РОДСТВЕННЫЕ ЗАЯВКИ
Данная заявка заявляет приоритет предварительной заявки на патент США № 61/234969, зарегистрированной 18 августа 2009, и предварительной заявки на патент США № 61/235586, зарегистрированной 20 августа 2009. Указанные заявки включены в данное описание в качестве ссылок.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Область техники, к которой относится изобретение
Данное изобретение относится к способам и композициям для модуляции иммунной функции. Конкретнее, данное изобретение относится к способам и композициям для модуляции TLR7- и/или TLR8-опосредуемой передачи сигнала.
Описание уровня техники
Стимуляция иммунной системы, которая включает стимуляцию или врожденного иммунитета или приобретенного иммунитета или того и другого, представляет собой сложное явление, которое может привести или к защитным или к вредным физиологическим последствиям для хозяина. В последние годы возрос интерес к механизмам, лежащим в основе врожденного иммунитета, который, как полагают, инициирует и поддерживает приобретенный иммунитет. Такой интерес частично подогревается недавним открытием семейства высококонсервативных белков рецепторов узнавания структуры, известных как Toll-подобные рецепторы (TLR), которые, как полагают, вовлекаются во врожденный иммунитет как рецепторы для патогенассоциированных молекулярных структур (РАМР). Поэтому композиции и способы, применимые для модуляции врожденного иммунитета, представляют большой интерес, так как они могут влиять на терапевтические подходы к состояниям, включая аутоиммунитет, воспаление, аллергию, астму, отторжение трансплантата, заболевание трансплантат против хозяина (CvHD), инфекцию, рак и иммунодефицит.
Toll-подобные рецепторы (TLR) представляют собой трансмембранные белки, которые позволяют организмам (в том числе, млекопитающим) обнаруживать микробы и инициировать иммунную реакцию (Beutler B., Nature, 2004, 430: 257-263). Они содержат гомологичные цитоплазматические домены и богатые лейцином внеклеточные домены и типично образуют гомодимеры, которые воспринимают внеклеточные (или интернализованные) сигналы и затем инициируют каскад сигнальной трансдукции через адаптерные молекулы, такие как MyD88 (миелоидный фактор дифференцировки 88). В цитоплазматических доменах такая высокая гомология, что сначала полагали, что подобные пути передачи сигнала существуют во всех TLR (Re F., Strominger J.L., Immunobiology, 2004, 209: 191-198). Действительно, все TLR могут активировать NF-kB и МАР-киназы; однако профили высвобождения цитокинов/хемокинов, полученные при активации TLR, оказываются уникальными для каждого TLR. Кроме того, путь передачи сигналов, который стимулируют TLR, очень схож с путем, который индуцирует цитокиновый рецептор IL-1R. Это может иметь место из-за гомологии, которую разделяют указанные рецепторы, т.е., домены TIR (гомология Toll/IL-1R). Как только TIR домен в TLR активируется и рекрутируется MyD88, происходит активация семейства IRAK серин/треонин-киназ, которая в конечном счете промотирует разрушение Ik-B и активацию NF-kB (Means T.K. et al., Life Sci., 2000, 68: 241-258). Хотя кажется, что такой каскад создается для возможности внеклеточной стимуляции для промотирования внутриклеточных событий, имеются данные, что некоторые TLR мигрируют в эндосомы, где также может инициироваться передача сигнала. Такой процесс может создать возможность для тесного контакта с поглощаемыми микробами и соответствует роли, которую указанные рецепторы играют во врожденной иммунной реакции (Underhill D.M. et al., Nature, 1999, 401: 811-815). Такой процесс также может давать возможность нуклеиновым кислотам хозяина, высвобождаемым поврежденными тканями (например, при воспалительном заболевании) или апоптозом, запускать реакцию через представление эндосом. У млекопитающих имеются 11 TLR, которые координируют такую быструю реакцию. Гипотеза, выдвинутая несколько лет назад (Janeway C.A., Jr., Cold Spring Harb. Syrup. Quant. Biol., 1989, 54: 1-13), что врожденная иммунная реакция инициирует адаптивную иммунную реакцию по образцу активации TLR, вызванной микробами, теперь обоснована. Таким образом, патогенассоциированные молекулярные структуры (РАМР), представленные смешанной группой вызывающих заражение микроорганизмов, приводят к врожденной иммунной реакции с участием некоторых цитокинов, хемокинов и факторов роста с последующей точной адаптивной иммунной реакцией, предназначенной для вызывающего заражение микроорганизма, через представление антигена, что приводит к выработке антител и генерации цитотоксичных Т-клеток.
Грамотрицательный бактериальный липополисахарид (ЛПС) давно признан как адъювант и иммуностимулятор и как фармакологический инструмент для индукции воспалительной реакции у млекопитающих, схожей с септическим шоком. С использованием генетического подхода идентифицирован TLR4 как рецептор ЛПС. Открытие того, что ЛПС является агонистом TLR4, иллюстрирует полезность модуляции TLR для вакцин и лечения болезней у человека (Aderem A., Ulevitch R.J., Nature, 2000, 406: 782-787). Теперь признано, что различные агонисты TLR могут активировать В-клетки, нейтрофилы, тучные клетки, эозинофилы, эндотелиальные клетки и некоторые типы эпителия в дополнение к регуляции пролиферации и апоптоза некоторых типов клеток.
На сегодняшний день TLR7 и TLR8, которые до некоторой степени похожи, характеризуются как рецепторы для одноцепочечной РНК, обнаруженной в эндосомных компартментах, и таким образом, как полагают, являются важными для иммунной реакции на заражение вирусами. Одобренное местное противовирусное/противораковое лекарственное средство имиквимод недавно описано как агонист TLR7, который показывает клиническую эффективность при некоторых кожных расстройствах (Miller R.L. et al., Int. J. Immunopharm., 1999, 21: 1-14). Это низкомолекулярное лекарственное средство описано как структурный миметик ссРНК. TLR8 впервые описан в 2000 (Du X. et al., European Cytokine Network, 2000 (Sept.), 11(3): 362-371), и вскоре ему было приписано участие во врожденной иммунной реакции на заражение вирусом (Miettinen M. et al., Genes and Immunity, 2001 (Oct.), 2(6): 349-355).
Недавно появилось сообщение, что некоторые имидазохинолиновые производные, обладающие противовирусной активностью, представляют собой лиганды TLR7 и TLR8 (Hemmi H. et al. (2002), Nat. Immunol., 3: 196-200; Jurk M. et al. (2002), Nat. Immunol., 3: 499). Имидазохинолины являются сильными синтетическими активаторами иммуноцитов с противовирусными и противоопухолевыми свойствами. На основании использования макрофагов от мышей дикого типа и с дефицитом MyD88 Hemmi H. et al. недавно сообщили, что два имидазохинолина имиквимод и резиквимод (R848) индуцируют фактор некроза опухоли (TNF) и интерлейкин-12 (IL-12) и активируют NF-icB только в клетках дикого типа, сообразно с активацией через TLR (Hemmi H. et al. (2002), Nat. Immunol., 3: 196-200). Макрофаги от мышей с дефицитом по TLR7, но не по другим TLR, не вырабатывают детектируемые цитокины в ответ на указанные имидазохинолины. Кроме того, имидазохинолины индуцируют зависимую от дозы пролиферацию селезеночных В-клеток и активацию внутриклеточных каскадов передачи сигналов в клетках мышей дикого типа, но не TLR7-/-. Анализ на люциферазу показывает, что экспрессия человеческого TLR7, но не TLR2 или TLR4, в человеческих эмбриональных клетках почки приводит к активации NF-KB в ответ на резиквимод. Таким образом, данные, полученные Hemmi H. et al., предполагают, что указанные производные имидазохинолина являются неприродными лигандами TLR7, которые могут индуцировать передачу сигнала через TLR7. Недавно появилось сообщение, что R848 также является лигандом TLR8 (Jurk M. et al. (2002), Nat. Immunol., 3: 499).
С учетом большого терапевтического потенциала соединений, которые модулируют Toll-подобные рецепторы, и несмотря на работу, которая уже сделана, существует значительная постоянная потребность в расширении их применения и терапевтического благоприятного действия.
Сущность изобретения
Композиции, описанные в данном описании, применимы для модуляции иммунных реакций in vitro и in vivo. Такие композиции найдут использование в ряде клинических применений, таких как способы лечения или предупреждения состояний с участием нежелательной иммунной активности, включая воспалительные и аутоиммунные расстройства.
Конкретно, изобретение относится к соединению формулы I
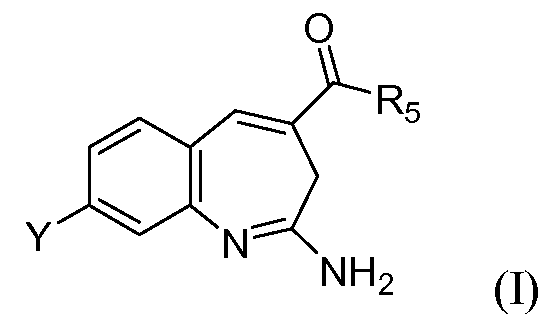
или его таутомеру, энантиомеру или соли, где в указанной формуле
Y представляет собой -(O)x(CH2)yR11;
х выбирают из 0 и 1;
у выбирают из 0, 1, 2 и 3;
R11 выбирают из арила, гетероарила и насыщенного или частично насыщенного гетероцикла, при этом когда х равен 0, указанный арил или гетероарил замещен -C(O)NR1R2 или Т;
R1 и R2 выбирают независимо из водорода и алкила, при этом указанный алкил необязательно замещен -C(O)О(CH2)tR12, или R1 и R2 вместе с атомом азота, к которому они присоединены, образуют насыщенный гетероцикл;
t выбирают из 0, 1, 2 и 3;
R12 выбирают из циклоалкила и арила;
Т выбирают из гетроцикла, -(CHR7)zOR9, -(O)u(CH2)sC(O)R8, -OSO2R13 и -СН(ОН)СН2ОН;
R7 выбирают из Н или -ОН;
R8 выбирают из -OR10 и алкила;
R9 выбирают из алкила и Н;
R10 выбирают из алкила, -(CH2)R12 и водорода, при этом указанный алкил необязательно замещен галогеном, амином, алкиламином или диалкиламином;
R13 выбирают из -ОН, алкила, CF3, циклоалкила, гетероцикла, арила и гетероарила;
u выбирают из 0 и 1;
z выбирают из 1, 2 и 3;
s выбирают из 1 и 2;
R5 выбирают из -NR3R4 и -OR10;
R3 и R4 выбирают независимо из Н, алкила, -(O)q(CH2)rP; при этом указанный алкил необязательно замещен одним или несколькими -ОН;
q выбирают из 0 и 1;
r выбирают из 0, 1, 2 и 3;
Р выбирают из арила, -SO2R6, -C(O)NH2 и гетероцикла; и
R6 выбирают из -NH2, -NH(алкила), -N(алкил)2,
при условии, что когда R11 представляет собой арил или гетероарил, тогда
а) x+y≥1;
или
b) R11 замещен Т;
или
с) R5 представляет собой NR3R4, и, по меньшей мере, один из R3 или R4 представляет собой [(O)q(CH2)rP], и q+r≥1;
или
d) по меньшей мере, один из R1 или R2 представляет собой алкил, замещенный -C(O)О(CH2)R12.
Изобретение также относится к соединению формулы II
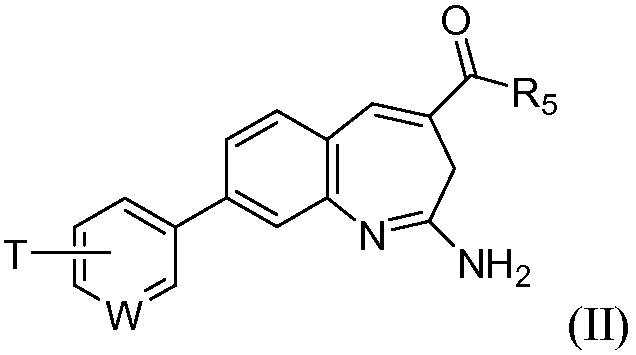
или его таутомеру, энантиомеру или соли, где в указанной формуле
W выбирают из N, C-T и СН; и Т и R5 имеют значения, указанные для формулы I.
Изобретение также относится к соединению формулы III
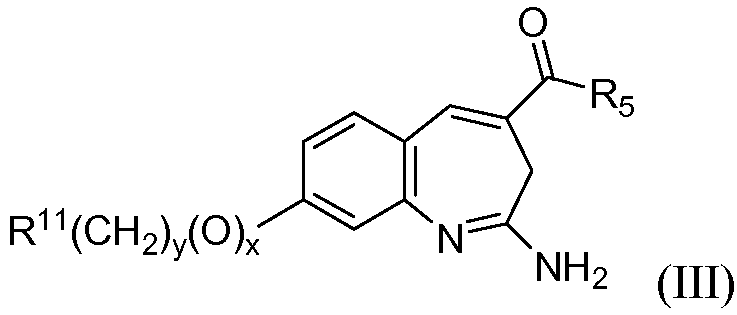
или его таутомеру, энантиомеру или соли, где в указанной формуле х, у, R11 и R5 имеют значения, указанные для формулы I;
при условии, что когда R11 представляет собой арил или гетероарил, тогда x+y≥1.
Изобретение также относится к соединению формулы IV
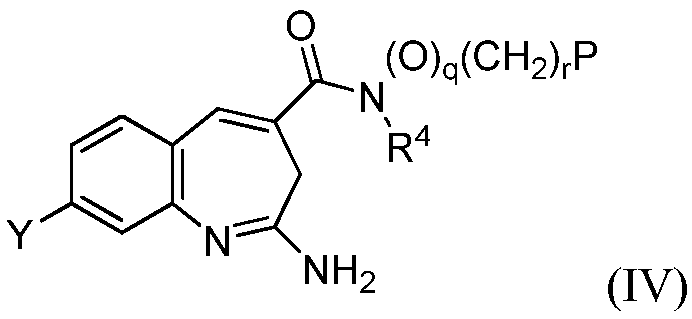
или его таутомеру, энантиомеру или соли, где в указанной формуле Y, R4, q, r и Р имеют значения, указанные для формулы I, и q+r≥1.
Изобретение также относится к соединению формулы V
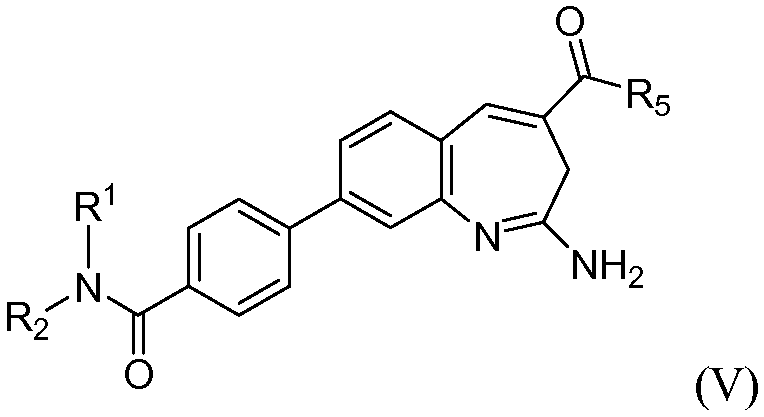
или его таутомеру, энантиомеру или соли, где в указанной формуле R1, R2 и R5 имеют значения, указанные для формулы I; и, по меньшей мере, один из R1 или R2 представляет собой алкил, замещенный -C(O)О(CH2)R12.
Изобретение также относится к соединению формулы VI
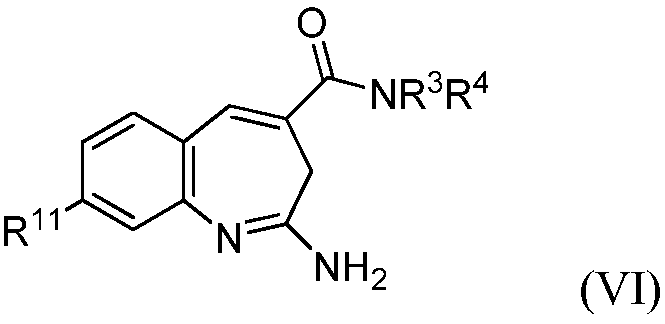
или его таутомеру, энантиомеру или соли, где в указанной формуле
R11 выбирают из арила и насыщенного или частично насыщенного гетероцикла, при этом указанный арил замещен Т;
Т выбирают из гетроцикла, -(O)u(CH2)sC(O)R8 и -СН(ОН)СН2ОН;
R8 выбирают из -OR10 и алкила;
R10 выбирают из алкила, -(CH2)R12 и водорода, при этом указанный алкил необязательно замещен галогеном, амином, алкиламином или диалкиламином;
R12 выбирают из циклоалкила и арила;
u выбирают из 0 и 1;
s выбирают из 1 и 2; и
R3 и R4 представляют собой независимо алкилы; при этом указанные алкилы необязательно замещены одним или несколькими -ОН.
Соединения по изобретению можно использовать в комбинации с другими известными терапевтическими средствами. Соответственно, данное изобретение также относится к фармацевтическим композициям, включающим терапевтически эффективное количество соединения по изобретению или его соли в комбинации со вторым терапевтическим средством.
Данное изобретение также относится к способам TLR7- и/или TLR8-опосредуемой модуляции передачи сигнала, включающим приведение в контакт клетки, экспрессирующей TLR7 и/или TLR8, с эффективным количеством соединения по изобретению или его соли. В одном аспекте способ ингибирует TLR7- и/или TLR8-опосредуемую передачу сигнала иммуностимуляции.
Данное изобретение также относится к способам модуляции TLR7- и/или TLR8-опосредуемой иммуностимуляции у субъекта, включающим введение пациенту, имеющему или находящемуся в опасности развития TLR7- и/или TLR8-опосредуемой иммуностимуляции, соединения по изобретению или его соли в количестве, эффективном для ингибирования TLR7- и/или TLR8-опосредуемой иммуностимуляции у субъекта.
Данное изобретение также относится к способам модуляции TLR7- и/или TLR8-опосредуемой иммуностимуляции у субъекта, включающим введение пациенту, имеющему или находящемуся в опасности развития TLR7- и/или TLR8-опосредуемой иммуностимуляции, соединения по изобретению или его соли в количестве, эффективном для промотирования TLR7- и/или TLR8-опосредуемой иммуностимуляции у субъекта.
Данное изобретение также относится к способам лечения или предупреждения заболевания или состояния путем модуляции TLR7- и/или TLR8-опосредуемых клеточных активностей, включающим введение теплокровному животному, такому как млекопитающее, например, человеку, имеющему или находящемуся в опасности развития указанного заболевания или состояния, соединения по изобретению или его соли.
Данное изобретение также относится к способам модуляции иммунной системы млекопитающего, включающим введение млекопитающему соединения по изобретению или его соли в количестве, эффективном для модуляции указанной иммунной системы.
Изобретение также относится к соединению или его соли для применения в качестве лекарственного средства при лечении заболеваний или состояний, описанных в данном описании (например, рака, аутоиммунного заболевания, инфекционного заболевания, воспалительного расстройства, отторжения трансплантата и болезни трансплантат против хозяина), у млекопитающего, например, человека, страдающего от такого заболевания или состояния. Изобретение также относится к применению соединения по изобретению или его соли при получении лекарственного средства для лечения заболеваний и состояний, описанных в данном описании (например, рака, аутоиммунного заболевания, инфекционного заболевания, воспалительного расстройства, отторжения трансплантата и болезни трансплантат против хозяина), у млекопитающего, например, человека, страдающего от такого заболевания или состояния.
Изобретение также относится к соединению или его соли для применения в качестве лекарственного средства при предупреждении заболеваний или состояний, описанных в данном описании (например, рака, аутоиммунного заболевания, инфекционного заболевания, воспалительного расстройства, отторжения трансплантата и болезни трансплантат против хозяина), у млекопитающего, например, человека, подверженного или предрасположенного к заболеванию или состоянию, но пока не испытывающего или не отображающего симптомы такого заболевания или состояния. Изобретение также относится к применению соединения по изобретению или его соли при получении лекарственного средства для лечения заболеваний и состояний, описанных в данном описании (например, рака, аутоиммунного заболевания, инфекционного заболевания, воспалительного расстройства, отторжения трансплантата и болезни трансплантат против хозяина), у млекопитающего, например, человека, страдающего от такого заболевания или состояния.
Заболевание или состояние выбирают из, например, рака, аутоиммунного заболевания, инфекционного заболевания, воспалительного расстройства, отторжения трансплантата и болезни трансплантат против хозяина.
Данное изобретение также относится к наборам, включающим одно или несколько соединений по изобретению или их солей. Набор также может включать второе соединение или препарат, включающий второе фармацевтическое средство.
Дополнительные преимущества и новые особенности данного изобретения будут частично представлены в описании, которое следует далее, и частично станут очевидными для специалистов в данной области техники после изучения последующего описания или могут быть выяснены путем практического осуществления изобретения. Преимущества изобретения могут быть реализованы и достигнуты посредством средств, комбинаций, композиций и способов, в особенности, указанных в прилагаемой формуле изобретения.
Подробное описание изобретения
В некоторых аспектах изобретение относится к композициям и способам, применимым для модуляции TLR7- и/или TLR8-опосредуемой передачи сигнала. Конкретнее, в одном аспекте данное изобретение относится к соединению формулы I
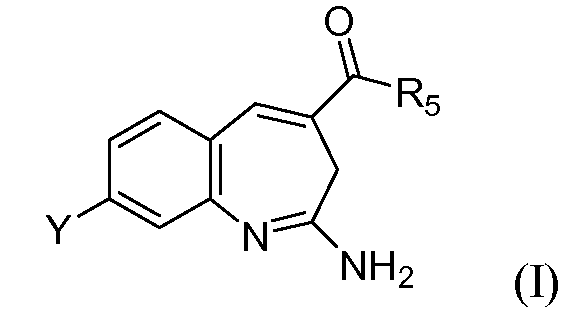
или его таутомеру, энантиомеру или соли, где в указанной формуле
Y представляет собой -(O)x(CH2)yR11;
х выбирают из 0 и 1;
у выбирают из 0, 1, 2 и 3;
R11 выбирают из арила, гетероарила и насыщенного или частично насыщенного гетероцикла, при этом когда х равен 0, указанный арил или гетероарил замещен -C(O)NR1R2 или Т;
R1 и R2 выбирают независимо из водорода и алкила, при этом указанный алкил необязательно замещен -C(O)О(CH2)tR12, или R1 и R2 вместе с атомом азота, к которому они присоединены, образуют насыщенный гетероцикл;
t выбирают из 0, 1, 2 и 3;
R12 выбирают из циклоалкила и арила;
Т выбирают из гетроцикла, -(CHR7)zOR9, -(O)u(CH2)sC(O)R8, -OSO2R13 и -СН(ОН)СН2ОН;
R7 выбирают из Н или -ОН;
R8 выбирают из -OR10 и алкила;
R9 выбирают из алкила и Н;
R10 выбирают из алкила, -(CH2)R12 и водорода, при этом указанный алкил необязательно замещен галогеном, амином, алкиламином или диалкиламином;
R13 выбирают из -ОН, алкила, CF3, циклоалкила, гетероцикла, арила и гетероарила;
u выбирают из 0 и 1;
z выбирают из 1, 2 и 3;
s выбирают из 1 и 2;
R5 выбирают из -NR3R4 и -OR10;
R3 и R4 выбирают независимо из Н, алкила, -(O)q(CH2)rP; при этом указанный алкил необязательно замещен одним или несколькими -ОН;
q выбирают из 0 и 1;
r выбирают из 0, 1, 2 и 3;
Р выбирают из арила, -SO2R6, -C(O)NH2 и гетероцикла; и
R6 выбирают из -NH2, -NH(алкила), -N(алкил)2,
при условии, что когда R11 представляет собой арил или гетероарил, тогда
а) x+y≥1;
или
b) R11 замещен Т;
или
с) R5 представляет собой NR3R4, и, по меньшей мере, один из R3 или R4 представляет собой [(O)q(CH2)rP], и q+r≥1;
или
d) по меньшей мере, один из R1 или R2 представляет собой алкил, замещенный -C(O)О(CH2)R12.
В одном воплощении изобретение относится к соединению формулы I, где R11 представляет собой арил или гетероарил, и x+y≥1.
В другом воплощении изобретение относится к соединению формулы I, где R11 представляет собой арил или гетероарил, и R11 замещен Т.
В другом воплощении изобретение относится к соединению формулы I, где R11 представляет собой арил или гетероарил, и R5 представляет собой NR3R4, и, по меньшей мере, один из R3 или R4 представляет собой [(O)q(CH2)rP], и q+r≥1.
В другом воплощении изобретение относится к соединению формулы I, где R11 представляет собой арил или гетероарил, и, по меньшей мере, один из R1 или R2 представляет собой алкил, замещенный -C(O)О(CH2)R12.
В другом воплощении изобретение относится к соединению формулы I, где R11 представляет собой арил или гетероарил, R11 замещен Т, Т представляет собой -(O)u(CH2)sC(O)R8, и u+s≥2.
В другом воплощении изобретение относится к соединению формулы I, где R11 представляет собой арил или гетероарил, R11 замещен Т, и Т выбирают из гетероцикла, -(CHR7)zOR9, -(O)u(CH2)sC(O)R8 и -OSO2R13.
В другом воплощении изобретение относится к соединению формулы I, где R11 выбирают из (С6-С10)-арила, гетероарила, включающего 1-4 гетероатома, выбранных из атомов N, O и S, и насыщенного или частично насыщенного гетероцикла, включающего 1-4 гетероатома, выбранных из атомов N, O и S, при этом когда х равен 0, указанный (С6-С10)-арил или гетероарил, включающий 1-4 гетероатома, выбранных из атомов N, O и S, замещен -C(O)NR1R2 или Т.
Один аспект изобретения относится к соединению формулы II
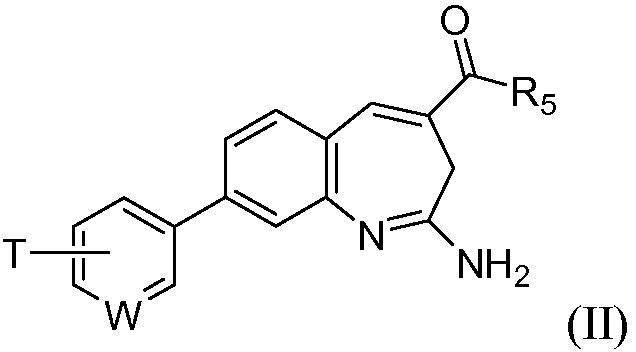
или его таутомеру, энантиомеру или соли, где в указанной формуле
W выбирают из N, C-T и СН; и Т и R5 имеют значения, указанные выше для формулы I.
В одном воплощении изобретение относится к соединению формулы IIa
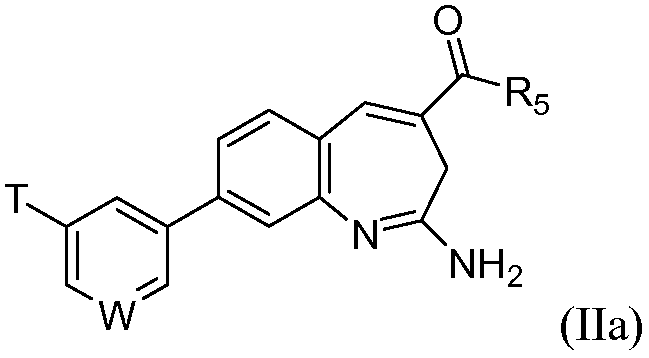
или его таутомеру, энантиомеру или соли. В другом воплощении изобретение относится к соединению формулы IIb
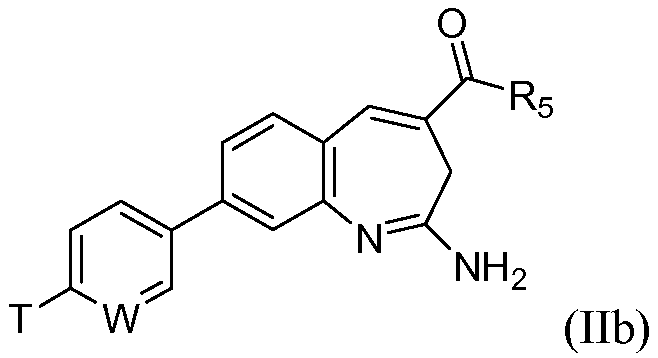
или его таутомеру, энантиомеру или соли, при этом
W выбирают из N, C-T и СН; и Т и R5 имеют значения, указанные выше для формулы I.
В одном воплощении изобретение относится к соединению формулы II, IIa или IIb или его соли, при этом W представляет собой СН.
В другом воплощении изобретение относится к соединению формулы I, II, IIa или IIb или его соли, при этом Т представляет собой -(O)u(CH2)sC(O)R8. В одном воплощении изобретение относится к соединению или его соли, при этом u равен 1, и s равен 1. В одном воплощении изобретение относится к соединению или его соли, при этом u равен 0, и s равен 1. В одном воплощении изобретение относится к соединению или его соли, при этом u равен 0, и s равен 2. В одном воплощении изобретение относится к соединению или его соли, при этом R8 представляет собой -О-алкил. В одном воплощении изобретение относится к соединению или его соли, при этом R8 представляет собой -О-алкил, и алкил выбирают из метила, этила, изопропила и изобутила.
В другом воплощении изобретение относится к соединению формулы I, II, IIa или IIb или его соли, при этом Т представляет собой гетероцикл. В одном воплощении изобретение относится к соединению или его соли, при этом гетероцикл выбирают из дигидрофуранона и диоксоланона. В одном воплощении изобретение относится к соединению или его соли, при этом гетероцикл выбирают из 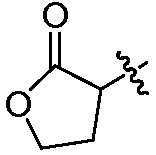 и
и 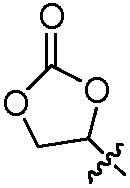 .
.
В другом воплощении изобретение относится к соединению формулы I, II, IIa или IIb или его соли, при этом Т представляет собой -(CHR7)zOR9.
В другом воплощении изобретение относится к соединению формулы I, II, IIa или IIb или его соли, при этом Т представляет собой -OSO2R13. В одном воплощении изобретение относится к соединению или его соли, при этом R13 представляет собой -CF3.
В другом воплощении изобретение относится к соединению формулы II, IIa или IIb или его соли, при этом W представляет собой N. В одном воплощении изобретение относится к соединению или его соли, при этом Т представляет собой -(CHR7)zOR9. В одном воплощении изобретение относится к соединению или его соли, при этом z равен 1, и R7 и R9, оба, представляют собой атомы водорода. В одном воплощении изобретение относится к соединению или его соли, при этом z равен 2, и R7 представляет собой ОН или Н, и R9 представляет собой Н.
Другой аспект изобретения относится к соединению формулы III
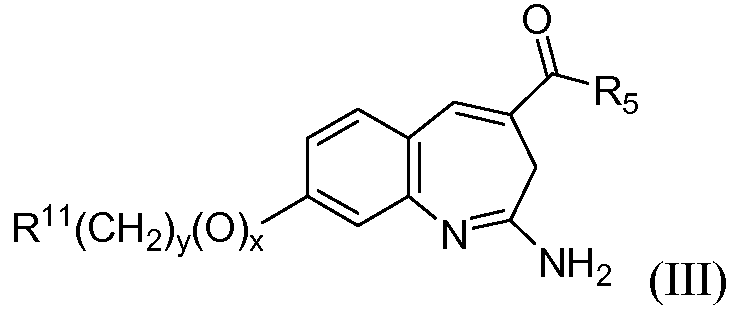
или его таутомеру, энантиомеру или соли, где в указанной формуле R5, R11, х и у имеют значения, указанные выше для формулы I; при условии, что когда R11 представляет собой арил или гетероарил, тогда x+y≥1.
В одном воплощении изобретение относится к соединению формулы IIIa
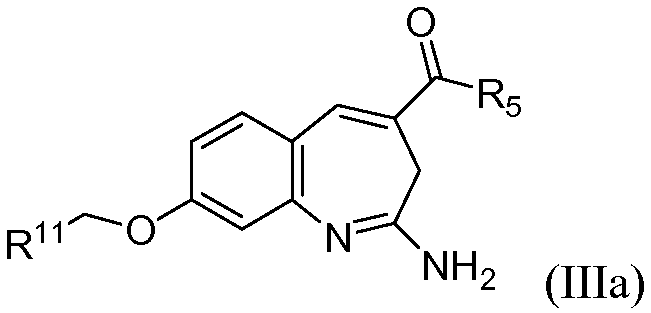
или его таутомеру, энантиомеру или соли.
В одном воплощении изобретение относится к соединению формулы IIIb
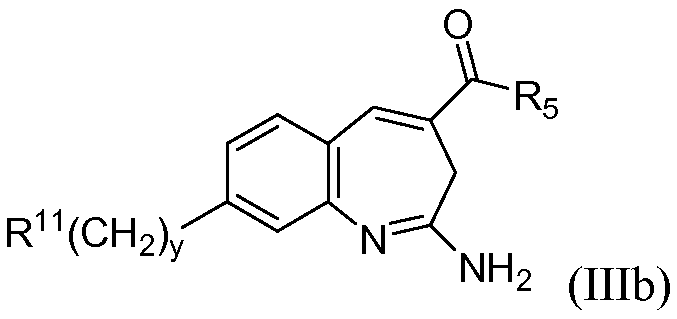
или его таутомеру, энантиомеру или соли, при этом у равен 1, 2 или 3.
В одном воплощении изобретение относится к соединению формулы IIIc
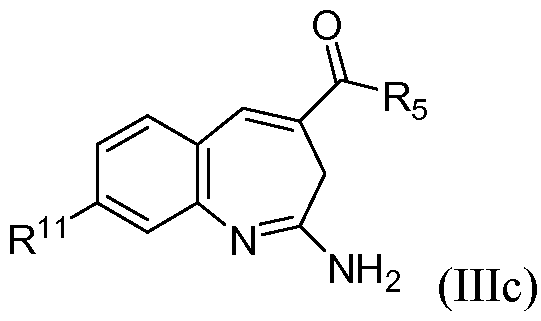
или его таутомеру, энантиомеру или соли, при этом R11 представляет собой насыщенный или частично насыщенный гетероцикл.
В одном воплощении изобретение относится к соединению формулы I, III, IIIa или IIIb или его соли, при этом х=0, и у=3.
В одном воплощении изобретение относится к соединению формулы I, III, IIIa или IIIb или его соли, при этом R11 представляет собой фенил.
В одном воплощении изобретение относится к соединению формулы I, III, IIIa или IIIb или его соли, при этом R11 представляет собой гетероцикл. В одном воплощении изобретение относится к соединению или его соли, при этом указанный гетероцикл частично замещен насыщенным гетероциклом. В одном воплощении изобретение относится к соединению или его соли, при этом указанный гетероцикл представляет собой морфолин. В одном воплощении изобретение относится к соединению или его соли, при этом указанный гетероцикл представляет собой изобензофуранон. В одном воплощении изобретение относится к соединению или его соли, при этом указанный изобензофуранон выбирают из 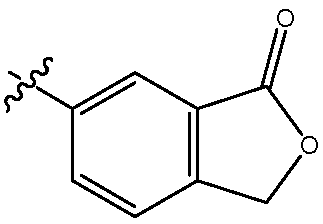 и
и 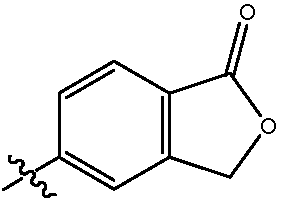 .
.
Другой аспект изобретения относится к соединению формулы I, II, IIa, IIb, III, IIIa или IIIb или его соли, при этом R5 представляет собой -OR10. В одном воплощении изобретение относится к соединению или его соли, при этом R10 представляет собой алкил. В одном воплощении изобретение относится к соединению или его соли, при этом R10 представляет собой этил.
Другой аспект изобретения относится к соединению формулы I, II, IIa, IIb, III, IIIa или IIIb или его соли, при этом R5 представляет собой -NR3R4. В одном воплощении изобретение относится к соединению или его соли, при этом R3 и R4, оба, представляют собой алкилы. В одном воплощении изобретение относится к соединению или его соли, при этом R3 и R4, оба, представляют собой пропил. В одном воплощении изобретение относится к соединению или его соли, при этом, по меньшей мере, один из R3 и R4 представляет собой алкил, замещенный одним -ОН. В одном воплощении изобретение относится к соединению или его соли, при этом, по меньшей мере, один из R3 и R4 представляет собой 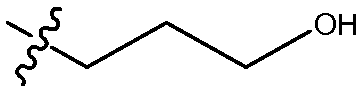 . В другом воплощении изобретение относится к соединению или его соли, при этом один из R3 и R4 представляет собой
. В другом воплощении изобретение относится к соединению или его соли, при этом один из R3 и R4 представляет собой 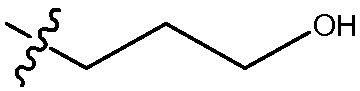 , и оставшийся R3 или R4 представляет собой пропил.
, и оставшийся R3 или R4 представляет собой пропил.
Другой аспект изобретения относится к соединению формулы IV
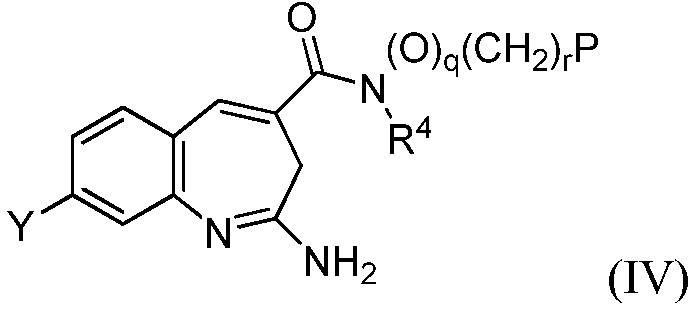
или его таутомеру, энантиомеру или соли, где в указанной формуле Y, R4, Р, q, r и имеют значения, указанные выше для формулы I, и q+r≥1.
В одном воплощении изобретение относится к соединению формулы IVa
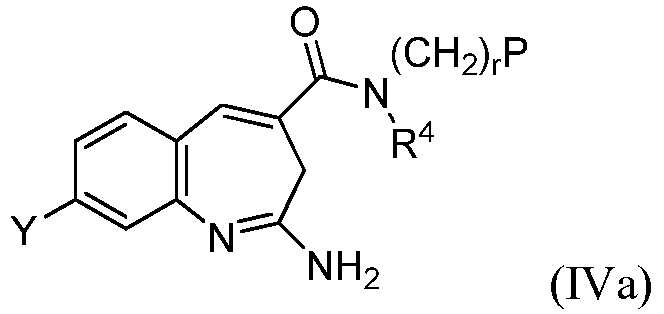
или его таутомеру, энантиомеру или соли, при этом r выбирают из 1, 2 или 3.
В одном воплощении изобретение относится к соединению формулы IVb
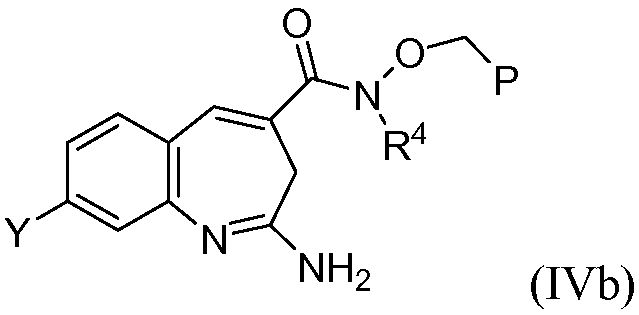
или его таутомеру, энантиомеру или соли.
В одном воплощении изобретение относится к соединению формулы I или IV или его соли, при этом t=3.
В одном воплощении изобретение относится к соединению формулы I, IV, IVa или IVb или его соли, при этом Р выбирают из арила, гетероцикла и -SO2R6.
В одном воплощении изобретение относится к соединению формулы I, IV, IVa или IVb или его соли, при этом Р представляет собой гетероцикл. В одном воплощении изобретение относится к соединению или его соли, при этом Р выбирают из пиперидина и пирролидина.
В одном воплощении изобретение относится к соединению формулы I, IV, IVa или IVb или его соли, при этом Р представляет собой арил.
В одном воплощении изобретение относится к соединению формулы I, IV, IVa или IVb или его соли, при этом Р представляет собой -SO2R6. В одном воплощении изобретение относится к соединению или его соли, при этом указанный R6 представляет собой -NH2.
В одном воплощении изобретение относится к соединению формулы I, IV, IVa или IVb или его соли, при этом х=0, и у=0. В одном воплощении изобретение относится к соединению или его соли, при этом R11 представляет собой арил. В одном воплощении изобретение относится к соединению или его соли, при этом указанный арил замещен -C(O)NR1R2. В одном воплощении изобретение относится к соединению или его соли, при этом указанные R1 и R2 вместе с атомом азота, к которому они присоединены, образуют насыщенный гетероцикл. В одном воплощении изобретение относится к соединению или его соли, при этом указанный насыщенный гетероцикл представляет собой пирролидиновый цикл.
Другой аспект изобретения относится к соединению формулы V
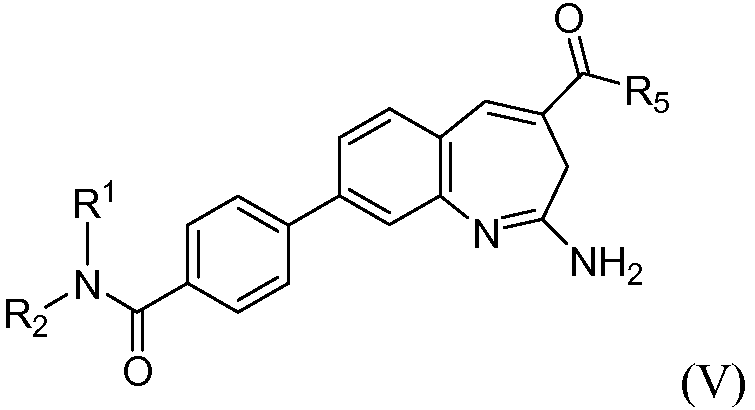
или его таутомеру, энантиомеру или соли, где в указанной формуле R1, R2 и R5 имеют значения, указанные выше для формулы I; и при этом также, по меньшей мере, один из R1 или R2 представляет собой алкил, замещенный -C(O)О(CH2)tR12.
В одном воплощении изобретение относится к соединению формулы V или его соли, при этом, по меньшей мере, один из R1 или R2 представляет собой алкил, замещенный -C(O)О(CH2)tR12. В одном воплощении изобретение относится к соединению или его соли, при этом указанный R12 представляет собой арил. В одном воплощении изобретение относится к соединению или его соли, при этом R1 или R2 представляет собой Н.
В другом воплощении изобретение относится к соединению формулы VI
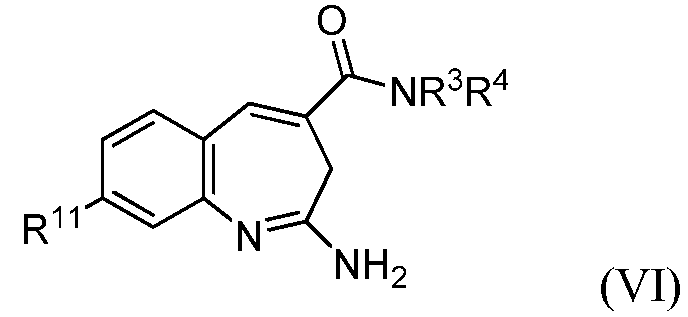
или его таутомеру, энантиомеру или соли, где в указанной формуле
R11 выбирают из арила и насыщенного или частично насыщенного гетероцикла, при этом указанный арил замещен Т;
Т выбирают из гетроцикла, -(O)u(CH2)sC(O)R8 и -СН(ОН)СН2ОН;
R8 выбирают из -OR10 и алкила;
R10 выбирают из алкила, -(CH2)R12 и водорода, при этом указанный алкил необязательно замещен галогеном, амином, алкиламином или диалкиламином;
R12 выбирают из циклоалкила и арила;
u выбирают из 0 и 1;
s выбирают из 1 и 2; и
R3 и R4 представляют собой независимо алкилы; при этом указанные алкилы необязательно замещены одним или несколькими -ОН.
В одном воплощении изобретение относится к соединению формулы VI или его соли, при этом R11 представляет собой арил, замещенный -(O)u(CH2)sC(O)R8. В другом воплощении указанный R8 представляет собой -OR10.
В другом воплощении изобретение относится к соединению формулы VI, при этом
R11 выбирают из арила и насыщенного или частично насыщенного гетероцикла, при этом указанный арил замещен Т;
Т выбирают из гетероцикла и -(O)u(CH2)sC(O)R8;
R8 выбирают из -OR10 и алкила;
R10 выбирают из алкила, -(CH2)R12 и водорода, при этом указанный алкил необязательно замещен галогеном, амином, алкиламином или диалкиламином;
R12 выбирают из циклоалкила и арила;
u выбирают из 0 и 1;
s выбирают из 1 и 2; и
R3 и R4 представляют собой независимо алкилы; при этом указанные алкилы необязательно замещены одним или несколькими -ОН.
В другом воплощении изобретение относится к соединению формулы VI, при этом
R11 представляет собой арил, замещенный Т;
Т выбирают из -(O)u(CH2)sC(O)R8 и -СН(ОН)СН2ОН;
R8 выбирают из -OR10 и алкила;
R10 выбирают из алкила, -(CH2)R12 и водорода, при этом указанный алкил необязательно замещен галогеном, амином, алкиламином или диалкиламином;
R12 выбирают из циклоалкила и арила;
u выбирают из 0 и 1;
s выбирают из 1 и 2; и
R3 и R4 представляют собой независимо алкилы; при этом указанные алкилы необязательно замещены одним или несколькими -ОН.
В другом воплощении изобретение относится к соединению формулы VI, при этом R3 и R4 представляют собой независимо алкилы; и также при этом один из R3 и R4 замещен одним или несколькими -ОН, в то время как другой является незамещенным. В другом воплощении изобретение относится к соединению формулы VI, при этом R3 и R4 представляют собой независимо незамещенные алкилы. В другом воплощении изобретение относится к соединению формулы VI, при этом R11 представляет собой арил. В другом воплощении изобретение относится к соединению формулы VI, при этом R11 представляет собой фенил. В другом воплощении изобретение относится к соединению формулы VI, при этом R11 представляет собой насыщенный или частично насыщенный гетероцикл.
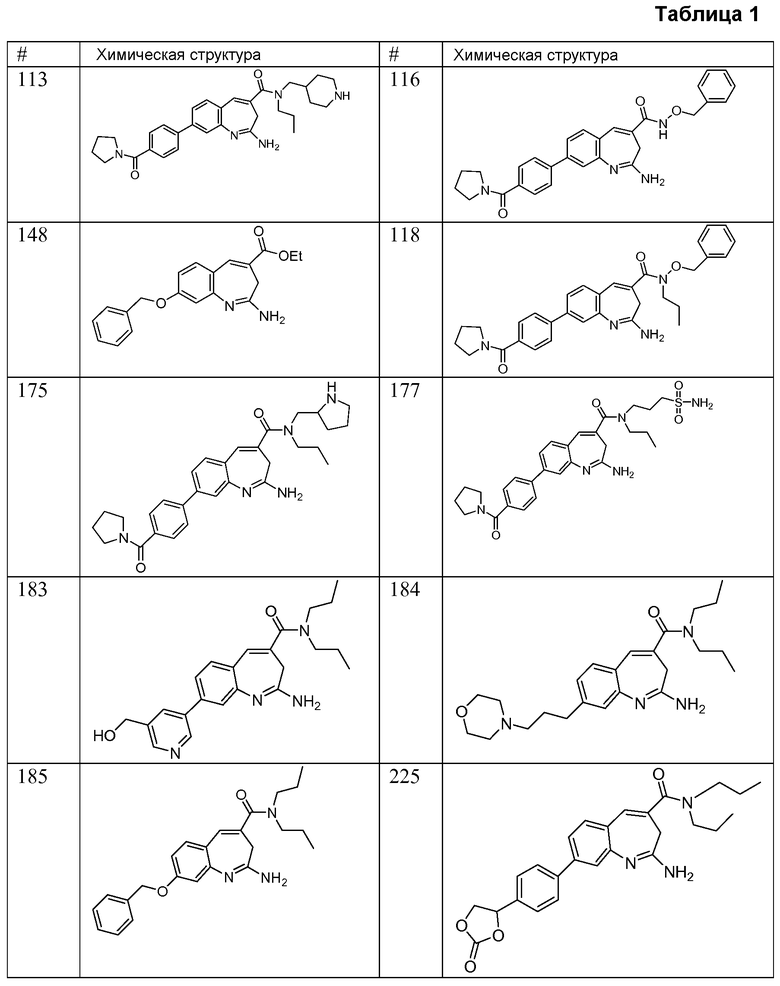
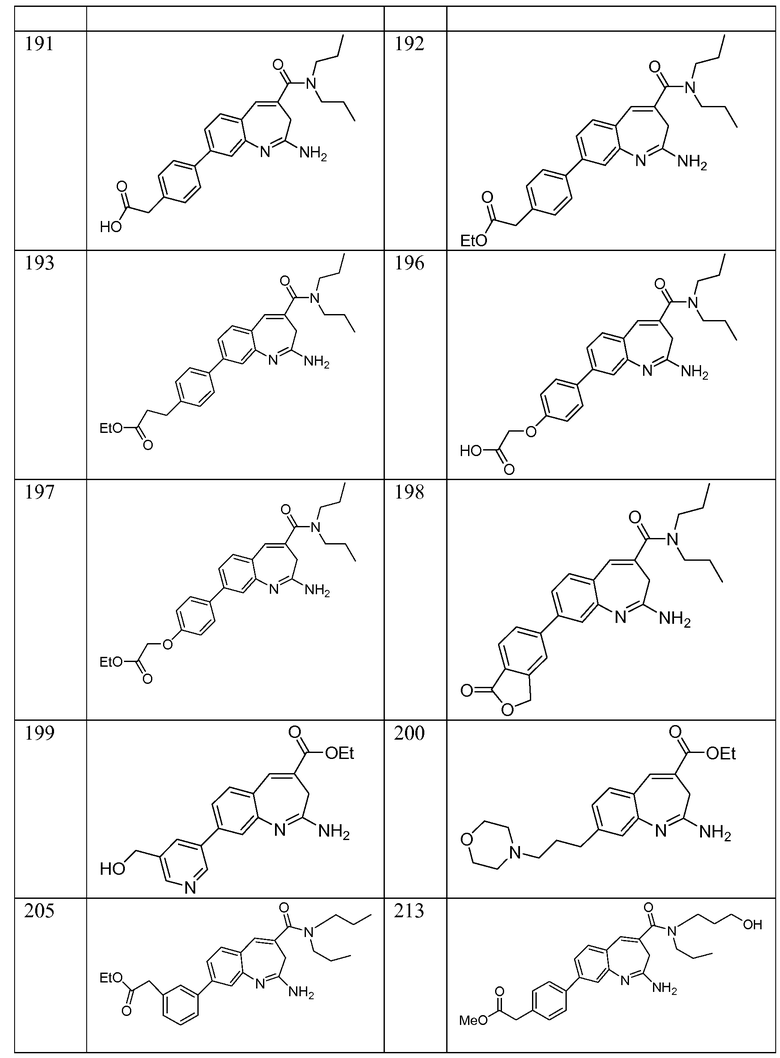
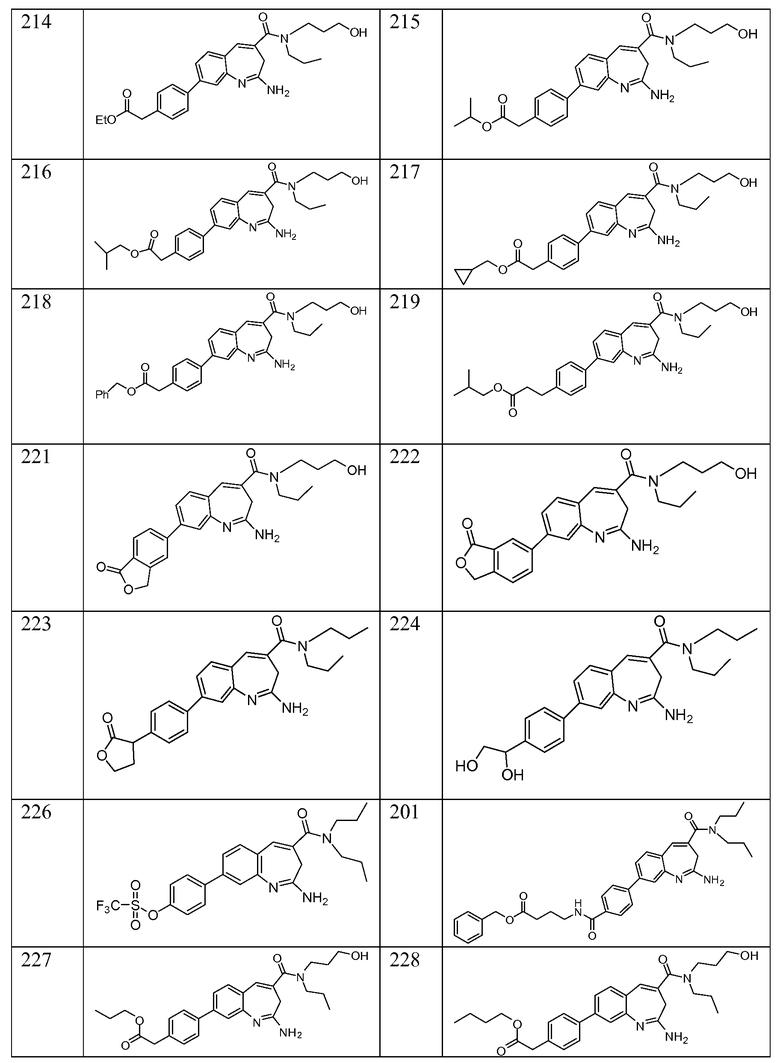
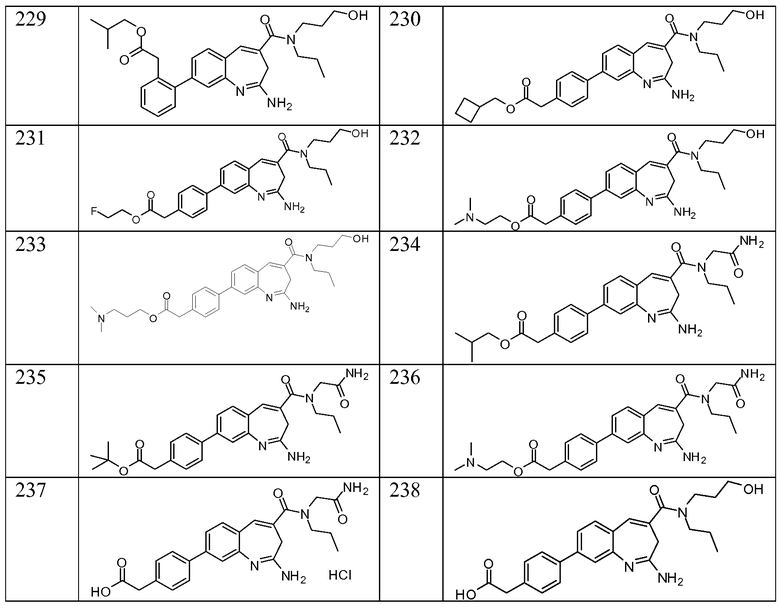
Другой аспект изобретения относится к соединению или его соли, выбранному из соединений в таблице 1. В одном воплощении изобретения относится к соединению, выбранному из соединений 196, 197, 183, 191, 192, 193, 199, 205, 213, 214, 215, 216, 217, 218, 219, 223, 224, 226 и 225, или его соли. В одном воплощении изобретение относится к соединению, выбранному из соединений 113, 175, 116, 118 и 177, или его соли. В одном воплощении изобретение относится к соединению, выбранному из соединений 148, 185, 184, 200, 198, 221 и 222, или его соли. В одном воплощении изобретение относится к соединению, выбранному из соединений 198, 225, 191, 192, 193, 196, 197, 221, 205, 213, 214, 215, 216, 217, 218, 219, 222, 223 и 224, или его соли. В одном воплощении изобретение относится к соединению, выбранному из соединений 198, 225, 191, 192, 193, 197, 221, 205, 213, 214, 215, 216, 217, 218, 219, 222 и 223, или его соли. В одном воплощении изобретение относится к соединению, выбранному из соединений 191, 196 и 224, или его соли.
В одном воплощении изобретение относится к соединению формулы I. В одном воплощении изобретение относится к соединению формулы II. В одном воплощении изобретение относится к соединению формулы IIa. В одном воплощении изобретение относится к соединению формулы IIb. В одном воплощении изобретение относится к соединению формулы III. В одном воплощении изобретение относится к соединению формулы IIIa. В одном воплощении изобретение относится к соединению формулы IIIb. В одном воплощении изобретение относится к соединению формулы IIIc. В одном воплощении изобретение относится к соединению формулы IV. В одном воплощении изобретение относится к соединению формулы IVa. В одном воплощении изобретение относится к соединению формулы IVb. В одном воплощении изобретение относится к соединению формулы V. В одном воплощении изобретение относится к соединению формулы VI.
Другой аспект изобретения относится к соединению по изобретению или его соли, при этом соль представляет собой фармацевтически приемлемую соль.
Другой аспект изобретения относится к набору для лечения TLR7- и/или TLR8-опосредуемого состояния, включающему
(а) первую фармацевтическую композицию, включающую соединение по изобретению или его соль; и (b) необязательно, инструкции по применению.
В одном воплощении набор также включает (с) вторую фармацевтическую композицию, при этом вторая фармацевтическая композиция включает второе соединение для лечения TLR7- и/или TLR8-опосредуемого состояния.
В одном воплощении набор также включает инструкции по одновременному, последовательному или раздельному введению указанных первой и второй фармацевтических композиций пациенту, нуждающемуся в этом.
Другой аспект изобретения относится к фармацевтической композиции, которая включает соединение по изобретению или его соль вместе с фармацевтически приемлемым разбавителем или носителем.
Другой аспект изобретения относится к соединению по изобретению или его соли для применения в качестве лекарственного средства для лечения TLR7- и/или TLR8-опосредуемого состояния у человека или животного. В одном воплощении изобретение относится к применению соединения по изобретению или его соли при получении лекарственного средства для лечения состояния анормального клеточного роста у человека или животного.
Другой аспект изобретения относится к способу лечения TLR7- и/или TLR8-опосредуемого состояния, включающему введение пациенту, нуждающемуся в этом, эффективного количества соединения по изобретению или его соли.
Другой аспект изобретения относится к способу модуляции иммунной системы пациента, включающему введение пациенту, нуждающемуся в этом, эффективного количества соединения по изобретению или его соли.
Изобретение включает соединение, выбранное из соединений, перечисленных в таблице 1.
В одном аспекте изобретение включает соединение или его соль с величиной МС50≤25000 нМ для TLR8. В другом аспекте изобретение включает соединение или его соль с величиной МС50≤10000 нМ для TLR8. В другом аспекте изобретение включает соединение или его соль с величиной МС50≤1000 нМ для TLR8. В другом аспекте изобретение включает соединение или его соль с величиной МС50≤100 нМ для TLR8. В другом аспекте изобретение включает соединение или его соль с величиной МС50≤25 нМ для TLR8.
В одном аспекте изобретение включает соединение или его соль с величиной МС50≤25000 нМ для TLR7. В другом аспекте изобретение включает соединение или его соль с величиной МС50≤10000 нМ для TLR7. В другом аспекте изобретение включает соединение или его соль с величиной МС50≤1000 нМ для TLR7. В другом аспекте изобретение включает соединение или его соль с величиной МС50≤100 нМ для TLR7. В другом аспекте изобретение включает соединение или его соль с величиной МС50≤25 нМ для TLR7.
В одном аспекте изобретение не включает соединение или его соль с величиной МС50>25000 нМ для TLR7. В одном аспекте изобретение не включает соединение или его соль с величиной МС50>25000 нМ для TLR8. В одном аспекте изобретение не включает соединение или его соль с величинами МС50>25000 нМ как для TLR7, так и для TLR8.
Другой аспект изобретения относится к мягким лекарственным средствам (также известным как «antedrugs»). «Мягкие лекарственные средства» могут быть определены как биологически активные химические соединения (лекарственные средства), которые метаболически дезактивируются после того, как они осуществили свою терапевтическую роль в установленнм для них месте действия. Использование мягких лекарственных средств вместо их недезактивируемых аналогов может привести к отсутствию нежелательного побочного действия. В одном аспекте метаболическое удаление мягких лекарственных средств происходит с поддающейся регулированию скоростью предсказуемым образом. Одно воплощение изобретения относится к соединениям, которые являются мягкими лекарственными средствами. Конкретно, изобретение относится к соединениям, которые созданы для расщепления in vivo после осуществления их терапевтического действия до менее активных частиц. Изобретение относится к соединениям, которые созданы для расщепления in vivo после осуществления их терапевтического действия до нетоксичных частиц. Мягкие лекарственные средства по изобретению включают такие соединения, как соединение 225, 192, 193, 197, 198, 205, 213, 214, 215, 216, 217, 218, 219, 221, 222 и 223.
Термин «соединение по изобретению» относится к соединениям, приведенным в качестве примеров, и соединениям, подпадающим в объем формул, описанных в данном описании.
Термин «замещенный», используемый в данном описании, означает, что один или больше атомов водорода у указанного атома заменены путем выбора из указанной группы, при условии, что главная валентность указанного атома не превышена, и что замещение приводит к устойчивому соединению. Когда заместителем является кетогруппа (т.е., =О), тогда заменяются 2 атома водорода у одного атома. Циклические двойные связи, как используется в данном описании, представляют собой двойные связи, которые образованы между двумя соседними атомами цикла (например, С=С, C=N или N=N).
Химическая структура с изображением химической связи пунктирной линией показывает, что связь присутствует необязательно. Например, пунктирная линия, подчеркнутая затем сплошной линией простой связи, показывает, что связь может быть или простой связью или двойной связью.
Когда связь с заместителем показана как пересекающая связь, соединяющая два атома в цикле, тогда такой заместитель может быть связан с любым атомом цикла.
Термин «алкил», используемый в данном описании, относится к насыщенному линейному или разветвленному одновалентному углеводородному радикалу с одним-двенадцатью, в том числе, одним-десятью атомами углерода (С1-С10), одним-шестью атомами углерода (С1-С6) и одним-четырьмя атомами углерода (С1-С4), при этом алкильный радикал может быть необязательно замещен независимо одним или несколькими заместителями, описанными ниже. Примеры алкильных радикалов включают углеводородные группы, такие как, но без ограничения, метил (Ме, -СН3), этил (Et, -CH2CH3), 1-пропил (н-Pr, н-пропил, -СН2СН2СН3), 2-пропил (изо-Pr, изопропил, -СН(СН3)2), 1-бутил (н-Bu, н-бутил, -СН2СН2СН2СН3), 2-метил-1-пропил (изо-Bu, изобутил, СН2СН(СН3)2), 2-бутил (втор-Bu, втор-бутил, -СН(СН3)СН2СН3), 2-метил-2-пропил (трет-Bu, трет-бутил, С(СН3)3), 1-пентил (н-пентил, -СН2СН2СН2СН2СН3), 2-пентил (-СН(СН3)СН2СН2СН3), 3-пентил (-СН(СН2СН3)2), 2-метил-2-бутил (-С(СН3)2СН2СН3), 3-метил-2-бутил (-СН(СН3)СН(СН3)2), 3-метил-1-бутил (-СН2СН2СН(СН3)2), 2-метил-1-бутил (-СН2СН(СН3)СН2СН3), 1-гексил (-СН2СН2СН2СН2СН2СН3), 2-гексил (-СН(СН3)СН2СН2СН2СН3), 3-гексил (-СН(СН2СН3)(СН2СН2СН3)), 2-метил-2-пентил (-С(СН3)2СН2СН2СН3), 3-метил-2-пентил (-СН(СН3)СН(СН3)СН2СН3), 4-метил-2-пентил (-СН(СН3)СН2СН(СН3)2), 3-метил-3-пентил (-С(СН3)(СН2СН3)2), 2-метил-3-пентил (-СН(СН2СН3)СН(СН3)2), 2,3-диметил-2-бутил (-С(СН3)2СН(СН3)2), 3,3-диметил-2-бутил (-СН(СН3)С(СН3)3, 1-гептил и 1-октил.
Термин «алкенил» относится к линейному или разветвленному одновалентному углеводородному радикалу с двумя-10 атомами углерода (С2-С10), в том числе, двумя-шестью атомами углерода (С2-С6) и двумя-четырьмя атомами углерода (С2-С4), и, по меньшей мере, одной двойной связью и включает, но не ограничивается указанным, этенил, пропенил, 1-бут-3-енил, 1-пент-3-енил, 1-гекс-5-енил и т.п., при этом алкенильный радикал может быть необязательно замещен независимо одним или несколькими заместителями, описанными ниже, и включает радикалы, имеющие «цис»- и «транс»-ориентации или, с другой стороны, «Е»- и «Z»-ориентации. Термин «алкенил» включает аллил.
Термин «алкинил» относится к линейному или разветвленному одновалентному углеводородному радикалу с двумя-двенадцатью атомами углерода (С2-С12), в том числе, двумя-10 атомами углерода (С2-С10), двумя-шестью атомами углерода (С2-С6) и двумя-четырьмя атомами углерода (С2-С4), содержащему, по меньшей мере, одну тройную связь. Примеры включают, но не ограничиваются указанным, этинил, пропинил, бутинил, пентин-2-ил и т.п., при этом алкинильный радикал может быть необязательно замещен независимо одним или несколькими заместителями, описанными ниже.
Термины «карбоцикл», «карбоциклил» или «циклоалкил» используются в данном описании как взаимозаменяемые и относятся к насыщенному или частично ненасыщенному циклическому углеводородному радикалу с тремя-двенадцатью атомами углерода (С3-С12), в том числе, тремя-десятью атомами углерода (С3-С10) и тремя-шестью атомами углерода (С3-С6). Термин «циклоалкил» включает моноциклические и полициклические (например, бициклические и трициклические) циклоалкильные структуры, при этом полициклические структуры необязательно включают насыщенный или частично ненасыщенный циклоалкил, конденсированный с насыщенным или частично ненасыщенным циклоалкилом или гетероциклоалкилом или арилом или гетероарилом. Примеры циклоалкильных групп включают, но не ограничиваются указанным, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и т.п. Бициклические карбоциклы содержат 7-12 циклических атомов, например, расположенных как система бицикло[4,5], [5,5], [5,6] или [6,6], или 9 или 10 циклических атомов, расположенных как системы бицикло[5,6] или [6,6], или как мостиковые системы, такие как бицикло[2.2.1]гептан, бицикло[2.2.2]октан и бицикло[3.2.2]нонан. Циклоалкил может быть необязательно замещен независимо в одном или нескольких возможных для замещения положениях одним или несколькими заместителями, описанными в данном описании. Такие циклоалкильные группы могут быть необязательно замещены, например, одной или несколькими группами, выбранными независимо из (С1-С6)-алкила, (С1-С6)-алкокси, галогена, гидрокси, циано, нитро, амино, моно((С1-С6)-алкил)амино, ди((С1-С6)-алкил)амино, (С2-С6)-алкенила, (С2-С6)-алкинила, (С1-С6)-галогеналкила, (С1-С6)-галогеналкокси, амино(С1-С6)-алкила, моно((С1-С6)-алкил)амино(С1-С6)-алкила и ди((С1-С6)-алкил)амино(С1-С6)-алкила.
Термин «циклоалкенил» относится к частично ненасыщенному циклическому углеводородному радикалу с тремя-десятью атомами углерода (С3-С10), в том числе, тремя-шестью атомами углерода (С3-С6), содержащему, по меньшей мере, одну двойную связь в карбоцикле.
Термин «гетероалкил» относится к насыщенному линейному или разветвленному одновалентному углеводородному радикалу с одним-двенадцатью атомами углерода (С1-С12), в том числе, одним-шестью атомами углерода (С1-С6) и одним-четырьмя атомами углерода (С1-С4), при этом, по меньшей мере, один из атомов углерода заменен гетероатомом, выбранным из атомов N, O или S, и при этом радикал может представлять собой радикал по атому углерода или радикал по гетероатому (т.е., гетероатом может находиться в середине или на конце радикала). Гетероалкильный радикал может быть необязательно замещен независимо одним или несколькими заместителями, описанными в данном описании. Термин «гетероалкил» охватывает радикалы алкокси и гетероалкокси.
Термины «гетероциклоалкил», «гетероцикл» и «гетероциклил» в данном описании используются как взаимозаменяемые и относятся к насыщенному или частично ненасыщенному карбоциклическому радикалу с 3-8 атомами в цикле, в котором, по меньшей мере, один из атомов цикла представляет собой гетероатом, выбранный из атомов азота, кислорода или серы, причем остальные атомы цикла являются С, где один или несколько атомов цикла могут быть необязательно замещены независимо одним или несколькими заместителями, описанными ниже. Радикал может представлять собой радикал по атому углерода или радикал по гетероатому. Термин «гетероцикл» включает гетероциклоалкокси. Термин также включает конденсированные циклические системы, которые включают гетроцикл, конденсированный с ароматической группой. «Гетероциклоалкил» также включает радикалы, в которых гетероциклические радикалы конденсированы с ароматическими или гетероароматическими циклами. Примеры гетероциклоалкильных циклов включают, но не ограничиваются указанным, пирролидинил, тетрагидрофуранил, дигидрофуранил, тетерагидротиенил, тетрагидропиранил, дигидропиранил, тетрагидротиопиранил, пиперидино, морфолино, тиоморфолино, тиоксанил, пиперазинил, гомопиперазинил, азетидинил, оксетанил, тиетанил, гомопиперидинил, оксепанил, тиепанил, оксазепинил, диазепенил, тиазепинил, 1,2,3,6-тетрагидропиридинил, 2-пирролинил, 3-пирролинил, индолинил, 2Н-пиранил, 4Н-пиранил, диоксанил, 1,3-диоксаланил, пиразолинил, дитианил, дитиоланил, дигидропиранил, дигидротиенил, дигидрофуранил, пиразолидинил, имидазолидинил, 3-азабицикло[3.1.0]гексанил, 3-азабицикло[4.1.0]гептанил, азабицикло[2.2.2]гексанил, 3Н-индолил, хинолизинил и N-пиридилмочевины. Спирогруппы также входят в объем данного определения. Вышеуказанные группы, как группы, происходящие от групп, перечисленных выше, могут быть С-присоединенными или N-присоединенными, когда такое возможно. Например, группа, образованная от пиррола, может представлять собой пиррол-1-ил (N-присоединенную) или пиррол-3-ил (С-присоединенную). Также группа, образованная от имидазола, может представлять собой имидазол-1-ил (N-присоединенную) или имидазол-3-ил (С-присоединенную). Примером гетероциклической группы, в которой 2 циклических атома углерода заменены группой оксо (=О), является 1,1-диоксотиоморфолинил. В данном случае гетероциклические группы являются незамещенными или, в определенных случаях, замещенными различными группами в одном или нескольких замещаемых положениях. Например, такие гетероциклические группы могут быть необязательно замещены, например, одной или несколькими группами, независимо выбранными из (С1-С6)-алкила, (С1-С6)-алкокси, галогена, гидрокси, циано, нитро, амино, моно((С1-С6)-алкил)амино, ди((С1-С6)-алкил)амино, (С2-С6)-алкенила, (С2-С6)-алкинила, (С1-С6)-галогеналкила, (С1-С6)-галогеналкокси, амино(С1-С6)-алкила, моно((С1-С6)-алкиламино(С1-С6)-алкила или ди(С1-С6)-алкиламино(С1-С6)-алкила.
Термин «арил» относится к одновалентному ароматическому карбоциклическому радикалу с одним циклом (например, фенил), несколькими циклами (например, бифенил) или несколькими конденсированными циклами, из которых, по меньшей мере, один цикл является ароматическим (например, 1,2,3,4-тетрагидронафтил, нафтил и т.д.), который необязательно замещен одним или несколькими заместителями, независимо выбранными из, например, галогена, низшего алкила, низшего алкокси, трифторметила, арила, гетероарила и гидрокси. В одном воплощении арил представляет собой 6-членный арил. Например, арил представляет собой фенил.
Термин «гетероарил» относится к одновалентному ароматическому радикалу с 5-, 6- или 7-членными циклами и включает конденсированные циклические системы (в которых, по меньшей мере, один цикл является ароматическим) из 5-10 атомов, содержащие, по меньшей мере, один и до четырех гетероатомов, выбранных из атомов азота, кислорода и серы. Примерами гетероарильных групп являются пиридинил, имидазолил, пиримидинил, пиразолил, триазолил, пиразинил, тетразолил, фурил, тиенил, изоксазолил, тиазолил, оксазолил, изотиазолил, пирролил, хинолинил, изохинолинил, индолил, бензимидазолил, бензофуранил, циннолинил, индазолил, индолизинил, фталазинил, пиридазинил, триазинил, изоиндолил, птеридинил, пуринил, оксадиазолил, тиадиазолил, фуразанил, бензофуразанил, бензотиофенил, бензотиазолил, бензоксазолил, хиназолинил, хиноксалинил, нафтиридинил, изобензофуран-1(3Н)-он и фуропиридинил. Спирогруппы также входят в объем данного определения. Гетероарильные группы являются необязательно замещенными одним или несколькими заместителями, независимо выбранными из, например, галогена, низшего алкила, низшего алкокси, галогеналкила, арила, гетероарила и гидрокси.
Термин «галоген» представляет собой фтор, бром, хлор и иод.
Термин «оксо» представляет собой =О.
Вообще, различные части или функциональные группы соединений по изобретению могут быть необязательно замещены одним или несколькими заместителями. Примеры заместителей, подходящих для целей изобретения, включают, но не ограничиваются указанным, оксо, галоген, циано, нитро, трифторметил, дифтометокси, трифторметокси, азидо, -NR”SO2R', -SO2NR'R”, -C(O)R', -C(O)OR', -OC(O)R, -NR”C(O)OR', -NR”C(O)R', -C(O)NR'R”, -NRC(O)NR”, -NRC(NCN)NR'R”, -OR', арил, гетероарил, арилалкил, гетероарилалкил, гетероциклил и гетероциклилалкил, где R', R” и R”' представляют собой независимо Н, алкил, гетероалкил, гетероциклоалкил, алкенил, алкинил, арил или гетероарил.
«(Алкил)арильная» группа, как используется в данном описании, представляет собой арильный заместитель, который присоединяется к соединению с помощью линейной или разветвленной алкильной группы с одним-двенадцатью атомами углерода. В одном аспекте арильный заместитель присоединяется к соединению с помощью линейной или разветвленной алкильной группы с 1-6 атомами углерода. Алкильная часть (алкил)арильной группы является необязательно замещенной. В одном воплощении арил представляет собой 6-членный арил. Например, арил представляет собой фенил.
«(Алкил)гетероциклоалкильная» группа, как используется в данном описании, представляет собой гетероциклический заместитель, который присоединяется к соединению с помощью линейной или разветвленной алкильной группы с одним-двенадцатью атомами углерода. В одном аспекте гетероциклический заместитель присоединяется к соединению с помощью линейной или разветвленной алкильной группы с 1-6 атомами углерода. Алкильная часть (алкил)гетероциклической группы является необязательно замещенной.
«(Алкил)циклоалкильная» группа, как используется в данном описании, представляет собой циклоалкильный заместитель, который присоединяется к соединению с помощью линейной или разветвленной алкильной группы с одним-двенадцатью атомами углерода. В одном аспекте циклоалкильный заместитель присоединяется к соединению с помощью линейной или разветвленной алкильной группы с 1-6 атомами углерода. Алкильная часть (алкил)циклоалкильной группы является необязательно замещенной.
«(Алкил)циклоалкенильная» группа, как используется в данном описании, представляет собой циклоалкенильный заместитель, который присоединяется к соединению с помощью линейной или разветвленной алкильной группы с одним-двенадцатью атомами углерода. В одном аспекте циклоалкенильный заместитель присоединяется к соединению с помощью линейной или разветвленной алкильной группы с 1-6 атомами углерода. Алкильная часть (алкил)циклоалкенильной группы является необязательно замещенной.
Соединения по данному изобретению могут иметь один или несколько асимметричных центров, поэтому такие соединения могут быть получены в виде отдельных (R)- или (S)-стереоизомеров или в виде их смеси. Если не указано иное, предполагается, что описание или название определенного соединения в описании и формуле изобретения включают оба отдельных энантиомера и рацемические или иные смеси его диастереомеров. Соответственно, данное изобретение также включает все такие изомеры, в том числе, смеси диастеремеров, чистые диастереомеры и чистые энантиомеры соединений и формул, описанных в данном описании.
Смеси диастеремеров можно разделить на ее отдельные диастереомеры на основе их физико-химических различий методами, известными специалистам в данной области техники, например, хроматографией или фракционной кристаллизацией. Энантиомеры можно разделить, превращая смесь энантиомеров в смесь диастереомеров взаимодействием с соответствующим оптически активным соединением (например, спиртом), разделяя диастереомеры и превращая (например, гидролизом) отдельные диастереомеры в соответствующие чистые энантиомеры. Энантиомеры также можно разделить, используя хиральную колонку для ВЭЖХ. Способы определения стереохимии и разделения стереоизомеров хорошо известны в технике (см. обсуждение в главе 4 «Advanced Organic Chemistry», 4th edition, J. March, John Wiley and Sons, New York, 1992).
В структурах, показанных в данном описании, когда стереохимия любого определенного хирального атома не конкретизируется, тогда все стереоизомеры рассматриваются и включаются как соединения по изобретению. Когда стереохимия конкретизируется темным клином или пунктирной линией, представляющими определенную конфигурацию, тогда такой стереоизомер является установленным и определенным таким образом.
Отдельный стереоизомер, например, энантиомер, по существу, свободный от своего стереоизомера, можно получить расщеплением рацемической смеси с использованием такого способа, как образование диастереомеров с использованием оптически активных агентов расщепления (Eliel E. and Wilen S., Stereochemistry of Organic Compounds, John Wiley & Sons, Inc., New York, 1994; Lochmuller C.H. (1975), J. Chromatogr., 113(3): 283-302). Рацемические смеси хиральных соединений по изобретению можно разделить и выделить любым подходящим способом, включая (1) образование ионных диастеромерных солей с хиральными соединениями и разделение фракционной кристаллизацией или другими способами, (2) образование диастереомерных соединений с хиральными дериватизирующими реагентами, разделение диастереомеров и превращение в чистые стреоизомеры, и (3) разделение, по существу, чистых или обогащенных стереоизомеров непосредственно в хиральных условиях. См. Drug Stereochemistry, Analytical Methods and Pharmacology, Irving W. Wainer, Ed., Marcel Dekker, Inc., New York (1993).
В способе (1) диастеромерные соли можно получить взаимодействием энантиомерно чистых хиральных оснований, таких как бруцин, хинин, эфедрин, стрихнин, а-метил-13-фенилэтиламин (амфетамин) и т.п., с асимметричными соединениями, содержащими кислотную функциональную группу, такими как карбоновая кислота и сульфоновая кислота. Диастеромерные соли можно побудить к разделению фракционной кристаллизацией или ионной хроматографией. В случае разделения оптических изомеров аминосоединений добавление хиральных карбоновых или сульфоновых кислот, таких как камфорсульфоновая кислота, винная кислота, миндальная кислота или молочная кислота, может привести к образованию диастеромерных солей.
С другой стороны, по способу (2) субстрат для расщепления вводят во взаимодействие с одним энантиомером хирального соединения с образованием диастереомерной пары (Eliel E. and Wilen S., «Stereochemistry of Organic Compounds», John Wiley & Sons, Inc., New York, 1994, р.322). Диастереомерные соединения можно получить взаимодействием асимметричных соединений с энантиомерно чистыми хиральными дериватизирующими реагентами, такими как производные ментола, с последующим разделением диастереомеров и гидролизом и получением чистого или обогащенного энантиомера. Способ определения оптической чистоты включает получение сложных хиральных эфиров, например, ментолового эфира, такого как (-)ментилхлорформиат, в присутствии основания, или эфира Mosher а-метокси-а-(трифторметил)фенилацетата (Jacob III (1982), J. Org. Chem., 47: 4165), из рацемической смеси и анализ спектра ЯМР на присутствие двух атропоизомерных энантиомеров или диастереомеров. Устойчивые диастереомеры атропоизомерных соединений можно разделить и выделить хроматографией с нормальной и обращенной фазой, следуя способам разделения атропоизомерных нафтилизохинолинов (WO 96/15111). По способу (3) рацемическую смесь двух энантиомеров можно разделить хроматографией с использованием хиральной неподвижной фазы (Chiral Liquid Chromatography (1989), W.J. Lough, Ed., Chapman and Hall, New York; Okamoto (1990), J. of Chromatogr., 513: 375-378). Обогащенные или очищенные энантиомеры можно различить способами, используемыми для распознавания других хиральных молекул с асимметричными атомами углерода, такими как оптическое вращение и круговой дихроизм.
Термин «таутомер» относится к соединению, структуры которого заметно различаются по расположению атомов, но которые существуют в состоянии легко и быстро устанавливающегося равновесия. Следует иметь в виду, что соединения по изобретению можно отобразить в виде различных таутомеров. Также следует иметь в виду, что когда соединения имеют таутомерные формы, предполагается, что все таутомерные формы входят в объем изобретения, и названия соединений не исключают какую-либо таутомерную форму.
Предполагается, что настоящее изобретение включает все изотопы атомов, встречающихся в соединениях по настоящему изобретению. Изотопы включают такие атомы, которые имеют один и тот же атомный номер, но различные массовые числа. Как общий пример, но без ограничения, изотопы водорода включают тритий и дейтерий, и изотопы углерода включают С-13 и С-14.
Кроме соединений по изобретению изобретение также включает фармацевтически приемлемые соли таких соединений.
«Фармацевтически приемлемая соль», если не указано иное, включает соли, которые сохраняют биологическую эффективность свободных кислот и оснований конкретного соединения, и которые не являются биологически или как-то иначе нежелательными. Соединение по изобретению может обладать достаточно кислотными, достаточно основными или теми и другими функциональными группами и соответственно взаимодействовать с любым основанием из числа неорганических или органических оснований и любой кислотой из числа неорганических или органических кислот с образованием фармацевтически приемлемой соли. Примеры фармацевтически приемлемых солей включают соли, полученные взаимодействием соединений по настоящему изобретению с минеральной или органической кислотой или неорганическим основанием, причем такие соли включают сульфаты, пиросульфаты, бисульфаты, фосфаты, моногидрофосфаты, дигидрофосфаты, метафосфаты, пирофосфаты, хлориды, бромиды, иодиды, ацетаты, пропионаты, деканоаты, каприлаты, акрилаты, формиаты, изобутираты, капроаты, гептаноаты, пропиолаты, оксалаты, малонаты, сукцинаты, субераты, себацаты, фумараты, малеаты, бутин-1,4-диоаты, гексин-1,6-диоаты, бензоаты, хлорбензоаты, метилбензоаты, динитробензоаты, гидроксибензоаты, метоксибензоаты, фталаты, сульфонаты, ксилолсульфонаты, фенилацетаты, фенилпропионаты, фенилбутираты, цитраты, лактаты, у-гидроксибутираты, гликоляты, тартраты, метансульфонаты, пропансульфонаты, нафталин-1-сульфонаты, нафталин-2-сульфонаты и манделаты. Так как отдельное соединение по настоящему изобретению может включать более одной кислотной или основной группы, соединения по настоящему изобретению могут включать моно-, ди- или трисоли одного соединения.
Если соединение по изобретению представляет собой основание, нужную фармацевтически приемлемую соль можно получить любым подходящим способом, доступным в технике, например, обработкой свободного основания кислотой, в частности, неорганической кислотой, такой как хлороводородная кислота, бромоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., или органической кислотой, такой как уксусная кислота, яблочная кислота, янтарная кислота, миндальная кислота, фумаровая кислота, малоновая кислота, пировиноградная кислота, щавелевая кислота, гликолевая кислота, салициловая кислота, пиранозидильная кислота, такая как глюкуроновая кислота или галактуроновая кислота, альфа-гидроксикислота, такая как лимонная кислота или винная кислота, аминокислота, такая как аспарагиновая кислота или глутаминовая кислота, ароматическая кислота, такая как бензойная кислота или коричная кислота, сульфоновая кислота, такая как п-толуолсульфоновая кислота или этансульфоновая кислота, или подобная кислота.
Если соединение по изобретению представляет собой кислоту, нужную фармацевтически приемлемую соль можно получить любым подходящим способом, например, обработкой свободной кислоты неорганическим или органическим основанием. Примеры подходящих неорганических солей включают соли, образованные щелочными и щелочноземельными металлами, такими как литий, натрий, калий, барий и кальций. Примеры подходящих солей органических оснований включают соли, например, соли аммония, дибензиламмония, бензиламмония, 2-гидроксиэтиламмония, бис(2-гидроксиэтил)аммония, фенилэтилбензиламина, дибензилэтилендиамина и подобные соли. Другие соли по кислотным группам могут включать, например, соли, образованные с прокаином, хинином и N-метилглюкозамином, и также соли, образованные с основными аминокислотами, такими как глицин, орнитин, гистидин, фенилглицин, лизин и аргинин.
Настоящее изобретение также относится к солям соединений по изобретению, которые не являются обязательно фармацевтически приемлемыми солями, но которые могут быть применимы в качестве промежуточных соединений для получения и/или очистки соединений по изобретению и/или разделения энантиомеров соединений по изобретению.
Соединения по изобретению можно получить с использованием реакций и схем синтеза, описанных на схеме I, с использованием методов, доступных в технике, с использованием исходных материалов, которые легко доступны.
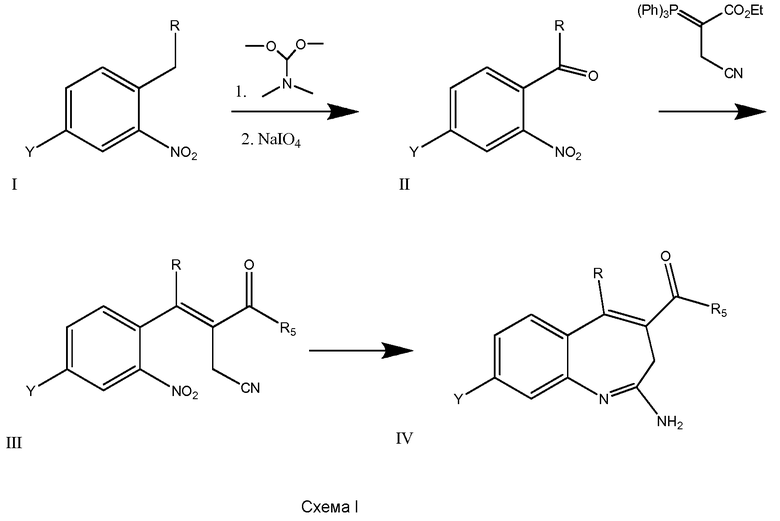
По схеме I соединения формулы II можно получить из алкиларена формулы I обработкой диметилацеталем диметилформамида с использованием пирролидина или без него (J. Org. Chem. (1986), 51(26), 5106-5110) в ДМФА при 70-90°С. Неочищенное промежуточное соединение (не показано) можно расщепить до альдегида формулы II NaClO4 в смеси ТГФ/фосфатный буфер, рН 7,2, при комнатной или при примерно комнатной температуре. Альдегид формулы II можно олифинировать с илидом фосфония в толуоле при температурах, коолеблющихся от 70 до 110°С (1-16 часов), и получить соединения формулы III. Соединения формулы IV можно получить из соединения формулы III с использованием порошка железа в уксусной кислоте. Реакцию можно проводить при температурах примерно 90°С в течение примерно 3-14 часов.
Следует отметить, что при получении некоторых соединений по изобретению, описанных в данном описании, может потребоваться защита функциональных групп, не участвующих в реакции. Необходимость такой защиты будет изменяться в зависимости от природы функциональной группы и условий, используемых в способах получения, и может быть легко определена специалистами в данной области техники. Такие способы введения/удаления защитной группы хорошо известны специалистам в данной области техники.
Соединения по изобретению находят использование в различных применениях. Например, в некоторых аспектах изобретение относится к способам модуляции TLR7- и/или TLR8-опосредуемой передачи сигнала. Способы по изобретению применимы, например, когда желательно изменить TLR7- и/или TLR8-опосредуемую передачу сигнала в ответ на подходящий лиганд TLR7 и/или TLR8 или агонист передачи сигнала TLR7 и/или TLR8.
Используемый в данном описании термины «лиганд TLR7 и/или TLR8», «лиганд для TLR7 и/или TLR8» и «агонист передачи сигнала TLR7 и/или TLR8» относятся к молекуле иной, чем соединение по изобретению, которая взаимодействует непосредственно или косвенно с TLR7 и/или TLR8 и индуцирует TLR7- и/или TLR8-опосредуемую передачу сигнала. В некоторых воплощениях лиганд TLR7 и/или TLR8 представляет собой природный лиганд, т.е., лиганд TLR7 и/или TLR8, который находят в природе. В некоторых воплощениях лиганд TLR7 и/или TLR8 имеет отношение к молекуле иной, чем природный лиганд TLR7 и/или TLR8, например, молекуле, полученной в результате деятельности человека.
Термин «модулировать», используемый в данном описании в отношении рецепторов TLR7 и/или TLR8, означает опосредование фармакодинамической реакции у субъекта путем (i) ингибирования или активации рецептора или (ii) прямого или косвенного воздействия на нормальную регуляцию рецепторной активности. Соединения, которые модулируют рецепторную активность, включают агонисты, антагонисты, смешанные агонисты/антагонисты и соединения, которые прямо или косвенно воздействуют на регуляцию рецепторной активности.
Термин «агонист» относится к соединению, которое в сочетании с рецептором (например, TLR) может вызывать клеточную реакцию. Агонист может представлять собой лиганд, который непосредственно связывается с рецептором. С другой стороны, агонист может объединяться с рецептором косвенно, путем, например, (а) образования комплекса с другой молекулой, которая непосредственно связывается с рецептором, или (b) иначе, что приводит к модификации другого соединения так, что другое соединение непосредственно связывается с рецептором. Агонист может быть отнесен к агонисту определенного TLR (например, агонисту TLR7 и/или TLR8). Термин «частичный агонист» относится к соединению, которое вызывает частичную, но не полную клеточную реакцию. Анализы, относящиеся к TLR7 и TLR8, известны в технике (например, Gorden et al., Journal of Immunology, 177, pp. 8164-8170 (2006), и Zhu et al., Molecular Immunology, vol.45 (11), pp. 3238-3242 (2008)).
Термин «антагонист», используемый в данном описании, относится к соединению, которое конкурирует с агонистом или частичным агонистом за связывание с рецептором, причем посредством этого блокируется действие агониста или частичного агониста на рецептор. Конкретнее, антагонист представляет собой соединение, которое ингибирует активность агониста TLR7 или TLR8 у рецептора TLR7 или TLR8, соответственно.
Термин «ингибировать» относится к любому снижению биологической активности, которое можно измерить. Таким образом, используемые в данном описании термины «ингибировать» или «ингибирование» можно отнести к проценту от нормального уровня активности.
В одном аспекте данного изобретения способ лечения или предупреждения состояния или расстройства, которое можно лечить модуляцией у субъекта TLR7- и/или TLR8-опосредуемых клеточных активностей, включает введение указанному субъекту композиции, включающей соединение по изобретению в количестве, эффективном для лечения или предупреждения состояния или расстройства. Термин «TLR7- и/или TLR8-опосредуемая» относится к биологической или химической активности, которая является результатом функции TLR7 и/или TLR8.
Состояния и расстройства, которые можно лечить способами по данному изобретению, включают, но не ограничиваются указанным, рак, ассоциированные с иммунным комплексом заболевания, аутоиммунные заболевания или расстройства, воспалительные расстройства, иммунодефицит, отторжение трансплантата, болезнь трансплантата против хозяина, аллергии, сердечно-сосудистую болезнь, фиброзное заболевание, астму, инфекцию и сепсис.
Конкретнее, в способах, применимых при лечении состояний, включая рак (терапевтическая или противораковая вакцина), аллергическое заболевание (например, атопический дерматит, аллергический ринит, астму), инфекционное заболевание (профилактика вакциной и противовирусная профилактика) и иммунодефицит, будут использоваться соединения по изобретению, которые ингибируют TLR7- и/или TLR8-опосредуемую передачу сигнала.
С другой стороны, в способах, применимых при лечении состояний, включая аутоиммунное заболевание, CF, сепсис, отторжение трансплантата и GVHD, как правило, будут использоваться соединения по изобретению, которые усиливают TLR7- и/или TLR8-опосредуемую передачу сигнала.
В некоторых случаях композиции могут быть использованы для ингибирования или промотирования TLR7- и/или TLR8-опосредуемой передачи сигнала в ответ на лиганд TLR7 и/или TLR8 или агонист передачи сигнала. В других случаях композиции могут быть использованы для ингибирования или промотирования TLR7- и/или TLR8-опосредуемой иммуностимуляции у субъекта.
Термин «лечение», используемый в данном описании, если не указано иное, обозначает, по меньшей мере, смягчение заболевания или состояния и включает, но не ограничивается указанным, модуляцию и/или ингибирование существующего заболевания или состояния и/или облегчение заболевания или состояния, к которому такой термин применяется, или одного или нескольких симптомов такого заболевания или состояния. Термин «лечение», используемый в данном описании, если не указано иное, относится к действию лечения, при этом «лечение» имеет значение, только что установленное выше. Терапевтическое лечение означает лечение, инициированное после наблюдения симптомов и/или предполагаемого воздействия причинного фактора заболевания или состояния. Как правило, терапевтическое лечение может уменьшать тяжесть и/или длительность симптомов, ассоциированных с заболеванием или состоянием.
Используемый в данном описании термин «предупреждение» означает препятствие развитию клинических симптомов заболевания или состояния, т.е., ингибирование начала заболевания или состояния у субъекта, которому он может подвергаться, или к которому он предрасположен, но пока не испытывает или не проявляет симптомы заболевания или состояния. Профилактическое лечение означает, что соединение по изобретению вводят субъекту до того, как наблюдают симптомы и/или предполагаемое воздействие причинного фактора состояния (например, патогенного микроорганизма или канцерогена). Как правило, профилактическое лечение может уменьшить (а) вероятность того, что у субъекта, который получает лечение, развивается такое состояние, и/или (b) длительность и/или тяжесть симптомов в случае, если у субъекта развивается такое состояние.
Используемые в данном описании термины «аутоиммунное заболевание», «аутоиммунное расстройство» и «аутоиммунитет» относятся к иммунологически опосредуемому острому или хроническому повреждению ткани или органа, исходящему от хозяина. Термины охватывают как клеточно-, так и антителоопосредованные аутоиммунные явления, а также органспецифический и органнеспецифический аутоиммунитет. Аутоиммунные заболевания включают инсулинзависимый сахарный диабет, ревматоидный артрит, системную красную волчанку, рассеяный склероз, атеросклкроз и воспалительное заболевание кишечника. Аутоиммунные заболевания также включают, без ограничения, анкилозирующий спондилит, аутоиммунную гемолитическую анемию, синдром Бехчета, синдром Гудпасчера, болезнь Грейвса, синдром Гийена-Барре, тиреоидит Хасимото, идиопатическую тромбоцитопению, тяжелую псевдопаралитическую миастению, пернициозную анемию, нодозный полиартериит, полимиозит/дерматомиозит, первичный билиарный склероз, псориаз, саркоидоз, склерозирующий холангит, синдром Шегрена, системный склероз (склеродерма и синдром CREST), артериит Токоясу, височный артериит и гранулематоз Вегенера. Аутоиммунные заболевания также включают некоторые заболевания, ассоциированные с иммунным комплексом.
Используемый в данном описании термин «фиброзное заболевание» относится к заболеваниям, включающим избыточное и персистентное образование рубцовой ткани, связанное с недостаточностью органа при различных хронических заболеваниях, поражающих легкие, почки, глаза, сердце, печень и кожу. Хотя ремоделирование ткани и рубцевание является частью нормального процесса заживления раны, повторное повреждение или инсульт могут привести к персистентному и избыточному рубцеванию и, в конечном итоге, к недостаточности органа.
Фиброзные состояния включают диффузную фиброзную болезнь легких, хроническую болезнь почек, включая диабетическую болезнь почек; фиброз печени (например, хроническую болезнь печени (CLD), вызванную непрерывными и повторяющимися инсультами печени в силу таких причин, как вирусный гепатит В и С, алкогольный цирроз или неалкогольная жирная печень (NAFLD) или первичный склерозирующий холангит (PSC) - редкое заболевание, характеризующееся фиброзным воспалительным разрушением желчных протоков внутри и на поверхности печени, ведущее к стазу желчи, фиброзу печени и, в конечном итоге, к циррозу и конечной стадии болезни печени); фиброз легких (например, идиопатический фиброз легких (IPF)); и системный склероз (дегенеративное расстройство, при котором избыточный фиброз имеет место в системе нескольких органов, включая кожу, кровеносные сосуды, сердце, легкие и почки).
Другие примеры включают кистозный фиброз поджелудочной железы и легких; инъекционный фиброз, который может иметь место как осложнение внутримышечных инъекций, в особенности у детей; эндомиокардиальный фиброз; медиастинальный фиброз, миелофиброз; ретроперитонеальный фиброз; прогрессирующий выраженный фиброз - осложнение пневмокониоза шахтеров; нефрогенный системный фиброз и осложнения некоторых типов операций по пересадке (например, появляющихся при попытках создания искусственной поджелудочной железы для лечения сахарного диабета).
Используемый в данном описании термин «сердечно-сосудистая болезнь» относится к заболеваниям или расстройствам сердечно-сосудистой системы, включающим воспалительную компоненту и/или накопление бляшек, включая, без ограничений, коронарную болезнь, болезнь мозгового кровообращения, периферическую артериальную болезнь, атеросклероз и артериосклероз.
Используемые в данном описании термины «рак» и «опухоль» относятся к состоянию, при котором у субъекта присутствуют анормально реплицирующиеся клетки хозяйского происхождения в детектируемом количестве. Рак может представлять собой злокачественный или незлокачественный рак. Онкозаболевания или опухоли включают, но не ограничиваются указанным, рак желчных протоков; рак головного мозга; рак молочной железы; цервикальный рак; хориокарциному; рак толстой кишки; эндометриальный рак; эзофагеальный рак; рак желудка; внутриэпителиальные неоплазмы; лейкозы; лимфомы; рак печени (например, мелкоклеточный и не-мелкоклеточный); меланому; нейробластомы; внутриротовой рак; ректальный рак; рак почек; саркомы; рак кожи; рак яичка; рак щитовидной железы, а также другие карциномы и саркомы. Онкозаболевания могут быть первичными и метаститическими.
Используемые в данном описании термины «воспалительное заболевание» и «воспалительное расстройство» относятся к состоянию, характеризующемуся воспалением, например, локализованной защитной реакции ткани на раздражение, повреждение или инфекцию, характеризующееся болью, покраснением, опуханием и иногда потерей функции. Воспалительные заболевания или расстройства включают, например, аллергию, астму и аллергическую сыпь.
Используемый в данном описании термин «заболевание, ассоциированное с иммунным комплексом» относится к любому заболеванию, характеризующемуся выработкой и/или отложением ткани иммунных комплексов (т.е., любого конъюгата, включая антитела и антигены, специфически связанные антителами), включая, но не ограничиваясь указанным, системную красную волчанку (SLE) и родственные заболевания соединительной ткани, ревматоидный артрит, иммуннокомплексное заболевание, родственное гепатиту С и гепатиту В (например, криоглобулинемию), синдром Бехчета, аутоиммунные гломерулонефриты и васкулопатию, ассоциированную с наличием LDL/анти-LDL иммунных комплексов.
Используемый в данном описании термин «иммунодефицит» относится к заболеванию или расстройству, при котором иммунная система пациента не функционирует в нормальном объеме, или при котором она не может быть применима для усиления иммунной реакции у субъекта, например, для устранения у субъекта опухоли или рака (например, опухолей головного мозга, легких (мелкоклеточный и не-мелкоклеточный рак), яичников, молочной железы, предстательной железы, толстой кишки, а также других карцином и сарком) или инфекции. Иммунодефицит может быть приобретенным или он может быть врожденным.
«Болезнь трансплантат против хозяина» (GvHD) представляет собой реакцию донорского костного мозга против собственной ткани пациента. GVHD наиболее часто проявляется в случаях, когда кровь донора костного мозга является неродственной пациенту, или когда донор родственен пациенту, но точного соответствия нет. Существуют две формы GVHD: ранняя форма, называемая острой GVHD, которая имеет место вскоре после пересадки, когда нарастает число белых клеток, и поздняя форма, называемая хронической GVHD.
Атопические заболевания, опосредуемые ТН2, включают, но не ограничиваются указанным, атопический дерматит или экзему, эозинофилию, астму, аллергию, аллергический ринит и синдром Омена.
Используемый в данном описании термин «аллергия» относится к приобретенной гиперчувствительности к веществу (аллергену). Аллергические состояния включают экзему, аллергический ринит или острый ринит, сенную лихорадку, астму, крапивницу (сыпь) и пищевые аллергии и другие атопические состояния.
Используемый в данном описании термин «астма» относится к расстройству дыхательной системы, характеризуемому воспалением, сужением дыхательных путей и повышенной реактивностью дыхательных путей на вдыхаемые вещества. Астма часто, хотя не исключительно, ассоциируется с атопическими или аллергическими симптомами. Например, астма может быть преципитирована воздействием аллергена, воздействием холодного воздуха, инфекцией дыхательных путей и напряжением.
Используемые в данном описании термины «инфекция» и равнозначный «инфекционное заболевание» относятся к состоянию, при котором инфекционный организм или агент присутствует в детектируемом количестве в крови или обычно стерильной ткани или обычно стерильном компартменте субъекта. Инфекционные организмы и агенты включают вирусы, бактерии, грибы и паразитов. Термины охватывают как острые, так и хронические инфекции, а также сепсис.
Используемый в данном описании термин «сепсис» относится к присутствию бактерий (бактеремия) или других инфекционных микроорганизмов или их токсинов в крови (септицемия) или другой ткани организма.
Изобретение также относится к соединению по изобретению или его соли для применения в качестве лекарственного средства при лечении заболеваний или состояний, описанных выше, у млекопитающего, например, человека, страдающего от такого заболевания или состояния. Изобретение также относится к применению соединения по изобретению или его соли для получения лекарственного средства для лечения заболеваний или состояний, описанных выше, у млекопитающего, например, человека, страдающего от такого расстройства.
Данное изобретение также включает фармацевтические композиции, содержащие соединение по изобретению, и способам лечения или предупреждения состояний и расстройств путем модуляции TLR7- и/или TLR8-опосредуемых клеточных активностей путем введения пациенту, нуждающемуся в этом, фармацевтической композиции, включающей соединение по изобретению или его соль.
Для того, чтобы использовать соединение по изобретению или его соль для терапевтического лечения (включая профилактическое лечение) млекопитающих, в том числе, людей, их обычно получают, согласно стандартной фармацевтической практике, в виде фармацевтической композиции.
Согласно такому аспекту изобретение относится к фармацевтической композиции, которая включает соединение по изобретению или его соль, определенные в данном описании выше, в ассоциации с фармацевтически приемлемым носителем или разбавителем.
Для того, чтобы получить фармацевтические композиции по изобретению, терапевтически или профилактически эффективное количество соединения по изобретению или его соли (одних или с дополнительным терапевтическим средством, как обсуждалось в данном описании) тщательно смешивают, например, с фармацевтически приемлемым носителем согласно обычным методам приготовления фармацевтических смесей для получения дозы. Носитель может принимать самые различные формы в зависимости от препарата, нужного для введения, например, перорального или парентерального. Примеры подходящих носителей включают любой и все растворители, дисперсионные среды, адъюванты, покрытия, антибактериальные и противогрибковые средства, вещества для регулирования тоничности и вещества, замедляющие поглощение, подслащивающие вещества, стабилизаторы (для промотирования длительного хранения), эмульгаторы, связующие вещества, загустители, соли, консерванты, корригенты и разнообразные материалы, такие как буферы и абсорбенты, которые могут потребоваться для того, чтобы получить определенную терапевтическую композицию. Использование таких сред и веществ с фармацевтически активными веществами хорошо известно в технике. За исключением случая, когда какие-либо обычные среды или вещества несовместимы с соединением по изобретению, их использование в терапевтических композициях и препаратах предполагается. В композиции и препараты, как описано в данном описании, также могут быть включены дополнительные активные ингредиенты.
Композиции по изобретению могут находиться в форме, подходящей для перорального применения (например, в виде таблеток, лепешек, твердых или мягких капсул, водных или масляных суспензий, эмульсий, диспрегируемых порошков или гранул, сиропов или эликсиров), для местного применения (например, в виде кремов, мазей, гелей или водных или масляных растворов или суспензий), для введения ингаляцией (например, в виде тонко измельченного порошка или жидкого аэрозоля), для введения инсуффляцией (например, в виде тонко измельченного порошка) или для парентерального введения (например, в виде стерильного водного или масляного раствора для внутривенного, подкожного или внутримышечного введения дозы или в виде суппозитория для ректального введения дозы). Например, композиции, предназначенные для перорального использования, могут содержать, например, один или несколько красителей, подслащивающих веществ, корригентов и/или консервантов.
Подходящие фармацевтически приемлемые эксципиенты для таблеток включают, например, инертные разбавители, такие как лактоза, карбонат натрия, фосфат кальция или карбонат кальция, вещества, способствующие образованию гранул и рассыпанию, такие как кукурузный крахмал или альгиновая кислота; связующие вещества, такие как крахмал; смазывающие вещества, такие как стеарат магния, стеариновая кислота или тальк; консерванты, такие как этил- или пропил-п-гидроксибензоат, и антиоксиданты, такие как аскорбиновая кислота. Таблетки могут быть без покрытия или с покрытием для модификации их рассыпания и последующего всасывания активного ингредиента в желудочно-кишечном тракте или для улучшения их устойчивости и/или внешнего вида, в любом случае, с использованием обычных веществ для покрытия и процедур, хорошо известных в технике.
Композиции для перорального использования могут находиться в форме твердых желатиновых капсул, в которых активный ингредиент смешан с инертным твердым разбавителем, например, карбонатом кальция, фосфатом кальция или каолином, или мягких желатиновых капсул, в которых активный ингредиент смешан с маслом, таким как арахисовое масло, жидкий парафин или оливковое масло.
Водные суспензии обычно содержат активный ингредиент в тонкоизмельченной форме вместе с одним или несколькими суспендирующими веществами, такими как натрийкарбоксиметилцеллюлоза, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, трагакантовая камедь и аравийская камедь; диспергирующими или смачивающими веществами, такими как лецитин или продукты конденсации алкиленоксида с жирными кислотами (например, полиоксиэтиленстеарат) или продукты конденсации этиленоксида с длинноцепными алифатическими спиртами, например, гептадекаэтиленоксицетанол, или продукты конденсации этиленоксида с неполными сложными эфирами, полученными из жирных кислот и гексита, такие как моноолеат полиоксиэтиленсорбита, или продукты конденсации этиленоксида с неполными сложными эфирами, полученными из жирных кислот и ангидридов гексита, например, моноолеат полиоксиэтиленсорбитана. Водные суспензии также могут содержать один или несколько консервантов, корригентов и/или подслащивающих веществ (таких как сахароза, сахарин или аспартам).
Масляные суспензии можно получить путем суспендирования активного ингредиента в растительном масле (таком как арахисовое масло, оливковое масло, сезамовое масло или кокосовое масло) или минеральном масле (таком как жидкий парафин). Масляные суспензии также могут содержать загуститель, такой как пчелиный воск, твердый парафин или цетиловый спирт. Подслащивающие вещества, такие как вещества, указанные выше, и корригенты могут быть добавлены для получения приятного перорального препарата. Такие композиции могут сохраняться за счет добавления антиоксиданта, такого как аскорбиновая кислота.
Диспергируемые порошки и гранулы, подходящие для получения водной суспензии путем добавления воды, как правило, содержат активный ингредиент вместе с диспергирующим или смачивающим веществом и одним или несколькими консервантами. Примерами подходящих диспергирующих и смачивающих веществ являются такие вещества, которые уже упоминались выше. Также могут присутствовать другие эксципиенты, такие как подслащивающие вещества, корригенты и красители.
Фармацевтические композиции по изобретению также могут находиться в форме эмульсий масло в воде. Масляная фаза может представлять собой растительное масло, такое как оливковое масло или арахисовое масло, или минеральное масло, такое как, например, жидкий парафин, или смесь любых указанных масел. Подходящими эмульгаторами могут являться, например, встречающиеся в природе смолы, такие как аравийская камедь или трагакантовая камедь, встречающиеся в природе фосфатиды, такие как соевый лецитин, эфиры или неполные эфиры, образованные из жирных кислот и ангидридов гексита (например, моноолеат сорбитана), и продукты конденсации указанных неполных эфиров с этиленоксидом, такие как моноолеат полиоксиэтиленсорбитана. Эмульсии также могут содержать подслащивающие вещества, корригенты и консерванты.
Сиропы и эликсиры могут быть получены с подслащивающими веществами, такими как глицерин, пропиленгликоль, сорбит, аспартам или сахароза; и также могут содержать средство, уменьшающее раздражение, консервант, корригент и/или краситель.
Фармацевтические композиции также могут находиться в форме стерильных водных или масляных суспензий для инъекции, которые можно получить согласно известным процедурам с использованием одного или нескольких соответствующих диспергирующих или смачивающих веществ и суспендирующих веществ, которые упомянуты выше. Для парентеральных композиций носитель, как правило, будет включать стерильную воду, водный раствор хлорида натрия, 1,3-бутандиол или любой другой подходящий нетоксичный парентерально приемлемый разбавитель или растворитель. Могут быть включены другие ингредиенты, которые способствуют диспергированию. Конечно, когда необходимо использовать стерильную воду и сохранять стерильность, композиции и носители также должны быть стерильными. Также можно получить суспензии для инъекций, и в таком случае могут быть использованы соответствующие жидкие носители, суспендирующие вещества и т.п.
Композиции-суппозитории можно получить путем смешивания активного ингредиента с подходящим не вызывающим раздражение эксципиентом, который является твердым при обычных температурах, но жидким при ректальной температуре, и поэтому будет плавиться в прямой кишке с высвобождением лекарственного средства. Подходящие эксципиенты включают, например, масло какао и полиэтиленгликоли.
Местные композиции, такие как кремы, мази, гели и водные или масляные растворы или суспензии, вообще можно получить, объединяя активный ингредиент с обычной приемлемой местно средой или разбавителем с использованием обычных процедур, хорошо известных в технике.
Композиции для введения инсуффляцией могут находиться в форме тонко измельченного порошка, содержащего частицы среднего диаметра, например, 30 микрон или меньше, причем сам порошок включает активный ингредиент или один или разбавленный одним или несколькими физиологически приемлемыми носителями, такими как лактоза. Порошок для инсуффляции также обычно сохраняют в капсуле, содержащей, например, 1-50 мг активного ингредиента, для использования с устройством для турбоингаляции, таким, какое используют для инсуффляции известного средства кромогликата натрия.
Композиции для введения ингаляцией могут находиться в форме обычного аэрозоля под давлением, приспособленного для диспергирования активного ингредиента или в виде аэрозоля, содержащего мелко измельченное твердое вещество, или капель жидкости. Могут использоваться обычные аэрозольные пропелленты, такие как летучие фторированные углеводороды или углеводороды, и аэрозольное устройство обычно приспособлено для диспергирования отмеренного количества активного ингредиента.
Композиции для трансдермального введения могут находиться в форме трансдермальных кожных пэтчей, которые хорошо известны специалистам в данной области техники. Другие системы доставки могут включать системы высвобождения со временем, отсроченным высвобождением или пролонгированным высвобождением. С такими системами можно избежать повторных введений соединений, что более удобно для субъекта и лечащего врача. Многие системы доставки с высвобождением доступны и известны специалистам в данной области техники. Они включают системы на основе полимеров, таких как полилактидгликолид, сополиоксалаты, поликапролактоны, полиэфирамиды, полиортоэфиры, полиоксимасляная кислота и полиангидриды. Микрокапсулы из вышеуказанных полимеров, содержащие лекарственные средства, описаны, например, в патенте США № 5075109. Системы доставки также включают неполимерные системы, которые представляют собой липиды, включая стерины, такие как холестерин, эфиры холестерина и жирных кислот, или нейтральные жиры, такие как моно-, ди- и триглицериды; системы высвобождения на основе гидрогелей; системы на основе силастиков; системы на основе пептидов; покрытия из воска; таблетки, прессованные с использованием обычных связующих веществ и эксципиентов; частично слившиеся имплантаты и подобные системы. Конкретные примеры включают, но не ограничиваются указанным, (а) эрозионные системы, в которых средство по изобретению содержится в форме, заключенной в матрицу, такие, как системы, описанные в патентах США №№ 4452775, 4675189 и 5736152, и (b) диффузные системы, в которых активный компонент выходит из полимера с регулируемой скоростью, такие, как описанные в патентах США №№ 3854480, 5133974 и 5407686. Кроме того, можно использовать насосные механические системы доставки, из которых некоторые адаптированы для имплантации.
Композиции можно вводить в форме раствора, например, водного или изотонического солевого, забуференного или незабуференного, или в виде суспензии, в виде капель или в виде спрея для интраназального введения. Предпочтительно, такие растворы или суспензии являются изотоничными относительно носовых выделений и с примерно тем же рН, колеблющемся, например, от примерно рН 4,0 до примерно рН 7,4 или от рН 6,0 до рН 7,0. Буферные растворы должны быть физиологически совместимыми и включают, просто для примера, фосфатные буферы. Например, характерное назальное противоотечное средство описано как забуференное до рН примерно 6,2 (Remington's Pharmaceutical Sciences, Ed. by Arthur Osol, p.1445 (1980)). Конечно, рядовой специалист может легко определить подходящее содержание физиологического раствора и рН для безвредного водного носителя для назального введения.
Другие неограничительные примеры интраназальных лекарственных форм, содержащих композицию, включают назальные гели, кремы, пасты или мази с вязкостью, например, от примерно 10 до примерно 3000 сП или от примерно 2500 до 6500 сП или выше, которые могут обеспечить более продолжительный контакт с поверхностями слизистой оболочки носовой полости. Такие вязкие препараты с носителями могут быть на основе, просто как пример, полимерных носителей, таких как алкилцеллюлозы, и/или других биосовместимых носителей высокой вязкости, хорошо известных в технике (см., например, Remington's, цитировано выше). Носителем, содержащим композицию, также может быть пропитана ткань, такая как марля, которую можно применить к поверхностям слизистой оболочки носовой полости для того, чтобы создать возможность активным веществам в изолированной фракции проникать в слизистую.
Другие ингредиенты, такие как известные в технике консерванты, красители, смазывающие вещества или вязкие минеральные или растительные масла, отдушки, природные или синтетические растительные экстракты, такие как ароматические масла, и влагоудерживающие вещества и усилители вязкости, такие как, например, глицерин, также могут быть включены для придания препарату дополнительной вязкости, свойства удерживать влагу и приятной текстуры и запаха.
Кроме того, для назального введения композиции в виде растворов или суспензий в технике доступны различные устройства для получения капель, капелек и спреев. Например, растворы, включающие изолированную фракцию, можно вводить в носовые проходы с помощью простой капельницы (или пипетки), которая включает стеклянную, пластиковую или металлическую распределяющую трубку, из которой содержимое выталкивается капля за каплей под давлением воздуха, обеспеченного сжатием пальцами, например, эластичного резинового шарика, присоединенного к одному концу. Мелкие капельки и спреи можно получить с помощью ручного или электрического нагнетательного распределяющего устройства или баллона под давлением, что хорошо известно в технике, например, созданных для вдувания смеси воздуха и мелких капель в носовые проходы.
Количество соединения по данному изобретению, которое объединяют с одним или несколькими эксципиентами для получения отдельной лекарственной формы, будет неизбежно изменяться в зависимости от субъекта, которого лечат, тяжести расстройства или состояния, способа введения, размещения соединения и предписания лечащего врача. Однако эффективная дозировка находится в интервале от примерно 0,001 до примерно 100 мг на кг массы тела в сутки, например, от примерно 0,05 до примерно 35 мг/кг/сутки, в одной или раздельных дозах. Например, дозировка составляет от примерно 0,0005 до примерно 2,5 г/сутки. Например, дозировка составляет от примерно 0,0005 до примерно 1 г/сутки в единой или раздельных дозировках. В некоторых случаях уровни дозировки ниже нижнего предела вышеуказанного интервала могут быть более, чем адекватными, в то время как в других случаях могут использоваться гораздо большие дозы без проявления вредного побочного действия, при условии, что такие большие дозы сначала разделят на несколько мелких доз для введения в течение суток.
Для дополнительной информации о способах введения и схемах лечения см. главу 25.3 в томе V Comprehensive Medicinal Chemistry (Corwin Hansch; Chairman of Editorial Board), Pergamon Press, 1990, включенной в данное описание в качестве ссылки.
Размер дозы соединения по изобретению для терапевтических или профилактических целей естественно будет изменяться в соответствии с природой и тяжестью состояний, возрастом и полом животного или пациента и способом введения, согласно хорошо известным принципам медицины. Следует иметь в виду, что конкретный уровень дозировки и частота дозировки для любого определенного субъекта может изменяться и будет зависеть от множества факторов, включая активность определенного соединения, вид, возраст, массу тела, общее состояние здоровья, пол и питание субъекта, способа и времени введения, скорости экскреции, комбинации лекарственных средств и тяжести определенного состояния, но тем не менее, могут быть повседневно определены специалистом в данной области техники.
В некоторых аспектах соединение по изобретению или его соль вводят субъекту в комбинации (например, в одной и той же композиции или в отдельных композициях) с другим терапевтическим средством («комбинированная терапия»). Соединение по изобретению вводят в смеси с другим терапевтическим средством или вводят в отдельной композиции. При введении в отдельной композиции соединение по изобретению и другое терапевтическое средство вводят, по существу, одновременно или последовательно. В одном аспекте соединение по изобретению вводят субъекту в комбинации с другим терапевтическим средством для лечения состояния или заболевания. В одном аспекте соединение по изобретению вводят субъекту в комбинации с другим терапевтическим средством для предупреждения состояния или заболевания. В одном аспекте соединение по изобретению вводят субъекту в комбинации с вакциной для предупреждения состояния или заболевания. В одном аспекте соединение по изобретению вводят субъекту в комбинации с вакциной против инфекционного заболевания. В одном аспекте соединение по изобретению вводят субъекту в комбинации с противораковой вакциной.
Соединение по изобретению может применяться в качестве адъюванта вакцины для применения в сочетании с любым материалом, который вызывает или гуморально- и/или клеточноопосредованную иммунную реакцию, таким как, например, иммуногены живых вирусов, бактерий или паразитов; инактивированные вирусные, полученные из опухолей, простейших, организмов, грибковые или бактериальные иммуногены, токсоиды, токсины; аутоантигены; полисахариды; белки; гликопротеины; пептиды; клеточные вакцины; ДНК-вакцины; рекомбинантные белки и т.п., для применения в сочетании с, например, вакцинами против БЦЖ, холеры, чумы, брюшного тифа, гепатита А, гепатита В, гепатита С, гриппа А, гриппа В, парагриппа, полиомиелита, бешенства, кори, свинки, краснухи, желтой лихорадки, столбняка, дифтерии, гриппа b, вызванного Hemophilus, туберкулеза, менингококковыми и пневмококковыми выкцинами, (вакцинами против) аденовируса, ВИЧ, ветряной оспы, цитомегаловируса, денге, лейкоза кошек, птичей чумы, HSV-1 и HSV-2, чумы свиней, японского энцефалита, респираторно-синцитиального вируса, ротавируса, вируса папилломы, желтой лихорадки и болезни Альцгеймера.
Соединение по изобретению также может быть полезно для индивидуумов с аномальной иммунной функцией. Например, соединение по изобретению можно использовать для лечения или предупреждения инфекций, вызванных оппортунистическими микроорганизмами, и опухолей, которые имеют место после подавления клеточноопосредованного иммунитета у, например, пациентов после трансплантации, онкобольных и больных ВИЧ.
Такое комбинированное лечение может включать, кроме соединения по изобретению, обычную операцию или лучевую терапию или химиотерапию. Такая химиотерапия может включать одну или больше следующих категорий противоопухолевых средств: (i) антипролиферативные/антинеопластические лекарственные средства и их комбинации; (ii) цитостатические средства; (iii) средства, которые ингибируют инвазию раковых клеток; (iv) ингибиторы функции факторов роста; (v) антиангиогенные средства; (vi) сосудоповреждающие средства; (vii) антисмысловая терапия; (viii) методы генной терапии; (ix) интерферон и (х) методы иммунотерапии.
Терапевтические средства для лечения или предупреждения респираторных заболеваний, которые можно вводить субъекту в комбинации с соединением по изобретению, включают, но не ограничиваются указанным, бета-адренергические средства, которые включают бронходилататоры, в том числе альбутерол, сульфат изопротеренола, сульфат метапротеренола, сульфат тербуталина, ацетат пирбутерола и сагнетерол формоторол; стероиды, в том числе дипропионат беклометазона, флунизолид, флутиказон, будезонид и триамцинолонацетонид. Противовоспалительные лекарственные средства, используемые в связи с лечением и предупреждением респираторных заболеваний, включают стероиды, такие как дипропионат беклометазона, триамцинолонацетонид, флунизолид и флутиказон. Другие противовоспалительные лекарственные средства включают кромогликаты, такие как кромолиннатрий. Другие противовоспалительные лекарственные средства, которые можно квалифицировать как бронходилататоры, включают антихолинергические средства, в том числе бромид ипратропия. Антигистаминные средства включают, но не ограничиваются перечисленным, дифенгидрамин, карбиноксамин, клемастин, дименгидринат, приламин, трипеленнамин, хлорфенирамин, бромфенирамин, гидроксизин, циклизин, меклизин, хлорциклизин, прометазин, доксиламин, лоратадин и терфенадин. Определенные антигистаминные средства включают риноласт (астелин®), кларатин (кларитин®), кларатин D (кларитин D®), тельфаст (аллегра®), циртек® и беконаз.
В некоторых воплощениях соединение по изобретению вводят как комбинированную терапию с интерфероном-гамма (IFN-гамма), кортикостероидом, таким как преднизон, преднизолон, метилпреднизолон, гидрокортизон, кортизон, дексаметазон, бетаметазон и т.д. или их сочетание, для лечения или предупреждения интерстициальной болезни легких, например, идиопатического фиброза легких.
В некоторых воплощениях соединение по изобретению вводят в комбинированной терапии с известным терапевтическим средством, используемым при лечении кистозного фиброза («CF»). Терапевтические средства, используемые при лечении CF, включают, но не ограничиваются указанным, антибиотики, противовоспалительные средства, ДНКазы (например, рекомбинантную человеческую ДНКазу, пульмозим, дорназ-альфа); миколитические средства (например, N-ацетилцистеин, мукомистТМ, мукозилТМ); противоотечные средства; бронходилататоры (например, теофидрин, бромид ипатропия), и подобные средства.
В некоторых воплощениях соединение по изобретению вводят профилактически для предупреждения сердечно-сосудистой болезни, например, атеросклероза.
В другом воплощении изобретение относится к изготовляемому изделию или «набору», содержащему материал, применимый для лечения или предупреждения заболеваний, описанных выше.
В одном воплощении набор включает контейнер, включающий композицию с соединением по изобретению или его фармацевтически приемлемой солью. В одном воплощении изобретение относится к набору для лечения или предупреждения TLR7- и/или TLR8-опосредуемого расстройства. В другом воплощении изобретение относится к набору для лечения состояния или расстройства, которое поддается лечению путем селективной модуляции у субъекта иммунной системы. Набор также может включать ярлык или вкладыш на или связанный с контейнером. Подходящие контейнеры включают, например, бутылки, флаконы, шприцы, блистеры и т.д. Контейнер содержит соединение по изобретению или фармацевтический препарат на его основе в количестве, эффективном для лечения или предупреждения состояния, и может иметь стерильное входное отверстие (например, контейнер может представлять собой мешок для внутривенного раствора или флакон с пробкой, которая протыкается гиподермической инъекционной иглой). Метка или вкладыш в упаковку показывает, что композицию используют для лечения или предупреждения выбранного состояния. В одном воплощении метка или вкладыш в упаковку показывает, что композицию, включающую соединение по изобретению, можно использовать, например, для лечения или предупреждения расстройства, поддающегося лечению путем модуляции TLR7- и/или TLR8-опосредуемых клеточных активностей. Ярлык или вкладыш в упаковку также может указывать, что композицию можно использовать для лечения или предупреждения других расстройств. С другой стороны, набор также может включать второй контейнер, включающий фармацевтически приемлемый буфер, такой как бактериостатическая вода для инъекции (BWFI), забуференный фосфатом физиологический раствор, раствор Рингера и раствор декстрозы. Он также может включать другие материалы, нужные с коммерческой или потребительской точки зрения, в том числе другие буферы, разбавители, фильтры, иглы и шприцы.
Набор также может включать указания по введению соединения по изобретению и второго фармацевтического препарата, если он присутствует. Например, если набор включает первую композицию, включающую соединение по изобретению, и второй фармацевтический препарат, набор также может включать указания для пациента, нуждающегося в этом, по одновременному, последовательному или раздельному введению первой и второй фармацевтических композиций.
В другом воплощении наборы подходят для доставки твердых пероральных форм соединения по изобретению, таких как таблетки или капсулы. Такой набор включает, например несколько стандартных лекарственных форм. Такие наборы включают карту с дозировками, установленными в порядке их предполагаемого использования. Примером такого набора является «блистерная упаковка». Блистерные упаковки хорошо известны в упаковочной индустрии и широко используются для упаковки фармацевтических стандартных лекарственных форм. При желании может быть представлена памятка, например, в форме чисел, букв или других маркировок, или с календарем-вкладышем с обозначением дней в схеме лечения, в которые можно вводить дозировки.
Согласно одному воплощению, набор может включать (а) первый контейнер с соединением по изобретению, содержащемся в нем; и, необязательно, (b) второй контейнер со вторым фармацевтическим препаратом, содержащемся в нем, при этом второй фармацевтический препарат включает второе соединение, которое может быть эффективным при лечении или предупреждении состояния или расстройства путем модуляции TLR7- и/или TLR8-опосредуемых клеточных активностей. С другой стороны или кроме того, набор также может содержать третий контейнер, включающий фармацевтически приемлемый буфер, такой как бактериостатическая вода для инъекции (BWFI), забуференный фосфатом физиологический раствор, раствор Рингера и раствор декстрозы. Он также может включать другие материалы, нужные с коммерческой и потребительской точки зрения, в том числе другие буферы, разбавители, фильтры, иглы и шприцы.
В некоторых других воплощениях, в которых набор включает фармацевтический препарат с соединением по изобретению и второй препарат, включающий второе терапевтическое средство, набор может включать контейнер, содержащий отдельные препараты, такой как разделенный пузырек или разделенный пакет из фольги; однако отдельные композиции также могут содержаться в едином неразделенном контейнере. Типично набор включает указания по введению отдельных компонентов. Форма набора особенно выгодна, когда отдельные компоненты вводят в различных лекарственных формах (например, пероральной и парентеральной), вводят с различными интервалами между дозами или когда желательно титрование отдельных компонентов по предписанию врача.
Активность соединений можно оценить согласно процедурам, описанным, например, в Gorden et al., Journal of Immunoligy, 177, pp. 8164-8170 (2006); и в Zhu et al., Molecular Immunoligy, vol. 45(11), pp. 3238-3242 (2008).
Величины МС50 для активности TLR8 являются, например, такими, какие указаны ниже.
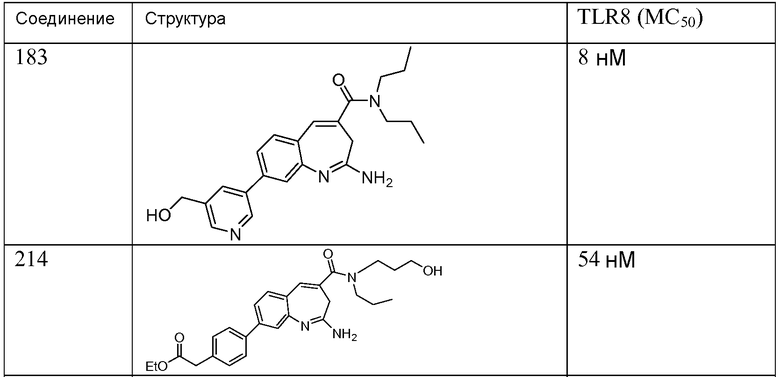
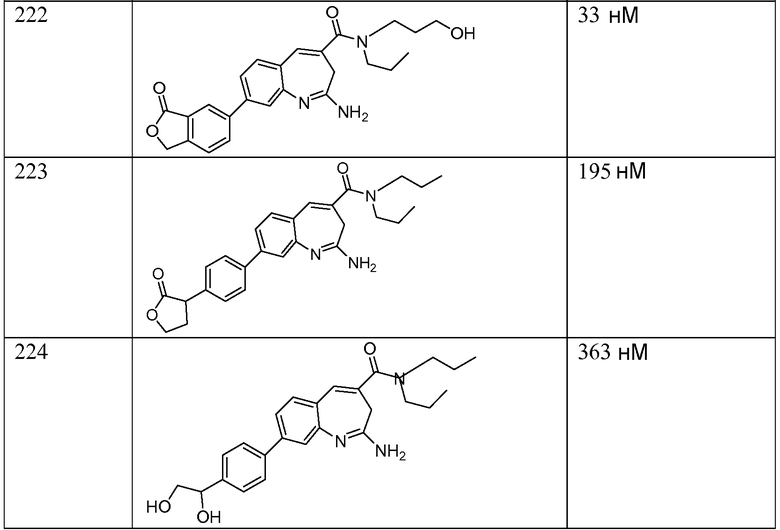
Величины МС50 для активности TLR7 являются, например, такими, какие указаны ниже.
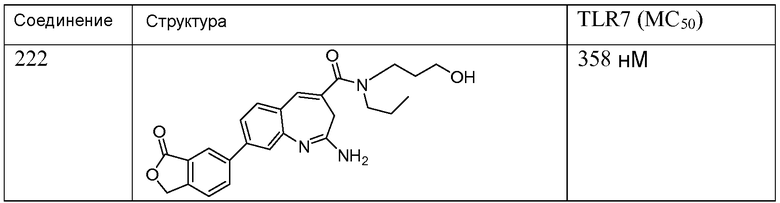
ПРИМЕРЫ
Для того, чтобы пояснить изобретение, включены примеры, приведенные далее. Однако следует иметь в виду, что указанные примеры не ограничивают изобретение и представляют только предложение способа практического осуществления изобретения. Специалистам в данной области техники будет понятно, что описанные химические реакции можно легко адаптировать для получения ряда других соединений по изобретению, и также предполагается, что другие способы получения соединений по данному изобретению входят в объем изобретения. Например, синтез соединений по изобретению, не приведенных в качестве примеров, можно успешно осуществить путем модификаций, очевидных для специалистов в данной области техники, например, путем соответствующей защиты мешающих групп, путем использования других подходящих реагентов, известных в технике, иных, чем описанные реагенты, и/или путем осуществления обычных модификаций условий реакций.
В примерах, описанных ниже, если не указано иное, все температуры представлены в градусах по Цельсию. Реагенты закупают у коммерческих поставщиков, таких как Aldrich Chemical Company, Lancaster, Acros, TCI, Alfa Aesar или Maybridge, и используют без дополнительной очистки, если не указано иное.
В примерах, описанных ниже, термин «Пример ###» относится к соединению ###. Например, пример 113 относится к соединению 113 и/или синтетическим процедурам, относящимся к соединению 113.
Реакции, представленные ниже, осуществляют, как правило, при положительном давлении азота или аргона или с трубкой с высушивающим веществом (если не указано иное) в безводных растворителях, и реакционные колбы типично снабжают резиновыми мембранами для ввода субстратов и реагентов через шприц. Стеклянную посуду сушат в шкафу и/или при нагревании.
Реакции под воздействием микроволнового излучения выполняют в системе Biotage Initiator.
Колоночную хроматографию осуществляют на системе Biotage или на колонке Isolute Flash Si SPE (изготовитель Biotage АВ) с колонкой с силикагелем или на картридже с диоксидом кремния SepPak (Waters). Спектры 1Н ЯМР записывают на приборе Varian, работающем при 400 МГц и 376 МГц, соответственно. Спектры 1Н ЯМР получают в растворах в CDCl3 или d6-ДМСО (приводятся в м.д.) с использованием хлороформа (7,26 м.д.) или тетраметилсилана (0 м.д.) в качестве эталонов. Когда приводятся мультиплетности пиков, используются следующие аббревиатуры: с (синглет), д (дублет), т (триплет), к (квартет), уш (уширенный), дд (дублет дублетов), дт (дублет триплетов), м (мультиплет).
Пример 1
Синтетические процедуры
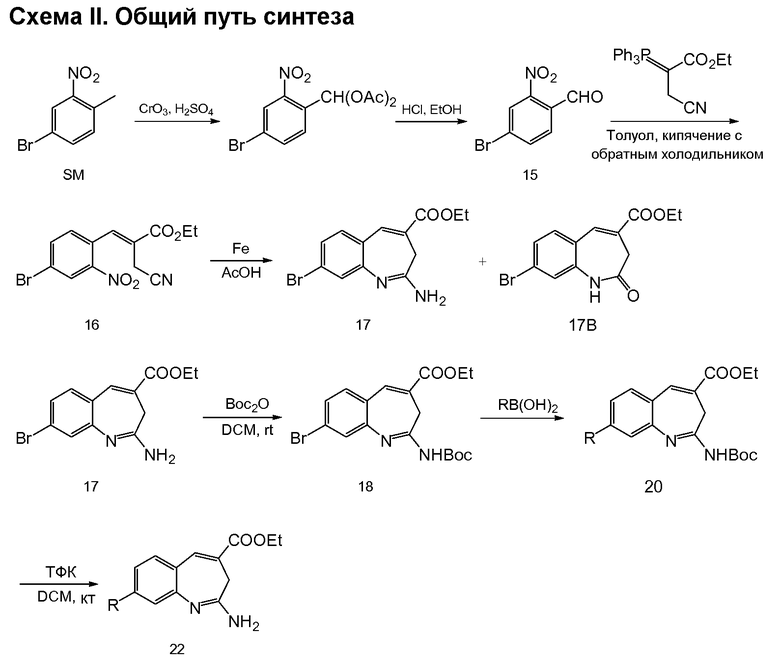
1. Синтез соединения 15
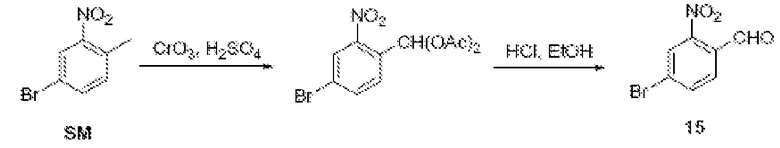
В трехгорлую колбу, снабженную механической мешалкой, капельной воронкой и термометром, размещенную на бане со смесью льда с солью, помещают 400 мл уксусного ангидрида и 50 г (0,23 моль) 4-бром-1-метил-2-нитробензола. К полученному раствору постепенно при перемешивании добавляют 54 мл концентрированной серной кислоты. Когда смесь охладится до 0°С, постепенно при перемешивании добавляют 64 г триоксида хрома в 360 мл уксусного ангидрида с такой скоростью, чтобы температура не превышала 10°С, и после того, как добавление завершится, продолжают перемешивание в течение 2 часов при 5-10°С на бане со смесью воды со льдом. Содержимое колбы выливают в смесь воды со льдом. Твердое вещество отфильтровывают и промывают водой до тех пор, пока промывная вода не станет бесцветной. Продукт суспендируют в 300 мл 2% водного раствора карбоната натрия и перемешивают. После перемешивания твердое вещество отфильтровывают, промывают водой и сушат.
Суспензию диацетата в смеси 272 мл концентрированной соляной кислоты, 250 мл воды и 80 мл этанола перемешивают и кипятят с обратным холодильником в течение 45 минут. Затем смесь охлаждают до комнатной температуры (RT), и твердое вещество отфильтровывают и промывают водой. Сырой продукт реакции очищают на колонке (22 г, 42%).
2. Синтез соединения 16
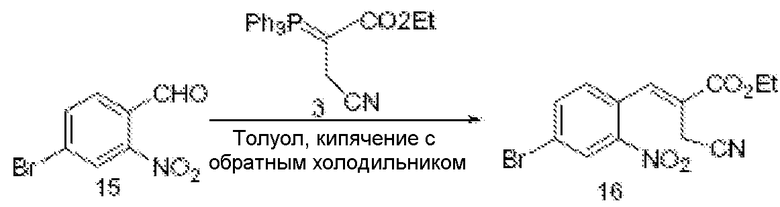
Смесь альдегида (0,73 г, 3,17 ммоль) и илида (1,42 г, 3,65 ммоль) в толуоле (8 мл) осторожно кипятят с обратным холодильником в течение 2,5 час. Реакционную смесь охлаждают до комнатной температуры и концентрируют при пониженном давлении, и получают сырой материал, который используют непосредственно без дополнительной очистки.
3. Синтез соединения 17 и 17В
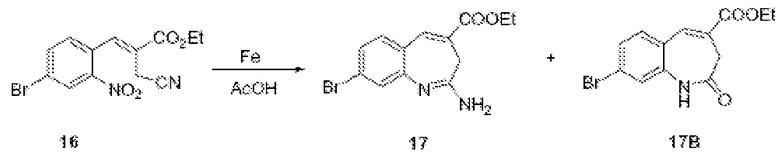
К раствору неочищенного нитрила в АсОН (25 мл) при комнатной температуре добавляют железо (1,15 г, 20,61 ммоль). Полученную смесь греют при 85°С в течение 4 час. Реакционную смесь охлаждают до комнатной температуры и разбавляют CH2Cl2 (8 мл). Полученную смесь фильтруют, и твердое вещество промывают CH2Cl2. Фильтрат концентрируют при пониженном давлении, и получают вязкое масло. К сырому материалу добавляют CH2Cl2 (8 мл). Постепенно при перемешивании добавляют водный раствор Na2CO3 и затем воду до тех пор, пока не получат рН=9-10. Твердое вещество отфильтровывают и промывают CH2Cl2. Органический слой отделяют. Водный слой экстрагируют CH2Cl2. Органический слой отделяют. Водный слой экстрагируют CH2Cl2. Объединенные органические слои промывают рассолом, сушат над Na2SO4, смесь концентрируют при пониженном давлении, получают сырой материал, который очищают колоночной флэш-хроматографией на силикагеле, и получают 0,329 г (33% за две стадии) нужного продукта, как показывает 1Н ЯМР.
4. Синтез соединения 18
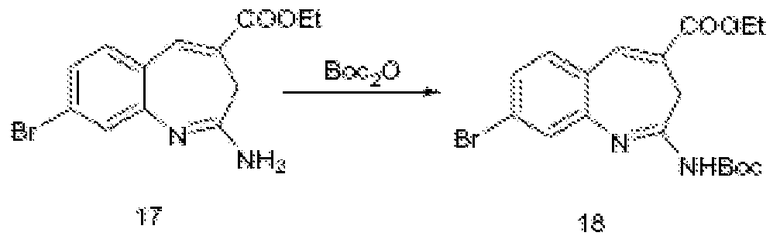
К бензазепину (2,34 г, 7,57 ммоль) в DCM (25 мл) при комнатной температуре добавляют Вос2О (2,06 г, 9,46 ммоль). Реакционную смесь перемешивают в течение 20 час. Полученную смесь последовательно промывают насыщенным водным раствором NaHCO3 и рассолом. Органический слой отделяют, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении, получают сырой продукт реакции, который очищают колоночной флэш-хроматографией на силикагеле (10% EtOAc в гексане), и получают 1,64 г (52,9%) нужного продукта.
5. Синтез соединений по изобретению
Пример 113
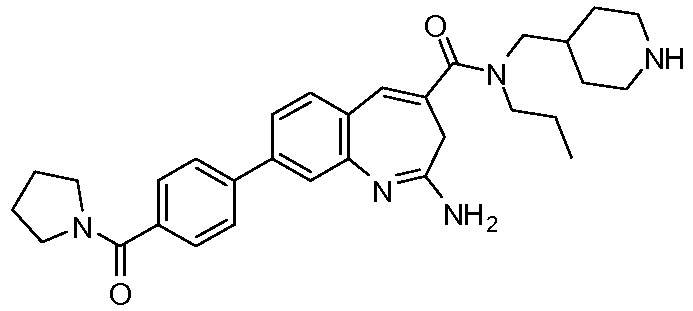
(1Е,4Е)-2-Амино-N-(пиперидин-4-илметил)-N-пропил-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоксамид
Стадия А. Получение трет-бутил-4-((бензилоксикарбониламино)метил)пиперидин-1-карбоксилата. трет-Бутил-4-(аминометил)пиперидин-1-карбоксилат (0,611 г, 2,851 ммоль) и диизопропилэтиламин (0,479 г, 3,706 ммоль) растворяют в 30 мл сухого дихлорметана. К полученной смеси добавляют бензилхлорформиат (0,552 г, 3,136 ммоль), и смесь перемешивают при комнатной температуре в течение 2 часов. Затем смесь разбавляют 50 мл дихлорметана, промывают один раз 1 N соляной кислотой, один раз насыщенным раствором бикарбоната натрия, сушат над сульфатом натрия и концентрируют при пониженном давлении до 950 мг (96%) названного в заголовке соединения, которое используют непосредственно без дополнительной очистки.
Стадия В. Получение трет-бутил-4-(((бензилоксикарбонил)(пропил)амино)метил)пиперидин-1-карбоксилата. трет-Бутил-4-((бензилоксикарбониламино)метил)пиперидин-1-карбоксилат (0,950 г, 2,726 ммоль) растворяют в сухом ДМФА (25 мл). К полученной смеси добавляют гидрид натрия (0,164 г, 4,090 ммоль, 60% дисперсия в минеральном масле), и реакционную смесь перемешивают при комнатной температуре в течение 30 минут. Затем добавляют пропилиодид (0,695 г, 4,090 ммоль), смесь перемешивают при комнатной температуре в течение 16 часов, затем разбавляют рассолом (200 мл), дважды экстрагируют EtOAc, и экстракты дважды промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Полученное масло очищают флэш-хроматографией (картридж, 100 г Biotage Snap, 30% EtOAc/гексан), и получают 0,280 г (26%) названного в заголовке соединения.
Стадия С. Получение трет-бутил-4-((пропиламино)метил)пиперидин-1-карбоксилата. К раствору трет-бутил-4-(((бензилоксикарбонил)(пропил)амино)метил)пиперидин-1-карбоксилата (0,280 г, 0,717 ммоль) в 7 мл метанола добавляют гидроксид палладия(II) (0,200 г, 20 мас.% Pd(OH)2 на угле, тип Degussa). Полученную смесь гидрируют водородом из баллона в течение 1,5 часов, затем фильтруют через фильтровальную бумагу GF/F, и фильтрат концентрируют. Получают 0,169 г (92%) названного в заголовке соединения, которое используют без дополнительной очистки.
Стадия D. Получение (1Е,4Е)-2-амино-N-(пиперидин-4-илметил)-N-пропил-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоксамида. Названное в заголовке соединение получают указанными процедурами с использованием (1Е,4Е)-2-(трет-бутоксикарбониламино)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоновой кислоты и трет-бутил-4-((пропиламино)метил)пиперидин-1-карбоксилата. Получение трет-бутил-(1Е,4Е)-4-(пропилкарбамоил)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбамата. Смесь (1Е,4Е)-2-(трет-бутоксикарбониламино)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоновой кислоты (200 мг, 0,42 ммоль), HOBt (114 мг, 0,84 ммоль) и EDCI (161 мг, 0,84 ммоль) в ДМФА (5 мл) перемешивают в течение 1 часа при комнатной температуре. К полученной смеси при комнатной температуре добавляют триэтиламин (0,12 мл, 0,84 ммоль) и пропан-1-амин (0,043 мл, 0,53 ммоль). Полученный раствор перемешивают еще в течение 2 час. Реакционную смесь разбавляют EtOAc (5 мл) и промывают насыщенным водным раствором NH4Cl. Водный слой отделяют и экстрагируют EtOAc (3×5 мл). Объединенные органические слои промывают рассолом (5 мл), насыщенным водным раствором NaHCO3 (5 мл) и рассолом (5 мл). Органический слой сушат над MgSO4, фильтруют и концентрируют при пониженном давлении, и получают сырой трет-бутил-(1Е,4Е)-4-(пропилкарбамоил)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-2-илкарбамат, который используют непосредственно без дополнительной очистки. Получение (1Е,4Е)-2-амино-N-пропил-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоксамида. К раствору трет-бутил-(1Е,4Е)-4-(пропилкарбамоил)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-2-илкарбамата (450 мг, 0,87 ммоль) в CH2Cl2 (5 мл) при 0°С добавляют 2,2,2-трифторуксусную кислоту (1,36 мл, 17,4 ммоль). Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 2 часов. Реакционную смесь концентрируют при пониженном давлении, и получают сырой материал, который снова разбавляют CH2Cl2 (10 мл) и насыщенным водным раствором NaHCO3 (15 мл). Полученную смесь перемешивают в течение 30 мин при комнатной температуре. Водный слой отделяют и экстрагируют CH2Cl2 (1×10 мл). Объединенные органические слои промывают насыщенным водным раствором NaHCO3 (2×10 мл) и рассолом (1×10 мл), сушат над MgSO4, фильтруют и концентрируют при пониженном давлении, и получают снова сырой материал, который очищают колоночной флэш-хроматографией на силикагеле (градиент 1-5% МеОН в CH2Cl2). m/z (APCI-полож.) М+1=514,3.
Соединение следующего примера 116 получают указанными процедурами с использованием (1Е,4Е)-2-(трет-бутоксикарбониламино)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоновой кислоты и соответствующих аминов (2-метил-1-(пропиламино)пропан-2-ол получают процедурой, описанной в J. Am. Chem. Soc., 1939, 61, 3562) или гидроксиламина. Получение трет-бутил-(1Е,4Е)-4-(пропилкарбамоил)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-2-илкарбамата. Смесь (1Е,4Е)-2-(трет-бутоксикарбониламино)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоновой кислоты (200 мг, 0,42 ммоль), HOBt (114 мг, 0,84 ммоль) и EDCI (161 мг, 0,84 ммоль) в ДМФА (5 мл) перемешивают в течение 1 часа при комнатной температуре. К полученной смеси при комнатной температуре добавляют триэтиламин (0,12 мл, 0,84 ммоль) и пропан-1-амин (0,043 мл, 0,53 ммоль). Полученный раствор перемешивают еще в течение 2 час. Реакционную смесь разбавляют EtOAc (5 мл) и промывают насыщенным водным раствором NH4Cl. Водный слой отделяют и экстрагируют EtOAc (3×5 мл). Объединенные органические слои промывают рассолом (5 мл), насыщенным водным раствором NaHCO3 (5 мл) и рассолом (5 мл). Органический слой сушат над MgSO4, фильтруют и концентрируют при пониженном давлении, и получают сырой трет-бутил-(1Е,4Е)-4-(пропилкарбамоил)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-2-илкарбамат, который используют непосредственно без дополнительной очистки. Получение (1Е,4Е)-2-амино-N-пропил-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоксамида. К раствору трет-бутил-(1Е,4Е)-4-(пропилкарбамоил)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-2-илкарбамата (450 мг, 0,87 ммоль) в CH2Cl2 (5 мл) при 0°С добавляют 2,2,2-трифторуксусную кислоту (1,36 мл, 17,4 ммоль). Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 2 часов. Реакционную смесь концентрируют при пониженном давлении, и получают сырой материал, который снова разбавляют CH2Cl2 (10 мл) и насыщенным водным раствором NaHCO3 (15 мл). Полученную смесь перемешивают в течение 30 мин при комнатной температуре. Водный слой отделяют и экстрагируют CH2Cl2 (1×10 мл). Объединенные органические слои промывают насыщенным водным раствором NaHCO3 (2×10 мл) и рассолом (1×10 мл), сушат над MgSO4, фильтруют и концентрируют при пониженном давлении, и получают снова сырой материал, который очищают колоночной флэш-хроматографией на силикагеле (градиент 1 - 5% МеОН в CH2Cl2).
Пример 116
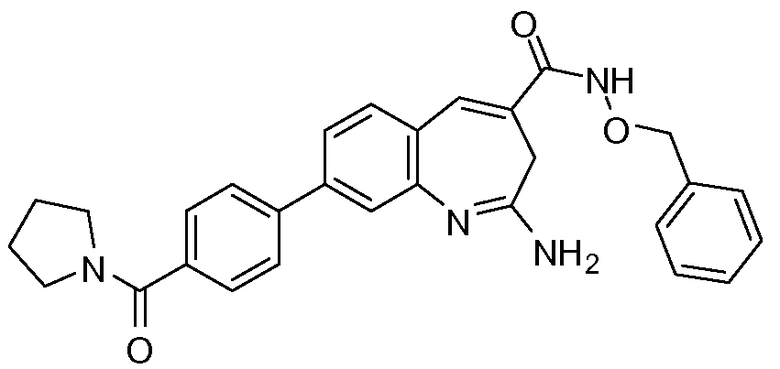
(1Е,4Е)-2-Амино-N-(бензилокси)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоксамид
1H-ЯМР (400 МГц, CDCl3) δ 11,59 (ушир.с, 1H), 7,74-7,78 (м, 2H), 7,61-7,65 (м, 2H), 7,33-7,53 (м, 8H), 4,91 (с, 2H), 3,40-3,53 (м, 4H), 3,03 (с, 2H), 1,80-1,92 (м, 4H); m/z (APCI-полож.) M+1=481,2.
Пример 118
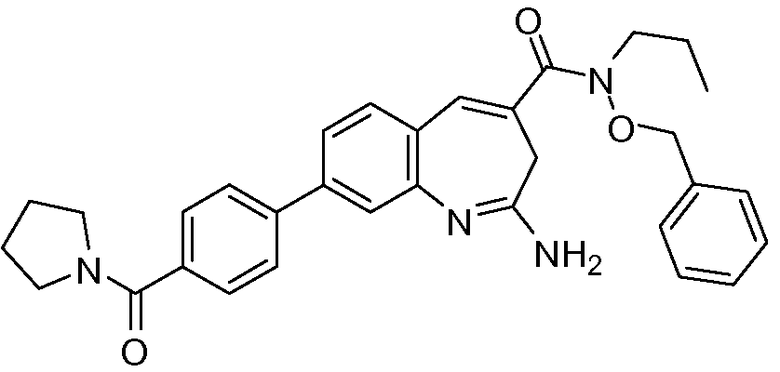
(1Е,4Е)-2-Амино-N-(бензилокси)-N-пропил-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоксамид
Стадия А. Получение О-бензил-N-пропилгидроксиламина. К раствору пропан-1-ола (6,25 мл, 83,2 ммоль) и 2,6-диметилпиридина (11,6 мл, 99,8 ммоль) в CH2Cl2 (500 мл) в атмосфере азота при -78°С добавляют по каплям трифторметансульфоновый ангидрид (14,0 мл, 83,2 ммоль). После перемешивания в течение 30 мин при -78°С добавляют по каплям раствор О-бензилгидроксиламина (10,7 мл, 91,5 ммоль) в CH2Cl2 (10 мл). Полученную смесь перемешивают при -78°С в течение 1 часа, затем нагревают до комнатной температуры и перемешивают еще в течение 2 час. Реакционную смесь разбавляют смесью воды со льдом (250 мл), и органический слой отделяют и промывают насыщенным водным раствором NaHCO3 (100 мл) и рассолом (100 мл). Водный слой снова экстрагируют EtOAc (1×200 мл). Объединенные органические слои сушат над MgSO4, фильтруют и концентрируют при пониженном давлении, и получают сырой материал, который очищают колоночной флэш-хроматографией на силикагеле (CH2Cl2). К фракции, содержащей продукт реакции и 2,6-диметилпиридин, добавляют 2 М водный раствор КОН (100 мл) и затем промывают МТВЕ. Затем водный слой доводят до рН ~5 1 М соляной кислотой, и затем экстрагируют МТВЕ (3×50 мл). Объединенные органические слои сушат над MgSO4, фильтруют и концентрируют при пониженном давлении, и получают 11,2 г (75%) О-бензил-N-пропилгидроксиламина.
Стадия В. Получение трет-бутил-(1Е,4Е)-4-(бензилокси(пропил)карбамоил)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-2-илкарбамата. Названное в заголовке соединение получают указанными процедурами с использованием (1Е,4Е)-2-(трет-бутоксикарбониламино)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоновой кислоты и О-бензил-N-пропилгидроксиламина. Получение трет-бутил-(1Е,4Е)-4-(пропилкарбамоил)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-2-илкарбамата. Смесь (1Е,4Е)-2-(трет-бутоксикарбониламино)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоновой кислоты (200 мг, 0,42 ммоль), HOBt (114 мг, 0,84 ммоль) и EDCI (161 мг, 0,84 ммоль) в ДМФА (5 мл) перемешивают в течение 1 часа при комнатной температуре. К полученной смеси при комнатной температуре добавляют триэтиламин (0,12 мл, 0,84 ммоль) и пропан-1-амин (0,043 мл, 0,53 ммоль). Полученный раствор перемешивают еще в течение 2 час. Реакционную смесь разбавляют EtOAc (5 мл) и промывают насыщенным водным раствором NH4Cl. Водный слой отделяют и экстрагируют EtOAc (3×5 мл). Объединенные органические слои промывают рассолом (5 мл), насыщенным водным раствором NaHCO3 (5 мл) и рассолом (5 мл). Органический слой сушат над MgSO4, фильтруют и концентрируют при пониженном давлении, и получают сырой трет-бутил-(1Е,4Е)-4-(пропилкарбамоил)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-2-илкарбамат, который используют непосредственно без дополнительной очистки. Получение (1Е,4Е)-2-амино-N-пропил-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоксамида. К раствору трет-бутил-(1Е,4Е)-4-(пропилкарбамоил)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-2-илкарбамата (450 мг, 0,87 ммоль) в CH2Cl2 (5 мл) при 0°С добавляют 2,2,2-трифторуксусную кислоту (1,36 мл, 17,4 ммоль). Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 2 часов. Реакционную смесь концентрируют при пониженном давлении, и получают сырой материал, который снова разбавляют CH2Cl2 (10 мл) и насыщенным водным раствором NaHCO3 (15 мл). Полученную смесь перемешивают в течение 30 мин при комнатной температуре. Водный слой отделяют и экстрагируют CH2Cl2 (1×10 мл). Объединенные органические слои промывают насыщенным водным раствором NaHCO3 (2×10 мл) и рассолом (1×10 мл), сушат над MgSO4, фильтруют и концентрируют при пониженном давлении, и получают снова сырой материал, который очищают колоночной флэш-хроматографией на силикагеле (градиент 1-5% МеОН в CH2Cl2). 1H-ЯМР (400 МГц, CDCl3) d 7,67-7,71 (м, 2H), 7,60-7,63 (м, 2H), 7,53-7,55 (м, 1H), 7,29-7,39 (м, 8H), 4,84 (с, 2H), 3,74-3,81 (м, 2H), 3,62-3,71 (м, 2H), 3,48-3,54 (м, 2H), 2,82 (с, 2H), 1,87-2,01 (м, 4H), 1,74-1,84 (м, 2H), 0,96-1,02 (м, 3H); m/z (APCI-полож.) M+1= 523,2.
Пример 148
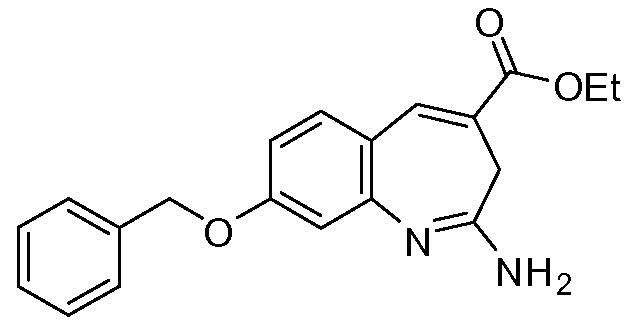
(1Е,4Е)-Этил-2-амино-8-(бензилокси)-3Н-бензо[b]азепин-4-карбоксилат
Названное в заголовке соединение получают указанными процедурами с использованием 4-(бензилокси)-1-метил-2-нитробензола. Получение (Е)-1-(4-бром-2-нитростирил)пирролидина. Раствор 4-бром-2-нитротолуола (100 г, 463 ммоль), пирролидина (46,2 мл, 565 ммоль) и диметилацеталя N,N-диметилформамида (75,6 мл, 565 ммоль) кипятят с обратным холодильником при 110°С в течение 4 часов. Реакционную смесь охлаждают до комнатной температуры и концентрируют при пониженном давлении, и получают сырой (Е)-1-(4-бром-2-нитростирил)пирролидин, который используют непосредственно без дополнительной очистки. Получение 4-бром-2-нитробензальдегида. К раствору периодата натрия (298 г, 1,40 моль) в смеси ТГФ-Н2О (4 л, 1:1) при 0°С добавляют (Е)-1-(4-бром-2-нитростирил)пирролидин (138 г, 464 ммоль). Смесь перемешивают в течение 15 час и затем фильтруют для удаления выпавшего в осадок твердого вещества. Водный слой фильтрата отделяют и экстрагируют EtOAc (4×200 мл). Объединенные органические слои промывают Н2О (2×200 мл). сушат над MgSO4, фильтруют и концентрируют при пониженном давлении, и получают сырой продукт реакции, который очищают колоночной флэш-хроматографией на силикагеле (5% EtOAc в гексане). Получение (Е)-этил-2-(цианометил)-3-(3-нитро-4'-(пирролидин-1-карбонил)бифенил-4-ил)акрилата. Смесь 3-нитро-4'-(пирролидин-1-карбонил)бифенил-4-карбальдегида (20,0 г, 61,7 ммоль) и α-цианометилкарбоэтоксиэтилидентрифенилфосфорана (26,3 г, 67,8 ммоль) в толуоле (200 мл) осторожно кипятят с обратным холодильником в течение 2,5 час. Реакционную смесь охлаждают до комнатной температуры и концентрируют при пониженном давлении, и получают сырой (Е)-этил-2-(цианометил)-3-(3-нитро-4'-(пирролидин-1-карбонил)бифенил-4-ил)акрилат, который используют непосредственно без дополнительной очистки. Получение (1Е,4Е)-этил-2-амино-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоксилата. К раствору неочищенного (Е)-этил-2-(цианометил)-3-(3-нитро-4'-(пирролидин-1-карбонил)бифенил-4-ил)акрилата в АсОН (650 мл) при комнатной температуре добавляют железо (29,1 г, 521 ммоль). Полученную смесь греют при 85°С в течение 4 час. Реакционную смесь охлаждают до комнатной температуры и разбавляют CH2Cl2 (250 мл). Твердое вещество отфильтровывают и промывают CH2Cl2 (200 мл). Фильтрат концентрируют при пониженном давлении, и получают сырой материал, который снова разбавляют CH2Cl2 (250 мл). К полученной смеси при энергичном перемешивании постепенно добавляют насыщенный водный раствор Na2CO3 (~330 мл) до тех пор, пока смесь не станет основной (рН ~9-10). Полученную смесь фильтруют, и остаток на фильтре промывают CH2Cl2 (~250 мл). Водный слой отделяют и экстрагируют CH2Cl2 (2×150 мл). Объединенные органические слои промывают рассолом, сушат над MgSO4 и фильтруют, и получают сырой материал, который разбавляют EtOAc (70 мл). Смесь выдерживают в течение 16 час при комнатной температуре. Суспензию фильтруют. Отфильтрованное твердое вещество промывают EtOAc (100 мл), и получают сырой продукт реакции, который промывают небольшим количеством CH2Cl2.
Пример 175
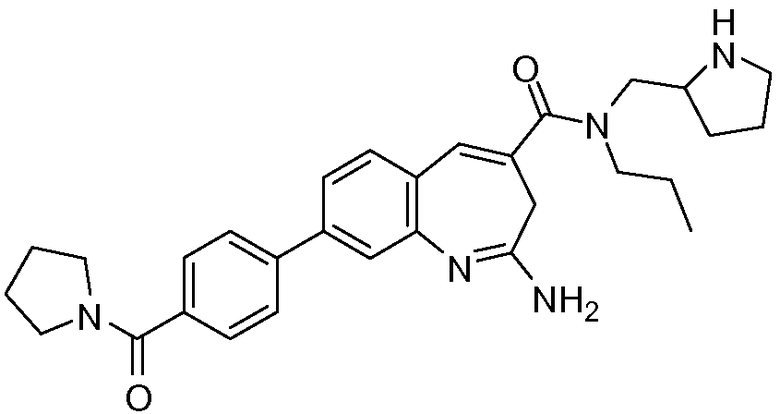
(1Е,4Е)-2-Амино-N-пропил-N-(пирролидин-2-илметил)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоксамид
Стадия А. Получение пирролидин-2-илметанола. К раствору DL-пролина (100 г, 869 ммоль) в МеОН (1500 мл) при 0°С постепенно добавляют SOCl2. Реакционную смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь концентрируют при пониженном давлении, и получают сырой материал, который снова растворяют в ТГФ (1700 мл). К полученной смеси при 0°С добавляют порциями LiAlH4 (132 г, 3,47 моль). Полученную смесь греют при 60°С в течение ночи. Избыток LiAlH4 гасят КОН. Реакционную смесь фильтруют, и твердое вещество промывают МеОН (1000 мл). Объединенные органические слои сушат, фильтруют и концентрируют при пониженном давлении, получают сырой материал, который очищают перегонкой, и получают 15,8 г (18%) пирролидин-2-илметанола.
Стадия В. Получение трет-бутил-2-(гидроксиметил)пирролидин-1-карбоксилата. К раствору пирролидин-2-илметанола (505 мг, 4,99 ммоль) и ТЕА (1,0 г, 9,9 ммоль) в CH2Cl2 (5 мл) добавляют раствор Вос2О (1,31 г, 6 ммоль) в CH2Cl2 (10 мл). Реакционную смесь перемешивают в течение 3 час при комнатной температуре. Реакцию гасят водой (100 мл), и смесь экстрагируют CH2Cl2. Органический слой промывают рассолом, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении, получают сырой материал, который очищают колоночной флэш-хроматографией на силикагеле (МеОН:CH2Cl2=1:100), и получают 14,2 г (48%) трет-бутил-2-(гидроксиметил)пирролидин-1-карбоксилата. ЖХМС ESI(+) детектирует m/z 202 (M+1).
Стадия С. Получение трет-бутил-2-((метилсульфонилокси)метил)пирролидин-1-карбоксилата. К раствору трет-бутил-2-(гидроксиметил)пирролидин-1-карбоксилата (1,0 г, 5,0 ммоль) и ТЕА (1,0 г, 9,9 ммоль) в CH2Cl2 (40 мл) постепенно добавляют MsCl (0,63 г, 5,5 ммоль). Реакционную смесь перемешивают в течение 30 мин при -20°С, и затем добавляют воду (60 мл). Водную фазу отделяют и экстрагируют CH2Cl2. Объединенные органические слои промывают рассолом, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении, и получают сырой трет-бутил-2-((метилсульфонилокси)метил)пирролидин-1-карбоксилат, который используют непосредственно без дополнительной очистки.
Стадия D. Получение трет-бутил-2-((пропиламино)метил)пирролидин-1-карбоксилата. Раствор трет-бутил-2-((метилсульфонилокси)метил)пирролидин-1-карбоксилата (5,0 г, 18 ммоль) и пропан-1-амин (20,0 г, 339 ммоль) в толуоле (50 мл) греют при 100°С в течение ночи. Реакционную смесь охлаждают до комнатной температуры и концентрируют при пониженном давлении, и получают сырой трет-бутил-2-((пропиламино)метил)пирролидин-1-карбоксилат, который используют непосредственно без дополнительной очистки. ЖХМС ESI(+) детектирует m/z 243 (M+1).
Стадия Е. Получение (1Е,4Е)-2-амино-N-пропил-N-(пирролидин-2-илметил)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоксамида. Названное в заголовке соединение получают указанными процедурами с использованием (1Е,4Е)-2-(трет-бутоксикарбониламино)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоновой кислоты и трет-бутил-2-((пропиламино)метил)пирролидин-1-карбоксилата. Получение трет-бутил-(1Е,4Е)-4-(пропилкарбамоил)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-2-илкарбамата. Смесь (1Е,4Е)-2-(трет-бутоксикарбониламино)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоновой кислоты (200 мг, 0,42 ммоль), HOBt (114 мг, 0,84 ммоль) и EDCI (161 мг, 0,84 ммоль) в ДМФА (5 мл) перемешивают в течение 1 часа при комнатной температуре. К полученной смеси при комнатной температуре добавляют триэтиламин (0,12 мл, 0,84 ммоль) и пропан-1-амин (0,043 мл, 0,53 ммоль). Полученный раствор перемешивают еще в течение 2 час. Реакционную смесь разбавляют EtOAc (5 мл) и промывают насыщенным водным раствором NH4Cl. Водный слой отделяют и экстрагируют EtOAc (3×5 мл). Объединенные органические слои промывают рассолом (5 мл), насыщенным водным раствором NaHCO3 (5 мл) и рассолом (5 мл). Органический слой сушат над MgSO4, фильтруют и концентрируют при пониженном давлении, и получают сырой трет-бутил-(1Е,4Е)-4-(пропилкарбамоил)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-2-илкарбамат, который используют непосредственно без дополнительной очистки. Получение (1Е,4Е)-2-амино-N-(2-амино-2-оксоэтил)-N-пропил-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоксамида. Через раствор неочищенного трет-бутил-(1Е,4Е)-4-((2-амино-2-оксоэтил)(пропил)карбамоил)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-2-илкарбамата в безводном CH2Cl2 (15 мл) при 0°С барботируют HCl (газ) в течение 4 час. Полученную смесь нагревают до комнатной температуры и перемешивают до тех пор, пока реакция не завершится. К полученной смеси при 0°С добавляют насыщенный раствор NaHCO3. Водный слой отделяют и экстрагируют CH2Cl2. Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении, и получают сырое соединение, которое очищают колоночной флэш-хроматографией на силикагеле (МеОН:CH2Cl2=1:50).
В данном случае продукт получают в виде соли HCl. МС APCI(+) детектирует m/z 536 (M+1). 1H-ЯМР (400 МГц, CDCl3) d 7,80 (с, 1H), 7,62-7,66 (м, 4H), 7,52 (д, 1H), 7,42 (д, 1H), 6,97 (с, 1H), 4,31 (ушир.с, 1H), 4,08 (ушир.с, 1H), 3,86 (ушир.с, 1H), 3,68 (т, 2H), 3,48 (ушир.с, 5H), 3,32 (м, 3H), 2,17 (ушир.с, 2H), 1,92-1,99 (м, 8H), 0,87 (т, 3H).
Пример 177
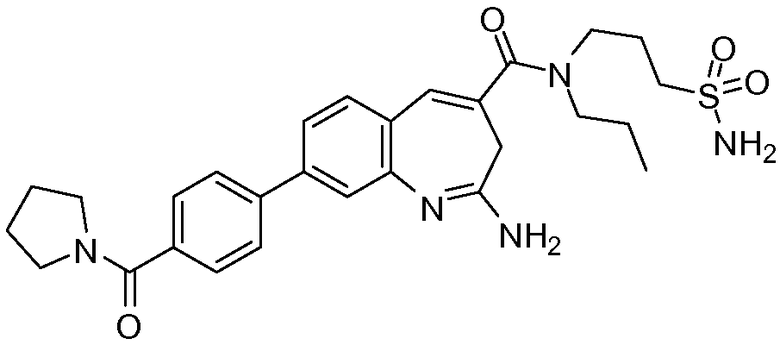
(1Е,4Е)-2-Амино-N-пропил-8-(4-(пирролидин-1-карбонил)фенил)-N-(3-сульфамоилпропил)-3Н-бензо[b]азепин-4-карбоксамид
МС APCI(+) детектирует m/z 538 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 7,65 (д, 2H), 7,59 (д, 2H), 7,45 (с, 1H), 7,32 (д, 1H), 7,28 (д, 1H), 6,80 (с, 1H), 3,66 (т, 2H), 3,55 (т, 2H), 3,49 (т, 2H), 3,42 (ушир.с, 2H), 3,14 (ушир.с, 2H), 2,80 (с, 2H), 2,14 (ушир.с, 2H), 1,87-1,99 (м, 4H), 1,59-1,64 (м, 2H), 0,87 (т, 3H).
Пример 183
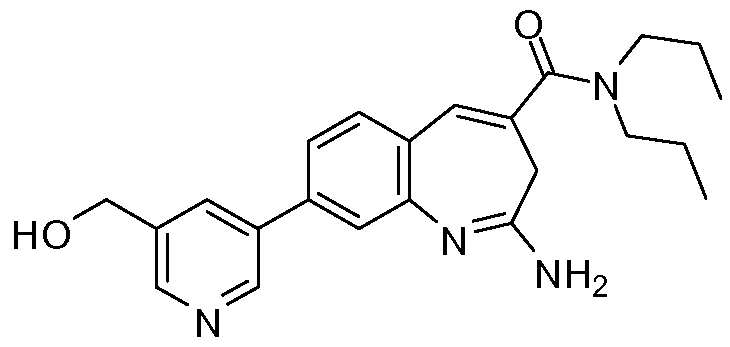
(1Е,4Е)-2-Амино-8-(5-(гидроксиметил)пиридин-3-ил)-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид
Названное в заголовке соединение получают указанными процедурами с использованием трет-бутил-(1Е,4Е)-8-бром-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-2-илкарбамата и 3-((трет-бутилдиметилсилилокси)метил)фенилбороновой кислоты. Получение 3-нитро-4'-(пирролидин-1-карбонил)бифенил-4-карбальдегида, за исключением того, что в данном случае в качестве основания используют Cs2CO3. К раствору 4-бром-2-нитробензальдегида (20,2 г, 87,9 ммоль), 4-(пирролидин-1-карбонил)фенилбороновой кислоты (21,2 г, 96,7 ммоль) и Pd(PPh3)4 (508 мг, 0,440 ммоль) в толуоле (200 мл) при комнатной температуре добавляют EtOH (40 мл), а затем Na2CO3 (2 М водный раствор, 70,0 мл, 140 ммоль). Полученную смесь греют при 100°С в течение 18 час. Реакционную смесь охлаждают до комнатной температуры, и органический слой отделяют. Водный слой экстрагируют EtOAc (300 мл). Объединенные органические слои промывают рассолом (500 мл), сушат над MgSO4, фильтруют и концентрируют при пониженном давлении, и получают сырой материал, который объединяют с другой партией сырого материала, полученной дополнительно такой же реакцией в том же масштабе. Объединенный сырой материал очищают колоночной флэш-хроматографией на силикагеле (от CH2Cl2 до 1% МеОН в CH2Cl2), и получают 51 г (90%) 3-нитро-4'-(пирролидин-1-карбонил)бифенил-4-карбальдегида. Получение трет-бутил-(1Е,4Е)-(дипропилкарбамоил)-8-(4-((R)-3-гидроксипирролидин-1-карбонил)фенил-3Н-бензо[b]азепин-2-илкарбамата. К раствору трет-бутил-(1Е,4Е)-8-(4-((R)-3-(трет-бутилдиметилсилилокси)пирролидин-1-карбонил)фенил-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-2-илкарбамата (225 мг, 0,327 ммоль) в ТГФ (4 мл) при 0°С добавляют раствор TBAF (0,34 мл, 0,34 ммоль, 1 М раствор в ТГФ). Полученную смесь нагревают до комнатной температуры и перемешивают в течение 1,5 час. Реакционную смесь разбавляют EtOAc и промывают рассолом (2×). Органический слой сушат над MgSO4, фильтруют и концентрируют при пониженном давлении, и получают сырой трет-бутил-(1Е,4Е)-(дипропилкарбамоил)-8-(4-((R)-3-гидроксипирролидин-1-карбонил)фенил-3Н-бензо[b]азепин-2-илкарбамат, который используют непосредственно без дополнительной очистки. Получение (1Е,4Е)-2-амино-N-(2-амино-2-оксоэтил)-N-пропил-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоксамида. Через раствор неочищенного трет-бутил-(1Е,4Е)-4-((2-амино-2-оксоэтил)(пропил)карбамоил)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-2-илкарбамата в безводном CH2Cl2 (15 мл) при 0°С барботируют HCl (газ) в течение 4 час. Полученную смесь нагревают до комнатной температуры и перемешивают до тех пор, пока реакция не завершится. К полученной смеси при 0°С добавляют насыщенный раствор NaHCO3. Водный слой отделяют и экстрагируют CH2Cl2. Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении, и получают сырое соединение, которое очищают колоночной флэш-хроматографией на силикагеле (МеОН:CH2Cl2=1:50).
МС APCI(+) детектирует m/z 393 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,77 (д, 1H), 8,55 (д, 1H), 7,95 (с, 1H), 7,41 (д, 1H), 7,36 (д, 1H), 7,25-7,28 (м, 1H), 6,82 (с, 1H), 4,80 (с, 2H), 3,47 (ушир.с, 4H), 2,80 (с, 2H), 1,63-1,72 (м, 4H), 0,94 (т, 6H).
Пример 184
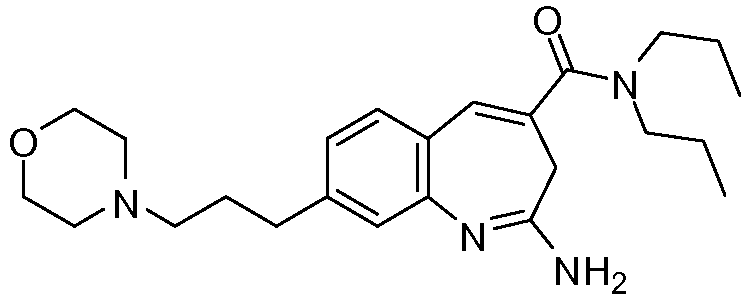
(1Е,4Е)-2-Амино-8-(3-морфолинопропил)-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид
Стадия А. Получение 4-(3-(9-борабицикло[3.3.1]нонан-9-ил)пропил)морфолина. К раствору 4-аллилморфолина (2,54 г, 20 ммоль) в ТГФ (40 мл) добавляют 9-BBN (2,44 г, 10 ммоль). Реакционную смесь кипятят с обратным холодильником до тех пор, пока реакция не завершится. После охлаждения до комнатной температуры смесь концентрируют при пониженном давлении, и получают сырой 4-(3-(9-борабицикло[3.3.1]нонан-9-ил)пропил)морфолин, который используют непосредственно без дополнительной очистки. ЖХМС ESI(+) детектирует m/z 250 (M+1).
Стадия В. Получение (1Е,4Е)-2-амино-8-(3-морфолинопропил)-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамида. Названное в заголовке соединение получают указанными процедурами с использованием (1Е,4Е)-8-бром-4-(дикарбамоил)-3Н-бензо[b]азепин-2-илкарбамата и 4-(3-(9-борабицикло[3.3.1]нонан-9-ил)пропил)морфолина. Получение 3-нитро-4'-(пирролидин-1-карбонил)бифенил-4-карбальдегида, за исключением того, что в данном случае используют сорастворитель EtOH/толуол/вода (2:1:1) и в качестве основания Cs2CO3. К раствору 4-бром-2-нитробензальдегида (20,2 г, 87,9 ммоль), 4-(пирролидин-1-карбонил)фенилбороновой кислоты (21,2 г, 96,7 ммоль) и Pd(PPh3)4 (508 мг, 0,440 ммоль) в толуоле (200 мл) при комнатной температуре добавляют EtOH (40 мл), а затем Na2CO3 (2 М водн. раствор, 70,0 мл, 140 ммоль). Полученную смесь греют при 100°С в течение 18 час. Реакционную смесь охлаждают до комнатной температуры, и органический слой отделяют. Водный слой экстрагируют EtOAc (300 мл). Объединенные органические слои промывают рассолом (500 мл), сушат над MgSO4, фильтруют и концентрируют при пониженном давлении, и получают сырой материал, который объединяют с другой партией сырого материала, полученной дополнительно такой же реакцией в том же масштабе. Объединенный сырой материал очищают колоночной флэш-хроматографией на силикагеле (от CH2Cl2 до 1% МеОН в CH2Cl2). Получение (1Е,4Е)-2-амино-N-(2-амино-2-оксоэтил)-N-пропил-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоксамида. Через раствор неочищенного трет-бутил-(1Е,4Е)-4-((2-амино-2-оксоэтил)(пропил)карбамоил)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-2-илкарбамата в безводном CH2Cl2 (15 мл) при 0°С барботируют HCl (газ) в течение 4 час. Полученную смесь нагревают до комнатной температуры и перемешивают до тех пор, пока реакция не завершится. К полученной смеси при 0°С добавляют насыщенный раствор NaHCO3. Водный слой отделяют и экстрагируют CH2Cl2. Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении, и получают сырое соединение, которое очищают колоночной флэш-хроматографией на силикагеле (МеОН:CH2Cl2=1:50).
МС APCI(+) детектирует m/z 413 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 7,20 (д, 1H), 7,07 (ушир.с, 1H), 6,90 (дд, 1H), 6,78 (с, 1H), 3,72 (т, 4H), 3,45 (ушир.с, 4H), 2,76 (с, 2H), 2,67 (т, 2H), 2,43 (ушир.с, 4H), 2,38 (т, 2H), 1,81-1,89 (м, 2H), 1,60-1,70 (м, 4H), 0,92 (т, 6H).
Пример 185
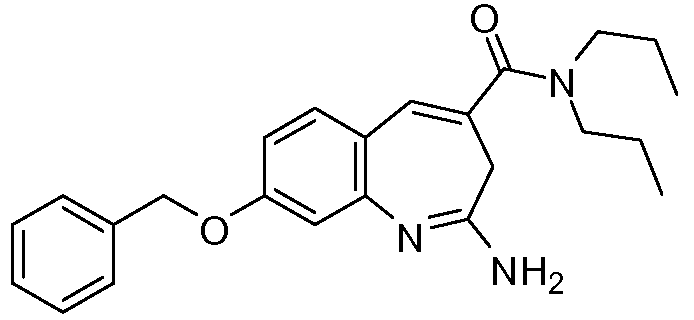
(1Е,4Е)-2-Амино-8-(бензилокси)-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид
Названное в заголовке соединение получают указанными процедурами с использованием (1Е,4Е)-этил-2-амино-8-(бензилокси)-3Н-бензо[b]азепин-4-карбоксилата и дипропиламина. Получение (1Е,4Е)-этил-2-(трет-бутоксикарбониламино)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоксилата. К смеси (1Е,4Е)-этил-2-амино-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоксилата (9,60 г, 23,8 ммоль) в CH2Cl2 (100 мл) при комнатной температуре добавляют Вос2О (5,97 мг, 27,4 ммоль). Реакционную смесь перемешивают в течение 3 суток. Полученную смесь промывают насыщенным водным раствором NaHCO3 и рассолом. Органический слой отделяют и сушат над MgSO4, фильтруют и концентрируют при пониженном давлении, и получают 12,7 г сырого (1Е,4Е)-этил-2-(трет-бутоксикарбониламино)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоксилата, который используют непосредственно без дополнительной очистки. Получение (1Е,4Е)-2-(трет-бутоксикарбониламино)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоновой кислоты. К раствору (1Е,4Е)-этил-2-(трет-бутоксикарбониламино)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоксилата (12,0 г, 23,8 ммоль) в смеси ТГФ-EtOH (60 мл/60 мл) при 0°С добавляют 4 N водный раствор LiOH (23,8 мл, 95,3 ммоль). Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 21 часа. Добавляют еще дважды 6 мл 4 N водного раствора LiOH через 21 час и 24 часа. После перемешивания еще в течение 6 час полученную смесь концентрируют при пониженном давлении, и получают сырой материал, который разбавляют водой (50 мл) и подкисляют до рН ~3,5 1 N водной фосфорной кислотой (~450 мл). Во время подкисления добавляют ~250 мл CH2Cl2 для экстрагирования сырого продукта реакции из клейкой суспензии. Твердые вещества, образовавшиеся во время подкисления, отфильтровывают с использованием стеклянного фильтра, заполненного целитом. Водный слой отделяют и экстрагируют CH2Cl2 (3×100 мл). Объединенные органические слои сушат над MgSO4, фильтруют и концентрируют при пониженном давлении, и получают сырую (1Е,4Е)-2-(трет-бутоксикарбониламино)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоновую кислоту, которую используют непосредственно без дополнительной очистки. Получение (1Е,4Е)-4-(пропилкарбамоил)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-2-илкарбамата. Смесь (1Е,4Е)-2-(трет-бутоксикарбониламино)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоновой кислоты (200 мг, 0,42 ммоль), HOBt (114 мг, 0,84 ммоль) и EDCI (161 мг, 0,84 ммоль) в ДМФА (5 мл) перемешивают в течение 1 часа при комнатной температуре. К полученной смеси при комнатной температуре добавляют триэтиламин (0,12 мл, 0,84 ммоль) и пропан-1-амин (0,043 мл, 0,53 ммоль). Полученный раствор перемешивают еще в течение 2 час. Реакционную смесь разбавляют EtOAc (5 мл) и промывают насыщенным водным раствором NH4Cl. Водный слой отделяют и экстрагируют EtOAc (3×5 мл). Объединенные органические слои промывают рассолом (5 мл), насыщенным водным раствором NaHCO3 (5 мл) и рассолом (5 мл). Органический слой сушат над MgSO4, фильтруют и концентрируют при пониженном давлении, и получают сырой трет-бутил-(1Е,4Е)-4-(пропилкарбамоил)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-2-илкарбамат, который используют непосредственно без дополнительной очистки. Получение (1Е,4Е)-2-амино-N-(2-амино-2-оксоэтил)-N-пропил-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоксамида. Через раствор неочищенного трет-бутил-(1Е,4Е)-4-((2-амино-2-оксоэтил)(пропил)карбамоил)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-2-илкарбамата в безводном CH2Cl2 (15 мл) при 0°С барботируют HCl (газ) в течение 4 час. Полученную смесь нагревают до комнатной температуры и перемешивают до тех пор, пока реакция не завершится. К полученной смеси при 0°С добавляют насыщенный раствор NaHCO3. Водный слой отделяют и экстрагируют CH2Cl2. Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении, и получают сырое соединение, которое очищают колоночной флэш-хроматографией на силикагеле (МеОН:CH2Cl2=1:50).
МС APCI(+) детектирует m/z 392 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 7,44-7,46 (м, 2H), 7,37-7,41 (м, 2H), 7,31-7,34 (м, 1H), 7,19 (д, 1H), 6,83 (д, 1H), 6,73-6,76 (м, 2H), 5,10 (с, 2H), 3,45 (ушир.с, 4H), 2,77 (с, 2H), 1,60-1,70 (м, 4H), 0,92 (т, 6H).
Пример 191
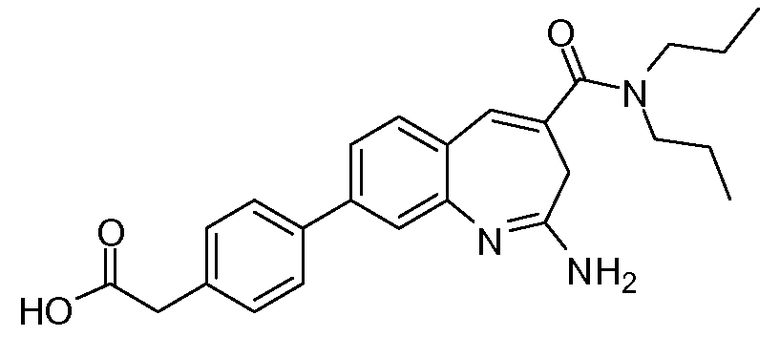
2-(4-((1Е,4Е)-2-Амино-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-8-ил)фенил)уксусная кислота
Стадия А. Бензил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)ацетат (41%) получают согласно описанному ниже примеру 192, стадия А, заменяя этанол на бензиловый спирт.
Стадия В. Бензил-2-(4-((1Е,4Е)-2-амино-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-6-ил)фенил)ацетат (33%) получают как описано далее, заменяя 4-(метоксикарбонил)фенилбороновую кислоту на бензил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)ацетат. (1Е,4Е)-2-Амино-8-бром-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид (75,0 мг, 0,206 ммоль), 4-(метоксикарбонил)фенилбороновую кислоту (55,6 мг, 0,309 ммоль), тетракис(трифенилфосфин)палладий(0) (23,8 мг, 0,021 ммоль) и 2 М водный раствор карбоната калия (0,309 мл, 0,618 ммоль) соединяют в 2 мл ацетонитрила во флаконе для реакции под действием микроволнового излучения. Полученную смесь греют микроволновым излучением при 100°С в течение 30 минут. Затем смесь разбавляют EtOAc, дважды промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Затем используют препаративную тонкослойную хроматографию (пластины 2×0,5 мм, 7% МеОН/DCM). m/z (APCI-полож.) М+1=510,3.
Стадия С. 2-(4-((1Е,4Е)-2-Амино-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-8-ил)фенил)уксусную кислоту (28%) получают как описано далее, заменяя бензил-4-((1Е,4Е)-2-амино-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-8-ил)бензоат на бензил-2-(4-((1Е,4Е)-2-амино-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат. Бензил-4-((1Е,4Е)-2-амино-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-8-ил)бензоат (0,025 г, 0,0504 ммоль) суспендируют в 1 мл метанола, добавляют 25 мг 10% Pd/C (тип Degussa), и смесь гидрируют водородом из баллона в течение одного часа. Затем полученную смесь фильтруют через фильтровальную бумагу GF/F, и фильтрат концентрируют. m/z (APCI-полож.) М+1=420,2.
Пример 192
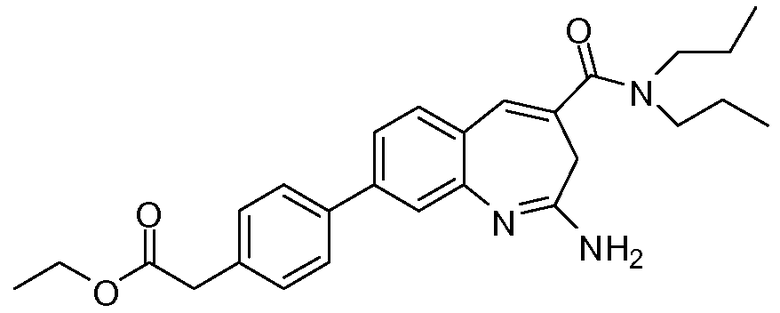
Этил-2-(4-((1Е,4Е)-2-амино-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат
Стадия А. 2-(4-(4,4,5,5-Тетраметил-1,3,2-диоксаборолан-2-ил)фенил)уксусную кислоту (0,18 мг, 0,67 ммоль) растворяют в сухом ТГФ (7 мл). К полученному раствору добавляют этанол (0,037 мл, 0,81 ммоль) и трифенилфосфин, связанный со смолой (0,93 г, 2,014 ммоль, 2,16 ммоль/г). Смесь осторожно перемешивают при комнатной температуре в течение 20 минут. Затем добавляют диизопропилазидодикарбоксилат (0,34 мл, 1,68 ммоль), и смесь перемешивают при комнатной температуре в течение одного часа, затем фильтруют, и смолу несколько раз промывают EtOAc. Фильтрат концентрируют, и полученный материал очищают на Flash 40 Biotage (картридж 40М, 30% EtOAc/гексан), и получают 137 мг (70%) этил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)ацетата в виде оранжевого масла.
Стадия В. Этил-2-(4-((1Е,4Е)-2-амино-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат (20%) получают как описано далее, заменяя 4-(метоксикарбонил)фенилбороновую кислоту на этил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)ацетат. (1Е,4Е)-2-Амино-8-бром-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид (75,0 мг, 0,206 ммоль), 4-(метоксикарбонил)фенилбороновую кислоту (55,6 мг, 0,309 ммоль), тетракис(трифенилфосфин)палладий(0) (23,8 мг, 0,021 ммоль) и 2 М водный раствор карбоната калия (0,309 мл, 0,618 ммоль) соединяют в 2 мл ацетонитрила во флаконе для реакции под действием микроволнового излучения. Полученную смесь греют микроволновым излучением при 100°С в течение 30 минут. Затем смесь разбавляют EtOAc, дважды промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Затем используют препаративную тонкослойную хроматографию (пластины 2×0,5 мм, 7% МеОН/DCM). 1H ЯМР (400 МГц, CDCl3) δ 7,59-7,65 (м, 2H), 7,48-7,53 (м, 1H), 7,30-7,39 (м, 4H), 6,84 (с, 1H), 5,50 (очень уширенный с, 1H), 4,14-4,23 (м, 2H), 3,66 (с, 2H), 3,38-3,52 (м, 4H), 2,86 (м, 2H), 1,59-1,72 (м, 4H), 1,22-1,32 (м, 3H), 0,88-0,97 (м, 6H); m/z (APCI-полож.) M+1=448,2.
Пример 193
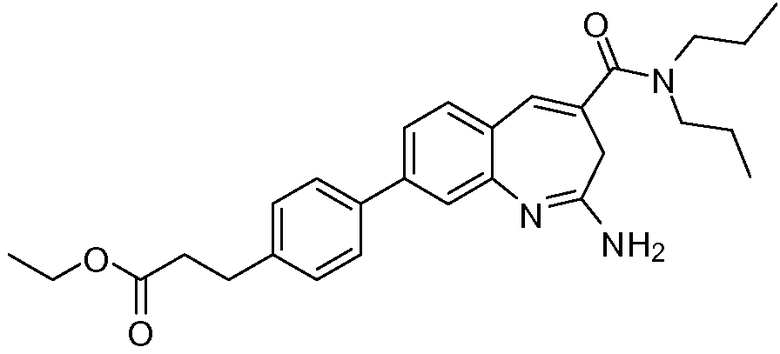
Этил-3-(4-((1Е,4Е)-2-амино-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-8-ил)фенил)пропаноат
Стадия А. Этил-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропаноат (24%) получают согласно описанному выше примеру 192, стадия А, заменяя 2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)уксусную кислоту на 3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропановую кислоту.
Стадия В. Этил-3-(4-((1Е,4Е)-2-амино-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-8-ил)фенил)пропаноат (27%) получают как описано далее, заменяя 4-(метоксикарбонил)фенилбороновую кислоту на этил-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропаноат. (1Е,4Е)-2-Амино-8-бром-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид (75,0 мг, 0,206 ммоль), 4-(метоксикарбонил)фенилбороновую кислоту (55,6 мг, 0,309 ммоль), тетракис(трифенилфосфин)палладий(0) (23,8 мг, 0,021 ммоль) и 2 М водный раствор карбоната калия (0,309 мл, 0,618 ммоль) соединяют в 2 мл ацетонитрила во флаконе для реакции под действием микроволнового излучения. Полученную смесь греют микроволновым излучением при 100°С в течение 30 минут. Затем смесь разбавляют EtOAc, дважды промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Затем используют препаративную тонкослойную хроматографию (пластины 2×0,5 мм, 7% МеОН/DCM). 1H ЯМР (400 МГц, CDCl3) δ 7,57-7,60 (м, 2H), 7,49-7,51 (м, 1H), 7,25-7,35 (м, 4H), 6,84 (с, 1H), 5,22 (очень уширенный синглет, 1H), 4,11-4,17 (м, 2H), 3,41-3,51 (м, 4H), 2,95-3,04 (м, 2H), 2,83 (с, 2H), 2,64-2,69 (м, 2H), 1,58-1,72 (м, 4H), 1,20-1,30 (м, 3H), 0,88-0,99 (м, 6H); m/z (APCI-полож.) M+1=462,3.
Пример 196
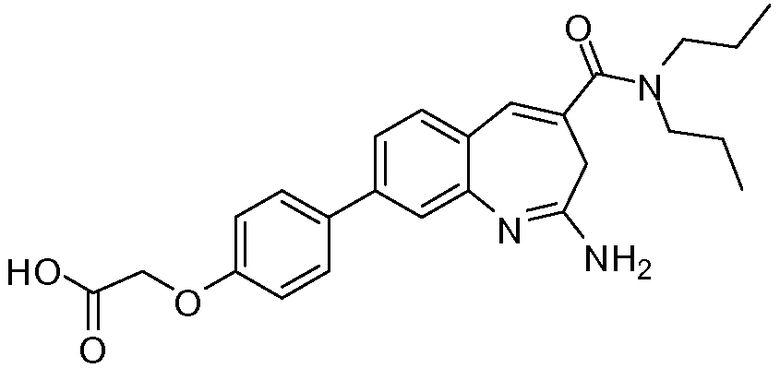
2-(4-((1Е,4Е)-2-Амино-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-8-ил)фенокси)уксусная кислота
Стадия А. (1Е,4Е)-2-Амино-8-(4-(бензилокси)фенил)-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид (36%) получают как описано далее, заменяя 4-(метоксикарбонил)фенилбороновую кислоту на 4-(бензилокси)фенилбороновую кислоту. (1Е,4Е)-2-Амино-8-бром-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид (75,0 мг, 0,206 ммоль), 4-(метоксикарбонил)фенилбороновую кислоту (55,6 мг, 0,309 ммоль), тетракис(трифенилфосфин)палладий(0) (23,8 мг, 0,021 ммоль) и 2 М водный раствор карбоната калия (0,309 мл, 0,618 ммоль) соединяют в 2 мл ацетонитрила во флаконе для реакции под действием микроволнового излучения. Полученную смесь греют микроволновым излучением при 100°С в течение 30 минут. Затем смесь разбавляют EtOAc, дважды промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Затем используют препаративную тонкослойную хроматографию (пластины 2×0,5 мм, 7% МеОН/DCM). m/z (APCI-полож.) М+1=468,2.
Стадия В. (1Е,4Е)-2-Амино-8-(4-(бензилокси)фенил)-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид (0,100 г, 0,214 ммоль), ди-трет-бутилдикарбонат (0,117 г, 0,535 ммоль) и диметиламинопиридин (0,026 г, 0,214 ммоль) соединяют в 3 мл сухого дихлорметана и перемешивают при комнатной температуре в течение 16 часов. Затем смесь помещают непосредственно в колонку Flash 40 Biotage (40М, EtOAc/гексан 3:1), и получают 0,056 г (40%) продукта. m/z (APCI-полож.) М+1=667,9.
Стадия С. Продукт со стадии В (0,050 г, 0,075 ммоль) растворяют в 1 мл EtOAc, и добавляют 0,050 г 10% Pd/C. Смесь гидрируют водородом из баллона в течение 1 часа. Добавляют еще 0,050 г катализатора, и смесь гидрируют еще в течение 1,5 часов и затем фильтруют через фильтровальную бумагу GF/F. Фильтрат концентрируют при пониженном давлении, и получают 37 мг (87%) продукта. m/z (APCI-полож.) М+1=577,9.
Стадия D. Продукт со стадии С (0,030 г, 0,519 ммоль) растворяют в 1 мл сухого ДМФА. К полученному раствору добавляют карбонат цезия (0,051 г, 0,156 ммоль) и бензил-2-бромацетат (0,012 мл, 0,078 ммоль), и смесь перемешивают при комнатной температуре в течение 16 часов. Затем реакционную смесь разбавляют EtOAc, промывают несколько раз рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Затем полученный остаток растворяют в 1 мл дихлорметана, и затем добавляют 1 мл ТФК. Через 1 час смесь концентрируют при пониженном давлении, и полученный остаток нейтрализуют концентрированным гидроксидом аммония и водой, экстрагируют дихлорметаном, и экстракты сушат над сульфатом натрия и концентрируют при пониженном давлении. Препаративная тонкослойная хроматография (пластины 2×0,5 мм, 7% МеОН/DCM) дает 0,015 г (55%) бензил-2-(4-((1Е,4Е)-2-амино-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-8-ил)фенокси)ацетата. m/z (APCI-полож.) М+1=526,2.
Стадия Е. 2-(4-((1Е,4Е)-2-Амино-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-8-ил)фенокси)уксусную кислоту (46%) получают как описано далее, заменяя бензил-4-((1Е,4Е)-2-амино-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-8-ил)бензоат на бензил-2-(4-((1Е,4Е)-2-амино-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-8-ил)фенокси)ацетат. Бензил-4-((1Е,4Е)-2-амино-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-8-ил)бензоат (0,025 г, 0,0504 ммоль) суспендируют в 1 мл метанола, добавляют 25 мг 10% Pd/C (тип Degussa), и смесь гидрируют водородом из баллона в течение одного часа. Затем полученную смесь фильтруют через фильтровальную бумагу GF/F, и фильтрат концентрируют. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,48-7,56 (м, 2H), 7,33-7,42 (м, 1H), 7,21-7,30 (м, 3H), 6,93-7,00 (м, 3H), 6,77 (с, 1H), 4,43 (с, 2H), 2,87 (с, 2H), 1,45-1,61 (м, 4H), 0,74-0,92 (м, 6H); m/z (APCI-отриц.) M-1=434,0.
Пример 197
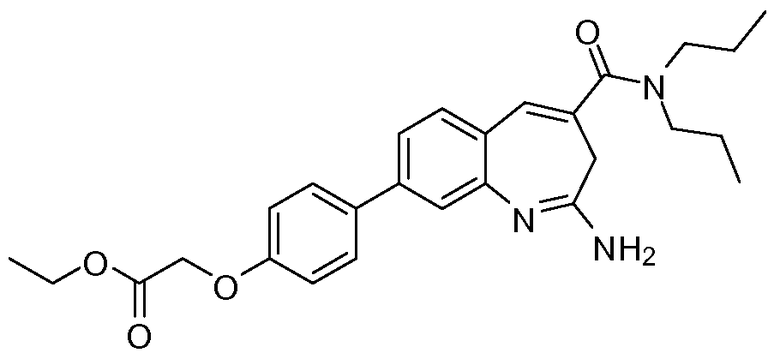
Этил-2-(4-((1Е,4Е)-2-амино-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-8-ил)фенокси)ацетат
Стадия А. Этил-2-(4-((1Е,4Е)-2-амино-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-8-ил)фенокси)ацетат (21%) получают как описано далее, заменяя 4-(метоксикарбонил)фенилбороновую кислоту на этил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)ацетат. (1Е,4Е)-2-Амино-8-бром-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид (75,0 мг, 0,206 ммоль), 4-(метоксикарбонил)фенилбороновую кислоту (55,6 мг, 0,309 ммоль), тетракис(трифенилфосфин)палладий(0) (23,8 мг, 0,021 ммоль) и 2 М водный раствор карбоната калия (0,309 мл, 0,618 ммоль) соединяют в 2 мл ацетонитрила во флаконе для реакции под действием микроволнового излучения. Полученную смесь греют микроволновым излучением при 100°С в течение 30 минут. Затем смесь разбавляют EtOAc, дважды промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Затем используют препаративную тонкослойную хроматографию (пластины 2×0,5 мм, 7% МеОН/DCM). 1H ЯМР (400 МГц, ДМСО-d6) δ 7,58-7,63 (м, 2H), 7,33-7,37 (м, 1H), 7,19-7,26 (м, 2H), 6,98-7,03 (м, 2H), 6,75 (с, 1H), 4,82 (с, 2H), 4,15-4,22 (м, 2H), 3,25-3,34 (м, 4H), 2,76 (с, 2H), 1,50-1,61 (м, 4H), 1,19-1,26 (м, 3H), 0,73-0,90 (м, 6H); m/z (APCI-полож.) M+1=464,3.
Пример 198
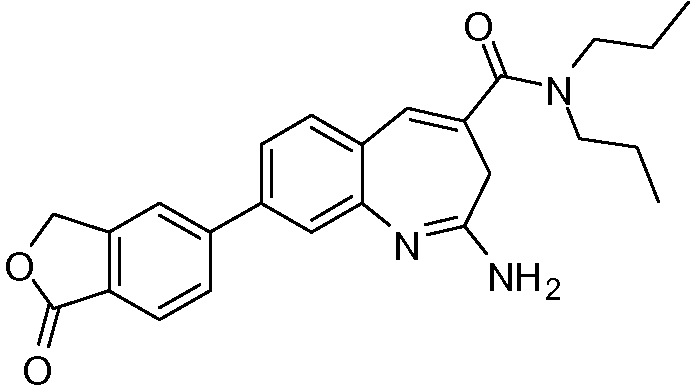
(1Е,4Е)-2-Амино-8-(1-оксо-1,3-дигидроизобензофуран-5-ил)-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид
Стадия А. 5-(4,4,5,5-Тетраметил-1,3,2-диоксаборолан-2-ил)изобензофуран-1(3Н)-он (57%) получают согласно описанному ниже примеру 224/225, стадия С.
Стадия В. (1Е,4Е)-2-Амино-8-(1-оксо-1,3-дигидроизобензофуран-5-ил)-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид получают как описано далее, заменяя 4-(метоксикарбонил)фенилбороновую кислоту на 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)изобензофуран-1(3Н)-он. (1Е,4Е)-2-Амино-8-бром-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид (75,0 мг, 0,206 ммоль), 4-(метоксикарбонил)фенилбороновую кислоту (55,6 мг, 0,309 ммоль), тетракис(трифенилфосфин)палладий(0) (23,8 мг, 0,021 ммоль) и 2 М водный раствор карбоната калия (0,309 мл, 0,618 ммоль) соединяют в 2 мл ацетонитрила во флаконе для реакции под действием микроволнового излучения. Полученную смесь греют микроволновым излучением при 100°С в течение 30 минут. Затем смесь разбавляют EtOAc, дважды промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Затем используют препаративную тонкослойную хроматографию (пластины 2×0,5 мм, 7% МеОН/DCM). 1H ЯМР (400 МГц, CDCl3) δ 7,96-8,00 (м, 1H), 7,78-7,82 (м, 1H), 7,72-7,74 (м, 1H), 7,52-7,55 (м, 1H), 7,38-7,42 (м, 1H), 7,30-7,35 (м, 1H), 6,85 (с, 1H), 5,37 (с, 2H), 3,60-3,71 (м, 2H), 3,39-3,53 (м, 4H), 2,85 (м, 2H), 1,59-1,75 (м, 4H), 0,90-0,99 (м, 6H); m/z (APCI-полож.) M+1=418,2.
Пример 199
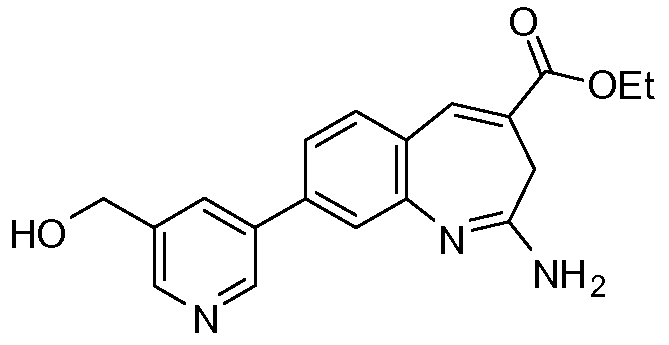
(1Е,4Е)-Этил-2-амино-8-(5-(гидроксиметил)пиридин-3-ил)-3Н-бензо[b]азепин-4-карбоксилат
Названное в заголовке соединение получают указанными процедурами с использованием (1Е,4Е)-этил-2-амино-8-бром-2-(трет-бутоксикарбониламино)-3Н-бензо[b]азепин-4-карбоксилата и 3-((трет-бутилдиметилсилилокси)метил)фенилбороновой кислоты. Получение 3-нитро-4'-(пирролидин-1-карбонил)бифенил-4-карбальдегида, за исключением того, что в данном случае используют Cs2CO3. К раствору 4-бром-2-нитробензальдегида (20,2 г, 87,9 ммоль), 4-(пирролидин-1-карбонил)фенилбороновой кислоты (21,2 г, 96,7 ммоль) и Pd(PPh3)4 (508 мг, 0,440 ммоль) в толуоле (200 мл) при комнатной температуре добавляют EtOH (40 мл), а затем Na2CO3 (2 М водный раствор, 70,0 мл, 140 ммоль). Полученную смесь греют при 100°С в течение 18 час. Реакционную смесь охлаждают до комнатной температуры, и органический слой отделяют. Водный слой экстрагируют EtOAc (300 мл). Объединенные органические слои промывают рассолом (500 мл), сушат над MgSO4, фильтруют и концентрируют при пониженном давлении, и получают сырой материал, который объединяют с другой партией сырого материала, полученной дополнительно такой же реакцией в том же масштабе. Объединенный сырой материал очищают колоночной флэш-хроматографией на силикагеле (от CH2Cl2 до 1% МеОН в CH2Cl2). Получение трет-бутил-(1Е,4Е)-(дипропилкарбамоил)-8-(4-((R)-3-гидроксипирролидин-1-карбонил)фенил-3Н-бензо[b]азепин-2-илкарбамата. К раствору трет-бутил-(1Е,4Е)-8-(4-((R)-3-(трет-бутилдиметилсилилокси)пирролидин-1-карбонил)фенил-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-2-илкарбамата (225 мг, 0,327 ммоль) в ТГФ (4 мл) при 0°С добавляют раствор TBAF (0,34 мл, 0,34 ммоль, 1 М раствор в ТГФ). Полученную смесь нагревают до комнатной температуры и перемешивают в течение 1,5 час. Реакционную смесь разбавляют EtOAc и промывают рассолом (2×). Органический слой сушат над MgSO4, фильтруют и концентрируют при пониженном давлении, и получают сырой трет-бутил-(1Е,4Е)-(дипропилкарбамоил)-8-(4-((R)-3-гидроксипирролидин-1-карбонил)фенил-3Н-бензо[b]азепин-2-илкарбамат, который используют непосредственно без дополнительной очистки. Получение (1Е,4Е)-2-амино-N-(2-амино-2-оксоэтил)-N-пропил-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоксамида. Через раствор неочищенного трет-бутил-(1Е,4Е)-4-((2-амино-2-оксоэтил)(пропил)карбамоил)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-2-илкарбамата в безводном CH2Cl2 (15 мл) при 0°С барботируют HCl (газ) в течение 4 час. Полученную смесь нагревают до комнатной температуры и перемешивают до тех пор, пока реакция не завершится. К полученной смеси при 0°С добавляют насыщенный раствор NaHCO3. Водный слой отделяют и экстрагируют CH2Cl2. Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении, и получают сырое соединение, которое очищают колоночной флэш-хроматографией на силикагеле (МеОН:CH2Cl2=1:50). MC APCI (+) детектирует m/z 338 (M+1); 1H-ЯМР (400 МГц, CDCl3) δ 8,78 (с, 1H), 8,57 (с, 1H), 7,96 (с, 1H), 7,84 (с, 1H), 7,49 (д, 1H), 7,42 (с, 1H), 7,31 (д, 1H), 4,80 (с, 2H), 4,33 (кв, 2H), 3,00 (с, 2H), 1,40 (т, 3H).
Пример 200
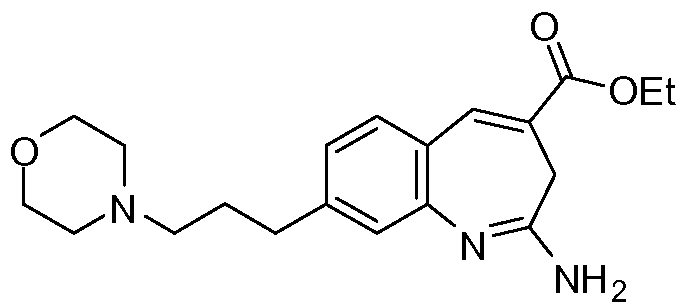
(1Е,4Е)-Этил-2-амино-8-(3-морфолинопропил)-3Н-бензо[b]азепин-4-карбоксилат
Названное в заголовке соединение получают указанными процедурами с использованием (1Е,4Е)-этил-8-бром-2-(трет-бутоксикарбониламино)-3Н-бензо[b]азепин-4-карбоксилата и 4-(3-(9-борабицикло[3.3.1]нонан-9-ил)пропил)морфолина. Получение 3-нитро-4'-(пирролидин-1-карбонил)бифенил-4-карбальдегида, за исключением того, что в данном случае используют сорастворитель EtOH/толуол/вода (2:1:1) и Cs2CO3. К раствору 4-бром-2-нитробензальдегида (20,2 г, 87,9 ммоль), 4-(пирролидин-1-карбонил)фенилбороновой кислоты (21,2 г, 96,7 ммоль) и Pd(PPh3)4 (508 мг, 0,440 ммоль) в толуоле (200 мл) при комнатной температуре добавляют EtOH (40 мл), а затем Na2CO3 (2 М водный раствор, 70,0 мл, 140 ммоль). Полученную смесь греют при 100°С в течение 18 час. Реакционную смесь охлаждают до комнатной температуры, и органический слой отделяют. Водный слой экстрагируют EtOAc (300 мл). Объединенные органические слои промывают рассолом (500 мл), сушат над MgSO4, фильтруют и концентрируют при пониженном давлении, и получают сырой материал, который объединяют с другой партией сырого материала, полученной дополнительно такой же реакцией в том же масштабе. Объединенный сырой материал очищают колоночной флэш-хроматографией на силикагеле (от CH2Cl2 до 1% МеОН в CH2Cl2). Получение (1Е,4Е)-2-амино-N-(2-амино-2-оксоэтил)-N-пропил-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоксамида. Через раствор неочищенного трет-бутил-(1Е,4Е)-4-((2-амино-2-оксоэтил)(пропил)карбамоил)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-2-илкарбамата в безводном CH2Cl2 (15 мл) при 0°С барботируют HCl (газ) в течение 4 час. Полученную смесь нагревают до комнатной температуры и перемешивают до тех пор, пока реакция не завершится. К полученной смеси при 0°С добавляют насыщенный раствор NaHCO3. Водный слой отделяют и экстрагируют CH2Cl2. Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении, и получают сырое соединение, которое очищают колоночной флэш-хроматографией на силикагеле (МеОН:CH2Cl2=1:50). МС APCI(+) детектирует m/z 358 (M+1); 1H-ЯМР (400 МГц, CDCl3) δ 7,78 (с, 1H), 7,30 (д, 1H), 7,06 (с, 1H), 6,92 (д, 1H), 4,30 (кв, 2H), 3,72 (т, 4H), 2,94 (с, 2H), 2,67 (т, 2H), 2,43 (ушир.с, 4H), 2,38 (т, 2H), 1,81-1,89 (м, 2H), 1,37 (т, 3H).
Пример 201
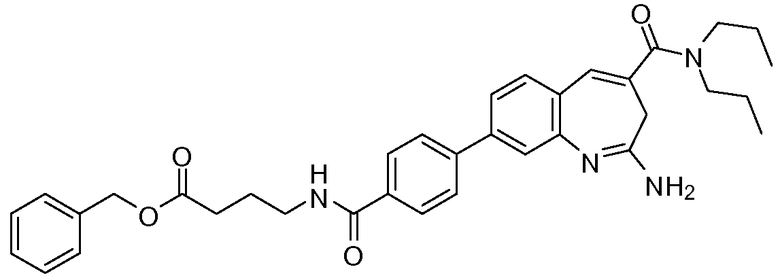
Бензил-4-(4-((1Е,4Е)-2-амино-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-8-ил)бензамидо)бутаноат
Стадия А. Получение бензил-4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензамидо)бутаноата. Названное в заголовке соединение получают указанными процедурами с использованием 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензойной кислоты и 4-метилбензолсульфоната бензил-4-аминобутаноата. Получение трет-бутил-(1Е,4Е)-4-(пропилкарбамоил)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-2-илкарбамата. Смесь (1Е,4Е)-2-(трет-бутоксикарбониламино)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоновой кислоты (200 мг, 0,42 ммоль), HOBt (114 мг, 0,84 ммоль) и EDCI (161 мг, 0,84 ммоль) в ДМФА (5 мл) перемешивают в течение 1 часа при комнатной температуре. К полученной смеси при комнатной температуре добавляют триэтиламин (0,12 мл, 0,84 ммоль) и пропан-1-амин (0,043 мл, 0,53 ммоль). Полученный раствор перемешивают еще в течение 2 час. Реакционную смесь разбавляют EtOAc (5 мл) и промывают насыщенным воднным раствором NH4Cl. Водный слой отделяют и экстрагируют EtOAc (3×5 мл). Объединенные органические слои промывают рассолом (5 мл), насыщенным водным раствором NaHCO3 (5 мл) и рассолом (5 мл). Органический слой сушат над MgSO4, фильтруют и концентрируют при пониженном давлении, и получают сырой трет-бутил-(1Е,4Е)-4-(пропилкарбамоил)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-2-илкарбамат, который используют непосредственно без дополнительной очистки. МС APCI(+) детектирует m/z 424 (M+1).
Стадия В. Получение бензил-4-(4-((1Е,4Е)-2-амино-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-8-ил)бензамидо)бутаноата. Названное в заголовке соединение получают указанными процедурами с использованием трет-бутил-(1Е,4Е)-8-бром-4-(дипропил-карбамоил)-3Н-бензо[b]азепин-2-илкарбамата и бензил-4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензамидо)-бутаноата. Получение трет-бутил-(1Е,4Е)-8-(4-(диметилкарбамоил)фенил)-4-(дипропилкарбамоил)-3Н-бензо[b]-азепин-2-илкарбамата, за исключением того, что в данном случае используют сорастворитель MeCN-вода (1,5:1). К Na2CO3 (129 мг, 1,214 ммоль) в 50-мл круглодонной колбе добавляют воду (3,7 мл), и барботируют N2 в течение 10 мин. К полученной смеси при комнатной температуре добавляют трет-бутил-(1Е,4Е)-8-бром-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-2-илкарбамат (200 мг, 0,40 ммоль) в EtOH (4,9 мл). Через полученную смесь барботируют N2 в течение 10 мин. Добавляют Pd(OAc)2 (9,3 мг, 0,040 ммоль) и гидрат дикалиевой соли 4,4'-(фенилфосфиниден)бисбензолсульфоновой кислоты (45 мг, 0,081 ммоль). Полученную смесь греют при 65°С при барботировании N2. К полученной смеси добавляют раствор 4-(диметилкарбамоил)фенилбороновой кислоты (97 мг, 0,49 ммоль) в EtOH (0,6 мл). Полученную смесь перемешивают при 65°С в течение 1 часа. Реакционную смесь охлаждают до комнатной температуры и концентрируют при пониженном давлении, и получают сырой материал, который разбавляют водой (5 мл) и EtOAc (10 мл). Смесь фильтруют через фильтр GF/F. Водный слой отделяют и экстрагируют EtOAc (10 мл). Объединенные органические слои промывают рассолом (10 мл), сушат над MgSO4, фильтруют и концентрируют при пониженном давлении, и получают сырой продукт, который очищают колоночной флэш-хроматографией на силикагеле (от CH2Cl2 до 2% МеОН в CH2Cl2). Получение (1Е,4Е)-2-амино-N-пропил-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоксамида. К раствору трет-бутил-(1Е,4Е)-4-(пропилкарбамоил)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-2-илкарбамата (450 мг, 0,87 ммоль) в CH2Cl2 (5 мл) при 0°С добавляют 2,2,2-трифторуксусную кислоту (1,36 мл, 17,4 ммоль). Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 2 час. Реакционную смесь концентрируют при пониженном давлении, и получают сырой материал, который снова разбавляют CH2Cl2 (10 мл) и насыщенным водным раствором NaHCO3 (15 мл). Полученную смесь перемешивают в течение 30 мин при комнатной температуре. Водный слой отделяют и экстрагируют CH2Cl2 (1×10 мл). Объединенные органические слои промывают насыщенным водным раствором NaHCO3 (2×10 мл) и рассолом (1×10 мл), сушат над MgSO4, фильтруют и концентрируют при пониженном давлении, и получают снова сырой материал, который очищают колоночной флэш-хроматографией на силикагеле (градиент 1-5% МеОН в CH2Cl2). МС APCI(+) детектирует m/z 581 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 7,82-7,84 (м, 2H), 7,70-7,72 (м, 2H), 7,51 (д, 1H), 7,30-7,38 (м, 7H), 6,83 (с, 1H), 6,53 (т, 1H), 5,13 (с, 2H), 3,54 (кв, 2H), 3,47 (ушир.с, 4H), 2,81 (с, 2H), 2,52 (т, 2H), 1,98-2,04 (м, 2H), 1,62-1,72 (м, 4H), 0,93 (т, 6H).
Пример 205
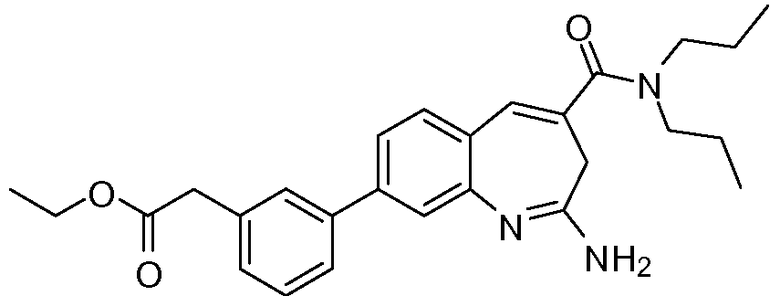
Этил-2-(3-((1Е,4Е)-2-амино-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат
Стадия А. К 2-(3-(4,4,5,5-тетраметил-1,3,2-диоксоборолан-2-ил)фенил)уксусной кислоте (0,50 г, 1,91 ммоль) в 20 мл сухого ДМФА добавляют карбонат цезия (1,24 г, 3,82 ммоль) и иодэтан (0,20 мл, 2,48 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 16 час, затем разбавляют рассолом, дважды экстрагируют EtOAc, экстракты промывают рассолом (2×), сушат над сульфатом натрия и концентрируют при пониженном давлении, и получают 360 мг (65%) этил-2-(3-(4,4,5,5-тетраметил-1,3,2-диоксоборолан-2-ил)фенил)ацетата в виде прозрачного масла.
Стадия В. Этил-2-(3-((1Е,4Е)-2-амино-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат получают как описано далее, заменяя 4-(метоксикарбонил)фенилбороновую кислоту на этил-2-(3-(4,4,5,5-тетраметил-1,3,2-диоксоборолан-2-ил)фенил)ацетат. (1Е,4Е)-2-Амино-8-бром-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид (75,0 мг, 0,206 ммоль), 4-(метоксикарбонил)фенилбороновую кислоту (55,6 мг, 0,309 ммоль), тетракис(трифенилфосфин)палладий(0) (23,8 мг, 0,021 ммоль) и 2 М водный раствор карбоната калия (0,309 мл, 0,618 ммоль) соединяют в 2 мл ацетонитрила во флаконе для реакции под действием микроволнового излучения. Полученную смесь греют микроволновым излучением при 100°С в течение 30 минут. Затем смесь разбавляют EtOAc, дважды промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Затем используют препаративную тонкослойную хроматографию (пластины 2×0,5 мм, 7% МеОН/DCM). 1H ЯМР (400 МГц, CDCl3) δ 7,49-7,60 (м, 3H), 7,26-7,42 (м, 4H), 6,83 (с, 1H), 5,20 (ушир.с, 1H), 4,12-4,21 (м, 2H), 3,67 (с, 2H), 3,42-3,52 (м, 4H), 2,83 (с, 2H), 1,56-1,74 (м, 4H), 1,23-1,30 (м, 3H), 0,90-0,97 (м, 6H); m/z (APCI-полож.) M+1=448,3.
Пример 213
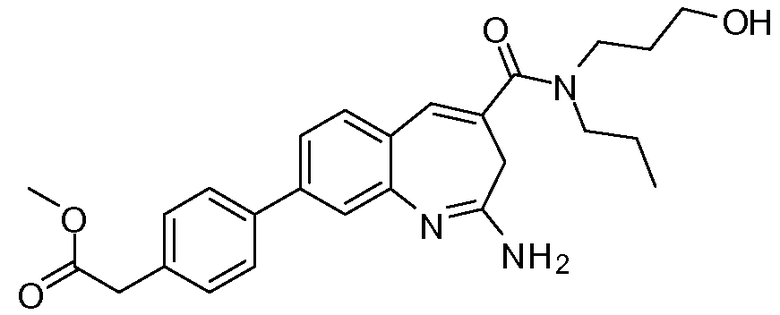
Метил-2-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат
Стадия А. Метил-2-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат получают как описано далее, заменяя 4-(этоксикарбонил)фенилбороновую кислоту на 4-(2-метокси-2-оксоэтил)фенилбороновую кислоту. Этил-4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)бензоат (44%) получают как описано далее, заменяя (1Е,4Е)-2-амино-8-бром-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид на трет-бутил-(1Е,4Е)-8-бром-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-2-илкарбамат и 4-(метоксикарбонил)фенилбороновую кислоту на 4-(этоксикарбонил)фенилбороновую кислоту. (1Е,4Е)-2-Амино-8-бром-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид (75,0 мг, 0,206 ммоль), 4-(метоксикарбонил)фенилбороновую кислоту (55,6 мг, 0,309 ммоль), тетракис(трифенилфосфин)палладий(0) (23,8 мг, 0,021 ммоль) и 2 М водный раствор карбоната калия (0,309 мл, 0,618 ммоль) соединяют в 2 мл ацетонитрила во флаконе для реакции под действием микроволнового излучения. Полученную смесь греют микроволновым излучением при 100°С в течение 30 минут. Затем смесь разбавляют EtOAc, дважды промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Затем используют препаративную тонкослойную хроматографию (пластины 2×0,5 мм, 7% МеОН/DCM). Этил-4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)бензоат (0,050 г, 0,075 ммоль) растворяют в 2 мл дихлорметана и 0,5 мл ТФК. Примерно через один час смесь концентрируют при пониженном давлении, и затем полученный остаток повторно растворяют в дихлорметане, добавляют 1 мл концентрированного гидроксида аммония, и смесь энергично перемешивают в течение 15 минут. Затем полученную смесь разбавляют водой, экстрагируют дихлорметаном (2×), и экстракты сушат над сульфатом натрия и концентрируют при пониженном давлении. Используют препаративную тонкослойную хроматографию (пластины 2×0,5 мм, 10% МеОН/DCM/0,5% NH4OH). 1H ЯМР (400 МГц, CDCl3) δ 7,59-7,66 (м, 2H), 7,48-7,51 (м, 1H), 7,28-7,39 (м, 4H), 6,87 (с, 1H), 3,72 (с, 3H), 3,57-3,70 (м, 6H), 3,45-3,53 (м, 2H), 2,83 (с, 2H), 1,79-1,88 (м, 2H), 1,65-1,75 (м, 2H), 0,90-0,96 (м, 3H); m/z (APCI-полож.) M+1=450,2.
Пример 214
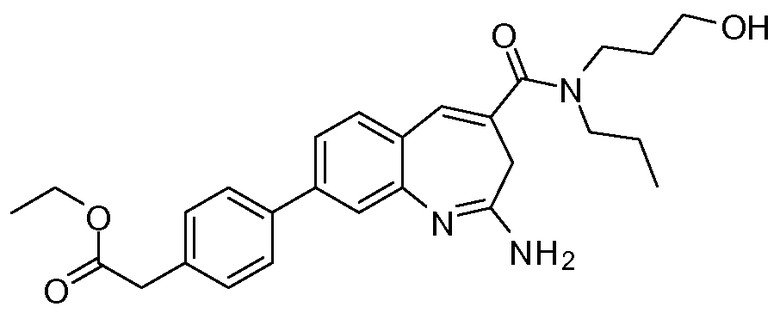
Этил-2-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат
Стадия А. Этил-2-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат (40%) получают как описано далее, заменяя 4-(этоксикарбонил)фенилбороновую кислоту на 4-(2-этокси-2-оксоэтил)фенилбороновую кислоту. Этил-4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)бензоат (44%) получают как описано далее, заменяя (1Е,4Е)-2-амино-8-бром-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид на трет-бутил-(1Е,4Е)-8-бром-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-2-илкарбамат и 4-(метоксикарбонил)фенилбороновую кислоту на 4-(этоксикарбонил)фенилбороновую кислоту. (1Е,4Е)-2-Амино-8-бром-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид (75,0 мг, 0,206 ммоль), 4-(метоксикарбонил)фенилбороновую кислоту (55,6 мг, 0,309 ммоль), тетракис(трифенилфосфин)палладий(0) (23,8 мг, 0,021 ммоль) и 2 М водный раствор карбоната калия (0,309 мл, 0,618 ммоль) соединяют в 2 мл ацетонитрила во флаконе для реакции под действием микроволнового излучения. Полученную смесь греют микроволновым излучением при 100°С в течение 30 минут. Затем смесь разбавляют EtOAc, дважды промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Затем используют препаративную тонкослойную хроматографию (пластины 2×0,5 мм, 7% МеОН/DCM). Этил-4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)бензоат (0,050 г, 0,075 ммоль) растворяют в 2 мл дихлорметана и 0,5 мл ТФК. Примерно через один час смесь концентрируют при пониженном давлении, и затем полученный остаток повторно растворяют в дихлорметане, добавляют 1 мл концентрированного гидроксида аммония, и смесь энергично перемешивают в течение 15 минут. Затем полученную смесь разбавляют водой, экстрагируют дихлорметаном (2×), и экстракты сушат над сульфатом натрия и концентрируют при пониженном давлении. Используют препаративную тонкослойную хроматографию (пластины 2×0,5 мм, 10% МеОН/DCM/0,5% NH4OH). 1H ЯМР (400 МГц, ДМСО-d6) δ 7,60-7,68 (м, 2H), 7,20-7,41 (м, 5H), 6,73-6,85 (м, 3H), 4,42-4,52 (м, 1H), 4,05-4,16 (м, 2H), 3,71 (с, 2H), 3,24-3,52 (м, 6H, частично затененный пиком воды), 2,75 (с, 2H), 1,66-1,78 (м, 2H), 1,50-1,65 (м, 2H), 1,15-1,23 (м, 3H), 0,75-0,92 (м, 3H); m/z (APCI-полож.) M+1=464,2.
Пример 215
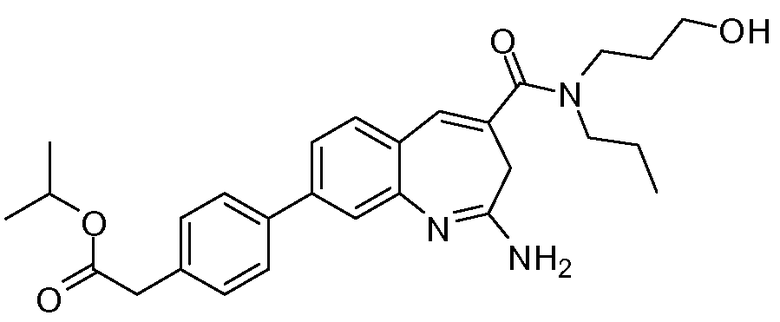
Изопропил-2-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат
Стадия А. Изопропил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)ацетат (59%) получают согласно описанному выше примеру 192, стадия А, заменяя этанол на 2-пропанол.
Стадия В. Изопропил-2-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат (42%) получают как описано далее, заменяя 4-(этоксикарбонил)фенилбороновую кислоту на изопропил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)ацетат. Этил-4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)бензоат (44%) получают как описано далее, заменяя (1Е,4Е)-2-амино-8-бром-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид на трет-бутил-(1Е,4Е)-8-бром-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-2-илкарбамат и 4-(метоксикарбонил)фенилбороновую кислоту на 4-(этоксикарбонил)фенилбороновую кислоту. (1Е,4Е)-2-Амино-8-бром-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид (75,0 мг, 0,206 ммоль), 4-(метоксикарбонил)фенилбороновую кислоту (55,6 мг, 0,309 ммоль), тетракис(трифенилфосфин)палладий(0) (23,8 мг, 0,021 ммоль) и 2 М водный раствор карбоната калия (0,309 мл, 0,618 ммоль) соединяют в 2 мл ацетонитрила во флаконе для реакции под действием микроволнового излучения. Полученную смесь греют микроволновым излучением при 100°С в течение 30 минут. Затем смесь разбавляют EtOAc, дважды промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Затем используют препаративную тонкослойную хроматографию (пластины 2×0,5 мм, 7% МеОН/DCM). Этил-4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)бензоат (0,050 г, 0,075 ммоль) растворяют в 2 мл дихлорметана и 0,5 мл ТФК. Примерно через один час смесь концентрируют при пониженном давлении, и затем полученный остаток повторно растворяют в дихлорметане, добавляют 1 мл концентрированного гидроксида аммония, и смесь энергично перемешивают в течение 15 минут. Затем полученную смесь разбавляют водой, экстрагируют дихлорметаном (2×), и экстракты сушат над сульфатом натрия и концентрируют при пониженном давлении. Используют препаративную тонкослойную хроматографию (пластины 2×0,5 мм, 10% МеОН/DCM/0,5% NH4OH). 1H ЯМР (400 МГц, CDCl3) δ 7,58-7,64 (м, 2H), 7,48-7,52 (м, 1H), 7,31-7,39 (м, 4H), 6,88 (с, 1H), 5,00-5,08 (м, 1H), 3,58-3,69 (м 6H), 3,44-3,51 (м, 2H), 2,85 (м, 2H), 1,79-1,88 (м, 2H), 1,65-1,75 (м, 2H), 1,23-1,28 (м, 6H), 0,90-0,97 (м, 3H); m/z (APCI-полож.) M+1=478,3.
Пример 216
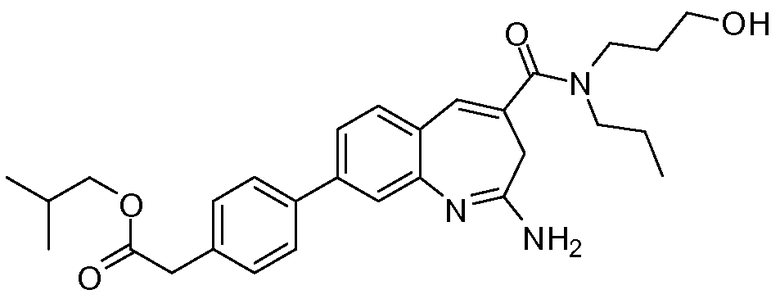
Изобутил-2-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат
Стадия А. Изобутил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)ацетат (54%) получают согласно описанному выше примеру 192, стадия А, заменяя этанол на 2-метилпропан-1-ол.
Стадия В. Изобутил-2-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат (40%) получают как описано далее, заменяя 4-(этоксикарбонил)фенилбороновую кислоту на изобутил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)ацетат. Этил-4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)бензоат (44%) получают как описано далее, заменяя (1Е,4Е)-2-амино-8-бром-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид на трет-бутил-(1Е,4Е)-8-бром-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-2-илкарбамат и 4-(метоксикарбонил)фенилбороновую кислоту на 4-(этоксикарбонил)фенилбороновую кислоту. (1Е,4Е)-2-Амино-8-бром-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид (75,0 мг, 0,206 ммоль), 4-(метоксикарбонил)фенилбороновую кислоту (55,6 мг, 0,309 ммоль), тетракис(трифенилфосфин)палладий(0) (23,8 мг, 0,021 ммоль) и 2 М водный раствор карбоната калия (0,309 мл, 0,618 ммоль) соединяют в 2 мл ацетонитрила во флаконе для реакции под действием микроволнового излучения. Полученную смесь греют микроволновым излучением при 100°С в течение 30 минут. Затем смесь разбавляют EtOAc, дважды промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Затем используют препаративную тонкослойную хроматографию (пластины 2×0,5 мм, 7% МеОН/DCM). Этил-4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)бензоат (0,050 г, 0,075 ммоль) растворяют в 2 мл дихлорметана и 0,5 мл ТФК. Примерно через один час смесь концентрируют при пониженном давлении, и затем полученный остаток повторно растворяют в дихлорметане, добавляют 1 мл концентрированного гидроксида аммония, и смесь энергично перемешивают в течение 15 минут. Затем полученную смесь разбавляют водой, экстрагируют дихлорметаном (2×), и экстракты сушат над сульфатом натрия и концентрируют при пониженном давлении. Используют препаративную тонкослойную хроматографию (пластины 2×0,5 мм, 10% МеОН/DCM/0,5% NH4OH). 1H ЯМР (400 МГц, CDCl3) δ 7,59-7,65 (м, 2H), 7,48-7,51 (м, 1H), 7,29-7,39 (м, 4H), 6,87 (с, 1H), 5,08 (ушир.с, 1H), 3,87-3,92 (м, 2H), 3,58-3,71 (м, 6H), 3,45-3,52 (м, 2H), 2,82 (с, 2H), 1,78-1,99 (м, 3H), 1,64-1,77 (м, 2H), 0,87-0,97 (м, 9H); m/z (APCI-полож.) M+1=492,2.
Пример 217
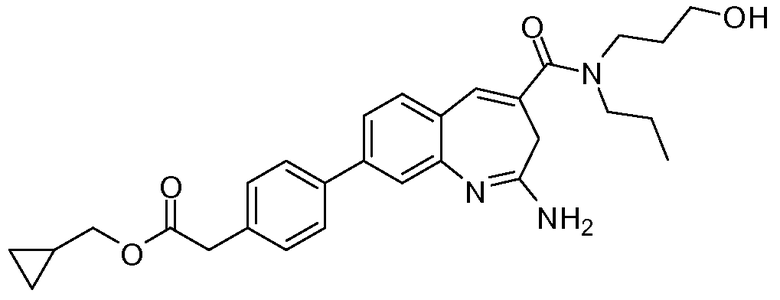
Циклопропилметил-2-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат
Стадия А. Циклопропилметил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)ацетат (47%) получают согласно описанному выше примеру 192, стадия А, заменяя этанол на циклопропилметанол.
Стадия В. Циклопропилметил-2-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат (31%) получают как описано далее, заменяя 4-(этоксикарбонил)фенилбороновую кислоту на циклопропилметил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)ацетат. Этил-4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)бензоат (44%) получают как описано далее, заменяя (1Е,4Е)-2-амино-8-бром-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид на трет-бутил-(1Е,4Е)-8-бром-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-2-илкарбамат и 4-(метоксикарбонил)фенилбороновую кислоту на 4-(этоксикарбонил)фенилбороновую кислоту. (1Е,4Е)-2-Амино-8-бром-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид (75,0 мг, 0,206 ммоль), 4-(метоксикарбонил)фенилбороновую кислоту (55,6 мг, 0,309 ммоль), тетракис(трифенилфосфин)палладий(0) (23,8 мг, 0,021 ммоль) и 2 М водный раствор карбоната калия (0,309 мл, 0,618 ммоль) соединяют в 2 мл ацетонитрила во флаконе для реакции под действием микроволнового излучения. Полученную смесь греют микроволновым излучением при 100°С в течение 30 минут. Затем смесь разбавляют EtOAc, дважды промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Затем используют препаративную тонкослойную хроматографию (пластины 2×0,5 мм, 7% МеОН/DCM). Этил-4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)бензоат (0,050 г, 0,075 ммоль) растворяют в 2 мл дихлорметана и 0,5 мл ТФК. Примерно через один час смесь концентрируют при пониженном давлении, и затем полученный остаток повторно растворяют в дихлорметане, добавляют 1 мл концентрированного гидроксида аммония, и смесь энергично перемешивают в течение 15 минут. Затем полученную смесь разбавляют водой, экстрагируют дихлорметаном (2×), и экстракты сушат над сульфатом натрия и концентрируют при пониженном давлении. Используют препаративную тонкослойную хроматографию (пластины 2×0,5 мм, 10% МеОН/DCM/0,5% NH4OH). 1H ЯМР (400 МГц, CDCl3) δ 7,59-7,65 (м, 2H), 7,47-7,51 (м, 1H), 7,28-7,40 (м, 4H), 6,88 (с, 1H), 5,18 (ушир.с, 1H), 3,92-3,99 (м, 2H), 3,58-3,72 (м, 6H), 3,43-3,53 (м, 2H), 2,83 (с, 2H), 1,79-1,88 (м, 2H), 1,65-1,77 (м, 2H), 1,08-1,20 (м, 1H), 0,90-0,98 (м, 3H), 0,53-0,60 (м, 2H), 0,24-0,31 (м, 2H); m/z (APCI-полож.) M+1=490,2.
Пример 218
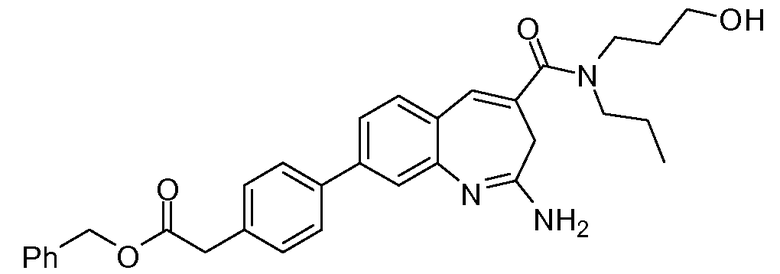
Бензил-2-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат
Стадия А. Бензил-2-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат (31%) получают как описано далее, заменяя 4-(этоксикарбонил)фенилбороновую кислоту на бензил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)ацетат. Этил-4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)бензоат (44%) получают как описано далее, заменяя (1Е,4Е)-2-амино-8-бром-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид на трет-бутил-(1Е,4Е)-8-бром-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-2-илкарбамат и 4-(метоксикарбонил)фенилбороновую кислоту на 4-(этоксикарбонил)фенилбороновую кислоту. (1Е,4Е)-2-Амино-8-бром-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид (75,0 мг, 0,206 ммоль), 4-(метоксикарбонил)фенилбороновую кислоту (55,6 мг, 0,309 ммоль), тетракис(трифенилфосфин)палладий(0) (23,8 мг, 0,021 ммоль) и 2 М водный раствор карбоната калия (0,309 мл, 0,618 ммоль) соединяют в 2 мл ацетонитрила во флаконе для реакции под действием микроволнового излучения. Полученную смесь греют микроволновым излучением при 100°С в течение 30 минут. Затем смесь разбавляют EtOAc, дважды промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Затем используют препаративную тонкослойную хроматографию (пластины 2×0,5 мм, 7% МеОН/DCM). Этил-4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)бензоат (0,050 г, 0,075 ммоль) растворяют в 2 мл дихлорметана и 0,5 мл ТФК. Примерно через один час смесь концентрируют при пониженном давлении, и затем полученный остаток повторно растворяют в дихлорметане, добавляют 1 мл концентрированного гидроксида аммония, и смесь энергично перемешивают в течение 15 минут. Затем полученную смесь разбавляют водой, экстрагируют дихлорметаном (2×), и экстракты сушат над сульфатом натрия и концентрируют при пониженном давлении. Используют препаративную тонкослойную хроматографию (пластины 2×0,5 мм, 10% МеОН/DCM/0,5% NH4OH). 1H ЯМР (400 МГц, CDCl3) δ 7,59-7,64 (м, 2H), 7,49-7,51 (м, 1H), 7,29-7,39 (м, 9H), 6,88 (с, 1H), 5,16 (с, 2H), 3,70 (с, 2H), 3,58-3,69 (м, 4H), 3,45-3,52 (м, 2H), 2,84 (с, 2H), 1,79-1,88 (м, 2H), 1,66-1,77 (м, 2H), 0,90-0,97 (м, 3H); m/z (APCI-полож.) M+1=526,2.
Пример 219
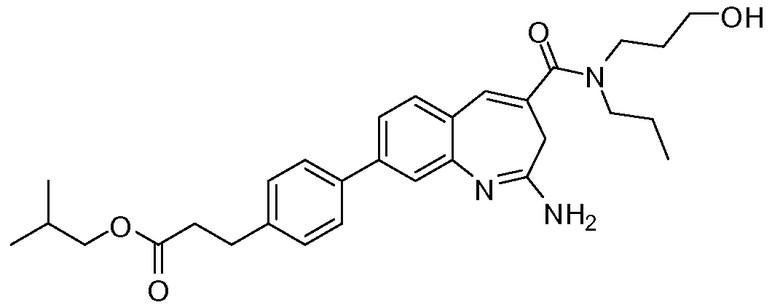
Изобутил-3-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)пропаноат
Стадия А. Изобутил-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропаноат (67%) получают согласно описанному выше примеру 192, стадия А, заменяя 2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)уксусную кислоту на 3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропановую кислоту и этанол на 2-метилпропан-1-ол.
Стадия В. Изобутил-3-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)пропаноат (31%) получают как описано далее, заменяя 4-(этоксикарбонил)фенилбороновую кислоту на изобутил-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропаноат. Этил-4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)бензоат (44%) получают как описано далее, заменяя (1Е,4Е)-2-амино-8-бром-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид на трет-бутил-(1Е,4Е)-8-бром-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-2-илкарбамат и 4-(метоксикарбонил)фенилбороновую кислоту на 4-(этоксикарбонил)фенилбороновую кислоту. (1Е,4Е)-2-Амино-8-бром-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид (75,0 мг, 0,206 ммоль), 4-(метоксикарбонил)фенилбороновую кислоту (55,6 мг, 0,309 ммоль), тетракис(трифенилфосфин)палладий(0) (23,8 мг, 0,021 ммоль) и 2 М водный раствор карбоната калия (0,309 мл, 0,618 ммоль) соединяют в 2 мл ацетонитрила во флаконе для реакции под действием микроволнового излучения. Полученную смесь греют микроволновым излучением при 100°С в течение 30 минут. Затем смесь разбавляют EtOAc, дважды промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Затем используют препаративную тонкослойную хроматографию (пластины 2×0,5 мм, 7% МеОН/DCM). Этил-4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)бензоат (0,050 г, 0,075 ммоль) растворяют в 2 мл дихлорметана и 0,5 мл ТФК. Примерно через один час смесь концентрируют при пониженном давлении, и затем полученный остаток повторно растворяют в дихлорметане, добавляют 1 мл концентрированного гидроксида аммония, и смесь энергично перемешивают в течение 15 минут. Затем полученную смесь разбавляют водой, экстрагируют дихлорметаном (2×), и экстракты сушат над сульфатом натрия и концентрируют при пониженном давлении. Используют препаративную тонкослойную хроматографию (пластины 2×0,5 мм, 10% МеОН/DCM/0,5% NH4OH). 1H ЯМР (400 МГц, CDCl3) δ 7,56-7,60 (м, 2H), 7,47-7,50 (м, 1H), 7,26-7,36 (м, 4H), 6,87 (с, 1H), 5,08 (ушир.с, 2H), 3,85-3,89 (м, 2H), 3,58-3,69 (м, 5H), 3,45-3,52 (м, 2H), 2,97-3,03 (м, 2H), 2,82 (с, 2H), 2,65-2,71 (м, 2H), 1,79-1,95 (м, 3H), 1,66-1,76 (м, 2H), 0,88-0,97 (м, 9H); m/z (APCI-полож.) M+1=506,3.
Пример 221
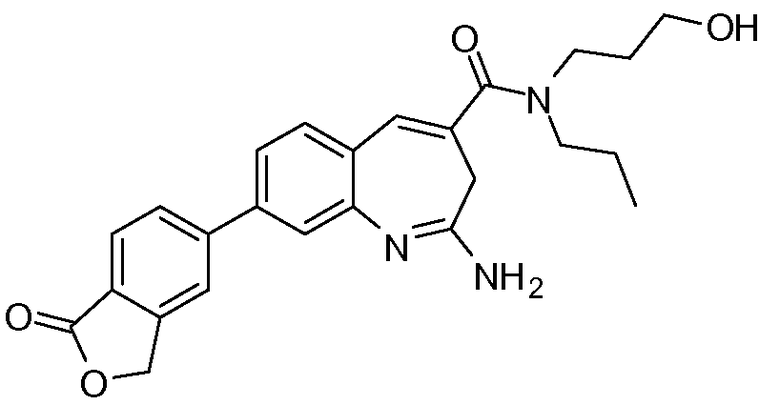
(1Е,4Е)-2-Амино-N-(3-гидроксипропил)-8-(1-оксо-1,3-дигидроизобензофуран-5-ил)-N-пропил-3Н-бензо[b]азепин-4-карбоксамид
Стадия А. 5-(4,4,5,5,-Тетраметил-1,3,2-диоксаборолан-2-ил)изобензофуран-1(3Н)-он (57%) получают согласно описанному ниже примеру 224/225, стадия С, заменяя 4-(4-бромфенил)-1,3-диоксолан-2-он на 5-бромизобензофуран-1(3Н)-он.
Стадия В. (1Е,4Е)-2-Амино-N-(3-гидроксипропил)-8-(1-оксо-1,3-дигидроизобензофуран-5-ил)-N-пропил-3Н-бензо[b]азепин-4-карбоксамид (33%) получают как описано далее, заменяя 4-(этоксикарбонил)фенилбороновую кислоту на 5-(4,4,5,5,-тетраметил-1,3,2-диоксаборолан-2-ил)изобензофуран-1(3Н)-он. Этил-4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)бензоат (44%) получают как описано далее, заменяя (1Е,4Е)-2-амино-8-бром-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид на трет-бутил-(1Е,4Е)-8-бром-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-2-илкарбамат и 4-(метоксикарбонил)фенилбороновую кислоту на 4-(этоксикарбонил)фенилбороновую кислоту. (1Е,4Е)-2-Амино-8-бром-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид (75,0 мг, 0,206 ммоль), 4-(метоксикарбонил)фенилбороновую кислоту (55,6 мг, 0,309 ммоль), тетракис(трифенилфосфин)палладий(0) (23,8 мг, 0,021 ммоль) и 2 М водный раствор карбоната калия (0,309 мл, 0,618 ммоль) соединяют в 2 мл ацетонитрила во флаконе для реакции под действием микроволнового излучения. Полученную смесь греют микроволновым излучением при 100°С в течение 30 минут. Затем смесь разбавляют EtOAc, дважды промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Затем используют препаративную тонкослойную хроматографию (пластины 2×0,5 мм, 7% МеОН/DCM). Этил-4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)бензоат (0,050 г, 0,075 ммоль) растворяют в 2 мл дихлорметана и 0,5 мл ТФК. Примерно через один час смесь концентрируют при пониженном давлении, и затем полученный остаток повторно растворяют в дихлорметане, добавляют 1 мл концентрированного гидроксида аммония, и смесь энергично перемешивают в течение 15 минут. Затем полученную смесь разбавляют водой, экстрагируют дихлорметаном (2×), и экстракты сушат над сульфатом натрия и концентрируют при пониженном давлении. Используют препаративную тонкослойную хроматографию (пластины 2×0,5 мм, 10% МеОН/DCM/0,5% NH4OH). 1H ЯМР (400 МГц, CDCl3) δ 7,95-8,00 (м, 1H), 7,78-7,82 (м, 1H), 7,70-7,73 (м, 1H), 7,50-7,53 (м, 1H), 7,37-7,42 (м, 1H), 7,28-7,33 (м, 1H), 6,89 (с, 1H), 5,37 (с, 2H), 5,12 (ушир.с, 1H), 3,58-3,71 (м, 4H), 3,43-3,55 (м, 3H), 2,82 (м, 2H), 1,79-1,88 (м, 2H), 1,64-1,77 (м, 2H), 0,90-0,99 (м, 3H); m/z (APCI-полож.) M+1=434,2.
Пример 222
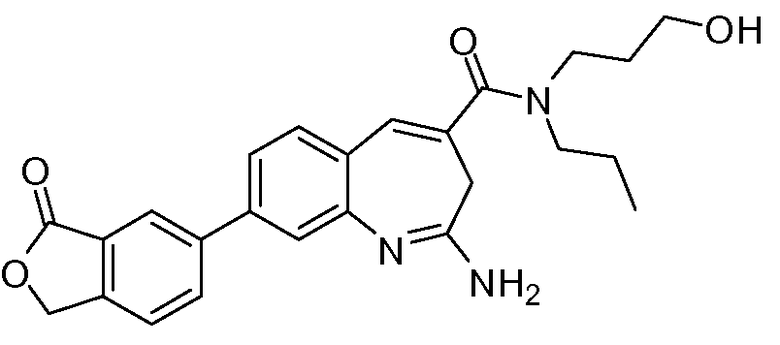
(1Е,4Е)-2-Амино-N-(3-гидроксипропил)-8-(3-оксо-1,3-дигидроизобензофуран-5-ил)-N-пропил-3Н-бензо[b]азепин-4-карбоксамид
Стадия А. 6-(4,4,5,5,-Тетраметил-1,3,2-диоксаборолан-2-ил)изобензофуран-1(3Н)-он (67%) получают согласно описанному ниже примеру 224/225, стадия С, заменяя 4-(4-бромфенил)-1,3-диоксолан-2-он на 6-бромизобензофуран-1(3Н)-он.
Стадия В. (1Е,4Е)-2-Амино-N-(3-гидроксипропил)-8-(3-оксо-1,3-дигидроизобензофуран-5-ил)-N-пропил-3Н-бензо[b]азепин-4-карбоксамид (36%) получают как описано далее, заменяя 4-(этоксикарбонил)фенилбороновую кислоту на 6-(4,4,5,5,-тетраметил-1,3,2-диоксаборолан-2-ил)изобензофуран-1(3Н)-он. Этил-4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)бензоат (44%) получают как описано далее, заменяя (1Е,4Е)-2-амино-8-бром-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид на трет-бутил-(1Е,4Е)-8-бром-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-2-илкарбамат и 4-(метоксикарбонил)фенилбороновую кислоту на 4-(этоксикарбонил)фенилбороновую кислоту. (1Е,4Е)-2-Амино-8-бром-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид (75,0 мг, 0,206 ммоль), 4-(метоксикарбонил)фенилбороновую кислоту (55,6 мг, 0,309 ммоль), тетракис(трифенилфосфин)палладий(0) (23,8 мг, 0,021 ммоль) и 2 М водный раствор карбоната калия (0,309 мл, 0,618 ммоль) соединяют в 2 мл ацетонитрила во флаконе для реакции под действием микроволнового излучения. Полученную смесь греют микроволновым излучением при 100°С в течение 30 минут. Затем смесь разбавляют EtOAc, дважды промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Затем используют препаративную тонкослойную хроматографию (пластины 2×0,5 мм, 7% МеОН/DCM). Этил-4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)бензоат (0,050 г, 0,075 ммоль) растворяют в 2 мл дихлорметана и 0,5 мл ТФК. Примерно через один час смесь концентрируют при пониженном давлении, и затем полученный остаток повторно растворяют в дихлорметане, добавляют 1 мл концентрированного гидроксида аммония, и смесь энергично перемешивают в течение 15 минут. Затем полученную смесь разбавляют водой, экстрагируют дихлорметаном (2×), и экстракты сушат над сульфатом натрия и концентрируют при пониженном давлении. Используют препаративную тонкослойную хроматографию (пластины 2×0,5 мм, 10% МеОН/DCM/0,5% NH4OH). 1H ЯМР (400 МГц, ДМСО-d6) δ 8,08-8,13 (м, 1H), 8,03-8,06 (м, 1H), 7,74-7,78 (м, 1H), 7,41-7,45 (м, 1H), 7,32-7,36 (м, 2H), 6,91 (ушир.с, 2H), 6,80 (с, 1H), 5,47 (с, 2H), 4,45-4,50 (м, 1H), 53,37-3,48 (м, 2H), 3,28-3,37 (м, 4H, затененный пиком воды), 2,75 (с, 2H), 1,66-1,77 (м, 2H), 1,51-1,62 (м, 2H), 0,72-0,91 (м, 3H); m/z (APCI-полож.) M+1=434,2.
Пример 223
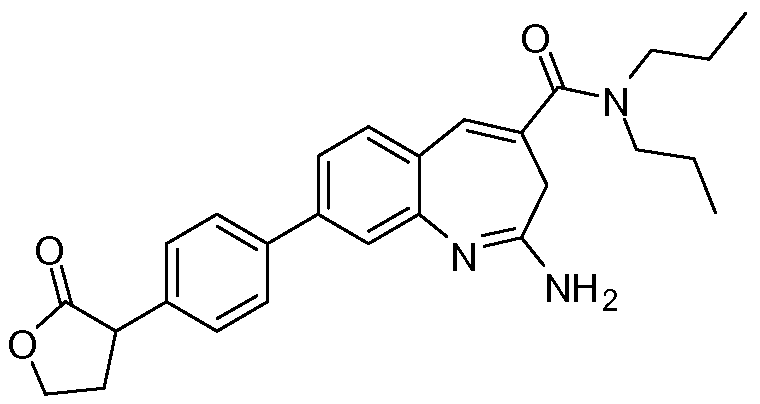
(1Е,4Е)-2-Амино-8-(4-(2-оксотетрагидрофуран-3-ил)фенил)-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид
Стадия А. 4-Бромфенилуксусную кислоту (1,00 г, 4,65 ммоль) растворяют в 45 мл сухого ТГФ и охлаждают до -78°С. К полученной смеси добавляют LDA (2 М раствор в смеси ТГФ/гептан, 9,30 мл, 18,6 ммоль), что приводит к потеменению смеси, и такую смесь перемешивают при -78°С в течение 20 минут. Затем добавляют через шприц (2-бромэтокси)(трет-бутил)диметилсилан (1,20 мл, 5,58 ммоль), затем охлаждающую баню убирают, и оставляют реакционную смесь нагреваться до комнатной температуры. Когда смесь нагреется, она становится янтарного цвета. Затем при комнатной температуре добавляют 50 мл 1 N соляной кислоты, и смесь энергично перемешивают в течение 16 час, затем экстрагируют EtOAc (2×), и экстракты промывают 2 М водным раствором карбоната натрия, сушат над сульфатом натрия и концентрируют до оранжевого масла. Обработка на Flash 40 Biotage (картридж 40М, EtOAc/гексан 3:1) дает 211 мг (19%) 3-(4-бромфенил)дигидрофуран-2(3Н)-она в виде оранжевого масла.
Стадия В. 3-(4-(4,4,5,5-Тетраметил-1,3,2-диоксаборолан-2-ил)фенил)дигидрофуран-2(3Н)-он (40%) получают согласно описанному ниже примеру 224/225, стадия С.
Стадия С. (1Е,4Е)-2-Амино-8-(4-(2-оксотетрагидрофуран-3-ил)фенил)-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид (9%) получают как описано далее, заменяя 4-(метоксикарбонил)фенилбороновую кислоту на 3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)дигидрофуран-2(3Н)-он. (1Е,4Е)-2-Амино-8-бром-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид (75,0 мг, 0,206 ммоль), 4-(метоксикарбонил)фенилбороновую кислоту (55,6 мг, 0,309 ммоль), тетракис(трифенилфосфин)палладий(0) (23,8 мг, 0,021 ммоль) и 2 М водный раствор карбоната калия (0,309 мл, 0,618 ммоль) соединяют в 2 мл ацетонитрила во флаконе для реакции под действием микроволнового излучения. Полученную смесь греют микроволновым излучением при 100°С в течение 30 минут. Затем смесь разбавляют EtOAc, дважды промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Затем используют препаративную тонкослойную хроматографию (пластины 2×0,5 мм, 7% МеОН/DCM). 1H ЯМР (400 МГц, CDCl3) δ 7,64-7,68 (м, 2H), 7,51-7,54 (м, 1H), 7,32-7,39 (м, 4H), 6,84 (с, 1H), 4,48-4,55 (м, 1H), 4,33-4,42 (м, 1H), 3,83-3,90 (м, 1H), 3,41-3,50 (м, 4H), 2,89 (с, 2H), 2,70-2,81 (м, 1H), 2,44-2,56 (м, 1H), 1,59-1,72 (м, 4H), 0,88-0,98 (м, 6H); m/z (APCI-полож.) M+1=446,2.
Пример 224
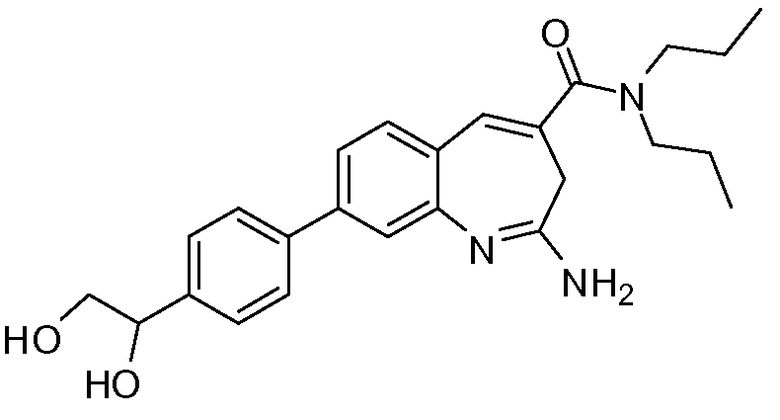
(1Е,4Е)-2-Амино-8-(4-(1,2-дигидроксиэтил)фенил)-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид
Пример 225
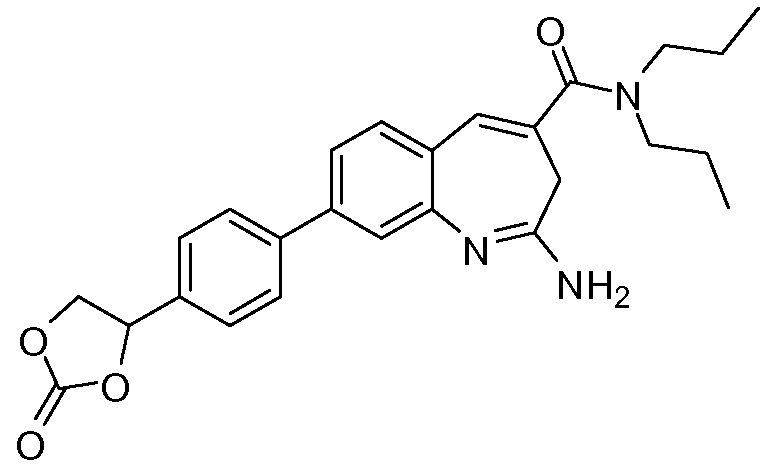
(1Е,4Е)-2-Амино-8-(4-(2-оксо-1,3-диоксолан-4-ил)фенил)-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид
(1Е,4Е)-2-Амино-8-(4-(1,2-дигидроксиэтил)фенил)-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид
Стадия А. 4-Бромстирол (1,00 г, 5,46 ммоль) растворяют в 25 мл смеси воды с ацетоном, 2:1. К полученному раствору добавляют N-оксид 4-метилморфолина (0,704 г, 6,01 ммоль) и тетроксид осмия (0,167 г, 0,061 ммоль, 2,5% раствор в трет-BuOH). Полученную смесь перемешивают при комнатной температуре в течение 16 часов, затем разбавляют EtOAc, промывают один раз 1 N соляной кислотой, один раз насыщенным раствором бикарбоната натрия, сушат над сульфатом натрия и концентрируют при пониженном давлении. Полученный сырой продукт очищают на Flash 40 Biotage (картридж 40М, 100% EtOAc в качестве элюента), и получают 335 мг (28%) 1-(4-бромфенил)этан-1,2-диола в виде белого твердого вещества.
Стадия В. 1-(4-Бромфенил)этан-1,2-диол (0,15 г, 0,69 ммоль) растворяют в 7 мл сухого ацетонитрила, и затем добавляют карбонилдиимидазол (0,17 г, 1,04 ммоль). Полученную смесь греют при 40°С в течение 1,5 часов. После охлаждения до комнатной температуры реакционную смесь разбавляют EtOAc и промывают насыщенным раствором хлорида аммония и водой, сушат над сульфатом натрия и концентрируют при пониженном давлении. Очистка на 10 г Sep Pak (EtOAc:гексан 1:1) дает 0,095 г 4-(4-бромфенил)-1,3-диоксолан-2-она (57%) в виде белого твердого вещества.
Стадия С. 4-(4-Бромфенил)-1,3-диоксолан-2-он (0,093 г, 0,383 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-ди(1,3,2-диксаборолан) (0,117 г, 0,459 ммоль), ацетат калия (0,113 г, 1,15 ммоль) и аддукт дихлор-1,1'-бис(дифенилфосфино)ферроценпалладия(II) и дихлорметана (0,0094 г, 0,012 ммоль) соединяют в диоксане и греют при 100°С в течение 3 часов. Затем смесь разбавляют водой, дважды экстрагируют EtOAc, и экстракты сушат над сульфатом натрия и концентрируют при пониженном давлении. Очистка на 10 г Sep Pak (100% DCM в качестве элюента) дает 0,070 г 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-1,3-диоксолан-2-она (63%) в виде белого твердого вещества.
Стадия D. Взаимодействие (1Е,4Е)-2-амино-8-бром-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамида и 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-1,3-диоксолан-2-она, как описано далее, дает (1Е,4Е)-2-амино-8-(4-(2-оксо-1,3-диоксолан-4-ил)фенил)-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид (9%). (1Е,4Е)-2-Амино-8-бром-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид (75,0 мг, 0,206 ммоль), 4-(метоксикарбонил)фенилбороновую кислоту (55,6 мг, 0,309 ммоль), тетракис(трифенилфосфин)палладий(0) (23,8 мг, 0,021 ммоль) и 2 М водный раствор карбоната калия (0,309 мл, 0,618 ммоль) соединяют в 2 мл ацетонитрила во флаконе для реакции под действием микроволнового излучения. Полученную смесь греют микроволновым излучением при 100°С в течение 30 минут. Затем смесь разбавляют EtOAc, дважды промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Затем используют препаративную тонкослойную хроматографию (пластины 2×0,5 мм, 7% МеОН/DCM). 1H ЯМР (400 МГц, ДМСО-d6) δ 7,74-7,80 (м, 2H), 7,55-7,60 (м, 2H), 7,26-7,42 (м, 3H), 6,78 (с, 1H), 5,89-5,96 (м, 1H), 4,88-4,95 (м, 1H), 4,45-4,52 (м, 1H), 2,78 (с, 2H), 1,50-1,63 (м, 4H), 0,71-0,93 (м, 6H); m/z (APCI-полож.) M+1=448,2. Как более полярный продукт получают (1Е,4Е)-2-амино-8-(4-(1,2-дигидроксиэтил)фенил)-N,N-дипропил-3Н-бензо[b]азепин-4-карбоксамид (19%). 1H ЯМР (400 МГц, ДМСО-d6) δ 7,59-7,64 (м, 2H), 7,39-7,44 (м, 2H), 7,33-7,38 (м, 1H), 7,26-7,28 (м, 1H), 7,20-7,24 (м, 1H), 6,84 (ушир.с, 1H), 6,74 (с, 1H), 5,23-5,27 (м, 1H), 4,71-4,75 (м, 1H), 4,54-4,62 (м, 1H), 3,44-3,50 (м, 2H), 3,28-3,35 (м, 4H), 2,74 (с, 2H), 1,51-1,61 (м, 4H), 0,73-0,90 (м, 6H); m/z (APCI-полож.) M+1=422,2.
Пример 226
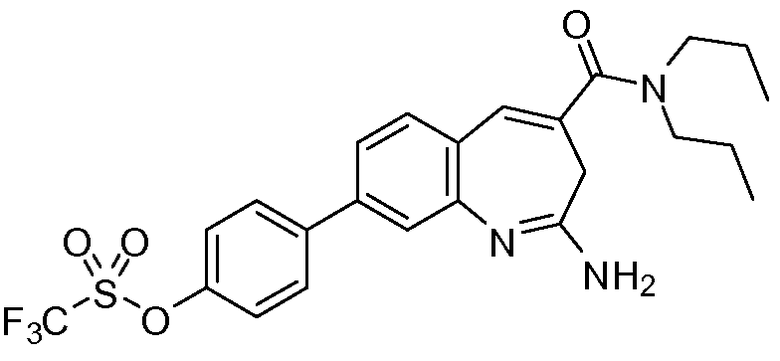
4-((1Е,4Е)-2-Амино-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-8-ил)фенилтрифторметансульфонат
Стадия А. Получение трет-бутил-(1Е,4Е)-4-(дипропилкарбамоил)-8-(4-гидроксифенил)-3Н-бензо[b]азепин-2-илкарбамата. Названное в заголовке соединение получают указанными процедурами с использованием трет-бутил-(1Е,4Е)-8-бром-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-2-илкарбамата и 4-гидроксифенилбороновой кислоты. Получение трет-бутил-(1Е,4Е)-8-(4-(дипропилкарбамоил)фенил)-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-2-илкарбамата. К Na2CO3 (129 мг, 1,214 ммоль) в 50-мл круглодонной колбе добавляют воду (3,7 мл), и барботируют N2 в течение 10 мин. К полученной смеси при комнатной температуре добавляют трет-бутил-(1Е,4Е)-8-бром-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-2-илкарбамат (200 мг, 0,40 ммоль) в EtOH (4,9 мл). Через полученную смесь барботируют N2 в течение 10 мин. Добавляют Pd(OAc)2 (9,3 мг, 0,040 ммоль) и гидрат дикалиевой соли 4,4'-(фенилфосфиниден)бисбензолсульфоновой кислоты (45 мг, 0,081 ммоль). Полученную смесь греют при 65°С при барботировании N2. К полученной смеси добавляют раствор 4-(диметилкарбамоил)фенилбороновой кислоты (97 мг, 0,49 ммоль) в EtOH (0,6 мл). Полученную смесь перемешивают при 65°С в течение 1 часа. Реакционную смесь охлаждают до комнатной температуры, концентрируют при пониженном давлении, и получают сырой материал, который разбавляют водой (5 мл) и EtOAc (10 мл). Смесь фильтруют через фильтр GF/F. Водный слой отделяют и экстрагируют EtOAc (10 мл). Объединенные органические слои промывают рассолом (10 мл), сушат над MgSO4, фильтруют и концентрируют при пониженном давлении, и получают сырой продукт, который очищают колоночной флэш-хроматографией на силикагеле (от CH2Cl2 до 2% МеОН в CH2Cl2). МС APCI(+) детектирует m/z 478 (М+1).
Стадия В. Получение 4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-8-ил)фенил)трифторметансульфоната. К раствору трет-бутил-(1Е,4Е)-4-(дипропилкарбамоил)-8-(4-(гидроксифенил)-3Н-бензо[b]азепин-2-илкарбамата (699 мг, 1,30 ммоль) и 1,1,1-трифтор-N-фенил-N-(трифторметилсульфонил)метансульфонамида (698 мг, 1,95 ммоль) в CH2Cl2 (7 мл) при комнатной температуре добавляют ТЕА (0,27 мл, 1,95 ммоль). После перемешивания в течение 23 часов при комнатной температуре реакционную смесь разбавляют CH2Cl2 (25 мл) и промывают насыщенным водным раствором NaHCO3 (15 мл), а затем рассолом (15 мл). Органический слой сушат над MgSO4, фильтруют и концентрируют при пониженном давлении, и получают сырой материал, который очищают колоночной флэш-хроматографией на силикагеле (градиент 10-30% EtOAc в гексане), и получают 526 мг (66%) 4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-8-ил)фенил)трифторметансульфоната. МС APCI(+) детектирует m/z 610 (М+1).
Стадия С. Получение 4-((1Е,4Е)-2-амино-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-8-ил)фенилтрифторметансульфоната. Названное в заголовке соединение получают указанными процедурами.
Получение (1Е,4Е)-2-амино-N-пропил-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-4-карбоксамида. К раствору трет-бутил-(1Е,4Е)-4-(пропилкарбамоил)-8-(4-(пирролидин-1-карбонил)фенил)-3Н-бензо[b]азепин-2-илкарбамата (450 мг, 0,87 ммоль) в CH2Cl2 (5 мл) при 0°С добавляют 2,2,2-трифторуксусную кислоту (1,36 мл, 17,4 ммоль). Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 2 час. Реакционную смесь концентрируют при пониженном давлении, и получают сырой материал, который снова разбавляют CH2Cl2 (10 мл) и насыщенным водным раствором NaHCO3 (15 мл). Полученную смесь перемешивают в течение 30 мин при комнатной температуре. Водный слой отделяют и экстрагируют CH2Cl2 (1×10 мл). Объединенные органические слои промывают насыщенным водным раствором NaHCO3 (2×10 мл) и рассолом (1×10 мл), сушат над MgSO4, фильтруют и концентрируют при пониженном давлении, и получают снова сырой материал, который очищают колоночной флэш-хроматографией на силикагеле (градиент 1-5% МеОН в CH2Cl2). МС APCI(+) детектирует m/z 510 (М+1). 1H-ЯМР (400 МГц, CDCl3) δ 7,69-7,72 (м, 2H), 7,45 (д, 1H), 7,33-7,37 (м, 3H), 7,24-7,26 (м, 1H), 6,83 (с, 1H), 3,47 (ушир.с, 4H), 2,81 (с, 2H), 1,62-1,72 (м, 4H), 0,94 (т, 6H); 19F-ЯМР (376 МГц, CDCl3) δ -73,2.
Пример 227
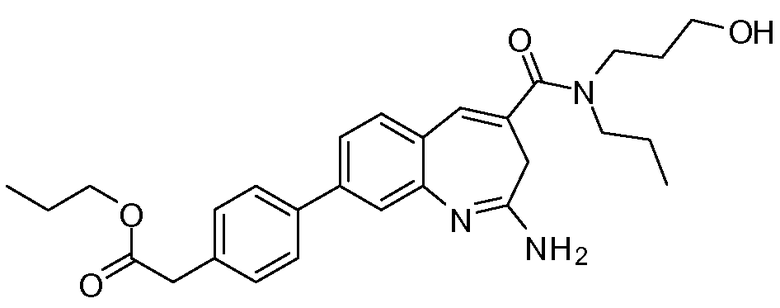
Пропил-2-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат
Стадия А. Пропил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)ацетат (43%) получают согласно примеру 192, стадия А, заменяя этанол на пропанол.
Стадия В. Пропил-2-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат (10%) получают как описано далее.
Стадия 1. трет-Бутил-(1Е,4Е)-8-бром-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-2-илкарбамат, пропил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)ацетат (1,5 эквив.), тетракис(трифенилфосфин)палладий(0) и 2 М водный раствор карбоната калия (3 эквив.) соединяют в 2 мл ацетонитрила во флаконе для реакции под действием микроволнового излучения. Полученную смесь греют микроволновым излучением при 100°С в течение 30 минут. Затем смесь разбавляют EtOAc, дважды промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Препаративная тонкослойная хроматография (пластины 2×0,5 мм, 7% МеОН/DCM) дает пропил-2-(4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат.
Стадия 2. Пропил-2-(4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат растворяют в 2 мл дихлорметана и 0,5 мл ТФК. Примерно через один час смесь концентрируют при пониженном давлении, и затем полученный остаток повторно растворяют в дихлорметане, добавляют 1 мл концентрированного гидроксида аммония, и смесь энергично перемешивают в течение 15 минут. Затем полученную смесь разбавляют водой, экстрагируют дихлорметаном (2×), и экстракты сушат над сульфатом натрия и концентрируют при пониженном давлении. Препаративная тонкослойная хроматография (пластины 2×0,5 мм, 10% МеОН/DCM/0,5% NH4OH) дает пропил-2-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,60-7,68 (м, 2H), 7,20-7,41 (м, 5H), 6,73-6,85 (м, 3H), 4,42-4,52 (м, 1H), 4,05-4,16 (м, 2H), 3,71 (с, 2H), 3,24-3,52 (м, 6H, частично затененный пиком воды), 2,75 (с, 2H), 1,66-1,78 (м, 2H), 1,50-1,65 (м, 2H), 1,15-1,23 (м, 3H), 0,75-0,92 (м, 3H); m/z (APCI-полож.) M+1=464,2.
Пример 228
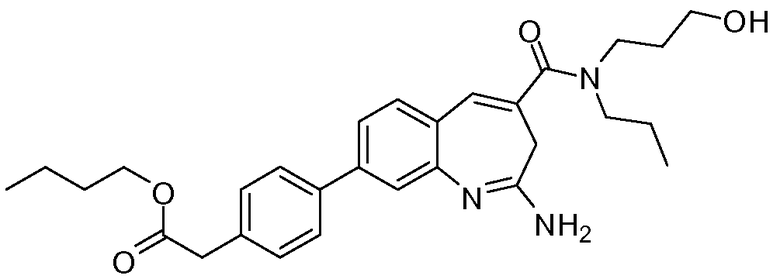
Бутил-2-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат
Стадия А. Бутил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)ацетат (40%) получают согласно примеру 192, стадия А, заменяя этанол на 1-бутанол.
Стадия В. Бутил-2-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат (8%) получают как описано далее.
Стадия 1. трет-Бутил-(1Е,4Е)-8-бром-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-2-илкарбамат, бутил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)ацетат (1,5 эквив.), тетракис(трифенилфосфин)палладий(0) и 2 М водный раствор карбоната калия (3 эквив.) соединяют в 2 мл ацетонитрила во флаконе для реакции под действием микроволнового излучения. Полученную смесь греют микроволновым излучением при 100°С в течение 30 минут. Затем смесь разбавляют EtOAc, дважды промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Препаративная тонкослойная хроматография (пластины 2×0,5 мм, 7% МеОН/DCM) дает бутил-2-(4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат.
Стадия 2. Бутил-2-(4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат растворяют в 2 мл дихлорметана и 0,5 мл ТФК. Примерно через один час смесь концентрируют при пониженном давлении, и затем полученный остаток повторно растворяют в дихлорметане, добавляют 1 мл концентрированного гидроксида аммония, и смесь энергично перемешивают в течение 15 минут. Затем полученную смесь разбавляют водой, экстрагируют дихлорметаном (2×), и экстракты сушат над сульфатом натрия и концентрируют при пониженном давлении. Препаративная тонкослойная хроматография (пластины 2×0,5 мм, 10% МеОН/DCM/0,5% NH4OH) дает бутил-2-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат. 1H ЯМР (400 МГц, CDCl3) δ 7,59-7,63 (м, 2H), 7,50-7,51 (м, 1H), 7,34-7,38 (м2Н), 7,32-7,34 (м, 2H), 6,88 (с, 2H), 4,09-4,14 (м, 2H), 3,58-3,68 (м, 6H), 3,45-3,51 (м, 2H), 2,87 (с, 2H), 1,79-1,88 (м, 2H), 1,57-1,76 (м, 4H), 1,30-1,42 (м, 2H), 0,89-0,96 (м, 6H); m/z (APCI-полож.) M+1=492,2.
Пример 229
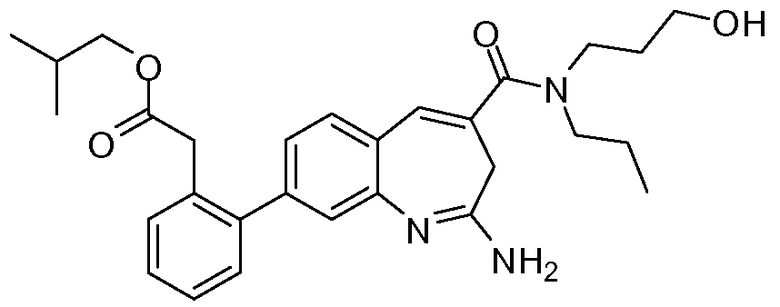
Изобутил-2-(2-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат
Стадия А. 2-(2-Бромфенил)уксусную кислоту (1,00 г, 4,65 ммоль) растворяют в 45 мл дихлорметана. К полученному раствору добавляют оксалилхлорид (0,609 мл, 6,98 ммоль), а затем каплю ДМФА. Полученную смесь перемешивают в течение двух часов при комнатной температуре и затем концентрируют при пониженном давлении. Затем полученный остаток повторно растворяют в сухом дихлорметане (45 мл), затем добавляют 2 мл 2-метилпропан-1-ола, и смесь перемешивают при комнатной температуре в течение 1,5 часов и затем концентрируют до изобутил-2-(2-бромфенил)ацетата (100%).
Стадия В. Изобутил-2-(2-(4,4,5,5,-тетраметил-1,3,2-диоксаборолан-2-ил)фенилацетат (49%) получают согласно примеру 224/225, стадия С, заменяя 4-(4-бромфенил)-1,3-диоксолан-2-он на изобутил-2-(2-бромфенил)ацетат.
Стадия С. Изобутил-2-(2-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат (30%) получают как описано далее.
Стадия 1. трет-Бутил-(1Е,4Е)-8-бром-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-2-илкарбамат, изобутил-2-(2-(бромфенил)ацетат (1,5 эквив.), тетракис(трифенилфосфин)палладий(0) и 2 М водный раствор карбоната калия (3 эквив.) соединяют в 2 мл ацетонитрила во флаконе для реакции под действием микроволнового излучения. Полученную смесь греют микроволновым излучением при 100°С в течение 30 минут. Затем смесь разбавляют EtOAc, дважды промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Препаративная тонкослойная хроматография (пластины 2×0,5 мм, 7% МеОН/DCM) дает изобутил-2-(4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат.
Стадия 2. Изобутил-2-(4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат растворяют в 2 мл дихлорметана и 0,5 мл ТФК. Примерно через один час смесь концентрируют при пониженном давлении, и затем полученный остаток повторно растворяют в дихлорметане, добавляют 1 мл концентрированного гидроксида аммония, и смесь энергично перемешивают в течение 15 минут. Затем полученную смесь разбавляют водой, экстрагируют дихлорметаном (2×), и экстракты сушат над сульфатом натрия и концентрируют при пониженном давлении. Препаративная тонкослойная хроматография (пластины 2×0,5 мм, 10% МеОН/DCM/0,5% NH4OH) дает изобутил-2-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат. 1H ЯМР (400 МГц, CDCl3) δ 7,27-7,39 (м, 5H), 7,19-7,21 (м, 1H), 7,01-7,06 (м, 1H), 6,87 (с, 1H), 3,80-3,85 (м, 2H), 3,58-3,69 (м, 6H), 3,46-3,53 (м, 2H), 2,82 (с, 2H), 1,80-1,89 (м, 3H), 1,66-1,75 (м, 2H), 0,90-0,98 (м, 3H), 0,81-0,87 (м, 6H); m/z (APCI-полож.) M+1=492,3.
Пример 230
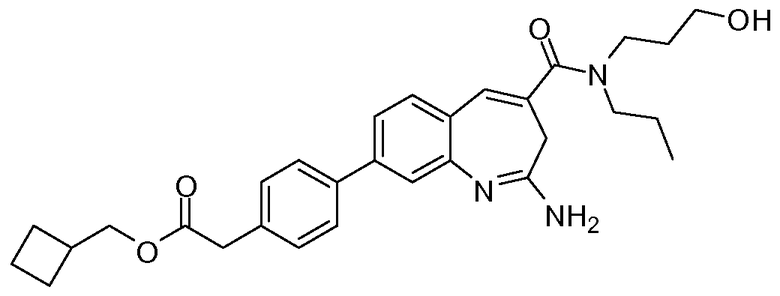
Циклобутилметил-2-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат
Стадия А. Циклобутилметил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)ацетат (100%) получают согласно примеру 192, стадия А, заменяя этанол на циклобутилметанол.
Стадия В. Циклобутилметил-2-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат (10%) получают как описано далее.
Стадия 1. трет-Бутил-(1Е,4Е)-8-бром-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-2-илкарбамат, циклобутилметил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)ацетат (1,5 эквив.), тетракис(трифенилфосфин)палладий(0) и 2 М водный раствор карбоната калия (3 эквив.) соединяют в 2 мл ацетонитрила во флаконе для реакции под действием микроволнового излучения. Полученную смесь греют микроволновым излучением при 100°С в течение 30 минут. Затем смесь разбавляют EtOAc, дважды промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Препаративная тонкослойная хроматография (пластины 2×0,5 мм, 7% МеОН/DCM) дает циклобутилметил-2-(4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат.
Стадия 2. Циклобутилметил-2-(4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат растворяют в 2 мл дихлорметана и 0,5 мл ТФК. Примерно через один час смесь концентрируют при пониженном давлении, и затем полученный остаток повторно растворяют в дихлорметане, добавляют 1 мл концентрированного гидроксида аммония, и смесь энергично перемешивают в течение 15 минут. Затем полученную смесь разбавляют водой, экстрагируют дихлорметаном (2×), и экстракты сушат над сульфатом натрия и концентрируют при пониженном давлении. Препаративная тонкослойная хроматография (пластины 2×0,5 мм, 10% МеОН/DCM/0,5% NH4OH) дает циклобутилметил-2-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат. 1H ЯМР (400 МГц, CDCl3) δ 7,59-7,64 (м, 2H), 7,49-7,51 (м, 1H), 7,29-7,39 (м, 4H), 6,87 (с, 1H), 4,06-4,12 (м, 2H), 3,57-3,70 (м, 6H), 3,44-3,53 (м, 2H), 2,83 (с, 2H), 2,56-2,67 (м, 1H), 1,96-2,07 (м, 2H), 1,66-1,92 (м, 8H), 0,91-0,97 (м, 3H); m/z (APCI-полож.) M+1=504,2.
Пример 231
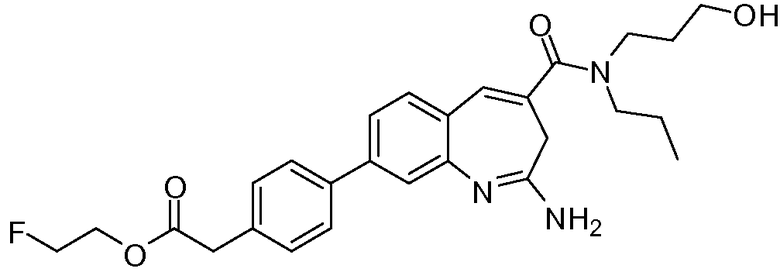
2-Фторэтил-2-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат
Стадия А. 2-Фторэтил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)ацетат (35%) получают согласно примеру 192, стадия А, заменяя этанол на 2-фторэтанол.
Стадия В. 2-Фторэтил-2-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат (25%) получают как описано далее.
Стадия 1. трет-Бутил-(1Е,4Е)-8-бром-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-2-илкарбамат, 2-фторэтил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)ацетат (1,5 эквив.), тетракис(трифенилфосфин)палладий(0) и 2 М водный раствор карбоната калия (3 эквив.) соединяют в 2 мл ацетонитрила во флаконе для реакции под действием микроволнового излучения. Полученную смесь греют микроволновым излучением при 100°С в течение 30 минут. Затем смесь разбавляют EtOAc, дважды промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Препаративная тонкослойная хроматография (пластины 2×0,5 мм, 7% МеОН/DCM) дает 2-фторэтил-2-(4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат.
Стадия 2. 2-Фторэтил-2-(4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат растворяют в 2 мл дихлорметана и 0,5 мл ТФК. Примерно через один час смесь концентрируют при пониженном давлении, и затем полученный остаток повторно растворяют в дихлорметане, добавляют 1 мл концентрированного гидроксида аммония, и смесь энергично перемешивают в течение 15 минут. Затем полученную смесь разбавляют водой, экстрагируют дихлорметаном (2×), и экстракты сушат над сульфатом натрия и концентрируют при пониженном давлении. Препаративная тонкослойная хроматография (пластины 2×0,5 мм, 10% МеОН/DCM/0,5% NH4OH) дает 0,012 г (35%) 2-фторэтил-2-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетата. 1H ЯМР (400 МГц, CDCl3) δ 7,60-7,65 (м, 2H), 7,49-7,51 (м, 1H), 7,29-7,39 (м, 4H), 6,87 (с, 1H), 4,66-4,69 (м, 1H), 4,54-4,58 (м, 1H), 4,39-4,43 (м, 1H), 4,32-4,36 (м, 1H), 3,73 (с, 2H), 3,58-3,68 (м, 4H), 3,43-3,52 (м, 2H), 1,78-1,87 (м, 2H), 1,65-1,75 (м, 2H), 0,90-0,96 (м, 3H); m/z (APCI-полож.) M+1=482,2.
Пример 232
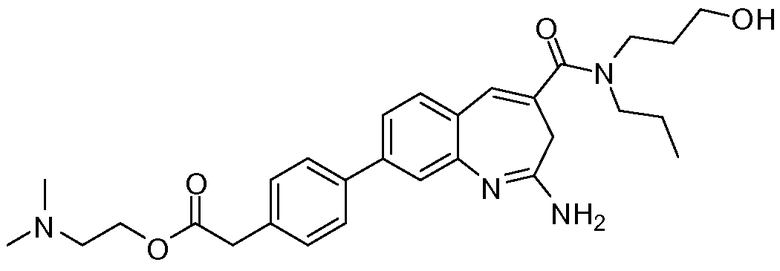
2-(Диметиламино)этил-2-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат
Стадия А. 2-(Диметиламино)этил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)ацетат (60%) получают согласно примеру 192, стадия А, заменяя этанол на 2-(диметиламино)этанол.
Стадия В. 2-(Диметиламино)этил-2-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат (5%) получают как описано далее.
Стадия 1. трет-Бутил-(1Е,4Е)-8-бром-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-2-илкарбамат, 2-(диметиламино)этил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)ацетат (1,5 эквив.), тетракис(трифенилфосфин)палладий(0) и 2 М водный раствор карбоната калия (3 эквив.) соединяют в 2 мл ацетонитрила во флаконе для реакции под действием микроволнового излучения. Полученную смесь греют микроволновым излучением при 100°С в течение 30 минут. Затем смесь разбавляют EtOAc, дважды промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Препаративная тонкослойная хроматография (пластины 2×0,5 мм, 7% МеОН/DCM) дает 2-(диметиламино)этил-2-(4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат.
Стадия 2. 2-(Диметиламино)этил-2-(4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат растворяют в 2 мл дихлорметана и 0,5 мл ТФК. Примерно через один час смесь концентрируют при пониженном давлении, и затем полученный остаток повторно растворяют в дихлорметане, добавляют 1 мл концентрированного гидроксида аммония, и смесь энергично перемешивают в течение 15 минут. Затем полученную смесь разбавляют водой, экстрагируют дихлорметаном (2×), и экстракты сушат над сульфатом натрия и концентрируют при пониженном давлении. Препаративная тонкослойная хроматография (пластины 2×0,5 мм, 10% МеОН/DCM/0,5% NH4OH) дает 2-(диметиламино)этил-2-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат. 1H ЯМР (400 МГц, CDCl3) δ 7,59-7,64 (м, 2H), 7,48-7,51 (м, 1H), 7,28-7,38 (м, 4H), 6,87 (с, 1H), 4,19-4,24 (м, 2H), 3,58-3,74 (м, 6H), 3,44-3,52 (м, 2H), 2,84 (с, 2H), 2,54-2,61 (м, 2H), 2,27 (с, 6H), 1,79-1,86 (м, 2H), 1,65-1,76 (м, 2H), 0,89-0,97 (м, 3H); m/z (APCI-полож.) M+1=507,2.
Пример 233
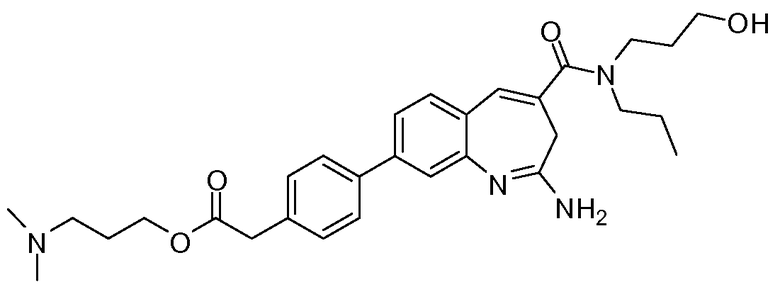
3-(Диметиламино)пропил-2-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат
Стадия А. 3-(Диметиламино)пропил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)ацетат (45%) получают согласно примеру 192, стадия А, заменяя этанол на 3-(диметиламино)пропан-1-ол.
Стадия В. 3-(Диметиламино)пропил-2-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат (6%) получают как описано далее.
Стадия 1. трет-Бутил-(1Е,4Е)-8-бром-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-2-илкарбамат, 3-(диметиламино)пропил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)ацетат (1,5 эквив.), тетракис(трифенилфосфин)палладий(0) и 2 М водный раствор карбоната калия (3 эквив.) соединяют в 2 мл ацетонитрила во флаконе для реакции под действием микроволнового излучения. Полученную смесь греют микроволновым излучением при 100°С в течение 30 минут. Затем смесь разбавляют EtOAc, дважды промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Препаративная тонкослойная хроматография (пластины 2×0,5 мм, 7% МеОН/DCM) дает 3-(диметиламино)пропил-2-(4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат.
Стадия 2. 3-(Диметиламино)пропил-2-(4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат растворяют в 2 мл дихлорметана и 0,5 мл ТФК. Примерно через один час смесь концентрируют при пониженном давлении, и затем полученный остаток повторно растворяют в дихлорметане, добавляют 1 мл концентрированного гидроксида аммония, и смесь энергично перемешивают в течение 15 минут. Затем полученную смесь разбавляют водой, экстрагируют дихлорметаном (2×), и экстракты сушат над сульфатом натрия и концентрируют при пониженном давлении. Препаративная тонкослойная хроматография (пластины 2×0,5 мм, 10% МеОН/DCM/0,5% NH4OH) дает 3-(диметиламино)пропил-2-(4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат. 1H ЯМР (400 МГц, CDCl3) δ 7,57-7,65 (м, 2H), 7,46-7,51 (м, 1H), 7,28-7,40 (м, 4H), 6,87 (с, 2H), 4,07-4,21 (м, 2H), 3,56-3,71 (м, 6H), 3,40-3,54 (м, 2H), 2,82 (с, 2H), 2,26-2,36 (м, 2H), 2,20 (с, 6H), 1,59-1,87 (м, 6H), 0,86-0,99 (м, 3H); m/z (APCI-полож.) M+1=521,3.
Пример 234
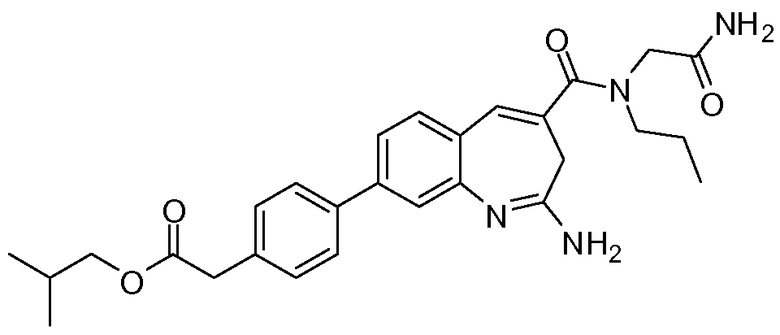
Изобутил-2-(4-((1Е,4Е)-2-амино-4-((2-амино-2-оксоэтил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат
Стадия А. Изобутил-2-(4-((1Е,4Е)-2-амино-4-((2-амино-2-оксоэтил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат (23%) получают так, как описано ниже на стадиях 1 и 2, за исключением того, что заменяют 4-(этоксикарбонил)фенилбороновую кислоту на изобутил-2-(4-(4,4,5,5,-тетраметил-1,3,2-диоксаборолан-2-ил)фенилацетат и трет-бутил-(1Е,4Е)-8-бром-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-2-илкарбамат на трет-бутил-(1Е,4Е)-4-((2-амино-2-оксоэтил)(пропил)карбамоил)-8-бром-3Н-бензо[b]азепин-2-илкарбамат.
Стадия 1. трет-Бутил-(1Е,4Е)-8-бром-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-2-илкарбамат, 4-(этоксикарбонил)фенилбороновую кислоту (1,5 эквив.), тетракис(трифенилфосфин)палладий(0) и 2 М водный раствор карбоната калия (3 эквив.) соединяют в 2 мл ацетонитрила во флаконе для реакции под действием микроволнового излучения. Полученную смесь греют микроволновым излучением при 100°С в течение 30 минут. Затем смесь разбавляют EtOAc, дважды промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Препаративная тонкослойная хроматография (пластины 2×0,5 мм, 7% МеОН/DCM) дает этил-4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)бензоат.
Стадия 2. Этил-4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)бензоат (0,050 г, 0,075 ммоль) растворяют в 2 мл дихлорметана и 0,5 мл ТФК. Примерно через один час смесь концентрируют при пониженном давлении, и затем полученный остаток повторно растворяют в дихлорметане, добавляют 1 мл концентрированного гидроксида аммония, и смесь энергично перемешивают в течение 15 минут. Затем полученную смесь разбавляют водой, экстрагируют дихлорметаном (2×), и экстракты сушат над сульфатом натрия и концентрируют при пониженном давлении. Препаративная тонкослойная хроматография (пластины 2×0,5 мм, 10% МеОН/DCM/0,5% NH4OH) дает 0,012 г (35%) этил-4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)бензоата. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,59-7,66 (м, 2H), 7,32-7,38 (м, 2H), 7,22-7,30 (м, 2H), 7,07-7,13 (м, 1H), 3,92-4,00 (м, 2H), 3,83-3,87 (м, 2H), 3,73 (с, 2H), 2,75 (с, 2H), 1,82-1,92 (м, 1H), 1,51-1,61 (м, 2H), 0,84-0,89 (м, 9H); m/z (APCI-полож.) M+1=491,2.
Пример 235
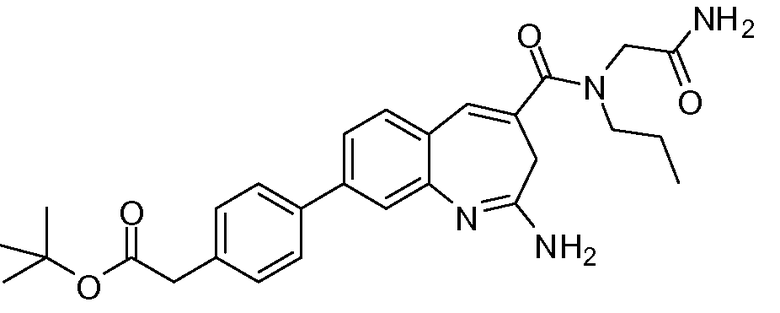
трет-Бутил-2-(4-((1Е,4Е)-2-амино-4-((2-амино-2-оксоэтил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат
Стадия А. 2-(4,4,5,5-Тетраметил-1,3,2-диоксаборолан-2-ил)фенил)уксусную кислоту (0,250 г, 0,954 ммоль) растворяют в 10 мл сухого ТГФ. К полученному раствору добавляют (Z)-трет-бутил-N,N'-диизопропилкарбамимидат (3,82 г, 19,08 ммоль), и смесь перемешивют при комнатной температуре в течение 16 часов. Затем смесь фильтруют, фильтрат концентрируют при пониженном давлении, остаток очищают на 50 г картридже Snap (Biotage, 10% EtOAc/гексан), и получают 280 мг (92%) трет-бутил-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)ацетата в виде густого масла.
Стадия В. трет-Бутил-2-(4-((1Е,4Е)-2-амино-4-((2-амино-2-оксоэтил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат (32%) получают так, как описано ниже на стадиях 1 и 2, за исключением того, что заменяют 4-(этоксикарбонил)фенилбороновую кислоту на трет-бутил-2-(4-(4,4,5,5,-тетраметил-1,3,2-диоксаборолан-2-ил)фенилацетат и трет-бутил-(1Е,4Е)-8-бром-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-2-илкарбамат на (1Е,4Е)-2-амино-N-(2-амино-2-оксоэтил)-8-бром-N-пропил-3Н-бензо[b]азепин-4-карбоксамид.
Стадия 1. трет-Бутил-(1Е,4Е)-8-бром-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-2-илкарбамат, 4-(этоксикарбонил)фенилбороновую кислоту (1,5 эквив.), тетракис(трифенилфосфин)палладий(0) и 2 М водный раствор карбоната калия (3 эквив.) соединяют в 2 мл ацетонитрила во флаконе для реакции под действием микроволнового излучения. Полученную смесь греют микроволновым излучением при 100°С в течение 30 минут. Затем смесь разбавляют EtOAc, дважды промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Препаративная тонкослойная хроматография (пластины 2×0,5 мм, 7% МеОН/DCM) дает этил-4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)бензоат.
Стадия 2. Этил-4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)бензоат (0,050 г, 0,075 ммоль) растворяют в 2 мл дихлорметана и 0,5 мл ТФК. Примерно через один час смесь концентрируют при пониженном давлении, и затем полученный остаток повторно растворяют в дихлорметане, добавляют 1 мл концентрированного гидроксида аммония, и смесь энергично перемешивают в течение 15 минут. Затем полученную смесь разбавляют водой, экстрагируют дихлорметаном (2×), и экстракты сушат над сульфатом натрия и концентрируют при пониженном давлении. Препаративная тонкослойная хроматография (пластины 2×0,5 мм, 10% МеОН/DCM/0,5% NH4OH) дает 0,012 г (35%) этил-4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)бензоата. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,50-7,65 (м, 2H), 7,23-7,36 (м, 5H), 7,09-7,14 (ушир.с, 1H), 6,84-6,91 (ушир.с, 1H), 3,92-4,00 (ушир.с, 2H), 3,61 (с, 2H), 3,51 (с, 1H), 2,77 (ушир.с, 2H), 1,51-1,61 (м, 2H), 1,42 (с, 9H), 0,73-0,91 (м, 3H); m/z (APCI-полож.) M+1=491,2.
Пример 236
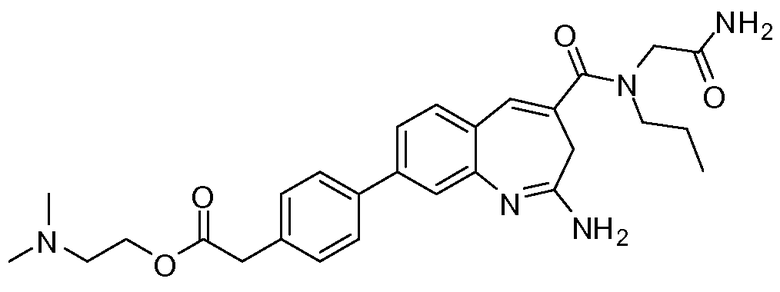
2-(Диметиламино)этил-2-(4-((1Е,4Е)-2-амино-4-((2-амино-2-оксоэтил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат
Стадия А. 2-(Диметиламино)этил-2-(4-((1Е,4Е)-2-амино-4-((2-амино-2-оксоэтил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат (19%) получают так, как описано ниже на стадиях 1 и 2, за исключением того, что заменяют 4-(этоксикарбонил)фенилбороновую кислоту на 2-(диметиламино)этил-2-(4-(4,4,5,5,-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)ацетат и трет-бутил-(1Е,4Е)-8-бром-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-2-илкарбамат на (1Е,4Е)-2-амино-N-(2-амино-2-оксоэтил)-8-бром-N-пропил-3Н-бензо[b]азепин-4-карбоксамид.
Стадия 1. трет-Бутил-(1Е,4Е)-8-бром-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-2-илкарбамат, 4-(этоксикарбонил)фенилбороновую кислоту (1,5 эквив.), тетракис(трифенилфосфин)палладий(0) и 2 М водный раствор карбоната калия (3 эквив.) соединяют в 2 мл ацетонитрила во флаконе для реакции под действием микроволнового излучения. Полученную смесь греют микроволновым излучением при 100°С в течение 30 минут. Затем смесь разбавляют EtOAc, дважды промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Препаративная тонкослойная хроматография (пластины 2×0,5 мм, 7% МеОН/DCM) дает этил-4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)бензоат.
Стадия 2. Этил-4-((1Е,4Е)-2-(трет-бутоксикарбониламино)-4-((3-(трет-бутилдиметилсилилокси)пропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)бензоат (0,050 г, 0,075 ммоль) растворяют в 2 мл дихлорметана и 0,5 мл ТФК. Примерно через один час смесь концентрируют при пониженном давлении, и затем полученный остаток повторно растворяют в дихлорметане, добавляют 1 мл концентрированного гидроксида аммония, и смесь энергично перемешивают в течение 15 минут. Затем полученную смесь разбавляют водой, экстрагируют дихлорметаном (2×), и экстракты сушат над сульфатом натрия и концентрируют при пониженном давлении. Препаративная тонкослойная хроматография (пластины 2×0,5 мм, 10% МеОН/DCM/0,5% NH4OH) дает 0,012 г (35%) этил-4-((1Е,4Е)-2-амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)бензоата. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,60-7,64 (м, 2H), 7,33-7,37 (м, 2H), 7,26-7,28 (м, 1H), 7,21-7,25 (м, 1H), 7,11 (ушир.с, 1H), 6,75-6,90 (ушир.м, 3H), 4,11-4,16 (м, 2H), 3,96 (ушир.с, 2H), 3,71 (с, 2H), 3,51 (с, 2H), 2,73 (с, 2H), 2,16 (с, 6H), 1,50-1,62 (м, 2H), 0,75-0,91 (м, 3H); m/z (APCI-полож.) M+1=506,2.
Пример 237
Гидрохлорид 2-(4-((1Е,4Е)-2-амино-4-((2-амино-2-оксоэтил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)уксусной кислоты
Стадия А. трет-Бутил-2-(4-((1Е,4Е)-2-амино-4-((2-амино-2-оксоэтил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат (0,037 г, 0,0754 ммоль) растворяют в 1 мл диоксана и охлаждают до 0°С. Через раствор барботируют газ HCl в течение 15 минут, реакционный сосуд плотно закрывают, и смесь оставляют на ночь нагреваться до комнатной температуры. Давление газа осторожно спускают, и смесь концентрируют до гидрохлорида 2-(4-((1Е,4Е)-2-амино-4-((2-амино-2-оксоэтил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)уксусной кислоты. m/z (APCI-полож.) М+1=435,1.
Пример 238
2-(4-((1Е,4Е)-2-Амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)уксусная кислота
Стадия А. 2-(4-((1Е,4Е)-2-Амино-4-((3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)уксусную кислоту (8%) получают так, как описано ниже на стадии 1, за исключением того, что заменяют бензил-4-((1Е,4Е)-2-амино-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-8-ил)бензоат на бензил-2-(4-((1Е,4Е)-2-амино-4-(3-гидроксипропил)(пропил)карбамоил)-3Н-бензо[b]азепин-8-ил)фенил)ацетат.
Стадия 1. Бензил-4-((1Е,4Е)-2-амино-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-8-ил)бензоат (0,025 г, 0,0504 ммоль) суспендируют в 1 мл метанола, добавляют 25 мг 10% Pd/C (тип Degussa), и смесь гидрируют водородом из баллона в течение одного часа. Затем полученную смесь фильтруют через фильтровальную бумагу GF/F, и фильтрат концентрируют до 16 мг 4-((1Е,4Е)-2-амино-4-(дипропилкарбамоил)-3Н-бензо[b]азепин-8-ил)бензойной кислоты (78%).
m/z (APCI-полож.) М+1=436,2.
Пример 2
Анализы HEK/TLR
Активность соединений по данному изобретению можно определить с помощью описанных далее анализов.
В анализе трансфектантов HEK-293 hTLR используют клетки HEK293, устойчиво трансфицированные различными hTLR и временно котрансфицированные плазмидой, содержащей управляемый NF-κB репортерный ген секретированной эмбриональной щелочной фосфатазы (SEAP). Стимуляция TLR активирует их пути передачи сигналов в прямом направлении и индуцирует ядерную транслокацию фактора транскрипции NF-κB. Затем измеряют активность репортерного гена с использованием спектрофотометрического анализа.
Для того, чтобы измерить активность агониста, получают эмбриональные клетки почки человека (НЕК), которые устойчиво экспрессируют гены различных TLR человека, включая TLR7 и TLR8, и репортерный ген NF-κB-люциферазы (например, клетки 293XL-hTLR8, доступные от InvivoGen, San Diego, CA) согласно инструкциям изготовителя и инкубируют с различными концентрациями испытываемого соединения в течение ночи. Количество индуцированной люциферазы измеряют, считывая поглощение при 650 нм. Соединения-агонисты по изобретению имеют МС50 25 мкМ или менее, при этом МС50 определяют как концентрацию, при которой наблюдают 50% индукцию от максимума.
Пример 3
Анализы РВМС на TLR7 и TLR8
Мононуклеарные клетки периферической крови (РВМС) из крови человека извлекают с использованием пробирок для получения клеток BD Vacutainer с цитратом натрия. Клетки инкубируют с соединением в течение ночи. Активность TLR8 оценивают, измеряя ELISA количество TNFα в супернатантах. Активность TLR7 оценивают, измеряя ELISA (R&D Systems) количество TNFα в супернатантах. Соединения по данному изобретению имеют МС50 25000 (нМ) или меньше, при этом МС50 представляет собой концентрацию, при которой отмечают 50% индукцию от максимума. Результаты такого анализа приводятся ниже в таблицах 2 и 3.
Величины МС50 представлены в виде показателей порядка, например, + показывает величину МС50×104 или величину в десятках тысяч наномолей (нМ интервал); ++ показывает величину МС50×103 или величину в тысячах; +++ показывает величину МС50×102 или величину в сотнях; и ++++ показывает величину МС50×101 или 100 или величину в десятках или единицах.
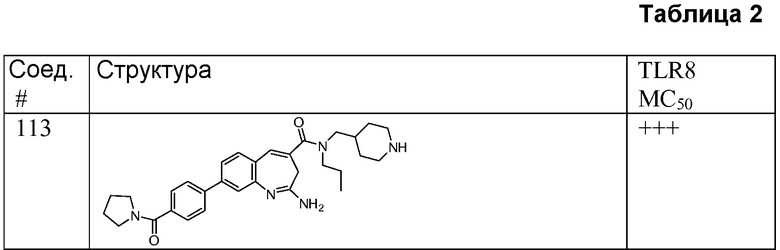
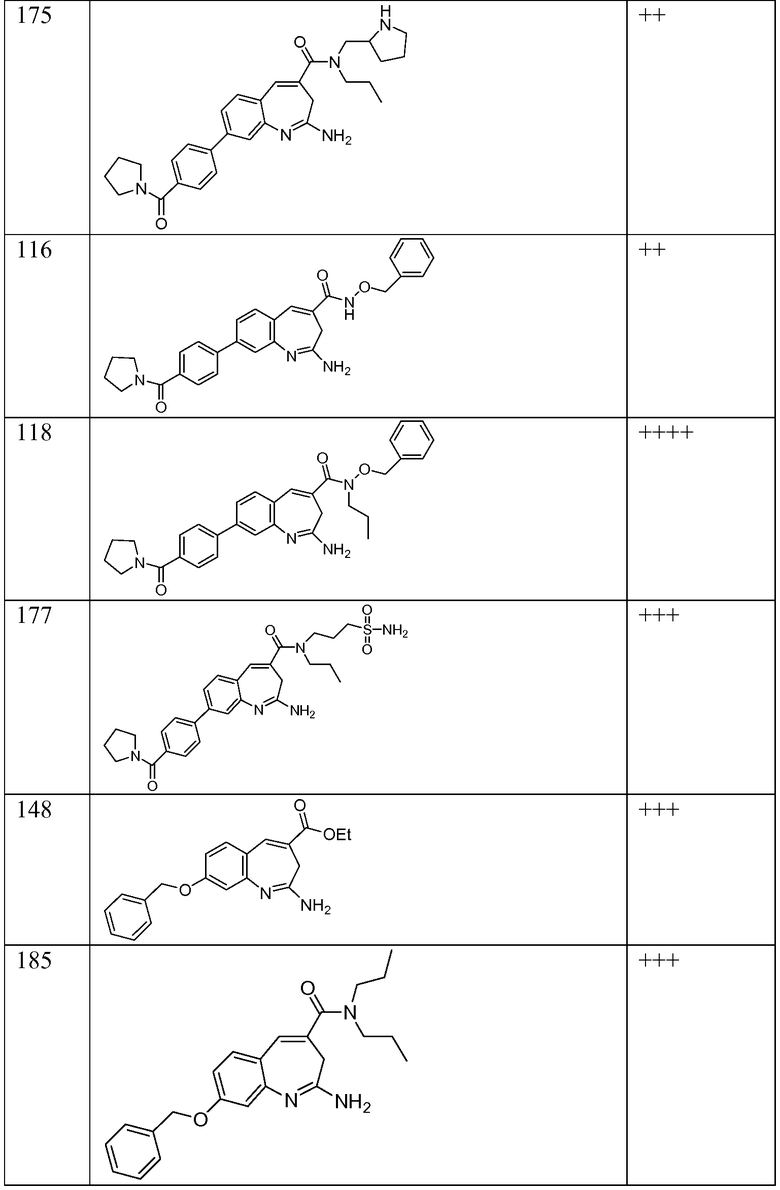
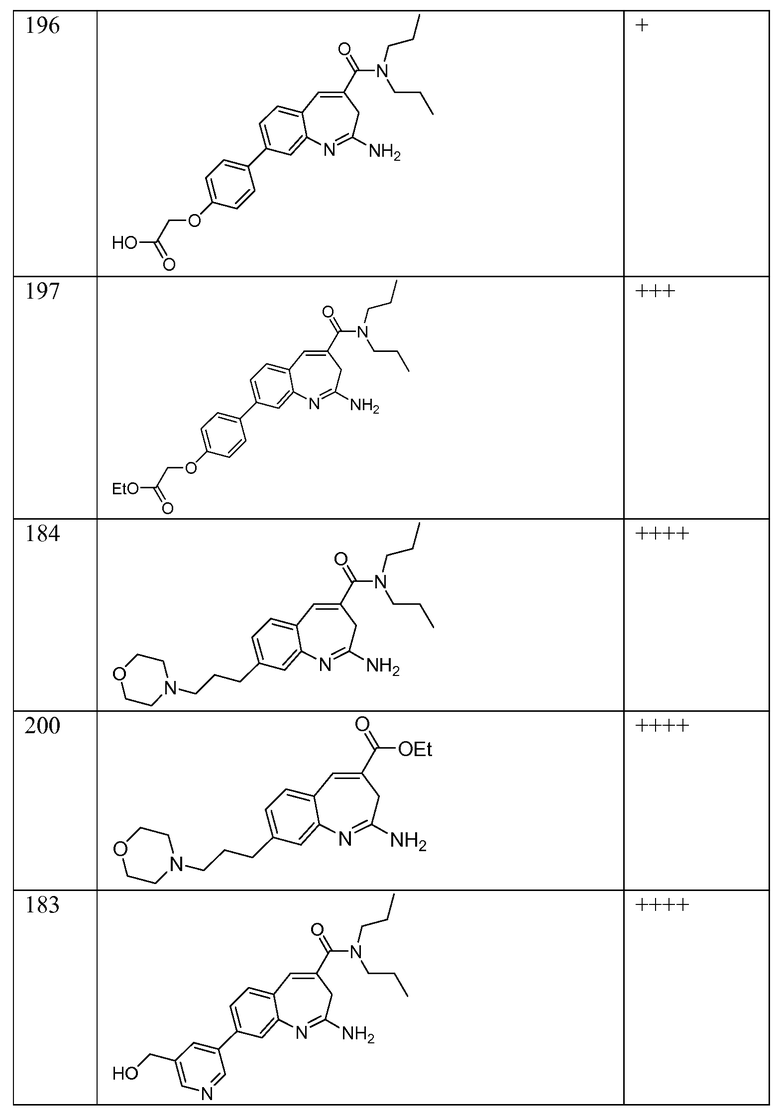
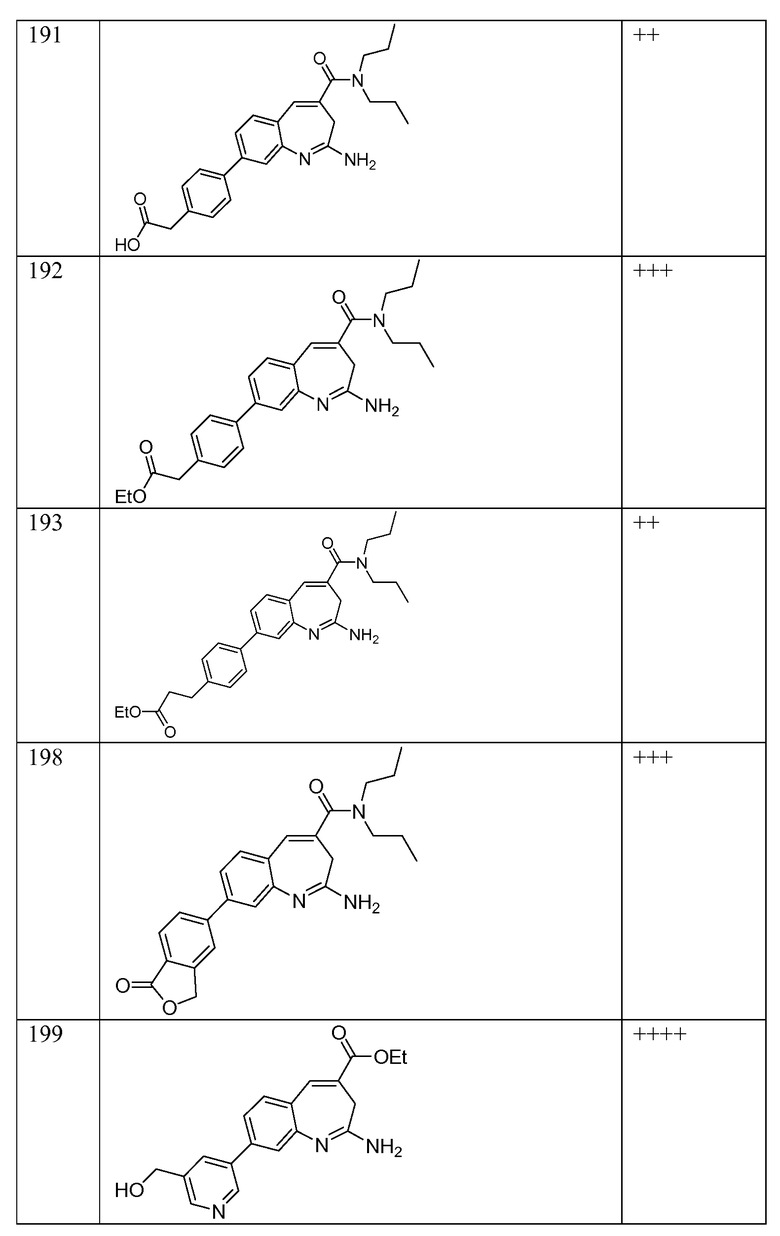
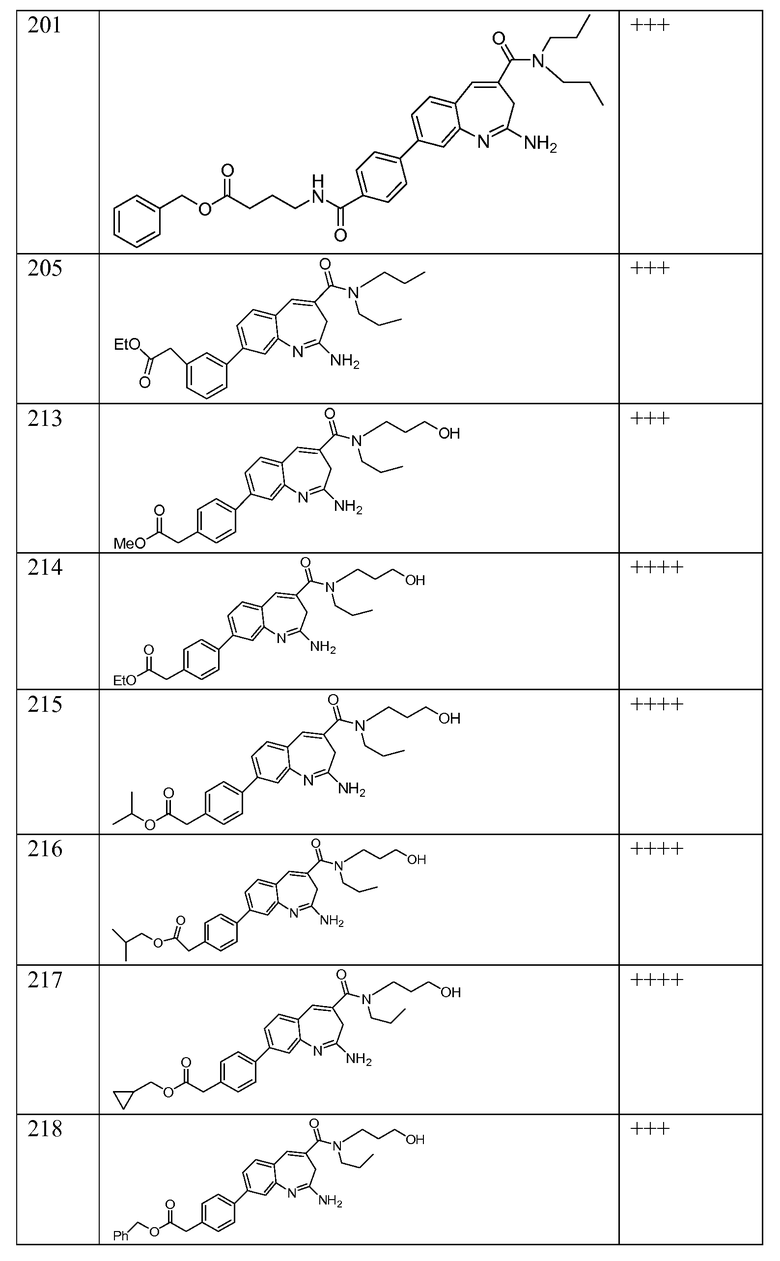
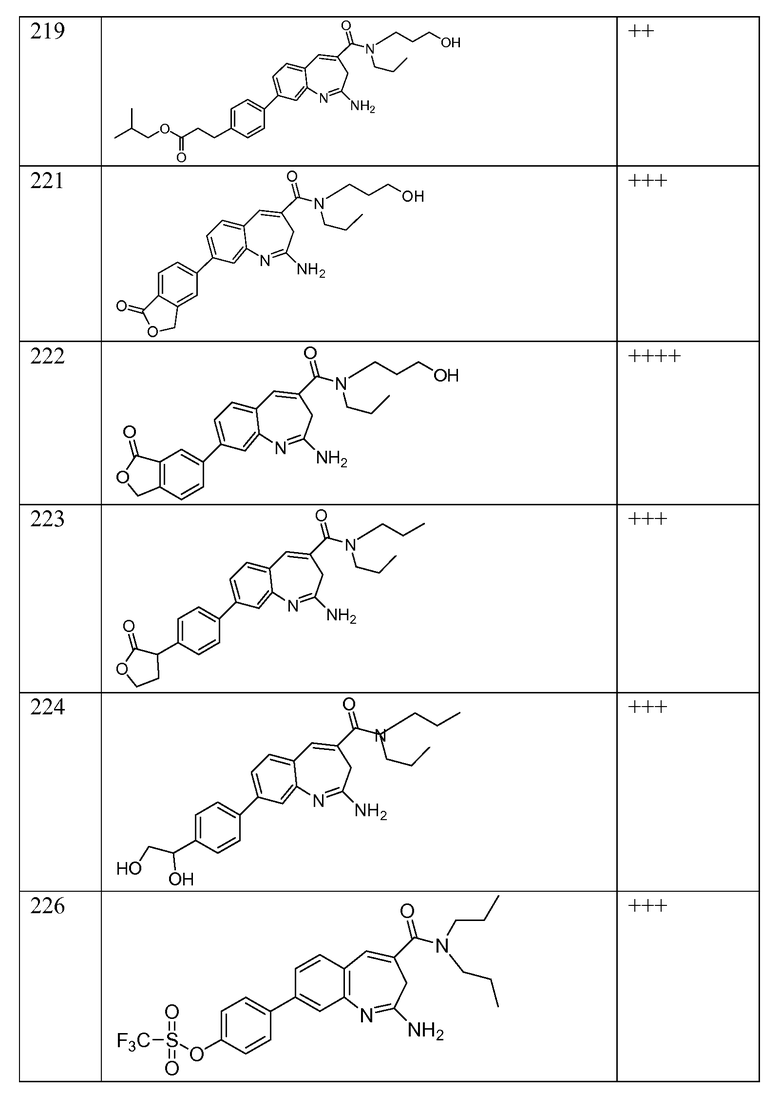
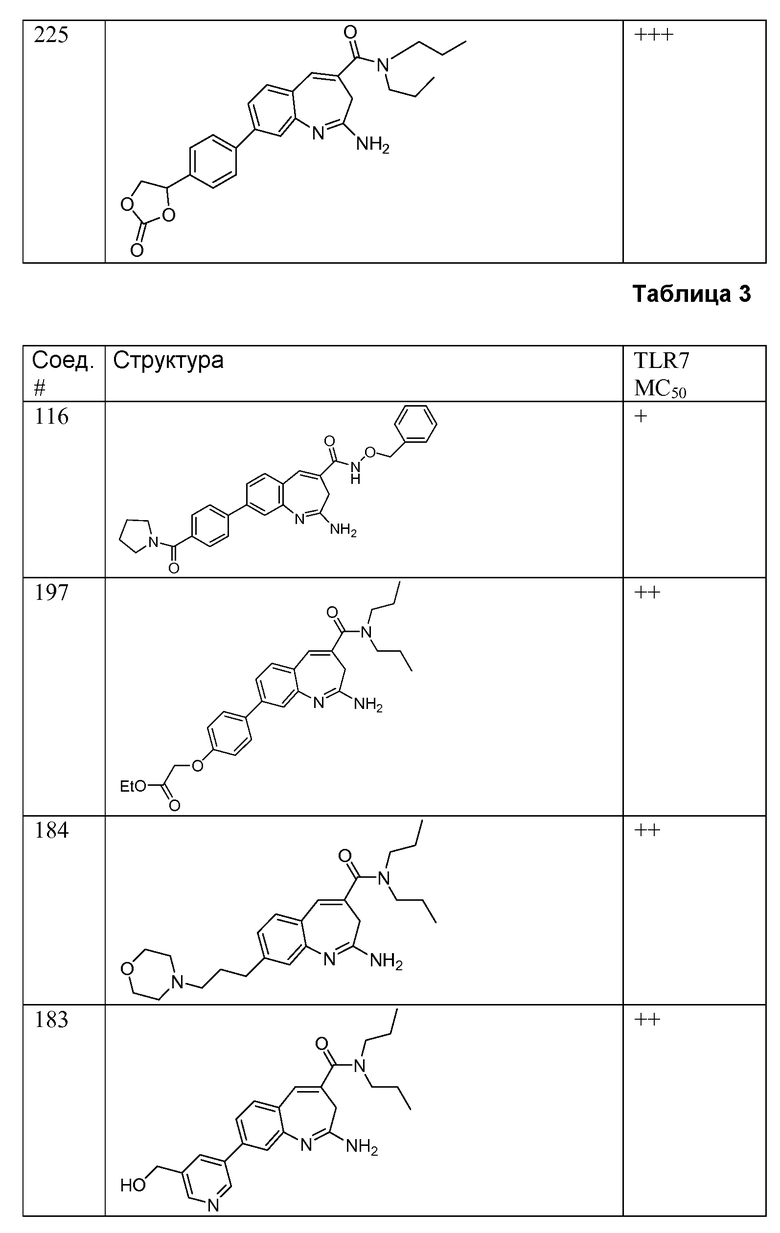
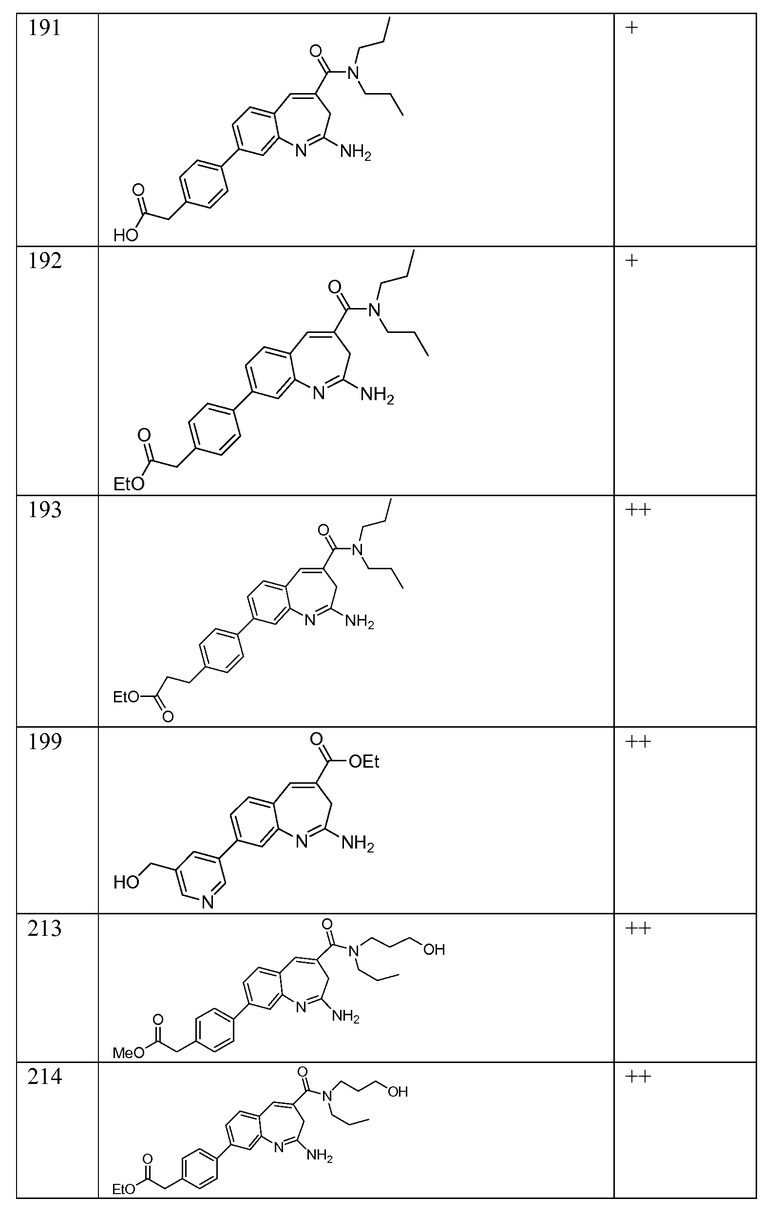
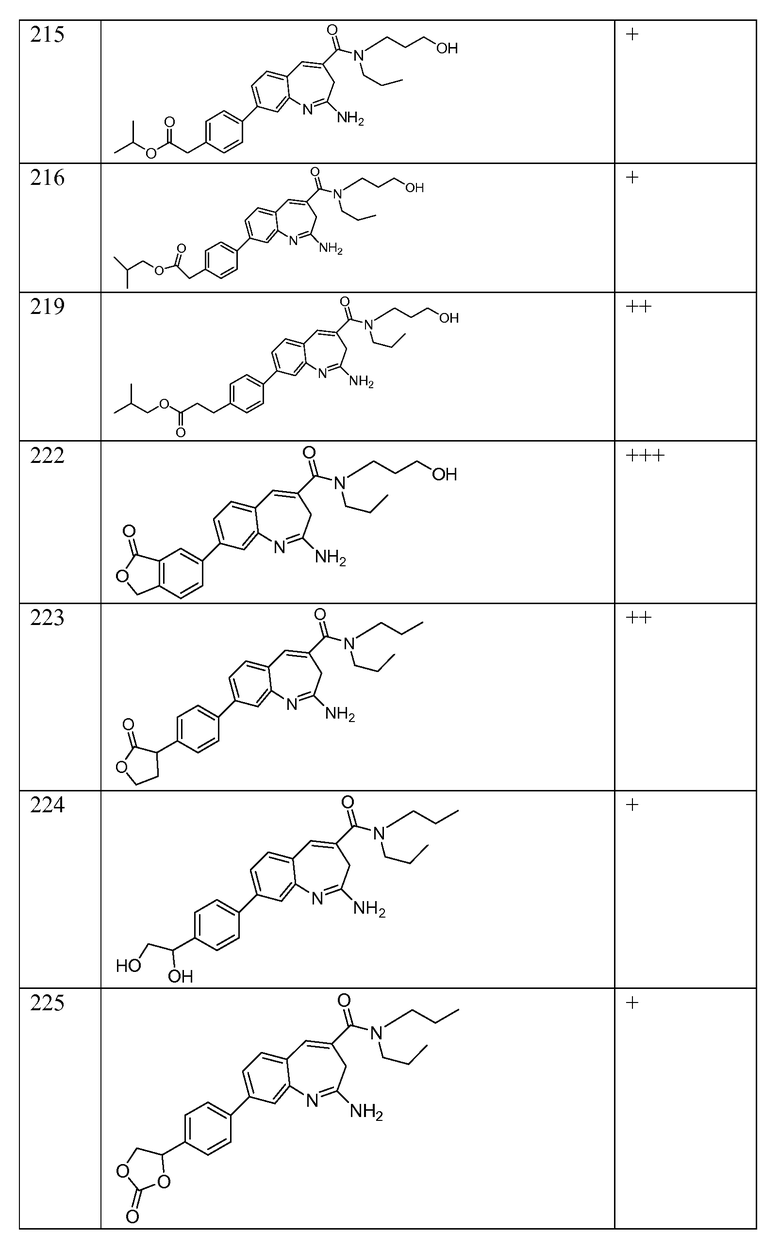
Приведенное выше описание рассматривается только как поясняющее принципы изобретения. Кроме того, так как многие модификации и изменения будут очевидны для специалистов в данной области техники, не следует ограничивать изобретение показанными точными конструкциями и способами, описанными выше. Соответственно, все подходящие модификации и эквиваленты могут быть отнесены к входящим в объем изобретения, определенный приведенной далее формулой изобретения.
Предполагается, что слова «включать», «включающий», «содержать», «содержащий» и «содержит», используемые в данном описании и в последующей формуле изобретения, определяют присутствие указанных признаков, целых, компонентов или стадий, но они не препятствуют присутствию или добавлению одного или нескольких других признаков, целых, компонентов стадий или их групп.
Если не указано иное, все ссылки, приведенные в данном описании, входят в него в качестве ссылок.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ БЕНЗОАЗЕПИНЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ TOLL-ПОДОБНОГО РЕЦЕПТОРА | 2010 |
|
RU2580320C2 |
| СОЕДИНЕНИЯ БЕНЗАЗЕПИНА ДИКАРБОКСАМИДА | 2016 |
|
RU2712248C2 |
| АНТАГОНИСТЫ РЕЦЕПТОРА CGRP | 2004 |
|
RU2308458C2 |
| 7-ЗАМЕЩЕННЫЕ СУЛЬФОНИМИДОИЛПУРИНОНЫ ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ВИРУСНОЙ ИНФЕКЦИИ | 2017 |
|
RU2838538C1 |
| 7-ЗАМЕЩЕННЫЕ СУЛЬФОНИМИДОИЛПУРИНОНЫ ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ВИРУСНОЙ ИНФЕКЦИИ | 2017 |
|
RU2751349C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2667498C2 |
| МОДУЛЯТОРЫ КИНАЗЫ, АССОЦИИРОВАННОЙ С РЕЦЕПТОРОМ ИНТЕРЛЕЙКИНА-1 | 2007 |
|
RU2459821C2 |
| ПРОИЗВОДНЫЕ 1,5-НАФТИРИДИНА И ИНГИБИТОРЫ MELK, СОДЕРЖАЩИЕ ИХ | 2012 |
|
RU2645339C1 |
| ИНГИБИТОРЫ СЕРИН/ТРЕОНИНОВЫХ КИНАЗ | 2013 |
|
RU2650501C2 |
| СОЕДИНЕНИЯ, КОМПОЗИЦИИ И СПОСОБЫ ПРЕДУПРЕЖДЕНИЯ МЕТАСТАЗОВ РАКОВЫХ КЛЕТОК | 2010 |
|
RU2519123C2 |
Изобретение относится к соединению формулы I или его таутомеру, энантиомеру или фармацевтически приемлемой соли, где в указанной формуле Y представляет собой -(О)х(СН2)yR11; х выбирают из 0 и 1; у выбирают из 0, 1, 2 и 3; R11 выбирают из фенила, пиридила, морфолинила, 1-оксо-1,3-дигидроизобензофуранила и 2-оксо-тетрагидрофуранила, при этом когда х равен 0, указанный фенил или пиридинил замещен -C(O)NR1R2 или Т; R1 и R2 выбирают независимо из водорода и С1-6алкила, при этом указанный алкил необязательно замещен -С(О)О(СН2)tR12, или R1 и R2 вместе с атомом азота, к которому они присоединены, образуют насыщенный 5-членный гетероцикл, содержащий 1-4 гетероатомов, выбранных из азота, кислорода и серы; t выбирают из 0, 1, 2 и 3; R12 выбирают из С3-6циклоалкила и фенила; Т выбирают из 2-оксо-1,3-диоксолана, 2-оксотетрагидрофурана, -(CHR7)zOR9, -(О)u(СН2)sC(О)R8, -OSO2R13 и -СН(ОН)СН2ОН; R7 представляет собой Н; R8 представляет собой -OR10; R9 представляет собой Н; R10 выбирают из С1-6алкила, -(CH2)R12 и водорода, при этом указанный алкил необязательно замещен галогеном или диС1-6алкиламином; R13 представляет собой CF3; u выбирают из 0 и 1; z выбирают из 1, 2 и 3; s выбирают из 1 и 2; R5 выбирают из -NR3R4 и -OR10; R3 и R4 выбирают независимо из Н, С1-12алкила, -(О)q(СН2)rP; при этом указанный алкил необязательно замещен -ОН; q выбирают из 0 и 1; r выбирают из 0, 1, 2 и 3; Р выбирают из фенила, -SO2R6, пиперидинила и пирролидинила; и R6 представляет собой -NH2, при условии, что когда R11 представляет собой фенил или пиридил, тогда a) х+у≥1; или b) R11 замещен Т; или c) R5 представляет собой NR3R4, и по меньшей мере один из R3 или R4 представляет собой -(О)q(СН2)rP-, и q+r≥1; или d) по меньшей мере один из R1 или R2 представляет собой алкил, замещенный -С(О)О(СН2)R12. Также изобретение относится к фармацевтической композиции, проявляющей TLR7- и/или TLR8 агонистическую активность, включающей эффективное количество соединения формулы I вместе с фармацевтически приемлемым разбавителем или носителем. Технический результат - замещенные бензоазепины, проявляющие TLR7- и/или TLR8 агонистическую активность. 4 н. и 13 з.п. ф-лы, 3 табл., 47 пр.

1. Соединение формулы I
или его таутомер, энантиомер или соль, где в указанной формуле
Y представляет собой -(О)х(СН2)yR11;
х выбирают из 0 и 1;
у выбирают из 0, 1, 2 и 3;
R11 выбирают из фенила, пиридила, морфолинила, 1-оксо-1,3-дигидроизобензофуранила и 2-оксо-тетрагидрофуранила, при этом когда х равен 0, указанный фенил или пиридинил замещен -C(O)NR1R2 или Т;
R1 и R2 выбирают независимо из водорода и С1-6алкила, при этом указанный алкил необязательно замещен -С(О)О(СН2)tR12, или R1 и R2 вместе с атомом азота, к которому они присоединены, образуют насыщенный 5-членный гетероцикл, содержащий 1-4 гетероатомов, выбранных из азота, кислорода и серы;
t выбирают из 0, 1, 2 и 3;
R12 выбирают из С3-6циклоалкила и фенила;
Т выбирают из 2-оксо-1,3-диоксолана, 2-оксотетрагидрофурана, -(CHR7)zOR9, -(О)u(СН2)sC(О)R8, -OSO2R13 и -СН(ОН)СН2ОН;
R7 представляет собой Н;
R8 представляет собой -OR10;
R9 представляет собой Н;
R10 выбирают из С1-6алкила, -(CH2)R12 и водорода, при этом указанный алкил необязательно замещен галогеном или диС1-6алкиламином;
R13 представляет собой CF3;
u выбирают из 0 и 1;
z выбирают из 1, 2 и 3;
s выбирают из 1 и 2;
R5 выбирают из -NR3R4 и -OR10;
R3 и R4 выбирают независимо из Н, С1-12алкила, -(О)q(СН2)rP; при этом указанный алкил необязательно замещен -ОН;
q выбирают из 0 и 1;
r выбирают из 0, 1, 2 и 3;
Р выбирают из фенила, -SO2R6, пиперидинила и пирролидинила;
и
R6 представляет собой -NH2,
при условии, что когда R11 представляет собой фенил или пиридил, тогда
a) х+у≥1;
или
b) R11 замещен Т;
или
c) R5 представляет собой NR3R4, и по меньшей мере один из R3 или R4 представляет собой -(О)q(СН2)rP-, и q+r≥1;
или
d) по меньшей мере один из R1 или R2 представляет собой алкил, замещенный -С(О)О(СН2)R12.
2. Соединение по п.1, имеющее формулу II
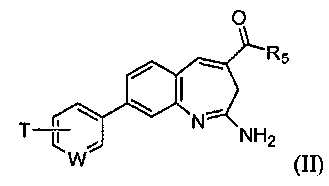
или его таутомер, энантиомер или соль, где в указанной формуле
W выбирают из N, С-Т и СН.
3. Соединение по п.2, имеющее формулу IIa
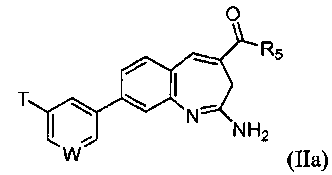
или его таутомер, энантиомер или соль.
4. Соединение по п.2, имеющее формулу IIb
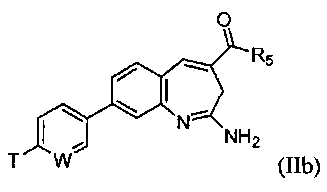
или его таутомер, энантиомер или соль.
5. Соединение по п.1, имеющее формулу III
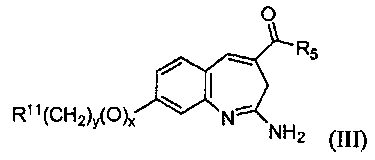
или его таутомер, энантиомер или соль, при условии, что когда R11 представляет собой фенил или пиридил, тогда х+у≥1.
6. Соединение по п.1, имеющее формулу IV
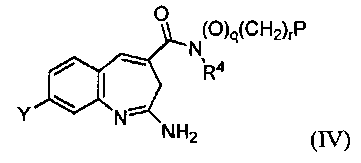
или его таутомер, энантиомер или соль, при этом q+r≥1.
7. Соединение по п.1, имеющее формулу V
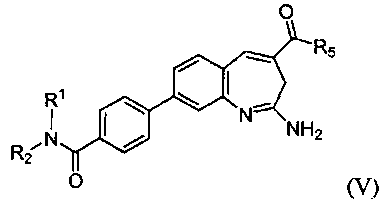
или его таутомер, энантиомер или соль, при этом по меньшей мере один из R1 или R2 представляет собой алкил, замещенный -C(O)O(CH2)R12.
8. Соединение по п.1, имеющее формулу VI
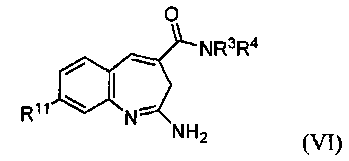
или его таутомер, энантиомер или соль, где в указанной формуле
R11 выбирают из фенила, морфолинила, 1-оксо-1,3-дигидроизобензофуранила и 2-оксо-тетрагидрофуранила, при этом указанный фенил замещен Т;
Т выбирают из 2-оксо-1,3-диоксолана,
2-оксотетрагидрофурана, -(О)u(СН2)sC(О)R8 и -СН(ОН) СН2ОН;
R8 представляет собой -OR10;
R10 выбирают из С1-6алкила, -(CH2)R12 и водорода, при этом указанный алкил необязательно замещен галогеном или ди(С1-6алкил)амином;
R12 выбирают из С3-6циклоалкила и фенила;
u выбирают из 0 и 1;
s выбирают из 1 и 2; и
R3 и R4 представляют собой независимо С1-6алкил; при этом указанный алкил необязательно замещен -ОН.
9. Соединение по п.8, при этом
R11 выбирают из фенила, морфолинила, 1-оксо-1,3-дигидроизобензофуранила и 2-оксо-тетрагидрофуранила, при этом указанный фенил замещен Т;
Т выбирают из 2-оксо-1,3-диоксолана, 2-оксотетрагидрофурана и -(O)u(CH2)sC(O)R8;
R8 представляет собой -OR10;
R10 выбирают из С1-6алкила, -(CH2)R12 и водорода, при этом указанный алкил необязательно замещен галогеном или ди(С1-6алкил)амином;
R12 выбирают из С3-6циклоалкила и фенила;
u выбирают из 0 и 1;
s выбирают из 1 и 2; и
R3 и R4 представляют собой независимо С1-6алкил; при этом указанный алкил необязательно замещен -ОН.
10. Соединение по п.8, при этом
R11 представляет собой фенил, замещенный Т;
Т выбирают из -(O)u(CH2)sC(O)R8 и -СН(ОН)СН2ОН;
R8 представляет собой -OR10;
R10 выбирают из C1-6алкила, -(CH2)R12 и водорода, при этом указанный алкил необязательно замещен галогеном или ди(С1-6алкил)амином;
R12 выбирают из С3-6циклоалкила и фенила;
u выбирают из 0 и 1;
s выбирают из 1 и 2; и
R3 и R4 представляют собой независимо С1-6алкил; при этом указанный алкил необязательно замещен -ОН.
11. Соединение, выбранное из группы, состоящей из:
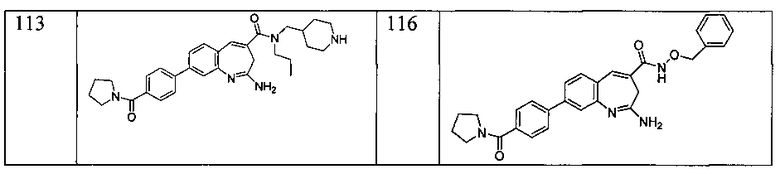
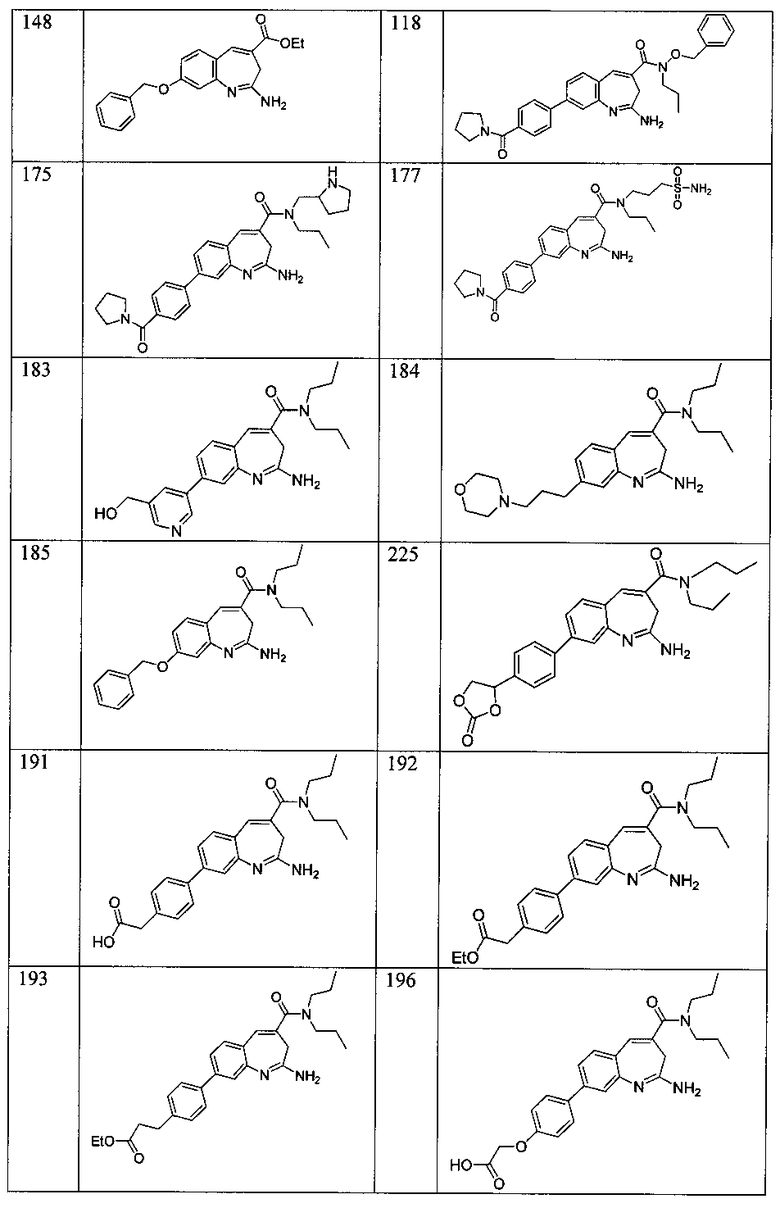
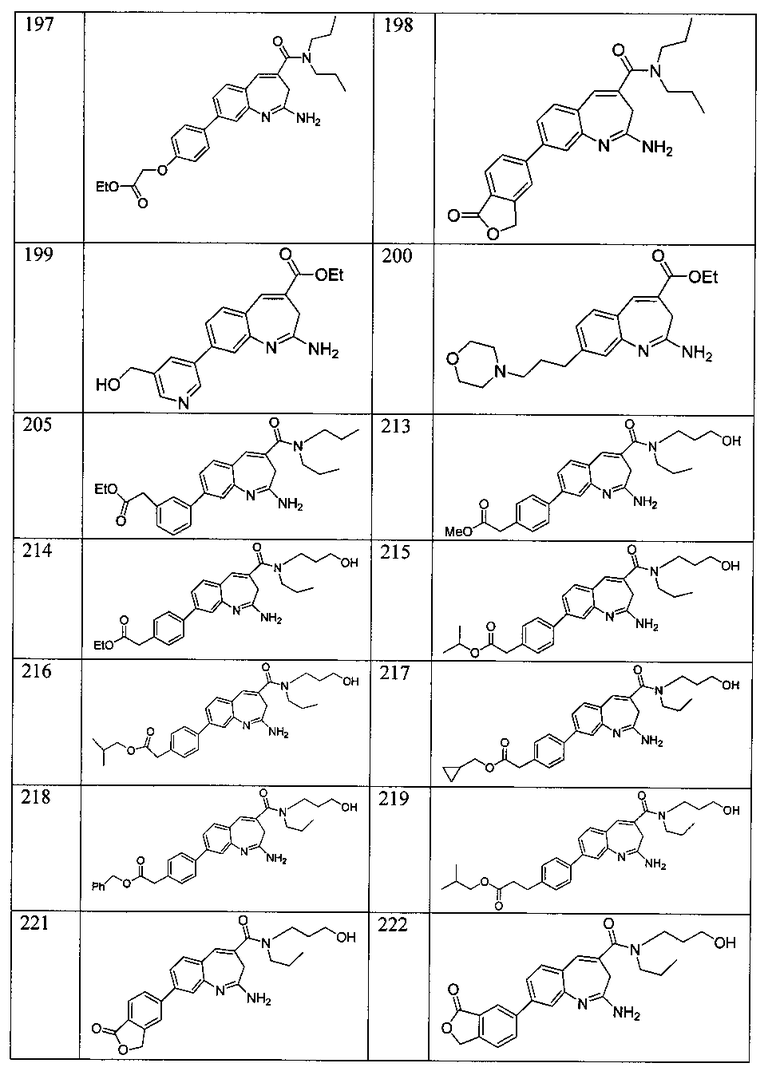
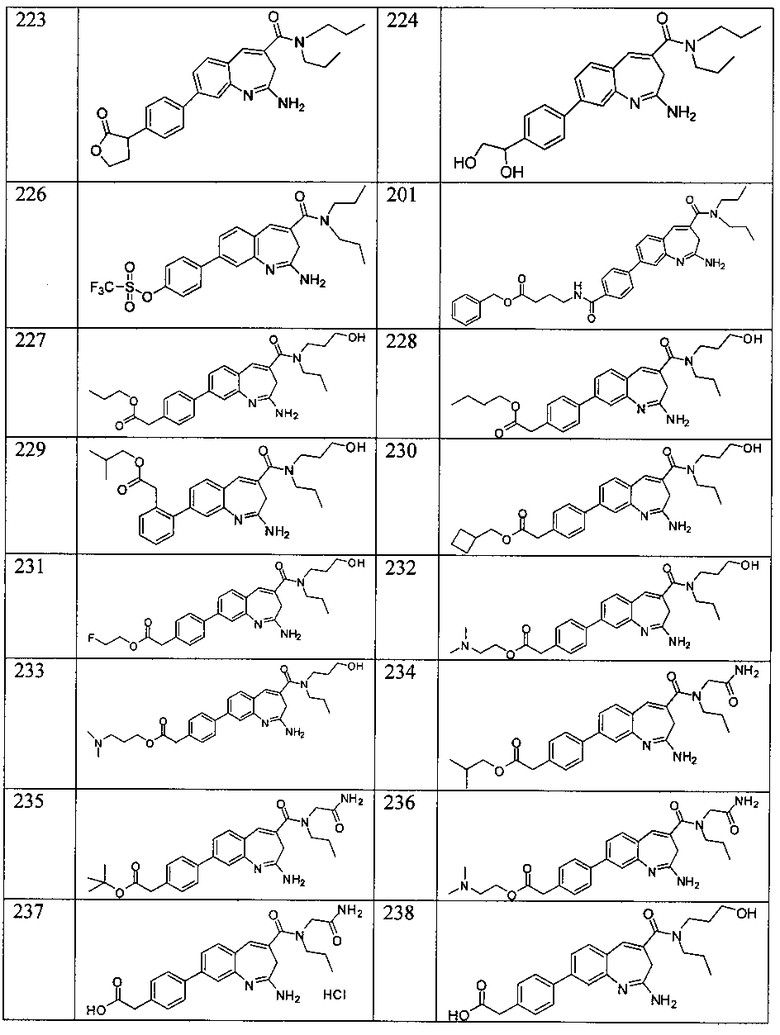
и их таутомеров, энантиомеров и солей.
12. Соединение по п.11, выбранное из группы, состоящей из:
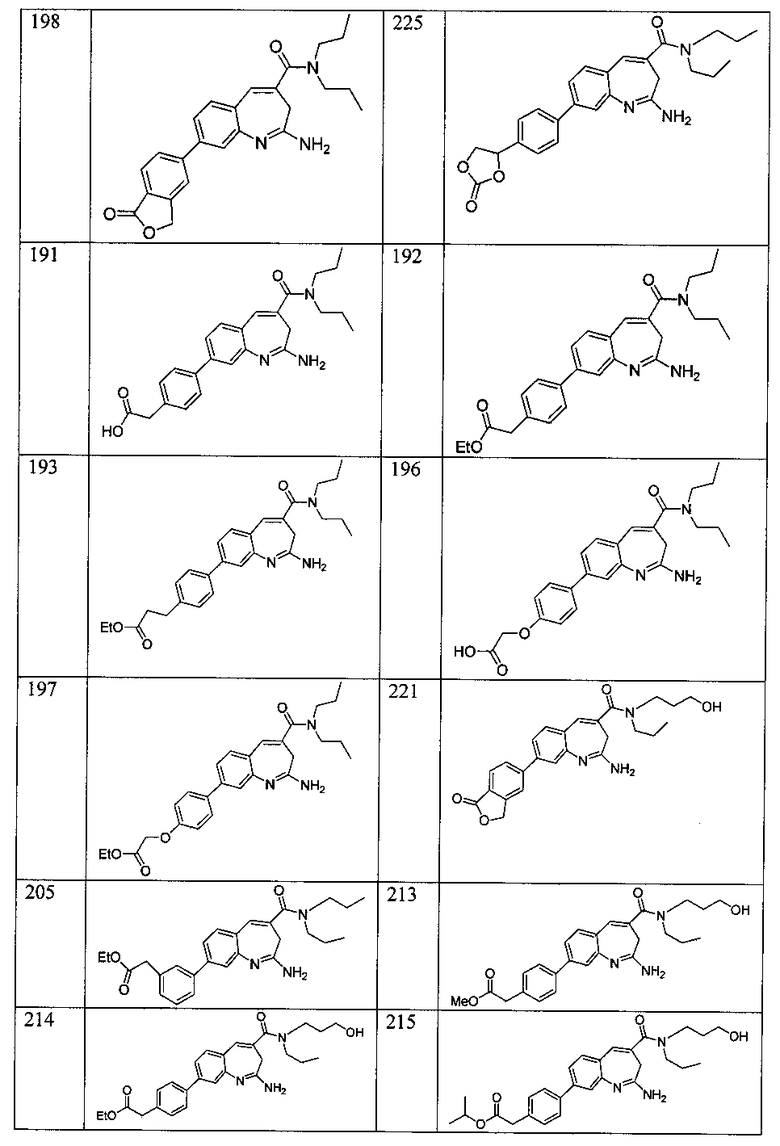
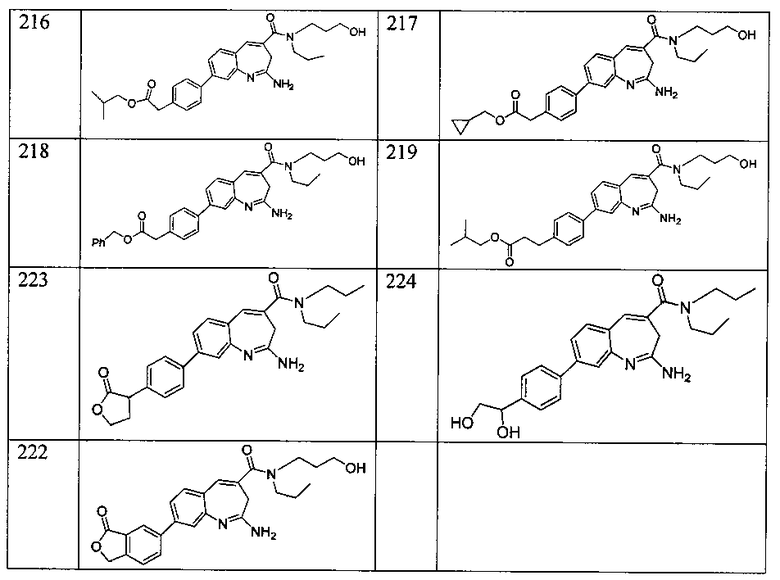
и их таутомеров, энантиомеров и солей.
13. Соединение по п.11, выбранное из группы, состоящей из
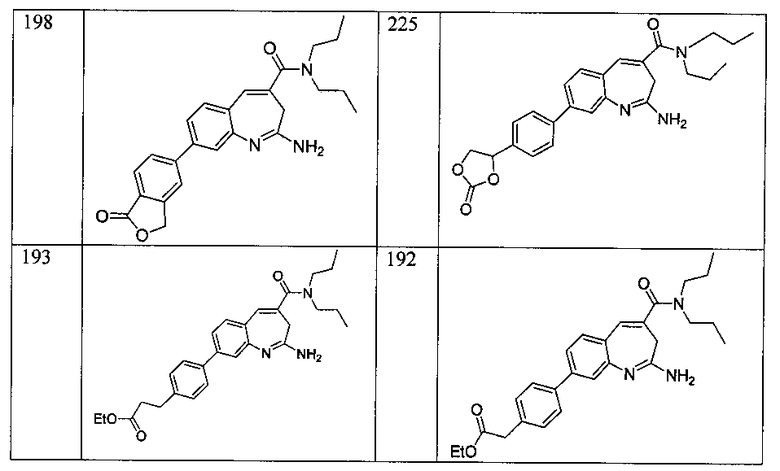
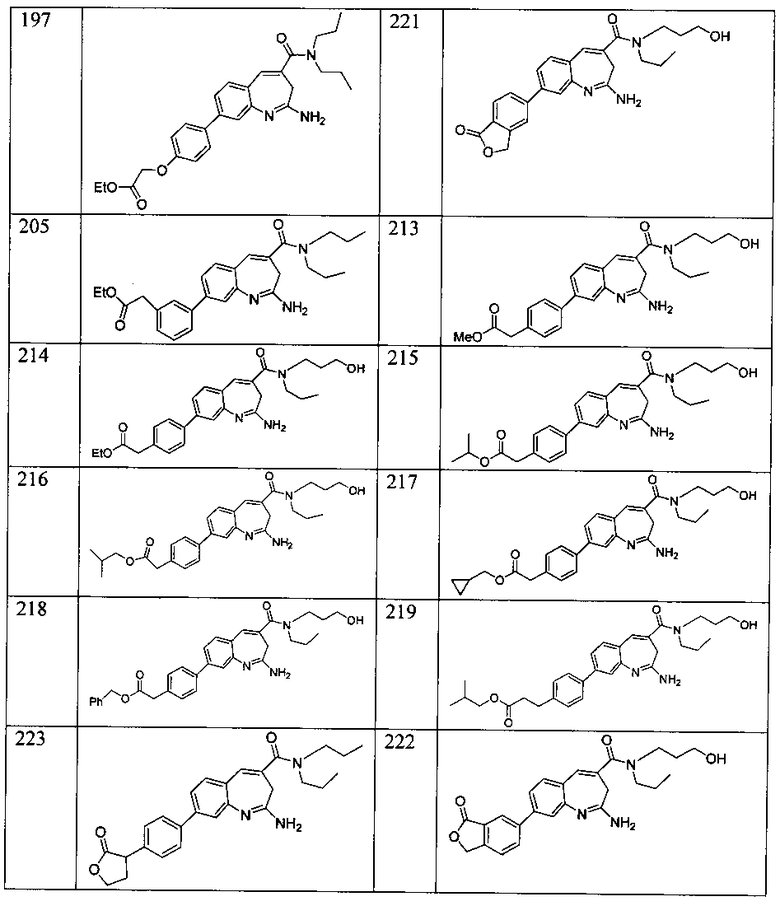
и их таутомеров, энантиомеров и солей.
14. Соединение по п.11, выбранное из группы, состоящей из
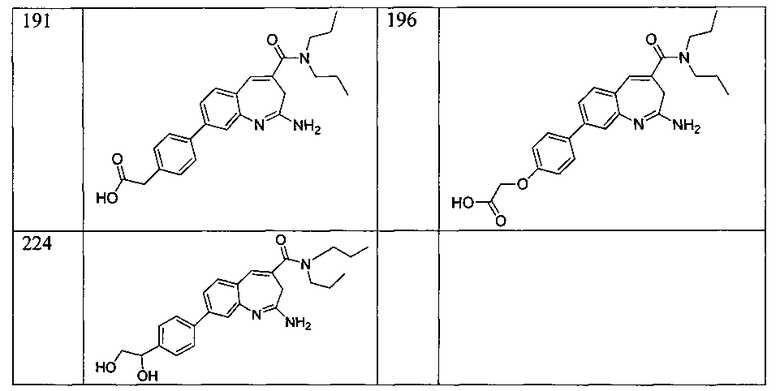
и их таутомеров, энантиомеров и солей.
15. Соединение по п.1, при этом соль представляет собой фармацевтически приемлемую соль.
16. Набор для лечения TLR7- и/или TLR8-опосредуемого состояния, включающий
a) фармацевтическую композицию, включающую соединение по любому из пп.1-14 или его таутомер, энантиомер или соль, и
b) необязательно инструкции по применению.
17. Фармацевтическая композиция, проявляющая TLR7- и/или TLR8 агонистическую активность, включающая эффективное количество соединения по любому из пп.1-14 или его таутомер, энантиомер или соль вместе с фармацевтически приемлемым разбавителем или носителем.
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Сито | 1979 |
|
SU825186A1 |
| Sudhanshu Agrawal et al.: "Cutting Edge: Different Toll-Like Receptor Agonists Instruct Dendritic Cells to Induce Distinct Th Responses via Differential Modulation of Extracellular Signal-Regulated Kinase-Mitogen-Activated Protein Kinase and c-Fos" THE JOURNAL OF IMMUNOLOGY, 2003, vol.171, | |||

