[Область техники]
[0001] Настоящее изобретение относится к новому способу получения соединения пиразоламида или его соли или гидрата, которое может быть использовано как ингибитор киназы пируватдегидрогеназы (в дальнейшем сокращенно PDHK), и к его промежуточному соединению.
[СУЩНОСТЬ ИЗОБРЕТЕНИЯ]
[0002] Задачей настоящего изобретения является разработка нового способа получения соединения пиразоламида или его соли или гидрата, которое может быть использовано для лечения или профилактики заболеваний, связанных с нарушением метаболизма глюкозы (например, диабета (диабет 1 типа, диабет 2 типа и т.д.)), синдрома инсулинорезистентности, метаболического синдрома, гипергликемии, гиперлактатацидоза, осложнений диабета (диабетическая невропатия, диабетическая ретинопатия, диабетическая нефропатия, катаракта и т.д.)), заболеваний, в которых ограничена доставка энергетического субстрата к тканям (например, сердечной недостаточности (острая сердечная недостаточность, хроническая сердечная недостаточность), кардиомиопатии, ишемии миокарда, инфаркта миокарда, стенокардии, дислипидемии, атеросклероза, заболевания периферических артерий, перемежающейся хромоты, хронических обструктивных болезней легких, ишемии головного мозга, кровоизлияния в мозг), митохондриального заболевания, митохондриальной энцефаломиопатии, рака, легочной гипертензии и т.п., и т.п.
Один вариант осуществления настоящего изобретения является таким, как показано в следующих пунктах [1] - [18a].
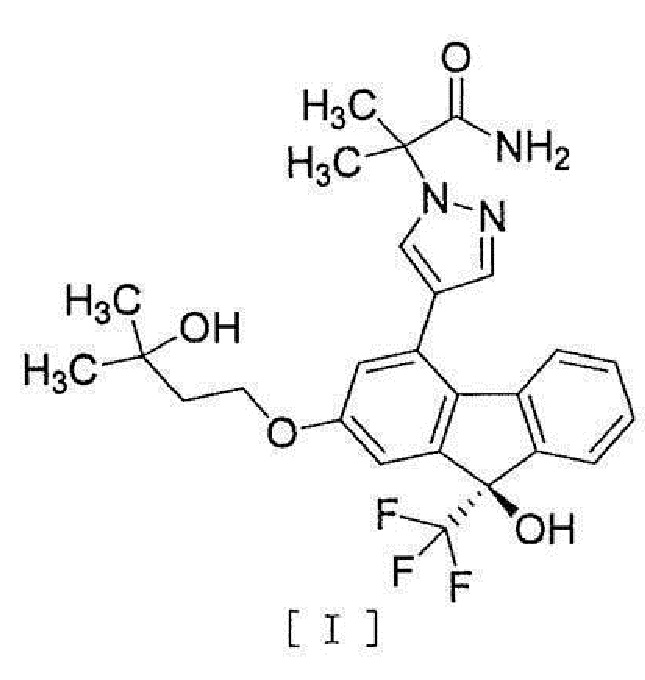
[1] Способ получения соединения, представленного формулой [I]:
[0003] 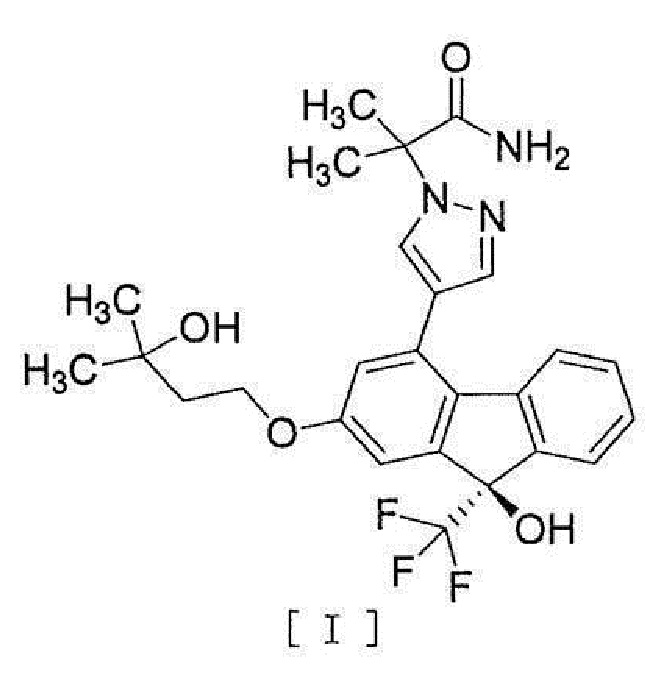
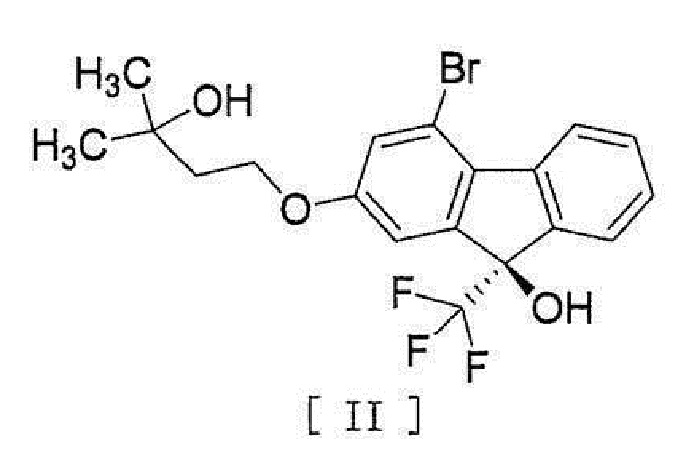
[0004] или его фармацевтически приемлемой соли или гидрата, включающий стадию превращения соединения, представленного формулой [II]:
[0005] 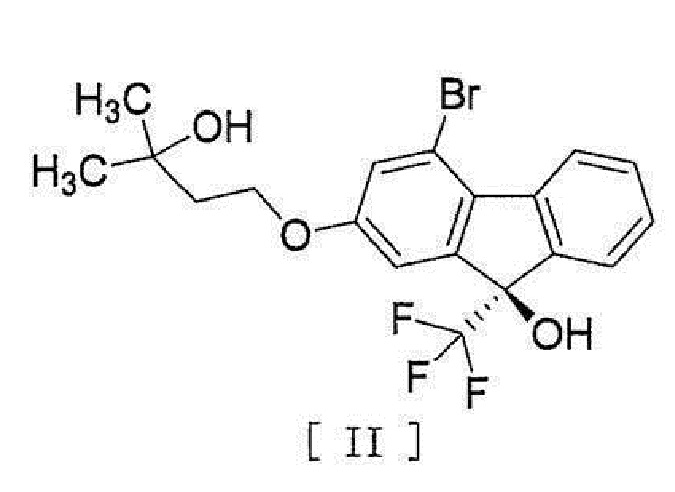
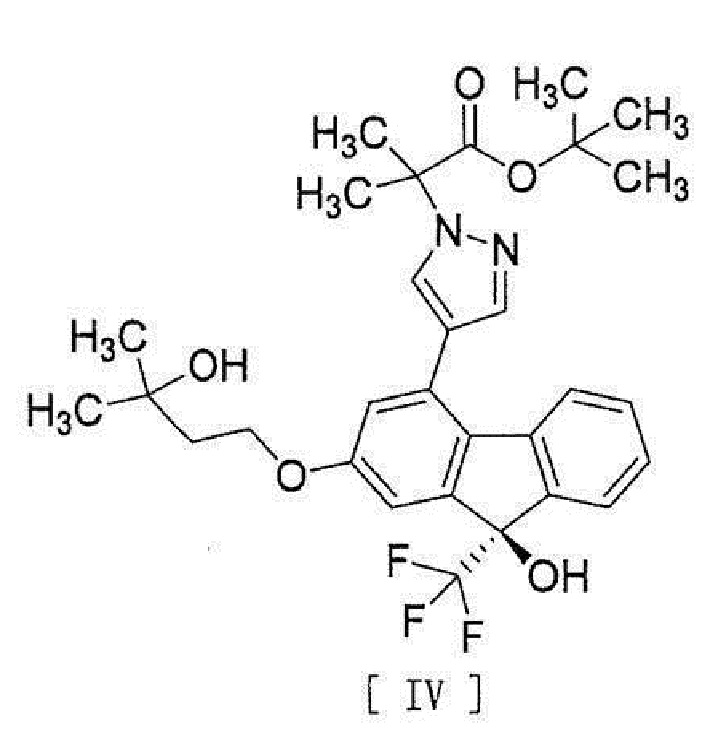
[0006] или его метанольного сольвата в соединение, представленное формулой [IV]:
[0007] 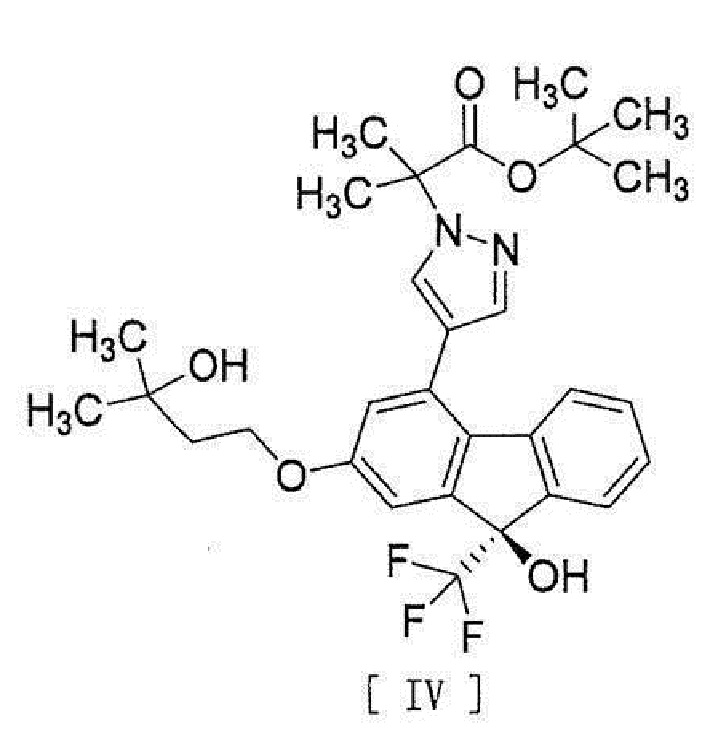
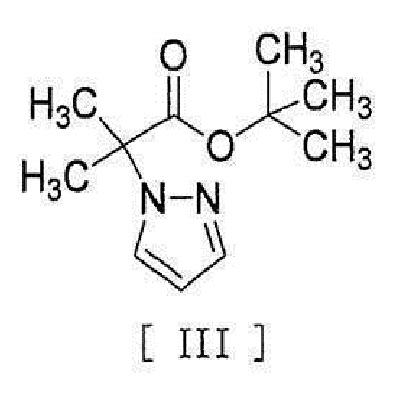
[0008] реакцией сочетания с соединением, представленным формулой [III]:
[0009] 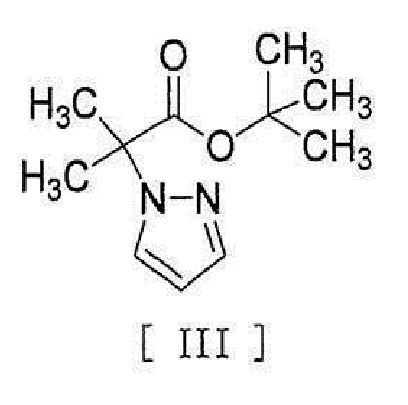
[0010] в присутствии металлического катализатора, основания и карбоновой кислоты.
[2] Способ согласно вышеописанному пункту [1], в котором металлический катализатор является палладиевым катализатором.
[3] Способ согласно вышеописанным пунктам [1] или [2], в котором основание является карбонатом щелочного металла или ацетатом щелочного металла.
[4] Способ согласно любому из вышеописанных пунктов [1] - [3], в котором карбоновая кислота является триметилуксусной кислотой, изомасляной кислотой, пропионовой кислотой или бензойной кислотой.
[5] Способ согласно любому из вышеописанных пунктов [1] - [4], в котором температура реакции реакции сочетания составляет от 80 до 150°C.
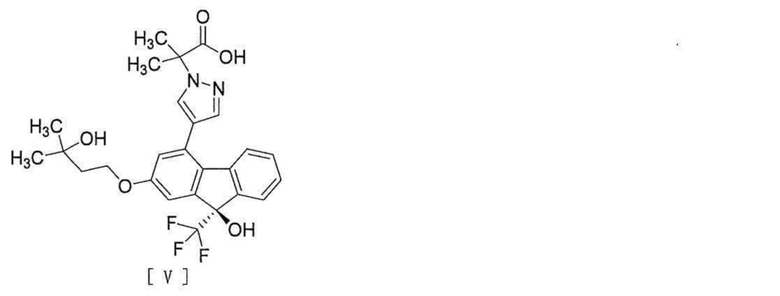
[6] Способ согласно любому из вышеописанных пунктов [1] - [5], дополнительно включающий стадию гидролиза соединения вышеописанной формулы [IV] для превращения его в соединение, представленное формулой [V]:
[0011]
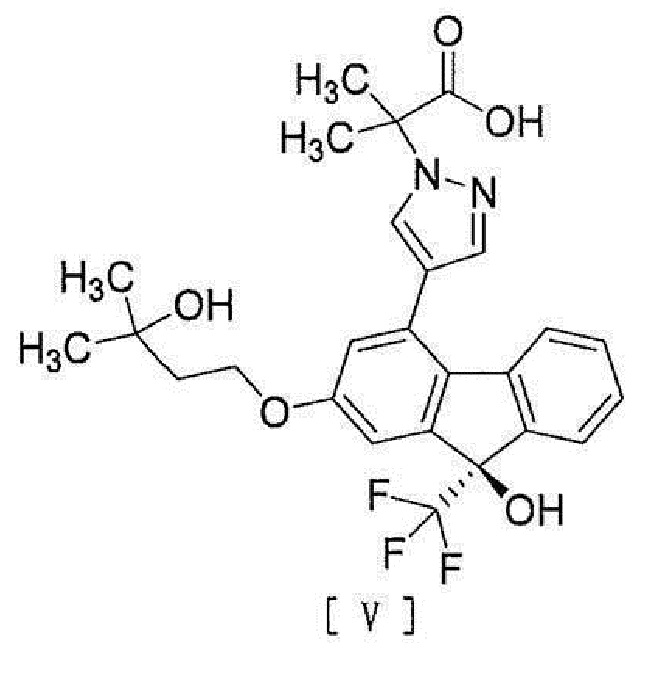
[0012] или его соль.
[7] Способ согласно вышеописанному пункту [6], дополнительно включающий стадию реакции соединения вышеописанной формулы [V] или его соли с аммиаком в присутствии конденсирующего агента для превращения его в соединение, представленное вышеописанной формулой [I], или его фармацевтически приемлемую соль или гидрат.
[8] Способ согласно любому из вышеописанных пунктов [1] - [7], в котором соединение вышеописанной формулы [II] получают способом, включающим стадию реакции соединения, представленного формулой [VI]:
[0013] 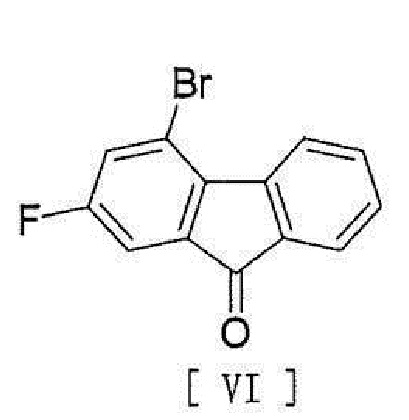
[0014] с соединением, представленным формулой [VII]:
[0015] 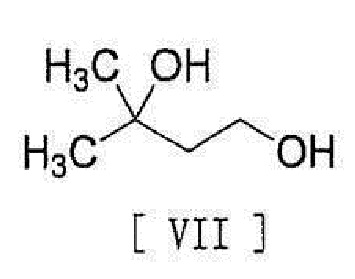
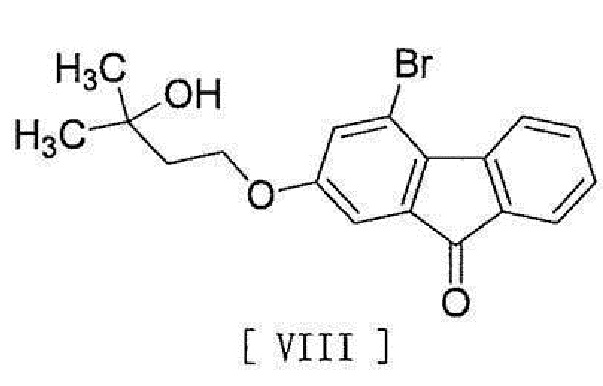
[0016] в присутствии основания для превращения его в соединение, представленное формулой [VIII]:
[0017] 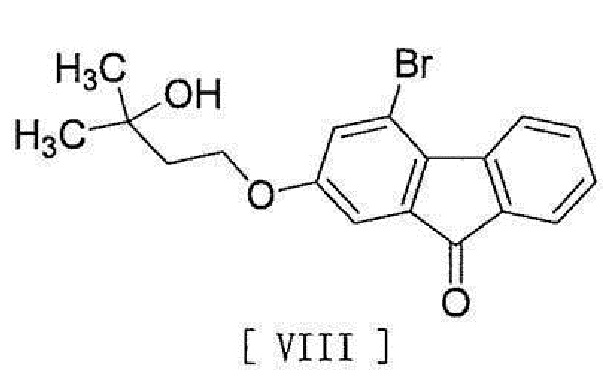
[0018] и стадию реакции соединения формулы [VIII] с (трифторметил)триметилсиланом в присутствии асимметрического органокатализатора и затем обработку кислотой.
[9] Способ согласно вышеописанному пункту [8], в котором асимметрический органокатализатор является солью цинхонидия.
[10] Способ согласно вышеописанному пункту [9], в котором солью цинхонидия является N-(4-трет-бутил-3-метоксибензил)цинхонидий бромид.
[10a] Способ согласно вышеописанному пункту [9], в котором вместе с солью цинхонидия используется добавка.
[10b] Способ согласно вышеописанному пункту [10a], в котором добавка является фенолятом натрия или трет-бутилалкоголятом натрия.
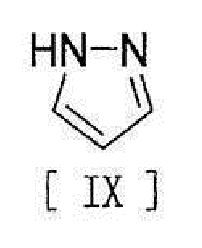
[11] Способ согласно любому из вышеописанных пунктов [1] - [10], в котором соединение вышеописанной формулы [III] получают путем реакции соединения, представленного формулой [IX]:
[0019] 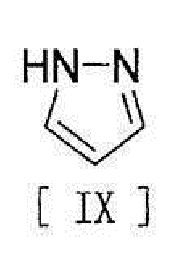
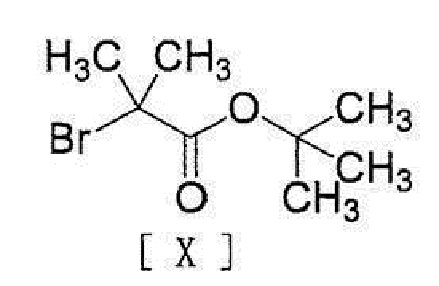
[0020] или его соли с соединением, представленным формулой [X]:
[0021] 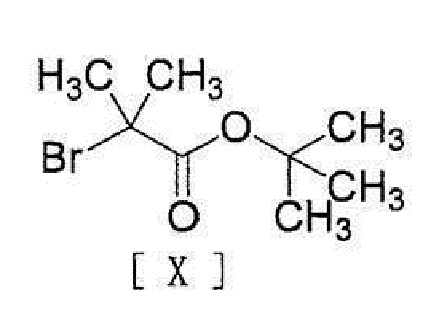
[0022] в присутствии основания.
[12] Способ получения соединения, представленного формулой [I]:
[0023]
[0024] или его фармацевтически приемлемой соли или гидрата, включающий стадию реакции соединения, представленного формулой [VI]:
[0025] 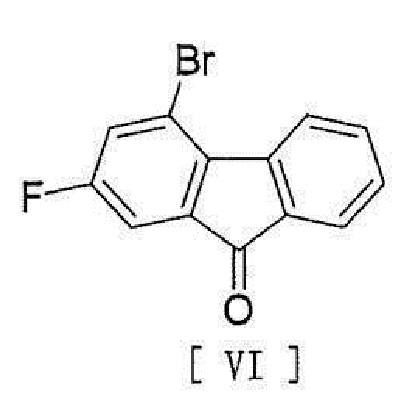
[0026] с соединением, представленным формулой [VII]:
[0027] 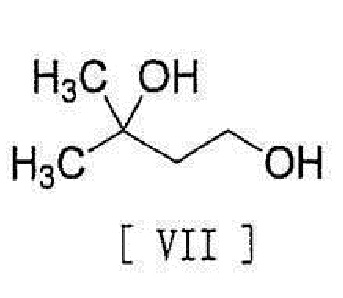

[0028] в присутствии основания для превращения его в соединение, представленное формулой [VIII]:
[0029] 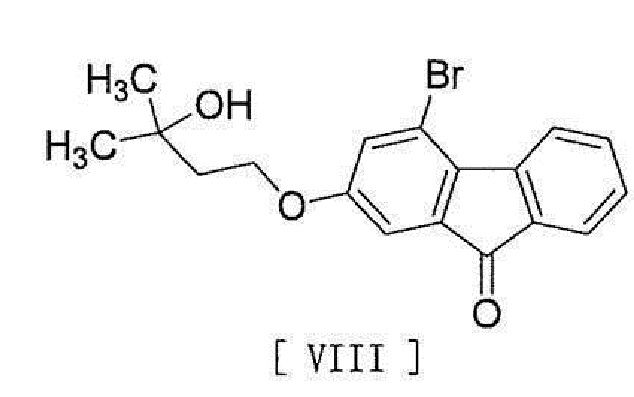
[0030] стадию реакции соединения формулы [VIII] с (трифторметил)триметилсиланом в присутствии асимметрического органокатализатора и обработку кислотой для получения соединения, представленного формулой [II]:
[0031] 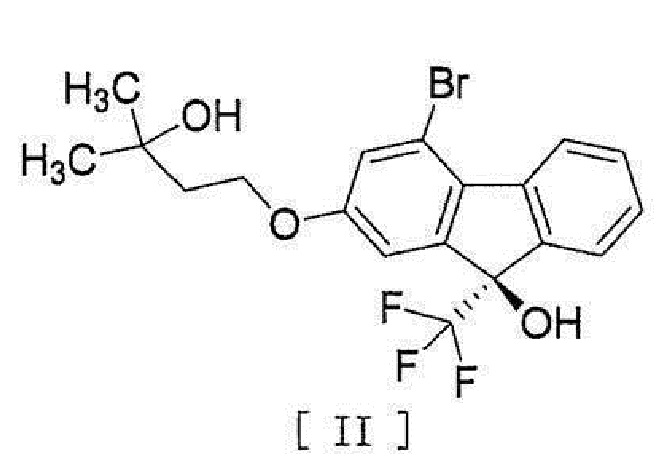
[0032] или его метанольного сольвата, стадию реакции соединения, представленного формулой [IX]:
[0033] 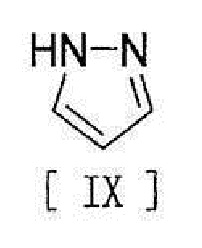
[0034] или его соли с соединением, представленным формулой [X]:
[0035] 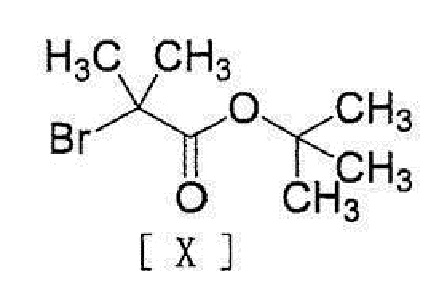
[0036] в присутствии основания для получения соединения, представленного формулой [III]:
[0037] 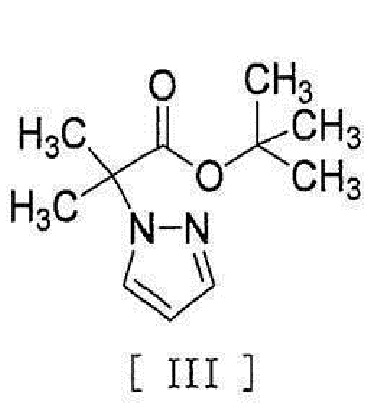
[0038], стадию введения соединения вышеописанной формулы [III] в реакцию сочетания с соединением вышеописанной формулы [II] или его метанольного сольвата в присутствии металлического катализатора, основания и карбоновой кислоты для превращения этого соединения в соединение, представленное формулой [IV]:
[0039] 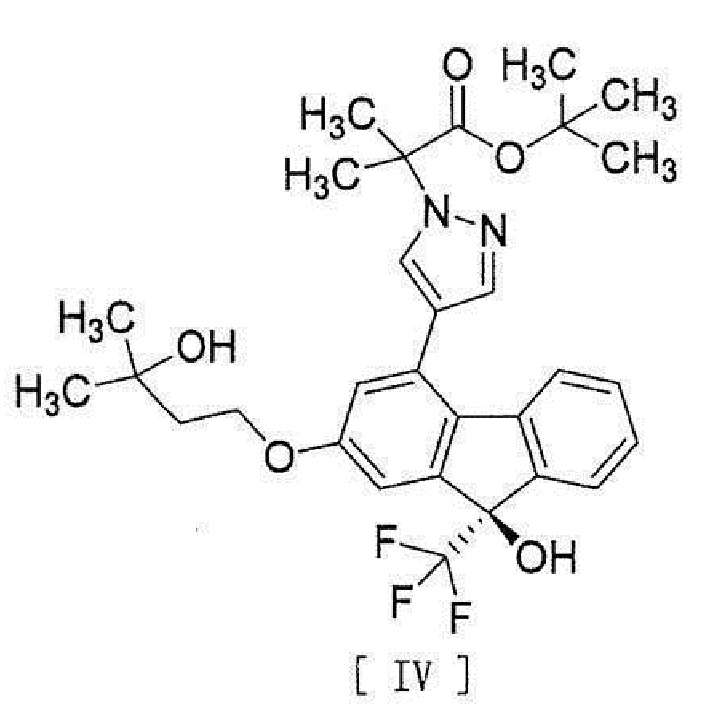
[0040], стадию гидролиза соединения вышеописанной формулы [IV] для превращения этого соединения в соединение, представленное формулой [V]:
[0041] 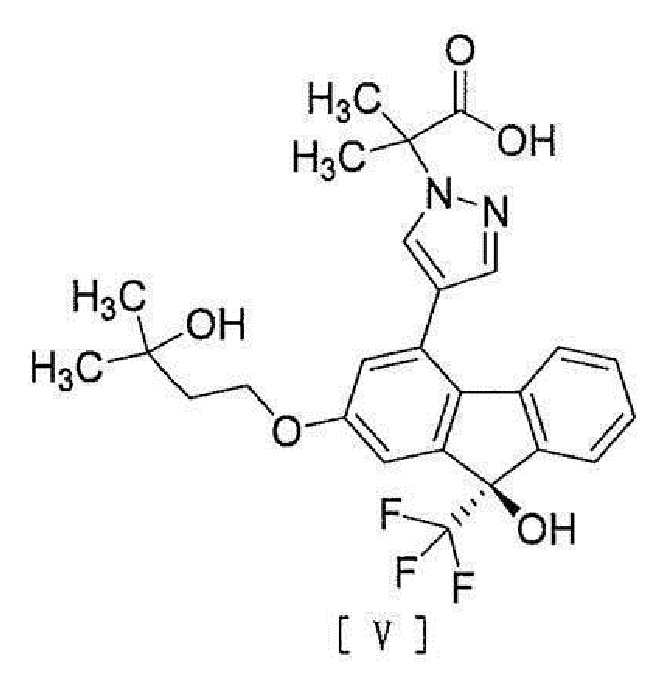
[0042] или его соль и стадию амидирования соединения вышеописанной формулы [V] или его соли путем реакции с аммиаком в присутствии конденсирующего агента.
[13] Способ получения соединения, представленного формулой [IV]:
[0043] 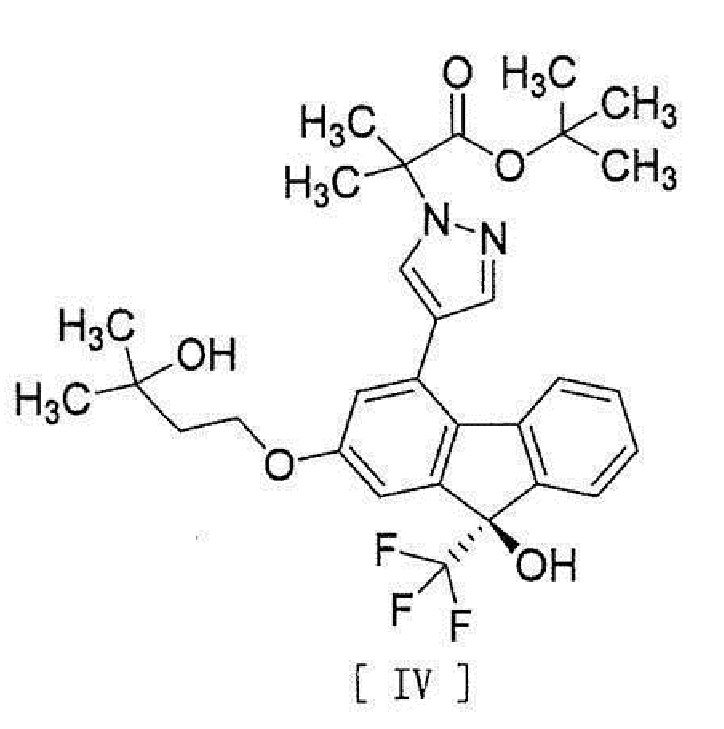
[0044] включающий введение соединения, представленного формулой [II]:
[0045] 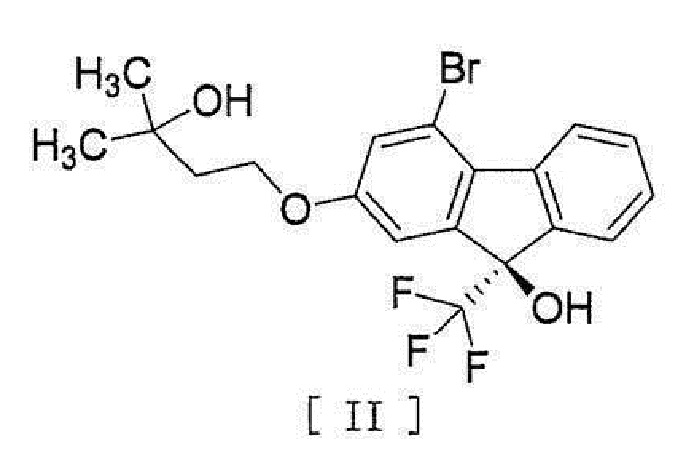
[0046] или его метанольного сольвата в реакцию сочетания с соединением, представленным формулой [III]:
[0047] 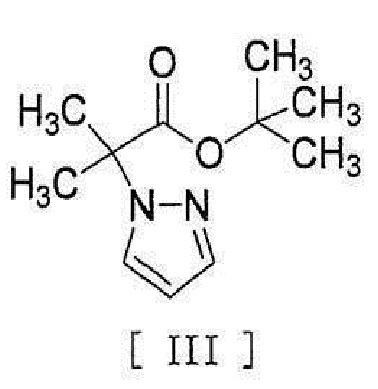
[0048] в присутствии металлического катализатора, основания и карбоновой кислоты.
[14] Соединение или его фармацевтически приемлемая соль, полученное способом согласно любому из вышеописанных пунктов [1] - [12].
[14a] Фармацевтическая композиция, включающая соединение или его фармацевтически приемлемую соль, полученное способом согласно вышеописанному пункту [1], и фармацевтически приемлемый носитель.
[15] Соединение, представленное формулой [IV]:
[0049] 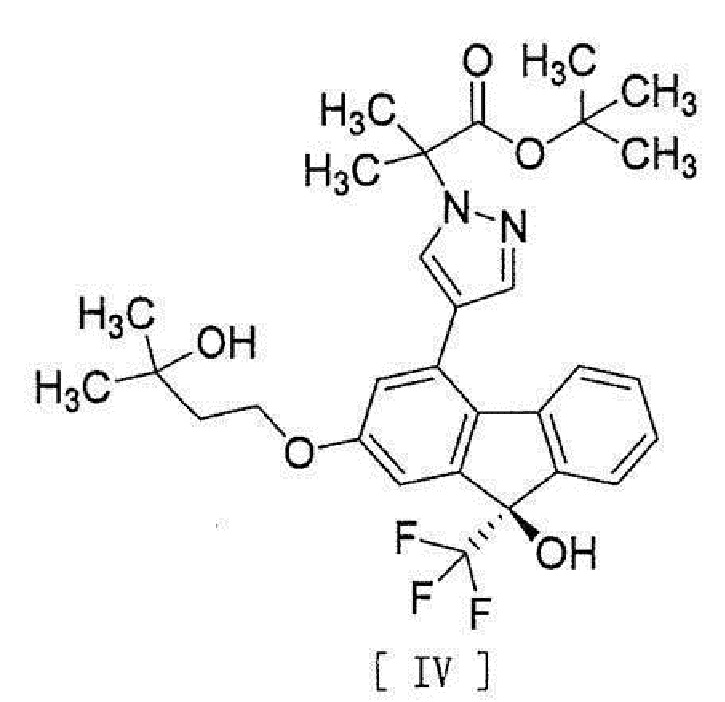
[0050] [16] Соединение, представленное формулой [II]:
[0051] 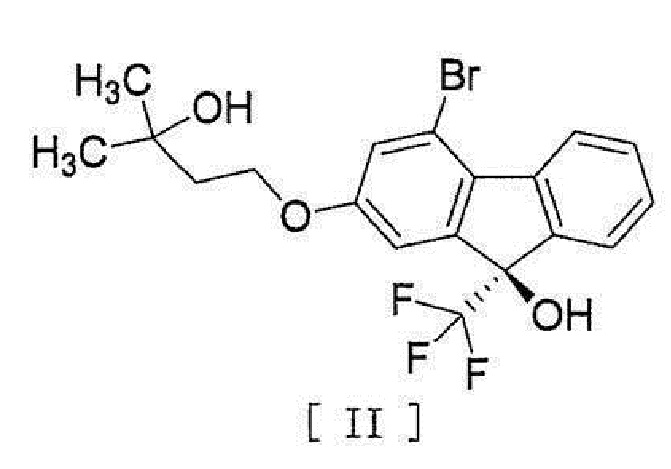
[0052] или его метанольный сольват.
[17] Соединение, представленное формулой [II]:
[0053]
[0054] или формулой [IIm]:
[0055]
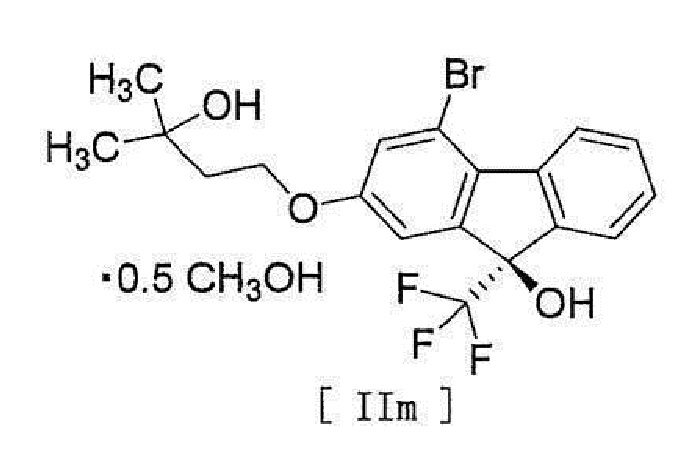
[0056] [18] Соединение, представленное формулой [III]:
[0057]
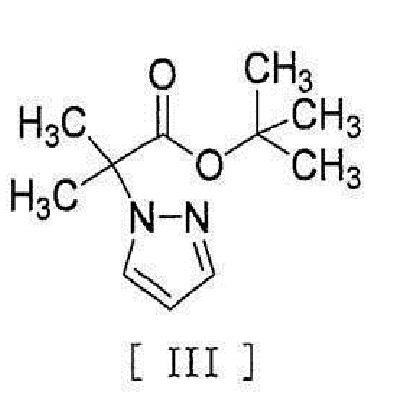
[0058] [18a] Соединение, представленное формулой [XV]:
[0059]
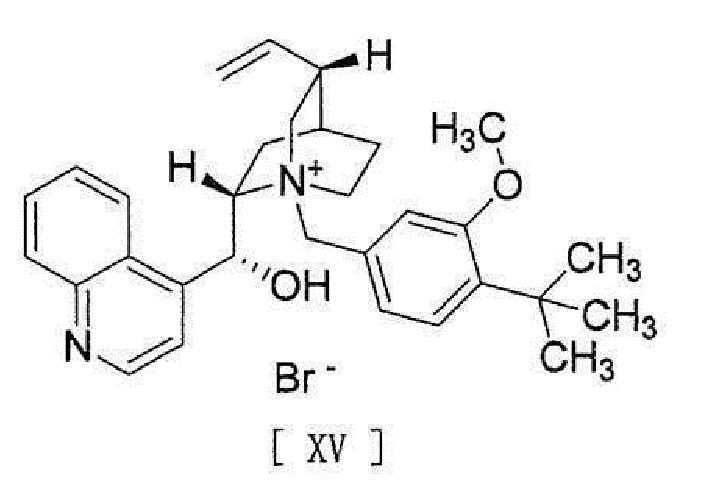
[0060]
[0061] Согласно способу получения согласно настоящему изобретению, соединение пиразоламида, имеющее ингибирующее действие в отношении PDHK и которое может быть использовано для лечения или профилактики заболеваний, связанных с нарушением метаболизма глюкозы, заболеваний, в которых ограничена доставка энергетического субстрата к тканям, митохондриального заболевания, митохондриальной энцефаломиопатии, рака, легочной гипертензии и т.п. может быть получено с высоким выходом обычной операцией через простые в обращении соединения. Способом по изобретению, кроме того, может быть получено новое промежуточное соединение для синтеза соединения пиразоламида.
[Описание вариантов осуществления]
[0062] Определения терминов в настоящем описании являются следующими.
ʺМеталлический катализаторʺ, используемый для реакции сочетания, должен быть катализатором на основе переходного металла, пригодным для реакции сочетания (реакция перекрестного связывания), и может быть упомянут, например, палладиевый катализатор и т.п. Среди них предпочтительны бис(трифенилфосфин)палладий (II) дихлорид, аддукт [1,1'-бис(дифенилфосфино)ферроцен]палладий (II) дихлорида и дихлорметана, ацетат палладия и ди(1-адамантил)-н-бутилфосфин, ацетат палладия и дициклогексил(2,2-дифенил-1-метилциклопропил)фосфин и т.п.
[0063] «Основание», используемое для реакции сочетания, может быть любым основанием при условии, что оно не препятствует прогрессу реакции сочетания, и может быть назван, например, карбонат щелочного металла, ацетат щелочного металла и т.п. Среди них карбонат калия является предпочтительным.
[0064] «Карбоновая кислота», используемая для реакции сочетания, может быть любой карбоновой кислотой, при условии, что она не препятствует прогрессу реакции сочетания, и может быть названа, например, триметилуксусная кислота, изомасляная кислота, пропионовая кислота, бензойная кислота и т.п. Среди них предпочтительными являются триметилуксусная кислотная или изомасляная кислота, и более предпочтительна триметилуксусная кислота.
[0065] «Конденсирующее средство», используемое для реакции соединения формулы [V] или его соли и аммиака, может быть любым конденсирующим средством, которое обычно используется для реакции амидирования карбоновой кислоты и амина, и могут быть названы, например, 1-этил-3-(3-диметиламинопропил)цианамид гидрохлорид, дициклогексилкарбодиимид (DCC), O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилуроний тетрафторборат (TBTU), 2-(1H-7-азабензотриазол-1-ил)-1,1,3,3-тетраметилуроний гексафторфосфат (HATU), 1,1'-карбонилдиимидазол и т.п. Среди них предпочтительным является 1-этил-3-(3-диметиламинопропил)цианамид гидрохлорид, который является водорастворимым конденсирующим средством (WSC). Более предпочтительно использовать конденсирующее средство вместе с обычной добавкой, такой как 1-гидроксибензотриазол моногидрат (HOBt), N-гидроксисукцинимид (HOSu), 6-хлор-1-гидроксибензотриазол (Cl-HOBt), 1-гидрокси-7-азабензотриазол (HOAt), 3-гидрокси-4-оксо-3,4-дигидро-1,2,3-бензотриазин и т.п. (предпочтительно, 1-гидроксибензотриазол моногидрат).
[0066] В качестве «аммиака» может использоваться (1) водный раствор аммиака или (2) аммиак, сгенерированный хлоридом аммония и триалкиламином (например, триэтиламином, диизопропилэтиламином и т.д.).
[0067] ʺАсимметрический органокатализаторʺ означает органическое соединение, которое является катализатором асимметрической реакции. Примеры асимметрического органокатализатора, пригодного для реакции превращения соединения формулы [VIII] в соединение формулы [II], включают соль цинхонидия и т.п., предпочтительно, N-(4-трет-бутил-3-метоксибензил)цинхонидий бромид.
[0068] ʺФармацевтически приемлемая сольʺ соединения может быть любой солью, при условии, что она формирует соль, не сопровождаемую чрезмерной токсичностью, известной в соответствующей области техники, с соединением формулы [I]. Примеры этого включают соли с неорганическими кислотами, соли с органическими кислотами, соли с аминокислотами и т.п.
[0069] Различные формы фармацевтически приемлемых солей известны в соответствующей области, и, например, они описаны в следующих справочных документах:
(a) Berge et al., J. Pharm. Sci., 66, p1-19(1977),
(b) Stahl et al., ʺHandbook of Pharmaceutical Salts: Properties, Selection, and Useʺ (Wiley-VCH, Weinheim, Germany, 2002),
(c) Paulekuhn et al., J. Med. Chem., 50, p6665-6672 (2007).
[0070] Каждая из них может быть получена путем реакции соединения формулы [I] с неорганической кислотой, органической кислотой или аминокислотой согласно способу, известному per se.
[0071] Примеры соли с неорганической кислотой включают соль с соляной кислотой, азотной кислотой, серной кислотой, фосфорной кислотой, бромистоводородной кислотой и т.п.
Примеры соли с органической кислотой включают соли с щавелевой кислотой, малеиновой кислотой, лимонной кислотой, фумаровой кислотой, молочной кислотой, яблочной кислотой, янтарной кислотой, винной кислотой, уксусной кислотой, трифторуксусной кислотой, глюконовой кислотой, аскорбиновой кислотой, метансульфоновой кислотой, бензолсульфоновой кислотой, п-толуолсульфоновой кислотой и т.п.
Примеры соли с аминокислотой включают соли с аспарагиновой кислотой, глутаминовой кислотой и т.п.
Фармацевтически приемлемая соль соединения согласно настоящему изобретению является предпочтительно солью с неорганической кислотой.
[0072] «Соль» соединения может быть любой солью, при условии, что она формирует соль с соединением согласно настоящему изобретению. Примеры этого включают соли с неорганическими кислотами, соли с органическими кислотами, соли с неорганическими основаниями, соли с органическими основаниями, соли с аминокислотами и т.п. Например, может быть названа вышеупомянутая ʺфармацевтически приемлемая сольʺ.
[0073] Примеры соли с неорганическим основанием включают соли с аммонием, алюминием, барием, висмутом, кальцием, литием, магнием, калием, натрием, цинком и т.п.
[0074] Примеры соли с органическим основанием включают соли с ареколином, клемизолью, этилендиамином, N-метилглюкамином, N-бензилфенэтиламином, трис(гидроксиметил)метиламином и т.п.
Примеры соли с аминокислотой включают соли с аргинином, лизином и т.п.
[0075] Каждая из них может быть получена путем реакции соединения согласно настоящему изобретению и неорганического основания, органического основания, неорганической кислоты, органической кислоты или аминокислоты согласно способу, известному per se.
[0076] Соединение согласно настоящему изобретению, его соль или фармацевтически приемлемая соль может существовать в форме сольвата. Термин «сольват» относится к соединению согласно настоящему изобретению, его соли или его фармацевтически приемлемой соли, с которой координируется молекула растворителя, и также включает гидраты. Такие сольваты являются предпочтительно фармацевтически приемлемыми сольватами. Такие сольваты включают, например, гидрат, метанольный сольват, этанольный сольват, диметилсульфоксидный сольват и т.п. соединения согласно настоящему изобретению, его соли или его фармацевтически приемлемой соли. ʺМетанольный сольватʺ относится к соединению согласно настоящему изобретению, с которым координируется молекула метанола. Например, может быть назван гемиметанольный сольват и т.п.
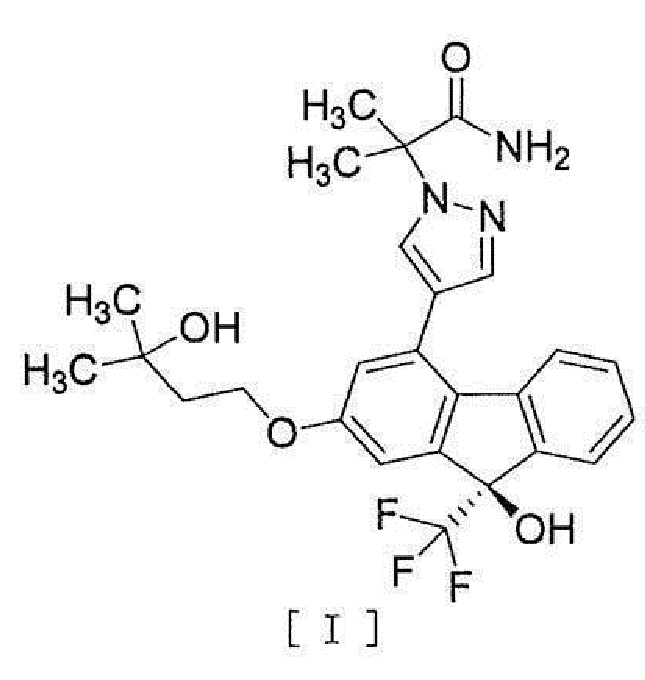
[0077] Примеры ʺсоединения, представленного формулой [I]:
[0078]
[0079] или его фармацевтически приемлемой соли или его гидрата ʺ включают следующие соединения
[0080] 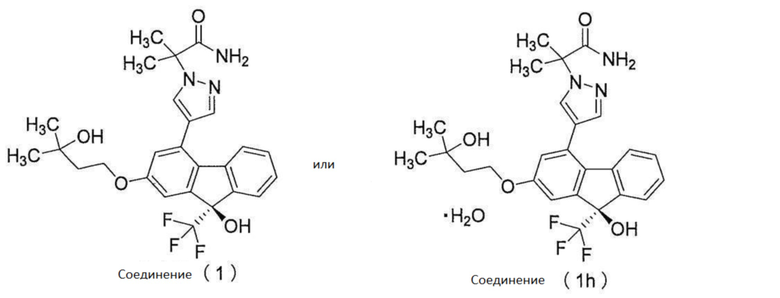
[0081]
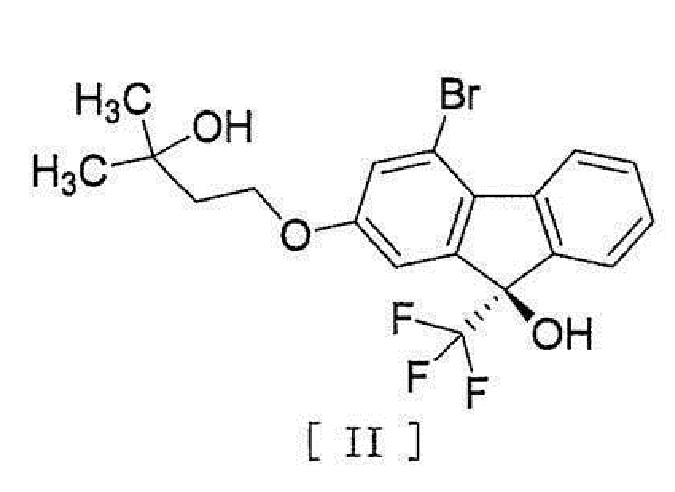
[0082] Примеры ʺсоединения, представленного формулой [II]:
[0083]
[0084] или его метанольного сольватаʺ включают следующие соединения
[0085] 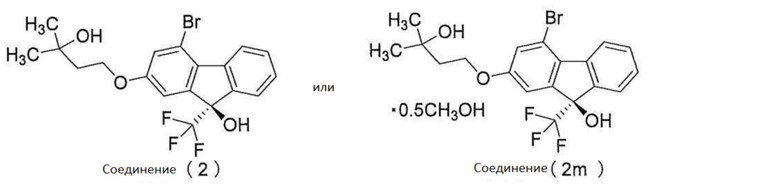
[0086]
[0087] Соединение формулы [I] или его фармацевтически приемлемая соль или его гидрат может быть мечено изотопным элементом (2H, 3H, 14C, 13C, 35S и т.д.).
[0088] Соединение формулы [I] или его фармацевтически приемлемая соль или его гидрат предпочтительно является по существу очищенным соединением формулы [I] или его гидратом. Предпочтительно, оно представляет собой соединение формулы [I] или его гидрат, очищенное до чистоты не менее чем 80%.
Более предпочтительно, оно представляет собой соединение формулы [I] или его гидрат, очищенное до чистоты не менее чем 90%.
[0089] Соединение согласно настоящему изобретению или его фармацевтически приемлемая соль или его гидрат может быть кристаллом, не-кристаллом (аморфным) или смесью этих форм.
[0090] Фармацевтическая композиция согласно настоящему изобретению может быть получена согласно способу, известному в области фармацевтических препаратов, путем смешивания соединения формулы [I] или его фармацевтически приемлемой соли, или соли его гидрата с подходящим количеством по меньшей мере одного вида фармацевтически приемлемого носителя и т.п.
[0091] Хотя содержание соединения формулы [I] или его фармацевтически приемлемой соли или соли его гидрата варьирует в зависимости от лекарственной формы, дозы и т.п., она может составлять, например, от 0,1 до 100 вес.% от общей массы композиции.
[0092] Примеры лекарственной формы соединения формулы [I] или его фармацевтически приемлемой соли или его гидрата включают пероральные препараты, такие как таблетка, капсула, гранула, порошок, пастилка, сироп, эмульсия, суспензия и т.п., или парентеральные средства, такие как наружный препарат, суппозиторий, инъекция, глазные капли, назальный препарат, ингаляционный препарат и т.п.
[0093] ʺТемпература реакцииʺ означает температуру в реакционном растворе, ʺвнутренняя температураʺ означает температуру в реакционной смеси, суспензии и т.п., и ʺвнешняя температураʺ означает температуру в масляной бане, водяной бане или сушильном шкафу.
[0094] Согласно определению, «приблизительно» означает ±5°C для температуры, ±10 минут для времени и ±10% для массы и объема.
[0095] Основные стадии способа получения согласно настоящему изобретению специфично объясняются следующим образом.
На каждой стадии реакция может быть осуществлена согласно обычно используемому способу. Полученный продукт может быть очищен путем соответствующего выбора стандартного способа, такого как перегонка, кристаллизация, перекристаллизация, колоночная хроматография, препаративная ВЭЖХ, промывка суспензии и т.п., или использования их в комбинации. Также возможно переходить к следующей стадии, не осуществляя выделение или очистку.
[0096] Стадия 4
[0097] 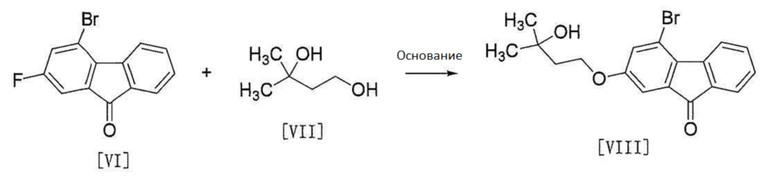
[0098] Соединение формулы [VIII] получают путем реакции соединения формулы [VI] с соединением формулы [VII].
Эту реакцию проводят в растворителе, не влияющем неблагоприятно на реакцию, и в присутствии основания.
Примеры растворителя включают углеводороды, такие как гексан, толуол и т.п., простые эфиры, такие как 1,4-диоксан, тетрагидрофуран, 1,2-диметоксиэтан и т.п., сульфоксиды, такие как диметил сульфоксид и т.п., амиды, такие как N, N-диметилацетамид, N-метил-2-пирролидон и т.п., воду и их смесь. Среди них предпочтительна смесь толуол/вода/тетрагидрофуран.
Примеры основания включают трикалий фосфат, карбонат цезия, трет-бутоксид калия, гидроксид натрия и т.п., предпочтительно, гидроксид натрия.
Количество используемого основания составляет от 2 до 20 моль, предпочтительно от 10 до 20 моль, более предпочтительно от 19 до 20 моль на 1 моль соединения формулы [VI].
Эту реакцию предпочтительно проводят в присутствии катализатора фазового переноса.
Примеры катализатора фазового переноса включают гидроксид тетра-н-бутиламмония, гидросульфат тетра-н-бутиламмония, фторид тетра-н-бутиламмония и т.п. Среди них предпочтительным является гидроксид тетра-н-бутиламмония. Количество используемого катализатора фазового переноса составляет от 0,1 до 1,5 моль, предпочтительно от 0,4 до 0,8 моль, более предпочтительно от 0,6 до 0,8 моль, на 1 моль соединения формулы [VI].
Количество используемого соединения формулы [VII] составляет от 1 до 4 моль, предпочтительно от 2 до 3 моль на 1 моль соединения формулы [VI].
Температура реакции и время реакции соответственно составляют от приблизительно 15°C до приблизительно 50°C и от приблизительно 1 часа до приблизительно 24 часов. Предпочтительная температура реакции является комнатной температурой, и предпочтительное время реакции составляет от приблизительно 4 часов до приблизительно 10 часов.
[0099] Стадия 5
[0100] 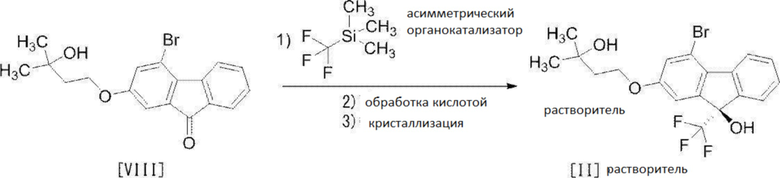
[0101] Эта стадия включает операцию 1 для реакции соединения формулы [VIII] с (трифторметил)триметилсиланом в присутствии асимметрического органокатализатора, операцию 2 для обработки триметилсилильной формы соединения, представленного формулой [II], полученной в операции 1, кислотой для удаления триметилсилильной группы и операции 3 для получения кристаллов сольвата соединения формулы [II] путем кристаллизации соединения, полученного в операции 2.
Подробности операций 1-3 объяснены ниже.
[0102] Операция 1
[0103] Операцию 1 осуществляют в растворителе, не влияющем неблагоприятно на реакцию, и в присутствии асимметрического органокатализатора. Асимметрический органокатализатор особенно не ограничен, и может быть упомянута, например, соль цинхонидия. Примеры соли цинхонидия включают N-(4-трет-бутил-3-метоксибензил)цинхонидий фторид, N-(4-трет-бутил-3-метоксибензил)цинхонидий бромид, N-(4-трет-бутил-3-метоксибензил)цинхонидий п-метоксифеноксид и т.п. Среди них N-(4-трет-бутил-3-метоксибензил)цинхонидий бромид является предпочтительным как соль цинхонидия.
Количество используемого асимметрического органокатализатора составляет от 0,005 до 0,3 моль, предпочтительно от 0,01 до 0,1 моль, особенно предпочтительно 0,05 моль на 1 моль соединения формулы [VIII].
Асимметрический органокатализатор в случае необходимости используется вместе с добавкой. Примеры добавки включают фенолят натрия, трет-бутилалкоголят натрия и т.п. Среди них эквимолярное количество фенолята натрия является предпочтительным как добавка.
Примеры растворителя включают ароматические углеводороды, такие как толуол, ксилол и т.п., простые эфиры, такие как тетрагидрофуран, простой диэтиловый эфир, 1,4-диоксан, 1,2-диметоксиэтан и т.п. и т.п. Среди них смешанный растворитель из толуола и тетрагидрофурана является предпочтительным.
Температура реакции составляет от приблизительно -78°C до приблизительно 0°C, предпочтительно от приблизительно -55°C до приблизительно-45°C.
Время реакции составляет от приблизительно 2 часов до приблизительно 8 часов, предпочтительно приблизительно 5 часов.
[0104] Операция 2
[0105] Операцию 2 осуществляют в растворителе, не влияющем неблагоприятно на реакцию, и в присутствии кислоты, и получают раствор, содержащий соединение формулы [II].
Примеры растворителя включают спирты, такие как метанол, этанол, 2-пропанол и т.п. Среди них предпочтителен метанол.
Хотя кислота особенно не ограничена, может быть названа, например, трифторуксусная кислота, соляная кислота и т.п., причем предпочтительной является соляная кислота.
Количество используемой кислоты составляет от 0,2 до 5,0 моль, предпочтительно от 0,3 до 2,0 моль на 1 моль соединения формулы [VIII].
Температура реакции является предпочтительно комнатной температурой.
Время реакции составляет от приблизительно 1 часа до приблизительно 6 часов, предпочтительно приблизительно 3 часа.
[0106] Операция 3
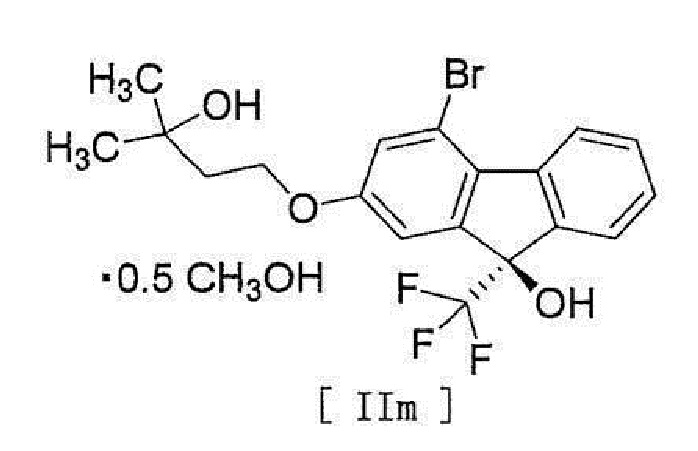
[0107] Кристаллизацию в операции 3 осуществляют в содержащем воду растворителе, и соединение формулы [II] получают как сольват (спиртовой сольват (предпочтительно, метанольный сольват)).
Примеры растворителя включают спирты, такие как метанол, этанол, 2-пропанол и т.п. Предпочтительным является метанол.
ʺМетанольный сольват соединения формулы [II]ʺ может быть смесью соединения формулы [II] и метанольного сольвата соединения формулы [II] в зависимости от условий высушивания и т.п.
[0108] Стадия 6
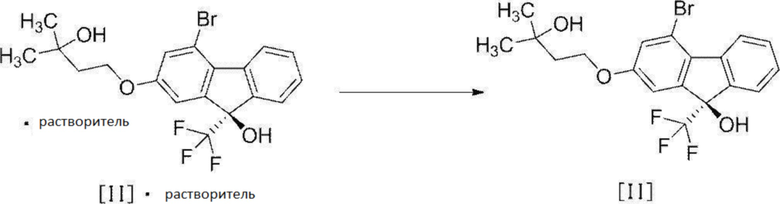 [0109] На Стадии 6 растворитель добавляют к сольвату соединения формулы [II], полученному на вышеописанной Стадии 5, полученную суспензия перемешивают при нагревании, растворитель выпаривают при пониженном давлении, остаток повторно суспендируют в растворителе, и суспензию перемешивают, фильтруют, промывают и высушивают, получая соединение формулы [II] в форме кристалла.
[0109] На Стадии 6 растворитель добавляют к сольвату соединения формулы [II], полученному на вышеописанной Стадии 5, полученную суспензия перемешивают при нагревании, растворитель выпаривают при пониженном давлении, остаток повторно суспендируют в растворителе, и суспензию перемешивают, фильтруют, промывают и высушивают, получая соединение формулы [II] в форме кристалла.
Примеры растворителя, используемого для получения суспензии сольвата соединения формулы [II], включают алифатические углеводороды, такие как гексан, гептан, октан и т.п., и/или ароматические углеводороды, такие как бензол, толуол, ксилол и т.п. Среди них предпочтительным является гептан.
Активная температура является внутренней температурой приблизительно от 60°C до температуры кипения растворителя, предпочтительно внутренняя температура составляет не менее чем 85°C.
Активное время составляет от приблизительно 1 часа до приблизительно 6 часов, предпочтительно от приблизительно 3 часов до приблизительно 5 часов.
[0110] Стадия 7
[0111] 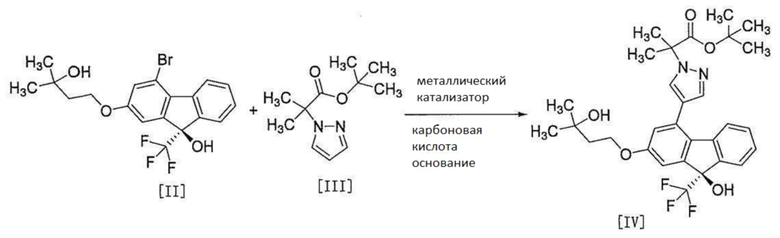
[0112] Соединение формулы [IV] получают путем введения соединения формулы [II] или его метанольного сольвата в реакцию сочетания с соединением формулы [III] в присутствии металлического катализатора. Для реакции сочетания соединение формулы [II] более предпочтительно, чем метанольный сольват соединения формулы [II].
Эту реакцию проводят в растворителе, не влияющем неблагоприятно на реакцию, и в присутствии карбоновой кислоты и основания.
Примеры растворителя включают простые эфиры, такие как тетрагидрофуран, 1,4-диоксан и т.п., нитрилы, такие как ацетонитрил и т.п., амиды, такие как N,N-диметилформамид, N-метилпирролидон, N,N-диметилацетамид и т.п., и углеводороды, такие как бензол, толуол и т.п. Среди них предпочтителен N,N-диметилацетамид.
Примеры металлического катализатора включают палладиевый катализатор и т.п. Среди них предпочтительны бис(трифенилфосфин)палладий (II) дихлорид, аддукт [1,1'-бис(дифенилфосфино)ферроцен]палладий (II) дихлорида и дихлорметана, ацетат палладия и ди(1-адамантил)-н-бутилфосфин, ацетат палладия и дициклогексил(2,2-дифенил-1-метилциклопропил)фосфин и т.п. Среди них бис(трифенилфосфин)палладий (II) дихлорид является более предпочтительным как металлический катализатор.
Примеры карбоновой кислоты включают триметилуксусную кислоту, изомасляную кислоту, пропионовую кислоту, бензойную кислоту и т.п. Карбоновая кислота является предпочтительно триметилуксусной кислотой или изомасляной кислотой, более предпочтительно триметилуксусной кислотой.
Примеры основания включают карбонат щелочного металла, ацетат щелочного металла и т.п., и предпочтительным является карбонат калия.
Количество используемого соединения формулы [III] составляет от 1,0 до 5,0 моль, предпочтительно от 1,6 до 2,0 моль на 1 моль соединения формулы [II].
Количество используемого металлического катализатора составляет от 0,005 до 0,2 моль, предпочтительно от 0,01 до 0,025 моль на 1 моль соединения формулы [II].
Количество используемой карбоновой кислоты составляет от 0,1 до 1,0 моль, предпочтительно от 0,2 до 0,5 моль на 1 моль соединения формулы [II].
Количество используемого основания составляет от 0,4 до 4,0 моль, предпочтительно от 0,6 до 1,8 моль на 1 моль соединения формулы [II].
Температура реакции составляет от приблизительно 80°C до приблизительно 150°C, предпочтительно от приблизительно 90°C до приблизительно 140°C, более предпочтительно от приблизительно 100°C до приблизительно 110°C.
Время реакции составляет от приблизительно 1 часа до приблизительно 6 часов, предпочтительно приблизительно 3 часа.
[0113] Стадия 8
[0114] 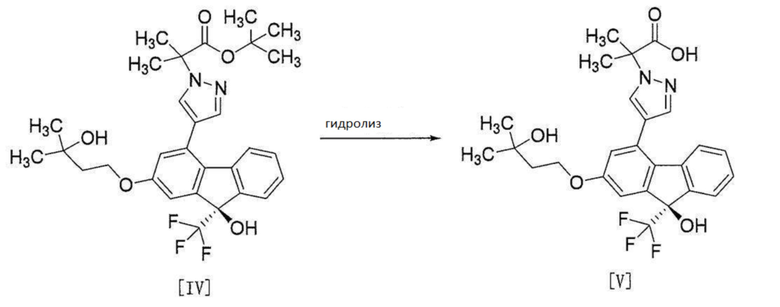
[0115] Соединение формулы [V] получают путем гидролиза соединения формулы [IV].
Эту реакцию проводят в растворителе, не влияющем неблагоприятно на реакцию, и в присутствии основания.
Примеры растворителя включают спирты, такие как метанол, этанол, 2-пропанол и т.п., воду и т.п. и их смесь. Среди них смешанный растворитель из этанола и воды является предпочтительным.
Примеры основания включают неорганические основания, такие как гидроксид натрия, гидроксид калия и т.п. Среди них предпочтителен гидроксид натрия.
Количество используемого основания составляет от 7 до 16 моль, предпочтительно от 10 до 13 моль на 1 моль соединения формулы [IV].
Температура реакции является внутренней температурой от приблизительно 25°C до температуры кипения растворителя, предпочтительно приблизительно 70°C.
Время реакции составляет от приблизительно 1 часа до приблизительно 8 часов, предпочтительно приблизительно 1,5 часа.
[0116] Стадия 9
[0117] 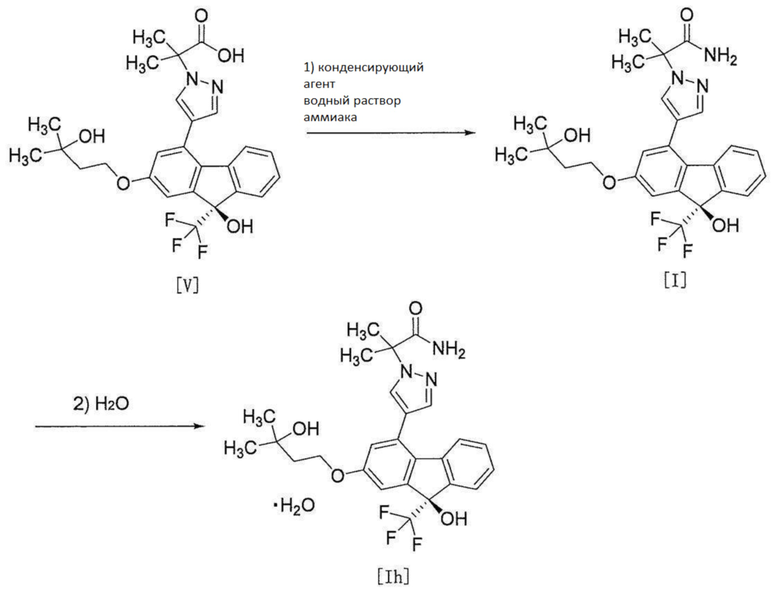
[0118] Соединение формулы [Ih] получают путем осуществления операции 1 для реакции соединения формулы [V] с аммиаком в присутствии конденсирующего агента, после чего проводят операцию 2 для кристаллизации в содержащем воду растворителе.
[0119] Операция 1
[0120] Реакцию операции 1 осуществляют в растворителе, не влияющем неблагоприятно на реакцию, и в присутствии конденсирующего агента.
Примеры конденсирующего агента включают 1-этил-3-(3-диметиламинопропил)цианамид гидрохлорид, дициклогексилкарбодиимид (DCC), O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилуроний тетрафторборат (TBTU), 2-(1H-7-азабензотриазол-1-ил)-1,1,3,3-тетраметилуроний гексафторфосфат (HATU), 1,1'-карбонилдиимидазол и т.п. Среди них предпочтительным является 1-этил-3-(3-диметиламинопропил)цианамид гидрохлорид. Они могут использоваться индивидуально или в комбинации с добавкой (например, 1-гидроксибензотриазол моногидрат (HOBt), N-гидроксисукцинимид (HOSu), 6-хлор-1-гидроксибензотриазол (Cl-HOBt), 1-гидрокси-7-азабензотриазол (HOAt) или 3-гидрокси-4-оксо-3,4-дигидро-1,2,3-бензотриазин и т.п., предпочтительно 1-гидроксибензотриазол моногидрат). Среди них особенно предпочтительно используется комбинация 1-этил-3-(3-диметиламинопропил)цианамид гидрохлорида и 1-гидроксибензотриазол моногидрата.
Количество используемого конденсирующего агента составляет от 1 до 10 моль, предпочтительно от 1 до 2 моль на 1 моль соединения формулы [V].
Количество используемой добавки составляет от 1 до 10 моль, предпочтительно от 1 до 2 моль на 1 моль соединения формулы [V].
Количество используемого аммиака, когда, например, используется водный раствор аммиака, составляет от 1 до 3 моль, предпочтительно от 1 до 2 моль, в пересчете на количество аммиака, на 1 моль соединения формулы [V].
Кроме того, хлорид аммония и основание (например, триалкил амин, в частности, триэтиламин, диизопропилэтиламин и т.д.) могут использоваться в качестве аммиака.
Примеры растворителя включают простые эфиры, такие как простой диэтиловый эфир, 1,4-диоксан, тетрагидрофуран и т.п., сложные эфиры, такие как этилацетат и т.п., галогензамещенные углеводороды, такие как хлороформ, дихлорметан и т.п., амиды, такие как N,N-диметилформамид и т.п., их смесь этого и т.п., и они могут быть смешаны. Среди них предпочтителен N,N-диметилформамид.
Температура реакции варьирует в зависимости от вида растворителя и составляет от приблизительно 0°C до приблизительно 40°C, предпочтительно от приблизительно 15°C до приблизительно 30°C. Время реакции составляет от приблизительно 0,5 часа до приблизительно 24 часов, предпочтительно от приблизительно 1,5 часов до приблизительно 8 часов.
[0121] Операция 2
[0122] В операции 2 спирт (например, этанол) добавляют в качестве растворителя к раствору соединения формулы [I], полученного в вышеописанной операции 1, и операцию азеотропной перегонки для выпаривания части растворителя при пониженном давлении повторяют. Спирт далее добавляют для получения раствора, его внутреннюю температуру повышают от приблизительно 40°C до приблизительно 50°C, воду добавляют по каплям при той же температуре, после перемешивания, внутреннюю температуру полученной суспензии повышают от приблизительно 55°C до приблизительно 65°C, суспензию перемешивают и дают постепенно остыть до комнатной температуры, и полученную смесь далее перемешивают, посредством чего соединение формулы [Ih] может быть осаждено в форме кристаллов.
Время перемешивания составляет от приблизительно 1 часа до приблизительно 7 часов, предпочтительно приблизительно 2 часа, при внутренней температуре от приблизительно 40°C до приблизительно 50°C, от приблизительно 1 часа до приблизительно 4 часов, предпочтительно от приблизительно 1 часа до приблизительно 2 часов, при внутренней температуре от приблизительно 55°C до приблизительно 65°C, и от приблизительно 8 часов до 24 часов, предпочтительно приблизительно 12 часов, при комнатной температуре.
В операции 2 операция кристаллизации может быть осуществлена путем добавления затравочного кристалла соединения формулы [Ih] после добавления по каплям воды.
[0123] Стадия 10
[0124] 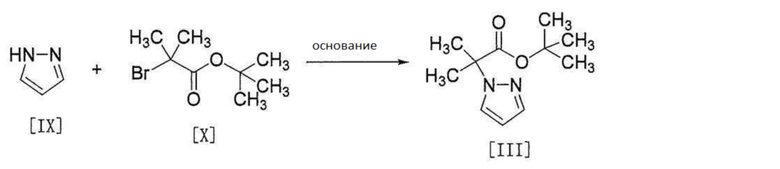
[0125] Соединение формулы [III] получают путем реакции соединения формулы [IX] с соединением формулы [X].
Эту реакцию проводят в растворителе, не влияющем неблагоприятно на реакцию, и в присутствии основания.
Примеры растворителя включают простые эфиры, такие как тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан и т.п., углеводороды, такие как гексан, толуол и т.п., амиды, такие как N,N-диметилформамид, N,N-диметилацетамид и т.п., и их смесь. Среди них предпочтителен тетрагидрофуран.
Примеры основания включают гидрид натрия, трет-бутоксид калия, метоксид натрия, бис(триметилсилил)амид натрия, карбонат цезия и т.п., предпочтительно гидрид натрия.
Количество используемого основания составляет от 1 до 2 моль, предпочтительно от 1 до 1,5 моль на 1 моль соединения формулы [IX].
Количество используемого соединения формулы [X] составляет от 1 до 1,5 моль, предпочтительно от 1 до 1,2 моль на 1 моль соединения формулы [IX].
Температура реакции является температурой от комнатной до температуры кипения растворителя, предпочтительно от приблизительно 50°C до приблизительно 65°C.
Время реакции составляет от приблизительно 1 часа до приблизительно 24 часов.
[0126] В качестве характеристик способа получения может быть упомянуто следующее.
1. Соединение (2) или соединение (2m)
[0127]
1-1.
Соединение (2) может быть очищено с получением соединения, имеющего хорошую химическую чистоту и хорошую оптическую чистоту (например, 95% э.и. или больше) единственной операцией путем превращения в метанольный сольват (соединение (2m)), имеющий хорошую кристалличность.
[0128]
2. Соединение (3)
[0129]
2-1.
Соединение (3) может быть очищено для достижения чистоты не менее чем 90% перегонкой (не менее чем 99% в зависимости от услвоий перегонки).
[0130]
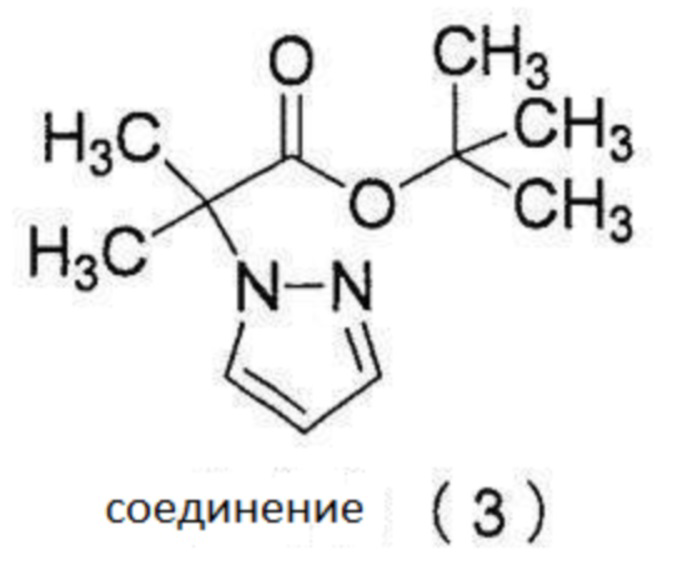
[0131]
3. Реакция сочетания
[0132]
3-1.
Когда соединение (2) и соединение (3) используют в определенных условиях реакции сочетания, побочная реакция реакции сочетания может быть подавлена больше, чем тогда, когда используют соединение (2) и соединение (101), упомянутые ниже.
[0133]
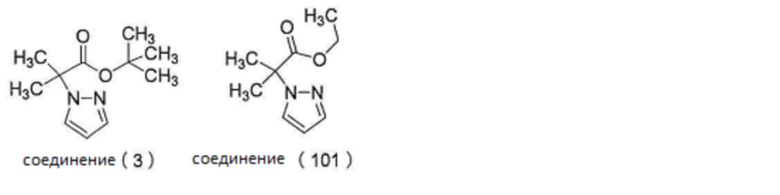
[0134]
3-2.
Когда соединение (3) и соединение (2) используют в определенных условиях реакции сочетания, целевой продукт сочетания может быть получен с более высоким выходом, чем тогда, когда используют соединение (3) и соединение (102) (хлор-соединение (рацемат), упомянутое ниже).
[0135]
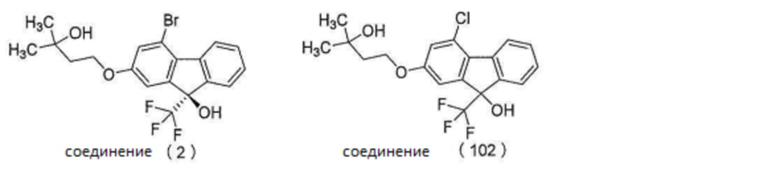
[Примеры]
[0136] Хотя настоящее изобретение объясняется подробно путем отсылки к следующим Примерам, настоящее изобретение ими не ограничено.
[0137] Даже если никакого описания не находится в способах получения в следующих Примерах, стадии могут быть изменены для эффективного получения, например, введением защитной группы в функциональную группу в случае необходимости с удалением защитной группы на последующей стадии, с использованием функциональной группы в качестве предшественника на каждой стадии с последующим превращением в желаемую функциональную группу на подходящей стадии, изменением порядка способов и стадий получения и т.п.
[0138] Реакция на каждой стадии может быть осуществлена обычным способом, где выделение и очистка могут быть осуществлены по мере необходимости согласно способу, соответствующим образом выбранному из обычных способов, таких как кристаллизация, перекристаллизация, перегонка, разделение, хроматография на силикагеле, препаративная ВЭЖХ и т.п. или их комбинация. Все реактивы и растворители имеют качество коммерчески доступных продуктов и использовались без очистки.
[0139] Следует отметить, что % означает % моль/моль для выхода и вес.% для других, если не указано иное. Кроме того, комнатная температура означает температуру от 15 до 30°C, если не указано иное. Другие сокращения, используемые в этом разделе в качестве примера, означают следующее.
с: синглет
д: дублет
т: триплет
к: квартет
м: мультиплет
ушир.: уширенный
дд: дублет дублетов
ддд: дублет дублетов дублетов
дддд: дублет дублетов дублетов дублетов
J: константа связывания
MeOH: метанол
DMSO-D6: дейтеродиметил сульфоксид
1H-ЯМР: протонный ядерный магнитный резонанс.
ВЭЖХ: высокоэффективная жидкостная хроматография
Спектр 1H-ЯМР измеряли в DMSO-D6, используя тетраметилсилан в качестве внутреннего стандарта, и все значения δ показаны в ppm.
[0140]
(Фосфатный буфер (pH 2,0))
Дигидрат дигидрофосфата натрия (4,68 г) растворяли в воде (3000 мл) и добавляли фосфорную кислоту (5 мл) для получения целевого буфера.
[0141]
Аналитические условия ВЭЖХ
В следующих аналитических условиях «%» означает объемный %. Градиент линейно изменяет отношение смешиваемых РАСТВОРА A и РАСТВОРА B.
Аналитическое условие 1
Устройство для измерения: система ВЭЖХ Waters Alliance
Колонка: Waters SunFire C18 3,5 мкм 4,6 мм х 150 мм
Температура колонки: 40°C
Подвижная фаза: (РАСТВОР A) фосфатный буфер (pH 2,0), (РАСТВОР B) ацетонитрил
Профиль градиента: время (мин) 0 30 35 40 45 (остановка)
А (%) 70 25 25 70 70
B (%) 30 75 75 30 30
Время анализа: 45 минут
Скорость потока: 1,0 мл/мин
Детекция: УФ (220 нм)
[0142] Аналитическое условие 2
Устройство для измерения: система ВЭЖХ Waters Alliance
Колонка: Waters SunFire C8 3,5 мкм 4,6 мм х 150 мм
Температура колонки: 40°C
Подвижная фаза: (РАСТВОР A) дистиллированная вода, (РАСТВОР B) ацетонитрил
Профиль градиента: время (мин) 0 20 35 36 40 (остановка)
А (%) 60 10 10 60 60
B (%) 40 90 90 40 40
Время анализа: 40 минут
Скорость потока: 1,0 мл/мин
Детекция: УФ (220 нм)
[0143] Аналитическое условие 3
Устройство для измерения: система ВЭЖХ Waters Alliance
Колонка: Daicel Corporation OD-3R CHIRALCEL 3 мкм 4,6 мм х 150 мм
Температура колонки: 40°C
Подвижная фаза: (РАСТВОР A) фосфатный буфер (pH 2,0), (РАСТВОР B) ацетонитрил
Профиль градиента: время (мин) 0 20 30 40 45 (остановка)
А (%) 70 40 40 70 70
B (%) 30 60 60 30 30
Время анализа: 45 минут
Скорость потока: 0,5 мл/мин
Детекция: УФ (254 нм)
[0144] Аналитическое условие 4
Устройство для измерения: система ВЭЖХ Waters Alliance
Колонка: Daicel Corporation CHIRALCEL OJ-3R 3 мкм 4,6 мм х 150 мм
Температура колонки: 40°C
Подвижная фаза: (РАСТВОР A) фосфатный буфер (pH 2,0), (РАСТВОР B) ацетонитрил
Состав подвижной фазы: РАСТВОР A:SOLUTION B=70:30
Время анализа: 15 минут
Скорость потока: 1,0 мл/мин
Детекция: УФ (220 нм)
[0145] Пример 1 Синтез 2-{4-[(9R)-9-гидрокси-2-(3-гидрокси-3-метилбутилокси)-9-(трифторметил)-9H-флуорен-4-ил]-1H-пиразол-1-ил}-2-метилпропанамид моногидрата (соединение (1h))
[0146] Стадия 1
(2-Амино-3-бром-5-фторфенил)(фенил)метанон (соединение (12))
[0147]
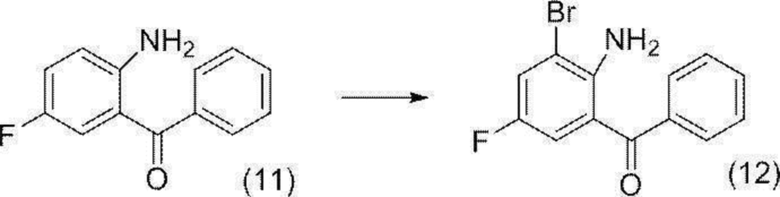
[0148] К суспензии (2-амино-5-фторфенил)(фенил)метанона (соединение (11)) (50,0 г) в уксусной кислоте (400 мл) добавляли по каплям при внутренней температуре от 21 до 30°C приблизительно 65% от общей массы раствор бром (40,8 г)/уксусная кислота (90 мл). После завершения добавления по каплям, добавляли затравочный кристалл (5 мг) гидробромида (2-амино-3-бром-5-фторфенил)(фенил)метанона (соединение (12)), и кристаллический осадок был визуально подтвержден. Полученную суспензию нагревали до внутренней температуры 41°C, и общее количество (приблизительно 35%) остальной части раствора бром (40,8 г)/уксусная кислота (90 мл) добавляли по каплям при внутренней температуре от 41 до 45°C. Воронку для добавления промывали уксусной кислотой (10 мл), и смесь перемешивали при внутренней температуре от 45 до 46°C в течение 17 минут. К полученной суспензии добавляли по каплям раствор сульфит натрия (4,39 г)/вода (45 мл) при внутренней температуре от 41 до 45°C, воронку для добавления промывали водой (5 мл), и смесь перемешивали при внутренней температуре от 41 до 46°C в течение 30 минут. К полученной суспензии добавляли по каплям воду (50 мл) при внутренней температуре от 43 до 46°C, добавляли затравочный кристалл (5 мг) (2-амино-3-бром-5-фторфенил)(фенил)метанона (соединение (12)), и смесь перемешивали в течение 5 минут. После того, как осаждение кристаллов было визуально подтверждено, добавляли по каплям воду (350 мл) при внутренней температуре от 47 до 51°C, и смесь перемешивали при внутренней температуре от 50 до 55°C в течение 1 часа. Полученную суспензию охлаждали до комнатной температуры и перемешивали в течение ночи. Осажденное твердое вещество собирали фильтрацией и промывали водой (100 мл) и высушивали при пониженном давлении с получением целевого соединения (65,2 г).
1H-ЯМР (400 МГц, DMSO-D6) δ: 7,78 (дд, 1H, J=7,8, 3,0 Гц), 7,67-7,61 (м, 3H), 7,58-7,51 (м, 2H), 7,11 (дд, 1H, J=9,3, 3,0 Гц), 6,73 (ушир., 2H).
[0149] Синтез затравочного кристалла гидробромида (2-амино-3-бром-5-фторфенил)(фенил)метанона (соединение (12)), используемого на Стадии 1:
К раствору (2-амино-3-бром-5-фторфенил)(фенил)метанона (соединение (12)) (1,00 г) в толуоле (25 мл) добавляли раствор 25%-й бромистоводородной кислоты в уксусной кислоты (0,87 мл) при комнатной температуре. Осажденное твердое вещество собирали фильтрацией и промывали толуолом. Полученное твердое вещество высушивали при пониженном давлении с получением затравочного кристалла (1,21 г) гидробромида (2-амино-3-бром-5-фторфенил)(фенил)метанона (соединение (12)).
[0150] Синтез затравочного кристалла (2-амино-3-бром-5-фторфенил)(фенил)метанона (соединение (12)), используемого на Стадии 1:
Затравочный кристалл целевого соединения (2-амино-3-бром-5-фторфенил)(фенил)метанона (соединение (12)) был получен тем же способом, как на вышеописанной Стадии 1 за исключением того, что не добавляли затравочный кристалл.
[0151] Стадия 2
4-Бром-2-фтор-9H-флуорен-9-он (соединение (6))
[0152]
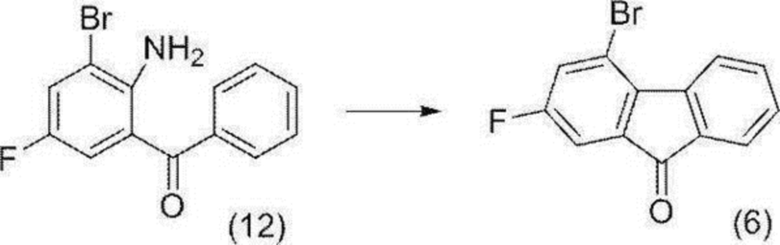
[0153] В атмосфере азота добавляли (2-амино-3-бром-5-фторфенил)(фенил)метанон (соединение (12)) (25,0 г), закись меди (7,30 г) и уксусную кислоту (150 мл), и добавляли 64%-ю серную кислоту (150 мл) при перемешивании смеси при комнатной температуре. Полученную суспензию нагревали до внутренней температуры 65°C, раствор нитрит натрия (8,80 г)/вода (87,5 мл) добавляли по каплям при внутренней температуре от 66 до 68°C, воронку для добавления промывали водой (13 мл), и смесь перемешивали при внутренней температуре от 65 до 70°C в течение 30 минут. Полученную реакционную смесь охлаждали до комнатной температуры, раствор сульфит натрия (8,57 г)/вода (62,5 мл) добавляли по каплям при внутренней температуре от 23 до 24°C, воронку для добавления промывали водой (13 мл), и смесь перемешивали при комнатной температуре в течение 30 минут. К полученной смеси добавляли толуол (375 мл), и смесь перемешивали в течение 30 минут. Нерастворимый материал отфильтровывали через целит и промывали толуолом (75 мл). Полученный фильтрат и смывы разделяли, водный слой удаляли, и органический слой промывали последовательно водой (125 мл х 2 раза), 5%-м водным раствором гидрокарбоната натрия (125 мл) и 1%-м солевым раствором (125 мл). Полученный органический слой концентрировали при пониженном давлении, и толуол (приблизительно 375 мл) выпаривали. К остатку добавляли 2-пропанол (250 мл), и смесь концентрировали при пониженном давлении до массы приблизительно 90 г. Подобную операцию проводили еще два раза, добавляли 2-пропанол (150 мл), и массу доводили до 207 г. Полученную суспензию нагревали с обратным холодильником в течение 30 минут, последовательно перемешивали при внешней температуре 75°C в течение 1 часа и внешней температуре 60°C в течение 1 часа, охлаждали до комнатной температуры, и перемешивали останавливали. После отстаивания при комнатной температуре в течение ночи, осажденное твердое вещество собирали фильтрацией и промывали 2-пропанолом (50 мл). Полученное твердое вещество высушивали при пониженном давлении с получением целевого соединения (15,6 г).
1H-ЯМР (400 МГц, DMSO-D6) δ: 8,27 (дд, 1H, J=8,2, 0,9 Гц), 7,79 (дд, 1H, J=8,8, 2,3 Гц), 7,72-7,67 (м, 2H), 7,51 (дд, 1H, J=6,8, 2,3 Гц), 7,46 (ддд, 1H, J=7,6, 7,6, 0,9 Гц).
[0154] Стадия 3 Синтез N-(4-трет-бутил-3-метоксибензил)цинхонидий бромида (соединение (15))
[0155]
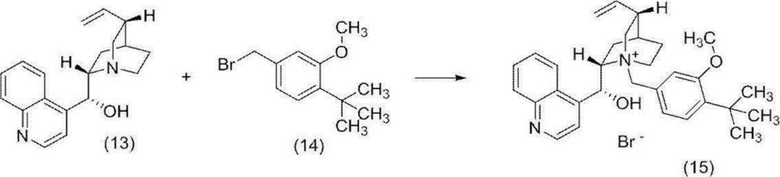
[0156] В атмосфере азота добавляли цинхонидин (соединение (13)) (2,50 г), тетрагидрофуран (43 мл) и 4-трет-бутил-3-метоксибензил бромид (соединение (14)) (2,29 г), и полученную смесь перемешивали при температуре от 61 до 62°C в течение 7 часов. Реакционную смесь охлаждали до комнатной температуры, и твердое вещество собирали фильтрацией и промывали тетрагидрофураном (10 мл). Полученное твердое вещество высушивали при пониженном давлении с получением целевого соединения (4,41 г).
1H-ЯМР (400 МГц, DMSO-D6) δ: 8,99 (d, 1H, J=4,4 Гц), 8,29 (д, 1H, J=8,3 Гц), 8,11 (дд, 1H, J=8,6, 1,4 Гц), 7,88-7,82 (м, 1H), 7,81 (д, 1H, J=4,4 Гц), 7,78-7,72 (м, 1H), 7,39 (д, 1H, J=8,1 Гц), 7,34 (д, 1H, J=1,8 Гц), 7,22 (дд, 1H, J=8,1, 1,8 Гц), 6,72 (д, 1H, J=4,6 Гц), 6,55 (д, 1H, J=4,6 Гц), 5,69 (ддд, 1H, J=17,4, 10,6, 6,5 Гц), 5,16 (дд, 1H, J=17,4, 1,4 Гц), 5,10 (д, 1H, J=12,2 Гц), 4,97 (д, 1H, J=12,2 Гц), 4,96 (дд, 1H, J=10,6, 1,4 Гц), 4,36-4,23 (м, 1H), 3,96-3,84 (м, 1H), 3,89 (с, 3H), 3,80-3,69 (м, 1H), 3,40-3,25 (м, 2H), 2,77-2,65 (м, 1H), 2,20-1,96 (м, 3H), 1,91-1,79 (м, 1H), 1,38 (с, 9H), 1,38-1,26 (м, 1H).
[0157] Стадия 4
4-Бром-2-(3-гидрокси-3-метилбутилокси)-9H-флуорен-9-он (соединение (8))
[0158] 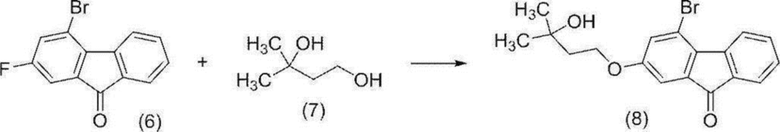
[0159] В атмосфере азота к 4-бром-2-фтор-9H-флуорен-9-ону (соединение (6)) (25,0 г) добавляли 3-метил-1,3-бутандиол (18,8 г) (соединение (7)), толуол (188 мл) и тетрагидрофуран (25 мл), и смесь охлаждали в ванне со льдом. К полученной смеси добавляли по каплям 55%-й водный раствор гидроксида тетра-н-бутиламмония (27,5 мл), 40%-й водный раствор гидроксида натрия (100 мл) и воду (10 мл) при внутренней температуре от 3,6 до 7,9°C, и смесь перемешивали при комнатной температуре в течение 9 часов 30 минут. К полученной реакционной смеси добавляли по каплям воду (125 мл) при внутренней температуре от 18 до 24°C, и смесь экстрагировали толуолом (188 мл). Водный слой удаляли. Органический слой промывали последовательно водой (125 мл), 5%-м солевым раствором (125 мл х 3 раза), 1M соляной кислотой (125 мл) и водой (125 мл) и концентрировали при пониженном давлении, пока масса не достигала 75 г или меньше. К остатку добавляли толуол (188 мл), и смесь концентрировали снова при пониженном давлении. Добавляли толуол (приблизительно 500 мл) с получением раствора целевого соединения в толуоле (481 г). Общее количество полученного раствора в толуоле использовали в качестве общего 100% выхода в следующем стадии 5.
1H-ЯМР (400 МГц, DMSO-D6) δ: 8,17 (дд, 1H, J=8,32, 0,92 Гц), 7,66-7,60 (м, 2H), 7,40-7,34 (м, 1H), 7,29 (д, 1H, J=2,3 Гц), 7,16 (д, 1H, J=2,3 Гц), 4,12 (с, 1H), 4,18 (т, 2H, J=7,2 Гц), 1,85 (т, 2H, J=7,2 Гц), 1,18 (с, 6H).
[0160] Стадия 5
(9R)-4-Бром-2-(3-гидрокси-3-метилбутилокси)-9-(трифторметил)-9H-флуорен-9-ол гемиметанольный сольват (соединение (2 m))
[0161]
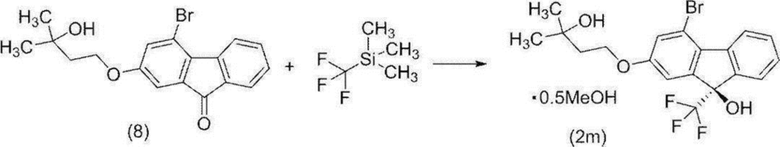
[0162] В атмосфере азота к раствору 4-бром-2-(3-гидрокси-3-метилбутилокси)-9H-флуорен-9-она (соединение (8)), полученного на предыдущей стадии, в толуоле (481 г) добавляли N-(4-трет-бутил-3-метоксибензил)цинхонидий бромид (соединение (15)) (2,49 г), фенолят натрия (524 мг) и тетрагидрофуран (130 мл), смесь охлаждали и (трифторметил)триметилсилан (11,4 г) добавляли по каплям за 3 минуты при внутренней температуре от -52 до -51°C. Полученную смесь перемешивали при внутренней температуреот -52 до -48°C в течение 30 минут, температуру повышали до приблизительно 0°C за 1 час и охлаждали снова до внутренней температуры -53°C. К реакционной смеси добавляли по каплям (трифторметил)триметилсилан (19,2 г) при внутренней температуре от -53 до -47°C за 2 часа, и смесь перемешивали при внутренней температуре приблизительно -50°C в течение 10 минут. Температуру полученной реакционной смеси повышали до комнатной температуры, и реакционную смесь концентрировали при пониженном давлении, пока масса не достигла 81,5 г или меньше, и добавляли метанол (130 мл). К полученному метанольному раствору добавляли по каплям при комнатной температуре 1M соляной кислоты (32,6 мл) и смесь перемешивали в течение 3 часов. Полученную реакционную смесь концентрировали при пониженном давлении, пока масса не достигла 97,8 г или меньше. Толуол (293 мл) и 10%-й солевой раствор (163 мл) добавляли к смеси, и смесь разделяли, и водный слой удаляли. Полученный органический слой промывали водой (163 мл) и концентрировали при пониженном давлении, пока масса не достигла 97,8 г или меньше. К полученному концентрату добавляли метанол (163 мл), и смесь концентрировали снова при пониженном давлении, пока масса не достигла 97,8 г или меньше, и добавляли метанол (163 мл) и активированный уголь (4,89 г). Полученную смесь перемешивали при комнатной температуре в течение 3 часов, фильтровали, и остаток промывали метанолом (98 мл). Полученный фильтрат и смывы концентрировали при пониженном давлении, пока масса не достигла 97,8 г или меньше, и добавляли метанол для получения метанольного раствора (130 г). К полученному метанольному раствору добавляли воду (57 мл) при внутренней температуре приблизительно 50°C, и затравочный кристалл (33 мг) сольвата (9R)-4-бром-2-(3-гидрокси-3-метилбутилокси)-9-(трифторметил)-9H-флуорен-9-ол гемиметанола (соединение (2m)) добавляли при внутренней температуре 45°C. Полученную суспензию перемешивали при внутренней температуре приблизительно 45°C в течение 2 часов, охлаждали до комнатной температуры и перемешивали в течение ночи. Осажденное твердое вещество собирали фильтрацией и промывали смешанным раствором метанол/вода (1,3об./0,7об.) (65 мл), охлажденным до 10°C или ниже. Полученное твердое вещество высушивали при пониженном давлении с получением целевого соединения (23,9 г).
1H-ЯМР (400 МГц, DMSO-D6) δ: 8,31 (ддд, 1H, J=7,8, 1,2, 0,7 Гц), 7,65 (дддд, 1H, J=7,8, 1,4, 0,9, 0,7 Гц), 7,54 (ддд, 1H, J=7,6, 7,6, 1,4 Гц), 7,40 (ддд, 1H, J=7,6, 7,6, 1,2 Гц), 7,40 (с, 1H), 7,29 (д, 1H, J=2,3 Гц), 7,19 (дд, 1H, J=2,3, 0,9 Гц), 4,42 (с, 1H), 4,18 (т, 2H, J=7,2 Гц), 4,08 (к, 0,49H, J=5,4 Гц), 3,17 (д, 1,42H, J=5,4 Гц), 1,86 (т, 2H, J=7,2 Гц), 1,18 (с, 6H).
(Термогравиметрический анализ)
Уменьшение массы по данным термогравиметрического анализа соответствовало теоретическому значению соединения (2m) (гемиметанольный сольват соединения (2)).
Вычислено: 3,58% (вычисленное значение для гемиметанольного сольвата)
Найдено: 3,56%
[0163] Синтез затравочного кристалла соединения (2m):
После обработки 1M соляной кислотой на Стадии 5, концентрация при пониженном давлении, разделение толуол-10% солевой раствор и промывка водой дали раствор в толуоле. Раствор в толуоле концентрировали, и соединение (2) отделяли (оптическая чистота 70,5% э.и.) колоночной хроматографией на силикагеле (элюент: гексан/этилацетат от (3об./2об.) до (5об./4об.)). Отделенное соединение (2) кристаллизовали из смешанного раствора MeOH/вода (3об./1,7об.) с получением затравочного кристалла соединения (2m) (оптическая чистота 98,9% э.и.). Оптическую чистоту определяли в аналитических условиях для ВЭЖХ 3. Время удерживания (R) формы составляло 24,5 минуты и время удерживания (S) формы составляло 23,2 минуты.
[0164] Стадия 6
(9R)-4-Бром-2-(3-гидрокси-3-метилбутилокси)-9-(трифторметил)-9H-флуорен-9-ол (соединение (2))
[0165] 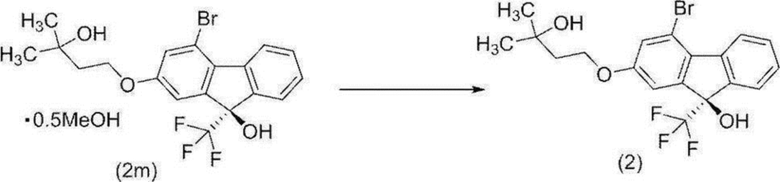
[0166] В атмосфере азота к гемиметанольному сольвату (9R)-4-бром-2-(3-гидрокси-3-метилбутилокси)-9-(трифторметил)-9H-флуорен-9-ола (соединение (2m)) (23,9 г), полученному на предыдущей стадии, добавляли н-гептан (120 мл), полученную суспензию перемешивали в течение 1 часа при поддержании внутренней температуры 85°C или выше, и приблизительно 25 мл растворителя удаляли при пониженном давлении при поддержании внутренней температуры приблизительно 70°C. К полученной суспензии добавляли н-гептан (25 мл) при внутренней температуре приблизительно 70°C, и приблизительно 25 мл растворителя удаляли снова при пониженном давлении при поддержании внутренней температуры приблизительно 70°C. К полученной суспензии добавляли н-гептан (25 мл) при внутренней температуре приблизительно 70°C и, после охлаждения до комнатной температуры, смесь перемешивали в течение приблизительно 2 часов. Твердое вещество собирали фильтрацией и промывали н-гептаном. Полученное твердое вещество высушивали при пониженном давлении при внешней температуре 60°C с получением целевого соединения (22,2 г, оптическая чистота 99,1% э.и.). Оптическую чистоту определяли в аналитических условиях для ВЭЖХ 3. Время удерживания (R) формы составляла 24,5 минуты и время удерживания (S) формы составляло 23,2 минуты.
Специфическое оптическое вращение было [α]D +11,4 ° (c=1,00 MeOH 25°C).
1H-ЯМР (DMSO-D6) δ: 8,30 (ддд, 1H, J=7,8, 1,2, 0,7 Гц), 7,64 (дддд, 1H, J=7,8, 1,4, 0,9, 0,7 Гц), 7,53 (ддд, 1H, J=7,6, 7,6, 1,4 Гц), 7,39 (ддд, 1H, J=7,6, 7,6, 1,2 Гц), 7,39 (с, 1H), 7,28 (д, 1H, J=2,3 Гц), 7,18 (дд, 1H, J=2,3, 0,9 Гц), 4,41 (с, 1H), 4,16 (т, 2H, J=7,2 Гц), 1,85 (т, 2H, J=7,2 Гц), 1,17 (с, 6H).
[0167] Кроме того, абсолютная конфигурация монокристалла соединения (2), полученного перекристаллизацией с использованием гептана, была определена рентгеноструктурным анализом монокристалла.
Соединение (2) было дериватизовано в нижеупомянутое соединение (1h), и специфическое оптическое вращение этого было измерено. В результате было показано специфическое оптическое вращение, эквивалентное таковому соединения (2-{4-[(9R)-9-гидрокси-2-(3-гидрокси-3-метилбутилокси)-9-(трифторметил)-9H-флуорен-4-ил]-1H-пиразол-1-ил}-2-метилпропанамид моногидрата, описанного в WO 2014/142290 (соединение (2h), описанное в WO 2014/142290)).
[0168] Стадия 7
Трет-бутил-2-{4-[(9R)-9-гидрокси-2-(3-гидрокси-3-метилбутилокси)-9-(трифторметил)-9H-флуорен-4-ил]-1H-пиразол-1-ил}-2-метилпропионат (соединение (4))
[0169] 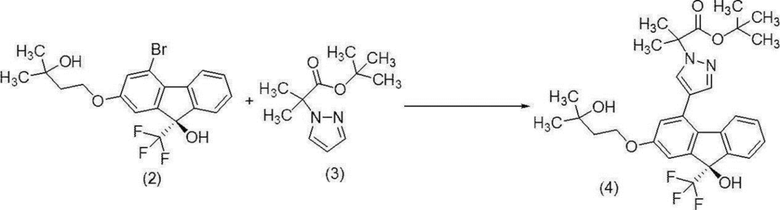
[0170] В атмосфере азота к бис(трифенилфосфин)палладий (II) дихлориду (2,44 г) добавляли N,N-диметилацетамид (420 мл), триметилуксусную кислоту (3,55 г) и карбонат калия (15,4 г). К полученной смеси добавляли (9R)-4-бром-2-(3-гидрокси-3-метилбутилокси)-9-(трифторметил)-9H-флуорен-9-ол (соединение (2)) (60,0 г), трет-бутил-2-метил-2-(1H-пиразол-1-ил)пропионат (соединение (3)) (46,8 г) и N,N-диметилацетамид (60 мл). Полученную смесь перемешивали при температуре от 104 до 107°C в течение 3 часов, реакционную смесь концентрировали при пониженном давлении и приблизительно 440 мл N,N-диметилацетамида выпаривали. Полученный концентрат, содержащий целевое соединение, использовали на следующей стадии с выходом 100%.
1H-ЯМР (400 МГц, DMSO-D6) δ: 8,16 (д, 1H, J=0,7 Гц), 7,68 (д, 1H, J=0,7 Гц), 7,62-7,57 (м, 1H), 7,32-7,18 (м, 3H), 7,23 (с, 1H), 7,15 (д, 1H, J=2,4 Гц), 6,84 (д, 1H, J=2,4 Гц), 4,41 (с, 1H), 4,16 (т, 2H, J=7,2 Гц), 1,87 (т, 2H, J=7,2 Гц), 1,81 (с, 6H), 1,38 (с, 9H), 1,18 (с, 6H).
[0171] Стадия 8
2-{4-[(9R)-9-Гидрокси-2-(3-гидрокси-3-метилбутилокси)-9-(трифторметил)-9H-флуорен-4-ил]-1H-пиразол-1-ил}-2-метилпропионовая кислота (соединение (5))
[0172] 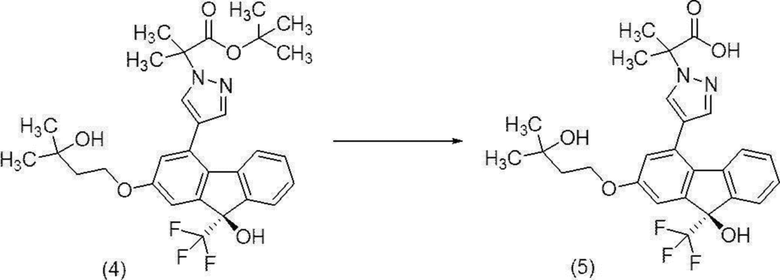
[0173] В атмосфере азота к концентрату трет-бутил-2-{4-[(9R)-9-гидрокси-2-(3-гидрокси-3-метилбутилокси)-9-(трифторметил)-9H-флуорен-4-ил]-1H-пиразол-1-ил}-2-метилпропионата (соединение (4)) полученного на Стадии 7, добавляли активированный уголь (6,00 г), 40%-й водный раствор гидроксида натрия (120 мл) и этанол (180 мл), и смесь перемешивали при внутренней температуре от 70 до 73°C в течение 1 часа 30 минут. После охлаждения до комнатной температуры добавляли воду (120 мл), полученную смесь фильтровали через целит, и остаток промывали смешанным раствором этанол/вода (1об./1об.) (180 мл). Полученный фильтрат и смывы концентрировали при пониженном давлении, пока масса не достигла 428 г, добавляли толуол (480 мл) и воду (180 мл), и смесь разделяли при внутренней температуре от 40 до 50°C. В другой сосуд добавляли 85%-ю фосфорную кислоту (228 г) и воду (180 мл), полученный водный раствор фосфорной кислоты охлаждали, и этилацетат (360 мл) добавляли при внутренней температуре приблизительно 0°C. К полученной смеси добавляли по каплям водный слой, полученный путем разделения, выполненного ранее, при внутренней температуре от -7 до 14°C. Смесь разделяли, и водный слой удаляли. Полученный органический слой промывали 4 раза водой (300 мл), концентрировали при пониженном давлении, пока масса не достигла 135 г, добавляли этилацетат (300 мл), и смесь концентрировали снова при пониженном давлении, пока масса жидкости не достигла 138 г. К полученному концентрату добавляли этилацетат (420 мл) и активированный уголь (3,00 г). Полученную смесь перемешивали при внутренней температуре от 23 до 26°C в течение 2 часов 20 минут, фильтровали, и остаток промывали этилацетатом (120 мл). Полученный фильтрат и смывы концентрировали при пониженном давлении при внешней температуре 50°C, пока масса не достигла 150 г, добавляли этилацетат (300 мл), и смесь концентрировали снова при пониженном давлении при внешней температуре 50°C, пока масса жидкости не достигла 150 г. Полученный раствор перемешивали при внутренней температуре от 45 до 50°C в течение 1 часа 30 минут, и осаждение кристаллов подтверждали. К полученной суспензии добавляли по каплям толуол (390 мл) при внутренней температуре от 46 до 49°C, и смесь перемешивали при внутренней температуре от 47 до 52°C в течение 1 часа 10 минут, охлаждали до комнатной температуры и перемешивали в течение ночи. Осажденное твердое вещество собирали фильтрацией и промывали смешанным раствором толуол/этилацетат (7об./1об.) (120 мл). Полученное твердое вещество высушивали при пониженном давлении с получением целевого соединения (58,9 г).
1H-ЯМР (400 МГц, DMSO-D6) δ: 13,09 (ушир., 1H), 8,17 (д, 1H, J =0 0,6 Гц), 7,64 (д, 1H, J=0,6 Гц), 7,61-7,56 (м, 1H), 7,29-7,18 (м, 4H), 7,15 (д, 1H, J=2,4 Гц), 6,86 (д, 1H, J=2,4 Гц), 4,40 (с, 1H), 4,16 (т, 2H, J=7,2 Гц), 1,87 (т, 2H, J=7,2 Гц), 1,84 (с, 3H), 1,83 (с, 3H), 1,18 (с, 6H).
[0174] Стадия 9
2-{4-[(9R)-9-Гидрокси-2-(3-гидрокси-3-метилбутилокси)-9-(трифторметил)-9H-флуорен-4-ил]-1H-пиразол-1-ил}-2-метилпропанамид моногидрат (соединение (1h))
[0175] 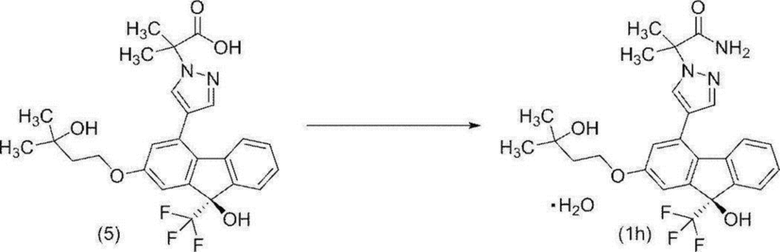
[0176] В атмосфере азота 2-{4-[(9R)-9-гидрокси-2-(3-гидрокси-3-метилбутилокси)-9-(трифторметил)-9H-флуорен-4-ил]-1H-пиразол-1-ил}-2-метилпропионовую кислоту (соединение (5)) (58,0 г) и 1-гидроксибензотриазол моногидрат (17,6 г) растворяли в N,N-диметилформамиде (174 мл). Полученный раствор охлаждали до 4°C, и 28%-й водный раствор аммиака (11,6 мл) добавляли по каплям при поддержании внутренней температуры менее 15°C. К полученному раствору при комнатной температуре добавляли 1-этил-3-(3-диметиламинопропил)цианамид гидрохлорид (6,60 г), и смесь перемешивали при комнатной температуре в течение 1 часа 40 минут. При комнатной температуре последовательно добавляли 1-этил-3-(3-диметиламинопропил)цианамид гидрохлорид (6,60 г), и смесь перемешивали при комнатной температуре в течение приблизительно 1 часа 30 минут. Эту операцию повторяли 3 раза. К полученной реакционной смеси добавляли этилацетат (580 мл), 20%-й солевой раствор (232 мл) и воду (58 мл) для разделения, и водный слой удаляли. Полученный органический слой промывали путем добавления этилацетата и 5%-ого водного раствора гидрокарбоната натрия (на 116 мл) (290 мл). Затем органический слой промывали последовательно водой (116 мл), 1M соляной кислотой (290 мл), 5%-м солевым раствором (290 мл) и водой (290 мл), и полученный органический слой концентрировали при пониженном давлении, пока масса не достигла 121 г. К полученному концентрату добавляли этанол (174 мл), и смесь концентрировали при пониженном давлении, пока масса не достигла 109 г. Добавляли этанол (174 мл), и смесь концентрировали снова при пониженном давлении, пока масса не достигла 112 г. К полученной концентрированной жидкости добавляли этанол (93 мл), и количество жидкости доводили до 200 мл. Полученный раствор фильтровали для удаления примесей, и остаток промывали этанолом (116 мл). Полученный фильтрат и смывы нагревали, добавляли по каплям воду (348 мл) при внутренней температуре от 46 до 50°C, добавляли затравочный кристалл (12 мг), и смесь перемешивали при внутренней температуре от 46 до 51°C в течение 2 часов. Полученную суспензию далее перемешивали при внутренней температуре от 55 до 62°C в течение 1 часа 10 минут, охлаждали до 30°C в течение приблизительно 4 часов и перемешивали при комнатной температуре в течение ночи. Осажденное твердое вещество собирали фильтрацией, последовательно промывали смешанным раствором этанол/вода (5об./7об.) (116 мл) и водой (116 мл). Полученное твердое вещество высушивали при пониженном давлении при внешней температуре 30°C, 1,3 кПа, с получением целевого соединения (54,6 г, оптическая чистота > 99,9% э.и.). Оптическую чистоту определяли в аналитических условиях для ВЭЖХ 4. Время удерживания (R) формы составляло 6,4 минут и время удерживания (S) формы составляло 9,2 минут.
Специфическое оптическое вращение было [α]D +39,1 ° (c=1,00 MeOH 25°C).
1H-ЯМР (400 МГц, DMSO-D6) δ: 8,09 (д, 1H, J=0,9 Гц), 7,68 (д, 1H, J=0,9 Гц), 7,62-7,56 (м, 1H), 7,36-7,30 (м, 1H), 7,30-7,22 (м, 3H), 7,21 (с, 1H), 7,14 (д, 1H, J=2,5 Гц), 6,97 (ушир., 1H), 6,88 (д, 1H, J=2,5 Гц), 4,40 (с, 1H), 4,16 (т, 2H, J=7,2 Гц), 1,87 (т, 2H, J=7,2 Гц), 1,80 (с, 3H), 1,80 (с, 3H), 1,18 (с, 6H).
(Определение содержания воды)
Количественное значение содержания воды, полученное методом титрования Карла Фишера (кулонометрическое титрование) соответствовало теоретическому значению для соединения (1h) (моногидрат соединения (1)).
Вычислено: 3,45% (вычисленное значение для моногидрата)
Найдено: 3,48%
[0177] Синтез затравочного кристалла соединения (1h):
Затравочный кристалл, используемый на Стадии 9, получали из смешанного растворителя из этанола и воды с использованием твердого вещества, полученного согласно Примеру (2-{4-[(9R)-9-гидрокси-2-(3-гидрокси-3-метилбутилокси)-9-(трифторметил)-9H-флуорен-4-ил]-1H-пиразол-1-ил}-2-метилпропанамид моногидрата/соединения (2h)), описанному в WO 2014/142290.
[0178] Альтернатива Стадии 9:
Даже когда затравочный кристалл не добавляли во время кристаллизации, кристалл целевого соединения (1h) получали способом, аналогичным таковому Стадии 9.
[0179] Стадия 10 Синтез трет-бутил-2-метил-2-(1H-пиразол-1-ил)пропионата (соединение (3))
[0180]
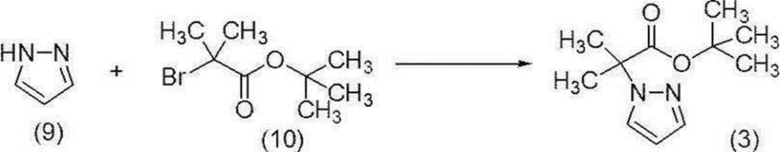
[0181] В атмосфере азота добавляли тетрагидрофуран (1250 мл) и 60%-й гидрид натрия (58,5 г) и охлаждали до внутренней температуры -9°C. К полученной суспензии добавляли по каплям раствор пиразол (соединение (9)) (100 г)/тетрагидрофуран (250 мл) при внутренней температуре от -9 до 5°C, и смесь перемешивали при температуре от -5 до 0°C в течение 30 минут. Температуру полученной смеси поднимали до внутренней температуры 14°C, трет-бутил-2-бром-2-метилпропионат (соединение (10)) (377 г) добавляли при внутренней температуре от 14 до 16°C, и смесь перемешивали при внутренней температуре 18°C в течение 15 минут и затем перемешивали при внутренней температуре от 47 до 51°C в течение 11 часов. Полученную реакционную смесь охлаждали до внутренней температуры 1°C, добавляли по каплям раствор трет-бутоксид калия (82,3 г)/тетрагидрофуран (300 мл) при внутренней температуре от 1 до 7°C, и смесь перемешивали при температуре от 10 до 12°C в течение 5 часов. К полученной реакционной смеси добавляли толуол (800 мл) и воду (800 мл) при комнатной температуре для разделения, и водный слой удаляли. Полученный органический слой промывали 1M соляной кислотой (800 мл) и водой (400 мл х 2 раза) и концентрировали при пониженном давлении. Полученный концентрат перегоняли при пониженном давлении при внешней температуре приблизительно 110°C при пониженном давлении приблизительно 0,6 кПа с получением целевого соединения (229 г).
1H-ЯМР (400 МГц, DMSO-D6) δ: 7,83 (дд, 1H, J=2,4, 0,7 Гц), 7,46 (дд, 1H, J=1,8, 0,7 Гц), 6,26 (дд, 1H, J=2,4, 1,8 Гц), 1,71 (с, 6H), 1,32 (с, 9H).
[Промышленная применимость]
[0182]
Настоящее изобретение относится к способу получения соединения формулы [I] или его фармацевтически приемлемой соли или его гидрата с хорошим выходом.
Кроме того, соединения формулы [II], формулы [IIm], формулы [III] и формулы [IV] согласно настоящему изобретению могут быть использованы как промежуточные соединения синтеза для получения соединения формулы [I] или его фармацевтически приемлемой соли или его гидрата.
Кроме того, способ получения согласно настоящему изобретению может быть использован как способ крупномасштабного промышленного синтеза, поскольку этот способ может быть осуществлен удобной операцией через простые в обращении соединения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТРИАЗОЛОПИРИДИНОВОЕ СОЕДИНЕНИЕ И ЕГО ДЕЙСТВИЕ В КАЧЕСТВЕ ИНГИБИТОРА ПРОЛИЛГИДРОКСИЛАЗЫ И ИНДУКТОРА ВЫРАБОТКИ ЭРИТРОПОЭТИНА | 2010 |
|
RU2538963C2 |
| 3,5-ДИЗАМЕЩЕННОЕ АЛКИНИЛБЕНЗОЛЬНОЕ СОЕДИНЕНИЕ И ЕГО СОЛЬ | 2013 |
|
RU2576384C1 |
| ПИРАЗОЛАМИДНОЕ СОЕДИНЕНИЕ И ЕГО ПРИМЕНЕНИЯ В МЕДИЦИНЕ | 2014 |
|
RU2664532C2 |
| ХИНОЛИЛПИРРОЛПИРИМИДИЛЬНОЕ КОНДЕНСИРОВАННОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ | 2013 |
|
RU2581039C1 |
| ПРОИЗВОДНОЕ ИНДОЛА, СОДЕРЖАЩЕЕ ПИПЕРИДИНОВЫЙ ЦИКЛ | 2005 |
|
RU2332413C1 |
| АЗОЛЗАМЕЩЕННОЕ СОЕДИНЕНИЕ ПИРИДИНА | 2017 |
|
RU2730498C2 |
| НОВОЕ ЦИАНОПИРИМИДИНОВОЕ ПРОИЗВОДНОЕ | 2010 |
|
RU2533115C2 |
| ПРОИЗВОДНОЕ БЕНЗОКСАЗОЛА, ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ЕГО ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ СОСТОЯНИЯ СИНДРОМА РАЗДРАЖЕННОГО КИШЕЧНИКА ИЛИ ФУНКЦИОНАЛЬНОГО РАССТРОЙСТВА ПИЩЕВАРИТЕЛЬНОГО ТРАКТА | 1999 |
|
RU2204558C2 |
| РЕЗИНОВАЯ СМЕСЬ | 2012 |
|
RU2605122C2 |
| НОВОЕ ПРОИЗВОДНОЕ ПИРАЗОЛ-3-КАРБОКСАМИДА, ОБЛАДАЮЩЕЕ АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ РЕЦЕПТОРА 5-НТ | 2009 |
|
RU2528406C2 |
Изобретение относится к вариантам способа получения соединения, представленного формулой [I], или его гидрата, которые находят применение в качестве ингибиторов пируватдегидрогеназы. В одном из вариантов способ включает стадию превращения соединения, представленного формулой [II], или его метанольного сольвата в соединение, представленное формулой [IV], реакцией сочетания с соединением, представленным формулой [III], в присутствии палладиевого катализатора, карбоната щелочного металла и карбоновой кислоты; стадию гидролиза соединения вышеописанной формулы [IV] для превращения его в соединение, представленное формулой [V], и стадию реакции соединения вышеописанной формулы [V] с аммиаком в присутствии конденсирующего агента. Изобретение также относится к промежуточным соединениям формул [IV], [II], [IIm], [III] и к способу получения соединения, представленного формулой [IV]. 7 н. и 6 з.п. ф-лы, 1 пр.
1. Способ получения соединения, представленного формулой [I]
 ,
,
или его гидрата, включающий стадию превращения соединения, представленного формулой [II]
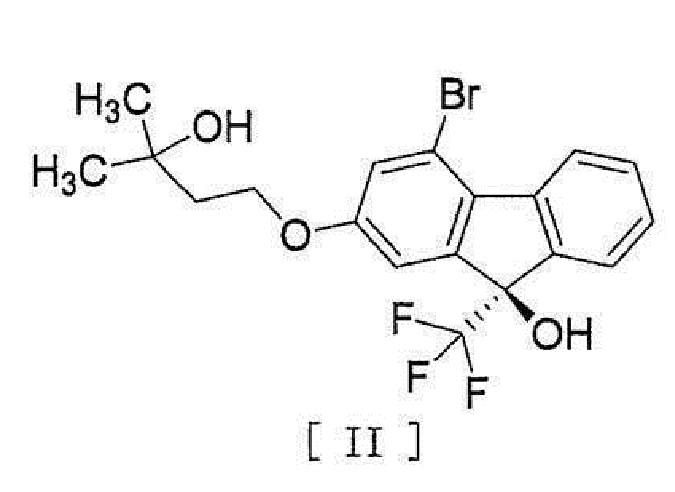 ,
,
или его метанольного сольвата в соединение, представленное формулой [IV]
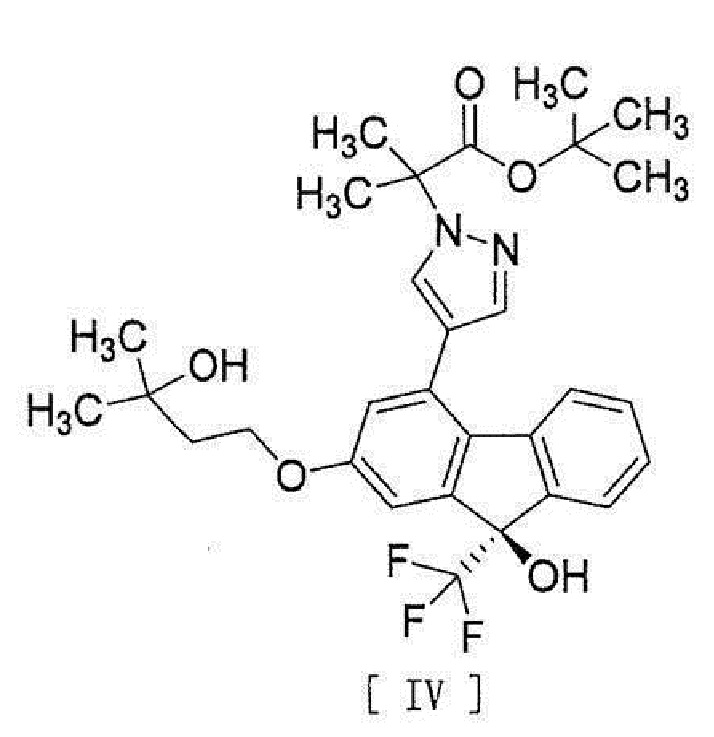 ,
,
реакцией сочетания с соединением, представленным формулой [III]
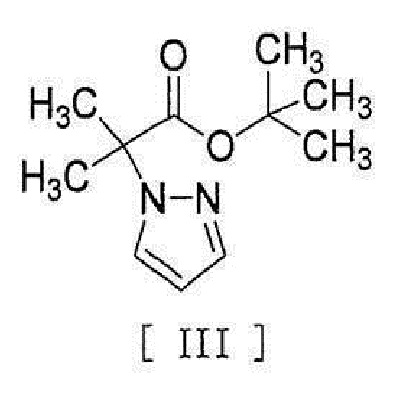 ,
,
в присутствии палладиевого катализатора, карбоната щелочного металла и карбоновой кислоты,
стадию гидролиза соединения вышеописанной формулы [IV] для превращения его в соединение, представленное формулой [V]
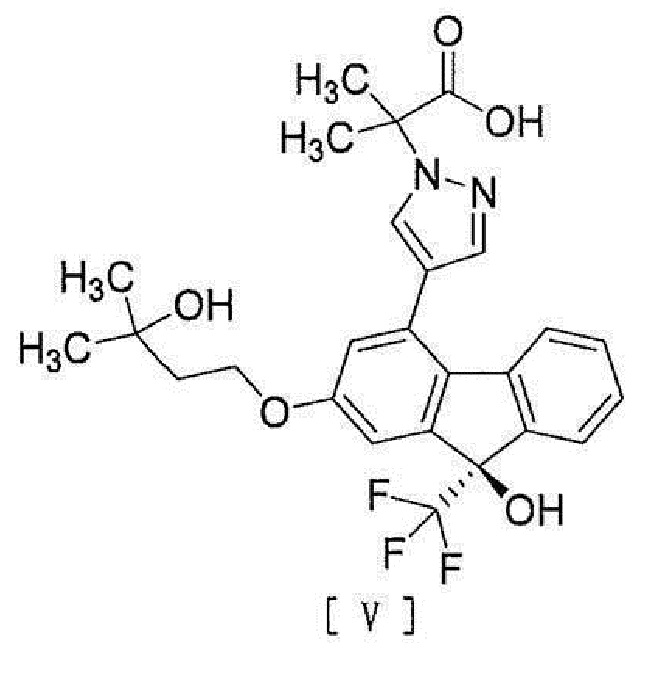 ,
,
и стадию реакции соединения вышеописанной формулы [V] с аммиаком в присутствии конденсирующего агента для превращения его в соединение, представленное вышеописанной формулой [I] или его гидрат.
2. Способ по п. 1, в котором карбоновая кислота является триметилуксусной кислотой, изомасляной кислотой, пропионовой кислотой или бензойной кислотой.
3. Способ по п. 1 или 2, в котором температура реакции сочетания составляет от 80 до 150°C.
4. Способ по любому из пп. 1-3, в котором соединение вышеописанной формулы [II] получают способом, включающим стадию реакции соединения, представленного формулой [VI]
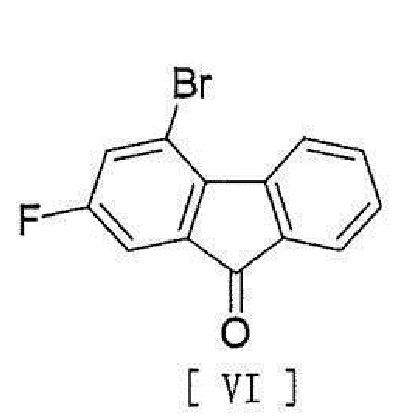 ,
,
с соединением, представленным формулой [VII]
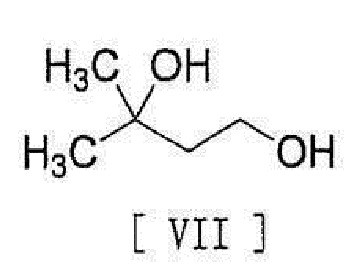 ,
,
в присутствии основания для превращения его в соединение, представленное формулой [VIII]
 ,
,
и стадию реакции соединения формулы [VIII] с (трифторметил)триметилсиланом в присутствии асимметрического органокатализатора и последующей обработки кислотой.
5. Способ по п. 4, в котором асимметрический органокатализатор представляет собой соль цинхонидия.
6. Способ по п. 5, в котором соль цинхонидия представляет собой N-(4-трет-бутил-3-метоксибензил)цинхонидий бромид.
7. Способ по любому из пп. 1-6, в котором соединение вышеописанной формулы [III] получают путем реакции соединения, представленного формулой [IX]
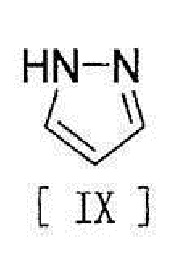 ,
,
с соединением, представленным формулой [X]
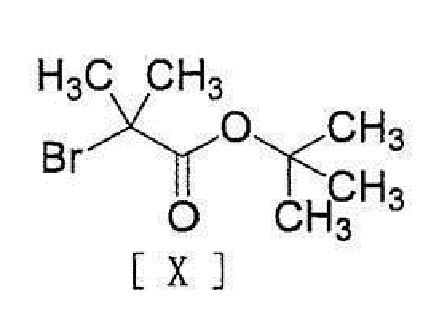 ,
,
в присутствии основания.
8. Способ получения соединения, представленного формулой [I]
,
или его гидрата, включающий стадию реакции соединения, представленного формулой [VI]
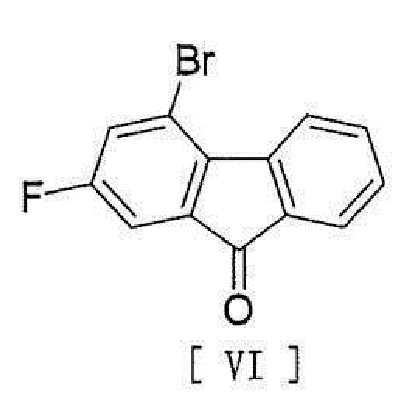 ,
,
с соединением, представленным формулой [VII]
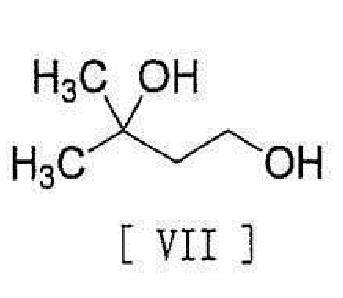 ,
,
в присутствии основания для превращения его в соединение, представленное формулой [VIII]
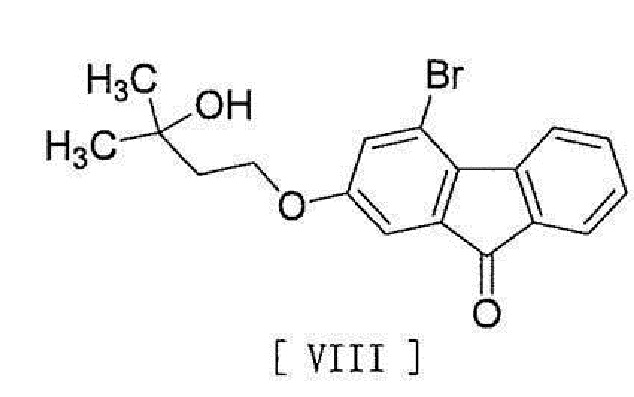 ,
,
стадию реакции соединения формулы [VIII] с (трифторметил)триметилсиланом в присутствии асимметрического органокатализатора и последующей обработки кислотой для получения соединения, представленного формулой [II]
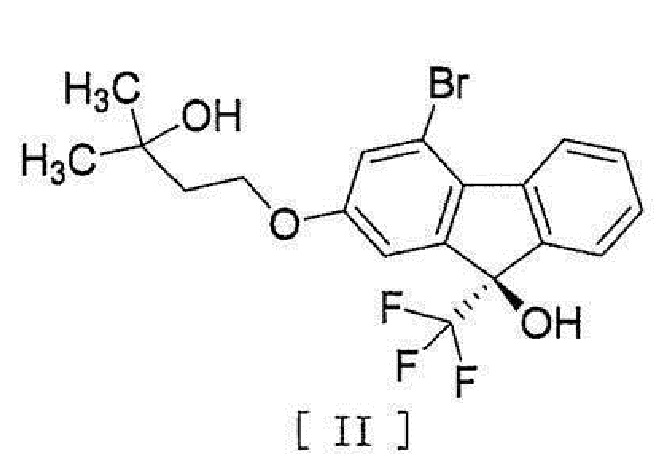 ,
,
или его метанольного сольвата, стадию реакции соединения, представленного формулой [IX]
 ,
,
с соединением, представленным формулой [X]
 ,
,
в присутствии основания для получения соединения, представленного формулой [III]
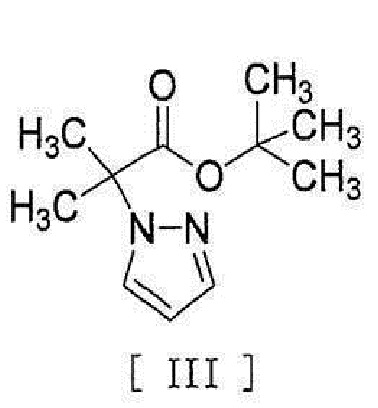 ,
,
стадию введения соединения вышеописанной формулы [III] в реакцию сочетания с соединением вышеописанной формулы [II] или его метанольным сольватом в присутствии палладиевого катализатора, карбоната щелочного металла и карбоновой кислоты для превращения этого соединения в соединение, представленное формулой [IV]
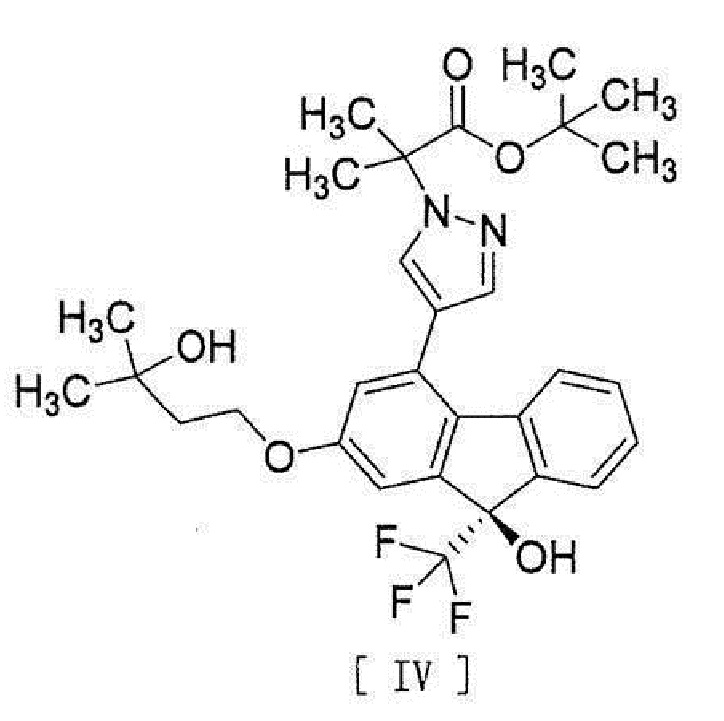 ,
,
стадию гидролиза соединения вышеописанной формулы [IV] для превращения этого соединения в соединение, представленное формулой [V]
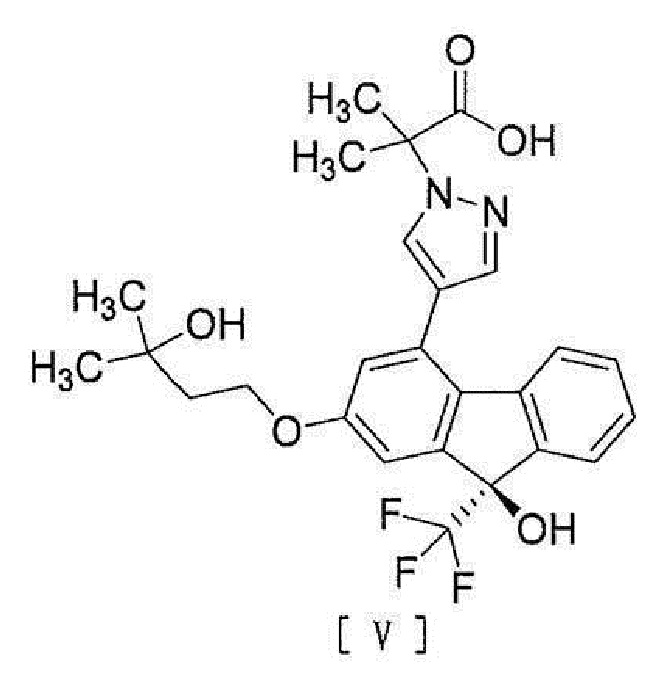 ,
,
и стадию амидирования соединения вышеописанной формулы [V] путем реакции с аммиаком в присутствии конденсирующего агента.
9. Способ получения соединения, представленного формулой [IV]
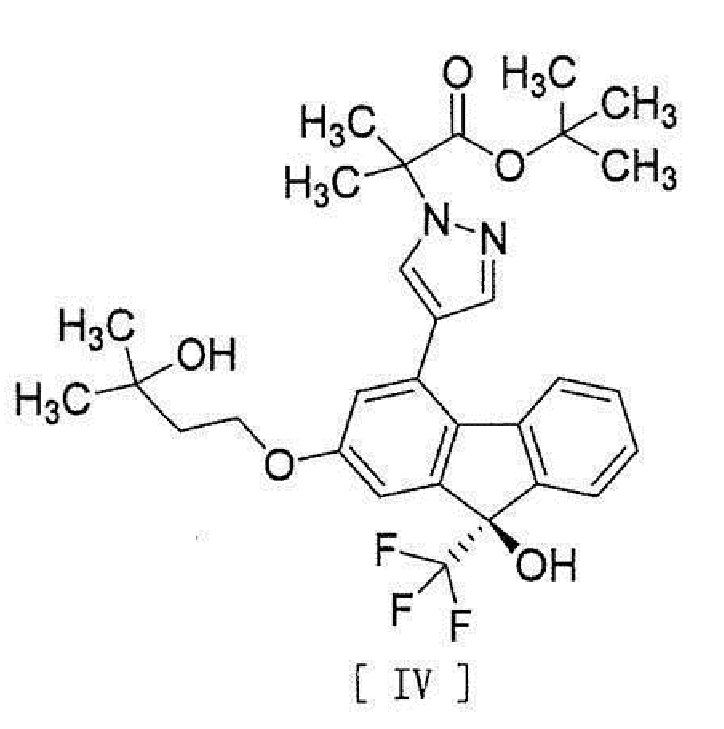 ,
,
включающий введение соединения, представленного формулой [II]
 ,
,
или его метанольного сольвата в реакцию сочетания с соединением, представленным формулой [III]
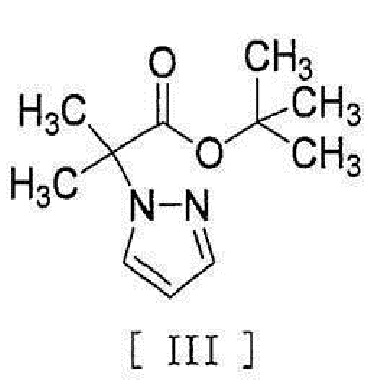 ,
,
в присутствии палладиевого катализатора, карбоната щелочного металла и карбоновой кислоты.
10. Соединение, представленное формулой [IV]
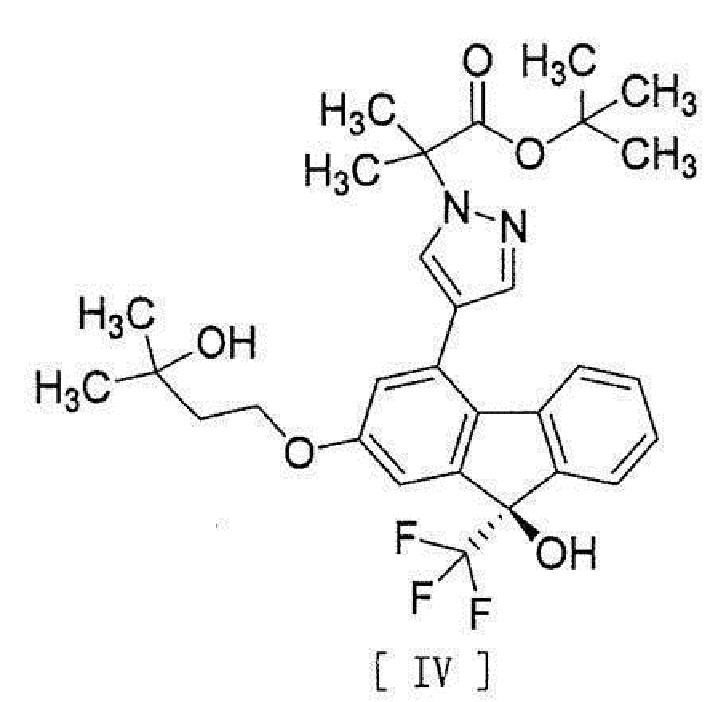 .
.
11. Соединение, представленное формулой [II]
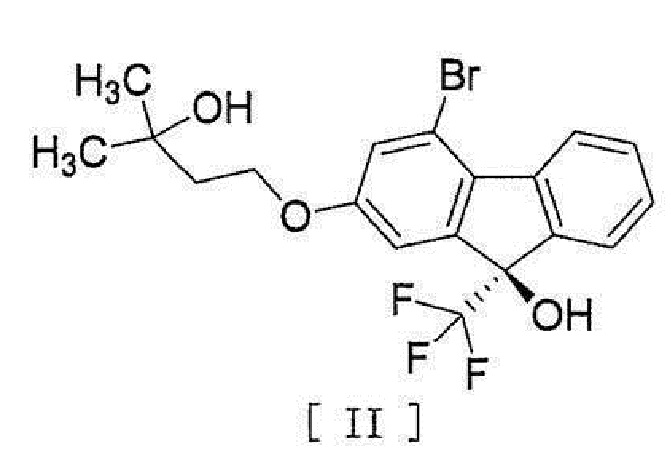 .
.
12. Соединение, представленное формулой [IIm]
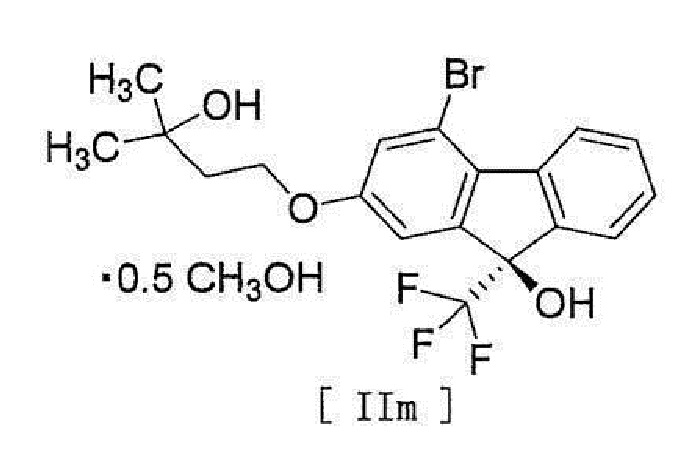 .
.
13. Соединение, представленное формулой [III]
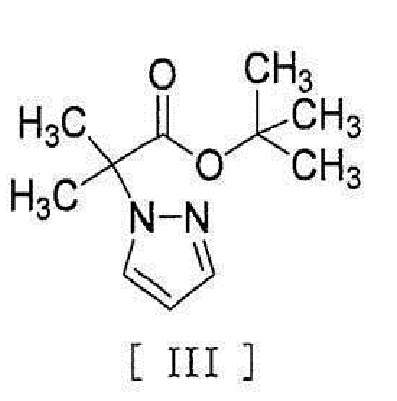 .
.
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| ТЕРАПЕВТИЧЕСКОЕ СРЕДСТВО ИЛИ ПРОФИЛАКТИЧЕСКОЕ СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ ФИБРОМИАЛГИИ | 2011 |
|
RU2560168C2 |

