Область техники, к которой относится изобретение
Настоящее изобретение относится к способу получения соединения бензо[b]тиофена.
Предшествующий уровень техники
Соединение 4-(1-пиперазинил)бензо[b]тиофена, представленное формулой (1):
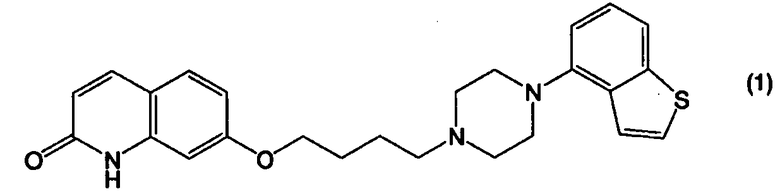
применимо для различных лекарственных препаратов, таких как антипсихотические препараты. Кроме того, соединение 4-(1-пиперазинил)бензо[b]тиофена, представленное формулой (4):
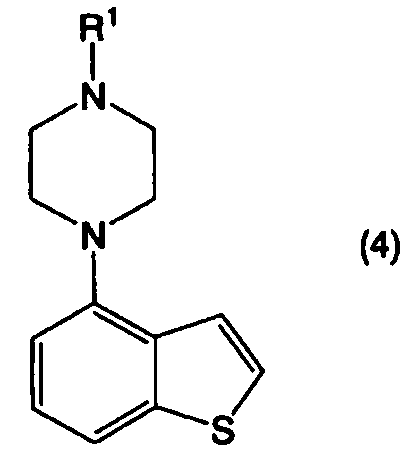
в котором R1 представляет собой атом водорода или защитную группу, применимо в качестве промежуточного соединения в синтезе соединения, представленного формулой (1).
В справочном примере 30 и примере 1 из PTL 1 конкретно описан способ получения соединение бензо[b]тиофена (схема реакции приведена ниже). В справочном примере 30, 4-(1-пиперазинил)бензо[b]тиофен получают кипячением смеси, содержащей 14,4 г 4-бромбензо[b]тиофена, 29,8 г безводного пиперазина, 9,3 г третбутилата натрия, 0,65 г (R)-(+)-2,2′-бис(дифенилфосфино)-1,1′-динафтила (BINAP), 0,63 г трис(дибензилиденацетон)дипалладия(0) и 250 мл толуола (стадия Х).
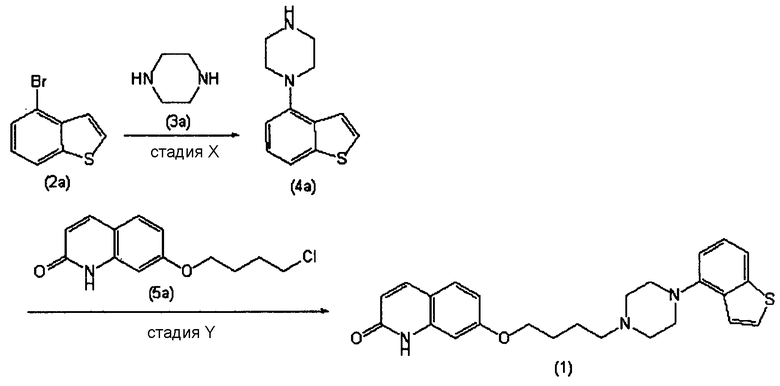
Однако в результате реакции на стадии Х образуется сравнительно большое количество трудноотделяемых побочных продуктов, и за счет этого чистота соединения (4а) неизбежно снижается. Кроме того, хотя для повышения чистоты соединения (4а) проводят очистку колоночной хроматографией, полностью удалить побочные продукты очень сложно, даже путем очистки колоночной хроматографией. Поэтому существует необходимость в новом способе получения соединения (4а) с высоким выходом и высокой чистотой.
Более того, побочные продукты, содержащиеся в соединении (4а), неизбежно снижают чистоту соединения (1) на последующей стадии Y. Поскольку в способе, описанном в PTL 1, для получения требуемого соединения (1) с высокой чистотой требуется очистка методом колоночной хроматографии, данный способ не подходит для промышленного способа крупномасштабного производства. Кроме того, согласно данному способу, неизбежно включение трудноотделяемых побочных продуктов, и продукты высокой чистоты, применимые в качестве активных фармацевтических ингредиентов, нельзя получить без очистки колоночной хроматографией.
Список ссылок
Патентная литература
PTL 1: нерассмотренная японская патентная публикация № 2006-316052
Непатентная литература
NPL 1: Tetrahedron Lett., 2004, 45, 9645
Сущность изобретения
Техническая проблема
Цель настоящего изобретения состоит в предоставлении нового способа получения, при помощи которого можно эффективно получать промышленным способом соединение, представленное формулой (1), или его соль. Другая цель настоящего изобретения заключается в предоставлении нового способа получения, при помощи которого можно эффективно получать промышленным способом соединение 4-(1-пиперазинил)бензо[b]тиофена, представленное формулой (4), которое является предшественником соединения, представленного формулой (1).
Решение проблемы
Для решения данной проблемы авторы настоящего изобретения провели подробное исследование, и обнаружили, что требуемые соединения можно получить при помощи определенных стадий с высокими выходами и высокой чистотой, подавляя, в то же время, образование побочных продуктов, без проведения очистки колоночной хроматографией. Настоящее изобретение было завершено на основе данных открытий.
В настоящем изобретении предоставлены способы согласно следующим пунктам с I-1 по I-19, пунктам с II-1 по II-21, и пунктам с III-1 по III-39.
Пункт I-1. Способ получения соединения, представленного формулой (4):
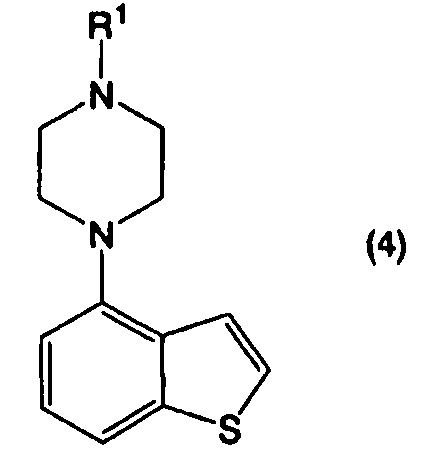
в котором R1 представляет собой атом водорода или защитную группу, или его соли, реакцией соединения, представленного формулой (2):
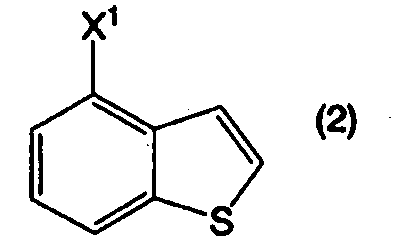
в котором Х1 представляет собой уходящую группу, с соединением, представленным формулой (3):
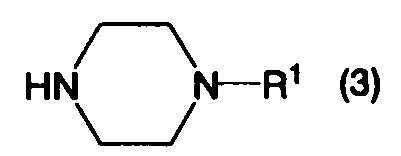
в котором R1 определен выше, или его солью, при этом данный способ включает:
стадию А: взаимодействие соединения, представленного формулой (2), с соединением, представленным формулой (3), или его солью, в присутствии (а) соединения палладия и третичного фосфина, или (b) карбенового комплекса палладия, в инертном растворителе или в отсутствие растворителя.
Пункт I-2. Способ по пункту I-1, в котором на стадии А соединение, представленное формулой (2), вводят во взаимодействие с соединением, представленным формулой (3), или его солью, в присутствии (а) соединения палладия и третичного фосфина, в инертном растворителе или в отсутствие растворителя.
Пункт I-3. Способ по пункту I-1, в котором на стадии А соединение, представленное формулой (2), вводят во взаимодействие с соединением, представленным формулой (3), или его солью, в присутствии (b) карбенового комплекса палладия, в инертном растворителе или в отсутствие растворителя.
Пункт I-4. Способ по любому из пунктов с I-1 по I-3, в котором третичный фосфин представляет собой, по меньшей мере, одно соединение, выбранное из группы, включающей три-третбутилфосфин, 2-(дитретбутилфосфино)-1,1′-дифенил, 2-(дитретбутилфосфино)-2′-метил-1,1′-дифенил, 2-(дитретбутилфосфино)-1,1′-динафтил, 2-дициклогексилфосфино-2′,6′-диметокси-1,1′-дифенил, 2-дициклогексилфосфино-2′,6′-диизопропокси-1,1′-дифенил, N-фенил-2-(дитретбутилфосфино)пиррол и 1-фенил-2-(дитретбутилфосфино)-1Н-инден.
Пункт I-5. Способ по любому из пунктов с I-1 по I-4, в котором карбеновый комплекс палладия представляет собой, по меньшей мере, один комплекс, выбранный из группы, включающей (1,4-нафтохинон)-[1,3-бис(2,6-диизопропилфенил)имидазол-2-илиден]палладий(0), аллилхлор-[1,3-бис(2,6-диизопропилфенил)имидазол-2-илиден]палладий(II), аллилхлор-[1,3-бис(2,6-диизопропилфенил)-4,5-дигидроимидазол-2-илиден]палладий(II) и (3-фенилаллилхлор)-[1,3-бис(2,6-диизопропилфенил)-4,5-дигидроимидазол-2-илиден]палладий(II).
Пункт I-6. Способ по любому из пунктов с I-1 по I-5, в котором третичный фосфин представляет собой три-третбутилфосфин и/или 2-дициклогексилфосфин-2′,6′-диизопропокси-1,1′-дифенил.
Пункт I-7. Способ по любому из пунктов с I-1 по I-6, в котором соединение палладия представляет собой, по меньшей мере, одно соединение, выбранное из группы, включающей тетрагидрат гексахлорпалладата(IV) натрия, гексахлорпалладат(IV) калия, хлорид палладия(II), бромид палладия(II), ацетат палладия(II), ацетилацетонат палладия(II), дихлорбис(бензонитрил) палладий(II), дихлорбис(ацетонитрил)палладий(II), дихлорбис(трифенилфосфин)палладий(II), дихлортетрааммиакат палладия(II), дихлор(циклоокта-1,5-диен)палладий(II), трифторацетат палладия(II), трис(дибензилиденацетон)дипалладий(0), комплекс трис(дибензилиденацетон)дипалладия(0) с хлороформом и тетракис(трифенилфосфин)палладий(0).
Пункт I-8. Способ по любому из пунктов с I-1 по I-7, в котором соединение палладия представляет собой ацетат палладия(II).
Пункт I-9. Способ по любому из пунктов с I-1 по I-8, в котором карбеновый комплекс палладия представляет собой аллилхлор-[1,3-бис(2,6-диизопропилфенил)-4,5-дигидроимидазол-2-илиден]палладий(II).
Пункт I-10. Способ по любому из пунктов с I-1 по I-9, в котором на стадии А инертный растворитель представляет собой ксилол и/или толуол.
Пункт I-11. Способ по любому из пунктов с I-1 по I-10, в котором уходящая группа, представленная Х1 в формуле (2), является атомом галогена.
Пункт I-12. Способ по пункту I-11, в котором уходящая группа, представленная Х1 в формуле (2), является атомом хлора.
Пункт I-13. Способ по любому из пунктов с I-1 по I-12, в котором соединение палладия используют в количестве от 0,01 до 5 мольных % из расчета на атом палладия, на моль соединения, представленного формулой (2), а третичный фосфин используют в количестве от 0,01 до 1000 моль на моль атома палладия в соединении палладия.
Пункт I-14. Способ по пункту I-13, в котором соединение палладия используют в количестве от 0,05 до 0,5 мольных % из расчета на атом палладия, на моль соединения, представленного формулой (2).
Пункт I-15. Способ по пункту I-13 или I-14, в котором третичный фосфин используют в количестве от 0,1 до 10 моль на моль атома палладия в соединении палладия.
Пункт I-16. Способ по любому из пунктов с I-13 по I-15, в котором третичный фосфин используют в количестве от 1 до 5 моль на моль атома палладия в соединении палладия.
Пункт I-17. Способ по любому из пунктов с I-1 по I-16, в котором карбеновый комплекс палладия используют в количестве от 0,001 до 5 мольных % из расчета на атом палладия, на моль соединения, представленного формулой (2).
Пункт I-18. Способ по пункту I-17, в котором карбеновый комплекс палладия используют в количестве от 0,01 до 0,5 мольных % из расчета на атом палладия, на моль соединения, представленного формулой (2).
Пункт I-19. Способ по любому из пунктов с I-1 по I-18, дополнительно включающий стадию получения соединения, представленного формулой (2), декарбоксилированием соединения, представленного формулой (6):
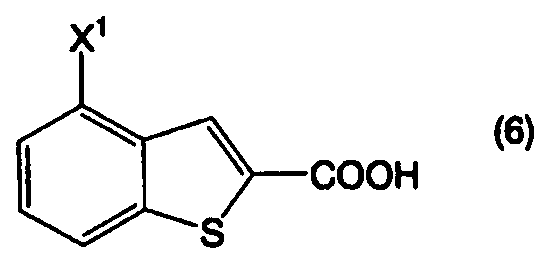
в котором Х1 определен выше, или его соли, в присутствии высококипящего основного соединения, в высококипящем растворителе или в отсутствии растворителя.
Пункт II-1. Способ получения соединения, представленного формулой (1):
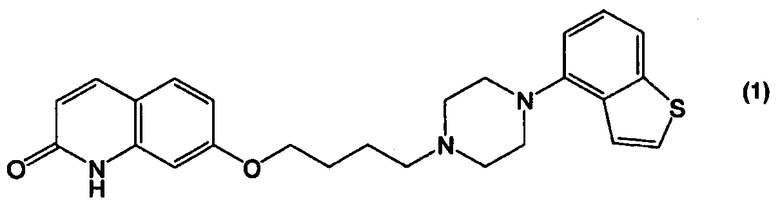
или его соли, взаимодействием соединения, представленного формулой (4а):
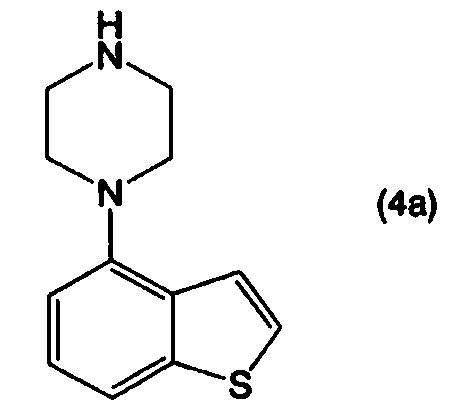
или его соли с соединением, представленным формулой (5):
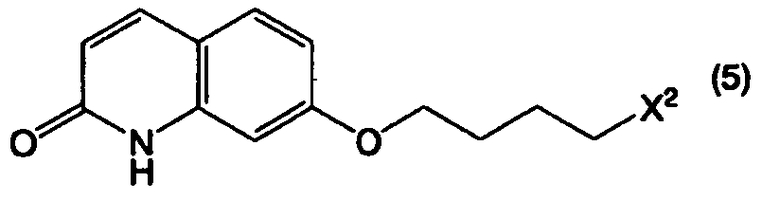
в котором Х2 представляет собой уходящую группу, или его солью, при этом данный способ включает в себя:
Стадию В: взаимодействие соединения, представленного формулой (4а), или его соли, с соединением, представленным формулой (5), или его солью, в присутствии основного соединения, в инертном растворителе или в отсутствие растворителя;
Стадию С: смешивание уксусной кислоты и спирта с продуктом реакции, полученным на стадии В; и
Стадию D: добавление хлористоводородной кислоты к смеси, полученной на стадии С, с получением гидрохлорида соединения, представленного формулой (1).
Пункт II-2. Способ по пункту II-1, дополнительно включающий:
Стадию Е: взаимодействие гидрохлорида соединения, представленного формулой (1), полученного на стадии D, в присутствии основного соединения, с получением соединения, представленного формулой (1).
Пункт II-3. Способ по пункту II-2, в котором на стадии Е основное соединение представляет собой гидроксид натрия.
Пункт II-4. Способ по пункту II-2 или II-3, дополнительно включающий:
Стадию F: перевод соединения, представленного формулой (1), полученного на стадии Е, в солевую форму.
Пункт II-5. Способ по любому из пунктов с II-1 по II-4, в котором на стадии С спирт представляет собой, по меньшей мере, один член, выбранный из группы, включающей в себя метанол, этанол и изопропиловый спирт.
Пункт II-6. Способ по пункту II-5, в котором на стадии С спирт представляет собой этанол.
Пункт II-7. Способ по любому из пунктов с II-1 по II-6, в котором на стадии В основное соединение используют в количестве от 0,3 до 5 моль на моль соединения, представленного формулой (5).
Пункт II-8. Способ по любому из пунктов с II-1 по II-7, в котором на стадии В реакцию проводят, кроме того, в присутствии галогенида щелочного металла.
Пункт II-9. Способ по пункту II-8, в котором на стадии В галогенид щелочного металла представляет собой йодид калия.
Пункт II-10. Способ по пункту II-8 или II-9, в котором на стадии В галогенид щелочного металла используют в количестве от 0,1 до 10 моль на моль соединения, представленного формулой (5).
Пункт II-11. Способ по любому из пунктов с II-1 по II-10, в котором на стадии С уксусную кислоту используют в количестве 0,1 мл или более на грамм соединения, представленного формулой (1), полученного на стадии В.
Пункт II-12. Способ по любому из пунктов с II-1 по II-11, в котором на стадии С уксусную кислоту используют в количестве 1 мл или более на грамм соединения, представленного формулой (1), полученного на стадии В.
Пункт II-13. Способ по любому из пунктов с II-1 по II-12, в котором на стадии С уксусную кислоту используют в количестве 1,5 мл или более на грамм соединения, представленного формулой (1), полученного на стадии В.
Пункт II-14. Способ по любому из пунктов с II-1 по II-13, в котором на стадии С уксусную кислоту используют в количестве 10 мл или менее на грамм соединения, представленного формулой (1), полученного на стадии В.
Пункт II-15. Способ по любому из пунктов с II-1 по II-14, в котором на стадии С спирт используют в количестве от 1 мл до 100 мл на грамм соединения, представленного формулой (1), полученного на стадии В.
Пункт II-16. Способ по любому из пунктов с II-1 по II-15, в котором на стадии D хлористоводородную кислоту используют в количестве 1 моль или более от молярного количества хлористого водорода в хлористоводородной кислоте, на моль соединения, представленного формулой (1), полученного на стадии В.
Пункт II-17. Способ по любому из пунктов с II-1 по II-16, в котором на стадии D хлористоводородную кислоту используют в количестве 10 моль или менее от молярного количества хлористого водорода в хлористоводородной кислоте, на моль соединения, представленного формулой (1), полученного на стадии В.
Пункт II-18. Способ по пункту II-17, в котором на стадии D хлористоводородную кислоту используют в количестве 2 моль или менее от молярного количества хлористого водорода в хлористоводородной кислоте, на моль соединения, представленного формулой (1), полученного на стадии В.
Пункт II-19. Способ по любому из пунктов с II-1 по II-18, в котором на стадии D гидрохлорид получают, добавляя хлористоводородную кислоту при температуре от 50°С до температуры кипения с обратным холодильником, и охлаждая смесь до температуры 20°С или менее.
Пункт II-20. Способ по пункту II-19, в котором на стадии D гидрохлорид получают, охлаждая смесь до температуры 10°С или менее.
Пункт II-21. Способ по любому из пунктов с II-1 по II-20, в котором хлористоводородная кислота представляет собой концентрированную хлористоводородную кислоту.
Пункт III-1. Способ получения соединения, представленного формулой (1):
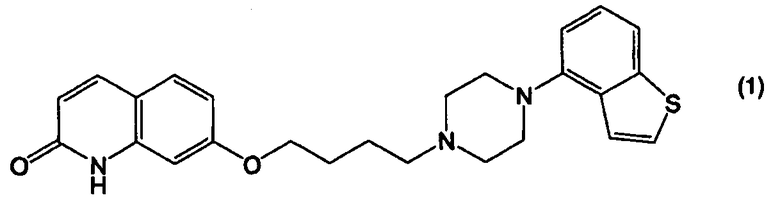
или его соли, взаимодействием соединения, представленного формулой (2):
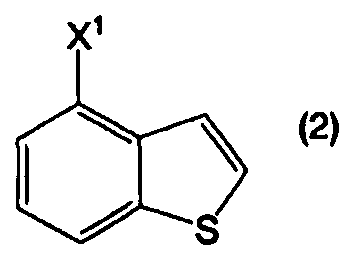
в котором Х1 представляет собой уходящую группу, с соединением, представленным формулой (3а):
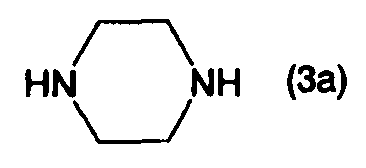
или его солью, и взаимодействием полученного соединения, представленного формулой (4а):
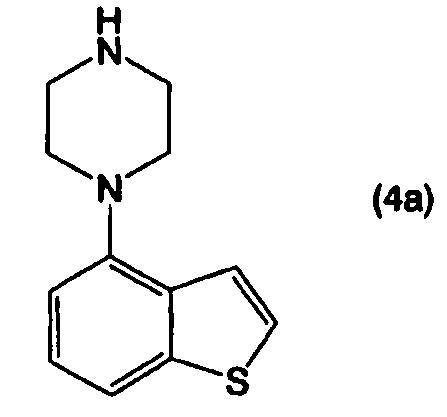
или его соли, с соединением, представленным формулой (5):
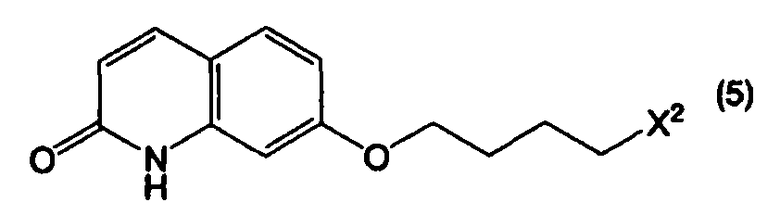
в котором Х2 представляет собой уходящую группу, или его солью, при этом данный способ включает в себя:
стадию А: взаимодействие соединения, представленного формулой (2), с соединением, представленным формулой (3а), или его солью, в присутствии (а) соединения палладия и третичного фосфина, или (b) карбенового комплекса палладия, в инертном растворителе или в отсутствие растворителя;
Стадию В: взаимодействие соединения, представленного формулой (4а), или его соли, с соединением, представленным формулой (5), или его солью, в присутствии основного соединения, в инертном растворителе или в отсутствие растворителя;
Стадию С: смешивание уксусной кислоты и спирта с продуктом реакции, полученным на стадии В; и
Стадию D: добавление хлористоводородной кислоты к смеси, полученной на стадии С, с получением гидрохлорида соединения, представленного формулой (1).
Пункт III-2. Способ по пункту III-1, в котором на стадии А соединение, представленное формулой (2), вводят во взаимодействие с соединением, представленным формулой (3) или его солью, в присутствии (а) соединения палладия и третичного фосфина, в инертном растворителе или в отсутствие растворителя.
Пункт III-3. Способ по пункту III-1, в котором на стадии А соединение, представленное формулой (2), вводят во взаимодействие с соединением, представленным формулой (3) или его солью, в присутствии (b) карбенового комплекса палладия, в инертном растворителе или в отсутствие растворителя.
Пункт III-4. Способ по любому из пунктов с III-1 по III-3, в котором на стадии А третичный фосфин представляет собой, по меньшей мере, один член, выбранный из группы, включающей в себя три-третбутилфосфин, 2-(дитретбутилфосфино)-1,1′-дифенил, 2-(дитретбутилфосфино)-2′-метил-1,1′-дифенил, 2-(дитретбутилфосфино)-1,1′-динафтил, 2-дициклогексилфосфино-2′,6′-диметокси-1,1′-дифенил, 2-дициклогексилфосфино-2′,6′-диизопропокси-1,1′-дифенил, N-фенил-2-(дитретбутилфосфино)пиррол и 1-фенил-2-(дитретбутилфосфино)-1Н-инден.
Пункт III-5. Способ по любому из пунктов с III-1 по III-4, в котором на стадии А карбеновый комплекс палладия представляет собой, по меньшей мере, один член, выбранный из группы, включающей в себя (1,4-нафтохинон)-[1,3-бис(2,6-диизопропилфенил)имидазол-2-илиден]палладий(0), аллилхлор-[1,3-бис(2,6-диизопропилфенил)имидазол-2-илиден]палладий(II), аллилхлор-[1,3-бис(2,6-диизопропилфенил)-4,5-дигидроимидазол-2-илиден]палладий(II) и (3-фенилаллилхлор)-[1,3-бис(2,6-диизопропилфенил)-4,5-дигидроимидазол-2-илиден]палладий(II).
Пункт III-6. Способ по любому из пунктов с III-1 по III-5, в котором на стадии А третичный фосфин представляет собой три-третбутилфосфин и/или 2-дициклогексилфосфино-2′,6′-диизопропокси-1,1′-дифенил.
Пункт III-7. Способ по любому из пунктов с III-1 по III-6, в котором на стадии А соединение палладия представляет собой, по меньшей мере, один член, выбранный из группы, включающей в себя тетрагидрат гексахлорпалладата(IV) натрия, гексахлорпалладат(IV) калия, хлорид палладия(II), бромид палладия(II), ацетат палладия(II), ацетилацетонат палладия(II), дихлорбис(бензонитрил)палладий(II), дихлорбис(ацетонитрил) палладий(II), дихлорбис(трифенилфосфин)палладий(II), дихлортетрааммиакат палладия(II), дихлор(циклоокта-1,5-диен) палладий(II), трифторацетат палладия(II), трис(дибензилиденацетон)дипалладий(0), комплекс трис(дибензилиденацетон)дипалладия(0) с хлороформом и тетракис(трифенилфосфин)палладий(0).
Пункт III-8. Способ по любому из пунктов с III-1 по III-7, в котором соединение палладия представляет собой ацетат палладия(II).
Пункт III-9. Способ по любому из пунктов с III-1 по III-8, в котором на стадии А карбеновый комплекс палладия представляет собой аллилхлор-[1,3-бис(2,6-диизопропилфенил)-4,5-дигидроимидазол-2-илиден]палладий(II).
Пункт III-10. Способ по любому из пунктов с III-1 по III-9, в котором на стадии А инертный растворитель представляет собой ксилол и/или толуол.
Пункт III-11. Способ по любому из пунктов с III-1 по III-10, в котором уходящая группа, представленная Х1 в формуле (2), является атомом галогена.
Пункт III-12. Способ по пункту III-11, в котором уходящая группа, представленная Х1 в формуле (2), является атомом хлора.
Пункт III-13. Способ по любому из пунктов с III-1 по III-12, в котором на стадии А соединение палладия используют в количестве от 0,01 до 5 мольных % из расчета на атом палладия, на моль соединения, представленного формулой (2), а третичный фосфин используют в количестве от 0,01 до 1000 моль на моль атома палладия в соединении палладия.
Пункт III-14. Способ по пункту III-13, в котором на стадии А соединение палладия используют в количестве от 0,01 до 0,5 мольных % из расчета на атом палладия, на моль соединения, представленного формулой (2).
Пункт III-15. Способ по пункту III-13 или III-14, в котором на стадии А третичный фосфин используют в количестве от 0,1 до 10 моль на моль атома палладия в соединении палладия.
Пункт III-16. Способ по любому из пунктов с III-13 по III-15, в котором на стадии А третичный фосфин используют в количестве от 1 до 5 моль на моль атома палладия в соединении палладия.
Пункт III-17. Способ по любому из пунктов с III-1 по III-16, в котором на стадии А карбеновый комплекс палладия используют в количестве от 0,001 до 5 мольных % из расчета на атом палладия, на моль соединения, представленного формулой (2).
Пункт III-18. Способ по пункту III-17, в котором карбеновый комплекс палладия используют в количестве от 0,01 до 0,5 мольных % из расчета на атом палладия, на моль соединения, представленного формулой (2).
Пункт III-19. Способ по любому из пунктов III-1 по III-18, дополнительно включающий:
Стадию Е: взаимодействие гидрохлорида соединения, представленного формулой (1), полученного на стадии D, в присутствии основного соединения, с получением соединения, представленного формулой (1).
Пункт III-20. Способ по пункту III-19, в котором на стадии Е основное соединение представляет собой гидроксид натрия.
Пункт III-21. Способ по пункту III-19 или III-20, дополнительно включающий:
Стадию F: перевод соединения, представленного формулой (1), полученного на стадии Е, в солевую форму.
Пункт III-22. Способ по любому из пунктов с III-1 по III-21, в котором на стадии С спирт представляет собой, по меньшей мере, один член, выбранный из группы, включающей в себя метанол, этанол и изопропиловый спирт.
Пункт III-23. Способ по пункту III-22, в котором спирт представляет собой этанол.
Пункт III-24. Способ по любому из пунктов с III-1 по III-23, в котором на стадии В основное соединение используют в количестве от 0,3 до 5 моль на моль соединения, представленного формулой (5).
Пункт III-25. Способ по любому из пунктов с III-1 по III-24, в котором на стадии В реакцию проводят, кроме того, в присутствии галогенида щелочного металла.
Пункт III-26. Способ по пункту III-25, в котором на стадии В галогенид щелочного металла представляет собой йодид калия.
Пункт III-27. Способ по пункту III-25 или III-26, в котором на стадии В галогенид щелочного металла используют в количестве от 0,1 до 10 моль на моль соединения, представленного формулой (5).
Пункт III-28. Способ по любому из пунктов с III-1 по III-27, в котором на стадии С уксусную кислоту используют в количестве 0,1 мл или более на грамм соединения, представленного формулой (1), полученного на стадии В.
Пункт III-29. Способ по любому из пунктов с III-1 по III-28, в котором на стадии С уксусную кислоту используют в количестве 1 мл или более на грамм соединения, представленного формулой (1), полученного на стадии В.
Пункт III-30. Способ по любому из пунктов с III-1 по III-29, в котором на стадии С уксусную кислоту используют в количестве 1,5 мл или более на грамм соединения, представленного формулой (1), полученного на стадии В.
Пункт III-31. Способ по любому из пунктов с III-1 по III-30, в котором на стадии С уксусную кислоту используют в количестве 10 мл или менее на грамм соединения, представленного формулой (1), полученного на стадии В.
Пункт III-32. Способ по любому из пунктов с III-1 по III-31, в котором на стадии С спирт используют в количестве от 1 мл до 100 мл на грамм соединения, представленного формулой (1), полученного на стадии В.
Пункт III-33. Способ по любому из пунктов с III-1 по III-32, в котором на стадии D хлористоводородную кислоту используют в количестве 1 моль или более от молярного количества хлористого водорода в хлористоводородной кислоте, на моль соединения, представленного формулой (1), полученного на стадии В.
Пункт III-34. Способ по любому из пунктов с III-1 по III-33, в котором на стадии D хлористоводородную кислоту используют в количестве 10 моль или менее от молярного количества хлористого водорода в хлористоводородной кислоте, на моль соединения, представленного формулой (1), полученного на стадии В.
Пункт III-35. Способ по пункту III-34, в котором на стадии D хлористоводородную кислоту используют в количестве 2 моль или менее от молярного количества хлористого водорода в хлористоводородной кислоте, на моль соединения, представленного формулой (1), полученного на стадии В.
Пункт III-36. Способ по любому из пунктов с III-1 по III-35, в котором на стадии D гидрохлорид получают, добавляя хлористоводородную кислоту при температуре от 50°С до температуры кипения, и охлаждая смесь до температуры 20°С или менее.
Пункт III-37. Способ по пункту III-36, в котором на стадии D гидрохлорид получают, охлаждая смесь до температуры 10°С или менее.
Пункт III-38. Способ по любому из пунктов с III-1 по III-37, в котором хлористоводородная кислота представляет собой концентрированную хлористоводородную кислоту.
Пункт III-39. Способ по любому из пунктов с III-1 по III-38, дополнительно включающий стадию получения соединения, представленного формулой (2), декарбоксилированием соединения, представленного формулой (6):
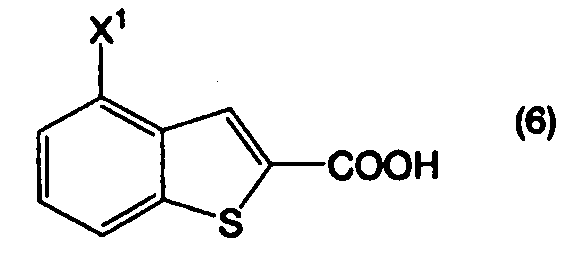
в котором Х1 определен выше, или его соли, в присутствии высококипящего основного соединения, в высококипящем растворителе или в отсутствие растворителя.
Полезные эффекты изобретения
При помощи способа получения по настоящему изобретению можно получать, с высокой чистотой и высоким выходом и по простой методике, соединение, представленное формулой (1), или его соль, применимое для различных лекарственных препаратов, таких как антипсихотические препараты. Кроме того, при помощи способа получения по настоящему изобретению можно получать, с высокой чистотой и высоким выходом и по простой методике, соединение, представленное формулой (4а), или его соль, которое является предшественником соединения, представленного формулой (1), или его соли, и применимо для различных лекарственных препаратов (например, антипсихотических препаратов) и пестицидов. То есть, при помощи способа получения по настоящему изобретению, соединение, представленное формулой (1), или его соль, и соединение, представленное формулой (4а), или его соль, можно получать с высокой чистотой и высоким выходом по простой методике, вместо колоночной хроматографии, которая представляет собой промышленно невыгодный способ.
Таким образом, способ получения согласно настоящему изобретению применим для промышленного использования.
Описание вариантов осуществления
В настоящем изобретении соединение, представленное формулой (1):
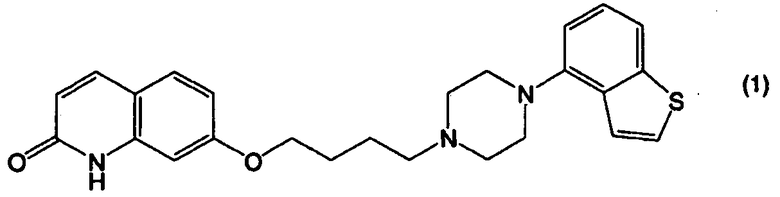
или его соль, можно получить из соединения бензо[b]тиофена, представленного формулой (2):
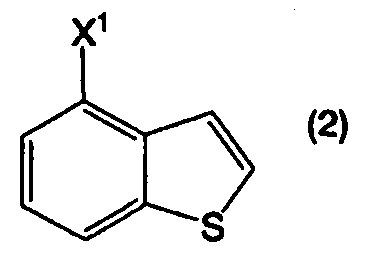
в котором Х1 представляет собой уходящую группу, или его соли, при помощи определенных стадий, приведенных ниже.
Схема реакции 1
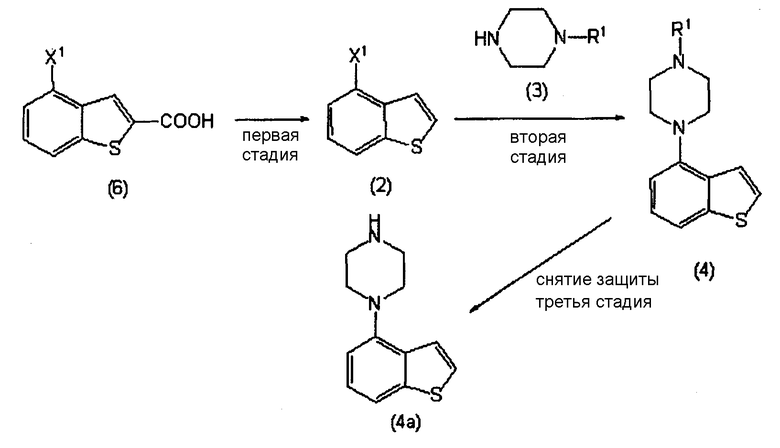
в котором R1 представляет собой атом водорода или защитную группу, а Х1 определен выше.
Соединение бензо[b]тиофена, представленное формулой (2), получают, как показано на приведенной выше схеме реакции 1, декарбоксилированием соединения, представленного формулой (6), или его соли, в присутствии высококипящего основного соединения (первая стадия).
Кроме того, соединение 4-(1-пиперазинил)бензо[b]тиофена, представленное формулой (4а), или его соль, получают взаимодействием соединения бензо[b]тиофена, представленного формулой (2), с соединением пиперазина, представленного формулой (3), или его солью (вторая стадия), и, необязательно, удаляя N-защищающую группу из полученного соединения (третья стадия). В настоящем изобретении вторую стадию называют также стадией А.
В формулах (6) и (2) примеры уходящей группы, представленной Х1, включают галоген, низший алкилсульфонилокси, перфторзамещенный низший алкилсульфонилокси, арилсульфонилокси, арилалкилсульфонилокси и так далее.
Примеры атома галогена, представленного Х1, включают фтор, хлор, бром и йод.
В настоящем изобретении «низший алкил», «низший алкокси» и «низший алканоил» включают «С1-6 линейный или разветвленный алкил», «С1-6 линейный или разветвленный алкокси» и «С1-6 линейный или разветвленный алканоил», соответственно.
Конкретные примеры низшей алкилсульфонилокси-группы, представленной Х1, включают в себя С1-6 линейные или разветвленные алкилсульфонилокси-группы, такие как метилсульфонилокси, этилсульфонилокси, н-пропилсульфонилокси, изопропилсульфонилокси, н-бутилсульфонилокси, третбутилсульфонилокси, н-пентилсульфонилокси и н-гексилсульфонилокси.
Конкретные примеры перфторзамещенной низшей алкилсульфонилокси-группы, представленной Х1, включают С1-6 линейные или разветвленные перфторалкилсульфонилокси-группы, такие как трифторметилсульфонилокси, 1,1,2,2,2-пентафтор-1-этилсульфонилокси, 1,1,2,2,3,3,3-гепта-1-пропилсульфонилокси и 1,1,2,2,3,3,4,4,4-нонафтор-1-бутилсульфонилокси.
Конкретные примеры арилсульфонилокси-группы, представленной Х1, включают фенилсульфонилокси-группы, необязательно содержащие в фенильном кольце от 1 до 3 заместителей, выбранных из группы, включающей С1-6 линейные или разветвленные алкильные группы, С1-6 линейные или разветвленные алкоксигруппы, нитрогруппы и атомы галогена, и нафтилсульфонилокси-группы. Конкретные примеры фенилсульфонилокси-группы, необязательно имеющей заместитель, включают фенилсульфонилокси, 4-метилфенилсульфонилокси, 2-метилфенилсульфонилокси, 4-нитрофенилсульфонилокси, 4-метоксифенилсульфонилокси, 2-нитрофенилсульфонилокси, 3-хлорфенилсульфонилокси и так далее. Конкретные примеры нафтилсульфонилокси-группы включают α-нафтилсульфонилокси, β-нафтилсульфонилокси и так далее.
Примеры арилалкилсульфонилокси-группы, представленной Х1, включают С1-6 линейные или разветвленные алкилсульфонилокси-группы, замещенные фенильной группой, необязательно содержащей, в фенильном цикле, от 1 до 3 заместителей, выбранных из группы, включающей С1-6 линейные или разветвленные алкильные группы, С1-6 линейные или разветвленные алкоксигруппы, нитрогруппы и атомы галогена, и С1-6 линейные или разветвленные алкилсульфонилокси-группы, замещенные нафтильной группой. Конкретные примеры алкилсульфонилокси-групп, замещенных фенильной группой, включают бензилсульфонилокси, 2-фенилэтилсульфонилокси, 4-фенилбутилсульфонилокси, 4-метилбензилсульфонилокси, 2-метилбензилсульфонилокси, 4-нитробензилсульфонилокси, 4-метоксибензилсульфонилокси, 3-хлорбензилсульфонилокси и так далее. Конкретные примеры алкилсульфонилокси-групп, замещенных нафтильной группой, включают α-нафтилметилсульфонилокси, β-нафтилметилсульфонилокси и так далее.
Первая стадия:
Соединение, представленное формулой (6), или его соль, подвергают декарбоксилированию в отсутствие растворителя или в высококипящем растворителе, в присутствии высококипящего основного соединения, получая в результате соединение, представленное формулой (2).
Примеры высококипящих растворителей включают простые эфиры, такие как диметиловый эфир и дибутиловый эфир диэтиленгликоля, ароматические углеводороды, такие как толуол, ксилол и мезитилен, спирты, такие как 1-гексанол, 2-гексанол, 3-гексанол, 1-гептанол, 2-гептанол, 3-гептанол, 1-октанол, 2-октанол, 3-октанол, 1-нонаол, 2-нонаол, 1-деканол, 2-деканол и 4-деканол, кетоны, такие как 2-октанон, 3-октанон, 2-нонанон, 5-нонанон, 2-деканон, 3-деканон и 4-деканон, и полярные растворители, такие как N,N-диметилформамид (ДМФА), диметилсульфоксид (ДМСО), N,N-диметилацетамид (DMA), 1,3-диметил-2-имидазолидинон (DMI), гексаметилфосфортриамид и трис(диметиламино)фосфин. В ряду данных высококипящих растворителей предпочтительны растворители с температурой кипения 160°С или выше, а особенно предпочтительным является DMI.
В качестве высококипящего основного соединения можно широко применять известные основные соединения с температурой кипения 200°С или выше. Их примеры включают органические основания, такие как 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU) и 1,5-диазабицикло[4.3.0]нон-5-ен (DBN). Данные высококипящие основные соединения можно использовать отдельно или в виде сочетания двух или более из них. Из данных высококипящих основных соединений предпочтительным является DBU.
Используемое количество высококипящего основного соединения обычно составляет от около 0,05 до 10 моль, а предпочтительно, от около 0,1 до 6 моль на моль соединения формулы (6).
Реакцию первой стадии можно осуществить при пониженном, нормальном или повышенном давлении, а также можно осуществить в атмосфере инертного газа, такого как азот или аргон.
Упомянутую выше реакцию обычно проводят при температуре от температуре окружающей среды до 300°С, предпочтительно, от 150 до 250°С, и, как правило, она завершается в течение от около 1 до 30 часов.
На первой стадии реакция декарбоксилирования протекает гладко без использования растворителя реакции, а количество используемого высококипящего основного соединения (например, DBU) является более низким. Таким образом, первая стадия имеет не только экономические преимущества, но также преимущества более простой обработки после реакции.
Вторая стадия (стадия А):
Соединение, представленное формулой (2), вводят во взаимодействие с соединением, представленным формулой (3), в отсутствие растворителя или в инертном растворителе, в присутствии или в отсутствие основного соединения, и в присутствии (а) катализатора, содержащего третичный фосфин и соединение палладия, или (b) карбеновый комплекс палладия, получая в результате соединение, представленное формулой (4).
Соединение, представленное формулой (3), является известным соединением, или может быть легко получено из известного соединения.
Примеры инертных растворителей включают воду, простые эфиры, такие как диоксан, тетрагидрофуран, диэтиловый эфир, диметиловый эфир диэтиленгликоля и диметиловый эфир этиленгликоля, ароматические углеводороды, такие как бензол, толуол и ксилол, низшие спирты, такие как метанол, этанол и изопропанол, кетоны, такие как ацетон и метилэтилкетон, и полярные растворители, такие как ДМФА, ДМСО, гексаметилфосфортриамид и ацетонитрил.
В качестве соединения палладия, применяемого в данной реакции, можно широко применять известные соединения палладия. Их примеры включают соединения четырехвалентного палладия, такие как тетрагидрат гексахлорпалладата(IV) натрия и гексахлорпалладат(IV) калия, соединения двухвалентного палладия, такие как хлорид палладия(II), бромид палладия(II), ацетат палладия(II), ацетилацетонат палладия(II), дихлорбис(бензонитрил)палладий(II), дихлорбис(ацетонитрил)палладий(II), дихлорбис(трифенилфосфин)палладий(II), дихлортетрааммиакат палладия(II), дихлор(циклоокта-1,5-диен)палладий(II) и трифторацетат палладия(II), соединения нульвалентного палладия, такие как трис(дибензилиденацетон)дипалладий(0), комплекс трис(дибензилиденацетон)дипалладия(0) с хлороформом и тетракис(трифенилфосфин)палладий(0), и так далее. Данные соединения палладия можно использовать по отдельности или в виде комбинации двух или более из них.
Используемое количество соединения палладия особенно не ограничено и может быть подходящим образом выбрано из широкого интервала. Например, соединение палладия обычно можно использовать в количестве от 0,000001 до 20 мольных %, из расчета на атом палладия, на моль соединения, представленного формулой (2). Когда количество соединения палладия находится в пределах данного интервала, соединение формулы (4) можно получить с высокой селективностью. С точки зрения получения требуемого соединения с высоким выходом в течение короткого промежутка времени, и с экономической точки зрения, предпочтительно, использовать соединение палладия в количестве от около 0,0001 до 20 мольных %, более предпочтительно, от около 0,0001 до 5 мольных %, еще более предпочтительно, от около 0,01 до 5 мольных %, а особенно предпочтительно, от около 0,01 до 0,5 мольных %, из расчета на атом палладия, на моль соединения, представленного формулой (2).
В качестве третичного фосфина, используемого в сочетании с соединением палладия в настоящем изобретении, можно широко применять известные третичные фосфины. Их конкретные примеры включают в себя триалкилфосфины, такие как триэтилфосфин, трициклогексилфосфин, триизопропилфосфин, три-н-бутилфосфин, триизобутилфосфин, три-вторбутилфосфин и три-третбутилфосфин, триарилфосфины, такие как трифенилфосфин, трипентафторфенилфосфин, три-о-толилфосфин, три-м-толилфосфин и три-п-толилфосфин, феноксифосфины, такие как три(2,6-диметилфенокси)фосфин, три(2-третбутилфенокси)фосфин, трифеноксифосфин, три(4-метилфенокси)фосфин и три(2-метилфенокси)фосфин, и диарилфосфины, такие как 2,2′-бис(дифенилфосфино)-1,1′-динафтил (рацемическое вещество), (R)-(+)-2,2′-бис(дифенилфосфино)-1,1′-динафтил и (S)-(+)-2,2′-бис(дифенилфосфино)-1,1′-динафтил, 2-(дитретбутилфосфино)-1,1′-дифенил (JohnPhos), 2-дициклогексилфосфино-2′-метилдифенил (MePhos), 2-дициклогексилфосфино-2′,6′-диметокси-1,1′-дифенил (SPhos), 2-(дициклогексилфосфино)-2′-(N,N-диметиламино)дифенил (DavePhos) и 2-(дициклогексилфосфино)-2′,6′-диизопропокси -1,1′-дифенил (RuPhos). Данные третичные фосфины можно использовать по отдельности или в виде комбинации двух или более из них. Более предпочтительными третичными фосфинами являются триалкилфосфины, такие как три-третбутилфосфин, и диарилфосфины, такие как 2-(дициклогексилфосфино)-2′,6′-диизопропокси-1,1′-дифенил (RuPhos).
Что касается подавления протекания побочных реакций, предпочтительными третичными фосфинами, предназначенными для применения в настоящем изобретении, являются три-третбутилфосфин и диарилфосфины, такие как 2-(дитретбутилфосфино)-1,1′-дифенил (в дальнейшем “L1”), 2-(дитретбутилфосфино)-2′-метил-1,1′-дифенил (в дальнейшем “L2”), 2-(дитретбутилфосфино)-1,1′-динафтил (в дальнейшем “L5”), 2-дициклогексилфосфино-2′,6′-диметокси-1,1′-дифенил (в дальнейшем “L10”), 2-дициклогексилфосфино-2′,6′-диизопропокси-1,1′-дифенил (RuPhos) (в дальнейшем “L12”), N-фенил-2-(дитретбутилфосфино)пиррол (в дальнейшем “L16”) и 1-фенил-2-(дитретбутилфосфино)-1Н-инден (в дальнейшем “L17”). Более предпочтительными из них являются три-третбутилфосфин и L12.
Кроме того, третичный фосфин, используемый в настоящем изобретении, можно предварительно получить в форме соли. Конкретные примеры подобных солей включают в себя тетрафенилборат три-третбутилфосфония и тетрафторборат три-третбутилфосфония.
Используемое количество третичного фосфина обычно составляет от около 0,01 до 10000 моль на моль атома палладия в соединении палладия. Предпочтительно, использовать третичный фосфин в количестве от около 0,1 до 1000 моль, более предпочтительно, от около 0,1 до 10 моль, и особенно предпочтительно, от около 1 до 5 моль на моль атома палладия в соединении палладия.
Подходящие комбинации соединения палладия и третичного фосфина включают в себя комбинации соединения палладия, выбранного из группы, включающей ацетат палладия, бис(дибензилиденацетон)палладий, трис(дибензилиденацетон)дипалладий, тетракис(трифенилфосфин)палладий, хлорид палладия, дихлорбис(трифенилфосфин)палладий и дихлорбис(три-о-толилфосфин)палладий, и третичного фосфина, выбранного из группы, включающей 2-(дитретбутилфосфино)-1,1′-дифенил, 2,8,9-триизобутил-2,5,8,9-тетрааза-1-фосфабицикло[3,3,3]ундекан, N-фенил-2-(дитретбутилфосфино)индол, N-фенил-2-(дитретбутилфосфино)пиррол, три-третбутилфосфин, 4,5-бис(дифенилфосфино)-9,9-диметилксантен, тетрафенилборат три-третбутилфосфония и тетрафторборат три-третбутилфосфония. Особенно предпочтительными комбинациями являются комбинация ацетата палладия и тетрафенилбората три-третбутилфосфония, и комбинация ацетата палладия и RuPhos.
Что касается подавления протекания побочных реакций, предпочтительными комбинациями соединения палладия и третичного фосфина являются комбинации ацетата палладия и три-третбутилфосфина, L1, L2, L5, L10, L12, L16 или L17, а более предпочтительными комбинациями являются комбинации ацетата палладия и три-третбутилфосфина или L12.
Соединение палладия и третичного фосфина можно предварительно получить в виде комплекса. При использовании соединения палладия и третичного фосфина в виде комплекса, можно использовать упомянутые выше комбинации соединения палладия и третичного фосфина. Количество комплекса предпочтительно является таким же, что и упомянутое выше количество соединения палладия.
Что касается карбенового комплекса палладия, можно широко применять известные карбеновые комплексы палладия. Их конкретные примеры включают (1,4-нафтохинон)-[1,3-бис(2,6-диизопропилфенил)имидазол-2-илиден]палладий(0) (в дальнейшем “СХ11”), (1,4-нафтохинон)-[1,3-бис(2,4,6-триметилфенил)имидазол-2-илиден]палладий(0) (в дальнейшем “СХ12”), аллилхлор-[1,3-бис(2,6-диизопропилфенил)имидазол-2-илиден]палладий(II) (в дальнейшем “СХ21”), аллилхлор-[1,3-бис(2,4,6-триметилфенил)имидазол-2-илиден]палладий(II) (в дальнейшем “СХ22”), аллилхлор-[1,3-бис(2,6-диизопропилфенил)-4,5-дигидроимидазол-2-илиден]палладий(II) (в дальнейшем “СХ23”), (3-фенилаллилхлор)-[1,3-бис(2,4,6-триметилфенил)имидазол-2-илиден]палладий(II) (в дальнейшем “СХ31”), (3-фенилаллилхлор)-[1,3-бис(2,6-диизопропилфенил)-4,5-дигидроимидазол-2-илиден]палладий(II) (в дальнейшем “СХ32”), димер дихлор-[1,3-бис(2,6-диизопропилфенил)имидазол-2-илиден]палладия(II) (в дальнейшем “СХ41”), и другие палладиевые комплексы N-гетероциклического карбена. Данные карбеновые комплексы палладия можно использовать по отдельности, или в виде комбинации двух или более из них. Что касается подавления протекания побочных реакций, предпочтительным карбеновым комплексом палладия для использования в настоящем изобретении является СХ11, СХ21, СХ23, СХ32 и другие палладиевые комплексы N-гетероциклического карбена. Более предпочтительными являются СХ11, СХ21, СХ23 и СХ32.
Используемое количество карбенового комплекса палладия особенно не ограничено и может быть подходящим образом выбрано из широкого интервала. Например, карбеновый комплекс палладия обычно можно использовать в количестве от около 0,0001 до 20 мольных % из расчета на атом палладия, на моль соединения, представленного формулой (2). Когда количество карбенового комплекса палладия находится в пределах данного интервала, соединение формулы (4) можно получать с высокой селективностью. С точки зрения получения требуемого соединения с высоким выходом за короткий промежуток времени, и с экономической точки зрения, предпочтительно использовать карбеновый комплекс палладия в количестве от около 0,001 до 5 мольных %, а более предпочтительно, от около 0,01 до 0,5 мольных % из расчета на атом палладия, на моль соединения, представленного формулой (2).
Карбеновый комплекс палладия можно получить предварительно в виде комплекса, или можно получить в реакционной системе из упомянутого выше соединения палладия и соответствующего предшественника карбена. Подходящие для использования предшественники карбена представляют собой N-гетероциклические галогенидные соли. Их конкретные примеры включают в себя хлорид 1,3-бис(2,6-диизопропилфенил)имидазолия, хлорид 1,3-бис(2,4,6-триметилфенил)имидазолия, хлорид 1,3-бис(2,6-диизопропилфенил)-4,5-дигидроимидазолия, хлорид 1,3-бис(2,4,6-триметилфенил)-4,5-дигидроимидазолия и другие соли 1,3-дизамещенного имидазолия или соли 1,3-дизамещенного дигидроимидазолия. Количество соединения палладия, используемое в данном случае, может быть таким же, как и количество упомянутого выше карбенового комплекса палладия из расчета на атом палладия. Кроме того, используемое количество карбенового комплекса палладия, как правило, находится в интервале от около 0,0001 до 20 мольных % на моль соединения, представленного формулой (2). С точки зрения получения требуемого соединения с высоким выходом за короткий промежуток времени, и с экономической точки зрения, предпочтительно использовать предшественник карбена в количестве от около 0,001 до 5 мольных %, а более предпочтительно, от около 0,01 до 0,5 мольных % на моль соединения, представленного формулой (2).
В качестве основного соединения можно широко применять известные основные соединения. Их примеры включают гидроксиды щелочных металлов, такие как гидроксид натрия, гидроксид калия, гидроксид цезия и гидроксид лития, карбонаты щелочных металлов, такие как карбонат натрия, карбонат калия, карбонат цезия и карбонат лития, гидрокарбонаты щелочных металлов, такие как гидрокарбонат лития, гидрокарбонат натрия и гидрокарбонат калия, щелочные металлы, такие как натрий и калия, неорганические основания, такие как амид натрия, гидрид натрия и гидрид калия, алкоголяты щелочных металлов, такие как метилат натрия, этилат натрия, метилат калия, этилат калия, третбутилат лития, третбутилат натрия и третбутилат калия, и органические основания, такие как триэтиламин, трипропиламин, пиридин, хинолин, пиперидин, имидазол, N-этилдиизопропиламин, диметиламинопиридин, триметиламин, диметиланилин, N-метилморфолин, DBN, DBU и 1,4-диазабицикло[2.2.2]октан (DABCO). Данные основные соединения можно использовать по отдельности или в виде комбинации двух или более из них. Предпочтительным основным соединением является алкоксид щелочного металла.
Основное соединение обычно используют в количестве от около 0,5 до 10 моль, а предпочтительно, от около 0,5 до 6 моль на моль соединения формулы (2).
Соединение формулы (3) обычно используют в количестве, по меньшей мере, примерно 0,5 моль, а предпочтительно, от около 0,5 до 5 моль на моль соединения формулы (2).
Реакцию второй стадии можно осуществить при нормальном или повышенном давлении, а также можно осуществить в атмосфере инертного газа, такого как азот или аргон.
Упомянутую выше реакцию, как правило, можно осуществить при температуре от температуры окружающей среды до 200°С, предпочтительно, от температуры окружающей среды до 150°С, и обычно она завершается в течение от 1 до 30 часов.
Третья стадия:
Для соединения, представленного формулой (4), или его соли, полученной на второй стадии, в случае, когда R1 представляет собой N-защищающую группу, данное соединение или его соль подвергают снятию N-защищающей группы, получая в результате соединение (4а) или его соль.
Предпочтительные примеры «N-защищающей группы» включают в себя моно-, ди- или трифенил(низший)алкил (например, бензил, фенетил, 1-фенилэтил, бензгидрил и тритил), низший алканоил (например, формил, ацетил, пропионил, гексаноил и пивалоил), моно- (или ди- или три) галоген(низший)алканоил (например, хлорацетил и трифторацетил), низший алкоксикарбонил (например, метоксикарбонил, этоксикарбонил и третбутоксикарбонил), моно- (или ди- или три-)галоген(низший)алкоксикарбонил (например, хлорметоксикарбонил, дихлорэтоксикарбонил и трихлорэтоксикарбонил), ароил (например, бензоил, толуоил, ксилоил и нафтоил), фенил(низший)алканоил (например, фенилацетил и фенилпропионил), фенил(низший)алкоксикарбонил, необязательно содержащий нитро или низший алкокси (например, бензилоксикарбонил, фенетилоксикарбонил, п-нитробензилоксикарбонил и п-метоксибензилоксикарбонил), низший алкилсульфонил (например, метилсульфонил, этилсульфонил, пропилсульфонил, изопропилсульфонил, пентилсульфонил и бутилсульфонил), арилсульфонил (например, фенилсульфонил, толилсульфонил, ксилилсульфонил и нафтилсульфонил), и фенил(низший)алкилсульфонил (например, бензилсульфонил, фенетилсульфонил и бензгидрилсульфонил).
Более предпочтительные примеры «N-защищающей группы» включают в себя трифенил (С1-4)алкил, (С1-4)алканоил и (С1-4)алкоксикарбонил, а особенно предпочтительно, третбутоксикарбонил.
Удаление N-защищающей группы (снятие защиты) проводят известным способом, таким как гидролиз или восстановление.
Гидролиз:
Гидролиз предпочтительно проводят в присутствии основания или кислоты, включая кислоту Льюиса.
Примеры подходящих оснований включают в себя неорганические основания, такие как гидроксиды щелочных металлов (например, гидроксид натрия и гидроксид калия), гидроксиды щелочноземельных металлов (например, гидроксид магния и гидроксид кальция), карбонаты щелочных металлов (например, карбонат натрия и карбонат калия), карбонаты щелочноземельных металлов (например, карбонат магния и карбонат кальция), и гидрокарбонаты щелочных металлов (например, гидрокарбонат натрия и гидрокарбонат калия), и органические основания, такие как триалкиламин (например, триметиламин и триэтиламин), пиколин, DBN, DBU и DABCO.
Подходящими кислотами являются органические кислоты (например, муравьиная кислота, уксусная кислота, пропионовая кислота, трихлоруксусная кислота и трифторуксусная кислота) и неорганические кислоты (например, хлористоводородная кислота, бромистоводородная кислота и серная кислота).
Снятие защиты при помощи кислоты Льюиса, такой как тригалогенуксусная кислота (например, трихлоруксусная кислота или трифторуксусная кислота), предпочтительно проводят в присутствии акцептора катионов (например, анизола или фенола).
Гидролиз осуществляют в обычно используемом растворителе, который не оказывает неблагоприятного влияния на реакцию. Его примеры включают в себя воду, спирты, такие как метанол, этанол, трифторэтанол и этиленгликоль, ацетон, простые эфиры, такие как диэтиловый эфир, диоксан и тетрагидрофуран, галогенсодержащие углеводороды, такие как хлороформ, метиленхлорид и этиленхлорид, сложные эфиры, такие как метилацетат и этилацетат, ацетонитрил и ДМФА, и их смеси. В случае, когда основание или кислоты является жидкостью, данное основание или кислоту можно также использовать в качестве растворителя.
Реакцию гидролиза обычно проводят при охлаждении или нагревании, или при температуре окружающей среды.
Восстановление:
Для восстановления можно использовать известную реакцию восстановления, такую как химическое восстановление и каталитическое восстановление.
Предпочтительные восстановители, используемые для химического восстановления, представляют собой, например, комбинации гидридов (например, алюмогидрида лития, боргидрида натрия, цианоборгидрида натрия и гидрида диизопропилалюминия), металлов (например, олова, цинка и железа), или соединений металлов (например, хлорида хрома и ацетата хрома) с органическими или неорганическими кислотами (например, муравьиной кислотой, уксусной кислотой, пропионовой кислотой, трифторуксусной кислотой, п-толуолсульфоновой кислотой, хлористоводородной кислотой и бромистоводородной кислотой).
Предпочтительные катализаторы, применяемые в каталитическом восстановлении, представляют собой платиновые катализаторы (например, платиновые пластины, платиновая губка, платиновая чернь, коллоидная платина, оксид платины и платиновая проволока), палладиевые катализаторы (например, палладиевая губка, палладиевая чернь, оксид палладия, палладий на угле, коллоидный палладий, палладий на сульфате бария и палладий на карбонате бария), никелевые катализаторы (например, восстановленный никель, оксид никеля и никель Рэнея), кобальтовые катализаторы (например, восстановленный кобальт и кобальт Рэнея), железные катализаторы (например, восстановленное железо и железо Рэнея) и медные катализаторы (например, восстановленная медь, медь Рэнея и медь Ульмана).
Реакцию восстановления проводят в обычно используемом растворителе, который не оказывает неблагоприятного влияния на реакцию. Его примеры включают в себя воду, спирты, такие как метанол, этанол, трифторэтанол и этиленгликоль, ацетон, простые эфиры, такие как диэтиловый эфир, диоксан и тетрагидрофуран, галогенсодержащие углеводороды, такие как хлороформ, метиленхлорид и этиленхлорид, сложные эфиры, такие как метилацетат и этилацетат, ацетонитрил, ДМФА и пиридин, и их смеси.
Реакцию восстановления обычно проводят при охлаждении или нагревании, или при температуре окружающей среды, предпочтительно, от температуры окружающей среды до 100°С, в течение от около 0,5 до 10 часов.
Кроме того, упомянутое выше снятие N-защищающей группы не ограничено указанными выше условиями реакции.
Например, на третьей стадии можно использовать реакцию, описанную у T. W. Green and P. G. M. Wuts, “Protective Groups in Organic Synthesis” 2nd ed., John Wiley & Sons; New York, 1991, p. 309.
Схема реакции 2
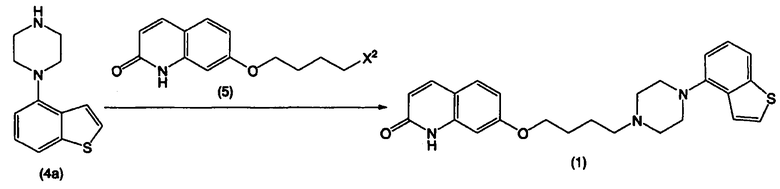
где Х2 представляет собой уходящую группу.
В формуле (5) примеры уходящей группы, представленной Х2, включают в себя уходящие группы, упомянутые в качестве примеров уходящей группы, представленной Х1, такие как галоген, низший алкилсульфонилокси, перфтор низший алкилсульфонилокси, арилсульфонилокси и арилалкилсульфонилокси.
Соединение, представленное формулой (5), является известным соединением, и может быть получено известным способом.
Стадия В:
Как показано на приведенной выше схеме реакции 2, соединение, представленное формулой (1), или его соль, можно получить взаимодействием соединения 4-(1-пиперазинил)бензо[b]тиофена, представленного формулой (4а), или его соли, с соединением, представленным формулой (5), или его солью, в отсутствие растворителя или в инертном растворителе, в присутствии основного соединения. На стадии В в реакционной системе предпочтительно присутствует галогенид щелочного металла.
Соединение формулы (4) или его соль используют в количестве от около 0,5 до 5 моль, предпочтительно, от около 0,9 до 2 моль, а более предпочтительно, от около 1 до 1,2 моль на моль соединения формулы (5) или его соли.
В качестве основного соединения можно широко применять известные основные соединения. Их примеры включают в себя гидроксиды щелочных металлов, такие как гидроксид натрия, гидроксид калия, гидроксид цезия и гидроксид лития, карбонаты щелочных металлов, такие как карбонат натрия, карбонат калия, карбонат цезия и карбонат лития, гидрокарбонаты щелочных металлов, такие как гидрокарбонат лития, гидрокарбонат натрия и гидрокарбонат калия, щелочные металлы, такие как натрий и калия, неорганические основания, такие как амид натрия, гидрид натрия и гидрид калия, алкоголяты щелочных металлов, такие как метилат натрия, этилат натрия, метилат калия, этилат калия, третбутилат лития, третбутилат натрия и третбутилат калия, органические основания, такие как триэтиламин, трипропиламин, пиридин, хинолин, пиперидин, пиперидин, имидазол, N-этилдиизопропиламин, диметиламинопиридин, триметиламин, диметиланилин, N-метилморфолин, DBN, DBU и DABCO. Данные основные соединения можно использовать по отдельности или в виде комбинации двух или более из них. Предпочтительные основные соединения включают карбонаты щелочных металлов.
Основное соединение обычно используют в количестве от около 0,3 до 5 моль, а предпочтительно, от около 1 до 2 моль на моль соединения формулы (5).
Примеры галогенидов щелочного металла включают в себя йодид калия и йодид натрия. Данные галогениды щелочных металлов можно использовать по отдельности или в виде комбинации двух или более из них. Предпочтительные галогениды щелочных металлов включают в себя йодид калия.
Галогенид щелочного металла обычно применяют в количестве от около 0,1 до 10 моль, а предпочтительно, от около 1 до 1,2 моль на моль соединения формулы (5).
Примеры инертных растворителей включают в себя воду, простые эфиры, такие как диоксан, тетрагидрофуран, диэтиловый эфир, диметиловый эфир диэтиленгликоля и диметиловый эфир этиленгликоля, ароматические углеводороды, такие как бензол, толуол и ксилол, низшие спирты, такие как метанол, этанол и изопропанол, кетоны, такие как ацетон и метилэтилкетон, и полярные растворители, такие как ДМФА, ДМСО, гексаметилфосфортриамид, и ацетонитрил.
Упомянутую выше реакцию обычно осуществляют при температуре от температуры окружающей среды до 200°С, предпочтительно, от температуры окружающей среды до 150°С, и она обычно завершается в течение от около 1 до 30 часов.
На схеме реакции 2 соединение, представленное формулой (1), или его соль, можно получить с высокой чистотой простым способом, который включает в себя следующие стадии С и D. То есть, соединение, представленное формулой (1), или его соль, которые достаточно применимы для медицинского использования, можно получить с высокой чистотой на стадиях С и D, без проведения очистки колоночной хроматографией.
Стадия С:
Раствор соединения, представленного формулой (1), можно получить смешиванием уксусной кислоты и спирта с соединением, представленным формулой (1), которое представляет собой продукт реакции, полученный на стадии В.
Примеры спиртов включают в себя метанол, этанол, изопропиловый спирт, н-пропиловый спирт, третбутиловый спирт и так далее. Предпочтительными являются метанол, этанол и изопропиловый спирт, а более предпочтительным является этанол. Данные спиртовые растворители можно использовать по отдельности или в виде комбинации двух или более из них.
На стадии С уксусную кислоту и спирт можно смешивать одновременно или по отдельности. Более конкретно, с соединением формулы (1) можно смешать растворитель, представляющий собой смесь уксусной кислоты и спирта, или же с соединением формулы (1) сначала смешивают что-то одно из уксусной кислоты или спирта, а затем смешивают другое.
Используемое количество уксусной кислоты может составлять 0,1 мл или более, предпочтительно, 1 мл или более, а более предпочтительно, 1,5 мл или более, на грамм соединения, представленного формулой (1), полученного на стадии В. Несмотря на то, что верхний предел количества не является ограничивающим, количество уксусной кислоты, используемой в настоящем описании составляет, например, 10 мл или ниже на грамм соединения, представленного формулой (1), полученного на стадии В.
Спиртовой растворитель можно использовать в количестве, позволяющем в достаточной степени растворить соединение, представленное формулой (1), и осадить гидрохлорид, представленный формулой (1), на стадии D, описанной далее. Например, указанный выше способ очистки можно осуществить, используя от 1 до 100 мл спиртового растворителя на грамм соединения, представленного формулой (1), полученного на стадии В.
На стадии С можно смешивать растворители, отличающиеся от уксусной кислоты и спирта. Примеры подобного растворителя включают в себя воду и другие растворители.
Стадия D:
Гидрохлорид соединения, представленного формулой (1), можно получить добавлением хлористоводородной кислоты к смеси, полученной на стадии С.
Примеры хлористоводородной кислоты включают в себя концентрированную хлористоводородную кислоту и хлористоводородные кислоты с нормальностью от 1 до 12. Можно также использовать смешанный раствор спирта (например, метанола, этанола или изопропилового спирта) и концентрированной хлористоводородной кислоты или хлористоводородной кислоты с нормальностью от 1 до 12. Кроме того, можно также использовать раствор, полученный при растворении хлористого водорода в органическом растворителе, таком как спирт (например, метанол, этанол или изопропиловый спирт), простой эфир (например, диоксан) или сложный эфир (например, этилацетат).
Что касается используемого количества хлористоводородной кислоты, молярное количество хлористого водорода в хлористоводородной кислоте предпочтительно составляет 1 моль или более на моль соединения, представленного формулой (1), полученного на стадии В. Несмотря на то, что верхний предел данного количества особенно не ограничен, молярное количество хлористого водорода в хлористоводородной кислоте составляет, например, 10 моль или менее, а предпочтительно, 2 моль или менее на моль соединения, представленного формулой (1), полученного на стадии В.
Температура, при которой осуществляют стадию D, особенно не ограничена, например, гидрохлорид можно получить добавлением хлористоводородной кислоты при температуре примерно от 50°С до температуры кипения, и охлаждением смеси до 20°С (предпочтительно, 10°С) или менее.
При использовании способа получения, включающего в себя указанные выше стадии В, С и D, трудноудаляемые побочные продукты можно удалить простым способом, без применения такого способа очистки, как колоночная хроматография, и соединение, представленное формулой (1), или его соль, можно получать с высоким выходом и высокой чистотой.
Стадия Е:
Соединение, представленное формулой (1), можно также получить дальнейшей реакцией гидрохлорида соединения, представленного формулой (1), полученного на стадии D, в смеси воды и спирта (например, метанола, этанола, изопропилового спирта) в присутствии основного соединения.
Смесь воды и спирта, используемую на стадии Е, можно получить, например, смешиванием спирта и воды в таком количестве, чтобы объемное соотношение воды и спирта составляло от 0,1 до 10.
В качестве основного соединения можно широко применять известные основные соединения. Их примеры включают в себя гидроксиды щелочных металлов, такие как гидроксид натрия, гидроксид калия, гидроксид цезия и гидроксид лития, карбонаты щелочных металлов, такие как карбонат натрия, карбонат калия, карбонат цезия и карбонат лития, гидрокарбонаты щелочных металлов, такие как гидрокарбонат лития, гидрокарбонат натрия и гидрокарбонат калия, щелочные металлы, такие как натрий и калий, неорганические основания, такие как амид натрия, гидрид натрия и гидрид калия, алкоголяты щелочных металлов, такие как метилат натрия, этилат натрия, метилат калия, этилат калия, третбутилат лития, третбутилат натрия и третбутилат калия, и органические основания, такие как триэтиламин, трипропиламин, пиридин, хинолин, пиперидин, имидазол, N-этилдиизопропиламин, диметиламинопиридин, триметиламин, диметиланилин, N-метилморфолин, DBN, DBU и DABCO. Данные основные соединения можно использовать по отдельности или в виде комбинации двух или более из них. Предпочтительным основным соединением является алкоголят щелочного металла. Предпочтительными основными соединениями являются гидроксиды щелочных металлов, такие как гидроксид натрия, гидроксид калия, гидроксид цезия и гидроксид лития, и гидроксиды щелочноземельных металлов, такие как гидроксид кальция. Более предпочтительным основным соединением является гидроксид натрия.
Температура, при которой осуществляют стадию Е, особенно не ограничена, например, соединение формулы (1) можно получить добавлением основного соединения при температуре примерно от 60°С до температуры кипения с обратным холодильником, и охлаждением смеси до 50°С (предпочтительно, 40°С) или менее.
Стадия F:
Соль соединения, представленного формулой (1), можно получить дальнейшим превращением соединения формулы (1), полученного на стадии Е, в солевую форму. Как правило, в качестве способа получения соли соединения формулы (1) можно использовать известные способы. Например, соединение, соответствующее требуемой соли (например, кислота, такая как хлористоводородная кислота) вводят во взаимодействие с раствором соединения, представленного формулой (1).
В способе, представленном на приведенной выше схеме реакции 1, соединение (6), используемое в качестве исходного вещества, может являться легкодоступным известным соединением, или может быть легко получено известным способом. Например, соединение (6) можно получить способом, представленным на следующей схеме реакции 3:
Схема реакции 3
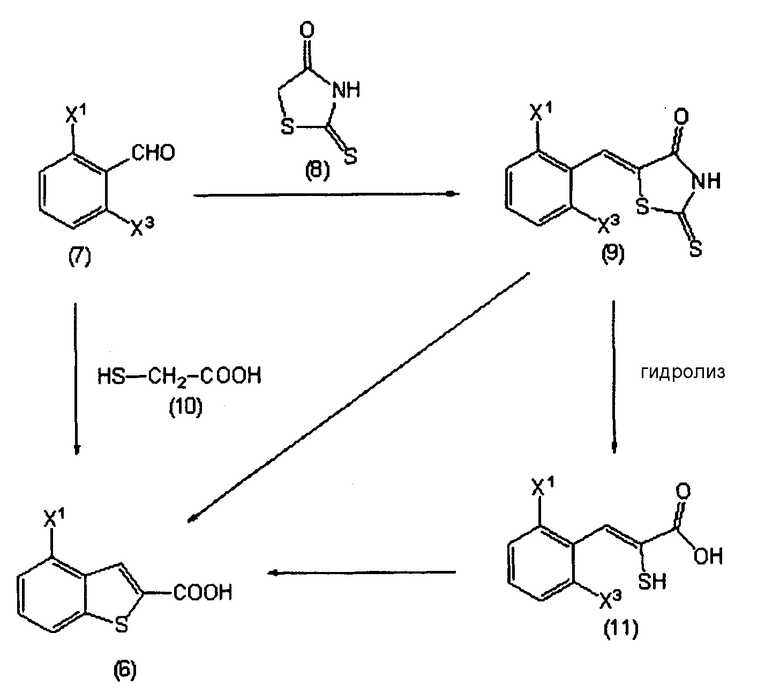
в котором Х1 определен выше, а Х3 представляет собой галоген (фтор, хлор, бром или йод).
Соединения, представленные формулами с (7) по (11), являются известными соединениями, или могут быть легко получены из известных соединений.
Данные последовательности реакций можно осуществить способами, описанными в справочных примерах с 1 по 8, или методами, сходными с данными способами.
Исходное соединение, используемое в каждой из приведенных выше схемах реакций, может быть предпочтительной солью. Кроме того, требуемое соединение, полученное в каждой реакции, может образовывать предпочтительную соль. Примеры каждой из предпочтительных солей соединений (1) и (4а) приведены далее.
Предпочтительные соли соединений (1) и (4а) представляют собой фармакологически приемлемые соли. Их примеры включают в себя соли металлов, таких как соли щелочных металлов (например, соли натрия и соли калия) и соли щелочноземельных металлов (например, соли кальция и соли магния), соли аммония, соли других неорганических оснований, такие как карбонаты щелочных металлов (например, карбонат лития, карбонат калия, карбонат натрия и карбонат цезия), гидрокарбонаты щелочных металлов (например, гидрокарбонат лития, гидрокарбонат натрия и гидрокарбонат калия), и гидроксиды щелочных металлов (например, гидроксид лития, гидроксид натрия, гидроксид калия и гидроксид цезия), соли органических оснований, таких как три(низшие)алкиламинов (например, триметиламина, триэтиламина и N-этилдиизопропиламина), пиридина, хинолина, пиперидина, имидазола, пиколина, диметиламинопиридина, диметиланилина, N-(низшего)алкилморфолина (например, N-метилморфолина), DBN, DBU и DABCO, соли неорганических кислот, такие как гидрохлорид, гидробромид, гидройодид, сульфат, нитрат и фосфат, соли органических кислот, такие как формиат, ацетат, пропионат, оксалат, малонат, сукцинат, фумарат, малеат, лактат, малат, цитрат, тартрат, цитрат, карбонат, пикрат, метансульфонат, этансульфонат, п-толуолсульфонат и глутамат, и так далее.
Каждое из целевых соединений, полученных по приведенным выше схемам реакций, можно выделить и очистить от реакционной смеси, например, охлаждением реакционной смеси с последующей процедурой выделения, такой как фильтрование, концентрирование или экстракция, для выделения сырого продукта реакции, а затем подверганием данного сырого продукта реакции обычному способу очистки, такому как перекристаллизация. С точки зрения промышленного получения, способ очистки предпочтительно представляет собой перекристаллизацию или тому подобное.
Согласно настоящему изобретению, соединения формул с (1) по (4а), безусловно, включают в себя геометрические изомеры, стереоизомеры, оптические изомеры и тому подобные изомеры.
Примеры
Настоящее изобретение описано далее более подробно со ссылкой на справочные примеры и примеры.
Справочный пример 1
- Синтез 2,6-дихлорбензилиденроданина
2,6-Дихлорбензальдегид (77,0 г), роданин (58,6 г) и уксусную кислоту (539 мл) суспендировали при перемешивании при комнатной температуре. К суспензии добавляли безводный ацетат натрия (116 г) и нагревали полученную смесь при кипении в течение 3 часов. Реакционную смесь охлаждали до 45°С и добавляли ледяную воду (700 мл). После перемешивания смеси в течение 0,2 часов, выпавшие кристаллы выделяли фильтрованием, промывали водой, а затем сушили, получая 2,6-дихлорбензилиденроданин. Даже в невысушенной форме, данный продукт можно было бы ввести на следующую стадию.
Выход: 125,4 г
1H-ЯМР (CDCl3) δ м.д. 7,30-7,44 (3H, м), 7,70 (1H, с), 9,6 (1H, шир.).
Справочный пример 2
- Синтез 2-хлор-6-фторбензилиденроданина
2-Хлор-6-фторбензальдегид (9,50 г), роданин (7,98 г) и уксусную кислоту (57 мл) перемешивали при комнатной температуре. К полученной суспензии прибавляли безводный ацетат натрия (14,0 г) и нагревали смесь при перемешивании в течение 2 часов. Реакционную смесь оставляли остывать до комнатной температуры и добавляли к ней ледяную воду (190 мл). Выпавшие кристаллы выделяли фильтрованием, промывали водой, а затем сушили, получая 2-хлор-6-фторбензилиденроданин.
Выход: 15,7 г
1H-ЯМР (CDCl3) δ м.д. 7,37-7,64 (4H, м), 13,9 (1H, шир.).
Справочный пример 3
- Синтез (Z)-3-(2,6-дихлорфенил)-2-меркапто-2-пропеновой кислоты
Суспензию 2,6-дихлорбензилиденроданина (160,4 г) и воды (800 мл) перемешивали при комнатной температуре и прибавляли гидроксид натрия (83,0 г) в течение 1 часа. Полученную смесь нагревали при перемешивании еще в течение 0,5 часов. Реакционную смесь охлаждали льдом (10°С) и добавляли концентрированную хлористоводородную кислоту (192 мл). После перемешивания смеси при охлаждении льдом в течение 0,5 часа, выпавшие кристаллы выделяли фильтрованием. Кристаллы, полученные фильтрованием, промывали водой, а затем сушили, получая эквивалентное количество (Z)-3-(2,6-дихлорфенил)-2-меркапто-2-пропеновой кислоты.
Выход: 138,9 г
1H-ЯМР (ДМСО-d6) δ м.д. 7,23-7,67 (4H, м), 3,5-5,7 (1H, шир.), 11,7-14,5 (1Н, шир.).
Справочный пример 4
- Синтез 2-карбокси-4-хлорбензо[b]тиофена
Суспензию (Z)-3-(2,6-дихлорфенил)-2-меркапто-2-пропеновой кислоты (72,4 г) и воды (362 г) перемешивали при комнатной температуре. Кроме того, добавляли гидроксид калия (40,8 г) и нагревали смесь при кипении в течение 4 часов. После остывания смеси, смесь перемешивали в течение 1 часа при охлаждении льдом. Выпавшие кристаллы (калиевая соль (Z)-3-(2,6-дихлорфенил)-2-меркапто-2-пропеновой кислоты) выделяли фильтрованием и промывали холодной водой. После суспендирования кристаллов в воде, добавляли 35%-ную концентрированную хлористоводородную кислоту (32 мл) (рН=1), и перемешивали смесь при комнатной температуре в течение 1 часа. Выпавшие кристаллы выделяли фильтрованием и сушили, получая 2-карбокси-4-хлорбензо[b]тиофен.
Выход: 48,8 г
1H-ЯМР (ДМСО-d6) δ м.д. 7,53 (1H, т, J=7,7 Гц), 7,58 (1H, дд, J=7,7, 1,3 Гц), 8,03 (1H, д, J=0,5 Гц), 8,07 (1H, д, J=7,6 Гц).
Справочный пример 5
- Синтез 4-хлорбензо[b]тиофен-2-карбоксилата натрия
После растворения гидроксида натрия (4,55 г) в воде (50 мл), прибавляли 2,6-дихлорбензилиденроданин (10,00 г). Смесь перемешивали при кипении в течение 5 часов, а затем охлаждали до комнатной температуры. Выпавшие кристаллы выделяли фильтрованием и промывали холодной водой, получая 4-хлорбензо[b]тиофен-2-карбоксилата натрия.
Выход: 7,24 г
1H-ЯМР (ДМСО-d6) δ м.д. 7,39 (т, J=7,7 Гц, 1H), 7,47 (дд, J=7,7, 1,0 Гц, 1H), 7,73 (д, J=0,8 Гц, 1H), 7,93 (дт, J=7,9, 0,9 Гц, 1H).
Справочный пример 6
- Синтез 2-карбокси-4-хлорбензо[b]тиофена
4-Хлорбензо[b]тиофен-2-карбоксилат натрия (2,40 г) растворяли в воде (33 мл) при 60°С. К раствору добавляли концентрированную хлористоводородную кислоту (1,3 мл) при той же температуре и перемешивали полученную смесь. Выпавшие кристаллы выделяли фильтрованием, промывали водой, а затем сушили, получая 2-карбокси-4-хлорбензо[b]тиофен.
Выход: 1,61 г
1H-ЯМР (ДСМО-d6) 7,53 (1H, т, J=7,7 Гц), 7,58 (1H, дд, J=7,7, 1,3 Гц), 8,03 (1H, д, J=0,5 Гц), 8,07 (1H, д, J=7,6 Гц).
Справочный пример 7
- Синтез 2-карбокси-4-хлорбензо[b]тиофена
После растворения гидроксида калия (30,8 г) в воде (179 мл) к раствору прибавляли тиогликолевую кислоту (19,4 г), а после этого прибавляли 2,6-дихлорбензальдегид (32,0 г). Полученную смесь нагревали при кипении в течение 2,5 часов. Смеси давали остыть, а затем оставляли при комнатной температуре на ночь. Выпавшие кристаллы (4-хлорбензо[b]тиофен-2-карбоксилата калия) выделяли фильтрованием и промывали холодной водой. Затем кристаллы диспергировали в воде (256 мл). После добавления к ним концентрированной хлористоводородной кислоты (20 мл) полученную смесь перемешивали в течение 1 часа. Выпавшие кристаллы выделяли фильтрованием и промывали водой. Сырые кристаллы данного 2-карбокси-4-хлорбензо[b]тиофена диспергировали в этилацетате (96 мл) и промывали при комнатной температуре. Выпавшие кристаллы промывали этилацетатом, а затем сушили, получая 29,12 г высушенного продукта. После этого высушенный продукт дополнительно промывали этилацетатом, а затем промывной раствор концентрировали до 70 мл. Вторую порцию выпавших кристаллов выделяли и сушили, получая 2-карбокси-4-хлорбензо[b]тиофен (1,35 г).
Выход: 30,5 г
Белые кристаллы
1H-ЯМР (ДМСО-d6) δ м.д. 7,53 (1H, т, J=7,7 Гц), 7,58 (1H, дд, J= 7,7, 1,3 Гц), 8,03 (1H, д, J=0,5 Гц), 8,07 (1H, д, J=7,6 Гц).
Справочный пример 8
- Синтез 4-хлорбензо[b]тиофена
Смесь 2-карбокси-4-хлорбензо[b]тиофена (7,24 г), хинолина (36 мл) и порошкообразной меди (1,45 г) перемешивали при температуре от 145 до 155°С в течение 1 часа. После остывания смеси до комнатной температуры смесь разбавляли диизопропиловым эфиром (145 мл), и удаляли нерастворимое вещество фильтрованием. Фильтрат промывали разбавленной хлористоводородной кислотой (40 мл 35%-ной концентрированной хлористоводородной кислоты + 200 мл холодной воды) и водой, затем сушили над сульфатом магния и концентрировали. Следовые количества осадка дополнительно удаляли из концентрированного раствора фильтрованием, получая 4-хлорбензо[b]тиофен.
Выход: 5,59 г
Светло-коричневое масло
1H-ЯМР (ДМСО-d6) δ м.д. 7,38 (1H, т, J=8,4 Гц), 7,51 (1H, дд, J=5,5, 0,8 Гц), 7,48 (1H, дд, J=7,7, 0,9 Гц), 7,94 (1H, дд, J=5,5, 0,4 Гц), 8,02(1H, дт, J=8,0, 0,9 Гц).
Пример 1
- Синтез 4-хлорбензо[b]тиофена
Смесь 2-карбокси-4-хлорбензо[b]тиофена (50,00 г), 1,3-диметил-2-имидазолидинона (DMI, 200 мл) и 1,8-диазабицикло[5.4.0]ундец-7-ена (140,7 мл) нагревали при температуре от 160 до 195°С при перемешивании в течение 6 часов. После охлаждения смеси до 10°С, смесь прибавляли к 3N хлористоводородной кислоте (350 мл), охлажденной до 10°С. После экстрагирования смеси толуолом (500 мл), слой толуола промывали 3N хлористоводородной кислотой, водой, водным раствором гидрокарбоната натрия, водой, насыщенным раствором соли, и водой в данном порядке, а затем концентрировали, получая 4-хлорбензо[b]тиофен.
Выход: 36,78 г
1H-ЯМР (ДМСО-d6) δ м.д. 7,38 (1H, т, J=8,4 Гц), 7,51 (1H, дд, J=5,5, 0,8 Гц), 7,48 (1H, дд, J=7,7, 0,9 Гц), 7,94 (1H, дд, J=5,5, 0,4 Гц), 8,02 (1H, дт, J=8,0, 0,9 Гц).
Пример 2
- Синтез гидрохлорида 4-(1-пиперазинил)бензо[b]тиофена
4-Хлорбензо[b]тиофен (5,00 г), пиперазин (5,11 г), ацетат паладия(II) (2,7 мг), тетрафенилборат три-третбутилфосфония (6,2 мг), третбутилат натрия (8,548 г) и ксилол (70 мл) перемешивали при температуре от 120 до 130°С в течение 5 часов. После охлаждения реакционной смеси до комнатной температуры к ней прибавляли воду и разделяли слои. Слой ксилола промывали водой, а затем насыщенным раствором соли. После добавления активированного угля смесь перемешивали при комнатной температуре в течение 30 минут. После фильтрования смеси к фильтрату прибавляли концентрированную хлористоводородную кислоту и перемешивали полученную смесь при комнатной температуре в течение 30 минут. Выпавшие кристаллы выделяли фильтрованием и сушили, получая гидрохлорид 4-(1-пиперазинил)бензо[b]тиофена.
Выход: 6,94 г
1H-ЯМР (ДМСО-d6) δ м.д. 3,30 (4H, шир. с), 3,61 (4H, шир. с), 6,97 (1H, д, J=7,8 Гц), 7,32 (1H, шир. дд, J=8,4, 7,8 Гц), 7,53 (1H, д, J=5,6 Гц), 7,70 (1H, д, J=8,4 Гц), 7,76 (1H, д, J=5,6 Гц), 9,37 (1H, шир. с).
Пример 3
- Синтез гидрохлорида 4-(1-пиперазинил)бензо[b]тиофена
4-Хлорбензо[b]тиофен (10,0 г) и ксилол (100 мл) помещали в реакционный сосуд. Реакционный сосуд вакуумировали, а затем продували аргоном. После этого прибавляли пиперазин (15,3 г), третбутилат натрия (17,1 г), ацетат палладия(II) (13,0 г) и 2-дициклогексилфосфино-2′,6′-диизопропокси-1,1′-дифенил (RuPhos) (69,0 мг). После вакуумирования и продувания аргоном, смесь нагревали при кипении в течение 2 часов. После охлаждения реакционной смеси примерно до 80°С, прибавляли воду (50 мл) и оксид кремния #600Н (0,65 г). Смесь перемешивали приблизительно при 60°С примерно в течение 10 минут, а затем фильтровали. После разделения фильтрата на два слоя, слой ксилола промывали водой. Затем слой ксилола снова помещали в реакционный сосуд. После добавления воды (200 мл) и концентрированной хлористоводородной кислоты (8,0 мл) смесь нагревали при перемешивании для растворения. Слои разделяли при 75°С или выше. После отделения водного слоя прибавляли толуол (150 мл) и 25%-ный водный раствор гидроксида натрия (16 мл), и перемешивали смесь. Слои разделяли и отделяли органический слой. Органический слой промывали водой и концентрировали на роторном испарителе. К концентрированному маслу прибавляли метанол (150 мл) для растворения масла, получая в результате раствор в метаноле. 2-Пропанол (150 мл) и концентрированную хлористоводородную кислоту (7 мл) помещали в другой реакционный сосуд и прибавляли по каплям раствор в метаноле в течение 15 минут или более. По окончании прибавления по каплям, смесь охлаждали и перемешивали при температуре 10°С или ниже примерно в течение 30 минут, а затем фильтровали (промывали смесью 5 мл метанола и 5 мл 2-пропанола). Кристаллы выделяли, а затем сушили, получая гидрохлорид 4-(1-пиперазинил)бензо[b]тиофена.
Выход: 11,61 г
1H-ЯМР (ДМСО-d6) δ м.д. 3,25-3,40 (8H, шир. с), 6,96 (1H, д, J=7,5 Гц), 7,32 (1H, дд, J=8,0, 7,5 Гц), 7,52 (1H, д, J=5,5 Гц), 7,70 (1H, д, J=8,0 Гц), 7,75 (1H, д, J=5,5 Гц), 9,35 (1H, шир. с).
Справочный пример 9
- Синтез 7-(4-хлорбутокси)-1Н-хинолин-2-она
После нагревания 7-гидрокси-1Н-хинолин-2-она (10 г) и ДМФА (50 мл) приблизительно до 30°С, прибавляли водный раствор карбоната калия (карбонат калия: 8,6 г, вода: 10 мл). После перемешивания смеси при температуре от 30 до 40°С примерно в течение 15 минут, прибавляли 1-бром-4-хлорбутан (14,3 мл) и перемешивали приблизительно при 40°С в течение 5 часов. Прибавляли по каплям воду (100 мл) в течение 30 минут или более, поддерживая температуру при 30°С или выше. После того, как смесь перемешивали приблизительно при 30°С в течение 30 минут, перемешивание продолжали при температуре от 10°С или ниже в течение 1 часа, после чего выпавшие кристаллы выделяли фильтрованием. После прибавления метанола (100 мл) к выпавшим кристаллам, смесь перемешивали при кипении, чтобы добиться растворения. Данный раствор охлаждали и перемешивали при температуре от 30 до 40°С в течение 30 минут, а затем при 5°С или ниже примерно в течение 1 часа, после чего выпавшие кристаллы выделяли фильтрованием. Кристаллы сушили при 60°С, получая 7-(4-хлорбутокси)-1Н-хинолин-2-он в виде порошка белого цвета.
Выход: 12,3 г
1H-ЯМР (300 МГц; CDCl3) δ м.д. 1,95-2,05 (4H, м), 3,64 (2H, т, J=6,0 Гц), 4,10 (2H, т, J=5,5 Гц), 6,56 (1H, д, J=9,5 Гц), 6,80 (1H, дд, J=9,0 Гц, 2,5 Гц), 6,84 (1H, д, J=2,5 Гц), 7,45 (1H, д, J=9,0 Гц), 7,73 (1H, д, J=9,5 Гц), 12,45 (1H, шир. с).
Пример 4
- Синтез 7-[4-(4-бензо[b]тиофен-4-ил-пиперазин-1-ил)бутокси]-1Н-хинолин-2-она
После перемешивания гидрохлорида 1-бензо[b]тиофен-4-ил-пиперазина (10,6 г), карбоната калия (5,8 г) и ДМФА (50 мл) при температуре от 30 до 40°С примерно в течение 30 минут, прибавляли 7-(4-хлорбутокси)-1Н-хинолин-2-он (10,0 г) и йодид калия (6,9 г). Смесь перемешивали при температуре от 90 до 100°С в течение 2 часов. Поддерживая температуру смеси при 60°С или выше, прибавляли по каплям воду (150 мл) в течение от 10 минут или более. После охлаждения смеси до 10°С или ниже, выпавшие кристаллы выделяли фильтрованием и промывали водой, а затем этанолом.
После добавления этанола (325 мл) и уксусной кислоты (25 мл) к выпавшим кристаллам, смесь перемешивали при кипении для растворения. Прибавляли концентрированную хлористоводородную кислоту (3,6 мл) примерно при 70°С, и охлаждали смесь. Убедившись в выпадении кристаллов, смесь вновь нагревали и перемешивали при кипении в течение 1 часа. После охлаждения смеси до 10°С или ниже, выпавшие кристаллы выделяли фильтрованием и промывали этанолом.
После добавления к выпавшим кристаллам этанола (191 мл) и воды (127 мл), смесь перемешивали при кипении для растворения. После добавления активированного угля (0,89 г), смесь перемешивали при кипении в течение 30 минут, а затем подвергали горячему фильтрованию. После удаления активированного угля, смесь снова нагревали для растворения. После прибавления 25%-ного водного раствора гидроксида натрия (5,8 мл) приблизительно при 70°С, смесь перемешивали при кипении в течение 30 минут, после чего прибавляли воду (64 мл) приблизительно при 70°С. После перемешивания смеси при 40°С в течение 30 минут, выпавшие кристаллы выделяли фильтрованием при температуре 40°С или менее, затем промывали водой и сушили, получая 7-[4-(4-бензо[b]тиофен-4-ил-пиперазин-1-ил)бутокси]-1Н-хинолин-2-он в виде кристаллов белого цвета.
Выход: 14,30 г
1H-ЯМР (ДМСО-d6) δ м.д. 1,6-1,75 (2H, м), 1,75-1,9 (2H, м), 2,44 (2H, т, J=7,0 Гц), 2,55-2,70 (4H, м), 3,00-3,15 (4H, м), 4,06 (2H, т, J=6,3 Гц), 6,30 (1H, д, J=9,5 Гц), 6,75-6,85 (2H, м), 6,88 (1H, д, J=7,5 Гц), 7,27 (1H, дд, J=8 Гц, 8 Гц), 7,40 (1H, д, J=5,5 Гц), 7,55 (1H, д, J=9,5 Гц), 7,61 (1H, д, J=8 Гц), 7,69 (1H, д, J=5,5 Гц), 7,80 (1H, д, J=9,5 Гц), 11,58 (1H, шир. с).
Пример 5
Исследовали соединение палладия и соединение третичного фосфина, или карбеновый комплекс палладия, использованный на второй стадии (стадии А).
Пример 5а
4-хлорбензо[b]тиофен (500 мг) и ксилол (5 мл) помещали в реакционный сосуд. Затем добавляли туда пиперазин (766 мг) и продували реакционный сосуд газообразным аргоном. После этого в реакционный сосуд помещали третбутилат натрия (855 мг), ацетат палладия(II) (6,6 мг, 1,0 мол.%) и 2-(дитретбутилфосфино)-1,1′-дифенил (17,6 мг, 2,0 мол.%). Реакционный сосуд вакуумировали, а затем продували аргоном. После этого реакцию оставляли протекать при кипении в течение 3 часов. После завершения реакции часть суспензии отбирали и анализировали непрореагировавшие вещества и продукт реакции, содержащиеся в реакционной суспензии, при помощи высокоэффективной жидкостной хроматографии (ВЭЖХ) (колонка: XBridge C8 (4,6 мм I.D. × 15 см), элюент: 10 ммоль/л водный раствор додецилсульфата натрия (SDS):ацетонитрил:уксусная кислота = 50:50:1, 290 нм, скорость потока: 1,0 мл/мин, температура: 30°С). В приведенной ниже таблице 1 представлены результаты.
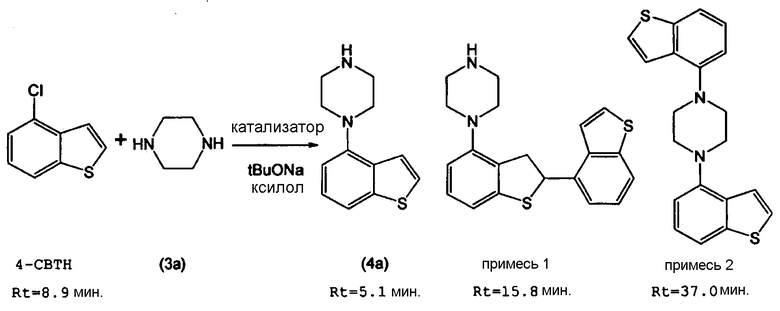
Rt представляет собой время удерживания в упомянутых выше условиях для ВЭЖХ, tBu представляет собой третбутил, а 4-СВТН означает 4-хлорбензо[b]тиофен.
Примеры с 5b по 5g
Продукт реакции, содержащийся в реакционной суспензии, анализировали так же, как и в примере 5а, за исключением того, что вместо 2-(дитретбутилфосфино)-1,1′-дифенила (L1) использовали соединения третичных фосфинов (упомянутые выше L2, L5, L10, L12, L16 и L17) (2,0 мол.%). В приведенной ниже таблице 1 представлены результаты.
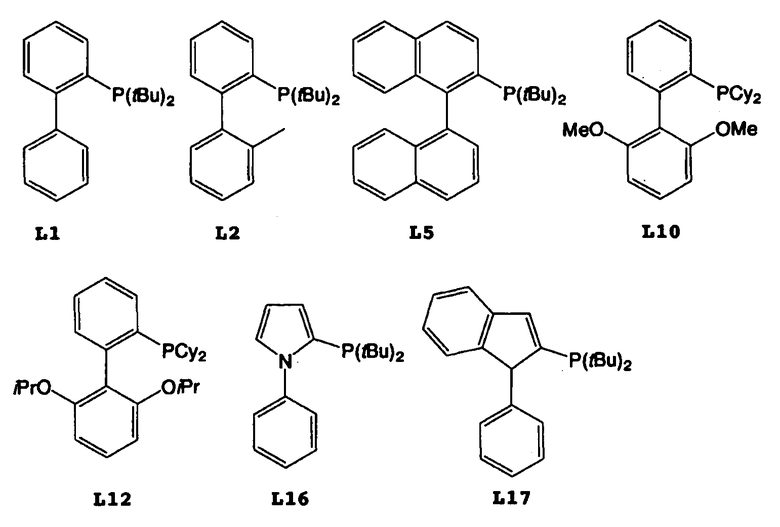
В приведенных выше формулах Ме представляет собой метил, tBu представляет собой третбутил, iPr представляет собой изопропил, а Cy представляет собой циклогексил.
Пример 6
Примеры с 6а по 6d
Продукт реакции, содержащийся в реакционной суспензии, анализировали так же, как и в примере 5а, за исключением того, что вместо ацетата палладия(II) и L1 использовали карбеновые комплексы палладия (упомянутые выше СХ11, СХ21, СХ23 и СХ32) (1,0 мол.%). В приведенной ниже таблице 1 представлены результаты.
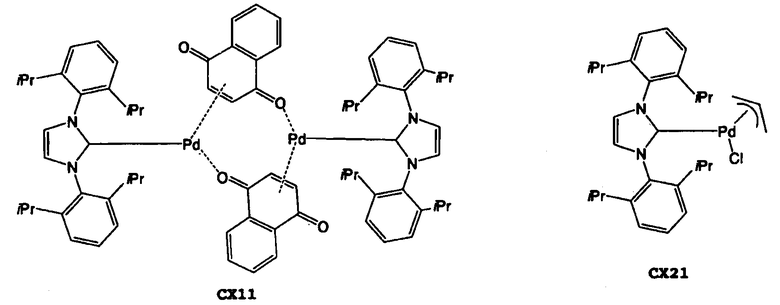
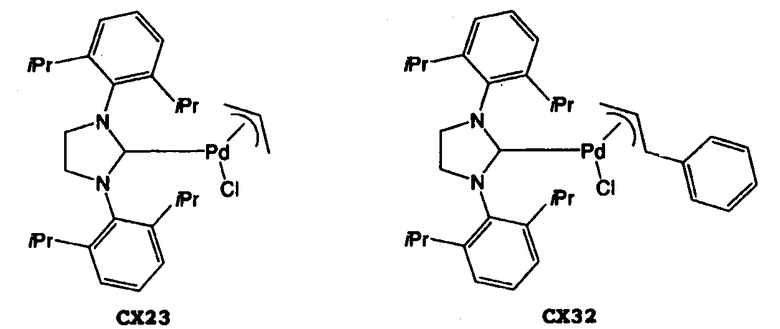
iPr определен выше.
Пример 7
Продукт реакции, содержащийся в реакционной суспензии, анализировали так же, как и в примере 5а, за исключением того, что количество ацетата палладия(II), используемое в примере 5а, изменяли на 0,08 мол.%, и вместо 2-(дитретбутилфосфино)-1,1′-дифенила использовали тетрафенилборат три-третбутилфосфония (0,12 мол.%). В приведенной ниже таблице 1 представлены результаты.
В таблице 1 каждую численную величину рассчитывали из площади пика по ВЭЖХ. TTBuP-K представляет собой тетрафенилборат три-третбутилфосфония, а 4-СВТН определен выше.
Промышленная применимость
В настоящем изобретении предоставлен промышленно эффективный способ получения соединения бензо[b]тиофена, которое применимо в качестве промежуточного соединения в синтезе различных фармацевтических препаратов, таких как антипсихотические лекарственные препараты, пестициды и так далее. В частности, данный способ можно эффективно применять в области производства фармацевтических препаратов.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ 5-ГИДРОКСИПИРИМИДИН-4-КАРБОКСАМИДА | 2010 |
|
RU2550693C2 |
| СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЯ ПИРАЗОЛАМИДА | 2017 |
|
RU2736722C2 |
| ГРИЗЕОФУЛЬВИНОВОЕ СОЕДИНЕНИЕ | 2017 |
|
RU2736208C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИНГИБИТОРОВ ГИРАЗЫ И ТОПОИЗОМЕРАЗЫ IV | 2012 |
|
RU2619116C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФЕНИЛЗАМЕЩЕННОГО ГЕТЕРОЦИКЛИЧЕСКОГО ПРОИЗВОДНОГО ПОСРЕДСТВОМ СОЧЕТАНИЯ С ИСПОЛЬЗОВАНИЕМ ПЕРЕХОДНОГО МЕТАЛЛА В КАЧЕСТВЕ КАТАЛИЗАТОРА | 2010 |
|
RU2510393C9 |
| СОЛЬ ГЕТЕРОЦИКЛИЧЕСКОГО СОЕДИНЕНИЯ, ЗАМЕЩЕННОГО ГАЛОГЕНОМ | 2015 |
|
RU2689315C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФЕНИЛЗАМЕЩЕННОГО ГЕТЕРОЦИКЛИЧЕСКОГО ПРОИЗВОДНОГО ПОСРЕДСТВОМ СПОСОБА СОЧЕТАНИЯ С ИСПОЛЬЗОВАНИЕМ СОЕДИНЕНИЯ ПАЛЛАДИЯ | 2011 |
|
RU2563459C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ ГЕТЕРОЦИКЛОМ ПРОИЗВОДНЫХ ПИРИДИНА | 2008 |
|
RU2474581C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ФЕНИЛАЛАНИНА И ИХ ПОЛУПРОДУКТОВ | 2003 |
|
RU2280641C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЭНАНТИОМЕРНО И ДИАСТЕРЕОМЕРНО ОБОГАЩЕННЫХ ЦИКЛОБУТАНАМИНОВ И -АМИДОВ | 2018 |
|
RU2793738C2 |
Изобретение относится к способу получения соединения, представленного формулой (4):
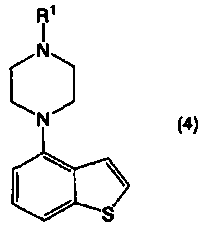
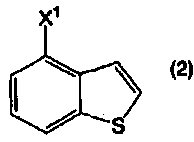
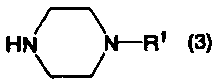
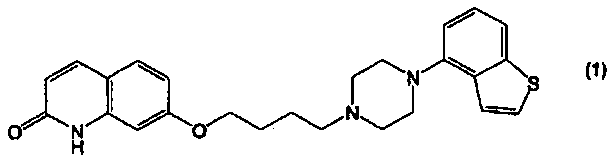
в котором R1 представляет собой атом водорода или защитную группу, или его соли, реакцией соединения, представленного формулой (2), в котором X1 представляет собой уходящую группу, с соединением, представленным формулой (3), в котором R1 определен выше, или его солью, при этом данный способ включает взаимодействие соединения, представленного формулой (2), с соединением, представленным формулой (3), или его солью, в присутствии (а) соединения палладия и третичного фосфина или (b) карбенового комплекса палладия, в инертном растворителе или в отсутствие растворителя; где третичный фосфин представляет собой по меньшей мере одно соединение, выбранное из группы, включающей три-третбутилфосфин, 2-(дитретбутилфосфино)-1,1′-дифенил, 2-(дитретбутилфосфино)-2′-метил-1,1′-дифенил, 2-(дитретбутилфосфино)-1,1′-динафтил, 2-дициклогексилфосфино-2′,6′-диметокси-1,1′-дифенил, 2-дициклогексилфосфино-2′,6′-диизопропокси-1,1′-дифенил, N-фенил-2-(дитретбутилфосфино)пиррол и 1-фенил-2-(дитретбутилфосфино)-1Н-инден. Изобретение также относится к способам получения соединения, представленного формулой (1). Технический результат: получение соединений формулы 4 с высоким выходом. 3 н. и 11 з.п. ф-лы, 1 табл., 16 пр.
1. Способ получения соединения, представленного формулой (4):
в котором R1 представляет собой атом водорода или защитную группу, или его соли, реакцией соединения, представленного формулой (2):
в котором X1 представляет собой уходящую группу, с соединением, представленным формулой (3):
в котором R1 определен выше, или его солью, при этом данный способ включает:
стадию А: взаимодействие соединения, представленного формулой (2), с соединением, представленным формулой (3), или его солью, в присутствии (а) соединения палладия и третичного фосфина или (b) карбенового комплекса палладия, в инертном растворителе или в отсутствие растворителя;
где третичный фосфин представляет собой по меньшей мере одно соединение, выбранное из группы, включающей три-третбутилфосфин, 2-(дитретбутилфосфино)-1,1′-дифенил, 2-(дитретбутилфосфино)-2′-метил-1,1′-дифенил, 2-(дитретбутилфосфино)-1,1′-динафтил, 2-дициклогексилфосфино-2′,6′-диметокси-1,1′-дифенил, 2-дициклогексилфосфино-2′,6′-диизопропокси-1,1′-дифенил, N-фенил-2-(дитретбутилфосфино)пиррол и 1-фенил-2-(дитретбутилфосфино)-1Н-инден.
2. Способ по п. 1, в котором стадию А осуществляют в присутствии (а) соединения палладия и третичного фосфина;
где третичный фосфин представляет собой по меньшей мере одно соединение, выбранное из группы, включающей три-третбутилфосфин, 2-(дитретбутилфосфино)-1,1′-дифенил, 2-(дитретбутилфосфино)-2′-метил-1,1′-дифенил, 2-(дитретбутилфосфино)-1,1′-динафтил, 2-дициклогексилфосфино-2′,6′-диметокси-1,1′-дифенил, 2-дициклогексилфосфино-2′,6′-диизопропокси-1,1′-дифенил, N-фенил-2-(дитретбутилфосфино)пиррол и 1-фенил-2-(дитретбутилфосфино)-1Н-инден.
3. Способ п. 1, в котором стадию А осуществляют в присутствии (b) карбенового комплекса палладия,
где карбеновый комплекс палладия представляет собой по меньшей мере одно соединение, выбранное из группы, включающей (1,4-нафтохинон)-[1,3-бис(2,6-диизопропилфенил)имидазол-2-илиден]палладий(0), аллилхлор-[1,3-бис(2,6-диизопропилфенил)имидазол-2-илиден]палладий(II), аллилхлор-[1,3-бис(2,6-диизопропилфенил)-4,5-дигидроимидазол-2-илиден]палладий(II) и (3-фенилаллилхлор)-[1,3-бис(2,6-диизопропилфенил)-4,5-дигидроимидазол-2-илиден]палладий(II).
4. Способ по любому из пп. 1-3, дополнительно включающий стадию получения соединения, представленного формулой (2), декарбоксилированием соединения, представленного формулой (6):
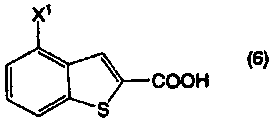
в котором X1 определен выше, или его соли, в присутствии высококипящего основного соединения, в высококипящем растворителе или в отсутствие растворителя.
5. Способ получения соединения, представленного формулой (1):
или его соли, включающий образование соединения, представленного формулой (4)
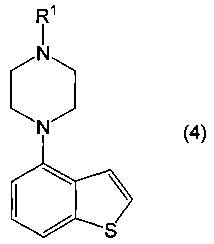
где R1 является защитной группой, или его соли согласно способу по п. 1;
получение соединения, представленного формулой (4а):
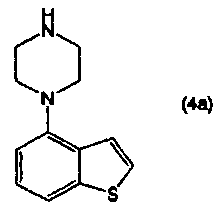
или его соли удалением защитной группы соединения, представленного формулой (4); и
стадию В: взаимодействие соединения, представленного формулой (4а), или его соли с соединением, представленным формулой (5):
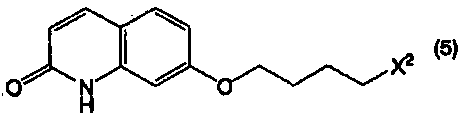
в котором X2 представляет собой уходящую группу, или его солью, в присутствии основного соединения, в инертном растворителе или в отсутствие растворителя.
6. Способ получения соединения, представленного формулой (1):
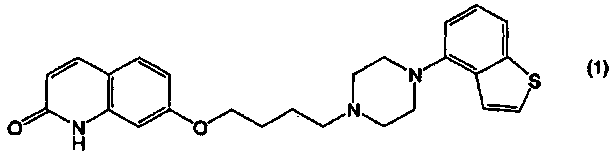
или его соли взаимодействием соединения, представленного формулой (2):
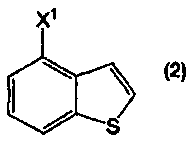
в котором X1 представляет собой уходящую группу, с соединением, представленным формулой (3а):
или его солью и взаимодействием полученного соединения, представленного формулой (4а):
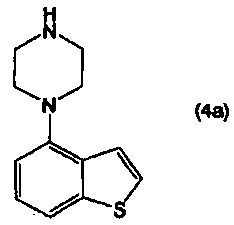
или его соли с соединением, представленным формулой (5):
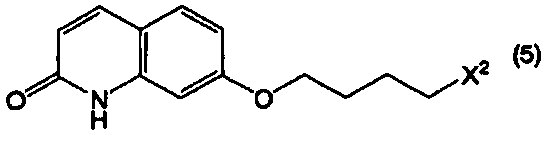
в котором X2 представляет собой уходящую группу, или его солью, при этом данный способ включает:
стадию А: взаимодействие соединения, представленного формулой (2), с соединением, представленным формулой (3а), или его солью, в присутствии (а) соединения палладия и третичного фосфина или (b) карбенового комплекса палладия, в инертном растворителе или в отсутствие растворителя; где третичный фосфин представляет собой по меньшей мере одно соединение, выбранное из группы, включающей три-третбутилфосфин, 2-(дитретбутилфосфино)-1,1′-дифенил, 2-(дитретбутилфосфино)-2′-метил-1,1′-дифенил, 2-(дитретбутилфосфино)-1,1′-динафтил, 2-дициклогексилфосфино-2′,6′-диметокси-1,1′-дифенил, 2-дициклогексилфосфино-2′,6′-диизопропокси-1,1′-дифенил, N-фенил-2-(дитретбутилфосфино)пиррол и 1-фенил-2-(дитретбутилфосфино)-1Н-инден; и
стадию В: взаимодействие соединения, представленного формулой (4а), или его соли с соединением, представленным формулой (5), или его солью, в присутствии основного соединения, в инертном растворителе или в отсутствие растворителя.
7. Способ по п. 5 или 6, в котором стадию А осуществляют в присутствии (а) соединения палладия и третичного фосфина;
где третичный фосфин представляет собой по меньшей мере одно соединение, выбранное из группы, включающей три-третбутилфосфин, 2-(дитретбутилфосфино)-1,1′-дифенил, 2-(дитретбутилфосфино)-2′-метил-1,1′-дифенил, 2-(дитретбутилфосфино)-1,1′-динафтил, 2-дициклогексилфосфино-2′,6′-диметокси-1,1′-дифенил, 2-дициклогексилфосфино-2′,6′-диизопропокси-1,1′-дифенил, N-фенил-2-(дитретбутилфосфино)пиррол и 1-фенил-2-(дитретбутилфосфино)-1Н-инден.
8. Способ по п. 5 или 6, в котором стадию А осуществляют в присутствии (b) карбенового комплекса палладия;
где карбеновый комплекс палладия представляет собой по меньшей мере один комплекс, выбранный из группы, включающей (1,4-нафтохинон)-[1,3-бис(2,6-диизопропилфенил)имидазол-2-илиден]палладий(0), аллилхлор-[1,3-бис(2,6-диизопропилфенил)имидазол-2-илиден]палладий(II), аллилхлор-[1,3-бис(2,6-диизопропилфенил)-4,5-дигидроимидазол-2-илиден]палладий(II) и (3-фенилаллилхлор)-[1,3-бис(2,6-диизопропилфенил)-4,5-дигидроимидазол-2-илиден]палладий(II).
9. Способ по п. 5 или 6, дополнительно включающий:
стадию С: смешивание уксусной кислоты и спирта с продуктом реакции, полученным на стадии В; и
стадию D: добавление хлористо-водородной кислоты к смеси, полученной на стадии С, с получением соединения гидрохлорида формулы (1).
10. Способ по п. 9, дополнительно включающий:
стадию Е: взаимодействие гидрохлорида соединения, представленного формулой (1), полученного на стадии D, в присутствии основного соединения, с получением соединения, представленного формулой (1).
11. Способ по п. 10, дополнительно включающий:
стадию F: перевод соединения, представленного формулой (1), полученного на стадии Е, в солевую форму.
12. Способ по п. 9, в котором спирт представляет собой по меньшей мере один спирт, выбранный из группы, включающей метанол, этанол и изопропиловый спирт.
13. Способ по п. 5 или 6, дополнительно включающий стадию получения соединения, представленного формулой (2), декарбоксилированием соединения, представленного формулой (6):
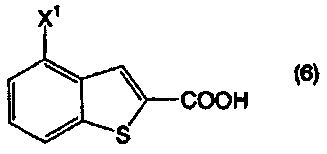
в котором X1 определен выше, или его соли, в присутствии высококипящего основного соединения, в высококипящем растворителе или в отсутствие растворителя.
14. Способ по п. 7, дополнительно включающий стадию получения соединения, представленного формулой (2), декарбоксилированием соединения, представленного формулой (6):
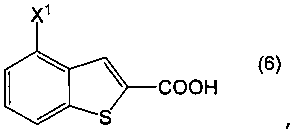
где X1 имеет значения, определенные выше, или его соли, в присутствии высококипящего основного соединения, в высококипящем растворителе или в отсутствие растворителя.
| ПИПЕРАЗИН-ЗАМЕЩЕННЫЕ БЕНЗОТИОФЕНЫ ДЛЯ ЛЕЧЕНИЯ ПСИХИЧЕСКИХ РАССТРОЙСТВ | 2006 |
|
RU2402549C2 |
| WO 2008047883 A1, 24.04.2008 | |||
| WO 9406789 A1, 31.03.1994 | |||
| D | |||
| Allen et al | |||
| "An improved synthesis of substituted benzo[b]thiophenes using microwave irradiation" TETRAHEDRON LETTERS, vol | |||
| Железобетонный фасонный камень для кладки стен | 1920 |
|
SU45A1 |

