Введение
Содержащие кетоны опиоиды подвержены неправильному использованию, злоупотреблению и передозировке. Следовательно, необходимо контролировать использование и доступ к этим лекарственным средствам. Контроль доступа к лекарственным средствам является дорогостоящим в осуществлении и может привести к отказу в лечении для пациентов, которые не способны к самостоятельному дозированию. Например, пациентам, страдающим от острой боли, может быть отказано в лечении опиоидом, если они не помещены в стационар. Кроме того, контроль за использованием часто неэффективен, что приводит к значительному уровню заболевания и опасным социальным последствиям.
Краткое описание
Варианты осуществления данного изобретения включают композиции, содержащие опиоидное пролекарство с модифицированным кетоном, где опиоидное пролекарство с модифицированным кетоном содержит опиоид с модифицированным кетоном, ковалентно связанный с фрагментом-предшественником, содержащим фрагмент, расщепляемый трипсином, где расщепление расщепляемого трипсином фрагмента промежуточными соединениями трипсина приводит к высвобождению кетон-содержащего опиоида; и трипсиновый ингибитор, который взаимодействует с трипсином, являющимся посредником в контролируемом ферментативном высвобождении кетон-содержащего опиоида из опиоидного пролекарства с модифицированным кетоном после приема внутрь композиции. Такое расщепление может инициировать, содействовать или влиять на высвобождение кетон-содержащего опиоида.
Варианты осуществления включают единицы дозы, содержащие композиции, содержащие опиоидное пролекарство с модифицированным кетоном и трипсиновый ингибитор, где опиоидное пролекарство с модифицированным кетоном и трипсиновый ингибитор присутствуют в единице дозы в эффективном количестве для того, чтобы обеспечить предварительно выбранную фармакокинетическую (ФК) кривую после принятия лекарства внутрь. В следующих вариантах осуществления предварительно выбранная ФК кривая содержит по меньшей мере одно значение ФК параметра, которое меньше значения ФК параметра кетон-содержащего опиоида, высвобождаемого после принятия внутрь эквивалентной дозы опиоидного пролекарства с модифицированным кетоном в отсутствие ингибитора. В дальнейших вариантах осуществления изобретения значение ФК параметра выбрано из значения Cmax кетон-содержащего опиоида, значения экспозиции кетон-содержащего опиоида и значения (1/Tmax кетон-содержащего опиоида).
В определенных вариантах осуществления изобретения единица дозы обеспечивает определенную заранее ФК кривую после принятия внутрь по меньшей мере двух единиц дозы. В родственных вариантах осуществления изобретения определенная заранее ФК кривая таких единиц дозы модифицирована по отношению к ФК кривой после принятия внутрь эквивалентной дозы опиоидного пролекарства с модифицированным кетоном без ингибитора. В родственных вариантах осуществления изобретения такая единица дозы обеспечивает то, что принятие внутрь увеличенного числа единиц дозы обеспечивает линейную ФК кривую. В родственных вариантах осуществления изобретения такая единица дозы обеспечивает то, что принятие внутрь увеличенного числа единиц дозы обеспечивает нелинейную ФК кривую. В родственных вариантах осуществления значение ФК параметра ФК кривой таких единиц дозы выбрано из значения Cmax кетон-содержащего опиоида, значения (1/Tmax кетон-содержащего опиоида) и значения экспозиции кетон-содержащего опиоида.
Варианты осуществления изобретения включают композиции, содержащие контейнер, пригодный для вмещения композиции для введения пациенту; и единицу дозы, описанную здесь, содержащуюся в контейнере.
Варианты осуществления включают единицы дозы опиоидного пролекарства с модифицированным кетоном и трипсинового ингибитора, где единица дозы имеет вес от 1 микрограмма до 2 граммов. Варианты осуществления изобретения включают фармацевтические композиции опиоидного пролекарства с модифицированным кетоном и трипсинового ингибитора, где общий вес опиоидного пролекарства с модифицированным кетоном и трипсинового ингибитора составляет от 0,1% до 99% при расчете на грамм композиции.
Варианты осуществления изобретения включают композиции и единицы дозы, где опиоидное пролекарство с модифицированным кетоном представляет собой вещество формулы KC-(Ia):
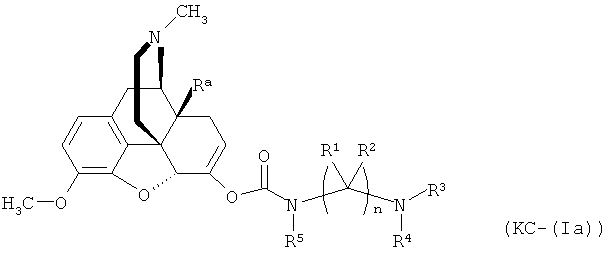
где:
Ra представляет собой водород или гидроксил;
R5 выбран из алкила, замещенного алкила, арилалкила, замещенного арилалкила, арила и замещенного арила;
каждый R1 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, ацила и аминоацила;
каждый R2 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, ацила и аминоацила;
или R1 и R2 вместе с углеродом, к которому они присоединены, образуют циклоалкильную, замещенную циклоалкильную, арильную или замещенную арильную группу, или две R1 или R2 группы на соседних атомах углерода вместе с атомами углерода, к которым они присоединены, образуют циклоалкильную, замещенную циклоалкильную, арильную или замещенную арильную группу;
n представляет собой целое число от 2 до 4;
R3 является водородом;
R4 представляет собой 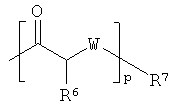 ;
;
каждый R6 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, арилалкила, замещенного арилалкила, гетероалкила, замещенного гетероалкила, гетероарила, замещенного гетероарила, гетероарилалкила и замещенного гетероарилалкила или, необязательно, R6 и R7 вместе с атомами, к которым они присоединены, образуют циклогетероалкильное или замещенное циклогетероалкильное кольцо;
каждый W независимо представляет собой -NR8-, -О- или -S-;
каждый R8 независимо выбран из водорода, алкила, замещенного алкила, арила и замещенного арила или, необязательно, каждый R6 и R8 независимо вместе с атомами, к которым они присоединены, образуют циклогетероалкильное или замещенное циклогетероалкильное кольцо;
p представляет собой целое число от 1 до 100; и
R7 выбран из водорода, алкила, замещенного алкила, ацила, замещенного ацила, алкоксикарбонила, замещенного алкоксикарбонила, арила, замещенного арила, арилалкила и замещенного арилалкила;
или его соль, гидрат или сольват.
Варианты осуществления изобретения включают композиции и единицы дозы, где опиоидное пролекарство с модифицированным кетоном представляет собой соединение формулы KC-(Ib):
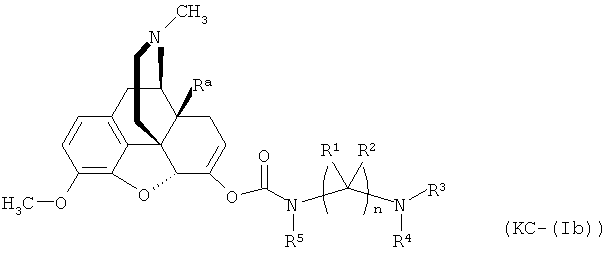
где:
Ra представляет собой водород или гидроксил;
R5 выбран из алкила, замещенного алкила, арилалкила, замещенного арилалкила, арила и замещенного арила;
каждый R0 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, ацила и аминоацила;
каждый R2 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, ацила и аминоацила;
или R1 и R2 вместе с углеродом, к которому они присоединены, образуют циклоалкильную или замещенную циклоалкильную группу, или две R1 или R2 группы на соседних атомах углерода вместе с атомами углерода, к которым они присоединены, образуют циклоалкильную или замещенную циклоалкильную группу;
n представляет собой целое число от 2 до 4;
R3 является водородом;
R4 представляет собой ;
каждый R независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, арилалкила, замещенного арилалкила, гетероалкила, замещенного гетероалкила, гетероарила, замещенного гетероарила, гетероарилалкила и замещенного гетероарилалкила или, необязательно, R6 и R7 вместе с атомами, к которым они присоединены, образуют циклогетероалкильное или замещенное циклогетероалкильное кольцо;
каждый W независимо представляет собой -NR8-, -О- или -S-;
каждый R8 независимо выбран из водорода, алкила, замещенного алкила, арила и замещенного арила или, необязательно, каждый R6 и R8 независимо вместе с атомами, к которым они присоединены, образуют циклогетероалкильное или замещенное циклогетероалкильное кольцо;
p представляет собой целое число от 1 до 100; и
R7 выбран из водорода, алкила, замещенного алкила, ацила, замещенного ацила, алкоксикарбонила, замещенного алкоксикарбонила, арила, замещенного арила, арилалкила и замещенного арилалкила;
или его соль, гидрат или сольват.
Варианты осуществления изобретения включают композиции и единицы дозы, где опиоидное пролекарство с модифицированным кетоном представляет собой соединение формулы KC-(II):
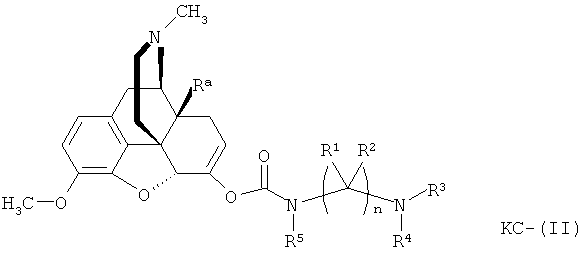
где:
Ra представляет собой водород или гидроксил;
R5 выбран из (1-6С) алкила, (1-6С) замещенного алкила, -(CH2)q(C6H4)-COOH, -(CH2)q(C3H4)-COOCH3, и -(CH2)q(C6H4)-COOCH2CH3, где q представляет собой целое число от 1 до 10;
каждый R1 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, ацила и аминоацила;
каждый R2 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, ацила и аминоацила;
или R1 и R2 вместе с углеродом, к которому они присоединены, образуют циклоалкильную или замещенную циклоалкильную группу, или две R1 или R2 группы на соседних атомах углерода вместе с атомами углерода, к которым они присоединены, образуют циклоалкильную или замещенную циклоалкильную группу;
n равно 2 или 3;
R3 является водородом;
R4 представляет собой остаток L-аминокислоты, выбранной из аланина, аргинина, аспарагина, аспарагиновой кислоты, цистеина, глицина, глутамина, глутаминовой кислоты, гистидина, изолейцина, лейцина, лизина, метионина, фенилаланина, пролина, серина, треонина, триптофана, тирозина и валина, или остатка N-ацильного производного любой из перечисленных аминокислот; или остаток пептида, состоящий из по меньшей мере двух остатков L-аминокислот, независимо выбранных из аланина, аргинина, аспарагина, аспарагиновой кислоты, цистеина, глицина, глутамина, глутаминовой кислоты, гистидина, изолейцина, лейцина, лизина, метионина, фенилаланина, пролина, серина, треонина, триптофана, тирозина и валина, или остатка N-ацильного производного их.
Варианты осуществления изобретения включают композиции и единицы дозы, где опиоидное пролекарство с модифицированным кетоном представляет собой соединение формулы KC-(IIIa):
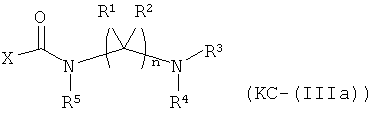
где:
Х представляет остаток кетон-содержащего опиоида, где атом водорода соответствующей енольной группы замещен ковалентной связью с -C(O)-NR5-(C(R1)(R2))n-NR3R4;
R5 выбран из алкила, замещенного алкила, арилалкила, замещенного арилалкила, арила и замещенного арила;
каждый R1 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, ацила и аминоацила;
каждый R2 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, ацила и аминоацила;
или R1 и R2 вместе с углеродом, к которому они присоединены, образуют циклоалкильную, замещенную циклоалкильную, арильную или замещенную арильную группу, или две R2 или R3 группы на соседних атомах углерода вместе с атомами углерода, к которым они присоединены, образуют циклоалкильную, замещенную циклоалкильную, арильную или замещенную арильную группу;
n представляет собой целое число от 2 до 4;
R3 является водородом;
R4 представляет собой ,
каждый R6 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, арилалкила, замещенного арилалкила, гетероалкила, замещенного гетероалкила, гетероарила, замещенного гетероарила, гетероарилалкила и замещенного гетероарилалкила или, необязательно, R6 и R7 вместе с атомами, к которым они присоединены, образуют циклогетероалкильное или замещенное циклогетероалкильное кольцо;
каждый W независимо представляет собой -NR8-, -О- или -S-;
каждый R8 независимо выбран из водорода, алкила, замещенного алкила, арила и замещенного арила или, необязательно, каждый R6 и R8 независимо вместе с атомами, к которым они присоединены, образуют циклогетероалкильное или замещенное циклогетероалкильное кольцо;
p представляет собой целое число от 1 до 100; и
R7 выбран из водорода, алкила, замещенного алкила, ацила, замещенного ацила, алкоксикарбонила, замещенного алкоксикарбонила, арила, замещенного арила, арилалкила и замещенного арилалкила;
или его соль, гидрат или сольват.
Варианты осуществления изобретения включают композиции и единицы дозы, где опиоидное пролекарство с модифицированным кетоном представляет собой соединение формулы KC-(IIIb):
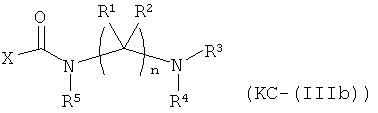
где:
Х представляет остаток кетон-содержащего опиоида, где атом водорода соответствующей енольной группы замещен ковалентной связью с -C(O)-NR5-(C(R1)(R2))n-NR3R4;
R5 выбран из алкила, замещенного алкила, арилалкила, замещенного арилалкила, арила и замещенного арила;
каждый R1 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, ацила и аминоацила;
каждый R2 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, ацила и аминоацила;
или R1 и R2 вместе с углеродом, к которому они присоединены, образуют циклоалкильную или замещенную циклоалкильную группу, или две R1 или R2 группы на соседних атомах углерода вместе с атомами углерода, к которым они присоединены, образуют циклоалкильную или замещенную циклоалкильную группу;
n представляет собой целое число от 2 до 4;
R3 является водородом;
R4 представляет собой ;
каждый R6 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, арилалкила, замещенного арилалкила, гетероалкила, замещенного гетероалкила, гетероарила, замещенного гетероарила, гетероарилалкила и замещенного гетероарилалкила или, необязательно, R6 и R7 вместе с атомами, к которым они присоединены, образуют циклогетероалкильное или замещенное циклогетероалкильное кольцо;
каждый W независимо представляет собой -NR8-, -О- или -S-;
каждый R8 независимо выбран из водорода, алкила, замещенного алкила, арила и замещенного арила или, необязательно, каждый R6 и R8 независимо вместе с атомами, к которым они присоединены, образуют циклогетероалкильное или замещенное циклогетероалкильное кольцо;
p представляет собой целое число от 1 до 100; и
R7 выбран из водорода, алкила, замещенного алкила, ацила, замещенного ацила, алкоксикарбонила, замещенного алкоксикарбонила, арила, замещенного арила, арилалкила и замещенного арилалкила;
или его соль, гидрат или сольват.
Варианты осуществления изобретения включают композиции и единицы дозы, где опиоидное пролекарство с модифицированным кетоном представляет собой соединение формулы KC-(IV):
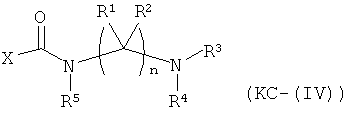
где:
Х представляет остаток кетон-содержащего опиоида, где атом водорода соответствующей енольной группы замещен ковалентной связью с -C(O)-NR5-(C(R1)(R2))n-NR3R4;
R5 выбран из (1-6С) алкила, (1-6С) замещенного алкила, -(СН2)q(C6H4)-COOH, -(CH2)q(C6H4)-COOCH3, и -(CH2)q(C6H4)-COOCH2CH3, где q представляет собой целое
число от 1 до 10;
каждый R1 независимо выбран из водорода, алкила, замещенного алкила, арила,
замещенного арила, ацила и аминоацила;
каждый R2 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, ацила и аминоацила;
или R1 и R2 вместе с углеродом, к которому они присоединены, образуют циклоалкильную или замещенную циклоалкильную группу, или две R1 или R2 группы на соседних атомах углерода вместе с атомами углерода, к которым они присоединены, образуют циклоалкильную или замещенную циклоалкильную группу;
n равно 2 или 3;
R3 является водородом;
R4 представляет собой остаток L-аминокислоты, выбранной из аланина, аргинина, аспарагина, аспарагиновой кислоты, цистеина, глицина, глутамина, глутаминовой кислоты, гистидина, изолейцина, лейцина, лизина, метионина, фенилаланина, пролина, серина, треонина, триптофана, тирозина и валина, или остатка N-ацильного производного любой из перечисленных аминокислот; или остаток пептида, состоящий из по меньшей мере двух остатков L-аминокислот, независимо выбранных из аланина, аргинина, аспарагина, аспарагиновой кислоты, цистеина, глицина, глутамина, глутаминовой кислоты, гистидина, изолейцина, лейцина, лизина, метионина, фенилаланина, пролина, серина, треонина, триптофана, тирозина и валина, или остатка N-ацильного производного их;
или его соль, гидрат или сольват.
Варианты осуществления изобретения включают композиции и единицы дозы, где опиоидное пролекарство с модифицированным кетоном представляет собой соединение формулы KC-(V):
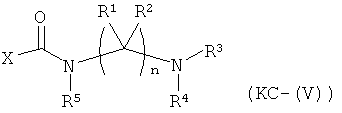
где:
Х представляет остаток кетон-содержащего опиоида, где атом водорода соответствующей енольной группы замещен ковалентной связью с -C(O)-NR5-(C(R1)(R2))n-NR3R4;
R5 выбран из алкила, замещенного алкила, арилалкила, замещенного арилалкила, арила и замещенного арила;
каждый R1 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, ацила и аминоацила;
каждый R2 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, ацила и аминоацила;
или R1 и R2 вместе с углеродом, к которому они присоединены, образуют циклоалкильную, замещенную циклоалкильную, арильную или замещенную арильную группу, или две R1 или R2 группы на соседних атомах углерода вместе с атомами углерода, к которым они присоединены, образуют циклоалкильную, замещенную циклоалкильную, арильную или замещенную арильную группу;
n представляет собой целое число от 2 до 4;
R3 является водородом;
R4 представляет собой расщепляемый трипсином фрагмент;
или его соль, гидрат или сольват.
Варианты осуществления включают способы лечения пациента, включающие введение любых композиций или единиц дозы, описанных в данном документе, нуждающемуся в них пациенту. Варианты осуществления включают способы уменьшения побочных эффектов лечения, включающие введение любых композиций или единиц дозы, описанных в данном документе, нуждающемуся в них пациенту. Варианты осуществления включают способы улучшения соблюдения пациентом лечения, предписанного врачом, включающие руководство введением любых композиций или единиц дозы, описанных в данном документе, нуждающемуся в них пациенту. Такие варианты осуществления могут обеспечить улучшение соблюдения пациентом предписанного лечения пролекарством с ингибитором по сравнению с соблюдением пациентом предписанного лечения с использованием лекарственного средства и/или пролекарства без ингибитора.
Варианты осуществления включают способы уменьшения риска непреднамеренной передозировки кетон-содержащим опиоидом, включающие руководство введением любых фармацевтических композиций или единиц дозы, описанных в данном документе, нуждающемуся в них пациенту.
Варианты осуществления изобретения включают способы изготовления единицы дозы, включающие объединение опиоидного пролекарства с модифицированным кетоном и трипсинового ингибитора в единице дозы, где опиоидное пролекарство с модифицированным кетоном и трипсиновый ингибитор присутствуют в единице дозы в количестве достаточном для уменьшения высвобождения кетон-содержащего опиоида из опиоидного пролекарства с модифицированным кетоном.
Варианты осуществления изобретения включают способы удерживания от злоупотребления и неправильного использования многократных единиц дозы опиоидного пролекарства с модифицированным кетоном, включающие объединение опиоидного пролекарства с модифицированным кетоном и трипсинового ингибитора в единице дозы, где опиоидное пролекарство с модифицированным кетоном и трипсиновый ингибитор присутствуют в единице дозы в количестве, достаточном для уменьшения высвобождения кетон-содержащего опиоида из опиоидного пролекарства с модифицированным кетоном таким образом, что введение внутрь многократных единиц дозы пациентом не приводит к пропорциональному высвобождению кетон-содержащего опиоида. В дополнительных вариантах осуществления высвобождение лекарственного препарата уменьшено по сравнению с высвобождением лекарственного препарата из эквивалентной дозы в отсутствие ингибитора.
Один вариант осуществления изобретения представляет собой способ идентифицирования пролекарства и ингибитора ЖК фермента, пригодный для приготовления единицы дозы. Такой способ может быть проведен, например, как in vitro количественный анализ, in vivo количественный анализ, или ex vivo количественный анализ.
Варианты осуществления данного изобретения включают способы идентифицирования опиоидного пролекарства с модифицированным кетоном и трипсинового ингибитора, пригодные для приготовления единицы дозы, включающие объединение опиоидного пролекарства с модифицированным кетоном, трипсинового ингибитора и трипсина в реакционной смеси и определение конверсии опиоидного пролекарства с модифицированным кетоном, где снижение конверсии опиоидного пролекарства с модифицированным кетоном в присутствии трипсинового ингибитора по сравнению с конверсией опиоидного пролекарства с модифицированным кетоном в отсутствие трипсинового ингибитора указывает на то, что опиоидное пролекарство с модифицированным кетоном и трипсиновый ингибитор пригодны для приготовления единицы дозы.
Варианты осуществления данного изобретения включают способы идентифицирования опиоидного пролекарства с модифицированным кетоном и трипсинового ингибитора пригодные для приготовления единицы дозы, включающие введение животному опиоидного пролекарства с модифицированным кетоном и трипсинового ингибитора и определение конверсии опиоидного пролекарства с модифицированным кетоном, где снижение конверсии кетон-содержащего опиоида в присутствии трипсинового ингибитора по сравнению с конверсией кетон-содержащего опиоида в отсутствие трипсинового ингибитора указывает на то, что кетон-содержащий опиоид и трипсиновый ингибитор пригодны для приготовления единицы дозы. В конкретных вариантах осуществления введение лекарства включает введение животному увеличенных доз ингибитора совместно с выбранной фиксированной дозой опиоидного пролекарства с модифицированным кетоном. Определение конверсии пролекарства может способствовать идентификации дозы ингибитора и дозы опиоидного пролекарства с модифицированным кетоном, которые обеспечивают предварительно выбранную фармакокинетическую (ФК) кривую. Такие способы могут быть проведены как, например, in vivo количественный анализ или ex vivo количественный анализ.
Варианты осуществления данного изобретения включают способы идентифицирования опиоидного пролекарства с модифицированным кетоном и трипсинового ингибитора пригодные для приготовления единицы дозы, включающие введение в ткани животного опиоидного пролекарства с модифицированным кетоном и трипсинового ингибитора и определение конверсии опиоидного пролекарства с модифицированным кетоном, где снижение конверсии опиоидного пролекарства с модифицированным кетоном в присутствии трипсинового ингибитора по сравнению с конверсией опиоидного пролекарства с модифицированным кетоном в отсутствие трипсинового ингибитора указывает на то, что опиоидное пролекарство с модифицированным кетоном и трипсиновый ингибитор пригодны для приготовления единицы дозы.
Краткое описание Графических материалов
На Фигуре 1 приведен график, изображающий влияние повышения уровня ингибитора ЖК фермента ("ингибитор", ось X) на ФК параметр (например, Cmax лекарственного средства) (ось Y) для фиксированной дозы пролекарства. Влияние ингибитора на ФК параметр пролекарства может меняться от неопределяемого, через среднее и до полного ингибирования (т.е. нет определяемого высвобождения лекарственного средства).
На Фигуре 2 представлены диаграммы зависимости концентрации лекарственного средства в плазме (ось Y) от времени (ось X). Секция А представляет собой диаграмму фармакокинетической (ФК) кривой после принятия внутрь пролекарства с ингибитором ЖК фермента (пунктирная линия), где Cmax лекарственного средства модифицирована относительно этого параметра пролекарства без ингибитора (сплошная линия). Секция В представляет собой диаграмму фармакокинетической (ФК) кривой после принятия внутрь пролекарства с ингибитором (пунктирная линия), где Cmax и Tmax лекарственного средства модифицированы относительно этих параметров пролекарства без ингибитора (сплошная линия). Секция С представляет собой диаграмму фармакокинетической (ФК) кривой после принятия внутрь пролекарства с ингибитором (пунктирная линия), где Tmax лекарственного средства модифицировано относительно этого параметра пролекарства без ингибитора (сплошная линия).
На Фигуре 3 представлены диаграммы характерных ФК кривых зависимости концентрация-доза, которые могут быть результатом принятия многократных единиц дозы (ось X) данного раскрытия. Различные ФК кривые (как показано здесь для типичного ФК параметра - Cmax лекарственного средства (ось Y)) могут быть обеспечены путем корректировки относительного количества пролекарства и ингибитора ЖК фермента, содержащихся в отдельной единице дозы, или путем использования другого пролекарства или ингибитора в единице дозы.
На Фигуре 4 показано изменение концентрации от времени продуцирования оксикодона после перорального (ПО) введения пролекарства оксикодона в крысах.
На Фигуре 5 показано изменение концентрации от времени продуцирования оксикодона после внутривенного (ВВ) введения пролекарства оксикодона в крысах.
На Фигуре 6 показано высвобождение оксикодона из пролекарства оксикодона под воздействием различных легкодоступных бытовых химикатов или препаратов ферментов.
На Фигуре 7 показано исчезновение пролекарства оксикодона и появление оксикодона после in vitro инкубации пролекарства и трипсина в присутствии и в отсутствие трипсинового ингибитора.
На Фигуре 8 приведено сравнение усредненных концентраций в плазме относительно времени высвобождения оксикодона после ПО введения крысам только пролекарства Соединения KC-2 и Соединения KC-2 совместно с трипсиновым ингибитором - Соединением 109.
На Фигуре 9 приведено сравнение усредненных концентраций в плазме относительно времени высвобождения оксикодона после ПО введения крысам увеличенных доз пролекарства Соединения KC-2.
На Фигуре 10 приведено сравнение усредненных концентраций в плазме относительно времени высвобождения оксикодона после ПО введения крысам пролекарства Соединения KC-2 совместно с увеличенными количествами трипсинового ингибитора - Соединения 109.
На Фигуре 11 приведено сравнение усредненных концентраций в плазме относительно времени высвобождения оксикодона после ПО введения крысам увеличенных доз Соединения KC-3.
На Фигуре 12 показано изменение концентрации в плазме от времени продуцирования оксикодона после внутривенного (ВВ) введения пролекарства Соединения KC-3 в крысах.
На Фигуре 13 приведено сравнение усредненных концентраций в плазме относительно времени высвобождения оксикодона после ПО введения крысам пролекарства Соединения KC-3 совместно с увеличенными количествами трипсинового ингибитора - Соединения 109.
На Фигуре 14 показано высвобождение оксикодона из пролекарства Соединения KC-3 под воздействием различных бытовых химикатов и препаратов ферментов.
На Фигуре 15 показано изменение концентрации в плазме от времени продуцирования оксикодона после внутривенного (ВВ) введения пролекарства Соединения KC-4 в крысах.
На Фигуре 16 приведено сравнение усредненных концентраций в плазме относительно времени высвобождения гидрокодона после ПО введения крысам пролекарства Соединения KC-4 без трипсинового ингибитора или совместно с ним.
На Фигуре 17 приведено сравнение усредненных концентраций в плазме относительно времени высвобождения оксикодона после ПО введения крысам Соединения KC-5.
На Фигуре 18 показано изменение концентрации в плазме от времени продуцирования оксикодона после внутривенного (ВВ) введения пролекарства - Соединения KC-5 в крысах.
На Фигуре 19 приведено сравнение усредненных концентраций в плазме относительно времени высвобождения оксикодона после ПО введения крысам Соединения KC-6.
На Фигуре 20 показано изменение концентрации в плазме от времени продуцирования оксикодона после внутривенного (ВВ) введения пролекарства - Соединения KC-6 в крысах.
Определения
Следующие выражения имеют следующие значения, если не указано иное. Любые неопределенные выражения имеют значения, принятые в области, к которой они принадлежат.
Как используется в данном документе, выражение "алкил" само по себе или как часть другого заместителя относится к насыщенному разветвленному или неразветвленному одновалентному углеводородному радикалу, полученному удалением одного атома водорода у одного атома углерода исходного алкана. Типичные алкильные группы включают, но без ограничения, метил; этил, пропилы, такие как пропан-1-ил или пропан-2-ил; и бутилы, такие как бутан-1-ил, бутан-2-ил, 2-метил-пропан-1-ил или 2-метил-пропан-2-ил. В некоторых вариантах осуществления алкильная группа содержит от 1 до 20 атомов углерода. В других вариантах осуществления алкильная группа содержит от 1 до 10 атомов углерода. В некоторых других вариантах осуществления алкильная группа содержит от 1 до 6 атомов углерода, например, от 1 до 4 атомов углерода.
"Алканил" сам по себе или как часть другого заместителя относится к насыщенному разветвленному, неразветвленному или циклическому алкильному радикалу, полученному удалением одного атома водорода у одного атома углерода исходного алкана. Типичные алканильные группы включают, но без ограничения, метанил; этанил; пропанилы, такие как пропан-1-ил, пропан-2-ил (изопропил), циклопропан-1-ил и т.д.; бутанилы, такие как бутан-1-ил, бутан-2-ил (втор-бутил), 2-метил-пропан-1-ил (изобутил), 2-метил-пропан-2-ил (трет-бутил), циклобутан-1-ил и т.д.; и т.п.
"Алкилен" относится к разветвленной или неразветвленной насыщенной углеводородной цепи, обычно имеющей в своем составе от 1 до 40 атомов углерода, более типично 1-10 атомов углерода и еще более типично 1-6 атомов углерода. Этот термин проиллюстрирован группами, такими как метилен (-СН2-), этилен (-CH2CH2-), изомерами пропилена (например, -CH2CH2CH2- и -СН(СН3)СН2-) и подобными.
"Алкенил" сам по себе или как часть другого заместителя относится к ненасыщенному разветвленному, неразветвленному или циклическому алкильному радикалу, имеющему по меньшей мере одну двойную связь углерод-углерод, полученному удалением одного атома водорода у одного атома углерода исходного алкена. Группа может иметь либо цис-, либо транс-конформацию относительно двойной связи(связей). Типичные алкенильные группы включают, но не ограничиваясь, этенил; пропенилы, такие как проп-1-ен-1-ил, проп-1-ен-2-ил, проп-2-ен-1-ил (аллил), проп-2-ен-2-ил, циклопроп-1-ен-1-ил; циклопроп-2-ен-1-ил; бутенилы, такие как бут-1-ен-1-ил, бут-1-ен-2-ил, 2-метил-проп-1-ен-1-ил, бут-2-ен-1-ил, бут-2-ен-1-ил, бут-2-ен-2-ил, бута-1,3-диен-1-ил, бута-1,3-диен-2-ил, циклобут-1-ен-1-ил, циклобут-1-ен-3-ил, циклобута-1,3-диен-1-ил и т.д.; и подобные.
"Алкинил" сам по себе или как часть другого заместителя относится к ненасыщенному разветвленному, неразветвленному или циклическому алкильному радикалу, имеющему по меньшей мере одну тройную связь углерод-углерод, полученному удалением одного атома водорода у одного атома углерода исходного алкина. Типичные алкинильные группы включают, но без ограничения, этинил; пропинилы, такие как проп-1-ин-1-ил, проп-2-ин-1-ил и т.д.; бутинилы, такие как бут-1-ин-1-ил, бут-1-ин-3-ил, бут-3-ин-1-ил и т.д.; и подобные.
"Ацил" сам по себе или как часть другого заместителя относится к радикалу -C(O)R30, где R30 представляет собой водород, алкил, циклоалкил, циклогетероалкил, арил, арилалкил, гетероалкил, гетероарил, гетероарилалкил, как определено в данном документе и их замещенные варианты. Типичные примеры включают, но без ограничения, формил, ацетил, циклогексилкарбонил, циклогексилметилкарбонил, бензоил, бензилкарбонил, пиперонил, сукцинил и малонил, и подобные.
Выражение "аминоацил" относится к группе -C(O)NR21R22, где R21 и R22 независимо выбирают из группы, которая включает водород, алкил, замещенный алкил, алкенил, замещенный алкенил, алкинил, замещенный алкинил, арил, замещенный арил, циклоалкил, замещенный циклоалкил, циклоалкенил, замещенный циклоалкенил, гетероарил, замещенный гетероарил, гетероцикл и замещенный гетероцикл, и где R21 и R22 факультативно вместе с азотом, к которому они присоединены, образуют гетероциклическую или замещенную гетероциклическую группу, и где алкил, замещенный алкил, алкенил, замещенный алкенил, алкинил, замещенный алкинил, циклоалкил, замещенный циклоалкил, циклоалкенил, замещенный циклоалкенил, арил, замещенный арил, гетероарил, замещенный гетероарил, гетероцикл и замещенный гетероцикл являются такими, как определено в данном документе.
"Алкокси" сам по себе или как часть другого заместителя относится к радикалу -OR31, где R31 представляет алкильную или циклоалкильную группу, как определено в данном документе. Типичные примеры включают, но без ограничения, метокси-, этокси-, пропокси-, бутокси-, циклогексилокси- и подобные.
"Алкоксикарбонил" сам по себе или как часть другого заместителя относится к радикалу -C(O)OR31, где R31 представляет алкильную или циклоалкильную группу, как определено в данном документе. Типичные примеры включают, но без ограничения, метоксикарбонил, этоксикарбонил, пропоксикарбонил, бутоксикарбонил, циклогексилоксикарбонил и подобные.
"Арил" сам по себе или как часть другого заместителя относится к одновалентному ароматическому углеводородному радикалу, полученному удалением одного атома водорода у одного атома углерода ароматической кольцевой системы. Типичные арильные группы включают, но без ограничения, группы, полученные из ацеантрилена, аценафтилена, ацефенантрилена, антрацена, азулена, бензола, хризена, коронена, флуорантена, флуорена, гексацена, гексафена, гексалена, as-индацена, s-индацена, индана, индена, нафталина, октацена, октафена, окталена, овалена, пента-2,4-диена, пентацена, пенталена, пентафена, перилена, феналена, фенантрена, пицена, плеядена, пирена, пирантрена, рубицена, трифенилена, тринафталина и подобных. В некоторых вариантах осуществления арильная группа содержит от 6 до 20 атомов углерода. В некоторых вариантах осуществления арильная группа содержит от 6 до 12 атомов углерода. Примерами арильной группы являются фенил и нафтил.
"Арилалкил" сам по себе или как часть другого заместителя относится к ациклическому алкильному радикалу, в котором один из атомов водорода связанный с атомом углерода, типично с концевым или sp3 атомом углерода, замещен арильной группой. Типичные арилалкильные группы включают, но без ограничения, бензил, 2-фенилэтан-1-ил,2-фенилэтен-1-ил, нафтилметил, 2-нафтилэтан-1-ил, 2-нафтилэтен-1-ил, нафтобензил, 2-нафтофенилэтан-1-ил и подобные. Если подразумеваются определенные алкильные части, используются номенклатурные названия арилалканил, арилалкенил и/или арилалкинил. В определенных вариантах осуществления арилалкильная группа представляет собой (С7-С30) арилалкил, например, алканильный, алкенильный или алкинильный фрагмент арилалкильной группы представляет собой (C1-С10), а арильный компонент представляет собой (С6-С20). В определенных вариантах осуществления арилалкильная группа представляет собой (С7-С30) арилалкил, например, алканильный, алкенильный или алкинильный фрагмент арилалкильной группы представляет собой (C1-C8), а арильный фрагмент представляет собой (С6-С12).
"Ариларил" сам по себе или как часть другого заместителя относится к одновалентной углеводородной группе, полученной путем удаления одного атома водорода у одного атома углерода кольцевой системы, в которой одна или более идентичных или неидентичных ароматических кольцевых систем связаны вместе напрямую одной связью, где количество таких прямых соединений на одно меньше, чем число соответствующих ароматических кольцевых систем. Типичные ариларильные группы включают, но без ограничения, бифенил, трифенил, фенил-нафтил, бинафтил, бифенил-нафтил и подобное. Когда число атомов углерода в ариларильной группе оговаривается, числа относятся к атомам углерода, включенным в каждое ароматическое кольцо. Например, (C5-C14) ариларил представляет собой ариларильную группу, в которой каждое ароматическое кольцо содержит от 5 до 14 углеродов, например, бифенил, трифенил, бинафтил, фенилнафтил, и т.д. В определенных вариантах осуществления каждая ароматическая кольцевая система ариларильной группы независимо представляет собой (C5-C14) ароматическое соединение. В некоторых вариантах осуществления каждая ароматическая кольцевая система ариларильной группы независимо представляет собой (С5-С10) ароматическое соединение. В некоторых вариантах осуществления каждая ароматическая кольцевая система является идентичной, например, бифенил, трифенил, бинафтил, тринафтил и др.
"Циклоалкил" сам по себе или как часть другого заместителя относится к насыщенному или ненасыщенному циклическому алкильному радикалу. Если подразумевают определенную степень насыщения, то используют номенклатуру "циклоалканил" или "циклоалкенил". Типичные циклоалкильные группы включают, но без ограничения, группы, полученные из циклопропана, циклобутана, циклопентана, циклогексана и подобного. В некоторых вариантах осуществления циклоалкильная группа представляет собой (С3-С10) циклоалкил. В некоторых вариантах осуществления циклоалкильная группа представляет собой (С3-C7) циклоалкил.
"Циклогетероалкил" или "гетероциклил" сам по себе или как часть другого заместителя относится к насыщенному или ненасыщенному циклическому алкильному радикалу, в котором один или более атомов углерода (и любые связанные атомы водорода) независимо замещены одинаковым или отличающимся гетероатомом. Типичные гетероатомы для замещения атома(ов) углерода включают, но без ограничения, N, Р, О, S, Si и др. Если предполагают определенную степень насыщения, используют номенклатуру "циклогетероалканил" или "циклогетероалкенил". Типичные циклогетероалкильные группы включают, но без ограничения, группы, полученные из эпоксидов, азиринов, тииранов, имидазолидина, морфолина, пиперазина, пиперидина, пиразолидина, пирролидина, хинуклидина и подобных.
"Гетероалкил, гетероалканил, гетероалкенил и гетероалкинил" сами по себе или как часть другого заместителя относятся к алкильной, алканильной, алкенильной и алкинильной группам, соответственно, в которых один или более атомов углерода (и любые связанные атомы водорода) независимо замещены одинаковыми или различными гетероатомными группами. Типичные гетероатомные группы, которые могут быть включены в эти группы, включают, но не ограничиваясь, -O-, -S-, -O-O-, -S-S-, -O-S-, -NR37R38-, =N-N=, -N=N-, -N=NR39R40, -PR41-, -P(O)2-, -POR42-, -O-P(O)2-, -S-O-, -S(O)-, -SO2-, -SnR43R44- и подобные, где R37, R38, R39, R40, R41, R42, R43 и R44 представляют собой независимо водород, алкил, замещенный алкил, арил, замещенный арил, арилалкил, замещенный арилалкил, циклоалкил, замещенный циклоалкил, Циклогетероалкил, замещенный Циклогетероалкил, гетероалкил, замещенный гетероалкил, гетероарил, замещенный гетероарил, гетероарилалкил или замещенный гетероарилалкил.
"Гетероарил" сам по себе или как часть другого заместителя относится к одновалентному гетероароматическому радикалу, полученному удалением одного атома водорода от одного атома гетероароматической кольцевой системы. Типичные гетероарильные группы включают, но без ограничения, группы, полученные из акридина, арсаиндола, карбазола, р-карболина, хромана, хромена, циннолина, фурана, имидазола, индазола, индола, индолина, индолизина, изобензофурана, изохромена, изоиндола, изоиндолина, изохинолина, изотиазола, изоксазола, нафтиридина, оксадиазола, оксазола, перимидина, фенантридина, фенантролина, феназина, фталазина, птеридина, пурина, пирана, пиразина, пиразола, пиридазина, пиридина, пиримидина, пиррола, пирролизина, хиназолина, хинолина, хинолизина, хиноксалина, тетразола, тиадиазола, тиазола, тиофена, триазола, ксантена, бензодиоксола и подобных. В определенных вариантах осуществления гетероарильная группа представляет собой 5-20-членный гетероарил. В определенных вариантах осуществления гетероарильная группа представляет собой 5-10-членный гетероарил. В определенных вариантах осуществления гетероарильные группы являются полученными из тиофена, пиррола, бензотиофена, бензофурана, индола, пиридина, хинолина, имидазола, оксазола и пиразина.
"Гетероарилалкил" сам по себе или как часть другого заместителя относится к ациклическому алкильному радикалу, в котором один из атомов водорода, связанный с атомом углерода, типично концевым или sp3 атомом углерода, замещен гетероарильной группой. Если подразумевают определенные алкильные части, то используют номенклатурные названия гетероарилалканил, гетероарилалкенил и/или гетероарилалкинил. В определенных вариантах осуществления гетероарилалкильная группа представляет собой 6-30-членный гетероарилалкил, например, алканильный, алкенильный или алкинильный фрагмент гетероарилалкила является 1-10-членным, а гетероарильный фрагмент является 5-20-членным гетероарилом. В определенных вариантах осуществления гетероарилалкильная группа представляет собой 6-20-членный гетероарилалкил, например, алканильный, алкенильный или алкинильный фрагмент гетероарилалкила является 1-8-членным, а гетероарильный фрагмент является 5-12-членным гетероарилом.
"Ароматическая кольцевая система" сама по себе или как часть другого заместителя относится к ненасыщенной циклической или полициклической системе колец, имеющей сопряженную систему л-электронов. В частности, включенная в определение "ароматическая кольцевая система" представляет собой конденсированные кольцевые системы, в которых одно или более из колец являются ароматическими, и одно или более из колец являются насыщенными или ненасыщенными, такие как, например, флуорен, индан, инден, фенален и т.д. Типичные ароматические кольцевые системы включают, но не ограничиваясь, ацеантрилен, аценафтилен, ацефенантрилен, антрацен, азулен, бензол, хризен, коронен, флуорантен, флуорен, гексацен, гексафен, гексален, as-индацен, s-индацен, индан, инден, нафталин, октацен, октафен, октален, овален, пента-2,4-диен, пентацен, пентален, пентафен, перилен, фенален, фенантрен, пицен, плеяден, пирен, пирантрен, рубицен, трифенилен, тринафталин и подобные.
"Гетероароматическая кольцевая система" сама по себе или как часть другого заместителя относится к ароматической кольцевой системе, в которой один или более атомов углерода (и любые связанные атомы водорода) независимо замещены одинаковым или разным гетероатомом. Типичные гетероатомы для замещения углеродных атомов включают, но без ограничения, N, Р, О, S, Si и др. Специально в определение "гетероароматической кольцевой системы" включены конденсированные кольцевые системы, в которых одно или более колец являются ароматическими и одно или более колец являются насыщенными или ненасыщенными, такими, как, например, арсаиндол, бензодиоксан, бензофуран, хроман, хромей, индол, индолин, ксантен и др. Типичные гетероароматические кольцевые системы включают, но без ограничения, арсаиндол, карбазол, β-карболин, хроман, хромен, циннолин, фуран, имидазол, индазол, индол, индолин, индолизин, изобензофуран, изохромен, изоиндол, изоиндолин, изохинолин, изотиазол, изоксазол, нафтиридин, оксадиазол, оксазол, перимидин, фенантридин, фенантролин, феназин, фталазин, птеридин, пурин, пиран, пиразин, пиразол, пиридазин, пиридин, пиримидин, пиррол, пирролизин, хиназолин, хинолин, хинолизин, хиноксалин, тетразол, тиадиазол, тиазол, тиофен, триазол, ксантен и подобные.
"Замещенный" относится к группе, в которой один или более атомов водорода независимо замещены одинаковым или разным заместителем(заместителями). Типичные заместители включают, но без ограничения, алкилендиокси (такой, как метилендиокси), -М, -R60, -O-, =O, -OR60, -SR60, -S-, =S, -NR60R61, =NR60, -CF3, -CN, -OCN, -SCN, -NO, -NO2, =N2, -N3, -S(O)2O-, -S(O)2OH, -S(O)2R60, -OS(O)2O-, -OS(O)2R60, -P(O)(O-)2, P(О)(OR60)(O-), -OP(O)(OR60)(OR61), -C(O)R60, -C(S)R60, -C(O)OR60, -C(O)NR60R61, -C(O)O-, -C(S)OR60, -NR62C(O)NR60R61, -NR62C(S)NR60R61, -NR62C(NR63)NR60R61 и -C(NR62)NR60R61, где М является галогеном; R60, R61, R62 и R63 независимо представляют собой водород, алкил, замещенный алкил, алкокси, замещенный алкокси, циклоалкил, замещенный циклоалкил, циклогетероалкил, замещенный циклогетероалкил, арил, замещенный арил, гетероарил или замещенный гетероарил, или необязательно R60 и R61 вместе с атомом азота, с которым они связаны, образуют циклогетероалкильное или замещенное циклогетероалкильное кольцо; и R64 и R65 независимо представляют собой водород, алкил, замещенный алкил, арил, циклоалкил, замещенный циклоалкил, циклогетероалкил, замещенный циклогетероалкил, арил, замещенный арил, гетероарил или замещенный гетероарил, или необязательно R64 и R65 вместе с атомом азота, с которым они связаны, образуют циклогетероалкильное или замещенное циклогетероалкильное кольцо. В определенных вариантах осуществления заместители включают -М, -R60, =O, -OR60, -SR60, -S-, =S-OS(O)2O-, -OS(O)2R60, -P(O)(O-)2, P(О)(OR60)(O-), -OP(O)(OR60)(OR61), -C(O)R60, -C(S)R60, -C(O)OR60, -C(O)NR60R61, -C(O)O-, -C(S)OR60, -NR62C(O)NR60R61. В некоторых вариантах осуществления заместители включают -M, -R, =O, -OR60, -SR60, -NR60R61, -CF3, -CN, -NO2, -S(O)2R60, P(О)(OR60)(O-), -OP(O)(OR60)(OR61), -C(O)R60, -C(O)OR60, -C(O)NR60R61, -C(O)O-. В некоторых вариантах осуществления заместители включают -М, -R60, =O, -OR60, -SR60, -NR60R61, -CF3, -CN, -NO2, -S(O)2R60, -OP(O)(OR60)(OR61), -C(O)R60, -C(O)OR60, -C(O)O-, где R60, R61 и R62 как определены выше. Например, замещенная группа может иметь метилендиокси-заместитель или один, два или три заместителя из атома галогена, (1-4С) алкильной группы и (1-4С) алкокси группы.
"Единица дозы", как употребляется в данном документе, относится к комбинации расщепляемого ЖК ферментами пролекарства (например, расщепляемого трипсином пролекарства) и ингибитора ЖК фермента (например, трипсинового ингибитора). "Одна единица дозы" - это одна единица комбинации расщепляемого ЖК ферментами пролекарства (например, расщепляемого трипсином пролекарства) и ингибитора ЖК фермента (например, трипсинового ингибитора), где одна единица дозы предоставляет терапевтически эффективное количество лекарственного средства (т.е. достаточное количество лекарства, чтобы вызвать терапевтический эффект, например, в пределах соответствующего терапевтического окна или терапевтического диапазона). "Многократные единицы дозы", или "множества единиц дозы", или "множество единиц дозы" относится по меньшей мере к двум отдельным единицам дозы.
"ФК кривая" относится к кривой концентрации лекарственного средства в крови или в плазме. Такая кривая может быть функцией концентрации лекарства от времени (т.е. "ФК кривая концентрация-время") или функция концентрации лекарственного средства от количества введенных доз (т.е. "ФК кривая концентрация-доза"). ФК кривая характеризуется ФК параметрами.
"ФК параметр" относится к показателю концентрации лекарственного средства в крови или в плазме такому, как: 1) "Cmax лекарственного препарата" - максимальная концентрация лекарственного средства, достигнутая в крови или плазме; 2) "Tmax лекарственного средства" - время, необходимое с момента принятия внутрь для достижения Cmax; и 3) "действие лекарственного средства" - общая концентрация лекарственного средства в крови или плазме в течение выбранного периода времени, которое может быть измерено в течение выбранного промежутка времени (t), используя площадь под кривой (ППК) в ходе высвобождения лекарственного средства. Изменение одного или более ФК параметров обусловливает изменение ФК кривой.
"Фармакодинамическая (ФД) кривая" относится к кривой эффективности лекарственного средства в пациенте (или субъекте, или потребителе), которая может быть охарактеризована ФД параметрами. "ФД параметры" включают "Emax лекарственного средства" (максимальная эффективность лекарственного средства), "ЕС50 лекарственного средства" (концентрация лекарственного средства при 50% Emax) и побочное действие.
"Желудочно-кишечный фермент" или "ЖК фермент" относится к ферменту, находящемуся в желудочно-кишечном (ЖК) тракте, который охватывает анатомические местоположения от рта до заднего прохода. Трипсин - это пример ЖК фермента.
"Расщепляемый желудочно-кишечным ферментом фрагмент" или "расщепляемый ЖК ферментом фрагмент" относится к группе, содержащей участок, подверженный расщеплению ЖК ферментом. Например, "расщепляемый трипсином фрагмент" относится к группе, содержащей участок, подверженный расщеплению трипсином.
"Ингибитор желудочно-кишечного фермента" или "ингибитор ЖК фермента" относится к любому средству, способному ингибировать действие желудочно-кишечного фермента на субстрат. Термин также включает соли ингибиторов желудочно-кишечных ферментов. Например, "ингибитор трипсина" относится к любому средству, способному ингибировать действие трипсина на субстрат.
"Фармацевтическая композиция" относится к по меньшей мере одному соединению и может дополнительно содержать фармацевтически приемлемый носитель, с которым соединение вводят пациенту.
"Фармацевтически приемлемая соль" относится к соли соединения, которая обладает желательной фармакологической активностью соединения. Такие соли включают: (1) кислотные аддитивные соли, образованные с неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и подобные; или образованные органическими кислотами, такими как уксусная кислота, пропионовая кислота, капроновая кислота, циклопентанпропионовая кислота, гликолевая кислота, пировиноградная кислота, молочная кислота, малоновая кислота, янтарная кислота, яблочная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, 3-(4-гидроксибензоил)бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, 1,2-этан-дисульфоновая кислота, 2-гидроксиэтансульфоновая кислота, бензолсульфоновая кислота, 4-хлорбензолсульфоновая кислота, 2-нафталинсульфоновая кислота, 4-толуолсульфоновая кислота, камфорсульфоновая кислота, 4-метилбицикло[2.2.2]-окт-2-ен-1-карбоновая кислота, глюкогептоновая кислота, 3-фенилпропионовая кислота, триметилуксусная кислота, третичная бутилуксусная кислота, лаурилсерная кислота, глюконовая кислота, глутаминовая кислота, гидроксинафтойная кислота, салициловая кислота, стеариновая кислота, муконовая кислота и подобные; или (2) соли, образованные, когда кислотный протон, присутствующий в соединении, замещен ионом металла, например, ионом щелочного металла, ионом щелочноземельного металла или ионом алюминия; или координируется с органическим основанием, таким как этаноламин, диэтаноламин, триэтаноламин, N-метилглюкамин и подобные.
Выражение "сольват", как используется в данном документе, относится к комплексу или агрегату, образованному одной или более молекулами растворенного вещества, например, пролекарством или его фармацевтически приемлемой солью и одной или более молекулой растворителя. Такие сольваты типично являются кристаллическими твердыми веществами, имеющими в основном установленное молярное соотношение растворенного вещества и растворителя. Типичные растворители включают, в качестве примера, воду, метанол, этанол, изопропанол, уксусную кислоту и подобное. Когда растворителем является вода, образованный сольват является гидратом.
"Фармацевтически приемлемый носитель" относится к разбавителю, вспомогательному веществу, наполнителю или носителю, с которым или в котором соединение вводится.
"Предотвращение", или "предупреждение", или "профилактика" относится к снижению риска возникновения состояния такого, как боль.
"Пролекарство" относится к производному активного средства, которое требует преобразования в организме для высвобождения действующего вещества. В определенных вариантах осуществления под преобразованием имеют в виду ферментативное преобразование. Пролекарства зачастую, хотя и необязательно, являются фармакологически неактивными до превращения в действующее вещество.
"Про-фрагмент" относится к форме защитной группы, которая при применении для маскировки функциональной группы в действующем веществе превращает действующее вещество в Пролекарство. Обычно, про-фрагмент присоединен к лекарственному средству посредством связи(связей), которые расщепляются ферментным или неферментным средством in vivo.
"Терапия" или "лечение" любого состояния, такого как боль, означает в определенных вариантах осуществления улучшение патологического состояния (т.е., купирование или снижение проявления указанного состояния). В определенных вариантах осуществления "терапия" или "лечение" относится к улучшению по меньшей мере одного физического параметра, который может незаметным для пациента. В определенных вариантах осуществления "терапия" или "лечение" относится к ингибированию состояния, либо физически (например, стабилизация заметного симптома), физиологически (например, стабилизация физического параметра), либо и тем, и другим образом. В определенных вариантах осуществления "терапия" или "лечение" относится к задерживанию появления патологического состояния.
"Терапевтически эффективное количество" означает количество соединения (например, пролекарства), которое при введении пациенту для предотвращения или терапии состояния такого, как боль, является достаточным для проведения такого лечения. "Терапевтически эффективное количество" будет варьироваться в зависимости от соединения, состояния и его тяжести, возраста, веса и т.д. пациента.
Подробное описание
Перед более подробным описанием данного изобретения следует понимать, что данное изобретение не ограничивается описанными конкретными вариантами осуществления, и, как таковое, может варьировать. Также следует понимать, что терминология, применяемая в данном документе, служит для описания только конкретных вариантов осуществления и не предназначена для ограничения, так как объем данного изобретения будет ограничен только приложенной формулой изобретения.
Следует отметить, что используемые в данном документе и в приложенной формуле изобретения единственные формы включают множественные, если контекст четко не диктует иное. Далее следует отметить, что формула изобретения может быть составлена таким образом, чтобы исключить любой необязательный элемент. В связи с этим, это изложение призвано служить предшествующей основой для применения такой исключительной терминологии, как "исключительно", "только" и подобное, в связи с перечислением заявленных элементов или применением "отрицательного" ограничения.
Следует понимать, что, как используется в данном документе, единственное число объекта подразумевает один или более таких объектов. Например, соединение означает одно или более соединения. По этой причине, выражения в единственном числе, "один или более" и "по меньшей мере один" могут использоваться взаимозаменяемо. Подобным образом, выражения "содержащий", "включающий" и "имеющий" могут использоваться взаимозаменяемо.
Публикации, раскрытые в данном документе, представлены исключительно для раскрытия известного уровня техники до даты подачи данной заявки. Изложенное в данном документе не может быть истолковано как признание того, что данное изобретение не имеет права предшествовать таким публикациям путем преимущества предшествующего изобретения. Кроме того, даты представленной публикации могут отличаться от фактических дат публикации, которые могут потребовать независимого подтверждения.
Если не указано иное, все технические и научные выражения, применяемые в данном документе, имеют то же значение, что обычно понимается специалистом в данной области, к которой относится данное изобретение. Хотя любые способы и материалы, подобные или эквивалентные описанным в данном документе, также могут быть применены при осуществлении или тестировании данного изобретения, теперь описываются предпочтительные способы и материалы. Все публикации, упомянутые в данном документе, включены в данный документ ссылкой на раскрытые и описанные способы и/или материалы, в связи с которыми публикации цитируются.
Если не указано иное, способы и методики данных вариантов осуществления обычно выполняют согласно общепринятым способам, хорошо известным в данном уровне техники, и как описано в различных общих и более конкретных ссылках, которые цитируются и обсуждаются во всем данном описании. Смотри, например, Loudon, Organic Chemistry, Fourth Edition, New York: Oxford University Press, 2002, pp.360-361, 1084-1085; Smith and March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Fifth Edition, Wiley-Interscience, 2001.
Номенклатура, применяемая в данном документе для именования объектов соединений, проиллюстрирована в Примерах в данном документе. Когда возможно, эти названия обычно получали с использованием коммерчески доступного программного обеспечения AutoNom (MDL, Сан Леандро, Калифорния).
Следует понимать, что определенные свойства изобретения, которые в целях ясности описаны в отдельных вариантах осуществления, могут также быть представлены в сочетании друг с другом в одном варианте изобретения. И наоборот, различные свойства изобретения, которые для краткости описаны в контексте одного варианта осуществления, могут быть также представлены отдельно или в любой подходящей суб-комбинации. Все комбинации вариантов осуществления, имеющие отношение к химическим группам, представленным переменными величинами, специально охвачены данным изобретением и раскрыты здесь также, как если бы каждая комбинация была индивидуально и подробно раскрыта до такой степени, что такие комбинации охватывали бы соединения, являющиеся стабильными соединениями (т.е. соединения, которые можно выделить, охарактеризовать и определить их биологическую активность). Кроме того, все суб-комбинации химических групп, перечисленных в описывающих такие переменные величины вариантах осуществления, также специально охвачены данным изобретением и раскрыты здесь также, как если бы каждая такая суб-комбинация химических групп была индивидуально и подробно раскрыта в данном документе.
Общие методики синтеза
Многие общие ссылки, дающие хорошо известные химические синтетические схемы и условия, используемые для синтеза раскрытых соединений широко доступны (смотри, например, Smith and March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Fifth Edition, Wiley-Interscience, 2001; или Vogel, A Textbook of Practical Organic Chemistry, Including Qualitative Organic Analysis, Fourth Edition, New York: Longman, 1978).
Описанные здесь соединения могут быть очищены любыми известными на этом уровне техники способом, включая хроматографию, как, например, высокоэффективная жидкостная хроматография (ВЭЖХ), препаративная тонкослойная хроматография, флэш-хроматография на колонке и ионообменная хроматография. Можно использовать любую подходящую стационарную фазу, включая прямую и обратную фазы, а также ионные смолы. Смотри, например. Introduction to Modem Liquid Chromatography, 2-е издание, под ред. L.R.Snyder и J.J.Kirki and, John Wiley and Sons, 1979; и Thin Layer Chromatography, ред. E.Stahl, Springer-Verlag, Нью Йорк, 1969.
В любом из процессов получения соединений данного раскрытия может быть необходимо и/или желательно защитить чувствительные или реакционно-способные группы в любой из молекул, к которой это имеет отношение. Это может быть достигнуто с помощью общепринятых защитных групп как описано в классических трудах таких, как Т.W.Greene and P.G.M. Wuts, "Protective Groups in Organic Synthesis", Fourth edition, Wiley, Нью Йорк 2006. Защитные группы могут быть удалены на удобной последующей стадии, используя известные в данном уровне техники способы.
Соединения, описанные в данном документе, могут включать один или более хиральных центров и/или двойных связей и, следовательно, могут существовать как стереоизомеры, такие как изомеры относительно двойной связи (т.е., геометрические изомеры), энантиомеры или диастереомеры. Соответственно, все возможные энантиомеры и стереоизомеры соединений, включая стереоизомерно чистую форму (например, геометрически чистую, энантиомерно чистую или диастереомерно чистую), энантиомерные и стереоизомерные смеси, включены в описание соединений в данном документе. Энантиомерные и стереоизомерные смеси могут быть разделены на составляющие их энантиомеры или стереоизомеры с использованием техник разделения или техник хирального синтеза, хорошо известных специалисту в данной области. Соединения также могут существовать в нескольких таутомерных формах, включая енольную форму, кето-форму и их смеси. Соответственно, химические структуры, описанные в данном документе, охватывают все возможные таутомерные формы иллюстрированных соединений. Описанные соединения также включают изотопно-меченные соединения, где один или более атомов имеют атомную массу, отличную от атомной массы, обычно встречающейся в природе. Примеры изотопов, которые могут быть включены в соединения, раскрытые в данном документе, включают, но без ограничения, 2H, 3H, 13C, 14C, 15C, 15N, 15O, 17O, и т.д. Соединения могут существовать как в несольватированных, так и в сольватированных формах, включая гидратированные формы. В общем, соединения могут быть гидратированными или сольватированными. Определенные соединения могут существовать в многочисленных кристаллических или аморфных формах. В основном, все физические формы являются эквивалентными для применений, предполагаемых в данном документе, и предназначены для включения в объем данного раскрытия.
Типичные варианты осуществления
Далее будут сделаны подробные ссылки на различные варианты осуществления изобретения. Следует понимать, что изобретение не ограничено данными вариантами осуществления. Напротив, они предназначены для того, чтобы охватить варианты, модификации и эквиваленты, которые могут охватываться объемом принятой формулы изобретения.
Данное раскрытие представляет фармацевтические композиции и способы их применения, где фармацевтические композиции содержат опиоидное пролекарство с модифицированным кетоном, которое обеспечивает ферментативно-регулируемое высвобождение кетон-содержащего опиоида и, по выбору, ингибитор фермента, который взаимодействует с ферментом(ферментами), который выступает посредником в ферментативно-регулируемом высвобождении кетон-содержащего опиоида из пролекарства с тем, чтобы уменьшить ферментативное расщепление пролекарства. Раскрытие предоставляет фармацевтические композиции, которые по выбору содержат ингибитор трипсина и опиоидное пролекарство с модифицированным кетоном, содержащее расщепляемый трипсином фрагмент, расщепление которого содействует высвобождению кетон-содержащего опиоида.
Согласно одному аспекту, варианты осуществления включают фармацевтические композиции, которые содержат расщепляемое трипсином опиоидное пролекарство с модифицированным кетоном и, по выбору, трипсиновый ингибитор. Примеры опиоидного пролекарства с модифицированным кетоном и трипсиновых ингибиторов описаны ниже.
Кетон-содержащие опиоиды
"Опиоид" относится к химическому веществу, которое оказывает свое фармакологическое действие путем взаимодействия с опиоидным рецептором. Опиоид может быть выделенным натуральным продуктом, синтетическим соединением или полусинтетическим соединением. "Кетон-содержащий опиоид" относится к подгруппе опиоидов, которые содержат группу кетона. Как используется в данном документе, кетон-содержащий опиоид является опиоидом, содержащим енолизируемую кетонную группу. Кетон-содержащий опиоид представляет собой соединение с фармакофором, который предоставляет опиоидному рецептору ароматическую группу и алифатическую аминную группу архитектурно обособленным образом. Смотри, например, Foye's Principles of Medicinal Chemistry, 6-е издание, ред. T.L.Lemke и D.A.Williams, Lippincott Williams & Wilkins, 2008, в особенности Главу 24, стр.653-678.
Например, кетон-содержащие опиоиды включают, но без ограничения, ацетилморфон, гидрокодон, гидроморфон, кетобемидон, метадон, налоксон, N-метилналоксон, налтрексон, N-метилналтрексон, оксикодон, оксиморфон и пентаморфон.
В определенных вариантах осуществления кетон-содержащий опиоид представляет собой гидрокодон или оксикодон.
Предполагается, что будут обнаружены опиоиды, несущие в себе по меньшей мере некоторые из описанных здесь функциональностей; такие опиоиды включены как часть данного раскрытия.
Опиоидные пролекарства с модифицированным кетоном
Данное раскрытие обеспечивает опиоидное пролекарство с модифицированным кетоном, которое обеспечивает регулируемое ферментами высвобождение кетон-содержащего опиоида. В опиоидном пролекарстве с модифицированным кетоном про-фрагмент присоединен к кетон-содержащему опиоиду по енольному атому кислорода кетонного фрагмента. В опиоидном пролекарстве с модифицированным кетоном атом водорода соответствующей енольной группы кетон-содержащего опиоида замещен ковалентной связью с про-фрагментом.
Как раскрыто в данном документе, расщепляемое трипсином опиоидное пролекарство с модифицированным кетоном представляет собой опиоидное пролекарство с модифицированным кетоном, содержащее расщепляемый трипсином фрагмент, т.е. фрагмент, имеющий поддающийся расщеплению трипсином участок. Такое пролекарство содержит кетон-содержащий опиоид, ковалентно связанный с про-фрагментом, содержащим расщепляемый трипсином фрагмент, где расщепление трипсином расщепляемого трипсином фрагмента способствует высвобождению лекарственного средства. Расщепление может инициировать, содействовать или влиять на высвобождение лекарственного средства.
Опиоидное пролекарство с модифицированным кетоном с про-фрагментом, содержащим циклизуемую спейсерную уходящую группу и расщепляемый фрагмент
Согласно определенным вариантам осуществления представлено опиоидное пролекарство с модифицированным кетоном, которое обеспечивает регулируемое ферментом высвобождение кетон-содержащего опиоида. Раскрытие обеспечивает кетон-содержащий опиоид, в котором про-фрагмент содержит циклизуемую спейсерную уходящую группу и расщепляемый фрагмент. В определенных вариантах осуществления кетон-содержащее опиоидное пролекарство является соответствующим соединением, в котором енольный атом кислорода имеет заместитель, являющийся спейсерной уходящей группой, несущей азотный нуклеофил, который защищен расщепляемым ферментом компонентом, причем конфигурация спейсерной уходящей группы и азотного нуклеофила является такой, чтобы при ферментативном расщеплении расщепляемого фрагмента азотный нуклеофил был способен образовывать циклическую мочевину, освобождая соединение от спейсерной уходящей группы, таким образом обеспечивая кетон-содержащий опиоид.
Соответствующие пролекарства обеспечивают следующую за введением активацию и регулируемое высвобождение кетон-содержащего опиоида. Пролекарство нуждается в ферментативном расщеплении для инициирования высвобождения кетон-содержащего опиоида, и, таким образом, скорость высвобождения кетон-содержащего опиоида зависит как от скорости ферментативного расщепления, так и от скорости циклизации. Соответственно, пролекарство обладает пониженной склонностью к непреднамеренной передозировке или злоупотреблению в результате преднамеренной передозировки, введения непредусмотренным образом, например, путем инъекции, или путем химической модификации с использованием легкодоступных бытовых химикатов. Пролекарство составлено таким образом, что оно не производит чрезмерно высокие уровни действующего вещества в плазме при непредусмотренном способе введения и не может быть легко разложено с образованием действующего вещества другим способом, кроме ферментативного расщепления с последующей регулируемой циклизацией.
Расщепляемый ферментом фрагмент, связанный с азотным нуклеофилом посредством амидной связи, может представлять собой, например, остаток аминокислоты или пептида, или (альфа)N-ацильное производное аминокислоты или пептида (например, N-ацильное производное фармацевтически приемлемой карбоновой кислоты). Пептид может содержать, например, вплоть до 100 аминокислотных остатков. Каждая аминокислота преимущественно может быть натуральной аминокислотой, как, например, L-аминокислотой. Примерами встречающихся в природе аминокислот являются аланин, аргинин, аспарагин, аспарагиновая кислота, цистеин, глицин, глутамин, глутаминовая кислота, гистидин, изолейцин, лейцин, лизин, метионин, фенилаланин, пролин, серии, треонин, триптофан, тирозин и валин. Соответственно, примеры расщепляемых ферментом фрагментов включают остатки L-аминокислот, перечисленных выше, и их N-ацильных производных и пептиды, образованные из по меньшей мере двух перечисленных выше L-аминокислот и их N-ацильных производных.
Циклическая группа, образующаяся при высвобождении кетон-содержащего опиоида, является фармацевтически приемлемой, в особенности фармацевтически приемлемой является циклическая мочевина. Специалистом будет оценено то, что циклические мочевины в основном являются весьма стабильными и малотоксичными.
Формулы KC-(I) и KC-(II)
Композиции настоящего раскрытия включают соединения формулы KC-(I) и KC-(II), показанные ниже. Соединения формул KC-(I) и KC-(II) представляют собой пролекарства оксикодон и гидрокодон. Фармацевтические композиции и способы настоящего раскрытия также подразумевают соединения формул KC-(I) и KC-(II).
Формула KC-(I)
В одном из композиционных аспектов настоящие варианты осуществления предоставляют соединение формулы KC-(Ia):
где:
Ra представляет собой водород или гидроксил;
R5 выбран из алкила, замещенного алкила, арилалкила, замещенного арилалкила, арила и замещенного арила;
каждый R1 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, ацила и аминоацила;
каждый R2 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, ацила и аминоацила;
или R1 и R2 вместе с углеродом, к которому они присоединены, образуют циклоалкильную, замещенную циклоалкильную, арильную или замещенную арильную группу, или две R1 или R2 группы на соседних атомах углерода вместе с атомами углерода, к которым они присоединены, образуют циклоалкильную, замещенную циклоалкильную, арильную или замещенную арильную группу;
n представляет собой целое число от 2 до 4;
R3 является водородом;
R4 представляет собой ;
каждый R6 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, арилалкила, замещенного арилалкила, гетероалкила, замещенного гетероалкила, гетероарила, замещенного гетероарила, гетероарилалкила и замещенного гетероарилалкила или, необязательно, R6 и R7 вместе с атомами, к которым они присоединены, образуют циклогетероалкильное или замещенное циклогетероалкильное кольцо;
каждый W независимо представляет собой -NR8-, -О- или -S-;
каждый R8 независимо выбран из водорода, алкила, замещенного алкила, арила и замещенного арила или, необязательно, каждый R6 и R8 независимо вместе с атомами, к которым они присоединены, образуют циклогетероалкильное или замещенное циклогетероалкильное кольцо;
p представляет собой целое число от 1 до 100; и
R выбран из водорода, алкила, замещенного алкила, ацила, замещенного ацила, алкоксикарбонила, замещенного алкоксикарбонила, арила, замещенного арила, арилалкила и замещенного арилалкила; или его соль, гидрат или сольват.
В одном из композиционных аспектов настоящие варианты осуществления предоставляют соединение формулы KC-(Ib):
где:
Ra представляет собой водород или гидроксил;
R5 выбран из алкила, замещенного алкила, арилалкила, замещенного арилалкила, арила и замещенного арила;
каждый R1 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, ацила и аминоацила;
каждый R2 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, ацила и аминоацила;
или R1 и R2 вместе с углеродом, к которому они присоединены, образуют циклоалкильную или замещенную циклоалкильную группу, или две R1 или R2 группы на соседних атомах углерода вместе с атомами углерода, к которым они присоединены, образуют циклоалкильную или замещенную циклоалкильную группу;
n представляет собой целое число от 2 до 4;
R3 является водородом;
R4 представляет собой R6 p;
каждый R6 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, арилалкила, замещенного арилалкила, гетероалкила, замещенного гетероалкила, гетероарила, замещенного гетероарила, гетероарилалкила и замещенного гетероарилалкила или, необязательно, R1 и R2 вместе с атомами, к которым они присоединены, образуют циклогетероалкильное или замещенное циклогетероалкильное кольцо;
каждый W независимо представляет собой -NR8-, -О- или -S-;
каждый R8 независимо выбран из водорода, алкила, замещенного алкила, арила и замещенного арила или, необязательно, каждый R6 и R8 независимо вместе с атомами, к которым они присоединены, образуют циклогетероалкильное или замещенное циклогетероалкильное кольцо;
p представляет собой целое число от 1 до 100; и
R7 выбран из водорода, алкила, замещенного алкила, ацила, замещенного ацила, алкоксикарбонила, замещенного алкоксикарбонила, арила, замещенного арила, арилалкила и замещенного арилалкила; или его соль, гидрат или сольват.
Ссылка на формулу KC-(I) призвана включать соединения формулы KC-(Ia) и KC-(Ib).
В формуле KC-(I) Ra может быть водородом или гидроксилом. В определенных случаях Ra представляет собой водород. В других случаях Ra представляет собой гидроксил.
В формуле KC-(I) R5 может быть выбран из алкила, замещенного алкила, арилалкила, замещенного арилалкила, арила и замещенного арила. В определенных случаях R5 является (1-6С)алкилом. В других случаях R5 представляет собой (1-4С)алкил. В других случаях R5 представляет собой метил или этил. В других случаях R представляет собой метил. В определенных случаях R5 представляет собой этил.
В определенных случаях R5 представляет собой замещенный алкил. В определенных случаях R5 представляет собой алкильную группу, замещенную карбоксильной группой такой, как карбоновая кислота, эфир карбоновой кислоты или амид карбоновой кислоты. В определенных случаях R5 представляет собой -(СН2)n-СООН, -(СН2)n-СООСН3, или -(CH2)n-COOCH2CH3, где n является числом от 1 до 10. В определенных случаях R1 представляет собой -(СН2)5-СООН, -(СН2)5-СООСН3 или -(СН2)5-СООСН2СН3.
В определенных случаях R5 в формуле KC-(I) представляет собой арилалкил или замещенный арилалкил. В определенных случаях R5 в формуле KC-(I) представляет собой арилалкил. В определенных случаях R5 представляет собой замещенный арилалкил. В определенных случаях R5 представляет собой арилалкильную группу, замещенную карбоксильной группой такой, как карбоновая кислота, эфир карбоновой кислоты или амид карбоновой кислоты. В определенных случаях R5 представляет собой -(CH2)q(C6H4)-COOH, -(СН2)q(С6Н4)-СООСН3 или -(CH2)q(C6H4)-COOCH2CH3, где q представляет собой целое число от 1 до 10; В определенных случаях R5 представляет собой -СН2(С6Н4)-СООН, -СН2(С6Н4)-СООСН3 или -СН2(С6Н4)-СООСН2СН3.
В определенных случаях R5 в формуле KC-(I) представляет собой арил. В определенных случаях R5 представляет собой замещенный арил. В определенных случаях R5 представляет собой арильную группу, замещенную в орто-, мета- или пара-положении на карбоксильную группу такую, как карбоновая кислота, эфир карбоновой кислоты или амид карбоновой кислоты. В определенных случаях R5 является -(С6Н4)-СООН, -(С6Н4)-СООСН3 или -(С6Н4)-СООСН2СН3.
В формуле KC-(I) каждый R1 может быть независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, ацила и аминоацила. В определенных случаях R1 представляет собой водород или алкил. В определенных случаях R1 является водородом. В определенных случаях R1 представляет собой алкил. В определенных случаях R1 представляет собой ацил. В определенных случаях R1 представляет собой аминоацил.
В формуле KC-(I) каждый R2 может быть независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, ацила и аминоацила. В определенных случаях R2 представляет собой водород или алкил. В определенных случаях R2 является водородом. В определенных случаях R2 представляет собой алкил. В определенных случаях R2 представляет собой ацил. В определенных случаях R2 представляет собой аминоацил.
В определенных случаях R1 и R2 являются водородом. В определенных случаях R1 и R2 оба на одном и том же углероде являются алкилом. В определенных случаях R1 и R2 на одном и том же углероде представляют собой метил. В определенных случаях R1 и R2 на одном и том же углероде представляют собой этил.
В определенных случаях, когда R1 и R2 являются соседними группами, они оба представляют собой алкил, и когда R2 и R2 являются соседними группами, они оба представляют собой водород. В определенных случаях, когда R1 и R1 являются соседними группами, они оба представляют собой этил, и когда R2 и R2 являются соседними группами, они оба представляют собой водород. В определенных случаях, когда R1 и R1 являются соседними группами, они оба представляют собой метил, и когда R2 и R2 являются соседними группами, они оба представляют собой водород.
В определенных случаях в цепи -[С(R1)(R2)]n- в Формуле KC-(I) замещены не все атомы углерода. В определенных случаях в цепи -[C(R1)(R2)]n- присутствует комбинация различных алкильных заместителей таких, как метил или этил.
В определенных случаях один из R1 и R2 представляют собой метил, этил или другой алкил, а R5 является алкилом. В определенных случаях, когда R1 и R1 являются соседними группами, они оба представляют собой алкил, и когда R2 и R2 являются соседними группами, они оба представляют собой водород, а R5 является алкилом. В определенных случаях, когда R1 и R1 являются соседними группами, они оба представляют собой этил, и когда R2 и R2 являются соседними группами, они оба представляют собой водород, а R5 является алкилом. В определенных случаях, когда R1 и R1 являются соседними группами, они оба представляют собой метил, и когда R2 и R2 являются соседними группами, они оба представляют собой водород, а R5 является алкилом.
В определенных случаях один из R1 и R2 представляет собой метил, этил или другой алкил, а R5 является замещенным алкилом. В определенных случаях один из R1 и R2 представляет собой метил, этил или другой алкил, а R5 представляет собой алкильную группу, замещенную карбоксильной группой такой, как карбоновая кислота, эфир карбоновой кислоты или амид карбоновой кислоты. В определенных случаях один из R1 и R2 является метилом, этилом или другим алкилом, а R5 представляет собой -(CH2)q(C6H4)-СООН, -(СН2)q(С6Н4)-СООСН3, или -(СН2)q(С6Н4)-СООСН2СН3, где q представляет собой целое число от 1 до 10; В определенных случаях один из R1 и R2 представляет собой метил, этил или другой алкил, а R5 является алкильной группой, замещенной карбоксамидом.
В формуле KC-(I) R1 и R2 вместе с углеродом, к которому они присоединены, могут образовать циклоалкильную или замещенную циклоалкильную группу, или две R1 или R2 группы на соседних атомах углерода вместе с атомами углерода, к которым они присоединены, могут образовать циклоалкильную или замещенную циклоалкильную группу. В определенных случаях R1 и R2 вместе с углеродом, к которому они присоединены, могут образовать циклоалкильную группу. Таким образом, в определенных случаях R1 и R2 на одном и том же углероде образуют спироцикл. В определенных случаях R1 и R2 вместе с углеродом, к которому они присоединены, могут образовать замещенную циклоалкильную группу. В определенных случаях две R1 или R2 группы на соседних атомах углерода вместе с атомами углерода, к которым они присоединены, могут образовать циклоалкильную группу. В определенных случаях две R1 или R2 группы на соседних атомах углерода вместе с атомами углерода, к которым они присоединены, могут образовать замещенную циклоалкильную группу.
В формуле KC-(I), R1 и R2 вместе с углеродом, к которому они присоединены, могут образовать ар ильную или замещенную арильную группу, или две R1 или R2 группы на соседних атомах углерода вместе с атомами углерода, к которым они присоединены, могут образовать арильную или замещенную арильную группу. В определенных случаях две R1 или R2 группы на соседних атомах углерода вместе с атомами углерода, к которым они присоединены, образуют фенильное кольцо. В определенных случаях две R1 или R2 группы на соседних атомах углерода вместе с атомами углерода, к которым они присоединены, образуют замещенное фенильное кольцо. В определенных случаях две R1 или R2 группы на соседних атомах углерода вместе с атомами углерода, к которым они присоединены, образуют нафталиновое кольцо.
В определенных случаях один из R1 и R2 представляет собой аминоацил.
В определенных случаях один или оба из R1 и R2 представляет собой аминоацил, содержащий фенилендиамин. В определенных случаях один из R1 и R2 представляет собой 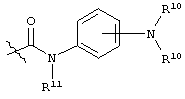 ; где каждый R10 независимо выбран из водорода, алкила, замещенного алкила и ацила, и R11 представляет собой алкил или замещенный алкил. В определенных случаях по меньшей мере один из R10 представляет собой ацил. В определенных случаях, по меньшей мере, один из R10 представляет собой алкил или замещенный алкил. В определенных случаях, по меньшей мере, один из R представляет собой водород. В определенных случаях оба R10 являются водородом.
; где каждый R10 независимо выбран из водорода, алкила, замещенного алкила и ацила, и R11 представляет собой алкил или замещенный алкил. В определенных случаях по меньшей мере один из R10 представляет собой ацил. В определенных случаях, по меньшей мере, один из R10 представляет собой алкил или замещенный алкил. В определенных случаях, по меньшей мере, один из R представляет собой водород. В определенных случаях оба R10 являются водородом.
В определенных случаях один из R1 и R2 представляет собой 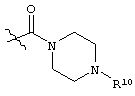 ; где R10 представляет собой водород, алкил, замещенный алкил или ацил. В определенных случаях R10 представляет собой ацил. В определенных случаях R10 представляет собой алкил или замещенный алкил. В определенных случаях R10 представляет собой водород.
; где R10 представляет собой водород, алкил, замещенный алкил или ацил. В определенных случаях R10 представляет собой ацил. В определенных случаях R10 представляет собой алкил или замещенный алкил. В определенных случаях R10 представляет собой водород.
В определенных случаях один из R1 и R2 представляет собой 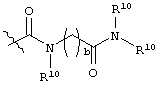 ; где каждый R10 независимо представляет собой водород, алкил, замещенный алкил или ацил, a b является числом от 1 до 5. В определенных случаях один из R1 и R2 представляет собой
; где каждый R10 независимо представляет собой водород, алкил, замещенный алкил или ацил, a b является числом от 1 до 5. В определенных случаях один из R1 и R2 представляет собой 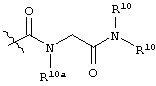 ; где R10a независимо представляет собой водород, алкил,
; где R10a независимо представляет собой водород, алкил,
замещенный алкил или ацил. В определенных случаях один из R1 и R2 представляет собой ; где R10a является алкилом, а каждый R10 независимо представляет собой водород, алкил, замещенный алкил или ацил.
В определенных случаях один из R1 и R2 представляет собой 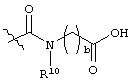 ; где R10 независимо представляет собой водород, алкил, замещенный алкил или ацил, а b является числом от 1 до 5. В определенных случаях один из R1 и R2 представляет собой ; где R10 независимо представляет собой водород, алкил, замещенный алкил или ацил.
; где R10 независимо представляет собой водород, алкил, замещенный алкил или ацил, а b является числом от 1 до 5. В определенных случаях один из R1 и R2 представляет собой ; где R10 независимо представляет собой водород, алкил, замещенный алкил или ацил.
В определенных случаях один из R1 и R2 представляет собой аминоацильную группу такую, как -C(O)NR10aR10b, где каждый R10a и R10b независимо выбран из водорода, алкила, замещенного алкила и ацила. В определенных случаях один из R1 и R2 представляет собой аминоацильную группу такую, как -C(O)NR10aR10b, где R10a является алкилом, и R10b представляет собой замещенный алкил. В определенных случаях один из R1 и R2 представляет собой аминоацильную группу такую, как -C(O)NR10aR10b, где R10a является алкилом, и R10b представляет собой алкил, замещенный карбоновой кислотой или эфиром карбоновой кислоты. В определенных случаях один из R1 и R2 представляет собой аминоацильную группу такую, как -C(O)NR10aR10b, где R10a является метилом, и R10b представляет собой алкил, замещенный карбоновой кислотой или эфиром карбоновой кислоты.
В определенных случаях R1 или R2 может изменять скорость внутримолекулярной циклизации. R1 или R2 может повышать скорость внутримолекулярной циклизации по сравнению с соответствующей молекулой, где R1 и R2 оба являются водородом. В определенных случаях R1 или R2 содержит электроноакцепторную группу или электронодонорную группу. В определенных случаях R1 или R2 содержит электроноакцепторную группу. В определенных случаях R1 или R2 содержит электронодонорную группу.
Атомы и группы, способные функционировать как электроноакцепторные заместители, хорошо известны в области органической химии. Они включают электроотрицательные атомы и группы, содержащие электроотрицательные атомы. Такие группы действуют путем понижения основности или состояния протонирования нуклеофильного азота в бета-положении через индуктивное оттягивание электронной плотности. Такие группы также могут быть расположены в других положениях вдоль алкиленовой цепи. Примеры включают атомы галогена (например, атом фтора), ацильные группы (например, алканоильная группа, ароильная группа, карбоксильная группа, алкоксикарбонильная группа, арилоксикарбонильная группа или аминокарбонильная группа (такая как карбамоильная, алкиламинокарбонильная, диалкиламинокарбонильная или ариламинокарбонильная группа)), оксо- (=O) заместитель, нитрильную группу, нитрогруппу, группы простого эфира (например, алкокси-группа) и фенильные группы, несущие заместитель в орто-положении, пара-положении или и в орто-, и в пара-положениях, каждый заместитель является независимо выбранным из атома галогена, фторалкильной группы (такой как трифторметил), нитрогруппы, цианогруппы и карбоксильной группы. Каждый из электроноакцепторных заместителей может быть независимо выбран из них.
В определенных случаях -[C(R1)R2)]n- выбирают из -CH(CH2F)CH(CH2F)-; -CH(CHF2)CH(CHF2)-; -СН(CF3)CH(CF3)-; -CH2CH(CF3)-; -CH2CH(CHF2)-; -CH2CH(CH2F)-; -CH2CH(F)CH2-; -CH2C(F2)CH2-; -CH2CH(C(O)NR20R21)-; -CH2CH(C(O)OR22)-; -СН2СН(С(O)ОН)-; -CH(CH2F)CH2CH(CH2F)-; -CH(CHF2)CH2CH(CHF2)-; СН(CF3)СН2СН(CF3)-; -СН2СН2СН(CF3)-; -CH2CH2CH(CHF2)-; -CH2CH2CH(CH2F)-; -CH2CH2CH(O)NR23R24)-; -CH2CH2CH(C(O)OR25)-; и -СН2СН2СН(С(O)ОН)-, в которых R20, R21, R22 и R23 каждый независимо представляет водород или (1-6С)алкил, и R24 и R25 каждый независимо представляет (1-6С)алкил.
В формуле KC-(I) n может быть целым числом от 2 до 4. В определенных случаях n равно 2. В других случаях n равно 3. В других случаях n равно 4.
В формуле KC-(I), R4 может быть остатком L-аминокислоты, выбранной из аланина, аргинина, аспарагина, аспарагиновой кислоты, цистеина, глицина, глутамина, глутаминовой кислоты, гистидина, изолейцина, лейцина, лизина, метионина, фенилаланина, пролина, серина, треонина, триптофана, тирозина и валина, или остатка N-ацильного производного любой из перечисленных аминокислот; или остаток пептида, состоящий из по меньшей мере двух остатков L-аминокислот, независимо выбранных из аланина, аргинина, аспарагина, аспарагиновой кислоты, цистеина, глицина, глутамина, глутаминовой кислоты, гистидина, изолейцина, лейцина, лизина, метионина, фенилаланина, пролина, серина, треонина, триптофана, тирозина и валина, или остатка их N-ацильного производного. Такой пептид может иметь длину от 2 до 100 аминокислот. Примеры N-ацильных производных включают ацетильные, бензоильные, малонильные, пиперонильные или сукцинильные производные.
В определенных случаях R4 представляет собой остаток L-аргинина или L-лизина, или остаток N-ацильного производного L-аргинина или L-лизина.
В определенных случаях в формуле KC-(I), когда p больше единицы, то R4, ближайший к азоту -N(R3)R4) группы, представляет собой остаток L-аргинина или L-лизина. В определенных случаях, когда p больше единицы, R4, ближайший к азоту в группе -N(R3)(R4), представляет собой остаток L-аргинина или L-лизина, а первый остаток присоединен к по меньшей мере одному дополнительному L-аминокислотному остатку, независимо выбранному из аланина, аргинина, аспарагина, аспарагиновой кислоты, цистеина, глицина, глутамина, глутаминовой кислоты, гистидина, изолейцина, лейцина, лизина, метионина, фенилаланина, пролина, серина, треонина, триптофана, тирозина и валина. Концевой остаток пептида может быть N-ацильным производным любой такой L-аминокислоты. В определенных случаях R4 представляет собой дипептид или его N-ацильное производное. В определенных случаях R представляет собой трипептид или его N-ацильное производное.
В формуле KC-(I), R4 представляет собой .
В формуле KC-(I) каждый R6 может быть независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, арилалкила, замещенного арилалкила, гетероалкила, замещенного гетероалкила, гетероарила, замещенного гетероарила, гетероарилалкила и замещенного гетероарилалкила или, необязательно, R6 и R7 вместе с атомами, к которым они присоединены, образуют циклогетероалкильное или замещенное циклогетероалкильное кольцо.
В определенных случаях в формуле KC-(I) R6 выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, арилалкила, замещенного арилалкила, гетероалкила, замещенного гетероалкила, гетероарила, замещенного гетероарила, гетероарилалкила и замещенного гетероарилалкила. В определенных случаях R6 выбран из водорода, алкила, замещенного алкила, арилалкила, замещенного арилалкила, гетероарилалкила и замещенного гетероарилалкила. В определенных случаях R6 является водородом. В определенных случаях R6 представляет собой алкил. В определенных случаях R представляет собой замещенный алкил. В определенных случаях R6 представляет собой арилалкил или замещенный арилалкил. В определенных случаях R6 представляет собой гетероарилалкил или замещенный гетероарилалкил.
В определенных случаях R6 представляет собой боковую цепь из аминокислоты такой, как аланин, аргинин, аспарагин, аспарагиновая кислота, цистеин, глицин, глутамин, глутаминовая кислота, гистидин, изолейцин, лейцин, лизин, метионин, фенилаланин, пролин, серии, треонин, триптофан, тирозин или валин. В определенных случаях R6 представляет собой боковую цепь из L-аминокислоты такой, как L-аланин, L-аргинин, L-аспарагин, L-аспарагиновая кислота, L-цистеин, L-глицин, L-глутамин, L-глутаминовая кислота, L-гистидин, L-изолейцин, L-лейцин, L-лизин, L-метионин, L-фенилаланин, L-пролин, L-серин, L-треонин, L-триптофан, L-тирозин или L-валин.
В определенных случаях R6 представляет собой -CH2CH2CH2NH(C=NH)NH2 или -CH2CH2CH2CH2NH2.
В формуле KC-(I), каждый W может быть независимо -NR8-, -О- или -S-. В определенных случаях W является -NR8-. В определенных случаях W является -O-. В определенных случаях W является -S-.
В формуле KC-(I) каждый R8 может быть независимо выбран из водорода, алкила, замещенного алкила, арила или замещенного арила или, необязательно, каждый R6 и R8 независимо вместе с атомами, к которым они присоединены, образуют циклогетероалкильное или замещенное циклогетероалкильное кольцо.
В определенных случаях R8 в формуле KC-(I) представляет собой водород или алкил. В определенных случаях R8 является водородом. В определенных случаях R8 представляет собой алкил. В определенных случаях R8 представляет собой арил. В определенных случаях R6 и R8 независимо вместе с атомами, к которым они связаны, могут образовать циклогетероалкильное или замещенное циклогетероалкильное кольцо.
В формуле KC-(I) p может быть целым числом от 1 до 100, а каждый R6 может быть выбран независимо из боковой цепи любой аминокислоты. В определенных случаях p может быть целым числом от 1 до 50. В определенных случаях p является целым числом от 1 до 90, 80, 70, 60, 50, 40, 30, 20, или 10. В определенных случаях p представляет собой примерно 100. В определенных случаях p представляет собой примерно 75. В определенных случаях p представляет собой примерно 50. В определенных случаях p представляет собой примерно 25. В определенных случаях p представляет собой примерно 20. В определенных случаях p представляет собой примерно 15. В определенных случаях p представляет собой примерно 10. В определенных случаях p представляет собой примерно 9. В определенных случаях p представляет собой примерно 8. В определенных случаях p представляет собой примерно 7. В определенных случаях p представляет собой примерно 6. В определенных случаях p представляет собой примерно 5. В определенных случаях p представляет собой примерно 4. В определенных случаях p представляет собой примерно 3. В определенных случаях p представляет собой примерно 2. В определенных случаях p представляет собой примерно 1.
В определенных случаях R6 в R4, расположенном рядом в азотом группы N-(R3)(R4), представляет собой -CH2CH2CH2NH(C=NH)NH2 или -CH2CH2CH2CH2NH2, а любой дополнительный R6 может быть боковой цепью из любой аминокислоты, независимо выбранной из аланина, аргинина, аспарагина, аспарагиновой кислоты, цистеина, глицина, глутамина, глутаминовой кислоты, гистидина, изолейцина, лейцина, лизина, метионина, фенилаланина, пролина, серина, треонина, триптофана, тирозина или валина.
В формуле KC-(I) R7 может быть выбран из водорода, алкила, замещенного алкила, ацила, замещенного ацила, алкоксикарбонила, замещенного алкоксикарбонила, арила, замещенного арила, арилалкила и замещенного арилалкила.
В определенных случаях R7 представляет собой водород, алкил, ацил или замещенный алкил. В определенных случаях R7 является водородом. В определенных случаях R7 представляет собой алкил. В определенных случаях R представляет собой ацил или замещенный ацил. В определенных случаях R7 представляет собой ацил. В определенных случаях R7 представляет собой замещенный ацил. В определенных случаях R7 может представлять собой ацетил, бензоил, малонил, пиперонил или сукцинил.
Формула KC-(II)
Соединения формулы KC-(II) являются соединениями формулы KC-(I), в которой R5 выбран из (1-6С)алкила, (1-6С) замещенного алкила, -(CH2)q(C6H4)-СООН, -(СН2)q(С6Н4)-СООСН3, и -(СН2)q(С6Н4)-СООСН2СН3, где q является целым числом от 1 до 10; n равно 2 или 3; R3 является водородом; R4 является L-аминокислотой или пептидом, где пептид может содержать L-аминокислоты. В одном из композиционных аспектов настоящие осуществления изобретения предоставляют соединение формулы KC-(II):
где:
Ra представляет собой водород или гидроксил;
R5 выбран из (1-6С)алкила, (1-6С) замещенного алкила, -(CH2)q(C6H4)-COOH, -(СН2)q(С6Н4)-СООСН3, и -(СН2)q(С6Н4)-СООСН2СН3, где q представляет собой целое число от 1 до 10;
каждый R1 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, ацила и аминоацила;
каждый R2 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, ацила и аминоацила;
или R1 и R2 вместе с углеродом, к которому они присоединены, образуют циклоалкильную или замещенную циклоалкильную группу, или две R1 или R2 группы на соседних атомах углерода вместе с атомами углерода, к которым они присоединены, образуют циклоалкильную или замещенную циклоалкильную группу;
n равно 2 или 3;
R3 является водородом;
R4 представляет собой остаток L-аминокислоты, выбранной из аланина, аргинина, аспарагина, аспарагиновой кислоты, цистеина, глицина, глутамина, глутаминовой кислоты, гистидина, изолейцина, лейцина, лизина, метионина, фенилаланина, пролина, серина, треонина, триптофана, тирозина и валина, или остатка N-ацильного производного любой из перечисленных аминокислот; или остаток пептида, состоящий из по меньшей мере двух остатков L-аминокислот, независимо выбранных из аланина, аргинина, аспарагина, аспарагиновой кислоты, цистеина, глицина, глутамина, глутаминовой кислоты, гистидина, изолейцина, лейцина, лизина, метионина, фенилаланина, пролина, серина, треонина, триптофана, тирозина и валина, или остатка N-ацильного производного их.
В определенных вариантах осуществления в Формуле KC-(II) R4 является остатком L-аминокислоты, выбранной из аргинина или лизина.
В определенных случаях в формуле KC-(II), когда R представляет собой пептид, то R4, ближайший к азоту -N(R3)(R4) группы, представляет собой остаток L-аргинина или L-лизина. В определенных случаях R4 представляет собой дипептид или его N-ацильное производное. В определенных случаях R представляет собой трипептид или его N-ацильное производное.
В определенных вариантах осуществления в Формуле KC-(II) R4 является его N-ацильным производным. В определенных случаях R4 является остатком его N-ацильного производного, где N-ацильное производное является замещенным таким, как, но без ограничения, малонил и сукцинил.
Формулы от KC-(III) до KC-(V)
Композиции настоящего раскрытия включают соединения формул от KC-(III) до KC-(V), показанные ниже. Фармацевтические композиции и способы настоящего раскрытия также подразумевают соединения формул от KC-(III) до KC-(V).
Формула KC-(III)
В одном из аспектов композиции настоящие осуществления изобретения предоставляют соединение формулы KC-(IIIa):
где:
Х представляет остаток кетон-содержащего опиоида, где атом водорода соответствующей енольной группы замещен ковалентной связью с -C(O)-NR5-(C(R1)(R2)n-NR3R4;
R5 выбран из алкила, замещенного алкила, арилалкила, замещенного арилалкила, арила и замещенного арила;
каждый R1 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, ацила и аминоацила;
каждый R2 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, ацила и аминоацила;
или R1 и R2 вместе с углеродом, к которому они присоединены, образуют циклоалкильную, замещенную циклоалкильную, арильную или замещенную арильную группу, или две R2 или R3 группы на соседних атомах углерода вместе с атомами углерода, к которым они присоединены, образуют циклоалкильную, замещенную циклоалкильную, арильную или замещенную арильную группу;
n представляет собой целое число от 2 до 4;
R3 является водородом;
R4 представляет собой ;
каждый R6 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, арилалкила, замещенного арилалкила, гетероалкила, замещенного гетероалкила, гетероарила, замещенного гетероарила, гетероарилалкила и замещенного гетероарилалкила или, необязательно, R6 и R7 вместе с атомами, к которым они присоединены, образуют циклогетероалкильное или замещенное циклогетероалкильное кольцо;
каждый W независимо представляет собой -NR8-, -О- или -S-;
каждый R8 независимо выбран из водорода, алкила, замещенного алкила, арила и замещенного арила или, необязательно, каждый R6 и R8 независимо вместе с атомами, к которым они присоединены, образуют циклогетероалкильное или замещенное циклогетероалкильное кольцо;
p представляет собой целое число от 1 до 100; и
R7 выбран из водорода, алкила, замещенного алкила, ацила, замещенного ацила, алкоксикарбонила, замещенного алкоксикарбонила, арила, замещенного арила, арилалкила и замещенного арилалкила; или его соль, гидрат или сольват.
В одном из аспектов композиции настоящие осуществления изобретения предоставляют соединение формулы KC-(IIIb):
где:
Х представляет остаток кетон-содержащего опиоида, где атом водорода соответствующей енольной группы замещен ковалентной связью с -C(O)-NR5-(C(R1)(R2)n-NR3R4;
R5 выбран из алкила, замещенного алкила, арилалкила, замещенного арилалкила, арила и замещенного арила;
каждый R1 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, ацила и аминоацила;
каждый R2 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, ацила и аминоацила;
или R1 и R2 вместе с углеродом, к которому они присоединены, образуют циклоалкильную или замещенную циклоалкильную группу, или две R1 или R2 группы на соседних атомах углерода вместе с атомами углерода, к которым они присоединены, образуют циклоалкильную или замещенную циклоалкильную группу;
n представляет собой целое число от 2 до 4;
R3 является водородом;
R4 представляет собой ;
каждый R независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, арилалкила, замещенного арилалкила, гетероалкила, замещенного гетероалкила, гетероарила, замещенного гетероарила, гетероарилалкила и замещенного гетероарилалкила или, необязательно, R6 и R7 вместе с атомами, к которым они присоединены, образуют циклогетероалкильное или замещенное циклогетероалкильное кольцо;
каждый W независимо представляет собой -NR8-, -О- или -S-;
каждый R8 независимо выбран из водорода, алкила, замещенного алкила, арила и замещенного арила или, необязательно, каждый R6 и R8 независимо вместе с атомами, к которым они присоединены, образуют циклогетероалкильное или замещенное циклогетероалкильное кольцо;
p представляет собой целое число от 1 до 100; и
R7 выбран из водорода, алкила, замещенного алкила, ацила, замещенного ацила, алкоксикарбонила, замещенного алкоксикарбонила, арила, замещенного арила, арилалкила и замещенного арилалкила; или его соль, гидрат или сольват.
Ссылка на формулу KC-(III) призвана включать соединения формулы KC-(IIIa) и KC-(IIIb).
В формуле KC-(III) R5 может быть выбран из алкила, замещенного алкила, арилалкила, замещенного арилалкила, арила и замещенного арила. В определенных случаях R5 является (1-6С)алкилом. В других случаях R5 представляет собой (1-4С)алкил. В других случаях R5 представляет собой метил или этил. В других случаях R представляет собой метил. В определенных случаях R5 представляет собой этил.
В определенных случаях R5 представляет собой замещенный алкил. В определенных случаях R5 представляет собой алкильную группу, замещенную карбоксильной группой такой, как карбоновая кислота, эфир карбоновой кислоты или амид карбоновой кислоты. В определенных случаях R5 представляет собой -(СН2)n-СООН, -(CH2)n-СООСН3, или -(CH2)n-COOH2CH3, где n является числом от 1 до 10. В определенных случаях R1 представляет собой -(CH2)5-СООН, -(СН2)5-СООСН3 или -(СН2)5-СООСН2СН3.
В определенных случаях R5 в формуле KC-(III) представляет собой арилалкил или замещенный арилалкил. В определенных случаях R5 в формуле KC-(III) представляет собой арилалкил. В определенных случаях R5 представляет собой замещенный арилалкил. В определенных случаях R5 представляет собой арилалкильную группу, замещенную карбоксильной группой такой, как карбоновая кислота, эфир карбоновой кислоты или амид карбоновой кислоты. В определенных случаях R5 представляет собой -(CH2)q(C6H4)-COOH, -(СН2)q(С6Н4)-СООСН3 или -(СН2)q(С6Н4)-СООСН2СН3, где q представляет собой целое число от 1 до 10. В определенных случаях R5 представляет собой -СН2(С6Н4)-СООН, -СН2(С6Н4)-СООСН3 или -СН2(С6Н4)-СООСН2СН3.
В определенных случаях R5 в формуле KC-(III) представляет собой арил. В определенных случаях R5 представляет собой замещенный арил. В определенных случаях R представляет собой арильную группу, замещенную в орто-, мета- или пара-положении на карбоксильную группу такую, как карбоновая кислота, эфир карбоновой кислоты или амид карбоновой кислоты. В определенных случаях R5 является -(C6H4)-COOH, -(С6Н4)-СООСН3 или -(С6Н4)-СООСН2СН3.
В формуле KC-(III) каждый R1 может быть независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, ацила и аминоацила. В определенных случаях R1 представляет собой водород или алкил. В определенных случаях R1 является водородом. В определенных случаях R1 представляет собой алкил. В определенных случаях R1 представляет собой ацил. В определенных случаях R1 представляет собой аминоацил.
В формуле KC-(III) каждый R2 может быть независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, ацила и аминоацила. В определенных случаях R2 представляет собой водород или алкил. В определенных случаях R2 является водородом. В определенных случаях R2 представляет собой алкил. В определенных случаях R2 представляет собой ацил. В определенных случаях R представляет собой аминоацил.
В определенных случаях R1 и R2 являются водородом. В определенных случаях R1 и R2 оба на одном и том же углероде являются алкилом. В определенных случаях R1 и R2 на одном и том же углероде представляют собой метил. В определенных случаях R1 и R2 на одном и том же углероде представляют собой этил.
В определенных случаях, когда R1 и R1 являются соседними группами, они оба представляют собой алкил, и когда R2 и R2 являются соседними группами, они оба представляют собой водород. В определенных случаях, когда R1 и R1 являются соседними группами, они оба представляют собой этил, и когда R2 и R2 являются соседними группами, они оба представляют собой водород. В определенных случаях, когда R1 и R1 являются соседними группами, они оба представляют собой метил, и когда R2 и R2 являются соседними группами, они оба представляют собой водород.
В определенных случаях в цепи -[C(R1)(R2)]n- в Формуле KC-(III) замещены не все атомы углерода. В определенных случаях в цепи -[C(R1)(R2)]n- присутствует комбинация различных алкильных заместителей таких, как метил или этил.
В определенных случаях один из R1 и R2 представляет собой метил, этил или другой алкил, а R5 является алкилом. В определенных случаях, когда R1 и R1 являются соседними группами, они оба представляют собой алкил, и когда R2 и R2 являются соседними группами, они оба представляют собой водород, а R5 является алкилом. В определенных случаях, когда R1 и R1 являются соседними группами, они оба представляют собой этил, и когда R2 и R2 являются соседними группами, они оба представляют собой водород, а R5 является алкилом. В определенных случаях, когда R1 и R1 являются соседними группами, они оба представляют собой метил, и когда R2 и R2 являются соседними группами, они оба представляют собой водород, а R5 является алкилом.
В определенных случаях один из R1 и R2 представляет собой метил, этил или другой алкил, a R5 является замещенным алкилом. В определенных случаях один из R и R2 представляет собой метил, этил или другой алкил, а R5 представляет собой алкильную группу, замещенную карбоксильной группой такой, как карбоновая кислота, эфир карбоновой кислоты или амид карбоновой кислоты. В определенных случаях один из R1 и R2 является метилом, этилом или другим алкилом, а R5 представляет собой -(СН2)q(С6Н4)-СООН, -(СН2)q(С6Н4)-СООСН3, или -(СН2)q(С6Н4)-СООСН2СН3, где q представляет собой целое число от 1 до 10; В определенных случаях один из R1 и R2 представляют собой метил, этил или другой алкил, а R5 является алкильной группой, замещенной карбоксамидом.
В формуле KC-(III) R1 и R2 вместе с углеродом, к которому они присоединены, могут образовать циклоалкильную или замещенную циклоалкильную группу, или две R1 или R2 группы на соседних атомах углерода вместе с атомами углерода, к которым они присоединены, могут образовать циклоалкильную или замещенную циклоалкильную группу. В определенных случаях R1 и R2 вместе с углеродом, к которому они присоединены, могут образовать циклоалкильную группу. Таким образом, в определенных случаях R1 и R2 на одном и том же углероде образуют спироцикл. В определенных случаях R1 и R2 вместе с углеродом, к которому они присоединены, могут образовать замещенную циклоалкильную группу. В определенных случаях две R1 или R2 группы на соседних атомах углерода вместе с атомами углерода, к которым они присоединены, могут образовать циклоалкильную группу. В определенных случаях две R1 или R2 группы на соседних атомах углерода вместе с атомами углерода, к которым они присоединены, могут образовать замещенную циклоалкильную группу.
В определенных случаях R1 и R2 вместе с углеродом, к которому они присоединены, могут образовать арильную или замещенную арильную группу, или две R1 или R2 группы на соседних атомах углерода вместе с атомами углерода, к которым они присоединены, могут образовать арильную или замещенную арильную группу. В определенных случаях две R1 или R2 группы на соседних атомах углерода вместе с атомами углерода, к которым они присоединены, образуют фенильное кольцо. В определенных случаях две R1 или R2 группы на соседних атомах углерода вместе с атомами углерода, к которым они присоединены, образуют замещенное фенильное кольцо. В определенных случаях две R1 или R2 группы на соседних атомах углерода вместе с атомами углерода, к которым они присоединены, образуют нафталиновое кольцо.
В определенных случаях один из R1 и R2 представляет собой аминоацил.
В определенных случаях один или оба из R1 и R2 представляют собой аминоацил, содержащий фенилендиамин. В определенных случаях один из R1 и R2 представляет собой ; где каждый R10 независимо выбран из водорода, алкила, замещенного алкила и ацила, и R11 представляет собой алкил или замещенный алкил. В определенных случаях по меньшей мере один из R10 представляет собой ацил. В определенных случаях, по меньшей мере, один из R10 представляет собой алкил или замещенный алкил. В определенных случаях, по меньшей мере, один из R10 представляет собой водород. В определенных случаях оба R10 являются водородом.
В определенных случаях один из R1 и R2 представляет собой ; где R10 представляет собой водород, алкил, замещенный алкил или ацил. В определенных случаях R10 представляет собой ацил. В определенных случаях R10 представляет собой алкил или замещенный алкил. В определенных случаях R10 представляет собой водород.
В определенных случаях один из R1 и R2 представляет собой ; где каждый R10 независимо представляет собой водород, алкил, замещенный алкил или ацил, a b является числом от 1 до 5. В определенных случаях один из R1 и R2 представляет собой ; где R10 независимо представляет собой водород, алкил,
замещенный алкил или ацил. В определенных случаях один из R1 и R2 представляет собой ; где R10a является алкилом, а каждый R10 независимо представляет собой водород, алкил, замещенный алкил или ацил.
В определенных случаях один из R1 и R2 представляет собой R10a; где R10 независимо представляет собой водород, алкил, замещенный алкил или ацил, а b является числом от 1 до 5. В определенных случаях один из R1 и R2 представляет собой ; где R10 независимо представляет собой водород, алкил, замещенный алкил или ацил.
В определенных случаях один из R1 и R2 представляет собой аминоацильную группу такую, как -C(O)NR10aR10b, где каждый R10a и R10b независимо выбран из водорода, алкила, замещенного алкила и ацила. В определенных случаях один из R1 и R2 представляет собой аминоацильную группу такую, как -C(O)NR10aR10b, где R10a является алкилом, и R10b представляет собой замещенный алкил. В определенных случаях один из R1 и R2 представляет собой аминоацильную группу такую, как -C(O)NR10aR10b, где R10a является алкилом, и R10b представляет собой алкил, замещенный карбоновой кислотой или эфиром карбоновой кислоты. В определенных случаях один из R1 и R2 представляет собой аминоацильную группу такую, как -C(O)NR10aR10b, где R10a является метилом, и R10b представляет собой алкил, замещенный карбоновой кислотой или эфиром карбоновой кислоты.
В определенных случаях R1 или R2 может изменять скорость внутримолекулярной циклизации. R1 или R2 может повышать скорость внутримолекулярной циклизации по сравнению с соответствующей молекулой, где R1 и R2 оба являются водородом. В определенных случаях R1 или R2 содержит электроноакцепторную группу или электронодонорную группу. В определенных случаях R или R содержит электроноакцепторную группу. В определенных случаях R или R содержит электронодонорную группу.
Атомы и группы, способные функционировать как электроноакцепторные заместители, хорошо известны в области органической химии. Они включают электроотрицательные атомы и группы, содержащие электроотрицательные атомы. Такие группы действуют путем понижения основности или состояния протонирования нуклеофильного азота в бета-положении через индуктивное оттягивание электронной плотности. Такие группы также могут быть расположены в других положениях вдоль алкиленовой цепи. Примеры включают атомы галогена (например, атом фтора), ацильные группы (например, алканоильная группа, ароильная группа, карбоксильная группа, алкоксикарбонильная группа, арилоксикарбонильная группа или аминокарбонильная группа (такая как карбамоильная, алкиламинокарбонильная, диалкиламинокарбонильная или ариламинокарбонильная группа)), оксо- (=O) заместитель, нитрильную группу, нитрогруппу, группы простого эфира (например, алкокси-группа) и фенильные группы, несущие заместитель в орто-положении, пара-положении или и в орто-, и в пара-положениях, каждый заместитель является независимо выбранным из атома галогена фторалкильной группы (такой как трифторметил), нитрогруппы, цианогруппы и карбоксильной группы. Каждый из электроноакцепторных заместителей может быть независимо выбран из них.
В определенных случаях -[C(R1)R2)]n- выбирают из -CH(CH2F)CH(CH2F)-; -CH(CHF2)CH(CHF2)-; -СН(CF3)CH(CF3)-; -CH2CH(CF3)-; -CH2CH(CHF2)-; -CH2CH(CH2F)-; -CH2CH(F)CH2-; -CH2C(F2)CH2-; -CH2CH(C(O)NR20R21)-; -CH2CH(C(O)OR22)-; -СН2СН(С(O)ОН)-; -CH(CH2F)CH2CH(CH2F)-; -CH(CHF2)CH2CH(CHF2)-; СН(CF3)СН2СН(CF3)-; -СН2СН2СН(CF3)-; -CH2CH2CH(CHF2)-; -CH2CH2CH(CH2F)-; -CH2CH2CH(O)NR23R24)-; -CH2CH2CH(C(O)OR25)-; и -СН2СН2СН(С(O)ОН)-, в которых R20, R21, R22 и R23 каждый независимо представляет водород или (1-6С)алкил, и R и R каждый независимо представляет (1-6С)алкил.
В формуле KC-(III) n может быть целым числом от 2 до 4. В определенных случаях n равно 2. В других случаях n равно 3. В других случаях n равно 4.
В формуле KС-(III), R4 может быть остатком L-аминокислоты, выбранной из аланина, аргинина, аспарагина, аспарагиновой кислоты, цистеина, глицина, глутамина, глутаминовой кислоты, гистидина, изолейцина, лейцина, лизина, метионина, фенилаланина, пролина, серина, треонина, триптофана, тирозина и валина, или остатка N-ацильного производного любой из перечисленных аминокислот; или остаток пептида, состоящий из по меньшей мере двух остатков L-аминокислот, независимо выбранных из аланина, аргинина, аспарагина, аспарагиновой кислоты, цистеина, глицина, глутамина, глутаминовой кислоты, гистидина, изолейцина, лейцина, лизина, метионина, фенилаланина, пролина, серина, треонина, триптофана, тирозина и валина, или остатка их N-ацильного производного. Такой пептид может иметь длину от 2 до 100 аминокислот. Примеры N-ацильных производных включают ацетильные, бензоильные, малонильные, пиперонильные или сукцинильные производные.
В определенных случаях R4 представляет собой остаток L-аргинина или L-лизина, или остаток N-ацильного производного L-аргинина или L-лизина.
В определенных случаях в формуле KC-(III), когда p больше единицы, то R4, ближайший к азоту в -N(R3)(R4) группе, представляет собой остаток L-аргинина или L-лизина. В определенных случаях, когда p больше единицы, R4, ближайший к азоту в группе -N(R3)(R4), представляет собой остаток L-аргинина или L-лизина, а первый остаток присоединен к по меньшей мере одному дополнительному L-аминокислотному остатку, независимо выбранному из аланина, аргинина, аспарагина, аспарагиновой кислоты, цистеина, глицина, глутамина, глутаминовой кислоты, гистидина, изолейцина, лейцина, лизина, метионина, фенилаланина, пролина, серина, треонина, триптофана, тирозина и валина. Концевой остаток пептида может быть N-ацильным производным любой такой аминокислоты. В определенных случаях R4 представляет собой дипептид или его N-ацильное производное. В определенных случаях R представляет собой трипептид или его N-ацильное производное.
В формуле KC-(III), R4 представляет собой .
В формуле KC-(III) каждый R6 может быть независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, арилалкила, замещенного арилалкила, гетероалкила, замещенного гетероалкила, гетероарила, замещенного гетероарила, гетероарилалкила и замещенного гетероарилалкила или, необязательно, R6 и R7 вместе с атомами, к которым они присоединены, образуют циклогетероалкильное или замещенное циклогетероалкильное кольцо;
В определенных случаях в формуле KC-(III) R6 выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, арилалкила, замещенного арилалкила, гетероалкила, замещенного гетероалкила, гетероарила, замещенного гетероарила, гетероарилалкила и замещенного гетероарилалкила. В определенных случаях R6 выбран из водорода, алкила, замещенного алкила, арилалкила, замещенного арилалкила, гетероарилалкила и замещенного гетероарилалкила. В определенных случаях R является водородом. В определенных случаях R6 представляет собой алкил. В определенных случаях R6 представляет собой замещенный алкил. В определенных случаях R6 представляет собой арилалкил или замещенный арилалкил. В определенных случаях R6 представляет собой гетероарилалкил или замещенный гетероарилалкил.
В определенных случаях R6 представляет собой боковую цепь из аминокислоты такой, как аланин, аргинин, аспарагин, аспарагиновая кислота, цистеин, глицин, глутамин, глутаминовая кислота, гистидин, изолейцин, лейцин, лизин, метионин, фенилаланин, пролин, серин, треонин, триптофан, тирозин или валин. В определенных случаях R6 представляет собой боковую цепь из L-аминокислоты такой, как L-аланин, L-аргинин, L-аспарагин, L-аспарагиновая кислота, L-цистеин, L-глицин, L-глутамин, L-глутаминовая кислота, L-гистидин, L-изолейцин, L-лейцин, L-лизин, L-метионин, L-фенилаланин, L-пролин, L-серин, L-треонин, L-триптофан, L-тирозин или L-валин.
В определенных случаях R6 представляет собой -CH2CH2CH2NH(C=NH)NH2 или -CH2CH2CH2CH2NH2.
В формуле KC-(III) каждый W может быть независимо -NR8-, -О- или -S-. В определенных случаях W является -NR8-. В определенных случаях W является -O-. В определенных случаях W является -S-.
В формуле KC-(III)каждый R8 может быть независимо выбран из водорода, алкила, замещенного алкила, арила или замещенного арила или, необязательно, каждый R6 и R8 независимо вместе с атомами, к которым они присоединены, образуют циклогетероалкильное или замещенное циклогетероалкильное кольцо.
В определенных случаях R8 в формуле KC-(III) представляет собой водород или алкил. В определенных случаях R8 является водородом. В определенных случаях R представляет собой алкил. В определенных случаях R8 представляет собой арил. В определенных случаях R6 и R8 независимо вместе с атомами, к которым они связаны, могут образовать циклогетероалкильное или замещенное циклогетероалкильное кольцо.
В формуле KC-(III) p может быть целым числом от 1 до 100, а каждый R6 может быть выбран независимо из боковой цепи любой аминокислоты. В определенных случаях p может быть целым числом от 1 до 50. В определенных случаях p является целым числом от 1 до 90, 80, 70, 60, 50, 40, 30, 20, или 10. В определенных случаях p представляет собой примерно 100. В определенных случаях p представляет собой примерно 75. В определенных случаях p представляет собой примерно 50. В определенных случаях p представляет собой примерно 25. В определенных случаях p представляет собой примерно 20. В определенных случаях p представляет собой примерно 15. В определенных случаях p представляет собой примерно 10. В определенных случаях p представляет собой примерно 9. В определенных случаях p представляет собой примерно 8. В определенных случаях p представляет собой примерно 7. В определенных случаях p представляет собой примерно 6. В определенных случаях p представляет собой примерно 5. В определенных случаях p представляет собой примерно 4. В определенных случаях p представляет собой примерно 3. В определенных случаях p представляет собой примерно 2. В определенных случаях p представляет собой примерно 1.
В определенных случаях R6 в R4, расположенном рядом в азотом группы -N(R3)(R4), представляет собой -CH2CH2CH2NH(C=NH)NH2 или -CH2CH2CH2CH2NH2, а любой дополнительный R6 может быть боковой цепью из любой аминокислоты, независимо выбранной из аланина, аргинина, аспарагина, аспарагиновой кислоты, цистеина, глицина, глутамина, глутаминовой кислоты, гистидина, изолейцина, лейцина, лизина, метионина, фенилаланина, пролина, серина, треонина, триптофана, тирозина или валина.
В формуле KC-(III) R7 может быть выбран из водорода, алкила, замещенного алкила, ацила, замещенного ацила, алкоксикарбонила, замещенного алкоксикарбонила, арила, замещенного арила, арилалкила и замещенного арилалкила.
В определенных случаях R7 представляет собой водород, алкил, ацил или замещенный алкил. В определенных случаях R7 является водородом. В определенных случаях R7 представляет собой алкил. В определенных случаях R7 представляет собой ацил или замещенный ацил. В определенных случаях R7 представляет собой ацил. В определенных случаях R7 представляет собой замещенный ацил. В определенных случаях R7 может представлять собой ацетил, бензоил, малонил, пиперонил или сукцинил.
Формула KC-(IV)
Соединения формулы KC-(IV) являются соединениями формулы KC-(III), в которой R5 выбран из (1-6С) алкила, (1-6С) замещенного алкила, -(CH2)q(C6H4)-COOH, -(CH2)q(C6H4)-COOCH3, и -(СН2)q(С6Н4)-СООСН2СН3, где q является целым числом от 1 до 10; n равно 2 или 3; R3 является водородом; R4 является L-аминокислотой или пептидом, где пептид может содержать L-аминокислоты. В одном из аспектов композиции настоящие осуществления изобретения предоставляют соединение формулы KC-(IV):
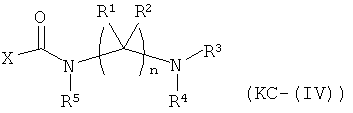
где:
Х представляет остаток кетон-содержащего опиоида, где атом водорода соответствующей енольной группы замещен ковалентной связью с --C(O)-NR5-(C(R1)(R2))n-NR3R4;
R5 выбран из (1-6С)алкила, (1-6С) замещенного алкила, -(СН2)q(C6H4)-COOH, -(CH2)q(C6H4)-COOCH3, и -(CH2)q(C6H4)-COOCH2CH3, где q представляет собой целое число от 1 до 10;
каждый R1 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, ацила и аминоацила;
каждый R2 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, ацила и аминоацила;
или R3 и R4 вместе с углеродом, к которому они присоединены, образуют циклоалкильную или замещенную циклоалкильную группу, или две R1 или R2 группы на соседних атомах углерода вместе с атомами углерода, к которым они присоединены, образуют циклоалкильную или замещенную циклоалкильную группу;
n равно 2 или 3;
R3 является водородом;
R4 представляет собой остаток L-аминокислоты, выбранной из аланина, аргинина, аспарагина, аспарагиновой кислоты, цистеина, глицина, глутамина, глутаминовой кислоты, гистидина, изолейцина, лейцина, лизина, метионина, фенилаланина, пролина, серина, треонина, триптофана, тирозина и валина, или остатка N-ацильного производного любой из перечисленных аминокислот; или остаток пептида, состоящий из по меньшей мере двух остатков L-аминокислот, независимо выбранных из аланина, аргинина, аспарагина, аспарагиновой кислоты, цистеина, глицина, глутамина, глутаминовой кислоты, гистидина, изолейцина, лейцина, лизина, метионина, фенилаланина, пролина, серина, треонина, триптофана, тирозина и валина, или остатка N-ацильного производного их;
или его соль, гидрат или сольват.
В определенных вариантах осуществления в Формуле KC-(IV) R является остатком L-аминокислоты, выбранной из аргинина или лизина.
В определенных случаях в формуле KC-(IV), когда R4 представляет собой пептид, то R4, ближайший к азоту в -N(R3)(R4) группе, представляет собой остаток L-аргинина или L-лизина. В определенных случаях R4 представляет собой дипептид или его N-ацильное производное. В определенных случаях R4 представляет собой трипептид или его N-ацильное производное.
В определенных вариантах осуществления в Формуле KC-(IM) R4 является его N-ацильным производным. В определенных случаях R является остатком его N-ацильного производного, где N-ацильное производное является замещенным таким, как, но без ограничения, малонил и сукцинил.
Формулы KC-(V)
Соединения формулы KC-(V) представляют собой соединения формулы KC-(III), в которых R4 является расщепляемым трипсином фрагментом.
В одном из аспектов композиции настоящие варианты осуществления изобретения обеспечивают соединение формулы KC-(V):
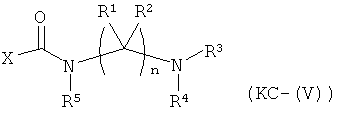
где:
X представляет остаток кетон-содержащего опиоида, где атом водорода соответствующей енольной группы замещен ковалентной связью с -C(O)-NR5-(C(R1)(R2))n-NR3R4;
R5 выбран из алкила, замещенного алкила, арилалкила, замещенного арилалкила, арила и замещенного арила;
каждый R1 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, ацила и аминоацила;
каждый R2 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, ацила и аминоацила;
или R1 и R2 вместе с углеродом, к которому они присоединены, образуют циклоалкильную, замещенную циклоалкильную, арильную или замещенную арильную группу, или две R1 или R2 группы на соседних атомах углерода вместе с атомами углерода, к которым они присоединены, образуют циклоалкильную, замещенную циклоалкильную, арильную или замещенную арильную группу;
n представляет собой целое число от 2 до 4;
R3 является водородом;
R4 представляет собой расщепляемый трипсином фрагмент;
или его соль, гидрат или сольват.
В формуле KC-(V) R4 представляет собой расщепляемый трипсином фрагмент. Расщепляемый трипсином фрагмент - это фрагмент структуры, поддающийся расщеплению трипсином. В определенных случаях расщепляемый трипсином фрагмент содержит заряженный фрагмент, который может разместиться в активном сайте трипсина и способен ориентировать пролекарство для расщепления по расщепляемой связи. Например, заряженный фрагмент может быть фрагментом основания, который существует в виде заряженного фрагмента при физиологическом pH.
В определенных вариантах осуществления в формуле KC-(V) R4 является -С(O)-CH(R6a)-NH(R7a), где R6a представляет собой аминокислотную боковую цепь или производное аминокислотной боковой цепи, что делает R4 способным к расщеплению трипсином фрагментом. Термин "производное" относится к веществу, которое было получено из другого вещества путем модификации, частичного замещения, гомологизации, усечения или изменения окислительного состояния.
Например, для образования расщепляемого трипсином фрагмента R6a может включать, но без ограничения, боковую цепь, состоящую из лизина (такого, как L-лизин), аргинина (такого, как L-аргинин), гомолизина, гомоаргинина и орнитина. Другие значения для R4 включают, но без ограничения, имитаторы аргинина, гомологи аргинина, усеченный аргинин, аргинин с различными окислительными состояниями (например, метаболиты), имитаторы лизина, гомологи лизина, усеченный лизин и лизин с различными окислительными состояниями (например, метаболиты). Примеры имитаторов аргинина и лизина включают арилгуанидины, ариламидины (замещенные бензамидины), бензиламины и (бицикло[2.2.2]октан-1-ил)метанамин и их производные.
В определенных случаях в формуле KC-(V) R6a представляет собой -CH2CH2CH2NH(C=NH)NH2 или -CH2CH2CH2CH2NH2, конфигурация атома углерода, к которому присоединен R4, соответствует конфигурации в L-аминокислоте.
В формуле KC-(V) R7a выбран из водорода, алкила, замещенного алкила, ацила, замещенного ацила, алкоксикарбонила, замещенного алкоксикарбонила, арила, замещенного арила, арилалкила и замещенного арилалкила. В определенных случаях R7a является аминокислотой или N-ацильным производным аминокислоты. В определенных случаях R7a является пептидом или N-ацильным производным такого пептида, где пептид содержит от 1 до 100 аминокислот и где каждая аминокислота может быть выбрана независимо. В определенных случаях пептид содержит от 1 до 50 аминокислот. В определенных случаях пептид содержит от 1 до 90, 80, 70, 60, 50, 40, 30, 20, или 10 аминокислот. В определенных случаях пептид содержит около 100 аминокислот. В определенных случаях пептид содержит около 75 аминокислот. В определенных случаях пептид содержит около 50 аминокислот. В определенных случаях пептид содержит около 25 аминокислот. В определенных случаях пептид содержит около 20 аминокислот. В определенных случаях пептид содержит около 15 аминокислот. В определенных случаях пептид содержит около 10 аминокислот. В определенных случаях пептид содержит около 9 аминокислот. В определенных случаях пептид содержит около 8 аминокислот. В определенных случаях пептид содержит около 7 аминокислот. В определенных случаях пептид содержит около 6 аминокислот. В определенных случаях пептид содержит около 5 аминокислот. В определенных случаях пептид содержит около 4 аминокислот. В определенных случаях пептид содержит около 3 аминокислот. В определенных случаях пептид содержит около 2 аминокислот. В определенных случаях пептид содержит около 1 аминокислоты.
Конкретные интересующие соединения и их соли, или сольваты, или стереоизомеры включают:
оксикодон 6-(N-метил-N-(2-N'-ацетиларгиниламино))этилкарбамат:
 ;
;
гидрокодон 6-(N-метил-N-(2-N'-ацетиларгиниламино))этилкарбамат:
оксикодон 6-(N-метил-N-(2-N'-малониларгиниламино))этилкарбамат:
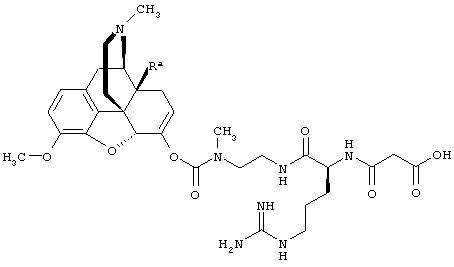 ;
;
оксикодон 6-(N-карбоксипентил-N(2-N'-ацетиларгиниламино))этилкарбамат:
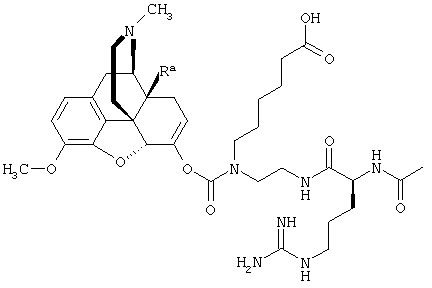 ;
;
гидрокодон 6-(N-метил-N-(2-N'-малониларгиниламино))этилкарбамат:
;
оксикодон 6-(N-метил-N-(2-N'-ацетиларгиниламино-2-(N-метил-N-карбоксиметил-ацетамидо))этилкарбамат:
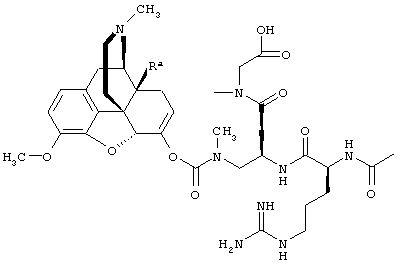 .
.
где аминокислотный остаток имеет L конфигурацию.
Варианты осуществления предоставляют фармацевтическую композицию, которая содержит соединение общей формулы от KC-(I) до KC-(II) или его фармацевтически приемлемую соль.
Варианты осуществления предоставляют фармацевтическую композицию, которая содержит соединение общей формулы от KC-(III) до KC-(V) или его фармацевтически приемлемую соль.
Варианты осуществления предоставляют фармацевтическую композицию, которая содержит раскрытое здесь соединение, отличное от соединения общей формулы от KC-(I) до KС-(II), или его фармацевтически приемлемую соль.
Общие методики синтеза для соединений формул от KC-(I) до KC-(VI)
Пример синтеза соединений формул KC-(I) и KC-(II) показан в следующей схеме. Соединения формул KC-(III)-KC-(VI) могут также быть синтезированы, используя раскрытые способы. Пример синтеза Соединения KC2O3 показан в Схеме KC-1. Значения R1, R2, R5, и n для Схемы KC-1 определены в этом документе. PG1 и PG2 обозначают защитные группы аминной функции.
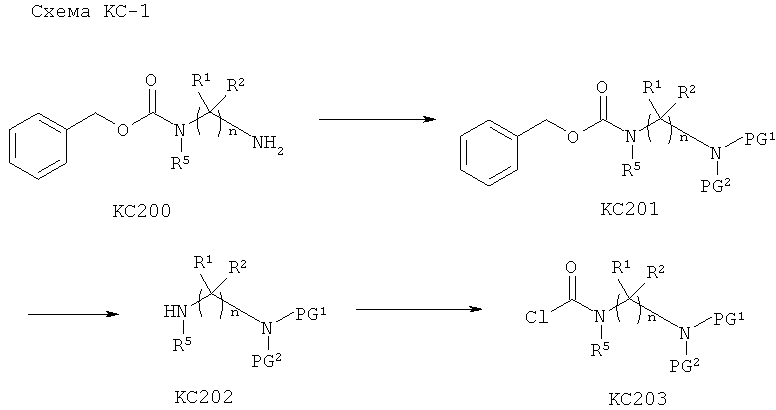
В Схеме КС-1 Соединение КС200 представляет собой коммерчески доступное исходное соединение. В качестве альтернативы Соединение КС200 может быть синтезировано путем различных синтезов с использованием коммерчески доступных исходных материалов и/или исходных материалов, полученных известными синтетическими способами.
Согласно Схеме KC-1, Соединение KC200 защищено по аминной группе с образованием Соединения KC201, где PG1 и PG2 являются защитными группами аминной функции. Защитные группы аминной функции могут быть найдены в монографии Т.W.Greene и Р.G.M. Wuts, "Protective Groups in Organic Synthesis", 4-е издание, Wiley, Нью Йорк, 2006. Типичные защитные группы аминной функции включают, но без ограничения, формильные группы; ацильные группы, например, алканоильные группы такие, как ацетил; алкоксикарбонильные группы такие, как трет-бутоксикарбонил (Boc); арилметоксикарбонильные группы такие, как бензилоксикарбонил (Cbz) и 9-флуоренилметоксикарбонил (Fmoc); арилметильные группы такие, как бензил (Bn), тритил (Tr)и 1,1-ди-(4'-метоксифенил)метил; силильные группы такие, как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS); и подобные.
В определенных вариантах осуществления PG1 и PG2 являются Вое группами. Условия формирования Вос групп на Соединении KC201 могут быть найдены в монографии Greene и Wuts. Один способ - это реакция Соединения KC200 с ди-трет-бутилдикарбонатом. Реакция может быть по выбору проведена в присутствии активирующего агента такого, как ДМАП.
Согласно Схеме KC-1, защитную карбоксибензильную группу в Соединении KC201 удаляют с образованием Соединения KC202. Условия для удаления карбоксибензильной группы могут быть найдены в монографии Greene и Wuts. Способы удаления карбоксибензильной группы включают гидрогенолиз Соединения KC201 или обработку Соединения KC201 с помощью HBr. Одним способом удаления карбоксибензильной группы является реакция Соединения KC201 с водородом и палладием.
Согласно Схеме KC-1, Соединение KC202 подвергают реакции с фосгеном с образованием Соединения KC203. Реакция с фосгеном образует ацилхлорид на аминогруппе Соединения KC202. Фосген может быть заменен другими реагентами такими, как дифосген или трифосген.
Пример синтеза Соединения KC302 показан в Схеме KC-2. Значения Ra, R1, R2, R5 и n для Схемы KC-2 определены в этом документе. PG1 и PG2 обозначают защитные группы аминной функции.
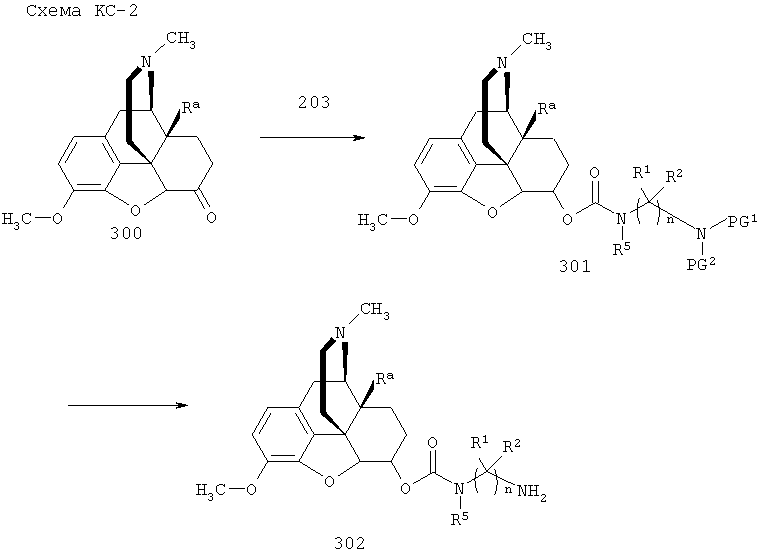
В Схеме KC-2 Соединение KC300 представляет собой коммерчески доступное исходное соединение. В качестве альтернативы Соединение KC300 может быть синтезировано путем различных синтезов с использованием коммерчески доступных исходных материалов и/или исходных материалов, полученных известными синтетическими способами.
Согласно Схеме KC-2, Соединение KC300 подвергают реакции с Соединением KC203 с образованием Соединения KC01. В этой реакции енолят Соединения KC300 реагирует с ацетилхлоридом Соединения KC203 с образованием карбамата.
Согласно Схеме KC-2, защитные группы PG1 и PG2 удаляют из Соединения KC301 с образованием Соединения KC302. Условия для удаления амино-групп могут быть найдены в монографии Greene и Wuts. Когда PG1 и PG2 представляют собой Вос группы, защитные группы могут быть удалены в кислотных условиях таких, как обработка трифторуксусной кислотой.
Пример синтеза Соединения KC402 показан в Схеме KC-3. Значения Ra, R1, R2, R5, R6, R7 и n для Схемы KC-3 определены в этом документе. PG3 представляет собой защитную группу аминной функции.
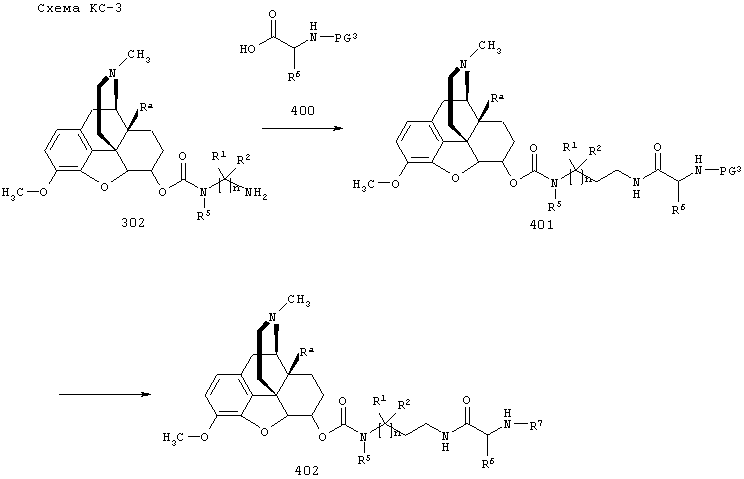
В Схеме KC-3 Соединение KC400 представляет собой коммерчески доступное исходное соединение. В качестве альтернативы Соединение KC400 может быть синтезировано путем различных синтезов с использованием коммерчески доступных исходных материалов и/или исходных материалов, полученных известными синтетическими способами.
Согласно Схеме KC-3, Соединение KC302 подвергают реакции с Соединением KC400 с образованием Соединения KC401 в реакции конденсации пептидов. Реакция конденсации пептидов типично вовлекает обычный конденсирующий агент пептидов и проходит в обычных для реакции конденсации условиях, обычно в присутствии триалкиламина такого, как этилдиизопропиламин или диизопропилэтиламин (ДИПЭА). Подходящие конденсирующие агенты для использования включают, в качестве примера, карбодиимиды такие, как этил-3-(3-диметиламино)пропилкарбодиимид (EDC), дициклогексилкарбодиимид (DCC), диизопропилкарбодиимид (DIC) и подобные, и другие хорошо известные конденсирующие агенты такие, как N,N'-карбонилдиимидозол, 2-этокси-1 -этоксикарбонил-1,2-дигидрохинолин (EEDQ), бензотриазол-1 -илокси-трис(диметиламино)фосфониум гексафторфосфат (ВОР), O-(7-азабензотриазол-1-ил)-N,N,N,N',N'-тетраметилурониум гексафторфосфат (HATU) и подобные. По выбору, в данной реакции могут быть использованы хорошо известные промоторы конденсации такие, как N-гидроксисукцинимид, 1-гидроксибензотриазол (НОВТ), 1-гидрокси-7-азабензотриазол (НОАТ), N,N-диметиламинопиридин (ДМАП) и подобные. Обычно эту реакцию конденсации проводят при комнатной температуре в диапазоне от примерно 0°С до примерно 60°С в течение от примерно 1 до примерно 72 часов в инертном разбавителе таком, как ТГФ или ДМФА. В определенных случаях Соединение KC302 реагирует с Соединением KC400 с образованием Соединения KC401 в присутствии HATU и ДИПЭА в ДМФА.
Согласно Схеме KC-3, Соединение KC401 переводят в Соединение KC402, удаляя защитную группу аминной функции и присоединяя группу R7. В определенных случаях защитной группой аминной функции является R7 и удаление защитной группы аминной функции необязательно.
Как раскрыто в данном документе, типичные защитные группы аминной функции включают, но без ограничения, формильные группы; ацильные группы, например, алканоильные группы такие, как ацетил; алкоксикарбонильные группы такие, как трет-бутоксикарбонил (Вос); арилметоксикарбонильные группы такие, как бензилоксикарбонил (Cbz) и 9-флуоренилметоксикарбонил (Fmoc); арилметильные группы такие, как бензил (Bn), тритил (Tr)и 1,1-ди-(4'-метоксифенил)метил; силильные группы такие, как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS); и подобные. В определенных вариантах осуществления PG3 является Вос группой. Когда PG3 представляет собой Вос группу, защитная группа может быть удалена в кислотных условиях таких, как обработка трифторуксусной кислотой.
В определенных случаях группу R7 присоединяют к Соединению KC401. Условия для присоединения R7 зависят от природы R7 и известны специалистам в данном уровне техники. В определенных случаях R7 представляет собой ацильную группу такую, как ацетил, бензоил, малонил, пиперонил или сукцинил.
N-ацетильные производные соединений формулы KC-(I) могут быть легко приготовлены путем ацилирования соответствующего соединения формулы KC-(I), используя подходящий ацилирующий агент, например, ангидрид, такой, как уксусный ангидрид (для приготовления N-ацетильного соединения) или галогенангидрид. Реакция легко проходит в присутствии не участвующего в реакции основания, например, третичного амина такого, как триэтиламин. Подходящие растворители включают амиды такие, как диметилформамид. Температура, при которой протекает реакция, находится в удобном интервале от 0 до 100°С, например, при комнатной температуре.
Согласно Схеме KC-3, удаление других защитных групп может быть проведено, если были использованы другие защитные группы такие, как защитные группы во фрагменте R6. Условия для удаления других защитных групп зависят от природы защитной группы и известны специалистам в данном уровне техники. Условия также могут быть найдены в монографии Greene и Wuts.
Как здесь более подробно описано, раскрытие обеспечивает способы и промежуточные соединения, пригодные для получения соединений данного раскрытия или его соли, или сольвата, или стереоизомера. Соответственно, настоящее раскрытие предоставляет способ получения соединения данного раскрытия, при этом способ включает: взаимодействие соединения формулы: R5 PG2 с соединением формулы  , где PG1 и PG2 являются защитными группами аминной функции.
, где PG1 и PG2 являются защитными группами аминной функции.
Соответственно, как здесь более подробно описано, настоящее раскрытие предоставляет способ получения соединения данного раскрытия, при этом способ включает:  взаимодействие соединения формулы:
взаимодействие соединения формулы: 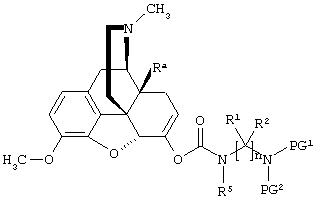 с соединением формулы
с соединением формулы 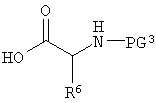 , где PG является защитной группой аминной функции.
, где PG является защитной группой аминной функции.
В одном случае вышеуказанный способ дополнительно включает стадию образования соли соединения настоящего раскрытия. Варианты осуществления направлены на другие способы, описанные здесь; а также на продукт, полученный любым из описанных здесь способов.
Ингибиторы трипсина
Фермент, способный расщеплять расщепляемый ферментом фрагмент опиоидного пролекарства с модифицированным кетоном, может быть протеазой. В определенных вариантах осуществления фермент является ферментом, локализованном в желудочно-кишечном (ЖК) тракте, т.е. желудочно-кишечным ферментом или ЖК ферментом. Фермент может быть пищеварительным ферментом таким, как фермент желудка, кишечника, поджелудочной железы или щеточной каймы, или ЖК микробной флоры такой, которая участвует в гидролизе пептидов. Примеры включают пепсин такой, как пепсин А или пепсин В; трипсин; химотрипсин; эластаза; карбоксипептидаза такая, как карбоксипептидаза А и карбоксипептидаза В; аминопептидаза такая, как аминопептидаза N или аминопептидаза А; эндопептидаза; экзопептидаза; дипептидиламинопептидаза такая, как дипептидиламинопептидаза IV; дипептидаза; трипептидаза; или энтеропептидаза. В определенных вариантах осуществления фермент представляет собой цитоплазматическую протеазу, локализованную на или в ЖК щеточной кайме. В определенных вариантах осуществления фермент представляет собой трипсин. Соответственно, в определенных осуществлениях соответствующая композиция вводится пациенту перорально.
Это раскрытие предоставляет композицию, содержащую ингибитор ЖК фермента. Такой ингибитор может ингибировать по меньшей мере один из любых ЖК ферментов, раскрытых здесь. Примером ингибитора ЖК фермента является ингибитор протеазы такой, как трипсиновый ингибитор.
Как используется в данном документе, выражение "трипсиновый ингибитор" относится к любому средству, способному к ингибированию действия трипсина на субстрат. Термин "трипсиновый ингибитор" также охватывает соли трипсинового ингибитора. Способность агента ингибировать трипсин может быть измерена, используя методы количественного анализа, известные в данном уровне техники. Например, в типичном количественном анализе одна единица соответствует количеству ингибитора, которое снижает активность трипсина на одну единицу этилового эфира бензоил-L-аргинина (BAEE-U). Одна BAEE-U представляет собой количество фермента, которое увеличивает поглощение при 253 нм на 0,001 в минуту при pH 7,6 и 25°C. Смотри, например, K.Ozawa, M. Laskowski, 1966, J. Biol. Chem. 241, 3955 и Y. Birk, 1976, Meth. Enzymol. 45, 700. В определенных случаях трипсиновый ингибитор может взаимодействовать с активным сайтом трипсина таким, как S 1 карман и S3/4 карман. S 1 карман содержит остаток аспартата, который обладает сродством к положительно заряженному фрагменту. S3/4 карман является гидрофобным карманом. Раскрытие обеспечивает специфические трипсиновые ингибиторы и неспецифические ингибиторы серин-протеазы.
Существует много трипсиновых ингибиторов, известных в данном уровне техники как специфичных к трипсину, так и таких, которые ингибируют трипсин и другие протеазы, например, химотрипсин. Раскрытие обеспечивает трипсиновые ингибиторы, которые представляют собой белки, пептиды и малые молекулы. Раскрытие обеспечивает трипсиновые ингибиторы, которые представляют собой необратимые или обратимые ингибиторы. Раскрытие обеспечивает трипсиновые ингибиторы, которые представляют собой конкурентные ингибиторы, неконкурентные ингибиторы или неконкурирующие ингибиторы. Раскрытие обеспечивает натуральные, синтетические или полусинтетические трипсиновые ингибиторы.
Трипсиновые ингибиторы могут быть получены из различных животных или растительных источников, таких как соя, кукуруза, фасоль лимская и другие бобы, тыква, подсолнечник, поджелудочных желез и легкого коровы и других животных, белка куриных и индюшиных яиц, детской смеси на основе сои и крови млекопитающих. Трипсиновые ингибиторы также могут быть микробного происхождения: например, антипаин; смотри, например, Н. Umezawa, 1976, Meth. Enzymol. 45, 678.
В одном варианте осуществления ингибитор трипсина получен из сои. Ингибиторы трипсина, полученные из сои (Glycine max), легкодоступны и считаются безопасными для потребления человеком. Они включают, но не ограничиваясь, SBTI, который ингибирует трипсин, и ингибитор Баумана-Бирка, который ингибирует трипсин и химотрипсин. Такие ингибиторы трипсина доступны, например, от Sigma-Aldrich, Сент-Луис, Миссури, США.
Трипсиновый ингибитор может представлять собой имитатор аргинина или имитатор лизина, как натуральное, так и синтетическое соединение. В определенных вариантах осуществления ингибитор трипсина представляет собой имитатор аргинина или имитатор лизина, где имитатором аргинина или имитатором лизина является синтетическое соединение. Как используется в данном документе, имитатор аргинина или лизина является соединением, которое способно связываться с Р1 карманом трипсина и/или мешать функционированию активного центра трипсина. Имитатор аргинина или лизина может быть расщепляемым или нерасщепляемым фрагментом.
Примеры трипсиновых ингибиторов, относящихся к имитаторам аргинина и/или имитаторам лизина, включают, но без ограничения, арилгуанидин, бензамидин, 3,4-дихлороизокумарин, диизопропилфторфосфат, габексата мезилат и фенилметансульфонил фторид или их замещенные варианты или аналоги. В определенных вариантах осуществления трипсиновые ингибиторы содержат группы, которые могут быть ковалентно модифицированы, как, например, хлоркетоновый фрагмент, альдегидный фрагмент или эпоксидный фрагмент. Другими примерами трипсиновых ингибиторов являются апротинин, камостат и пентамидин.
Другие примеры трипсиновых ингибиторов включают соединения формулы:
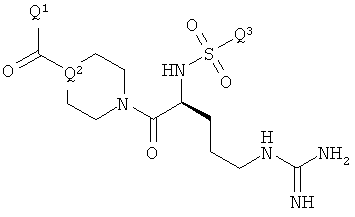
где:
Q1 выбран из -O-Q4 или -Q4-COOH, где Q4 представляет собой C1-C4алкил;
Q2 представляет собой N или СН; и
Q3 представляет собой арил или замещенный арил.
Определенные трипсиновые ингибиторы включают соединения формулы:
где:
Q5 представляет собой -С(O)-СООН или -NH-Q6-Q7-SO2-C6H5, где
Q6 представляет собой -(СН2)p-СООН;
Q7 представляет собой -(СH2)r-C6H5;
Q8 представляет собой NH;
n является числом от 0 до 2;
o равно 0 или 1;
p представляет собой целое число от 1 до 3; и
r представляет собой целое число от 1 до 3.
Определенные ингибиторы трипсина включают соединения формулы:
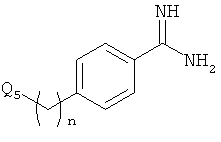 ,
,
где:
Q5 представляет собой -С(O)-СООН или -NH-Q6-Q7-SO2-C6H5, где
Q6 представляет собой -(СН2)p-СООН;
Q7 представляет собой -(СН2)r-С6Н5; и
p представляет собой целое число от одного до трех; и
r представляет собой целое число от одного до трех.
Определенные ингибиторы трипсина включают следующие:
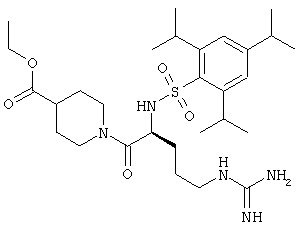
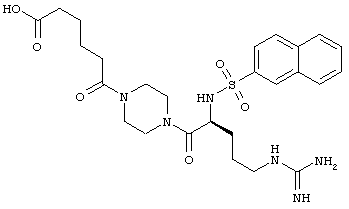
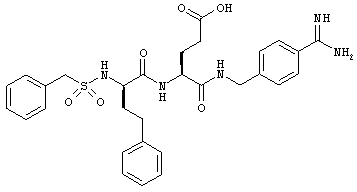
В определенных вариантах осуществления трипсиновым ингибитором является SBTI, BBSI, Соединение 101, Соединение 106, Соединение 108, Соединение 109 или Соединение 110. В определенных вариантах осуществления трипсиновый ингибитор представляет собой камостат.
В определенных вариантах осуществления трипсиновый ингибитор представляет собой соединение формулы T-I.
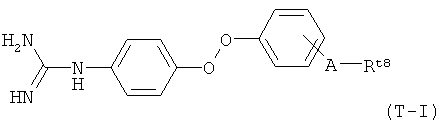
где:
А представляет собой группу следующей формулы:
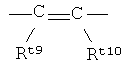 ,
,
Rt9 и Rt10 каждый независимо представляет собой атом водорода или группу
С1-4алкила,
Rt8 представляет собой группу, выбранную из следующих формул:
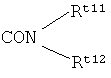 ,
, 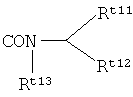 , или
, или 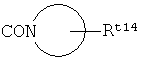
где Rt11, Rt12 и Rt13 каждый независимо представляет собой
(1) атом водорода,
(2) фенильную группу,
(3) группу C1-4алкила, замещенного фенильной группой,
(4) группу C1-10алкила,
(5) группу C1-10алкоксила,
(6) группу C2-10алкенила, имеющую от 1 до 3 двойных связей,
(7) группу C2-10алкинила, имеющую от 1 до 2 тройных связей,
(8) группу формулы: Rt15-C(O)XRt16,
где Rt15 представляет собой одинарную связь или группу C1-8алкилена,
X представляет собой атом кислорода или NH-группу, и
Rt16 представляет собой атом водорода, группу C1-4алкила, фенильную группу или группу C1-4алкила, замещенного фенильной группой, или
(9) группу C3-7циклоалкила,
структура  представляет собой моноциклическое гетероатомное кольцо с 4-7 членами, содержащее от 1 до 2 атомов азота или кислорода,
представляет собой моноциклическое гетероатомное кольцо с 4-7 членами, содержащее от 1 до 2 атомов азота или кислорода,
Rt14 представляет собой атом водорода, группу C1-4алкила, замещенного фенильной группой или группу формулы COORt17, где Rt17 представляет собой атом водорода, группу C1-4алкила или группу C1-4алкила, замещенного фенильной группой;
при условии, что Rt11, Rt12 и Rt13 одновременно не являются атомами водорода;
или нетоксичные соли, соли кислотного присоединения или их гидраты.
В определенных вариантах осуществления трипсиновый ингибитор представляет собой соединение, выбранное из следующего:
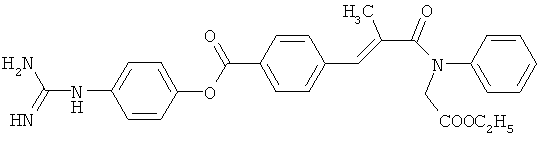 ,
,
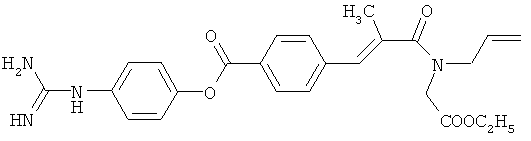 ,
,
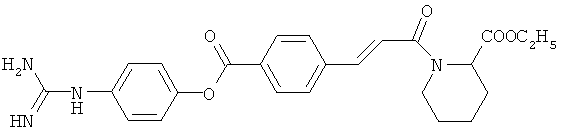 , или
, или
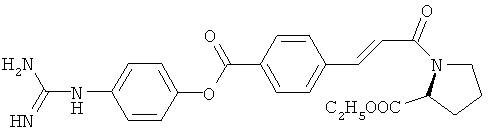 .
.
В определенных вариантах осуществления трипсиновый ингибитор представляет собой соединение формулы T-II.
где:
X является NH;
n равно 0 или 1; и
Rt1 выбран из водорода, галогена, нитро-группы, алкила, замещенного алкила, алкокси, карбоксила, алкоксикарбонила, ацила, аминоацила, гуанидина, амидино-группы, карбамида, амино-группы, замещенной амино-группы, гидроксила, циано-группы и -(СН2)m-С(O)-O-(CH2)m-C(O)-N-Rn1Rn2, где каждый m независимо равен числу от 0 до 2; и R"1 и R"2 независимо выбраны из водорода и С1-4алкила.
В определенных вариантах осуществления Rt1 в формуле T-II представляет собой гуанидино- или амидино-группы.
В определенных вариантах осуществления Rt1 в формуле T-II представляет собой -(СН2)m-С(O)-O-(CH2)m-C(O)-N-Rn1Rn2, где m равно 1 и Rn1 и Rn2 представляют собой метил.
В определенных вариантах осуществления трипсиновый ингибитор представляет собой соединение формулы T-III:
В определенных вариантах осуществления трипсиновый ингибитор представляет собой соединение формулы T-III:
где:
Х является NH;
n равно 0 или 1;
Lt1 выбран из -С(O)-O-; -О-С(О)-; -O-(СН2)m-O-; -OCH2-Art2-CH2O-; -C(O)-NRt3-; и - NRt3-C(O)-;
Rt3 выбран из водорода, C1-6алкила и замещенного C1-6алкила;
Art1и Art2 независимо представляют собой замещенную или незамещенную группу арила;
m представляет собой число от 1 до 3; и
Rt2 выбран из водорода, галогена, нитро-группы, алкила, замещенного алкила, алкокси, карбоксила, алкоксикарбонила, ацила, аминоацила, гуанидина, амидино-группы, карбамида, амино-группы, замещенной амино-группы, гидроксила, пиано-группы и -(-(СН2)m-С(O)-O-(CH2)m-C(O)-N-Rn1Rn2, где каждый m независимо равен числу от 0 до 2; и R"' и R"2 независимо выбраны из водорода и С1-4алкила.
В определенных вариантах осуществления R1 в формуле T-III представляет собой гуанидино- или амидино-группы.
В определенных вариантах осуществления Rt2 в формуле T-III представляет собой -(СН2)m-С(O)-O-(CH2)m-C(O)-N-Rn1Rn2, где m равно 1 и Rn1 и Rn2 представляют собой метил.
В определенных вариантах осуществления трипсиновый ингибитор представляет собой соединение формулы T-IV.
где:
каждый Х является NH;
каждое n независимо равно 0 или 1;
Lt1 выбран из -С(O)-O-; -О-С(О)-; -O-(СН2)m-O-; -OCH2-Art2-CH2O-; -C(O)-NRt3-; и -NRt3-C(O)-;
Rt3 выбран из водорода, C1-6алкила и замещенного C1-6алкила;
Art1 и Art2 независимо представляют собой замещенную или незамещенную группу арила; и
m представляет собой число от 1 до 3.
В определенных вариантах осуществления Art1 или Art2 в формуле T-IV представляет собой фенил.
В определенных вариантах осуществления Art1 или Art2 в формуле T-IV представляет собой нафтил.
В определенных вариантах осуществления трипсиновый ингибитор представляет собой Соединение 109.
В определенных вариантах осуществления трипсиновый ингибитор представляет собой
В определенных вариантах осуществления трипсиновый ингибитор представляет собой Соединение 110 или его бис-ариламидиновый вариант; см., например, J.D.Geratz, M.C.-F. Cheng и R.R.Tidwell (1976) J Med. Chem. 19, 634-639.
Следует иметь в виду, что фармацевтические композиции согласно вариантам осуществления могут, кроме того, включать один или более других трипсиновых ингибиторов.
Следует иметь в виду, что данное изобретение также включает ингибиторы других ферментов, вовлеченных в ассимиляцию белка, которые могут быть использованы в комбинации с пролекарством, раскрытым здесь, содержащим аминокислоту аланин, аргинин, аспарагин, аспарагиновую кислоту, цистеин, глутаминовую кислоту, глутамин, глицин, гистидин, изолейцин, лейцин, лизин, метионин, фенилаланин, пролин, серин, треонин, триптофан, тирозин или валин или их варианты. Термин "вариант аминокислоты" относится к аминокислоте, которая была модифицирована из встречающейся в природе аминокислоты, но по-прежнему содержит функциональность схожую с таковой натуральной кислоты.
Комбинации пролекарства и трипсинового ингибитора
Как обсуждалось выше, данное раскрытие предоставляет фармацевтические композиции, которые содержат трипсиновый ингибитор и опиоидное пролекарство с модифицированным кетоном, которое содержит расщепляемый трипсином фрагмент, который при расщеплении содействует высвобождению кетон-содержащего опиоида. Примеры композиций, содержащих опиоидное пролекарство с модифицированным кетоном и трипсиновый ингибитор, описаны ниже.
Комбинации Формул KC-(I)-KC-(II) и трипсинового ингибитора
Варианты осуществления предоставляют фармацевтическую композицию, которая содержит трипсиновый ингибитор и соединение общей формулы от KC-(I) до KC-(II) или его фармацевтически приемлемую соль. Варианты осуществления предоставляют фармацевтическую композицию, которая содержит соединение формулы от T-I до Т-VI и соединение общей формулы от KC-(I) до KC-(II) или его фармацевтически приемлемую соль. Варианты осуществления предоставляют фармацевтическую композицию, которая содержит Соединение 109 и соединение общей формулы от KC-(I) до KC-(II) или его фармацевтически приемлемую соль.
Определенные варианты осуществления обеспечивают комбинацию соединения Формулы KC-(I) и трипсинового ингибитора, в которой кетон-содержащий опиоид формулы KC-(I) и трипсиновый ингибитор показаны в таблице ниже. Определенные варианты осуществления обеспечивают комбинацию соединения Формулы KC-(II) и трипсинового ингибитора, в которой кетон-содержащий опиоид формулы KC-(II) и трипсиновый ингибитор также показаны в таблице ниже.
Комбинации Формул KC-(III)-KC-(V) и трипсинового ингибитора
Варианты осуществления предоставляют фармацевтическую композицию, которая содержит трипсиновый ингибитор и соединение общей Формулы от KC-(III) до KC-(V) или его фармацевтически приемлемую соль. Варианты осуществления предоставляют фармацевтическую композицию, которая содержит соединение формулы от T-I до Т-VI и соединение общей Формулы от KC-(III) до KC-(V) или его фармацевтически приемлемую соль. Варианты осуществления предоставляют фармацевтическую композицию, которая содержит Соединение 109 и соединение общей Формулы от KC-(III) до KC-(V) или его фармацевтически приемлемую соль.
Варианты осуществления предоставляют фармацевтическую композицию, которая содержит трипсиновый ингибитор и раскрытое здесь соединение, отличное от соединения общей Формулы от KC-(I) до KC-(II), или его фармацевтически приемлемую соль.
Определенные варианты осуществления обеспечивают комбинацию соединения Формулы KC-(III) и трипсинового ингибитора, в которой кетон-содержащий опиоид формулы KC-(III) и трипсиновый ингибитор показаны в таблице ниже. Определенные варианты осуществления обеспечивают комбинацию соединения Формулы KC-(IV) и трипсинового ингибитора, в которой кетон-содержащий опиоид Формулы KC-(IV) и трипсиновый ингибитор показаны в таблице ниже. Определенные варианты осуществления обеспечивают комбинацию соединения Формулы KC-(V) и трипсинового ингибитора, в которой кетон-содержащий опиоид формулы KC-(V) и трипсиновый ингибитор показаны в таблице ниже.
Комбинации Соединения KC-2 и трипсинового ингибитора
Определенные варианты осуществления обеспечивают комбинацию Соединения KC-2 и трипсинового ингибитора, в которой трипсиновый ингибитор показан в следующей таблице.
Комбинации опиоидного пролекарства с модифицированным кетоном и других лекарственных средств
Раскрытие предоставляет опиоидное пролекарство с модифицированным кетоном и дополнительные пролекарства или лекарственные средства, включенные в фармацевтическую композицию. Такое пролекарство или лекарственное средство обеспечивают дополнительное обезболивание или другие преимущества. Примеры включают опиоиды, ацетаминофен, нестероидные противовоспалительные средства (НПВС) и другие анальгетики. В одном варианте осуществления пролекарство опиоидного агониста или лекарственного средства могут быть объединены с пролекарством опиоидного антагониста или лекарственным средством. Другие примеры включают лекарственные средства или пролекарства, которые оказывают благоприятное воздействие, отличное от болеутоляющего или дополнительное к нему. Варианты осуществления предоставляют фармацевтическую композицию, которая содержит опиоидное пролекарство с модифицированным кетоном и ацетаминофен и необязательно содержит трипсиновый ингибитор. Также включены его фармацевтически приемлемые соли.
В определенных вариантах осуществления опиоидное пролекарство с модифицированным кетоном представляет собой соединение общей Формулы KC-(I)-KC-(V).
В определенных вариантах осуществления трипсиновый ингибитор выбирают из SBTI, BBSI, Соединения 101, Соединения 106, Соединения 108, Соединения 109 и Соединения 110. В определенных вариантах осуществления трипсиновый ингибитор представляет собой камостат.
В определенных вариантах осуществления фармацевтическая композиция может содержать опиоидное пролекарство с модифицированным кетоном, неопиоидное лекарственное средство и по меньшей мере один опиоид или опиоидное пролекарство. Фармацевтические композиции и способы их использования Фармацевтическая композиция согласно вариантам осуществления может дополнительно содержать фармацевтически приемлемый носитель. Композиция составлена в удобной форме, подходящей для перорального (включая буккальное и подъязычное) введения, например, в виде таблетки, капсулы, тонкой пленки, порошка, суспензии, раствора, сиропа, дисперсии или эмульсии. Композиция может содержать компоненты, общепринятые в фармацевтических препаратах, например, один или более носителей, связующих веществ, смазочных средств, наполнителей (например, для обеспечения характеристик контролируемого высвобождения), модификаторов pH, подсластителей, объемообразующих средств, красящих веществ или других активных средств.
"Пациент" включает людей, а также других млекопитающих таких, как домашний скот, животные в зоопарке и животные-компаньоны такие, как кошка, собака или лошадь.
В другом аспекте варианты осуществления предоставляют фармацевтическую композицию, как описано в данном документе выше, для применения в лечении боли. Фармацевтическая композиция согласно вариантам осуществления применима, например, в лечении пациента, страдающего от или с риском страдания от боли. Соответственно, настоящее раскрытие предоставляет способы лечения или предупреждения боли в субъектах, способы, включающие введение субъекту раскрываемой композиции. Настоящее раскрытие предоставляет раскрываемую композицию для использования в лечении, или предупреждении, или как лекарственный препарат. Настоящее раскрытие также предоставляет использование раскрываемой композиции в производстве лекарственных препаратов, особенно в производстве лекарственных препаратов для лечения или предупреждения боли.
Композиции данного раскрытия могут быть использованы в лечении или предупреждении боли, включая, но без ограничения, острую боль, хроническую боль, невропатическую боль, острую травматическую боль, боль в суставах, боль при остеоартрите, ревматоидно-артритную боль, мышечно-скелетную боль, боль после стоматологических операций, зубную боль, миофасциальную боль, боль при раке, висцеральную боль, боль при диабете, мышечную боль, неврологическую боль после герпеса, хроническую тазовую боль, боль при эндометриозе, боль при воспалении органов таза и боль при родах. Острая боль включает, но без ограничения, острую травматическую боль или послеоперационную боль. Хроническая боль включает, но без ограничения, невропатическую боль, артритную боль, боль при остеоартрите, ревматоидно-артритную боль, мышечно-скелетную боль, зубную боль, миофасциальную боль, боль при раке, боль при диабете, висцеральную боль, мышечную боль, неврологическую боль после герпеса, хроническую тазовую боль, боль при эндометриозе, боль при воспалении органов таза и боль в спине.
Данное раскрытие представляет применение опиоидного пролекарства с модифицированным кетоном и трипсинового ингибитора в лечении боли. Данное раскрытие представляет применение опиоидного пролекарства с модифицированным кетоном и трипсинового ингибитора в предупреждении боли.
Данное раскрытие представляет применение опиоидного пролекарства с модифицированным кетоном и трипсинового ингибитора в изготовлении лекарственного препарата для лечения боли. Данное раскрытие представляет применение опиоидного пролекарства с модифицированным кетоном и трипсинового ингибитора в изготовлении лекарственного препарата для предупреждения боли.
В другом аспекте варианты осуществления предоставляют способ лечения боли у пациента, нуждающегося в таком лечении, который включает введение эффективного количества фармацевтической композиции, как описано в данном документе выше. В другом аспекте варианты осуществления представляют способ предупреждения боли у пациента, нуждающегося в таком лечении, который включает введение эффективного количества фармацевтической композиции, как описано в данном документе выше.
Эффективное количество раскрываемой здесь композиции, подлежащей введению пациенту (т.е. количество, достаточное для обеспечения уровня кетон-содержащего опиоида в крови, достаточного для эффективного лечения или профилактики боли), будет зависеть от биодоступности конкретной композиции, чувствительности конкретной композиции к ферментной активации в кишечнике, количества и активности трипсинового ингибитора, присутствующего в композиции, а также других факторов, таких как вид, возраст, вес, пол и состояние пациента, путь введения и назначение, прописанное врачом. Обычно доза опиоидного пролекарства с модифицированным кетоном может быть такой, чтобы находиться в диапазоне от 0,01 до 20 миллиграмм на килограмм (мг/кг) веса тела. Например, пролекарство, содержащее остаток оксикодона или гидрокодона может вводиться в дозе, эквивалентной введению свободного оксикодона или гидрокодона в диапазоне от 0,02 до 0,5 мг/кг веса тела, или от 0,01 мг/кг до 10 мг/кг веса тела, или от 0,01 до 2 мг/кг веса тела. В одном варианте осуществления, где композиция содержит пролекарство оксикодона или гидрокодона, композиция может быть введена в такой дозе, чтобы достигаемый в крови уровень оксикодона или гидрокодона находился в пределах от 0,5 нг/мл до 10 нг/мл.
Эффективное количество трипсинового ингибитора, подлежащего введению пациенту (для уменьшения высвобождения кетон-содержащего опиоида, поскольку назначение только соединения раскрытого в настоящем документе привело бы к передозировке кетон-содержащего опиоида) будет зависеть от эффективной дозы конкретного пролекарства и действенности конкретного ингибитора, а также других факторов, таких как вид, возраст, вес, пол и состояние пациента, путь введения и назначение, прописанное врачом. В основном, доза ингибитора может быть в диапазоне от 0,05 до 50 мг на мг пролекарства, раскрытого здесь. В определенном варианте осуществления доза ингибитора может быть в диапазоне от 0,001 до 50 мг на мг пролекарства, раскрытого здесь. В одном варианте осуществления, доза ингибитора может быть в диапазоне от 0,01 наномолей до 100 микромолей на микромоль пролекарства, раскрытого здесь.
Единицы дозы пролекарства и ингибитора, имеющие желаемую фармакокинетическую кривую
Настоящее раскрытие предоставляет единицы дозы пролекарства и ингибитора, которые могут обеспечить желаемую фармакокинетическую (ФК) кривую. Единицы дозы могут обеспечить модифицированную по сравнению с ФК кривой сравнения ФК кривую согласно настоящему раскрытию. Следует понимать, что модифицированная ФК кривая может обеспечить модифицированную фармакодинамическую (ФД) кривую. Введение внутрь множества таких единиц дозы также может обеспечить желаемую ФК кривую.
Если специально не указано другое, "единица дозы", как употребляется в данном документе, относится к комбинации расщепляемого ЖК ферментами пролекарства (например, расщепляемого трипсином пролекарства) и ингибитора ЖК фермента (например, трипсинового ингибитора). "Одна единица дозы" - это одна единица комбинации расщепляемого ЖК ферментами пролекарства (например, расщепляемого трипсином пролекарства) и ингибитора ЖК фермента (например, трипсинового ингибитора), где одна единица дозы предоставляет терапевтически эффективное количество лекарства (т.е. достаточное количество лекарства, чтобы вызвать терапевтический эффект, например, в пределах соответствующего терапевтического окна или терапевтического диапазона). "Многократные единицы дозы", или "множества единиц дозы", или "множество единиц дозы" относится по меньшей мере к двум отдельным единицам дозы.
Как используется здесь, "ФК кривая" относится к кривой концентрации лекарственного препарата в крови или в плазме. Такая кривая может быть функцией концентрации лекарства от времени (т.е. "ФК кривая концентрация-время") или функцией концентрации лекарственного препарата от количества введенных доз (т.е. "ФК кривая концентрация-доза"). ФК кривая характеризуется ФК параметрами.
Как используется здесь, "ФК параметр" относится к показателю концентрации лекарственного средства в крови или в плазме такому, как: 1) "Cmax лекарственного препарата" - максимальная концентрация лекарственного средства, достигнутая в крови или плазме; 2) "Tmax лекарственного средства" - время, необходимое с момента принятия внутрь для достижения Cmax; и 3) "действие лекарственного средства" - общая концентрация лекарственного средства в крови или плазме в течение выбранного периода времени, которое может быть измерено в течение выбранного промежутка времени (t), используя площадь под кривой (ППК) в ходе высвобождения лекарственного средства. Изменение одного или более ФК параметров обусловливает изменение ФК кривой.
В целях описания особенностей единиц дозы в настоящем раскрытии "значения ФК параметра", которые определяют ФК кривую, включают Cmax лекарственного средства (например, Cmax кетон-содержащего опиоида), общее действие лекарственного средства (например, площадь под кривой) (например, действие кетон-содержащего опиоида) и 1/(Tmax лекарственного средства) (таким образом, что уменьшенный 1/Tmax говорит о задержке Tmax по отношению к Tmax сравнения) (например, 1/Tmax кетон-содержащего опиоида). Так уменьшение значения ФК параметра относительно значения ФК параметра сравнения может означать, например, снижение в Cmax лекарственного средства, снижение действия лекарственного средства и/или задержку Tmax.
Единицы дозы настоящего раскрытия могут быть приспособлены для обеспечения модифицированных ФК кривых, например, ФК кривой, которая отличается от таковой, полученной введением данной дозы пролекарства в отсутствие ингибитора (т.е. без ингибитора). Например, единицы дозы могут обеспечить по меньшей мере одно из сниженного Cmax лекарственного средства, замедленного Tmax и/или пониженное действие лекарственного средства по сравнению с введением внутрь дозы пролекарства в том же количестве, но в отсутствие ингибитора. Такая модификация происходит благодаря включению ингибитора в единицу дозы.
Как используется здесь, "фармакодинамическая (ФД) кривая" относится к кривой эффективности лекарственного средства в пациенте (или субъекте, или потребителе), которая может быть охарактеризована ФД параметрами. "ФД параметры" включают "Emax лекарственного средства" (максимальная эффективность лекарственного средства), "ЕС50 лекарственного средства" (концентрация лекарственного средства при 50% Emax) и побочное действие.
Фигура 1 представляет схематическую иллюстрацию примера влияния увеличения концентраций ингибитора на ФК параметр Cmax лекарственного средства при фиксированной дозе пролекарства. При низких концентрациях ингибитора может не наблюдаться заметного влияния на высвобождение лекарственного средства, о чем свидетельствует плоский участок графика Cmax лекарственного средства (ось Y) относительно концентрации ингибитора (ось X). По мере увеличения концентрации ингибитора достигается концентрация, при которой высвобождение лекарственного средства из пролекарства истощается, вызывая снижение или подавление Cmax лекарственного средства. Таким образом, влияние ингибитора на ФК параметр пролекарства в единице дозы данного раскрытия может меняться от неопределяемого, через среднее и до полного ингибирования (т.е. нет определяемого высвобождения лекарственного средства).
Единица дозы может быть приспособлена для обеспечения желаемой ФК кривой (например, ФК кривой концентрация-время) после приема внутрь одной дозы. Единица дозы может быть приспособлена для обеспечения желаемой ФК кривой (например, ФК кривой концентрация-время) после введения многократных единиц дозы (например, по меньшей мере 2, по меньшей мере 3, по меньшей мере 4 или более единиц дозы).
Единицы дозы, обеспечивающие модифицированную ФК кривую
Комбинация пролекарства и ингибитора в единице дозы может обеспечить желаемую (или "заранее выбранную") ФК кривую (например, ФК кривую концентрация-время) после принятия внутрь одной дозы. ФК кривая такой единицы дозы может быть охарактеризована одной или более заранее выбранной Cmax лекарственного средства, заранее выбранным Tmax или заранее выбранным действием лекарственного средства. ФК кривая единицы дозы может быть модифицирована относительно ФК кривой, полученной от эквивалентной дозы пролекарства в отсутствие ингибитора (т.е. дозы, такой же, как единица дозы, за исключением того, что в ней нет ингибитора).
Модифицированная ФК кривая может иметь сниженное значение ФК параметра по отношению к значению ФК параметра сравнения (т.е. значение ФК параметра ФК кривой после приема внутрь дозы пролекарства, которая эквивалентна единице дозы за исключением отсутствия ингибитора). Например, единица дозы может обеспечить сниженную Cmax лекарственного средства, сниженное действие лекарственного средства и/или замедленное Tmax.
Фигура 2 представляет схематические графики, показывающие примеры модифицированных ФК кривых концентрация-время одной единицы дозы. Секция А представляет собой диаграмму концентрации лекарственного средства в крови или плазме (ось Y) за период времени (ось X) после приема внутрь пролекарства в отсутствие или в присутствии ингибитора. Сплошная верхняя линия Секции А представляет пример концентрации лекарственного средства после приема внутрь пролекарства без ингибитора. Пунктирная нижняя линия Секции А представляет концентрацию лекарственного средства после приема внутрь такой же дозы пролекарства с ингибитором. Прием внутрь ингибитора вместе в пролекарством обеспечивает сниженную Cmax лекарственного средства по сравнению с Cmax лекарственного средства, которая является результатом приема внутрь такого же количества пролекарства в отсутствие ингибитора. Секция А также показывает, что полное действие лекарственного средства после приема внутрь пролекарства с ингибитором также снижается по отношению к приему внутрь такого же количества пролекарства без ингибитора.
Секция В Фигуры 2 приводит другой пример единицы дозы, имеющей модифицированную ФК кривую концентрация-время. Как и в Секции А, сплошная верхняя линия представляет собой концентрацию лекарственного средства от времени в крови или плазме после приема внутрь пролекарства без ингибитора, в то время как пунктирная нижняя линия представляет собой концентрацию лекарственного средства после приема такого же количества пролекарства с ингибитором. В этом примере единица дозы обеспечивает ФК кривую, имеющую сниженную Стах лекарственного средства, сниженное действие лекарственного средства, замедленное Tmax лекарственного средства (т.е. сниженное (1/Tmax лекарственного средства)) по отношению к введению внутрь такой же дозы пролекарства без ингибитора.
Секция С Фигуры 2 приводит другой пример единицы дозы, имеющей модифицированную ФК кривую концентрация-время. Как и в Секции А, сплошная линия представляет собой концентрацию лекарственного средства от времени в крови или плазме после приема внутрь пролекарства без ингибитора, в то время как пунктирная линия представляет собой концентрацию лекарственного средства после приема такого же количества пролекарства с ингибитором. В этом примере единица дозы обеспечивает ФК кривую, имеющую замедленное Tmax лекарственного средства (т.е. сниженное (1/Tmax лекарственного средства)) по отношению к введению внутрь такой же дозы пролекарства без ингибитора.
Единицы дозы, которые обеспечивают модифицированную ФК кривую (т.е. сниженную Cmax лекарственного средства и/или замедленное Tmax лекарственного средства по сравнению с ФК кривой лекарственного средства или ФК кривой пролекарства без ингибитора), находят применение при индивидуальном подборе дозы согласно потребностям пациента (например, путем выбора определенной единицы дозы и/или выбора режима приема), в уменьшении побочного действия и/или для улучшения соблюдения пациентом предписанного лечения (по сравнению с побочным действием или соблюдением пациентом предписанного лечения с использованием лекарственного средства или пролекарства без ингибитора). Как используется в данном документе, "соблюдение пациентом предписанного лечения" относится к тому, соблюдает ли пациент предписания врача (например, терапевта), включая прием дозы, которая значительно не отличается в обе стороны от предписанной. Такие единицы дозы также снижают риск неправильного использования, злоупотребления или передозировки пациентом по сравнению с аналогичным риском(ами), связанным с лекарственным средством или пролекарством без ингибитора. Например, единицы дозы со сниженным Cmax лекарственного средства обеспечивают меньший эффект на прием, чем доза такого же количества лекарственного средства и/или такого же количества пролекарства без ингибитора.
Единицы дозы, обеспечивающие модифицированные ФК кривые при приеме многократных единиц дозы
Единица дозы данного изобретения может быть приспособлена для обеспечения желаемой ФК кривой (например, ФК кривой концентрация-время или ФК кривой концентрация-доза) после введения многократной единицы дозы (например, по меньшей мере 2, по меньшей мере 3, по меньшей мере 4 или более единиц дозы). ФК кривая концентрация-доза относится к отношению между выбранным ФК параметром и числом принятых внутрь однократных единиц дозы. Такая кривая может быть пропорциональна дозе, линейной (линейная ФК кривая) или нелинейной (нелинейная ФК кривая). Модифицированная ФК кривая концентрация-доза может быть обеспечена путем подбора относительных количеств пролекарства и ингибитора, содержащихся в однократной единице дозы и/или путем использования другого пролекарства и/или ингибитора.
На Фигуре 3 представлены диаграммы примеров ФК кривых концентрация-доза (проиллюстрированных Cmax лекарственного средства, ось Y), которые могут быть результатом принятия многократной единицы дозы (ось X) данного раскрытия. Каждую кривую можно сравнить с ФК кривой концентрация-доза, полученной путем увеличения доз только одного лекарственного средства, где количество лекарственного средства в крови или плазме от одной дозы представляет терапевтически эффективное количество, эквивалентное количеству лекарственного средства, высвобожденного в кровь или плазму одной единицей дозы данного раскрытия. Такая ФК кривая "только одного лекарственного средства" обычно пропорциональна дозе, при этом она имеет положительный уклон при угле сорок пять градусов. Также следует понимать, что ФК кривую концентрация-доза, полученную при приеме многократной единицы дозы данного изобретения, можно сравнить с другими эталонами, как, например, ФК кривой концентрация-доза, полученной при приеме увеличенного числа доз пролекарства без ингибитора, где количество лекарственного средства, высвобожденного в кровь или плазму одной дозой пролекарства в отсутствие ингибитора представляет собой терапевтически эффективное количество, эквивалентное количеству лекарственного средства, выделенного в кровь или плазму одной единицей дозы данного изобретения.
Как проиллюстрировано отношением между концентрацией пролекарства и ингибитора на Фигуре 2, единица дозы может содержать ингибитор в количестве, влияние которого на высвобождение лекарственного средства после приема внутрь невозможно обнаружить. Прием такой многократной единицы дозы может произвести ФК кривую концентрация-доза, где соотношение между числом принятых единиц доз и значением ФК параметра является линейным и имеет положительный уклон, сходный, например, с ФК кривой, пропорциональной дозе увеличенного количества только одного пролекарства. Секция А Фигуры 3 представляет такую кривую. Единицы дозы, которые обеспечивают ФК кривую концентрация-доза с таким необнаруживаемым по сравнению с кривой одного только пролекарства изменением в Cmax лекарственного средства in vivo, могут найти применение в пресечении ферментной переработки пролекарства из единицы дозы, которая содержит достаточно ингибитора для снижения или предотвращения расщепления in vitro расщепляемого ферментом пролекарства соответствующим ферментом.
Секция В Фигуры 3 представляет ФК кривую концентрация-доза, в которой соотношение между числом принятых внутрь единиц дозы и значением ФК параметра является линейным и имеет положительный уклон, где кривая демонстрирует меньший уклон по сравнению с Секцией А. Такая единица дозы обеспечивает кривую, имеющую сниженное значение ФК параметра (например, Cmax лекарственного средства) по отношению к значению ФК параметра сравнения, демонстрирующего пропорциональность дозе.
ФК кривая концентрация-доза после приема многократной единицы дозы может быть нелинейной. Секция С Фигуры 3 представляет пример нелинейной двухфазной ФК кривой концентрация-доза. В этом примере двухфазная ФК кривая концентрация-доза содержит первую фазу, в которой ФК кривая концентрация-доза имеет положительный подъем, а затем вторую фазу, в которой зависимость между числом принятых внутрь единиц дозы и значением ФК параметра (например, Cmax лекарственного средства) остается практически неизменяемой (в основном линейной с нулевым уклоном). Для такой единицы дозы, например, Cmax лекарственного средства может быть увеличена для выбранного числа единиц дозы (например, 2, 3, или 4 единиц дозы). Однако прием дополнительных единиц дозы не приводит к существенному повышению Cmax лекарственного средства.
Секция D Фигуры 3 представляет другой пример нелинейной двухфазной ФК кривой концентрация-доза. В этом примере двухфазная ФК кривая концентрация-доза характеризуется первой фазой, в которой ФК кривая концентрация-доза имеет положительный подъем, и второй фазой, в которой зависимость между числом принятых внутрь единиц дозы и значением ФК параметра (например, Cmax лекарственного средства) является убывающей. Единицы дозы, которые обеспечивают такую ФК кривую концентрация-доза, обеспечивают увеличение Cmax лекарственного средства для выбранного числа принятых единиц дозы (например, 2, 3 или 4 единиц дозы). Однако прием последующих дополнительных единиц дозы не приводит к существенному повышению Cmax лекарственного средства и, напротив, обеспечивает понижение Cmax лекарственного средства.
Секция Е Фигуры 3 представляет ФК кривую концентрация-доза, в которой зависимость между числом принятых внутрь единиц дозы и ФК параметром (например, Cmax лекарственного средства) является линейной с нулевым уклоном. Такие единицы дозы не производят значительного увеличения или снижения Cmax лекарственного средства при приеме многократных единиц дозы.
Секция F Фигуры 3 представляет ФК кривую концентрация-доза, в которой зависимость между числом принятых внутрь единиц дозы и значением ФК параметра (например, Cmax лекарственного средства) является линейной с отрицательным уклоном. Так, Cmax лекарственного средства уменьшается при увеличении принятых внутрь единиц дозы.
Единицы дозы, которые обеспечивают ФК кривую концентрация-доза в случаях приема внутрь многократной единицы дозы, находят применение при индивидуальном подборе режима дозирования для обеспечения терапевтического уровня высвобождаемого лекарственного средства при одновременном снижении риска передозировки, неправильного применения и злоупотребления. Такое снижение риска может быть сравнено с эталоном, например, с приемом только одного лекарственного средства или только одного пролекарства. В одном варианте осуществления риск снижен по сравнению с приемом лекарственного средства или пролекарства, который обеспечивает пропорциональную ФК кривую концентрация-доза. Единица дозы, которая обеспечивает ФК кривую концентрация-доза, может снизить риск передозировки пациентом из-за небрежного приема единиц дозы сверх предписанной дозировки. Такая единица дозы может снизить риск неправильного использования пациентом (например, в результате самолечения). Такая единица дозы может препятствовать злоупотреблению в результате преднамеренного приема многократных единиц дозы. Например, единица дозы, обеспечивающая двухфазную ФК кривую концентрация-доза, может позволить увеличить высвобождение лекарственного средства для ограниченного числа принятых внутрь единиц дозы, после чего увеличение высвобождения лекарственного средства с приемом дополнительных единиц дозы не наступает. В другом примере единица дозы, обеспечивающая ФК кривую концентрация-доза с нулевым уклоном, может позволить сохранять одинаковую кривую высвобождения лекарственного средства независимо от числа принятых единиц дозы.
Прием внутрь многократной единицы дозы может привести к корректировке значения ФК параметра относительно приема многократного количества одной и той же дозы (либо только одного лекарственного средства, либо пролекарства) в отсутствие ингибитора таким образом, что, например, прием выбранного числа (например, 2, 3, 4 или более) одиночных единиц дозы приводит к уменьшению значения ФК параметра по сравнению с приемом такого же числа доз в отсутствие ингибитора.
Фармацевтические композиции включают таковые, имеющие ингибитор, для обеспечения защиты лечащего вещества от разложения в ЖК тракте. Ингибитор может быть совмещен с лекарственным средством (т.е. не пролекарством) для обеспечения защиты лекарственного средства от разложения в ЖК системе. В этом примере композиция ингибитора и лекарственного средства обеспечивает модифицированную ФК кривую путем увеличения ФК параметра. Ингибитор может также быть совмещен с пролекарством, склонным к разложению ЖК ферментами, и имеет зону действия вне ЖК тракта. В этой композиции ингибитор защищает принятое внутрь пролекарство в ЖК тракте до его доставки за пределы ЖК тракта и расщепления в предназначенной зоне действия.
Способы, используемые для определения относительных количеств пролекарства и ингибитора в единице дозы
Единицы дозы, обеспечивающие желаемую ФК кривую такую, как ФК желаемая кривая концентрация-время и/или желаемая ФК кривая концентрация-доза, могут быть получены путем объединения пролекарства и ингибитора в единице дозы в относительных количествах, эффективных для обеспечения высвобождения лекарственного средства, которое обеспечивает желаемую ФК кривую лекарственного средства после приема внутрь пациентом.
Пролекарство может быть выбрано как пригодное для использования в единице дозы путем определения способности пролекарства высвобождать лекарственное средство под действием ЖК фермента. Это может быть выполнено in vitro, in vivo или ex vivo.
Количественный анализ in vitro может быть проведен путем объединения пролекарства с ЖК ферментом (например, трипсином) в реакционной смеси. ЖК фермент может быть обеспечен в реакционной смеси в количестве, достаточном для каталитического расщепления пролекарства. Количественный анализ проводят в подходящих условиях и необязательно в условиях, которые имитируют таковые в ЖК тракте субъекта, например, человека. "Конверсия пролекарства" относится к высвобождению лекарственного средства из пролекарства. Конверсия пролекарства может быть оценена путем определения уровня продукта конверсии пролекарства (например, высвобожденного лекарственного средства) и/или путем определения уровня пролекарства, которое поддерживается в присутствии ЖК фермента. Конверсия пролекарства может также быть определена путем определения скорости образования продукта конверсии пролекарства или скорости, с которой расходуется пролекарство. Увеличение содержания высвобожденного лекарственного средства или уменьшение содержания пролекарства свидетельствуют о том, что произошла конверсия пролекарства. Пролекарства, демонстрирующие приемлемый уровень конверсии пролекарства в присутствии ЖК фермента в течение приемлемого периода времени, являются пригодными для использования в единице дозы в сочетании с ингибитором ЖК фермента, который способен регулировать конверсию пролекарства.
Количественный анализ in vivo оценивает пригодность пролекарства для использования в единице дозы путем введения пролекарства животному (например, человеку или животному, например, крысе, собаке, свинье и т.д.). Такое введение может быть энтеральным (например, пероральным введением). Конверсия пролекарства может быть определена путем, например, определения продукта конверсии пролекарства (например, высвобождение лекарственного средства или метаболита высвобождаемого лекарственного средства) или определения пролекарства в крови или плазме животного в желаемый момент(ы) времени после введения.
Количественный анализ ех vivo такой, как анализ в петле кишки или обратный анализ в петле кишки, может оценить пригодность пролекарства для использования в единице дозы путем, например, введения пролекарства в замкнутую петлю кишки животного. Конверсия пролекарства может быть определена путем, например, определения продукта конверсии пролекарства (например, высвобождение лекарственного средства или метаболита высвобождаемого лекарственного средства) или определения пролекарства в замкнутой петле кишки животного в желаемый момент(ы) времени после введения.
Ингибиторы в общем выбирают на основании, например, активности взаимодействия с ЖК ферментом(ами), который является посредником в высвобождении лекарственного средства из пролекарства, совместно с которым дозируют ингибитор. Такие количественные определения могут быть проведены в присутствии фермента с пролекарством или без него. Ингибиторы также могут быть выбраны согласно их свойствам таким, как период полужизни в ЖК системе, активность, авидность, сродство, молекулярный размер и/или кривая ингибирования фермента (например, крутизна кривой ингибирования в анализе активности фермента, скорость инициирования ингибирования). Ингибиторы для использования в комбинации пролекарство-ингибитор могут быть выбраны путем количественных определений in vitro, in vivo и/или ex vivo.
Один вариант осуществления - это способ определения пролекарства и ингибитора ЖК фермента, подходящих для препарата в единице дозы, где способ включает объединение пролекарства (например, опиоидного пролекарства с модифицированным кетоном), ингибитора ЖК фермента (например, трипсинового ингибитора) и ЖК фермента (например, трипсина) в реакционной смеси и определение конверсии пролекарства. Такую комбинацию тестируют на взаимодействие между пролекарством, ингибитором и ферментом, например, тестируют с целью определить, как ингибитор будет взаимодействовать с ферментом, который выступает посредником в контролируемом ферментом высвобождении лекарственного средства из пролекарства. В одном примере осуществления, снижение конверсии пролекарства в присутствии ингибитора ЖК фермента по сравнению с конверсией пролекарства в отсутствие ингибитора ЖК фермента указывает на то, что пролекарство и ингибитор ЖК фермента пригодны для приготовления единицы дозы. Такой способ может быть in vitro анализом.
Один вариант осуществления - это способ определения пролекарства и ингибитора ЖК фермента, подходящих для препарата в единице дозы, где способ включает ввод животному пролекарства и ингибитора ЖК фермента и определение конверсии пролекарства. В одном примере осуществления, снижение конверсии пролекарства в присутствии ингибитора ЖК фермента по сравнению с конверсией пролекарства в отсутствие ингибитора ЖК фермента указывает на то, что пролекарство и ингибитор ЖК фермента пригодны для приготовления единицы дозы. Такой способ может быть in vivo анализом; например, пролекарство и ингибитор ЖК фермента могут быть введены перорально. Такой способ может также быть ех vivo анализом; например, пролекарство и ингибитор ЖК фермента могут быть введены перорально или в ткани такие, как кишка, которую по меньшей мере временно подвергают воздействию. Определение может происходить в крови или плазме или в соответствующих тканях. Как используется здесь, термин "ткани" относится как к самим тканям, так и к содержимому тканей.
Один вариант осуществления - это способ определения пролекарства и ингибитора ЖК фермента, подходящих для препарата в единице дозы, где способ включает ввод пролекарства и ингибитора желудочно-кишечного (ЖК) фермента в ткани животного, которые были удалены из животного, и определения конверсии пролекарства. В одном примере осуществления, снижение конверсии пролекарства в присутствии ингибитора ЖК фермента по сравнению с конверсией пролекарства в отсутствие ингибитора ЖК фермента указывает на то, что пролекарство и ингибитор ЖК фермента пригодны для приготовления единицы дозы.
Количественный анализ in vitro может быть проведен путем объединения пролекарства с ингибитором и ЖК ферментом в реакционной смеси. ЖК фермент может быть обеспечен в реакционной смеси в количестве, достаточном для каталитического расщепления пролекарства, и анализы следует проводить в подходящих условиях, по выбору - в условиях, имитирующих условия в ЖК тракте субъекта, например, человека. Конверсия пролекарства может быть оценена путем определения уровня продукта конверсии пролекарства (например, высвобожденного лекарственного средства) и/или путем определения уровня пролекарства, поддерживаемого в присутствии ЖК фермента. Конверсия пролекарства может также быть определена путем определения скорости образования продукта конверсии пролекарства или скорости, с которой расходуется пролекарство. Конверсия пролекарства, модифицированная присутствием ингибитора, по сравнению с уровнем конверсии пролекарства в отсутствие ингибитора, указывает на пригодность ингибитора для ослабления конверсии пролекарства и использования в единице дозы. Реакционные смеси, имеющие постоянное количество пролекарства и увеличиваемые количества ингибитора или постоянное количество ингибитора и увеличиваемые количества пролекарства, могут быть использованы для определения относительных количеств пролекарства и ингибитора, которые обеспечивают желаемую модификацию конверсии пролекарства.
In vivo анализ может определить комбинации пролекарства и ингибитора путем совместного дозирования пролекарства и ингибитора животному. Такое совместное дозирование может быть энтеральным. "Совместное дозирование" относится к введению пролекарства и ингибитора в виде отдельных доз или в виде комбинированных доз (т.е. в одном препарате). Конверсия пролекарства может быть определена путем, например, определения продукта конверсии пролекарства (например, высвобождение лекарственного средства или метаболита лекарственного средства) или определения пролекарства в крови или плазме животного в желаемый момент(ы) времени после введения. Можно определить комбинации пролекарства и ингибитора, которые обеспечивают уровень конверсии пролекарства, дающие желаемую ФК кривую по сравнению, например, с пролекарством без ингибитора.
Комбинации относительных количеств пролекарства и ингибитора, которые обеспечивают желаемую ФК кривую, могут быть идентифицированы путем дозирования животным постоянного количества пролекарства и увеличиваемых количеств ингибитора или постоянного количества ингибитора и увеличиваемых количеств пролекарства. Затем можно определять один или более ФК параметров, например, Cmax лекарственного средства, Tmax лекарственного средства, действие лекарственного средства. Относительные количества пролекарства и ингибитора, которые обеспечивают желаемую ФК кривую, могут быть идентифицированы в виде количеств пролекарства и ингибитора для использования в единице дозы. ФК кривую комбинации пролекарства и ингибитора можно, например, охарактеризовать сниженным значением ФК параметра по сравнению с пролекарством без ингибитора. Снижение значения ФК параметра комбинации ингибитор-пролекарство (например, снижение Cmax лекарственного средства, снижение 1/(Tmax лекарственного средства) (т.е. замедление Tmax лекарственного средства) или снижение действия лекарственного средства) по сравнению с соответствующим значением ФК параметра после введения пролекарства без ингибитора может говорить о том, что комбинация ингибитор-пролекарство может обеспечить желаемую ФК кривую. Количественную оценку можно проводить с различными количествами ингибитора и пролекарства.
In vivo анализ можно использовать для определения комбинаций пролекарства, которые обеспечивают единицы дозы, обеспечивающие желаемые ФК кривые концентрация-доза после приема многократной единицы дозы (например, по меньшей мере 2, по меньшей мере 3, по меньшей мере 4 или более). Ех vivo анализ можно использовать при прямом введении пролекарства и ингибитора в ткани и/или содержимое тканей животного, как, например, кишечник, включая введение путем инъекции в полость замкнутого кишечника (например, анализ в петле кишки или в петле кишечника или обратный анализ в петле кишки). Ех vivo анализ также можно проводить путем иссечения ткани и/или их содержимого из животного и введения пролекарства и ингибитора в эти ткани и/или их содержимое.
Например, выбирают дозу пролекарства, которая необходима для однократной единицы дозы (например, количество, которое обеспечивает эффективный уровень лекарственного средства в плазме). Затем выбирают множество однократных единиц дозы, для которого необходимо протестировать взаимосвязь между этим множеством и ФК параметром. Например, если необходимо составить ФК кривую концентрация-доза для приема внутрь 2, 3, 4, 5, 6, 7, 8, 9 или 10 единиц дозы, то определяют количество пролекарства, эквивалентное приему внутрь такого же числа единиц дозы (далее - "высокая доза"). Множество единиц дозы можно выбрать на основании числа принятых внутрь таблеток, при котором Cmax лекарственного средства модифицирована по отношению к приему однократной единицы дозы. Если, например, кривая должна обеспечить предотвращение злоупотребления, то можно выбрать множество равное 10. Можно протестировать разнообразие различных ингибиторов (например, из перечня ингибиторов), используя различные относительные количества ингибитора и пролекарства. Можно использовать количественные анализы для определения подходящей комбинации(й) ингибитора и пролекарства для того, чтобы получить терапевтически эффективную однократную единицу дозы, где такая комбинация при приеме в виде множества единиц дозы обеспечивает модифицированный ФК параметр по сравнению с приемом внутрь такого же количества одного только лекарственного средства или пролекарства (где однократная доза как одного только лекарственного средства, так и пролекарства, высвобождает в кровь или плазму такое же количество лекарственного средства, как и высвобождаемое однократной единицей дозы).
Затем животным дозируют одновременно повышенные количества ингибитора с высокой дозой пролекарства. Определяют уровень дозы ингибитора, который обеспечивает желаемую Cmax лекарственного средства после приема внутрь высокой дозы пролекарства, и устанавливают получающееся в результате отношение ингибитора к пролекарству.
Затем дозируют одновременно пролекарство и ингибитор в количествах, эквивалентных отношению ингибитора к пролекарству, которое обеспечило желаемый результат при высокой дозе пролекарства. Затем оценивают значение интересующего ФК параметра (например, Cmax лекарственного средства). Если результатом является желаемое значение ФК параметра после приема эквивалента одной единицы дозы, то однократные единицы дозы, обеспечивающие желаемую ФК кривую концентрация-доза, считают установленными. Например, в случаях, где нужна нулевая линейная кривая, Cmax лекарственного средства после приема однократной единицы дозы значительно не увеличивается после приема многократного числа однократных единиц дозы.
Способ производства, составления и упаковки единиц дозы
Единицы дозы настоящего раскрытия могут быть изготовлены, используя способы производства, доступные в данном уровне техники, и могут представлять собой различные формы, пригодные для энтерального (включая перороальное, буккальное и подъязычное) введение, например, в виде таблетки, капсулы, тонкой пленки, порошка, суспензии, раствора, сиропа, дисперсии или эмульсии. Единица дозы может содержать компоненты, общепринятые в фармацевтических препаратах, например, один или более носителей, связующих веществ, смазочных средств, наполнителей (например, для обеспечения характеристик контролируемого высвобождения), модификаторов pH, вкусовых добавок (например, подсластителей), объемообразующих средств, красящих веществ или других активных средств. Единицы дозы настоящего раскрытия могут включать энтеросолюбильное покрытие или иной компонент(ы) для обеспечения защиты от кислоты в желудке, если необходимо.
Единицы дозы могут быть любого размера или формы. Единица дозы может быть любой формы, пригодной для энтерального введения, например, эллипсоидной, линзообразной, круглой, прямоугольной, цилиндрической и подобной.
Единицы дозы, обеспеченные в виде сухих единиц дозы могут иметь общий вес от примерно 1 микрограмма до примерно 1 грамма, и могут быть от примерно 5 микрограммов до 1,5 грамма, от примерно 50 микрограммов до примерно 1 грамма, от примерно 100 микрограммов до примерно 1 грамма, от примерно 100 микрограммов до примерно 750 миллиграммов и может быть от примерно 1 микрограмма до 2 граммов.
Единицы дозы могут включать компоненты в любых относительных количествах. Например, единицы дозы могут содержать от примерно 0,1% (вес.) до 99% (вес.) активных ингредиентов (например, пролекарства и ингибитора) от общего веса единицы дозы (0,1% до 99% объединенного веса пролекарства и ингибитора от общего веса однократной единицы дозы). В некоторых вариантах осуществления единицы дозы могут содержать от 10% до 50%, от 20% до 40% или от примерно 30% (вес.) активных ингредиентов от общего веса единицы дозы.
Единицы дозы могут быть обеспечены в разнообразных формах и по выбору обеспечены в пригодном для хранения виде. Например, единицы дозы могут быть помещены в контейнере, пригодном для содержания фармацевтической композиции. Контейнер может быть, например, бутылкой (например, с закрывающим устройством таким, как крышка), блистерной упаковкой (например, которая может обеспечить одну или более единиц дозы в упаковке), пузырьком, мягкой упаковкой (например, запечатанные мешки из майлара или пластика), ампулой (для однократных единиц дозы в растворе), капельницей, тонкой пленкой, трубкой и подобным.
Контейнеры могут включать съемную крышку (например, навинчивающуюся крышку), которая присоединена к контейнеру по открытому концу, через который единицы дозы помещают внутрь контейнера и через который можно получить доступ к ним.
Контейнеры могут включать уплотнение с контролем вскрытия и/или устойчивый к взлому элемент, уплотнение которого нарушается при получении доступа к единице дозы, размещенной в контейнере. Такой запечатывающий элемент может быть, например, хрупким элементом, который ломается или изменяется любым другим образом при получении доступа к единице дозы, размещенной в контейнере. Примеры такого хрупкого запечатывающего элемента включают запечатывающий элемент, расположенный над открытым концом контейнера таким образом, что доступ к единице дозы в контейнере требует нарушения запечатывающего элемента (например, путем отрывания и/или прокалывания запечатывающего элемента). Примеры хрупкого запечатывающего элемента включают хрупкое кольцо, размещенное вокруг открытого конца контейнера и связанное с крышкой таким образом, чтобы это кольцо ломалось при открывании крышки для получения доступа к единицам дозы в контейнере.
Сухие и жидкие единицы дозы могут быть размещены в контейнере (например, бутылке или упаковке, например, мягком мешке) размером и конфигурацией приспособленном для поддержания стабильности единиц дозы в течение времени, необходимого для отпуска единиц дозы согласно предписанию. Например, контейнеры могут иметь размер и конфигурацию для размещения 10, 20, 30, 40, 50, 60, 70, 80, 90, 100 или более однократных сухих или жидких единиц дозы. Контейнеры могут быть запечатанными или допускать повторное запечатывание. Контейнеры могут быть упакованы в тару (например, для перевозки от производителя в аптеку или другой пункт раздачи). Такая тара может быть коробкой, тубусом или иметь другую конфигурацию и может быть изготовлена из любого материала (например, картона, пластика и подобное). Упаковочная система и/или размещенные в ней контейнеры могут иметь одну или более наклеенных этикеток (например, для предоставления информации такой, как номер партии, тип единицы дозы, производитель и подобное).
Контейнер может включать защищающий от влаги вкладыш и/или светозащитный барьер, например, для обеспечения поддержания стабильности активных ингредиентов в единицах дозы, содержащихся в нем. В случае, если единица дозы является сухой единицей дозы, контейнер может включать пакет с осушителем, размещенным внутри контейнера. Контейнер может быть приспособлен для содержания однократной единицы дозы или многократных единиц доз. Контейнер может включать контролирующий раздачу механизм такой, как закрывающий механизм, который обеспечивает поддерживание режима дозирования.
Единицы дозы могут быть представлены в твердой или полутвердой форме и могут быть сухой единицей дозы. "Сухая единица дозы" относится к единице дозы, которая находится в форме, отличающейся от полностью жидкой формы. Примеры сухих единиц дозы включают, например, таблетки, капсулы (например, твердые капсулы, капсулы, содержащие жидкость), тонкую пленку, микрочастицы, гранулы, порошок и подобное. Единицы дозы могут быть представлены как жидкие единицы дозы, когда единицы дозы могут быть обеспечены как однократные или многократные дозы препарата, содержащего пролекарство и ингибитор в жидкой форме. Однократные дозы сухой или жидкой единицы дозы могут быть размещены внутри запечатанного контейнера, а запечатанные контейнеры по выбору предоставлены в упаковочной системе, например, для обеспечения предписанного числа доз, для обеспечения перевозки единиц дозы и подобного.
Единицы дозы могут быть сформулированы таким образом, чтобы пролекарство и ингибитор были представлены в одном носителе, например, солюбилизированы или суспендированы в одной и той же матрице. Альтернативно, единицы дозы могут состоять из двух или более частей, где пролекарство и ингибитор могут быть обеспечены в одной или разных частях, и могут быть обеспечены в соседних или разделенных частях.
Можно обеспечить единицы дозы в контейнере, в котором они размещены, и представить их как часть упаковочной системы (необязательно с инструкцией по пользованию). Например, единицы дозы, содержащие различные количества пролекарства можно предусмотреть в различных контейнерах, которые можно разместить в более крупных контейнерах (например, для обеспечения защиты единиц дозы во время перевозки). Например, одна или более единиц дозы, как описаны здесь, могут быть предусмотрены в разных контейнерах, где единицы дозы различного состава находятся в разных контейнерах, а разные контейнеры размещены в упаковке для раздачи.
В другом примере, единицы дозы могут быть обеспечены в двухкамерных диспенсерах, где первая камера содержит препарат пролекарства, а вторая камера содержит препарат ингибитора. Диспенсер можно приспособить для обеспечения перемешивания препарата пролекарства и препарата ингибитора перед приемом внутрь. Например, две камеры диспенсера могут быть разделены удаляемой перегородкой (например, хрупкой перегородкой), которую ломают или удаляют перед вводом, чтобы осуществить смешивание препаратов двух камер. Первая и вторая камеры могут иметь выход в диспенсирующий выход, через необязательно общую камеру. Препараты могут быть обеспечены в сухой или жидкой форме, или их комбинации. Например, препарат в первой камере может быть жидким и препарат во второй камере может быть сухим, оба могут быть сухими или оба могут быть жидкими.
В данном раскрытии рассмотрены единицы дозы, обеспечивающие контролируемое высвобождение пролекарства, ингибитора или как пролекарства, так и ингибитора, где под "контролируемым высвобождением" понимают высвобождение как пролекарства, так и ингибитора из единицы дозы за выбранный промежуток времени и/или предварительно определенным способом.
Способы использования единиц дозы
Единицы дозы предпочтительны, поскольку они находят использование в способах уменьшения побочных эффектов и/или улучшают переносимость лекарственных средств пациентами, нуждающимися в них, например, ограничивая ФК параметр, как описано здесь. Настоящее раскрытие обеспечивает способы уменьшения побочных эффектов путем введения единицы дозы настоящего раскрытия пациенту, нуждающемуся в ней, таким образом, чтобы обеспечить уменьшение побочных эффектов по сравнению с таковыми, вызванными введением лекарственного средства и/или по сравнению с введением пролекарства без ингибитора. Настоящее раскрытие также обеспечивает способы улучшения переносимости лекарственного средства путем введения единицы дозы настоящего раскрытия пациенту, нуждающемуся в нем, таким образом, чтобы обеспечить улучшение переносимости по сравнению с введением лекарственного средства и/или по сравнению с введением пролекарства без ингибитора.
Единицы дозы находят применение в способах улучшения соблюдения пациентом лечения, предписанного врачом, где такие способы включают управление введением описанной здесь единицы дозы пациенту, нуждающемуся в лечении, таким образом, чтобы обеспечить улучшенное соблюдение лечения пациентом по сравнению с лечением, включающем в себя введение лекарственного средства и/или по сравнению с введением пролекарства без ингибитора. Такие способы могут помочь увеличению вероятности того, что определенное доктором лечение проходит согласно предписанию.
Единицы дозы могут обеспечить повышенное соблюдение пациентом лечения и клинический контроль. Например, путем ограничения ФК параметра (например, такого, как Cmax лекарственного средства или действия лекарственного средства) в случае приема многократных (например, двух или более, трех или более или четырех или более) единиц дозы пациент, нуждающийся в высокой дозе лекарственного средства, должен обратиться к врачу. Единицы дозы могут обеспечить контроль уровня, до которого пациент может легко проводить "самолечение", и далее может помочь пациенту регулировать дозу в пределах разрешенного диапазона. Единицы дозы могут обеспечить уменьшение побочных эффектов путем, например, обеспечения доставки лекарственного препарата в эффективной дозе, но с модифицированной ФК кривой за период лечения, например, как определено сниженными ФК параметрами (например, сниженной Cmax лекарственного средства, сниженным действием лекарственного средства).
Единицы дозы находят применение в способах уменьшения риска непреднамеренной передозировки лекарственного средства после введения многократных доз, которые были приняты одновременно или за короткий период времени. Такие способы настоящего раскрытия могут обеспечить уменьшение риска непреднамеренной передозировки по сравнению с риском непреднамеренной передозировки лекарственного средства и/или по сравнению с риском непреднамеренной передозировки пролекарства без ингибитора. Такие способы включают управление введением описанной здесь дозировки пациенту, нуждающемуся в лекарственном средстве, высвобождаемом путем конверсии пролекарства. Такие способы могут помочь избежать непреднамеренной передозировки из-за преднамеренного или непреднамеренного неправильного использования единицы дозы.
Настоящее раскрытие обеспечивает способы снижения неправильного использования и злоупотребления лекарственным средством, а также уменьшение риска передозировки, которая может возникнуть при приеме многократных доз лекарственного средства, например, принятых одновременно. Такие способы в общем включают объединение в единице дозы пролекарства и ингибитора ЖК фермента, который является посредником высвобождения лекарственного средства из пролекарства, где ингибитор присутствует в единице дозы в количестве, эффективном для ослабления высвобождения лекарственного средства из пролекарства, например, после приема внутрь пациентом многократных единиц дозы. Такие способы обеспечивают модифицированную ФК кривую концентрация-доза, при этом обеспечивая терапевтически эффективные уровни однократной единицы дозы, как предписано лечащим врачом. Такие способы могут обеспечить, например, уменьшение рисков, сопутствующих неправильному использованию и/или злоупотреблению пролекарства, особенно там, где конверсия пролекарства обеспечивает высвобождение наркотика или другого наркотического лекарственного средства (например, опиоида). Например, когда пролекарство обеспечивает высвобождение наркотического лекарственного средства, единицы дозы могут обеспечить снижение наркотического эффекта, который может последовать за приемом многократных единиц дозы наркотического лекарственного средства.
Единицы дозы могут обеспечить врачам большую свободу в предписании лекарственного средства. Например, врач может предписать режим приема, включающий дозы с различной силой, которые могут включать две или более различные единицы дозы пролекарства и ингибитора, имеющие различные относительные количества пролекарства, различные количества ингибитора или различные количества как пролекарства, так и ингибитора. Такие единицы дозы с различной силой могут обеспечить доставку лекарственного средства согласно различным ФК параметрам (например, действию лекарственного средства, Cmax лекарственного средства и подобное, как описано выше). Например, первая единица дозы может обеспечить доставку первой дозы лекарственного средства после приема внутрь, а вторая единица дозы может обеспечить доставку второй дозы после приема внутрь. Первая и вторая дозы пролекарства единиц дозы могут иметь различную силу, например, вторая доза может быть больше, чем первая доза. Доктор может таким образом предписать набор двух или более, трех или более единиц дозы различной силы, что может сопровождаться инструкциями для обеспечения некоторой степени самолечения, например, для увеличения доставки опиоидного лекарства согласно потребностям пациента для лечения боли.
Препятствование несанкционированному использованию путем использования опосредованного трипсином высвобождения кетон-содержащего опиоида из пролекарства
Раскрытие представляет композицию, содержащую раскрываемое здесь соединение и трипсиновый ингибитор, что снижает потенциальное злоупотребление лекарственным средством. Трипсиновый ингибитор может препятствовать возможности потребителя применять трипсин для влияния на высвобождение кетон-содержащего опиоида из кетон-содержащего опиоидного пролекарства in vitro. Например, если злоупотребляющее лицо пытается выдерживать трипсин с композицией вариантов осуществления, которая включает кетон-содержащее опиоидное пролекарство и трипсиновый ингибитор, трипсиновый ингибитор может снизить действие добавленного трипсина, тем самым, препятствуя попыткам высвобождения кетон-содержащего опиоида для целей злоупотребления.
Примеры
Следующие примеры приведены для того, чтобы представить специалистам в данной области полное раскрытие и описание осуществления и применения вариантов осуществления, и не предназначены для ограничения объема того, что изобретатели считают собственным изобретением, не предназначены для представления, что эксперименты ниже являются всеми возможными или единственными выполненными экспериментами. Предприняты усилия, чтобы обеспечить точность в отношении используемых количественных характеристик (например, количеств, температур и т.д.), но должны быть учтены некоторые экспериментальные ошибки и отклонения. Если не указано иное, части являются весовыми частями, молекулярный вес является средневесовым молекулярным весом, температура измеряется в градусах Цельсия, а давление соответствует или приближено к атмосферному. Могут применяться стандартные аббревиатуры.
Синтез малых молекул трипсиновых ингибиторов
Пример 1 Синтез (S)-этил-4-(5-гуанидино-2-(нафталин-2-сульфонамидо)-пентаноил)пиперазин-1-карбоксилата (Соединение 101)
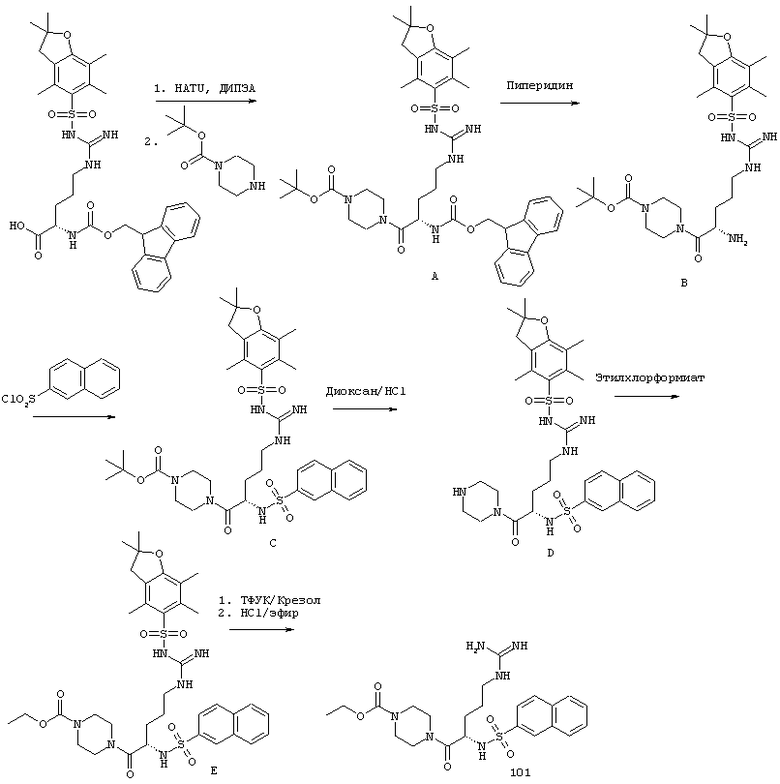
Методика 1
Синтез трет-бутилового эфира 4-[(S)-5-({амино-[(Е)-2,2,4,6,7-пентаметил-2,3-дигидро-бензофуран-5-сульфонилимино]-метил}-амино)-2-(9Н-флуорен-9-илметоксикарбониламино)-пентаноил]-пиперазин-1-карбоновой кислоты (А).
К раствору Fmoc-Arg(Pbf)-OH 1 (25,0 г, 38,5 ммоль) в ДМФА (200 мл) при комнатной температуре добавили ДИПЭА (13,41 мл, 77,1 ммоль). После перемешивания при комнатной температуре в течение 10 минут реакционную смесь охладили до ~5°C. К реакционной смеси добавили HATU (16,11 г, 42,4 ммоль) порциями, перемешивали 20 минут и добавили по каплям раствор трет-бутил-1-пиперазина карбоксилата (7,18 г, 38,5 ммоль) в ДМФА (50 мл). Реакционную смесь перемешивали при ~5°C 5 минут. Затем реакционной смеси дали нагреться до комнатной температуры и перемешивали 2 часа. Растворитель удалили в вакууме и остаток растворили в этилацетате (ЭА) (500 мл), промыли водой (2×750 мл), 1% H2SO4 (300 мл) и солевым раствором (750 мл). Органический слой отделили, высушили над Na2SO4 и растворитель удалили в вакууме до общего объема 100 мл. Соединение А взяли для следующего этапа в виде раствора в ЭА (100 мл). ЖХ-МС [М+Н] 817,5 (C43H56N6O8S+H, рассчит.: 817,4).
Методика 2
Синтез трет-бутилового эфира 4-[(S)-2-амино-5-({амино-[(Е)-2,2,4,6,7-пентаметил-2,3-дигидро-бензофуран-5-сульфонилимино]-метил}-амино)-пентаноил]-пиперазин-1-карбоновой кислоты (В).
К раствору соединения А (46,2 ммоль) в этилацетате (175 мл) при комнатной температуре добавили пиперидин (4,57 мл, 46,2 ммоль) и реакционную смесь перемешивали 18 часов при комнатной температуре. Затем растворитель удалили в вакууме и образовавшийся остаток растворили в минимальном количестве этилацетата (~50 мл) и добавили гексан (~1 л). Осажденный неочищенный продукт отфильтровали и снова перекристаллизовали из этилацетата (~30 мл) и гексана (~750 мл). Осадок отфильтровали, промыли гексаном и высушили в вакууме с получением соединения В (28,0 г, 46,2 ммоль). ЖХ-МС [М+Н] 595,4 (C28H46N6O6S+H, рассчит.: 595,3). Соединение В применяли без дополнительной очистки.
Методика 3
Синтез трет-бутилового эфира 4-[(S)-5-({амино-[(Е)-2,2,4,6,7-пентаметил-2,3-дигидро-бензофуран-5-сулъфонилимино]-метил}-амино)-2-(нафталин-2-сульфаниламино)-пентаноил]-пиперазин-1-карбоновой кислоты (С).
К раствору соединения В (28,0 г, 46,2 ммоль) в ТГФ (250 мл) добавили водный 1 н NaOH (171 мл). Реакционную смесь охладили до ~5°C, раствор 2-нафталин-сульфонилхлорида (26,19 г, 115,6 ммоль) в ТГФ (125 мл) добавили по каплям. Реакционную смесь перемешивали при ~5°C 10 минут, перемешивание продолжали при комнатной температуре 2 часа. Реакционную смесь разбавили этилацетатом (1 л), промыли водным 1 н NaOH (1 л), водой (1 л) и солевым раствором (1 л). Органический слой отделили, высушили над Na2SO4 и удалили растворитель в вакууме с получением соединения С (36,6 г, 46,2 ммоль). ЖХ-МС [М+Н] 785,5 (C38H52N6O8S+H, рассчит.: 785,9).
Соединение С применяли без дополнительной очистки.
Методика 4
Синтез 2,2,4,6,7-Пентаметил-2,3-дигидро-бензофуран-5-сульфоновой кислоты 1-амино-1-[(S)-4-(нафталин-2-сульфониламино)-5-оксо-5-пиперазин-1-ил-пентиламино]-мет-(E)-илиденамида (D).
К раствору соединения С (36,6 г, 46,2 ммоль) в диоксане (60 мл) добавили 4 М раствор HCl в диоксане (58 мл) по каплям. Реакционную смесь перемешивали при комнатной температуре 1,5 часа. К реакционной смеси добавили диэтиловый эфир (ДЭ) (600 мл), осажденный продукт отфильтровали, промыли диэтиловым эфиром и окончательно высушили под вакуумом с получением соединения D (34,5 г, 46,2 ммоль). ЖХ-МС [М+Н] 685,4 (C33H44H6O6S+Н, рассчит.: 685,9). Соединение D применяли без дополнительной очистки.
Методика 5
Синтез этилового эфира 4-[(S)-5-({амино-[(Е)-2,2,4,6,7-пентаметил-2,3-дигидро-бензофуран-5-сульфонилимино]-метил}-амино)-2-(нафталин-2-сульфаниламино)-пентаноил]-пиперазин-1-карбоновой кислоты (Е).
К раствору соединения D (8,0 г, 11,1 ммоль) в CHCl3 (50 мл) добавили ДИПЭА (4,1 мл, 23,3 ммоль) при комнатной температуре и перемешивали 15 минут. Смесь охладили до ~5°C, по каплям добавили этилхлорформиат (1,06 мл, 11,1 ммоль). После перемешивания при комнатной температуре в течение ночи (~18 часов) растворитель удалили в вакууме. Остаток растворили в метаноле (~25 мл) и добавили ДЭ (~500 мл). Осажденный неочищенный продукт отфильтровали, промыли диэтиловым эфиром и высушили в вакууме с получением соединения Е (8,5 г, 11,1 ммоль). ЖХ-МС [М+Н] 757,6 (C36H48N6O8S+H, рассчит.: 757,9). Соединение Е применяли без дополнительной очистки.
Синтез (S)-этил-4-(5-гуанидино-2-(нафталин-2-сульфонамидо)пентаноил)пиперазин-1-карбоксилата (Соединение 101)
5% Раствор м-крезола в трифторуксусной кислоте (ТФУК) (50 мл) добавили к соединению Е (8,5 г, 11,1 ммоль) при комнатной температуре. После перемешивания в течение 1 часа реакционную смесь осадили диэтиловым эфиром (~500 мл). Осадок отфильтровали, промыли диэтиловым эфиром, и высушили в вакууме с получением неочищенного продукта. Неочищенный продукт очистили препаративной обращенно-фазовой ВЭЖХ. [Колонка: VARIAN, LOAD & LOCK, L&L 4002-2, заполнение: Microsorb 100-10 Cl 8, объем впрыска: ~15 мл×2, скорость введенного потока: 20 мл/мин, 100% А, (вода/О, 1% ТФУК), скорость потока: 100 мл/мин, фракция: 30 с (50 мл), способ: 0% В (ацетонитрил (АН)/0,1% ТФУК)-60% В/60 мин/ 100 мл/мин/ 254 нм]. Растворители удалили из чистых фракций в вакууме. Следы воды удалили совместным выпариванием с изопропанолом (ИПС)(2×50 мл). Остаток растворили в минимальном количестве ИПС и продукт осадили 2 М раствором HCl в ДЭ. Продукт отфильтровали, промыли диэтиловым эфиром и высушили в вакууме с получением Соединения 101 в виде гидрохлорида 7 (3,78 г, выход 63%, чистота 99,4%). ЖХ-МС [М+Н] 505,4 (C38H52N6O8S+H, рассчит.: 505,6).
Пример 2
Синтез (S)-этил-4-(5-гуанидино-2-(2,4,6-триизопропилфенилсульфонамидо)-пентаноил)пиперазин-1-карбоксилата (Соединение 102)
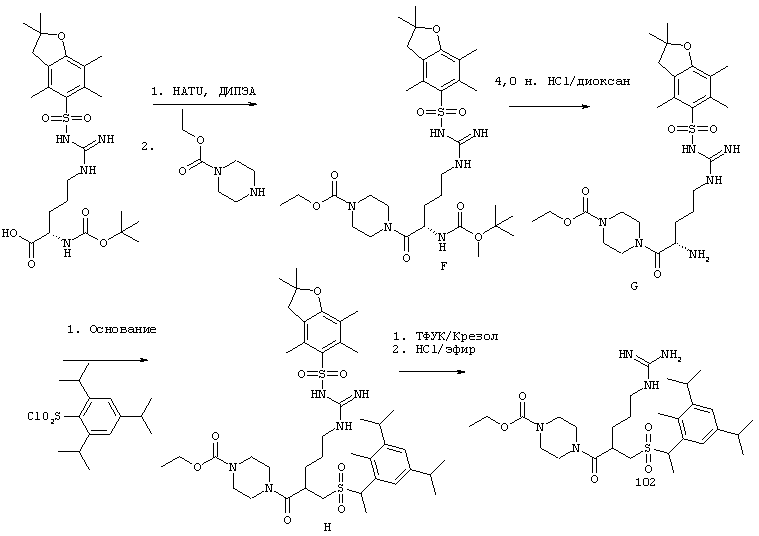
Методика 6 Синтез этилового эфира 4-[(S)-5-({амино-[(Е)-2,2,4,6,7-пентаметил-2,3-дигидро-бензофуран-5-сульфонилимино]-метил}-амино)-2-трет-бутилкарбониламино-пентаноил]-пиперазин-1-карбоновой кислоты (F).
К раствору Boc-Arg(Pbf)-OH (13,3 г, 25,3 ммоль) в ДМФА (10 мл) добавили ДИПЭА (22,0 мл, 126,5 ммоль) при комнатной температуре и перемешивали 15 минут. Затем реакционную смесь охладили до ~5°C, порциями добавили HATU (11,5 г, 30,3 ммоль) и перемешивали 30 минут с последующим добавлением по каплям этил-1-пиперазин-карбоксилата (4,0 г, 25,3 ммоль) в ДМФА (30 мл). Через 40 минут реакционную смесь разбавили этилацетатом (400 мл) и влили в H2O (1 л). Экстрагировали этилацетатом (2×400 мл) и промыли водой (800 мл), 2% H2SO4 (500 мл), H2O (2×800 мл) и солевым раствором (800 мл). Органический слой отделили, высушили над MgSO4 и растворитель удалили в вакууме. Образованный маслянистый остаток высушили в вакууме с получением соединения F (16,4 г, 24,5 ммоль) в виде пенистого твердого вещества. ЖХ-МС [М+Н] 667,2 (C31H50N6O8S+H, рассчит.: 667,8). Соединение F применяли без дополнительной очистки.
Методика 7
Синтез этилового эфира 4-[(S)-2-амино-5-({амино-[(Е)-2,2,4,6,7-пентаметил-2,3-дигидро-бензофуран-5-сульфонилимино]-метил}-амино)-пентаноил]-пиперазин-1-карбоновой кислоты (G).
Раствор соединения F (20,2 г, 30,2 ммоль) в дихлорметане (90 мл) обработали 4,0 н раствором HCl в 1,4-диоксане (90 мл, 363,3 ммоль) и перемешивали при комнатной температуре 2 ч. Затем основное количество дихлорметана (~90%) удалили в вакуумеи добавили ДЭ (~1 л). Образованный осадок отфильтровали, промыли диэтиловым эфиром и высушили в вакууме с получением соединения G (17,8 г, 30,2 ммоль). ЖХ-МС [М+Н] 567,8 (C26H42N6O6S+H, рассчит.: 567,8). Соединение G применяли без дополнительной очистки.
Методика 8
Синтез этилового эфира 4-[(S)-5-({амино-[(Е)-2,2,4,6,7-пентаметил-2,3-дигидро-бензофуран-5-сульфонилимино]-метил}-амино)-2-(2,4,6-триизопропил-бензолсульфаниламино)-пентаноил]-пиперазин-1-карбоновой кислоты (Н).
К раствору соединения G (1,0 г, 1,8 ммоль) в ТГФ (7 мл) добавили 3,1 н водного NaOH (4,0 мл) и перемешивали 5 минут. Реакционную смесь охладили до ~5°C, а затем по каплям добавили раствор трипсилхлорида (2,2 г, 7,3 ммоль) в ТГФ (5 мл) и перемешивали при комнатной температуре в течение ночи (-18 часов). Реакционную смесь разбавили H2O (130 мл), подкислили 2% H2SO4 (15 мл) и экстрагировали этилацетатом (3×80 мл). Органический слой объединили и промыли водой (2×400 мл), насыщенным NaHCO3 (100 мл), H2O (200 мл) и солевым раствором (200 мл). Органический слой отделили, высушили над MgSO4 и растворитель удалили в вакууме с получением (2,9 г) неочищенного продукта. Его очистили нормально-фазовой флэш-хроматографией (5-10% метанол/дихлорметан (ДХМ)) с получением соединения Н (0,52 г, 1,0 ммоль). ЖХ-МС [М+Н] 833,8 (C41H64N6O8S+H, рассчит.: 834,1).
Синтез (S)-этил-4-(5-гуанидино-2-(2,4,6-триизопропилфенилсулъфонамидо)-пентаноил)пиперазин-1-карбоксилата (Соединение 102)
5% Раствор м-крезола в ТФУК (40 мл) добавили к соединению Н (3,73 г, 3,32 ммоль) при комнатной температуре. После перемешивания в течение 45 минут растворители удалили в вакууме. Остаток растворили в дихлорметане (100 мл), промыли водой (3×200 мл) и солевым раствором (200 мл). Органический слой отделили, высушили над MgSO4, а затем растворитель удалили в вакууме. Остаток растворили в дихлорметане (~5 мл), а затем добавили гексан (~250 мл) и получили осадок. Его промыли гексаном и высушили в вакууме с получением неочищенного продукта (1,95 г). Неочищенный продукт очистили обращенно-фазовой ВЭЖХ [колонка: VARIAN, LOAD & LOCK, L&L 4002-2, заполнение: Microsorb 100-10 С 18, объем впрыска: ~15 мл, скорость введенного потока: 20 мл/мин, 100% А, (вода/0,1% ТФУК), скорость потока: 100 мл/мин, фракция: 30 с (50 мл), способ: 25% В (АН/0,1% ТФУК/70% В/98 мин/ 100 мл/мин/ 254 нм]. Растворители удалили из чистых фракций в вакууме. Следы воды удалили совместным выпариванием с ИПС (2×50 мл). Остаток растворили в минимальном количестве ИПС и продукт осадили 2 М раствором HCl в ДЭ. Продукт отфильтровали, промыли диэтиловым эфиром и высушили в вакууме с получением продукта в виде гидрохлорида Соединения 102 (0,72 г, выход 35%, чистота 99,8%). ЖХ-МС [М+Н] 581,6 (C28H48N6O5S+Н, рассчит.: 581,7).
Пример 3
Синтез гидрохлорида (S)-этил-1-(5-гуанидино-2-(нафталин-2-сульфонамидо)пентаноил)пиперидин-4-карбоксилата (Соединение 103)
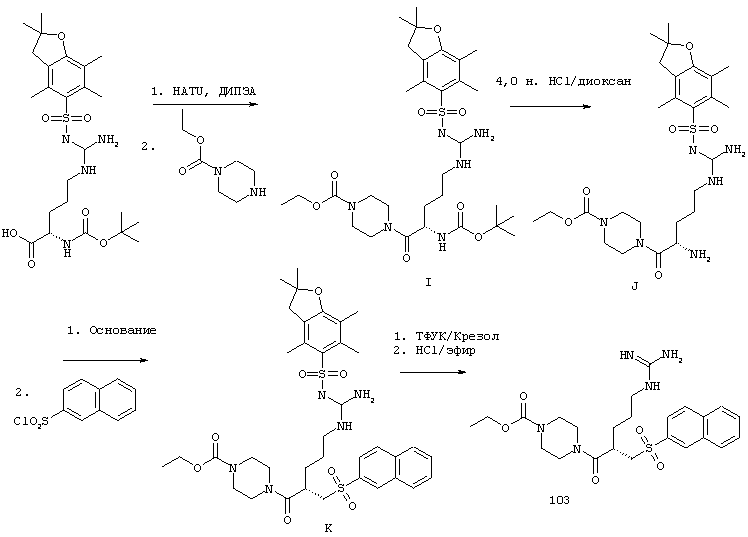
Методика 9 Синтез этилового эфира 1-[boc-Arg(Pbf)]-пиперидин-4-карбоновой кислоты (I)
К раствору Boc-Arg(Pbf)-OH (3,4 г, 6,36 ммоль) и HATU (2,9 г, 7,63 ммоль) в ДМФА (15 мл) добавили ДИПЭА (7,4 мл, 42,4 ммоль) и реакционную смесь перемешивали 10 минут при комнатной температуре. Раствор этилизонипекотата (1,0 г, 6,36 ммоль) в ДМФА (6 мл) добавили в реакционную смесь по каплям. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, затем разбавили этилацетатом (150 мл) и вылили в воду (500 мл). Продукт экстрагировали этилацетатом (2×100 мл). Органический слой промыли водной 0,1 н HCl (200 мл), 2% водным бикарбонатом натрия (200 мл), водой (200 мл) и солевым раствором (200 мл). Органический слой высушили над сульфатом натрия, отфильтровали, а затем выпарили в вакууме. Образованный маслянистый продукт высушили в вакууме в течение ночи с получением соединения I (3,7 г, 5,57 ммоль) в виде вязкого твердого вещества. ЖХ-МС [М+Н] 666,5 (C32H51N5O8S+H, рассчит.: 666,7). Соединение I применяли без дополнительной очистки.
Методика 10 Синтез гидрохлорида этилового эфира 1-[Arg(Pbf)]-пиперидин-4-карбоновой кислоты (J)
К раствору соединения I (4,7 г, 7,07 ммоль) в дихлорметане (25 мл) добавили 4 н HCl в диоксане (25,0 мл, 84,84 ммоль) и реакционную смесь перемешивали при комнатной температуре 2 часа. Реакционную смесь концентрировали в вакууме до ~20 мл растворителя, а затем разбавили диэтиловым эфиром (250 мл) с получением белого тонкого осадка. Реакционную смесь перемешивали в течение 1 часа и твердое вещество промыли эфиром (50 мл) и высушили в вакууме в течение ночи с получением соединения J (4,3 г, 7,07 ммоль) в виде тонкого порошка. ЖХ-МС [М+Н] 566,5 (C27H43N5O6S+H, рассчит.: 566,7). Соединение J применяли без дополнительной очистки.
Методика 11
Синтез этилового эфира 1-[5(S)-(N'-Pbf-гуанидино)-2-(нафталин-2-сулъфониламино)-пентаноил]-пиперидин-4-карбоновой кислоты (K)
К раствору соединения J (1,1 г, 1,6 ммоль) и NaOH (260 мг, 5,9 ммоль) в смеси ТГФ (5 мл) и воды (3 мл) добавили раствор 2-нафталосульфонилхлорида (0,91 г, 2,5 ммоль) в ТГФ (10 мл) по каплям с перемешиванием при ~5°C. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, затем разбавили водой (5 мл). Водную 1 н HCl (5 мл) добавили для достижения pH ~3. Дополнительно добавили воду (20 мл) и экстрагировали продукт этилацетатом (3×50 мл). Органический слой отделили, а затем промыли 2% водным бикарбонатом натрия (50 мл), водой (50 мл) и солевым раствором (50 мл). Экстракт высушили над безводным сульфатом натрия, отфильтровали и выпарили в вакууме. Образованный маслянистый продукт высушили в вакууме в течение ночи с получением соединения K (1,3 г, 1,6 ммоль) в виде маслянистого пенистого твердого вещества. ЖХ-МС [М+Н] 756,5 (C37H49N5O8S+H, рассчит.: 756,7). Соединение K применяли без дополнительной очистки.
Синтез гидрохлорида (S)-этил-1-(5-гуанидино-2-(нафталин-2-сульфонамидо)пентаноил)пиперидин-4-карбоксилата (Соединение 103)
В колбу добавили соединение K (1,3 г, 1,6 ммоль), а затем обработали 5% раствором м-крезола в ТФУК (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Затем реакционную смесь концентрировали в вакууме до объема ~5 мл. Затем добавили диэтиловый эфир (200 мл) к остатку и получили тонкий белый осадок. Осадок отфильтровали и промыли эфиром (2×25 мл). Образованное твердое вещество высушили в вакууме в течение ночи с получением неочищенного материала, который очистили препаративной обращенно-фазовой ВЭЖХ. [колонка Nanosyn-Pack Microsorb (100-10) С-18 (50×300 мм); скорость потока: 100 мл/мин; объем впрыска 12 мл (ДМСО-вода, 1:1, об./об.); подвижная фаза А: 100% вода, 0,1% ТФУК; подвижная фаза В: 100% ACN, 0,1% ТФУК; градиентное элюирование от 25% В до 55% В за 90 минут, детектирование при 254 нм]. Фракции, содержащие желаемое соединение, объединили и концентрировали в вакууме. Остаток растворили в ИПС (100 мл) и выпарили в вакууме (повторили дважды). Затем остаток растворили в ИПС (5 мл) и обработали 2 н раствором НСl в эфире (100 мл, 200 ммоль) с получением белого осадка. Его высушили в вакууме в течение ночи с получением Соединения 103 (306 мг, выход 31%, чистота 95,7%) в виде белого твердого вещества. ЖХ-МС [М+Н] 504,5 (C24H33N5O5S+H, рассчит.: 504,6).
Пример 4 Синтез гидрохлорида (S)-этил-1-(5-гуанидино-2-(2,4,6-триизопропилфенилсульфонамидо)пентаноил)пиперидин-4-карбоксилата (Соединение 104)
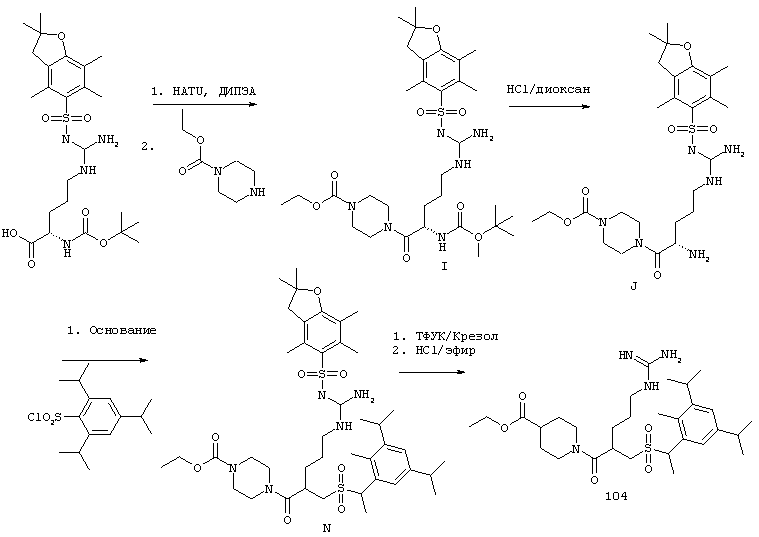
Методика 12
Синтез этилового эфира 1-[5(S)-(N'-Pbf-гуанидино)-2-(2,4,6-триизопромил-бензолсуль(рониламино)-пентаноил]-пиперидин-4-карбоновой кислоты (N)
К раствору соединения J (1,0 г, 1,6 ммоль) и NaOH (420,0 мг, 10,4 ммоль) в смеси ТГФ (5 мл) и воды (4 мл) добавили по каплям раствор 2,4,6-триизопропил-бензолсульфонилхлорида (2,4 г, 8,0 ммоль) при перемешивании и выдерживали при ~5°C. Реакционную смесь затем перемешивали при комнатной температуре 1 ч, наблюдая за протеканием реакции, затем разбавили водой (20 мл), и подкислили водной 1 н HCl (5 мл) до pH ~3. Дополнительно добавили воду (30 мл) и экстрагировали продукт этилацетатом (3×50 мл). Органический слой промыли 2% водным бикарбонатом натрия (50 мл), водой (50 мл) и солевым раствором (50 мл). Органический слой высушили над безводным сульфатом натрия, отфильтровали и выпарили в вакууме. Образованный маслянистый остаток высушили в вакууме в течение ночи с получением соединения N (1,0 г, 1,2 ммоль) в виде маслянистого материала. ЖХ-МС [М+Н] 832,8 (C42H65N5O8S+H, рассчит.: 832,7). Соединение N применяли без дополнительной очистки.
Синтез гидрохлорида (3)-этил-1-(5-гуанидино-2-(2,4,6-триизопропилфенилсульфонамидо)пентаноил)пиперидин-4-карбоксилата (Соединение 104)
В колбу добавили соединение N (2,3 г, 2,8 ммоль), а затем обработали 5% раствором м-крезола в ТФУК (16 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Затем реакционную смесь концентрировали в вакууме до объема 5 мл. Добавили гексан (200 мл) к остатку и декантировали с получением маслянистого осадка. Продукт очистили препаративной обращенно-фазовой ВЭЖХ. [колонка Nanosyn-Pack Microsorb (100-10) С-18 (50×300 мм); скорость потока: 100 мл/мин; объем впрыска 15 мл (ДМСО-вода, 1:1, об./об.); подвижная фаза А: 100% вода, 0,1% ТФУК; подвижная фаза В: 100% ACN, 0,1% ТФУК; градиентное элюирование от 35% В до 70% B в течение 90 минут, детектирование при 254 нм]. Фракции, содержащие желаемое соединение, объединили и концентрировали в вакууме. Остаток растворили в ИПС (100 мл) и выпарили в вакууме (повторили дважды). Остаток растворили в ИПС (5 мл) и обработали 2 н раствором HCl в эфире (100 мл, 200 ммоль) с получением белого осадка. Его сушили в вакууме в течение ночи с получением Соединения 104 (1,08 г, 62,8%) в виде вязкого твердого вещества. ЖХ-МС [М+Н] 580,6 (C29H49N5O5S+H, рассчит.: 580,8).
Пример 5
Синтез (S)-6-(4-(5-гуанидино-2-(нафталин-2-сульфонамидо)пентаноил)-пиперазин-1-ил)-6-оксогексановой кислоты (Соединение 105)
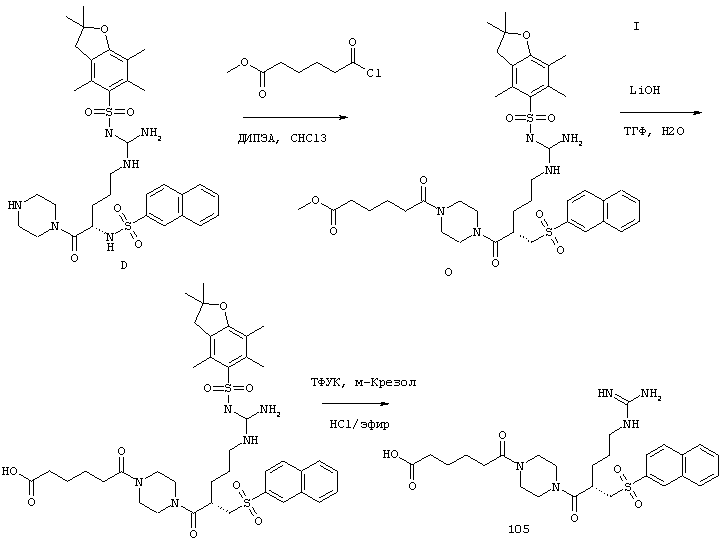
Методика 13
Синтез метилового эфира 6-{4-[(S)-5-({амино-[(Е)-2,2,4,6,7-пентаметил-2,3-дигидро-бензофуран-5-сульфонилимино]-метил}-амино)-2-(нафталин-2-сульфаниламино)-пентаноил]-пипвразин-1-ил}-6-оксогексановой кислоты (О)
К раствору соединения D (1,5 г, 2,08 ммоль) в хлороформе (50 мл) добавили ДИПЭА (1,21 мл, 4,16 ммоль), а затем адипоилхлорид (0,83 мл, 6,93 ммоль) по каплям. Реакционную смесь перемешивали при комнатной температуре в течение ночи (~18 часов). Растворители удалили в вакууме, а остаток высушили в вакууме с получением соединения О (2,1 г, превышает количественный). ЖХ-МС [М+Н] 827,5 (C40H54N6O9S+H, рассчит.: 827,3). Соединение О применяли без дополнительной очистки.
Методика 14
Синтез 6-{4-[(S)-5-({амино-[(Е)-2,2,4,6,7-пентаметил-2,3-дигидро-бензофуран-5-сульфонилимино]-метил}-амино)-2-(нафталин-2-сульфаниламино)-пентаноил]-пиперазин-1-ил}-6-оксогексановой кислоты (Р)
К раствору соединения О (2,1 г, 2,08 ммоль) в ТГФ (5 мл), Н20 (5 мл) добавили 2 молярный водный LiOH (6 мл). Реакционную смесь перемешивали при комнатной температуре 2 часа. Растворители удалили в вакууме, затем остаток растворили в воде (~50 мл), подкислили насыщенным водным NaHSO4 (~100 мл) и экстрагировали этилацетатом (2×100 мл). Органический слой высушили над Na2SO4 и, удалив растворитель, получили соединение Р (1,72 г, 2,08 ммоль). ЖХ-МС [М+Н] 813,5 (C39H52H6O9S+Н, рассчит.: 813,3). Соединение Р применяли без дополнительной очистки.
Синтез (S)-6-(4-(5-гуанидино-2-(нафталин-2-сульфонамидо)пентаноил)пиперазин-1-ил)-6-оксогексановой кислоты (Соединение 105)
Раствор 5% м-крезола в ТФУК (25 мл) добавили к соединению Р (1,72 г, 2,08 ммоль) при комнатной температуре. После перемешивания в течение 30 минут реакционную смесь осадили добавлением ДЭ (~200 мл). Осадок отфильтровали, и промыли диэтиловым эфиром, и высушили в вакууме с получением неочищенного продукта. Неочищенный продукт очистили препаративной обращенно-фазовой ВЭЖХ [колонка: VARIAN, LOAD&LOCK, L&L 4002-2, заполнение: Microsorb 100-10 С 18, объем впрыска: ~25 мл, скорость введенного потока: 20 мл/мин, 95% А, (вода/О, 1% ТФУК), скорость потока: 100 мл/мин, фракция: 30 с (50 мл), способ: 5% В (ацетонитрил/0,1% ТФУК)/5 мин/ 25% В/20 мин/25% В/15 мин/50% В/25 мин/100 мл/мин/254 нм]. Растворители удалили из чистых фракций в вакууме. Следы воды удалили совместным выпариванием с ИПС (25 мл) (повторили дважды). Остаток растворили в минимальном количестве ИПС, затем добавили 2 М раствор HCl в ДЭ (~50 мл) и разбавили диэтиловым эфиром (~250 мл). Образованный осадок отфильтровали, промыли диэтиловым эфиром и высушили в вакууме с получением продукта в виде гидрохлорида Соединения 105 (0,74 г, выход 59%, чистота 98,9%). ЖХ-МС [М+Н] 561,4 (C26H36N6O6S+Н, рассчит.: 561,2).
Пример 6
Синтез 3-(4-карбамимидоилфенил)-2-оксопропановой кислоты (Соединение 107)
Соединение 107, т.е. 3-(4-карбамимидоилфенил)-2-оксопропановая кислота, может быть получено с использованием способов, известных специалистам данной области техники, таких как описанные в публикации Richter Р и др., Pharmazie, 1977, 32, 216-220 и в содержащихся там ссылках. Чистоту Соединения 107, примененного в Примере 7, оценили как 76%, оценка обусловлена низким УФ-поглощением этого соединения посредством ВЭЖХ. Данные масс-спектрометрии: ЖХ-МС [М+Н] 207,0 (C10H10N2O3+Н, рассчит.: 207,1).
Пример 7
Синтез (S)-5-(4-карбамимидоилбензиламино)-5-оксо-4-((R)-4-фенил-2-(фенилметилсульфонамидо)бутанамидо)пентановой кислоты (Соединение 108)
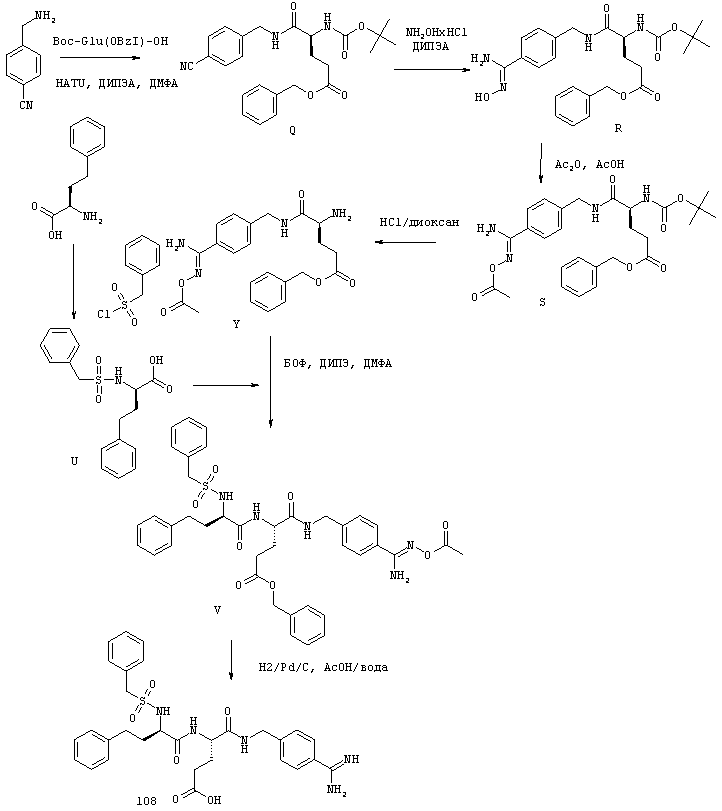
Методика 15
Синтез бензинового эфира (8)-4-трет-бутоксикарбониламино-4-(4-циано-бензилкарбамоил)-масляной кислоты (Q).
Раствор Boc-Glu(OBzl)-OH (7,08 г, 21,0 ммоль), ВОР (9,72 г, 22,0 ммоль) и ДИПЭА (12,18 мл, 70,0 ммоль) в ДМФА (50 мл) выдерживали при комнатной температуре 20 минут с последующим добавлением 4-(аминометил)бензонитрила гидрохлорида (3,38 г, 20,0 ммоль). Реакционную смесь перемешивали при комнатной температуре еще 1 час и разбавили этилацетатом (500 мл). Полученный раствор экстрагировали водой (100 мл), водным 5% NaHCO3 (100 мл) и водой (2×100 мл). Органический слой высушили над MgSO4, выпарили и высушили в вакууме с получением соединения Q (9,65 г, выход превысил количественный) в виде желтоватого масла. ЖХ-МС [М+Н] 452,0 (C25H29N3O5S+H, рассчит.: 452,4). Соединение Q применяли без дополнительной очистки.
Методика 16
Синтез бензинового эфира (S)-4-трет-бутоксикарбониламино-4-[4-(N-гидроксикарбамимидоил)-бензил-карбамоил]-масляной кислоты (R).
Раствор соединения Q (9,65 г, 20,0 ммоль), гидроксиламина гидрохлорида (2,10 г, 30,0 ммоль) и ДИПЭА (5,22 мл, 30,0 ммоль) в этаноле (абсолютный, 150 мл) нагревали с обратным холодильником 6 часов. Реакционной смеси дали охладиться до комнатной температуры и перемешивали еще 16 часов, затем растворители выпарили в вакууме. Образованный остаток высушили в вакууме с получением соединения R (14,8 г, выход превысил количественный) в виде желтоватого масла. ЖХ-МС [М+Н] 485,5 (C25H32N4O6S+H, рассчит.: 485,8). Соединение R применяли без дополнительной очистки.
Методика 17
Синтез бензинового эфира (S)-4-трет-бутоксикарбониламино-4-[4-(N-ацетилгидроксикарбамимидоил)-бензил-карбамоил]-масляной кислоты (S).
Раствор соединения R (14,8 г, 20,0 ммоль) и уксусного ангидрида (5,7 мл, 60,0 ммоль) в уксусной кислоте (100 мл) перемешивали при комнатной температуре 45 минут, а затем растворитель выпарили в вакууме. Образованный остаток растворили в ЭА (300 мл) и экстрагировали водой (2×75 мл) и солевым раствором (75 мл). Затем органический слой высушили над MgSO4, выпарили и высушили в вакууме с получением соединения S (9,58 г, 18,2 ммоль) в виде желтоватого твердого вещества. ЖХ-МС [М+Н] 527,6 (C27H34N4O7S+H, рассчит.: 527,9). Соединение S применяли без дополнительной очистки.
Методика 18
Синтез бензилового эфира (S)-4-[4-(N-ацетилгидроксикарбамимидоил)-6ензил-карбамоил]-масляной кислоты (Т).
Соединение S (9,58 г, 18,2 ммоль) растворили в 1,4-диоксане (50 мл) и обрабатывали 4 н раствором НС1 в диоксане (50 мл, 200 ммоль) при комнатной температуре в течение 1 часа. Затем растворитель выпарили в вакууме. Образованный остаток растерли с эфиром (200 мл). Полученный осадок отфильтровали, промыли эфиром (100 мл) и гексаном (50 мл) и высушили в вакууме с получением соединения Т (9,64 г, выход превысил количественный) в виде грязно-белого твердого вещества. ЖХ-МС [М+Н] 426,9 (C22H26N4O5S+H, рассчит.: 427,3). Соединение Т применяли без дополнительной очистки.
Методика 19
Синтез (R)-4-фенил-2-фенилметансульфониламино-масляной кислоты (U)
Раствор D-гомо-фенилаланина (10,0 г, 55,9 ммоль) и NaOH (3,35 г, 83,8 ммоль) в смеси 1,4-диоксана (80 мл) и воды (50 мл) охладили до ~5°C, затем поочередно добавляли а-толуолсульфонил хлорид (16,0 г, 83,8 ммоль; 5 порций по 3,2 г) и 1,12 М раствор NaOH (50 мл, 55,9 ммоль; 5 порций по 10 мл), поддерживая pH>10. Реакционную смесь затем подкислили 2% водн. H2SO4 до pH приблизительно pH 2. Продукт экстрагировали этилацетатом (2×200 мл). Полученный органический слой промыли водой (3×75 мл), высушили над MgSO4, а затем растворитель выпарили в вакууме. Полученный остаток высушили в вакууме с получением соединения U (12,6 г, 37,5 ммоль) в виде белого твердого вещества. ЖХ-МС [М+Н] 334,2 (C17H19NO4S+H, рассчит.: 333,4). Соединение U применяли без дополнительной очистки.
Методика 20
Синтез бензинового эфира (S)-4-[4-(N-ацетилгидроксикарбамимидоил)-бензилкарбамоил]-4-((R)-4-фенил-2-фенилметансульфониламино-бутириламино)-масляной кислоты (V).
Раствор соединения U (5,9 г, 17,8 ммоль), дигидрохлорида соединения Т (18,0 ммоль), ВОР (8,65 г, 19,6 ммоль) и ДИПЭА (10,96 мл, 19,6 ммоль) в ДМФА (250 мл) перемешивали при комнатной температуре 2 часа. Реакционную смесь разбавили этилацетатом (750 мл) и экстрагировали водой (200 мл). Образованный осадок отфильтровали, промыли этилацетатом (200 мл) и водой (200 мл) и высушили при комнатной температуре в течение ночи (~18 часов) с получением соединения V (8,2 г, 11,0 ммоль) в виде грязно-белого твердого вещества. ЖХ-МС [М+Н] 743,6 (C39H43N5O8S+H, рассчит.: 743,9). Соединение V применяли без дополнительной очистки.
Синтез (S)-5-(4-карбамимидоилбензиламино)-5-оксо-4-((R)-4-фенил-2-(фенилметилсульфонамидо)бутанамидо)пентановой кислоты (Соединение 108)
Соединение V (8,0 г, 10,77 ммоль) растворили в уксусной кислоте (700 мл) с последующим добавлением Pd/C (5% (вес.), 3,0 г) в виде суспензии в воде (50 мл). Реакционную смесь подвергли гидрированию (аппарат Парра, 50 psi H2) при комнатной температуре в течение 3 часов. Катализатор отфильтровали через подушку целита на фильтре из спеченного стекла и промыли метанолом. Фильтрат выпарили в вакууме с получением Соединения 108 в виде бесцветного масла. ЖХ-МС [М+Н] 594,2 (C30H35N5O6S+Н, рассчит.: 594). Полученное масло растворили в воде (150 мл) и подвергли очистке с помощью ВЭЖХ. [колонка Nanosyn-Pack Microsorb (100-10) С-18 (75×300 мм); скорость потока: 250 мл/мин; объем впрыска 150 мл; подвижная фаза А: 100% вода, 0,1% ТФУК; подвижная фаза В: 100% ацетонитрил, 0,1% ТФУК; изократическое элюирование при 10% В за 4 мин, градиентное элюирование до 24% В за 18 мин, изократическое элюирование при 24% В за 20 мин, градиентное элюирование от 24% В до 58% В за 68 мин; определение при 254 нм]. Фракции, содержащие желаемое соединение, объединили и концентрировали в вакууме. Остаток растворили в ИПС (75 мл) и выпарили в вакууме (процедуру повторили дважды) с получением Соединения 108 (4,5 г, выход 70%, чистота 98,0%) в виде белого твердого вещества. ЖХ-МС [М+Н] 594,2 (C30H35N5O6S+H, рассчит.: 594). Время удерживания*: 3,55 мин. * - [Колонка Chromolith SpeedRod RP-18e С 18 (4,6×50 мм); скорость потока 1,5 мл/мин; подвижная фаза А: 0,1% ТФУК/вода; подвижная фаза В 0,1% ТФУК/ацетонитрил; градиентное элюирование от 5% В до 100% В за 9,6 мин, определение 254 нм.]
Синтез опиоидного пролекарства с модифицированным кетоном Пример 8: Синтез N,N'-бис(трет-бутил) N'-2-(хлоркарбонил(метил)амино)этилкарбамата
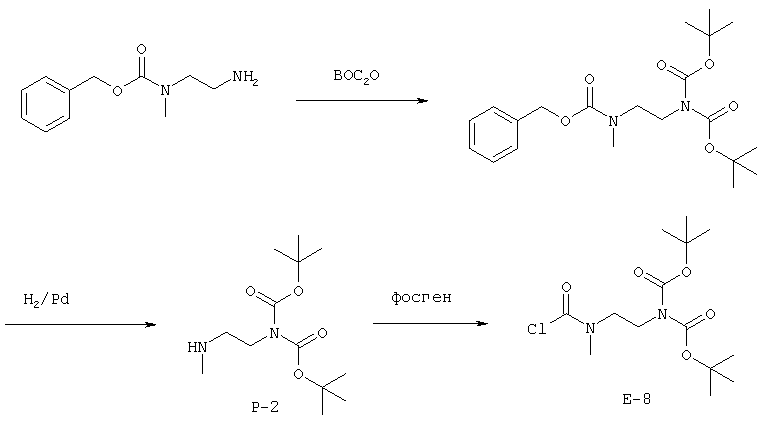
Методика 21: Синтез трет-бутилового эфира [2-(бензилокарбонил-метил-амино)-этил]-дикарбаминовой кислоты (Р-1)
Бензиловый эфир 2-(аминоэтил)-метил-карбаминовой кислоты (2,0 г, 9,6 ммоль) растворили в дихлорэтене (20 мл) при комнатной температуре. Добавили триэтиламин (NEt3) (1,40 мл, 11,5 ммоль), а затем ди-трет-бутилдикарбонат (ВОС2О) (10,5 г, 48 ммоль) и диметиламинопиридин (ДМАП) (120 мг). Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода (N2) в течение 2 ч, а затем нагревали при 60°C в течение 16 ч. Затем реакционную смесь концентрировали. Остаток очищали с помощью хроматографии на силикагеле, используя смесь гексаны-ЭА 4:1, с получением Р-1 с выходом 86% (3,4 г, 8,3 ммоль). МС: (масса/заряд) рассчит.: 408,2, наблюдаемый пик (M+Na+)431,9.
Методика 22: Синтез N1,N1-бис-ВОС-N2-метилэтан-1,2-диамина (Р-2)
Р-1 (1,3 г, 3,18 ммоль) растворили в метаноле/ЭА (10 мл/3 мл соответственно). Смесь дегазировали и насыщали N2. Добавили палладий на угле (Pd/C) (330 мг, 5% на угле). Смесь встряхивали в колбе аппарата Парра (50 psi Н2) в течение 4 ч. Затем смесь фильтровали через подушку целита и фильтрат концентрировали с образованием Р-2 (1,08 г, выход превысил расчетный). Соединение Р-2 применяли без дополнительной очистки.
Синтез N,N-бис(трет-бутил) N'-2-(хлоркарбонил(метил)амино)этилкар6амата (Е-8)
Р-2 (500 мг, 1,82 ммоль) и NEt3 (0,4 мл, 2,74 ммоль) смешали в дихлорметане(4 мл). Смесь добавили к предварительно охлажденному до 0°C раствору фосгена (5,5 мл, 0,5 мол. в толуоле). Реакционную смесь затем перемешивали при 0°C в течение 1 ч с последующим разбавлением эфиром (20 мл) и отфильтровывали на фильтровальной бумаге. Фильтрат концентрировали и пропустили через короткую колонку с силикагелем (10 см×3 см), элюировали смесью гексаны-ЭА 3:1. Фракции концентрировали с получением N,N-бис(трет-бутил) N'-2-(хлоркарбонил(метил)амино)этилкарбамата (Е-8) в виде бесцветного твердого вещества с количественным выходом (615 мг, 1,82 ммоль). МС: (масса/заряд) рассчит.: 336,1, наблюдаемый пик (M+Na+) 359,8.
Пример 9: Синтез бис-трифторацетата 6-(N-метил-N-(2-амино)этилкарбамата оксикодона
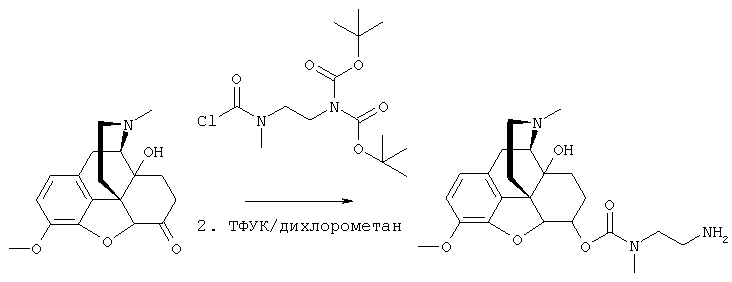
Синтез бис-трифторацетата 6-(N-метил-N-(2-амино)этилкарбамата оксикодона Свободное основание оксикодона (6,5 г, 20,6 ммоль) растворили в сухом дегазированном тетрагидрофуране (120 мл) и смесь охладили до -10°C, используя охлаждающую баню с сухим льдом/ацетоном. Бис(триметилсилил)амид калия (KHMDS) (103,0 мл, 51,6 ммоль, 0,5 мол. в толуоле) был добавлен через полую иглу. Смесь перемешивали в атмосфере N2 при температуре ниже -5°С в течение 30 мин. N,N-бис(трет-бутил) N'-2-(хлоркарбонил(метил)амино)этилкарбамат (8,0 г, 23,7 ммоль) (Е-8), приготовленный как описано в Примере 8, в ТГФ (30 мл) вводили через полую иглу в течение 15 мин. Смесь перемешивали при -5°С в течение 30 мин. Добавили другую порцию карбамоилхлорида (4,0 г, 11,9 ммоль) в ТГФ (10 мл). Реакционную смесь затем перемешивали при комнатной температуре в течение 2 ч. Добавили бикарбонат натрия (10 мл, насыщ. водн. раствор). Смесь концентрировали в вакууме до половины первоначального объема. Добавили ЭА (50 мл) и слои разделили. Далее органическую фазу промывали водой (3×20 мл), солевым раствором (40 мл) и затем концентрировали. Остаток очистили хроматографией на силикагеле, используя дихлорметан/метанол (градиент от 100/1 до 100/15) с получением белой пены с выходом 55% (7,0 г, 13,4 ммоль). Этот материал растворили в смеси дихлорметан-трифторуксусная кислота (ТФУК) 1:1 (20 мл/20 мл) при комнатной температуре и перемешивали в течение 1 ч. Раствор затем концентрировали в вакууме с получением бис-трифторацетата 6-(N-метил-N-(2-амино)этилкарбамата оксикодона в виде вязкого масла (7,3 г, 11,4 ммоля, с чистотой 99%). МС: (масса/заряд) рассчит.: 415,2, наблюдаемый пик (M+Na+) 416,5. Бис-трифторацетат 6-(N-метил-N-(2-амино)этилкарбамата оксикодона (Е-9) был затем использован без дальнейшей очистки.
Пример 10: Синтез 6-(N-метил-N-(2-N'-ацетиларгиниламино))этилкарбамата оксикодона (Соединение KC-2)
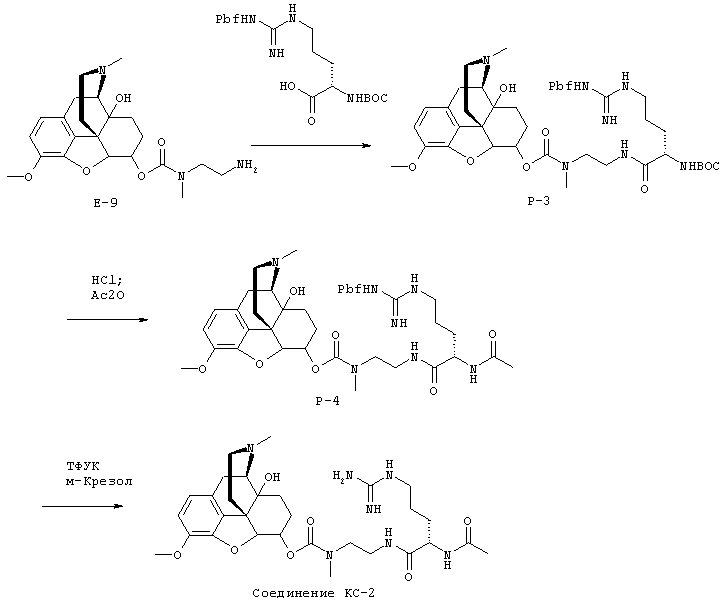
Методика 23: Синтез 6-(N-метил-N-(2-N'-Boc-аргинил(Pbf)амино))этилкарбамата оксикодона (Р-3)
Бис-трифторацетат 6-(N-метил-N-(2-амино)этилкарбамата оксикодона (7,3 г, 11,4 ммоль), приготовленный как описано в Примере 9, растворили в диметилформамиде (ДМФА) (60 мл). Boc-Arg(Pbf)-OH (6,0 г, 11,4 ммоль), HATU (4,75 г, 12,5 ммоль) и диизопропилэтиламин (ДИПЭА) (6,0 мл, 34,4 ммоль) добавили в указанном порядке. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Смесь затем концентрировали в вакууме и остаток распределили между ЭА и водой (100 мл/ 60 мл). Органический слой промыли водой (60 мл), солевым раствором (50 мл), высушили над Na2SO4, концентрировали с получением неочищенного Р-3 (11,0 г). Соединение Р-2 применяли без дополнительной очистки.
Методика 24: Синтез 6-(N-метил-N-(2-N'-ацетиларгинил(Pbf)амино))этилкарбамата оксикодона (Р-4)
Р-3 (11,0 г), приготовленный как описано выше, растворили в диоксане (10 мл) и охладили до 0°C. Добавили раствор соляной кислоты (HCl) в диоксане (4 н, 30 мл). Реакционную смесь перемешивали при комнатной температуре еще 3 час и разбавили этилацетатом (500 мл). 10 г неочищенной смеси растворили в смеси ДИПЭА (5,0 мл, 28,5 ммоль) в дихлорметане (60 мл). Уксусный ангидрид (1,4 мл, 14,3 ммоль) добавили по каплям. Реакционную смесь затем перемешивали при комнатной температуре в течение 2 ч. Затем добавили NaHCO3 (30 мл, насыщ. водн. раствор). Слои разделили и слой дихлорметана высушили над Na2SO4, отфильтровали и концентрировали с получением Р-4 (8,5 г). Соединение Р-4 применяли без дополнительной очистки.
Синтез 6-(N-метил-N-(2-N'-ацетиларгининамино))этилкарбамата оксикодона в виде бис-трифторацетата (Соединение KC-2)
Р-4 (8,5 г) растворили в смеси м-крезола (3 мл) в ТФУК (30 мл). Реакционную смесь перемешивали при комнатной температуре еще 3 час. ТФУК затем удалили под вакуумом. Остаток растворили в метаноле (10 мл) и по каплям добавили к перемешиваемому раствору HCl в эфире (40 мл, 2 мол.) Осадок отфильтровали и промыли этиловым эфиром (4×30 мл). Белое твердое вещество далее очищали препаративной ВЭЖХ (*колонка RP-18e С 18 (4,6×50 мм); скорость потока 1,5 мл/мин; подвижная фаза А: 0,1% ТФУ К/вода; подвижная фаза В 0,1% ТФУК/ацетонитрил (CH3CN); градиентное элюирование), с получением Соединения KC-2 (3,5 г, 4,1 ммоль, с чистотой 96,6%). МС: (масса/заряд) рассчит.: 613,7, наблюдаемый пик (M+Na+) 614,5.
Пример 11: Синтез N-{(S)-4-гуанидино-1-(2-(метил-((5R,9R,13S,14S)-4,5а-эпокси-6,7-дидегидро-14-гидрокси-3-метокси-17-метилморфинан-6-окси]карбонил-амино)-этилкарбамоил]-бутил}-малонамовой кислоты (Соединение KC-3)
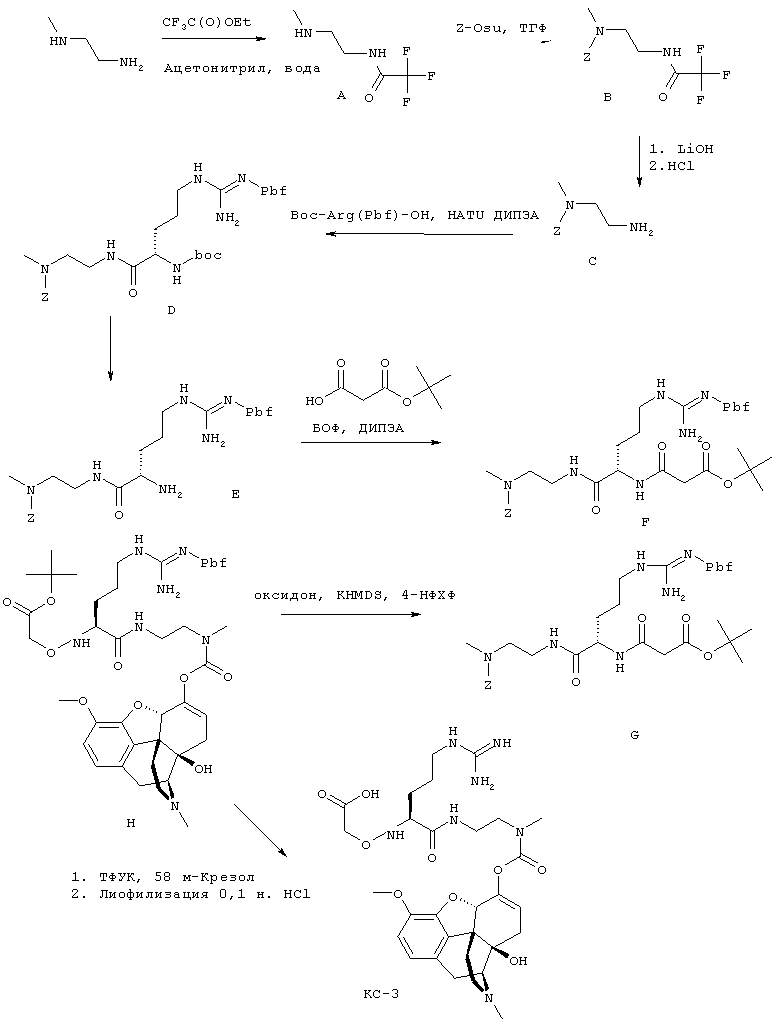
Методика 25: Синтез 2,2,2-трифтор-М-(2-метиламино-этил)-ацетамида (А)
Раствор N-метилэтилендиамина (27,0 г, 364,0 ммоль) и этилтрифторацетата (96,6 мл, 838,0 ммоль) в смеси ацетонитрила (350 мл) и воды (7,8 мл, 436 ммоль) нагревали с обратным холодильником в течение ночи с перемешиванием. Растворители выпарили в вакууме. Остаток повторно испарили с ИПС (3×100 мл) с последующей кристаллизацией путем нагрева и охлаждения из дихлорметана (500 мл). Образовавшиеся кристаллы отфильтровали, промыли дихлорметаном и высушили в вакууме с получением соединения А (88,3 г, 85%) в виде белого твердого порошка.
Методика 26: Синтез бензинового эфира метил-[2-(2,2,2-трифтор-ацетиламино)-этил]-карбаминовой кислоты (В).
Раствор соединения А (88,2 г, 311 ммоль) и ДИПЭА (54,1 мл, 311 ммоль) в ТГФ (100 мл) охладили в ледяной бане до ~5°C с последующим добавлением раствора N-(бензилоксикарбонил)сукцинимида (76,6 г, 307 ммоль) в ТГФ (150 мл) по каплям за 20 минут. Температуру реакционной смеси повысили до комнатной температуры и перемешивание продолжали еще 30 минут. Растворители выпарили и остаток растворили в ЭА (600 мл). Органический слой экстрагировали 5% водн. NaHCO3 (2 мл) и солевым раствором (150 мл). Органический слой выпарили с получением соединения B в виде желтоватого масла. ЖХ-МС [М+Н] 305,1 (C13H15F3N2O3+H, рассчит.: 305,3). Соединение В использовали сразу в следующей реакции без очистки в виде раствора в метаноле.
Методика 27: Синтез бензинового эфира (2-амино-этил)-метил-карбаминовой кислоты (С).
К раствору соединения В (~311 ммоль) в метаноле (1,2 л) добавили раствор LiOH (14,9 г, 622 ммоль) в воде (120 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Растворители выпарили до 75% начального объема с последующим разбавлением водой (400 мл). Продукт экстрагировали этилацетатом (2×300 мл). Органический слой промыли солевым раствором (200 мл), высушили над MgSO4 и выпарили в вакууме. Остаток растворили в эфире (200 мл) и обработали 2 н раствором HCl в эфире (200 мл). Образованный осадок отфильтровали, промыли эфиром и высушили в вакууме с получением хлористоводородной соли соединения С (67,8 г, 89%) в виде белого твердого вещества. ЖХ-МС [М+Н] 209,0 (C11H16N2O2+H, рассчит.: 209,3). Соединение С использовали сразу в следующей реакции без очистки в виде раствора в ДМФА.
Методика 28: Синтез бензинового эфира {2-[boc-Arg(Pbf)]-аминоэтил}-метил-карбаминовой кислоты (D).
Раствор Boc-Arg(Pbf)-OH (16,0 г, -30,4 ммоль), гидрохлорид соединения С (8,2 г, 33,4 ммоль) и ДИПЭА (16,9 мл, 97,2 ммоль) в ДМФА (150 мл) охладили в ледяной бане с последующим добавлением по каплям раствора HATU (13,8 г, 36,4 ммоль) в течение 20 мин. Температуру реакционной смеси повысили до комнатной и продолжали перемешивание еще 1 ч. Реакционную смесь разбавили этилацетатом (1 л) и экстрагировали водой (3×200 мл) и солевым раствором (200 мл). Затем органический слой высушили над MgSO4 и выпарили с получением соединения D (24,4 г, выход превысил количественный) в виде желтоватого масла. ЖХ-МС [М+Н] 717,4 (C35H52N6O8S+H, рассчит.: 717,9). Соединение D использовали сразу в следующей реакции без очистки в виде раствора в диоксане.
Методика 29: Синтез бензинового эфира {2-[H-Arg(Pbf)]- аминоэтил}-метил-карбаминовой кислоты (Е).
Соединение D (24,4 г, ~30,4 ммоль) растворили в диоксане (150 мл) и обрабатывали 4 н раствором HCl в диоксане (150 мл, 600 ммоль) при комнатной температуре в течение 1 часа. Затем растворитель выпарили. Остаток растворили в ИПС (100 мл) и выпарили в вакууме (процедуру повторили дважды). Затем остаток высушили в вакууме с получением соединения Е (21,1 г, выход превысил количественный) в виде желтоватого твердого вещества. ЖХ-МС [М+Н] 617,5 (C30H44N6O6S+H, рассчит.: 617,8). Соединение Е использовали сразу в следующей реакции без очистки в виде раствора в ДМФА.
Методика 30: Синтез бензинового эфира {2-[2-трет-бутилмалонил-Arg(Pbf)]-аминоэтил}-метил-карбаминовой кислоты (F).
Раствор соединения Е (21,1 г, -30,4 ммоль), моно-трет-бутил малоната (5,9 мл, 36,7 ммоль), ВОР (16,2 г, 36,7 ммоль) и ДИПЭА (14,9 мл, 83,5 ммоль) в ДМФА (100 мл) перемешивали при комнатной температуре 1 час. Реакционную смесь разбавили этилацетатом (1 мл) и экстрагировали водой (500 мл), 5% водн. NaHCO3 (500 мл), водой (3×500 мл) и солевым раствором (500 мл). Органический слой высушили над MgSO4, отфильтровали, а затем выпарили с получением соединения F (24,5 г, 97%) в виде желтоватого аморфного твердого вещества. ЖХ-МС [М+Н] 759,6 (C37H54N6O9S+H, рассчит.: 759,9). Соединение F применяли без дополнительной очистки.
Методика 31: Синтез N-{2-[2-трет-бутилмалонил-Arg(Pfb)]}-N'-метил-этан-1,2-диамина (G).
Соединение F (12,3 г, 16,7 ммоль) растворили в метаноле (100 мл) с последующим добавлением Pd/C (5% (вес.), 2 г) суспензии в воде (2 мл). Реакционную смесь подвергли гидрированию (аппарат Парра, 70 psi H2) при комнатной температуре в течение 1 часов. Катализатор затем отфильтровали и промыли метанолом. Фильтрат выпарили в вакууме с
получением соединения G (10,0 г, 99%) в виде бесцветного аморфного твердого вещества. ЖХ-МС [М+Н] 625,5 (C29H48H6O7S+Н, рассчит.: 625,8). Соединение G применяли без дополнительной очистки.
Методика 32: Свободное основание оксикодона
Гидрохлорид оксикодона (10,0 г, 28,5 ммоль) растворили в хлороформе (150 мл) и промыли 5% водн. NaHCO3 (50 мл). Органический слой высушили над MgSO4 и выпарили. Остаток сушили в вакууме в течение ночи с получением свободного основания оксикодона (8,3 г, 93%) в виде белого твердого вещества.
Методика 33: Синтез трет-бутилового эфира N-{(S)-4-(2,2,4,6,7-пентаметилдигидробензофуран-5-сульфонил-гуанидино)-1-[2-(метил-[(5R,9R,13S,14S)-4,5a-эпокси-6,7-дидегидро-14-гидрокси-3-метокси-17-метилморфинан-6-окси]карбонил-амино)-этилкарбамоил]-бутил}-малонамовой кислоты (Н).
Раствор свободного основания оксикодона (6,6 г, 21,0 ммоль) в ТГФ (400 мл) охладили до -20°C, затем добавили 0,5 мол. раствор KHMDS в толуоле (46,3 мл, 23,1 ммоль). Полученный раствор по каплям добавили к раствору 4-нитро-фенилхлорформиата (4,3 г, 21,0 ммоль) в ТГФ (100 мл) в течение 20 мин при -20°C. Реакцию проводили при -20°C в течение еще 1 часа с последующей добавкой раствора соединения G (10,0 г, 16,1 ммоль) в ТГФ (200 мл) при -20°C. Реакционной смеси дали нагреться до комнатной температуры и перемешивали в течение ночи. Растворители выпарили в вакууме. Полученный остаток растворили в ЭА (20 мл) и осадили эфиром (1 л). Образовавшийся осадок отфильтровали, промыли эфиром и высушили в вакууме с получением соединения Н (13,6 г, 87%) в виде грязно-белого твердого вещества. ЖХ-МС [М+Н] 966,9 (C48H67N7O12S+H, рассчит.: 966,2).
Синтез N-{(S)-4-гуанидино-1-[2-(метил-[(5R,9R,13S,14S)-4,5а-эпокси-6,7-дидегидро-14-гидрокси-3-метокси-17-метилморфинан-6-окси]карбонил-амино)-этилкарбамоил]-бутил}-малонамовой кислоты (Соединение KC-3)
Соединение Н (13,6 мг, 14,1 ммоль) растворили в смеси 5% м-крезола в ТФУК (100 мл). Реакционную смесь выдерживали при комнатной температуре в течение 1 часа с последующим разбавлением этиловым эфиром (1 л). Образованный осадок отфильтровали, промыли эфиром и гексаном, и высушили в вакууме с получением трифторацетата Соединения КС-3 (11,4 г, 81%) в виде грязно-белого твердого вещества. ЖХ-МС [М+Н] 658,6 (C31H43N7O9S+H, рассчит.: 658,7).
Трифторацетат неочищенного Соединения KC-3 (11,4 г, 11,4 ммоль) растворили в воде (50 мл). Полученный раствор очистили с использованием ВЭЖХ. [колонка Nanosyn-Pack YMC-GEL-ODS A (100-10) С-18 (75×500 мм); скорость потока: 250 мл/мин; объем впрыска 50 мл; подвижная фаза А: 100% вода, 0,1% ТФУК; подвижная фаза В: 100% ацетонитрил, 0,1% ТФУК; изократическое элюирование при 0% В за 4 мин, градиентное элюирование от 0% до 10% В за 20 мин, изократическое элюирование при 10% В за 30 мин, градиентное элюирование от 10% В до 30% В за 41 мин; определение при 254 нм]. Фракции, содержащие Соединение KC-3, объединили и концентрировали в вакууме. Противоион ТФУК в последнем соединении заменили противоионом HCl путем лиофилизации с использованием 0,1 н HCl с получением гидрохлорида Соединения KC-3 (4,2 г, выход 41%) в виде белового твердого вещества. ЖХ-МС [М+Н] 658,6 (C31H43N7O9S+H, рассчит.: 658,7).
Пример 12: Синтез N-((S)-1-{2-[(дигидрокодеин-6-енилоксикарбонил)-метиламино]-этилкарбамоил-4-гуанидино}-бутил-малонамовой кислоты (Соединение KC-4)
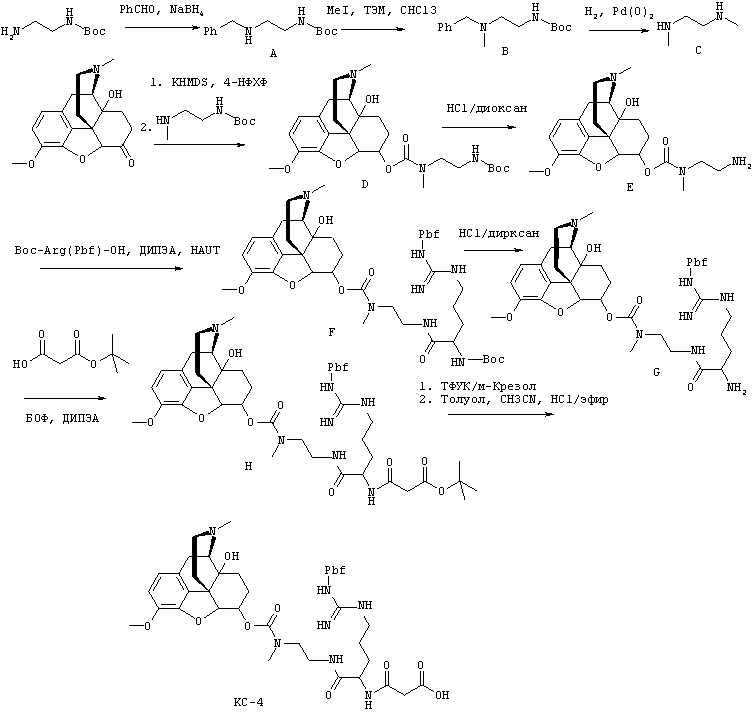
Методика 34: Синтез трет-бутил 2-(бензиламино)этилкарбамата (А)
К раствору трет-бутил 2-аминоэтилкарбамата (6,4 г, 40,0 ммоль) в метаноле (60 мл) добавили бензальдегид (4,7 г, 44,0 ммоль) и молекулярные сита 3Å. После перемешивания при комнатной температуре в течение ночи смесь охладили примерно до -10°С (баня со льдом и солью) и порциями обрабатывали с применением NaBH4 (9,1 г, 240,0 ммоль) в течение 30 мин. После добавления всего раствора баню убрали и реакционную смесь перемешивали при комнатной температуре 16 ч. Раствор выпарили, остаток перенесли в ЭА (150 мл) и вылили в воду (100 мл). Органический слой экстрагировали 0,5 н раствором HCl (3 х 100 мл). Объединенные водные растворы охладили до 0°С, защелочили насыщенным NaHCO3 и экстрагировали хлороформом (3×100 мл). Объединенные органические слои промыли солевым раствором (200 мл). После высушивания над MgSO4 и фильтрования растворитель выпарили под вакуумом с получением Соединения А (9,2 г, 36,8 ммоль, 92%) в виде бесцветного масла. ЖХ-МС [М+Н] 251,2 (C14H22N2O2S+H, рассчит.: 251,3). ТСХ Rf (дихлорметан/метанол 9:1): 0,30. Соединение А применяли без дополнительной очистки.
Методика 35: Синтез трет-бутил 2-(N-бензил-N-метиламино)этилкарбамата (В)
К охлажденному (~5°С) раствору Соединения А (6,2 г, 25,0 ммоль) и ТЭА (3,0 г, 29,7 ммоль, 4,13 мл) в хлороформе (50 мл) добавили иодметан (4,2 г, 29,7 ммоль, 1,85 мл). Сосуд для работы под давлением закрыли и смесь перемешивали при комнатной температуре 20 ч. Затем осадок из смеси высадили эфиром (300 мл); белое твердое вещество отфильтровали и промыли эфиром (50 мл). Фильтрат сконцентрировали и полученное желтое масло (5,2 г) очистили на хроматографической колонке с силикагелем (2-10% градиент метанола в дихлорметане) с получением соединения В (3,3 г, 12,5 ммоль, 50%) в виде бесцветного масла. ТСХ Rf (дихлорметан/метанол 9:1); 0,55. ЖХ-МС [М+Н] 264,3 (C15H24N2O2S+H, рассчит.: 264,4).
Методика 36: Синтез трет-бутил 2-(метиламино)этилкарбамата (С)
В колбу добавили 20% Pd(OH)2 на угле (3,1 г), соединение В (3,3 г, 12,5 ммоль) в метаноле (200 мл) и воду (10 мл), и гидрировали в атмосфере НЬ под давлением (40 psi). Через 2,5 часа реакционную смесь отфильтровали через целит и сконцентрировали в вакууме. Затем добавили воду (50 мл) и смесь довели до pH 12 (добавлением 1 н NaOH) и экстрагировали дихлорметаном (3×50 мл). Объединенные органические слои высушили над MgSO4, отфильтровали и сконцентрировали в вакууме с получением соединения С (2,0 г, 11,7 ммоль, 94%) в виде бесцветного масла. ЖХ-МС [М+Н] 686,5 (C35H51N5O7S+H, рассчит.: 685,9). Соединение С применяли без дополнительной очистки.
Методика 37: Синтез трет-бутилового эфира [2-(N-дигидрокодеин-6-енилоксикарбонил-N-метиламино)-этил]-карбаминовой кислоты (D)
К охлажденному (~5°С) раствору гидрокодона (2,9 г, 9,8 ммоль, свободное основание) в безводном ТГФ (150 мл) по каплям добавили 0,5 мол. раствор KHMDS в толуоле (11,6 ммоль, 23,3 мл) в течение 20 мин. Желтый раствор перемешивали при этой температуре 30 мин. Раствор через полую иглу добавили к охлажденному раствору (-30°С) 4-нитрофенилхлорформиата (1,9 г, 9,5 ммоль) в безводном ТГФ (40 мл) в течение 15 мин. Баню убрали и смесь перемешивали при комнатной температуре 15 мин, после чего по каплям добавляли раствор соединения С (2,3 г, 11,6 ммоль) в безводном ТГФ (15 мл) в течение 10 мин. После перемешивания при комнатной температуре в течение 18 ч реакционную смесь погасили насыщенным раствором NaHCO3 (7 мл). Полученный осадок отфильтровали, промыли этилацетатом (30 мл) и фильтрат концентрировали в вакууме. Остаток перенесли в ЭА (300 мл) и промыли смесью воды (100 мл) и 2% водн. H2SO4 (30 мл). Водный слой обработали 2 н раствором NaOH до pH 12 и экстрагировали этилацетатом (2×200 мл). Объединенные органические слои промыли водой (2×400 мл) и солевым раствором (300 мл), высушили над MgSO4, отфильтровали и концентрировали в вакууме с получением желтоватого пенообразного твердого вещества (5,9 г), которое очистили с помощью ВЭЖХ. [колонка Nanosyn-Pack Microsorb (100-10) С-18 (50×300 мм); скорость потока: 100 мл/мин; объем впрыска: 65 мл; подвижная фаза А: 100% вода, 0,1% ТФУК; подвижная фаза В: 100% ацетонитрил, 0,1% ТФУК; изократическое элюирование при 10% В за 5 мин, градиентное элюирование до 18% В за 8 мин, изократическое элюирование при 18% В за 20 мин, градиентное элюирование от 18% В до 40% В за 44 мин; определение при УФ 254 нм]. Фракции, содержащие желаемое соединение, объединили и концентрировали в вакууме. Следы воды удалили обработкой остатка толуолом (30 мл) с последующим выпариванием в вакууме (процедуру повторили дважды). Выделенные фракции представляют собой смесь 1:1 соединения D и соединения Е с удаленной boc защитой (4,37 г, 7,85 ммоль, 83%). ЖХ-МС [М+Н] 500,2 (C27H37N3O6+H, рассчит.: 500,6). Время удерживания [колонка Chromolith SpeedRod RP-18е С 18 (4,6×50 мм); скорость потока: 1,5 мл/мин; подвижная фаза А: 0,1% ТФУК/вода; подвижная фаза В 0,1% ТФУК/ацетонитрил; градиентное элюирование от 5% В до 100% В за 9,6 мин, определение 254 нм]. 3,52 мин (соединение D), 1,82 (соединение Е).
Методика 38: Синтез дигидрокодеин-6-енил-2-аминоэтилметилкарбамата (Е)
Раствор соединения D (4,4 г, 8,8 ммоль) в дихлорметане (40 мл) обработали 4 мол. раствором HCl в диоксане (105 ммоль, 26 мл), что привело к образованию небольшого осадка. Смесь гомогенизировали добавлением ацетонитрила (20 мл) и перемешивали при комнатной температуре 45 мин. Добавили эфир (400 мл) и образовавшийся белый осадок отфильтровали, промыли эфиром (50 мл) и гексаном (50 мл) и затем высушили в вакууме с образованием соединения Е в виде грязно-белого твердого вещества (2,4 г, 4,7 ммоль, 58%). ЖХ-МС [М+Н] 400,3 (C22H29N3O4+H, рассчит.: 400,5). Соединение Е применяли без дополнительной очистки.
Методика 39: Синтез гидрокодонового эфира {2-[boc-Arg(Pbf)]-аминометил}-этил-карбаминовой кислоты (F)
Соединение Е (2,0 г, 4,0 ммоль). Boc-Arg(Pbf)-OH (2,0 г, 3,8 ммоль) и HATU (1,7 г, 4,3 ммоль) растворили в ДМФА (40 мл), довели до ~5°С и по каплям обработали ДИПЭА (3,2 мл, 18,1 ммоль) в течение 10 мин. Реакционную смесь перемешивали при ~5°С еще 10 мин и затем нагрели до комнатной температуры с последующим перемешиванием 30 мин. Реакционную смесь затем разбавили этилацетатом (200 мл) и вылили в воду (250 мл). Слои разделили, водный экстрагировали этилацетатом (2×150 мл), и объединенные органические слои промыли 2% водн. H2SO4 (30 мл), водой (2×250 мл) и солевым раствором (250 мл). Органический слой высушили над MgSO4, отфильтровали, а затем концентрировали в вакууме с получением соединения F (3,0 г, 3,2 ммоль, 83%) в виде желтоватого пенообразного твердого вещества. ЖХ-МС [М+Н] 908,7 (C46H65N7O10S+Н, рассчит.: 909,1). Соединение F применяли без дополнительной очистки.
Методика 40: Синтез гидрокодонового эфира {2-[H-ArG(Pbf]-аминометил}-этил-карбаминовой кислоты (G)
Раствор соединения F (3,0 г, 3,3 ммоль) в дихлорметане (20 мл) обработали 4 мол. раствором HCl в диоксане (39 ммоль, 9,8 мл) и перемешивали при комнатной температуре 30 мин. Добавили эфир (500 мл) и образовавшийся белый осадок отфильтровали, промыли эфиром (50 мл) и гексаном (50 мл) и затем высушили в вакууме с образованием соединения G в виде грязно-белого твердого вещества (2,7 г, 3,0 ммоль, 93%). ЖХ-МС [М+Н] 808,7 (C41H57N7O8S+H, рассчит.: 809,0). Соединение G применяли без дополнительной очистки.
Методика 41: Синтез трет-бутилового эфира N-((S)-1-{2-[(дигидрокодеин-6-енилоксикарбонил)-метиламино]-этилкарбамоил-4-гуанидино}-бутил-малонамовой кислоты (Н)
К охлажденному (~5°С) раствору соединения G (2,7 г, 3,0 ммоль) добавили моно (трет-бутил)малонат (474 мг, 3,0 ммоль, 438 мкл) в ДМФА (25 мл), а затем ВОР (1,4 г, 3,2 ммоль) в течение 5 мин и, наконец, по каплям ДИПЭА (1,6 г, 12,1 ммоль, 2,1 мл) в течение 10 мин. После дополнительных 15 мин ледяную баню убрали и смесь перемешивали при комнатной температуре. Через 45 минут реакционную смесь разбавили этилацетатом (300 мл) и вылили в Н2О (200 мл). Слои разделили и водный слой экстрагировали этилацетатом (2×250 мл). Объединенные органические слои промыли водой (500 мл), 2% водн. H2SO4 (100 мл), водой (3×500 мл) и солевым раствором (2×500 мл). После высушивания над MgSO4 растворитель выпарили в вакууме и остаток высушили под высоким вакуумом с получением Соединения Н (1,7 г, 1,8 ммоль, 58%) в виде желтоватого твердого вещества. ЖХ-МС [М+Н] 950,8 (C48H67N7O11S+H. рассчит.: 951,2). Соединение Н применяли без дополнительной очистки.
Синтез N-((S)-1-{2-[(дигидрокодеин-6-внилоксикарбонил)-метиламино]-этилкар6амоил-4-гуанидино}-бутил-малонамовой кислоты (Соединение KC-4)
Раствор соединения Н (1,7 г, 1,8 ммоль) в 5% растворе м-крезола в ТФУК (45 мл) перемешивали при комнатной температуре. Через 1 час смесь разбавили эфиром (300 мл). Полученную тонкую суспензию отфильтровали, осадок промыли эфиром (30 мл) и гексаном (30 мл) и сушили под вакуумом 15 мин. Неочищенный материал растворили в воде (35 мл) и очистили, используя ВЭЖХ [колонка Nanosyn-Pack Microsorb (100-10) С-18 (50×300 мм); скорость потока: 100 мл/мин; объем впрыска: 35 мл; подвижная фаза А: 100% вода, 0,1% ТФУК; подвижная фаза В: 100% ацетонитрил, 0,1% ТФУК; градиентное элюирование от 0 до 10% В за 10 мин, изократическое элюирование при 10% В за 20 мин, градиентное элюирование от 10% В до 42% В за 60 мин; определение при УФ 254 нм]. Фракции, содержащие желаемое соединение, объединили и концентрировали в вакууме. Остаток обработали толуолом (50 мл) для удаления следов воды и совместно выпарили в вакууме (процедуру повторили дважды). Остаток растворили в ацетонитриле (5 мл), обработали 2,0 М раствором HCl в эфире (20 мл) и затем разбавили эфиром (100 мл). Полученное твердое вещество отфильтровали, промыли эфиром(20 мл) и гексаном (20 мл) и сушили под вакуумом в течение ночи с получением Соединения КС-4 (1,1 г, выход 86%) в виде белого твердого вещества - гидрохлорида. ЖХ-МС [М+Н] 642,5 (C31H43N7O8+H, рассчит.: 642,7). Чистота >95% (УФ/254 нм). Время удерживания [колонка Chromolith SpeedRod RP-18e С 18 (4,6×50 мм); скорость потока: 1,5 мл/мин; подвижная фаза А: 0,1% ТФУК/вода; подвижная фаза В 0,1% ТФУК/ацетонитрил; градиентное элюирование от 5% В до 100% В за 9,6 мин, определение 254 нм]: 2,24 мин.
Пример 13: Синтез 6-{[2-(2-ацетиламино-5-гуанидино-пентаноиламино)-этил]-[(5R,9R,13S,14S)-4,5а-эпокси-6,7-дидегидро-14-гидрокси-3-метокси-17-метилморфинан-6-окси]-1-енилоксикарбонил-амино}-капроновой кислоты (Соединение KC-5)
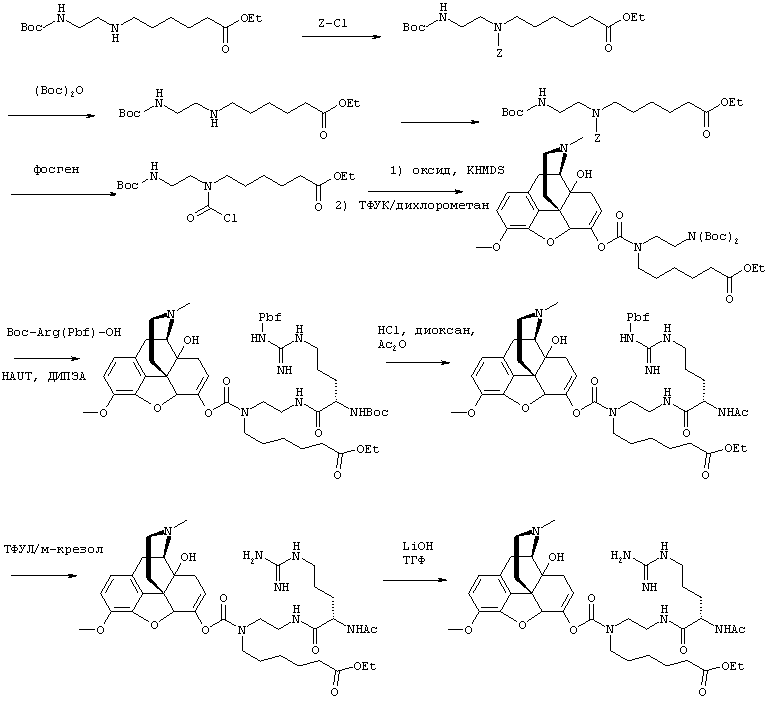
Методика 42: Синтез этилового эфира 6-[бензилоксикарбонил-(2-трет-бутоксикарбониламино-этил)-амино]-капроновой кислоты (В)
Соединение А (26,8 г, 88,6 ммоль) растворили в дихлорметане (200 мл) при комнатной температуре. Добавили NEt3 (12,5 мл, 88,6 ммоль), а затем Cbz-Cl (Z-Cl) (12,5 мл, 88,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в атмосфере N2 в течение 2 часов. Реакционную смесь обработали NaHCO3 (30 мл, водн. насыщ.). Слои разделили и органический слой высушили над MgSO4, отфильтровали и сконцентрировали. Остаток очистили с помощью хроматографии на силикагеле, используя смесь гексаны-ЭА 4:1, с получением Соединения B в виде бесцветного масла (22,5 г, 66,5 ммоль, 75%).
Методика 43: Синтез промежуточного продукта (С)
Соединение В (22,0 г, 50,4 ммоль) растворили в дихлорэтане (100 мл) при комнатной температуре. Добавили NEt3 (8,5 мл, 61 ммоль), затем (Boc)2О (33,0 г, 151,2 ммоль) и ДМАП (615 мг, 5,0 ммоль). Реакционную смесь перемешивали при комнатной температуре в атмосфере N2 в течение 2 часов, а затем нагревали при 60°С 16 часов.
Реакционную смесь упарили и остаток очистили хроматографией на силикагеле, используя смесь гексаны-ЭА 4:1, с получением соединения С в виде бесцветного масла (23,2 г, 41,9 ммоль, 86%). МС: (масса/заряд) рассчит.: 536,6, наблюдаемый пик (M+Na+) 560,1.
Методика 44: Синтез промежуточного продукта (D)
Соединение С (22,5 г, 41,9 ммоль) растворили в этаноле (50 мл). Смесь дегазировали и насыщали N2. Добавили Pd/C (500 мг, 5% на угле). Смесь встряхивали в колбе аппарата Парра (2 psi H2) в течение 2 ч. Затем смесь отфильтровали через подушку целита и фильтрат концентрировали с образованием соединения D в виде бесцветного масла(21,0 г, 52,2 ммоль, неочищенное). Этот материал применяли без дополнительной очистки.
Методика 45: Синтез промежуточного продукта (Е)
Соединение D (21,0 мг, 52,2 ммоль) и NEt3 (11,0 мл, 78,3 ммоль) смешали в дихлорметане (150 мл). Смесь добавили к предварительно охлажденному (водяная баня со льдом) раствору фосгена в толуоле (41,2 мл, 20% (вес.) в толуоле, ~83,3 ммоль). Реакционную смесь перемешивали при 0°С в течение 2 ч. Затем ее сконцентрировали до одной трети ее первоначального объема и разбавили эфиром (50 мл). Смесь отфильтровали на бумажном фильтре. Фильтрат сконцентрировали с получением соединения Е в виде белого твердого вещества (20,0 г, 43,1 ммоль, 82%) МС: (масса/заряд) рассчит.: 464,2, наблюдаемый пик (M+Na+) 487,7. Соединение Е применяли без дополнительной очистки.
Методика 46: Синтез промежуточного продукта (F)
Свободное основание оксикодона (1,0 г, 3,2 ммоль) растворили в сухом ТГФ (15 мл) и смесь охладили до -10°С, используя охлаждающую баню с сухим льдом/ацетоном. Бис(триметилсилил)амид калия (KHMDS) (7,6 мл, 3,8 ммоль, 0,5 мол. в толуоле) был добавлен из шприца. Смесь перемешивали в атмосфере N2 при температуре -5°С в течение 30 мин. Соединение Е (1,5 г, 3,2 ммоль) в ТГФ (10 мл) затем добавили из шприца в течение 5 мин. Смесь перемешивали при -5°С в течение 30 мин. Реакцию продолжали при комнатной температуре в течение 2 ч. Добавили NaHCO3 (10 мл, насыщ. водн.). Смесь концентрировали в вакууме до половины первоначального объема. Добавили ЭА (20 мл) и слои разделили. Органическую фазу дополнительно промыли водой (20 мл) и солевым раствором (20 мл), затем сконцентрировали и полученный остаток очистили хроматографией на силикагеле (дихлорметан/метанол (градиент от 100/1 до 100/15)) с получением бесцветного масла (~1,7 г, 3,1 ммоль, 97%). Этот материал растворили в смеси дихлорметан/ТФУК (5 мл/5 мл) при комнатной температуре и перемешивали в течение 1 ч. Затем концентрировали в вакууме с получением соединения F в виде его трифторацетата (1,8 г, 2,7 ммоль, 88%). МС: (масса/заряд) рассчит.: 543,7, наблюдаемый пик (M+Na+) 545,2. Соединение F применяли без дополнительной очистки.
Методика 47: Синтез промежуточного продукта (G)
Соединение F (1,8 г, 2,6 ммоль) растворили в ДМФА (20 мл) при перемешивании. Boc-Arg(Pbf)-OH (1,4 г, 11,4 ммоль), HATU (1,1 г, 2,9 ммоль) и ДИПЭА (1,4 мл, 8,0 ммоль) добавили при перемешивании. Реакцию продолжали при комнатной температуре в течение 2 ч. Смесь затем концентрировали и остаток распределили между ЭА и водой (30 мл/ 20 мл). Органический слой отделили, промыли водой (20 мл), солевым раствором (20 мл), высушили над Na2SO4, концентрировали с получением неочищенного соединения G (1,5 г, 1,4 ммоль, 54%). МС: (масса/заряд) рассчит.: 1052,3, наблюдаемый пик (M+Na+) 1053,9. Соединение G применяли без дополнительной очистки.
Методика 48: Синтез промежуточного продукта (Н)
Неочищенное соединение G (1,5 г, 1,4 ммоль) перенесли в диоксан (3 мл) и охладили в водяной бане со льдом. Добавили раствор HCl в диоксане (4 н, 10 мл, 40 ммоль) и смесь перемешивали при комнатной температуре 3 ч. и затем концентрировали в вакууме с получением белой пены. Этот материал растворили в смеси ДИПЭА (0,8 мл, 4,3 ммоль) в дихлорметане (20 мл). Уксусный ангидрид (0,2 мл, 2,1 ммоль) добавили по каплям. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Затем добавили NaHCO3 (20 мл, насыщ. водн. раствор). Слои разделили и слой дихлорметана высушили над Na2S04, отфильтровали и концентрировали с получением промежуточного соединения Н (0,85 г, неочищенный). Соединение Н применяли без дополнительной очистки.
Синтез 6-{[2-(2-ацетиламино-5-гуанидино-пентаноиламино)-этил]-[(5R,9R,13S,14S)-4,5а-эпокси-6,7-дидегидро-14-гидрокси-3-метокси-17-метилморфинан-6-окси]-1-енилоксикарбонил-амино}-капроновой кислоты (Соединение KC-5)
Соединение Н (0,85 г, неочищенное) растворили в смеси м-крезола (0,5 мл) в ТФУК (20 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Смесь концентрировали под вакуумом. Остаток перенесли в метанол (3 мл) и по каплям добавили к перемешиваемому раствору HCl в эфире (20 мл, 2 М, 40 ммоль). Полученное белое твердое вещество (соединение I) отфильтровали и промыли этиловым эфиром (3×10 мл). Соединение I растворили в смеси ТГФ/вода (2 мл/ 2 мл) при комнатной температуре. LiOH (41 мг, 1,7 ммоль) добавили одной порцией. Смесь перемешивали 4 ч. Затем смесь подкислили добавлением уксусной кислоты до pH ~6. Смесь затем концентрировали и остаток очистили с помощью препаративной ВЭЖХ, используя колонку RP-18e С 18 (4,6×50 мм); скорость потока: 1,5 мл/мин; подвижная фаза А: 0,1% ТФУК/вода; подвижная фаза В 0,1% ТФУК/ацетонитрил; градиентное элюирование. Лиофилизацией собранных фракций получили Соединение КС-5 (трифторацетат) в виде белого твердого вещества. Твердое вещество обработали 0,1 н раствором HCl (водн.) и лиофилизировали с образованием соответствующего гидрохлорида Соединения KC-5 в виде белой пены (406 мг, 38% на соединение Е, 100% чистота) МС: (масса/заряд) рассчит.: 713,8, наблюдаемый пик (M+Na+) 714,5.
Пример 14: ({(S)-2-((S)-2-ацетиламино-5-гуанидино-пентаноиламино)-3-[(оксикодон-енилоксикарбонил)-метил-амино]-пропионил}-метил-амино)-уксусная кислота (Соединение KC-6)
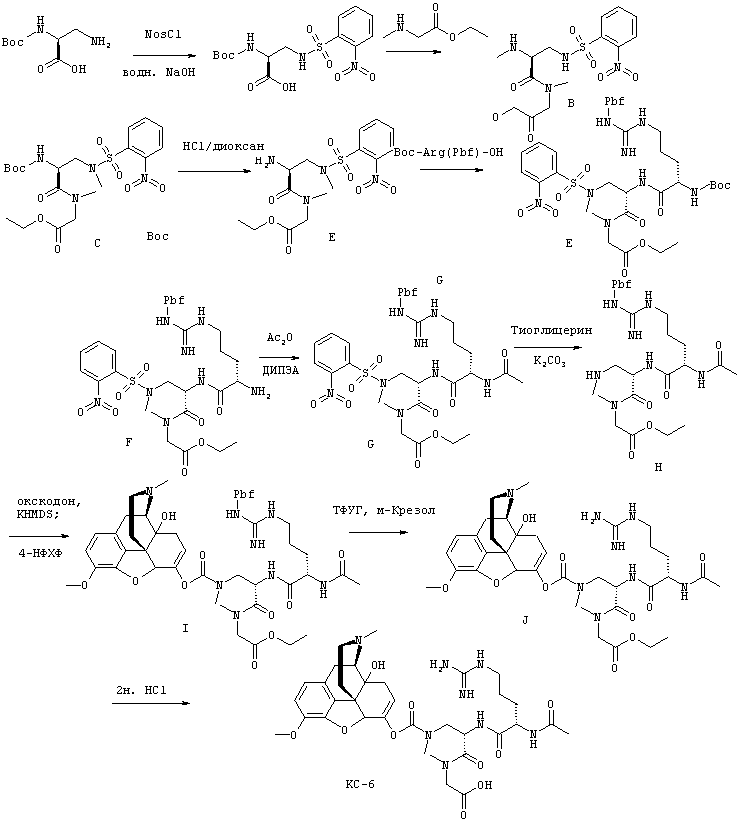
Методика 49: (8)-2-трет-бутоксикарбониламино-3-(2-нитро-6ензолсулъфониламино)-пропионовая кислота (А).
(8)-2-трет-бутоксикарбониламино-3-пропионовую кислоту (14,9 г, 73,2 ммоль) растворили в смеси ТГФ (45 мл) и 3 н водном растворе NaOH (45 мл). Реакционную смесь охладили до -10°С и по каплям добавили нозилхлорид (17,9 г, 80,5 ммоль) в виде раствора в ТГФ в течение 30 мин. Реакционную смесь перемешивали при -10°С в течение 45 мин с последующим перемешиванием при комнатной температуре в течение 30 мин. Реакционную смесь разбавили водой (150 мл), подкислили 2% водной H2SO4 (до pH ~2) и дополнительно разбавили водой (450 мл). Продукт экстрагировали этилацетатом (общий объем 600 мл) и промыли водой (3×400 мл) и солевым раствором (100 мл). Органический слой отделили, высушили над Na2SO4, отфильтровали и сконденсировали в вакууме с получением соединения А (20,0 г, 70% выход) в виде твердого вещества кремового цвета. ЖХ-МС [М+Н] 290,3 (C14H19N3O8S+H, рассчит.: 390,4). Чистота >95% (УФ/254 нм). Соединение А применяли без дополнительной очистки.
Методика 50: Этиловый эфир{[(S)-2-трет-бутоксикарбониламино-3-(2-нитро-бензолсулъфониламино)-пропионил]-метил-амино}-уксусной кислоты (В).
Метод получения свободного основания из этилового эфира саркозина. Гидрохлорид этилового эфира саркозина (39,3 г, 256,8 ммоль) растворили в воде (300 мл), промыли диэтиловым эфиром (2×100 мл), pH довели до ~8, экстрагировали хлороформом (3×100 мл), высушили над Na2SO4 и в конце отфильтровали.
К раствору соединения А (10,0 н, 25,7 ммоль) в ДМФА (100 мл) добавили HOBt (5,2 г, 38,5 ммоль) и реакционную смесь охладили до -10°С. К этой реакционной смеси порциями в течение 10 мин. добавили EDC-HCl (5,4 г, 28,2 ммоль) и перемешивали при -10°С в течение 20 мин. К реакционной смеси по каплям в течение 30 мин. добавили этиловый эфир саркозина (256,8 ммоль) в хлороформе (300 мл). Реакционную смесь перемешивали при этой температуре 30 мин., а затем при комнатной температуре в течение ночи. Затем растворители удалили в вакууме и остаток растворили в ЭА (500 мл), промыли водой (3×300 мл), насыщенным водным NaHCO3 (2×300 мл) и солевым раствором (100 мл). Органический слой отделили, высушили над Na3SO4 и сконцентрировали в вакууме с получением соединения В (11,5 г, 91% выход) в виде твердого вещества кремового цвета. ЖХ-МС [М+Н] 489,5 (C19H28N4O9S+H, рассчит.: 489,3). Чистота >95% (УФ/254 нм). Соединение В применяли без дополнительной очистки.
Методика 51: Этиловый эфир ({(S)-2-трет-бутоксикарбониламино-3-[метил-(2-нитро-бензолсульфонил)-амино)-пропионил]-метил-амино}-уксусной кислоты (С).
Соединение В (8,0 г, 16,3 ммоль) растворили в ДМФА (40 мл) и реакционную смесь охладили до -10°С. К реакционной смеси добавили K2CO3 (6,8 г, 49,1 ммоль), затем по каплям метилиодид (5,1 мл, 81,9 ммоль) и перемешивали при 0°С в течение 1 ч. Реакционную смесь отфильтровали и промыли этилацетатом. Растворители удалили в вакууме, и остаток растворили в ЭА (250 мл) и вылили в воду (500 мл), экстрагировали этилацетатом (2×250 мл), промыли водой (250 мл) и солевым раствором (100 мл). Органический слой высушили над Na2SO4, отфильтровали и затем сконцентрировали в вакууме с получением соединения С (8,1 г, 98% выход) в виде твердого вещества кремового цвета. ЖХ-МС [М+Н] 503,1 (C20H30H4O9S+Н, рассчит.: 503,5). Чистота >95% (УФ/254 нм). Соединение С применяли без дополнительной очистки.
Методика 52: Этиловый эфир ({(S)-2-амино-3-[метил-(2-нитро-бензолсульфонил)-амино]-пропионил}-метил-амино)-уксусной кислоты (D).
Соединение С (6,9 г, 13,8 ммоль) растворили в дихлорметане (45 мл) и обработали 4 М раствором HCl в диоксане (40 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 90 мин. Смесь сконцентрировали в вакууме до общего объема -25 мл, и добавили ДЭ (400 мл). Выпавший в осадок продукт отфильтровали, промыли диэтиловым эфиром (250 мл) и гексаном (250 мл) и окончательно высушили в вакууме с получением соединения D (6,3 г, выход 100%) в виде твердого вещества кремового цвета. ЖХ-МС [М+Н] 403,3 (C15H22N4O7S+H, рассчит.: 403,4). Чистота >95% (УФ/254 нм). Соединение D применяли без дополнительной очистки.
Методика 53: Этиловый эфир ({(S)-2-[(S)-5-({амино-[(Z)-2,2,4,6,7-пентаметил1-2,3-дигидро-бензофуран-5-сульфонилимино]-метил}-амино)-2-трет-бутоксикарбониламино-пентаноиламино]-3-[метил-(2-нитро-бензолсульфонил)-амино]-пропионил}-метил-амино)-уксусной кислоты (Е).
К раствору Boc-Arg(Pbf)-OH (7,3 г, 13,8 ммоль), ДИПЭА (7,7 мл, 44,2 ммоль) в ДМФА (35 мл) добавили HATU (5,8 г, 15,2 ммоль) и перемешивали при 5°С в течение 15 мин. К этой реакционной смеси добавили соединение D (6,3 г, 13,8 ммоль) и перемешивали при комнатной температуре 1 ч. Затем ДМФА удалили в вакууме до общего объема ~15 мл. Реакционную смесь разбавили этилацетатом (250 мл) и вылили в воду (500 мл), экстрагировали этилацетатом (2×250 мл) и промыли 2% водной H2SO4 (150 мл), водой (150 мл) и солевым раствором (150 мл). Органический слой высушили над безводным Na2SO4,отфильтровали и затем выпарили с получением маслянистого остатка, который сушили в течение ночи в высоком вакууме с получением соединения Е (7,4 г, 59%) в виде грязно-белого твердого вещества. ЖХ-МС [М+Н] 911,5 (C39H58N8O13S+H, рассчит.: 912,05). Чистота >95% (УФ/254 нм). Соединение Е применяли без дополнительной очистки.
Методика 54: Этиловый эфир ({(S)-2-[(S)-2-амино-5-({амино-[(2)-2,2,4,6,7-пентаметил-2,3-дигидро-бензофуран-5-сульфонилимино]-метил}-амино)-пентаноиламино]-3-[метил-(2-нитро-бензолсульфонил)-амино]-пропионил}-метил-амино)-уксусной кислоты (F).
Соединение Е (7,4 г, 8,2 ммоль) в дихлорметане (24 мл) обработали 4 М раствором HCl в диоксане (24 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Дихлорметан и основную часть диоксана удалили в вакууме до общего объема -15 мл, и добавили ДЭ (300 мл). Выпавший в осадок продукт отфильтровали, промыли диэтиловым эфиром (150 мл) и гексаном и окончательно высушили в вакууме с получением соединения F (6,3 г, выход 100%) в виде твердого вещества кремового цвета. ЖХ-МС [М+Н] 811,4 (C34H50N8O11S+H, рассчит.: 811,94). Чистота >95% (УФ/254 нм). Соединение F применяли без дополнительной очистки.
Методика 55: Этиловый эфир ({(S)-2-[(8)-2-ацетиламино-5-({амино-[(Z)-2,2,4,6,7-пентаметил-2,3-дигидро-бензофуран-5-сульфонилимино]-метил}-амино)-пентаноиламино]-3-[метил-(2-нитро-бензолсульфонил)-амино]-пропионил}-метиламино)-уксусной кислоты (G).
К раствору соединения F (6,6 г, 7,8 ммоль) в хлороформе (50 мл) при 5°С добавили ДИПЭА (4,8 мл, 27,4 ммоль) и затем уксусный ангидрид (0,9 мл, 9,4 ммоль). Реакционную смесь перемешивали при комнатной температуре 30 мин. Растворители удалили в вакууме, а затем остаток разбавили водой (500 мл) и этилацетатом (500 мл). Органический слой отделили и промыли водой (300 мл), 2% водной H2SO4 (200 мл), водой (2×300 мл) и солевым раствором (100 мл). Органический слой отделили, высушили над Na2SO4 и растворитель удалили в вакууме с получением соединения G (5,5 г, 82%). ЖХ-МС [М+Н] 853,4 (C36H52N8O12S+Н, рассчит.: 853,9). Чистота >95% (УФ/254 нм). Соединение G применяли без дополнительной очистки.
Методика 56: Этиловый эфир ({(S)-2-[(S)-2-ацетиламино-5-({амино-[(2)-2,2,4,б,7-пентаметил-2,3-дигидро-бензофуран-5-сульфонилимино]-метил}-амино)-пентаноиламино]-3-метиламино-пропионил}-метиламино)-уксусной кислоты (Н).
К раствору соединения G (5,5 г, 6,5 ммоль) в ДМФА (21 мл) при комнатной температуре добавили K2CO3 (8,9 г, 64,5 ммоль) и затем тиоглицерин (5,6 мл, 64,5 ммоль). Реакционную смесь перемешивали при комнатной температуре 1 час, отфильтровали и удалили ДМФА под вакуумом. Образовавшийся остаток растворили в воде (500 мл) и экстрагировали этилацетатом (2×300 мл) и хлороформом (2×300 мл). Объединенные органические слои высушили, и растворители удалили в вакууме с получением неочищенного продукта. Неочищенный продукт был очищен с помощью флэш-хроматографии элюированием этилацетатом, а затем смесью 10% метанола в хлороформе с получением соединения Н (1,3 г, 39%). ЖХ-МС [М+Н] 668,3 (C30H49N7O8S+H, рассчит.: 667,8). Чистота >95% (УФ/254 нм).
Методика 57: Этиловый эфир ({(S)-2-[(S)-2-ацетиламино-5-({амино-[(2)-2,2,4,6,7-пентаметил-2,3-дигидро-бензофуран-5-сульфонилимино]-метил}-амино)-пентаноиламино]-3-[(оксикодон-енилоксикарбонил)-метиламино]-пропионил}-метиламино)-уксусной кислоты (I).
К раствору свободного основания оксикодона (2,0 г, 6,3 ммоль) в ТГФ (100 мл) при -60°С по каплям добавили 0,5 М раствор KHMDS (13,9 мл, 7,0 ммоль). Реакционную смесь перемешивали 30 мин. и затем перенесли в раствор 4-нитрофенилхлорформиата (1,3 г) в ТГФ (100 мл) при -60°С и перемешивали 30 мин. Раствор амина соединения Н (3,2 г, 4,9 ммоль) добавили в виде раствора в ТГФ (20 мл) к реакционной смеси. После перемешивания при -60°С в течение 15 мин. охлаждающую баню убрали и реакционную смесь перемешивали при комнатной температуре в течение ночи. Еще одну порцию (1,0 г, 3,2 ммоль) свободного основания оксикодона активировали, используя описанную выше процедуру, и добавили к реакционной смеси, как описано выше, и продолжили перемешивание в течение ночи. Конец реакции определили с помощью ЖХ-МС. Растворители удалили и остаток растворили в метаноле (-25 мл) и осадили диэтиловым эфиром (400 мл). Осадок промыли диэтиловым эфиром и гексаном и высушили в вакууме. Продукт растворили в воде и ДМСО и очистили с помощью ВЭЖХ. [колонка Nanosyn-Pack Microsorb (100-10) С-18 (50×300 мм); скорость потока: 100 мл/мин; объем впрыска 15 мл; подвижная фаза А: 100% вода, 0,1% ТФУК; подвижная фаза В: 100% ацетонитрил, 0,1% ТФУК; градиентное элюирование от 0% до 33% В за 30 мин, изократическое элюирование при 33% В за 30 мин, градиентное элюирование от 33% В до 50% В за 33 мин; определение при 254 нм]. Целевые фракции объединили и высушили в вакууме с получением соединения I (5 г, выход 92%). ЖХ-МС [М+Н] 979,6 (C48H66N8O12S+H, рассчит.: 980,15). Чистота >95% (УФ/254 нм).
Методика 58: Этиловый эфир ({(S)-2-((S)-2-ацетиламино-5-гуанидино-пентаноиламино)-3-[(оксикодон-енилоксикарбонил)-метил-амино]-пропионил}-метиламино)-уксусной кислоты (J)
Соединение I (5 г, 4,5 ммоль) обработали 5% раствором м-крезола в ТФУК (25 мл). Через 1 час к реакционной смеси добавили эфир (400 мл). Осажденный продукт отфильтровали, промыли диэтиловым эфиром и гексаном и высушили в вакууме с получением соединения J (3,2 г, выход 65%). ЖХ-МС [М+Н] 757,7 (C36H52N8O10+Н, рассчит.: 757,9). Чистота >95% (УФ/254 нм). Соединение J применяли без дополнительной очистки.
({(S)-2-(S)-2-Ацетиламино-5-гуанидино-пентаноиламино)--3-[(оксикодон-енилоксикарбонил)-метил-амино]-пропионил}-метиламино)-уксусная кислота (Соединение KC-6).
Соединение J обработали 2 н водным раствором HCl (75 мл) и нагревали при 55°С в течение 6,5 ч. Нагрев убрали, реакционную смесь охладили до 5°С и pH довели до ~6 водным насыщенным раствором NaHCO3. Основное количество воды удалили под вакуумом до общего объема -50 мл. Полученный раствор очистили с использованием ВЭЖХ. [колонка Nanosyn-Pack Microsorb (100-10) С-18 (50×300 мм); скорость потока: 100 мл/мин; объем впрыска 15 мл; подвижная фаза А: 100% вода, 0,1% ТФУК; подвижная фаза В: 100% ацетонитрил, 0,1% ТФУК; изократическое элюирование при 0% В за 2 мин, градиентное элюирование от 0% до 8% В за 14 мин, изократическое элюирование при 8% В за 30 мин, градиентное элюирование от 8% В до 33% В за 55 мин; определение при 254 нм]. Целевые фракции объединили и высушили в вакууме, затем провели лиофилизацию, используя 0,1 н HCl, с получением Соединения KC-6 в виде гидрохлорида (1,5 г, выход 48%). ЖХ-МС [М+Н] 729,6 (C34H48N8O10+H, рассчит.: 729,8). Чистота >95% (УФ/254 нм).
Биологические данные
Пример 15: Фармакокинетика пролекарства оксикодона после ПО введения крысам
В этом Примере сравнивают концентрации оксикодона в плазме крыс после перорального (ПО) введения 6-(N-метил-N-(2-N'-ацетиларгиниламино)) этилкарбамата оксикодона (полученного как описано в Примере 10, а также упомянутого здесь как Соединение KC-2) или оксикодона.
Каждое из Соединения KC-2 и оксикодона растворили в физиологическом растворе и дозировали в эквимолярных дозах (20 мг/кг и 10 мг/кг соответственно) принудительным кормлением через желудочный зонд самцам крысы Спраг-Доули с введенным в яремную вену катетером; дозирование проводили в группе из четырех крыс. Через определенные промежутки времени брали пробы крови, выделяли плазму путем центрифугирования при 5400 об/мин при 4°С в течение 5 мин, и 100 микролитров (мкл) плазмы переносили из каждого образца в чистую пробирку, содержащую 1 мкл муравьиной кислоты. Пробирки встряхивали на шейкере типа Vortex в течение 5-10 секунд, немедленно помещали на сухой лед, а затем хранили до проведения анализа с помощью высокоэффективной жидкостной хроматографии / масс спектрометрии (ВЭЖХ/МС).
В Таблице 1 приведены значения Cmax в плазме (максимальной концентрация в плазме) и Tmax (время после введения, после которого концентрация в плазме максимальна) оксикодона (среднее ± стандартное отклонение) для каждой группы из 4-х крыс. Также приведены значения Сmax и Тmax для оксиморфона - метаболита оксикодона.
Фигура 4 сравнивает средние концентрации оксикодона в плазме (± стандартные отклонения) от времени высвобождения после ПО введения крысам 20 мг/кг Соединения KC-2 (сплошная линия) или 10 мг/кг оксикодона (пунктирная линия).
Результаты в Таблице 1 и на Фигуре 4 указывают на то, что введение Соединения KC-2 дает концентрации оксикодона в плазме, которые демонстрируют пониженное Cmax и замедленное Tmax по сравнению с введением оксикодона.
Пример 16: Фармакокинетика пролекарства оксикодона после ВВ введения крысам
В этом Примере сравнивают концентрации пролекарства и оксикодона в плазме крыс после внутривенного (ВВ) введения 6-(N-метил-N-(2-N'-ацетиларгиниламино)) этилкарбамата оксикодона (полученного как описано в Примере 10, а также упомянутого здесь как Соединение KC-2).
Соединение KC-2 растворили в физиологическом растворе и дозу 2 мг/кг вводили в хвостовую вену 4-м самцам крысы Спраг-Доули с введенным в яремную вену катетером. Через определенные промежутки времени брали пробы крови, выделяли плазму путем центрифугирования при 5400 об/мин при 4°С в течение 5 мин, и 100 микролитров (мкл) плазмы переносили из каждого образца в чистую пробирку, содержащую 1 мкл муравьиной кислоты. Пробирки встряхивали на шейкере типа Vortex в течение 5-10 секунд, немедленно помещали на сухой лед, а затем хранили до проведения анализа с помощью высокоэффективной жидкостной хроматографии / масс спектрометрии (ВЭЖХ/МС).
В Таблице 2 приведены значения Cmax в плазме (среднее ± стандартное отклонение)
Соединения КС-2, оксикодона и оксиморфона (метаболита оксикодона).
На Фигуре 5 приведено сравнение средних концентраций (± стандартное отклонение) от времени для Соединения КС-2 (сплошная линия) и оксикодона (пунктирная линия) после ВВ введения крысам 2 мг/кг Соединения KC-2. Значения на оси Y также показывают значения Cmax для Соединения KC-2 и оксикодона соответственно.
Таблица 2 и Фигура 5 показывают, что концентрация оксикодона в плазме крыс, которым ВВ ввели Соединение KC-2, составляет только 0,03% от концентрации Соединения KC-2 в плазме, что говорит о том, что ВВ введение Соединения KC-2 не приводит к значительному высвобождению оксикодона.
Пример 17: In vitro стабильность пролекарства оксикодона
Этот Пример демонстрирует стабильность 6-(N-метил-N-(2-N'-ацетиларгиниламино)) этилкарбамата оксикодона (полученного, как описано в Примере 10, а также упомянутого здесь как Соединение КС-2) в отношении ряда легкодоступных бытовых химикатов и препаратов ферментов.
Соединение КС-2 при комнатной температуре (КТ) или 80°С в течение 1 часа или 24 часов подвергали действию следующих бытовых химикатов: водка (40% спирта), питьевая сода (насыщенный раствор бикарбоната натрия, pH 9), WINDEX® с Ammonia-D (pH 11) и уксус (5% уксусной кислоты). Соединение KC-2 также подвергали действию следующих содержащих ферменты композиций при КТ в течение 1 часа или 24 часов: GNC® Super Digestive (2 капсулы ферментов GNC® Super Digestive растворяли в 5 мл воды), тендеризатор (Adolfs тендеризатор мяса, в основном папаин, растворяли в воде до концентрации 0,123 г/мл до приблизительной концентрации маринада, приведенной на этикетке), и субтилизин (8 таблеток чистящего средства для глазных линз ULTRAZYME® (Advanced Medical Optics) растворяли в 4 мл воды). Образцы инкубировали, как описано, брали аликвоты через 1 час и 24 часа и стабилизировали добавлением раствора 50% или 100% 85%-ной фосфорной кислоты для получения конечного pH меньше или равного 4. Стабилизированные аликвоты затем разбавили путем добавления 4- или 6-кратного количества воды, встряхивали на шейкере типа Vortex и анализировали с использованием ВЭЖХ.
Фигура 6 демонстрирует высвобождение оксикодона при воздействии ряда бытовых химикатов и содержащих ферменты композиций на Соединение KC-2, как описано выше. Процент оставшегося Соединения KC-2 после воздействия показан в виде сплошных черных столбцов, а процент конвертированного в оксикодон Соединения KC-2 показан в виде столбцов более светлого цвета с черным контуром. Эти результаты показывают, что воздействие на Соединение KC-2 в этих различных условиях приводит к конверсии в оксикодон, значительно более низкой чем 10%.
Пример 18: In vitro IC50 данные по некоторым возможным трипсиновым ингибиторам
Несколько возможных трипсиновых ингибиторов, а именно Соединения 101-105, 107 и 108, получили, как описано здесь в Примерах. Соединение 106 (также известное как 4-аминобензамидин), Соединение 109 и Соединение 110 можно приобрести у Sigma-Aldrich (Сент-Луис, Миссури, США).
Значения половины максимальной ингибирующей концентрации (IC50 или IC50) каждого из Соединений 101-110, а также SBTI и BBSI, определили с использованием модифицированного анализа трипсина, как описано в монографии Bergmeyer, HU и др., 1974, Methods of Enzymatic Analysis Volume 1, 2-е изд., 515-516, Bergmeyer, HU, ред., Academic Press, Inc.New York, Нью Йорк.
Таблица 3 показывает значения IC50 для каждого из установленных ингибиторов трипсина.
Результаты Таблицы 3 показывают, что каждое из Соединений 101-110 проявляет ингибирующую трипсин активность.
Пример 19: Влияние ингибирования трипсина на in vitro опосредованное трипсином высвобождение оксикодона из Соединения КС-2
Соединение KC-2 (которое может быть получено, как описано в Примере 10) инкубировали с трипсином из бычьей поджелудочной железы (Кат. №Т8003, тип I, ~10000 ВАЕЕ единиц/мг белка, Sigma-Aldrich) в отсутствие или в присутствии Соединения 109 (Кат. №N0289, Sigma-Aldrich). В случаях, когда Соединение 109 является частью инкубированной смеси, Соединение KC-2 добавляли через 5 мин после других компонентов инкубирования. В частности, реакционные смеси включали 0,523 мг/мл (0,761 ммол.) Соединения KC-2·2HCl, 0,0228 мг/мл трипсина, 22,5 ммол. хлорида кальция, 172 ммол. Буфера Tris (pH 8) и 0,00108 мг/мл (2 мкмол.) Соединения 109 или 0,25% ДМСО в зависимости от того, был ли ингибитор включен для инкубации. Реакции проводили при 37°С в течение 24 часов. Образцы отбирали в определенные моменты времени, переносили в 0,5% муравьиную кислоту в ацетонитриле для прекращения активности трипсина и хранили при температуре ниже -70°С до анализа с помощью ЖХ-МС/МС (жидкостная хроматография тандемная масс-спектрометрия).
На фигуре 7 показаны результаты воздействия трипсина на Соединение KC-2 в отсутствие каких-либо трипсиновых ингибиторов (закрашенные символы) или в присутствие Соединения 109 (незакрашенные символы). Квадратные символы показывают исчезновение Соединения KC-2, и треугольные символы показывают появление оксикодона от времени в условиях, описанных в этом Примере.
Результаты на Фигуре 7 показывают, что трипсиновый ингибитор вариантов осуществления может снижать опосредованное трипсином высвобождение оксикодона из Соединения KC-2. В дополнение к этому, такой трипсиновый ингибитор может препятствовать использованию пользователем трипсина для воздействия на высвобождение оксикодона из Соединения KC-2.
Таблица 4 содержит результаты воздействия трипсина на Соединение KC-2 в отсутствие и в присутствии Соединения 109. Результаты приведены в виде полураспада пролекарства при воздействии трипсина (т.е. полураспад пролекарства под действием трипсина) в часах и скорости образования оксикодона на единицу трипсина.
Результаты в Таблице 4 показывают, что трипсин может действовать на высвобождение оксикодона из пролекарства вариантов осуществления, и что трипсиновый ингибитор вариантов осуществления может снижать опосредованное трипсином высвобождение оксикодона.
Пример 20: Пероральное введение Соединения KC-2 и трипсинового ингибитора Соединения 109 крысам
Растворы Соединения KC-2 в физиологическом растворе (которые можно приготовить, как описано в Примере 10) дозировали при 8,7 мкмоль/кг (6 мг/кг) с или без одновременного дозирования 55 мкмоль/кг (30 мг/кг) Соединения 109 (Кат. №3081, Tocris Bioscience, Ellisville, Миссури, США или Кат. No. WS38665, Waterstone Technology, Cannel, Индиана, США), как показано в Таблице 5, путем принудительного желудочного введения через зонд самцам крысы Спраг-Доули (4 в группе) с введенным в яремную вену катетером, которых подвергли голоданию за 16-18 часов до перорального введения. Через определенные промежутки времени брали пробы крови, выделяли плазму путем центрифугирования при 5400 об/мин при 4°С в течение 5 мин, и 100 микролитров (мкл) плазмы переносили из каждого образца в чистую пробирку, содержащую 2 мкл 50%-ной муравьиной кислоты. Пробирки встряхивали на шейкере типа Vortex в течение 5-10 секунд, немедленно помещали на сухой лед, а затем хранили при -80°С в морозильной камере до анализа с помощью ВЭЖХ/МС.
Таблица 5 и Фигура 8 представляют результаты воздействия оксикодона на крыс, которым ввели Соединение KC-2 в отсутствие или в присутствии трипсинового ингибитора. Результаты в Таблице 5 представлены в виде (а) максимальной концентрации в плазме (Cmax) оксикодона (ОС) (среднее ± стандартное отклонение) и (b) времени после введения Соединения KC-2, необходимого для достижения максимальной концентрации оксикодона (Tmax) (среднее ± стандартное отклонение).
На Фигуре 8 приведено сравнение усредненных концентраций в плазме относительно времени высвобождения оксикодона после ПО введения крысам Соединения KC-2 без трипсинового ингибитора или совместно с ним.
Результаты в Таблице 5 и на Фигуре 8 показывают, что Соединение 109 ослабляет способность Соединения KC-2 высвобождать оксикодон, при этом как снижая Cmax, так и замедляя Tmax.
Пример 21: Фармакокинетика Соединения KC-2 после ПО введения крысам
Растворы Соединения 2 в физиологическом растворе (которые можно приготовить, как описано в Примере 10) дозировали, как показано в Таблице 6, принудительным кормлением через желудочный зонд самцам крыс Спраг-Доули (4 на группу) с введенным в яремную вену катетером, которых подвергли голоданию за 16-18 часов до перорального введения. Через определенные промежутки времени брали пробы крови, выделяли плазму путем центрифугирования при 5400 об/мин при 4°С в течение 5 мин., и 100 микролитров (мкл) плазмы переносили из каждого образца в чистую пробирку, содержащую 2 мкл 50%-ной муравьиной кислоты. Пробирки встряхивали на шейкере типа Vortex в течение 5-10 секунд, немедленно помещали на сухой лед, а затем хранили при -80° в морозильной камере до анализа с помощью ВЭЖХ/МС.
Таблица 6 и Фигура 9 представляют результаты воздействия оксикодона на крыс, которым ввели различные дозы Соединения KC-2. Результаты в Таблице 6 представлены для каждой группы крыс в виде (а) максимальной концентрации оксикодона (ОС) в плазме (Cmax) (среднее ± стандартное отклонение), (b) времени после введения Соединения KC-2, необходимого для достижения максимальной концентрации оксикодона (Tmax) (среднее ± стандартное отклонение) и (с) площади под кривой (AUC) от 0 до 24 часов (среднее±стандартное отклонение).
Нижний предел определения концентрации составлял 0,0500 нг/мл, кроме 20 мг/кг, где он составлял 0,0250 нг/мл
На Фигуре 9 приведено сравнение усредненных концентраций в плазме относительно времени высвобождения оксикодона после ПО введения крысам увеличенных доз Соединения KC-2.
Результаты на Фигуре 6 и 9 говорят о том, что концентрации оксикодона в плазме повышаются пропорционально дозе Соединения KC-2.
Пример 22: Пероральное введение Соединения KC-2 совместно с трипсиновым ингибитором Соединения 109 крысам
Растворы Соединения KC-2 в физиологическом растворе дозировали при 7,3 мкмоль/кг (5 мг/кг) и 73 мкмоль/кг (50 мг/кг) Более высокие концентрации вводили совместно с более высокими концентрациями Соединения 109 (Кат.№3081, Tocris Bioscience или Кат. №WS38665, Waterstone Technology), как показано в Таблице 7, принудительным кормлением через желудочный зонд самцам крыс Спраг-Доули (4 в группе) с введенным в яремную вену катетером, которых подвергли голоданию за 16-18 часов до перорального введения. Через определенные промежутки времени брали пробы крови, выделяли плазму путем центрифугирования при 5400 об/мин при 4°С в течение 5 мин., и 100 микролитров (мкл) плазмы переносили из каждого образца в чистую пробирку, содержащую 2 мкл 50%-ной муравьиной кислоты. Пробирки встряхивали на шейкере типа Vortex в течение 5-10 секунд, немедленно помещали на сухой лед, а затем хранили при -80°С в морозильной камере до анализа с помощью ВЭЖХ/МС.
Таблица 7 и Фигура 10 представляют результаты воздействия оксикодона на крыс, которым ввели различные дозы Соединения KC-2. Результаты в Таблице 7 представлены для каждой группы крыс в виде (а) максимальной концентрации оксикодона (ОС) в плазме (Cmax) (среднее ± стандартное отклонение), (b) времени после введения Соединения KC-2, необходимого для достижения максимальной концентрации оксикодона (Tmax) (среднее ± стандартное отклонение) и (с) площади под кривой (AUC) от 0 до 24 часов (среднее ± стандартное отклонение).
Нижний предел определения составил 0,0250 нг/мл
На Фигуре 10 приведено сравнение усредненных концентраций в плазме относительно времени высвобождения оксикодона после ПО введения крысам Соединения KC-2 совместно с увеличенными количествами трипсинового ингибитора -Соединения 109.
Результаты в Таблице 7 и на Фигуре 10 показывают способность Соединения 109 ослаблять способность Соединения KC-2 высвобождать оксикодон в зависимости от дозы, при этом как снижая Cmax и AUC, так и замедляя Tmax.
Пример 23: Анализ связывания in vitro человеческого рецептора µ-опиоида
В этом примере измеряют сродство соединения KC-2 к µ (мю)-опиоидному рецептору, экпрессированному в рекомбинантных НЕК-293 клетках.
Общая процедура следует протоколу, описанному у Wang, J.-B., Johnson, P.S., Perscio, A.M., Hawkins, A.L., Griffin, C.A. и Uhl, G.R. (1994). FEBS Lett., 338: 217-222. Более конкретно, анализ включает, соответственно, оксикодон или Соединение KC-2 (которые могут быть приготовлены как описано в Примере 10) также, как и рекомбинантные НЕК-293 клетки, экспрессирующие человеческий µ-опиоидный рецептор на своей клеточной поверхности, соединение для сравнения [d-Ala2, N-Me-Phe4, Gly5-ol]-энкефалин (DAMGO), радиолиганд [3H]DAMGO (0,5 нмол.) и неспецифический лиганд налоксон (10 мкмол.). Реакции проводили при 22°С в течение 2 часов. В образцах затем проводили измерение активности сцинтилляционным методом.
В этих количественных анализах специфическое связывание тестируемого соединения с рецепторами определяли как разницу между полным связыванием и неспецифическим связыванием, определяемым в присутствии избытка немеченного лиганда. Результаты выразили как процент контрольного специфического связывания и как процент ингибирования контрольного специфического связывания, полученного в присутствии тестируемой композиции. Значения IC50 (концентрация конкурентного лиганда, необходимая для 50% ингибирования [3H]DAMGO связывания) и параметры Хилла (nH) определяли путем нелинейного регрессионного анализа конкурентных кривых, используя уравнение Хилла для аппроксимации кривой.
В Таблице 8 показаны значения IC50 для оксикодона и Соединения KC-2.
Эти значения демонстрируют то, что Соединение КС-2 связывается с µ-опиоидным рецептором со сродством приблизительно в 2 раза меньшим, чем к оксикодону.
Пример 24: Клеточный функциональный анализ in vitro агониста человеческого рецептора µ-опиоида
В этом Примере измеряют способность определенных соединений данного раскрытия влиять на ответ агонистов под воздействием рекомбинантного человеческого рецептора µ-опиоида, экспрессированного в СНО клетках.
Общая процедура следует протоколу, описанному у Wang, J.-B., Johnson, P.S., Perscio, A.M., Hawkins, A.L., Griffin, C.A. и Uhl, G.R. (1994). FEBS Lett., 338: 217-222. Более конкретно анализ включает каждое из соединений, показанных в Таблице 9, и рекомбинантные клетки яичников китайского хомяка (СНО), экспрессирующие человеческий рецептор µ-опиоида на поверхности своих клеток. Контрольная реакция включает 1 мкмол. DAMGO. Реакционные смеси инкубировали при 37°С в течение 10 мин, и продуктом реакции являлся циклический AMP (cAMP). Образцы подвергли гомогенной флуоресценции с разрешением во времени (HTRF®). Значения ЕС50 (концентрация, вызывающая половину максимального специфического ответа) были определены с помощью программы Hillplot нелинейной регрессионной аппроксимации.
В Таблице 9 приведены результаты 3-х отдельных экспериментов. Значения EC50 привели для Соединения KC-2, Соединения KC-3, Соединения KC-5 и Соединения KC-6 (каждое из которых может быть получено как описано в Примерах 10, 11, 13 и 14 соответственно) и сравнили со значениями ЕС50 для оксикодона, измеренными в таких же соответствующих экспериментах. Также показаны значения ЕС50 для Соединения KC-4 (которое может быть получено, как описано в Примере 12) и гидрокодона, измеренные в том же эксперименте. В Таблице 9 приведены отношение силы лекарственного средства к пролекарству (лекарственное средство/пролекарство) (т.е. ЕС50 в человеческом рецепторе µ-опиоида) оксикодона или гидрокодона к пролекарству соответствующего лекарственного средства.
Результаты в Таблице 9 говорят о том, что пролекарства вариантов осуществления демонстрируют относительную силу лекарственное средство/пролекарство больше 1; таким образом, пролекарства вариантов осуществления являются менее сильным в отношении человеческого рецептора µ-опиоида, чем соответствующие высвобождаемые им лекарственные средства.
Пример 25: Фармакокинетика после ВВ введения Соединения KC-2 или оксикодона крысам: проникновение в плазму и спинномозговую жидкость
В этом примере сравнивают концентрации пролекарства - Соединения KC-2 и оксикодона в плазме и спинномозговой жидкости (СМЖ) после внутривенного (ВВ) введения соответствующего соединения крысам. Коэффициент распределения плазма/СМЖ предсказывает способность соединения проникать сквозь гематоэнцефалический барьер.
Соединение KC-2 (которое может быть приготовлено, как описано в Примере 10) в дозе 10 мг/кг или в эквимолярной дозе оксикодона каждую растворили в физиологическом растворе и ввели в хвостовую вену 4-м самцам крысы Спраг-Даули. Через 15 минут крыс анастезировали диоксидом углерода путем асфиксии и брали пробы крови, выделяли плазму путем центрифугирования при 5400 об/мин при 4°С в течение 5 мин., и 100 микролитров (мкл) плазмы переносили из каждого образца в чистую пробирку, содержащую 2 мкл 50%-ной муравьиной кислоты. СМЖ брали, используя иглу размером 22×1 дюйм, присоединенную к полиуретановому катетеру с трубкой типа MRE-040 (Braintree Scientific, Inc., Braintree, Массачусетс). Иглу вводили сразу ниже затылочного гребня в области большого затылочного отверстия; прозрачную СМЖ собирали в катетер и переносили в сборную пробирку. Образцы СМЖ центрифугировали при 5400 об/мин при 4°С в течение 5 мин., и 100 мкл СМЖ из каждого образца переносили в чистую пробирку. Образцы плазмы и СМЖ немедленно помещали на сухой лед, а затем хранили при -80°С в морозильной камере до проведения анализа с помощью высокоэффективной жидкостной хроматографии / масс спектрометрии (ВЭЖХ/МС).
Результаты в Таблице 10 представлены для каждой группы из 4-х крыс в виде усредненных концентраций указанных соединений в плазме или СМЖ. В Таблице 10 также представлен коэффициент распределения между плазмой и СМЖ (плазма/СМЖ), т.е. отношение концентрации в плазме к концентрации в СМЖ для указанных соединений.
Результаты в Таблице 10 говорят о том, что относительный коэффициент распределения плазма/СМЖ Соединения KC-2 и оксикодона составляет примерно 116 (т.е. 439/3,8); это означает, что Соединение КС-2 в 116 раз хуже проникает в СМЖ, чем оксикодон. Кроме того, как показано в Примере 24, относительная сила лекарственное средство/пролекарство Соединения KC-2 составляет приблизительно 4,1. Таким образом, ожидается, что Соединение KC-2 при введении внутривенно в эквимолярных количествах будет приблизительно в 475 раз (т.е. 116×4,1) менее эффективно для СМЖ рецепторов µ-опиоида, чем оксикодон.
Пример 26: Фармакокинетика Соединения KC-3 после ПО введения крысам
В этом примере сравнивают фармакокинетику нескольких концентраций Соединения KC-3, введенного крысам перорально (ПО).
Растворы Соединения KC-3 в физиологическом растворе (которые можно приготовить, как описано в Примере 11) дозировали, как показано в Таблице 11, принудительным кормлением через желудочный зонд самцам крыс Спраг-Доули (4 в группе) с введенным в яремную вену катетером, которых подвергли голоданию за 16-18 часов до перорального введения. Через определенные промежутки времени брали пробы крови, выделяли плазму путем центрифугирования при 5400 об/мин при 4°С в течение 5 мин., и 100 мкл плазмы переносили из каждого образца в чистую пробирку, содержащую 2 мкл 50%-ной муравьиной кислоты. Пробирки встряхивали на шейкере типа Vortex в течение 5-10 секунд, немедленно помещали на сухой лед, а затем хранили при -80° в морозильной камере до анализа с помощью ВЭЖХ/МС.
Таблица 11 и Фигура 11 представляют результаты воздействия оксикодона на крыс, которым ввели различные дозы Соединения KC-3. Результаты в Таблице 11 представлены для каждой группы крыс в виде (а) максимальной концентрации оксикодона (ОС) в плазме (Cmax) (среднее ± стандартное отклонение), (b) времени после введения Соединения KC-3, необходимого для достижения максимальной концентрации оксикодона (Tmax) (среднее ± стандартное отклонение) и (с) площади под кривой (AUC) от 0 до 24 часов (среднее ± стандартное отклонение) для всех доз, кроме доз 1,4 мг/кг и 22 мг/кг, где значения AUC рассчитывали для интервала от 0 до 8 часов (среднее ± стандартное отклонение).
Нижний предел количественного определения составлял 0,05 нг/мл На Фигуре 11 приведено сравнение усредненных концентраций в плазме относительно времени высвобождения оксикодона после ПО введения крысам увеличенных доз Соединения KC-3.
Результаты в Таблице 11 и Фигуре 11 говорят о том, что концентрации оксикодона в плазме повышаются пропорционально дозе Соединения KC-3.
Пример 27: Фармакокинетика Соединения KC-3 после ВВ введения крысам
В этом Примере сравнивают концентрации пролекарства и оксикодона в плазме крыс после внутривенного (ВВ) введения Соединения KC-3.
Соединение KC-3 (которое можно приготовить, как описано в Примере 11) растворили в физиологическом растворе и дозу 2 мг/кг вводили в хвостовую вену 4-м самцам крысы Спраг-Доули с введенным в яремную вену катетером. Через определенные промежутки времени брали пробы крови, выделяли плазму путем центрифугирования при 5400 об/мин при 4°С в течение 5 мин., и 100 мкл плазмы переносили из каждого образца в чистую пробирку, содержащую 2 мкл 50%-ной муравьиной кислоты. Пробирки встряхивали на шейкере типа Vortex в течение 5-10 секунд, немедленно помещали на сухой лед, а затем хранили при -80°С в морозильной камере до проведения анализа с помощью высокоэффективной жидкостной хроматографии / масс спектрометрии (ВЭЖХ/МС).
Таблица 12 и Фигура 12 представляют результаты воздействия Соединения KC-3 и оксикодона на группу крыс, которым внутривенно вводили Соединение KC-3. Результаты в Таблице 12 представлены в виде максимальной концентрации в плазме (Cmax) Соединения KC-3 и оксикодона (ОС) соответственно (среднее ± стандартное отклонение).
Нижний предел количественного определения составлял 0,05 нг/мл
Таблица 12 и Фигура 12 показывают, что концентрации оксикодона в плазме крыс, которым внутривенно ввели Соединение KC-3, составляет только 0,04% от концентрации Соединения KC-3 в плазме, что говорит о том, что ВВ введение Соединения KC-2 не приводит к значительному высвобождению оксикодона в плазму.
Пример 28: Влияние ингибирования трипсина на in vitro опосредованное трипсином трипсиновое высвобождение оксикодона из опиоидного пролекарства с модифицированным кетоном.
В этом примере демонстрируют способность трипсина расщеплять пролекарство вариантов осуществления и влияние трипсиновых ингибиторов на данное расщепление.
Соединение KC-3, Соединение KC-4, Соединение KC-5 и Соединение KC-6 каждое инкубировали с трипсином из бычьей поджелудочной железы (Кат. №Т8003, Тип I, ~10.000 ВАЕЕ единиц/мг белка, Sigma-Aldrich). В частности, рекционные смеси включали 0,761 ммол. Соединения KC-3·2HCl, Соединения KC-5·2HCl, Соединения KC-4·2HCl или Соединения KC-6·2HCl, 22,5 ммол. хлорида кальция, от 40 до 172 ммол. Буфера Tris (pH 8) и 0,25% ДМСО с различной активностью трипсина, как приведено в Таблице 13А. Реакции проводили при 37°С в течение 24 часов. Образцы отбирали в определенные моменты времени, переносили в 0,5% муравьиную кислоту в ацетонитриле для прекращения активности трипсина и хранили при температуре ниже -70°С до анализа с помощью ЖХ-МС/МС (жидкостная хроматография тандемная масс-спектрометрия).
Соединение KC-3 также инкубировали в присутствии 2 мкмол. (мкМ) трипсинового ингибитора Соединения 109. В этом случае Соединение KC-3 добавили через 5 мин после других инкубируемых компонентов. Другая реакция и обработка образцов были проведены, как описано выше.
В Таблицах 13А и 13В показаны результаты воздействия трипсина на тестируемые вещества в отсутствие или в присутствии трипсинового ингибитора. Результаты приведены в виде времени полураспада пролекарства при воздействии трипсина (т.е. полураспада пролекарства под действием трипсина) в часах и скорость образования оксикодона или гидрокодона в мкмоль в час на единицу ВАЕЕ трипсина (мкмоль/ч/BAEEU).
Результаты в Таблице 13А показывают, что трипсин может быть посредником высвобождения оксикодона или гидрокодона из пролекарства вариантов осуществления. Результаты в Таблице 13 В показывают, что трипсиновый ингибитор вариантов осуществления может снижать опосредованное трипсином высвобождение лекарственного средства из опиоидного пролекарства с модифицированным кетоном вариантов осуществления.
Пример 29: Пероральное введение Соединения KC-3 и трипсинового ингибитора Соединения 109 крысам
В этом Примере демонстрируют способность трипсинового ингибитора вариантов осуществления влиять на высвобождение лекарственного средства из Соединения KC-3 в плазму после перорального введения.
Растворы Соединения KC-3 в физиологическом растворе (которые можно приготовить, как описано в Примере 11) дозировали при 6,8 мкмоль/кг (5 мг/кг) и 68 мкмоль/кг (50 мг/кг) Соединения KC-3 с или без одновременного дозирования увеличивающихся концентраций Соединения 109 (Кат.№3081, Tocris Bioscience или Кат.No. WS38665, Waterstone Technology), как показано в Таблице 14, путем принудительного желудочного введения через зонд самцам крысы Спраг-Доули (4 в группе) с введенным в яремную вену катетером, которых подвергли голоданию за 16-18 часов до перорального введения. Через определенные промежутки времени брали пробы крови, выделяли плазму путем центрифугирования при 5400 об/мин при 4°С в течение 5 мин, и 100 мкл плазмы переносили из каждого образца в чистую пробирку, содержащую 2 мкл 50%-ной муравьиной кислоты. Пробирки встряхивали на шейкере типа Vortex в течение 5-10 секунд, немедленно помещали на сухой лед, а затем хранили при -80°С в морозильной
камере до анализа с помощью ВЭЖХ/МС.
Таблица 14 и Фигура 13 представляют результаты воздействия оксикодона на крыс, которым ввели Соединение KC-3 в отсутствие или в присутствии трипсинового ингибитора. Результаты в Таблице 14 представлены для каждой группы крыс в виде (а) максимальной концентрации оксикодона (ОС) в плазме (Cmax) (среднее ± стандартное отклонение), (b) времени после введения Соединения КС-3, необходимого для достижения максимальной концентрации оксикодона (Tmax) (среднее ± стандартное отклонение) и (с) площади под кривой (AUC) от 0 до 24 часов (среднее±стандартное отклонение).
Нижний предел количественного определения составлял 0,025 нг/мл
На Фигуре 13 приведено сравнение усредненных концентраций в плазме относительно времени высвобождения оксикодона после ПО введения крысам Соединения KC-3 без трипсинового ингибитора или совместно с ним.
Результаты в Таблице 14 и на Фигуре 13 показывают, что Соединение 109 ослабляет способность Соединения KC-3 высвобождать оксикодон, при этом как снижая Cmax и AUC, так и замедляя Tmax.
Пример 30: Фармакокинетика после ВВ введения Соединения KC-3 или оксикодона крысам: проникновение в плазму и спинномозговую жидкость
В этом Примере сравнивают концентрации Соединения KC-3 и оксикодона в плазме и спинномозговой жидкости (СМЖ) после внутривенного (ВВ) введения соответствующего соединения крысам. Коэффициент распределения плазма/СМЖ предсказывает способность соединения проникать через гематоэнцефалический барьер.
Соединение KC-3 (которое может быть приготовлено, как описано в Примере 11) в дозе 10 мг/кг или в эквимолярной дозе оксикодона каждую растворили в физиологическом растворе и ввели в хвостовую вену 4-м самцам крысы Спраг-Даули. Через 2 минут крыс анастезировали диоксидом углерода путем асфиксии и брали пробы крови, выделяли плазму путем центрифугирования при 5400 об/мин при 4°С в течение 5 мин, и 100 мкл плазмы переносили из каждого образца в чистую пробирку, содержащую 2 мкл 50%-ной муравьиной кислоты. СМЖ брали, используя иглу размером 22×1 дюйм, присоединенную к полиуретановому катетеру с трубкой типа MRE-040 (Braintree Scientific, Inc.). Иглу вводили сразу ниже затылочного гребня в области большого затылочного отверстия; прозрачную СМЖ собирали в катетер и переносили в сборную пробирку. Образцы СМЖ центрифугировали при 5400 об/мин при 4°С в течение 5 мин, и 100 мкл СМЖ из каждого образца переносили в чистую пробирку. Образцы плазмы и СМЖ немедленно помещали на сухой лед, а затем хранили при -80°С в морозильной камере до проведения анализа с помощью высокоэффективной жидкостной хроматографии / масс спектрометрии (ВЭЖХ/МС). С целью изучения зависимости проникновения Соединения KC-3 и оксикодона в плазму и СМЖ от времени, дополнительным группам из 4-х крыс вводили соединения, как описано выше, и анестезировали в определенные моменты времени. Плазму и СМЖ отбирали и анализировали, как описано выше. Результаты, полученные на этих крысах, показывают, что равновесие в плазме и отделениях СМЖ быстро устанавливается после дозировки, и что коэффициент распределения между СМЖ и плазмой был постоянным во всех временных точках измерения. Таким образом, в Таблице 15 приведены данные только для 2-минутных временных точек измерения.
Результаты в Таблице 15 представлены для каждой группы из 4-х крыс в виде усредненных концентраций указанного соединения в плазме или СМЖ. В Таблице 15 также представлены коэффициенты распределения между плазмой и СМЖ (плазма/СМЖ), т.е. отношение концентрации в плазме к концентрации в СМЖ для указанных соединений.
Результаты в Таблице 15 говорят о том, что относительный коэффициент распределения плазма/СМЖ Соединения KC-3 и оксикодона составляет примерно 364 (т.е. 1 737/4,8); это означает, что Соединение KC-3 в 364 раза хуже проникает в СМЖ, чем оксикодон. Кроме того, как показано в Примере 24, относительная сила лекарственное средство/пролекарство Соединения KC-3 составляет приблизительно 40. Таким образом, ожидается, что Соединение KC-3 при введении внутривенно в эквимолярных количествах будет приблизительно в 14 500 раз (т.е. 364×40) менее эффективно для СМЖ рецепторов µ-опиоида, чем оксикодон.
Пример 31: In vivo переносимость Соединения KC-3 в крысах
В этом Примере показано, что Соединение КС-3 переносится крысами при внутривенном введении.
В этом исследовании использовали самцов крысы Спраг-Даули, 4 на дозу. Крыс взвешивали и помещали под нагревающую лампу на 15-20 минут с целью расширить поперечные хвостовые вены. Объемы доз рассчитывали на основании веса тела (1 мл/кг);
дозирование Соединения KC-3 (которое можно приготовить, как описано в Примере 11) приведено в Таблице 16. Перед дозированием крыс помещали в ограничители Брума, и лекарственное средство вводили в одну из хвостовых вен, используя шприц с иглой. После дозирования установили таймер и наблюдали за крысами с целью детектирования клинических признаков. Образцы крови брали из подкожной вены через 5 минут после дозирования. За крысами наблюдали в течение вплоть до 24-х часов. Результаты приведены в Таблице 16.
Результаты Таблицы 16 показывают, что крысы переносят дозу 97 мкмоль/кг Соединения KC-3, и их нормальная активность восстанавливается в течение 2 минут.
Пример 32: In vitro стабильность пролекарства оксикодона Соединения KC-3
Этот пример демонстрирует стабильность Соединения KC-3 по отношению к различным легкодоступным бытовым химикатам и препаратам ферментов.
Соединение KC-3 (которое можно получить как описано в Примере 11) при комнатной температуре (КТ) или 80°С в течение 1 часа или 24 часов подвергали действию следующих бытовых химикатов: водка (40% спирта), питьевая сода (насыщенный раствор бикарбоната натрия, pH 9), WINDEX® с Ammonia-D (pH 11)и уксус (5% уксусной кислоты). Соединение КС-3 также подвергали действию следующих содержащих ферменты композиций при КТ в течение 1 часа или 24 часов: GNC® Super Digestive (2 капсулы ферментов GNC® Super Digestive растворяли в 5 мл воды), тендеризатор (Adolfs тендеризатор мяса, в основном папаин, растворяли в воде до концентрации 0,123 г/мл до приблизительной концентрации маринада, приведенной на этикетке), и субтилизин (8 таблеток чистящего средства для глазных линз ULTRAZYME® (Advanced Medical Optics) растворяли в 4 мл воды). Образцы инкубировали как описано. Аликвоты брали после 1 часа и 24 часов и стабилизировали добавлением раствора 50% или 100% 85%-ой фосфорной кислоты для получения конечного pH меньше или равного 4. Стабилизированные аликвоты затем разбавляли путем добавления 4- или 6-кратного количества воды, встряхивали на шейкере типа Vortex и анализировали с использованием ВЭЖХ.
Фигура 14 демонстрирует высвобождение оксикодона при воздействии ряда бытовых химикатов и содержащих ферменты композиций на Соединение KC-3, как описано выше. Процент оставшегося Соединения KC-3 после воздействия показан в виде сплошных черных столбцов, а процент конвертированного в оксикодон Соединения KC-3 показан в виде столбцов более светлого цвета с черным контуром. Эти результаты показывают, что воздействие на Соединение KC-3 в этих различных условиях приводит к конверсии в оксикодон, значительно более низкой чем 10%.
Пример 33: Фармакокинетика Соединения KC-4 после ПО введения крысам
Этот пример демонстрирует высвобождение гидрокодона в плазму при пероральном (ПО) введении Соединения KC-4 крысам.
Растворы Соединения KC-4 в физиологическом растворе (которые можно приготовить, как описано в Примере 12) дозировали, как показано в Таблице 17, принудительным кормлением через желудочный зонд самцам крыс Спраг-Доули (4 на группу) с введенным в яремную вену катетером, которых подвергли голоданию за 16-18 часов до перорального введения. Через определенные промежутки времени брали пробы крови, выделяли плазму путем центрифугирования при 5400 об/мин при 4°С в течение 5 мин, и 100 мкл плазмы переносили из каждого образца в чистую пробирку, содержащую 2 мкл 50%-ной муравьиной кислоты. Пробирки встряхивали на шейкере типа Vortex в течение 5-10 секунд, немедленно помещали на сухой лед, а затем хранили при -80°С в морозильной камере до анализа с помощью ВЭЖХ/МС.
В Таблице 17 представлены результаты воздействия гидрокодона на крыс, которым перорально ввели Соединение KC-4. Результаты в Таблице 17 представлены для каждой группы крыс в виде (а) максимальной концентрации гидрокодона (ОС) в плазме (Cmax) (среднее ± стандартное отклонение), (b) времени после введения Соединения KC-4, необходимого для достижения максимальной концентрации гидрокодона (Tmax) (среднее ± стандартное отклонение) и (с) площади под кривой (AUC) от 0 до 24 часов (среднее ± стандартное отклонение).
Нижний предел количественного определения составлял 0,025 нг/мл
Результаты в Таблице 17 показывают, что пероральное введение Соединения KC-4 приводит к высвобождению гидрокодона пролекарством гидрокодона вариантов осуществления.
Пример 34: Фармакокинетика Соединения КС-4 после ВВ введения крысам
В этом Примере сравнивают концентрации пролекарства и гидрокодона в плазме крыс после внутривенного (ВВ) введения Соединения KC-4.
Соединение KC-4 (которое можно приготовить, как описано в Примере 14) растворили в физиологическом растворе, и дозу 2 мг/кг вводили в хвостовую вену 4-м самцам крысы Спраг-Доули с введенным в яремную вену катетером. Через определенные промежутки времени брали пробы крови, выделяли плазму путем центрифугирования при 5400 об/мин при 4°С в течение 5 мин, и 100 мкл плазмы переносили из каждого образца в чистую пробирку, содержащую 2 мкл 50%-ной муравьиной кислоты. Пробирки встряхивали на шейкере типа Vortex в течение 5-10 секунд, немедленно помещали на сухой лед, а затем хранили при -80°С в морозильной камере до проведения анализа с помощью высокоэффективной жидкостной хроматографии / масс спектрометрии (ВЭЖХ/МС).
Таблица 18 и Фигура 15 представляют результаты воздействия Соединения KC-4 и гидрокодона на группу крыс, которым внутривенно вводили Соединение KC-4. Результаты в Таблице 18 представлены в виде максимальной концентрации в плазме (Cmax) Соединения KC-4 и гидрокодона (НС) соответственно (среднее ± стандартное отклонение).
Таблица 18 и Фигура 15 показывают, что концентрация гидрокодона в плазме крыс, которым внутривенно ввели Соединение KC-4, составляет только 0,006% от концентрации Соединения KC-4 в плазме, что говорит о том, что ВВ введение Соединения KC-4 не приводит к значительному высвобождению гидрокодона в плазму.
Пример 35: Пероральное введение Соединения KC-4 и трипсинового ингибитора Соединения 109 крысам
В этом Примере демонстрируют способность трипсинового ингибитора вариантов осуществления влиять на высвобождение лекарственного средства из Соединения KC-4 в плазму после перорального введения.
Растворы Соединения KC-4 в физиологическом растворе (которые можно приготовить, как описано в Примере 12) дозировали при 8,4 мкмоль/кг (6 мг/кг) с или без одновременного дозирования 55 мкмоль/кг (30 мг/кг) Соединения 109 (Кат. №3081, Tocris Bioscience или Кат. No. WS38665, Waterstone Technology), как показано в Таблице 19, путем принудительного желудочного введения через зонд самцам крысы Спраг Доули (4 в группе) с введенным в яремную вену катетером, которых подвергли голоданию за 16-18 часов до перорального введения. Через определенные промежутки времени брали пробы крови, выделяли плазму путем центрифугирования при 5400 об/мин при 4°С в течение 5 мин, и 100 мкл плазмы переносили из каждого образца в чистую пробирку, содержащую 2 мкл 50%-ной муравьиной кислоты. Пробирки встряхивали на шейкере типа Vortex в течение 5-10 секунд, немедленно помещали на сухой лед, а затем хранили при -80° в морозильной камере до анализа с помощью ВЭЖХ/МС.
Таблица 19 и Фигура 16 представляют результаты воздействия гидрокодона на крыс, которым ввели Соединение KC-4 в отсутствие или в присутствии трипсинового ингибитора. Результаты в Таблице 19 представлены для каждой группы крыс в виде (а) максимальной концентрации гидрокодона (НС) в плазме (Cmax) (среднее ± стандартное отклонение), (b) времени после введения Соединения KC-4, необходимого для достижения максимальной концентрации гидрокодона (Tmax) (среднее ± стандартное отклонение) и (с) площади под кривой от 0 до 24 часов (среднее ± стандартное отклонение).
Нижний предел количественного определения составлял 0,025 нг/мл
На Фигуре 16 приведено сравнение усредненных концентраций в плазме относительно времени высвобождения гидрокодона после ПО введения крысам Соединения КС-4 без трипсинового ингибитора или совместно с ним.
Результаты в Таблице 19 и на Фигуре 16 показывают, что Соединение 109 ослабляет способность Соединения KC-4 высвобождать гидрокодон, при этом как снижая Cmax и AUC, так и замедляя Tmax.
Пример 36: Фармакокинетика Соединения KC-5 после ПО введения крысам
Этот пример демонстрирует высвобождение оксикодона в плазму при пероральном (ПО) введении Соединения KC-5 крысам.
Растворы Соединения KC-5 в физиологическом растворе (которые можно приготовить, как описано в Примере 13) дозировали, как показано в Таблице 20, принудительным кормлением через желудочный зонд самцам крыс Спраг-Доули (4 на группу) с введенным в яремную вену катетером, которых подвергли голоданию за 16-18 часов до перорального введения. Через определенные промежутки времени брали пробы крови, выделяли плазму путем центрифугирования при 5400 об/мин при 4°С в течение 5 мин, и 100 мкл плазмы переносили из каждого образца в чистую пробирку, содержащую 2 мкл 50%-ной муравьиной кислоты. Пробирки встряхивали на шейкере типа Vortex в течение 5-10 секунд, немедленно помещали на сухой лед, а затем хранили при -80°С в морозильной камере до анализа с помощью ВЭЖХ/МС.
Таблица 20 и Фигура 17 представляют результаты воздействия оксикодона на группу крыс, которым внутривенно вводили Соединение KC-5. Результаты в Таблице 20 представлены для каждой группы крыс в виде (а) максимальной концентрации оксикодона (ОС) в плазме (Cmax) (среднее ± стандартное отклонение), (b) времени после введения Соединения KC-5, необходимого для достижения максимальной концентрации оксикодона (Tmax) (среднее ± стандартное отклонение) и (с) площади под кривой (AUC) (нг×ч)/мл от 0 до 8 часов (среднее ± стандартное отклонение).
Нижний предел количественного определения составлял 0,025 нг/мл
На Фигуре 17 приведено сравнение усредненных концентраций в плазме относительно времени высвобождения оксикодона после ПО введения Соединения KC-5.
Результаты в Таблице 20 и на Фигуре 17 указывают на то, что введение Соединения KC-5 создает концентрации оксикодона в плазме, которые демонстрируют пониженное Cmax и AUC и замедленное Tmax по сравнению с введением оксикодона (см. Пример 15).
Пример 37:
Фармакокинетика Соединения KC-5 после ВВ введения крысам
В этом Примере сравнивают концентрации пролекарства и оксикодона в плазме крыс после внутривенного (ВВ) введения Соединения KC-5.
Соединение KC-5 (которое можно приготовить, как описано в Примере 13), растворили в физиологическом растворе, и дозу 2 мг/кг вводили в хвостовую вену 4-м самцам крысы Спраг-Доули с введенным в яремную вену катетером. Через определенные промежутки времени брали пробы крови, выделяли плазму путем центрифугирования при 5400 об/мин при 4°С в течение 5 мин, и 100 мкл плазмы переносили из каждого образца в чистую пробирку, содержащую 2 мкл 50%-ной муравьиной кислоты. Пробирки встряхивали на шейкере типа Vortex в течение 5-10 секунд, немедленно помещали на сухой лед, а затем хранили при -80°С в морозильной камере до проведения анализа с помощью высокоэффективной жидкостной хроматографии / масс спектрометрии (ВЭЖХ/МС).
Таблица 21 и Фигура 18 представляют результаты воздействия Соединения KC-5 и оксикодона на группу крыс, которым внутривенно вводили Соединение KC-5. Результаты в Таблице 21 представлены в виде максимальной концентрации в плазме (Cmax) Соединения KC-5 и оксикодона (ОС) соответственно (среднее ± стандартное отклонение).
∧Нижний предел количественного определения составлял 0,0125 нг/мл.
Таблица 21 и Фигура 18 показывают, что концентрации оксикодона в плазме крыс, которым внутривенно ввели Соединение KC-5, составляет только 0,028% от концентрации Соединения KC-5 в плазме, что говорит о том, что ВВ введение Соединения KC-5 не приводит к значительному высвобождению оксикодона в плазму.
Пример 38: Фармакокинетика после ВВ введения Соединения KC-5 или оксикодона крысам: проникновение в плазму и спинномозговую жидкость
В этом примере сравнивают концентрации пролекарства - Соединения KC-5 и оксикодона в плазме и спинномозговой жидкости (СМЖ) после внутривенного (ВВ) введения соответствующего соединения крысам. Коэффициент распределения плазма/СМЖ предсказывает способность соединения проникать сквозь гематоэнцефалический барьер.
Соединение KC-5 (которое может быть приготовлено, как описано в Примере 13) в дозе 10 мг/кг или в эквимолярной дозе оксикодона каждую растворили в физиологическом растворе и ввели в хвостовую вену 4-м самцам крысы Спраг-Даули. Через 2 минут крыс анастезировали диоксидом углерода путем асфиксии и брали пробы крови, выделяли плазму путем центрифугирования при 5400 об/мин при 4°С в течение 5 мин, и 100 мкл плазмы переносили из каждого образца в чистую пробирку, содержащую 2 мкл 50%-ной муравьиной кислоты. СМЖ брали, используя иглу размером 22×1 дюйм, присоединенную к полиуретановому катетеру с трубкой типа MRE-040 (Braintree Scientific, Inc.). Иглу вводили сразу ниже затылочного гребня в области большого затылочного отверстия, и прозрачную СМЖ собирали в катетер и переносили в сборную пробирку. Образцы СМЖ центрифугировали при 5400 об/мин при 4°С в течение 5 мин, и 100 мкл СМЖ из каждого образца переносили в чистую пробирку. Образцы плазмы и СМЖ немедленно помещали на сухой лед, а затем хранили при -80°С в морозильной камере до проведения анализа с помощью высокоэффективной жидкостной хроматографии / масс-спектрометрии (ВЭЖХ/МС). С целью изучения зависимости проникновения Соединения KC-5 и оксикодона в плазму и СМЖ от времени, дополнительным группам из 4-х крыс вводили соединения, как описано выше, и анестезировали в определенные моменты времени. Плазму и СМЖ отбирали и анализировали, как описано выше. Результаты, полученные на этих крысах, показывают, что равновесие в плазме и отделениях СМЖ быстро устанавливается после дозировки, и что коэффициент распределения между СМЖ и плазмой был постоянным во всех временных точках измерения. Таким образом, в Таблице 22 приведены данные только для 2-минутных временных точек измерения.
Результаты в Таблице 22 представлены для каждой группы из 4-х крыс в виде усредненных концентраций указанных соединений в плазме или СМЖ. В Таблице 22 также представлен коэффициент распределения между плазмой и СМЖ (плазма/СМЖ), т.е. отношение концентрации в плазме к концентрации в СМЖ для указанных соединений.
Результаты в Таблице 22 говорят о том, что относительный коэффициент распределения между плазмой и СМЖ Соединения KC-5 и оксикодона составляет примерно 316 (т.е. 1 508/4,8); это означает, что Соединение KC-5 в 316 раз хуже проникает в СМЖ, чем оксикодон. Кроме того, как показано в Примере 24, относительная сила лекарственное средство/пролекарство Соединения KC-5 составляет приблизительно 50. Таким образом, ожидается, что Соединение KC-5 при введении внутривенно в эквимолярных количествах будет приблизительно в 15800 раз (т.е. 316×50) менее эффективно для СМЖ рецепторов µ-опиоида, чем оксикодон.
Пример 39: Фармакокинетика Соединения KC-6 после ПО введения крысам
Этот пример демонстрирует высвобождение оксикодона в плазму при пероральном (ПО) введении Соединения KC-6 крысам.
Растворы Соединения KC-6 в физиологическом растворе (которые можно приготовить, как описано в Примере 14) дозировали, как показано в Таблице 23, принудительным кормлением через желудочный зонд самцам крыс Спраг-Доули (4 на группу) с введенным в яремную вену катетером, которых подвергли голоданию за 16-18 часов до перорального введения. Через определенные промежутки времени брали пробы крови, выделяли плазму путем центрифугирования при 5400 об/мин при 4°С в течение 5 мин, и 100 мкл плазмы переносили из каждого образца в чистую пробирку, содержащую 2 мкл 50%-ной муравьиной кислоты. Пробирки встряхивали на шейкере типа Vortex в течение 5-10 секунд, немедленно помещали на сухой лед, а затем хранили при -80°С в морозильной камере до анализа с помощью ВЭЖХ/МС.
Таблица 23 и Фигура 19 представляют результаты воздействия оксикодона на группу крыс, которым перорально вводили Соединение КС-6. Результаты в Таблице 23 представлены для каждой группы крыс в виде (а) максимальной концентрации оксикодона (ОС) в плазме (Cmax) (среднее ± стандартное отклонение), (b) времени после введения Соединения KC-6, необходимого для достижения максимальной концентрации оксикодона (Tmax) (среднее ± стандартное отклонение) и (с) площади под кривой (AUC) (нг×ч)/мл от 0 до 8 часов (среднее ± стандартное отклонение).
Нижний предел количественного определения составлял 0,025 нг/мл
На Фигуре 19 приведено сравнение усредненных концентраций в плазме относительно времени высвобождения оксикодона после ПО введения Соединения KC-6.
Результаты в Таблице 23 и на Фигуре 19 указывают на то, что введение Соединения KC-6 создает концентрации оксикодона в плазме, которые демонстрируют пониженное Cmax и AUC и замедленное Tmax по сравнению с введением оксикодона (см. Пример 15).
Пример 40: Фармакокинетика Соединения KC-6 после ВВ введения крысам
В этом Примере сравнивают концентрации пролекарства и оксикодона в плазме крыс после внутривенного (ВВ) введения Соединения KC-6.
Соединение KC-6 (которое можно приготовить, как описано в Примере 14) растворили в физиологическом растворе и дозу 2 мг/кг вводили в хвостовую вену 4-м самцам крысы Спраг-Доули с введенным в яремную вену катетером. Через определенные промежутки времени брали пробы крови, выделяли плазму путем центрифугирования при 5400 об/мин при 4°С в течение 5 мин, и 100 мкл плазмы переносили из каждого образца в чистую пробирку, содержащую 2 мкл 50%-ной муравьиной кислоты. Пробирки встряхивали на шейкере типа Vortex в течение 5-10 секунд, немедленно помещали на сухой лед, а затем хранили при -80°С в морозильной камере до проведения анализа с помощью высокоэффективной жидкостной хроматографии/масс спектрометрии (ВЭЖХ/МС).
Таблица 24 и Фигура 20 представляют результаты воздействия Соединения KC-6 и оксикодона на группу крыс, которым внутривенно вводили Соединение KC-6. Результаты в Таблице 24 представлены в виде максимальной концентрации в плазме (Cmax) Соединения KC-6 и оксикодона (ОС) соответственно (среднее ± стандартное отклонение).
Таблица 24 и Фигура 20 показывают, что концентрации оксикодона в плазме крыс, которым внутривенно ввели Соединение KC-6, составляет только 0,015% от концентрации Соединения KC-6 в плазме, что говорит о том, что ВВ введение Соединения KC-6 не приводит к значительному высвобождению оксикодона в плазму.
Пример 41; Фармакокинетика после ВВ введения Соединения KC-6 крысам: проникновение в плазму и спинномозговую жидкость
В этом примере сравнивают концентрации пролекарства - Соединения KC-6 и оксикодона в плазме и спинномозговой жидкости (СМЖ) после внутривенного (ВВ) введения соответствующего соединения крысам. Коэффициент распределения между плазмой и СМЖ предсказывает способность соединения проникать сквозь гематоэнцефалический барьер.
Соединение KC-6 (которое может быть приготовлено, как описано в Примере 14) в дозе 7,5 мг/кг и оксикодон в дозе 7,5 мг/кг, каждый растворили в физиологическом растворе и ввели в хвостовую вену 4-м самцам крысы Спраг-Даули. Через 2 минут крыс анастезировали диоксидом углерода путем асфиксии, и брали пробы крови, выделяли плазму путем центрифугирования при 5400 об/мин при 4°С в течение 5 мин, и 100 (мкл) плазмы переносили из каждого образца в чистую пробирку, содержащую 2 мкл 50%-ной муравьиной кислоты. СМЖ брали, используя иглу размером 22×1 дюйм, присоединенную к полиуретановому катетеру с трубкой типа MRE-040 (Braintree Scientific, Inc.). Иглу вводили сразу ниже затылочного гребня в области большого затылочного отверстия, и прозрачную СМЖ собирали в катетер и переносили в сборную пробирку. Образцы СМЖ центрифугировали при 5400 об/мин при 4°С в течение 5 мин, и 100 мкл СМЖ из каждого образца переносили в чистую пробирку. Образцы плазмы и СМЖ немедленно помещали на сухой лед, а затем хранили при -80°С в морозильной камере до проведения анализа с помощью высокоэффективной жидкостной хроматографии/масс спектрометрии (ВЭЖХ/МС). С целью изучения зависимости проникновения Соединения KC-6 и оксикодона в плазму и СМЖ от времени, дополнительным группам из 4-х крыс вводили соединения, как описано выше, и анестезировали в определенные моменты времени. Плазму и СМЖ отбирали и анализировали, как описано выше. Результаты, полученные на этих крысах, показывают, что равновесие в плазме и отделениях СМЖ быстро устанавливается после дозировки, и что коэффициент распределения между СМЖ и плазмой был постоянным во всех временных точках измерения. Таким образом, в Таблице 25 приведены данные только для 2-минутных временных точек измерения.
Результаты в Таблице 25 представлены для каждой группы из 4-х крыс в виде усредненных концентраций указанных соединений в плазме или СМЖ. В Таблице 25 также представлен коэффициент распределения между плазмой и СМЖ (плазма/СМЖ), т.е. отношение концентрации в плазме к концентрации в СМЖ для указанных соединений.
Результаты в Таблице 25 говорят о том, что относительный коэффициент распределения между плазмой и СМЖ Соединения KC-6 и оксикодона составляет примерно 171 (т.е. 815/4,8); это означает, что Соединение KC-6 в 171 раз хуже проникает в СМЖ, чем оксикодон. Кроме того, как показано в Примере 24, относительная сила лекарственное средство/пролекарство Соединения KC-6 составляет приблизительно 23. Таким образом, ожидается, что Соединение KC-6 при введении внутривенно в эквимолярных количествах будет приблизительно в 3 940 раз (т.е. 171×23) менее эффективно для СМЖ рецепторов µ-опиоида, чем оксикодон.
Хотя данное изобретение описано со ссылкой на его специфические варианты осуществления, специалисту в данном уровне техники понятно, что могут быть сделаны различные изменения, и эквиваленты могут быть замещены без отступления от истинной сущности и объема данного изобретения. К тому же могут быть сделаны многочисленные модификации для приспосабливания конкретной ситуации, материала, композиции материала, процесса, этапа или этапов процесса к цели, сущности и объему данного изобретения. Предполагается, что все эти модификации охвачены объемом приложенной формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОЛЕКАРСТВА ДЕЙСТВУЮЩИХ ВЕЩЕСТВ ГЕТЕРОЦИКЛИЧЕСКИМИ ЛИНКЕРАМИ | 2012 |
|
RU2608305C2 |
| КОМПОЗИЦИИ, СОДЕРЖАЩИЕ РАСЩЕПЛЯЕМОЕ ФЕРМЕНТАМИ ПРОЛЕКАРСТВО ОКСИКОДОНА | 2012 |
|
RU2609412C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ С УМЕНЬШЕННЫМ ВЫСВОБОЖДЕНИЕМ ФЕНОЛЬНЫХ ОПИОИДОВ | 2009 |
|
RU2562583C2 |
| КОНЪЮГАТЫ ОКСИКОДОНА С БЕНЗОЙНОЙ КИСЛОТОЙ, ПРОИЗВОДНЫМИ БЕНЗОЙНОЙ КИСЛОТЫ И ГЕТЕРОАРИЛКАРБОНОВОЙ КИСЛОТОЙ | 2015 |
|
RU2683317C2 |
| БЕНЗОЙНАЯ КИСЛОТА, ПРОИЗВОДНЫЕ БЕНЗОЙНОЙ КИСЛОТЫ И КОНЪЮГАТЫ ГЕТЕРОАРИЛКАРБОНОВОЙ КИСЛОТЫ С ГИДРОМОРФОНОМ, ПРОЛЕКАРСТВА, СПОСОБЫ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2573388C2 |
| ПРОЛЕКАРСТВА МЕТИЛГИДРОФУМАРАТА, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ С НИМИ И СПОСОБЫ ПРИМЕНЕНИЯ | 2009 |
|
RU2554347C2 |
| КОНЪЮГАТЫ ГИДРОКОДОНА С БЕНЗОЙНОЙ КИСЛОТОЙ, ПРОИЗВОДНЫМИ БЕНЗОЙНОЙ КИСЛОТЫ И ГЕТЕРОАРИЛКАРБОНОВОЙ КИСЛОТОЙ, ПРОЛЕКАРСТВА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2010 |
|
RU2505541C2 |
| ИНГИБИТОРЫ GSK-3 | 2005 |
|
RU2379300C2 |
| ПРОЛЕКАРСТВА ЛЕВОДОПА, КОМПОЗИЦИИ НА ИХ ОСНОВЕ И ИХ ПРИМЕНЕНИЯ | 2005 |
|
RU2365580C2 |
| КОМБИНИРОВАНИЕ ИНГИБИТОРОВ FBP-АЗЫ И АНТИДИАБЕТИЧЕСКИХ СРЕДСТВ ДЛЯ ЛЕЧЕНИЯ ДИАБЕТА | 2001 |
|
RU2328308C2 |
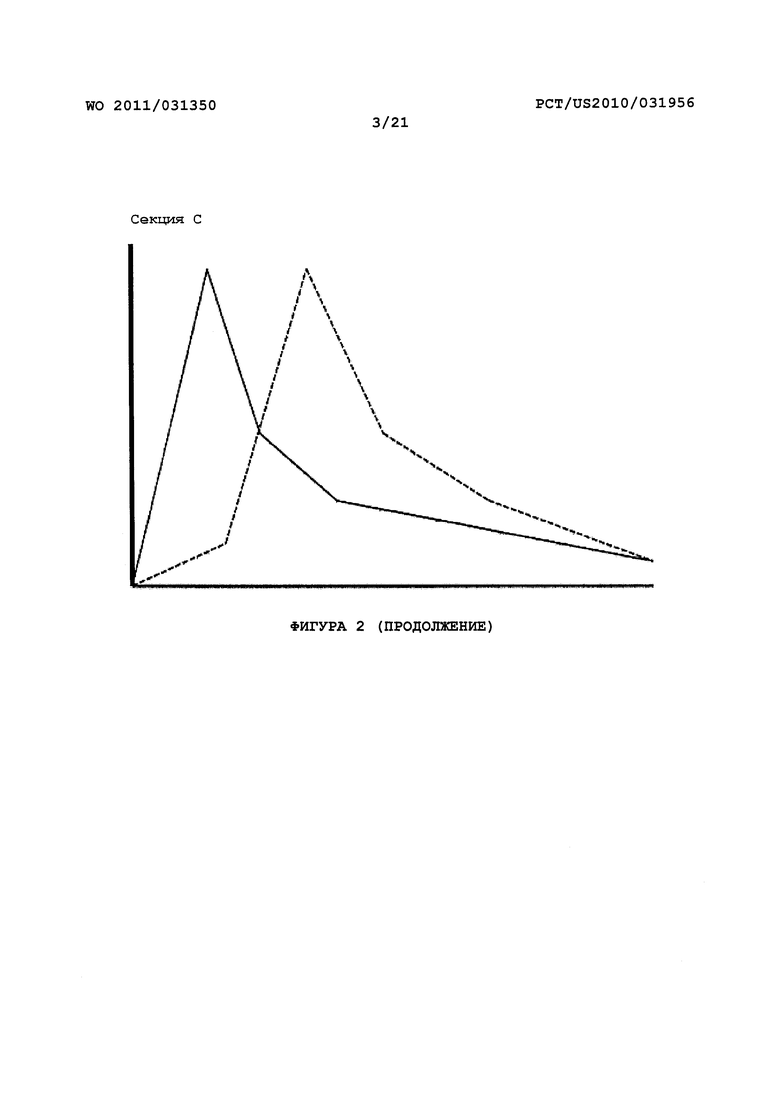
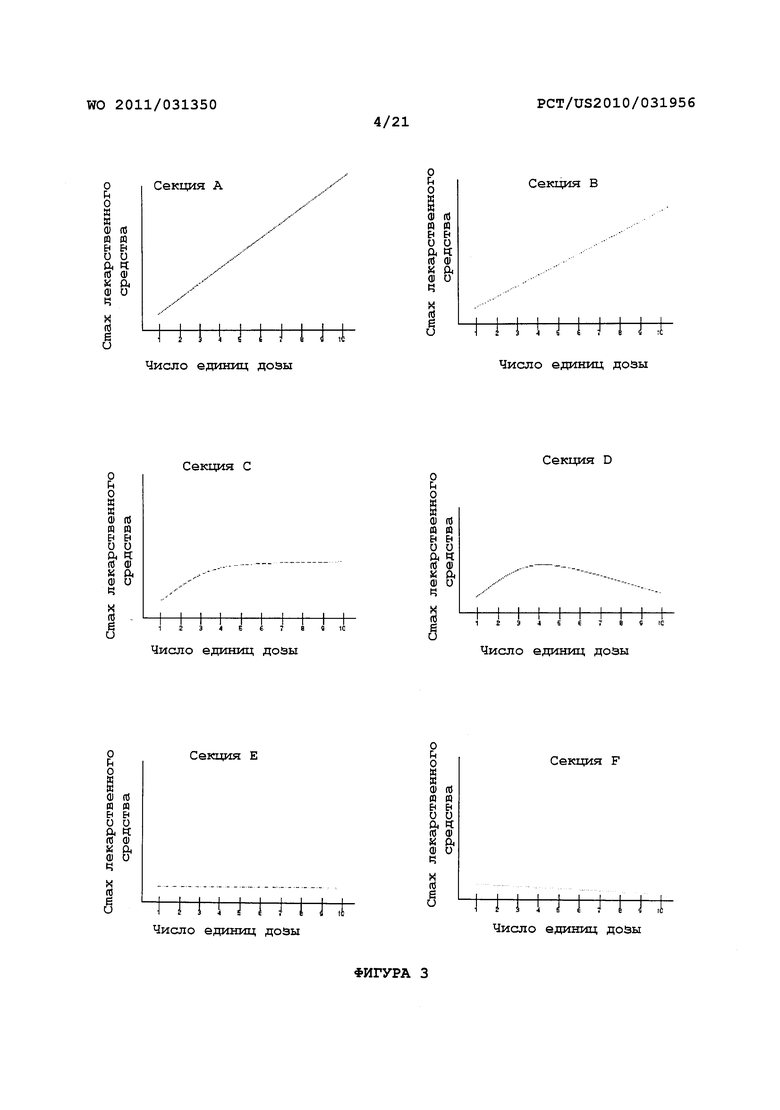
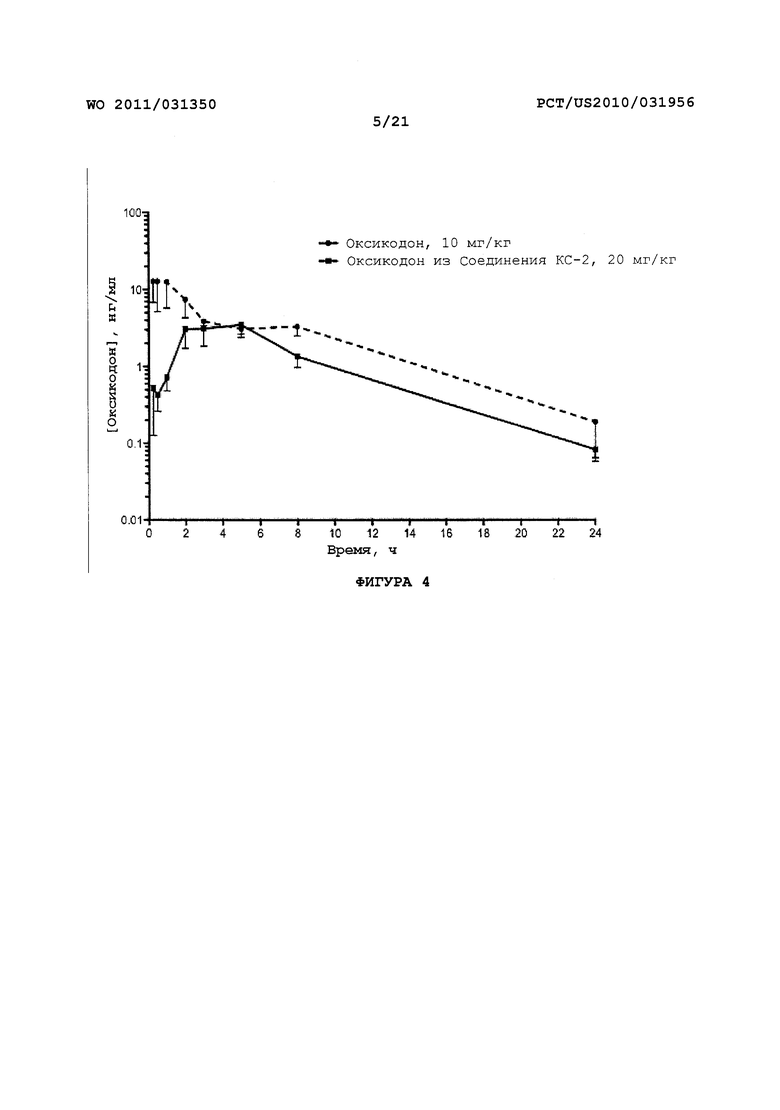
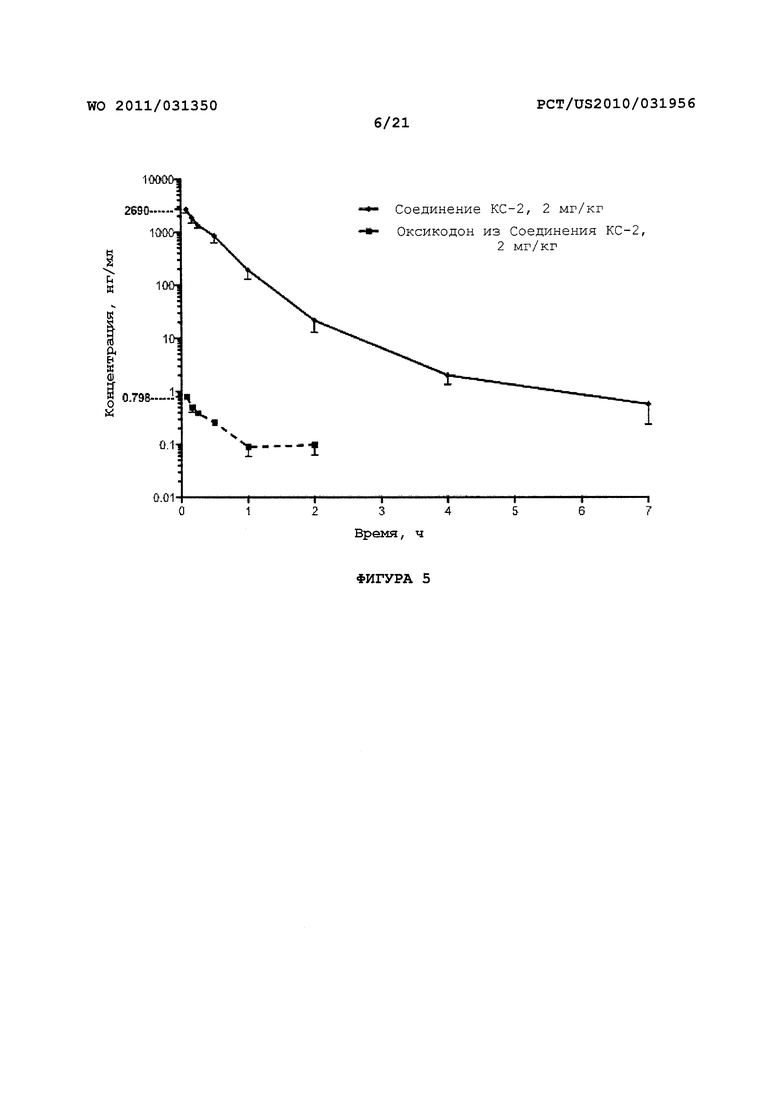
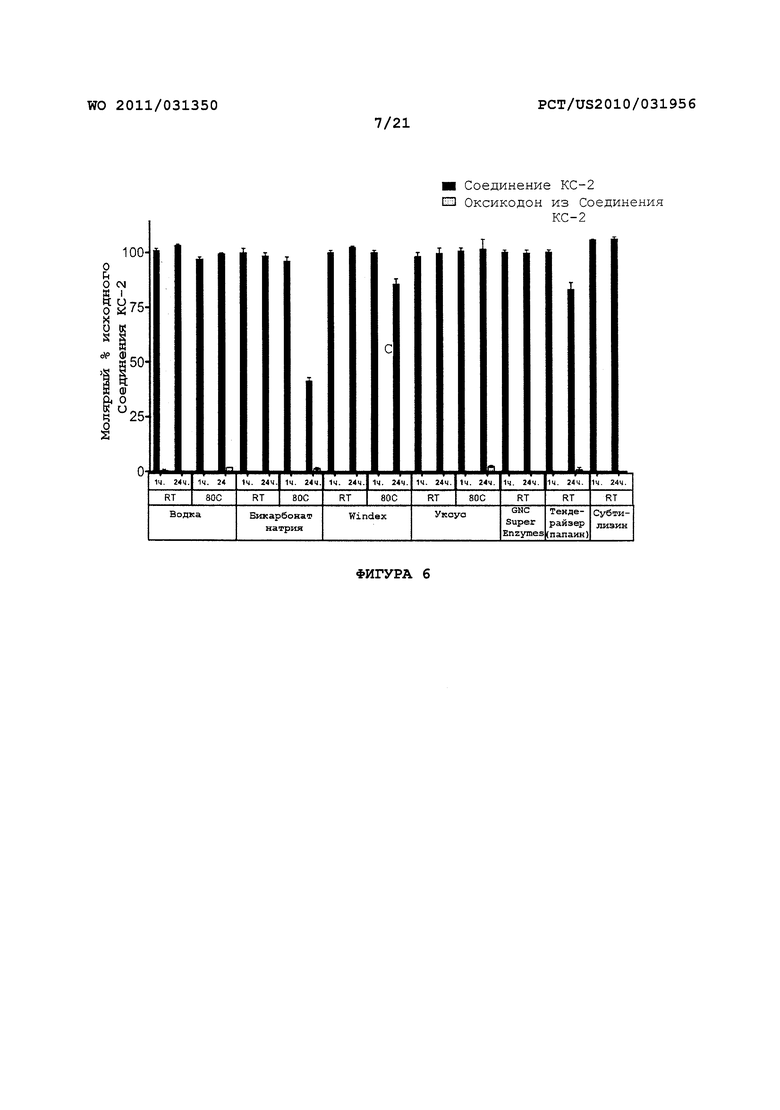
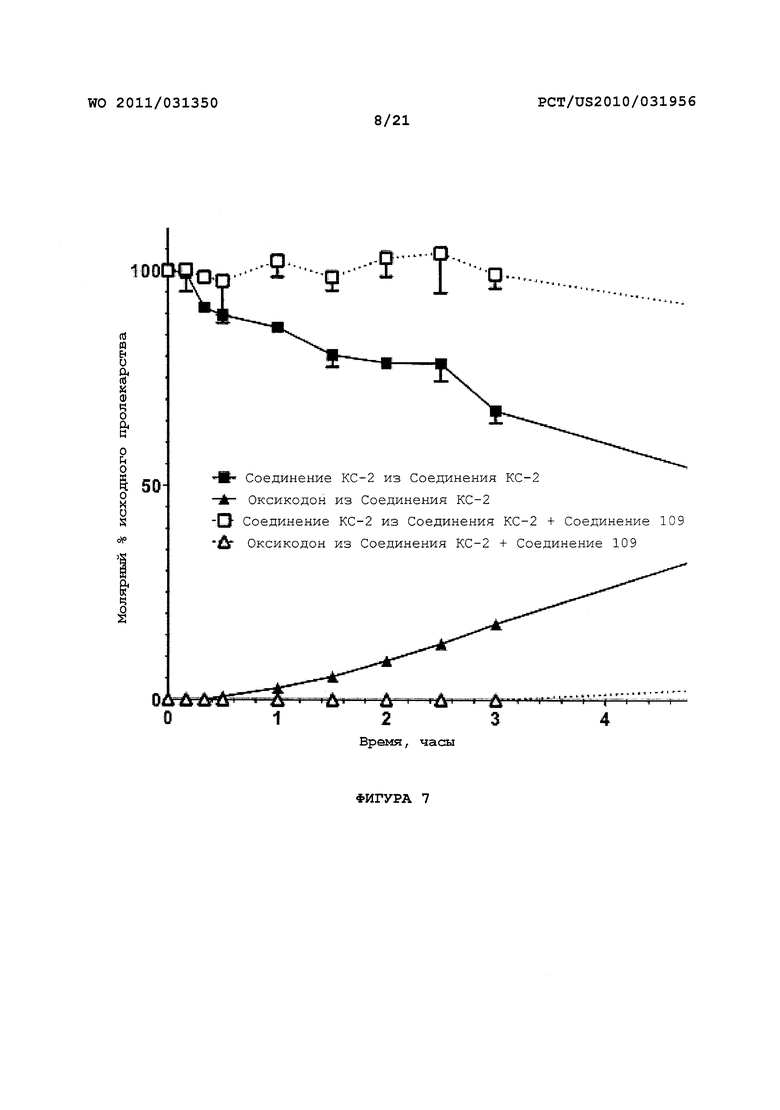
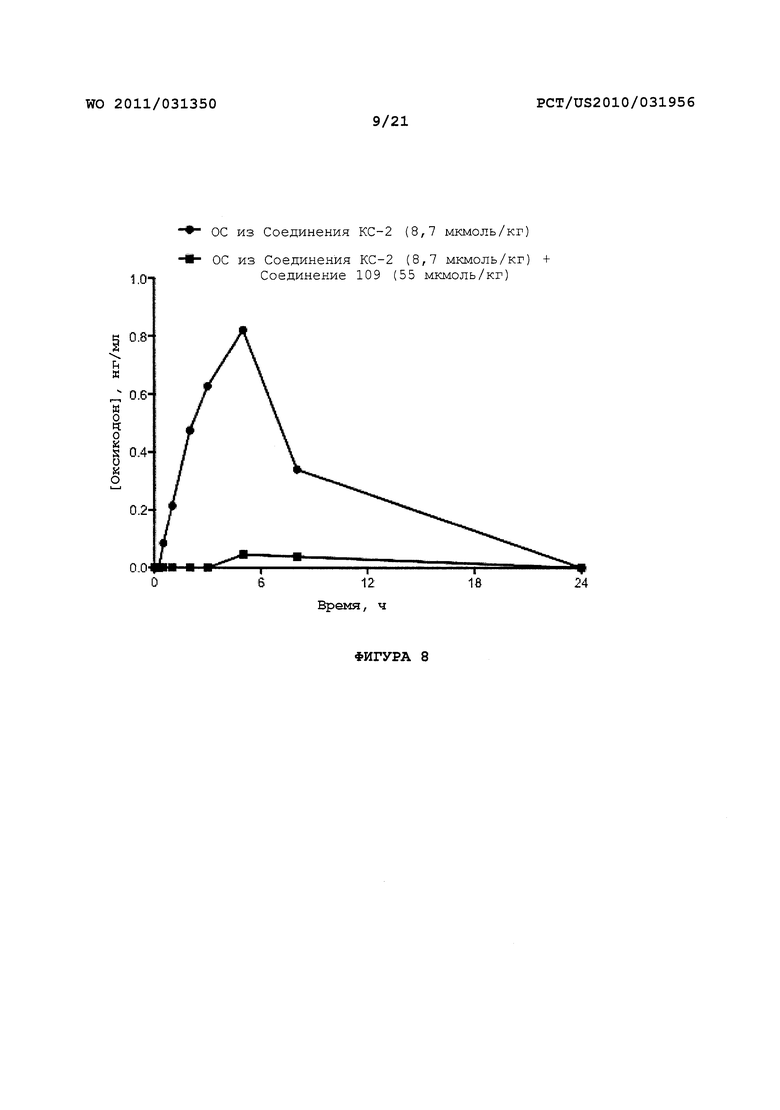
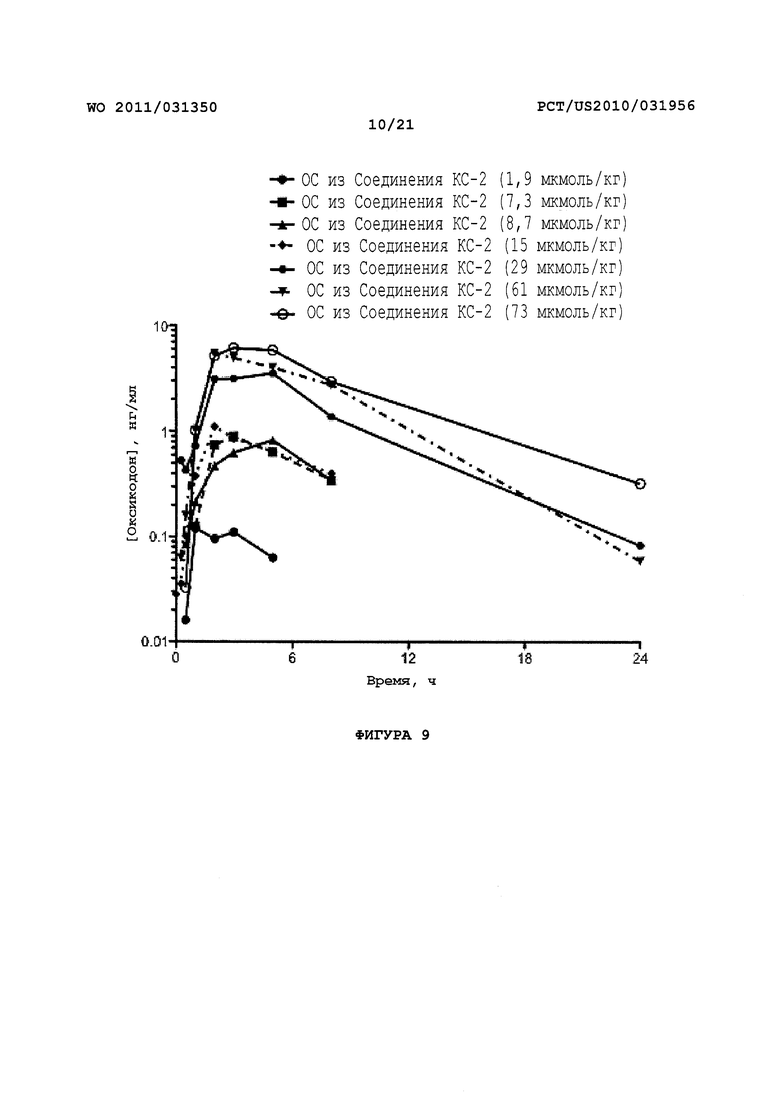
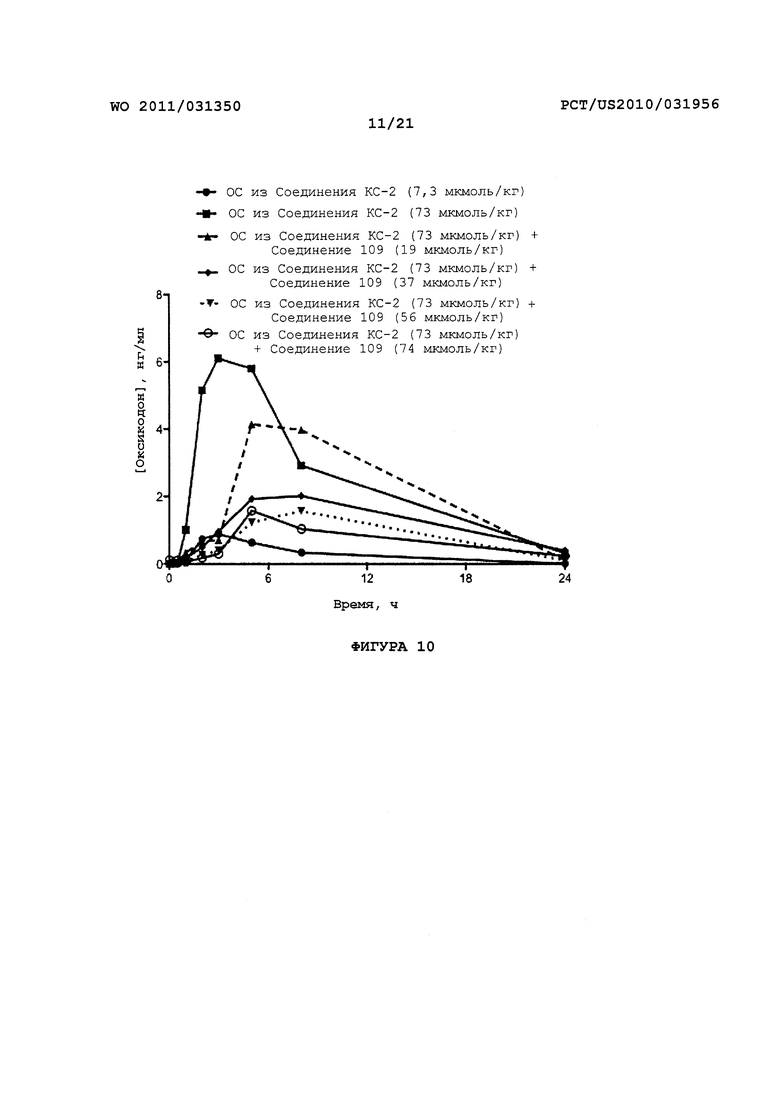
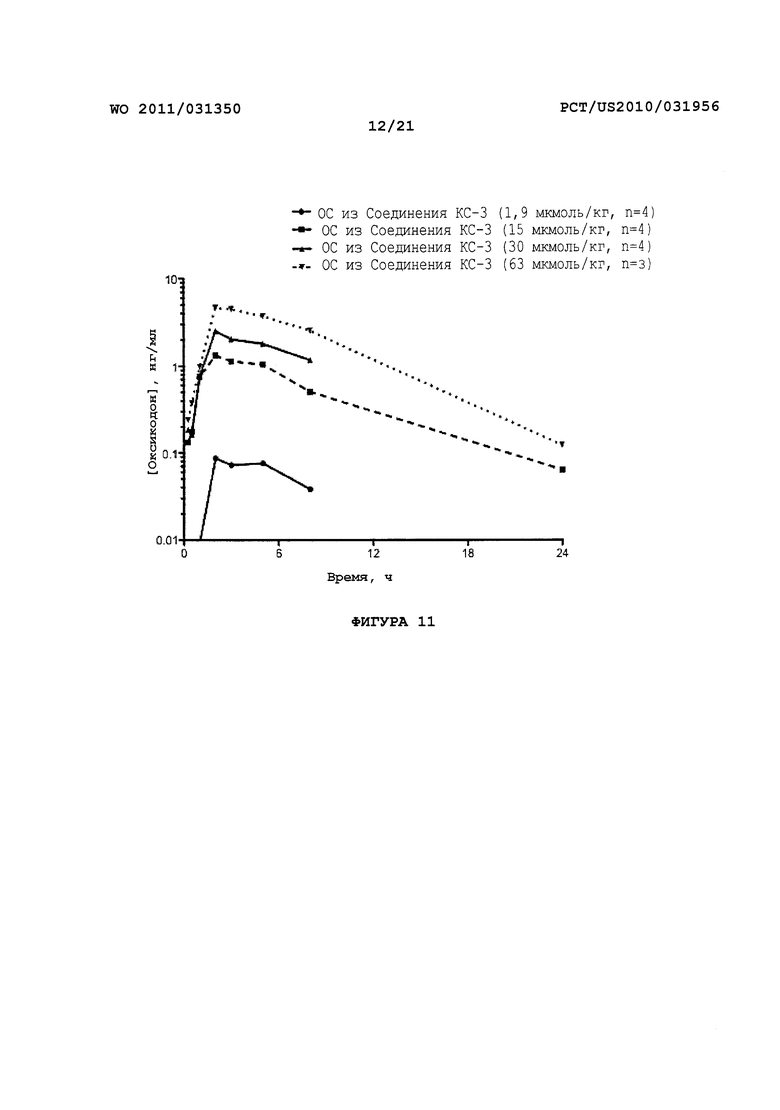
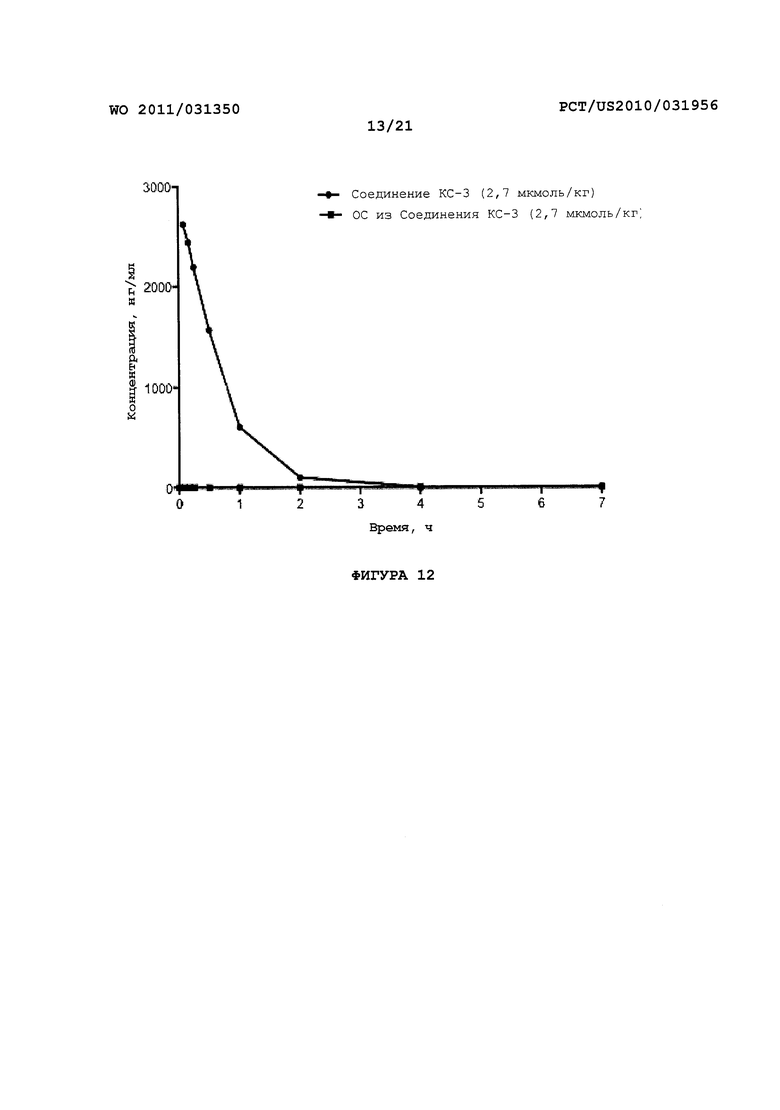
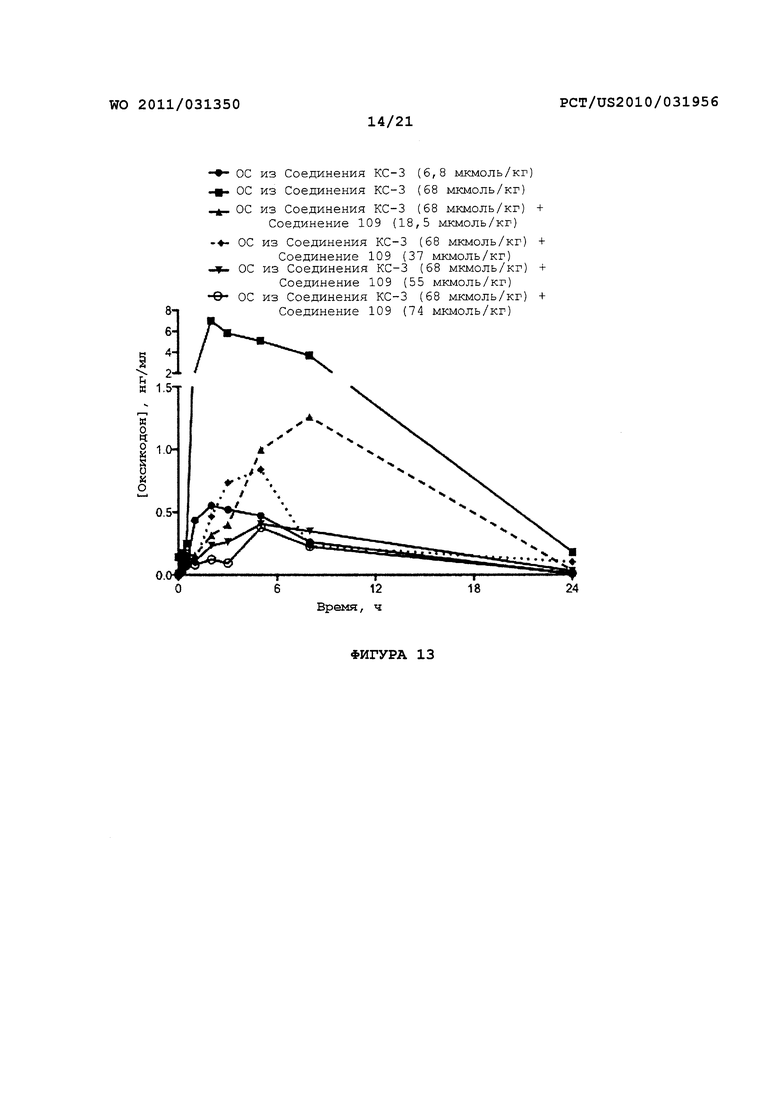
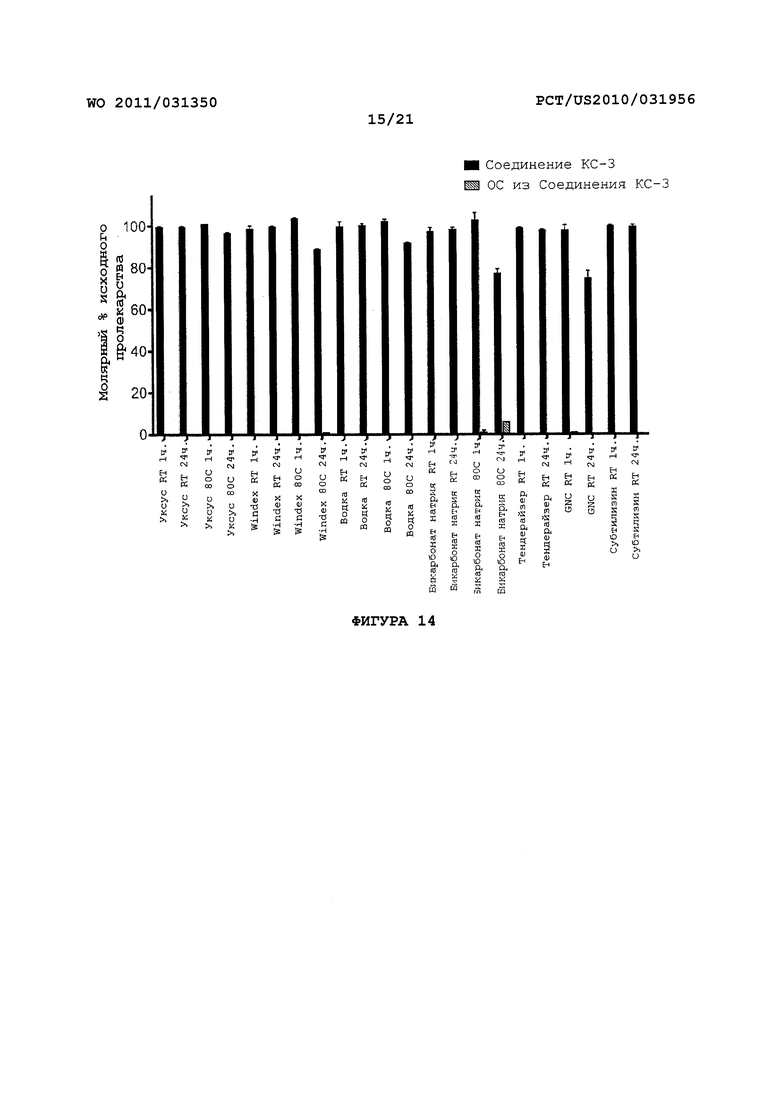
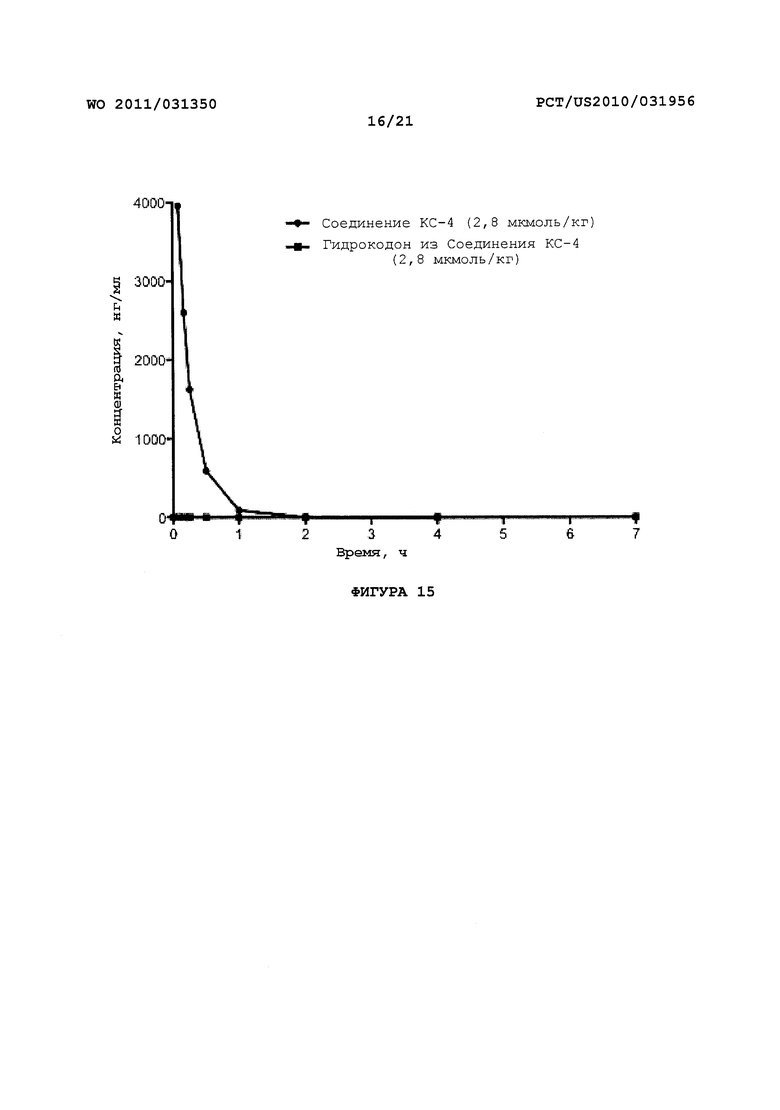
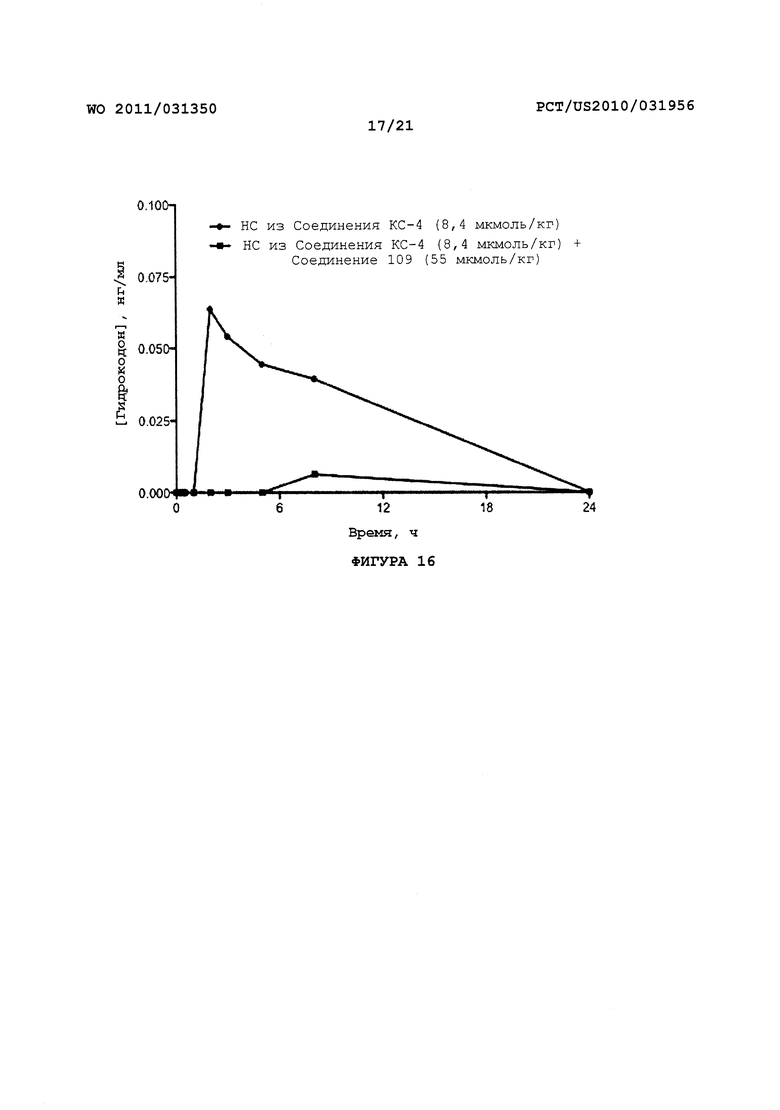
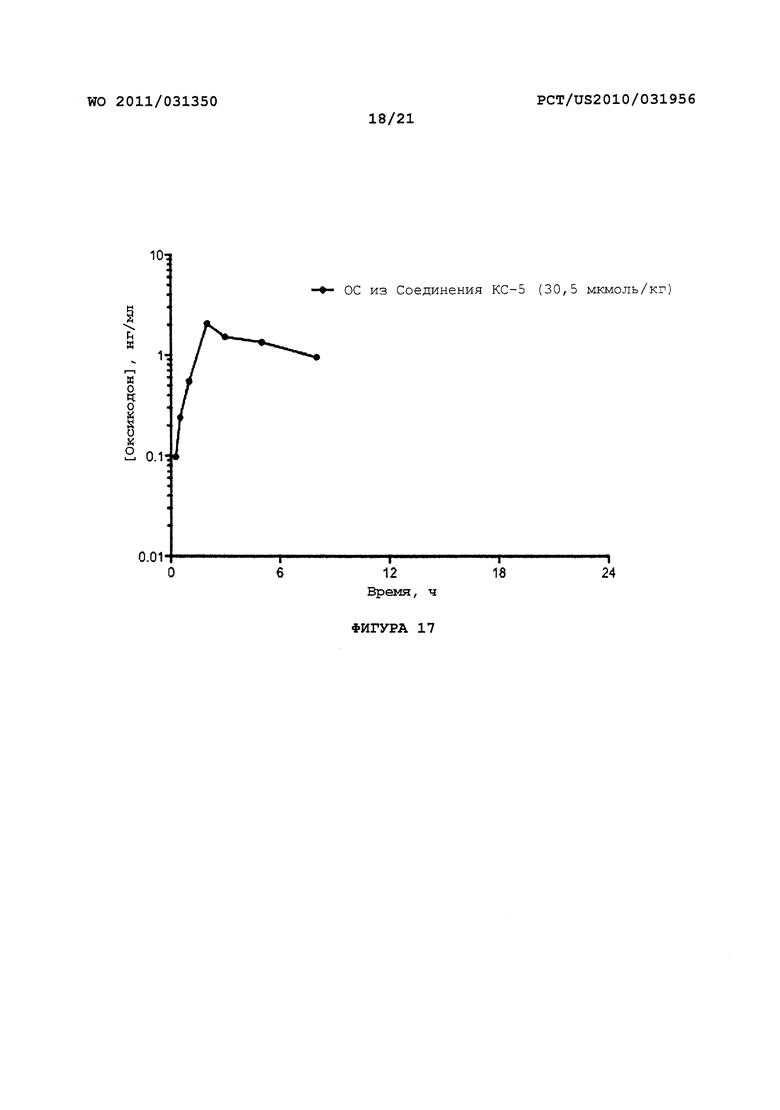
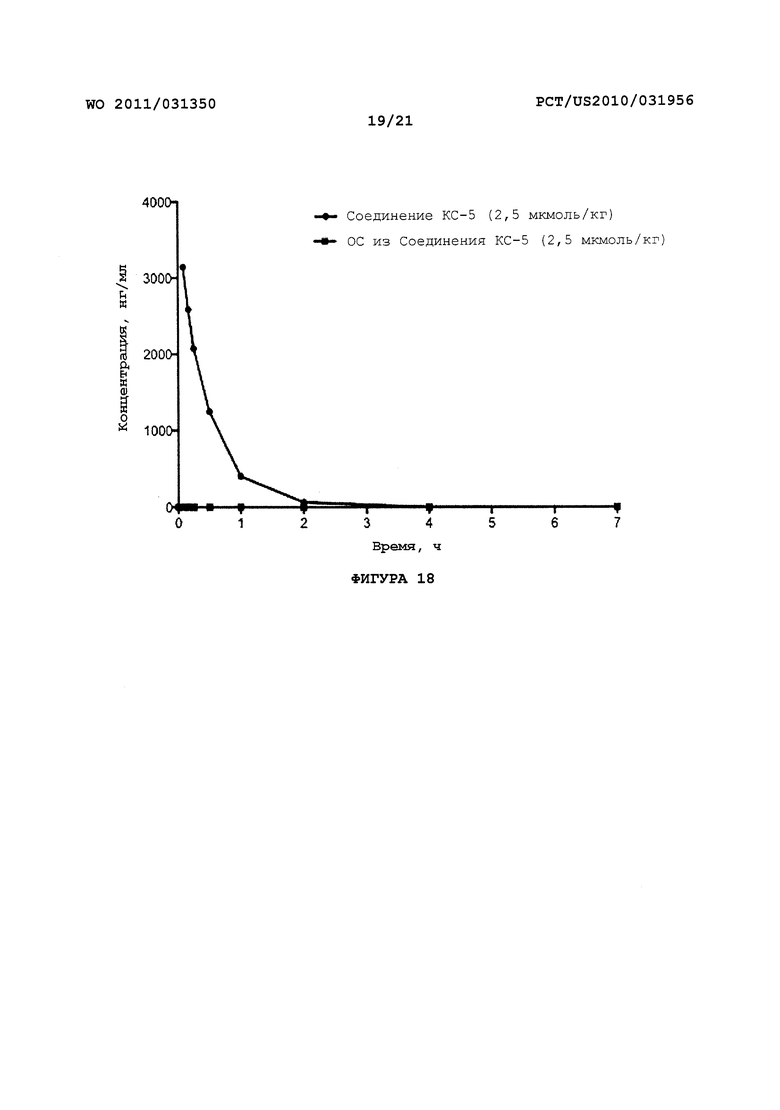
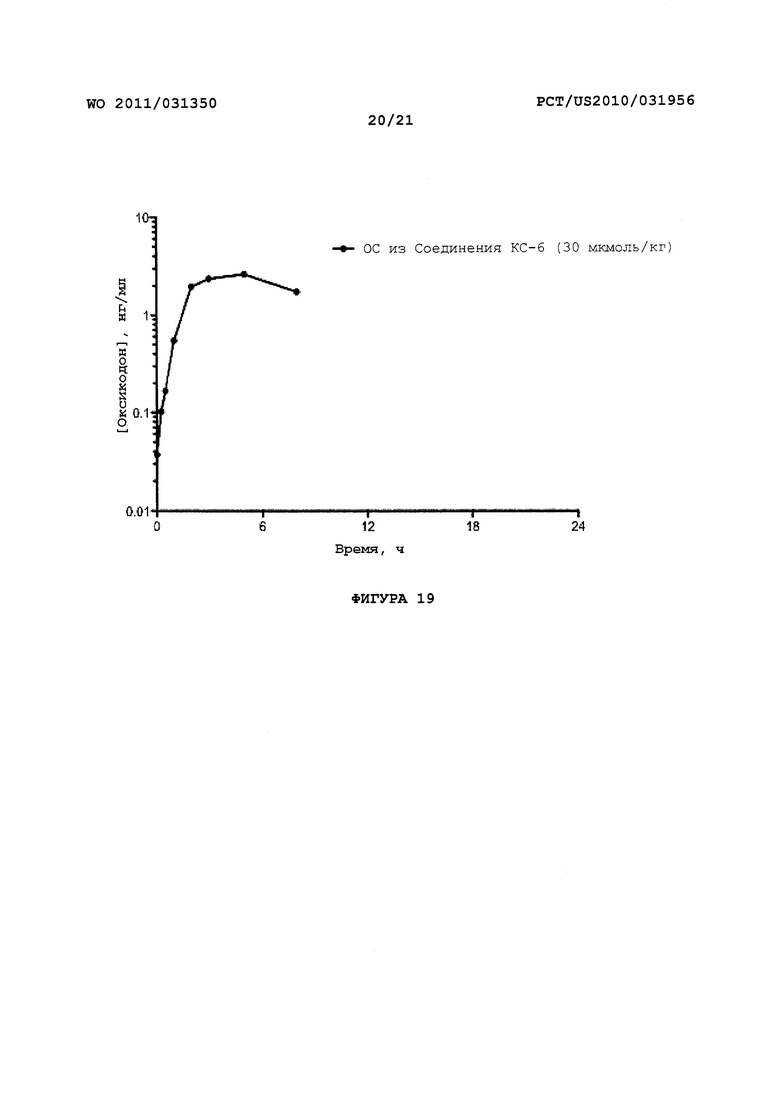
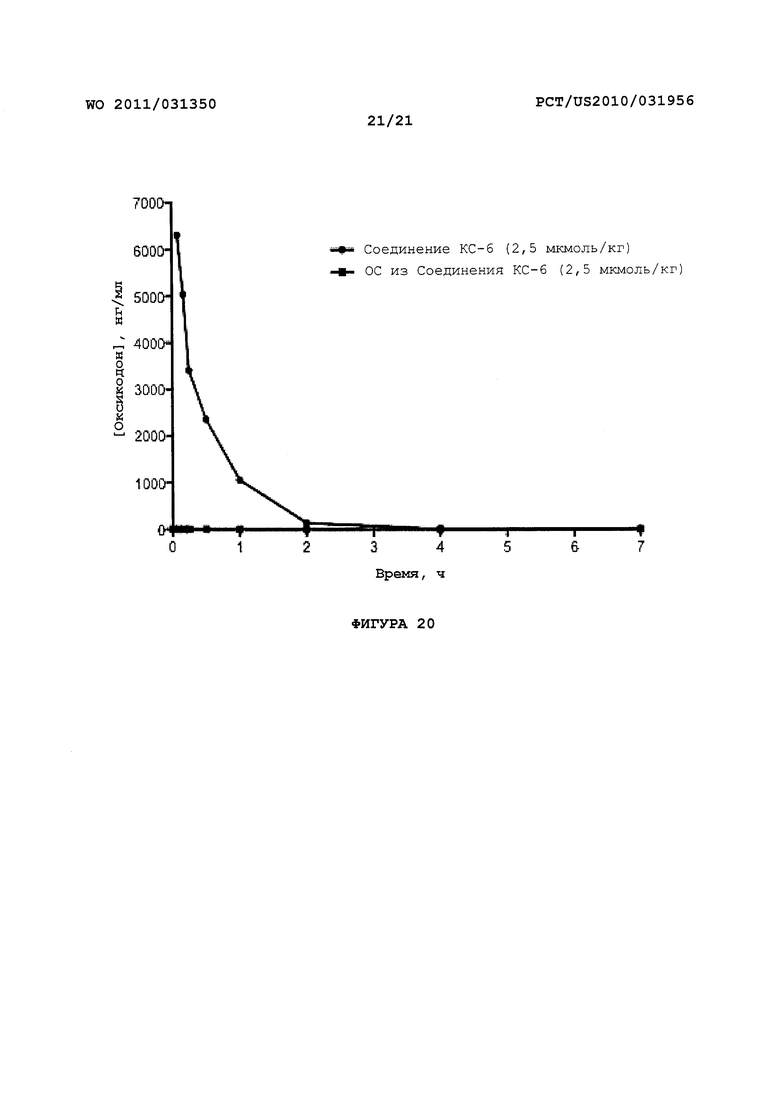
Изобретение относится к соединениям формулы КС-(II), их применению лечения или предупреждения боли, способу их получения, фармацевтическим композициям на их основе, единичной дозе их содержащей, способу ее получения, способу идентификации соединения формулы КС-(II), способу уменьшения потенциальной возможности злоупотребления композицией, содержащей соединение формулы КС-(II). В общей формуле КС-(II) Ra представляет собой водород или гидроксил; R5 выбран из (1-6С)алкила и (1-6С)алкила, замещенного (1-6С)алкоксикарбонильной группой; каждый R1 независимо выбран из водорода и (1-6С)алкила; каждый R2 независимо выбран из водорода, (1-6С)алкила, и -C(O)NR21R22, где R21 и R22 независимо выбраны из группы, состоящей из водорода, (1-6С)алкила и (1-6С)алкила, замещенного -C(O)OR60, где R60 означает водород; n равно целому числу от 2 до 4; R3 является водородом; R4 представляет собой 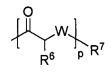 , где каждый R6 независимо выбран из водорода и (1-6С)алкила, замещенного гуанидильной группой или аминогруппой; W представляет собой -NR8-; R8 представляет собой водород; p равно 1 или 2; и R7 выбран из ацила и ацила, замещенного -СООН или -NHCOCH3. 10 н. и 8 з.п. ф-лы, 20 ил., 25 табл., 41 пр.
, где каждый R6 независимо выбран из водорода и (1-6С)алкила, замещенного гуанидильной группой или аминогруппой; W представляет собой -NR8-; R8 представляет собой водород; p равно 1 или 2; и R7 выбран из ацила и ацила, замещенного -СООН или -NHCOCH3. 10 н. и 8 з.п. ф-лы, 20 ил., 25 табл., 41 пр.
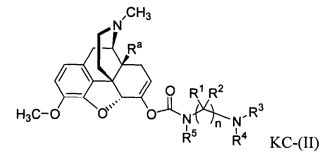
1. Соединение формулы KC-(II):
в которой:
Ra представляет собой водород или гидроксил;
R5 выбран из (1-6С)алкила и (1-6С)алкила, замещенного (1-6С)алкоксикарбонильной группой;
каждый R1 независимо выбран из водорода и (1-6С)алкила;
каждый R2 независимо выбран из водорода, (1-6С)алкила и -C(O)NR21R22, где R21 и R22 независимо выбраны из группы, состоящей из водорода, (1-6С)алкила и (1-6С)алкила, замещенного -C(O)OR60, где R60 означает водород;
n равно целому числу от 2 до 4;
R3 является водородом;
R4 представляет собой ,
где каждый R6 независимо выбран из водорода и (1-6С)алкила, замещенного гуанидильной группой или аминогруппой;
W представляет собой -NR8-;
R8 представляет собой водород;
p равно 1 или 2; и
R7 выбран из ацила и ацила, замещенного -СООН или -NHCOCH3;
или его соль.
2. Соединение по п. 1, в котором R6 выбирают из боковых цепей лизина (например, L-лизина), аргинина (например, L-аргинина), гомолизина, гомоаргинина и орнитина.
3. Соединение по п. 1, в котором R6 представляет собой боковую цепь L-лизина.
4. Соединение по п. 1, в котором R6 представляет собой боковую цепь L-аргинина.
5. Способ получения соединения по п. 1, включающий:
взаимодействие соединения формулы:  с соединением формулы
с соединением формулы  , где PG1 и PG2 являются защитными группами для амина, и
, где PG1 и PG2 являются защитными группами для амина, и
удаление защитных групп PG1 и PG2 с получением соединения формулы
 ;
;
приведение в контакт соединения формулы 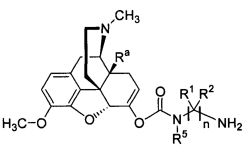 с соединением формулы
с соединением формулы 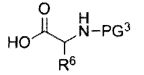 , где PG3 является защитной группой аминной функции; и снятие защитной группы PG3.
, где PG3 является защитной группой аминной функции; и снятие защитной группы PG3.
6. Фармацевтическая композиция для лечения или предупреждения боли, содержащая
соединение по любому из пп. 1-4; и
фармацевтически приемлемый носитель.
7. Фармацевтическая композиция для лечения или предупреждения боли, содержащая соединение по любому из пп. 1-4, которое обеспечивает ферментативно-регулируемое высвобождение кетон-содержащего опиоида, и ингибитор трипсина.
8. Фармацевтическая композиция для лечения или предупреждения боли, содержащая:
соединение по любому из пп. 1-4, содержащее кетон-содержащий опиоид, ковалентно связанный с про-фрагментом, содержащим расщепляемый трипсином фрагмент, где расщепление трипсином расщепляемого трипсином фрагмента способствует высвобождению кетон-содержащего опиоида; и
трипсиновый ингибитор, который взаимодействует с трипсином, выступающим посредником в ферментативно-регулируемом высвобождении кетон-содержащего опиоида из указанного соединения после приема композиции внутрь.
9. Единичная доза, содержащая композицию по п. 8, где указанное соединение и трипсиновый ингибитор присутствуют в количествах, эффективных для обеспечения предварительно выбранной фармакокинетической (ФК) кривой после приема внутрь.
10. Единичная доза по п. 9, где единичная доза обеспечивает выбранную заранее ФК кривую после приема внутрь по меньшей мере двух единичных доз.
11. Способ получения единичной дозы по п. 9, включающий:
объединение соединения по любому из пп. 1-4 и трипсинового ингибитора;
где указанное соединение и трипсиновый ингибитор присутствуют в единичной дозе в количествах, эффективных для снижения высвобождения кетон-содержащего опиоида из указанного соединения таким образом, что прием внутрь пациентом множества единичных доз не приводит к пропорциональному высвобождению кетон-содержащего опиоида.
12. Способ идентификации соединения по любому из пп. 1-4 и трипсинового ингибитора, пригодных для составления единичной дозы, включающий:
объединение соединения по любому из пп. 1-4, трипсинового ингибитора и трипсина в реакционной смеси или
введение соединения по любому из пп. 1-4 и трипсинового ингибитора животному или в ткани животного,
где указанное соединение содержит кетон-содержащий опиоид, ковалентно связанный с про-фрагментом, содержащим расщепляемый трипсином фрагмент, где расщепление трипсином расщепляемого трипсином фрагмента способствует высвобождению кетон-содержащего опиоида; и
определение конверсии указанного соединения,
где снижение конверсии указанного соединения в присутствии трипсинового ингибитора по сравнению с конверсией соединения в отсутствие трипсинового ингибитора указывает на то, что данное соединение и трипсиновый ингибитор пригодны для составления единичной дозы.
13. Соединение по любому из пп. 1-4 или композиция по любому из пп. 6-8, или единичная доза по любому из пп. 9 и 10, для использования с целью лечения или предупреждения боли у нуждающегося в этом пациента.
14. Соединение по любому из пп. 1-4 для использования с целью лечения или предупреждения боли у нуждающегося в этом пациента.
15. Применение соединения по любому из пп. 1-4 или композиции по любому из пп. 6-8, или единичной дозы по любому из пп. 9 и 10 для получения лекарственного средства, предназначенного для лечения или предупреждения боли у нуждающихся в этом пациентов.
16. Способ уменьшения потенциальной возможности злоупотребления композицией, содержащей соединение по любому из пп. 1-4, включающий объединение соединения по любому из пп. 1-4 с трипсиновым ингибитором, где трипсиновый ингибитор уменьшает возможность потребителя высвобождать кетон-модифицированный опиоид из указанного соединения путем добавления трипсина.
17. Соединение по п. 1, в котором n равно 3, каждый R1 независимо выбран из водорода и (1-6С)алкила.
18. Соединение по п. 1, в котором каждый R2 независимо выбран из водорода и (1-6С)алкила.
| US 20090137618 A1, 28.05.2009 | |||
| US 20090209569 A1, 20.08.2009 | |||
| US 20050176644 A1, 11.08.2005 | |||
| СЛОЖНЫЕ ЭФИРЫ N-ЗАМЕЩЕННЫХ 14-ГИДРОКСИМОРФИНАНОВ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2002 |
|
RU2215741C1 |

