Область техники
Варианты предлагаемого изобретения в целом относятся к способу и системе выявления вариации числа копий в образце генома, и к использованию с этой целью машиночитаемого носителя.
Предшествующий уровень
В научно-исследовательской и прикладной областях, обычно возникают проблемы анализа одной клетки, группы клеток, или следов нуклеиновой кислоты. Например, преимплантационная генетическая диагностика (ПГД) и преимплантационный генетический скрининг (ПГС) в области вспомогательной репродуктивной технологии включает анализ одиночных половых клеток, отдельных клеток бластомера или эмбриональных клеток; область технологии неинвазивного пренатального анализа включает проблему определения следов фетальных клеток в материнской периферической крови. Метагеномика включает анализ одиночных клеток или следов биологических клеток в окружающей среде. Медицинские или физические исследования включают анализ одной клетки в ткани или жидкости тела.
Однако, в настоящее время способ определения вариации числа копий все еще требует совершенствования.
Краткое описание изобретения
Предлагаемое изобретение направлено на решение, по крайней мере, частичное, одной из существующих проблем.
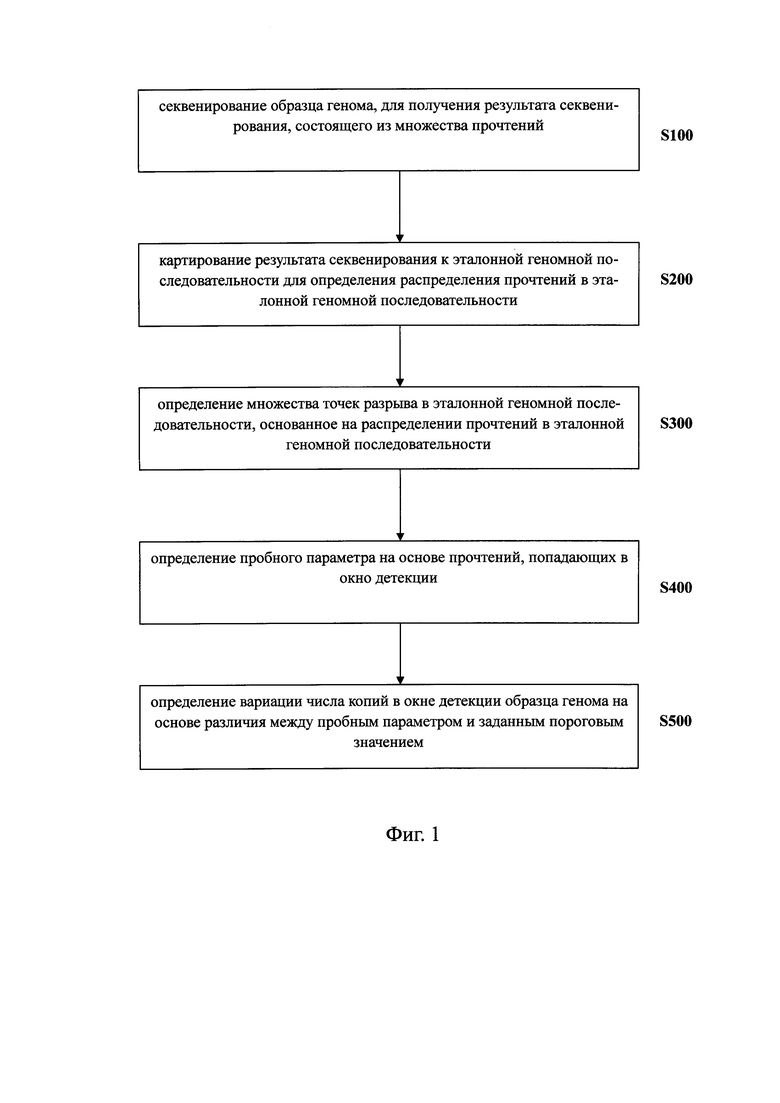
Первым объектом предлагаемого изобретения в широком смысле является способ выявления вариации числа копий в образце генома. Согласно вариантам предлагаемого изобретения, способ может включать следующие этапы: секвенирование образца генома для получения в результате множества прочтений; картирование результата секвенирования к последовательности референсного генома (эталонной геномной последовательности) для определения распределения прочтений в последовательности референсного генома; определение множества точек разрыва в последовательности референсного генома на основании распределения прочтений в последовательности референсного генома, в котором число прочтений является значимым по обе стороны от точек разрыва; определение окна детекции в референсном геноме на основании множества точек разрыва; определение пробного параметра на основании прочтений, попадающих в окно детекции; и определение вариации числа копий в окне детекции образца генома на основе различия между пробным параметром и заданным пороговым значением. Используя способ выявления вариации числа копий в образце генома согласно вариантам предлагаемого изобретения, можно эффективно выявлять разные вариации числа копий в образце генома, в частности, анеуплоидию хромосомы, делецию хромосомы и также вставку, микроделецию и микродупликацию участков хромосомы.
Вторым объектом предлагаемого изобретения в широком смысле является система выявления вариации числа копий в образце генома. Согласно вариантам предлагаемого изобретения система может содержать:
секвенатор, выполняющий секвенирование образца генома для получения в результате множества прочтений; анализатор, соединенный с секвенатором и определяющий вариации числа копий в образце генома на основе результатов секвенирования; при этом анализатор содержит: блок картирования для сравнения результата секвенирования с последовательностью референсного генома (эталонной геномной последовательностью) для определения распределения прочтений в последовательности референсного генома; блок определения точек разрыва, соединенный с блоком картирования для определения множества точек разрыва в последовательности референсного генома на основании распределения прочтений в последовательности референсного генома, при том, что число прочтений является значимым с обеих сторон от точек разрыва; блок определения окна детекции, соединенный с блоком определения точек разрыва, способный определять окно детекции в референсном геноме на основании множества точек разрыва; блок определения параметров, соединенный с блоком определения окна детекции и предназначенный для определения пробного параметра на основании прочтений, попадающих в окно детекции; и блок обнаружения, соединенный с блоком определения параметров, предназначенный для выявления вариации числа копий в образце генома в окне детекции, на основании различия между пробным параметром и заданным пороговым значением. Используя систему выявления вариации числа копий в образце генома, согласно вариантам предлагаемого изобретения, можно эффективно осуществить способ выявления вариации числа копий в образце генома, в частности, анеуплоидии хромосомы, делеции хромосомы, а также вставку, микроделецию и микродупликацию участков хромосомы.
Третьим объектом предлагаемого изобретения в широком смысле является машиночитаемый носитель. Согласно вариантам предлагаемого изобретения, выявление вариации числа копий в образце генома осуществляется машиночитаемым носителем с помощью процессора и включает следующие этапы: картирование результата секвенирования к последовательности референсного генома для определения распределения прочтений в последовательности референсного генома; определение множества точек разрыва в последовательности референсного генома на основании распределения прочтений в последовательности референсного генома, в котором число прочтений является значимым по обе стороны от точек разрыва; определение окна детекции в референсном геноме на основании множества точек разрыва; определение пробного параметра на основании прочтений, попадающих в окно детекции; и выявление вариации числа копий в образце генома в окне детекции на основе различия между пробным параметром и заданным пороговым значением. Используя машиночитаемый носитель, можно эффективно осуществлять способ выявления вариации числа копий в образце генома согласно вариантам предлагаемого изобретения, для того чтобы эффективно выявлять разные вариации числа копий в образце генома, в частности, анеуплоидию хромосомы, делецию хромосомы, а также вставку, микроделецию и микродупликацию участков хромосомы.
Другие особенности и преимущества вариантов предлагаемого изобретения частично будут даны в дальнейшем описании, или будут видны из описания, или могут быть установлены при осуществлении предлагаемого изобретения.
Краткое описание чертежей
Эти и другие особенности и преимущества предлагаемого изобретения будут более понятны и наглядны из последующего описания с отсылками к прилагаемым чертежам, на которых:
Фиг. 1 - блок-схема, показывающая способ выявления вариации числа копий в образце генома; согласно варианту предлагаемого изобретения;
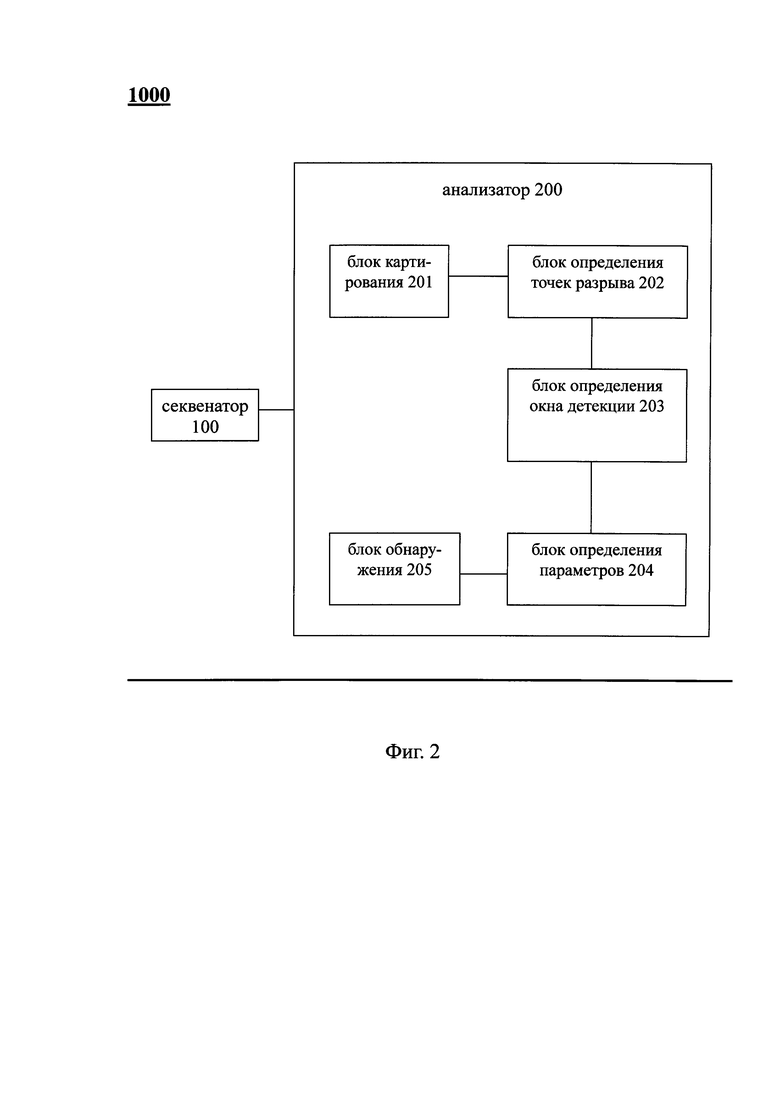
Фиг. 2 - схема, показывающая систему для выявления вариации числа копий в образце генома; согласно варианту предлагаемого изобретения;
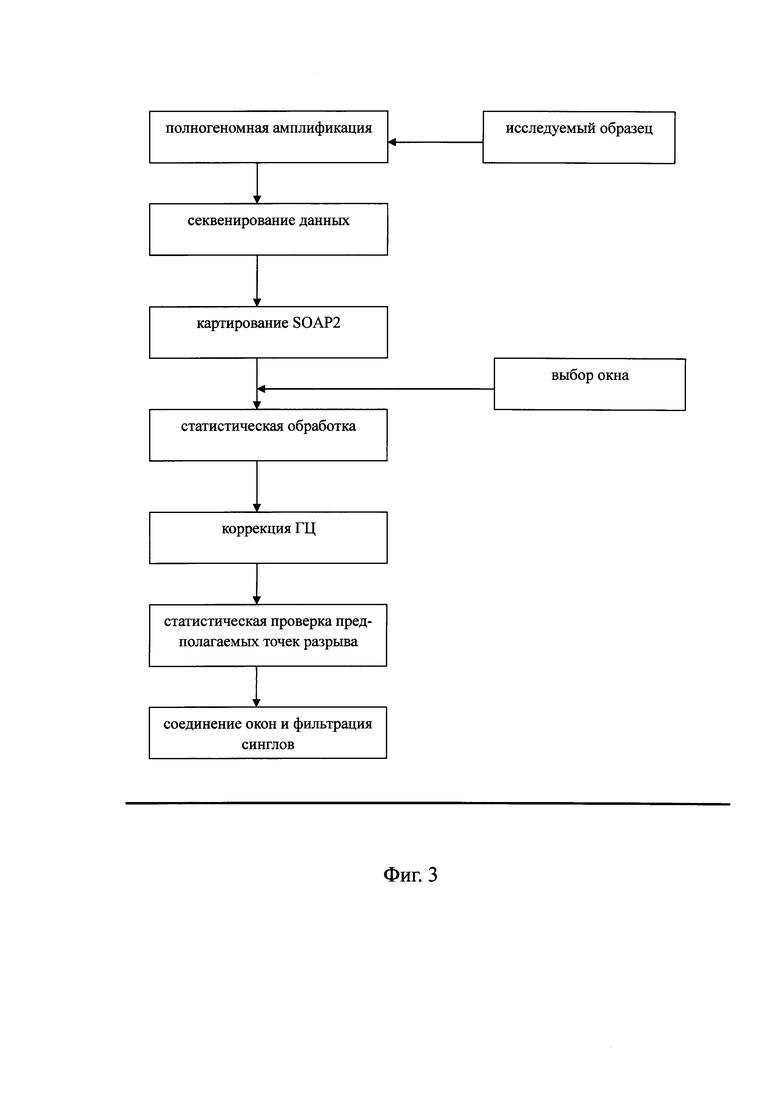
Фиг. 3 - блок-схема, показывающая способ выявления вариации числа копий в образце генома; согласно другому варианту предлагаемого изобретения;
Фиг. 4 - изображение анализа кариотипа хромосомы из образца S1 согласно вариантам предлагаемого изобретения, на которой на левой панели показан результат, полученный способом выявления вариации числа копий согласно варианту предлагаемого изобретения, для одиночной эмбриональной клетки, подвергнутой полногеномной амплификации; на правой панели показан результат, который был получен при прямом секвенировании (без предварительной полногеномной амплификации) ДНК, выделенной из той же одиночной эмбриональной клетки; и
Фиг. 5 - изображение анализа кариотипа хромосомы из образца S2 согласно вариантам предлагаемого изобретения, на которой на левой панели показан результат, полученный способом выявления вариации числа копий согласно варианту предлагаемого изобретения с одиночной эмбриональной клеткой, подвергнутой полногеномной амплификации; на правой панели показан результат, который был получен при прямом секвенировании (без предварительной полногеномной амплификации) ДНК, выделенной из той же одиночной эмбриональной клетки.
Подробное описание
Детали изобретения будут раскрыты при описании отдельных вариантов. Одни и те же или аналогичные элементы и элементы, имеющие те же или такие же функции, обозначены одинаковыми числовыми позициями по всему описанию. Описываемые варианты со ссылками на чертежи являются иллюстративными, пояснительными и используются для общего понимания предлагаемого изобретения. Они не должны рассматриваться как ограничивающие данное изобретение.
Термины «первый» и «второй» используются в настоящем документе с описательной целью и не указывают и не подразумевают относительную важность или значение признаков. Таким образом, признаки, определенные как «первый» или «второй» могут явно или неявно включать один или несколько упомянутых признаков. Кроме того, в описании предлагаемого изобретения термин «множество» означает «два или более», если не указано иное. Если не указанное иное, в используемых в данном документе формулах или знаках одинаковые символы имеют одинаковое значение.
I. Способ выявления вариации числа копий в образце генома
Первым объектом предлагаемого изобретения является способ выявления вариации числа копий в образце генома. Используемый в документе термин «вариация числа копий» - ВЧК (CNV) относится к аномальному числу хромосом или участков хромосом, в частности, к анеуплоидии хромосомы, делеции участка хромосомы, вставке, микроделеции и микродупликации участка хромосомы.
На Фиг. 1 показан способ выявления вариации числа копий в образце генома согласно вариантам предлагаемого изобретения, включающий:
S100: секвенирование образца генома для получения в результате множества прочтений.
Согласно вариантам предлагаемого изобретения, типы образцов генома, в отношении которых отсутствуют специальные ограничения, могут представлять собой полный геном или часть генома, например, хромосому или участок хромосомы. Кроме того, согласно вариантам предлагаемого изобретения, способ выявления вариации числа копий в образце генома может предварительно включать этап выделения образца генома из биологического образца. Соответственно, биологический образец может быть непосредственно использован в качестве исходного материала для получения информации о возможной вариации числа копий в биологическом образце, отражающей состояние здоровья организмов. Согласно вариантам предлагаемого изобретения, в отношении используемого биологического образца отсутствуют специальные ограничения. В некоторых конкретных примерах предлагаемого изобретения, биологический образец выбирают из группы, включающей кровь, мочу, слюну, ткань, половые клетки, оосперму, бластомер или эмбрион. Специалисты высоко оценят возможность использования разных биологических образцов для анализа различных заболеваний. Соответственно, эти образцы легко получить из организмов. При конкретных заболеваниях могут быть направленно использованы разные образцы, для целенаправленного выбора конкретных средств для анализа определенных заболеваний. Например, у пациента с предполагаемым определенном типом рака может быть получен образец раковой ткани или предположительно раковой ткани с последующим выделением из образца клеток для анализа, чтобы точно определить, является ли данная ткань раковой, на как можно более ранней стадии. Согласно одному из примеров предлагаемого изобретения, в качестве биологического образца может быть использована одиночная клетка. В предлагаемом изобретении в отношении способов и устройств для выделения одиночной клетки из биологического образца специальные ограничения отсутствуют. Согласно некоторым конкретным примерам предлагаемого изобретения, для выделения одиночной клетки можно использовать, по крайней мере, один из следующих приемов и средств: разбавление, обычная пипетка, микроманипуляции (предпочтительно, микродиссекция), выделение методом проточной цитометрии, микроструйные методы. Эти подходы позволяют эффективно и легко получать одиночные клетки из биологического образца для использования их на следующих этапах. Далее эффективность выявления вариации числа копий в образце генома может быть еще усовершенствована.
Кроме того, согласно вариантам предлагаемого изобретения, в отношении способов секвенирования образца генома специальные ограничения отсутствуют. Согласно одному из вариантов предлагаемого изобретения, этап секвенирования образца генома дополнительно включает следующие подэтапы: 1) амплификацию образца генома для получения образца с амплифицированным геномом; 2) создание библиотеки фрагментов ДНК с секвенированнным образцом амплифицированного генома; и 3) секвенирование созданной библиотеки фрагментов ДНК с получением результата секвенирования, включающего множество прочтений. Соответственно, секвенирование образца генома позволяет успешно получать информацию о полном геноме, и образец, представляющий собой геном единичной клетки или следы нуклеиновой кислоты, может быть эффективно секвенирован для более надежного выявления вариации числа копий в образце генома. Специалисты могут выбирать разные способы конструирования библиотеки фрагментов ДНК в зависимости от конкретных решений, используемых в методике секвенирования генома. С процессом конструирования библиотеки фрагментов ДНК можно подробно ознакомиться в спецификации производителя секвенатора, например, Illumina Company - «Руководство по мультиплексированной подготовке образца» (Часть №1005063; февраль 2010 г.), включенной в настоящий документ посредством ссылки.
Факультативно, на этапе выделения образца генома из биологического образца, являющегося одиночной клеткой, согласно вариантам предлагаемого изобретения, способ может дополнительно включать этап лизиса одиночной клетки, приводящий к высвобождению полного генома одиночной клетки. Согласно некоторым примерам предлагаемого изобретения, в отношении способов лизиса одиночной клетки с высвобождением полного генома специальные ограничении отсутствуют, поскольку при лизисе одиночной клетки преимущественно происходит полный лизис. В конкретных примерах предлагаемого изобретения, для высвобождения полного генома одиночной клетки для лизиса одиночной клетки используют щелочной лизат. Авторы предлагаемого изобретения обнаружили, что этап лизирования одиночной клетки может эффективно обеспечивать лизис одиночной клетки с выделением полного генома, и при секвенировании высвободившегося полного генома может достигаться более высокая точность, что дополнительно повышает эффективность выявления вариаций числа копий в образце генома. Согласно вариантам предлагаемого изобретения, в отношении способов амплификации полного генома одиночной клетки отсутствуют какие-либо особые ограничения и могут быть использованы разновидности ПЦР, например, PEP-PCR (полимеразная цепная реакция с удлинением продукта предыдущего цикла амплификации, DOP-PCR (полимеразная цепная реакция с использованием частично вырожденных праймеров) и полногеномный анализ (WGA) с использованием наборов OmniPlex. Также могут быть использованы методы, не связанные с ПЦР, например, амплификация с множественным замещением цепи (MDA). Согласно конкретным примерам предлагаемого изобретения, предпочтительным являются разновидности ПЦР, например OmniPlex WGA. Могут быть использованы коммерческие наборы, в частности GenomePlex компании Sigma Aldrich, PicoPlex компании Rubicon Genomics, REPLI-g компании Qiagen, illustra GenomiPhi компании GE Healthcare и т.д. Согласно конкретным примерам предлагаемого изобретения, перед подэтапом конструирования библиотеки фрагментов ДНК для секвенирования, может быть выполнена полногеномная амплификация одиночной клетки с использованием набора OmniPlex WGA. Соответственно, эффективная полногеномная амплификация может дополнительно повысить эффективность выявления вариации числа копий в образце генома. Согласно вариантам предлагаемого изобретения, на подэтапе секвенирования полного генома для конструирования библиотеки фрагментов ДНК используется, по крайней мере, одна технология, выбранная из технологий секвенирования следующего поколения, включающих систему Hiseq (Illumina Company), систему Miseq (Illumina Company), систему Genome Analyzer (GA) (Illumina Company), 454 FLX (Roche Company), систему SOLiD (Applied Biosystems Company), систему Ion Torrent (Life Technologie Company). Соответственно, могут быть использованы характеристики высокоскоростного и глубокого секвенирования, обеспечиваемого этими аппаратами, что дополнительно повышает эффективность выявления вариации числа копий в образце генома. Несомненно, специалисты поймут, что для секвенирования полного генома могут применяться также и другие методы секвенирования и оборудование для секвенирования, например, технология секвенирования третьего поколения (т.е., технология секвенирования на одномолекулярном уровне), например, любая система HeliScope (Helicos BioSciences Company), система RS (PacBio Company) и т..д. и также более передовые технологии секвенирования, которые могут быть разработаны в будущем. Согласно вариантам предлагаемого изобретения, в отношении длины секвенированных последовательностей полного генома специальные ограничения отсутствуют. В конкретных примерах предлагаемого изобретения, средняя длина множества полученных при секвенировании данных составляет приблизительно 50 п.н. Авторы предлагаемого изобретения неожиданно обнаружили, что определенные последовательности длиной приблизительно 50 п.н. могут значительно облегчить анализ результатов секвенирования, повышая эффективность анализа и существенно снижая стоимость анализа и, тем самым, дополнительно повышая эффективность выявления анеуплоидности хромосом одиночной клетки и снижая стоимость выявления анеуплоидности хромосом в одиночной клетке. Термин «средняя длина» в данном описании относится к среднему значению длины каждой определенной последовательности.
S200: картирование результата секвенирования к последовательности референсного генома для определения распределения прочтений в последовательности референсного генома
После завершения этапа секвенирования образца генома полученная в результате последовательность включает множество данных секвенирования. Полученный результат секвенирования картируют к последовательности референсного генома, чтобы определить расположение полученного при секвенировании результата в последовательности референсного генома. Согласно вариантам предлагаемого изобретения, для того чтобы рассчитать общее число этих определенных последовательностей, могут быть использованы любые известные методы. Например, для анализа можно использовать программное обеспечение производителя секвенатора. Предпочтительно применяются Пакет программ для анализа коротких олигонуклеотидов (SOAP) и Burrows-Wheeler Aligner (BWA), позволяющие картировать прочтения к последовательности референсного генома и определить расположение этих прочтений в последовательности референсного генома. При совмещении последовательностей может быть использован параметр, предоставляемый программным обеспечением по умолчанию, либо соответствующий параметр может быть выбран специалистом. В варианте предлагаемого изобретения в качестве программного обеспечения картирования используют SOAPaligner/soap2.
В вариантах предлагаемого изобретения, последовательность референсного генома может представлять собой эталонную последовательность генома человека из базы данных Национального центра биотехнологической информации (NCBI) (например, hg18, NCBI Build 36); или часть последовательности известного генома, например, по крайней мере, одну последовательность, выбранную из группы, включающей 21-ю хромосому, 18-ю хромосому, 13-ю хромосому, Х-хромосому и Y-хромосому человека.
Согласно вариантам предлагаемого изобретения, на этапе картирования результата секвенирования к последовательности референсного генома для последующего анализа могут быть выбраны последовательности, имеющие однозначное соответствие последовательности референсного генома. Соответственно, можно избежать вызванного повторами искажения результатов анализа вариации числа копий и, тем самым, дополнительно повысить эффективность выявления вариации числа копий в образце генома.
S300: определение множества точек разрыва в последовательности референсного генома на основании распределения прочтений в последовательности референсного генома
В данном описании термин «точки разрыва» относится к таким участкам генома, в которых число прочтений с каждой стороны этого участка в этих двух областях значимо различается. Поскольку прочтения получены из образца генома, в том случае, когда определенная область содержит вариацию числа копий в образце генома, число соответствующих прочтений в области также значимо изменяется. Соответственно, после определения множества точек разрывов можно предварительно установить вероятное присутствие вариации числа копий в области между двумя последовательными точками разрыва;
Согласно вариантам предлагаемого изобретения, этап определения множества точек разрыва в последовательности референсного генома дополнительно включает следующие подэтапы:
На первом подэтапе последовательность референсного генома делят на множество первичных окон заданной длины и определяют прочтения, попадающие в каждое из множества первичных окон. Согласно конкретным примерам предлагаемого изобретения, типовые программы картирования позволяют соотнести прочтения, содержащиеся в полученном результате секвенирования, с последовательностью референсного генома, что позволяет определить прочтения в каждом из множества первичных окон; например, это может быть выполнено на описанном выше этапе S200. В конкретных примерах предлагаемого изобретения, прочтения, попадающие в каждое из множества первичных окон, являются однозначно картированными прочтениями. Соответственно, можно избежать вызванного повторами искажения результатов анализа вариации числа копий и, тем самым, дополнительно повысить эффективность выявления вариации числа копий в образце генома.
На втором подэтапе, по крайней мере, для одного участка в последовательности референсного генома определяют число прочтений, попадающих в одинаковое число первичных окон по обе стороны участка. Согласно вариантам предлагаемого изобретения, может быть проведен корреляционный анализ всех участков последовательности референсного генома или интересующей хромосомы; например, проводится такой корреляционный анализ всех участков, по крайней мере, на одной из следующих хромосом человека: 21-й хромосомы, 18-й хромосомы, 13-й хромосомы, Х-хромосомы и Y-хромосомы. В вариантах предлагаемого изобретения, первичные области могут быть одинаковыми или разными по длине, возможно частичное перекрытие первичных окон при условии, что известна информация о каждой первичной области. Предпочтительна одинаковая длина всех первичных окон. В вариантах предлагаемого изобретения, длина каждой из множества первичных областей может составлять от 100 до 200 тысяч п.н., предпочтительно 150 тысяч п.н. Согласно вариантам предлагаемого изобретения, в отношении количества первичных окон, расположенных по обе стороны участка, какие-либо ограничения отсутствуют. В конкретных примерах предлагаемого изобретения, может быть выбрано 100 первичных окон на каждой стороне участка соответственно.
На третьем подэтапе посредством статистического анализа может быть определена величина p участка, при этом величина p означает, что число прочтений по обе стороны участка является значимым. Если величина p участка меньше конечной величины p, это означает, что участок является точкой разрыва. В вариантах предлагаемого изобретения, диапазон конечной величины p может быть определен посредством параллельного анализа образца с известной последовательностью. Согласно конкретным примерам предлагаемого изобретения, конечная величина p составляет 1.1X10-50.
Согласно вариантам предлагаемого изобретения, подэтап определения величины p дополнительно включает:
Для выбранного участка выбирают первичные окна с одинаковыми порядковыми номерами с обеих сторон участка, подсчитывают относительное число прочтений, попадающих в каждое первичное окно Ri, где i - порядковый номер первичного окна,
для того, чтобы определить величину p участка, к числу прочтений, попадающих в каждое первичное окно Ri, применяют критерий серий, при этом
относительное число прочтений определяется следующей формулой:
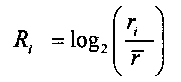
где ri - число прочтений, попадающих в i-тое первичное окно,
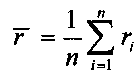 ,
,
n - общее число первичных окон.
Более подробно тестирование - применение критерия серий ко всему относительному числу прочтений, расположенных в каждом из множества первичных окон, дополнительно включает: коррекцию относительного числа прочтений, попадающих в каждое из множества первичных окон Ri, на содержание ГЦ-пар, чтобы получить уточненное относительное число прочтений 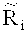
Более конкретно, уточненное относительное число прочтений 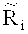
На первом этапе подсчитывают содержание ГЦ-пар в каждом первичном окне;
На втором этапе содержание ГЦ-пар делят на множество областей в соответствии с заданной величиной, и подсчитывают среднюю величину Ms относительного числа прочтений в каждой из множества областей, при этом s - номер области во множестве областей. Согласно вариантам предлагаемого изобретения, заданная величина может быть любым числом от 0.0005 до 0.01 при длине соответствующей области от 50 тысяч п.н. до 300 тысяч п.н., предпочтительно 0.001, при которой возможна корреляция с оптимальной возможностью.
На третьем этапе определяют уточненное относительное число прочтений 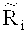
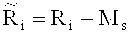
Наконец, среднеарифметическое число прочтений Zi определяют по формуле
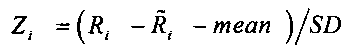 , в которой
, в которой
mean 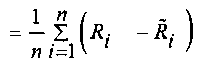
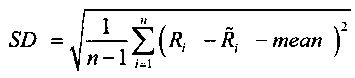 .
.
(mean=среднее значение; SD=СКО=среднеквадратичное отклонение)
Соответственно, может быть выполнена корреляция между числом прочтений и содержанием ГЦ-пар. Таким образом, можно устранить влияние системной ошибки, связанной с амплификацией генома, и, тем самым, дополнительно повысить эффективность выявления вариации числа копий в образце генома.
После определения множества точек разрыва можно предварительно определить возможность вариации числа копий в области между двумя последовательными точками разрыва. Соответственно, при дальнейшем выявлении вариации числа копий такие области могут рассматриваться в качестве окон детекции. Если при предварительном определении было получено относительно больше точек разрыва, далее может быть проведен скрининг полученных точек разрыва. Соответственно, согласно вариантам предлагаемого изобретения, этап определения окна детекции в референсном геноме на основании множества точек разрыва включает:
1) определение множества предполагаемых точек разрыва, когда существуют другие точки разрыва до и после предполагаемых точек разрыва;
2) определение величины p каждой предполагамой точки разрыва и исключение предполагаемой точки разрыва, имеющей максимальную величину p;
3) выполнение этапа 2) с остальными предполагаемыми точками разрыва до тех пор, пока все величины p остальных предполагаемых точек разрыва не окажутся меньше конечной величины p, при этом остальные предполагаемые точки разрыва рассматриваются как «прошедшие скрининг» предполагаемые точки разрыва; и
4) определение области между двумя прошедшими скрининг последовательными предполагаемыми точками разрыва в качестве окна детекции.
Согласно вариантам предлагаемого изобретения, получение величины p предполагаемой точки разрыва включает следующие этапы:
выбор области между предполагаемой точкой разрыва и предыдущей предполагаемой точкой разрыва в качестве первой предполагаемой области, и выбор области между предполагаемой точкой разрыва и следующей предполагаемой точкой разрыва в качестве второй предполагаемой области;
применение критерия серий к среднеарифметическому числу прочтений Zi, попадающих в первичные окна, которые включены и в первую предполагаемую область, и во вторую предполагаемую область, для определения величины p предполагаемых точек разрыва (критерий серий - непараметрический критерий для оценки статистически значимого различия между популяциями при использовании равномерно распределенного в двух популяциях статуса смешанных элементов. Этот критерий подробно описан в публикации Wald A. WJ. «Критерий для оценки принадлежности двух выборок к одной и той же популяции». The Annals of Mathematical Statistics 1940; 11:147-162, включенной в данный документ посредством ссылки).
Согласно вариантам предлагаемого изобретения, получение конечной величины p предполагаемой точки разрыва включает следующие этапы:
на основании результата секвенирования контрольного образца, повторение этапа определения окна детекции в референсном геноме и регистрация величин p точек разрыва, удаляемых каждый раз до тех пор, пока число точек разрыва не станет равным нулю, Использованный термин «контрольный образец» означает образец, у которого в известной нуклеотидной последовательности отсутствует вариация числа копий; и
на основании распределения величины p удаленных точек разрыва определяют конечную величину p, например, посредством построения диаграммы распределения величины p удаленных точек разрыва, и в качестве конечной величины p (pfinal) рассматривают величину p с максимальным трендом к изменению.
Согласно конкретным примерам предлагаемого изобретения, конечная величина p составляет 1,1×10-50.
S400: определение пробного параметра на основании прочтений, попадающих в окно детекции
После определения окон детекции, прочтения, содержащиеся в окнах детекции могут быть подвергнуты статистическому анализу, чтобы выявить возможные вариации числа копий в окнах детекции. Согласно вариантам предлагаемого изобретения, этап определения пробного параметра на основании прочтений, попадающих в окна детекции, включает: определение среднего величины среди всех среднеарифметических чисел прочтений, попадающих в каждое из множества первичных окон 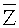
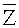
S500: выявление вариации числа копий в образце генома, в окне детекции, на основании различия между пробным параметром и заданным пороговым значением
Согласно вариантам предлагаемого изобретения, может быть выполнено сравнение определенного пробного параметра с заданным пороговым значением. Затем по разнице между пробным параметром и заданным пороговым значением выявляется возможное присутствие вариации числа копий в образце генома применительно к конкретному окну детекции. Исходя из результата секвенирования образца генома число прочтений, попадающих в определенное окно, прямо пропорционально емкости определенного окна в хромосоме или геноме. Соответственно, проводя статистический анализ прочтений из определенного окна результата секвенирования, можно эффективно выявить вариацию числа копий в образце генома, применительно к определенному окну. В настоящем документе термин «заданное пороговое значение» относится к относительному параметру в определенном окнеи, полученному при повторении операций и анализе в перечисленных выше вариантах изобретения и при использовании эталонного образца генома с известной последовательностью. Следует понимать, что относительный параметр в определенном окне и относительный параметр нормальных клеток могут быть получены в таких же условиях секвенирования и теми же математическими методами. В настоящем описании в качестве заданного порогового значения может быть использован относительный параметр нормальных клеток. Кроме того, использованный в настоящем документе термин «заданный» рассматривается в широком смысле, и может быть определен экспериментально или получен в параллельных экспериментах при анализе биологического образца. Термин «параллельный эксперимент» рассматривается в широком смысле и может относиться к секвенированию и анализу одновременно известных и неизвестных образцов либо может относиться к проведению этапов секвенирования и анализа последовательно и в одинаковых условиях. Согласно вариантам предлагаемого изобретения, заданное пороговое значение включает первое пороговое значение и второе пороговое значение; при сравнении первого параметра 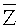
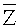
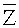
Способ выявления вариации числа копий в образце генома, согласно вариантам предлагаемого изобретения? позволяет эффективно определить присутствие вариации числа копий в образце генома и приемлем для разнообразных вариаций, в частности, анеуплодности хромосомы, делеции фрагмента хромосомы, добавления фрагмента хромосомы, добавления, микроделеции и микродупликации фрагмента хромосомы. Вариация числа копий - важный фактор, вызывающий пороки развития, и также очень часто встречается в in vitro культуре эмбрионов, являясь важнейшей причиной, приводящей к неудачам при использовании экстракорпоральных методов репродукции. Вариация числа копий также является патогенным фактором многих заболеваний, включая онкологические. Метод полногеномной амплификации заключается в амплификации целого генома в одиночной клетке, множестве клеток или следов нуклеиновой кислоты и позволяет увеличить количества образца, исходя из сохранения репрезентативности целого генома, для достижения требуемого количества образца. Однако для метода полногеномной амплификации, в целом, характерна проблема систематической ошибки, связанной с амплификацией, которая становится источником отклонения при последующем анализе. В соответствии со способом выявления вариации числа копий в образце генома, согласно вариантам предлагаемого изобретения5 после полногеномной амплификации в образце, содержащем одиночную клетку или следы нуклеиновой кислоты, данные для анализа вариации числа копий получают при использовании технологий секвенирования. С одной стороны, полногеномная амплификация решает проблемы, связанные со сложностью анализа образца, содержащего одиночную клетку или следы нуклеиновой кислоты. С другой стороны, удается избежать систематической ошибки при анализе вариации числа копий, вызванной полногеномной амплификацией, вследствие чего обнаружение становится более точным и всеобъемлющим; в частности, эффективность обнаружения может быть дополнительно повышена посредством корреляции с содержанием ГЦ. Кроме того, согласно вариантам изобретения, на подэтапе конструирования библиотеки фрагментов ДНК из различных образцов, вводят разные коэффициенты, что позволяет одновременно анализировать несколько образцов, и, тем самым, дополнительно повысить эффективность выявления вариации числа копий в образце генома. Способ выявления числа копий в образце генома, согласно вариантам предлагаемого изобретения, может быть использован для скрининга и диагностики вариации числа копий перед имплантацией эмбриона или неинвазивного скрининга вариации числа копий у плода, которые целесообразно проводить в рамках генетического консультирования и которые могут являться основной для клинического решения; с помощью пренатальной диагностики, можно эффективно предотвратить имплантацию дефектных эмбрионов, приводящую к рождению детей с патологией.
II. Система выявления вариации числа копий в образце генома
Вторым объектом предлагаемого изобретения является система выявления вариации числа копий в образце генома. Использование системы позволяет эффективно осуществить вышеописанный способ выявления вариации числа копий в образце генома, для того чтобы устанавливать присутствие вариации числа копий в образце генома.
Как показано на Фиг. 2, согласно вариантам предлагаемого изобретения, система 1000 для выявления вариации числа копий в образце генома включает: секвенатор 100 и анализатор 200.
Согласно вариантам предлагаемого изобретения, секвенатор 100 выполняет секвенирование образца генома с получением результата секвенирования, состоящего из множества прочтений. В вариантах предлагаемого изобретения, система 1000 для выявления вариации числа копий в образце генома может дополнительно включать устройство для выделения генома (не показано на чертежах). Устройство для выделения генома осуществляет выделение образца генома из биологического образца, и соединено с секвенатором 100 для предоставления образца генома. Соответственно, биологический образец может быть непосредственно использован в качестве исходного материала для получения информации о возможной вариации копий в биологическом образце, отражающей состояние здоровья организмов. Согласно вариантам предлагаемого изобретения, секвенатор 100 может дополнительно включать: блок амплификации генома, блок построения библиотеки секвенирования и блок секвенирования, при этом блок амплификации генома осуществляет амплификацию образце генома; блок построения библиотеки секвенирования, соединенный с блоком амплификации генома, создает библиотеку секвенирования амплифицированного образца генома, а блок секвенирования соединенный с блоком построения библиотеки секвенирования определяет молекулярную последовательность для библиотеки секвенирования. В вариантах предлагаемого изобретения, на подэтапе секвенирования полного генома для построения библиотеки секвенирования используется, по крайней мере, одна технология, выбранная из технологий секвенирования следующего поколения, включающих систему Hiseq (Illumina Company), систему Miseq (Illumina Company), систему Genome Analyzer (GA) (Illumina Company), 454 FLX (Roche Company), систему SOLiD (Applied Biosystems Company), систему Ion Torrent (Life Technologie Company) и секвенатор отдельных молекул. Соответственно, могут быть использованы характеристики высокоскоростного и глубокого секвенирования, обеспечиваемого этими аппаратами, что дополнительно повышает эффективность выявления вариации числа копий в образце генома.
Согласно вариантам предлагаемого изобретения, анализатор 200 соединен с секвенатором 100 для выявления вариации числа копий в образце генома на основании результата секвенирования. В вариантах предлагаемого изобретения, анализатор 200 далее включает: блок 201 картирования, блок 202 определения точки разрыва, блок 203 определения окна детекции, блок 204 определения параметра и блок 205 обнаружения, при этом блок 201 картирования выполняет функцию картирования результата секвенирования к последовательности референсного генома для определения распределения прочтений в последовательности референсного генома. В вариантах предлагаемого изобретения, в качестве последовательности референсного генома в блоке 201 картирования сохранена известная последовательность генома человека; факультативно, в качестве последовательности референсного генома может быть выбрана, по крайней мере, одна из следующей группы: 21-ая хромосома человека, 18-ая хромосома, 13-ая хромосома, Х-хромосома и Y-хромосома. Блок 202 определения точки разрыва, соединенный с блоком 201 картирования, выполняет функцию определения множества точек разрыва в последовательности референсного генома на основании распределения прочтений в последовательности референсного генома, как описано выше, при этом число прочтений должно быть значимым по обе стороны от точек разрыва. Блок 203 определения окна детекции, соединенный с блоком 202 определения точки разрыва, выполняет функцию определения окна детекции в референсном геноме на основании множества точек разрыва. Блок 204 определения параметра, соединенный с блоком определения окна детекции, выполняет функцию определения пробного параметра на основании прочтений, попадающих в окно детекции. Блок 205 обнаружения, соединенный с блоком 204 определения параметра, предназначен для выявления вариации числа копий в окне детекции образца генома на основе различия между пробным параметром и заданным пороговым значением.
Согласно вариантам изобретения, блок 202 определения точки разрыва включает модуль, выполняющий следующие подэтапы:
деление последовательности референсного генома на множество первичных окон заданной длины и определение прочтений, попадающих в каждое из множества первичных окон;
Вначале последовательность референсного генома делят на множество первичных областей заданной длины и определяют прочтения, попадающие в каждую из множества первичных окон. Согласно конкретным примерам предлагаемого изобретения, типовые программы картирования позволяют соотнести прочтения, содержащиеся в полученном результате секвенирования, с последовательностью референсного генома, что позволяет определить прочтения, попадающие в каждое из множества первичных окон. В вариантах предлагаемого изобретения, первичные окна могут иметь разную или одинаковую длину; первичные области могут частично перекрываться, при условии известности информации, закодированной в каждой первичном окне. В предпочтительном варианте все первичные окна имеют одинаковую длину. Согласно вариантам предлагаемого изобретения, длина каждой из множества первичных окон может составлять от 100 до 200 тысяч п.н., предпочтительно 150 тысяч п.н. Согласно изобретению, в отношении числа первичных окон, расположенных по обе стороны участка (разрыва), какие-либо ограничения отсутствуют., и в конкретных примерах предлагаемого изобретения, с каждой стороны участка может быть выбрано 100 первичных окон.
Далее определяют величину p участка; которая может отражать значимое различие между числом прочтений между двумя сторонами участка. Кроме того, если величина p участка меньше конечной величины p, участок определяют, как точку разрыва. Согласно вариантам предлагаемого изобретения, диапазон конечной величины p может быть определен посредством параллельного анализа образца с известной последовательностью. В примерах предлагаемого изобретения, конечная величина р составляет 1,1×10-50.
Согласно предлагаемому изобретению, блок 202 определения точек разрыва дополнительно включает модуль для осуществления следующих подэтапов:
для выбранного участка выбирают одинаковое число первичных окон по обе стороны участка, соответственно, и подсчитывают относительное число прочтений, попадающих в каждое первичное окно Ri, где i - номер первичного окна,
для того чтобы определить величину p участка, к числу прочтений, попадающих в каждое первичное окно Ri, применяют критерий серий, при этом относительное число прочтений определяется следующей формулой:
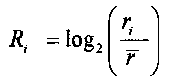
где ri - число прочтений, попадающих в i-тое первичное окно,
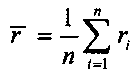 ,
,
n - общее число первичных окон.
Согласно вариантам предлагаемого изобретения, блок 202 определения точек разрыва дополнительно включает модуль для выполнения операций по применению статистического критерия серий ко всем относительным числам прочтений, попадающих в каждое из множества первичных окон:
для того, чтобы получить уточненное относительное число прочтений 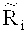
определяют среднеарифметическое число прочтений Zi, попадающих в каждое из множества первичных окон на основе уточненного относительного числа прочтений; и
применяют критерий серий ко всем из среднеарифметического числа прочтений Zi, попадающих в каждое из множества первичных окон.
Согласно вариантам предлагаемого изобретения, уточненное относительное число прочтений 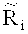
расчета содержания ГЦ-пар в каждом из множества первичных окон;
деления содержания ГЦ-пар на множество областей с точностью до 0.001 и расчета средней величины Ms среди всех относительных чисел прочтений, попадающих в каждую из множества областей, где s - номер области во множестве областей;
определения уточненного относительного число прочтений 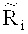
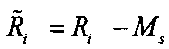 ;
;
определения среднеарифметического числа прочтений Zi по следующей формуле:
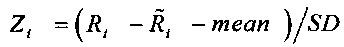 , где
, где
mean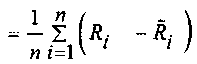 ,
,
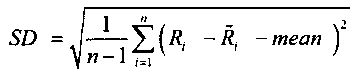 .
.
(mean=среднее значение; SD=СКО=среднеквадратичное отклонение)
После определения множества точек разрыва можно предварительно определить возможность вариации числа копий в области между двумя последовательными точками разрыва. Соответственно, при дальнейшем выявлении вариации числа копий такие области могут рассматриваться в качестве окон детекции. Если при предварительном определении было получено относительно больше точек разрыва, далее может быть проведен скрининг полученных точек разрыва. Согласно вариантам предлагаемого изобретения, на основании множества точек разрыва блок определения окна детекции включает модуль для выполнения следующих операций:
1) определение множества предполагаемых точек разрыва, где существуют другие точки разрыва как до, так и после предполагаемых точек разрыва;
2) определение величины p каждой предполагаемой точки разрыва и исключение предполагаемой точки разрыва с максимальной величиной p;
3) выполнение этапа 2) с остальными предполагаемыми точками разрыва до тех пор, пока все величины p остальных предполагаемых точек разрыва не окажутся меньше конечной величины p, при этом остальные предполагаемые точки разрыва рассматриваются как «прошедшие скрининг» предполагаемые точки разрыва; и
4) определение области между двумя прошедшими скрининг последовательными предполагаемыми точками разрыва, как окна детекции.
Согласно вариантам предлагаемого изобретения, получение величины p предполагаемой точки разрыва включает следующие этапы:
выбор области между предполагаемой точкой разрыва и предыдущей предполагаемой точкой разрыва в качестве первой предполагаемой области, и выбор области между предполагаемой точкой разрыва и следующей предполагаемой точкой разрыва в качестве второй предполагаемой области;
применение критерия серий к среднеарифметическому числу прочтений Zi, попадающих в первичные окна, которые включены и в первую предполагаемую область, и во вторую предполагаемую область, для определения величины p предполагаемых точек разрыва.
Согласно вариантам предлагаемого изобретения, получение конечной величины p предполагаемой точки разрыва включает следующие этапы:
на основании результата секвенирования контрольного образца, повторение этапа определения окна детекции в референсном геноме и регистрация величин p точек разрыва, удаляемых каждый раз до тех пор, пока число точек разрыва не станет равным нулю, и
на основании распределения величины p удаленных точек разрыва определяют конечную величину p, например, посредством построения диаграммы распределения величины p удаленных точек разрыва, и в качестве конечной величины p (pfinal) рассматривают величину p с максимальным трендом к изменению.
Согласно конкретным примерам предлагаемого изобретения, конечная величина р составляет 1,1×10-50.
Согласно вариантам предлагаемого изобретения, блок 204 определения параметров включает модуль для выполнения следующих операций: определения среднего значения для всех среднеарифметических чисел прочтений 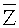
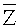
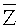
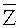
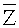
Соответственно, система и способ выявления вариации числа копий в образце генома согласно вариантам предлагаемого изобретения, позволяют эффективно определять присутствие вариации числа копий в образце генома и приемлем для разных вариаций, в частности, анеуплоидии хромосомы, делеции хромосомы и вставки, микроделеции и микродупликации фрагментов хромосомы.
Специалистам следует принять во внимание, что описанные выше характеристики и преимущества способа выявления вариации числа копий в образце генома относятся также к применению системы для выявления вариации числа копий в образце генома, и не включены в данное описание в целях удобства и краткости изложения.
III. Машиночитаемый носитель
Третьим объектом предлагаемого изобретения является машиночитаемый носитель. Согласно вариантам предлагаемого изобретения, на машиночитаемом носителе хранится алгоритм, предназначенный для выполнения процессором с целью выявления вариации числа копий в образце генома, и включает следующие этапы: картирование результата секвенирования к последовательности референсного генома для определения распределения прочтений в последовательности референсного генома; определение множества точек разрыва в последовательности референсного генома на основании распределения прочтений в последовательности референсного генома при значимом числе прочтений по обе стороны точек разрыва; определение окна детекции в референсном геноме на основании множества точек разрыва; определение пробного параметра на основании прочтений, попадающих в окно детекции; и выявление вариации числа копий в образце генома в окне детекцмм на основании различия между пробным параметром и заданным пороговым значением. Используя машиночитаемый носитель, можно эффективно осуществлять способ выявления вариации числа копий в образце генома согласно вариантам предлагаемого изобретения, в частности, выявлять анеуплоидию хромосомы, делецию хромосомы и также вставку, микроделецию и микродупликацию фрагментов хромосомы.
Специалистам следует принять во внимание, что описанные выше характеристики и преимущества способа выявления вариации числа копий в образце генома относятся также к применению машиночитаемого носителя, и не включены в данный документ в целях удобства и краткости изложения.
Будет приведена подробная ссылка на примеры предлагаемого изобретения. Специалистам следует принять во внимание, что описанные ниже примеры представлены с разъяснительной целью и не должны ограничивать объем предлагаемого изобретения. В тех случаях, когда в примерах отсутствует указание на конкретную технологию или условия, при воспроизведении такого примера следует использовать технологии и условия, описанные в литературе или известные из уровня техники (например, см. публикацию J. Sambrook, et al. (в переводе Huang РТ), Молекулярное клонирование: Руководство по лабораторным исследованиям, 3-е издание, Science Press), или руководствоваться инструкциями на продукт. Если производители реактивов или оборудования не указаны, можно использовать серийно выпускаемые товары, например, компанией Illumina.
Общий подход
На Фиг. 3 показано, что способ выявления вариации числа копий в образце генома, использованном в примерах, включает:
На первом этапе проведение полногеномной амплификация образца с последующим секвенированием полного генома и получением результата секвенирования в виде прочтений;
На втором этапе выполняют картирование полученных прочтений путем сравнения их с эталонной последовательностью референсного генома человека из базы данных NCBI с использованием программного обеспечения SOAP2, для того, чтобы получить информацию о расположении прочтений в геноме. Для того чтобы избежать вызванного повторами искажения результатов анализа вариации числа копий, для последующего анализа выбирают только те прочтения, для которых установлено однозначное соответствие при совмещении с последовательностью референсного генома человека.
На третьем этапе находят участок, по обе стороны которого присутствует статистически значимое число прочтений, осуществляя следующие операции:
a) расчет относительного числа прочтений в исследуемом образце (одновременно возможен анализ нескольких образцов):
в последовательности референсного генома человека выбирают окно длиной w (где w - целое число больше 1, например, от 10 тысяч до 10 миллионов п. н., предпочтительно -от 50 тысяч до 1 миллиона п.н.; боле предпочтительно, от 100 тысяч до 300 тысяч п.н., например, 150 тысяч п.н.); для всех полученных последовательностей рассчитывают число прочтений в каждом окне ri,j, где подстрочный индекс i указывает на номер окна, а подстрочный индек j - на номер образца; также рассчитывают содержание ГЦ в каждом окне GCi,j затем рассчитывают относительное число прочтений по формуле 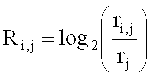
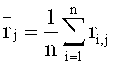
b) корреляция и нормализация данных
В системе координат по оси абсцисс (X) откладывают содержание ГЦ, по оси ординат (Y) - относительное число прочтений (R). Отрезок по оси X делят на множество областей, содержащих одинаковое количество единиц измерения, для каждой области рассчитывают среднее значение Ms для R (s - номер области ГЦ);
для каждого окна в образце рассчитывают относительное число прочтений 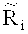
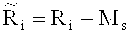
для каждого окна в образце рассчитывают нормализованное относительное число прочтений Zi,j по формуле:
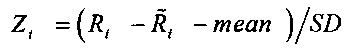 ,
,
где
mean 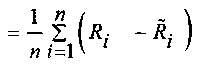 ,
,
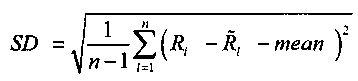 ,
,
mean=среднее; SD=СКО
с) определение и скрининг точек разрыва
определение точек разрыва: для каждого участка в последовательности референсного генома выбирают n окон (например, 100 окон), соответственно, с двух сторон участка, в качестве 2 популяций для статистической проверки; получают одну величину p, соответствующую каждому участку, рассчитывая разницу между двумя сторонами участка. m участков (например, 3000 участков), имеющих минимальную величину p, является точкой разрыва.
Скрининг точек разрыва: все упорядоченные точки разрыва записывают следующим образом: Bc={b1,b2,…,bs}, каждая точка разрыва находится между двумя последовательными фрагментами, где такие два фрагмента являются областями между предыдущей точкой разрыва и рассматриваемой точкой разрыва, и между рассматриваемой точкой разрыва и следующей точкой разрыва; в отношении всех Zi,j в таких двух фрагментах проводят статистическую проверку (например, с использованием критерия серий, представляющего собой непараметрический критерий, для оценки значимого различия между двумя популяциями при использовании равномерно распределенного в двух популяциях статуса смешанных элементов). Полученную величину p (pk) записывают как ″bk и рассматривают как показатель значимости точки разрыва". Предполагаемую точку разрыва, для которой максимальная величина p составляет pk, удаляют, и эту операцию повторяют до тех пор, пока все оставшиеся величины p не будут меньше конечной величины p pfinal для такой хромосомы;
получение конечной величины p: во время обнаружения выполняют вышеописанный этап определения множества точек разрыва, используя для анализа контрольный образец. Все упорядоченные предполагаемые точки разрыва в полном геноме записывают следующим образом Bc={b1,b2,…,bs}, каждая предполагаемая точка разрыва bk расположена между двумя последовательными фрагментами. В отношении всех Zi,j в таких двух фрагментах проводят статистическую проверку. Полученная величина p (pk) записывают как ″bk и принимают как показатель значимости точки разрыва". Предполагаемая точка разрыва, имеющая миниимальную величину p pk, удаляется, и эту операцию повторяют до тех пор, пока число предполагаемых точек рараыва не станет равным нулю. Строят диаграмму распределения с удаленной предполагаемой точкой разрыва, причем величину p с максимальным трендом к изменению принимают в качестве конечной величины p (pfinal);
определение окна детекции и подтверждение окна детекции: после получения точек разрыва, прошедших скрининг, определяют окно детекции. Для того чтобы далее определить окно детекции, рассчитывают среднее значение Zi,j в таком фрагменте и обозначают его 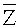
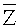
для каждого фрагмента после комбинации окон рассчитывают среднее значение и среднеквадратическую ошибку нормализованного числа анализируемых областей Zi,j, расположенных во всех контрольных образцах. Поскольку 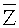
Пример 1. Выявление вариации числа копий во фрагментах плода при анализе образца одиночной эмбриональной клетки и выявление анеуплоидии хромосомы в образце одиночной эмбриональной клетки
1. Полногеномная амплификация: Для полногеномной амплификации двух образцов одиночной эмбриональной клетки, описанных в данном примере, использовали набор для полногеномной амплификации одиночной клетки GenomePlex® (Sigma Aldrich Company). Образец одиночной эмбриональной клетки представлял собой клетку трофобласта 5-дневной бластоцисты, которая была получены из бластеи методом лазерной захватной микродиссекции. После лизирования двух образцов одиночной эмбриональной клетки была выполнена полногеномная амплификация в соответствии с инструкциями производителя набора для анализа.
2. Секвенирование: В данном примере для секвенирования ДНК из образца одиночной эмбриональной клетки после полногеномной амплификации использовали метод секвенирования Hiseq2000 (Illumina Company). В соответствии с инструкциями Illumina Company было проведено конструирование библиотеки фрагментов ДНК для секвенирования и компьютерное секвенирование. В результате из каждого образца были получены данные объемом приблизительно 0,36 Г; различавшиеся индексными последовательностями. Прочтения, полученные при секвенировании, были соотнесены с помощью программы картирования SOAP2 с референсной последовательностью генома человека, хранящейся в базе данных NCBI, Build 36, для определения местонахождения полученных прочтений в референсной последовательности генома человека.
3. Анализ данных
а) расчет относительного числа анализируемых последовательностей в исследуемом образце и контрольном образце (термин «контрольный образец» применяется в отношении образца с нормальным кариотипом)
Референсная последовательность генома человека была разделена на множество окон длиной 150 тысяч п.н. Рассчитывали число прочтений, полученных на 2-м этапе, попадающих в каждое окно ri,j, где подстрочный индекс i - номер множества областей, индекс j - номер образца. Также рассчитывали содержание ГЦ в каждом окне. Относительное число прочтений рассчитывали по формуле из раздела Общий подход.
b) коррекция и нормализация данных
В системе координат откладывают содержание ГЦ по оси X и относительное число прочтений по оси Y. Отрезок по оси X делят на множество областей, содержащих одинаковое количество единиц измерения, где единица измерения составляет 0,001. В каждой области рассчитывают среднюю величину Ms для R; s - номер области ГЦ; эти значения показаны в Таблице 1. Коррекцию и нормализацию полученных прочтений осуществляли по формуле из раздела Общий подход.
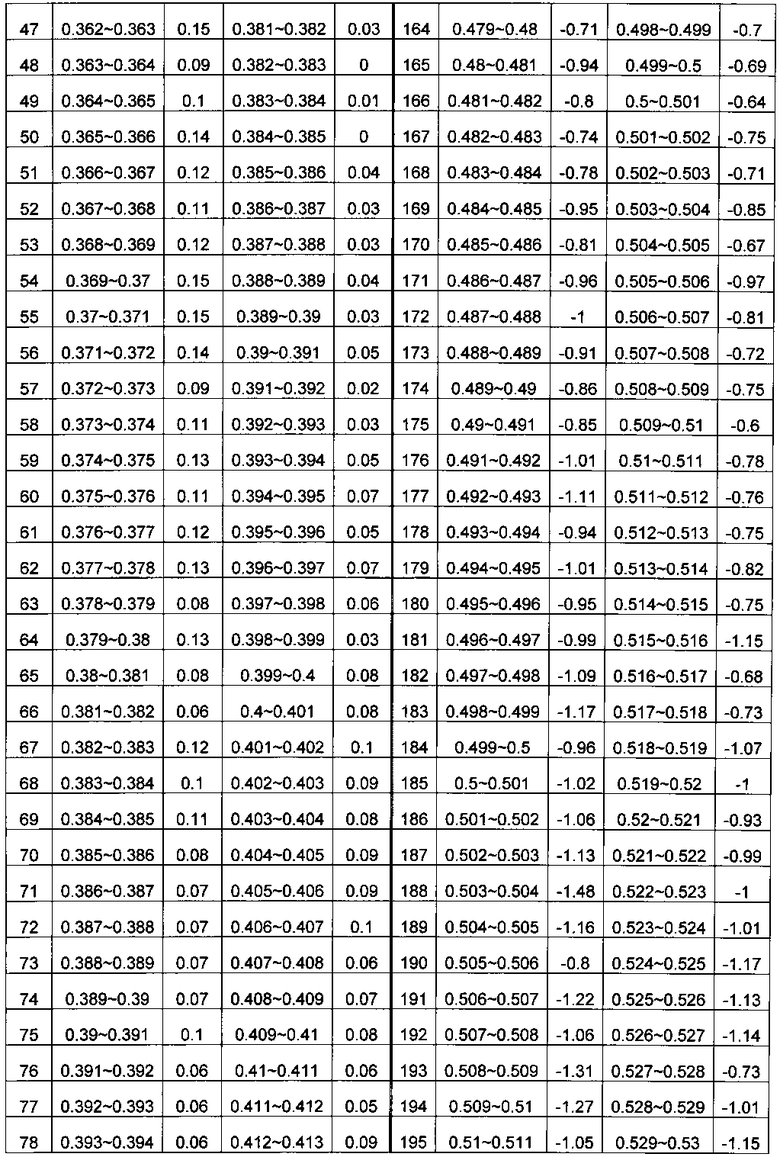
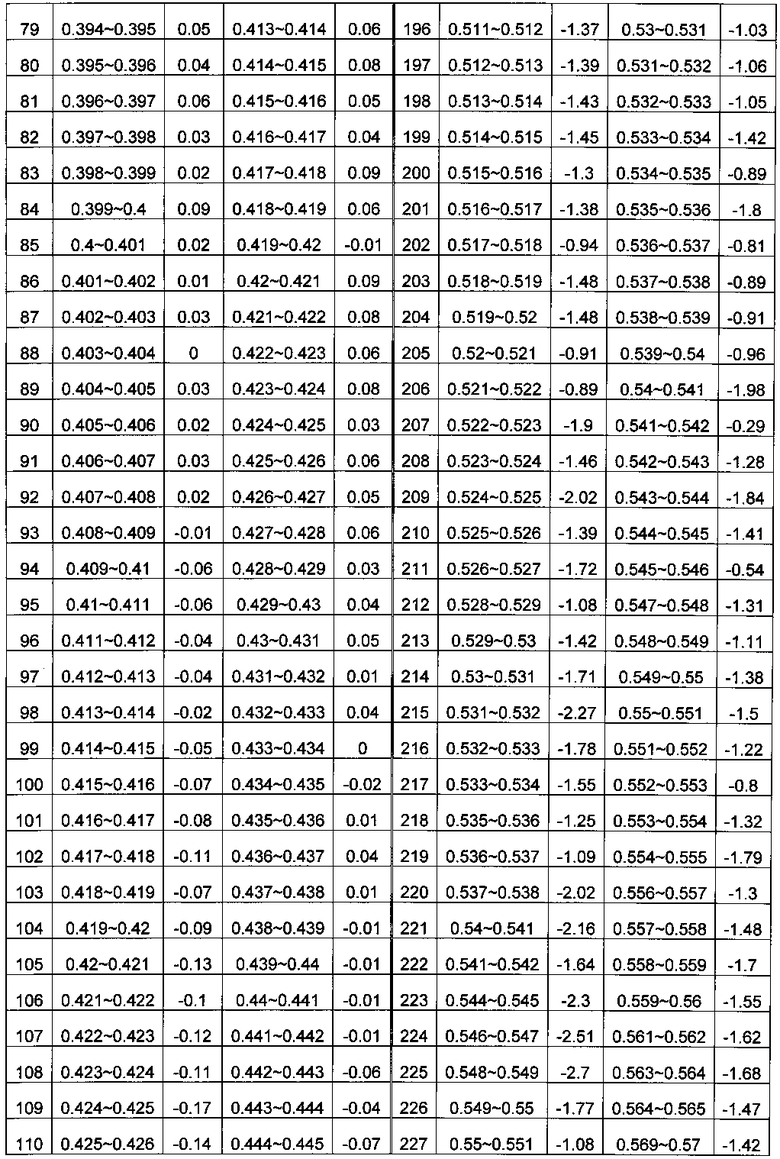
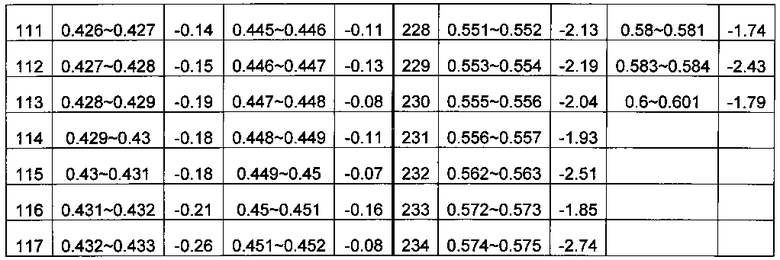
c) Комбинирование окон
Определение точки разрыва. Для каждого участка в последовательности референсного генома выбирали по 100 окон, расположенных по обе стороны участка, соответственно, в качестве двух популяций, в отношении которых проводилась проверка с использованием критерия серий. В результате расчета различия между двумя сторонами участка была получена одна величина p, соответствующая каждому участку; рассматривали 3000 участков, участки с минимальной величиной p рассматривали в качестве точки разрыва.
Скрининг точек разрыва: все упорядоченные точки разрыва записывают следующим образом: Bc={b1,b2,…,bs}, каждая точка разрыва находится между двумя последовательными фрагментами, где такие два фрагмента являются областями между предыдущей точкой разрыва и рассматриваемой точкой разрыва, и между рассматриваемой точкой разрыва и следующей точкой разрыва. В отношении всех Zi,j в таких двух фрагментах проводили статистическую проверку с использованием критерия серий. Полученную величину p (pk) записывали как ″bk и рассматривали как показатель значимости точки разрыва". Предполагаемую точку разрыва, для которой максимальноая величина p составляла pk, удаляли, и эту операцию повторяли до тех пор, пока все оставшиеся величины p не оказывались меньше конечной величины p pfinal для такой хромосомы, равной 1,1×10-50.
d) После скрининга точек разрыва область между двумя последовательными точками разрыва определяли как окно детекции, для комбинирования окон. Для того чтобы дополнительно фильтровать фрагменты, полученные при комбинировании окон, рассчитывали среднее значение Zi,j в таком фрагменте, записывая результат как 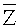
4. Результат
В Таблице 2 показан результат анализа - список вариаций числа копий после полногеномной амплификации образца одиночной эмбриональной клетки, изученного в данном примере.
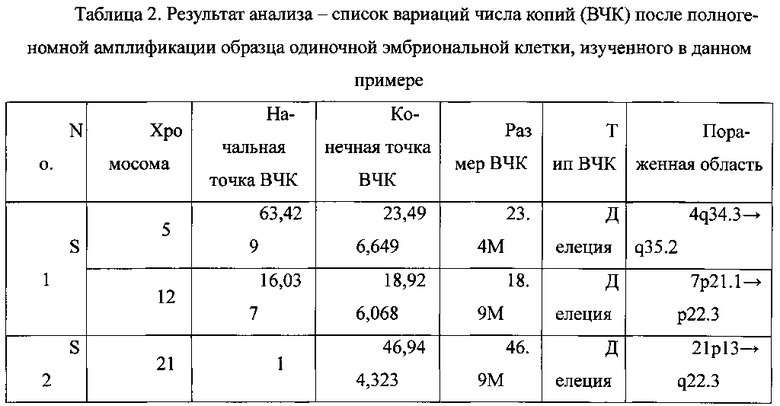
Как видно из Таблицы 2, использование способа выявления вариации числа копий в образце генома, согласно вариантам предлагаемого изобретения, позволяет эффективно определять разнообразные типы вариаций числа копий.
Пример 2
Использовали образец одиночной эмбриональной клетки, такой же, как в Примере 1; этапы анализа были такими же, как в Примере 1, но геномную ДНК не подвергали полногеномной амплификации, а сразу секвениировали. В Таблице 3, Фиг. 4 и Фиг. 5 представлено сравнение результатов, полученных в Примере 1 и Примере 2.

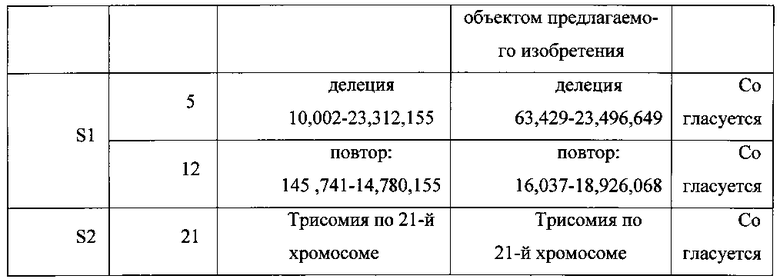
Как видно из Таблицы 3 и изображений кариотипа хромосомы на Фиг. 4 и Фиг. 5, анализ вариации числа копий в прочтениях в образце геномной ДНК, подвергнутой полногеномной амплификации, и образце геномной ДНК, не подвергавшейся полногеномной амплификации, приводит к одинаковым результатам. В отношении различия начальной и конечной точек делеции и повтора в Таблице 3, в связи с трудностью точного определения границы вариации числа копий, в целом, для первичной области длиной около 150 тысяч п.н., две границы, различавшиеся на 100-300 тысяч п.н. были определены как полностью согласующиеся, две границы, различавшиеся на 300 тысяч-1 млн п.н. - как вполне согласующиеся. Поскольку различие между границами вариации числа копий, определенными двумя методами (Таблица 3), составляло 100-300 тысяч п.н. или 300 тысяч - 1 млн п.н., сделано заключение о согласованности границ вариации числа копий, определенных двумя методами.
Промышленная применимость
Предложенные согласно изобретению способ, система, используемая для осуществления способа, и машиночитаемый носитель могут эффективно применяться для выявления вариации числа копий в образце генома.
Термины по всему описанию "вариант", "некоторые варианты", "один конструктивный вариант", "пример", "другой пример", или "некоторые примеры" означают, что отдельные признаки, структуры, материалы или параметры, приведенные для примера или конструктивного варианта относятся, по крайней мере, к одному из вариантов или примеров изобретения. Словесные обороты, такие как "в некоторых конструктивных вариантах", "Согласно одному из вариантов изобретения", "в конструктивном варианте", "в примере", "в отдельных примерах", или "в некоторых примерах" в разных местах описания не обязательно означают отсылку только к одному конкретному примеру воплощения изобретения. Более того, отдельные признаки, структуры, материалы или параметры могут сочетаться любым подходящим образом в одном или более вариантов или примеров.
Хотя были представлены и описаны поясняющие примеры, специалисту должно быть понятно, что конструктивные варианты не ограничивают объем изобретения. Могут существовать изменения, варианты и модификации, не отклоняющиеся от духа и смысла изобретения, поэтому объем испрашиваемой защиты определяется формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОБНАРУЖЕНИЯ ВАРИАЦИЙ ЧИСЛА КОПИЙ (CNV) ПО ДАННЫМ СЕКВЕНИРОВАНИЯ ПОЛНОГО ЭКЗОМА ЧЕЛОВЕКА И ГЕНОМА С НИЗКИМ ПОКРЫТИЕМ | 2023 |
|
RU2822040C1 |
| СПОСОБ НЕИНВАЗИВНОЙ ДИАГНОСТИКИ АНЕУПЛОИДИЙ ПЛОДА МЕТОДОМ СЕКВЕНИРОВАНИЯ | 2014 |
|
RU2543155C1 |
| СПОСОБ НЕИНВАЗИВНОЙ ПРЕНАТАЛЬНОЙ ДИАГНОСТИКИ АНЕУПЛОИДИЙ ПЛОДА | 2015 |
|
RU2627673C2 |
| СПОСОБ НЕИНВАЗИВНОЙ ПРЕНАТАЛЬНОЙ ДИАГНОСТИКИ АНЕУПЛОИДИЙ ПЛОДА | 2014 |
|
RU2583830C2 |
| СПОСОБЫ И СИСТЕМЫ МОНИТОРИНГА СОСТОЯНИЯ ЗДОРОВЬЯ И ПАТОЛОГИИ ОРГАНОВ | 2020 |
|
RU2818052C2 |
| ТЕХНОЛОГИЯ ОПРЕДЕЛЕНИЯ АНЕУПЛОИДИИ МЕТОДОМ СЕКВЕНИРОВАНИЯ | 2012 |
|
RU2529784C2 |
| НЕИНВАЗИВНЫЙ ДИАГНОСТИЧЕСКИЙ ТЕСТ ДНК ДЛЯ ОБНАРУЖЕНИЯ АНЕУПЛОИДИИ | 2012 |
|
RU2638456C2 |
| Система обработки данных полногеномного секвенирования | 2023 |
|
RU2804535C1 |
| СПОСОБЫ И СИСТЕМЫ ДЛЯ ОПРЕДЕЛЕНИЯ ТОГО, ЯВЛЯЕТСЯ ЛИ ГЕНОМ АНОМАЛЬНЫМ | 2011 |
|
RU2599419C2 |
| Способ выявления вариаций и изменений числа копий в генах BRCA1 и BRCA2 по данным таргетного массового параллельного секвенирования генома | 2020 |
|
RU2759953C2 |


Группа изобретений относится к области геномики. Предложены способ выявления вариации числа копий в образце генома и система, используемая для осуществления способа. Способ выявления вариации числа копий в образце генома, включает этапы: секвенирования образца генома, для получения результата секвенирования в виде множества прочтений; картирования результата секвенирования к эталонной геномной последовательности для установления распределения прочтений в эталонной геномной последовательности; определения множества точек разрыва в эталонной геномной последовательности, основанное на распределении прочтений в эталонной геномной последовательности, где число прочтений значимо в обеих сторонах от точек разрыва; определения окна детекции в эталонной геномной последовательности на основе множества точек разрыва; определения пробного параметра на основе прочтений, попадающих в окно детекции; и определения вариации числа копий в окне детекции образца генома на основе различия между пробным параметром и заданным пороговым значением. Система содержит секвенатор, анализатор, блок картирования, блок определения точек разрыва, блок определения окна детекции, блок определения параметров, блок обнаружения. Использование данных изобретений позволяет эффективно определить присутствие разнообразных вариаций числа копий в образце генома. 2 н. и 17 з.п. ф-лы, 5 ил., 3 табл., 2 пр.
1. Способ определения вариации числа копий в образце генома, включающий:
секвенирование образца генома, для получения результата секвенирования, состоящего из множества прочтений;
картирование результата секвенирования к эталонной геномной последовательности для определения распределения прочтений в эталонной геномной последовательности;
определение множества точек разрыва в эталонной геномной последовательности, основанное на распределении прочтений в эталонной геномной последовательности, где число прочтений значимо в обеих сторонах от точек разрыва;
определение окна детекции в эталонной геномной последовательности на основе множества точек разрыва;
определение пробного параметра на основе прочтений, попадающих в окно детекции; и определение вариации числа копий в окне детекции образца генома на основе различия между пробным параметром и заданным пороговым значением.
2. Способ по п. 1, отличающийся тем, что образец генома получают из отдельной клетки, и способ включает:
лизис отдельной клетки для выделения целого генома отдельной клетки; и амплификацию целого генома для получения образца генома.
3. Способ по п. 1, отличающийся тем, что вариация числа копий представляет собой по крайней мере одну из группы, включающей анеуплоидность хромосомы, делецию хромосомы, вставку участка хромосомы, микроделецию участка хромосомы и микродупликацию участка хромосомы.
4. Способ по п. 1, отличающийся тем, что этап определения множества точек разрыва в эталонной геномной последовательности включает:
деление эталонной геномной последовательности на множество первичных окон, имеющих заданную длину, и определение прочтений, попадающих в первичные окна;
определение по крайней мере для одного участка в эталонной геномной последовательности числа прочтений, попадающих в первичные окна с тем же порядковым номером с обеих сторон участка;
определение величины p сайта, где величина p показывает, какое число прочтений, попадающих на каждую сторону сайта, имеет значимость, и
в котором участок представляет точку разрыва, если величина p участка меньше, чем конечная величина p.
5. Способ по п. 4, отличающийся тем, что прочтения, попадающие в каждое из первичных окон, являются однозначно картированными.
6. Способ по п. 4, отличающийся тем, что 100 из первичных окон выбраны с каждой стороны участка.
7. Способ по п. 4, отличающийся тем, что первичные окна имеют длину от 100 до 200 тысяч пар оснований.
8. Способ по п. 4, в котором конечная величина p составляет 1,1·10-50 или меньше.
9. Способ по п. 4, отличающийся тем, что стадия определения величины p участка включает:
выбор для участка первичных окон с одинаковыми порядковыми номерами с обеих сторон участка соответственно, и вычисление относительного числа прочтений, попадающих в каждое первичное окно Ri, где i представляет порядковый номер первичного окна, и
тестирование относительного числа прочтений, попадающих во все первичные окна Ri, для определения таким образом величины p участка,
где относительное число прочтений определяют по следующей формуле:
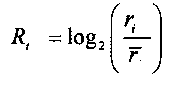
где ri представляет число прочтений, попадающих в i-тое первичное окно, и
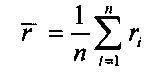 ,
,
где n представляет общее число первичных окон.
10. Способ по п. 9, отличающийся тем, что тестирование относительного числа прочтений, попадающих во все первичные окна, включает:
коррекцию относительного числа прочтений, попадающих в каждое первичное окно Ri с учетом содержания ГЦ-пар для получения уточненного относительного числа прочтений 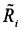 ;
;
определение среднеарифметического числа прочтений Zi, попадающих в каждое из первичных окон на основе уточненного относительного числа прочтений; и
тестирование всех среднеарифметических чисел прочтений, попадающих в каждое первичное окно Zi.
11. Способ по п. 10, отличающийся тем, что уточненное относительное число прочтений получают, осуществляя следующие этапы:
подсчет содержания ГЦ в каждом первичном окне;
деление содержания ГЦ на множество областей с точностью до 0.001, и подсчет средней величины Ms относительного числа прочтений, попадающих в каждую из множества областей, где s показывает номер областей во множестве областей;
определение уточненного относительного числа прочтений по следующей формуле:
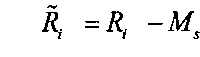 ; и
; и
определение среднеарифметического числа прочтений Z по следующей формуле:
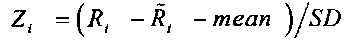 , где
, где
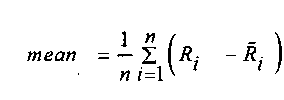 , и
, и
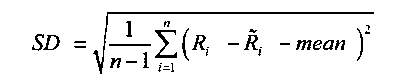
и mean - означает среднее.
12. Способ по п. 11, отличающийся тем, что этап определения окна детекции в эталонной геномной последовательности на основе множества точек разрывов включает:
1) определение множества предполагаемых точек разрыва, где существуют другие точки разрыва, как перед, так и после предполагаемой точки разрыва;
2) определение величины p каждой предполагаемой точки разрыва, и исключение предполагаемой точки разрыва, имеющей конечную величину p;
3) выполнение этапа 2) с оставшимися предполагаемыми точками разрыва до тех пор, пока величина p оставшихся предполагаемых точек разрыва остается меньше, чем конечная величина p, в котором оставшиеся предполагаемые точки разрывы считаются прошедшими скрининг предполагаемыми точками разрыва; и
4) определение области между двумя последовательными прошедшими скриниг предполагаемыми точками разрыва в качестве окна детекции,
где значение p предполагаемой точки разрыва получают, осуществляя следующие этапы:
выбор области между предполагаемыми точками разрыва и предшествующей предполагаемой точкой разрыва в качестве первой области-кандидата, и выбор области между предполагаемыми точками разрыва и следующей предполагаемой точкой разрыва в качестве второй области-кандидата;
тестирование среднеарифметического числа прочтений Zi, попадающих в первичные окна, которые включены и в первую, и во вторую области- кандидаты, для определения таким образом величины p предполагаемых точек разрыва.
13. Способ по п. 12, отличающийся тем, что этап определения пробного параметра на основе прочтений, попадающих в окно детекции, включает определение среднего показателя среднеарифметических чисел прочтений 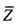 , попадающих во все первичные окна,
, попадающих во все первичные окна,
которые включены в окна детекции, где средний показатель среднеарифметических чисел прочтений принимается в качестве пробного параметра.
14. Способ по п. 1, отличающийся тем, что заданные пороговые значения составляют -1.645 для первого порога и 1.645 для второго порога.
15. Способ по п. 1, отличающийся тем, что эталонная геномная последовательность представляет собой по крайней мере одну, выбранную из группы, включающей последовательность человеческой хромосомы 21, человеческой хромосомы 18, человеческой хромосомы 13, человеческой хромосомы X и человеческой хромосомы Y.
16. Система определения вариации числа копий в образце генома, содержащая: секвенатор, выполняющий секвенирование образца генома для получения в результате множества прочтений;
анализатор, соединенный с секвенатором и определяющий вариации числа копий в образце генома, основываясь на результатах секвенирования, при этом анализатор содержит:
блок картирования, выполняющий функцию картирования результата секвенирования к эталонной геномной последовательности для определения распределения прочтений в эталонной геномной последовательности;
блок определения точек разрыва, соединенный с блоком картирования, выполняющий функцию определения множества точек разрыва в эталонной геномной последовательности, основанное на распределении прочтений в эталонной геномной последовательности, где число прочтений значимо с обеих сторон от точек разрыва;
блок определения окна детекции, соединенный с блоком определения точек разрыва, выполняющий функцию определения окна детекции в эталонной геномной последовательности на основе множества точек разрыва;
блок определения параметров, соединенный с блоком определения окна детекции, выполняющий функцию определения пробного параметра на основе прочтений, попадающих в окно детекции; и
блок обнаружения, соединенный с блоком определения параметров и выполняющий функцию определения вариации числа копий в образце генома в окне детекции на основе различия между пробным параметром и заданным пороговым значением.
17. Система по п. 16, отличающаяся тем, что содержит устройство для выделения генома из биологического образца.
18. Система по п. 16, отличающаяся тем, что секвенатор содержит:
блок амплификации образца генома;
блок построения библиотеки секвенирования, соединенный с блоком амплификации генома, и выполняющий функцию построения библиотеки секвенирования амплифицированного образца генома;
и
блок секвенирования, соединенный с блоком построения библиотеки секвенирования, выполняющий функцию определения молекулярной последовательности для библиотеки секвенирования.
19. Система по п. 18, отличающаяся тем, что блок секвенирования содержит по крайней мере один аппарат, выбранный из группы, включающей систему Hiseq, систему Miseq, систему Genome Analyzer, 454 FLX, систему SOLiD, систему Ion Torrent и одномолекулярный секвенатор.
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Способ приготовления лака | 1924 |
|
SU2011A1 |

