Изобретение относится к экспериментальной медицине, фармакологии и клинической фармакологии и предназначено для изучения принадлежности лекарственных препаратов к субстратам эффлюксного белка-транспортера гликопротеина-Р (P-gp, АВСВ1 белок), а также использования в качестве контроля ингибирующей активности P-gp при установлении веществ аналогичного типа действия. Для этого моделируют в эксперименте состояние ингибирования функциональной активности АВСВ1 белка. В качестве препарата-ингибитора используют блокатор дипептидилпептидазы 4 (DPP-4), предпочтительно вилдаглиптин или его фармацевтически приемлемые соли, а в качестве маркерного субстрата P-gp - фексофенадин.
В последнее время все большее значение в фармакокинетике лекарственных веществ придается лекарственным транспортерам, так как для многих лекарственных препаратов существует вероятность фармакокинетических, лекарственно-опосредованных взаимодействий. В ряде случаев они клинически значимы и возникает необходимость коррекции доз и даже введение запрета на совместное использование лекарственных препаратов в практике. Лекарственно-опосредованные взаимодействия возникают, когда два (или более) совместно вводимых лекарственных препарата взаимодействуют на этапах фармакокинетики, что приводит к увеличению или снижению системных эффектов одного или более препаратов (объектов взаимодействия). Взаимодействия считаются клинически значимыми, когда концентрация/эффекты препарата превышают безопасный уровень или происходит снижение концентрации/эффектов препарата до субтерапевтического уровня.
Большинство лекарственно-опосредованных взаимодействий связаны с изменениями со стороны ферментных систем, но все больше признается участие в их реализации белков-транспортеров. Установлено, что транспортные белки оказывают влияние на абсорбцию лекарственных препаратов при их пероральном введении (дигоксин, сульфасалазин, фексофенадин), пресистемный метаболизм (статины), распределение в тканях (метотрексат), экскрецию с желчью и мочой (дигоксин, метформин, пенициллины, противовирусные препараты). Многие лекарственные препараты способны модулировать функциональную активность и/или уровень экспрессии транспортеров, что приводит к клинически значимым лекарственным взаимодействиям. Таким образом, есть два различных аспекта рисков лекарственно-опосредованных взаимодействий, которые необходимо учитывать, в том числе и при разработке новых потенциальных лекарственных препаратов. Во-первых будет ли иметь место конкуренция за белок-транспортер между совместно используемыми лекарственными препаратами/потенциальным лекарственным препаратом и совместно используемыми с ним лекарственными препаратами. Во-вторых, не оказывает ли влияние лекарственный препарат/потенциальный лекарственный препарат на фармакокинетику используемых совместно с ним лекарственных средств. Рассмотрение каждого из аспектов необходимо для мотивированной и комплексной оценки рисков нежелательных лекарственных реакций в клинической практике (Ayrton A. et al., 2008). Потенциал лекарственных взаимодействий, как правило, оценивается с помощью исследований in vitro с последующим исследованиями in vivo (European Medicines Agency ((EMEA) Европейское агентство лекарственных средств), Guideline on the Investigation of Drug Interactions, 2012)
Наиболее клинически значимым переносчиком лекарственных веществ является - гликопротеин-Р (P-gp, АВСВ1 белок, MDR1), что определяется его широкой субстратной специфичностью и локализацией в организме. Гликопротеин-Р (P-gp) осуществляет транспортировку липофильных соединений против градиента концентрации за счет гидролиза АТФ (Hennessy M. et al., 2007).
Наиболее известна изоформа, кодируемая генном MDR1, которая связана с фенотипом множественной лекарственной устойчивости (MDR/МЛУ) (Hennessy M. et al., 2007). Однако P-gp имеет большое клиническое значение не только в противоопухолевой терапии. АВСВ1 белок участвует в процессах всасывания, распределения и выделения широкого спектра лекарственных веществ, являющихся его субстратами (Zhou S.F., 2008). P-gp обнаружен в тонком и толстом кишечнике, в печени (Thiebaut F et al., 1987), в почках (Schinkel А.Н. et al., 2003; Tramonti G. et al., 2006), в плаценте (Cordon-Cardo С.et al., 1990), в гематоэнцефалическом барьере (Zhou S.F., 2008). P-gp осуществляет выделение ряда физиологических субстратов (стероидные гормоны) (Ueda K. et al., 1992), а также ксенобиотиков, в желудочно-кишечный тракт, желчь и мочу. P-gp транспортирует разнообразные по структуре соединения от небольших молекул, таких как органические катионы, углеводы и аминокислоты, до макромолекул, таких как белки и полисахариды (Zhou S.F., 2008), 50% существующих препаратов являются его субстратами или ингибиторами (Food and Drug Administration ((FDA) Управление по надзору за качеством пищевых продуктов и медикаментов), Guidance for Industry Drug Interaction Studies - Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations, 2012; John P. et al., 2006).
Признанием важности данного белка-транспортера является разработка FDA и ЕМЕА рекомендаций по выявлению отношения потенциальных лекарственных субстанций к гликопротеину-Р, для фармацевтических компаний, регистрирующих новые препараты.
Однако высока вероятность совпадений субстратной специфичности и свойств ингибиторов и индукторов P-gp и CYP3A4. Последний является одной из наиболее важных изоформ цитохрома Р450, участвующей в метаболизме ксенобиотиков в организме человека, доля которой среди всех CYP450 составляет около 50% (Кукес В.Г. и соавт. 2013). Более 60% применяемых в настоящее время лекарственных препаратов метаболизируются при участии CYP3A4 (Li А.Р. et al., 1995).
Примером перекрестной чувствительности может быть влияние итраконазола, ингибирующего CYP3A и P-gp, рифампицина, индуцирующего CYP3A и P-gp. Тем не менее, ингибирующий потенциал по отношению к CYP3A и P-gp не обязательно одинаково выражен (табл. №1).
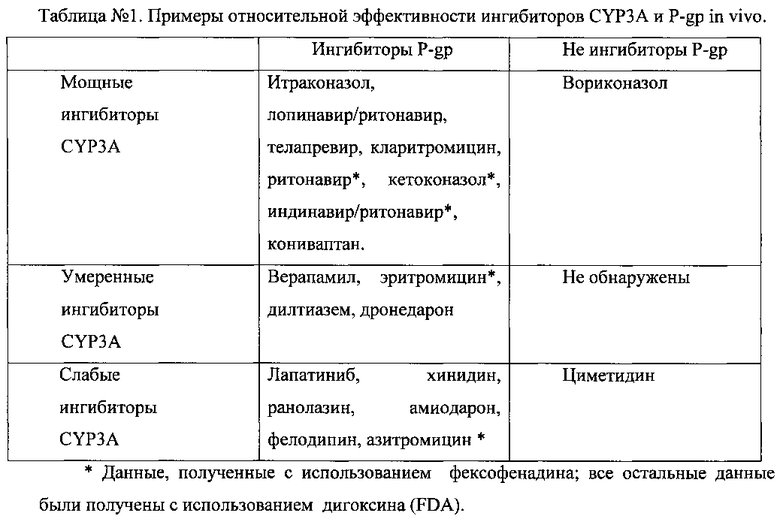
Например, сильный ингибитор CYP3A-вориконазол не вызывает значительных изменений транспорта субстратов P-gp, таких как дигоксин или фексофенадин. Кроме того, некоторые мощные ингибиторы P-gp, такие как амиодарон и хинидин (изменяющие AUC дигоксина или фексофенадина ≥1,5 раза), являются слабыми ингибиторами CYP3A (FDA, Guidance for Industry Drug Interaction Studies - Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations, 2012).
Согласно рекомендациям FDA и ЕМЕА для установления in vivo принадлежности лекарственного препарата к субстратам P-gp, необходимо использовать мощный селективный ингибитор данного белка-транспортера (FDA, Guidance for Industry Drug Interaction Studies - Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations, 2012; EMEA Guideline on the Investigation of Drug Interactions, 2012). Однако, до сих пор не удалось найти клинически подходящий, селективный ингибитор гликопротеина-Р, который не являлся бы также ингибитором CYP3A4 (Keogh J.P., 2012). Кроме того, очевидно, что в случае использования ингибитора in vivo для целей клинической фармакологии и персонализированной медицины, в отношении него должны предъявляться такие требования, как безопасность и минимальное влияние на гемодинамику, чтобы исключить изменения фармакокинетических показателей, не опосредованных изменением функциональной активности и/или экспрессией гликопротеина-Р.
В связи с вышеизложенным перспективными препаратами, которые могут быть использованы с данной целью являются глиптины, предпочтительно вилдаглиптин. Глиптины относятся к новому классу оральных гипогликемических препаратов, используемых для фармакотерапии сахарного диабета 2-го типа, и представляют собой селективные и обратимые ингибиторы дипептидилпептидазы 4 (DPP-4), фермента, который инактивирует инкретиновые гормоны: глюкагон-подобный пептид-1 (ГПП-1(GLP-1), а также глюкозоинсулинотропный полипептид (ГИП(GIP)), которые вносят существенный вклад в поддержание гомеостаза глюкозы (ЕМЕА, 2007). Принципиально важной особенностью влияния инкретинов на функцию панкреатических α- и β-клеток является его глюкозозависимый характер. Это означает, что инкретины стимулируют секрецию инсулина и, напротив, подавляет продукцию глюкагона лишь в условиях гипергликемии. Как только уровень глюкозы плазмы снижается до нормального уровня, вышеуказанные эффекты инкретинов нивелируются, что является надежным физиологическим механизмом, предотвращающим развитие гипогликемических состояний (Hoist J. et al., 2008). Согласно имеющимся данным на фоне монотерапии вилдаглиптином (Галвус 50 мг; производитель Novartis Pharma AG, Швейцария) в дозе 50 мг 1 или 2 раза в сутки частота развития гипогликемии без увеличения степени тяжести состояния составляет 0,5% (2 человека из 409) или 0,3% (4 из 1082), что сопоставимо с препаратами сравнения и плацебо (0,2%). При применении вилдаглиптина (Галвуса) в виде монотерапии не отмечалось повышения массы тела пациентов (Novartis Pharma AG). Вилдаглиптин имеет высокую степень безопасности, хорошо переносится, обладает глюкозозависимым действием и лишен серьезных побочных эффектов (Wilhauer Ε., 2010).
Отсутствуют существенные различия в фармакокинетических параметрах вилдаглиптина на этапе абсорбции между животными различных видов (крысой, кроликом, собакой) и человеком (ЕМЕА, 2007). Биотрансформация является основным путем инактивации вилдаглиптина. Около 60% от введенной дозы вилдаглиптина подвергается метаболической трансформации за счет гидролиза. Окислению подвергается лишь 1,6% препарата. На долю конъюгации с глюкуроновой кислотой приходится 4,4%. Изоферментные системы CYP450 участвуют в метаболизме вилдаглиптина лишь в незначительной степени. In vitro определялся низкий потенциал взаимодействия с изоферментами CYP450. И согласно инструкции по применению препарата вилдаглиптин не ингибирует и не индуцирует ферментные системы цитохрома Р450, при одновременном применении не влияет на скорость метаболизма препаратов, являющихся субстратами ферментов: CYP1A2, 2С8, 2С9, 2С19, 2D6, 2Е1, 3А4/5 (ЕМЕА, 2007; Novartis).
Целью изобретения являлось создание такой модели ингибирования функциональной активности P-gp, которая проявляла бы селективный эффект в отношении P-gp, не влияя на CYP3A4, и не сопровождалась возникновением клинически значимых побочных эффектов, а при изучении на животных была бы методически обоснована согласно международным требованиям по изучению субстратов P-gp.
Поставленная задача достигается тем, что в качестве ингибитора P-gp выбран блокатор ДПП-4 вилдаглиптин, безопасный и экономически доступный препарат.
Описание способа
В качестве экспериментальной модели использовали кроликов, которые являются адекватной трансляционной моделью для изучения гликопротеина-Р (Колхир П.В., 2007). Эксперимент выполнен на 21 половозрелом кролике-самце породы Шиншилла, средней массой 3500-4500 г. Вилдаглиптин вводили животным в течение 14 дней внутрижелудочно в дозе 5 мг/кг массы тела. Функциональную активность P-gp определяли по анализу динамики плазменной концентрации фексофенадина, маркерного субстрата белка-транспортера. Фексофенадин был выбран в качестве специфического субстрата P-gp, с низкой биодоступностью при пероральном введении, более чувствительного к снижению функциональной активности и/или экспрессии P-gp в кишечнике, чем пероральный дигоксин (ЕМЕА, Guideline on the Investigation of Drug Interactions. 27 стр). Фексофенадин (Препарат Телфаст 180 мг; производитель: Aventis Pharma, Италия) вводился однократно внутрижелудочно через зонд в дозе 67,5 мг/кг массы тела животного до и после 14-дневного введения вилдаглиптина. Пробы крови отбирали в объеме 3-5 мл из краевой вены уха кролика в гепаринизированные пробирки через 1, 2, 3, 4, 5, 6, 8, 12 и 24 часа после однократного внутрижелудочного введения фексофенадина, центрифугировали 10 минут при 3000 об/мин, плазму хранили при -28°С до анализа (Колхир С.В, 2007).
Содержание фексофенадина в плазме крови определяли методом ВЭЖХ на хроматографе «Стайер» (Россия) с ультрафиолетовым детектором и обращенно-фазовой колонке «Beckman Coulter» 4,6·250 мм, зернением 5 мкм. Экстракцию и хроматографирование маркерного субстрата осуществляли по методу Раменской Г.В. с соавт. в собственной модификации. Анализ выполняли при длине волны 220 нм и скорости подвижной фазы 1 мл/мин.
Элюирование выполняли подвижной фазой следующего состава (на 200 мл): 133,7 мл бидистиллированной воды, содержащей 2,33 мл ледяной уксусной кислоты и 0,936 мл триэтиламипа, доведенной триэтиламином до рН 4,3 и 64 мл ацетонитрила. Время удерживания пика фексофенадина составило 12,31 мин.
В качестве экстрагентов для жидкостной экстракции фексофенадина использовали дихлорметан, этилацетат и диэтиловый эфир. Коэффициент экстракции фексофенадина из плазмы крови составил 64%.
Полученные экспериментальные данные были подвергнуты математико-статистической обработке с использованием офисного пакета «Microsoft Office ХР» и программ Statistica 8.0. и IBM SPSS Satistics 20. Характер распределения данных оценивали по критерию Шапиро-Уилка. Для исследования статистической значимости показателей, имеющих нормальное распределение, использовали тест ANOVA повторных измерений. Для оценки статистической значимости показателей, распределение которых отличалось от нормального, использовали критерий Фридмана. Наличие достоверных различий определяли по параметрическому и не параметриескому критерию Ньюмена-Кейлса, соответственно. Для данных, имеющих нормальное распределение, рассчитывали среднее арифметическое значение (Mean) и стандартное отклонение (SD). Для данных, имеющих распределение, отличное от нормального, рассчитывали медиану (Median), верхний и нижний квартили (lq; uq).
Фармакокинетические параметры фексофенадина рассчитывали при помощи программы «Kinetica 5.0». Полученные данные представлены в табл. №2.

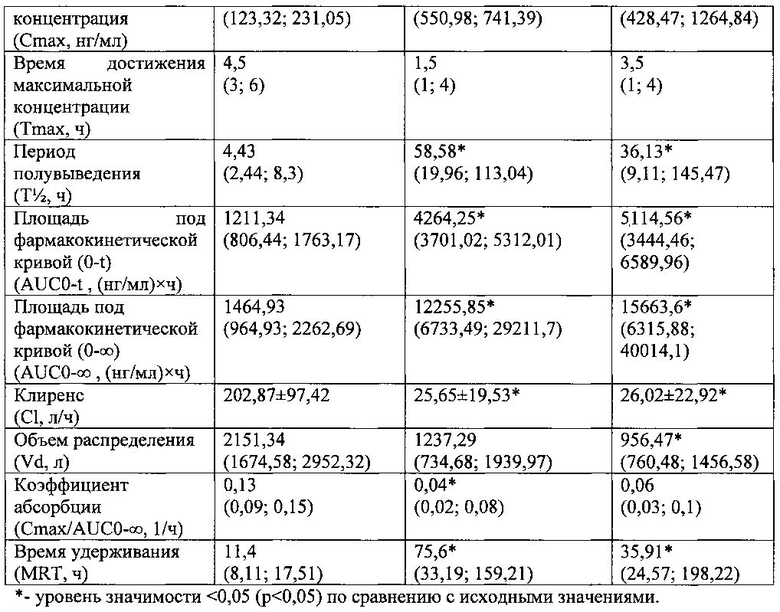
При введении вилдаглиптина в дозе 5 мг/кг массы курсом 14 дней по сравнению с исходными значениями выявлены следующие изменения фармакокинетики маркерного субстрата P-gp - фексофенадина: достоверное увеличение медиан значений Cmax после 14 дней введения на 204,5% (р<0,05) и на 5-й день отмены на 239,58% (р<0,05), медиан значений Т½ после 14 дней введения на 1222,35% (р<0,05) и на 5-й день отмены на 715,57% (р<0,05), медиан значений AUCO-t после 14 дней введения на 252,03% (р<0,05) и на 5-й день отмены на 322,22% (р<0,05), медиан значений AUCO-∞ после 14 дней введения на 736,62% (р<0,05) и на 5-й день отмены на 969,24% (р<0,05), медиан значений MRT после 14 дней введения на 563,16% (р<0,05) и на 5-й день отмены на 215% (р<0,05), снижение средних значений С1 после 14 дней введения на 87,36% (р<0,05) и на 5-й день отмены на 87,17% (р<0,05), медиан значений Vd после 14 дней введения на 42,49% (р<0,05) и на 5-й день отмены на 55,54% (р<0,05), медиан значений Смах/AUCO-∞ после 14 дней введения на 69,23% (р<0,05) и на 5-й день отмены на 53,85% (р<0,05).
Указанные изменения свидетельствуют об увеличении концентрации фексофенадина в крови за счет увеличения абсорбции и замедления выведения маркерного субстрата. В соответствии с рекомендациями FDA ингибитором P-gp признаются вещества, увеличивающие AUC фексофенадина более чем на 25%, что может служить доказательством ингибирующего влияния вилдаглиптина на функциональную активность P-gp.
Поскольку глюкоза и инсулин способны регулировать активность гликопротеина-Р (Yeh S.Y. et al., 2012), у интактных животных после 14 дней введения вилдаглиптина и на 5-й день его отмены изучали уровни инсулина натощак и на 45 минуту после глюкозной нагрузки (3 г/кг), а также уровень глюкозы до и через 90 минут после глюкозной нагрузки. Рассчитывали гликемический и инсулиногенный индексы (табл. №3) (указанные сроки были выбраны в связи с тем, что в данные промежутки времени наблюдаются максимальные отличия от нормы уровня инсулина, глюкозы и инсулиногенного индекса при введении вилдаглиптина) (Burkey B.F. et al., 2005; Руководство по проведению доклинических исследований лекарственных средств, 2012). Уровень инсулина определяли радиоиммунным методом, концентрацию глюкозы - глюкозоксидазным методом в центральной научно-исследовательской лаборатории РязГМУ.
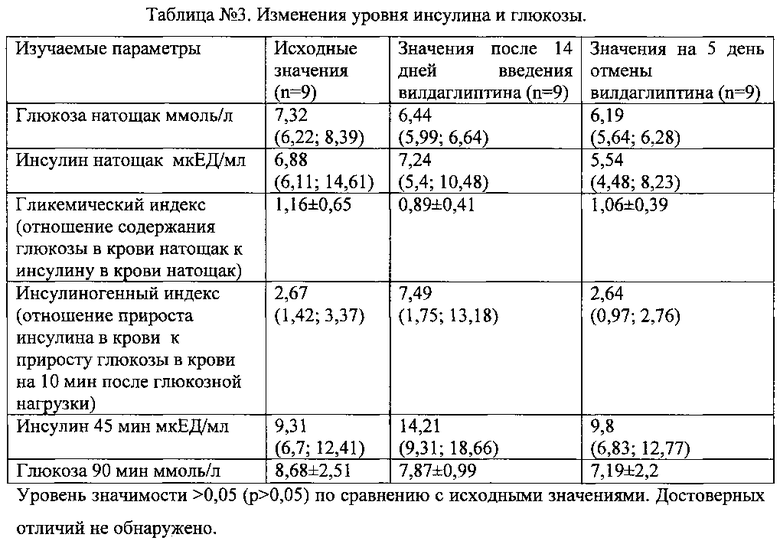
Изученные показатели представлены в табл. №3. Достоверных различий в уровнях глюкозы и инсулина натощак, показателях гликемического и инсулиногенного индекса до и после 14 дней введения вилдаглиптина, а также на 5-й день его отмены не обнаружено. Таким образом изменения функциональной активности P-gp не могут быть связаны с уровнем глюкозы и/или инсулина.
Использование предлагаемого способа моделирования состояния ингибирования функциональной активности P-gp позволяет применять вилдаглиптин в качестве положительного контроля пониженной функциональной активности белка-транспортера при поиске веществ аналогичного действия, а также для прогнозирования потенциальных субстратов P-gp среди лекарственных и/или потенциальных лекарственных веществ на этапе доклинических исследований.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ МОДЕЛИРОВАНИЯ СОСТОЯНИЯ ИНДУКЦИИ ФУНКЦИОНАЛЬНОЙ АКТИВНОСТИ ГЛИКОПРОТЕИНА-Р ФИНАСТЕРИДОМ В ЭКСПЕРИМЕНТЕ | 2012 |
|
RU2504018C1 |
| СПОСОБ МОДЕЛИРОВАНИЯ СОСТОЯНИЯ ИНГИБИРОВАНИЯ ФУНКЦИОНАЛЬНОЙ АКТИВНОСТИ ГЛИКОПРОТЕИНА-Р ЛИНЕСТРЕНОЛОМ В ЭКСПЕРИМЕНТЕ | 2014 |
|
RU2553362C1 |
| СРЕДСТВО ДЛЯ СНИЖЕНИЯ ФУНКЦИОНАЛЬНОЙ АКТИВНОСТИ И ЭКСПРЕССИИ ГЛИКОПРОТЕИНА-Р | 2017 |
|
RU2649134C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ФУНКЦИОНАЛЬНОЙ АКТИВНОСТИ ГЛИКОПРОТЕИНА-Р | 2015 |
|
RU2587780C1 |
| СПОСОБ ИНГИБИРОВАНИЯ АВСВ1-БЕЛКА В ЭКСПЕРИМЕНТЕ IN VITRO | 2023 |
|
RU2811993C1 |
| СПОСОБ ОЦЕНКИ ФУНКЦИОНАЛЬНОЙ АКТИВНОСТИ ГЛИКОПРОТЕИНА-P В ГЕМАТОЭНЦЕФАЛИЧЕСКОМ БАРЬЕРЕ | 2018 |
|
RU2677286C1 |
| ГИДРОЛИЗАТ ПОЛИСАХАРИДНОГО КОМПЛЕКСА ЦВЕТКОВ ПИЖМЫ ОБЫКНОВЕННОЙ КАК ИНГИБИТОР БЕЛКА-ТРАНСПОРТЕРА ГЛИКОПРОТЕИНА-Р | 2019 |
|
RU2699042C1 |
| СПОСОБ ПОВЫШЕНИЯ БЕЗОПАСНОСТИ ПРИМЕНЕНИЯ ЛЕКАРСТВЕННЫХ СРЕДСТВ ПРИ ЛЕЧЕНИИ ПАЦИЕНТОВ С СИНДРОМОМ ОТМЕНЫ АЛКОГОЛЯ | 2018 |
|
RU2690522C1 |
| СПОСОБ ИНГИБИРОВАНИЯ ГЛИКОПРОТЕИНА-Р В ЭКСПЕРИМЕНТЕ IN VITRO | 2021 |
|
RU2779177C1 |
| Применение 2-метил-N-(2-метилфенил)-4-(2-фурил)-5-циано-6-({ 2-[(4-этоксифенил)амино]-2-оксоэтил} тио)-1,4-дигидропиридин-3-карбоксамида в качестве гипогликемического, гиполипидемического средства, способствующего снижению массы тела | 2023 |
|
RU2812569C1 |
Изобретение относится к экспериментальной медицине, фармакологии и предназначено для изучения принадлежности лекарственных препаратов к субстратам эффлюксного белка-транспортера гликопротеина-Р (P-gp, АВСВ1 белок), а также использования в качестве контроля ингибирующей активности P-gp при установлении веществ аналогичного типа действия. Для этого моделируют в эксперименте состояние ингибирования функциональной активности P-gp. В качестве препарата-ингибитора используют ингибитор дипептидилпептидазы-4 вилдаглиптин, который вводят кролику внутрижелудочно в дозе 5 мг/кг массы тела в течение 14 дней. При этом в качестве маркерного субстрата P-gp используют фексофенадин, который вводят животному внутрижелудочно в дозе 67,5 мг/кг до и после 14-дневного введения вилдаглиптина с последующей оценкой содержания фексофенадина в плазме крови. Способ обеспечивает создание такой модели, при которой проявляется селективный эффект в отношении P-gp в отсутствии влияния на CYP3A4 без возникновения клинически значимых побочных эффектов. 3 табл.
Способ моделирования состояния ингибирования функциональной активности эффлюксного белка-транспортера гликопротеина-P в эксперименте, включающий введение препарата-ингибитора, отличающийся тем, что в качестве такого препарата используют ингибитор дипептидилпептидазы-4 вилдаглиптин, который вводят кролику внутрижелудочно в дозе 5 мг/кг массы тела в течение 14 дней и в качестве маркерного субстрата гликопротеина-P используют фексофенадин, который вводят животному внутрижелудочно в дозе 67,5 мг/кг до и после 14-дневного введения вилдаглиптина с последующей оценкой содержания фексофенадина в плазме крови.
| СПОСОБ МОДЕЛИРОВАНИЯ СОСТОЯНИЯ ИНДУКЦИИ ФУНКЦИОНАЛЬНОЙ АКТИВНОСТИ ГЛИКОПРОТЕИНА-Р ФИНАСТЕРИДОМ В ЭКСПЕРИМЕНТЕ | 2012 |
|
RU2504018C1 |
| СОСТАВ С МОДИФИЦИРОВАННЫМ ВЫСВОБОЖДЕНИЕМ, СОДЕРЖАЩИЙ 1-[(3-ГИДРОКСИАДАМАНТ-1-ИЛАМИНО)АЦЕТИЛ]ПИРРОЛИДИН-2(S)-КАРБОНИТРИЛ | 2006 |
|
RU2423124C2 |
| ЯКУШЕВА Е | |||
| Н | |||
| и др | |||
| Дозозависимое влияние тироксина на функциональную активность гликопротеина-Р в эксперименте Биомедицина, 2012, Выпуск N 2, том 1 http://cyberleninka.ru/article/n/dozozavisimoe-vliyanie-tiroksina-na-funktsionalnuyu-aktivnost-glikoproteina-r-v-eksperimente | |||
| ISHIGURO N et al. | |||

