Настоящее изобретение относится главным образом к локализованной или пространственной детекции нуклеиновой кислоты в образце ткани. Нуклеиновая кислота может представлять собой РНК или ДНК. Таким образом, согласно настоящему изобретению предложены способы детекции и/или анализа РНК, например РНК-транскриптов, или геномной ДНК для получения пространственной информации о локализации, распределении или экспрессии генов или, фактически, о локализации или распределении любой геномной вариации (необязательно в гене) в образце ткани, например в отдельной клетке. Настоящее изобретение, таким образом, обеспечивает условия для развития пространственной геномики и пространственной транскриптомики.
Более конкретно, настоящее изобретение относится к способу определения и/или анализа транскриптома или генома и особенно общего транскриптома или генома образца ткани. В частности, данный способ относится к количественному и/или качественному методу анализа распределения, локализации или экспрессии геномных последовательностей в образце ткани, при этом картина пространственной экспрессии или распределения либо локализации в образце ткани сохраняется. Таким образом, этот новый способ представляет собой способ осуществления "пространственной транскриптомики" или "пространственной геномики", который дает возможность пользователю одновременно определять картину экспрессии или картину локализации/распределения экспрессированных генов, либо генов или геномных локусов, присутствующих в образце ткани.
В основе данного изобретения лежит, в частности, технология чипов в сочетании с высокопроизводительными технологиями секвенирования ДНК, которая дает возможность для захвата и мечения позиционной меткой молекулы нуклеиновой кислоты (например, молекул РНК или ДНК) в образце ткани, в частности, мРНК или ДНК. За этой стадией следует синтез молекул ДНК, которые подвергают секвенированию и анализу, чтобы определить, какие гены экспрессируются в какой-либо части и во всех частях образца ткани. Преимущественно, что индивидуальный, отдельный и специфический транскриптом каждой клетки в образце ткани можно получить одновременно. Таким образом, можно сказать, что способы по изобретению обеспечивают получение высокопараллельных исчерпывающих отличительных характеристик транскриптома индивидуальных клеток в образце ткани без потери пространственной информации в указанном исследованном образце ткани. Согласно изобретению также предложен чип для осуществления способа по изобретению и способы изготовления чипов по изобретению.
Организм человека содержит свыше 100 триллионов клеток и состоит более чем из 250 разных органов и тканей. Развитие и организация таких сложных органов, как головной мозг, далеки от понимания, и существует потребность в анализе экспрессии генов в таких тканях с использованием количественных способов для изучения и определения генов, которые регулируют развитие и функционирование таких тканей. Сами эти органы представляют собой смесь дифференцированных клеток, которые дают возможность всем функциям организма, таким как транспорт питательных веществ, защита и т.д., быть согласованными и поддерживаться. Следовательно, функционирование клеток зависит от расположения клетки внутри конкретной тканевой структуры и взаимодействий с другими клетками в ткани, в которых она участвует как непосредственно, так и опосредованно. Таким образом, существует потребность в том, чтобы определить на транскрипционном уровне как эти взаимодействия влияют на каждую клетку в ткани.
Полученные недавно данные с использованием глубокого секвенирования РНК продемонстрировали, что большинство транскриптов можно детектировать в линии клеток человека, и что большая часть (75%) генов человека, кодирующих белки, экспрессируется в большинстве тканей. Аналогичным образом, детальное изучение 1% человеческого генома показало, что хромосомы транскрибируются повсеместно и что большая часть всех оснований включается в первичные транскрипты. Ввиду этого, аппарат транскрипции на общем уровне может быть описан как случайный.
Хорошо известно, что транскрипты являются всего лишь показателем присутствия белка, поскольку скорости трансляции, деградации и т.д. РНК будут оказывать влияние на количество белка, произведенного из одного транскрипта. В этом отношении, недавний основанный на использовании антител анализ человеческих органов и тканей подтверждает, что тканевая специфичность достигается за счет точной регуляции уровней белка в пространстве и времени, и что разные ткани в организме приобретают свои уникальные характеристики не посредством контроля того, какие белки экспрессируются, а посредством контроля того, сколько каждого из них производится.
Однако в последующих общих исследованиях при сравнении корреляций между транскриптомом и протеомом было показано, что экспрессируется большая часть всех генов. Интересно, что была показана строгая корреляция между изменениями в уровнях РНК и белка для продуктов индивидуальных генов, что указывает на биологическую бесполезность изучения транскриптома в отдельных клетках в контексте функциональной роли белков.
Действительно, анализ гистологии и картины экспрессии в тканях является краеугольным камнем в области биомедицинских исследований и диагностики. Гистология более чем столетие назад с использованием разных методов окрашивания первой выявила основы структурной организации здоровых органов и те изменения, которые происходят при общих патологиях. Результатом разработок в этой области явилась возможность изучения распределения белков с помощью иммуногистохимии, а также экспрессии генов посредством гибридизации in situ.
Однако параллельное развитие более современных методов гистологии и экспрессии генов привело к разделению визуализации и транскриптомного анализа, и до появления способов по настоящему изобретению не существовало никакого подходящего способа, пригодного для общего транскриптомного анализа с пространственным разрешением.
В качестве альтернативы или в дополнение к методам in situ были разработаны способы для анализа белков и нуклеиновых кислот in vitro, т.е. в результате выделения молекул из образцов целой ткани, отдельных типов клеток или даже из единственной клетки и количественного определения конкретного типа молекул в указанных экстрактах, например с использованием ELISA, количественной ПЦР (полимеразной цепной реакции) и т.д.
Недавние достижения в анализе экспрессии генов привели к тому, что появилась возможность оценки полного транскриптома ткани с использованием микрочипов или секвенирования РНК, и такое развитие событий сыграло важную роль в понимании биологических процессов и для диагностики. Однако транскриптомный анализ в типичном случае осуществляют на мРНК, выделенной из целых тканей (или даже целых организмов), и способы отбора небольших участков тканей или индивидуальных клеток для транскриптомного анализа как правило являются трудоемкими, дорогостоящими и характеризуются низкой точностью.
Следовательно, в большинстве исследований генной экспрессии, основанных на использовании микрочипов или секвенировании РНК следующего поколения, применяется репрезентативный образец, содержащий большое количество клеток. Таким образом, результаты представляют собой средние уровни экспрессии исследуемых генов. Разделение клеток, являющихся фенотипически различными, использовалось в некоторых случаях вместе с платформами для исследования общей экспрессии генов (Tang F. et al., Nat. Protoc., 2010, 5: 516-35; Wang D. & Bodovitz S., Trends Biotechnol., 2010, 28: 281-90) и привело к получению очень точной информации о межклеточных вариациях. Однако, высокопроизводительные способы изучения транскрипционной активности с высоким разрешением в интактных тканях до сих пор недоступны.
Таким образом, существующие методы анализа картин экспрессии генов дают информацию о пространственной локализации транскрипции только для одного гена или небольшого числа генов одновременно либо предоставляют информацию о транскрипции для всех генов в образце за счет потери информации о положении. Поэтому очевидно, что существует необходимость в способах одновременного, раздельного и специфического определения транскриптома каждой клетки в образце, т.е. способах, позволяющих осуществлять общий анализ экспрессии генов в образцах ткани, результатом применения которых является получение информации о транскриптоме с пространственным разрешением, и настоящее изобретение направлено на решение этой задачи.
В новом подходе, относящемся к способам и продуктам по настоящему изобретению, используется твердо установившаяся в настоящее время технология чипов и секвенирования, дающая возможность получения информации о транскрипции для всех генов в образце с сохранением информации о положении каждого транскрипта. Специалисту в данной области будет очевидно, что это является ключевой точкой в области наук о живой природе. Эта новая технология открывает новую область так называемой "пространственной транскриптомики", которая по всей вероятности будет иметь глубокие последствия для нашего понимания развития тканей и функционирования тканей и клеток во всех многоклеточных организмах. Очевидно, что такие методики будут особенно полезны для нашего понимания причин и развития болезненных состояний и для разработки эффективных методов лечения таких заболеваний, как например рак. Способы по изобретению также найдут применение в диагностике многочисленных медицинских состояний.
Несмотря на то что первоначально они задуманы с целью проведения транскриптомного анализа, как подробно описано ниже, принципы и способы по настоящему изобретению могут быть применены также для анализа ДНК и, следовательно, также для геномных анализов ("пространственной геномики"). Соответственно, в самом широком его понимании настоящее изобретение относится к детекции и/или анализу нуклеиновых кислот в целом.
Технология чипов, в частности микрочипов, возникла в результате исследований в Стэндфордском университете, когда небольшие количества олигонуклеотидов ДНК были успешно присоединены к поверхности стекла в упорядоченном расположении, на так называемом "чипе", и использованы для мониторинга транскрипции 45 генов (Schena M. et al., Science, 1995, 270: 368-9, 371).
С тех пор исследователями во всем мире опубликовано более 30000 научных работ с использованием технологии микрочипов. Были разработаны многочисленные типы микрочипов для различных применений, например для детекции однонуклеотидных полиморфизмов (SNP), или для генотипирования или повторного секвенирования мутантных геномов, и важным применением технологии микрочипов является ее использование для исследования экспрессии генов. Действительно, микрочип для генной экспрессии был создан как средство для анализа уровня экспрессированного генетического материала в конкретном образце с реальной возможностью использования для сравнения уровней экспрессии многих генов одновременно. Имеется несколько коммерческих платформ микрочипов для экспериментов такого типа, но также есть возможность создания чипов на заказ для анализа экспрессии генов.
Несмотря на то, что применение микрочипов в исследованиях экспрессии генов сейчас широко распространено, очевидно, что набирают ход новые и более совершенные, так называемые технологии "секвенирования ДНК следующего поколения" (NGS), для замены ДНК-микрочипов во многих практических применениях, например при использовании в глубоком транскриптомном анализе.
Разработка технологий NGS для ультра-быстрого секвенирования генома является ключевой точкой в области наук о живой природе (Petterson E. et at., Genomics, 2009, 93: 105-11). Эти новые технологии существенно снизили стоимость секвенирования ДНК и сделали возможным определение генома высших организмов с беспрецедентной скоростью, включая геномы конкретных индивидуумов (Wade CM. et al., Science, 2009, 326: 865-7; Rubin J. et al., Nature, 2010, 464: 587-91). Эти новые достижения высокопроизводительной геномики изменили ландшафт биологических исследований и, в дополнение к полному определению характеристик геномов, также появилась возможность для исследования полного транскриптома в цифровом и количественном отношении. Средства биоинформатики для визуализации и объединения этих обширных наборов данных были также значительно улучшены в последние годы.
Однако неожиданно оказалось, что уникальная комбинация гистологических методов, методов с использованием микрочипов и NGS-методов может обеспечить получение обширной транскрипционной или геномной информации из многочисленных клеток в образце ткани, информации характеризующейся двумерным пространственным разрешением. Так, в одном предельном случае способы по настоящему изобретению могут быть использованы для анализа экспрессии единичного гена в одной клетке образца с сохранением клетки в пределах ее окружения в образце ткани. В другом предельном случае и в предпочтительном аспекте изобретения данные способы могут быть использованы для определения экспрессии каждого из генов в каждой клетке или по существу во всех клетках в образце одновременно, т.е. для определения картины общей пространственной экспрессии в образце ткани. Очевидно, что способы по изобретению также позволяют проводить промежуточные анализы.
В своей простейшей форме изобретение может быть проиллюстрировано следующим кратким изложением. Изобретение предусматривает наличие праймеров для обратной транскрипции (ОТ), которые также содержат уникальные позиционные метки (области), которые должны быть расположены на целевой подложке, например стеклянном слайде, с целью создания "чипа". Уникальные позиционные метки соответствуют локализации ОТ-праймеров на чипе (элементы чипа). На данный чип помещают тонкие срезы ткани и проводят реакцию обратной транскрипции в срезе ткани на целевом слайде. ОТ-праймеры, с которыми РНК из образца ткани связывается (или гибрид изуется), удлиняют, используя связавшуюся РНК в качестве матрицы, с получением кДНК, которая ввиду этого оказывается связанной с поверхностью чипа. Как следствие наличия уникальных позиционных меток в ОТ-праймерах, каждая нить кДНК несет информацию о расположении матричной РНК в срезе ткани. Срез ткани может быть визуализирован, или может быть получено его изображение, например, срез ткани может быть окрашен и сфотографирован до или после стадии синтеза кДНК, чтобы позиционная метка в молекуле кДНК коррелировала с расположением в образце ткани. Проводят секвенирование кДНК, в результате чего получают транскриптом с полной информацией о положении. Схема данного способа показана на Фиг.1. Данные о последовательностях затем могут быть сопоставлены с расположением в образце ткани, что дает возможность осуществить визуализацию, например с использованием компьютера, данных о последовательностях вместе со срезом ткани, например для отображения картины экспрессии какого-либо представляющего интерес гена в целом по ткани (Фиг.2). Подобным же образом можно отметить разные области среза ткани на экране компьютера и получить информацию о дифференциально экспрессируемых генах между любыми представляющими интерес выбранными областями. Очевидно, что применение способов по изобретению приводит к получению данных, которые резко контрастируют с данными, полученными с использованием современных способов исследования популяций мРНК. Например, способы, основанные на гибридизации in situ, обеспечивают получение только относительной информации в виде транскриптов единичной мРНК. Таким образом, способы по настоящему изобретению обладают очевидными преимуществами по сравнению с современными технологиями in situ. Информация об общей экспрессии генов, которую можно получить с использованием способов по изобретению, также дает возможность получения информации о совместной экспрессии и количественных оценок присутствия транскриптов. Очевидно, что это будет широко применимая стратегия, подходящая для анализа любой ткани из любых видов, например животного, растения, гриба.
Из того, что отмечалось выше и описано более подробно ниже, будет очевидно, что эта основная методология без труда может быть распространена на анализ геномной ДНК, например для идентификации клеток в образце ткани, которые содержат одну или более чем одну конкретную мутацию. Например, геномную ДНК можно фрагментировать и предоставить ей возможность гибридизоваться с праймерами (эквивалентными ОТ-праймерам, описанным выше), которые способны захватывать фрагментированную ДНК (например, с фрагментированной ДНК может быть лигирован адаптер, имеющий последовательность, которая комплементарна праймеру, или фрагментированная ДНК может быть удлинена, например с использованием фермента, для встраивания дополнительных нуклеотидов на конце последовательности, например поли(А)-хвоста, для создания последовательности, которая комплементарна праймеру), и праймировать синтез нитей, комплементарных захватывающим молекулам. Остальные стадии анализа могут быть такими, как описано выше. Таким образом, конкретные воплощения изобретения, описанные ниже в случае транскриптомного анализа, также могут быть применены в способах анализа геномной ДНК там, где это целесообразно.
Из приведенных выше объяснений будет очевидно, что огромное значение имеет сочетание информации о положении с информацией о транскриптоме или геноме. Например, это дает возможность проведения общего картирования генной экспрессии с высоким разрешением, что будет полезно в многочисленных практических применениях, включая, например, исследования рака и диагностику.
Кроме того, очевидно, что способы, изложенные в данном описании, значительно отличаются от описанных ранее способов анализа общего транскриптома образца ткани, и эти отличия дают многочисленные преимущества. Настоящее изобретение основывается на неожиданном открытии того, что применение срезов тканей не нарушает синтеза ДНК (например, кДНК), праймированного праймерами (например, праймерами обратной транскрипции), которые присоединены к поверхности чипа.
Таким образом, в его первом и наиболее широком аспекте согласно настоящему изобретению предложен способ локализованной детекции нуклеиновой кислоты в образце ткани, включающий:
(а) предоставление чипа, содержащего подложку, на которой непосредственно или опосредованно иммобилизованы многочисленные разновидности захватывающих зондов, так что каждая разновидность занимает определенное положение на чипе и ориентирована таким образом, что имеет свободный 3′-конец, позволяющий указанному зонду действовать в качестве праймера в реакции удлинения или лигирования праймера, где каждая разновидность указанного захватывающего зонда содержит молекулу нуклеиновой кислоты, имеющую в направлении от 5′ к 3′:
(1) позиционную область, которая соответствует положению захватывающего зонда на чипе, и
(2) захватывающую область;
(б) приведение указанного чипа в контакт с образцом ткани таким образом, что положение захватывающего зонда на чипе может быть сопоставлено с положением в образце ткани, и предоставление возможности нуклеиновой кислоте образца ткани гибридизоваться с захватывающей областью в указанных захватывающих зондах;
(в) синтез молекул ДНК на основе захваченных молекул нуклеиновой кислоты с использованием указанных захватывающих зондов в качестве праймеров удлинения или лигирования, где указанные удлиненные или лигированные молекулы ДНК являются мечеными благодаря позиционной области;
(г) возможно синтез комплементарной нити указанной меченой ДНК и/или возможно амплификацию указанной меченой ДНК;
(д) высвобождение по меньшей мере части меченых молекул ДНК и/или их комплементов или ампликонов с поверхности чипа, где указанная часть включает позиционную область или ее комплемент;
(е) прямой или опосредованный анализ последовательности высвобожденных молекул ДНК.
Способы по изобретению представляют собой значительный шаг вперед по сравнению с другими способами пространственной транскриптомики, известными в данной области. Например, способы, изложенные в данном описании, позволяют получить общий и пространственный профиль всех транскриптов в образце ткани. Кроме того, может быть осуществлено количественное определение экспрессии каждого гена для каждого положения или элемента на чипе, что дает возможность осуществления большого числа анализов на основе данных одного анализа. Таким образом, способы по настоящему изобретению дают возможность детекции и/или количественного определения пространственной экспрессии всех генов в единичном образце ткани. Кроме того, поскольку визуализация присутствия транскриптов не производится непосредственно, например, с использованием флуоресценции, как в случае стандартного микрочипа, то возможно одновременное измерение экспрессии генов в единичном образце даже в том случае, когда указанные транскрипты присутствуют в том же образце в очень разных концентрациях.
Соответственно, можно отметить, что во втором и более конкретном аспекте согласно настоящему изобретению предложен способ определения и/или анализа транскриптома образца ткани, включающий:
(а) предоставление чипа, содержащего подложку, на которой непосредственно или опосредованно иммобилизованы многочисленные разновидности захватывающих зондов, так что каждая разновидность занимает определенное положение на чипе и ориентирована таким образом, что имеет свободный 3′-конец, позволяющий указанному зонду действовать в качестве праймера обратной транскриптазы (ОТ), где каждая разновидность указанного захватывающего зонда содержит молекулу нуклеиновой кислоты, имеющую в направлении от 5′ к 3′:
(1) позиционную область, которая соответствует положению захватывающего зонда на чипе, и
(2) захватывающую область;
(б) приведение указанного чипа в контакт с образцом ткани таким образом, что положение захватывающего зонда на чипе может быть сопоставлено с положением в образце ткани, и предоставление возможности РНК образца ткани гибридизоваться с захватывающей областью в указанных захватывающих зондах;
(в) синтез молекул кДНК на основе захваченных молекул РНК с использованием указанных захватывающих зондов в качестве ОТ-праймеров и возможно амплификацию указанных молекул кДНК;
(г) высвобождение по меньшей мере части молекул кДНК и/или возможно их ампликонов с поверхности чипа, где указанная высвобожденная молекула может представлять собой первую нить и/или вторую нить молекулы кДНК или ее ампликон, и где указанная часть включает позиционную область или ее комплемент;
(д) прямой или опосредованный анализ последовательности высвобожденных молекул.
Как описано более подробно ниже, на стадии анализа может быть использован любой метод анализа нуклеиновой кислоты. В типичном случае он может включать секвенирование, но нет необходимости проводить фактическое определение последовательности. Например, можно использовать специфичные к последовательности методы анализа. Например, может быть осуществлена специфичная к последовательности реакция амплификации, например с использованием праймеров, специфичных к позиционной области и/или к специфической целевой последовательности, например конкретной целевой ДНК, детекцию которой необходимо осуществить (т.е. соответствующей конкретной кДНК/РНК или гену и т.д.). Типичным методом анализа является специфичная к последовательности ПЦР-реакция.
Информация из анализа последовательности на стадии (д) может быть использована для получения пространственной информации об РНК в образце. Другими словами, информация из анализа последовательности может обеспечить получение информации о локализации РНК в образце. Эта пространственная информация может вытекать из природы информации, полученной из анализа последовательности, например, она может отражать наличие конкретной РНК, которая сама может быть информативна в пространственном отношении в случае используемого образца ткани, и/или пространственная информация (например, пространственная локализация) может вытекать из положения образца ткани на чипе, в сочетании с информацией, полученной в результате секвенирования. Таким образом, способ может включать только сопоставление информации из анализа последовательностей с положением в образце ткани, например, на основании позиционной метки и ее корреляции с положением в образце ткани. Однако, как описано выше, пространственная информация соответственно может быть получена посредством сопоставления данных из анализа последовательности с изображением образца ткани, и это представляет собой одно из предпочтительных воплощений изобретения. Соответственно, в предпочтительном воплощении способ также включает стадию:
(е) сопоставления информации из анализа указанной последовательности с изображением указанного образца ткани, где изображение образца ткани получают до или после стадии (в).
В своем самом широком смысле способ по изобретению может быть использован для локализованной детекции нуклеиновой кислоты в образце ткани. Таким образом, в одном из воплощений способ по изобретению может быть использован для определения и/или анализа всего транскриптома или генома образца ткани, например общего транскриптома образца ткани. Однако, способ этим не ограничен и охватывает определение и/или анализ всего или части транскриптома или генома. Таким образом, способ может включать в себя определение и/или анализ части или подмножества транскриптома или генома, например транскриптома, соответствующего подмножеству генов, например набору конкретных генов, например имеющих отношение к конкретному заболеванию или состоянию, типу ткани и т.д.
С точки зрения другого аспекта, изложенные выше стадии способа можно рассматривать в качестве предложения способа получения определяемого в пространстве транскриптома или генома и в частности определяемого в пространстве общего транскриптома или генома образца ткани.
С альтернативной точки зрения способ по изобретению можно рассматривать как способ локализованной или пространственной детекции нуклеиновой кислоты, ДНК или РНК, в образце ткани либо локализованного или пространственного определения и/или анализа нуклеиновой кислоты (ДНК или РНК) в образце ткани. В частности, данный способ можно использовать для локализованной или пространственной детекции либо определения и/или анализа экспрессии генов или геномной вариации в образце ткани. Термин "локализованная/пространственная детекция/определение/анализ" означает, что РНК или ДНК может быть локализована в соответствии с ее природным положением или природной локализацией внутри клетки или ткани в образце ткани. Так, например, РНК или ДНК может быть локализована в клетке, или группе клеток, или типе клеток в образце либо в конкретных участках областей в образце ткани. Можно определить природную локализацию или природное положение РНК или ДНК (или другими словами, локализацию или положение РНК или ДНК в образце ткани), например экспрессированного гена или геномного локуса.
Также можно отметить, что согласно изобретению предложен чип для применения в способах по изобретению, содержащий подложку, на которой непосредственно или опосредованно иммобилизованы многочисленные разновидности захватывающих зондов, так что каждая разновидность занимает определенное положение на чипе и ориентирована таким образом, что имеет свободный 3′-конец, позволяющий указанному зонду действовать в качестве праймера обратной транскриптазы (ОТ), где каждая разновидность указанного захватывающего зонда содержит молекулу нуклеиновой кислоты, имеющую в направлении от 5′ к 3′:
(1) позиционную область, которая соответствует положению захватывающего зонда на чипе, и
(2) захватывающую область для захвата РНК образца ткани, который контактирует с указанным чипом.
В родственном аспекте согласно настоящему изобретению также предложено применение чипа, содержащего подложку, на которой непосредственно или опосредованно иммобилизованы многочисленные разновидности захватывающего зонда, так что каждая разновидность занимает определенное положение на чипе и ориентирована таким образом, что имеет свободный 3′-конец, позволяющий указанному зонду действовать в качестве праймера обратной транскриптазы (ОТ), где каждая разновидность указанного захватывающего зонда содержит молекулу нуклеиновой кислоты, имеющую в направлении от 5′ к 3′:
(1) позиционную область, которая соответствует положению захватывающего зонда на чипе, и
(2) захватывающую область;
для захвата РНК образца ткани, который контактирует с указанным чипом.
Предпочтительно, указанное применение предназначено для определения и/или анализа транскриптома и, в частности, общего транскриптома образца ткани, и дополнительно содержит стадии:
(а) синтеза молекул кДНК на основе захваченных молекул РНК с использованием указанных захватывающих зондов в качестве ОТ-праймеров и возможно амплификации указанных молекул кДНК;
(б) высвобождения по меньшей мере части молекул кДНК и/или возможно их ампликонов с поверхности чипа, где указанная высвобожденная молекула может представлять собой первую нить и/или вторую нить молекулы кДНК или ее ампликон, и где указанная часть включает позиционную область или ее комплемент;
(в) прямого или опосредованного анализа последовательности высвобожденных молекул; и возможно
(г) сопоставления информации, полученной из указанного анализа последовательности, с изображением указанного образца ткани, при этом изображение образца ткани получают до или после стадии (а).
В силу вышесказанного можно отметить, что чип по настоящему изобретению может быть использован для захвата РНК, например мРНК, образца ткани, который контактирует с указанным чипом. Данный чип также можно использовать для определения и/или анализа частичного или общего транскриптома образца ткани или для получения определяемого в пространстве частичного или общего транскриптома образца ткани. Способы по данному изобретению могут, таким образом, рассматриваться как способы количественного определения пространственной экспрессии одного или более генов в образце ткани. Выражаясь по-другому, способы по настоящему изобретению могут быть использованы для детекции пространственной экспрессии одного или более генов в образце ткани. Иными словами, способы по настоящему изобретению могут быть использованы для одновременного определения экспрессии одного или более генов в одном или более положениях в образце ткани. Более того, данные способы можно рассматривать как способы частичного или общего транскриптомного анализа образца ткани с двумерным пространственным разрешением.
РНК может представлять собой любую молекулу РНК, которая может присутствовать в клетке. Так, она может представлять собой мРНК, тРНК (транспортная РНК), рРНК (рибосомальная РНК), вирусную РНК, малую ядерную РНК (мяРНК), малую ядрышковую РНК (мякРНК), микроРНК, малую интерферирующую РНК (миРНК), РНК, взаимодействующую по piwi-типу (piRNA), РНК рибозимов, антисмысловую РНК или некодирующую РНК. Однако предпочтительно, чтобы она представляла собой мРНК.
Считается, что стадия (в) приведенного выше способа (соответствующая стадии (а) в формулировке предпочтительного применения, изложенного выше) синтеза кДНК на основании захваченной РНК относится к синтезу кДНК. Она будет включать в себя стадию обратной транскрипции захваченной РНК, удлинения захватывающего зонда, действующего как ОТ-праймер, использование захваченной РНК в качестве матрицы. В результате осуществления этой стадии синтезируется так называемая первая нить кДНК. Как будет описано более подробно ниже, синтез второй нити кДНК возможно может происходить на чипе или может происходить как отдельная стадия после высвобождения первой нити кДНК с чипа. Так же, как более подробно описано ниже, в некоторых воплощениях синтез второй нити может происходить на первой стадии амплификации высвобожденной первой нити молекулы кДНК.
Ниже рассмотрены и описаны чипы для применения в случае анализа нуклеиновых кислот в целом и анализа ДНК в частности. Конкретные детали и воплощения, изложенные в данном описании в отношении чипов и захватывающих зондов для применения в случае РНК, в равной степени применимы (там, где это целесообразно) ко всем подобным чипам, включая таковые для применения с ДНК.
Как использовано в данном описании, термин "многочисленный" означает два или более, либо по меньшей мере два, например 3, 5, 10, 15, 20, 30, 40, 50, 60, 70, 80, 90, 100, 150, 200, 400, 500, 1000, 2000, 5000, 10000 или более и т.д. Так, например, число захватывающих зондов может представлять собой любое целое число в любом диапазоне между любыми двумя вышеупомянутыми числами. Однако, следует иметь в виду, что предполагается возможность использования чипов традиционного типа со многими сотнями, тысячами, десятками тысяч, сотнями тысяч или даже миллионами захватывающих зондов.
Таким образом, в способах, изложенных в данном описании, используют чипы с высокой плотностью нуклеиновых кислот, содержащие "захватывающие зонды" для захвата и мечения транскриптов из всех одиночных клеток в образце ткани, например в тонком слое образца ткани или "срезе". Образцы ткани или срезы для анализа получают строго параллельным образом, так что пространственная информация в срезе сохраняется. Захваченные молекулы РНК (предпочтительно мРНК) для каждой клетки или "транскриптомы" транскрибируются в кДНК, и полученные молекулы кДНК анализируют, например, с использованием высокопроизводительного секвенирования. Полученные данные могут быть сопоставлены с изображениями исходных образцов ткани, например срезов, с использованием так называемых штрихкодовых последовательностей (или ID-меток, определенных в данном описании как позиционные области), введенных в образующие чип нуклеиновокислотные зонды.
Чипы с высокой плотностью нуклеиновых кислот или микрочипы являются основным компонентом способа пространственного мечения транскриптома, изложенного в данном описании. Технология микрочипов представляет собой мультиплексную технологию, используемую в молекулярной биологии. Типичный микрочип состоит из образующей чип серии микроскопических пятен олигонуклеотидов (в один чип могут быть введены сотни тысяч пятен, как правило, десятки тысяч). Определенное положение каждого пятна нуклеиновой кислоты (олигонуклеотида) (каждой разновидности олигонуклеотида/молекулы нуклеиновой кислоты) известно как "элемент" (и, следовательно, в изложенных выше способах каждая разновидность захватывающего зонда может рассматриваться как специфический элемент чипа; каждый элемент занимает определенное положение на чипе) и в типичном случае каждый отдельный элемент содержит в пикомольном диапазоне (10-12 моль) молекулы ДНК с конкретной последовательностью ("разновидности"), которые известны как "зонды" (или "репортеры"). В типичном случае они могут представлять собой короткий участок гена или другой элемент нуклеиновой кислоты, с которым образец кДНК или кРНК (или "мишень") может гибридизоваться в строгих условиях. Однако, как описано ниже, зонды по настоящему изобретению отличаются от зондов стандартных микрочипов.
В микрочипах, предназначенных для анализа экспрессии генов, гибридизацию зонд-мишень как правило детектируют и количественно определяют путем детекции оптического сигнала, например от флуорофора, иона серебра или хемилюминесцентной метки, которые были введены во все мишени. Интенсивность оптического сигнала коррелирует с относительным содержанием каждой целевой нуклеиновой кислоты в образце. Поскольку чип может содержать десятки тысяч зондов, в эксперименте с использованием микрочипов можно выполнять много генетических тестов параллельно.
В стандартных микрочипах зонды присоединены к твердой поверхности или подложке посредством ковалентной связи с химическим матриксом, например эпоксисиланом, аминосиланом, лизином, полиакриламидом и т.д. Подложка обычно представляет собой стеклянный, пластмассовый или кремниевый чип или слайд, хотя известны и другие платформы для микрочипов, например микроскопические гранулы.
Зонды могут быть присоединены к чипу по изобретению с использованием любых подходящих способов. В предпочтительном воплощении зонды иммобилизуют на подложке чипа путем химической иммобилизации. Она может быть результатом взаимодействия между подложкой (материалом подложки) и зондом, основанного на химической реакции. Подобная химическая реакция обычно не зависит от поступления энергии в виде тепла или света, но может быть ускорена либо в результате использования тепла, например при определенной оптимальной для химической реакции температуре, либо использования света определенной длины волны. Например, химическая иммобилизация может происходить в результате взаимодействия между функциональными группами на подложке и соответствующими функциональными элементами зондов. Такие соответствующие функциональные элементы зондов могут либо представлять собой собственную химическую группу зонда, например гидроксильную группу, либо могут быть введены дополнительно. Примером такой функциональной группы является аминогруппа. Обычно иммобилизуемый зонд содержит функциональную аминогруппу или химически модифицирован так, чтобы содержать функциональную аминогруппу. Средства и способы для такой химической модификации хорошо известны.
Локализация указанной функциональной группы в предназначенном для иммобилизации зонде может быть использована для того, чтобы регулировать и формировать протекание процесса связывания зонда и/или ориентацию зонда, например, функциональная группа может быть расположена на 5′- или 3′-конце зонда или внутри последовательности зонда. Типичная подложка для иммобилизации зонда содержит группировки, которые способны связываться с такими зондами, например нуклеиновыми кислотами с функционализированными аминогруппами. Примерами таких подложек являются карбокси-, альдегид- или эпокси-содержащие подложки. Подобные материалы известны специалисту в данной области. Функциональные группы, обеспечивающие протекание реакции присоединения между зондами, которые являются химически реакционноспособными благодаря введению аминогруппы, и подложками чипов, известны специалисту в данной области.
Альтернативные подложки, на которых могут быть иммобилизованы зонды, можно химически активировать, например, посредством активации функциональных групп, доступных на подложке чипа. Термин "активированная подложка" относится к материалу, в котором взаимодействующие или реакционноспособные химические функциональные группы были созданы или в который они были включены с использованием методик химической модификации, известных специалисту в данной области. Например, подложка, содержащая карбоксильные группы, должна быть активирована перед применением. Кроме того, имеются доступные подложки, содержащие функциональные группы, которые могут реагировать со специфическими группировками, уже имеющимися в нуклеиновокислотных зондах.
Альтернативно, зонды могут быть синтезированы непосредственно на подложке. Подходящие способы для такого подхода известны специалисту в данной области. Примерами являются производственные методы, разработанные в Agilent Inc., Affymetrix Inc., Roche Nimblegen Inc. или Flexgen BV. В типичном случае используют лазеры и набор зеркал, которые специфически активируют пятна, в которые должны быть добавлены нуклеотиды. Применение такого подхода может обеспечить, например, получение пятен с размером (т.е. с размером элемента) примерно 30 мкм или больше.
Таким образом, подложкой может являться любая подходящая подложка, известная специалисту в данной области. Подложка может иметь любую подходящую форму или формат, например она может быть плоской, изогнутой, например выпуклой или вогнутой по отношению к области, где происходит взаимодействие между образцом ткани и подложкой. Особенно предпочтительно, когда подложка является плоской, т.е. плоским чипом или слайдом.
В типичном случае подложка представляет собой твердую подложку, и это дает возможность осуществлять точное и контролируемое расположение зондов на подложке. Примером подложки является твердый материал или подложка, содержащая функциональные химические группы, например аминогруппы или амин-функционализированные группы. Подложка, предусмотренная настоящим изобретением, представляет собой непористую подложку. Предпочтительными непористыми подложками являются стекло, кремний, покрытый поли-L-лизином материал, нитроцеллюлоза, полистирол, сополимеры циклических олефинов (СОС), полимеры циклических олефинов (СОР), полипропилен, полиэтилен и поликарбонат.
Может быть использован любой подходящий материал, известный специалисту в данной области. В типичном случае используют стекло или полистирол. Полистирол является гидрофобным материалом, подходящим для связывания отрицательно заряженных макромолекул, поскольку он в норме содержит небольшое количество гидрофильных групп. Для нуклеиновых кислот, иммобилизованных на стеклянных слайдах, также известно, что их иммобилизация может быть улучшена в результате увеличения гидрофобности поверхности стекла. Подобная оптимизация условий может позволить получить относительно более плотную упаковку. В дополнение к нанесению покрытия или обработке поверхности поли-L-лизином, подложка, в частности стеклянная, может быть обработана посредством силанирования, например, обработана эпоксисиланом или аминосиланом, или посредством силинирования, или обработана полиакриламидом.
В продаже имеется ряд стандартных чипов и как количество элементов, так и размер элемента может варьировать. Согласно настоящему изобретению расположение элементов может быть изменено в соответствии с размером и/или плотностью клеток, присутствующих в разных тканях или организмах. Например, животные клетки в типичном случае характеризуются поперечным сечением в диапазоне 1-100 мкм, в то время как размер поперечного сечения растительных клеток в типичном случае может лежать в диапазоне 1-10000 мкм. Следовательно, имеющиеся в продаже чипы от Nimblegen®, содержащие до 2,1 миллиона элементов, или до 4,2 миллиона элементов, и с размерами элемента 13 микрометров, могут быть предпочтительными для образцов ткани из животного или гриба, в то время как другие форматы, например содержащие 8×130 т (тысяч) элементов, могут быть достаточными для образцов растительной ткани. Кроме этого, в продаже имеются выпускаемые промышленностью чипы, которые используют в области анализа последовательностей и, в частности, в области NGS-технологий. Такие чипы также могут быть использованы в качестве поверхностей чипов по настоящему изобретению, например, может быть использован чип фирмы Illumina на основе гранул. В дополнение к имеющимся в продаже чипам, которые сами по себе могут быть изготовлены на заказ, на заказ можно изготовить нестандартные чипы "собственной разработки" и способы создания чипов хорошо известны. В способах по изобретению можно использовать как стандартные, так и нестандартные чипы, содержащие зонды, как определено ниже.
Зонды на микрочипе могут быть иммобилизованы, т.е. присоединены к чипу или связаны с чипом предпочтительно через 5′- или 3′-конец в зависимости от химического матрикса чипа. В типичном случае, для имеющихся в продаже чипов, зонды присоединяют связью через 3′-конец, при этом 5′-конец остается свободным. Однако также доступны чипы, содержащие зонды, присоединенные к подложке связью через 5′-конец, при этом свободным остается 3′-конец, и они могут быть синтезированы с использованием стандартных методов, которые хорошо известны в данной области и изложены в другом месте данного описания.
Ковалентную связь, используемую для присоединения нуклеиновокислотного зонда к подложке чипа, можно рассматривать как в качестве непосредственной, так и в качестве опосредованной связи, в том смысле, что, несмотря на то, что зонд соединен "непосредственной" ковалентной связью, может существовать химическая группировка или линкер, отделяющие "первый" нуклеотид нуклеиновокислотного зонда от, например, стеклянной или кремниевой подложки, т.е. в этом случае связь является опосредованной. Для решения задач настоящего изобретения зонды, которые иммобилизованы на подложке с использованием ковалентной связи и/или химического линкера, в целом рассматриваются как иммобилизованные или присоединенные непосредственно к подложке.
Как будет описано более подробно ниже, захватывающие зонды по изобретению могут быть иммобилизованы на чипе или могут взаимодействовать с чипом непосредственно или опосредованно. Так, нет необходимости в непосредственном связывании захватывающих зондов с чипом, но они могут взаимодействовать опосредованно, например, в результате связывания с молекулой, которая сама связывается непосредственно или опосредованно с чипом (например, захватывающий зонд может взаимодействовать (например, связываться или гибридизоваться) с партнером захватывающего зонда по связыванию, т.е. поверхностным зондом, который сам связан с чипом непосредственно или опосредованно). В целом, однако, захватывающий зонд будет непосредственно или опосредованно (через одно или более чем одно промежуточное соединение) связан с чипом или иммобилизован на чипе.
Применение, способ и чип по изобретению могут содержать зонды, которые иммобилизованы через свой 5′- или 3′-конец. Однако, в тех случаях когда захватывающий зонд иммобилизован непосредственно на подложке чипа, он может быть иммобилизован только таким образом, что 3′-конец захватывающего зонда является свободным для того, чтобы было возможным его удлинение, например иммобилизован через свой 5′-конец. Захватывающий зонд может быть иммобилизован опосредованно, таким образом, чтобы он имел свободный, т.е. удлиняемый 3′-конец.
Под удлиненным или удлиняемым 3′-концом понимают то, что дополнительные нуклеотиды могут быть добавлены к последнему 3′-нуклеотиду молекулы нуклеиновой кислоты, например к захватывающему зонду, для увеличения длины молекулы нуклеиновой кислоты, т.е. для удлинения молекул нуклеиновой кислоты может быть использована стандартная реакция полимеризации, например матричной полимеризации, катализируемая полимеразой.
Так, в одном из воплощений чип содержит зонды, которые иммобилизованы непосредственно через свой 3′-конец, так называемые поверхностные зонды, которые определены ниже. Каждая разновидность поверхностного зонда содержит область, комплементарную каждой разновидности захватывающего зонда, так что захватывающий зонд может гибридизоваться с поверхностным зондом, в результате получается захватывающий зонд, содержащий свободный удлиняемый 3′-конец. В предпочтительном аспекте изобретения, когда чип содержит поверхностные зонды, захватывающие зонды синтезируют in situ на данном чипе.
Зонды чипа могут быть составлены из рибонуклеотидов и/или дезоксирибонуклеотидов, а также из остатков синтетических нуклеотидов, способных участвовать во взаимодействиях пар оснований по типу Уотсона-Крика или аналогичных им. Так, нуклеиновокислотная область может представлять собой ДНК или РНК или любую их модификацию, например ПНК (пептидо-нуклеиновая кислота) или другие производные, содержащие ненуклеотидые остовы. Однако, в случае транскриптомного анализа, захватывающая область захватывающего зонда должна быть способна праймировать реакцию обратной транскрипции с образованием кДНК, которая комплементарна захваченным молекулам РНК. Как более подробно описано ниже, в случае анализа генома, захватывающая область захватывающего зонда должна быть способна связываться с фрагментами ДНК, что может включать в себя связывание со связывающей областью, которая была добавлена к фрагментированной ДНК. В некоторых воплощениях захватывающая область захватывающего зонда может праймировать реакцию удлинения ДНК (полимераза) с образованием ДНК, которая комплементарна захваченным молекулам ДНК. В других воплощениях захватывающая область может служить матрицей для реакции лигирования между захваченными молекулами ДНК и поверхностным зондом, который непосредственно или опосредованно иммобилизован на подложке. В других воплощениях захватывающая область может быть лигирована с одной нитью захваченных молекул ДНК.
В предпочтительном воплощении изобретения по меньшей мере захватывающая область захватывающего зонда содержит дезоксирибонуклеотиды (dNTP) или состоит из дезоксирибонуклеотидов. В особо предпочтительном воплощении весь захватывающий зонд содержит дезоксирибонуклеотиды или полностью состоит из дезоксирибонуклеотидов.
В предпочтительном воплощении изобретения захватывающие зонды иммобилизованы на подложке чипа непосредственно, т.е. через свой 5′-конец, в результате чего они имеют свободный удлиняемый 3′-конец.
Захватывающие зонды по изобретению содержат по меньшей мере две области, захватывающую область и позиционную область (метку или область идентификации элемента; позиционная область может альтернативно быть определена как идентификационная (ID) область или метка или как позиционная метка). Захватывающий зонд может дополнительно содержать универсальную область, как определено далее ниже. Когда захватывающий зонд опосредованно присоединяется к поверхности чипа в результате гибридизации с поверхностным зондом, необходимо, чтобы захватывающий зонд содержал последовательность (например, часть или область), которая комплементарна последовательности поверхностного зонда. Такая комплементарная последовательность может быть комплементарной к позиционной/идентификационной области и/или универсальной области поверхностного зонда. Другими словами, позиционная область и/или универсальная область могут составлять участок или часть зонда, которая комплементарна последовательности поверхностного зонда. Однако захватывающий зонд может также содержать дополнительную область (или участок, часть или последовательность), которая комплементарна последовательности поверхностного зонда. Как описано более подробно ниже, такой участок, комплементарный последовательности поверхностного зонда, может быть предусмотрен для простоты синтеза, как часть или как результат удлинения захватывающей области (как часть или результат удлинения, которые не будут использоваться для связывания с целевой нуклеиновой кислотой, например РНК, или не способны к такому связыванию).
Захватывающая область в типичном случае локализована на 3′-конце захватывающего зонда и содержит свободный 3′-конец, который может быть удлинен, например, посредством матричной полимеризации. Захватывающая область содержит нуклеотидную последовательность, способную гибридизоваться с нуклеиновой кислотой, например РНК (предпочтительно мРНК), присутствующей в клетках образца ткани, контактирующего с чипом.
Предпочтительно, захватывающая область может быть выбрана или сконструирована так, чтобы связываться (или, в более общем смысле, чтобы быть способной к связыванию) избирательно или специфически с конкретной нуклеиновой кислотой, например РНК, которую желательно детектировать или анализировать. Например, захватывающая область может быть выбрана или синтезирована для избирательного захвата мРНК. Как хорошо известно в данной области, это можно осуществить на основе гибридизации с поли(А)-хвостом мРНК. Таким образом, в предпочтительном воплощении захватывающая область содержит поли(Т)-ДНК-олигонуклеотид, т.е. серии следующих один за другим остатков дезокситимидина, соединенных фосфодиэфирными связями, который способен гибридизоваться с поли(А)-хвостом мРНК. Альтернативно, захватывающая область может содержать нуклеотиды, которые являются функционально или структурно аналогичными поли(Т), т.е. способными избирательно связываться с поли(А), например поли(U)-олигонуклеотид или олигонуклеотид, содержащий аналоги дезокситимидина, при этом указанный олигонуклеотид сохраняет функциональное свойство связываться с поли(А). В конкретном предпочтительном воплощении захватывающая область или более конкретно, поли(Т)-элемент захватывающей области, содержит по меньшей мере 10 нуклеотидов, предпочтительно по меньшей мере 11, 12, 13, 14, 15, 16, 17, 18, 19 или 20 нуклеотидов. В следующем воплощении, захватывающая область или более конкретно, поли(Т)-элемент захватывающей области содержит по меньшей мере 25, 30 или 35 нуклеотидов.
Для захвата нуклеиновой кислоты также могут быть использованы случайные последовательности, как известно в данной области, например случайные гексамеры или сходные последовательности и, следовательно, такие случайные последовательности могут быть использованы для формирования всей захватывающей области или ее части. Например, случайные последовательности могут быть использованы вместе с поли(Т)-(или аналогичными поли(Т)- и т.д.) последовательностями. Так, когда захватывающая область содержит поли(Т)- (или "поли(Т)-подобный") олигонуклеотид, она также может содержать случайную олигонуклеотидную последовательность. Она может, например, быть расположена в 5′- или 3′-направлении по отношению к поли(Т)-последовательности, например на 3′-конце захватывающего зонда, но точное местоположение такой случайной последовательности не является критическим. Подобная конструкция может облегчать захват начальной части поли(А)-хвоста мРНК. Альтернативно, захватывающая область может целиком представлять собой случайную последовательность. Кроме этого могут быть использованы вырожденные захватывающие области, в соответствии с принципами, известными в данной области.
Захватывающая область может быть способна к избирательному связыванию с желаемым подтипом или подклассом нуклеиновой кислоты, например РНК, например конкретным типом РНК, таким как мРНК или рРНК и т.д., как приведено выше, или с конкретным подклассом данного типа РНК, например, конкретной разновидностью мРНК, например соответствующей конкретному гену или группе генов. Такой захватывающий зонд может быть выбран или синтезирован на основании последовательности РНК, которую желательно захватить. Так, он может представлять собой специфичный к последовательности захватывающий зонд, специфичный к конкретной РНК-мишени или группе мишеней (целевой группе и т.п.). Так, в его основе может лежать последовательность конкретного гена или последовательность конкретного мотива или общая/консервативная последовательность и т.д., в соответствии с принципами, хорошо известными в данной области.
В тех воплощениях, где захватывающий зонд иммобилизован на подложке чипа опосредованно, например в результате гибридизации с поверхностным зондом, захватывающая область может дополнительно содержать расположенную "вверх по течению" последовательность (в направлении 5′ по отношению к последовательности, которая гибридизуется с нуклеиновой кислотой, например с РНК образца ткани), которая способна гибридизоваться с 5′-концом поверхностного зонда. Саму по себе захватывающую область захватывающего зонда можно рассматривать как олигонуклеотид захватывающей области, который может быть использован в синтезе захватывающего зонда в воплощениях, когда захватывающий зонд иммобилизован на чипе опосредованно.
Позиционная область (область идентификации элемента или метка) захватывающего зонда локализована непосредственно или опосредованно "вверх по течению", т.е. ближе к 5′-концу захватывающего зонда - молекулы нуклеиновой кислоты, по отношению к захватывающей области. Предпочтительно, позиционная область непосредственно примыкает к захватывающей области, т.е. между захватывающей областью и позиционной областью нет промежуточной последовательности. В некоторых воплощениях позиционная область формирует 5′-конец захватывающего зонда, который может быть иммобилизован непосредственно или опосредованно на подложке чипа.
Как обсуждалось выше, каждый элемент (определенное положение) чипа содержит пятно с разновидностью нуклеиновокислотного зонда, при этом позиционная область каждого элемента является уникальной. Таким образом, "разновидность" захватывающего зонда определяется со ссылкой на его позиционную область; одна и та же разновидность захватывающего зонда будет иметь одну и ту же позиционную область. Однако, не требуется, чтобы каждый представитель разновидности захватывающего зонда имел одну и ту же последовательность на всей своей протяженности. В частности, поскольку захватывающая область может представлять собой или может содержать случайную или вырожденную последовательность, захватывающие области индивидуальных зондов в пределах разновидности могут варьировать. Соответственно, в некоторых воплощениях, в которых захватывающие области захватывающих зондов являются одинаковыми, каждый элемент содержит одну последовательность зонда. Однако в других воплощениях, в которых последовательность захватывающего зонда варьирует, представители разновидности зонда не будут иметь в точности одинаковую последовательность, хотя последовательность позиционной области каждого представителя разновидности будет одной и той же. Требуется, чтобы каждый элемент или каждое положение чипа содержал(о) захватывающий зонд одной разновидности (в частности, чтобы каждый элемент или каждое положение содержал(о) захватывающий зонд, имеющий идентичную позиционную метку, т.е. чтобы каждому элементу или положению соответствовала одна позиционная область). Каждая разновидность имеет свою позиционную область, которая определяет принадлежность к данной разновидности. Однако, каждый представитель разновидности может в некоторых случаях, которые изложены более подробно в настоящем описании, иметь свою захватывающую область, поскольку захватывающая область может быть случайной или вырожденной, либо содержать случайный или вырожденный компонент. Это означает, что в пределах данного элемента или положения, захватывающие области зондов могут отличаться друг от друга.
Таким образом, в некоторых, но необязательно во всех воплощениях, нуклеотидная последовательность любой молекулы зонда, иммобилизованой в конкретном элементе, будет такой же, как и у молекул других зондов, иммобилизованных в том же элементе, но нуклеотидная последовательность зондов каждого элемента отличается, не совпадает или отличима от нуклеотидной последовательности зондов, иммобилизованых в каждом другом элементе. Предпочтительно, каждый элемент содержит свою разновидность зонда. Тем не менее, в некоторых воплощениях может быть преимущественным для группы элементов содержать одну и ту же разновидность зонда, т.е. эффективным является получение элемента, перекрывающего область чипа, превышающую область, перекрываемую одним элементом, например с целью уменьшения разрешения чипа. В других воплощениях чипа, нуклеотидная последовательность позиционной области любой молекулы зонда, иммобилизованной в конкретном элементе, может быть такой же, как и у других молекул зонда, иммобилизованных в этом же элементе, но захватывающая область может отличаться. Захватывающая область может, однако, быть синтезирована так, чтобы захватывать один и тот же тип молекул, например мРНК в целом.
Позиционная область (или метка) захватывающего зонда содержит последовательность, которая является уникальной для каждого элемента и выполняет функцию позиционного или пространственного маркера (идентификационной метки). Таким образом, каждый участок или каждую область образца ткани, например каждую клетку в ткани, можно будет идентифицировать в результате пространственного разрешения по всему чипу, связывающему нуклеиновую кислоту, например РНК (например, транскрипты) из определенной клетки, используя уникальную последовательность позиционной области в захватывающем зонде. Благодаря позиционной области, положение захватывающего зонда на чипе может быть сопоставлено с положением в образце ткани, например оно может быть сопоставлено с положением клетки в образце. Таким образом, позиционную область захватывающей области можно рассматривать как нуклеиновокислотную метку (идентификационную метку).
Любая подходящая последовательность может быть использована в качестве позиционной области в захватывающих зондах по изобретению. Под подходящей последовательностью понимают то, что позиционная область не должна мешать взаимодействию между РНК образца ткани и захватывающей областью захватывающего зонда (т.е. ингибировать или искажать это взаимодействие). Например, позиционная область должна быть синтезирована таким образом, чтобы молекулы нуклеиновой кислоты в образце ткани не гибридизовались специфически с позиционной областью. Предпочтительно, нуклеиновокислотная последовательность позиционной области захватывающих зондов имеет менее 80% идентичности последовательности с нуклеиновокислотными последовательностями в образце ткани. Предпочтительно, позиционная область захватывающего зонда имеет менее 70%, 60%, 50% или менее 40% идентичности последовательности с последовательностями значительной части молекул нуклеиновых кислот в образце ткани. Идентичность последовательностей может быть определена с использованием любого соответствующего способа, известного в данной области, например, с использованием выравнивания с применением алгоритма BLAST.
В предпочтительном воплощении позиционная область каждой разновидности захватывающего зонда содержит уникальную штрихкодовую последовательность. Штрихкодовые последовательности могут быть получены с использованием генератора случайных последовательностей. Генерированные случайным образом последовательности могут далее быть подвергнуты строгому фильтрованию посредством сравнения с картами геномов всех общих эталонных видов и при заранее заданных интервалах Tm, содержании GC-пар и с определенной дистанцией отличия от других штрихкодовых последовательностей, чтобы гарантировать, что данные штрихкодовые последовательности не будут мешать захвату нуклеиновой кислоты, например РНК, из образца ткани и будут различимы между собой без затруднений.
Как упоминалось выше, и в предпочтительном воплощении, захватывающий зонд содержит также универсальную область (или линкерную область или метку). Универсальная область захватывающего зонда локализована непосредственно или опосредованно "вверх по течению", т.е. ближе к 5′-концу захватывающего зонда - молекулы нуклеиновой кислоты, по отношению к позиционной области. Предпочтительно, универсальная область непосредственно примыкает к позиционной области, т.е. между позиционной областью и универсальной областью нет промежуточной последовательности. В воплощениях, где захватывающий зонд содержит универсальную область, данная область будет формировать 5′-конец захватывающего зонда, который может быть иммобилизован непосредственно или опосредованно на подложке чипа.
Универсальная область может быть использована различным образом в способах и применениях по изобретению. Например, способы по изобретению включают стадию высвобождения (например, удаления) по меньшей мере части синтезированных (т.е. удлиненных или лигированных) молекул нуклеиновой кислоты, например кДНК, с поверхности чипа. Как изложено в другом месте данного описания, это может быть достигнуто с использованием ряда способов, один из которых состоит в отщеплении молекулы нуклеиновой кислоты, например кДНК, от поверхности чипа. Таким образом, универсальная область может сама содержать область расщепления, т.е. последовательность, которая может быть расщеплена специфически, либо химически, либо, предпочтительно, ферментативно.
Таким образом, область расщепления может содержать последовательность, которая узнается одним или более чем одним ферментом, способным расщеплять молекулу нуклеиновой кислоты, т.е. способным разрывать фосфодиэфирную связь между двумя или более чем двумя нуклеотидами. Например, область расщепления может содержать последовательность, распознаваемую эндонуклеазой рестрикции (рестрикционным ферментом). Рестрикционные ферменты разрезают двунитевую или однонитевую ДНК в специфически распознаваемых нуклеотидных последовательностях, известных как сайты рестрикции, и подходящие ферменты хорошо известны в данной области. Например, особые преимущества дает применение редкощепящих рестрикционных ферментов, т.е. ферментов с протяженным сайтом распознавания (длиной по меньшей мере 8 пар оснований), для снижения вероятности расщепления в другом месте молекулы нуклеиновой кислоты, например кДНК. В этом отношении будет очевидно, что для удаления или высвобождения по меньшей мере части молекулы нуклеиновой кислоты, например кДНК, требуется высвобождение части, содержащей позиционную область нуклеиновой кислоты, например кДНК, и всей последовательности, расположенной "вниз по течению" по отношению к данной области, т.е. всей последовательности, которая расположена в 3′-направлении по отношению к позиционной области. Следовательно, расщепление молекулы нуклеиновой кислоты, например кДНК, должно осуществляться в 5′-направлении по отношению к позиционной области.
В качестве примера, область расщепления может содержать поли(U)-последовательность, которая может быть расщеплена в результате действия смеси урацил-ДНК-гликозилазы (UDG) и ДНК-гликозилазы-лиазы эндонуклеазы VIII, известной под торговым названием как USER™ enzyme.
Следующий пример области расщепления может быть использован в воплощениях, когда захватывающий зонд иммобилизован на подложке чипа опосредованно, т.е. через поверхностный зонд. Область расщепления может содержать один или более чем один ошибочно спаренный нуклеотид, т.е. когда комплементарные части поверхностного зонда и захватывающего зонда не являются комплементарными на 100%. Такое ошибочное спаривание узнается, например, ферментами MutY и Т7 эндонуклеазой I, что приводит к расщеплению молекулы нуклеиновой кислоты в положении этого ошибочного спаривания.
В некоторых воплощениях изобретения позиционная область захватывающего зонда содержит область расщепления, при этом указанная область расщепления локализована на 5′-конце позиционной области.
Универсальная область может содержать также область амплификации. Она может содержаться в дополнение к области расщепления или вместо области расщепления. В некоторых воплощениях изобретения, как изложено в другом месте данного описания, может иметь преимущество амплифицирование молекул нуклеиновой кислоты, например кДНК, например после того, как они будут высвобождены (например, удалены или отщеплены) с подложки чипа. Следует иметь в виду, однако, что начальный цикл амплификации или, в действительности, любой или все последующие циклы амплификации, также могут происходить in situ на чипе. Область амплификации содержит определенную последовательность, с которой может гибридизоваться амплификационный праймер. Область амплификации универсальной области захватывающего зонда предпочтительно одинакова для каждой разновидности захватывающего зонда. Следовательно, одной реакции амплификации будет достаточно для амплифицирования всей нуклеиновой кислоты, например молекул кДНК (которые могут быть высвобождены с подложки чипа, либо могут не быть высвобождены с подложки чипа перед амплификацией).
Любая подходящая последовательность может быть использована в качестве области амплификации в захватывающих зондах по изобретению. Под подходящей последовательностью понимают то, что область амплификации не должна мешать взаимодействию между нуклеиновой кислотой, например РНК образца ткани, и захватывающей областью захватывающего зонда (т.е. ингибировать или искажать это взаимодействие). Кроме того, область амплификации должна содержать последовательность, которая не является одинаковой или по существу одинаковой с любой последовательностью в нуклеиновой кислоте, например РНК образца ткани, так, чтобы праймер, используемый в реакции амплификации, мог бы гибридизоваться только с областью амплификации в условиях реакции амплификации.
Например, область амплификации должна быть синтезирована так, чтобы молекулы нуклеиновой кислоты в образце ткани не гибридизовались специфически с областью амплификации или ее комплементом. Предпочтительно, нуклеиновокислотная последовательность области амплификации захватывающих зондов и ее комплемент имеют менее 80% идентичности с нуклеиновокислотными последовательностями в образце ткани. Предпочтительно, позиционная область захватывающего зонда имеет менее 70%, 60%, 50% или менее 40% идентичности последовательности с последовательностями значительной части молекул нуклеиновых кислот в образце ткани. Идентичность последовательностей может быть определена с использованием любого подходящего способа, известного в данной области, например с использованием выравнивания с применением алгоритма BLAST.
Таким образом, саму по себе универсальную область захватывающего зонда можно рассматривать как олигонуклеотид универсальной области, который может быть использован в синтезе захватывающего зонда в воплощениях, когда захватывающий зонд иммобилизован на чипе опосредованно.
В одном из репрезентативных воплощений изобретения только позиционная область каждой разновидности захватывающего зонда является уникальной. Поэтому захватывающие области и универсальные области (если они присутствуют) являются в одном из воплощений одинаковыми для каждой разновидности захватывающего зонда для любого конкретного чипа для обеспечения того, чтобы захват нуклеиновой кислоты, например РНК из образца ткани, происходил равномерно по всему чипу. Однако, как обсуждалось выше, в некоторых воплощениях захватывающие области могут отличаться вследствие включения в них случайных или вырожденных последовательностей.
В тех воплощениях, когда захватывающий зонд иммобилизован на подложке чипа опосредованно, например, в результате гибридизации с поверхностным зондом, захватывающий зонд может быть синтезирован на чипе, как описано ниже.
Поверхностные зонды иммобилизованы на подложке чипа непосредственно, например, через свой 3′-конец или на своем 3′-конце. Каждая разновидность поверхностного зонда является уникальной по отношению к каждому элементу (определенному положению) чипа и частично комплементарной захватывающему зонду, определенному выше.
Следовательно, поверхностный зонд содержит на своем 5′-конце область (комплементарную захватывающую область), комплементарную части захватывающей области, которая не связывается с нуклеиновой кислотой, например РНК образца ткани. Другими словами, он содержит область, которая может гибридизоваться по меньшей мере с частью олигонуклеотида захватывающей области. Поверхностный зонд дополнительно содержит область (комплементарную позиционную область или комплементарную область идентификации элемента), которая является комплементарной позиционной области захватывающего зонда. Комплементарная позиционная область локализована непосредственно или опосредованно "вниз по течению" (т.е. на 3′-конце) по отношению к комплементарной захватывающей области, т.е. может иметься промежуточная или линкерная последовательность, разделяющая комплементарную позиционную область и комплементарную захватывающую область. В воплощениях, когда захватывающий зонд синтезируют на поверхности чипа, поверхностные зонды чипа всегда содержат область (комплементарную универсальную область) на 3′-конце поверхностного зонда, т.е. непосредственно или опосредованно "вниз по течению" по отношению к позиционной области, которая комплементарна универсальной области захватывающего зонда. Другими словами, он содержит область, которая может гибридизоваться по меньшей мере с частью олигонуклеотида универсальной области.
В некоторых воплощениях изобретения последовательность поверхностного зонда демонстрирует 100% комплементарности или идентичности последовательности с позиционной и универсальной областями и частью захватывающей области, которая не связывается с нуклеиновой кислотой, например РНК образца ткани. В других воплощениях, последовательность поверхностного зонда может демонстрировать менее 100% идентичности с последовательностями областей захватывающего зонда, например, менее 99%, 98%, 97%, 96%, 95%, 94%, 93%, 92%, 91% или 90%. В особо предпочтительном воплощении изобретения, комплементарная универсальная область имеет менее 100% идентичности последовательности с универсальной областью захватывающего зонда.
В одном из воплощений изобретения захватывающий зонд синтезируют или создают на подложке чипа. В репрезентативном воплощении (см. Фиг.3) данный чип содержит поверхностные зонды, как определено выше. Олигонуклеотиды, которые соответствуют захватывающей области и универсальной области захватывающего зонда, приводят в контакт с чипом и дают им возможность гибридизоваться с комплементарными областями поверхностных зондов. Избыток олигонуклеотидов может быть удален посредством промывки чипа в стандартных условиях гибридизации. Полученный чип содержит частично однонитевые зонды, причем оба конца, 5′-и 3′-конец, поверхностного зонда являются двунитевыми, а комплементарная позиционная область является однонитевой. Данный чип может быть обработан ферментом полимеразой для удлинения 3′-конца олигонуклеотида универсальной области зависимым от матрицы образом, с тем, чтобы синтезировать позиционную область захватывающего зонда. 3′-конец синтезированной позиционной области затем лигируют, например, используя фермент лигазу, с 5′-концом олигонуклеотида захватывающей области, чтобы синтезировать захватывающий зонд. В этом отношении будет очевидно, что 5′-конец олигонуклеотида захватывающей области является фосфорилированным, чтобы дать возможность осуществиться лигированию. Поскольку каждая разновидность поверхностного зонда содержит уникальную комплементарную позиционную область, каждая разновидность захватывающего зонда будет содержать уникальную позиционную область.
Термин "гибридизация" или "гибридизуется", как он использован в данном описании, относится к образованию дуплекса между нуклеотидными последовательностями, которые являются в достаточной степени комплементарными для образования дуплексов посредством взаимодействия пар оснований по типу Уотсона-Крика. Две нуклеотидные последовательности являются "комплементарными" одна другой, когда эти молекулы обладают гомологией организации пар оснований. "Комплементарные" нуклеотидные последовательности в силу специфичности будут объединяться с образованием стабильного дуплекса в соответствующих условиях гибридизации. Например, две последовательности являются комплементарными, когда участок первой последовательности может связываться с участком второй последовательности в антипараллельном направлении, причем 3′-конец каждой из последовательностей связывается с 5′-концом другой последовательности и каждый из A, T(U), G и С одной последовательности затем выстраивается напротив T(U), А, С и G, соответственно, другой последовательности. РНК-последовательности могут также включать комплементарные пары оснований G=U или U=G. Таким образом, две последовательности не обязательно должны иметь идеальную гомологию, для того, чтобы быть "комплементарными" согласно изобретению. Обычно две последовательности являются в достаточной мере комплементарными, когда по меньшей мере примерно 90% (предпочтительно по меньшей мере примерно 95%) нуклеотидов обладают соответствующей друг другу организацией пар оснований на протяжении заданной длины молекулы. Области захватывающих и поверхностных зондов, таким образом, содержат участок комплементарности. Кроме того, захватывающая область захватывающего зонда содержит участок комплементарности нуклеиновой кислоте, например РНК (предпочтительно мРНК) образца ткани.
Захватывающий зонд также может быть синтезирован на подложке чипа с использованием удлинения полимеразой (аналогично тому, как описано выше) и с использованием фермента концевой трансферазы для добавления "хвоста", который может составлять захватывающую область. Это описано далее в примере 7, ниже. Применение концевых трансфераз для добавления нуклеотидных последовательностей к концу олигонуклеотида известно в данной области, например с целью введения гомополимерного хвоста, например поли(Т)-хвоста. Соответственно, при проведении такого синтеза, олигонуклеотид, который соответствует универсальной области захватывающего зонда, может быть приведен в контакт с чипом, и ему дают возможность гибридизоваться с комплементарной областью поверхностных зондов. Избыток олигонуклеотидов может быть удален посредством промывки чипа в стандартных условиях гибридизации. Полученный чип содержит частично однонитевые зонды, причем 5′-концы поверхностных зондов являются двунитевыми, а комплементарная позиционная область является однонитевой. Данный чип может быть обработан ферментом полимеразой для удлинения 3′-конца олигонуклеотида универсальной области зависимым от матрицы образом, с тем, чтобы синтезировать позиционную область захватывающего зонда. Для синтеза захватывающего зонда затем с использованием концевой трансферазы, добавляющей поли(Т)-хвост, может быть введена захватывающая область, например содержащая поли(Т) последовательность.
Типичный чип по изобретению и для применения в способах по изобретению может содержать многочисленные пятна или "элементы". Элемент может быть определен как область или определенное положение на подложке чипа, в которой(м) иммобилизована единственная разновидность захватывающего зонда. Следовательно, каждый элемент будет содержать большое количество молекул зонда одной и той же разновидности. В данном контексте следует понимать, что, несмотря на то, что считается, что каждый захватывающий зонд одной и той же разновидности может иметь одну и ту же последовательность, это необязательно соответствует действительности. Каждая разновидность захватывающего зонда будет иметь одну и ту же позиционную область (т.е. каждый представитель разновидности и, следовательно, каждый зонд в элементе, будут одинаково "помеченными"), но последовательность каждого представителя элемента (разновидности) может иметь отличия, поскольку последовательность захватывающей области может варьировать. Как описано выше, могут быть использованы случайные или вырожденные захватывающие области. Таким образом, захватывающие зонды внутри элемента могут содержать различные случайные или вырожденные последовательности. Количество и плотность элементов чипа будет определять разрешение чипа, т.е. уровень детализации, с которой транскриптом или геном образца ткани может быть проанализирован. Следовательно, более высокая плотность элементов будет, как правило, увеличивать разрешение чипа.
Как обсуждено выше, размер и количество элементов чипа по изобретению будут зависеть от природы образца ткани и требуемого разрешения. Так, если желательно определение транскриптома или генома только для участков клеток в образце ткани (или если образец содержит большие клетки), то количество и/или плотность элементов чипа могут быть уменьшены (т.е. меньше, чем возможное максимальное количество элементов) и/или размер элементов может быть увеличен (т.е. площадь каждого элемента может быть больше, чем площадь наименьшего из возможных элементов), например как на чипе, содержащем небольшое количество больших элементов. Альтернативно, если желательно определение транскриптома или генома индивидуальных клеток в образце, может понадобиться применение максимально возможного количества элементов, что потребует использования минимально возможного размера элемента, например как на чипе, содержащем много небольших элементов.
Хотя разрешение на уровне одной клетки может представлять собой предпочтительный и преимущественный признак настоящего изобретения, его достижение не является необходимым, и разрешение на уровне группы клеток также представляет интерес, например для детекции или распознавания конкретного типа клеток или участка ткани, например для установления различий между нормальными и опухолевыми клетками.
В репрезентативных воплощениях изобретения чип может содержать по меньшей мере 2, 5, 10, 50, 100, 500, 750, 1000, 1500, 3000, 5000, 10000, 20000, 40000, 50000, 75000, 100000, 150000, 200000, 300000, 400000, 500000, 750000, 800000, 1000000, 1200000, 1500000, 1750000, 2000000, 2100000, 3000000, 3500000, 4000000 или 4200000 элементов. Хотя 4200000 представляет собой максимальное количество элементов, доступное в настоящее время на выпускаемых промышленностью чипах, предполагается, что могут быть изготовлены чипы с количеством элементов, превышающим это число, и такие чипы представляют интерес с точки зрения настоящего изобретения. Как отмечено выше, размер элемента может быть уменьшен, и это может позволить размещать большее количество элементов в пределах одной и той же или аналогичной площади. В качестве примера, эти элементы могут быть размещены в пределах площади менее, чем примерно 20 см2, 10 см2, 5 см2, 1 см2, 1 мм2 или 100 мкм2.
Таким образом, в некоторых воплощениях изобретения площадь каждого элемента может быть от примерно 1 мкм2, 2 мкм2, 3 мкм2, 4 мкм2, 5 мкм2, 10 мкм2, 12 мкм2, 15 мкм2, 20 мкм2, 50 мкм2, 75 мкм2, 100 мкм2, 150 мкм2, 200 мкм2, 250 мкм2, 300 мкм2, 400 мкм2 или 500 мкм2.
Будет очевидно, что в способах по изобретению можно использовать образец ткани из любого организма, например растения, животного или гриба. Чип по изобретению позволяет захватывать любую нуклеиновую кислоту, например молекулы мРНК, которые имеются в клетках, способных к транскрипции и/или трансляции. Чипы и способы по изобретению, в частности, подходят для выделения и анализа транскриптома или генома клеток в образце, когда желательным является получение пространственного разрешения транскриптомов или геномов, например, когда клетки являются взаимосвязанными или непосредственно контактируют с соседними клетками. Однако специалисту в данной области будет очевидно, что способы по изобретению также могут быть полезными для анализа транскриптома или генома различных клеток или типов клеток в образце, даже когда указанные клетки не взаимодействуют непосредственно, например в образце крови. Другими словами, нет необходимости, чтобы клетки находились в составе ткани, и они могут быть нанесены на чип в виде отдельных клеток (например, клеток, выделенных из нефиксированной ткани). Такие отдельные клетки, необязательно фиксированные в определенном положении в ткани, тем не менее, могут быть нанесены в определенное положение на чипе и индивидуально идентифицированы. Таким образом, в случае анализа клеток, которые не взаимодействуют непосредственно или не находятся в составе ткани, могут быть применены пространственные параметры описанных способов для получения или считывания уникальной или независимой информации о транскриптоме или геноме индивидуальных клеток.
Таким образом, образец может представлять собой отобранный на анализ или взятый на биопсию образец ткани или, возможно, образец культивируемой ткани. Репрезентативные образцы включают клинические образцы, например цельную кровь или извлеченные из крови продукты, клетки крови, ткани, биоптаты или культивируемые ткани или клетки и т.д., включая клеточные суспензии. Искусственные ткани, например, могут быть получены из клеточной суспензии (включая, например, клетки крови). Клетки можно заключить в матрикс (например, гелевый матрикс, например агар, агарозу и т.д.) и далее традиционным образом можно приготовить их срезы. Такие методики известны в области иммуногистохимии (см., например, Andersson et al., 2006, J. Histochem. Cytochem. 54(12): 1413-23. Epub 2006 Sep 6).
Способ приготовления ткани и обработки полученного образца может влиять на транскриптомный или геномный анализ в способах по изобретению. Кроме того, различные образцы ткани будут иметь разные физические характеристики, и от квалификации специалиста в данной области зависит выполнение необходимых манипуляций для получения образца ткани для применения в способах по изобретению. Однако, из описаний, изложенных в данном изобретении, очевидно, что для получения образца ткани, подходящего для применения в способах по изобретению, может быть использован любой способ приготовления образца. Например, в способах по изобретению можно использовать любой слой клеток толщиной приблизительно в 1 клетку или меньше. В одном из воплощений толщина образца ткани может составлять менее 0,9, 0,8, 0,7, 0,6, 0,5, 0,4, 0,3, 0,2 или 0,1 части поперечного сечения клетки. Однако, поскольку, как отмечалось выше, настоящее изобретение не ограничено разрешением на уровне единичной клетки, то следовательно нет необходимости в том, чтобы образец ткани имел толщину, равную диаметру одной клетки или меньше; при желании можно использовать образцы ткани большей толщины. Например, можно использовать криостатные срезы, толщина которых может составлять, например 10-20 мкм.
Приготовление образца ткани может быть осуществлено любым подходящим или желаемым способом, и изобретение не ограничено каким-либо конкретным типом приготовления ткани. Можно использовать свежие, замороженные, фиксированные или нефиксированные ткани. Для фиксации или заливки образца ткани может быть использована любая желаемая подходящая методика, которая описана и известна в данной области. Следовательно, могут быть использованы любые известные фиксаторы или вещества для заливки тканей.
В качестве первого репрезентативного примера образца ткани для применения в данном изобретении, приготовление ткани можно осуществить посредством глубокого замораживания при температуре, подходящей для поддержания или сохранения целостности (т.е. физических характеристик) структуры ткани, например, менее -20°С и предпочтительно менее -25, -30, -40, -50, -60, -70 или -80°С. Можно приготовить срезы, т.е. тонко нарезанные слои, замороженного образца ткани на поверхности чипа с использованием любого подходящего средства. Например, образец ткани можно приготовить, используя охлажденный микротом, криостат с установленной температурой, подходящей для поддержания как структурной целостности образца ткани, так и химических свойств нуклеиновых кислот в этом образце, например, до температуры менее -15°С и предпочтительно менее -20 или -25°С. Таким образом, образец должен быть обработан таким образом, чтобы свести к минимуму разрушение или деградацию нуклеиновой кислоты, например РНК, в данной ткани. Такие условия хорошо известны в данной области и степень какой-либо деградации можно контролировать посредством выделения нуклеиновой кислоты, например, выделения суммарной РНК, и последующего качественного анализа на различных стадиях приготовления образца ткани.
Во втором репрезентативном примере приготовление ткани можно осуществить с использованием стандартных методов фиксации в формалине и заливки парафином (FFPE), которые хорошо известны в данной области. После фиксации образца ткани и заливки в парафиновый блок или блок из смолы можно приготовить срезы, т.е. тонко нарезанные слои, образцов ткани на чипе. Как отмечалось выше, могут быть использованы другие фиксаторы и/или вещества для заливки тканей.
Очевидно, что срез образца ткани необходимо будет обработать для удаления используемого для заливки вещества, например с целью депарафинизации, т.е. удаления парафина или смолы, из образца перед осуществлением способов по изобретению. Этого можно достичь любым подходящим способом, и способы удаления парафина или смолы или другого вещества из образцов ткани хорошо известны в данной области, например посредством инкубации образца (на поверхности чипа) в соответствующем растворителе, например ксилоле, например, дважды в течение 10 минут, затем промывки этанолом, например 99,5%-ным этанолом в течение 2 минут, 96%-ным этанолом в течение 2 минут и 70%-ным этанолом в течение 2 минут.
Специалисту будет очевидно, что РНК в срезах ткани, полученных с использованием метода FFPE или других методов фиксации и заливки, с большей вероятностью будет частично разрушена по сравнению со случаем использования замороженной ткани. Однако, без связи с какой-либо конкретной теорией, полагают, что это может выгодно в способах по изобретению. Например, если РНК в образце частично разрушена, средняя длина полинуклеотидов РНК будет меньше и в большей степени рандомизирована, чем в образце, не подвергшемся разрушению. Ввиду этого постулируется, что наличие частично разрушенной РНК будет приводить к более низкой систематической ошибке на различных стадиях обработки, изложенных в другом месте данного описания, например при лигировании адаптеров (областей амплификации), амплификации молекул кДНК и их секвенировании.
Таким образом, в одном из воплощений изобретения образец ткани, т.е. срез образца ткани, контактирующего с чипом, получают, используя метод FFPE или другие методы фиксации и заливки. Другими словами, можно осуществить фиксацию образца, например, фиксацию и заливку. В альтернативном воплощении изобретения образец ткани получают, используя глубокое замораживание. В другом воплощении можно использовать контактный отпечаток ткани в соответствии с методиками, известными в данной области. В других воплощениях может быть использован нефиксированный образец.
Толщина среза образца ткани для применения в способах по изобретению может зависеть от использованного способа приготовления образца и физических характеристик ткани. Таким образом, в способах по изобретению может быть использована любая подходящая толщина среза. В репрезентативных воплощениях изобретения толщина среза образца ткани будет составлять по меньшей мере 0,1 мкм, более предпочтительно, по меньшей мере 0,2, 0,3, 0,4, 0,5, 0,7, 1,0, 1,5, 2, 3, 4, 5, 6, 7, 8, 9 или 10 мкм. В других воплощениях толщина среза образца ткани составляет по меньшей мере 10, 12, 13, 14, 15, 20, 30, 40 или 50 мкм. Однако толщина не является критичной, и эти значения приведены только в качестве примера. Можно использовать образцы большей толщины, если это желательно или удобно, например 70 или 100 мкм или более. В типичном случае толщина среза образца ткани находится в диапазоне 1-100 мкм, 1-50 мкм, 1-30 мкм, 1-25 мкм, 1-20 мкм, 1-15 мкм, 1-10 мкм, 2-8 мкм, 3-7 мкм или 4-6 мкм, но, как упомянуто выше, можно использовать образцы большей толщины.
При контакте среза образца ткани с чипом, например, после удаления вещества заливки, например депарафинизации, молекулы нуклеиновой кислоты, например РНК, в образце ткани будут связываться с иммобилизованными на чипе захватывающими зондами. В некоторых воплощениях может оказаться выгодным облегчение гибридизации молекул нуклеиновой кислоты, например РНК, с захватывающими зондами. Как правило, облегчение гибридизации включает в себя изменение условий, в которых проходит гибридизация. Основными условиями, которые могут быть изменены, являются продолжительность и температура инкубации среза ткани на чипе перед проведением стадии обратной транскрипции, которая изложена в другом месте данного описания.
Например, при контакте среза образца ткани с чипом можно проводить инкубацию чипа в течение по меньшей мере 1 часа, чтобы обеспечить возможность гибридизации нуклеиновой кислоты, например РНК, с захватывающими зондами. Предпочтительно чип можно инкубировать в течение по меньшей мере 2, 3, 5, 10, 12, 15, 20, 22 или 24 часов или до момента высыхания среза образца ткани. Продолжительность инкубации чипа не является критичной, и можно использовать любую удобную или желаемую продолжительность. Типичная продолжительность инкубации чипа может составлять до 72 часов включительно. Так, инкубацию можно осуществлять при любой подходящей температуре, например при комнатной температуре, хотя в предпочтительном воплощении срез образца ткани инкубируют на чипе при температуре по меньшей мере 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36 или 37°С. Температура инкубации до 55°С включительно является обычной в данной области техники. В особо предпочтительном воплощении срез образца ткани оставляют высыхать на чипе при 37°С в течение 24 часов. После высыхания среза образца ткани чип можно хранить при комнатной температуре до проведения стадии обратной транскрипции. Очевидно, что если срез образца ткани оставляют высыхать на поверхности чипа, то будет необходимо провести его регидратацию перед проведением дальнейших манипуляций с захваченной нуклеиновой кислотой, например перед стадией обратного транскрибирования захваченной РНК.
Таким образом, способ по изобретению может включать дополнительную стадию регидратации образца ткани после приведения образца в контакт с чипом.
В некоторых воплощениях может оказаться выгодным блокировать (например, маскировать или модифицировать) захватывающие зонды перед приведением в контакт образца ткани с чипом, в частности, когда нуклеиновая кислота в образце ткани является предметом способа модификации до ее захвата на чипе. В частности, может оказаться выгодным блокировать или модифицировать свободный 3′-конец захватывающего зонда. В конкретном воплощении нуклеиновая кислота в образце ткани, например фрагментированная геномная ДНК, может быть модифицирована таким образом, чтобы она могла быть захвачена захватывающим зондом. В качестве примера и как описано более подробно ниже, к концу нуклеиновой кислоты, например фрагментированной геномной ДНК, может быть добавлена адаптерная последовательность (содержащая связывающую область, способную к связыванию с захватывающей областью захватывающего зонда). Этого можно достичь, например, посредством лигирования адаптера или удлинения нуклеиновой кислоты, например с использованием фермента для встраивания дополнительных нуклеотидов в конец последовательности, например поли(А)-хвоста. Перед приведением в контакт образца ткани с чипом необходимо осуществить блокирование или модификацию захватывающих зондов, в частности, свободного 3′-конца захватывающего зонда, чтобы избежать модификации захватывающих зондов, например избежать добавления поли(А)-хвоста к 3′-концу захватывающих зондов. Предпочтительно, чтобы встраивание блокирующей области в захватывающий зонд могло быть осуществлено в процессе его синтеза. Однако блокирующую область можно встраивать в захватывающий зонд после его синтеза.
В некоторых воплощениях захватывающие зонды могут быть блокированы любым подходящим и обратимым способом, который будет препятствовать модификации захватывающих областей в процессе модификации нуклеиновой кислоты образца ткани, который происходит после того, как образец ткани приведен в контакт с чипом. Другими словами, можно обратимо маскировать или модифицировать захватывающие зонды таким образом, чтобы захватывающая область захватывающего зонда не содержала свободного 3′-конца, т.е. таким образом, чтобы 3′-конец был удален или модифицирован либо сделан недоступным, чтобы захватывающий зонд стал невосприимчивым к способу, который используют для модификации нуклеиновой кислоты образца ткани, например лигированию или удлинению, или можно удалить дополнительные нуклеотиды, чтобы демаскировать и/или восстановить 3′-конец захватывающей области захватывающего зонда.
Например, с захватывающими зондами могут быть гибридизованы блокирующие зонды, чтобы маскировать свободный 3′-конец захватывающей области, например зонды в форме шпилек или частично двунитевые зонды, подходящие примеры которых известны в данной области. Блокирование свободного 3′-конца захватывающей области можно осуществить посредством химической модификации, например путем добавления азидометильной группы в качестве химически обратимой кэппирующей группировки, с тем, чтобы захватывающие зонды не содержали свободного 3′-конца. Подходящие альтернативные кэппирующие группировки хорошо известны в данной области, например, концевой нуклеотид захватывающей области может представлять собой обратимый терминирующий нуклеотид, который может быть включен в захватывающий зонд во время или после синтеза зонда.
Альтернативно или в дополнение к этому, захватывающая область захватывающего зонда может быть модифицирована с возможностью последующего удаления любых модификаций захватывающего зонда, например дополнительных нуклеотидов, которые присутствуют, когда молекулы нуклеиновой кислоты образца ткани модифицируют. Например, захватывающие зонды могут содержать дополнительную последовательность, расположенную "вниз по течению" по отношению к захватывающей области, т.е. в 3′-направлении по отношению к захватывающей области, а именно, блокирующую область. Она может быть в форме, например последовательности, распознаваемой эндонуклеазой рестрикции, или последовательности нуклеотидов, расщепляемой под действием ферментов, обладающих специфической активностью, например, быть в форме урацил-содержащей последовательности. После модификации нуклеиновой кислоты образца ткани захватывающие зонды могут быть подвергнуты ферментативному расщеплению, что позволит осуществить удаление блокирующей области и любых дополнительных нуклеотидов, добавленных к 3′-концу захватывающего зонда во время осуществления процесса модификации. Удаление блокирующей области будет демаскировать и/или восстанавливать свободный 3′-конец захватывающей области захватывающего зонда. Блокирующую область можно синтезировать как часть захватывающего зонда или можно добавлять к захватывающему зонду in situ (т.е. в качестве модификации существующего чипа), например путем лигирования блокирующей области.
Блокирование захватывающих зондов можно осуществить, используя любую комбинацию описанных выше механизмов блокирования.
После того, как нуклеиновая кислота образца ткани, например фрагментированная геномная ДНК, модифицирована с целью придания ей способности гибридизоваться с захватывающей областью захватывающего зонда, захватывающий зонд должен быть разблокирован, например посредством диссоциации блокирующего олигонуклеотида, удаления кэппирующей группировки и/или блокирующей области.
Чтобы сопоставить результаты анализа последовательности или информацию о транскриптоме или геноме, полученную от каждого элемента чипа, с участком (т.е. областью или клеткой) образца ткани, образец ткани ориентируют по отношению к элементам чипа. Другими словами, образец ткани помещают на чип таким образом, чтобы положение захватывающего зонда на чипе можно было сопоставить с положением в образце ткани. Посредством этого можно определить, какому положению в образце ткани соответствует положение каждой разновидности захватывающего зонда (или каждого элемента чипа). Другими словами, можно идентифицировать, какому месту в образце ткани соответствует положение каждой разновидности захватывающего зонда. Это может быть выполнено благодаря позиционным маркерам, присутствующим на чипе, как описано ниже. В целях удобства, но это необязательно, можно осуществить визуализацию образца ткани после приведения его в контакт с чипом. Это можно выполнить до или после обработки нуклеиновой кислоты образца ткани, например, до или после стадии синтеза кДНК в соответствии со способом, в частности стадии синтеза первой нити кДНК с использованием обратной транскрипции. В предпочтительном воплощении визуализацию образца ткани осуществляют до высвобождения захваченной и синтезированной (т.е. удлиненной или лигированной) ДНК, например кДНК, с чипа. В особо предпочтительном воплощении визуализацию ткани осуществляют после обработки нуклеиновой кислоты образца ткани, например, после стадии обратной транскрипции и удаления любой оставшейся ткани (например, промывки) с чипа перед высвобождением молекул, например кДНК, с чипа. В некоторых воплощениях стадия обработки захваченной нуклеиновой кислоты, например стадия обратной транскрипции, может способствовать удалению оставшейся ткани с поверхности чипа, например при использовании ткани, приготовленной путем глубокого замораживания. В таком случае визуализацию образца ткани можно осуществлять перед проведением стадии обработки, например стадии синтеза кДНК. Вообще говоря, визуализацию можно осуществлять в любой момент времени после приведения образца ткани в контакт с поверхностью, но до проведения любой стадии, на которой может произойти разрушение или удаление образца ткани. Как отмечалось выше, это может зависеть от образца ткани.
Предпочтительно, чип может содержать маркеры для облегчения ориентации образца ткани или его изображения относительно элементов чипа. Для маркировки чипа можно использовать любые подходящие средства, которые поддаются детектированию при визуализации образца ткани. Например, можно иммобилизовать молекулы, например флуоресцентные молекулы, генерирующие сигнал, предпочтительно видимый сигнал, непосредственно или опосредованно на поверхности чипа. Предпочтительно, чтобы чип содержал по меньшей мере два маркера в разных положениях на поверхности чипа, также предпочтительно по меньшей мере 3, 4, 5, 6, 7, 8, 9, 10, 12, 15, 20, 30, 40, 50, 60, 70, 80, 90 или 100 маркеров. Для удобства можно использовать несколько сотен или даже несколько тысяч маркеров. Может быть предусмотрена схема размещения маркеров, например, они могут быть размещены на внешней кромке чипа, например по всему внешнему ряду элементов чипа. Могут быть использованы другие информативные схемы размещения, например линии, разделяющие чип на секции. Это может способствовать выравниванию изображения образца ткани по отношению к чипу или даже в целом сопоставлению элементов чипа с образцом ткани. Таким образом, маркер может представлять собой иммобилизованную молекулу, с которой дающая сигнал молекула может взаимодействовать для генерирования сигнала. В репрезентативном примере чип может содержать маркерный элемент, например нуклеиновокислотный зонд, иммобилизованный на подложке чипа, с которым может гибридизоваться меченая нуклеиновая кислота. Например, молекула меченой нуклеиновой кислоты или маркерной нуклеиновой кислоты может быть соединена или связана с химической группировкой, способной флуоресцировать, когда ее подвергают воздействию света определенной длины волны (или диапазона длин волн), т.е. возбуждают. Такая молекула маркерной нуклеиновой кислоты может быть приведена в контакт с чипом до окрашивания, одновременно с окрашиванием или после окрашивания образца ткани, чтобы осуществить визуализацию или получить изображение образца ткани. Однако маркер должен быть детектируемым при осуществлении визуализации образца ткани. Таким образом, в предпочтительном воплощении детекцию маркера можно осуществить, применяя те же условия визуализации, которые используются для визуализации образца ткани.
В особо предпочтительном воплощении изобретения чип содержит маркерные элементы, с которыми гибридизуется меченая, предпочтительно меченная флуоресцентной меткой, молекула маркерной нуклеиновой кислоты, например олигонуклеотид.
На стадии визуализации ткани можно применять любые подходящие гистологические способы, известные в данной области, например световую, светлопольную, темнопольную, фазово-контрастную, флуоресцентную, отражательную, интерференционную, конфокальную микроскопию или их комбинацию. В типичном случае образец ткани окрашивают перед визуализацией для обеспечения контраста между разными участками, например клеток, образца ткани. Тип используемого красящего средства будет зависеть от типа ткани и участка клеток, подлежащих окрашиванию. Такие протоколы окрашивания известны в данной области. В некоторых воплощениях может быть использовано более чем одно красящее средство для визуализации (получения изображения) разных аспектов образца ткани, например разных участков образца ткани, специфических клеточных структур (например, органелл) или разных типов клеток. В других воплощениях можно осуществить визуализацию или получить изображение образца ткани без окрашивания образца, например, если образец ткани уже содержит пигменты, обеспечивающие получение достаточного контраста, или если используются конкретные типы микроскопии.
В предпочтительном воплощении визуализацию или получение изображения образца ткани осуществляют, используя флуоресцентную микроскопию.
Образец ткани, т.е. любую оставшуюся ткань, которая сохраняется в контакте с подложкой чипа после стадии обратной транскрипции и, возможно, визуализации, если визуализация желательна и не была осуществлена перед проведением обратной транскрипции, предпочтительно удаляют перед стадией высвобождения молекул кДНК с чипа. Таким образом, способы по изобретению могут включать стадию промывки чипа. Удаление оставшегося образца ткани может быть осуществлено с использованием любых подходящих способов и будет зависеть от образца ткани. В самом простом воплощении чип можно промывать водой. Вода может содержать различные вспомогательные вещества, например поверхностно-активные вещества (например, детергенты), ферменты и т.д., для облегчения удаления ткани. В некоторых воплощениях чип промывают раствором, содержащим фермент протеазу (и подходящий буфер), например протеазу К. В других воплощениях этот раствор также или альтернативно может содержать ферменты целлюлазу, гемицеллюлазу или хитиназу, например, если источником для образца ткани являются растения или грибы. В следующих воплощениях температура раствора, используемого для промывки чипа, может составлять, например, по меньшей мере 30°С, предпочтительно по меньшей мере 35, 40, 45, 50 или 55°С. Очевидно, что при применении промывочного раствора следует свести к минимуму разрушение иммобилизованных молекул нуклеиновой кислоты. Например, в некоторых воплощениях молекулы нуклеиновой кислоты могут быть иммобилизованы на подложке чипа опосредованно, например, путем гибридизации захватывающего зонда и РНК и/или захватывающего зонда и поверхностного зонда, поэтому стадия промывки не должна мешать взаимодействию между молекулами, иммобилизованными на чипе, т.е. не должна вызывать денатурации молекул нуклеиновой кислоты.
После стадии приведения чипа в контакт с образцом ткани в условиях, достаточных для осуществления гибридизации между нуклеиновой кислотой, например РНК (предпочтительно мРНК), образца ткани с захватывающими зондами, осуществляют стадию закрепления (связывания) гибридизованной нуклеиновой кислоты. Закрепление или связывание захваченной нуклеиновой кислоты включает в себя ковалентное присоединение комплементарной нити гибридизованной нуклеиновой кислоты к захватывающему зонду (т.е. посредством межнуклеотидной связи, фосфодиэфирной связи между расположенными рядом 3′-гидроксильным и 5′-фосфатным концами двух непосредственно примыкающих друг к другу нуклеотидов), тем самым захваченная нуклеиновая кислота метится или маркируется позиционной областью, специфичной к элементу, на котором нуклеиновая кислота захвачена.
В некоторых воплощениях закрепление гибридизованной нуклеиновой кислоты, например однонитевой нуклеиновой кислоты, может включать удлинение захватывающего зонда с получением копии захваченной нуклеиновой кислоты, например синтез кДНК на основе захваченной (гибридизованной) РНК. Понятно, что речь идет о синтезе комплементарной нити гибридизованной нуклеиновой кислоты, например синтезе кДНК на основе захваченной РНК-матрицы (РНК, гибридизованной с захватывающей областью захватывающего зонда). Таким образом, на начальной стадии удлинения захватывающего зонда, например синтеза кДНК, захваченная (гибридизованная) нуклеиновая кислота, например РНК, действует в качестве матрицы на стадии удлинения, например, обратной транскрипции. В других воплощениях, как описано ниже, закрепление гибридизованной нуклеиновой кислоты, например, частично двунитевой ДНК, может включать ковалентное связывание гибридизованной нуклеиновой кислоты, например фрагментированной ДНК, с захватывающим зондом, например лигирование с захватывающим зондом комплементарной нити нуклеиновой кислоты, гибридизованной с захватывающим зондом, в реакции лигирования.
Термин "обратная транскрипция" относится к стадии синтеза кДНК (комплементарной или копийной ДНК) по РНК, предпочтительно мРНК (матричной (или информационной) РНК), с использованием обратной транскриптазы. Таким образом, кДНК можно рассматривать как копию РНК, присутствующей в клетке на момент отбора образца ткани, т.е. она соответствует всем или некоторым генам, которые были экспрессированы в указанной клетке на момент выделения.
Захватывающий зонд, конкретно, захватывающая область захватывающего зонда, действует в качестве праймера для получения комплементарной нити нуклеиновой кислоты, гибридизованной с захватывающим зондом, например праймера для обратной транскрипции. Таким образом, молекулы нуклеиновой кислоты, например кДНК, синтезированные в реакции удлинения, например реакции обратной транскрипции, включают в себя последовательность захватывающего зонда, т.е. реакция удлинения, например реакция обратной транскрипции, может рассматриваться как путь опосредованного мечения нуклеиновой кислоты, например транскриптов, образца ткани, который находится в контакте с каждым элементом чипа. Как упомянуто выше, каждая разновидность захватывающего зонда содержит позиционную область (идентификационную метку элемента), которая представляет собой уникальную последовательность для каждого элемента чипа. Таким образом, все молекулы нуклеиновой кислоты, например кДНК, синтезированные в пределах конкретного элемента, будут содержать одну и ту же нуклеиновокислотную "метку".
Молекулы нуклеиновой кислоты, например кДНК, синтезированные в пределах каждого элемента чипа, могут представлять геном участка или области образца ткани, находящейся в контакте с этим элементом, например ткани или типа клеток либо их группы или подгруппы, либо гены, экспрессируемые в этом участке или области образца ткани, и также могут представлять гены, экспрессируемые в конкретных условиях, например в конкретный момент времени, в определенном окружении, на определенной стадии развития или в ответ на стимул и т.д. Таким образом, кДНК в пределах любого единичного элемента может представлять гены, экспрессируемые в одной клетке, или, если этот элемент находится в контакте с образцом в области межклеточных контактов, кДНК может представлять гены, экспрессируемые более чем в одной клетке. Аналогично, если одна клетка находится в контакте со многими элементами, то каждый элемент может представлять часть генов, экспрессируемых в указанной клетке. Аналогично, в тех воплощениях, в которых захваченная нуклеиновая кислота представляет собой ДНК, любой единичный элемент может быть типичным представителем генома одной клетки или более чем одной клетки. Альтернативно, геном одной клетки может быть представлен многими элементами.
Стадия удлинения захватывающего зонда, например стадия обратной транскрипции, может быть проведена с использованием любых подходящих ферментов, и в данной области существует много протоколов, как подробно описано ниже. Однако, будет очевидно, что нет необходимости в предоставлении праймера для синтеза первой нити нуклеиновой кислоты, например кДНК, поскольку захватывающая область захватывающего зонда действует в качестве праймера, например праймера обратной транскрипции.
Предпочтительно, что в контексте настоящего изобретения закрепленную нуклеиновую кислоту (т.е. нуклеиновую кислоту, ковалентно присоединенную к захватывающему зонду), например кДНК, обрабатывают таким образом, чтобы она содержала двунитевую ДНК. Однако, в некоторых воплощениях, захваченная ДНК может уже содержать двунитевую ДНК, например, когда с захватывающим зондом лигируют частично двунитевую фрагментированную ДНК. Обработка захваченной нуклеиновой кислоты для получения двунитевой ДНК может быть осуществлена в одной реакции с синтезом только второй нити ДНК, например кДНК, т.е. с получением двунитевых молекул ДНК без увеличения числа двунитевых молекул ДНК, либо в реакции амплификации с получением большого числа копий второй нити, которые могут быть в форме однонитевой ДНК (например, при линейной амплификации) или двунитевой ДНК, например кДНК (например, при экспоненциальной амплификации).
Стадия синтеза второй нити ДНК, например кДНК, может протекать in situ на чипе либо в виде отдельной стадии синтеза второй нити, например с использованием случайных праймеров, как описано более подробно ниже, либо на начальной стадии реакции амплификации. Альтернативно, первая нить ДНК, например кДНК, (нить, содержащая, т.е. включающая в себя захватывающий зонд) может быть высвобождена с чипа, а синтез второй нити, независимо от того, происходит ли он в виде отдельной стадии или в реакции амплификации, может осуществляться позже, например в реакции, проводимой в растворе.
В том случае, когда синтез второй нити протекает на чипе (т.е. in situ), данный способ может включать возможную стадию удаления захваченной нуклеиновой кислоты, например РНК, перед синтезом второй нити, например с использованием расщепляющего РНК фермента (РНКазы), например РНКазы Н. Применяемые для этого методики хорошо известны и описаны в данной области. Однако обычно в этом нет необходимости, и в большинстве случаев РНК разрушается естественным образом. Удаление образца ткани с чипа обычно будет приводить к удалению РНК с чипа. При желании для повышения надежности удаления РНК можно использовать РНКазу Н.
Например, в образцах ткани, которые содержат большие количества РНК, на стадии синтеза двунитевой кДНК можно получить достаточное количество кДНК, которая может быть секвенирована непосредственно (после высвобождения с чипа). В этом случае синтез второй нити кДНК может быть выполнен любым способом, известным в данной области, и как описано ниже. Реакция синтеза второй нити может быть осуществлена на чипе непосредственно, т.е. пока кДНК иммобилизована на чипе, или, предпочтительно, после высвобождения кДНК с подложки чипа, как описано ниже.
В других воплощениях потребуется увеличить, т.е. амплифицировать, количество закрепленной нуклеиновой кислоты, например синтезированной кДНК, чтобы получить количества, достаточные для секвенирования ДНК. В этом воплощении первая нить закрепленных молекул нуклеиновой кислоты, например кДНК, которые также содержат захватывающий зонд элементов чипа, действует в качестве матрицы для реакции амплификации, например полимеразной цепной реакции. Первый продукт реакции амплификации будет представлять собой вторую нить ДНК, например кДНК, которая сама будет действовать в качестве матрицы для следующих циклов реакции амплификации.
В любом из описанных выше воплощений вторая нить ДНК, например кДНК, будет содержать участок, комплементарный последовательности захватывающего зонда. Если захватывающий зонд содержит универсальную область и, в частности, область амплификации в пределах данной универсальной области, то это можно использовать для последующей амплификации ДНК, например кДНК, например подвергаемая амплификации реакционная смесь может содержать праймер с той же последовательностью, что и область амплификации, т.е. праймер, который является комплементарным участку, комплементарному области амплификации, т.е. гибридизуется с ним. Учитывая тот факт, что область амплификации расположена "вверх по течению" по отношению к позиционной области захватывающего зонда (в закрепленной нуклеиновой кислоте, например первой нити кДНК), данный участок, комплементарный позиционной области, будет встроен во вторую нить молекул ДНК, например кДНК.
В тех воплощениях, где вторую нить ДНК, например кДНК, синтезируют в одной реакции, синтез второй нити может быть осуществлен любым подходящим способом. Например, первая нить кДНК предпочтительно, но необязательно, высвобожденная с подложки чипа, может быть инкубирована со случайными праймерами, например гексамерными праймерами, и ДНК-полимеразой, предпочтительно полимеразой, обладающей активностью замещения нити, например фрагментом Кленова (экзо), в условиях, достаточных для осуществления матричного синтеза ДНК. В результате этого процесса будут получены двунитевые молекулы кДНК различной длины, и маловероятно получение полноразмерных молекул кДНК, т.е. молекул кДНК, которые соответствуют целой мРНК, на основе которой они были синтезированы. Случайные праймеры будут гибридизоваться с первой нитью молекул кДНК в случайном положении, т.е. скорее в пределах последовательности, чем на конце последовательности.
Если желательно синтезировать полноразмерные молекулы ДНК, например кДНК, т.е. молекулы, которые соответствуют целой захваченной молекуле нуклеиновой кислоты, например молекуле РНК (если нуклеиновая кислота, например РНК, была частично разрушена в образце ткани, то захваченные молекулы нуклеиновой кислоты, например РНК, не будут "полноразмерными" транскриптами или не будут иметь ту же длину, что и начальные фрагменты геномной ДНК), то 3′-конец закрепленных молекул нуклеиновой кислоты, например первой нити кДНК, может быть модифицирован. Например, с 3′-концом молекул кДНК может быть лигирован линкер или адаптер. Этого можно достичь, используя ферменты, осуществляющие лигирование одной нити, такие как Т4 РНК-лигаза или Circligase™ (Epicentre Biotechnologies).
Альтернативно, вспомогательный зонд (частично двунитевая молекула ДНК, способная гибридизоваться с 3′-концом первой нити молекулы кДНК), может быть лигирован с 3′-концом закрепленной молекулы нуклеиновой кислоты, например первой нитью кДНК, с использованием фермента, осуществляющего лигирование двух нитей, такого как Т4 ДНК-лигаза. Другие ферменты, подходящие для стадии лигирования, известны в данной области и включают, например Tth ДНК-лигазу, Taq ДНК-лигазу, ДНК-лигазу Thermococcus sp.(штамм 9°N) (ДНК-лигазу 9°N™, New England Biolabs) и Ampligase™ (Epicentre Biotechnologies). Вспомогательный зонд также содержит специфическую последовательность, которая может праймировать синтез второй нити ДНК, например кДНК, с использованием праймера, комплементарного части вспомогательного зонда, лигированного с закрепленной нуклеиновой кислотой, например первой нитью кДНК. Другая альтернатива заключается в применении фермента с активностью концевой трансферазы для встраивания полинуклеотидного хвоста, например поли(А)-хвоста, по 3′-концу закрепленных молекул нуклеиновой кислоты, например первой нити кДНК. Синтез второй нити можно праймировать, используя поли(Т)-праймер, который также может содержать специфическую область амплификации для последующей амплификации. Другие способы синтеза молекул "полноразмерной" двунитевой ДНК, например кДНК (или синтеза второй нити максимальной длины), хорошо известны в данной области.
В некоторых воплощениях для синтеза второй нити можно использовать метод переключения матрицы, например, применяя технологию SMART™ от Clontech®. Технология SMART (Switching Mechanism at 5′ End of RNA Template, механизм переключения на 5′-конце матрицы РНК) хорошо известна в данной области и основана на открытии того, что ферменты обратные транскриптазы, например Superscript® II (Invitrogen), способны добавлять несколько нуклеотидов к 3′-концу удлиняемой молекулы кДНК, т.е. создавать гибрид ДНК/РНК с однонитевым выступом ДНК на 3′-конце. Наличие выступа ДНК может обеспечить получение целевой последовательности, с которой может гибридизоваться олигонуклеотидный зонд, создавая дополнительную матрицу для дальнейшего удлинения молекулы кДНК. Предпочтительно, олигонуклеотидный зонд, который гибридизуется с выступом кДНК, содержит последовательность области амплификации, комплемент которой встроен в синтезированный продукт, первую нить кДНК. Праймеры, содержащие последовательность области амплификации, которые будут гибридизоваться с последовательностью, комплементарной области амплификации, встроенной в первую нить кДНК, могут быть добавлены к реакционной смеси с целью праймирования синтеза второй нити с использованием подходящего фермента полимеразы и первой нити кДНК в качестве матрицы. Этот способ позволяет избежать необходимости лигирования адаптеров с 3′-концом первой нити кДНК. Несмотря на то, что метод переключения матрицы первоначально был разработан для полноразмерных мРНК, которые имеют 5′-кэппированную структуру, позже было продемонстрировано, что он одинаково хорошо работает в случае укороченных мРНК, не имеющих кэппированной структуры. Таким образом, в способах по изобретению можно использовать метод переключения матрицы для синтеза полноразмерных и/или неполных или укороченных молекул кДНК. Таким образом, в предпочтительном воплощении изобретения для синтеза второй нити можно применить метод переключения матрицы или синтеза второй нити можно добиться, применяя метод переключения матрицы. В особо предпочтительном воплощении реакцию переключения матрицы, т.е. последующее удлинение первой нити кДНК для встраивания участка, комплементарного области амплификации, осуществляют in situ (пока захватывающий зонд все еще присоединен, непосредственно или опосредованно, к чипу). Предпочтительно, реакцию синтеза второй нити также проводят in situ.
В тех воплощениях, когда может оказаться необходимым или выгодным увеличить количество, обогатить или амплифицировать молекулы ДНК, например кДНК, в молекулы ДНК, например кДНК, могут быть встроены области амплификации. Как обсуждалось выше, первая область амплификации может быть встроена в закрепленные молекулы нуклеиновой кислоты, например первую нить молекул кДНК, когда захватывающий зонд содержит универсальную область, содержащую область амплификации. В этих воплощениях в процессе синтеза второй нити может быть встроена вторая область амплификации. Например, праймеры, используемые для синтеза второй нити кДНК, например случайные гексамерные праймеры, поли(Т)-праймер, праймер, комплементарный вспомогательному зонду, могут содержать на своем 5′-конце область амплификации, т.е. нуклеотидную последовательность, с которой может гибридизоваться амплифицирующий праймер. Таким образом, полученная двунитевая ДНК может содержать область амплификации при каждом 5′-конце или в направлении каждого 5′-конца двунитевых молекул ДНК, например кДНК. Эти области амплификации можно использовать в качестве мишеней для праймеров, применяемых в реакции амплификации, например ПЦР. Альтернативно, линкер или адаптер, лигированный с 3′-концом молекул закрепленной нуклеиновой кислоты, например молекул первой нити кДНК, может содержать вторую универсальную область, содержащую вторую область амплификации. Аналогично, вторую область амплификации можно встроить в первую нить молекул кДНК, используя метод переключения матрицы.
В тех воплощениях, где захватывающий зонд не содержит универсальной области, в частности, содержащей область амплификации, вторая нить молекул кДНК может быть синтезирована в соответствии с приведенным выше описанием. Полученные молекулы двунитевой ДНК могут быть модифицированы с целью встраивания области амплификации по 5′-концу первой нити ДНК, например кДНК, (первая область амплификации) и, если она не встроена на стадии синтеза второй нити ДНК, например кДНК, то по 5′-концу второй нити ДНК, например кДНК, (вторая область амплификации). Такие области амплификации могут быть встроены, например, путем лигирования двунитевых адаптеров с концами молекул ДНК, например кДНК. Ферменты, подходящие для стадии лигирования, известны в данной области и включают, например Tth ДНК-лигазу, Taq ДНК-лигазу, ДНК-лигазу из Thermococcus sp.(штамм 9°N) (ДНК-лигазу 9°N™, New England Biolabs), Ampligase™ (Epicentre Biotechnologies) и Т4 ДНК-лигазу. В предпочтительном воплощении первая и вторая области амплификации содержат разные последовательности.
Таким образом, на основании изложенного выше очевидно, что универсальные области, которые могут содержать область амплификации, можно добавить к закрепленным (т.е. удлиненным или лигированным) молекулам ДНК, например, к молекулам кДНК, или к их комплементам (например, второй нити), используя различные способы и методики и комбинации таких методик, известные в данной области, например посредством применения праймеров, включающих в себя такую область, лигирования адаптеров, применения ферментов-концевых трансфераз, и/или посредством применения методов переключения матрицы. Как очевидно из приведенного в данном описании обсуждения, такие области могут быть добавлены до или после высвобождения молекул ДНК с чипа.
На основании изложенного выше описания будет очевидно, что все молекулы ДНК, например кДНК, из одного чипа, которые синтезированы способами по изобретению, могут содержать одни и те же первую и вторую области амплификации. Следовательно, одной реакции амплификации, например ПЦР, может быть достаточно, чтобы амплифицировать все молекулы ДНК, например кДНК. Таким образом, в предпочтительном воплощении способ по изобретению может включать стадию амплификации молекул ДНК, например кДНК. В одном из воплощений стадию амплификации осуществляют после высвобождения молекул ДНК, например кДНК, с подложки чипа. В других воплощениях амплификация может быть осуществлена на чипе (т.е. in situ на чипе). В данной области известно, что реакции амплификации можно проводить на чипах, и для проведения таких реакций имеются термоциклеры для чипов. Так, в одном из воплощений в качестве основных чипов по настоящему изобретению могут быть использованы чипы, которые известны в данной области как платформы для секвенирования или для применения в каком-либо виде анализа последовательностей (например, в технологиях или с использованием технологий секвенирования следующего поколения) (например, чипы на основе гранул от Illumina и т.д.).
Для синтеза второй нити ДНК, например кДНК, предпочтительно использовать полимеразу замещения нити (например, Ф29 ДНК-полимеразу, Bst ДНК-полимеразу (экзо-), ДНК-полимеразу Кленова (экзо-)), если высвобожденная с подложки чипа кДНК содержит молекулу частично двунитевой нуклеиновой кислоты. Например, высвобожденные нуклеиновые кислоты будут по меньшей мере частично двунитевыми (например, гибрид ДНК:ДНК, ДНК:РНК или ДНК:ДНК/РНК) в тех воплощениях, где захватывающий зонд иммобилизован опосредованно на подложке чипа через поверхностный зонд, и стадия высвобождения молекул ДНК, например кДНК, включает стадию отщепления. Полимераза замещения нити необходима для обеспечения того, чтобы при синтезе второй нити кДНК во вторую нить ДНК, например кДНК, был встроен участок, комплементарный позиционной области (область идентификации элемента).
Очевидно, что стадия высвобождения по меньшей мере части молекул ДНК, например кДНК, или их ампликонов с поверхности или подложки чипа может быть осуществлена с использованием ряда способов. Первоочередной задачей стадии высвобождения является получение молекул, в которых позиционная область захватывающего зонда (или его комплемента) встроена (или включена) таким образом, чтобы молекулы ДНК, например кДНК, или их ампликоны были бы "мечены" соответственно их элементу (или положению) на чипе. Таким образом, в результате осуществления стадии высвобождения, молекулы ДНК, например кДНК, или их ампликоны, удаляются с чипа, причем молекулы ДНК, например кДНК, или ампликоны включают в себя позиционную область или ее комплемент (посредством которых они встроены в закрепленную нуклеиновую кислоту, например первую нить кДНК, в результате, например удлинения захватывающего зонда, и, возможно, скопированы во вторую нить ДНК, если синтез второй нити происходит на чипе, или скопированы в ампликоны, если амплификация происходит на чипе). Следовательно, чтобы получить данные из анализа последовательностей, которые могут быть соотнесены с различными участками в образце ткани, необходимо, чтобы высвобожденные молекулы содержали позиционную область захватывающего зонда (или ее комплемент).
Поскольку высвобожденная молекула может представлять собой молекулу первой и/или второй нити ДНК, например кДНК, или ампликон, и поскольку захватывающий зонд может быть иммобилизован на чипе опосредованно, очевидно, что хотя стадия высвобождения может включать стадию отщепления молекулы ДНК, например кДНК, от чипа, необходимости в стадии отщепления для стадии высвобождения нуклеиновой кислоты нет; молекула ДНК, например кДНК, или ампликон может просто высвобождаться в результате денатурации двунитевой молекулы, например в результате отсоединения второй нити кДНК от первой нити кДНК, или отсоединения ампликона от его матрицы, или отсоединения молекулы первой нити кДНК (т.е. удлиненного захватывающего зонда) от поверхностного зонда. Соответственно, молекула ДНК, например кДНК, может высвобождаться с чипа в результате отщепления нуклеиновой кислоты и/или в результате денатурации (например, под действием нагревания для денатурации двунитевой молекулы). В том случае, когда амплификацию осуществляют in situ на чипе, это несомненно будет относиться к высвобождению ампликонов в результате денатурации в циклически повторяющейся реакции.
В некоторых воплощениях, молекулы ДНК, например кДНК, высвобождаются в результате ферментативного расщепления области расщепления, которая может быть локализована в универсальной области или позиционной области захватывающего зонда. Как упомянуто выше, область расщепления должна быть локализована "вверх по течению" (на 5′-конце) относительно позиционной области, чтобы высвобожденные молекулы ДНК, например кДНК, содержали позиционную (идентификационную) область. Подходящие ферменты для расщепления нуклеиновых кислот включают эндонуклеазы рестрикции, например Rsal. Другие ферменты, например смесь урацил-ДНК-гликозилазы (UDG) и ДНК-гликозилазы-лиазы и эндонуклеазы VIII (USER™ enzyme) или комбинация ферментов MutY и Т7 эндонуклеазы I, относятся к предпочтительным воплощениям способов по изобретению.
В альтернативном воплощении, молекулы ДНК, например кДНК, могут высвобождаться с поверхности или с подложки чипа физическим способом. Например, в тех воплощениях, где захватывающий зонд иммобилизован на подложке чипа опосредованно, например, в результате гибридизации с поверхностным зондом, может быть достаточно разрушить взаимодействие между молекулами нуклеиновой кислоты. Способы разрушения взаимодействия между молекулами нуклеиновой кислоты, например денатурация двунитевых молекул нуклеиновой кислоты, хорошо известны в данной области. Эффективный способ высвобождения молекул ДНК, например кДНК, (т.е. отделения от чипа синтезированных молекул ДНК, например кДНК) заключается в использовании раствора, который служит помехой водородным связям в двунитевых молекулах. В предпочтительном воплощении изобретения, молекулы ДНК, например кДНК, могут высвобождаться посредством применения нагретой воды, например воды или буфера, нагретых по меньшей мере до 85°С, предпочтительно по меньшей мере до 90, 91, 92, 93, 94, 95, 96, 97, 98, 99°С. Как альтернатива или в дополнение к применению температуры, достаточной для разрушения водородных связей, раствор может содержать соли, поверхностно-активные вещества и т.д., которые также могут дестабилизировать взаимодействие между молекулами нуклеиновой кислоты, приводя к высвобождению молекул ДНК, например кДНК.
Очевидно, что применение раствора с повышенной температурой, например воды при 90-99°С, может оказаться достаточным, чтобы разрушить ковалентную связь, использованную для иммобилизации захватывающего зонда или поверхностного зонда на подложке чипа. Таким образом, в предпочтительном воплощении, молекулы ДНК, например кДНК, могут высвобождаться в результате подведения горячей воды к чипу с целью разрушения ковалентно иммобилизованных захватывающих или поверхностных зондов.
Подразумевается, что высвобожденные молекулы ДНК, например кДНК (раствор, содержащий высвобожденные молекулы ДНК, например кДНК) собирают для дальнейшей работы, например, для синтеза и/или амплификации второй нити. Тем не менее, можно отметить, что способ по изобретению включает стадию сбора или извлечения высвобожденных молекул ДНК, например кДНК. Как указано выше, в случае амплификации in situ высвобожденные молекулы могут включать ампликоны закрепленной нуклеиновой кислоты, например кДНК.
В воплощениях способов по изобретению может оказаться желательным удалить любые неудлиненные или нелигированные захватывающие зонды. Это можно сделать, например, после стадии высвобождения молекул ДНК с чипа. Для такого удаления можно использовать любой желаемый или подходящий способ, включая, например, применение фермента для разрушения неудлиненных или нелигированных зондов, например экзонуклеазы.
Молекулы ДНК, например кДНК, или ампликоны, высвобожденные с чипа, которые могут быть модифицированы, как обсуждалось выше, анализируют исследовательских целях (например, чтобы определить их последовательность, хотя, как указано выше, необходимости в фактическом определении последовательности нет, можно использовать любой метод анализа последовательности). Таким образом, можно использовать любой метод анализа нуклеиновых кислот. На стадии анализа последовательности можно идентифицировать позиционную область и вследствие этого осуществить локализацию положения анализируемой молекулы в образце ткани. Аналогично, можно определить основные свойства или особенности анализируемой молекулы. Таким образом можно определить нуклеиновую кислоту, например РНК, в заданном положении на чипе и вследствие этого в образце ткани. Следовательно, стадия анализа может включать или на стадии анализа можно использовать любой метод идентификации анализируемой молекулы (и вследствие этого "целевой молекулы") и ее позиционной области. В общем случае таким методом будет специфичный к последовательности метод. Например, в данном способе можно использовать специфичные к последовательности праймеры или зонды, в частности, праймеры или зонды, специфичные к позиционной области и/или к молекуле конкретной нуклеиновой кислоты, детекцию или анализ которой необходимо провести, например к молекуле ДНК, которая соответствует молекуле нуклеиновой кислоты, например РНК или кДНК, детекцию которой необходимо провести. Обычно в таком методе могут быть использованы специфичные к последовательности амплификационные праймеры, например ПЦР-праймеры.
В некоторых воплощениях может оказаться желательным провести анализ подкласса или семейства молекул, родственных целевым молекулам, например всех последовательностей, которые кодируют конкретную группу белков, характеризующихся сходством последовательности и/или наличием консервативных доменов, например семейства рецепторов. Следовательно, в способах амплификации и/или анализа, изложенных в данном описании, можно применять вырожденные или специфичные к семейству генов праймеры или зонды, которые гибридизуются с подклассом захваченных нуклеиновых кислот или происходящих из них нуклеиновых кислот, например с ампликонами. В особо предпочтительном воплощении в способах амплификации и/или анализа можно применять универсальный праймер (т.е. праймер, общий для всех захваченных последовательностей) в комбинации с вырожденным или специфичным к семейству генов праймером, специфичным для подкласса целевых молекул.
Таким образом, в одном из воплощений используют основанные на амплификации, в частности, основанные на ПЦР, способы анализа последовательностей.
Однако, на стадиях модифицикации и/или амплификации высвобожденных молекул ДНК, например кДНК, в образец могут вводиться дополнительные компоненты, например ферменты, праймеры, нуклеотиды и т.д. Таким образом, способы по изобретению могут дополнительно включать стадию очистки образца, содержащего высвобожденные молекулы ДНК, например кДНК, или ампликоны, перед проведением анализа последовательностей, например для удаления олигонуклеотидных праймеров, нуклеотидов, солей и т.д., которые могут препятствовать реакциям секвенирования. Можно использовать любой подходящий метод очистки молекул ДНК, например молекул кДНК.
Как указано выше, анализ последовательностей высвобожденных молекул ДНК может быть непосредственным или опосредованным. Так, объектом для анализа последовательности (который можно рассматривать как молекулу, которую подвергают стадии или процессу анализа последовательности) может непосредственно быть молекула, которая высвобождается с чипа, или это может быть происходящая из нее молекула. Таким образом, например, в случае стадии анализа последовательности, которая включает в себя реакцию секвенирования, матрицей для секвенирования может быть молекула, которая высвобождается с чипа, или это может быть происходящая из нее молекула. Например, первая и/или вторая нить молекулы ДНК, например кДНК, высвобожденной с чипа, может быть непосредственно подвергнута анализу последовательности (например, секвенированию), т.е. может непосредственно принимать участие в реакции или процессе анализа последовательности (например, в реакции секвенирования или процессе секвенирования, или быть молекулой, которую секвенируют или идентифицируют иным способом). В случае амплификации in situ высвобожденная молекула может представлять собой ампликон. Альтернативно, высвобожденная молекула может быть подвергнута стадии синтеза или амплификации второй нити перед проведением анализа последовательности (например, перед секвенированием или идентификацией другими способами). Таким образом, объект анализа последовательности (например, матрица) может представлять собой ампликон или вторую нить молекулы, которая непосредственно высвобождается с чипа.
Анализу последовательности (например, секвенированию) можно подвергнуть обе нити двунитевой молекулы, но данное изобретение этим не ограничивается, и можно провести анализ однонитевых молекул (например, кДНК) (например, секвенировать их). Например, можно использовать различные технологии секвенирования для секвенирования отдельной молекулы, например технологии Helicos или Pacbio либо разрабатываемые технологии секвенирования на основе нанопор. Так, в одном из воплощений секвенированию может быть подвергнута первая нить ДНК, например кДНК. Для первой нити ДНК, например кДНК, может потребоваться модификация по 3′-концу, дающая возможность осуществлять секвенирование одинарной молекулы. Это может быть выполнено с использованием методик, аналогичных таковым для обработки второй нити ДНК, например кДНК. Подобные методики известны в данной области.
В предпочтительном аспекте изобретения, с использованием анализа последовательности будет идентифицирована или раскрыта часть последовательности захваченной нуклеиновой кислоты, например последовательности РНК, и последовательность позиционной области. Последовательность позиционной области (или метки) будет определять элемент, которым молекула нуклеиновой кислоты, например мРНК, была захвачена. Последовательность захваченной молекулы нуклеиновой кислоты, например РНК, можно сравнить с базой данных последовательностей того организма, из которого происходит образец, чтобы определить ген, которому она соответствует. В результате установления того, какой участок (например, клетка) образца ткани был в контакте с данным элементом, можно определить, в каком участке образца ткани происходила экспрессия указанного гена (или содержался данный ген, например, в случае пространственной геномики). Такой анализ может быть проведен для всех молекул ДНК, например кДНК, синтезированных способами по изобретению, с получением данных о пространственном транскриптоме или геноме образца ткани.
В качестве репрезентативного примера можно провести анализ данных секвенирования с целью сортировки последовательностей по конкретным разновидностям захватывающего зонда, т.е. в соответствии с последовательностью позиционной области. Это можно осуществить, например, используя штрихкодовый сплиттер с применением формата FASTQ пакета FastX-toolkit (FastX toolkit FASTQ Barcode splitter tool) с целью сортировки последовательностей по отдельным файлам для соответствующих последовательностей позиционных областей (меток) захватывающих зондов. Для определения идентичности транскриптов можно провести анализ последовательностей каждой разновидности, т.е. из каждого элемента. Например, последовательности могут быть идентифицированы с использованием, например, программного обеспечения Blastn для сравнения последовательностей с одной или более чем одной из баз данных по геномам, предпочтительно с базой данных для того организма, из которого образец ткани был получен. Идентичность последовательности из базы данных с наибольшим сходством с последовательностью, синтезированной способами по изобретению, будет отнесена к указанной последовательности. В общем случае, только совпадения с достоверностью по меньшей мере 1е-6, предпочтительно 1е-7, 1е-8 или 1е-9, будут считаться успешно идентифицированными.
Очевидно, что в способах по изобретению может быть использован любой метод секвенирования нуклеиновых кислот. Однако, в настоящем изобретении особое применение найдут так называемые методы "секвенирования следующего поколения". Высокопроизводительное секвенирование особенно полезно в способах по изобретению, поскольку оно позволяет провести частичное секвенирование большого количества нуклеиновых кислот за очень короткий промежуток времени. С учетом недавнего быстрого роста числа полностью или частично секвенированных геномов нет необходимости в секвенировании полной последовательности синтезированных молекул ДНК, например кДНК, чтобы установить ген, которому каждая молекула соответствует. Например, первых 100 нуклеотидов с каждого конца молекул ДНК, например кДНК, должно быть достаточно для идентификации как элемента, которым нуклеиновая кислота, например мРНК, была захвачена (т.е. ее локализации на чипе), так и экспрессированного гена. В результате проведения реакции секвенирования начиная с "конца захватывающего зонда" молекул ДНК, например кДНК, получают данные о последовательности позиционной области и данные по меньшей мере о примерно 20 основаниях, предпочтительно 30 или 40 основаниях специфичной к транскрипту последовательности. В результате проведения реакции секвенирования с "конца незахватывающего зонда" можно получить данные о примерно 70 основаниях, предпочтительно 80, 90 или 100 основаниях специфичной к транскрипту последовательности.
В качестве репрезентативного примера, реакция секвенирования может быть основана на применении таких обратимых красителей-терминаторов, которые использованы в технологии Illumina™. Например, сначала молекулы ДНК присоединяют к праймерам, например на стеклянном или кремниевом слайде, и амплифицируют, в результате чего образуют локальные кластерные колонии (мостиковая амплификация). Добавляют четыре типа ddNTP и не встроившиеся нуклеотиды отмывают. В отличие от пиросеквенирования, ДНК может быть удлинена за один раз только на один нуклеотид. С помощью камеры регистрируют флуоресцентно меченные нуклеотиды, затем из ДНК удаляют химическим способом краситель вместе с блокатором 3′-конца, давая возможность протеканию следующего цикла. Это можно повторять до тех пор, пока не будут получены необходимые данные о последовательности. С использованием этой технологии можно секвенировать одновременно на одном слайде тысячи нуклеиновых кислот.
Для способов по изобретению в равной степени могут быть приемлемы другие высокопроизводительные методы секвенирования, например пиросеквенирование. В этом способе ДНК амплифицируют внутри капель воды в масляном растворе (эмульсионная ПЦР), при этом каждая капля содержит единственную матрицу ДНК, присоединенную к одной покрытой праймером грануле, которая затем образует клональную колонию. Установка для секвенирования содержит много лунок пиколитрового объема, каждая из которых содержит одну гранулу и ферменты, используемые для секвенирования. В пиросеквенировании используют люциферазу для генерирования света с целью детекции индивидуальных нуклеотидов, добавляемых к растущей ДНК, и объединенные данные используют для считывания последовательности.
Один из примеров разрабатываемой технологии основан на детекции ионов водорода, которые высвобождаются в процессе полимеризации ДНК. В микролунку, содержащую в качестве матрицы нить ДНК, подлежащую секвенированию, помещают нуклеотид одного типа. Если введенный нуклеотид комплементарен первому нуклеотиду матрицы, он встраивается в растущую комплементарную нить. Это приводит к высвобождению иона водорода, который запускает гиперчувствительный ионный сенсор, и это указывает на то, что реакция осуществилась. Если в последовательности матрицы присутствуют гомополимерные повторы, то за один цикл будут встраиваться несколько нуклеотидов. Это приводит к высвобождению соответствующего количества ионов водорода и пропорционально более высокому электронному сигналу.
Таким образом, ясно, что постепенно становятся доступными перспективные форматы секвенирования с более короткой продолжительностью рабочего цикла, как одной из основных особенностей этих платформ, и очевидно, что в способах по изобретению будут полезны другие технологии секвенирования.
Существенным признаком настоящего изобретения, как описано выше, является стадия закрепления комплементарной нити захваченных молекул нуклеиновой кислоты на захватывающем зонде, например стадия обратного транскрибирования захваченных молекул РНК. Реакция обратной транскрипции хорошо известна в данной области, и в репрезентативных реакциях обратной транскрипции реакционная смесь включает в себя обратную транскриптазу, dNTP и подходящий буфер. Реакционная смесь может содержать другие компоненты, например ингибитор(ы) РНКаз. Праймеры и матрица представляют собой захватывающую область захватывающего зонда, а захваченные молекулы РНК описаны выше. В способах, являющихся объектом изобретения, каждый dNTP обычно будет представлен в концентрации, находящейся в пределах от примерно 10 до 5000 мкМ, обычно от примерно 20 до 1000 мкМ. Очевидно, что для синтеза комплементарной нити захваченной молекулы ДНК можно провести аналогичную реакцию, используя фермент с ДНК-полимеразной активностью. Реакции такого типа хорошо известны в данной области и описаны более подробно ниже.
Желаемую активность обратной транскриптазы можно обеспечить путем использования одного или более чем одного из различных ферментов, подходящими примерами которых являются: ферменты M-MLV (вирус мышиного лейкоза Молони), MuLV (вирус лейкоза мышей), AMV (вирус миелобластоза птиц), ВИЧ (вирус иммунодефицита человека), ArrayScript™, MultiScribe™, ThermoScript™ и Superscript® I, II и III.
Реакция с использованием обратной транскриптазы может быть проведена при любой подходящей температуре, которая будет зависеть от свойств фермента. Обычно реакции с использованием обратной транскриптазы проводят при 37-55°С, хотя также могут быть целесообразны температуры за пределами этого диапазона. Продолжительность реакции может составлять от всего 1, 2, 3, 4 или 5 минут до 48 часов включительно. Обычно реакцию проводят в течение 5-120 минут, предпочтительно 5-60, 5-45 или 5-30 минут либо 1-10 или 1-5 минут согласно выбору. Продолжительность реакции не является критичной, и может быть использована любая желаемая продолжительность реакции.
Как указано выше, некоторые воплощения способов включают стадию амплификации, на которой количество копий синтезированных молекул ДНК, например кДНК, увеличивают, например, для обогащения образца для получения улучшенного представления нуклеиновых кислот, например транскриптов, захваченных из образца ткани. По желанию, амплификация может быть линейной или экспоненциальной, при этом представляющие интерес репрезентативные протоколы амплификации включают, без ограничения ими: полимеразную цепную реакцию (ПЦР); изотермальную амплификацию и т.д.
Полимеразная цепная реакция (ПЦР) хорошо известна в данной области, причем она описана в патентах США №: 4683202, 4683195, 4800159, 4965188 и 5512462, описания которых включены сюда посредством ссылки. В репрезентативных реакциях амплификации методом ПЦР реакционную смесь, которая включает в себя описанные выше высвобожденные молекулы ДНК, например кДНК, с чипа, объединяют с одним или более праймерами, которые применяют в реакции удлинения праймеров, например, ПЦР-праймерами, которые гибридизуются с первой и/или второй областями амплификации (такими как прямой и обратный праймеры, используемые в геометрической (или экспоненциальной) амплификации, или единственный праймер, используемый в линейной амплификации). Олигонуклеотидные праймеры, с которыми контактируют высвобожденные молекулы ДНК, например кДНК (далее для удобства называемые как матричная ДНК), будут иметь достаточную длину для обеспечения гибридизации с комплементарной матричной ДНК в условиях отжига (описанных более подробно ниже). Длина праймеров будет зависеть от длины областей амплификации, но как правило будет составлять по меньшей мере 10 п.о. в длину, обычно по меньшей мере 15 п.о. в длину и чаще всего по меньшей мере 16 п.о. в длину и может составлять до 30 п.о. в длину или более, при этом длина праймеров как правило будет находиться в диапазоне от 18 до 50 п.о. в длину, обычно от примерно 20 до 35 п.о. в длину. Матричная ДНК может быть приведена в контакт с одним праймером или набором из двух праймеров (прямого и обратного праймеров), в зависимости от того, что является желательным: удлинение праймера либо линейная или экспоненциальная амплификация матричной ДНК.
Помимо указанных выше компонентов реакционная смесь, приготовленная в способах, являющихся объектом изобретения, обычно включает в себя полимеразу и дезоксирибонуклеозидтрифосфаты (dNTP). Желаемую полимеразную активность можно обеспечить путем использования одного или более чем одного из различных ферментов полимераз. Во многих воплощениях реакционная смесь включает в себя по меньшей мере полимеразу семейства А, при этом репрезентативные представляющие интерес полимеразы семейства А включают, без ограничения ими: полимеразы Thermus aquaticus, в том числе природную полимеразу (Taq) и ее производные и гомологи, такие как Klentaq (как описано в Barnes et al., Proc. Natl. Acad. Sci. USA (1994) 91: 2216-2220); полимеразы Thermus thermophilus, в том числе природную полимеразу (Tth) и ее производные и гомологи, и тому подобное. В некоторых воплощениях, где проводимая реакция амплификации представляет собой реакцию с высокой точностью воспроизведения, реакционная смесь может дополнительно включать в себя фермент полимеразу, обладающую 3′-5′-экзонуклеазной активностью, например, что можно обеспечить путем использования полимеразы семейства В, при этом представляющие интерес полимеразы семейства В включают, без ограничения ими: ДНК-полимеразу Thermococcus litoralis (Vent), которая описана в Perler et al., Proc. Natl. Acad. Sci. USA (1992) 89: 5577-5581; Pyrococcus species GB-D (Deep Vent); ДНК-полимеразу Pyrococcus furiosus (Pfu), которая описана в Lundberg et al., Gene (1991) 108: 1-6; Pyrococcus woesei (Pwo) и тому подобные. Если реакционная смесь включает в себя полимеразу как семейства А, так и семейства В, то полимераза семейства А может быть представлена в реакционной смеси в большем количестве, чем полимераза семейства В, причем разница в активности обычно будет по меньшей мере 10-кратной и чаще всего по меньшей мере примерно 100-кратной. Обычно реакционная смесь будет включать в себя четыре разных типа dNTP, т.е. dATP, dTTP, dCTP и dGTP, соответствующих имеющимся четырем природным основаниям. В способах, являющихся объектом изобретения, каждый dNTP обычно будет представлен в концентрации, находящейся в пределах от примерно 10 до 5000 мкМ, обычно от примерно 20 до 1000 мкМ.
Реакционные смеси, приготовленные на стадиях с использованием обратной транскриптазы и/или амплификации в способах, являющихся объектом изобретения, могут дополнительно включать водную буферную среду, в состав которой входят источник одновалентных ионов, источник двухвалентных катионов и буферный агент. Можно использовать любой подходящий источник одновалентных ионов, такой как KCL, K-ацетат, NH4-ацетат, K-глутамат, NH4Cl, сульфат аммония и тому подобные. Двухвалентным катионом может быть катион магния, марганца, цинка и тому подобных, причем обычно катионом будет магний. Можно использовать любой подходящий источник катиона магния, в том числе MgCl2, Mg-ацетат и тому подобные. Концентрация Mg2+, присутствующего в буфере, может находиться в пределах от 0,5 до 10 мМ, однако предпочтительно будет находиться в пределах от примерно 3 до 6 мМ и в идеальном случае будет составлять примерно 5 мМ. Репрезентативные буферные агенты или соли, которые могут присутствовать в буфере, включают трис, трицин, HEPES (N-2-гидроксиэтил-пиперазин-N-2-этансульфоновая кислота), MOPS (3-(N-морфолино)пропансульфоновая кислота) и тому подобное, где концентрация буферного агента в типичном случае будет находиться в пределах от примерно 5 до 150 мМ, обычно от примерно 10 до 100 мМ и чаще всего от примерно 20 до 50 мМ, причем в некоторых предпочтительных воплощениях буферный агент будет присутствовать в количестве, достаточном для получения рН в диапазоне от примерно 6,0 до 9,5, где наиболее предпочтительным является рН 7,3 при 72°С. Другие агенты, которые могут присутствовать в буферной среде, включают хелатирующие агенты, такие как EDTA (этилендиаминтетрауксусная кислота), EGTA (этиленгликоль-тетрауксусная кислота) и тому подобное.
При приготовлении реакционной смеси для обратной транскриптазы, удлинения или амплификации ДНК на стадиях способов, являющихся предметом изобретения, различные составляющие компоненты могут быть объединены в любом удобном порядке. Например, буфер в реакции амплификации можно объединить с праймером, полимеразой и затем матричной ДНК или можно единовременно объединить все разнообразные составляющие компоненты с получением реакционной смеси.
Как обсуждалось выше, в предпочтительном воплощении изобретения, молекулы ДНК, например кДНК, могут быть модифицированы посредством добавления областей амплификации к концам молекул нуклеиновой кислоты, что может включать реакцию лигирования. Реакция лигирования также необходима для синтеза in situ захватывающего зонда на чипе, когда захватывающий зонд иммобилизуют на поверхности чипа опосредованно.
Как известно в данной области, лигазы катализируют образование фосфодиэфирной связи между расположенными рядом З′-гидроксильным и 5′-фосфатным концами двух непосредственно примыкающих друг к другу нуклеиновых кислот. Можно использовать любую подходящую лигазу, при этом представляющие интерес репрезентативные лигазы включают, без ограничения ими, термочувствительные и термостабильные лигазы. Термочувствительные лигазы включают, без ограничения ими, ДНК-лигазу бактериофага Т4, лигазу бактериофага Т7 и лигазу Е. coli. Термостабильные лигазы включают, без ограничения ими, Taq-лигазу, Tth-лигазу и Pfu-лигазу. Термостабильная лигаза может быть получена из термофильных или гипертермофильных организмов, включая, без ограничения ими, прокариотические организмы, эукариотические организмы или археи. В способах по изобретению также могут быть применены некоторые РНК-лигазы.
На этой стадии лигирования подходящую лигазу и все необходимые и/или желательные реагенты объединяют с реакционной смесью и поддерживают в условиях, достаточных для того, чтобы произошло лигирование релевантных олигонуклеотидов. Условия реакции лигирования хорошо известны специалистам в данной области. Во время лигирования реакционную смесь в некоторых воплощениях можно поддерживать при температуре в диапазоне от примерно 4°С до примерно 50°С, как например от примерно 20°С до примерно 37°С, в течение периода времени в диапазоне от примерно 5 секунд до примерно 16 часов, как например от примерно 1 минуты до примерно 1 часа. В других воплощениях реакционную смесь можно поддерживать при температуре в диапазоне от примерно 35°С до примерно 45°С, как например от примерно 37°С до примерно 42°С, например, при температуре, равной или составляющей примерно 38°С, 39°С, 40°С или 41°С, в течение периода времени в диапазоне от примерно 5 секунд до примерно 16 часов, как например от примерно 1 минуты до примерно 1 часа, в том числе от примерно 2 минут до примерно 8 часов. В репрезентативном воплощении смесь для проведения реакции лигирования включает в себя 50 мМ Трис рН 7,5, 10 мМ MgCl2, 10 мМ DTT (дитиотреит), 1 мМ АТФ (аденозинтрифосфат), BSA (бычий сывороточный альбумин) (25 мг/мл), ингибитор РНКаз (0,25 единицы/мл) и Т4 ДНК-лигазу (0,125 единицы/мл). В еще одном репрезентативном воплощении применяют ионы магния (2,125 мМ), ингибитор РНКаз (0,2 единицы/мл) и ДНК-лигазу (0,125 единицы/мл). Количество адаптера в реакции будет зависеть от концентрации ДНК, например кДНК, в образце и, как правило, будет находиться в диапазоне 10-100-кратного молярного избытка по отношению к ДНК, например кДНК.
В качестве репрезентативного примера способ по изобретению может включать следующие стадии:
(а) приведение чипа в контакт с образцом ткани, где чип содержит подложку, на которой непосредственно или опосредованно иммобилизованы многочисленные разновидности захватывающих зондов, так что каждая разновидность занимает определенное положение на чипе и ориентирована таким образом, что имеет свободный 3′-конец, позволяющий указанному зонду действовать в качестве праймера обратной транскриптазы (ОТ), где каждая разновидность указанного захватывающего зонда содержит молекулу нуклеиновой кислоты, имеющую в направлении от 5′ к 3′:
(1) позиционную область, которая соответствует положению захватывающего зонда на чипе, и
(2) захватывающую область;
так что РНК образца ткани гибридизуется с указанными захватывающими зондами;
(б) визуализация образца ткани на чипе;
(в) обратное транскрибирование захваченных молекул мРНК для синтеза молекул кДНК;
(г) промывка чипа для удаления оставшейся ткани;
(д) высвобождение по меньшей мере части молекул кДНК с поверхности чипа;
(е) проведение синтеза второй нити кДНК на высвобожденных молекулах кДНК;
(ж) анализ последовательности (например, секвенирование) молекул кДНК.
В качестве альтернативного репрезентативного примера способ по изобретению может включать следующие стадии:
(а) приведение чипа в контакт с образцом ткани, где чип содержит подложку, на которой непосредственно или опосредованно иммобилизованы по меньшей мере две разновидности захватывающих зондов, так что каждая разновидность занимает определенное положение на чипе и ориентирована таким образом, что имеет свободный 3′-конец, позволяющий указанному зонду действовать в качестве праймера обратной транскриптазы (ОТ), где каждая разновидность указанного захватывающего зонда содержит молекулу нуклеиновой кислоты, имеющую в направлении от 5′ к 3′:
(1) позиционную область, которая соответствует положению захватывающего зонда на чипе, и
(2) захватывающую область;
так что РНК образца ткани гибридизуется с указанными захватывающими зондами;
(б) возможно регидратация образца ткани;
(в) обратное транскрибирование захваченных молекул мРНК для синтеза первых нитей молекул кДНК и возможно синтез вторых нитей молекул кДНК;
(г) визуализация образца ткани на чипе;
(д) промывка чипа для удаления оставшейся ткани;
(е) высвобождение по меньшей мере части молекул кДНК с поверхности чипа;
(ж) амплификация высвобожденных молекул кДНК;
и
(з) анализ последовательности (например, секвенирование) амплифицированных молекул кДНК.
В качестве еще одного репрезентативного примера способ по изобретению может включать следующие стадии:
(а) приведение чипа в контакт с образцом ткани, где чип содержит подложку, на которой непосредственно или опосредованно иммобилизованы многочисленные разновидности захватывающих зондов, так что каждая разновидность занимает определенное положение на чипе и ориентирована таким образом, что имеет свободный 3′-конец, позволяющий указанному зонду действовать в качестве праймера обратной транскриптазы (ОТ), где каждая разновидность указанного захватывающего зонда содержит молекулу нуклеиновой кислоты, имеющую в направлении от 5′ к 3′:
(1) позиционную область, которая соответствует положению захватывающего зонда на чипе, и
(2) захватывающую область;
так что РНК образца ткани гибридизуется с указанными захватывающими зондами;
(б) возможно визуализация образца ткани на чипе;
(в) обратное транскрибирование захваченных молекул мРНК для синтеза молекул кДНК;
(г) возможно визуализация образца ткани на чипе, если она еще не проведена, как на стадии (б);
(д) промывка чипа для удаления оставшейся ткани;
(е) высвобождение по меньшей мере части молекул кДНК с поверхности чипа;
(ж) проведение синтеза второй нити кДНК на высвобожденных молекулах кДНК;
(з) амплификация двунитевых молекул кДНК;
(и) возможно очистка молекул кДНК для удаления компонентов, которые могут служить помехой проведению реакции секвенирования;
и
(к) анализ последовательности (например, секвенирование) амплифицированных молекул кДНК.
Настоящее изобретение включает любую подходящую комбинацию стадий в вышеописанных способах. Очевидно, что данное изобретение также охватывает вариации этих способов, например те, где амплификацию осуществляют на чипе in situ. Также охвачены способы, в которых пропущена стадия визуализации.
Также можно отметить, что данное изобретение включает способ изготовления или получения чипа (1) для применения в захвате мРНК из образца ткани, который приводят в контакт с указанным чипом; или (2) для применения в определении и/или анализе (например, частичного или общего) транскриптома образца ткани, причем указанный способ включает иммобилизацию многочисленных разновидностей захватывающего зонда непосредственно или опосредованно на подложке чипа, где каждая разновидность указанного захватывающего зонда содержит молекулу нуклеиновой кислоты, имеющую в направлении от 5′ к 3′:
(1) позиционную область, которая соответствует положению захватывающего зонда на чипе; и
(2) захватывающую область.
Кроме того, способ изготовления чипа по изобретению можно определить как способ, где каждая разновидность захватывающего зонда иммобилизована в виде элемента на чипе.
Способ иммобилизации захватывающих зондов на чипе может быть осуществлен с использованием любого подходящего средства, как изложено в данном описании. В том случае, когда захватывающие зонды иммобилизованы на чипе опосредованно, захватывающий зонд может быть синтезирован на чипе. Указанный способ может включать любую одну или более чем одну из следующих стадий:
(а) иммобилизация многочисленных поверхностных зондов непосредственно или опосредованно на подложке чипа, где поверхностные зонды содержат:
(1) область, способную к гибридизации с частью олигонуклеотида захватывающей области (частью, не вовлеченной в захват нуклеиновой кислоты);
(2) комплементарную позиционную область; и
(3) комплементарную универсальную область;
(б) гибридизация олигонуклеотидов захватывающих областей и олигонуклеотидов универсальных областей с поверхностными зондами, иммобилизованными на чипе;
(в) удлинение олигонуклеотидов универсальных областей, посредством матричной полимеризации, для синтеза позиционной области захватывающего зонда;и
(г) лигирование позиционной области с олигонуклеотидом захватывающей области для получения захватывающего олигонуклеотида.
Лигирование на стадии (г) может происходить одновременно с удлинением на стадии (в). Таким образом, нет необходимости проводить его в виде отдельной стадии, хотя при желании такое, несомненно, осуществляют.
Элементы чипа, изготовленного указанным выше способом изготовления чипа по изобретению, также могут быть определены в соответствии с приведенным выше описанием.
Несмотря на то, что данное изобретение описано выше со ссылкой на детекцию или анализ РНК и транскриптомный анализ или детекцию, будет очевидно, что описанные принципы аналогичным образом могут быть приемлемы в детекции или анализе ДНК в клетках и в геномных исследованиях. Таким образом, при более широком рассмотрении можно считать, что данное изобретение главным образом применимо для детекции нуклеиновых кислот в целом и в другом более конкретном аспекте, как предоставляющее способы для анализа или детекции ДНК. Пространственная информация может быть полезной также в случае геномики, т.е. детекции и/или анализа молекул ДНК с пространственным разрешением. Этого можно достичь, используя геномное мечение по настоящему изобретению. Такие способы локализованной или пространственной детекции могут быть полезны, например, в случае изучения геномных вариаций в различных клетках или участках ткани, например при сравнении нормальных и больных клеток или тканей (например, нормальных и опухолевых клеток или тканей) или при изучении изменений в геноме при прогрессировании заболеваний и т.д. Например, опухолевые ткани могут содержать гетерогенную популяцию клеток, которые могут отличаться вариантами генома, которые они содержат (например, мутациями и/или другими генетическими аберрациями, например хромосомными перестройками, хромосомными амплификациями/делециями/инсерциями и т.д.). Детекция геномных вариаций или разных геномных локусов в различных клетках по месту локализации может быть полезна в этом контексте, например для изучения пространственного распределения геномных вариаций. Принципиальным было бы применение такого способа в анализе опухолей. В контексте настоящего изобретения может быть получен чип, сконструированный, например, для захвата генома целой клетки на одном элементе. Таким образом можно сравнивать разные клетки в образце ткани. Несомненно, данное изобретение не ограничено такой конструкцией, и возможны другие варианты, где детекцию ДНК осуществляют по месту локализации и положение захваченной на чипе ДНК сопоставляют с положением или локализацией в образце ткани.
Соответственно, можно отметить, что в более общем аспекте согласно настоящему изобретению предложен способ локализованной детекции нуклеиновой кислоты в образце ткани, включающий:
(а) предоставление чипа, содержащего подложку, на которой непосредственно или опосредованно иммобилизованы многочисленные разновидности захватывающих зондов, так что каждая разновидность занимает определенное положение на чипе и ориентирована таким образом, что имеет свободный 3′-конец, позволяющий указанному зонду действовать в качестве праймера в реакции удлинения или лигирования праймера, где каждая разновидность указанного захватывающего зонда содержит молекулу нуклеиновой кислоты, имеющую в направлении от 5′ к 3′:
(1) позиционную область, которая соответствует положению захватывающего зонда на чипе, и
(2) захватывающую область;
(б) приведение указанного чипа в контакт с образцом ткани таким образом, что положение захватывающего зонда на чипе может быть сопоставлено с положением в образце ткани, и предоставление возможности нуклеиновой кислоте образца ткани гибридизоваться с захватывающей областью в указанных захватывающих зондах;
(в) синтез молекул ДНК на основе захваченных молекул нуклеиновой кислоты с использованием указанных захватывающих зондов в качестве праймеров удлинения или лигирования, где указанные удлиненные или лигированные молекулы ДНК являются мечеными благодаря позиционной области;
(г) возможно синтез комплементарной нити указанной меченой ДНК и/или возможно амплификацию указанной меченой ДНК;
(д) высвобождение по меньшей мере части меченых молекул ДНК и/или их комплементов или ампликонов с поверхности чипа, где указанная часть включает позиционную область или ее комплемент; и
(е) прямой или опосредованный анализ последовательности (например, секвенирование) высвобожденных молекул ДНК.
Как описано более подробно ниже, на стадии анализа может быть использован любой метод анализа нуклеиновой кислоты. В типичном случае он может включать секвенирование, но необходимости в фактическом определении последовательности нет. Например, можно использовать специфичные к последовательности методы анализа. Например, может быть осуществлена специфичная к последовательности реакция амплификации, например с использованием праймеров, специфичных к позиционной области и/или к специфической целевой последовательности, например конкретной целевой ДНК, детекцию которой необходимо осуществить (т.е. соответствующей конкретной кДНК/РНК или гену, или варианту гена, или геномному локусу, или геномному варианту и т.д.). Типичным методом анализа является специфичная к последовательности ПЦР-реакция.
Информация из анализа последовательности (например, секвенирования) на стадии (е) может быть использована для получения пространственной информации о нуклеиновой кислоте в образце. Другими словами, информация из анализа последовательности может обеспечить получение информации о локализации нуклеиновой кислоты в образце. Эта пространственная информация может вытекать из природы информации, полученной из анализа последовательности, например, из определенной и идентифицированной последовательности, например, она может отражать наличие конкретной молекулы нуклеиновой кислоты, которая сама может быть информативна в пространственном отношении в случае используемого образца ткани, и/или пространственная информация (например, пространственная локализация) может вытекать из положения образца ткани на чипе, в сочетании с информацией, полученной в результате секвенирования. Однако, как описано выше, пространственная информация соответственно может быть получена посредством сопоставления данных из анализа последовательности с изображением образца ткани, и это представляет собой одно из предпочтительных воплощений изобретения.
Соответственно, в предпочтительном воплощении данный способ также включает стадию:
(ж) сопоставления информации из анализа указанной последовательности с изображением указанного образца ткани, где изображение образца ткани получают до или после стадии (в).
Реакция удлинения праймера, упомянутая на стадии (а), может быть определена как катализируемая полимеразой реакция удлинения нити, и ее результатом является получение комплементарной нити молекулы захваченной нуклеиновой кислоты, которая присоединена ковалентно к захватывающему зонду, т.е. путем синтеза комплементарной нити с использованием захватывающего зонда в качестве праймера и захваченной нуклеиновой кислоты в качестве матрицы. Другими словами, это может быть любая реакция удлинения праймеров, осуществляемая любым ферментом полимеразой. Нуклеиновой кислотой может быть РНК или ей может быть ДНК. Соответственно, полимеразой может быть любая полимераза. Это может быть обратная транскриптаза или это может быть ДНК-полимераза. Реакция лигирования может быть проведена с помощью любой лигазы, и ее результатом является закрепление комплементарной нити захваченной молекулы нуклеиновой кислоты на захватывающем зонде, т.е. когда захваченная молекула нуклеиновой кислоты (гибридизованной с захватывающим зондом) является частично двунитевой и комплементарная нить лигирована с захватывающим зондом.
Одним из предпочтительных воплощений такого способа является способ, описанный выше для определения и/или анализа транскриптома или для детекции РНК. В альтернативном предпочтительном воплощении детектируемой молекулой нуклеиновой кислоты является ДНК. В таком воплощении согласно изобретению предложен способ локализованной детекции ДНК в образце ткани, включающий:
(а) предоставление чипа, содержащего подложку, на которой непосредственно или опосредованно иммобилизованы многочисленные разновидности захватывающих зондов, так что каждая разновидность занимает определенное положение на чипе и ориентирована таким образом, что имеет свободный 3′-конец, позволяющий указанному зонду действовать в качестве праймера в реакции удлинения или лигирования праймера, где каждая разновидность указанного захватывающего зонда содержит молекулу нуклеиновой кислоты, имеющую в направлении от 5′ к 3′:
(1) позиционную область, которая соответствует положению захватывающего зонда на чипе, и
(2) захватывающую область;
(б) приведение указанного чипа в контакт с образцом ткани таким образом, что положение захватывающего зонда на чипе может быть сопоставлено с положением в образце ткани, и предоставление возможности ДНК образца ткани гибридизоваться с захватывающей областью в указанных захватывающих зондах;
(в) фрагментацию ДНК в указанном образце ткани, которую осуществляют до, в процессе или после приведения чипа в контакт с образцом ткани на стадии (б);
(г) удлинение указанных захватывающих зондов в реакции удлинения праймеров с использованием захваченных фрагментов ДНК в качестве матриц для синтеза удлиненных молекул ДНК или лигирование захваченных фрагментов ДНК с захватывающими зондами в реакции лигирования для синтеза лигированных молекул ДНК, где указанные удлиненные или лигированные молекулы ДНК являются мечеными благодаря позиционной области;
(д) возможно синтез комплементарной нити указанной меченой ДНК и/или возможно амплификацию указанной меченой ДНК;
(е) высвобождение по меньшей мере части меченых молекул ДНК и/или их комплементов и/или ампликонов с поверхности чипа, где указанная часть включает позиционную область или ее комплемент;
(ж) прямой или опосредованный анализ последовательности высвобожденных молекул ДНК.
Данный способ может дополнительно включать стадию:
(з) сопоставления информации из анализа указанной последовательности с изображением указанного образца ткани, где изображение образца ткани получают до или после стадии (г).
В случае пространственной геномики, где целевой нуклеиновой кислотой является ДНК, предпочтительным в некоторых обстоятельствах может быть включение стадий визуализации и сопоставления изображений.
В тех воплощениях, где осуществляется захват ДНК, эта ДНК может представлять собой любую молекулу ДНК, которая может встречаться в клетке. Таким образом, это может быть геномная, т.е. ядерная, ДНК, митохондриальная ДНК или пластидная ДНК, например ДНК хлоропластов. В предпочтительном воплощении ДНК представляет собой геномную ДНК.
Очевидно, что если фрагментацию осуществляют после приведения в контакт на стадии (б), т.е. после помещения образца ткани на чип, то фрагментация происходит до того, как ДНК гибридизуется с захватывающей областью. Другими словами, с захватывающей областью в указанных захватывающих зондах гибридизуются фрагменты ДНК (или более конкретно, имеют возможность гибридизоваться).
Предпочтительно, но необязательно, в конкретном воплощении этого аспекта изобретения во фрагментах ДНК образца ткани может быть предусмотрена связывающая область, чтобы обеспечить или облегчить их захват захватывающими зондами на чипе. Соответственно, эта связывающая область способна гибридизоваться с захватывающей областью захватывающего зонда. Поэтому такую связывающую область можно рассматривать как комплемент захватывающей области (т.е. она может считаться комплементарной захватывающей областью), хотя абсолютной комплементарности между захватывающей и связывающей областями не требуется, необходимо лишь, чтобы связывающая область была комплементарна в достаточной степени для осуществления продуктивной гибридизации, т.е. чтобы фрагменты ДНК в образце ткани были способны гибридизоваться с захватывающей областью захватывающих зондов. Обеспечение такой связывающей области может гарантировать, что ДНК, находящаяся в образце, не свяжется с захватывающими зондами до окончания стадии фрагментации. Снабжение фрагментов ДНК связывающей областью можно осуществить с использованием методик, хорошо известных в данной области, например, путем лигирования с адаптерной или линкерной последовательностями, которые могут содержать связывающую область. Например, можно использовать линкерную последовательность с выступающим концом. Связывающая область может находиться в однонитевой части такого линкера, так что после лигирования линкера с фрагментами ДНК такая однонитевая часть, содержащая связывающую область, будет доступна для гибридизации с захватывающей областью захватывающих зондов. Альтернативно и в предпочтительном воплощении связывающую область можно ввести, используя фермент концевую трансферазу, применяемую для введения полинуклеотидного хвоста, например гомополимерного хвоста, такого как поли(А)-область. Это может быть осуществлено с использованием методики, аналогичной описанной выше для введения универсальной области в случае методов с использованием РНК. Таким образом, в предпочтительных воплощениях можно ввести общую связывающую область. Другими словами, связывающую область, которая является общей для всех фрагментов ДНК и которая может быть использована для достижения захвата фрагментов на чипе.
Если реакцию наращивания хвоста проводят для введения (общей) связывающей области, то захватывающие зонды на чипе могут быть защищены от реакции наращивания хвоста, т.е. захватывающие зонды могут быть блокированы или замаскированы, как описано выше. Этого можно достичь, например, посредством гибридизации блокирующего олигонуклеотида с захватывающим зондом, например с выступающим концом (например, однонитевой частью) захватывающего зонда. Когда захватывающая область содержит, например, поли(Т)-последовательность, таким блокирующим олигонуклеотидом может быть поли(А)-олигонуклеотид. Блокирующий олигонуклеотид может иметь блокированный 3′-конец (т.е. конец, который невозможно удлинить или нарастить). Захватывающие зонды также можно защитить, т.е. блокировать, посредством химических и/или ферментативных модификаций, как подробно описано выше.
Если обеспечение связывающей области осуществляют посредством лигирования линкера, как описано выше, будет очевидно, что вместо удлинения захватывающего зонда для синтеза комплементарной копии захваченного фрагмента ДНК, который содержит позиционную метку праймера захватывающего зонда, фрагмент ДНК можно лигировать с 3′-концом захватывающего зонда. Как указано выше, для осуществления лигирования необходимо, чтобы подлежащий лигированию 5′-конец был фосфорилирован. Соответственно, в одном из воплощений 5′-конец добавленного линкера, а именно, подлежащий лигированию с захватывающим зондом конец (т.е. невыступающий конец линкера, добавленного к фрагментам ДНК), будет фосфорилирован. В таком воплощении с использованием лигирования, соответственно, будет видно, что линкер может быть лигирован с фрагментами двунитевой ДНК, причем указанный линкер имеет однонитевой выступающий 3′-конец, который содержит связывающую область. При контакте с чипом выступающий конец гибридизуется с захватывающей областью захватывающих зондов. В результате этой гибридизации 3′-конец захватывающего зонда приходит в непосредственную необходимую для лигирования близость с 5′-концом (невыступающим концом) добавленного линкера. Таким образом, захватывающий зонд и, следовательно, позиционная область встраивается в захваченный фрагмент ДНК в результате этого лигирования. Подобное воплощение показано схематично на Фиг.21.
Таким образом, способ по этому аспекту изобретения в более конкретном воплощении может включать:
(а) предоставление чипа, содержащего подложку, на которой непосредственно или опосредованно иммобилизованы многочисленные разновидности захватывающих зондов, так что каждая разновидность занимает определенное положение на чипе и ориентирована таким образом, что имеет свободный 3′-конец, позволяющий указанному зонду действовать в качестве праймера в реакции удлинения или лигирования праймера, где каждая разновидность указанного захватывающего зонда содержит молекулу нуклеиновой кислоты, имеющую в направлении от 5′ к 3′:
(1) позиционную область, которая соответствует положению захватывающего зонда на чипе, и
(2) захватывающую область;
(б) приведение указанного чипа в контакт с образцом ткани таким образом, что положение захватывающего зонда на чипе может быть сопоставлено с положением в образце ткани;
(в) фрагментацию ДНК в указанном образце ткани, которую осуществляют до, в процессе или после приведения чипа в контакт с образцом ткани на стадии (б);
(г) обеспечение указанных фрагментов ДНК связывающей областью, которая способна гибридизоваться с указанной захватывающей областью;
(д) предоставление возможности указанным фрагментам ДНК гибридизоваться с захватывающей областью в указанных захватывающих зондах;
(е) удлинение указанных захватывающих зондов в реакции удлинения праймеров с использованием захваченных фрагментов ДНК в качестве матриц для синтеза удлиненных молекул ДНК или лигирование захваченных фрагментов ДНК с захватывающими зондами в реакции лигирования для синтеза лигированных молекул ДНК, где указанные удлиненные или лигированные молекулы ДНК являются мечеными благодаря позиционной области;
(ж) возможно синтез комплементарной нити указанной меченой ДНК и/или возможно амплификацию меченой ДНК;
(з) высвобождение по меньшей мере части меченых молекул ДНК и/или их комплементов и/или ампликонов с поверхности чипа, где указанная часть включает позиционную область или ее комплемент;
(и) прямой или опосредованный анализ последовательности высвобожденных молекул ДНК.
Данный способ возможно может включать дополнительную стадию:
(к) сопоставления информации из анализа указанной последовательности с изображением указанного образца ткани, где изображение образца ткани получают до или после стадии (е).
В приведенных выше способах детекции нуклеиновой кислоты или ДНК возможная стадия синтеза комплементарной копии меченой нуклеиновой кислоты/ДНК или амплификации меченой ДНК может включать в себя применение фермента полимеразы замещения нити согласно принципам, разъясненным выше в отношении способов анализа/детекции РНК/транскриптома. Подходящие полимеразы замещения нити рассмотрены выше. Это необходимо для того, чтобы гарантировать копирование позиционной области с образованием комплементарной копии или ампликона. В частности, это будет в том случае, когда захватывающий зонд иммобилизован на чипе посредством гибридизации с поверхностным зондом.
Однако, применение полимеразы замещения нити на этой стадии не является обязательным. Например, вместе с лигированием олигонуклеотида, который гибридизуется с позиционной областью, можно использовать полимеразу, не обладающую активностью замещения нити. Такая методика аналогична методике, описанной выше для синтеза захватывающих зондов на чипе.
В одном из воплощений способ по изобретению может быть использован для определения и/или анализа всего генома образца ткани, например общего генома образца ткани. Однако, способ этим не ограничивается и охватывает определение и/или анализ всего или части генома. Таким образом, способ может включать в себя определение и/или анализ части или подмножества генома, например частичного генома, соответствующего подмножеству или группе генов или хромосом, например набору конкретных генов или хромосом либо конкретному участку или части генома, например имеющих отношение к конкретному заболеванию или состоянию, типу ткани и т.д. Таким образом, данный способ может быть использован для детекции или анализа геномных последовательностей или геномных локусов из опухолевой ткани в сравнении с нормальной тканью или даже в пределах разных типов клеток в образце ткани. Можно исследовать наличие или отсутствие либо распределение или локализацию разных геномных вариантов или локусов в различных клетках, группах клеток, тканях или частях или типах ткани.
С точки зрения другого аспекта, изложенные выше стадии способа можно рассматривать в качестве предложения способа получения информации о пространственной локализации, касающейся нуклеиновых кислот, например геномных последовательностей, вариантов или локусов образца ткани. Другими словами, способы по изобретению могут быть использованы для введения метки в геномы (или мечения геномов), в частности, отдельные или пространственно-распределенные геномы.
С альтернативной точки зрения способ по изобретению можно рассматривать как способ пространственной детекции ДНК в образце ткани, или способ детекции ДНК с пространственным разрешением, или способ локализованного или пространственного определения и/или анализа ДНК в образце ткани. В частности, данный способ можно использовать для локализованной или пространственной детекции либо определения и/или анализа генов или геномных последовательностей либо геномных вариантов или локусов (например, распределения геномных вариантов или локусов) в образце ткани. Термин "локализованная/пространственная детекция/определение/анализ" означает, что ДНК может быть локализована в соответствии с ее природным(ой) положением или локализацией внутри клетки или ткани в образце ткани. Так например, ДНК может быть локализована в клетке, или группе клеток, или типе клеток в образце либо в конкретных участках областей в образце ткани. Можно определить природную(ое) локализацию или положение ДНК (или другими словами, локализацию или положение ДНК в образце ткани), например геномного варианта или локуса.
В силу вышесказанного можно отметить, что чип по настоящему изобретению может быть использован для захвата нуклеиновой кислоты, например ДНК, образца ткани, который приводят в контакт с указанным чипом. Данный чип также можно использовать для определения и/или анализа частичного или общего генома образца ткани или для получения определяемого в пространстве частичного или общего генома образца ткани. Таким образом, способы по изобретению можно рассматривать как способы количественного определения пространственного распределения одной или более геномных последовательностей (или вариантов или локусов) в образце ткани. Выражаясь по-другому, способы по настоящему изобретению могут быть использованы для детекции пространственного распределения одной или более геномных последовательностей либо одного или более геномных вариантов или геномных локусов в образце ткани. Иначе, способы по настоящему изобретению могут быть использованы для одновременного определения локализации или распределения одной или более геномных последовательностей либо одного или более геномных вариантов или геномных локусов в одном или более положениях в образце ткани. Более того, данные способы можно рассматривать как способы частичного или общего анализа нуклеиновой кислоты, например ДНК, образца ткани с пространственным разрешением, например, с двумерным пространственным разрешением.
Также можно отметить, что согласно изобретению предложен чип для применения в способах по изобретению, содержащий подложку, на которой непосредственно или опосредованно иммобилизованы многочисленные разновидности захватывающих зондов, так что каждая разновидность занимает определенное положение на чипе и ориентирована таким образом, что имеет свободный 3′-конец, позволяющий указанному зонду действовать в качестве праймера удлинения или лигирования, где каждая разновидность указанного захватывающего зонда содержит молекулу нуклеиновой кислоты, имеющую в направлении от 5′ к 3′:
(1) позиционную область, которая соответствует положению захватывающего зонда на чипе, и
(2) захватывающую область для захвата нуклеиновой кислоты образца ткани, который приводят в контакт с указанным чипом.
В одном из аспектов подлежащей захвату молекулой нуклеиновой кислоты является ДНК. Захватывающая область может быть специфичной к конкретной подлежащей детекции ДНК или к конкретному классу или группе ДНК, например вследствие специфичной гибридизации со специфической последовательностью мотива в целевой ДНК, например консервативной последовательностью, по аналогии со способами, описанными выше в случае детекции РНК. Альтернативно, подлежащая захвату ДНК может быть снабжена связывающей областью, например общей связывающей областью, как описано выше, при этом связывающая область может распознаваться захватывающей областью захватывающих зондов. Так, как указано выше, связывающая область может представлять собой, например, гомополимерную последовательность, например поли(А). Опять же, такая связывающая область может быть предусмотрена согласно или аналогично принципам и способам, описанным выше в отношении способов для анализа или детекции РНК/транскриптома. В этом случае захватывающая область может быть комплементарна связывающей области, введенной в молекулы ДНК образца ткани.
Так же, как описано выше в случае РНК, захватывающая область может представлять собой случайную или вырожденную последовательность. Таким образом, ДНК может быть захвачена неспецифично посредством связывания со случайной или вырожденной захватывающей областью или с захватывающей областью, которая содержит по меньшей мере частично случайную или вырожденную последовательность.
В родственном аспекте согласно настоящему изобретению также предложено применение чипа, содержащего подложку, на которой непосредственно или опосредованно иммобилизованы многочисленные разновидности захватывающего зонда, так что каждая разновидность занимает определенное положение на чипе и ориентирована таким образом, что имеет свободный 3′-конец, позволяющий указанному зонду действовать в качестве праймера удлинения или лигирования, где каждая разновидность указанного захватывающего зонда содержит молекулу нуклеиновой кислоты, имеющую в направлении от 5′ к 3′:
(1) позиционную область, которая соответствует положению захватывающего зонда на чипе; и
(2) захватывающую область;
для захвата нуклеиновой кислоты, например ДНК или РНК, образца ткани который приводят в контакт с указанным чипом.
Предпочтительно, указанное применение относится к локализованной детекции нуклеиновой кислоты в образце ткани и дополнительно включает стадии:
(а) синтеза молекул ДНК на основе захваченных молекул нуклеиновой кислоты с использованием указанных захватывающих зондов в качестве праймеров удлинения или лигирования, где указанные удлиненные или лигированные молекулы являются мечеными благодаря позиционной области;
(б) возможно синтеза комплементарной нити указанной меченой нуклеиновой кислоты и/или амплификации указанной меченой нуклеиновой кислоты;
(в) высвобождения по меньшей мере части меченых молекул ДНК и/или их комплементов или ампликонов с поверхности чипа, где указанная часть включает позиционную область или ее комплемент;
(г) прямого или опосредованного анализа последовательности высвобожденных молекул ДНК; и возможно
(д) сопоставления информации, полученной из указанного анализа последовательности, с изображением указанного образца ткани, где изображение образца ткани получают до или после стадии (а).
Стадия фрагментирования ДНК в образце ткани может быть проведена с использованием любой желаемой методики, известной в данной области. Так, можно использовать физические методы фрагментации, например разрушение ультразвуком или обработку ультразвуком. Также известны химические методы. Помимо этого можно использовать ферментативные методы фрагментации, например с применением эндонуклеаз, например, ферментов рестрикции. Опять же, используемые для этого методы и ферменты хорошо известны в данной области. Фрагментация может быть осуществлена до, во время или после приготовления образца ткани для размещения его на чипе, например, приготовления среза ткани. В целях удобства фрагментация может быть осуществлена на стадии фиксации ткани. Так, например, фиксация формалином может приводить к фрагментации ДНК. Применение других фиксаторов может давать аналогичные результаты.
Что касается подробностей приготовления и использования чипов в соответствии с этими аспектами изобретения, то будет понятно, что описание и подробности, приведенные выше в случае способов для РНК, точно так же применимы к более общим способам детекции нуклеиновых кислот и ДНК, изложенным в данном описании. Таким образом, все аспекты и подробности, рассмотренные выше, применимы аналогично. Например, обсуждение, касающееся праймеров для обратной транскриптазы и реакций с ее участием и т.д. аналогичным образом может быть применено к любому аспекту в отношении праймеров удлинения, полимеразной реакции и т.д., упомянутых выше. Подобным же образом, ссылки на синтез первой и второй нити «ДНК могут быть применены аналогично к меченой молекуле ДНК и ее комплементу. Можно использовать способы анализа последовательности, которые рассмотрены выше.
В качестве примера, захватывающая область может быть такой, как она описана выше для захватывающих зондов. Например, можно использовать захватывающую область в виде поли(Т) или области, содержащей поли(Т), когда фрагменты ДНК снабжены связывающей областью, содержащей поли(А)-последовательность.
Захватывающие зонды/меченые молекулы ДНК (т.е. меченые удлиненные или лигированные молекулы) могут быть снабжены универсальными областями, как описано выше, например, для амплификации и/или расщепления.
Далее изобретение будет описано следующими неограничивающими примерами со ссылкой на приведенные ниже графические материалы, в которых:
на Фиг.1 показана общая концепция использования организованных в виде чипа "штрихкодовых" олиго(dT)-зондов для захвата мРНК из срезов тканей для проведения транскриптомного анализа;
на Фиг.2 показана схема визуализации присутствия транскриптов для соответствующих срезов ткани;
на Фиг.3 показано строение ориентированного от 3′ к 5′ поверхностного зонда и синтез ориентированных от 5′ к 3′ захватывающих зондов, которые иммобилизованы на поверхности чипа опосредованно;
на Фиг.4 показана гистограмма, демонстрирующая эффективность высвобождения под действием ферментов (USER или Rsal) с чипов собственной разработки и под действием воды (99°С) с чипов производства Agilent, по результатам измерений гибридизации зондов, меченных флуоресцентной меткой, с поверхностью чипов после высвобождения зондов;
на Фиг.5 показано флуоресцентное изображение после опосредованного водой (99°С) высвобождения ДНК поверхностных зондов с промышленных чипов производства Agilent. Гибридизацию флуоресцентного детектирующего зонда осуществляли после обработки горячей водой. Верхний чип представляет собой не подвергнутый обработке контроль;
на Фиг.6 показан фиксированный срез ткани головного мозга мыши на поверхности захватывающего чипа для анализа транскриптома после синтеза кДНК и обработки красителями, окрашивающими цитоплазму (вверху) и ядра (посередине), соответственно, и совмещенное изображение, демонстрирующее оба типа окрашивания (внизу);
на Фиг.7 показана таблица, в которой перечислены результаты считываний, отсортированных относительно их исходного положения, по всему ДНК-захватывающему чипу низкой плотности собственной разработки, как показано схематическим представлением;
на Фиг.8 показана FFPE-ткань головного мозга мыши со специфичными к ядрам и Мар2 (белок, ассоциированный с микротрубочками) красителями с использованием штрихкодового микрочипа;
на Фиг.9 показана FFPE-ткань обонятельной луковицы головного мозга мыши с окрашиванием ядер (белый цвет) и различимой морфологией;
на Фиг.10 показана FFPE-ткань обонятельной луковицы головного мозга мыши (прибл. 2×2 мм) с окрашиванием ядер (белый цвет), с наложением полученной теоретически картины расположения пятен для чипа низкого разрешения;
на Фиг.11 показана FFPE-ткань обонятельной луковицы головного мозга мыши (прибл. 2×2 мм) с окрашиванием ядер (белый цвет), с наложением полученной теоретически картины расположения пятен для чипа умеренно высокого разрешения;
на Фиг.12 показана FFPE-ткань обонятельной луковицы головного мозга мыши при увеличении клубочковой зоны (правый верхний угол Фиг.9);
на Фиг.13 показан продукт, полученный в результате высвобождения под действием USER в процессе амплификации с использованием случайного гексамерного праймера (R6), соединенного с "В_ручкой", (B_R6); продукт, зарегистрированный на биоанализаторе;
на Фиг.14 показан продукт, полученный в результате высвобождения под действием USER в процессе амплификации с использованием случайного октамерного праймера (R8), соединенного с "В_ручкой", (B_R8); продукт, зарегистрированный на биоанализаторе;
на Фиг.15 показаны результаты эксперимента, проведенного на FFPE-ткани головного мозга, покрывающей весь чип.ID5 (слева) и ID20 (справа) амплифицированы с использованием ID-специфичных и ген-специфичных праймеров (экзон 4 гена В2М) после синтеза и высвобождения кДНК с поверхности; ID5 и ID20 амплифицированы;
на Фиг.16 показана схематическая иллюстрация принципа способа, описанного в примере 4, т.е. применения микрочипов с иммобилизованными ДНК-олигонуклеотидами (захватывающими зондами), несущими последовательности с маркирующими метками для пространственной локализации (позиционные области). Каждый элемент из олигонуклеотидов микрочипа несет 1) уникальную маркирующую метку (позиционную область) и 2) захватывающую последовательность (захватывающую область);
на Фиг.17 показаны результаты описанного в примере 5 протокола пространственной геномики, выполненного с использованием геномной ДНК, предварительно фрагментированной до среднего размера 200 п.о. Внутренние продукты амплифицированы на меченной и синтезированной на чипе ДНК. Зарегистрированный пик соответствует ожидаемому размеру;
на Фиг.18 показаны результаты описанного в примере 5 протокола пространственной геномики, выполненного с использованием геномной ДНК, предварительно фрагментированной до среднего размера 700 п.о. Внутренние продукты амплифицированы на меченной и синтезированной на чипе ДНК. Зарегистрированный пик соответствует ожидаемому размеру;
на Фиг.19 показаны результаты описанного в примере 5 протокола пространственной геномики, выполненного с использованием геномной ДНК, предварительно фрагментированной до среднего размера 200 п.о. Продукты амплифицированы с использованием одного внутреннего праймера и одной универсальной последовательности, содержащейся в поверхностном олигонуклеотиде. Амплификацию проводили на меченной и синтезированной на чипе ДНК. Изображение ожидаемого продукта является размытым ввиду того, что в результате случайной фрагментации и мечения геномной ДНК концевой трансферазой будет образовываться пул разнообразных образцов;
на Фиг.20 показаны результаты описанного в примере 5 протокола пространственной геномики, выполненного с использованием геномной ДНК, предварительно фрагментированной до среднего размера 700 п.о. Продукты амплифицированы с использованием одного внутреннего праймера и одной универсальной последовательности, содержащейся в поверхностном олигонуклеотиде. Амплификацию проводили на меченной и синтезированной на чипе ДНК. Изображение ожидаемого продукта является размытым ввиду того, что в результате случайной фрагментации и мечения геномной ДНК концевой трансферазой будет образовываться пул разнообразных образцов;
на Фиг.21 показана схематическая иллюстрация лигирования линкера с фрагментом ДНК для введения связывающей области для гибридизации с поли(Т)-содержащей захватывающей областью и последующего лигирования с захватывающим зондом;
на Фиг.22 показано строение ориентированных от 5′ к 3′ захватывающих зондов, использованных на захватывающих чипах высокой плотности;
на Фиг.23 показана рамка чипов высокой плотности, которую используют для ориентации образца ткани, с визуализацией посредством гибридизации флуоресцентных маркерных зондов;
на Фиг.24 показаны захватывающие зонды, отщепленные и неотщепленные от чипа высокой плотности, при этом рамочные зонды не отщеплены, поскольку не содержат урациловых оснований. Захватывающие зонды метили флуорофорами, присоединенными к поли(А)-олигонуклеотидам;
на Фиг.25 показано зарегистрированное с использованием биоанализатора изображение полученной библиотеки для секвенирования, содержащей транскрипты, захваченные из обонятельной луковицы мыши;
на Фиг.26 показана визуализация захваченных транскриптов из суммарной РНК, выделенной из обонятельной луковицы мыши, полученная с использованием Matlab;
на Фиг.27 показаны транскрипты Olfr (обонятельного рецептора; англ. olfactory receptor), визуализированные по всему захватывающему чипу с применением Matlab-визуализации после захвата из ткани обонятельной луковицы мыши;
на Фиг.28 показана картина печати для содержащих 41-ID-метку микрочипов собственной разработки;
на Фиг.29 показана библиотека пространственной геномики, созданная на основе специфической транслокации А431 после захвата геномных фрагментов с поли(А)-хвостом на захватывающем чипе;
на Фиг.30 показана детекция специфической транслокации А431 после захвата перенесенных 10% и 50% геномных фрагментов А431 с поли(А)-хвостами в геномные фрагменты U2OS с поли(А)-хвостами на захватывающем чипе;
на Фиг.31 показана Matlab-визуализация захваченных ID-меченных транскриптов из ткани обонятельной луковицы мыши на содержащих 41-ID-метку чипах собственной разработки с наложением изображения ткани. Для наглядности, конкретные элементы, на которых были идентифицированы конкретные гены, обведены кружком.
Пример 1
Изготовление чипа
В приведенных далее экспериментах демонстрируется, как можно присоединить олигонуклеотидные зонды к подложке чипа через 5′- или 3′-конец, чтобы получить чип с захватывающими зондами, способными гибридизоваться смРНК.
Изготовление печатного микрочипа собственной разработки с ориентированными от 5′ к 3′ зондами
На стеклянные слайды наносили в виде пятен 20 РНК-захватывающих олигонуклеотидов с содержащими индивидуальные метки последовательностями (метки 1-20, Таблица 1) в качестве захватывающих зондов. Зонды синтезировали таким образом, чтобы они содержали на 5′-конце аминолинкер с С6-спейсером. Все зонды были синтезированы в Sigma-Aldrich (St. Louis, МО, USA). РНК-захватывающие зонды суспендировали в концентрации 20 мкМ в 150 мМ фосфате натрия, рН 8,5, и наносили в виде пятен, используя наноплоттер (nanoplotter) NP2.1/E (Gesim, Grosserkmannsdorf, Germany), на активированные слайды для микрочипов CodeLink™ (7,5 см×2,5 см; Surmodics, Eden Prairie, MN, USA). После печати осуществляли блокирование поверхности в соответствии с инструкциями производителя. Печать зондов выполняли в виде 16 идентичных чипов на слайде, и каждый чип имел предварительно определенную картину печати. Эти 16 подчипов были отделены друг от друга в процессе гибридизации посредством состоящего из 16 ячеек экрана (ChipClip™, Schleicher & Schuell BioScience, Keene, NH, USA).




Изготовление печатного микрочипа собственной разработки с ориентированными от 3′ к 5′ зондами и синтез ориентированных от 5′ к 3′ захватывающих зондов
Печать олигонуклеотидов поверхностного зонда осуществляли, как и в приведенном выше случае с ориентированными от 5′ к 3′ зондами, используя линкер амино-С7 на 3′-конце, как показано в Таблице 1.
Чтобы осуществить гибридизацию праймеров для синтеза захватывающих зондов, раствор для гибридизации, содержащий 4×SSC (раствор хлорида и цитрата натрия) и 0,1% SDS (додецилсульфат натрия), 2 мкМ праймер удлинения (олигонуклеотид универсальной области) и 2 мкМ праймер для соединения нитей (олигонуклеотид захватывающей области) инкубировали в течение 4 мин при 50°С. За это время прикрепляли чип собственной разработки к ChipClip (Whatman). Далее этот чип инкубировали при 50°С в течение 30 мин на шейкере при 300 об./мин вместе с 50 мкл раствора для гибридизации на одну лунку.
После инкубации чип извлекали из ChipClip и промывали, используя 3 приведенные ниже стадии: 1) 50°С, раствор 2×SSC с 0,1% SDS в течение 6 мин на шейкере при 300 об./мин; 2) 0,2×SSC в течение 1 мин на шейкере при 300 об./мин; и 3) 0,1×SSC в течение 1 мин на шейкере при 300 об./мин. Затем чип сушили в центрифуге и помещали обратно в ChipClip.
Для осуществления реакции удлинения и лигирования (с целью создания позиционной области захватывающего зонда) 50 мкл ферментативной смеси, содержащей 10× буфер для лигазы Ampligase, 2,5 ед. фрагмента Стоффеля AmpliTaq ДНК-полимеразы (Applied Biosystems), Юед. лигазы Ampligase (Epicentre Biotechnologies), dNTP (2 мМ каждый; Fermentas) и воду, вносили в каждую лунку с помощью пипетки. Затем этот чип инкубировали при 55°С в течение 30 мин. После инкубации чип промывали в соответствии с описанным ранее способом промывки чипа, но продолжительность первой стадии составляла 10 мин вместо 6 мин.
Способ проиллюстрирован на Фиг.3.
Подготовка ткани
Приведенные далее эксперименты демонстрируют, как можно изготовить срезы образцов ткани для применения в способах по изобретению.
Подготовка свежезамороженной ткани и изготовление срезов для нанесения на чипы. содержащие захватывающие зонды
Свежую нефиксированную ткань головного мозга мыши обрезали, если необходимо, и замораживали в охлажденном до -40°С изопентане и после этого монтировали на криостате для приготовления срезов толщиной 10 мкм. Срез ткани помещали на поверхность каждого предназначенного для использования чипа, содержащего захватывающие зонды.
Приготовление фиксированной в формалине залитой парафином (FFPE) ткани
Ткань головного мозга мыши фиксировали в 4%-ном формалине при 4°С в течение 24 ч. После этого ее инкубировали следующим образом: 3× инкубация в 70%-ном этаноле в течение 1 часа; 1х инкубация в 80%-ном этаноле в течение 1 часа; 1× инкубация в 96%-ном этаноле в течение 1 часа; 3× инкубация в 100%-ном этаноле в течение 1 часа; и 2× инкубация в ксилоле при комнатной температуре в течение 1 ч.
Дегидратированные образцы далее инкубировали в жидком низкоплавком парафине при 52-54°С в течение не более 3 часов включительно, за это время один раз проводили замену парафина, чтобы отмыть от оставшегося ксилола. Готовые блоки ткани далее хранили при КГ (комнатная температура). Затем, используя микротом, делали срезы в парафине толщиной 4 мкм для нанесения на поверхность каждого предназначенного для использования чипа, содержащего захватывающие зонды.
Срезы сушили при 37°С на слайдах чипов в течение 24 часов и хранили при КГ.
Депарафинизация FFPE-ткани
Фиксированные в формалине парафинизированные срезы головного мозга мыши толщиной Юмкм, нанесенные на поверхность слайдов CodeLink, дважды подвергали депарафинизации в ксилоле в течение 10 мин; в 99,5%-ном этаноле в течение 2 мин; в 96%-ном этаноле в течение 2 мин; в 70%-ном этаноле в течение 2 мин; и затем сушили на воздухе.
Синтез кДНК
Приведенные далее эксперименты демонстрируют, что мРНК, захваченная на чипе из срезов образцов ткани, может быть использована в качестве матрицы для синтеза кДНК.
Синтез кДНК на чипе
16-Луночный экран и держатель слайдов Chip Clip от Whatman прикрепляли к слайду CodeLink. При осуществлении синтеза кДНК использовали систему одностадийной ОТ-ПЦР Superscript™ III с Platinum® Taq ДНК-полимеразой от Invitrogen. Для проведения каждой реакции смешивали 25 мкл 2× реакционной смеси (система одностадийной ОТ-ПЦР Superscript™ III с Platinum® Taq ДНК-полимеразой, Invitrogen), 22,5 мкл НзО и 0,5 мкл 100×BSA и нагревали до 50°С. В реакционную смесь добавляли смесь ферментов Superscript Ill/Platinum Taq, по 2 мкл на одну реакцию, и по 50 мкл этой реакционной смеси добавляли в каждую лунку на чипе. Чип инкубировали при 50°С в течение 30 мин (термосмеситель Comfort, Eppendorf).
Реакционную смесь удаляли из лунок и слайд промывали, используя: 2×SSC, 0,1% SDS при 50°С в течение 10 мин; 0,2×SSC при комнатной температуре в течение 1 мин и 0,1×SSC при комнатной температуре в течение 1 мин. Затем этот чип сушили в центрифуге.
В случае срезов FFPE-ткани эти срезы теперь можно было окрашивать и визуализировать, после чего удалить ткань, см. ниже раздел, касающийся визуализации.
Визуализация
Гибридизация флуоресцентных маркерных зондов перед окрашиванием
Перед наложением ткани флуоресцентные маркерные зонды гибридизовали с элементами, содержащими маркерные олигонуклеотиды, отпечатанные на чипе, содержащем захватывающие зонды. Флуоресцентные маркерные зонды способствуют ориентации полученного изображения после визуализации ткани, давая возможность комбинировать это изображение с полученными профилями экспрессии для индивидуальных, несущих "метку" (позиционную область) захватывающих зондов, последовательностей, полученных после секвенирования. Чтобы осуществить гибридизацию флуоресцентных зондов, раствор для гибридизации, содержащий 4×SSC и 0,1% SDS, 2 мкМ детектирующий зонд (Р), инкубировали в течение 4 мин при 50°С. В это время прикрепляли чип собственной разработки к ChipClip (Whatman). Далее этот чип инкубировали при 50°С в течение 30 мин на шейкере при 300 об./мин вместе с 50 мкл раствора для гибридизации на одну лунку.
После инкубации чип извлекали из ChipClip и промывали, используя 3 приведенные ниже стадии: 1) 50°С, раствор 2×SSC с 0,1% SDS в течение 6 мин на шейкере при 300 об/мин, 2) 0,2×SSC в течение 1 мин на шейкере при 300 об./мин и 3) 0,1×SSC в течение 1 мин на шейкере при 300 об./мин. Затем чип сушили в центрифуге.
Общее гистологическое окрашивание срезов FFPE-ткани до или после синтеза кДНК
Срезы FFPE-ткани, иммобилизованные на чипах, содержащих захватывающие зонды, промывали и регидратировали после депарафинизации перед синтезом кДНК, как описано ранее, или промывали после синтеза кДНК, как описано ранее. Далее их обрабатывали, как приведено ниже: инкубировали в течение 3 минут в гематоксилине; промывали деионизованной водой; инкубировали в течение 5 минут в водопроводной воде; быстро окунали от 8 до 12 раз в подкисленный этанол; промывали 2 раза по 1 минуте в водопроводной воде; промывали 2 минуты в деонизованной воде; инкубировали 30 секунд в эозине; промывали 3х5 минут в 95%-ном этаноле; промывали 3×5 минут в 100%-ном этаноле; промывали 3×10 минут в ксилоле (может быть выполнено в течение ночи); помещали покровное стекло на слайды, используя DPX (ди-н-бутилфталат в ксилоле); сушили слайды в вытяжном шкафу в течение ночи.
Общее иммуногистохимическое окрашивание целевого белка в срезах FFPE-ткани до или после синтеза кДНК
Срезы FFPE-ткани, иммобилизованные на чипах, содержащих захватывающие зонды, промывали и регидратировали после депарафинизации перед синтезом кДНК, как описано ранее, или промывали после синтеза кДНК, как описано ранее. Далее их обрабатывали, как приведено ниже, не позволяя высыхать в течение всего процесса окрашивания: срезы инкубировали с первичным антителом (разбавленным первичным антителом в блокирующем растворе, содержащем 1×забуференный трисом физиологический раствор (TBS) (50 мМ Трис, 150 мМ NaCl, pH 7,6), 4% сыворотки осла и 0,1% тритона-х) 103 в камере с увлажнением в течение ночи при КГ; промывали три раза 1×TBS; инкубировали срез с соответствующим вторичным антителом, конъюгированным с флуорохромом (FITC (флуоресцеинизотиоцианат), Cy3 или Cy5) в камере с увлажнением при КГ в течение 1 часа. Промывали 3 раза, используя 1×TBS, удаляли TBS в той степени, насколько это возможно, заключали срез в ProLong Gold+DAPI (4′,6-диамидино-2-фенилиндол) (Invitrogen) и анализировали с использованием флуоресцентного микроскопа и подбора подходящих наборов фильтров.
Удаление оставшейся ткани
Замороженная ткань
Чтобы полностью удалить ткань в случае свежезамороженной ткани головного мозга мыши, было достаточно стадии промывки сразу после синтеза кДНК.
FFPE-ткань
Слайды с фиксированными в формалине парафинизированными срезами ткани головного мозга мыши прикрепляли к держателям слайдов ChipClip и 16-луночным экранам (Whatman). На каждые 150 мкл буфера для переваривания протеиназой К из набора RNeasy FFPE (Qiagen) добавляли по 10 мкл раствора протеиназы К (Qiagen). В каждую лунку добавляли по 50 мкл конечной смеси, и слайд инкубировали при 56°С в течение 30 мин.
Высвобождение захватывающих зондов (кДНК)
Высвобождение захватывающих зондов с использованием смеси расщепляющего по урацилам Фермент USER enzyme с буфером для ПНР (ковалентно присоединенных зондов)
16-Луночный экран и слайд CodeLink прикрепляли к держателю ChipClip (Whatman). 50 мкл смеси, содержащей 1× реакционный буфер FastStart High Fidelity (HiFi) (для быстрых реакций с высокой точностью) с 1,8 мМ MgCl2 (Roche), 200 мкМ dNTP (New England Biolabs) и USER Enzyme (0,1 ед./1 мкл; New England Biolabs), нагревали до 37°С, добавляли в каждую лунку и инкубировали при 37°С в течение 30 мин с перемешиванием (3 секунды при 300 об./мин, 6 секунд в неподвижном состоянии) (термосмеситель Comfort; Eppendorf). Реакционную смесь, содержащую высвобожденные кДНК и зонды, затем извлекали из лунок с помощью пипетки.
Высвобождение захватывающих зондов с использованием смеси расщепляющего по урацилам USER enzyme с буфером для TdT (концевой трансферазы) (ковалентно присоединенных зондов)
50 мкл смеси, содержащей: 1× буфер для TdT (20 мМ Трис-ацетат (рН 7,9), 50 мМ ацетат калия и 10 мМ ацетат магния) (New England Biolabs, www.neb.com); BSA (0,1 мкг/мкл) (New England Biolabs); и USER Enzyme (0,1 ед./мкл; New England Biolabs), нагревали до 37°С, добавляли в каждую лунку и инкубировали при 37°С в течение 30 мин с перемешиванием (3 секунды при 300 об./мин, 6 секунд в неподвижном состоянии) (термосмеситель Comfort; Eppendorf). Реакционную смесь, содержащую высвобожденные кДНК и зонды, затем извлекали из лунок с помощью пипетки.
Высвобождение захватывающих зондов с использованием кипящей воды (ковалентно присоединенных зондов)
16-Луночный экран и слайд CodeLink прикрепляли к держателю ChipClip (Whatman). В каждую лунку с помощью пипетки вносили по 50 мкл воды (99°С). Эту воду (99°С) оставляли для взаимодействия в течение 30 минут. Реакционную смесь, содержащую высвобожденные кДНК и зонды, затем извлекали из лунок с помощью пипетки.
Высвобождение захватывающих зондов с использованием нагретого буфера для ПЦР (захватывающих зондов, синтезированных посредством гибридизации in situ, т.е. захватывающих зондов, гибридизованных с поверхностными зондами)
50 мкл смеси, содержащей: 1х буфер для TdT (20 мМ Трис-ацетат (рН 7,9), 50 мМ ацетат калия и 10 мМ ацетат магния) (New England Biolabs, www.neb.com); BSA (0,1 мкг/мкл) (New England Biolabs); и USER Enzyme (0,1 ед./мкл; New England Biolabs), предварительно нагревали до 95°С. Затем эту смесь добавляли в каждую лунку и инкубировали в течение 5 минут при 95°С с перемешиванием (3 секунды при 300 об./мин, 6 секунд в неподвижном состоянии) (термосмеситель Comfort; Eppendorf). Реакционную смесь, содержащую высвобожденные зонды, затем извлекали из лунок.
Высвобождение захватывающих зондов с использованием нагретого буфера для TdT (концевой трансферазы) (захватывающих зондов, синтезированных посредством гибридизации in situ, т.е. захватывающих зондов, гибридизованных с поверхностными зондами)
50 мкл смеси, содержащей: 1× буфер для TdT (20 мМ Трис-ацетат (рН 7,9), 50 мМ ацетат калия и 10 мМ ацетат магния) (New England Biolabs, www.neb.com); BSA (0,1 мкг/мкл) (New England Biolabs); и USER Enzyme (0,1 ед./мкл; New England Biolabs), предварительно нагревали до 95°С. Затем эту смесь добавляли в каждую лунку и инкубировали в течение 5 минут при 95°С с перемешиванием (3 секунды при 300 об./мин, 6 секунд в неподвижном состоянии) (термосмеситель Comfort; Eppendorf). Реакционную смесь, содержащую высвобожденные зонды, затем извлекали из лунок.
Эффективность обработки чипа с использованием USER enzyme и воды, нагретой до 99°С, можно увидеть на Фиг.3. Ферментативное расщепление с применением USER enzyme и фермента Rsal проводили, используя чипы "собственной разработки", описанные выше (Фиг.4). Опосредованное горячей водой высвобождение ДНК поверхностных зондов проводили, используя промышленные чипы производства Agilent (см. Фиг.5).
Сбор зондов и введение линкеров
Результаты экспериментов показывают, что первая нить кДНК, высвобожденная с поверхности чипа, может быть модифицирована с получением двунитевой ДНК и в дальнейшем амплифицирована.
Амплификация целого транскриптома с использованием набора для амплификации целого генома Picoplex (последовательности захватывающих зондов, включающие последовательности позиционных областей (меток), не сохранены на конце полученной днДНК (двунитевая ДНЮ)
Захватывающие зонды высвобождали с использованием смеси расщепляющего по урацилам USER enzyme с буфером для ПЦР (ковалентно присоединенные захватывающие зонды) или с использованием нагретого буфера для ПЦР (захватывающие зонды, синтезированные посредством гибридизации in situ, т.е. захватывающие зонды, гибридизованные с поверхностными зондами).
Высвобожденную кДНК амплифицировали, применяя метод амплификации целого генома с использованием случайных праймеров Picoplex (Rubicon Genomics), который осуществляли в соответствии с инструкциями производителя.
Амплификация целого транскриптома посредством наращивания (dA)-хвоста с использованием концевой трансферазы (TdT) (последовательности захватывающих зондов, включающие последовательности позиционных областей (меток), сохранены на конце полученной днДНК)
Захватывающие зонды высвобождали с использованием смеси расщепляющего по урацилам USER enzyme с буфером для концевой трансферазы (TdT) (ковалентно присоединенные захватывающие зонды) или нагретого буфера для TdT (концевой трансферазы) (захватывающие зонды, синтезированные посредством гибридизации in situ, т.е. захватывающие зонды, гибридизованные с поверхностными зондами).
38 мкл смеси после отщепления помещали в чистую пробирку для ПЦР емкостью 0,2 мл. Смесь содержала: 1× буфер для TdT (20 мМ Трис-ацетат (рН 7,9), 50 мМ ацетат калия и 10 мМ ацетат магния) (New England Biolabs, www.neb.com), BSA (0,1 мкг/мкл) (New England Biolabs); USER Enzyme (0,1 ед./мкл; New England Biolabs) (не для высвобождения нагреванием); высвобожденную кДНК (удлиненную с поверхностных зондов); и высвобожденные поверхностные зонды. В пробирку для ПЦР добавляли 0,5 мкл РНКазы Н (5 ед./мкл, конечная концентрация 0,06 ед./мкл), 1 мкл TdT (20 ед./мкл, конечная концентрация 0,5 ед./мкл) и 0,5 мкл dATP (100 мМ, конечная концентрация 1,25 мМ). Для наращивания (dA)-хвоста пробирку инкубировали в термоциклере (Applied Biosystems) при 37°С в течение 15 мин, после чего инактивировали TdT при 70°С в течение 10 мин. После наращивания (dA)-хвоста готовили мастер-микс (master mix) для ПЦР. Смесь содержала: 1× буфер для ПЦР Faststart HiFi (рН 8,3) с 1,8 мМ MgCl2 (Roche); dNTP (0,2 мМ каждый; Fermentas); праймеры (0,2 мМ каждый): А (комплементарный области амплификации захватывающего зонда) и B_(dT)24 (Eurofins MWG Operon) (комплементарный поли(А)-хвосту, который должен быть добавлен к 3′-концу первой нити кДНК); и ДНК-полимеразу (0,1 ед./мкл) Faststart HiFi (Roche). По 23 мкл мастер-микс для ПЦР помещали в девять чистых пробирок для ПЦР емкостью 0,2 мл. В восемь пробирок добавляли по 2 мкл смеси для наращивания (dA)-хвоста, тогда как в последнюю пробирку добавляли 2 мкл воды (без РНКазы/ДНКазы) (отрицательный контроль). ПЦР-амплификацию проводили по следующей программе: горячий старт при 95°С в течение 2 минут, синтез второй нити при 50°С в течение 2 минут и при 72°С в течение 3 минут, амплификация за 30 циклов ПЦР при 95°С в течение 30 секунд, 65°С в течение 1 минуты, 72°С в течение 3 минут и окончательное удлинение при 72°С в течение 10 минут.
Очистка и анализ после проведения реакции
Четыре продукта амплификации объединяли вместе, обрабатывали с использованием колонки Qiaquick для очистки после ПЦР (Qiagen) и элюировали в 30 мкл ЕВ (элюирующего буфера) (10 мМ Трис-Cl, рН 8,5). Продукт анализировали на биоанализаторе (Agilent). Использовали набор DNA 1000 в соответствии с инструкциями производителей.
Секвенирование
Секвенирование по Illumina
Секвенирование по Illumina библиотеки днДНК с использованием индексирования образцов осуществляли в соответствии с инструкциями производителей. Секвенирование проводили на платформе HiSeq2000 (Illumina).
Биоинформатика
Получение цифровой информации о транскриптоме из данных секвенирования библиотек для целого транскриптома. полученных амплификацией с применением подхода с наращиванием (dA)-хвоста концевой трансферазой
Данные секвенирования сортировали, используя штрихкодовый сплиттер с применением формата FASTQ пакета FastX-toolkit, по отдельным файлам для соответствующих последовательностей позиционных областей (меток) захватывающих зондов. Данные по меченным индивидуальными метками последовательностям затем анализировали посредством сравнения с картами генома мыши, используя программу для картирования Tophat. Полученный SAM-файл обрабатывали для подсчета транскриптов посредством программного обеспечения HTseq-count.
Получение цифровой информации о транскриптоме из данных секвенирования библиотек для целого транскриптома. полученных амплификацией с применением подхода с использованием набора для амплификации целого генома Picoplex
Данные секвенирования преобразовывали из формата FASTQ в формат FASTA, используя конвертер FASTQ-to-FASTA пакета FastX-toolkit. Проводили выравнивание прочтений последовательностей с последовательностями позиционных областей (меток) захватывающих зондов, используя Blastn, и прочтения с совпадениями лучшими, чем 1е-6 в отношении одной из содержащих метку последовательностей отсортировывали в индивидуальные файлы для каждой меченой последовательности, соответственно. Затем с использованием Blastn проводили выравнивание прочтений содержащих метку последовательностей из данного файла с транскриптомом мыши и отбирали совпадения.
Совмещение данных визуализации и профилей экспрессии
Проводили совмещение профилей экспрессии для индивидуальных последовательностей позиционных областей (меток) захватывающих зондов с пространственной информацией, полученной из срезов тканей посредством окрашивания. Тем самым можно путем прямого сравнения проанализировать данные о транскриптоме из клеточных компартментов среза ткани, что дает возможность устанавливать различия между особенностями экспрессии для разных клеточных подтипов в данном структурном контексте.
Пример 2
На Фиг.8-12 показаны результаты успешной визуализации окрашенных срезов FFPE-ткани головного мозга мыши (обонятельной луковицы) на поверхности штрихкодового захватывающего чипа для анализа транскриптома согласно общей методике, описанной в примере 1. По сравнению с экспериментом со свежезамороженной тканью, приведенном в примере 1, на Фиг.8 показана более хорошая морфология при использовании FFPE-ткани. На Фиг.9 и 10 показано, как ткань может располагаться на чипах с различной плотностью зондов.
Пример 3
Амплификация целого транскриптома посредством синтеза второй нити с использованием случайных праймеров с последующей амплификацией с применением универсальной ручки (последовательности захватывающих зондов, включающие меченые последовательности, сохранены на конце полученной днДНК)
- после высвобождения захватывающих зондов с использованием смеси расщепляющего по урацилам USER enzyme с буфером для ПЦР (ковалентно присоединенных зондов)
или
- после высвобождения захватывающих зондов с использованием нагретого буфера для ПЦР (захватывающих зондов, синтезированных посредством гибридизации in situ).
1 мкл РНКазы Н (5 ед./мкл) добавляли в каждую из двух пробирок (конечная концентрация 0,12 ед./мкл), содержащих по 40 мкл 1х буфера для ПЦР Faststart HiFi (pH 8,3) с 1,8 мМ MgCl2 (Roche, www.roche-applied-science.com), dNTP (0,2 мМ каждый) (Fermentas, www.fermentas.com), BSA (0,1 мкг/мкл) (New England Biolabs, www.neb.com), USER Enzyme (0,1 ед./мкл; New England Biolabs), высвобожденую кДНК (удлиненную с поверхностных зондов) и высвобожденные поверхностные зонды. Пробирки инкубировали при 37°С в течение 30 мин, затем при 70°С в течение 20 мин в термоциклере (Applied Biosystems, www.appliedbiosystems.com). В эти две пробирки добавляли по 1 мкл фрагмента Кленова (от 3′ к 5′ экзо-минус) (Illumina, www.illumina.com) и по 1 мкл соединенного с "ручкой" случайного праймера (10 мкМ) (Eurofins MWG Operon, www.eurofinsdna.com) (B_R8 (октамер) в одну из пробирок и B_R6 (гексамер) в другую пробирку), конечная концентрация 0,23 мкМ. Эти две пробирки инкубировали при 15°С в течение 15 мин, 25°С в течение 15 мин, 37°С в течение 15 мин и в конце при 75°С в течение 20 мин в термоциклере (Applied Biosystems). После инкубации в обе пробирки добавляли по 1 мкл каждого праймера, А_Р и В (10 мкМ) (Eurofins MWG Operon), конечная концентрация каждого 0,22 мкМ. В обе пробирки также добавляли по 1 мкл (5 ед./мкл) ДНК-полимеразы Faststart HiFi (Roche), конечная концентрация 0,11 ед./мкл. ПЦР-амплификацию осуществляли в термоциклере (Applied Biosystems) в соответствии со следующей программой: горячий старт при 94°С в течение 2 мин, затем 50 циклов при 94°С в течение 15 секунд, 55°С в течение 30 секунд, 68°С в течение 1 минуты и окончательное удлинение при 68°С в течение 5 минут. После амплификации очищали по 40 мкл из каждой из этих двух пробирок, используя колонку Qiaquick для очистки после ПЦР (Qiagen, www.qiagen.com), и элюировали в 30 мкл ЕВ (10 мМ Трис-Cl, рН 8,5). Очищенные продукты анализировали на биоанализаторе (Agilent, www.home.agilent.com), с использованием набора DNA 7500. Результаты показаны на Фиг.13 и 14.
Этот пример демонстрирует применение синтеза второй нити с использованием случайного гексамера и случайного октамера с последующей амплификацией с целью создания популяции из высвобожденных молекул кДНК.
Пример 4
Амплификация IP-специфичных и ген-специфичных продуктов после синтеза кДНК и сбора зондов
- после высвобождения захватывающих зондов с использованием смеси расщепляющего по урацилам USER enzyme с буфером для ПЦР (ковалентно присоединенных зондов).
Отщепленную кДНК амплифицировали в конечных реакционных объемах 10 мкл. 7 мкл отщепленной матрицы, 1 мкл ID-специфичного прямого праймера (2 мкМ), 1 мкл ген-специфичного обратного праймера (2 мкМ) и 1 мкл ферментативной смеси FastStart High Fidelity в 1,4х реакционном буфере FastStart High Fidelity с 1,8 мМ MgCl2 смешивали, получая конечную реакционную смесь объемом 10 мкл с 1× реакционным буфером FastStart High Fidelity с 1,8 мМ MgCl2 и 1 ед. ферментативной смеси FastStart High Fidelity. ПЦР-амплификацию осуществляли в термоциклере (Applied Biosystems) в соответствии со следующей программой: горячий старт при 94°С в течение 2 мин, затем 50 циклов при 94°С в течение 15 секунд, 55°С в течение 30 секунд, 68°С в течение 1 минуты и окончательное удлинение при 68°С в течение 5 минут.
Последовательности праймеров, использованные для получения продукта длиной приблизительно 250 п.о.:
бета-2-микроглобулиновый праймер (В2М):
5′-TGGGGGTGAGAATTGCTAAG-3′ (SEQ ID NO:43),
ID-1 праймер:
5′-ССТТСТССТТСТССТТСАСС-3′ (SEQ ID NO:44),
ID-5 праймер:
5′-GTCCTCTATTCCGTCACCAT-3′ (SEQ ID NO:45),
ID-20 праймер:
5′-CTGCTTCTTCCTGGAACTCA-3′ (SEQ ID NO:46).
Результаты показаны на Фиг.15. Показана успешно проведенная амплификация ID-специфичных и ген-специфичных продуктов из ткани головного мозга, покрывающей все эти зонды, с использованием двух разных ID-содержащих праймеров (т.е. праймера, специфичного к ID-меткам, расположенным в разных местах на микрочипе, и равным образом ген-специфичного праймера). Соответственно, в этом эксперименте установлено, что продукты могут быть идентифицированы по реакции амплификации, специфичной в отношении ID-метки или в отношении целевой нуклеиновой кислоты. Также установлено, что можно провести различение разных ID-меток. Во втором эксперименте с использованием ткани, покрывающей только половину ID-содержащих зондов (т.е. захватывающих зондов) на чипе, положительный результат (продукт ПЦР) получили для пятен, которые были покрыты тканью.
Пример 5
Пространственная геномика
Общие положения. Задачей этого способа является захват молекул ДНК из образца ткани с сохранением пространственного разрешения, что дает возможность определить, из какой части ткани конкретный фрагмент ДНК происходит.
Способ. Принцип данного способа заключается в применении микрочипов с иммобилизованными ДНК-олигонуклеотидами (захватывающими зондами), несущими последовательности с маркирующими метками для пространственной локализации (позиционные области). Каждый элемент из олигонуклеотидов микрочипа несет 1) уникальную маркирующую метку (позиционную область) и 2) захватывающую последовательность (захватывающую область). Отслеживание того, в каком месте и какая маркирующая метка размещается на поверхности чипа, дает возможность извлечь информацию о положении в двух измерениях от каждой маркирующей метки. Фрагментированную геномную ДНК добавляют к микрочипу, например путем добавления тонкого среза FFPE-обработанной ткани. Геномная ДНК в этом срезе ткани предварительно фрагментирована в результате фиксирующей обработки.
Как только срез ткани помещен на чип, проводят универсальную реакцию наращивания хвоста, применяя фермент концевую трансферазу. В результате реакции наращивания хвоста к выступающим 3′-концам фрагментов геномной ДНК в ткани добавляются поли(dA)-хвосты. Олигонуклеотиды на поверхности блокируют от наращивания хвоста под действием концевой трансферазы посредством использования гибридизованного и Заблокированного nonn(dA)-зонда.
После наращивания хвоста под действием концевой трансферазы фрагменты геномной ДНК получают способность гибридизоваться с олигонуклеотидами, несущими пространственную метку близко друг к другу, в результате встречи поли(dA)-хвоста с поли(dT)-содержащей захватывающей последовательностью поверхностных олигонуклеотидов. По завершении гибридизации полимераза замещения нити такая как фрагмент Кленова (экзо-), может использовать олигонуклеотид на поверхности в качестве праймера для создания новой нити ДНК, комплементарной гибридизованному фрагменту геномной ДНК. Новая нить ДНК будет теперь также содержать информацию о положении маркирующей метки олигонуклеотида на поверхности.
На последней стадии вновь синтезированные меченые нити ДНК отщепляют от поверхности, используя или ферментативные методы, или денатурацию, или физические методы. Затем нити собирают, и весь набор нитей может быть подвергнут амплификации в направлении "вниз по течению" с использованием введения универсальных "ручек", амплификации специфических ампликонов и/или подвергнут секвенированию.
Фиг.16 представляет собой схематическую иллюстрацию этого способа.
Материалы и методы
Изготовление печатного микрочипа собственной разработки с от ориентированными 5′ к 3′ зондами
На стеклянные слайды наносили в виде пятен 20 ДНК-захватывающих олигонуклеотидов с содержащими индивидуальные метки последовательностями (Таблица 1) в качестве захватывающих зондов. Зонды синтезировали таким образом, чтобы они содержали на 5′-конце аминолинкер с С6-спейсером. Все зонды были синтезированы в Sigma-Aldrich (St. Louis, МО, USA). ДНК-захватывающие зонды суспендировали в концентрации 20 мкМ в 150 мМ фосфате натрия, рН 8,5, и наносили в виде пятен, используя наноплоттер (nanoplotter) NP2.1/E (Gesim, Grosserkmannsdorf, Germany), на активированные слайды для микрочипов CodeLink™ (7,5 см×2,5 см; Surmodics, Eden Prairie, MN, USA). После печати осуществляли блокирование поверхности в соответствии с инструкциями производителя. Печать зондов выполняли в виде 16 идентичных чипов на слайде, и каждый чип имел предварительно определенную картину печати. Эти 16 подчипов были отделены друг от друга в процессе гибридизации посредством состоящего из 16 ячеек экрана (ChipClip™, Schleicher & Schuell BioScience, Keene, NH, USA).
Изготовление печатного микрочипа собственной разработки с ориентированными от 3′ к 5′ зондами и синтез ориентированных от 5′ к 3′ захватывающих зондов
Печать олигонуклеотидов осуществляли, как и в приведенном выше случае с ориентированными от 5′ к 3′ зондами.
Чтобы осуществить гибридизацию праймеров для синтеза захватывающих зондов, раствор для гибридизации, содержащий 4×SSC и 0,1% SDS, 2 мкМ праймер удлинения (А_праймер) и 2 мкМ праймер для соединения нитей (P_поли_dT) инкубировали в течение 4 мин при 50°С. За это время прикрепляли чип собственной разработки к ChipClip (Whatman). Далее этот чип инкубировали при 50°С в течение 30 мин на шейкере при 300 об./мин вместе с 50 мкл раствора для гибридизации на одну лунку.
После инкубации чип извлекали из ChipClip и промывали, используя 3 приведенные ниже стадии: 1) 50°С, раствор 2×SSC с 0,1% SDS в течение 6 мин на шейкере при 300 об./мин; 2) 0,2×SSC в течение 1 мин на шейкере при 300 об./мин; и 3) 0,1×SSC в течение 1 мин на шейкере при 300 об./мин. Затем чип сушили в центрифуге и помещали обратно в ChipClip.
Для осуществления реакции удлинения и лигирования 50 мкл ферментативной смеси, содержащей 10×буфер для лигазы Ampligase, 2,5 ед. фрагмента Стоффеля AmpliTaq ДНК-полимеразы (Applied Biosystems), 10 ед. лигазы Ampligase (Epicentre Biotechnologies), dNTP 2 мМ каждый (Fermentas) и воду, вносили в каждую лунку с помощью пипетки. Затем этот чип инкубировали при 55°С в течение 30 мин. После инкубации чип промывали в соответствии с описанным ранее способом промывки чипа, но продолжительность первой стадии составляла 10 мин вместо 6 мин.
Гибридизация поли(dA)-зонда для защиты захватывающих последовательностей поверхностных олигонуклеотидов от наращивания (dA)-хвоста
Чтобы осуществить гибридизацию 3′-биотин-блокированного поли(с)А)-содержащего зонда для защиты захватывающих последовательностей поверхностных олигонуклеотидов, раствор для гибридизации, содержащий 4×SSC и 0,1% SDS, 2 мкМ 3′-биотин-поли(dA), инкубировали в течение 4 мин при 50°С. За это время прикрепляли чип собственной разработки к ChipClip (Whatman). Далее этот чип инкубировали при 50°С в течение 30 мин на шейкере при 300 об./мин вместе с 50 мкл раствора для гибридизации на одну лунку.
После инкубации чип извлекали из ChipClip и промывали, используя 3 приведенные ниже стадии: 1) 50°С, раствор 2×SSC с 0,1% SDS в течение 6 мин на шейкере при 300 об./мин; 2) 0,2×SSC в течение 1 мин на шейкере при 300 об./мин; и 3) 0,1×SSC в течение 1 мин на шейкере при 300 об./мин. Затем чип сушили в центрифуге и помещали обратно в ChipClip.
Приготовление фиксированной в формалине залитой парафином (FFPE) ткани
Ткань головного мозга мыши фиксировали в 4%-ном формалине при 4°С в течение 24 ч. После этого ее инкубировали следующим образом: 3× инкубация в 70%-ном этаноле в течение 1 часа; 1× инкубация в 80%-ном этаноле в течение 1 часа; 1× инкубация в 96%-ном этаноле в течение 1 часа; 3× инкубация в 100%-ном этаноле в течение 1 часа; и 2х инкубация в ксилоле при комнатной температуре в течение 1 ч.
Дегидратированные образцы далее инкубировали в жидком легкоплавком парафине при 52-54°С в течение до 3 часов включительно, за это время один раз проводили замену парафина, чтобы отмыть от оставшегося ксилола. Готовые блоки ткани далее хранили при КГ. Затем, используя микротом, делали срезы в парафине толщиной 4 мкм для нанесения на поверхность каждого предназначенного для использования чипа, содержащего захватывающие зонды.
Срезы сушили при 37°С на слайдах чипов в течение 24 часов и хранили при КТ.
Депарафинизация FFPE-ткани
Фиксированные в формалине парафинизированные срезы головного мозга мыши толщиной 10 мкм, нанесенные на поверхность слайдов CodeLink, дважды подвергали депарафинизации в ксилоле в течение 10 мин, в 99,5%-ном этаноле в течение 2 мин, в 96%-ном этаноле в течение 2 мин, в 70%-ном этаноле в течение 2 мин и затем сушили на воздухе.
Наращивание универсального хвоста в геномную ДНК
Для наращивания (dA)-хвоста готовили 50 мкл реакционной смеси, содержащей 1х буфер для TdT (20 мМ Трис-ацетат (рН 7,9), 50 мМ ацетат калия и 10 мМ ацетат магния) (New England Biolabs, www.neb.com), BSA (0,1 мкг/мкл) (New England Biolabs), 1 мкл TdT (20 ед./мкл) и 0,5 мкл dATP (100 мМ). Смесь добавляли на поверхность чипа и чип инкубировали в термоциклере (Applied Biosystems) при 37°С в течение 15 мин, после чего инактивировали TdT при 70°С в течение 10 мин. После этого температуру снова понижали до 50°С, чтобы обеспечить возможность гибридизации геномных фрагментов, содержащих (dA)-хвосты, с захватывающими областями поверхностных олигонуклеотидов.
После инкубации чип извлекали из ChipClip и промывали, используя 3 приведенные ниже стадии: 1) 50°С, раствор 2×SSC с 0,1% SDS в течение 6 мин на шейкере при 300 об./мин; 2) 0,2×SSC в течение 1 мин на шейкере при 300 об./мин; и 3) 0,1×SSC в течение 1 мин на шейкере при 300 об./мин. Затем чип сушили в центрифуге.
Удлинение меченой ДНК
Реакционную смесь объемом 50 мкл, содержащую 1х буфер Кленова, 200 мкМ dNTP (New England Biolabs) и 1 мкл фрагмента Кленова (от 3′ к 5′ экзо-минус), нагревали до 37°С, добавляли в каждую лунку и инкубировали при 37°С в течение 30 мин с перемешиванием (3 секунды при 300 об./мин; 6 секунд в неподвижном состоянии) (термосмеситель Comfort; Eppendorf).
После инкубации чип извлекали из ChipClip и промывали, используя 3 приведенные ниже стадии: 1) 50°С, раствор 2×SSC с 0,1% SDS в течение 6 мин на шейкере при 300 об./мин, 2) 0,2×SSC в течение 1 мин на шейкере при 300 об./мин и 3) 0,1×SSC в течение 1 мин на шейкере при 300 об./мин. Затем чип сушили в центрифуге.
Удаление оставшейся ткани
Слайды с фиксированными в формалине парафинизированными срезами ткани головного мозга мыши прикрепляли к держателям слайдов ChipClip и 16-луночным экранам (Whatman). На каждые 150 мкл буфера для переваривания протеиназой К из набора RNeasy FFPE (Qiagen) добавляли по 10 мкл раствора протеиназы К (Qiagen). В каждую лунку добавляли по 50 мкл конечной смеси и слайд инкубировали при 56°С в течение 30 мин.
Высвобождение захватывающих зондов с использованием смеси расщепляющего по урацилам USER enzyme с буфером для ПЦР (ковалентно присоединенных зондов)
16-луночный экран и слайд CodeUnk прикрепляли к держателю ChipClip (Whatman). 50 мкл смеси, содержащей 1× реакционный буфер FastStart High Fidelity с 1,8 мМ MgCl2 (Roche), 200 мкМ dNTP (New England Biolabs) и USER Enzyme (0,1 ед./1 мкл; New England Biolabs), нагревали до 37°С, добавляли в каждую лунку и инкубировали при 37°С в течение 30 мин с перемешиванием (3 секунды при 300 об./мин, 6 секунд в неподвижном состоянии) (термосмеситель Comfort; Eppendorf). Реакционную смесь, содержащую высвобожденные кДНК и зонды, затем извлекали из лунок с помощью пипетки.
Амплификация ID-специсЬичных и ген-специфичных продуктов после синтеза кДНК и сбора зондов
- после высвобождения захватывающих зондов с использованием смеси расщепляющего по урацилам USER enzyme с буфером для ПЦР (ковалентно присоединенных зондов).
Отщепленную кДНК амплифицировали в конечных реакционных объемах 10 мкл. 7 мкл отщепленной матрицы, 1 мкл ID-специфичного прямого праймера (2 мкМ), 1 мкл ген-специфичного обратного праймера (2 мкМ) и 1 мкл ферментативной смеси FastStart High Fidelity в 1,4х реакционном буфере FastStart High Fidelity с 1,8 мМ MgCl2 смешивали, получая конечную реакционную смесь объемом 10 мкл с 1× реакционным буфером FastStart High Fidelity с 1,8 мМ MgCl2 и 1 ед. ферментативной смеси FastStart High Fidelity. ПЦР-амплификацию осуществляли в термоциклере (Applied Biosystems) в соответствии со следующей программой: горячий старт при 94°С в течение 2 мин, затем 50 циклов при 94°С в течение 15 секунд, 55°С в течение 30 секунд, 68°С в течение 1 минуты и окончательное удлинение при 68°С в течение 5 минут.
Амплификация целого генома посредством синтеза второй нити с использованием случайных праймеров с последующей амплификацией с применением универсальной ручки (последовательности захватывающих зондов, включающие меченые последовательности, сохранены на конце полученной днДНК)
- после высвобождения захватывающих зондов с использованием смеси расщепляющего по урацилам USER enzyme с буфером для ПЦР (ковалентно присоединенных зондов).
Готовили реакционную смесь, содержащую 40 мкл 1× буфера для ПЦР (рН 8,3) FastStart HiFi с 1,8 мМ MgCl2 (Roche, www.roche-applied-science.com), dNTP (0,2 мМ каждый) (Fermentas, www.fermentas.com), BSA (0,1 мкг/мкл) (New England Biolabs, www.neb.com), USER Enzyme (0,1 ед./мкл; New England Biolabs), высвобожденную ДНК (удлиненную с поверхностных зондов) и высвобожденные поверхностные зонды. Пробирку инкубировали при 37°С в течение 30 мин, затем при 70°С в течение 20 мин в термоциклере (Applied Biosystems, www.appliedbiosystems.com). В эту пробирку добавляли 1 мкл фрагмента Кленова (от 3′ к 5′ экзо-минус) (Illumina, www.illumina.com) и 1 мкл соединенного с "ручкой" случайного праймера (10 мкМ) (Eurofins MWG Орегоп, www.eurofinsdna.com). Пробирку инкубировали при 15°С в течение 15 мин, при 25°С в течение 15 мин, при 37°С в течение 15 мин и в конце при 75°С в течение 20 мин в термоциклере (Applied Biosystems). После инкубации в пробирку добавляли по 1 мкл каждого праймера, А_Р и В (10 мкМ) (Eurofins MWG Operon). В пробирку также добавляли 1 мкл (5 ед./мкл) ДНК-полимеразы FastStart HiFi (Roche). ПЦР-амплификацию осуществляли в термоциклере (Applied Biosystems) в соответствии со следующей программой: горячий старт при 94°С в течение 2 мин, затем 50 циклов при 94°С в течение 15 секунд, 55°С в течение 30 секунд, 68°С в течение 1 минуты и окончательное удлинение при 68°С в течение 5 минут. После амплификации проводили очистку 40 мкл из пробирки, используя колонки Qiaquick для очистки после ПЦР (Qiagen, www.qiagen.com), и элюировали в 30 мкл ЕВ (10 мМ Трис-Cl, рН 8,5). Очищенный продукт анализировали на биоанализаторе (Agilent, www.home.agilent.com), с использованием набора DNA 7500.
Визуализация
Гибридизация флуоресцентных маркерных зондов перед окрашиванием
Перед наложением ткани флуоресцентные маркерные зонды гибридизовали с предусмотренными маркерными последовательностями, отпечатанными на чипе, содержащем захватывающие зонды. Флуоресцентные маркерные зонды способствуют ориентации полученного изображения после визуализации ткани, давая возможность комбинировать это изображение с полученными профилями экспрессии для индивидуальных, несущих "метку" захватывающих зондов, последовательностей, полученных после секвенирования. Чтобы осуществить гибридизацию флуоресцентных зондов, раствор для гибридизации, содержащий 4xSSC и 0,1% SDS, 2 мкМ детектирующий зонд (Р), инкубировали в течение 4 мин при 50°С. За это время прикрепляли чип собственной разработки к ChipClip (Whatman). Далее этот чип инкубировали при 50°С в течение 30 мин на шейкере при 300 об./мин вместе с 50 мкл раствора для гибридизации на одну лунку.
После инкубации чип извлекали из ChipClip и промывали, используя 3 приведенные ниже стадии: 1) 50°С, раствор 2×SSC с 0,1% SDS в течение 6 мин на шейкере при 300 об./мин; 2) 0,2×SSC в течение 1 мин на шейкере при 300 об./мин; и 3) 0,1×SSC в течение 1 мин на шейкере при 300 об./мин. Затем чип сушили в центрифуге.
Общее гистологическое окрашивание срезов FFPE-ткани до или после синтеза меченой ДНК
Срезы FFPE-ткани, иммобилизованные на чипах, содержащих захватывающие зонды, промывали и регидратировали после депарафинизации перед синтезом меченой ДНК, как описано ранее, или промывали после синтеза меченой ДНК, как описано ранее. Далее их обрабатывали, как приведено ниже: инкубировали в течение 3 минут в гематоксилине; промывали деионизованной водой; инкубировали в течение 5 минут в водопроводной воде; быстро окунали от 8 до 12 раз в подкисленный этанол; промывали 2 раза по 1 минуте в водопроводной воде; промывали 2 минуты в деонизованной воде; инкубировали 30 секунд в эозине; промывали 3×5 минут в 95%-ном этаноле; промывали 3×5 минут в 100%-ном этаноле; промывали 3×10 минут в ксилоле (может быть выполнено в течение ночи); помещали покровное стекло на слайды, используя DPX; сушили слайды в вытяжном шкафу в течение ночи.
Общее иммуногистохимическое окрашивание целевого белка в срезах FFPE-ткани до или после синтеза меченой ДНК
Срезы FFPE-ткани, иммобилизованные на чипах, содержащих захватывающие зонды, промывали и регидратировали после депарафинизации перед синтезом меченой ДНК, как описано ранее, или промывали после синтеза меченой ДНК, как описано ранее. Далее их обрабатывали, как приведено ниже, не позволяя высыхать в течение всего процесса окрашивания:
разбавляли первичное антитело в блокирующем растворе (1×TBS (забуференный трисом физиологический раствор (50 мМ Трис, 150 мМ NaCl, рН 7,6), 4% сыворотки осла и 0,1% тритона-х), инкубировали срезы с первичным антителом в камере с увлажнением в течение ночи при КГ, промывали 3 раза, используя 1×TBS; инкубировали срез с соответствующим вторичным антителом, конъюгированным с флуорохромом (FITC, Cy3 или Cy5) в камере с увлажнением при KT в течение 1 часа, промывали 3 раза, используя 1×TBS, удаляли TBS в той степени, насколько это возможно, заключали срез в Prolong Gold+DAPI (Invitrogen) и анализировали с использованием флуоресцентного микроскопа и подбора подходящих наборов фильтров.
Пример 6
Этот эксперимент проводили, следуя принципам примера 5, но с использованием на чипе фрагментированной геномной ДНК, а не ткани. Геномную ДНК предварительно фрагментировали до среднего размера 200 п.о. и 700 п.о. соответственно. Этот эксперимент показывает, что данный принцип работает. Фрагментированная геномная ДНК в данном случае очень похожа на FFPE-ткань.
Амплификация ген-специфичных внутренних продуктов после синтеза меченой ДНК и сбора зондов
- после высвобождения захватывающих зондов с использованием смеси расщепляющего по урацилам USER enzyme с буфером для ПЦР (ковалентно присоединенных зондов), содержащей 1× реакционный буфер FastStart High Fidelity с 1,8 мМ MgCl2 (Roche), 200 мкМ dNTP (New England Biolabs) и USER Enzyme (0,1 ед./1 мкл; New England Biolabs).
Отщепленную ДНК амплифицировали в конечном реакционном объеме 50 мкл. К 47 мкл отщепленной матрицы добавляли 1 мкл ID-специфичного прямого праймера (10 мкМ), 1 мкл ген-специфичного обратного праймера (10 мкМ) и 1 мкл ферментативной смеси FastStart High Fidelity. ПЦР-амплификацию осуществляли в термоциклере (Applied Biosystems) в соответствии со следующей программой: горячий старт при 94°С в течение 2 мин, затем 50 циклов при 94°С в течение 15 секунд, 55°С в течение 30 секунд, 68°С в течение 1 минуты и окончательное удлинение при 68°С в течение 5 минут.
Амплификация специфичных к метке и ген-специсЬичных продуктов после синтеза меченой ДНК и сбора зондов
- после высвобождения захватывающих зондов с использованием смеси расщепляющего по урацилам USER enzyme с буфером для ПЦР (ковалентно присоединенных зондов), содержащей 1× реакционный буфер FastStart High Fidelity с 1,8 мМ MgCl2 (Roche), 200 мкМ dNTP (New England Biolabs) и USER Enzyme (0,1 ед./1 мкл; New England Biolabs).
Отщепленную ДНК амплифицировали в конечном реакционном объеме 50 мкл. К 47 мкл отщепленной матрицы добавляли 1 мкл специфичного к метке прямого праймера (10 мкМ), 1 мкл ген-специфичного обратного праймера (10 мкМ) и 1 мкл ферментативной смеси FastStart High Fidelity. ПЦР-амплификацию осуществляли в термоциклере (Applied Biosystems) в соответствии со следующей программой: горячий старт при 94°С в течение 2 мин, затем 50 циклов при 94°С в течение 15 секунд, 55°С в течение 30 секунд, 68°С в течение 1 минуты и окончательное удлинение при 68°С в течение 5 минут.
Прямой праймер - праймер для геномной ДНК человека:
B′-GACTGCTCTTTTCACCCATC-S′ (SEQ ID NO:47);
Обратный праймер - праймер для геномной ДНК человека:
5′-GGAGCTGCTGGTGCAGGG-3′ (SEQ ID NO:48);
P - праймер, специфичный к метке:
5′-ATCTCGACTGCCACTCTGAA-3′ (SEQ ID NO:49).
Результаты показаны на Фиг.17-20. На этих рисунках показаны внутренние продукты, амплифицированные на чипе, - зарегистрированные пики на Фиг.17 и 18 соответствуют ожидаемому размеру. Таким образом, этот результат демонстрирует, что существует возможность захвата и амплификации геномной ДНК. На Фиг.19 и 20 изображение ожидаемого продукта является размытым ввиду того, что в результате случайной фрагментации и мечения геномной ДНК концевой трансферазой будет образовываться пул очень разнообразных образцов.
Пример 7
Альтернативный синтез ориентированных от 5′ к 3′ захватывающих зондов с использованием удлинения под действием полимеразы и наращивания хвоста под действием концевой трансферазы
Чтобы осуществить гибридизацию праймеров для синтеза захватывающих зондов, раствор для гибридизации, содержащий 4×SSC и 0,1% SDS и 2 мкМ праймер для удлинения (А_праймер) инкубировали в течение 4 мин при 50°С. За это время прикрепляли чип собственной разработки (см. пример 1) к ChipClip (Whatman). Далее этот чип инкубировали при 50°С в течение 30 мин на шейкере при 300 об./мин вместе с 50 мкл раствора для гибридизации на одну лунку.
После инкубации чип извлекали из ChipClip и промывали, используя 3 приведенные ниже стадии: 1) 50°С, раствор 2×SSC с 0,1% SDS в течение 6 мин на шейкере при 300 об./мин, 2) 0,2×SSC в течение 1 мин на шейкере при 300 об./мин и 3) 0,1×SSC в течение 1 мин на шейкере при 300 об./мин. Затем чип сушили в центрифуге и помещали обратно в ChipClip.
1 мкл фрагмента Кленова (от 3′ к 5′ экзо-минус) (Illumina, www.illumina.com) вместе с 10× буфером Кленова, dNTP (2 мМ каждый; Fermentas) и водой смешивали в 50 мкл реакционной смеси и вносили с помощью пипетки в каждую лунку.
Чип инкубировали при 15°С в течение 15 мин, при 25°С в течение 15 мин, при 37°С в течение 15 мин и в конце при 75°С в течение 20 мин в термосмесителе от Eppendorf.
После инкубации чип извлекали из ChipClip и промывали, используя 3 приведенные ниже стадии: 1) 50°С, раствор 2×SSC с 0,1% SDS в течение 6 мин на шейкере при 300 об./мин, 2) 0,2×SSC в течение 1 мин на шейкере при 300 об./мин и 3) 0,1×SSC в течение 1 мин на шейкере при 300 об./мин. Затем чип сушили в центрифуге и помещали обратно в ChipClip.
Для наращивания (dT)-хвоста готовили 50 мкл реакционной смеси, содержащей 1× буфер для TdT (20 мМ Трис-ацетат (рН 7,9), 50 мМ ацетат калия и 10 мМ ацетат магния) (New England Biolabs, www.neb.com), BSA (0,1 мкг/мкл) (New England Biolabs), 0,5 мкл РНКазы Н (5 ед./мкл), 1 мкл TdT (20 ед./мкл) и 0,5 мкл dTTP (100 мМ). Смесь добавляли на поверхность чипа и чип инкубировали в термоциклере (Applied Biosystems) при 37°С в течение 15 мин, после чего инактивировали TdT при 70°С в течение 10 мин.
Пример 8
Пространственная транскриптомика с использованием ориентированных от 5′ к 3′ чипов с высокой плотностью зондов и фиксированной в формалине замороженной (FF-замороженной) ткани с применением системы расщепления USER и амплификации с использованием концевой трансферазы
Изготовление чипов
Стандартные микрочипы высокой плотности были заказаны у Roche-Nimblegen (Madison, WI, USA). Каждый использованный чип с захватывающими зондами содержал 135000 элементов, из которых 132640 содержали уникальную меченую ID-меткой последовательность (позиционную область) и захватывающий участок (захватывающую область). Размер каждого элемента составлял 13×13 мкм. Захватывающие зонды имели направление от 56 к 3′ и состояли из универсальной области, содержащей пять dUTP оснований, (области расщепления) и общей области амплификации, ID-метки (позиционной области) и захватывающего участка (захватывающей области) (Фиг.22 и Таблица 2). Каждый чип также был снабжен рамкой маркерных зондов (Фиг.23), несущих характерную последовательность из 30 п.о. (Таблица 2), дающую возможность гибридизоваться флуоресцентным зондам для помощи в ориентации во время визуализации чипа.
Подготовка ткани: приготовление фиксированной в формалине замороженной ткани
Животное (мышь) перфузировали, используя 50 мл PBS (забуференный фосфатом физиологический раствор) и 100 мл 4%-ного раствора формалина. После иссечения обонятельной луковицы эту ткань помещали в баню с 4%-ным формалином для последующей фиксации в течение 24 ч. Затем ткань обрабатывали сахарозой, используя 30%-ную сахарозу, растворенную в PBS, в течение 24 ч для стабилизации морфологической картины и для удаления избытка формалина. Ткань замораживали до -40°С при регулируемой скорости и хранили при -20°С между экспериментами. Аналогичным образом готовили препарат ткани с последующей фиксацией в течение 3 ч или без последующей фиксации для получения параллельного контрольного образца. Кроме того, успешно использовали перфузию 2%-ным формалином без последующей фиксации. Точно так же может быть опущена стадия обработки сахарозой. Ткань закрепляли в криостате для приготовления срезов толщиной 10 мкм. Срез ткани помещали на поверхность каждого предназначенного для использования чипа, содержащего захватывающие зонды. Для лучшей адгезии ткани чип с чипом возможно помещали на 50°С на 15 минут.
Возможный контроль: получение суммарной РНК из срезов ткани
Суммарную РНК выделяли из одного среза ткани (10 мкм), используя набор RNeasy FFPE (Qiagen) в соответствии с инструкциями производителя. Эту суммарную РНК, полученную из среза ткани, использовали в контрольных экспериментах для сравнения с экспериментами, в которых РНК была захвачена на чипе непосредственно из среза ткани. Соответственно, в том случае, когда на чип наносили суммарную РНК, стадии окрашивания, визуализации и деградации ткани опускали.
Реакции на чипе
Гибридизацию маркерного зонда с рамочными зондами, обратную транскрипцию, окрашивание ядер, переваривание ткани и расщепление зондов - все эти реакции проводили в 16-луночной кассете с силиконовой прокладкой (Arraylt, Sunnyvale, CA, USA) в реакционном объеме 50 мкл на одну лунку. Чтобы предотвратить испарение, кассеты покрывали приспособлением для запечатывания планшетов (In Vitro AB, Stockholm, Sweden).
Необязательная стадия: пермеабилизация ткани перед синтезом кДНК
Для пермеабилизации с использованием протеиназы К выполняли разбавление протеиназы К (Qiagen, Hilden, Germany) до 1 мкг/мкл в PBS. Раствор добавляли в лунки, и слайд инкубировали при комнатной температуре в течение 5 минут, затем температуру постепенно повышали до 80°С в течение 10 минут. Слайд быстро промывали в PBS перед проведением реакции обратной транскрипции.
Альтернативно, что касается пермеабилизации с использованием микроволнового излучения, то после прикрепления ткани слайд помещали на дно стеклянного сосуда, содержащего 50 мл 0,2×SSC (Sigma-Aldrich), и нагревали в микроволновой печи в течение 1 минуты при 800 Вт. Сразу после обработки микроволновым облучением слайд помещали на бумажную ткань и сушили в течение 30 минут в камере, защищенной от нежелательного воздействия воздуха. По окончании сушки слайд быстро погружали в воду (не содержащую РНКаз/ДНКаз) и окончательно сушили в центрифуге перед инициацией синтеза кДНК.
Синтез кДНК
Для проведения реакции обратной транскрипции использовали систему одностадийной ОТ-ПЦР Superscript™ III с Platinum® Taq ДНК-полимеразой (Life Technologies/lnvitrogen, Carlsbad, CA, USA). Реакционные смеси для обратной транскрипции содержали реакционную смесь (1×), 1× BSA (New England Biolabs, Ipswich, MA, USA) и 2 мкл смеси Superscript III RT/Platinum Taq в конечном объеме 50 мкл. Этот раствор нагревали до 50°С, после чего наносили на срезы ткани и проводили реакцию при 50°С в течение 30 минут. Раствор для проведения обратной транскрипции затем удаляли из лунок, и слайд оставляли высыхать на воздухе в течение 2 часов.
Визуализация ткани
После синтеза кДНК проводили одновременно окрашивание ядер и гибридизацию маркерного зонда с рамочными зондами (зондами, присоединенными к подложке чипа, чтобы иметь возможность провести ориентацию образца ткани на чипе). Готовили раствор с DAPI в концентрации 300 нМ и маркерным зондом в концентрации 170нМ в PBS. Этот раствор добавляли в лунки, и слайд инкубировали при комнатной температуре в течение 5 минут, затем быстро промывали в PBS и сушили в центрифуге.
Альтернативно, перед тем, как поместить ткань на чип, проводили гибридизацию маркерного зонда с рамочными зондами. Затем маркерный зонд разбавляли до 170 нМ в буфере для гибридизации (4×SSC; 0,1% SDS). Этот раствор нагревали до 50°С, после чего наносили на чип и проводили гибридизацию при 50°С в течение 30 минут при 300 об./мин. По окончании гибридизации слайд промывали в 2×SSC с 0,1% SDS при 50°С и 300 об./мин в течение 10 минут, 0,2×SSC при 300 об./мин в течение 1 минуты и 0,1×SSC при 300 об./мин в течение 1 минуты. В этом случае окрашивающий раствор после синтеза кДНК содержал только краситель клеточных ядер DAPI, разбавленный до 300 нМ в PBS. Раствор вносили в лунки, и слайд инкубировали при комнатной температуре в течение 5 минут с последующей быстрой промывкой в PBS и сушкой в центрифуге.
Срезы изучали под микроскопом, используя Zeiss Axio Imager Z2, и обрабатывали, применяя программное обеспечение MetaSystems.
Удаление ткани
Срезы тканей переваривали с использованием протеиназы K, разбавленной до 1,25 мкг/мкл в буфере PKD (Proteinase K Digestion Buffer; буфер для переваривания протеиназой K) из набора RNeasy FFPE (оба от Qiagen), при 56°С в течение 30 минут с интервальным перемешиванием при 300 об./мин в течение 3 секунд, затем 6 секунд в неподвижном состоянии. Слайд последовательно промывали в 2×SSC с 0,1% SDS при 50°С и 300 об./мин в течение 10 минут, 0,2×SSC при 300 об./мин в течение 1 минуты и 0,1×SSC при 300 об./мин в течение 1 минуты.
Высвобождение зондов
16-Луночную кассету для гибридизации с силиконовой прокладкой (Arraylt) предварительно нагревали до 37°С и присоединяли к слайду Nimblegen. В каждую из лунок, содержащих иммобилизованную на поверхности кДНК, добавляли смесь для расщепления объемом 50 мкл, предварительно нагретую до 37°С, состоящую из лизирующего буфера неизвестной концентрации (Takara), USER Enzyme (0,1 ед./мкл; NEB) и BSA (0,1 мкг/мкл). После удаления пузырьков слайд герметично закрывали и инкубировали при 37°С в течение 30 минут в термосмесителе Comfort в режиме периодического встряхивания при 300 об./мин в течение 3 секунд и перерывами в 6 секунд между встряхиваниями. По окончании инкубации из каждой используемой лунки отбирали по 45 мкл смеси после отщепления и помещали в пробирки для ПЦР объемом 0,2 мл (Фиг.24).
Создание библиотеки
Обработка экзонуклеазой
После охлаждения растворов на льду в течение 2 минут добавляли экзонуклеазу I (NEB) до конечного объема 46,2 мкл и конечной концентрации 0,52 ед./мкл, чтобы удалить неудлиненные кДНК-зонды. Пробирки инкубировали в термоциклере (Applied Biosystems) при 37°С в течение 30 минут, затем проводили инактивацию экзонуклеазы при 80°С в течение 25 минут.
Наращивание (аА)-хвоста с использованием концевой трансферазы
После стадии обработки экзонуклеазой к каждому из образцов добавляли по 45 мкл смеси для наращивания поли(А)-хвоста, состоящей в соответствии с инструкциями производителя из буфера для TdT (Takara), 3 мМ dATP (Takara) и TdT-содержащей ферментативной смеси от производителя (TdT и РНКаза Н) (Takara). Смеси инкубировали в термоциклере при 37°С в течение 15 минут, затем проводили инактивацию TdT при 70°С в течение 10 минут.
Синтез второй нити и ПЦР-амплификация
После наращивания (dA)-хвоста в четыре новые пробирки для ПЦР объемом 0,2 мл помещали по 23 мкл мастер-микс для ПЦР из расчета на один образец, в каждую пробирку добавляли по 2 мкл образца в качестве матрицы. Конечные ПЦР-смеси содержали: 1× буфер для Ex Taq (Takara), dNTP (200 мкМ каждый, Takara), 600 нМ А_праймер (MWG), 600 нМ B_dT20VN_праймер (MWG) и E× Taq-полимеразу (0,025 ед./мкл; Takara) (Таблица 2). Образование второй нити кДНК осуществлялось в результате прохождения одного цикла в термоциклере при 95°С в течение 3 минут, 50°С в течение 2 минут и 72°С в течение 3 минут. Далее образцы амплифицировали, проводя 20 циклов (для создания библиотеки) или 30 циклов (для подтверждения присутствия кДНК) при 95°С в течение 30 секунд, 67°С в течение 1 минуты и 72°С в течение 3 минут, с последующим окончательным удлинением при 72°С в течение 10 минут.
Очистка библиотеки
После проведения амплификации четыре ПЦР-смеси (100 мкл) смешивали с 500 мкл буфера для связывания (Qiagen), наносили на колонку Qiaquick для очистки после ПЦР (Qiagen) и центрифугировали в течение 1 минуты при 17900хд для связывания амплифицированной кДНК с мембраной. Мембрану затем промывали буфером для промывки (Qiagen), содержащим этанол, и окончательно элюировали в 50 мкл 10 мМ Трис-Cl, рН 8,5.
Очищенный и концентрированный образец дополнительно очищали и концентрировали, используя СА-очистку (очистку на суперпарамагнитных гранулах, конъюгированных с карбоновой кислотой) с применением робота MBS (Magnetic Biosolutions). Для удаления фрагментов меньше 150-200 п.о. использовали ПЭГ (полиэтиленгликоль) в конечной концентрации 10%. Амплифицированную кДНК оставляли связываться с СА-гранулами (Invitrogen) в течение 10 мин и затем элюировали в 15 мкл 10 мМ Трис-Cl, рН 8,5.
Анализ качества библиотеки
Образцы, амплифицированные в результате проведения 30 циклов, анализировали с использованием биоанализатора Agilent (Agilent) для подтверждения наличия библиотеки амплифицированной кДНК; в зависимости от количества материала использовали набор DNA High Sensitivity или набор DNA1000.
Создание библиотеки для секвенирования
Индексирование библиотеки
Образцы, амплифицированные в результате проведения 20 циклов, использовали далее для создания библиотек для секвенирования. Для каждого образца готовили помеченный индексом мастер-микс для ПЦР и по 23 мкл помещали в шесть пробирок объемом 0,2 мл. В каждую из шести ПЦР-смесей добавляли по 2 мкл амплифицированной и очищенной кДНК в качестве матрицы, в результате ПЦР-смеси содержали 1× мастер-микс Phusion (Fermentas), 500 нМ InPE1.0 (Illumina), 500 нМ Index 1-12 (IIIumina) и 0,4 нМ InPE2.0 (Illumina). Образцы амплифицировали в термоциклере, проводя 18 циклов при 98°С в течение 30 секунд, 65°С в течение 30 секунд и 72°С в течение 1 минуты, с последующим окончательным удлинением при 72°С в течение 5 минут.
Очистка библиотеки для секвенирования
После проведения амплификации шесть ПЦР-смесей (150 мкл) смешивали с 750 мкл буфера для связывания, наносили на колонку Qiaquick для очистки после ПЦР (Qiagen) и центрифугировали в течение 1 минуты при 17900хд для связывания амплифицированной кДНК с мембраной (ввиду большого объема образца (900 мкл), образец центрифугировали в два приема (каждый раз по 450 мкл), и связывание осуществляли за две отдельные стадии). Мембрану затем промывали буфером для промывки, содержащим этанол, и окончательно элюировали в 50 мкл 10 мМ Трис-Cl, рН 8,5.
Очищенный и концентрированный образец дополнительно очищали и концентрировали, используя СА-очистку с применением робота MBS. Для удаления фрагментов меньше 300-350 п.о. использовали ПЭГ в конечной концентрации 7,8%. Амплифицированную кДНК оставляли связываться с СА-гранулами в течение 10 мин и затем элюировали в 15 мкл 10 мМ Трис-Cl, рН 8,5. Образцы анализировали с использованием биоанализатора Agilent с целью подтверждения наличия и размера полученных библиотек; в зависимости от количества материала в соответствии с инструкциями производителя использовали набор DNA High Sensitivity или набор DNA 1000 (Фиг.25).
Секвенирование
Библиотеки секвенировали на Hiseq2000 или Miseq от Illumina в зависимости от желаемой пропускной способности данных в соответствии с инструкциями производителя. В некоторых случаях для прочтения 2 использовали изготовленный на заказ секвенирующий праймер В_r2, чтобы избежать секвенирования посредством гомополимерного удлинения 20Т.
Анализ данных
В прочтении 1 были удалены 42 основания с 5′-конца. В прочтении 2 были удалены 25 оснований с 5′-конца (в результате прочтения 2 при применении изготовленного на заказ праймера возможно отсутствие какого-либо удаления оснований). Затем эти прочтения сопоставляли с картой целого генома мыши Mus musculus 9 с маскированными повторами, используя программу bowtie, и выходные данные форматировали в формате SAM-файлов. Получали картированные прочтения и аннотировали их, используя аннотации генов UCSC refGene. Индексы считывали с использованием "indexFinder" (программного обеспечения собственной разработки для считывания индексов). Затем создавали базу данных Mongo DB, содержащую информацию обо всех обнаруженных транскриптах и их положении на чипе соответственно индексу.
Применение matlab сочеталось с базой данных и создавало возможность для пространственной визуализации и анализа данных (Фиг.26).
По желанию, проводили сопоставление полученных визуализацией данных и полученного с помощью микроскопа изображения с использованием меченных флуоресцентной меткой рамочных зондов для строгого выравнивания и обеспечения условий получения данных относительно пространственной транскриптомики.
Пример 9
Пространственная транскриптомика с использованием ориентированных от 3′ к 5′ чипов с высокой плотностью зондов и FFPE-ткани вместе с расщеплением системой MutY и амплификацией с использованием TdT
Изготовление чипов
Стандартные микрочипы высокой плотности были заказаны у Roche-Nimblegen (Madison, WI, USA). Каждый использованный чип с захватывающими зондами содержал 72 тыс.элементов, из которых 66022 содержали уникальную меченую ID-меткой комплементарную последовательность. Размер каждого элемента составлял 16×16 мкм. Захватывающие зонды имели направление от 3′ к 5′, так же как зонды, использованные на печатных ориентированных от 3′ к 5′ чипах собственной разработки, за исключением 3 дополнительных оснований, которые добавляют к верхней (Р′) общей "ручке" зонда, чтобы получить длинную версию Р′, LP′ (Таблица 2). Каждый чип также был снабжен рамкой зондов, несущих стандартную последовательность из 30 п.о., дающую возможность гибридизоваться флуоресцентным зондам для помощи в ориентации во время визуализации чипа.
Синтез ориентированных от 5′ к 3′ захватывающих зондов
Синтез ориентированных от 5′ к 3′ захватывающих зондов на чипах высокой плотности проводили так же, как в случае печатных чипов собственной разработки, за исключением того, что стадии удлинения и лигирования осуществляли при 55°С в течение 15 мин, затем при 72°С в течение 15 мин. Зонд А-"ручка" (Таблица 2) включал ошибочное спаривание A/G, позволяющее осуществить описанное ниже последующее высвобождение зондов с использованием ферментативной системы MutY. Р-зонд был заменен на более длинную версию LP для соответствия более длинным зондам на поверхности.
Приготовление фиксированной в формалине залитой парафином ткани и депарафинизация
Эту процедуру проводили, как описано выше, в соответствии с протоколом собственной разработки.
Синтез кДНК и окрашивание
Синтез кДНК и окрашивание проводили так же, как описано в протоколе для ориентированных от 5′ к 3′ чипов высокой плотности Nimblegen, за исключением того, что меченные биотином dCTP и dATP добавляли во время синтеза кДНК вместе с четырьмя обычными dNTP (каждый из которых присутствовал в 25-кратном избытке по сравнению с таковым, меченным биотином).
Удаление ткани
Удаление ткани проводили таким же образом, как описано в протоколе для ориентированных от 5′ к 3′ чипов высокой плотности Nimblegen, приведенном в примере 8.
Отщепление зондов с использованием MutY
16-Луночную кассету для инкубации с силиконовой прокладкой (Arraylt) предварительно нагревали до 37°С и присоединяли к слайду CodeLink. В каждую из лунок, содержащих иммобилизованную на поверхности кДНК, добавляли смесь для расщепления объемом 50 мкл, предварительно нагретую до 37°С, состоящую из буфера для эндонуклеазы VIII (NEB), MutY (10 ед./мкл) (Trevigen), эндонуклеазы VIII (10 ед./мкл; NEB) и BSA (0,1 мкг/мкл). После удаления пузырьков слайд герметично закрывали и инкубировали при 37°С в течение 30 минут в термосмесителе Comfort в режиме периодического встряхивания при 300 об./мин в течение 3 секунд и с перерывами в 6 секунд между встряхиваниями. По окончании инкубации приспособление для запечатывания планшетов удаляли, и из каждой используемой лунки отбирали по 40 мкл смеси после отщепления и помещали в планшет для ПЦР.
Создание библиотеки
Биотин-стрептавидин-опосредованная очистка библиотеки
Для удаления неудлиненных кДНК зондов и для замены буфера образцы очищали посредством связывания меченной биотином кДНК с покрытыми стрептавидином С1-гранулами (Invitrogen) и промывки гранул 0,1 М NaOH (свежеприготовленным). Очистку проводили с применением робота MBS (Magnetic Biosolutions); меченную биотином кДНК оставляли связываться с С1-гранулами в течение 10 мин и затем элюировали в 20 мкл воды посредством нагревания суспензии гранулы-вода до 80°С для нарушения связывания биотин-стрептавидин.
Наращивание (dA)-хвоста с использованием концевой трансферазы
После проведения стадии очистки по 18 мкл каждого образца помещали в новые пробирки для ПЦР объемом 0,2 мл и смешивали с 22 мкл мастер-микс для наращивания поли(А)-хвоста, получая в результате 40 мкл реакционной смеси, в соответствии с инструкциями производителя состоящей из лизирующего буфера (набор для амплификации целого транскриптома Cellamp, Takara), буфера для TdT (Takara), 1,5 мМ dATP (Takara) и TdT-содержащей ферментативной смеси (TdT и РНКаза Н) (Takara). Смеси инкубировали в термоциклере при 37°С в течение 15 минут, затем проводили инактивацию при 70°С в течение 10 минут.
Синтез второй нити и ПЦР-амплификация
После наращивания (dA)-хвоста в четыре новые пробирки для ПЦР объемом 0,2 мл помещали по 23 мкл мастер-микс для ПЦР из расчета на один образец, в каждую пробирку добавляли по 2 мкл образца в качестве матрицы. Конечные ПЦР-смеси состояли из: 1× буфера для ExTaq (Takara), dNTP (200 мкМ каждый; Takara), 600 нМ А_праймера (MWG), 600 нМ B_dT20VN_праймера (MWG) и E× Taq-полимеразы (0,025 ед./мкл; Takara). Образование второй нити кДНК осуществлялось в результате прохождения одного цикла в термоциклере при 95°С в течение 3 минут, 50°С в течение 2 минут и 72°С в течение 3 минут. Далее образцы амплифицировали, проводя 20 циклов (для создания библиотеки) или 30 циклов (для подтверждения наличия кДНК) при 95°С в течение 30 секунд, 67°С в течение 1 минуты и 72°С в течение 3 минут, с последующим окончательным удлинением при 72°С в течение 10 минут.
Очистка библиотеки
После проведения амплификации четыре ПЦР-смеси (100 мкл) смешивали с 500 мкл буфера для связывания (Qiagen), наносили на колонку Qiaquick для очистки после ПЦР (Qiagen) и центрифугировали в течение 1 минуты при 17900×g для связывания амплифицированной кДНК с мембраной. Мембрану затем промывали буфером для промывки (Qiagen), содержащим этанол, и окончательно элюировали в 50 мкл 10 мМ Трис-Cl, рН 8,5.
Очищенный и концентрированный образец дополнительно очищали и концентрировали, используя СА-очистку (очистку на суперпарамагнитных гранулах, конъюгированных с карбоновой кислотой) с применением робота MBS (Magnetic Biosolutions). Для удаления фрагментов меньше 150-200 п.о. использовали ПЭГ в конечной концентрации 10%. Амплифицированную кДНК оставляли связываться с СА-гранулами (Invitrogen) в течение 10 мин и затем элюировали в 15 мкл 10 мМ Трис-Cl, рН 8,5.
Вторая ПЦР-амплиФикация
Конечные ПЦР-смеси состояли из: 1х буфера для ExTaq (Takara), dNTP (200 мкМ каждый; (Takara), 600 нМ А_праймера (MWG), 600 нМ В_праймера (MWG) и E× Taq-полимеразы (0,025 ед./мкл; Takara). Образцы нагревали до 95°С в течение 3 минут и затем амплифицировали, проводя 10 циклов при 95°С в течение 30 секунд, 65°С в течение 1 минуты и 72°С в течение 3 минут, с последующим окончательным удлинением при 72°С в течение 10 минут.
Вторая очистка библиотеки
После проведения амплификации четыре ПЦР-смеси (100 мкл) смешивали с 500 мкл буфера для связывания (Qiagen), наносили на колонку Qiaquick для очистки после ПЦР (Qiagen) и центрифугировали в течение 1 минуты при 17900×g для связывания амплифицированной кДНК с мембраной. Мембрану затем промывали буфером для промывки (Qiagen), содержащим этанол, и окончательно элюировали в 50 мкл 10 мМ Трис-Cl, рН 8,5.
Очищенный и концентрированный образец дополнительно очищали и концентрировали, используя СА-очистку (очистку на суперпарамагнитных гранулах, конъюгированных с карбоновой кислотой) с применением робота MBS (Magnetic Biosolutions). Для удаления фрагментов меньше 150-200 п.о. использовали ПЭГ в конечной концентрации 10%. Амплифицированную кДНК оставляли связываться с СА-гранулами (Invitrogen) в течение 10 мин и затем элюировали в 15 мкл 10 мМ Трис-Cl, рН 8,5.
Создание библиотеки для секвенирования
Индексирование библиотеки
Образцы, амплифицированные в результате проведения 20 циклов, использовали далее для создания библиотек для секвенирования. Для каждого образца готовили помеченный индексом мастер-микс для ПЦР и по 23 мкл помещали в шесть пробирок объемом 0,2 мл. В каждую из шести ПЦР-смесей добавляли по 2 мкл амплифицированной и очищенной кДНК в качестве матрицы, в результате ПЦР-смеси содержали 1× мастер-микс Phusion (Fermentas), 500 нМ InPE1.0 (Illumina), 500 нМ Index 1-12 (Illumina) и 0,4 нМ InPE2.0 (Illumina). Образцы амплифицировали в термоциклере, проводя 18 циклов при 98°С в течение 30 секунд, 65°С в течение 30 секунд и 72°С в течение 1 минуты, с последующим окончательным удлинением при 72°С в течение 5 минут.
Очистка библиотеки для секвенирования
После проведения амплификации образцы очищали и концентрировали, используя СА-очистку с применением робота MBS. Для удаления фрагментов меньше 300-350 п.о. использовали ПЭГ в конечной концентрации 7,8%. Амплифицированную кДНК оставляли связываться с СА-гранулами в течение 10 мин и затем элюировали в 15 мкл 10 мМ Трис-Cl, рН 8,5.
По 10 мкл амплифицированных и очищенных образцов помещали на чип Caliper XT и вырезали фрагменты от 480 п.о. до 720 п.о., используя Caliper XT (Caliper). Образцы анализировали с использованием биоанализатора Agilent с целью подтверждения наличия и размера полученных библиотек; использовали набор DNA High Sensitivity.
Секвенирование и анализ данных
Секвенирование и биоинформатический анализ данных проводили таким же образом, как описано в протоколе для ориентированных от 5′ к 3′ чипов высокой плотности Nimblegen, как описано в примере 8. Однако, при анализе данных прочтение 1 в картировании транскриптов не использовали. С использованием Matlab-визуализации можно было отсортировать специфичные транскрипты Olfr (Фиг.27).
Пример 10
Пространственная транскриптомика с использованием печатного содержащего 41 метку микрочипа собственной разработки с ориентированными от 5′ к 3′ зондами и фиксированной в формалине замороженной (FF-замороженной) ткани с применением пермеабилизации с использованием протеиназы К или микроволнового излучения, с применением расщепления системой USER и амплификации с использованием TdT
Изготовление чипов
Чипы собственной разработки печатали, как описано ранее, но с включением содержащих 41 уникальную ID-метку зондов того же состава, что и зонды на ориентированном от 5′ к 3′ чипе высокой плотности в примере 8 (Фиг.28).
Все другие стадии осуществляли таким же образом, как описано в протоколе, приведенном в примере 8.
Пример 11
Альтернативный способ проведения стадии синтеза кДНК
Синтез кДНК на чипе, который описан выше, также можно объединить со стадией переключения матрицы с целью создания второй нити путем добавления праймера переключения матрицы к реакционной смеси для синтеза кДНК (Таблица 2). Вторую область амплификации вводят посредством связывания ее с концевыми основаниями, добавленными с использованием обратной транскриптазы на 3′-конец первой нити кДНК, и инициируют синтез второй нити. Эта библиотека может быть легко амплифицирована сразу после высвобождения двунитевого комплекса с поверхности чипа.
Пример 12
Пространственная геномика с использованием печатного содержащего 41 метку микрочипа собственной разработки с ориентированными от 5′ к 3′ зондами и фрагментированной гДНК с наращенным поли(А)-хвостом, с применением отщепления системой USER и амплификации посредством наращивания хвоста с использованием TdT или специфических для транслокации праймеров
Изготовление чипов
Чипы собственной разработки печатали, используя слайд CodeLink (Surmodics), как описано ранее, но с включением содержащих 41 уникальную ID-метку зондов того же состава, что и зонды на ориентированном от 5′ к 3′ чипе высокой плотности в примере 8.
Получение суммарной ДНК из клеток
Фрагментация ДНК
Геномную ДНК (гДНК) из клеточных линий А431 и U2OS выделяли, используя набор DNeasy (Qiagen), в соответствии с инструкциями производителя. ДНК фрагментировали до 500 п.о. на ультразвуковом аппарате Covaris (Covaris) в соответствии с инструкциями производителя.
Образец очищали и концентрировали, используя СА-очистку (очистку на суперпарамагнитных гранулах, конъюгированных с карбоновой кислотой) с применением робота MBS (Magnetic Biosolutions). Для удаления фрагментов меньше 150-200 п.о. использовали ПЭГ в конечной концентрации 10%. Фрагментированную ДНК оставляли связываться с СА-гранулами (Invitrogen) в течение 10 мин и затем элюировали в 15 мкл 10 мМ Трис-Cl, рН 8,5.
Возможный контроль: перенос разных клеточных линий
Посредством переноса ДНК А431 в ДНК U2OS можно измерить разные уровни чувствительности захвата, например, в зависимости от переноса 1%, 10% или 50% ДНК А431.
Наращивание (dA)-XBOCTa с использованием концевой трансферазы
45 мкл смеси для наращивания поли(А)-хвоста, состоящей в соответствии с инструкциями производителя из буфера для TdT (Такага), 3 мМ dATP (Такага) и TdT-содержащей ферментативной смеси (TdT и РНКаза Н) (Такага), добавляли к 0,5 мкг фрагментированной ДНК. Смеси инкубировали в термоциклере при 37°С в течение 30 минут, затем проводили инактивацию TdT при 80°С в течение 20 минут.Фрагменты с наращенными (dA)-хвостами затем очищали на колонке Qiaquick (Qiagen) в соответствии с инструкциями производителя и концентрацию измеряли, используя систему Qubit (Invitcogen) в соответствии с инструкциями производителя.
Эксперименты на чипе
Реакции гибридизации, синтеза второй нити и отщепления проводили на чипе в 16-луночной кассете с силиконовой прокладкой (Arraylt, Sunnyvale, CA, USA). Чтобы предотвратить испарение, кассеты покрывали приспособлением для запечатывания планшетов (In Vitro AB, Stockholm, Sweden).
Гибридизация
117 нг ДНК вносили в лунку на предварительно нагретый чип (50°С) в общем объеме 45 мкл, состоящем из 1× буфера от NEB (New England Biolabs) и 1×BSA. Смесь инкубировали в течение 30 мин при 50°С в термосмесителе Comfort (Eppendorf), оснащенном блоком МТР (титрационные микропланшеты), со встряхиванием при 300 об./мин.
Синтез второй нити
Не удаляя смесь после гибридизации, в лунку добавляли 15 мкл смеси для реакции удлинения с использованием фрагмента Кленова, состоящей из 1,5 мкл полимеразы Кленова в 1× буфере от NEB и 3,75 мкл dNTP (по 2 мМ каждого). Реакционную смесь инкубировали в термосмесителе Comfort (Eppendorf) при 37°С в течение 30 мин без встряхивания.
Затем слайд промывали в 2×SSC с 0,1% SDS при 50°С и 300 об./мин в течение 10 минут, 0,2×SSC при 300 об./мин в течение 1 минуты и 0,1×SSC при 300 об./мин в течение 1 минуты.
Высвобождение зондов
Смесь объемом 50 мкл, содержащую 1× реакционный буфер FastStart High Fidelity с 1,8 мМ MgCl2 (Roche), 200 мкМ dNTP (New England Biolabs), 1×BSA и USER Enzyme (0,1 ед.Л мкл; New England Biolabs), нагревали до 37°С, добавляли в каждую лунку и инкубировали при 37°С в течение 30 мин с перемешиванием (3 секунды при 300 об./мин, 6 секунд в неподвижном состоянии) (термосмеситель Comfort; Eppendorf). Реакционную смесь, содержащую высвобожденную кДНК, затем извлекали из лунок с помощью пипетки.
Создание библиотеки
Реакция амплификации
Амплификацию проводили в реакционных смесях по Юмкл, состоящих из 7,5 мкл высвобожденного образца, 1 мкл каждого праймера и 0,5 мкл фермента (Roche, ПЦР-система FastStart HiFi). Реакцию проводили в следующем циклическом режиме: 94°С в течение 2 мин, один цикл при 94°С в течение 15 с, 55°С в течение 2 мин, 72°С в течение 2 мин, 30 циклов при 94°С в течение 15 с, 65°С в течение 30 с, 72°С в течение 90 с и окончательное удлинение при 72°С в течение 5 мин.
При создании библиотеки для секвенирования использовали два праймера, состоящие из А-"ручки" поверхностного зонда и либо специфического для транслокации праймера (для А431), либо специфического для SNP праймера, связанного с Выручкой" (Таблица 2).
Очистка библиотеки
Очищенный и концентрированный образец дополнительно очищали и концентрировали, используя СА-очистку (очистку на суперпарамагнитных гранулах, конъюгированных с карбоновой кислотой) с применением робота MBS (Magnetic Biosolutions). Для удаления фрагментов меньше 150-200 п.о. использовали ПЭГ в конечной концентрации 10%. Амплифицированную кДНК оставляли связываться с СА-гранулами (Invitrogen) в течение 10 мин и затем элюировали в 15 мкл 10 мМ Трис-Cl, рН 8,5.
Анализ качества библиотеки
Образцы анализировали с использованием биоанализатора Agilent (Agilent) с целью подтверждения наличия библиотеки амплифицированной ДНК; в зависимости от количества материала использовали набор DNA High Sensitivity или набор DNA 1000.
Индексирование библиотеки
Образцы, амплифицированные в результате проведения 20 циклов, использовали далее для создания библиотек для секвенирования. Для каждого образца готовили помеченный индексом мастер-микс для ПЦР и по 23 мкл помещали в шесть пробирок объемом 0,2 мл. В каждую из шести ПЦР-смесей добавляли по 2 мкл амплифицированной и очищенной кДНК в качестве матрицы, в результате ПЦР-смеси содержали 1× мастер-микс Phusion (Fermentas), 500 нМ InPE1.0 (Illumina), 500 нМ Index 1-12 (Illumina) и 0,4 нМ InPE2.0 (Illumina). Образцы амплифицировали в термоциклере, проводя 18 циклов при 98°С в течение 30 секунд, 65°С в течение 30 секунд и 72°С в течение 1 минуты, с последующим окончательным удлинением при 72°С в течение 5 минут.
Очистка библиотеки для секвенирования
Очищенный и концентрированный образец дополнительно очищали и концентрировали, используя СА-очистку с применением робота MBS. Для удаления фрагментов меньше 300-350 п.о. использовали ПЭГ в конечной концентрации 7,8%. Амплифицированную ДНК оставляли связываться с СА-гранулами (Invitrogen) в течение 10 мин и затем элюировали в 15 мкл 10 мМ Трис-Cl, рН 8,5. Образцы анализировали с использованием биоанализатора Agilent с целью подтверждения наличия и размера полученных библиотек; в зависимости от количества материала в соответствии с инструкциями производителя использовали набор DNA High Sensitivity или набор DNA 1000 (Фиг.29).
Секвенирование
Секвенирование проводили тем же способом, что и в протоколе для ориентированных от 5′ к 3′ чипов высокой плотности от Nimblegen, описанных в примере 8.
Анализ данных
Анализ данных проводили с целью определения чувствительности захвата организованных в чип ID-содержащих захватывающих зондов. Прочтение 2 сортировали на основе содержания в нем праймеров, специфических либо для транслокации, либо для SNP. Эти прочтения далее сортировали по их ID, содержащихся в прочтении 1.
Возможный контроль: прямая амплификация специфических для клеточных линий транслокаций
Это использовали для измерения чувствительности захвата перенесенных клеточных линий непосредственно с применением ПЦР. Прямой и обратный праймеры (Таблица 2) для транслокаций А431 использовали для исследования и детекции наличия транслокации во второй нити скопированного и высвобожденного материала (Фиг.30).




| название | год | авторы | номер документа |
|---|---|---|---|
| ПАНЕЛЬ И СПОСОБ ПОЛУЧЕНИЯ ПРОСТРАНСТВЕННОЙ ИНФОРМАЦИИ О НУКЛЕИНОВЫХ КИСЛОТАХ | 2020 |
|
RU2803202C2 |
| ПРЯМОЙ ЗАХВАТ, АМПЛИФИКАЦИЯ И СЕКВЕНИРОВАНИЕ ДНК-МИШЕНИ С ИСПОЛЬЗОВАНИЕМ ИММОБИЛИЗИРОВАННЫХ ПРАЙМЕРОВ | 2011 |
|
RU2565550C2 |
| ДИАГНОСТИКА И ЛЕЧЕНИЕ РАКА МОЛОЧНОЙ ЖЕЛЕЗЫ | 2011 |
|
RU2550925C2 |
| ПРОСТРАНСТВЕННОЕ КАРТИРОВАНИЕ МОЛЕКУЛЯРНЫХ ПРОФИЛЕЙ ОБРАЗЦОВ БИОЛОГИЧЕСКИХ ТКАНЕЙ | 2016 |
|
RU2733545C2 |
| СИСТЕМА АНАЛИЗА ДЛЯ ОРТОГОНАЛЬНОГО ДОСТУПА К БИОМОЛЕКУЛАМ И ИХ МЕЧЕНИЯ В КЛЕТОЧНЫХ КОМПАРТМЕНТАХ | 2017 |
|
RU2771892C2 |
| СПОСОБ ИДЕНТИФИКАЦИИ НАТИВНЫХ ПАР ФРАГМЕНТОВ ДНК ИЛИ РНК, ПРИСУТСТВУЮЩИХ В ОДНИХ И ТЕХ ЖЕ ЖИВЫХ КЛЕТКАХ | 2013 |
|
RU2578009C2 |
| СПОСОБ ИДЕНТИФИКАЦИИ НАТИВНЫХ ПАР ФРАГМЕНТОВ ДНК ИЛИ РНК, ПРИСУТСТВУЮЩИХ В ОДНИХ И ТЕХ ЖЕ ЖИВЫХ КЛЕТКАХ | 2015 |
|
RU2600873C1 |
| АНАЛИЗ МНОЖЕСТВА АНАЛИТОВ С ИСПОЛЬЗОВАНИЕМ ОДНОГО АНАЛИЗА | 2019 |
|
RU2824049C2 |
| СПОСОБ ДИАГНОСТИКИ НЕОПЛАЗМ-II | 2008 |
|
RU2565540C2 |
| СПОСОБ АМПЛИФИКАЦИИ | 1989 |
|
RU2107729C1 |





























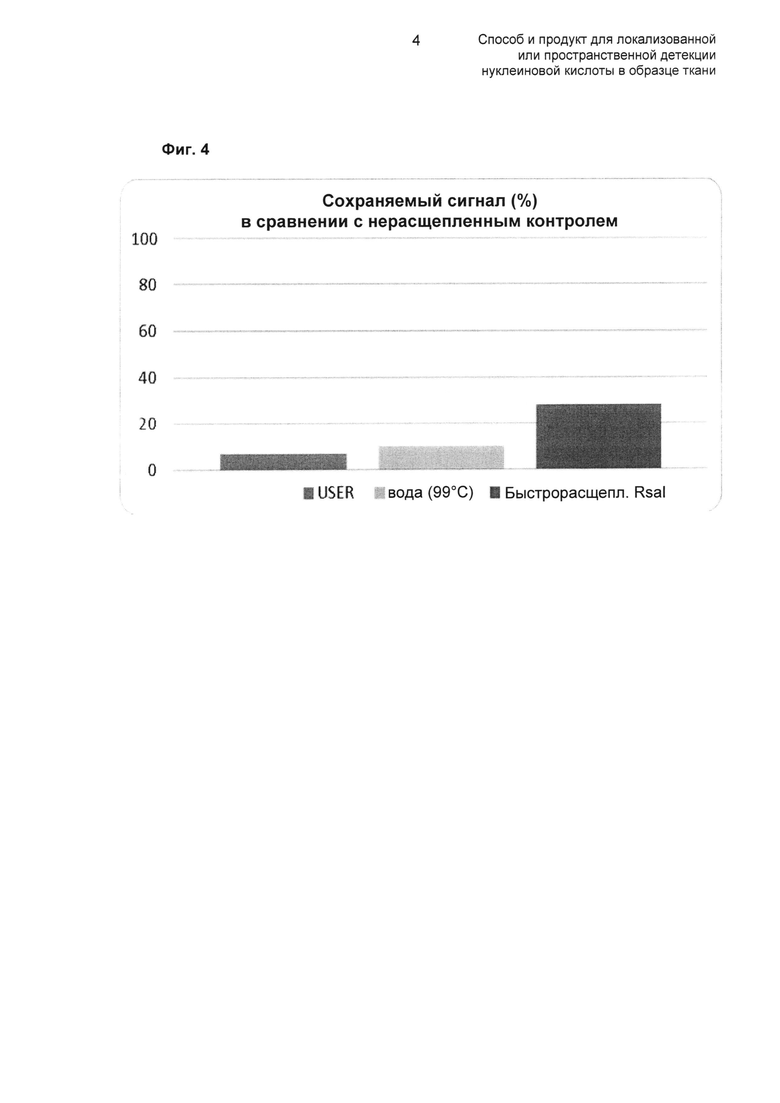
























Настоящее изобретение относится к способам и продуктам для локализованной или пространственной детекции нуклеиновой кислоты в образце ткани и, в частности, к способу локализованной детекции нуклеиновой кислоты в образце ткани, включающему: (а) предоставление чипа, содержащего подложку, на которой непосредственно или опосредованно иммобилизованы многочисленные разновидности захватывающих зондов, так что каждая разновидность занимает определенное положение на чипе и ориентирована таким образом, что имеет свободный 3′-конец, позволяющий указанному зонду действовать в качестве праймера в реакции удлинения или лигирования праймера, где каждая разновидность указанного захватывающего зонда содержит молекулу нуклеиновой кислоты, имеющую в направлении от 5′ к 3′: (1) позиционную область, которая соответствует положению захватывающего зонда на чипе, и (2) захватывающую область; (б) приведение указанного чипа в контакт с образцом ткани таким образом, что положение захватывающего зонда на чипе может быть сопоставлено с положением в образце ткани, и предоставление возможности нуклеиновой кислоте образца ткани гибридизоваться с захватывающей областью в указанных захватывающих зондах; (в) синтез молекул ДНК на основе захваченных молекул нуклеиновой кислоты с использованием указанных захватывающих зондов в качестве праймеров удлинения или лигирования, где указанные удлиненные или лигированные молекулы ДНК являются мечеными благодаря позиционной области; (г) возможно синтез комплементарной нити указанной меченой ДНК и/или возможно амплификацию указанной меченой ДНК; (д) высвобождение по меньшей мере части меченых молекул ДНК и/или их комплементов или ампликонов с поверхности чипа, где указанная часть включает позиционную область или ее комплемент; и (е) прямой или опосредованный анализ последовательности высвобожденных молекул ДНК. 3 н. и 52 з.п. ф-лы, 31 ил., 2 табл., 12 пр.
1. Способ локализованной детекции нуклеиновой кислоты в образце ткани, включающий:
(а) предоставление чипа, содержащего подложку, на которой непосредственно или опосредованно иммобилизованы многочисленные разновидности захватывающих зондов, так что каждая разновидность занимает определенное положение на чипе и ориентирована таким образом, что имеет свободный 3′-конец, позволяющий указанному зонду действовать в качестве праймера в реакции удлинения или лигирования праймера, где каждая разновидность указанного захватывающего зонда содержит молекулу нуклеиновой кислоты, имеющую в направлении от 5′ к 3′:
(1) позиционную область, содержащую штрихкодовую последовательность, которая соответствует положению захватывающего зонда на чипе, и
(2) захватывающую область для захвата нуклеиновой кислоты образца ткани, который приводят в контакт с указанным чипом;
(б) приведение указанного чипа в контакт с образцом ткани таким образом, что положение захватывающего зонда на чипе может быть сопоставлено с положением в образце ткани, и предоставление возможности нуклеиновой кислоте образца ткани гибридизоваться с захватывающей областью в указанных захватывающих зондах;
(в) синтез молекул ДНК на основе захваченных молекул нуклеиновой кислоты с использованием указанных захватывающих зондов в качестве праймеров удлинения или лигирования, где указанные удлиненные или лигированные молекулы ДНК являются мечеными благодаря позиционной области;
(г) возможно синтез комплементарной нити указанной меченой ДНК и/или возможно амплификацию указанной меченой ДНК;
(д) высвобождение по меньшей мере части меченых молекул ДНК и/или их комплементов или ампликонов с поверхности чипа, где указанная часть включает позиционную область или ее комплемент;
(е) прямой или опосредованный анализ последовательности высвобожденных молекул ДНК; и
(ж) сопоставление информации из анализа последовательности с положением в образце ткани.
2. Способ по п. 1, где стадия (ж) включает сопоставление информации из анализа указанной последовательности с изображением указанного образца ткани, где изображение образца ткани получают до или после стадии (в).
3. Способ по п. 1, представляющий собой способ определения и/или анализа транскриптома образца ткани, включающий:
(а) предоставление чипа, содержащего подложку, на которой непосредственно или опосредованно иммобилизованы многочисленные разновидности захватывающих зондов, так что каждая разновидность занимает определенное положение на чипе и ориентирована таким образом, что имеет свободный 3′-конец, позволяющий указанному зонду действовать в качестве праймера обратной транскриптазы (ОТ), где каждая разновидность указанного захватывающего зонда содержит молекулу нуклеиновой кислоты, имеющую в направлении от 5′ к 3′:
(1) позиционную область, содержащую штрихкодовую последовательность, которая соответствует положению захватывающего зонда на чипе; и
(2) захватывающую область для захвата нуклеиновой кислоты образца ткани, который приводят в контакт с указанным чипом;
(б) приведение указанного чипа в контакт с образцом ткани таким образом, что положение захватывающего зонда на чипе может быть сопоставлено с положением в образце ткани, и предоставление возможности РНК образца ткани гибридизоваться с захватывающей областью в указанных захватывающих зондах;
(в) синтез молекул кДНК на основе захваченных молекул РНК с использованием указанных захватывающих зондов в качестве ОТ-праймеров и возможно амплификацию указанных молекул кДНК;
(г) высвобождение по меньшей мере части молекул кДНК и возможно их ампликонов с поверхности чипа, где указанная высвобожденная молекула может представлять собой первую нить и/или вторую нить молекулы кДНК или ее ампликон и где указанная часть включает позиционную область или ее комплемент;
(д) прямой или опосредованный анализ последовательности высвобожденных молекул ДНК; и
(е) сопоставление информации из анализа последовательности с положением в образце ткани, предпочтительно где информацию из анализа указанной последовательности сопоставляют с изображением указанного образца ткани, где изображение образца ткани получают до или после стадии (в).
4. Способ по п. 1, где захватывающие зонды представляют собой молекулы ДНК.
5. Способ по п. 1, где разновидность захватывающего зонда содержит захватывающие области с отличающимися последовательностями.
6. Способ по п. 1, где захватывающие зонды дополнительно содержат универсальную область, которая расположена ближе к 5′-концу относительно позиционной области, где указанная универсальная область содержит:
(1) область амплификации для амплифицирования синтезированных молекул ДНК; и/или
(2) область расщепления для высвобождения синтезированных молекул ДНК с поверхности чипа.
7. Способ по п. 1, где позиционная область каждой разновидности захватывающего зонда содержит уникальную штрихкодовую последовательность.
8. Способ по п. 1, где захватывающая область содержит поли(Т)-ДНК-олигонуклеотид, содержащий по меньшей мере 10 остатков дезокситимидина, и/или случайную или вырожденную олигонуклеотидную последовательность.
9. Способ по п. 1, где захватывающая область содержит последовательность, специфичную к конкретному целевому гену или группе генов.
10. Способ по п. 1, где захватывающие зонды иммобилизованы непосредственно на поверхности чипа через их 5′-конец.
11. Способ по п. 1, где захватывающие зонды иммобилизованы опосредованно на поверхности чипа в результате гибридизации с поверхностным зондом, где захватывающая область захватывающих зондов содержит расположенную "вверх по течению" последовательность, способную гибридизоваться с 5′-концом поверхностных зондов, которые иммобилизованы на чипе.
12. Способ по п. 11, где поверхностные зонды иммобилизованы на поверхности чипа через их 3′-конец.
13. Способ по п. 11, где поверхностные зонды содержат последовательность, комплементарную:
(1) по меньшей мере части захватывающей области;
(2) позиционной области; и
(3) по меньшей мере части универсальной области амплификации.
14. Способ по п. 1, где указанную комплементарную нить или указанную вторую нить молекулы кДНК синтезируют до или после высвобождения указанных меченых молекул ДНК или указанных молекул кДНК с поверхности чипа.
15. Способ по п. 1, где для синтеза нити, комплементарной второй нити кДНК, используют полимеразу замещения нити.
16. Способ по п. 1, где для синтеза комплементарной нити или второй нити кДНК используют случайные праймеры.
17. Способ по п. 16, где случайные праймеры содержат область амплификации.
18. Способ по п. 17, где область амплификации случайных праймеров состоит из нуклеотидной последовательности, отличающейся от области амплификации захватывающего зонда.
19. Способ по п. 1, где к одному или обоим концам меченых молекул или их комплементов или молекул кДНК лигируют адаптеры.
20. Способ по п. 1, дополнительно включающий стадию амплификации меченых молекул или молекул кДНК до проведения анализа последовательности.
21. Способ по п. 20, где стадию амплификации осуществляют после высвобождения меченых молекул ДНК или кДНК с подложки чипа или где стадию амплификации осуществляют на чипе in situ.
22. Способ по п. 20, где стадия амплификации включает ПЦР (полимеразную цепную реакцию).
23. Способ по п. 1, где молекулы высвобождают с поверхности чипа посредством:
(1) отщепления нуклеиновой кислоты;
(2) денатурации; и/или
(3) физического способа.
24. Способ по п. 23 (1), где молекулы высвобождают посредством ферментативного расщепления области расщепления, которая локализована в универсальной области или позиционной области захватывающего зонда.
25. Способ по п. 23 (2) или (3), где молекулы высвобождают посредством подведения горячей воды или буфера к чипу.
26. Способ по п. 1, дополнительно включающий стадию очистки высвобожденных молекул до секвенирования.
27. Способ по п. 1, где подложка выбрана из группы, состоящей из стекла, кремния, покрытого поли-L-лизином материала, нитроцеллюлозы, полистирола, сополимеров циклических олефинов (СОС), полимеров циклических олефинов (СОР), полипропилена, полиэтилена и поликарбоната.
28. Способ по п. 1, где образец ткани представляет собой срез ткани.
29. Способ по п. 28, где срез ткани получают с использованием фиксированной ткани, например фиксированной в формалине залитой парафином (FFPE) ткани, или ткани после глубокого замораживания.
30. Способ по п. 1, дополнительно включающий стадию регидратации образца ткани после приведения образца в контакт с чипом и до стадии (в).
31. Способ по п. 1, где образец ткани происходит из растения, животного или гриба.
32. Способ по п. 1, дополнительно включающий стадию промывки чипа для удаления оставшейся ткани, где указанную стадию осуществляют до высвобождения молекул с чипа.
33. Способ по п. 1, где чип содержит по меньшей мере один позиционный маркер, позволяющий осуществлять ориентацию образца ткани на чипе.
34. Способ по п. 33, где чип содержит по меньшей мере 10, 50, 100, 200, 500, 1000 или 2000 позиционных маркеров в определенных положениях на поверхности чипа.
35. Способ по п. 33, где позиционные маркеры способны гибридизоваться с меченой, предпочтительно меченной флуоресцентной меткой, маркерной молекулой нуклеиновой кислоты.
36. Способ по п. 2, где изображение образца ткани получают с использованием световой, светлопольной, темнопольной, фазово-контрастной, флуоресцентной, отражательной, интерференционной или конфокальной микроскопии или их комбинации.
37. Способ по п. 36, где изображение образца ткани получают с использованием флуоресцентной микроскопии.
38. Способ по п. 1, где стадия анализа последовательности включает стадию секвенирования и, предпочтительно, где стадия секвенирования включает реакцию секвенирования, основанную на обратимых красителях-терминаторах.
39. Способ по п. 1, где подложка чипа является подходящей для применения в качестве платформы для секвенирования.
40. Способ по п. 39, где подложка чипа является подходящей для применения в технологиях секвенирования следующего поколения.
41. Способ по п. 1, где захватывающие зонды иммобилизованы на чипе посредством мостиковой амплификации.
42. Способ по п. 1, где каждая разновидность захватывающего зонда иммобилизована на подложке чипа посредством мостиковой амплификации с образованием кластерной колонии захватывающего зонда, так что каждая разновидность захватывающего зонда занимает определенное положение на подложке чипа.
43. Способ по п. 1, где чип представляет собой чип на основе гранул.
44. Способ по п. 43, где каждая разновидность захватывающего зонда иммобилизована на отличной грануле, так что каждая разновидность захватывающего зонда занимает определенное положение на подложке чипа.
45. Способ изготовления или получения чипа для применения в локализованной детекции нуклеиновой кислоты в образце ткани, включающий иммобилизацию многочисленных разновидностей захватывающего зонда непосредственно или опосредованно на подложке чипа, где каждая разновидность захватывающего зонда занимает определенное положение на чипе и ориентирована таким образом, что имеет свободный 3′-конец, позволяющий указанному зонду действовать в качестве праймера в реакции удлинения или лигирования праймера, где каждая разновидность указанного захватывающего зонда содержит молекулу нуклеиновой кислоты, имеющую в направлении от 5′ к 3′:
(1) позиционную область, содержащую штрихкодовую последовательность, которая соответствует положению захватывающего зонда на чипе; и
(2) захватывающую область для захвата нуклеиновой кислоты образца ткани, содержащую:
а) поли(Т)-ДНК-олигонуклеотид, содержащий по меньшей мере 10 остатков дезокситимидина, и/или случайную или вырожденную олигонуклеотидную последовательность; или
б) последовательности, специфичные к группе генов.
46. Способ по п. 45, дополнительно включающий любую одну или более чем одну из следующих стадий:
(а) иммобилизация многочисленных поверхностных зондов непосредственно или опосредованно на подложке чипа, где поверхностные зонды содержат:
(1) область, способную гибридизоваться с частью олигонуклеотида захватывающей области (частью, не вовлеченной в захват нуклеиновой кислоты);
(2) комплементарную позиционную область; и
(3) комплементарную универсальную область;
(б) гибридизация олигонуклеотидов захватывающих областей и олигонуклеотидов универсальных областей с поверхностными зондами, иммобилизованными на чипе;
(в) удлинение олигонуклеотидов универсальных областей посредством матричной полимеризации для синтеза позиционной области захватывающего зонда; и
(г) лигирование позиционной области с олигонуклеотидом захватывающей области для получения захватывающего олигонуклеотида.
47. Чип для применения в локализованной детекции нуклеиновой кислоты в образце ткани, содержащий подложку, на которой непосредственно или опосредованно иммобилизованы многочисленные разновидности захватывающих зондов, так что каждая разновидность занимает определенное положение на чипе и ориентирована таким образом, что имеет свободный 3′-конец, позволяющий указанному зонду действовать в качестве праймера удлинения или лигирования, где каждая разновидность указанного захватывающего зонда содержит молекулу нуклеиновой кислоты, имеющую в направлении от 5′ к 3′:
(1) позиционную область, содержащую штрихкодовую последовательность, которая соответствует положению захватывающего зонда на чипе, и
(2) захватывающую область для захвата нуклеиновой кислоты образца ткани, содержащую:
а) поли(Т)-ДНК-олигонуклеотид, содержащий по меньшей мере 10 остатков дезокситимидина, и/или случайную или вырожденную олигонуклеотидную последовательность; или
б) последовательности, специфичные к группе генов.
48. Чип по п. 47, где подложка чипа является подходящей для применения в качестве платформы для секвенирования.
49. Чип по п. 48, где подложка чипа является подходящей для применения в технологиях секвенирования следующего поколения.
50. Чип по п. 47, где захватывающие зонды иммобилизованы на чипе посредством мостиковой амплификации.
51. Чип по п. 47, где каждая разновидность захватывающего зонда иммобилизована на подложке чипа посредством мостиковой амплификации с образованием кластерной колонии захватывающего зонда, так что каждая разновидность захватывающего зонда занимает определенное положение на подложке чипа.
52. Чип по п. 47, где чип представляет собой чип на основе гранул.
53. Чип по п. 52, где каждая разновидность захватывающего зонда иммобилизована на отличной грануле, так что каждая разновидность захватывающего зонда занимает определенное положение на подложке чипа
54. Способ по п. 1, представляющий собой способ локализованной детекции ДНК в образце ткани, включающий:
(а) предоставление чипа, содержащего подложку, на которой непосредственно или опосредованно иммобилизованы многочисленные разновидности захватывающих зондов, так что каждая разновидность занимает определенное положение на чипе и ориентирована таким образом, что имеет свободный 3′-конец, позволяющий указанному зонду действовать в качестве праймера в реакции удлинения или лигирования праймера, где каждая разновидность указанного захватывающего зонда содержит молекулу нуклеиновой кислоты, имеющую в направлении от 5′ к 3′:
(1) позиционную область, содержащую штрихкодовую последовательность, которая соответствует положению захватывающего зонда на чипе, и
(2) захватывающую область для захвата нуклеиновой кислоты образца ткани, который приводят в контакт с указанным чипом;
(б) приведение указанного чипа в контакт с образцом ткани таким образом, что положение захватывающего зонда на чипе может быть сопоставлено с положением в образце ткани, и предоставление возможности ДНК образца ткани гибридизоваться с захватывающей областью в указанных захватывающих зондах;
(в) фрагментацию ДНК в указанном образце ткани, которую осуществляют до, в процессе или после приведения чипа в контакт с образцом ткани на стадии (б);
(г) удлинение указанных захватывающих зондов в реакции удлинения праймеров с использованием захваченных фрагментов ДНК в качестве матриц для синтеза удлиненных молекул ДНК или лигирование захваченных фрагментов ДНК с захватывающими зондами в реакции лигирования для синтеза лигированных молекул ДНК, где указанные удлиненные или лигированные молекулы ДНК являются мечеными благодаря позиционной области;
(д) возможно синтез комплементарной нити указанной меченой ДНК и/или возможно амплификацию указанной меченой ДНК;
(е) высвобождение по меньшей мере части меченых молекул ДНК и/или их комплементов и/или ампликонов с поверхности чипа, где указанная часть включает позиционную область или ее комплемент;
(ж) прямой или опосредованный анализ последовательности (например, секвенирование) высвобожденных молекул ДНК; и
(з) сопоставление информации из анализа последовательности с положением в образце ткани, предпочтительно где информацию из анализа указанной последовательности сопоставляют с изображением указанного образца ткани, где изображение образца ткани получают до или после стадии (г).
55. Способ по п. 54, включающий:
(а) предоставление чипа, содержащего подложку, на которой непосредственно или опосредованно иммобилизованы многочисленные разновидности захватывающих зондов, так что каждая разновидность занимает определенное положение на чипе и ориентирована таким образом, что имеет свободный 3′-конец, позволяющий указанному зонду действовать в качестве праймера в реакции удлинения или лигирования праймера, где каждая разновидность указанного захватывающего зонда содержит молекулу нуклеиновой кислоты, имеющую в направлении от 5′ к 3′:
(1) позиционную область, содержащую штрихкодовую последовательность, которая соответствует положению захватывающего зонда на чипе, и
(2) захватывающую область для захвата нуклеиновой кислоты образца ткани, который приводят в контакт с указанным чипом;
(б) приведение указанного чипа в контакт с образцом ткани таким образом, что положение захватывающего зонда на чипе может быть сопоставлено с положением в образце ткани;
(в) фрагментацию ДНК в указанном образце ткани, которую осуществляют до, в процессе или после приведения чипа в контакт с образцом ткани на стадии (б);
(г) обеспечение указанных фрагментов ДНК связывающей областью, которая способна гибридизоваться с указанной захватывающей областью;
(д) предоставление возможности указанным фрагментам ДНК гибридизоваться с захватывающей областью в указанных захватывающих зондах;
(е) удлинение указанных захватывающих зондов в реакции удлинения праймеров с использованием захваченных фрагментов ДНК в качестве матриц для синтеза удлиненных молекул ДНК или лигирование захваченных фрагментов ДНК с захватывающими зондами в реакции лигирования для синтеза лигированных молекул ДНК, где указанные удлиненные или лигированные молекулы ДНК являются мечеными благодаря позиционной области;
(ж) возможно синтез комплементарной нити указанной меченой ДНК и/или возможно амплификацию меченой ДНК;
(з) высвобождение по меньшей мере части меченых молекул ДНК и/или их комплементов и/или ампликонов с поверхности чипа, где указанная часть включает позиционную область или ее комплемент;
(и) прямой или опосредованный анализ последовательности (например, секвенирование) высвобожденных молекул ДНК; и
(к) сопоставление информации из анализа последовательности с положением в образце ткани, предпочтительно где информацию из анализа указанной последовательности сопоставляют с изображением указанного образца ткани, где изображение образца ткани получают до или после стадии (е).
| US 20020168645 A1, 14.11.2002 | |||
| US 20060211001 A1, 21.09.2006.WO 2007030373 A2, 15.03.2007.RU 2270254 C2, 20.02.2006.RU 2145635 C1, 20.02.2000. |

