ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Данное изобретение касается новых пиразол-3-карбоксамидных производных. Соединения по данному изобретению являются соединениями, полезными в качестве селективных антагонистов рецептора 5-HT2B и полезными для профилактики или лечения различных заболеваний, связанных с указанным рецептором. Данное изобретение также касается фармацевтической композиции, содержащей вышеуказанные производные.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Серотонин (5-гидрокситриптамин), впервые открытый в 1948, является одним из нейромедиаторов и одним из триптаминовых производных, распространенных, в высокой концентрации, в гипоталамической области, базальном ганглии, серотонинергических нейронах ядер шва в мозге и так далее. Серотонин является химическим веществом, содержащимся в организме животных, включая человека, и биосинтезируется из триптофана. Около 10 мг серотонина обнаружено в организме человека, большая часть которого распределена в хромаффинной клетке слизистой оболочки тонкой кишки. Серотонин, синтезируемый здесь, действует на мышцы, такие как мышцы кишечника, и в значительной степени связан с двигательной активностью желудочно-кишечного тракта. Серотонин обнаружен также в центральной нервной системе и вносит вклад в психическую активность человека. Значительное внимание уделяется влиянию серотонина в повседневной жизни на психические расстройства, как было замечено, такие как депрессия и невроз. В последние годы были разработаны лекарственные препараты против указанных заболеваний с использованием лекарственных средств, оказывающих влияние на серотонин.
С другой стороны, серотонин является одним из сопряженных с G-белком рецепторов, преимущественно, в центральной нервной системе. Серотонин подразделяется на 7 семейств, от 5-HT1 до 5-HT7, и распознаны 14 подтипов. Хотя продолжаются фармакологические исследования относительно каждого подтипа (непатентная литература 1), три подтипа, 5-HT2A, 5-HT2B и 5-HT2C, установлены в семействе 5-HT2. Кроме того, сообщается о различных фармакологических воздействиях на рецептор 5-HT2B, полезных для профилактики и лечения различных заболеваний.
В целом, установлено, что антагонисты рецептора 5-HT2B полезны для профилактики или лечения различных заболеваний, включая такие, как мигрень, боль при воспалении, ноцицептивная боль, фибромиалгия, хроническая поясничная боль, висцеральная боль, гастроэзофагеальная рефлюксная болезнь (GERD), констипация, диарея, функциональное желудочно-кишечное расстройство, синдром раздраженного кишечника (сокращенно называемый здесь IBS. Определение и критерии описаны в ROME III, непатентная литература, 2), астма, остеоартрит, ревматоидный артрит, болезнь Крона, неспецифический язвенный колит, гломерулонефрит, нефрит, дерматит, гепатит, васкулит, почечная ишемия, мозговой инсульт, инфаркт миокарда, ишемия головного мозга, болезнь Альцгеймера, обратимая обструкция дыхательных путей, синдром респираторного заболевания взрослых, хроническое обструктивное заболевание легких (COPD), легочная гипертензия (PH), идиопатическая интерстициальная пневмония, бронхит, фиброз печени, криптогенный фиброзирующий альвеолит, рассеянный склероз, депрессия, тревога и ожирение (непатентная литература 3-7).
Кроме того, что касается рецепторов 5-HT2B, взаимосвязь указанного рецептора с пищеварительным аппаратом и легочной артерией изучена на основе экспериментов с использованием селективных ингибиторов 5-HT2B.
Что касается роли пищеварительного аппарата, антагонисты рецептора 5-HT2B полезны при IBS, основанном на подавлении сокращения кишечника человека путем электростимуляции (патентная литература 1). Описано, что антагонисты 5-HT2B эффективны при лечении функционального расстройства кишечника, основанного на сокращении кишечника крыс путем стимуляции серотонином (патентная литература 2). Кроме того, сообщается о снижении порога болевой чувствительности при вздутии толстой кишки у крыс, обработанных 2,4,6-тринитробензолсульфоновой кислотой (здесь далее обозначенной TNBS), что рассматривается как модель висцеральной гиперчувствительности (непатентная литература 8).
Вдобавок, антагонисты 5-HT2B снижают увеличение дефекационной массы при стрессе в модели, вызванной стрессом дефекации на крысах, обычно рассматриваемой в качестве модели IBS, что подтверждает полезность указанных антагонистов при IBS с доминирующей диареей. Вдобавок, когда крысы подвергаются стрессу, усиливается болевая реакция на вздутие толстой кишки, агонисты 5-HT2B подавляют усиление болевой реакции.
Что касается роли антагонистов в легочной артерии, описано, что установлена взаимосвязь между рецептором 5-HT2B и выздоровлением на модели легочной гипертензии у мышей с хронической гипоксией, соединения-антагонисты 5-HT2B эффективны при лечении легочной гипертензии (непатентная литература 9). Сообщается, что селективные антагонисты 5-HT2B снижают кровяное давление в исследовании на ранней фазе II пациентов с легочной гипертензией, а также с хроническим обструктивным заболеванием легких (COPD), в двойном контрольном испытании с использованием плацебо в качестве эталона (непатентная литература 10), что подтверждает безопасность и полезность для человека селективных антагонистов 5-HT2B.
СПИСОК ЦИТИРОВАННЫХ МАТЕРИАЛОВ
ПАТЕНТНАЯ ЛИТЕРАТУРА
Патентная литература 1: международная публикация 02/056010, брошюра
Патентная литература 2: публикация нерассмотренной патентной заявки Японии (перевод PCT заявки) № 1997-510216
НЕПАТЕНТНАЯ ЛИТЕРАТУРА
Непатентная литература 1: Pharmacol.Rev., 1994, 46, 157-203
Непатентная литература 2: Drossman et al., Journal of Gastrointestinal and Liver Diseases (2006) Vol.15 (3), 237-241
Непатентная литература 3: Johnson KwCephalalgia 23(2): 117-23(2003)
Непатентная литература 4: Allman JM et al, TRENDS In Cognitive Sciences 9(8): 367-373(2005)
Непатентная литература 5: Borman RA et al, Br J Pharmacol. 135 (5):114, 4-51(2002)
Непатентная литература 6: Beattie DT et al, Br J Pharmacol. 143(5):549-60(2004)
Непатентная литература 7: Kubera M et al, Psychiatry Res. 30; 134(3):251-8(2005)
Непатентная литература 8: The Journal of Pharmacology and Experimental Therapeutics, Vol.302, No.3, 1013-1022 (2002); Pharmacology (2008), 81(2), 144-150
Непатентная литература 9: Nature Medicine, 8(10): 1129-1135, 2002
Непатентная литература 10: PRX-08066: EPIX Pharmaceuticals
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
ТЕХНИЧЕСКАЯ ПРОБЛЕМА
Цель данного изобретения состоит в разработке лекарственного средства или фармацевтической композиции, содержащей в качестве активных ингредиентов соединения с селективной антагонистической активностью в отношении рецептора 5-HT2B. Кроме того, целью данного изобретения является также снижение различных неблагоприятных воздействий, к которым склонен антагонист рецептора 5-HT2B, за счет высокой селективной аффинности к рецептору и снижения взаимодействий с другими рецепторами.
РЕШЕНИЕ ПРОБЛЕМЫ
Авторы данного изобретения при решении вышеуказанной проблемы обнаружили, что новые пиразол-3-карбоксамидные производные с уникальной химической структурой проявляют селективную и сильную антагонистическую активность в отношении рецептора 5-HT2B, относящегося к числу подтипов серотониновых рецепторов. Кроме того, авторы подтвердили, что новые пиразол-3-карбоксамидные производные эффективно улучшают порог чувствительности висцеральной боли в TNBS-индуцированной модели IBS на крысах. Таким образом, новые 5-замещенные-1H-пиразол-3-карбоксамидные производные полезны для профилактики или лечения болезненных состояний, опосредованных стимуляцией вышеуказанного рецептора, таких как мигрень, боль при воспалении, ноцицептивная боль, фибромиалгия, хроническая поясничная боль, висцеральная боль, гастроэзофагеальная рефлюксная болезнь (GERD), констипация, диарея, функциональное желудочно-кишечное расстройство, синдром раздраженного кишечника (IBS), астма, остеоартрит, ревматоидный артрит, болезнь Крона, неспецифический язвенный колит, гломерулонефрит, нефрит, дерматит, гепатит, васкулит, почечная ишемия, мозговой инсульт, инфаркт миокарда, ишемия головного мозга, болезнь Альцгеймера, обратимая обструкция дыхательных путей, синдром респираторного заболевания взрослых, хроническое обструктивное заболевание легких (COPD), легочная гипертензия (PH), идиопатическая интерстициальная пневмония, бронхит, фиброз печени, криптогенный фиброзирующий альвеолит, рассеянный склероз, депрессия, тревога и ожирение.
В связи с вышеизложенным, данное изобретение завершено и предусматривает нижеперечисленные соединения или соответствующие фармацевтически приемлемые соли, указанные соединения или соответствующие фармацевтически приемлемые соли в качестве активных ингредиентов средств профилактики или лечения болезни, связанной с рецептором 5-HT2B, фармацевтические композиции, содержащие указанные соединения или соответствующие фармацевтически приемлемые соли, или способ лечения с использованием указанных соединений или соответствующих фармацевтически приемлемых солей.
А именно, данное изобретение является следующим:
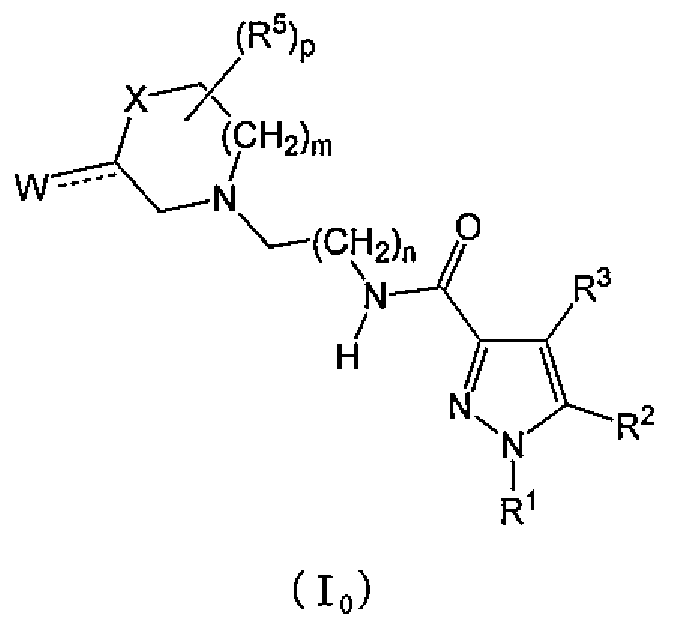
[1] Соединение следующей общей формулы (I0) или его фармацевтически приемлемая соль,
 ,
,
где
R1 означает линейную, разветвленную или циклическую низшую алкильную группу с 1-6 атомами углерода или линейную, разветвленную или циклическую галогеналкильную группу с 1-6 атомами углерода;
R2 означает (гетеро)арильную циклическую группу следующей общей формулы (Ar);
 ,
,
R3 означает водород или атом галогена;
R4 означает линейную, разветвленную или циклическую низшую алкильную группу с 1-6 атомами углерода, линейную, разветвленную или циклическую галогеналкильную группу с 1-6 атомами углерода; OH, OR1A, галоген, -(CH2)aOH, CO2H, CONH2, CONHR1A, CONR1AR1A, CN, COR1A, NH2, NHR1A, NR1AR1A, NHCOR1A, SR1A, SOR1A, SO2R1A, SO2NH2, SO2NHR1A, SO2NR1AR1A или NHSO2R1A, когда q является множественным числом, R4 может быть одинаковым или различным; когда R4 имеет два R1A, указанные заместители могут быть одинаковыми или различными, или R1A может быть объединен с другим R1A;
R5 означает линейную, разветвленную или циклическую низшую алкильную группу с 1-6 атомами углерода, -(CH2)aOH, -(CH2)aOR1B, галоген, CONH2, CONR1BR1B, COR1B, SO2R1B, -OCH2CH2NR1BR1B или линейную, разветвленную или циклическую галогеналкильную группу с 1-6 атомами углерода; когда p является множественным числом, R5 может быть одинаковым или различным, или R5 может быть объединен с другим R5;
каждый из R1A и R1B независимо означает линейную, разветвленную или циклическую низшую алкильную группу с 1-6 атомами углерода, или линейную, разветвленную или циклическую галогеналкильную группу с 1-6 атомами углерода;
a равно 0, 1 или 2;
m равно 0, 1 или 2;
n равно 1, или 2;
p равно 0, 1, 2, 3, 4 или 5 и
q равно 0, 1, 2 или 3;
X означает CH2, NH, O, S, SO, SO2, CHR5, CR5R5 (R5 является таким же, как указано выше, и может быть одинаковым или различным) или NR5 (R5 является таким же, как указано выше);
W означает атом кислорода, (H, H), (H, R5) или (R5, R5), когда X означает CH2, NH, O, CHR5, CR5R5 или NR5, или W означает (H, H), (H, R5) или (R5, R5), когда X означает S, SO или SO2; где (H, H), (H, R5) или (R5, R5) означает, что W представляет собой две одновалентные группы и указанными двумя одновалентными группами являются H и H, H и R5, R5 и R5;
Y означает NH, NR1, O или S;
каждый из Z1, Z2, Z3, Z4, Z5 и Z6 независимо означает N, C, CH или CR4 (R4 является таким же, как указано выше, и 1, 2 или 3 из Z1-Z6 могут означать атом азота).
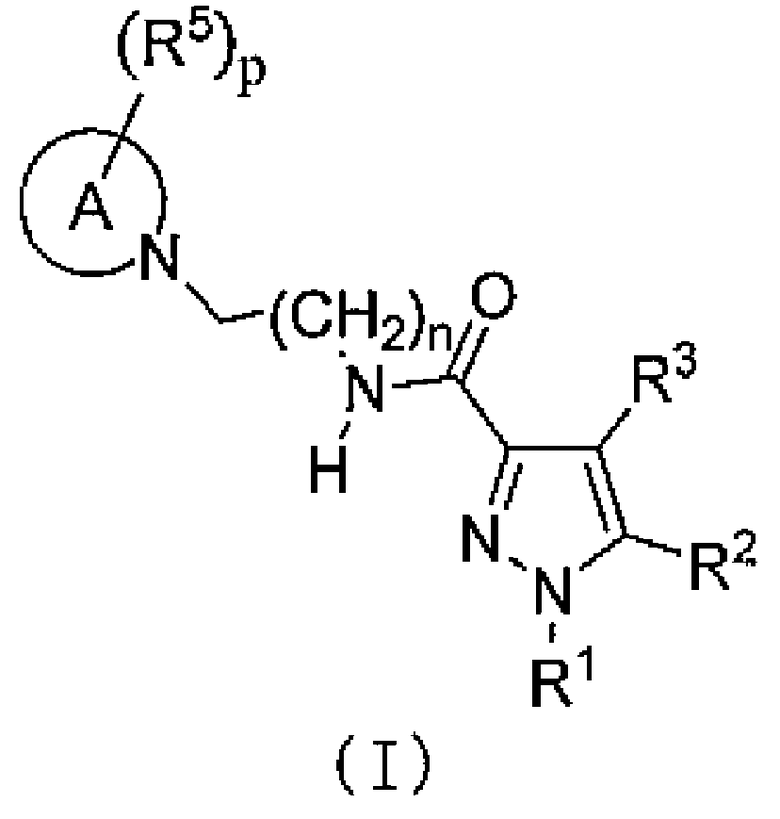
[2] Соединение следующей общей формулы (I) или его фармацевтически приемлемая соль,
 ,
,
где
A означает 3-8-членный цикл и может содержать 0-4 гетероатомов, выбранных из O, S и N;
R1 означает C1-C6-алкильную группу или C1-C6-галогеналкильную группу;
R2 означает насыщенную или частично или полностью ненасыщенную моноциклическую или бициклическую арильную группу, которая может быть замещена R4;
R3 означает водород или атом галогена;
R4 означает C1-C6-алкильную группу, C1-C6-галогеналкильную группу, OH, OR1A, галоген, -(CH2)aOH, CO2H, CONH2, CONHR1A, CONR1AR1A, CN, COR1A, NH2, NHR1A, NR1AR1A, NHCOR1A, SR1A, SOR1A, SO2R1A, SO2NH2, SO2NHR1A, SO2NR1AR1A или NHSO2R1A, когда q является множественным числом, R4 может быть одинаковым или различным; когда R4 имеет два R1A, указанные заместители могут быть одинаковыми или различными, или R1A может быть объединен с другим R1A;
R5 означает C1-C6-алкильную группу, -(CH2)aOH, -(CH2)aOR1B, галоген, CONH2, CONR1BR1B, COR1B, SO2R1B, -OCH2CH2NR1BR1B или C1-C6-галогеналкильную группу; когда p является множественным числом, R5 может быть одинаковым или различным, или R5 может быть объединен с другим R5;
каждый из R1A и R1B независимо означает C1-C6-алкильную группу или C1-C6-галогеналкильную группу;
a равно 0, 1 или 2;
m равно 0, 1 или 2;
n равно 1 или 2;
p равно 0, 1, 2, 3, 4 или 5 и
q равно 0, 1, 2 или 3.
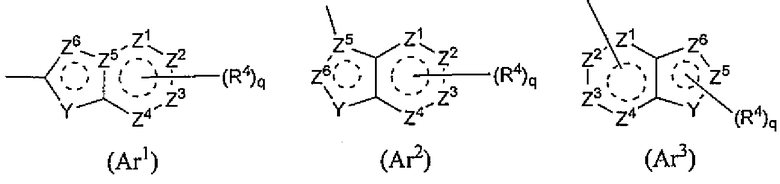
[3] Соединение или его фармацевтически приемлемая соль, как описано в приведенном выше пункте [2], где R2 означает следующие Ar1, Ar2, Ar3 или Ar4,
 ,
,
где,
R4 и q такие же, как указано в приведенном выше пункте [2];
Y означает NH, NR6, O или S;
каждый из Z1, Z2, Z3, Z4, Z5 и Z6 независимо означает N, C, CH или CR4 (1, 2 или 3 из Z1-Z6 могут представлять собой атом азота); и
R6 означает водород, C1-C6-алкильную группу, C1-C6-галогеналкильную группу, C1-C6-алкокси-C1-C6-алкильную группу, гидроксил-C1-C6-алкильную группу, галоген-C1-C6-алкокси-C1-C6-алкильную группу, ди-C1-C6-алкиламино-C1-C6-алкильную группу, моно-C1-C6-алкиламино-C1-C6-алкильную группу, амино-C1-C6-алкильную группу, C3-C8-цикло-C1-C6-алкильную группу (указанная C3-C8-цикло-C1-C6-алкильная группа может быть замещена 1 или 2 группами, каждую из которых независимо выбирают из гидрокси, C1-C6-алкокси и C1-C6-ацилокси, и может содержать S (серу), O (кислород) или NR1), аминокарбонил-C1-C6-алкильную группу, моно-C1-C6-алкиламинокарбонил-C1-C6-алкильную группу, ди-C1-C6-алкиламинокарбонил-C1-C6-алкильную группу, гидроксикарбонил-C1-C6-алкильную группу или C1-C6-алкилсульфонильную группу,
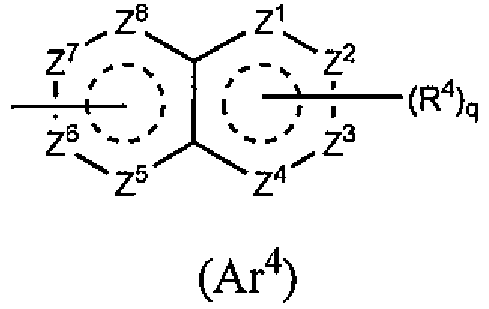 ,
,
где
R4 и q такие же, как указано в приведенном выше пункте [2]; и
каждый из Z1, Z2, Z3, Z4, Z5, Z6, Z7 и Z8 независимо означает N, C, CH или CR4 (1, 2 или 3 из Z1-Z8 могут представлять собой атом азота).
[4] Соединение или его фармацевтически приемлемая соль, как описано в приведенном выше пункте [3], где Ar1, Ar2, Ar3 или Ar4 представлены следующей общей формулой:
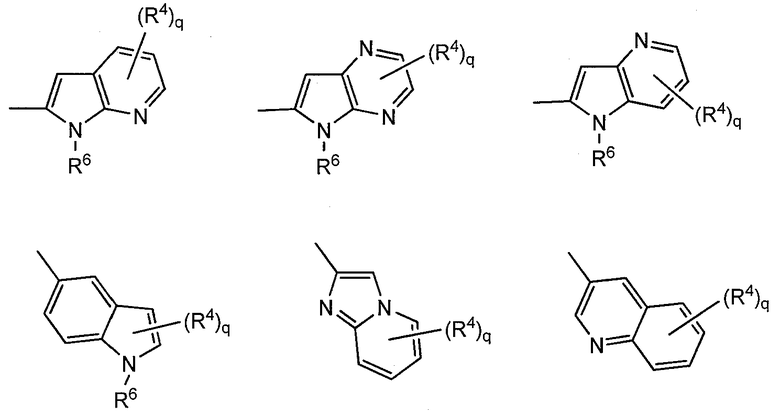 ,
,
где
R4 и q такие же, как указано в приведенном выше пункте [2];
R6 означает водород или C1-C6-алкильную группу и
(R4)q может замещать один из двух циклов или оба цикла.
[5] Соединение или его фармацевтически приемлемая соль, как описано в приведенном выше пункте [2], где цикл A представляет собой морфолин, пиперидин, пирролидин или азетидин, который связан по N;
n равно 1;
p равно 0, 1 или 2 и
q равно 0, 1 или 2.
[6] Соединение или его фармацевтически приемлемая соль, как описано в приведенном выше пункте [2], где соединение, представленное общей формулой (I), выбирают из группы, включающей
1-метил-N-[2-(морфолин-4-ил)этил]-5-(хинолин-3-ил)-1H-пиразол-3-карбоксамид;
1-метил-5-{5-метил-1H-пирроло[3,2-b]пиридин-2-ил}-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид;
1-метил-N-[2-(морфолин-4-ил)этил]-5-{1H-пирроло[2,3-b]пиридин-2-ил}-1H-пиразол-3-карбоксамид;
1-метил-N-[2-(морфолин-4-ил)этил]-5-{7H-пирроло[2,3-d]пиримидин-6-ил}-1H-пиразол-3-карбоксамид;
1-метил-N-[2-(морфолин-4-ил)этил]-5-[5-(трифторметил)-1H-пирроло[3,2-b]пиридин-2-ил]-1H-пиразол-3-карбоксамид;
1-метил-5-{5-метил-1H-пирроло[2,3-b]пиридин-2-ил}-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид;
5-{5-фтор-1H-пирроло[2,3-b]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид;
5-{5-циано-1H-пирроло[3,2-b]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид;
5-{6-фтор-1H-пирроло[3,2-b]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид;
1-метил-N-[2-(морфолин-4-ил)этил]-5-{5H-пирроло[2,3-b]пиразин-6-ил}-1H-пиразол-3-карбоксамид;
5-{5-циано-1H-пирроло[2,3-b]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид;
5-{5-фтор-1-метил-1H-пирроло[2,3-b]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид;
N-[2-(3,3-дифторазетидин-1-ил)этил]-5-(5-фтор-1H-индол-2-ил)-1-метил-1H-пиразол-3-карбоксамид;
N-[2-(азетидин-1-ил)этил]-5-(5-фтор-1H-индол-2-ил)-1-метил-1H-пиразол-3-карбоксамид;
1-метил-5-(2-метил-1H-индол-5-ил)-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид;
5-(1,2-диметил-1H-индол-5-ил)-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид;
5-[1-(2-метоксиэтил)-1H-индол-3-ил]-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид;
5-(4-ацетамидо-1H-индол-2-ил)-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид;
5-{имидазо[1,2-a]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид;
5-{6-фторимидазо[1,2-a]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид;
5-{7-фторимидазо[1,2-a]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид;
5-{6-цианоимидазо[1,2-a]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид;
N-[2-(3,3-дифторазетидин-1-ил)этил]-1-метил-5-(хинолин-3-ил)-1H-пиразол-3-карбоксамид;
N-[2-(3,3-дифторазетидин-1-ил)этил]-1-метил-5-{1H-пирроло[2,3-b]пиридин-2-ил}-1H-пиразол-3-карбоксамид и
5-{7-цианоимидазо[1,2-a]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид.
[7] Промежуточное соединение для соединения, описанного в приведенном выше в пункте [2], представленное общей формулой (1A):
 ,
,
где каждое определение такое же, как указано в приведенном выше пункте [2].
[8] Промежуточное соединение для соединения, описанного в приведенном выше в пункте [2], представленное общей формулой (IB):
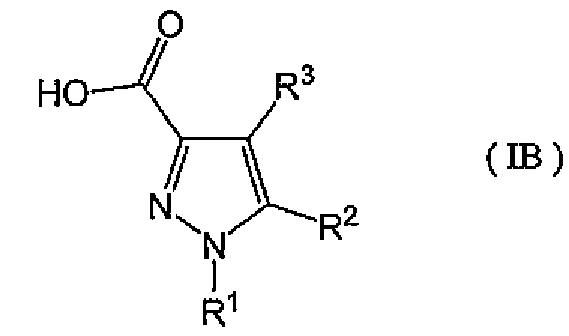 ,
,
где R1, R2, R3 такие же, как указаны для формулы (I), и OH карбоновой кислоты может быть заменен удаляемым заместителем.
[9] Профилактическое или терапевтическое средство от заболеваний, в которые вовлечены рецепторы 5-HT2B, где эффективным ингредиентом является соединение или его фармацевтически приемлемая соль по любому из п.п. [2]-[6].
[10] Фармацевтическая композиция, содержающая соединение или его фармацевтически приемлемую соль, по любому из п.п. [2]-[6], и фармацевтически приемлемый носитель.
[11] Фармацевтическая композиция для профилактики или лечения болезненного состояния, опосредованного рецепторами 5-HT2B, у млекопитающего, содержащая эффективное количество соединения или его фармацевтически приемлемой соли, по любому из п.п. [2]-[5], и фармацевтически приемлемый носитель.
[12] Фармацевтическая композиция, содержащая соединение по любому из п.п. [2]-[6] и дополнительно содержащая другой фармакологически активный агент.
[13] Соединение или его фармацевтически приемлемая соль по любому из п.п. [2]-[6] применяемая в профилактике или лечении болезненного состояния, опосредованного рецепторами 5-HT2B.
[14] Применение соединения или его фармацевтически приемлемой соли по любому из п.п. [2]-[6], в изготовлении лекарственного средства для профилактики или лечения состояния, опосредованного рецепторами 5-HT2B.
[15] Способ профилактики или лечения таких заболеваний, как мигрень, боль при воспалении, ноцицептивная боль, невропатическая боль, фибромиалгия, хроническая поясничная боль, висцеральная боль, гастроэзофагеальная рефлюксная болезнь (GERD), констипация, диарея, функциональное желудочно-кишечное расстройство, синдром раздраженного кишечника, астма, остеоартрит, ревматоидный артрит, болезнь Крона, неспецифический язвенный колит, гломерулонефрит, нефрит, дерматит, гепатит, васкулит, почечная ишемия, мозговой инсульт, инфаркт миокарда, ишемия головного мозга, болезнь Альцгеймера, обратимая обструкция дыхательных путей, синдром респираторного заболевания взрослых, хроническое обструктивное заболевание легких (COPD), легочная гипертензия (PH), идиопатическая интерстициальная пневмония, бронхит, фиброз печени, криптогенный фиброзирующий альвеолит, рассеянный склероз, депрессия, тревога или ожирение, включающий введение человеку или млекопитающему эффективного количества фармацевтической композиции, содержащий соединение или его фармацевтически приемлемую соль по любому из п.п.[2]-[6] и фармацевтически приемлемый носитель.
ПОЛЕЗНЫЕ ЭФФЕКТЫ ИЗОБРЕТЕНИЯ
Эффективный ингредиент, пиразол-3-карбоксамидные производные по данному изобретению, имеет новые ядра и ингибирует сильно и селективно функцию рецептора 5-HT2B. Сильная антагонистическая активность в отношении рецептора 5-HT2B лекарственного средства по данному изобретению проявляется в терапевтических действиях, основанных на превосходных фармацевтических эффектах. Вдобавок, высокая селективность лекарственного средства по данному изобретению полезна для ослабления широкого ряда побочных эффектов, основанных на активностях в отношении рецепторов, иных, чем рецептор 5-HT2B.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг. 1 представляет собой график результатов исследования вздутия толстой кишки с использованием модели индуцированного TNBS IBS на крысах с помощью соединения по примеру 24.
ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
Соединение по данному изобретению отличается специфическими активностями связывания рецептора 5-HT2B. Соединение по данному изобретению селективно ингибирует активности рецептора 5-HT2B путем связывания антагонистически с рецептором 5-HT2B, что полезно для лечения или предварительного лечения млекопитающего, от заболевания, связанного с указанным рецептором.
Термин “антагонистический агент" также означает антагонист и подразумевает, что лекарственное средство действует антагонистически против агониста и снижает эффекты. Способность к частичному связыванию таких антагонистов и агонистов называют аффинностью связывания, и оценку аффинности связывания, как в описанных ниже примерах, проводят путем сравнения величины Ki, рассчитанной при изучении рецепторного связывания in vitro, или величин IC50, рассчитанных при проведении исследования рецепторного связывания в тех же условиях, в некоторых случаях.
При изучении рецепторного связывания, когда IC50 не может быть рассчитано из-за непроявления достаточных антагонистических активностей, IC50 соединения можно расценивать как превышающее указанную концентрацию.
Соединение по данному изобретению обладает аффинностью связывания с рецептором 5-HT2B, и величина IC50, которая демонстрирует активность ингибирования серотонина (ингибирующая активность), предпочтительно составляет ниже 1000 нМ, более предпочтительно, ниже 100 нМ, еще более предпочтительно, ниже 10 нМ и, наиболее предпочтительно, ниже 1 нМ.
Согласно ингибирующей активности в отношении 5-HT2B по сравнению с другими рецепторами соединение по данному изобретению или его фармацевтически приемлемая соль подходит в качестве “селективного”. “Селективный” означает, что ингибирующая активность в отношении указанного рецептора выше, чем ингибирующие активности в отношении “других рецепторов”. “Селективный” по данному изобретению означает, что величина IC50 активности ингибирования указанного рецептора составляет одну десятую или менее, предпочтительно, одну сотую или менее и, более предпочтительно, одну тысячную или менее, по сравнению с величиной IC50 для “других рецепторов”.
“Другие рецепторы“ означает здесь остальные рецепторы, указанные для имеющихся неселективных серотониновых антагонистов. В частности, после определения селективностей в отношении 5-HT2A, 5-HT2C, оценка характерных соединений по влиянию на имеющийся рецептор и ферменты является положительной.
Ингибирующие активности или рецепторные антагонистические активности селективных в отношении 5-HT2B антагонистов по данному изобретению легко можно оценить по известным методикам, упомянутым ниже.
В данном контексте, термин "C1-C6", как указано в вышеупомянутой общей формуле, если не указано иное, означает линейную или разветвленную углеродную цепь с 1-6 атомами углерода. Таким образом, " C1-C6-алкильная группа" означает алкильную группу с 1-6 атомами углерода, включающую, предпочтительно, метил (здесь далее иногда обозначаемый Me), этил (здесь далее иногда обозначаемый Et), пропил, изопропил, бутил, изобутил, трет-бутил.
"Галоген" означает 17 группу периодической таблицы, включая, предпочтительно, F, Cl, Br или I.
"Галогеналкильная группа" означает C1-C6-алкильную группу, замещенную 1-5 атомом (атомами) галогена.
"Арильный цикл" означает моно- или бициклическое кольцо, которое может быть насыщенным или частично или полностью ненасыщенным. Арил означает заместитель, который связан по месту одного ушедшего от арильного цикла атома водорода, включая, предпочтительно, Ar1, Ar2, Ar3 и Ar4.
Ненасыщенная моноциклическая кольцевая группа включает, например, как упомянуто, фенил, пиразолил, фурил, тиенил, оксазолил, тетразолил, тиазолил, имидазолил, тиадиазолил, пиридил, пиримидинил, пирролил, тиофенил, пиразинил, пиридазинил, изоксазолил, изотиазолил, триазолил, фуразанил.
Ненасыщенная бициклическая кольцевая группа включает, например, как упомянуто, нафтил, бензофуранил, изобензофуранил, бензотиофенил, индолил, изоиндолил, бензоксазолил, бензотиазолил, индазолил, бензимидазолил, хинолил, изохинолил, циннолинил, фталазинил, хиназолинил, хиноксалинил.
Пример насыщенной циклической группы включает цикл, являющийся частично насыщенным или полностью насыщенным в ненасыщенной части вышеуказанных моно- или бициклических кольцевых групп.
“R1A может быть объединен с другим R1A” означает, что NR1AR1A, такой как NR1AR1A, CONR1AR1A и SO2NR1AR1A, может обозначать указанной комбинацией 3-13-членную углерод-содержащую циклическую группу (напр., r равно 1-12 в нижеследующей схеме (IIa)).
Из числа указанных групп удобной является 3-8-членная углерод-содержащая циклическая группа (напр., r равно 1-6 в нижеследующей схеме (IIa)). Фактически, CONR1AR1A и NR1AR1A в R4 могут быть предусмотрены нижеследующей схемой (IIa). Тем не менее, вид связывания не ограничен только в нижеследующей схеме.
“R1B может быть объединен с другим R1B” имеет то же самое значение, что указано выше при замене R1A на R1B.
Удаляемые заместители иллюстрируются такими группами, как этокси, фенокси, галоген, алкоксикарбонилокси, арилоксикарбонилокси, имидазол-1-ил, 4-нитрофенокси, но не ограничиваются только перечисленными группами.

Соли соединения формулы (I) являются фармацевтически приемлемыми солями и включают кислотно-аддитивные и основно-аддитивные соли (включая дикислотные соли и диосновную соль).
Как правило, подходящие кислотно-аддитивные соли получают из кислот, которые образуют нетоксичные соли. Примеры включают такие соли, как ацетат, аспартат, бензоат, безилат, бикарбонат/карбонат, бисульфат/сульфат, борат, камзилат, цитрат, эдизилат, эзилат, формиат, фумарат, глюцептат, глюконат, гексафторфосфат, гибензат, гидрохлорид, гидробромид, гидроиодид, изетионат, лактат, малат, малеат, малонат, мезилат, метилсульфат, нафтилат, 2-напсилат, никотинат, нитрат, оротат, оксалат, пальмитат, памоат, фосфат/гидрофосфат/дигидрофосфат, сахарат, стеарат, сукцинат, тартрат, тозилат и трифторацетат.
Подходящие основные соли получают из оснований, которые образуют нетоксичные соли. Примеры основных солей включают соли алюминия, аргинина, бензатина, кальция, холина, диэтиламина, диоламина, глицина, лизина, магния, меглумина, оламина, калия, натрия, трометамина и цинка. Смотри, при необходимости, Handbook of Pharmaceutical Salts: Properties, Selection, and Use by Stahl and Wermuth (Wiley-VCH, Weinheim, Germany, 2002).
Фармацевтически приемлемые соли соединений формулы (I) легко могут быть получены смешением раствора требуемой кислоты или требуемого основания. Образующиеся соли могут быть осаждены и собраны фильтрованием, или могут быть выделены выпариванием растворителя. Степень ионизации в полученных солях может изменяться от полностью ионизованной до почти неионизованной.
Соединения по изобретению могут существовать как в несольватированной, так и сольватированной формах. Термин “сольват” используется здесь для обозначения молекулярного комплекса, включающего соединение по изобретению и стехиометрическое количество, одну или несколько молекул, фармацевтически приемлемого растворителя, например, этанола. Термин “гидрат” используется, когда указанным растворителем является вода.
Фармацевтически приемлемые сольваты по изобретению включают те сольваты, в которых растворитель кристаллизации может быть изотопно замещенным, напр., D2O, d6-ацетон, d6-диметилсульфоксид.
В объем изобретения включены комплексы, такие как клатраты, комплексы включения лекарственное вещество-хозяин, где, в противоположность вышеупомянутым сольватам, лекарственное вещество и хозяин присутствуют в стехиометрических или нестехиометрических количествах. Кроме того, в объем изобретения входят комплексы лекарственного вещества, содержащие два или несколько органических и/или неорганических компонента, которые могут быть в стехиометрических или нестехиометрических количествах. Образующиеся комплексы могут быть ионизованными, частично ионизованными или неионизованными. Смотри, при необходимости, J Pharm Sci, 64 (8), 1269-1288, by Haleblian (August 1975).
Здесь далее все ссылки на соединения формулы (I) включают ссылки на соответствующие соли, сольваты и комплексы указанных соединений и сольваты и комплексы указанных солей.
Соединения по изобретению включают соединения формулы (I), как определено выше, включая полиморфы и габитусы кристаллов, пролекарства и изомеры (включая оптические, геометрические и таутомерные изомеры), как определено ниже, и изотопно-меченные соединения формулы (I).
Как указано, так называемые “пролекарства” соединений формулы (I) или соответствующих солей также входят в объем изобретения. При этом некоторые производные соединений формулы I, которые могут обладать низкой фармакологической активностью или не обладать фармакологической активностью сами по себе, могут, при введении в организм, превращаться в соединения формулы (I), обладающие требуемой активностью, например, путем гидролитического расщепления. Такие производные носят название “пролекарства”. Дополнительную информацию по применению пролекарств можно найти в Pro-drugs as Novel Delivery Systems, Vol. 14, ACS Symposium Series (T. Higuchi and W. Stella) и Bioreversible Carriers in Drug Design, Pergamon Press, 1987 (ed. E. B. Roche, American Pharmaceutical Association).
Пролекарства по данному изобретению могут быть, например, получены заменой соответствующих функциональностей, присутствующих в соединениях формулы (I), некоторыми группами, известными специалистам в данной области под названием “прогруппы”, как указано, например, в Design of Prodrugs by H. Bundgaard (Elsevier, 1985).
Некоторые примеры пролекарств по изобретению включают:
когда соединение формулы (I) или соль указанного соединения содержит функциональность карбоновой кислоты (-COOH), соответствующий сложный эфир и, например, этиловый эфир, фениловый эфир, карбоксиметиловый эфир, диметиламинометиловый эфир, пивалоилоксиметиловый эфир, этоксикарбонилоксиэтиловый эфир, фталидиловый эфир, (5-метил-2-оксо-1,3-диоксолен-4-ил)метиловый эфир, 1-(циклогексилоксикарбонилокси)этиловый эфир, метиламид) и тому подобное;
когда соединение формулы (I) или соль указанного соединения содержит спиртовую функциональность (-OH), соединение, в котором гидрокси-функциональность подвергнута ацилированию, алкилированию, фосфорилированию и борированию, например, ацетил-соединение, пальмитоил-соединение, пропаноил-соединение, пивалоил-соединение, сукцинил-соединение, аланил-соединение, диметиламинометилкарбонил-соединение и тому подобное. Вдобавок, в зависимости от заместителей, пролекарство может образовывать N-оксид. Такие N-оксиды также включены в рамки объема изобретения;
когда соединение формулы (I) или соль указанного соединения содержит амино-функциональность, соответствующий амид, например, соединение, в котором, в зависимости от конкретного случая, один или оба водорода амино-функциональности подвергнуты ацилированию, алкилированию и фосфорилированию, например, эйкозанонил-соединение, аланил-соединение, пентиламинокарбонил-соединение, (5-метил-2-оксо-1,3-диоксолен-4-ил)метоксикарбонил-соединение, тетрагидрофуранил-соединение, пирролидинилметил-соединение, трет-бутил-соединение и тому подобное.
Дополнительные примеры замещающих групп, в соответствии с вышеупомянутыми примерами, и примеры других типов пролекарств могут быть найдены в вышеприведенных ссылках. Кроме того, некоторые соединения формулы I могут сами по себе действовать как пролекарства других соединений формулы (I).
Соединения формулы (I), содержащие один или несколько асимметрических атомов углерода, могут существовать в виде двух или нескольких стереоизомеров. Когда соединение общей формулы (I) содержит алкенильную или алкениленовую группу, возможны геометрические цис/транс- (или Z/E-) изомеры. Когда соединение содержит, например, кето- или оксимгруппу или ароматическую группу, может возникать таутомерный изомеризм (“таутомеризм”). Отсюда следует, что отдельное соединение может обладать несколькими типами изомеризма.
В объем данного изобретения включены все стереоизомеры, геометрические изомеры и таутомерные формы соединений общей формулы (I), включая соединения, обладающие более чем двумя типами изомеризма, и смеси одной или нескольких изомерных форм. Также включены кислотно-аддитивные или основно-аддитивные соли, в которых противоин является оптически активным, например, D-лактат или L-лизин, или рацемическим, например, DL-тартрат или DL-аргинин.
Цис/транс-изомеры могут быть разделены общепринятыми методами, хорошо известными специалистам в данной области, например, хроматографией или фракционированной кристаллизацией. Общепринятые способы получения/выделения индивидуальных энантиомеров включают хиральный синтез из подходящего оптически чистого предшественника или разделение рацемата (либо рацемата соли или производного) с применением, например, хиральной высокоэффективной жидкостной хроматографии (ВЭЖХ).
Альтернативно, рацемат (или рацемический предшественник) может быть подвергнут взаимодействию с подходящим оптически активным соединением, например, спиртом, или, в случае, когда соединение общей формулы (I) содержит кислотную или основную группу, с кислотой или основанием, такими как винная кислота или 1-фенилэтиламин. Образующаяся диастереомерная смесь может быть разделена хроматографией и/или фракционированной кристаллизацией, и один или оба диастереоизомера превращены в соответствующий чистый энантиомер(ы) хорошо известными специалисту способами.
Хиральные соединения по изобретению (и соответствующие хиральные предшественники) могут быть получены в энантиомерно-обогащенной форме с применением хроматографии, обычно ВЭЖХ, на асимметрической смоле с подвижной фазой, состоящей из углеводорода, как правило, гептана или гексана, содержащего от 0 до 50 (масс./масс.)% изопропанола, обычно, от 2 до 20 (масс./масс.)%, и от 0 до 5 (масс./масс.)% алкиламина, обычно, 0,1 (масс./масс.)% диэтиламина. Концентрация элюата дает обогащенную смесь.
Стереоизомерные конгломераты могут быть разделены общепринятыми способами, известными специалисту в данной области - смотри, например, Stereochemistry of Organic Compounds by E L Eliel (Wiley, New York, 1994).
Данное изобретение включает все фармацевтически приемлемые изотопно-меченные соединения общей формулы (I), в которых один или несколько атомов заменены атомами, имеющими тот же самый атомный номер, но атомную массу или массовое число, отличные от атомной массы или массового числа, обычно существующих в природе.
Примеры изотопов, пригодных для включения в соединения по изобретению, включают изотопы водорода, такие как 2H и 3H, углерода, такие как 11C, 13C и 14C, хлора, такие как 36Cl, фтора, такие как 18F, иода, такие как 123I и 125I, азота, такие как 13N и 15N, кислорода, такие как 15O, 17O и 18O, фосфора, такие как 32P, и серы, такие как 35S.
Замещение более тяжелыми изотопами, такими как дейтерий, т.е., 2H, может давать определенные терапевтические преимущества, возникающие вследствие большей метаболической стабильности, например, увеличенный период полураспада in vivo или пониженная требуемая дозировка, и, следовательно, в некоторых случаях, может быть более предпочтительно, чем 1H- нормальное соединение.
Замещение позитронно-активными изотопами, такими как 11C, 18F, 15O и 13N, может быть использовано в позитронно-эмиссионных томографических (PET) исследованиях для оценки степени занятости рецептора субстратом.
Отдельные изотопно-меченные соединения формулы (I), например, соединения, включающие радиоактивный изотоп, полезны в исследованиях распределения лекарственного вещества и/или субстрата в тканях. Радиоактивные изотопы трития, т.е., 3H, и углерода-14, т.е., 14C, в особенности полезны для указанной цели в связи с легкостью включения и легких способов детектирования.
Все соединения общей формулы (I) могут быть получены способами, описанными в приведенных ниже общих способах, или конкретными способами, описанными в разделе Примеры и разделе Получения, или определенными модификациями соответствующих способов. Данное изобретение также охватывает любой отдельный или ряд таких способов получения соединений общей формулы (I) и, кроме того, любых используемых здесь новых промежуточных соединений.
Соединение общей формулы (I) по данному изобретению может быть получено известным способом получения или может быть получено по общей методике или способу получения, представленным следующей реакционной схемой. Если не указано иное, R1-R5 и X, Y и Z в приведенных ниже способах имеют вышеуказанные значения. Термин “защитная группа”, как использован ниже, означает гидроксил- или амино-защитную группу, выбранную из типичных гидрокси-, ацетилен- или амино-защитных групп, описанных в Protective Groups in Organic Synthesis edited by T. W. Greene et al. (John Wiley & Sons, 1999). Кроме того, каждое соединение, указанное в реакционной схеме, кроме тех случаев, когда соединение ингибирует взаимодействие, может образовывать соль, которая включает такую же соль, как соединение (I). Пролекарство по данному изобретению может быть получено введением специфической группы на стадии промежуточного соединения или при осуществлении взаимодействия с использованием полученного соединения, сходного с вышеуказанной защитной группой. Взаимодействие, такое как этерификация, амидирование и дегидратация, может быть выполнено с применением стандартных методик, хорошо известных специалисту в данной области.
Получение лекарственных составов для парентерального введения в стерильных условиях, например, лиофилизацией, легко может быть выполнено с использованием стандартных фармацевтических методик, хорошо известных специалисту в данной области.
Получение соединения формулы IA из соединения формулы II по процессу A-2 (способ 1) и получение соединения формулы IA из соединения формулы II по процессу A-3 (Способ 2) представлено ниже.
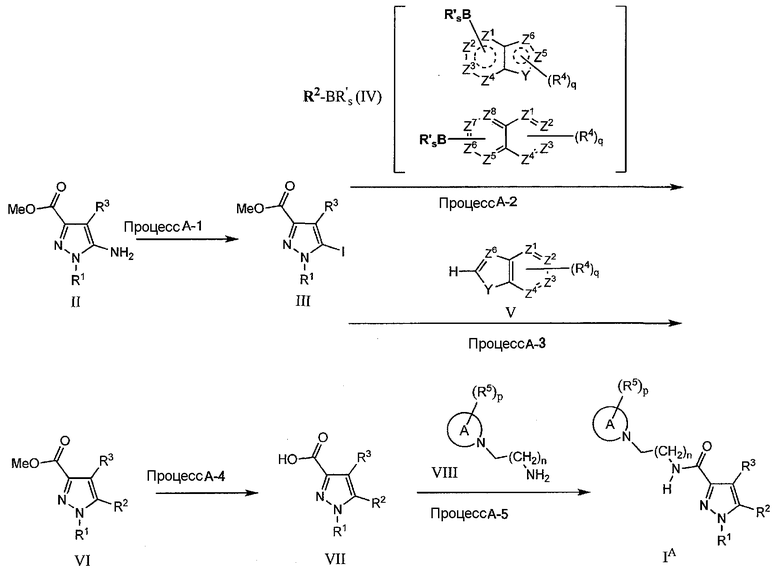
В условном обозначении R'sB, R' означает OH, O-низший алкил, низший алкил или фтор, и s равно 2 или 3, B означает атом бора. Как конкретное изображение заместителя, указаны (OH)2B, (O-низший алкил)2B, (низший алкил)2B, трифторборат калия (BF3 -)(BF3K), но когда в случае (O-низший алкил)2B между низшими алкильными группами может образовываться циклическое кольцо.
Процесс A-1
На данной стадии, содержащие иод соединения формулы III могут быть получены однореакторным синтезом в присутствии подходящих агентов иодирования через соли диазония, либо после образования солей диазония указанные соединения могут быть получены присоединением подходящих агентов иодирования. Получение солей диазония может быть произведено по известной методике. Согласно типичной методике, получение диазония осуществляют с использованием нитрита натрия в кислотном растворе. В качестве кислотного раствора, может быть использован, например, раствор уксусной кислоты, соляной кислоты, муравьиной кислоты или серной кислоты, где предпочтительной является уксусная кислота. Взаимодействие протекает от 10 минут до 12 часов, но, как правило, от 30 минут до 6 часов. Реакционная температура отвечает диапазону приблизительно от -20°C до 30°C, но, как правило, от -10°C до 5°C. Подходящим агентом иодирования является иодид калия, иодид натрия или иодид, где иодид калия является предпочтительным. В реакционной схеме, Me означает метильную группу (то же самое ниже).
Процесс A-2
На данной стадии, соединение (VI) может быть получено с использованием реакции кросс-сочетания арила с соединением (III), полученным в процессе A-1. Указанное соединение (VI) может быть получено в условиях сочетания в присутствии подходящего катализатора на основе переходного металла и основания (или без основания) в смеси вода-органический растворитель. В качестве подходящего R'sB заместителя в реагенте на основе арилметалла, упомянуты, например, (OH)2B, (O-низший алкил)2B, (низший алкил)2B, калиевая соль (BF3K) трифторбората(BF3 -), но в случае (O-низший алкил)2B, между низшими алкильными группами может быть образовано циклическое кольцо.
В качестве катализатора на основе переходного металла подходящим является, например, тетракис(трифенилфосфин)палладий, бис(трифенилфосфин)палладий(II)хлорид, медь(0), ацетат меди(I), бромид меди(I), хлорид меди(I), иодид меди(I), оксид меди(I), трифторметансульфонат меди(I), ацетат меди(II), бромид меди(II), хлорид меди(II), иодид меди(II), оксид меди(II), медь(II)трифторметансульфонат(II), ацетат палладия(II), хлорид палладия(II), бис(ацетонитрил)дихлорпалладий(II), бис(дибензилиденацетон)палладий(0), трис(дибензилиденацетон)дипалладий(0), [1,1'-бис(дифенилфосфино)ферроцен]палладий(II)дихлорид и тому подобное. В особенности подходящими являются тетракис(трифенилфосфин)палладий, бис(трифенилфосфин)палладий(II)хлорид, ацетат палладия(II), бис(ацетонитрил)дихлорпалладий(II), трис(дибензилиденацетон)дипалладий(0), [1,1'-бис(дифенилфосфино)ферроцен]палладий(II)дихлорид. В качестве реагента на основе арилметалла упомянуты, например, но не в порядке ограничения, реагенты на основе бороновой кислоты, такие как производное 2-индолилбороновой кислоты, и реагенты на основе эфиров бороновой кислоты, такие как производное эфира 2-индолилбороновой кислоты. В качестве подходящего органического растворителя в смешанном водно-органическом растворе, например, в присутствии или отсутствии водорастворимого основания, такого как раствор гидроксида калия, гидроксида натрия, гидроксида лития и карбоната калия, упомянуты тетрагидрофуран, 1,4-диоксан, N,N-диметилформамид (ДМФА), ацетонитрил, спирты, такие как метанол и этанол, галогенированные углеводороды, такие как дихлорметан, 1,2-дихлорэтан, хлороформ, тетрахлорид углерода; или диэтиловый эфир. Указанное взаимодействие может быть выполнено при наличии соответствующих дополнительных факторов. В качестве такого дополнительного фактора упомянуты, например, трифенилфосфин, тритрет-бутилфосфин, 1,1'-бис(дифенилфосфино)ферроцен, три-2-фурилфосфин, 2-(дихлоргексилфосфино)бифенил, трифениларсин, хлорид тетрабутиламмония, фторид тетрабутиламмония, уксуснокислый литий, хлорид лития, триэтиламин, метилат калия (или натрия), гидроксид натрия, карбонат натрия, фосфат калия, карбонат цезия, бикарбонат натрия или иодид натрия. Данное взаимодействие осуществляют приблизительно при 0°C-200°C и, как правило, приблизительно при 20°C-120°C. Период взаимодействия составляет примерно от 5 минут до 96 часов и, как правило, примерно от 30 минут до 24 часов. Кроме того, во время взаимодействия может быть использован микроволновый реактор. Вдобавок, когда Y означает NH, атом азота может быть защищен низшей алкоксикарбонильной группой (напр., группой Boc) и (п-алкил)бензолсульфонильной группой (напр., бензолсульфонильной и п-толуолсульфонильной группой).
Может быть использована иная, чем вышеуказанная реакция перекрестного сочетания Сузуки-Мияура, реакция перекрестного сочетания Стилле, с использованием триалкилолова вместо R'sB заместителя, и реакция перекрестного сочетания Негиши, с использованием цинк-галогена, где в качестве галогена упомянуты хлор, бром, иод, вместо R'sB заместителя.
Процесс A-3
На данной стадии, гетероциклическое соединение (VI), отвечающее общей формуле R2, может быть получено превращением с образованием сложного арилборонатного эфира по реакции C-H борилирования между пинаколбораном (HBpin) или бис(пинаколат)дибораном (B2pin2, pin=Me4C2O2) и гетероциклическим соединением (V) в присутствии подходящего катализатора на основе переходного металла (напр., иридия) и соответствующего органического растворителя. (C-H borylation; T. Ishiyama et al., Organic Synthesis (2005), 82, 126-133.) Продукт сочетания-соединение (VI) может быть получен по Сузуки-Мияура взаимодействием образовавшегося сложного арилборонатного эфира с соединением (III). Указанные взаимодействия могут быть выполнены по одностадийной или двухстадийной реакционной методике.
В качестве катализатора на основе переходного металла могут быть упомянуты, например, [Ir(OMe)(COD)]2 (COD означает 1,5-циклооктадиен), Cp*Rh(η4-C6Me6) (Cp* означает C5Me5), Ir(η5-C9H7)(COD), [IrCl(COD)]2, [IrCl(COE)2]2 или RhCl{P(i-Pr)3}(N2). В качестве вспомогательного компонента, например, 1,2-бис(диметилфосфино)этан(dmpe), 2,2'-бипиридин-(dpy), 4,4'-ди-трет-бутил-2,2'-бипиридин-(dtbpy) или dppe. В качестве подходящего органического растворителя упомянуты, например, углеводороды, такие как н-гексан, или циклогексан. Удобный вариант получения состоит в использовании в качестве катализатора комбинации 1/2[IrCl(COD)]2 и 4,4'-ди-трет-бутил-2,2'-бипиридина-(dtbpy) в гексане при взаимодействии пинаколборана или бис(пинаколат)диборана с арильным соединением. После чего реакционные сложные арилборонатные эфиры, полученные как указано выше с соединением (III), переносят на соединение (VI) по реакции Сузуки-Мияура. Указанное взаимодействие является, по существу, таким же, как описано в процессе A-2. Могут быть использованы те же реагенты и реакционные условия в процессе A-3, который подобен вышеуказанному процессу A-2. При условии, что данное взаимодействие осуществляют как одностадийный процесс по реакции Сузуки-Мияура, целесообразной является комбинация N,N-диметилформамида (ДМФА) или 1,4-диоксана в качестве растворителя, твердого фосфата калия (K3PO4) в качестве основания, [1,1'-бис(дифенилфосфино)ферроцен]палладий(II)дихлорида (PdCl2(dppf)) в качестве палладиевого катализатора.
Описанное выше C-H борилирование, сопровождаемое реакцией прямого введения бициклической гетероарильной группы (V), что сходно с реакцией Сузуки-Мияура, может быть заменено реакцией прямого арилирования, опосредуемой палладием (непатентная литература 10), родием (непатентная литература 11) и медью (непатентная литература 12).
Непатентная литература 11: Aldrichimica Acta Vol.40, No.2-(2007) 35-41.
Непатентная литература 12: Tetrahedron Letter 49 (2008) 1598-1600.
Процесс A-4
На данной стадии, карбокислотное соединение (VII) может быть получено гидролизом сложноэфирного соединения (VI) в реакционном растворителе.
Гидролиз может быть проведен по известной из публикации методике. Согласно типичной методике, гидролиз может быть проведен в щелочных условиях, таких как гидроксид натрия, гидроксид калия или гидроксид лития. В качестве подходящего растворителя упомянуты, например, спирты, такие как метанол, этанол, пропанол, бутанол, 2-метоксиметанол или этиленгликоль; простые эфиры, такие как тетрагидрофуран (ТГФ), 1,2-диметоксиэтан (DME) или 1,4-диоксан; амиды, такие как N,N-диметилформамид (ДМФА) или гексаметилфосфортриамид; сульфоксиды, такие как диметилсульфоксид (ДМСО), или вода. Реакционный период составляет приблизительно от 30 минут до 48 часов и, как правило, приблизительно от 60 минут до 30 часов. Реакционная температура отвечает диапазону приблизительно от -20°C до 100°C, и, как правило, составляет приблизительно от 20°C до 75°C.
Гидролиз может быть проведен в кислотной среде, например, упомянуты гидрогалогенид, такой как гидрохлорид или гидробромид; сульфоновые кислоты, такие как п-толуолсульфоновая кислота или бензолсульфоновая кислота; пиридий-п-толуолсульфонат; и карбоновые кислоты, такие как уксусная кислота или трифторуксусная кислота. В качестве подходящего растворителя упомянуты, например, спирты, такие как метанол, этанол, пропанол, бутанол, 2-метоксиметанол или этиленгликоль; простые эфиры, такие как тетрагидрофуран (ТГФ), 1,2-диметоксиэтан (DME) или 1,4-диоксан; или галогенированный углеводород, такой как 1,2-дихлорэтан; или амиды, такие как N,N-диметилформамид (ДМФА) или гексаметилфосфортриамид; сульфоксиды, такие как диметилсульфоксид (ДМСО), или вода. Реакционный период составляет приблизительно от 30 минут до 24 часов и, как правило, приблизительно от 60 минут до 10 часов. Реакционная температура отвечает диапазону приблизительно от -20°C до 100°C, и, как правило, составляет приблизительно от 0°C до 65°C.
Процесс A-5
На данной стадии, амидное соединение (IA) может быть получено в присутствии или отсутствии связующего реагента в инертном растворителе по реакции сочетания аминового соединения (VIII) с карбокислотным соединением (VII) в реакционном растворителе. Кроме того, данное взаимодействие может быть проведено в присутствии или отсутствии вспомогательных компонентов, таких как 1-гидроксибензотриазол (HOBt) или 1-гидроксиазабензотриазол. В качестве подходящего растворителя упомянуты, например, ацетон, нитрометан, N,N-диметилформамид (ДМФА), сульфолан, диметилсульфоксид (ДМСО), 1-метил-2-пирролидинон (NMP), 2-бутанон, ацетонитрил; галогенированные углеводороды, такие как дихлорметан, 1,2-дихлорэтан, хлороформ; простые эфиры, такие как тетрагидрофуран и 1,4-диоксан. Реакционный период составляет приблизительно от 5 минут до 1 недели и, как правило, приблизительно от 30 минут до 24 часов. Реакционная температура отвечает диапазону приблизительно от -20°C до 100°C, и, как правило, составляет приблизительно от 0°C до 60°C. В качестве подходящего связующего агента, может быть использован агент, обычно применяемый в пептидном синтезе, например, дициклогексилкарбодиимид (DCC), водорастворимый карбодиимид (WSC), гексафторфосфат-O-бензотриазол-1-ил-N,N,N',N'-тетраметилуроний (HBTU), 2-этокси-N-этоксикарбонил-1,2-дигидрохинолин, 2-бром-1-этилпиридиний тетрафторборной кислоты (BEP), 2-хлор-1,3-диметилимидазолинийхлорид, бензотриазол-1-илокситрис(диметиламино)фосфонийгексафторфосфат (BOP), диэтилазодикарбоксилат-трифенилфосфин, диэтилцианофосфат, диэтилфосфорилазид, 2-хлор-1-метилпиридинийиодид, N,N'-карбонилдиимидазол, бензотриазол-1-илдиэтилфосфат, этилхлорформиат или изобутилхлорформиат. Вдобавок, желательно проводить взаимодействие в присутствии оснований, таких как N,N-диизопропилэтиламин, N-метилморфолин, 4-(диметиламино)пиридин или триэтиламин. Амидное соединение (IA) может быть получено через соответствующий ацилгалогенид, получаемый при взаимодействии с галогенирующим агентом, таким как оксалилхлорид, оксихлорид фосфора или тионилхлорид. Полученный ацилгалогенид может быть превращен в соответствующее амидное соединение (IA) обработкой аминовым соединением (VIII) без использования реагентов конденсации, указанных для данной стадии.
Синтез азаиндольного цикла (способ 3) с использованием реакции образования цикла в процессе B-6 показан ниже.
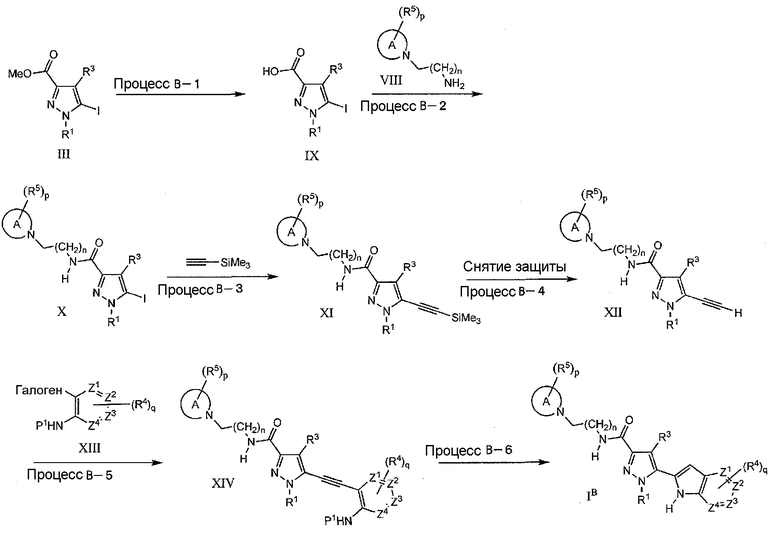
Процесс B-1
На данной стадии, соединение IX может быть получено гидролизом сложноэфирных соединений (III). Указанное взаимодействие является, по-существу, таким же, как в процессе A-4, и те же самые реагенты и реакционные условия, как в процессе A-4, могут быть использованы аналогичным процессу A-4 образом.
Процесс B-2
На данной стадии, соединение X может быть получено по реакции амидирования карбокислотного соединения (IX) аминовым соединением (VIII). Указанное взаимодействие является, по-существу, таким же, как в процессе A-5, и те же самые реагенты и реакционные условия, как в процессе A-5, могут быть использованы аналогичным процессу A-5 образом.
Процесс B-3
На данной стадии, соединение (XI) может быть получено по реакции кросс-сочетания соединения (X) с ацетиленовым соединением, защищенным триалкилсилильной группой, такой как триметилсилильная группа, в присутствии каталитического количества палладиевого реагента и соли меди(I) или палладиевого реагента и фосфинового лиганда, в подходящем растворителе, включающем основание, или с использованием только основания самого по себе в качестве растворителя. В качестве предпочтительного примера палладиевого реагента, упомянуты тетракис(трифенилфосфин)палладий и бис(трифенилфосфин)палладий(II)хлорид. В качестве предпочтительного примера соли меди(I) упомянуты иодид меди(I) и бромид меди(I). В качестве фосфинового лиганда упомянут, например, бис(дифенилфосфино)бутан (DPPB). В качестве примера основания упомянуты, например, диэтиламин, триэтиламин, диизопропилэтиламин, дициклогексиламин, карбонат калия и карбонат натрия. Вдобавок, в качестве реакционного растворителя, упомянуты, например, тетрагидрофуран, 1,4-диоксан, N,N-диметилформамид (ДМФА), ацетонитрил, этил ацетат, углеводороды, такие как н-гексан, циклогексан, бензол, толуол, и диэтиловый эфир. Реакционный период составляет приблизительно от 5 минут до 96 часов и, как правило, приблизительно от 30 минут до 24 часов. Реакционная температура отвечает диапазону приблизительно от -78°C до 200°C, и, как правило, составляет приблизительно от -20°C до 80°C. Кроме того, во время взаимодействия может быть использован микроволновый реактор.
Процесс B-4
На данной стадии, соединение (XII) может быть получено снятием триалкилсилильной защитной группы с применением общепринятого, в целом известного способа, такого как способ, описанный в John wiley & Sons, Protecting Groups in Organic Synthesis (1999). В качестве общепринятого способа, снятие защиты может быть выполнено в присутствии основания, такого как карбонат калия и карбонат натрия, в спиртовом растворителе, таком как метиловый спирт и этиловый спирт.
Процесс B-5
Данное взаимодействие является, по-существу, таким же, как в процессе B-3, и соединение (XIV) может быть получено по реакции Соногашира сочетанием ацетиленового соединения (XII) и арилгалогенидного соединения (XIII), где на схеме P1 означает водород, трет-бутоксикарбонильную группу или амино-защитную группу, с использованием тех же самых реагентов и реакционных условий, как в процессе B-3, которые могут быть использованы аналогичным процессу B-3 образом.
Процесс B-6
На данной стадии, соединение (IB) может быть получено по реакции внутримолекулярного циклоприсоединения ацетиленового соединения (XIV) с применением подходящего основания. В качестве подходящего основания используют трет-бутилат калия, трет-бутилат натрия, трет-бутилат цезия, гидроксид цезия, 1,8-диазабицикло[5.4.0]ундека-7-ен (DBU), 1,1,3,3-тетраметилгуанидин, триэтиламин и тому подобное, и взаимодействие проводят в подходящем растворителе. В качестве подходящего растворителя упомянуты N,N-диметилформамид (ДМФА), N-метилпирролидинон (NMP), толуол, 1,4-диоксан, спирты, такие как метанол и этанол. Реакционный период составляет приблизительно от 5 минут до 96 часов и, как правило, приблизительно от 30 минут до 24 часов. Реакционная температура отвечает диапазону приблизительно от -78°C до 150°C, и, как правило, составляет приблизительно от -20°C до 150°C. Предпочтительно, взаимодействие проводят с использованием трет-бутилата калия в ДМФА в диапазоне температур от комнатной температуры до 80°C. В другом способе, внутримолекулярное циклоприсоединение может быть выполнено с использованием палладиевого катализатора, где дихлорбис(трифенилфосфин)палладий(II), иодид меди(I), триэтиламин, ДМФА упомянуты в качестве характерной комбинации. Вдобавок, может быть осуществлен катализатор на основе металла или комплексы металлов, включающие медь, золото, иридий, ртуть, молибден, платину и родий. Кроме того, когда NHP1 заместитель означает фенольную или тиольную группу, внутримолекулярное циклоприсоединение может быть выполнено в вышеуказанных условиях, приводящих к получению соответствующих бензотиофеновых и бензофурановых производных. Кроме того, после реакции циклизации, когда остается защитная группа (P1), снятие защитной группы может быть проведено при соответствующем условии.
Ниже представлен синтез имидазо[1,2-a]пиридинового цикла (способ 4) с использованием реакции образования цикла, согласно процессу C-3.
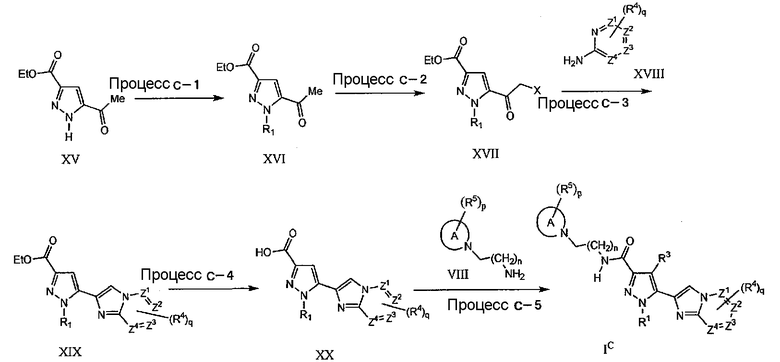
Процесс C-1
На данной стадии, соединение XVI может быть получено по реакции N-алкилирования соединения XV, которое легко может быть получено, согласно литературе, с использованием подходящего основания и алкилгалогенида. В качестве подходящего основания упомянуты, но не в порядке ограничения, например, этилат натрия, трет-бутилат калия, гидрид калия, гидрид натрия, бис(триметилсилил)амид натрия, карбонат калия, карбонат натрия, карбонат цезия, гидроксид натрия. Кроме того, в качестве подходящего органического растворителя упомянуты, например, тетрагидрофуран, N,N-диметилформамид (ДМФА), диэтиловый эфир, ацетонитрил. Реакционный период составляет приблизительно от 5 минут до 96 часов и, как правило, приблизительно от 30 минут до 24 часов. Реакционная температура отвечает диапазону приблизительно от -78°C до 250°C и, как правило, составляет приблизительно от -20°C до 150°C.
Процесс C-2
На данной стадии, соединение XVII может быть получено по реакции альфа-галогенирования (X=Cl, Br, I) соединения (XVI) с использованием подходящего реагента галогенирования. В качестве подходящего реагента галогенирования упомянуты, например, бром, хлор, сульфурилхлорид, бромводород, N-бромсукцинимид (NBS), 5,5-дибром-2,2-диметил-4,6-диоксо-1,3-диоксан, фенилтриметиламмонийтрибромид. В качестве подходящего органического растворителя могут быть использованы, например, уксусная кислота, сероуглерод, простой эфир, тетрагидрофуран, N,N-диметилформамид (ДМФА), галогенированный углеводород, такой как дихлорметан, 1,2-дихлорэтан, хлороформ, тетрахлорид углерода. Реакционный период составляет приблизительно от 5 минут до 96 часов и, как правило, приблизительно от 30 минут до 24 часов. Реакционная температура отвечает диапазону приблизительно от -78°C до 250°C, и, как правило, составляет приблизительно от -20°C до 150°C.
Процесс C-3
На данной стадии, соединение XIX может быть получено по реакции циклической конденсации альфа-галогенкетонового соединения (XVII) с соответствующим аминовым соединением в присутствии подходящего растворителя, при нагревании. В качестве подходящего растворителя упомянуты, например, тетрагидрофуран, 1,4-диоксан, N,N-диметилформамид (ДМФА), ацетонитрил, спирты, такие как метанол и этанол. Реакционный период составляет приблизительно от 5 минут до 96 часов и, как правило, приблизительно от 30 минут до 24 часов. Реакционная температура отвечает диапазону приблизительно от 0°C до 250°C и, как правило, составляет приблизительно от 30°C до 150°C.
Процесс C-4
На данной стадии, соединение XX может быть получено гидролизом сложноэфирного соединения (XIX). Указанное взаимодействие является, по-существу, таким же, как в процессе A-4, и те же самые реагенты и реакционные условия, как в процессе A-4, могут быть использованы аналогичным процессу A-4 образом.
Процесс C-5
На данной стадии, соединение IC может быть получено по реакции амидирования карбокислотного соединения (XX) аминовым соединением (VIII). Указанное взаимодействие является, по-существу, таким же, как в процессе A-5, и те же самые реагенты и реакционные условия, как в процессе A-5, могут быть использованы аналогичным процессу A-5 образом.
Изменение аминовой боковой цепи (способ 5) с использованием процесса D-3 представлено ниже.
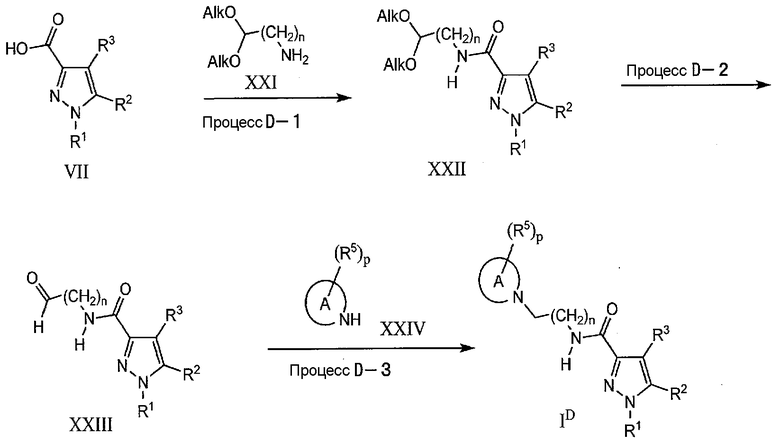
Процесс D-1
На данной стадии, соединение XXII может быть получено по реакции амидирования карбокислотного соединения (VII) аминовым соединением (XXI). Указанное взаимодействие является, по-существу, таким же, как в процессе A-5, и те же самые реагенты и реакционные условия, как в процессе A-5, могут быть использованы аналогичным процессу A-5 образом.
Процесс D-2
На данной стадии, соединение (XXIII) может быть получено снятием ацетальной защитной группы с применением общепринятого, в целом известного способа, такого как способ, описанный в John wiley & Sons, Protecting Groups in Organic Synthesis (1999). В качестве общепринятого способа, снятие защиты может быть выполнено в присутствии кислоты, такой как разбавленная соляная кислота, п-толуолсульфоновая кислота, или в кислотной среде в обычном органическом растворителе.
Процесс D-3
На данной стадии, соединение (ID) может быть получено по реакции восстановительного аминирования альдегидного соединения (XXIII) аминовым соединением XXIV с использованием подходящего восстанавливающего агента. В качестве подходящего восстанавливающего агента упомянуты, например, боргидрид натрия (NaBH4), цианоборгидрид натрия (NaBH3CN), триацетоксиборгидрид натрия [NaBH(OAc)3]. В качестве подходящего растворителя могут быть использованы, например, уксусная кислота, тетрагидрофуран, галогенированные углеводороды, такие как дихлорметан, 1,2-дихлорэтан, хлороформ, тетрахлорид углерода и, если понадобится, каталитическое количество уксусной кислоты или кислот Льюиса, таких как тетрахлорид титана, тетраизопропоксититан [Ti(O-iPr)4]. Когда используют цианоборгидрид (NaBH3CN), взаимодействие может также быть проведено в кислотной среде. Реакционный период составляет приблизительно от 5 минут до 96 часов и, как правило, приблизительно от 30 минут до 24 часов. Реакционная температура отвечает диапазону приблизительно от 0°C до 250°C, и, как правило, составляет приблизительно от 30°C до 100°C.
Изменение аминовой боковой цепи (способ 6) с использованием процесса E представлено ниже.
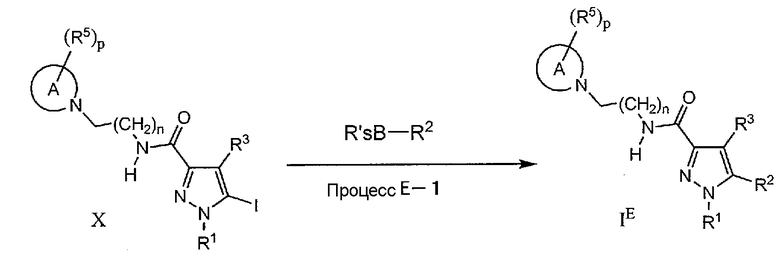
Процесс E-1
На данной стадии, соединение IE может быть получено по реакции связывания галогенированного соединения (X) с производным арилбороновой кислоты (или сложным эфиром). Указанное взаимодействие является, по-существу, таким же, как в процессе A-2, и те же самые реагенты и реакционные условия, как в процессе A-2, могут быть использованы аналогичным процессу A-2 образом.
Изменение боковой цепи R2 (способ 7) с использованием процесса F представлено ниже. В приведенном ниже соединении XXVI, P2 означает защитную группу, выбранную из низшей алкоксикарбонильной группы, бензилоксикарбонильной группы, бензолсульфонильной и 4-алкилбензолсульфонильной группы.
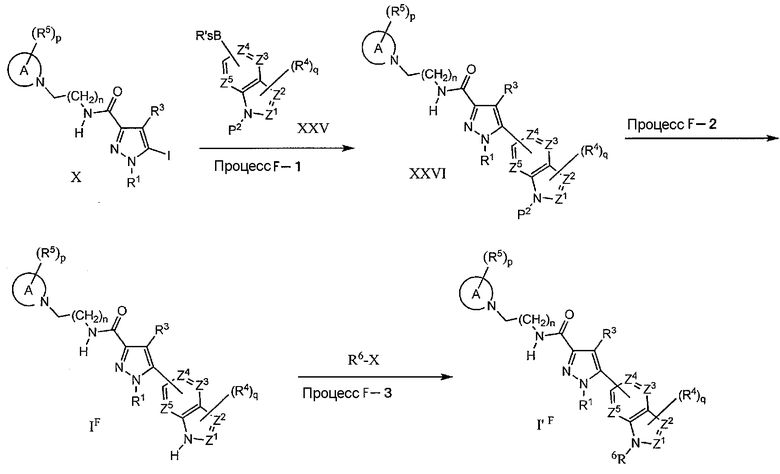
Процесс F-1
На данной стадии, соединение XXVI может быть получено по реакции связывания галогенированного соединения (X) с производным гетероарилбороновой кислоты (или сложным эфиром) XXV, которое может быть защищено трет-бутоксикарбонильной группой или бензолсульфонильной или 4-алкилбензолсульфонильной группой. Указанное взаимодействие является, по-существу, таким же, как в процессе A-2, и те же самые реагенты и реакционные условия, как в процессе A-2, могут быть использованы аналогичным процессу A-2 образом.
Процесс F-2
На данной стадии, соединение (IF) может быть получено снятием защитной триалкилсилильной группы и арилсульфонильной группы, с применением общепринятого, в целом известного способа, такого как способ, описанный в John Wiley & Sons, Protecting Groups in Organic Synthesis (1999). В качестве общепринятого способа, снятие бутоксикарбонильной защитной группы может быть выполнено в присутствии кислотного катализатора, такого как разбавленная соляная кислота, п-толуолсульфоновая кислота, или в кислотной среде в обычном органическом растворителе. Снятие защитной бензолсульфонильной или 4-алкилбензолсульфонильной группы может быть выполнено в присутствии щелочного реагента, такого как карбонат калия, карбонат натрия, карбонат цезия и гидроксид натрия, в комбинации с обычным органическим растворителем.
Процесс F-3
На данной стадии, соединение (I'F) может быть получено превращением N-H-связи на гетероарильном цикле IF в N-R6-связь. Когда R6-X реагент представляет собой алкилгалогенид, указанное взаимодействие является, по-существу, таким же, как в процессе C-1, и взаимодействие может быть проведено в тех же условиях, как в процессе C-1. Кроме того, O-тозилат, O-мезилат и O-трифлат, содержащие уходящую группу на гидроксильной группе (-OH), могут быть взаимозаменяемыми. Далее, когда R6 означает алкилсульфонильную группу, взаимодействие является, по-существу, таким же, как в процессе C-1, где алкилсульфонилхлорид используют в таких же условиях, как в процессе C-1.
Промежуточное соединение (IA) полезно для получения соединения по данному изобретению. Например, промежуточное соединение X, указанное в процессе B, в общем синтезе, эффективно для использования в получении соединения по данному изобретению.
Промежуточное соединение (IB) полезно для получения соединения по данному изобретению. Например, промежуточные соединения VI и VII в процессе A, XIX и XX в C-процессе синтеза, XXI в D-процессе синтеза, в общем синтезе, эффективны для использования в получении соединения по данному изобретению.
Фармакологические действия соединений по данному изобретению, как антагонистов 5-HT2B, могут быть оценены измерением положительной динамики в повышении легочного кровяного давления в модели на животных (крысы, мыши), подвергнутых хронической гипоксии. Существующие лекарства от легочной артериальной гипертензии (напр., препараты силденафил и простагландин) и RS-127445, известный как селективный антагонист 5-HT2B, могут быть использованы в качестве соединений сравнения.
Другие фармакологические действия соединений по данному изобретению, как антагонистов 5-HT2B, могут быть оценены измерением противодиарейных эффектов в модели на животных (крысы, мыши), подвергнутых действию лекарственных веществ или стрессу. Существующие противодиарейные средства (напр., лоперамид и берберин) и RS-127445, известный как селективный антагонист 5-HT2B, могут быть использованы в качестве соединений сравнения.
Таким образом, полученные соединения могут быть выделены и очищены в свободной форме или в форме соли в результате общепринятой обработки, приводящей к образованию соли. Выделение и очистка могут быть достигнуты применением обычного химического метода, такого как экстракция, концентрирование, разбавление, кристаллизация, фильтрование, перекристаллизация, различная хроматография и так далее.
Различные изомеры могут быть выделены общепринятым способом, использующим различие в физикохимических свойствах между изомерами. Например, оптические изомеры могут быть разделены и очищены путем образования диастереомерных солей из рацематов с оптически активными органическими кислотами (напр., винной кислотой) и последующей фракционированной кристаллизацией, или колоночной хроматографией с использованием хиральной неподвижной фазы. Кроме того, оптически активные соединения могут быть получены с применением подходящего оптически активного соединения в качестве исходного вещества. В указанном случае, смесь диастереомеров может также быть разделена фракционированной кристаллизацией или хроматографией на соответствующие чистые энантиомеры (энантиомер).
ОРАЛЬНОЕ ВВЕДЕНИЕ
Соединения по изобретению могут быть введены орально. Оральное введение может включать проглатывание, с тем, чтобы соединение поступало в желудочно-кишечный тракт, либо может быть использовано буккальное или сублингвальное введение при котором соединение поступает в кровоток прямо изо рта.
Композиции, пригодные для перорального введения, включают твердые композиции, такие как, например, таблетки, содержащие частицы, жидкости или порошки, капсулы, пастилки (включая наполненные жидкостью), жевательные таблетки, мультичастицы, наночастицы, гели, твердые растворы, липосомы, пленки (включая мукоадгезивные), суппозитории, спреи и жидкие композиции.
Жидкие композиции включают, например, суспензии, растворы, сиропы и эликсиры. Такие композиции могут быть использованы как наполнители в мягких или твердых капсулах и, как правило, содержат носитель, например, такой как вода, этанол, полиэтиленгликоль, пропиленгликоль, метилцеллюлоза или подходящее масло, и эмульгирующих веществ и/или суспендирующих веществ. Жидкие композиции могут также быть получены ресуспендированием твердого вещества, например, из саше, в воде и тому подобном.
Соединения по изобретению могут также быть использованы в быстрорастворимых, быстрораспадающихся дозированных формах, таких как описаны в Expert Opinion in Therapeutic Patents, 11 (6), 981-986 by Liang and Chen (2001).
Для таблетированных дозированных форм, в зависимости от дозы, лекарственное средство может составлять приблизительно от 1 мас.% до 80 мас.% от дозированной формы, в основном, приблизительно от 5 мас.% до 60 мас.% от дозированной формы.
В дополнение к лекарственному средству в качестве активного ингредиента, таблетки обычно содержат дезинтегрант. Примеры дезинтегрантов включают такие вещества, как натриевая соль гликолята крахмала, карбоксиметилцеллюлоза натрия, карбоксиметилцеллюлоза кальция, кроскармеллоза натрия, кросповидон, поливинилпирролидон, метилцеллюлоза, микрокристаллическая целлюлоза, замещенная низшими алкилами гидроксипропилцеллюлоза, крахмал, прежелатинизированный крахмал и альгинат натрия. Обычно дезинтегрант составляет приблизительно от 1 мас.% до 25 мас.%, предпочтительно, приблизительно от 5 мас.% до 20 мас.% от дозированной формы.
Связующие вещества обычно используют для придания липких свойств таблетированным композициям.
Таблетированная композиция может содержать связующие вещества для наделения когезивными свойствами, иными, чем у лекарственного средства в качестве активного ингредиента. Подходящие связующие вещества включают такие, как микрокристаллическая целлюлоза, желатин, лактоза (моногидрат, высушенный распылением моногидрат, безводный и тому подобное), маннит, ксилит, декстроза, сахароза, сорбит, полиэтиленгликоль, природные и синтетические смолы, поливинилпирролидон, прежелатинизированный крахмал и гидроксипропилцеллюлоза, дегидрат дикальцийортофосфата и гидроксипропилметилцеллюлоза, и тому подобное.
Таблетки могут также, необязательно, включать поверхностно-активные вещества, такие как лаурилсульфат натрия и полисорбат 80, и вещества, способствующие скольжению, такие как диоксид кремния и тальк. Если присутствуют, поверхностно-активные вещества могут составлять приблизительно от 0,2 мас.% до 5 мас.% таблетки и вещества, способствующие скольжению, могут составлять приблизительно от 0,2 мас.% до 1 мас.% таблетки.
Таблетки также обычно содержат лубриканты, такие как стеарат магния, стеарат кальция, стеарат цинка, стеарилфумарат натрия и смеси стеарата магния с лаурилсульфатом натрия. Лубриканты обычно составляют приблизительно от 0,25 мас.% до 10 мас.%, предпочтительно, приблизительно от 0,5 мас.% до 3 мас.% таблетки.
Другие возможные ингредиенты включают антиоксиданты, красители, ароматизаторы, консерванты, вещества, исправляющие вкус и запах лекарственного средства, и тому подобное.
Типичные таблетки содержат приблизительно до 80 мас.% лекарственного вещества, приблизительно от 10 мас.% до 90 мас.% связующего вещества, приблизительно от 0 мас.% до 85 мас.% разбавителя, приблизительно от 2 мас.% до 10 мас.% дезинтегранта и приблизительно от 0,25 мас.% до 10 мас.% лубриканта.
Способы получения таблеток не ограничены, хотя могут быть соответственно использованы общепринятые способы получения таблеток. Например, таблеточные смеси можно прессовать непосредственно или с помощью ротационной машины с получением таблеток. Таблеточные смеси или порции смесей, альтернативно, могут быть гранулированы мокрым, сухим способом или из расплава, охлаждены до затвердевшего расплава или экструдированы перед таблетированием. Конечная таблетка может содержать один или несколько слоев и может быть с оболочкой или без оболочки; указанная композиция может даже быть инкапсулированной.
Что касается составления таблеток, можно сослаться на материалы, содержащиеся в "Pharmaceutical Dosage Forms: Tablets Vol. 1", by H. Lieberman and L. Lachman, Marcel Dekker, N.Y., 1980 (ISBN 0-8247-6918-X).
Твердые композиции для перорального введения могут быть составлены для немедленного и/или модифицированного высвобождения. Композиции модифицированного высвобождения включают, например, отсроченное, замедленное, прерывистое, контролируемое, нацеленное и программируемое высвобождение.
Подходящие композиции модифицированного высвобождения для целей по изобретению описаны в патенте США № 6106864. Подробные описания других подходящих технологий высвобождения, таких как макроэргические дисперсии и осмотические частицы и частицы с оболочкой, можно найти в Verma et al, Pharmaceutical Technology On-line, 25(2), 1-14 (2001). Применение жевательных резинок для достижения контролируемого высвобождения описано в WO 00/35298.
ПАРЕНТЕРАЛЬНОЕ ВВЕДЕНИЕ
Соединения по изобретению могут также быть введены непосредственно в кровоток, в мышцу или во внутренний орган. Подходящие способы парентерального введения включают внутривенный, внутриартериальный, внутрибрюшинный, подоболочечный, внутрижелудочковый, внутриуретральный, внутригрудинный, внутричерепной, внутримышечный и подкожный. Подходящие устройства для парентерального введения включают игольные (включая микроигольные) инъекторы, безигольные инъекторы и инфузионное оборудование.
Композиции парентерального введения могут содержать эксципиенты, такие как соли, карбогидраты и буферные вещества (предпочтительно с pH приблизительно от 3 до 9). Указанные композиции, как правило, могут являться водными растворами, но, для некоторых применений, такие композиции могут быть составлены в более удобные формы, такие как стерильный неводный раствор или в виде высушенной формы, используемой в сочетании с подходящим разбавителем, таким как стерильная, апирогенная вода.
Получение композиций для парентерального введения в стерильных условиях, например, лиофилизацией, легко может быть выполнено с применением стандартных фармацевтических методик, хорошо известных специалисту в данной области.
Растворимость соединений формулы (I), используемых в получении парентеральных растворов, может быть повышена путем применения соответствующих технологий составления, таких как включение усилителей растворимости.
Композиции для парентерального введения могут быть составлены для немедленного и/или модифицированного высвобождения. Композиции модифицированного высвобождения включают, например, отсроченное, замедленное, прерывистое, контролируемое, нацеленное и программируемое высвобождение. Такие соединения по изобретению могут быть составлены как твердое вещество, полутвердое вещество или тиксотропная жидкость для введения в качестве имплантированного депо, обеспечивающего модифицированное высвобождение активного соединения. Примеры таких композиций включают стенты с лекарственным покрытием и микросферы из PGLA.
МЕСТНОЕ ВВЕДЕНИЕ
Соединения по изобретению могут также быть введены местно через кожу или слизистую оболочку, то есть, дермально или трансдермально. Типичные композиции для указанной цели включают, например, гели, гидрогели, лосьоны, растворы, кремы, мази, присыпки, повязки, пены, пленки, трансдермальные пластыри, прокладки, имплантанты, салфетки, нити, перевязочные материалы и микроэмульсии. Могут также быть использованы липосомы. Типичные носители включают, например, спирт, воду, минеральное масло, вазелиновое масло, белый вазелин, глицерин, полиэтиленгликоль и пропиленгликоль или воду, и тому подобное. Могут быть включены вещества, способствующие проникновению, - смотри, например, J Pharm Sci, 88 (10), 955-958 by Finnin and Morgan (October 1999).
Другие способы местного введения включают, например, доставку электропорацией, ионтофорезом, фонофорезом, сонофорезом и с помощью микроигольной или безигольной (напр. Powderject (зарегистрированный товарный знак), Bioject (зарегистрированный товарный знак) и проч.) инъекции.
Композиции для местного введения могут быть составлены для немедленного и/или модифицированного высвобождения. Композиции модифицированного высвобождения включают, например, отсроченное, пролонгированное, прерывистое, контролируемое, направленное и программируемое высвобождение.
ДРУГИЕ ТЕХНОЛОГИИ
Соединения по данному изобретению можно комбинировать с растворимыми макромолекулярными соединениями, такими как циклодекстрин и подходящие производные циклодекстрина или содержащие полиэтиленгликоль полимеры, для улучшения растворимости, скорости растворения, маскировки вкуса, улучшения биологической доступности и/или и стабильности при использовании в любых вышеуказанных способах введения.
Например, установлено, что комплексы лекарственное вещество-циклодекстрин обычно полезны для большинства дозированных форм и способов введения. Могут быть использованы как комплексы включения, так и без включения. В качестве альтернативы прямого комплексообразования с лекарственным веществом, циклодекстрин может быть использован как вспомогательное вещество, т.е. в качестве носителя, разбавителя или солюбилизатора. Чаще всего для данных целей используют альфа-, бета- и гамма-циклодекстрины, примеры которых могут быть найдены в WO 91/11172, WO 94/02518 и WO 98/55148.
Наборы, состоящие из нескольких компонентов
Объемом данного изобретения предусмотрено, что две или несколько фармацевтических композиций, по меньшей мере, одна из которых содержит соединение по изобретению, целесообразно объединять в форме набора, удобного для введения комбинации, например, совместного введения композиций.
Таким образом, набор по изобретению включает две или несколько отдельных фармацевтических композиций, по меньшей мере, одна из которых содержит соединение формулы (I) по изобретению, и средства раздельного хранения указанных композиций, такие как контейнер, разделенная бутылка или разделенный пакет из фольги. Примером такого набора служит обычная блистерная упаковка, используемая для упаковки таблеток, капсул и тому подобного.
Набор по изобретению в особенности удобен для введения различных дозированных форм, например, для орального и парентерального введения, для введения отдельных композиций с различными интервалами между введением или для титрования отдельных композиций другой композицией. Согласно законодательству, набор обычно включает инструкцию по применению и может быть обеспечен так называемым мнемоническим представлением информации.
ДОЗИРОВКА
Для введения пациентам-людям, из расчета, что среднестатистический человек имеет вес приблизительно от 65 кг до 70 кг, общая суточная доза соединений по изобретению соответствует обычно диапазону, приблизительно, от 0,05 мг до 1000 мг, предпочтительно, диапазону, приблизительно, от 0,1 мг до 100 мг и, более предпочтительно, диапазону, приблизительно, от 0,5 мг до 20 мг. Конечно, в зависимости от способа введения, например, пероральное введение может требовать общей суточной дозы приблизительно от 1 мг до 500 мг, тогда как внутривенная доза может требоваться лишь в диапазоне приблизительно от 0,5 мг до 250 мг. Общая суточная доза может быть введена в виде одной дозы или разделенной общей дозы. Указанные дозировки могут соответственно изменяться в зависимости от пола, возраста или болезненных состояний субъектов-людей.
Как описано выше, соединение по изобретению обладает антагонистической активностью в отношении 5-HT2B. Антагонист 5-HT2B по данному изобретению полезно комбинировать с другим фармакологически активным соединением или с двумя или несколькими другими фармакологически активными соединениями, в особенности, при лечении рака, воспалительных заболеваний, иммуномодулируемых заболеваний и желудочно-кишечного расстройства, например, нарушения моторики желудочно-кишечного тракта и сенсорной ирритации, или для регулирования кровяного давления в легких и восстановления артерий.
Например, антагонист 5-HT2B, в частности, соединение формулы (I) или фармацевтически приемлемую соль указанного соединения, как определено выше, можно вводить одновременно, последовательно или раздельно в комбинации с одним или несколькими средствами, выбираемыми из группы, включающей:
[Перечень 1]
Легкие слабительные средства: напр., упомянуты регулон (зарегистрированный товарный знак) и целевак (зарегистрированный товарный знак);
Противосудорожное средство: упомянуты, напр., мебеверин, пинаверин, отилоний бромид и тримебутин, обладающие эффектом релаксации гладких мышц; напр., дицикловерин, гиосциамин и циметропий, обладающие антимускариновыми действиями.
Опиоидное/цетрального действия лекарственное средство: упомянуты, напр., лоперамид, налтрексон, метилналтрексон, модулон (зарегистрированный товарный знак) и алвимопан, являющиеся MOR-агонистами; напр., федотозин и азимадолин, являющиеся KOR-агонистами; напр., имипрамин, амитриптилин, кломипрамин, дезипрамин и лофепрамин, являющиеся трициклическими антидепрессантами; напр., сертралин, пароксетин, флуоксетин и эсциталопрам, являющиеся селективными ингибиторами обратного захвата серотонина; напр., венлафаксин и дулоксетин, являющиеся селективными ингибиторами обратного захвата серотонина-норадреналина; напр., моклобемид, являющийся обратимо действующим ингибитором моноаминооксидазы; напр., диазепам, празепам, клоназепам и декстрофизопам, являющиеся агонистами бензодиазепинов; напр., оксиморфон ER и трамадол, являющиеся анальгетиками центрального действия; напр., изокарбоксазид, фенелзин, траниципромин и селегилин, являющиеся ингибиторами моноаминооксидазы.
Серотонинергические рецепторные модуляторы: упомянуты, напр., алозетрон, ондансетрон, трописетрон, палоносетрон, рамосетрон, митразапин, индисетрон, цилансетрон, гранисетрон и доласетрон, являющиеся антагонистами 5-HT3; напр., тегасерод и мозаприд, являющиеся агонистами 5-HT4; напр., MKC-733, являющийся агонистом 5-HT3; напр., рензаприд, являющийся агонистом 5-HT4/5-HT3; напр., индисетрон, являющийся антагонистом 5-HT3/5-HT4; напр., DR-4004, SB-269970, SB-258719 и SB-258741, являющиеся антагонистом 5-HT7; напр., буспирон и эпирон, являющиеся агонистами или антагонистами 5-HT1A; напр., буспирон, являющийся агонистом 5-HT1A/1B/D; напр., эрготамин, суматриптан и ризатриптан, являющиеся противомигреневыми средствами.
Средство улучшения моторики желудочно-кишечного тракта: упомянуты, напр., маропитант, апрепитант и эзлопитант, являющиеся антагонистами NK1; напр., непадутант и саредутант, являющиеся антагонистами NK2; напр., талнетант, являющийся антагонистом NK3; напр., CP-154526, NBI-35965 и CRA-1000, являющиеся антагонистами рецептора CRF1; напр., декслоксиглумид, являющийся антагонистом рецептора CCK-A; напр., митемцинал и PF-4548043, являющиеся мотилиновыми агонистами; напр., лубипростон, являющийся агонистом натриевых каналов (тип 2); напр., линаклотид, являющийся агонистом гуанилатциклаза; напр., GTP-010, являющийся агонистом глюкагоноподобного пептида-1; напр., ибутаморен и капроморелин, являющиеся агонистами грелинового рецептора.
Антибиотики: упомянуты, напр., сульфацетамид, эритромицин, рифаксимин, тобрамицин и ципрофлоксацин.
Пробиотические бактерии: упомянуты, напр., непатогенные бифидобактерии bifidobacterium infantis 35624 и кишечная палочка E.coli.
Антианальгетические средства: упомянуты, напр., клонидин, медетомидин, лофексидин, дексмедетомидин и AGN-2-3818, являющиеся альфа-2-адренергетиками; напр., солабегрон, являющийся бета-3-адренергетиком; напр., GRC-10622, GW842166 и S-777469, являющиеся каннабиноидными -1 или -2 агонистами; напр., целекоксиб, рофекоксиб, валдекоксиб, эторикоксиб и люмиракоксиб, являющиеся селективными ингибиторами COX-2; напр., пироксикам, напроксен, ибупрофен, диклофенак и индометацин, являющиеся нестероидными противовоспалительными средствами (NSAIDs); напр., дизоцилпин, являющийся NMDA-антагонистом; напр., ресинифератоксин и капсазепин, являющиеся модуляторами TRPs (подтипов V1, V3, V4, M8, A1); напр., габапентин, прегабалин и 3-метилгабапентин, являющиеся альфа-2-дельта- лигандами; напр., топирамат, цинолазепам и клоназепам, являющиеся GABA-агонистами.
Противовоспалительные средства: упомянуты, напр., дексаметазон, преднизолон, циклезонид и будезонид, являющиеся синтетическим гормоном коры надпочечников; напр., анакинра, атлизумаб и меполизумаб, являющиеся терапевтическими средствами на основе интерлейкина.
Противоаллергическое средство: упомянуты, напр., монтелукаст, зафирлукаст и пранлукаст, являющиеся антагонистами лейкотриенов; напр., альбутерол, левалбутерол, салметерол, формотерол и арформотерол, являющиеся бета-2- агонистами; напр., рофлумиласт, тиотропий и израпафант, используемые при астме и/или хронической обструктивной болезни легких.
Другие терапевтические средства: упомянуты, напр., полифул (зарегистрированный товарный знак), метамуцил (зарегистрированный товарный знак), клофелемер и шелуха семян подорожника.
Ассоциированная легочная гипертензия: упомянуты, напр., берапрост, являющийся производным простагландина; напр., силденафил, являющийся ингибитором PDE5; напр., бозентан, являющийся антагонистом эндотелина-1.
ПРИМЕРЫ
Далее настоящее изобретение поясняется подробно с помощью примеров, но приведенные ниже примеры никоим образом не ограничивают данное изобретение, и могут быть использованы различные изменения, не выходящие за рамки объема изобретения. Такие различные изменения, не выходящие за рамки объема изобретения, также входят в объем изобретения.
Изобретение иллюстрируется следующими неограничивающими примерами, в которых, если не указано иное: все операции осуществляют при комнатной температуре или температуре окружающей среды, то есть, в диапазоне приблизительно 18-25°C; выпаривание растворителя производят, используя ротационный испаритель, при пониженном давлении и температуре бани около 60°C; мониторинг взаимодействий осуществляют с помощью тонкослойной хроматографии (ТСХ) и время взаимодействия приводится лишь для наглядности; температуры плавления (т.пл.) приведены без учета поправки (полиморфизм может приводить к различным температурам плавления); структуру и чистоту всех выделенных соединений подтверждают, по меньшей мере, одним из следующих способов: ТСХ (пластины для ТСХ F254, предварительно покрытые силикагелем 60, Merck, или пластины для ВЭТСХ F254s, с предварительно нанесенным NH2, Merck), масс-спектрометрией, ядерным магнитным резонансом (ЯМР), инфракрасными спектрами поглощения (ИК) или микроанализом. Выходы приведены исключительно с целью иллюстрации. Флэш-колоночную хроматографию проводят, используя силикагель 300HG, WAKO (40-60 мкм) или Fuji Silysia Chromatorex (зарегистрированный товарный знак) DU3050 (амино-тип, 30-50 мкм), или диоксид кремния Biotage (32-63 мм, KP-Sil), или амино-связанный диоксид кремния Biotage (35-75 мм, KP-NH).
Используемой для взаимодействия микроволновой аппаратурой является оптимизатор Emrys (Personal chemistry) или Initiator (зарегистрированный товарный знак) Sixty (Biotage). Используемой для взаимодействия ультразвуковой аппаратурой является Ultra Sonic Cleaner SINGLE Frequency (AS ONE). Сокращения для обозначения реакционных растворителей следующие: тетрагидрофуран (ТГФ), диметилсульфоксид (ДМСО) и диметилформамид (ДМФА). Кроме того, гексафторфосфат 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраэтилурония (HBTU).
Очистку конечного соединения с применением ВЭЖХ выполняют на следующей аппаратуре при указанных ниже условиях.
Аппаратура: МС-триггерная система автоочистки MS-trigger AutoPurification (зарегистрированный товарный знак) system, waters (называемая здесь далее устройство для очистки A)
Колонка: XTerra C18, 19×50 мм, частицы 5 мкм;
Способ A: метанол или ацетонитрил/0,05% (об./об.), водный раствор муравьиной кислоты или
способ B: метанол или ацетонитрил/0,01% (об./об.), водный раствор аммиака.
Подтверждение химической чистоты в способе очистки с использованием устройства для очистки A осуществляют с помощью следующей аппаратуры при указанных ниже условиях.
Аппаратура: детектор Acquity Ultra Parformance LC on TUV Detector и масс-спектрометр ZQ mass spectrometer, waters
Колонка: waters ACQUITY C18, 2,1×50 мм, частицы 7 мкм
Температура колонки: 60°C, скорость потока: 10 мл/мин, УФ-детектор: 210 нм,
МС-детектирование: ESI- режим определения положительных ионов, метод: QC_neutral_full_1pt5min.
Элюент: ацетонитрил/10 мМ раствор ацетата аммония,
Градиент: 5% (0-0,1 мин), 5-95% (0,10-8 мин), 95% (0,8-1 мин), время анализа: 1,5 мин.
Очистку с применением ВЭЖХ выполняют на следующей аппаратуре при указанных ниже условиях.
Аппаратура: УФ- триггерная препаративная ВЭЖХ система UV-trigger system, waters (называемая здесь далее устройство для очистки B).
Колонка: XTerra C18, 5 мкм, 19×50 мм или 30×50 мм,
Детектор: УФ 254 нм,
Скорость потока: 20 мл/мин (19×50 мм) или 40 мл/мин (30×50 мм) при комнатной температуре.
Данные масс-спектров низкого разрешения (EI) получают на масс-спектрометре Integrity (waters) или масс-спектрометре Automass 120 (JEOL) или 6890GC/5793MSD (ГХ-МС технология от Agilent).
Данные масс-спектров низкого разрешения (ESI) получают на следующей аппаратуре при указанных ниже условиях.
Аппаратура: система для ВЭЖХ Alliance waters на масс-спектрометре ZQ или ZMD и УФ-детекторе.
Когда в молекуле содержится несколько атомов брома, с учетом относительного изотопного состава, могут быть указаны два или несколько численных значений величин в зависимости от числа атомов брома.
Колонка: waters Xterra (зарегистрированный товарный знак) C18, 2,1×30 мм, частицы 3,5 микрометра,
Градиент: 4-96% (0-2 мин), 96% (2-4 мин), скорость потока: 0,5 мл/мин,
УФ-детектирование: 254 нм,
МС-детектирование: ESI-метод posi/nega,
Элюент: ацетонитрил/0,025% (об./об.) водный раствор формиата аммония (полный нейтральный диапазон), ацетонитрил/0,05% водный раствор муравьиной кислоты (полный кислотный диапазон), ацетонитрил/0,01% водный раствор аммиака (полный основной диапазон).
Данные ЯМР получают при 270 МГц (спектрометр JEOL JNM-LA 270 spectrometer) или 300 МГц (JEOL JNM-LA300), используя в качестве растворителя дейтерированный хлороформ (99,8% D) или диметилсульфоксид (99,9% D), если не указано иное, относительно тетраметилсилана (ТМС) в качестве внутреннего стандарта, в миллионных долях (м.д.).
Использованы общепринятые сокращения: с=синглет (с), д=дублет (д), т=триплет (т), кв=квартет (кв), м=мультиплет (м), уш.=уширенный и т.д. ИК спектр получают на инфракрасном спектрометре Shimadzu infrared spectrometer (IR-470). Оптическое вращение измеряют, используя поляриметр JASCO DIP-370 Digital Polarimeter (Japan Spectroscopic Co., Ltd.). Химические символы имеют общепринятые значения; т.кип. (температура кипения), т.пл. (температура плавления), л (литр(ы)), мл (миллилитр(ы)), г (грамм(ы)), мг (миллиграмм(ы)), моль (моли), ммоль (миллимоли). Кроме того, защитная группа Ts означает п-толуолсульфоновую кислоту.
Соединение по примеру 1: синтез 5-(1H-индол-2-ил)-1-метил-N-[2-(пиперидин-1-ил)этил]-1H-пиразол-3-карбоксамида
Промежуточное соединение 2: синтез метил-3-циано-2-натрийокси-2-пропеноата
Небольшой кусочек металлического натрия (75,87 г, 2,79 моль) добавляют небольшими порциями к охлажденному льдом метанолу (1,2 л). После исчезновения металлического натрия, смесь диметилщавелевой кислоты (300 г, 2,541 моль) и ацетонитрила (114,72 г, 2,79 моль) в метаноле добавляют по каплям к метанольному раствору полученного ранее метилата натрия. После чего концентрирование смеси в вакууме дает желтое промежуточное соединение 2 (377,4 г, 99,5% выход). Промежуточное соединение 2 используют на следующей стадии без дополнительной очистки.
Промежуточное соединение 3: синтез метилгидразинсульфата
Сульфоновую кислоту (23,4 г, 0,238 моль) добавляют по каплям к 40% раствору метилгидразина (25 г, 0,217 моль). После завершения добавления реакционную смесь выдерживают 4 часа. Полученную смесь сушат сублимацией и получают промежуточное соединение 3.
Промежуточное соединение 4: синтез метил-5-амино-1-метил-1H-пиразол-3-карбоксилат
Промежуточное соединение 3 (280 г, 1,94 моль) добавляют к метанольной суспензии (1,5 л) промежуточного соединения 2 (263,39 г, 1,77 моль). Полученную смесь выдерживают 48 часов при комнатной температуре. После чего к реакционной смеси добавляют 2 М гидроксид натрия (200 мл) и дихлорметан (1 л), и органический слой отделяют. Затем полученный органический слой промывают насыщенным хлоридом натрия и сушат над безводным сульфатом натрия. После фильтрования осушающего вещества, концентрирование полученного фильтрата в вакууме дает желтые кристаллы сырого промежуточного соединения 4 (82,20 г, 30% выход). Полученное промежуточное соединение 4 используют на следующей стадии без дополнительной очистки.
Промежуточное соединение 5: синтез метил-5-иод-1-метил-1H-пиразол-3-карбоксилата
Раствор сульфита натрия (17,06 г, 0,247 моль) добавляют осторожно по каплям к раствору промежуточного соединения 4 (32,0 г, 0,206 моль) и иодида калия (342,4 г, 2,06 моль) в уксусной кислоте-воде (3/1 (об./об.), 300 мл). После добавления по каплям, реакционную смесь выдерживают на 3 часа при 0°C. После подтверждения израсходованности промежуточного соединения 4 методом ТСХ (смесь этил ацетат: гексан=1:4 (об./об.)), полученную смесь доводят до pH 10-pH 11 добавлением твердого гидрокарбоната натрия. Водный слой трижды экстрагируют 1100 мл этилацетата. Затем собранный органический слой сушат над безводным сульфатом натрия. После фильтрования, концентрированный при пониженном давлении фильтрат дает коричневое, сиропообразное, сырое промежуточное соединение 5. Полученное сырое промежуточное соединение 5 очищают колоночной хроматографией, используя силикагель (смесь этилацетат/петролейный эфир, (0/1-1/3 (об./об.)) и получают промежуточное соединение 5 (16,4 г, 30% выход) в виде белых кристаллов.
1H-ЯМР (270 МГц, CDCl3) δ 6,98 (с, 1H), 4,01 (с, 3H), 3,92 (с, 3H).
МС(ИЭР)m/z: [M+H]+267.
Промежуточное соединение 7: синтез 5-(1H-индол-2-ил)-1-метил-1-1H-пиразол-3-карбоновой кислоты
Синтез 2-(4,4,5,5-тетраметил-1,3,2-диоксаборан-2-ил)-1H-индола методом C-H- борилирования
Раствор (25 мл) смеси 1H-индола (2,50 г, 21,3 ммоль), [Ir(OMe)(COD)]2 (28,4 мг, 0,042 ммоль), 4,4'-ди-трет-бутил-2,2'-бипиридила (dtbpy) (22,9 мг, 0,085 ммоль) и бис(пинаколят)диборана (3,25 г, 12,8 ммоль) в абсолютном диоксане выдерживают при 80°C в течение 1 часа. Указанный реакционный раствор используют на следующей стадии без очистки.
Промежуточное соединение 6, полученное сочетанием по Сузуки: синтез метил-5-(1H-индол-2-ил)-1-метил-1H-пиразол-3-карбоксилата
Промежуточное соединение 5 (3,73 г, 14,0 ммоль), трис(дибензилиденацетон)дипалладий(0) (128 мг, 0,14 ммоль), раствор фосфата калия (4,88 г, 23 ммоль), трициклогексилфосфин (78,5 мг, 0,28 ммоль) и воду (3 мл) добавляют к вышеуказанному раствору реакционной смеси.
1H-ЯМР (270 МГц, CDCl3) δ 8,40 (уш.с, 1H), 7,68 (д, J=7,3 Гц, 1H), 7,44 (д, J=6,6 Гц, 1H), 7,31-7,25 (м, 1H), 7,21-7,15 (м, 1H), 7,04 (с, 1H), 6,75 (с, 1H), 4,15 (с, 3H), 3,97 (с, 3H).
Промежуточное соединение 7: синтез 5-(1H-индол-2-ил)-1-метил1-1H-пиразол-3-карбоновой кислоты
Реакционный раствор промежуточного соединения 6 (742 мг, 2,91 ммоль) и 2М раствора гидроксида натрия (5 мл, 10 ммоль) в смеси метанол (15 мл) - ТГФ (5 мл) выдерживают при 70°C в течение 1 часа. После охлаждения до комнатной температуры, реакционную смесь доводят до pH 3 с помощью 2М раствора HCl и разбавляют насыщенным хлоридом натрия. Смесь экстрагируют дихлорметаном, объединенный органический слой сушат над безводным сульфатом магния. После фильтрования, органический слой концентрируют при пониженном давлении, и сырой продукт-промежуточное соединение 7 (673 мг, 96%), выделяют в виде белого твердого вещества.
1H-ЯМР (300 МГц, ДМСО-d6) δ 11,58 (с, 1H), 7,61 (д, J=8,0 Гц, 1H), 7,42 (д, J=8,0 Гц, 1H), 7,22-7,10 (м, 2H), 7,09-7,02 (м, 1H), 6,88 (с, 1H), 4,12 (с, 3H).
Отсутствует пик, обусловленный NH.
МС(ИЭР)m/z: [M+H]+242, [M-H]-240.
Промежуточное соединение 7: альтернативный синтез 5-(1H-индол-2-ил)-1-метил-1-1H-пиразол-3-карбоновой кислоты
Промежуточное соединение 8: синтез трет-бутил-2-[3-(метоксикарбонил)-1-метил-1H-пиразол-5-ил]-1H-индол-1-карбоксилата
Промежуточное соединение 5 (3,14 г, 11,8 ммоль), ацетат палладия (265 мг, 1,18 ммоль) и трифенилфосфин (1,24 г, 4,71 ммоль) растворяют в растворе диоксан/толуол (3,5/1 (об./об.), 27 мл). Полученный раствор выдерживают при комнатной температуре 10 минут. После чего, к реакционному раствору добавляют трет-бутиловый эфир 2-(дигидроксиборанил)-1H-индол-1-карбоновой кислоты (4,00 г, 15,3 ммоль), воду (3 мл) и карбонат натрия (3,12 г, 29,5 ммоль). Раствор нагревают до температуры кипения с обратным холодильником 1,5 часа. После охлаждения, реакционный раствор добавляют к воде (150 мл). Затем водный слой экстрагируют этилацетатом (150 мл ×2). После чего полученный органический слой сушат над сульфатом магния, осушители фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток предварительно обрабатывают методом колоночной хроматографии (этилацетат), используя силикагель, пропитанный амином. Затем промежуточное соединение 8 (1,72 г, 41% выход) получают в виде белого твердого вещества очисткой с применением колоночной хроматографии на силикагеле (смесь гексан-диэтиловый эфир (1,5/1-1/1) (об./об.)).
1H-ЯМР (300МГц, CDCl3) δ 8,29 (д, J=8,1 Гц, 1H), 7,61 (д, J=8,0 Гц, 1H), 7,42 (т, J=7,3 Гц, 1H), 7,31 (т, J=7,3 Гц, 1H), 6,88 (с, 1H), 6,71 (с, 1H), 3,96 (с, 3H), 3,79 (с, 3H), 1,39 (с, 9H).
МС(ИЭР)m/z: [M+H]+356.
Промежуточное соединение 7: Синтез 5-(1H-индол-2-ил)-1-метил-1-1H-пиразол-3-карбоновой кислоты
2М раствор гидроксида натрия (12,1 мл, 24,2 ммоль) добавляют к метанольному раствору промежуточного соединения 8 (1,72 г, 4,84 ммоль) при 50°C за 7 часов. После охлаждения, полученную смесь добавляют к реакционному раствору до pH 3. затем к смеси добавляют воду. Образующийся осадок фильтруют и промывают небольшим количеством воды. Получают промежуточное соединение 7 (106 г, 90% выход) в виде белого твердого вещества.
1H-ЯМР (300МГц, ДМСО-d6) δ 11,59 (с, 1H), 7,61 (д, J=7,3 Гц, 1H), 7,42 (д, J=8,1 Гц, 1H), 7,18 (т, J=8,0 Гц, 1H), 7,14 (с, 1H), 7,06 (т, J=8,1 Гц, 1H), 6,88 (с, 1H), 4,12 (с, 3H).
ОТСУТСТВУЕТ ПИК, ОБУСЛОВЛЕННЫЙ NH.
МС(ИЭР)m/z: [M+H]+242, [M-H]-240.
Промежуточное соединение 10: Синтез 5-(5-фтор-1H-индол-2-ил)-1-метил-1H-пиразол-3-карбоновой кислоты
Промежуточное соединение 9: Синтез трет-бутил-5-фтор-2-[3-(метоксикарбонил)-1-метил-1H-пиразол-5-ил]-1H-индол-1-карбоксилата
Карбонат натрия (2,53 г, 23,89 ммоль) и воду (3 мл) добавляют к промежуточному соединению 5 (2,50 г, 9,56 ммоль), ацетату палладия(II) (215 мг, 0,96 ммоль), трифенилфосфину (100 г, 3,82 ммоль) и трет-бутиловому эфиру 2-(дигидроксиборанил)-5-фтор-1H-индол-1-карбоновой кислоты (3,20 г, 11,47 ммоль) в смеси диоксана (20 мл) и толуола (10 мл). Полученную смесь нагревают до температуры кипения с обратным холодильником 1,5 часа. После охлаждения до комнатной температуры, реакционный раствор добавляют к воде (80 мл) и экстрагируют этилацетатом (100 мл ×2). Объединенный экстракт сушат над безводным сульфатом магния. Осушитель фильтруют. Фильтрат концентрируют при пониженном давлении. Полученный остаток очищают колоночной хроматографией (смесь гексан/этилацетат=2:1 (об./об.)), используя аминосиликагель. При этом промежуточное соединение 10 (990 мг, 37% выход) получают в виде белого твердого вещества.
МС(ИЭР)m/z: [M+H]+374.
Промежуточное соединение 10: синтез 5-(5-фтор-1H-индол-2-ил)-1-метил-1H-пиразол-3-карбоновой кислоты
2М раствор гидроксида натрия (6,6 мл, 13,26 ммоль) добавляют к раствору (10 мл) промежуточного соединения 9 (990 мг, 2,65 ммоль) в ТГФ. Полученный раствор перемешивают при 45°C в течение 2 часов и перемешивают при комнатной температуре в течение ночи. Реакционный раствор концентрируют при пониженном давлении. Затем, 2M раствор HCl (8 мл) добавляют к полученному остатку. Образующийся раствор экстрагируют этилацетатом (120 мл×2), и объединенный экстракт сушат над безводным сульфатом магния. После фильтрования осушителя, фильтрат концентрируют при пониженном давлении. При этом получают промежуточное соединение 10 (680 мг, 100% выход) в виде светло-коричнево-желтого твердого вещества.
1H-ЯМР (300 МГц, CDCl3/ДМСО-d6 (1 капля)) δ 10,49 (уш.с, 1H), 7,40-7,25 (м, 2H)7,13 (с, 1H), 6,66 (с, 1H), 7,00-6,94 (м, 1H), 4,15 (с, 3H).
МС(ИЭР)m/z: [M+H]+260, [M-H]-258.
Соединение по примеру 1: синтез 5-(1H-индол-2-ил)-1-метил-N-[2-(пиперидин-1-ил)этил]-1H-пиразол-3-карбоксамида
Раствор (0,5 мл) гексафторфосфата O-бензотриазол-1-ил-N,N,N',N'-тетраметилурония (36 мг, 1,5 эквивалент) в ДМФА добавляют к раствору (0,5 мл) промежуточного соединения 7 (15 мг), амина (1,1 эквивалент, 14 мг в виде 2-(пиперидин-1-ил)этан-1-амина), триэтиламина (0,026 мл, 3 эквивалент) в ДМФА при комнатной температуре. Полученный раствор перемешивают при 50°C в течение 2 часов. Затем полученный раствор концентрируют при пониженном давлении, к остатку добавляют 1М раствор гидроксида натрия (0,5 мл), и смесь дважды экстрагируют этилацетатом (1 мл). Остаток объединенного органического слоя растворяют в небольшом количестве метанола. Раствор загружают в SCX картридж (сильный катионообменный картридж), с последующей промывкой метанолом (10 мл), и, наконец, элюируют 1М раствором аммиак-метанол (8 мл). Сырой продукт, полученный концентрацией, очищают препаративной ВЭЖХ (устройство для очистки A, описанное в начале раздела {примеры}).
МС(ИЭР)m/z: [M+H]+352.
Ниже приведены примеры соединений, синтезированных с использованием взаимодействия, аналогичного описанному выше:
Соединение по примеру 2:
5-(1H-индол-2-ил)-N-[2-(4-метоксипиперидин-1-ил)этил]-1-метил-1H-пиразол-3-карбоксамид
Соединение по примеру 3:
N-[2-(4-гидроксипиперидин-1-ил)этил]-5-(1H-индол-2-ил)-1-метил-1H-пиразол-3-карбоксамид
Соединение по примеру 4:
N-[2-(4-этилпиперидин-1-ил)этил]-5-(1H-индол-2-ил)-1-метил-1H-пиразол-3-карбоксамид
Соединение по примеру 5:
N-[2-(3-гидроксипиперидин-1-ил)этил]-5-(1H-индол-2-ил)-1-метил-1H-пиразол-3-карбоксамид
Соединение по примеру 6:
1-(2-{[5-(1H-индол-2-ил)-1-метил-1H-пиразол-3-ил]формамид}этил)пиперидин-3-карбоксамид
Соединение по примеру 7:
5-(1H-индол-2-ил)-1-метил-N-{2-[4-(пропан-2-ил)пиперидин-1-ил]этил}-1H-пиразол-3-карбоксамид
Соединение по примеру 8:
5-(1H-индол-2-ил)-1-метил-N-[2-(4-метилпиперидин-1-ил)этил]-1H-пиразол-3-карбоксамид
Соединение по примеру 9:
5-(1H-индол-2-ил)-1-метил-N-[2-(пирролидин-1-ил)этил]-1H-пиразол-3-карбоксамид
Соединение по примеру 10:
5-(1H-индол-2-ил)-1-метил-N-[2-(2-метилпиперидин-1-ил)этил]-1H-пиразол-3-карбоксамид
Соединение по примеру 11:
N-[2-(4,4-дифторпиперидин-1-ил)этил]-5-(1H-индол-2-ил)-1-метил-1H-пиразол-3-карбоксамид
Соединение по примеру 12:
N-[2-(2,6-диметилморфолин-4-ил)этил]-5-(1H-индол-2-ил)-1-метил-1H-пиразол-3-карбоксамид
Соединение по примеру 13:
5-(1H-индол-2-ил)-1-метил-N-[3-(морфолин-4-ил)пропил]-1H-пиразол-3-карбоксамид
Соединение по примеру 14:
5-(5-фтор-1H-индол-2-ил)-N-{2-[(2S)-2-(метоксиметил)пирролидин-1-ил]этил}-1-метил-1H-пиразол-3-карбоксамид
Соединение по примеру 15:
5-(5-фтор-1H-индол-2-ил)-N-{2-[(2R)-2-(метоксиметил)пирролидин-1-ил]этил}-1-метил-1H-пиразол-3-карбоксамид
Соединение по примеру 16:
5-(5-фтор-1H-индол-2-ил)-N-{2-[(3S)-3-гидроксипирролидин-1-ил]этил}-1-метил-1H-пиразол-3-карбоксамид
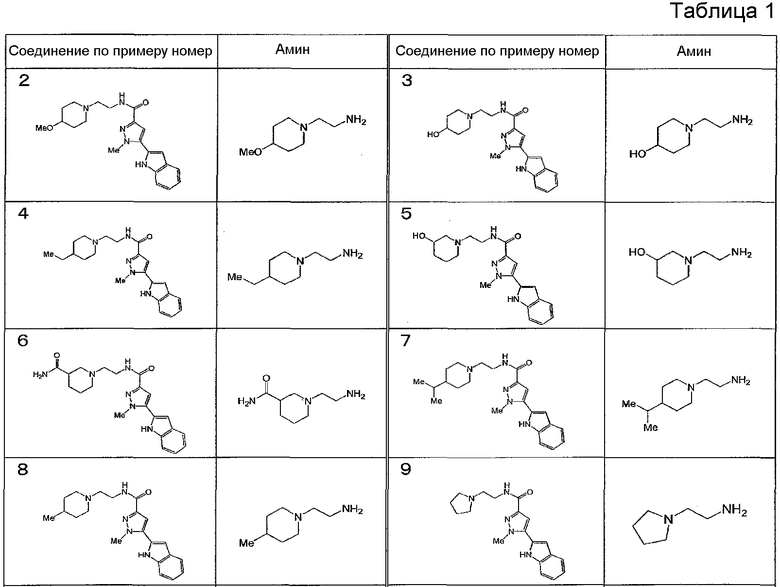
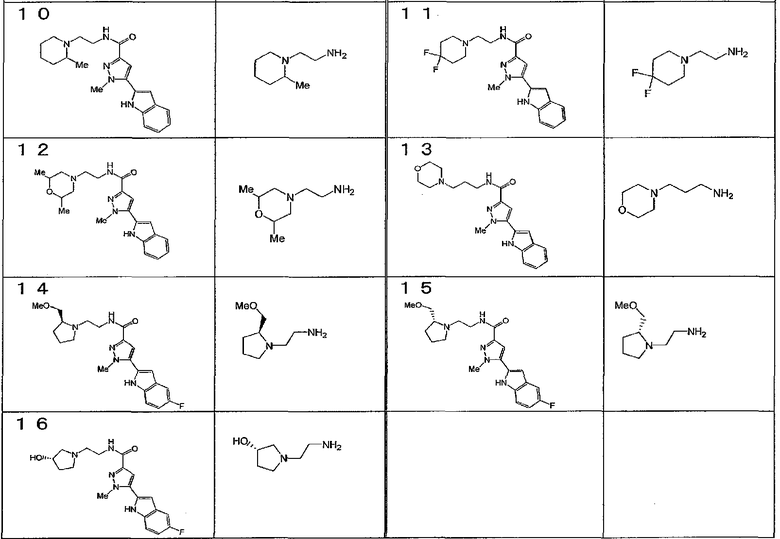
Способ синтеза соединения по примеру 17: синтез 1-метил-N-[2-(морфолин-4-ил)этил]-5-(хинолин-3-ил)-1H-пиразол-3-карбоксамида
Промежуточное соединение 11: синтез метил-1-метил-5-(хинолин-3-ил)-1H-пиразол-3-карбоксилата
Трис(дибензилиденацетон)дипалладий(0) (540 мг, 0,59 ммоль) добавляют к смешанному раствору промежуточного соединения 5 (1,57 г, 5,90 ммоль), 3-хинолинбороновой кислоты (102 г, 5,90 ммоль), фосфата калия (1,88 г, 8,85 ммоль) и 1,4-диоксана (65 мл) с три(циклогексил)фосфином (165 мг, 0,59 ммоль) и водой (15 мл). Реакционную смесь перемешивают при 100°C в течение ночи (15 часов). После охлаждения до комнатной температуры, смешанный раствор разбавляют этилацетатным растворителем, и фильтруют через целит для удаления катализатора. Затем органический слой фильтрата отделяют, водный слой вновь экстрагируют этилацетатным растворителем. После чего объединенный органический слой промывают насыщенным раствором хлорида натрия, сушат над сульфатом натрия. После фильтрования получают остаток. Очистка остатка колоночной хроматографией (смесь гексан-этилацетат=(1/1) (об./об.) - (2/3) (об./об.)), используя силикагель, что дает промежуточное соединение 11 (873 мг, 55% выход) в виде слегка желтоватых кристаллов.
1H-ЯМР (300 МГц, CDCl3) δ 8,99 (д, J=2,2 Гц, 1H), 8,23 (д, J=2,2 Гц, 1H), 8,18 (д, J=8,8 Гц, 1H), 7,79-7,75 (м, 2H), 7,54-7,52 (м, 1H), 7,04 (с, 1H), 4,05 (с, 3H), 3,98 (с, 3H).
МС(ИЭР)m/z; [M+H]+268.
Промежуточное соединение 12: синтез 1-метил-5-(хинолин-3-ил)-1H-пиразол-3-карбоновой кислоты
Метанольный раствор (30 мл) промежуточного соединения 11 (870 мг, 3,25 ммоль) и 2M раствор гидроксида натрия (4,20 мл, 8,20 ммоль) перемешивают при 75°C в течение 2 часов. После удаления растворителя, оставшийся раствор доводят до PH 6-7 раствором соляной кислоты. Осажденное твердое вещество фильтруют с отсасыванием, сушат в присутствии пентаоксида фосфора в вакууме и получают промежуточное соединение 12 (790 мг, 96% выход) в виде светло-коричневых кристаллов.
1H-ЯМР (300 МГц, ДМСО-d6) δ 12,8 (уш.с, 1H), 9,11 (д, J=2,2 Гц, 1H), 8,69 (д, J=2,2 Гц, 1H), 8,1,3-8,04 (м, 2H), 7,90-7,81 (м, 1H), 7,75-7,66 (м, 1H), 7,13 (с, 1H), 4,05 (с, 3H).
МС(ИЭР)m/z; [M+H]+254, [M-H]-252.
Способ синтеза соединения по примеру 17: синтез 1-метил-N-[2-(морфолин-1-ил)этил]-5-(хинолин-3-ил)-1H-пиразол-3-карбоксамида
Согласно подобному способу для соединения по примеру 1, соединение по примеру 17 (628 мг, 86% выход) получают в виде белых кристаллов из промежуточного соединения 12 (506 мг, 2,00 ммоль) и 4-(2-аминоэтил)морфолина (286 мг, 2,20 ммоль).
1H-ЯМР (300 МГц, CDCl3) δ 8,99 (д, J=2,2 Гц, 1H), 8,21 (д, J=2,2 Гц, 1H), 8,17 (д, J=8,1 Гц, 1H), 7,90 (д, J=8,1 Гц, 1H), 7,94-7,76 (м, 1H), 7,70-7,60 (м, 1H), 7,34-7,22 (м, 1H), 7,01 (с, 1H), 4,00 (с, 3H), 3,79-3,72 (м, 4H), 3,63-3,54 (м, 2H), 2,66-2,48 (м, 6H).
МС(ИЭР)m/z; [M+H]+366.
Ниже приведены примеры соединений, синтезированных с использованием взаимодействия, аналогичного описанному выше:
Соединение по примеру 18:
1-метил-N-[2-(пиперидин-1-ил)этил]-5-(хинолин-3-ил)-1H-пиразол-3-карбоксамид
Соединение по примеру 19:
1-метил-N-[2-(пирролидин-1-ил)этил]-5-(хинолин-3-ил)-1H-пиразол-3-карбоксамид
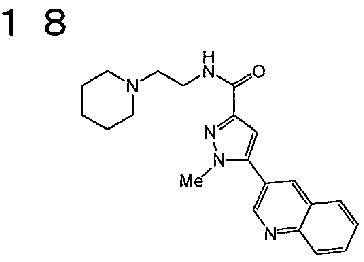
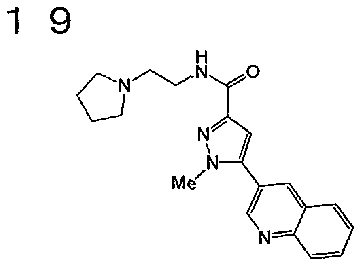

Соединение по примеру 20: синтез 1-метил-N-[2-(пирролидин-1-ил)этил]-5-{1H-пирроло[3,2-b]пиридин-2-ил}-1H-пиразол-3-карбоксамида
Промежуточное соединение 13: Синтез трет-бутил-2-(дигидроксиборанил)-1H-пирроло[3,2-b]-пиридин-1-карбоксилата
н-Бутиллитий (8,3 мл, 1,65 М, раствор в гексане) добавляют по каплям к ТГФ-раствору (10 мл) диизопропиламина (1,39 г, 13,8 ммоль) в условиях охлаждения льдом, в атмосфере азота, за 5 минут. Перемешивание полученной смеси в условиях охлаждения льдом в течение 20 минут дает ТГФ-раствор диизопропиламида лития.
ТГФ-раствор диизопропиламида лития, полученный выше, добавляют по каплям к смеси (20 мл) трет-бутил-1H-пирроло[3,2-b]пиридин-1-карбоксилата 12 (2,00 г, 9,16 ммоль) и триизопропилового эфира борной кислоты (2,76 г, 14,66 ммоль) в ТГФ при -20°C, в атмосфере азота, за 1 час. После перемешивания полученной смеси при -10°C в течение 3 часов, к реакционному раствору добавляют 10% раствор гидросульфата калия, и полученную смесь экстрагируют этилацетатом. Полученный слой экстракта промывают насыщенным раствором хлорида натрия и сушат над безводным сульфатом магния. После фильтрования, фильтрат концентрируют при пониженном давлении, выделяя сырое промежуточное соединение 13. Промежуточное соединение 13 суспендируют в диизопропиловом эфире. Образовавшийся осадок фильтруют, и получают кристаллическое промежуточное соединение 13 (1,90 г, 79% выход).
1H-ЯМР (300 МГц, ДМСО-d6) δ 8,45 (1H, д, J=4,4 Гц), 8,36 (2H, с), 8,33 (1H, д, J=8,8 Гц), 7,28 (1H, дд, J=8,1, 5,1 Гц), 6,72 (1H, с), 1,61 (9H, с).
МС(ИЭР)m/z: [M+H]+263.
Промежуточное соединение 14: синтез метил-5-{1-[(трет-бутокси)карбонил]-1H-пирроло[3,2-b]пиридин-2-ил}-1-метил-1H-пиразол-3-карбоксилата
Согласно способу получения промежуточного соединения 8, промежуточное соединение 14 (0,37 г, 27% выход) синтезируют из промежуточного соединения 13 (1,3 г, 5,0 ммоль) и промежуточного соединения 5 (10 г, 3,8 ммоль).
1H-ЯМР (300 МГц, ДМСО-d6) δ 8,61 (1H, д, J=5,1 Гц), 8,53 (1H, д, J=8,8 Гц), 7,34 (1H, дд, J=8,8, 5,1 Гц), 6,93 (2H, с), 3,96 (3H, с), 3,82 (3H, с), 1,41 (9H, с).
МС(ИЭР)m/z: [M+H]+357.
Промежуточное соединение 15: синтез 1-метил-5-{1H-пирроло[3,2-b]пиридин-2-ил}-1H-пиразол-3-карбоновой кислоты
Согласно подобному способу (гидролиз) альтернативного синтеза, описанного для промежуточного соединения 7, промежуточное соединение 15 (87 мг) синтезируют из промежуточного соединения 14 (210 мг, 0,59 ммоль) с 61% выходом.
1H-ЯМР (300 МГц, ДМСО-d6) δ 11,83 (1H, уш.с), 8,38 (1H, д, J=2,9 Гц), 7,80 (1H, д, J=8,1 Гц), 7,21 (1H, с), 7,18 (1H, дд, J=4,4, 8,1 Гц), 7,04 (1H, с), 4,15 (3H, с).
Отсутствует пик, обусловленный NH.
МС(ИЭР)m/z: [M+H]+243.
Соединение по примеру 20: синтез 1-метил-N-[2-(пирролидин-1-ил)этил]-5-{1H-пирроло[3,2-b]пиридин-2-ил}-1H-пиразол-3-карбоксамида
Промежуточное соединение 15 (120 мг, 0,5 ммоль) и N-(2-аминоэтил)пирролидин (59 мг, 0,5 ммоль) растворяют в ДМФА (4 мл). Триэтиламин (260 мг, 2,6 ммоль) и N-(3-диметиламинопропил)-N'-этилкарбодиимидгидрохлорид (98 мг, 0,5 ммоль), и 1-гидроксибензотриазолмоногидрат (39 мг, 0,3 ммоль) последовательно добавляют к полученной смеси при комнатной температуре. Полученную смесь перемешивают при комнатной температуре 2 дня. Растворитель удаляют при пониженном давлении, и полученный сырой продукт очищают на препаративной ВЭЖХ системе (устройство для очистки B), получая кристаллический продукт (15 мг, 9% выход).
МС(ИЭР)m/z: [M+H]+339.
Для дополнительной очистки используют препаративную ВЭЖХ систему (устройство для очистки A), которая была использована для очистки соединения по примеру 1.
Соединение по примеру 21: синтез N-[2-(3-гидроксипиперидин-1-ил)этил]-1-метил-5-{1H-пирроло[3,2-b]пиридин-2-ил}-1H-пиразол-3-карбоксамида
Согласно способу для соединения по примеру 20, соединение по примеру 21 (16 мг, 34% выход) получают из промежуточного соединения 15 (30 мг, 0,12 ммоль) и 1-(2-аминоэтил)пиперидин-3-ола (27 мг, 0,19 ммоль).
МС(ИЭР)m/z: [M+H]+369.
Для дополнительной очистки используют препаративную ВЭЖХ систему (устройство для очистки A), которая была использована для очистки соединения по примеру 1.
Соединение по примеру 22: Синтез 1-метил-5-{5-метил-1H-пирроло[3,2-b]пиридин-2-ил}-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Промежуточное соединение 16: синтез 5-иод-1-метил-1H-пиразол-3-карбоновой кислоты
2М раствор гидроксида натрия (56,4 ммоль, 28,2 мл) добавляют к метанольному раствору (100 мл) промежуточного соединения 5 (6,0 г, 22,6 ммоль), и смесь нагревают до 50°C. Спустя два часа, метанольный растворитель удаляют при пониженном давлении, и оставшийся раствор доводят до pH 2-3 с помощью 2М раствора HCl, в условиях охлаждения льдом. Потом полученные осажденные кристаллы растворяют в этилацетате, органический слой отделяют, и затем водный слой дважды экстрагируют этилацетатом. Затем объединенный слой органического экстракта промывают насыщенным раствором хлорида натрия и сушат над безводным сульфатом натрия. После фильтрования, фильтрат концентрируют при пониженном давлении, получая сырое промежуточное соединение 16 (5,68 г, количественный выход) в виде светло-желтого твердого вещества.
1H-ЯМР (270 МГц, ДМСО-d6) δ 12,81 (уш.с, 1H), 6,89 (с, 1H), 3,92 (с, 3H).
МС(ИЭР)m/z; [M+H]+253, [M-H]-251.
Промежуточное соединение 17: синтез 5-иод-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
4-(2-аминоэтил)морфолин (4,15 г, 31,9 ммоль), триэтиламин (12,1 мл, 86,9 ммоль), 1-гидроксибензотриазолмоногидрат (8,9 г, 57,9 ммоль) и N-(3-диметиламинопропил)-N'-этилкарбодиимидгидрохлорид (11,1 г, 57,9 ммоль) последовательно добавляют к раствору (150 мл) промежуточного соединения 16 (7,3 г, 29,0 ммоль) в дихлорметане, при комнатной температуре. Затем полученную смесь перемешивают при комнатной температуре в течение 20 часов, к реакционному раствору добавляют насыщенный раствор бикарбоната натрия, и водный слой экстрагируют дихлорметаном. Полученный органический слой сушат над безводным сульфатом натрия. После фильтрования, фильтрат концентрируют при пониженном давлении, и выделяют сырое промежуточное соединение 17. Очистка колоночной хроматографией (смесь этилацетат/метанол =9/1 (об./об.)) с использованием силикагеля дает промежуточное соединение 15 (9,8 г, 93% выход) в виде белых кристаллов.
1H-ЯМР (270МГц, CDCl3) δ 6,95 (с, 1H), 3,95 (с, 3H), 3,80-3,64 (м, 4H), 3,62-3,40 (м, 2H), 2,57 (т, J=6,3 Гц, 2H), 2,54-2,37 (м, 4H).
Отсутствует пик, обусловленный NH.
МС(ИЭР)m/z; [M+H]+365.
Промежуточное соединение 18: синтез 1-метил-N-[2-(морфолин-4-ил)этил]-5-[2-(триметилсилил)этинил]-1H-пиразол-3-карбоксамида
Триэтиламин (6,12 мл, 43,9 ммоль) добавляют к ТГФ-раствору (45 мл) промежуточного соединения 17 (4,00 г, 10,98 ммоль), иодида меди(I) (209 мг, 1,10 ммоль), триметилсилилацетилена (2,33 мл, 16,5 ммоль) и дихлорбис(ацетонитрил)палладий(II)хлорида (770 мг, 1,10 ммоль) в атмосфере азота, и реакционную смесь перемешивают при комнатной температуре 2 часа. Затем, полученную смесь фильтруют через целит, и полученный фильтрат концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией (смесь дихлорметан/метанол=30/1 (об./об.)), используя силикагель, что дает промежуточное соединение 18 (3,67 г, 100% выход) в виде желто-коричневого твердого вещества.
1H-ЯМР (300 МГц, CDCl3) δ 7,22-7,14 (уш.с, 1H), 6,90 (с, 1H), 3,93 (с, 3H), 3,78-3,70 (м, 4H), 3,58-3,48 (м, 2H), 2,62-2,45 (м, 6H), 0,28 (с, 9H).
МС(ИЭР)m/z; [M+H]+335.
Промежуточное соединение 19: синтез 5-этинил-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Метанольный раствор (60 мл) промежуточного соединения 18 (3,43 г, 10,3 ммоль) и карбоната калия (2,13 г, 15,4 ммоль) перемешивают при комнатной температуре 1,5 часа. Затем, полученную смесь фильтруют через целит, и полученный фильтрат концентрируют при пониженном давлении. Остаток разбавляют насыщенным раствором хлорид натрия/вода (1/1 (об./об.))(40 мл) и дихлорметаном (200 мл). После процесса экстракции, органический слой отделяют. Затем полученный органический слой сушат над безводным сульфатом натрия, осушитель фильтруют и фильтрат концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией (смесь дихлорметан/метанол=30/1-20/1 (об./об.)), используя силикагель, что дает промежуточное соединение 19 (2,23 г, 83% выход) в виде светло-коричневого твердого вещества.
1H-ЯМР (270МГц, CDCl3) δ 7,20 (уш.с, 1H), 6,96 (с, 1H), 3,96 (с, 3H), 3,77-3,70 (м, 4H), 3,58-3,48 (м, 3H), 2,62-2,46 (м, 6H).
МС(ИЭР)m/z; [M+H]+263.
Промежуточное соединение 20: Синтез 5-[2-(3-амино-6-метилпиридин-2-ил)этинил]-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Ацетат палладия (19 мг, 0,027 ммоль) добавляют к ацетонитрильному раствору (5,0 мл) промежуточного соединения 19 (308 мг, 1,18 ммоль), 3-амино-2-бром-6-метилпиридина (200 мг, 107 ммоль), 1,4-бис(дифенилфосфино)бутана (dppb) (18,3 мг, 0,043 ммоль) и карбоната калия (444 мг, 3,21 ммоль), в атмосфере азота. Полученную смесь перемешивают при 80°C в течение 15 часов. Затем, полученную смесь фильтруют через целит, и полученный фильтрат концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией (смесь дихлорметан/метанол=25/1-10/1 (об./об.)), используя силикагель, что дает промежуточное соединение 20 (36,9 мг, 9,4% выход) в виде темно-желтых кристаллов.
1H-ЯМР (300 МГц, CDCl3) δ 7,28-7,18 (уш.с, 1H), 7,03-6,98 (м, 3H), 4,14 (уш.с, 2H), 4,04 (с, 3H), 3,78-3,71 (м, 4H), 3,59-3,50 (м, 2H), 2,63-2,45 (м, 6H), 2,47 (с, 3H).
МС(ИЭР)m/z; [M+H]+369.
Соединение по примеру 22: синтез 1-метил-5-{5-метил-1H-пирроло[3,2-b]пиридин-2-ил}-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Трет-бутилат калия (56 мг, 0,5 ммоль) добавляют к ДМФА- (1 мл) раствору промежуточного соединения 20 (36,9 мг, 0,1 ммоль) при комнатной температуре, за один раз. Полученную смесь нагревают при перемешивании, при 35°C в течение 2 часов. После завершения взаимодействия, полученную смесь обрабатывают водой (0,5 мл) и растворитель удаляют концентрированием при пониженном давлении. Полученный остаток очищают колоночной хроматографией (смесь дихлорметан/метанол=25/1-10/1 (об./об.)), используя силикагель, что дает соединение по примеру 22 (31,3 мг, 84% выход) в виде желтых кристаллов.
1H-ЯМР (300 МГц, CDCl3) δ 10,4 (уш.с, 1H), 7,70 (д, J=8,8 Гц, 1H), 7,50-7,40 (м, 1H), 7,42 (с, 1H), 7,06 (д, J=8,8 Гц, 1H), 6,87 (с, 1H), 4,15 (с, 3H), 3,80-3,69 (м, 4H), 3,67-3,55 (м, 2H), 2,68 (с, 3H), 2,66-2,57 (м, 2H), 2,55-2,44 (м, 4H).
МС(ИЭР)m/z: [M+H]+369, [M-H]-367.
Соединение по примеру 23: Синтез 5-{5,7-диметил-1H-пирроло[3,2-b]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Промежуточное соединение 21: синтез 5-[2-(3-амино-4,6-диметилпиридин-2-ил)этинил]-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Согласно способу для соединения по примеру 20, промежуточное соединение 21 (46,8 мг, 13,3% выход) получают из промежуточного соединения 19 (240 мг, 0,915 ммоль) и 3-амино-2-бром-4,6-диметилпиридина (184 мг, 0,915 ммоль).
1H-ЯМР (300 МГц, CDCl3) δ 7,30-7,20 (м, 1H), 7,02 (с, 1H), 6,91 (с, 1H), 4,11 (уш.с, 2H), 4,04 (с, 3H), 3,80-3,71 (м, 4H), 3,60-3,50 (м, 2H), 2,65-2,48 (м, 6H), 2,43 (с, 3H), 2,19 (с, 3H).
МС(ИЭР)m/z: [M+H]+383, [M+HCO2]-427.
Соединение по примеру 23: синтез 5-{5,7-диметил-1H-пирроло[3,2-b]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Согласно подобному способу синтеза соединения по примеру 22, соединение по примеру 23 (31,3 мг, 84% выход) получают в виде светло-желтых кристаллов из промежуточного соединения 21 (46,8 мг, 0,122 ммоль).
1H-ЯМР (300 МГц, CDCl3) δ 9,75 (уш.с, 1H), 7,40-7,35 (м, 1H), 7,18 (с, 1H), 6,88 (с, 1H), 6,82 (с, 1H), 4,09 (с, 3H), 3,80-3,67 (м, 4H), 3,57-3,44 (м, 2H), 2,62 (с, 3H), 2,60-2,40 (м, 6H), 2,54 (с, 3H).
МС(ИЭР)m/z: [M+H]+383, [M-H]-381.
Соединение по примеру 24: синтез 1-метил-N-[2-(морфолин-4-ил)этил]-5-{1H-пирроло[2,3-b]пиридин-2-ил}-1H-пиразол-3-карбоксамида
Промежуточное соединение 22: синтез 5-[2-(2-аминопиридин-3-ил)этинил]-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Триэтиламин (404 мг, 4,0 ммоль) добавляют к ТГФ-раствору (6 мл) промежуточного соединения 19 (262 мг, 1,00 ммоль), 2-амино-3-иодпиридина (264 мг, 1,20 ммоль), дихлорбис(ацетонитрил)палладий(II)хлорида (70,1 мг, 0,1 ммоль) и меди(I) (19 мг, 0,1 ммоль) в атмосфере азота. Реакционный раствор перемешивают при комнатной температуре 7,5 часов. Затем, полученную смесь фильтруют через целит, и полученный фильтрат концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией (смесь дихлорметан/метанол=20/1 (об./об.)), используя силикагель, что дает промежуточное соединение 22 (207 мг, 58,4% выход) в виде желтых кристаллов.
1H-ЯМР (300 МГц, CDCl3) δ 8,14-8,08 (м, 1H), 7,65-7,60 (м, 1H), 7,27-7,20 (м, 1H), 6,99 (с, 1H), 6,73-6,65 (м, 1H), 5,06 (уш.с, 2H), 4,01 (с, 3H), 3,78-3,71 (м, 4H), 3,59-3,50 (м, 2H), 2,63-2,47 (м, 6H).
МС(ИЭР)m/z: [M+H]+355, [M-H]-353.
Соединение по примеру 24: синтез 1-метил-N-[2-(морфолин-4-ил)этил]-5-{1H-пирроло[2,3-b]пиридин-2-ил}-1H-пиразол-3-карбоксамида
Согласно подобному способу синтеза соединения по примеру 22, соединение по примеру 24 (51,5 мг, 26% выход) получают в виде желтых кристаллов из промежуточного соединения 22 (200 мг, 0,56 ммоль).
1H-ЯМР (300 МГц, CDCl3) δ 12,2 (уш.с, 1H), 8,45-8,38 (м, 1H), 8,04-7,97 (м, 1H), 7,36 (с, 1H), 7,40-7,30 (м, 1H), 7,23-7,14 (м, 1H), 6,69 (с, 1H), 4,16 (с, 3H), 3,82-3,73 (м, 4H), 3,71-3,62 (м, 2H), 2,70-2,61 (м, 2H), 2,60-2,50 (м, 4H).
МС(ИЭР)m/z: [M+H]+355, [M-H]-353.
Соединение по примеру 25: синтез 1-метил-N-[2-(морфолин-4-ил)этил]-5-{5H-пирроло[3,2-d]пиримидин-6-ил}-1H-пиразол-3-карбоксамида
Промежуточное соединение 23: синтез 5-[2-(5-аминопиридин-4-ил)этинил]-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Согласно способу синтеза промежуточного соединения 22, промежуточное соединение 23 (112 мг, 41% выход) получают в виде темно-коричневого твердого вещества нагреванием при перемешивании, при 100°C, в течение 16 часов, из соединения 19 (200 мг, 0,762 ммоль) и 4-бромпиримидин-5-амина (159 мг, 0,914 ммоль).
1H-ЯМР (300 МГц, CDCl3) δ 8,64 (с, 1H), 8,34 (с, 1H), 7,09 (с, 1H), 4,41 (уш.с, 2H), 4,06 (с, 3H), 3,82-3,69 (м, 4H), 3,64-3,51 (м, 2H), 2,62-2,46 (м, 6H).
Отсутствует пик, обусловленный NH.
МС(ИЭР)m/z: [M+H]+356, [M-H]-354.
Соединение по примеру 25: синтез 1-метил-N-[2-(морфолин-4-ил)этил]-5-{5H-пирроло[3,2-d]пиримидин-6-ил}-1H-пиразол-3-карбоксамида
Согласно подобному способу синтеза соединения по примеру 22, сырое соединение по примеру 25 (41 мг, выход 37%) получают в виде светло-коричневого твердого вещества из промежуточного соединения 23 (112 мг, 0,315 ммоль).
1H-ЯМР (300 МГц, CDCl3) δ 11,51 (уш.с, 1H), 9,03 (с, 1H), 9,00 (с, 1H), 7,65 (с, 1H), 6,94 (с, 1H), 4,22 (с, 3H), 3,80-3,62 (м, 6H), 2,67-2,42 (м, 6H).
Отсутствует пик, обусловленный NH.
МС(ИЭР)m/z: [M+H]+356, [M-H]-354.
Для дополнительной очистки используют препаративную ВЭЖХ систему (устройство для очистки A), которая была использована для очистки соединения по примеру 1.
Соединение по примеру 26: синтез 1-метил-N-[2-(морфолин-4-ил)этил]-5-{7H-пирроло[2,3-d]пиримидин-6-ил}-1H-пиразол-3-карбоксамида
Промежуточное соединение 24: синтез 5-[2-(4-аминопиридин-5-ил)этинил]-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Согласно подобному способу синтеза промежуточного соединения 22, соединение 19 (200 мг, 0,762 ммоль) и 5-иодпиримидин-4-амин (253 мг, 1,14 ммоль) нагревают при перемешивании, при 90°C, в течение 16 часов, получая сырое промежуточное соединение 24 (129 мг) в виде светло-желтого сиропа.
1H-ЯМР (300 МГц, CDCl3) δ 8,56 (с, 1H), 8,44 (с, 1H), 7,32-7,28 (м, 1H), 7,00 (с, 1H), 6,08 (уш.с, 2H), 4,00 (с, 3H), 3,76-3,73 (м, 4H), 3,57-3,51 (м, 2H), 2,63-2,49 (м, 6H).
МС(ИЭР)m/z: [M+H]+356, [M-H]-354.
Соединение по примеру 26: синтез 1-метил-N-[2-(морфолин-4-ил)этил]-5-{7H-пирроло[2,3-d]пиримидин-6-ил}-1H-пиразол-3-карбоксамида
Согласно подобному способу синтеза промежуточного соединения 22, промежуточное соединение 24 (115 мг, 0,324 ммоль) нагревают при перемешивании, при 70°C, в течение 1 часа. После чего, сырое соединение по примеру 26 (41 мг, 37% выход) получают в виде светло-коричневого твердого вещества.
1H-ЯМР (300 МГц, CDCl3) δ 12,01 (уш.с, 1H), 9,03 (с, 1H), 8,90 (с, 1H), 7,89 (с, 1H), 7,55-7,51 (м, 1H), 6,77 (с, 1H), 4,22 (с, 3H), 3,96-3,90 (м, 2H), 3,78-3,75 (м, 4H), 2,72-2,51 (м, 6H).
МС(ИЭР)m/z: [M+H]+356, [M-H]-354.
Для дополнительной очистки используют препаративную ВЭЖХ систему (устройство для очистки A), которая была использована для очистки соединения по примеру 1.
Соединение по примеру 27: синтез 1-метил-N-[2-(морфолин-4-ил)этил]-5-[5-(трифторметил)-1H-пирроло[3,2-b]пиридин-2-ил]-1H-пиразол-3-карбоксамида
Промежуточное соединение 25: синтез 5-{2-[3-амино-6-(трифторметил)пиридин-2-ил]этинил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Согласно подобному способу синтеза промежуточного соединения 20, промежуточное соединение 19 (262 мг, 1,00 ммоль) и 3-амино-2-иод-6-трифторметилпиридин (288 мг, 1,00 ммоль) нагревают при перемешивании, при 80°C, в течение 14 часов и получают сырой продукт. Продукт очищают колоночной хроматографией (смесь дихлорметан/метанол=30/1-20/1 (об./об.)), используя силикагель, что дает промежуточное соединение 25 (245 мг, 58% выход) в виде белых кристаллов.
1H-ЯМР (300 МГц, CDCl3) δ 7,48 (д, J=8,8 Гц, 1H), 7,28-7,20 (м, 1H), 7,15 (д, J=8,8 Гц, 1H), 7,05 (с, 1H), 4,67 (уш.с, 2H), 4,05 (с, 3H), 3,80-3,69 (м, 4H), 3,59-3,49 (м, 2H), 2,63-2,45 (м, 6H).
МС(ИЭР)m/z: [M+H]+423, [M-H]-421.
Соединение по примеру 27: синтез 1-метил-N-[2-(морфолин-4-ил)этил]-5-[5-(трифторметил)-1H-пирроло[3,2-b]пиридин-2-ил]-1H-пиразол-3-карбоксамида
Согласно подобному способу синтеза соединения по примеру 22, сырой продукт получают в виде белых кристаллов из промежуточного соединения 25 (240 мг, 0,578 ммоль) и трет-бутилата калия (324 мг, 2,89 ммоль). Продукт очищают колоночной хроматографией (смесь дихлорметан/метанол=15/1 (об./об.)), используя силикагель, что дает промежуточное соединение 27 (187 мг, 78% выход) в виде белых кристаллов.
1H-ЯМР (300 МГц, ДМСО-d6) δ 12,3 (уш.с, 1H), 8,17-8,08 (м, 1H), 8,03 (д, J=8,8 Гц, 1H), 7,64 (д, J=8,8 Гц, 1H), 7,23 (с, 1H), 7,21 (с, 1H), 4,17 (с, 3H), 3,62-3,52 (м, 4H), 3,4, 4-3,30 (м, 2H), 2,54-2,36 (м, 6H).
МС(ИЭР)m/z: [M+H]+383, [M-H]-381.
Соединение по примеру 28: синтез 1-метил-5-{5-метил-1H-пирроло[2,3-b]пиридин-2-ил}-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Промежуточное соединение 26: синтез 5-[2-(2-амино-5-метилпиридин-3-ил)этинил]-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Согласно подобному способу синтеза промежуточного соединения 20, промежуточное соединение 19 (393 мг, 1,50 ммоль) и 2-амино-3-иод-5-метилпиридин (421 мг, 1,80 ммоль) нагревают при перемешивании, при 80°C, в течение 14 часов и получают сырой продукт. Продукт очищают колоночной хроматографией (смесь дихлорметан/метанол=30/1-20/1 (об./об.)), используя силикагель, что дает промежуточное соединение 26 (245 мг, 58% выход) в виде белых кристаллов.
1H-ЯМР (300 МГц, CDCl3) δ 7,94 (д, J=2,2 Гц, 1H), 7,46 (д, J=2,2 Гц, 1H), 7,30-7,18 (м, 1H), 6,98 (с, 1H), 4,88 (уш.с, 2H), 4,00 (с, 3H), 3,78-3,70 (м, 4H), 3,60-3,50 (м, 2H), 2,64-2,46 (м, 6H), 2,21 (с, 3H).
МС(ИЭР)m/z: [M+H]+369.
Соединение по примеру 28: синтез 1-метил-5-{5-метил-1H-пирроло[2,3-b]пиридин-2-ил}-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Согласно подобному способу синтеза соединения по примеру 22, сырой продукт получают из промежуточного соединения 26 (460 мг, 1,25 ммоль) и трет-бутилата калия (1200 мг, 10,7 ммоль). Продукт очищают колоночной хроматографией (смесь дихлорметан/метанол=50/1 (об./об.)), используя аминосиликагель, и перекристаллизовывают из смеси метанола и диизопропилового эфира, что дает соединение по примеру 28 (229 мг, 50% выход) в виде белых кристаллов.
1H-ЯМР (300 МГц, CDCl3) δ 11,8 (уш.с, 1H), 8,25 (д, J=1,5 Гц, 1H), 7,78 (уш.с, 1H), 7,38-7,27 (м, 1H), 7,34 (с, 1H), 6,59 (с, 1H), 4,14 (с, 3H), 3,80-3,62 (м, 6H), 2,68-2,50 (м, 6H), 2,47 (с, 3H).
МС(ИЭР)m/z: [M+H]+369.
Соединение по примеру 29: синтез 5-{5-фтор-1H-пирроло[2,3-b]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Промежуточное соединение 27: 5-[2-(2-амино-5-фторпиридин-3-ил)этинил]-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Триэтиламин (309 мг, 3,05 ммоль) добавляют к ТГФ- (10 мл) раствору промежуточного соединения 19 (200 мг, 0,76 ммоль), 5-фтор-3-иодпиридин-2-амина (218 мг, 0,915 ммоль), дихлорбис(трифенилфосфин)палладий(II)дихлорида (54 мг, 0,076 ммоль), иодида меди(I) (14,5 мг, 0,076 ммоль) и смесь перемешивают при комнатной температуре в течение двух дней. Реакционный раствор концентрируют при пониженном давлении, и полученный маслянистый коричневый остаток очищают колоночной хроматографией (этилацетат в качестве растворителя), используя аминосиликагель, что дает промежуточное соединение 27 (187 мг, 66% выход) в виде белого твердого вещества.
1H-ЯМР (300 МГц, CDCl3) δ 8,00 (д, J=2,9 Гц, 1H), 7,39 (дд, J=2,9, 8,0 Гц, 1H), 7,01 (с, 1H), 4,91 (уш.с, 2H), 4,01 (с, 3H), 3,74 (т, J=4,4 Гц, 4H), 3,54 (дд, J=5,9, 11,7 Гц, 2H), 2,59 (т, J=6,6 Гц, 2H), 2,50-2,20 (м, 4H).
Отсутствует пик, обусловленный NH амида.
МС(ИЭР)m/z: [M+H]+373.
Соединение по примеру 29: синтез 5-{5-фтор-1H-пирроло[2,3-b]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Согласно подобному способу синтеза соединения по примеру 22, сырой продукт получают из промежуточного соединения 27 (180 мг, 0,48 ммоль) и трет-бутилата калия (271 мг, 2,41 ммоль). Продукт очищают колоночной хроматографией (смесь дихлорметан/метанол=10/1 (об./об.)), используя силикагель, и перекристаллизовывают из смеси метанола и диизопропилового эфира, что дает промежуточное соединение 29 (110 мг, 61% выход) в виде белых кристаллов.
1H-ЯМР (300 МГц, CDCl3) δ 11,30 (уш.с, 1H), 8,23 (д, J=2,2 Гц, 1H), 7,64 (дд, J=2,9, 8,8 Гц, 1H), 7,58 (с, 1H), 7,41 (уш.с, 1H), 6,66 (д, J=2,2 Гц, 1H), 4,17 (с, 3H), 3,83-3,70 (м, 6H), 2,66 (т, J=5,9 Гц, 2H), 2,56-2,50 (м, 4H).
МС(ИЭР)m/z: [M+H]+373, [M-H]-371.
Соединение по примеру 30: синтез 5-{5-циано-1H-пирроло[3,2-b]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Промежуточное соединение 28: 5-[2-(3-амино-6-цианопиридин-2-ил)этинил]-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Согласно подобному способу синтеза соединения по примеру 20, промежуточное соединение 19 (262 мг, 1,00 ммоль) и 3-амино-2-иод-6-цианопиридин (245 мг, 1,00 ммоль) нагревают при перемешивании, при 80°C, в течение 14 часов и получают сырой продукт. Продукт очищают колоночной хроматографией (смесь дихлорметан/метанол=20/1 (об./об.)), используя силикагель, что дает промежуточное соединение 28 (231 мг, 61% выход) в виде светло-коричневого твердого вещества.
1H-ЯМР (300 МГц, ДМСО-d6) δ 8,20-8,12 (м, 1H), 7,68 (д, J=8,8 Гц, 1H), 7,18 (д, J=8,8 Гц, 1H), 7,11 (с, 1H), 6,83 (уш.с, 2H), 4,02 (с, 3H), 3,60-3,53 (м, 4H), 3,40-3,30 (м, 2H), 2,48-2,35 (м, 6H).
МС(ИЭР)m/z: [M+H]+380.
Соединение по примеру 30: 5-{5-циано-1H-пирроло[3,2-b]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Согласно подобному способу синтеза соединения по примеру 22, сырой продукт получают из промежуточного соединения 28 (227 мг, 0,60 ммоль) и трет-бутилата калия (674 мг, 6,00 ммоль). Продукт очищают колоночной хроматографией (смесь дихлорметан/метанол=15/1 (об./об.)), используя силикагель, и перекристаллизовывают из смеси метанола и диизопропилового эфира, что дает соединение по примеру 30 (229 мг, 50% выход) в виде белых кристаллов.
1H-ЯМР (300 МГц, ДМСО-d6) δ 12,5 (уш.с, 1H), 8,20-8,12 (м, 1H), 8,00 (д, J=8,8 Гц, 1H), 7,76 (д, J=8,8 Гц, 1H), 7,22 (общий 2H в результате наложения различных (с, 1H) пиков), 4,17 (с, 3H), 3,62-3,52 (м, 4H), 3,43-3,30 (м, 2H), 2,55-2,35 (м, 6H).
МС(ИЭР)m/z: [M+H]+380, [M-H]-378.
Соединение по примеру 31: синтез 5-{6-фтор-1H-пирроло[3,2-b]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Промежуточное соединение 29: 5-[2-(5-фторпиридин-3-нитропиридин-2-ил)этинил]-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Согласно подобному способу синтеза соединения по примеру 29, промежуточное соединение 29 (307 мг, 50% выход) получают в виде коричневато-желтого твердого вещества из промежуточного соединения 19 (400 мг, 1,53 ммоль), 2-хлор-5-фтор-3-нитропиридина (323 мг, 1,83 ммоль), бис(трифенилфосфин)палладий(II)дихлорида (176 мг, 0,15 ммоль), иодида меди(I) (29 мг, 0,15 ммоль) и триэтиламина (617 мг, 6,10 ммоль).
1H-ЯМР (300 МГц, CDCl3) δ 8,81 (д, J=2,2 Гц, 1H), 8,24 (дд, J=2,2, 7,3 Гц, 1H), 7,18 (с, 1H), 4,13 (с, 3H), 3,75 (т, J=5,1 Гц, 4H), 3,58-3,52 (м, 2H), 2,59 (т, J=5,9 Гц, 2H), 2,58-2,49 (м, 4H).
Отсутствует пик, обусловленный NH амида.
Промежуточное соединение 30: 5-[2-(3-амино-5-фторпиридин-2-ил)этинил]-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Метанольный раствор промежуточного соединения 29 (690 мг, 1,72 ммоль), хлорид олова(II) (4877 мг, 25,7 ммоль), хлорид аммония (1376 мг, 25,7 ммоль) и воду (927 мг, 51,4 ммоль) перемешивают при 80°C в течение 16 часов. После охлаждения до комнатной температуры, смесь доводят до pH 12 с помощью 2M раствора гидроксида натрия. Полученную смесь фильтруют через целит и промывают метанолом. Фильтрат концентрируют при пониженном давлении, и остаток разбавляют дихлорметаном, и промывают насыщенным раствором хлорида натрия, и сушат над безводным сульфатом натрия. После фильтрования осушителя, полученный остаток концентрируют при пониженном давлении, получая сырое промежуточное соединение 30 (405 мг, 63%) в виде желтого твердого вещества.
МС(ИЭР)m/z: [M+H]+373, [M-H]-371.
Соединение по примеру 31: 5-{6-фтор-1H-пирроло[3,2-b]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Согласно подобному способу синтеза соединения по примеру 22, сырой продукт получают из промежуточного соединения 30 (400 мг, 107 ммоль) и трет-бутилата калия (603 мг, 5,37 ммоль). Продукт очищают колоночной хроматографией (смесь дихлорметан/метанол=20/1 (об./об.)), используя силикагель, и перекристаллизовывают из метанола, что дает соединение по примеру 31 (71 мг, 18% выход) в виде белого твердого вещества.
1H-ЯМР (300 МГц, CDCl3) δ 11,23 (уш.с, 1H), 8,36 (с, 1H), 7,46 (д, J=8,8 Гц, 1H), 7,22 (с, 1H), 6,87 (с, 1H), 4,14 (с, 3H), 3,77-3,73 (м, 4H), 3,60-3,50 (м, 2H), 2,63-2,50 (м, 6H).
Отсутствует пик, обусловленный NH пирроло[3,2-b]пиридина.
МС(ИЭР)m/z: [M+H]+373, [M-H]-371.
Соединение по примеру 32: синтез 1-метил-N-[2-(морфолин-4-ил)этил]5-{5H-пирроло[2,3-b]пиразин-6-ил}-1H-пиразол-3-карбоксамида
Промежуточное соединение 31: 5-[2-(3-амино-пиразин-2-ил)этинил]-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Согласно подобному способу синтеза соединения по примеру 20, промежуточное соединение 19 (393 мг, 1,50 ммоль) и 2-амино-3-хлорпиразин (233 мг, 1,80 ммоль) нагревают при перемешивании, при 80°C, в течение 14 часов и получают сырой продукт. Продукт очищают колоночной хроматографией (смесь дихлорметан/метанол=20/1-15/1 (об./об.)), используя силикагель, что дает промежуточное соединение 31 (272 мг, 51% выход) в виде светло-желтого твердого вещества.
1H-ЯМР (300 МГц, CDCl3) δ 8,04 (д, J=2,2 Гц, 1H), 7,99 (д, J=2,2 Гц, 1H), 7,30-7,20 (м, 1H), 7,07 (с, 1H), 5,13 (уш.с, 2H), 4,05 (с, 3H), 3,80-3,70 (м, 4H), 3,60-3,50 (м, 2H), 2,65-2,45 (м, 6H).
МС(ИЭР)m/z: [M+H]+380, [M-H]-378.
Соединение по примеру 32: 1-метил-N-[2-(морфолин-4-ил)этил]5-{5H-пирроло[2,3-b]пиразин-6-ил}-1H-пиразол-3-карбоксамид
Согласно подобному способу синтеза соединения по примеру 22, сырой продукт получают из промежуточного соединения 31 (267 мг, 0,75 ммоль) и трет-бутилата калия (420 мг, 3,76 ммоль). Продукт очищают колоночной хроматографией (смесь дихлорметан/метанол=20/1-15/1 (об./об.)), используя силикагель, и перекристаллизовывают из метанола и диизопропилового эфира, что дает соединение по примеру 32 (183 мг, выход 69%) в виде белых кристаллов.
1H-ЯМР (300 МГц, ДМСО-d6) δ 12,6 (уш.с, 1H), 8,46 (д, J=2,9 Гц, 1H), 8,31 (д, J=2,9 Гц, 1H), 8,18-8,09 (м, 1H), 7,25 (с, 1H), 7,13 (с, 1H), 4,17 (с, 3H), 3,62-3,53 (м, 4H), 3,43-3,30 (м, 2H), 2,55-2,35 (м, 6H).
МС(ИЭР)m/z: [M+H]+356, [M-H]-354.
Соединение по примеру 33: синтез 5-{5-циано-1H-пирроло[2,3-b]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Промежуточное соединение 32: 5-[2-(2-амино-5-цианопиридин-3-ил)этинил]-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Согласно подобному способу синтеза соединения по примеру 20, промежуточное соединение 19 (393 мг, 1,50 ммоль) и 2-амино-3-иод-5-цианопиридин (441 мг, 1,80 ммоль) нагревают при перемешивании, при 80°C, в течение 14 часов и получают сырой продукт. Продукт очищают колоночной хроматографией (смесь дихлорметан/метанол=15/1 (об./об.)), используя силикагель, что дает промежуточное соединение 32 (122 мг, 21% выход) в виде светло-желтого твердого вещества.
1H-ЯМР (300 МГц, ДМСО-d6) δ 8,43 (д, J=2,2 Гц, 1H), 8,16 (д, J=2,2 Гц, 1H), 8,18-8,08 (м, 1H), 7,50 (уш.с, 2H), 7,02 (с, 1H), 4,00 (с, 3H), 3,60-3,53 (м, 4H), 3,40-3,30 (м, 2H), 2,48-2,35 (м, 6H).
МС(ИЭР)m/z: [M+H]+380, [M-H]-378.
Соединение по примеру 33: 5-{5-циано-1H-пирроло[2,3-b]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Согласно подобному способу синтеза соединения по примеру 22, сырой продукт получают из промежуточного соединения 32 (122 мг, 0,32 ммоль) и трет-бутилата калия (360 мг, 3,20 ммоль). Продукт очищают колоночной хроматографией (смесь дихлорметан/метанол=15/1 (об./об.)), используя силикагель, и перекристаллизовывают из метанола и диизопропилового эфира, что дает соединение по примеру 33 (57 мг, 47% выход) в виде белых кристаллов.
1H-ЯМР (300 МГц, ДМСО-d6) δ 12,9 (уш.с, 1H), 8,68 (д, J=2,2 Гц, 1H), 8,60 (д, J=2,2 Гц, 1H), 8,16-8,08 (м, 1H)7,22 (с, 1H), 7,04 (с, 1H)4,14 (с, 3H), 3,62-3,53 (м, 4H), 3,42-3,30 (м, 2H), 2,55-2,35 (м, 6H).
МС(ИЭР)m/z: [M+H]+380, [M-H]-378.
Соединение по примеру 34: синтез 5-{имидазо[1,2-a]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Промежуточное соединение 33: синтез этил-5-ацетил-1-метил-1H-пиразол-3-карбоксилата
ДМФА-раствор (10 мл) этил-5-ацетил-1H-пиразол-3-карбоксилата (1,50 г, 8,23 ммоль, ссылка: US5470862) добавляют по каплям к ДМФА-суспензии (10 мл) 60% (масс./масс.; в масле) гидрида натрия (442 мг, 11,5 ммоль) в условиях охлаждения льдом. Полученную смесь перемешивают при комнатной температуре в течение 10 минут, и к реакционному раствору добавляют иодметан (103 мл, 16,5 ммоль) и перемешивают при комнатной температуре 30 минут. После чего, реакционный раствор добавляют к воде (20 мл) и водный слой экстрагируют диэтиловым эфиром (80 мл × 2). Затем полученный органический слой сушат над сульфатом магния, осушитель фильтруют и фильтрат концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией (смесь гексан/этилацетат=10/1-6/1 (об./об.)), используя силикагель, что дает промежуточное соединение 33 (180 мг, 11% выход) в виде белых твердых кристаллов.
1H-ЯМР (300 МГц, CDCl3) δ 7,36 (с, 1H), 4,43 (кв, J=7,3 Гц, 2H), 4,24 (с, 3H), 2,56 (с, 3H), 1,42 (т, J=7,3 Гц, 3H).
МС(ИЭР)m/z: [M+H]+197.
Промежуточное соединение 34: синтез этил-5-(2-бромацетил)-1-метил-1H-пиразол-3-карбоксилата
Фенилтриметиламмонийтрибромид (148 мг, 0,40 ммоль) добавляют к ТГФ-раствору (1 мл) промежуточного соединения 33 (79 мг, 0,40 ммоль), и смесь перемешивают при комнатной температуре 1,5 часа. Полученный реакционный раствор добавляют к воде (20 мл). Водный слой экстрагируют диэтиловым эфиром (20 мл ×2). Затем полученный органический слой сушат над сульфатом магния, осушитель фильтруют и фильтрат концентрируют при пониженном давлении. Остаточное твердое вещество промывают небольшим количеством смеси изопропиловый эфир/гексан=1/2 (об./об.), и промежуточное соединение 34 (114 мг, количественный выход) получают в виде белых кристаллов.
1H-ЯМР (300 МГц, CDCl3) δ 7,45 (с, 1H), 4,44 (кв, J=7,3 Гц, 2H), 4,31 (с, 2H), 4,26 (с, 3H), 1,42 (т, J=7,3 Гц, 3H).
МС(ИЭР)m/z: [M+H]+275 и 277.
Промежуточное соединение 35: синтез этил-5-{имидазо[1,2-a]пиридин-2-ил}-1-метил-1H-пиразол-3-карбоксилата
Метанольный раствор (2 мл) промежуточного соединения 34 (114 мг, 0,41 ммоль) и 2-аминопиридина (39 мг, 0,41 ммоль) нагревают до температуры кипения с обратным холодильником в течение 20 часов. После охлаждения, реакционный раствор добавляют к насыщенному раствору бикарбоната натрия (20 мл) и водный слой экстрагируют дихлорметаном (20 мл ×2). Полученный органический слой сушат над сульфатом магния. Затем осушители фильтруют, фильтрат концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией (смесь гексан/этилацетат=1/3 (об./об.)), используя силикагель, что дает промежуточное соединение 35 (64 мг, 57% выход) в виде светло-желтого твердого вещества.
1H-ЯМР (300 МГц, CDCl3) δ 8,16 (д, J=6,6 Гц, 1H), 7,81 (с, 1H), 7,65 (д, J=8,8 Гц, 1H), 7,28-7,20 (м, 1H), 7,09 (с, 1H), 6,87 (т, J=7,3 Гц, 1H), 4,43 (кв, J=6,6 Гц, 2H), 4,34 (с, 3H), 1,43 (т, J=6,6 Гц, 3H).
МС(ИЭР)m/z: [M+H]+271.
Промежуточное соединение 36: синтез 5-{имидазо[1,2-a]пиридин-2-ил}-1-метил-1H-пиразол-3-карбоновой кислоты
2М раствор гидроксида натрия (0,25 мл, 0,50 ммоль) добавляют к метанольному раствору (1 мл) промежуточного соединения 35 (60 мг, 0,22 ммоль), и смесь перемешивают при 70°C в течение 1 часа. После охлаждения, к реакционному раствору добавляют 2М раствор HCl (0,25 мл, 0,50 ммоль), и полученную смесь концентрируют при пониженном давлении, что дает промежуточное соединение 36 (54 мг) в виде белых кристаллов, представляющих собой смесь хлорида натрия. Промежуточное соединение используют для последующего взаимодействия без дополнительной очистки.
1H-ЯМР (300 МГц, CDCl3) δ 8,57 (д, J=7,3 Гц, 1H), 8,42 (с, 1H), 7,64 (д, J=10,2 Гц, 1H), 7,33 (т, 10,3 Гц, 1H), 7,00 (с, 1H), 6,98 (т, J=7,3 Гц, 1H), 4,25 (с, 3H).
Отсутствует пик, обусловленный COOH.
МС(ИЭР)m/z: [M+H]+243, [M-H]-241.
Соединение по примеру 34: синтез 5-{имидазо[1,2-a]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Гексафторфосфат-O-бензотриазол-1-ил-N,N,N',N'-тетраметилуроний (126 мг, 0,33 ммоль) добавляют к ацетонитрильному раствору (1 мл) промежуточного соединения 36 (54 мг), 4-(2-аминоэтил)морфолина (32 мг, 0,24 ммоль) и триэтиламина (0,09 мл, 0,67 ммоль), и смесь перемешивают при комнатной температуре. После чего полученный раствор концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией (смесь дихлорметан/метанол=30/1 (об./об.)), используя силикагель, обработанный амином, что дает сырой бесцветный маслянистый продукт (79 мг).
1H-ЯМР (300 МГц, CDCl3) δ 8,17 (д, J=7,3 Гц, 1H), 7,83 (с, 1H), 7,63 (д, J=9,5 Гц, 1H), 7,30-7,20 (м, 1H), 7,04 (с, 1H), 6,86 (т, J=6,6 Гц, 1H), 6,15 (уш.с, 1H), 4,30 (с, 3H), 3,78-3,68 (м, 4H), 3,61-3,53 (м, 2H), 2,65-2,45 (м, 6H).
МС(ИЭР)m/z: [M+H]+355.
Для дополнительной очистки используют препаративную ВЭЖХ систему (устройство для очистки A), которая была использована для очистки соединения по примеру 1.
Соединение по примеру 35: синтез 5-(1,3-бензотиазол-2-ил)-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
ДМСО-раствор (0,7 мл) промежуточного соединения 17 (100 мг, 0,275 ммоль), бензотиазола (31 мг, 0,229 ммоль), иодида меди(I) (44 мг, 0,229 ммоль), трифенилфосфина (12 мг, 0,046 ммоль) и трикалийфосфата (97 мг, 0,458 ммоль) перемешивают при 160°C в течение 1 часа, в атмосфере азота. Полученный остаток очищают колоночной хроматографией (смесь дихлорметан/метанол=10/1 (об./об.)) и обрабатывают на SCX (сильный катионообменный картридж) способом, аналогичным описанному для соединения по примеру 1, что дает сырой продукт 35 (100 мг).
Для дополнительной очистки используют препаративную ВЭЖХ систему (устройство для очистки A), которая была использована для очистки соединения по примеру 1.
МС(ИЭР)m/z: [M+H]+372.
Соединение по примеру 36: Синтез 5-{5-фтор-1-метил-1H-пирроло[2,3-b]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Метилиодид (14,2 мг, 0,10 ммоль) добавляют к ацетонитрильному раствору (5 мл) соединения по примеру 29 (30 мг, 0,08 ммоль) и карбоната цезия (52 мг, 0,16 ммоль). Реакционный раствор перемешивают при 60°C в течение 3 часов. После охлаждения до комнатной температуры, реакционный раствор разбавляют дихлорметаном. Затем полученный раствор промывают водой и насыщенным раствором хлорида натрия, и сушат над сульфатом натрия. Затем осушитель фильтруют, фильтрат концентрируют при пониженном давлении, и сырой продукт (32 мг) получают в виде желтого аморфного вещества. Для дополнительной очистки используют препаративную ВЭЖХ систему (устройство для очистки A), которая была использована для очистки соединения по примеру 1.
МС(ИЭР)m/z: [M+H]+387.
Соединение по примеру 37: 5-(5-фтор-1H-индол-2-ил)-N-{2-[(2R)-2-(гидроксиметил)пирролидин-1-ил]этил}-1-метил-1H-пиразол-3-карбоксамид
Промежуточное соединение 37: синтез N-(2,2-диметоксиэтил)-5-(5-фтор-1H-индол-2-ил)-1-метил-1H-пиразол-3-карбоксамида
Гексафторфосфат-O-бензотриазол-1-ил-N,N,N',N'-тетраметилуроний (505 мг, 1,33 ммоль) добавляют к ацетонитрильному раствору (10 мл) промежуточного соединения 10 (230 мг, 0,89 ммоль), диэтилацеталя аминоацетальдегида (93 мг, 0,89 ммоль), триэтиламина (0,50 мл, 3,55 ммоль) при комнатной температуре. Реакционный раствор перемешивают при комнатной температуре 16 часов. Реакционный раствор разбавляют этилацетатом. Затем полученный раствор промывают водой, органический слой сушат над сульфатом магния. Затем осушитель фильтруют, фильтрат концентрируют при пониженном давлении. остаток очищают колоночной хроматографией (смесь гексан/этилацетат=1/2 (об./об.)), используя силикагель, что дает промежуточное соединение 37 (307 мг, количественный выход) в виде белого твердого вещества.
1H-ЯМР (300 МГц, CDCl3) δ 9,28 (с, 1H), 7,42-7,39 (м, 1H), 7,32-7,26 (м, 1H), 7,20-7,12 (м, 2H), 7,05-6,98 (м, 1H), 6,68 (д, J=1,5 Гц, 1H), 4,50 (т, J=5,9 Гц, 1H), 4,11 (с, 3H), 3,64 (т, J=5,9 Гц, 2H), 3,42 (с, 6H).
МС(ИЭР)m/z: [M-H]-345.
Промежуточное соединение 38: синтез 5-(5-фтор-1H-индол-2-ил)-1-метил-N-(2-оксоэтил)-1H-пиразол-3-карбоксамида
2М раствор HCl (5 мл) добавляют к ТГФ-раствору (5 мл) промежуточного соединения 37 (294 мг, 0,85 ммоль) и полученный раствор перемешивают при 50°C в течение 2 часов. После охлаждения, реакционный раствор нейтрализуют, добавляя 2М раствор гидроксида натрия (5 мл), и экстрагируют этилацетатом. Полученный органический слой промывают насыщенным раствором хлорида натрия и сушат над сульфатом магния. Затем осушитель фильтруют, фильтрат концентрируют при пониженном давлении, получая промежуточное соединение 38 (300 мг, количественный выход) в виде белого твердого вещества. Промежуточное соединение используют для последующего взаимодействия без дополнительной очистки.
МС(ИЭР)m/z: [M+H]+301.
Соединение по примеру 37: 5-(5-фтор-1H-индол-2-ил)-N-{2-[(2R)-2-(гидроксиметил)пирролидин-1-ил]этил}-1-метил-1H-пиразол-3-карбоксамид
ТГФ-раствор (4,2 мл) промежуточного соединения 38 (30 мг, 0,10 ммоль), (R)-(-)-2-пирролидинметанола (15 мг, 0,15 ммоль) и уксусной кислоты (0,15 мл) перемешивают при комнатной температуре в течение 10 минут. После чего к раствору добавляют ТГФ-раствор (0,2 мл) триацетоксиборгидрида натрия (63 мг, 0,30 ммоль) и образовавшийся раствор перемешивают в течение ночи при комнатной температуре. Затем реакционный раствор концентрируют, остаток полностью растворяют добавлением этилацетата (0,7 мл) и 2М раствора гидроксида натрия (0,5 мл). Затем полученный органический слой загружают на SCX картридж (сильный катионообменный картридж), промывают метанолом (5 мл) и, наконец, элюируют смесью аммиак-метанол (1M, 4 мл). Сырой продукт, полученный концентрированием, очищают препаративной ВЭЖХ (устройство для очистки A, описанное в начале раздела ПРИМЕРЫ).
МС(ИЭР)m/z: [M+H]+386.
Ниже приведены примеры соединений, синтезированных с использованием взаимодействия, аналогичного описанному выше:
Соединение по примеру 38:
5-(5-фтор-1H-индол-2-ил)-N-{2-[(3R)-3-(гидроксипирролидин-1-ил]этил}-1-метил-1H-пиразол-3-карбоксамид
Соединение по примеру 39:
5-(5-фтор-1H-индол-2-ил)-N-{2-[(3S)-3-(гидроксипиперидин-1-ил]этил}-1-метил-1H-пиразол-3-карбоксамид
Соединение по примеру 40:
5-(5-фтор-1H-индол-2-ил)-1-метил-N-{2-[(1S,4S)-2-окса-5-азабицикло[2.2.1]гептан-5-ил]этил}-1H-пиразол-3-карбоксамид
Соединение по примеру 41:
N-[2-(3,3-дифторазетидин-1-ил)этил]-5-(5-фтор-1H-индол-2-ил)-1-метил-1H-пиразол-3-карбоксамид
Соединение по примеру 42:
5-(5-фтор-1H-индол-2-ил)-N-{2-[(2S)-2-(гидроксиметил)пирролидин-1-ил]этил}-1-метил-1H-пиразол-3-карбоксамид
Соединение по примеру 43:
5-(5-фтор-1H-индол-2-ил)-N-{2-[(3R)-3-фторпирролидин-1-ил]этил}-1-метил-1H-пиразол-3-карбоксамид
Соединение по примеру 44:
5-(5-фтор-1H-индол-2-ил)-N-{2-[(3S)-3-фторпирролидин-1-ил]этил}-1-метил-1H-пиразол-3-карбоксамид
Соединение по примеру 45:
5-(5-фтор-1H-индол-2-ил)-N-[2-(4-фторпиперидин-1-ил]этил}-1-метил-1H-пиразол-3-карбоксамид
Соединение по примеру 46:
5-(5-фтор-1H-индол-2-ил)-1-метил-N-[2-(1,4-оксазепам-4-ил]этил}-1H-пиразол-3-карбоксамид
Соединение по примеру 47:
N-[2-(азетидин-1-ил)этил]-5-(5-фтор-1H-индол-2-ил)-1-метил-1H-пиразол-3-карбоксамид
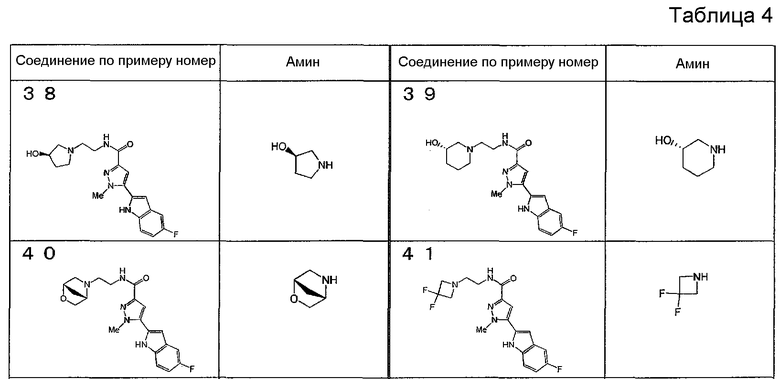
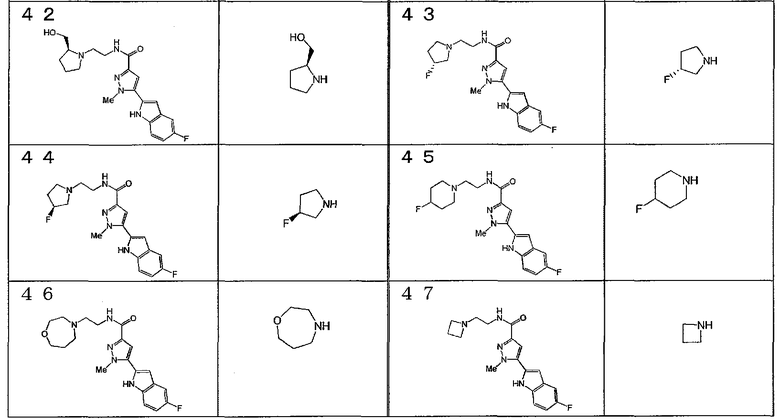
Альтернативный способ синтеза соединения по примеру 17: синтез 1-метил-N-[2-(морфолин-4-ил)этил]-5-(хинолин-3-ил)-1H-пиразол-3-карбоксамида
Согласно приведенному выше подобному способу для промежуточного соединения 11, трис(дибензилиденацетон)дипалладий(0) (66 мг, 0,0723 ммоль) добавляют к смешанному раствору, 1,4-диоксан (20 мл)-вода(3 мл), промежуточного соединения 17 (526 мг, 1,44 ммоль), 3-хинолинбороновой кислоты (250 мг, 1,44 ммоль), фосфата калия (458 мг, 2,16 ммоль) и трициклогексилфосфина (40,4 мг, 0,14 ммоль). Реакционный раствор перемешивают при 100°C в течение ночи (15 часов). Полученный остаток очищают колоночной хроматографией (смесь дихлорметан/метанол=20/1 (об./об.)), используя силикагель, что дает белые кристаллы. Затем соединение по примеру 17 (204 мг, 39% выход) получают в виде белых кристаллов перекристаллизацией из смеси гексан-этилацетат.
Ниже приведены примеры соединений, синтезированных с использованием взаимодействия, аналогичного описанному выше, или с использованием реакционных условий для промежуточного соединения 8:
Соединение по примеру 48:
1-метил-N-[2-(морфолин-4-ил)этил]-5-(хинолин-6-ил)-1H-пиразол-3-карбоксамид
Соединение по примеру 49:
1-метил-N-[2-(морфолин-4-ил)этил]-5-(хинолин-7-ил)-1H-пиразол-3-карбоксамид
Соединение по примеру 50:
1-метил-N-[2-(морфолин-4-ил)этил]-5-(нафталин-2-ил)-1H-пиразол-3-карбоксамид
Соединение по примеру 51:
5-(6-метоксинафталин-2-ил)-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Соединение по примеру 52:
5-(7-метоксинафталин-2-ил)-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Соединение по примеру 53:
5-(1-бензотиофен-3-ил)-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Соединение по примеру 54:
1-метил-N-[2-(морфолин-4-ил)этил]-5-(хиноксалин-6-ил)-1H-пиразол-3-карбоксамид
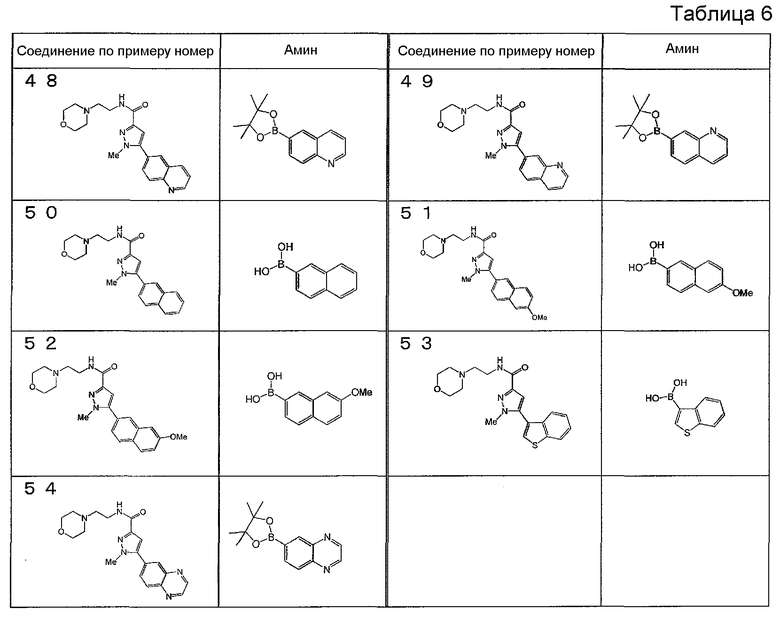
Синтез соединения по примеру 55: синтез 5-(1H-индол-3-ил)-1-метил-N-[2-(морфолин-4-ил)этил)-1H-пиразол-3-карбоксамида
Промежуточное соединение 39: синтез 5-[1-(бензолсульфонил)-1H-индол-3-ил]-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Промежуточное соединение 17 (200 мг, 0,55 ммоль), палладий ацетат (25 мг, 0,11 ммоль) и трифенилфосфин (58 мг, 0,22 ммоль) растворяют в растворе диоксан/толуол (3,3/1 (об./об.), 5,2 мл) и полученную смесь перемешивают при комнатной температуре 10 минут. Затем, к реакционному раствору добавляют 1-(фенилсульфонил)-3-индолбороновую кислоту (215 мг, 0,71 ммоль), воду (1,2 мл) и карбонат натрия (233 мг, 2,20 ммоль), и полученную смесь нагревают до температуры кипения с обратным холодильником в течение 16 часов. После охлаждения, реакционный раствор разбавляют этилацетатом. К образовавшемуся раствору добавляют сульфат натрия для удаления воды и полученную смесь фильтруют. Затем фильтрат концентрируют при пониженном давлении, остаток предварительно обрабатывают колоночной хроматографией (этилацетат), используя силикагель. После чего, полученный органический слой загружают на SCX картридж (сильный катионообменный картридж), промывают метанолом и, наконец, элюируют смесью аммиак-метанол (1М). Элюат концентрируют при пониженном давлении, получая промежуточное соединение 39 (271 мг, количественный выход) в виде сырого продукта. Промежуточное соединение используют для последующего взаимодействия без дополнительной очистки.
МС(ИЭР)m/z: [M+H]+494.
Соединение по примеру 55: синтез 5-(1H-индол-3-ил)-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Промежуточное соединение 39 (271 мг) растворяют в метаноле (5 мл), и к раствору добавляют 2М раствор гидроксида натрия (5 мл), полученный раствор перемешивают при 70°C в течение 30 минут. После охлаждения, к реакционному раствору добавляют воду (10 мл), и полученную смесь трижды экстрагируют дихлорметаном (50 мл). Затем объединенный органический слой сушат над сульфатом магния, осушитель фильтруют и фильтрат концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией (смесь дихлорметан/метанол=10/1 (об./об.)), используя силикагель, что дает сырой продукт (169 мг) в виде светло-коричневого твердого вещества. Для дополнительной очистки используют препаративную ВЭЖХ систему (устройство для очистки A), которая была использована для очистки соединения по примеру 1.
МС(ИЭР)m/z: [M+H]+354.
Синтез соединения по примеру 56: 1-метил-5-(1-метил-1H-индол-3-ил)-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Соединение по примеру 55 (50 мг) растворяют в ацетонитриле (1 мл) и к полученному раствору добавляют карбонат цезия (138 мг, 0,42 ммоль) и метилиодид (0,013 мл, 0,21 ммоль), и смесь нагревают до температуры кипения с обратным холодильником 1 час. После охлаждения, нерастворимые вещества удаляют фильтрованием, и фильтрат концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией (смесь дихлорметан/метанол=10/1 (об./об.)), используя силикагель, что дает сырой продукт (45 мг) в виде бесцветного сиропа. Для дополнительной очистки используют препаративную ВЭЖХ систему (устройство для очистки A), которая была использована для очистки соединения по примеру 1.
МС(ИЭР)m/z: [M+H]+368.
Ниже приведены промежуточные соединения и соединения по примерам, синтезированные с использованием взаимодействия, аналогичного описанному выше:
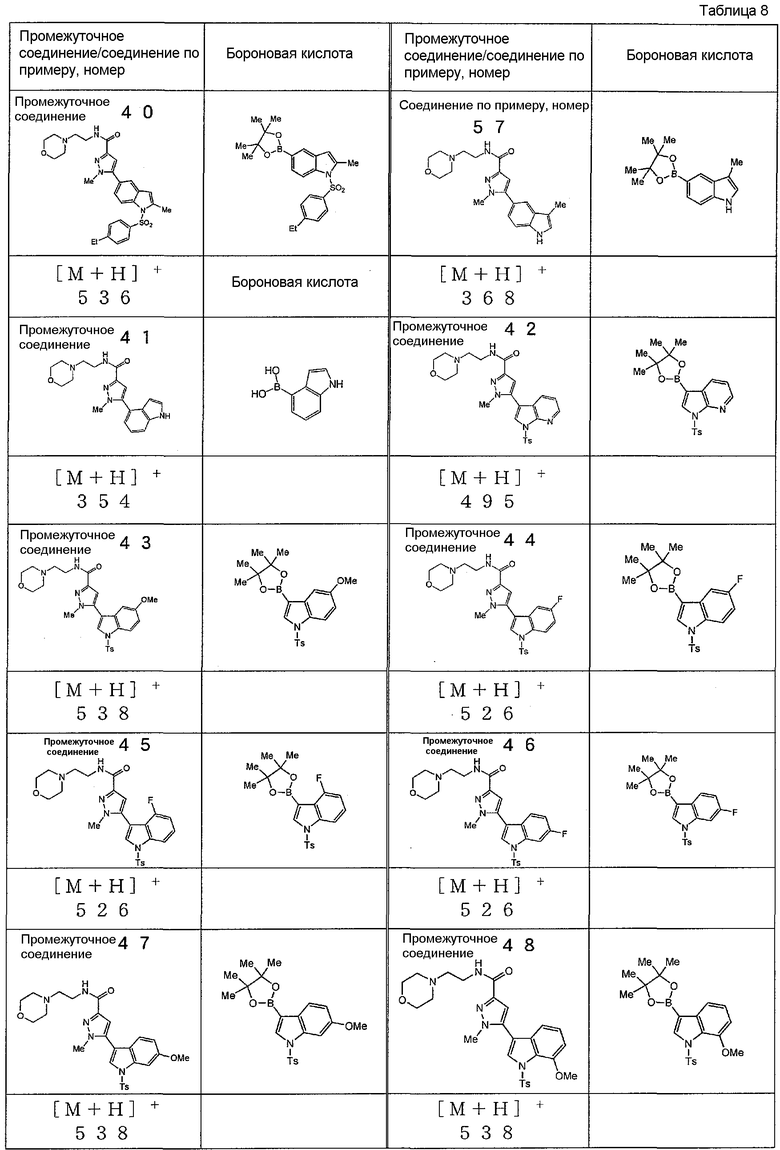
Соединение по примеру 57:
1-метил-5-(3-метил-1H-индол-5-ил)-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Соединение по примеру 58:
1-метил-5-(2-метил-1H-индол-5-ил)-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Соединение по примеру 59:
1-метил-N-[2-(морфолин-4-ил)этил]-5-{1H-пирроло[2,3-b]пиридин-3-ил}-1H-пиразол-3-карбоксамид
Соединение по примеру 60:
5-(5-метокси-1H-индол-3-ил)-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Соединение по примеру 61:
5-(5-фтор-1H-индол-3-ил)-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Соединение по примеру 62:
5-(1,2-диметил-1H-индол-5-ил)-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Соединение по примеру 63:
5-(1,3-диметил-1H-индол-5-ил)-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Соединение по примеру 64:
1-метил-5-{1-метил-1H-пирроло[2,3-b]пиридин-3-ил}-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Соединение по примеру 65:
5-(5-метокси-1-метил-1H-индол-3-ил)-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Соединение по примеру 66:
5-(5-фтор-1-метил-1H-индол-3-ил)-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Соединение по примеру 67:
1-метил-5-(1-метил-1H-индол-4-ил)-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Соединение по примеру 68:
5-[1-(2-метоксиэтил)-1H-индол-3-ил]-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Соединение по примеру 69:
5-(1-этил-1H-индол-3-ил)-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Соединение по примеру 70:
5-(4-фтор-1H-индол-3-ил)-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Соединение по примеру 71:
5-(6-фтор-1H-индол-3-ил)-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Соединение по примеру 72:
5-(6-метокси-1H-индол-3-ил)-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Соединение по примеру 73:
5-(7-метокси-1H-индол-3-ил)-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Соединение по примеру 74:
5-[1-(2-метоксиэтил)-2-метил-1H-индол-5-ил]-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Соединение по примеру 75:
5-[1-(2-гидроксиэтил)-2-метил-1H-индол-5-ил]-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Ниже приведены примеры соединений, синтезированных с использованием взаимодействия, аналогичного описанному выше: где, как легко понять специалисту в данной области, вместо метилиодида могут быть использованы соответствующие примерам алкилгалогениды.

Соединение по примеру 76: синтез 5-(4-метансульфонил-1H-индол-2-ил)-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Промежуточное соединение 49: синтез 1-(бензолсульфонил)-4-(метилсульфанил)-1H-индола
Метантиолат натрия (1300 мг, 18,16 ммоль) и 1н раствор HCl-диэтиловый эфир (20 мл) добавляют, на ледяной бане, к ТГФ- (50 мл) раствору 1-(бензолсульфонил)-4,5,6,7-тетрагидро-1H-индол-4-она (1250 мг, 4,54 ммоль), полученного из 4,5,6,7-тетрагидро-1H-4-она по ссылке на патентную литературу (JP-7-247263A), и смесь перемешивают при комнатной температуре в течение ночи. Затем к смеси добавляют диэтиловый эфир (30 мл). Полученную смесь промывают насыщенным раствором бикарбоната натрия (50 мл) и сушат над безводным сульфатом магния. Потом осушитель фильтруют, полученный фильтрат концентрируют при пониженном давлении, что дает маслянистый остаток. Маслянистый остаток растворяют в толуоле (15 мл) и к смеси добавляют DDQ (1500 мг, 6,81 ммоль). Смесь нагревают до температуры кипения с обратным холодильником в течение 2 часов. После охлаждения, образовавшуюся смесь концентрируют при пониженном давлении, получая сырое маслянистое промежуточное соединение 49. Полученное сырое промежуточное соединение 49 очищают колоночной хроматографией (смесь гексан-этилацетат=10:1 (об./об.)), используя силикагель, что дает промежуточное соединение 49 (690 мг, 50% выход) в виде белого полутвердого вещества.
1H-ЯМР (300 МГц, CDCl3) δ 7,88-7,79 (м, 3H), 7,58 (д, J=4,0 Гц, 1H), 7,57-7,50 (м, 1H), 7,46-7,41 (м, 2H), 7,43-7,24 (м, 1H), 7,08 (д, J=7,3 Гц, 1H), 6,78 (д, J=3,7 Гц, 1H), 3,58 (с, 3H).
МС(ИЭР)m/z: [M+H]+304.
Промежуточное соединение 50: синтез [1-(бензолсульфонил)-4-(метилсульфанил)-1H-индол-2-ил]борандиола
н-Бутиллитий (2,5 мл, 4,12 ммоль, 1,65М циклогексановый раствор) добавляют по каплям к ТГФ-раствору (5 мл) диизопропиламина (417 мг, 0,58 ммоль), на ледяной бане, и смесь перемешивают 20 минут, что дает литийдиизопропиламид.
Полученный выше литийдиизопропиламид добавляют, на ледяной бане, по каплям к ТГФ-раствору (25 мл) промежуточного соединения 49 (833 мг, 2,75 ммоль) и эфира триизопропилбороновой кислоты (620 мг, 3,29 ммоль), полученному отдельно. Спустя два часа, дополнительно добавляют по каплям такое же количество литийдиизопропиламида, на ледяной бане. Затем смесь перемешивают, на ледяной бане, в течение 1 часа, к смеси добавляют 1М раствор HCl и доводят раствор до pH 3. Полученный раствор экстрагируют этилацетатом (25 мл ×2), и объединенную смесь сушат над безводным сульфатом магния. Потом осушитель фильтруют, полученный фильтрат концентрируют при пониженном давлении. Полученный остаток очищают колоночной хроматографией (смесь гексан/этилацетат), используя силикагель, что дает сырое промежуточное соединение 50 (953 мг, 100% выход) в виде желтого полутвердого вещества.
Промежуточное соединение 51: синтез 5-[1-(бензолсульфонил)-4-(метилсульфанил)-1H-индол-2-ил]-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Согласно подобному способу синтеза промежуточного соединения 8 по примеру 1, промежуточное соединение 51 (390 мг, 29% выход) получают в виде белого полутвердого вещества из промежуточного соединения 17 (909 мг, 2,50 ммоль), промежуточного соединения 50 (953 мг, 2,74 ммоль), ацетата палладия(II) (56 мг, 0,25 ммоль), трифенилфосфина (262 мг, 1,00 ммоль) и карбоната натрия (661 мг, 6,24 ммоль).
МС(ИЭР)m/z: [M+H]+539.
Промежуточное соединение 52: синтез 5-[1-(бензолсульфонил)-4-(метансульфонил)-1H-индол-2-ил]-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Оксон-персульфатное соединение (683 мг, 1,11 ммоль) добавляют к раствору промежуточного соединения 51 (200 мг, 0,37 ммоль) в смеси диоксан/вода (8 мл/3 мл), при комнатной температуре. Реакционный раствор перемешивают при комнатной температуре в течение 2,5 часов. Реакционный раствор сушат над безводным сульфатом магния. Затем осушитель фильтруют, полученный фильтрат концентрируют при пониженном давлении, что дает желтое, маслянистое, промежуточное соединение 52 (61 мг, 29% выход).
МС(ИЭР)m/z: [M+H]+572.
Соединение по примеру 76: синтез 5-(4-метансульфонил-1H-индол-2-ил)-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Моногидрат тетрабутиламмонийфторида (140 мг, 0,53 ммоль) добавляют к ТГФ-раствору (5 мл) промежуточного соединения 52 (61 мг, 0,11 ммоль), и смесь нагревают до температуры кипения с обратным холодильником в течение 3 часов. После охлаждения до комнатной температуры, остаток, полученный при пониженном давлении, очищают на SCX картридже (сильный катионообменный картридж) совершенно аналогично соединению по примеру 1, что дает сырой продукт 76 в виде желтого твердого вещества. Дополнительная очистка колоночной хроматографией (смесь дихлорметан/метанол=10/1 (об./об.)) с использованием силикагеля дает соединение по примеру 76 (17 мг, 37% выход) в виде желтого твердого вещества.
МС(ИЭР)m/z: [M+H]+432.
Соединение по примеру 77: синтез 5-(4-ацетамидо-1H-индол-2-ил)-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Соединение по примеру 78: синтез 5-(4-метансульфонамидо-1H-индол-2-ил)-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Промежуточное соединение 55: Синтез 5-(1H-4-амино-индол-2-ил)-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамиддигидрохлорид
Промежуточное соединение 53: синтез 1-(трет-бутил)-4-(ди-трет-бутил)амино-2-индолбороновой кислоты
н-Бутиллитий (0,63 мл, 1,65М, раствор в гексане) добавляют по каплям к охлажденному ТГФ-раствору (5 мл) диизопропиламина (105 мг, 104 ммоль), в условиях охлаждения льдом, в течение 5 минут, в атмосфере азота. Полученную смесь перемешивают 20 минут, что дает ТГФ-раствор литийдиизопропиламида.
ТГФ-раствор литийдиизопропиламида, полученный выше, добавляют, на ледяной бане, по каплям к ТГФ-раствору (5 мл) 1-(трет-бутил)-4-(ди-трет-бутил)аминоиндола (300 мг, 0,694 ммоль) и триизопропилбората (157 мг, 0,832 ммоль), полученному отдельно. Затем реакционную смесь перемешивают, на ледяной бане, в течение 1 часа, раствор доводят до pH 3 добавлением 1М раствора HCl. Полученный раствор экстрагируют этилацетатом (15 мл ×2), и объединенный раствор сушат над безводным сульфатом магния, получая сырой продукт 53, 1-(трет-бутил)-4-(ди-трет-бутил)амино-2-индолбороновую кислоту. Сырой продукт используют для последующего взаимодействия без дополнительной очистки.
Промежуточное соединение 54: синтез 5-[1-(трет-бутил)-4-(ди-трет-бутил)аминоиндол-2-ил]-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Промежуточное соединение 17 (178 мг, 0,49 ммоль), ацетат палладия (14 мг, 0,063 ммоль) и трифенилфосфин (52 мг, 0,20 ммоль) растворяют в растворе диоксан/толуол (3,3/1 (об./об.), 5,2 мл) и смесь перемешивают при комнатной температуре в течение 10 минут. Затем к реакционному раствору добавляют 1-(трет-бутил)-4-(ди-трет-бутил)амино-2-индолбороновую кислоту 53 (256 мг, 0,54 ммоль), воду (5 мл) и карбонат натрия (129 мг, 1,22 ммоль), и смесь нагревают до температуры кипения с обратным холодильником в течение 1,5 часов. После охлаждения, реакционный раствор разбавляют этилацетатом и добавляют к раствору сульфат натрия для удаления воды. Потом полученный раствор фильтруют. Затем фильтрат концентрируют при пониженном давлении, продукт очищают колоночной хроматографией (этилацетат), используя силикагель с амино-слоем, что дает 5-{1-(трет-бутил)-4-(ди-трет-бутил)аминоиндол-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид 54 и 5-{-4-(ди-трет-бутил)аминоиндол-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид, полученные в виде смеси (127 мг, бесцветное твердое вещество). Указанную смесь не подвергают какому-либо дополнительному разделению и используют для последующего взаимодействия.
Промежуточное соединение 55: синтез 5-(1H-4-амино-индол-2-ил)-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид дигидрохлорида
Смесь (127 мг), содержащую вышеуказанное промежуточное соединение 54 и 10% раствор HCl-метанол (15 мл) в колбе на 100 мл выдерживают в атмосфере азота при комнатной температуре 16 часов. Полученный раствор концентрируют при пониженном давлении, и получают соль 55 - 5-(1H-4-аминоиндол-2-ил)-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид дигидрохлорид, (99 мг, количественный выход).
1H-ЯМР (300 МГц, CD3OD) δ 7,50-7,47 (м, 1H), 7,23-7,08 (м, 4H), 4,10 (с, 3H), 4,03-3,99 (м, 2H), 3,87-3,76 (м, 4H), 3,67-3,63 (м, 2H), 3,43-3,40 (м, 2H), 3,29-3,19 (м, 2H).
Другие протоны не наблюдаются по причине перекрывания с пиками растворителя, основанного на CD3OD.
МС(ИЭР)m/z: [M+H]+369.
Приведенные ниже соединения по примерам получены, по-существу, тем же способом, что в указанных выше для процесса F-3, и в условиях, выбранных из указанных для процесса F-3.
Соединение по примеру 77: синтез 5-(4-ацетамидо-1H-индол-2-ил)-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Соединение по примеру 78: 5-(4-метансульфонамидо-1H-индол-2-ил)-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид
Соединение по примеру 77 и соединение по примеру 78 в приведенной ниже таблице получают в присутствии основания (триэтиламин) в 1,2-дихлорэтановом растворе с использованием каждого реагента (ацетилхлорид или уксусный ангидрид, метансульфонилхлорид) и промежуточного соединения 55. Для дополнительной очистки используют препаративную ВЭЖХ систему (устройство для очистки A), которая была использована для очистки соединения по примеру 1.
Соединение по примеру 79: синтез 5-(1-метансульфонил-1H-индол-3-ил)-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Соединение по примеру 80: синтез 5-(1-метансульфонил-3-метил-1H-индол-5-ил)-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Соединение по примеру 79 и соединение по примеру 80 в приведенной ниже таблице получают в присутствии основания (гидрид натрия) в ДМФА-растворе с использованием каждого реагента (метансульфонилхлорид) и промежуточного соединения 55. Для дополнительной очистки используют препаративную ВЭЖХ систему (устройство для очистки A), которая была использована для очистки соединения по примеру 1.
Реагенты и примеры приведены ниже.
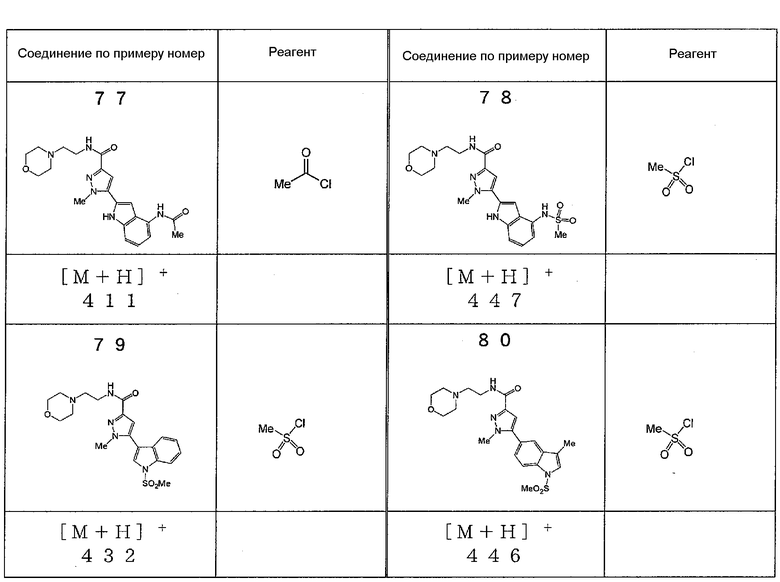
Соединение по примеру 81: синтез 1-метил-5-(3-метил-1H-индол-1-ил)-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
3-Метилиндол (27 мг, 0,21 ммоль), карбонат цезия (179 мг, 0,55 ммоль) и иодид меди(I) (52 мг, 0,28 ммоль) добавляют к раствору промежуточного соединения 17 (50 мг, 0,14 ммоль), растворенного в ДМФА (1 мл), и смесь перемешивают при 110°C 20 часов. После охлаждения, нерастворимые вещества удаляют фильтрованием, фильтрат концентрируют при пониженном давлении. Остаток предварительно обрабатывают колоночной хроматографией (смесь дихлорметан/этанол (10/1) (об./об.)), используя обработанный амином силикагель, и затем загружают на SCX картридж (сильный катионообменный картридж), промывают метанолом (10 мл) и, наконец, элюируют смесью аммиак-метанол (1M, 8 мл). Сырой продукт, полученный концентрацией, очищают препаративной ВЭЖХ (устройство для очистки A, описанное в начале раздела ПРИМЕРЫ).
МС(ИЭР)m/z: [M+H]+368.
Соединение по примеру 82: синтез 5-{5-циано-1H-пирроло[2,3-b]пиридин-2-ил}-N-[2-(3,3-дифторазетидин-1-ил)этил]-1-метил-1H-пиразол-3-карбоксамида
Промежуточное соединение 56: синтез N-(2,2-диэтоксиэтил)-5-иод -1-метил-1H-пиразол-3-карбоксамида
Согласно подобному способу синтеза соединения по примеру 1, триэтиламин (3,32 мл, 23,80 ммоль) добавляют к раствору смеси в безводном ДМФА (50 мл) промежуточного соединения 16 (2,00 г, 7,94 ммоль), диэтилацеталя аминоацетальдегида (1,27 г, 9,52 ммоль) и HBTU (4,52 г, 11,9 ммоль), и смесь перемешивают при комнатной температуре. Полученный остаток очищают колоночной хроматографией (смесь гексан/этилацетат 2:1 (об./об.)), используя силикагель, что дает промежуточное соединение 56 (3,84 г) в виде светло-желтого масла.
1H-ЯМР (300 МГц, CDCl3) δ 6,99 (уш.с, 1H), 6,94 (с, 1H), 4,61-4,55 (м, 1H), 3,94 (с, 3H), 3,80-3,68 (м, 2H), 3,64-3,50 (м, 4H), 1,23 (т, J=7,0 Гц, 6H).
Промежуточное соединение 57: синтез N-[2-(3,3-дифторазетидин-1-ил)этил]-5-иод-1-метил-1H-пиразол-3-карбоксамида
Реакционную смесь из ТГФ-раствора (50 мл) промежуточного соединения 56 (3,84 г, 10,45 ммоль) и 2М раствора HCl (25 мл) перемешивают при 50°C в течение 2 часов. После охлаждения до комнатной температуры, реакционную смесь доводят до pH выше 10 насыщенным раствором бикарбоната натрия и дважды экстрагируют этилацетатом. Полученный органический слой промывают насыщенным раствором хлорида натрия и сушат над гидрохлоридом натрия. Затем осушитель фильтруют и фильтрат концентрируют при пониженном давлении, получая светло-желтое твердое вещество. Триацетоксиборгидрид натрия (6,64 г, 31,35 ммоль) добавляют, за несколько порций, к смеси в 1,2-дихлорэтане (50 мл) сырого альдегидного промежуточное соединения и 3,3-дифторазетидинмоногидрохлорида (1.35 г, 10,45 ммоль) и этилацетата (10 мл). Затем полученную смесь перемешивают при комнатной температуре 15 часов, реакционную смесь доводят до pH выше 10 насыщенным раствором бикарбоната натрия и раствор трижды экстрагируют дихлорметаном. Полученный органический слой промывают насыщенным раствором хлорида натрия и сушат над сульфатом натрия. Затем осушитель фильтруют, фильтрат концентрируют при пониженном давлении. Полученный остаток очищают колоночной хроматографией (смесь дихлорметан/метанол=40/1-30/1 (об./об.)), используя силикагель, что дает промежуточное соединение 57 (2,93 г, 76% выход) в виде бесцветного масла.
1H-ЯМР (300 МГц, CDCl3) δ 7,05 (уш.с, 1H), 6,94 (с, 1H), 3,95 (с, 3H), 3,70-3,58 (м, 4H), 3,47-3,39 (м, 2H), 2,80-2,74 (м, 2H).
МС(ИЭР)m/z: [M+H]+371.
Промежуточное соединение 58: синтез N-[2-(3,3-дифторазетидин-1-ил)этил]-1-метил-5-[2-(триметилсилил)этинил]-1H-пиразол-3-карбоксамида
Согласно подобному способу синтеза промежуточного соединения 18, описанному для соединения по примеру 22, триэтиламин (3,46 мл, 24,86 ммоль) добавляют к раствору (30 мл) в безводном ТГФ промежуточного соединения 57 (2,30 г, 6,21 ммоль), триметилсилилацетилена (1,32 мл, 9,32 ммоль), иодида меди(I) (118 мг, 0,621 ммоль) и дихлорбис(ацетонитрил)палладий(II)хлорида (436 мг, 0,621 ммоль), и полученную смесь перемешивают при комнатной температуре в течение 1,5 часов. После обычной обработки, остаток очищают колоночной хроматографией (смесь гексан/этилацетат 3:2-1:1 (об./об.)), используя силикагель, что дает промежуточное соединение 58 (1,76 г, 83% выход) в виде желтовато-коричневого масла. И полученное промежуточное соединение 58 используют на следующей стадии, без определения каких-либо физических характеристик.
Промежуточное соединение 59: синтез N-[2-(3,3-дифторазетидин-1-ил)этил]-5-этинил-1-метил-1H-пиразол-3-карбоксамида
Метанольную смесь (30 мл) промежуточного соединения 58 (1,76 г, 5,17 ммоль) и карбоната калия (107 г, 7,75 ммоль) перемешивают при комнатной температуре в течение 2 часов. После завершения взаимодействия, карбонат калия фильтруют. Фильтрат концентрируют при пониженном давлении. Полученный остаток вновь разбавляют дихлорметаном и промывают водой и потом насыщенным раствором хлорида натрия. Затем раствор сушат над сульфатом натрия. После чего осушитель фильтруют и фильтрат концентрируют при пониженном давлении. Полученный остаток очищают колоночной хроматографией (смесь гексан/этилацетат 1:2 (об./об.)), используя силикагель, что дает промежуточное соединение 59 (1,32 г, 95% выход) в виде желтого твердого вещества.
1H-ЯМР (300 МГц, CDCl3) δ 7,12 (уш.с, 1H), 6,95 (с, 1H), 3,96 (с, 3H), 3,70-3,54 (м, 5H), 3,48-3,40 (м, 2H), 2,82-2,74 (м, 2H).
МС(ИЭР)m/z: [M+H]+269.
Промежуточное соединение 60: синтез 5-[2-(2-амино-5-цианопиридин-3-ил)этинил]-N-[2-(3,3-дифторазетидин-1-ил)этил]-1-метил-1H-пиразол-3-карбоксамида
Согласно способу синтеза промежуточного соединения 20, промежуточное соединение 59 (300 мг, 1,12 ммоль) и 2-амино-3-иод-5-цианопиридин (329 мг, 1,34 ммоль) нагревают при перемешивании, при 85°C, 14 часов, и получают промежуточное соединение 60 (29,0 мг, 6,7% выход) в виде светло-желтого твердого вещества.
МС(ИЭР)m/z: [M+H]+386, [M-H]-384.
Соединение по примеру 82: синтез 5-{5-циано-1H-пирроло[2,3-b]пиридин-2-ил}-N-[2-(3,3-дифторазетидин-1-ил)этил]-1-метил-1H-пиразол-3-карбоксамида
Согласно подобному способу синтеза соединения по примеру 22, соединение по примеру 82 (21 мг, 72% выход) получают в виде светло-желтого твердого вещества из промежуточного соединения 60 (29 мг, 0,075 ммоль).
1H-ЯМР (300 МГц, ДМСО-d6) δ 12,85 (уш.с, 1H), 8,68 (д, J=2,2 Гц, 1H), 8,59 (д, J=2,2 Гц, 1H), 8,20-8,12 (м, 1H), 7,23 (с, 1H), 7,04 (с, 1H), 4,14 (с, 3H), 3,68-3,53 (м, 4H), 3,31-3,20 (м, 2H), 2,73-2,63 (м, 2H).
МС(ИЭР)m/z: [M+H]+355, [M-H]-353.
Соединение по примеру 83: синтез N-[2-(3,3-дифторазетидин-1-ил)этил]-1-метил-5-{1H-пирроло[2,3-b]пиридин-2-ил}-1H-пиразол-3-карбоксамида
Промежуточное соединение 61: синтез 5-[2-(2-аминопиридин-3-ил)этинил]-N-[2-(3,3-дифторазетидин-1-ил)этил]-1-метил-1H-пиразол-3-карбоксамида
Согласно способу синтеза промежуточного соединения 18, промежуточное соединение 59 (250 мг, 1,12 ммоль) и 3-иод-2-аминопиридин (226 мг, 1,03 ммоль) перемешивают при комнатной температуре в течение 3 часов, что дает промежуточное соединение 61 (50,3 мг, 15% выход) в виде светло-желтого твердого вещества.
МС(ИЭР)m/z: [M+H]+361.
Соединение по примеру 83: синтез N-[2-(3,3-дифторазетидин-1-ил)этил]-1-метил-5-{1H-пирроло[2,3-b]пиридин-2-ил}-1H-пиразол-3-карбоксамида
Согласно подобному способу синтеза соединения по примеру 22, соединение по примеру 83 (37,9 мг, 75% выход) получают в виде светло-желтого твердого вещества из промежуточного соединения 61 (50 мг, 0,075 ммоль).
1H-ЯМР (300 МГц, ДМСО-d6) δ 12,19 (уш.с, 1H), 8,31-8,26 (м, 1H), 8,15-8,08 (м, 1H), 8,05-7,98 (м, 1H), 7,18-7,08 (м, 2H), 6,89 (с, 1H), 4,12 (с, 3H), 3,66-3,54 (м, 4H), 3,30-3,20 (м, 2H), 2,72-2,63 (м, 2H).
МС(ИЭР)m/z: [M+H]+361, [M-H]-359.
Соединение по примеру 84: синтез 5-{6-фторимидазо[1,2-a]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Промежуточное соединение 62: синтез этил-5-{6-фторимидазо[1,2-a]пиридин-2-ил}-1-метил-1H-пиразол-3-карбоксилата
Согласно подобному способу синтеза промежуточного соединения 35 для соединения по примеру 34, метанольный раствор (2 мл) промежуточного соединения 34 (114 мг, 0,41 ммоль) и 2-амино-5-фторпиридина (39 мг, 0,41 ммоль) нагревают при перемешивании 20 часов. Остаток очищают колоночной хроматографией (смесь гексан/этилацетат 1/3 (об./об.)), используя силикагель, что дает промежуточное соединение 62 (101 мг, 48% выход) в виде светло-желтого твердого вещества.
1H-ЯМР (300 МГц, CDCl3) δ 8,11-8,09 (м, 1H), 7,81 (с, 1H), 7,65-7,60 (м, 1H), 7,21-7,14 (м, 1H), 7,07 (с, 1H), 4,43 (т, J=7,3 Гц, 2H), 4,33 (с, 3H), 1,42 (т, J=7,3 Гц, 3H).
МС(ИЭР)m/z: [M+H]+289.
Промежуточное соединение 63: синтез 5-{6-фторимидазо[1,2-a]пиридин-2-ил}-1-метил-1H-пиразол-3-карбоновой кислоты
Согласно подобному способу синтеза промежуточного соединения 36 для соединения по примеру 34, 2М раствор гидроксида натрия (0,175 мл, 0,350 ммоль) добавляют к метанольному раствору (5 мл) промежуточного соединения 62 (101 мг, 0,350 ммоль), и полученную смесь перемешивают при 70°C в течение 1 часа. После нейтрализации 2М раствором HCl, промежуточное соединение 63 (67 мг, 73% выход) получают в виде белых кристаллов. Промежуточное соединение используют для последующего взаимодействия без дополнительной очистки.
1H-ЯМР (300 МГц, ДМСО-d6) δ 8,75 (уш.с, 1H), 8,38 (с, 1H), 8,23 (с, 1H), 7,70-7,65 (м, 1H), 7,39-7,33 (м, 1H), 7,06 (с, 1H), 4,24 (с, 3H).
МС(ИЭР)m/z: [M+H]+261.
Соединение по примеру 84: синтез 5-{6-фторимидазо[1,2-a]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Согласно подобному способу синтеза соединения по примеру 1, сырое соединение по примеру 84 (52,3 мг, 54% выход) получают в форме белых кристаллов из промежуточного соединения 63 (67 мг, 0,257 ммоль) и 4-(2-аминоэтил)морфолина (36,9 мг, 0,283 ммоль). Для дополнительной очистки используют препаративную ВЭЖХ систему (устройство для очистки A), которая была использована для очистки соединения по примеру 1.
МС(ИЭР)m/z: [M+H]+373.
Соединение по примеру 85: синтез 5-{7-фторимидазо[1,2-a]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Промежуточное соединение 64: синтез этил-5-{7-фторимидазо[1,2-a]пиридин-2-ил}-1-метил-1H-пиразол-3-карбоксилата
Согласно подобному способу синтеза промежуточного соединения 35 для соединения по примеру 34, метанольный раствор (2 мл) промежуточного соединения 34 (114 мг, 0,41 ммоль) и 2-амино-4-фторпиридин (39 мг, 0,41 ммоль) нагревают при перемешивании 20 часов. Затем остаток очищают колоночной хроматографией (смесь гексан/этилацетат=1/3 (об./об.)), используя силикагель, что дает промежуточное соединение 64 (120 мг, 57% выход) в виде светло-желтого твердого вещества.
1H-ЯМР (300 МГц, CDCl3) δ 8,10 (уш.с, 1H), 7,81 (с, 1H), 7,65-7,60 (м, 1H), 7,22-7,15 (м, 1H), 7,07 (с, 1H), 4,46-4,40 (т, J=7,3 Гц, 2H), 4,33 (с, 3H), 1,42 (т, J=7,3 Гц, 3H).
МС(ИЭР)m/z: [M+H]+289.
Промежуточное соединение 65: синтез 5-{7-фторимидазо[1,2-a]пиридин-2-ил}-1-метил-1H-пиразол-3-карбоновой кислоты
Согласно подобному способу синтеза промежуточного соединения 36 для соединения по примеру 34, 2M раствор гидроксида натрия (0,21 мл, 0,416 ммоль) добавляют к метанольному раствору (5 мл) промежуточного соединения 64 (120 мг, 0,416 ммоль), и полученную смесь перемешивают при 70°C в течение 1 часа. После нейтрализации 2М раствором HCl, промежуточное соединение 65 (67 мг) получают в виде белых кристаллов. Промежуточное соединение используют для последующего взаимодействия без дополнительной очистки.
1H-ЯМР (300 МГц, ДМСО-d6) δ 8,81-8,79 (м, 1H), 8,41 (с, 1H), 7,75-7,70 (м, 1H), 7,46-7,39 (м, 1H), 7,07 (с, 1H), 4,23 (с, 3H).
МС(ИЭР)m/z: [M+H]+261.
Соединение по примеру 85: синтез 5-{7-фторимидазо[1,2-a]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Согласно подобному способу синтеза соединения по примеру 1, сырое соединение по примеру 85 (102 мг) получают в форме белых кристаллов из промежуточного соединения 65 (67 мг, 0,257 ммоль) и 4-(2-аминоэтил)морфолина (36,9 мг, 0,283 ммоль). Для дополнительной очистки используют препаративную ВЭЖХ систему (устройство для очистки A), которая была использована для очистки соединения по примеру 1.
МС(ИЭР)m/z: [M+H]+373.
Соединение по примеру 86: синтез 5-{6-цианоимидазо[1,2-a]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Промежуточное соединение 66: синтез этил-5-{6-бромимидазо[1,2-a]пиридин-2-ил}-1-метил-1H-пиразол-3-карбоксилата
Согласно подобному способу синтеза промежуточного соединения 35 для соединения по примеру 34, этанольный раствор (10 мл) промежуточного соединения 34 (235 мг, 0,854 ммоль) и 2-амино-5-бромпиридина (148 мг, 0,854 ммоль) нагревают до температуры кипения с обратным холодильником в течение 20 часов, и затем остаток очищают колоночной хроматографией (смесь гексан/этилацетат 1/4 (об./об.), дихлорметан/метанол=30/1 (об./об.)), используя силикагель, что дает промежуточное соединение 66 (106 мг, 36% выход) в виде светло-желтого твердого вещества.
1H-ЯМР (300 МГц, CDCl3) δ 8,32 (с, 1H), 7,77 (с, 1H), 7,56-7,53 (м, 1H), 7,32-7,27 (м, 1H), 7,09-7,08 (м, 1H), 4,4, 4-4,39 (м, 2H), 4,34-4,33 (м, 3H), 1,42 (т, J=7,3 Гц, 3H).
МС(ИЭР)m/z: [M+H]+349 и 351.
Промежуточное соединение 67: синтез 5-{6-бромимидазо[1,2-a]пиридин-2-ил}-1-метил-1H-пиразол-3-карбоновой кислоты
Согласно подобному способу синтеза промежуточного соединения 36 для соединения по примеру 34, 2М раствор гидроксида натрия (0,304 мл, 0,608 ммоль) добавляют к метанольному раствору (15 мл) промежуточного соединения 66 (106 мг, 0,304 ммоль), и полученную смесь перемешивают при 70°C в течение 1 часа. После нейтрализации 2М раствором HCl, промежуточное соединение 67 (66,7 мг) получают в виде белого твердого вещества. Промежуточное соединение используют для последующего взаимодействия без дополнительной очистки.
1H-ЯМР (300 МГц, ДМСО-d6) δ 8,93 (с, 1H), 8,38 (с, 1H), 7,65 (д, J=10,4 Гц, 1H), 7,50 (д, J=10,4 Гц, 1H), 7,10 (с, 1H), 4,23 (с, 3H).
МС(ИЭР)m/z: [M+H]+321 и 323.
Промежуточное соединение 68: синтез 5-{6-бромимидазо[1,2-a]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Согласно подобному способу синтеза соединения по примеру 1, сырое промежуточное соединение 68 (77 мг, 0,078 ммоль) получают из промежуточного соединения 67 (67 мг, 0,209 ммоль) и 4-(2-аминоэтил)морфолина (29,9 мг, 0,23 ммоль). Затем остаток очищают SCX, и промежуточное соединение 68 (74,9 мг, 83% выход) получают в виде белого твердого вещества.
МС(ИЭР)m/z: [M+H]+433 и 435.
Соединение по примеру 86: синтез 5-{6-цианоимидазо[1,2-a]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
В безводном ДМФА смесь (4 мл) промежуточного соединения 68 (74,9 мг, 0,173 ммоль) и цианида цинка (12,5 мг, 0,107 ммоль), и тетракис(трифенилфосфин)палладия (20,0 мг, 0,017 ммоль) перемешивают при 100°C 20 часов. После охлаждения до комнатной температуры к реакционному раствору добавляют воду, и полученную смесь экстрагируют раствором этилацетат/толуол (9/1). Поскольку большая часть соединений переходит в водный слой, водный слой вновь концентрируют при пониженном давлении. Полученный твердый остаток фракционируют небольшим количеством метанола. Полученное твердое вещество сушат при пониженном давлении, что дает сырое соединение по примеру 86 (58,0 мг, 88% выход) в виде белого твердого вещества. Для дополнительной очистки используют препаративную ВЭЖХ систему (устройство для очистки A), которая была использована для очистки соединения по примеру 1.
МС(ИЭР)m/z: [M+H]+380, [M-H]-378.
Соединение по примеру 87: синтез N-[2-(3,3-дифторазетидин-1-ил)этил]-1-метил-5-(хинолин-3-ил)-1H-пиразол-3-карбоксамида
Согласно подобному способу синтеза промежуточного соединения 11 для соединения по примеру 17, 1,4-диоксановый раствор (10 мл) промежуточного соединения 57 (224 мг, 0,605 ммоль) и 3-хинолинбороновой кислоты (122 мг, 0,666 ммоль) нагревают при перемешивании и 100°C в течение 15 часов, полученный остаток очищают колоночной хроматографией (смесь дихлорметан/метанол=30/1-20/1 (об./об.)), используя силикагель, что дает соединение по примеру 81 (181 мг, 80% выход) в виде светло-коричневого твердого вещества. Соединение по примеру (181 мг) очищают перекристаллизацией из раствора этилацетата и гексана, что дает соединение по примеру 87 (121 мг).
1H-ЯМР (300 МГц, CDCl3) δ 9,00-8,97 (м, 1H), 8,23-8,14 (м, 2H), 7,93-7,77 (м, 2H), 7,69-7,60 (м, 1H), 7,19 (уш.с, 1H), 7,01 (с, 1H), 4,00 (с, 3H), 3,73-3,61 (м, 4H), 3,54-3,45 (м, 2H), 2,86-2,78 (м, 2H).
МС(ИЭР)m/z: [M+H]+372.
Соединение по примеру 88: синтез 5-{7-цианоимидазо[1,2-a]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Промежуточное соединение 69: синтез 5-ацетил-1-метил-1H-пиразол-3-карбоновой кислоты
Согласно подобному способу синтеза промежуточного соединения 16 для соединения по примеру 22, 2M раствор гидроксида натрия (2,55 мл, 5,10 ммоль) добавляют к метанольному раствору (12 мл) промежуточного соединения 33 (500 мг, 2,55 ммоль), и полученную смесь перемешивают при комнатной температуре 16 часов. После нейтрализации 2M раствором HCl, остаток промывают холодной водой, что дает промежуточное соединение 69 (220 мг, 51% выход) в виде белого твердого вещества.
МС(ИЭР)m/z: [M+H]+169, [M-H]-167.
Промежуточное соединение 70: синтез 5-ацетил-N-(2,2-диэтоксиэтил)-1-метил-1H-пиразол-3-карбоксамид
Согласно подобному способу синтеза промежуточного соединения 56 для соединения по примеру 82, триэтиламин (397 мг, 3,93 ммоль) добавляют к смешанному раствору (5 мл), в безводном ДМФА, промежуточного соединения 69 (220 мг, 1,31 ммоль) и диэтилацеталя аминоацетальдегида (192 мг, 1,44 ммоль), HBTU (595 мг, 1,57 ммоль). Полученную смесь перемешивают при комнатной температуре 3 часа. После обычной обработки, полученный остаток очищают колоночной хроматографией (смесь гексан/этилацетат 2/1 (об./об.)), используя силикагель, что дает промежуточное соединение 70 (351 мг, 95% выход) в виде белого твердого вещества.
МС(ИЭР)m/z: [M-H]-282,27.
Промежуточное соединение 71: синтез 5-(2-бромацетил)-N-(2,2-диэтоксиэтил)-1-метил-1H-пиразол-3-карбоксамида
Согласно подобному способу синтеза промежуточного соединения 34 для соединения по примеру 34, фенилтриметиламмонийбромид (466 мг, 1,24 ммоль) добавляют к ТГФ-раствору (5 мл) промежуточного соединения 70 (351 мг, 1,24 ммоль), полученную смесь перемешивают при комнатной температуре. После обычной обработки, полученный остаток очищают колоночной хроматографией (смесь гексан/этилацетат 3/1 (об./об.)), используя силикагель, что дает промежуточное соединение 71 (223 мг, 50% выход) в виде белого твердого вещества.
МС(ИЭР)m/z: [M+H]+362 и 364, [M-H]-360 и 362.
Промежуточное соединение 72: синтез 5-{7-цианоимидазо[1,2-a]пиридин-2-ил}-N-(2,2-диэтоксиэтил)-1-метил-1H-пиразол-3-карбоксамида
Согласно подобному способу синтеза промежуточного соединения 35 для соединения по примеру 34, этанольный раствор (10 мл) промежуточного соединения 71 (222,9 мг, 0,615 ммоль) и 2-амино-4-цианопиридина (73,3 мг, 0,615 ммоль) нагревают при перемешивании 15 часов. Остаток очищают колоночной хроматографией (смесь дихлорметан/метанол 30/1 (об./об.)), используя силикагель, что дает промежуточное соединение 72 (174,1 мг, 74% выход) в виде светло-желтого твердого вещества.
МС(ИЭР)m/z: [M+H]+337.
Соединение по примеру 88: синтез 5-{7-цианоимидазо[1,2-a]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамида
Согласно подобному способу синтеза промежуточного соединения 57 для соединения по примеру 82, реакционную смесь ТГФ-раствора (5 мл) промежуточного соединения 72 (174,1 мг, 0,455 ммоль) и 2М раствора HCl (2,5 мл) перемешивают при 50°C 1 час. После охлаждения до комнатной температуры, реакционный раствор доводят до pH свыше 10 с помощью 2М раствора гидроксида натрия (2,5 мл), и полученный раствор экстрагируют этилацетатом. Полученный органический слой промывают насыщенным раствором хлорида натрия, и раствор сушат над сульфатом натрия. Затем осушитель фильтруют, фильтрат концентрируют при пониженном давлении и получают светло-желтое твердое вещество. Триацетоксиборгидрид натрия (289,3 мг, 1,365 ммоль) добавляют к смеси сырого промежуточного альдегидного соединения и 1,2-дихлорэтанового раствора (5 мл) морфолина (39,2 мг, 0,455 ммоль) и уксусной кислоты (1 мл) при комнатной температуре. Затем полученную смесь перемешивают при комнатной температуре 15 часов, реакционную смесь доводят до pH выше 10 раствором бикарбоната натрия и раствор трижды экстрагируют дихлорметаном, полученный органический слой промывают насыщенным раствором хлорида натрия и сушат над сульфатом натрия. Затем осушитель фильтруют, фильтрат концентрируют при пониженном давлении. Полученный остаток очищают колоночной хроматографией (смесь дихлорметан/метанол=30/1-10/1 (об./об.)), используя силикагель. После чего соединение по примеру 88 (7 мг, 4% выход) получают в виде светло-оранжевого твердого вещества.
МС(ИЭР)m/z: [M+H]+380.
Перечень промежуточных соединений для синтеза соединений по примерам приведен ниже в таблицах 11-1 - 11-4.
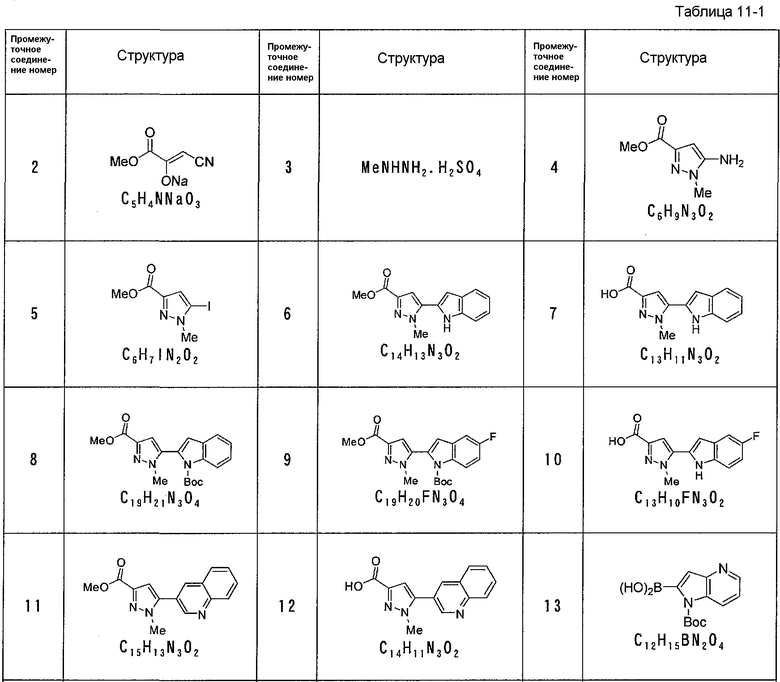

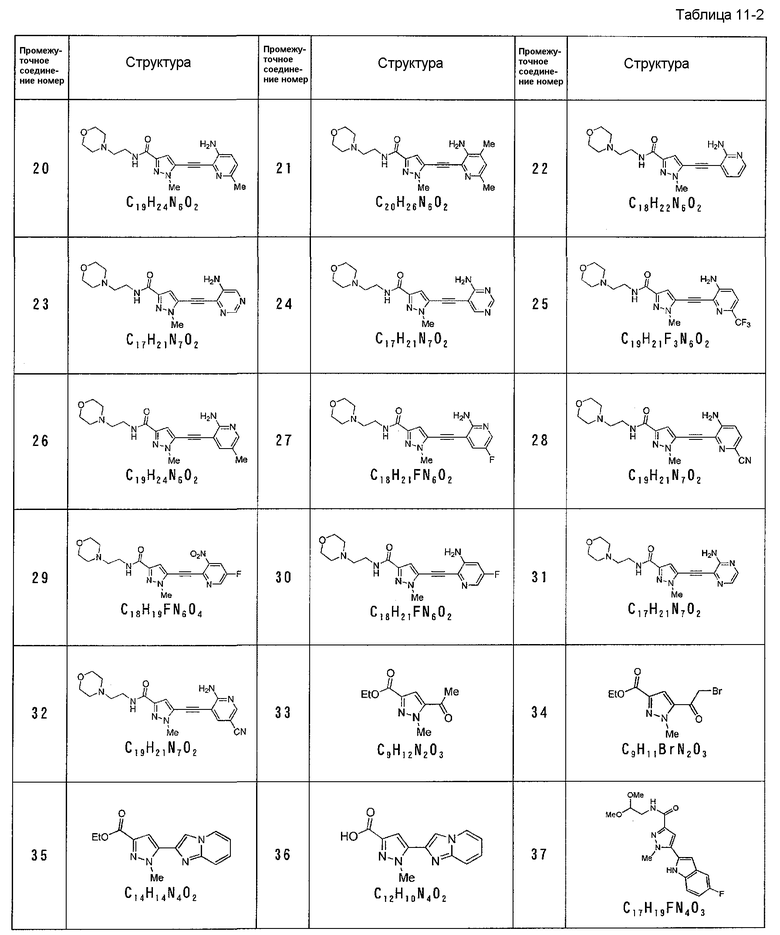
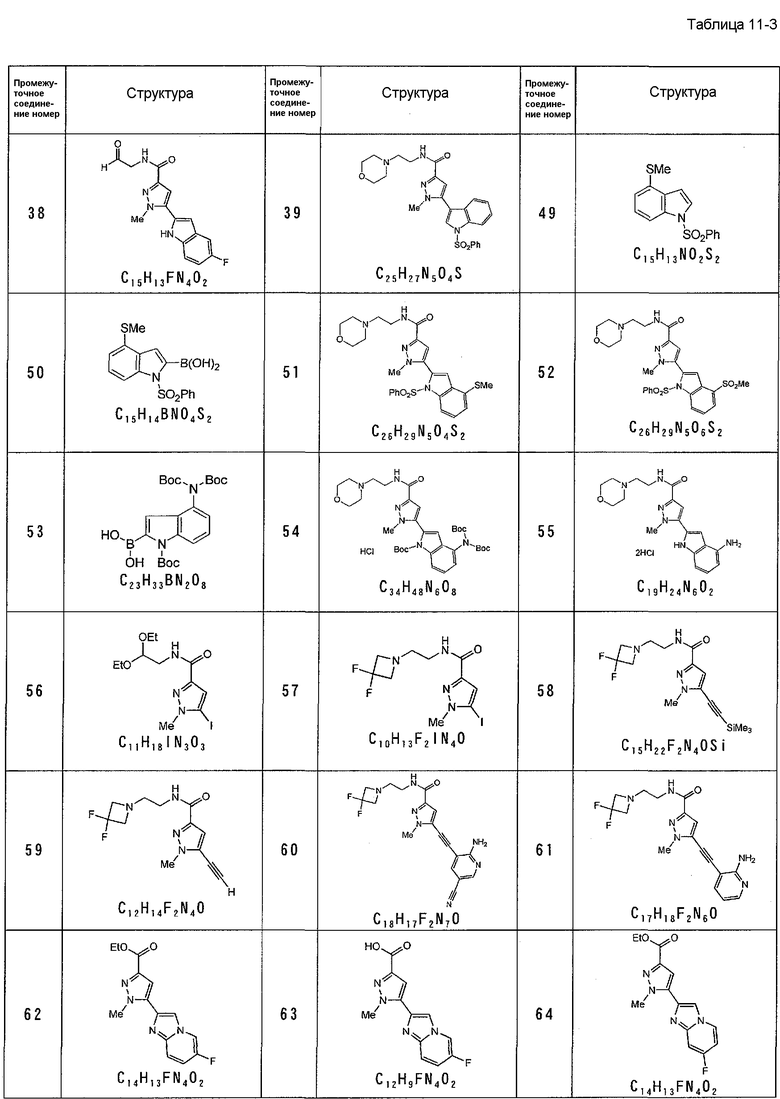
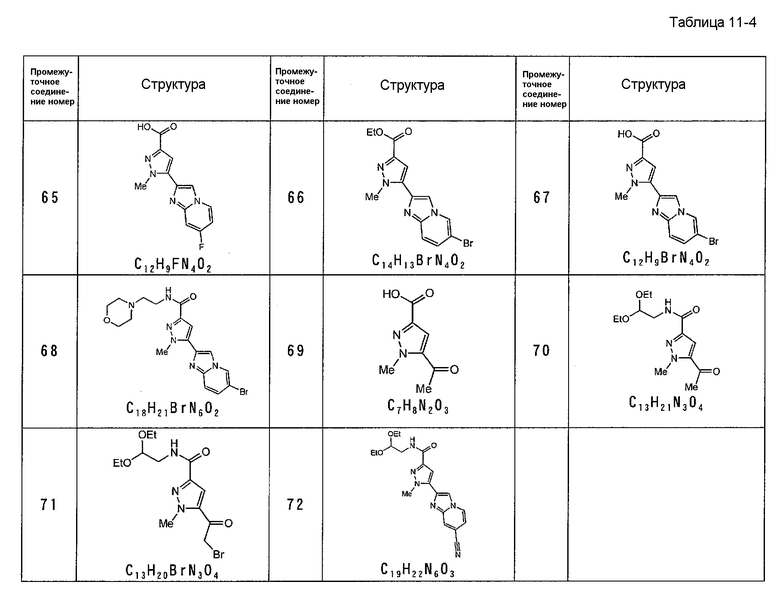
Испытание на связывание 5-HT2B человека
Аффинность связывания с рецептором 5-HT2B соединений по данному изобретению демонстрируется следующими методиками.
Клетки CHO-K1, трансфицированные 5-HT2B человека, приобретают у Euroscreen (№ по кат.: ES-314-F) и выращивают на месте. Собранные клетки суспендируют в 50 мМ HEPES (pH 7,4), с добавлением смеси ингибиторов протеаз (SIGMA, разведение 1:100) и 1 мМ EDTA, и гомогенизируют, используя дезинтегратор Polytron PT1200, поставленный на полную мощность, за 30 секунд, на льду. Гомогенаты центрифугируют при 1000 об/мин и 4°C в течение 5 мин, и супернатанты замораживают при -80°C в течение 10 мин. Затем замороженные супернатанты ресуспендируют в 50 мМ HEPES (pH 7,4), гомогенизируют и центрифугируют еще раз тем же способом. Супернатанты центрифугируют при 25000 об/мин и 4°C в течение 60 мин. После чего осадок в пробирке после центрифугирования ресуспендируют в 50 мМ HEPES (pH 7,4), гомогенизируют, разделяют на части и хранят при -80°C до употребления. Аликвотную долю мембранных фракций используют для определения концентрации белка с применением набора для анализа на содержание белка с помощью BCA (PIERCE) и планшет-ридер ARVOsx (Wallac).
Для экспериментов по рецепторному связыванию, 20 мкл испытуемых соединений инкубируют с 100 мкл [3H]-мезулергина (GE healthcare, 10 нМ) и 80 мкл мембранного гомогената (20 мкг белка) в течение 120 мин при комнатной температуре. Неспецифическое связывание определяют с помощью 10 мкМ миансерина (SIGMA) при конечной концентрации. Все инкубации завершают быстрым вакуумным фильтрованием через бумагу на стеклянном фильтре с пропиткой из 0,2 (об./об.)% PEI, применяя харвестер Filtermate (PerkinElmer), с последующей пятикратной промывкой 50 мМ HEPES (pH 7,4). Рецепторно-связанную радиоактивность определяют подсчетом импульсов в жидкой фазе, используя TopCount (PerkinElmer).
По результатам эксперимента, все соединения примеров проявляют аффинность к рецептору 5-HT2B человека.
Испытание на кальциевый инфлюкс с использованием клеток CHO-K1, трансфицированных 5-HT2B человека
Аффинность связывания с рецептором 5-HT2B соединений по данному изобретению демонстрируется следующими методиками.
Клетки CHO-K1, трансфицированные 5-HT2B человека, приобретают у Euroscreen и выращивают. Клетки выращивают при 37°C и 5% CO2 в среде UltraCHO (Cambrex) с добавлением 400 мкг/мл G418, 250 мкг/мл зеоцина, 100 Ед/мл пенициллина, 100 мкг/мл стрептомицина и 1(об/об)% диализованной FBS (фетальной телячьей сыворотки). После выращивания до 60-80% слияния, культуральную среду клеток заменяют KRH-буфером (1,8 мМ CaCl2, 1 мМ MgSO4, 115 мМ NaCl, 5,4 мМ KCl, 11 мМ D-глюкозы, 0,96 мМ NaH2PO4, 25 мМ HEPES, доведенным до pH 7,4 с помощью NaOH), включающим 5 мкМ Fura-2 AM. Клетки инкубируют 120 мин при комнатной температуре. После инкубации, клетки разделяют смесью 0,05 (вес/вес)% трипсин/1 мМ EDTA и промывают PBS. Затем указанные клетки суспендируют в KRH-буфере, получая концентрацию 1,0×106 клетки/мл.
384-луночные планшеты предварительно обрабатывают соединениями по данному изобретению (50 мкл/лунка). По 34 мкл клеточной суспензии (3,4×104 клеток) распределяют в каждую ячейку 384-луночного черного аналитического планшета с прозрачным дном. Аналитические планшеты помещают на FDSS6000 (Hamamatsu Photonics) и начинают мониторинг сигналов. Спустя тридцать секунд в каждую лунку автоматически добавляют по 6 мкл последовательных разведений соединений, и продолжают мониторинг на FDSS6000 еще 4,5 мин для исследования антагонистической активности. Затем клетки инкубируют в течение 10 мин при комнатной температуре, в темноте. Аналитические планшеты вновь помещают на FDSS6000 и начинают мониторинг сигналов. Спустя тридцать секунд в каждую лунку автоматически добавляют по 20 мкл 9 нМ 5-HT, и продолжают мониторинг на FDSS6000 еще 4,5 мин для изучения величин IC50 испытуемых соединений. Ссылкой на данный эксперимент является Br. J. Pharmacol., 1999 September; 128(1): 13-20.
Антагонистические активности в отношении рецепторов 5-HT2B (IC50, нМ) всех 88 соединений по примерам, приведенные в нижеследующих таблицах 12-15, составляют от 0,1 нМ до 100 нМ.
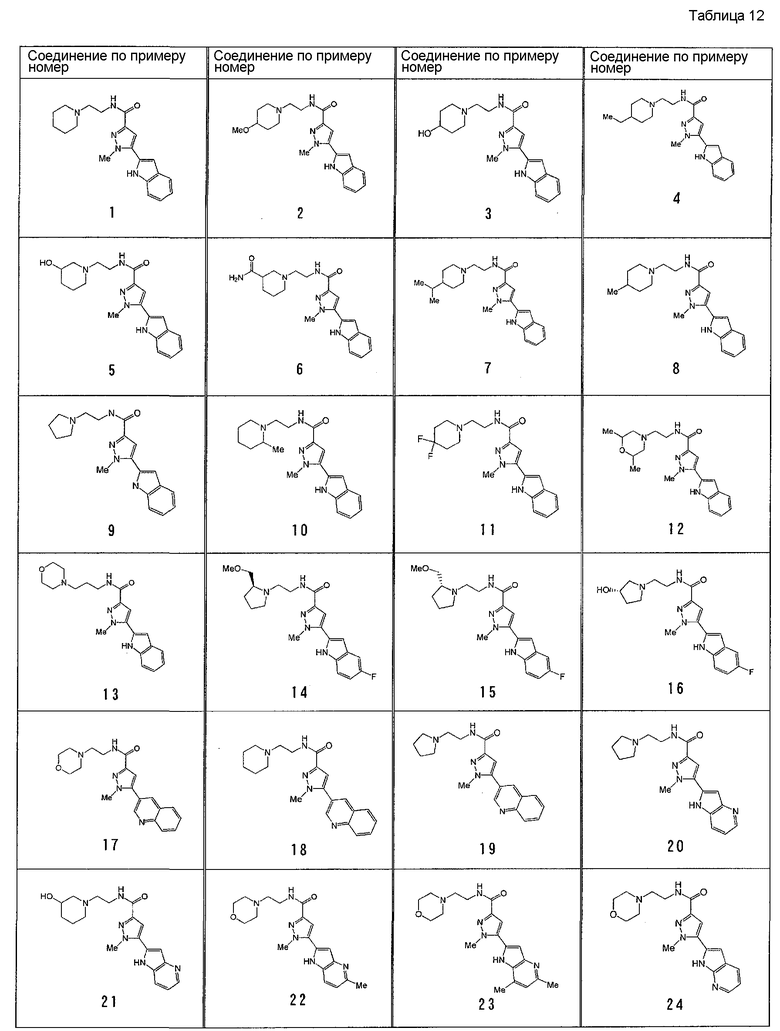
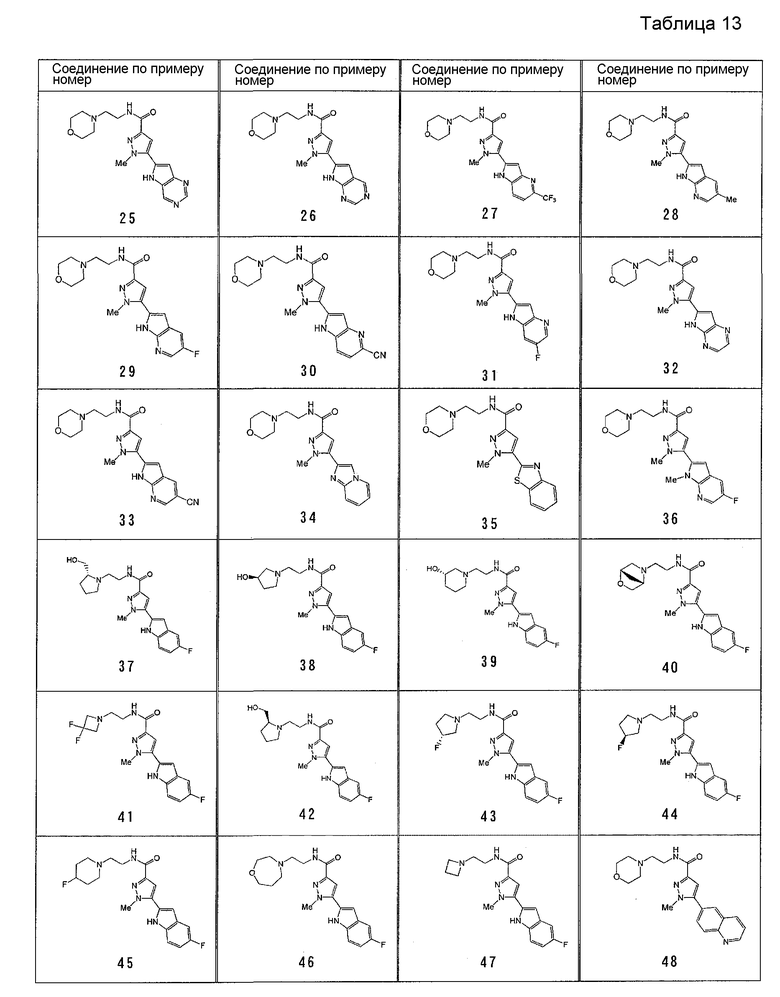
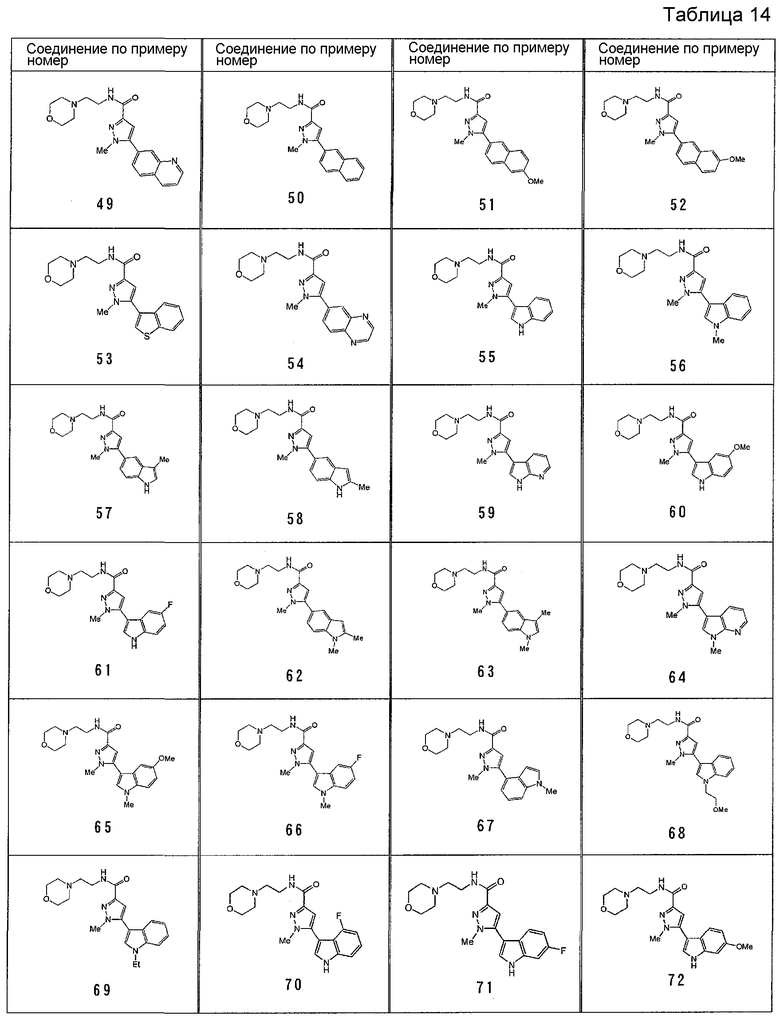

Испытание на приток кальция с использованием 3T3 клеток, трансфицированных 5-HT2B человека
Аффинность связывания с рецептором 5-HT2B соединений по данному изобретению определяют по следующим методикам.
3T3 клетки, трансфицированные 5-HT2B человека, получают на месте. Клетки выращивают при 37°C и 5% CO2 в среде DMEM (Invitrogen) с добавлением 400 мкг/мл G418, 100 Ед/мл пенициллина, 100 мкг/мл стрептомицина и 10 (об/об)% FBS. После выращивания до 60-80% слияния, культуральную среду клеток заменяют KRH-буфером (1,8 мМ CaCl2, 1 мМ MgSO4, 115 мМ NaCl, 5,4 мМ KCl, 11 мМ D-глюкозы, 0,96 мМ NaH2PO4, 25 мМ HEPES, доведенным до pH 7,4 с помощью NaOH), включающим 5 мкМ Fura-2 AM. Клетки инкубируют 120 мин при комнатной температуре. После инкубации, клетки разделяют смесью 0,05% трипсин/1 мМ EDTA и промывают PBS. Затем указанные клетки суспендируют в KRH-буфере, получая концентрацию 0,3×106 клетки/мл.
384-луночные планшеты предварительно обрабатывают соединениями по данному изобретению (50 мкл/лунка). По 34 мкл клеточной суспензии (1,0×104 клеток) распределяют в каждую ячейку 384-луночного черного аналитического планшета с прозрачным дном. Аналитические планшеты помещают на FDSS6000 (Hamamatsu Photonics) и начинают мониторинг сигналов. Спустя тридцать секунд в каждую лунку автоматически добавляют по 6 мкл последовательных разведений соединений, и продолжают мониторинг на FDSS6000 еще 4,5 мин для исследования антагонистической активности. Затем клетки инкубируют в течение 10 мин при комнатной температуре, в темноте. Аналитические планшеты вновь помещают на FDSS6000 и начинают мониторинг сигналов. Спустя тридцать секунд в каждую лунку автоматически добавляют по 20 мкл 90 нМ 5-HT, и продолжают мониторинг на FDSS6000 еще 4,5 мин для изучения величин IC50 испытуемых соединений. Ссылкой на данный эксперимент является Br. J. Pharmacol., 1999 September; 128(1): 13-20.
Испытание на приток кальция с использованием 3T3 клеток, трансфицированных 5-HT2C человека
Аффинность связывания с рецептором 5-HT2C соединений по данному изобретению определяют по следующим методикам.
3T3 клетки, трансфицированные 5-HT2C человека, получают на месте. Клетки выращивают при 37°C и 5% CO2 в среде DMEM (Invitrogen) с добавлением 20 мкг/мл G418, 100 Ед/мл пенициллина, 100 мкг/мл стрептомицина и 10 (об/об)% FBS. После выращивания до 60-80% слияния, культуральную среду клеток заменяют KRH-буфером (1,8 мМ CaCl2, 1 мМ MgSO4, 115 мМ NaCl, 5,4 мМ KCl, 11 мМ D-глюкозы, 0,96 мМ NaH2PO4, 25 мМ HEPES, доведенным до pH 7,4 с помощью NaOH), включающим 5 мкМ Fura-2 AM. Клетки инкубируют 120 мин при комнатной температуре. После инкубации, клетки разделяют смесью 0,05% трипсин/1 мМ EDTA и промывают PBS. Указанные клетки суспендируют в KRH-буфере, получая концентрацию 0,45×106 клетки/мл.
384-луночные планшеты предварительно обрабатывают соединениями по данному изобретению (50 мкл/лунка). По 34 мкл клеточной суспензии (1,5×104 клеток) распределяют в каждую ячейку 384-луночного черного аналитического планшета с прозрачным дном. Аналитические планшеты помещают на FDSS6000 (Hamamatsu Photonics) и начинают мониторинг сигналов. Спустя тридцать секунд в каждую лунку автоматически добавляют по 6 мкл последовательных разведений соединений, и продолжают мониторинг на FDSS6000 еще 4,5 мин для исследования антагонистической активности. Затем клетки инкубируют в течение 10 мин при комнатной температуре, в темноте. Аналитические планшеты вновь помещают на FDSS6000 и начинают мониторинг сигналов. Спустя тридцать секунд в каждую лунку автоматически добавляют по 20 мкл 3 нМ 5-HT, и продолжают мониторинг на FDSS6000 еще 4,5 мин для изучения величин IC50 испытуемых соединений. Ссылкой на данный эксперимент является Br.J. Pharmacol., 1999 September; 128(1): 13-20.
Оценка терапевтических действий на IBS у крыс
Фармакологические действия испытуемых соединений по данному изобретению оценивают, определяя положительные эффекты против снижения порога болевой чувствительности при стимуляции вытяжения толстой кишки в модели TNBS-индуцированного IBS.
Для уточнения деталей, просим ознакомиться с литературным источником, Katsuyo Ohashi et al., Pharmacology, 81(2): 144-150(2008).
Экспериментальная методика
Производят срединный разрез при анестезии животных, самцов крыс SD, 240-270 г. Раствором TNBS (50 мг/кг, 30% метанол) обрабатывают начало прямой кишки крыс. После обработки, слепую кишку возвращают в брюшную полость. Затем мышечную стенку сшивают. После операции животных содержат в обычных условиях и используют для фармакологической оценки после 7 дней с момента хирургического вмешательства. Стимуляцию вытяжения прямой кишки используют для оценки соединений, Diop L. et al., J Pharmacol Exp Ther.302(3): 1013-22(2002). Баллон (длиной 5 см) вводят через анус и удерживают на месте (верхушка баллона в 5 см от ануса). Затем баллон постепенно пошагово накачивают, от 0 до 70 мм Hg, используя баростат (Barostat DISTENDER II R, G & J, CANADA). Порог болевой чувствительности определяется по давлению, вызывающему первое абдоминальное сокращение (абдоминальная судорога: Wesselmann U et al., (1998) Neurosci Lett 246: 73-76).
Результаты для соединения по примеру 24 представлены на фигуре 1. Число животных в каждой группе равно 8. На графике приведены средние данные. Столбец диаграммы показывает величины 25% и 75%. Статистический анализ проводят по замкнутому критерию, используя критерий Манна-Уитни.
Вертикальная ось указывает давление порога болевой чувствительности. В данном случае, p.o. 10 мг/кг дает положительные эффекты против снижения порога болевой чувствительности при TNBS. Таким образом, новые пиразол-3-карбоксамидные производные могут быть полезны для лечения IBS.
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
Соединение по данному изобретению полезно в качестве селективного антагониста рецептора 5-HT2B и полезно для предварительного лечения или профилактики различных заболеваний, связанных с рецептором 5-HT2B.
| название | год | авторы | номер документа |
|---|---|---|---|
| СИНТЕЗ НОВОГО АНТАГОНИСТА EP4 И ПРИМЕНЕНИЕ ПРИ РАКЕ И ВОСПАЛЕНИИ | 2021 |
|
RU2804153C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2734390C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛХИНАЗОЛИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2011 |
|
RU2652638C2 |
| МОДУЛЯТОРЫ КАЛЬПАИНА И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2773288C2 |
| ИНГИБИТОРЫ HPK1 И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2839132C2 |
| ПРОИЗВОДНЫЕ БЕНЗОДИАЗЕПИНА КАК ИНГИБИТОРЫ RSV | 2016 |
|
RU2738232C2 |
| ПЕРВИЧНЫЕ КАРБОКСАМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ BТK | 2014 |
|
RU2708395C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2716136C2 |
| Селективный ингибитор киназы JAK1 | 2020 |
|
RU2818002C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛОПИРИМИДИНЫ И ЗАМЕЩЕННЫЕ ПУРИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ УБИКВИТИН-СПЕЦИФИЧЕСКОЙ ПРОЦЕССИРУЮЩЕЙ ПРОТЕАЗЫ 1 (USP1) | 2019 |
|
RU2833222C2 |
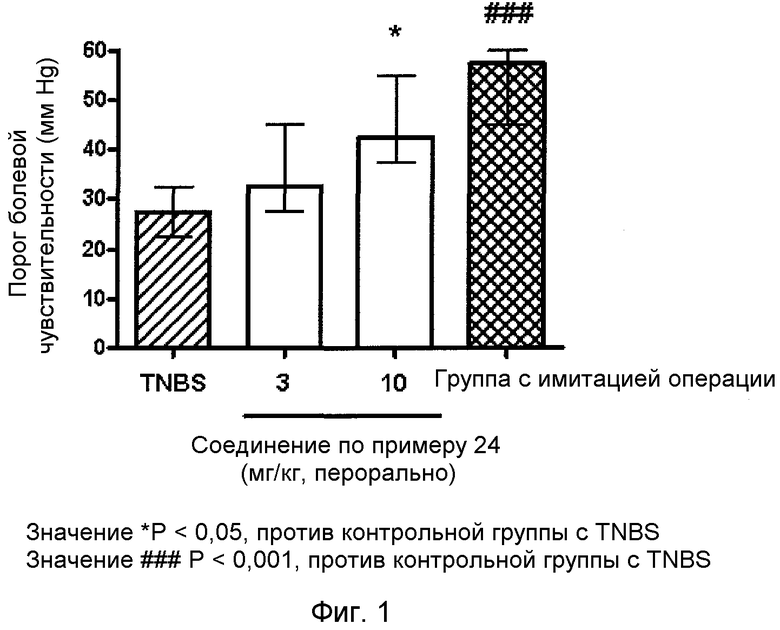
Изобретение относится к соединениям формулы (I), где A означает морфолинил, 1,4-оксазепамил, пиперидинил, пирролидинил или азетидинил, который связан по N; R1 означает C1-C6-алкильную группу; R2 означает бициклическую арильную группу, выбранную из 1H-индолила, 1H-пирроло[3,2-b]пиридила, хинолила, нафтила, 1H-пирроло[2,3-b]пиридила, 5H-пирроло[3,2-d]пиримидинила, 7H-пирроло[2,3-d]пиримидинила, бензо[b]тиофенила, имидазо[1,2-а]пиридила, бензо[b]тиазолила, 5Н-пирроло[2,3-b]пиразинила и хиноксалинила, которая может быть замещена R4; R3 означает водород или атом галогена; R4 означает C1-C6-алкильную группу, C1-C6-галогеналкильную группу, OR1A, галоген, -(CH2)aOH, CN, NHCOR1A, SO2R1A или NHSO2R1A; R5 означает C1-C6-алкильную группу, -(CH2)aOH, -(CH2)aOR1B, галоген или CONH2; когда p является множественным числом, R5 может быть одинаковым или различным, или R5 может быть объединен с другим R5; каждый из R1A и R1B независимо означает C1-C6-алкильную группу; a равно 0, 1 или 2; n равно 1 или 2; p равно 0, 1, 2, 3, 4 или 5. Также изобретение относится к промежуточным соединениям формул (IA) и (IB) для получения соединений формулы (I), к профилактическому или терапевтическому средству, содержащему соединения формулы (I), фармацевтическим композициям, применению соединений формулы (I) и к способу профилактики или лечения заболеваний. Технический результат - соединения формулы (I) в качестве селективных антагонистов 5-HT2B рецептора. 8 н. и 3 з.п. ф-лы, 1 ил., 18 табл., 88 пр.
,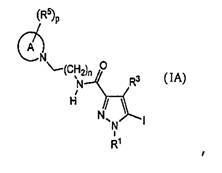

1. Соединение следующей общей формулы (I) или его фармацевтически приемлемая соль
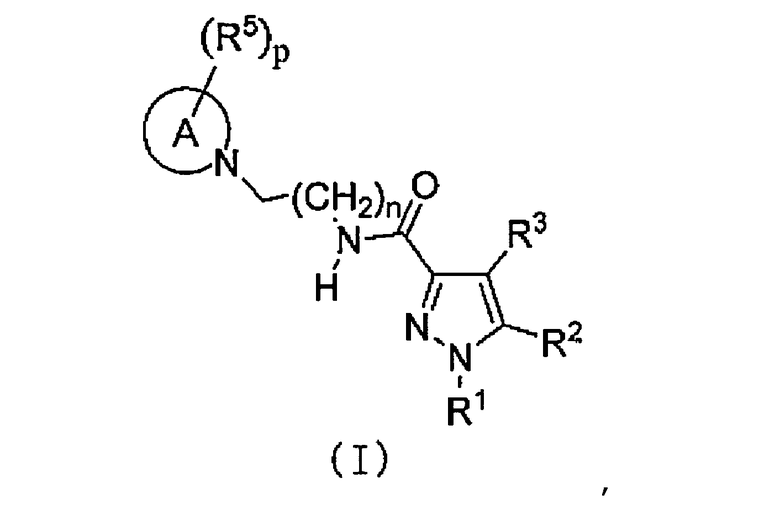
где
A означает морфолинил, 1,4-оксазепамил, пиперидинил, пирролидинил или азетидинил, который связан по N;
R1 означает C1-C6-алкильную группу;
R2 означает бициклическую арильную группу, выбранную из 1H-индолила, 1H-пирроло[3,2-b]пиридила, хинолила, нафтила, 1H-пирроло[2,3-b]пиридила, 5H-пирроло[3,2-d]пиримидинила, 7H-пирроло[2,3-d]пиримидинила, бензо[b]тиофенила, имидазо[1,2-а]пиридила, бензо[b]тиазолила, 5Н-пирроло[2,3-b]пиразинила и хиноксалинила, которая может быть замещена R4,
R3 означает водород или атом галогена;
R4 означает C1-C6-алкильную группу, C1-C6-галогеналкильную группу, OR1A, галоген, -(CH2)aOH, CN, NHCOR1A, SO2R1A или NHSO2R1A;
R5 означает C1-C6-алкильную группу, -(CH2)aOH, -(CH2)aOR1B, галоген или CONH2; когда p является множественным числом, R5 может быть одинаковым или различным, или R5 может быть объединен с другим R5;
каждый из R1A и R1B независимо означает C1-C6-алкильную группу;
a равно 0, 1 или 2;
n равно 1 или 2 и
p равно 0, 1, 2, 3, 4 или 5.
2. Соединение или его фармацевтически приемлемая соль по п.1, где цикл A означает морфолинил, пиперидинил, пирролидинил или азетидинил, который связан по N;
n равно 1;
p равно 0, 1 или 2.
3. Соединение или его фармацевтически приемлемая соль по п.1, где соединение, представленное общей формулой (I), выбрано из группы, включающей
1-метил-N-[2-(морфолин-4-ил)этил]-5-(хинолин-3-ил)-1H-пиразол-3-карбоксамид;
1-метил-5-{5-метил-1H-пирроло[3,2-b]пиридин-2-ил}-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид;
1-метил-N-[2-(морфолин-4-ил)этил]-5-{1H-пирроло[2,3-b]пиридин-2-ил}-1H-пиразол-3-карбоксамид;
1-метил-N-[2-(морфолин-4-ил)этил]-5-{7H-пирроло[2,3-d]пиримидин-6-ил}-1H-пиразол-3-карбоксамид;
1-метил-N-[2-(морфолин-4-ил)этил]-5-[5-(трифторметил)-1H-пирроло[3,2-b]пиридин-2-ил]-1H-пиразол-3-карбоксамид;
1-метил-5-{5-метил-1H-пирроло[2,3-b]пиридин-2-ил}-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид;
5-{5-фтор-1H-пирроло[2,3-b]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид;
5-{5-циано-1H-пирроло[3,2-b]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид;
5-{6-фтор-1H-пирроло[3,2-b]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид;
1-метил-N-[2-(морфолин-4-ил)этил]-5-{5H-пирроло[2,3-b]пиразин-6-ил}-1H-пиразол-3-карбоксамид;
5-{5-циано-1H-пирроло[2,3-b]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид;
5-{5-фтор-1-метил-1H-пирроло[2,3-b]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид;
N-[2-(3,3-дифторазетидин-1-ил)этил]-5-(5-фтор-1H-индол-2-ил)-1-метил-1H-пиразол-3-карбоксамид;
N-[2-(азетидин-1-ил)этил]-5-(5-фтор-1H-индол-2-ил)-1-метил-1H-пиразол-3-карбоксамид;
1-метил-5-(2-метил-1H-индол-5-ил)-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид;
5-(1,2-диметил-1H-индол-5-ил)-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид;
5-(4-ацетамидо-1H-индол-2-ил)-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид;
5-{имидазо[1,2-a]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид;
5-{6-фторимидазо[1,2-a]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид;
5-{7-фторимидазо[1,2-a]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид;
5-{6-цианоимидазо[1,2-a]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид;
N-[2-(3,3-дифторазетидин-1-ил)этил]-1-метил-5-(хинолин-3-ил)-1H-пиразол-3-карбоксамид;
N-[2-(3,3-дифторазетидин-1-ил)этил]-1-метил-5-{1H-пирроло[2,3-b]пиридин-2-ил}-1H-пиразол-3-карбоксамид и
5-{7-цианоимидазо[1,2-a]пиридин-2-ил}-1-метил-N-[2-(морфолин-4-ил)этил]-1H-пиразол-3-карбоксамид.
4. Промежуточное соединение для соединения по п.1, представленное общей формулой (IA):
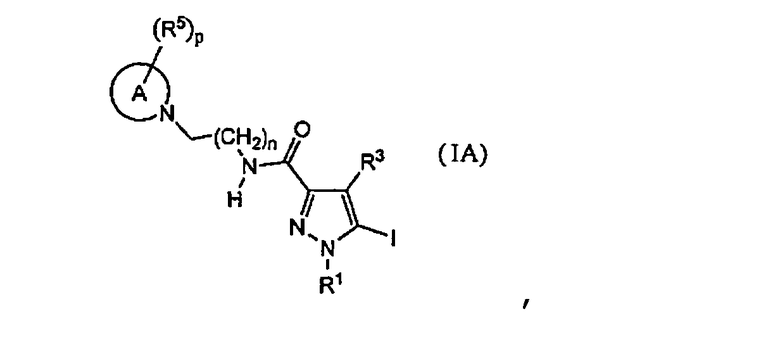
где A означает морфолинил или азетидинил;
R1 означает C1-C6-алкильную группу;
R3 означает водород;
R5 означает галоген;
n равно 1 или 2;
p равно 0, 1, 2, 3, 4 или 5.
5. Промежуточное соединение для соединения по п.1, представленное общей формулой (IB):

где R1 означает C1-C6-алкильную группу;
R2 означает бициклическую арильную группу, выбранную из 1H-индолила, 1H-пирроло[3,2-b]пиридила, хинолила, имидазо[1,2-a]пиридила, которая может быть замещена R4,
R3 означает водород,
R4 означает C1-C6-алкильную группу или галоген.
6. Профилактическое или терапевтическое средство от заболеваний, в которые вовлечены рецепторы 5-HT2B, где эффективным ингредиентом является соединение или его фармацевтически приемлемая соль по любому из пп.1-3.
7. Фармацевтическая композиция, обладающая антагонистической активностью в отношении рецепторов 5-HT2B, содержащая эффективное количество соединения или его фармацевтически приемлемой соли по любому из пп.1-3, и фармацевтически приемлемый носитель.
8. Фармацевтическая композиция для профилактики или лечения болезненного состояния, опосредованного рецепторами 5-HT2B, у млекопитающего, содержащая эффективное количество соединения или его фармацевтически приемлемой соли по любому из пп.1-3, и фармацевтически приемлемый носитель.
9. Соединение или его фармацевтически приемлемая соль по любому из пп.1-3, применимые в профилактике или лечении болезненного состояния, опосредованного рецепторами 5-HT2B.
10. Применение соединения или его фармацевтически приемлемой соли по любому из пп.1-3 в изготовлении лекарственного средства для профилактики или лечения состояния, опосредованного рецепторами 5-HT2B.
11. Способ профилактики или лечения таких заболеваний, как висцеральная боль, гастроэзофагеальная рефлюксная болезнь (GERD), констипация, диарея, функциональное желудочно-кишечное расстройство, синдром раздраженного кишечника, болезнь Крона или неспецифический язвенный колит, включающий введение человеку или млекопитающему эффективного количества фармацевтической композиции, содержащей соединение или его фармацевтически приемлемую соль по любому из пп.1-3 и фармацевтически приемлемый носитель.
| EP 1935881 А1, 25.06.2008 | |||
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| EP 1988075 B1, 25.01.2012 | |||
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Ветряная турбина | 1926 |
|
SU3925A1 |

