Область техники
[0001] Данная заявка относится к области фармацевтической биохимической инженерии, в частности, относится к способу получения промежуточного соединения для получения авибактама, и более конкретно относится к простому способу получения соли ({[(2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]окт-6-ил]окси}сульфонил)тетрабутиламмония.
Уровень техники
[0002] Авибактам, который является одним из соединений диазабициклооктанона, поскольку он представляет собой ингибитор не на основе β-лактама, может ингибировать β-лактамазы типа A (в том числе ESBL и KPC) и типа C. При совместном введении с различными типами антибиотиков, представляющих собой цефалоспорины и карбапенем, авибактам обладает активностью широкого спектра действия в отношении бактерий, в частности, обладает значительной активностью в отношении Escherichia coli и Klebsiella pneumoniae, содержащих широкий спектр β-лактамаз, Escherichia coli, содержащей избыточное количество фермента AmpC, и Escherichia coli, содержащей как AmpC, так и широкий спектр β-лактамаз. Авибактам (I), представляющий собой [(1R,2S,5R)-2-(аминокарбонил)-7-оксо-1,6-диазабицикло[3.2.1]окт-6-ил]сульфат натрия, с номером CAS 1192491-61-4, характеризуется структурной формулой, представленной формулой I:

[0003] В патентной литературе CN 103649051 A, CN 105294690 A, CN 106866668 A, WO 2012086241, US 8148540, US 9284273 и US 9567335 раскрыто получение (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-формамида с применением 5R-[(бензилокси)амино]пиперидин-2S-карбоксилата оксалата (III) в качестве исходного материала, с помощью пути амидирования с последующей циклизацией мочевины или пути циклизации мочевины с последующим амидированием, и при этом полученный после этого (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-формамид подвергали дебензилированию посредством каталитического гидрогенолиза с помощью палладия на угле с последующим сульфатированием комплекса триоксида серы, преобразованием в солевую форму аммония и ионным обменом с получением, тем самым, авибактама (I), в соответствии со схемой 1.

III

Схема 1
[0004] 1. В способе амидирования с последующей циклизацией мочевины сначала проводят амидирование 5R-[(бензилокси)амино]пиперидин-2S-карбоксилата оксалата (III) с помощью раствора аммиака в метаноле или раствора гидроксида аммония в спирте с получением (2S,5R)-5-[(бензилокси)амино]пиперидин-2-формамида; затем получают (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-формамид (с общим выходом, составляющим 61,2% – 89,1%) посредством проведения стадий введения защиты для аминогруппы в пиперидиновом кольце с помощью 9-фторенилметил-хлорформиата (FMOC-CL) или ди-трет-бутилдикарбоната, осуществления реакции карбонилирования карбонилдиимидазола с бензилоксиламином, удаления защитной группы с пиперидинового кольца с помощью диэтиламина, и затем осуществляют циклизацию мочевины.
[0005] В данном способе после амидирования получают амидную группу, таким образом оно является необходимым в реакции циклизации мочевины, в которой нельзя применять недорогие и легкодоступные трифосген или дифосген. Это связано с тем, что под действием трифосгена или дифосгена амидные группы легко дегидратируются с получением цианида; это обуславливает высокое содержание побочных продуктов и сложную очистку, в соответствии со схемой 2.

Схема 2
[0006] Даже если применяют карбонилдиимидазол (CDI) с высокой селективностью в качестве реагента для циклизации мочевины, 9-фторенилметил-хлорформиат (FMOC-CL) или ди-трет-бутилдикарбонат по-прежнему является необходимым для защиты аминогрупп в пиперидиновом кольце; в противном случае, поскольку карбонилдиимидазол и две аминогруппы (аминогруппа в пиперидиновом кольце и в бензилоксиламино) характеризуются близкими значениями активности реакции, образуются производные, в которых имидазолкарбонил вводится при двух атомах азота, при этом молярное соотношение двух производных составляет приблизительно 1:1, тогда как имидазолкарбонил при пиперидиновом кольце предпочтительно вступает в реакцию с карбоксамидной группой в орто-положении, целевой продукт не может быть получен, и выход продукта составляет менее 50%, в соответствии со схемой 3.

Схема 3
[0007] Защитное средство, представляющее собой 9-фторенилметил-хлорформиат (FMOC-CL) или ди-трет-бутилдикарбонат, характеризуется высокой стоимостью; кроме того, они обеспечивают только одну карбонильную группу; следовательно, атомная эффективность реакции является низкой, и осуществление способа является трудоемким.
[0008] 2. В способе циклизации мочевины с последующим амидированием получают (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновую кислоту посредством проведения стадий циклизации мочевины 5R-[(бензилокси)амино]пиперидин-2S-карбоксилата оксалата (III) с помощью реагента для циклизации мочевины (органического основания на основе трифосгена, карбонилдиимидазола или другого средства для карбонилирования) с последующим гидролизом в щелочной среде, такой как водный раствор гидроксида лития; и затем получают (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-формамид посредством проведения стадий активирования карбоксильной группы с образованием ангидридов с помощью триметилацетилхлорида или другого реагента, с последующим амидированием с помощью гидроксида аммония, при этом общий выход составляет 34,5-65,5%. Бензил-(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксилат, полученный после циклизации мочевины, характеризуется низкой активностью, вследствие чего он не может быть амидирован в растворе аммиака в метаноле; вместо этого эффективное амидирование может быть реализовано только путем сначала гидролиза сложноэфирной группы с образованием карбоксильной группы и последующего активирования карбоксильной группы с образованием ангидридов; следовательно, при этом задействуют больше стадий операций.
[0009] Следовательно, ни один из вышеперечисленных способов не обеспечивает простое промышленное производство промежуточного соединения для получения авибактама, а именно соли ({[(2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]окт-6-ил]окси}сульфонил)тетрабутиламмония (II).
Краткое описание
[0010] Чтобы устранить недостатки предшествующего уровня техники, в данной заявке предусмотрен простой способ получения промежуточного соединения для получения авибактама, а именно соли ({[(2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]окт-6-ил]окси}сульфонил)тетрабутиламмония (II), который характеризуется легко управляемыми условиями реакции, высоким удобством использования, оптимизированным процессом, низкой стоимостью, меньшим количеством побочных продуктов, высокой атомной эффективностью реакции, а также высокой степенью чистоты и высоким выходом продукта (II). Авибактам (I) можно получать посредством реакции ионного обмена полученного продукта (II).
[0011] Определение терминов
[0012] Соединение формулы II: промежуточное соединение для получения авибактама, а именно соль ({[(2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]окт-6-ил]окси}сульфонил)тетрабутиламмония, где -Bu4 в структурной формуле означает тетрабутил;
[0013] соединение формулы III: 5R-[(бензилокси)амино]пиперидин-2S-карбоксилата оксалат, где -Bn означает бензил;
[0014] соединение формулы V: N,N-двузамещенный бензил-5R-бензилоксиаминопиперидин-2S-формамид, где -Bn означает бензил;
[0015] соединение формулы VI: N,N-двузамещенный бензил-(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окто-2-формамид; где -Bn означает бензил.
[0016] Номера соединений в описании полностью соответствуют номерам их структурных формул, и они имеют одни и те же ссылки.
[0017] Техническое решение в соответствии с данной заявкой представлено ниже.
[0018] Способ получения промежуточного соединения для получения авибактама, предусматривающий следующие стадии:
[0019] 1) осуществление реакции амидирования, в соответствии с которой соединение формулы III вводят в реакцию с амидом формулы IV в растворителе A и в присутствии основания A с получением соединения формулы V;

[0020] где в соединении формулы III R означает C1-6-алифатическую группу или C1-6алкилзамещенный фенил; предпочтительно R выбран из группы, состоящей из метила, этила, н-пропила, изопропила, н-бутила, изобутила, трет-бутила, н-пентила, изопентила, трет-амила, гексила, бензила, о-метилбензила и п-метилбензила;
[0021] в соединении формулы IV R’ выбран из группы, состоящей из водорода, о-метокси, о-метила, п-метокси и п-метила;
[0022] R’ в соединении формулы V является идентичным R’ в соединении формулы IV;
[0023] 2) циклизация мочевины, в соответствии с которой соединение формулы V вводят в реакцию с реагентом для карбонилирования в растворителе B и в присутствии основания B с получением соединения формулы VI;

VI,
где R’ в соединении формулы VI является идентичным R’ в соединении формулы IV;
[0024] 3) каталитическое гидрирование, в соответствии с которым бензил или замещенный бензил в соединении формулы VI удаляют в растворителе C и в присутствии основания C, затем полученное соединение сульфатируют с помощью комплекса триоксида серы с получением продукта; затем осуществляют преобразование продукта в солевую форму тетрабутиламмония с получением соединения формулы II:
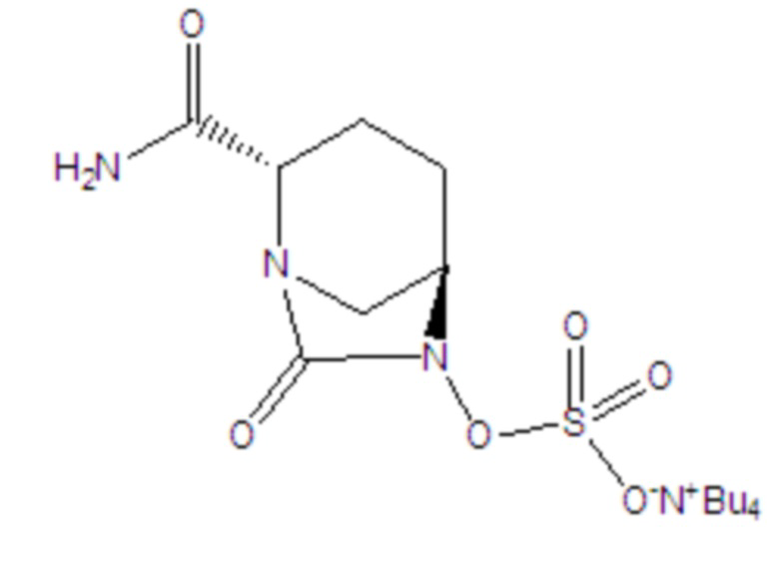
II.
[0025] Предпочтительно, в соответствии с данной заявкой, растворитель A на стадии (1) выбран из группы, состоящей из дихлорметана, 1,2-дихлорэтана, трихлорметана, тетрахлорметана, тетрагидрофурана, 2-метилтетрагидрофурана, метоксициклопентана, метилбензола и комбинаций из двух или более из них.
[0026] Предпочтительно, в соответствии с данной заявкой, на стадии (1) массовое соотношение растворителя A и соединения формулы III составляет 4-20:1.
[0027] Предпочтительно, в соответствии с данной заявкой, на стадии (1) основание A представляет собой неорганическое основание или органическое основание; предпочтительно неорганическое основание представляет собой одно из карбоната калия, или карбоната натрия, или смеси двух из них, и органическое основание представляет собой дибензиламин.
[0028] Предпочтительно, в соответствии с данной заявкой, на стадии (1) молярное соотношение основания A и соединения формулы III составляет 2,0-5,0:1.
[0029] Предпочтительно, в соответствии с данной заявкой на стадии (1) амид формулы IV выбран из группы, состоящей из дибензиламина, ди(о-метокси)бензиламина, ди(п-метокси)бензиламина, ди(о-метил)бензиламина и ди(п-метил)бензиламина.
[0030] Предпочтительно, в соответствии с данной заявкой, на стадии (1) молярное соотношение амида формулы IV и соединения формулы III составляет 1-4:1.
[0031] Предпочтительно, в соответствии с данной заявкой, на стадии (1) реакцию амидирования проводят при температуре от 0°C до 100°C; предпочтительно реакцию амидирования проводят при температуре от 30°C до 80°C. Продолжительность реакции находится в диапазоне от 1 до 8 часов.
[0032] Предпочтительно, в соответствии с данной заявкой, на стадии (2) растворитель B выбран из группы, состоящей из дихлорметана, 1,2-дихлорэтана, трихлорметана, тетрахлорметана, тетрагидрофурана, 2-метилтетрагидрофурана, метоксициклопентана, н-бутанола, трет-бутанола или метилбензола и комбинаций из двух или более из них.
[0033] Предпочтительно, в соответствии с данной заявкой на стадии (2) массовое соотношение растворителя B и соединения формулы V составляет 4-27:1.
[0034] Предпочтительно, в соответствии с данной заявкой, на стадии (2) основание B представляет собой органическое основание; предпочтительно органическое основание представляет собой триэтиламин или три-н-бутиламин.
[0035] Предпочтительно, в соответствии с данной заявкой, на стадии (2) молярное соотношение основания B и соединения формулы V составляет 2-6:1.
[0036] Предпочтительно, в соответствии с данной заявкой, на стадии (2) молярное соотношение реагента для карбонилирования и соединения формулы V составляет 0,3-3:1.
[0037] Предпочтительно, в соответствии с данной заявкой, на стадии (2) реагент для карбонилирования выбран из группы, состоящей из трифосгена, дифосгена, карбонилдиимидазола и ди-трет-бутилдикарбоната.
[0038] Предпочтительно молярное соотношение трифосгена и соединения формулы V составляет 0,3-1,5:1; молярное соотношение дифосгена и соединения формулы V составляет 0,5-2,0:1; молярное соотношение карбонилдиимидазола или ди-трет-бутилдикарбоната и соединения формулы V составляет 1,0-3,0:1.
[0039] Предпочтительно, в соответствии с данной заявкой, на стадии (2) температура реакции циклизации мочевины находится в диапазоне от −20°C до 100°C; предпочтительно температура реакции циклизации мочевины находится в диапазоне от 10 до 40°C. Продолжительность реакции находится в диапазоне от 4 часов до 10 часов.
[0040] Предпочтительно, в соответствии с данной заявкой, на стадии (3) растворитель C выбран из группы, состоящей из изопропанола, н-бутанола, трет-бутанола, тетрагидрофурана, N,N-диметилформамида, воды и смесей из двух или более из них.
[0041] Предпочтительно, в соответствии с данной заявкой, на стадии (3) массовое соотношение растворителя C и соединения формулы VI составляет 4-20:1.
[0042] Предпочтительно, в соответствии с данной заявкой, на стадии (3) основание C представляет собой органическое основание; предпочтительно органическое основание представляет собой триэтиламин.
[0043] Предпочтительно, в соответствии с данной заявкой, на стадии (3) молярное соотношение основания C и соединения формулы VI составляет 0,1-0,3:1.
[0044] Предпочтительно, в соответствии с данной заявкой, на стадии (3) катализатор для каталитического гидрогенолиза представляет собой палладий на угле с массовым содержанием палладия 5% или палладий на угле с массовым содержанием палладия 10%; при этом палладий на угле характеризуется содержанием воды, составляющим 5-55 вес. %.
[0045] Предпочтительно, в соответствии с данной заявкой, на стадии (3) масса катализатора для каталитического гидрогенолиза составляет 1,0-20,0% от массы соединения формулы VI; давление водорода, применяемого при каталитическом гидрогенолизе, составляет 0,05-0,30 МПа.
[0046] Предпочтительно, в соответствии с данной заявкой, на стадии (3) комплекс триоксида серы представляет собой один из триоксида триметиламина и серы, триоксида пиридина и серы или триоксида триэтиламина и серы.
[0047] Предпочтительно, в соответствии с данной заявкой, молярное соотношение комплекса триоксида серы и соединения формулы VI составляет 1,0-2,0:1.
[0048] Предпочтительно, в соответствии с данной заявкой, на стадии (3) источник тетрабутиламмония, применяемый при преобразовании в солевую форму тетрабутиламмония, представляет собой гидроксид тетрабутиламмония или ацетат тетрабутиламмония.
[0049] Предпочтительно, в соответствии с данной заявкой, на стадии (3) молярное соотношение источника тетрабутиламмония, применяемого при преобразовании в солевую форму тетрабутиламмония, и соединения формулы VI составляет 0,8-1,2:1.
[0050] Предпочтительно, в соответствии с данной заявкой, на стадии (3) значения температуры реакции каталитического гидрогенолиза и реакции сульфатирования находятся в диапазоне от −10°C до 60°C; еще более предпочтительно значения температуры реакции каталитического гидрогенолиза и реакции сульфатирования находятся в диапазоне от 10°C до 40°C. Каталитический гидрогенолиз и сульфатирование являются реакциями для "однореакторного способа".
[0051] Предпочтительно, в соответствии с данной заявкой, на стадии (3), значения продолжительности реакции каталитического гидрогенолиза и реакции сульфатирования составляют 1-6 часов.
[0052] Предпочтительно, в соответствии с данной заявкой, на стадии (3) температура реакции преобразования в солевую форму тетрабутиламмония находится в диапазоне от 0°C до 50°C; предпочтительно температура реакции преобразования в солевую форму тетрабутиламмония находится в диапазоне от 10°C до 30°C.
[0053] Предпочтительно, в соответствии с данной заявкой, на стадии (3) продолжительность реакции преобразования в солевую форму тетрабутиламмония составляет 1-15 часов; предпочтительно продолжительность реакции преобразования в солевую форму тетрабутиламмония составляет 2-4 часа.
[0054] В данной заявке N,N-двузамещенный бензил-5R-бензилоксиаминопиперидин-2S-формамид (V) получают посредством проведения стадии амидирования 5R-[(бензилокси)амино]пиперидин-2S-карбоксилата оксалата (III) в качестве исходного материала с помощью амида формулы IV; осуществляют циклизацию мочевины полученного соединения V с помощью реагента для карбонилирования с получением N,N-двузамещенного бензил-(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окто-2-формамида (VI); посредством проведения стадий удаления бензила или замещенного бензила из соединения VI посредством каталитического гидрогенолиза с последующим сульфатированием с помощью комплекса триоксида серы и преобразованием в солевую форму тетрабутиламмония, получают соль ({[(2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]окт-6-ил]окси}сульфонил)тетрабутиламмония (II) (схема 4); посредством реакции ионного обмена полученного соединения формулы II может быть получен авибактам (I).

Схема 4
[0055] В данной заявке предусмотрены следующие преимущественные эффекты.
[0056] 1. Полученный N,N-двузамещенный бензил-5R-бензилоксиаминопиперидин-2S-формамид (V), полученный в соответствии с данной заявкой характеризуется высокой стабильностью, и способ в соответствии с данной заявкой обеспечивает предотвращение протекания побочной реакции при его карбоксамиде в положении 2, исходя из разработанной реакции; кроме того, не требуется защита пиперидинового кольца, и при этом реагент для карбонилирования, который является недорогим и легкодоступным, можно применять непосредственно для циклизации мочевины; условия реакции являются легко управляемыми; способ получения характеризуется высоким удобством проведения, простотой способа, высокой атомной эффективностью реакции и низкой стоимостью.
[0057] 2. Дополнительно, а также неожиданно, N,N-двузамещенный бензил-(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окто-2-формамид (VI) характеризуется подходящей активностью гидрогенолиза, которая обеспечивает уменьшение накопления в системе продукта гидрогенолиза, представляющего собой (2S,5R)-6-гидроксил-7-оксо-1,6-диазабицикло[3.2.1]окто-2-формамид; как только образуется продукт гидрогенолиза, сульфатирование можно проводить своевременно; кроме того, полученный продукт (II) характеризуется высокой степенью чистоты и высоким выходом.
Примеры
[0058] Далее в данном документе данная заявка будет подробно проиллюстрирована со ссылкой на примеры; однако данная заявка не ограничена ими.
[0059] Если не указано иное, все значения процентного содержания в примерах означают массовые процентные доли.
[0060] Исходный материал, представляющий собой 5R-[(бензилокси)амино]пиперидин-2S-карбоксилата оксалат (III), является легкодоступным на рынке (от фармацевтической компании Jinan Qinsi), при этом он представляет собой белый порошок с оптической чистотой, составляющей 99,6%.
[0061] Процесс протекания реакции и чистоту продукта контролировали с помощью жидкостного хроматографа. Жидкостный хроматограф, оснащенный хиральной колонкой (ES-OVS, 150 мм x 4,6 мм, Agilent), применяют для определения оптической чистоты (отношение значений площади %) и расчета выхода, а также % значения энантиомерной чистоты.
[0062] Пример 1. Получение N,N-дибензил-5R-[(бензилокси)амино]пиперидин-2S-формамида (V1)
[0063] В 4-горлую колбу объемом 500 мл, оснащенную мешалкой и термометром, загружали 250 г тетрагидрофурана, 28,0 г карбоната калия, 43,0 г (0,1 моль) бензил-5R-[(бензилокси)амино]пиперидин-2S-карбоксилата оксалата (III) и 30 г (0,15 моль) дибензиламина и затем реакционную смесь перемешивали в течение 5 часов при 40°C – 45°C, затем охлаждали до 20°C – 25°C и фильтровали. Осадок на фильтре дважды промывали тетрагидрофураном, каждый раз по 30 г. Органические фазы объединяли и перегоняли с извлечением тетрагидрофурана. Загружали 40 г метил-трет-бутилового сложного эфира в остаток, затем растирали и промывали, и фильтровали с получением 41,1 г N,N-дибензил-5R-[(бензилокси)амино]пиперидин-2S-формамида с выходом 95,8% и чистотой 99,92%, определенными с помощью HPLC.
[0064] Данные ЯМР (ядерного магнитного резонанса) полученного продукта приведены ниже: 1H-ЯМР (400 МГц, DMSO-d6)
1H-ЯМР (400 МГц, DMSO-d6) δ: 1,12 (1H, q), 1,29 (1H, q), 1,86 (2H, d), 2,29 (1H, t), 2,76 (1H, m), 2,95 (1H, d), 3,18 (1H, d), 4,62 (4H, s), 4,83 (2H, s), 6,50 (1H, d), 7,28-7,47 (15H, m).
[0065] Пример 2. Получение N,N-дибензил-5R-[(бензилокси)амино]пиперидин-2S-формамида (V1)
[0066] В 4-горлую колбу объемом 500 мл, оснащенную мешалкой и термометром, загружали 300 г 1,2-дихлорэтана, 43,0 г (0,1 моль) бензил-5R-[(бензилокси)амино]пиперидин-2S-карбоксилата оксалата (III) и 60 г (0,30 моль) дибензиламина и затем реакционную смесь перемешивали в течение 4 часов при 50°C – 55°C, затем охлаждали до 20°C – 25°C и фильтровали. Осадок на фильтре дважды промывали 1,2-дихлорэтаном, каждый раз по 30 г. Органические фазы объединяли и перегоняли с извлечением 1,2-дихлорэтана. Загружали 50 г метил-трет-бутилового сложного эфира в остаток, затем растирали и промывали, и фильтровали с получением 40,5 г N,N-дибензил-5R-[(бензилокси)амино]пиперидин-2S-формамида с выходом 94,4% и чистотой 99,86%, определенными с помощью HPLC.
[0067] Пример 3. Получение N,N-ди(п-метоксибензил)-5R-[(бензилокси)амино]пиперидин-2S-формамида (V2)
[0068] В 4-горлую колбу объемом 500 мл, оснащенную мешалкой и термометром, загружали 250 г тетрагидрофурана, 28,0 г карбоната калия, 37,0 г (0,1 моль) этил-5R-[(бензилокси)амино]пиперидин-2S-карбоксилата оксалата (III) и 40 г (0,16 моль) ди(п-метокси)бензиламина и затем реакционную смесь перемешивали в течение 4 часов при 50°C – 55°C, затем охлаждали до 20°C – 25°C и фильтровали. Осадок на фильтре дважды промывали тетрагидрофураном, каждый раз по 30 г. Органические фазы объединяли и перегоняли с извлечением тетрагидрофурана. Загружали 40 г метил-трет-бутилового сложного эфира в остаток, затем растирали и промывали, и фильтровали с получением 45,7 г N,N-ди(п-метоксибензил)-5R-[(бензилокси)амино]пиперидин-2S-формамида с выходом 93,5% и чистотой 99,93%, определенными с помощью HPLC.
[0069] Данные ЯМР (ядерного магнитного резонанса) полученного продукта приведены ниже: 1H-ЯМР (400 МГц, DMSO-d6)
1H-ЯМР (400 МГц, DMSO-d6) δ: 1,15 (1H, q), 1,34 (1H, q), 1,88 (2H, d), 2,30 (1H, t), 2,90 (1H, m), 3,01 (1H, d), 3,21 (1H, d), 3,80 (6H, s), 4,46 (4H, s), 4,76 (2H, s), 6,48 (1H, d), 6,90 (4H, d), 7,25 (4H, d), 7,55 (5H, m).
[0070] Пример 4. Получение N,N-ди(п-метилбензил)-5R-[(бензилокси)амино]пиперидин-2S-формамида (V3)
[0071] В 4-горлую колбу объемом 500 мл, оснащенную мешалкой и термометром, загружали 250 г 2-метилтетрагидрофурана, 30,0 г карбоната калия, 39,5 г (0,1 моль) трет-бутил-5R-[(бензилокси)амино]пиперидин-2S-карбоксилата оксалата (III) и 40 г (0,18 моль) ди(п-метил)бензиламина и затем реакционную смесь перемешивали в течение 4 часов при 60°C – 65°C, затем охлаждали до 20°C – 25°C и фильтровали. Осадок на фильтре дважды промывали 2-метилтетрагидрофураном, каждый раз по 30 г. Органические фазы объединяли и перегоняли с извлечением 2-метилтетрагидрофурана. Загружали 40 г метил-трет-бутилового сложного эфира в остаток, затем растирали и промывали, и фильтровали с получением 43,6 г N,N-ди(п-метилбензил)-5R-[(бензилокси)амино]пиперидин-2S-формамида с выходом 95,5% и чистотой 99,89%, определенными с помощью HPLC.
[0072] Данные ЯМР (ядерного магнитного резонанса) полученного продукта приведены ниже: 1H-ЯМР (400 МГц, DMSO-d6)
1H-ЯМР (400 МГц, DMSO-d6) δ: 1,13 (1H, q), 1,31 (1H, q), 1,85 (2H, d), 2,06 (6H, s), 2,27 (1H, t), 2,85 (1H, m), 2,91 (1H, d), 3,17 (1H, d), 4,60 (4H, s), 4,78 (2H, s), 6,54 (1H, d), 7,08 (4H, d), 7,19 (4H, d), 7,48 (5H, m).
[0073] Структурные формулы соединений V1, V2 и V3 являются следующими:

[0074] Пример 5. Получение N,N-дибензил-(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окто-2-формамида (VI1)
[0075] В 4-горлую колбу объемом 1000 мл, оснащенную мешалкой и термометром, загружали 250 г тетрагидрофурана, 43 г (0,1 моль) N,N-дибензил-5R-[(бензилокси)амино]пиперидин-2S-формамида (V1), полученного в соответствии с примером 1, и 50 г триэтиламина и затем охлаждали до −10°C – 0°C, затем загружали 30 г (0,1 моль) трифосгена и 100 г раствора тетрагидрофурана и затем реакционную смесь перемешивали в течение 8 часов при 10-20°C. Реакционную смесь выливали в 400 г смеси льда и воды и разделяли и водную фазу трижды экстрагировали дихлорметаном, каждый раз по 100 г. Органические фазы объединяли и затем дважды промывали насыщенным водным раствором хлорида натрия, каждый раз по 50 г; после того, как растворитель извлекали из полученной органической фазы, к остатку добавляли 60 г метил-трет-бутилового сложного эфира; остаток растирали, промывали и затем фильтровали с получением 42,1 г N,N-дибензил-(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окто-2-формамида с выходом 92,5% и чистотой 99,96%, определенными с помощью HPLC.
[0076] Данные ЯМР (ядерного магнитного резонанса) полученного продукта приведены ниже: 1H-ЯМР (400 МГц, DMSO-d6) δ: 1,65 (2H, m), 1,84 (1H, br), 2,06 (1H, m), 2,90 (2H, s), 3,62 (1H, s), 4,58 (4H, s), 4,93 (2H, dd), 7,28-7,47 (15H, m).
[0077] Пример 6. Получение N,N-дибензил-(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окто-2-формамида (VI1)
[0078] В 4-горлую колбу объемом 500 мл, оснащенную мешалкой и термометром, загружали 60 г тетрагидрофурана, 4,3 г (0,01 моль) N,N-дибензил-5R-[(бензилокси)амино]пиперидин-2S-формамида (V1), полученного в соответствии с примером 1, и 5,0 г три-н-бутиламина и затем охлаждали до −10°C – 0°C, затем загружали 3,0 г (0,015 моль) дифосгена и 20 г раствора тетрагидрофурана и затем реакционную смесь перемешивали в течение 8 часов при 10-20°C. Реакционную смесь выливали в 200 г смеси льда и воды и разделяли и водную фазу трижды экстрагировали дихлорметаном, каждый раз по 50 г. Органические фазы объединяли и затем дважды промывали насыщенным водным раствором хлорида натрия, каждый раз по 20 г; после того, как растворитель извлекали из полученной органической фазы, к остатку добавляли 20 г метил-трет-бутилового сложного эфира; остаток растирали, промывали и затем фильтровали с получением 4,15 г N,N-дибензил-(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окто-2-формамида с выходом 91,2% и чистотой 99,9%, определенными с помощью HPLC.
[0079] Пример 7. Получение N,N-ди(п-метокси)бензил-(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окто-2-формамида (VI2)
[0080] В 4-горлую колбу объемом 500 мл, оснащенную мешалкой и термометром, загружали 100 г дихлорметана, 4,9 г (0,01 моль) N,N-ди(п-метоксибензил)-5R-[(бензилокси)амино]пиперидин-2S-формамида (V2), полученного в соответствии с примером 3, и 5,0 г три-н-бутиламина и затем охлаждали до −10°C – 0°C, затем загружали 3,0 г (0,01 моль) трифосгена и 20 г раствора дихлорметана и затем реакционную смесь перемешивали в течение 8 часов при 10-20°C. Реакционную смесь выливали в 200 г смеси льда и воды и разделяли и водную фазу трижды экстрагировали дихлорметаном, каждый раз по 50 г. Органические фазы объединяли и затем дважды промывали насыщенным водным раствором хлорида натрия, каждый раз по 20 г; после того, как растворитель извлекали из полученной органической фазы, к остатку добавляли 20 г метил-трет-бутилового сложного эфира; остаток растирали, промывали и затем фильтровали с получением 4,75 г N,N-ди(п-метокси)бензил-(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окто-2-формамида с выходом 92,2% и чистотой 99,92%, определенными с помощью HPLC.
[0081] Данные ЯМР (ядерного магнитного резонанса) полученного продукта приведены ниже: 1H-ЯМР (400 МГц, DMSO-d6) δ: 1,60 (2H, m), 1,81 (1H, br), 2,02 (1H, m), 2,88 (2H, s), 3,59 (1H, s), 3,62 (1H, d), 3,78 (6H, s), 4,55 (4H, s), 4,85 (2H, dd), 6,82 (4H, d), 7,16 (4H, d), 7,47 (5H, m).
[0082] Пример 8. Получение N,N-ди(п-метил)бензил-(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окто-2-формамида (VI3)
[0083] В 4-горлую колбу объемом 500 мл, оснащенную мешалкой и термометром, загружали 100 г дихлорметана, 4,6 г (0,01 моль) N,N-ди(п-метилбензил)-5R-[(бензилокси)амино]пиперидин-2S-формамида (V3), полученного в соответствии с примером 4, и 4,0 г триэтиламина и затем охлаждали до 0°C – 10°C, затем загружали 3,0 г (0,01 моль) трифосгена и 20 г раствора дихлорметана и затем реакционную смесь перемешивали в течение 6 часов при 20-30°C. Реакционную смесь выливали в 200 г смеси льда и воды и разделяли и водную фазу трижды экстрагировали дихлорметаном, каждый раз по 50 г. Органические фазы объединяли и затем дважды промывали насыщенным водным раствором хлорида натрия, каждый раз по 20 г; после того, как растворитель извлекали из полученной органической фазы, к остатку добавляли 20 г метил-трет-бутилового сложного эфира; остаток растирали, промывали и затем фильтровали с получением 4,47 г N,N-ди(п-метил)бензил-(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окто-2-формамида с выходом 92,5% и чистотой 99,96%, определенными с помощью HPLC.
[0084] Данные ЯМР (ядерного магнитного резонанса) полученного продукта приведены ниже: 1H-ЯМР (400 МГц, DMSO-d6) δ: 1,63 (2H, m), 1,85 (1H, br), 2,05 (1H, m), 2,10 (6H, s), 2,91 (2H, s), 3,62 (1H, s), 3,68 (1H, d), 3,83 (6H, s), 4,61 (4H, s), 4,88 (2H, dd), 6,84 (4H, d), 7,19 (4H, d) и 7,51 (5H, m).
[0085] Структурные формулы соединений VI1, VI2 и VI3 являются следующими:

[0086] Пример 9. Получение соли ({[(2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]окт-6-ил]окси}сульфонил)тетрабутиламмония
[0087] В реактор из нержавеющей стали загружали 14 г изопропанола, 17 г воды, 4,9 г (0,01 моль) N,N-дибензил-(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окто-2-формамида (VI1), полученного в соответствии со способом из примера 5, 1,56 г (0,0112 моль) комплекса триоксида серы и триметиламина, 0,2 г (0,002 моль) триэтиламина и 0,11 г палладия на угле (с содержанием воды, составляющим 55 вес. %) с массовым содержанием палладия, составляющим 10%; после того, как реактор закрывали, вводили защитную атмосферу газообразного азота. В реактор периодически вводили водород для поддержания давления газообразного водорода на уровне 0,07-0,13 МПа; реактор изолировали при комнатной температуре в течение 1 часа, пока исходный материал VI1 полностью не прореагировал (в данный момент давление повышалось). После замещения с помощью газообразного азота реактор продолжали изолировать при комнатной температуре в течение еще 1,5 ч. После того, как загружали 0,16 г (0,0026 моль) уксусной кислоты для нейтрализации, палладий на угле отфильтровали и осадок на фильтре промывали с помощью 8,6 г воды. Фильтрат промывали с помощью 26 мл н-бутилацетата и разделяли на слои и затем отделяли водную фазу.
[0088] Предварительно растворяли 3,68 г (0,0122 моль) ацетата тетрабутиламмония и 0,06 г (0,001 моль) уксусной кислоты в 6,5 г воды с получением раствора ацетата тетрабутиламмония. Загружали 70 вес. % раствор ацетата тетрабутиламмония в промытую и отделенную водную фазу и осуществляли преобразование в солевую форму при комнатной температуре в течение 1-2 ч. Применяли 26 мл дихлорметана для экстрагирования органических фаз; затем для получения органических фаз раствор разделяли на слои. Оставшийся 30 вес. % раствор ацетата тетрабутиламмония загружали в органическую фазу и осуществляли преобразование в солевую форму при комнатной температуре в течение 1-2 ч. с последующим экстрагированием с помощью 9 мл дихлорметана. Затем органические фазы объединяли и концентрировали до 20 мл. Затем загружали 50 мл метил-изобутилкетона, после чего смесь концентрировали до 40 мл, охлаждали до 0°C и фильтровали. Затем осадок на фильтре промывали с помощью 10 мл метил-изобутилкетона, затем высушивали в вакууме и получали в результате 4,5 г соли ({[(2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]окт-6-ил]окси}сульфонил)тетрабутиламмония (II) с выходом 88,7% и чистотой 99,3%, определенными с помощью HPLC.
[0089] Данные ЯМР (ядерного магнитного резонанса) полученного продукта приведены ниже: 1H-ЯМР (400 МГц, CDCl3) δ: 1,00 (12H, t), 1,45 (8H, m), 1,67 (9H, m), 1,87 (1H, m), 2,16 (1H, m), 2,37 (1H, dd), 2,87 (1H, d), 3,31 (9H, m), 3,91 (1H, d), 4,33 (1H, s), 5,87 (1H, s), 6,69 (1H, s).
[0090] Пример 10. Получение соли ({[(2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]окт-6-ил]окси}сульфонил)тетрабутиламмония (II)
[0091] В реактор для гидрогенизации загружали 16 г изопропанола, 20 г воды, 4,9 г (0,01 моль) N,N-ди(п-метокси)бензил-(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окто-2-формамида (VI2), полученного в соответствии с примером 7, 1,56 г (0,0112 моль) триоксида серы и триметиламина, 0,2 г (0,002 моль) триэтиламина и 0,12 г палладия на угле (с содержанием воды, составляющим 55 вес. %) с массовым содержанием палладия, составляющим 10%; после того, как реактор закрывали, применяли защитную атмосферу газообразного азота. В реактор периодически вводили водород для поддержания давления газообразного водорода на уровне 0,07-0,13 МПа; реактор изолировали при комнатной температуре в течение 1 часа, пока исходный материал VI2 полностью не прореагировал (в данный момент давление повышалось). После замещения с помощью газообразного азота реактор продолжали изолировать при комнатной температуре в течение еще 1,5 ч. После того, как загружали 0,16 г (0,0026 моль) уксусной кислоты для нейтрализации, палладий на угле отфильтровали и осадок на фильтре промывали с помощью 10 г воды. Фильтрат промывали с помощью 30 мл н-бутилацетата и разделяли на слои и затем отделяли водную фазу.
[0092] Предварительно растворяли 3,68 г (0,0122 моль) ацетата тетрабутиламмония и 0,06 г (0,001 моль) уксусной кислоты в 7,5 г воды с получением раствора ацетата тетрабутиламмония. Загружали 70 вес. % раствор ацетата тетрабутиламмония в промытую и отделенную водную фазу и осуществляли преобразование в солевую форму при комнатной температуре в течение 1-2 ч. Применяли 30 мл дихлорметана для экстрагирования органических фаз; затем для получения органических фаз раствор разделяли на слои. Оставшийся 30 вес. % раствор ацетата тетрабутиламмония загружали в органическую фазу и осуществляли преобразование в солевую форму при комнатной температуре в течение 1-2 ч, после чего смесь экстрагировали с помощью 10 мл дихлорметана, органические фазы объединяли и концентрировали до 20 мл. Затем загружали 50 мл метил-изобутилкетона, после чего смесь концентрировали до 40 мл, охлаждали до 0°C и фильтровали. Затем осадок на фильтре промывали с помощью 10 мл метил-изобутилкетона, затем высушивали в вакууме и получали в результате 4,7 г соли ({[(2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]окт-6-ил]окси}сульфонил)тетрабутиламмония (II) с выходом 92,7% и чистотой 99,1%, определенными с помощью HPLC.
[0093] Пример 11. Получение соли ({[(2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]окт-6-ил]окси}сульфонил)тетрабутиламмония (II)
[0094] В реактор для гидрогенизации загружали 15 г изопропанола, 18 г воды, 4,6 г (0,01 моль) N,N-ди(п-метил)бензил-(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окто-2-формамида (VI3), полученного в соответствии с примером 8, 1,56 г (0,0112 моль) триоксида серы и триметиламина, 0,2 г (0,002 моль) триэтиламина и 0,11 г палладия на угле (с содержанием воды, составляющим 55 вес. %) с массовым содержанием палладия, составляющим 10%; после того, как реактор закрывали, применяли защитную атмосферу газообразного азота. В реактор периодически вводили водород для поддержания давления газообразного водорода на уровне 0,07-0,13 МПа; реактор изолировали при комнатной температуре в течение 1 часа, пока исходный материал VI3 полностью не прореагировал (в данный момент давление повышалось). После замещения с помощью газообразного азота реактор продолжали изолировать в течение 1,5 ч при комнатной температуре. После того, как загружали 0,16 г (0,0026 моль) уксусной кислоты для нейтрализации, палладий на угле отфильтровали и осадок на фильтре промывали с помощью 8,6 г воды. Фильтрат промывали с помощью 26 мл н-бутилацетата и разделяли на слои и затем отделяли водную фазу.
[0095] Предварительно растворяли 3,68 г (0,0122 моль) ацетата тетрабутиламмония и 0,06 г (0,001 моль) уксусной кислоты в 6,5 г воды с получением раствора ацетата тетрабутиламмония. Загружали 70 вес. % раствор ацетата тетрабутиламмония в промытую и отделенную водную фазу и осуществляли преобразование в солевую форму при комнатной температуре в течение 1-2 ч. Применяли 26 мл дихлорметана для экстрагирования органических фаз; затем для получения органических фаз раствор разделяли на слои. Оставшийся 30 вес. % раствор ацетата тетрабутиламмония загружали в органическую фазу и осуществляли преобразование в солевую форму при комнатной температуре в течение 1-2 ч, после чего смесь экстрагировали с помощью 9 мл дихлорметана, органические фазы объединяли и концентрировали до 20 мл. Затем загружали 50 мл метил-изобутилкетона, после чего смесь концентрировали до 40 мл, охлаждали до 0°C и фильтровали. Затем осадок на фильтре промывали с помощью 10 мл метил-изобутилкетона, затем высушивали в вакууме с получением в результате 4,4 г соли ({[(2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]окт-6-ил]окси}сульфонил)тетрабутиламмония (II) с выходом 86,8% и чистотой 99,4%, определенными с помощью HPLC.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОСТОЙ СПОСОБ ПОЛУЧЕНИЯ АВИБАКТАМА | 2018 |
|
RU2711358C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ АВИБАКТАМА | 2018 |
|
RU2722932C1 |
| НОВЫЙ ИНГИБИТОР бета-ЛАКТАМАЗЫ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2013 |
|
RU2693898C2 |
| НОВЫЙ ИНГИБИТОР β-ЛАКТАМАЗЫ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2019 |
|
RU2800050C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2695219C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2801220C2 |
| СПОСОБ ПОЛУЧЕНИЯ 5R-[(БЕНЗИЛОКСИ)АМИНО]ПИПЕРИДИН-2S-КАРБОНОВОЙ КИСЛОТЫ ИЛИ ЕЁ ПРОИЗВОДНОГО | 2018 |
|
RU2730006C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, ВКЛЮЧАЯ ТРАНС-7-ОКСО-6-(СУЛЬФОКСИ)-1,6-ДИАЗАБИЦИКЛО[3.2.1]ОКТАН-2-КАРБОКСАМИД И ЕГО СОЛИ | 2012 |
|
RU2769076C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2023 |
|
RU2838718C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2023 |
|
RU2840014C2 |
Изобретение относится к способу получения промежуточного соединения формулы II для получения авибактама, предусматривающему следующие стадии: (1) амидирование, в соответствии с которым соединение формулы III вводят в реакцию с амидом формулы IV в растворителе A, выбранном из дихлорэтана, тетрагидрофурана и метилтетрагидрофурана, и в присутствии основания A, выбранного из карбоната калия и дибензиламина с получением соединения формулы V; (2) циклизация мочевины, в соответствии с которой соединение формулы V вводят в реакцию с реагентом для карбонилирования, выбранным из трифосгена и дифосгена, в растворителе B, выбранном из дихлорметана и тетрагидрофурана, и в присутствии основания B, выбранного из триэтиламина и трибутиламина, с получением соединения формулы VI; (3) каталитическое гидрирование, в соответствии с которым бензил или замещенный бензил в соединении формулы VI удаляют в растворителе C, представляющем собой смесь изопропанола и воды, и в присутствии основания C, представляющего собой триэтиламин, затем полученное соединение сульфатируют с помощью комплекса триоксида серы с получением продукта; затем осуществляют преобразование продукта в солевую форму тетрабутиламмония с получением соединения формулы II. Способ получения характеризуется простотой способа, низкой стоимостью, меньшим количеством побочных продуктов и высокой эффективностью реакции; полученный продукт (II) характеризуется высокой степенью чистоты и высоким выходом. В общих формулах R выбран из группы, состоящей из этила, трет-бутила и бензила; R' выбран из группы, состоящей из водорода, п-метокси и п-метила. 9 з.п. ф-лы.


1. Способ получения промежуточного соединения формулы II для получения авибактама,

при этом способ предусматривает следующие стадии:
(1) осуществление реакции амидирования, в соответствии с которой соединение формулы III вводят в реакцию с амидом формулы IV в растворителе A, выбранном из дихлорэтана, тетрагидрофурана и метилтетрагидрофурана, и в присутствии основания A, выбранного из карбоната калия и дибензиламина с получением соединения формулы V:

где в соединении формулы III R выбран из группы, состоящей из этила, трет-бутила и бензила;
в соединении формулы IV R' выбран из группы, состоящей из водорода, п-метокси и п-метила;
R' в соединении формулы V является идентичным R' в соединении формулы IV;
(2) циклизация мочевины, в соответствии с которой соединение формулы V вводят в реакцию с реагентом для карбонилирования, выбранным из трифосгена и дифосгена, в растворителе B, выбранном из дихлорметана и тетрагидрофурана, и в присутствии основания B, выбранного из триэтиламина и трибутиламина, с получением соединения формулы VI:
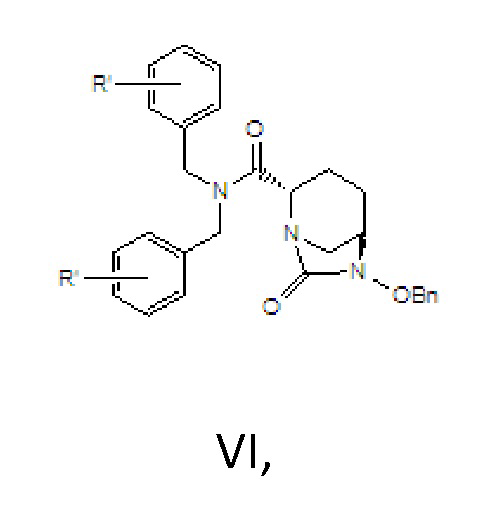
где R' в соединении формулы VI является идентичным R' в соединении формулы IV;
(3) каталитическое гидрирование, в соответствии с которым бензил или замещенный бензил в соединении формулы VI удаляют в растворителе C, представляющем собой смесь изопропанола и воды, и в присутствии основания C, представляющего собой триэтиламин, затем полученное соединение сульфатируют с помощью комплекса триоксида серы с получением продукта; затем осуществляют преобразование продукта в солевую форму тетрабутиламмония с получением соединения формулы II.
2. Способ по п. 1, где на стадии (1) массовое соотношение растворителя A и соединения формулы III составляет 4-20:1; и
предпочтительно на стадии (1) молярное соотношение основания A и соединения формулы III составляет 2,0-5,0:1.
3. Способ по п. 1, где на стадии (1) амид формулы IV выбран из группы, состоящей из дибензиламина, ди(п-метокси)бензиламина, и ди(п-метил)бензиламина;
предпочтительно на стадии (1) молярное соотношение амида формулы IV и соединения формулы III составляет 1-4:1.
4. Способ по п. 1, где на стадии (1) реакцию амидирования проводят при температуре от 0°C до 100°C; предпочтительно реакцию амидирования проводят при температуре от 30°C до 80°C.
5. Способ по п. 1, где на стадии (2) массовое соотношение растворителя B и соединения формулы V составляет 4-27:1;
предпочтительно на стадии (2) молярное соотношение основания B и соединения формулы V составляет 2-6:1.
6. Способ по п. 1, где на стадии (2) молярное соотношение реагента для карбонилирования и соединения формулы V составляет 0,3-3:1.
7. Способ по п. 1, где на стадии (2) молярное соотношение трифосгена и соединения формулы V составляет 0,3-1,5:1; или молярное соотношение дифосгена и соединения формулы V составляет 0,5-2,0:1.
8. Способ по п. 1, где на стадии (2) температура реакции циклизации мочевины находится в диапазоне от −20°C до 100°C; предпочтительно температура реакции циклизации мочевины находится в диапазоне от 1°C до 40°C.
9. Способ по п. 1, где на стадии (3) массовое соотношение растворителя C и соединения формулы VI составляет 4-20:1;
предпочтительно на стадии (3) молярное соотношение основания C и соединения формулы VI составляет 0,1-0,3:1;
предпочтительно на стадии (3) катализатор для каталитического гидрогенолиза представляет собой палладий на угле с массовым содержанием палладия, составляющим 5%, или палладий на угле с массовым содержанием палладия, составляющим 10%; при этом палладий на угле характеризуется содержанием воды, составляющим 5-55 вес. %; на стадии (3) масса катализатора для каталитического гидрогенолиза составляет 1,0-20,0% от массы соединения формулы VI; и давление водорода, применяемого при каталитическом гидрогенолизе, составляет 0,05-0,30 МПа;
предпочтительно на стадии (3) комплекс триоксида серы представляет собой один из триоксида триметиламина и серы, триоксида пиридина и серы или триоксида триэтиламина и серы; предпочтительно молярное соотношение комплекса триоксида серы и соединения формулы VI составляет 1,0-2,0:1;
предпочтительно на стадии (3) источник тетрабутиламмония, применяемый при преобразовании в солевую форму тетрабутиламмония, представляет собой гидроксид тетрабутиламмония или ацетат тетрабутиламмония; и на стадии (3) молярное соотношение источника тетрабутиламмония, применяемого при преобразовании в солевую форму тетрабутиламмония, и соединения формулы VI составляет 0,8-1,2:1.
10. Способ по п. 1, где на стадии (3) значения температуры реакции каталитического гидрогенолиза и реакции сульфатирования находятся в диапазоне от −10°C до 60°C; предпочтительно значения температуры реакции каталитического гидрогенолиза и реакции сульфатирования находятся в диапазоне от 10°C до 40°C;
предпочтительно на стадии (3) температура реакции преобразования в солевую форму тетрабутиламмония находится в диапазоне от 0°C до 50°C; еще более предпочтительно температура реакции преобразования в солевую форму тетрабутиламмония находится в диапазоне от 10°C до 30°C.
| WO 2012172368 A1, 20.12.2012 | |||
| US 8772490 B2, 08.07.2014 | |||
| WO 2014135930 A1, 12.09.2014 | |||
| WO 2012133486 A1, 04.10.2012 | |||
| СПОСОБ ПОЛУЧЕНИЯ НАТРИЕВОЙ СОЛИ (2S,5R)-2-КАРБОКСАМИДО-7-ОКСО-6-СУЛЬФООКСИ-1,6-ДИАЗАБИЦИКЛО[3.2.1]ОКТАНА | 2013 |
|
RU2632192C2 |

