Настоящее изобретение относится к новым производным сульфонимидоилпуринонов, обладающим агонистической активностью in vivo в отношении Toll-подобных рецепторов, а также к их получению, к содержащим их фармацевтическим композициям и их возможному применению в качестве лекарственных средств.
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям формулы (I),
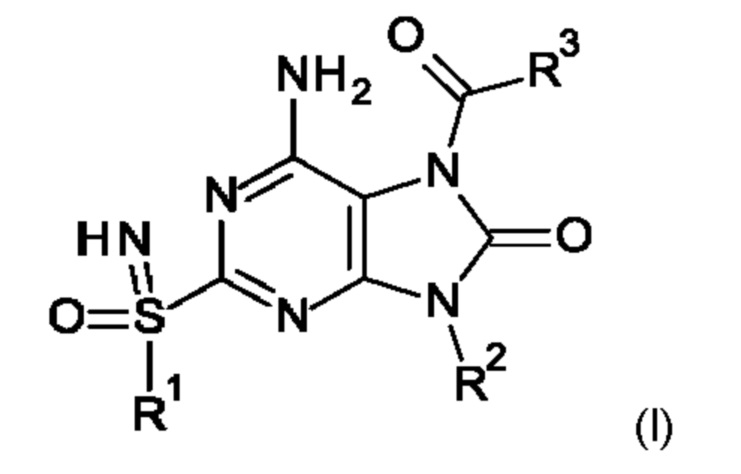
где R1-R3 являются такими, как определено ниже, или к их фармацевтически приемлемым соли, энантиомеру или диастереомеру.
Toll-подобные рецепторы распознают (TLR) широкий ряд консервативных патоген-ассоциированных молекулярных паттернов (РАМР). Они играют важную роль в обнаружении инвазивных патогенов и последующем инициировании врожденных иммунных ответов. У людей известны 10 представителей TLR семейства, которые являются трансмембранными белками I типа, характерной чертой которых считается наличие внеклеточного обогащенного остатками лейцина домена и цитоплазматического «хвоста», содержащего консервативный домен Toll-подобного рецептора/рецептора интерлейкина (IL)-1 (TIR). Такие члены этого семейства, как TLR3, TLR7, TLR8 и TLR9, располагаются в эндосомах. TLR7 может активироваться посредством связывания со специфическим низкомолекулярным лигандом (например, агонистом TLR7) или его нативным лигандом (например, одноцепочечной РНК, оцРНК). Считается, что после того, как оцРНК связывается с TLR7, рецептор, находящийся в своей димеризованной форме, подвергается структурному изменению, приводящему к последующему рекрутменту адаптерных белков к его цитоплазматическому домену, включая белок 88 миелоидной дифференцировки первичного генного ответа (MyD88). После инициации сигнального каскада с участием рецепторов MyD88-опосредованного пути цитоплазматические транскрипционные факторы, такие как регуляторный фактор 7 интерферонов (IRF-7) и ядерный фактор каппа В (NF-κВ), активируются. Затем эти транскрипционные факторы транслоцируются в ядро и инициируют транскрипцию различных генов, например, генов интерферона-альфа (IFN-α) и других противовирусных цитокинов. В большинстве случаев TLR7 экспрессируется на плазмоцитоидных клетках и также на В-клетках. Изменение восприимчивости иммунных клеток может способствовать ослаблению врожденных иммунных ответов при хронических вирусных инфекциях. Поэтому агонист-индуцированная активация TLR7 может представлять новый подход к лечению хронических вирусных инфекций (D.J. Connolly and L. AJ  , Current Opinion in Pharmacology, 2012, 12: 510-518, P.A. Roethle et al., J. Med. Chem., 2013, 56, 7324-7333).
, Current Opinion in Pharmacology, 2012, 12: 510-518, P.A. Roethle et al., J. Med. Chem., 2013, 56, 7324-7333).
Современная терапия хронической инфекции вирусом гепатита В (HBV) основывается на применении двух разных типов лекарственных средств: на традиционных противовирусных нуклеоз(т)идных аналогах и разработанном за последние годы ПЭГилированном IFN-α (полиэтиленгликоль(ПЭГ)-IFN-α). Действие пероральных нуклеоз(т)идных аналогов заключается в подавлении репликации HBV. Это относится к пожизненно принимаемому курсу лечения, в ходе которого часто имеет место лекарственная устойчивость. В качестве альтернативного варианта для лечения некоторых пациентов с хронической HBV-инфекцией в рамках определенной по продолжительности терапии был использован ПЭГилированный IFN-α (ПЭГ-IFN-α). Несмотря на достижение сероконверсии по антигену "е" HBV (HBeAg) по меньшей мере у небольшой процентной доли пациентов с HBV-инфекцией, неблагоприятные эффекты делают его плохо переносимым. Примечательно, что в обоих случаях применения существующей в настоящее время терапии функциональное излечение, определяемое как сероконверсия по поверхностному антигену HBV (HBsAg), наблюдается очень редко. Поэтому разработка терапевтического препарата нового поколения для лечения пациентов с HBV-инфекцией с целью функционального излечения представляет собой острую потребность. Лечение пероральным агонистом TLR7 представляет собой перспективное решение для обеспечения более значительной эффективности с лучшей переносимостью. В настоящее время для лечения хронической HBV-инфекции используется ПЭГилированный IFN-α (ПЭГ-IFN-α), что представляет собой альтернативный вариант возможно пожизненно получаемому лечению противовирусными нуклеоз(т)идными аналогами. В подгруппе пациентов с хронической HBV-инфекцией терапия с использованием ПЭГ-IFN-α может вызывать устойчивый иммунологический контроль вируса после определенной по продолжительности терапии. Однако, процентная доля пациентов с HBV-инфекцией, которые достигают сероконверсии в случае терапии интерферонами, низка (до 27% включительно для HBeAg-положительных пациентов), и такое лечение обычно является плохо переносимым. Кроме того, функциональное излечение (определяемое как утрата HBsAg и сероконверсия по нему) также очень нечасто наблюдается при лечении как ПЭГ-IFN-α, так и нуклеоз(т)идами. Принимая во внимание эти ограничения, можно сказать, что имеется неудовлетворенная потребность в улучшенных терапевтических вариантах лечения и в достижении функционального излечения хронической HBV-инфекции. Лечение пероральным низкомолекулярным агонистом TLR7 является перспективным подходом с возможностью обеспечения более значительной эффективности и переносимости (Т. Asselah et al., Clin. Liver Dis., 2007, 11, 839-849).
На самом деле, рассмотрение некоторых идентифицированных агонистов TLR7 на предмет терапевтических применений проведено. К настоящему времени имиквимод (ALDARA™) представляет собой являющееся агонистом TLR7 лекарственное средство, одобренное Управлением по контролю качества пищевых продуктов и лекарственных средств (FDA) США для местного применения с целью лечения поражений кожи, вызываемых папиллома в и русом человека. Проведена оценка агониста TLR7/8 двойного действия резиквимода (R-848) и агониста TLR7 852А в отношении лечения у человека генитального герпеса и плохо поддающейся химиотерапевтическому лечению метастазирующей меланомы, соответственно. ANA773 представляет собой пероральное пролекарство агониста TLR7, разработанное для лечения пациентов с хронической инфекцией вирусом гепатита С (HCV) и хронической инфекцией вирусом гепатита В. GS-9620 представляет собой агонист TLR7 для перорального приема. На фазе Ib испытания было продемонстрировано, что лечение препаратом GS-9620 оказалось безопасным, хорошо переносимым и приводило к дозозависимому индуцированию мРНК ISG15 (интерферон-стимулированный ген 15) у пациентов с хроническим гепатитом В (E.J. Gane ег al., Annu. Meet. Am. Assoc. Study Liver Dis. (November 1-5, Washington, D.C.) 2013, Abst. 946). Таким образом, имеется огромная неудовлетворенная клиническая потребность в разработке сильнодействующих и безопасных агонистов TLR7 в качестве новых средств для лечения HBV-инфекции с целью предоставления дополнительных терапевтических средств или замены существующей частично эффективной схемы лечения.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Согласно настоящему изобретению предложен ряд новых соединений 6-амино-2-сульфонимидоил-9-замещенных-7-замещенных-пурин-8-онов, обладающих агонистической активностью в отношении Toll-подобных рецепторов, и их пролекарств. Согласно изобретению также обеспечивается биологическая активность таких соединений с целью индуцирования повышения уровня SEAP (секретируемая эмбриональная щелочная фосфатаза) посредством активации Toll-подобных рецепторов, таких как рецептор TLR7, метаболическое превращение пролекарств в исходные соединения в присутствии гепатоцитов человека и терапевтическое или профилактическое применение таких соединений и фармацевтических композиций, содержащих эти соединения и их пролекарства, для лечения или предупреждения инфекционного заболевания типа инфекции HBV или HCV. Согласно настоящему изобретению также предложены соединения с очень высокой активностью. Помимо этого, соединения формулы (I) также демонстрируют хорошие профили растворимости и фармакокинетические (ФК) профили.
Настоящее изобретение относится к новым соединениям формулы (I),

где
R1 представляет собой С1-6алкил;
R2 представляет собой бензил, при этом указанный бензил не замещен или замещен одним, двумя или тремя заместителями, независимо выбранными из галогена и С1-6алкила;
R3 представляет собой -NR4R5, где
R4 представляет собой С1-6алкил или С1-6алкоксиС1-6алкил;
R5 представляет собой (С1-6алкил)2NCOOC1-6алкил, С1-6алкоксиС1-6алкил, С1-6алкоксикарбонил(С1-6алкил)аминоС1-6алкил, С1-6алкоксикарбонил(фенил)С1-6алкил, С1-6алкоксикарбонилС1-6алкил, С1-6алкоксикарбонилоксиС1-6алкил, C1-6алкил, С1-6алкилкарбонил(С1-6алкил)аминоС1-6алкил или пирролидинилкарбамоилоксиС1-6алкил; или
R4 и R5 вместе с атомом азота, к которому они присоединены, образуют гетероцикпил;
или к их фармацевтически приемлемым соли, энантиомеру или диастереомеру; при условии, что исключены
6-амино-9-бензил-2-(пропилсульфонимидоил)-7-(пирролидин-1-карбонил)пурин-8-он;
6-амино-9-бензил-7-(пиперидин-1-карбонил)-2-(пропилсульфонимидоил)пурин-8-он;
6-амино-9-бензил-7-(морфолин-4-карбонил)-2-(пропилсульфонимидоил)пурин-8-он;
6-амино-9-бензил-7-(3,3-диметилпирролидин-1-карбонил)-2-(пропилсульфонимидоил)пурин-8-он;
этил-1-[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]пирролидин-2-карбоксилат;
6-амино-7-(2-азаспиро[3.3]гептан-2-карбонил)-9-бензил-2-(пропилсульфонимидоил)пурин-8-он;
6-амино-9-бензил-7-(2-окса-6-азаспиро[3.3]гептан-6-карбонил)-2-(пропилсульфонимидоил)пурин-8-он;
6-амино-9-бензил-7-(3,3-дифторпирролидин-1-карбонил)-2-(пропилсульфонимидоил)пурин-8-он;
6-амино-9-бензил-7-(3-фтор-3-метил-пирролидин-1-карбонил)-2-(пропилсульфонимидоил)пурин-8-он;
и их энантиомеры или диастереомеры.
Данное изобретение также относится к их получению, лекарственным средствам на основе соединения по изобретению и их приготовлению, а также к применению таких соединений формулы (I) в качестве агониста TLR7. Соответственно, соединения формулы (I) являются полезными для лечения или профилактики HBV- и/или HCV-инфекции путем агонизма в отношении Toll-подобных рецепторов.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Если не указано иное, все технические и научные термины, использованные в данном описании, имеют точно такое же значение которое обычно понимается специалистом средней квалификации в области техники, к которой принадлежит данное изобретение. Кроме того, следующие далее определения приведены для иллюстрации и определения значения и объема различных терминов, использованных для описания данного изобретения.
ОПРЕДЕЛЕНИЯ
Термин "С1-6алкил" означает алкильную группу с насыщенной линейной или разветвленной цепью, содержащую 1-6, в частности, 1-4 атома углерода, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил и тому подобное. Особыми "С1-6алкильными" группами являются метил, этил и н-пропил.
Термин "С1-6алкокси" означает группу формулы С1-6алкил-О-. Примеры С1-6алкоксигруппы включают, но не ограничиваются этим, метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси и трет-бутокси. Особыми группами "С1-6алкокси" являются метокси, этокси и изопропокси. Более конкретной С1-6алкоксигруппой является этокси.
Термины "галоген" и "атом галогена" используются в данном описании взаимозаменяемо и означают фтор, хлор, бром или йод.
Термин "гетероциклил" относится к одновалентной насыщенной или частично ненасыщенной моно- или бициклической кольцевой системе из 3-10 атомов в кольце, содержащей 1-5 гетероатомов в кольце, выбранных из N, О и S, причем остальные атомы в кольце представляют собой атомы углерода. В конкретных воплощениях гетероциклил представляет собой одновалентную насыщенную моноциклическую кольцевую систему из 4-7 атомов в кольце, содержащую 1, 2 или 3 гетероатома в кольце, выбранных из N, О и S, причем остальные атомы в кольце представляют собой атомы углерода. Примерами моноциклического насыщенного гетероциклила являются азиридинил, оксиранил, азетидинил, оксетанил, пирролидинил, диметилпирролидинил, этоксикарбонилпирролидинил, тетрагидрофуранил, тетрагидро-тиенил, пиразолидинил, имидазолидинил, оксазолидинил, изоксазолидинил, тиазолидинил, пиперидинил, тетрагидропиранил, тетрагидротиопиранил, пиперазинил, морфолинил, тиоморфолинил, диоксотиоморфолинил, азепанил, диазепанил, гомопиперазинил или оксазепанил. Моноциклический насыщенный гетероциклил может быть дополнительно замещен одним-тремя заместителями, независимо выбранными из галогена, С1-6алкила и С1-6алкоксикарбонила. Примерами замещенного моноциклического насыщенного гетероциклила являются 4-метилпиперазинил, диметилпирролидинил, этоксикарбонилпирролидинил, дифторпирролидинил, фтор(метил)пирролидинил. Примерами бицикпического насыщенного гетероциклила являются азабицикло[3.2.1]октил, хинуклидинил, оксаазабицикло[3.2.1]октил, азабицикло[3.3.1]нонил, оксаазабицикло[3.3.1]нонил, тиаазабицикло[3.3.1]нонил, азаспиро[3.3]гептанил и оксаазаспиро[3.3]гептанил. Примерами частично ненасыщенного гетероциклила являются дигидрофурил, имидазолинил, дигидрооксазолил, тетрагидропиридинил и дигидропиранил.
Термин "карбонил" по отдельности или в комбинации относится к группе -С(О)-.
Термин "С1-6алкилкарбонил" относится к группе С1-6алкил-С(O)-, где "С1-6алкил" является таким, как определено выше. Особой "С1-6алкилкарбонильной" группой является ацетил.
Термин "энантиомер" обозначает два стереоизомера соединения, которые не совпадают с зеркальными отображениями друг друга.
Термин "диастереомер" обозначает стереоизомеры с двумя или несколькими центрами хиральности, и их молекулы не являются зеркальными отображениями друг друга. Диастереомеры имеют другие физические свойства, например, точки плавления, точки кипения, спектральные свойства и реакционные способности.
Термин "фармацевтически приемлемые соли" означает соли, которые не являются биологически или иным образом нежелательными. Фармацевтически приемлемые соли включают соли присоединения как кислоты, так и основания.
Термин "фармацевтически приемлемая соль присоединения кислоты" означает такие фармацевтически приемлемые соли, которые образуются из неорганических кислот, таких как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, угольная кислота, фосфорная кислота, и органических кислот, выбранных из кислот классов алифатических, циклоалифатических, ароматических, аралифатических, гетероциклических, карбоновых и сульфоновых органических кислот, таких как муравьиная кислота, уксусная кислота, пропионовая кислота, гликолевая кислота, глюконовая кислота, молочная кислота, пировиноградная кислота, щавелевая кислота, яблочная кислота, малеиновая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, винная кислота, лимонная кислота, аспарагиновая кислота, аскорбиновая кислота, глутаминовая кислота, антраниловая кислота, бензойная кислота, коричная кислота, миндальная кислота, эмбоновая кислота, фенилуксусная кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота и салициловая кислота.
Термин "фармацевтически приемлемая соль присоединения основания" означает такие фармацевтически приемлемые соли, которые образуются из органического или неорганического основания. Примеры приемлемых неорганических оснований включают соли натрия, калия, аммония, кальция, магния, железа, цинка, меди, марганца и алюминия. Соли, происходящие из фармацевтически приемлемых органических нетоксичных оснований, включают соли первичных, вторичных и третичных аминов, замещенных аминов, включая природные замещенные амины, циклических аминов и основных ионообменных смол, таких как изопропиламин, триметиламин, диэтиламин, триэтиламин, трипропиламин, этаноламин, 2-диэтиламиноэтанол, триметамин, дициклогексиламин, лизин, аргинин, гистидин, кофеин, прокаин, гидрабамин, холин, бетаин, этилендиамин, глюкозамин, метилглюкамин, теобромин, пурины, пиперазин, пиперидин, N-этилпиперидин и полиаминные смолы.
Соединения общей формулы (I) и их пролекарства, которые содержат один или несколько хиральных центров, могут быть представлены в виде или рацематов, или диастереомерных смесей, или оптически активных индивидуальных изомеров. Рацематы могут быть разделены на энантиомеры в соответствии с известными методами. В частности, соли диастереомеров, которые могут быть разделены путем кристаллизации, образуются из рацемических смесей в результате взаимодействия с оптически активной кислотой, такой как, например, D- или L-винная кислота, миндальная кислота, яблочная кислота, молочная кислота или камфорсульфоновая кислота.
Термин "пролекарство" означает форму или производное соединения, которая(ое) метаболизируется in vivo, например, под действием биологических жидкостей или ферментов субъекта после введения, в фармакологически активную форму соединения с целью оказания желаемого фармакологического эффекта. Пролекарства описаны, например, в "The Organic Chemistry of Drug Design and Drug Action", под редакцией Richard В. Silverman, Academic Press, San Diego, 2004, Chapter 8. Prodrugs and Drug Delivery Systems, pp. 497-558.
Подразумевается, что термин "фармацевтически активный метаболит" означает фармакологически активный продукт, образующийся в организме в ходе метаболизма конкретного соединения или его соли. После попадания в организм большая часть лекарственных средств становится субстратами для химических реакций, которые могут изменить их физические свойства и биологические эффекты. Такие метаболические превращения, обычно затрагивающие полярность соединений по изобретению, изменяют путь, согласно которому лекарственные средства распределяются в организме и выводятся из него. Однако, в некоторых случаях необходимым условием для проявления терапевтического эффекта является метаболизм лекарственного средства.
Термин "терапевтически эффективное количество" означает количество соединения или молекулы по настоящему изобретению, которое, при введении субъекту, (1) лечит или предотвращает конкретное заболевание, состояние или расстройство, (2) ослабляет, уменьшает интенсивность симптомов или устраняет один или более симптомов конкретного заболевания, состояния или расстройства или (3) предотвращает или задерживает начало появления одного или более симптомов конкретного заболевания, состояния или расстройства, описанного в данной заявке. Терапевтически эффективное количество будет варьировать в зависимости от соединения, подлежащего лечению болезненного состояния, тяжести подвергаемого лечению заболевания, возраста и относительного состояния здоровья субъекта, пути и формы введения, компетентности лечащего врача или ветеринара и других факторов.
Термин "фармацевтическая композиция" означает смесь или раствор, содержащую(ий) терапевтически эффективное количество активного фармацевтического ингредиента вместе с фармацевтически приемлемыми эксципиентами, подлежащую(ий) введению млекопитающему, например, человеку, нуждающийся в этом.
АГОНИСТ TLR7 И ПРОЛЕКАРСТВО
Настоящее изобретение относится к соединению формулы (I),
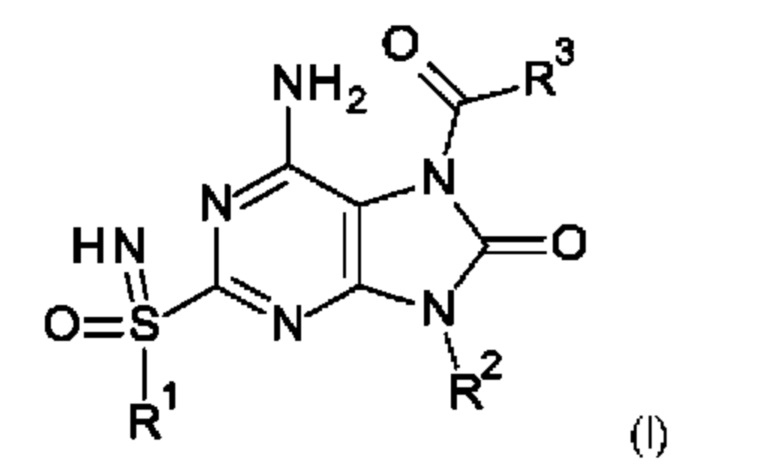
где
R1 представляет собой С1-6алкил;
R2 представляет собой бензил, при этом указанный бензил не замещен или замещен одним, двумя или тремя заместителями, независимо выбранными из галогена и С1-6алкила;
R3 представляет собой -NR4R5, где
R4 представляет собой С1-6алкил или С1-6алкоксиС1-6алкил;
R5 представляет собой (С1-6алкил)2NCOOC1-6алкил, С1-6алкоксиС1-6алкил, C1-6алкоксикарбонил(С1-6алкил)аминоС1-6алкил, С1-6алкоксикарбонил(фенил)С1-6алкил, С1-6алкоксикарбонилС1-6алкил, С1-6алкоксикарбонилоксиС1-6алкил, С1-6алкил, C1-6алкилкарбонил(С1-6алкил)аминоС1-6алкил или пирролидинилкарбамоилоксиС1-6алкил; или
R4 и R5 вместе с атомом азота, к которому они присоединены, образуют гетероциклил;
или к его фармацевтически приемлемым соли, энантиомеру или диастереомеру;
при условии, что исключены
6-амино-9-бензил-2-(пропилсульфонимидоил)-7-(пирролидин-1-карбонил)пурин-8-он;
6-амино-9-бензил-7-(пиперидин-1-карбонил)-2-(пропилсульфонимидоил)пурин-8-он;
6-амино-9-бензил-7-(морфолин-4-карбонил)-2-(пропилсульфонимидоил)пурин-8-он;
6-амино-9-бензил-7-(3,3-диметилпирролидин-1-карбонил)-2-(пропилсульфонимидоил)пурин-8-он;
этил-1-[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]пирролидин-2-карбоксилат;
6-амино-7-(2-азаспиро[3.3]гептан-2-карбонил)-9-бензил-2-(пропилсульфонимидоил)пурин-8-он;
6-амино-9-бензил-7-(2-окса-6-азаспиро[3.3]гептан-6-карбонил)-2-(пропилсульфонимидоил)пурин-8-он;
6-амино-9-бензил-7-(3,3-дифторпирролидин-1-карбонил)-2-(пропилсульфонимидоил)пурин-8-он;
6-амино-9-бензил-7-(3-фтор-3-метил-пирролидин-1-карбонил)-2-(пропилсульфонимидоил)пурин-8-он;
и их энантиомеры или диастереомеры.
Следующим воплощением настоящего изобретения является (2) соединение формулы (I), где
R1 представляет собой С1-6алкил;
R2 представляет собой бензил, при этом указанный бензил не замещен или замещен галогеном и С1-6алкилом;
R3 представляет собой азетидинил;
пиперазинил, замещенный С1-6алкилом;
пиперидинил, замещенный пиперидинилом;
пирролидинил; или
-NR4R5, где
R4 представляет собой С1-6алкил или С1-6алкоксиС1-6алкил;
R5 представляет собой (С1-6алкил)2NCOOC1-6алкил, С1-6алкоксиС1-6алкил, C1-6алкоксикарбонил(C1-6алкил)аминоС1-6алкил, С1-6алкоксикарбонил(фенил)С1-6алкил, С1-6алкоксикарбонилС1-6алкил, С1-6алкоксикарбонилоксиС1-6алкил, С1-6алкил, С1-6алкилкарбонил(C1-6алкил)аминоС1-6алкил или пирролидинилкарбамоилоксиС1-6алкил;
или его фармацевтически приемлемые соль, энантиомер или диастереомер.
Следующим воплощением настоящего изобретения является (3) соединение формулы (I), где
R1 представляет собой этил или пропил;
R2 представляет собой бензил, бромбензил, хлорбензил, фторбензил или метилбензил;
R3 представляет собой азетидинил;
4-метилпиперазинил;
пиперидинилпиперидинил;
пирролидинил; или
-NR4R5, где
R4 представляет собой метил, этил, пропил или метоксиэтил;
R5 представляет собой ацетил(метил)аминоэтил, бутил, бутил(метил)карбамоилоксиэтил, диэтилкарбамоилоксиэтил, этоксикарбонил(метил)аминоэтил, этоксикарбонилэтил, этоксикарбонилизобутил, этоксикарбонилизопентил, этоксикарбонилметил, этоксикарбонилоксиэтил, этоксикарбонил(фенил)этил, этил, изобутил, изопропоксикарбонилизопентил, изопропоксикарбонил(фенил)этил, изопропил, метоксикарбонил(метил)аминоэтил, метоксиэтил, метоксипропил, пропил, пропил(метил)карбамоилоксиэтил, пирролидинилкарбамоилоксиэтил, трет-бутоксикарбонил(метил)аминоэтил, трет-бутоксикарбонилэтил, трет-бутоксикарбонилизопентил или трет-бутоксикарбонил(фенил)этил;
или его фармацевтически приемлемые соль, энантиомер или диастереомер.
Следующим воплощением настоящего изобретения является (3-1) соединение формулы (I), где
R1 представляет собой этил или пропил;
R2 представляет собой бензил, хлорбензил, фторбензил или метилбензил;
R3 представляет собой азетидинил;
4-метилпиперазинил;
пиперидинилпиперидинил;
пирролидинил; или
-NR4R5, где
R4 представляет собой метил, этил, пропил или метоксиэтил;
R5 представляет собой ацетил(метил)аминоэтил, бутил, бутил(метил)карбамоилоксиэтил, диэтилкарбамоилоксиэтил, этоксикарбонил(метил)аминоэтил, этоксикарбонилэтил, этоксикарбонилизобутил, этоксикарбонилизопентил, этоксикарбонилметил, этоксикарбонилоксиэтил, этоксикарбонил(фенил)этил, этил, изобутил, изопропоксикарбонилизопентил, изопропоксикарбонил(фенил)этил, изопропил, метоксикарбонил(метил)аминоэтил, метоксиэтил, метоксипропил, пропил, пропил(метил)карбамоилоксиэтил, пирролидинилкарбамоилоксиэтил, трет-бутоксикарбонил(метил)аминоэтил, трет-бутоксикарбонилэтил, трет-бутоксикарбонилизопентил или трет-бутоксикарбонил(фенил)этил;
или его фармацевтически приемлемые соль, энантиомер или диастереомер.
Следующим воплощением настоящего изобретения является (4) соединение формулы (I), где R3 представляет собой азетидинил, 4-метилпиперазинил, пиперидинилпиперидинил, пирролидинил, ацетил(метил)аминоэтил(метил)амино, бис(метоксиэтил)амино, бутил(этил)амино, бутил (метил)амино, бутил(метил)карбамоилоксиэтил(метил)амино, диэтилкарбамоилоксиэтил(метил)амино, этоксикарбонил(метил)аминоэтил(метил)амино, этоксикарбонилэтил(метил)амино, этоксикарбонилизобутил(метил)амино, этоксикарбонилизопентил(метил)амино, этоксикарбонилметил(метил)амино, этоксикарбонилоксиэтил(метил)амино, этоксикарбонил(фенил)этил(метил)амино, этил(метил)амино, изобутил(метил)амино, изопропоксикарбонилизопентил(метил)амино, изопропоксикарбонил(фенил)этил(метил)амино, изопропил(метил)амино, метоксикарбонил(метил)аминоэтил(метил)амино, метоксиэтил(этил)амино, метоксиэтил(метил)амино, метоксиэтил(пропил)амино, метоксипропил(метил)амино, пропил(этил)амино, пропил(метил)амино, пропил(метил)карбамоилоксиэтил(метил)амино, пирролидинилкарбамоилоксиэтил(метил)амино, трет-бутоксикарбонил(метил)аминоэтил(метил)амино, трет-бутоксикарбонилэтил(метил)амино, трет-бутоксикарбонилизопентил(метил)амино или трет-бутоксикарбонил(фенил)этил(метил)амино; или его фармацевтически приемлемые соль, энантиомер или диастереомер.
Следующим воплощением настоящего изобретения является (5) соединение формулы (I), где R1 представляет собой этил.
Следующим воплощением настоящего изобретения является (6) соединение формулы (I), где R2 представляет собой бензил, замещенный галогеном или С1-6алкилом.
Следующим воплощением настоящего изобретения является (7) соединение формулы (I), где R2 представляет собой бромбензил, хлорбензил, фторбензил или метилбензил.
Следующим воплощением настоящего изобретения является (7-1) соединение формулы (I), где R2 представляет собой хлорбензил, фторбензил или метилбензил.
Следующим воплощением настоящего изобретения является (8) соединение формулы (I), где R2 представляет собой бромбензил, хлорбензил или фторбензил.
Следующим воплощением настоящего изобретения является (8-1) соединение формулы (I), где R2 представляет собой хлорбензил или фторбензил.
Следующим воплощением настоящего изобретения является (9) соединение формулы (I), где R3 представляет собой -NR4R5, где R4 представляет собой С1-6алкил, R5 представляет собой С1-6алкил.
Следующим воплощением настоящего изобретения является (10) соединение формулы (I), где R3 представляет собой пропил(метил)амино или этил(метил)амино.
Следующим воплощением настоящего изобретения является (11) соединение формулы (I), где
R1 представляет собой С1-6алкил;
R2 представляет собой бензил, при этом указанный бензил замещен галогеном или С1-6алкилом;
R3 представляет собой -NR4R5, где R4 представляет собой С1-6алкил, R5 представляет собой С1-6алкил;
или его фармацевтически приемлемые соль, энантиомер или диастереомер.
Следующим воплощением настоящего изобретения является (12) соединение формулы (I), где
R1 представляет собой этил;
R2 представляет собой метилбензил, бромбензил, хлорбензил или фторбензил;
R3 представляет собой пропил(метил)амино или этил(метил)амино;
или его фармацевтически приемлемые соль, энантиомер или диастереомер.
Следующим воплощением настоящего изобретения является (12-1) соединение формулы (I), где
R1 представляет собой этил;
R2 представляет собой метилбензил, хлорбензил или фторбензил;
R3 представляет собой пропил(метил)амино или этил(метил)амино;
или его фармацевтически приемлемые соль, энантиомер или диастереомер.
Другое воплощение настоящего изобретения заключается в том, что (13) конкретными соединениями формулы (I) являются следующие:
6-амино-9-бензил-N-метил-8-оксо-N-пропил-2-(пропилсульфонимидоил)пурин-7-карбоксамид;
6-амино-9-бензил-N-(2-метоксиэтил)-N-метил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамид;
6-амино-9-бензил-N-этил-8-оксо-N-пропил-2-(пропилсульфонимидоил)пурин-7-карбоксамид;
6-амино-9-бензил-7-[4-(1-пиперидил)пиперидин-1-карбонил]-2-(пропилсульфонимидоил)пурин-8-он;
6-амино-9-бензил-N-этил-N-(2-метоксиэтил)-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамид;
6-амино-9-бензил-N-бутил-N-этил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамид;
6-амино-9-бензил-N-(2-метоксиэтил)-8-оксо-N-пропил-2-(пропилсульфонимидоил)пурин-7-карбоксамид;
6-амино-9-бензил-N,N-бис(2-метоксиэтил)-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамид;
6-амино-7-(азетидин-1-карбонил)-9-бензил-2-(пропилсульфонимидоил)пурин-8-он;
6-амино-9-бензил-N-изопропил-N-метил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамид;
6-амино-9-бензил-7-(4-метилпиперазин-1-карбонил)-2-(пропилсульфонимидоил)пурин-8-он;
6-амино-9-бензил-N-(3-метоксипропил)-N-метил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамид;
6-амино-9-бензил-N-изобутил-N-метил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамид;
этил-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]ацетат;
этил-3-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]пропаноат;
трет-бутил-3-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]пропаноат;
этил-(2S)-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]пропаноат;
трет-бутил-(2S)-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]-4-метил-пентаноат;
изопропил-(2S)-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]-4-метил-пентаноат;
этил-(2S)-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]-3-метил-бутаноат;
этил-(2S)-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]-4-метил-пентаноат;
этил-(2S)-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]-3-фенил-пропаноат;
изопропил-(2S)-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]-3-фенил-пропаноат;
трет-бутил-(2S)-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]-3-фенил-пропаноат;
N-[2-[ацетил(метил)амино]этил]-6-амино-9-бензил-N-метил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамид;
метил-N-[2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]этил]-N-метил-карбамат;
трет-бутил-N-[2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]этил]-N-метил-карбамат;
этил-N-[2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]этил]-N-метил-карбамат;
2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]этил-N-бутил-N-метил-карбамат;
2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]этил-пирролидин-1-карбоксилат;
2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]этил-N-метил-N-пропил-карбамат;
2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]этил-N,N-диэтилкарбамат;
2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]этил-этилкарбонат;
6-амино-N-бутил-9-[(4-хлорфенил)метил]-N-метил-8-оксо-2-[S(S)-пропилсульфонимидоил]пурин-7-карбоксамид;
6-амино-N-бутил-9-[(4-хлорфенил)метил]-N-метил-8-оксо-2-[S(S)-пропилсульфонимидоил]пурин-7-карбоксамид;
6-амино-9-[(4-хлорфенил)метил]-N-этил-N-метил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамид;
6-амино-N-метил-8-оксо-N-пропил-2-[S(S)-пропилсульфонимидоил]-9-(п-толилметил)пурин-7-карбоксамид;
6-амино-N-метил-8-оксо-N-пропил-2-[S(R)-пропилсульфонимидоил]-9-(п-толилметил)пурин-7-карбоксамид;
6-амино-2-[S(S)-пропилсульфонимидоил]-9-(п-толилметил)-7-(пирролидин-1-карбонил)пурин-8-он;
6-амино-2-[S(R)-пропилсульфонимидоил]-9-(п-толилметил)-7-(пирролидин-1-карбонил)пурин-8-он;
6-амино-N-(2-метоксиэтил)-N-метил-8-оксо-2-[S(S)-пропилсульфонимидоил]-9-(п-толилметил)пурин-7-карбоксамид;
6-амино-N-(2-метоксиэтил)-N-метил-8-оксо-2-[S(R)-пропилсульфонимидоил]-9-(п-толилметил)пурин-7-карбоксамид;
6-амино-N-этил-N-метил-8-оксо-2-(пропилсульфонимидоил)-9-(п-толилметил)пурин-7-карбоксамид;
6-амино-N-бутил-N-метил-8-оксо-2-(пропилсульфонимидоил)-9-(п-толилметил)пурин-7-карбоксамид;
6-амино-9-[(4-хлорфенил)метил]-2-[S(R)-этилсульфонимидоил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамид;
6-амино-9-[(4-хлорфенил)метил]-2-[S(S)-этилсульфонимидоил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамид;
6-амино-9-[(4-хлорфенил)метил]-N-этил-2-[S(S)-этилсульфонимидоил]-N-метил-8-оксо-пурин-7-карбоксамид;
6-амино-9-[(4-хлорфенил)метил]-N-этил-2-[S(R)-этилсульфонимидоил]-N-метил-8-оксо-пурин-7-карбоксамид;
6-амино-2-[S(S)-этилсульфонимидоил]-N-метил-8-оксо-N-пропил-9-(п-толилметил)пурин-7-карбоксамид;
6-амино-2-[S(R)-этилсульфонимидоил]-N-метил-8-оксо-N-пропил-9-(п-толилметил)пурин-7-карбоксамид;
6-амино-N-этил-2-[S(S)-этилсульфонимидоил]-N-метил-8-оксо-9-(п-толилметил)пурин-7-карбоксамид;
6-амино-N-этил-2-[S(R)-этилсульфонимидоил]-N-метил-8-оксо-9-(п-толилметил)пурин-7-карбоксамид;
6-амино-2-[S(S)-этилсульфонимидоил]-9-[(4-фторфенил)метил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамид;
6-амино-2-[S(R)-этилсульфонимидоил]-9-[(4-фторфенил)метил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамид;
6-амино-N-этил-2-(этилсульфонимидоил)-9-[(4-фторфенил)метил]-N-метил-8-оксо-пурин-7-карбоксамид;
6-амино-N-этил-2-[S(S)-(этилсульфонимидоил)]-9-[(4-фторфенил)метил]-N-метил-8-оксо-пурин-7-карбоксамид;
6-амино-N-этил-2-[S(R)-(этилсульфонимидоил)]-9-[(4-фторфенил)метил]-N-метил-8-оксо-пурин-7-карбоксамид;
6-амино-9-[(4-бромфенил)метил]-2-(этилсульфонимидоил)-N-метил-8-оксо-N-пропил-пурин-7-карбоксамид;
6-амино-2-[S(R)-этилсульфонимидоил]-9-[(4-бромфенил)метил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамид;
6-амино-2-[S(S)-этилсульфонимидоил]-9-[(4-бромфенил)метил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамид;
6-амино-9-[(4-бромфенил)метил]-N-этил-2-(этилсульфонимидоил)-N-метил-8-оксо-пурин-7-ка рбоксамид;
6-амино-9-[(4-бромфенил)метил]-N-этил-2-[S(S)-(этилсульфонимидоил)]-N-метил-8-оксо-пурин-7-карбоксамид; и
6-амино-9-[(4-бромфенил)метил]-N-этил-2-[S(R)-(этилсульфонимидоил)]-N-метил-8-оксо-пурин-7-карбоксамид;
или их фармацевтически приемлемые соль, энантиомер или диастереомер.
Другое воплощение настоящего изобретения заключается в том, что (14) более конкретными соединениями формулы (I) являются следующие:
6-амино-9-бензил-N-метил-8-оксо-N-пропил-2-(пропилсульфонимидоил)пурин-7-карбоксамид;
6-амино-9-[(4-хлорфенил)метил]-2-[S(R)-этилсульфонимидоил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамид;
6-амино-9-[(4-хлорфенил)метил]-2-[S(S)-этилсульфонимидоил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамид;
6-амино-9-[(4-хлорфенил)метил]-N-этил-2-[S(S)-этилсульфонимидоил]-N-метил-8-оксо-пурин-7-карбоксамид;
6-амино-9-[(4-хлорфенил)метил]-N-этил-2-[S(R)-этилсульфонимидоил]-N-метил-8-оксо-пурин-7-карбоксамид;
6-амино-2-[S(S)-этилсульфонимидоил]-N-метил-8-оксо-N-пропил-9-(п-толилметил)пурин-7-карбоксамид;
6-амино-2-[S(R)-этилсульфонимидоил]-N-метил-8-оксо-N-пропил-9-(п-толилметил)пурин-7-карбоксамид;
6-амино-N-этил-2-[S(S)-этилсульфонимидоил]-N-метил-8-оксо-9-(п-толилметил)пурин-7-карбоксамид;
6-амино-N-этил-2-[S(R)-этилсульфонимидоил]-N-метил-8-оксо-9-(п-толилметил)пурин-7-карбоксамид;
6-амино-2-(этилсульфонимидоил)-9-[(4-фторфенил)метил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамид;
6-амино-2-[S(S)-этилсульфонимидоил]-9-[(4-фторфенил)метил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамид;
6-амино-2-[S(R)-этилсульфонимидоил]-9-[(4-фторфенил)метил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамид;
6-амино-N-этил-2-(этилсульфонимидоил)-9-[(4-фторфенил)метил]-N-метил-8-оксо-пурин-7-карбоксамид;
6-амино-N-этил-2-[S(R)-(этилсульфонимидоил)]-9-[(4-фторфенил)метил]-N-метил-8-оксо-пурин-7-карбоксамид;
6-амино-N-этил-2-[S(R)-(этилсульфонимидоил)]-9-[(4-фторфенил)метил]-N-метил-8-оксо-пурин-7-карбоксамид;
6-амино-9-[(4-бромфенил)метил]-2-(этилсульфонимидоил)-N-метил-8-оксо-N-пропил-пурин-7-карбоксамид;
6-амино-2-[S(R)-этилсульфонимидоил]-9-[(4-бромфенил)метил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамид;
6-амино-2-[S(S)-этилсульфонимидоил]-9-[(4-бромфенил)метил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамид;
6-амино-9-[(4-бромфенил)метил]-N-этил-2-(этилсульфонимидоил)-N-метил-8-оксо-пурин-7-карбоксамид;
6-амино-9-[(4-бромфенил)метил]-N-этил-2-[S(S)-(этилсульфонимидоил)]-N-метил-8-оксо-пурин-7-карбоксамид; и
6-амино-9-[(4-бромфенил)метил]-N-этил-2-[S(R)-(этилсульфонимидоил)]-N-метил-8-оксо-пурин-7-карбоксамид;
или их фармацевтически приемлемые соль, энантиомер или диастереомер.
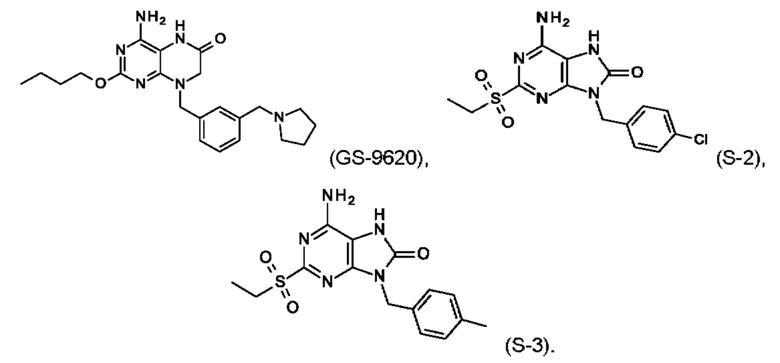
В некоторых воплощениях тестировали соединения по настоящему изобретению и сравнивали с приведенными ниже референсными соединениями. Как и большинство успешных биофармацевтических компаний, специализирующихся на открытии и разработке агонистов TLR7 для лечения заболеваний печени, Gilead располагает ассортиментом самых передовых агонистов TLR7, среди которых имеются такие ведущие соединения, как GS-9620, для которого начата фаза II испытаний. Предложенное Gilead соединение GS-9620, раскрытое в US 20100143301 как соединение примера 49, соединение S-2 и соединение S-3, раскрытые в JP 1999193282, все были выбраны в этой заявке в качестве референсных соединений:
СИНТЕЗ
Соединения по настоящему изобретению могут быть получены любым традиционным способом. Подходящие методы синтеза этих соединений, а также исходные вещества для их получения приведены ниже на схемах и в примерах. Все заместители, в частности, от R1 до R14, являются такими, как определено выше, если не указано иное. Кроме того и если прямо не указано иное, все реакции, реакционные условия, сокращения и символы имеют значения, хорошо известные специалисту средней квалификации в органической химии.
Схема 1
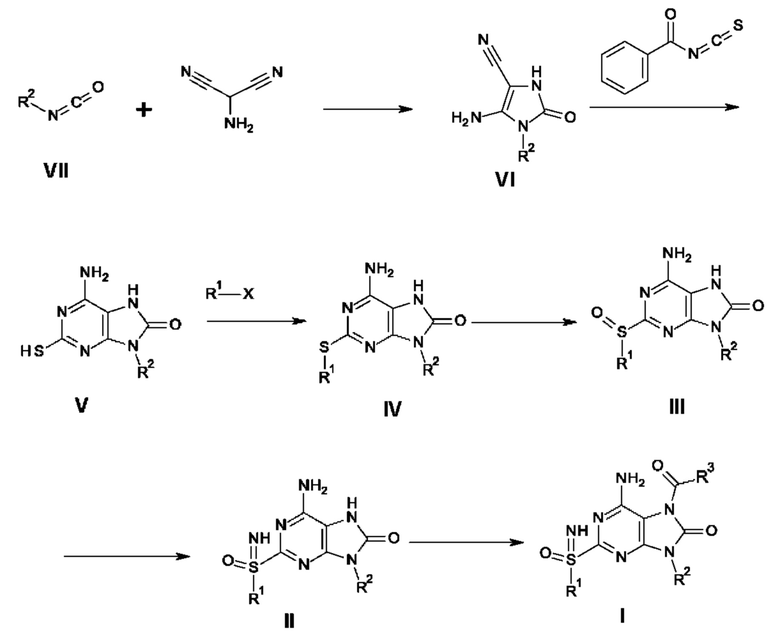
Соединение формулы VI получают путем циклизации изоцианата VII с использованием аминомалононитрил-п-толуолсульфоната. Затем синтезируют бицикл V, проводя взаимодействие соединения формулы VI с бензоилизотиоцианатом в присутствии неорганического основания, такого как NaOH или KOH. После алкилирования бицикла V алкилгалогенидом в присутствии основания, такого как K2CO3, NaH или Cs2CO3, получают соединение формулы IV. Соединение формулы III получают путем окисления соединения формулы IV, используя окислитель, такой как мета-хлорпероксибензойная кислота, аддукт перекиси водорода с мочевиной и HIO4. Соединение формулы II получают путем иминирования соединения формулы III с использованием иминирующего реагента, такого как азид натрия в кислоте, причем указанной кислотой является, например, реагент Итона или РРА (полифосфорная кислота). Соединение формулы I получают путем взаимодействия соединения формулы II с карбамоилхлоридом в присутствии смеси оснований, таких как пиридин и триэтиламин, пиридин и DIPEA (N,N-диизопропилэтиламин), DMAP (4-диметиламинопиридин) и триэтиламин или DMAP и DIPEA.
Схема 2
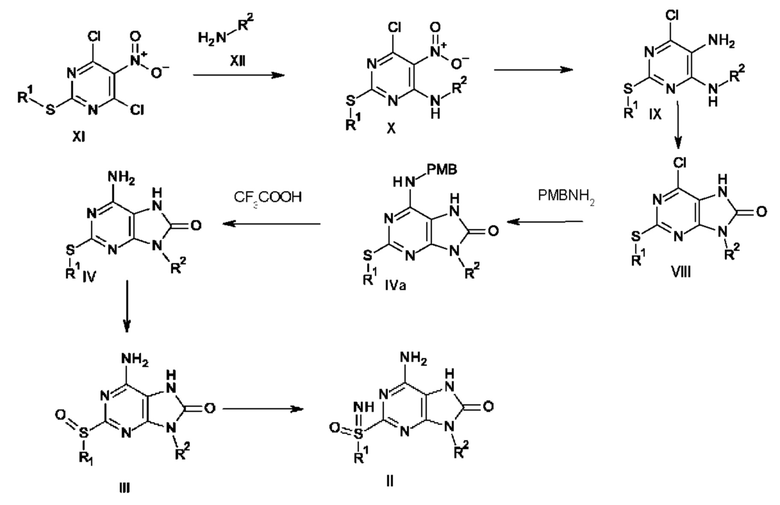
Кроме того, соединение формулы II может быть получено так, как на схеме 2.
Соединение формулы X получают путем взаимодействия соединения формулы XI с R2NH2. После восстановления соединения X с использованием восстанавливающего реагента, такого как порошок цинка или железа в АсОН, получают соединение формулы IX. После циклизации соединения формулы IX с использованием циклизующих реагентов, таких как фосген, карбонилдиимидазол, диэтилкарбонат и трифосген, получают соединение формулы VIII. Соединение формулы IVa получают путем обработки соединения формулы VIII пара-метоксибензиламином (PMBNH2). Соединение формулы III получают путем удаления защитной группы с соединения формулы IVa, используя кислоту, такую как CF3COOH, с последующим окислением с применением окислителя, такого как мета-хлорпероксибензойная кислота, аддукт перекиси водорода с мочевиной и HIO4. Соединение формулы II получают путем иминирования соединения формулы III с использованием иминирующего реагента, такого как азид натрия в кислоте, причем указанной кислотой является, например, реагент Итона или РРА.
Данное изобретение также относится к способу получения соединения формулы (I), включающему приведение во взаимодействие:
соединения формулы (II)
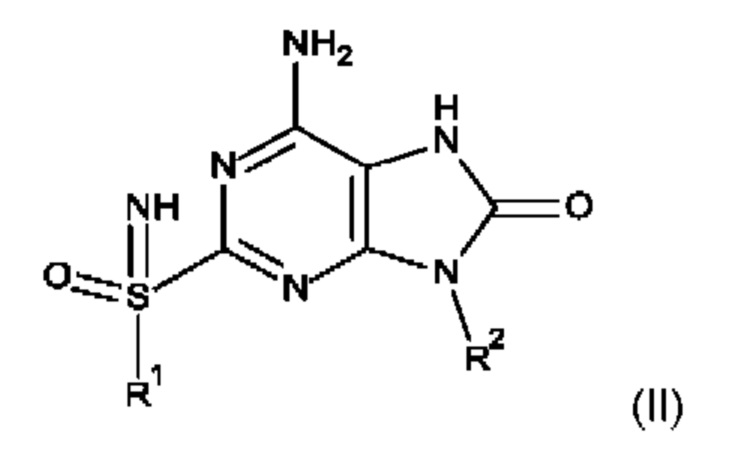
с карбамоилхлоридом в присутствии смеси оснований;
где R1 и R2 определены выше.
На вышеуказанной стадии смесью оснований может быть, например, пиридин и триэтиламин, пиридин и DIPEA, DMAP и триэтиламин или DMAP и DIPEA.
Соединение формулы (I), полученное согласно упомянутому выше способу, также составляет предмет данного изобретения.
ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ВВЕДЕНИЕ
В другом воплощении предложены фармацевтические композиции или лекарственные средства, содержащие соединения по изобретению и терапевтически инертный носитель, разбавитель или эксципиент, а также способы применения соединений по изобретению для приготовления таких композиций и лекарственных средств. В одном из примеров на основе соединений формулы (I) могут быть приготовлены композиции путем смешивания их при температуре окружающей среды, соответствующем значении рН и желаемой степени чистоты с физиологически приемлемыми носителями, т.е. носителями, которые являются нетоксичными для реципиентов, в дозировках и концентрациях, применяемых в форме галенова препарата для введения. Значение рН композиции зависит гланым образом от конкретного применения и концентрации соединения, но предпочтительно везде изменяется в диапазоне от примерно 3 до примерно 8. В одном из примеров на основе соединения формулы (I) готовят композицию в ацетатном буфере, при рН 5. В другом воплощении соединения формулы (I) являются стерильными. Соединение можно хранить, например, в виде твердой или аморфной композиции, в виде лиофилизированной композиции или в виде водного раствора.
Данные композиции готовят, дозируют и вводят способом, соответствующим надлежащей медицинской практике. Факторы, рассматриваемые в этом контексте, включают конкретное подвергаемое лечению расстройство, конкретное подвергаемое лечению млекопитающее, клиническое состояние отдельного пациента, причину расстройства, место доставки агента, способ введения, схему введения и другие факторы, известные врачам. "Эффективное количество" подлежащего введению соединения будет обусловлено такого рода соображениями и составлять минимальное количество, необходимое для активации рецептора TLR7 и вызывающее продуцирование INF-α и других цитокинов, которые могут быть использованы, без ограничения, для лечения пациентов с инфекцией вирусом гепатита В и/или С либо предупреждения у них данной инфекции.
В одном из примеров фармацевтически эффективное количество вводимого парентерально соединения по изобретению из расчета на одну дозу будет находиться в диапазоне примерно 0,1-50 мг/кг, альтернативно примерно от 0,1-30 мг/кг массы тела пациента в сутки, причем типичный начальный диапазон для используемого соединения составляет 0,3-15 мг/кг/сутки. В другом воплощении пероральные стандартные лекарственные формы, такие как таблетки и капсулы, предпочтительно содержат от примерно 20 до примерно 1000 мг соединения по изобретению.
Соединения по изобретению можно вводить любым подходящим способом, включая пероральное, местное (в том числе трансбуккальное и сублингвальное), ректальное, вагинальное, трансдермальное, парентеральное, подкожное, внутрибрюшинное, внутрилегочное, интрадермальное, интратекальное, эпидуральное и интраназальное введение, и, если необходимо для местного лечения, введение в область поражения. Парентеральные инфузии включают внутримышечное, внутривенное, интраартериальное, внутрибрюшинное или подкожное введение.
Соединения по настоящему изобретению можно вводить в любой форме, удобной для введения, например, в таблетках, порошках, капсулах, растворах, дисперсиях, суспензиях, сиропах, спреях, суппозиториях, гелях, эмульсиях, пластырях и так далее. Такие композиции могут содержать компоненты, традиционно используемые в фармацевтических препаратах, например, разбавители, носители, модификаторы рН, подсластители, объемообразующие агенты и другие активные агенты.
Типичную композицию готовят путем смешивания соединения по настоящему изобретению и носителя или эксципиента. Подходящие носители и эксципиенты хорошо известны специалистам в данной области техники и описаны подробно, например, в Ansel, Howard С., et al., Ansel's Pharmaceutical Dosage Forms and Drug Delivery Systems. Philadelphia: Lippincott, Williams & Wilkins, 2004; Gennaro, Alfonso R., et al., Remington: The Science and Practice of Pharmacy. Philadelphia: Lippincott, Williams & Wilkins, 2000; и Rowe, Raymond C. Handbook of Pharmaceutical Excipients. Chicago, Pharmaceutical Press, 2005. Композиции также могут включать одно или более чем одно из следующего: буферы, стабилизирующие агенты, поверхностно-активные вещества, увлажняющие агенты, смазывающие вещества, эмульгаторы, суспендирующие агенты, консерванты, антиоксиданты, придающие непрозрачность агенты, глиданты, технологические добавки, красители, подсластители, отдушки, корригенты, разбавители и другие известные вспомогательные вещества для обеспечения наилучшей презентации лекарственного средства (т.е. соединения по настоящему изобретению или фармацевтической композиции на его основе) или содействия производству фармацевтического продукта (т.е. лекарственного средства).
Примером подходящей пероральной лекарственной формы является таблетка, содержащая примерно от 20 до 1000 мг соединения по изобретению в смеси с примерно 30-90 мг безводной лактозы, примерно 5-40 мг натриевой соли кроскармелозы, примерно 5-30 мг поливинилпирролидона (PVP) K30 и примерно 1-10 мг стеарата магния. Измельченные ингредиенты сначала смешивают вместе и затем перемешивают с раствором PVP. Полученную композицию можно сушить, подвергать гранулированию, смешивать со стеаратом магния и подвергать прессованию в форму таблетки, используя традиционное оборудование Типичная аэрозольная композиция может быть приготовлена путем растворения соединения по изобретению, например 20-1000 мг, в подходящем буферном растворе, например фосфатном буфере, добавления регулирующего тоничность вещества, например, соли, такой как хлорид натрия, если это желательно. Можно выполнить фильтрование раствора, например, используя фильтр (0,2 микрона), чтобы удалить примеси и загрязняющие вещества.
Таким образом, одно из воплощений включает фармацевтическую композицию, содержащую соединение формулы (I) или его фармацевтически приемлемые соли либо их энантиомеры или диастереомеры.
Следующее воплощение включает фармацевтическую композицию, содержащую соединение формулы (I) или его фармацевтически приемлемые соли либо их энантиомеры или диастереомеры вместе с фармацевтически приемлемым носителем или эксципиентом.
Другое воплощение включает фармацевтическую композицию, содержащую соединение формулы (I) или его фармацевтически приемлемые соли либо их энантиомеры или диастереомеры для применения в лечении инфекции вирусом гепатита В.
ПОКАЗАНИЯ И СПОСОБЫ ЛЕЧЕНИЯ
Согласно настоящему изобретению предложены способы лечения или предупреждения инфекции вирусом гепатита В и/или инфекции вирусом гепатита С у пациента, нуждающегося в этом.
Согласно настоящему изобретению дополнительно предложены способы введения терапевтически эффективного количества соединения формулы (I) или других соединений по изобретению в кровоток пациента для лечения и/или предупреждения инфекции вирусом гепатита В и/или С.
Способы по настоящему изобретению особенно хорошо подходят для являющихся людьми пациентов. В частности, способы и дозы по настоящему изобретению могут быть полезны для HBV- и/или HCV-инфицированных пациентов, но ими не ограничиваются. Способы и дозы по настоящему изобретению также полезны для пациентов, проходящих лечение другими противовирусными препаратами. Способы предупреждения по настоящему изобретению особенно полезны для пациентов с риском вирусной инфекции. Эти пациенты включают, но не ограничиваются этим, работников здравоохранения, например, врачей, медсестер, лиц, обеспечивающих уход в хосписах; военнослужащих; учителей; работников по уходу за детьми; пациентов, путешествующих по зарубежным странам или проживающих в них, в частности, в странах третьего мира, включая работников социальных служб, миссионеров и иностранных дипломатов. И наконец, данные способы и композиции относятся к лечению плохо поддающихся лечению пациентов или пациентов, не реагирующих на лечение, например, с устойчивостью к ингибиторам обратной транскриптазы, ингибиторам протеаз и так далее.
Другое воплощение включает способ лечения или предупреждения инфекции вирусом гепатита В и/или инфекции вирусом гепатита С у млекопитающего, нуждающегося в таком лечении, при этом способ включает введение указанному млекопитающему терапевтически эффективного количества соединения формулы (I) или его энантиомеров, диастереомеров, пролекарств либо их фармацевтически приемлемых солей.
КРАТКОЕ ОПИСАНИЕ ФИГУР
Фиг. 1. Дифракция рентгеновских лучей на монокристалле соединения из примера 41-В.
Фиг. 2. Дифракция рентгеновских лучей на монокристалле соединения из примера 42-А.
Фиг. 3. Дифракция рентгеновских лучей на монокристалле соединения из примера 43-В.
На Фиг. 4 показан уровень ДНК HBV, уровень HBsAg и уровень анти-HBs-антител у инфицированных аденоассоциированным вирусом (AAV)-HBV мышей, обработанных разбавителем, соединением из примера 43-А в дозе 10 мг/кг один раз в двое суток (QOD) и один раз в неделю (QW) в течение 42 суток. Результаты представлены в виде среднего значения ± SEM (стандартная ошибка среднего). LLOQ: нижний предел количественного определения.
На Фиг. 5 показаны уровни ДНК HBV, уровни HBsAg и уровни анти-HBs-антител у AAV-HBV-инфицированных мышей, обработанных разбавителем, соединением из примера 41-А в дозе 1, 3, 10 мг/кг QOD и в дозе 10 мг/кг QW в течение 42 суток. Результаты представлены в виде среднего значения ± SEM. LLOQ: нижний предел количественного определения.
На Фиг. 6 показаны уровни ДНК HBV, уровни HBsAg и уровни анти-HBs-антител у AAV-HBV-инфицированных мышей, обработанных разбавителем, соединением из примера 42-А в дозе 1, 3 и 10 мг/кг QOD в течение 42 суток. Результаты представлены в виде среднего значения ± SEM. LLOQ: нижний предел количественного определения.
На Фиг. 7 показаны уровни ДНК HBV, уровни HBsAg и уровни анти-HBs-антител у AAV-HBV-инфицированных мышей, обработанных разбавителем, соединением из примера 41-В в дозе 1, 3 и 10 мг/кг QOD в течение 42 суток. Результаты представлены в виде среднего значения ± SEM. LLOQ: нижний предел количественного определения.
ПРИМЕРЫ
Изобретение будет более полно обосновано со ссылкой на следующие далее примеры. Однако их не следует истолковывать в качестве ограничения объема данного изобретения.
СОКРАЩЕНИЯ

ОБЩИЕ ЭКСПЕРИМЕНТАЛЬНЫЕ УСЛОВИЯ
Промежуточные соединения и конечные соединения очищали флэш-хроматографией, используя один из следующих приборов: 1) SP1-систему и модуль картриджей Quad 12/25 от Biotage; 2) прибор для комби-флэш-хроматографии от ISCO. Марка и размер пор силикагеля: 1) KP-SIL 60  , размер частиц: 40-60 мкм; 2) регистрационный № в CAS (Химическая реферативная служба): силикагель: 63231-67-4, размер частиц силикагеля: 47-60 микрон; 3) ZCX от Qingdao Haiyang Chemical Co., Ltd; размер пор: 200-300 или 300-400.
, размер частиц: 40-60 мкм; 2) регистрационный № в CAS (Химическая реферативная служба): силикагель: 63231-67-4, размер частиц силикагеля: 47-60 микрон; 3) ZCX от Qingdao Haiyang Chemical Co., Ltd; размер пор: 200-300 или 300-400.
Промежуточные соединения и конечные соединения очищали препаративной HPLC на колонке для обращенно-фазовой хроматографии, используя препаративную С18-колонку XBridge™ (5 мкм, OBD™ 30×100 мм) или препаративную С18-колонку SunFire™ (5 мкм, OBD™ 30×100 мм).
LC/MS-спектры получали, используя одноквадрупольный детектор (SQD) масс для сверхэффективной жидкостной хроматографии (UPLC) от Waters. Применяли приведенные ниже стандартные условия LC/MS (продолжительность испытания 3 минуты).
Кислотные условия: А: 0,1% муравьиной кислоты и 1% ацетонитрила в H2O; В: 0,1% муравьиной кислоты в ацетонитриле;
основные условия: А: 0,05% NH3⋅H2O в H2O; В: ацетонитрил.
Масс-спектры (MS): обычно приводятся значения только для ионов, соответствующих исходной массе, и, если не указано иное, приведенная масса иона представляет собой массу положительно заряженного иона (М+Н)+.
Спектры ядерного магнитного резонанса (ЯМР) получали, используя прибор Avance 400 МГц от Bruker.
Все реакции с участием чувствительных к воздуху реагентов проводили в атмосфере аргона. Реагенты использовали по получении от коммерческих поставщиков без дополнительной очистки, если не указано иное.
ПОДГОТОВИТЕЛЬНЫЕ ПРИМЕРЫ
Получение промежуточных соединений
Промежуточное соединение АА
N-Метил-N-пропил-карбамоилхлорид
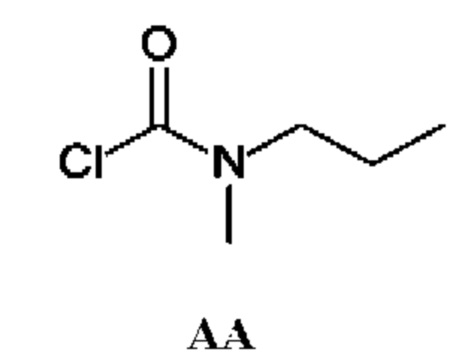
К смеси N-метилпропан-1-амина (5 г; 68,4 ммоль) и гидрокарбоната натрия (11,5 г; 137 ммоль) в дихлорметане (DCM) (70 мл) при 0°С по каплям добавляли бис(трихлорметил)карбонат (8,11 г; 27,3 ммоль) в DCM (30 мл). Смесь перемешивали при комнатной температуре в течение 2 ч и фильтровали. Фильтрат концентрировали в вакууме. Полученный N-метил-N-пропил-карбамоилхлорид (7,2 г; промежуточное соединение АА) использовали на следующей стадии без дополнительной очистки.
Промежуточное соединение АВ
N-(2-Метоксиэтил)-N-метил-карбамоилхлорид
Промежуточное соединение АВ получали по аналогии с промежуточным соединением АА, используя 2-метокси-N-метилэтанамин вместо N-метилпропан-1-амина. Получали N-(2-метоксиэтил)-N-метил-карбамоилхлорид (8 г; промежуточное соединение АВ) и использовали на следующей стадии без дополнительной очистки.
Промежуточное соединение АС
N-Этил-N-пропил-карбамоилхлорид
Промежуточное соединение АС получали по аналогии с промежуточным соединением АА, используя N-этилпропан-1-амин вместо метилпропан-1-амина. N-Этил-N-пропил-карбамоилхлорид (12,6 г; промежуточное соединение АС) получали в виде желтого масла и использовали на следующей стадии без дополнительной очистки.
Промежуточное соединение AD
N-Этил-N-(2-метоксиэтил)карбамоилхлорид
Промежуточное соединение AD получали по аналогии с промежуточным соединением АА, используя N-этил-2-метоксиэтанамин вместо N-метилпропан-1-амина. Неочищенный N-этил-N-(2-метоксиэтил)карбамоилхлорид (2,5 г; промежуточное соединение AD) получали в виде светло-желтого масла и использовали на следующей стадии без дополнительной очистки.
Промежуточное соединение АЕ
N-Бутил-N-этил-карбамоилхлорид
Промежуточное соединение АЕ получали по аналогии с промежуточным соединением АА, используя N-этилбутан-1-амин (5 г) вместо N-метилпропан-1-амина. Неочищенный N-бутил-N-этил-карбамоилхлорид (6,3 г; промежуточное соединение АЕ) получали в виде светло-желтого масла и использовали на следующей стадии без дополнительной очистки.
Промежуточное соединение AF
N-(2-Метоксиэтил)-N-пропил-карбамоилхлорид
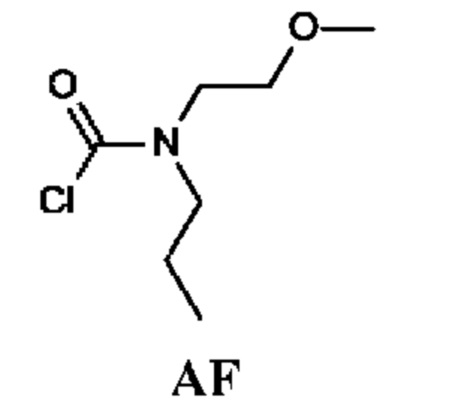
Промежуточное соединение AF получали по аналогии с промежуточным соединением АА, используя N-(2-метоксиэтил)пропан-1-амин (2 г; 17,1 ммоль) вместо N-метилпропан-1-амина. Неочищенный N-(2-метоксиэтил)-N-пропил-карбамоилхлорид (2,5 г; промежуточное соединение AF) получали в виде светло-желтого масла и использовали на следующей стадии без дополнительной очистки.
Промежуточное соединение AG
N,N-Бис(2-метоксиэтил)карбамоилхлорид
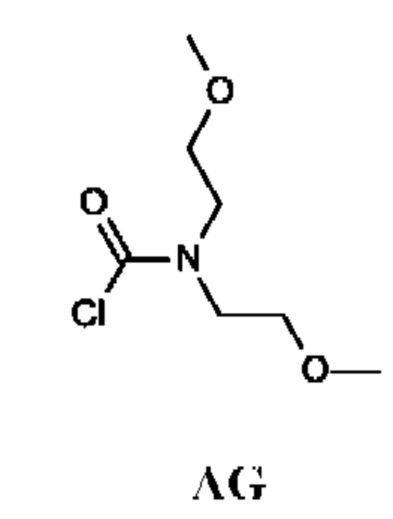
Промежуточное соединение AG получали по аналогии с промежуточным соединением АА, используя бис(2-метоксиэтил)амин (2 г; 15 ммоль) вместо N-метилпропан-1-амина. Неочищенный продукт N,N-бис(2-метоксиэтил)карбамоилхлорид (2,6 г; промежуточное соединение AG) получали в виде светло-желтого масла и использовали на следующей стадии без дополнительной очистки.
Промежуточное соединение АН
Азетидин-1-карбонилхлорид
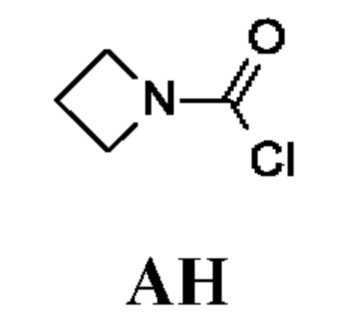
Промежуточное соединение АН получали по аналогии с промежуточным соединением АА, используя азетидина гидрохлорид (10,7 г; 107 ммоль) и бикарбонат натрия (3 экв.) вместо N-метилпропан-1-амина и бикарбоната натрия (2 экв.). Неочищенный азетидин-1-карбонилхлорид (1,5 г; промежуточное соединение АН) получали в виде светло-желтого масла и использовали на следующей стадии без дополнительной очистки.
Промежуточное соединение AI
N-Изопропил-N-метил-карбамоилхлорид
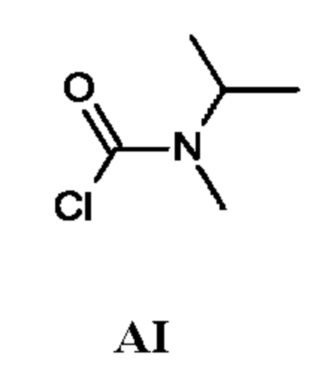
Промежуточное соединение AI получали по аналогии с промежуточным соединением АА, используя N-метилпропан-2-амин (5 г; 19,4 ммоль) вместо N-метилпропан-1-амина. Неочищенный N-изопропил-N-метил-карбамоилхлорид (8,6 г; промежуточное соединение AI) получали в виде желтого масла и использовали на следующей стадии без дополнительной очистки.
Промежуточное соединение AL
N-Изобутил-N-метил-карбамоилхлорид
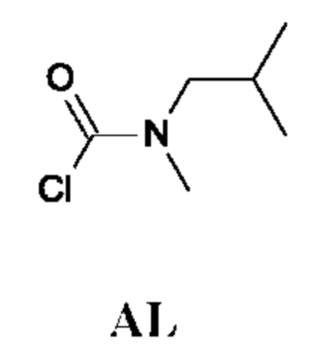
Промежуточное соединение AL получали по аналогии с промежуточным соединением АА, используя N-2-диметилпропан-1-амин (4,8 г) вместо N-метилпропан-1-амина. Неочищенный N-изобутил-N-метил-карбамоилхлорид (8,1 г; промежуточное соединение AL) получали в виде светло-желтого масла и использовали на следующей стадии без дополнительной очистки.
Промежуточное соединение АР
Этил-2-[хлоркарбонил(метил)амино]ацетат
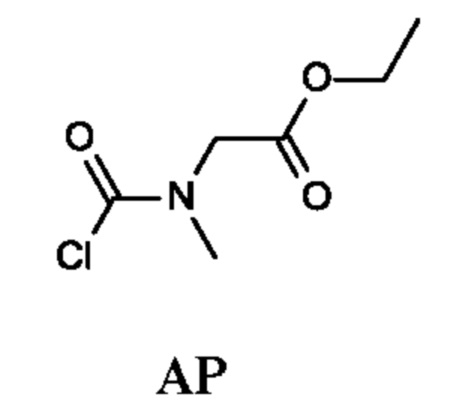
К раствору трифосгена (728 мг; 2,45 ммоль) в DCM (5 мл) по каплям при 0°С добавляли раствор этил-2-(метиламино)ацетата гидрохлорида (1,3 г; 8,46 ммоль) и пиридина (1 мл) в DCM (5 мл). Реакционная смесь становилась оранжевой и появлялся желтый осадок, затем смесь оставляли нагреваться до комнатной температуры. После перемешивания в течение 1 ч к реакционной смеси добавляли водный раствор HCl (0,1 н.; 25 мл), органический слой отделяли, дважды промывали 0,1 н. раствором HCl (10 мл), рассолом (10 мл), сушили над Na2SO4 и концентрировали в вакууме, получая неочищенный этил-2-[хлоркарбонил(метил)амино]ацетат (2,0 г; промежуточное соединение АР) в виде светло-желтого масла, и использовали на следующей стадии без дополнительной очистки.
Промежуточное соединение AR
трет-Бутил-3-[хлоркарбонил(метил)амино]пропаноат
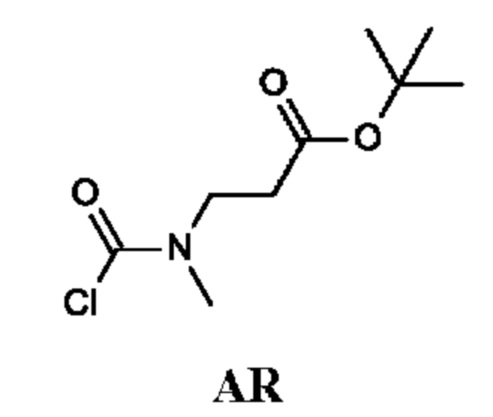
Стадия 1. Получение трет-бутил-3-(метиламино)пропаноата (соединения AR-1)
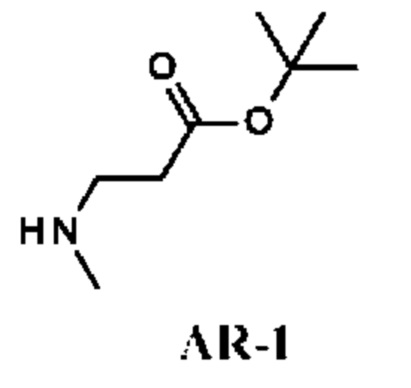
К раствору трет-бутилакрилата (3 г) в диметилформамиде (DMF) (40 мл) при -45°С добавляли метиламина гидрохлорид (4,74 г; 70 ммоль) и DBU (21,4 г; 140 ммоль). Затем температуру реакционной смеси повышали до -10°С. Реакционную смесь перемешивали при этой же температуре в течение 2,5 ч. Добавляли Et2O (200 мл) и полученную смесь четыре раза промывали рассолом (50 мл). Отделенный органический слой сушили над Na2SO4 и концентрировали в вакууме, получая трет-бутил-3-(метиламино)пропаноат (3,5 г; соединение AR-1) в виде светло-желтого масла.
Стадия 2. Получение трет-бутил-3-[хлоркарбонил(метил)амино]-пропаноата (промежуточного соединения AR)
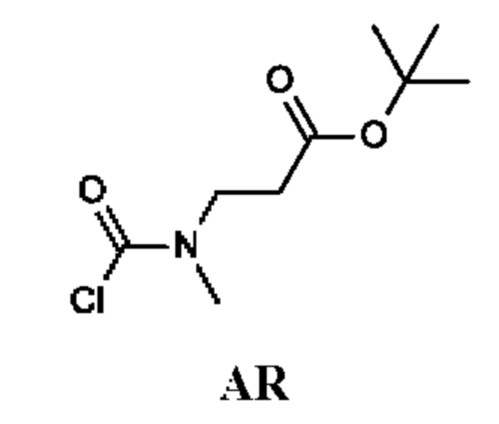
Промежуточное соединение AR получали по аналогии с промежуточным соединением АР, используя трет-бутил-3-(метиламино)пропаноат (3,4 г; соединение AR-1) вместо этил-2-(метиламино)ацетата гидрохлорида. Получали неочищенный трет-бутил-3-[хлоркарбонил(метил)амино]пропаноат (3,5 г; промежуточное соединение AR) и использовали на следующей стадии без дополнительной очистки.
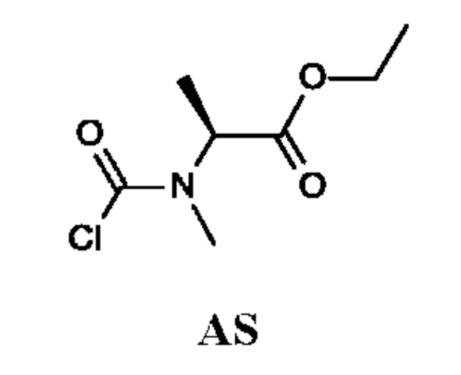
Промежуточное соединение AS
Этил-(2S)-2-[хлоркарбонил(метил)амино]пропаноат
Стадия 1. Получение этил-(2S)-2-(метиламино)пропаноата гидрохлорида (соединения AS-1)
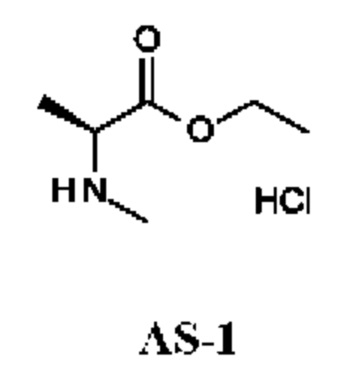
К раствору (2S)-2-(метиламино)пропановой кислоты (1 г; 9,70 ммоль) в EtOH (10 мл) по каплям при 0°С в течение 0,5 ч добавляли SOCl2 (1,50 г; 12,61 ммоль). Реакционную смесь перемешивали при 25°С в течение 15,5 ч, затем разбавляли ЕА (20 мл), промывали H2O (5 мл) и рассолом (5 мл). Органический слой сушили над Na2SO4 и концентрировали в вакууме. Получали этил-(2S)-2-(метиламино)пропаноата гидрохлорид (1,8 г; соединение AS-1) в виде желтого масла и использовали на следующей стадии без дополнительной очистки.
Стадия 2. Получение этил-(2S)-2-(метиламино)пропаноата (соединения AS-2)
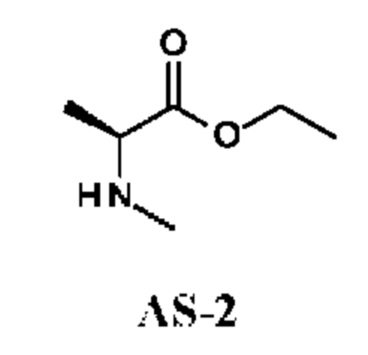
Значение рН раствора этил-(2S)-2-(метиламино)пропаноата гидрохлорида (1,8 г; соединение AS-1) в ЕА (10 мл) подводили до 8, используя 10 масс. %-ный водный раствор NaHCO3. Реакционную смесь перемешивали при комнатной температуре в течение 0,5 ч. Органический слой промывали рассолом (5 мл), сушили над Na2SO4 и концентрировали в вакууме. Получали этил-(2S)-2-(метиламино)пропаноат (620 мг; соединение AS-2) в виде желтого масла и использовали на следующей стадии без дополнительной очистки.
Стадия 3. Получение этил-(2S)-2-[хлоркарбонил(метил)амино]-пропаноата (промежуточного соединения AS)
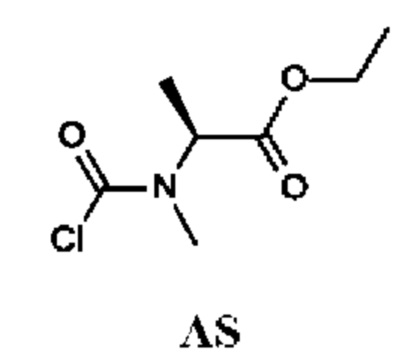
Промежуточное соединение AS получали по аналогии с промежуточным соединением АР, используя этил-(2S)-2-(метиламино)пропаноат (260 мг; соединение AS-2) вместо этил-2-(метиламино)ацетата гидрохлорида. Неочищенный этил-(2S)-2-[хлоркарбонил(метил)амино]пропаноат (200 мг; промежуточное соединение AS) получали в виде желтого масла и использовали на следующей стадии без дополнительной очистки.
Промежуточное соединение AT
трет-Бутил-(2S)-2-[хлоркарбонил(метил)амино]-4-метил-пентаноат
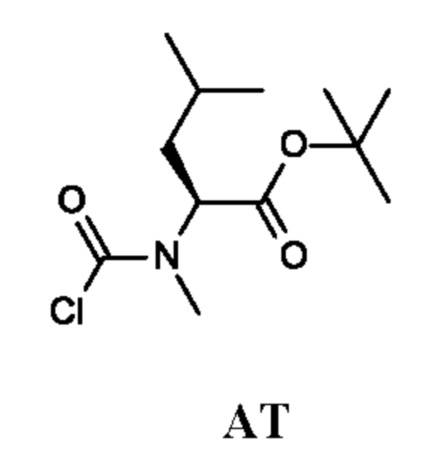
Стадия 1. Получение трет-бутил-(2S)-4-метил-2-(метиламино)пентаноата (соединения АТ-1)
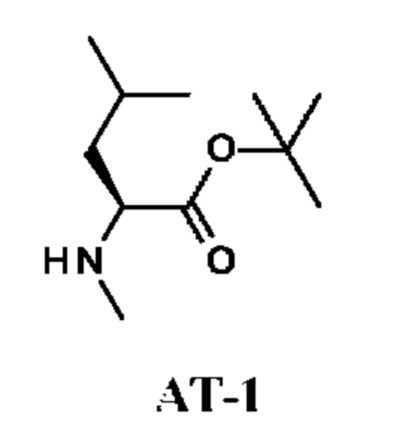
2-Метилпропен (25 г; 446 ммоль) барботировали через DCM (50 мл) при -78°С. Затем к раствору (S)-4-метил-2-(метиламино)пентановой кислоты гидрохлорида (500 мг; 2,75 ммоль) и H2SO4 (3,68 г; 2 мл; 37,5 ммоль) в диоксане (20 мл) при 0°С добавляли раствор 2-метилпропена. Реакционную смесь перемешивали при комнатной температуре в течение 18 ч в герметично закрытой пробирке. Реакционный раствор выливали в охлажденный во льду водный раствор KOH (8,4 г в воде (30 мл)) и полученную смесь дважды экстрагировали DCM (50 мл). Объединенный органический слой дважды промывали рассолом (30 мл), сушили над Na2SO4 и концентрировали в вакууме, получая неочищенный продукт, трет-бутил-(2S)-4-метил-2-(метиламино)пентаноат (соединение АТ-1), в виде светло-желтого масла.
Стадия 2. Получение трет-бутил-(2S)-2-[хлоркарбонил(метил)амино]-4-метил-пентаноата (промежуточного соединения AT)
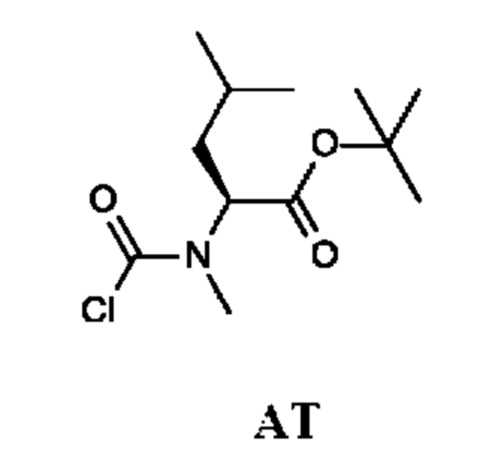
Промежуточное соединение AT получали по аналогии с промежуточным соединением АР, используя трет-бутил-(2S)-4-метил-2-(метиламино)пентаноат (300 мг; соединение АТ-1) вместо этил-2-(метиламино)ацетата гидрохлорида. Неочищенный трет-бутил-(2S)-2-[хлоркарбонил(метил)амино]-4-метил-пентаноат (350 мг; промежуточное соединение AT) получали в виде светло-желтого масла и использовали на следующей стадии без дополнительной очистки.
Промежуточное соединение AU
Изопропил-(2S)-2-[хлоркарбонил(метил)амино]-4-метил-пентаноат
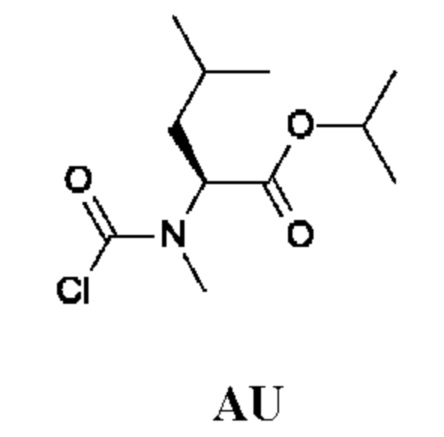
Стадия 1. Получение изопропил-(2S)-4-метил-2-(метиламино)пентаноата гидрохлорида (соединения AU-1)
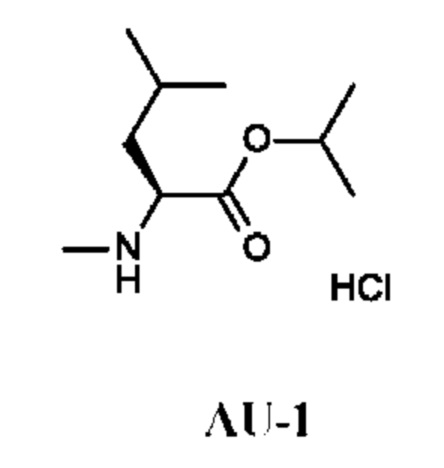
К раствору (S)-4-метил-2-(метиламино)пентановой кислоты гидрохлорида (0,5 г) в i-PrOH (7,8 г; 10 мл) по каплям при комнатной температуре добавляли тионилхлорид (655 мг; 402 мкл). Полученную смесь перемешивали и кипятили с обратным холодильником в течение 16 ч и затем концентрировали в вакууме. Остаток подщелачивали насыщенным водным раствором NaHCO3 (30 мл) и экстрагировали DCM (50 мл). Органический слой промывали рассолом, сушили над Na2SO4 и концентрировали в вакууме. Остаток переводили в форму соли, используя HCl/EtOAc (10 мл; 1 ммоль/мл), и концентрировали, получая изопропил-(2S)-4-метил-2-(метиламино)пентаноата гидрохлорид (510 мг; соединение AU-1) в виде белого твердого вещества.
Стадия 2. Получение изопропил-(2S)-2-[хлоркарбонил(метил)амино]-4-метил-пентаноата (промежуточного соединения AU)
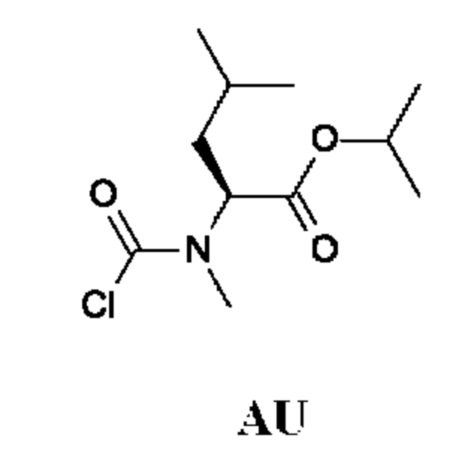
Промежуточное соединение AU получали по аналогии с промежуточным соединением АР, используя изопропил-(2S)-4-метил-2-(метиламино)пентаноата гидрохлорид (500 мг; соединение AU-1) вместо этил-2-(метиламино)ацетата гидрохлорида. Неочищенный изопропил-(2S)-2-[хлоркарбонил(метил)амино]-4-метил-пентаноат (650 мг; промежуточное соединение AU) получали в виде светло-желтого масла и использовали на следующей стадии без дополнительной очистки.
Промежуточное соединение AV
Этил-(2S)-2-[хлоркарбонил(метил)амино]-3-метил-бутаноат
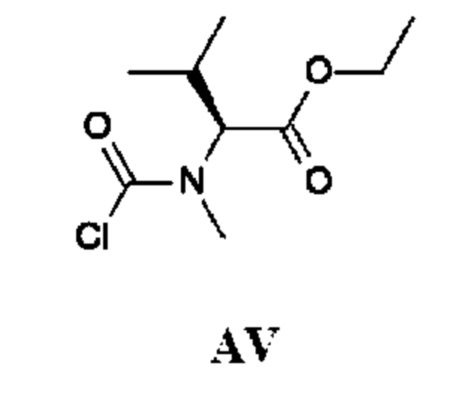
Стадия 1. Получение этил-(2S)-3-метил-2-(метиламино)бутаноата гидрохлорида (соединения AV-1)
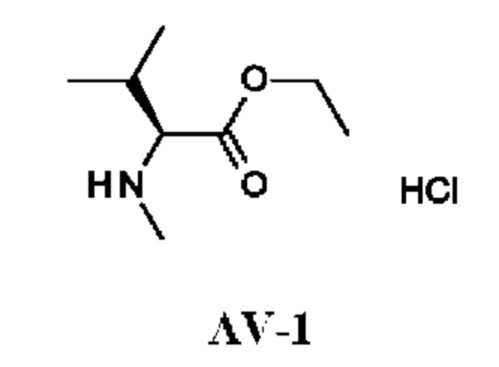
К раствору (2S)-3-метил-2-(метиламино)бутановой кислоты (1,0 г; 7,6 ммоль) в EtOH (10 мл) по каплям при комнатной температуре добавляли тионилхлорид (2,45 г; 21 ммоль). Полученную смесь перемешивали и кипятили с обратным холодильником в течение 16 ч и затем концентрировали в вакууме. Остаток подщелачивали насыщенным водным раствором NaHCO3 (30 мл) и дважды экстрагировали DCM (50 мл). Объединенный органический слой промывали рассолом, сушили над Na2SO4 и концентрировали в вакууме. Остаток растворяли в HCl/EtOAc (10 мл; 1 М) и концентрировали, получая этил-(2S)-3-метил-2-(метиламино)бутаноата гидрохлорид (1,9 г; соединение AV-1) в виде белого твердого вещества.
Стадия 2. Получение этил-(2S)-2-[хлоркарбонил(метил)амино]-3-метил-бутаноата (промежуточного соединения AV)
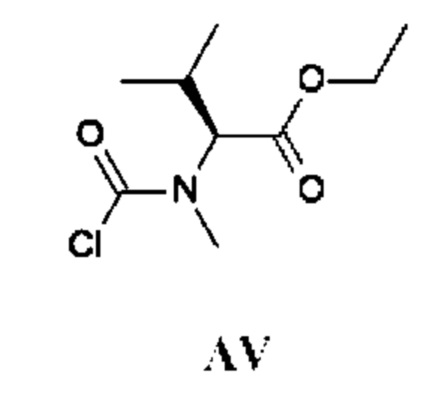
Промежуточное соединение AV получали по аналогии с промежуточным соединением АР, используя этил-(2S)-3-метил-2-(метиламино)бутаноата гидрохлорид (500 мг; соединение AV-1) вместо этил-2-(метиламино)ацетата гидрохлорида. Неочищенный этил-(2S)-2-[хлоркарбонил(метил)амино]-3-метил-бутаноат (600 мг; промежуточное соединение AV) получали в виде светло-желтого масла и использовали на следующей стадии без дополнительной очистки.
Промежуточное соединение AW
Этил-(2S)-2-[хлоркарбонил(метил)амино]-4-метил-пентаноат
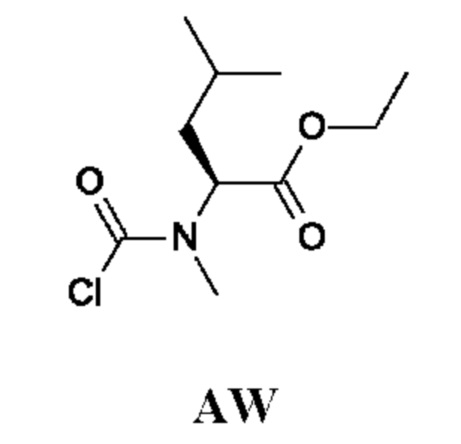
Стадия 1. Получение этил-(2S)-4-метил-2-(метиламино)пентаноата гидрохлорида (соединения AW-1)
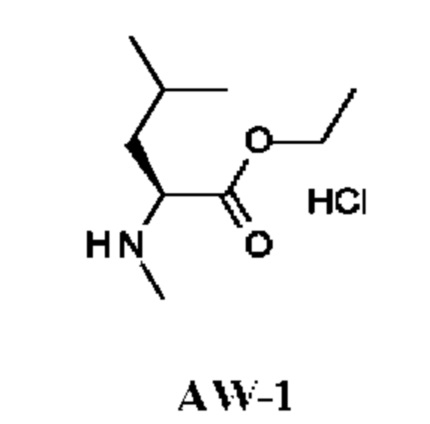
К раствору (2S)-4-метил-2-(метиламино)пентановой кислоты (1 г; 6,9 ммоль) в EtOH (10 мл) по каплям при комнатной температуре добавляли тионилхлорид (1,07 г; 8,3 ммоль). Полученную смесь перемешивали при температуре дефлегмации в течение 16 ч и затем концентрировали в вакууме. Остаток подщелачивали насыщенным водным раствором NaHCO3 (30 мл) и экстрагировали DCM (50 мл). Органический слой промывали рассолом, сушили над Na2SO4 и концентрировали в вакууме. Остаток переводили в форму соли, используя HCl/EtOAc (10 мл; 1 ммоль/мл), и концентрировали, получая этил-(2S)-4-метил-2-(метиламино)пентаноата гидрохлорид (1,8 г; соединение AW-1) в виде белого твердого вещества.
Стадия 2. Получение этил-(2S)-2-[хлоркарбонил(метил)амино]-4-метил-пентаноата (промежуточного соединения AW)
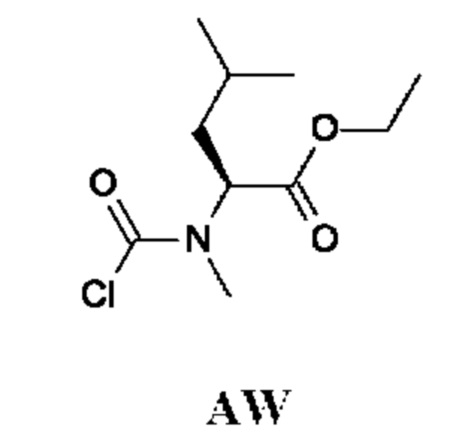
Промежуточное соединение AW получали по аналогии с промежуточным соединением АР, используя этил-(2S)-4-метил-2-(метиламино)пентаноата гидрохлорид (610 мг; AW-1) вместо этил-2-(метиламино)ацетата гидрохлорида. Неочищенный этил-(2S)-2-[хлоркарбонил(метил)амино]-4-метил-пентаноат (280 мг; промежуточное соединение AW) получали в виде светло-желтого масла и использовали на следующей стадии без дополнительной очистки.
Промежуточное соединение АХ
Этил-(2S)-2-[хлоркарбонил(метил)амино]-3-фенил-пропаноат
Хиральное
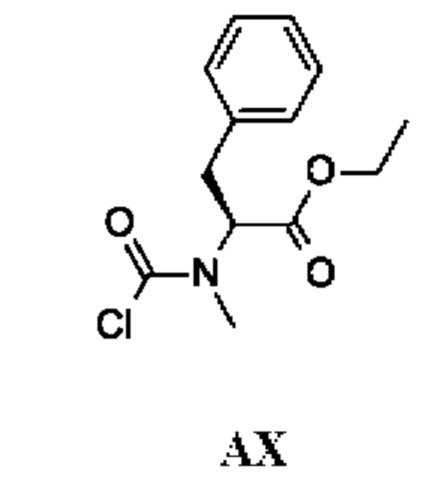
Промежуточное соединение АХ получали по аналогии с промежуточным соединением АР, используя (S)-этил-2-(метиламино)-3-фенилпропаноат вместо этил-2-(метиламино)ацетата гидрохлорида. Неочищенный этил-(2S)-2-[хлоркарбонил(метил)амино]-3-фенил-пропаноат (200 мг; промежуточное соединение АХ) получали в виде светло-желтого масла и использовали на следующей стадии без дополнительной очистки.
Промежуточное соединение AY
Изопропил-(2S)-2-[хлоркарбонил(метил)амино]-3-фенил-пропаноат
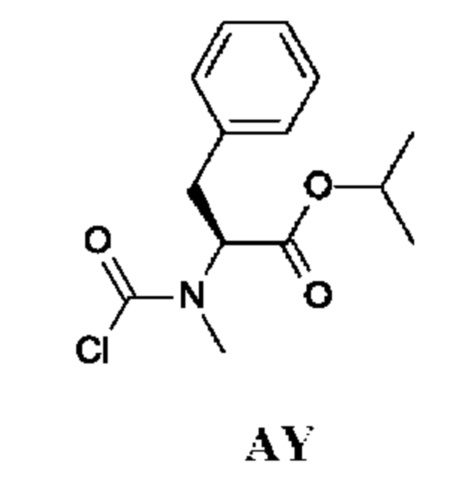
Промежуточное соединение AY получали по аналогии с промежуточным соединением АР, используя изопропил-(2S)-2-(метиламино)-3-фенил-пропаноат (190 мг) вместо этил-2-(метиламино)ацетата гидрохлорида. Неочищенный изопропил-(2S)-2-[хлоркарбонил(метил)амино]-3-фенил-пропаноат (220 мг; промежуточное соединение AY) получали в виде светло-коричневого масла и использовали на следующей стадии без дополнительной очистки.
Промежуточное соединение AZ
(S)-трет-Бутил-2-((хлоркарбонил)(метил)амино)-3-фенилпропаноат
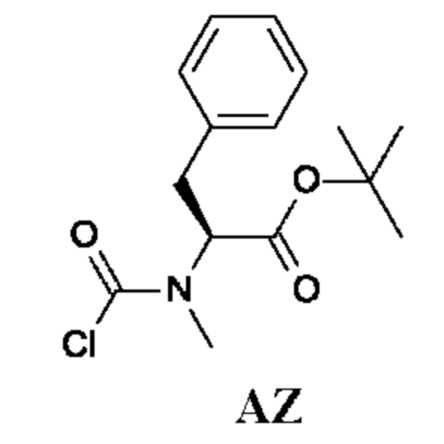
Стадия 1. Получение трет-бутил-(2S)-2-(метиламино)-3-фенил-пропаноата (соединения AZ-1)
2-Метилпропен (25 г; 446 ммоль) барботировали через DCM (50 мл) при -78°С. Затем к раствору (S)-2-(метиламино)-3-фенилпропановой кислоты (500 мг) и H2SO4 (3,68 г; 2 мл) в диоксане (20 мл) при 0°С добавляли раствор 2-метилпропена. Реакционную смесь перемешивали при комнатной температуре в течение 18 ч в герметично закрытой пробирке. Реакционную смесь выливали в охлажденный во льду водный раствор KOH (8,4 г в воде (30 мл)) и полученную смесь дважды экстрагировали DCM (50 мл). Органический слой 2 раза промывали рассолом (30 мл), сушили над Na2SO4 и концентрировали в вакууме, получая трет-бутил-(2S)-2-(метиламино)-3-фенилпропаноат (710 мг; соединение AZ-1) в виде светло-желтого масла.
Стадия 2. Получение (S)-трет-бутил-2-((хлоркарбонил)(метил)амино)-3-фенилпропаноата (промежуточного соединения AZ)
Промежуточное соединение AZ получали по аналогии с промежуточным соединением АР, используя трет-бутил-(2S)-2-(метиламино)-3-фенил-пропаноат (соединение AZ-1) вместо этил-2-(метиламино)ацетата гидрохлорида. Неочищенный трет-бутил-(2S)-2-[хлоркарбонил(метил)амино]-3-фенил-пропаноат (360 мг; промежуточное соединение AZ) получали в виде светло-желтого масла и использовали на следующей стадии без дополнительной очистки
Промежуточное соединение ВА
N-[2-[Ацетил(метил)амино]этил]-N-метил-карбамоилхлорид
Стадия 1. Получение трет-бутил-N-[2-[ацетил(метил)амино]этил]-N-метил-карбамата (соединения ВА-1)
К раствору трет-бутил-метил(2-(метиламино)этил)карбамата (1,13 г; 6 ммоль) в пиридине (10 мл) по каплям при 0°С добавляли уксусный ангидрид (3,06 г; 30 ммоль). Затем раствор перемешивали при комнатной температуре в течение 0,5 ч. Растворитель удаляли в вакууме и остаток распределяли между EtOAc (50 мл) и насыщенным водным раствором NaHCO3 (25 мл). Органический слой отделяли, промывали рассолом (20 мл), сушили над Na2SO4 и концентрировали в вакууме, получая трет-бутил-N-[2-[ацетил(метил)амино]этил]-N-метил-карбамат (1,28 г; соединение ВА-1) в виде желтого масла.
Стадия 2. Получение N-метил-N-(2-(метиламино)этил)ацетамида гидрохлорида (соединения ВА-2)
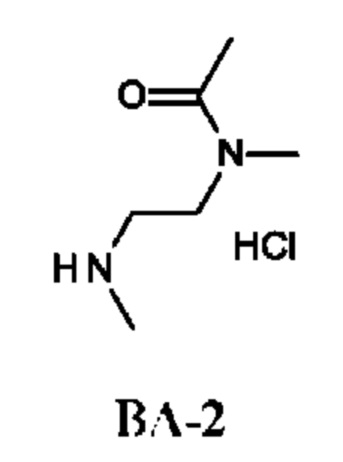
Смесь трет-бутил-N-[2-[ацетил(метил)амино]этил]-N-метил-карбамата (1,1 г; соединения ВА-1) в HCl/EtOAc (10 мл; 1 н. HCl в EtOAc) перемешивали при комнатной температуре в течение 2 ч, затем смесь фильтровали. Собранное твердое вещество три раза промывали EtOAc (5 мл) и сушили в вакууме, получая неочищенный N-метил-N-(2-(метиламино)этил)ацетамида гидрохлорид (460 мг; соединение ВА-2) в виде белого твердого вещества.
Стадия 3. Получение N-[2-[ацетил(метил)амино]этил]-N-метил-карбамоилхлорида (промежуточного соединения ВА)
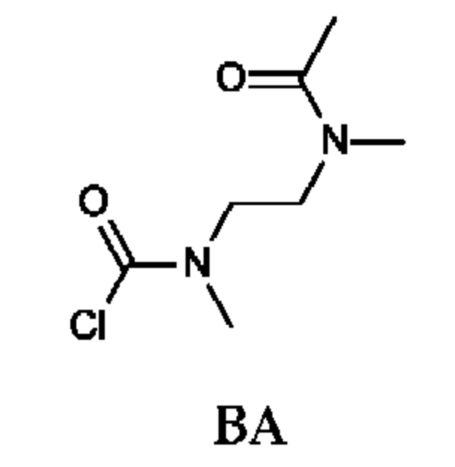
Промежуточное соединение ВА получали по аналогии с промежуточным соединением АР, используя N-метил-N-(2-(метиламино)этил)ацетамида гидрохлорид (200 мг; соединение ВА-2) вместо этил-2-(метиламино)ацетата гидрохлорида. Получали неочищенный N-[2-[ацетил(метил)амино]этил]-N-метил-карбамоилхлорид (300 мг; промежуточное соединение ВА) и использовали на следующей стадии без дополнительной очистки.
Промежуточное соединение ВВ
Метил-N-[2-[хлоркарбонил(метил)амино]этил]-N-метил-карбамат
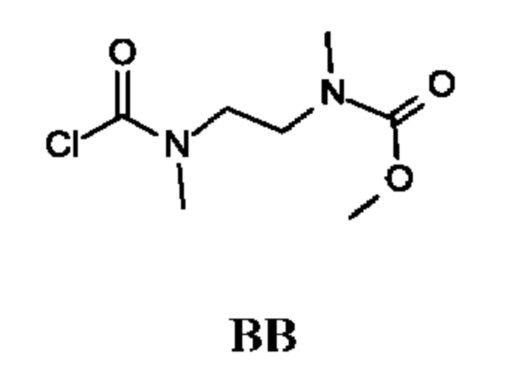
Стадия 1. Получение метил-N-метил-N-[2-(метиламино)этил]карбамата (соединения ВВ-1)
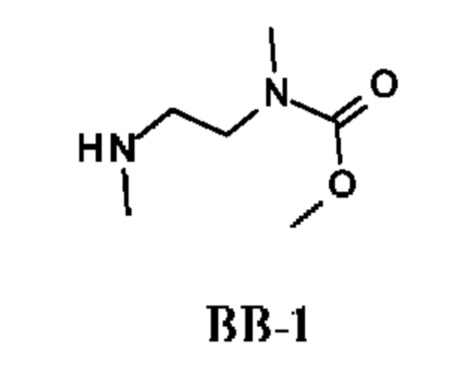
К раствору N,N'-диметилэтан-1,2-диамина (10 г) в THF (40 мл) по каплям при -70°С в течение 1 ч добавляли метилхлорформиат (1,92 г). Смесь перемешивали при 25°С в течение 15 ч, затем фильтровали и промывали водой и рассолом. Органический слой сушили и концентрировали, получая желтый остаток, который очищали колоночной хроматографией, получая метил-N-метил-N-[2-(метиламино)этил]карбамат (2 г; соединение ВВ-1) в виде бесцветного масла.
Стадия 2. Получение метил-N-[2-[хлоркарбонил(метил)амино]этил]-N-метил-карбамата (промежуточного соединения ВВ)
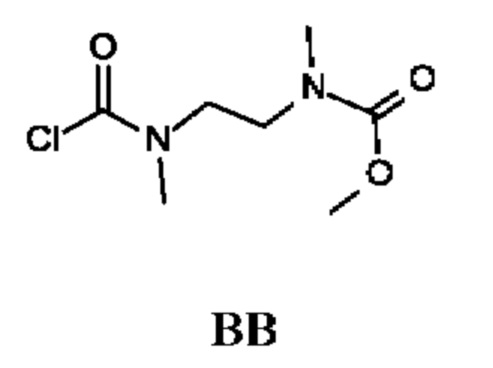
Промежуточное соединение ВВ получали по аналогии с промежуточным соединением АР, используя метил-N-метил-N-[2-(метиламино)этил]карбамат (2,0 г; соединение ВВ-1) вместо этил-2-(метиламино)ацетата гидрохлорида. Получали неочищенный метил-N-[2-[хлоркарбонил(метил)амино]этил]-N-метил-карбамат (2,2 г; промежуточное соединение ВВ) и использовали на следующей стадии без дополнительной очистки.
Промежуточное соединение ВС
трет-Бутил-N-[2-[хлоркарбонил(метил)амино]этил]-N-метил-карбамат
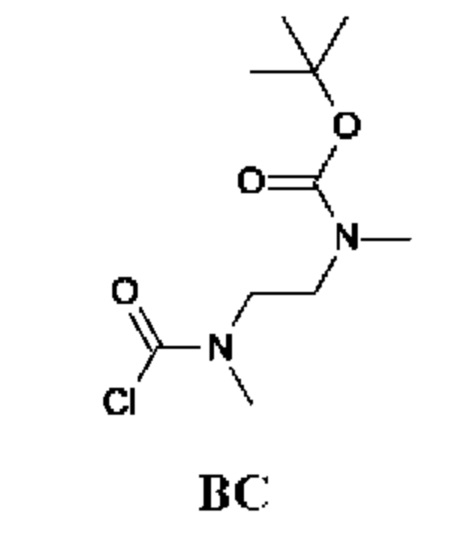
Стадия 1. Получение трет-бутил-N-метил-N-[2-(метиламино)этил]карбамата (соединения ВС-1)
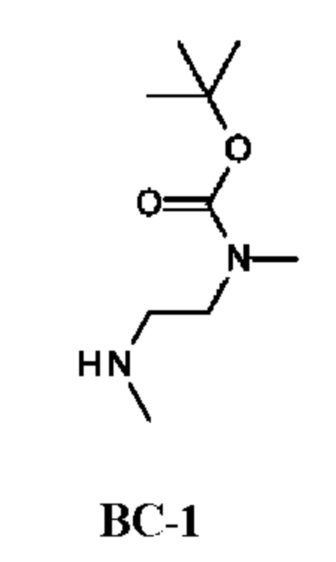
К раствору N,N'-диметилэтан-1,2-диамина (40,4 г) в DCM (300 мл) по каплям при 0°С в течение 1 ч добавляли раствор Boc2O (10 г; 10,6 мл; 45,8 ммоль) в DCM (100 мл). Реакционную смесь перемешивали при комнатной температуре в течение 18 ч. Органический слой промывали насыщенным водным раствором NaHCO3 (50 мл), рассолом (50 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали колоночной хроматографией, получая трет-бутил-N-метил-N-[2-(метиламино)этил]карбамат (6,8 г; соединение ВС-1) в виде желтого масла. 1Н-ЯМР (400 МГц, CDCl3) δ млн-1: 3.34 (br. s., 2Н), 2.89 (s, 3Н), 2.74 (t, J=6,7 Гц, 2Н), 2.46 (s, 3Н), 1.47 (s, 9Н).
Стадия 2. Получение трет-бутил-N-[2-[хлоркарбонил(метил)амино]-этил]-N-метил-карбамата (промежуточного соединения ВС)
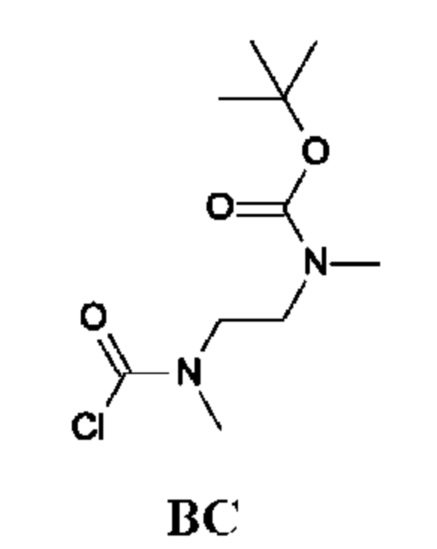
Промежуточное соединение ВС получали по аналогии с промежуточным соединением АР, используя трет-бутил-N-метил-N-[2-(метиламино)этил]карбамат (1,15 г; соединение ВС-1) вместо этил-2-(метиламино)ацетата гидрохлорида. Получали неочищенный трет-бутил-N-[2-[хлоркарбонил(метил)амино]этил]-N-метил-карбамат (1,3 г; промежуточное соединение ВС) и использовали на следующей стадии без дополнительной очистки.
Промежуточное соединение BD
Этил-N-[2-[хлоркарбонил(метил)амино]этил]-N-метил-карбамат
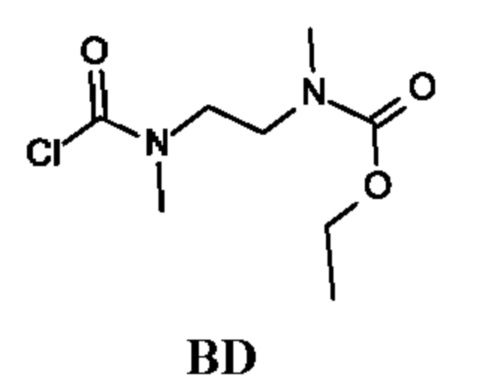
Стадия 1. Получение этил-N-метил-N-[2-(метиламино)этил]карбамата (соединения BD-1)
К раствору N,N'-диметилэтан-1,2-диамина (10 г) в DCM (40 мл) по каплям при -70°С в течение 1 ч добавляли этилхлорформиат (2,58 г). Реакционную смесь перемешивали при 25°С в течение 15 ч, затем фильтровали и промывали водой и рассолом. Органический слой сушили и концентрировали в вакууме. Желтый остаток очищали колоночной хроматографией, получая этил-N-метил-N-[2-(метиламино)этил]карбамат (2 г; соединение BD-1) в виде бесцветного масла.
Стадия 2. Получение этил-N-[2-[хлоркарбонил(метил)амино]этил]-N-метил-карбамата (промежуточного соединения BD)
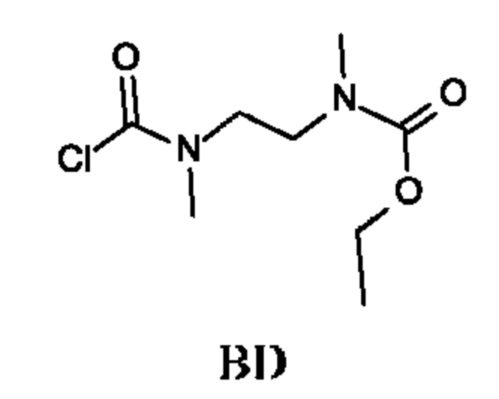
Промежуточное соединение BD получали по аналогии с промежуточным соединением АА, используя этил-N-метил-N-[2-(метиламино)этил]карбамат (соединение BD-1) вместо этил-2-(метиламино)ацетата гидрохлорида. Получали неочищенный этил-N-[2-[хлоркарбонил(метил)амино]этил]-N-метил-карбамат (2,2 г; промежуточное соединение BD) и использовали на следующей стадии без дополнительной очистки.
Промежуточное соединение BE
2-[Хлоркарбонил(метил)амино]этил-N-бутил-N-метил-карбамат
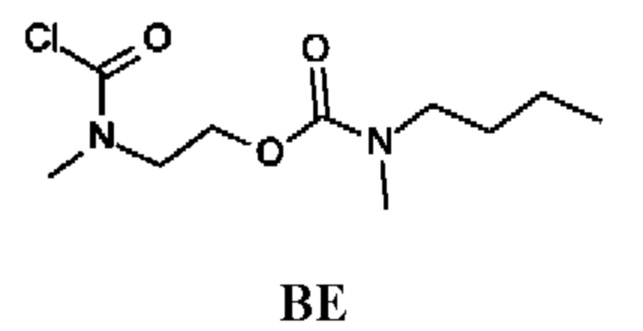
Стадия 1. Получение трет-бутил-N-(2-гидроксиэтил)-N-метил-карбамата (соединения ВЕ-1)
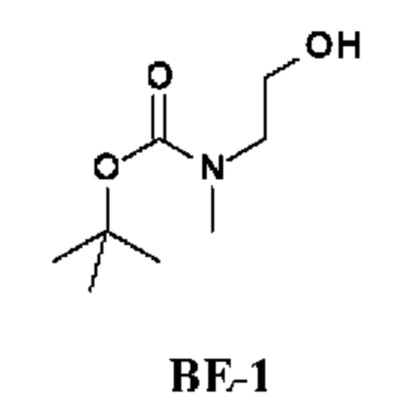
К раствору 2-(метиламино)этанола (10 г; 133,14 ммоль) в DCM (10 мл) добавляли Boc2O (34,87 г; 159,77 ммоль) при 25°С. Смесь перемешивали при 25°С в течение 16 ч и затем концентрировали. Остаток очищали колоночной хроматографией, получая трет-бутил-N-(2-гидроксиэтил)-N-метил-карбамат (20 г; соединение ВЕ-1) в виде бесцветного масла.
Стадия 2. Получение 2-[трет-бутоксикарбонил(метил)амино]этил-N-бутил-N-метил-карбамата (соединения ВЕ-2)
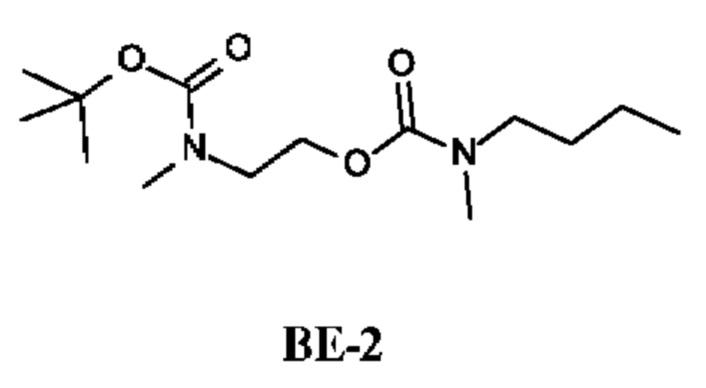
К раствору трет-бутил-N-(2-гидроксиэтил)-N-метил-карбамата (880 мг; соединения ВЕ-1) и Et3N (1 г; 10,08 ммоль) в DCM (10 мл) по каплям при -10°С в течение 1 ч добавляли N-бутил-N-метил-карбамоилхлорид (903 мг; 7,04 ммоль). Реакционную смесь перемешивали при 25°С в течение 15 ч, затем фильтровали и промывали водой и рассолом. Органический слой сушили и концентрировали, получая 2-[трет-бутоксикарбонил(метил)амино]этил-N-бутил-N-метил-карбамат (2 г; соединение ВЕ-2) в виде бесцветного масла.
Стадия 3. Получение 2-(метиламино)этил-N-бутил-N-метил-карбамата гидрохлорида (соединения ВЕ-3)

К раствору 2-[трет-бутоксикарбонил(метил)амино]этил-N-бутил-N-метил-карбамата (1 г; соединения ВЕ-2) добавляли HCl/EA (40 мл; 1 М). Реакционную смесь перемешивали при 0°С в течение 0,5 ч, нагревали до 25°С и перемешивали в течение еще 15,5 ч. Реакционную смесь концентрировали, получая 2-(метиламино)этил-N-бутил-N-метил-карбамата гидрохлорид (400 мг; соединение ВЕ-3) в виде бесцветного масла.
Стадия 4. Получение 2-[хлоркарбонил(метил)амино]этил-N-бутил-N-метил-карбамата (промежуточного соединения BE)
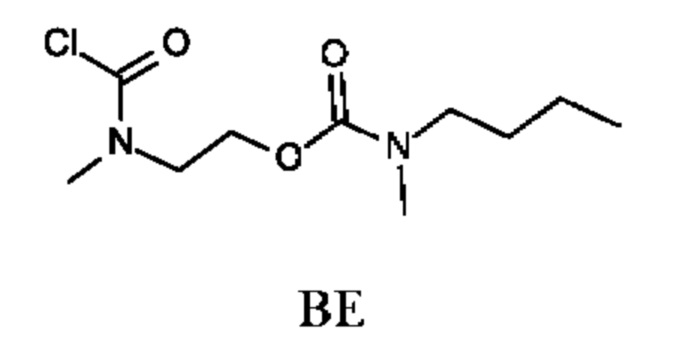
Промежуточное соединение BE получали по аналогии с промежуточным соединением АР, используя 2-(метиламино)этил-N-бутил-N-метил-карбамата гидрохлорид (374 мг; соединение ВЕ-3) вместо этил-2-(метиламино)ацетата гидрохлорида. Получали неочищенный 2-[хлоркарбонил(метил)амино]этил-N-бутил-N-метил-карбамат (330 мг; промежуточное соединение BE) и использовали на следующей стадии без дополнительной очистки.
Промежуточное соединение BF
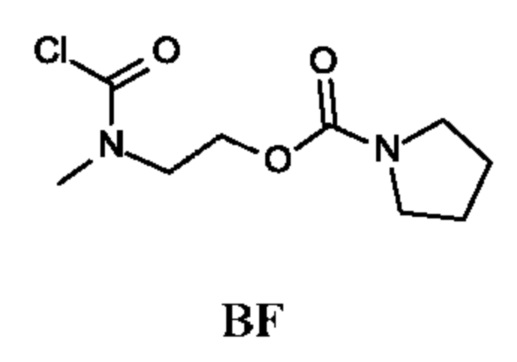
2-[Хлоркарбонил(метил)амино]этил-пирролидин-1-карбоксилат
Стадия 1. Получение трет-бутил-N-(2-гидроксиэтил)-N-метил-карбамата (соединения BF-1)
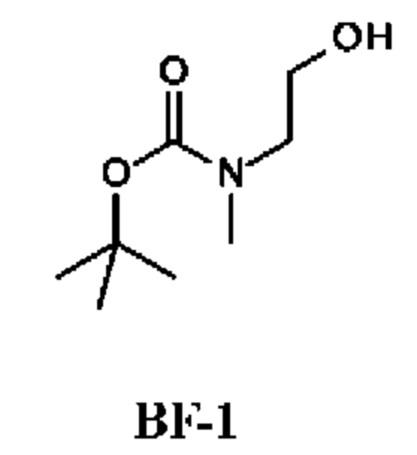
К раствору 2-(метиламино)этанола (10 г; 133,14 ммоль) в DCM (10 мл) добавляли Boc2O (34,87 г; 159,77 ммоль) при 25°С. Смесь перемешивали при 25°С в течение 16 ч. Реакционную смесь концентрировали с получением остатка, который очищали колоночной хроматографией, получая трет-бутил-N-(2-гидроксиэтил)-N-метил-карбамат (20 г; соединение BF-1) в виде бесцветного масла.
Стадия 2. Получение 2-[трет-бутоксикарбонил(метил)амино]этил-пирролидин-1-карбоксилата (соединения BF-2)
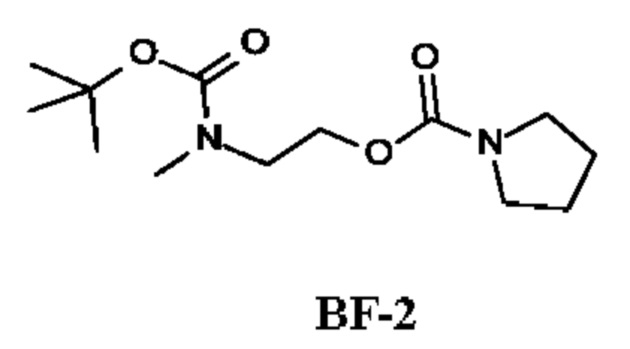
К раствору трет-бутил-N-(2-гидроксиэтил)-N-метил-карбамата (300 мг; 1,71 ммоль; соединения BF-1) и Et3N (578 мг; 5,71 ммоль) в DCM (5 мл) по каплям при 0°С в течение 0,5 ч добавляли пирролидин-1-карбонилхлорид (458 мг; 3,4 ммоль) и затем перемешивали при 25°С в течение 15,5 ч. После фильтрования фильтрат промывали водой и рассолом. Органический слой сушили и концентрировали, получая 2-[трет-бутоксикарбонил(метил)амино]этил-пирролидин-1-карбоксилат (335 мг; соединение BF-2) в виде бесцветного масла.
Стадия 3. Получение 2-(метиламино)этил-пирролидин-1-карбоксилата гидрохлорида (соединения BF-3)
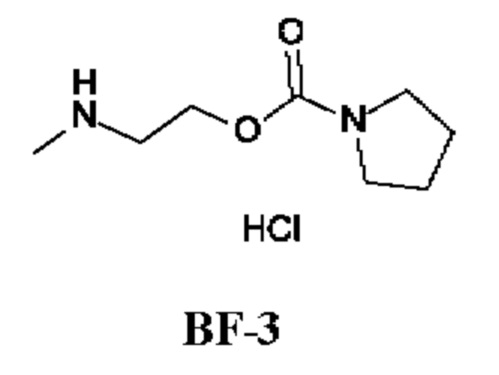
2-[трет-Бутоксикарбонил(метил)амино]этил-пирролидин-1-карбоксилат (335 мг; соединение BF-2) добавляли к раствору HCl в ЕА (12,3 мл; 1 М), смесь перемешивали при 0°С в течение 0,5 ч и затем при 25°С в течение еще 15,5 ч. Реакционную смесь концентрировали, получая 2-(метиламино)этил-пирролидин-1-карбоксилата гидрохлорид (300 мг; соединение BF-3) в виде бесцветного масла.
Стадия 4. Получение 2-[хлоркарбонил(метил)амино]этил-пирролидин-1-карбоксилата (промежуточного соединения BF)

Промежуточное соединение BF получали по аналогии с промежуточным соединением АР, используя 2-(метиламино)этил-пирролидин-1-карбоксилата гидрохлорид (299 мг; соединение BF-3) вместо этил-2-(метиламино)ацетата гидрохлорида. Получали неочищенный 2-[хлоркарбонил(метил)амино]этил-пирролидин-1-карбоксилат (230 мг; промежуточное соединение BF) и использовали на следующей стадии без дополнительной очистки.
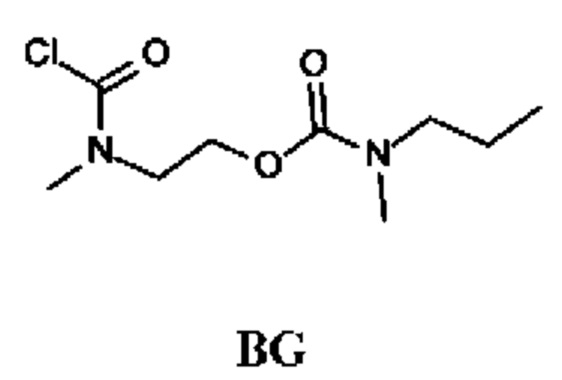
Промежуточное соединение BG
2-[Хлоркарбонил(метил)амино]этил-N-метил-N-пропил-карбамат
Стадия 1. Получение трет-бутил-N-(2-гидроксиэтил)-N-метил-карбамата (соединения BG-1)
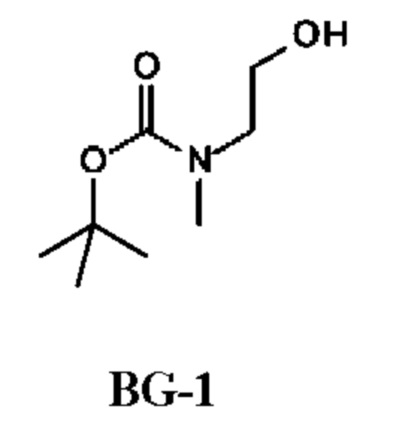
К раствору 2-(метиламино)этанола (10 г; 133,14 ммоль) в DCM (10 мл) добавляли Boc2O (34,87 г; 159,77 ммоль) при 25°С. Реакционную смесь перемешивали при 25°С в течение 16 ч, затем концентрировали с получением остатка, который очищали колоночной хроматографией, получая трет-бутил-N-(2-гидроксиэтил)-N-метил-карбамат (20 г; соединение BG-1) в виде бесцветного масла.
Стадия 2. Получение трет-бутил-N-метил-N-[2-[метил(пропил)карбамоил]оксиэтил]карбамата (соединения BG-2)

К раствору трет-бутил-N-(2-гидроксиэтил)-N-метил-карбамата (265 мг; соединения BG-1) и Et3N (1 мл; 5,71 ммоль) в DCM (5 мл) по каплям при 0°С в течение 0,5 ч добавляли N-метил-N-пропил-карбамоилхлорид (410 мг; 1,83 ммоль). Реакционную смесь перемешивали при 25°С в течение 15,5 ч, затем фильтровали и фильтрат промывали водой и рассолом. Органический слой сушили и концентрировали, получая трет-бутил-N-метил-N-[2-[метил(пропил)карбамоил]оксиэтил]карбамат (380 мг; соединение BG-2) в виде бесцветного масла.
Стадия 3. Получение 2-(метиламино)этил-N-метил-N-пропил-карбамата гидрохлорида (соединения BG-3)

трет-Бутил-N-метил-N-[2-[метил(пропил)карбамоил]оксиэтил]карбамат (380 мг; соединение BG-2) добавляли к раствору HCl в ЕА (13,7 мл; 1 М). Смесь перемешивали при 0°С в течение 0,5 ч. Затем смесь перемешивали при 25°С в течение еще 15,5 ч и концентрировали, получая 2-(метиламино)этил-N-метил-N-пропил-карбамата гидрохлорид (300 мг; соединение BG-3) в виде бесцветного масла.
Стадия 4. Получение 2-[хлоркарбонил(метил)амино]этил-N-метил-N-пропил-карбамата (промежуточного соединения BG)
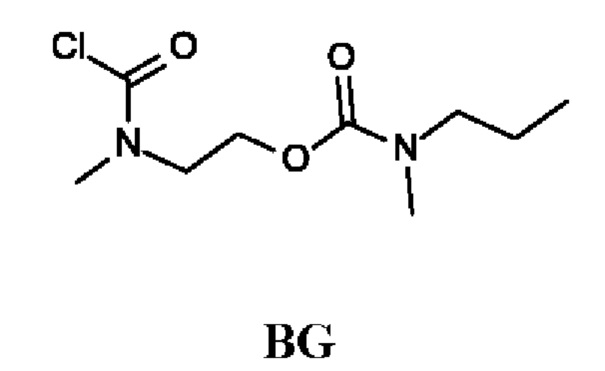
Промежуточное соединение BG получали по аналогии с промежуточным соединением АР, используя 2-(метиламино)этил-N-метил-N-пропил-карбамата гидрохлорид (330 мг; соединение BG-3) вместо этил-2-(метиламино)ацетата гидрохлорида. Получали 2-[хлоркарбонил(метил)амино]этил-N-метил-N-пропил-карбамат (300 мг; промежуточное соединение BG) и использовали на следующей стадии без дополнительной очистки.
Промежуточное соединение ВН
2-[Хлоркарбонил(метил)амино]этил-N,N-диэтилкарбамат
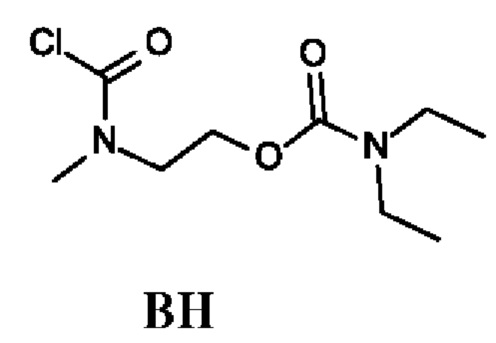
Стадия 1. Получение трет-бутил-N-(2-гидроксиэтил)-N-метил-карбамата (соединения ВН-1)

К раствору 2-(метиламино)этанола (10 г; 133,14 ммоль) в DCM (10 мл) добавляли Boc2O (34,87 г; 159,77 ммоль) при 25°С. Смесь перемешивали при 25°С в течение 16 ч и затем концентрировали, остаток очищали колоночной хроматографией, получая трет-бутил-N-(2-гидроксиэтил)-N-метил-карбамат (20 г; соединение ВН-1) в виде бесцветного масла.
Стадия 2. Получение 2-[трет-бутоксикарбонил(метил)амино]этил-N,N-диэтилкарбамата (соединения ВН-2)
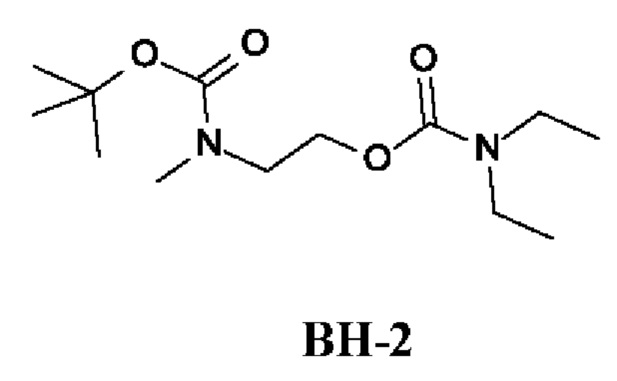
К раствору трет-бутил-N-(2-гидроксиэтил)-N-метил-карбамата (200 мг; 1,14 ммоль; соединения ВН-1) и Et3N (578 мг; 5,71 ммоль) в DCM (5 мл) по каплям при 0°С в течение 0,5 ч добавляли N,N-диэтилкарбамоилхлорид (248 мг; 1,83 ммоль) и перемешивали при 25°С в течение 15,5 ч. После фильтрования фильтрат промывали водой и рассолом. Органический слой сушили и концентрировали, получая 2-[трет-бутоксикарбонил(метил)амино]этил-N,N-диэтилкарбамат (313 мг; соединение ВН-2) в виде бесцветного масла.
Стадия 3. Получение 2-(метиламино)этил-N,N-диэтилкарбамата гидрохлорида (соединения ВН-3)
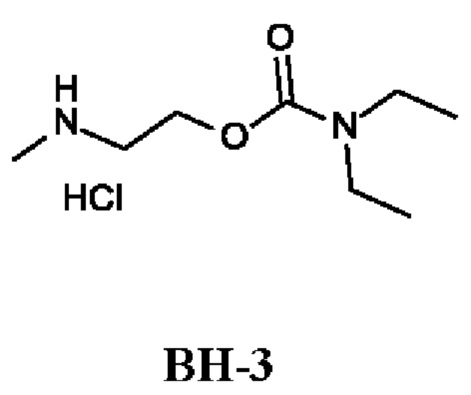
2-[трет-Бутоксикарбонил(метил)амино]этил-N,N-диэтилкарбамат (436 мг; 1,77 ммоль; соединение ВН-2) добавляли к раствору HCl в ЕА (17 мл; 1 М). Смесь перемешивали при 0°С в течение 0,5 ч. Затем смесь перемешивали при 25°С в течение еще 15,5 ч и концентрировали, получая 2-(метиламино)этил-N,N-диэтилкарбамата гидрохлорид (230 мг; соединение ВН-3) в виде бесцветного масла.
Стадия 4. Получение 2-[хлоркарбонил(метил)амино]этил-N,N-диэтилкарбамата (промежуточного соединения ВН)
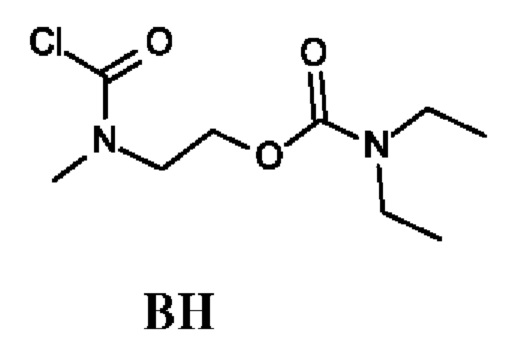
Промежуточное соединение ВН получали по аналогии с промежуточным соединением АР, используя 2-(метиламино)этил-N,N-диэтилкарбамата гидрохлорид (274 мг; соединение ВН-3) вместо этил-2-(метиламино)ацетата гидрохлорида. Получали неочищенный 2-[хлоркарбонил(метил)амино]этил-N,N-диэтилкарбамат (250 мг; промежуточное соединение ВН) и использовали на следующей стадии без дополнительной очистки.
Промежуточное соединение BI
2-[Хлоркарбонил(метил)амино]этил-этилкарбонат
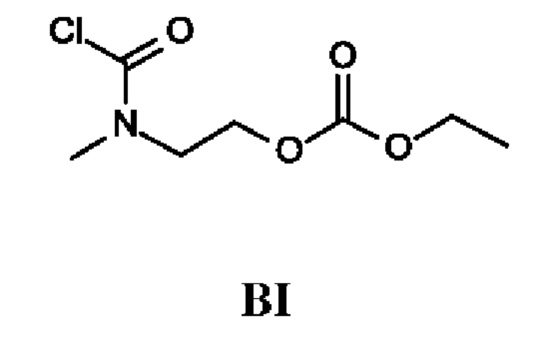
Стадия 1. Получение трет-бутил-N-(2-гидроксиэтил)-N-метил-карбамата (соединения BI-1)

К раствору 2-(метиламино)этанола (1 г; 13,31 ммоль) в DCM (10 мл) добавляли Boc2O (3,49 г; 15,98 ммоль) при 25°С. Реакционную смесь перемешивали при 25°С в течение 16 ч, затем концентрировали, получая неочищенный продукт, который очищали колоночной хроматографией, получая трет-бутил-N-(2-гидроксиэтил)-N-метил-карбамат (1,6 г; соединение BI-1) в виде бесцветного масла.
Стадия 2. Получение 2-[трет-бутоксикарбонил(метил)амино]этил-метилкарбоната (соединения BI-2)
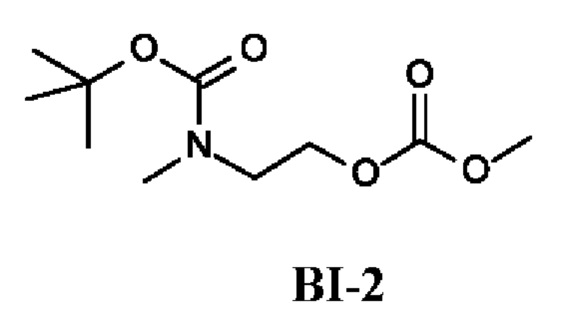
К раствору трет-бутил-N-(2-гидроксиэтил)-N-метил-карбамата (1 г; соединения BI-1), DMAP (0,1 г) и пиридина (1,15 г; 11,41 ммоль) в ЕА (20 мл) по каплям при -10°С добавляли метилхлорформиат (1,21 г; 11,15 ммоль). Смесь перемешивали при -10°С в течение 1 ч. Реакционную смесь фильтровали и фильтрат промывали 5%-ным раствором лимонной кислоты и рассолом. Органический слой сушили и концентрировали, получая 2-[трет-бутоксикарбонил(метил)амино]этил-метилкарбонат (1,22 г; соединение BI-2) в виде бесцветного масла.
Стадия 3. Получение этил-2-(метиламино)этилкарбоната гидрохлорида (соединения BI-3)
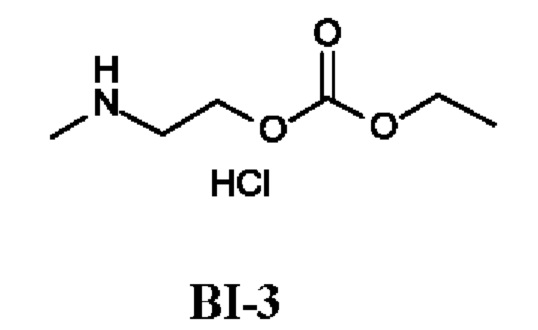
2-[трет-Бутоксикарбонил(метил)амино]этил-метилкарбонат (1,22 г; 4,94 ммоль; соединение BI-2) добавляли к раствору HCl в ЕА (10 мл; 40 ммоль) и смесь перемешивали при 0°С в течение 0,5 ч и при 25°С в течение еще 15,5 ч. Реакционную смесь концентрировали, получая этил-2-(метиламино)этилкарбоната гидрохлорид (1,06 г; соединение BI-3).
Стадия 4. Получение 2-[хлоркарбонил(метил)амино]этил-этилкарбоната (промежуточного соединения BI)
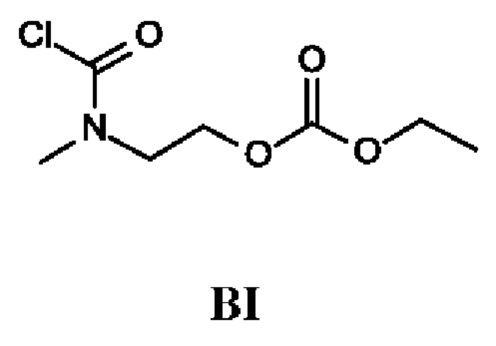
Промежуточное соединение BI получали по аналогии с промежуточным соединением АР, используя этил-2-(метиламино)этилкарбоната гидрохлорид (150 мг; промежуточное соединение BI-3) вместо этил-2-(метиламино)ацетата гидрохлорида. Получали неочищенный 2-[хлоркарбонил(метил)амино]этил-этилкарбонат (145 мг; промежуточное соединение BI) и использовали на следующей стадии без дополнительной очистки.
ПОДГОТОВИТЕЛЬНЫЕ ПРИМЕРЫ
Пример 1
6-Амино-9-бензил-N-метил-8-оксо-N-пропил-2-(пропилсульфонимидоил)пурин-7-карбоксамид
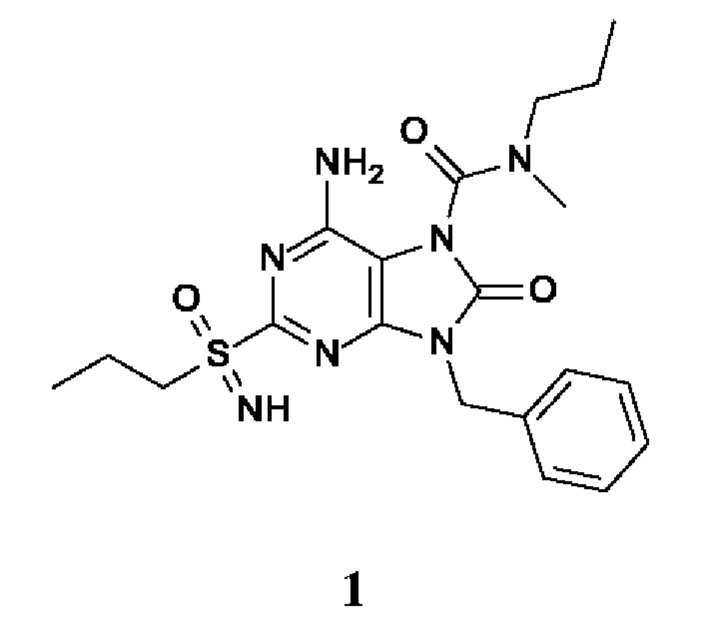
Способ А
Стадия 1. Получение 4-амино-3-бензил-2-оксо-1Н-имидазол-5-карбонитрила (соединения 1а)
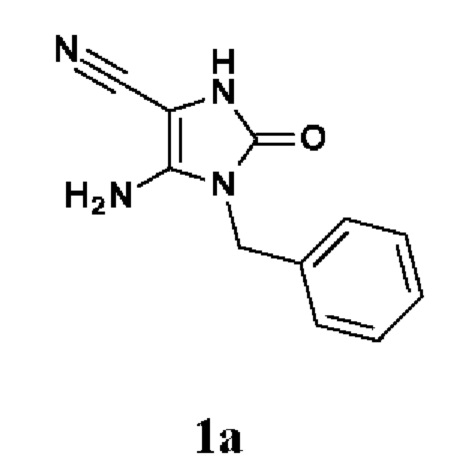
К раствору аминомалононитрил-п-толуолсульфоната (25 г; 98,5 ммоль; TCI, номер по каталогу: A1119-25G) в безводном THF (100 мл) добавляли бензилизоцианат (13,2 г; 98,5 ммоль) и TEA (10,2 г; 79,0 ммоль) при КТ. После перемешивания при КТ в течение 24 ч реакционную смесь концентрировали в вакууме и остаток распределяли между EtOAc (500 мл) и водой (250 мл). Отделенный органический слой дважды промывали рассолом (50 мл) и дважды экстрагировали раствором гидроксида натрия (50 мл; 1 н.). Объединенный слой в растворе гидроксида натрия нейтрализовали 10 масс. %-ным раствором гидросульфата натрия и экстрагировали EtOAc. Отделенный органический слой промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток растирали в 2-изопропоксипропане и затем суспензию фильтровали, получая 4-амино-3-бензил-2-оксо-1Н-имидазол-5-карбонитрил (15 г; соединение 1а) в виде желтого твердого вещества. Продукт использовали на следующей стадии без дополнительной очистки. MS: обнаружено (ESI+) [(М+Н)+]: 215.
Стадия 2. Получение 6-амино-9-бензил-2-сульфанил-7Н-пурин-8-она (соединения 1b)
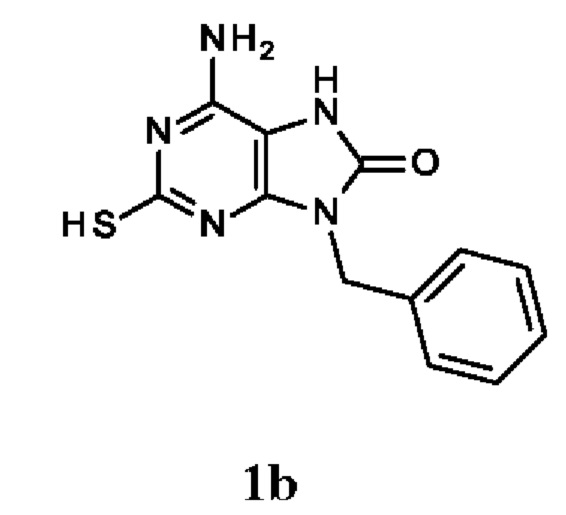
К раствору 4-амино-3-бензил-2-оксо-1Н-имидазол-5-карбонитрила (15,0 г; 70,0 ммоль; соединения 1а) в THF (700 мл) по каплям добавляли бензоилизотиоцианат (28,6 г; 175,1 ммоль; TCI, номер по каталогу: А11596-100G). После перемешивания при КТ в течение 12 ч реакционную смесь концентрировали в вакууме. Остаток растирали в диэтиловом эфире (100 мл) и полученный осадок собирали фильтрованием.
К раствору полученного осадка в THF (700 мл) добавляли раствор гидроксида натрия (70 мл; 2 н.). Смесь кипятили с обратным холодильником в течение 50 ч и затем подкисляли до рН 3, используя 10 масс. %-ный водный раствор гидросульфата натрия. Полученный осадок собирали фильтрованием, получая неочищенный 6-амино-9-бензил-2-сульфанил-7Н-пурин-8-он (8,1 г; соединение 1b) в виде желтого твердого вещества. Продукт использовали на следующей стадии без дополнительной очистки. MS: обнаружено (ESI+) [(М+Н)+]: 274.
Стадия 3. Получение 6-амино-9-бензил-2-(2-пропилсульфанил)-7Н-пурин-8-она (соединения 1с)
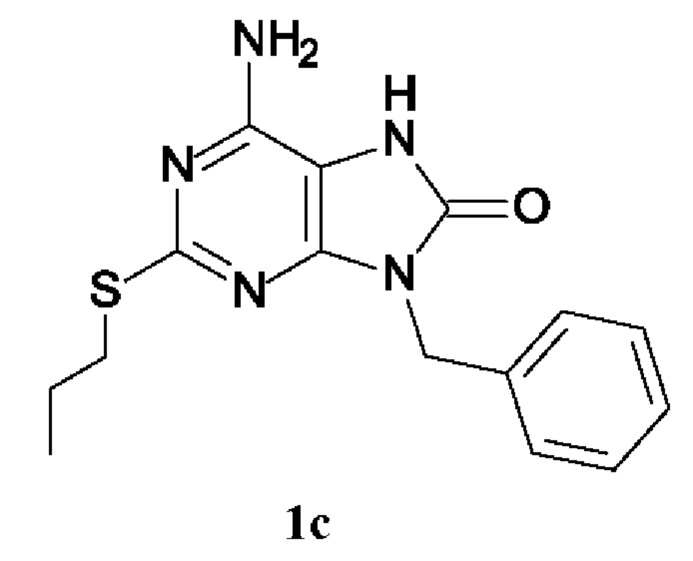
К раствору 6-амино-9-бензил-2-сульфанил-7Н-пурин-8-она (5,46 г; 20,0 ммоль; соединения 1b) в DMF добавляли карбонат калия (2,76 г; 20,0 ммоль). И затем к предыдущему раствору медленно добавляли 1-бромпропан (2,44 г; 20,0 ммоль; TCI, номер по каталогу: B0638-500G) в DMF (5,0 мл). После перемешивания при КТ в течение 12 ч реакционную смесь выливали в воду (200 мл), затем подкисляли, используя 10 масс. %-ный водный раствор гидросульфата натрия, и дважды экстрагировали EtOAc (100 мл). Органический слой промывали рассолом, сушили над Na2SO4 и концентрировали в вакууме, получая неочищенный продукт, который очищали флэш-хроматографией на силикагеле, получая 6-амино-9-бензил-2-(2-пропилсульфанил)-7Н-пурин-8-он (4,8 г; соединение 1с) в виде белого твердого вещества. MS: обнаружено (ESI+) [(М+Н)+]: 316.
Стадия 4. Получение 6-амино-9-бензил-2-пропилсульфинил-7Н-пурин-8-она (соединения 1d)
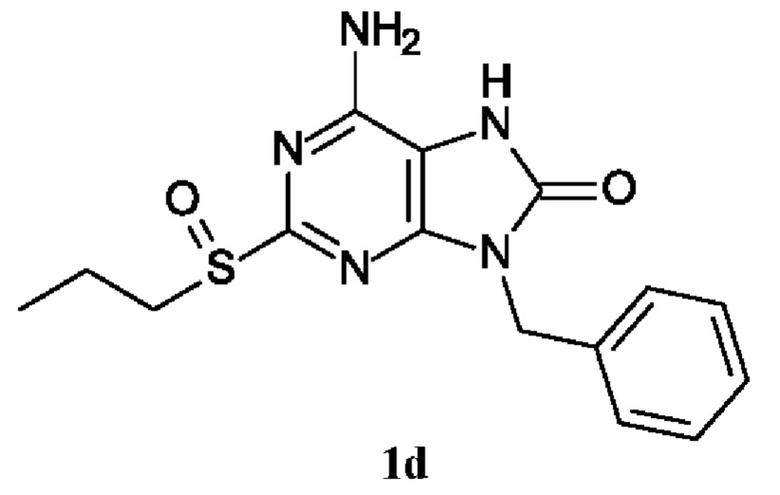
К суспензии соединения 6-амино-9-бензил-2-(2-пропилсульфанил)-7Н-пурин-8-она (2,7 г; 8,7 ммоль; соединения 1с) в DCM/MeOH (500 мл; об./об. = 1:1) добавляли 3-хлорпербензойную кислоту (2,15 г; 8,7 ммоль; чистота 70%, Aldrich, номер по каталогу: 273031-100G). После перемешивания реакционной смеси в течение 2 ч объем реакционной смеси уменьшали в вакууме примерно до 50 мл. Полученный осадок собирали фильтрованием, промывали метанолом и сушили, получая 6-амино-9-бензил-2-пропилсульфинил-7Н-пурин-8-он (1,0 г; соединение 1d) в виде белого твердого вещества. Продукт использовали на следующей стадии без дополнительной очистки. MS: обнаружено (ESI+) [(М+Н)+]: 332.
Стадия 5. Получение 6-амино-9-бензил-2-(пропилсульфонимидоил)-7Н-пурин-8-она (соединения 1е)
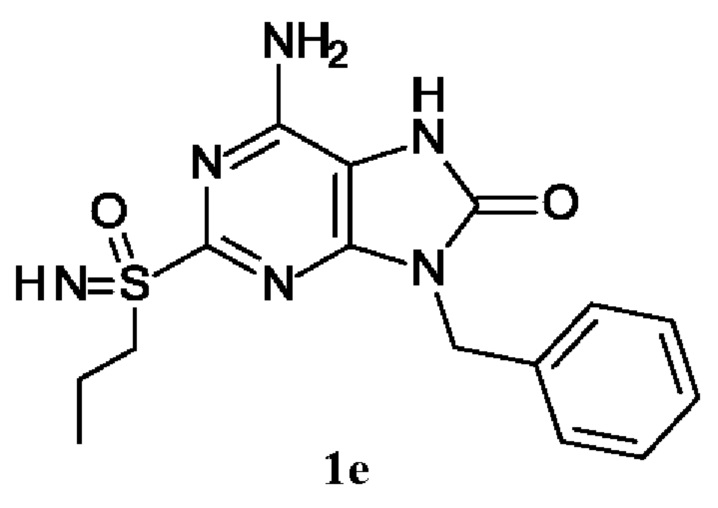
К раствору 6-амино-9-бензил-2-пропилсульфинил-7Н-пурин-8-она (1,52 г; 4,6 ммоль; соединения 1d) в реагенте Итона (40 мл; пятиокись фосфора; 7,5 масс. %-ная в метансульфоновой кислоте; Aldrich, номер по каталогу: 380814-100ML) добавляли азид натрия (360 мг; 5,5 ммоль) при 50°С. После перемешивания при этой температуре в течение 30 минут реакционную смесь охлаждали до КТ и выливали в насыщ. водный раствор бикарбоната натрия. Реакционную смесь дважды экстрагировали н-BuOH (100 мл) и органическую фазу концентрировали в вакууме. Остаток подвергали очистке препаративной HPLC, получая 6-амино-9-бензил-2-(пропилсульфонимидоил)-7Н-пурин-8-он (1,2 г; соединение 1е) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, диметилсульфоксид (DMSO)-d6) δ млн-1: 10.65 (br. s., 1Н), 7.26-7.37 (m, 5Н), 6.98 (br. s., 2H), 4.97 (s, 2H), 4.02 (s, 1H), 3.33 (t, J=7,53 Гц, 2H), 1.55-1.74 (m, 2H), 0.92 (t, J=7,53 Гц, 3H). MS: обнаружено (ESI+) [(M+H)+]: 347.
После разделения соединения 1е хиральной HPLC получали соединение 1е-А (медленнее элюирующееся; 500 мг) и соединение 1е-В (быстрее элюирующееся; 490 мг) в виде белого твердого вещества (условия разделения: 5%→40% метанола с (0,05% диэтиламина (DEA))/CO2 на колонке ChiralPak AS-3).
Соединение 1е-А: 1Н-ЯМР (DMSO-d6, 400 МГц) δ млн-1: 10.56 (s, 1Н), 7.21-7.46 (m, 5Н), 7.03 (s, 2Н), 4.96 (s, 2Н), 4.04 (s, 1Н), 3.25-3.33 (m, 2Н), 1.59-1.67 (m, 2Н), 0.92 (t, J=7,4 Гц, 3Н).
Соединение 1е-В: 1Н-ЯМР (DMSO-d6, 400 МГц) δ млн-1: 10.57 (s, 1Н), 7.23-7.39 (m, 5Н), 6.97 (s, 2Н), 4.96 (s, 2Н), 4.05 (s, 1Н), 3.31-3.30 (m, 2Н), 1.49-1.74 (m, 2Н), 0.91 (t, J=7,4 Гц, 3Н).
Стадия 6. Получение 6-амино-9-бензил-N-метил-8-оксо-N-пропил-2-(пропилсульфонимидоил)пурин-7-карбоксамида (соединения из примера 1)
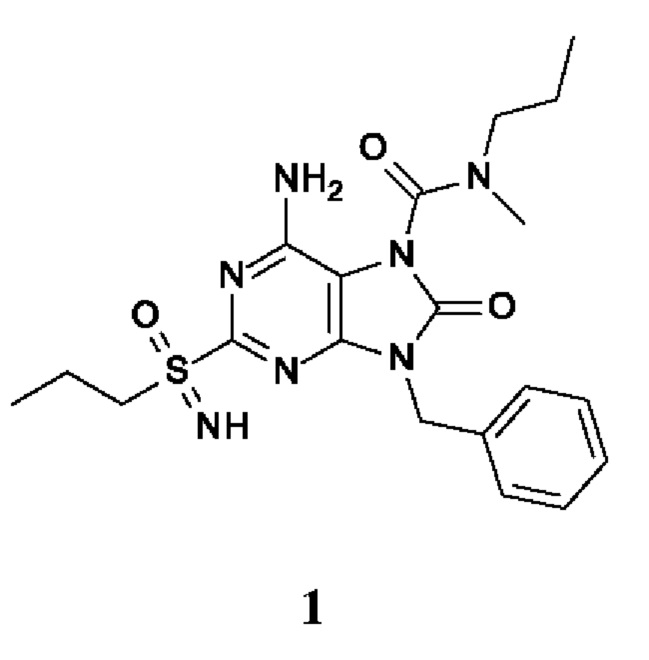
К раствору 6-амино-9-бензил-2-(пропилсульфонимидоил)-7Н-пурин-8-она (300 мг; соединения 1е), пиридина (329 мг; 4,2 ммоль) и DIPEA (538 мг; 4,2 ммоль) в NMP (5 мл) добавляли N-метил-N-пропил-карбамоилхлорид (564 мг; 4,2 ммоль; промежуточного соединения АА) при КТ. Смесь перемешивали при КТ в течение 10 ч. Реакционную смесь концентрировали и остаток очищали препаративной HPLC, получая 6-амино-9-бензил-N-метил-8-оксо-N-пропил-2-(пропилсульфонимидоил)пурин-7-карбоксамид (108 мг; соединение из примера 1) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.45-7.24 (m, 5Н), 6.89 (s, 2Н), 5.01 (s, 2Н), 4.17 (s, 1Н), 3.44-3.34 (m, 2Н), 3.36-3.34 (m, 2Н), 3.10-3.00 (m, 3Н), 1.74-1.52 (m, 4Н), 1.01-0.72 (m, 6Н). MS: обнаружено (ESI+) [(М+Н)+]: 446.
После разделения соединения из примера 1 хиральной HPLC получали соединение из примера 1-А (медленнее элюирующееся; 50 мг) и соединение из примера 1-В (быстрее элюирующееся; 40 мг) в виде белого твердого вещества, используя 5%→40% изопропанола с (0,05% DEA)/CO2 на колонке ChiralPak AD-3.
Соединение из примера 1-А: 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.44-7.24 (m, 5Н), 6.89 (s, 2Н), 5.01 (s, 2Н), 4.17 (s, 1Н), 3.44-3.37 (m, 2Н), 3.37-3.35 (m, 2Н), 3.10-3.00 (m, 3Н), 1.74-1.52 (m, 4Н), 1.00-0.72 (m, 6Н). MS: обнаружено (ESI+) [(М+Н)+]: 446.
Соединение из примера 1-В: 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.45-7.26 (m, 5Н), 6.88 (s, 2Н), 5.01 (s, 2Н), 4.15 (s, 1Н), 3.44-3.36 (m, 2Н), 3.34 (s, 2Н), 3.10-3.01 (m, 3Н), 1.77-1.52 (m, 4Н), 1.02-0.67 (m, 6Н). MS: обнаружено (ESI+) [(М+Н)+]: 446.
Способ В. Альтернативный способ получения 6-амино-9-бензил-2-(пропилсульфонимидоил)-7Н-пурин-8-она (соединения 1е)
Стадия 1. Получение N-бензил-6-хлор-5-нитро-2-пропилсульфанил-пиримидин-4-амина (соединения 1f)
К раствору 4,6-дихлор-5-нитро-2-пропилсульфанилпиримидина (150,0 г; 559,5 ммоль) и DIPEA (108,5 г; 839,2 ммоль) в THF (1,5 л) медленно при -78°С добавляли фенилметанамин (60,0 г; 559,5 ммоль) в THF (200 мл). По окончании добавления смесь нагревали до 25°С и перемешивали при этой температуре в течение 16 ч. Полученную смесь разбавляли ЕА (1 л), три раза промывали водой (400 мл) и рассолом (500 мл). Отделенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали в вакууме, получая N-бензил-6-хлор-5-нитро-2-пропилсульфанил-пиримидин-4-амин (180,0 г; соединение 1f) в виде желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки. MS: обнаружено (ESI+) [(М+Н)+]: 339,1.
Стадия 2. Получение N4-бензил-6-хлор-2-пропилсульфанил-пиримидин-4,5-диамина (соединения 1g)
К раствору N-бензил-6-хлор-5-нитро-2-пропилсульфанил-пиримидин-4-амина (180 г; соединения 1f) и НОАс (319 г; 5,31 моль) в THF (3,0 л) медленно при 25°С добавляли Zn (174 г; 2,66 моль). По окончании добавления смесь перемешивали при 25°С в течение 16 ч. Реакционную смесь фильтровали и фильтрат подщелачивали насыщенным водным раствором NaHCO3 (800 мл), три раза экстрагировали ЕА (400 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали хроматографией на силикагеле, получая N4-бензил-6-хлор-2-пропилсульфанил-пиримидин-4,5-диамин (125 г; соединение 1g) в виде коричневого твердого вещества. MS: обнаружено (ESI+) [(М+Н)+]: 309,1.
Стадия 3. Получение 9-бензил-6-хлор-2-пропилсульфанил-7Н-пурин-8-она (соединения 1h)
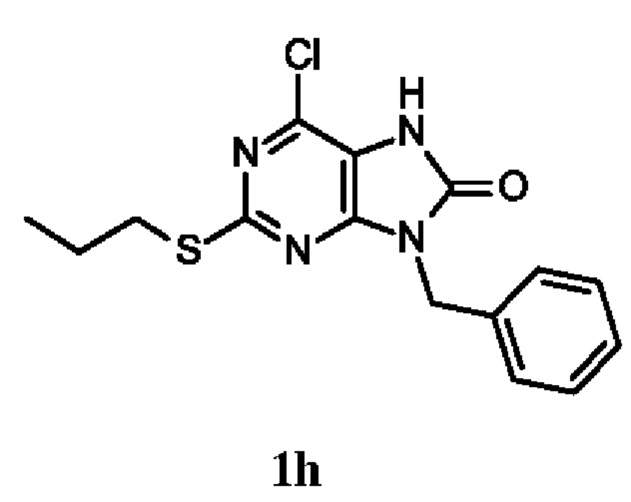
Раствор N-бензил-6-хлор-2-(пропилсульфанил)пиримидин-4,5-диамина (72,0 г; 233,1 ммоль; соединения 1g) и CDI (75,2 г; 233,1 ммоль) в THF (800 мл) перемешивали при 80°С в течение 16 ч. Полученную смесь разбавляли ЕА (400 мл), дважды промывали водой (200 мл) и рассолом (200 мл). Отделенный органический слой сушили над Na2SO4, концентрировали в вакууме. Остаток промывали МТВЕ (200 мл), получая 9-бензил-6-хлор-2-пропилсульфанил-7Н-пурин-8-он (58,0 г; соединение 1h) в виде белого твердого вещества, которое использовали на следующей стадии без дополнительной очистки. MS: обнаружено (ESI+) [(М+Н)+]: 335,1.
Стадия 4. Получение 9-бензил-6-[(4-метоксифенил)метиламино]-2-пропилсульфанил-7Н-пурин-8-она (соединения 1i)
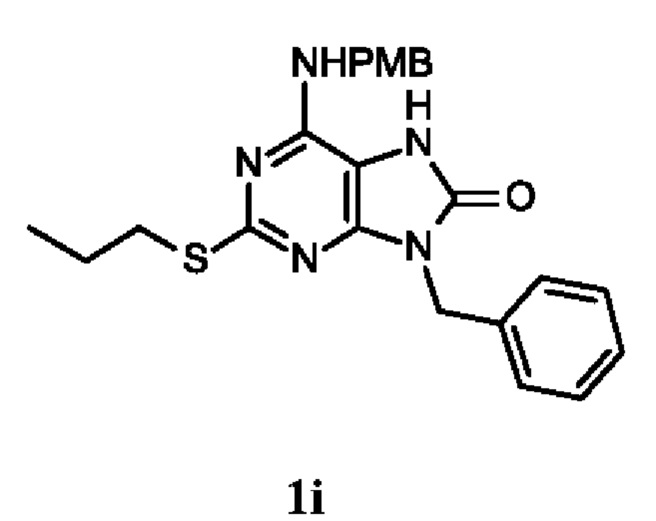
Раствор 9-бензил-6-хлор-2-пропилсульфанил-7Н-пурин-8-она (58,0 г; соединения 1h) и PMBNH2 (54,7 г; 398,42 ммоль) в н-BuOH (600 мл) перемешивали при 120°С в течение 20 ч. Реакционную смесь концентрировали и остаток промывали МТВЕ (400 мл), получая 9-бензил-6-[(4-метоксифенил)метиламино]-2-пропилсульфанил-7Н-пурин-8-он (75 г; соединение 1i) в виде белого твердого вещества, которое использовали на следующей стадии без дополнительной очистки. MS: обнаружено (ESI+) [(М+Н)+]: 436,2.
Стадия 5. Получение 6-амино-9-бензил-2-пропилсульфанил-7Н-пурин-8-она (соединения 1с)

9-Бензил-6-[(4-метоксифенил)метиламино]-2-пропилсульфанил-7Н-пурин-8-он (87,0 г; соединение 1i) в TFA (200 мл) перемешивали при 80°С в течение 16 ч. Полученную реакционную смесь концентрировали, подщелачивали насыщенным водным раствором NaHCO3 (600 мл). Полученный осадок собирали фильтрованием и промывали (PE/DCM = 2:1, 400 мл), получая 6-амино-9-бензил-2-пропилсульфанил-7Н-пурин-8-он (38,0 г; соединение 1с) в виде белого твердого вещества. MS: обнаружено (ESI+) [(М+Н)+]: 316,1.
Стадия 6. Получение 6-амино-9-бензил-2-пропилсульфинил-7Н-пурин-8-она (соединения 1d)
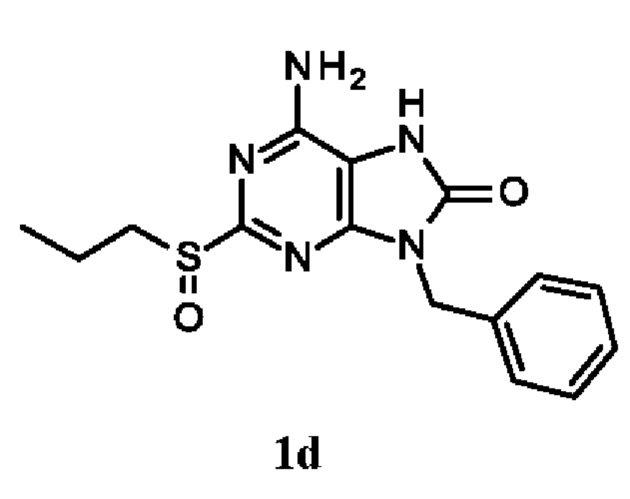
Раствор m-СРВА (22,98 г; 113,2 ммоль) в THF (50 мл) по каплям добавляли к суспензии 6-амино-9-бензил-2-пропилсульфанил-7Н-пурин-8-она (35,0 г; соединения 1с) в THF (200 мл) при 0°С. По окончании добавления реакционную смесь перемешивали при 25°С в течение 0,5 ч. Смесь фильтровали и промывали MeCN (400 мл), МТВЕ (500 мл), получая 6-амино-9-бензил-2-пропилсульфинил-7Н-пурин-8-он (35,1 г; соединение 1d) в виде белого твердого вещества, которое использовали на следующей стадии без дополнительной очистки. MS: обнаружено (ESI+) [(М+Н)+]: 332,1.
Стадия 7. Получение 6-амино-9-бензил-2-(пропилсульфонимидоил)-7Н-пурин-8-она (соединения 1е)
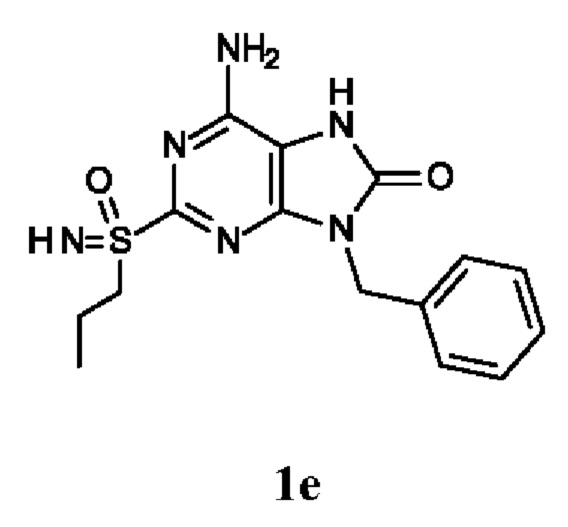
К раствору 6-амино-9-бензил-2-пропилсульфинил-7Н-пурин-8-она (34,0 г; соединения 1d) в реагенте Итона (170,0 мл; 7,5 масс. %-ной пятиокиси фосфора в метансульфоновой кислоте) медленно добавляли NaN3 (15,34 г; 253,97 ммоль) при 60°С. Затем смесь перемешивали при 60°С в течение 30 мин. Полученную реакционную смесь охлаждали до 25°С, выливали в охлажденный во льду NH3⋅H2O (500 мл; 1 моль/л), четыре раза экстрагировали н-BuOH (100 мл) и концентрировали в вакууме. Остаток очищали препаративной HPLC, получая 6-амино-9-бензил-2-(пропилсульфонимидоил)-7Н-пурин-8-он (10 г; соединение 1е). 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 10.65 (br. s., 1Н), 7.26-7.37 (m, 5Н), 6.98 (br. s., 2Н), 4.97 (s, 2Н), 4.02 (s, 1Н), 3.33 (t, J=7,53 Гц, 2H), 1.55-1.74 (m, 2Н), 0.92 (t, J=7,53 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 347.
Пример 2
6-Амино-9-бензил-N-(2-метоксиэтил)-N-метил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамид
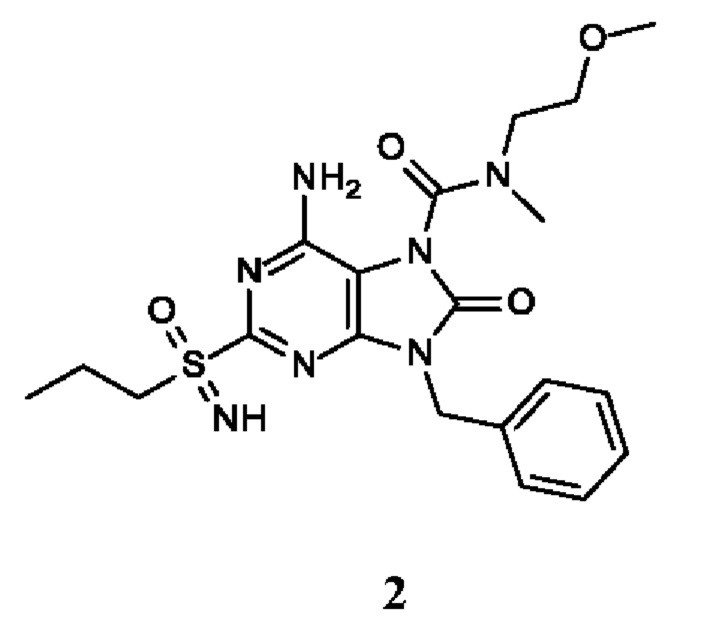
Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя N-(2-метоксиэтил)-N-метил-карбамоилхлорид (промежуточное соединение АВ) вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). 6-Амино-9-бензил-N-(2-метоксиэтил)-N-метил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамид (120 мг; соединение из примера 2) получали в виде белого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.27-7.39 (m, 5Н), 6.89 (br. s., 1Н), 6.78 (br. s., 1Н), 5.00 (s, 2Н), 4.16 (br. d, J=4 Гц, 1Н), 3.62 (br. dd, J=4; 12 Гц, 2Н), 3.28-3.42 (m, 6Н), 3.12 (d, J=12 Гц, 3Н), 3.05 (s, 1Н), 1.58-1.72 (m, 2Н), 0.93 (t, J=8 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 462.
После разделения соединения из примера 2 хиральной HPLC получали соединение из примера 2-А (быстрее элюирующееся; 33 мг) и соединение из примера 2-В (медленнее элюирующееся; 46 мг) в виде белого твердого вещества, используя 5%→40% метанола с (0,05% DEA)/CO2 на колонке ChiralPak OJ-3.
Соединение из примера 2-А: 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.27-7.39 (m, 5Н), 6.89 (br. s., 1Н), 6.78 (br. s., 1Н), 5.00 (s, 2Н), 4.16 (br. d, J=4 Гц, 1Н), 3.62 (br. dd, J=4; 12 Гц, 2Н), 3.28-3.42 (m, 6Н), 3.12 (d, J=12 Гц, 3Н), 3.05 (s, 1Н), 1.58-1.72 (m, 2Н), 0.93 (t, J=8 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 462.
Соединение из примера 2-В: 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.27-7.39 (m, 5Н), 6.89 (br. s., 1Н), 6.78 (br. s., 1Н), 5.00 (s, 2Н), 4.16 (br. d, J=4 Гц, 1Н), 3.62 (br. dd, J=4; 12 Гц, 2Н), 3.28-3.42 (m, 6Н), 3.12 (d, J=12 Гц, 3Н), 3.05 (s, 1Н), 1.58-1.72 (m, 2Н), 0.93 (t, J=8 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 462.
Пример 3
б-Амино-9-бензил-N-этил-8-оксо-N-пропил-2-(пропилсульфонимидоил)пурин-7-карбоксамид
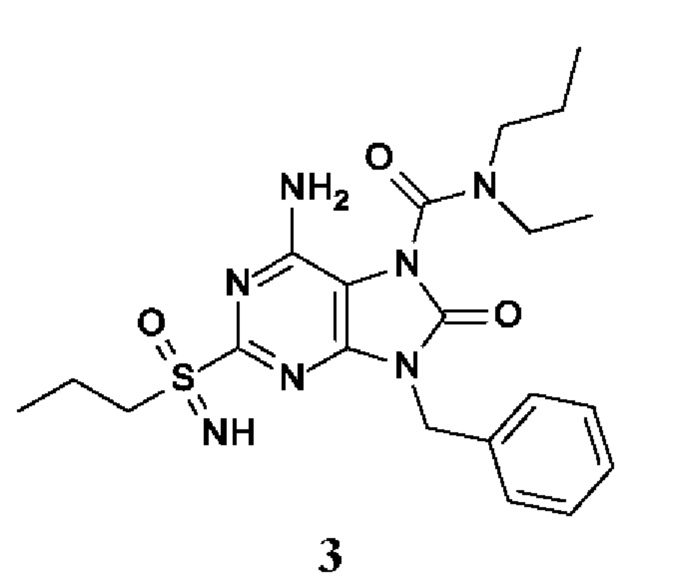
Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя N-этил-N-пропил-карбамоилхлорид (промежуточное соединение АС) вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). 6-Амино-9-бензил-N-этил-8-оксо-N-пропил-2-(пропилсульфонимидоил)пурин-7-карбоксамид (51 мг; соединение из примера 3) получали в виде белого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.27-7.39 (m, 5Н), 6.85 (br. s., 2Н), 4.99 (s, 2Н), 4.20 (br. d, J=8,0 Гц, 1Н), 3.13-3.54 (m, 4Н), 1.46-1.72 (m, 4Н), 1.30-1.39 (m, 1Н), 1.00-1.26 (m, 6Н), 0.81-0.95 (m, 5Н), 0.73 (t, J=8 Гц, 1Н). MS: обнаружено (ESI+) [(М+Н)+]: 474.
Пример 4
6-Амино-9-бензил-7-[4-(1-пиперидил)пиперидин-1-карбонил]-2-(пропилсульфонимидоил)пурин-8-он
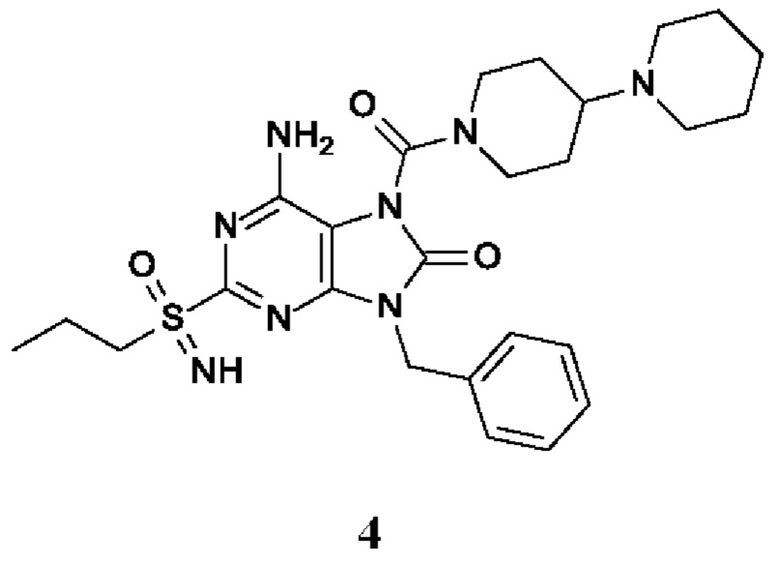
Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя (1,4'-бипиперидин)-1'-карбонилхлорид вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). 6-Амино-9-бензил-7-[4-(1-пиперидил)пиперидин-1-карбонил]-2-(пропилсульфонимидоил)пурин-8-он (55 мг; соединение из примера 4) получали в виде белого порошка. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.39-7.27 (m, 5Н), 6.97 (br. s., 2Н), 4.99 (s, 2Н), 4.20 (br. s., 2Н), 3.85 (d, J=12,5 Гц, 1Н), 3.43-3.15 (m, 3Н), 2.96 (t, J=12,3 Гц, 2Н), 2.56 (m, 4Н), 1.83 (m, 1Н), 1.79-1.54 (m, 4Н), 1.50 (br. s., 4Н), 1.45-1.33 (m, 3Н), 0.93 (t, J=7,4 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 541,2.
Пример 5
6-Амино-9-бензил-N-этил-N-(2-метоксиэтил)-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамид
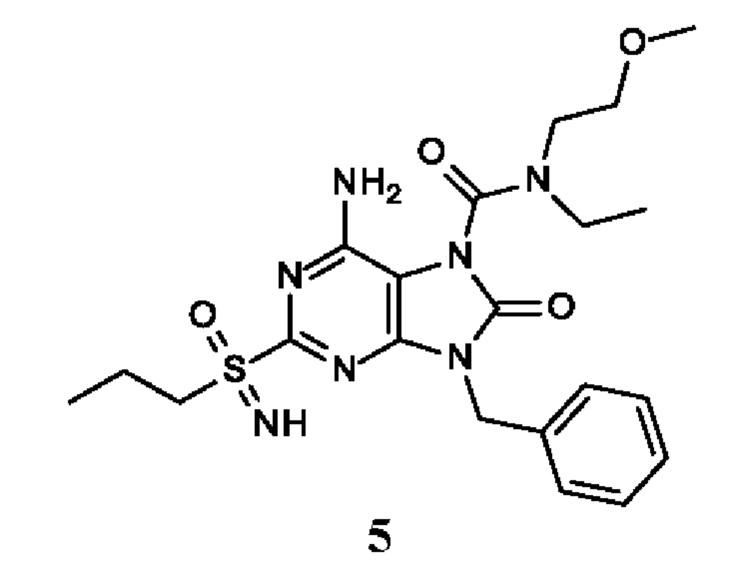
Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя N-этил-N-(2-метоксиэтил)карбамоилхлорид (промежуточное соединение AD) вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). 6-Амино-9-бензил-N-этил-N-(2-метоксиэтил)-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамид (34 мг; соединение из примера 5) получали в виде белого порошка. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.39-7.28 (m, 5Н), 6.89 (br. s., 1Н), 6.74 (br. s., 1Н), 4.99 (s, 2Н), 4.17 (d, J=8,1 Гц, 1H), 3.67 (br. s., 2H), 3.63-3.51 (m, 2H), 3.50-3.34 (m, 4H), 3.29 (s, 1H), 3.11 (s, 2H), 1.73-1.59 (m, 2H), 1.23-1.07 (m, 3Н), 0.93 (t, J=7,5 Гц, 3Н). MS: обнаружено (ESI+) [(M+H)+]: 476,3.
Пример 6
6-Амино-9-бензил-N-бутил-N-этил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамид
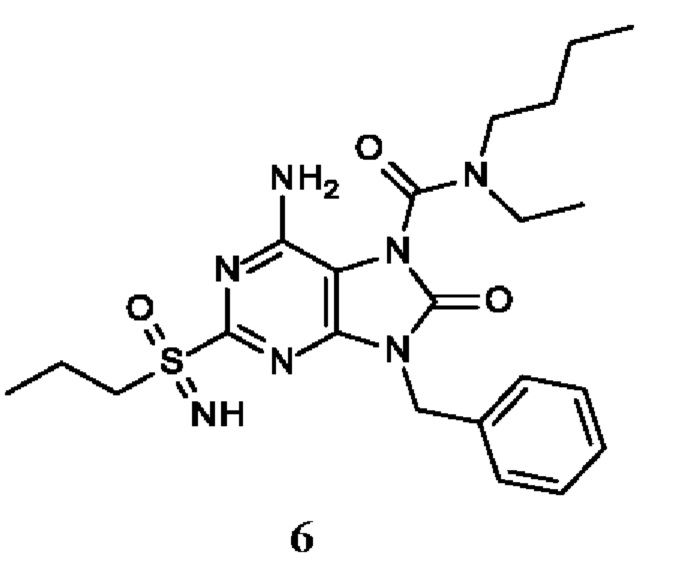
Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя N-бутил-N-этил-карбамоилхлорид (промежуточное соединение АЕ) вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). 6-Амино-9-бензил-N-бутил-N-этил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамид (51 мг; соединение из примера 6) получали в виде белого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.27-7.39 (m, 5Н), 6.85 (br. s., 2Н), 4.99 (s, 2Н), 4.20 (br. d, J=8,0 Гц, 1H), 3.13-3.54 (m, 4Н), 1.46-1.72 (m, 4Н), 1.30-1.39 (m, 1Н), 1.00-1.26 (m, 6Н), 0.81-0.95 (m, 5Н), 0.73 (t, J=8 Гц, 1Н). MS: обнаружено (ESI+) [(М+Н)+]: 474.
Пример 7
б-Амино-9-бензил-N-(2-метоксиэтил)-8-оксо-N-пропил-2-(пропилсульфонимидоил)пурин-7-карбоксамид
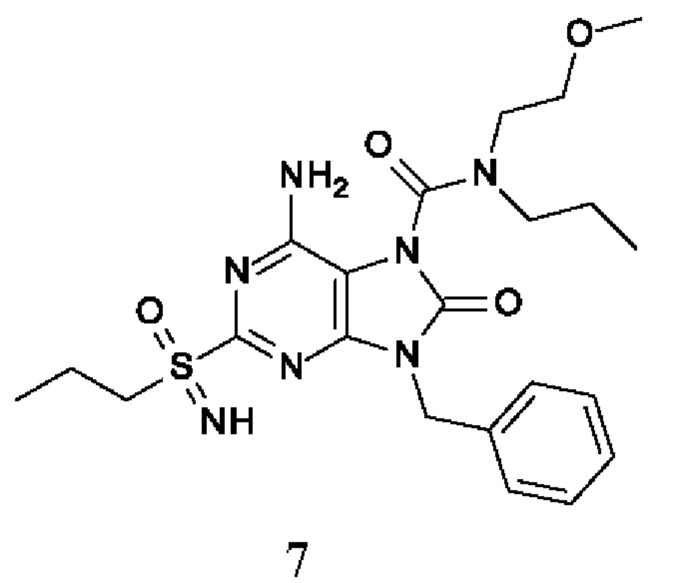
Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя N-этил-N-(2-метоксиэтил)карбамоилхлорид (промежуточное соединение AF) вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). 6-Амино-9-бензил-N-(2-метоксиэтил)-8-оксо-N-пропил-2-(пропилсульфонимидоил)пурин-7-карбоксамид (35 мг; соединение из примера 7) получали в виде белого порошка. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.40-7.28 (m, 5Н), 6.89 (br. s., 1Н), 6.75 (br. s., 1Н), 5.00 (d, J=5,5 Гц, 2H), 4.24-4.16 (m, 1Н), 3.77 (br. s., 1Н), 3.67 (br. s., 1Н), 3.62-3.53 (m, 1Н), 3.42-3.27 (m, 5Н), 3.23-3.02 (m, 3Н), 1.66-1.38 (m, 4Н), 0.96-0.70 (m, 6Н). MS: обнаружено (ESI+) [(М+Н)+]: 490,5.
Пример 8
6-Амино-9-бензил-N,N-бис(2-метоксиэтил)-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамид
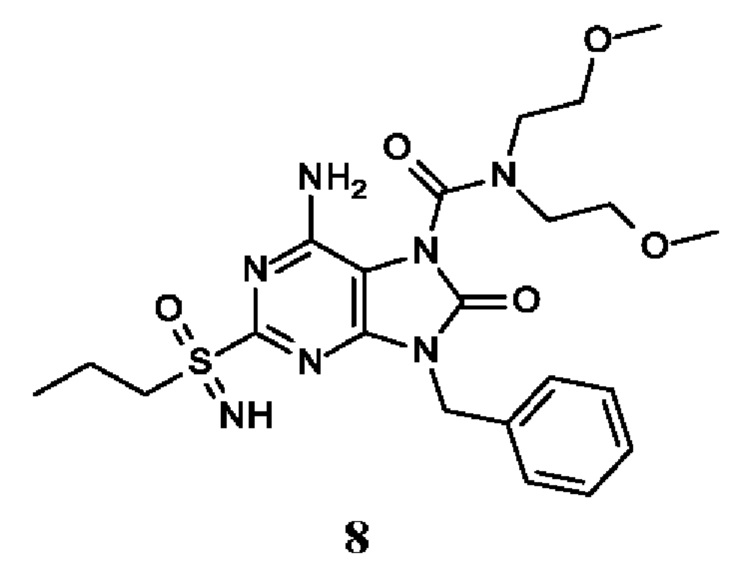
Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя бис(2-метоксиэтил)карбамоилхлорид (промежуточное соединение AG) вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). 6-Амино-9-бензил-N,N-бис(2-метоксиэтил)-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамид (35 мг; соединение из примера 8) получали в виде белого порошка. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.40-7.28 (m, 5Н), 6.83 (br. s., 2Н), 4.99 (s, 2Н), 3.71 (br. s., 3Н), 3.52-3.27 (m, 11Н), 3.09 (s, 3Н), 1.73-1.59 (m, 2Н), 0.93 (t, J=7,5 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 506.
Пример 9
6-Амино-7-(азетидин-1-карбонил)-9-бензил-2-(пропилсульфонимидоил)пурин-8-он
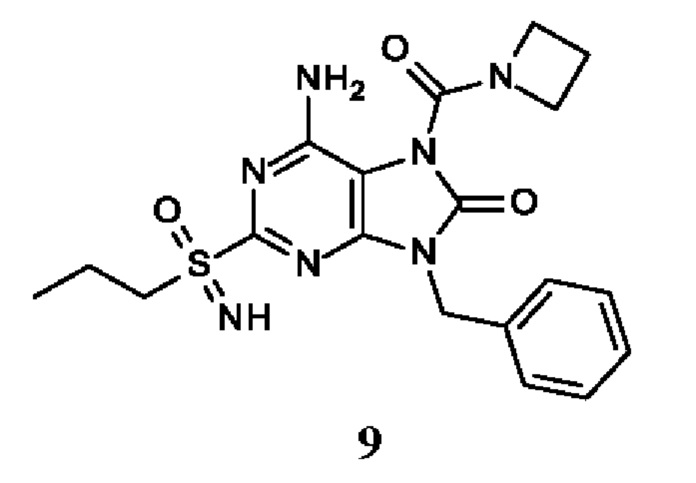
Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя азетидин-1-карбонилхлорид (промежуточное соединение АН) вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). 6-Амино-7-(азетидин-1-карбонил)-9-бензил-2-(пропилсульфонимидоил)пурин-8-он (120 мг; соединение из примера 9) получали в виде белого порошка. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.02-7.43 (m, 7Н), 4.99 (s, 2H), 4.31 (t, J=7,65 Гц, 2H), 4.08-4.23 (m, 3Н), 3.34-3.41 (m, 2H), 2.28 (m, 2H), 1.56-1.73 (m, 2H), 0.93 (t, J=7,40 Гц, 3Н). MS: обнаружено (ESI+) [(M+H)+]: 430.
Пример 10
6-Амино-9-бензил-N-изопропил-N-метил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамид

Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя N-изопропил-N-метил-карбамоилхлорид (промежуточное соединение AI) вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). 6-Амино-9-бензил-N-изопропил-N-метил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамид (97 мг; соединение из примера 10) получали в виде белого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.27-7.39 (m, 5Н), 6.87 (br. s., 2Н), 4.99 (s, 2Н), 4.38-4.45 (m, 1Н), 4.09-4.21 (m, 1Н), 3.29-3.43 (m, 2Н), 2.89-2.95 (m, 3Н), 1.58-1.73 (m, 2Н), 1.21 (br d, J=8 Гц, 6Н), 0.93 (t, J=8 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 446.
Пример 11
6-Амино-9-бензил-7-(4-метилпиперазин-1-карбонил)-2-(пропилсульфонимидоил)пурин-8-он
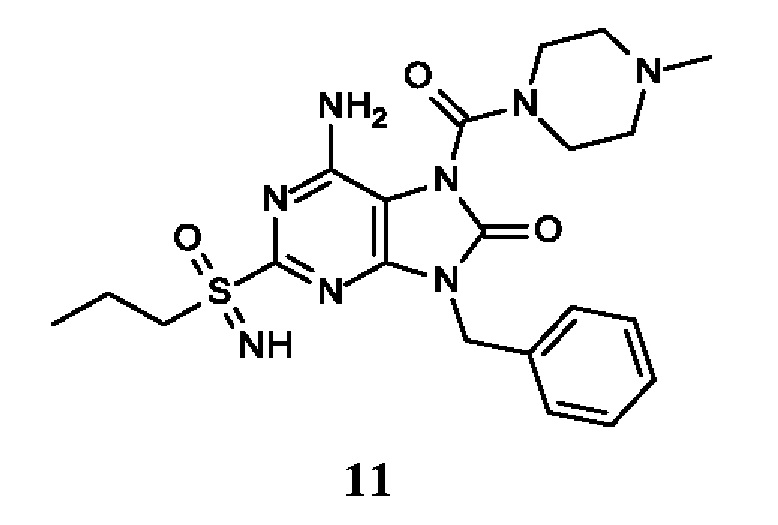
Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя 4-метилпиперазин-1-карбонилхлорид вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). 6-Амино-9-бензил-7-(4-метилпиперазин-1-карбонил)-2-(пропилсульфонимидоил)пурин-8-он (59,5 мг; соединение из примера 11) получали в виде желтого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.39-7.31 (m, 5Н), 6.99 (s, 2Н), 4.98 (s, 2Н), 4.18 (s, 1Н), 3.58-3.49 (m, 6Н), 2.42 (m, 4Н), 2.22 (s, 3Н), 1.66-1.61 (m, 2Н), 0.95-0.91 (t, J=7,2 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 473.
Пример 12
6-Амино-9-бензил-N-(3-метоксипропил)-N-метил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамид
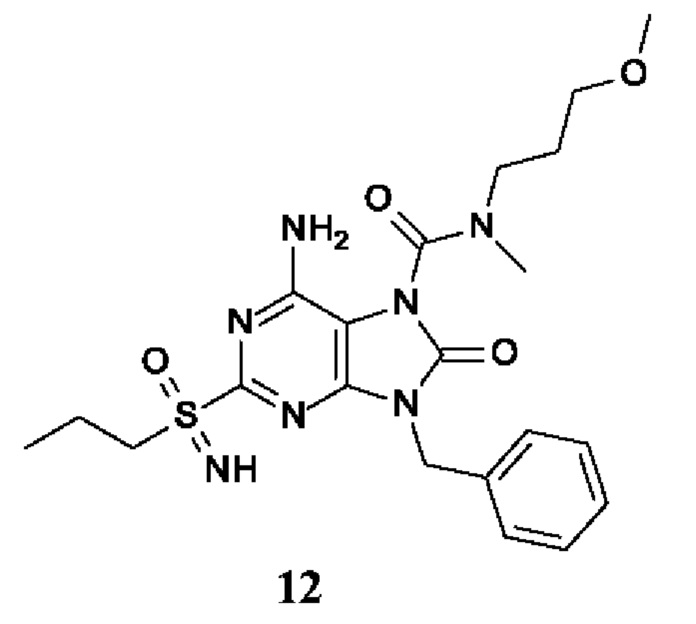
Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя N-(3-метоксипропил)-N-метил-карбамоилхлорид вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). 6-Амино-9-бензил-N-(3-метоксипропил)-N-метил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамид (92,2 мг; соединение из примера 12) получали в виде белого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.23-7.45 (m, 5Н), 6.94 (s., 2Н), 4.93-5.08 (m, 2Н), 4.19 (s, 1Н), 3.30-3.62 (m, 6Н), 3.25 (s, 3Н), 3.02-3.10 (m, 3Н), 1.74-1.90 (m, 2Н), 1.55-1.77 (m, 2Н), 0.98-0.82 (m, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 476,3.
Пример 13
6-Амино-9-бензил-N-изобутил-N-метил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамид
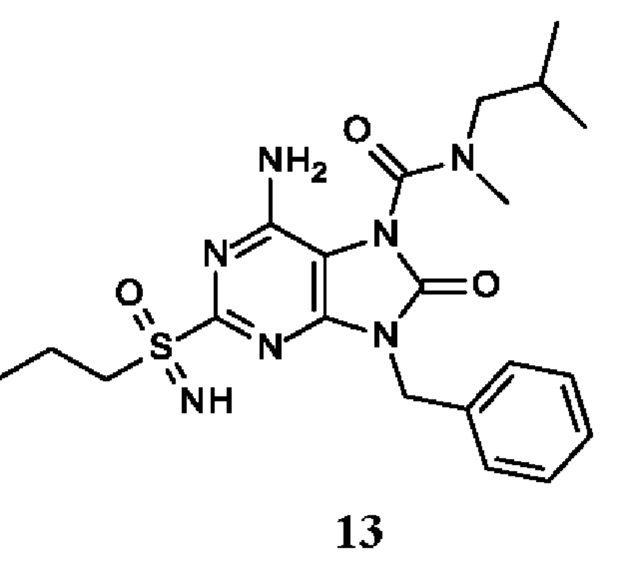
Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя N-изобутил-N-метил-карбамоилхлорид (промежуточное соединение AL) вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). 6-Амино-9-бензил-N-изобутил-N-метил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамид (64 мг; соединение из примера 13) получали в виде белого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.27-7.40 (m, 5Н), 6.89 (br. s., 2Н), 5.00 (s, 2Н), 4.16 (br. s., 1Н), 3.25-3.44 (m, 4Н), 3.07 (s, 2Н), 3.03 (s, 1Н), 1.87-2.09 (m, 1Н), 1.57-1.74 (m, 2Н), 0.75-0.99 (m, 9Н). MS: обнаружено (ESI+) [(М+Н)+]: 460.
Пример 14
Этил-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]ацетат
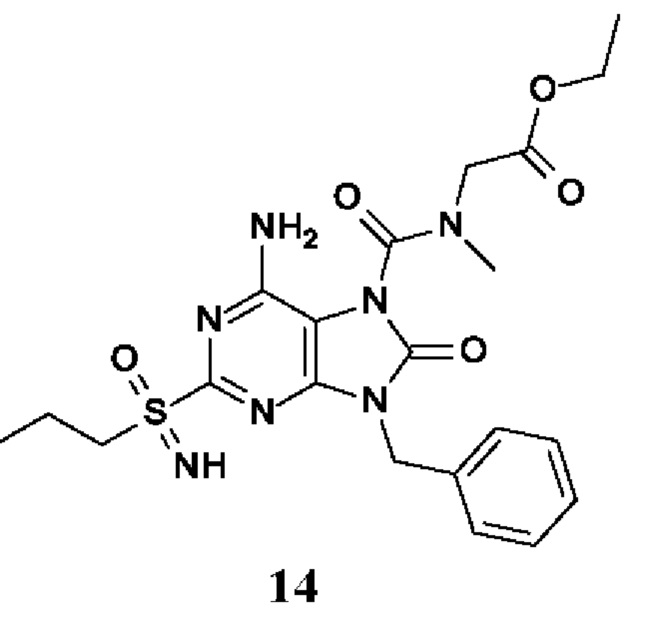
Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя этил-2-((хлоркарбонил)(метил)амино)ацетат (промежуточное соединение АР) вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). Этил-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]ацетат (38 мг; соединение из примера 14) получали в виде светло-желтого порошка. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.41-7.27 (m, 5Н), 6.82 (br. s., 1Н), 5.04-4.95 (m, 2Н), 4.35 (br. s., 1Н), 4.28 (br. s., 1Н), 4.23-4.16 (m, 2Н), 4.08 (q, J=7,2 Гц, 1Н), 3.43-3.28 (m, 3Н), 3.15 (s, 2Н), 3.08 (s, 1Н), 1.71-1.58 (m, 2Н), 1.24 (t, J=7,0 Гц, 2Н), 1.12 (t, J=7,0 Гц, 1Н), 0.93 (t, J=7,4 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 490.
Пример 15
Этил-3-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]пропаноат
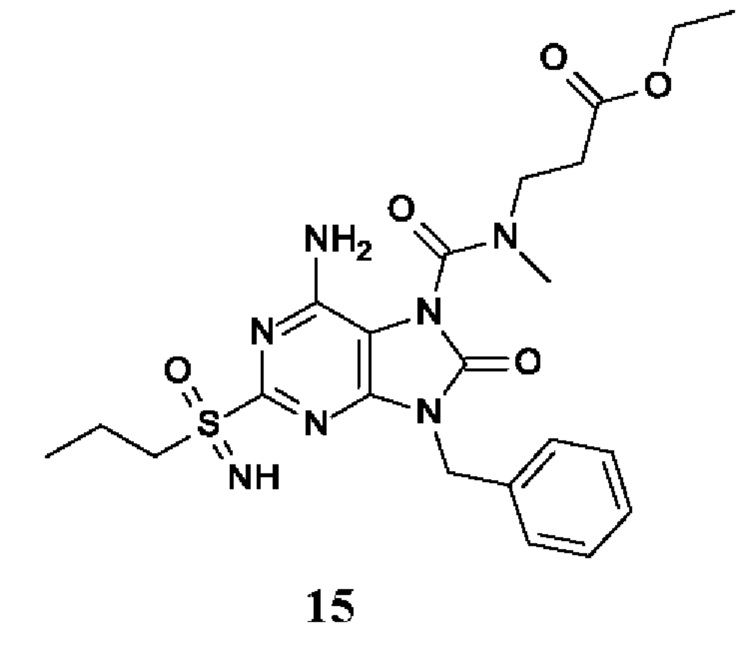
Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя этил-3-((хлоркарбонил)(метил)амино)пропаноат вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). Этил-3-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]пропаноат (35 мг; соединение из примера 15) получали в виде белого порошка. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.43-7.26 (m, 5Н), 6.93 (br. s., 2Н), 4.99 (s, 2Н), 4.16 (s, 1Н), 4.08 (q, J=7,1 Гц, 1Н), 3.99 (d, J=7,0 Гц, 1Н), 3.67 (br. s., 2Н), 3.40-3.29 (m, 2Н), 3.08 (s, 2Н), 2.99 (s, 1Н), 2.71 (t, J=6,4 Гц, 2Н), 1.74-1.56 (m, 2Н), 1.27-1.05 (m, 3Н), 0.93 (t, J=7,5 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 504.
Пример 16
трет-Бутил-3-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]пропаноат
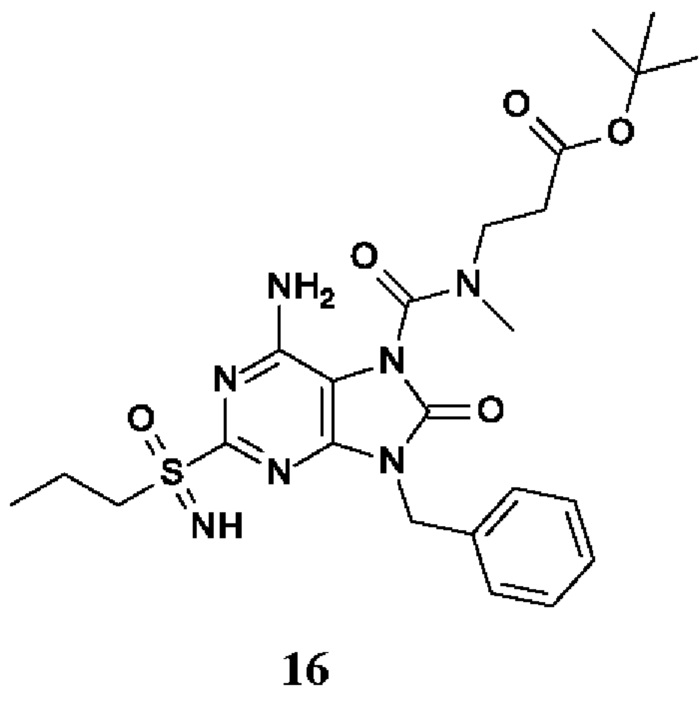
Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя трет-бутил-3-[хлоркарбонил(метил)амино]пропаноат (промежуточное соединение AR) вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). трет-Бутил-3-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]пропаноат (60 мг; соединение из примера 16) получали в виде белого порошка. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.41-7.27 (m, 5Н), 6.93 (br. s., 2Н), 4.99 (s, 2Н), 4.15 (s, 1Н), 3.64 (br. s., 2Н), 3.51-3.33 (m, 2Н), 3.08 (s, 2Н), 2.98 (s, 1Н), 2.62 (t, J=6,9 Гц, 2Н), 1.71-1.57 (m, 2Н), 1.41 (s, 6Н), 1.34 (s, 3Н), 0.93 (t, J=7,4 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 532.
Пример 17
Этил-(2S)-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]пропаноат
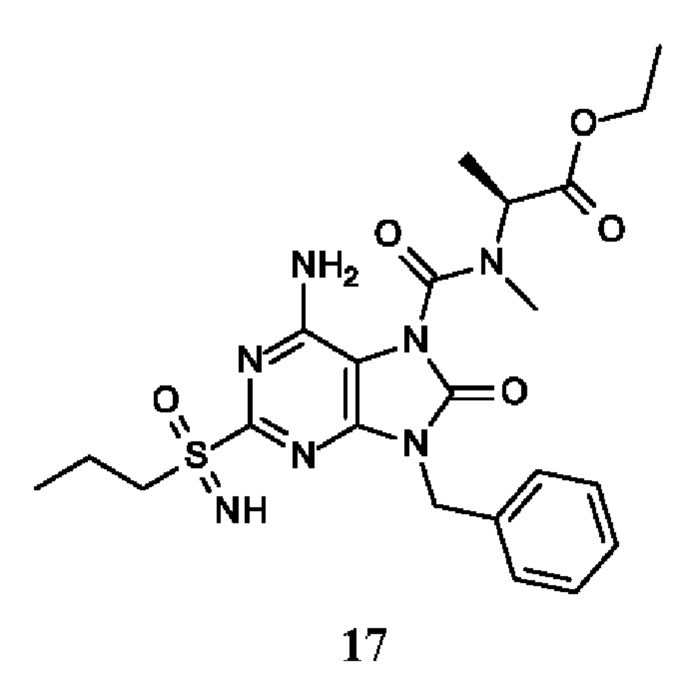
Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя этил-(2S)-2-[хлоркарбонил(метил)амино]пропаноат (промежуточное соединение AS) вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). Этил-(2S)-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]пропаноат (34,1 мг; соединение из примера 17) получали в виде желтого твердого вещества. 1Н-ЯМР (300 МГц, DMSO-d6) δ млн-1: 7.22-7.49 (m, 5Н), 6.78 (br. s., 2Н), 4.93-5.08 (m, 2Н), 4.75 (br. s., 1Н), 3.96-4.29 (m, 3Н), 3.30-3.46 (m, 2Н), 3.09 (s, 2Н), 2.93 (br. s., 1Н), 1.55-1.77 (m, 2Н), 1.48 (d, J=7,16 Гц, 3Н), 1.09-1.29 (m, 3Н), 0.94 (t, J=7,44 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 504,2.
Пример 18
трет-Бутил-(2S)-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]-4-метилпентаноат

Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя трет-бутил-(2S)-2-[хлоркарбонил(метил)амино]-4-метилпентаноат (промежуточное соединение AT) вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). трет-Бутил-(2S)-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]-4-метилпентаноат (22 мг; соединение из примера 18) получали в виде белого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.42-7.27 (m, 5Н), 6.78 (br. s., 2Н), 5.05-4.96 (m, 2Н), 4.78 (br. s., 1Н), 4.33 (br. s., 1Н), 3.51-3.37 (m, 2Н), 3.01 (s, 3Н), 1.75-1.54 (m, 4Н), 1.44 (s, 8Н), 1.33-1.11 (m, 2Н), 0.99-0.82 (m, 9Н). MS: обнаружено (ESI+) [(М+Н)+]: 574,3.
Пример 19
Изопропил-(2S)-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]-4-метилпентаноат
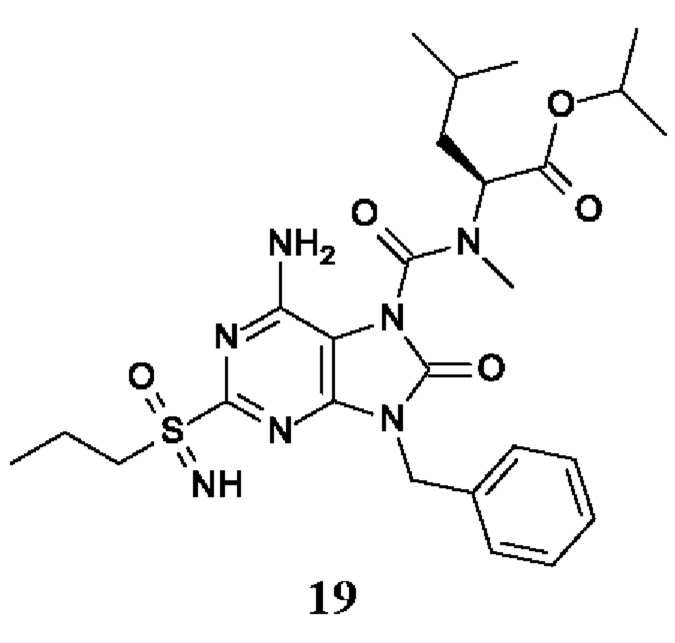
Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя изопропил-(2S)-2-[хлоркарбонил(метил)амино]-4-метилпентаноат (промежуточное соединение AU) вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). Изопропил-(2S)-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]-4-метилпентаноат (43 мг; соединение из примера 19) получали в виде белого порошка. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.43-7.27 (m, 5Н), 6.75 (br. s., 2Н), 5.05-4.94 (m, 3Н), 4.88 (br. s., 1Н), 4.19 (br. s., 1Н), 3.43-3.34 (m, 2Н), 3.01 (s, 3Н), 1.91 (br. s., 1Н), 1.77-1.56 (m, 4Н), 1.25-1.16 (m, 6Н), 0.99-0.83 (m, 9Н). MS: обнаружено (ESI+) [(М+Н)+]: 560,3.
Пример 20
Этил-(2S)-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]-3-метилбутаноат
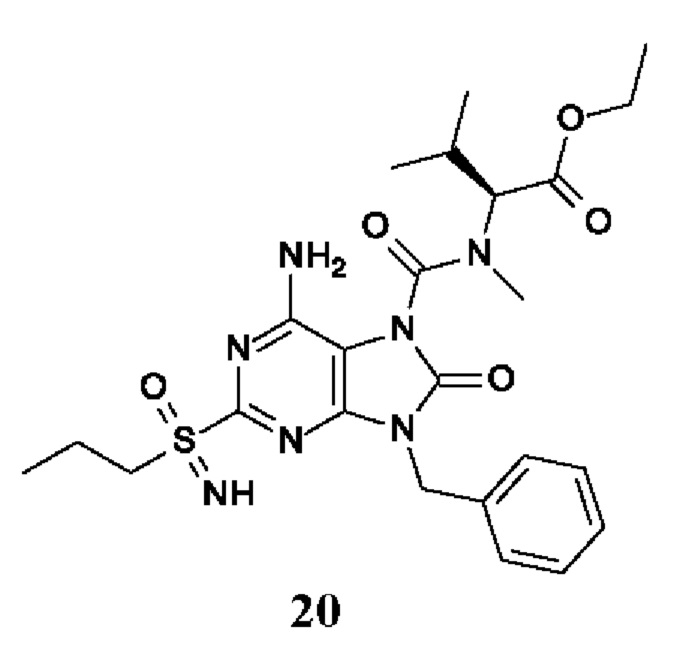
Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя этил-(2S)-2-[хлоркарбонил(метил)амино]-3-метилбутаноат (промежуточное соединение AV) вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). Этил-(2S)-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]-3-метилбутаноат (51,5 мг; соединение из примера 20) получали в виде белого порошка. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.23-7.51 (m, 5Н), 6.76 (br. s., 2Н), 5.01 (br. s., 2Н), 4.42 (br. s., 1H), 3.97-4.26 (m, 3Н), 3.34-3.45 (m, 2H), 3.12 (br. s., 3Н), 2.24 (br. s., 1H), 1.65 (br. s., 2H), 1.13-1.29 (m, 3H), 0.88-1.10 (m, 9H). MS: обнаружено (ESI+) [M+H+]: 532,2.
Пример 21
Этил-(2S)-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]-4-метилпентаноат
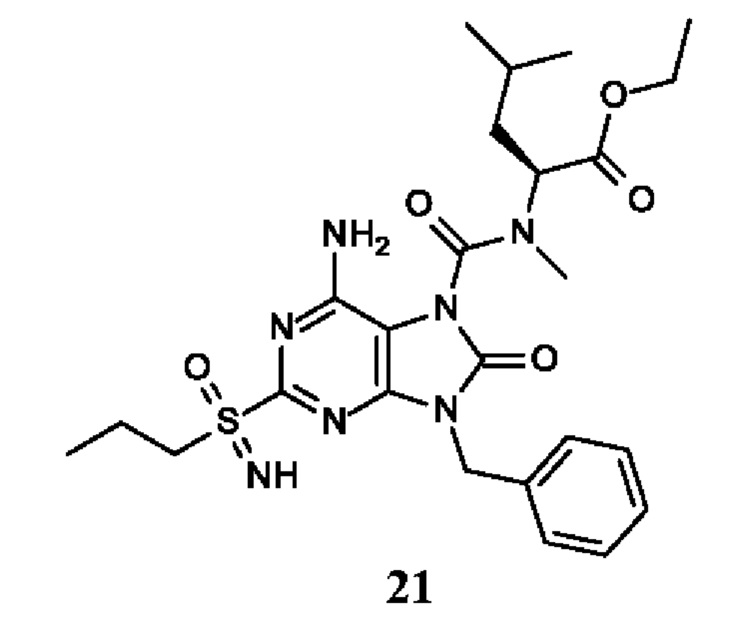
Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя этил-(2S)-2-[хлоркарбонил(метил)амино]-4-метилпентаноат (промежуточное соединение AW) вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). Этил-(2S)-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]-4-метилпентаноат (17,3 мг; соединение из примера 21) получали в виде белого порошка. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.26-7.45 (m, 5Н), 6.73 (br. s., 2Н), 4.91-5.09 (m, 3Н), 4.06-4.25 (m, 3Н), 3.34-3.45 (m, 2Н), 3.04 (br. s., 3Н), 1.93 (br. s., 1Н), 1.54-1.78 (m, 4Н), 1.22 (t, J=7,09 Гц, 3Н), 0.77-1.01 (m, 9Н). MS: обнаружено (ESI+) [(М+Н)+]: 546,3.
Пример 22
Этил-(2S)-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]-3-фенилпропаноат

Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя этил-(2S)-2-[хлоркарбонил(метил)амино]-3-фенилпропаноат (промежуточное соединение АХ) вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). Этил-(2S)-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]-3-фенилпропаноат (30 мг; соединение из примера 22) получали в виде белого порошка. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.42-7.16 (m, 10Н), 4.97 (s, 3Н), 4.19 (q, J=7,1 Гц, 2Н), 3.35-3.15 (m, 6Н), 3.10-2.90 (m, 3Н), 1.71-1.46 (m, 2Н), 1.28-1.18 (m, 4Н), 0.97-0.85 (m, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 580.
Пример 23
Изопропил-(2S)-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)-пурин-7-карбонил]-метил-амино]-3-фенилпропаноат
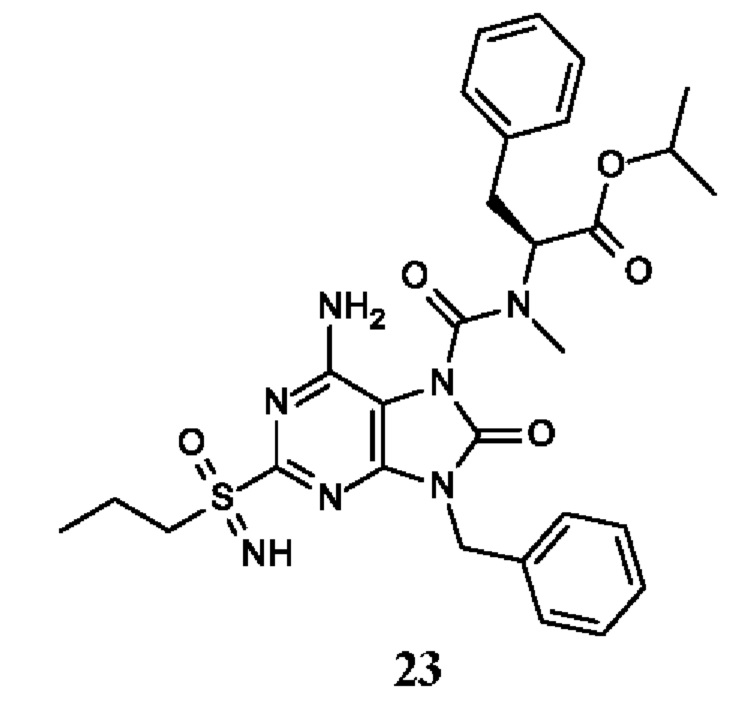
Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя изопропил-(2S)-2-[хлоркарбонил(метил)амино]-3-фенилпропаноат (промежуточное соединение AY) вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). Изопропил-(2S)-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]-3-фенилпропаноат (22 мг; соединение из примера 23) получали в виде белого порошка. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.35-7.01 (m, 10Н), 5.02-4.89 (m, 3Н), 3.37-3.17 (m, 3Н), 3.02-3.09 (m, 3Н), 3.10-2.90 (m, 3Н), 1.66-1.62 (m, 2Н), 1.22-1.11 (m, 8Н), 0.92 (t, J=7,4 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 594.
Пример 24
трет-Бутил-(2S)-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)-пурин-7-карбонил]-метил-амино]-3-фенилпропаноат
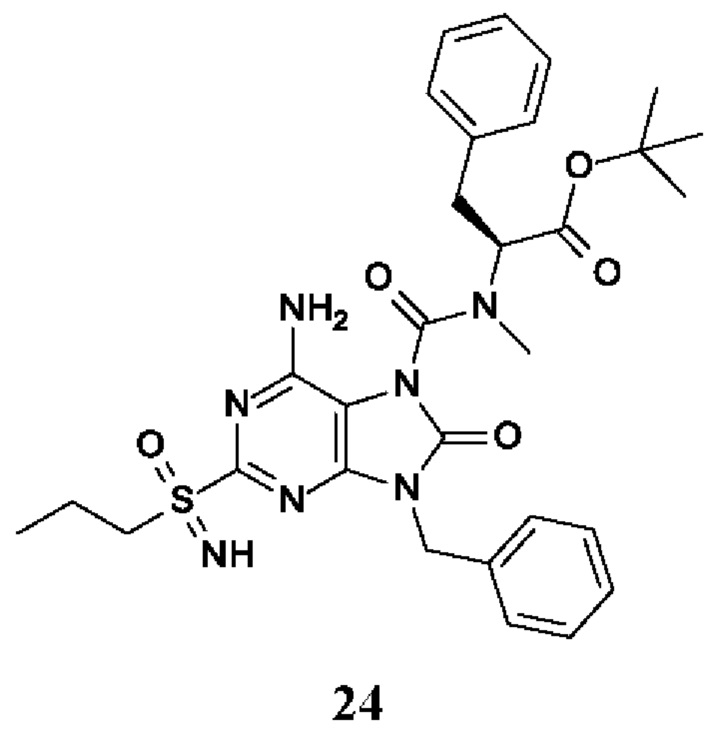
Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя трет-бутил-(2S)-2-[хлоркарбонил(метил)амино]-3-фенилпропаноат (промежуточное соединение AZ) вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). трет-Бутил-(2S)-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]-3-фенилпропаноат (34 мг; соединение из примера 24) получали в виде белого порошка. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.42-7.16 (m, 10Н), 5.03-4.90 (m, 3Н), 3.68-3.24 (m, 5Н), 3.24-3.09 (m, 2Н), 3.01 (s, 3Н), 1.68-1.57 (m, 2Н), 1.43 (s, 9Н), 0.99-0.85 (m, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 608,3.
Пример 25
N-[2-[Ацетил(метил)амино]этил]-6-амино-9-бензил-N-метил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамид
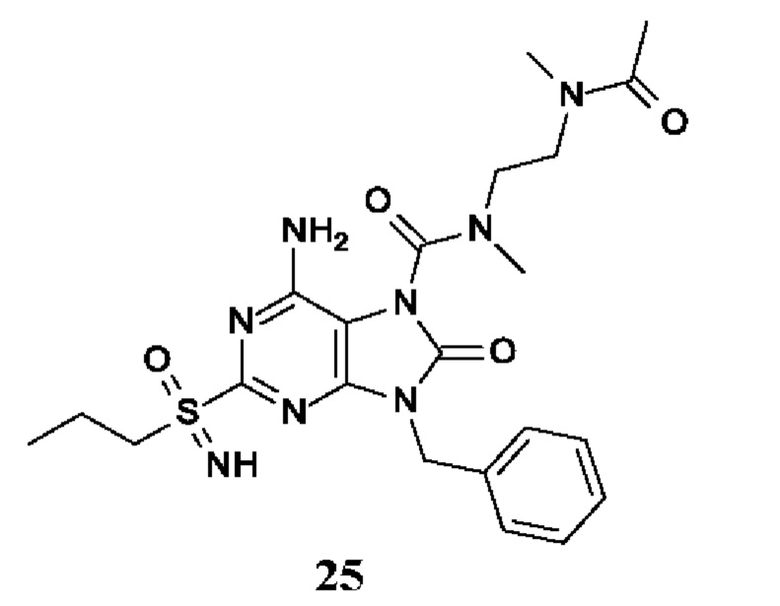
Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя N-[2-[ацетил(метил)амино]этил]-N-метил-карбамоилхлорид (промежуточное соединение ВА) вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). N-[2-[Ацетил(метил)амино]этил]-6-амино-9-бензил-N-метил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамид (26,1 мг; соединение из примера 25) получали в виде белого порошка. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.43-7.27 (m, 5Н), 7.02 (br, 2Н), 5.04-4.97 (m, 2Н), 4.19-4.13 (m, 1Н), 3.57 (d, J=5,5 Гц, 2Н), 3.49-3.34 (m, 2Н), 3.14 (s, 1Н), 3.12-3.02 (m, 4Н), 2.86 (d, J=7,5 Гц, 2Н), 2.69-2.64 (m, 1Н), 2.05 (s, 1Н), 1.99 (s, 1Н), 1.91-1.83 (m, 1Н), 1.70-1.59 (m, 2Н), 0.97-0.90 (m, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 503,2.
Пример 26
Метил-N-[2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]этил]-N-метилкарбамат
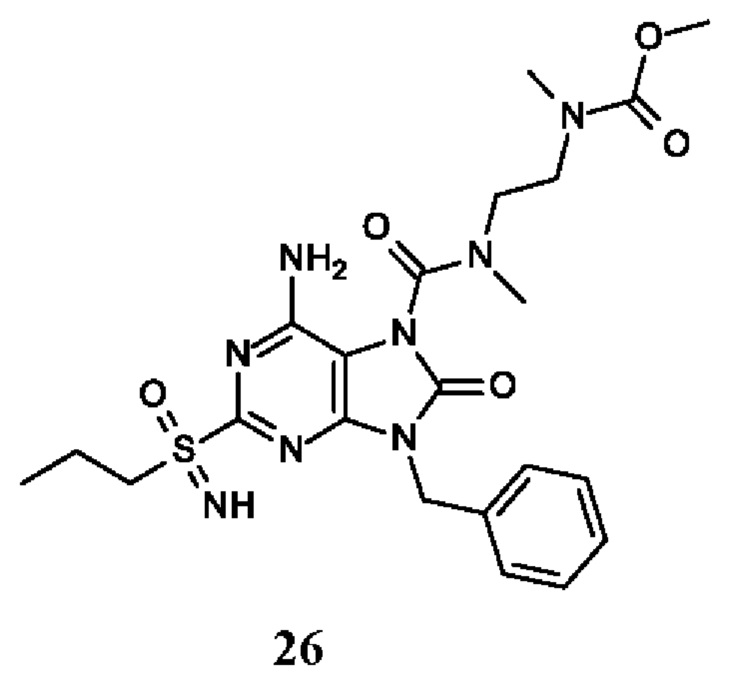
Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя метил-N-[2-[хлоркарбонил(метил)амино]этил]-N-метилкарбамат (промежуточное соединение ВВ) вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). Метил-N-[2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]этил]-N-метилкарбамат (65 мг; соединение из примера 26) получали в виде желтого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3) δ млн-1: 7.29-7.49 (m, 5Н), 5.63-5.92 (m, 2Н), 5.03-5.17 (m, 2Н), 3.43-3.69 (m, 8Н), 3.13-3.27 (m, 3Н), 2.96-3.05 (m, 2Н), 2.72 (br. s., 1Н), 1.05 (t, J=7,40 Гц, 3Н), 1.87 (dd, J=14,12; 6,96 Гц, 2Н). MS: обнаружено (ESI+) [(М+Н)+]: 519,2.
Пример 27
трет-Бутил-N-[2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)-пурин-7-карбонил]-метил-амино]этил]-N-метилкарбамат
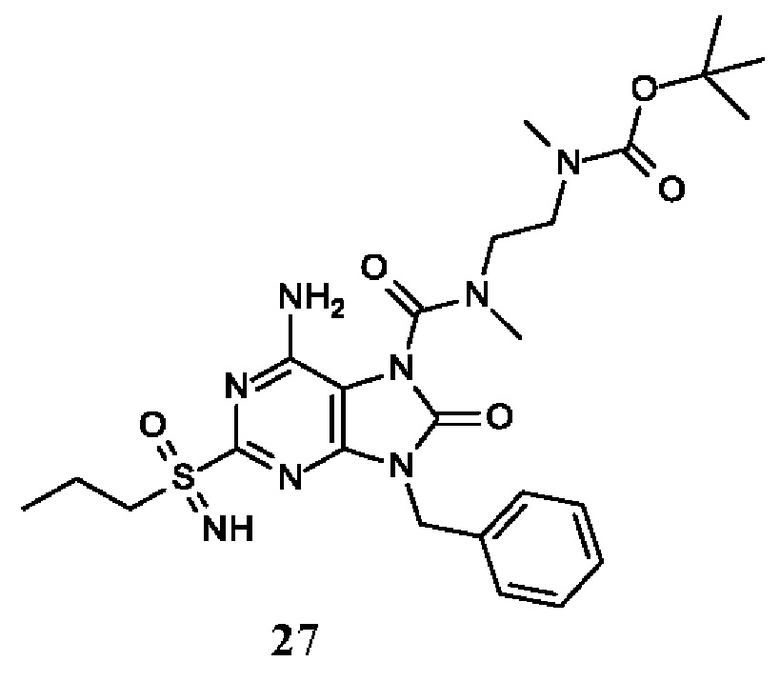
Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя трет-бутил-N-[2-[хлоркарбонил(метил)амино]этил]-N-метил-карбамат (промежуточное соединение ВС) вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). трет-Бутил-N-[2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]этил]-N-метил-карбамат (32 мг; соединение из примера 27) получали в виде белого порошка. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.43-7.26 (m, 5Н), 6.89 (br. s., 2Н), 4.99 (d, J=5,0 Гц, 2Н), 4.16 (s, 1Н), 3.55 (br. s., 2Н), 3.48-3.34 (m, 2Н), 3.10 (s, 2Н), 3.07 (s, 1Н), 2.86 (d, J=12,8 Гц, 2Н), 2.74 (d, J=9,5 Гц, 1Н), 2.70-2.60 (m, 1Н), 1.72-1.54 (m, 2Н), 1.39 (s, 6Н), 1.23 (s, 2Н), 1.13 (s, 2Н), 0.93 (t, J=7,4 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 562.
Пример 28
Этил-N-[2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]этил]-N-метилкарбамат
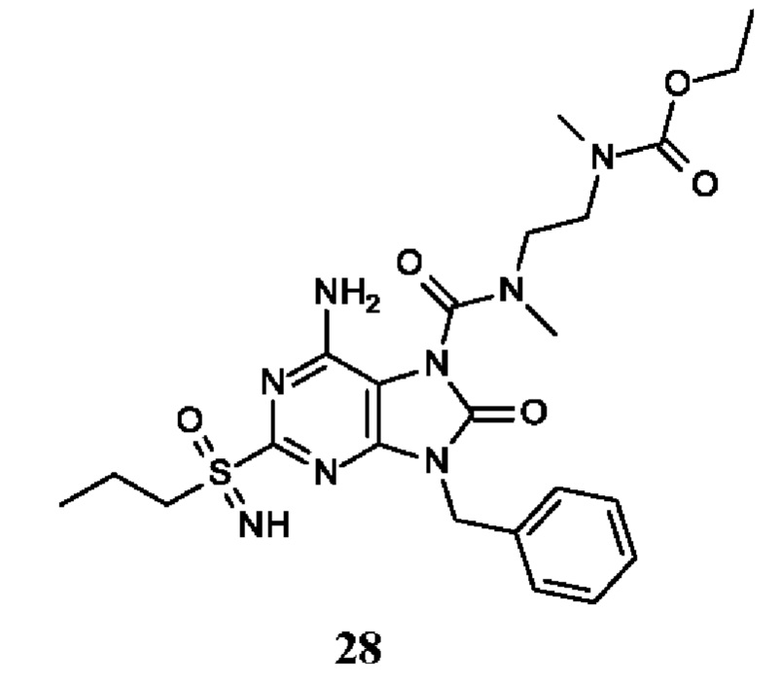
Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя этил-N-[2-[хлоркарбонил(метил)амино]этил]-N-метилкарбамат (промежуточное соединение BD) вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). Этил-N-[2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]этил]-N-метилкарбамат (87 мг; соединение из примера 28) получали в виде желтого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3) δ млн-1: 7.29-7.53 (m, 5Н), 5.65-5.90 (m, 2Н), 5.02-5.14 (m, 2Н), 3.38-4.21 (m, 9Н), 3.14-3.26 (m, 3Н), 3.00 (br. s., 2Н), 2.73 (s, 1Н), 1.76-1.99 (m, 2Н), 1.22-1.31 (m, 3Н), 1.05 (s, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 533,2.
Пример 29
2-[[6-Амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]этил-N-бутил-N-метилкарбамат
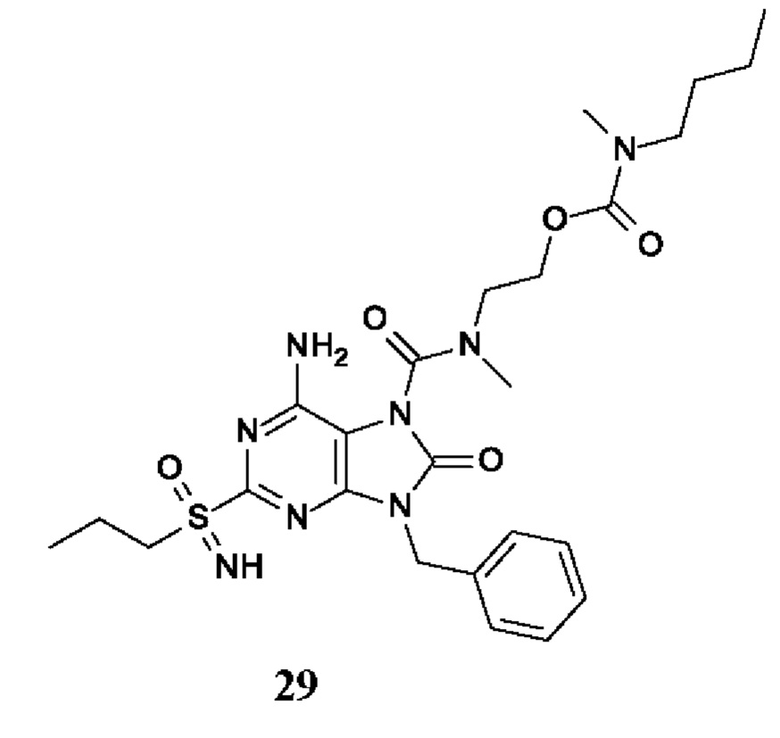
Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя 2-[хлоркарбонил(метил)амино]этил-N-бутил-N-метилкарбамат (промежуточное соединение BE) вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). 2-[[6-Амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]этил-N-бутил-N-метилкарбамат (19 мг; соединение из примера 29) получали в виде желтого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.25-7.48 (m, 5Н), 6.96 (br. s., 2Н), 4.99 (s, 2Н), 4.06-4.36 (m, 3Н), 3.59-3.83 (m, 1Н), 3.33-3.49 (m, 3Н), 3.07-3.21 (m, 4Н), 2.79 (s, 2Н), 1.65 (br. s., 2Н), 1.05-1.47 (m, 6Н), 0.93 (t, J=7,40 Гц, 3Н), 0.70-0.87 (m, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 561,2.
Пример 30
2-[[6-Амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]этил-пирролидин-1-карбоксилат

Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя 2-[хлоркарбонил(метил)амино]этил-пирролидин-1-карбоксилат (промежуточное соединение BF) вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). 2-[[6-Амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]этил-пирролидин-1-карбоксилат (10,0 мг; соединение из примера 30) получали в виде желтого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.26-7.41 (m, 5Н), 6.96 (br. s., 2Н), 4.99 (s, 2Н), 4.01-4.35 (m, 4Н), 3.29-3.47 (m, 3Н), 3.23 (br. s., 3Н), 3.03-3.17 (m, 4Н), 1.52-1.84 (m, 6Н), 0.90-0.96 (m, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 545,2.
Пример 31
2-[[6-Амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]этил-N-метил-N-пропилкарбамат
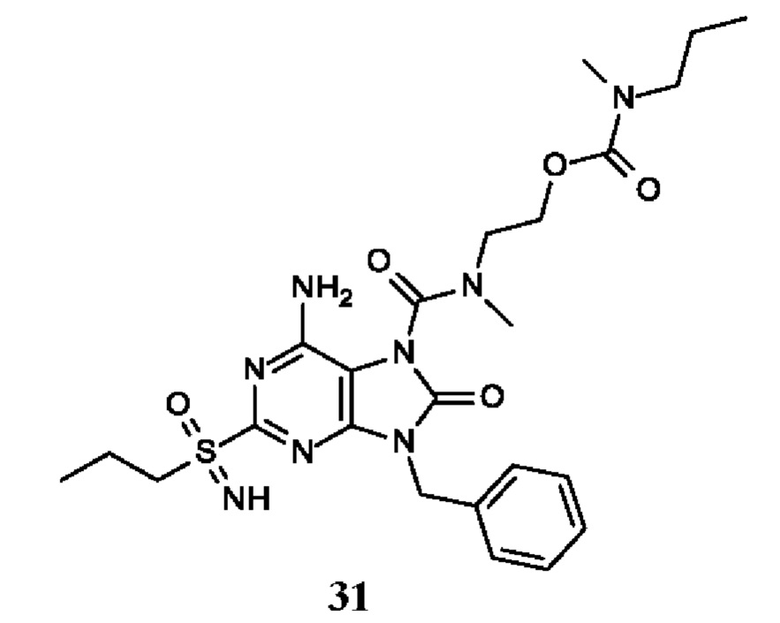
Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя 2-[хлоркарбонил(метил)амино]этил-N-метил-N-пропилкарбамат (промежуточное соединение BG) вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). 2-[[6-Амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]этил-N-метил-N-пропил-карбамат (3,7 мг; соединение из примера 31) получали в виде желтого твердого вещества. 1Н-ЯМР (400 МГц, CD3OD) δ млн-1: 7.22-7.48 (m, 5Н), 5.09-5.22 (m, 4Н), 4.55 (s, 2Н), 3.38-3.57 (m, 4Н), 3.13 (s, 3Н), 1.61-1.85 (m, 4Н), 1.22-1.41 (m, 3Н), 0.88-1.13 (m, 6Н). MS: обнаружено (ESI+) [(М+Н)+]: 547,2.
Пример 32
2-[[6-Амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]этил-N,N-диэтилкарбамат
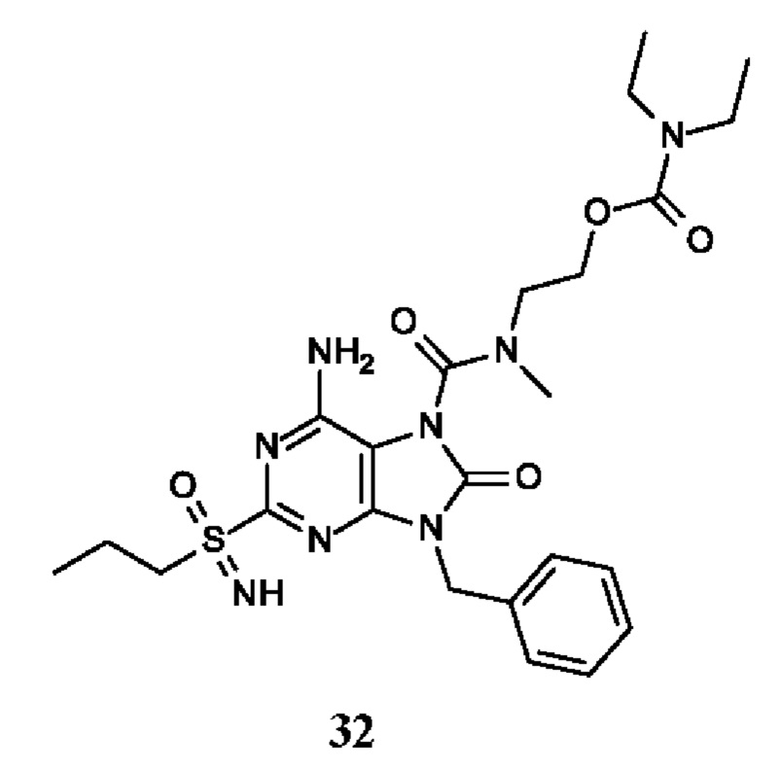
Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя 2-[хлоркарбонил(метил)амино]этил-N,N-диэтилкарбамат (промежуточное соединение ВН) вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). 2-[[6-Амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]этил-N,N-диэтилкарбамат (21,7 мг; соединение из примера 32) получали в виде желтого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.25-7.41 (m, 5Н), 6.96 (br. s., 2Н), 4.99 (s, 2Н), 4.08-4.36 (m, 3Н), 3.70 (br, 1Н), 3.33-3.46 (m, 3Н), 3.01-3.24 (m, 7Н), 1.55-1.74 (m, 2Н), 0.86-1.05 (m, 9Н). MS: обнаружено (ESI+) [(М+Н)+]: 547,2.
Пример 33
2-[[6-Амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]этил-этил карбонат
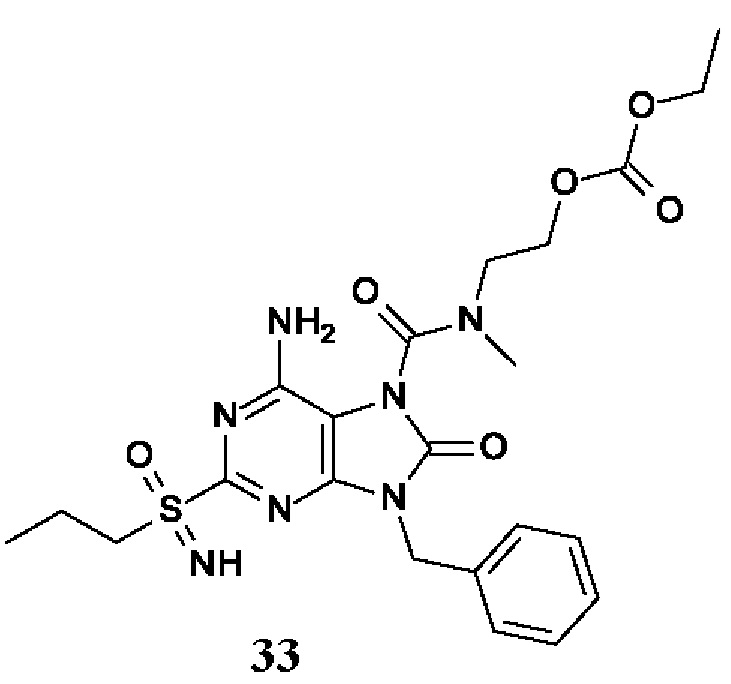
Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя 2-[хлоркарбонил(метил)амино]этил-этилкарбонат (промежуточное соединение BI) вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). 2-[[6-Амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]этил-этилкарбонат (46 мг; соединение из примера 33) получали в виде желтого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 0.82-0.99 (m, 3Н), 1.02-1.28 (m, 3Н), 1.56-1.76 (m, 2Н), 3.05-3.18 (m, 3Н), 3.35-3.48 (m, 3Н), 3.73 (t, J=5,08 Гц, 2Н), 4.08-4.27 (m, 3Н), 4.37 (br. s., 1Н), 5.00 (s, 2Н), 6.76-7.11 (m, 2Н), 7.22-7.45 (m, 5Н). MS: обнаружено (ESI+) [(М+Н)+]: 520.
Пример 34-А и пример 34-В
6-Амино-N-бутил-9-[(4-хлорфенил)метил]-N-метил-8-оксо-2-[S(S)-пропилсульфонимидоил]пурин-7-карбоксамид и 6-амино-N-бутил-9-[(4-хлорфенил)метил]-N-метил-8-оксо-2-[S(S)-пропилсульфонимидоил]пурин-7-карбоксамид
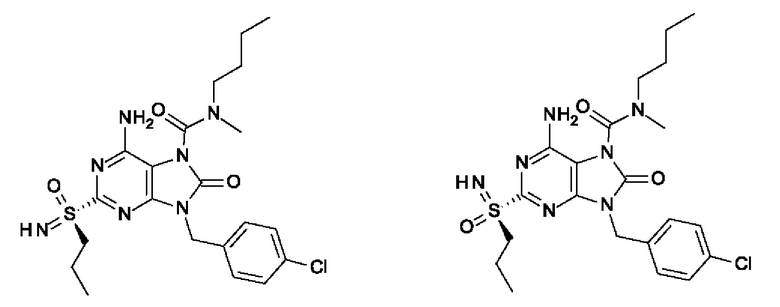
Стадия 1. Получение 4-амино-3-[(4-хлорфенил)метил]-2-оксо-1Н-имидазол-5-карбонитрила (соединения 34а)
Соединение 34а получали по аналогии со стадией 1 способа А из примера 1, используя 4-хлорбензилизоцианат вместо бензилизоцианата. 4-Амино-3-[(4-хлорфенил)метил]-2-оксо-1Н-имидазол-5-карбонитрил (8,0 г; соединение 34а) получали в виде желтого твердого вещества. MS: обнаружено (ESI+) [(М+Н)+]: 249.
Стадия 2. Получение 6-амино-9-[(4-хлорфенил)метил]-2-сульфанил-7Н-пурин-8-она (соединения 34b)
Соединение 34b получали по аналогии со стадией 2 способа А из примера 1, используя 4-амино-3-[(4-хлорфенил)метил]-2-оксо-1Н-имидазол-5-карбонитрил (соединение 34а) вместо 4-амино-3-фенилметил-2-оксо-1Н-имидазол-5-карбонитрила (соединения 1а). 6-Амино-9-[(4-хлорфенил)метил]-2-сульфанил-7Н-пурин-8-он (6,4 г; соединение 34b) получали в виде желтого твердого вещества и использовали на следующей стадии без дополнительной очистки. MS: обнаружено (ESI+) [(М+Н)+]: 308.
Стадия 3. Получение 6-амино-9-[(4-хлорфенил)метил]-2-пропилсульфанил-7Н-пурин-8-она (соединения 34с)
Соединение 34с получали по аналогии со стадией 3 способа А из примера 1, используя 6-амино-9-[(4-хлорфенил)метил]-2-сульфанил-7Н-пурин-8-он (соединение 34b) вместо 6-амино-9-фенилметил-2-сульфанил-7Н-пурин-8-она (соединения 1b). 6-Амино-9-[(4-хлорфенил)метил]-2-пропилсульфанил-7Н-пурин-8-он (800 мг; соединение 34с) получали в виде белого твердого вещества. MS: обнаружено (ESI+) [(М+Н)+]: 350.
Стадия 4. Получение 6-амино-9-[(4-хлорфенил)метил]-2-пропилсульфинил-7Н-пурин-8-она (соединения 34d)

Соединение 34d получали по аналогии со стадией 4 способа А из примера 1, используя 6-амино-9-[(4-хлорфенил)метил]-2-пропилсульфанил-7Н-пурин-8-он (соединение 34с) вместо 6-амино-9-бензил-2-пропилсульфанил-7Н-пурин-8-она (соединения 1с). 6-Амино-9-[(4-хлорфенил)метил]-2-пропилсульфинил-7Н-пурин-8-он (150 мг; соединение 34d) получали в виде белого твердого вещества. MS: обнаружено (ESI+) [(М+Н)+]: 366.
Стадия 5. Получение 6-амино-9-[(4-хлорфенил)метил]-2-(пропилсульфонимидоил)-7Н-пурин-8-она (соединения 34е), 6-амино-9-[(4-хлорфенил)метил]-2-[S(S)-пропилсульфонимидоил)-7Н-пурин-8-она и 6-амино-9-[(4-хлорфенил)метил]-2-[S(S)-пропилсульфонимидоил)-7Н-пурин-8-она (соединения 34е-А и соединения 34е-В)
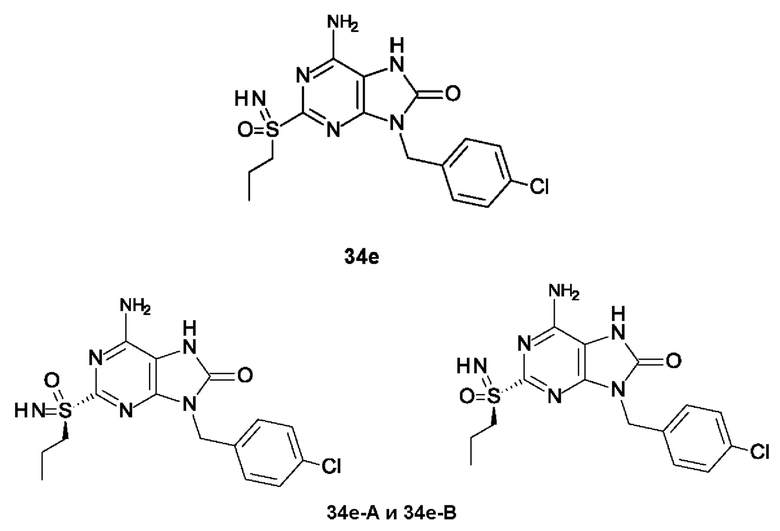
Соединение 34е получали по аналогии со стадией 5 способа А из примера 1, используя 6-амино-9-[(4-хлорфенил)метил]-2-пропилсульфинил-7Н-пурин-8-он (соединение 34d) вместо 6-амино-9-бензил-2-(2-пропилсульфинил)-7Н-пурин-8-она (соединения 1d). 6-Амино-9-[(4-хлорфенил)метил]-2-(пропилсульфонимидоил)-7Н-пурин-8-он (250 мг; соединение 34е) получали в виде белого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 10.60 (br. s, 1Н), 7.32-7.42 (m, 4Н), 6.98 (br. s, 2Н), 4.96 (s, 2Н), 4.03 (s, 1Н), 3.25-3.41 (m, 2Н), 1.56-1.68 (m, 2Н), 0.91 (t, J=8 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 381.
После разделения хиральной HPLC смеси, представляющей собой соединение 34е, получали соединение 34е-А (быстрее элюирующееся, 110 мг) и соединение 34е-В (медленнее элюирующееся, 100 мг) в виде белого твердого вещества, используя 5%→40% метанола с (0,05% DEA)/CO2 на колонке ChiralPak OJ-3.
Соединение 34е-А: 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 10.63 (br. s, 1Н), 7.33-7.42 (m, 4Н), 6.99 (br. s, 2Н), 4.96 (s, 2Н), 4.05 (br. s, 1 H), 3.26-3.39 (m, 2H), 1.53-1.69 (m, 2H), 0.91 (t, J=7,4 Гц, 3Н). MS: обнаружено (ESI+) [(M+H)+]: 381.
Соединение 34е-В: 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 10.63 (br. s, 1Н), 7.33-7.42 (m, 4Н), 6.99 (br. s, 2Н), 4.96 (s, 2Н), 4.05 (br. s, 1 H), 3.26-3.40 (m, 2H), 1.54-1.69 (m, 2H), 0.91 (t, J=7,5 Гц, 3Н). MS: обнаружено (ESI+) [(M+H)+]: 381.
Стадия 6. 6-Амино-N-бутил-9-[(4-хлорфенил)метил]-N-метил-8-оксо-2-[S(S)-пропилсульфонимидоил]пурин-7-карбоксамид и 6-амино-N-бутил-9-[(4-хлорфенил)метил]-N-метил-8-оксо-2-[S(S)-пропилсульфонимидоил]пурин-7-карбоксамид (соединение из примера 34-А и соединение из примера 34-В)
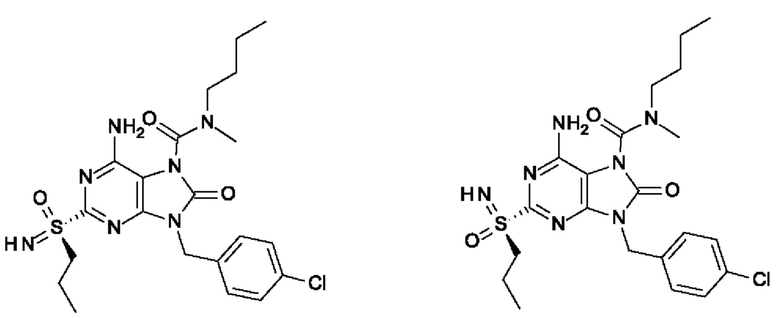
Соединение из примера 34-А получали по аналогии со стадией 6 способа А из примера 1, используя соединение 34е-А и N-бутил-N-метил-карбамоилхлорид вместо 6-амино-9-бензил-2-(пропилсульфонимидоил)-7Н-пурин-8-она (соединения 1е) и N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА).
Соединение из примера 34-А (160 мг): 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.37-7.45 (m, 4Н), 6.91 (br. s., 2Н), 4.99 (s, 2Н), 4.17 (s, 1Н), 3.28-3.40 (m, 4Н), 3.05 (s, 2Н), 3.02 (s, 1Н), 1.49-1.70 (m, 4Н), 1.15-1.37 (m, 2Н), 0.89-0.94 (m, 5Н), 0.76 (t, J=8 Гц, 1Н). MS: обнаружено (ESI+) [(М+Н)+]: 494.
Соединение из примера 34-В (167 мг) получали по аналогии с соединением из примера 34-А, используя соединение 34е-В вместо соединения 34е-А.
Соединение из примера 34-В: 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.36-7.45 (m, 4Н), 6.91 (br. s., 2Н), 4.99 (s, 2Н), 4.17 (s, 1Н), 3.28-3.41 (m, 4Н), 3.05 (s, 2Н), 3.02 (s, 1Н), 1.50-1.71 (m, 4Н), 1.15-1.37 (m, 2Н), 0.89-0.94 (m, 5Н), 0.76 (t, J=7,4 Гц, 1Н). MS: обнаружено (ESI+) [(М+Н)+]: 494.
Пример 35
6-Амино-9-[(4-хлорфенил)метил]-N-этил-N-метил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамид
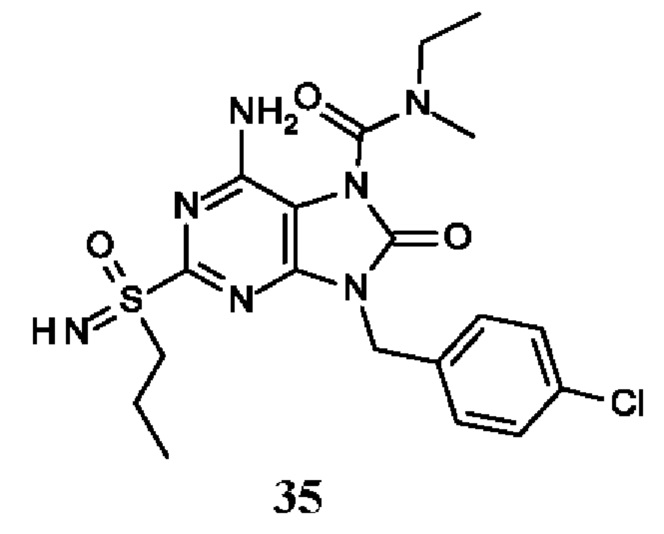
Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя 6-амино-9-[(4-хлорфенил)метил]-2-(пропилсульфонимидоил)-7Н-пурин-8-он (соединение 34е) и N-этил-N-метил-карбамоилхлорид вместо 6-амино-9-бензил-2-(пропилсульфонимидоил)-7Н-пурин-8-она (соединения 1е) и N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). 6-Амино-9-[(4-хлорфенил)метил]-N-этил-N-метил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамид (60 мг; соединение из примера 35) получали в виде белого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.40 (s, 4Н), 6.91 (br s, 2Н), 4.99 (s, 2Н), 4.16 (s, 1Н), 3.34-3.44 (m, 4Н), 3.05 (s, 2Н), 3.01 (s, 1Н), 1.58-1.67 (m, 2Н), 1.18 (t, J=8,0 Гц, 3Н), 0.92 (t, J=8,0 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 466.
Пример 36-А и пример 36-В
6-Амино-N-метил-8-оксо-N-пропил-2-[S(S)-пропилсульфонимидоил]-9-(п-толилметил)пурин-7-карбоксамид и 6-амино-N-метил-8-оксо-N-пропил-2-[S(R)-пропилсульфонимидоил]-9-(п-толилметил)пурин-7-карбоксамид
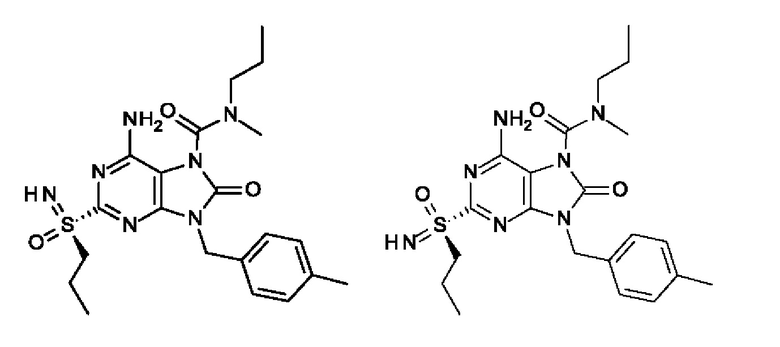
Стадия 1. Получение 6-хлор-5-нитро-2-пропилсульфанил-N-(п-толилметил)пиримидин-4-амина (соединения 36а)
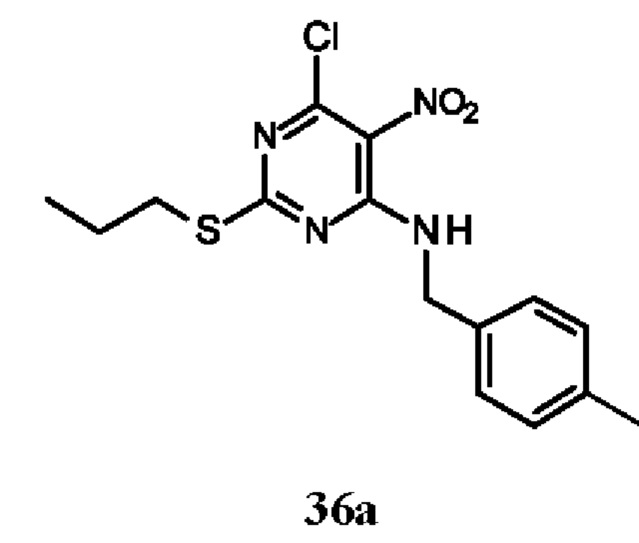
Соединение 36а получали по аналогии со стадией 1 способа В из примера 1, используя п-толилметиламин вместо фенилметанамина. 6-Хлор-5-нитро-2-пропилсульфанил-N-(п-толилметил)пиримидин-4-амин (3,9 г; соединение 36а) получали в виде белого твердого вещества. MS: обнаружено (ESI+) [(М+Н)+]: 353.
Стадия 2. Получение 6-хлор-2-пропилсульфанил-N-4-(п-толилметил)пиримидин-4,5-диамина (соединения 36b)
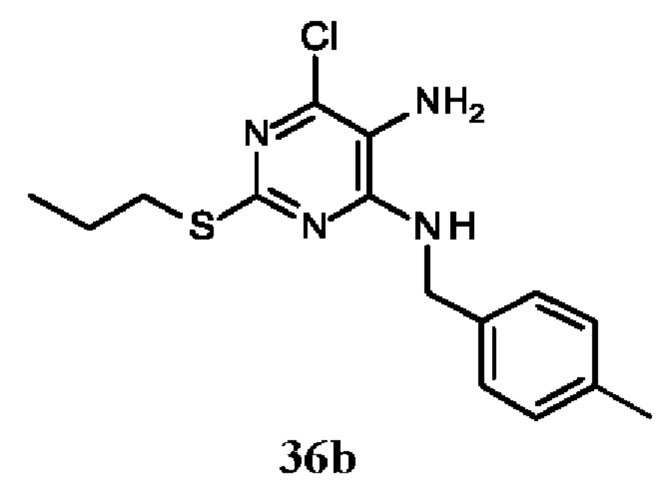
Соединение 36b получали по аналогии со стадией 2 способа В из примера 1, используя 6-хлор-5-нитро-2-пропилсульфанил-N-(п-толилметил)пиримидин-4-амин (соединение 36а) вместо N-бензил-6-хлор-5-нитро-2-пропилсульфанил-пиримидин-4-амина (соединения 1f). 6-Хлор-2-пропилсульфанил-N4-(п-толилметил)пиримидин-4,5-диамин (2,2 г; соединение 36b) получали в виде белого твердого вещества. MS: обнаружено (ESI+) [(М+Н)+]: 323.
Стадия 3. Получение 6-хлор-2-пропилсульфанил-9-(п-толилметил)-7Н-пурин-8-она (соединения 36с)
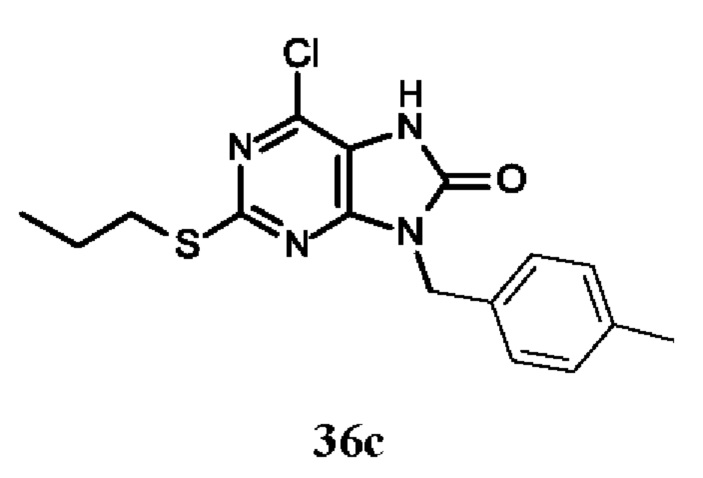
Соединение 36с получали по аналогии со стадией 3 способа В из примера 1, используя 6-хлор-2-пропилсульфанил-N-4-(п-толилметил)пиримидин-4,5-диамин (соединение 36b) вместо N-бензил-6-хлор-2-(пропилсульфанил)пиримидин-4,5-диамина (соединения 1g). 6-Хлор-2-пропилсульфанил-9-(п-толилметил)-7Н-пурин-8-он (2,2 г; соединение 36с) получали в виде белого твердого вещества. MS: обнаружено (ESI+) [(М+Н)+]: 349.
Стадия 4. Получение 6-[(4-метоксифенил)метиламино]-2-пропилсульфанил-9-(п-толилметил)-7Н-пурин-8-она (соединения 36d)
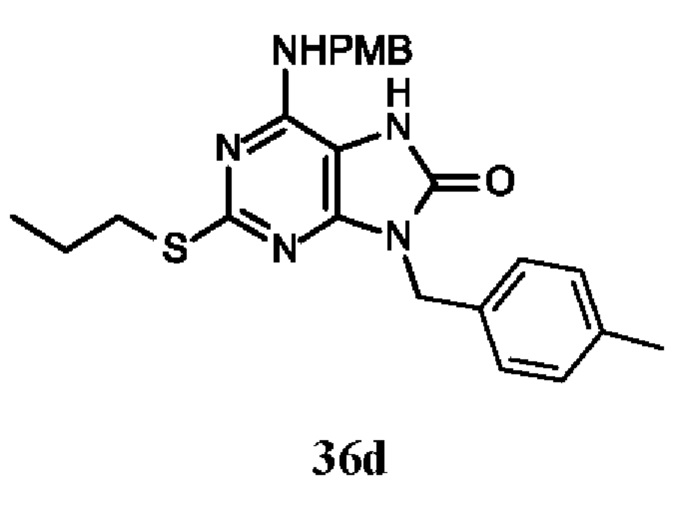
Соединение 36d получали по аналогии со стадией 4 способа В из примера 1, используя 6-хлор-2-пропилсульфанил-9-(п-толилметил)-7Н-пурин-8-он (соединение 36с) вместо 9-бензил-6-хлор-2-пропилсульфанил-7Н-пурин-8-она (соединения 1h). 6-[(4-Метоксифенил)метиламино]-2-пропилсульфанил-9-(п-толилметил)-7Н-пурин-8-он (2,0 г; соединение 36d) получали в виде белого твердого вещества. MS: обнаружено (ESI+) [(М+Н)+]: 450.
Стадия 5. Получение 6-амино-2-пропилсульфанил-9-(п-толилметил)-7Н-пурин-8-она (соединения 36е)
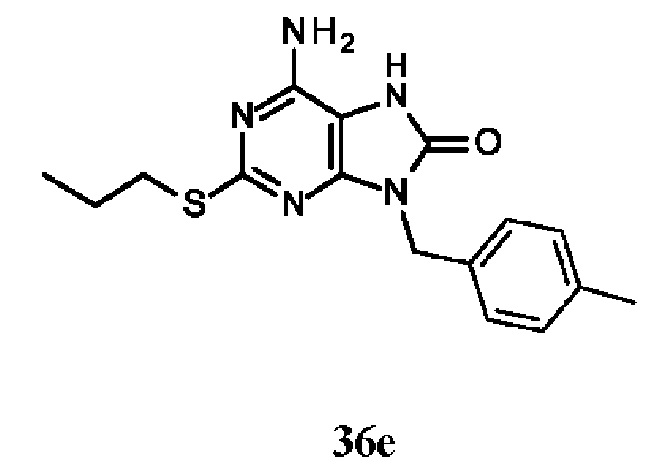
Соединение 36е получали по аналогии со стадией 5 способа В из примера 1, используя 6-[(4-метоксифенил)метиламино]-2-пропилсульфанил-9-(п-толилметил)-7Н-пурин-8-он (соединение 36d) вместо 6-амино-9-бензил-2-пропилсульфанил-7Н-пурин-8-она (соединения 1i). 6-Амино-2-пропилсульфанил-9-(п-толилметил)-7Н-пурин-8-он (1,0 г; соединение 36е) получали в виде белого твердого вещества. MS: обнаружено (ESI+) [(М+Н)+]: 330.
Стадия 6. Получение 6-амино-2-пропилсульфинил-9-(п-толилметил)-7Н-пурин-8-она (соединения 36f)
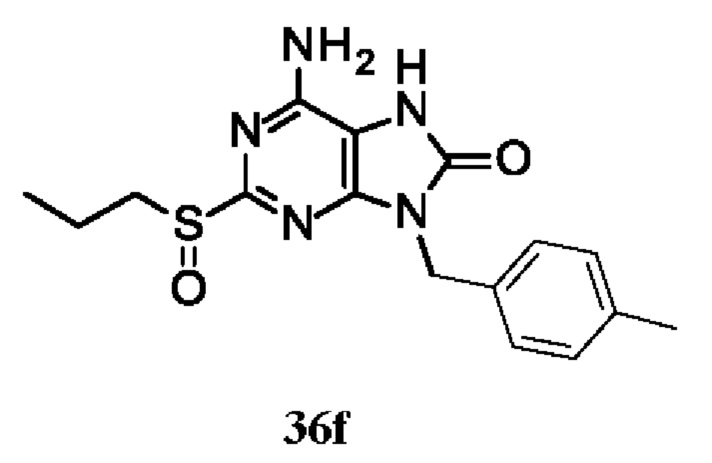
Соединение 36f получали по аналогии со стадией 6 способа В из примера 1, используя 6-амино-2-пропилсульфанил-9-(п-толилметил)-7Н-пурин-8-он (соединение 36е) вместо 6-амино-9-бензил-2-(2-пропилсульфанил)-7Н-пурин-8-она (соединения 1с). 6-Амино-2-пропилсульфинил-9-(п-толилметил)-7Н-пурин-8-он (220 мг; соединение 36f) получали в виде белого твердого вещества. MS: обнаружено (ESI+) [(М+Н)+]: 345.
Стадия 7. Получение 6-амино-2-(пропилсульфонимидоил)-9-(п-толилметил)-7Н-пурин-8-она (соединения 36g)
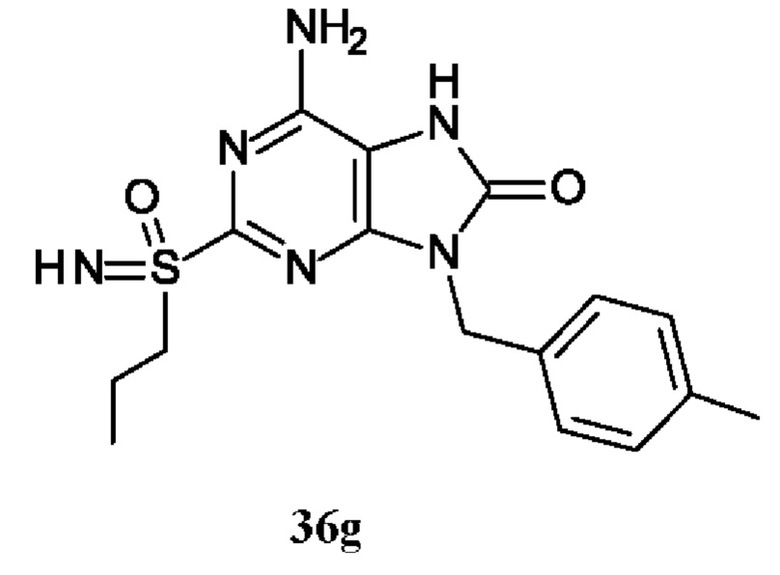
Соединение 36g получали по аналогии со стадией 7 способа В из примера 1, используя 6-амино-2-пропилсульфинил-9-(п-толилметил)-7Н-пурин-8-он (соединение 36f) вместо 6-амино-9-бензил-2-пропилсульфинил-7Н-пурин-8-она (соединения 1d). 6-Амино-2-(пропилсульфонимидоил)-9-(п-толилметил)-7Н-пурин-8-он (127 мг; соединение 36g) получали в виде белого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 10.67 (br. s., 1Н), 7.23 (d, J=8,0 Гц, 2Н), 7.13 (d, J=8,0 Гц, 2Н), 6.98 (br. s., 2Н), 4.91 (s, 2Н), 4.05 (s, 1Н), 3.34-3.27 (m, 2Н), 2.26 (s, 3Н), 1.67-1.62 (m, 2Н), 0.92 (t, J=8,0 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 361.
После разделения соединения 36g хиральной HPLC получали соединение 36g-А (быстрее элюирующееся; 50 мг) и соединение 36g-В (медленнее элюирующееся; 49 мг) в виде белого твердого вещества, используя 30%-ный изопропанол с (0,05% DEA)/CO2 на колонке ChiralPak AD-3.
Соединение 36g-А: 1Н-ЯМР: (400 МГц, DMSO-d6) δ млн-1: 10.51 (s, 1Н), 7.22 (d, J=8,0 Гц, 2Н), 7.12 (d, J=8,0 Гц, 2Н), 7.00 (s, 2 Н), 4.91 (s, 2Н), 4.03 (s, 1Н), 3.35-3.31 (m, 2Н), 2.26 (s, 3Н), 1.70-1.58 (m, 2Н), 0.93 (t, J=7,40 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 361.
Соединение 36g-В: 1Н-ЯМР: (400 МГц, DMSO-d6) δ млн-1: 10.54 (s, 1Н), 7.23 (d, J=8,0 Гц, 2Н), 7.13 (d, J=8,0 Гц, 2Н), 6.97 (s, 2Н), 4.91 (s, 2Н), 4.04 (s, 1Н), 3.34-3.30 (m, 2Н), 2.26 (s, 3Н), 1.72-1.57 (m, 2Н), 0.93 (t, J=7,40 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 361.
Стадия 8. Получение 6-амино-N-метил-8-оксо-N-пропил-2-[S(S)-пропилсульфонимидоил]-9-(п-толилметил)пурин-7-карбоксамида и 6-амино-N-метил-8-оксо-N-пропил-2-[S(R)-пропилсульфонимидоил]-9-(п-толилметил)пурин-7-карбоксамида (соединения из примера 36-А и соединения из примера 36-В)
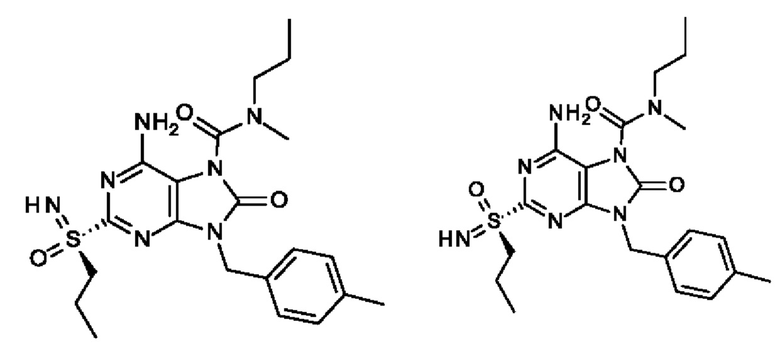
Соединение из примера 36-А получали по аналогии со стадией 6 способа А из примера 1, используя соединение 36g-А вместо 6-амино-9-бензил-2-(пропилсульфонимидоил)-7Н-пурин-8-она (соединения 1е). Соединение из примера 36-А (108 мг) получали в виде белого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.27 (d, J=8 Гц, 2Н), 7.14 (d, J=8 Гц, 2Н), 6.87 (br. s., 2Н), 4.95 (s, 2Н), 4.15 (s, 1Н), 3.33-3.57 (m, 4Н), 3.05 (s, 2Н), 3.02 (s, 1Н), 2.26 (s, 3Н), 1.52-1.73 (m, 4Н), 0.75-0.97 (m, 6Н). MS: обнаружено (ESI+) [(М+Н)+]: 460.
Соединение из примера 36-В получали по аналогии со стадией 6 способа А из примера 1, используя соединение 36g-В вместо 6-амино-9-бензил-2-(пропилсульфонимидоил)-7Н-пурин-8-она (соединения 1е). Соединение из примера 36-В (125 мг): 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.27 (d, J=8 Гц, 2Н), 7.14 (d, J=8 Гц, 2Н), 6.87 (br. s., 2Н), 4.95 (s, 2Н), 4.15 (s, 1Н), 3.33-3.57 (m, 4Н), 3.05 (s, 2Н), 3.02 (s, 1Н), 2.26 (s, 3Н), 1.52-1.73 (m, 4Н), 0.75-0.97 (m, 5Н). MS: обнаружено (ESI+) [(М+Н)+]: 460.
Пример 37-А и пример 37-В
6-Амино-2-[S(S)-пропилсульфонимидоил]-9-(п-толилметил)-7-(пирролидин-1-карбонил)пурин-8-он и 6-амино-2-[S(R)-пропилсульфонимидоил]-9-(п-толилметил)-7-(пирролидин-1-карбонил)пурин-8-он
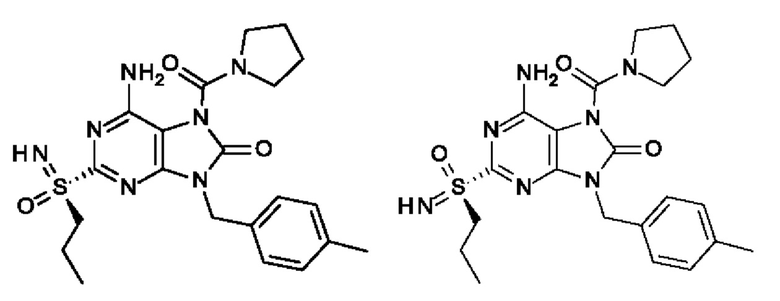
Соединение из примера 37-А получали по аналогии со стадией 6 способа А из примера 1, используя соединение 36g-А и пирролидин-1-карбонилхлорид вместо 6-амино-9-бензил-2-(пропилсульфонимидоил)-7Н-пурин-8-она (соединения 1е) и N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА).
Соединение из примера 37-А (390 мг) получали в виде белого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.31-7.11 (m, 4Н), 7.04 (s, 2Н), 4.95 (s, 2Н), 4.15 (s, 1Н), 3.65-3.47 (m, 4Н), 3.37 (m, 2Н), 2.27 (s, 3Н), 1.97-1.81 (m, 4Н), 1.71-1.59 (m, 2Н), 0.94 (t, J=7,4 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 458,2.
Соединение из примера 37-В (125 мг) получали по аналогии с соединением из примера 37-А, используя соединение 36g-В вместо соединения 36g-А. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.28-7.14 (m, 4Н), 7.04 (s, 2Н), 4.95 (s, 2Н), 4.15 (s, 1Н), 3.65-3.47 (m, 4Н), 3.37 (m, 2Н), 2.27 (s, 3Н), 1.93-1.84 (m, 4Н), 1.65-1.60 (m, 2Н), 0.95 (t, J=7,4 Гц, 3Н). MS: обнаружено ESI+ [(М+Н)+]: 458,3.
Пример 38-А и пример 38-В
6-Амино-N-(2-метоксиэтил)-N-метил-8-оксо-2-[S(S)-пропилсульфонимидоил]-9-(п-толилметил)пурин-7-карбоксамид и 6-амино-N-(2-метоксиэтил)-N-метил-8-оксо-2-[S(R)-пропилсульфонимидоил]-9-(п-толилметил)пурин-7-карбоксамид
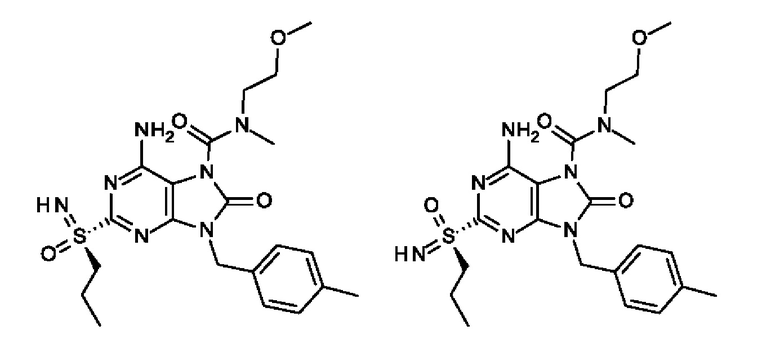
Соединение из примера 38-А получали по аналогии со стадией 6 способа А из примера 1, используя соединение 36g-А и N-(2-метоксиэтил)-N-метил-карбамоилхлорид (промежуточное соединение АВ) вместо 6-амино-9-бензил-2-(пропилсульфонимидоил)-7Н-пурин-8-она (соединения 1е) и N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА).
Соединение из примера 38-А (57,8 мг) получали в виде белого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.26 (d, J=7,6 Гц, 2Н), 7.14 (d, J=7,6 Гц, 2Н), 6.89-6.78 (m, 2Н), 4.95 (s, 2Н), 4.18 (s, 1Н), 3.62-3.58 (m, 2Н), 3.43-3.37 (m, 2Н), 3.30-3.10 (m, 3Н), 3.09-3.08 (m, 3Н), 3.08-3.05 (m, 2Н), 2.27 (s, 3Н), 1.77-1.54 (m, 2Н), 0.95 (t, J=7,4 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 476,3.
Соединение из примера 38-В (46,6 мг) получали по аналогии с соединением из примера 38-А, используя соединение 36g-В вместо соединения 36g-А. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.26 (d, J=7,6 Гц, 2Н), 7.14 (d, J=7,6 Гц, 2Н), 6.89-6.78 (m, 2Н), 4.95 (s, 2Н), 4.18 (s, 1Н), 3.62-3.58 (m, 2Н), 3.43-3.37 (m, 2Н), 3.30-3.10 (m, 3Н), 3.09-3.08 (m, 3Н), 3.08-3.05 (m, 2Н), 2.27 (s, 3Н), 1.77-1.54 (m, 2Н), 0.95 (t, J=7,4 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 476,3.
Пример 39
6-Амино-N-этил-N-метил-8-оксо-2-(пропилсульфонимидоил)-9-(п-толилметил)пурин-7-карбоксамид
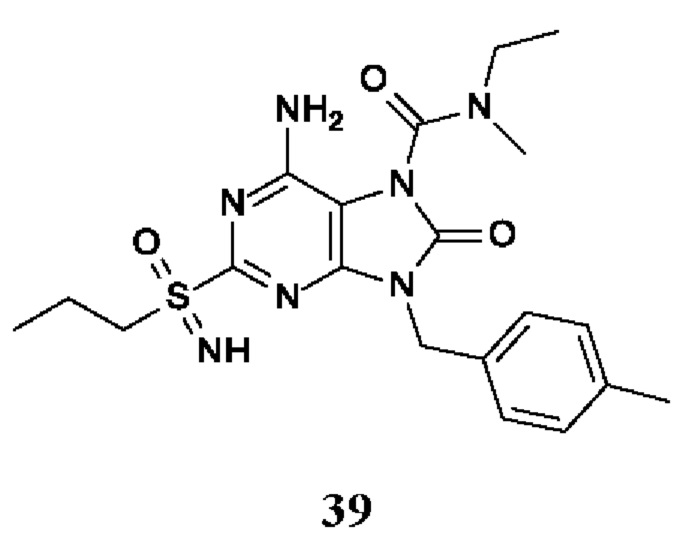
Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя N-этил-N-метил-карбамоилхлорид и 6-амино-2-(пропилсульфонимидоил)-9-(п-толилметил)-7Н-пурин-8-он (соединение 36g) вместо N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА) и 6-амино-9-бензил-2-(пропилсульфонимидоил)-7Н-пурин-8-она (соединения 1е). 6-Амино-N-этил-N-метил-8-оксо-2-(пропилсульфонимидоил)-9-(п-толилметил)-пурин-7-карбоксамид (141,8 мг; соединение из примера 39) получали в виде светло-желтого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.26 (d, J=7,9 Гц, 2Н), 7.15 (d, J=7,9 Гц, 2Н), 6.89 (s, 2Н), 4.95 (s, 2Н), 4.24-4.07 (m, 1Н), 3.52-3.35 (m, 4H), 3.10-2.95 (m, 3Н), 2.26 (s, 3Н), 1.77-1.55 (m, 2H), 1.24-1.10 (m, 3Н), 0.95 (t, J=7,4 Гц, 3Н). MS: обнаружено ESI+ [(M+H)+]: 446,1.
Пример 40
6-Амино-N-бутил-N-метил-8-оксо-2-(пропилсульфонимидоил)-9-(п-толилметил)пурин-7-карбоксамид
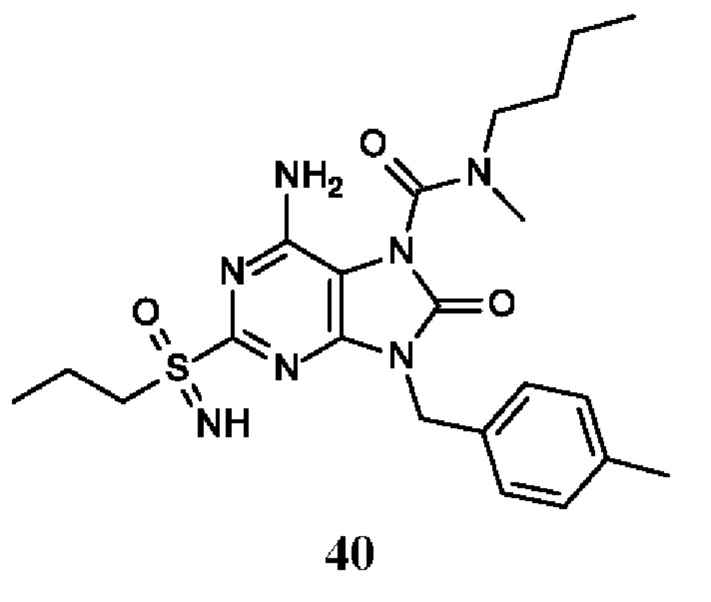
Указанное в заголовке соединение получали по аналогии со стадией 6 способа А из примера 1, используя 6-амино-2-(пропилсульфонимидоил)-9-(п-толилметил)-7Н-пурин-8-он (соединение 36g) и N-бутил-N-метил-карбамоилхлорид вместо 6-амино-9-бензил-2-(пропилсульфонимидоил)-7Н-пурин-8-она (соединения 1е) и N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА).
6-Амино-N-бутил-N-метил-8-оксо-2-(пропилсульфонимидоил)-9-(п-толилметил)-пурин-7-карбоксамид (32 мг; пример 40) получали в виде белого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.28-7.14 (m, 4Н), 6.88 (s, 2Н), 4.95 (s, 2Н), 4.16 (s, 1Н), 3.41-3.36 (m, 2Н), 3.10-2.99 (m, 3Н), 2.53-2.51 (m, 2Н), 2.27 (s, 3Н), 1.71-1.63 (m, 2Н), 1.62-1.51 (m, 2Н), 1.42-1.26 (m, 2Н), 0.97-0.74 (m, 6Н). MS: обнаружено (ESI+) [(М+Н)+]: 474,3.
Пример 41-А и пример 41-В
6-Амино-9-[(4-хлорфенил)метил]-2-[S(R)-этилсульфонимидоил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамид (соединение из примера 41-А) и 6-амино-9-[(4-хлорфенил)метил]-2-[S(S)-этилсульфонимидоил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамид (соединение из примера 41-В)
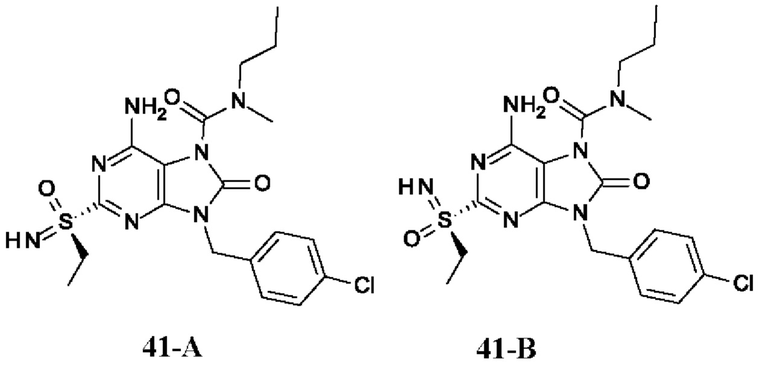
Стадия 1. Получение 6-амино-9-[(4-хлорфенил)метил]-2-этилсульфанил-7Н-пурин-8-она (соединения 41а)
Соединение 41а получали по аналогии со стадией 3 способа А из примера 1, используя иодэтан и 6-амино-9-[(4-хлорфенил)метил]-2-сульфанил-7Н-пурин-8-он (соединение 34b) вместо бромпропана и 6-амино-9-фенилметил-2-сульфанил-7Н-пурин-8-она (соединения 1b). 6-Амино-9-[(4-хлорфенил)метил]-2-этилсульфанил-7Н-пурин-8-он (2,5 г; соединение 41а) получали в виде белого твердого вещества. MS: обнаружено (ESI+) [(М+Н)+]: 336.
Стадия 2. Получение 6-амино-9-(4-хлорбензил)-2-этилсульфинил-7Н-пурин-8-она (соединения 41b)
Соединение 41b получали по аналогии со стадией 4 способа А из примера 1, используя 6-амино-9-[(4-хлорфенил)метил]-2-этилсульфанил-7Н-пурин-8-он (соединение 41а) вместо 6-амино-9-бензил-2-пропилсульфанил-7Н-пурин-8-она (соединения 1с). 6-Амино-9-(4-хлорбензил)-2-этилсульфинил-7Н-пурин-8-он (1,94 г; соединение 41b) получали в виде белого твердого вещества. MS: обнаружено (ESI+) [(М+Н)+]: 352.
Стадия 3. Получение 6-амино-9-[(4-хлорфенил)метил]-2-(этилсульфонимидоил)-7Н-пурин-8-она (соединения 41с)
Соединение 41с получали по аналогии со стадией 4 способа А из примера 1, используя 6-амино-9-(4-хлорбензил)-2-этилсульфинил-7Н-пурин-8-он (соединение 41b) вместо 6-амино-9-бензил-2-(2-метилсульфинил)-7Н-пурин-8-она (соединения 1d). 6-Амино-9-[(4-хлорфенил)метил]-2-(этилсульфонимидоил)-7Н-пурин-8-он (217 мг; соединение из примера 41с) получали в виде белого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 10.61 (s, 1Н), 7.42-7.35 (m, 4Н), 6.98 (s, 2Н), 4.96 (s, 2Н), 4.05 (s, 1Н), 3.42-3.37 (m, 2Н), 1.16 (t, J=7,4 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 367,0.
После разделения хиральной HPLC смеси, представляющей собой соединение 41с, получали соединение 41с-А (быстрее элюирующееся; 31,8 мг) и соединение 41с-В (медленнее элюирующееся; 10 мг) в виде белого твердого вещества, используя 5%→40% метанола с (0,05% DEA)/CO2 на колонке ChiralPak IC-3.
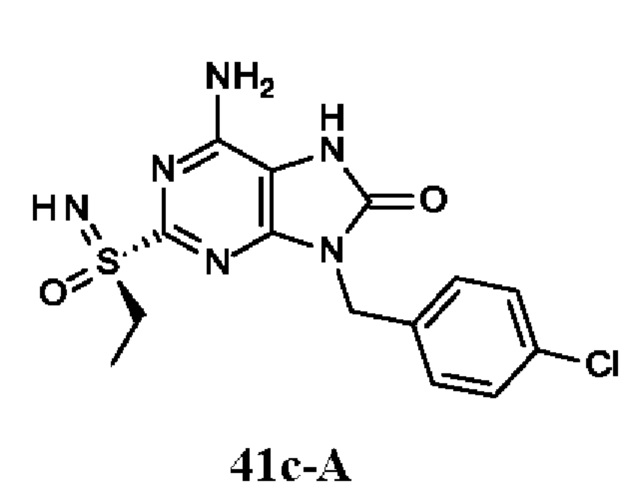
Соединение 41с-А: 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 10.76 (s, 1Н), 7.45-7.33 (m, 4Н), 7.01 (s, 2Н), 4.96 (s, 2Н), 4.03 (s, 1Н), 3.40-3.34 (m, 2Н), 1.17 (t, J=7,4 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 367,0.
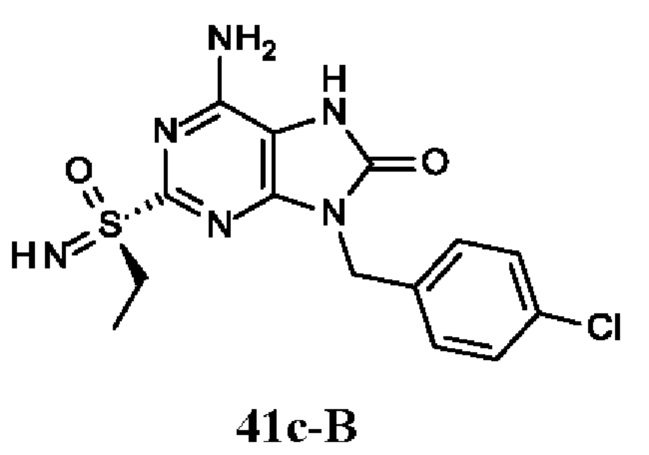
Соединение 41с-В: 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 10.70 (s, 1Н), 7.46-7.28 (m, 4Н), 7.01 (s, 2Н), 4.96 (s, 2Н), 4.03 (s, 1Н), 3.44-3.36 (m, 2Н), 1.17 (t, J=7,4 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 367,0.
Стадия 4. 6-Амино-9-[(4-хлорфенил)метил]-2-[S(R)-этилсульфонимидоил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамид (соединение из примера 41-А) и 6-амино-9-[(4-хлорфенил)метил]-2-[S(S)-этилсульфонимидоил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамид (соединение из примера 41-В)
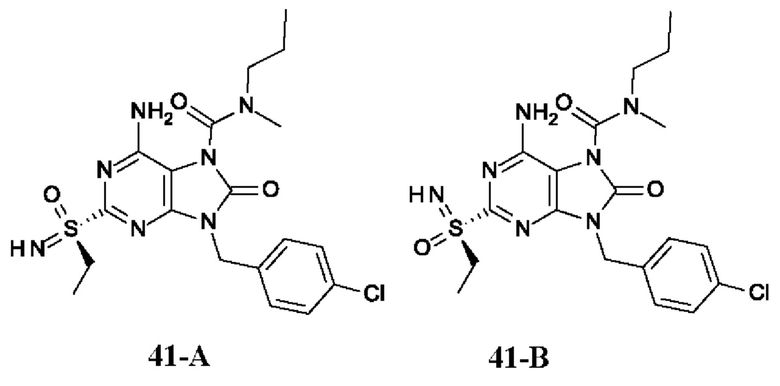
Соединение из примера 41-А получали по аналогии со стадией 6 способа А из примера 1, используя соединение 41с-В вместо 6-амино-9-бензил-2-(пропилсульфонимидоил)-7Н-пурин-8-она (соединения 1е). 6-Амино-9-[(4-хлорфенил)метил]-2-[S(R)-этилсульфонимидоил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамид (соединение из примера 41-А, 78 мг) получали в виде белого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.43-7.41 (m, 4Н), 6.90 (s, 2Н), 5.00 (s, 2Н), 4.19 (s, 1Н), 3.46-3.39 (m, 2Н), 3.39-3.38 (m, 2Н), 3.09-2.99 (m, 3Н), 1.69-1.52 (m, 2Н), 1.19 (t, J=7,28 Гц, 3Н), 0.95-0.66 (m, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 466,1.
Соединение из примера 41-В (125 мг) получали по аналогии со стадией 6 способа А из примера 1, используя соединение 41с-А вместо 6-амино-9-бензил-2-(пропилсульфонимидоил)-7Н-пурин-8-она (соединения 1е). 6-Амино-9-[(4-хлорфенил)метил]-2-[S(S)-этилсульфонимидоил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамид (соединение из примера 41-В, 38 мг) получали в виде белого твердого вещества. 1H-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.43-7.41 (m, 4H), 6.90 (s, 2H), 5.00 (s, 2H), 4.20 (s, 1H), 3.46-3.41 (m, 2H), 3.40-3.39 (m, 2H), 3.10-3.00 (m, 3Н), 1.69-1.50 (m, 2H), 1.24-1.12 (m, 3Н), 0.93-0.73 (m, 3Н). (MS: обнаружено (ESI+) [(М+Н)+]: 466,2.
Стереохимическую конформацию для соединения из примера 41-В определяли с использованием дифракции рентгеновских лучей на монокристалле, как показано на Фиг. 1.
Пример 42-А и пример 42-В
6-Амино-9-[(4-хлорфенил)метил]-N-этил-2-[S(S)-этилсульфонимидоил]-N-метил-8-оксо-пурин-7-карбоксамид (соединение из примера 42-А) и 6-амино-9-[(4-хлорфенил)метил]-N-этил-2-[S(R)-этилсульфонимидоил]-N-метил-8-оксо-пурин-7-карбоксамид (соединение из примера 42-В)
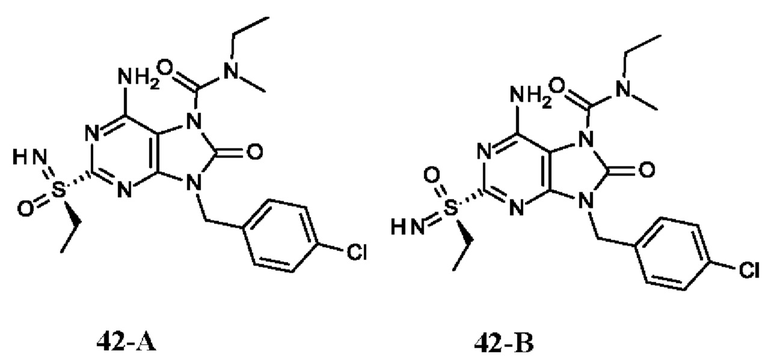
Соединение из примера 42-А получали по аналогии со стадией 6 способа А из примера 1, используя соединение 41с-А и N-этил-N-метил-карбамоилхлорид вместо 6-амино-9-бензил-2-(пропилсульфонимидоил)-7H-пурин-8-она (соединения 1е) и N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). 6-Амино-9-[(4-хлорфенил)метил]-N-этил-2-[S(S)-этилсульфонимидоил]-N-метил-8-оксо-пурин-7-карбоксамид (соединение из примера 42-А, 40 мг) получали в виде белого твердого вещества. 1H-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.43-7.41 (m, 4H), 6.90 (s, 2H), 4.99 (s, 2H), 4.18 (s, 1H), 3.48-3.40 (m, 2H), 3.39 (s, 2H), 3.05-3.01 (m, 3Н), 1.20-1.14 (m, 6H). MS: обнаружено (ESI+) [(М+Н)+]: 452,2.
Соединение из примера 42-В получали по аналогии со стадией 6 способа А из примера 1, используя соединение 41с-В и N-этил-N-метил-карбамоилхлорид вместо 6-амино-9-бензил-2-(пропилсульфонимидоил)-7Н-пурин-8-она (соединения 1е) и N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). 6-Амино-9-[(4-хлорфенил)метил]-N-этил-2-[S(R)-этилсульфонимидоил]-N-метил-8-оксо-пурин-7-карбоксамид (соединение из примера 42-В, 38 мг) получали в виде белого твердого вещества. 1H-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.43-7.41 (m, 4H), 6.91 (s, 2Н), 4.98 (s, 2H), 4.19 (s, 1H), 3.48-3.40 (m, 2H), 3.39 (s, 2H), 3.09-2.97 (m, 3Н), 1.23-1.11 (m, 6H). MS: обнаружено (ESI+) [(М+Н)+]: 452,2.
Стереохимическую конформацию для соединения из примера 42-А определяли с использованием дифракции рентгеновских лучей на монокристалле, как показано на Фиг. 2.
Пример 43-А и пример 43-В
6-Амино-2-[S(R)-этилсульфонимидоил]-N-метил-8-оксо-N-пропил-9-(п-толилметил)пурин-7-карбоксамид (соединение из примера 43-А) и 6-амино-2-[S(S)-этилсульфонимидоил]-N-метил-8-оксо-N-пропил-9-(п-толилметил)пурин-7-карбоксамид (соединение из примера 43-В)
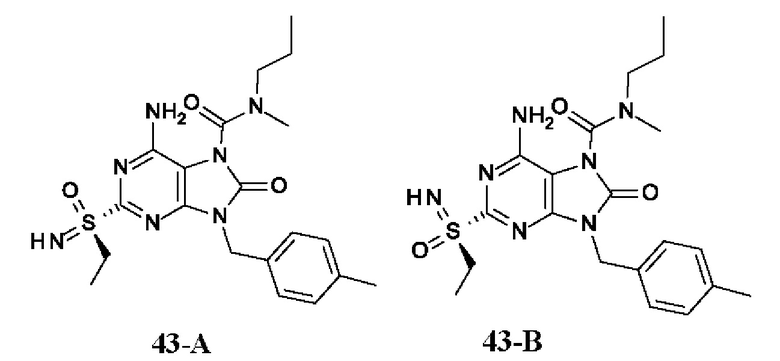
Стадия 1. Получение 4-амино-2-оксо-3-(п-толилметил)-1Н-имидазол-5-карбонитрила (соединения 43а)
Соединение 43а получали по аналогии со стадией 1 способа А из примера 1, используя 4-метилбензилизоцианат вместо бензилизоцианата. 4-Амино-2-оксо-3-(п-толилметил)-1H-имидазол-5-карбонитрил (26,6 г; соединение 43а) получали в виде серого твердого вещества и использовали непосредственно на следующей стадии без дополнительной очистки. MS: обнаружено (ESI+) [(М+Н)+]: 229,2.
Стадия 2. Получение 6-амино-9-(п-толилметил)-2-сульфанил-7Н-пурин-8-она (соединения 43b)
Соединение 43b получали по аналогии со стадией 2 способа А из примера 1, используя 4-амино-2-оксо-3-(п-толилметил)-1Н-имидазол-5-карбонитрил (соединение 43а) вместо 4-амино-3-бензил-2-оксо-1H-имидазол-5-карбонитрила (соединения 1а). 6-Амино-9-(п-толилметил)-2-сульфанил-7H-пурин-8-он (20,0 г; соединение 43b) получали в виде желтого твердого вещества. MS: обнаружено (ESI+) [(М+Н)+]: 288.
Стадия 3. Получение 6-амино-2-этилсульфанил-9-(п-толилметил)-7Н-пурин-8-она (соединения 43 с)

Соединение 43с получали по аналогии со стадией 3 способа А из примера 1, используя 6-амино-9-(п-толилметил)-2-сульфанил-7Н-пурин-8-он (соединение 43b) и иодэтан вместо 6-амино-9-бензил-2-сульфанил-7H-пурин-8-она (соединения 1b) и бромпропана. 6-Амино-2-этилсульфанил-9-(п-толилметил)-7Н-пурин-8-он (13 г; соединение 43с) получали в виде желтого твердого вещества. MS: обнаружено (ESI+) [(М+Н)+]: 316.
Стадия 4. Получение 6-амино-2-этилсульфинил-9-(п-толилметил)-7Н-пурин-8-она (соединения 43d)
Соединение 43d получали по аналогии со стадией 4 способа А из примера 1, используя 6-амино-2-этилсульфанил-9-(п-толилметил)-7Н-пурин-8-он (соединение 43с) вместо 6-амино-9-бензил-2-метилсульфанил-7Н-пурин-8-она (соединения 1с). 6-Амино-2-этилсульфинил-9-(п-толилметил)-7H-пурин-8-он (3,5 г; соединение 43d) получали в виде желтого твердого вещества. MS: обнаружено (ESI+) [(М+Н)+]: 332.
Стадия 5. Получение 6-амино-2-(этилсульфонимидоил)-9-(п-толилметил)-7Н-пурин-8-она (соединения 43е)
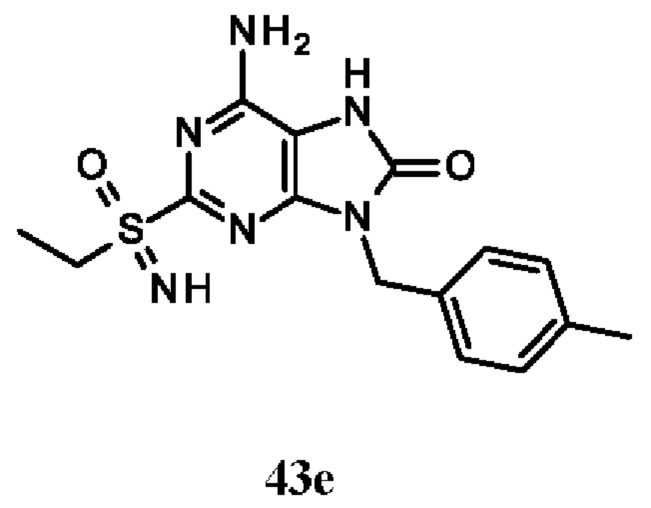
Соединение 43е получали по аналогии со стадией 4 способа А из примера 1, используя 6-амино-2-этилсульфинил-9-(п-толилметил)-7H-пурин-8-он (соединение 43d) вместо 6-амино-9-бензил-2-метилсульфинил-7Н-пурин-8-она (соединения 1d). 6-Амино-2-(этилсульфонимидоил)-9-(п-толилметил)-7Н-пурин-8-он (530 мг; соединение 43е) получали в виде желтого твердого вещества. 1H-ЯМР (400 МГц, DMSO-d6) δ млн-1: 10.53 (s, 1Н), 7.24 (d, J=8,03 Гц, 2H), 7.13 (d, J=8,03 Гц, 2H), 6.94 (br. s., 2H), 4.91 (s, 2H), 4.03 (s, 1Н), 3.36-3.41 (m, 2H), 2.26 (s, 3Н), 1.18 (t, J=7,28 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 347.
После разделения хиральной HPLC смеси, представляющей собой соединение 43е, получали соединение 43е-А (быстрее элюирующееся; 56,8 мг) и соединение 43е-В (медленнее элюирующееся; 56,7 мг) в виде белого твердого вещества, используя 5%→40% метанола с (0,05% DEA)/CO2 на колонке ChiralPak AD-3.
Соединение 43е-А: 1H-ЯМР (400 МГц, DMSO-d6) δ млн-1: 10.52 (br. s., 1H), 7.23 (d, J=8,0 Гц, 2H), 7.13 (d, J=7,9 Гц, 2H), 6.94 (br. s., 2H), 4.90 (s, 2H), 4.03 (s, 1H), 3.42-3.33 (m, 2H), 2.25 (s, 3H), 1.17 (t, J=7,3 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 347.
Соединение 43е-В: 1H-ЯМР (400 МГц, DMSO-d6) δ млн-1: 10.56 (br. s., 1H), 7.23 (d, J=8,0 Гц, 2H), 7.13 (d, J=8,0 Гц, 2H), 6.95 (br. s., 2H), 4.90 (s, 2H) 4.03 (s, 1H), 3.44-3.29 (m, 2H), 2.25 (s, 3Н), 1.17 (t, J=7,3 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 347.
Стадия 6. Получение 6-амино-2-[S(R)-этилсульфонимидоил]-N-метил-8-оксо-N-пропил-9-(п-толилметил)пурин-7-карбоксамида (соединения из примера 43-А) и 6-амино-2-[S(S)-этилсульфонимидоил]-N-метил-8-оксо-N-пропил-9-(п-толилметил)пурин-7-карбоксамида (соединения из примера 43-В)
Соединение из примера 43-А получали по аналогии со стадией 6 способа А из примера 1, используя соединение 43е-А вместо 6-амино-9-бензил-2-(пропилсульфонимидоил)-7Н-пурин-8-она (соединения 1е). 6-Амино-2-[S(R)-этилсульфонимидоил]-N-метил-8-оксо-N-пропил-9-(п-толилметил)пурин-7-карбоксамид (соединение из примера 43-А; 58,1 мг; быстрее элюирующееся; изопропанол от 5% до 40% с (0,05% DEA)/CO2 на колонке ChiralPak AD-3) получали в виде белого твердого вещества. 1H-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.28 (d, J=7,8 Гц, 2Н), 7.15 (d, J=7,8 Гц, 2H), 6.88 (br. s., 2H), 5.03-4.87 (m, 2H), 4.19 (s, 1H), 3.61-3.36 (m, 4H), 3.11-2.96 (m, 3H), 2.26 (s, 3H), 1.72-1.45 (m, 2H), 1.20 (t, J=7,2 Гц, 3Н), 0.97-0.65 (m, 3H). MS: обнаружено (ESI+) [(М+Н)+]: 446.
Соединение из примера 43-В получали по аналогии со стадией 6 способа А из примера 1, используя соединение 43е-В вместо 6-амино-9-бензил-2-(пропилсульфонимидоил)-7H-пурин-8-она (соединения 1е). 6-Амино-2-[S(S)-этилсульфонимидоил]-N-метил-8-оксо-N-пропил-9-(п-толилметил)пурин-7-карбоксамид (соединение из примера 43-В; 40,1 мг; медленнее элюирующееся, изопропанол от 5% до 40% с (0,05% DEA)/CO2 на колонке ChiralPak AD-3) получали в виде белого твердого вещества: 1H-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.28 (d, J=7,5 Гц, 2H), 7.15 (d, J=7,5 Гц, 2H), 6.89 (br. s., 2H), 5.03-4.86 (m, 2H), 4.19 (s, 1H), 3.49-3.37 (m, 4H), 3.08-3.00 (m, 3H), 2.27 (s, 3H), 1.70-1.48 (m, 2H), 1.20 (t, J=7,2 Гц, 3Н), 0.95-0.71 (m, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 446,3.
Стереохимическую конформацию для соединения из примера 43-В определяли с использованием дифракции рентгеновских лучей на монокристалле, как показано на Фиг. 3.
Пример 44-А и пример 44-В
6-Амино-N-этил-2-[S(S)-этилсульфонимидоил]-N-метил-8-оксо-9-(п-толилметил)пурин-7-карбоксамид (соединение из примера 44-А) и 6-амино-N-этил-2-[S(R)-этилсульфонимидоил]-N-метил-8-оксо-9-(п-толилметил)пурин-7-карбоксамид (соединение из примера 44-В)

Соединение из примера 44-А получали по аналогии со стадией 6 способа А из примера 1, используя соединение 43е-В и N-этил-N-метил-карбамоилхлорид вместо 6-амино-9-бензил-2-(пропилсульфонимидоил)-7H-пурин-8-она (соединения 1е) и N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). 6-Амино-N-этил-2-[S(S)-этилсульфонимидоил]-N-метил-8-оксо-9-(п-толилметил)-пурин-7-карбоксамид (соединение из примера 44-А; 73,1 мг) получали в виде белого твердого вещества. 1H-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.28 (d, J=7,8 Гц, 2Н), 7.15 (d, J=7,8 Гц, 2Н), 6.90 (br. s., 2H), 4.95 (s, 2H), 4.19 (br. s., 1H), 3.48-3.39 (m, 4H), 3.06-3.00 (m, 3H), 2.27 (s, 3H), 1.29-1.04 (m, 6H). MS: обнаружено (ESI+) [(М+Н)+]: 432.
Соединение из примера 44-В получали по аналогии со стадией 6 способа А из примера 1, используя соединение 43е-А и N-этил-N-метил-карбамоилхлорид вместо 6-амино-9-бензил-2-(пропилсульфонимидоил)-7H-пурин-8-она (соединения 1е) и N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). 6-Амино-N-этил-2-[S(R)-этилсульфонимидоил]-N-метил-8-оксо-9-(п-толилметил)-пурин-7-карбоксамид (соединение из примера 44-В; 46,7 мг) получали в виде белого твердого вещества: 1H-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.28 (d, J=7,9 Гц, 2H), 7.15 (d, J=7,9 Гц, 2H), 6.90 (br. s., 2H), 4.95 (s, 2H), 4.19 (br. s., 1H), 3.50-3.39 (m, 4H), 3.10-2.96 (m, 3H), 2.27 (s, 3H), 1.27-1.10 (m, 6H). MS: обнаружено (ESI+) [(М+Н)+]: 432.
Пример 45-А и пример 45-В
6-Амино-2-[S(R)этилсульфонимидоил]-9-[(4-фторфенил)метил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамид и 6-амино-2-[S(S)-этилсульфонимидоил]-9-[(4-фторфенил)метил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамид
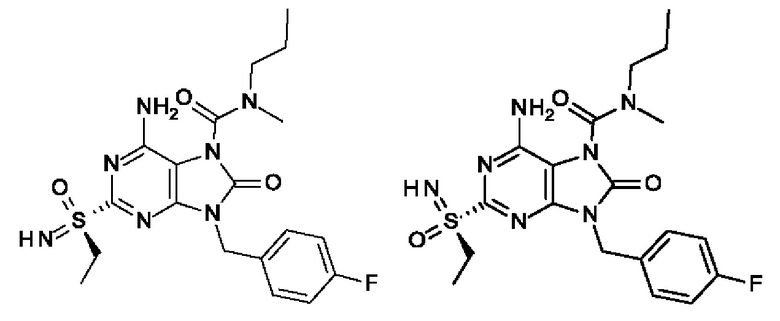
Стадия 1. Получение 4-амино-3-[(4-фторфенил)метил]-2-оксо-1Н-имидазол-5-карбонитрила (соединения 45а)
Соединение 45а получали по аналогии со стадией 1 способа А из примера 1, используя 4-фторбензилизоцианат вместо бензилизоцианата. 4-Амино-3-[(4-фторфенил)метил]-2-оксо-1Н-имидазол-5-карбонитрил (48 г; соединение 45а) получали в виде светло-желтого твердого вещества и использовали непосредственно на следующей стадии без дополнительной очистки. MS: обнаружено (ESI+) [(М+Н)+]: 233.
Стадия 2. Получение 6-амино-9-[(4-фторфенил)метил]-2-сульфанил-7Н-пурин-8-она (соединения 45b)
Соединение 45b получали по аналогии со стадией 2 способа А из примера 1, используя 4-амино-3-[(4-фторфенил)метил]-2-оксо-1H-имидазол-5-карбонитрил (соединение 45а) вместо 4-амино-3-фенилметил-2-оксо-1H-имидазол-5-карбонитрила (соединения 1а). 6-Амино-9-[(4-фторфенил)метил]-2-сульфанил-7Н-пурин-8-он (32,0 г; соединение 45b) получали в виде желтого твердого вещества. MS: обнаружено (ESI+) [(М+Н)+]: 292.
Стадия 3. Получение 6-амино-2-этилсульфанил-9-[(4-фторфенил)метил]-7Н-пурин-8-она (соединения 45с)

Соединение 45 с получали по аналогии со стадией 3 способа А из примера 1, используя 6-амино-9-[(4-фторфенил)метил]-2-сульфанил-7H-пурин-8-он (соединение 45b) и иодэтан вместо 6-амино-9-бензил-2-сульфанил-7H-пурин-8-она (соединения 1b) и бромпропана. 6-Амино-2-этилсульфанил-9-[(4-фторфенил)метил]-7Н-пурин-8-он (5,6 г; соединение 45с) получали в виде желтого твердого вещества. MS: обнаружено (ESI+) [(М+Н)+]: 320.
Стадия 5. Получение 6-амино-2-этилсульфинил-9-[(4-фторфенил)метил]-7Н-пурин-8-она (соединения 45d)
Соединение 45d получали по аналогии со стадией 4 способа А из примера 1, используя 6-амино-2-этилсульфанил-9-[(4-фторфенил)метил]-7Н-пурин-8-он (соединение 45с) вместо 6-амино-9-бензил-2-пропилсульфанил-7Н-пурин-8-она (соединения 1с). 6-Амино-2-этилсульфинил-9-[(4-фторфенил)метил]-7H-пурин-8-он (4,8 г; соединение 45d) получали в виде желтого твердого вещества. MS: обнаружено (ESI+) [(М+Н)+]: 332.
Стадия 6. Получение 6-амино-2-(этилсульфонимидоил)-9-[(4-фторфенил)метил]-7Н-пурин-8-она (соединения 45е)
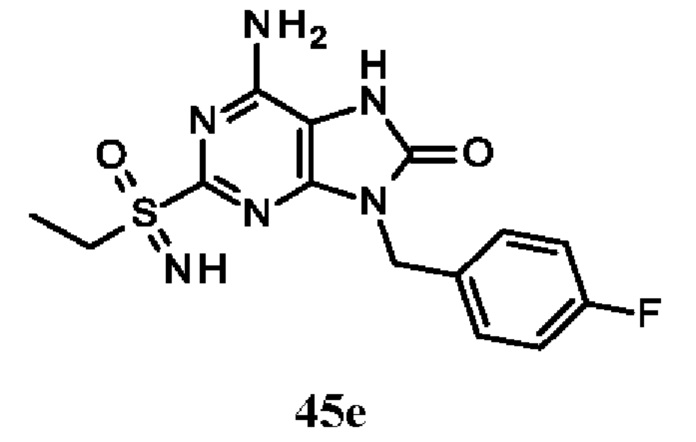
Соединение 45е получали по аналогии со стадией 4 способа А из примера 1, используя 6-амино-2-этилсульфинил-9-[(4-фторфенил)метил]-7Н-пурин-8-он (соединение 45d) вместо 6-амино-9-бензил-2-пропилсульфинил-7Н-пурин-8-она (соединения 1d). 6-Амино-2-(этилсульфонимидоил)-9-[(4-фторфенил)метил]-7Н-пурин-8-он (2,9 г; соединение 45е) получали в виде желтого твердого вещества. 1H-ЯМР (400 МГц, DMSO-d6) δ млн-1: 10.57 (br. s., 1H), 7.40 (dd, J=8,5; 5,5 Гц, 2Н), 7.16 (t, J=8,9 Гц, 2Н), 6.97 (br. s., 2H), 4.94 (s, 2H), 4.07 (s, 1H), 3.43-3.36 (m, 2H), 1.17 (t, J=7,4 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 351.
После разделения хиральной HPLC смеси, представляющей собой соединение 45е, получали соединение 45е-А (быстрее элюирующееся; 85,4 мг) и соединение 45е-В (медленнее элюирующееся; 36,4 мг) в виде белого твердого вещества, используя 5%→40% метанола с (0,05% DEA)/CO2 на колонке ChiralPak AD-3.
Соединение 45е-А: 1H-ЯМР (400 МГц, DMSO-d6) δ млн-1: 10.53 (br. s., 1H), 7.41 (dd, J=8,5; 5,5 Гц, 2Н), 7.17 (t, J=8,9 Гц, 2H), 6.98 (br. s., 2H), 4.95 (s, 2H), 4.07 (s, 1H), 3.45-3.36 (m, 2H), 1.17 (t, J=7,3 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 351.
Соединение 45е-В: 1H-ЯМР (400 МГц, DMSO-d6) δ млн-1: 10.53 (br. s., 1H), 7.41 (dd, J=8,5; 5,5 Гц, 2H), 7.17 (t, J=8,9 Гц, 2H), 6.98 (br. s., 2H), 4.95 (s, 2H), 4.07 (s, 1H), 3.44-3.37 (m, 2H) 1.17 (t, J=7,3 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 351.
Стадия 7. Получение 6-амино-2-(этилсульфонимидоил)-9-[(4-фторфенил)метил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамида (соединения из примера 45), 6-амино-2-[S(R)этилсульфонимидоил]-9-[(4-фторфенил)метил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамида и 6-амино-2-[S(S)-этилсульфонимидоил]-9-[(4-фторфенил)метил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамида (соединения из примера 45-А и соединения из примера 45-В)
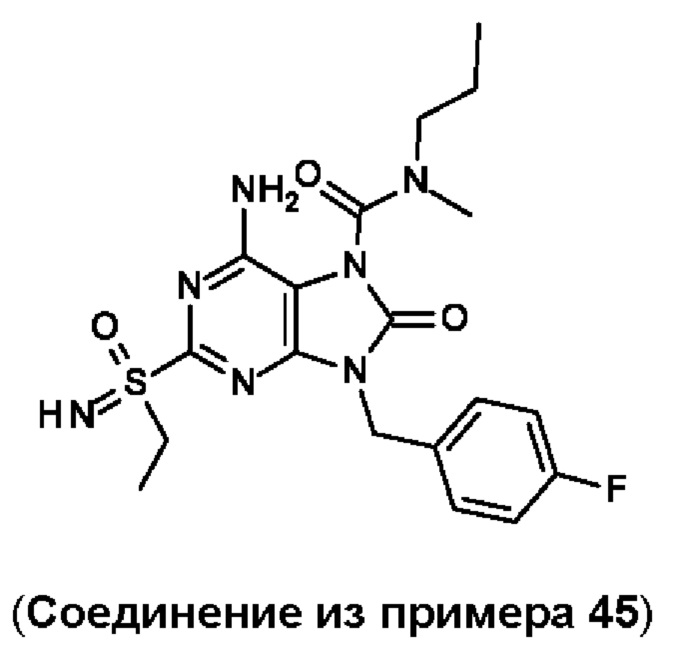
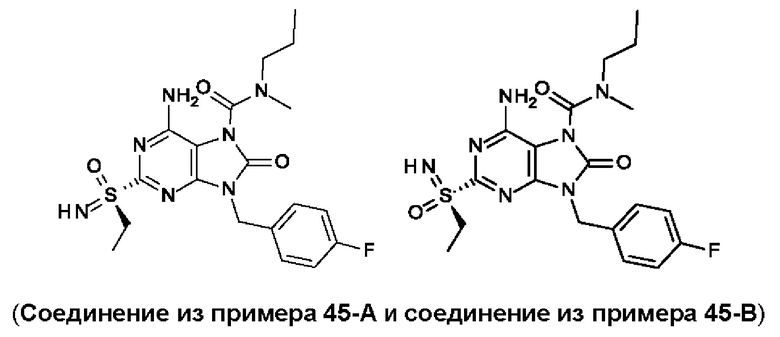
Соединение из примера 45 получали по аналогии со стадией 6 способа А из примера 1, используя 6-амино-2-(этилсульфонимидоил)-9-[(4-фторфенил)метил]-7H-пурин-8-он (соединение 45е) вместо 6-амино-9-бензил-2-(пропилсульфонимидоил)-7Н-пурин-8-она (соединения 1е). 6-Амино-2-(этилсульфонимидоил)-9-[(4-фторфенил)метил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамид (162,4 мг; соединение из примера 45) получали в виде белого твердого вещества.
После разделения хиральной HPLC смеси, представляющей собой соединение из примера 45, получали соединение из примера 45-А (быстрее элюирующееся; 85,3 мг) и соединение из примера 45-В (медленнее элюирующееся; 52 мг) в виде белого твердого вещества, используя 5%→40% метанола с (0,05% DEA)/CO2 на колонке ChiralPak AD-3.
Соединение из примера 45-А: 1H-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.53-7.38 (m, 2Н), 7.18 (t, J=8,9 Гц, 2H), 6.90 (br. s., 2H), 4.99 (s, 2H), 4.21 (s, 1H), 3.48-3.37 (m, 4H), 3.10-3.01 (m, 3H), 1.69-1.49 (m, 2H), 1.25-1.14 (m, 3H), 0.94-0.72 (m, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 450.
Соединение из примера 45-В: 1H-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.54-7.38 (m, 2H), 7.18 (t, J=8,9 Гц, 2H), 7.01-6.72 (m, 2H), 4.99 (s, 2H), 4.21 (s, 1H), 3.46-3.38 (m, 4H), 3.10-3.01 (m, 3H), 1.76-1.50 (m, 2H), 1.25-1.16 (m, 3H), 0.99-0.69 (m, 3H). MS: обнаружено (ESI+) [(М+Н)+]: 450.
Пример 46-А и пример 46-В
6-Амино-N-этил-2-(этилсульфонимидоил)-9-[(4-фторфенил)метил]-N-метил-8-оксо-пурин-7-карбоксамид (соединение из примера 46), 6-амино-N-этил-2-[S(S)-(этилсульфонимидоил)]-9-[(4-фторфенил)метил]-N-метил-8-оксо-пурин-7-карбоксамид и 6-амино-N-этил-2-[S(R)-(этилсульфонимидоил)]-9-[(4-фторфенил)метил]-N-метил-8-оксо-пурин-7-карбоксамид (соединение из примера 46-А и соединение из примера 46-В)
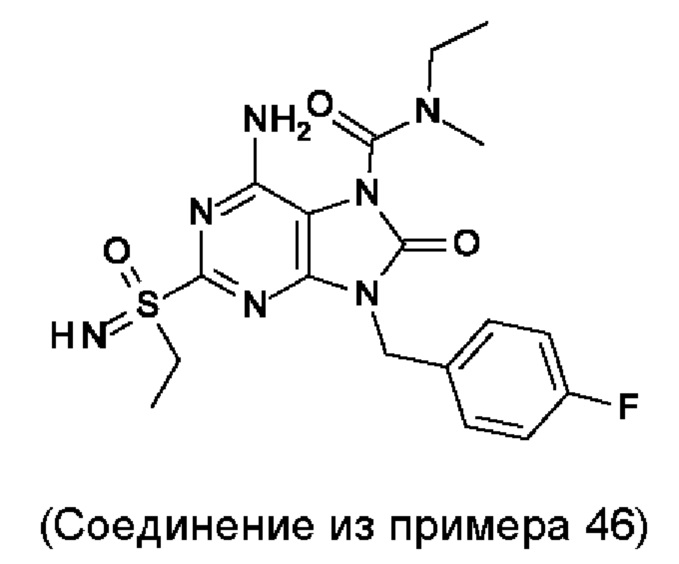
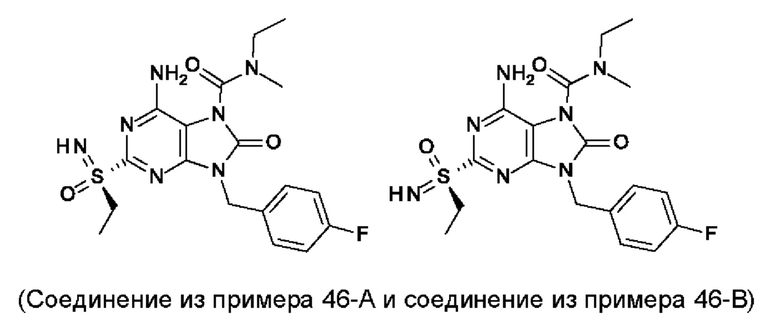
Соединение из примера 46 получали по аналогии со стадией 6 способа А из примера 1, используя 6-амино-2-(этилсульфонимидоил)-9-[(4-фторфенил)метил]-7Н-пурин-8-он (соединение 45е) и N-этил-N-метил-карбамоилхлорид вместо 6-амино-9-бензил-2-(пропилсульфонимидоил)-7H-пурин-8-она (соединения 1е) и N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). 6-Амино-N-этил-2-(этилсульфонимидоил)-9-[(4-фторфенил)метил]-N-метил-8-оксо-пурин-7-карбоксамид (51 мг; соединение из примера 46) получали в виде белого твердого вещества. 1H-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.46-7.43 (m, 2Н), 7.20-7.15 (m, 2Н), 6.90 (br. s., 2H), 4.98 (s, 2H), 4.18 (s, 1H), 3.47-3.32 (m, 4H), 3.05-3.01 (m, 3H), 1.21-1.14 (m, 6H). MS: обнаружено (ESI+) [(М+Н)+]: 436.
После разделения хиральной HPLC смеси, представляющей собой соединение из из примера 46, получали соединение из примера 46-А (быстрее элюирующееся; 72 мг) и соединение из примера 46-В (медленнее элюирующееся; 45 мг) в виде белого твердого вещества, используя 5%→40% метанола с (0,05% DEA)/CO2 на колонке ChiralPak AD-3.
Соединение из примера 46-А: 1H-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.46-7.43 (m, 2H), 7.20-7.16 (m, 2H), 6.90 (br. s., 2H), 4.98 (s, 2H), 4.18 (s, 1H), 3.47-3.32 (m, 4H), 3.05-3.01 (m, 3H), 1.21-1.14 (m, 6H). MS: обнаружено (ESI+) [(М+Н)+]: 436.
Соединение из примера 46-В: 1H-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.46-7.43 (m, 2H), 7.20-7.14 (m, 2H), 6.92 (br. s., 2H), 4.98 (s, 2H), 4.20 (br. s., 1H), 3.47-3.32 (m, 4H), 3.05-3.01 (m, 3H), 1.23-1.19 (m, 6H). MS: обнаружено (ESI+) [(М+Н)+]: 436.
Пример 47-А и пример 47-В
6-Амино-9-[(4-бромфенил)метил]-2-(этилсульфонимидоил)-N-метил-8-оксо-N-пропил-пурин-7-карбоксамид (соединение из примера 47), 6-амино-2-[S(R)-этилсульфонимидоил]-9-[(4-бромфенил)метил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамид и 6-амино-2-[S(S)-этилсульфонимидоил]-9-[(4-бромфенил)метил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамид
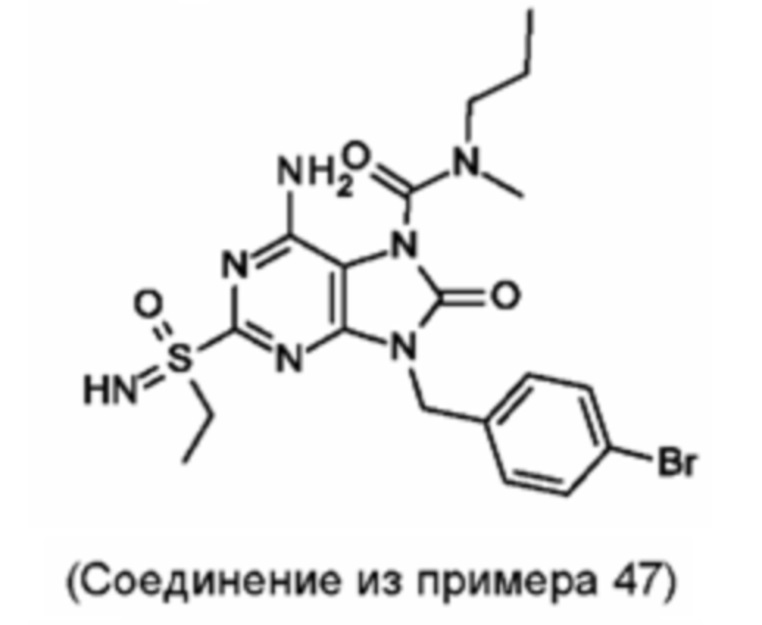
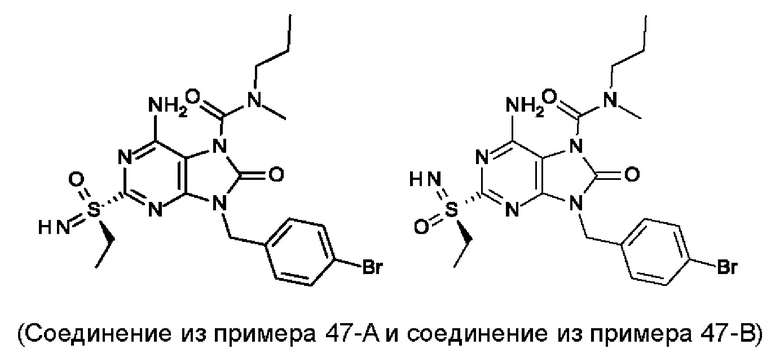
Стадия 1. Получение 4-амино-3-[(4-бромфенил)метил]-2-оксо-1Н-имидазол-5-карбонитрила (соединения 47а)
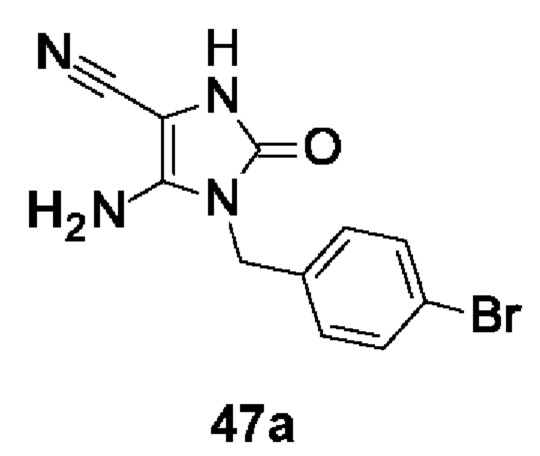
Соединение 47а получали по аналогии со стадией 1 способа А из примера 1, используя 4-бромбензилизоцианат вместо бензилизоцианата. 4-Амино-3-[(4-бромфенил)метил]-2-оксо-1Н-имидазол-5-карбонитрил (500 мг; соединение 47а) получали в виде светло-желтого твердого вещества и использовали непосредственно на следующей стадии без дополнительной очистки. 1H-ЯМР (400 МГц, DMSO-d6) δ млн-1: 9.94 (S, 1Н), 7.55-7.53 (d, J=8,0 Гц, 2Н), 7.20-7.18 (d, J=8,0 Гц, 2Н), 6.52 (br. s., 2Н), 4.74 (s, 2Н). MS: обнаружено (ESI+) [(М+Н)+]: 293.
Стадия 2. Получение 6-амино-9-[(4-бромфенил)метил]-2-сульфанил-7Н-пурин-8-она (соединения 47b)

Соединение 47b получали по аналогии со стадией 2 способа А из примера 1, используя 4-амино-3-[(4-бромфенил)метил]-2-оксо-1H-имидазол-5-карбонитрил (соединение 47а) вместо 4-амино-3-фенилметил-2-оксо-1H-имидазол-5-карбонитрила (соединения 1а). 6-Амино-9-[(4-бромфенил)метил]-2-сульфанил-7Н-пурин-8-он (300 мг; соединение 47b) получали в виде желтого твердого вещества. MS: обнаружено (ESI+) [(М+Н)+]: 352.
Стадия 3. Получение 6-амино-2-этилсульфанил-9-[(4-бромфенил)метил]-7Н-пурин-8-она (соединения 47с)
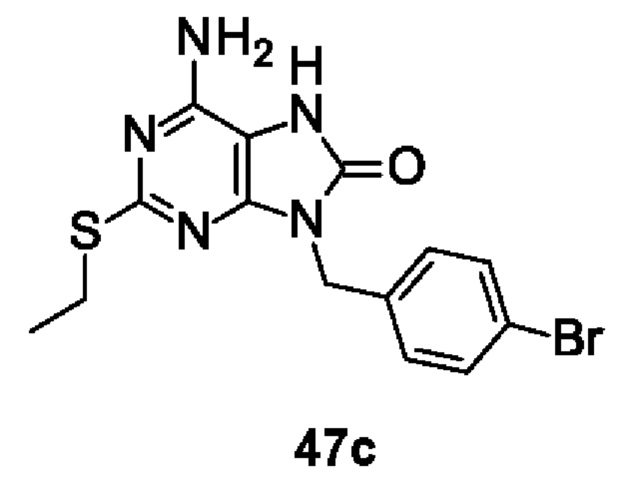
Соединение 47с получали по аналогии со стадией 3 способа А из примера 1, используя 6-амино-9-[(4-бромфенил)метил]-2-сульфанил-7Н-пурин-8-он (соединение 45b) и иодэтан вместо 6-амино-9-бензил-2-сульфанил-7Н-пурин-8-она (соединения 1b) и бромпропана. 6-Амино-2-этилсульфанил-9-[(4-бромфенил)метил]-7H-пурин-8-он (5,6 г; соединение 47с) получали в виде желтого твердого вещества. MS: обнаружено (ESI+) [(М+Н)+]: 380.
Стадия 4. Получение 6-амино-9-[(4-бромфенил)метил]-2-этилсульфинил-7Н-пурин-8-она (соединения 47d)
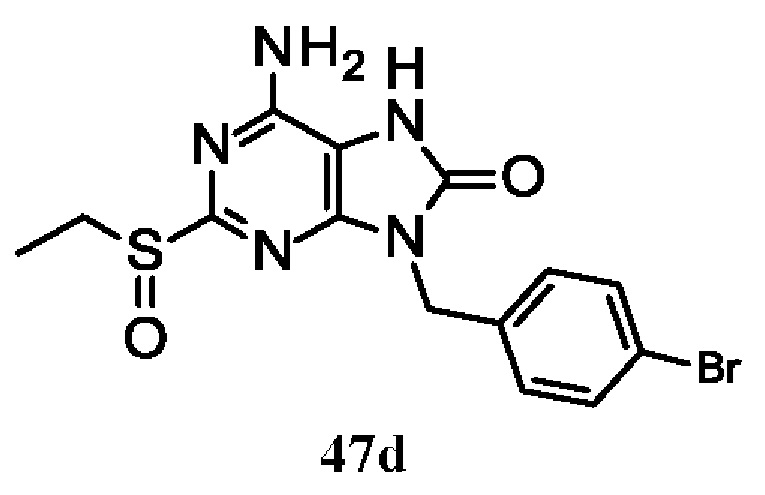
Соединение 47d получали по аналогии со стадией 6 способа В из примера 1, используя 6-амино-9-[(4-бромфенил)метил]-2-этилсульфанил-7Н-пурин-8-он (соединение 47с) вместо 6-амино-9-бензил-2-(2-пропилсульфанил)-7H-пурин-8-она (соединения 1с). 6-Амино-9-[(4-бромфенил)метил]-2-этилсульфинил-7H-пурин-8-он (3,2 г; соединение 47d) получали в виде белого твердого вещества. MS: обнаружено (ESI+) [(М+Н)+]: 396.
Стадия 5. Получение 6-амино-9-[(4-бромфенил)метил]-2-(этилсульфонимидоил)-7Н-пурин-8-она (соединения 47е)
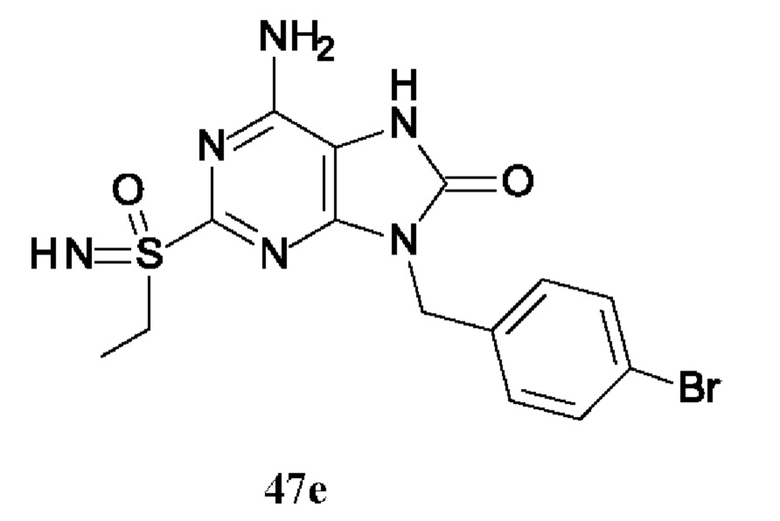
Соединение 47е получали по аналогии со стадией 7 способа В из примера 1, используя 6-амино-9-[(4-бромфенил)метил]-2-этилсульфинил-7Н-пурин-8-он (соединение 47d) вместо 6-амино-9-бензил-2-пропилсульфинил-7H-пурин-8-она (соединения 1d). 6-Амино-9-[(4-бромфенил)метил]-2-(этилсульфонимидоил)-7H-пурин-8-он (4,0 г; соединение 47е) получали в виде белого твердого вещества. MS: обнаружено (ESI+) [(М+Н)+]: 411.
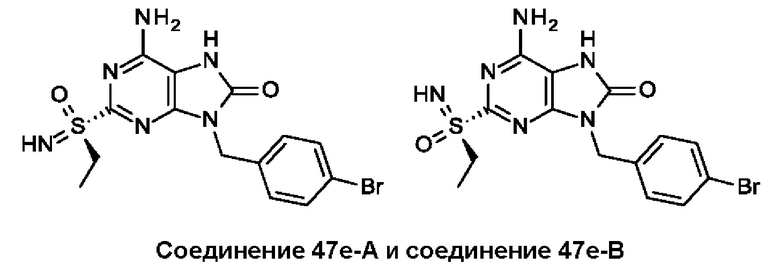
После разделения хиральной HPLC смеси, представляющей собой соединение 47е, получали соединение 47е-А (быстрее элюирующееся; 112 мг) и соединение 47е-В (медленнее элюирующееся; 99 мг) в виде белого твердого вещества, используя 5%→40% метанола с (0,05% DEA)/CO2 на колонке ChiralPak AD-3.
Соединение 47е-А: 1H-ЯМР (400 МГц, DMSO-d6) δ млн-1: 10.58 (br. s., 1H), 7.52-7.54 (d, J=8,0 Гц, 2H), 7.31-7.29 (t, J=8,0 Гц, 2H), 6.54 (br. s., 2H), 4.93 (s, 2H), 4.05 (s, 1H), 3.42-3.31 (m, 2H), 1.15 (t, J=7,3 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 411.
Соединение 47е-В: 1H-ЯМР (400 МГц, DMSO-d6) δ млн-1: 10.58 (br. s., 1H), 7.54-7.52 (d, J=8,0 Гц, 2H), 7.31-7.29 (t, J=8,0 Гц, 2H), 6.98 (br. s., 2H), 4.93 (s, 2H), 4.06 (s, 1H), 3.40-3.37 (m, 2H), 1.15 (t, J=7,3 Гц, 3Н). MS: обнаружено (ESI+) [(М+Н)+]: 411.
Стадия 6. Получение 6-амино-9-[(4-бромфенил)метил]-2-(этилсульфонимидоил)-N-метил-8-оксо-N-пропил-пурин-7-карбоксамида (соединения из примера 47), 6-амино-9-[(4-бромфенил)метил]-2-[S(R)-этилсульфонимидоил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамида и 6-амино-9-[(4-бромфенил)метил]-2-[S(S)-этилсульфонимидоил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамида (соединения из примера 47-А и соединения из примера 47-В)
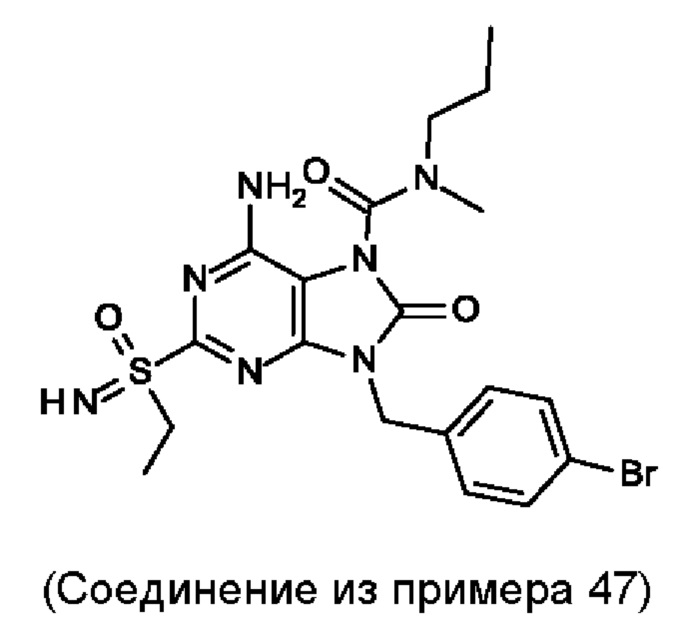
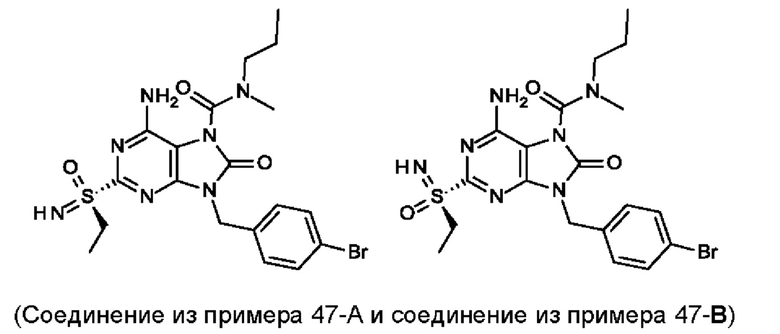
Соединение из примера 47 получали по аналогии со стадией 6 способа А из примера 1, используя 6-амино-9-[(4-бромфенил)метил]-2-(этилсульфонимидоил)-7H-пурин-8-он (соединение 47е) вместо 6-амино-9-бензил-2-(пропилсупьфонимидоил)-7H-пурин-8-она (соединения 1е). 6-Амино-9-[(4-бромфенил)метил]-2-(этилсульфонимидоил)-N-метил-8-оксо-N-пропил-пурин-7-карбоксамид (570 мг; соединение из примера 47) получали в виде белого твердого вещества. 1H-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.56-7.53 (m, 2H), 7.36-7.34 (m, 2H), 6.92 (br. s., 2H), 4.97 (s, 2H), 4.18 (s, 1H), 3.45-3.38 (m, 4H), 3.05-3.02 (m, 3H), 1.65-1.56 (m, 2H), 1.19 (t, J=8,0 Гц, 3H), 0.93-0.75 (m, 3H). MS: обнаружено (ESI+) [(М+Н)+]: 510.
После разделения хиральной HPLC смеси, представляющей собой соединение из примера 47, получали соединение из примера 47-А (быстрее элюирующееся; 260 мг) и соединение из примера 47-В (медленнее элюирующееся; 266 мг) в виде белого твердого вещества, используя 5%→40% метанола с (0,05% DEA)/CO2 на колонке ChiralPak AD-3.
Соединение из примера 47-А: 1H-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.56-7.54 (d, J=8,0 Гц, 2Н), 7.36-7.33 (d, J=8,0 Гц, 2Н), 6.90 (br. s., 2H), 4.97 (s, 2H), 4.21 (s, 1H), 3.46-3.41 (m, 4H), 3.05-3.02 (m, 3H), 1.65-1.54 (m, 2H), 1.24-1.16 (m, 3H), 0.93-0.75 (m, 3H). MS: обнаружено (ESI+) [(М+Н)+]: 510.
Соединение из примера 47-В: 1H-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.54-7.53 (d, J=8,0 Гц, 2H), 7.36-7.33 (d, J=8,0 Гц, 2H), 6.90 (br. s., 2H), 4.97 (s, 2H), 4.21 (s, 1H), 3.46-3.41 (m, 4H), 3.06-3.02 (m, 3H), 1.65-1.54 (m, 2H), 1.20-1.16 (m, 3H), 0.93-0.75 (m, 3H). MS: обнаружено (ESI+) [(М+Н)+]: 510.
Пример 48-А и пример 48-В
6-Амино-9-[(4-бромфенил)метил]-N-этил-2-(этилсульфонимидоил)-N-метил-8-оксо-пурин-7-карбоксамид (соединение из примера 48), 6-амино-9-[(4-бромфенил)метил]-N-этил-2-[S(S)-(этилсульфонимидоил)]-N-метил-8-оксо-пурин-7-карбоксамид и 6-амино-9-[(4-бромфенил)метил]-N-этил-2-[S(R)-(этилсульфонимидоил)]-N-метил-8-оксо-пурин-7-карбоксамид (соединение из примера 48-А и соединение из примера 48-В)
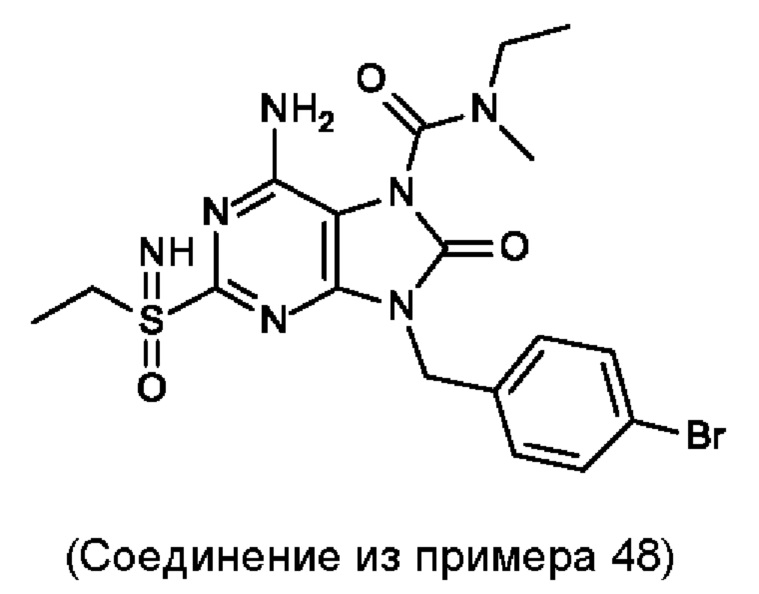
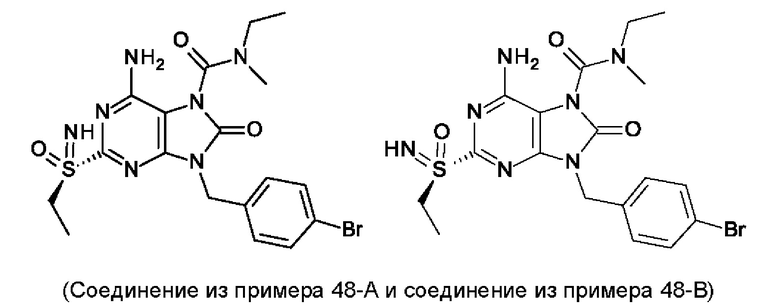
Соединение из примера 48 получали по аналогии со стадией 6 способа А из примера 1, используя 6-амино-9-[(4-бромфенил)метил]-2-(этилсульфонимидоил)-7Н-пурин-8-он (соединение 47е) и N-этил-N-метил-карбамоилхлорид вместо 6-амино-9-бензил-2-(пропилсульфонимидоил)-7Н-пурин-8-она (соединения 1е) и N-метил-N-пропил-карбамоилхлорида (промежуточного соединения АА). 6-Амино-9-[(4-бромфенил)метил]-2-(этилсульфонимидоил)-N-метил-8-оксо-N-пропил-пурин-7-карбоксамид (469 мг; соединение из примера 48) получали в виде белого твердого вещества. 1H-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.56-7.54 (d, J=8,0 Гц, 2Н), 7.36-7.34 (d, J=8,0 Гц, 2H), 6.98 (br. s., 2H), 4.97 (s, 2H), 3.53-3.46 (m, 4H), 3.05-3.01 (m, 3H), 1.22-1.16 (m, 6H). MS: обнаружено (ESI+) [(М+Н)+]: 496.
После разделения хиральной HPLC смеси, представляющей собой соединение из примера 48, получали соединение из примера 48-А (быстрее элюирующееся; 198 мг) и соединение из примера 48-В (медленнее элюирующееся; 202 мг) в виде белого твердого вещества, используя 5%→40% метанола с (0,05% DEA)/CO2 на колонке ChiralPak AD-3.
Соединение из примера 48-А: 1H-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.56-7.54 (d, J=8,0 Гц, 2H), 7.36-7.34 (d, J=8,0 Гц, 2H), 6.92 (br. s., 2H), 4.97 (s, 2H), 4.19-4.18 (m, 1H), 3.46-3.41 (m, 4H), 3.05-3.01 (m, 3H), 1.20-1.14 (m, 6H). MS: обнаружено (ESI+) [(М+Н)+]: 496.
Соединение из примера 48-В: 1H-ЯМР (400 МГц, DMSO-d6) δ млн-1: 7.56-7.54 (d, J=8,0 Гц, 2H), 7.36-7.34 (d, J=8,0 Гц, 2H), 6.92 (br. s., 2H), 4.97 (s, 2H), 4.24 (br. s., 1H), 3.58-3.41 (m, 4H), 3.05-3.01 (m, 3H), 1.26-1.01 (m, 6H). MS: обнаружено (ESI+) [(М+Н)+]: 496.
Пример 49
Активность соединений и соединений из примеров в анализе с использованием несущих TLR-7 человека клеток почки человеческого эмбриона (HEK293-hTLR-7)
Анализ с использованием клеток HEK293-Blue-hTLR-7
Стабильную клеточную линию HEK293-Blue-hTLR-7 приобретали у InvivoGen (№ по каталогу: hkb-htlr7; Сан-Диего, Калифорния, США). Эти клетки предназначены для изучения стимуляции TLR7 человека путем мониторинга активации NF-κВ. Репортерный ген SEAP (секретируемой эмбриональной щелочной фосфатазы) помещали под контроль минимального промотора IFN-β, слитого с пятью сайтами связывания с NF-κВ и АР-1 (активаторный белок 1). SEAP индуцировали активацией NF-κВ и АР-1 посредством стимуляции клеток HEK-Blue-hTLR7 лигандами к TLR7. Ввиду этого экспрессия репортера регулировалась промотором NF-κВ после стимуляции TLR7 человека в течение 20 ч. Активность репортера SEAP в супернатанте клеточной культуры определяли, используя набор QUANTIBlue™ (№ по каталогу: rep-qb1; Invivogen, Сан-Диего, Калифорния, США) на длине волны 640 нм, содержащий среду для обнаружения, которая становилась пурпурной или синей в присутствии щелочной фосфатазы.
Клетки HEK293-Blue-hTLR7 инкубировали в течение 24 ч при плотности 250000-450000 кпеток/мл в объеме 180 мкл в 96-луночном планшете в модифицированной Дульбекко среде Игла (DMEM), содержащей глюкозу (4,5 г/л), пенициллин (50 ед./мл), стрептомицин (50 мг/мл), нормоцин (100 мг/мл), 2 мМ L-глутамин, 10% (об./об.) инактивированной нагреванием фетальной телячьей сыворотки. Затем клетки HEK293-Blue-hTLR-7 инкубировали с добавлением 20 мкл раствора тестируемого соединения, взятого в серийном разведении в присутствии DMSO в конечной концентрации 1%, и осуществляли инкубирование при 37°С в CO2-инкубаторе в течение 20 ч. Затем по 20 мкл супернатанта из каждой лунки инкубировали с 180 мкл раствора субстрата Quantiblue при 37°С в течение 2 ч и измеряли поглощение при 620-655 нм, используя спектрофотометр. Путь передачи сигнала, при котором активация TLR7 приводит к последующей активации NF-κВ, является общепризнанным, и поэтому для оценки агониста TLR7 также широко использовали аналогичный анализ репортерного гена (Tsuneyasu Kaisho and Takashi Tanaka, Trends in Immunology, volume 29, issue 7, July 2008, pages 329.sci; Hiroaki Hemmi et al., Nature Immunology, 3, 196-200 (2002)).
Соединения и соединения из примеров по настоящему изобретению тестировали в анализе с использованием клеток HEK293-hTLR-7 на предмет их агонистической активности в отношении TLR7, как изложено в данном описании, и результаты приведены в Таблице 1. Было обнаружено, что пролекарства из примеров имеют ЕС50 от примерно 2,1 мкМ до примерно 1000 мкМ, что активные формы соединений имеют ЕС50 меньше 0,2 мкМ. Рассчитанные соотношения ЕС50(пролекарство)/ЕС50(активная форма) находились в диапазоне от 32 до примерно 7600.

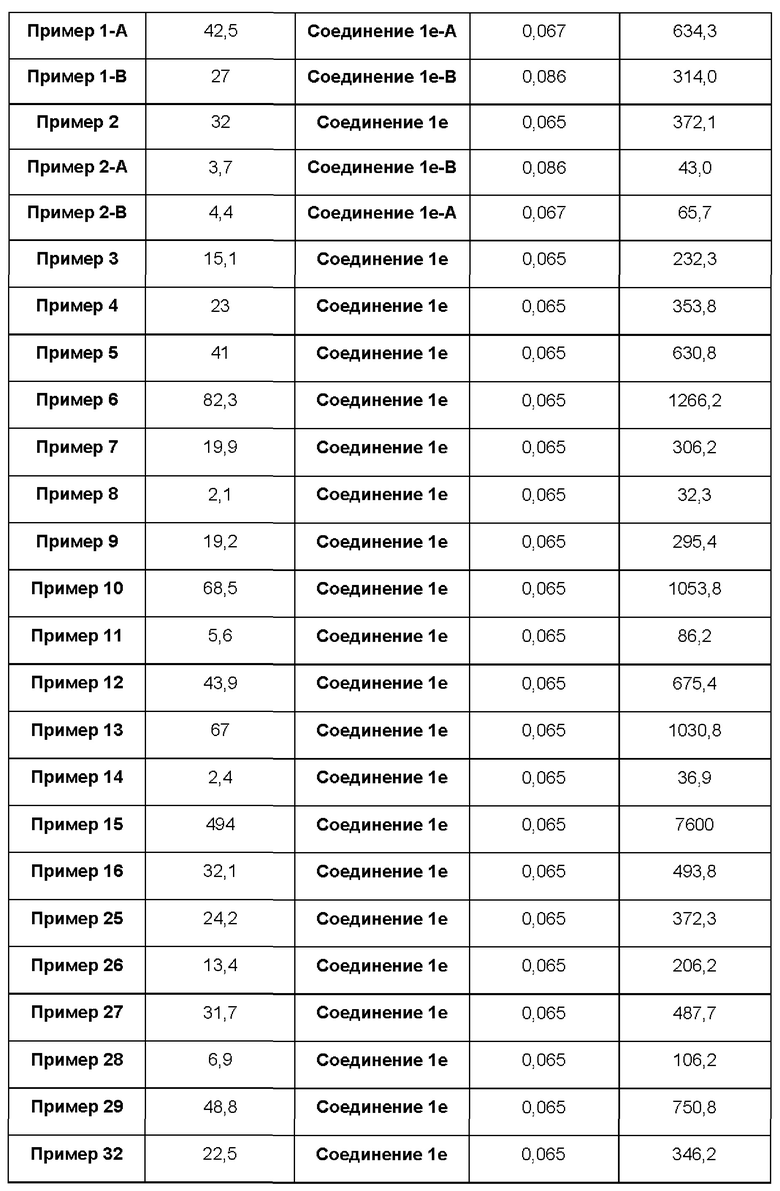

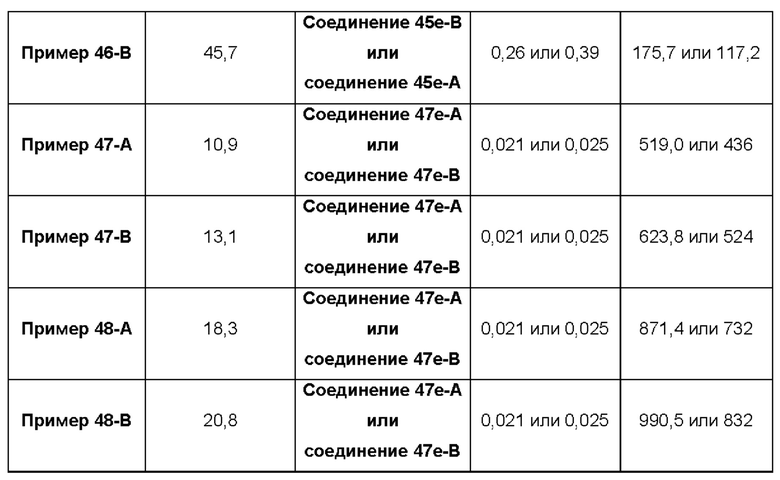
Пример 50
Метаболизм пролекарств соединения формулы (I)
Проводили исследование для оценки метаболического превращения пролекарств соединения формулы (I) в их соответствующую активную форму. Соединения формулы (I), при условии их функционирования в качестве пролекарств, могут метаболизироваться в организме до активного соединения или других соединений по изобретению. Для оценки степени метаболического превращения пролекарств в организме животного или человека часто используют микросомы печени человека.
Материалы
Систему кофактора NADPH (восстановленная форма NADP), включающую в себя β-никотинамидаденин-динуклеотидфосфат (NADP), изолимонную кислоту и изоцитратдегидрогеназу, приобретали у Sigma-Aldrich Co. (Сент-Луис, Миссури, США). Микросомы печени человека (№ по каталогу 452117, партия №38290) получали от Coming (Уоберн, Массачусетс, США). Микросомы печени мыши (№ по каталогу М1000, партия №1310028) получали от Xenotech.
Рабочий раствор соединений и другой раствор
Соединения растворяли в DMSO, готовя концентрированные 10 мМ растворы. 10 мкл концентрированного раствора разбавляли ацетонитрилом (990 мкл), получая 100 мкМ рабочий раствор.
Инкубирование
Предварительно микросомы инкубировали с тестируемым соединением в течение 10 мин при 37°С в 100 мМ калийфосфатном буфере с рН 7,4. Реакции инициировали добавлением NADPH-регенерирующей системы, получая конечный объем инкубируемой смеси 200 мкл, и смеси встряхивали в водяной бане при 37°С. Инкубируемые смеси содержали микросомы печени (0,5 мг микросомального белка/мл), субстраты (1,0 мкМ) и NADP (1 мМ), изоцитратдегидрогеназу (1 единица/мл), изолимонную кислоту (6 мМ).
Приготовление образцов для анализа
Через 30 мин реакцию гасили, добавляя 600 мкл холодного ацетонитрила (содержащего толбутамид (100 нг/мл) и лабеталол (100 нг/мл) в качестве внутреннего стандарта). Образцы центрифугировали при 4000 об./мин в течение 20 минут и полученные супернатанты подвергали LC-MS/MS-анализу.
Образцы для калибровочной кривой готовили следующим образом. В 96-луночный планшет следует внести микросомы печени (100 мкл/лунка) и раствор NADPH-регенерирующей системы (98 мкл/лунка). Сначала добавить по 600 мкл раствора для гашения, а затем по 2 мкл раствора для построения стандартной кривой и рабочего QC-раствора (рабочего раствора для контроля качества).
Биологический анализ
Количественное определение соединений выполняли на приборе API4000 для LC-MS/MS с ESI в режиме положительных ионов с применением мониторинга множественных реакций (MRM).
Проводили исследование для оценки метаболического превращения пролекарств (1 мкМ), а именно, соединений из примера 1, примера 1-А, примера 1-В, примера 2, примера 2-А, примера 2-В, примера 3, примера 4, примера 5, примера 6, примера 7, примера 8, примера 9, примера 10, примера 11, примера 12, примера 13, примера 14, примера 15, примера 16, примера 17, примера 21, примера 22, примера 23, примера 25, примера 26, примера 27, примера 28, примера 29, примера 30, примера 31, примера 32, примера 33, примера 34-А, примера 34-В, примера 36-А, примера 36-В, примера 37-А, примера 37-В, примера 38-А, примера 38-В, примера 39, примера 40, примера 41, примера 41-А, примера 41-В, примера 42, примера 42-А, примера 42-В, примера 43, примера 43-А, примера 43-В, примера 44, примера 44-А, примера 44-В и примера 45-А, примера 46-А, примера 46-В, примера 47-А, примера 47-В, примера 48-А, примера 48-В, в соответствующие активные формы, а именно, соединение 1е, соединение 1е-А, соединение 1е-В, соединение 34е-А, соединение 34е-В, соединение 36g-А, соединение 36g-В, соединение 36g, соединение 41с, соединение 41с-В, соединение 41с-А, соединение 43е, соединение 43е-А, соединение 43е-В, соединение 45е-А, соединение 45е-В, соединение 47е-А и соединение 47е-В, в присутствии микросом печени человека. Результаты суммированы и приведены в Таблице 2.
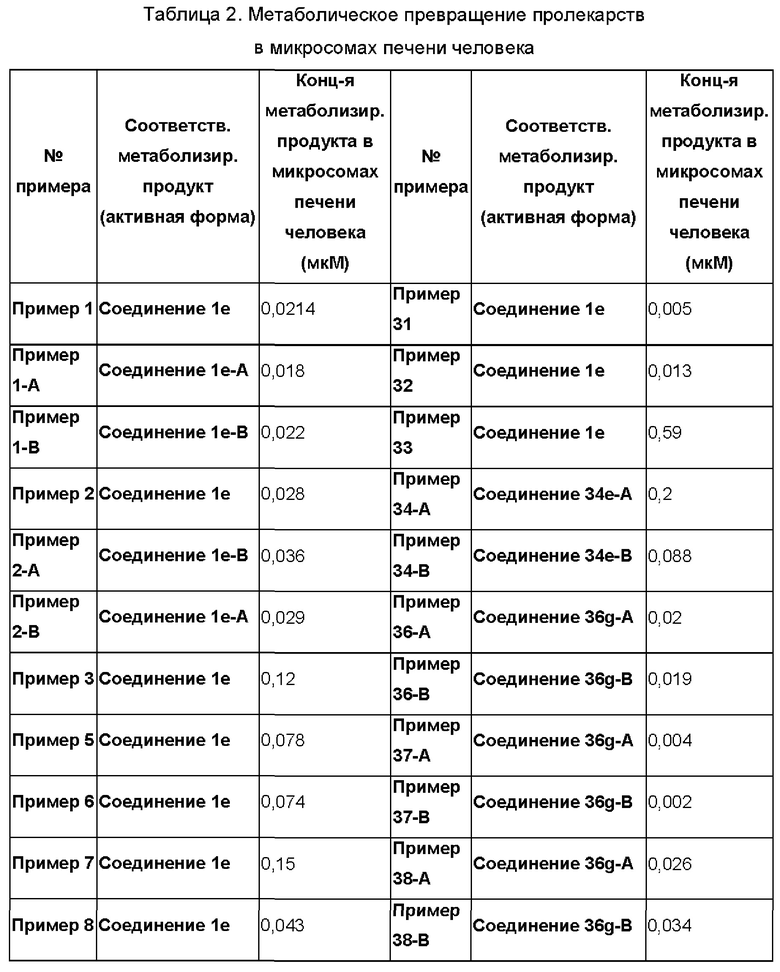
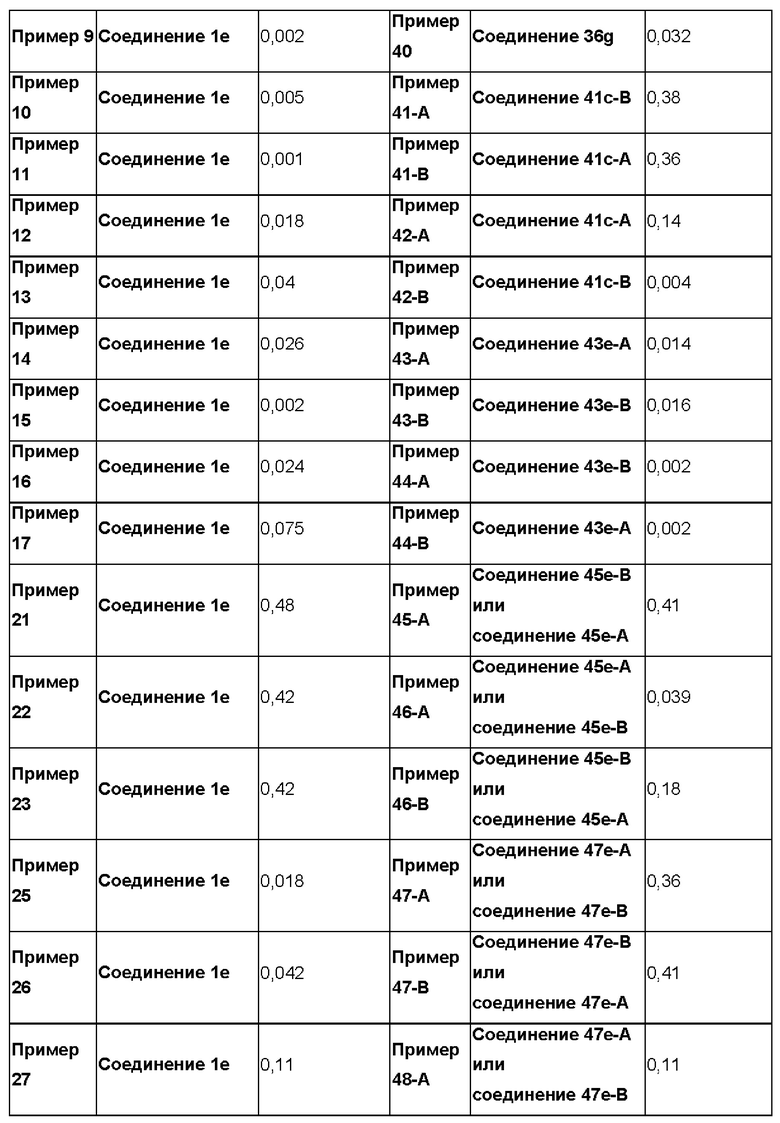

Пример 51
Противовирусная эффективность in vivo соединения из примера 43-А в AAV-HBV мышиной модели
Животная модель
Беспатогенных самцов мышей C57BL/6 в возрасте 4-6 недель приобретали у Шанхайского центра лабораторных животных Китайской академии наук (SLAC) и размещали в помещении для ухода за животными в по отдельности вентилируемых клетках в условиях регулируемых температуры и освещения, следуя указаниям Комитета по содержанию и уходу за животными. Вирус AAV-HBV получали из Пекинского института молекулярной медицины FivePlus (Beijing FivePlus Molecular Medicine Institute) (Пекин, Китай). Рекомбинантный вирус несет 1,3 копии генома HBV, упакованные в капсиды AAV, серотипа 8 (AAV8). Мышам C57BL/6 в хвостовую вену вводили по 200 мкл препарата рекомбинантного вируса, разбавленного в забуференном физиологическом растворе. На 14-е сутки у мышей отбирали кровь для измерения поверхностного антигена HBV (HBsAg) и геномной ДНК HBV в сыворотке крови и затем животных рандомизировали по группам в соответствии с этими биомаркерами HBV.
Измерение биомаркеров HBV
Уровни HBsAg и HBeAg в сыворотке крови измеряли, используя наборы для хемилюминесцентного иммунохимического анализа (CLIA) (Autobio Diagnostics Co., Ltd., Чжэнчжоу, Китай) в соответствии с инструкциями производителя. Нижний предел обнаружения для HBsAg составлял 0,05 международных единиц (МЕ)/мл. Для получения значений в пределах линейного диапазона стандартной кривой использовали разведение сыворотки крови в 500 раз.
ДНК HBV из сыворотки крови экстрагировали, используя набор для определения ДНК и вирусных нуклеиновых кислот (NA) на приборе MagNA Pure 96 в пробах малого объема (MagNA Pure 96 DNA and Viral NA Small Volume Kit) (Roche), следуя инструкциям производителя. Анализ образцов ДНК проводили с использованием количественной ПЦР (кПЦР) в режиме реального времени, применяя набор HBV-специфичных праймеров и зонда для специфичной амплификации и детекции участка генома HBV длиной 128 пар оснований (п.о.), начиная от нуклеотида 2969 до 3096. Последовательности праймеров и зонда:
прямой праймер: AAGAAAAACCCCGCCTGTAA;
обратный праймер: CCTGTTCTGACTACTGCCTCTCC;
HBV-зонд: 5'TAMRA-CCTGATGTGATGTTCTCCATGTTCAGC-BHQ2-3'.
Антитела к HBsAg в сыворотке крови тестировали, используя наборы для определения антител к HBsAg посредством CLIA (antiHBs CLIA) (Autobio Diagnostics Co., Ltd., Чжэнчжоу, Китай) и мышиное антитело к иммуноглобулину G (IgG), конъюгированное с биотином (0,5 мг/мл), из набора для определения методом иммуноферментных пятен (Elispot) Mabtech В. Конъюгат (антитело к IgG)-биотин разбавляли в PBS (забуференный фосфатом физиологический раствор) до конечной концентрации 1 мкг/мл. По 25 мкл раствора мышиного антитела к IgG смешивали с образцами сыворотки крови в лунках планшета из набора antiHBs CLIA для проведения 1-часовой инкубации. Затем следует промыть планшет и добавить стрептавидин-HRP (пероксидаза хрена) для проведения 1-часовой инкубации при комнатной температуре. После повторения стадии промывки смешать субстрат А и В из набора CLIA и добавить по 50 мкл этой смеси в каждую лунку. Через 5 минут инкубирования при комнатной температуре планшет прочитывали на планшетном ридере Envision (PerkinElmer) для измерения люминесценции.
План исследования и результаты
Мышиную модель с высоким уровнем экспрессии как ДНК HBV, так и HBsAg создавали посредством введения мышам C57BL/6 рекомбинантного аденоассоциированного вируса (AAV), несущего реплицируемый геном HBV (AAV-HBV). В случае длительной виремии HBV и полностью компетентной иммунной системы AAV-HBV мышиную модель использовали для оценки противовирусной эффективности агонистов TLR7, следуя плану исследования, показанному в Таблице 3.

ПО означает перорально.
Конкретно, группам 2 и 3 перорально вводили соединение из примера 43-А в дозе 10 мг/кг один раз в двое суток (QOD) и один раз в неделю (QW), соответственно, а контрольная группа 1 получала только разбавитель. При вводимом объеме 10 мл/кг образец на основе соединения из примера 43-А (1 мг/мл) готовили в виде аддукта, содержащего 2% Klucel марки LF, 0,5% TPGS (токоферил-полиэтилен-гликоль-1000-сукцинат), 0,09% метилпарабенов, 0,01% пропилпарабенов в воде. На протяжении всего исследования животные получали лечение в общей сложности в течение 42 суток и у них два раза в неделю отбирали кровь из поднижнечелюстной вены для получения сыворотки. Образцы сыворотки крови подвергали анализу на предмет биомаркеров HBV.
Как показано на Фиг. 4, лечение соединением из примера 43-А в дозе 10 мг/кг QOD приводила к резкому снижению уровней ДНК HBV (>3 log) и HBsAg (>2,8 log). К окончанию длившегося 42 суток лечения уровни этих вирусных маркеров не детектировались и становились меньше нижнего предела количественного определения (LLOQ). Даже при менее частом дозировании в режиме QW введение соединения из примера 43-А значительно снижало уровни как ДНК HBV (>2 log), так и HBsAg (>2,8 log). Кроме того, лечение соединением из примера 43-А в дозе 10 мг/кг, независимо от введения в режиме QOD и QW, индуцировала выработку антител к HBsAg до значительного уровня. В заключение отметим, что соединение из примера 43-А демонстрировало хорошую антиHBV-активность in vivo, снижая уровень вирусных маркеров HBV и стимулируя выработку HBV-специфичных антител.
Пример 52
Противовирусная эффективность in vivo соединения из примера 41-А в AAV-HBV мышиной модели
Противовирусную эффективность соединения из примера 41-А оценивали в той же самой AAV-HBV модели, следуя плану исследования, приведенному в Таблице 4, с теми же методами измерения уровня биомаркеров HBV, которые описаны в примере 51.
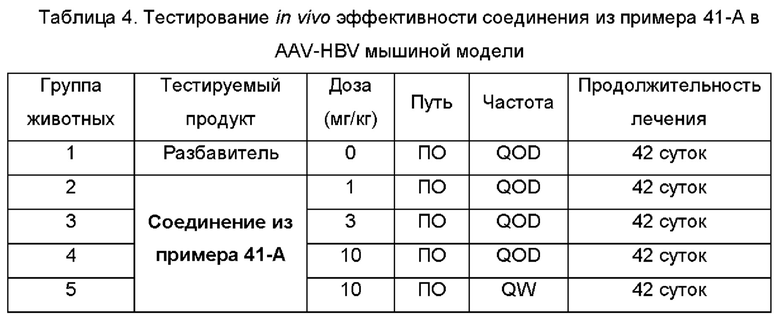
Конкретно, группам 2, 3 и 4 перорально вводили соединение из примера 41-А в дозах 1, 3 и 10 мг/кг QOD, соответственно. Группа 5 получала лечение в дозе 10 мг/кг QW, в то время как группа 1 получала только разбавитель. При вводимом объеме 10 мл/кг образец на основе соединения из примера 41-А (0,1; 0,3 и 1 мг/мл) готовили в виде аддукта, содержащего 2% Klucel марки LF, 0,5% TPGS, 0,09% метилпарабенов, 0,01% пропилпарабенов в воде. На протяжении всего исследования животные получали лечение в общей сложности в течение 42 суток и у них два раза в неделю отбирали кровь из поднижнечелюстной вены для получения сыворотки. Образцы сыворотки крови подвергали анализу на предмет биомаркеров HBV.
Как показано на Фиг. 5, лечение соединением из примера 41-А в дозах 1, 3 и 10 мг/кг QOD дозозависимым образом снижала уровни ДНК HBV и HBsAg. К окончанию длившегося 42 суток лечения при применении всех трех доз удалось снизить содержание этих вирусных маркеров до более низких или близких к LLOQ уровней. Даже при менее частом дозировании в режиме QW введение соединения из примера 41-А в дозе 10 мг/кг также снижало содержание ДНК HBV и HBsAg до недетекгируемых уровней к окончанию лечения. Кроме того, лечение соединением из примера 41-А индуцировало выработку антител к HBsAg до значительно более высоких уровней, чем обработка разбавителем. В заключение отметим, что соединение из примера 41-А демонстрировало хорошую антиHBV-активность in vivo, снижая уровень вирусных маркеров HBV и стимулируя выработку HBV-специфичных антител.
Пример 53
Противовирусная эффективность in vivo соединения из примера 42-А в AAV-HBV мышиной модели
Противовирусную эффективность соединения из примера 42-А оценивали в той же самой AAV-HBV модели, следуя плану исследования, приведенному в Таблице 5, с теми же методами измерения уровня биомаркеров HBV, которые описаны в примере 51.
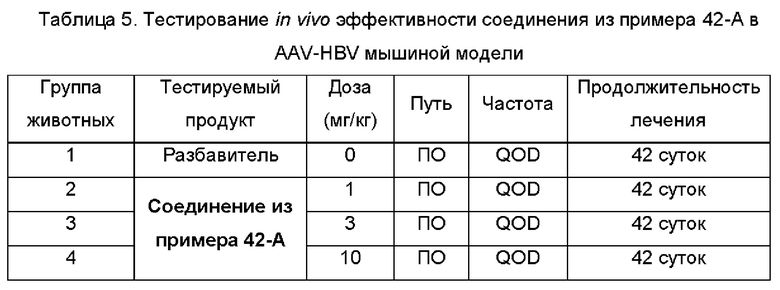
Конкретно, группам 2, 3 и 4 перорально вводили соединение из примера 42-А в дозах 1, 3 и 10 мг/кг QOD, соответственно, в то время как группа 1 получала только разбавитель. При вводимом объеме 10 мл/кг образец на основе соединения из примера 42-А (0,1; 0,3 и 1 мг/мл) готовили в виде аддукта, содержащего 2% Klucel марки LF, 0,5% TPGS, 0,09% метилпарабенов, 0,01% пропилпарабенов в воде. На протяжении всего исследования животные получали лечение в общей сложности в течение 42 суток и у них два раза в неделю отбирали кровь из поднижнечелюстной вены для получения сыворотки. Образцы сыворотки крови подвергали анализу на предмет биомаркеров HBV.
Как показано на Фиг. 6, лечение соединением из примера 42-А в дозах 1, 3 и 10 мг/кг QOD во всех случаях оказалась эффективной в отношении снижения уровней ДНК HBV и HBsAg. Несмотря на то, что более высокие дозы способствовали более быстрому выведению ДНК HBV и HBsAg, к окончанию длившегося 42 суток лечения при применении всех трех доз удалось снизить содержание этих вирусных маркеров до более низких или близких к LLOQ уровней. Во всех группах, получавших лечение соединением из примера 42-А, наблюдали значительно высокие уровни антител к HBsAg. В заключение отметим, что соединение из примера 42-А демонстрировало хорошую антиHBV-активность in vivo, снижая уровень вирусных маркеров HBV и стимулируя выработку HBV-специфичных антител.
Пример 54
Противовирусная эффективность in vivo соединения из примера 41-В в AAV-HBV мышиной модели
Противовирусную эффективность соединения из примера 41-В оценивали в той же самой AAV-HBV модели, следуя плану исследования, приведенному в Таблице 6, с теми же методами измерения уровня биомаркеров HBV, которые описаны в примере 51.
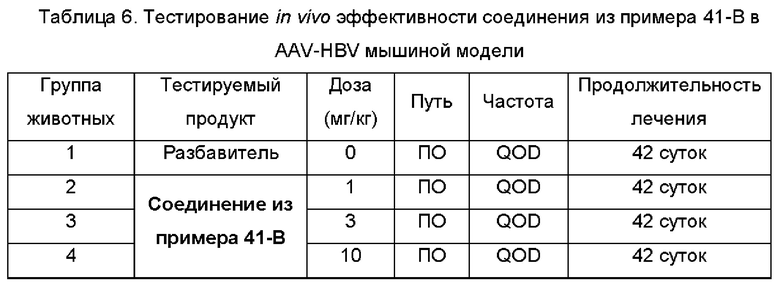
Конкретно, группам 2, 3 и 4 перорально вводили соединение из примера 41-В в дозах 1, 3 и 10 мг/кг QOD, соответственно, в то время как группа 1 получала только разбавитель. При вводимом объеме 10 мл/кг образец на основе соединения из примера 42-А (0,1; 0,3 и 1 мг/мл) готовили в виде аддукта, содержащего 2% Klucel марки LF, 0,5% TPGS, 0,09% метилпарабенов, 0,01% пропилпарабенов в воде. На протяжении всего исследования животные получали лечение в общей сложности в течение 42 суток и у них два раза в неделю отбирали кровь из поднижнечелюстной вены для получения сыворотки. Образцы сыворотки крови подвергали анализу на предмет биомаркеров HBV.
Как показано на Фиг. 7, обработка соединением из примера 41-В в дозах 1, 3 и 10 мг/кг QOD во всех случаях оказалась эффективной в отношении снижения уровней ДНК HBV и HBsAg. К окончанию длившегося 42 суток лечения при применении всех трех доз удалось снизить содержание этих вирусных маркеров до уровней ниже LLOQ. Во всех группах, получавших лечение соединением из примера 41-В, наблюдали высокие уровни антител к HBsAg по сравнению с группой, получавшей разбавитель. В заключение отметим, что соединение из примера 41-В демонстрировало антиHBV-активность in vivo, снижая уровень вирусных маркеров HBV и стимулируя выработку HBV-специфичных антител.
Пример 55
ФК исследование разовой дозы на самцах крыс Wister-Han
Для оценки фармакокинетических свойств тестируемых соединений на самцах крыс Wister-Han проводили ФК исследование разовой дозы. Двум группам животных через желудочный зонд (РОЕ) вводили соответствующее соединение. Образцы крови (приблизительно по 20 мкл) отбирали через яремную вену или из альтернативного места через 15 мин, 30 мин, 1 ч, 2 ч, 4 ч, 7 ч и 24 ч после введения дозы группам. Образцы крови помещали в пробирки, содержащие антикоагулянт EDTA(этилендиаминтетрауксусная кислота)-K2, и центрифугировали при 5000 об./мин в течение 6 мин при 4°С для получения плазмы крови из образцов. После центрифугирования полученную плазму переносили в чистые пробирки для проведения биоанализа как пролекарства, так и активной формы посредством LC/MS/MS. В группах, которым вводили пролекарство, концентрация пролекарств в образцах плазмы крови была ниже предела обнаружения "Тестируемое соединение" в Таблице 8 использовали в качестве внутреннего стандарта для исследования in vivo метаболита (активной формы) "вводимого соединения". Фармакокинетические параметры рассчитывали, используя модуль для некомпартментного анализа из программного обеспечения WinNonlin® Professional 6,2. Максимальную концентрацию (Cmax) регистрировали непосредственно из экспериментальных наблюдений. Площадь под кривой зависимости концентрации в плазме крови от времени (AUC0-t) рассчитывали, используя линейное правило трапеции до последней детектируемой концентрации включительно.
Cmax и AUC0-last (AUC в интервале от нулевой отметки до последней временной точки) являются двумя важными ФК параметрами, связанными с эффективностью in vivo тестируемого соединения. Соединения с более высокими значениями Cmax и AUC0-last будут обладать более хорошей эффективностью in vivo. Результаты, полученные для ФК параметров после перорального введения активных форм и соединений-конкурентов, приведены в Таблице 7. ФК параметры для пролекарств сведены в Таблицу 8.
После перорального введения пролекарств в плазме крови наблюдали присутствие активных форм и поэтому проводили их тестирование. Приведенные в качестве примеров пролекарства по настоящему изобретению (соединения из примеров 41-В, 42-А, 42-В, 43-А, 45-А и 45-В) неожиданным образом продемонстрировали во много раз более высокие значения Cmax (увеличение в 5-175 раз) и AUC0-last (увеличение в 2,5-56 раз) по сравнению с референсными соединениями (GS9620, S-2 и S-3) и соединениями, упомянутыми в настоящем изобретении (соединениями 41с-А, 41с-В и 43е-А), все они представляют собой активные формы. Эти результаты четко демонстрировали неожиданное превосходство пролекарств над активными формами по ФК параметрам, что приводило к лучшей эффективности in vivo.
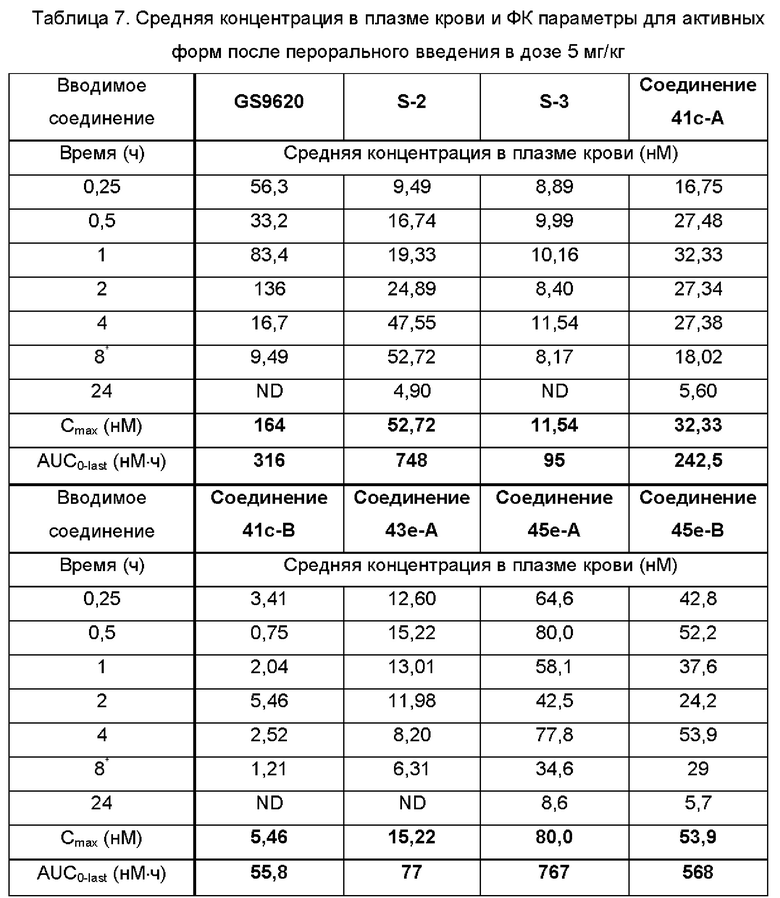
* 7 ч для соединения 41с-А, соединения 41с-В и соединения 43е-А.
ND означает "не определено".
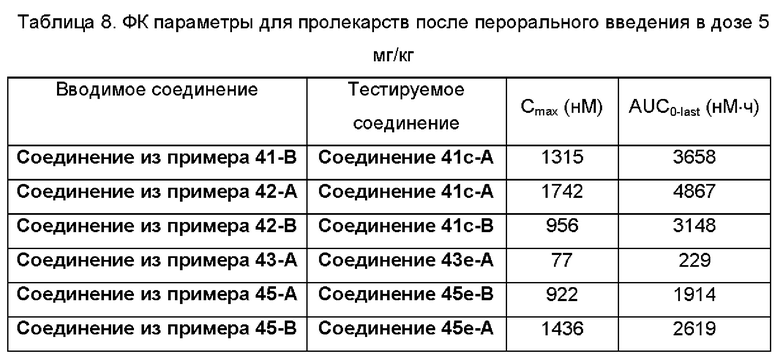
Пример 56
Исследование растворимости с применением LYSA
Исследование с применением LYSA (анализ растворимости лиофилизованных препаратов) используют для определения растворимости в воде тестируемых соединений. Образцы готовили в двух повторах, исходя из 10 мМ концентрированного раствора в DMSO. После выпаривания DMSO с использованием центрифужного вакуумного испарителя соединения растворяли в 0,05 М фосфатном буфере (рН 6,5), перемешивали в течение одного часа и встряхивали в течение двух часов. По прошествии одной ночи растворы фильтровали, используя микротитрационный фильтровальный планшет. Затем фильтрат и его же в разведении 1/10 анализировали с применением HPLC с детекцией в ультрафиолете (HPLC-UV). Помимо этого из 10 мМ концентрированных растворов готовили разведения для получения калибровочной кривой по четырем точкам и использовали для определения растворимости соединений. Результаты выражали в мкг/мл. В том случае, когда величина процентного содержания образца, измеренного в растворе после выпаривания, деленного на рассчитанное максимальное количество образца, превышала 80%, указывали, что растворимость превышает это значение.
Результаты LYSA показаны в Таблице 9. Было ясно, что растворимость активных форм неожиданным образом повышалась в число раз от 10 до более 200 в случае превращения их в различные пролекарства.
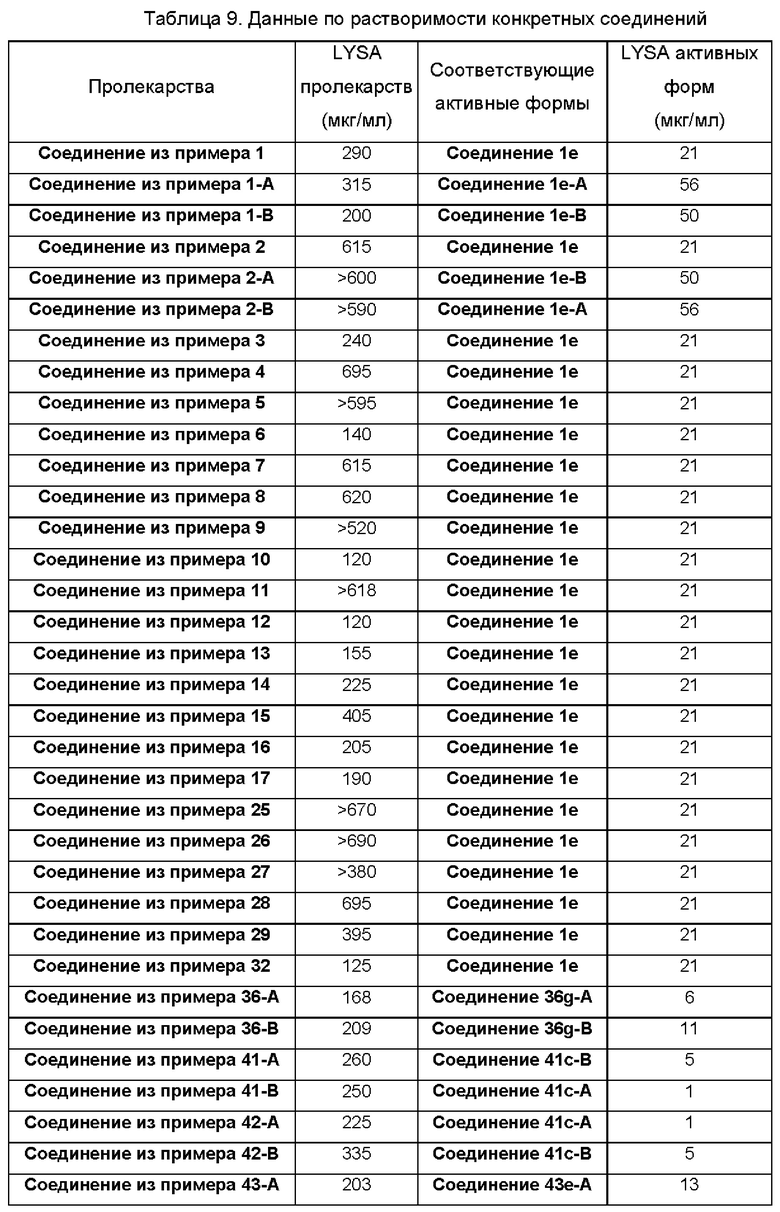
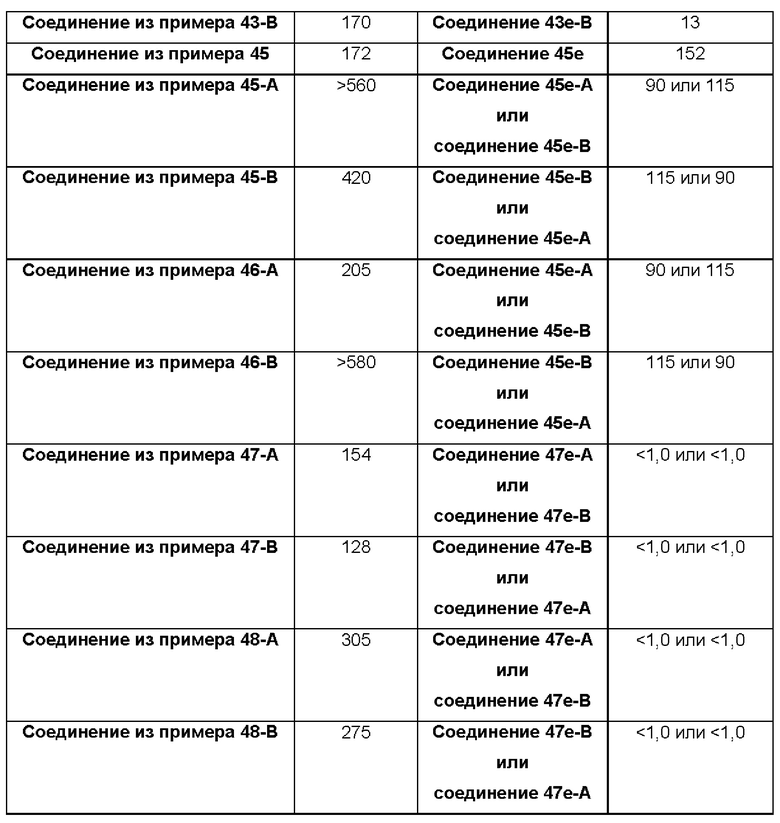
Пример 57
Исследование с использованием воротной вены
Цель этого исследования состояла в том, чтобы понять, остается ли пролекарство неизменным при всасывании через кишечник в воротную систему кровообращения и продемонстрировать первичное место, где происходит превращение.
Хирургическая процедура канюлирования воротной вены (PVC) и канюлирования сонной артерии (САС)
Операцию проводили под анестезией пентобарбиталом/изофлураном. Кратко, после дезинфекции области брюшины бетадином и 70%-ным изопропиловым спиртом делали небольшой разрез по средней линии живота. Вытягивали слепую кишку, идентифицировали мезентериальную вену и обособляли примерно 5 мм сосуда. Свободную лигатуру располагали проксимально и дистальный конец вены лигировали. Следует сделать небольшой разрез (ровно такой, чтобы выполнить введение катетера) в обособленной вене и ввести полиуретановый (PU) катетер в направлении печени на соответствующую длину. Катетер закрепляли на месте путем обвязывания свободной лигатуры вокруг канюлированного сосуда. Слепую кишку помещали обратно в брюшную полость. В правой стенке брюшной полости делали отверстие, чтобы свободно проходил конец катетера. Катетер прикрепляли шовным материалом к стенке брюшной полости. Разрез в брюшной мышце зашивали, используя шовный материал. В области лопатки делали небольшой разрез, служащий местом выхода катетера. Катетер туннелировали под кожу и выводили через разрез в области лопатки. В области лопатки накладывали фиксирующий шов. Проверяли проходимость катетера и затем его выводили из подкожного пространства в дорсальную область шеи. После осторожного протирания поверхности брюшную полость зашивали. Затем канюлировали левую сонную артерию, вводя полиэтиленовый катетер марки РЕ50. Оба временно выведенных на поверхность тела катетера прочно закрепляли в дорсальной области шеи и фиксировали. Затем животных оставляли в своих клетках для восстановления и использовали для исследования в течение по меньшей мере 3-х суток после операции. Для поддержания проходимости все катетеры промывали один раз в сутки сильной струей, используя гепаринизированный физиологический раствор.
ФК исследование в случае перорального введения на крысах с двойным PVC/CAC канюлированием
Животным не давали корма в течение ночи (n=3) и затем перорально через желудочный зонд вводили соединения (10 мг/кг, 10 мл/кг). Образцы крови (по 60 мкл) отбирали одновременно из катетеров воротной вены и сонной артерии в моменты времени 0,083; 0,25; 0,5; 1, 2, 4, 7, 24 ч. Все образцы крови должны быть перенесены в микроцентрифужные пробирки, содержащие 2 мкл K2-EDTA (0,5 М) в качестве антикоагулянта, и помещены на натуральный лед. Затем образцы крови для получения плазмы должны быть обработаны центрифугированием при температуре приблизительно 4°С, 3000 g в пределах получаса. Образцы плазмы крови должны храниться в полипропиленовых пробирках в быстро замороженном на сухом льду состоянии и при -70±10°С до проведения LC/MS/MS анализа.
Определяли и анализировали фармакокинетические параметры (среднее значение ± стандартное отклонение (SD), n=3) для пролекарств и активных форм в образцах из воротной вены и сонной артерии после перорального введения пролекарств (10 мг/кг) крысам с канюлированной воротной веной. Результаты тестирования соединений из примеров 1-В, 41-А, 41-В, 42-А и 43-А суммированы ниже.
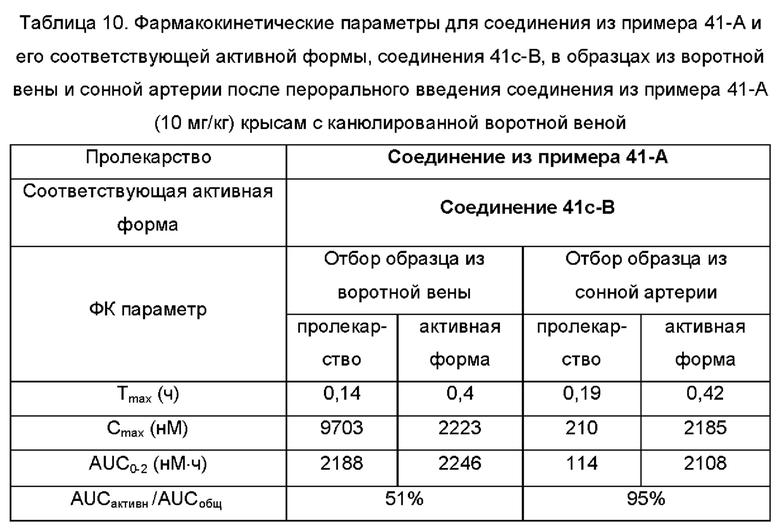


На основании приведенных выше результатов был сделан вывод, что первичным местом, где происходит превращение пролекарства, скорее была печень, а не кишечник, поскольку значение соотношения AUCактивн./AUCобщ. было выше в образцах из сонной артерии по сравнению с AUCактивн./AUCобщ. в образцах из воротной вены.
--->
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> Ф. ХОФФМАНН-ЛЯ РОШ АГ
<120> 7-замещенные сульфонимидоилпуриноны для лечения и профилактики
вирусной инфекции
<130> P33761
<160> 3
<170> PatentIn version 3.5
<210> 1
<211> 20
<212> ДНК
<213> Вирус гепатита B
<400> 1
aagaaaaacc ccgcctgtaa 20
<210> 2
<211> 23
<212> ДНК
<213> Вирус гепатита B
<400> 2
cctgttctga ctactgcctc tcc 23
<210> 3
<211> 27
<212> ДНК
<213> Вирус гепатита B
<400> 3
cctgatgtga tgttctccat gttcagc 27
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| 7-ЗАМЕЩЕННЫЕ СУЛЬФОНИМИДОИЛПУРИНОНЫ ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ВИРУСНОЙ ИНФЕКЦИИ | 2017 |
|
RU2838538C1 |
| НОВЫЕ СОЕДИНЕНИЯ И ПРОИЗВОДНЫЕ СУЛЬФОНИМИДОИЛПУРИНОНА ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ВИРУСНОЙ ИНФЕКЦИИ | 2016 |
|
RU2745269C2 |
| ГИДРОКСИПУРИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2685417C1 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ, ИХ КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2007 |
|
RU2474582C2 |
| Новые соединения пиридопиримидинона для модулирования каталитической активности гистонлизиндеметилаз (KDMS) | 2015 |
|
RU2684396C2 |
| ПРОИЗВОДНЫЕ ДИГИДРОПТЕРИДИНОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2010 |
|
RU2559881C2 |
| МОДУЛЯТОРЫ РЕЦЕПТОРА НМДА И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2621049C2 |
| ПРОИЗВОДНЫЕ АЗАСПИРОАЛКАНОВ В КАЧЕСТВЕ ИНГИБИТОРОВ МЕТАЛЛОПРОТЕАЗ | 2004 |
|
RU2379303C2 |
| Гетероциклические соединения в качестве ингибиторов PAD | 2018 |
|
RU2764243C2 |
| МОДУЛЯТОРЫ ПРОТЕОЛИЗА И СООТВЕТСТВУЮЩИЕ СПОСОБЫ ПРИМЕНЕНИЯ | 2019 |
|
RU2805511C2 |

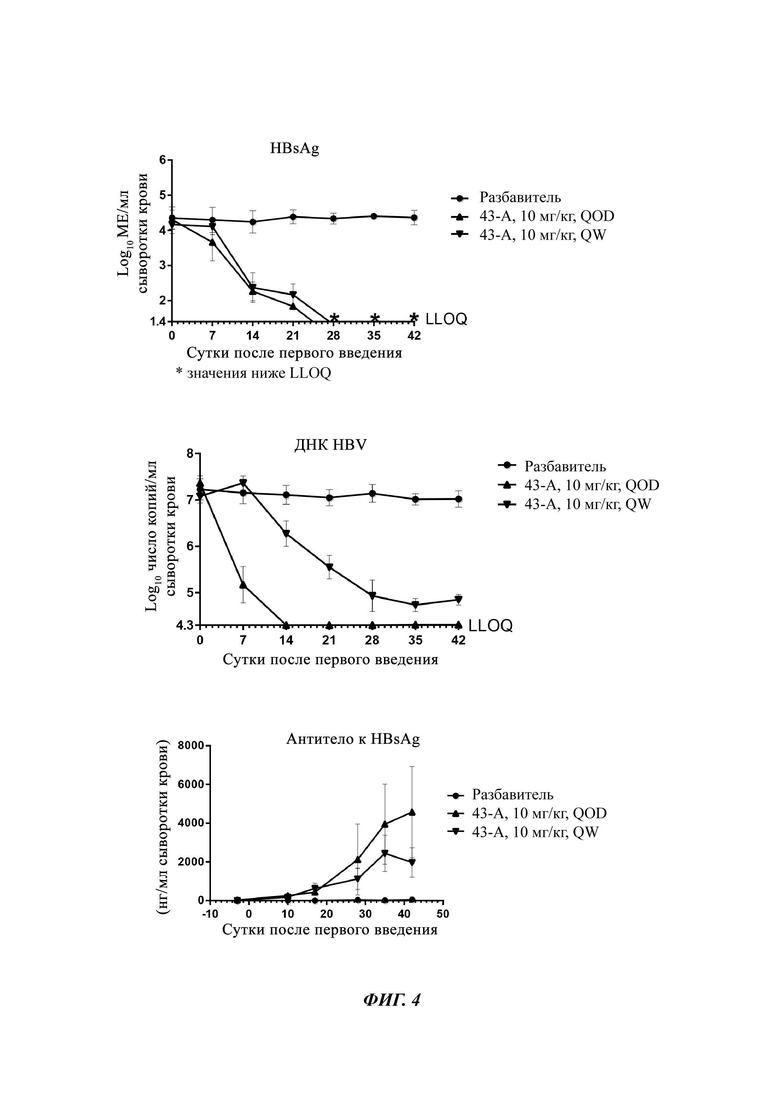
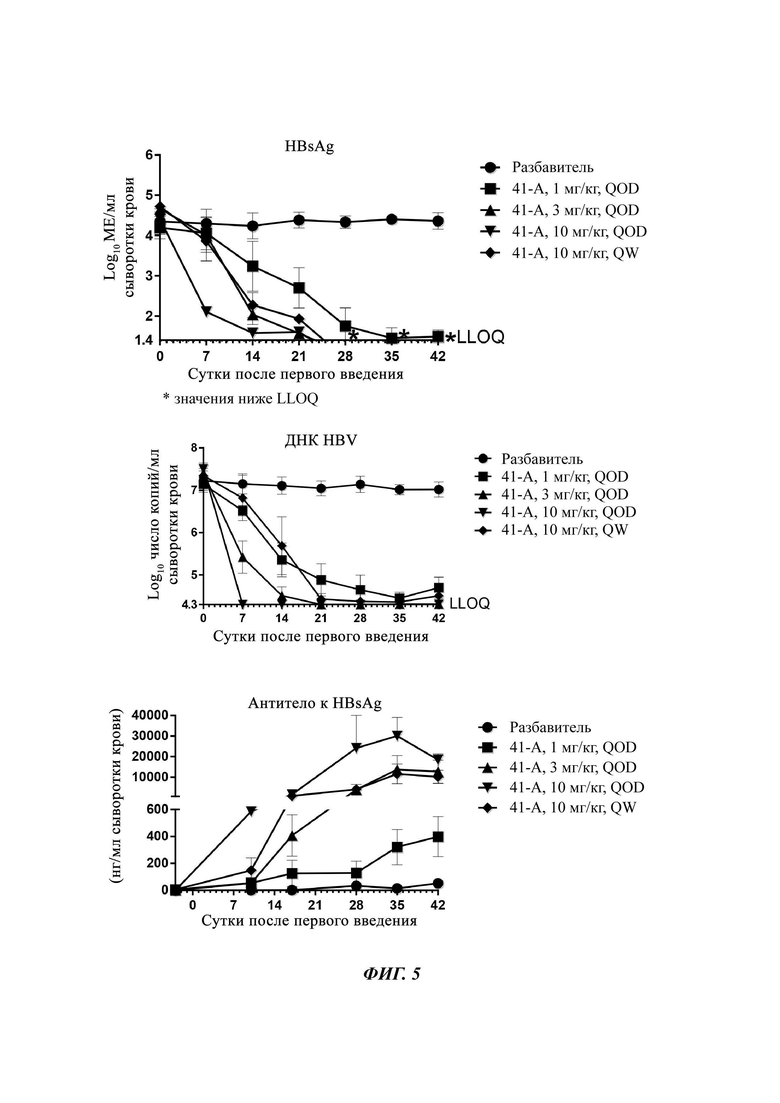
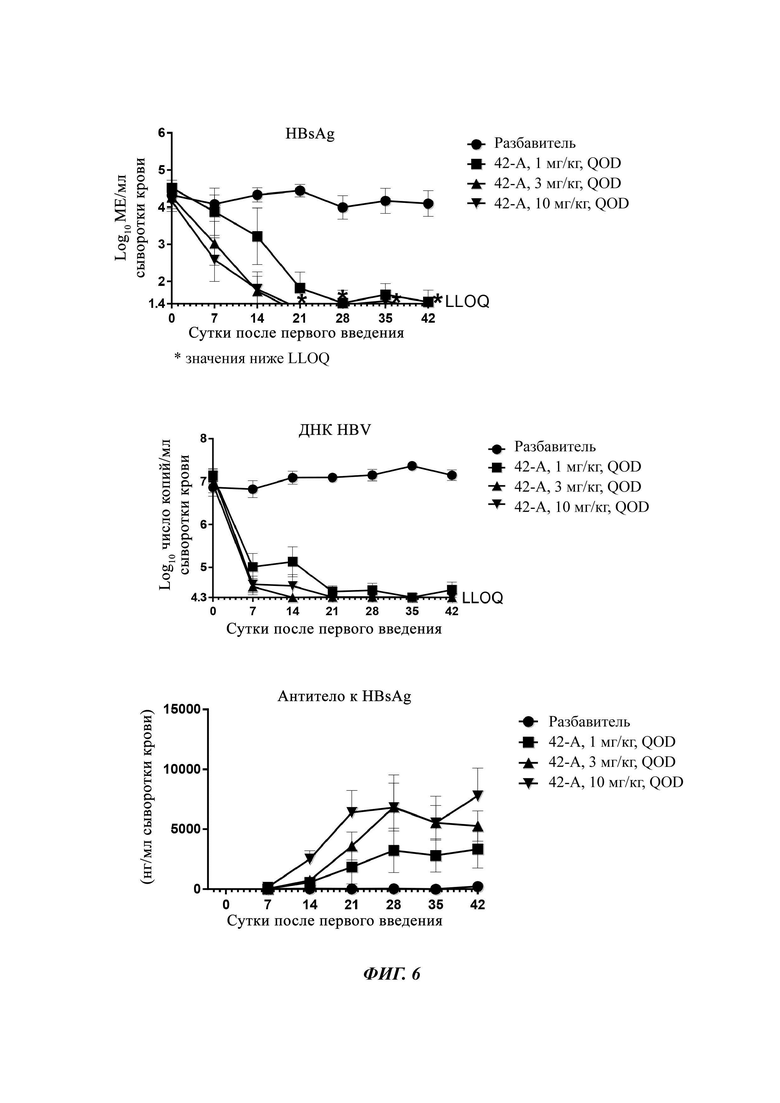
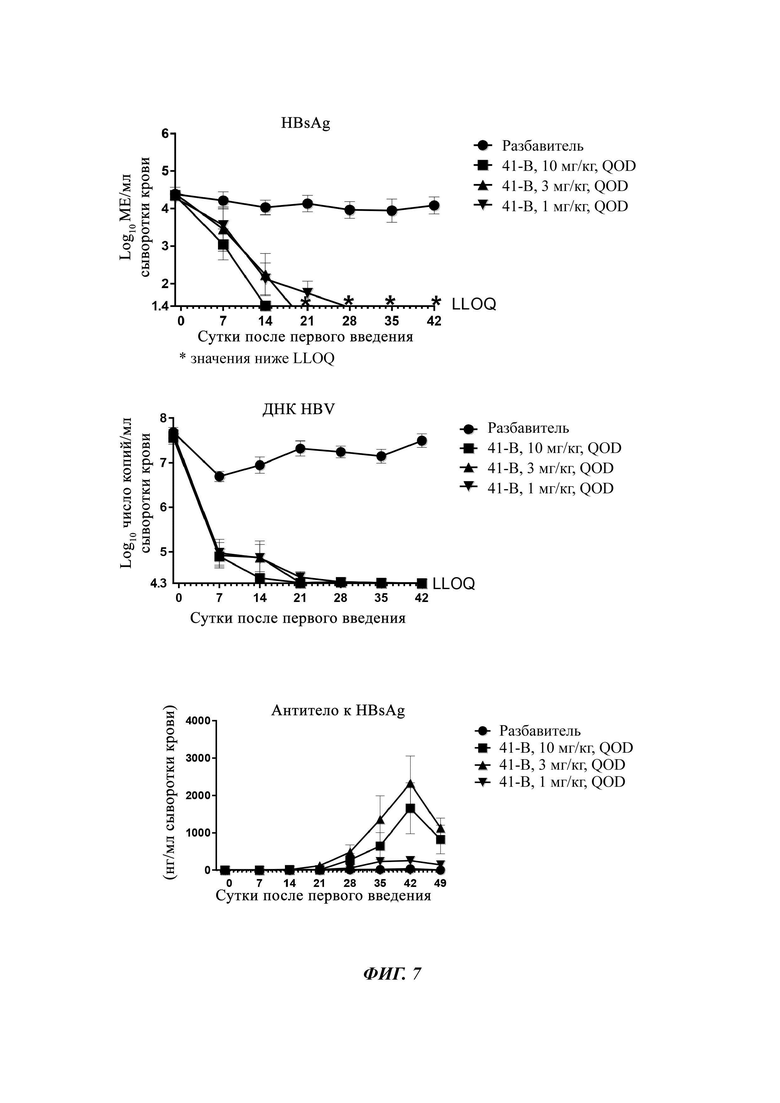
Изобретение относится к соединению формулы (I)
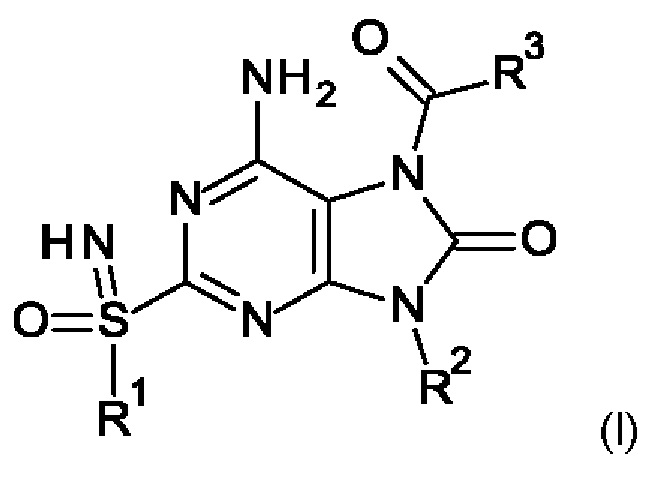 ,
,
где R1 представляет собой С1-6алкил; R2 представляет собой бензил, при этом указанный бензил не замещен или замещен одним, двумя или тремя заместителями, независимо выбранными из галогена и С1-6алкила; R3 представляет собой -NR4R5, где R4 представляет собой С1-6алкил или С1-6алкоксиС1-6алкил; R5 представляет собой (С1-6алкил)2NCOOC1-6алкил, С1-6алкоксиС1-6алкил, С1-6алкоксикарбонил(С1-6алкил)аминоС1-6алкил, С1-6алкоксикарбонил(фенил)С1-6алкил, С1-6алкоксикарбонилС1-6алкил, С1-6алкоксикарбонилоксиС1-6алкил, C1-6алкил, С1-6алкилкарбонил(С1-6алкил)аминоС1-6алкил или пирролидинилкарбамоилоксиС1-6алкил или R4 и R5 вместе с атомом азота, к которому они присоединены, образуют гетероциклил; или его фармацевтически приемлемой соли, энантиомеру или диастереомеру; к композициям, содержащим данные соединения, и к способам применения этих соединений в качестве агонистов Toll-подобного рецептора 7 (TLR7). 9 н. и 16 з.п. ф-лы, 7 ил., 14 табл., 57 пр.
1. Соединение формулы (I),
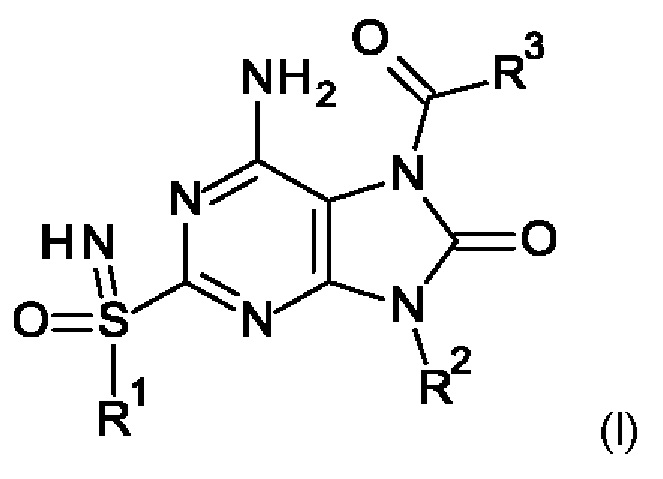
где
R1 представляет собой С1-6алкил;
R2 представляет собой бензил, при этом указанный бензил не замещен или замещен одним, двумя или тремя заместителями, независимо выбранными из галогена и С1-6алкила;
R3 представляет собой -NR4R5, где
R4 представляет собой С1-6алкил или С1-6алкоксиС1-6алкил;
R5 представляет собой (С1-6алкил)2NCOOC1-6алкил, С1-6алкоксиС1-6алкил, С1-6алкоксикарбонил(С1-6алкил)аминоС1-6алкил, С1-6алкоксикарбонил(фенил)С1-6алкил, С1-6алкоксикарбонилС1-6алкил, С1-6алкоксикарбонилоксиС1-6алкил, C1-6алкил, С1-6алкилкарбонил(С1-6алкил)аминоС1-6алкил или пирролидинилкарбамоилоксиС1-6алкил; или
R4 и R5 вместе с атомом азота, к которому они присоединены, образуют гетероциклил;
или его фармацевтически приемлемые соль, энантиомер или диастереомер; при условии, что исключены
6-амино-9-бензил-2-(пропилсульфонимидоил)-7-(пирролидин-1-карбонил)пурин-8-он;
6-амино-9-бензил-7-(пиперидин-1-карбонил)-2-(пропилсульфонимидоил)пурин-8-он;
6-амино-9-бензил-7-(морфолин-4-карбонил)-2-(пропилсульфонимидоил)пурин-8-он;
6-амино-9-бензил-7-(3,3-диметилпирролидин-1-карбонил)-2-(пропилсульфонимидоил)пурин-8-он;
этил-1-[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]пирролидин-2-карбоксилат;
6-амино-7-(2-азаспиро[3.3]гептан-2-карбонил)-9-бензил-2-(пропилсульфонимидоил)пурин-8-он;
6-амино-9-бензил-7-(2-окса-6-азаспиро[3.3]гептан-6-карбонил)-2-(пропилсульфонимидоил)пурин-8-он;
6-амино-9-бензил-7-(3,3-дифторпирролидин-1-карбонил)-2-(пропилсульфонимидоил)пурин-8-он;
6-амино-9-бензил-7-(3-фтор-3-метил-пирролидин-1-карбонил)-2-(пропилсульфонимидоил)пурин-8-он;
и их энантиомеры или диастереомеры.
2. Соединение по п. 1, где
R1 представляет собой С1-6алкил;
R2 представляет собой бензил, при этом указанный бензил не замещен или замещен галогеном или С1-6алкилом;
R3 представляет собой азетидинил;
пиперазинил, замещенный С1-6алкилом;
пиперидинил, замещенный пиперидинилом;
пирролидинил; или
-NR4R5, где
R4 представляет собой С1-6алкил или С1-6алкоксиС1-6алкил;
R5 представляет собой (С1-6алкил)2NCOOC1-6алкил, С1-6алкоксиС1-6алкил, С1-6алкоксикарбонил(С1-6алкил)аминоС1-6алкил, С1-6алкоксикарбонил(фенил)С1-6алкил, С1-6алкоксикарбонилС1-6алкил, C1-6алкоксикарбонилоксиС1-6алкил, С1-6алкил, С1-6алкилкарбонил(C1-6алкил)аминоС1-6алкил или пирролидинилкарбамоилоксиС1-6алкил.
3. Соединение по п. 1 или 2, где
R1 представляет собой этил или пропил;
R2 представляет собой бензил, бромбензил, хлорбензил, фторбензил или метилбензил;
R3 представляет собой азетидинил;
4-метилпиперазинил;
пиперидинилпиперидинил;
пирролидинил; или
-NR4R5, где
R4 представляет собой метил, этил, пропил или метоксиэтил;
R5 представляет собой ацетил(метил)аминоэтил, бутил, бутил(метил)карбамоилоксиэтил, диэтилкарбамоилоксиэтил, этоксикарбонил(метил)аминоэтил, этоксикарбонилэтил, этоксикарбонилизобутил, этоксикарбонилизопентил, этоксикарбонилметил, этоксикарбонилоксиэтил, этоксикарбонил(фенил)этил, этил, изобутил, изопропоксикарбонилизопентил, изопропоксикарбонил(фенил)этил, изопропил, метоксикарбонил(метил)аминоэтил, метоксиэтил, метоксипропил, пропил, пропил(метил)карбамоилоксиэтил, пирролидинилкарбамоилоксиэтил, трет-бутоксикарбонил(метил)аминоэтил, трет-бутоксикарбонилэтил, трет-бутоксикарбонилизопентил или трет-бутоксикарбонил(фенил)этил.
4. Соединение по п. 3, где R3 представляет собой азетидинил, 4-метилпиперазинил, пиперидинилпиперидинил, пирролидинил, ацетил(метил)аминоэтил(метил)амино, бис(метоксиэтил)амино, бутил(этил)амино, бутил(метил)амино, бутил(метил)карбамоилоксиэтил(метил)амино, диэтилкарбамоилоксиэтил(метил)амино, этоксикарбонил(метил)аминоэтил(метил)амино, этоксикарбонилэтил(метил)амино, этоксикарбонилизобутил(метил)амино, этоксикарбонилизопентил(метил)амино, этоксикарбонилметил(метил)амино, этоксикарбонилоксиэтил(метил)амино, этоксикарбонил(фенил)этил(метил)амино, этил(метил)амино, изобутил(метил)амино, изопропоксикарбонилизопентил (метил)амино, изопропоксикарбонил(фенил)этил(метил)амино, изопропил(метил)амино, метоксикарбонил(метил)аминоэтил(метил)амино, метоксиэтил(этил)амино, метоксиэтил(метил)амино, метоксиэтил(пропил)амино, метоксипропил(метил)амино, пропил(этил)амино, пропил(метил)амино, пропил(метил)карбамоилоксиэтил(метил)амино, пирролидинилкарбамоилоксиэтил(метил)амино, трет-бутоксикарбонил(метил)аминоэтил(метил)амино, трет-бутоксикарбонилэтил(метил)амино, трет-бутоксикарбонилизопентил(метил)амино или трет-бутоксикарбонил(фенил)этил(метил)амино.
5. Соединение по любому из пп. 1-4, где R1 представляет собой этил.
6. Соединение по п. 1 или 2, где R2 представляет собой бензил, замещенный галогеном или С1-6алкилом.
7. Соединение по любому из пп. 2-6, где R2 представляет собой бромбензил, хлорбензил, фторбензил или метилбензил.
8. Соединение по п. 7, где R2 представляет собой бромбензил, хлорбензил или фторбензил.
9. Соединение по п. 1 или 2, где R3 представляет собой -NR4R5, при этом R4 представляет собой С1-6алкил, R5 представляет собой С1-6алкил.
10. Соединение по п. 9, где R3 представляет собой пропил(метил)амино или этил(метил)амино.
11. Соединение по любому из пп. 1, 2, 6 и 9, где
R1 представляет собой С1-6алкил;
R2 представляет собой бензил, при этом указанный бензил замещен галогеном или С1-6алкилом;
R3 представляет собой -NR4R5, где R4 представляет собой С1-6алкил, R5 представляет собой С1-6алкил.
12. Соединение по п. 11, где
R1 представляет собой этил;
R2 представляет собой метилбензил, бромбензил, хлорбензил или фторбензил;
R3 представляет собой пропил(метил)амино или этил(метил)амино.
13. Соединение, выбранное из:
6-амино-9-бензил-N-метил-8-оксо-N-пропил-2-(пропилсульфонимидоил)пурин-7-карбоксамида;
6-амино-9-бензил-N-(2-метоксиэтил)-N-метил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамида;
6-амино-9-бензил-N-этил-8-оксо-N-пропил-2-(пропилсульфонимидоил)пурин-7-карбоксамида;
6-амино-9-бензил-7-[4-(1-пиперидил)пиперидин-1-карбонил]-2-(пропилсульфонимидоил)пурин-8-она;
6-амино-9-бензил-N-этил-N-(2-метоксиэтил)-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамида;
6-амино-9-бензил-N-бутил-N-этил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамида;
6-амино-9-бензил-N-(2-метоксиэтил)-8-оксо-N-пропил-2-(пропилсульфонимидоил)пурин-7-карбоксамида;
6-амино-9-бензил-N,N-бис(2-метоксиэтил)-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамида;
6-амино-7-(азетидин-1-карбонил)-9-бензил-2-(пропилсульфонимидоил)пурин-8-она;
6-амино-9-бензил-N-изопропил-N-метил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамида;
6-амино-9-бензил-7-(4-метилпиперазин-1-карбонил)-2-(пропилсульфонимидоил)пурин-8-она;
6-амино-9-бензил-N-(3-метоксипропил)-N-метил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамида;
6-амино-9-бензил-N-изобутил-N-метил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамида;
этил-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]ацетата;
этил-3-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]пропаноата;
трет-бутил-3-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]пропаноата;
этил-(2S)-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]пропаноата;
трет-бутил-(2S)-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]-4-метил-пентаноата;
изопропил-(2S)-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]-4-метил-пентаноата;
этил-(2S)-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]-3-метил-бутаноата;
этил-(2S)-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]-4-метил-пентаноата;
этил-(2S)-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]-3-фенил-пропаноата;
изопропил-(2S)-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]-3-фенил-пропаноата;
трет-бутил-(2S)-2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]-3-фенил-пропаноата;
N-[2-[ацетил(метил)амино]этил]-6-амино-9-бензил-N-метил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамида;
метил-N-[2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]этил]-N-метил-карбамата;
трет-бутил-N-[2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]этил]-N-метил-карбамата;
этил-N-[2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]этил]-N-метил-карбамата;
2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]этил-N-бутил-N-метил-карбамата;
2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]этил-пирролидин-1-карбоксилата;
2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]этил-N-метил-N-пропил-карбамата;
2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]этил-N,N-диэтилкарбамата;
2-[[6-амино-9-бензил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбонил]-метил-амино]этил-этил-карбоната;
6-амино-N-бутил-9-[(4-хлорфенил)метил]-N-метил-8-оксо-2-[S(R)-пропилсульфонимидоил]пурин-7-карбоксамида;
6-амино-N-бутил-9-[(4-хлорфенил)метил]-N-метил-8-оксо-2-[S(S)-пропилсульфонимидоил]пурин-7-карбоксамида;
6-амино-9-[(4-хлорфенил)метил]-N-этил-N-метил-8-оксо-2-(пропилсульфонимидоил)пурин-7-карбоксамида;
6-амино-N-метил-8-оксо-N-пропил-2-[S(S)-пропилсульфонимидоил]-9-(п-толилметил)пурин-7-карбоксамида;
6-амино-N-метил-8-оксо-N-пропил-2-[S(R)-пропилсульфонимидоил]-9-(п-толилметил)пурин-7-карбоксамида;
6-амино-2-[S(S-пропилсульфонимидоил]-9-(п-толилметил)-7-(пирролидин-1-карбонил)пурин-8-она;
6-амино-2-[S(R-пропилсульфонимидоил]-9-(п-толилметил)-7-(пирролидин-1-карбонил)пурин-8-она;
6-амино-N-(2-метоксиэтил)-N-метил-8-оксо-2-[S(S)-пропилсульфонимидоил]-9-(п-толилметил)пурин-7-карбоксамида;
6-амино-N-(2-метоксиэтил)-N-метил-8-оксо-2-[S(R)-пропилсульфонимидоил]-9-(п-толилметил)пурин-7-карбоксамида;
6-амино-N-этил-N-метил-8-оксо-2-(пропилсульфонимидоил)-9-(п-толилметил)пурин-7-карбоксамида;
6-амино-N-бутил-N-метил-8-оксо-2-(пропилсульфонимидоил)-9-(п-толилметил)пурин-7-карбоксамида;
6-амино-N-[(4-хлорфенил)метил]-2-[S(R)-этилсульфонимидоил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамида;
6-амино-9-[(4-хлорфенил)метил]-2-[S(S)-этилсульфонимидоил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамида;
6-амино-9-[(4-хлорфенил)метил]-N-этил-2-[S(S)-этилсульфонимидоил]-N-метил-8-оксо-пурин-7-карбоксамида;
6-амино-9-[(4-хлорфенил)метил]-N-этил-2-[S(R)-этилсульфонимидоил]-N-метил-8-оксо-пурин-7-карбоксамида;
6-амино-2-[S(S)-этилсульфонимидоил]-N-метил-8-оксо-N-пропил-9-(п-толилметил)пурин-7-карбоксамида;
6-амино-2-[S(R)-этилсульфонимидоил]-N-метил-8-оксо-N-пропил-9-(п-толилметил)пурин-7-карбоксамида;
6-амино-N-этил-2-[S(S)-этилсульфонимидоил]-N-метил-8-оксо-9-(п-толилметил)пурин-7-карбоксамида;
6-амино-N-этил-2-[S(R)-этилсульфонимидоил]-N-метил-8-оксо-9-(п-толилметил)пурин-7-карбоксамида;
6-амино-2-[S(S)-этилсульфонимидоил]-9-[(4-фторфенил)метил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамида;
6-амино-2-[S(R)-этилсульфонимидоил]-9-[(4-фторфенил)метил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамида;
6-амино-N-этил-2-(этилсульфонимидоил)-9-[(4-фторфенил)метил]-N-метил-8-оксо-пурин-7-карбоксамида;
6-амино-N-этил-2-[S(S)-(этилсульфонимидоил)]-9-[(4-фторфенил)метил]-N-метил-8-оксо-пурин-7-карбоксамида;
6-амино-N-этил-2-[S(R)-(этилсульфонимидоил)]-9-[(4-фторфенил)метил]-N-метил-8-оксо-пурин-7-карбоксамида;
6-амино-9-[(4-бромфенил)метил]-2-(этилсульфонимидоил)-N-метил-8-оксо-N-пропил-пурин-7-карбоксамида;
6-амино-2-[S(R)-этилсульфонимидоил]-9-[(4-бромфенил)метил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамида;
6-амино-2-[S(S)-этилсульфонимидоил]-9-[(4-бромфенил)метил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамида;
6-амино-9-[(4-бромфенил)метил]-N-этил-2-(этилсульфонимидоил)-N-метил-8-оксо-пурин-7-карбоксамида;
6-амино-9-[(4-бромфенил)метил]-N-этил-2-[S(S)-(этилсульфонимидоил)]-N-метил-8-оксо-пурин-7-карбоксамида; и
6-амино-9-[(4-бромфенил)метил]-N-этил-2-[S(R)-(этилсульфонимидоил)]-N-метил-8-оксо-пурин-7-карбоксамида;
или его фармацевтически приемлемые соль, энантиомер или диастереомер.
14. Соединение по п. 13, выбранное из:
6-амино-9-бензил-N-метил-8-оксо-N-пропил-2-(пропилсульфонимидоил)пурин-7-карбоксамида
6-амино-9-[(4-хлорфенил)метил]-2-[S(R)-этилсульфонимидоил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамида
6-амино-9-[(4-хлорфенил)метил]-2-[S(S)-этилсульфонимидоил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамида
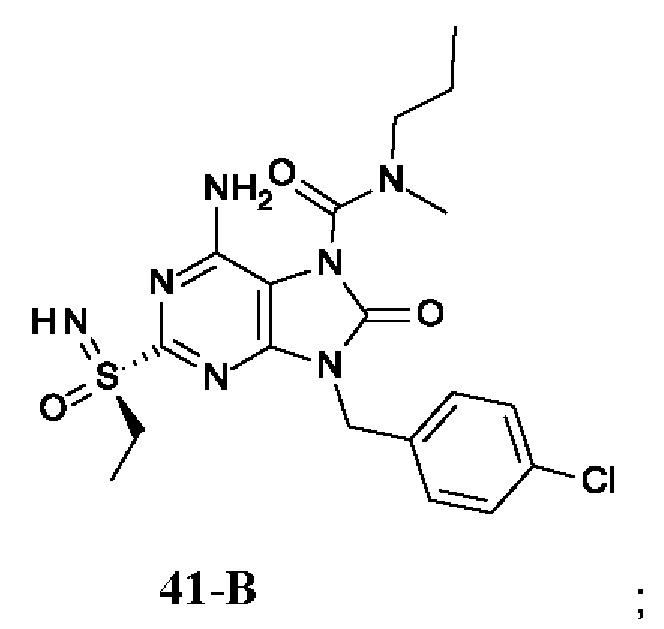
6-амино-9-[(4-хлорфенил)метил]-N-этил-2-[S(S)-этилсульфонимидоил]-N-метил-8-оксо-пурин-7-карбоксамида
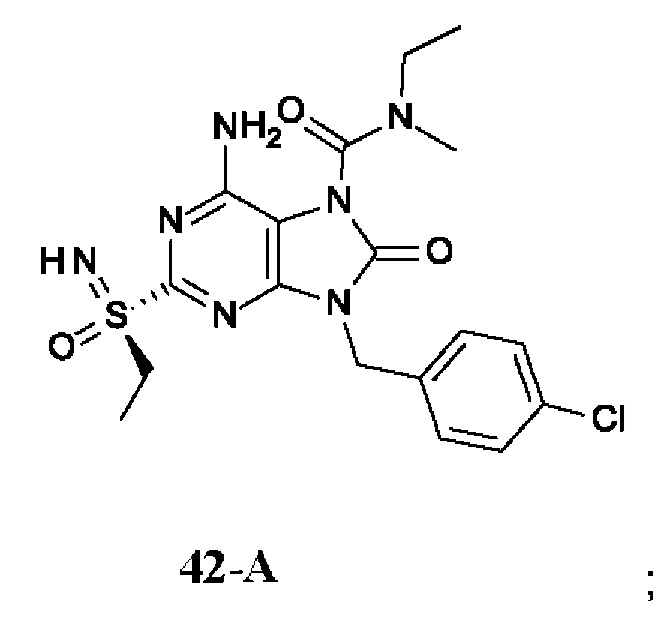
6-амино-9-[(4-хлорфенил)метил]-N-этил-2-[S(R)-этилсульфонимидоил]-N-метил-8-оксо-пурин-7-карбоксамида

6-амино-2-[S(S)-этилсульфонимидоил]-N-метил-8-оксо-N-пропил-9-(п-толилметил)пурин-7-карбоксамида
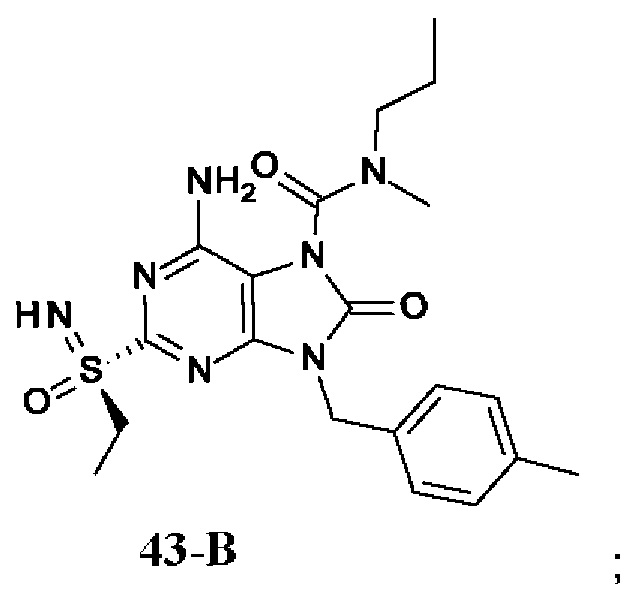
6-амино-2-[S(R)-этилсульфонимидоил]-N-метил-8-оксо-N-пропил-9-(п-толилметил)пурин-7-карбоксамида
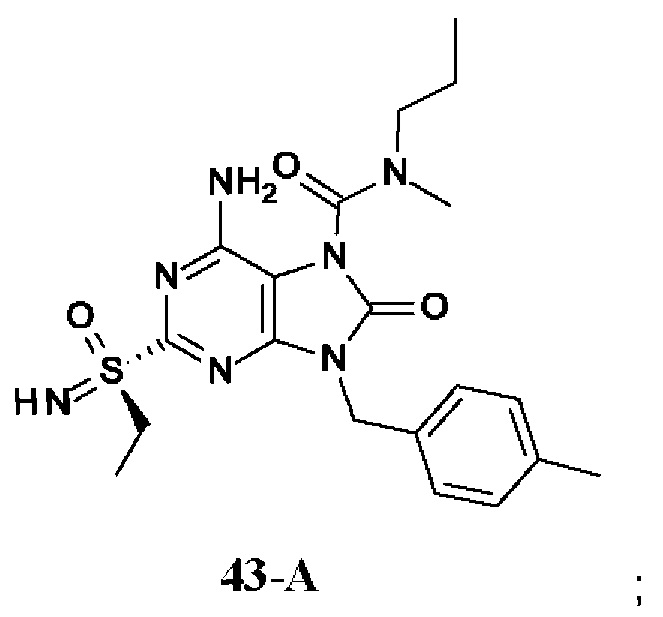
6-амино-N-этил-2-[S(S)-этилсульфонимидоил]-N-метил-8-оксо-9-(п-толилметил)пурин-7-карбоксамида
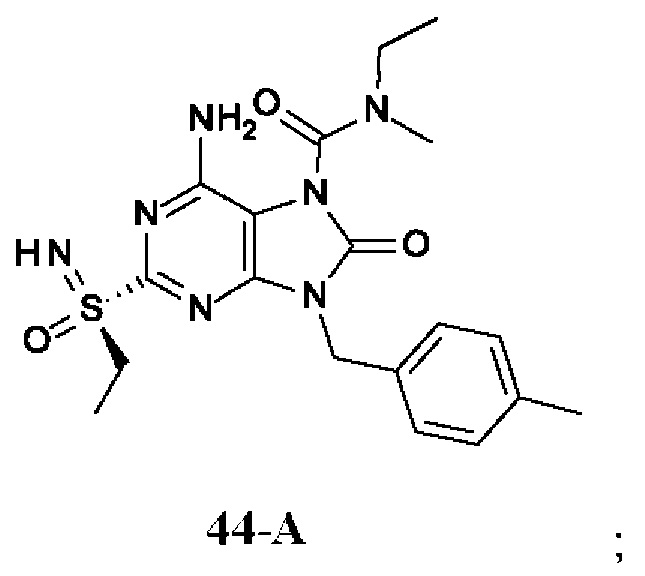
6-амино-N-этил-2-[S(R)-этилсульфонимидоил]-N-метил-8-оксо-9-(п-толилметил)пурин-7-карбоксамида
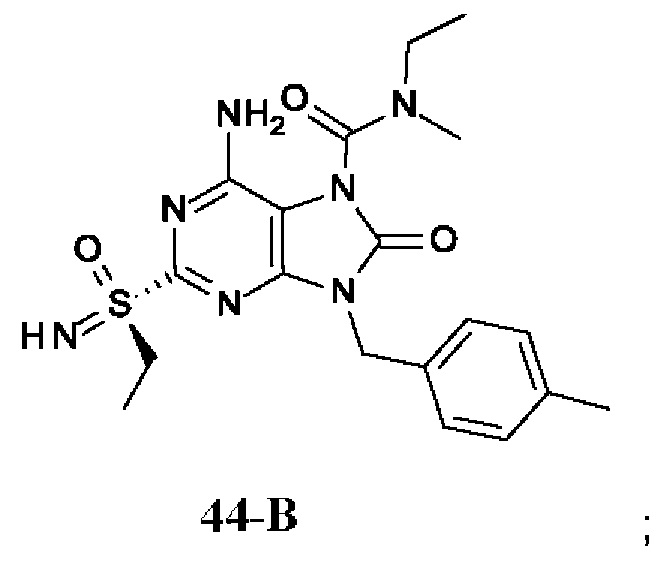
6-амино-2-(этилсульфонимидоил)-9-[(4-фторфенил)метил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамида
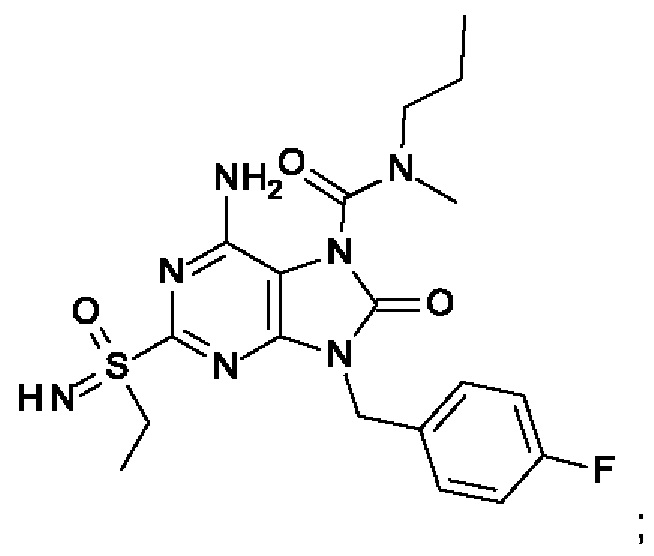
6-амино-2-[S(S)-этилсульфонимидоил]-9-[(4-фторфенил)метил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамида
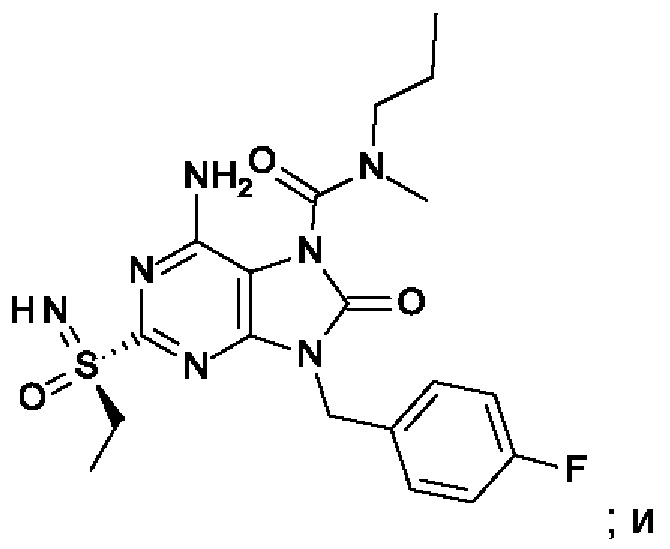
6-амино-2-[S(R)-этилсульфонимидоил]-9-[(4-фторфенил)метил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамида
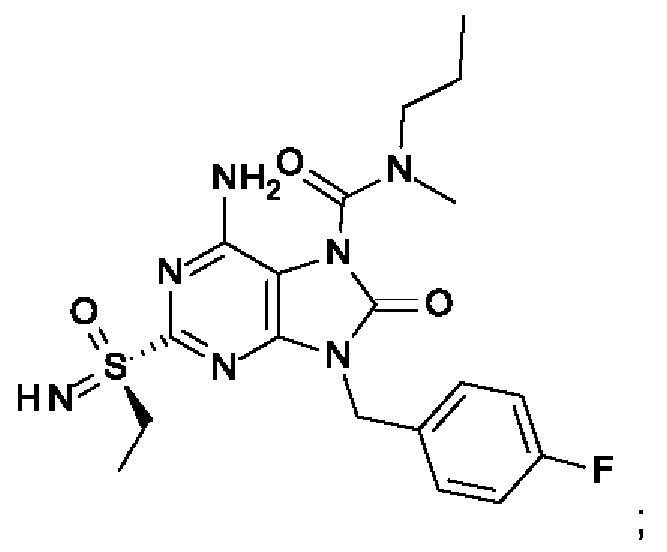
или его фармацевтически приемлемые соль, энантиомер или диастереомер.
15. Соединение по п. 13, выбранное из:
6-амино-9-[(4-бромфенил)метил]-2-(этилсульфонимидоил)-N-метил-8-оксо-N-пропил-пурин-7-карбоксамида
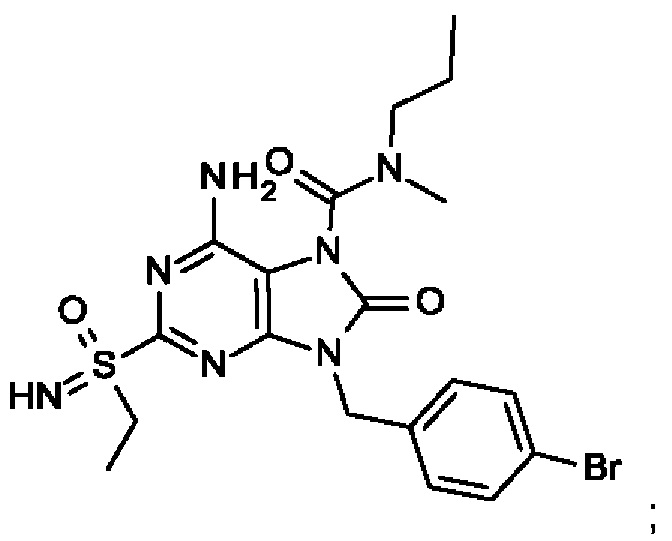
6-амино-2-[S(R)-этилсульфонимидоил]-9-[(4-бромфенил)метил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамида
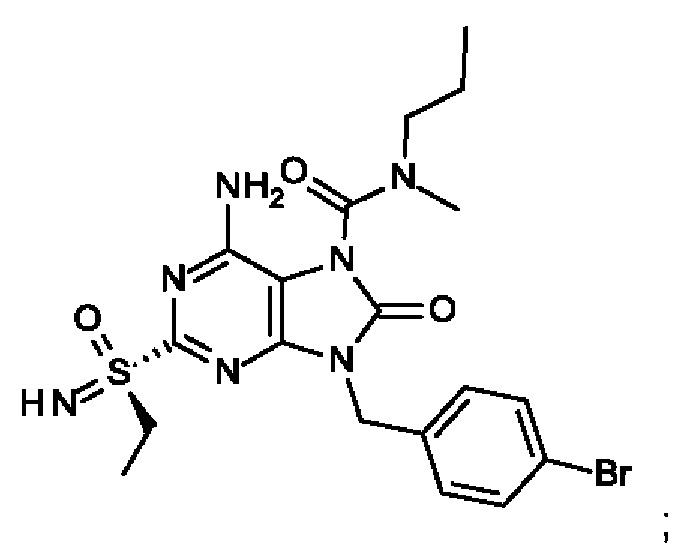
6-амино-2-[S(S)-этилсульфонимидоил]-9-[(4-бромфенил)метил]-N-метил-8-оксо-N-пропил-пурин-7-карбоксамида
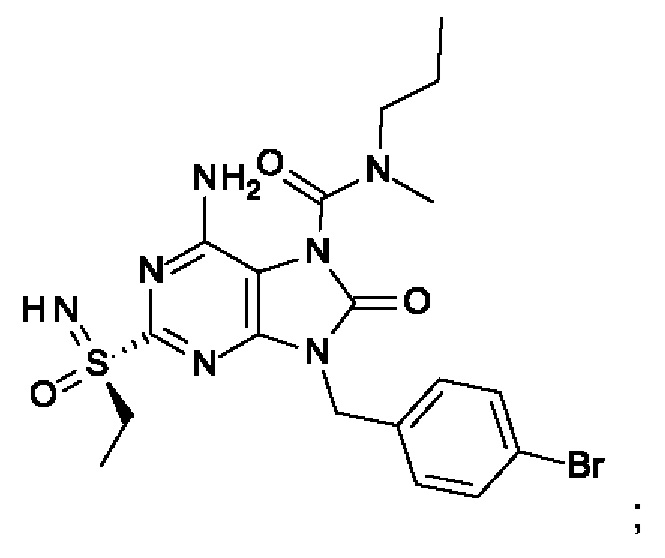
6-амино-9-[(4-бромфенил)метил]-N-этил-2-(этилсульфонимидоил)-N-метил-8-оксо-пурин-7-карбоксамида
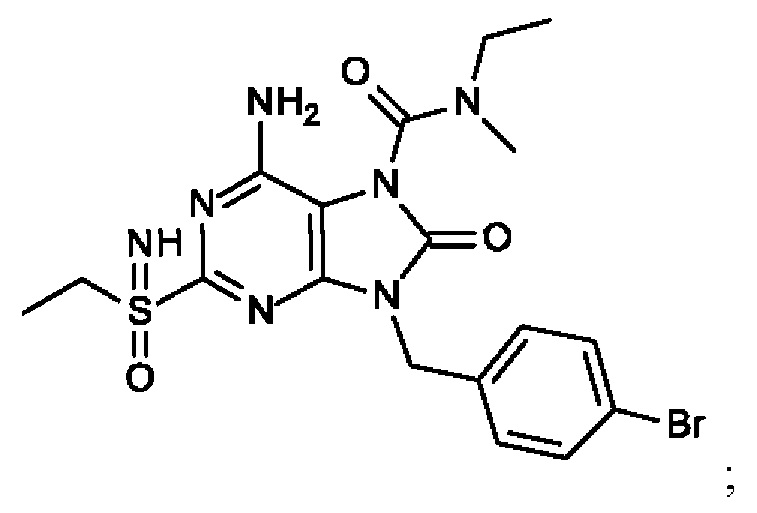
6-амино-9-[(4-бромфенил)метил]-N-этил-2-[S(S)-(этилсульфонимидоил)]-N-метил-8-оксо-пурин-7-карбоксамида
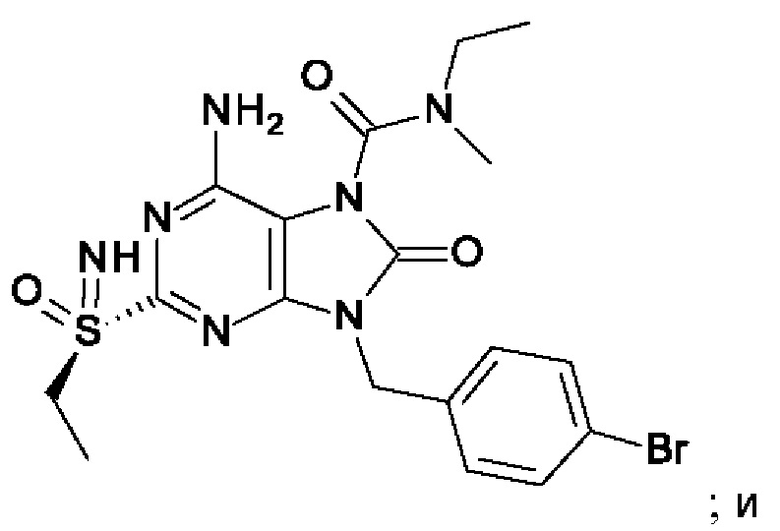
6-амино-9-[(4-бромфенил)метил]-N-этил-2-[S(R)-(этилсульфонимидоил)]-N-метил-8-оксо-пурин-7-карбоксамида
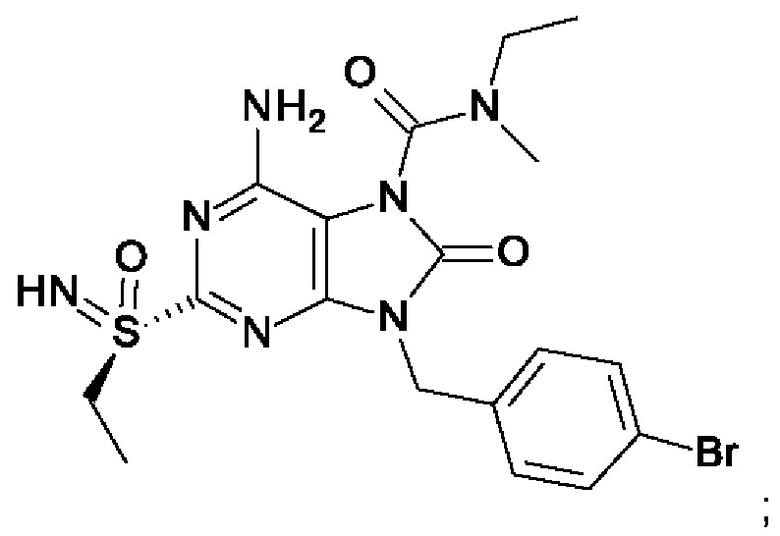
или его фармацевтически приемлемые соль, энантиомер или диастереомер.
16. Способ получения соединения по любому из пп. 1-15, включающий следующую стадию:
приведение во взаимодействие соединения формулы (II)
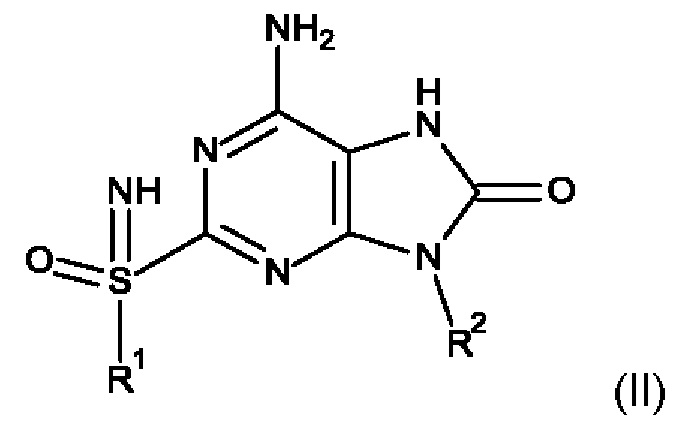
с карбамоилхлоридом в присутствии смеси оснований;
где смесью оснований является пиридин и триэтиламин, пиридин и DIPEA (диизопропилэтиламин), DMAP (4-диметиламинопиридин) и триэтиламин или DMAP и DIPEA; R1 и R2 являются такими, как определено в любом из пп. 1-12.
17. Соединение или фармацевтически приемлемые соль, энантиомер или диастереомер по любому из пп. 1-15 для применения в качестве терапевтически активного вещества, обладающего агонистической активностью в отношении Toll-подобного рецептора 7 (TLR7).
18. Фармацевтическая композиция для лечения или профилактики инфекции, вызванной вирусом гепатита В, содержащая эффективное количество соединения по любому из пп. 1-15 и терапевтически инертный носитель.
19. Применение соединения по любому из пп. 1-15 для лечения или профилактики инфекции, вызванной вирусом гепатита В.
20. Применение соединения по любому из пп. 1-15 для приготовления лекарственного средства для лечения или профилактики инфекции, вызванной вирусом гепатита В.
21. Применение соединения по любому из пп. 1-15 в качестве агониста Toll-подобного рецептора 7 (TLR7).
22. Применение соединения по любому из пп. 1-15 для индуцирования образования интерферона-α.
23. Соединение или фармацевтически приемлемые соль, энантиомер или диастереомер по любому из пп. 1-15 для лечения или профилактики инфекции, вызванной вирусом гепатита В.
24. Соединение или фармацевтически приемлемые соль, энантиомер или диастереомер по любому из пп. 1-15, полученные согласно способу по п. 16.
25. Способ лечения или профилактики инфекции, вызванной вирусом гепатита В, включающий введение терапевтически эффективного количества соединения, которое определено в любом из пп. 1-15.
| US 20100143301 A1, 10.06.2010 | |||
| Микрометр | 1930 |
|
SU21463A1 |
| US 20060052403 A1, 09.03.2006. | |||

