ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новому производному транс-2-деценовой кислоты или его фармацевтически приемлемой соли, и фармацевтическому средству, содержащему соединение (в настоящем изобретении, в случае, когда оно просто указано как “соединение”, оно может включать в себя его фармацевтически приемлемую соль) в качестве активного ингредиента. Более конкретно, настоящее изобретение относится к производному транс-2-деценовой кислоты или его фармацевтически приемлемой соли, обладающему профилактическими или терапевтическими действиями для расстройств периферической нервной системы, вызванных введением противораковых средств, нейротрофической фактор-подобной активностью, такой как фактор роста нервов (NGF) или нейротрофический фактор головного мозга (BDNF), и фармакологическим действием, таким как обезболивающее действие, и к фармацевтическому средству, содержащему соединение в качестве активного ингредиента.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
На сегодняшний день, при лечении рака (злокачественной опухоли) используют хирургическое вмешательство, облучение или химиотерапию по отдельности или в любой их комбинации в соответствии с требованиями. Из таких средств, противораковые средства (средства против злокачественных опухолей), используемые в химиотерапии, по своей природе являются цитотоксичными и повреждают не только раковые клетки, но также и нормальные клетки человека, вызывая побочные эффекты. Таким образом, важно, чтобы противораковые средства вводили пациентам, обеспечивая при этом предотвращение или лечение таких побочных эффектов насколько это возможно, с обеспечением достаточных противораковых эффектов.
Примеры побочных эффектов, вызванных введением противораковых средств, включают различные заболевания крови, желудочно-кишечные расстройства, нервные расстройства и т.д. и, в частности, в качестве последней тенденции увеличилось количество острых или хронических нервных расстройств. Эта тенденция, как полагают, обусловлена следующими факторами: частое возникновение нервных расстройств в качестве основного побочного эффекта новых противораковых средств, обеспечивающих замечательные противораковые эффекты, эффекты полилекарственной терапии как основной используемой современной терапии, и относительно улучшенная тенденция развития побочных эффектов, таких как заболевания крови и желудочно-кишечные расстройства. Таким образом, не существует никаких эффективных контрмер против нервных расстройств, которые являются основным побочным эффектом используемой в настоящее время противораковой химиотерапии, когда уже произошло развитие расстройств, из-за трудности регенерации нервных клеток. Таким образом, серьезные симптомы или необратимые нарушения могут развиваться таким образом, что введение противораковых средств не может быть продолжено в некоторых случаях. Соответственно, нервные расстройства являются важной терапевтической проблемой для рака.
Нервные расстройства, вызванные введением противораковых средств, наблюдаются, помимо центральной нервной системы, в вегетативной нервной системе и в периферической нервной системе, органах чувств, таких как чувство вкуса. Среди них, нервные расстройства в периферической нервной системе, которые возникают со сравнительно высокой частотой и поэтому являются проблемами, представляют собой боли, такие как чувство жжения и жгучая боль, парестезию, такую как онемение конечностей и жжение, гиперестезию, такую как повышенная чувствительность к холодным стимулам, дизестезию, такую как потеря чувствительности, сенсорный паралич и чувство дискомфорта, сенсорная атаксия, мышечная слабость, или подобные. В настоящем изобретении противораковое средство, индуцирующее периферическое нервное расстройство, может относиться к любому типу таких средств, и считают, что поражения в периферической нервной системе, вызванные введением противораковых средств, в основном, являются результатом нейроаксональной дегенерации. Микротрубочки в аксоне играют важную роль в поддержании нормальной функции клеток, например, образуя веретенообразную структуру в процессе клеточного деления, и участвуя в размещении субклеточных органелл и транспортировки веществ. Таксановое лекарственное средство, такое как паклитаксел и доцетаксел, и лекарственное средство, содержащее алкалоиды барвинка, такое как винкристин, винбластин, виндезин и винорелбин, нацелены на микротрубочки для ингибирования пролиферации раковых клеток. Таким образом, считается, что микротрубочки в нормальных нервных клетках также более вероятно являются поврежденными, вызывая нервные расстройства. Кроме того, считается, что препараты платины, такие как оксалиплатин, карбоплатин, цисплатин и недаплатин, непосредственно повреждают нервные клетки и, следовательно, вторично приводят к аксонопатии, поэтому полагают, что они вызывают большинство нервных расстройств.
Несмотря на описанную выше ситуацию, нейротоксичность противораковых средств подробно не изучена, и надлежащие способы профилактики и терапевтического лечения расстройства периферической нервной системы, которое является побочным эффектом, вызванным таксановыми противораковыми лекарственными средствами и другими, описанными выше, до сих пор не установлены. Таким образом, в настоящее время, для облегчения симптомов онемения используют препараты витамина B12, такие как мекобаламин, и препарат китайской травяной медицины Gosha-jinki-gan. При болях используют антидепрессант (амитриптилингидрохлорид), противоэпилептическое средство (карбамазепин), средство от антиаритмии (мексилетингидрохлорид), адренокортикостероид и подобные. Однако эти терапии имеют ограниченную эффективность. Остановка введения противоракового средства является единственным надежным методом для предотвращения развития расстройства периферической нервной системы, но даже после прекращения введения расстройства периферической нервной системы могут продолжиться или ухудшиться. Требуется введение противораковых средств для лечения рака, но значительные расстройства периферической нервной системы могут затруднить продолжение введения важных противораковых средств, которые являются весьма эффективными против рака, и это становится серьезной проблемой для лечения рака. С учетом изложенных выше проблем, более эффективные профилактические или терапевтические средства против расстройства периферической нервной системы, вызываемого противораковыми средствами, крайне необходимы в клинической практике.
Кроме того, нервная клетка представляет собой клетку, обладающую функцией передачи информации, и ее повреждение проявляется как серьезная потеря функции черепно-мозгового нерва. Регенерацию аксона вряд ли можно ожидать в центральных нервах головного мозга и спинного мозга, и таким образом, когда обнаружено, что имеет место повреждение или дегенерация нервных клеток, необходимо защитить и активировать нервные клетки. В качестве указанного выше биологического защитного механизма, роли нейротрофических факторов в дифференциации нервных клеток, поддержании выживаемости, увеличении функций синапсов и регенерации или восстановлении поврежденного нервного аксона являются незаменимыми.
Среди нейротрофических факторов, фактор роста нервов (NGF), нейротрофический фактор головного мозга (BDNF), нейротрофин-3 (NT-3), нейротрофин-4/5 (NT-4/5) и подобные составляют нейротрофиновое семейство, имеющее 50% или более гомологии последовательности с NGF в качестве прототипа. Когда внеклеточно секретируемый нейротрофин связан с высокоаффинным рецептором (Trks) на мембране нервной клетки, сигналы передаются по трем направлениям в нервных клетках. Фактор транскрипции CREB (белок, связывающий с элементом сАМР-ответа) активируется посредством активации пути передачи информации МАР-киназы (митоген-активируемые протеинкиназы (МАП)/внеклеточный сигнал-регулируемые протеинкиназы 1/2 (ERK1/2)), включая активацию (фосфорилирование) МАР-киназы, которая является одним из высокоаффинных рецепторов, тем самым регулируя множество генных экспрессий. Поэтому, как только могут быть активированы передачи сигналов через путь передачи информации MAP киназы, появляется возможность клинических применений к нервному расстройству, вызванному дегенерацией нервных клеток или гибелью клеток. Кроме того, имеется сообщение о связи между BDNF и некоторыми заболеваниями.
Согласно исследованиям генных полиморфизмов BDNF, сообщалось, что конкретные полиморфизмы BDNF связаны с болезнью Паркинсона, болезнью Альцгеймера, депрессией, биполярной депрессией, тревожными расстройствами, расстройством аутистического спектра, глаукомой и подобными. Кроме того, есть несколько сообщений о том, что снижение функции синапсов у генетически преобразованной мыши с болезнью Гентингтона лечат введением BDNF, и что введение ингибитора фосфорилирования MAP киназы провоцирует антидепрессивное состояние.
Нейротрофические факторы, как видно в примерах BDNF, указанных выше, демонстрируют терапевтический эффект против конкретных нервных заболеваний и обладают некоторыми действиями, связанными с развитием аксона и увеличением его длины. Однако, поскольку нейротрофические факторы представляют собой белки с высокой молекулярной массой, они имеют проблемы из-за трудностей достижения головного мозга, поскольку они не могут проходить через гематоэнцефалический барьер, даже при введении из периферии. Таким образом, была сделана попытка поиска фармацевтического средства, обладающего нейротрофической фактор-подобной активностью, которое активирует нервные клетки при помощи низкомолекулярного соединения, и фармацевтического средства, стимулирующего продукцию и секрецию нейротрофических факторов.
Патентная публикация 1 раскрывает, что жирная кислота или сложный эфир жирной кислоты, содержащие 8 атомов углерода или от 10 до 12 атомов углерода, обладают нейротрофической фактор-подобной активностью. Однако соединения, описанные в Патентной публикации 1, которые представляют собой обычную жирную кислоту или ее сложный эфир, четко отличаются по структуре в группе карбоновой кислоты от соединений по настоящему изобретению. Кроме того, соединения по настоящему изобретению 1-((Е)-2-деценоил)пирролидин (Соединение 1), 1-((Е)-2-деценоил)пиперидин (Соединение 5) и 1-((Е)-2-деценоил)азепан (Соединение 32) раскрыты в Патентной публикации 2, и соединение по настоящему изобретению (4-((Е)-2-деценоил)морфолин (Соединение 17) раскрыто в Патентной публикации 3 или Непатентной публикации 1, соответственно. Однако Патентная публикация 2 относится к токсическому действию против моллюсков, и Патентная публикация 3 относится к ингибирующим рост действиям против болезнетворных микробов или плесени, и Непатентная публикация 1 представляет собой публикацию, относящуюся к репелленту против домашних мух. В заключение, ни одна из этих публикаций не включает фармацевтические применения соединений по настоящему изобретению и какие-либо описания, предлагающие применения.
ССЫЛКА НА ИЗВЕСТНЫЙ УРОВЕНЬ ТЕХНИКИ
ПАТЕНТНЫЕ ПУБЛИКАЦИИ
Патентная публикация 1: WO 2009/038110.
Патентная публикация 2: WO 2010/123894.
Патентная публикация 3: Патент США № 3294794.
НЕПАТЕНТНЫЕ ПУБЛИКАЦИИ
Непатентная публикация 1: Journal of Economic Entomology, 1970, 63(6), 1752-1755.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
ПРОБЛЕМЫ, РЕШАЕМЫЕ НАСТОЯЩИМ ИЗОБРЕТЕНИЕМ
Целью настоящего изобретения является обеспечение нового производного транс-2-деценовой кислоты или его фармацевтически приемлемой соли и фармацевтического средства, содержащего соединение в качестве активного ингредиента, и, более конкретно, обеспечение фармацевтического средства, обладающего профилактическим или терапевтическим действием против расстройства периферической нервной системы, индуцированного противораковым средством, нейротрофической фактор-подобной активностью или фармакологическим действием, таким как обезболивающее действие, где в настоящем изобретении “расстройства периферической нервной системы” можно заменить синонимом “невропатия”; кроме того, “предотвращение” и “лечение” включают “улучшение” или “облегчение”, и “профилактическое средство” и терапевтическое средство” включают “средство для улучшения” или “средство для облегчения (симптомов).”
СРЕДСТВА ДЛЯ РЕШЕНИЯ ПРОБЛЕМ
В результате интенсивных исследований для решения вышеуказанных проблем, авторы настоящего изобретения обнаружили, что производное транс-2-деценовой кислоты, представленное общей формулой (I), определенной ниже, или его фармацевтически приемлемая соль обладает превосходным фармакологическим действием, таким как действие, направленное на профилактику или лечение расстройств периферической нервной системы, индуцируемых противораковыми средствами, нейротрофическая фактор-подобная активность или анальгетическое действие. Авторы настоящего изобретения провели дальнейшие исследования на основе этих открытий, и, таким образом, было создано настоящее изобретение.
В частности, настоящее изобретение обеспечивает следующие соединения и фармацевтические средства, содержащие соединения.
(1) Производное транс-2-деценовой кислоты, представленное следующей общей формулой (I'), или его фармацевтически приемлемая соль:
 ,
,
где X′ представляет собой:
(a) 1-пирролидил, замещенный карбоксилом или алкоксикарбонилом,
(b) 3-тиазолидил,
(c) пиперидино, замещенный алкилом, оксо, гидрокси, алкокси, карбоксилом, алкоксикарбонилом, алкиламино, алкиламиноалкилом, фенилом, карбоксиалкилом, алкоксикарбонилалкилом, циано или галогенофенилом,
(d) тиоморфолино,
(e) 1-пиперазил, который может быть замещен алкилом, карбоксиалкилом, алкоксикарбонилалкилом, алкиламиноалкилом, циклоалкилом, пиперидиноалкилом, фенилалкилом, пиридилом, пиримидилом, карбоксифенилалкилом или алкоксикарбонилфенилалкилом,
(f) 1-пиперазил, замещенный фенилом, который может быть замещен алкиламино, галогеном, алкокси, алкилом, гидрокси, карбоксиалкокси или алкоксикарбонилалкокси,
(g) 1,4-диазепанил, который может быть замещен алкилом или алкиламиноалкилом, или
(h) карбоксиморфолино.
(2) Производное транс-2-деценовой кислоты или его фармацевтически приемлемая соль в соответствии с описанным выше пунктом (1), где X′ представляет собой 1-пирролидил, замещенный карбоксилом или алкоксикарбонилом.
(3) Производное транс-2-деценовой кислоты или его фармацевтически приемлемая соль в соответствии с описанным выше пунктом (1), где X′ представляет собой 3-тиазолидил.
(4) Производное транс-2-деценовой кислоты или его фармацевтически приемлемая соль в соответствии с описанным выше пунктом (1), где X′ представляет собой пиперидино, замещенный алкилом, оксо, гидрокси, алкокси, карбоксилом, алкоксикарбонилом, алкиламино, алкиламиноалкилом, фенилом, карбоксиалкилом, алкоксикарбонилалкилом, циано или галогенофенилом.
(5) Производное транс-2-деценовой кислоты или его фармацевтически приемлемая соль в соответствии с описанным выше пунктом (1), где X′ представляет собой тиоморфолино.
(6) Производное транс-2-деценовой кислоты или его фармацевтически приемлемая соль в соответствии с описанным выше пунктом (1), где X′ представляет собой 1-пиперазил, который может быть замещен алкилом, карбоксиалкилом, алкоксикарбонилалкилом, алкиламиноалкилом, циклоалкилом, пиперидиноалкилом, фенилалкилом, пиридилом, пиримидилом, карбоксифенилалкилом или алкоксикарбонилфенилалкилом.
(7) Производное транс-2-деценовой кислоты или его фармацевтически приемлемая соль в соответствии с описанным выше пунктом (1), где X′ представляет собой 1-пиперазил, замещенный фенилом, который может быть замещен алкиламино, галогеном, алкокси, алкилом, гидрокси, карбоксиалкокси или алкоксикарбонилалкокси.
(8) Производное транс-2-деценовой кислоты или его фармацевтически приемлемая соль в соответствии с описанным выше пунктом (1), где X′ представляет собой 1,4-диазепанил, который может быть замещен алкилом или алкиламиноалкилом.
(9) Производное транс-2-деценовой кислоты или его фармацевтически приемлемая соль в соответствии с описанным выше пунктом (1), где X′ представляет собой карбоксиморфолино.
(10) Фармацевтическое средство, содержащее в качестве активного ингредиента, по меньшей мере, один член, выбранный из производного транс-2-деценовой кислоты, представленного следующей общей формулой (I), и его фармацевтически приемлемой соли:
 ,
,
где X представляет собой:
(a) 1-пирролидил, который может быть замещен карбоксилом или алкоксикарбониломом,
(b) 3-тиазолидил,
(c) пиперидино, который может быть замещен алкилом, оксо, гидрокси, алкокси, карбоксилом, алкоксикарбонилом, алкиламино, алкиламиноалкилом, фенилом, карбоксиалкилом, алкоксикарбонилалкилом, циано или галогенофенилом,
(d) морфолино,
(e) тиоморфолино, который может быть замещен карбоксилом,
(f) 1-пиперазил, который может быть замещен алкилом, карбоксиалкилом, алкоксикарбонилалкилом, алкиламиноалкилом, циклоалкилом, пиперидиноалкилом, фенилалкилом, пиридилом, пиримидилом, карбоксифенилалкилом или алкоксикарбонилфенилалкилом,
(g) 1-пиперазил, замещенный фенилом, который замещен алкиламино, галогеном, алкокси, алкилом, гидрокси, карбоксиалкокси или алкоксикарбонилалкокси,
(h) 1-азепанил или
(i) 1,4-диазепанил, который может быть замещен алкилом или алкиламиноалкилом.
(11) Фармацевтическое средство в соответствии с описанным выше пунктом (10), где X представляет собой 1-пирролидил, который может быть замещен карбоксилом или алкоксикарбонилом.
(12) Фармацевтическое средство в соответствии с описанным выше пунктом (10), где X представляет собой 3-тиазолидил.
(13) Фармацевтическое средство в соответствии с описанным выше пунктом (10), где X представляет собой пиперидино, который может быть замещен алкилом, оксо, гидрокси, алкокси, карбоксилом, алкоксикарбонилом, алкиламино, алкиламиноалкилом, фенилом, карбоксиалкилом, алкоксикарбонилалкилом, циано или галогенофенилом.
(14) Фармацевтическое средство в соответствии с описанным выше пунктом (10), где X представляет собой морфолино, который может быть замещен карбоксилом.
(15) Фармацевтическое средство в соответствии с описанным выше пунктом (10), где X представляет собой тиоморфолино.
(16) Фармацевтическое средство в соответствии с описанным выше пунктом (10), где X представляет собой 1-пиперазил, который может быть замещен алкилом, карбоксиалкилом, алкоксикарбонилалкилом, алкиламиноалкилом, циклоалкилом, пиперидиноалкилом, фенилалкилом, пиридилом, пиримидилом, карбоксифенилалкилом или алкоксикарбонилфенилалкилом.
(17) Фармацевтическое средство в соответствии с описанным выше пунктом (10), где X представляет собой 1-пиперазил, замещенный фенилом, который может быть замещен алкиламино, галогеном, алкокси, алкилом, гидрокси, карбоксиалкокси или алкоксикарбонилалкокси.
(18) Фармацевтическое средство в соответствии с описанным выше пунктом (10), где X представляет собой 1-азепанил.
(19) Фармацевтическое средство в соответствии с описанным выше пунктом (10), где X представляет собой 1,4-диазепанил, который может быть замещен алкилом или алкиламиноалкилом.
(20) Фармацевтическое средство по любому из описанных выше пунктов (10)-(19), где фармацевтическое средство представляет собой профилактическое средство или терапевтическое средство для расстройств периферической нервной системы, вызванных введением лекарственного средства, содержащего, по меньшей мере, одно противораковое средство.
(21) Фармацевтическое средство в соответствии с описанным выше пунктом (20), где противораковое средство представляет собой ингибитор микротрубочек.
(22) Фармацевтическое средство в соответствии с описанным выше пунктом (21), где ингибитор микротрубочек представляет собой таксановое лекарственное средство.
(23) Фармацевтическое средство в соответствии с описанным выше пунктом (22), где таксановое лекарственное средство представляет собой паклитаксел или доцетаксел.
(24) Фармацевтическое средство в соответствии с описанным выше пунктом (23), где таксановое лекарственное средство представляет собой паклитаксел.
(25) Фармацевтическое средство в соответствии с описанным выше пунктом (21), где ингибитор микротрубочек представляет собой лекарственное средство на основе алкалоида барвинка.
(26) Фармацевтическое средство в соответствии с описанным выше пунктом (25), где лекарственное средство на основе алкалоида барвинка представляет собой винкристин.
(27) Фармацевтическое средство в соответствии с описанным выше пунктом (20), где противораковое средство представляет собой платиновое лекарственное средство.
(28) Фармацевтическое средство в соответствии с описанным выше пунктом (27), где платиновое лекарственное средство представляет собой оксалиплатин или цисплатин.
(29) Фармацевтическое средство по любому из описанных выше пунктов (20)-(28), где расстройства периферической нервной системы, индуцированные противораковым средством, представляют собой острую или хроническую боль, онемение, парестезию, гиперестезию или дизестезию.
(30) Фармацевтическое средство по любому из описанных выше пунктов (10)-(19), где фармацевтическое средство представляет собой средство, обладающее нейротрофической фактор-подобной активностью.
(31) Фармацевтическое средство в соответствии с описанным выше пунктом (30), где средство, обладающее нейротрофической фактор-подобной активностью, представляет собой профилактическое или терапевтическое средство для нейродегенеративных заболеваний или психических заболеваний.
(32) Фармацевтическое средство в соответствии с описанным выше пунктом (31), где средство, обладающее нейротрофической фактор-подобной активностью, представляет собой профилактическое или терапевтическое средство для нейродегенеративных заболеваний.
(33) Фармацевтическое средство в соответствии с описанным выше пунктом (32), где нейродегенеративное заболевание представляет собой деменцию, болезнь Альцгеймера, болезнь Паркинсона, амиотрофический боковой склероз (ALS), болезнь Гентингтона, прогрессирующий супрануклеарный паралич (PSP), диабетическую невропатию или глаукому.
(34) Фармацевтическое средство в соответствии с описанным выше пунктом (31), где средство, обладающее нейротрофической фактор-подобной активностью, представляет собой профилактическое или терапевтическое средство для психических заболеваний.
(35) Фармацевтическое средство в соответствии с описанным выше пунктом (34), где психические заболевания представляют собой депрессию, тревожные расстройства (неврозы) или расстройства аутистического спектра.
(36) Фармацевтическое средство по любому из описанных выше пунктов (10)-(19), где фармацевтическое средство представляет собой средство для лечения или восстановления повреждения спинного мозга.
(37) Фармацевтическое средство по любому из описанных выше пунктов (10)-(19), где фармацевтическое средство представляет собой анальгетик против болевых синдромов.
(38) Фармацевтическое средство в соответствии с описанным выше пунктом (37), где анальгетик против болевых синдромов представляет собой терапевтическое средство против боли в суставах.
(39) Фармацевтическое средство в соответствии с описанным выше пунктом (38), где боль в суставах представляет собой боль, вызванную остеоартритом.
(40) Фармацевтическое средство в соответствии с описанным выше пунктом (39), где остеоартрит представляет собой остеоартрит колена или остеоартрит бедра.
(41) Фармацевтическое средство в соответствии с любым из описанных выше пунктов (10)-(40), где фармацевтическое средство представляет собой препарат для инъекций.
(42) Фармацевтическое средство в соответствии с любым из описанных выше пунктов (10)-(40), где фармацевтическое средство представляет собой пероральный препарат.
(43) Фармацевтическое средство в соответствии с описанным выше пунктом (41) или (42), где препарат для инъекций или пероральный препарат представляет собой комплекс включения циклодекстрина.
(44) Фармацевтическое средство в соответствии с любым из описанных выше пунктов (10)-(40), где фармацевтическое средство представляет собой препарат для наружного применения.
(45) Фармацевтическое средство в соответствии с описанным выше пунктом (44), где препарат для наружного применения представляет собой пластырь.
(46) Соединение или его фармацевтически приемлемая соль в соответствии с любым из описанных выше пунктов (10)-(19) для применения в профилактике или лечении заболевания, как определено в любом из описанных выше пунктов (20)-(29) и (31)-(40).
(47) Соединение или его фармацевтически приемлемая соль в соответствии с любым из описанных выше пунктов (10)-(19) для применения в профилактике или лечении расстройств периферической нервной системы, вызванных введением лекарственного средства, содержащего, по меньшей мере, одно противораковое средство.
(48) Соединение или его фармацевтически приемлемая соль в соответствии с любым из описанных выше пунктов (10)-(19) для применения в профилактике или лечении нейродегенеративного заболевания или психического заболевания.
(49) Соединение или его фармацевтически приемлемая соль в соответствии с любым из описанных выше пунктов (10)-(19) для применения в профилактике или лечении болевых синдромов.
(50) Способ профилактики или лечения заболевания, определенного в любом из описанных выше пунктов (20)-(29) и (31)-(40), отличающийся тем, что такой способ включает введение соединения или его фармацевтически приемлемой соли в соответствии с любым из описанных выше пунктов (10)-(19) в эффективном количестве пациенту с заболеванием, определенным в любом из описанных выше пунктов (20)-(29) и (31)-(40).
(51) Способ профилактики или лечения расстройств периферической нервной системы, вызванных введением лекарственного средства, содержащего, по меньшей мере, одно противораковое средство, отличающийся тем, что такой способ включает введение соединения или его фармацевтически приемлемой соли в соответствии с любым из описанных выше пунктов (10)-(19) в эффективном количестве пациенту с расстройством периферической нервной системы, вызванным введением лекарственного средства, содержащего, по меньшей мере, одно противораковое средство.
(52) Способ профилактики или лечения нейродегенеративного заболевания или психического заболевания, отличающийся тем, что такой способ включает введение соединения или его фармацевтически приемлемой соли в соответствии с любым из описанных выше пунктов (10)-(19) в эффективном количестве пациенту с нейродегенеративным заболеванием или психическим заболеванием.
(53) Способ профилактики или лечения болевого синдрома, отличающийся тем, что такой способ включает введение соединения или его фармацевтически приемлемой соли в соответствии с любым из описанных выше пунктов (10)-(19) в эффективном количестве пациенту с болевым синдромом.
(54) Применение соединения или его фармацевтически приемлемой соли в соответствии с любым из описанных выше (10)-(19) для получения фармацевтического средства для лечения заболевания, определенного в любом из описанных выше пунктов (20)-(29) и (31)-(40).
(55) Применение соединения или его фармацевтически приемлемой соли в соответствии с любым из описанных выше (10)-(19) для получения фармацевтического средства для профилактики или лечения расстройств периферической нервной системы, вызванных введением лекарственного средства, содержащего, по меньшей мере, одно противораковое средство.
(56) Применение соединения или его фармацевтически приемлемой соли в соответствии с любым из описанных выше (10)-(19) для получения фармацевтического средства для профилактики или лечения нейродегенеративного заболевания или психического заболевания.
(57) Применение соединения или его фармацевтически приемлемой соли в соответствии с любым из описанных выше (10)-(19) для получения анальгетика.
ЭФФЕКТЫ ИЗОБРЕТЕНИЯ
Соединение по настоящему изобретению эффективно в качестве лекарственного средства для профилактики или лечения нервных расстройств периферической нервной системы, индуцированных противораковыми средствами, таких как парестезия, например, онемение конечностей человека и животных, и гипералгезия, например, боли.
Кроме того, соединение по настоящему изобретению обладает превосходной нейротрофической фактор-подобной активностью, таким образом, соединение можно использовать в качестве средства, обладающего нейротрофической фактор-подобной активностью. Это средство, обладающее нейротрофической фактор-подобной активностью, полезно в качестве профилактического средства или терапевтического средства от нервных расстройств, поскольку передачи сигналов активируются через путь передачи информации MAP киназы с нейротрофической фактор-подобной активностью. Среди нервных расстройств, средство особенно полезно в качестве профилактического средства или терапевтического средства для нейродегенеративного заболевания, такого как деменция, болезнь Альцгеймера, болезнь Паркинсона, амиотрофический боковой склероз (ALS), болезнь Гентингтона, прогрессирующий супрануклеарный паралич (PSP), диабетическая невропатия или глаукома. Кроме того, среди нервных расстройств, средство также полезно в качестве улучшающего состояние средства для профилактики психических заболеваний. Среди психических заболеваний, средство особенно полезно в качестве средства для профилактики или лечения депрессии, тревожных расстройств (неврозов) или расстройств аутистического спектра, и особенно полезно в качестве средства для профилактики или лечения депрессии и тревожных расстройств (неврозов).
Кроме того, соединение по настоящему изобретению полезно в качестве терапевтического средства при повреждении спинного мозга (в случае повреждения спинного мозга, его иногда называют как "восстанавливающее средство").
Далее, соединение по настоящему изобретению представляет собой соединение, демонстрирующее превосходное анальгетическое действие, которое полезно в качестве лекарственного средства для профилактики или лечения различных болевых синдромов, таких как боли, вызванные болями в суставах, такие как остеоартрит.
СПОСОБЫ ОСУЩЕСТВЛЕНИЯ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к производному транс-2-деценовой кислоты, представленному следующей общей формулой (I'), или его фармацевтически приемлемой соли:
 ,
,
где X′ представляет собой:
(a) 1-пирролидил, замещенный карбоксилом или алкоксикарбонилом,
(b) 3-тиазолидил,
(c) пиперидино, замещенный алкилом, оксо, гидрокси, алкокси, карбоксилом, алкоксикарбонилом, алкиламино, алкиламиноалкилом, фенилом, карбоксиалкилом, алкоксикарбонилалкилом, циано или галогенофенилом,
(d) тиоморфолино,
(e) 1-пиперазил, который может быть замещен алкилом, карбоксиалкилом, алкоксикарбонилалкилом, алкиламиноалкилом, циклоалкилом, пиперидиноалкилом, фенилалкилом, пиридилом, пиримидилом, карбоксифенилалкил или алкоксикарбонилфенилалкилом,
(f) 1-пиперазил, замещенный фенилом, который может быть замещен алкиламино, галогеном, алкокси, алкилом, гидрокси, карбоксиалкокси или алкоксикарбонилалкокси,
(g) 1,4-диазепанил, который может быть замещен алкилом или алкиламиноалкилом или
(h) карбоксилморфолино.
Кроме того, настоящее изобретение относится к фармацевтическому средству, такому как профилактическое средство или терапевтическое средство от расстройств периферической нервной системы, вызванных противораковым средством, средству, обладающему нейротрофической фактор-подобной активностью, или анальгетику, содержащему, в качестве активного ингредиента, по меньшей мере, один член, выбранный из производного транс-2-деценовой кислоты, представленного следующей общей формулой (I), и его фармацевтически приемлемой соли:
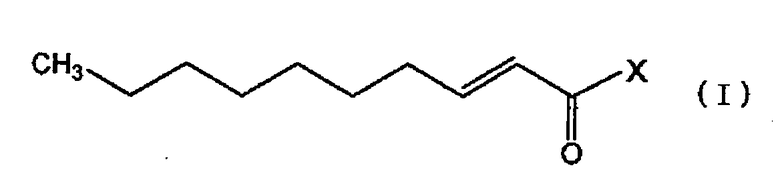 ,
,
где X представляет собой:
(a) 1-пирролидил, который может быть замещен карбоксилом или алкоксикарбонилом,
(b) 3-тиазолидил,
(c) пиперидино, который может быть замещен алкилом, оксо, гидрокси, алкокси, карбоксилом, алкоксикарбонилом, алкиламино, алкиламиноалкилом, фенилом, карбоксиалкилом, алкоксикарбонилалкилом, циано или галогенофенилом,
(d) морфолино, который может быть замещен карбоксилом,
(e) тиоморфолино,
(f) 1-пиперазил, который может быть замещен алкилом, карбоксиалкилом, алкоксикарбонилалкилом, алкиламиноалкилом, циклоалкилом, пиперидиноалкилом, фенилалкилом, пиридилом, пиримидилом, карбоксифенилалкилом или алкоксикарбонилфенилалкилом,
(g) 1-пиперазил, замещенный фенилом, который может быть замещен алкиламино, галогеном, алкокси, алкилом, гидрокси, карбоксиалкокси или алкоксикарбонилалкокси,
(h) 1-азепанил или
(i) 1,4-диазепанил, который может быть замещен алкилом или алкиламиноалкилом. Соединение, представленное общей формулой (I), охватывает соединение, представленное общей формулой (I′), указанной выше.
В заместителях в общих формулах (I) и (I′), указанных выше, алкил (включая “алкил” в алкиламино, алкиламиноалкиле, карбоксиалкиле, алкоксикарбонилалкиле, пиперидиноалкиле, фенилалкиле, карбоксифенилалкиле или алкоксикарбонилфенилалкиле) может быть любым, и алкил представляет собой линейные или разветвленные алкильные группы, содержащие от 1 до 4 атомов углерода, предпочтительно включающие метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил и т.д. Также, алкиламино (включая “алкиламино” в алкиламиноалкиле) относится к аминогруппе, замещенной 1 или 2 алкилами.
Алкокси (включая “алкокси” в алкоксикарбониле, карбоксиалкокси или алкоксикарбонилалкокси) может быть любым, и алкокси представляет собой линейную или разветвленную алкоксигруппу, содержащую от 1 до 4 атомов углерода, предпочтительно включающую метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси, трет-бутокси и т.д.
Циклоалкил может быть любым, и циклоалкил представляет собой циклоалкильную группу, содержащую от 3 до 8 атомов углерода, предпочтительно включающую циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил и т.д., и более предпочтительно, циклоалкильную группу, содержащую 5 или 6 атомов углерода.
Галоген (включая “галогено” в галогенофениле) представляет собой фтор, хлор, бром, йод и т.д.
Соединение, представленное общей формулой (I) (включая соединение, представленное формулой (I′); далее в описании обозначающие одно и то же) по настоящему изобретению может быть получено с использованием транс-2-деценовой кислоты в качестве исходного вещества способом, например, как показано на следующей схеме реакций:
Формула реакции
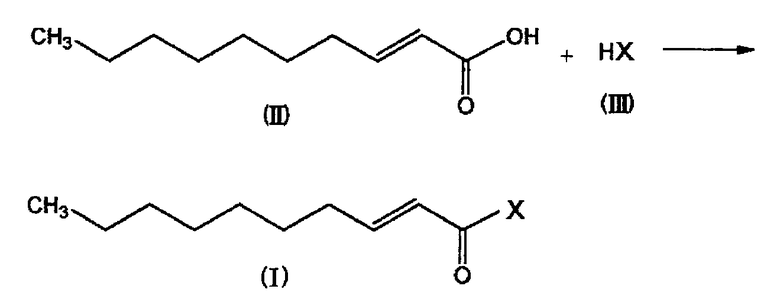 ,
,
где X имеет значение, определенное выше.
Соединение, представленное общей формулой (I) можно получить, подвергая соединение, представленное общей формулой (II), и соединение, представленное общей формулой (III), содержащее активный водород, дегидратационной конденсации. В качестве реакции дегидратационной конденсации может быть использован известный способ. Например, соединение, представленное общей формулой (II), и соединение, представленное общей формулой (III), можно подвергнуть взаимодействию в присутствии соответствующего агента конденсации, включающего, например, дициклогексилкарбодиимид (DCC) или N-(3-диметиламинопропил)-N′-этилкарбодиимид∙HCl. Реакцию обычно можно осуществлять в растворителе, включающем, например, дихлорметан. Количество соединения, представленного общей формулой (III), обычно составляет от 0,5 до 2 моль, и предпочтительно от 1 до 1,5 моль, на один моль соединения, представленного общей формулой (II).
Альтернативно, например, соединение, представленное общей формулой (II), преобразуют в галогенид карбоновой кислоты и затем подвергают взаимодействию с соединением, представленным общей формулой (3), в присутствии или в отсутствие основания. Преобразование в галогенид карбоновой кислоты можно осуществить, например, с использованием галогенирующего агента, такого как тионилхлорид, сульфурилхлорид, трихлорид фосфора, пентахлорид фосфора, оксалилхлорид или трихлорид фосфорной кислоты. Примеры основания включают триэтиламин и пиридин. Количество соединения, представленного общей формулой (III), обычно составляет от 0,5 до 2 моль, и предпочтительно от 1 до 1,5 моль, на один моль соединения, представленного общей формулой (II). При использовании основания, количество основания обычно составляет от 1 до 5 моль или около того на один моль соединения, представленного общей формулой (II).
После завершения указанной выше реакции используют известные процедуры очистки и выделения, включая, например, экстракцию, хроматографию, дистилляцию, или перекристаллизацию, в результате чего может быть получено требуемое соединение.
Примеры полученных таким образом соединений представлены в Таблицах 1-5. При указании далее в настоящей заявке каждого соединения, используют номер соединения, указанный в таблицах.




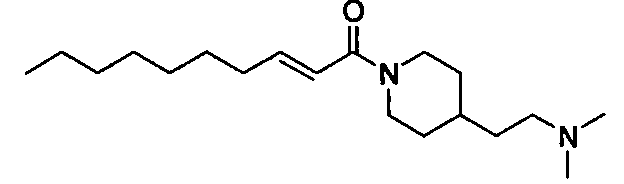


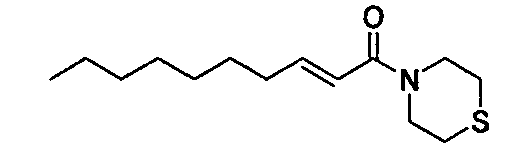
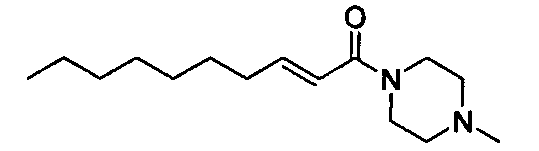


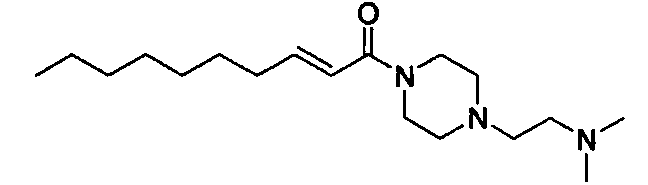












Соединение, представленное общей формулой (I) по настоящему изобретению, включает не только указанные выше свободные формы, но также и формы солей, сольватов и пролекарств. При образовании соли, в случае использования соединения в качестве фармацевтического средства, предпочтительной является форма фармацевтически приемлемой соли. Примеры солей включают соли присоединения кислот, таких как фосфорная кислота, хлористоводородная кислота, серная кислота, азотная кислота, бромистоводородная кислота, перхлорная кислота, тиоциановая кислота, борная кислота, муравьиная кислота, уксусная кислота, галогенуксусная кислота, пропионовая кислота, лимонная кислота, винная кислота, молочная кислота, гликолевая кислота, янтарная кислота, глюконовая кислота, малоновая кислота, фумаровая кислота, антраниловая кислота, бензойная кислота, коричная кислота, пара-толуолсульфоновая кислота, нафталинсульфоновая кислота, или сульфаниловая кислота. Кроме того, эти соли включают соли с металлами, такими как щелочные металлы, например, натрий и калий, или щелочноземельные металлы, такие как кальций или магний, или алюминий; и соли с основанием, таким как аммиак или органический амин. Эти соли могут быть получены из каждого соединения в свободной форме, или преобразованы
обратимо, в соответствии с известным способом. Сольваты включают гидраты, алкоголяты и т.д. Кроме того, в случае, когда соединение, представленное общей формулой (I) по настоящему изобретению, содержит асимметричный атом углерода, соединение также охватывает различные изомеры, такие как оптические изомеры, рацемические смеси и диастереомеры. В случае, когда соединение по настоящему изобретению получают в виде кристаллов, кристаллы также охватывают различные виды кристаллических форм (кристаллический полиморфизм), которые могут быть образованы таким образом.
Соединение, представленное общей формулой (I) по настоящему изобретению, является эффективным в качестве средства, обладающего профилактическим или терапевтическим действием против расстройств периферической нервной системы, вызванных введением противораковых средств. Одно из противораковых средств, развивающих расстройство периферической нервной системы, представляет собой противораковое средство, которое повреждает микротрубочки, что вызывает расстройство периферической нервной системы. Примеры такого лекарственного средства включают таксановое лекарственное средство, такое как паклитаксел или доцетаксел, и лекарственное средство на основе алкалоида барвинка, такое как винкристин, винбластин, виндезин или винорелбин. Другое противораковое средство представляет собой противораковое средство, которое повреждает нервные клетки, что вызывает аксонопатию и затем вызывает расстройства периферической нервной системы. Примеры такого средства включают препарат на основе платины, такой как оксалиплатин, карбоплатин, цисплатин или недаплатин.
Примеры расстройства периферической нервной системы, вызванного этими противораковыми средствами, включают боли, такие как жгучая боль и жжение, онемение конечностей, парестезию, такую как чувство жжения, гиперестезию, такую как повышенная чувствительность к холодным стимулам, дизестезию, такую как потеря чувствительности, сенсорный паралич и чувство дискомфорта, сенсорную атаксию и мышечную слабость. Расстройство периферической нервной системы, вызванное противораковым средством, на профилактику или лечение которого направленно действие соединений по настоящему изобретению, включает расстройство периферической нервной системы, вызванное монотерапией с использованием одного из противораковых средств, а также расстройство периферической нервной системы, вызванное полилекарственной терапией, в которой несколько лекарственных средств, имеющих различные механизмы действия, вводят совместно или путем биохимической модуляции, в которой комбинация лекарственных средств и способ введения рассчитаны таким образом, чтобы лекарственные средства, имеющие различные механизмы действия, могли обеспечить максимальную эффективность.
Поскольку соединение, представленное общей формулой (I) по настоящему изобретению, обладает нейротрофической фактор-подобной активностью, соединение является полезным в качестве средства, обладающего нейротрофической фактор-подобной активностью, и является полезным для профилактики и лечения нервных расстройств. Нервное расстройство относится к клиническому состоянию повреждения функций нервных клеток вследствие дегенерации или клеточной гибели нервных клеток, и включает нейродегенеративные заболевания и психические заболевания. Нейродегенеративные заболевания относятся к деменции, болезни Альцгеймера, болезни Паркинсона, амиотрофическому боковому склерозу (ALS), болезни Гентингтона, прогрессирующему супрануклеарному параличу (PSP), диабетической невропатии, глаукомы зрительного нерва и т.д. Психические заболевания относятся к депрессии (включая биполярную депрессию), тревожному расстройству (невроз), синдрому интеграционного расстройства, расстройству аутистического спектра и т.д. Когда средство используют от депрессии, требуется по меньшей мере от 3 до 4 недель до проявления эффектов традиционных терапевтических средств для лечения депрессии, таких как трициклический антидепрессант, тетрациклический антидепрессант, селективный ингибитор повторного поглощения серотонина (SSRI), ингибитор повторного поглощения норадреналина и серотонина (SNRI) и подобные, и эти средства следует принимать периодически в течение этого срока; однако можно ожидать, что фармацевтическое средство, содержащее соединение по настоящему изобретению, обладает незамедлительной аффективностью по сравнению с существующими медицинскими препаратами.
Соединение, представленное общей формулой (I), является полезным в качестве терапевтического средства (восстанавливающего средства) при повреждении спинного мозга. Не существует эффективных способов лечения поражения спинного мозга в результате физических повреждений, вызванных дорожно-транспортными происшествиями, спортивными травмами или компрессионными переломами у пожилых людей, и были проведены различные исследования, касающиеся способов лечения с использованием восстанавливающей терапии. Ожидается, что фармацевтическое средство, содержащее соединение по настоящему изобретению, может лечить (восстанавливать) повреждения спинного мозга при введении путем инъекции, с использованием лекарственных препаратов для внутреннего применения и наружного применения.
Кроме того, соединение, представленное общей формулой (I) по настоящему изобретению, является полезным в качестве средства для профилактики или лечения различных болевых синдромов. Примеры болевых синдромов включают боли в суставах, такие, как боли при остеоартрите (например, остеоартрит колена и остеоартрит шейки бедра) или боли при ревматоидном артрите.
Соединения по настоящему изобретению можно использовать для получения фармацевтического препарата в различных лекарственных формах (таких как препараты для перорального применения, препараты для инъекций и препараты для наружного применения) путем объединения с соответствующим фармацевтическим носителем или разбавителем. Фармацевтическое средство по настоящему изобретению может представлять собой композицию, в которой соединение по настоящему изобретению используют вместе с другим фармацевтически активным ингредиентом(ингредиентами). Кроме того, фармацевтическое средство по настоящему изобретению можно сформулировать в препарат в виде комплекса включения с циклодекстрином и т.д. Как результат, могут быть случаи, когда можно получить усиление фармакологической активности, улучшение стабильности, пролонгированное действие, удобство использования и т.д. Комплекс включения может быть образован путем, например, смешения соединения по настоящему изобретению с α-, β- или γ-циклодекстрином в соответствии с обычным способом.
Когда соединение по настоящему изобретению формулируют в виде перорального препарата, препарат в форме таблеток, порошка, гранул или капсул можно получить путем формулирования подходящих комбинаций соответствующих добавок, включающих, например, эксципиент, связующее, разрыхлитель, смазывающее вещество, наполнитель, увлажнитель, буфер, консервант и ароматизатор. Когда соединение по настоящему изобретению формулируют в виде препарата для инъекций, препарат для инъекций можно сформулировать путем добавления стабилизатора, консерванта, изотонического вещества или подобных к раствору или суспензии, содержащим сложный эфир C10 жирной кислоты. Когда соединение по настоящему изобретению формулируют в виде препарата для наружного применения, препарат для наружного применения можно сформулировать в виде пластыря, геля, мази, крема или т.п. Таким образом, соединение по настоящему изобретению, например, смешивают с, расплавляют в, или эмульгируют в подходящей основе для получения препарата, и, в случае препарата в виде пластыря, полученную композицию распределяют и наносят на подложку. В случае препарата в виде пластыря, гелевого препарата или подобных, можно получить, например, композицию с использованием органического гелеобразующего агента. Также, в зависимости от используемой лекарственной формы каждого препарата для наружного применения, можно выбрать подходящие обычно используемые консервант, антиоксидант, отдушку, адгезив или т.п. и добавить к композиции.
Желаемая доза соединения по настоящему изобретению может быть соответствующим образом увеличена или уменьшена, принимая во внимание схему введения лекарственного средства, возраст, пол, симптом у пациента и т.д., и соединение, как правило, можно вводить в количестве от 1 до 1000 мг, и предпочтительно от 5 до 300 мг, для взрослых, в виде разового введения или нескольких отдельных введений в день.
ПРИМЕРЫ
Настоящее изобретение будет далее объяснено при помощи Примеров, без намерения ограничить настоящее изобретение этими Примерами.
Пример 1
Получение 1-((Е)-2-Деценоил)пирролидина [Соединение 1]
Тетрагидрофурановый раствор (20 мл) пирролидина (0,71 г, 0,01 моль), содержащий пиридин (0,79 г, 0,01 моль), добавляли к тетрагидрофурановому раствору (20 мл) хлорангидрида (Е)-2-деценовой кислоты (1,9 г, 0,01 моль), синтезированного с использованием (E)-2-деценовой кислоты и тионилхлорида, и смесь нагревали при кипячении с обратным холодильником в теплой водяной бане в течение 3 часов. После отгонки избытка тетрагидрофурана к жидкой реакционной смеси добавляли воду и жидкую смесь экстрагировали этилацетатом, промывали водой и этилацетат отгоняли. Остаток очищали при помощи колоночной хроматографии на силикагеле (проявляющий растворитель: гексан:этилацетат = 1:3), с получением требуемого соединения (1,4 г) в форме бесцветного маслянистого вещества.
Бесцветное маслянистое вещество, C14H25NO MW 223, ESIMS (режим положительных ионов: относительный интервал): m/z 246,184 [M+Na]+ (Рассчитано для C14H25NONa, 246,1834), 1H-ЯМР (400 МГц, CDCl3) δ: 0,88 (3H, т, J=6,8 Гц), 1,25-1,32 (8H, м), 1,45 (2H, м), 1,86 (2H, м), 1,96 (2H, м), 2,20 (2H, м), 3,518 (2H, т, J=7,2 Гц), 3,524 (2H, т, J=7,2 Гц), 6,09 (1H, д, J=15,4 Гц), 6,91 (1H, дт, J=15,4, 6,8 Гц).
Пример 2
Получение метилового эфира (S)-1-((Е)-2-Деценоил)-пирролидин-2-карбоновой кислоты [Соединение 3]
Триэтиламин (0,84 мл, 6 ммоль) и l-этил-3-(3-диметиламинопропил)карбодиимидгидрохлорид (WSC∙HCl) (1,05 г, 5,5 ммоль) добавляли к дихлорметановому раствору (50 мл), содержащему (Е)-2-деценовую кислоту (0,92 мл, 5 ммоль) и гидрохлорид метилового эфира L-пролина (0,91 г, 5,5 ммоль), при комнатной температуре, и смесь перемешивали в течение 24 часов. Полученный реакционный раствор промывали водой и насыщенным раствором хлорида натрия и затем сушили над безводным сульфатом натрия. Остаток очищали при помощи колоночной хроматографии на силикагеле (гексан:этилацетат = 7:3), с получением требуемого соединения (1,23 г, 87%) в форме бесцветного маслянистого вещества. Здесь, в случае, когда исходный амин не является гидрохлоридом, триэтиламин можно не добавлять.
Бесцветное маслянистое вещество, C16H27NO3 MW 281,4, EIMS: m/z 282 [M+H]+, 1H-ЯМР (500 МГц, ДМСО-d6) δ: 0,86 (т, J=7,0 Гц, 3H), 1,21-1,31 (м, 8H), 1,34-1,44 (м, 2H), 1,70-1,97 (м, 3H), 2,02-2,27 (м, 3H), 3,38-3,65 (м, 5H), 4,31-4,35 и 4,70-4,74 (м, 1H), 5,97 и 6,25 (м, 1H), 6,61-6,71 (м, 1H).
Пример 3
Получение (S)-1-((E)-2-деценоил)пирролидин-2-карбоновой кислоты [Соединение 2]
Соединение 3 (0,84 г, 3 ммоль) растворяли в метаноле (40 мл) и к этой смеси при комнатной температуре добавляли 1 моль/л водный раствор гидроксида натрия (4 мл, NaOH 4 ммоль) и смесь перемешивали в течение 20 часов. Растворитель отгоняли при пониженном давлении и остаток затем растворяли в воде и к этой смеси добавляли 10% водный раствор лимонной кислоты до доведения pH до приблизительно 4. Жидкую смесь экстрагировали дихлорметаном, органический слой промывали водой и насыщенным раствором хлорида натрия и промытый экстракт затем сушили над безводным сульфатом натрия, с получением требуемого соединения (0,72 г, 90%) в форме бесцветного маслянистого вещества.
Бесцветное маслянистое вещество, C15H25NO3 MW 267,4, EIMS: m/z 268 [M+H]+, 1H-ЯМР (500 МГц, ДМСО-d6) δ: 0,86 (т, J=7,0 Гц, 3Н), 1,20-1,45 (м, 10Н), 1,70-2,27 (м, 6Н), 3,37-3,63 (м, 2Н), 4,23-4,28 и 4,55-4,59 (м, 1H), 6,25 и 5,99 (м, 1H), 6,62-6,71 (м, 1H), 12,30-12,70 (уш., 1H).
Пример 4
Получение 3-((E)-2-деценоил)тиазолидина [Соединение 4]
Те же процедуры, которые использовали в Примере 1, осуществляли с использованием (E)-2-деценовой кислоты и тиазолидина в качестве исходных веществ, с получением целевого соединения.
Бесцветное маслянистое вещество, C13H23NOS MW 241, HR-ESIMS (режим положительных ионов): m/z 264,1443 [M+Na]+ (Рассчитано для C13H23NOSNa, 264,1398), 1H-ЯМР (400 МГц, CDCl3) δ: 0,88 (3H, т, J=6,8 Гц), 1,22-1,34 (8H, м), 1,46 (2H, м), 2,22 (2H, м), 3,01 (1H, т, J=6,1 Гц), 3,11 (1H, т, J=6,1 Гц), 3,83 (1H, т, J=6,1 Гц), 3,91 (1H, уш. т), 4,58 (1H, с), 4,65 (1H, с), 6,11 (1H, д, J=15,0 Гц) , 6,96 (1H, дт, J=15,0, 7,3 Гц).
Пример 5
Получение 1-((Е)-2-деценоил)пиперидина [Соединение 5]
Те же процедуры, которые использовали в Примере 1, осуществляли с использованием (E)-2-деценовой кислоты и пиперидина в качестве исходных веществ, с получением целевого соединения.
Бесцветное маслянистое вещество, C15H27NO MW 237, EIMS: m/z 237 [M]+, 1H-ЯМР (500 МГц, CDCl3) δ: 0,88 (3H, т, J=7,0 Гц), 1,22-1,34 (8H, м), 1,42-1,47 (2H, м), 1,54-1,59 (4H, м), 1,62-1,68 (2H, м), 2,16-2,21 (2H, м), 3,44-3,65 (4H, м), 6,22-6,26 (1H, м), 6,83 (1H, дт, J=15,1, 7,1 Гц).
Пример 6
Получение 1-((E)-2-деценоил)-4-метилпиперидина [Соединение 6]
Те же процедуры, которые использовали в Примере 2, осуществляли с использованием (E)-2-деценовой кислоты и 4-метилпиперидина в качестве исходных веществ, с получением целевого соединения.
Бесцветное маслянистое вещество, C15H29NO MW 251,4, EIMS: m/z 252 [M+H]+, 1H-ЯМР (500 МГц, ДМСО-d6) δ: 0,93-1,11 (м, 8H), 1,20-1,32 (м, 8H), 1,35-1,44 (м, 2Н), 1,54-1,67 (м, 3Н), 2,12-2,19 (м, 2Н), 2,51-2,60 (м, 1H), 2,92-3,11 (м, 1H), 3,96-4,15 (м, 1H), 4,32-4,41 (м, 1H), 6,43 (д, J=15,0 Гц, 1H), 6,61 (дт, J=15,0, 7,0 Гц, 1H).
Пример 7
Получение 1-((Е)-2-деценоил)пиперидин-4-она [Соединение 7]
Те же процедуры, которые использовали в Примере 1, осуществляли с использованием (E)-2-деценовой кислоты и пиперидин-4-она в качестве исходных веществ, с получением целевого соединения.
Бледно-желтое маслянистое вещество, C15H25NO2 MW 251, HR-ESIMS (режим положительных ионов): m/z 274,1824 [M+Na] (Рассчитано для C15H25NO2Na, 274,1783), 1H-ЯМР (400 МГц, CDCl3) δ: 0,88 (3H, т, J=6,8 Гц), 1,22-1,35 (8H, м), 1,47 (2H, м), 2,23 (2H, м), 2,51 (4H, т, J=6,4 Гц) 3,82-3,97 (4H, м), 6,31 (1H, д, J=15,2 Гц), 6,97 (1H, дт, J=15,2, 7,3 Гц).
Пример 8
Получение 1-((Е)-2-деценоил)-4-гидроксипиперидина [Соединение 8]
Те же процедуры, которые использовали в Примере 2, осуществляли с использованием (E)-2-деценовой кислоты и 4-гидроксипиперидина в качестве исходных веществ, с получением целевого соединения.
Бесцветное маслянистое вещество, C15H27NO2 MW 253,4, EIMS: m/z 254 [M+H]+, 1H-ЯМР (500 МГц, ДМСО-d6) δ: 0,86 (т, J=7,0 Гц, 3H), 1,20-1,32 (м, 10H), 1,36-1,44 (м, 2H), 1,66-1,76 (м, 2H), 2,13-2,20 (м, 2H), 3,00-3,09 (м, 1H), 3,16-3,25 (м, 1H), 3,65-3,73 (м, 1H), 3,76-3,84 (м, 1H), 3,88-3,96 (м, 1H), 4,74 (д, J=4,1 Гц, 1H), 6,45 (д, J=15,0 Гц, 1H), 6,63 (дт, J=15,0, 7,0 Гц, 1H).
Пример 9
Получение 1-((Е)-2-деценоил)-4-метоксипиперидина [Соединение 9]
Соединение 8 (0,62 г, 2,4 ммоль) растворяли в THF (10 мл) и к этой смеси при охлаждении льдом добавляли гидрид натрия (60% в минеральном масле) (0,10 г, 2,6 ммоль). После перемешивания в течение 15 минут при охлаждении льдом к этой смеси добавляли метилйодид (0,16 мл, 2,6 ммоль) и перемешивали при комнатной температуре в течение 20 часов. К жидкой реакционной смеси добавляли воду, смесь экстрагировали этилацетатом, органический слой промывали водой и насыщенным раствором хлорида натрия и промытый экстракт затем сушили над безводным сульфатом натрия. Остаток очищали при помощи колоночной хроматографии на силикагеле (проявляющий растворитель: гексан:этилацетат = 3:2), с получением целевого соединения (0,42 г, 65%) в форме бледно-желтого маслянистого вещества.
Бледно-желтое маслянистое вещество, C16H29NO2 MW 267,4, EIMS: m/z 268 [M+H]+, 1H-ЯМР (500 МГц, ДМСО-d6) δ: 0,86 (т, J=7,0 Гц, 3H), 1,20-1,43 (м, 12H), 1,75-1,85 (м, 2H), 2,12-2,19 (м, 2H), 3,08-3,17 (м, 1H), 3,22-3,30 (м, 1H), 3,25 (с, 3H), 3,33-3,42 (м, 1H), 3,70-3,88 (м, 2H), 6,45 (д, J=15,0 Гц, 1H), 6,63 (дт, J=15,0, 7,0 Гц, 1H).
Пример 10
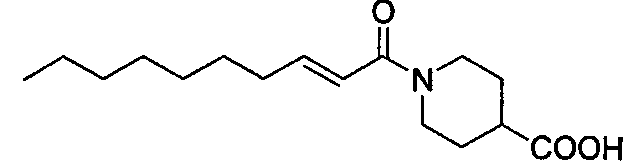
Получение 1-((Е)-2-деценоил)пиперидин-4-карбоновой кислоты [Соединение 10]
Те же процедуры, которые использовали в Примере 3, осуществляли с использованием Соединения 11 в качестве исходного вещества, с получением целевого соединения.
Белые кристаллы, т.пл. 88-89°С, C16H27NO3 MW 281,4, EIMS: m/z 281 [M]+, 1H-ЯМР (500 МГц, ДМСО-d6) δ: 0,86 (т, J=7,0 Гц, 3H), 1,19-1,31 (м, 8H), 1,32-1,46 (м, 4H), 1,77-1,87 (м, 2H), 2,12-2,20 (м, 2H), 2,46-2,54 (м, 1H), 2,71-2,82 (м, 1H), 3,03-3,16 (м, 1H), 3,90-4,00 (м, 1H), 4,18-4,28 (м, 1Н), 6,44 (д, J=15,0 Гц, 1H), 6,63 (дт, J=15,0, 7,0 Гц, 1H), 12,28 (уш.с, 1H).
Пример 11
Получение этилового эфира 1-((Е)-2-деценоил)пиперидин-4-карбоновой кислоты [Соединение 11]
Те же процедуры, которые использовали в Примере 2, осуществляли с использованием (E)-2-деценовой кислоты и этилизонипекотата в качестве исходных веществ, с получением целевого соединения.
Бесцветное маслянистое вещество, C18H31NO3 MW 309,4, EIMS: m/z 309 [M]+, 1H-ЯМР (500 МГц, ДМСО-d6) δ: 0,86 (т, J=7,0 Гц, 3H), 1,18 (т, J=7,0 Гц, 3H), 1,20-1,32 (м, 8H), 1,33-1,49 (м, 4H), 1,80-1,88 (м, 2H), 2,13-2,19 (м, 2H), 2,56-2,64 (м, 1H), 2,71-2,81 (м, 1H), 3,05-3,16 (м, 1H), 3,92-4,07 (м, 1H), 4,07 (кв., J=7,0 Гц, 2H), 4,20-4,28 (м, 1H), 6,45 (д, J=15,0 Гц, 1H), 6,63 (дт, J=15,0, 7,0 Гц, 1H).
Пример 12
Получение 1-((E)-2-деценоил)-4-диметиламинопиперидина [Соединение 12]
Те же процедуры, которые использовали в Примере 2, осуществляли с использованием (Е)-2-деценовой кислоты и 4-диметиламинопиперидина в качестве исходных веществ, с получением целевого соединения.
Бледно-желтое маслянистое вещество, C17H32N2O MW 280,5, EIMS: m/z 281 [M+H]+, 1H-ЯМР (500 МГц, ДМСО-d6) δ: 0,86 (т, J=7,0 Гц, 3H), 1,13-1,31 (м, 10H), 1,36-1,44 (м, 2H), 1,70-1,79 (м, 2H), 2,12-2,20 (м, 7H), 2,26-2,34 (м, 1H), 2,56-2,65 (м, 1H), 2,93-3,04 (м, 1H), 3,99-4,07 (м, 1H), 4,33-4,41 (м, 1H), 6,45 (д, J= 5,0 Гц, 1H), 6,63 (дт, J=15,0, 7,0 Гц, 1H).
Кроме того, маслянистый продукт Соединения 12 растворяли в метиленхлориде и обрабатывали смесью хлористый водород-диоксан, с получением 1-((Е)-2-деценоил)-4-диметиламинопиперидин гидрохлорида (кристаллы, т.пл. 185°-188°С).
Пример 13
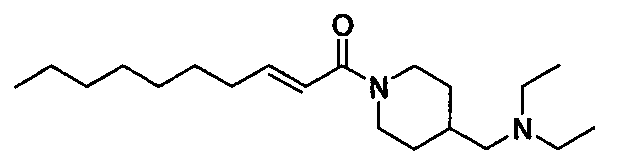
Получение 1-((E)-2-деценоил)-4-диэтиламинопиперидина [Соединение 13]
Те же процедуры, которые использовали в Примере 2, осуществляли с использованием (E)-2-деценовой кислоты и 4-диэтиламинопиперидина в качестве исходных веществ, с получением целевого соединения.
Бледно-желтое маслянистое вещество, C19H36N2O MW 308,5, EIMS: m/z 309 [M+H]+, 1H-ЯМР (500 МГц, ДМСО-d6) δ: 0,86 (т, J=7,0 Гц, 3H), 0,95 (т, J=7,0 Гц, 6H), 1,15-1,31 (м, 10H), 1,36-1,43 (м, 2H), 1,63-1,72 (м, 2H), 2,13-2,20 (м, 2H), 2,45 (кв., J=7,0 Гц, 4H), 2,50-2,60 (м, 1H), 2,65-2,73 (м, 1H), 2,90-3,02 (м, 1H), 4,01-4,10 (м, 1H), 4,39-4,47 (м, 1H), 6,44 (д, J=15,0 Гц, 1H), 6,61 (дт, J=15,0, 7,0 Гц, 1H).
Пример 14
Получение 1-((Е)-2-деценоил)-4-диэтиламинометилпиперидина [Соединение 14]
Те же процедуры, которые использовали в Примере 2, осуществляли с использованием (Е)-2-деценовой кислоты и 4-(диэтиламинометил)пиперидина в качестве исходных веществ, с получением целевого соединения.
Бледно-красное маслянистое вещество, C20H38N2O MW 322,5, EIMS: m/z 323 [М+Н]+, 1Н-ЯМР (500 МГц, ДМСО-d6) δ: 0,86 (т, J=7,0 Гц, 3Н), 0,93 (т, J=7,0 Гц, 6Н), 0,90-0, 97 (м, 2Н), 1,20-1,30 (м, 8Н), 1,35-1,43 (м, 2Н), 1,60-1,80 (м, 3Н), 2,10-2,19 (м, 4Н), 2,41 (кв., J=7,0 Гц, 4Н), 2,51-2,60 (м, 1Н), 2,92-3,02 (м, 1Н), 3,97-4, 06 (м, 1Н), 4,33-4,42 (м, 1Н), 6,43 (д, J=15,0 Гц, 1Н), 6,61 (дт, J=15,0, 7,0 Гц, 1Н).
Пример 15
Получение 1-((Е)-2-деценоил)-4-(2-диметиламиноэтил)пиперидина [Соединение 15]
Те же процедуры, которые использовали в Примере 2, осуществляли с использованием (Е)-2-деценовой кислоты и 4-(2-диметиламиноэтил)пиперидина в качестве исходных веществ, с получением целевого соединения.
Бледно-желтое маслянистое вещество, C19H36N2O MW 308,5, EIMS: m/z 309 [M+H]+, 1Н-ЯМР (500 МГц, ДМСО-d6) δ: 0,86 (т, J=7,0 Гц, 3Н), 0,90-1,15 (м, 2Н), 1,20-1,34 (м, 10Н), 1,35-1,43 (м, 2Н), 1,48-151,59 (м, 1Н), 1,62-1,71 (м, 2Н), 2,09 (с, 6Н), 2,13-2,22 (м, 4Н), 2,50-2,58 (м, 1Н), 2,90-3,01 (м, 1Н), 3,95-4,05 (м, 1Н), 4,32-4,41 (м, 1Н), 6,43 (д, J=15,0 Гц, 1Н), 6,60 (дт, J=15,0, 7,0 Гц, 1Н).
Пример 16
Получение 1-((Е)-2-деценоил)-4-фенилпиперидина [Соединение 16]
Те же процедуры, которые использовали в Примере 1, осуществляли с использованием (Е)-2-деценовой кислоты и 4-фенилпиперидина в качестве исходных веществ, с получением целевого соединения.
Бесцветное маслянистое вещество, C21H31NO MW 313, HR-ESIMS (режим положительных ионов): m/z 336,2330 [M+Na]+ (Рассчитано для C21H31NONa, 336,2303), 1H-ЯМР (400 МГц, CDCl3) δ: 0,88 (3H, т, J=7,2 Гц), 1,22-1,34 (8H, м), 1,46 (2Н, м), 1,66 (2Н, м), 1,91 (2Н, уш.д), 2,21 (2Н, м), 2,66-2,80 (2Н, м), 3,15 (1H, уш. т), 4,14 (1H, уш. д), 4,84 (1H, уш. д), 6,29 (1H, д, J=15,0 Гц), 6,89 (1H, дт, J=15,0, 7,3 Гц), 7,18-7,24 (3H, м), 7,32 (2H, т, J=7,4 Гц).
Пример 17
Получение 4-((E)-2-деценоил)морфолин [Соединение 17]
Те же процедуры, которые использовали в Примере 1, осуществляли с использованием (E)-2-деценовой кислоты и морфолина в качестве исходных веществ, с получением целевого соединения.
Бесцветное маслянистое вещество, C14H25NO2 MW 239, HR-ESIMS (режим положительных ионов): m/z 262,1827 [M+Na]+ (Рассчитано для C14H25NO2Na, 262,1783), 1H-ЯМР (400 МГц, CDCl3) δ: 0,88 (3H, т, J=7,2 Гц), 1,24-1,33 (8H, м), (2H, м), 2,20 (2H, м), 3,57 (2H, уш.с), 3,69 (6H, уш.с), 6,20 (1H, д, J=15,2 Гц), 6,91 (1H, дт, J=15,2, 7,1 Гц).
Пример 18
Получение 4-((E)-2-деценоил)тиоморфолина [Соединение 18]
Те же процедуры, которые использовали в Примере 1, осуществляли с использованием (Е)-2-деценовой кислоты и тиоморфолина в качестве исходных веществ, с получением целевого соединения.
Бесцветное маслянистое вещество, C14H25NOS MW 255, HR-ESIMS (режим положительных ионов): m/z 278,1575 [M+Na]+ (Рассчитано для C14H25NOSNa, 278,1555), 1H-ЯМР (400 МГц, CDCl3) δ: 0,88 (3H, т, J=7,2 Гц), 1,23-1,33 (8H, м), 1,45 (2H, м), 2,20 (2H, м), 2,63 (4H, уш.с), 3,83 (2H, уш.с), 3,92 (2H, уш.с), 6,20 (1H, д, J=15,2 Гц), 6,87 (1H, дт, J=15,2, 7,3 Гц).
Пример 19
Получение 1-[(Е)-2-деценоил)-4-метилпиперазина [Соединение 19]
Те же процедуры, которые использовали в Примере 2. осуществляли с использованием (E)-2-деценовой кислоты и 1-метилпиперазина в качестве исходных веществ, с получением целевого соединения.
Бледно-желтое маслянистое вещество, C15H28N2O MW 252,4, EIMS: 1H-ЯМР (500 МГц, ДМСО-d6) δ: 0,86 (т, J=7,0 Гц, 3H), 1,20-1,31 (м, 8H), 1,35-1,44 (м, 2H), 2,13-2,19 (м, 2H), 2,17 (с, 3H), 2,22-2,30 (м, 4H), 3,45-3,55 (м, 4H), 6,44 (дт, Jd=14,0 Гц, Jt=1,1 Гц, 1H), 6,65 (дт, J=14,0, 7,2 Гц, 1H).
Пример 20
Получение 1-((E)-2-деценоил-4-изопропилпиперазин [Соединение 20]
Те же процедуры, которые использовали в Примере 2. осуществляли с использованием (Е)-2-деценовой кислоты и 1-изопропилпиперазина в качестве исходных веществ, с получением целевого соединения.
Бледно-желтое маслянистое вещество, C17H32N2O MW 280,5, EIMS: m/z 280 [M]+, 1H-ЯМР (500 МГц, ДМСО-d6) δ: 0,86 (т, J=7,0 Гц, 3H), 0,96 (д, J=6,5 Гц, 6H), 1,20-1,31 (м, 8H), 1,36-1,42 (м, 2H), 2,11-2,20 (м, 2H), 2,35-2,42 (м, 4H), 2,65 (кв.кв., J=6,5, 6,5 Гц, 1H), 3,44-3,54 (м, 4H), 6,46 (дт, J=15,0, 1,1 Гц, 1H), 6,63 (дт, J=15,0, 7,0 Гц, 1H).
Пример 21
Получение 3-[4-((E)-2-деценоил)пиперазин-1-ил]пропионовой кислоты [Соединение 21]
Те же процедуры, которые использовали в Примере 2, осуществляли с использованием (Е)-2-деценовой кислоты и этил 3-пиперазин-1-илпропионата дигидрохлорида (этиловый эфир соединения 21) в качестве исходных веществ, с получением этил 3-[4-((E)-2-деценоил)пиперазин-1-ил]пропионата, и подвергали щелочному омылению, с получением целевого соединения в виде белых кристаллов.
Белые кристаллы, т.пл. 53-55°C, C17H30N2O3 MW 310,4, EIMS: m/z 311 [M+H]+, 1H-ЯМР (500 МГц, ДМСО-d6) δ: 0,86 (т, J=7,0 Гц, 3H), 1,19-1,31 (м, 8H), 1,35-1,44 (м, 2H), 2,12-2,20 (м, 2H), 2,32-2,43 (м, 6H), 2,55-2,61 (м, 2H), 3,43-3,57 (м, 4H), 6,45 (д, J=15,0 Гц, 1H), 6,65 (дт, J=15,0, 7,0 Гц, 1H), 11,70-12,70 (уш.с, 1H).
Кроме того, низкоплавкие кристаллы Соединения 21 растворяли в метиленхлориде и обрабатывали смесью хлористый водород-диоксан, с получением гидрохлорида 3-[4-((Е)-2-деценоил)пиперазин-1-ил]пропионовой кислоты (кристаллы, т.пл. 201°-203°C).
Пример 22
Получение 1-((Е)-2-деценоил)-4-[2-(диметиламино)этил]пиперазина [Соединение 22]
Те же процедуры, которые использовали в Примере 1, осуществляли с использованием (E)-2-деценовой кислоты и 1-[2-(диметиламино)этил]пиперазина в качестве исходных веществ, с получением целевого соединения.
Маслянистое вещество, C18H35N3O MW 309, HR-ESIMS (режим положительных ионов): m/z 310,2868 [M+H]+ (Рассчитано для C18H36N3O, 310,2858), 1H-ЯМР (500 МГц, CDCl3) δ: 0,88 (3H, т, J=7,4 Гц), 1,27-1,31 (8H, м), 1,45 (2H, уш.т, J=7,5 Гц), 2,19 (2H, дт, J=6,9, 8,0 Гц), 2,32 (6H, с), 2,41 (4H, уш.с), 2,53 (4H, с), 3,56 (2H, уш.с), 3,68 (2H, уш.с), 6,21 (1H, д, J=15,2 Гц), 6,86 (1H, дт, J=6,9, 15,2 Гц).
Пример 23
Получение 4-циклогексил-1-((E)-2-деценоил)пиперазина [Соединение 23]
Те же процедуры, которые использовали в Примере 2, осуществляли с использованием (E)-2-деценовой кислоты и 1-циклогексилпиперазина в качестве исходных веществ, с получением целевого соединения.
Белые кристаллы, т.пл. 33-34°C, C20H36N2O MW 320,5, EIMS: m/z 320 [M]+, 1H-ЯМР (500 МГц, ДМСО-d6) δ: 0,85 (т, J=7,0 Гц, 3H), 1,01-1,11 (м, 1H), 1,11-1,21 (м, 4H), 1,21-1,31 (м, 8H), 1,36-1,43 (м, 2H), 1,53-1,59 (м, 1H), 1,68-1,76 (м, 4H), 2,13-2,28 (м, 3H), 2,40-2,48 (м, 4H), 3,41-3,53 (м, 4H), 6,43 (д, J=15,0 Гц, 1H), 6,64 (дт, J=15,0, 7,0 Гц, 1H).
Пример 24
Получение 1-((Е)-2-деценоил)-4-(2-пиперидин-1-илэтил)пиперазина [Соединение 24]
Те же процедуры, которые использовали в Примере 2, осуществляли с использованием (E)-2-деценовой кислоты и 1-[2-(l-пиперидинил)этил]пиперазина в качестве исходных веществ, с получением целевого соединения.
Белые кристаллы, C21H39N3O MW 349,6, EIMS: m/z 350 [M+H]+, 1H-ЯМР (500 МГц, ДМСО-d6) δ: 0,86 (т, J=6,7 Гц, 3H), 1,20-1,31 (м, 8H), 1,32-1,42 (м, 4H), 1,43-1,50 (м, 4H), 2,11-2,20 (м, 2H), 2,27-2,43 (м, 12H), 3,42-3,53 (м, 4H), 6,43 (д, J=15,0 Гц, 1H), 6,64 (дт, J=15,0, 7,0 Гц, 1H).
Пример 25
Получение 1-((Е)-2-деценоил)-4-фенилпиперазина [Соединение 25]
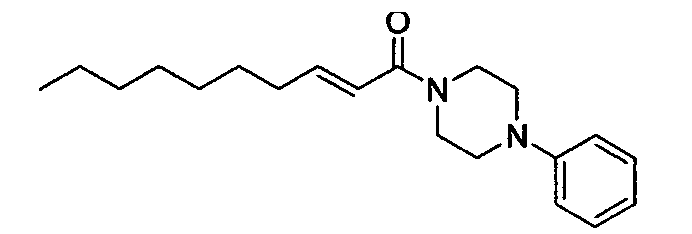
Те же процедуры, которые использовали в Примере 1, осуществляли с использованием (Е)-2-деценовой кислоты и 1-фенилпиперазина в качестве исходных веществ, с получением целевого соединения.
Розовое маслянистое вещество, C20H30N2O MW 314, HR-ESIMS (режим положительных ионов): m/z 337,2244 [M+Na]+, (Рассчитано для C20H30N2ONa, 337,2256), 1Н-ЯМР (400 МГц, CDCl3) δ: 0,88 (3Н, т, J=6,8 Гц), 1,21-1,36 (8H, м), 1,45 (2H, м), 2,22 (2H, м), 3,18 (4H, т, J=5,4 Гц), 3,71 (2H, уш.с), 3,83 (2H, уш.с), 6,27 (1H, д, J=15,6 Гц), 6,88-6,96 (4H, м), 7,26-7,30 (2H, м).
Пример 26
Получение 4-Бензил-1-((Е)-2-деценоил)пиперазина [Соединение 26]
Те же процедуры, которые использовали в Примере 2, осуществляли с использованием (E)-2-деценовой кислоты и 1-бензилпиперазин дигидрохлорида в качестве исходных веществ, с получением целевого соединения.
Белые кристаллы, т.пл. 62-63°С, C21H32N2O MW 328,5, EIMS: m/z 2329 [M+H]+, 1H-ЯМР (500 МГц, ДМСО-d6) δ: 0,86 (т, J=7,0 Гц, 3H), 1,19-1,31 (м, 8H), 1,35-1,42 (м, 2H), 2,11-2,19 (м, 2H), 2,28-2,38 (м, 4H), 3,48 (с, 2H), 3,48-3,57 (м, 4H), 6,43 (д, J=15,0 Гц, 1H), 6,65 (дт, J=15,0, 7,0 Гц, 1H), 7,22-7,36 (м, 5H).
Пример 27
Получение 1-((Е)-2-деценоил)-4-(2-фенилэтил)пиперазина [Соединение 27]
Те же процедуры, которые использовали в Примере 2, осуществляли с использованием (E)-2-деценовой кислоты и 1-(2-фенэтил)пиперазин дигидрохлорида в качестве исходных веществ, с получением целевого соединения.
Белые кристаллы, т.пл. 33-34°С, C22H34N2O MW 342,5, EIMS: m/z 343 [M+H]+, 1H-ЯМР (500 МГц, ДМСО-d6) δ: 0,86 (т, J=7,0 Гц, 3H), 1,19-1,31 (м, 8H), 1,36-1,44 (м, 2H), 2,11-2,21 (м, 2H), 2,36-2,45 (м, 4H), 2,52 (дд, J=6,5, 8,3 Гц, 2H), 2,74 (дд, J=6,5, 8,3 Гц, 2H), 3,46-3,56 (м, 4Н), 6,45 (д, J=15,0 Гц, 1H), 6,66 (дт, J=15,0, 7,0 Гц, 1H), 7,15-7,31 (м, 5H).
Пример 28
Получение 1-((Е)-2-деценоил)-4-(4-диметиламинофенил)пиперазина [Соединение 28]
Те же процедуры, которые использовали в Примере 2, осуществляли с использованием (Е)-2-деценовой кислоты и 1-(4-диметиламинофенил)пиперазин дигидрохлорида в качестве исходных веществ, с получением целевого соединения.
Бледно-коричневые кристаллы, т.пл. 73-74°С, C22H35N3O MW 357,5, EIMS: 1H-ЯМР (500 МГц, ДМСО-d6) δ: 0,85 (т, J=7,0 Гц, 3H), 1,20-1,31 (м, 8H), 1,36-1,46 (м, 2H), 2,15-2,21 (м, 2H), 2,78 (с, 6H), 2,87-2-2,96 (м, 4H), 3,55-3,74 (м, 4H), 6,49 (д, J=15,0 Гц, 1H), 6,64-6,72 (м, 3H), 6,86 (д, J=8,7 Гц, 2H).
Пример 29
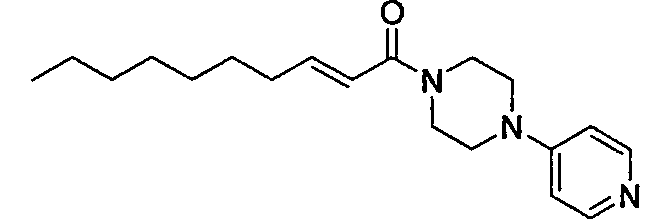
Получение 1-((Е)-2-деценоил)-4-(пиридин-4-ил)пиперазина [Соединение 29]
Те же процедуры, которые использовали в Примере 1, осуществляли с использованием (E)-2-деценовой кислоты и 1-(4-пиридил)пиперазина в качестве исходных веществ, с получением целевого соединения.
Бледно-коричневое маслянистое вещество, C19H29N3O MW 315, HR-ESIMS (режим положительных ионов): m/z 316,2349 [M+H]+ (Рассчитано для C19H30N3O, 316,2383), 1H-ЯМР (400 МГц, CDCl3) δ: 0,89 (3H, т, J=7,2 Гц), 1,19-1,35 (l0H, м), 1,47 (2H, м), 2,23 (2H, м), 2,49 (1H, уш.с), 3,39 (4H, т, J=5,4 Гц), 3,70-3,88 (4H, м), 6,25 (1H, д, J=15,0 Гц), 6,67 (2H, д, J=6,4 Гц), 6,95 (1H, дт, J=15,0, 7,3 Гц), 8,31 (2H, д, J=6,4 Гц).
Пример 30
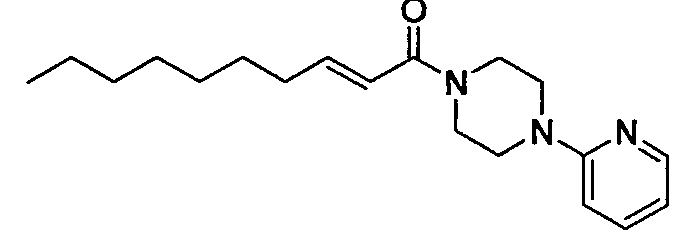
Получение 1-((Е)-2-деценоил)-4-(пиридин-2-ил)пиперазина [Соединение 30]
Те же процедуры, которые использовали в Примере 1, осуществляли с использованием (E)-2-деценовой кислоты и 1-(2-пиридил)пиперазина в качестве исходных веществ, с получением целевого соединения.
Бесцветные кристаллы, т.пл. 59-61°С, C19H29N3O MW 315, HR-ESIMS (режим положительных ионов): m/z 316,2346 [M+H]+ (Рассчитано для C19H29N3O, 316,2383), 1H-ЯМР (400 МГц, CDCl3) δ: 0,88 (3H, т, J=7,2 Гц), 1,22-1,35 (8H, м), 1,47 (2H, м), 2,22 (2H, м), 3,53 (2H, уш.с), 3,60-3,71 (4H, м), 3,81 (2H, уш.с), 6,27 (1H, д, J=15,2 Гц), 6,64-6,69 (2H, т, м), 6,93 (1H, дт, J=15,2, 7,3 Гц), 7,51 (1H, м), 8,20 (1H, м).
Пример 31
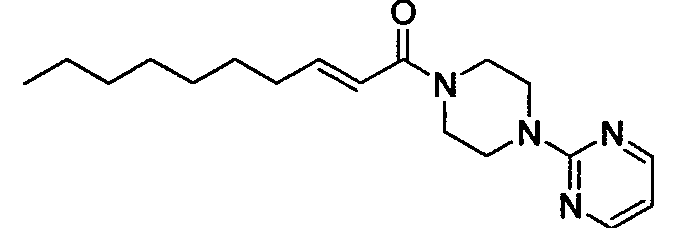
Получение 1-((Е)-2-деценоил)-4-(пиримидин-2-ил)пиперазина [Соединение 31]
Те же процедуры, которые использовали в Примере 1, осуществляли с использованием (E)-2-деценовой кислоты и 1-(2-пиримидил)пиперазина в качестве исходных веществ, с получением целевого соединения.
Бесцветные кристаллы, т.пл. 93-94°С, C18H28N4O MW 316, HR-ESIMS (режим положительных ионов): m/z 339,2155 [M+Na]+ (Рассчитано для C18H29N4ONa, 339,2161), 1H-ЯМР (400 МГц, CDCl3) δ: 0,88 (3H, т, J=7,2 Гц), 1,22-1,35 (8H, м), 1,47 (2H, м), 2,23 (2H, м), 3,64 (2H, уш.с), 3,76 (2H, уш.с), 3,82-3,88 (4H, м), 6,28 (1H, д, J=15,2 Гц), 6,54 (1H, т, J=4,9 Гц), 6,93 (1H, дт, J=15,2, 7,4 Гц), 8,33 (2H, д, J=4,9 Гц).
Пример 32
Получение 1-((Е)-2-деценоил)азепана [Соединение 32]
Те же процедуры, которые использовали в Примере 1, осуществляли с использованием (E)-2-деценовой кислоты и гексаметиленимина в качестве исходных веществ, с получением целевого соединения.
Бледно-коричневое маслянистое вещество, C16H29NO MW 251, HR-ESIMS (режим положительных ионов): m/z 274,2145 [M+Na] (Рассчитано для C16H29NONa, 274,2147), 1H-ЯМР (400 МГц, CDCl3) δ: 0,88 (3H, т, J=6,8 Гц), 1,24-1,33 (8H, м), 1,45 (2H, м), 1,53-1,60 (4H, м), 1,69-1,77 (4H, м), 2,20 (2H, м), 3,50 (2H, т, J=6,0 Гц), 3,58 (2H, т, J=6,2 Гц), 6,22 (1H, д, J=15,6 Гц), 6,90 (1H, дт, J=15,6, 7,4 Гц).
Пример 33
Получение 1-((Е)-2-деценоил)-4-метил-[1,4]диазепана [Соединение 33]
Те же процедуры, которые использовали в Примере 2, осуществляли с использованием (E)-2-деценовой кислоты и 1-метил-1,4-диазепана в качестве исходных веществ, с получением целевого соединения.
Бледно-желтое маслянистое вещество, C16H30N2O MW 266,4, EIMS: m/z 267 [M+H]+, 1H-ЯМР (500 МГц, ДМСО-d6) δ: 0,86 (т, J=7,0 Гц, 3H), 1,19-1,31 (м, 8H), 1,36-1,45 (м, 2H), 1,71-1,81 (м, 2H), 2,13-2,20 (м, 2H), 2,23 и 2,24 (с 2, 3H), 2,39-2,55 (м, 4H), 3,46-3,58 (м, 4H), 6,34-6,40 (м, 1H), 6,61-6,69 (м, 1H).
Пример 34
Получение 1-((Е)-2-деценоил)-4-(2-диметиламиноэтил)-[1,4]диазепана [Соединение 34]
Те же процедуры, которые использовали в Примере 2, осуществляли с использованием (E)-2-деценовой кислоты и 1-(2-диметиламиноэтил)-1,4-диазепана в качестве исходных веществ, с получением целевого соединения.
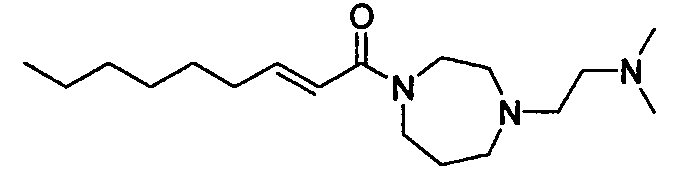
Бледно-желтое маслянистое вещество, C19H37N3O MW 323,5, EIMS: m/z 324 [M+H], 1H-ЯМР (500 МГц, ДМСО-d6) δ: 0,86 (т, J=7,0 Гц, 3H), 1,20-1,30 (м, 8H), 1,36-1,44 (м, 2H), 1,67-1,75 (м, 2H), 2,11 (с, 6H), 2,13-2,20 (м, 2H), 2,26-2,31 (м, 2H), 2,48-2,59 (м, 4H), 2,60-2,67 (м, 2H), 3,44-3,57 (м, 4H), 6,33-6,40 (м, 1H), 6,60-6,69 (м, 1H).
Пример 35
Получение 2-(1-((Е)-2-деценоил)-4-пиперидил)уксусной кислоты [Соединение 35]
(1) Те же процедуры, которые использовали в Примере 2, осуществляли с использованием (E)-2-деценовой кислоты и этил 2-(4-пиперидил)ацетат гидрохлорида в качестве исходных веществ, с получением этил 2-(1-((E)-2-деценоил)-4-пиперидил)ацетата (этиловый эфир соединения 35).
(2) Этил 2-(1-((Е)-2-деценоил)-4-пиперидил)ацетат подвергали щелочному омылению таким же образом, как в Примере 3, с получением целевого соединения.
Белые кристаллы, т.пл. 65-66°С, C17H29NO3 MW 295,4, EIMS: m/z 296 [M+H]+, 1H-ЯМР (500 МГц, ДМСО-d6) δ: 0,87 (т, J=7,1 Гц, 3H), 0,96-1,10 (м, 2H), 1,20-1,31 (м, 8H), 1,36-1,43 (м, 2H), 1,63-1,72 (м, 2H), 1,85-1,94 (м, 1H), 2,11-2,18 (м, 4H), 2,53-2,62 (м, 1H), 2,95-3,05 (м, 1H), 3,98-4,05 (м, 1H), 4,32-4,41 (м, 1H), 6,43 (д, J=15,0 Гц, IG), 6,61 (дт, Jd=15,0 Гц, Jt=7,0 Гц, 1H).
Пример 36
Получение 3-(1-((Е)-2-деценоил)-4-пиперидил)пропановой кислоты [Соединение 36]
(1) Те же процедуры, которые использовали в Примере 2, осуществляли с использованием (E)-2-деценовой кислоты и этил 3-(4-пиперидил)пропионат гидрохлорида в качестве исходных веществ, с получением этил 3-(l-((Е)-2-деценоил)-4-пиперидил)пропионата (этиловый эфир соединения 36).
(2) Этил 3-(1-((Е)-2-деценоил)-4-пиперидил)пропионат подвергали щелочному омылению таким же образом, как в Примере 3, с получением целевого соединения.
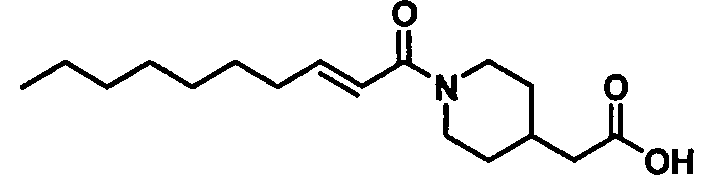
Бесцветное маслянистое вещество, C18H31NO3 MW 309,4, EIMS: m/z 310 [M+H]+, 1H-ЯМР (500 МГц, ДМСО-d6) δ: 0,86 (т, J=6,5 Гц, 3H), 0,90-1,02 (м, 2H), 1,20-1,31 (м, 8H), 1,36-1,51 (м, 5H), 1,63-1,71 (м, 2H), 2,13-2,18 (м, 2H), 2,23 (т, J=7,5 Гц, 2H), 2,50-2,58 (м, 1H), 2,90-3,00 (м, 1H), 3,98-4,06 (м, 1H), 4,35-4,43 (м, 1H), 6,43 (д, J=15,0 Гц, 1H), 6,61 (дт, Jd=15,0 Гц, Jt=7,0 Гц, 1H), 12,03 (уш., 1H).
Пример 37
Получение 1-((E)-2-деценоил)-4-цианопиперидина (1-((E)-дец-2-еноил)-4-цианопиперидина) [Соединение 37]
Те же процедуры, которые использовали в Примере 2, осуществляли с использованием (E)-2-деценовой кислоты и пиперидин-4-карбонитрила в качестве исходных веществ, с получением целевого соединения.
Бесцветное маслянистое вещество, C16H26N2O MW 262,4, EIMS: m/z 263 [M+H]+, 1H-ЯМР (500 МГц, ДМСО-d6) δ: 0,87 (т, J=6,5 Гц, 3H), 1,20-1,23 (м, 8H), 1,37-1,43 (м, 2H), 1,56-1,69 (м, 2H), 1,81-1,90 (м, 2H), 2,14-2,18 (м, 2H), 3,08-3,14 (м, 1H), 3,20-3,28 (м, 1H), 3,35-3,43 (м, 1H), 3,70-3,83 (м, 2H), 6,45 (д, J=15,0 Гц, 1H), 6,65 (дт, Jd=15,0 Гц, Jt=7,0 Гц, 1Н).
Пример 38
Получение 1-((Е)-2-деценоил)-4-(4-хлорфенил)пиперидина (1-((Е)-дец-2-еноил)-4-(4-хлорфенил)пиперидина) [Соединение 38]
Те же процедуры, которые использовали в Примере 2, осуществляли с использованием (E)-2-деценовой кислоты и 4-(4-хлорфенил)пиперидина в качестве исходных веществ, с получением целевого соединения.
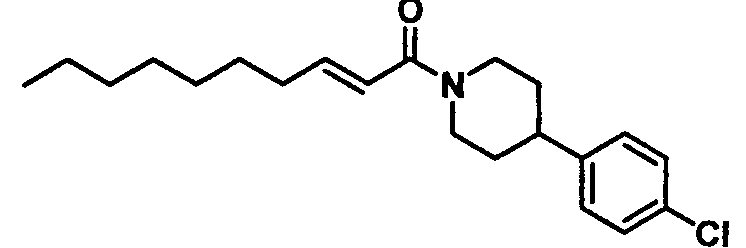
Бледно-желтое маслянистое вещество, C21H30ClNO MW 347,9, EIMS: m/z 348 [M+H]+, 1H-ЯМР (500 МГц, ДМСО-d6) δ: 0,86 (т, J=7,0 Гц, 3H), 1,22-1,32 (м, 8H), 1,32-1,54 (м, 4H), 1,74-1,83 (м, 2H), 2,15-2,21 (м, 2H), 2,26-2,69 (м, 1H), 2,75-2,83 (м, 1H), 3,05-3,15 (м, 1H), 4,12-4,21 (м, 1H), 4,54-4,62 (м, 1H), 6,48 (д, J=15,0 Гц, 1H), 6,65 (дт, Jd=15,0 Гц, Jt=7,0 Гц, 1H), 7,27 (д, J=8,5 Гц, 2H), 7,34 (д, J=8,5 Гц, 2H).
Пример 39
Получение гидрохлорида [4-[(Е]-2-деценоил]пиперазин-1-ил]уксусной кислоты
[Соединение 39]
(1) Те же процедуры, которые использовали в Примере 2, осуществляли с использованием (Е)-2-деценовой кислоты и этил 2-(1-пиперазинил)ацетат дигидрохлорида в качестве исходных веществ, с получением этил [4-[(Е)-2-деценоил)пиперазин-1-ил)ацетата (этиловый эфир свободной формы соединения 39).
(2) Этил [4-[(Е)-2-деценоил)пиперазин-1-ил)ацетат подвергали щелочному омылению таким же образом, как в Примере 3, с получением [4-[(Е)-2-деценоил)пиперазин-1-ил]уксусной кислоты (свободная форма соединения 39).
Полученное соединение растворяли в метиленхлориде и затем обрабатывали смесью хлористый водород-диоксан, с получением целевого соединения.
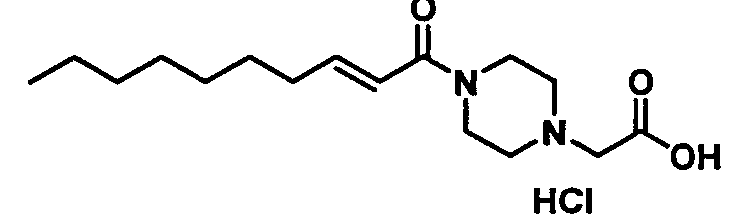
Белые кристаллы, т.пл. 195°С (разложение), C16H29ClN2O3 MW 332,9, EIMS: m/z 297 [M+H]+, 1H-ЯМР (500 МГц, ДМСО-d6) δ: 0,87 (т, J=7,0 Гц, 3Н), 1,20-1,33 (м, 8Н), 1,38-1,46 (м, 2Н), 2,15-2,21 (м, 2Н), 3,20-3,53 (м, 4Н), 3,70-4,10 (м, 4Н), 4,12 (с, 2Н), 6,48 (д, J=15,0 Гц, 6,73 (дт, Jd=15,0 Гц, Jt=7,0 Гц, 1Н).
Пример 40
Получение гидрохлорида 4-[4-[(Е)-2-деценоил)пиперазин-1-ил]бутановой кислоты
[Соединение 40]
(1) Те же процедуры, которые использовали в Примере 2, осуществляли с использованием (Е)-2-деценовой кислоты и этил 4-(1-пиперазинил)бутират дигидрохлорида в качестве исходных веществ, с получением этил 4-[4-((Е)-2-деценоил)пиперазин-1-ил)бутаноата (этиловый эфир свободной формы соединения 40).
(2) Этил 4-[4-((Е)-2-деценоил)пиперазин-1-ил)бутаноат подвергали щелочному омылению таким же образом, как в Примере 3, с получением 4-[4-[(Е)-2-деценоил)пиперазин-1-ил]бутановой кислоты (свободная форма соединения 40).
(3) Полученное соединение растворяли в метиленхлориде и затем обрабатывали смесью хлористый водород-диоксан, с получением целевого соединения.
Белые кристаллы, т.пл. 203-205°С, C18H33ClN2O3 MW 360,9, EIMS: m/z 325 [М+Н]+, 1H-ЯМР (500 МГц, ДМСО-d6) δ: 0,86 (т, J=7,0 Гц, 3Н), 1,20-1,31 (м, 8Н), 1,35-1,43 (м, 2Н), 1,61-1,69 (м, 2Н), 2,13-2,18 (м, 2Н), 2,23 (т, J=7,2 Гц, 2Н), 2,26-2,35 (м, 6Н), 3,45-3,55 (м, 4Н), 6,43 (д, J=15,0 Гц, 1Н), 6,64 (дт, Jd=15,0 Гц, Jt=7,0 Гц, 1Н).
Пример 41
Получение 1-((Е)-2-деценоил)-4-(4-хлорфенил)пиперазин [Соединение 41]
Те же процедуры, которые использовали в Примере 2, осуществляли с использованием (Е)-2-деценовой кислоты и 1-(4-хлорфенил)пиперазина в качестве исходных веществ, с получением целевого соединения.
Бледно-желтое маслянистое вещество, C20H29ClN2O MW 348,9, EIMS: m/z 348 [M]+, 1Н-ЯМР (500 МГц, ДМСО-d6) δ: 0,86 (т, J=6,5 Гц, 3Н), 1,20-1,31 (м, 8Н), 1,37-1,45 (м, 2Н), 2,15-2,22 (м, 2Н), 3,07-3,18 (м, 4Н), 3,60-3,70 (м, 4Н), 6,51 (д, J=15,0 Гц, 1Н), 6,70 (дт, Jd=15,0 Гц, Jt=7,0 Гц, 1Н), 6,90 (д, J=9,0 Гц, 2Н), 7,25 (J=9,0 Гц, 2Н).
Пример 42
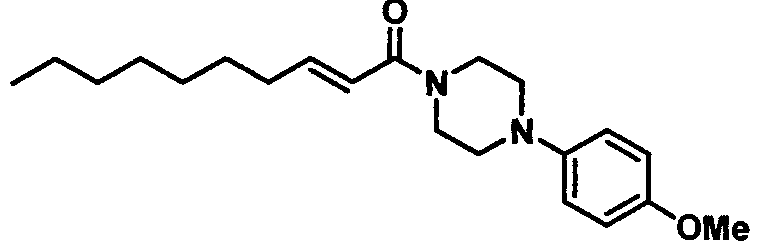
Получение 1-((Е)-2-деценоил)-4-(4-метоксифенил)пиперазина [Соединение 42]
Те же процедуры, которые использовали в Примере 2, осуществляли с использованием (Е)-2-деценовой кислоты и 1-(4-метоксифенил)пиперазина в качестве исходных веществ, с получением целевого соединения.
Белые кристаллы, т.пл. 50-52°С, C21H32N2O2 MW 344,5, EIMS: m/z 344 [M]+, 1Н-ЯМР (500 МГц, ДМСО-dg) δ: 0,86 (т, J=6,8 Гц, 3Н), 1,20-1,31 (м, 8Н), 1,38-1,45 (м, 2Н), 2,15-2,21 (м, 2Н), 2,94-3,01 (м, 4Н), 3,60-3,67 (м, 4Н), 3,68 (с, 3Н), 6,50 (д, J=15,0 Гц, 1Н), 6,68 (дт, Jd=15,0 Гц, Jt=7,0 Гц, 1Н), 6,83 (д, J=9,l Гц, 2Н), 6,91 (д, J=9,l Гц).
Пример 43
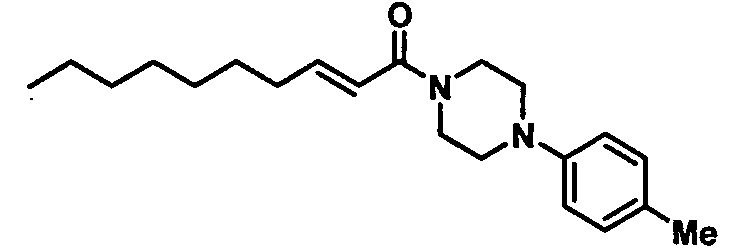
Получение 1-((Е)-2-деценоил)-4-(4-метилфенил)пиперазина [Соединение 43]
Те же процедуры, которые использовали в Примере 2, осуществляли с использованием (Е)-2-деценовой кислоты и 1-(пара-толил)пиперазина в качестве исходных веществ, с получением целевого соединения.
Белые кристаллы, т.пл. 45-46°С, C21H32N2O MW 328,5, EIMS: m/z 328 [М]+, 1Н-ЯМР (500 МГц, ДМСО-d6) δ: 0,85 (т, J=6,9 Гц, 3Н), 1,20-1,30 (м, 8Н), 1,36-1,45 (м, 2Н), 2,20 (с, 3Н), 2,15-2,23 (м, 2Н), 3,00-3,08 (м, 4Н), 3,60-3,70 (м, 4Н), 6,50 (д, J=15,0 Гц, 1Н), 6,69 (дт, Jd=15,0 Гц, Jt=7,0 Гц, 1Н), 6,85 (д, J=8,5 Гц, 2Н), 7,03 (д, J=8,5 Гц, 2Н).
Пример 44
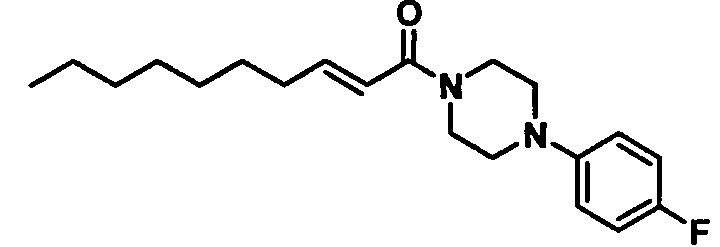
Получение 1-((Е)-2-деценоил)-4-(4-фторфенил)пиперазина [Соединение 44]
Те же процедуры, которые использовали в Примере 2, осуществляли с использованием (Е)-2-деценовой кислоты и 1-(4-фторфенил)пиперазина в качестве исходных веществ, с получением целевого соединения.
Бледно-желтое маслянистое вещество, C20H29FN2O MW 332,5, EIMS: m/z 332 [M]+, 1H-ЯМР (500 МГц, ДМСО-d6) δ: 0,86 (т, J=6,5 Гц, 3Н), 1,20-1,31 (м, 8Н), 1,37-1,45 (м, 2Н), 2,16-2,22 (м, 2Н), 3,00-3,09 (м, 4Н), 3,62-3,72 (м, 4Н), 6,50 (д, J=15,0 Гц, 1Н), 6,69 (дт, Jd=15,0 Гц, Jt=7,0 Гц, 1Н), 6,95-7,00 (м, 2Н), 7,03-7,09 (м, 2Н).
Пример 45
Получение 1-((Е)-2-деценоил)-4-(2-хлорфенил)пиперазина
[Соединение 45]
Те же процедуры, которые использовали в Примере 2, осуществляли с использованием (Е)-2-деценовой кислоты и 1-(2-хлорфенил)пиперазина в качестве исходных веществ, с получением целевого соединения.
Бледно-желтое маслянистое вещество, C20H29ClN2O MW 348,9, EIMS: m/z 348 [M]+, 1H-ЯМР (500 МГц, ДМСО-d6) δ: 0,86 (т, J=6,5 Гц, 3Н), 1,21-1,31 (м, 8Н), 1,38-1,45 (м, 2Н), 2,15-2,21 (м, 2Н), 2,90-2,98 (м, 4Н), 3,65-3,75 (м, 4Н), 6,50 (д, J=15,0 Гц, 1Н), 6,70 (дт, Jd=15,0 Гц, Jt=7,0 Гц, 1Н), 7,04-7,08 (м, 1Н), 7,13-7,17 (м, 1Н), 7,28-7,32 (м, 1Н), 7,41-7,45 (м, 1Н).
Пример 46
Получение 1-((Е)-2-деценоил)-4-(4-гидроксифенил)пиперазина [Соединение 46]
Те же процедуры, которые использовали в Примере 2, осуществляли с использованием (Е)-2-деценовой кислоты и 4-(1-пиперазинил)фенола в качестве исходных веществ, с получением целевого соединения.
Бледно-красные кристаллы, т.пл. 85-87°С, C20H30N2O2 MW 330,5, EIMS: m/z 330 [М+Н]+ 1Н-ЯМР (500 МГц, ДМСО-d6) δ: 0,86 (т, J=7,0 Гц, 3Н), 1,20-1,30 (м, 8Н), 1,38-1,40 (м, 2Н), 2,15-2,21 (м, 2Н), 2,87-2,96 (м, 4Н), 3,59-3,69 (м, 4Н), 6,49 (д, J=15,0 Гц, 1Н), 6,63-6,71 (м, 3Н), 6,80 (д, J=6,8 Гц, 2Н), 8,88 (с, 1Н).
Пример 47
Получение 2-[4-[4-((Е) -2-деценоил) пиперазин-1-ил]фенокси]уксусной кислоты [Соединение 47]
(1) Те же процедуры, которые использовали в Примере 2, осуществляли с использованием (Е)-2-деценовой кислоты и этил 2-[4-(1-пиперазинил)фенокси]ацетат дигидрохлорида в качестве исходных веществ, с получением этил 2-[4-[4-((Е)-2-деценоил)пиперазин-1-ил]фенокси]ацетата (этиловый эфир соединения 47).
(2) Этил 2-[4-[4-((Е)-2-деценоил)пиперазин-1-ил]фенокси]ацетат подвергали щелочному омылению таким же образом, как в Примере 3, с получением целевого соединения.
Белые кристаллы, т.пл. 210°С (разложение), C22H32N2O4 MW 388,5, EIMS: m/z 388 [M]+, 1Н-ЯМР (500 МГц, ДМСО-d6) δ: 0,86 (т, J=7,0 Гц, 3Н), 1,21-1,32 (м, 8Н), 1,37-1,45 (м, 2Н), 2,15-2,20 (м, 2Н), 2,91-3,00 (м, 4Н), 3,60-3,71 (м, 4Н), 6,50 (д, J=15,0 Гц, 1Н), 6,68 (дт, Jd=15,0 Гц, Jt=7,0 Гц, 1Н), 6,76 (д, J=7,1 Гц, 2Н), 6,87 (д, J=7,1 Гц, 2Н).
Пример 48
Получение 4-[[4-((Е)-2-деценоил)пиперазин-1-ил]метил]бензойной кислоты [Соединение 48]
(1) Те же процедуры, которые использовали в Примере 2, осуществляли с использованием (Е)-2-деценовой кислоты и метил 4-[1-пиперазинилметил)бензоат дигидрохлорида в качестве исходных веществ, с получением метил 4-[[4-((Е)-2-деценоил)пиперазин-1-ил]метил]бензоата (метиловый эфир соединения 48).
(2) Метил 4-[[4-((Е)-2-деценоил)пиперазин-1-ил]метил]бензоат подвергали щелочному омылению таким же образом, как в Примере 3, с получением целевого соединения.
Белые кристаллы, т.пл. 114-116°С, C22H32N2O3 MW 372,5, EIMS: m/z 373 [М+Н]+, 1Н-ЯМР (500 МГц, ДМСО-d6) δ: 0,86 (т, J=7,0 Гц, 3Н), 1,20-1, 30 (м, 8Н), 1,35-1,43 (м, 2Н), 2,12-2,18 (м, 2Н), 2,30-2,38 (м, 4Н), 3,45-3, 55 (м, 4Н), 3,55 (с, sH), 6,43 (д, J=15,0 Гц, 1Н), 6,64 (дт, Jd=15,0 Гц, Jt=7,0 Гц, 1Н), 7,44 (д, J=8,2 Гц, 2Н), 12,8 (уш., 1Н),
Пример 4 9
Получение 4-[(Е)-2-деценоил]морфолин-3-карбоновой кислоты [Соединение 49]
(1) Те же процедуры, которые использовали в Примере 2, осуществляли с использованием (Е)-2-деценовой кислоты и метил морфолин-3-карбоксилат гидрохлорида в качестве исходных веществ, с получением метил 4-[(Е)-2-деценоил)морфолин-3-карбоксилата (метиловый эфир соединения 49).
(2) Метил 4-[(Е)-2-деценоил)морфолин-3-карбоксилат подвергали щелочному омылению таким же образом, как в Примере 3, с получением целевого соединения.
Бледно-желтое маслянистое вещество, C15H25NO4 MW 283,4, EIMS: m/z 284 [M+H]+, 1H-ЯМР (500 МГц, CDCl3) δ: 0,88 (т, J=7,0 Гц, 3Н), 1,22-1,35 (м, 8Н), 1,40-1, 50 (м, 2Н), 2,17-2,26 (м, 2Н), 3,10-4,00 (м, 5Н), 4,45-4,40 и 5,15-5,18 (м, 2Н), 6,05 и
6,23 (д × 2, J=15,0 Гц, 1H), 6,88-7,02 (м, 1H), 9,00 (уш., 1H).
Пример испытания 1 (Оценка действия против паклитаксел-индуцированного расстройства периферической нервной системы у крыс)
Эффекты соединений по настоящему изобретению оценивали в отношении расстройства периферической нервной системы, которое является побочным эффектом, вызванным введением противоракового средства паклитаксела, таким как гиперестезия, включая аллодинию, индуцируемую механическими стимулами, которая представляет собой сильную боль, индуцируемую такими тактильными стимулами, которые, как правило, не вызывают боли. Соединение по настоящему изобретению вводили интраперитонеально крысам в качестве испытываемого лекарственного средства для осуществления тест фон Фрея.
(1) Подготовка крыс с паклитаксел-индуцированным расстройством периферической нервной системы и введение исследуемых лекарственных средств
Самцов крыс линии SD шестинедельного возраста по шесть крыс в группе) использовали в качестве экспериментальных животных, и паклитаксел (2 мг/кг) вводили интраперитонеально через день четыре раза в общей сложности для подготовки крыс с паклитаксел-индуцированным расстройством периферической нервной системы. В течение периода от 18 до 25 дней или в течение периода от 20 до 27 дней после начала введения паклитаксела исследуемое лекарственное средство вводили интраперитонеально в виде однократной дозы 300 мкг/кг, и осуществляли следующий тест фон Фрея.
(2) Тест фон Фрея
Крыс со стадии (1) выше помещали в прозрачную акриловую клетку с полом, представляющим собой проволочную решетку, и приучали в течение примерно трех минут и измеряли 50% порог реакции к механическим стимулам правой задней конечности перед введением испытываемого лекарственного средства и через 1 и 5 часов после начала введения.
Измерение осуществляли с использованием нити фон Фрея (производства North Coast Medical Inc.) в соответствии с методами Chaplan, et al. (Journal of Neuroscience Methods, 53(1), 55-63, 1994) и Dixon, et al. (Annual Review of Pharmacology and Toxicology, 20, 441-462, 1980). Из восьми нитей [нагрузки стимула (г): 0,4, 0,6, 1,0, 2,0,4,0, 6,0, 8,0 и 15,0], испытание начинали с нити с нагрузкой в 2,0 г, нить вертикально прилагали к подошве на 2-3 секунды с такой силой, чтобы нить была слегка изогнута, и случай, когда задние конечности показали реакцию избегания, называли положительной реакцией. Случай, когда крыса демонстрировала избегание при удалении нити, также называли положительным. Когда обнаруживали положительную реакцию, прилагали стимул аналогичным образом с использованием нити одним рангом слабее, и при отсутствии реакции, прилагали стимул аналогичным образом с нитью одним рангом сильнее, и точку, в которой реакция изменялась с отрицательной на положительную или с положительной на отрицательную, называли первыми двумя реакциями. Затем стимулы применяли непрерывно четыре раза тем же способом, вверх-вниз. 50% порог реакции к механическим стимулам измеряли с использованием реакции на шесть стимулов в общей сложности и затем рассчитывали (среднее значение)±(стандартная ошибка) для каждой группы. В этой связи, когда стимул достигал нагрузки в 15,0 г без положительной реакции, или когда положительная реакция продолжалась до нагрузки 0,4 г, 15,0 г или 0,25 г принимали в качестве каждого из 50% порогов реакции. Что касается более высокого значения 50% порога реакции между 50% пороговыми значениями через 1 час и 5 часов после введения испытываемого лекарственного средства, скорость восстановления (%) 50% порога реакции рассчитывали при помощи следующего выражения, где 15 принимали в качестве нормального порогового значения. Один пример приведенных выше результатов испытаний показан в Таблицах 6 и 7.
Скорость восстановления (%) значения 50% порога реакции = [(50% порог реакции через 1 час и 5 часов после введения испытываемого лекарственного средства)-(50% порог реакции до введения испытываемого лекарственного средства)]/[(нормальное пороговое значение)-(50% порог реакции до введения испытываемого лекарственного средства)]×100.
Таблицы 6 и 7 показывают, что соединения по настоящему изобретению проявляют отличный эффект улучшения состояния при гиперестезии, вызванной введением паклитаксела и, таким образом, обладают действием облегчения побочных эффектов, вызванных введением противоракового средства. Кроме того, соединения по настоящему изобретению также показывают превосходное действие по облегчению гиперестезии, когда оксалиплатин из платиновых лекарственных средств используют таким же образом, как используют паклитаксел из таксановых лекарственных средств.
Пример испытания 2 [Оценка активации (фосфорилирования) MAP киназы)
Что касается соединений по настоящему изобретению, активацию MAP киназы (ERK 1/2) измеряли в соответствии с вестерн-иммуноблоттингом следующим образом.
Нервные клетки диспергировали из коры головного мозга плода крысы 17 дневного возраста, и нервные клетки культивировали в течение одного дня в модифицированной Дульбекко среде Игла (DMEM), содержащей 5% фетальную бычью сыворотку. Культуральную среду заменяли бессывороточной средой (B27 дополненная Neurobasal; Invitrogen) и нервные клетки культивировали при плотности 20000-40000 клеток/см2 в покрытой полиорнитином чашке для культивирования.
Через три дня добавляли соединение по настоящему изобретению и культивирование продолжали в течение 30 минут. После этого клетки извлекали на лед с использованием раствора, содержащего ингибитор фосфатазы, где в качестве основания использовали буфер Tris-HCl. Концентрацию белка в полученном клеточном экстракте определяли количественно при помощи BCA набора для определения количества белка (Takara Bio Co., Ltd.), и некоторое количество белка (3 мкг для измерения MAP киназы и 5 мкг для измерения фосфорилированной MAP киназы) подвергали электрофорезу в полиакриламидном геле. Белок переносили из геля для электрофореза на PVDF мембрану и осуществляли вестерн-иммуноблоттинг с импользованием каждого из анти-MAP киназа антитела (Cell Signaling Technology) и анти-фосфорилированная MAP киназа антитела (Cell Signaling Technology) первичного антитела.
Затем осуществляли реакцию с меченным щелочной фосфатазой анти-кроличьим IgG антителом (Promega) вторичного антитела, и в результате ферментативной активности проявлялся цвет, что давало возможность осуществить измерения MAP киназы (MAPK) и фосфорилированной MAP киназы (pMAPK).
В этой связи, каждое из соединений по настоящему изобретению растворяли в 0,1% ДМСО для доведения его концентрации до 250 мкг/мл. В качестве контроля добавляли 0,1% раствор ДМСО.
Интенсивность концентрации полос геля для электрофореза рассчитывали и количественно оценивали с использованием Image J (K.K. Bioarts). Каждое из численных значений для MAPK при использовании соединений по настоящему изобретению делили на численное значение для MAPK контроля, и численное значение для pMAPK при использовании соединений по настоящему изобретению делили на численное значение для pMAPK контроля, таким образом получали отношение МАПК при использовании каждого из соединений по настоящему изобретению к контролю и отношение pMAPK при использовании каждого из соединений по настоящему изобретению к контролю. Далее полученное отношение pMAPK к контролю делили на отношение MAPK к контролю с получением отношения pMAPK к MAPK. Один пример результатов показан в Таблицах 8-10.
Таблицы 8-10 показывают, что соединения по настоящему изобретению демонстрируют высокую активацию (фосфорилирование) MAP киназы по сравнению с контролем, что дает основание предположить, что они обладают нейротрофической фактор-подобной активностью.
Пример испытания 3 Оценка активации (фосфорилирования) MAP киназы при легких стрессовых нагрузках
Что касается соединений по настоящему изобретению, уровень активации MAP киназы (ERK1/2) измеряли с использованием мышиной модели хронической индуцированной легким стрессом депрессии следующим образом.
(1) Подготовка мышиной модели хронической индуцированной легким стрессом депрессии и введение испытываемого лекарственного средства
Самой мышей ddY семинедельного возраста (n=8-12) подвергали следующим процедурам: (A) Мышей подвергали принудительному плаванию в течение 15 минут и затем условиям нормального разведения в течение двух дней. (B) Мышей содержали в клетке с наклоном в течение двух дней и затем в нормальных условиях в течение одного дня. (C) Мышей содержали в течение одного дня в клетке, коврик на полу которой смачивали, и затем в нормальных условиях в течение одного дня. (D) Мышей содержали в течение одного дня клетке, вращающейся со скоростью 180 оборотов/минута, и затем в нормальных условиях в течение одного дня. Далее, после осуществления одного цикла (A)-(D), (B)-(D) повторяли в течение двух циклов. Таким образом, стресс прилагали в течение трех недель в общей сложности для подготовки мышиной модели хронической индуцированной легким стрессом депрессии. В течение этого времени каждое из соединений по настоящему изобретению, растворенное в PBS или в PBS растворителе, содержащем ДМСО, и т.д., вводили перорально один раз в день в течение трех недель.
(2) Измерения MAP киназы и фосфорилированной MAP киназы
После завершения поведенческих анализов после вышеуказанных хронических легких стрессовых нагрузок (испытание с подвешиванием за хвост и испытание свет-темнота) гиппокамп вырезали из головного мозга. Гиппокамп и дезинтегрирующий раствор (20 мМ Tris-HCl буфера (pH 7,4): RIPA буфер, содержащий 1% Nonidet P40 [зарегистрированный товарный знак], 1% дезоксихолата натрия, 2 мМ EDTA, 0,1% додецилсульфата натрия (SDS), 0,15 MNaCl, 10 мг/мл апротинина, 10 мг/мл лейпептина, 50 мМ NaF, 1 мМ ортованадата натрия, 1 мМ фенилметилсульфонилфторида (PMSF)) в количестве в 19 раз большем сырого веса гиппокампа помещали в микропробирку (1,5 мл) и смесь дезинтегрировали ультразвуком. После этого, микропробирку оставляли на льду в течение 30 минут, центрифугировали при 1400×g в течение 15 минут с получением супернатанта в виде белкового экстракта. К белковому экстракту добавляли буфер для образца для электрофореза (0,2 M буфера Tris-HCl (pH 7,2), 8% SDS, 40% глицерина, бромфенол голубой (BPB)) в количестве 1/3 от количества белкового экстракта, и 2-меркаптоэтанол в количестве 1/10 от количества белкового экстракта. Смесь подвергали термообработке при 95C в течение 5 минут и подвергали SDS электрофорезу с 10% полиакриламидным гелем.
Каждую из MAP киназ (МАПК) и фосфорилированных MAP киназ (pMAPK) подвергали иммуноблот-анализу, и 2 мкг или 5 мкг каждого из экстрагированных на ткани белков подвергали электрофорезу. После этого белок переносили из геля на PVDF мембрану. PVDF мембрану блокировали раствором, содержащим 5% снятое молоко, и давали возможность прореагировать с каждым из 1000-кратных разведений анти-MAPK антитела и анти-pMAPK антитела в течение ночи при 4°C. Далее, реакционной смеси давали прореагировать с вторичным антителом (меченным щелочной фосфатазой анти-мышиным IgG антителом (5000-кратное разведение)), и в конце инкубировали в растворе с добавлением субстрата щелочной фосфатазы в течение нескольких минут для проявления цвета. Интенсивность окраски каждой полоски оценивали количественно и получали отношение pMAPK к MAPK (n=4-7). В данном случае, критерий значимости определяли в соответствии с критерием Даннетта.
Как результат описанного выше теста, отношение pMAPK к MAPK контрольной группы нормальных животных составило 1,26±0,11, и то же самое отношение для контрольной группы животных с мягкой стрессовой нагрузкой было значительно снижено до 0,56±0,04. Те же отношения в группе, которой вводили соединения по настоящему изобретению, были такими, что группа введения Соединения 10 (1000 мкг/кг) имела значение 1,00±0,04, а группа введения Соединения 12 (500 мкг/кг) имела значение 1,09±0,04, тем самым показывая, что соединения по настоящему изобретению значительно увеличивали фосфорилирование MAPK, которое было снижено в контрольной группе животных с мягкой стрессовой нагрузкой, и подтверждая нейротрофическую фактор-подобную активность.
Пример испытания 4 Оценка активации (фосфорилирования) CREB при легких нагрузках
В отношении соединений по настоящему изобретению, уровень активации фактора транскрипции CREB (отношение фосфорилированного CREB (pCREB) к CREB), который играет ключевую роль в функциях нервных клеток и способностях к запоминанию и обучению, путь передачи сигналов которого находится ниже от MAPK (ERK 1/2), измеряли с использованием мышиной модели хронической индуцированной легким стрессом депрессии таким же способом, как в описанном выше Примере испытания 3, в соответствии с вестерн-блоттинг анализом (n=3-8). Однако первичным антителам (анти-CREB антитело и анти-pCREB антитело) давали прореагировать в 1000-кратных разведениях, и вторичным антителам давали прореагировать в 10000-кратных разведениях.
Как результат описанного выше испытания, отношение pCREB к CREB контрольной группы нормальных животных составило 0,95±0,09, и то же самое отношение для животных контрольной группы с мягкой стрессовой нагрузкой было значительно снижено до 0,50±0,07. Те же отношения в группе введения соединений по настоящему изобретению были такими, что группа введения Соединения 10 (1,000 мкг/кг) имела значение 0,94±0,10, а группа введения Соединения 12 (500 мкг/кг) имела значение 1,08±0,14, тем самым показывая, что соединения по настоящему изобретению значительно увеличивали отношение pCREB, которое было снижено в контрольной группе животных с легкой стрессовой нагрузкой, таким же образом, как и в случае фосфорилирования MAPK, и подтверждая нейротрофическую фактор-подобную активность.
Пример испытания 5 Оценка эффектов подавления симптомов депрессии, сопровождающих легкие стрессовые нагрузки
Соединения по настоящему изобретению вводили в качестве испытываемого лекарственного средства мышиной модели хронической индуцированной легким стрессом депрессии из описанного выше Примера испытаний 3(1), и используемых в качестве модели мышей затем подвергали испытанию подвешивания за хвост для оценки эффектов подавления симптомов депрессии. Конкретно, мышей крепко удерживали рукой на расстоянии 1 см от кончика хвоста мыши и держали на высоте 10 см от пола. Наблюдения осуществляли в течение 6 минут и измеряли продолжительность периода неподвижности, служащую в качестве показателя симптомов депрессии. В данном случае, критерий значимости определяли в соответствии с критерием Даннетта.
Как результат описанного выше испытания подвешивания за хвост, период неподвижности для контрольной группы нормальных животных составил 73,16±9,20 секунд, а период неподвижности для подверженных мягкому стрессу контрольных животных был значительно продлен до 157,01±11,97 секунд. Период неподвижности группы введения Соединения 12 по настоящему изобретению в дозе 500 мкг/кг составил 69,26±16,41 секунд, тем самым ярко демонстрируя действие подавления симптомов депрессии. Кроме того, период неподвижности группы введения Соединения 10 по настоящему изобретению в дозе 1000 мкг/кг составил 96,03±25,24 секунд, таким образом, было обнаружено некоторое подавление симптомов депрессии.
Пример испытания 6 Оценка эффектов подавления симптомов тревоги, сопровождающей легкие стрессовые нагрузки
Соединения по настоящему изобретению вводили в качестве испытываемого лекарственного средства мышиной модели хронической индуцированной легким стрессом депрессии из указанного выше Примера испытаний 3(1), и используемых в качестве модели мышей затем подвергали испытанию свет-темнота для оценки эффекты подавления симптомов тревоги. В частности, использовали деревянный прямоугольный аппарат, состоящий из светлой камеры и темной камеры, имеющих высоту 50 см, длину 50 см, ширину 25 см, мышь помещали в темную камеру и давали возможность свободного поиска, и затем количество раз забегания в светлую камеру и время пребывания в светлой камере записывали в течение 5 минут. Это испытание представляет собой испытание для оценки уровня беспокойства у мыши, используя такую характерную особенность мышей, как тенденция выбирать темные места с меньшей вероятностью оставаться в светлых местах, но с вероятностью проникновения в светлую камеру из-за поискового поведения из любопытства. В данном случае, критерий значимости определяли в соответствии с критерием Даннетта.
Как результат описанного выше испытания свет-темнота, в отличие от времени пребывания в светлой камере контрольной группы нормальных животных 110,74±5,29 секунд, время пребывания в светлой камере контрольной группы, подверженной легкому стрессу, было значительно снижено до 74,72±4,09 секунд. Время пребывания в светлой камере группы введения Соединения 12 по настоящему изобретению в дозе 500 мкг/кг составило 108,70±12,06 секунд, демонстрируя значительные эффекты подавления тревожных симптомов. Кроме того, время пребывания в светлой камере группы введения Соединения 10 по настоящему изобретению в дозе 1000 мкг/кг, составило 107,05±8,29 секунд, таким образом, было обнаружено подавление симптомов тревоги.
Пример испытания 7 Оценка анальгетического действия против крысиной модели остеоартрита
Следующие эксперименты осуществляли для изучения анальгетического действия анальгетика по настоящему изобретению с использованием крыс с остеоартритом (OA), индуцированным монойодацетатом натрия (MIA), которые были модельными животными для ОА.
(1) Подготовка крыс с MIA-индуцированным ОА
Измеряли 50% порог реакции на механический стимул у самцов крыс Wistar шестинедельного возраста (метод измерения будет указан позже), и выбирали контрольную группу нормальных животных. MIA, полученный с физиологическим раствором, вводили в разовой дозе 300 мкг/50 мкл в правый коленный сустав крыс, за исключением контрольной группы нормальных животных, в то время как 50 мкл физиологического раствора вводили в левый коленный сустав, таким образом, получали крыс с MIA-индуцированным ОА. Нормальной контрольной группе вводили 50 мкл Физиологического раствора в суставы обоих колен.
(2) Распределение на группы
Что касается самцов крыс Wistar шестинедельного возраста, используемых в качестве экспериментальных животных, за исключением контрольной группы нормальных животных, их значения 50% порога реакции на механический стимул (метод измерения будет указан позже) и массу тела измеряли через 24 дня после введения MIA, указанного в (1), и затем были организованы 3 группы, то есть, контрольная группа нормальных животных, начальная контрольная группа и группа введения испытываемого лекарственного средства, по шесть крыс в группе.
(3) Введение испытываемого лекарственного средства
Раствор испытываемого лекарственного средства (100 мкг/мл) с использованием каждого из соединений по настоящему изобретению в качестве испытываемого лекарственного средства получали с использованием фосфатно-буферного физиологического раствора (PBS), содержащего 0,1 об. % диметилсульфоксида (ДМСО).
Сразу после разделения на группы (через 14 дней после введения MIA), раствор испытываемого лекарственного средства вводили интраперитонеально в виде однократной дозы 500 мкг/кг группе введения испытываемого лекарственного средства. Кроме того, в нормальной контрольной группе и начальной контрольной группе вводили PBS, содержащий 0,1 об. % ДМСО, интраперитонеально в виде однократной дозы.
(4) Результаты измерения значений 50% порога реакции на механический стимул (тест фон Фрея)
Тест фон Фрея осуществляли таким же образом, как и в Примере испытаний 1 (2), и была рассчитана скорость восстановления (%) значений 50% порога реакции против гипералгезии OA, индуцированного введением MIA. Один пример результатов показан в таблице 11.
Как видно из таблицы 11, соединения по настоящему изобретению показали супрессорные эффекты против гипералгезии OA, индуцированного введением MIA, тем самым подтверждая их полезность в качестве обезболивающего средства.
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
Как видно из вышеуказанных результатов фармакологических испытаний, было обнаружено, что соединения по настоящему изобретению обладают превосходными терапевтическими эффектами против вызываемой механическим стимулом гипералгезии у крыс, индуцированной введением противоракового средства. В этой связи, соединения по настоящему изобретению являются эффективными в качестве средства для профилактики или лечения расстройств периферической нервной системы, индуцированных противораковыми средствами, таких как парестезия, такая как онемение конечностей человека и животных, и гипералгезия, такая как боли, таким образом, соединения являются чрезвычайно полезными.
Кроме того, при оценках с использованием культивированных нервных клеток коры головного мозга крыс и гиппокампа мышей мышиной модели хронической индуцированной легким стрессом депрессии, соединения показали превосходное фосфорилирующее действие в отношении MAP киназы и фосфорилирующее действие в отношении CREB (нейротрофическая фактор-подобная активность). Кроме того, в различных испытаниях с использованием мышиной модели хронической индуцированной легким стрессом депрессии были показаны действия улучшения симптомов депрессии или симптомов тревоги. Таким образом, ожидается, что соединения по настоящему изобретению будут полезными в качестве профилактического или терапевтического средства от деменции, болезни Альцгеймера, болезни Паркинсона, диабетической невропатии, депрессии, глаукомы, расстройства аутистического спектра или подобных, и восстанавливающего средства при повреждении спинного мозга.
Кроме того, соединения по настоящему изобретению обладают отличными обезболивающими действиями и эффектами подавления гипералгезии в экспериментах на животных с использованием крыс с MIA-индуцированным ОА, которые являются моделями OA. Поэтому соединения по настоящему изобретению являются чрезвычайно полезными в качестве средства для профилактики или лечения различных болевых синдромов, включая, например, боли, вызванные OA, или подобные.
Настоящее изобретение относится к новому производному транс-2-деценовой кислоты формулы (I') или его фармацевтически приемлемой соли
 , обладающему профилактическим или терапевтическим действием для расстройств периферической нервной системы, вызванных противораковыми средствами, и/или нейротрофической фактор-подобной активностью и/или анальгитическим действием, а также к фармацевтическую средству на его основе. 2 н. и 12 з.п. ф-лы, 11 табл., 56 пр.
, обладающему профилактическим или терапевтическим действием для расстройств периферической нервной системы, вызванных противораковыми средствами, и/или нейротрофической фактор-подобной активностью и/или анальгитическим действием, а также к фармацевтическую средству на его основе. 2 н. и 12 з.п. ф-лы, 11 табл., 56 пр.
1. Производное транс-2-деценовой кислоты, представленное следующей общей формулой (I′) или его фармацевтически приемлемая соль:
,
где X′ представляет собой:
(a) 1-пирролидил, замещенный карбоксилом или C1-С4алкоксикарбонилом,
(b) 3-тиазолидил,
(c) пиперидино, замещенный С1-С4алкилом, оксо, гидрокси, С1-С4алкокси, карбоксилом, С1-С4алкоксикарбонилом, диС1-С4алкиламино, диС1-С4алкиламиноС1-С4алкилом, фенилом, карбоксиС1-С4алкилом, циано или галогенофенилом,
(d) тиоморфолино,
(e) 1-пиперазил, который может быть замещен С1-С4алкилом, карбоксиС1-С4алкилом, диС1-С4алкиламиноС1-С4алкилом, С3-С8циклоалкилом, пиперидиноС1-С4алкилом, фенилС1-С4алкилом, пиридилом, пиримидилом или карбоксифенилС1-С4алкилом,
(f) 1-пиперазил, замещенный фенилом, который может быть замещен диС1-С4алкиламино, галогеном, С1-С4алкокси, С1-С4алкилом, гидрокси или карбоксиС1-С4алкокси,
(g) 1,4-диазепанил, который может быть замещен С1-С4алкилом или диС1-С4алкиламиноС1-С4алкилом, или
(h) карбоксиморфолино.
2. Производное транс-2-деценовой кислоты или его фармацевтически приемлемая соль по п. 1, где X′ представляет собой 1-пирролидил, замещенный карбоксилом или С1-С4алкоксикарбонилом.
3. Производное транс-2-деценовой кислоты или его фармацевтически приемлемая соль по п. 1, где X′ представляет собой 3-тиазолидил, тиоморфолино или карбоксиморфолино.
4. Производное транс-2-деценовой кислоты или его фармацевтически приемлемая соль по п. 1, где X′ представляет собой пиперидино, замещенный С1-С4алкилом, оксо, гидрокси, С1-С4алкокси, карбоксилом, С1-С4алкоксикарбонилом, диС1-С4алкиламино, диС1-С4алкиламиноС1-С4алкилом, фенилом, карбоксиС1-С4алкилом, циано или галогенофенилом.
5. Производное транс-2-деценовой кислоты или его фармацевтически приемлемая соль по п. 1, где X′ представляет собой 1-пиперазил, который может быть замещен С1-С4алкилом, карбоксиС1-С4алкилом, диС1-С4алкиламиноС1-С4алкилом, С3-С8циклоалкилом, пиперидиноС1-С4алкилом, фенилС1-С4алкилом, пиридилом, пиримидилом, карбоксифенилС1-С4алкилом.
6. Производное транс-2-деценовой кислоты или его фармацевтически приемлемая соль по п. 1, где X′ представляет собой 1-пиперазил, замещенный фенилом, который может быть замещен диС1-С4алкиламино, галогеном, С1-С4алкокси, С1-С4алкилом, гидрокси или карбоксиС1-С4алкокси.
7. Производное транс-2-деценовой кислоты или его фармацевтически приемлемая соль по п. 1, где X′ представляет собой 1,4-диазепанил, который может быть замещен С1-С4алкилом или диС1-С4алкиламиноС1-С4алкилом.
8. Фармацевтическое средство, обладающее профилактическим или терапевтическим действием для расстройств периферической нервной системы, вызванных противораковыми средствами, и/или нейротрофической фактор-подобной активностью и/или анальгитическим действием, содержащее, в качестве активного ингредиента, по меньшей мере один член, выбранный из производного транс-2-деценовой кислоты, представленного следующей общей формулой (I), и его фармацевтически приемлемой соли:
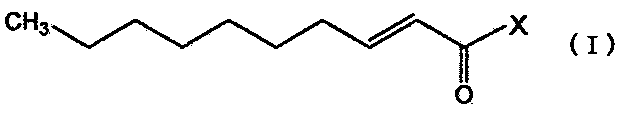 ,
,
где X представляет собой:
(a) 1-пирролидил, замещенный карбоксилом или С1-С4алкоксикарбонилом,
(b) 3-тиазолидил,
(c) пиперидино, замещенный С1-С4алкилом, оксо, гидрокси, С1-С4алкокси, карбоксилом, С1-С4алкоксикарбонилом, диС1-С4алкиламино, диС1-С4алкиламиноС1-С4алкилом, фенилом, карбоксиС1-С4алкилом, циано или галогенофенилом,
(d) тиоморфолино,
(e) 1-пиперазил, который может быть замещен С1-С4алкилом, карбоксиС1-С4алкилом, диС1-С4алкиламиноС1-С4алкилом, С3-С8циклоалкилом, пиперидино С1-С4алкилом, фенилС1-С4алкилом, пиридилом, пиримидилом или карбоксифенилС1-С4алкилом,
(f) 1-пиперазил, замещенный фенилом, который может быть замещен диС1-С4алкиламино, галогеном, С1-С4алкокси, С1-С4алкилом, гидрокси или карбоксиС1-С4алкокси,
(g) 1,4-диазепанил, который может быть замещен С1-С4алкилом или диС1-С4алкиламиноС1-С4алкилом, или
(h) карбоксиморфолино.
9. Фармацевтическое средство по п. 8, где фармацевтическое средство представляет собой профилактическое средство или терапевтическое средство от расстройств периферической нервной системы, вызванных введением лекарственного средства, включающего по меньшей мере одно противораковое средство.
10. Фармацевтическое средство по п. 8, где фармацевтическое средство представляет собой профилактическое средство или терапевтическое средство от нейродегенеративных заболеваний.
11. Фармацевтическое средство по п. 8, где фармацевтическое средство представляет собой профилактическое средство или терапевтическое средство от психических заболеваний.
12. Фармацевтическое средство по п. 11, где психические заболевания представляют собой депрессию, тревожные расстройства (неврозы) или расстройства аутистического спектра.
13. Фармацевтическое средство по любому одному из пп. 8-12, где фармацевтическое средство представляет собой препарат для инъекций.
14. Фармацевтическое средство по любому одному из пп. 8-12, где фармацевтическое средство представляет собой пероральный препарат.
| J.P.Ley et al, Structure-activity relationships of trigeminal effects for artificial and naturally occurring alkamides related to spilanthol | |||
| Developments in Food Science, 2006, 43, 21-24;WO 2010123894A2, 28.10.2010 ;US 3294794A1, 27.12.1966 ;RU 2227794C2, 27.04.2004. |

