Настоящее изобретение относится к области синтеза гетероциклических соединений, конкретно - к способу получения 4′-фторспиро[циклопропан-1,3′-индол]-2′(1′H)-она, являющегося ценным полупродуктом в синтезе ряда фармацевтиков.
Известен способ получения 4′-фторспиро[циклопропан-1,3′-индол]-2′(1′H)-она путем алкилирования 4-фтор-1,3-дигидро-2H-индол-2-она с использованием 1,2-дибромэтана в абсолютном тетрагидрофуране, в присутствии полученного in situ из бутиллития и диизопропиламина диизопропиламида лития при температуре -40°C. Выход технического продукта составляет 93% [US 2008051585, C07D 209/04, 28.02.2008].
Недостатками данного способа являются необходимость проведения реакции при очень низкой температуре и использование легковоспламеняющегося основания, что требует дополнительного лабораторного оформления процесса. Кроме этого, в способе описано получение технического продукта, а дополнительная его очистка для достижения фармацевтической чистоты приводит к ощутимому снижению выхода 4′-фторспиро[циклопропан-1,3′-индол]-2′(1′H)-она.
Известен способ получения 4′-фторспиро[циклопропан-1,3′-индол]-2′(1′H)-она путем внутримолекулярного N-арилирования 1-(2,6-дифторфенил)циклопропан-1-карбоксамида с использованием гидрида лития в абсолютном диметилформамиде (ДМФА) с выходом 82% и чистотой 98.6% [US 20060247441, C07D 209/54; C07D 403/02, 02.11.2006]. Исходный 1-(2,6-дифторфенил)циклопропан-1-карбоксамид, в свою очередь, получается путем частичного окислительного гидролиза 1-(2,6-дифторфенил)циклопропан-1-карбонитрила с выходом 84%. Последний получается путем алкилирования 2-(2,6-дифторфенил)ацетонитрила 1,2-дибромэтаном в присутствии водного раствора гидроксида калия и тетрабутиламмония бромида с выходом 65% [US 4859232, A01N 25/32, С07С 205/55, С07С 233/58, С07С 255/46, С07С 255/51, С07С 259/16, С07С 61/39, С07С 61/40, 22.08.1989]. Суммарный выход 4′-фторспиро[циклопропан-1,3′-индол]-2′(1′H)-она в расчете на 2-(2,6-дифторфенил)ацетонитрил составляет 45%.
Основным недостатком способа является его многостадийность при небольшом выходе целевого продукта реакции. Кроме этого, использование в синтезе токсичного растворителя - ДМФА, требует дополнительных операций по его полному удалению, использование пожароопасного основания - гидрида лития - требует дополнительного лабораторного оформления процесса.
Наиболее близким является способ получения 4′-фторспиро[циклопропан-1,3′-индол]-2′(1′H)-она, основанный на алкилировании 2-(2,6-дифторфенил)ацетонитрила 1,2-дибромэтаном в присутствии водного раствора гидроксида натрия и тетрабутиламмония бромида, с последующим частичным гидролизом технического продукта реакции под действием гидроксида калия в среде трет-пентилового спирта до 1-(2,6-дифторфенил)циклопропан-1-карбоксамида, который подвергается внутримолекулярному N-арилированию в присутствии трет-пентоксида натрия в абсолютном 1-метилпирролидин-2-оне [US 20080319204, C07D 209/96, 25.12.2008].
Недостатками данного способа является его многостадийность, использование в синтезе токсичного растворителя и пожароопасного, чувствительного к влаге воздуха основания - трет-пентоксида натрия. Кроме этого, в ходе реализации данного синтеза образуется значительное количество трудно регенерируемых стоков, а 1-метилпирролидин-2-он - плохо поддается рекуперации.
Задачей настоящего технического решения является разработка нового технологичного способа получения 4′-фторспиро [циклопропан-1,3′-индол]-2′(1′H)-она, с использованием дешевых и доступных исходных реагентов.
Техническим результатом является упрощение процесса получения 4′-фторспиро[циклопропан-1,3′-индол]-2′(1′H)-она и повышение чистоты образующегося продукта.
Предлагаемый технический результат достигается в способе получения 4′-фторспиро[циклопропан-1,3′-индол]-2′(1′H)-она путем алкилирования 2-(2,6-дифторфенил)ацетонитрила 1,2-дибромэтаном, с последующим частичным гидролизом полученного продукта реакции до 1-(2,6-дифторфенил)циклопропан-1-карбоксамида и его внутримолекулярным N-арилированием, при этом внутримолекулярное N-арилирование проводят обработкой полученного in situ 1-(2,6-дифторфенил)циклопропан-1-карбоксамида раствором гидроксида калия в кипящем этиленгликоле.
Сущностью предлагаемого способа является однореакторная схема синтеза, при которой продукт алкилирования 2-(2,6-дифторфенил)ацетонитрила обрабатывается раствором гидроксида калия в кипящем этиленгликоле. При этом происходит реакция внутримолекулярного N-арилирования, образующегося in situ 1-(2,6-дифторфенил)циклопропан-1-карбоксамида. Гидроксид калия в процессе реакции используется в качестве основания.
Главным преимуществом данного способа является упрощение способа за счет сокращения числа стадий синтеза и получение целевого продукта более высокой степени чистоты.
Кроме этого, способ реализуется при минимальном лабораторном оформлении процесса, не используются пожароопасные и дорогостоящие основания и не требуется предварительной подготовки растворителей. Все перечисленное за счет повышения технологичности реакции позволяет упростить способ получения 4′-фторспиро[циклопропан-1,3′-индол]-2′(1′H)-она.
Параллельно с основным процессом происходит образование небольшого количества продукта полного гидролиза - 1-(2,6-дифторфенил)циклопропан-1-карбоновой кислоты (в виде калиевой соли). Образующаяся в качестве побочного продукта реакции 1-(2,6-дифторфенил)циклопропан-1-карбоновая кислота, находит свое применение при получении средств химической защиты растений [US 4859232, A01N 25/32, С07С 205/55, С07С 233/58, С07С 255/46, С07С 255/51, С07С 259/16, С07С 61/39, С07С 61/40, 22.08.1989].
Способ осуществляется следующим образом.
В водном растворе гидроксида натрия в присутствии каталитических количеств тетрабутиламмония бромида алкилируют 1,2-дибромэтаном 2-(2,6-дифторфенил)ацетонитрил [US 20080319204]. К полученному техническому продукту реакции добавляют гидроксид калия и этиленгликоль. Реакционную смесь кипятят при перемешивании с защитой от углекислоты воздуха до полного прекращения выделения аммиака. Затем выделяют целевой продукт экстракцией метил трет-бутиловым эфиром. Органический экстракт сушат, фильтруют и упаривают досуха. Кубовой остаток перекристаллизовывают из изооктана. В результате получают целевой продукт с содержанием основного вещества 99.99%.
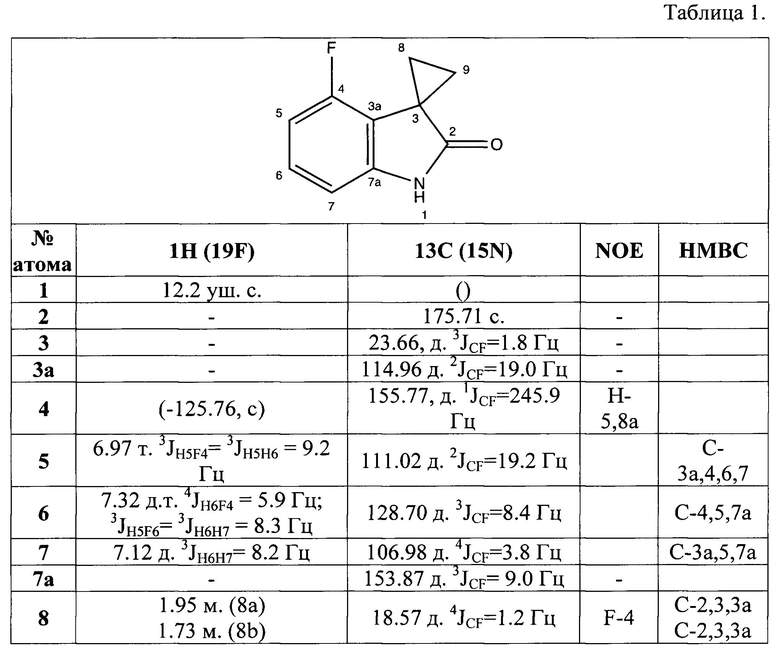
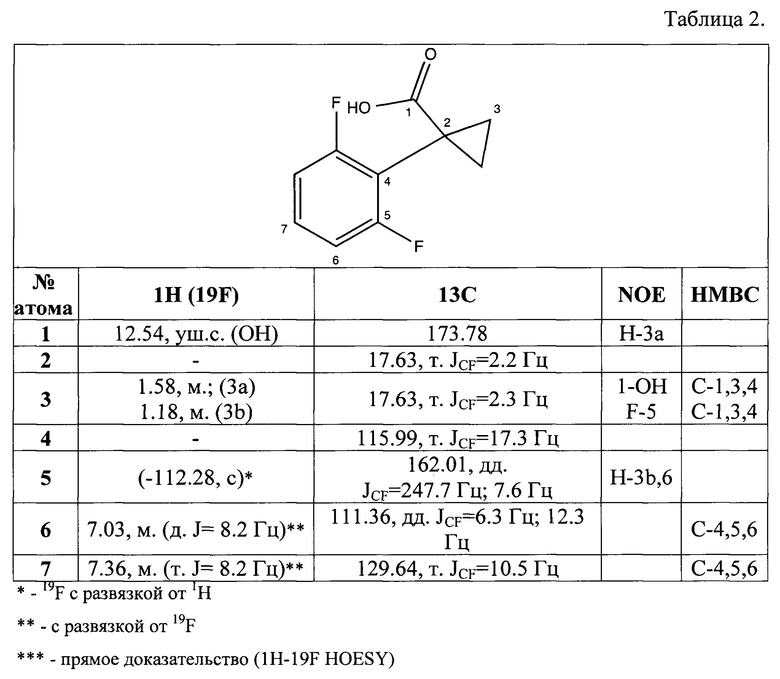
Структура полученного вещества подтверждена данными спектроскопии ЯМР 1H, 13C, 15N, 19F, с использованием корреляционных методов НМВС (Heteronuclear multiple bond correlation - двумерная гетероядерная корреляционная спектроскопия - корреляция между протонами и ядрами 13C или 15N, разделенными двумя или тремя связями) и NOESY (Nuclear overhauser effect spectroscopy - спектроскопия ядерного эффекта Оверхаузера - метод корреляции протонов, близко расположенных в пространстве).
Спектры ЯМР на протонах и ядрах 13C, 19F записывались на приборе Bruker Avance III 400, оснащенном широкополосным датчиком с Z-градиентом в DMSO-d6 при 30°C. Химические сдвиги 1H и 19F калибровались по сигналам атомов углерода (39.50 м.д.) и остаточных протонов (2.50 м.д.) диметилсульфоксида. Химические сдвиги 19F калибровались по сигналу внешнего стандарта - CFCl3 (0.0 м.д.). Спектр 19F был записан с широкополосной развязкой от протонов, спектры 1H были записан как с развязкой от ядра фтора-19, так и без нее. Двумерные эксперименты были поставлены по стандартным методикам фирмы Bruker с использованием Z-градиентных импульсов. Время смешивания в спектре NOESY составило 0.7 с. Спектр 1H-13C НМВС был оптимизирован для константы спин-спинового взаимодействия 10 Гц. Спектры 19F-1H HOESY были записаны в режиме наблюдения протонов, время смешивания 0.5 с.
Пример 1
К техническому продукту алкилирования 15.3 г (12.4 мл, 0.1 моль) (2-(2,6-дифторфенил)ацетонитрила 1,2-дибромэтаном прибавляют гидроксид калия (22.2 г, 0.33 моль; содержание основного вещества ~84.5%) и перегнанный этиленгликоль (150 мл). Полученную смесь кипятят при перемешивании с защитой от углекислоты воздуха до полного прекращения выделения аммиака.
Реакционную массу разбавляют водой (300 мл) и извлекают целевой продукт метил трет-бутиловым эфиром (3×100 мл). Органический экстракт сушат путем непрерывной азеотропной отгонки воды, фильтруют и выпаривают досуха, а кубовой остаток перекристаллизовывают из изооктана.
В результате получают целевой продукт с выходом 11.1 г (63% в расчете на исходный 2-(2,6-дифторфенил)ацетонитрил)) и содержанием основного вещества 99.99% (подтверждается результатами газо-жидкостной хроматографии и масс-спектроскопическими исследованиями). Т. пл. 72.5-74.5°C. Результаты ЯМР-исследований суммированы в таблице 1.
Пример 2
После извлечения 4′-фторспиро[циклопропан-1,3′-индол]-2′(1′H)-она метил трет-бутиловым эфиром, оставшийся водно-гликолевый раствор с помощью концентрированной соляной кислоты при перемешивании и охлаждении доводят до рН 1. Выделившийся осадок отфильтровывают, промывают водой, сушат непрерывной азеотропной отгонкой воды с толуолом и перекристаллизовывают из толуола. В результате выделяют 1-(2,6-дифторфенил)циклопропан-1-карбоновую кислоту с выходом 4.0 г (20% в расчете на исходный 2-(2,6-дифторфенил)ацетонитрил)) и т. пл. 156.5-157.5°C. Содержание основного вещества - 99.0% (подтверждается результатами газо-жидкостной хроматографии и масс-спектроскопическими исследованиями).
Структура полученного вещества была доказана данными спектроскопии ЯМР 1H, 13C, 19F, с использованием корреляционных методов НМВС и NOESY. Результаты ЯМР-исследований суммированы в таблице 2.
Таким образом, новый технологичный способ получения 4′-фторспиро[циклопропан-1,3′-индол]-2′(1′H)-она внутримолекулярным N-арилированием полученного in situ 1-(2,6-дифторфенил)циклопропан-1-карбоксамида раствором гидроксида калия в кипящем этиленгликоле позволяет получать целевой продукт с высокой чистотой и в одну стадию.
| название | год | авторы | номер документа |
|---|---|---|---|
| Соединения, полезные в качестве пестицидов, и промежуточные соединения, композиции и способы, связанные с ними | 2016 |
|
RU2821715C2 |
| МОЛЕКУЛЫ С ПЕСТИЦИДНОЙ ФУНКЦИЕЙ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, КОМПОЗИЦИИ И СПОСОБЫ, СВЯЗАННЫЕ С НИМИ | 2016 |
|
RU2735602C2 |
| МОЛЕКУЛЫ С ПЕСТИЦИДНОЙ ФУНКЦИЕЙ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, КОМПОЗИЦИИ И СПОСОБЫ, СВЯЗАННЫЕ С НИМИ | 2016 |
|
RU2742119C2 |
| Спироконденсированные производные 2,3-дигидроиндола, их применение в офтальмологии | 2017 |
|
RU2712039C2 |
| СПОСОБ ПОЛУЧЕНИЯ 3,5-ДИФТОР-БЕНЗИЛАМИДА (S)-3-ГИДРОКСИ-1-(1H-ИНДОЛ-5-ИЛ)-2-ОКСО-ПИРРОЛИДИН-3-КАРБОНОВОЙ КИСЛОТЫ | 2020 |
|
RU2826010C2 |
| СПОСОБ ПОЛУЧЕНИЯ 3,5-ДИФТОР-БЕНЗИЛАМИДА (S)-3-ГИДРОКСИ-1-(1H-ИНДОЛ-5-ИЛ)-2-ОКСО-ПИРРОЛИДИН-3-КАРБОНОВОЙ КИСЛОТЫ | 2020 |
|
RU2813291C2 |
| Способ получения бензанилида | 2019 |
|
RU2712053C1 |
| Способ получения ароматических амидов 1-адамантанкарбоновой кислоты | 2019 |
|
RU2698193C1 |
| НОВОЕ ПРОИЗВОДНОЕ ПИРАЗОЛ-3-КАРБОКСАМИДА, ОБЛАДАЮЩЕЕ АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ РЕЦЕПТОРА 5-НТ | 2009 |
|
RU2528406C2 |
| МОДУЛЯТОРЫ ЭСТРОГЕНОВЫХ РЕЦЕПТОРОВ | 2017 |
|
RU2738646C2 |
Изобретение относится к способу получения 4′-фторспиро[циклопропан-1,3′-индол]-2′(1′H)-она путем алкилирования 2-(2,6-дифторфенил)ацетонитрила 1,2-дибромэтаном, с последующим частичным гидролизом полученного продукта реакции до 1-(2,6-дифторфенил)циклопропан-1-карбоксамида и его внутримолекулярным N-арилированием, при этом внутримолекулярное N-арилирование проводят обработкой полученного in situ 1-(2,6-дифторфенил)циклопропан-1-карбоксамида раствором гидроксида калия в кипящем этиленгликоле. Техническим результатом является упрощение процесса получения 4′-фторспиро[циклопропан-1,3′-индол]-2′(1′H)-она и повышение чистоты образующегося продукта. 2 табл., 2 пр.
Способ получения 4′-фторспиро[циклопропан-1,3′-индол]-2′(1′H)-она путем алкилирования 2-(2,6-дифторфенил)ацетонитрила 1,2-дибромэтаном, с последующим частичным гидролизом полученного продукта реакции до 1-(2,6-дифторфенил)циклопропан-1-карбоксамида и его внутримолекулярным N-арилированием, отличающийся тем, что внутримолекулярное N-арилирование проводят обработкой полученного in situ 1-(2,6-дифторфенил)циклопропан-1-карбоксамида раствором гидроксида калия в кипящем этиленгликоле.
| RU 2007101303 A, 20.09.2008 | |||
| US 20080319204 A1, 25.12.2008 | |||
| WO 2006118955 A2, 09.11.2006 | |||
| US 20080051585 A1, 28.02.2008 | |||
| WO 2014184163 A1, 20.11.2014. |

