R2 представляет собой 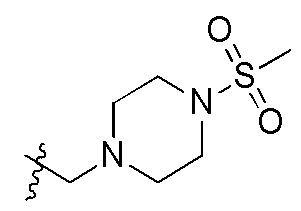 ,
, 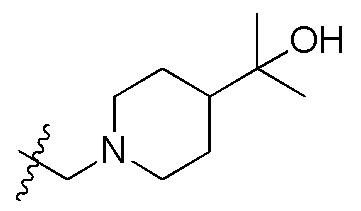 ,
, 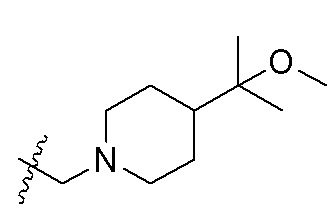 ,
, 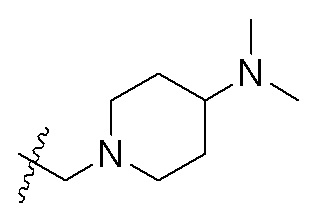 ,
, 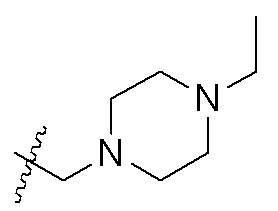 ,
, 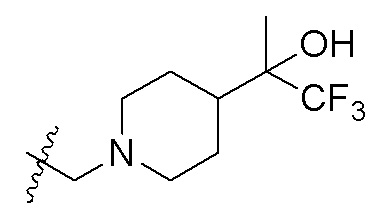 ,
, 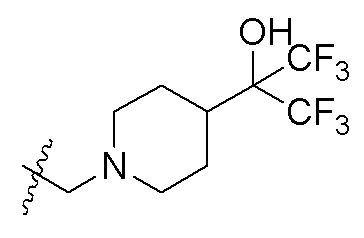 или
или 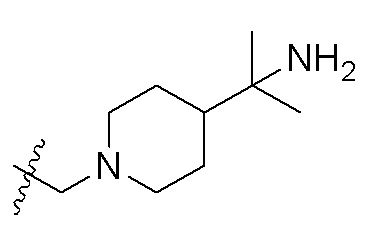 .
.
8. Соединение на основе 7-замещенного пирроло[2,1-f][1,2,4]триазина, представленное общей формулой I, по п. 1 или его фармацевтически приемлемая соль, где R3 представляет собой 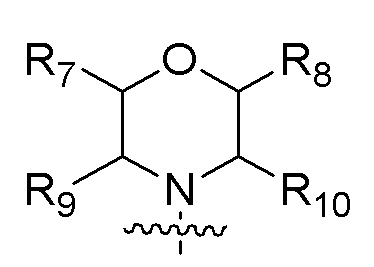 , и где каждый из R7, R8, R9 и R10 независимо представляет собой водород или C1-4алкил.
, и где каждый из R7, R8, R9 и R10 независимо представляет собой водород или C1-4алкил.
9. Соединение на основе 7-замещенного пирроло[2,1-f][1,2,4]триазина, представленное общей формулой I, по п. 8 или его фармацевтически приемлемая соль, где R3 представляет собой морфолинил или (S)-3-метилморфолинил.
10. Соединение на основе 7-замещенного пирроло[2,1-f][1,2,4]триазина, представленное общей формулой I, по п. 1 или его фармацевтически приемлемая соль, где R4 представляет собой -NH2, -NHCONHR11 или -NHCO2R12, где каждый из R11 и R12 независимо представляет собой C1-4алкил, C3-6циклоалкил или фенил, который является незамещенным или замещен одним заместителем, выбранным из фтора, хлора, брома и -C(O)OR13, где R13 представляет собой C1-4алкил.
11. Соединение на основе 7-замещенного пирроло[2,1-f][1,2,4]триазина, представленное общей формулой I, по п. 10 или его фармацевтически приемлемая соль, где каждый из R11 и R12 независимо представляет собой метил, этил, изопропил, циклопропил, фенил, -Ph-CO2Et-p или 4-фторфенил.
12. Соединение на основе 7-замещенного пирроло[2,1-f][1,2,4]триазина, представленное общей формулой I, по п. 1 или его фармацевтически приемлемая соль, где соединение характеризуется структурой, представленной одной из следующих общих формул:
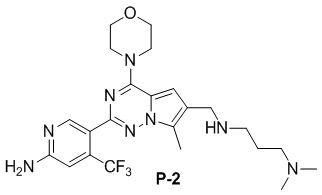
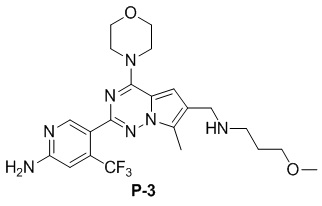
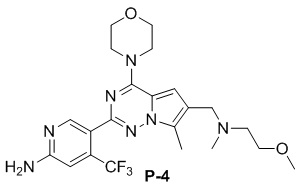
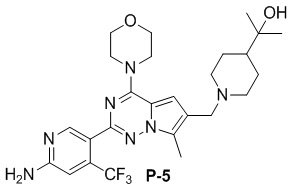
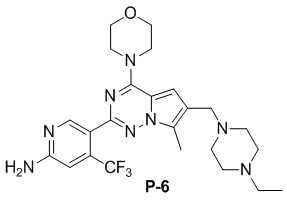
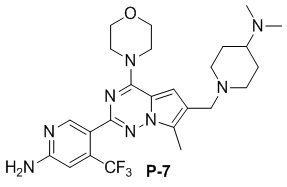
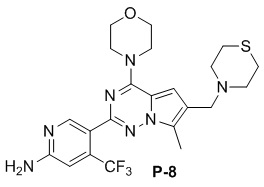
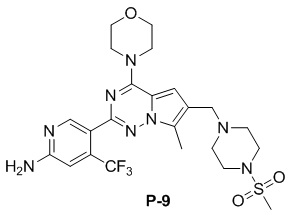
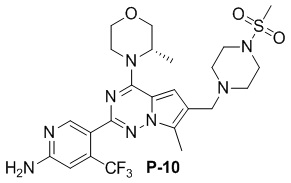
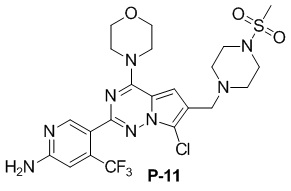
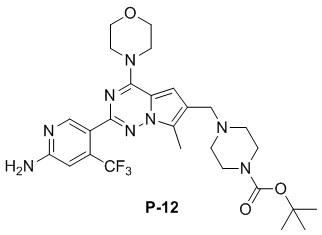
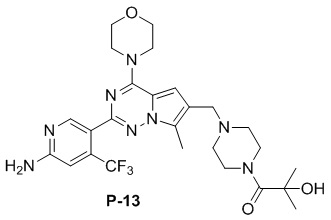
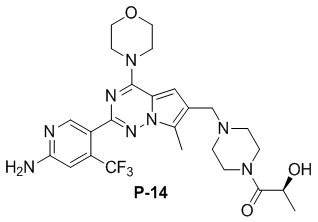
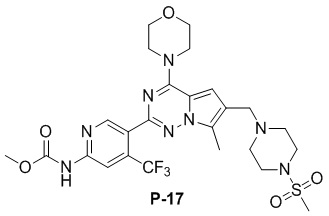
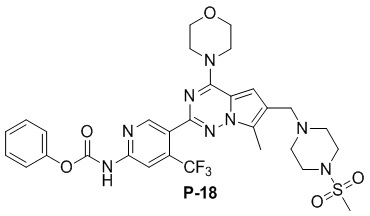
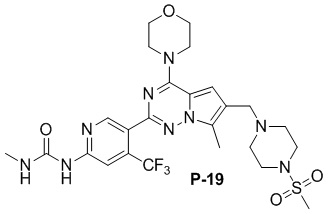
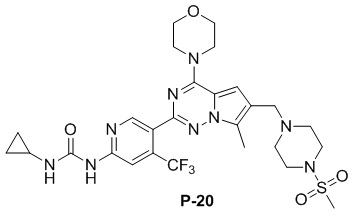
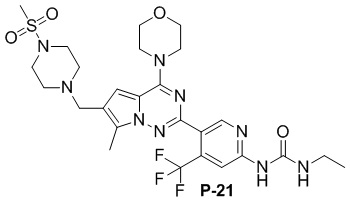
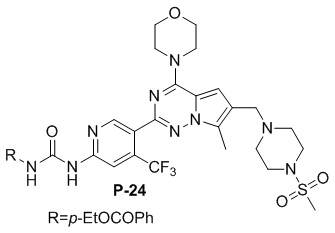
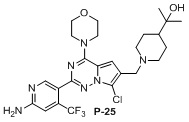
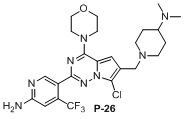
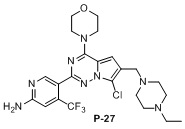
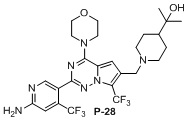
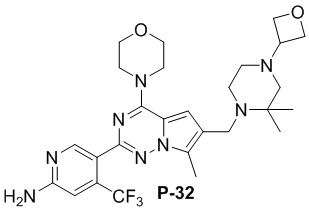
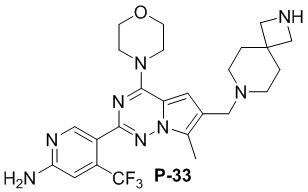
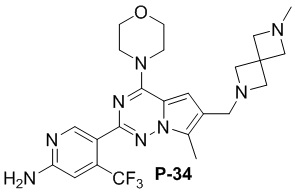
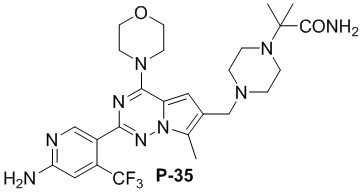
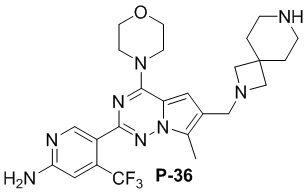
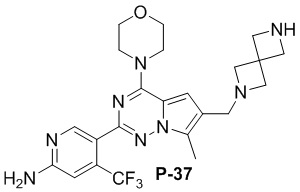
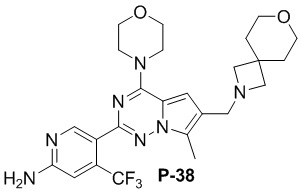
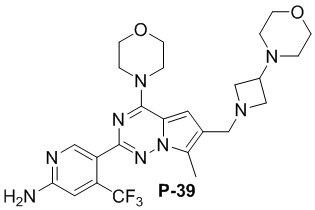
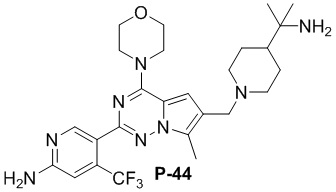
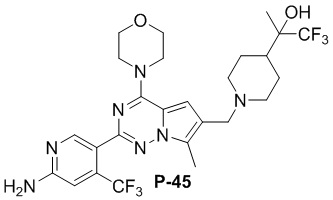
13. Применение соединения на основе 7-замещенного пирроло[2,1-f][1,2,4]триазина, представленного общей формулой I, по любому из пп. 1-12 или его фармацевтически приемлемой соли в получении ингибитора PI3K.
14. Применение соединения на основе 7-замещенного пирроло[2,1-f][1,2,4]триазина, представленного общей формулой I, или его фармацевтически приемлемой соли в получении ингибитора PI3K по п. 13, где ингибитор PI3K характеризуется селективным ингибирующим эффектом в отношении PI3Kδ.
15. Применение соединения на основе 7-замещенного пирроло[2,1-f][1,2,4]триазина, представленного общей формулой I, или его фармацевтически приемлемой соли в получении ингибитора PI3K по п. 13, где ингибитор PI3K применяется в лекарственном препарате для лечения заболевания, связанного с путем PI3K.
16. Применение соединения на основе 7-замещенного пирроло[2,1-f][1,2,4]триазина, представленного общей формулой I, или его фармацевтически приемлемой соли в получении ингибитора PI3K по п. 15, где заболевание, связанное с путем PI3K, представляет собой по меньшей мере одно из опухолей, видов лейкоза и аутоиммунных заболеваний.
17. Фармацевтическая композиция для лечения заболевания, связанного с путем PI3K, содержащая терапевтически эффективное количество соединения на основе 7-замещенного пирроло[2,1-f][1,2,4]триазина, представленного общей формулой I, по любому из пп. 1-12 или его фармацевтически приемлемой соли.
| ЕР 2857403 А1, 08.04.2015 | |||
| Dugar, S | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Приспособление к эксцентриковому прессу двойного действия, предназначенного для увеличения его рабочего хода | 1929 |
|
SU23775A1 |

