Изобретение относится к области молекулярной биологии и диагностической медицины и может быть использовано для определения крупных перестроек в генах BRCA1 и BRCA2.
Мутации в генах могут иметь различные последствия для функционирования организма человека. В случае герминальных мутаций, возникших в клетках зародышевых путей предков человека, может развиваться наследственное заболевание. Примерами могут служить фенилкетонурия, галактоземия, нейрофиброматоз, семейные формы рака. Другой тип мутаций - соматические - имеет значение только для данного человека и могут вести к развитию многих видов заболеваний, включая все виды опухолей. По другой классификации все мутации можно разделить на точечные и протяженные. При этом современные методы секвенирования ДНК, в первую очередь, массовое параллельное секвенирование (MPS, или NGS), позволяют выявлять их с высокой скоростью и точностью. Особо высокой эффективностью по соотношению цена/количество полезной информации обладают методы таргетного NGS, когда изучаются выбранные исследователем районы генома пациента, конкретные гены и/или их экзоны.
Для обработки получаемых данных и выявления точечных мутаций разработано и протестировано большое число алгоритмов и программ: samtools (http://samtools.sourceforge.net) [1], GenomeAnalysisToolkit [2], BWA [3] и другие. В то же время, несмотря на то, что на сегодняшний день опубликовано и разработано большое число программ, направленных на выявление крупных перестроек и вариаций/изменений в числе копий (CNV – герминальные мутации и CNA – соматические), нет программ, которые бы с высокой точностью могли бы предсказывать CNV- и CNA-мутации по NGS-данным. Одним из наиболее удобных и часто используемых подходов является анализ покрытия прочтениями целевых фрагментов генома пациента. Таких программ уже разработано более 15 [4]: SegSeq [5], RDXplorer [6], CNAseg [7], cnv-seq [8] и другие. Кроме того, в литературе представлено изобретение способа выявления вариаций числа копий по данным полногеномного секвенирования [9]. Большинство из описанных изобретений могут проводить анализ только с использованием референсных образцов с известным статусом наличия или отсутствия CNV-мутации [4], что повышает стоимость такого исследования для одного пациента. При этом остальные программы из-за амплификации фрагментов при приготовлении библиотеки имеют трудности при обработке данных таргетного NGS-секвенирования, которое, благодаря своей стоимости и количеству получаемой клинически важной информации, имеет высокую применимость в медицине. Золотым стандартом выявления CNV остается метод мультиплексной амплификации лигазо-связанных проб (MLPA) [10].
Таким образом, разработка нового способа выявления вариаций и изменений числа копий по данным таргетного NGS позволит повысить эффективность использования данной технологии в медицине благодаря детекции герминальных и соматических крупных перестроек (CNV и CNA, соответственно), который ранее не выявлялся существующими алгоритмами.
Наиболее близким к заявленному способу - прототипом, является способ выявления герминальных крупных перестроек, предложенный Márton Zsolt Enyedi и соавт. [11], где также использовались таргетное массовое параллельное секвенирование и вычисление значения покрытия полученными прочтениями ампликонов. Сначала проводится картирование полученных после секвенирования прочтений и вычисляются значения покрытия для каждого из ампликонов всех пациентов. После этого осуществляются нормализация этих значений и определение герминальных крупных перестроек. В данном способен используют три референсных образца, для каждого из которых заранее было показано отсутствие CNV-мутаций.
Недостатками известного способа являются необходимость в референсных образцах, что значительно увеличивает себестоимость такого анализа, а также неприменимость для выявления соматических крупных перестроек (CNA). Кроме того, известный способ применим только для библиотеки, приготовленной с помощью амлификации целевых фрагментов и дальнейшей их гибридизацией с адаптерными последовательностями, что значительно сужает его применимость на практике.
Задачей изобретения является создание нового способа выявления герминальных и соматических вариаций и изменений числа копий путем таргетного массового параллельного секвенирования экзонов генов BRCA1 и BRCA2 и обработки получаемых данных.
Технический результат: снижение себестоимости анализа и повышение клинической чувствительности способа.
Поставленная задача достигается предлагаемым способом, заключающимся в следующем.
Образцы геномной ДНК, выделенные из лейкоцитов крови пациентов или гистологических блоков, амплифицируют, используя коммерческие или собственные панели для таргетного секвенирования одного или многих генов или множества экзонов различных генов. Преимущественно используют набор праймеров, представленных в таблице 1, однако он может быть заменен на любой другой, который позволяет выявлять точечные мутации в генах BRCA1 и BRCA2. В случае необходимости, на данном этапе проводят очистку полученных ампликонов, после чего их лигируют с адаптерными последовательностями, необходимыми для проведения секвенирования, либо присоединяют их с помощью еще одной стадии полимеразной цепной реакции (ПЦР). Далее проводят секвенирование полученной библиотеки на приборе для проведения NGS (например, MiniSeq, MiSeq, NextSeq, HiSeq Illumina или Ion Torrent PGM). Полученные после секвенирования прочтения картируют на последовательность референсного генома человека или последовательности отдельных хромосом. Перед картированием каждую референсную последовательность индексируют программой samtools (http://samtools.sourceforge.net) [1]. Картирование осуществляют функцией mem программы BWA (http://bio-bwa.sourceforge.net) [3]. Далее полученный SAM-файл конвертируют в BAM-файл функцией view программы samtools, а последний сортируют и индексируют функциями sort и index samtools, соответственно. После этого могут быть проведены дополнительные обработки, например, перевыравнивание областей с инсерциями/делециями, рекалибровка и другие. Однако эти процедуры необязательны.
Полученные сортированные BAM-файлы обрабатывают с помощью команды mpileup программы samtools или любой другой аналогичной, которая может посчитать число прочтений, картированных на выбранный район. При этом для каждого выбранного района рассчитывается среднее значение покрытия и записывается в один файл-таблицу. Далее значения для всех образцов делят на такие группы, в каких осуществлялось приготовление библиотеки. Если все образцы готовились одновременно и единообразно, данный этап может быть пропущен. Затем оставляют только те образцы, для которых медианное значение покрытия составило более 50 прочтений, и для оставшихся образцов каждое значение в таблице нормируют в три этапа. На первом этапе каждое значение нормируют на медианное значение для всех ампликонов данного пациента. На втором - на медиану нормированных на первом этапе значений для каждого пациента данного ампликона. На третьем - на медиану дважды нормированных значений для каждого мультиплекса данного пациента. Последний этап могут не проводить, если информация о составе мультиплексов отсутствует. В конечном итоге, все значения для удобства умножают на два, что также может быть опущено.
После этого проводят поиск рядов значений, все элементы которого не более 1,8 (потенциальные делеции), а также рядов, все элементы которого не менее 2,4 (потенциальные инсерции). Далее из всех найденных потенциальных делеций оставляют те, у которых присутствует хотя бы один элемент со значением не более 1,3. Для всех оставшихся потенциальных делеций рассчитывается показатель (score):
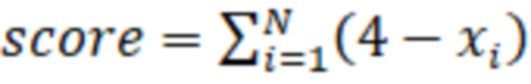 ,
,
где xi - значение элемента потенциальной делеции, N - длина потенциальной делеции.
Из потенциальных инсерций оставляют те, у которых присутствует хотя бы один элемент со значением не менее 2,7. Для всех оставшихся потенциальных инсерций также рассчитывается показатель (score):
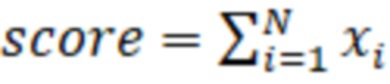 ,
,
где xi - значение элемента потенциальной инсерции, N - длина потенциальной инсерции.
Следующая процедура учитывает тот факт, что по крайней мере для большинства крупных перестроек границы мутации находятся в интронах, а, значит, CNV-мутация не может затрагивать только часть экзона. Поэтому для всех потенциальных инсерций и делеций определяется, покрывают ли затронутые ампликоны экзон(-ы) полностью или только его часть. Если полностью не покрыт ни один из экзонов, такая CNV-мутация далее не рассматривается. Если часть ампликонов покрывает один экзон целиком, а остальные - только часть экзона(-ов), то показатель такой CNV-мутации умножается на 0,75, и далее рассматривается только целиком удаленный(-ые) экзон(-ы). Для дальнейшего анализа оставляют только те потенциальные инсерции и делеции, которые имеют показатель не ниже 2.
На следующем этапе проводят статистический анализ вероятности наблюдения данной CNV-мутации при случайном перемешивании нормированных значений каждого ампликона пациента и случайном перемешивании рядов ампликонов, составляющих потенциальную CNV-мутацию, всех пациентов. Для этого при каждом перемешивании оценивают число наблюдений потенциальной CNV-мутации равной протяженности и с показателем не более, чем на 10% выше, чем без перемешивания. Процент таких наблюдений от общего числа перемешиваний составляет вероятность того, что данная CNV-мутация ложноположительная. Оставляют только те CNV-мутации, для которых данная вероятность составляет не более 0,05. Оставшиеся мутации считаются выявленными и проверяются с помощью MLPA.
Определяющими отличиями заявляемого способа, по сравнению с прототипом, являются применение для нормализации медианных значений покрытия вместо средних, трехэтапная нормализация вместо двух этапов, использование информации об экзонно-интронной структуре гена, а также вычисление оценочного значения (score) для каждой потенциальной крупной перестройки (CNV или CNA) и вероятности того, что данная мутация ложно-положительна. заявляемый. При этом используют экспериментально подобранный оптимальный набор праймеров для обогащении библиотеки целевыми последовательностями, представленный в таблице 1. Заявляемый способ подходит для библиотеки, приготовленной как с помощью амплификации и гибридизации, так и с помощью только амплификации.
Предлагаемый способ обладает высокой специфичностью и чувствительностью при анализе ДНК, выделенной из крови (для поиска CNV) или гистологических блоков (для поиска CNA). По сравнению с золотым стандартом (MLPA) разработанный способ обладает высокой чувствительностью (100%) и специфичностью (94%), что дает возможность его использования для скрининга пациентов на протяженные делеции и инсерции в генах BRCA1 и BRCA2.
Способ апробирован на представительной выборке в 127 образцов ДНК пациентов клинической лаборатории больных раком яичников. Разработанный подход может быть использован клиническими лабораториями как для выявления уже известных CNV- и CNA-мутаций, так и для еще ранее не выявленных, обладает наиболее низкой стоимостью анализа среди описанных к настоящему времени альтернативных подходов. Данный метод хорошо адаптируем к инструментальным возможностям современных клинико-диагностических лабораторий и, таким образом, уже в настоящее время может быть использован в онкологических диспансерах и лабораториях.
Пример 1. Выявление герминальных вариаций числа копий в генах BRCA1 и BRCA2 в образцах ДНК из лейкоцитов крови.
Образцы геномной ДНК выделяют из лейкоцитов крови пациентов любым из возможных способов, например, путём лизиса клеток с использованием 10% SDS и протеиназы K, очистки от белков с помощью фенола и хлороформа и осаждения ДНК в этаноле. Далее проводят амплификацию с использованием праймеров, представленных в таблице 1. К полученным ампликонам пришивают адаптеры, содержащие индексирующие последовательности и последовательности для проведения секвенирования, с помощью лигирования или второго этапа амплификации. После этого полученную библиотеку очищают от невключившихся адаптеров и коротких ампликонов и подвергают массовому параллельному секвенированию на подходящем приборе, в зависимости от используемых адаптеров. Полученные прочтения картируют на референсный геном человека и определяют среднее покрытие для каждого из целевых регионов. Далее проводят обработку и анализ полученных значений, как описано выше.
Пример 2. Выявление соматических изменений числа копий в генах BRCA1 и BRCA2 в образцах ДНК из парафиновых гистологических блоков.
Все процедуры проводятся таким же образом, как и в Примере 1, за исключением того, что ДНК выделяют из гистологических блоков соответствующим способом, например, с помощью щелочного лизиса с последующим выделением ДНК из осадка.
Пример 3. Выявление соматических изменений числа копий в генах BRCA1 и BRCA2 в образцах ДНК из парафиновых гистологических блоков с включением адаптеров с помощью лигирования.
Все процедуры проводятся таким же образом, как и в Примере 1, за исключением того, что полученные после ПЦР с праймерами из таблицы 1 ампликоны очищают с помощью магнитных частиц и адаптеры включаются с помощью лигирования и повторных очисток. Примером такого способа приготовления библиотеки может быть GeneRead BRCA panel v2 (Qiagen).
Таким образом, разработанный новый способ выявления герминальных и соматических вариаций и изменений числа копий по данным таргетного NGS позволит повысить эффективность использования данной технологии в медицине благодаря детекции крупных перестроек, которые ранее не выявлялись существующими способами.
Перечень последовательностей праймеров
Таблица 1.
Источники информации
[1] Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009 Aug 15;25(16):2078-2079.
[2] McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010 Sep;20(9):1297-1303.
[3] Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009 Jul 15;25(14):1754-1760.
[4] Zhao M, Wang Q, Wang Q, Jia P, Zhao Z. Computational tools for copy number variation (CNV) detection using next-generation sequencing data: features and perspectives. BMC Bioinformatics. 2013;14.
[5] Chiang DY, Getz G, Jaffe DB, O'Kelly MJ, Zhao X, Carter SL, Russ C, Nusbaum C, Meyerson M, Lander ES. High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat Methods. 2009 Jan;6(1):99-103.
[6] Yoon S, Xuan Z, Makarov V, Ye K, Sebat J. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res. 2009 Sep;19(9):1586-1592.
[7] Ivakhno S, Royce T, Cox AJ, Evers DJ, Cheetham RK, Tavaré S. CNAseg - a novel framework for identification of copy number changes in cancer from second-generation sequencing data. Bioinformatics. 2010 Dec 15;26(24):3051-3058.
[8] Xie C, Tammi MT. CNV-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinformatics. 2009 Mar 6;10:80.
[9] Инь С., Чжан Ч., Чэнь Ш., Чжан Ч., Пань С., Цзян Х., Чжан С. Способ и система выявления вариации числа копий в геноме // Патент РФ № 2593708 от 10.08.2016 г., заявка № 2014134175/10 от 20.01.2012 г.
[10] Anna Samelak-Czajka, Malgorzata Marszalek-Zenczak, Malgorzata Marcinkowska-Swojak, Piotr Kozlowski, Marek Figlerowicz, and Agnieszka Zmienko. MLPA-Based Analysis of Copy Number Variation in Plant Populations. Front Plant Sci. 2017; 8:222.
[11] Márton Zsolt Enyedi, Gábor Jaksa, Lajos Pintér,2 Farkas Sükösd, Zoltán Gyuris, Adrienn Hajdu, Erika Határvölgyi, Katalin Priskin, and Lajos Haracska. Simultaneous detection of BRCA mutations and large genomic rearrangements in germline DNA and FFPE tumor samples. Oncotarget. 2016 Sep 20;7(38):61845-61859.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ определения нуклеотидных последовательностей экзонов генов BRCA1 и BRCA2 | 2015 |
|
RU2612894C1 |
| СПОСОБ ОБНАРУЖЕНИЯ ВАРИАЦИЙ ЧИСЛА КОПИЙ (CNV) ПО ДАННЫМ СЕКВЕНИРОВАНИЯ ПОЛНОГО ЭКЗОМА ЧЕЛОВЕКА И ГЕНОМА С НИЗКИМ ПОКРЫТИЕМ | 2023 |
|
RU2822040C1 |
| Панель для первичного генетического тестирования наследственной формы рака молочной железы | 2024 |
|
RU2833074C1 |
| Способ создания таргетной панели для исследования геномных регионов для выявления терапевтических биомаркеров ингибиторов иммунных контрольных точек (ИКТ) | 2023 |
|
RU2818360C1 |
| Способ обработки данных полногеномного секвенирования | 2023 |
|
RU2806429C1 |
| Способ диагностики предрасположенности к раку молочной железы в русской популяции на основе ПЦР-ПДРФ | 2018 |
|
RU2723585C2 |
| Набор высокоспецифичных олигонуклеотидных праймеров для детекции генетических вариантов методом высокопроизводительного секвенирования ДНК | 2024 |
|
RU2833429C1 |
| СПОСОБ ПРОГНОЗИРОВАНИЯ РИСКА ЗЛОКАЧЕСТВЕННЫХ ЗАБОЛЕВАНИЙ МОЛОЧНОЙ ЖЕЛЕЗЫ И/ИЛИ ЯИЧНИКОВ У ПАЦИЕНТОВ ПОСЛЕ ТРАНСПЛАНТАЦИИ ПОЧКИ | 2023 |
|
RU2821583C1 |
| Система обработки данных полногеномного секвенирования | 2023 |
|
RU2804535C1 |
| ИНДИВИДУАЛИЗИРОВАННЫЕ ПРОТИВООПУХОЛЕВЫЕ ВАКЦИНЫ | 2012 |
|
RU2670745C9 |
Изобретение относится к биотехнологии. Предложен способ выявления вариаций и изменений числа копий в генах BRCA1 и BRCA2. Изобретение обеспечивает более точное выявление не только герминальных вариаций числа копий в генах BRCA1 и BRCA2, но и соматических изменений числа копий, а также снижение стоимости анализа. 3 з.п. ф-лы, 1 табл., 3 пр.
1. Способ выявления вариаций и изменений числа копий в генах BRCA1 и BRCA2 по данным таргетного массового параллельного секвенирования генома, включающий выделение образца геномной ДНК, амплификацию экзонов исследуемых генов с использованием набора олигонуклеотидных праймеров, включение адаптеров, секвенирование полученных ампликонов с помощью технологии массового параллельного секвенирования и анализ полученных данных c выявлением вариаций и изменения числа копий в генах BRCA1 и BRCA2, отличающийся тем, что протокол обработки данных секвенирования включает в себя использование медианных значений, трехэтапную нормализацию данных, использование информации об экзонно-интронной структуре гена, а также вычисление оценочного значения для каждой потенциальной крупной перестройки и вероятности того, что данная мутация ложноположительна.
2. Способ определения по п.1, отличающийся тем, что образец геномной ДНК выделяют из лейкоцитов крови пациентов или гистологических блоков.
3. Способ определения по п.1, отличающийся тем, что для амплификации ДНК используются праймеры для генов BRCA1 и BRCA2, представленные в таблице 1.
4. Способ определения по п.1, отличающийся тем, что включение адаптеров проводят с помощью лигирования или амплификации.
| Narod S | |||
| A., Foulkes W | |||
| D., BRCA1 and BRCA2: 1994 and beyond, Nature Reviews Cancer, 2004, Т | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Разборный с внутренней печью кипятильник | 1922 |
|
SU9A1 |
| Приспособление для разгонки рельсов ударами | 1923 |
|
SU665A1 |
| Chen S., Parmigiani G., Meta-analysis of BRCA1 and BRCA2 penetrance, Journal of clinical oncology: official journal of the American Society of Clinical Oncology, 2007, Т | |||
| Видоизменение пишущей машины для тюркско-арабского шрифта | 1923 |
|
SU25A1 |
| Походная разборная печь для варки пищи и печения хлеба | 1920 |
|
SU11A1 |
| Штемпельный прибор | 1920 |
|
SU1329A1 |
| Батенева Е | |||
| и др | |||
| Частота одиннадцати | |||

