Задачей изобретения было обнаружение новых соединений, обладающих ценными свойствами, в частности тех, которые можно использовать для приготовления лекарственных средств.
Настоящее изобретение относится к способу получения ингибитора MetAP-2 3,5-дифтор-бензиламида (S)-3-гидрокси-1-(1Н-индол-5-ил)-2-оксо-пирролидин-3-карбоновой кислоты ("S-9"), который синтезируют на ключевой стадии с помощью асимметричного окисляющего агента: 'оксазиридин Дэвиса'.
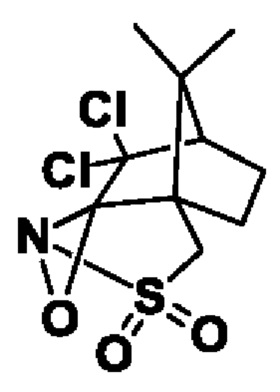
Оксазиридин Дэвиса: (+)-(2R,4aS,7S,8aR)-4H-4a,7-метанооксазирино[3,2-i][2,1]бензизотиазол, 8,8-дихлортетрагидро-9,9-диметил-3,3-диоксид
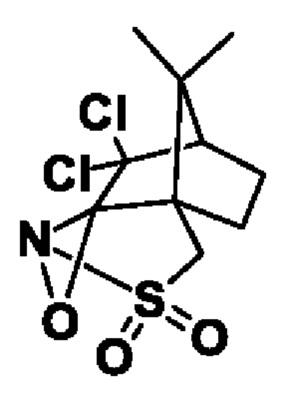
Этот способ получения «S-9» является неизвестным.
Ссылки на предшествующий уровень техники WO 2012048775, WO 2013149704 и WO 2016020031 раскрывают рацемический синтез для получения рацемического соединения с последующим хиральным разделением.
Анализ/сравнение обоих способов, рацемического синтеза и заявленного асимметричного пути, ясно демонстрирует, что асимметричное окисление превосходит уровень техники.
Асимметричный способ требует меньше стадий и более высокого выхода.
Схема 1 дает обзор различий путей.
Обычным промежуточным соединением является соединение номер 5. Исходя из этого соединения 5 установленный путь требует пяти дополнительных стадий синтеза и хиральной хроматографии для получения желаемого энантиомера S-9 с общим выходом 15%.
С помощью 3 дополнительных стадий (R-9 - 10: активация спирта; от 10 до 11: инверсия; 11 - S-9: гидролиз) выход может быть увеличен, но и значительно увеличивается количество работы.
Этот новый способ дает S-9 в три дополнительных стадии из соединения 5 с общим выходом 15% 27%.
Ключевой стадией является энантиоселективное окисление 1-[1-(бензолсульфонил)-1Н-индол-5-ил]-N-[(3,5-дифторфенил)метил]-2-оксопирролидин-3-карбоксамида (12) с получением (3S)-1-[1-(бензолсульфонил)-1Н-индол-5-ил]-N-[(3,5-дифторфенил)метил]-3-гидрокси-2-оксопирролидин-3-карбоксамида (13).
УРОВЕНЬ ТЕХНИКИ
Ссылки на известный уровень техники WO 2012/048775, WO 2013/149704 и WO 2016020031 раскрывают рацемический синтез для получения рацемического соединения с последующим хиральным разделением.
3,5-Дифтор-бензиламид (S)-3-гидрокси-1-(1Н-индол-5-ил)-2-оксо-пирролидин-3-карбоновой кислоты описан как "В8" в WO 2013/149704.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Изобретение относится к способу получения 3,5-дифтор-бензиламида (S)-3-гидрокси-1-(1Н-индол-5-ил)-2-оксо-пирролидин-3-карбоновой кислоты ("S-9"), который отличается тем, что:
a) 2-оксо-1-(1-(фенилсульфонил)-1Н-индол-5-ил)пирролидин-3-карбоновую кислоту ("5") вводят в реакцию с 3,5-дифторбензиламином с получением 1-[1-(бензолсульфонил)-1Н-индол-5-ил]-N-[(3,5-дифторфенил)метил]-2-оксопирролидин-3-карбоксамида ("12"),
b) затем "12" энантиоселективно окисляют с получением (3S)-1-[1-(бензолсульфонил)-1Н-индол-5-ил]-N-[(3,5-дифторфенил)метил]-3-гидрокси-2-оксопирролидин-3-карбоксамида ("13"),
c) и потом фенилсульфонильную группу отщепляют от "13" с получением 3,5-дифтор-бензиламида (S)-3-гидрокси-1-(1Н-индол-5-ил)-2-оксо-пирролидин-3-карбоновой кислоты ("S-9").
Предпочтительно изобретение относится к способу получения 3,5-дифтор-бензиламида (S)-3-гидрокси-1-(1Н-индол-5-ил)-2-оксо-пирролидин-3-карбоновой кислоты ("S-9"), который отличается тем, что:
2-оксо-1-(1-(фенилсульфонил)-1Н-индол-5-ил)пирролидин-3-карбоновую кислоту ("5") вводят в реакцию с 3,5-дифторбензиламином с получением 1-[1-(бензолсульфонил)-1Н-индол-5-ил]-N-[(3,5-дифторфенил)метил]-2-оксопирролидин-3-карбоксамида ("12"),
затем "12" вводят в реакцию с (+)-(2R,4aS,7S,8aR)-4H-4a,7-метанооксазирино[3,2-i][2,1]бензизотиазолом, 8,8-дихлортетрагидро-9,9-диметил-3,3-диоксидом с получением (3S)-1-[1-(бензолсульфонил)-1Н-индол-5-ил]-N-[(3,5-дифторфенил)метил]-3-гидрокси-2-оксопирролидин-3-карбоксамида ("13"),
и потом фенилсульфонильную группу отщепляют от "13" с получением 3,5-дифтор-бензиламида (S)-3-гидрокси-1-(1Н-индол-5-ил)-2-оксо-пирролидин-3-карбоновой кислоты ("S-9").
Реакцию соединения 5 с 3,5-дифторбензиламином на стадии 4С как правило осуществляют в присутствии органического основания, такого как DIPEA, триэтиламин, диметиланилин, пиридин, хинолин, диазабициклоундецен (DBU) или диизопропилэтиламин (основание Хюнига). Наиболее предпочтительно реакцию осуществляют в присутствии триэтиламина, DBU или диизопропилэтиламина.
В зависимости от применяемых условий время реакции составляет от нескольких минут до 14 дней, температура реакции составляет от приблизительно -30°С до 140°С, как правило от -10°С до 90°С, в частности от приблизительно 0°С до приблизительно 40°С.
Реакцию предпочтительно осуществляют в инертном растворителе.
Примеры подходящих инертных растворителей представляют собой углеводороды, такие как гексан, петролейный эфир, бензол, толуол или ксилол; хлорированные углеводороды, такие как трихлорэтилен, 1,2-дихлорэтан, тетрахлорид углерода, хлороформ или дихлорметан; спирты, такие как метанол, этанол, изопропанол, н-пропанол, н-бутанол или трет-бутанол; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран (ТГФ) или диоксан; простые гликолевые эфиры, такие как монометиловый или моноэтиловый эфир этиленгликоля, диметиловый эфир этиленгликоля (диглим); кетоны, такие как ацетон или бутанон; амиды, такие как ацетамид, диметилацетамид или диметилформамид (ДМФА); нитрилы, такие как ацетонитрил; сульфоксиды, такие как диметилсульфоксид (ДМСО); сероуглерод; карбоновые кислоты, такие как муравьиная кислота или уксусная кислота; нитросоединения, такие как нитрометан или нитробензол; сложные эфиры, такие как этилацетат, или смеси указанных растворителей.
Особое предпочтение отдается ацетонитрилу, дихлорметану и/или ДМФА, особенно предпочтительным является дихлорметан.
Амидное сочетание соединения 5 с 3,5-дифторбензиламином предпочтительно осуществляют в присутствии Т3Р (ангидрид пропанфосфоновой кислоты).
Другими предпочтительными соединениями, активирующими кислоту, являются следующие, такие как:
карбодиимиды:
EDCI (1-этил-3-(3-диметиламинопропил)карбодиимид),
DCC (дициклогексилкарбодиимид);
фосфониевые соли:
ВОР (бензотриазолилокситрис(диметиламино)-фосфония гексафторфосфат),
РуВОР (бензотриазол-1-ил-окситрипирролидинофосфоний-гексафторфосфат);
иммониевые соли описаны So-Yeop Han, Young-Ah Kim: Recent development of peptide coupling reagents in organic synthesis: Tetrahedron 60, 2004, S. 2447;
аминиевые соли:
HATU: O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний-гексафторфосфат;
HBTU (2-(1Н-Бензотриазол -1-ил)-1,1,3,3-тетраметилуроний-гексафторфосфат);
урониевые соли:
COMU ((1-циано-2-этокси-2-оксоэтилиденаминоокси)диметиламино-морфолино-карбений-гексафторфосфат);
имидазолиевые соли: имидазолиевые соли описаны So-Yeop Han, Young-Ah Kim: Recent development of peptide coupling reagents in organic synthesis: Tetrahedron 60, 2004, S. 2447;
HObt (Гидроксибензотриазол).
Окисление соединения 12 до соединения 13 на стадии С5 предпочтительно осуществляют в органическом растворителе, таком как ТГФ или диэтиловый эфир.
Реакцию, как правило, осуществляют с использованием основания, такого как NaHMDS (гексаметилдисилазан натрия), LiHMDS (гексаметилдисилазан лития), KHMDS (гексаметилдисилазан калия), LDA (диизопропиламид лития), BuLi (бутиллитий) или трет-бутилат калия. Особое предпочтение отдается NaHMDS.
Реакцию предпочтительно осуществляют с асимметричным окислительным реагентом (+)-(2R,4aS,7S,8aR)-4H-4a,7-метанооксазирино[3,2-i][2,1]бензизотиазол, 8,8-дихлортетрагидро-9,9-диметил-3,3-диоксид
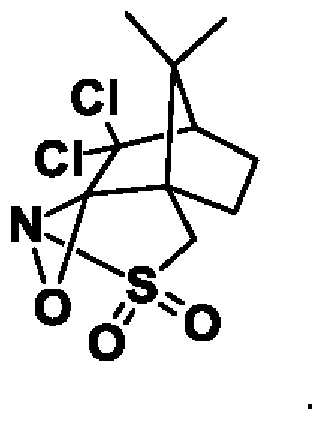
Вместо дихлористого соединения предпочтительными являются дибромистые или дифтористые соединения.
Реакцию соединения 13 до соединения S-9 (отщепление фенилсульфонильной группы) на стадии С6 наиболее предпочтительно осуществляют с гидроксидом щелочных или щелочно-земельных металлов, карбонатом или бикарбонатом или другой солью слабой кислоты щелочных или щелочно-земельных металлов, предпочтительно калия, натрия, кальция или цезия.
Реакцию предпочтительно осуществляют в инертном растворителе.
В зависимости от применяемых условий время реакции составляет от нескольких минут до 14 дней, температура реакции составляет от приблизительно -30°С до 140°С, как правило от -10°С до 90°С, в частности от приблизительно 0°С до приблизительно 70°С.
Примеры подходящих инертных растворителей представляют собой углеводороды, такие как гексан, петролейный эфир, бензол, толуол или ксилол; хлорированные углеводороды, такие как трихлорэтилен, 1,2-дихлорэтан, тетрахлорид углерода, хлороформ или дихлорметан; спирты, такие как метанол, этанол, изопропанол, н-пропанол, н-бутанол или трет-бутанол; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран (ТГФ) или диоксан; простые гликолевые эфиры, такие как монометиловый или моноэтиловый эфир этиленгликоля, диметиловый эфир этиленгликоля (диглим); кетоны, такие как ацетон или бутанон; амиды, такие как ацетамид, диметилацетамид или диметилформамид (ДМФА); нитрилы, такие как ацетонитрил; сульфоксиды, такие как диметилсульфоксид (ДМСО); сероуглерод; карбоновые кислоты, такие как муравьиная кислота или уксусная кислота; нитросоединения, такие как нитрометан или нитробензол; сложные эфиры, такие как этилацетат, или смеси указанных растворителей.
Особое предпочтение отдается этанолу и или ТГФ.
Реакцию соединения 13 до соединения S-9 (отщепление фенилсульфонильной группы) на стадии С6 наиболее предпочтительно осуществляют с NaOH в смеси этанол/ТГФ.
Более предпочтительно, отщепление фенилсульфонильной группы от индольного кольца осуществляют следующим образом:
с фторидом тетрабутиламмония в ТГФ;
с трет-бутоксидом магния или лития в ТГФ;
с трет-бутилатом натрия в диоксане;
с 1-(N,N-диметиламино)пиреном, триэтиламином в ацетонитриле;
с изопропилатом титана(IV), хлортриметилсиланом, магнием в ТГФ.
Примеры
Стадия-1: 3
[5-нитро-1-(фенилсульфонил)-1H-индол]
Схема реакции:
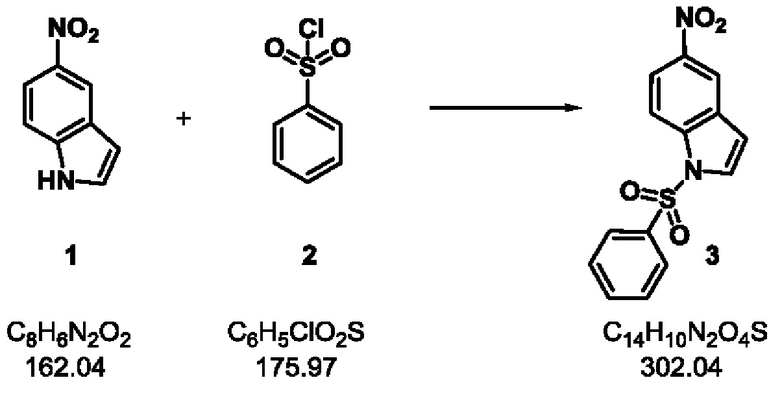
Экспериментальная методика:
5-нитроиндол 1 (500 г, 3.08 моль) растворяли в ТГФ (5 л) и смесь охлаждали до 0°С и перемешивали в течение 20 минут. Гидрид натрия (140 г, 3.5 моль) добавляли порциями и смесь перемешивали в течение еще 30 минут при 15°С. Бензолсульфонилхлорид 2 (475 мл, 3.7 моль) вводили через дополнительную воронку в течение 30 минут при перемешивании. После завершения добавления смесь перемешивали в течение 4 часов. После завершения реакции, реакционную массу охлаждали до 0°С и гасили льдом (3 л).
Добавляли этилацетат (5 л) и воду (2.5 л). После разделения фаз водный слой повторно экстрагировали этилацетатом (5 л). Объединенный органический слой сушили над сульфатом натрия и концентрировали при пониженном давлении при 55°С. Смесь этилацетат/петролейный эфир (8%, 5 л) добавляли к сырой массе и смесь перемешивали в течение 20 мин при комнатной температуре. Продукт фильтровали и промывали смесью этилацетата и петролейного эфира (5%, 2 л). Продукт сушили в вакууме с получением соединения 3 в виде желтого твердого вещества.
Выход 890 г (95%).
1H ЯМР (400 МГц, ДМСО-d6) δ 8.63-8.55 (m, 1Н), 8.26-8.14 (m, 2 Н), 8.13-8.02 (m, 3Н), 7.79-7.70 (m, 1Н), 7.69-7.59 (m, 2Н), 7.10 (d, J=3.7 Гц, 1Н); молекулярная формула: C14H10N2O4S; чистота ВЭЖХ: 99.92%; ожидаемая масса ЖХМС: 302.0; наблюдаемая: 161.2 (М-141).
Стадия-2: 4
1-(фенилсульфонил)-1H-индол-5-амин
Схема реакции:
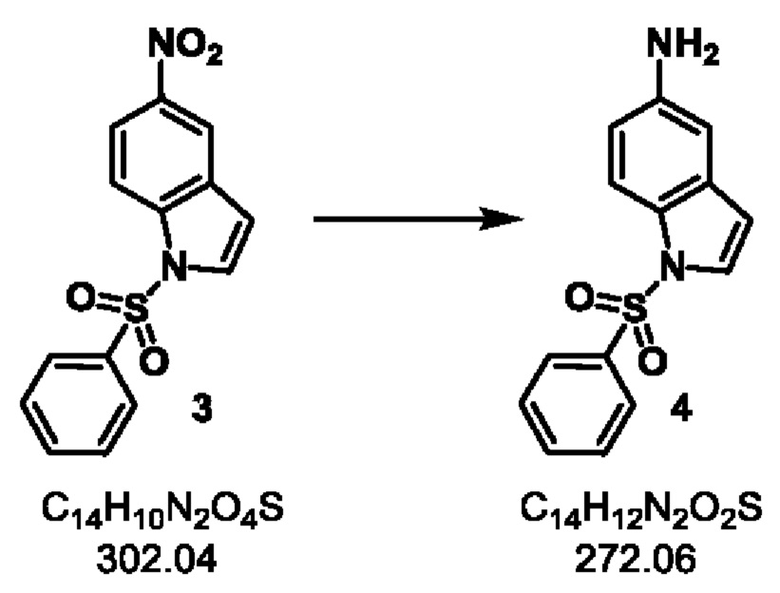
Экспериментальная методика:
Продукт 3 Стадии-1 (500 г, 1.65 моль) растворяли в этаноле (7 л). Добавляли железный порошок (500 г, 8.95 моль) и смесь нагревали до 50°С. Через 15 минут, раствор NH4Cl (1 кг, 18.69 моль) в воде (3.1 л) добавляли к реакционной смеси через дополнительную воронку в течение 1 часа. Реакционная смесь нагревали до 80°С в течение 2 часов. После завершения реакции, реакционную массу охлаждали до 40°С, фильтровали через цеолит и концентрировали при пониженном давлении при 50°С. Этилацетат и воду (5 л каждого) добавляли и слои разделяли. Водн. слой повторно экстрагировали этилацетатом (5 л). Объединенный органический слой сушили над сульфатом натрия и концентрировали при пониженном давлении при 50°С. Оставшееся суспендировали в смеси этилацетат/петролейный эфир (5%, 5 л) и затем охлаждали до комнатной температуры. Продукт фильтровали и промывали смесью этилацетат/петролейный эфир (5%, 5 л). Продукт сушили в вакууме с получением продукта 4 в виде коричневого твердого вещества.
Выход 400 г (89%).
1Н ЯМР (300 МГц, ДМСО-d6) δ 7.86 (d, J=7.5 Гц, 2 Н), 7.70-7.42 (m, 5 Н), 6.67-6.48 (m, 3 Н), 4.97 (s, 2 Н); молекулярная формула: C14H12N2O2S; чистота ВЭЖХ: 97.25%; ожидаемая масса ЖХМС: 272.1; наблюдаемая: 273.0 (М+1).
Стадия-3: 5
2-оксо-1-(1-(фенилсульфонил)-1Н-индол-5-ил)пирролидин-3-карбоновая кислота.
Схема реакции:
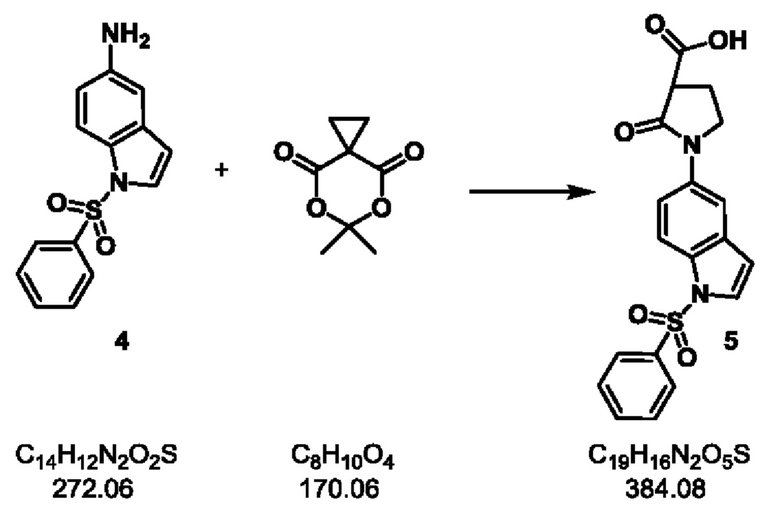
Экспериментальная методика:
Продукт 4 Стадии-2 (1.6 кг, 5.87 моль) и циклопропиловую кислоту Мельдрума (1.2 кг, 7.05 моль) подавали в реактор с последующим добавлением ацетонитрила (5.5 л) и ДМФА (1.9 л). Смесь нагревали до 70°С в течение 16 часов в атмосфере азота. После завершения реакции, реакционную массу концентрировали при пониженном давлении при 50-55°С. Остаток охлаждали и обрабатывали водой и этилацетатом (10 л каждого). После разделения фаз органический слой промывали соляным раствором (5 л), сушили над сульфатом натрия и концентрировали при пониженном давлении при 40-45°С. Полученное сырое твердое вещество промывали смесью этилацетат/петролейный эфир (5%, 2 л) с получением продукта 5 в виде коричневого твердого вещества.
Выход: 1.8 кг (80%).
1Н ЯМР (300 МГц, ДМСО-d6) δ 12.80 (br. s., 1 Н), 8.02-7.85 (m, 3 Н), 7.85 -7.74 (m, 2 Н), 7.71-7.47 (m, 4 Н), 6.84 (d, J=3.6 Гц, 1 Н), 3.94-3.76 (m, 2 Н), 3.57 (t, J=8.5 Гц, 1 Н), 2.37-2.20 (m, 2 Н); молекулярная формула: C19H16N2O5S; чистота ВЭЖХ: 91.51%; ожидаемая масса ЖХМС: 384.08; наблюдаемая: 385.0 (М+1).
Продукт 5 является отправной точкой для обеих методик, рацемического и асимметричного синтезов.
Стадия-4: 6
3-гидрокси-2-оксо-1-(1-(фенилсульфонил)-1Н-индол-5-ил)пирролидин-3-карбоновая кислота.
Схема реакции:
Экспериментальная методика:
Продукт 5 Стадии-3 (1.0 кг, 2.60 моль) обрабатывали ДМФА (8.5 л) и гексагидратом монопероксифталата магния 80% (1.9 кг, 3.84 моль). Смесь нагревали до 60°С в течение 2 часов в атмосфере азота. После завершения реакции, реакционную массу концентрировали при пониженном давлении при 50-55°С. Остаток вносили в воду (5 л) и этилацетат (3 л) и перемешивали в течение 12 часов при комнатной температуре. Продукт фильтровали и промывали водой и этилацетатом (3 л каждого). Продукт сушили в вакууме при 65°С с получением продукта 6 в виде грязно-белого твердого вещества.
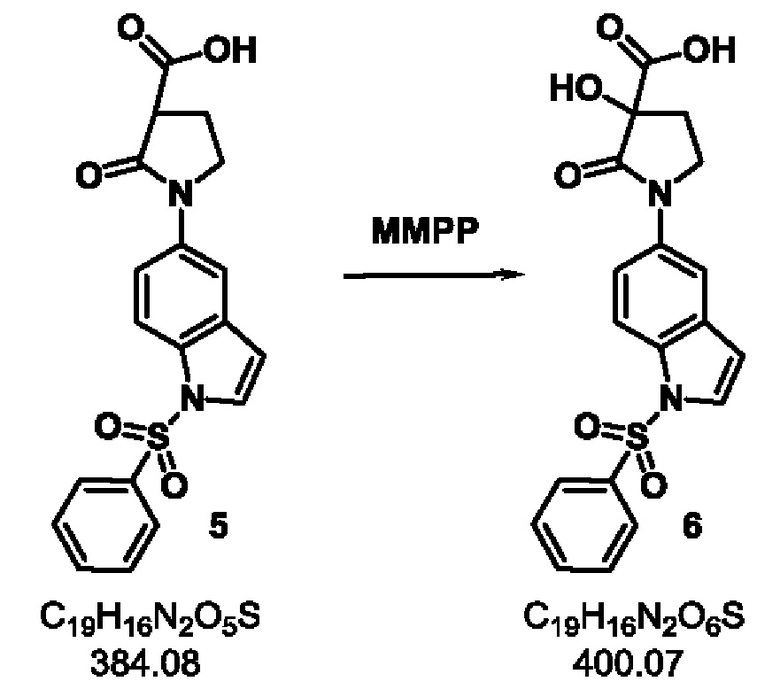
Выход: 700 г (67%).
Примечание: содержание влаги в продукте стадии 4 должно быть менее 0,5%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 7.97-7.94 (m, 3 Н), 7.87 (s, 1 Н), 7.81-7.80 (d, J=3.4 Гц, 1Н), 7.73-7.66 (m, 2 Н), 7.60-7.56 (m, 2 Н), 7.16 (brs, 1Н), 6.87 (d, J=3.4 Гц, 1 Н), 3.92 (q, J=8.4 Гц, 1 Н), 3.75 (t, J=8.7 Гц, 1 Н), 3.45-3.42 (m, 1Н), 2.43-2.38 (m, 1 Н), 2.03-1.96 (m, 1 Н); молекулярная формула: C19H16N2O6S; чистота ВЭЖХ: 96.12%; ожидаемая масса ЖХМС: 400.07; наблюдаемая: 401.0 (М+1).
Стадия-5: 7
3-ацетокси-2-оксо-1-(1-(фенилсульфонил)-1Н-индол-5-ил)пирролидин-3-карбоновая кислота.
Схема реакции:
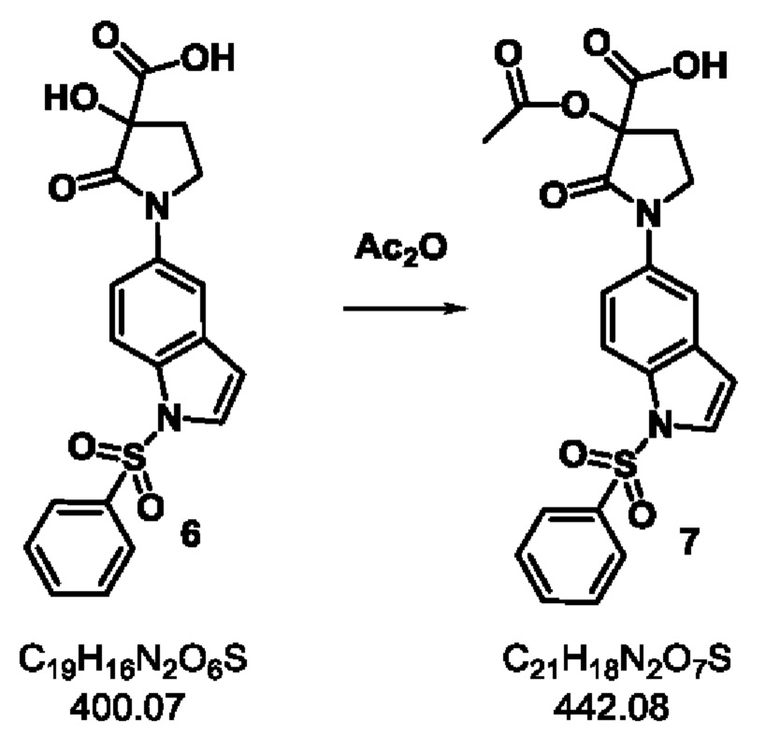
Экспериментальная методика:
Продукт 6 Стадии-4 (1.0 кг, 2.5 моль) и ДМФА (8 л) подавали в реактор при комнатной температуре и перемешивали в течение 10 минут. Ангидрид уксусной кислоты (355 мл, 3.75 моль) добавляли медленно у массе и смесь перемешивали в течение 12 часов. После завершения реакции, реакционную массу концентрировали при пониженном давлении при 50-55°С. Остаток охлаждали до 0°С, суспендировали водой (5 л) и перемешивали в течение ночи при комнатной температуре. Осадок фильтровали, промывали водой (3 л) и затем суспендировали в ацетоне (3 л) в течение 1 часа. Фильтрация давала продукт 7 в виде белого твердого вещества, которое сушили в вакууме при 65°С.
Выход: 940 г (85%).
Примечание: содержание влаги в продукте стадии 5 должно быть менее 0.5%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 7.98-7.89 (m, 3 Н), 7.86-7.77 (m, 2 Н), 7.73-7.62 (m, 2 Н), 7.60-7.51 (m, 2 Н), 6.85 (d, J=3.8 Гц, 1 Н), 3.98 (q, J=8.1 Гц, 1 Н), 3.72 (t, J=9.0 Гц, 1 Н), 2.79 (dd, J=7.3, 12.1 Гц, 1 Н), 2.22-2.09 (m, 1 Н), 2.01 (s, 3 Н); молекулярная формула: C21H18N2O7S; чистота ВЭЖХ: 97.83%; ожидаемая масса ЖХМС: 442.08; наблюдаемая: 443.0 (М+1).
NB:
Реакционную смесь необходимо концентрировать ниже 55°С и выпаривание должно завершиться в течение 2 часов. Более высокая температура и длительное нагревание приводит к декарбоксилированию продукта.
Стадия-6: 8
3-((3,5-дифторбензил)карбамоил)-2-оксо-1-(1-(фенилсульфонил)-1Н-индол-5-ил)пирролидин-3-илацетат.
Схема реакции:
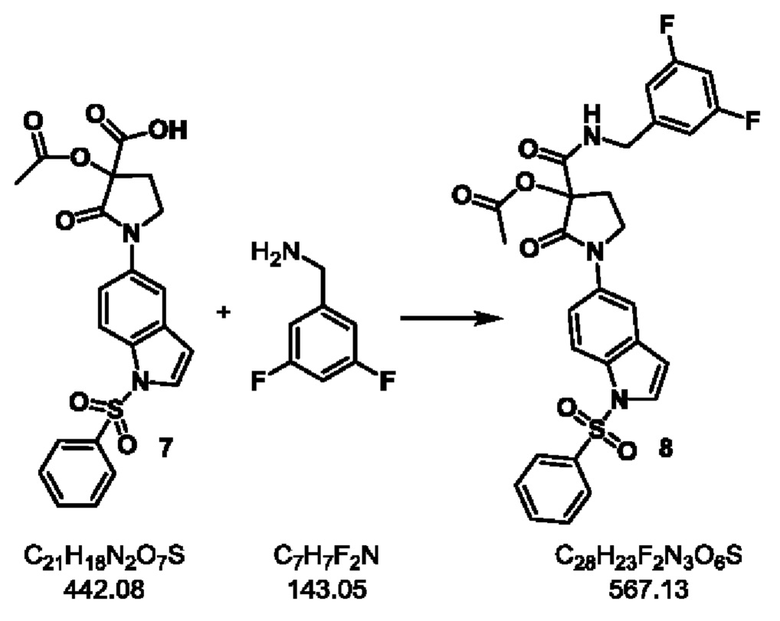
Экспериментальная методика:
Продукт 7 Стадии-5 (1.0 кг, 2.26 моль) растворяли в СН2С12 (10 л) при комнатной температуре в течение 10 минут и затем охлаждали до 0°С. Добавляли триэтиламин (690 мл, 4.95 моль), 3,5-дифторбензиламин (405 г, 2.83 моль) и 2,4,6-трипропил-[1,3,5,2,4,6]триоксатрифосфинан 2,4,6-триоксид (2.15 L, 3.38 моль) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. После завершения реакции, реакционную массу разбавляли водой (5 л) и перемешивали в течение 10 минут. Водный слой удаляли сверху. Это водное промывание повторяли 3 раза. Органический слой фильтровали и осадок промывали CH2Cl2 (1 л) и ацетоном (0.5 л) с получением продукта 8 в виде грязно-белого твердого вещества. Выход: 1.07 кг (83%).
1H ЯМР (400 МГц, ДМСО-d6) δ 8.99 (t, J=6.2 Гц, 1 Н), 7.97 (d, J=8.1 Гц, 3 Н), 7.83 (dd, J=2.8, 5.2 Гц, 2 Н), 7.72-7.63 (m, 2 Н), 7.62-7.53 (m, 2 Н), 7.06 (t, J=9.4 Гц, 1 Н), 6.93 (d, J=7.0 Гц, 2 Н), 6.87 (d, J=3.5 Гц, 1 Н), 4.33 (dq, J=6.0, 16.1 Гц, 2 Н), 3.99-3.83 (m, 2 Н), 2.88 (ddd, J=2.6, 7.9, 13.2 Гц, 1 Н), 2.43-2.28 (m, 1 Н), 2.18 (s, 3 Н); молекулярная формула: C28H23F2N3O6S; чистота ВЭЖХ: 99.88%; ожидаемая масса ЖХМС: 567.13; наблюдаемая: 568.0 (М+1).
Стадия-7: 9
N-(3,5-дифторбензил)-3-гидрокси-1-(1Н-индол-5-ил)-2-оксопирролидин-3-карбоксамид
Схема реакции:
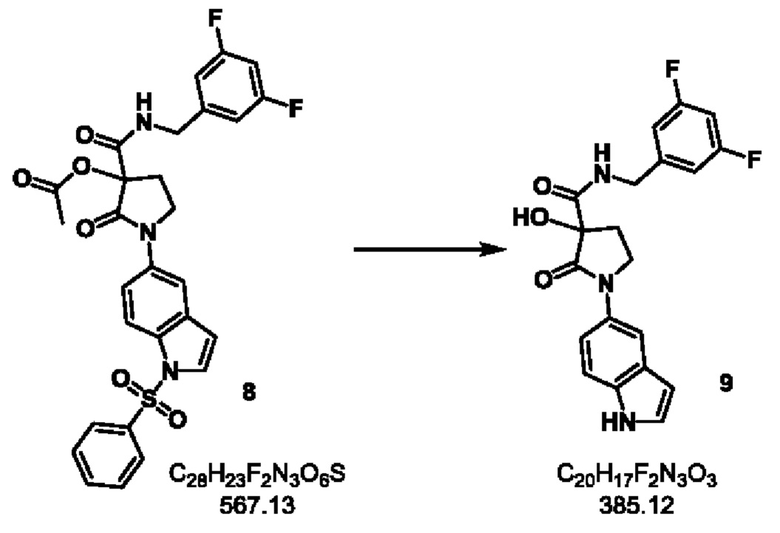
Экспериментальная методика:
Стадия-6 продукт 8 (1.2 кг, 2.11 моль) растворяли в этаноле (5 л) и ТГФ (10 л) и перемешивали при комнатной температуре в течение 10 минут. Добавляли гидроксид натрия (422 г, 10.55 моль) и перемешивали в течение 2 часов при 50°С. После завершения реакции, реакционную массу концентрировали при пониженном давлении при 45°С. Остаток растворяли в этилацетат (10 л) и воде (5 л). После разделения фаз органический слой промывали водой (2×5 л) и соляным раствором (5 л). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении при 45-50°С. CH2Cl2 (1 л) добавляли к оставшемуся и осадок фильтровали и промывали CH2Cl2 (1.0 л) с получением продукта 9 в виде грязно-белого твердого вещества.
Выход: 700 г (86%).
1Н ЯМР (400 МГц, ДМСО-d6) δ 11.12 (br. s., 1 Н), 8.69 (t, J=6.4 Гц, 1 Н), 7.70 (s, 1 Н), 7.46-7.32 (m, 3 Н), 7.12-6.94 (m, 3 Н), 6.69 (s, 1 Н), 6.47-6.38 (m, 1 Н), 4.41 (dd, J=6.9, 16.0 Гц, 1 Н), 4.25 (dd, J=5.9, 15.8 Гц, 1 Н), 3.94-3.81 (m, 2 Н), 2.66-2.54 (m, 1 Н), 2.13 (td, J=7.6, 13.0 Гц, 1 Н); молекулярная формула: C20H17F2N3O3; чистота ВЭЖХ: 98.11%; ожидаемая масса ЖХМС: 385.12; наблюдаемая: 386.0 (М+1).
Разделение SFC:
Изомеры продукта 9 (5.20 кг) разделяли с помощью разделения SFC.

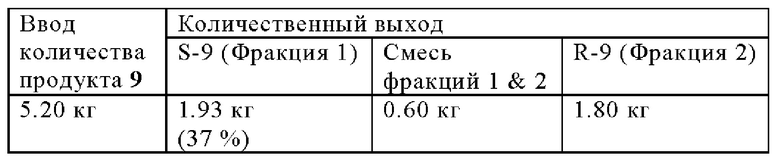
Фракция 1 (S-9): 1H ЯМР (400 МГц, ДМСО-d6) δ 11.12 (br. s., 1 Н), 8.69 (t, J=6.4 Гц, 1 H), 7.70 (s, 1 Н), 7.46-7.32 (m, 3 Н), 7.12-6.94 (m, 3 Н), 6.69 (s, 1 H), 6.47-6.38 (m, 1 Н), 4.41 (dd, J=6.9, 16.0 Гц, 1 Н), 4.25 (dd, J=5.9, 15.8 Гц, 1 Н), 3.94-3.81 (m, 2 Н), 2.66-2.54 (m, 1 Н), 2.13 (td, J=7.6, 13.0 Гц, 1 Н); молекулярная формула: C20H17F2N3O3; чистота ВЭЖХ: 99.46%; чистота хиральной ВЭЖХ: 100%; ожидаемая масса ЖХМС: 385.12; наблюдаемая: 386.0 (М+1); SOR: +14.69.
Фракция 2 (R-9): 1Н ЯМР (400 МГц, ДМСО-d6) δ 11.12 (br. s., 1 Н), 8.69 (t, J=6.3 Гц, 1 H), 7.73-7.68 (m, 1 Н), 7.45-7.31 (m, 3 H), 7.11-6.94 (m, 3 Н), 6.69 (s, 1 H), 6.46-6.38 (m, 1 Н), 4.41 (dd, J=6.7, 15.8 Гц, 1 Н), 4.25 (dd, J=5.9, 15.8 Гц, 1 Н), 3.94-3.79 (m, 2 Н), 2.66-2.56 (m, 1 Н), 2.13 (td, J=7.6, 13.0 Гц, 1 Н);
молекулярная формула: C20H17F2N3O3; чистота ВЭЖХ: 97.20%; чистота хиральной ВЭЖХ: 98.17%; ожидаемая масса ЖХМС: 385.12; наблюдаемая: 386.2 (М+1); SOR: -13.49.
Смесь фракций (1 & 2): чистота ВЭЖХ: 98.90%; чистота хиральной ВЭЖХ: 32.99% (фракция 1) & 67.01 (фракция 2).
Оптимизация выхода может быть достигнута путем инверсии хирального центра. Изобретенная методика приведена.
Стадия 8: 10
(R)-3-((3,5-дифторбензил)карбамоил)-1-(1Н-индол-5-ил)-2-оксопирролидин-3-ил метансульфонат Схема реакции:
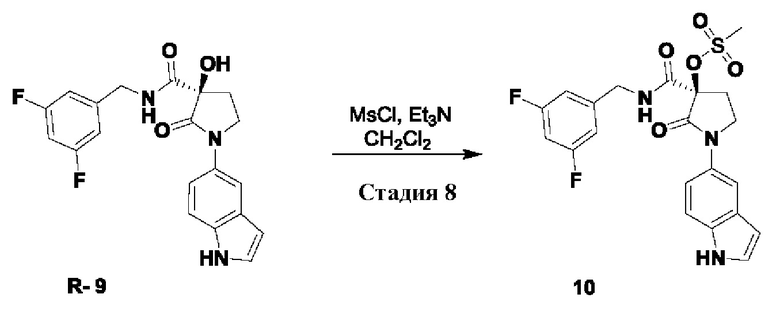
Экспериментальная методика:
к охлажденному льдом раствору R-9 (200 г, 0.52 моль) в сухом CH2Cl2 (2 л), добавляли Et3N (252 мл, 1.81 моль), с последующим добавлением по каплям мезилхлорида (80.4 мл, 1.03 моль). Реакционную смесь перемешивали при такой же температуре в течение еще 2 часов. После завершения реакционную смесь промывали водой (1.5 л), 5% лимонной кислотой (1 л) и насыщенным водным раствором NaHCO3 (1×5 л). Объединенные органические экстракты промывали соляным раствором (1 л), сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением продукта 10 (260 г). Сырой продукт брали непосредственно на следующую стадию без дополнительной очистки.
Молекулярная формула: C21H19F2N3O5S; чистота ВЭЖХ: 81.76%; ожидаемая масса ЖХМС: 463.10; наблюдаемая: 464.0 (М+1).
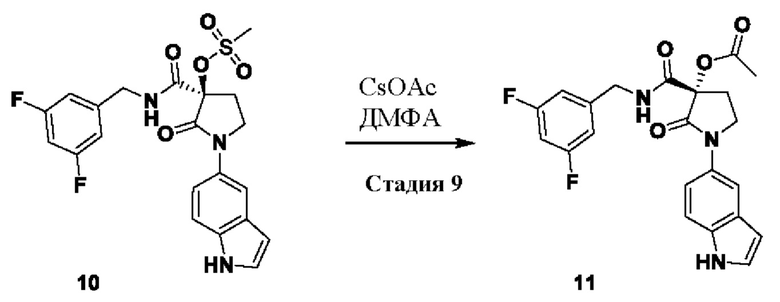
Стадия 9: 11
(S)-3-((3,5-дифторбензил)карбамоил)-1-(1Н-индол-5-ил)-2-оксопирролидин-3-ил ацетат
Схема реакции:
Экспериментальная методика:
к раствору ацетата цезия (214 г, 1.12 моль) в сухом ДМФА (1.2 л) при 100°С добавляли раствор продукта 10 (260 г, 0.560 моль) в ДМФА (1.0 л) по каплям в течение 20 минут через дополнительную воронку. Нагревание продолжали в течение еще 1.5 часов. После завершения, реакционную массу концентрировали в вакууме. Сырую массу растворяли в этилацетате (2 л) и промывали водой (2X2 л). Объединенные органические экстракты промывали соляным раствором (1 л), сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением продукта 11 (250 г). Сырой продукт брали непосредственно на следующую стадию без дополнительной очистки.
Молекулярная формула: C22H19F2N3O4; чистота ВЭЖХ: 45.64%; ожидаемая масса ЖХМС 427.13; наблюдаемая: 428.3 (М+1).
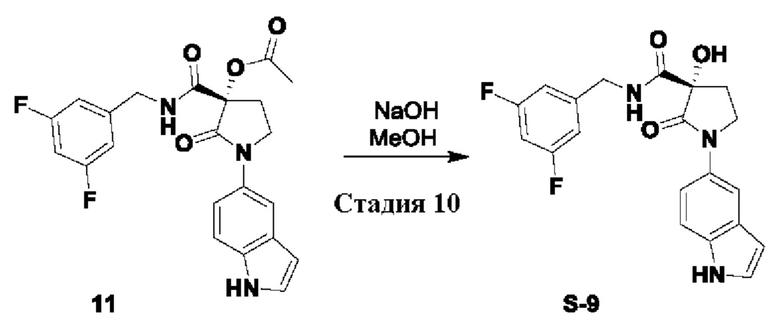
Стадия 10: S-9
(S)-N-(3,5-дифторбензил)-3-гидрокси-1-(1Н-индол-5-ил)-2-оксопирролидин-3-карбоксамид
Схема реакции:
Экспериментальная методика:
к охлажденному льдом раствору сырого продукта 11 (250 г 0.52 моль) в метаноле (2.5 л) добавляли пеллеты NaOH (63 г, 1.56 моль). Реакционную смесь перемешивали при КТ в течение 2 часов. После завершения метанол концентрировали в вакууме при <55°С. Сырую массу растворяли в этилацетате (2 л) и промывали водой (3X2 л). Объединенные органические экстракты промывали соляным раствором (1 л), сушили над Na2SO4, фильтровали и концентрировали в вакууме. Полученное пропускали через промывочную колонку (Silica 60-120) и неполярные примеси удаляли с использованием 30-40% этилацетата/петролейного эфира. Продукт элюировали посредством 3-5% МеОН/CH2Cl2. Выделенный продукт растворяли в минимальном количестве этилацетата и оставляли в холодильной камере на 16 часов. Образовавшееся твердое вещество фильтровали через воронку Бюхнера, промывали этилацетатом (3×100 мл) с получением продукта S-9 (56 г, 28% в три стадии) в виде грязно-белого твердого вещества.
Высушенное твердое вещество перемалывали в штифтовой мельнице (6000 об/мин, 30 мин) с получением в конце API.
1H ЯМР (400 МГц, ДМСО-d6) δ 11.12 (br. s., 1 Н), 8.69 (t, J=6.4 Гц, 1 Н), 7.70 (s, 1 Н), 7.46-7.32 (m, 3 Н), 7.12-6.94 (m, 3 Н), 6.69 (s, 1 Н), 6.47-6.38 (m, 1 Н), 4.41 (dd, J=6.9, 16.0 Гц, 1 Н), 4.25 (dd, J=5.9, 15.8 Гц, 1 Н), 3.94-3.81 (m, 2Н), 2.66-2.54 (m, 1 Н), 2.13 (td, J=7.6, 13.0 Гц, 1 Н); молекулярная формула: C20H17F2N3O3; чистота ВЭЖХ: 98.72%; чистота хиральной ВЭЖХ: 98.68%; (ее: 97.36%); ожидаемая масса ЖХМС: 385.12; наблюдаемая: 386.2 (М+1).



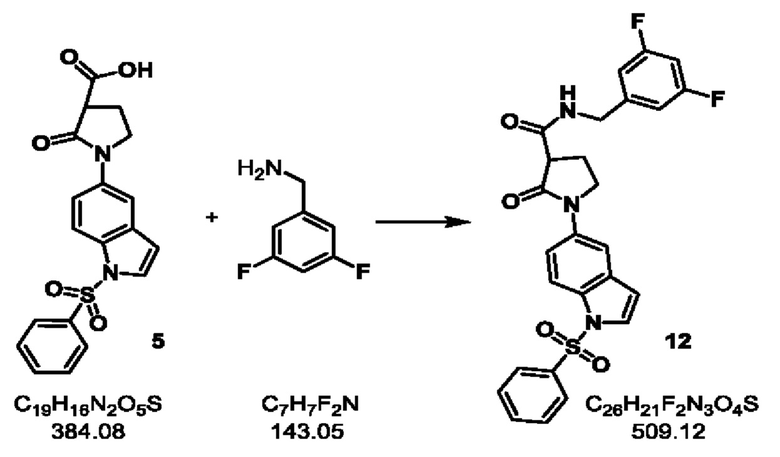
Стадия-4С: 12
1-[1-(бензолсульфонил)-1Н-индол-5-ил]-N-[(3,5-дифторфенил)метил]-2-оксопирролидин-3-карбоксамид
Экспериментальная методика:
к охлажденному льдом раствору продукта 5 (750 г, 1.95 моль) в CH2Cl2 (7 л), добавляли Et3N (600 мл, 4.29 моль) с последующим добавлением 3,5-дифторбензиламина (363 г, 2.53 моль). В реакционную смесь по каплям медленно добавляли Т3Р (ангидрид пропанфосфоновой кислоты) (1.86 л (50% ЭА [этилацетат] раствор), 3.0 моль) и реакционную смесь перемешивали при КТ в течение 2 ч. После завершения реакции, реакционную массу гасили водой (3 л) и перемешивали в течение 10 минут. Органический слой разделяли и промывали 10% раствором NaHCO3 (2 л), а с последующей промывкой водой (3 л × 3). Органическую фазу в конце промывали соляным раствором (3 л), сушили над безводным сульфатом натрия и концентрировали с получением сырого продукта в виде светло-коричневого твердого вещества. Готовили суспензию с минимальным количеством этилацетата (1.5 л) и фильтровали. Осадок на фильтре промывали ледяным этилацетатом (1 л × 2) с получением чистого продукта в виде грязно-белого твердого вещества с ВЭЖХ>99%.
Полученное количество: 720 г; выход: 72%;
1Н-ЯМР (400 МГц, ДМСО-d6): δ 8.82 (t, J=5.60 Гц, 1H), 7.99-7.94 (m, 3Н), 7.83 (d, J=2.80 Гц, 2Н), 7.71-7.67 (m, 2Н), 7.61-7.57 (m, 2Н), 7.12-7.05 (m, 3Н), 6.87 (d, J=3.60 Гц, 1H), 4.49 (dd, J=6.80, 16.20 Гц, 1H), 4.27 (dd, J=5.20, 16.00 Гц, 1H), 3.90-3.85 (m, 2H), 3.63 (t, J=8.80 Гц, 1H), 2.40-2.27 (m, 2H).
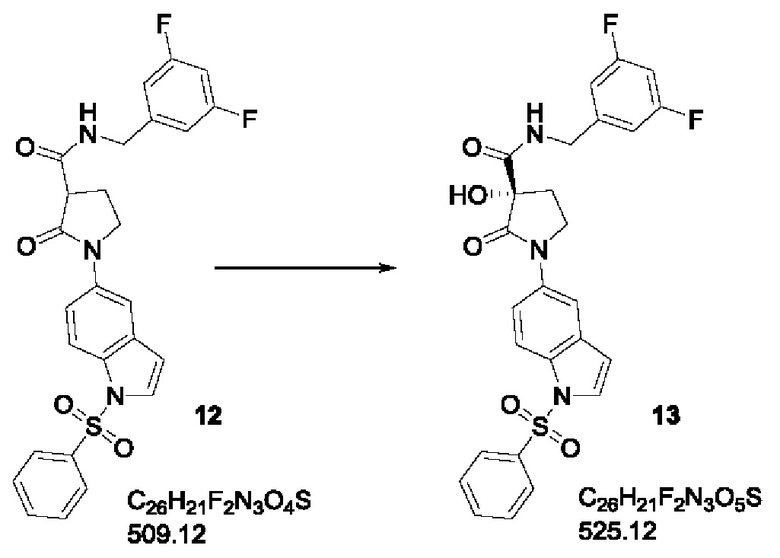
Стадия C5: 13
(3S)-1-[1-(бензолсульфонил)-1Н-индол-5-ил]-N-[(3,5-дифторфенил)метил]-3-гидрокси-2-оксопирролидин-3-карбоксамид
Экспериментальная методика:
суспензию продукта 12 (500 г, 0.98 моль) в сухом ТГФ (7.5 л) охлаждали до -68°С (внутренняя температура). По каплям добавляли раствор NaHMDS (1078 мл, 1М в ТГФ, 1.078 моль) в течение 1.5 ч при поддержании того же диапазона температур. После завершения добавления температуре реакционной смеси давали подняться до -55°С в течение еще 1 ч и затем повторно охлаждали до внутренней температуры -68°С. К желтой реакционной смеси по каплям добавляли раствор ((+)-(2R,4aS,7S,8aR)-4H-4a,7-метанооксазирино[3,2-i][2,1]бензизотиазола, 8,8-дихлортетрагидро-9,9-диметил-3,3-диоксида (365 г, 1.22 моль) в ТГФ (1.1 л) в течение 1.3 ч при такой же температуре. Реакционной массе давали достичь температуры -25°С в течение еще 1.5 ч. После полного превращения систему гасили при -15°С смесью лед/вода (2 л). Этилацетат (5 л) добавляли к реакционной смеси, органический слой разделяли и промывали водой (3 л). Водный слой насыщали NaCl и повторно экстрагировали этилацетатом (1 л). Объединенный органический слой в конце промывали соляным раствором, сушили над Na2SO4 и концентрировали с получением сырого продукта, который очищали с помощью колоночной хроматографии (SiO2 230-400 меш). Продукт элюировали в 50-60% этилацетате и получали в виде грязно-белого твердого вещества.
Полученное количество: 375 г; выход: 73%.
1H ЯМР (500 МГц, ДМСО-d6) м.д.=8.67 (t, J=6.4, 1H), 7.98-7.92 (m, 3Н), 7.86 (d, J=2.2, 1H), 7.82 (d, J=3.7, 1H), 7.75-7.65 (m, 2Н), 7.61-7.55 (m, 2Н), 7.05 (tt, J=9.3, 2.4, 1Н), 7.01-6.93 (m, 2Н), 6.89-6.84 (m, 1Н), 6.72 (s, 1H), 4.39 (dd, J=15.8, 6.8, 1Н), 4.25 (dd, J=15.8, 6.0, 1H), 3.90-3.83 (m, 2H), 2.65-2.54 (m, 1H), 2.17-2.09 (m, 1H). ЖХМС система A: H2O+0,05% HCOOH | система В: MeCN+0,04% HCOOH; T: 30°C | Поток: 2,4 мл/мин | Колонка: Chromolith RP-18e 100-3 | MC: 85-800 а.е.м. Градиент: 4% -->100% (В) 0 -->2,8 мин | 100% (В) 2,8-3,3 мин. Время удержания: 2.376 мин (М+Н+): 526.1.
Примечание: средняя хиральная чистота, достигнутая на этой стадии, составляла 85%, а максимальная - 87%. Важно, что любая промывка сырого вещества приведет к значительному снижению хиральной чистоты.
Структура оксазиридина Дэвиса:
Стадия С6: S-9
3,5-Дифтор-бензиламид (S)-3-гидрокси-1-(1Н-индол-5-ил)-2-оксо-пирролидин-3-карбоновой кислоты
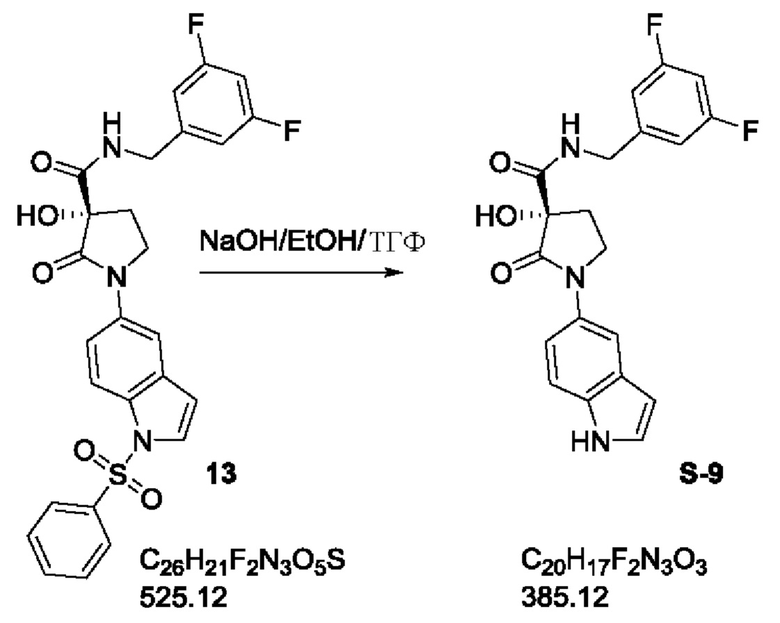
Экспериментальная методика:
пеллеты гидроксида натрия (140 г, 3.56 моль) добавляли к перемешиваемой суспензии продукта 13 (375 г, 0.71 моль) в смеси этанол/ТГФ (3 л/1 л) при КТ. Реакционную смесь нагревали при 50°С в течение 2 ч. После полного превращения реакционную смесь концентрировали с получением сырой массы. Добавляли воду (4 л) и перемешивали в течение 1 ч при КТ. Образовавшееся твердое вещество фильтровали через воронку Бюхнера, нейтрализовали промывкой 1.5 н. HCl, а затем водой (1 л × 3). Остаток в конце промывали эфиром (2 л) с получением сырого продукта. На этой стадии проверяли хиральную чистоту соединения и устанавливали, что она составляла 95.5%. Для повышения хиральной чистоты твердое вещество растворяли в минимальном количестве смеси ТГФ/этилацетат (9:1) и нагревали с обратным холодильником при 60°С в течение 30 мин. Раствор фильтровали через воронку Бюхнера и прозрачный фильтрат охлаждали льдом в течение 2-3 ч и образовавшееся твердое вещество фильтровали. Фильтрат снова охлаждали льдом в течение 2 ч и твердое вещество отдельно фильтровали. Проверяли хиральную чистоту каждого твердого вещества и все фракции смешивали с э.и. > 98.7% и в конце очищали с помощью колоночной хроматографии (SiO2 230-400 меш) с использованием DCM/MeOH в качестве элюента. Чистый продукт элюировали посредством 2% метанола, концентрировали при пониженном давлении с получением желаемого продукта S-9 в виде грязно-белого твердого вещества. Продукт сушили при 60°С в течение 12 ч.
Полученное количество: 140 г; выход: 51%.
Примечание 01: если хиральная чистота сырого продукта S-9 составляла > 97%, массу обрабатывали смесью этилацетат/ТГФ в минимальном объеме (3 В, 9:1), перемешивали в течение 30 мин при КТ и фильтровали с получением желаемой хиральной чистоты > 98.7%.
Примечание 02: собранный основной водный слой подкисляли посредством 2н. HCl и образовавшееся твердое вещество фильтровали. Осадок на фильтре нейтрализовали путем промывки водой и в конце промывали холодным этилацетатом с получением оставшегося соединения с хиральной чистотой 60%.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 3,5-ДИФТОР-БЕНЗИЛАМИДА (S)-3-ГИДРОКСИ-1-(1H-ИНДОЛ-5-ИЛ)-2-ОКСО-ПИРРОЛИДИН-3-КАРБОНОВОЙ КИСЛОТЫ | 2020 |
|
RU2826010C2 |
| Новые 3-индол замещенные производные, фармацевтические композиции и способы применения | 2016 |
|
RU2672252C1 |
| (АЗА)ИНДОЛ-, БЕНЗОТИОФЕН- И БЕНЗОФУРАН-3-СУЛЬФОНАМИДЫ | 2017 |
|
RU2767904C2 |
| ПИРИДИНИЛ- И ПИРАЗИНИЛ(АЗА)ИНДОЛСУЛЬФОНАМИДЫ | 2019 |
|
RU2799321C2 |
| Соединения формул (I) и (A), фармацевтическая композиция, лекарственное средство, применение и способ получения соединения формулы (I) | 2018 |
|
RU2822758C2 |
| ПРОИЗВОДНЫЕ ПИРИДИНА И ИХ ПРИМЕНЕНИЕ ПРОТИВ МИКОБАКТЕРИЙ | 2015 |
|
RU2664587C1 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ОБЕСПЕЧИВАЮЩИЕ РАЗРУШЕНИЕ БЕЛКА IKAROS И БЕЛКА AIOLOS | 2020 |
|
RU2833608C2 |
| МОДУЛЯТОРЫ РЕЦЕПТОРА НМДА И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2621049C2 |
| СПИРОСОЕДИНЕНИЯ В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРОВ МЕЛАНОКОРТИНА 4 И ИХ ПРИМЕНЕНИЯ | 2021 |
|
RU2813541C1 |
| ПРОИЗВОДНЫЕ 2-ФЕНОКСИ И 2-ФЕНИЛСУЛЬФОНАМИДА С ССR3 АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ ДЛЯ ЛЕЧЕНИЯ АСТМЫ И ДРУГИХ ВОСПАЛИТЕЛЬНЫХ ИЛИ ИММУНОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2004 |
|
RU2380356C2 |
Настоящее изобретение относится к области тонкого органического синтеза, конкретно к методу получения 3,5-дифтор-бензиламида (S)-3-гидрокси-1-(1Н-индол-5-ил)-2-оксо-пирролидин-3-карбоновой кислоты (соединение S-9), являющегося ингибитором MetAP-2. Способ получения S-9 характеризуется тем, что 2-оксо-1-(1-(фенилсульфонил)-1Н-индол-5-ил)пирролидин-3-карбоновую кислоту (соединение 5) вводят в реакцию с 3,5-дифторбензиламином с получением 1-[1-(бензолсульфонил)-1Н-индол-5-ил]-N-[(3,5-дифторфенил)метил]-2-оксопирролидин-3-карбоксамида (соединение 12). Затем, на ключевой стадии, соединение 12 энантиоселективно, с помощью асимметричного окисляющего агента «оксазиридина Дэвиса», окисляют с получением (3S)-1-[1-(бензолсульфонил)-1Н-индол-5-ил]-N-[(3,5-дифторфенил)метил]-3-гидрокси-2-оксопирролидин-3-карбоксамида (соединение 13). После этого фенилсульфонильную группу соединения 13 отщепляют с получением 3,5-дифтор-бензиламида (S)-3-гидрокси-1-(1Н-индол-5-ил)-2-оксо-пирролидин-3-карбоновой кислоты. Техническим результатом изобретения является обеспечение способа получения целевого продукта с меньшим количеством стадий синтеза. 7 з.п. ф-лы, 12 пр.
1. Способ получения 3,5-дифтор-бензиламида (S)-3-гидрокси-1-(1Н-индол-5-ил)-2-оксо-пирролидин-3-карбоновой кислоты ("S-9"), который отличается тем, что:
a) 2-оксо-1-(1-(фенилсульфонил)-1Н-индол-5-ил)пирролидин-3-карбоновую кислоту ("5") вводят в реакцию с 3,5-дифторбензиламином с получением 1-[1-(бензолсульфонил)-1Н-индол-5-ил]-N-[(3,5-дифторфенил)метил]-2-оксопирролидин-3-карбоксамида ("12"),
b) затем "12" энантиоселективно окисляют с получением (3S)-1-[1-(бензолсульфонил)-1Н-индол-5-ил]-N-[(3,5-дифторфенил)метил]-3-гидрокси-2-оксопирролидин-3-карбоксамида ("13"),
c) и потом фенилсульфонильную группу отщепляют от "13" с получением 3,5-дифтор-бензиламида (S)-3-гидрокси-1-(1Н-индол-5-ил)-2-оксо-пирролидин-3-карбоновой кислоты ("S-9").
2. Способ по п. 1, где стадию а) осуществляют в присутствии основания, выбранного из триэтиламина, DBU или диизопропилэтиламина.
3. Способ по п. 1 или 2, где стадию а) осуществляют в дихлорметане.
4. Способ по пп. 1, 2 или 3, где стадию а) осуществляют в присутствии ангидрида пропанфосфоновой кислоты.
5. Способ по п. 1, где стадию b) осуществляют в присутствии окисляющего реагента (+)-(2R,4aS,7S,8aR)-4H-4a,7-метанооксазирино[3,2-i][2,1]бензизотиазола, 8,8-дихлортетрагидро-9,9-диметил-3,3-диоксида.
6. Способ по п. 1 или 5, где стадию b) осуществляют в присутствии ТГФ или диэтилового эфира.
7. Способ по пп. 1, 5 или 6, где стадию b) осуществляют в присутствии NaHMDS.
8. Способ по п. 1, где стадию с) осуществляют в присутствии NaOH.
| RU 2017107089 A3, 21.01.2019 | |||
| WO 2013149704 A1, 10.10.2013 | |||
| WO 2012048775 A1, 19.04.2012. |

