Область техники
Настоящее изобретение относится к способу простого и удобного получения ваборбактама, поэтому оно относится к области медицинской биохимической промышленности.
Уровень техники
Ваборбактам (I, Vaborbactam) представляет собой новый не-β-лактамный ингибитор β-лактамазы на основе фармакофора циклической борной кислоты и представляет собой ингибитор β-лактамазы типа A и C широкого применения. Разработчиком этого продукта являются копания «Medicines Company» и Министерство здравоохранения и социального обеспечения США, а исходной исследовательской компанией является компания «Rempex Pharmaceuticals».
В августе 2017 года Управление по контролю качества продуктов и лекарств США одобрило многокомпонентный препарат «Vabomere», который состоит из ваборбактама и меропенема, для лечения сложных инфекций мочевыводящих путей (cUTI) у взрослых, в том числе пиелонефрита, вызванного чувствительными энтеробактериями.
Номером CAS ваборбактама (I) является 1360457-46-0, а химическим названием является (3R,6S)-{2-гидрокси-3-[2-(тиофен-2-ил)ацетамидо]-[1,2]-оксаборинан-6-ил}уксусная кислота, структурная формула которой следующая:
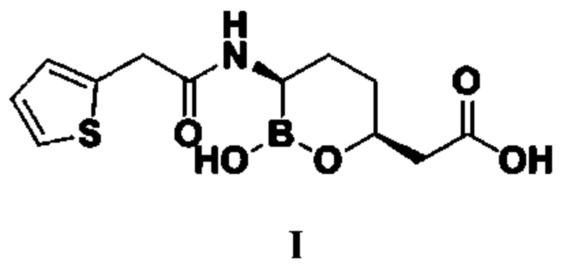
В документе «Journal of Medicinal Chemistry», 2015 г., 58, 3682-3692 и в патентном документе № CN103180328A говорится об использовании сложного трет-бутилового эфира R-3-гидрокси-4-пентеновой кислоты в качестве исходного сырья для изготовления ваборбактама (I). Сложный трет-бутиловый эфир R-3-гидрокси-4-пентеновой кислоты сначала вступает в реакцию с диметил-трет-бутилхлорсиланом для получения защищенного гидроксилом трет-бутилпентеноата под действием имидазола, а затем под каталитическим действием металлического иридия реагирует с пинакол-бораном с получением промежуточного соединения 4 из концевого присоединяемого бора; это промежуточное соединение 4 реагирует с (1S,2S,3R,5S)-(+)-2,3-пинандиолом с получением промежуточного соединения 5, а затем в условиях низкой температуры -95°C и под действием н-бутиллития реагирует с дихлорметаном с получением промежуточного соединения 6, чтобы увеличивалась хиральная углеродная цепь; к промежуточному соединению 6 при низкой температуре -78°C капельно добавляют бис(триметилсилил)амид лития, а затем оставляют реагировать при комнатной температуре в течение 16 часов; затем при 0°C под действием карбоксильного активатора EDCI и HOBt конденсируют с 2-тиофенуксусной кислотой с получением промежуточного соединения 7; под действием соляной кислоты 3N промежуточное соединение 7 подвергается циклизации и гидролизу с получением целевого продукта I. Подробнее см. схему реакции 1.
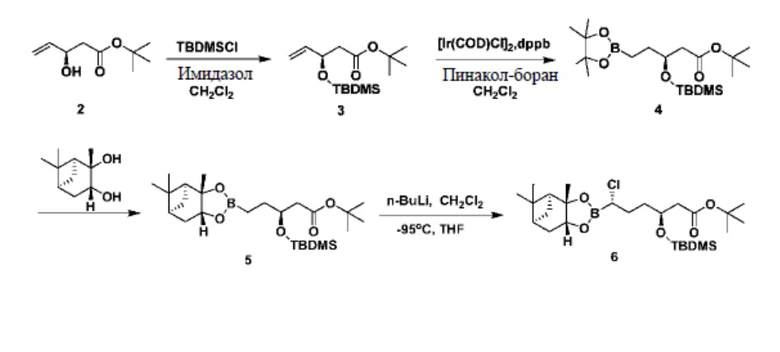
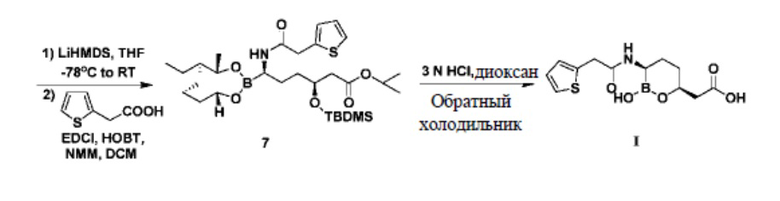
Схема реакции 1
Однако используемое в вышеуказанной схеме реакции 1 сырье нелегко получить, и его цена очень высокая; условия реакции жестокие, температура реакции на двух этапах очень низкая, а также нужно использовать литиевые реагенты, безопасность применения которых очень низкая, что является неблагоприятным для промышленного производства.
В патентном документе № US20170057979 описан способ синтеза, схожий с вышеуказанной схемой реакции 1; разница заключается в том, что в этом способе сначала гидролизуется сложный пинаколовый эфир бороновой кислоты, потом после использования этаноламина для образования спиросоединения обеспечивается реакция с (1S,2S,3R,5S)-(+)-2,3-пинандиолом; последующие реакции являются аналогичными; в конце с использованием разбавленной серной кислоты и борной кислоты проводятся циклизация и гидролиз с получением целевого продукта I. Подробнее см. схему реакции 2.
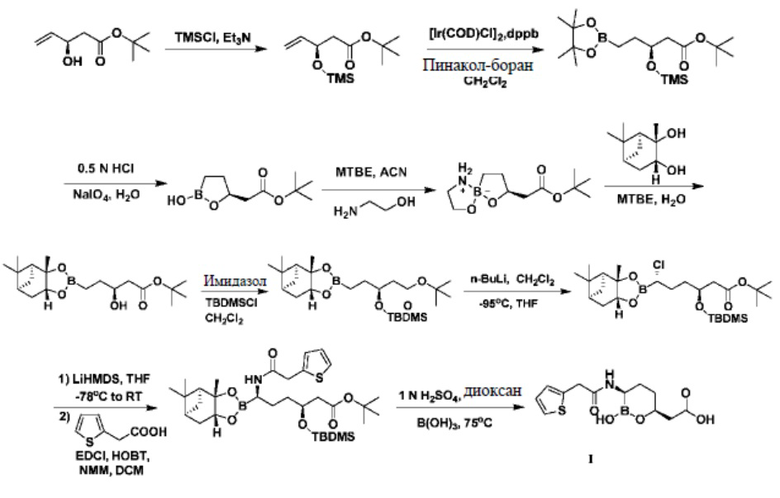
Схема реакции 2
Однако используемое в вышеуказанной схеме реакции 2 сырье по-прежнему дорогое и его нелегко получить; условия реакции жестокие, температура реакции на двух этапах очень низкая, а также нужно использовать литиевые реагенты, безопасность применения которых очень низкая, что является неблагоприятным для промышленного производства.
Суть изобретения
С учетом недостатков, известных из уровня техники, согласно настоящему изобретению предложен способ простого и удобного получения ваборбактама, то есть (3R,6S)-{2-гидрокси-3-[2-(тиофен-2-ил)ацетамидо]-[1,2]-оксаборинан-6-ил}уксусной кислоты. Сырье для настоящего изобретения легко приобрести по умеренной цене, поэтому себестоимость низкая; технологический процесс является безопасным, простым и удобным для осуществления и не требует жестких условий реакции, поэтому процесс реакции безопасен для окружающей среды и подходит для целей промышленного производства.
Описание терминов:
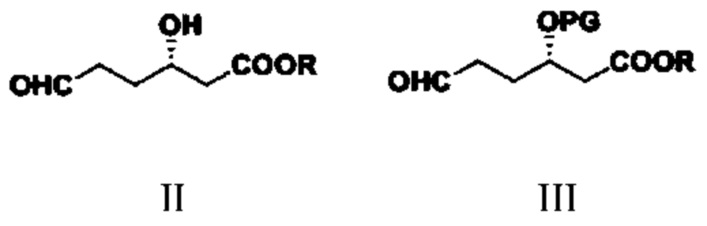
Соединение типа II: S-3-гидрокси-6-оксогексаноат;
соединение типа III: S-3-(алкокси-защитная-группа)-6-оксогексаноат; в структурной формуле PG представляет собой защитную группу;
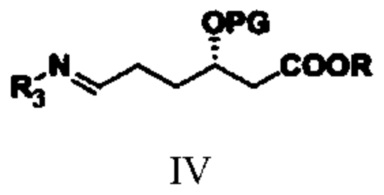
соединение типа IV: S-3-(алкокси-защитная-группа)-6-(N-амино-заместитель)гексаноат;
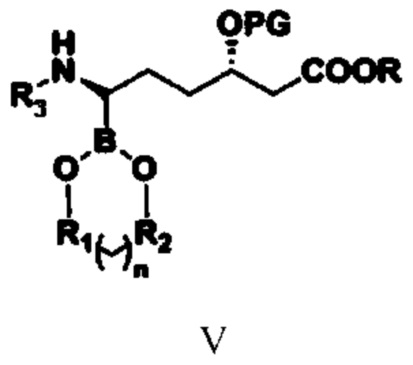
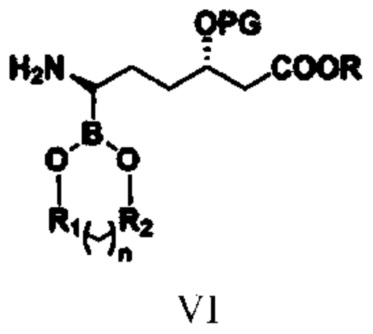
соединение типа V: (3S,6R)-3-(алкокси-защитная-группа)-6-(N-амино-заместитель)-6-боратгексаноат;
соединение типа VI: (3S,6R)-3-(алкокси-защитная-группа)-6-амино-6-боратгексаноат;
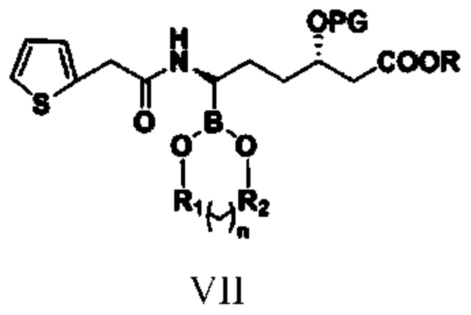
соединение типа VII: (3S,6R)-3-(алкокси-защитная-группа)-6-борат-6-[2-(2-тиофен)ацетамидо]гексаноат.
Номера соединений в этом описании полностью совпадают с номерами структурных формул, и они имеют одинаковое ссылочное отношение, при этом основой служит структурная формула.
Техническое решение согласно настоящему изобретению следующее:
Способ получения ваборбактама, включающий этапы, на которых:
(1) путем обеспечения реакции соединения типа II и реагента для введения защитной группы гидроксигруппы получают соединение типа III;
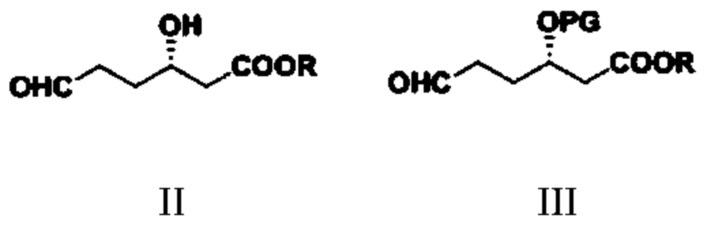
при этом в структурных формулах соединений типа II и III R представляет собой метил, этил, изопропил, н-пропил, н-бутил, изобутил, втор-бутил или трет-бутил; в структурной формуле соединения типа III защитная группа PG представляет собой триметилсилил (TMS), трет-бутил-диметилсилил (TBDMS), бензил (Bn), метилсульфонил (Ms), п-толуолсульфонил (Ts), трифторацетил (TFA) или ацетил (Ac);
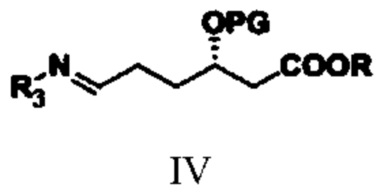
(2) путем обеспечения реакции амидирования соединения типа III с аминным соединением получают соединение типа IV;
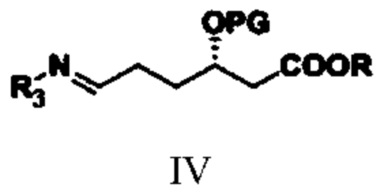
при этом в структурной формуле соединения типа IV R3 представляет собой гидроксил, бензоил, фенилацетил, 2-тиофенацетил или алкилсульфинил; R и PG означают то же, что R и PG в структурной формуле соединения типа III;
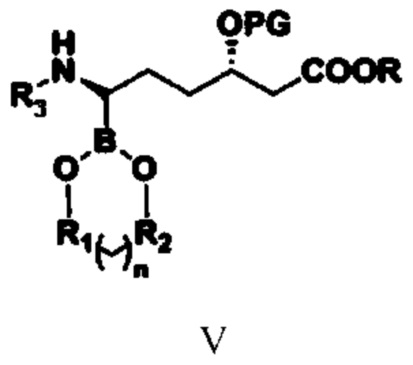
(3) путем обеспечения реакции соединения типа IV с бораном или боратным соединением получают соединение типа V;
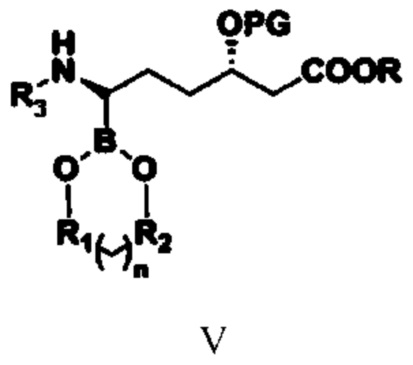
при этом в структурной формуле соединения типа V n равняется 0, 1, 2 или 3; если n равняется 0, то R1 и R2 представляют собой алкильные группы или арильные группы, предпочтительно R1 и R2 представляют собой фенил, метилфенил, хлорфенил или алкил, содержащий от 1 до 4 атомов углерода; если n равняется 1, 2 или 3, то R1 и R2 представляют собой алкильные группы, содержащие от 1 до 4 атомов углерода; предпочтительно R1 и R2 представляют собой этиленовую группу (-CH2CH2-) или замещенную этиленовую группу; R1 и R2 являются одинаковыми или разными; R, PG и R3 означают то же, что и R, PG и R3 в структурной формуле соединения типа IV;
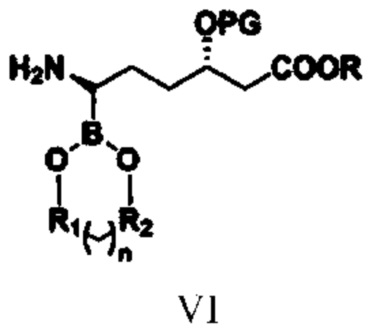
(4) путем проведения в отношении соединения типа V реакции удаления защитной группы удаляют защитную группу для аминогруппы с получением соединения типа VI;
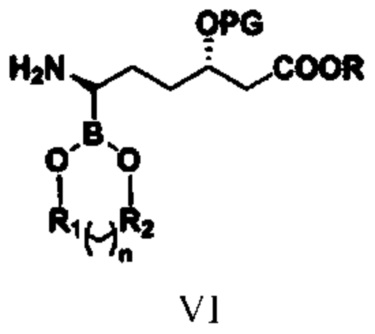
при этом R1, R2, n, R и PG в структурной формуле соединения типа VI означают то же, что и R1, R2, n, R и PG в структурной формуле соединения типа V;
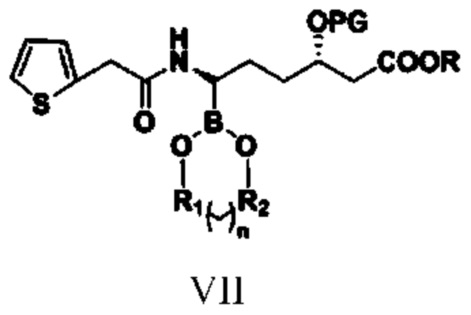
(5) путем обеспечения реакции амидирования соединения типа VI с реагентом амидирования получают соединение типа VII;
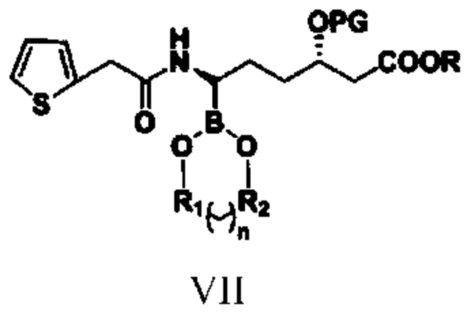
при этом R1, R2, n, R и PG в структурной формуле соединения типа VII означают то же, что и R1, R2, n, R и PG в структурной формуле соединения типа V;
(6) путем осуществления циклизации и гидролиза соединения типа VII получают ваборбактам I.
Согласно настоящему изобретению на этапе (1) соединение типа II может быть куплено или же получено согласно решениям, известным из уровня техники.
Согласно настоящему изобретению реакцию на этапе (1) соединения типа II и реагента для введения защитной группы для гидроксигруппы предпочтительно проводят в растворителе А в присутствии основания.
Предпочтительно указанный растворитель A представляет собой растворитель, не содержащий спирта, более предпочтительно выбран в виде одного из этилацетата, бутилацетата, ацетона, метилизобутилкетона, ацетонитрила, тетрагидрофурана, 2-метилтетрагидрофурана, 2,4-эпоксициклогексана, метоксициклопентана, метил-трет-бутилового эфира, растворителя на основе галогенированных углеводородов или бензольного растворителя или в виде комбинации из двух или более из этого; массовое соотношение между указанным растворителем A и соединением типа II составляет (4–20):1; предпочтительно массовое соотношение между растворителем A и соединением типа II составляет (5–10):1.
Предпочтительно указанное основание представляет собой органическое основание или неорганическое основание; более предпочтительно органическое основание представляет собой триметиламин, триэтиламин, три-н-бутиламин, диизопропилэтиламин или имидазол; более предпочтительно неорганическое основание представляет собой один из карбоната калия, карбоната натрия или карбоната кальция либо комбинацию из двух или более из этого; молярное соотношение между указанным основанием и соединением типа II составляет (1,0–2,0):1; более предпочтительно молярное соотношение между основанием и соединением типа II составляет (1,1–1,5):1.
Предпочтительно указанный реагент для введения защитной группы для гидроксигруппы представляет собой триметилхлорсилан, иодотриметилсилан, диметил-трет-бутилхлорсилан, диметил-трет-бутилиодосилан, метилсульфонилхлорид, п-толуолсульфонилхлорид, бензилхлорид, бензилбромид, трифторуксусную кислоту или ангидрид уксусной кислоты; молярное соотношение между реагентом для введения защитной группы для гидроксигруппы и соединением типа II составляет (1,0–2,0):1, более предпочтительно молярное соотношение между реагентом для введения защитной группы для гидроксигруппы и соединением типа II составляет (1,1–1,5):1.
Предпочтительно температура реакции указанного соединения типа II с реагентом для введения защитной группы для гидроксигруппы составляет 0–60°C, более предпочтительно составляет 20–40°C; время реакции соединения типа II с реагентом для введения защитной группы для гидроксигруппы составляет 1–7 часов, более предпочтительно составляет 2–5 часов.
Согласно настоящему изобретению на этапе (2) реакцию амидирования соединения типа III с аминным соединением предпочтительно проводят в растворителе B с участием катализатора C.
Предпочтительно указанный растворитель B представляет собой одно из метанола, этанола, изопропанола, ацетонитрила, тетрагидрофурана, 2-метилтетрагидрофурана, 2,4-эпоксициклогексана, метоксициклопентана, метил-трет-бутилового эфира, растворителя на основе галогенированных углеводородов или бензольного растворителя либо комбинацию из двух или более из этого; массовое соотношение между растворителем B и соединением типа III составляет (1–20):1; более предпочтительно массовое соотношение между растворителем B и соединением типа III составляет (2–10):1.
Предпочтительно указанный катализатор C представляет собой одно или комбинацию из двух или более из уксусной кислоты, метансульфоновой кислоты, бензолсульфоновой кислоты, п-толуолсульфоновой кислоты, пиридин-п-толуолсульфоната, сульфата меди, ацетата меди, хлорида меди или хлорида железа; молярное соотношение между катализатором C и соединением типа III составляет 1–3:1.
Предпочтительно указанное аминное соединение представляет собой гидроксиламин, бензамид, фенилацетамид, тиофенацетамид или алкилсульфонамид; молярное соотношение между аминным соединением и соединением типа III составляет (1,0–2,0):1.
Предпочтительно температура реакции амидирования составляет от -10 до 100°C, более предпочтительно составляет от 0 до 70°C и особенно предпочтительно составляет от 30 до 50°C.
Согласно настоящему изобретению на этапе (3) реакцию соединения типа IV с бораном или боратным соединением предпочтительно проводят в растворителе D в присутствии катализатора E и лиганда.
Предпочтительно указанный растворитель D представляет собой одно из метанола, этанола, изопропанола, ацетонитрила, тетрагидрофурана, 2-метилтетрагидрофурана, 1,3-эпоксициклогексана, метоксициклопентана, метил-трет-бутилового эфира, воды, растворителя на основе галогенированных углеводородов или бензольного растворителя либо комбинацию из двух или более из этого; массовое соотношение между растворителем D и соединением типа IV составляет (2–20):1; более предпочтительно массовое соотношение между растворителем D и соединением типа IV составляет (2–10):1.
Предпочтительно указанный катализатор E представляет собой сульфат меди, хлорид меди, хлористую медь, хлорид палладия, ацетат палладия, три(трифенилфосфин)родия хлорид, катализатор Граббса, оксид иридия/алюминия или (1,5-циклооктадиен)(пиримидин)(трициклогексилфосфин)иридий(I) гексафторфосфат; количество молей указанного катализатора E составляет 1,0–10,0% от количества молей соединения типа IV; более предпочтительно количество молей указанного катализатора E составляет 1,0–5,0% от количества молей соединения типа IV.
Предпочтительно указанный лиганд представляет собой один из азотного лиганда или фосфорного лиганда либо комбинацию из этих двух, более предпочтительно указанный лиганд представляет собой комбинацию из азотного лиганда и фосфорного лиганда; более предпочтительно указанный азотный лиганд представляет собой замещенный имидазол или бензиламин, а указанный фосфорный лиганд представляет собой трифенилфосфин или оксид трифенилфосфина; количество молей указанного лиганда составляет 1,0–10,0% от количества молей соединения типа IV; более предпочтительно количество молей лиганда составляет 1,0–7,0% от количества молей соединения типа IV.
Предпочтительно указанный боран представляет собой диалкоксиборан или пинаколборан; указанное боратное соединение представляет собой триалкилборат или биборат; молярное соотношение между бораном или боратным соединением и соединением типа IV составляет 1,0–2,0:1.
Предпочтительно температура реакции соединения типа IV с бораном или боратным соединением составляет 20–120°C, более предпочтительно составляет 20–40°C.
Согласно настоящему изобретению на этапе (4) реакцию удаления защитной группы в отношении соединения типа V предпочтительно проводят в растворителе F в присутствии реагента для реакции удаления защитной группы.
Предпочтительно указанный растворитель F представляет собой одно из метанола, этанола, изопропанола, ацетонитрила, тетрагидрофурана, 2-метилтетрагидрофурана, 2,4-эпоксициклогексана, метоксициклопентана, метил-трет-бутилового эфира, воды, растворителя на основе галогенированных углеводородов или бензольного растворителя либо комбинацию из двух или более из этого; массовое соотношение между растворителем F и соединением типа V составляет 1–20:1.
Предпочтительно указанный реагент для реакции удаления защитной группы представляет собой кислоту или основание; указанная кислота представляет собой хлористый водород, серную кислоту или фосфорную кислоту, при этом концентрация ионов водорода в указанной кислоте составляет 3–8 моль/л; указанное основание представляет собой гидроксид натрия, гидроксид калия, гидроксид бария, карбонат натрия или карбонат калия; молярное соотношение между реагентом для реакции удаления защитной группы и соединением типа V составляет 3–7:1.
Предпочтительно температура реакции удаления защитной группы составляет от -10 до 100°C; более предпочтительно температура реакции удаления защитной группы составляет от -5 до 30°C.
Согласно настоящему изобретению на этапе (5) реакцию амидирования соединения типа VI и реагента амидирования предпочтительно проводят в растворителе G и под действием основания H.
Предпочтительно указанный растворитель G представляет собой одно из метанола, этанола, изопропанола, ацетонитрила, тетрагидрофурана, 2-метилтетрагидрофурана, 2,4-эпоксициклогексана, 1,4-диоксана, метоксициклопентана, метил-трет-бутилового эфира, растворителя на основе галогенированных углеводородов или бензольного растворителя либо комбинацию из двух или более из этого; массовое соотношение между растворителем G и соединением типа VI составляет 4–20:1; более предпочтительно массовое соотношение между растворителем G и соединением типа VI составляет 4–10:1.
Предпочтительно указанный реагент амидирования представляет собой 2-тиофенацетилхлорид или 2-тиофенуксусную кислоту; молярное соотношение между указанным реагентом амидирования и соединением типа VI составляет 1–2,0:1.
Более предпочтительно, если реагентом амидирования является 2-тиофенуксусная кислота, то нужно использовать средство для конденсации с дегидратацией; указанное средство для конденсации с дегидратацией представляет собой один или комбинацию двух или более из дициклогексилкарбодиимида (DCC), 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорида (EDCI) или 1-гидроксибензотриазола (HOBt); молярное соотношение между указанным средством для конденсации с дегидратацией и соединением типа VI составляет 1,0–3,0:1.
Предпочтительно указанное основание H представляет собой органическое основание или неорганическое основание; указанное органическое основание представляет собой триметиламин, триэтиламин, три-н-бутиламин, диизопропилэтиламин, имидазол, морфолин или N-метилморфолин; указанное неорганическое основание представляет собой один или комбинацию двух или более из карбоната калия, карбоната натрия или карбоната кальция; молярное соотношение между указанным основанием H и соединением типа VI составляет (1,0–3,0):1.
Предпочтительно температура реакции амидирования составляет 0–120°C; более предпочтительно температура реакции амидирования составляет 15–80°C.
Согласно настоящему изобретению на этапе (6) реакцию циклизации и гидролиза в отношении соединения типа VII предпочтительно проводят в растворителе J в присутствии кислоты.
Предпочтительно указанный растворитель J представляет собой одно из метанола, этанола, изопропанола, ацетонитрила, тетрагидрофурана, 2-метилтетрагидрофурана, 1,4-диоксана, метоксициклопентана, метил-трет-бутилового эфира, растворителя на основе галогенированных углеводородов или бензольного растворителя либо комбинацию из двух или более из этого; массовое соотношение между растворителем J и соединением типа VII составляет 4–20:1; более предпочтительно массовое соотношение между растворителем J и соединением типа VI составляет 2–10:1.
Предпочтительно указанная кислота представляет собой одну или комбинацию двух или более из соляной кислоты, серной кислоты, борной кислоты или трифторуксусной кислоты; молярное соотношение между указанной кислотой и соединением типа VII составляет 5–7:1.
Предпочтительно температура реакции циклизации и гидролиза составляет 10–100°C; более предпочтительно температура реакции циклизации и гидролиза составляет 70–95°C. Время реакции циклизации и гидролиза составляет 1–10 часов.
Процесс реакции согласно настоящему изобретению описан с применением представленной ниже схемы реакции 3:
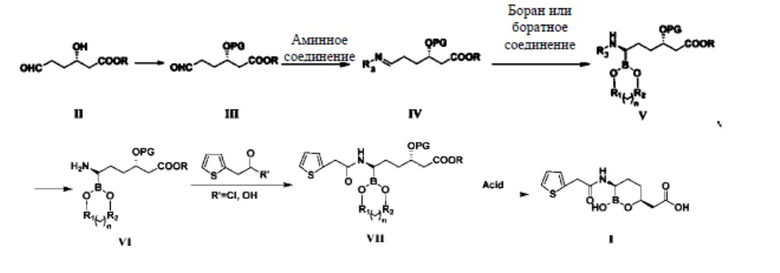
Схема реакции 3
где R представляет собой метил, этил, изопропил, н-пропил, н-бутил, изобутил, втор-бутил или трет-бутил; PG представляет собой триметилсилил (TMS), трет-бутил-диметилсилил (TBDMS), бензил (Bn), метилсульфонил (Ms), п-толуолсульфонил (Ts), трифторацетил (TFA) или ацетил (Ac); n равняется 0, 1, 2 или 3; если n равняется 0, то R1 и R2 представляют собой алкильные группы или арильные группы; если n равняется 1, 2 или 3, то R1 и R2 представляют собой алкильные группы, содержащие от 1 до 4 атомов углерода; R1 и R2 являются одинаковыми или разными; R3 представляет собой гидроксил, бензоил, фенилацетил, 2-тиофенацетил или алкилсульфинил.
Технические особенности и полезные эффекты настоящего изобретения:
Сырье для настоящего изобретения легко приобрести по умеренной цене, при этом не используется сложный трет-бутиловый эфир R-3-гидрокси-4-пентеновой кислоты и другое сырье, поэтому себестоимость низкая; применяемый согласно настоящему изобретению тип реакции является стандартным, условия реакции легко контролируются и легко обеспечиваются, а также можно избежать жестких условий реакции при низкой температуре и снизить потребление энергии; кроме того настоящее изобретение позволяет отказаться от использования литиевых реагентов, а также является безопасным, простым и удобным для осуществления, при этом процесс реакции безопасен для окружающей среды, что способствует осуществлению экологичного промышленного производства ваборбактама.
Типы реакции на всех этапах согласно настоящему изобретению являются стандартными, при этом, без применения жестких условий для целей настоящего изобретения, реакции на всех этапах могут обеспечивать высокий выход продукта; степень чистоты и выход продукта реакции на каждом этапе относительно высокие, что выгодно для целей последующего промышленного производства.
Конкретный способ осуществления
Ниже настоящее изобретение описано подробно посредством примеров осуществления, но при этом настоящее изобретение ими не ограничивается.
Используемые в примерах сырье и реагенты представляют собой продукты, имеющиеся в продаже, при этом оптическая чистота S-3-гидрокси-6-оксогексаноата (II) составляет 99,6%.
Если не указано иное, то все % в примерах осуществления представляют собой проценты по массе; все описанные выходы представляют собой молярный выход.
Посредством газового или жидкостного хроматографа осуществляется контроль процесса реакции и чистоты продукта; посредством жидкостного хроматографа с хиральной колонкой (ES-OVS, 150 мм × 4,6 мм, компания Agilent) определяется оптическая чистота (соотношение площадей, %); и вычисляется выход и значение ee.
Пример осуществления 1. Получение сложного трет-бутилового эфира S-3-триметилсилилокси-6-оксогексановой кислоты (III1)
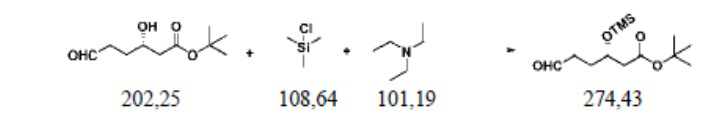
В защитной атмосфере азота в колбу на четыре горлышка объемом 250 мл, выполненную с возможностью перемешивания и снабженную термометром, добавляли 120 г дихлорметана, 20,2 г (0,1 моль) сложного трет-бутилового эфира S-3-гидрокси-6-оксогексановой кислоты (II1) и 12,1 г (0,12 моль) триэтиламина; при внутренней температуре 15–20°C капельно добавляли раствор 13,1 г (0,12 моль) триметилхлорсилана и 20 г дихлорметана, при этом капельное добавление происходило 30 минут. После этого перемешивают при температуре 20–25°C и оставляют прореагировать на 3 часа. Реакционный раствор добавляли в 50 г воды для расслоения и водный слой два раза экстрагировали дихлорметаном, каждый раз 30 г. Объединяли органические фазы; два раза промывали насыщенным раствором хлористого натрия, каждый раз 20 г; после восстановления растворителя из полученной органической фазы получали 26,0 г сложного трет-бутилового эфира S-3-триметилсилилокси-6-оксогексановой кислоты (III1), при этом чистота жидкой фазы составляла 99,9%, а выход составлял 94,9%.
Пример осуществления 2. Получение сложного трет-бутилового эфира S-3-диметил-трет-бутилсилилокси-6-оксогексановой кислоты (III2)
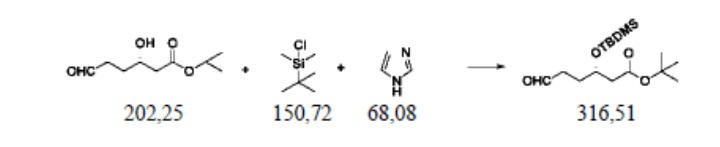
В защитной атмосфере азота в колбу на четыре горлышка объемом 250 мл, выполненную с возможностью перемешивания и снабженную термометром, добавляли 120 г дихлорметана, 20,2 г (0,1 моль) сложного трет-бутилового эфира S-3-гидрокси-6-оксогексановой кислоты (II1) и 8,2 г (0,12 моль) имидазола; при внутренней температуре 15–20°C капельно добавляли раствор 16,6 г (0,11 моль) диметил-трет-бутилхлорсилана и 30 г дихлорметана, при этом капельное добавление происходило 30 минут. После этого перемешивают при температуре 20–25°C и оставляют прореагировать на 4 часа. Полученный реакционный раствор добавляли в 50 г воды для расслоения и водный слой два раза экстрагировали дихлорметаном, каждый раз 30 г. Объединяли органические фазы; два раза промывали насыщенным раствором хлористого натрия, каждый раз 20 г; после восстановления растворителя из полученной органической фазы получали 30,4 г сложного трет-бутилового эфира S-3-диметил-трет-бутилсилилокси-6-оксогексановой кислоты (III2), при этом чистота жидкой фазы составляла 99,9%, а выход составлял 96,0%.
Пример осуществления 3. Получение сложного трет-бутилового эфира S-3-триметилсилилокси-6-(N-(R-1-трет-бутилсульфинил)имино)гексановой кислоты (IV1)
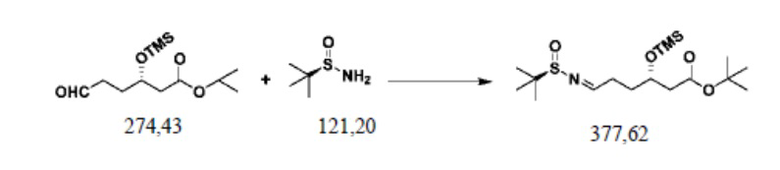
В защитной атмосфере азота в колбу на четыре горлышка объемом 250 мл, выполненную с возможностью перемешивания и перегонки, а также снабженную термометром, добавляли 120 г дихлорметана, 27,4 г (0,1 моль) полученного согласно примеру осуществления 1 сложного трет-бутилового эфира S-3-триметилсилилокси-6-оксогексановой кислоты (III1), 2,5 г (0,01 моль) пиридин-п-толуолсульфоната, 31,9 грамм (0,2 моль) сульфата меди и 13,3 г (0,11 моль) R-трет-бутилсульфинамида; нагревали с использованием обратного холодильника до 40–45°C до завершения реакции, выявляемого посредством HPLC. Фильтровали с удалением нерастворимых веществ. Путем перегонки под уменьшенным давлением рекуперировали дихлорметан; остаток отделяли и очищали с помощью колоночной хроматографии (РЕ/ЕА=10:1), или применяли ректификацию, с получением 36,0 г сложного трет-бутилового эфира S-3-триметилсилилокси-6-(N-(R-1-трет-бутилсульфинил)имино)гексановой кислоты (IV1), при этом чистота жидкой фазы составляла 99,9%, а выход составлял 95,3%.
Пример осуществления 4. Получение сложного трет-бутилового эфира S-3-диметил-трет-бутилсилилокси-6-(N-(R-1-трет-бутилсульфинил)имино)гексановой кислоты (IV2)
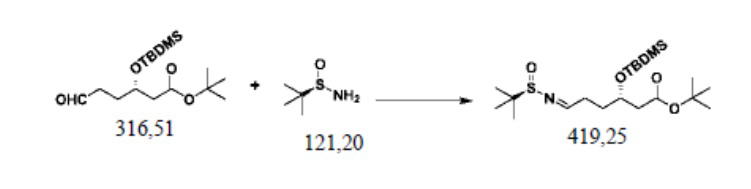
В защитной атмосфере азота в колбу на четыре горлышка объемом 250 мл, выполненную с возможностью перемешивания и перегонки, а также снабженную термометром, добавляли 120 г дихлорметана, 31,7 г (0,1 моль) полученного согласно примеру осуществления 2 сложного трет-бутилового эфира S-3-диметил-трет-бутилсилилокси-6-оксогексановой кислоты (III2), 2,5 г (0,01 моль) пиридин-п-толуолсульфоната, 31,9 грамм (0,2 моль) сульфата меди и 13,3 г (0,11 моль) R-трет-бутилсульфинамида; нагревали с использованием обратного холодильника до 40–45°C до завершения реакции, выявляемого посредством HPLC. Фильтровали с удалением нерастворимых веществ. Путем перегонки под уменьшенным давлением рекуперировали дихлорметан; остаток отделяли и очищали с помощью колоночной хроматографии (РЕ/ЕА=10:1), или применяли ректификацию, с получением 40,7 г сложного трет-бутилового эфира S-3-диметил-трет-бутилсилилокси-6-(N-(R-1-трет-бутилсульфинил)имино)гексановой кислоты (IV2), при этом чистота жидкой фазы составляла 99,9%, а выход составлял 97,0%.
Пример осуществления 5. Получение сложного трет-бутилового эфира (3S,6R)-3-триметилсилилокси-6-(N-(R-1-трет-бутилсульфинил)амино)-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)гексановой кислоты (V1)
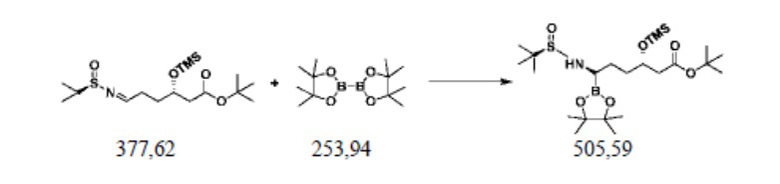
В колбу на четыре горлышка объемом 250 мл, выполненную с возможностью перемешивания и снабженную термометром, добавляли 100 г толуола, 20 г воды, 0,2 г (1,2 ммоль) сульфата меди, 0,37 г (1,2 ммоль) оксида трифенилфосфина, 0,54 г (5 ммоль) бензиламина и перемешивали. Добавляли 37,7 г (0,1 ммоль) полученного согласно примеру осуществления 3 сложного трет-бутилового эфира S-3-триметилсилилокси-6-(N-(R-1-трет-бутилсульфинил)имино)гексановой кислоты (IV1) и 30,5 г (0,12 ммоль) сложного пинаколового эфира бороновой кислоты и перемешивали при комнатной температуре до завершения реакции, выявляемого посредством HPLC. В систему добавляли 50 г этилацетата; после фильтрования концентрировали путем перегонки под уменьшенным давлением; остаток отделяли и очищали с помощью колоночной хроматографии (EA/DCM=10:90), или применяли ректификацию, с получением 39,2 г сложного трет-бутилового эфира (3S,6R)-3-триметилсилилокси-6-(N-(R-1-трет-бутилсульфинил)амино)-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)гексановой кислоты (V1), при этом чистота жидкой фазы составляла 98,6%, а выход составлял 77,6%.
Пример осуществления 6. Получение сложного трет-бутилового эфира (3S,6R)-3-диметил-трет-бутилсилилокси-6-(N-(R-1-трет-бутилсульфинил)амино)-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)гексановой кислоты (V2)
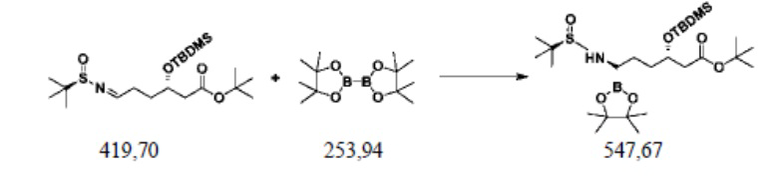
В реакционную колбу объемом 250 мл, выполненную с возможностью перемешивания и снабженную термометром, добавляли 100 г толуола, 20 г воды, 0,2 г (1,2 ммоль) сульфата меди, 0,37 г (1,2 ммоль) оксида трифенилфосфина, 0,54 г (5 ммоль) бензиламина и перемешивали. Добавляли 42,0 (0,1 ммоль) полученного способом согласно примеру осуществления 4 сложного трет-бутилового эфира S-3-диметил-трет-бутилсилилокси-6-(N-(R-1-трет-бутилсульфинил)имино)гексановой кислоты (IV2) и 30,5 г (0,12 ммоль) сложного пинаколового эфира бороновой кислоты и перемешивали при комнатной температуре до завершения реакции, выявляемого посредством HPLC. В систему добавляли 50 г этилацетата; после фильтрования концентрировали путем перегонки под уменьшенным давлением; остаток отделяли и очищали с помощью колоночной хроматографии (EA/DCM=10:90), или применяли ректификацию, с получением 44,7 г сложного трет-бутилового эфира (3S,6R)-3-диметил-трет-бутилсилилокси-6-(N-(R-1-трет-бутилсульфинил)амино)-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)гексановой кислоты (V2), при этом чистота жидкой фазы составляла 98,4%, а выход составлял 81,6%.
Пример осуществления 7. Получение гидрохлорида сложного трет-бутилового эфира (3S,6R)-3-диметил-трет-бутилсилилокси-6-амино-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)гексановой кислоты (VI)
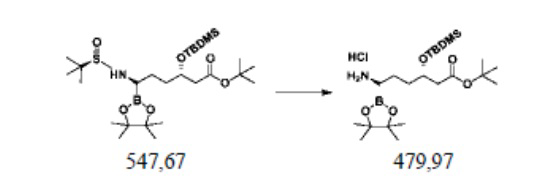
В реакционную колбу объемом 250 мл, выполненную с возможностью перемешивания и снабженную термометром, добавляли 100 г 20%-ного метанольного раствора хлороводорода и понижали температуру до 0°C. Добавляли 54,8 г (0,1 моль) полученного согласно примеру осуществления 6 сложного трет-бутилового эфира (3S,6R)-3-диметил-трет-бутилсилилокси-6-(N-(R-1-трет-бутилсульфинил)амино)-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)гексановой кислоты (V2) и перемешивали при 0°C до завершения реакции, выявляемого посредством HPLC. После концентрирования путем перегонки под уменьшенным давлением получали 47,3 г гидрохлорида сложного трет-бутилового эфира (3S,6R)-3-диметил-трет-бутилсилилокси-6-амино-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)гексановой кислоты, при этом чистота жидкой фазы составляла 99,2%, а выход составлял 98,6%.
Пример осуществления 8. Получение сложного трет-бутилового эфира (3S,6R)-3-диметил-трет-бутилсилилокси-6-[2-(2-тиофен)ацетамидо]-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)гексановой кислоты (VII)
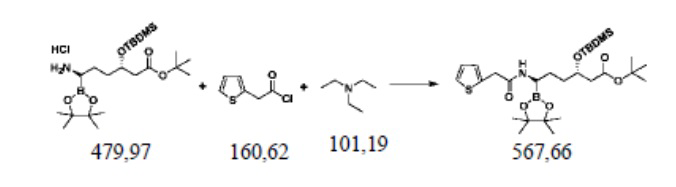
В реакционную колбу объемом 250 мл, выполненную с возможностью перемешивания и снабженную термометром, добавляли 100 г тетрагидрофурана, 24,0 г (0,05 моль) полученного способом согласно примеру осуществления 7 гидрохлорида сложного трет-бутилового эфира (3S,6R)-3-диметил-трет-бутилсилилокси-6-амино-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)гексановой кислоты (VI), 9,6 г (0,06 моль) 2-тиофенацетилхлорида, 11,1 г (0,11 моль) триэтиламина и перемешивали при комнатной температуре до завершения реакции, выявляемого посредством HPLC. Добавляли 80 г воды и перемешивали полчаса. Добавляли 100 г этилацетата, помещали в делительную воронку и оставляли для отделения жидкости. Промывали водой органическую фазу два раза, каждый раз 30 г, объединяли органические фазы и высушивали посредством безводного сульфата натрия. После отфильтровывания сульфата натрия фильтрат подвергали перегонке под уменьшенным давлением для удаления растворителя с получением светло-желтого неочищенного продукта. Осуществляли перекристаллизацию с использованием метил-трет-бутилового эфира и н-гексана с получением 21,7 г сложного трет-бутилового эфира (3S,6R)-3-диметил-трет-бутилсилилокси-6-[2-(2-тиофен)ацетамидо]-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)гексановой кислоты (VII), представляющего собой белое твердое вещество, при этом чистота жидкой фазы составляла 99,3%, а выход составлял 76,4%.
Пример осуществления 9. Получение сложного трет-бутилового эфира (3S,6R)-3-диметил-трет-бутилсилилокси-6-[2-(2-тиофен)ацетамидо]-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)гексановой кислоты (VII)
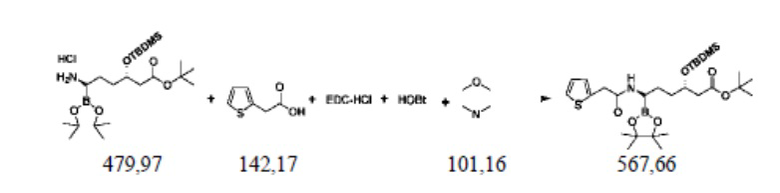
В защитной атмосфере азота в реакционную колбу объемом 250 мл, выполненную с возможностью перемешивания и снабженную термометром, добавляли 100 г дихлорметана, 8,5 г (0,06 моль) тиофенуксусной кислоты, 14,8 г (0,075 моль) EDC-HCl (EDCI), 8,1 г (0,06 моль) 1-гидроксибензотриазола (HOBt), понижали температуру до 0 ℃ и перемешивали. 24,0 г полученного способом согласно примеру осуществления 7 гидрохлорида сложного трет-бутилового эфира (3S,6R)-3-диметил-трет-бутилсилилокси-6-амино-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)гексановой кислоты (VI) растворяли в 60 г дихлорметана, добавляли в систему и добавляли 11,1 г (0,11 моль) N-метилморфолина. После добавления температуру повышали до комнатной и перемешивали до завершения реакции, выявляемого посредством HPLC. Добавляли 80 г воды, помещали в делительную воронку и оставляли для отделения жидкости, потом промывали органическую фазу с использованием 30 г воды и высушивали органическую фазу безводным сульфатом натрия. Концентрировали путем перегонки под уменьшенным давлением; из полученного осадка путем перекристаллизации с использованием метил-трет-бутилового эфира и н-гексана получали 23,3 г сложного трет-бутилового эфира (3S,6R)-3-диметил-трет-бутилсилилокси-6-[2-(2-тиофен)ацетамидо]-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)гексановой кислоты (VII), представляющего собой белое твердое вещество, при этом чистота жидкой фазы составляла 99,4%, а коэффициент выхода составлял 82,1%.
Пример осуществления 10. Получение ваборбактама (I)
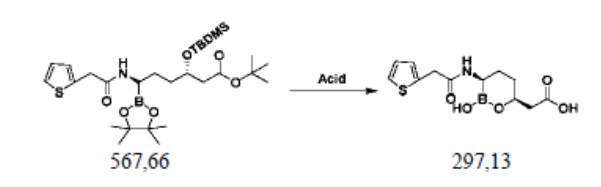
В реакционную колбу объемом 250 мл, выполненную с возможностью перемешивания и снабженную термометром, добавляли 80 г 1,4-диоксана, 28,4 г (0,05 моль) полученного способом согласно примеру осуществления 9 сложного трет-бутилового эфира (3S,6R)-3-диметил-трет-бутилсилилокси-6-[2-(2-тиофен)ацетамидо]-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)гексановой кислоты (VII) и перемешивали. Добавляли 100 мл соляной кислоты 3 моль/л; смесь нагревали до 90-95°C и перемешивали при обратном потоке в течение 2 часов. Охлаждали до комнатной температуры, добавляли 100 г воды и 150 г метил-трет-бутилового эфира, помещали в делительную воронку и оставляли для отделения жидкости. Путем перегонки под уменьшенным давлением концентрировали водную фазу. К полученному остатку добавляли ацетонитрил и трижды проводили азеотропную перегонку под уменьшенным давлением, при этом каждый раз применяли 80 г ацетонитрила. Полученный остаток растворяли в 20%-ном растворе 1,4-диоксана/воды и проводили лиофильную сушку с получением 14,3 г белого порошка. В полученный белый порошок добавляли 200 г этилацетата и 60 г воды, перемешивали при комнатной температуре в течение 1 часа и оставляли, чтобы образовался белый осадок. В отношении осадка осуществляли фильтрование с отсасыванием, а затем дважды промывали этилацетатом, каждый раз 15 г. Проводили вакуумную сушку с получением 10,5 г продукта I, представляющего собой белое твердое вещество, при этом чистота жидкой фазы составляла 99,6%, а выход составлял 71,0%.
Полученные данные ЯМР, касающиеся продукта, следующие:
1H ЯМР (CD3OD) ppm: δ 7,35 (dd, 1H), 7,05 (dd, 1H), 7,0 (dd, 1H), 4,15-4,05 (m, 1H), 3,98 (s, 2H), 2,61 (br d,1H), 2,37 (dd, 1H), 2,24 (dd, 1H), 1,74 (br d, 1H), 1,66-1,52 (m, 2H), 1,03 (br q, 1H).
Сравнительный пример 1. Получение сложного трет-бутилового эфира (3S,6R)-3-диметил-трет-бутилсилилокси-6-(N-(R-1-трет-бутилсульфинил)амино)-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)гексановой кислоты (V2)
В реакционную колбу объемом 250 мл, выполненную с возможностью перемешивания и снабженную термометром, добавляли 10 г толуола, 2 г воды, 0,02 г (0,12 ммоль) сульфата меди, 0,037 г (0,12 ммоль) оксида трифенилфосфина и перемешивали. Добавляли 4,2 г (10 ммоль) полученного согласно примеру осуществления 4 сложного трет-бутилового эфира S-3-диметил-трет-бутилсилилокси-6-(N-(R-1-трет-бутилсульфинил)имино)гексановой кислоты (IV2) и 3,1 г (12 ммоль) сложного пинаколового эфира бороновой кислоты и перемешивали при комнатной температуре до завершения реакции, выявляемого посредством HPLC. В течение двух дней отслеживали реакции путем контроля с применением HPLC; было преобразовано лишь менее 5% исходного материала IV2. Температуру реакции повышали до 100°C и в течение двух дней продолжали отслеживать реакции путем контроля с применением HPLC; было преобразовано лишь менее 10% исходного материала IV2.
Сравнительный пример 1 говорит о том, что в реакции с присоединением амида присутствие бензиламина является очень важным, а отсутствие бензиламина может заметно повлиять на время и температуру реакции, и в конечном итоге целевой продукт почти невозможно получить.
Сравнительный пример 2. Получение сложного трет-бутилового эфира (3S,6R)-3-диметил-трет-бутилсилилокси-6-(N-(R-1-трет-бутилсульфинил)амино)-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)гексановой кислоты (V2)
В реакционную колбу объемом 250 мл, выполненную с возможностью перемешивания и снабженную термометром, добавляли 10 г толуола, 2 г воды, 0,37 г (0,12 ммоль) оксида трифенилфосфина, 0,054 г (0,5 ммоль) бензиламина и перемешивали. Добавляли 4,2 г (10 ммоль) полученного согласно примеру осуществления 4 сложного трет-бутилового эфира S-3-диметил-трет-бутилсилилокси-6-(N-(R-1-трет-бутилсульфинил)имино)гексановой кислоты (IV2) и 3,1 г (12 ммоль) сложного пинаколового эфира бороновой кислоты и перемешивали при комнатной температуре до завершения реакции, выявляемого посредством HPLC. В течение двух дней отслеживали реакции путем контроля с применением HPLC; целевой продукт не был обнаружен и сырье IV2 не изменилось.
Сравнительный пример 2 говорит о том, что в реакции с присоединением амида присутствие катализатор является важным, и при отсутствии катализатора реакция не происходит.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОСТОЙ СПОСОБ ПОЛУЧЕНИЯ АВИБАКТАМА | 2018 |
|
RU2711358C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ АВИБАКТАМА | 2018 |
|
RU2722625C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ АВИБАКТАМА | 2018 |
|
RU2722932C1 |
| СПОСОБЫ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ У МЛЕКОПИТАЮЩИХ | 2004 |
|
RU2344121C2 |
| СПОСОБ ПОЛУЧЕНИЯ 5R-[(БЕНЗИЛОКСИ)АМИНО]ПИПЕРИДИН-2S-КАРБОНОВОЙ КИСЛОТЫ ИЛИ ЕЁ ПРОИЗВОДНОГО | 2018 |
|
RU2730006C1 |
| МАКРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ МОЧЕВИНЫ И СУЛЬФАМИДА В КАЧЕСТВЕ ИНГИБИТОРОВ TAFIa | 2009 |
|
RU2502736C2 |
| СПОСОБ ПОЛУЧЕНИЯ СТЕРЕОСПЕЦИФИЧЕСКОГО СОЕДИНЕНИЯ | 1989 |
|
RU2024528C1 |
| СПОСОБ ПОЛУЧЕНИЯ (3S,4S)-4-((R)-2-(БЕНЗИЛОКСИ)ТРИДЕЦИЛ)-3-ГЕКСИЛ-2-ОКСЕТАНОНА И ИСПОЛЬЗУЕМОГО ДЛЯ ЭТОГО НОВОГО ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ | 2009 |
|
RU2471790C1 |
| СПОСОБ ПОЛУЧЕНИЯ 6-(1-АКРИЛОИЛПИПЕРИДИН-4-ИЛ)-2-(4-ФЕНОКСИФЕНИЛ)НИКОТИНАМИДА | 2020 |
|
RU2810260C2 |
| НОВЫЕ ПОЛИНЕНАСЫЩЕННЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ | 2006 |
|
RU2466988C2 |
Изобретение относится к способу получения ваборбактама I. Способ включает этапы (1)-(6). На этапе (1) путем обеспечения реакции соединения типа II и реагента для введения защитной группы гидроксигруппы получают соединение типа III. На этапе (2) путем обеспечения реакции соединения типа III с аминным соединением получают соединение типа IV. На этапе (3) путем обеспечения реакции соединения типа IV с бораном или боратным соединением в присутствии катализатора E и лиганда получают соединение типа V, при этом указанный лиганд представляет собой комбинацию из бензиламина и оксида трифенилфосфина. На этапе (4) путем проведения в отношении соединения типа V реакции удаления защитной группы удаляют защитную группу для аминогруппы с получением соединения типа VI. На этапе (5) путем обеспечения реакции амидирования соединения типа VI с реагентом амидирования получают соединение типа VII, при этом указанный реагент амидирования представляет собой 2-тиофенацетилхлорид или 2-тиофенуксусную кислоту. На этапе (6) путем осуществления циклизации и гидролиза соединения типа VII получают ваборбактам I. В указанных формулах R представляет собой метил, этил, изопропил, н-пропил, н-бутил, изобутил, втор-бутил или трет-бутил; защитная группа PG представляет собой триметилсилил (TMS), трет-бутил-диметилсилил (TBDMS), бензил (Bn), метилсульфонил (Ms), п-толуолсульфонил (Ts), трифторацетил (TFA) или ацетил (Ac); R3 представляет собой гидроксил, бензоил, фенилацетил, 2-тиофенацетил или алкилсульфинил; n равняется 0, 1, 2 или 3; если n равняется 0, то R1 и R2 представляют собой алкильные группы или арильные группы; если n равняется 1, 2 или 3, то R1 и R2 представляют собой алкильные группы, содержащие от 1 до 4 атомов углерода. Предлагаемый способ является безопасным, простым, удобным, не требует жестких условий и обладает низкой себестоимостью, поскольку исходное сырье имеет умеренную цену. 7 з.п. ф-лы, 10 пр.
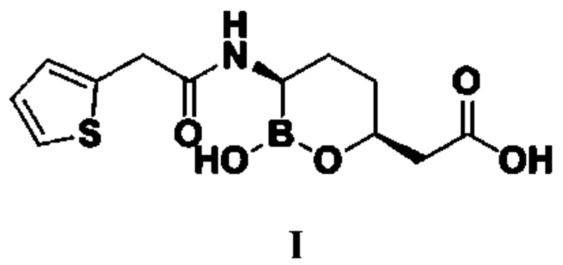
1. Способ получения ваборбактама
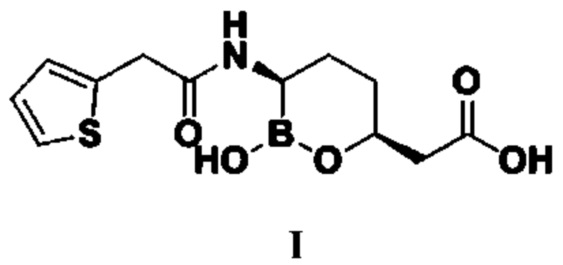 ,
,
включающий этапы, на которых:
(1) путем обеспечения реакции соединения типа II и реагента для введения защитной группы гидроксигруппы получают соединение типа III
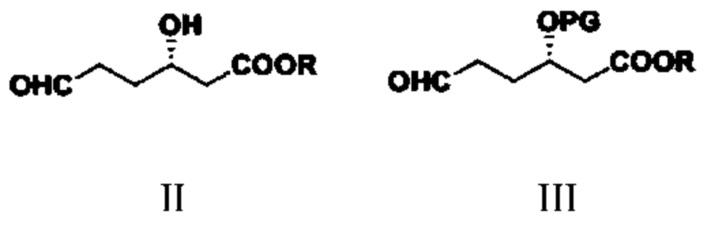 ,
,
при этом в структурных формулах соединений типа II и III R представляет собой метил, этил, изопропил, н-пропил, н-бутил, изобутил, втор-бутил или трет-бутил; в структурной формуле соединения типа III защитная группа PG представляет собой триметилсилил (TMS), трет-бутил-диметилсилил (TBDMS), бензил (Bn), метилсульфонил (Ms), п-толуолсульфонил (Ts), трифторацетил (TFA) или ацетил (Ac);
(2) путем обеспечения реакции соединения типа III с аминным соединением получают соединение типа IV
 ,
,
при этом в структурной формуле соединения типа IV R3 представляет собой гидроксил, бензоил, фенилацетил, 2-тиофенацетил или алкилсульфинил; R и PG означают то же, что R и PG в структурной формуле соединения типа III;
(3) путем обеспечения реакции соединения типа IV с бораном или боратным соединением в присутствии катализатора E и лиганда получают соединение типа V
 ,
,
при этом в структурной формуле соединения типа V n равняется 0, 1, 2 или 3; если n равняется 0, то R1 и R2 представляют собой алкильные группы или арильные группы; если n равняется 1, 2 или 3, то R1 и R2 представляют собой алкильные группы, содержащие от 1 до 4 атомов углерода; R1 и R2 являются одинаковыми или разными; R, PG и R3 означают то же, что и R, PG и R3 в структурной формуле соединения типа IV, и при этом указанный лиганд представляет собой комбинацию из бензиламина и оксида трифенилфосфина;
(4) путем проведения в отношении соединения типа V реакции удаления защитной группы удаляют защитную группу для аминогруппы с получением соединения типа VI
 ,
,
при этом в структурной формуле соединения типа VI R1, R2, n, R и PG означают то же, что и R1, R2, n, R и PG в структурной формуле соединения типа V;
(5) путем обеспечения реакции амидирования соединения типа VI с реагентом амидирования получают соединение типа VII
 ,
,
при этом в структурной формуле соединения типа VII R1, R2, n, R и PG означают то же, что и R1, R2, n, R и PG в структурной формуле соединения типа V, и при этом указанный реагент амидирования представляет собой 2-тиофенацетилхлорид или 2-тиофенуксусную кислоту;
(6) путем осуществления циклизации и гидролиза соединения типа VII получают ваборбактам I.
2. Способ получения ваборбактама по п. 1, отличающийся тем, что предусматривает одно или более из следующих условий:
a) на этапе (1) реакцию соединения типа II и реагента для введения защитной группы для гидроксигруппы проводят в растворителе А в присутствии основания;
b) на этапе (2) реакцию амидирования соединения типа III с аминным соединением проводят в растворителе B с участием катализатора C;
c) на этапе (3) реакцию соединения типа IV с бораном или боратным соединением проводят в растворителе D;
d) на этапе (4) реакцию удаления защитной группы в отношении соединения типа V проводят в растворителе F в присутствии реагента для реакции удаления защитной группы;
e) на этапе (5) реакцию амидирования соединения типа VI и реагента амидирования проводят в растворителе G и под действием основания H;
f) на этапе (6) реакцию циклизации и гидролиза в отношении соединения типа VII проводят в растворителе J в присутствии кислоты.
3. Способ получения ваборбактама по п. 2, отличающийся тем, что на этапе (1) при выполнении условия а) предусматривает одно или более из следующих условий:
i) указанный растворитель A представляет собой растворитель, не содержащий спирта, более предпочтительно выбран в виде одного из этилацетата, бутилацетата, ацетона, метилизобутилкетона, ацетонитрила, тетрагидрофурана, 2-метилтетрагидрофурана, 2,4-эпоксициклогексана, метоксициклопентана, метил-трет-бутилового эфира, растворителя на основе галогенированных углеводородов или бензольного растворителя или в виде комбинации из двух или более из этого; массовое соотношение между указанным растворителем A и соединением типа II составляет (4-20):1;
ii) указанное основание представляет собой органическое основание или неорганическое основание; предпочтительно органическое основание представляет собой триметиламин, триэтиламин, три-н-бутиламин, диизопропилэтиламин или имидазол; предпочтительно неорганическое основание представляет собой одно из карбоната калия, карбоната натрия или карбоната кальция либо комбинацию из двух или более из этого; молярное соотношение между указанным основанием и соединением типа II составляет (1,0-2,0):1;
iii) указанный реагент для введения защитной группы для гидроксигруппы представляет собой триметилхлорсилан, иодотриметилсилан, диметил-трет-бутилхлорсилан, диметил-трет-бутилиодосилан, метилсульфонилхлорид, п-толуолсульфонилхлорид, бензилхлорид, бензилбромид, трифторуксусную кислоту или ангидрид уксусной кислоты; молярное соотношение между реагентом для введения защитной группы для гидроксигруппы и соединением типа II составляет (1,0-2,0):1;
iv) температура реакции указанного соединения типа II с реагентом для введения защитной группы для гидроксигруппы составляет 0-60°C, более предпочтительно составляет 20-40°C.
4. Способ получения ваборбактама по п. 2, отличающийся тем, что на этапе (2) при выполнении условия b) предусматривает одно или более из следующих условий:
i) указанный растворитель B представляет собой одно из метанола, этанола, изопропанола, ацетонитрила, тетрагидрофурана, 2-метилтетрагидрофурана, 2,4-эпоксициклогексана, метоксициклопентана, метил-трет-бутилового эфира, растворителя на основе галогенированных углеводородов или бензольного растворителя либо комбинацию из двух или более из этого; массовое соотношение между растворителем B и соединением типа III составляет (1-20):1;
ii) указанный катализатор C представляет собой одно или комбинацию из двух или более из уксусной кислоты, метансульфоновой кислоты, бензолсульфоновой кислоты, п-толуолсульфоновой кислоты, пиридин-п-толуолсульфоната, сульфата меди, ацетата меди, хлорида меди или хлорида железа; молярное соотношение между катализатором C и соединением типа III составляет 1-3:1;
iii) указанное аминное соединение представляет собой гидроксиламин, бензамид, фенилацетамид, тиофенацетамид или алкилсульфонамид; молярное соотношение между аминным соединением и соединением типа III составляет (1,0-2,0):1;
iv) температура реакции амидирования составляет от -10 до 100°C, более предпочтительно составляет от 0 до 70°C и особенно предпочтительно составляет от 30 до 50°C.
5. Способ получения ваборбактама по п. 2, отличающийся тем, что на этапе (3) при выполнении условия c) предусматривает одно или более из следующих условий:
i) указанный растворитель D представляет собой одно из метанола, этанола, изопропанола, ацетонитрила, тетрагидрофурана, 2-метилтетрагидрофурана, 1,3-эпоксициклогексана, метоксициклопентана, метил-трет-бутилового эфира, воды, растворителя на основе галогенированных углеводородов или бензольного растворителя либо комбинацию из двух или более из этого; массовое соотношение между растворителем D и соединением типа IV составляет (2-20):1;
ii) указанный катализатор E представляет собой сульфат меди, хлорид меди, хлористую медь, хлорид палладия, ацетат палладия, три(трифенилфосфин)родия хлорид, катализатор Граббса, оксид иридия/алюминия или (1,5-циклооктадиен)(пиримидин)(трициклогексилфосфин)иридий(I) гексафторфосфат; количество молей указанного катализатора E составляет 1,0-10,0% от количества молей соединения типа IV;
iii) количество молей указанного лиганда составляет 1,0-10,0% от количества молей соединения типа IV;
iv) указанный боран представляет собой диалкоксиборан или пинаколборан; указанное боратное соединение представляет собой триалкилборат или биборат; молярное соотношение между бораном или боратным соединением и соединением типа IV составляет 1,0-2,0:1;
v) температура реакции соединения типа IV с бораном или боратным соединением составляет 20-120°C, более предпочтительно составляет 20-40°C.
6. Способ получения ваборбактама по п. 2, отличающийся тем, что на этапе (4) при выполнении условия d) предусматривает одно или более из следующих условий:
i) указанный растворитель F представляет собой одно из метанола, этанола, изопропанола, ацетонитрила, тетрагидрофурана, 2-метилтетрагидрофурана, 2,4-эпоксициклогексана, метоксициклопентана, метил-трет-бутилового эфира, воды, растворителя на основе галогенированных углеводородов или бензольного растворителя либо комбинацию из двух или более из этого; массовое соотношение между растворителем F и соединением типа V составляет 1-20:1;
ii) указанный реагент для реакции удаления защитной группы представляет собой кислоту или основание; указанная кислота представляет собой хлористый водород, серную кислоту или фосфорную кислоту, при этом концентрация ионов водорода в указанной кислоте составляет 3-8 моль/л; указанное основание представляет собой гидроксид натрия, гидроксид калия, гидроксид бария, карбонат натрия или карбонат калия; молярное соотношение между реагентом для реакции удаления защитной группы и соединением типа V составляет 3-7:1;
iii) температура реакции удаления защитной группы составляет от -10 до 100°C; предпочтительно температура реакции удаления защитной группы составляет от -5 до 30°C.
7. Способ получения ваборбактама по п. 2, отличающийся тем, что на этапе (5) при выполнении условия e) предусматривает одно или более из следующих условий:
i) указанный растворитель G представляет собой одно из метанола, этанола, изопропанола, ацетонитрила, тетрагидрофурана, 2-метилтетрагидрофурана, 2,4-эпоксициклогексана, 1,4-диоксана, метоксициклопентана, метил-трет-бутилового эфира, растворителя на основе галогенированных углеводородов или бензольного растворителя либо комбинацию из двух или более из этого; массовое соотношение между растворителем G и соединением типа VI составляет 4-20:1;
ii) молярное соотношение между указанным реагентом амидирования и соединением типа VI составляет 1-2,0:1;
предпочтительно, если реагентом амидирования является 2-тиофенуксусная кислота, то нужно использовать средство для конденсации с дегидратацией; указанное средство для конденсации с дегидратацией представляет собой один или комбинацию двух или более из дициклогексилкарбодиимида (DCC), 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорида (EDCI) или 1-гидроксибензотриазола (HOBt); молярное соотношение между указанным средством для конденсации с дегидратацией и соединением типа VI составляет 1,0-3,0:1;
iii) указанное основание H представляет собой органическое основание или неорганическое основание; указанное органическое основание представляет собой триметиламин, триэтиламин, три-н-бутиламин, диизопропилэтиламин, имидазол, морфолин или N-метилморфолин; указанное неорганическое основание представляет собой один или комбинацию двух или более из карбоната калия, карбоната натрия или карбоната кальция; молярное соотношение между указанным основанием H и соединением типа VI составляет (1,0-3,0):1;
iv) температура реакции амидирования составляет 0-120°C; предпочтительно температура реакции амидирования составляет 15-80°C.
8. Способ получения ваборбактама по п. 2, отличающийся тем, что на этапе (6) при выполнении условия f) предусматривает одно или более из следующих условий:
i) указанный растворитель J представляет собой одно из метанола, этанола, изопропанола, ацетонитрила, тетрагидрофурана, 2-метилтетрагидрофурана, 1,4-диоксана, метоксициклопентана, метил-трет-бутилового эфира, растворителя на основе галогенированных углеводородов или бензольного растворителя либо комбинацию из двух или более из этого; массовое соотношение между растворителем J и соединением типа VII составляет 4-20:1;
ii) указанная кислота представляет собой одну или комбинацию двух или более из соляной кислоты, серной кислоты, борной кислоты или трифторуксусной кислоты; молярное соотношение между указанной кислотой и соединением типа VII составляет 5-7:1;
iii) температура реакции циклизации и гидролиза составляет 10-100°C; предпочтительно температура реакции циклизации и гидролиза составляет 70-95°C.
| ЦИКЛИЧЕСКИЕ БОРОНОВЫЕ КИСЛОТНО-ЭФИРНЫЕ ПРОИЗВОДНЫЕ И ИХ ИСПОЛЬЗОВАНИЕ В ТЕРАПИИ | 2011 |
|
RU2599791C2 |
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| Способ получения цианистых соединений | 1924 |
|
SU2018A1 |

