Настоящее изобретение относится к конденсированному производному аминодигидротиазина и его фармацевтическому применению. Конкретнее, настоящее изобретение относится к конденсированному производному аминодигидротиазина, которое оказывает ингибирующий эффект на продукцию белка амилоида-β (далее именуемый Aβ) или ингибирующий эффект на фермент 1 расщепления белка-предшественника амилоида-β (далее именуемый BACE1 или бета-секретаза) и эффективен для лечения нейродегенеративного заболевания, вызванного белком Aβ, в частности, деменции типа Альцгеймера, синдрома Дауна или тому подобных, и к фармацевтической композиции, содержащей конденсированное производное аминодигидротиазина в качестве активного ингредиента.
Болезнь Альцгеймера представляет собой заболевание, характеризуемое дегенерацией и утратой нейронов, а также образованием сенильных бляшек и нейроволоконных сплетений. В настоящее время лечат только симптомы болезни Альцгеймера с использованием облегчающего симптомы средства, характерным примером которого является ингибитор ацетилхолинэстеразы, а фундаментальное лекарственное средство для ингибирования прогрессирования заболевания еще не было разработано. Необходимо разработать способ для борьбы с причиной начала патологии для создания фундаментального лекарственного средства для лечения болезни Альцгеймера.
Предполагают, что белки Aβ в качестве продуктов распада белков-предшественников амилоида (далее именуемые APP) активно участвуют в дегенерации и утрате нейронов и начале симптомов деменции. Белки Aβ имеют в качестве основных компонентов Aβ40, состоящий из 40 аминокислот, и Aβ42 с двумя аминокислотами, добавленными на C-конце. Известно, что Aβ40 и Aβ42 имеют высокую склонность к агрегации и являются основными компонентами сенильных бляшек. Кроме того, известно, что уровень Aβ40 и Aβ42 повышается мутациями в APP и генов пресенилина, что наблюдается при семейной болезни Альцгеймера. Соответственно, ожидается, что соединение, которое снижает продукцию Aβ40 и Aβ42, является ингибитором прогрессирования заболевания или профилактическим средством при болезни Альцгеймера.
Aβ продуцируется расщеплением APP бета-секретазой (BACE1) и в последующем гамма-секретазой. По этой причине, предпринимали попытки для создания ингибиторов гамма-секретазы и бета-секретазы для ингибирования продукции Aβ.
В опубликованной заявке на Международный патент WO2011/005738 (Eli Lilly and Company) описаны соединения формулы (A) и их применение в качестве ингибиторов BACE:

где R1, R2, R3, X, m, n и p определены в настоящем описании.
Соединения конденсированного аминодигидротиазина формулы (B) уже были описаны в опубликованной заявке на Международный патент WO2009/091016 (Eisai R&D Management Co., Ltd.):
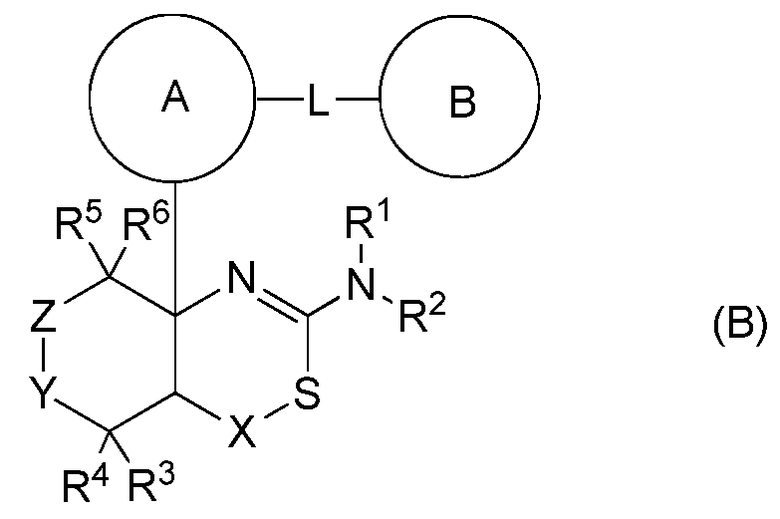
где кольцо A представляет C6-14арильную группу или тому подобную; L представляет -NReCO- [где Re представляет атом водорода или тому подобный] или тому подобные; кольцо B представляет C6-14арильную группу или тому подобную; X представляет C1-3алкиленовую группу или тому подобную; Y представляет одиночную связь или тому подобную; Z представляет C1-3алкиленовую группу или тому подобную; R1 и R2 независимо представляют атом водорода или тому подобный; и R3, R4, R5 и R6 независимо представляют атом водорода, атом галогена или тому подобный.
Другие конденсированные аминодигидротиазиновые соединения формулы (C) были описаны в опубликованной заявке на Международный патент WO2010/038686 (Eisai R&D Management Co., Ltd.):

где кольцо A представляет C6-14арильную группу или тому подобную; L представляет -NReCO- [где Re представляет атом водорода или тому подобный] или тому подобные; кольцо B представляет C6-14арильную группу или тому подобную; X представляет C1-3алкиленовую группу или тому подобную; Y представляет одиночную связь и тому подобную; Z представляет атом кислорода или тому подобный; R1 и R2 каждый независимо представляет атом водорода или тому подобный; и R3, R4, R5 и R6 каждый независимо представляет атом водорода, атом галогена или тому подобный.
Настоящее изобретение представляет выбор из рода соединений, описанных в опубликованной заявке на Международный патент WO2009/091016.
Целью настоящего изобретения является получение дополнительных соединений, которые оказывают ингибирующий эффект на продукцию Aβ или ингибирующий эффект на BACE1, и могут применяться в качестве профилактических или терапевтических средств при вызванном Aβ нейродегенеративном заболевании, характерным примером которого является деменция, причем указанные соединения представляют собой конденсированные производные аминодигидротиазина.
Таким образом, настоящее изобретение относится к соединению формулы (I):

где
X обозначает водород или фтор;
A обозначает CH или N;
Y обозначает метил, этил, монофторметил, дифторметил, трифторметил, дифторэтил, метокси, этокси, метоксиметил или -C≡N;
и его фармацевтически приемлемым солям.
В одном варианте осуществления настоящего изобретения, X обозначает водород.
В другом варианте осуществления настоящего изобретения, A обозначает N.
В другом варианте осуществления настоящего изобретения, Y обозначает метил, монофторметил, дифторметил, трифторметил или метокси.
Одна предпочтительная группа соединений по настоящему изобретению представляет собой соединение формулы (Ia) и его фармацевтически приемлемые соли:
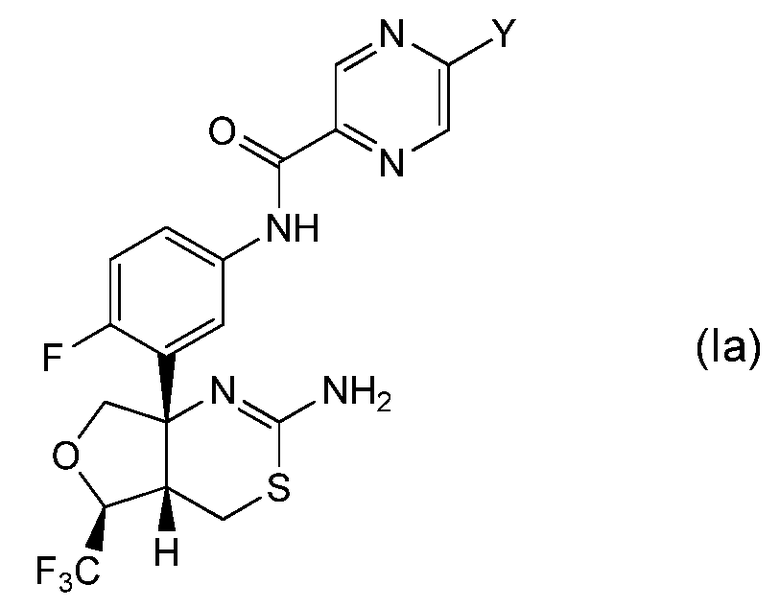
где Y обозначает, как определено выше в настоящем описании. Предпочтительно, Y обозначает метил, монофторметил, дифторметил, трифторметил, дифторэтил, метокси, этокси или метоксиметил.
В одном варианте осуществления настоящее изобретение относится к соединению формулы (Ia), где Y обозначает метокси или монофторметил.
Другая предпочтительная группа соединений по настоящему изобретению представляет собой соединение формулы (Ib) и его фармацевтически приемлемые соли:
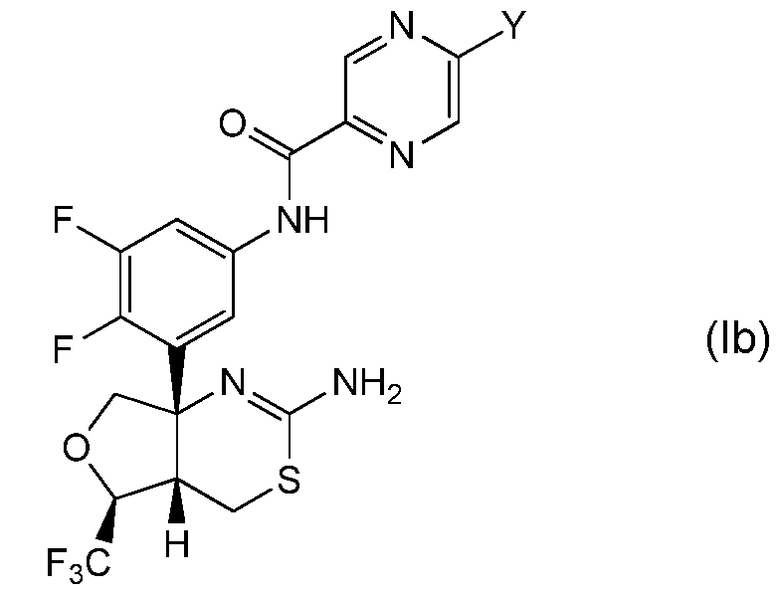
где Y обозначает, как определено выше в настоящем описании. Предпочтительно, Y обозначает метил, монофторметил, дифторметил или метокси.
Другая предпочтительная группа соединений по настоящему изобретению представляет собой соединение формулы (Ic) и его фармацевтически приемлемые соли:
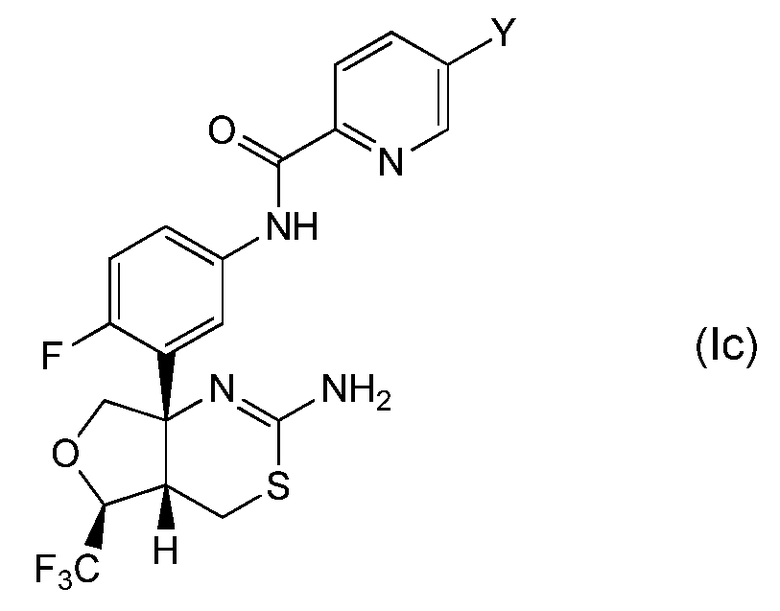
где Y обозначает, как определено выше в настоящем описании. Предпочтительно, Y обозначает метил, этил, трифторметил, метокси или -C≡N.
Предпочтительными соединениями по настоящему изобретению являются:
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-метоксипиразин-2-карбоксамид:
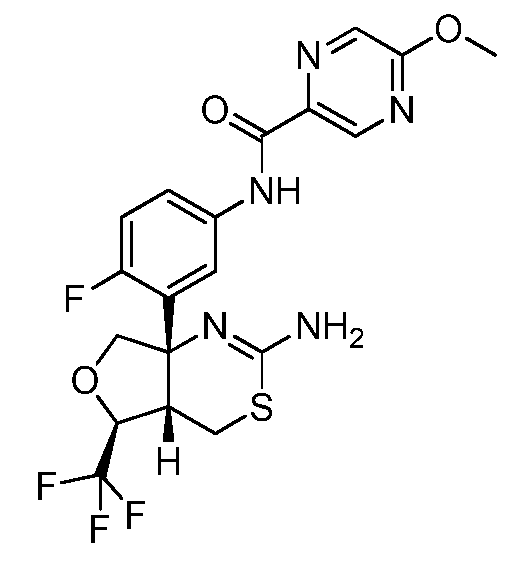 ;
;
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-цианопиколинамид:
 ;
;
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-(дифторметил)пиразин-2-карбоксамид:
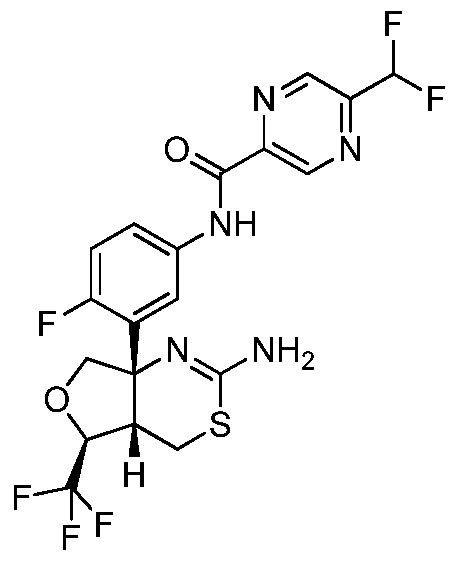 ;
;
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-(трифторметил)пиколинамид:
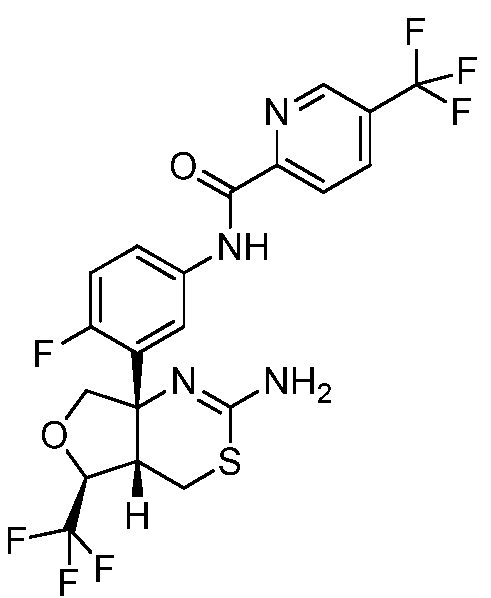 ;
;
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-метилпиразин-2-карбоксамид:
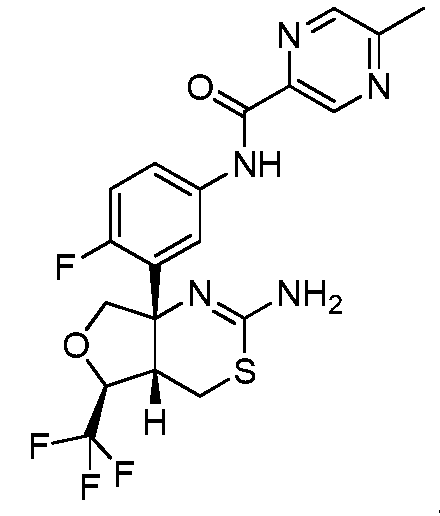 ;
;
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-метилпиколинамид:
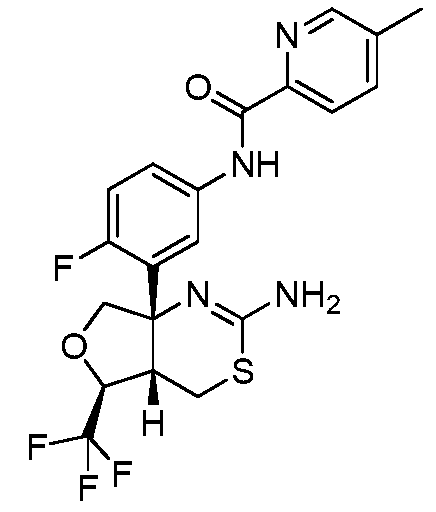 ;
;
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-этилпиколинамид:
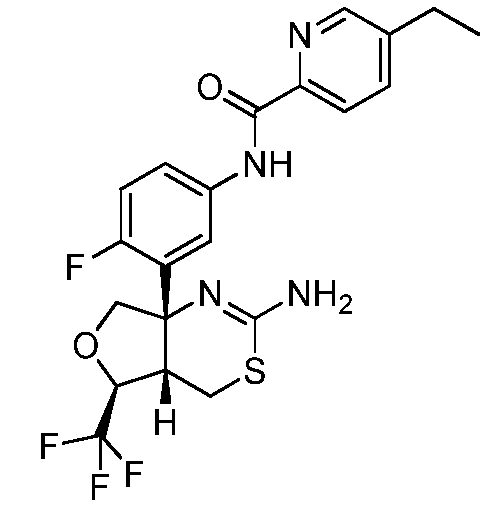 ;
;
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-(фторметил)пиразин-2-карбоксамид:
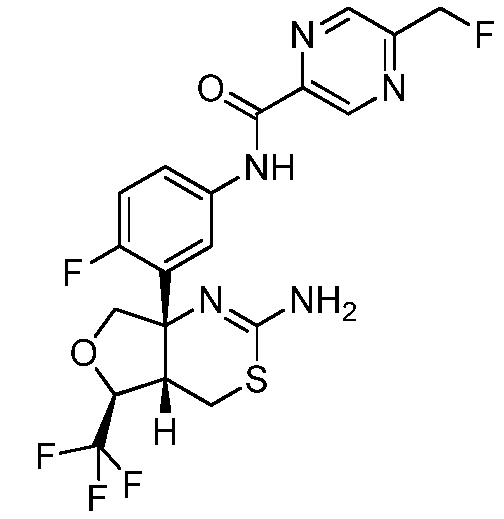 ;
;
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-метоксипиколинамид:
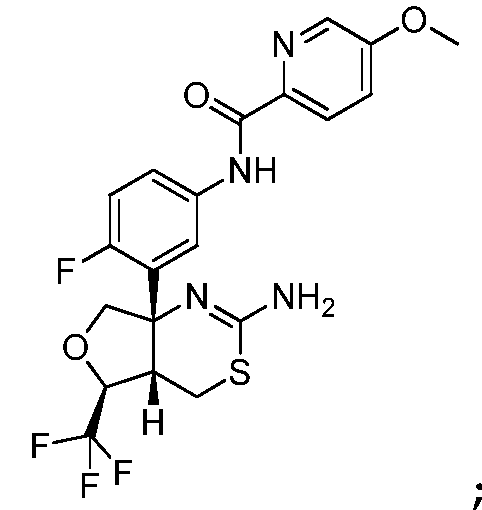
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-этоксипиразин-2-карбоксамид:
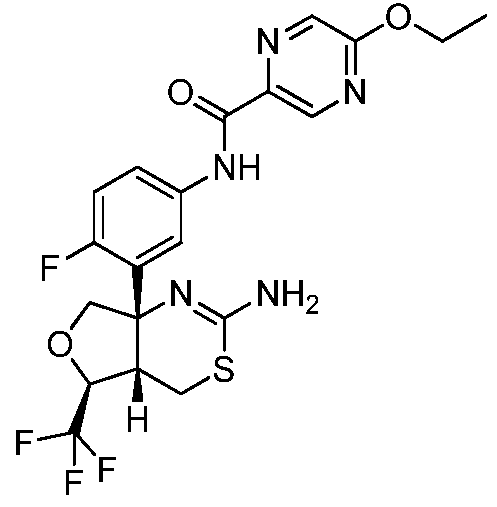 ;
;
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-(1,1-дифторэтил)пиразин-2-карбоксамид:
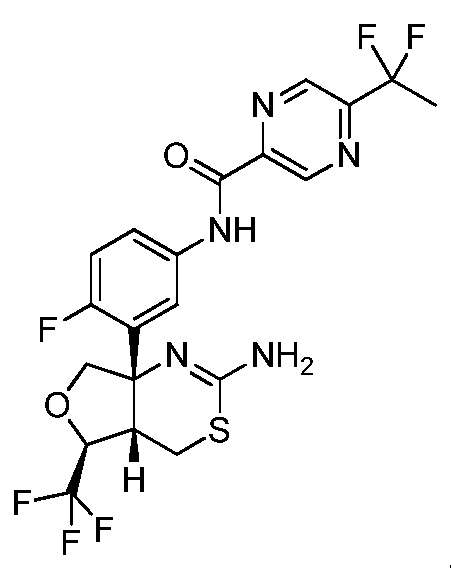 ;
;
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-(трифторметил)пиразин-2-карбоксамид:
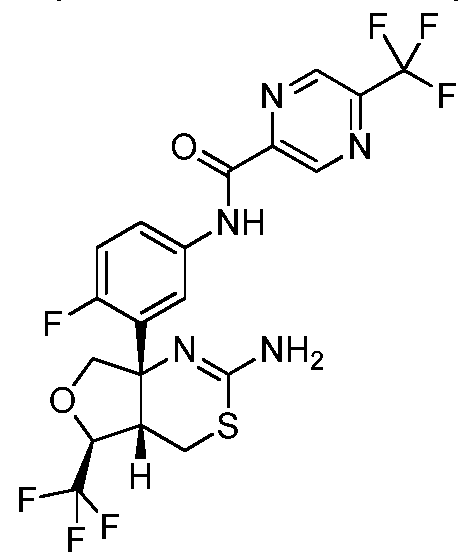 ;
;
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-(метоксиметил)пиразин-2-карбоксамид:
 ;
;
N-{3-[(4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5-дигидро-4H-фуро[3,4d][1,3]тиазин-7a(7H)-ил]-4-фторфенил}-5-[(2H3)метилокси]пиразин-2-карбоксамид:
 ;
;
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4,5-дифторфенил)-5-(дифторметил)пиразин-2-карбоксамид:
 ;
;
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4,5-дифторфенил)-5-метоксипиразин-2-карбоксамид:
 ;
;
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4,5-дифторфенил)-5-метилпиразин-2-карбоксамид:
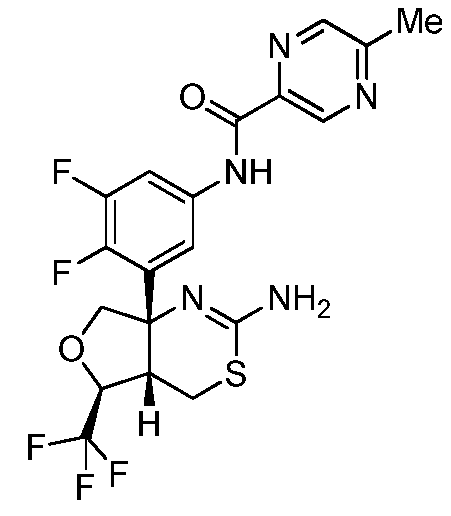 ;
;
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4,5-дифторфенил)-5-(фторметил)-пиразин-2-карбоксамид:
 ;
;
и их фармацевтически приемлемые соли.
В одном варианте осуществления настоящее изобретение относится к соединению, которое представляет собой N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-метоксипиразин-2-карбоксамид или его фармацевтически приемлемую соль.
В другом варианте осуществления настоящее изобретение относится к соединению, которое представляет собой N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-(фторметил)пиразин-2-карбоксамид или его фармацевтически приемлемую соль.
Определенные соединения в пределах объема настоящего изобретения включают соединения, названные ниже в разделе «Примеры», и их фармацевтически приемлемые соли.
Используемый в настоящем описании термин «дифторэтил» относится к алкильной группе, имеющей два атома углерода и замещенной двумя атомами фтора. Примерами данной группы являются CH3-CF2-, CH2F-CHF- и CHF2-CH2-. В настоящем изобретении группа предпочтительно представляет собой CH3-CF2-.
Соединение формулы (I) не ограничивается определенным изомером и включает все возможные изомеры (такие как кето-енольный изомер, имин-енаминовый изомер и ротамер) и их смеси. Например, соединение формулы (I) включает следующие таутомеры:
Соединения по настоящему изобретению содержат три хиральных центра, локализующихся на тетрагидрофуро-тиазинильном кольце внутри формулы (I). Стереохимическая конфигурация в каждом из этих хиральных центров представляет собой предпочтительно S, т.е., они представляют собой (4aS,5S,7aS) стереоизомеры. Во избежание сомнений, (4aS,5S,7aS) стереоизомеры по настоящему изобретению могут присутствовать в виде смеси с одним или более других возможных стереоизомеров, например, в рацемической смеси.
В одном варианте осуществления настоящее изобретение относится к соединению формулы (I), которое является стереохимически чистым в хиральных центрах (4aS,5S,7aS). В контексте настоящего описания, термин «стереохимически чистое» обозначает соединение, которое имеет 80% масс. или более стереоизомера (4aS,5S,7aS), и 20% масс. или менее других стереоизомеров. В еще одном варианте осуществления, соединение формулы (I) имеет 90% масс. или более стереоизомера (4aS,5S,7aS) и 10% масс. или менее других стереоизомеров. В еще одном варианте осуществления, соединение формулы (I) имеет 95% масс. или более стереоизомера (4aS,5S,7aS) и 5% масс. или менее других стереоизомеров. В еще одном варианте осуществления, соединение формулы (I) имеет 97% масс. или более стереоизомера (4aS,5S,7aS) и 3% масс. или менее других стереоизомеров.
Хотя в настоящем описании могут присутствовать кристаллические полиморфы соединения, соединение аналогичным образом не ограничивается им и может присутствовать в форме одиночного кристалла или в формах смеси одиночных кристаллов. Соединение может представлять собой ангидрид или гидрат. Любая из этих форм включена в притязания настоящего описания изобретения.
Настоящее изобретение также включает изотопно меченные соединения, которые идентичны соединениям формулы (I), за исключением того, что один или несколько атомов заменены атомом, имеющим атомную массу или массовое число, отличное от атомной массы или массового числа, обнаруживаемых в природе. Примеры изотопов, которые могут быть включены в соединения по изобретению, включают изотопы водорода, углерода, азота, кислорода, фтора, фосфора, хлора, технеция и йода, такие как 2H, 3H, 11C, 14C, 13N, 15O, 18F, 32P, 99mTc, 123I и 131I.
Соединения по настоящему изобретению и фармацевтически приемлемые производные (например, соли) указанных соединений, которые содержат указанные выше изотопы и/или другие изотопы других атомов, находятся в пределах объема настоящее изобретения. Изотопно меченные соединения по настоящему изобретению, например, те, в которые включены радиоактивные изотопы, такие как 3H и/или 14C, используются в анализах лекарственных средств и/или распределения в ткани субстрата. Изотопы 3H и 14C считаются полезными вследствие легкости их получения и возможности выявления. Изотопы 11C, 15O и 18F считаются полезными при PET (позитронной эмиссионной томографии), и изотопы 99mTc, 123I и 131I считаются полезными при SPECT (одиночнофотонной эмиссионной компьютерной томографии), все полезны при визуализации мозга. Замещение более тяжелыми изотопами, такими как 2H, могут обеспечить определенные терапевтические преимущества в результате большей метаболической устойчивости, например, увеличенного периода полувыведения in vivo или сниженных потребностей в дозировке, и, следовательно, считаются полезными в некоторых ситуациях. Изотопно меченные соединения формулы (I) по настоящему изобретению могут быть в целом получены проведением процедур, описанных в «Схемах», и/или ниже в разделе «Примеры», замещением легкодоступным изотопно меченным реагентом не-изотопно меченным реагентом.
Конденсированное производное аминодигидротиазина формулы (I) в соответствии с настоящим изобретением может представлять собой фармацевтически приемлемую соль. Фармацевтически приемлемые соли включают соли, описанные в публикации Berge, Bighley and Monkhouse, J. Pharm. Sci., 1977, 766, 1-19. Определенные примеры фармацевтически приемлемой соли включают соли неорганических кислот (такие как сульфаты, нитраты, перхлораты, фосфаты, карбонаты, бикарбонаты, гидрофториды, гидрохлориды, гидробромиды и гидройодиды), органические карбоксилаты (такие как ацетаты, оксалаты, малеаты, тартраты, фумараты, цитраты, малонаты и лактаты), органические сульфонаты (такие как метансульфонаты, трифторметансульфонаты, этансульфонаты, бензолсульфонаты, толуолсульфонаты и камфорсульфонаты), соли аминокислот (такие как аспартаты и глутаматы), соли четвертичных аминов, соли щелочных металлов (такие как соли натрия и соли калия) и соли щелочноземельных металлов (такие как соли магния и соли кальция).
Соединение формулы (I) в соответствии с настоящим изобретением может быть при необходимости превращено в фармацевтически приемлемую соль обычным способом. Соль может быть получена способом, в котором соответствующим образом комбинируются способы, обычно используемые в области органической синтетической химии, и тому подобные. Определенные примеры способа включают нейтрализационное титрование свободного раствора соединения по настоящему изобретению раствором кислоты.
Конденсированное производное аминодигидротиазина формулы (I) или фармацевтически приемлемая соль в соответствии с настоящим изобретением могут представляет собой его сольват. Примеры сольвата включают гидрат.
Соединение формулы (I) в соответствии с настоящим изобретением может быть при необходимости превращено в сольват подверганием соединения по существу известной реакции образования сольвата.
Настоящее изобретение, кроме того, относится к соединению формулы (I) или его фармацевтически приемлемой соли для применения при лечении.
Конденсированное производное аминодигидротиазина или его фармацевтически приемлемая соль или сольват в соответствии с настоящим изобретением оказывает превосходный ингибирующий эффект на продукцию Aβ или ингибирующий эффект на BACE1 и полезно в качестве профилактического или терапевтического средства при нейродегенеративном заболевании, вызванном Aβ, характерным примером которого является деменция типа Альцгеймера. Соединения по изобретению снижают уровень и Aβ40, и Aβ42. Кроме того, соединения по настоящему изобретению могут оказывать ингибирующий эффект на BACE 2.
Таким образом, в другом аспекте настоящее изобретение относится к соединению формулы (I), как определено выше, или его фармацевтически приемлемой соли для ингибирования продукции белка амилоида-β.
В еще одном аспекте настоящее изобретение относится к соединению формулы (I), как определено выше, или его фармацевтически приемлемой соли для ингибирования бета-сайта фермента 1, расщепляющего белок-предшественник амилоида-β (BACE 1).
В еще одном аспекте настоящее изобретение относится к соединению формулы (I), как определено выше, или его фармацевтически приемлемой соли для лечения нейродегенеративного заболевания. Примеры нейродегенеративных заболеваний включают деменцию типа Альцгеймера (AD), синдром Дауна, цереброваскулярную амилоидную ангиопатию (CAA), легкое когнитивное нарушение (MCI), потерю памяти, пресенильную деменцию, сенильную деменцию, наследственное церебральное кровоизлияние с амилоидозом и другие дегенеративные деменции, такие как деменции смешанного сосудистого и дегенеративного происхождения, деменция, связанная с супрануклеарным параличом, деменция, связанная с кортикальной базальной дегенерацией, деменция, связанная с болезнью Паркинсона (PD) и деменция, связанная с типом AD с диффузными тельцами Леви. В одном варианте осуществления нейродегенеративное заболевание представляет собой деменцию типа Альцгеймера (AD).
В еще одном аспекте настоящее изобретение относится к применению соединения формулы (I), как определено выше, или его фармацевтически приемлемой соли для получения лекарственного средства для лечения или прифилактики нейродегенеративного заболевания, такого как деменция типа Альцгеймера (AD), синдром Дауна, цереброваскулярная амилоидная ангиопатия (CAA), легкое когнитивное нарушение (MCI), потеря памяти, пресенильная деменция, сенильная деменция, наследственное церебральное кровоизлияние с амилоидозом и другие дегенеративные деменции, такие как деменции смешанного сосудистого и дегенеративного происхождения, деменция, связанная с супрануклеарным параличом, деменция, связанная с кортикальной базальной дегенерацией, деменция, связанная с болезнью Паркинсона (PD) и деменция, связанная с типом AD с диффузными тельцами Леви. В одном варианте осуществления нейродегенеративное заболевание представляет собой деменцию типа Альцгеймера (AD).
В еще одном аспекте настоящее изобретение относится к способу ингибирования продукции белка амилоида-β и/или лечения или профилактики нейродегенеративного заболевания, такого как деменция типа Альцгеймера (AD), синдром Дауна, цереброваскулярная амилоидная ангиопатия (CAA), легкое когнитивное нарушение (MCI), потеря памяти, пресенильная деменция, сенильная деменция, наследственное церебральное кровоизлияние с амилоидозом и другие дегенеративные деменции, такие как деменции смешанного сосудистого и дегенеративного происхождения, деменция, связанная с супрануклеарным параличом, деменция, связанная с кортикальной базальной дегенерацией, деменция, связанная с болезнью Паркинсона (PD) и деменция, связанная с типом AD с диффузными тельцами Леви. В одном варианте осуществления нейродегенеративное заболевание представляет собой деменцию типа Альцгеймера (AD). «Эффективное количество» означает количество, достаточное для обеспечения благоприятного воздействия на индивида, или по меньшей мере для вызова изменения состояния индивида.
Дополнительные состояния, при которых могут быть введены соединения настоящего изобретения, включают диабет 2 типа, болезнь Крейтцфельдта-Якоба (CJD), повреждение периферического нерва, периферическую нейропатию, прогрессирующий надъядерный паралич, удар, амиотрофический латеральный склероз (ALS), аутоиммунные заболевания, воспаление, артериальный тромбоз, тревожные расстройства, психические расстройства, эпилепсию, эпилептические припадоки, конвульсии, стрессовые состояния, сосудистый амилоидоз, Синдром Герстмана-Штраусслера-Шейнкера, почесуху, энцефалопатию, спинально-церебеллярную атаксию, Вильсона болезнь, базедову болезнь, болезнь Хантингтона, синдром Уипла, болезнь Костманна, глаукому, наследственный геморрагический инсульт с амилоидозом, геморрагический инсульт с амилоидозом, сосудистый амилоидоз, черепно-мозговое воспаление, синдром умственной отсталости, синдром Туретта, миозит с включёнными тельцами, депрессия, биполярное расстройство и обсессивно-компульсивное расстройство.
В еще одном аспекте настоящее изобретение дополнительно относится к соединению формулы (I), как определено выше, или его фармацевтически приемлемой соли для лечения диабета 2 типа. В еще одном аспекте настоящее изобретение дополнительно относится к применению соединения формулы (I), как определено выше, или его фармацевтически приемлемой соли для получения лекарственного средства для лечения или профилактики диабета 2 типа.
В еще одном аспекте настоящее изобретение относится к применению соединения формулы (I), как определено выше, или его фармацевтически приемлемой соли для получения лекарственного средства для лечения или профилактики диабета 2 типа.
В еще одном аспекте настоящее изобретение, кроме того, относится к способу ингибирования продукции белка амилоида-β и/или лечения или профилактики диабета 2 типа, включающему введение нуждающемуся в нем человеку терапевтически или профилактически эффективного количества соединения формулы (I) ли его фармацевтически приемлемой соли.
Еще один аспект изобретения относится к фармацевтической композиции, содержащей соединение формулы (I), как определено выше, или его фармацевтически приемлемой соли в качестве активного ингредиента в ассоциации с фармацевтически приемлемым носителем. Композиция может быть представлена в любой подходящей форме в зависимости от предполагаемого способа введения. Она может, например, быть в форме таблетки, капсулы или жидкости для перорального введения или раствора или суспензии для парентерального введения.
Конденсированное производное аминодигидротиазина или его фармацевтически приемлемая соль в соответствии с настоящим изобретением может быть составлено обычным способом. Предпочтительные примеры лекарственной формы включают таблетки, покрытые таблетки, такие как таблетки с пленочным покрытием или таблетки с сахарным покрытием, мелкие гранулы, гранулы, порошки, капсулы, сиропы, пастилки, ингаляционные средства, суппозитории, инъекционные препараты, мази, глазные капли, нозальные капли, ушные капли, горячие компрессы и лосьоны.
Эти твердые препараты, такие как таблетки, капсулы, гранулы и порошки, могут содержать в целом от 0,01 до 100% масс., и предпочтительно, от 0,1 до 100% масс. конденсированного производного аминодигидротиазина или его фармацевтически приемлемой соли в соответствии с настоящим изобретением качестве активного ингредиента.
Активный ингредиент включают в состав смешиванием ингредиентов, в целом используемых в качестве материалов для фармацевтического препарата, и добавлением эксципиента, разрыхлителя, связывающего агента, смазывающего вещества, красителя и обычно используемого корригента и добавлением стабилизатора, эмульгатора, усилителя поглощения, поверхностно-активного вещества, регулятора pH, консерванта и при необходимости антиоксиданта, например, с использованием обычного способа. Примеры таких ингредиентов включают животные и растительные масла, такие как соевое масло, говяжье сало и синтетический глицерид; углеводороды, такие как жидкий парафин, сквален и твердый парафин; сложноэфирные масла, такие как октилдодецил миристат и изопропил миристат; высшие спирты, такие как цетостеариловый спирт и бегениловый спирт; кремнийорганическую смолу; силиконовое масло; поверхностно-активные вещества, такие как сложный эфир жирной кислоты полиоксиэтилена, сложный эфир жирной кислоты и сорбитана, полиоксиэтилен-гидрогенизированное касторовое масло и блок сополимер полиоксиэтилена-полиоксипропилена; растворимые в воде полимеры, такие как гидроксиэтилцеллюлоза, полиакриловая кислота, карбоксивиниловый полимер, полиэтиленгликоль, поливинилпирролидон и метилцеллюлоза; низшие спирты, такие как этанол и изопропанол; полиатомные спирты, такие как глицерин, пропиленгликоль, дипропиленгликоль и сорбит; сахара, такие как глюкоза и сахароза; неорганические порошки, такие как кремневый ангидрид, силикат магния алюминия и силикат алюминия; и очищенную воду. Примеры эксципиента включают лактозу, кукурузный крахмал, сахарозу, глюкозу, маннит, сорбит, кристаллическую целлюлозу и диоксид кремния. Примеры используемого связывающего агента включают поливиниловый спирт, поливиниловый простой эфир, метилцеллюлозу, этилцеллюлозу, аравийскую камедь, трагакант, желатин, шеллак, гидроксипропилметилцеллюлозу, гидроксипропилцеллюлозу, поливинилпирролидон, блок сополимер полипропиленгликоля-полиоксиэтилена и меглумин. Примеры используемого разрыхлителя включают крахмал, агар, желатиновый порошок, кристаллическую целлюлозу, карбонат кальция, бикарбонат натрия, цитрат кальция, декстрин, пектин и карбоксиметилцеллюлозу кальция. Примеры используемого смазывающего вещества включают стеарат магния, тальк, полиэтиленгликоль, диоксид кремния и гидрогенизированное растительное масло. Примеры используемого красителя включают те, которые разрешены для добавления в фармацевтические средства. Примеры используемого корригента включают порошок какао, ментол, эмпазм, мятное масло, борнеол и коричный порошок. Очевидно, что ингредиенты не ограничиваются указанными выше дополнительными ингредиентами.
Например, пероральный препарат получают добавлением конденсированного производного аминодигидротиазина или его фармацевтически приемлемой соли в соответствии с настоящим изобретением в качестве активного ингредиента, эксципиента и при необходимости связывающего агента, разрыхлителя, смазывающего вещества, красителя, корригента и тому подобных, и затем формованием смеси в порошок, мелкие гранулы, гранулы, таблетки, покрытые таблетки, капсулы или тому подобные обычным способом. Очевидно, что таблетки или гранулы могут быть соответствующим образом покрыты, например, при необходимости покрыты сахаром.
Например, сироп или инъекционный препарат получают обычным способом добавления регулятора pH, солюбилизатора, изотонизирующего агент и тому подобных, и при необходимости солюбилизирующего агента, стабилизатора и тому подобных. Инъекционный препарат может представлять собой предварительно полученный раствор или может представлять собой сам порошок или порошок, содержащий подходящую добавку, который растворяют перед применением. Инъекционный препарат может обычно содержать от 0,01 до 100% масс., и предпочтительно, от 0,1 до 100% масс. активного ингредиента. Кроме того, жидкий препарат для перорального введения, такой как суспензия или сироп, может содержать обычно от 0,01 до 100% масс., и предпочтительно, от 0,1 до 100% масс. активного ингредиента.
Например, наружный препарат может быть получен любым обычным способом без определенных ограничений. В качестве материала основы может использоваться любой из различных материалов, обычно используемых для фармацевтического средства, препарат общего воздействия, косметический препарат или тому подобные. Примеры материала основы включают материалы, такие как животные и растительные масла, минеральные масла, сложноэфирные масла, воски, высшие спирты, жирные кислоты, силиконовые масла, поверхностно-активные вещества, фосфолипиды, спирты, полиатомные спирты, растворимые в воде полимеры, минералы глины и очищенная вода. При необходимости может добавляться регулятор pH, антиоксидант, хелатообразующий агент, консервант и фунгицид, краситель, отдушка и тому подобные. Кроме того, при необходимости могут смешиваться ингредиенты, такие как ингредиент, оказывающий эффект индукции дифференциации, усилитель кровотока, бактерицидное средство, противовоспалительное средство, клеточный активатор, витамин, аминокислота, увлажнитель и кератолитический агент.
Доза конденсированного производного аминодигидротиазина или его фармацевтически приемлемой соли в соответствии с настоящим изобретением варьируется, например, в соответствии со степенью симптомов, возрастом, полом, массой тела, способом введения, типом соли и определенным типом заболевания. Обычно, активный ингредиент перорально вводится взрослому в количестве примерно от 30 мкг до 10 г, предпочтительно, от 100 мкг до 5 г, и предпочтительнее, от 100 мкг до 1 г в день, или вводится взрослому инъекцией в количестве примерно от 30 мкг до 1 г, предпочтительно, от 100 мкг до 500 мг, и предпочтительнее, от 100 мкг до 300 мг в день, соответственно одной или несколькими дозами.
Соединения формулы (I) могут использоваться в комбинации с другими терапевтическими средствами, например, лекарственными средствами, заявленными как полезные или в качестве средств лечения, модифицирующих заболевание, или симптоматических средств, нейродегенеративного заболевания, такого как болезнь Альцгеймера. Таким образом, в еще одном аспекте настоящее изобретение относится к фармацевтическому продукту, содержащему в комбинации первый активный ингредиент, который представляет собой соединение формулы (I) или его фармацевтически приемлемую соль, и по меньшей мере еще один активный ингредиент, полезный при лечении нейродегенеративного заболевания. В одном варианте осуществления изобретения, нейродегенеративное заболевание представляет собой деменцию типа Альцгеймера (AD). Подходящими примерами такого дополнительного активного ингредиента могут быть симптоматические средства, например, те, которые, как известно, модифицируют холинергическую передачу, такие как агонисты M1 и M3 мускариновых рецепторов или аллостерические модуляторы, M2 мускариновые антагонисты, M4 агонисты или положительные аллостерические модуляторы (PAM), ингибиторы ацетилхолинэстеразы (такие как тетрагидроаминоакридин, донепезил гидрохлорид и ривастигмин), агонисты никотиновых рецепторов или аллостерические модуляторы (такие как α7 агонисты или аллостерические модуляторы или агонисты α4β2 или аллостерические модуляторы), агонисты PPAR (такие как агонисты PPARγ), агонисты или частичные агонисты 5-HT4 рецепторов или, антагонисты H3-гистаминовых рецепторов, антагонисты 5-HT6 рецепторов или лиганды 5HT1A рецепторов и антагонисты или модуляторы NMDA (N-Метил-D-Аспартата) рецепторов, 5-HT2A антагонисты, 5-HT7 антагонисты, агонисты D1 допаминовых рецепторов или PAM, агонисты D4 допаминовых рецепторов или РАМы, агонисты D5 допаминовых рецепторов или PAM (положительные аллостерические модуляторы), инверсионные агонисты или отрицательные аллостерические модуляторы GABA-A α5 (NAM), агонисты GABA-A α2/3 или PAM, модуляторы mGluR2 (PAM или NAM), mGluR3 PAM, mGluR5 PAM, ингибиторы PDE (фосфодиэстеразы) 1, ингибиторы PDE 2, ингибиторы PDE 4, ингибиторы PDE 5, ингибиторы PDE 9, ингибиторы PDE 10, ингибиторы GlyT1, ингибиторы DAAO (оксидазы D-аминокислот), ингибиторы ASC1, модуляторы AMPA, активаторы или ингибиторы SIKT1, антагонисты AT4, антагонисты GalR1, лиганды GalR3, антагонисты аденозиновых A1-рецепторов, антагонисты аденозиновых A2a рецепторов, антагонисты или агонисты адренергических α2A-рецепторов, селективные и неселективные ингибиторы обратного захвата норэпинефрина (SNRI), или потенциальное модифицирующее течение заболевания, такие как ингибиторы или модуляторы гамма-секретазы, активаторы или модуляторы альфа-секретазы, ингибиторы агрегации амилоида, антитела к амилоиду, ингибиторы агрегации тау или ингибиторы фосфорилирования/киназы тау, активаторы дефосфорилирования тау/фосфатазы, ингибиторы митоген-активированной протеинкиназы киназы 4 (MKK4/MEK4/MAP2K4), ингибиторы c-Jun N-концевой киназы (JNK), ингибиторы казеинкиназы, ингибиторы MK2 (митоген-активированной активированной протеинкиназой протеинкиназы 2), ингибиторы MARK (киназы, регулирующей аффинитет микротрубочек), ингибиторы CDK5 (циклин-зависимой киназы 5), ингибиторы GSK-3 (киназы-3 гликогенсинтазы) и ингибиторы тау-тубулин киназы-1 (TTBK1). Дополнительными примерами таких других терапевтических средств могут быть блокаторы кальциевых каналов, ингибиторы HMG-CoA (3-гидрокси-3-метил-глутарил-CoA) редуктазы (статины) и средства, снижающие уровень липидов, имитаторы NGF (фактора роста нервов), антиоксиданты, лиганды GPR3, активаторы плазмина, активаторы неприлизина (NEP), активаторы IDE (инсулин-разрушающего фермента), агонисты мелатонина MT1 и/или MT2, лиганды TLX/NR2E1 (рецептора бесхвостой X), лиганды GluR1, антагонисты RAGE (рецептора для конечных продуктов поздних стадий глицирования), ингибиторы EGFR (рецепторов эпидермального фактора роста), лиганды FPRL-1 (формилпептид-подобного рецептора-1), антагонисты GABA (гамма-аминомасляной кислоты) и ингибиторы MICAL (молекулы, взаимодействующей с casL), например, ингибиторы оксоредуктазы, антагонисты/инверсионные агонисты CB1, нестероидные противовоспалительные препараты (NSAID), противовоспалительные средства (например, средства, которые можно применять для лечения нейровоспаления или усилением, или снижением нейровоспаления), лиганды белка-предшественника амилоида (APP), вакцины и/или антитела к амилоиду, средства, которые стимулируют или усиливают отток и/или клиренс амилоида, ингибиторы гистондеацетилазы (HDAC), антагонисты EP2, ингибиторы 11-бета HSD1 (гидроксистероид-дегидрогеназы), агонисты печеночных X рецепторов (LXR) PAM, имитаторы и/или лиганды и/ил энхансеры и/или ингибиторы связанного с рецепторами липопротеина белка (LRP), ингибиторы бутирилхолинэстеразы, антагонисты кинуриновой кислоты и/или ингибиторы кинуренинаминотрансферазы (KAT), антагонисты орфанина FQ/ноцицептина (NOP)/опиоид-подобного рецептора 1 (ORL1), лиганды транспортера возбуждающих аминокислот (EAAT) (активаторы или ингибиторы) и ингибиторы ингибитора-1 активатора плазминогена (PAI-1), агонисты ниацина и/или GPR109 или PAM в комбинации со средствами, снижающим уровень холестерина и/или ингибиторами HMGCoA редуктазы (статинами), димеболин или аналогичные средства, антигистаминные препараты, средства, связывающие металлы/хелатирующие агенты, антибиотики, стимуляторы секреции гормона роста, средства, снижающие уровень холестерина, витамин E, ингибиторы всасывания холестерина, стимуляторы и/или активаторы оттока холестерина и средства, повышающие секрецию инсулина.
В одном варианте осуществления настоящее изобретение относится к фармацевтическому продукту, содержащему, в комбинации, первый активный ингредиент, который представляет собой соединение формулы (I) или его фармацевтически приемлемую соль, и по меньшей мере один дополнительный активный ингредиент, выбранный из:
ингибиторов холинэстеразы, например, донепезила, галантамина, ривастигамина, тетрагидроаминоакридина и их фармацевтически приемлемых солей,
антагонистов 5-HT6 рецепторов, например, SB-742457 и его фармацевтически приемлемой соли,
ингибиторов HMGCoA редуктазы, например, ловастатина, росувастатина, аторвастатина, симвастатина, флувастатина, питавастатина, правастатина и их фармацевтически приемлемых солей.
Отдельные компоненты таких комбинаций могут вводиться или последовательно, или одновременно в отдельных или комбинированных фармацевтических препаративных формах. Таким образом, фармацевтический продукт может, например, представлять собой фармацевтическую композицию, содержащую первый и дополнительные активные ингредиенты в смеси. Альтернативно, фармацевтический продукт может, например, содержать первый и дополнительные активные ингредиенты в отдельных фармацевтических препаратах, подходящих для одновременного, последовательного или отдельного введения нуждающемуся в них пациенту.
Указанные выше комбинации могут для удобства быть представлены для применения в форме фармацевтической препаративной формы и, таким образом, фармацевтические препаративные формы, содержащие определенную выше комбинацию вместе с фармацевтически приемлемым носителем или эксципиентом, составляют еще один аспект изобретения.
Когда соединение формулы (I) или его фармацевтически приемлемая соль применяется в комбинации со вторым терапевтическим активным средством, то доза каждого соединения может отличаться от дозы, когда соединение применяется отдельно. Специалисты в данной области легко определят соответствующие дозы.
Таким образом, дополнительный аспект изобретения относится к способу получения фармацевтической композиции, включающему смешивание по меньшей мере одного соединения формулы (I), как определено выше, или его фармацевтически приемлемой соли, с одним или более фармацевтически приемлемыми адъювантами, разбавителями или носителями и/или с одним или более другими терапевтически или профилактически активными средствами.
В одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, одно или несколько других средств для лечения болезни Альцгеймера, таких как агонист или аллостерический модулятор M1 и M3 мускариновых рецепторов, антагонист M2 мускариновых рецепторов, ингибитор ацетилхолинэстеразы, агонист или аллостерический модулятор никотиновых рецепторов, агонист PPAR, агонист или частичный агонист 5-HT4 рецепторов, антагонист гистаминовых H3 рецепторов, антагонист 5-HT6 рецепторов, лиганд 5HT1A рецепторов, антагонист или модулятор NMDA рецепторов, антагонист 5-HT2A рецепторов, антагонист 5-HT7 рецепторов, агонист или положительный аллостерический модулятор (PAM) D1 допаминовых рецепторов, агонист или PAM D4 допаминовых рецепторов или инверсионный агонист или отрицательный аллостерический модулятор (NAM) GABA-A α5, агонист или PAM GABA-A α2/3, модулятор mGluR2 (PAM или NAM), PAM mGluR3, PAM mGluR5, ингибитор PDE 1, ингибитор PDE 2, ингибитор PDE 4, ингибитор PDE 5, ингибитор PDE 9, ингибитор PDE 10, ингибитор GLyT1, ингибитор DAAO, ингибитор ASC1, модулятор AMPA, активатор или ингибитор SIKT1, антагонист AT4, антагонист GalR1, лиганд GalR3, антагонист аденозиновых рецепторов A1, антагонист аденозиновых рецепторов A2a, антагонист или агонист рецепторов α2A, селективный или не селективный ингибитор обратного захвата норэпинефрина (SNRI), ингибитор или модулятор гамма-секретазы, активатор или модулятор альфа-секретазы, ингибитор агрегации амилоида, антитело к амилоиду, ингибитор агрегации тау-белка, ингибитор фосфорилирования тау-белка, ингибитор MK2 (митоген-активированной протеинкиназы-активированной протеинкиназы 2), ингибитор MARK (киназы, регулирующей аффинитет микротрубочек), ингибитор CDK5 (циклин-зависимой киназы-5), ингибитор GSK-3 (киназы-3 гликогенсинтазы), блокатор кальциевых каналов, ингибитор HMG-CoA (3-гидрокси-3-метил-глутарил-CoA) редуктазы (статин) и средство, снижающее уровень липидов, имитатор NGF (фактора роста нервов), антиоксидант, лиганд GPR3, активатор плазмина, активатор неприлизина (NEP), активатор IDE (инсулин-разрушающего фермента), агонист мелатонина MT1 и/или MT2, лиганд TLX (рецептора бесхвостой X), лиганд GluR1, антагонист RAGE (рецептора для конечных продуктов поздних стадий глицирования), ингибитор EGFR (рецепторов эпидермального фактора роста), лиганд FPRL-1 (формилпептид-подобного рецептора-1), антагонист GABA (гамма-аминомасляной кислоты) или ингибитор MICAL (молекулы, взаимодействующей с casL), такой как ингибитор оксоредуктазы, в ассоциации с фармацевтически приемлемым носителем. В еще одном варианте осуществления настоящее изобретение относится к комбинации, включающей соединение формулы (I) или его фармацевтически приемлемую соль, вместе с дополнительным терапевтическим средством, как описано выше в настоящей заявке, для последовательного или одновременного введения в отдельных или комбинированных фармацевтических препаративных формах.
В дополнительном аспекте изобретение относится к способу ингибирования продукции белка амилоида-β и/или лечения или профилактики нейродегенеративного заболевания, такого как деменция типа Альцгеймера (AD), синдром Дауна, цереброваскулярная амилоидная ангиопатия (CAA), легкое когнитивное нарушение (MCI), потерю памяти, пресенильную деменцию, сенильную деменцию, наследственное церебральное кровоизлияние с амилоидозом и другие дегенеративные деменции, такие как деменции смешанного сосудистого и дегенеративного происхождения, деменция, связанная с супрануклеарным параличом, деменция, связанная с кортикальной базальной дегенерацией, деменция, связанная с болезнью Паркинсона (PD), и деменция, связанная с типом AD с диффузными тельцами Леви, причем способ включает введение человеку, страдающему указанным состоянием, терапевтически или профилактически эффективного количества описанной выше фармацевтической композиции или определенного выше соединения формулы (I) или его фармацевтически приемлемой соли.
«Эффективное количество» означает количество, достаточное для обеспечения благоприятного воздействия на индивида, или по меньшей мере для вызова изменения состояния индивида.
Болезнь Альцгеймера (AD) патоморфологически характеризуется присутствием нейроволоконных сплетений (NFT) и бляшек, состоящих из β-амилоидных пептидов (Aβ) варьирующейся длины, например, из 42 аминокислот (Aβ42) и 40 аминокислот (Aβ40). В дополнение к этим патологическим маркерам, очевидна также атрофия мозга. Считают, что образование бляшек связано с агрегацией пептидов Aβ. Пептиды Aβ образуются в мозге последовательным расщеплением белка-предшественника амилоида (APP) β-секретазой (BACE-1) и γ-секретазой. Поэтому, потенциальные лекарственные средства при AD, нацеленные на ингибирование образования амилоида ингибированием BACE-1 или γ-секретазы, должны быть способны достичь адекватного воздействия в мозге для оказания эффекта на AD.
Хотя BACE-1 представляет собой привлекательную мишень для остановки или замедления продукции амилоидных пептидов, различные группы нашли проблематичной идентификацию ингибиторов BACE-1, которые могут проникать в центральную нервную систему (ЦНС) и, таким образом, ингибировать фермент в участке действия.
Мозг защищен несколькими барьерами, включая гематоэнцефалический барьер (BBB) и транспортеры (Hitchcock and Pennington, J Med Chem 2006, 29, 7559; Ueno, Curr. Med. Chem. 2007, 14, 1199; Gloor et al., Brain Res. Rev. 2001, 36, 258). Были охарактеризованы несколько транспортеров оттока, которые предотвращают поступление соединений в мозг. Одним из лучше всех охарактеризованных и наиболее значимых в предотвращении проникновения в ЦНС ксенобиотиков является P-гликопротеин (Pgp) (Kusuhara and Sugiyama, Drug Discovery Today, 2001, 6, 150; Mahar Doan et al., J. Pharm. Expt. Ther. 2002, 303, 1029; Lin, Drugs of Today 2004, 40, 5; Lin & Yamazaki, Clin Pharmacokinet. 2003, 42, 59; Schinkel, Adv. Drug Deliv. Rev. 1999, 36, 179). Было показано, что отток Pgp важен для ингибиторов BACE-1 (Hussain et al., J. Neurochem. 2007, 100, 802). Таким образом, важным является преодоление оттока Pgp.
Специалистам в данной области понятно, что существует несколько путей измерения или прогноза проникновения в ЦНС in vitro или in vivo. Потенциал проникновения в ЦНС можно оценить in vitro определением того, может ли соединение подвергаться оттоку Pgp, т.е., проведением анализа Pgp in vitro. Специалистам в данной области понятно, что может использоваться ряд клеточных линий, и что эти клеточные линии могут воздействовать или не воздействовать на результаты анализа. Один такой анализ описан ниже (Cyprotex UK).
Следующий анализ MDR-1 MDCK использовали для оценки оттока Pgp. Анализ проводили в компании Cyprotex Discovery Ltd. 15 Beech Lane, Macclesfield, Cheshire, UK, SK10 2DR
Способность проникновения MDR1-MDCK (Двунаправленная; pH 7,4/pH 7,4)
Краткое изложение протокола
Клетки MDCK представляют собой линию эпителиальных клеток, происходящих из почек собак. Эти клетки могут быть трансфецированы для устойчивой экспрессии активного P-гликопротеина (MDR1-MDCK) и являются идеальными для изучения оттока лекарственных средств. Тестируемое соединение добавляли или на апикальную, или базолатеральную сторону конфлюэнтного монослоя клеток MDR1-MDCK, и проницаемость измеряли путем мониторинга появления тестируемого соединения на противоположной стороне мембраны, используя LC-MS/MS (жидкостную хроматографию с масс-спектрометрией/масс-спектрометрию). По этим данным измеряли/рассчитывали коэффициент видимой проницаемости (Papp) и отношение оттока.
Цель
Измерить проницаемость тестируемого соединения в направлении от апикального к базолатеральному (A-B) и от базолатерального к апикальному (B-A) через клетки MDR1-MDCK. Рассчитывали отношение величин проницаемости B-A и A-B (отношение оттока) для того, чтобы показать, подвергается ли соединение оттоку P-гликопротеина.
Соединения поступали в виде 200 мкл раствора 10 мМ тестируемого соединения в ДМСО (диметилсульфоксиде).
Экспериментальная процедура
Использовали клетки MDR1-MDCK, полученные из NIH (Национального Института Здоровья) (Rockville, MD, USA). После культивирования до слияния, монослои получали промыванием и базолатеральной, и апикальной поверхностей дважды буфером с pH 7,4 при 37°C. Затем клетки инкубировали с буфером при pH 7,4 и в апикальном, и в базолатеральном компартментах в течение 40 мин для стабилизации физиологических параметров.
Затем буфер при pH 7,4 удаляли из апикального компартмента и заменяли растворами для введения тестируемого соединения. Растворы получали разбавлением 10 мМ тестируемого соединения в ДМСО буфером для получения конечной концентрации тестируемого соединения 10 мкМ (конечная концентрация ДМСО, доведенная до 1%). В раствор для введения также включали флуоресцентный маркер целостности люцифер желтый. Затем вставки апикального компартмента помещали в «парные» планшеты, содержащие свежий буфер при pH 7,4. Аналитические стандарты готовили из растворов для введения.
Для экспериментов в направлении от базолатеральной к апикальной стороне (B-A), эксперимент начинали заменой буфера во вставках, затем помещением их в парные планшеты, содержащие растворы для введения. Инкубации проводили в атмосфере 5% CO2 при относительной влажности 95% при 37°C в течение 60 минут.
После периода инкубации, парный планшет удаляли, и апикальный и базолатеральный образцы разбавляли для анализа LC-MS/MS. Проницаемость тестируемого соединения оценивали в двух повторениях. В качестве контролей, на каждом планшете исследовали соединения с известными характеристиками проницаемости.
Тестируемые и контрольные соединения количественно определяли кассетным анализом LC-MS/MS c использованием 5-точечной калибровки при соответствующем разбавлении образцов. Использовали генерические аналитические условия Cyprotex. Исходную концентрацию (C0) определяли по раствору для введения и экспериментальному выходу, рассчитанному по C0 и обеим концентрациям в апикальном и базолатеральном компартментах.
Целостность монослоев в течение всего эксперимента проверяли мониторингом проникновения люцифера желтого с использованием флуориметрического анализа. Проникновение люцифера желтого низкое, если монослой не был поврежден. Если величина Papp люцифера желтого была выше пределов QC (количественного определения) в одной отдельной лунке с тестируемым соединением, то регистрировали результат n=1. Если величина Papp люцифера желтого была выше пределов QC в обеих копиях лунок для тестируемого соединения, то проводили повторное тестирование соединения. Если после повторения наблюдали высокое проникновение люцифера желтого в обеих лунках, то предполагали токсичность эндогенной флуоресценции тестируемого соединения. В этом случае дальнейшие эксперименты не выполняли.
Анализ данных
Коэффициент проницаемости для каждого соединения (Papp) рассчитывали по следующему уравнению:
Papp=(dQ÷dt)÷(C0×A)
где dQ/dt обозначает скорость проникновения лекарственного средства через клетки, C0 обозначает концентрацию в донорском компартменте в нулевое время, и A обозначает площадь клеточного монослоя. C0 получали по анализу раствора для введения в начале эксперимента.
Кроме того, отношение оттока (ER) рассчитывали по средним данным A-B и B-A. Его выводят из уравнения:
ER=((Papp (B-A))÷((Papp (A-B))
Наряду с тестируемыми соединениями, проводили скрининг контрольных соединений, пропранолола (высокопроницаемого) и празосина (субстрата для P-гликопротеина).
К удивлению, было обнаружено, что соединения по настоящему изобретению проявляют более низкое отношение оттока Pgp, чем соединения, иллюстрируемые в опубликованной заявке на Международный патент WO2009/091016, указывая на то, что они могут проявлять более высокое проникновение в ЦНС. Данные по выбранным примерам показаны ниже в таблице 1.
Данные анализа MDR-1 MDCK Pgp
Сравнительные примеры с 1 по 6 описаны в опубликованной заявке на Международный патент WO2009/091016; Сравнительные примеры с 1 по 4 в частности описаны в заявке WO2009/091016 соответственно в виде примеров 32, 35, 54 и 73.
Сравнительные примеры 5 и 6 соответственно представляют
N-(3-((4aS,5S,7aS)-2-амино-5-(фторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-этоксипиколинамид и
N-(3-((4aS,5R,7aS)-2-амино-5-метил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-этоксипиколинамид.
Эти данные демонстрируют, что соединения по настоящему изобретению и конкретные примеры 1-13 и 15-18 имеют более низкий отток Pgp, и поэтому потенциально более высокое проникновение в ЦНС, чем репрезентативные примеры из заявки WO2009/091016 при использовании указанного выше признанного способа оценки проникновения в ЦНС. Например, в сравнительных примерах с 1 по 6 имеются более высокие отношения оттока Pgp, чем соединения по настоящему изобретению. Кроме того, в сравнительных примерах 5 и 6 имеются более высокие отношения оттока Pgp, чем у близких аналогов, в примере 10 настоящего изобретения, который ясно демонстрирует благоприятный эффект, который оказывает трифторметильная группа на кольце тетрагидрофурана на отток Pgp, т.е., трифторметильная группа снижает отток Pgp.
Специалистам в данной области понятно, что описанный выше анализ Pgp in vitro является прогностическим анализом проникновения в ЦНС in vivo. Таким образом, также очень желательно, чтобы уменьшенный опосредованный Pgp отток транслировался на ситуацию in vivo. Специалистам в данной области понятно, что существует много путей оценки проникновения в ЦНС соединений in vivo. Например, можно количественно определить концентрацию соединения в крови или плазме и мозге рассчитать отношение мозг:кровь (Br:Bl) или мозг:плазма (Br:Pl). Этот способ использовался исторически, и были широко принят как способ определения проникновения в ЦНС (Summerfield et al., J Pharmacol. Expt. Ther. 2007, 322, 205). Специалистам в данной области понятно, что этот тип анализа может проводиться в стационарном состоянии, в одну точку времени, множественные точки времени или может проводиться воспроизведением отношений площади под кривой концентрация-время (AUC). Все способы одинаково обоснованы, но каждый может иметь определенные предосторожности, известные специалистам в данной области. В недавних публикациях было высказано предположение, что важно учитывать свободные концентрации in vivo, и что когда происходит отток из мозга, то свободная концентрация в плазме должна быть такой же или эквивалентной свободной концентрации в мозге (Kalvass and Maurer, Biopharmaceutics & Drug Disposition 2002, 23, 327; Mauer et al, Drug Metab. Disposition 2005, 33, 175; Trainor ExpeKT Opin. Drug Discov. 2007, 2, 51). Таким образом, соединение, которое может свободно проникать в ЦНС, и не подвергается активному оттоку, например, Pgp или другим транспортером, должно демонстрировать отношение свободная концентрация в мозге: свободная концентрация в плазме (Brfr : Plfr) или несвязанная концентрация в мозге: несвязанная концентрация в плазме (Bru:Plu) приблизительно 1:1. Специалистам в данной области понятно, что свободную или несвязанную концентрации можно рассчитать умножением общей концентрации в мозге или общей концентрации в плазме на фракцию, не связанную в ткани мозга или плазме, которую можно измерить описанным ниже анализом. Специалистам в данной области понятно, что несвязанная фракция может изменяться в зависимости от экспериментальных факторов, например, концентрации или температуры и т.д. Специалисты в данной области смогут оценить и выбрать самый целесообразный набор условий. Специалистам в данной области также понятно, что пока условия являются одинаковыми для каждого соединения, подвергаемого скринингу, то анализ даст согласованные данные для диапазона тестируемых соединений, таким образом, сводя к минимуму любые несоответствия. Было также предложено, чтобы концентрации лекарственного средства в спинномозговой жидкости (CSF) были эквивалентны свободным концентрациям в мозге для соединений, которые не подвергаются активному оттоку из мозга (He et al., Xenobiotica 2009, 39, 687). Таким образом, другим способом определения проникновения в ЦНС была бы оценка отношения концентрация в CSF:свободная концентрация в плазме (CSF:Plfr) или концентрация в CSF: несвязанная концентрация в плазме (CSF:Plu). Если свободный лекарственный препарат в плазме способен проникать в ЦНС и не подвергается активному притоку или оттоку, то CSF:Plfr или CSF:Plu должны составлять приблизительно 1:1. Специалистам в данной области понятны проблемы, связанные с определением концентраций лекарственных средств в CSF и взятием CSF, например, CSF может быть загрязнена кровью, в зависимости от способа взятия, а также, в зависимости от используемой дозы, величины концентрации в CSF могут быть менее точными.
Так, было показано, что ингибитор BACE, выпускаемый компанией GlaxoSmithKline (GSK188909), BACE-1 IC50 5нМ, который подвергается низкому воздействию CNS, был неэффективен при снижении продукции Aβ40 в мозге мышей TASTPM (у которых имеется сверхэкспрессия человеческих и APPsweK595N/M596I, и PS-1M146V) после болюсного введения (Hussain et al., J. Neurochem. 2007, 100, 802-809). После пероральной дозы 250 мг/кг концентрация в мозге GSK188909 у мышей TASTPM составила 0,62 мкМ. Когда ингибитор Pgp (GF120918) вводили за 5 часов до перорального введения GSK188909, было обнаружено, что концентрация GSK188909 в мозге составляла 5,43 мкМ после пероральной дозы 250 мг/кг, т.е., совместное введение ингибитора Pgp вызывало почти 9-кратное увеличение проникновения в ЦНС, показывая, что отток Pgp является важным механизмом предотвращения проникновения ингибиторов BACE в ЦНС. Кроме того, в отсутствие ингибитора Pgp, пероральная доза 250 мг/кг GSK188909 не оказывала никакого эффекта на уровне Aβ40 в мозге у мышей TASTPM, в то время как, когда совместно вводился ингибитор Pgp (за 5 часов до введения GSK188909), наблюдалось снижение на 68% уровней Aβ40 в мозге относительно мышей, получавших носитель.
В другой статье сообщалось о подобном эффекте, полученном тремя ингибиторами BACE-1, выпускаемыми компанией Bristol-Myers Squibb (Meredith et al., J. Pharm. Expt. Ther. 2008, 326, 502-513). Было обнаружено, что три описанных соединения являются субстратами Pgp in vitro. При введении мышам, эти три соединения проявили низкое проникновение в ЦНС и не снижали уровни амилоида в мозге, но были способны снижать уровни амилоида в плазме. Когда те же три соединения вводили мышам с нокаутом Pgp (KO), уровень проникновения в ЦНС увеличивался, и соединения были способны снижать уровни амилоида в мозге.
Исследователи в компании Schering-Plough также опубликовали статью (Iserloh et al., Bioorg. Med. Chem. Lett. 2008, 18, 418), показывающую, что ингибиторы BACE-1 из их серии (см. пример 11 из указанной выше ссылки), подвергаются оттоку Pgp, в результате которого было обнаружено, что соединение проявляет низкое отношение Br:Pl (<0,1) у крыс.
В приведенных выше публикациях подчеркиваются трудности идентификации ингибиторов BACE-1, которые не подвергаются оттоку Pgp. Такие ингибиторы были бы очень желательны, и многие исследовательские группы предпринимали безуспешные попытки обнаружить такие соединения. Таким образом, были бы желательны ингибиторы BACE-1, которые не являются субстратами Pgp, и поэтому могут легко проникать в ЦНС и снижать уровень амилоида в мозге.
Позднее, исследователи в компании Wyeth сообщили об обширной работе по преодолению оттока Pgp у серии циклических ацилгуанидиновых ингибиторов BACE-1 (Malamas et al, Bioorg. Med. Chem. Lett. 2010, 20, 6597). Были обнаружены соединения, которые были слабыми субстратами Pgp и с Br:Pl, приближающимся к 1:1. Однако в двух основных примерах со сниженным оттоком Pgp (84 и 89 из указанной выше ссылки), уровень Aβ40 в мозге мышей Tg2576 не снижался через 8 часов после пероральной дозы 30 мг/кг. Отсутствие эффективности относили на счет того, что соединения проявляли высокое связывание с тканью мозга. Таким образом, важно обнаружить ингибиторы BACE-1, которые не являются субстратами Pgp, но при этом имеют целесообразную несвязанную фракцию в ткани мозга и способны снижать уровень амилоида в мозге.
Было также показано, что ингибиторы BACE, которые не являются субстратами Pgp in vitro, могут проникать в ЦНС (например, TC-1 от компании Merck), и могут снижать уровни Aβ40 в мозге мышей APP-YAC и обезьян (Sankaranarayanan et al., J. Pharmacol. Expt. Ther. 2009, 328, 131-140). Так, анализы Pgp in vitro показал, что TC-1 не является субстратом Pgp, и когда TC-1 вводили мышам APP-YAC (100 мг/кг внутрибрюшинно), он был способен незначительно проникать в ЦНС, как показано концентрациями в мозге и отношением мозг:плазма, и эта способность привела к умеренному снижению уровня амилоида в мозге.
(мкМ)
(мкМ)
Концентрация TC-1 в мозге и плазме после внутрибрюшинного введения дозы 100 мг/кг и соответствующие воздействия на уровни Aβ40 в мозге у мышей APP-YAC.
В отдельных экспериментах было показано, что TC-1 может проникать в ЦНС обезьян при совместном введении с ингибитором CYP3A4 (ритонавиром). В этих экспериментах было обнаружено, что средняя концентрация TC-1 в плазме составила 2,7 мкМ, в то время как было обнаружено, что концентрация в ЦНС составляла 0,025 мкМ. Однако поскольку TC-1 на ~99% связан с белками плазмы, то свободная концентрация в плазме по расчетам составляет ~0,027 нМ. Было обнаружено, что уровни Aβ40 в ЦНС проявили снижение на 42% относительно контрольной группы, получавшей носитель. Таким образом, ожидалось, что ингибитор BACE, который может свободно проникать в ЦНС, способен снижать уровни амилоида в ЦНС. Было бы благоприятным его совместное введение с ингибитором CYP3A4.
Было показано, что соединения по настоящему изобретению снижают продукцию Aβ в клеточных анализах, что коррелируется с их способностью снижать продукцию Aβ у животных. Таким образом, соединения по настоящему изобретению могут применяться при снижении продукции Aβ у людей, и, следовательно, полезны при лечении нейродегенеративных заболеваний, таких как болезнь Альцгеймера.
Проникновение в ЦНС крыс in vivo
Самцов крыс Sprague Dawley приобретали у компании Charles River UK Ltd. (Margate, UK) и содержали в соответствии с руководствами UK Home Office. Концентрации лекарственных средств доводили до соответствующего уровня в 0,5% метилцеллюлозе. Животным через внутрижелудочный зонд перорально вводили соединения (2 мл/кг) в дозах, указанных ниже в таблицах 2-4.
В точки времени после введения, определенные ниже в таблицах 2-4, животным внутрибрюшинно инъецировали пентобарбитон натрия (приблизительно 330 мг/кг для конечной анестезии).
С использование гильотины, животных декапитировали, и кровь туловища собирали в пробирки типа Falcon емкостью 15 мл, содержащие 100 МЕ гепарина. Кровь перемешивали в вихревой мешалке с последующим центрифугированием при 6000 об/мин, 4°C в течение 5 минут. Плазму собирали для анализов DMPK (гена миотонической протеинкиназы) и ELISA (иммуноферментного анализа) и хранили при -80°C до использования. Мозг извлекали и делили по средней линии, взвешивали и хранили при -80°C до дальнейшего использования.
Способ анализа образцов плазмы, мозга и CSF
Получение рабочих растворов ацетонитрила
Тестируемое соединение получали в виде 1 мг свободного основания/мл раствора в ДМСО, смешивали в вихревой мешалке и обрабатывали ультразвуком в течение 5 мин. Раствор в ДМСО 1 мг/мл разбавляли для получения основных растворов, содержащих 10 и 30 мкг/мл ацетонитрила, добавлением соответственно 10 мкл к 990 мкл ацетонитрила и 30 мкл к 970 мкл ацетонитрила. Затем основные растворы, содержащие 10 и 30 мкг/мл ацетонитрила, затем серийно разбавляли 1:9 (об./об.) (100 мкл основного раствора в 900 мкл ацетонитрила) для получения следующих растворов: 0,003, 0,01, 0,03, 0,1, 0,3, 1, 3, 10 и 30 мкг/мл ацетонитрила.
Получение стандартов плазмы, контролей и образцов
Образцы контрольной плазмы самцов крыс Sprague Dawley и исследуемой плазмы хранили при -80°C до дня анализа, когда их оттаивали при комнатной температуре. Контрольную плазму центрифугировали (2000g в течение 10 мин) и распределяли аликвотами (90 мкл) в пробирки Eppendorf для получения стандартов и контрольных образцов. Исследуемые образцы предварительно распределяли аликвотами (100 мкл) в пробирки Eppendorf сразу после сбора плазмы.
Аликвот (10 мкл) соответствующего основного раствора ацетонитрила добавляли к контрольной плазме (для получения конечного объема 100 мкл) для получения требуемых калибровочных стандартов, охватывающих диапазон от 1 до 3000 нг/мл. Двойные контрольные и контрольные образцы получали добавлением 10 мкл ацетонитрила к 90 мкл контрольной плазмы.
Получение стандартов мозга, контролей и образцов
Контрольные и исследуемые образцы мозга самцов крыс Sprague Dawley взвешивали после взятия и хранили при -80°C до дня анализа, когда их оттаивали при комнатной температуре. После оттаивания, мозги разбавляли водой (4 мл на грамм ткани) и гомогенизировали, используя механический гомогенизатор. Аликвот (100 мкл) каждого исследуемого образца брали в пробирки Micronics, получали готовые для анализа и достаточные аликвоты (90 мкл) гомогената контрольного мозга, полученного для приготовления стандартов и контролей.
Аликвот (10 мкл) соответствующих основных растворов ацетонитрила добавляли к контрольному гомогенату мозга (для получения конечного объема 100 мкл) для получения требуемых калибровочных стандартов, охватывающих диапазон от 1,5 до 5000 нг/г. Двойные контрольные и контрольные образцы получали добавлением 10 мкл ацетонитрила к 90 мкл контрольного гомогената мозга.
Взятие образцов плазмы и мозга, стандартов и контролей
Каждый образец плазмы и гомогената мозга, стандарта и контроля (100 мкл) экстрагировали аликвотом (300 мкл) ацетонитрила (содержащего 0,1% муравьиную кислоту и 100 нг/мл соответствующего внутреннего стандарта). Двойные контроли экстрагировали аликвотом (300 мкл) ацетонитрила (содержащего 0,1% муравьиную кислоту). Затем все образцы, стандарты и контроли смешивали в вихревой мешалке и центрифугировали (2000 g в течение 15 мин). Затем аликвот (50 мкл) полученного супернатанта помещали в 96-глубоколуночный планшет емкостью 2 мл и разбавляли ацетонитрилом:водой (50:50 об./об.) (150 мкл), готовый для анализа специальным способом LC-MS/MS.
Получение образцов CSF, стандартов и контролей
Контрольные и исследуемые образцы CSF самцов крыс Sprague Dawley хранили при -80°C до дня анализа, когда их оттаивали при комнатной температуре. Аликвот (50 мкл) каждого исследуемого образца помещали в пробирки Micronics, готовые для анализа, и достаточные аликвоты (45 мкл) контрольной CSF, полученной для приготовления стандартов и контролей.
Аликвот (5 мкл) соответствующих основных растворов ацетонитрила добавляли к контрольной CSF (для получения конечного объема 50 мкл) для получения требуемых калибровочных стандартов, охватывающих диапазон от 1 до 1000 нг/мл. Двойные контрольные и контрольные образцы получали добавлением 5 мкл ацетонитрила к 45 мкл контрольной CSF.
Взятие образцов CSF, стандартов и контролей
Каждый образец CSF, стандарт и контроль (50 мкл) экстрагировали аликвотом (150 мкл) ацетонитрила (содержащего 0,1% муравьиную кислоту и 100 нг/мл соответствующего внутреннего стандарта). Двойные контроли экстрагировали аликвотом (150 мкл) ацетонитрила, содержащего 0,1% муравьиную кислоту. Затем все образцы смешивали в вихревой мешалке, и затем аликвот (50 мкл) каждого дополнительно разбавляли в 150 мкл ацетонитрила:воды (50/50 об./об.) в 96-глубоколуночном планшете емкостью 2 мл, готовом для анализа LC-MS/MS.
Все образцы затем анализировали, используя сверхэффективный жидкостный хроматограф Waters Acquity UPLC, соединенный с масс-спектрометром Waters Xevo TQ.
Условия жидкостной хроматографии:
Колонка: Acquity UPLC BEH C18, 1,7 мкм, 2,1×50 мм, поддерживаемая при 40°C
Подвижная фаза: A = 95% воды : 5% MeOH, содержащая 0,01M ацетат аммония
B = 5% воды : 95% MeOH, содержащая 0,01M ацетат аммония
Скорость потока: 0,6 мл/мин; объем инжекции 5 мкл; температура автосамплера 6°C
Поток ЖХ отводили в сток в течение первых 0,3 мин каждой инжекции
MS/MS переходы автоматически оптимизировали программным обеспечением Waters QuanOptimise.
Выявление амилоида
Экстракция DEA/NaCl пептидов Aβ из мозга крыс:
Готовили 100 мл свежего охлажденного 0,2% диэтиламина (DEA) в 50 мМ NaCl (pH 10), и 1 мл/25 мг ткани мозга добавляли в каждое полушарие (т.е., 40x объем мозга). Мозги сразу гомогенизировали, используя гомогенизатор Polytron PT 1200, в течение 1,5 минут, и образцы оставляли для инкубации на льду в течение одного часа после гомогенизации. 3 мл гомогената переносили в полиалломерную пробирку (Beckman #362333) центрифугировали при 133000×g (55,000 об/мин) в течение 45 мин при температуре 4°C. Затем супернатант нейтрализовали до pH 8-8,3 добавлением 1/10 объема 0,5M Tris/HCl, pH 6,8. Образцы могут использоваться свежими или быстрозамороженными на сухом льду и хранили при -80°C, пока они не требовались для анализа.
ELISA человеческого/крысиного βАмилиоида (40) (Набор Wako)
В наборе ELISA Wako Aβ40 (Code No. 294-62501) используется моноклональное антитело BNT77, выработанное против эпитопа Aβ(11-28), и моноклональное антитело BA27, которое специфически выявляет C-концевую часть Aβ40. Этот набор используется для количественного определения человеческого или крысиного Aβ(1-40), а также усеченных на N-конце видов Aβ40 (Aβ(x-40)) в биологических матрицах, таких как среда для культуры ткани, гомогенат ткани, CSF и плазма.
Для анализа образцы плазмы и мозга разбавляют 1:1 стандартным разбавителем, содержащимся в наборе, и образцы CSF разводят 1:8 стандартным разбавителем, содержащимся в наборе. Анализ проводят в соответствии с инструкциями производителя, и образцы анализируют в двух повторениях. Данные анализируют, используя Microsoft Excel 2003, и статистический анализ проводят, используя 9-е издание программного обеспечения Genstat.
Так, когда в сравнительном примере 4 введение производили в дозе 10 мг/кг перорально, и образцы плазмы, мозга и CSF брали через 2, 4, 6 и 8 часов после введения, то измеряли следующие концентрации (таблица 2):
Данные сравнительного примера 4
2 Рассчитано умножением [Br] на Br Fu.
По описанному выше исследованию, сравнительный пример 4 показал снижение на 59% и 64% Aβ40 в мозге соответственно через 4 и 6 часов; и снижение на 76% и 70% Aβ40 в CSF соответственно через 4 и 6 часов.
Определенные соединения по настоящему изобретению оценивали in vivo у крыс для подтверждения уровней проникновения в CNS; эти данные представлены ниже в таблицах.
К удивлению, было обнаружено, что соединения по настоящему изобретению проявляют увеличенное проникновение в CNS у крыс относительно соединений из заявки WO2009/091016, по данным любого из указанных выше признанных способов определения проникновения в CNS. Таким образом, соединения по настоящему изобретению могут проявлять улучшенные профили в том, что они легче нацеливаются на участок действия, мозг, и поэтому могут проявить улучшенную эффективность или эффективность при более низких концентрациях или дозах, или сниженные периферически опосредованные побочные эффекты путем предпочтительного разделения CNS, или комбинацией любого или всех из этих аспектов.
Так, когда в сравнительном примере 8 введение производили в дозе 10 мг/кг перорально, и образцы плазмы, мозга и CSF брали через 2, 4, 6 и 8 часов после введения, то измеряли следующие концентрации (таблица 3):
Данные по примеру 8
(нМ)
2. Рассчитано умножением [Br] на Br Fu.
3. Количественно не определяемо. Концентрации, близкие или ниже нижнего предела количественного определения, и их невозможно точно количественно определить.
4. Не определялось.
Из описанного выше исследования, пример 8 показал снижение на 68% и 72% Aβ40 в мозге соответственно через 4 и 6 часов; и снижение на 82% и 74% Aβ40 в CSF соответственно через 4 и 6 часов. Таким образом, соединения по настоящему изобретению проявляют уменьшенный отток Pgp относительно предыдущих описаний, в то же время, демонстрируя эффективность в CNS. Таким образом, эффективность достигается при более низких концентрациях в циркулирующей плазме.
Когда в примере 1 настоящего изобретения введение производили в дозе 10 мг/кг перорально, и образцы плазмы, мозга и CSF брали через 2, 4, 6 и 8 часов после введения, то измеряли следующие концентрации (таблица 4):
Данные по примеру 1
Из описанного выше исследования, пример 1 показал снижение на 64% и 70% Aβ40 в мозге соответственно через 4 и 6 часов; и снижение на 80% и 85% Aβ40 в CSF соответственно через 4 и 6 часов. Таким образом, соединения по настоящему изобретению проявляют уменьшенный отток Pgp относительно предыдущих изобретений, в то же время, демонстрируя эффективность в CNS. Таким образом, эффективность достигается при более низких концентрациях в циркулирующей плазме.
Способ определения связывания с белком плазмы (PPB) и связывания с мозговой тканью (BTB)
Подготовка соединения
Соединения растворяли в ДМСО для получения раствора 1 мг свободного основания/мл раствора перед дальнейшим разбавлением до 100 мкг/мл в ацетонитриле (100 мкл 1 мг/мл в 900 мкл ацетонитрила).
Получение матрицы
В утро диализа, контрольную плазму и мозг самцов крыс Sprague Dawley, ранее хранившиеся при -80°C, оттаивали при комнатной температуре. Проверяли pH плазмы, и при необходимости доводили до 7,4 1M HCl. Затем плазму центрифугировали (2000 g в течение 10 мин), и мозги разводили 2 мл солевого раствора с фосфатным буфером (pH 7,4) на грамм ткани и гомогенизировали, используя механический гомогенизатор. Затем аликвот (10 мкл) раствора в ацетонитриле 100 мкг/мл соединения добавляли к 1 мл плазмы и гомогенату ткани мозга и смешивали в вихревой мешалке для получения конечной концентрации соединения 1 мкг/мл в матрице.
Получение планшета RED
Планшет быстрого равновесного диализа (RED) (Thermo Scientific) получали в соответствии с инструкциями производителя, т.е., основной планшет пропитывали 20% (об./об.) этанолом в течение 10 мин и затем дважды споласкивали деионизированной водой перед предоставлением возможности высыхания. Затем основной планшет заполняли соответствующим числом одноразовых вставок (n=3 на соединение) (Thermo Scientific), и матрицу, содержащую 1 мкг/мл соединения, добавляли в матричную камеру вставок (200 мкл), и в камеру буфера добавляли аликвот (350 мкл) PBS. Затем планшет покрывали клеящим агентом и инкубировали на воздухе при 37°C в течение 6 часов при перемешивании со скоростью 130 об/мин.
Взятие проб
После 6 часов инкубации, уплотнитель удаляли, брали аликвот (50 мкл), взятый из камер PBS, и подавали в пробирки Micronics. Также, аликвот (50 мкл) удаляли из матричной камеры и помещали в отдельные пробирки Micronics. Затем плазму и мозг подгоняли к матрице 50 мкл не содержащего лекарственное средство PBS, и образцам PBS с 50 мкл соответствующей не содержащей лекарственное средство матрицы, для получения равных конечных композиций и объемов (100 мкл).
Анализ образцов
Образцы смешивали в вихревой мешалке и добавляли аликвот (300 мкл) ацетонитрила, содержащего 0,1% муравьиную кислоту и 100 нг/ил соответствующего внутреннего стандарта. Затем образцы смешивали и центрифугировали (2000 g в течение 15 мин), и алкивот супернатанта (100 мкл) удаляли в 96-глубоколуночный планшет и разбавляли равным объемом воды, готовый для анализа LC-MS/MS. Были получены следующие данные для следующих соединений в описанном выше анализе (таблица 5).
fu = несвязанная фракция
Из представленных выше данных, для специалистов в данной области очевидно, что соединения примеров 1 и 8 достигают такого же снижения Aβ40 в мозге как снижение в сравнительном примере 4, но при более низкой концентрации в плазме и свободной концентрации в плазме. Это имеет преимущества и указывает на то, что соединения по изобретению имеют такую же или лучшую эффективность при более низких концентрациях, чем соединения заявки WO2009/091016, и, следовательно, менее вероятно вызовут нежелательные периферически опосредованные побочные эффекты, такие как сердечнососудистые эффекты, фосфолипидоз, печеночная токсичность, почечная токсичность и желудочно-кишечная токсичность.
Оценка воздействий на интервал QTc у морских свинок
Самцов морских свинок Dunkin-HaKTley взвешивали и наркотизировали 4% изфолюраном в карбогене. Анестезию поддерживали при 1,5% изофлюрана, и животных держали в условиях анестезии в течение всего исследования. Ксилазин в дозе 2 мг/кг вводили внутримышечно в заднюю лапу в качестве вызывающего брадикардию средства для обеспечения возможности выявления удлинения интервала QTc программным обеспечением.
Сонную артерию и яремную вену канюлировали магистралями, содержащими гепаринизированный солевой раствор, подсоединяли электроды ЭКГ в трех отведениях, и мониторинг проводили, используя программное обеспечение LabChaKT Pro. Животным давали возможность стабилизироваться в течение 30 минут после завершения хирургической процедуры, перед началом внутривенной инфузии носителя (5% ДМСО/90% MilliQ/5% 0,1н HCl) от нулевого времени (скорость инфузии = 0,2 мл/кг/мин). Через 10 мин, брали образец артериальной крови для анализа PK (фармакокинетики) (150 мкл; все шпицы для взятия проб были гепаринизированы). Через 12 мин начинали внутривенную инфузию лекарственного средства со скоростью 2,0 мг/кг/10 мин. Дозу увеличивали до 6,0 мг/кг/10 мин, затем до 20 мг/кг/10 мин, при 10-минутном периоде инфузии и двух минутах взятия образца крови при каждой дозе. После конечной дозы брали образец крови и начинали инфузию второго носителя. Через 8 минут брали конечный образец крови для анализа PK в плазме, и животных умерщвляли способом схемы 1.
Изменения интервала QTc (Bazett's) анализировали, используя программное обеспечение LabChaKT Pro. QTc оставался неизменным до самой высокой тестированной дозы/концентрации, которая соответствовала несвязанной концентрации в плазме 9503 нМ для примера 8 и 296 нМ для примера 1.
Оценка воздействий на интервал QTc у собак-биглей
Самцов собак-биглей взвешивали, и им инъецировали тиопентал натрия для вводной анестезии. Анестезию поддерживали смесью 1-1,5% изофлюрана и кислорода, и животных держали в условиях искусственной вентиляции легких и анестезии с использованием изофлюрана в течение всего исследования.
Сонную артерию и большую подкожную вену бедра канюлировали магистралями, содержащими гепаринизированный солевой раствор, подсоединяли электрод ЭКГ в отведении LII, и мониторинг проводили, используя систему полиграфа. Животным давали возможность стабилизироваться в течение 30 минут перед началом инфузии через канюлю от нулевого времени при скорости 1 мг/кг/10 мин. Дозу увеличивали до 3 мг/кг/10 мин, затем до 10 мг/кг/10 мин. Образец артериальной крови брали после каждого введения для анализа PK.
Интервал QTc оставался неизменным до самой высокой тестированной дозы/концентрации, которая соответствовала несвязанной концентрации в плазме 4128 нМ для примера 8 и 1329 нМ для примера 1.
Далее будут описаны способы получения соединения формулы (I) или его фармацевтически приемлемой соли в соответствии с настоящим изобретением.
A. Общий способ получения A:


В формуле X, Y и A обозначают, как определено выше.
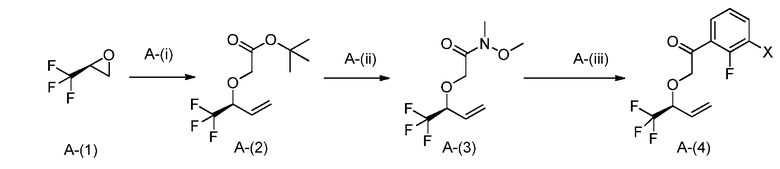
Общий способ получения A представляет собой способ получения соединения A-(15), которое соответствует соединению (I) в соответствии с настоящим изобретением, из соединения A-(1) в качестве сырьевого материала посредством множественных стадий со стадии A-(i) до стадии A-(xiv).
Соединение A-(1) имеется в продаже.
Стадия A-(i):
Данная стадия представляет собой стадию получения соединения A-(2) открытием эпоксида A-(1) сульфоний-илидом для получения промежуточного соединения аллильного алкоксида, который затем алкилируют для получения соединения A-(2). Специалистам в данной области понятно, что эта трансформация может проводиться одностадийной реакцией или в виде двух отдельных реакций. Специалистам в данной области понятны преимущества и недостатки одностадийной реакции, по сравнению с проведением двух отдельных реакций, и они в соответствии со своими потребностями выберут наилучший способ.
В частности, эпоксид A-(1) может быть открыт анионом триметилсульфония йодида и итоговой потерей диметилсульфида для получения соответствующего аллильного алкоксида. Триметилсульфоний йодид может быть депротонирован подходящим основанием, например, бутиллитием. Растворитель, используемый при реакции, конкретно не ограничивается, пока он не препятствует реакции. Примеры подходящих растворителей включают THF. Специалистам в данной области понятно, что слово растворитель в данном случае используется для обозначения жидкость, в которой осуществляется реакция, и что реагенты могут быть не растворены. Предпочтительно, реакция должна проводиться при температуре ниже комнатной, предпочтительно -30-20°C. После добавления реакционная смесь может нагреваться до комнатной температуры для содействия реакции. Время реакции конкретно не ограничивается и обычно составляет от 5 минут до 24 часов, предпочтительно, 1-6 часов.
Специалистам в данной области понятно, что алкоксид, полученный в результате этой реакции, может непосредственно взаимодействовать с алкилирующим агентом, таким как трет-бутилбромацетат, и что эта реакция может протекать с дополнительными растворителями и без них. Если требуются дополнительные растворители для содействия реакции, то подходят такие растворители как DMF или NMP. Температура реакции конкретно не ограничивается. Подходящие температуры реакции включают от комнатной температуры до 80°C, предпочтительно, комнатную температуру. Время реакции конкретно не ограничивается и обычно составляет от 5 минут до 1 недели, предпочтительно, 1-48 часов.
Специалистам в данной области понятно, что промежуточный алкоксид может гаситься, выделяться и очищаться, а затем подвергаться условиям независимого алкилирования. Эта реакция может выполняться в таких же условиях, как условия, обычно используемые при реакции O-алкилирования спиртового соединения (таких как условия, описанные в публикации Tetrahedron Lett. 46 (2005) 45, 7751-7755). В этой реакции соединение A-(2) может быть получено добавлением основания, такого как гидрид натрия, к раствору промежуточного спирта в THF для получения алкоксида, и затем взаимодействием алкоксида, например, с трет-бутилбромацетатом. Растворитель, используемый в реакции, конкретно не ограничивается, пока он не ингибирует реакцию, и обеспечивает возможность растворения в нем исходного материала в определенной степени. Примеры растворителя включают такие растворители как THF, DMF и диметилсульфоксид. Реакция может выполняться вызовом действия от 1 до 3 эквивалентов соответствующего основания в присутствии такого растворителя. Примеры используемого основания включают гидрид натрия, гидрид калия и т-бутоксикалий. Время реакции конкретно не ограничивается и обычно составляет от 0,5 до 72 часов, и предпочтительно, от 0,5 до 12 часов. Температура реакции составляет обычно от -20°C до 50°C.
Более предпочтительный результат, такой как улучшенный выход, может быть достигнут добавлением соли, такой как тетрабутиламмоний йодид, при этой реакции.
Стадия A-(ii):
Эта стадия представляет собой двухстадийную последовательную реакцию для получения соединения A-(3) из соединения A-(2) снятием защиты сложноэфирной группы, а затем образованием амида Вайнреба.
В частности, защита сложного трет-бутилового эфира соединения A-(2) может быть снята в тих же условиях, как те, которые в целом используются при снятии защиты соединения сложного трет-бутилового эфира (таких как условия, описанные в документе, таком как T. W. Greene and P. G. M. Wuts, "Protective Groups in Organic Chemistry, Third Edition", John Wiley & Sons (1999), p. 404-408). При этой реакции соединение A-(2) может взаимодействовать с соответствующей кислотой в подходящем растворителе, таком как муравьиная кислота, как, например, растворитель и кислота. Растворитель, используемый в реакции, конкретно не ограничивается, пока он не ингибирует реакцию и обеспечивает возможность растворения в нем исходного материала в определенной степени. Время реакции конкретно не ограничивается и обычно составляет от 0,5 до 72 часов, и предпочтительно, от 0,5 до 24 часов. Температура реакции составляет обычно от температуры 0°С до 60°C.
Затем промежуточная кислота может быть трансформирована в амид Вайнреба (Tetrahedron Lett. 1981, 22, 3815) реакцией гидрохлорид N,O-диметилгидроксиламина в стандартных условиях образования амида, т.е., конденсацией промежуточной кислоты с гидрохлорид N,O-диметилгидроксиламином с использованием конденсирующего агента. Альтернативно, эта стадия представляет собой стадию получения соединения A-(3) конденсацией промежуточной кислоты с гидрохлоридом N,O-диметилгидроксиламина реакцией ацилирования.
Реакция конденсации промежуточной кислоты с гидрохлоридом N,O-диметилгидроксиламина с использованием конденсирующего агента может быть выполнена в таких же условиях как условия, обычно используемые и описанные в следующих документах. Примеры известных способов включают способы, описанные в публикациях Rosowsky et al.; J. Med. Chem., 34 (1), 227-234 (1991), Brzostwska et al.; Heterocycles, 32 (10), 1968-1972 (1991) и Romero et al.; J. Med. Chem., 37 (7), 998-1014 (1994).
Гидрохлорид N,O-диметилгидроксиламина может представлять собой свободную форму или соль.
Растворитель, используемый в реакции, конкретно не ограничивается, пока он не ингибирует реакцию. Примеры растворителя включают тетрагидрофуран, 1,4-диоксан, этилацетат, метилацетат, дихлорметан, хлороформ, N,N-диметилформамид, толуол, ацетонитрил и ксилол. Примеры конденсирующего агента включают CDI (N,N'-карбонилдиимидазол), Bop (1H-1,2,3-бензотриазол-1-илокси(три(диметиламино))фосфоний гексафторфосфат), WSC (1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид), DCC (N,N-дициклогексилкарбодиимид), диэтилфосфорил цианид, PyBOP (бензотриазол-1-илокситрис(пирролидино)фосфоний гексафторфосфат) и EDC·HCl гидрохлорид (1-этил-3-(3-диметиламиопропил)карбодиимид). Подходящие условия включают агент для активации кислоты, такой как N,N'-карбонилдиимидазол. Используется от одного эквивалента до большого избытка гидрохлорида N,O-диметилгидроксиламина в отношении промежуточной кислоты. При необходимости, может добавляться от одного эквивалента до большого избытка органического основания, такого как триээтиламин.
Время реакции конкретно не ограничивается и обычно составляет от 0,5 до 72 часов, и предпочтительно, от 0,5 до 24 часов. Температура реакции варьируется в соответствии с используемым сырьевым материалом, растворителем и тому подобным и конкретно не ограничивается. Приемлемая температура от °С до температуры кипения растворителя, предпочтительная температура от 0°С до комнатной.
Стадия A-(iii):
Эта стадия представляет собой стадию получения соединения A-(4) взаимодействием металлорганического (ариллитиевого реагента или реагента Гриньяра) реагента с соединением A-(3), как описано в публикации Tetrahedron Lett. 1981, 22, 3815.
Реакция на этой стадии может выполняться в таких же условиях как условия, описанные, например, в публикации Tetrahedron Lett. 1981, 22, 3815.
Ариллитиевый реагент (включая гетероциклический) или реагент Гриньяра (включая гетероциклический) может быть получен способом, известным специалисту в данной области. В частности, соответствующий фениллитиевый реагент или фенилмагниевый (Гриньяра) реагент может быть получен обменом галогена-металла между галоген арильным соединением и имеющимся в продаже металлоорганическим реагентом, таким как алкиллитиевый реагент, такой как н-, втор- или трет-бутиллитий или реагент Гриньяра, такой как изопропилмагний бромид, или, например, металлический магний.
Растворитель, используемый на этой стадии, варьируется в соответствии с используемым исходным материалом и реагентом и конкретно не ограничивается, пока он не ингибирует реакцию, обеспечивает возможность растворения в нем исходного материала в определенной степени и является всегда инертным во время реакции. Предпочтительные примеры растворителя включают органические растворители, такие как диэтиловый простой эфир, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, бензол и толуол и их смешанные растворители. Время реакции конкретно не ограничивается и обычно составляет от 0,1 до 48 часов, и предпочтительно от 0,1 до 12 часов. Температура реакции варьируется в соответствии с исходным материалом, используемым реагентом и тому подобным и предпочтительно поддерживается на низком уровне, например, при -78 - -60°C.
Стадия A-(iv):
Эта стадия представляет собой стадию получения соединения A-(5) оксимацией соединения A-(4).
Реакция на этой стадии может выполняться в таких же условиях как условия, обычно используемые при реакции оксимации карбонильного соединения, такие как условия, описанные в публикациях Org. Lett. 9 (2007) 5, 753-756, Tetrahedron: Asymmetry 5 (1994) 6, 1018-1028 и Tetrahedron 54 (1998) 22, 5868-5882.
В частности, соединение A-(5) может быть получено взаимодействием соединения A-(4) с гидроксиламином или солью гидроксиламина (такой как гидрохлорид гидроксиламина или гидроксиламинсульфат) в присутствии основания или, например, в отсутствие основания.
Растворитель, используемый на этой стадии, варьируется в соответствии с используемым исходным материалом и реагентом и конкретно не ограничивается, пока он не ингибирует реакцию. Предпочтительные примеры растворителя включают органические растворители, такие как этанол, метанол, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан и дихлорметан и смеси этих растворителей и воды. Примеры используемого основания включают ацетат натрия, пиридин, гидроксид натрия, гидроксид цезия, гидроксид бария и 2,6-лутидин. Время реакции конкретно не ограничивается и обычно составляет от 5 минут до 24 часов, и предпочтительно от 5 минут до 12 часов. Температура реакции обычно составляет от -20°C до температуры кипения растворителя, а предпочтительнее, от 0°C до температуры кипения растворителя.
Стадия A-(v):
Эта стадия представляет собой стадию получения соединения A-(6) термическим внутримолекулярным циклоприсоединением алкенилоксима A-(5).
Реакция проводится в присутствии добавки, например, гидрохинона.
Растворитель, используемый в этой реакции, конкретно не ограничивается, пока он не ингибирует реакцию. Подходящие реакционные растворители включают ксилены с высокой точкой кипения. Температура реакции конкретно не ограничивается и обычно составляет 80-200°C или температуру кипения растворителя. Время реакции конкретно не ограничивается и составляет обычно от 0,5 до 48 часов, и предпочтительно от 0,5 до 24 часов.
Стадия A-(vi):
Эта стадия представляет собой стадию подвергания соединения A-(6) восстановительному расщеплению соединения N-O.
Реакция восстановительного расщепления связи N-O может выполняться в условиях с использованием цинк-уксусной кислоты, металлического катализатора, такого как, например, гидроксид платины или литий алюминий гидрид.
Реакция с использованием цинка, такого как цинк-уксусная кислота, может выполняться в таких же условиях как условия, описанные, например, в публикациях J. Org. Chem. 2003, 68, 1207-1215 и Org. Lett. 7 (2005) 25, 5741-5742. Примеры используемой кислоты включают уксусную кислоту, муравьиную кислоту и хлористоводородную кислоту. Растворитель, используемый в этой реакции, конкретно не ограничивается, пока он не ингибирует реакцию и обеспечивает возможность растворения в нем исходного материала в определенной степени. Примеры растворителя включают метанол, этанол, 1,4-диоксан, THF и воду. Указанная выше кислота может также использоваться в качестве растворителя. Температура реакции составляет обычно от -20°C до температуры кипения растворителя, и предпочтительно, от температуры 0°С до температуры кипения растворителя. Время реакции конкретно не ограничивается и составляет обычно от 5 минут до 48 часов и предпочтительно, от 5 минут до 24 часов.
Реакция с использованием металлического катализатора, такого как гидроксид платины, может выполняться в таких же условиях как условия, описанные, например, в публикациях Tetrahedron: Asymmetry 5 (1994) 6, 1018-1028 и Tetrahedron, Vol. 53, No. 16, pp 5752-5746, 1997. Соединение A-(7) может быть получено гидрогенизацией соединения A-(6) с использованием оксида платины в качестве катализатора в растворителе, таком как, например, метанол.
Реакция с использованием гидрида лития алюминия может выполняться в таких же условиях как условия, описанные, например, в публикации Bull. Chem. Soc. Jpn., 66, 2730-2737 (1993). Соединение A-(7) может быть получено восстановлением соединения A-(6) с использованием гидрида лития алюминия в растворителе, таком как, например, простой эфир.
Стадия A-(vii):
Эта стадия представляет собой стадию получения соединения A-(8) из соединения A-(7). Производное тиомочевины A-(8) может быть получено из соединения A-(7) способом, известным специалисту в данной области.
Соединение A-(8) может быть получено на этой стадии взаимодействием соединения A-(7) с бензоилизотиоцианатом в растворителе, таком как дихлорметан или толуол. Эта реакция может выполняться в таких же условиях как условия, описанные, например, в публикации J. Med. Chem. 1990, 33, 2393-2407. Растворитель, используемый в реакции, конкретно не ограничивается, пока он не ингибирует реакцию и обеспечивает возможность растворения в нем исходного материала в определенной степени. Примеры растворителя включают дихлорметан, хлороформ, толуол, 1,4-диоксан и THF. Температура реакции составляет обычно от -20°C до температуры кипения растворителя, и предпочтительно, от 0°С до температуры кипения растворителя. Время реакции конкретно не ограничивается и составляет обычно от 5 минут до 48 часов, и предпочтительно, от 5 минут до 24 часов.
Стадия A-(viii):
Эта стадия представляет собой способ получения соединения A-(9) циклизацией соединения A-(8).
В этой реакции соединение A-(8) может быть циклизовано в различных условиях для получения соединения A-(9) активацией спирта соединения A-(8).
Например, соединение A-(9) может быть получено в этой реакции нагреванием соединения A-(8) в растворителе, таком как метанол, в присутствии кислоты, такой как, например, концентрированная хлористоводородная кислота. Растворитель, используемый в реакции, конкретно не ограничивается, пока он не ингибирует реакцию и обеспечивает возможность растворения в нем исходного материала в определенной степени. Примеры растворителя включают растворители, такие как метанол, этанол, 1-пропанол и вода, их смешанные растворители и кислоты, используемые в качестве растворителя. Реакция может выполняться использованием от одного эквивалента до большого избытка соответствующей кислоты для действия в присутствии или в отсутствие такого растворителя. Примеры используемой кислоты включают концентрированную хлористоводородную кислоту, бромистоводородную кислоту, серную кислоту, трифторуксусную кислоту, метансульфоновую кислоту, трифторметансульфоновую кислоту и их смеси. Время реакции конкретно не ограничивается и обычно составляет от 0,5 до 72 часов, и предпочтительно, от 0,5 до 24 часов. Температура реакции составляет обычно от 0°С до температуры кипения растворителя.
Альтернативно, соединение A-(9) может быть получено взаимодействием соединения A-(8) с трифторметансульфоновым ангидридом в растворителе, таком как дихлорметан, в присутствии основания, такого как пиридин. Растворитель, используемый в реакции, конкретно не ограничивается, пока он не ингибирует реакцию и обеспечивает возможность растворения в нем исходного материала в определенной степени. Специалистам в данной области понятно, что растворитель не всегда требуется, и реакция может также проводиться в отсутствие растворителя, например, когда основание представляет собой пиридин. Примеры растворителя включают растворители, такие как дихлорметан, хлороформ, 1,2-дихлорэтан, THF, 1,2-диметоксиэтан и толуол, и их смешанные растворители. Реакция может выполняться с использованием от 1 до 20 эквивалентов соответствующего основания в таком растворителе. Примеры используемого основания включают пиридин, 2,6-лутидин, карбонат натрия, карбонат калия и их смеси. Время реакции конкретно не ограничивается и составляет обычно от 0,5 до 24 часов и предпочтительно, от 0,5 до 12 часов. Температура реакции составляет обычно от -78°C до комнатной температуры.
Стадия A-(ix):
Эта стадия представляет собой способ получения соединения A-(10) снятием защиты защитной группы соединения A-(9). Соединение A-(10) может быть получено в условиях снятия защиты, известных специалисту в данной области.
Когда защитная группа представляет собой бензоильную группу, то соединение A-(10) может быть получено в этой реакции нагреванием соединения A-(9) в растворителе, таком как метанол, в присутствии основания, такого как, например, DBU. Эта реакция может выполняться в таких же условиях как условия, описанные, например, в публикации Synth. Commun. 32 (2), 265-272 (2002). Растворитель, используемый в реакции, конкретно не ограничивается, пока он не ингибирует реакцию и обеспечивает возможность растворения в нем исходного материала в определенной степени. Примеры растворителя включают растворители, такие как метанол, этанол и 1-пропанол. Реакция может выполняться с использованием от 1 до 20 эквивалентов соответствующего основания в таком растворителе. Примеры используемого основания включают DBU. Время реакции конкретно не ограничивается и составляет обычно от 0,5 до 24 часов, и предпочтительно, от 0,5 до 12 часов. Температура реакции составляет обычно от комнатной температуры до температуры кипения растворителя.
Альтернативно, соединение A-(10) может быть получено в этой реакции нагреванием соединения A-(9) с неорганическим основанием, таким как, например, карбонат калия. Растворитель, используемый в реакции, конкретно не ограничивается, пока он не ингибирует реакцию и обеспечивает возможность растворения в нем исходного материала в определенной степени. Примеры растворителя включают растворители, такие как метанол, этанол и 1-пропанол. Реакция может выполняться с использованием от 1 до 20 эквивалентов соответствующего основания в таком растворителе, и предпочтительно используется небольшой избыток. Примеры используемого основания включают карбонат калия. Время реакции конкретно не ограничивается и составляет обычно от 0,5 до 24 часов и предпочтительно, от 0,5 до 12 часов. Температура реакции составляет обычно от комнатной температуры до температуры кипения растворителя и предпочтительно, 50-100°C. Специалистам в данной области понятно, что выбранный растворитель ограничивает температуру реакции температурой его кипения. Примеры подходящих растворителей включают кипящий метанол.
Стадия A-(x):
Эта стадия представляет собой стадию получения соединения A-(11) реакцией нитрации соединения A-(10). В этой реакции нитрации соединение A-(11) может быть получено из соединения A-(10) способом, известным специалисту в данной области. Примеры агента нитрации, используемого в реакции, включают нитрат калия/концентрированную серную кислоту, дымящуюся азотную кислоту/концентрированную серную кислоту и дымящуюся азотную кислоту/уксусный ангидрид. Подходящие растворители для реакции включают трифторуксусную кислоту. Температура реакции конкретно не ограничивается и составляет обычно от -20°C до комнатной температуры, и предпочтительные температуры реакции включают от 0°С до 10°C.
Стадия A-(xi):
Эта стадия представляет собой стадию получения соединения A-(12) т-бутоксикарбонилированием аминогруппы соединения A-(11).
Реакция может выполняться в таких же условиях как условия, в целом используемые при т-бутоксикарбонилировании аминосоединения, такие как условия, описанные в документе, таком как T. W. Greene and P. G. M. Wuts, "Protective Groups in Organic Chemistry, Third Edition", John Wiley & Sons (1999), P. 518-525. Соединение A-(12) может быть получено взаимодействием соединения A-(11) с ди-трет-бутилдикарбонатом с использованием растворителя, такого как, например, тетрагидрофуран. Альтернативные растворители включают ацетонитрил и DMF. Специалистам в данной области понятно, что основание может также добавляться к реакционной смеси, хотя это не существенно. Подходящие примеры основания включают без ограничения триэтиламин и диизопропилэтиламин. Температура реакции конкретно не ограничивается и обычно составляет от комнатной температуры до температуры кипения и предпочтительно, от комнатной температуры до 60°C.
Стадия A-(xii):
Эта стадия представляет собой стадию получения соединения A-(13) из соединения A-(12).
Соединение A-(13) синтезируют восстановлением нитро соединения A-(12) способом синтеза, известным специалисту в данной области. Примеры способа включают восстановление каталитическим гидрогенизированием с использованием катализатора из благородного металла, такого как никель Ренея, палладий, рутений, родий или платина. Другие восстанавливающие агенты включают, например, хлорид олова. Примеры растворителя включают спиртовые растворили, такие как метанол, этанол и 1-пропанол, предпочтительно этанол. Время реакции конкретно не ограничивается и составляет обычно от 0,5 до 24 часов, и предпочтительно, от 0,5 до 18 часов. Температурой реакции обычно является комнатная температура. Альтернативные условия реакции восстановления включают реакцию с железом с добавкой, такой как хлорид аммония или хлористоводородная кислота, в спиртовом растворителе, таком как этанол, при соответствующей температуре реакции, например, 65°C.
Стадия A-(xiii):
Эта стадия представляет собой стадию получения соединения A-(14) из соединения A-(13) конденсацией соединения A-(13) с карбоновой кислотой и конденсирующим агентом. Реакция конденсации может выполняться в таких же условиях как условия, обычно используемые и описанные в следующих документах. Примеры известного способа включают те, которые описаны в публикациях Rosowsky et al.; J. Med. Chem., 34 (1), 227-234 (1991), Brzostwska et al.; Heterocycles, 32 (10), 1968-1972 (1991) и Romero et al.; J. Med. Chem., 37 (7), 998-1014 (1994).
Соединение A-(13) может представлять собой свободную форму или соль.
Растворитель в этой реакции конкретно не ограничивается, пока он не ингибирует реакцию. Примеры растворителя включают тетрагидрофуран, 1,4-диоксан, этилацетат, метилацетат, дихлорметан, хлороформ, N,N-диметилформамид, толуол, ацетонитрил и ксилол. Примеры конденсирующего агента включают CDI (N,N'-карбонилдиимидазол), Bop (1H-1,2,3-бензотриазол-1-илокси(три(диметиламино))фосфоний гексафторфосфат), WSC гидрохлорид (1-этил-3-(3-диметиламиопропил)карбодиимида), DCC (N,N-дициклогексилкарбодиимид), диэтилфосфорил цианид, PyBOP (бензотриазол-1-илокситри(пирролидино)фосфоний гексафторфосфат) и EDC·HCl гидрохлорид (1-этил-3-(3-диметиламиопропил)карбодиимида). Подходящие условия включают агент для активации кислоты, такой как N,N'-карбонилдиимидазол. Может использоваться от одного эквивалента до большого избытка кислоты относительно соединения A-(13). При необходимости может добавляться от одного эквивалента до большого избытка органического основания, такого как триэтиламин или N,N-диизопропилэтиламин.
Время реакции конкретно не ограничивается и составляет обычно от 0,5 до 72 часов и предпочтительно, от 0,5 до 24 часов. Температура реакции варьируется в соответствии с используемым сырьевым материалом, растворителем и тому подобным и конкретно не ограничивается. Приемлемая температура от 0°С до температуры кипения растворителя, предпочтительная температура от 0°С до комнатной.
Альтернативно, соединение A-(14) может быть получено превращением желательной карбоновой кислоты в соответствующий хлорид кислоты и затем взаимодействием хлорида кислоты с соединением A-(13). Хлорид кислоты может быть синтезирован способом, известным специалисту в данной области. Например, желательная карбоновая кислота может быть превращена в соответствующий хлорид кислоты взаимодействием с тионилхлоридом в присутствии или в отсутствие растворителя, например, дихлорметана, N,N'-диметилимидазолин-2-она, NMP или DMF. Могут использоваться от одного до двух эквивалентов или большой избыток тионилхлорида относительно желательной карбоновой кислоты. Температура реакции составляет от -30°C до температуры кипения и предпочтительно, от -10°C до комнатной температуры. Хлорид кислоты может быть также образован обработкой кислоты оксалилхлоридом в растворителе, таком как дихлорметан, в присутствии DMF. Температура реакции составляет от -30°C до комнатной температуры и предпочтительно, от -10°C до комнатной температуры.
Альтернативно, соединение A-(14) может быть получено превращением желательной карбоновой кислоты в смешанный ангидрид кислоты и затем взаимодействием смешанного ангидрида кислоты с соединением A-(13). Смешанный ангидрид кислоты может быть синтезирован средством, известным специалисту в данной области. Синтез выполняют взаимодействием желательной карбоновой кислоты с хлорформиатом, таким как этилхлорформиат, в присутствии основания, такого как, например, триэтиламин. От одного до двух эквивалентов хлорформиата и основания используют относительно желательной карбоновой кислоты. Температура реакции составляет от -30°C до комнатной температуры и предпочтительно, от -20°C до комнатной температуры.
Стадию конденсации смешанного ангидрида кислоты с соединением 1-(13) выполняют взаимодействием смешанного ангидрида кислоты с соединением 1-(13) в растворителе, таком как, например, дихлорметан, тетрагидрофуран или N,N-диметилформамид. Используют от одного эквивалента до большого избытка желательной карбоновой кислоты относительно соединения A-(13).
Время реакции конкретно не ограничивается и составляет обычно от 0,5 до 48 часов и предпочтительно, от 0,5 до 12 часов. Температура реакции составляет от -20°C до 50°C и предпочтительно, от -20°C до комнатной температуры.
Альтернативно, соединение A-(14) может быть получено превращением желательной карбоновой кислоты в активный сложный эфир и затем взаимодействием активного сложного эфира с соединением A-(13). Стадию получения активного сложного эфира выполняют взаимодействием желательной карбоновой кислоты с реагентом синтеза активного сложного эфира в растворителе, таком как 1,4-диоксан, тетрагидрофуран или N,N-диметилформамид, в присутствии конденсирующего агента, такого как, например, DCC. Примеры реагента синтеза активного сложного эфира включают N-гидроксисукцинимид. Используют от одного до 1,5 эквивалентов реагента синтеза активного сложного эфира относительно соединения A-(13). Время реакции конкретно не ограничивается и составляет обычно от 0,5 до 48 часов и предпочтительно, от 0,5 до 24 часов.
Температура реакции составляет от -20°C до 50°C и предпочтительно, от -20°C до комнатной температуры.
Стадию конденсации активного сложного эфира с соединением A-(13) выполняют взаимодействием активного сложного эфира с соединением A-(13) в растворителе, таком как, например, дихлорметан, тетрагидрофуран или N,N-диметилформамид. Используют от одного эквивалента до большого избытка активного сложного эфира относительно соединения A-(13). Время реакции конкретно не ограничивается и составляет обычно от 0,5 до 48 часов и предпочтительно, от 0,5 до 24 часов. Температура реакции составляет от -20°C до 50°C и предпочтительно, от -20°C до комнатной температуры.
Стадия A-(xiv):
Эта стадия представляет собой стадию получения соединения A-(15) снятием защиты т-бутоксикарбонильной группы соединения A-(14).
Реакция может выполняться в таких же условиях как условия, в целом используемые при реакции снятия защиты т-бутоксикарбонильной группы, такие как условия, описанные в документе, таком как T. W. Greene and P. G. M. Wuts, "Protective Groups in Organic Chemistry, Third Edition", John Wiley & Sons (1999), P. 518-525. Соединение A-(15) может быть получено взаимодействием соединения 1-(14) с сильной кислотой, например, трифторуксусной кислотой, в присутствии или в отсутствие растворителя. Подходящие растворители включают дихлорметан. Альтернативные кислоты включают хлористоводородную кислоту в подходящих растворителях, таких как, например, дихлорметан или диоксан.
Температура реакции обычно составляет от 0°С до 80°C, предпочтительно, комнатная температура. Время реакции конкретно не ограничивается и составляет обычно от 5 минут до 48 часов и предпочтительно, от 5 минут до 12 часов.
B. Общий способ получения B:
В формуле, X, Y и A обозначают, как определено выше.
Общий способ получения B представляет собой альтернативный способ получения соединения A-(15), которое соответствует соединению (I) в соответствии с настоящим изобретением, из соединения A-(11) в качестве сырьевого материала посредством множественных стадий со стадии B-(i) до стадии B-(ii).
Соединение A-(11) может быть получено, как описано в Общем способе получения A или примерах.

Стадия B-(i):
Эта стадия представляет собой стадию получения соединения A-(16) из соединения A-(11).
Соединение A-(16) синтезируют восстановлением нитро соединения A-(11) способом синтеза, известным специалисту в данной области. Примеры способа включают восстановление каталитическим гидрогенизированием с использованием катализатора в виде благородного металла, такого как никель Ренея, палладий, рутений, родий или платина. Другие восстанавливающие реагенты включают, например, хлорид олова. Примеры растворителя включают спиртовые растворители, такие как метанол, этанол и 1-пропанол, предпочтительно этанол. Время реакции конкретно не ограничивается и составляет обычно от 0,5 до 24 часов и предпочтительно, от 0,5 до 18 часов. Температура реакции обычно представляет собой комнатную температуру. Альтернативные условия реакции восстановления включают взаимодействие с железом с добавкой, такой как хлорид аммония или хлористоводородная кислота, в спиртовом растворителе, таком как этанол, при соответствующей температуре реакции, например, 65°C.
Стадия B-(ii):
Эта стадия представляет собой стадию получения соединения A-(15) из соединения A-(16) конденсацией соединения A-(13) с карбоновой кислотой и конденсирующим агентом. Реакция конденсации может выполняться в таких же условиях как условия, обычно используемые и описанные в следующих документах. Примеры известного способа включают те, которые описаны в публикациях Rosowsky et al.; J. Med. Chem., 34 (1), 227-234 (1991), Brzostwska et al.; Heterocycles, 32 (10), 1968-1972 (1991) и Romero et al.; J. Med. Chem., 37 (7), 998-1014 (1994).
Соединение A-(15) может быть получено превращением желательной карбоновой кислоты в соответствующий хлорид кислоты и затем взаимодействием хлорида кислоты с соединением A-(16). Хлорид кислоты может быть синтезирован средством, известным специалисту в данной области. Например, желательная карбоновая кислота может превращаться в соответствующий хлорид кислоты взаимодействием с тионилхлоридом в присутствии или в отсутствие растворителя, например, дихлорметана, N,N'-диметилимидазолин-2-она, NMP или DMF. От одного до двух эквивалентов или большой избыток тионилхлорида может использоваться относительно желательной карбоновой кислоты. Специалистам в данной области понятно, что выбор используемых условий реакции может воздействовать на исход реакции, например, условия могут воздействовать на то, будет ли хлорид кислоты взаимодействовать с анилиновой или изотиомочевинной составляющими. Специалистам в данной области понятно, что взаимодействие тионилхлорида с карбоновой кислотой приводит к сопутствующему образованию 1 эквивалента хлористоводородной кислоты в дополнение к образованию желательного хлорида кислоты. Специалистам в данной области понятно, что в современных условиях не используется способ удаления образованной таким образом хлористоводородной кислоты. Хлористоводородная кислота, образованная в этой реакции, может воздействовать или не воздействовать на избирательность реакции, которые могут привести или не привести к благоприятному исходу. Время реакции конкретно не ограничивается и составляет обычно от 0,5 до 48 часов и предпочтительно, от 0,5 до 12 часов. Температура реакции составляет от -30°C до дефлегмации и предпочтительно, от -10°C до комнатной температуры. Хлорид кислоты может быть также образован обработкой кислоты оксалилхлоридом в растворителе, таком как дихлорметан, в присутствии DMF. Температура реакции составляет от -30°C до комнатной температуры и предпочтительно, от -10°C до комнатной температуры.
C. Общий способ получения C:
Общий способ получения C представляет собой альтернативный способ получения соединения A-(2), которое представляет собой синтетическое промежуточное соединение соединения (I) в соответствии с настоящим изобретением из соединения A-(17) в качестве сырьевого материала посредством стадии C-(i).
Соединение A-(17) имеется в продаже.
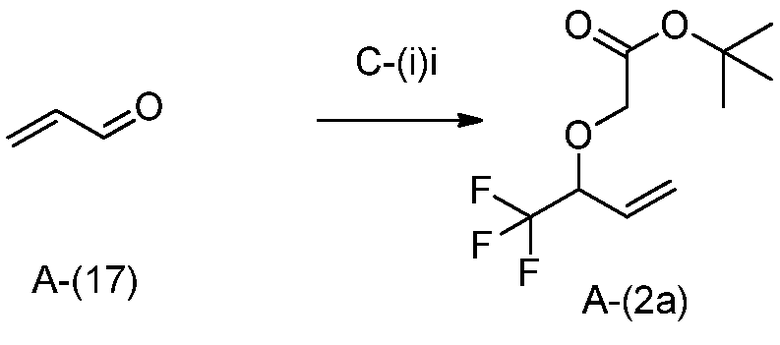
Стадия i:
Эта стадия представляет собой стадию получения соединения A-(2a) из A-(17) добавлением трифторметильного аниона к соединению A-(17) для получения промежуточного аллильного алкоксида или промежуточного простого триметилсилильного эфира, который затем алкилируется для получения соединения A-(2a). Специалистам в данной области понятно, что эта трансформация может проводиться одностадийной реакцией или в виде двух отдельных реакций. Специалистам в данной области понятны преимущества и недостатки одностадийной реакции, по сравнению с проведением отдельных реакций, и, соответственно, выбор наилучшего способа для их потребностей.
В частности, акролеин A-(17) может взаимодействовать с трифторметильным анионом, который может быть получен действием фторида на реагенты, такие как (трифторметил)триметилсилан, для получения соответствующего аллильного алкоксида или аллильного простого триметилсилильного эфира. Растворитель, используемый в реакции, конкретно не ограничивается, пока он не препятствует реакции. Примеры подходящих растворителей включают THF. Приемлемые диапазоны температуры для реакции включают от -10°C до температуры кипения растворителя, предпочтительно, ниже комнатной температуры. Специалистам в данной области понятно, что определенные химические реакции могут быть экзотермическими, и что следует применять меры по контролю над этими экзотермами. Специалистам в данной области понятно, что контроль над экзотермой реакции возможен предоставлением растворителю возможности возвращать обратно флегму. Подходящие предшественники для получения трифторметильного аниона включают без ограничения (трифторметил)триметилсилан (реагент Руперта, Chem Rev 1997, 97, 757), и подходящие источники фторида включают без ограничения тетрабутиламмоний фторид (TBAF), тетрабутиламмоний дифтортрифенилсиликат (TBAT) и фторид цезия. Хотя начальная температура реакции может быть ниже комнатной температуры, приемлемо допущение достижения температуры кипения растворителя во время хода реакции для содействия реакции. Время реакции конкретно не ограничивается и составляет обычно от 5 минут до 24 часов, предпочтительно, 1-6 часов.
Специалистам в данной области понятно, что алкоксид, полученный в результате этой реакции, может непосредственно взаимодействовать с алкилирующим агентом, таким как трет-бутилбромацетат, и что эта реакция может протекать с дополнительными растворителями или без них. Если требуются дополнительные растворители для содействия реакции, то подходят такие растворители как DMF или NMP. Температура реакции конкретно не ограничивается. Подходящие температуры реакции включают от комнатной температуры до 80°C, предпочтительно, комнатную температуру. Время реакции конкретно не ограничивается и составляет обычно от 5 минут до 1 недели, предпочтительно, от 1 до 48 часов. Альтернативно, реакция алкилирования может проводиться в условиях фазового перехода, например, добавлением водного основания, например, водного гидроксида натрия. Специалистам в данной области понятно, что когда применяются эти условия, то требуется использование катализатора фазового перехода. Подходящие катализаторы фазового перехода включают без ограничения гидросульфат тетрабутиламмония.
Специалистам в данной области понятно, что промежуточный алкоксид может быть погашен, выделен и очищен, затем подвергнут условиям независимого алкилирования. Эта реакция может выполняться в таких же условиях как условия, обычно используемые при реакции O-алкилирования спиртового соединения (такие как условия, описанные в публикации Tetrahedron Lett. 46 (2005) 45, 7751-7755). При этой реакции соединение A-(2a) может быть получено добавлением основания, такого как гидрид натрия, к раствору промежуточного спирта в THF для получения алкоксида, и затем взаимодействием алкоксида, например, с трет-бутилбромацетатом. Растворитель, используемый в реакции, конкретно не ограничивается, пока он не ингибирует реакцию и обеспечивает возможность растворения в нем исходного материала в определенной степени. Примеры растворителя включают растворители, такие как THF, DMF и диметилсульфоксид. Реакция может выполняться воздействием от 1 до 3 эквивалентов соответствующего основания в присутствии такого растворителя. Примеры используемого основания включают гидрид натрия, гидрид калия и т-бутоксикалий. Время реакции конкретно не ограничивается и составляет обычно от 0,5 до 72 часов и предпочтительно, от 0,5 доо 12 часов. Температура реакции составляет обычно от -20°C до 50°C.
Более предпочтительный результат, такой как улучшенный выход, может быть достигнут добавлением соли, такой как тетрабутиламмоний йодид, при этой реакции.
Специалистам в данной области понятно, что эта реакция создает новый хиральный центр в соединении A-(2a), и что соединение A-(2a) является таким же как соединение A-(2), за исключением того, что соединение A-(2) является энантиомерно чистым, тогда как соединение A-(2a) является рацемическим. Специалистам в данной области понятно, что энантиомерно чистое соединение A-(2) и рацемическое соединение A-(2a) не различаемы аналитическими методиками, такими как ЯМР и жидкостная хроматография, однако они могут различаться хиральной ВЭЖХ. Специалистам в данной области известны наиболее целесообразные способы для получения желательного энантиомера из соединения A-(2a) или более продвинутого синтетического промежуточного соединения или конечного соединения. Соответствующие способы и соответствующие стадии энантиомерной очистки включают те, которые подробно описаны в примерах.
В еще одном аспекте, настоящее изобретение относится к способу получения соединения формулы (I), который включает взаимодействие соединения формулы A-(16), где X определен в формуле (I), с соединением формулы (II), где A и Y обозначают, как определено в формуле (I), или его сложный C1-6 алкиловый эфир, ангидрид кислоты или галоидангидрид, для выхода соединения формулы (I), и, необязательно, превращение соединения в дополнительное соединение формулы (I) или образование его фармацевтически приемлемой соли.

Реакция соединений A-(16) и (II) может для удобства проводиться в растворителе (таком как тетрагидрофуран, 1,4-диоксан, этилацетат, метилацетат, дихлорметан, хлороформ, N,N-диметилформамид, толуол, ацетонитрил или ксилол) при температуре в диапазоне от -30°C до 100°C. В одном варианте осуществления изобретения соединение (II) может для удобства принимать форму галоидангидрида, (например, хлорида), который может быть получен взаимодействием кислоты с подходящим реагентом (например, тионилхлоридом).
Специалистам в данной области понятно, что в способе по настоящему изобретению может потребоваться защита защитными группами определенных функциональных групп, таких как гидроксильные, карбоксильные или аминогруппы в исходных реагентах. Таким образом, получение соединений формулы (I) может дополнительно вовлекать включение и удаление одной или более защитных групп. Защита и снятие защиты функциональных групп, например, описано в руководстве T. W. Greene and P. G. M. Wuts, "Protective Groups in Organic Chemistry, Third Edition", John Wiley & Sons (1999).
Настоящее изобретение будет более конкретно описано ниже со ссылкой на Примеры, Примеры получения и Пример тестирования. Однако настоящее изобретение не ограничивается ими. Аббревиатуры, используемые в Примерах, представляют собой обычные аббревиатуры, известные специалисту в данной области. Некоторые аббревиатуры показаны ниже:
LCMS, LC/MS и LC-MS (жидкостная хроматография/масс спектрометрия); MS (масс спектрометрия); MDAP (масс-направленная автоочистка; ЯМР (ядерный магнитный резонанс); с, д, т, дд, м, шир. (синглет, дублет, триплет, дублет дублетов, мультиплет, широкий); Ph, Me, Et, Pr, Bu, Bn (фенил, метил, этил, пропил, бутил, бензил); THF (тетрагидрофуран); DCM (дихлорметан); DMF (N,N-диметилформамид); h, hr, hrs (часы); EDC и EDAC гидрохлорид (N-3-(диметиламиопропил)-N'этилкарбодиимида); диметилсульфоксид); UV (ультрафиолетовое излучение); КТ и кт (комнатная температура); ТК (время удерживания); мин (минуты); EtOAc (этилацетат); Et2O (простой диэтиловый эфир); MeCN (ацетонитрил); EtOH (этанол); MeOH (метанол); PhCH3 и PhMe (толуол); tlc (тонкослойная хроматография); TFA (трифторуксусная кислота); NaOH (гидроксид натрия); HCl (хлористоводородная кислота); NMP (N-метилпирролидинон или 1-метил-2-пирролидинон); HPLC (ВЭЖХ, высокоэффективная жидкостная хроматография); TBAF (тетрабутиламмоний фторид); BuLi (n-бутиллитий); PyBOP: бензотриазол-1-илокситрис(пирролидино)фосфоний гексафторфосфат; Pd2dba3: трис(дибензилиденацетон)дипалладий; Pd(t-Bu3P)2: бис(три-т-буилфосфин)палладий; TFA: трифторуксусная кислота; pTLC: препаративная тонкослойная хроматография; HRMS (масс спектрометрия с высоким разрешением); Tr или TKT (тритил или трифенилметил).
1H-ЯМР спектры регистрировали на спектрометре Bruker серии AM, работающем при (представленной) частоте 400 МГц. Химические сдвиги в спектрах протонного ядерного магнитного резонанса регистрируются в единицах δ (м.д.) относительно тетраметилсилана, а константы соединения (J) регистрируются в герцах (Hz, Гц). Типы обозначаются как с: синглет, д: дублет, т; триплет, шир.; широкий.
«Комнатная температура» в следующих Примерах и Примерах получения обычно относится к диапазону от примерно 10°C до примерно 35°C. Пока нет других определений, «%» указывает % масс.
Химические названия были образованы из химических структур с использованием программного обеспечения ChemBioDraw Ultra 11.0 и 12.0.
Описание чертежей:
Фиг. 1 представляет типичную хроматограмму по хиральному ВЭЖХ выделению соединения 1-(20).
Пример получения 1
Синтез трет-бутил-((4aS,5S,7aS)-7a-(5-амино-2-фторфенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил)карбамата 1-(13)
1-(2) Синтез трет-бутил-{[(2S)-1,1,1-трифторбут-3-ен-2-ил]окси}ацетата
К суспензии триметилсульфония йодида (110 г) в THF (500 мл) при -30°C порциями добавляли гексаметилдисилазид лития (530 мл, 1н в THF) в течение 45 мин. После перемешивания при -20°C в течение 20 мин, добавляли (S)-2-трифторметилоксиран (37,97 г) при такой же температуре в течение 15 мин, и смеси давали возможность нагреться до КТ и перемешивали в течение 3 час. Затем кашицеобразную суспензию порциями добавляли к охлажденному до 0°С раствору трет-бутил бромацетата (105,68 г) в NMP (200 мл). Полученной смеси давали возможность нагреться до КТ и перемешивали в течение 2 дней перед разбавлением EtOAc (1 л). Органический слой промывали бакарбонатом натрия (насыщ., водн., 4×400 мл), сушили над MgSO4 и выпаривали. Остаток очищали колоночной хроматографией на силикагеле (5% EtOAc в гексанах) для получения указанного в заголовке соединения (70,1 г), которое использовали на последующей стадии без очистки. 1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,30 (с, 9 H) 3,83-3,96 (м, 2 H) 4,14-4,21 (м, 1 H) 5,34-5,48 (м, 2 H) 5,56-5,71 (м, 1 H).


1-(3) Синтез (S)-N-метокси-N-метил-2-((1,1,1-трифторбут-3-ен-2-ил)окси)ацетамида
Трет-бутил-{[(2S)-1,1,1-трифторбут-3-ен-2-ил]окси}ацетат (70,1 г, неочищенный) растворяли в охлажденной до 0°С муравьиной кислоте (200 мл). Смеси давали возможность нагреться до КТ и перемешивали в течение ночи. Реакционную смесь затем концентрировали под пониженным давлением, добавляли толуол (200 мл), смесь концентрировали перед вторым добавлением толуола (200 мл) и концентрацией в масло. Остаток растворяли в DCM (600 мл), охлаждали в бане со льдом и порциями добавляли N,N'-карбонилдиимидазол (35 г) в течение 20 мин. После перемешивания в течение 45 мин, добавляли гидрохлорид N,O-диметилгидроксиламина (22 г), и реакционной смеси давали возможность нагреться до КТ и перемешивали в течение ночи. Затем добавляли насыщенный NaHCO3 (500 мл) и рассол (250 мл), и смесь экстрагировали EtOAc (3×750 мл). Объединенные органические части сушили над MgSO4 и выпаривали. Остаток очищали колоночной хроматографией на силикагеле (1% до 30% EtOAc в гексанах) для получения указанного в заголовке соединения (25,17 г). 1H-ЯМР (400 МГц, CDCl3) δ (м.д.) 3,21 (с, 3 H), 3,71 (м, 3 H), 4,36-4,51 (м, 3 H), 5,54-5,69 (м, 2 H), 5,84 (ддд, J=17,7, 10,4, 7,3 Гц, 1 H).
1-(4) Синтез (S)-1-(2-фторфенил)-2-((1,1,1-трифторбут-3-ен-2-ил)окси)этанона
Раствор н-бутиллития в гексане (2,50 M; 90 мл) добавляли по каплям в течение 25 мин к раствору 2-бромфторбензол (40,35 г) в THF (250 мл) в атмосфере N2 при -78°C. Реакционному раствору давали возможность нагреться до -60°C и перемешивали в течение 60 мин. К реакционному раствору по каплям добавляли (S)-N-Метокси-N-метил-2-((1,1,1-трифторбут-3-ен-2-ил)окси)ацетамид (40 г) в THF (25 мл), и после перемешивания при -60°C в течение 2 часов, к реакционному раствору добавляли водный NH4Cl (100 мл) с последующим нагреванием до КТ. К реакционному раствору добавляли рассол (200 мл), и смесь экстрагировали EtOAc (3×400 мл). Объединенные органические части сушили над MgSO4, выпаривали, и остаток очищали колоночной хроматографией на силикагеле (от 1% до 10% EtOAc в гексанах) для получения указанного в заголовке соединения (33,59 г). 1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 4,40 (пентет, J=6,3 Гц, 1 H) 4,81-4,87 (м, 2 H), 5,54-5,69 (м, 2 H), 5,86 (ддд, J 17,4, 10,4, 7,3 Гц, 1 H) 7,12-7,22 (м, 1 H) 7,24-7,34 (м, 1 H) 7,54-7,63 (м, 1 H) 7,94-8,02 (м, 1 H).
1-(5) Синтез (S)-1-(2-фторфенил)-2-((1,1,1-трифторбут-3-ен-2-ил)окси)этаноноксима
(S)-1-(2-Фторфенил)-2-((1,1,1-трифторбут-3-ен-2-ил)окси)этанон (41,22 г) растворяли в безводном метаноле (400 мл) и добавляли гидрохлорид гидроксиламина (14,0 г) и ацетат натрия (19,0 г). Реакционную смесь нагревали до 50°C в течение 90 мин, затем охлаждали до КТ, концентрировали в вакууме и остаток очищали хроматографией на силикагеле (от 2% до 15% EtOAc в гексанах) для получения указанного в заголовке соединения в виде смеси геометрических изомеров (40,54 г). 1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 4,04-4,15 (м, 0,8 H), 4,18-4,26 (м, 0,2 H), 4,44-4,57 (м, 0,4 H) 4,79-4,90 (м, 1,6 H) 5,37-5,56 (м, 2 H) 5,64-5,78 (м, 1 H) 7,03-7,26 (м, 2 H) 7,33-7,54 (м, 2 H), 7,90 (шир. с, 0,2 H), 8,51 (шир. с, 0,8 H).
1-(6) Синтез (3aR,4S,6aS)-6a-(2-фторфенил)-4-(трифторметил)гексагидрофуро[3,4-c]изоксазола
(S)-1-(2-фторфенил)-2-((1,1,1-трифторбут-3-ен-2-ил)окси)этаноноксим (40,54 г) растворяли в ксилолах (400 мл) и добавляли гидрохинон (4,0 г). Реакционную смесь нагревали в сосуде с обратным холодильником (температура нагревателя 140°C) в течение 22 часов, затем охлаждали и выпаривали. Остаток очищали колоночной хроматографией на силикагеле (от 1% до 30% EtOAc в гексанах) для получения указанного в заголовке соединения (28,76 г). 1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 3,71-3,81 (м, 1 H), 4,04-4,35 (м, 4 H), 4,51-4,62 (м, 1 H), 5,38-5,54 (м, 1 H), 7,07-7,26 (м, 2 H), 7,32-7,42 (м, 1 H), 7,54-7,67 (м, 1 H).
1-(7) ((2S,3R,4S)-4-амино-4-(2-фторфенил)-2-(трифторметил)-тетрагидрофуран-3-ил)метанол
(3aR,4S,6aS)-6a-(2-фторфенил)-4-(трифторметил)-гексагидрофуро[3,4-c]изоксазол (28,76 г) растворяли в уксусной кислоте (200 мл), и раствор охлаждали до 0°C. Добавляли цинк (50 г), и реакционной смеси давали возможность нагреться и перемешивали при КТ в течение 16 часов. Затем реакционную смесь разбавляли EtOAc (500 мл) и фильтровали через целит, промывая дополнительными 500 мл EtOAc. Объединенные органические части выпаривали, растворяли в хлороформе (200 мл) и медленно добавляли аммиак (28% водн., 250 мл). Слои разделяли, и водную часть дополнительно экстрагировали хлороформом (2×250 мл). Объединенные органические экстракты сушили над безводным MgSO4 и выпаривали для получения указанного в заголовке соединения (31,12 г), которое использовали на последующей стадии без дополнительной очистки. 1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,93 (ддд, J=7,7, 4,9, 2,5 Гц, 1 H), 3,84 (дд, J=12,4, 4,8 Гц, 1 H), 4,05 (дд, J=9,2, 3,2 Гц, 1 H), 4,17 (дд, J=12,4, 2,3 Гц, 1 H), 4,31 (д, J=9,3 Гц, 1 H), 4,72 (квин., J=7,3 Гц, 1 H), 7,13 (ддд, J=13,1, 8,8, 1,3 Гц, 1 H), 7,22 (тд, J=7,6, 1,3 Гц, 1 H), 7,31-7,40 (м, 1 H), 7,51 (тд, J=8,0, 1,6 Гц, 1 H).
1-(8) Синтез N-(((3S,4R,5S)-3-(2-фторфенил)-4-(гидроксиметил)-5-(трифторметил)тетрагидрофуран-3-ил)карбамотиоил)бензамида
Бензоилизотиоцианат (19,0 мл) добавляли к раствору, содержащему ((2S,3R,4S)-4-амино-4-(2-фторфенил)-2-(трифторметил)тетрагидрофуран-3-ил)метанол (28,72 г) в DCM (150 мл), и смесь перемешивали при КТ в течение 18 ч. Затем добавляли бикарбонат натрия (насыщ., водн., 200 мл), смесь экстрагировали EtOAc (3×300 мл), сушили над MgSO4 и концентрировали под пониженным давлением. Остаток очищали колоночной хроматографией на силикагеле (от 5% до 30% EtOAc в гексанах) для получения указанного в заголовке соединения (37,07 г). 1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 3,22 (дд, J=8,1, 4,5 Гц, 1 H), 3,31 (тд, J=8,0, 3,0 Гц, 1 H), 3,94-4,07 (м, 1 H), 4,31-4,46 (м, 1 H), 4,53 (д, J=9,9 Гц, 1 H), 4,83 (д, J=9,9 Гц, 1 H), 6,97-7,14 (м, 1 H), 7,22 (тд, J=7,7, 1,3 Гц, 1 H), 7,31-7,45 (м, 1 H), 7,49-7,61 (м, 2 H), 7,61-7,70 (м, 1 H), 7,75 (тд, J=8,1, 1,5 Гц, 1 H), 7,79-7,93 (м, 2 H), 8,90 (с, 1 H), 11,85 (с, 1 H).
1-(9) Синтез N-((4aS,5S,7aS)-7a-(2-фторфенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил)бензамида
N-(((3S,4R,5S)-3-(2-Фторфенил)-4-(гидроксиметил)-5-(трифторметил)тетрагидрофуран-3-ил)карбамотиоил)бензамид (31,1 г) растворяли в пиридине (150 мл), и смесь охлаждали до -20°C. Трифторметансульфоновый ангидрид (14,0 мл) по каплям добавляли в течение 30 мин, и реакционной смеси давали возможность нагреться до 0°C. После перемешивания в течение 2 часов, реакцию гасили добавлением аммония хлорида (насыщенного, водного, 400 мл) и экстрагировали EtOAc (3×500 мл). Объединенные органические экстракты сушили над MgSO4, концентрировали в вакууме и очищали колоночной хроматографией на силикагеле (от 2% до 30% EtOAc/гексаны) для получения указанного в заголовке соединения (18,50 г). 1H-ЯМР (400 МГц, CDCl3) δ м.д. 2,86 (дд, J=13,9, 3,5 Гц, 1H), 3,25 (д, J=13,6 Гц, 1 H), 3,61 ((шир. с, 1 H), 4,00-4,10 (м, 1 H), 4,66 (д, J=8,8 Гц, 1 H), 4,78-4,87 (м, 1 H), 7,12-7,60 (м, 6 H), 7,68-7,73 (м, 1 H), 7,99-8,16 ((шир. с, 2 H), 8,62-8,66 (м, 1 H).
1-(10) Синтез N-((4aS,5S,7aS)-7a-(2-фторфенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил)бензамида
N-((4aS,5S,7aS)-7a-(2-Фторфенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил)бензамид (21,5 г) растворяли в метаноле (160 мл), добавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (16,29 г), и раствор нагревали в сосуде с обратным холодильником (температура нагревателя 80°C). Через 16 часов реакционную смесь концентрировали под пониженным давлением, и остаток очищали колоночной хроматографией на силикагеле (от 10% до 60% EtOAc в гексанах) для получения указанного в заголовке соединения (13,82 г). 1H-ЯМР (400 МГц, CDCl3) δ м.д. 2,85 (дд, J=13,6, 3,8 Гц, 1 H), 3,14 (дд, J=13,5, 3,2 Гц, 1 H), 3,33-3,45 (м, 1 H), 3,92 (дд, J=8,1, 2,0 Гц, 1 H), 4,49 ((шир. с, 2 H), 4,63-4,76 (м, 2 H), 7,08 (ддд, J=12,6, 8,1, 1,0 Гц, 1 H), 7,13-7,22 (м, 1 H), 7,25-7,36 (м, 1 H), 7,44 (тд, J=8,0, 1,9 Гц, 1 H).
1-(11) Синтез (4aS,5S,7aS)-7a-(2-фтор-5-нитрофенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-амина
N-((4aS,5S,7aS)-7a-(2-Фторфенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил)бензамид (5,15 г) растворяли в TFA (75 мл), и раствор охлаждали до 0°C. Добавляли серную кислоту (концентрированную, 20 мл) с последующим добавлением по каплям дымящейся азотной кислоты (2 мл) в течение 20 мин. После перемешивания при 0°C в течение 90 мин, реакционную смесь выливали в лед (200 г) и повышали основность до pH 12 6н NaOH (водн.). После предоставления возможности льду растаять, смесь экстрагировали EtOAc (3×500 мл), и объединенные органические части сушили над MgSO4 и выпаривали для получения указанного в заголовке соединения (22,1 г, чистота приблизительно 71%), которое использовали на последующей стадии без очистки. 1H-ЯМР (400 МГц, CDCl3) δ м.д. 2,89 (д, J=3,8 Гц, 1 H), 3,09 ((шир. с, 1 H), 3,28-3,54 (м, 1 H), 3,80-4,03 (м, 1 H), 4,50-4,70 (м, 3 H), 4,71-4,86 (м, 1 H), 7,21-7,30 (м, 1 H), 8,18-8,28 (м, 1 H), 8,45 (дд, J=6,8, 2,8 Гц, 1 H).
1-(12) Синтез трет-бутил-((4aS,5S,7aS)-7a-(2-фтор-5-нитрофенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил)карбамата
(4aS,5S,7aS)-7a-(2-Фтор-5-нитрофенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-амин (20,6 г, неочищенный) растворяли в THF (300 мл), порциями добавляли ди-трет-бутил дикарбонат (12 г) в течение 20 мин, и реакционную смесь нагревали до 60°C. Добавляли дополнительные порции ди-трет-бутил дикарбоната (10 г) до тех пор, пока исходный материал не расходовался TLC. Реакционную смесь охлаждали и добавляли бикарбонат натрия (насыщенный, водный, 200 мл) и рассол (200 мл). Затем смесь экстрагировали EtOAc (3×500 мл) и объединенные органические порции сушили над MgSO4 и выпаривали. Остаток очищали колоночной хроматографией на силикагеле (от 10% до 25% EtOAc в гексанах) для получения указанного в заголовке соединения (16,62 г). 1H-ЯМР (400 МГц, CDCl3) δ м.д. 1,55 (с, 9 H), 2,73-2,84 (м, 1 H), 2,92-3,05 (м, 1 H), 3,43-3,55 (м, 1 H), 3,81-3,94 (м, 1 H), 4,57 (д, J=8,3 Гц, 1 H), 4,73-4,83 (м, 1 H), 7,19-7,39 (м, 2 H), 8,20-8,29 (м, 1 H), 8,32 (д, J=6,8 Гц, 1 H).
1-(13) Синтез трет-бутил-((4aS,5S,7aS)-7a-(5-амино-2-фторфенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил)карбамата
Трет-бутил-((4aS,5S,7aS)-7a-(2-фтор-5-нитрофенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил)карбамат (16,61 г) растворяли в этаноле (250 мл) и добавляли дигидрохлорид олова (25,0 г). После перемешивания при КТ в течение 18 часов раствор выливали на NaOH (2н водн., 300 мл) и добавляли целит® (~50 г). Полученную смесь фильтровали еще через целит® и экстрагировали EtOAc (2×500 мл). Объединенные органические порции сушили над MgSO4 и выпаривали для получения указанного в заголовке соединения (15,52 г). Этот материал можно было использовать неочищенным, но порцию очищали колоночной хроматографией на силикагеле (от 20% до 50% EtOAc в гексанах) для получения чистого материала (извлечение 79%). 1H-ЯМР (400 МГц, CDCl3) δ м.д. 1,53 (с, 9H), 2,77 (д, J=14,4 Гц, 1H), 3,09 ((шир. с, 1H), 3,46 ((шир. с, 1H), 3,62 ((шир. с, 2H), 3,87 ((шир. с, 1H), 4,61 (д, J=8,6 Гц, 1H), 4,71 ((шир. с, 1H), 6,61 ((шир. с, 2H), 6,85-6,95 (м, 1H).
Пример получения 2
Синтез трет-бутил-((4aS,5S,7aS)-7a-(5-амино-2-фторфенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил)карбамата 2-(10)

2-(1) Синтез (S)-1-(2,3-дифторфенил)-2-((1,1,1-трифторбут-3-ен-2-ил)окси)этанона
Раствор н-бутиллития в гексане (2,50 M, 13,5 мл) по каплям добавляли в течение 20 мин к раствору, содержащему 1-бром-2,3-дифторбензол (6,50 г) в Et2O (50 мл) в атмосфере N2 при -78°C. Реакционному раствору давали возможность перемешиваться в течение 60 мин. Затем к реакционному раствору по каплям добавляли (S)-N-Метокси-N-метил-2-((1,1,1-трифторбут-3-ен-2-ил)окси)ацетамид) (5,20 г) в Et2O (10 мл), и после перемешивания при -78°C в течение 1 часа, к реакционному раствору добавляли водный NH4Cl (50 мл) с последующим нагреванием до КТ. К реакционному раствору добавляли NaHCO3 (насыщенный водный, 100 мл), и смесь экстрагировали EtOAc (3×100 мл). Объединенные органические порции сушили над MgSO4, выпаривали, и остаток очищали колоночной хроматографией на силикагеле (от 1% до 10% EtOAc в гексанах) для получения указанного в заголовке соединения (3,91 г). 1H-ЯМР (400 МГц, CDCl3) δ м.д.: 4,33-4,43 (м, 1 H), 4,80-4,84 (м, 2 H), 5,55-5,67 (м, 2 H), 5,76-5,94 (м, 1 H), 7,18-7,28 (м, 1 H), 7,37-7,47 (м, 1 H), 7,70 (ддт, J=7,9, 6,0, 1,7 Гц, 1 H).
2-(2) Синтез (S)-1-(2,3-дифторфенил)-2-((1,1,1-трифторбут-3-ен-2-ил)окси)этаноноксима
(S)-1-(2,3-дифторфенил)-2-((1,1,1-трифторбут-3-ен-2-ил)окси)этанон (3,91 г) растворяли в безводном метаноле (40 мл) и добавляли гидрохлорид гидроксиламина (1,25 г) и ацетат натрия (1,68 г). Реакционную смесь нагревали до 50°C в течение 90 мин, затем охлаждали до КТ, концентрировали в вакууме, и остаток очищали хроматографией на силикагеле (от 2% до 20% EtOAc в гексанах) для получения указанного в заголовке соединения в виде смеси геометрических изомеров (4,10 г). 1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 4,04-4,26 (м 1 H), 4,43-4,55 (м, 0,4 H) 4,80-4,89 (м, 1,6 H) 5,39-5,55 (м, 2 H) 5,64-5,80 (м, 1 H) 7,05-7,30 (м, 3 H), 7,76 (шир. с, 0,2 H), 8,30 (шир. с, 0,8 H).
2-(3) Синтез (4S)-6a-(2,3-дифторфенил)-4-(трифторметил)-гексагидрофуро[3,4-c]изоксазола
(S)-1-(2,3-дифторфенил)-2-((1,1,1-трифторбут-3-ен-2-ил)окси)этаноноксим (4,10 г) растворяли в ксилолах (40 мл) и добавляли гидрохинон (380 мг). Реакционную смесь нагревали в сосуде с обратным холодильником (температура нагревателя 140°C) в течение 20 часов, затем охлаждали и выпаривали. Остаток очищали колоночной хроматографией на силикагеле (от 1% до 25% EtOAc в гексанах) для получения указанного в заголовке соединения (3,16 г). 1H-ЯМР (400 МГц, CDCl3) δ м.д. 3,77 ((шир. с, 1H), 3,99-4,16 (м, 1H), 4,16-4,22 (м, 1H), 4,22-4,44 (м, 2H), 4,51 (д, J=9,9 Гц, 1H), 5,44 (с, 1H), 7,07-7,24 (м, 2H), 7,38 ((шир. с, 1H).
2-(4) Синтез ((2S,3R,4S)-4-амино-4-(2,3-дифторфенил)-2-(трифторметил)тетрагидрофуран-3-ил)метанола
(4S)-6a-(2,3-дифторфенил)-4-(трифторметил)гексагидрофуро-[3,4-c]изоксазол (3,16 г) растворяли в уксусной кислоте (20 мл), и реакционную смесь охлаждали до 0°C. Добавляли цинк (5,0 г), и реакционной смеси давали возможность нагреться и перемешивали при КТ в течение 20 ч. Затем реакционную смесь разбавляли EtOAc (50 мл) и фильтровали через целит®, промывая дополнительными 100 мл EtOAc. Объединенные органические порции выпаривали, растворяли в CHCl3 (20 мл) и медленно добавляли аммиак (28% водн., 25 мл). Слои разделяли, и водную часть дополнительно экстрагировали CHCl3 (2×25 мл). Объединенные органические экстракты сушили над безводным MgSO4 и выпаривали для получения указанного в заголовке соединения (3,12 г), которое использовали на последующей стадии без дополнительной очистки. 1H-ЯМР (400 МГц, CDCl3) δ м.д. 2,93 (ддд, J=7,8, 5,1, 2,8 Гц, 1 H), 3,85 (дд, J=12,4, 5,1 Гц, 1 H), 4,03 (дд, J=9,1, 2,8 Гц, 1 H), 4,14 (дд, J=12,3, 2,7 Гц, 1 H), 4,35 (д, J=9,1 Гц, 1 H), 4,68 (квин., J=7,3 Гц, 1 H), 7,09-7,25 (м, 2 H), 7,25-7,34 (м, 1 H).
2-(5) Синтез N-(((3S,4R,5S)-3-(2,3-дифторфенил)-4-(гидроксиметил)-5-(трифторметил)тетрагидрофуран-3-ил)карбамотиоил)бензамида
Бензоилизотиоцианат (2,0 мл) добавляли к раствору, содержащему ((2S,3R,4S)-4-амино-4-(2,3-дифторфенил)-2-(трифторметил)тетрагидрофуран-3-ил)метанол (3,12 г) в DCM (20 мл), и смесь перемешивали при КТ в течение 18 ч. Затем добавляли бикарбонат натрия (насыщ., водн., 50 мл), смесь экстрагировали EtOAc (3×75 мл), сушили над MgSO4 и концентрировали под пониженным давлением. Остаток очищали колоночной хроматографией на силикагеле (от 5% до 40% EtOAc в гексанах) для получения указанного в заголовке соединения (4,18 г). 1H-ЯМР (400 МГц, CDCl3) δ м.д. 3,12 (дд, J=7,6, 4,3 Гц, 1 H), 3,18-3,29 (м, 1 H), 4,03 (ддд, J=12,3, 7,2, 4,5 Гц, 1 H), 4,35-4,49 (м, 1 H), 4,59 (д, J=9,9 Гц, 1 H), 4,81 (д, J=9,6 Гц, 1 H), 7,07-7,23 (м, 2 H), 7,49 (т, J=7,2 Гц, 1 H), 7,56 (т, J=7,7 Гц, 2 H), 7,67 (т, J=7,5 Гц, 1 H), 7,88 (д, J=7,1 Гц, 2 H), 8,92 (с, 1 H), 11,89 (с, 1 H).
2-(6) Синтез N-((4aS,5S,7aS)-7a-(2,3-дифторфенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил)бензамида
N-(((3S,4R,5S)-3-(2,3-Дифторфенил)-4-(гидроксиметил)-5-(трифторметил)тетрагидрофуран-3-ил)карбамотиоил)бензамид (2,99 г) растворяли в пиридине (14 мл), и смесь охлаждали до -20°C. Трифторметансульфоновый ангидрид (1,55 мл) по каплям добавляли в течение 10 мин, и реакционной смеси давали возможность нагреться до -10°C. После перемешивания в течение 2 часов, дополнительную порцию трифторметансульфонового ангидрида (1,0 мл) по каплям добавляли в течение 10 мин, реакционную смесь перемешивали в течение еще 2 часа и затем гасили добавлением хлорида аммония (насыщ., водн., 50 мл) и экстрагировали EtOAc (3×100 мл). Объединенные органические экстракты сушили над MgSO4, концентрировали в вакууме и очищали колоночной хроматографией на силикагеле (от 5% до 20% EtOAc/гексаны) для получения указанного в заголовке соединения (1,20 г). 1H-ЯМР (400 МГц, CDCl3) δ м.д. 2,86 (д, J=10,6 Гц, 1 H), 3,20 ((шир. с, 1 H), 3,55 ((шир. с, 1 H), 4,04 ((шир. с, 1 H), 4,65 (д, J=8,8 Гц, 1 H), 4,81 ((шир. с, 1 H), 7,06-7,24 (м, 3 H), 7,40-7,64 (м, 3 H), 7,82-8,21 (м, 2 H).
2-(7) Синтез (4aS,5S,7aS)-7a-(2,3-дифторфенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-амина
N-((4aS,5S,7aS)-7a-(2,3-Дифторфенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил)бензамид (2,00 г) растворяли в метаноле (250 мл), добавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (1,53 г), и раствор нагревали в сосуде с обратным холодильником (температура нагревателя 80°C). Через 3 часа реакционную смесь концентрировали под пониженным давлением, разбавляли водой (100 мл) и экстрагировали EtOAc (3×100 мл). Объединенные органические порции сушили над MgSO4, выпаривали, и остаток очищали колоночной хроматографией на силикагеле (от 0% до 50% EtOAc в гексанах) для получения указанного в заголовке соединения (1,42 г). 1H-ЯМР (400 МГц, CDCl3) δ м.д. 2,86 (дд, J=13,6, 3,8 Гц, 1 H), 3,15 (дд, J=13,8, 3,2 Гц, 1 H), 3,27-3,42 (м, 1 H), 3,93 (дд, J=8,2, 1,9 Гц, 1 H), 4,39-4,78 (м, 4 H), 6,96-7,25 (м, 3 H).
2-(8) Синтез (4aS,5S,7aS)-7a-(2,3-дифтор-5-нитрофенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-амина
(4aS,5S,7aS)-7a-(2,3-Дифторфенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-амин (1,42 г) растворяли в TFA (6 мл), и раствор охлаждали до 0°C. Добавляли серную кислоту (концентрированную, 1 мл) с последующим добавлением по каплям дымящейся азотной кислоты (0,30 мл) в течение 20 мин. После перемешивания при 0°C в течение 1 часа реакционную смесь выливали в лед (50 г) и повышали основность до pH 12 2н NaOH (водным). После предоставления возможности льду растаять, смесь экстрагировали EtOAc (3×75 мл), и объединенные органические порции сушили над MgSO4 и выпаривали для получения указанного в заголовке соединения (1,91 г, чистота приблизительно 80%), которое использовали на последующей стадии без очистки. 1H-ЯМР (400 МГц, CDCl3) δ м.д. 2,88 (дд, J=13,8, 3,9 Гц, 1 H), 3,11 (дд, J=13,6, 2,8 Гц, 1 H), 3,37 (дт, J=7,4, 3,5 Гц, 1 H), 3,93 (д, J=7,8 Гц, 1 H), 4,53-4,83 (м, 4 H), 8,09 (ддд, J=9,0, 6,3, 2,9 Гц, 1 H), 8,27 (дт, J=5,2, 2,6 Гц, 1 H).
2-(9) Синтез трет-бутил-((4aS,5S,7aS)-7a-(2,3-дифтор-5-нитрофенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил)карбамата
(4aS,5S,7aS)-7a-(2,3-Дифтор-5-нитрофенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-амин (1,91 г, неочищенный) растворяли в THF (10 мл), порциями добавляли ди-трет-бутил дикарбонат (1,3 г) в течение 20 мин, и реакционную смесь нагревали до 65°C. Через 3 часа реакционную смесь охлаждали и добавляли бикарбонат натрия (насыщенный, водный, 50 мл). Затем смесь экстрагировали EtOAc (3×750 мл), и объединенные органические порции сушили над MgSO4 и выпаривали. Остаток очищали колоночной хроматографией на силикагеле (от 0% до 20% EtOAc в гексанах) для получения указанного в заголовке соединения (1,43 г неочищенного в смеси с вариантом бис-бутоксикарбонил).
2-(10) Синтез трет-бутил-((4aS,5S,7aS)-7a-(5-амино-2,3-дифторфенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил)карбамата
Неочищенную смесь, полученную в примере получения 2-(9), содержащую трет-бутил-((4aS,5S,7aS)-7a-(2,3-дифтор-5-нитрофенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил)карбамат) (1,43 г), растворяли в этаноле (25 мл) и добавляли дигидрохлорид олова (2,50 г). После перемешивания в течение 18 часов раствор выливали на NaOH (2н водн., 100 мл) и экстрагировали EtOAc (3×100 мл). Объединенные органические части сушили над MgSO4, выпаривали и очищали колоночной хроматографией на силикагеле (от 0% до 30% EtOAc в гексанах) для получения, во-первых, бис-бутоксикарбонильного продукта (730 мг), и, во-вторых, указанного в заголовке соединения (300 мг). 1H-ЯМР (400 МГц, CDCl3) δ м.д. 1,53 (с, 9 H), 2,76 (дд, J=13,9, 3,8 Гц, 1 H), 3,08 (д, J=13,6 Гц, 1 H), 3,36-3,45 (м, 1 H), 3,71 ((шир. с, 2 H), 3,90 (д, J=8,3 Гц, 1 H), 4,57 (д, J=8,6 Гц, 1 H), 4,68-4,79 (м, 1 H), 6,30-6,37 (м, 1 H), 6,42-6,49 (м, 1 H).
Пример получения 3: Синтез 5-этоксипиразин-2-карбоновой кислоты (3-(2))
Синтез этил 5-этоксипиразин-2-карбоксилата 3-(1)
Перемешанный раствор метил-5-хлорпиразин-2-карбоксилата (0,50 г) в этаноле (10 мл) охлаждали до 0°C, и добавляли этоксид натрия (21% масс./масс. раствор в этаноле, 1 мл) в течение 10 мин. После предоставления возможности нагревания до КТ и перемешивания в течение 2 часов добавляли воду (100 мл), и смесь экстрагировали EtOAc (2×150 мл). Объединенные органические порции сушили над MgSO4 и выпаривали для получения указанного в заголовке соединения (0,65 г, чистота приблизительно 85%). 1H-ЯМР (400 МГц, CDCl3) δ м.д. 1,45 (т, J=7,1 Гц, 3 H), 1,46 (т, J=7,1 Гц, 3 H), 4,48 (кв., J=7,1 Гц, 2 H), 4,49 (кв., J=7,1 Гц, 2 H), 8,28 (д, J=1,3 Гц, 1 H), 8,88 (д, J=1,3 Гц, 1 H).
Синтез 5-этоксипиразин-2-карбоновой кислоты 3-(2)
Этил-5-этоксипиразин-2-карбоксилат (0,65 г, чистота приблизительно 85%) растворяли в диоксане (3 мл) и добавляли воду (3 мл) с последующим добавлением гидроксида моногидрата лития (255 мг, порциями в течение 10 мин). После перемешивания при КТ в течение 24 часов добавляли Et2O (25 мл) и NaHCO3 (насыщ., водн., 25 мл). Слои разделяли, и органический слой экстрагировали NaOH (1N, водн., 25 мл). Объединенные водные порции подкисляли 6н HCl до pH 2, и смесь экстрагировали EtOAc (3×40 мл). Объединенные экстракты EtOAc сушили над MgSO4 и выпаривали для получения указанного в заголовке соединения в виде не совсем белого порошка. 1H-ЯМР (400 МГц, CDCl3) δ м.д. 1,47 (т, J=7,1 Гц, 3 H), 4,53 (кв., J=7,1 Гц, 2 H), 8,16 (д, J=1,2 Гц, 1 H), 8,98 (д, J=1,2 Гц, 1 H).
Пример получения 4: Синтез 5-этоксипиразин-2-карбоновой кислоты (4-(3))

Метил-5-ацетилпиразин-2-карбоксилата 4-(1)
5-Ацетилпиразин-2-карбоксамид (3,275 г) растворяли в метаноловом растворе HCl (1,25н, 150 мл) и реакционную смесь нагревали в сосуде с обратным холодильником и перемешивали в течение ночи. После охлаждения добавляли бикарбонат натрия, и смесь экстрагировали EtOAc. Слой EtOAc сушили над MgSO4 и выпаривали для получения указанного в заголовке соединения (3,79 г, чистота приблизительно 90%). 1H-ЯМР (400 МГц, CDCl3) δ м.д. 2,78 (с, 3 H), 4,10 (с, 3 H), 9,33 (д, J=1,5 Гц, 1 H), 9,36 (д, J=1,5 Гц, 1 H).
Метил-5-(1,1-дифторэтил)пиразин-2-карбоксилат 4-(2)
Метил-5-ацетилпиразин-2-карбоксилат (300 мг, чистота приблизительно 90%) растворяли в DCM (15 мл) и охлаждали до 0°C в атмосфере азота. По каплям добавляли трифторид бис(2-метоксиэтил)аминосеры (0,61 мл), и реакционной смеси давали возможность нагреться до КТ и перемешиваться в течение ночи. Осторожно добавляли бикарбонат натрия (насыщенный, водный), и смесь экстрагировали DCM. Органические порции сушили над MgSO4, выпаривали и очищали хроматографией на силикагеле (35% EtOAc в гексане) для получения указанного в заголовке соединения (155 мг) в виде белого твердого вещества. 1H-ЯМР (400 МГц, CDCl3) δ м.д. 2,00 (т, J=18,8 Гц, 3 H), 4,01 (с, 3 H), 8,98 (д, J=1,5 Гц, 1 H), 9,24 (д, J=1,5 Гц, 1 H).
5-(1,1-Дифторэтил)пиразин-2-карбоновая кислота 4-(3)
Метил-5-(1,1-дифторэтил)пиразин-2-карбоксилат (0,65 г, чистота приблизительно 85%) растворяли в диоксане (2 мл) и добавляли воду (2 мл) с последующим добавлением гидроксида моногидрата лития (54 мг, порциями). После перемешивания при КТ в течение 90 мин, смесь концентрировали до 2 мл и добавляли Et2O (20 мл). Затем смесь экстрагировали NaOH (1н, водным, 20 мл), и водные порции подкисляли 6н HCl до pH 2. Затем водную порцию экстрагировали EtOAc, сушили над MgSO4 и выпаривали для получения указанного в заголовке соединения в виде белого твердого вещества (119 мг). 1H-ЯМР (400 МГц, CDCl3) δ м.д. 2,11 (т, J=18,8 Гц, 3 H), 9,01 (д, J=1,3 Гц, 1 H), 9,47 (д, J=1,3 Гц, 1 H).
Пример получения 5: Синтез 5-(фторметил)пиразин-2-карбоновой кислоты (5-(3))
Метил-5-(гидроксиметил)пиразин-2-карбоксилат 5-(1)
К раствору метил-5-формилпиразин-2-карбоксилата (2,47 г) в THF (20 мл) порциями добавляли боргидрид натрия (170 мг) в течение 10 мин. После перемешивания в течение 1 часа добавляли метанол (10 мл). Реакционную смесь перемешивали в течение еще 20 мин, и затем добавляли HCl (1н, водную, 20 мл) и рассол (20 мл). Смесь экстрагировали EtOAc (3×40 мл), и объединенные органические порции сушили над MgSO4 и выпаривали для получения указанного в заголовке соединения (1,31 г). 1H-ЯМР (400 МГц, CDCl3) δ м.д. 4,07 (с, 3 H), 4,98 ((шир. с, 2 H), 8,80 (с, 1 H), 9,27 (с, 1 H).
Метил-5-(фторметил)пиразин-2-карбоксилат 5-(2)
К раствору метил-5-(гидроксиметил)пиразин-2-карбоксилата (0,64 г) в THF (20 мл) добавляли триэтиламин (2,30 г) и раствор охлаждали до 0°C. Затем добавляли триэтиламин тригидрофторида (1,22 г) с последующим добавлением по каплям нонафторбутансульфонилфторида (2,28 г) в течение 5 мин. После нагревания до КТ и перемешивания в течение 2 часов добавляли NaHCO3 (насыщенный, водный, 100 мл), и смесь экстрагировали EtOAc (2×50 мл). Объединенные органические порции сушили над MgSO4, выпаривали, и очищали хроматографией на силикагеле (от 5% до 50% EtOAc в гексане) для получения указанного в заголовке соединения (94 мг). 1H-ЯМР (400 МГц, CDCl3) δ м.д. 4,07 (с, 3 H), 5,67 (д, J=46,5 Гц, 2 H), 8,89 (с, 1 H), 9,28 (с, 1 H).
5-(Фторметил)пиразин-2-карбоновая кислота 5-(3)
Метил-5-(фторметил)пиразин-2-карбоксилат (94 мг) растворяли в диоксане (1 мл) и добавляли воду (1 мл) с последующим добавлением гидроксида моногидрата лития (60 мг). После перемешивания при КТ в течение 18 часов добавляли Et2O (20 мл) и затем смесь экстрагировали NaOH (1н, водн., 2×20 мл). Водные порции подкисляли 6н HCl до pH 1, экстрагировали EtOAc (2×40 мл), объединенные органические порции сушили над MgSO4 и выпаривали для получения указанного в заголовке соединения в виде белого твердого вещества (71 мг). 1H-ЯМР (400 МГц, CDCl3) δ м.д. 5,70 (д, J=46,2 Гц, 2 H), 8,85 (с, 1 H), 9,40 (с, 1 H).
Пример получения 6: Синтез 5-дифторметилпиразин-2-карбоновой кислоты (6-(5)
Синтез т-бутил-5-метилпиразин-2-карбоксилата 6-(1)
Комплекс трифторида бора с диэтиловым эфиром (91,7 мкл) по каплям добавляли к суспензии 2-метилпиразин-5-карбоновой кислоты (1 г) и трет-бутил-2,2,2-трихлорацетимидата (4,75 г в THF (20 мл) при охлаждении льдом. Реакционный раствор нагревали до КТ с последующим перемешиванием в течение 2 ч. К реакционному раствору добавляли насыщенный раствор NaCl и EtOAc, и органический слой отделяли. Органический слой сушили над безводным MgSO4, и нерастворимое вещество отделяли фильтрацией. фильтрат концентрировали и очищали колоночной хроматографией на силикагеле для получения указанного в заголовке соединения (1,4 г). 1H-ЯМР (CDCl3) δ (м.д.): 1,65 (с, 9H), 2,65 (с, 3H), 8,57 (д, J=1,2 Гц, 1H), 9,10 (д, J=1,6 Гц, 1H).
Синтез трет-бутил-5-((E)-2-диметиламиновинил)пиразин-2-карбоксилата 6-(2)
Смесь трет-бутил-5-метилпиразин-2-карбоксилата (1,35 г), DMF (25 мл) и N,N-диметилформамида диметилацеталя (25 мл) перемешивали при 130°C в течение 5 ч. Реакционный раствор охлаждали до КТ и разбавляли EtOAc. Смесь три раза промывали насыщенным раствором NaCl. Органический слой сушили над безводным MgSO4, и нерастворимое вещество отделяли фильтрацией. Фильтрат концентрировали, и остаток очищали колоночной хроматографией на силикагеле для получения указанного в заголовке соединения (648 мг). 1H-ЯМР (CDCl3) δ (м.д.): 1,63 (с, 9H), 3,00 (с, 6H), 5,16 (д, J=12,8 Гц, 1H), 7,72 (д, J=12,8 Гц, 1H), 8,16 (д, J=1,2 Гц, 1H), 8,81 (д, J=1,6 Гц, 1H).
Синтез трет-бутил-5-формилпиразин-2-карбоксилата 6-(3)
Периодат натрия (1,67 г) добавляли к раствору трет-бутил-5-((E)-2-диметиламиновинил)пиразин-2-карбоксилата (645 мг) в 50% THF-воде (26 мл), и смесь перемешивали при КТ в течение 4 ч. К реакционному раствору добавляли насыщенный раствор NaHCO3 и EtOAc, и органический слой отделяли. Органический слой промывали насыщенным раствором NaCl и сушили над безводным MgSO4. Нерастворимое вещество отделяли фильтрацией, и фильтрат концентрировали. Остаток очищали колоночной хроматографией на силикагеле для получения указанного в заголовке соединения (249 мг). 1H-ЯМР (CDCl3) δ (м.д.): 1,68 (с, 9H), 9,25 (д, J=1,2 Гц, 1H), 9,36 (д, J=1,6 Гц, 1H), 10,2 (с, 1H).
Синтез т-бутил-5-дифторметилпиразин-2-карбоксилата 6-(4)
Трифторид [бис(2-метоксиэтил)амино]серы (662 мкл) по каплям добавляли к раствору трет-бутил-5-формилпиразин-2-карбоксилата (249 мг) в CH2Cl2 (12 мл) в атмосфере N2 при охлаждении льдом. Реакционный раствор перемешивали в течение 2 часов при постепенном возврате к КТ. К реакционному раствору добавляли насыщенный раствор NaHCO3 и EtOAc, и органический слой отделяли. Органический слой промывали насыщенным раствором NaCl и сушили над безводным MgSO4. Нерастворимое вещество отделяли фильтрацией и фильтрат концентрировали. Остаток очищали колоночной хроматографией на силикагеле для получения указанного в заголовке соединения (175 мг). 1H-ЯМР (CDCl3) δ (м.д.): 1,67 (с, 9H), 6,75 (т, J=54,4 Гц, 1H), 9,02 (д, J=0,8 Гц, 1H), 9,25 (д, J=0,8 Гц, 1H).
Синтез 5-дифторметилпиразин-2-карбоновой кислоты 6-(5)
Трифторуксусную кислоту (1 мл) добавляли к раствору трет-бутил-5-дифторметилпиразин-2-карбоксилата (175 мг) в дихлорметане (1 мл), и смесь перемешивали при КТ в течение 5 ч. К реакционному раствору добавляли простой эфир и 5н NaOH. Водный слой отделяли и делали кислотным 5н хлористоводородной кислотой. К водному слою добавляли EtOAc, и органический слой отделяли. Органический слой сушили над безводным MgSO4, и нерастворимое вещество отделяли фильтрацией. Фильтрат концентрировали для получения указанного в заголовке соединения (100 мг). 1H-ЯМР (CDCl3) δ (м.д.): 6,80 (т, J=54,4 Гц, 1H), 9,02 (с, 1H), 9,47 (с, 1H).
Пример получения 7: Синтез 5-цианопиридин-2-карбоновой кислоты (7-(2))
Синтез метил 5-цианопиридин-2-карбоксилата 7 (1)
Смесь метил-5-бромпиридин-2-карбоксилата (2,8 г) и цианида меди (3,6 г) в NMP (30 мл) нагревали с перемешиванием при 170°C в течение 1,5 ч. К реакционному раствору добавляли воду при КТ, и нерастворимое вещество удаляли фильтрацией. Фильтрат экстрагировали EtOAc. Экстракт промывали насыщенным раствором NaCl и затем сушили над безводным MgSO4. Сушащий агент удаляли фильтрацией и фильтрат концентрировали под пониженным давлением. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (система EtOAc-гептан) для получения указанного в заголовке соединения (920 мг). 1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 4,06 (с, 3H), 8,16 (дд, J=2,0, 8,0 Гц, 1H), 8,27 (д, J=8,0 Гц, 1H), 9,01 (д, J=2,0 Гц, 1H).
Синтез 5-цианопиридин-2-карбоновой кислоты 7-(2)
Раствор соединения примера получения 13-(1) (920 мг) и 5н раствор NaOH (2,26 мл) в этаноле (30 мл) перемешивали при КТ в течение 10 мин. К реакционному раствору добавляли 5н хлористоводородную кислоту (5,2 мл) при КТ с последующей экстракцией EtOAc. Экстракт сушили над безводным MgSO4. Сушащий агент удаляли фильтрацией, и фильтрат концентрировали под пониженным давлением для получения указанного в заголовке соединения (800 мг). 1H-ЯМР (400 МГц, ДМСОd6) δ (м.д.): 8,18 (д, J=8,0 Гц, 1H), 8,51 (дд, J=2,0, 8,0 Гц, 1H), 9,12-9,18 (м, 1H).
Пример получения 8: Синтез 5-(метоксиметил)пиразин-2-карбоновой кислоты (8-(2))
Синтез метил 5-(метоксиметил)пиразин-2-карбоксилата 8-(1)
Соединение, полученное в примере получения 5-(1) (279 мг) растворяли в DMF, и раствор охлаждали до 0°C. Добавляли гидрид натрия (60% в минеральном масле, 70 мг) с последующим добавлением иодометана (250 мг). Через 2 дня добавляли воду (25 мл), и раствор экстрагировали EtOAc (100 мл). Водный слой насыщали NaCl и дополнительно экстрагировали EtOAc (2×50 мл). Объединенные органические части сушили над MgSO4, выпаривали, и очищали хроматографией на силикагеле (от 30% до 50% EtOAc в гексанах) для получения указанного в заголовке соединения (55 мг, чистота приблизительно 65%). 1H-ЯМР (400 МГц, CDCl3) δ м.д. 3,55 (с, 3 H), 4,05 (с, 3 H), 4,72 (с, 2 H), 8,84 (д, J=1,0 Гц, 1 H), 9,25 (д, J=1,0 Гц, 1 H).
Синтез 5-(метоксиметил)пиразин-2-карбоновой кислоты 8-(2)
Метил-5-(метоксиметил)пиразин-2-карбоксилат 8-(1) (55 мг, неочищенный) растворяли в 1,4-диоксане (1 мл) и добавляли воду (1 мл) с последующим добавлением гидроксида моногидрата лития (50 мг). После перемешивания при КТ в течение 1 часа, добавляли воду (20 мл), и смесь экстрагировали простым эфиром (20 мл). Водную порцию подкисляли до pH 2 и экстрагировали EtOAc (2×25 мл). Объединенные EtOAc слои сушили над MgSO4 и выпаривали для получения указанного в заголовке соединения (19 мг). 1H-ЯМР (400 МГц, CDCl3) δ м.д. 3,58 (с, 3 H), 4,77 (с, 2 H), 8,80 ((шир. с, 1 H), 9,38 ((шир. с, 1 H).
Пример получения 9: 5-Метоксипиразин-2-карбоновая кислота
5-Хлорпиразин-2-карбоновую кислоту (5,0 г, 0,032 моль) загружали в круглодонную колбу, оборудованную термопарой, подвесной мешалкой и обратным холодильником. Загружали метанол (37,5 мл, 0,926 моль) с последующей загрузкой концнтрированной серной кислоты (0,2 мл, 0,004 моль). 3-горловую колбу оборудовали нагревающим кожухом, и затем реакционную смесь нагревали до приблизительно 65,0°C (внутренняя температура). Реакционную смесь продолжали перемешивать приблизительно при 65,0°C (внутренняя температура) в течение приблизительно 4 ч. Реакционную смесь охлаждали приблизительно до 25,8°C (внутренняя температура). Загружали метанол (12 мл, 0,31 моль) и кашицеобразную суспензию продолжали перемешивать приблизительно при 22,3°C (внутренняя температура) в течение приблизительно 15 мин, затем охлаждали до приблизительно 10,0°C (внутренняя температура) в атмосфере азота. В колбу загружали 25% метоксид натрия в метаноле (1:3, метоксид натрия:метанол, 7,7 мл), в то время как температура оставалась ниже 30,0°C (внутренняя температура). Температуру реакционной смеси доводили до 20,4°C (внутренняя температура). Через 30 мин, объединяли гидроксид натрия (2,0 г, 0,04 моль) и воду (37,5 мл, 2,08 моль) для образования раствора, и затем раствор загружали в реакционную смесь. Загружали воду (50,0 мл, 2,78 моль) и затем реакционную смесь нагревали до 40,0°C (внутренняя температура) в течение приблизительно 60 мин. Нагревающий кожух удаляли, и затем реакционную смесь охлаждали приблизительно до 25,4°C (внутренняя температура). Добавляли 38% водный раствор HCl (38:62, гидрохлорид:вода, 4,0 мл) со скоростью (приблизительно 5 мин) с тем, чтобы температура оставалась ниже 30,0°C (внутренняя температура). Густую кашицеобразную суспензию перемешивали в течение 1 часа приблизительно при 21,4°C (внутренняя температура) и затем фильтровали через спеченную воронку. Твердые вещества промывали водой (10,0 мл, 0,555 моль) и сушили в вакууме в течение ночи для получения 5-метоксипиразин-2-карбоновой кислоты (3,59 г). 1H-ЯМР (500 МГц, ДМСО) δ 13,24 (1H, шир. с), 8,79 (1H, д, J=1,2 Гц), 8,37 (1H, д, J=1,2 Гц), 3,98 (с, 3H); 13C ЯМР (125 МГц, ДМСО) δ 165,36, 161,88, 143,88, 136,82, 135,55, 54,69.
Общая процедура для соединения анилинов, полученных в Примерах получения 1-(13) и 2-(10), с арилкарбоновыми кислотами: Получение N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-метоксипиразин-2-карбоксамида (Пример 1)

Трет-бутил-((4aS,5S,7aS)-7a-(5-амино-2-фторфенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил)карбамат (Пример получения 1, 56 мг) растворяли в DCM (2 мл) и добавляли 5-метоксипиразин-2-карбоновую кислоту (40 мг), N,N-диизопропилэтиламин (80 мг) и (1H-бензотриазол-1-илокси)трипирролидин-1-ил)фосфоний гексафторфосфат (135 мг). Реакционную смесь перемешивали при КТ в течение 18 часов и добавляли бикарбонат натрия (насыщ., водн., 25 мл). Смесь экстрагировали EtOAc (2×40 мл), объединенные органические порции сушили над MgSO4, выпаривали и очищали хроматографией на силикагеле (градиент EtOAc/гексанов) для получения амида (40 мг) в виде белого твердого вещества. Амид растворяли в DCM (2 мл) и добавляли TFA (1 мл). После перемешивания при КТ в течение 2 часов, реакционную смесь выпаривали и добавляли бикарбонат натрия (насыщенный, водный, 25 мл). Смесь экстрагировали EtOAc (2×25 мл), и объединенные органические порции сушили над MgSO4 и выпаривали для получения указанного в заголовке соединения в виде белого твердого вещества (34 мг). 1H-ЯМР (400 МГц, CDCl3) δ м.д. 2,78-2,87 (м, 1 H), 3,11-3,21 (м, 1 H), 3,38-3,48 (м, 1 H), 3,82-4,06 (м, 4 H), 4,48-4,72 (м, 2 H), 6,96-7,10 (м, 1 H), 7,52 (д, J=4,8 Гц, 1 H), 7,87 (д, J=8,8 Гц, 1 H), 8,09 (с, 1 H), 8,95 (с, 1 H), 9,46 ((шир. с, 1 H).
Пример 2: N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-цианопиколинамид
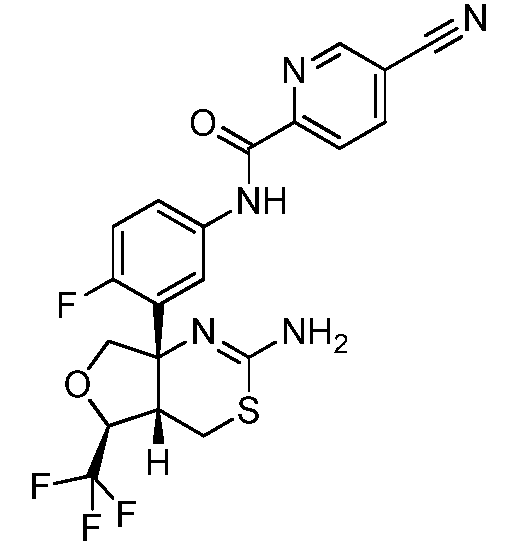
Синтезировали из трет-бутил-[(4aS,5S,7aS)-7a-(5-амино-2-фторфенил)-5-трифторметил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата и 5-цианопиразин-2-карбоновой кислоты в соответствии с общей процедурой. 1H-ЯМР (400 МГц, CDCl3) δ м.д. 2,82 (дд, J=13,6, 3,5 Гц, 1 H), 3,14-3,21 (м, 1 H), 3,37-3,45 (м, 1 H), 3,91 (д, J=9,1 Гц, 1 H), 4,50-4,71 (м, 2 H), 7,08 (дд, J=11,9, 8,8 Гц, 1 H), 7,46-7,57 (м, 1 H), 7,92 (дт, J=8,8, 3,4 Гц, 1 H), 8,17 (дд, J=8,3, 2,0 Гц, 1 H), 8,34 (д, J=8,1 Гц, 1 H), 8,81-8,92 (м, 1 H).
Пример 3: N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-(дифторметил)пиразин-2-карбоксамид

Синтезировали из трет-бутил-[(4aS,5S,7aS)-7a-(5-амино-2-фторфенил)-5-трифторметил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата и 5-дифторметил-пиразин-2-карбоновой кислоты в соответствии с общей процедурой. 1H-ЯМР (400 МГц, CDCl3) δ м.д. 2,80 (дд, J=13,8, 3,7 Гц, 1 H), 3,11 (дд, J=13,6, 2,8 Гц, 1 H), 3,31-3,41 (м, 1 H), 3,86 (д, J=8,3 Гц, 1 H), 4,57 (д, J=8,3 Гц, 1 H), 4,64 (дт, J=14,7, 7,1 Гц, 1 H), 4,75 (шир. с, 2 H), 6,69 (т, J=56,3 Гц, 1 H), 7,07 (дд, J=11,6, 8,8 Гц, 1 H), 7,57 (дд, J=7,1, 2,8 Гц, 1 H), 7,87 (дт, J=8,5, 3,6 Гц, 1 H), 8,85 (с, 1 H), 9,45 (с, 1 H), 9,58 ((шир. с, 1 H).
Пример 4: N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-(трифторметил)пиколинамид
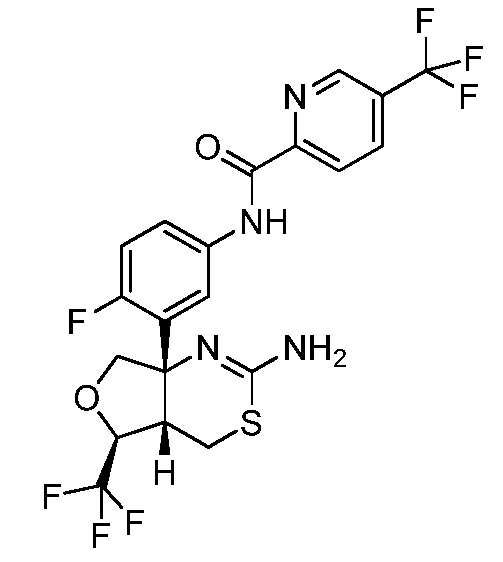
Синтезируют из трет-бутил-[(4aS,5S,7aS)-7a-(5-амино-2-фторфенил)-5-трифторметил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата и 5-трифторметил-пиридин-2-карбоновой кислоты в соответствии с общей процедурой. 1H-ЯМР (400 МГц, CDCl3) δ м.д. 2,88 (дд, J=13,6, 3,8 Гц, 1 H), 3,20 (дд, J=13,6, 3,0 Гц, 1 H), 3,37-3,53 (м, 1 H), 3,94 (дд, J=8,3, 2,3 Гц, 1 H), 4,40-4,89 (м, 4 H), 7,14 (дд, J=11,9, 8,8 Гц, 1 H), 7,66 (дд, J=6,8, 2,8 Гц, 1 H), 7,98 (ддд, J=8,8, 4,0, 3,0 Гц, 1 H), 8,20 (дд, J=8,2, 1,6 Гц, 1 H), 8,45 (д, J=8,3 Гц, 1 H), 8,90 (с, 1 H), 9,95 (с, 1 H).
Пример 5: N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-метилпиразин-2-карбоксамид

Ситезировали из трет-бутил-[(4aS,5S,7aS)-7a-(5-амино-2-фторфенил)-5-трифторметил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата и 5-метил-пиразин-2-карбоновой кислоты в соответствии с общей процедурой. 1H-ЯМР (400 МГц, CDCl3) δ м.д. 2,72 (с, 3 H), 2,88 (дд, J=13,6, 3,8 Гц, 1 H), 3,21 (дд, J=13,6, 3,0 Гц, 1 H), 3,42-3,49 (м, 1 H), 3,94 (д, J=8,3 Гц, 1 H), 4,39-4,80 (м, 4 H), 7,14 (дд, J=11,9, 8,8 Гц, 1 H), 7,62 (дд, J=7,1, 2,8 Гц, 1 H), 7,93-7,99 (м, 1 H), 8,46 (д, J=1,0 Гц, 1 H), 9,39 (д, J=1,3 Гц, 1 H), 9,65 (с, 1 H).
Пример 6: N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-метилпиколинамид

Синтезировали из трет-бутил-[(4aS,5S,7aS)-7a-(5-амино-2-фторфенил)-5-трифторметил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата и 5-метил-пиридин-2-карбоновой кислоты в соответствии с общей процедурой. 1H-ЯМР (400 МГц, CDCl3) δ м.д. 2,37 (с, 3H), 2,78 (дд, J=13,6, 3,8 Гц, 1H), 3,12 (дд, J=13,6, 2,8 Гц, 1H), 3,28-3,40 (м, 1H), 3,87 (д, J=8,1 Гц, 1H), 4,28-5,02 (м, 4H), 7,02 (дд, J=11,9, 8,8 Гц, 1H), 7,55 (дд, J=7,1, 2,8 Гц, 1H), 7,63 (дд, J=8,0, 1,4 Гц, 1H), 7,88 (ддд, J=8,8, 4,0, 2,9 Гц, 1H), 8,10 (д, J=8,1 Гц, 1H), 8,31-8,40 (м, 1H), 9,90 (с, 1H).
Пример 7: N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-этилпиколинамид

Синтезировали из трет-бутил-[(4aS,5S,7aS)-7a-(5-амино-2-фторфенил)-5-трифторметил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата и 5-этил-пиридин-2-карбоновой кислоты в соответствии с общей процедурой. 1H-ЯМР (400 МГц, CDCl3) δ м.д. 1,33 (т, J=7,6 Гц, 3 H), 2,78 (кв., J=7,6 Гц, 2 H), 2,88 (дд, J=13,5, 3,7 Гц, 1 H), 3,22 (дд, J=13,6, 2,8 Гц, 1 H), 3,42-3,48 (м, 1 H), 3,96 (д, J=7,3 Гц, 1 H), 4,44-4,95 (м, 4 H), 7,12 (дд, J=11,6, 8,8 Гц, 1 H), 7,62 (дд, J=6,9, 2,7 Гц, 1 H), 7,74 (дд, J=8,1, 1,8 Гц, 1 H), 7,98 (дт, J=8,7, 3,5 Гц, 1 H), 8,21 (д, J=8,1 Гц, 1 H), 8,44-8,48 (м, 1 H), 10,00 (с, 1 H).
Пример 8: N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-(фторметил)пиразин-2-карбоксамид

Синтезировали из трет-бутил-[(4aS,5S,7aS)-7a-(5-амино-2-фторфенил)-5-трифторметил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата и 5-фторметил-пиразин-2-карбоновой кислоты в соответствии с общей процедурой. Подробности действительного получения следующие: трет-бутил-((4aS,5S,7aS)-7a-(5-амино-2-фторфенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил)карбамат (500 мг) растворяли в DCM (10 мл) и добавляли 5-фторметил-пиразин-2-карбоновую кислоту (223 мг), N,N-диизопропилэтиламин (521 мг) и (1H-бензотриазол-1-илокси)трипирролидин-1-ил)фосфоний гексафторфосфат (750 мг). Реакционную смесь перемешивали при КТ в течение 1 часа и добавляли бикарбонат натрия (насыщенный, водный, 50 мл). Смесь экстрагировали EtOAc (2×75 мл), объединенные органические части сушили над MgSO4, выпаривали и очищали хроматографией на силикагеле (градиент от 0% до 30% EtOAc/гаксаны) для получения амида (613 мг) в виде белого твердого вещества. Амид растворяли в DCM (2 мл) и добавляли TFA (1 мл). После перемешивания при КТ в течение 2 часов реакционную смесь выпаривали и добавляли бикарбонат натрия (насыщенный, водный, 25 мл). Смесь экстрагировали EtOAc (3×25 мл), и объединенные органические порции сушили над MgSO4 и выпаривали для получения указанного в заголовке соединения в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ м.д. 2,89 (дд, J=13,8, 3,7 Гц, 1 H), 3,21 (дд, J=13,9, 2,5 Гц, 1 H), 3,40-3,51 (м, 1 H), 3,96 (д, J=7,3 Гц, 1 H), 4,42-4,85 (м, 4 H), 5,69 (д, J= 46,5 Гц, 2 H), 7,15 (дд, J=11,9, 8,8 Гц, 1 H), 7,64 (дд, J=7,1, 2,8 Гц, 1 H), 7,94-8,00 (м, 1 H), 8,77 (с, 1 H), 9,47 (с, 1 H), 9,68 (с, 1 H).
Пример 9: N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-метоксипиколинамид
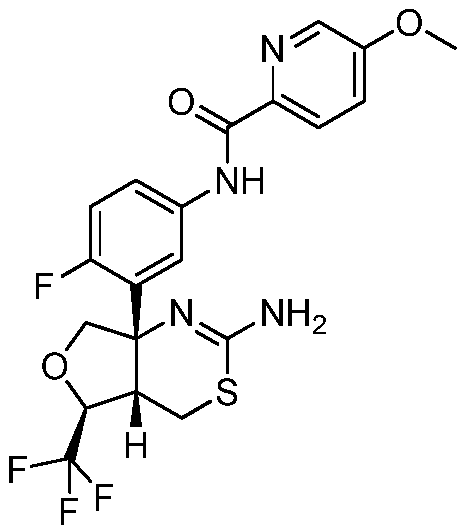
Синтезировали из трет-бутил-[(4aS,5S,7aS)-7a-(5-амино-2-фторфенил)-5-трифторметил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата и 5-метоксипиридин-2-карбоновой кислоты в соответствии с общей процедурой. 1H-ЯМР (400 МГц, CDCl3) δ м.д. 2,88 (дд, J=13,8, 3,7 Гц, 1 H), 3,23 (дд, J=13,8, 2,4 Гц, 1 H), 3,46 (д, J=7,3 Гц, 1 H), 3,89-4,02 (м, 4 H), 4,54-5,00 (м, 4 H), 7,11 (дд, J=11,9, 8,8 Гц, 1 H), 7,36 (дд, J=8,8, 2,8 Гц, 1 H), 7,61 (дд, J=7,1, 2,8 Гц, 1 H), 7,97 (дт, J=8,5, 3,6 Гц, 1 H), 8,23-8,30 (м, 2 H), 9,86 (с, 1 H).
Пример 10: N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-этоксипиразин-2-карбоксамид
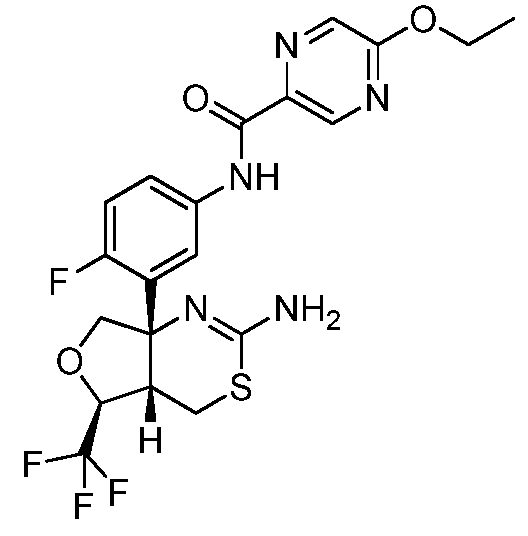
Синтезировали из трет-бутил-[(4aS,5S,7aS)-7a-(5-амино-2-фторфенил)-5-трифторметил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата и 5-этоксипиримидин-2-карбоновой кислоты в соответствии с общей процедурой. 1H-ЯМР (400 МГц, CDCl3) δ м.д. 1,47 (т, J=7,1 Гц, 3 H), 2,87 (дд, J=13,6, 3,5 Гц, 1 H), 3,20 (д, J=13,6 Гц, 1 H), 3,40-3,50 (м, 1 H), 3,94 (д, J=7,8 Гц, 1 H), 4,46-4,95 (м, 6 H), 7,11 (дд, J=11,5, 9,0 Гц, 1 H), 7,54-7,63 (м, 1 H), 7,90-8,01 (м, 1 H), 8,13 (с, 1 H), 9,00 (с, 1 H), 9,52 ((шир. с, 1 H).
Пример 11: N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-(1,1-дифторэтил)пиразин-2-карбоксамид
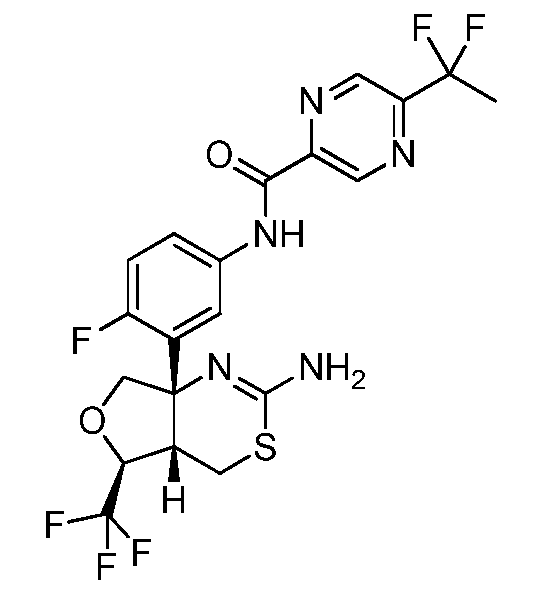
Синтезировали из трет-бутил-[(4aS,5S,7aS)-7a-(5-амино-2-фторфенил)-5-трифторметил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата и 5-(1,1-дифторэтил)пиразин-2-карбоновой кислоты в соответствии с общей процедурой. 1H-ЯМР (600 МГц, CDCl3) δ м.д. 2,03 (т, J=18,8 Гц, 3 H), 2,82 (д, J=12,0 Гц, 1 H), 3,14 (д, J=12,4 Гц, 1 H), 3,35-3,46 (м, 1 H), 3,77-4,07 (м, 1 H), 4,20-4,93 (м, 4 H), 7,08 (дд, J=11,7, 9,0 Гц, 1 H), 7,56 (д, J=4,5 Гц, 1 H), 7,80-7,93 (м, 1 H), 8,87 (с, 1 H), 9,43 (с, 1 H), 9,59 ((шир. с, 1 H).
Пример 12: N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-(трифторметил)пиразин-2-карбоксамид
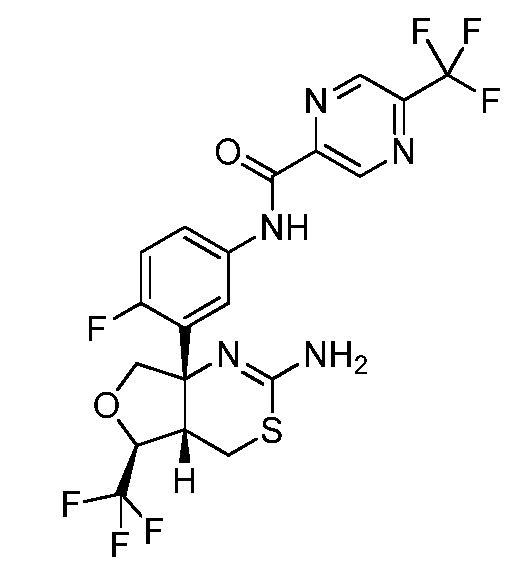
Синтезировали из трет-бутил-[(4aS,5S,7aS)-7a-(5-амино-2-фторфенил)-5-трифторметил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата и 5-трифторметилпиразин-2-карбоновой кислоты в соответствии с общей процедурой. 1H-ЯМР (400 МГц, CDCl3) δ м.д. 2,80 (дд, J=13,6, 3,8 Гц, 1 H), 3,11 (дд, J=13,8, 2,9 Гц, 1 H), 3,30-3,44 (м, 1 H), 3,87 (д, J=8,3 Гц, 1 H), 4,25-5,14 (м, 4 H), 7,07 (дд, J=11,9, 8,8 Гц, 1 H), 7,57 (дд, J=6,8, 2,8 Гц, 1 H), 7,86 (дт, J=8,4, 3,6 Гц, 1 H), 8,89 (с, 1 H), 9,53 (с, 2 H).
Пример 13: N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-(метоксиметил)пиразин-2-карбоксамид
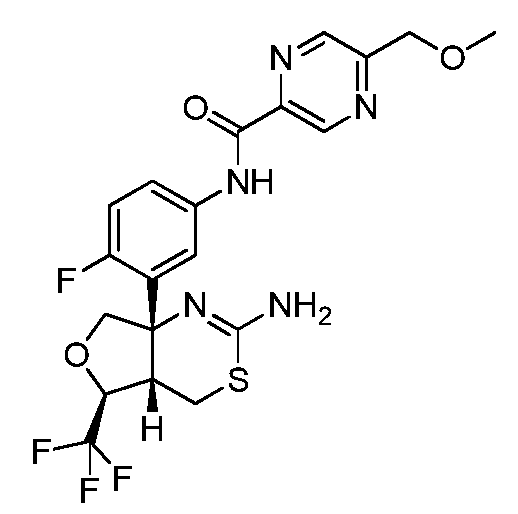
Синтезировали из трет-бутил-[(4aS,5S,7aS)-7a-(5-амино-2-фторфенил)-5-трифторметил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата и 5-(метоксиметил)пиримидин-2-карбоновой кислоты в соответствии с общей процедурой. 1H-ЯМР (400 МГц, CDCl3) δ м.д. 2,79 (дд, J=13,6, 3,8 Гц, 1 H), 3,11 (дд, J=13,6, 2,8 Гц, 1 H), 3,32-3,39 (м, 1 H), 3,49 (с, 3 H), 3,84 (д, J=8,3 Гц, 1 H), 4,34-4,73 (м, 6 H), 7,05 (дд, J=11,6, 8,8 Гц, 1 H), 7,55 (дд, J=6,8, 2,8 Гц, 1 H), 7,88 (дт, J=8,4, 3,6 Гц, 1 H), 8,63 (с, 1 H), 9,34 (д, J=1,0 Гц, 1 H), 9,60 (с, 1 H).
Пример 14: N-{3-[(4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5-дигидро-4H-фуро[3,4d][1,3]тиазин-7a(7H)-ил]-4-фторфенил}-5-[(2H3)метилокси]пиразин-2-карбоксамид
Синтез 14-(2) 5-[(2H3)метилокси]пиразин-2-карбоновой кислоты

Часть (I): (2H3)метил-5-[(2H3)метилокси]пиразин-2-карбоксилат
Свежеотрезанный металлический натрий (160 мг) порциями добавляли в течение 10 мин к (2H3)метан(2H)олу (5 мл) и раствор перемешивали до растворения натрия. Этот раствор затем добавляли к метил-5-хлорпиразин-2-карбоксилату (1,02 г) в (2H3)метан(2H)оле (5 мл) и раствору давали возможность перемешиваться при КТ в течение 1 часа. Затем раствор концентрировали под пониженным давлением до объема примерно 2 мл и добавляли воду (50 мл). Смесь экстрагировали EtOAc (2×50 мл), объединенные органические части сушили над MgSO4 и выпаривали для получения указанного в заголовке соединения (745 мг). 1H-ЯМР (400 МГц, CDCl3) δ м.д. 8,30 (д, J=1,3 Гц, 1 H), 8,91 (д, J=1,3 Гц, 1 H).
Часть (II): 5-[(2H3)метилокси]пиразин-2-карбоновая кислота
К перемешанному раствору (2H3)метил-5-[(2H3)метилокси]пиразин-2-карбоксилата в 1,4-диоксане (5 мл) добавляли воду (5 мл) с последующим добавлением гидроксида моногидрата лития (300 мг). После перемешивания в течение 1 часа реакционную смесь концентрировали под пониженным давлением до примерно 5 мл и экстрагировали простым диэтиловым эфиром (25 мл). Органический слой экстрагировали 1н NaOH (водн., 10 мл), и объединенные водные порции подкисляли до pH 2 6н хлористоводородной кислотой. После охлаждения в холодильнике смесь фильтровали для получения указанного в заголовке соединения в виде бледно-коричневого порошка (660 мг). 1H-ЯМР (400 МГц, CDCl3) δ м.д. 8,21 (д, J=1,3 Гц, 1 H), 9,01 (д, J=1,3 Гц, 1 H), 10,12 (шир. с, 1 H).
Часть (III): N-{3-[(4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5-дигидро-4H-фуро[3,4d][1,3]тиазин-7a(7H)-ил]-4-фторфенил}-5-[(2H3)метилокси]пиразин-2-карбоксамид
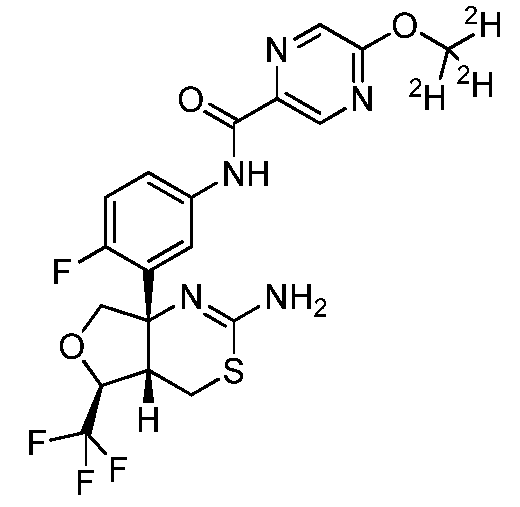
Трет-бутил-((4aS,5S,7aS)-7a-(5-амино-2-фторфенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил)карбамат (100 мг) растворяли в DCM (2 мл) и добавляли 5-[(2H3)метилокси]пиразин-2-карбоновую кислоту (55 мг), N,N-диизопропилэтиламин (112 мг) и (1H-бензотриазол-1-илокси)трипирролидин-1-ил)фосфоний гексафторфосфат (180 мг). Реакционную смесь перемешивали при КТ в течение 18 часов и добавляли бикарбонат натрия (насыщенный, водный, 25 мл). Смесь экстрагировали EtOAc (2×40 мл), объединенные органические части сушили над MgSO4, выпаривали и очищали хроматографией на силикагеле (от 2% до 25% EtOAc в гексанах) для получения амида (127 мг) в виде белого твердого вещества. Амид растворяли в DCM (2 мл) и добавляли TFA (1 мл). После перемешивания при КТ в течение 2 часов реакционную смесь выпаривали и добавляли бикарбонат натрия (насыщенный, водный, 25 мл). Смесь экстрагировали EtOAc (2×40 мл), и объединенные органические порции сушили над MgSO4 и выпаривали для получения указанного в заголовке соединения в виде белого твердого вещества (104 мг). 1H-ЯМР (400 МГц, CDCl3) δ м.д. 2,87 (дд, J=13,6, 3,8 Гц, 1 H), 3,21 (дд, J=13,6, 2,8 Гц, 1 H), 3,39-3,53 (м, 1 H), 3,95 (д, J=8,3 Гц, 1 H), 4,65 (д, J=8,3 Гц, 1 H), 4,72 (квин., J=7,2 Гц, 1 H), 4,87 (шир. с, 2 H), 7,12 (дд, J=11,9, 8,8 Гц, 1 H), 7,60 (дд, J=6,9, 2,7 Гц, 1 H), 7,95 (дт, J=8,5, 3,6 Гц, 1 H), 8,16 (д, J=1,0 Гц, 1 H), 9,02 (д, J=1,0 Гц, 1 H), 9,52 (с, 1 H).
Пример 15: N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4,5-дифторфенил)-5-(дифторметил)пиразин-2-карбоксамид

Синтезировали из трет-бутил-[(4aS,5S,7aS)-7a-(5-амино-2,3-дифторфенил)-5-трифторметил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата и 5-дифторметил-пиразин-2-карбоновой кислоты в соответствии с общей процедурой. 1H-ЯМР (400 МГц, CDCl3) δ м.д. 2,90 (дд, J=13,8, 3,7 Гц, 1 H), 3,19 (дд, J=13,8, 2,7 Гц, 1 H), 3,31-3,49 (м, 1 H), 3,95 (д, J=7,6 Гц, 1 H), 4,44-5,15 (м, 4 H), 6,81 (т, J=55,8 Гц, 4 H), 7,22-7,35 (м, 1 H), 8,08 (ддд, J=11,2, 6,8, 2,7 Гц, 1 H), 8,94 (с, 1 H), 9,53 (с, 1 H), 9,67 (с, 1 H).
Пример 16: N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4,5-дифторфенил)-5-метоксипиразин-2-карбоксамид

Синтезировали из трет-бутил-[(4aS,5S,7aS)-7a-(5-амино-2,3-дифторфенил)-5-трифторметил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата и 5-метоксипиразин-2-карбоновой кислоты в соответствии с общей процедурой. 1H-ЯМР (400 МГц, CDCl3 + MeOD) δ м.д. 2,83 (дд, J=13,9, 3,8 Гц, 1 H), 3,14 (дд, J=13,9, 3,0 Гц, 1 H), 3,29-3,39 (м, 1 H), 3,87 (д, J=8,3 Гц, 1 H), 4,04 (с, 3 H), 4,60 (д, J=8,3 Гц, 1 H), 4,67 (квин., J=6,3 Гц, 1 H), 7,11-7,21 (м, 1 H), 8,03 (ддд, J=11,6, 6,9, 2,7 Гц, 1 H), 8,15 (д, J=1,3 Гц, 1 H), 8,96 (д, J=1,3 Гц, 1 H).
Пример 17: N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4,5-дифторфенил)-5-метилпиразин-2-карбоксамид

Синтезировали из трет-бутил-[(4aS,5S,7aS)-7a-(5-амино-2,3-дифторфенил)-5-трифторметил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата и 5-метилпиразин-2-карбоновой кислоты в соответствии с общей процедурой. 1H-ЯМР (400 МГц, CDCl3 + MeOD) δ м.д. 2,68 (с, 3 H), 2,84 (дд, J=13,6, 3,8 Гц, 1 H), 3,15 (дд, J=13,9, 3,0 Гц, 1 H), 3,30-3,42 (м, 1 H), 3,88 (д, J=10,4 Гц, 1 H), 4,61 (д, J=8,6 Гц, 1 H), 4,68 (квин., J=7,2 Гц, 1 H), 7,13-7,25 (м, 1 H), 8,05 (ддд, J=11,6, 6,8, 2,8 Гц, 1 H), 8,46 (с, 1 H), 9,30 (д, J=1,3 Гц, 1 H).
Пример 18: N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4,5-дифторфенил)-5-(фторметил)-пиразин-2-карбоксамид

Синтезировали из трет-бутил-[(4aS,5S,7aS)-7a-(5-амино-2,3-дифторфенил)-5-трифторметил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата и 5-(фторметил)пиразин-2-карбоновой кислоты в соответствии с общей процедурой. 1H-ЯМР (400 МГц, CDCl3) δ м.д. 2,89 (дд, J=13,6, 3,8 Гц, 1 H), 3,19 (дд, J=13,6, 3,0 Гц, 1 H), 3,43 (дд, J=7,5, 3,4 Гц, 1 H), 3,86-3,99 (м, 1 H), 4,39-4,67 (м, 3 H), 4,74 (квин., J=7,1 Гц, 1 H), 5,76 (д, J=45,5 Гц, 2 H), 8,10 (ддд, J=11,4, 6,8, 2,8 Гц, 1 H), 8,77 (с, 1 H), 9,46 (с, 1 H), 9,69 (с, 1 H).
Альтернативные способы получения соединений примеров 1-8 описаны ниже в настоящей заявке. Для этих альтернативных способов получения 1H-ЯМР и 13C-ЯМР спектры регистрировали на приборе Varian 400 МГц или 500 МГц с использованием программного обеспечения vNMR 6.1C.
Альтернативное получение N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-метоксипиразин-2-карбоксамида (Пример 1)
1-(14) Синтез трет-бутил-2-(1,1,1-трифторбут-3-ен-2-илокси)ацетата
В реакционный сосуд загружали толуол (3,2 л), THF (0,60 л) и акролеин (0,40 л, 5,985 моль) при КТ в атмосфере азота. Добавляли (трифторметил)триметилсилан (1,003 кг, 7,059 моль) при 17°C. Реакционную смесь охлаждали до 2,5°C и добавляли TBAF (0,01M в THF, 0,400 л, 0,004 моль) в течение 2 часов. Во время добавления TBAF температура реакционной смеси увеличилась до 65°C. Реакционную смесь охлаждали до 0°C, и через 2 часа добавляли гидросульфат тетра-н-бутиламмония (0,171 кг, 0,503 моль) с последующим добавлением трет-бутил бромацетата (0,987 кг, 5,064 моль). Добавляли гидроксид натрия (50% масс. в воде, 4,2 кг, 52,6 моль) в течение 2 часов, поддерживая температуру ниже 10°C. Через 2 часа при 0-5°C к реакционной смеси добавляли воду (2,9 л) и метил-трет-бутиловый эфир (6,0 л). Водную фазу экстрагировали один или более раз метил-трет-бутиловым эфиром (6,0 л). Органические фазы объединяли и промывали 14% водным NaCl (3×1,6 л). Органические вещества концентрировали в вакууме для получения указанного в заголовке соединения в виде масла (1,150 кг, 94,5%), которое использовали на последующей стадии без дополнительной очистки. 1H-ЯМР (500 МГц, CDCl3) δ м.д. 5,86-5,74 (м, 1H), 5,59 (д, J=17,5 Гц, 1H), 5,56 (д, J=10,9 Гц, 1H), 4,37-4,30 (м, 1H), 4,11 (д, J=16,5 Гц, 1H), 4,06 (д, J=16,4 Гц, 1H), 1,40 (с, 9H); 13C ЯМР (125 МГц, CDCl3) δ м.д. 168,51, 128,49 (д, J=1,7 Гц), 123,86, 123,71 (кв., J=281,8 Гц), 82,22, 78,67 (кв., J=31,5 Гц), 66,60, 28,02.
1-(15) Синтез N-Метокси-N-метил-2-(1,1,1-трифторбут-3-ен-2-илокси)ацетамида

В реактор, содержащий трет-бутил-2-(1,1,1-трифторбут-3-ен-2-илокси)ацетат (1,150 кг, 4,788 моль), добавляли муравьиную кислоту (6,2 кг) при КТ. Реакционную смесь нагревали до 55-60°C в течение 4-5 ч. Муравьиную кислоту выпаривали в вакууме (T=40-45°C) и вытесняли толуолом (2×3,0 л). К остатку добавляли CH2Cl2 (2,0 л) и дополнительно концентрировали в вакууме. К полученному остатку добавляли CH2Cl2 (4,6 л), и раствор охлаждали до 0°C с последующим добавлением N,N-карбонилдиимидазола (1,05 кг, 6,49 моль) пятью порциями. Смесь перемешивали в течение 30 мин, и порциями добавляли N,O-диметилгидроксиламин гидрохлорид (0,67 кг, 6,72 моль) при поддержании температуры ниже 10°C. Реакционную смесь нагревали до КТ и перемешивали в течение 14 часов. Реакционную смесь охлаждали до 3,2°C и двумя порциями загружали имидазол (100,7 г, 1,48 моль). Реакционную смесь нагревали до КТ и добавляли воду с последующим добавлением метил-трет-бутилового эфира (14,0 л). Органическую фазу промывали 2,0н водной HCl (1,0 л и 0,7 л) с последующим промыванием насыщенным водным NaHCO3 (1,2 л) и насыщенной водной NaCl (1,20 л). Органические вещества концентрировали для получения указанного в заголовке соединения в виде масла (0,95 кг, 87,2%). 1H-ЯМР (500 МГц, CDCl3) δ м.д. 5,85-5,76 (м, 1H), 5,62 (д, J=17,2 Гц, 1H), 5,56 (д, J=10,4 Гц, 1H), 4,49-4,34 (м, 3H), 3,68 (с, 3H), 3,67 (с, 1H), 3,18 (с, 3H), 3,08 (с, 1H); 13C ЯМР (126 МГц, CDCl3) δ м.д. 169,9*, 163,4*, 128,61, 123,87 (д, J=282,0 Гц), 123,82, 78,54 (кв., J=31,3 Гц), 66,12, 61,52, 60,56, 36,20, 32,24. Примечание: это соединение представляет собой смесь 3:1 ротамеров амидной связи. *Карбонильные химические сдвиги, косвенно оцененные посредством 1H-13C HMBC (гетеронуклеарной корреляции кратной связи).
HRMS, рассчитанная для C8H12F3NO3 [M+H]+ 228,0848; найденная 228,0836.
1-(16) Синтез 1-(2-Фторфенил)-2-(1,1,1-трифторбут-3-ен-2-илокси)этанона

К раствору 1-бром-2-фторбензола (0,967 кг, 5,527 моль) в THF (6,2 L) при -75°C добавляли н-бутиллитий (2,50 M в гексане, 2,09 л, 5,22 моль) при поддержании температуры ниже -65°C (приблизительно 100 мин). Через 15 мин добавляли раствор N-метокси-N-метил-2-(1,1,1-трифторбут-3-ен-2-илокси)ацетамид (0,949 кг, 4,178 моль) в THF (1,6 л) при поддержании температуры ниже -65°C (приблизительно 70 мин). Через 2,5 часа при -78°C реакцию гасили добавлением насыщенного водного NH4Cl (3,0 л) и метил-трет-бутилового эфира (9,0 л). Реакционную смесь нагревали до КТ, водную фазу снова экстрагировали метил-трет-бутиловым эфиром (2,5 л). Органические фазы объединяли, промывали насыщенным водным NaCl (2×0,3 л) и концентрировали в вакууме для получения указанного в заголовке соединения в виде масла (1,007 кг, 80,0%). 1H-ЯМР (500 МГц, CDCl3) δ м.д. 7,96 (тд, J=7,6, 1,8 Гц, 1H), 7,62-7,54 (м, 1H), 7,33-7,25 (м, 1H), 7,20-7,12 (м, 1H), 5,86 (ддд, J=17,5, 10,4, 7,3 Гц, 1H), 5,60 (дд, J=20,5, 13,8 Гц, 2H), 4,91-4,76 (м, 2H), 4,39 (д кв., J=12,8, 6,4 Гц, 1H); 13C ЯМР (125 МГц, CDCl3) δ м.д. 193,55, 162,14 (д, JCF=254,1 Гц), 135,36 (д, JCF=9,2 Гц), 130,62 (д, JCF=3,2 Гц), 128,49, 124,85 (д, JCF=3,3 Гц), 123,89, 122,93, 122,72 (д, JCF=24,5 Гц), 116,50 (д, JCF=23,7 Гц), 78,97 (кв., JCF=31,4 Гц), 74,56 (д, JCF=12,4 Гц).
HRMS, рассчитанная для C12H10F4O2 [M+H]+ 263,0695; найденная 263,0709.
1-(17) Синтез 1-(2-Фторфенил)-2-(1,1,1-трифторбут-3-ен-2-илокси)этаноноксима

В реактор добавляли гидрохлорид гидроксиламина (0,34 кг, 4,95 моль), ацетат натрия (0,47 кг, 5,70 моль) и MeOH (2,68 л). В эту суспензию загружали раствор 1-(2-фторфенил)-2-(1,1,1-трифторбут-3-ен-2-илокси)этанона (0,998 кг, 3,806 моль) в MeOH (1,8 л), и реакционную смесь нагревали до 40-50°C. После завершения (приблизительно 2 часа), реакционную смесь охлаждали до КТ и фильтровали через целит (0,5 масс.) и промывали EtOAc (3,0 л). Фильтрат концентрировали в вакууме, и к полученному остатку добавляли метил-трет-бутиловый эфир (6,3 л), воду (0,94 л) и насыщенный водный NaHCO3 (2,5 л). Органическую фазу промывали однократно водой (1,6 л) и насыщенной водной NaCl (0,1 л). Органическую фазу концентрировали в вакууме для получения указанного в заголовке соединения в виде масла (1,03 кг, 95,0%). 1H-ЯМР (500 МГц, CDCl3) δ м.д. 7,49-7,35 (м, 2H), 7,24-7,06 (м, 2H), 5,78-5,65 (м, 1H), 5,54-5,40 (м, 2H), 4,89-4,81 (м, 1H), 4,53 (д, J=12,6 Гц, 1H), 4,47 (д, J=12,6 Гц, 0,5H), 4,27-4,18 (м, 1H), 4,13-4,05 (м, 0,5H).
HRMS, рассчитано для C12H11F4NO2 [M+H]+ 278,0804; найдено 278,0780.
Примечание: 1-(2-Фторфенил)-2-(1,1,1-трифторбут-3-ен-2-илокси)этаноноксим существует в виде равновесия структурных изомеров, который составляет менее чем общее число интегральных величин.
1-(18) Синтез (3aR*,4S*,6aS*)-6a-(2-фторфенил)-4-(трифторметил)гексагидрофуро[3,4-c]изоксазола

К раствору 1-(2-фторфенил)-2-(1,1,1-трифторбут-3-ен-2-илокси)этаноноксима (1,085 кг, 3,328 моль) в ксилолах (6,9 л) добавляли гидрохинон (86,2 г, 0,8 моль) при КТ. Раствор нагревали до 128°C (внутренняя температура) в течение 18 ч. Раствор охлаждали до КТ и добавляли гексаны (7,0 л) с последующим добавлением 4,0M водной HCl (2,4 л). Реакционную смесь перемешивали в течение 1 часа и фильтровали. К твердому веществу добавляли воду (2,0 л), метил-трет-бутиловый эфир (7,0 л) и 25% масс. водн. NaOH (0,4 л). Водный слой однократно экстрагировали метил-трет-бутиловым эфиром (7,0 л), органические вещества объединяли, промывали 27% водной NaCl (2,0 л) и концентрировали в вакууме до черного масла (512,0 г, 56%). 1H-ЯМР (500 МГц, CDCl3) δ м.д. 7,64-7,52 (м, 1H), 7,39-7,31 (м, 1H), 7,19 (тд, J=7,7, 1,2 Гц, 1H), 7,11 (ддд, J=11,9, 8,2, 1,0 Гц, 1H), 4,54 (д, J=10,1 Гц, 1H), 4,34-4,23 (м, 1H), 4,26-4,17 (м, 1H), 4,16 (д, J=10,2 Гц, 1H), 4,10 (д, J=8,5 Гц, 1H), 3,71 (д, J=20,2 Гц, 1H); 13C ЯМР (125 МГц, CDCl3) δ м.д. 160,59 (д, JCF=247,0 Гц), 130,50 (д, JCF=8,7 Гц), 128,72, 124,69 (д, JCF=3,3 Гц), 124,45 (кв., JCF=281,8 Гц), 124,43 (д, JCF=11,9 Гц), 116,66 (д, JCF=22,7 Гц), 83,70 (кв., JCF=32,1 Гц), 78,17 (д, JCF=3,1 Гц), 77,63, 54,53.
HRMS, рассчитано для C12H11F4NO2 [M+H]+ 278,0804; найдено 278,0802.
1-(19) Синтез ((2S*,3R*,4S*)-4-амино-4-(2-фторфенил)-2-(трифторметил)тетрагидрофуран-3-ил)метанола

Цинк (389,2 г, 5,95 моль) помещали в реакционный сосуд и добавляли воду (893 мл). Добавляли уксусную кислоту (135 мл, 2,38 моль) при поддержании температуры ниже 10°C. Через 15 мин добавляли 6a-(2-фторфенил)-4-(трифторметил)гексагидрофуро[3,4-c]изоксазол (550,0 г, 1,98 моль) в виде раствора в THF (665 мл). Реакционную смесь перемешивали в течение 16 часов при КТ. Добавляли метиленхлорид (1,89 л) с последующим добавлением 28% водного NH4OH (552 мл) при поддержании температуры ниже 30°C. Смесь перемешивали в течение 30 мин и затем фильтровали через целит (80 г), промывали метиленхлоридом (378 мл). Водный слой экстрагировали метиленхлоридом (1,89 л). Органические вещества объединяли, промывали насыщенным, водным NaCl (1,0 л) и концентрировали в вакууме для получения масла (502 г, 90,6%). Неочищенный остаток использовали на следующей стадии без дополнительной очистки.
HRMS, рассчитано для C12H13F4NO2 [M+H]+ 280,0961; найдено 280,0972.
1-(20) Синтез ((2S,3R,4S)-4-Амино-4-(2-фторфенил)-2-(трифторметил)тетрагидрофуран-3-ил)метанол (2S,3S)-2,3-бис(бензоилокси)сукцината

К раствору 4-амино-4-(2-фторфенил)-2-(трифторметил)тетрагидрофуран-3-ил)метанола (0,502 кг, 1,798 моль) в этаноле (4,865 л) добавляли дибензоил-D-винную кислоту (0,642 кг, 1,798 моль). Полученную суспензию нагревали до 67°C. Добавляли воду (94,0 мл, 5,2 моль) в течение 15 мин при поддержании температуры >66°C. Полученный раствор охлаждали до 45°C, пока происходило осаждение. Кашицеобразную суспензию повторно нагревали до 60°C и затем охлаждали до окружающей температуры при 5°C/час. Кашицеобразную суспензию фильтровали, и твердое вещество промывали предварительно смешанным и охлажденным раствором этанола (950 мл) и водой (20 мл). Твердое вещество сушили в вакууме до постоянной массы (370 г, 97,6% ee). 1H-ЯМР (500 МГц, CD3OD) δ м.д. 8,13 (д, J=7,2 Гц, 4H), 7,66-7,58 (м, 3H), 7,54-7,45 (м, 5H), 7,36-7,20 (м, 2H), 5,92 (с, 2H), 4,79-4,66 (м, 1H), 4,40-4,28 (м, 1H), 4,04 (дд, J=12,1, 3,4 Гц, 1H), 3,92 (дд, J=12,1, 5,4 Гц, 1H), 3,30-3,24 (м, 1H); 13C ЯМР (125 МГц, ДМСО) δ м.д. 169,61, 165,81, 160,23 (д, J=246,1 Гц), 133,00, 131,34 (д, J=9,1 Гц), 129,65, 129,55, 128,08, 127,97 (д, J=3,5 Гц), 124,95 (д, J=3,3 Гц), 116,56 (д, J=23,5 Гц), 77,48 (кв., JCF=31,0 Гц), 76,33, 73,20, 65,61 (д, J=3,1 Гц), 57,11.
HRMS, рассчитано для C12H13F4NO2 [M+H]+ 280,0961; найдено 280,0967 (для аминоспирта).
Абсолютную стереохимию указанного в заголовке соединения определяли сравнением с образцом, полученным из энантиообогащенного (S)-2-(трифторметил)оксирана.
Параметры хиральной ВЭЖХ:
Оборудование, реагенты и подвижная фаза:
Подвижная фаза:
Добавить 70 мл 2-пропанола и 930 мл гептана (отмеренных отдельно градуированными цилиндрами емкостью 100 мл и 1000-мл) и 1,0 мл триэтиламина (отмеренного мерной стеклянной пипеткой) в соответствующую колбу и смешать. Дегазировать ин-лайн во время использования.
Разбавляющий раствор: - 2-Пропанол
(RKT 1,00)
(Энантиомер)
(RKT 0,93)
Типичная хроматограмма по выделению хиральной ВЭЖХ Соединения 1-(20) представлена на фиг 1.
1-(21) Синтез N-((3S,4R,5S)-3-(2-фторфенил)-4-(гидроксиметил)-5-(трифторметил)-тетрагидрофуран-3-илкарбамотиоил)бензамида
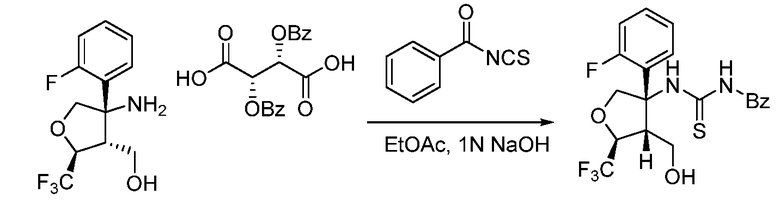
К хиральной соли ((2S,3R,4S)-4-Амино-4-(2-фторфенил)-2-(трифторметил)тетрагидрофуран-3-ил)метанол (2S,3S)-2,3-бис(бензоилокси)сукцинат (0,361 кг, 0,556 моль) добавляли EtOAc (1,08 л), и суспензию охлаждали до -3°C. 1,0н водный NaOH (1,30 л) добавляли в течение 20 мин при поддержании T <5°C. Через 5 мин добавляли бензоилизотиоцианат (80,0 мл, 594 ммоль) в течение 8 мин при поддержании T <5°C. Через 1 часа загружали EtOAc (722 мл). Водный слой удаляли, и органические вещества промывали насыщенным водным NaHCO3 (361 мл) и насыщенным водным NaCl (361 мл). Органические вещества фильтровали через целит (90 г) и промывали EtOAc (360 мл). Органические вещества концентрировали в вакууме для получения остатка, который повторно растворяли в CH2Cl2 (1,1 л) и концентрировали для получения указанного в заголовке соединения в виде желтой пены (261 г, выход 99%, обеспечиваемый остаточными растворителями), которое использовали на следующей стадии. 1H-ЯМР (500 МГц, ДМСО) δ м.д. 12,04 (с, 2H), 11,20 (с, 2H), 7,95 (д, J=7,4 Гц, 2H), 7,69-7,60 (м, 1H), 7,56-7,42 (м, 2H), 7,37-7,28 (м, 1H), 7,24-7,12 (м, 2H), 5,59 (т, J=4,5 Гц, 1H), 5,03 (д, J=9,7 Гц, 1H), 4,92 (д, J=9,7 Гц, 1H), 4,75-4,63 (м, 1H), 3,92-3,74 (м, 2H), 2,77-2,66 (м, 1H); 13C ЯМР (125 МГц, ДМСО) δ м.д. 179,98, 167,85, 159,75 (д, JCF=245,0 Гц), 133,44, 132,58, 129,88, 129,81, 129,04, 128,85, 126,31 (д, JCF=9,8 Гц), 124,36, 116,83 (д, JCF=23,4 Гц), 76,11 (кв., JCF=31,0 Гц), 74,37 (д, JCF=6,1 Гц), 68,77 (д, JCF=3,4 Гц), 57,03, 52,23.
HRMS, рассчитано для C20H18F4N2O3S [M+H]+ 441,0896; найдено 441,0818.
1-(22) Синтез N-((4aS,5S,7aS)-7a-(2-фторфенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил)бензамида
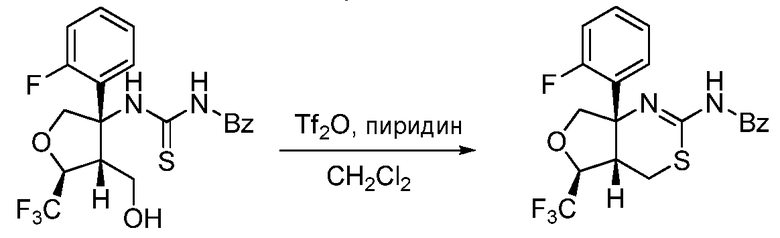
Раствор N-((3S,4R,5S)-3-(2-фторфенил)-4-(гидроксиметил)-5-(трифторметил)-тетрагидрофуран-3-илкарбамотиоил)бензамида (258,3 г, 583,8 ммоль) в CH2Cl2 (1,55 л) охлаждали до -19,4°C. Добавляли пиридин (118 мл, 1,46 моль) при поддержании температуры на уровне -20°C, и затем реакционную смесь охлаждали до -24°C. В другой продуваемый азотом сосуд добавляли CH2Cl2 (258 мл) с последующим добавлением трифторметансульфонового ангидрида (108,0 мл, 642,2 ммоль). Полученный раствор добавляли к реакционной смеси в течение 30 мин при поддержании температуры на уровне <-19,7°C. После завершения добавления, реакционную смесь перемешивали в течение 30 мин при температуре от -20°C до -15°C, и затем нагревали до -11°C в течение 20 мин. Добавляли насыщенный водный NH4Cl (646 мл) и воду (390 мл). Смесь нагревали до окружающей температуры и водный слой удаляли. Органические вещества промывали предварительно смешанным водным NH4Cl (646 мл) и водой (390 мл). Водные слои объединяли и однократно экстрагировали CH2Cl2 (520 мл). Органические вещества объединяли и концентрировали в вакууме для получения светло-оранжевой пены (250 г, 100%). Остаток использовали на следующей стадии без очистки. 1H-ЯМР (500 МГц, CDCl3) δ м.д. 8,03 (д, J=6,7 Гц, 2H), 7,52 (т, J=7,0 Гц, 1H), 7,48-7,31 (м, 4H), 7,20 (т, J=7,4 Гц, 1H), 7,12 (дд, J=12,0, 8,4 Гц, 1H), 4,82-4,73 (м, 1H), 4,60 (д, J=8,9 Гц, 1H), 4,03 (д, J=8,3 Гц, 1H), 3,57 (д, J=2,7 Гц, 1H), 3,20 (д, J=13,6 Гц, 1H), 2,81 (дд, J=13,8, 2,5 Гц, 1H); 13C ЯМР (125 МГц, CDCl3) δ м.д. 171,50, 159,57 (д, JCF=247,2 Гц), 134,62, 132,49, 130,65 (д, JCF J=8,8 Гц), 129,77, 128,51, 128,45, 125,14 (кв., JCF=281,8 Гц), 124,97 (д, JCF=3,0 Гц), 124,66 (д, JCF=10,3 Гц), 117,05 (д, JCF=23,5 Гц), 66,81 (д, JCF=5,2 Гц), 38,90, 23,20.
HRMS, рассчитано для C20H16F4N2O2S [M+H]+ 425,0947; найдено 425,0945.
1-(23) Синтез (4aS,5S,7aS)-7a-(2-фторфенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-амина
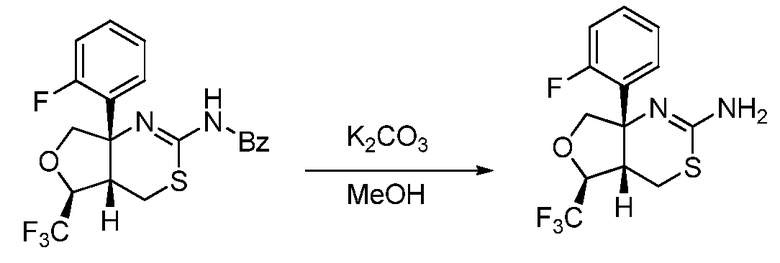
К раствору N-((4aS,5S,7aS)-7a-(2-фторфенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил)бензамида (250,2 г, 589,5 ммоль) в метаноле (1,25 ) добавляли K2CO3 (81,5 г, 590,0 ммоль). Суспензию нагревали до 65°C в течение 6 часов. После охлаждения до окружающей температуры растворитель выпаривали в вакууме. К полученному остатку добавляли 1,0н водный NaOH (1,18 л) и THF (502 мл). Гетерогенную смесь нагревали до 45°C в течение 1 часов. Смесь охлаждали до окружающей температуры и добавляли EtOAc (1,38 л). Водный слой экстрагировали EtOAc (0,75 л). Органические вещества объединяли, промывали насыщенным водным NaHCO3 (500 мл) и насыщенным водным NaCl (500 мл). Органические вещества концентрировали в вакууме для получения указанного в заголовке соединения в виде масла коричневого цвета (184,1 г, выход 91,6%, обеспечиваемый остаточными растворителями). 1H-ЯМР (500 МГц, ДМСО) δ м.д. 7,49-7,42 (м, 1H), 7,40-7,33 (м, 1H), 7,26-7,15 (м, 2H), 6,26 (с, 2H), 4,77-4,54 (м, 1H), 4,40 (д, J=8,0 Гц, 1H), 3,80 (дд, J=7,9, 2,3 Гц, 1H), 3,24-3,17 (м, 1H), 3,00 (дд, J=13,9, 3,2 Гц, 1H), 2,85 (дд, J=13,9, 3,9 Гц, 1H); 13C ЯМР (125 МГц, ДМСО) δ м.д. 159,75 (д, JCF=245,1 Гц), 149,51, 131,31 (д, JCF=3,9 Гц), 130,13 (д, JCF=8,8 Гц), 128,08 (д, JCF=10,4 Гц), 128,28 (кв., JCF=282,1 Гц), 124,87 (д, JCF=3,0 Гц), 116,80 (д, J=23,8 Гц), 78,77, 76,80 (кв., JCF=30,8 Гц), 66,31, 36,37, 23,27.
HRMS, рассчитано для C13H12F4N2OS [M+H]+ 321,0685; найдено 321,0677.
1-(24) Синтез гидрохлорида (4aS,5S,7aS)-7a-(2-фтор-5-нитрофенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-амина

В охлажденный сосуд, содержащий (4aS,5S,7aS)-7a-(2-фторфенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-амин (184,1 г, 574,8 ммоль) порциями добавляли трифторуксусную кислоту (0,954 кг) при поддержании температуры ниже 20°C. Смесь охлаждали до 3,5°C и добавляли серную кислоту (146 мл, 2,73 моль) в течение 20 мин при поддержании температуры ниже 5°C. Дымящуюся азотную кислоту (39,8 мл, 0,948 моль) добавляли в течение 30 мин при поддержании температуры ниже 10°C. Через 1,5 часа при 0-10°C реакционную смесь медленно гасили переносом в водный раствор NaOH (575 г, 14,4 моль) в воде (4,6 л), охлажденный до 5°C. Полученную суспензию перемешивали в течение 1 часа при 21°C. Затем суспензию фильтровали, и твердое вещество промывали холодной водой (920 мл). Твердое вещество сушили в вакууме до постоянной массы и затем растворяли в этаноле (1,05 л). Раствор нагревали до 35°C и добавляли концентрированную HCl (55,6 мл, 0,690 моль) при поддержании температуры ниже 40°C. Суспензию затем охлаждали до -5°C, выдерживали в течение 1 часа и фильтровали. Твердое вещество промывали холодным этанолом (420 мл) и сушили до постоянной массы для получения указанного в заголовке соединения (185,0 г, 87,3%). 1H-ЯМР (500 МГц, ДМСО) δ м.д. 11,80 (с, 2H), 8,45-8,36 (м, 1H), 8,31 (дд, J=6,6, 2,5 Гц, 1H), 7,66 (дд, J=11,1, 9,3 Гц, 1H), 4,96-4,72 (м, 1H), 4,58 (д, J=10,0 Гц, 1H), 4,27 (д, J=9,9 Гц, 1H), 3,76-3,66 (м, 1H), 3,39 (дд, J=14,9, 3,6 Гц, 1H), 3,24 (дд, J=14,3, 4,6 Гц, 1H); 13C ЯМР (125 МГц, ДМСО) δ м.д. 168,34, 163,33 (д, JCF=257,8 Гц), 144,58, 127,61 (д, JCF=11,6 Гц), 125,84, 124,10, 119,28 (д, JCF=26,5 Гц), 77,38 (кв., JCF=31,5 Гц), 75,99, 65,88 (д, JCF=4,8 Гц), 40,36, 23,98.
HRMS, рассчитано для C13H11F4N3O3S [M+H]+ 366,0536; найдено 366,0523.
1-(25) Синтез (4aS,5S,7aS)-7a-(5-амино-2-фторфенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-амина
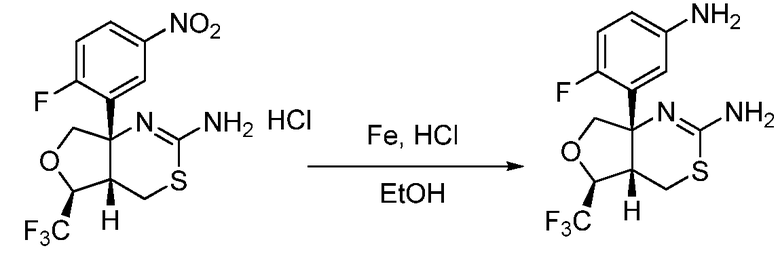
Этанол (0,975 л) добавляли к порошку железа (62,5 г, 1,12 моль) в атмосфере азота. Добавляли концентрированную HCl (9,03 мл) при окружающей температуре и суспензию нагревали до 65°C в течение 1,5 ч. Суспензию затем охлаждали до 50°C и добавляли насыщенный водный NH4Cl (299 г). Температуре реакционной смеси давали возможность достичь 50°C и порциями добавляли гидрохлорид (4aS,5S,7aS)-7a-(2-фтор-5-нитрофенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-амина (75,0 г, 187,0 моль) при поддержании температуры ниже 68°C. Через 30 мин добавляли этанол (0,45 л) и реакционную смесь охлаждали до 20-25°C в течение 1 ч. Суспензию перемешивали в течение 2 часов и фильтровали через целит (75 г), промывая этанолом (0,972 л). Раствор концентрировали в вакууме до твердого вещества коричневого цвета. Добавляли воду (0,9 л) с последующим добавлением 3,0н NaOH (0,187 л, 560 ммоль) при поддержании температуры ниже 35°C. Полученную суспензию перемешивали в течение 1 часа при 20-25°C. Суспензию фильтровали, и твердое вещество промывали холодной водой (0,38 л). Твердое вещество сушили в вакууме при 40-45°C в течение 24 часов для получения указанного в заголовке соединения (57,7 г, 95,5 %). 1H-ЯМР (500 МГц, ДМСО) δ м.д. 6,81 (дд, J=12,5, 8,6 Гц, 1H), 6,62 (дд, J=7,0, 2,9 Гц, 1H), 6,50-6,42 (м, 1H), 6,16 (с, 2H), 4,96 (с, 2H), 4,72-4,54 (м, 1H), 4,35 (д, J=7,8 Гц, 1H), 3,74 (дд, J=7,8, 2,5 Гц, 1H), 3,18-3,08 (м, 1H), 3,01 (дд, J=13,9, 3,0 Гц, 1H), 2,84 (дд, J=13,8, 3,8 Гц, 1H); 13C ЯМР (125 МГц, ДМСО) δ м.д. 156,20 (д, JCF=243,0 Гц), 148,73, 145,49, 127,86 (д, JCF=11,0 Гц), 116,79 (д, JCF=24,8 Гц), 116,10 (д, JCF=3,3 Гц), 114,10 (д, JCF=8,0 Гц), 78,89, 76,57 (кв., JCF=31,0 Гц), 66,35, 36,35, 23,11.
HRMS, рассчитано для C13H13F4N3OS [M+H]+ 336,0794; найдено 336,0789.
Указанное в заголовке соединение подвергали тесту Ames (Штаммы тестирования Salmonella typhimurium TA98, TA100, TA1535 и TA1537 и штамм тестирования Escherichia coli WP2 uv. Mutation Research 1975, 31, 347; Mutation Research 1976, 38, 3; Proc. Nat. Acad. Sci. USA 1976, 73, 950; Proc. Nat. Acad. Sci. USA 1975, 72, 5135) в отсутствие и в присутствии крысиной печени S9. Соединение дало отрицательный результат до самой высокой тестированной дозы/концентрации (5000 мкг/планшет).
1-(26) Синтез N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-метоксипиразин-2-карбоксамида
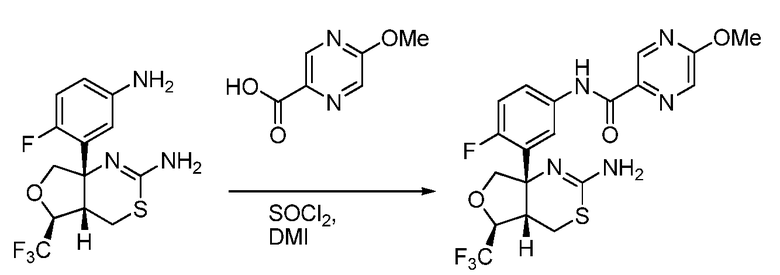
Суспензию 5-метоксипиразин-2-карбоновой кислоты (26,29 г, 0,17 моль) в N,N'-диметилимидазолин-2-оне (160 мл) перемешивали при окружающей температуре в течение 15 мин, затем охлаждали до 2,2°C. Добавляли тионилхлорид (14,7 мл, 0,202 моль) при поддержании температуры ниже 5°C. Полученную суспензию перемешивали при 0-10°C в течение 2 часов при его превращении в прозрачный раствор. В другом сосуде (4aS,5S,7aS)-7a-(5-амино-2-фторфенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-амин (52,0 г, 0,155 моль) растворяли в N,N'-диметилимидазолин-2-оне (160 мл). Полученный раствор добавляли к раствору ацилхлорида при поддержании температуры ниже 10°C. Реакционную смесь перемешивали в течение 30 мин. Загружали воду (780 мл) при поддержании температуры ниже 30°C. Полученную смесь перемешивали в течение 30 мин и затем добавляли EtOAc (780 мл). К этой смеси добавляли 50% водный NaOH (84,8 г) до тех пор, пока pH водного слоя достигал 11. Водный слой экстрагировали EtOAc (260 мл). Органические вещества объединяли, промывали насыщенным водным NaCl (260 мл) и водой (260 мл). Органические вещества фильтровали через прокладку целита (26 г) и промывали EtOAc (260 мл). Органические вещества концентрировали в вакууме для получения твердого вещества. К твердому веществу добавляли 1-пропанол (728 мл), и суспензию нагревали до 75°C до образования прозрачного раствора. Раствор охлаждали до -10°C и держали в течение 1 ч. Твердое вещество фильтровали, промывали холодным 1-пропанолом (104 мл) и сушили в вакууме (35°C) до постоянной массы для получения указанного в заголовке соединения (62,1 г, 84,9 %). 1H-ЯМР (500 МГц, ДМСО) δ м.д. 10,56 (с, 2H), 8,88 (д, J=1,2 Гц, 1H), 8,39 (д, J=1,2 Гц, 1H), 7,95-7,83 (м, 2H), 7,18 (дд, J=12,0, 8,8 Гц, 1H), 6,25 (с, 2H), 4,76-4,60 (м, 1H), 4,36 (д, J=8,1 Гц, 1H), 4,01 (с, 3H), 3,88 (дд, J=7,9, 2,3 Гц, 1H), 3,23-3,11(м, 2H), 2,91 (дд, J=13,8, 3,6 Гц, 1H); 13C ЯМР (125 МГц, ДМСО) δ м.д. 162,11, 161,93, 156,13 (д, JCF=242,9 Гц), 149,38, 142,01, 138,35, 135,09, 133,98, 128,53 (д, JCF=11,6 Гц), 126,06 (кв., JCF=282,0 Гц), 123,32, 121,93 (д, JCF=8,6 Гц), 116,76 (д, JCF=25,1 Гц), 78,86 (д, JCF=6,9 Гц), 76,94 (кв., JCF=30,5 Гц), 66,37, 54,75, 36,44, 23,53.
HRMS, рассчитано для C19H17F4N5O3S [M+H]+ 472,1066; найдено 472,1052.
Удельное оптическое вращение [α]D+110,5 (c 0,519, MeOH)
Параметры удельного оптического вращения:
Параметры прибора:
Альтернативное получение N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-(фторметил)пиразин-2-карбоксамида (Пример 8)
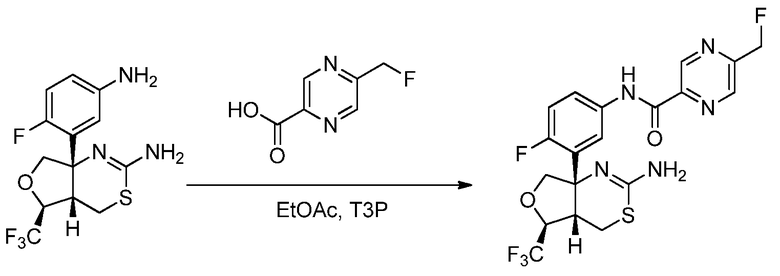
5-(Фторметил)пиразин-2-карбоновую кислоту (32,6 г, 1,05 эквивалента) и (4aS,5S,7aS)-7a-(5-амино-2-фторфенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-амин (70,0 г, 1,0 эквивалент)1 загружали в реактор и к смеси добавляли этилацетат (EtOAc, 630 мл) для получения суспензии. Добавляли раствор ангидрида н-пропанфосфоновой кислоты (T3P, 146 г, 1,10 эквивалента, 50 масс.% в EtOAc) при окружающей температуре при поддержании внутренней температуры ниже 30°C. Реакционную смесь перемешивали при 40-45°C >3 часов и проводили мониторинг ВЭЖХ. Реакционную смесь охлаждали до 15-20°C и загружали воду (140 мл). Через 10-15 минут загружали 28% гидроксид аммония (175 мл) при поддержании внутренней температуры ниже 30°C. Добавляли EtOAc (245 мл) и реакционную смесь перемешивали в течение 30 минут при окружающей температуре. Водную фазу отделяли и обратно экстрагировали в EtOAc (490 мл). Органические фазы объединяли и промывали 15% водной NaCl (140 мл) и водой (140 мл). Органический слой фильтровали через целит (1,0 масс.) и промывали EtOAc (140 мл). Раствор концентрировали в вакууме для получения твердого вещества бежевого цвета (количественный выход неочищенного продукта), которое перекристаллизовывали из 1-пропанола для получения N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-(фторметил)пиразин-2-карбоксамида в виде белого твердого вещества (70,0 г).
1H-ЯМР (500 МГц, ДМСО) δ 10,89 (с, 1H), 9,30 (с, 1H), 8,89 (с, 1H), 7,95 (дд, J=7,3, 2,7 Гц, 1H), 7,94-7,89 (м, 1H), 7,21 (дд, J=12,0, 8,8 Гц, 1H), 6,22 (с, 2H), 5,71 (д, J=46,3 Гц, 2H), 4,77-4,61 (м, 1H), 4,37 (д, J=8,1 Гц, 1H), 3,87 (дд, J=8,0, 2,7 Гц, 1H), 3,20 (дт, J=7,0, 3,5 Гц, 1H), 3,15 (дд, J=13,9, 3,1 Гц, 1H), 2,91 (дд, J=13,8, 3,8 Гц, 1H),13C ЯМР (126 МГц, ДМСО) δ 161,32 (с), 155,82 (д, J=243,4 Гц), 153,71 (д, J=18,7 Гц), 148,77 (с), 144,71 (д, J=1,9 Гц), 143,30 (с), 141,01 (д, J=5,6 Гц), 134,36 (д, J=2,0 Гц), 128,20 (д, J=12,1 Гц), 125,57 (кв., J=283,0 Гц), 123,12 (д, J=3,6 Гц), 121,64 (д, J=8,6 Гц), 116,35 (д, J=25,2 Гц), 82,55 (д, J=165,8 Гц), 78,37 (с), 76,44 (кв., J=30,6 Гц), 65,89 (д, J=5,3 Гц), 35,89 (с), 23,01 (с).
HRMS, рассчитано для C19H17F5N5O2S [M+H]+ 474,1023; найдено 474,1032.
Удельное оптическое вращение: [α]D 20=+102,4
1Получение (4aS,5S,7aS)-7a-(5-амино-2-фторфенил)-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-амина описано выше в настоящей заявке на стадии 1-(25) в Альтернативном получении N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил)-5-метоксипиразин-2-карбоксамида (Пример 1).
Клеточный анализ in vitro:
Количественное определение пептида Aβ в культуре нейронов из мозга крысиного плода
(1) Первичная нейронная культура крыс
Первичные нейронные культуры получали из коры головного мозга 18-дневных эмбрионов крыс Wistar (Charles River, UK). В частности, эмбрионов в асептических условиях удаляли у беременных крыс под эфирной анестезией. Мозг выделяли из эмбриона и погружали в HBSS (сбалансированный солевой раствор Хенкса) (Sigma Aldrich #H9269), содержащий 10 мМ HEPES (N-2-Гидроксиэтилпиперазин-N'-2этансульфоновой кислоты) (Gibco #15630-056). Кору головного мозга брали из изолированного мозга под стереоскопическим микроскопом. Взятые фрагменты коры головного мозга ферментативно обрабатывали в ферментном растворе, содержащем 0,05% раствор трипсина-EDTA (GIBCO, #25300) при 37°C в течение 20 минут для диспергирования клеток. Затем клетки дважды промывали и затем осторожно ресуспендировали в Нейробазальной среде (Gibco #21103) с добавлением добавки 2% B27 (GIBCO #17504-044), 0,5 мМ L-глутамина (GIBCO #25030), 1x N2 (GIBCO #17502-048), 100 мкг/мл пенициллина/стрептомицина (GIBCO 15140-122) и 5% инактивированной FCS (фетальной телячьей сыворотки) (PAA #A15-701). Клеточную дисперсию фильтровали через нейлоновую сетку с размером пор 40 мкм (BD Falcon #352340) для удаления остающейся клеточной массы, и, таким образом, получали суспензию нейронных клеток. Суспензию нейронных клеток разбавляли указанной выше средой и затем высевали в объеме 100 мкл/лунку при первоначальной плотности клеток 3,25×105 клеток/мл в покрытом поли-D-лизином 96-луночном культуральном планшете (Greiner #655940). Высеянные клетки культивировали в культуральном планшете при 37°C в 5% CO2-95% воздухе в течение 24 ч. Общее количество среды замещали «аналитической Нейробазальной средой» (как указано выше, исключая инактивированную нагреванием FCS), и затем клетки культивировали в течение еще пяти дней.
(2) Добавление соединения
Лекарственное средство добавляли в культуральный планшет на 6-ой день культивирования следующим образом. Получали 8 уровней серийных разведений в ДМСО при концентрации x1000 конечной аналитической концентрации (FAC). Затем растворы соединения получали добавлением 999 мкл «аналитической Нейробазальной среды» (как описано в предыдущем разделе) к 1 мкл основного раствора соединения в ДМСО. Общее количество среды удаляли из каждой лунки планшета и добавляли 140 мкл/лунку «аналитической Нейробазальной среды» с последующим добавлением 60 мкл раствора соединения. Конечная концентрация ДМСО составляла 0,1%.
(3) Взятие проб
Клетки культивировали в течение или 1, или 3 дней после добавления соединения для анализов ABx-40 и ABx-42 соответственно. Брали 150 мкл среды образца и использовали в качестве образца для ELISA (иммуноферментного анализа).
(4) Оценка выживания клеток
Выживание клеток оценивали, используя анализ Alamar в соответствии со следующей процедурой. После взятия образца, подлежащего использованию при анализе ELISA, 50 мкл 20% раствора Alamar синего (Invitrogen #DAL1100) в аналитической Нейробазальной среде добавляли к 50 мкл остающегося образца внутри каждой лунки. Затем клетки инкубировали при 37°C в 5% CO2-95% воздуха в течение 1 ч.
Измерение интенсивности флуоресценции для каждой лунки проводили при 540/590 нм, используя планшетное считывающее устройство Pherastar plus (BMG labtech). После измерения, лунки, не имеющие клеток, и содержащие только среду и раствор Alamar, устанавливали в качестве фона (bkg).
(5) ELISA Aβ
Набор для определения человеческого/крысиного β Амилоида (42) ELISA Kit Wako (#290-62601) и набор для определения человеческого/крысиного β Амилоида (40) ELISA Kit Wako (#294-62501), выпускаемые компанией Wako Pure Chemical Industries, Ltd., использовали для ELISA Aβ. ELISA Aβ проводили в соответствии с протоколами, рекомендуемыми производителями, описанными в документах, сопровождающих наборы. Результаты были показаны в виде процентов относительно контрольных групп, и величины IC50 для каждого соединения определяли, используя четырехпараметрическую модель подгонки к логистической кривой с использованием пакета программного обеспечения XLFIT5 (IDBS).
Соединения по настоящему изобретению обладают эффектом снижения продукции Aβ42.
Соединение общей формулы (I) или его фармацевтически приемлемая соль в соответствии с настоящим изобретением обладают эффектом снижения продукции Aβ42. Таким образом, настоящее изобретение может в частности обеспечить получение профилактического или терапевтического средства по поводу нейродегенеративного заболевания, вызванного Aβ, такого как деменция типа Альцгеймера или синдром Дауна.
По данным измерения описанным выше анализом in vitro, Примеры с 1 по 18 соединения показали величины IC50 менее чем 0,1 мкМ, как показано в таблице 5:
| название | год | авторы | номер документа |
|---|---|---|---|
| КОНДЕНСИРОВАННОЕ ПРОИЗВОДНОЕ АМИНОДИГИДРОТИАЗИНА | 2009 |
|
RU2476431C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ОСНОВЕ КОНДЕНСИРОВАННОГО ПРОИЗВОДНОГО АМИНОДИГИДРОТИАЗИНА | 2015 |
|
RU2710227C2 |
| АМИДЫ КОНДЕНСИРОВАННОГО ПИПЕРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ ИОННЫХ КАНАЛОВ | 2014 |
|
RU2741810C2 |
| КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ АНТИТЕЛО ПРОТИВ ПРОТОФИБРИЛЛ АБЕТА И ИНГИБИТОР БЕТА-СЕКРЕТАЗЫ ВАСЕ1 ДЛЯ ЛЕЧЕНИЯ БОЛЕЗНИ АЛЬЦГЕЙМЕРА | 2017 |
|
RU2786476C2 |
| РЕАКЦИИ МАКРОЦИКЛИЗАЦИИ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И ДРУГИЕ ФРАГМЕНТЫ, ПРИГОДНЫЕ В ПОЛУЧЕНИИ АНАЛОГОВ ХАЛИХОНДРИНА B | 2014 |
|
RU2710545C2 |
| МОДУЛЯТОРЫ SHIP1 И ОТНОСЯЩИЕСЯ К НИМ СПОСОБЫ | 2014 |
|
RU2679805C2 |
| ФЕНИЛОКСАЗОЛИДИНОНЫ, ИМЕЮЩИЕ С-С-СВЯЗЬ С 4-8-ЧЛЕННЫМИ ГЕТЕРОЦИКЛИЧЕСКИМИ КОЛЬЦАМИ | 1996 |
|
RU2175324C2 |
| N-[3-(5-АМИНО-3,3А,7,7А-ТЕТРАГИДРО-1Н-2,4-ДИОКСА-6-АЗА-ИНДЕН-7-ИЛ)-ФЕНИЛ]-АМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ВАСЕ1 И(ИЛИ) ВАСЕ2 | 2012 |
|
RU2597308C2 |
| ИНГИБИТОРЫ RMT5 | 2019 |
|
RU2814198C2 |
| ИНГИБИТОРЫ ПРОТЕИНКИНАЗ ДЛЯ УСИЛЕНИЯ РЕГЕНЕРАЦИИ ПЕЧЕНИ ИЛИ СНИЖЕНИЯ ИЛИ ПРЕДОТВРАЩЕНИЯ ГИБЕЛИ ГЕПАТОЦИТОВ | 2019 |
|
RU2822216C2 |
Изобретение относится к соединениям формулы (I), обладающим свойством ингибитора ВАСЕ1, их применению и фармацевтической композиции для лечения заболеваний, опосредованных активностью ВАСЕ1, в том числе нейродегенеративных заболеваний, таких как деменция типа Альцгеймера или синдром Дауна. В общей формуле (I) X обозначает водород или фтор; A обозначает CH или N; Y обозначает метил, этил, монофторметил, дифторметил, трифторметил, дифторэтил, метокси, этокси, метоксиметил или -C≡N. 3 н. и 9 з.п. ф-лы, 5 табл., 1 ил., 18 пр.

1. Соединение формулы (I):

или его фармацевтически приемлемая соль,
где
X обозначает водород или фтор;
А обозначает СН или N;
Y обозначает метил, этил, монофторметил, дифторметил, трифторметил, дифторэтил, метокси, этокси, метоксиметил или C≡N.
2. Соединение по п. 1, где X обозначает водород, или его фармацевтически приемлемая соль.
3. Соединение по п. 1 или 2, где А обозначает N, или его фармацевтически приемлемая соль.
4. Соединение по п. 1 или 2, где Y обозначает метил, монофторметил, дифторметил, трифторметил или метокси, или его фармацевтически приемлемая соль.
5. Соединение по п. 1, где соединение выбрано из:
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-d][1,3]тиазин-7а-ил)-4-фторфенил)-5-метоксипиразин-2-карбоксамида;
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4Н-фуро[3,4-d][1,3]тиазин-7а-ил)-4-фторфенил)-5-цианопиколинамида;
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4Н-фуро[3,4-d][1,3]тиазин-7а-ил)-4-фторфенил)-5-(дифторметил)пиразин-2-карбоксамида;
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4Н-фуро[3,4-d][1,3]тиазин-7а-ил)-4-фторфенил)-5-(трифторметил)пиколинамида;
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4Н-фуро[3,4-d][1,3]тиазин-7а-ил)-4-фторфенил)-5-метилпиразин-2-карбоксамида;
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4Н-фуро[3,4-d][1,3]тиазин-7а-ил)-4-фторфенил)-5-метилпиколинамида;
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4Н-фуро[3,4-d][1,3]тиазин-7а-ил)-4-фторфенил)-5-этилпиколинамида;
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4Н-фуро[3,4-d][1,3]тиазин-7а-ил)-4-фторфенил)-5-(фторметил)пиразин-2-карбоксамида;
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-d][1,3]тиазин-7а-ил)-4-фторфенил)-5-метоксипиколинамида;
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-d][1,3]тиазин-7а-ил)-4-фторфенил)-5-этоксипиразин-2-карбоксамида;
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4a,5,7,7a-тетрагидро-4Н-фуро[3,4-d][1,3]тиазин-7а-ил)-4-фторфенил)-5-(1,1-дифторэтил)пиразин-2-карбоксамида;
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-d][1,3]тиазин-7а-ил)-4-фторфенил)-5-(трифторметил)пиразин-2-карбоксамида;
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-d][1,3]тиазин-7а-ил)-4-фторфенил)-5-(метоксиметил)пиразин-2-карбоксамида;
N-{3-[(4aS,5S,7aS)-2-амино-5-(трифторметил)-4а,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7а(7Н)-ил]-4-фторфенил}-5-[(2Н3)метилокси]пиразин-2-карбоксамида;
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-d][1,3]тиазин-7а-ил)-4,5-дифторфенил)-5-(дифторметил)пиразин-2-карбоксамида;
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-d][1,3]тиазин-7а-ил)-4,5-дифторфенил)-5-метоксипиразин-2-карбоксамида;
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-d][1,3]тиазин-7а-ил)-4,5-дифторфенил)-5-метилпиразин-2-карбоксамида;
N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-d][1,3]тиазин-7а-ил)-4,5-дифторфенил)-5-(фторметил)-пиразин-2-карбоксамида;
или его фармацевтически приемлемая соль.
6. Соединение по п. 1, которое представляет собой N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-d][1,3]тиазин-7а-ил)-4-фторфенил)-5-метоксипиразин-2-карбоксамид, или его фармацевтически приемлемая соль.
7. Соединение по п. 1, которое представляет собой N-(3-((4aS,5S,7aS)-2-амино-5-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-d][1,3]тиазин-7а-ил)-4-фторфенил)-5-(фторметил)пиразин-2-карбоксамид, или его фармацевтически приемлемая соль.
8. Соединение по п. 1 или его фармацевтически приемлемая соль для применения при лечении заболеваний, опосредованных активностью ВАСЕ1.
9. Соединение по п. 1 или его фармацевтически приемлемая соль для ингибирования бета-сайта фермента 1 расщепления белка-предшественника амилоида-β (ВАСЕ1).
10. Соединение по п. 1 или его фармацевтически приемлемая соль для лечения нейродегенеративного заболевания, такого как деменция типа Альцгеймера (AD) или синдром Дауна.
11. Применение соединения по любому из пп. 1-7 или его фармацевтически приемлемой соли для получения лекарственного средства для лечения или профилактики нейродегенеративного заболевания, такого как деменция типа Альцгеймера (AD) или синдром Дауна.
12. Фармацевтическая композиция, обладающая свойством ингибитора ВАСЕ1, содержащая соединение из пп. 1-7 или его фармацевтически приемлемую соль, в качестве активного ингредиента в сочетании с фармацевтически приемлемым носителем.
| СПОСОБ И СИСТЕМА ЗАКАЗА, ЗАГРУЗКИ И ИСПОЛЬЗОВАНИЯ ВХОДНЫХ КАРТОЧЕК | 1999 |
|
RU2233474C2 |
| WO 2011005738 A1, 13.01.2011 | |||
| КОНДЕНСИРОВАННЫЙ ПИРИДАЗИН ИЛИ ЕГО ФАРМАКОЛОГИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, СРЕДСТВО, ПРОЯВЛЯЮЩЕЕ ИНГИБИРУЮЩУЮ АКТИВНОСТЬ В ОТНОШЕНИИ ЦИКЛИЧЕСКОЙ ГМФ- ФОСФОДИЭСТЕРАЗЫ | 1995 |
|
RU2128175C1 |

