Область технического применения изобретения
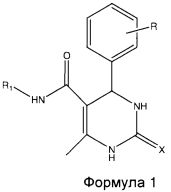
Настоящее изобретение относится к соединениям формулы 1, проявляющим противотуберкулезную активность. В частности, это изобретение относится к соединениям, которые проявляют противотуберкулезную активность в отношении микобактерий, находящихся в стадии покоя, и к способу их получения.
 ,
,
где R представляет собой Н, галоген, дигалоген, О-алкил, ди-О-алкил, R1 представляет собой фенил, хлорфенил, нитрофенил, дихлорфенил, циклоалкил, предпочтительно циклогексил, X представляет собой О или S.
Предшествующий уровень техники
Известно, что туберкулез (ТВ) поражает приблизительно 8 миллионов человек ежегодно, что приводит к 2 миллионам смертей в год. Известно, что Индия характеризуется наибольшей частотой возникновения данного заболевания среди своего населения, при этом число пораженных составляет до 1,8 миллиона включительно. Увеличение числа случаев ВИЧ-инфекции и пренебрежение программами по контролю за ТВ спровоцировали всплеск туберкулеза. Появление лекарственно-устойчивых штаммов также внесло вклад в эту новую эпидемическую ситуацию, при которой с 2000 по 2004 г.г. наблюдалось 20% случаев ТВ, устойчивого к стандартному лечению, и 2% случаев туберкулеза, устойчивого к лекарственным препаратам второй линии. На фоне этого мрачного сценария необходимо проведение постоянных исследований для отыскания все более новых и новых лекарственных средств, с помощью которых можно эффективно бороться с данным заболеванием.
Большинство доступных противотуберкулезных лекарственных средств и некоторые новые соединения, которые были предложены в качестве активного начала против данных бацилл, действуют на бактерии, находящиеся в фазе роста. V.Virsodia и др. в статье с названием "Synthesis, screening for antitubercular activity and 3D-QSAR studies of substituted N-phenyl-6-methyl-2-oxo-4-phenyl-1,2,3,4-tetrahydropyrimidine-5-carboxamide" в European Journal of Medicinal Chemistry, 43, 2103-2115 (2008) сообщают о синтезе и оценке замещенных N-фенил-6-метил-2-оксо-4-фенил-1,2,3,4-тетрагидропиримидин-5-карбоксамидов в качестве противотуберкулезных средств. Во всяком случае, было обнаружено, что эти средства активны против протестированного штамма микобактерий в активной фазе или фазе роста.
В статье Akshay М. Pansuriya и др., озаглавленной "One-pot synthesis of 5-carboxanilide-dihydropyrimidinones using etidronic Acid", опубликованной в General Papers ARKIVOC, 2009 (vii) 79-85 (ISSN 1551-7012), описывается синтез 5-карбоксанилид-4-замещенных дигидропиримидинонов с использованием реакции циклоконденсации 1,3-дикетона, альдегида и мочевины. Указанные производные 5-карбоксанилид-4-замещенных дигидропиримидинонов демонстрируют широкий спектр биологических эффектов, включая антивирусную, противораковую, антибактериальную, противовоспалительную активности. Тем не менее, противотуберкулезной активности в отношении штамма микобактерий в активной фазе или фазе роста либо в фазе покоя не установлено.
В данной области техники существует необходимость в соединениях (molecules), которые могут действовать против бацилл в фазе покоя, для эффективного снижения частоты возникновения туберкулеза, а также снижения заболеваемости и смертности от этого заболевания. Кроме того, это может заложить основы стратегии борьбы с бациллами с множественной лекарственной устойчивостью, которые все чаще делают обычные схемы лечения неэффективными.
Задача настоящего изобретения
Настоящим изобретением решается главная задача получения соединений формулы 1, проявляющих противотуберкулезную активность.
Таким образом, задача данного изобретения состоит в получении соединений, активных в отношении микобактерий в фазе покоя.
Другая задача изобретения заключается в разработке способа синтеза указанных соединений.
Сущность изобретения
Таким образом, настоящее изобретение относится к соединениям формулы 1, проявляющим противотуберкулезную активность в стадии покоя микобактерий, и кроме того согласно настоящему изобретению предложен способ их получения.
 ,
,
где R представляет собой Н, галоген, дигалоген, О-алкил, ди-О-алкил, R1 представляет собой фенил, хлорфенил, нитрофенил, дихлорфенил, циклоалкил, предпочтительно циклогексил, X представляет собой О или S.
В одном из воплощений настоящего изобретения общая формула 1 представлена следующими соединениями:
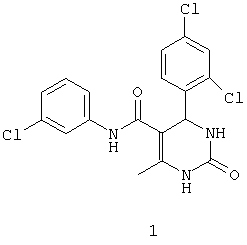
N-(3-хлорфенил)-4-(2,4-дихлорфенил)-6-метил-2-оксо-1,2,3,4-тетрагидропиримидин-5-карбоксамидом (1),
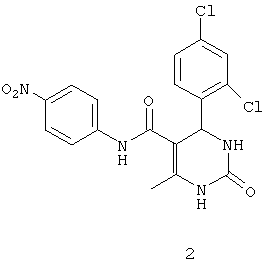
N-(4-нитрофенил)-4-(2,4-дихлорфенил)-6-метил-2-оксо-1,2,3,4-тетрагидропиримидин-5-карбоксамидом (2),
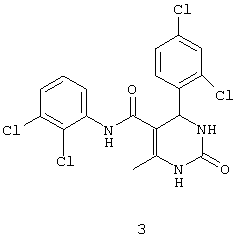
N-(2,3-дихлорфенил)-4-(2,4-дихлорфенил)-6-метил-2-оксо-1,2,3,4-тетрагидропиримидин-5-карбоксамидом (3),
N-циклогексил-4-фенил-6-метил-2-оксо-1,2,3,4-тетрагидропиримидин-5-карбоксамидом (4),
N-(3-хлорфенил)-4-фенил-6-метил-2-оксо-1,2,3,4-тетрагидропиримидин-5-карбоксамидом (5),
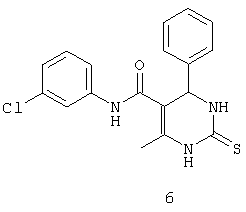
N-(3-хлорфенил)-4-фенил-6-метил-2-тиоксо-1,2,3,4-тетрагидропиримидин-5-карбоксамидом (6),
N-(4-нитрофенил)-4-фенил-6-метил-2-оксо-1,2,3,4-тетрагидропиримидин-5-карбоксамидом (7),
N-(3-хлорфенил)-4-(3-хлорфенил)-6-метил-2-оксо-1,2,3,4-тетрагидропиримидин-5-карбоксамидом (8),
N-(2,3-дихлорфенил)-4-(3-хлорфенил)-6-метил-2-оксо-1,2,3,4-тетрагидропиримидин-5-карбоксамидом (9),
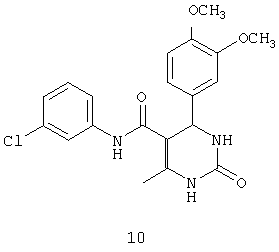
N-(3-хлорфенил)-4-(3,4-диметоксифенил)-6-метил-2-оксо-1,2,3,4-тетрагидропиримидин-5-карбоксамидом (10),
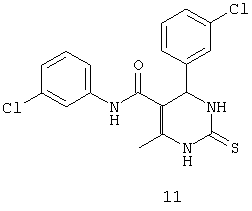
N,4-бис(3-хлорфенил)-6-метил-2-тиоксо-1,2,3,4-тетрагидропиримидин-5-карбоксамидом (11),
N-(2,3-дихлорфенил)-4-(3-хлорфенил)-6-метил-2-тиоксо-1,2,3,4-тетрагидропиримидин-5-карбоксамидом (12),
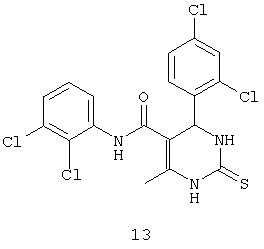
N-(2,3-дихлорфенил)-4-(2,4-дихлорфенил)-6-метил-2-тиоксо-1,2,3,4-тетрагидропиримидин-5-карбоксамидом (13),
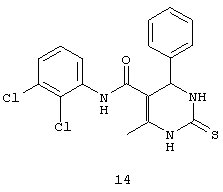
N-(2,3-дихлорфенил)-6-метил-4-фенил-2-тиоксо-1,2,3,4-тетрагидропиримидин-5-карбоксамидом (14),
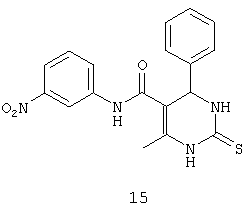
N-(3-нитрофенил)-6-метил-4-фенил-2-тиоксо-1,2,3,4-тетрагидропиримидин-5-карбоксамидом (15),
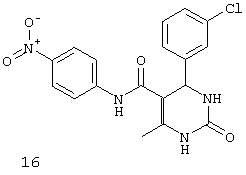
N-(4-нитрофенил)-4-(3-хлорфенил)-6-метил-2-оксо-1,2,3,4-тетрагидропиримидин-5-карбоксамидом (16),
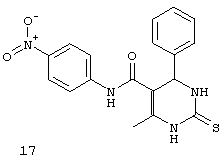
N-(4-нитрофенил)-6-метил-4-фенил-2-тиоксо-1,2,3,4-тетрагидропиримидин-5-карбоксамидом (17),
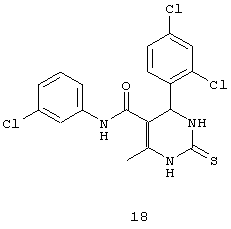
N-(3-хлорфенил)-4-(2,4-дихлорфенил)-6-метил-2-тиоксо-1,2,3,4-тетрагидропиримидин-5-карбоксамидом (18),
N-циклогексил-6-метил-4-фенил-2-тиоксо-1,2,3,4-тетрагидропиримидин-5-карбоксамидом (19),
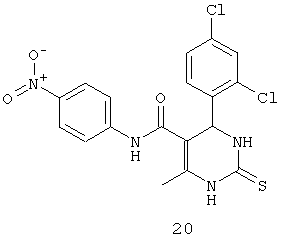
N-(4-нитрофенил)-4-(2,4-дихлорфенил)-6-метил-2-тиоксо-1,2,3,4-тетрагидропиримидин-5-карбоксамидом (20),
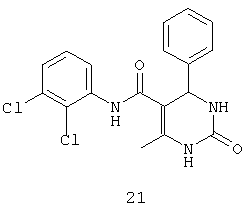
N-(2,3-дихлорфенил)-6-метил-2-оксо-4-фенил-1,2,3,4-тетрагидропиримидин-5-карбоксамидом (21),
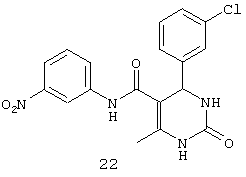
N-(3-нитрофенил)-4-(3-хлорфенил)-6-метил-2-оксо-1,2,3,4-тетрагидропиримидин-5-карбоксамидом (22),
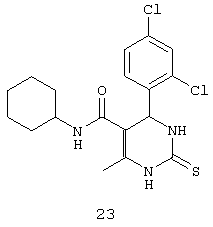
N-циклогексил-4-(2,4-дихлорфенил)-6-метил-2-тиоксо-1,2,3,4-тетрагидропиримидин-5-карбоксамидом (23),
N-циклогексил-4-(2,4-дихлорфенил)-6-метил-2-оксо-1,2,3,4-тетрагидропиримидин-5-карбоксамидом (24).







В другом воплощении настоящего изобретения описаны соединения общей формулы 1, проявляющие противотуберкулезную активность.
В другом воплощении настоящего изобретения указанное соединение используют против микобактерий, находящейся в фазе покоя.
В другом воплощении настоящего изобретения предложена фармацевтическая композиция, содержащая фармацевтически приемлемый эксципиент и эффективное количество соединения формулы 1, заявленного в п.1 формулы изобретения.
В другом воплощении настоящего изобретения предложена фармацевтическая композиция, содержащая фармацевтически приемлемые эксципиенты и эффективное количество соединения формулы 1, заявленного в п.1 формулы изобретения.
В другом воплощении настоящего изобретения предложен способ получения соединений формулы 1, заявленных в п.1 формулы изобретения, при этом указанный способ включает:
(a) смешивание реагента, выбранного из замещенного ацетоацетанилида или бензилацетоацетата, замещенного альдегида и мочевины, в растворителе, предпочтительно абсолютном этаноле;
(b) нагревание реакционной смеси, полученной на стадии (а), при температуре в диапазоне 60-100°C в течение периода времени в диапазоне 4-8 часов;
(c) добавление p-TSA (п-толуолсульфоновой кислоты) или конц. HCl в реакционную смесь, полученную на стадии (b), с последующим кипячением с обратным холодильником при температуре в диапазоне 60-100°C в течение периода времени в диапазоне 4-8 часов;
(d) охлаждение реакционной смеси, полученной на стадии (с), с последующим отделением твердого вещества фильтрацией, промывку спиртом с получением соединений №№1-3, 5-18 и 20-22 или бензилового эфира тетрагидропиримидона;
(e) синтез тетрагидропиримидон-5-карбоновой кислоты из бензилового эфира тетрагидропиримидона, полученного на стадии (d), либо посредством смешивания бензилового эфира тетрагидропиримидона, полученного на стадии (d), 5% Pd/C и формиата аммония в метаноле в атмосфере азота в течение периода времени в диапазоне 8-10 ч при температуре в диапазоне 55-65°C, затем подведения до pH 9 раствором щелочи, предпочтительно водн. KOH, фильтрования реакционной смеси с получением фильтрата, затем подведения до рН 4 раствором кислоты, предпочтительно водн. HCl, с получением тетрагидропиримидон-5-карбоновой кислоты, либо посредством гидролиза бензилового эфира тетрагидропиримидона, полученного на стадии (d), с использованием NaOH или KOH;
(f) перемешивание тетрагидропиримидон-5-карбоновой кислоты, полученной на стадии (е), и N,N′-дициклогексилкарбодиимида (DCC) в течение периода времени в диапазоне 6-12 часов при температуре в диапазоне 50-65°C, и последующее добавление к этой смеси раствора соответствующего амина в DMF (диметилформамиде), предпочтительно циклогексиламина, и продолжение перемешивания при температуре в диапазоне 55-65°C с последующим добавлением метанола в эту реакционную смесь;
(g) фильтрование реакционной смеси, полученной на стадии (f), для удаления дициклогексилмочевины, затем удаление метанола с получением соединений №№4, 19, 23 и 24.
В другом воплощении настоящего изобретения замещенный ацетоацетанилид, используемый на стадии (а), выбран из группы, состоящей из 3-хлорацетоацетанилида, 4-нитроацетоацетанилида, 2,3-дихлорацетоацетанилида и 3-нитроацетоацетанилида.
В другом воплощении настоящего изобретения замещенный альдегид, используемый на стадии (а), выбран из группы, состоящей из 2,4-дихлорбензальдегида, бензальдегида, 3-хлорбензальдегида и 3,4-диметоксибензальдегида.
В другом воплощении настоящего изобретения молярное соотношение замещенного ацетоацетанилида, замещенного альдегида и мочевины составляет 1:1:1,5.
В другом воплощении настоящего изобретения выход соединения общей формулы 1 находится в диапазоне 42-60%.
Краткое описание графических материалов
Схема 1. Способ получения соответствующих тетрагидропиримидонов/тетрагидротиопиримидона.
Подробное описание изобретения
Теперь изобретение будет описано подробно, чтобы можно было более полно понять и оценить различные его аспекты.
Согласно настоящему изобретению предложены новые противотуберкулезные соединения формулы 1, эффективные против микобактерий (Mycobacterium bacilli), находящихся в стадии покоя.
,
где R представляет собой Н, галоген, дигалоген, О-алкил, ди-О-алкил, R1 представляет собой фенил, хлорфенил, нитрофенил, дихлорфенил, циклоалкил, предпочтительно циклогексил, X представляет собой О или S.
В предпочтительном аспекте согласно настоящему изобретению предложены 5-(замещенный)-фенилкарбамоил-4-(замещенный)-фенил-6-метил-1,2,3,4-тетрагидропиримид-2-он и -2-тион в качестве противотуберкулезных соединений формулы 1, которые приводятся ниже в Таблице 1.
Еще в одном другом предпочтительном аспекте согласно настоящему изобретению предложены соединения 5-(замещенный)-циклоалкил-4-(замещенный)-фенил-6-метил-1,2,3,4-тетрагидропиримидин-2-он и -2-тион формулы 1.
Соединения формулы 1 получают способом, в котором используется модифицированная реакции Биджинелли. Данный способ включает в себя взаимодействие замещенных ацетоацетанилидов (1 моль-экв.) с мочевиной/тиомочевиной (1,5 моль-экв.) и замещенными бензальдегидами (1 моль-экв.) в присутствии каталитического количества п-толуолсульфоновой кислоты (p-TSA) с использованием абсолютного этанола в качестве растворителя для получения соответствующих тетрагидропиримидонов/тетрагидротио-пиримидона.
Так, смесь замещенных ацетоацетанилидов, мочевины/тиомочевины и замещенных бензальдегидов в молярном соотношении 1:1,5:1 растворяют в абсолютном этаноле и нагревают до образования прозрачного раствора, затем добавляют каталитическое количество p-TSA и далее кипятят с обратным холодильником в течение приблизительно 3-5 ч. Отделенное твердое вещество промывают спиртом и сушат с получением соответствующих тетрагидропиримидонов/тетрагидротиопиримидона.
Замещенные ацетоацетанилиды получают традиционным способом, включающим в себя приведение во взаимодействие замещенного ароматического амина с этил/трет-бутил-ацетоацетатом в растворителе.
Приведенные выше синтезированные соединения по настоящему изобретению дополнительно охарактеризованы своими спектральными данными (данными инфракрасных спектров/спектров ядерного магнитного резонанса (ИК/ЯМР)).
Производные тетрагидропиримидонов/тетрагидротиопиримидона по настоящему изобретению могут быть использованы предпочтительно для лечения патологических состояний или заболеваний, вызываемых микобактериями.
В одном из аспектов противотуберкулезный потенциал новых соединений по настоящему изобретению тестируют в анализе, основанном на использовании целых клеток, проводимом в формате микропланшетов. В ходе выполнения протокола скрининга для подтверждения стадии покоя используют нитратредуктазную активность, тогда как для подтверждения активной фазы роста бацилл используют оптическую плотность культуры при 620 нм.
Активность соединений по настоящему изобретению против микобактерий, находящихся в стадии покоя, тестировали по их (соединений) активности в фазе роста, а также в фазе покоя, и данные ингибирующей активности приводятся ниже в Таблице 1 и Таблице 2.
Согласно Filippini et al. "ANTIMICROBIAL AGENTS AND CHEMOTHERAPY, June 2010, p.2712-2715 микобактерий могут активно расти в присутствии кислорода в среде. Предполагают, что у людей с латентным туберкулезом (ТВ), группы, составляющей приблизительно одну треть мирового населения, Mycobacterium tuberculosis находятся в нереплицирующемся (покоящемся) состоянии в очагах казеозных поражений легких с ограниченным доступом к кислороду или в располагающихся вне легких участках, содержащих жировую ткань. Нереплицирующиеся М. tuberculosis могут получаться в результате адаптации реплицирующихся культур к гипоксии посредством самогенерируемого формирования градиента кислорода (модель Уэйна) или внутри адипоцитов.
V.Virsodia и др. сообщали о синтезе и оценке замещенных N-фенил-6-метил-2-оксо-4-фенил-1,2,3,4-тетрагидропиримидин-5-карбоксамидов в качестве противотуберкулезных средств. Но было обнаружено, что эти средства активны против протестированного штамма микобактерий в активной фазе или фазе роста (EJMC, соединения (01-05), Таблица 3).
Противобактериальная активность соединения 8 для стадии покоя наблюдается для концентрации менее 4,5 мкг/мл.
В предпочтительном аспекте согласно настоящему изобретению предложен N,4-бис(3-хлорфенил)-6-метил-2-oкco-1,2,3,4-тетрагидропиримидин-5-карбоксамид, который эффективен против микобактерий в стадии покоя.
В одном из аспектов данное изобретение относится к фармацевтической композиции, содержащей активный ингредиент формулы 1, который определен выше, либо как таковой, либо в виде своих солей вместе с фармацевтически приемлемыми эксципиентами.
Фармацевтическая композиция по изобретению может быть представлена в форме твердого вещества, например порошков, гранул, таблеток, капсул, или может быть представлена в жидкой форме, такой как растворы, эмульсии, суспензии и так далее, или в форме композиции для инъекций.
Согласно данному изобретению предложено применение соединений формулы 1 и/или их производных против активных микобактерий, находящихся в стадии покоя.
Согласно одному из аспектов изобретения предложено применение соединения формулы 1 и/или его производного в изготовлении лекарственного средства или фармацевтической композиции против микобактерий в стадии покоя.
Производные тетрагидропиримидонов/тетрагидротиопиримидона формулы 1 и содержащие их фармацевтические композиции могут, согласно данному изобретению, быть введены с использованием любого количества, любой формы фармацевтической композиции и любого пути введения, эффективного для данного лечения. После изготовления лекарственной формы вместе с соответствующим фармацевтически приемлемым носителем в желаемой дозировке, как известно специалистам в данной области, фармацевтические композиции по данному изобретению могут быть введены любым способом, посредством которого активный фармацевтический ингредиент(ы) можно доставить в участок организма, в соответствии с чем он может оказывать терапевтический эффект на пациента.
Следующие далее примеры, которые включают предпочтительные воплощения, предназначаются для иллюстрации практического применения данного изобретения, при этом следует понимать, что указанные подробные данные приведены только в качестве примеров и с целью иллюстрации обсуждения предпочтительных воплощений изобретения, и они не ограничивают объем данного изобретения.
Примеры
Экспериментальная часть
Реакция Биджинелли: общая методика
СПОСОБА

Смесь замещенного ацетоацетанилида (1 мэкв.), замещенного бензальдегида (1 мэкв.) и мочевины (1,5 мэкв.) в 2 мл абсолютного спирта нагревали до получения прозрачного раствора в течение одного часа и затем в нее добавляли при 78°C каталитическое количество p-TSA (п-толуолсульфоновой кислоты) (15 мг). Смесь кипятили с обратным холодильником в течение 4,5 часа (или до тех пор, пока по данным TCL (тонкослойной хроматографии) не оставалось исходного вещества). По истечении некоторого времени продукт выпадал в осадок. Продукт отделяли фильтрацией и промывали спиртом (3x5 мл). Продукт сушили в сушильном шкафу при 60°C. Ту же методику можно использовать для синтеза всех производных. Для получения некоторых производных необходимо п-толуолсульфоновой кислоты (p-TSA) в количестве, превышающем каталитическое.
СПОСОБ В. Синтез тетрагидропиримидонов через промежуточную карбоновую кислоту (Соединения 4, 19, 23, 24)
Стадия 1. Синтез бензилацетоацетата

Бензилацетоацетат синтезировали путем кипячения с обратным холодильником трет-бутил-ацетоацетата и бензилового спирта в толуоле в течение 9 ч.
Стадия 2. Синтез бензилового эфира тетрагидропиримидона
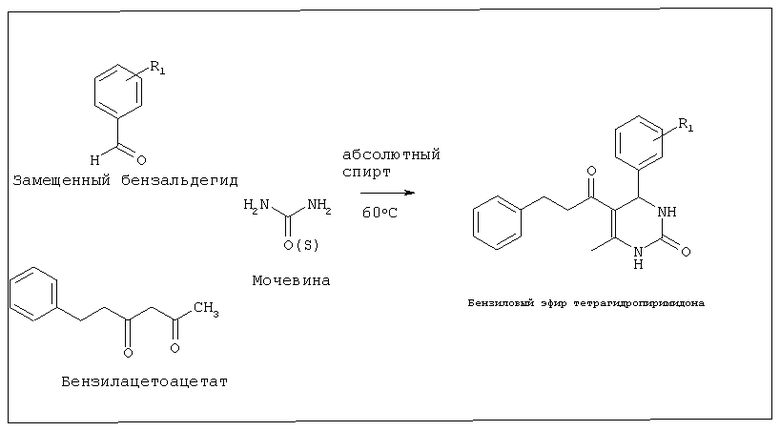
Смесь 1 эквивалента каждого из бензилацетоацетата и бензальдегида, 1,5 эквивалента мочевины, 10 мл абсолютного спирта и 5 капель конц. HCl кипятили с обратным холодильником в течение 4 часов. После завершения реакции (контролируемой с использованием TCL) смесь слегка охлаждали и затем опрокидывали в смесь льда и воды. Получившийся осадок отфильтровывали и перекристаллизовывали из кипящего спирта.
Стадия 3. Синтез тетрагидропиримидон-5-карбоновой кислоты

В атмосфере азота 1 эквивалент бензилового эфира тетрагидропиримидона, 5% Pd/C (10%-ного, масс/масс), формиат аммония (10 эквивалентов) и 5 мл безводного метанола перемешивали в течение 8 часов при 60°C(или до тех пор, пока по данным TCL не оставалось эфира). Добавляли 0,5 М раствор KOH до pH 9. Реакционную смесь фильтровали через целит. Остаток промывали метанолом и pH фильтрата подводили до 4, используя 2 М HCl. Остаток отфильтровывали и промывали водой для получения тетрагидропиримидон-5-карбоновой кислоты. Эту реакцию не использовали для продуктов конденсации тиомочевины, поскольку сера вызывает дезактивацию Pd/C. Кислоту тиооксопиримидонов следует синтезировать путем гидролиза этилового эфира соответствующих сложноэфирных производных с использованием NaOH или КОН.
Стадия 4. Синтез тетрагидропиримидон-5-карбоксамида
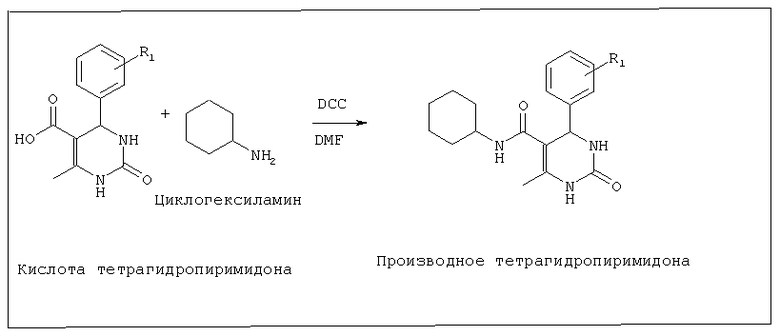
В атмосфере азота 1 эквивалент тетрагидропиримидон-5-карбоновой кислоты и 1,2 эквивалента дициклогексилкарбодиимида (DCC) в 5 мл безводного DMF перемешивали в течение 0,5 часа при 45°C и туда добавляли раствор предварительно взвешенного амина в безводном DMF (1 эквивалент). Перемешивание продолжали в течение 24 часов или до тех пор, пока вся кислота не израсходуется. Через 24 часа к реакционной смеси добавляли метанол и фильтровали для удаления дициклогексилмочевины. Метанол удаляли для получения тетрагидропиримидон-5-карбоксамида.
Примеры соединений, синтезированных с использованием вышеупомянутых двух способов, приведены ниже.
Пример 1: N-(3-хлорфенил)-4-(2,4-дихлорфенил)-6-метил-2-оксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид (1)
Синтезировали с использованием способа "А" из раздела экспериментальных методик (3-хлорацетоацетанилид; 2,4-дихлорбензальдегид).
Выход: 46%; т.пл. 201-202°C; ИК (KBr, см-1): 3227, 2953, 1703, 1680, 1472, 1236, 772; 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 1.71 (s, 3Н, СН3), 5.49 (s, 1Н, CH), 7.02-7.53 (m, 7Н, Ar-H), 7.6 (s, 1Н, NH), 8.95 (s, 1Н, NH), 9.45 (s, 1Н, NH).
Пример 2: N-(4-нитрофенил)-4-(2,4-дихлорфенил)-6-метил-2-оксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид (2)
Синтезировали с использованием способа "А" из раздела экспериментальных методик (4-нитроацетоацетанилид; 2,4-дихлорбензальдегид).
Выход: 55%; т.пл. 225-227°C; ИК (KBr, см-1): 3375, 2924, 1693, 1545, 1237, 756; 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 2.05 (s, 3H, CH3), 5.78 (s, 1Н, CH), 7.42-8.17 (m, 7Н, Ar-H), 7.69 (s, 1Н, NH), 9.03 (s, 1Н, NH), 10.32 (s, 1Н, NH).
Пример 3: N-(2,3-дихлорфенил-4-(2,4-дихлорфенил)-6-метил-2-оксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид (3)
Синтезировали с использованием способа "А" из раздела экспериментальных методик (2,3-дихлорацетоацетанилид; 2,4-дихлорбензальдегид).
Выход: 60%; т.пл. 264-266°C; ИК (KBr, см-1): 3286, 2928, 1707, 1458, 1214, 765; 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 2.14 (s, 3H, СН3), 5.74 (s, 1Н, CH), 7.36-7.70 (m, 6H, Ar-H), 7.46 (s, 1Н, NH), 8.95 (s, 1Н, NH), 9.45 (s, 1Н, NH).
Пример 4: N-циклогексил-4-фенил-6-метил-2-оксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид (4)
Синтезировали с использованием способа "В" из раздела экспериментальных методик (циклогексиламин; бензальдегид).
Выход: 54%; т.пл. 192-194°C; ИК (KBr, см-1): 3281, 2924, 1710, 1568, 1254, 756; 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 1.1 (m, 6Н, циклогексил), 1.61 (m, 5Н, циклогексил), 1.94 (s, 3H, СН3), 5.28 (s, 1Н, CH), 7.22-7.3 (m, 5Н, Ar-H), 7.45 (m, 2Н, NH), 8.5 (s, 1Н, NH).
Пример 5: N-(3-хлорфенил)-4-фенил-6-метил-2-оксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид (5)
Синтезировали с использованием способа "А" из раздела экспериментальных методик (3-хлорацетоацетанилид; бензальдегид).
Выход: 51%; т.пл. 236-237°C; ИК (KBr, см-1): 3281, 2924, 1710, 1566, 1248, 775; 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 1.7 (s, 3H, СН3), 5.06 (s, 1Н, CH), 6.69-6.73 (d, 1Н, Ar-H), 6.88-6.95 (m, 6Н, Ar-H), 7.3 (s, 1Н, Ar-H), 7.58 (d, 1Н, Ar-H), 7.1 (s, 1Н, NH), 8.47 (s, 1Н, NH), 9.41 (s, 1Н, NH).
Пример 6: N-(3-хлорфенил)-4-фенил-6-метил-2-тиоксо-1,2,3,4-
тетрагидропиримидин-5-карбоксамид (6)
Синтезировали с использованием способа "А" из раздела экспериментальных методик (3-хлорацетоацетанилид; бензальдегид).
Выход: 42%; т.пл. 140-142°C; ИК (KBr, см-1): 3176, 2923, 1670, 1570, 1502, 1121; 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 2.07 (s, 3H, СН3), 5.4 (s, 1Н, СН), 7.09 (d, 1Н, Ar-H), 7.24-7.36 (m, 6Н, Ar-H), 7.46 (d, 1Н, Ar-H), 7.74 (s, 1Н, Ar-H), 9.49 (s, 1Н, NH), 9.93 (s, 1Н, NH), 10.08 (s, 1Н, NH).
Пример 7: N-(4-нитрофенил)-4-фенил-6-метил-2-оксо-1,2,3,4- тетрагидропиримидин-5-карбоксамид (7)
Синтезировали с использованием способа "А" из раздела экспериментальных методик (4-нитроацетоацетанилид; бензальдегид).
Выход: 42%; т.пл. 217-219°C; ИК (KBr, см-1): 3520, 3361, 1605, 1570, 1502, 1121; 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 2.04 (s, 3H, СН3), 5.41 (s, 1Н, СН), 7.2-7.33 (m, 5Н, Ar-H), 7.75-7.8 (d, 2Н, Ar-H), 8.11-8.16 (d, 2Н, Ar-H), 8.89 (s, 1Н, NH), 8.90 (s, 1Н, NH), 10.12 (s, 1Н, NH).
Пример 8: N-(3-хлорфенил)-4-(3-хлорфенил)-6-метил-2-оксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид (8)
Синтезировали с использованием способа "А" из раздела экспериментальных методик (3-хлорацетоацетанилид; 3-хлорбензальдегид).
Выход: 45%; т.пл. 180-182°C; ИК (KBr, см-1): 3240, 1633, 1597, 1502, 1111; 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 2.05 (s, 3H, СН3), 5.39 (s, 1Н, CH), 7.08 (m, 1Н, Ar-H), 7.29-7.38 (m, 6Н, Ar-H), 7.72 (s, 1Н, NH), 7.73 (s, 1Н, Ar-H), 8.89 (s, 1Н, NH), 9.49 (s, 1Н, NH).
Пример 9: N-(2,3-дихлорфенил)-4-(3-хлорфенил)-6-метил-2-тиоксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид (9)
Синтезировали с использованием способа "А" из раздела экспериментальных методик (2,3-дихлорацетоацетанилид; 3-хлорбензальдегид).
Выход: 52%; т.пл. 182-183°C; ИК (KBr, см-1): 3232, 2953, 1685, 1580, 1502, 1121, 771; 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 2.13 (s, 3H, СН3), 5.34 (s, 1Н, CH), 7.31 (m, 8Н, Ar-H), 7.69 (s, 1Н, NH), 8.89 (s, 1Н, NH), 9.3 (s, 1Н, NH).
Пример 10: N-(3-хлорфенил)-4-(3,4-диметоксифенил)-6-метил-2-оксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид (10)
Синтезировали с использованием способа "А" из раздела экспериментальных методик (3-хлорацетоацетанилид; 3,4-диметокси-бензальдегид).
Выход: 58%; т.пл. 259-262°C; ИК (KBr, см-1): 3247, 2953, 1630, 1580, 1502, 721; 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 2.04 (s, 3H, СН3), 3.66 (s, 3H, ОСН3), 3.71 (s, 3H, OCH3), 5.36 (s, 1Н, CH), 6.8 (dd, 1Н, СН, 2 Гц, 8 Гц), 6.8 (d, 1Н, Ar-H, 8 Гц) 6.9 (d, 1Н, Ar-H, 8 Гц), 7.07 (dd, 1Н, Ar-H, 2 Гц, 8 Гц), 7.28 (t, 1Н, Ar-H), 7.44 (dd, 1Н, Ar-H, 2 Гц, 8 Гц), 7.75 (s, 1Н, Ar-H), 7.57 (s, 1Н, NH), 8.75 (s, 1Н, NH), 9.75 (s, 1Н, NH).
Пример 11: N,4-бис(3-хлорфенил)-6-метил-2-тиоксо-1,2,3,4- тетрагидропиримидин-5-карбоксамид (11)
Синтезировали с использованием способа "А" из раздела экспериментальных методик (3-хлорацетоацетанилид; 3-хлорбензальдегид).
Выход: 55%; т.пл. 157-159°C; ИК (KBr, см-1): 3232, 2953, 1685, 1580, 1502, 1121, 771; 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 2.08 (s, 3H, СН3), 5.39 (s, 1Н, CH), 7.08 (s, 1Н, Ar-H), 7.2-7.4 (m, 6Н, Ar-Н), 7.73 (s, 1Н, Ar-H), 9.52 (s, 1Н, NH), 9.58 (s, 1Н, NH), 10.14 (s, 1Н, NH).
Пример 12: N-(2,3-дихлорфенил)-4-(3-хлорфенил)-6-метил-2-тиоксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид (12)
Синтезировали с использованием способа "А" из раздела экспериментальных методик (2,3-дихлорацетоацетанилид; 3-хлорбензальдегид).
Выход: 51%; т.пл. 194-196°C; ИК (KBr, см-1): 3232, 2953, 1685, 1565, 1517, 1182, 774; 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 2.21 (s, 3H, СН3), 5.42 (s, 1Н, CH), 7.3-7.49 (m, 7Н, Ar-H), 9.62 (s, 1Н, NH), 9.64 (s, 1Н, NH), 10.21 (s, 1Н, NH).
Пример 13: N-(2,3-дихлорфенил)-4-(2,4-дихлорфенил)-6-метил-2-тиоксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид (13)
Синтезировали с использованием способа "А" из раздела экспериментальных методик (2,3-дихлорацетоацетанилид; 2,4-дихлор-бензальдегид).
Выход: 51%; т.пл. 236-237°C; ИК (KBr, см-1): 3400, 3238, 2953, 1685, 1567, 1516, 1181, 773; 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 2.14 (s, 3H, CH3), 5.74 (s, 1Н, CH), 7.31 (t, 1Н, Ar-H), 7.36 (dd, 1Н, Ar-H, 8 Гц), 7.4 (s, 1Н, Ar-H), 7.46 (dd, 1Н, Ar-H, 2 Гц, 8 Гц), 7.51 (dd, 1Н, Ar-H, 2 Гц, 8 Гц), 9.41 (s, 1Н, NH), 9.71 (s, 1Н, NH), 10.16 (s, 1Н, NH).
Пример 14: N-(2,3-дихлорфенил)-6-метил-4-фенил-2-тиоксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид (14)
Синтезировали с использованием способа "А" из раздела экспериментальных методик (2,3-дихлорацетоацетанилид; бензальдегид).
Выход: 52%; т.пл. 208-209°C; ИК (KBr, см-11): 3387, 3251, 2953, 1633, 1567, 1516, 1271, 773; 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 2.18 (s, 3H, СН3), 5.4 (s, 1Н, CH), 7.31-7.4 (m, 7Н, Ar-H), 9.49 (s, 1Н, NH), 9.5 (s, 1Н, NH), 10.08 (s, 1Н, NH).
Пример 15: N-(3-нитрофенил)-6-метил-4-фенил-2-тиоксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид (15)
Синтезировали с использованием способа "А" из раздела экспериментальных методик (3-нитроацетоацетанилид; бензальдегид).
Выход: 40%; т.пл. 154-156°C; ИК (KBr, см-1): 3362, 3251, 2953, 1531, 1633, 1567, 1245, 1205, 752; 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 2.11 (s, 3H, СН3), 5.4 (s, 1Н, CH), 7.25-7.38 (m, 5Н, Ar-H), 7.56 (t, 1Н, Ar-H, 8 Гц), 7.86 (dd, 1Н, Ar-H, 2 Гц, 8 Гц), 7.92 (dd, 1Н, Ar-H, 2 Гц, 8 Гц), 8.58 (s, 1Н, Ar-H), 9.5 (s, 1Н, NH), 10.1 (s, 1Н, NH), 10.18 (s, 1Н, NH).
Пример 16: N-(4-нитрофенил)-4-(3-хлорфенил)-6-метил-2-оксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид (16)
Синтезировали с использованием способа "А" из раздела экспериментальных методик (4-нитроацетоацетанилид; 3-хлорбензальдегид).
Выход: 48%; т.пл. 275-276°C; ИК (KBr, см-1): 3362, 3251, 2953, 1531, 1633, 1559, 1245, 1205, 752; 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 2.11 (s, 3H, СН3), 5.4 (s, 1Н, CH), 7.31-7.36 (m, 4Н, Ar-H), 7.80 (s, 1Н, NH), 7.82 (d, 2Н, Ar-H, 8 Гц), 8.22 (d, 2Н, Ar-H, 8 Гц), 9.03 (s, 1Н, NH), 10.02 (s, 1Н, NH).
Пример 17: N-(4-нитрофенил)-6-метил-4-фенил-2-тиоксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид (17)
Синтезировали с использованием способа "А" из раздела экспериментальных методик (4-нитроацетоацетанилид, бензальдегид).
Выход: 48%; т.пл. 231-233°C; ИК (KBr, см-1): 3468, 3369, 3238, 2953, 1685, 1540, 1516, 1203, 1181, 746; 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 2.08 (s, 3H, СН3), 5.39 (s, 1Н, CH), 7.08-7.43 (m, 7Н, Ar-H), 9.52 (s, 1Н, NH), 9.96 (s, 1Н, NH), 10.14 (s, 1Н, NH).
Пример 18: N-(3-хлорфенил)-4-(2,4-дихлорфенил)-6-метил-2-тиоксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид (18)
Синтезировали с использованием способа "А" из раздела экспериментальных методик (3-хлорацетоацетанилид, 2,4-дихлорбензальдегид).
Выход: 58%; т.пл. 215-217°C; ИК (KBr, см-1): 3403, 3232, 2953, 1685, 1581, 1181, 773; 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 2.03 (s, 3H, СН3), 5.74 (s, 1Н, CH), 7.07 (d, 1Н, Ar-H, 8 Гц), 7.28 (d, 1Н, Ar-H, 8 Гц), 7.39-7.46 (m, 2Н, Ar-H и s, 1Н, NH), 7.54 (s, 1Н, Ar-H), 7.61 (s, 1Н, Ar-H), 7.71 (s, 1Н, Ar-H), 8.92 (s, 1Н, NH), 9.89 (s, 1Н, NH).
Пример 19: N-циклогексил-6-метил-4-фенил-2-тиоксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид (19)
Синтезировали с использованием способа "А" из раздела экспериментальных методик (циклогексиламин, бензальдегид).
Выход: 48%; т.пл. 261-262°C; ИК (KBr, см-1): 3381, 3285, 2924, 1710, 1568, 1254, 756; 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 1.1 (m, 6Н, циклогексил), 1.61 (m, 5Н, циклогексил), 1.97 (s, 3H, СН3), 5.28 (s, 1Н, CH), 7.19-7.23 (m, 5Н, Ar-H), 7.55 (s, 1Н, NH), 9.28 (s, 1Н, NH), 9.8 (s, 1Н, NH).
Пример 20: N-(4-нитрофенил)-4-(2,4-дихлорфенил)-6-метил-2-тиоксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид (20)
Синтезировали с использованием способа "А" из раздела экспериментальных методик (4-нитроацетоацетанилид, 2,4-дихлорбензальдегид).
Выход: 54%; т.пл. 183-185°C; ИК (KBr, см-1): 3395, 3238, 2953, 1685, 1567, 1516, 1161, 773; 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 2.05 (s, 3H, СН3), 5.79 (s, 1Н, CH), 7.31 (t, 1Н, Ar-H), 7.42-7.44 (dd, 1Н, Ar-H, 8 Гц), 7.46-7.48 (dd, 1Н, Ar-H, 8 Гц), 7.54 (s, 1Н, Ar-H), 7.61 (s, 1Н, Ar-H), 7.7 (s, 1Н, NH), 7.74-7.76 (d, 1Н, Ar-H, 8 Гц), 8.15-8.17 (d, 1Н, Ar-H, 8 Гц), 9.04 (s, 1Н, NH), 10.32 (s, 1Н, NH).
Пример 21: N-(2,3-дихлорфенил)-6-метил-2-оксо-4-фенил-1,2,3,4-тетрагидропиримидин-5-карбоксамид (21)
Синтезировали с использованием способа "А" из раздела экспериментальных методик (2,3-дихлорацетоацетанилид, бензальдегид).
Выход: 52%; т.пл. 261-263°C; ИК (KBr, см-1): 3402, 3228, 2953, 1633, 1271, 773; 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 2.16 (s, 3H, СН3), 5.38 (s, 1Н, CH), 7.28-7.45 (m, 7Н, Ar-H), 7.67 (s, 1Н, NH), 8.86 (s, 1Н, NH), 9.21 (s, 1Н, NH).
Пример 22: N-(3-нитрофенил)-4-(3-хлорфенил)-6-метил-2-оксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид (22)
Синтезировали с использованием способа "А" из раздела экспериментальных методик (3-нитроацетоацетанилид, 3-хлорбензальдегид).
Выход: 52%; т.пл. 221-223°C; ИК (KBr, см-1): 3402, 3228, 2953, 1633, 1567, 1516, 1271, 773; 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 2.07 (s, 3H, СН3), 5.47 (s, 1Н, CH), 7.20-7.9 (m, 8Н, Ar-H), 8.57 (s, 1Н, NH), 8.95 (s, 1Н, NH), 10.41 (s, 1Н, NH).
Пример 23: N-циклогексил-4-(2,4-дихлорфенил)-6-метил-2-тиоксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид (23)
Синтезировали с использованием способа "А" из раздела экспериментальных методик (циклогексиламин, 2,4-дихлорбензальдегид).
Выход: 58%; т.пл. 255-257°C; ИК (KBr, см-1): 3281, 2924, 1710, 1568, 1254, 756; 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 1.1 (m, 6Н, циклогексил), 1.61 (m, 5Н, циклогексил), 2.16 (s, 3H, СН3), 5.38 (s, 1Н, CH), 7.31 (d, 1Н, Ar-H, 8 Гц), 7.47 (dd, 1Н, Ar-H, 2 Гц, 8 Гц), 7.56 (d, 1Н, Ar-H, 8 Гц), 7.68 (s, 1Н, NH), 9.18 (s, 1Н, NH), 9.9 (s, 1Н, NH).
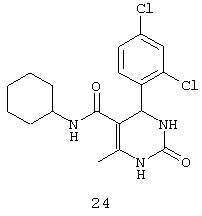
Пример 24: N-циклогексил-4-(2,4-дихлорфенил)-6-метил-2-оксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид (24)
Синтезировали с использованием способа "В" из раздела экспериментальных методик (циклогексиламин, 2,4-дихлорбензальдегид).
Выход: 58%; т.пл. 235-237°C; ИК (KBr, см-1): 3281, 2924, 1710, 1568, 1254, 756; 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 1.1 (m, 6Н, циклогексил), 1.61 (m, 5Н, циклогексил), 1.93 (s, 3H, СН3), 5.6 (s, 1Н, CH), 7.39 (d, 2Н, Ar-H, 8 Гц), 7.43 (dd, 1Н, Ar-H, 2 Гц, 8 Гц), 7.53 (d, 1Н, Ar-H, 8 Гц), 7.37 (s, 1Н, NH), 7.47 (s, 1Н, NH), 8.93 (s, 1Н, NH).
Пример 25
Для скрининга соединений использовали протокол, позволяющий идентифицировать ингибиторы активных, а также покоящихся туберкулезных бацилл. Для подтверждения стадии покоя использовали нитратредуктазную активность, тогда как для подтверждения активной фазы роста бацилл в этом протоколе скрининга использовали оптическую плотность культуры при 620 нм. По 2,5 мкл раствора соединений в концентрации 10 мг/мл в DMSO (диметилсульфоксиде) вносили в стерильных условиях в отдельные лунки стерильных 96-луночных планшетов. В каждую лунку в стерильных условиях вносили по 247,5 мкл культуры BCG (бациллы Кальметта-Герена) штамма М. bovis (АТСС (Американская коллекция типовых культур) 35745; получена из подразделения по R&D (R&D (research and development) Division) компании M/S Astrazeneca в Бангалоре), содержащей ~105 клеток/мл, дополненной 40 мМ NaNO3, с получением конечного объема 250 мкл и планшет герметично закрывали. В каждой лунке оставляли по 125 мкл пространства, чтобы отношение свободного пространства над культурой к объему культуры составляло точно 0,5. После герметичного закрывания планшеты с культурами инкубировали при 37°C. По окончании 8 суток инкубации проводили измерение OD (оптической плотности) при 620 нм. Затем из каждой лунки отбирали по 80 мкл культуры и переносили в отдельный 96-луночный планшет. Далее в каждую лунку добавляли по 80 мкл 1%-ной сульфаниловой кислоты и по 80 мкл 0,1%-ного раствора N-(1-нафтил)-этилендиамина дигидрохлорида и планшет инкубировали в течение 15 минут при комнатной температуре для развития розовой окраски. Интенсивность окраски измеряли, используя Spectramaxplus 384 (Molecular Devices, USA), при 540 нм с целью определения нитратредуктазной активности.
Пример 26
Соединения по изобретению тестировали по их активности в фазе роста бацилл, и результаты сведены в данном описании в таблицу. В Таблицу 1 включены величины IC50 и кривые зависимости ответа от дозы для соединений.
ингибиторов для фазы роста
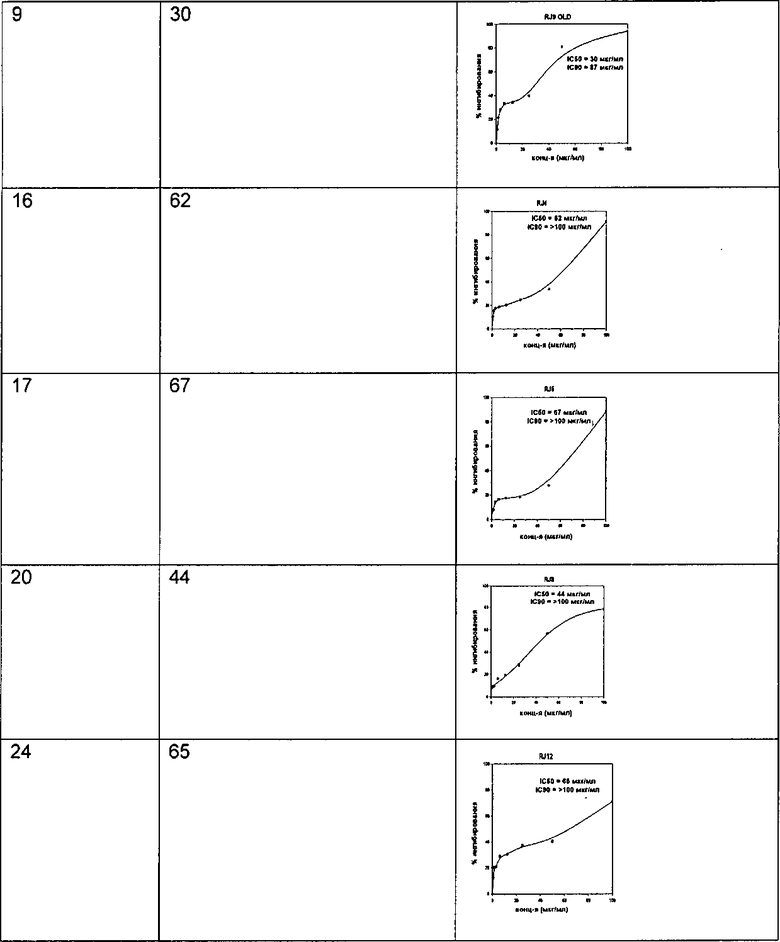
Пример 27
Соединения по изобретению тестировали по их активности в фазе покоя бацилл, и результаты сведены в данном описании в таблицу. В Таблицу 2 включены величины IC50 и кривые зависимости (ответа от дозы для соединений.

Пример 28. Данные по ингибированию для соединений (01-05) из статьи V. Virsodia и др. в European Journal of Medicinal Chemistry (EJMC).
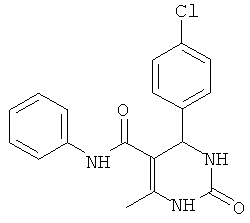
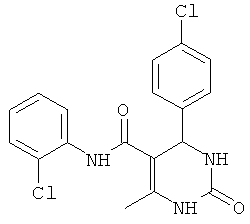


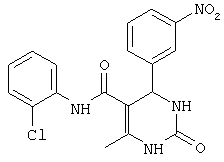
Установлено, что средства, известные из предшествующего уровня техники, активны против протестированного штамма микобактерий только в их активной фазе или фазе роста.
Теперь согласно настоящему изобретению решена проблема, связанная с туберкулезом, путем использования новых производных тетрагидропиримидонов/тетрагидротиопиримидона формулы 1, эффективных против микобактерий, находящихся в стадии покоя.
| название | год | авторы | номер документа |
|---|---|---|---|
| Применение 1,6-диметил-N-фенил-4-(4-фторфенил)-2-тиоксо-1,2,3,4-тетрагидропиримидин-5-карбоксамида в качестве средства, обладающего анальгетической активностью | 2022 |
|
RU2796070C1 |
| Применение 4-(4-бромфенил)-6-метил-N-(2-хлорфенил)-2-тиоксо-1,2,3,4-тетрагидропиримидин-5-карбоксамида в качестве средства, обладающего анальгетической активностью | 2022 |
|
RU2793550C1 |
| ПРИМЕНЕНИЕ 4-(4-БРОМФЕНИЛ)-6-МЕТИЛ-2-ТИОКСО-N-(о-ТОЛИЛ)-1,2,3,4-ТЕТРАГИДРОПИРИМИДИН-5-КАРБОКСАМИДА В КАЧЕСТВЕ СРЕДСТВА, ОБЛАДАЮЩЕГО АНАЛЬГЕТИЧЕСКОЙ АКТИВНОСТЬЮ | 2024 |
|
RU2835601C1 |
| БИЦИКЛИЧЕСКИЕ ТЕТРАГИДРОТИАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ НЕОПЛАСТИЧЕСКИХ И/ИЛИ ИНФЕКЦИОННЫХ ЗАБОЛЕВАНИЙ | 2016 |
|
RU2730524C2 |
| ГИБРИДНЫЕ СОЕДИНЕНИЯ НА ОСНОВЕ ФУРАН-2(3Н)-ОНА И ХРОМЕН-4(4Н)-ТИОНА, ОБЛАДАЮЩИЕ АНТИБАКТЕРИАЛЬНОЙ АКТИВНОСТЬЮ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2024 |
|
RU2830172C1 |
| ПРОИЗВОДНЫЕ ДИАЗЕПИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ АРИТМИИ | 1994 |
|
RU2155587C2 |
| СПОСОБ ПОЛУЧЕНИЯ ДИСПИРОИНДОЛИНОНОВ | 2018 |
|
RU2682678C1 |
| 2-{ [3-циано-4-R-5,6,7,8-тетрагидрохинолин-2-ил]тио} -N[(2Z)-3-Ar-4-фенил-1,3-тиазол-2(3Н)-илиден]ацетамиды, способ их получения и применение в качестве антидотов 2,4-Д на подсолнечнике | 2022 |
|
RU2786234C1 |
| ПРОИЗВОДНЫЕ ИНДОЛИН-2-ОНА В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗ | 2013 |
|
RU2627706C2 |
| БИОЛОГИЧЕСКИ АКТИВНЫЕ ВЕЩЕСТВА, ПОДАВЛЯЮЩИЕ ПАТОГЕННЫЕ БАКТЕРИИ, И СПОСОБ ИНГИБИРОВАНИЯ СЕКРЕЦИИ III ТИПА У ПАТОГЕННЫХ БАКТЕРИЙ | 2012 |
|
RU2495036C1 |

Изобретение относится к новым соединениям общей формулы 1 обладающим противотуберкулезной активностью, и способу их получения. Соединения настоящего изобретения проявляют противотуберкулезную активность в отношении микобактерий, находящихся как в активной фазе, так и в состоянии покоя, что повышает эффективность снижения частоты возникновения туберкулеза и позволяет снизить заболеваемость и смертность от этого заболевания. В общей формуле 1
R представляет собой Н, галоген, дигалоген, R1 представляет собой хлорфенил, нитрофенил, дихлорфенил, циклогексил, X представляет собой О или S, при этом соединения выбраны из группы: N-(3-хлорфенил)-4-(3-хлорфенил)-6-метил-2-оксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид, N-(2,3-дихлорфенил)-4-(3-хлорфенил)-6-метил-2-оксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид, N-(4-нитрофенил)-4-(3-хлорфенил)-6-метил-2-оксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид), N-(4-нитрофенил)-6-метил-4-фенил-2-тиоксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид, N-(4-нитрофенил)-4-(2,4-дихлорфенил)-6-метил-2-тиоксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид, N-циклогексил-4-(2,4-дихлорфенил)-6-метил-2-оксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид. Способ получения соединений формулы 1 включает а) смешивание замещенного ацетоацетанилида, замещенного альдегида и мочевины, в молярном соотношении 1:1:1,5 в растворителе, предпочтительно абсолютном этаноле; b) нагревание реакционной смеси, полученной на стадии (а), при температуре в диапазоне 60-100°С в течение периода времени в диапазоне 4-8 часов; c) добавление p-TSA (п-толуолсульфоновой кислоты) в реакционную смесь, полученную на стадии (b), с последующим кипячением с обратным холодильником при температуре в диапазоне 60-100°С в течение периода времени в диапазоне 4-8 часов; d) охлаждение реакционной смеси, полученной на стадии (с), с последующим отделением твердого вещества фильтрацией, промывкой спиртом. 5 н. и 4 з.п. ф-лы, 1 ил., 3 табл., 27 пр.
1. Соединения общей формулы 1,
где R представляет собой Н, галоген, дигалоген, R1 представляет собой хлорфенил, нитрофенил, дихлорфенил, циклогексил, X представляет собой О или S, где соединения выбраны из группы:
N-(3-хлорфенил)-4-(3-хлорфенил)-6-метил-2-оксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид (8),
N-(2,3-дихлорфенил)-4-(3-хлорфенил)-6-метил-2-оксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид (9),
N-(4-нитрофенил)-4-(3-хлорфенил)-6-метил-2-оксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид (16),
N-(4-нитрофенил)-6-метил-4-фенил-2-тиоксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид (17),
N-(4-нитрофенил)-4-(2,4-дихлорфенил)-6-метил-2-тиоксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид (20),
N-циклогексил-4-(2,4-дихлорфенил)-6-метил-2-оксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид (24).


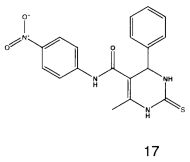
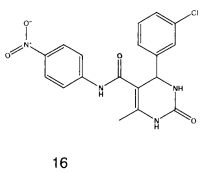

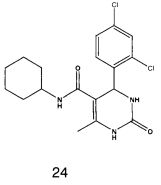
2. Соединения общей формулы 1 по п. 1, проявляющие противотуберкулезную активность.
3. Соединения формулы 1 по п. 1, причем указанное соединение используют против микобактерий, находящихся в фазе покоя.
4. Фармацевтическая композиция, обладающая противотуберкулезной активностью, содержащая эффективное количество соединения формулы 1 по п. 1 и фармацевтически приемлемый эксципиент.
5. Способ получения соединений формулы 1 по п. 1, включающий:
а) смешивание замещенного ацетоацетанилида, замещенного альдегида и мочевины, в молярном соотношении 1:1:1,5 в растворителе, предпочтительно абсолютном этаноле;
b) нагревание реакционной смеси, полученной на стадии (а), при температуре в диапазоне 60-100°С в течение периода времени в диапазоне 4-8 часов;
c) добавление p-TSA (п-толуолсульфоновой кислоты) в реакционную смесь, полученную на стадии (b), с последующим кипячением с обратным холодильником при температуре в диапазоне 60-100°С в течение периода времени в диапазоне 4-8 часов;
d) охлаждение реакционной смеси, полученной на стадии (с), с последующим отделением твердого вещества фильтрацией, промывкой спиртом с получением соединений формулы
 ,
,
где X представляет собой О, a R1 и R являются такими, как определено в п. 1.
6. Способ по п. 5, где замещенный ацетоацетанилид, используемый на стадии (а), выбран из группы, состоящей из 3-хлорацетоацетанилида, 4-нитроацетоацетанилида, 2,3-дихлорацетоацетанилида и 3-нитроацетоацетанилида.
7. Способ по п. 5, где замещенный альдегид, используемый на стадии (а), выбран из группы, состоящей из 2,4-дихлорбензальдегида, бензальдегида, 3-хлорбензальдегида.
8. Применение соединения по любому из пп. 1-3 против микобактерий в стадии покоя.
9. Применение соединения по любому из пп. 1-3 в изготовлении лекарственного средства или фармацевтической композиции против микобактерий в стадии покоя.
| VISORDIA V;et al., Synthesis, screening for antitubercular activity and 3D-QSAR studies of substituted N-phenyl-6-methyl-2-oxo-4-phenyl-1,2,3,4-tetrahydro-pyrimidine-5-carboxamides, EUROPEAN JOURNAL OF MEDICINAL CHEMISTRY, 2008, Vol:43, Nr:10, Page(s):2103 - 2115 | |||
| AKSHAY M PANSURIYA et al., Cation Exchange Resin (Indion 130): An Efficient, |

