Область техники
Настоящее изобретение относится к сульфату 5-гидрокси-1H-имидазол-4-карбоксамида.
Предшествующий уровень техники
5-гидрокси-1H-имидазол-4-карбоксамид (в дальнейшем также обозначаемый как «соединение A») является соединением, пригодным в качестве средства против злокачественных опухолей (патентный документ 1). Для того чтобы предоставить стабильное соединение A, было предложено превратить соединение A в органическую соль сульфоновой кислоты (патентный документ 2).
Ссылки на известный уровень техники
Патентные документы
Патентный документ 1: непроверенная публикация японского патента (Kokai) № 53-32124
Патентный документ 2: Международная публикация WO 2009/035168
Сущность изобретения
Цель, которую надо достичь посредством данного изобретения
Поскольку, например, соединение A легко окисляется и легко окрашивается в синий цвет, нельзя сказать, что соединение A демонстрирует достаточную стабильность при хранении. Однако очень важно, что активные фармацевтические ингредиенты являются чрезвычайно чистыми и стабильными при комнатной температуре или дополнительно при более высокой температуре, чтобы сохранить их качество и продемонстрировать низкую гигроскопичность.
Целью настоящего изобретения является предоставить соль соединения A, демонстрирующую ослабленное окрашивание в синий цвет, высокую степень чистоты, низкую гигроскопичность и превосходную стабильность хранения.
Средства для достижения цели
При данных обстоятельствах авторы настоящего изобретения провели различные исследования, и в результате обнаружили, что сульфат (соль серной кислоты) соединения A демонстрирует сниженное окрашивание в синий цвет, низкую гигроскопичность и превосходную стабильность при хранении, и его можно получать с высокой степенью чистоты. Они создали настоящее изобретение на основе этого открытия. Специфические средства для достижения цели, описанной выше, изложены далее.
Сульфат соединения A
Кристалл сульфата соединения A, который демонстрирует дифракционные пики при дифракционных углах 2θ 5,0, 20,3 и 29,6° порошковой рентгенограммы.
Кристалл сульфата соединения A, который демонстрирует дифракционные пики при дифракционных углах 2θ 5,0, 10,1, 13,6, 14,3, 14,7, 20,3, и 29,6° порошковой рентгенограммы.
Кристалл сульфата соединения A, который демонстрирует дифракционные пики при дифракционных углах 2θ 5,0, 10,1, 13,6, 14,3, 14,7, 20,3, 27,4, 28,0, 28,8 и 29,6° порошковой рентгенограммы (в дальнейшем также обозначаемый как «кристалл в β-форме»).
Кристалл в α-форме можно отличить от других кристаллических форм на основе указанных выше дифракционных пиков при дифракционных углах 2θ 5,0, 20,3 и 29,6°, или дифракционных пиков при дифракционных углах 2θ 5,0, 10,1, 13,6, 14,3, 14,7, 20,3 и 29,6°. Эти значения являются характеристическими значениями этой кристаллической формы.
Фармацевтическая композиция, содержащая кристалл сульфата соединения A, который демонстрирует дифракционные пики при дифракционных углах 2θ 5,0, 20,3 и 29,6° порошковой рентгенограммы; кристалл сульфата соединения A, который демонстрирует дифракционные пики при дифракционных углах 2θ 5,0, 10,1, 13,6, 14,3, 14,7, 20,3 и 29,6° порошковой рентгенограммы; или кристалл сульфата соединения A, который демонстрирует дифракционные пики при дифракционных углах 2θ 5,0, 10,1, 13,6, 14,3, 14,7, 20,3, 27,4, 28,0, 28,8 и 29,6° порошковой рентгенограммы.
Кристалл сульфата соединения A, который демонстрирует дифракционные пики при дифракционных углах 2θ 13,7, 17,0, 18,0 и 20,1° порошковой рентгенограммы.
Кристалл сульфата соединения A, который демонстрирует дифракционные пики при дифракционных углах 2θ 10,0, 13,7, 14,6, 17,0, 18,0, 20,1, 24,4 и 25,2° порошковой рентгенограммы.
Кристалл сульфата соединения A, который демонстрирует дифракционные пики при дифракционных углах 2θ 10,0, 13,7, 14,6, 17,0, 18,0, 20,1, 24,4, 25,2 и 28,1° порошковой рентгенограммы (в дальнейшем также обозначаемый как «кристалл в α-форме»).
Кристалл в α-форме можно отличить от других кристаллических форм на основе указанных выше дифракционных пиков при дифракционных углах 2θ 13,7, 17,0, 18,0 и 20,1°, или дифракционные пики при дифракционных углах 2θ 10,0, 13,7, 14,6, 17,0, 18,0, 20,1, 24,4 и 25,2°. Эти значения являются характеристическими значениями этой кристаллической формы.
Фармацевтическая композиция, содержащая кристалл сульфата соединения A, которая демонстрирует дифракционные пики при дифракционных углах 2θ 13,7, 17,0, 18,0 и 20,1° порошковой рентгенограммы; кристалл сульфата соединения A, который демонстрирует дифракционные пики при дифракционных углах 2θ 10,0, 13,7, 14,6, 17,0, 18,0, 20,1, 24,4 и 25,2°; или кристалл сульфата соединения A, который демонстрирует дифракционные пики при дифракционных углах 2θ 10,0, 13,7, 14,6, 17,0, 18,0, 20,1, 24,4, 25,2 и 28,1°.
Кристалл сульфата соединения A, который демонстрирует дифракционные пики при дифракционных углах 2θ 10,3, 20,7, 20,9 и 31,2° порошковой рентгенограммы.
Кристалл сульфата соединения A, который демонстрирует дифракционные пики при дифракционных углах 2θ 10,3, 20,7, 20,9, 28,0 и 31,2° порошковой рентгенограммы (в дальнейшем также обозначаемый как «γ-форма кристалла»).
Кристалл в γ-форме можно отличить от других кристаллических форм на основе указанного выше дифракционного пика при дифракционных углах 2θ 10,3, 20,7, 20,9 и 31,2°. Эти значения являются характерными значениями этой кристаллической формы.
Фармацевтическая композиция, содержащая кристалл сульфата соединения A, который демонстрирует дифракционные пики при дифракционных углах 2θ 10,3, 20,7, 20,9 и 31,2° порошковой рентгенограммы; или кристалл сульфата соединения A, который демонстрирует дифракционные пики при дифракционных углах 2θ 10,3, 20,7, 20,9, 28,0 и 31,2° порошковой рентгенограммы.
Способ получения кристалла в соответствии с [2] или [3], который содержит этап добавления водного раствора серной кислоты к кристаллу в соответствии с [5] или [6] для суспендирования кристалла и помешивание суспензии при от 10 до 65°C, и где менее чем 4 моль серной кислоты предпочтительно используют для 1 моль соединения A или его гидрата.
Способ получения кристалла в соответствии с [5] или [6], который содержит этап растворения соединения A или его гидрата в водном растворе серной кислоты при нагревании, и постепенное охлаждение раствора, чтобы обеспечить осаждение кристалла.
Способ получения кристалла в соответствии с [8] или [9], который содержит этап добавления водного раствора серной кислоты к кристаллу в соответствии с [5] или [6] для суспендирования кристалла, и помешивание суспензии при от 10 до 65°C, и где 4 моль или более серной кислоты предпочтительно используют для 1 моль соединения A или его гидрата.
Эффект изобретения
По настоящему изобретению, может быть представлен сульфат соединения A, имеющий такие превосходные качества, как ослабленное синее окрашивание, высокая степень чистоты и/или низкая гигроскопичность, и демонстрирующий высокую стабильность хранения.
Кроме того, по настоящему изобретению, может быть представлен кристалл сульфата соединения A, имеющий такие превосходные качества, как ослабленное синее окрашивание, высокая степень чистоты и/или низкая гигроскопичность, и демонстрирующий высокую стабильность хранения.
Краткое описание чертежей
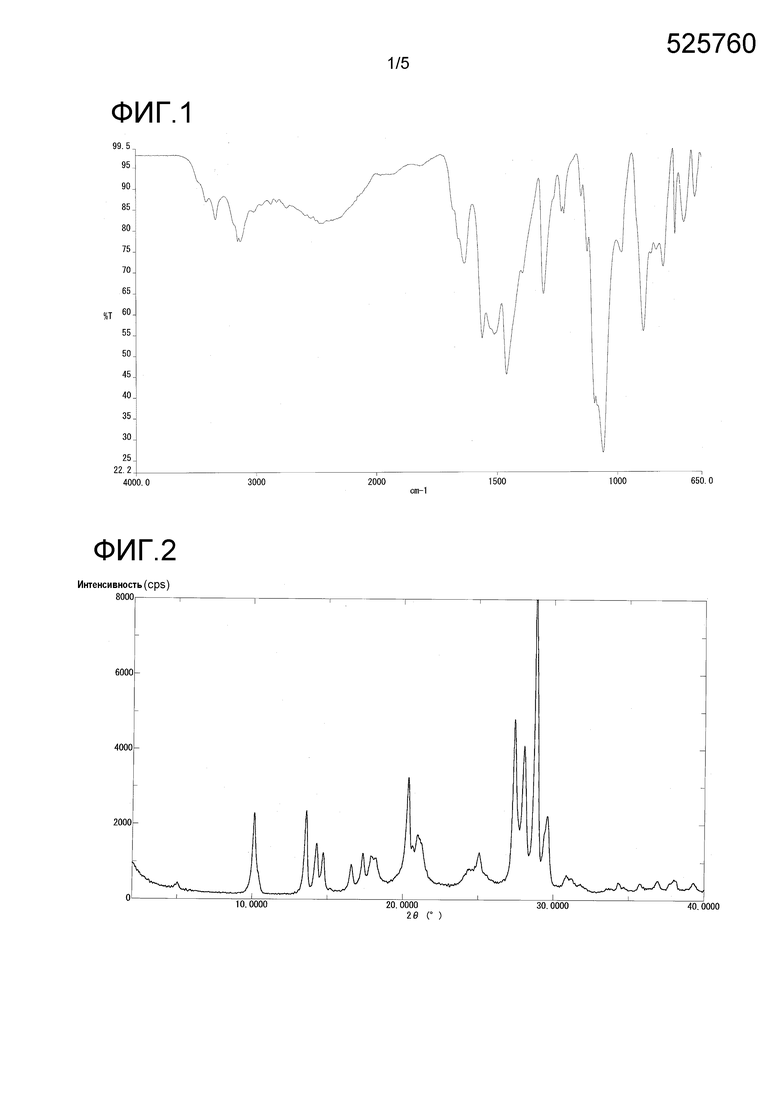
На фиг. 1 представлен пример инфракрасного спектра поглощения (ATR способ) кристалла в β-форме.
На фиг. 2 представлен пример порошковой рентгенограммы кристалла в β-форме.
На фиг. 3 представлен пример инфракрасного спектра поглощения (ATR способ) кристалла в α-форме.
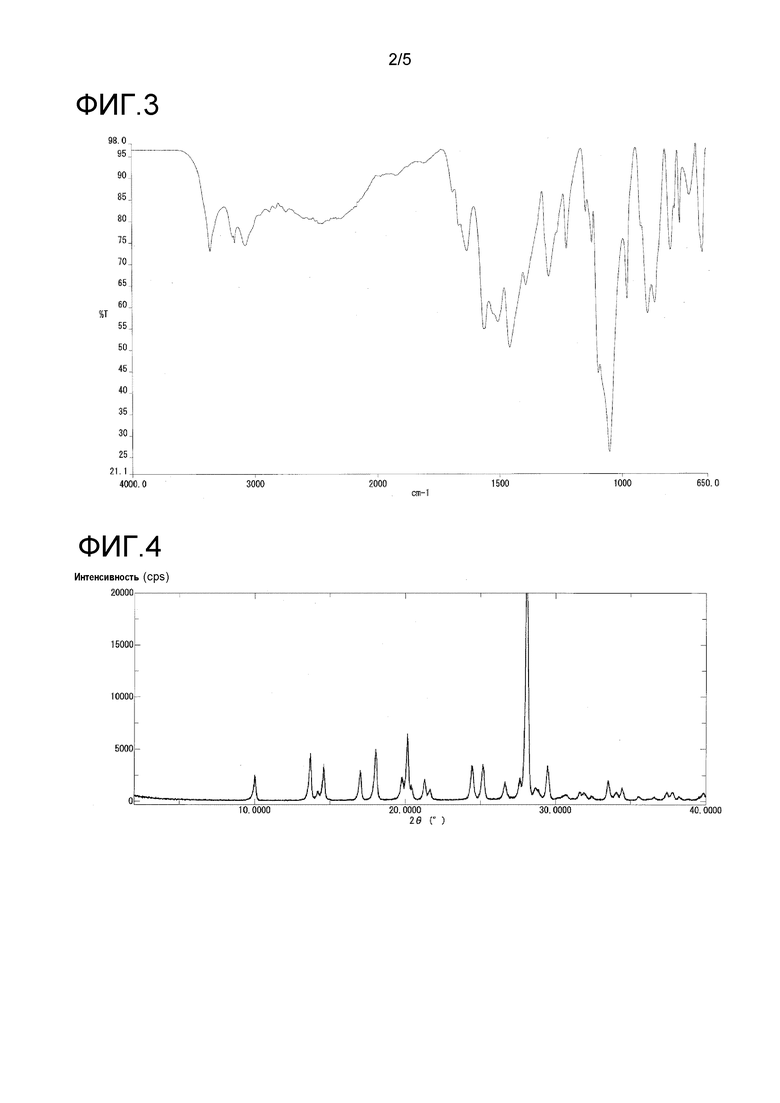
На фиг. 4 представлен пример порошковой рентгенограммы кристалла в α-форме.
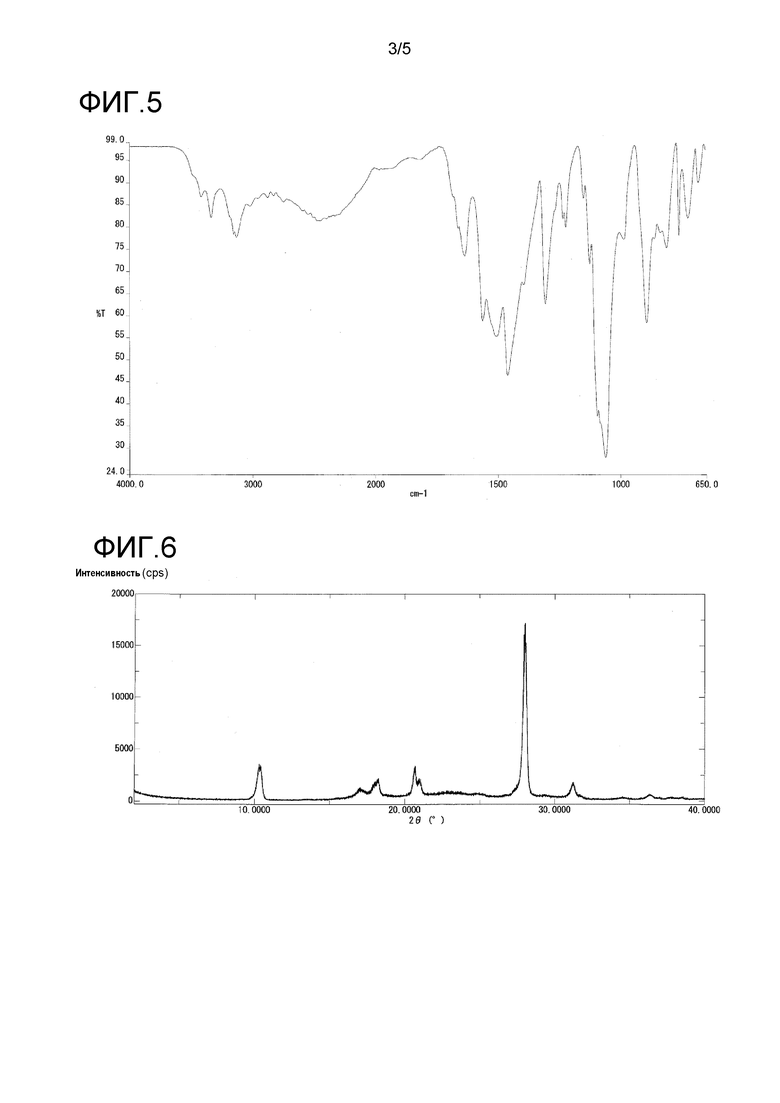
На фиг. 5 представлен пример инфракрасного спектра поглощения (ATR способ) кристалла в γ-форме.
На фиг. 6 представлен пример порошковой рентгенограммы кристалла в γ-форме.
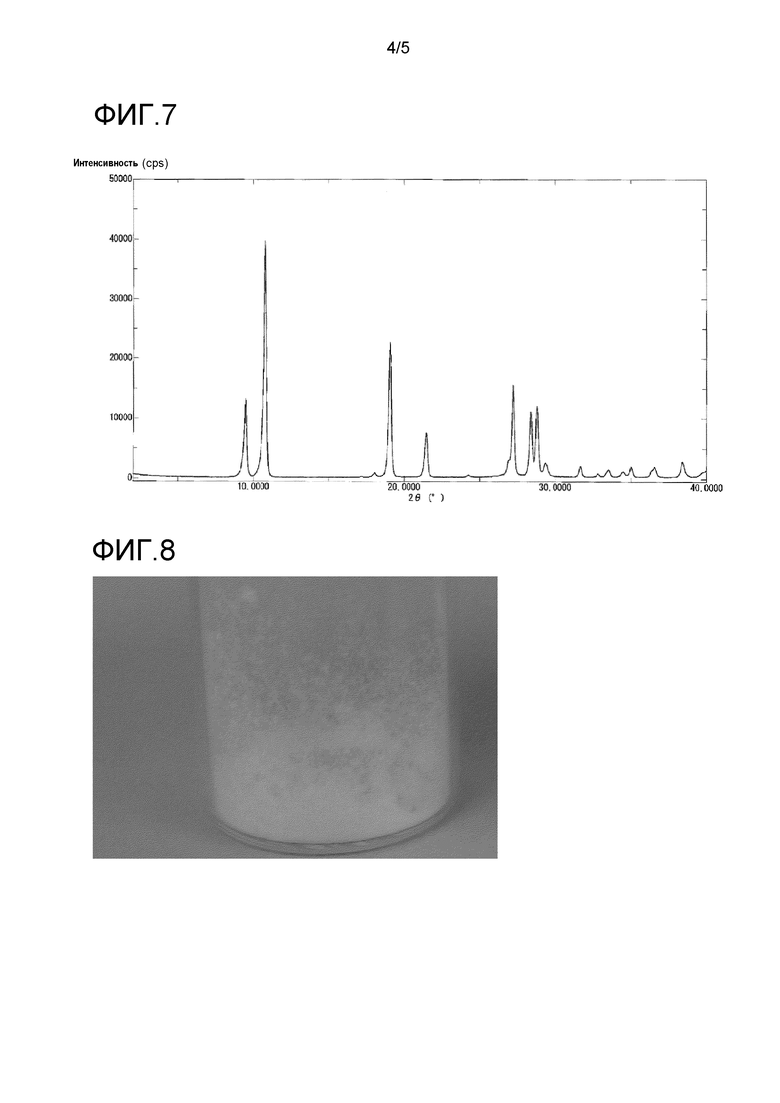
На фиг. 7 представлен пример порошковой рентгенограммы кристалла соединения A, полученного в сравнительном примере 1.
Фиг. 8 является фотографией, на которой представлены кристаллы в α-форме сульфата соединения A после хранения в течение двух недель в условиях 60°C и относительной влажности 75%.
Фиг. 9 является фотографией, на которой представлены кристаллы в β-форме сульфата соединения A после хранения в течение двух недель в условиях 60°C и относительной влажности 75%.
Фиг. 10 является фотографией, на которой представлены кристаллы соединения A после хранения в течение двух недель в условиях 60°C и относительной влажности 75%.
Способы осуществления изобретения
В дальнейшем, настоящее изобретение объясняется в подробностях. В настоящем изобретении, область числовых значений, обозначенная с помощью «to», означает диапазон, включающий значения, упомянутые до и после «to» в качестве максимального и минимального значения диапазона. В настоящем изобретении, когда композиция содержит множество видов веществ, соответствующих одному компоненту композиции, количество компонентов означает общее количество множества видов веществ, содержащихся в композиции, если не указано иначе.
Соль по настоящему изобретению включает ангидрид и его гидрат.
Кристалл по настоящему изобретению относится к кристаллу, содержащему ангидрид или гидрат соли и кристалл, имеющий воду адгезии.
Термин «дифракционный угол 2θ X°», применяемый для порошковой рентгенограммы кристалла по настоящему изобретению, означает «дифракционный угол 2θ (от (X - 0,2) до (X + 0,2))°», если не указано иначе.
Соль и ее кристалл по настоящему изобретению демонстрируют любую одну (предпочтительно все) характеристики из (1) ослабленное синее окрашивание, (2) высокая степень чистоты, (3) низкая гигроскопичность, и (4) превосходная стабильность хранения, и пригодны в качестве активных фармацевтических ингредиентов.
Способ получения кристалла в α-форме
Кристалл в α-форме можно получать посредством растворения соединения A или его гидрата в водном растворе серной кислоты при нагревании, и постепенно охлаждая раствор, чтобы обеспечить осаждение кристалла. Соединение A или его гидрат можно получать, например, по способу, описанному в непроверенной публикации японского патента (Kokai) № 58-24569, или по способу, описанному в Примере получения 1, приведенном ниже.
Количество серной кислоты, содержащейся в водном растворе серной кислоты, может составлять от 0,5 до 1,4 моль, предпочтительно от 1,0 до 1,2 моль, для 1 моль соединения A или его гидрата.
Объем водного раствора серной кислоты, применяемый для растворения соединения A или его гидрата, конкретно не ограничен. Например, он составляет предпочтительно от 20- до 100-кратных объемов (v/w), более предпочтительно от 40- до 60-кратных объемов (v/w), соединения A или его гидрата.
Температура нагревания для растворения соединения A или его гидрата в водном растворе серной кислоты конкретно не ограничена. Например, она может составлять от 40 до 60°C, предпочтительно от 50 до 60°C, более предпочтительно от 50 до 55°C.
Температура для осаждения кристалла конкретно не ограничена. Например, она может составлять от 0 до 40°C, предпочтительно от 0 до 20°C, более предпочтительно от 0 до 10°C.
Время для осаждения кристалла конкретно не ограничено. Например, оно составляет предпочтительно от 0,5 до 24 часов, более предпочтительно от 0,5 до 6 часов.
Способ получения кристалла в β-форме
Кристалл в β-форме можно получать посредством, например, добавления водного раствора серной кислоты к кристаллу в α-форме для суспендирования кристалла и перемешивания суспензии при температуре от 10 до 65°C. Посредством обеспечения осаждения кристалла выход кристалла может быть увеличен.
Количество серной кислоты, содержащейся в водном растворе серной кислоты, составляет предпочтительно от 2 до 3 моль для 1 моль соединения A в форме кристалла в α-форме.
Применяемый объем водного раствора серной кислоты конкретно не ограничен. Например, он составляет предпочтительно от 1- до 50-кратного объема (v/w), более предпочтительно от 5- до 20-кратного объема (v/w), кристалла в α-форме.
Время перемешивания суспензии конкретно не ограничено. Например, оно составляет предпочтительно от 0,5 до 5 часов, более предпочтительно от 2 до 5 часов.
Время для обеспечения осаждения кристалла конкретно не ограничено. Например, оно составляет предпочтительно от 12 до 24 часов.
Способ получения кристалла в γ-форме
Кристалл в γ-форме можно получать посредством, например, добавления водного раствора серной кислоты к кристаллу в α-форме для суспендирования кристалла и перемешивания суспензии при температуре от 10 до 65°C. Посредством обеспечения осаждения кристалла выход кристалла может быть увеличен.
Количество серной кислоты, содержащейся в водном растворе серной кислоты, составляет предпочтительно от 4 до 10 моль для 1 моль соединения A в форме кристалла в α-форме.
Применяемый объем водного раствора серной кислоты конкретно не ограничен. Например, он составляет предпочтительно от 5- до 20-кратного объема (v/w), более предпочтительно от 10- до 15-кратного объема (v/w), кристалла в α-форме.
Время для перемешивания суспензии конкретно не ограничено. Например, оно составляет предпочтительно от 0,5 до 5 часов, более предпочтительно от 2 до 5 часов.
Время для обеспечения осаждения кристалла конкретно не ограничено. Например, оно составляет предпочтительно от 12 до 24 часов.
Во время получения кристалла в α-форме, предпочтительно применяют затравочный кристалл. Таким образом можно контролировать, чтобы кристаллическая форма была более однородной.
В качестве затравочного кристалла можно использовать кристалл, полученный при предшествующем получении, или часть осажденных кристаллов можно получить посредством фильтрации в начале получения и использовать в качестве затравочного кристалла.
Хотя температура для сбора кристаллов в α-, β- или γ-форме, полученных посредством осаждения кристалла или перемешивания суспензии посредством фильтрации конкретно не ограничена, она составляет предпочтительно от 0 до 25°C.
Осаждение кристалла или перемешивание суспензии можно проводить в атмосфере воздуха или инертного газа, и его предпочтительно проводят в атмосфере инертного газа. Примеры атмосферы инертного газа включают атмосферу аргона, атмосферу азота и т.д.
Фармацевтическая композиция
Фармацевтическая композиция по настоящему изобретению содержит один или несколько видов кристаллов, выбранных из кристаллов в α-, β- и γ-форме. Фармацевтическая композиция по настоящему изобретению демонстрирует превосходную стабильность хранения, так как она содержит кристалл в α-, β- и/или γ-форме.
Когда кристалл в α-, β- и/или γ-форме применяют в качестве фармацевтической композиции, фармацевтические вспомогательные средства, как правило, применяемые для производства фармацевтических препаратов, такие как эксципиенты, носители и разбавители, могут быть соответствующим образом смешаны. Композицию можно вводить перорально или парентерально общепринятым способом в форме таблетки, капсулы, порошка, сиропа, гранулы, пилюли, суспензии, эмульсии, раствора, порошкового препарата, суппозиториев, глазных капель, носовых капель, ушных капель, пластыря, мази, инъекции или т.п. Способ введения, доза и частота могут быть соответствующим образом выбраны в соответствии с возрастом, массой и симптомами пациента. Для взрослого, от 0,01 до 2000 мг/кг в сутки соли может быть, как правило, введено перорально или парентерально (посредством, например, инъекции, капельной инфузии, введение в ректальную область или т.п.) раз в сутки или несколько раз в сутки посредством деления указанной выше дозы.
Далее, настоящее изобретение будет объяснено в подробностях со ссылками на примеры, примеры получения и примеры фармацевтических препаратов. Однако настоящее изобретение не ограничено этим. Если не указано иначе, % означает % по массе.
Значение аббревиатуры, применяемой в следующих описаниях, представлено ниже.
ВЭЖХ: Высокоэффективная жидкостная хроматография
Порошковую рентгеновскую дифракцию измеряли с использованием UltimaIV (Rigaku) при следующих условиях.
(Режим измерений)
Измерительный катод: Cu
Рабочее напряжение разрядной лампы: 40 кВ
Ток трубки: 40 мА
Ось сканирования: 2θ
ИК-спектры поглощения измеряли с использованием Spectrum One (PerkinElmer) в соответствии с описаниями Японской фармакопеи, Методами проведения общего тестирования, Способом инфракрасной спектроскопии с фурье-преобразованием нарушенного полного отражения (ATR способ).
Абсолютную влажность измеряли с применением влагомера Карла Фишера.
Степени чистоты выражены в единицах измерения ВЭЖХ % по площади. ВЭЖХ измерение проводили с применением Prominence (Shimadzu) при следующих условиях.
(Режим измерений)
Длина волны измерения: 210 нм
Колонка: Hydrosphere C18 (внутренний диаметр 6,0 мм × длина 250 мм, диаметр частиц 5 мкм)
Температура колонки, 40°C
Скорость потока: 0,8 мл/минута
Раствор A для подвижной фазы: вода (950 мл) и 0,4 моль/л фосфатный/триэтиламиновый буфер (pH 3, 50 мл)
Раствор B для подвижной фазы: вода (50 мл), ацетонитрил (900 мл), и 0,4 моль/л фосфат/триэтиламин буфер (pH 3, 50 мл)
В качестве подвижной фазы применяли смеси растворов A и B для подвижной фазы как следующий линейный градиент.
(Пример получения 1)
(1) В атмосфере азота, 2-аминомалонамид (30 г, Tateyama Kasei) и щавелевую кислоту (115 мг) добавляли к 2-пропанолу (600 мл), смесь нагревали до 82°C, а затем триэтил ортоформиат (106 мл, степень чистоты 99,5%, Nippoh Chemicals) добавляли капельно к смеси через 10 минут. Затем, реакционную смесь перемешивали при 84°C в течение 7 часов и 30 минут. Реакционную смесь охлаждали до 57°C, а затем воду (30 мл) и концентрированную соляную кислоту (24 мл) последовательно добавляли к реакционной смеси. Реакционную смесь охлаждали до 5°C, и кристаллы собирали посредством фильтрации, и промывали с применением ацетона (120 мл) для получения дигидрата 5-гидрокси-1H-имидазол-4-карбоксамид гидрохлорида в виде бледно-желтых кристаллов (49 г).
(2) В атмосфере азота дигидрат 5-гидрокси-1H-имидазол-4-карбоксамид гидрохлорида (20,0 г) добавляли к 0,45 моль/л соляной кислоты (240 мл) и растворяли посредством нагревания смеси до 50°C. К этому раствору, через 33 минуты добавляли капельно раствор натрий формиата (14,3 г) в воде (40 мл). Реакционную смесь охлаждали до 5°C, и кристаллы собирали посредством фильтрации, промывали с применением смеси ацетона (20 мл) и воды (40 мл), а затем промывали с применением ацетона (60 мл) для получения 3/4 гидрата 5-гидрокси-1H-имидазол-4-карбоксамида в виде бледно-желтых кристаллов (12,8 г).
(Пример 1)
К 3/4 гидрату 5-гидрокси-1H-имидазол-4-карбоксамида (1,5 г) добавляли воду (30 мл) и концентрированную серную кислоту (625 мкл), смесь нагревали до 50-55°C, затем дополнительно добавляли воду (45 мл) для растворения соединения, и смесь перемешивали при той же температуре в течение 30 минут. Реакционную смесь постепенно охлаждали до комнатной температуры. После того как было подтверждено осаждение кристаллов, смесь охлаждали на льду, и кристаллы собирали посредством фильтрации. Кристаллы отмывали водой и высушивали на воздухе для получения кристаллов в α-форме (1,8 г).
Кристаллы в α-форме продемонстрировали характеристические пики при дифракционных углах 2θ 10,0, 13,7, 14,6, 17,0, 18,0, 20,1, 24,4, 25,2, и 28,1° порошковой рентгенограммы.
Кристаллы в α-форме также продемонстрировали характеристические пики при 1563, 1504, 1456, 1390, 1050 и 863 см-1 в инфракрасном спектре поглощения.
Инфракрасный спектр поглощения (ATR способ) кристаллов в α-форме продемонстрирован на фиг. 3, и их порошковая рентгенограмма продемонстрирована на фиг. 4 и в таблице 2.
Абсолютная влажность кристаллов в α-форме составляла 9,1%.
(Пример 2)
К кристаллам в α-форме (4,0 г), полученным в соответствии со способом, описанным в примере 1, добавляли 1 моль/л водный раствор серной кислоты (40 мл), и смесь перемешивали при комнатной температуре в течение 2 часов и 45 минут в атмосфере азота. К реакционной смеси добавляли 1 моль/л водный раствор серной кислоты (20 мл), затем смесь оставляли на ночь, и в дальнейшем перемешивали в течение 1 часа и 30 минут, и кристаллы собирали посредством фильтрации. Кристаллы промывали с применением 1 моль/л водного раствора серной кислоты и ацетона, и высушивали на воздухе для получения кристаллов в β-форме (3,7 г).
Кристаллы в β-форме продемонстрировали характеристические пики при дифракционных углах 2θ 5,0, 10,1, 13,6, 14,3, 14,7, 20,3, 27,4, 28,0, 28,8 и 29,6° порошковой рентгенограммы.
Кристаллы в β-форме также продемонстрировали характеристические пики при 1560, 1508, 1459 и 1059 см-1 в инфракрасном спектре поглощения.
Инфракрасный спектр поглощения (ATR способ) кристаллов в β-форме продемонстрирован на фиг. 1, и их порошковая рентгенограмма продемонстрирована на фиг. 2 и в таблице 3.
Абсолютная влажность кристаллов в β-форме составляла 8,1%.
(Пример 3)
К кристаллам в α-форме (0,5 г), полученным в соответствии со способом, описанным в примере 1, добавляли 2 моль/л водного раствора серной кислоты (5 мл), и смесь перемешивали при комнатной температуре в течение 3 часов и 20 минут. К реакционной смеси дополнительно добавляли концентрированную серную кислоту (1 мл), а затем смесь перемешивали в течение 4 часов и 40 минут. Смесь оставляли на ночь, а затем кристаллы собирали посредством фильтрации. Кристаллы отмывали с применением ацетона, и высушивали на воздухе для получения кристаллов в γ-форме (0,5 г).
Кристаллы в γ-форме продемонстрировали характеристические пики при дифракционных углах 2θ 10,3, 20,7, 20,9, 28,0, и 31,2° порошковой рентгенограммы.
Кристаллы в γ-форме также продемонстрировали характеристические пики при 1560, 1503, 1459, 1304, 1061 и 892 см-1 в инфракрасном спектре поглощения.
Инфракрасный спектр поглощения (ATR способ) кристаллов в γ-форме продемонстрирован на фиг. 5, и их порошковая рентгенограмма продемонстрирована на фиг. 6 и в таблице 4.
Абсолютная влажность кристаллов в γ-форме составляла 4,1%.
(Сравнительный пример 1)
5-Гидрокси-1H-имидазол-4-карбоксамид получали в виде бледно-желтого порошка в соответствии со способом, описанным в примере 6 международной публикации WO 2009/035168.
В результате высокоэффективной жидкостной хроматографии, полученный 5-гидрокси-1H-имидазол-4-карбоксамид содержал приблизительно 0,15% бензойной кислоты.
Его порошковая рентгенограмма продемонстрирована на фиг. 7 и в таблице 5.
(Сравнительный пример 2)
Метансульфонат 5-гидрокси-1H-имидазол-4-карбоксамида получали, как описано ниже со ссылкой на способ, описанный в примере 3 международной публикации WO2009/035168.
К 3/4 гидрату 5-гидрокси-1H-имидазол-4-карбоксамида (1,4 г) добавляли воду (30 мл) и метансульфоновую кислоту (1,0 г), смесь нагревали при 50°C для растворения соединений, а затем раствор перемешивали при той же температуре в течение 30 минут. После охлаждения реакционной смеси до комнатной температуры, добавляли толуол, воду удаляли посредством азеотропии при пониженном давлении, и осажденное твердое вещество собирали посредством фильтрации. Твердое вещество отмывали этилацетатом и высушивали на воздухе для получения метансульфоната 5-гидрокси-1H-имидазол-4-карбоксамида (1,7 г).
Абсолютная влажность метансульфоната 5-гидрокси-1H-имидазол-4-карбоксамида составляла 7,8%.
(Тестовый пример 1)
Тест стабильности хранения (1) в условиях 60°C и относительной влажности 75%
В качестве тестируемых веществ были выбраны кристаллы, полученные в примерах 1 и 2 и сравнительном примере 1.
Каждое тестируемое вещество (приблизительно 0,3 г) помещали в стеклянную бутылку и хранили в условиях 60°C и относительной влажности 75% в течение 2 недель.
Кристаллы из примеров 1 и 2 после теста продемонстрированы на фиг. 8 и 9, соответственно. Кристаллы из сравнительного примера 1 после теста продемонстрированы на фиг. 10.
Кристаллы, полученные в примерах 1 и 2, продемонстрировали окрашивание в очевидно меньшей степени по сравнению с кристаллами, полученными в сравнительном примере 1, и таким образом продемонстрировали превосходную стабильность хранения.
(Тестовый пример 2)
Тест стабильности хранения (2) в условиях 60°C и относительной влажности 75%
В качестве тестируемого вещества были выбраны кристаллы, полученные в примере 1.
Тестируемое вещество (приблизительно 0,3 г) помещали в стеклянную бутылку и хранили в условиях 60°C и относительной влажности 75% в течение 2 недель. После завершения теста, степень чистоты вещества измеряли посредством ВЭЖХ. В результате было выявлено, что степень чистоты тестируемого вещества составляла 99,97%.
Кристаллы, полученные в примере 1, продемонстрировали высокую степень чистоты даже после хранения в течение 2 недель, и таким образом продемонстрировали превосходную стабильность хранения.
Кристаллы, полученные в примере 2, также продемонстрировали высокую степень чистоты даже после хранения в течение 2 недель при анализе, произведенном тем же способом, как и тестовый пример 2, и таким образом продемонстрировали превосходную стабильность хранения.
(Тестовый пример 3)
Тест стабильности хранения (3) в условиях 60°C и относительной влажности 75%
В качестве тестируемых веществ были выбраны кристаллы, полученные в примере 2 и сравнительном примере 2.
Каждое тестируемое вещество (приблизительно 0,3 г) помещали в стеклянную бутылку и хранили в условиях 60°C и относительной влажности 75% в течение 2 недель, а затем измеряли массу. Соотношение изменения массы каждого тестируемого вещества рассчитывали в соответствии со следующим уравнением.
Соотношение изменения массы (%) = {(A1 - A0)/A0} × 100
A0: Масса (г) тестируемого вещества до теста
A1: Масса (г) тестируемого вещества после теста
Изменение цветового признака тестируемых веществ также было визуально оценено.
Результаты представлены в таблице 6.
Кристаллы, полученные в примере 2, продемонстрировали меньшую гигроскопичность по сравнению с кристаллами, полученными в сравнительном примере 2, и не продемонстрировали никакого изменения цветового признака.
Дополнительно, когда эти кристаллы хранили в течение 2 недель в условиях 25°C и относительной влажности 97%, кристаллы, полученные в примере 2, не продемонстрировали изменения признака, и таким образом были стабильны, в то время как кристаллы, полученные в сравнительном примере 2, растворились, и таким образом были нестабильными.
(Пример фармацевтического препарата 1)
Фармацевтическую композицию, содержащую соль по настоящему изобретению или ее кристаллы, можно получать, например, в следующем составе.
Ингредиенты (1), (2) и (3) смешивают, ингредиент (4) распыляют на порошковую смесь, смесь гранулируют, ингредиенты (5) и (6) смешивают с гранулированной смесью, и смесь превращают в таблетки.
Промышленная применимость
Соль и ее кристалл по настоящему изобретению демонстрируют такие характеристики, как (1) сниженное синее окрашивание, (2) высокая степень чистоты, (3) низкая гигроскопичность и (4) превосходная стабильность при хранении, и пригодны в качестве фармацевтических ингредиентов лекарственных средств.

Изобретение относится к области органической химии, а именно к сульфату 5-гидрокси-1Н-имидазол-4-карбоксамида и его кристаллическим формам. Также изобретение относится к фармацевтической композиции на основе α-формы, β-формы или γ-формы сульфата 5-гидрокси-1Н-имидазол-4-карбоксамида. Технический результат: получены кристаллические формы сульфата 5-гидрокси-1Н-имидазол-4-карбоксамида, обладающие полезной биологической активностью, а также такими характеристиками, как сниженное синее окрашивание, высокая степень чистоты, низкая гигроскопичность и превосходная стабильность при хранении. 10 н.п. ф-лы, 10 ил., 6 табл, 9 пр.
1. Сульфат 5-гидрокси-1Н-имидазол-4-карбоксамида.
2. Кристалл сульфата 5-гидрокси-1Н-имидазол-4-карбоксамида в β-форме, который демонстрирует дифракционные пики при дифракционных углах 2θ 5,0, 20,3 и 29,6° порошковой рентгенограммы.
3. Кристалл сульфата 5-гидрокси-1Н-имидазол-4-карбоксамида в β-форме, который демонстрирует дифракционные пики при дифракционных углах 2θ 5,0, 10,1, 13,6, 14,3, 14,7, 20,3, 27,4, 28,0, 28,8 и 29,6° порошковой рентгенограммы.
4. Фармацевтическая противораковая композиция, содержащая:
кристалл сульфата 5-гидрокси-1Н-имидазол-4-карбоксамида в β-форме, который демонстрирует дифракционные пики при дифракционных углах 2θ 5,0, 20,3, и 29,6° порошковой рентгенограммы; или
кристалл сульфата 5-гидрокси-1Н-имидазол-4-карбоксамида в β-форме, который демонстрирует дифракционные пики при дифракционных углах 2θ 5,0, 10,1, 13,6, 14,3, 14,7, 20,3, 27,4, 28,0, 28,8 и 29,6° порошковой рентгенограммы.
5. Кристалл сульфата 5-гидрокси-1Н-имидазол-4-карбоксамида в α-форме, который демонстрирует дифракционные пики при дифракционных углах 2θ 13,7, 17,0, 18,0 и 20,1° порошковой рентгенограммы.
6. Кристалл сульфата 5-гидрокси-1Н-имидазол-4-карбоксамида в α-форме, который демонстрирует дифракционные пики при дифракционных углах 2θ 10,0, 13,7, 14,6, 17,0, 18,0, 20,1, 24,4, 25,2 и 28,1° порошковой рентгенограммы.
7. Фармацевтическая противораковая композиция, содержащая:
кристалл сульфата 5-гидрокси-1Н-имидазол-4-карбоксамида в α-форме, который демонстрирует дифракционные пики при дифракционных углах 2θ 13,7, 17,0, 18,0 и 20,1° порошковой рентгенограммы; или
кристалл сульфата 5-гидрокси-1Н-имидазол-4-карбоксамида в α-форме, который демонстрирует дифракционные пики при дифракционных углах 2θ 10,0, 13,7, 14,6, 17,0, 18,0, 20,1, 24,4, 25,2 и 28,1°.
8. Кристалл сульфата 5-гидрокси-1Н-имидазол-4-карбоксамида в γ-форме, который демонстрирует дифракционные пики при дифракционных углах 2θ 10,3, 20,7, 20,9 и 31,2° порошковой рентгенограммы.
9. Кристалл сульфата 5-гидрокси-1Н-имидазол-4-карбоксамида в γ-форме, который демонстрирует дифракционные пики при дифракционных углах 2θ 10,3, 20,7, 20,9, 28,0, и 31,2° порошковой рентгенограммы.
10. Фармацевтическая противораковая композиция, содержащая:
кристалл сульфата 5-гидрокси-1Н-имидазол-4-карбоксамида в γ-форме, который демонстрирует дифракционные пики при дифракционных углах 2θ 10,3, 20,7, 20,9 и 31,2° порошковой рентгенограммы; или
кристалл сульфата 5-гидрокси-1Н-имидазол-4-карбоксамида в γ-форме, который демонстрирует дифракционные пики при дифракционных углах 2θ 10,3, 20,7, 20,9, 28,0 и 31,2° порошковой рентгенограммы.
| СПОСОБ ПРОИЗВОДСТВА ПИТАТЕЛЬНОЙ СМЕСИ ДЛЯ ДЕТСКОГО, ДИЕТИЧЕСКОГО ИЛИ ВНУТРИКИШЕЧНОГО ЗОНДОВОГО ПИТАНИЯ | 2001 |
|
RU2196460C1 |
| US 4181731, 01.01.1980 A1 | |||
| Прибор для выпускания воздуха из спринклерной сети | 1927 |
|
SU12267A1 |

