Область техники, к которой относится изобретение
[0001] Настоящее изобретение касается 5-гидрокси-1Н-имидазол-4-карбоксамид.гидрата. Кроме того, настоящее изобретение касается кристалла 5-гидрокси-1Н-имидазол-4-карбоксамид·3/4 гидрата и способа его получения.
[0002] 5-Гидрокси-1Н-имидазол-4-карбоксамид (здесь далее также обозначен как «соединение А») представляет собой соединение, которое полезно в качестве ингибитора рака (см., например, патентный документ 1)
Соединение А получают, например, из 2-аминомалонамида (см., например, непатентный документ 1 и патентные документы 1 и 2).
В непатентном документе 1 раскрыто, что соединение можно получить взаимодействием 2-аминомалонамида с этилформимидатом. Однако из-за низкого выхода этого способа получения он не является вполне удовлетворительным.
[0003] В патентном документе 1 раскрыто, что бензолсульфонат соединения A можно получить взаимодействием бензолсульфоната 2-аминомалонамида с триметилортоформиатом в присутствии бензолсульфоновой кислоты. Раскрыто также, что соединение A возможно получить нейтрализацией бензолсульфоната соединения A гидрокарбонатом натрия.
Однако этот способ получения имеет недостатки, состоящие в том, что получают эфир бензолсульфоновой кислоты, имеющий генетическую токсичность, и требуется триметилортоформиат в больших количествах. Следовательно, трудно сказать, что данный способ получения действительно представляет собой превосходный с промышленной точки зрения способ производства. Кроме того, получаемое соединение A является окрашенным и имеет плохую стабильность при хранении. В экспериментальных примерах 1 и 2 патентного документа 1 раскрыто, что даже несмотря на то, что сульфонат соединения A и соль соляной кислоты соединения A являются стабильными, цвет самого соединения A изменяется до темно-синего или голубого. В патентном документе 1 раскрыто также, что для получения соединения A, обладающего превосходной стабильностью при хранении, требуется включить в соединение A следовое количество кислоты. В примере 6 описано соединение A, содержащее около 2,5% бензойной кислоты. Однако отсутствует конкретное описание стабильности.
[0004] В патентном документе 2 раскрыто, что сырой кристалл соединения можно получить взаимодействием 2-аминомалонамида с триэтилортоформиатом в присутствии серной кислоты. Однако этот способ получения имеет недостатки, состоящие в том, что требуется большое количество триэтилортоформиата и также требуется большое количество активированного угля. Таким образом, трудно сказать, что способ получения действительно представляет собой способ получения, подходящий с промышленной точки зрения. В патентном документе 2 также описано, что можно получить соединение A взаимодействием сырого кристалла соединения A с кислотой и нейтрализацией аммиаком. Однако конкретное описание стабильности отсутствует.
[0005] В технологии составления препаратов соединения A известно, что синее окрашивание можно предотвратить, включая кислотный материал (см., например, патентный документ 3). В патентном документе 3 описано, что "настоящее соединение имеет свойство проявлять цвет само по себе или под воздействием кислорода, тепла или света, и в случае, когда настоящее соединение применяют, например, в качестве перорального агента, наблюдается тенденция демонстрировать более существенное окрашивание на основе более сложных реакционных путей в результате взаимодействия с сосуществующими наполнителями".
Патентные документы
[0006] Патентный документ 1: международная публикация № 2009/035168
Патентный документ 2: выложенная патентная заявка Японии (JP-A) № 58-24569.
Патентный документ 3: JP-A № 60-185727.
Непатентный документ
[0007] Непатентный документ 1: Journal of American Chemical Society (J. Am. Chem. Soc), т. 74, стр. 2892-2894, 1952.
КРАТКОЕ СОДЕРЖАНИЕ ИЗОБРЕТЕНИЯ
Техническая проблема
[0008] До настоящего времени считается, что из-за синего окрашивания соединение A имеет проблему в смысле стабильности при хранении. Полагают также, что для получения соединения A, обладающего превосходной стабильностью при хранении, требуется следующее: (1) получение соли кислоты соединения A, (2) совместное присутствие кислотного материала или (3) содержание следового количества кислотного материала. Кроме того, до настоящего времени едва ли известно соединение A, обладающее превосходной стабильностью при хранении без использования добавок в комбинации. Между тем, существует строгое требование, чтобы исходное соединение, применяемое для лекарственных препаратов, представляло собой стабильное индивидуальное соединение, а не смесь.
[0009] Целью изобретения является получение кристалла соединения A, которое имеет превосходную стабильность при хранении. Другой целью данного изобретения является получение кристалла соединения A, имеющего немного примесей, имеющего небольшое цветовое различие между кристаллом до хранения и кристаллом после хранения и превосходную стабильность при хранении, и способ его получения.
Решение проблемы
[0010] При этих обстоятельствах авторы настоящего изобретения провели интенсивные исследования и обнаружили в результате следующее: пункты с [1] по [24]. Таким образом, выполнено изобретение. Далее представлены конкретные средства решения проблемы.
[0011] [1] Способ получения гидрата соединения A, включающий взаимодействие 2-аминомалонамида с соединением, представленным следующей формулой [1], в присутствии карбоновой кислоты с получением соединения A,
[0012]
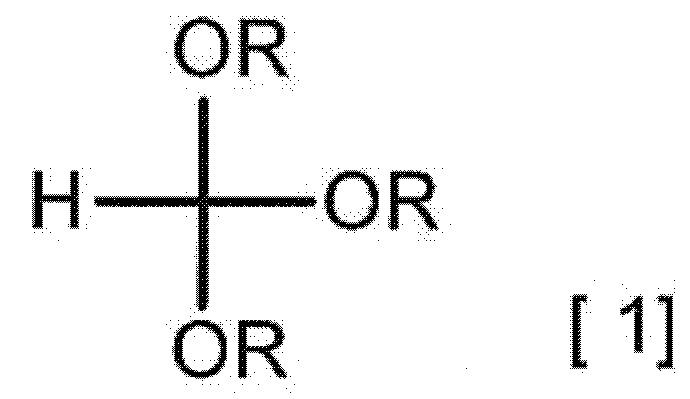
[0013] (в формуле [1] каждый R независимо представляет собой C1-3 алкильную группу), взаимодействие полученного соединения A с кислотным соединением с получением соли кислоты соединения A или ее гидрата и взаимодействие полученной соли кислоты соединения A или ее гидрата с солью в присутствии кислотного растворителя с получением гидрата соединения A.
[0014] [2] Способ получения гидрата соединения A, включающий взаимодействие 2-аминомалонамида с соединением, представленным следующей формулой [1], в отсутствие минеральной кислоты и в отсутствие сульфоновой кислоты с получением соединения A
[0015]
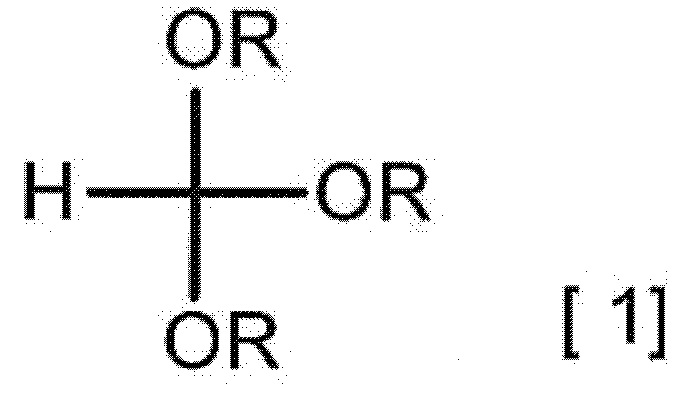
[0016] (в формуле каждый R независимо представляет собой C1-3 алкильную группу), взаимодействие полученного соединения A с кислотным соединением с получением соли кислоты соединения A или ее гидрата и взаимодействие полученной соли кислоты соединения A или ее гидрата с солью в присутствии кислотного растворителя с получением гидрата соединения A.
[0017] [3] Способ получения гидрата соединения A по п.[2], где получение соединения A выполняют в присутствии карбоновой кислоты.
[0018] [4] Способ получения гидрата соединения A по п.[1] или [3], где карбоновая кислота включает муравьиную кислоту или щавелевую кислоту.
[0019] [5] Способ получения гидрата соединения A по любому из п.п.[1], [3] и [4], где карбоновая кислота представляет собой щавелевую кислоту.
[0020] [6] Способ получения гидрата соединения A по любому из п.п.[1] и [3]-[5], где используемое количество карбоновой кислоты составляет от 0,001 до 0,05 молярного количества 2-аминомалонамида.
[0021] [7] Способ получения гидрата соединения A по любому из п.п.[1]-[6], где кислотное соединение представляет собой соляную кислоту, и соль кислоты представляет собой солянокислую соль.
[0022] [8] Способ получения гидрата соединения A по любому из п.п.[1]-[7], где кислотный растворитель представляет собой соляную кислоту.
[0023] [9] Способ получения гидрата соединения A по любому из п.п.[1]-[8], где кислотный растворитель представляет собой соляную кислоту с концентрацией от 0,3 моль/л до 0,8 моль/л.
[0024] [10] Способ получения гидрата соединения A по любому из п.п.[1]-[9], где соль представляет собой соль карбоновой кислоты.
[0025] [11] Способ получения гидрата соединения A по любому из п.п.[1]-[10], где соль представляет собой соль щелочного металла карбоновой кислоты.
[0026] [12] Способ получения гидрата соединения A по любому из п.п.[1]-[11], где соль представляет собой соль щелочного металла карбоновой кислоты, которая имеет первую константу диссоциации кислоты (pKa1) от 2 до 4.
[0027] [13] Способ получения гидрата соединения A по любому из п.п.[1]-[12], где соль представляет собой соль щелочного металла карбоновой кислоты, которая имеет первую константу диссоциации кислоты (pKa1) от 3 до 4.
[0028] [14] Кристалл 5-гидрокси-1H-имидазол-4-карбоксамид·3/4 гидрата (здесь далее, также обозначаемого как "гидрат соединения A"), имеющий содержание кислотного соединения 0,1% масс. или менее, где, в случае хранения кристалла в условиях температуры 40°C и относительной влажности 75% в течение 2 недель, кристалл демонстрирует цветовое различие (ΔE), равное 6 или менее, между состояниями до хранения и после хранения.
[0029] [15] Кристалл гидрата соединения A по п.[14], где цветовое различие (ΔE) равно 3 или менее.
[0030] [16] Кристалл гидрата соединения A по п.[14], где цветовое различие (ΔE) равно 3 или менее, и содержание кислотного соединения составляет 0,05% масс. или менее.
[0031] [17] Кристалл гидрата соединения A по любому из п.п.[14]-[16], где кристалл является бесцветным, бледно-желтым или желтым до хранения кристалла в условиях температуры 40°C и относительной влажности 75% в течение 2 недель.
[0032] [18] Кристалл гидрата соединения A по любому из п.п.[14]-[16], где кристалл является бесцветным или имеет цветовой тон (H) от 1Y до 6Y по цветовой системе Манселла до хранения кристалла в условиях температуры 40°C и относительной влажности 75% в течение 2 недель.
[0033] [19] Гидрат соединения A, полученный способом по любому из п.п.[1]-[13].
[0034] [20] Кристалл гидрата соединения A, который получен способом по любому из п.п.[1]-[13] и имеет содержание кислотного соединения 0,1% масс. или менее, где, в случае хранения кристалла в условиях температуры 40°C и относительной влажности 75% в течение 2 недель, кристалл демонстрирует цветовое различие (ΔE), равное 6 или менее, между состояниями до хранения и после хранения.
[0035] [21] Кристалл гидрата соединения A по п.[20], где цветовое различие (ΔE) равно 3 или менее.
[0036] [22] Кристалл гидрата соединения A по п.[20], где цветовое различие (ΔE) равно 3 или менее, и содержание кислотного соединения составляет 0,05% масс. или менее.
[0037] [23] Кристалл (здесь далее также обозначен как "кристалл 3 типа") гидрата соединения A, где кристалл имеет дифракционные пики при углах дифракции 8,1, 12,6, 17,1, 19,3, 20,3 и 21,6°, представленных как 2θ на картине порошковой рентгеновской дифракции.
[0038] [24] Фармацевтическая композиция, содержащая кристалл гидрата соединения A, где кристалл имеет дифракционные пики при углах дифракции 8,1, 12,6, 17,1, 19,3, 20,3 и 21,6°, представленных как 2θ на картине порошковой рентгеновской дифракции.
Полезные эффекты изобретения
[0039] Согласно настоящему изобретению, можно получить кристалл соединения A, обладающий превосходной стабильностью при хранении. Согласно настоящему изобретению, также можно получить кристалл соединения A, имеющий немного примесей, имеющий небольшое цветовое различие между кристаллом до хранения и кристаллом после хранения и обладающий превосходной стабильностью при хранении, и обеспечить способ его получения.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
[0040] На фиг.1 представлен график для иллюстрации одного примера спектра инфракрасного поглощения (ATR-метод) кристалла β-типа.
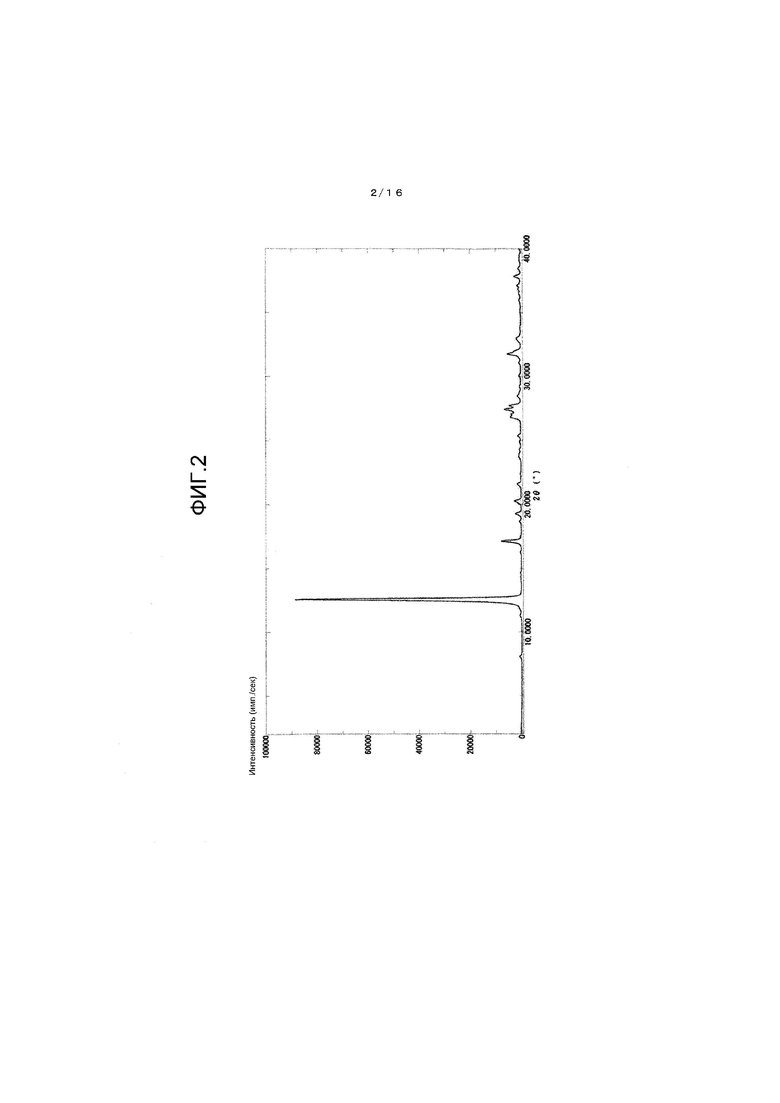
На фиг.2 представлен график для иллюстрации одного примера картины порошковой рентгеновской дифракции кристалла β-типа.
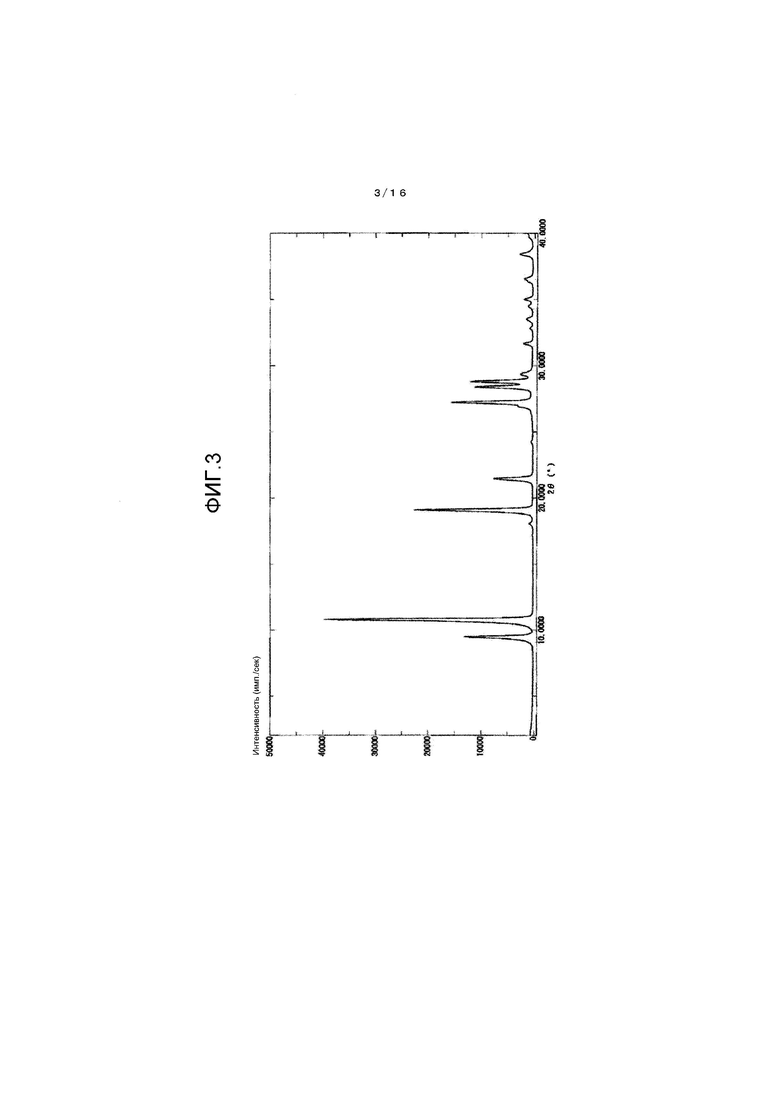
На фиг.3 представлен график для иллюстрации одного примера картины порошковой рентгеновской дифракции кристалла соединения A, который получен в сравнительном примере 1.
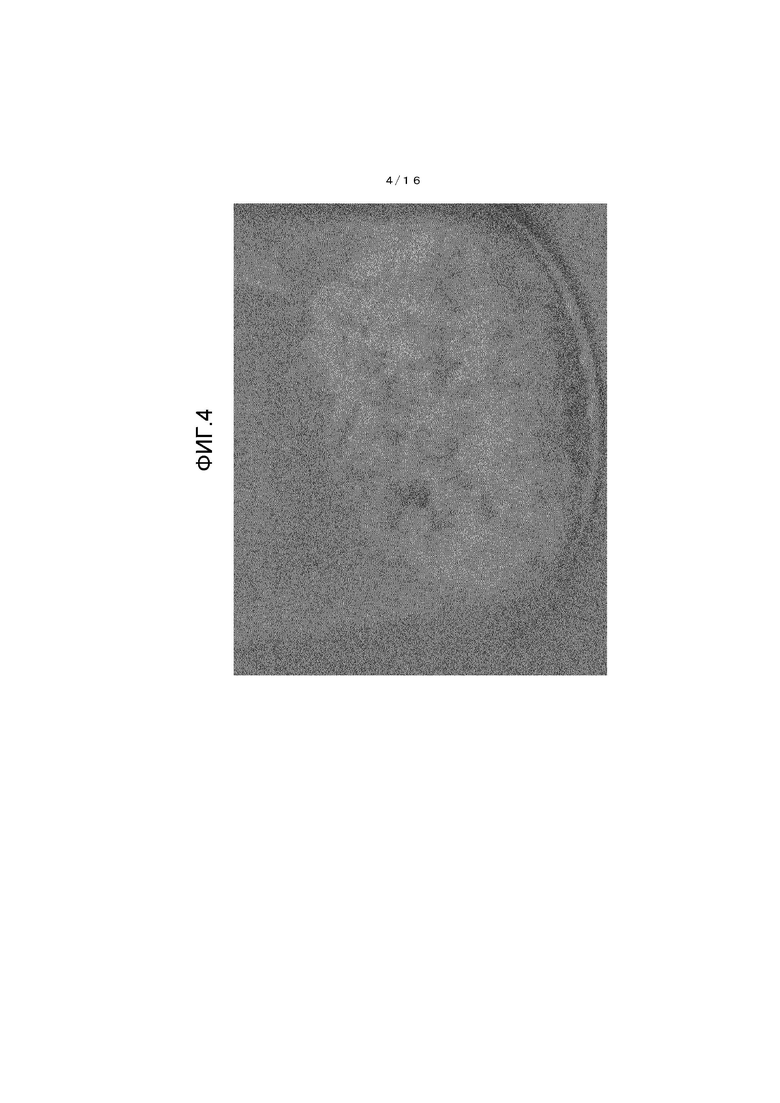
На фиг.4 представлено фотоизображение, иллюстрирующее один пример кристалла β-типа.
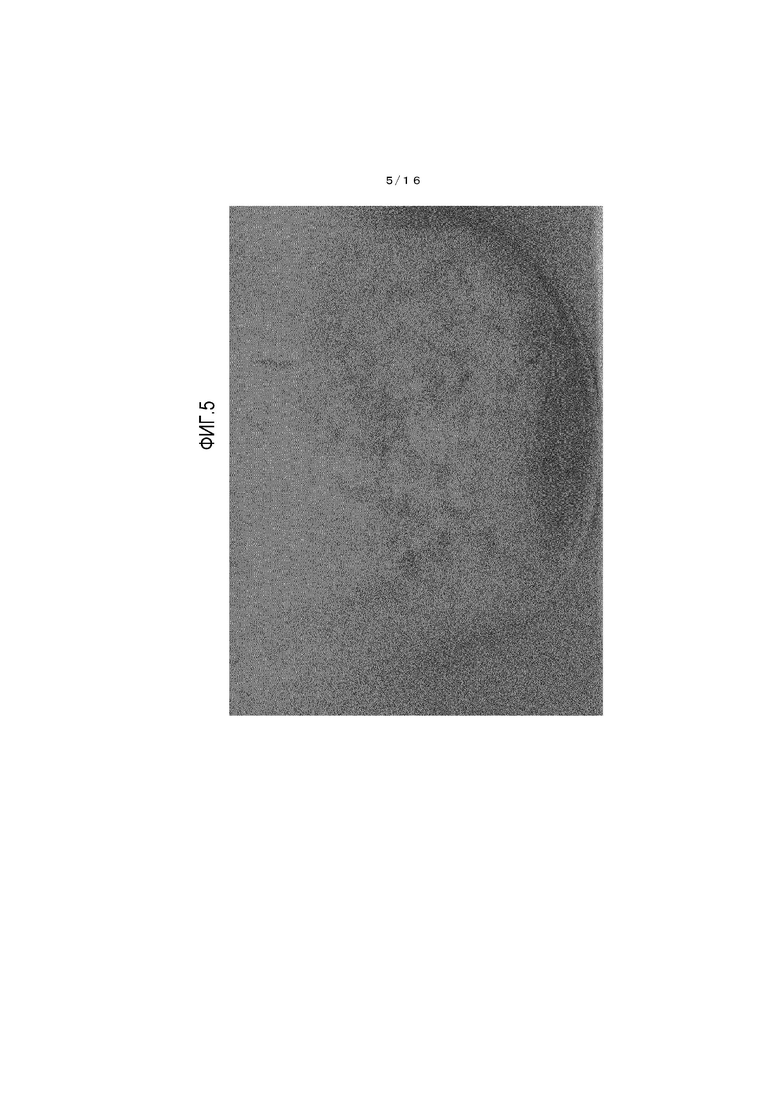
На фиг.5 представлено фотоизображение, иллюстрирующее состояние кристалла β-типа после хранения в течение одной недели в условиях температуры 60°C и относительной влажности 75%.
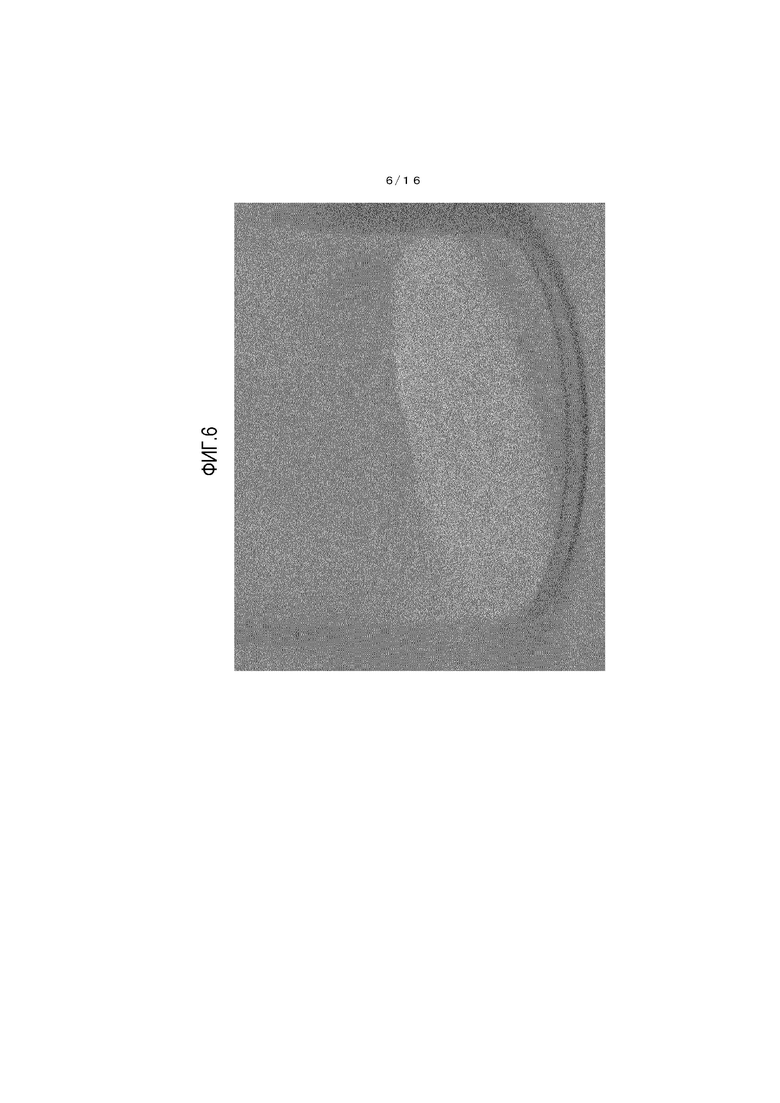
На фиг.6 представлено фотоизображение, иллюстрирующее один пример кристалла соединения A, который получен в сравнительном примере 1.
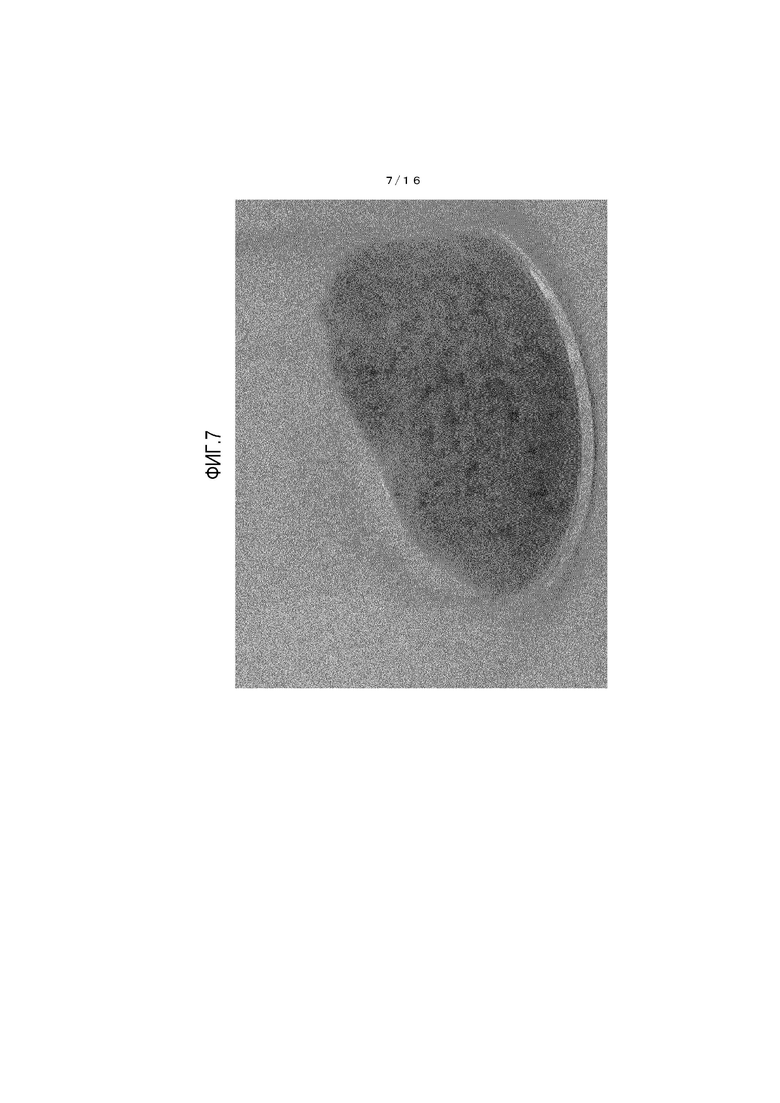
На фиг.7 представлено фотоизображение, иллюстрирующее состояние кристалла 5-гидрокси-1H-имидазол-4-карбоксамида после хранения в течение одной недели в условиях температуры 60°C и относительной влажности 75%.
На фиг.8 представлен график для иллюстрации одного примера картины порошковой рентгеновской дифракции гидрата соединения A.
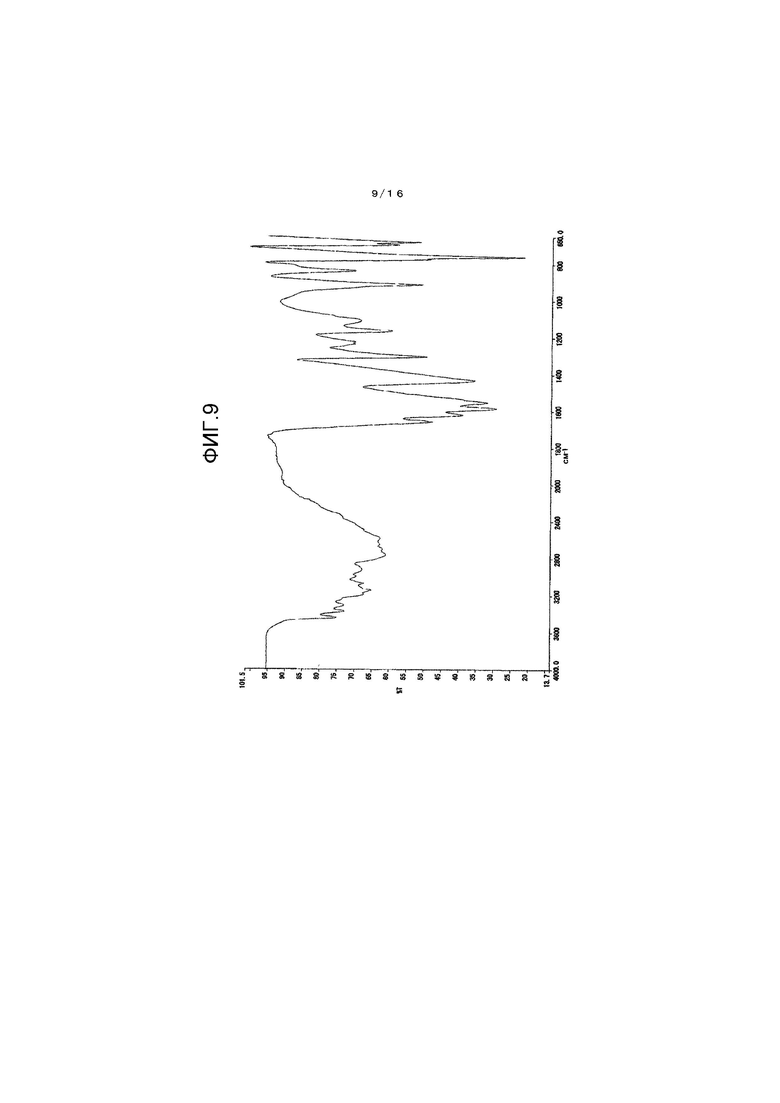
На фиг.9 представлен график для иллюстрации одного примера спектра инфракрасного поглощения (ATR-метод) гидрата соединения A.
На фиг.10 представлено фотоизображение гидрата соединения A.
На фиг.11 представлено фотоизображение, иллюстрирующее состояние гидрата соединения A после хранения в течение 2 недель в условиях температуры 60°C и относительной влажности 75%.
На фиг.12 представлено фотоизображение, иллюстрирующее состояние реакционной жидкости, когда взаимодействие по справочному примеру 2 завершено.
На фиг.13 представлено фотоизображение, иллюстрирующее состояние реакционной жидкости, когда взаимодействие по справочному примеру 3 завершено.
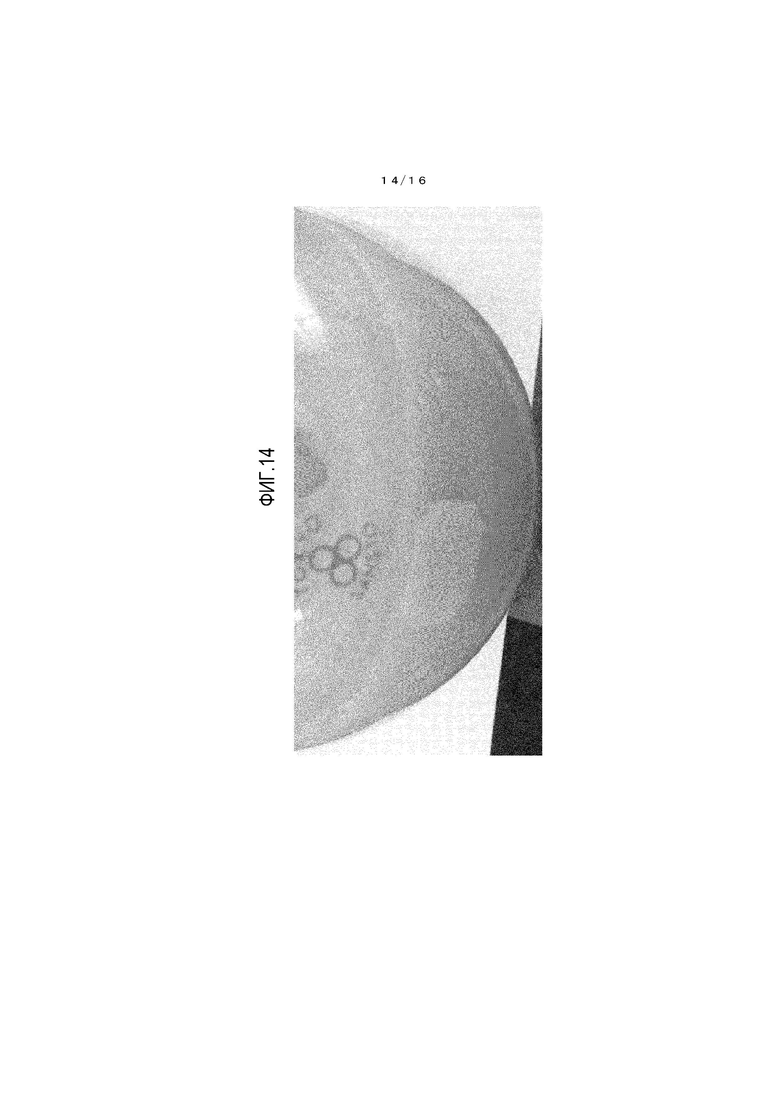
На фиг.14 представлено фотоизображение, иллюстрирующее состояние реакционной жидкости, когда взаимодействие по справочному примеру 4 завершено.
На фиг.15 представлено фотоизображение, иллюстрирующее состояние реакционной жидкости, когда взаимодействие по сравнительному примеру 2 завершено.
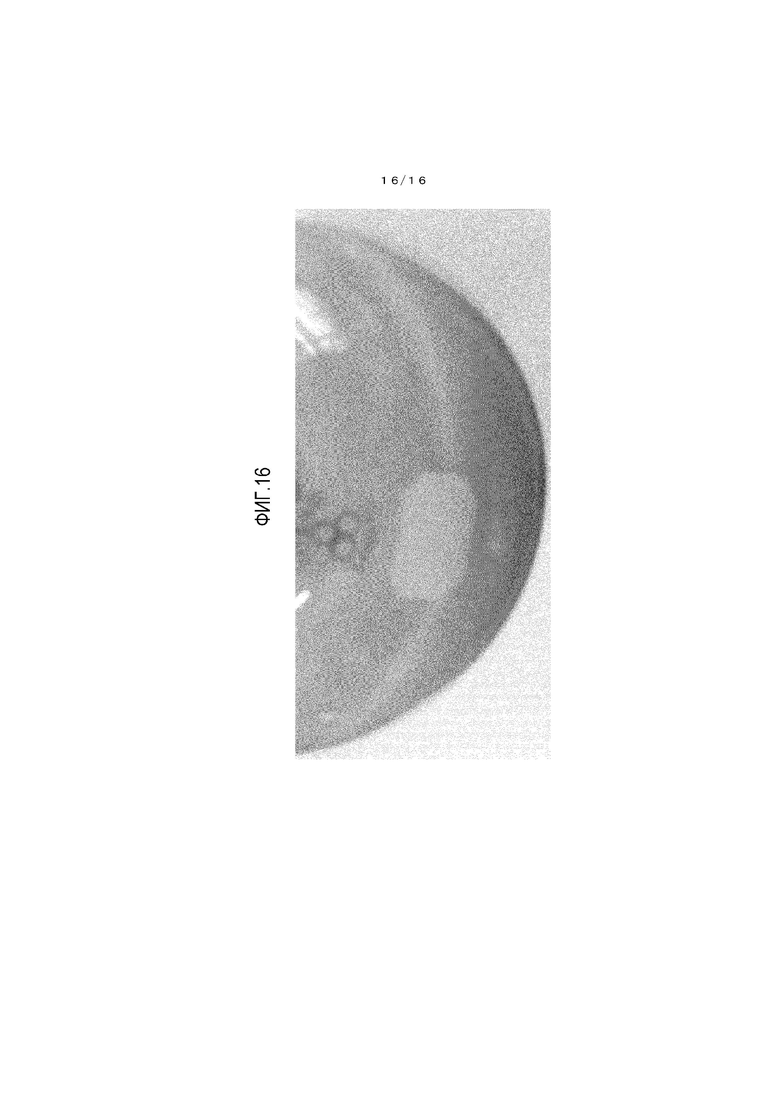
На фиг.16 представлено фотоизображение, иллюстрирующее состояние реакционной жидкости, когда взаимодействие по сравнительному примеру 3 завершено.
ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
[0041] Здесь далее изобретение объясняется более подробно. В спецификации числовые диапазоны, описанные как "от A до B", указывают диапазон, который включает каждое из числовых значений A и B как минимальную или максимальную величину. Кроме того, как здесь описано, количество каждого компонента в композиции обозначает общее количество соответствующих многочисленных материалов, которые присутствуют в композиции в случае, когда в композиции представлен один или более материалов, соответствующих каждому компоненту, если конкретно не указано иное.
[0042] Далее определены используемые здесь термины, если конкретно не указано иное.
Термин «C1-3 алкильная группа» обозначает метильную группу, этильную группу, пропильную группу или изопропильную группу.
Под галогенированными углеводородами подразумевают метиленхлорид, хлороформ или дихлорэтан.
Под спиртами подразумевают метанол, этанол, пропанол, 2-пропанол, бутанол или 2-метил-2-пропанол.
Под простыми эфирами подразумевают диэтиловый эфир, диизопропиловый эфир, диоксан, тетрагидрофуран, анизол, диметиловый эфир этиленгликоля, диметиловый эфир диэтиленгликоля или диэтиловый эфир диэтиленгликоля.
Под кетонами подразумевают ацетон, 2-бутанон или 4-метил-2-пентанон.
Под сложными эфирами подразумевают метилацетат, этилацетат, пропилацетат или бутилацетат.
Под амидами подразумевают N,N-диметилформамид, N,N-диметилацетамид или 1-метил-2-пирролидон.
Под солями щелочных металлов подразумевают соль лития, соль натрия или соль калия.
[0043] Цветовая система Манселла представляет собой цветовую систему, разработанную Альбертом Г. Манселлом. В данной системе цвет определяют с точки зрения трех признаков: тона, яркости и насыщенности. Тон (H) представляет виды цветов, и цвет определяют по цветовой системе Манселла, используя 10 тонов, включающих 5 основных тонов красный (R), желтый (Y), зеленый (G), синий (B) и фиолетовый (P) и 5 промежуточных тонов желто-красный (YR), желто-зеленый (YG), сине-зеленый (BG), сине-фиолетовый (BP) и красно-фиолетовый (RP). Кроме того, каждый тон из 10 тонов дополнительно подразделяют, применяя равноконтрастные цветовые шкалы, так что максимальная классификация соответствует 10. Тон (H) можно определить, применяя измерительное устройство, например, колориметр или дифференциальный колориметр.
[0044] <Кристалл гидрата соединения A - первый вариант осуществления>
Кристалл гидрата соединения A по настоящему изобретению характеризуется тем, что имеет дифракционные пики при углах дифракции 8,1, 12,6, 17,1, 19,3, 20,3 и 21,6°, представленных как 2θ на картине порошковой рентгеновской дифракции.
Кроме того, кристалл по настоящему изобретению имеет по меньшей мере одну из следующих характеристик: (1) он не содержит добавок, (2) его можно хранить в течение длительного периода времени, так как не происходит ухудшения качества, например, окрашивания, даже в условиях высокой температуры и высокой влажности, (3) он имеет мало примесей, (4) он легко поддается обработке, (5) его получают, используя растворитель, безопасный для человеческого организма, (6) его получают в условиях с низкими экологическими проблемами, (7) его можно получать в крупном масштабе, и он пригоден в качестве исходного лекарственного вещества для фармацевтического препарата.
[0045] Упоминаемый выше кристалл β-типа не был известен до настоящего времени и является новым кристаллом, который никогда не был описан в брошюре международной публикации № 2009/035168 или подобном. Между тем, характеристические пики при рентгеновской дифракции порошка могут отличаться в зависимости от условий измерения. Вообще, имеет место ошибка 2θ в диапазоне ±0,2°. Таким образом, выражение "угол дифракции X°, представленный как 2θ" обозначает "угол дифракции от ((X-0,2) до (X+0,2))°, представленный как 2θ".
Рентгеновскую дифракцию можно измерять на основании Японского промышленного стандарта JIS K 9131 или подобного.
[0046] Один пример результата измерений порошковой рентгеновской дифракции для кристалла β-типа проиллюстрирован на фиг.2. Также условия измерения порошковой рентгеновской дифракции описаны здесь ниже.
(Условия измерения)
Антикатод: Cu
Напряжение на лампе: 40 кВ
Ток на лампе: 40 мА
Ось сканирования: 2θ
[0047] Кроме того, кристалл β-типа характеризуется также длиной волны поглощения в инфракрасном абсорбционном спектре, определяемой в следующих условиях.
Один пример измерения результирующего спектра инфракрасного поглощения для кристалла β-типа показан на фиг.1. Между тем, инфракрасный абсорбционный спектр измеряют согласно Фармакопее Японии, общими способами тестирования и ИК абсорбционным методом затухающего полного отражения (ATR-метод).
Как проиллюстрировано на фиг.1, кристалл β-типа имеет характеристические пики при 1614 см-1, 1576 см-1 и 1559 см-1.
[0048] Далее объясняется способ получения кристалла β-типа. Кристалл β-типа можно получить, например, следующим способом.
[0049] [Способ получения]
Кристалл β-типа можно получить нагреванием и растворением соединения A в водном растворе кислоты с последующим медленным охлаждением с целью кристаллизации. Здесь можно получить соединение А способом, описанным, например, в брошюре международной публикации № 2009/035168.
[0050] Примеры кислоты включают органическую кислоту, такую как уксусная кислота и щавелевая кислота, и неорганическую кислоту, такую как соляная кислота. Кислоту можно использовать или отдельно, или в комбинации кислот двух или более типов. Предпочтительные примеры кислоты включают органическую кислоту, такую как уксусная кислота и щавелевая кислота. Уксусная кислота является более предпочтительной.
Концентрация кислоты в водном растворе кислоты не имеет особых ограничений и может быть выбрана в зависимости от типа предполагаемой к использованию кислоты. Концентрация кислоты предпочтительно составляет, например, от 1% масс. до 10% масс. и более предпочтительно от 3% масс. до 7% масс.
[0051] Температура нагревания и растворения соединения A в водном растворе кислоты не имеет особых ограничений и может быть выбрана в зависимости от типа или количества водного раствора кислоты. Например, температура может составлять от 50°C до 100°C. Она предпочтительно составляет от 75°C до 100°C и более предпочтительно от 80°C до 98°C.
Температура кристаллизации не имеет особых ограничений. Она предпочтительно составляет от 50°C до 100°C, более предпочтительно от 50°C до 80°C и еще предпочтительнее от 60°C до 80°C.
[0052] Количество водного раствора кислоты, используемое для растворения соединения A, не имеет особых ограничений. Оно предпочтительно составляет, например, от 10-кратного (об./масс.) до 50-кратного и более предпочтительно от 15-кратного (об./масс.) до 40-кратного количества соединения A.
[0053] Для упоминаемого выше способа получения в процессе кристаллизации предпочтительно присутствует соль. Выполняя медленное охлаждение в присутствии соли, можно эффективно получить кристалл с более высокой стабильностью.
Примеры соли, которую используют, когда требуется, включают соль щелочного металла карбоновой кислоты. А именно, предпочтительной является по меньшей мере одна соль, выбранная из ацетата натрия, ацетата калия, формиата натрия, формиата калия, бензоата натрия, цитрата натрия, малата натрия, фумарата натрия и сукцината натрия. Более предпочтительной является по меньшей мере одна соль, выбранная из формиата натрия, ацетата натрия, цитрата натрия, малата натрия, фумарата натрия и сукцината натрия. Еще более предпочтительной является по меньшей мере одна соль, выбранная из формиата натрия и ацетата натрия.
[0054] В упоминаемом выше способе получения время, необходимое для кристаллизации, не имеет особых ограничений. Предпочтительно оно составляет, например, от 0,5 ч до 48 ч и более предпочтительно от 0,5 ч до 6 ч.
[0055] Кроме того, предпочтительно применять затравку для кристаллизации кристалла β-типа. Таким образом, можно регулировать получение кристалла более однородного типа.
Между тем, что касается затравочного кристалла, можно использовать кристаллы, полученные при предыдущем получении, или можно заблаговременно отфильтровывать и собирать часть кристаллизованных затравочных кристаллов и использовать в качестве затравки.
[0056] Температура фильтрования и сбора кристаллизованных кристаллов β-типа, хотя и не имеет особых ограничений, предпочтительно равна температуре кристаллизации.
Кроме того, хотя атмосфера для кристаллизации не имеет особых ограничений, предпочтительно выполнять кристаллизацию в атмосфере инертного газа. Примеры атмосферы инертного газа включают атмосферу аргона и атмосферу азота.
[0057] <Фармацевтическая композиция>
Фармацевтическая композиция по настоящему изобретению содержит кристалл β-типа, и он, если необходимо, включает другие составляющие компоненты. Фармацевтическая композиция, содержащая кристалл β-типа, имеет превосходную стабильность при хранении.
[0058] В случае использования гидрата β-типа в фармацевтической композиции можно соответствующим образом смешивать вспомогательные добавки, которые обычно применяют для получения составов, например, наполнители, носители или разбавители. Их можно вводить обычным способом или перорально, или парентерально в виде таблетки, капсулы, порошка, сиропа, гранулы, пилюли, суспензии, эмульсии, жидкости, порошкового препарата, суппозитория, глазных капель, назального состава, ушных капель, пластыря, мази или раствора для инъекций. Кроме того, способ введения, дозировку и количество введений можно соответствующим образом выбирать в зависимости от возраста, массы тела и симптомов субъекта, подвергаемого лечению. В общем, взрослым можно вводить от 0,01 мг/кг до 1000 мг/кг в день, за один раз или поделив на несколько порций, пероральным или парентеральным способом (например, посредством инъекции, в виде жидких капель или ректальным способом введения).
Применимость данного изобретения объясняют, принимая во внимание данные ниже примеры.
[0059] <Кристалл гидрата соединения A - второй аспект>
Кристалл гидрата соединения A по настоящему изобретению имеет цветовое различие (ΔE), равное 6 или менее, между кристаллом до хранения и кристаллом после хранения в течение 2 недель в условиях температуры 40°C и относительной влажности 75%, и кристалл имеет содержание кислотного соединения 0,1% масс. или менее.
[0060] Кристалл гидрата соединения A имеет цветовое различие (ΔE), равное 6 или менее, между кристаллом до хранения и кристаллом после хранения в течение 2 недель в условиях температуры 40°C и относительной влажности 75%. Однако с точки зрения стабильности при хранении предпочтительным является различие 3 или менее. Как здесь описано, цветовое различие кристалла определяют отражательным методом, используя спектроколориметр.
[0061] Кристалл гидрата соединения A до хранения кристалла в условиях температуры 40°C и относительной влажности 75% в течение 2 недель предпочтительно является бесцветным, бледно-желтым или желтым, более предпочтительно бесцветным или бледно-желтым.
[0062] Кристалл гидрата соединения A до хранения кристалла в условиях температуры 40°C и относительной влажности 75% в течение 2 недель предпочтительно имеет тон (H) от 1Y до 6Y по цветовой системе Манселла.
[0063] Кроме того, содержание кислотного соединения в кристалле гидрата соединения A, предпочтительно составляет 0,1% масс. или менее. Однако с точки зрения стабильности при хранении более предпочтительно содержание 0,05% масс. или менее. Кроме того, содержание кислотного соединения в кристалле гидрата соединения A определяют обычным аналитическим способом, который выбирают соответственно в зависимости от типа кислотного соединения. Например, содержание кислотного соединения в кристалле гидрата соединения можно определить методом высокоэффективной жидкостной хроматографии (ВЭЖХ), ионной хроматографии или газовой хроматографии. Например, содержание кислотного соединения в кристалле гидрата, полученном в примере 9, определяют методом ионной хроматографии. Результат определения показывает, что содержание кислотного соединения составляет 0,05% масс. или менее (ниже предела определения).
Примеры кислотного соединения, содержащегося в кристалле гидрата соединения A, включают минеральную кислоту, сульфоновую кислоту или карбоновую кислоту, которые описаны ниже. В случае, когда в кристалле гидрата соединения A содержится кислотное соединение, предпочтительной является сульфоновая кислота или карбоновая кислота.
[0064] <Способ получения гидрата соединения A>
Первый способ получения по настоящему изобретению включает следующие стадии: взаимодействие 2-аминомалонамида с соединением, представленным следующей формулой [1], в присутствии карбоновой кислоты с получением соединения A,
[0065]
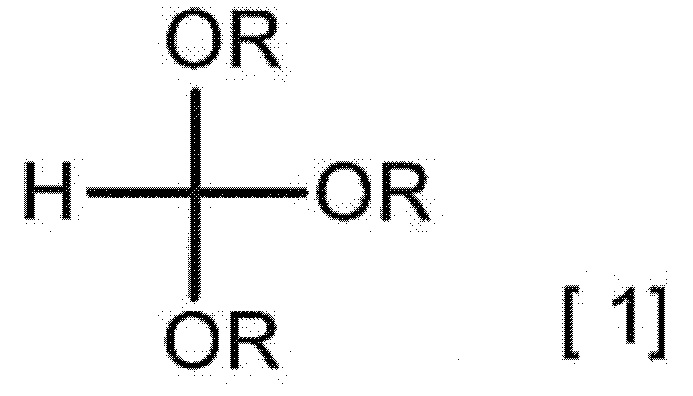
[0066] (в формуле каждый R независимо представляет C1-3 алкильную группу), взаимодействие полученного соединения A с кислотным соединением с получением соли кислоты или гидрата соли кислоты, взаимодействие полученной соли кислоты или полученного гидрата соли кислоты с солью в присутствии кислотного растворителя с получением гидрата соединения A и, если необходимо, другие стадии.
[0067] Кроме того, второй способ получения по настоящему изобретению включает стадии: взаимодействие 2-аминомалонамида с соединением, представленным следующей формулой [1], в отсутствие минеральной кислоты и в отсутствие сульфоновой кислоты с получением соединения A
[0068]
[0069] (в формуле каждый R независимо представляет C1-3 алкильную группу), взаимодействие полученного соединения A с кислотным соединением с получением соли кислоты или гидрата соли кислоты, взаимодействие полученной соли кислоты или полученного гидрата соли кислоты с солью в присутствии кислотного растворителя с получением гидрата соединения A и, если необходимо, другие стадии.
[0070] Гидрат соединения A, который получают способом получения по настоящему изобретению, имеет следующие характеристики: (1) он не содержит добавок, (2) цветовое различие между кристаллом до хранения и кристаллом после хранения мало, (3) он имеет превосходную стабильность при хранении, и (4) он имеет мало примесей. Кроме того, если отсутствует необходимость включения следовых количеств кислоты для повышения стабильности, то гидрат соединения A является высокочистым. Кроме того, если отсутствует необходимость регулирования содержания кислоты на постоянном уровне, то возможно получить большое количество гидрата соединения A с превосходной производительностью.
Способ получения гидрата соединения A по настоящему изобретению также имеет следующие характеристики: (5) не генерируется эфир бензолсульфоновой кислоты, имеющий генетическую токсичность, и (6) не требуется большой избыток триэфира ортомуравьиной кислоты.
Таким образом, способ получения по настоящему изобретению пригоден в качестве способа промышленного производства гидрата соединения A.
[0071] Кроме того, согласно другому варианту осуществления настоящего изобретения, способ получения гидрата соединения A характеризуется тем, что можно регулировать образование однородного кристалла даже при меньшей температуре.
Согласно способу получения по настоящему изобретению, становится возможным, во-первых, получать гидрат соединения A, который (1) не содержит добавок, (2) для которого цветовое различие между кристаллом до хранения и кристаллом после хранения мало, (3) который имеет превосходную стабильность при хранении, и (4) он имеет мало примесей.
Таким образом, способ получения по настоящему изобретению пригоден в качестве способа промышленного производства гидрата соединения A.
[0072] Гидрат соединения A, полученный способом по настоящему изобретению, предпочтительно является бесцветным, бледно-желтым или желтым и более предпочтительно бесцветным или бледно-желтым. [0073] Гидрат соединения A, полученный способом по настоящему изобретению, предпочтительно имеет тон (H) от 1Y до 6Y по цветовой системе Манселла.
[0074] (Первая стадия)
Соединение A можно получить способом (A), включающим взаимодействие 2-аминомалонамида с соединением, представленным формулой [1], в присутствии карбоновой кислоты, или способом (B), включающим взаимодействие 2-аминомалонамида с соединением, представленным формулой [1], в отсутствие минеральной кислоты и в отсутствие сульфоновой кислоты. В этой формуле R имеет такое же определение, как приведено выше.
[0075]
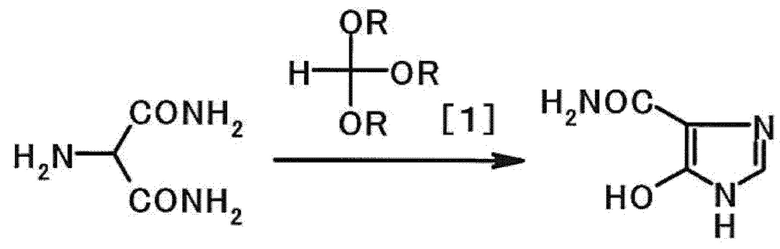
[0076] Примеры соединения, представленного формулой [1], включают триметилортоформиат и триэтилортоформиат.
[0077] Коммерчески доступное соединение, представленное формулой [1], может содержать в качестве примеси основное соединение, такое как триазин. Для такого случая предпочтительно проводить взаимодействие в присутствии карбоновой кислоты.
В случае, когда соединение, представленное формулой [1], не содержит основного соединения, такого как триазин, взаимодействие можно проводить без использования карбоновой кислоты.
[0078] Взаимодействие предпочтительно проводить в присутствии растворителя. Растворитель, который можно использовать, не имеет особых ограничений до тех пор, пока он является обычно применяемым растворителем. Примеры растворителя включают галогенированные углеводороды; спирты; простые эфиры; кетоны; сложные эфиры; амиды; ацетонитрил и диметилсульфоксид. Его можно использовать отдельно или в виде комбинации двух или более веществ. Кроме того, соединение, представленное формулой [1], можно использовать в качестве растворителя.
Предпочтительные примеры растворителя включают спирты. Более предпочтительными являются этанол и 2-пропанол. Еще более предпочтителен 2-пропанол.
Используемое количество растворителя не имеет особых ограничений. Предпочтительно оно равно 1-100-кратному (об./масс.) количеству 2-аминомалонамида. Более предпочтительно оно равно 1-30-кратному (об./масс.) количеству. Еще более предпочтительно оно равно 1-25-кратному (об./масс.) количеству.
[0079] Для первой стадии предпочтительно использовать соединение, в котором R представляет метильную группу или этильную группу. Более предпочтительно использовать соединение, в котором R представляет этильную группу.
Используемое количество соединения, представленного формулой [1], предпочтительно равно 1-10-кратному молярному количеству 2-аминомалонамида. Более предпочтительно оно равно 1-5-кратному молярному количеству. Еще более предпочтительно 2-3-кратному молярному количеству.
[0080] Примеры минеральной кислоты включают соляную кислоту, азотную кислоту, фосфорную кислоту и серную кислоту. Кроме того, примеры сульфоновой кислоты включают органическую сульфоновую кислоту, такую как метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота и толуолсульфоновая кислота.
[0081] Примеры карбоновой кислоты включают алифатические карбоновые кислоты, такие как муравьиная кислота и уксусная кислота; гидроксикислоты, такие как молочная кислота, яблочная кислота, лимонная кислота и винная кислота; ароматические карбоновые кислоты, такие как бензойная кислота и фталевая кислота; и дикарбоновые кислоты, такие как щавелевая кислота, фумаровая кислота и малеиновая кислота. Предпочтительными являются муравьиная кислота или щавелевая кислота. Более предпочтительна щавелевая кислота.
Используемое количество карбоновой кислоты предпочтительно составляет от 0,001 до 0,05 молярного количества 2-аминомалонамида. Более предпочтительно количество составляет от 0,001 до 0,01 его молярного количества. Еще более предпочтительно составляет от 0,002 до 0,01 его молярного количества.
[0082] Реакционная температура предпочтительно составляет от 0°C до 150°C, более предпочтительно от 70°C до 100°C и еще более предпочтительно от 75°C до 85°C.
Время взаимодействия предпочтительно составляет от 5 ч до 50 ч. Более предпочтительно составляет от 5 ч до 10 ч.
Кроме того, реакционная атмосфера не имеет особых ограничений, но взаимодействие предпочтительно проводить в атмосфере инертного газа. Примеры атмосферы инертного газа включают атмосферу аргона и атмосферу азота.
[0083] Соединение A, получаемое на первой стадии, после выделения можно использовать для следующей стадии. Однако предпочтительно использовать его на следующей стадии без выделения.
В случае, когда присутствует сольват, гидрат и кристаллы различных форм соединения A, получаемого на первой стадии, они также включены в изобретение.
[0084] Способ получения соединения A уже известен. Например, в JP-A № 58-24569 раскрыто, что сырой кристалл соединения A можно получить взаимодействием 2-аминомалонамида с триэтилортоформиатом в присутствии серной кислоты.
Авторы настоящего изобретения пытались получить соединение A со ссылкой на способ получения, описанный в JP-A № 58-24569. Однако реакционная жидкость окрашивается в глубокий синий цвет. Взаимодействие также выполняют, используя пара-толуолсульфоновую кислоту вместо серной кислоты. Однако реакционная жидкость также окрашивается в глубокий синий цвет.
С другой стороны, согласно первой стадии способа получения соединения A, окрашивание реакционной жидкости ингибируют.
[0085] (Вторая стадия)
При взаимодействии соединения A, полученного на первой стадии, с кислотным соединением, можно получить соль кислоты или его гидрат соли кислоты.
[0086]
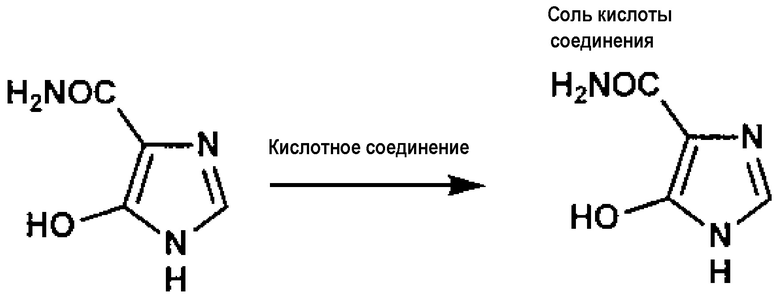
[0087] Способ взаимодействия соединения A с кислотным соединением не имеет особых ограничений и может быть подходящим образом выбран из обычно практикуемых способов. А именно, при добавлении водного раствора, содержащего кислотное соединение, к раствору или суспензии, содержащей соединение A, соединение A может взаимодействовать с кислотным соединением.
Кислотное соединение не имеет особых ограничений. Его примеры включают минеральную кислоту, такую как соляная кислота и серная кислота; органическую сульфоновую кислоту, такую как метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота и толуолсульфоновая кислота; и щавелевую кислоту. Предпочтительной является минеральная кислота. Более предпочтительной является соляная кислота.
Примеры соли кислоты включают соль с минеральной кислотой, например, соляной кислотой и серной кислотой; соль с органическими сульфоновыми кислотами, такими как метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота и толуолсульфоновая кислота; и соль со щавелевой кислотой. Предпочтительной является соль с минеральной кислотой. Более предпочтительной является солянокислая соль.
[0088] Реакционная температура и время взаимодействия для второй стадии не имеют особых ограничений и могут быть подходящим образом выбраны в зависимости от типа используемого кислотного соединения.
Также реакционная атмосфера не имеет особых ограничений, но взаимодействие предпочтительно проводить в атмосфере инертного газа. Примеры атмосферы инертного газа включают атмосферу аргона и атмосферу азота.
[0089] Соль кислоты соединения A, полученную на второй стадии, можно использовать на следующей стадии без выделения. Однако предпочтительно использовать соль на следующей стадии после выделения.
В случае, когда присутствует сольват, гидрат и кристаллы различных форм соли кислоты соединения A, получаемой на второй стадии, они также включены в изобретение.
[0090] (Третья стадия)
Взаимодействием соли кислоты или гидрата соли кислоты, полученной на второй стадии, с солью в присутствии кислотного растворителя можно получить гидрат соединения.
[0091]
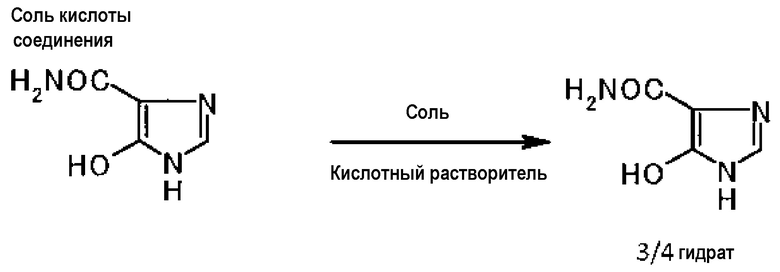
[0092] Примеры кислотного растворителя включают водный раствор минеральной кислоты. Их конкретные примеры включают соляную кислоту, серную кислоту и азотную кислоту. Предпочтительной является соляная кислота. Используя соляную кислоту, можно получить однородный кристалл гидрата соединения даже при условии меньшей температуры.
Используемое количество кислотного растворителя не имеет особых ограничений. Оно может составлять (5-50)-кратное (об./масс.) количество соли кислоты соединения A.
Концентрация кислотного растворителя не имеет особых ограничений. В случае, когда используют, например, соляную кислоту, концентрация соляной кислоты предпочтительно составляет от 0,3 моль/л до 0,8 моль/л и более предпочтительно от 0,4 моль/л до 0,5 моль/л.
[0093] Предпочтительно использовать в качестве соли соль органической кислоты. Примеры соли органической кислоты включают соль карбоновой кислоты. Предпочтительной является соль щелочного металла карбоновой кислоты. Предпочтительна соль щелочного металла карбоновой кислоты, имеющей первую константу диссоциации кислоты (pKa1) от 2 до 4. Более предпочтительна соль щелочного металла карбоновой кислоты, имеющей первую константу диссоциации кислоты (pKa1) от 3 до 4.
Примеры соли щелочного металла карбоновой кислоты включают ацетат натрия, ацетат калия, формиат лития, формиат натрия, формиат калия, бензоат натрия, цитрат натрия, малат натрия, фумарат натрия и сукцинат натрия. Предпочтительной является по меньшей мере одна соль, выбранная из формиата натрия, формиата калия, цитрата натрия, малата натрия или фумарата натрия. Более предпочтительной является по меньшей мере одна соль, выбранная из формиата натрия, малата натрия или цитрата натрия. Еще более предпочтителен формиат натрия.
Примеры соли щелочного металла карбоновой кислоты, имеющей первую константу диссоциации кислоты (pKa1) от 2 до 4, включают малат натрия, тартрат натрия, тартрат калия, гидротартрат калия, гидротартрат натрия, формиат лития, формиат натрия, формиат калия, цитрат натрия, малат натрия и фумарат натрия.
Примеры соли щелочного металла карбоновой кислоты, имеющей первую константу диссоциации кислоты (pKa1) от 3 до 4, включают формиат лития, формиат натрия, формиат калия, цитрат натрия, малат натрия и фумарат натрия. Предпочтительной является по меньшей мере одна соль, выбранная из формиата натрия, малата натрия или цитрата натрия. Более предпочтителен формиат натрия.
[0094] Используемое количество соли можно выбирать подходящим образом в зависимости от типа соли кислоты, типа соли и типа и концентрации кислотного растворителя. Например, используемое количество соли предпочтительно регулируют таким образом, что pH суспензии или раствора соединения A после добавления соли составляет от 1 до 4. Более предпочтительно регулировать используемое количество соли таким образом, чтобы pH суспензии или раствора составлял от 1,5 до 2,5.
Конкретно, в случае, когда соль соляной кислоты соединения A растворяется в от 0,4 моль/л до 0,5 моль/л соляной кислоты и взаимодействует с формиатом натрия, предпочтительное используемое количество формиата натрия равно 1,8-3,0-кратному молярному количеству солянокислой соли соединения A.
[0095] Третью стадию можно выполнять, добавляя соль к суспензии или раствору соли кислоты соединения A. Способ добавления соли к раствору соли кислоты соединения A является предпочтительным.
В частности, можно добавлять соль кислоты соединения A к кислотному растворителю с последующим нагреванием, если необходимо, получая раствор соли кислоты соединения A, и добавлять к раствору соль.
[0096] Кислоту и основание, которые составляют соль, предполагаемую к использованию на третьей стадии, можно добавлять таким образом, при котором каждую кислоту и основание добавляют отдельно в таком порядке к суспензии или раствору соли кислоты соединения A.
Например, вместо использования в качестве соли формиата натрия, после добавления муравьиной кислоты к суспензии или раствору соли кислоты соединения A можно добавить туда гидроксид натрия или подобное.
[0097] Реакционная температура на третьей стадии предпочтительно составляет от комнатной температуры до 60°C и более предпочтительно от 40°C до 50°C.
Время взаимодействия может составлять, например, от 1 мин до 24 часов.
Кроме того, реакционная атмосфера не имеет особых ограничений, но взаимодействие предпочтительно проводить в атмосфере инертного газа. Примеры атмосферы инертного газа включают атмосферу аргона и атмосферу азота.
[0098] Способ получения гидрата соединения A уже известен. Например, в брошюре международной публикации № 2009/035168 раскрыто, что гидрат соединения А можно получить взаимодействием бензолсульфоната соединения A с гидрокарбонатом натрия. Кроме того, в JP-A № 58-24569 раскрыт способ использования аммиачной воды.
Авторы настоящего изобретения пытались получить гидрат соединения A описанными выше способами. Однако получаемый гидрат соединения A был окрашен в синий цвет, если добавляемое количество гидрокарбоната натрия или аммиачной воды немного превышает эквивалентную точку. С другой стороны, гидрат соединения A, который получают, используя соль карбоновой кислоты, не окрашивается, даже если добавляемое количество сильно превышает эквивалентную точку.
[0099] В случае, когда в качестве фармацевтического препарата используют гидрат соединения A, полученный способом получения по настоящему изобретению, вспомогательное средство, которое обычно используют для приготовления состава, например, наполнитель, носитель или разбавитель, можно смешивать соответствующим образом. Состав можно вводить обычным способом или перорально, или парентерально в виде таблетки, капсулы, порошка, сиропа, гранулы, пилюли, суспензии, эмульсии, жидкости, порошкового состава, суппозитория, глазных капель, назального состава, ушных капель, пластыря, мази или раствора для инъекций. Кроме того, способ введения, дозировку и количество введений можно выбрать соответствующим образом в зависимости от возраста, массы тела и симптомов субъекта, подвергаемого лечению. Вообще, взрослым можно вводить от 0,01 мг/кг до 1000 мг/кг в день, или за один раз, или поделив на несколько порций, пероральным или парентеральным способом (например, посредством инъекции, жидких капель или ректального введения).
Применимость данного изобретения объясняют, принимая во внимание приведенные ниже примеры.
ПРИМЕРЫ
[0100] Здесь далее настоящее изобретение объясняют, принимая во внимание справочные примеры, примеры и сравнительные примеры. Однако очевидно, что изобретение не ограничено ими. Кроме того, если конкретно не указано иное, "%" означает "% масс."
[0101] Здесь далее будут описаны примеры, относящиеся к кристаллу гидрата 5-гидрокси-1H-имидазол-4-карбоксамида по настоящему изобретению.
(Пример 1)
В атмосфере азота к 5,0 г 5-гидрокси-1H-имидазол-4-карбоксамида добавляли 150 мл 5% водного раствора уксусной кислоты, и растворяли в ней при нагревании до 82°C с последующим медленным охлаждением. После подтверждения осаждения кристаллов при 50°C смесь нагревали при внутренней температуре 75°C и перемешивали при той же температуре в течение 15 мин. Раствор медленно охлаждали. Кристаллы, выпавшие в осадок при 50°C, собирали фильтрованием, затем промывали три раза 10 мл ацетона, сушили на воздухе и, таким образом, получали 1,7 г кристаллов β-типа.
На фиг.1 показан инфракрасный абсорбционный спектр (ATR-метод) полученного кристалла β-типа.
ИК (ATR) 1614 см-1, 1576 см-1, 1550 см-1.
Кроме того, картина порошковой рентгеновской дифракции полученного кристалла β-типа аналогична картине описанного ниже примера 2.
[0102] (Пример 2)
В атмосфере азота к 10,0 г 5-гидрокси-1H-имидазол-4-карбоксамида и 7,04 г щавелевой кислоты добавляли 200 мл воды и растворяли в ней при нагревании до 95°C. К смеси добавляли 5,54 г ацетата натрия, медленно охлаждали и добавляли при 85°C 0,1 г кристалла, полученного в примере 1. Выполняли перемешивание при 65°C в течение 40 мин, и выпавшие в осадок кристаллы при той же температуре собирали фильтрованием. После промывания 30 мл 5% водного раствора уксусной кислоты с последующим двукратным промыванием 30 мл ацетона и сушкой на воздухе получали 4,65 г кристалла β-типа.
Картина порошковой рентгеновской дифракции полученного кристалла β-типа показана на фиг.2 и в таблице 1.
Кроме того, инфракрасный абсорбционный спектр (ATR-метод) аналогичен спектру примера 1.
[0103]
[0104] (Пример 3)
В атмосфере азота к 5,0 г 5-гидрокси-1H-имидазол-4-карбоксамида добавляли 150 мл 5% водного раствора уксусной кислоты и растворяли в нем при нагревании до 85°C. Затем смесь медленно охлаждали до начала осаждения кристаллов. После осаждения кристаллов раствор нагревали до 83°C и затем снова охлаждали до температуры раствора 50°C. Выпавшие в осадок кристаллы собирали фильтрованием и затем промывали три раза 5 мл ацетона с последующей сушкой на воздухе и получением 2,3 г кристалла β-типа.
Картина порошковой рентгеновской дифракции полученного кристалла β-типа аналогична картине из примера 2, и инфракрасный абсорбционный спектр (ATR-метод) аналогичен спектру из примера 1.
[0105] (Сравнительный пример 1)
Что касается способа, описанного в примере 6 брошюры международной публикации № 2009/035168, получали 5-гидрокси-1H-имидазол-4-карбоксамид в виде бледно-желтого порошка.
По результатам анализа методом высокоэффективной жидкостной хроматографии полученный 5-гидрокси-1H-имидазол-4-карбоксамид содержит бензойную кислоту в количестве примерно 0,15%.
Картина порошковой рентгеновской дифракции показана на фиг.3 и в таблице 2.
[0106]
[0107] (Пример исследования 1) Тест на стабильность при хранении в условиях высокой температуры и высокой влажности
В качестве испытываемого материала выбирали кристаллы, полученные в примере 3 и сравнительном примере 1.
Каждый из испытываемых материалов (около 0,2 г) добавляли в стеклянную пробирку и хранили в течение одной недели в условиях температуры 60°C и относительной влажности 75%.
Состояние кристалла из примера 3 до начала теста показано на фиг.4, и состояние кристалла из примера 3 по завершении теста показано на фиг.5. Также состояние кристалла из сравнительного примера 1 до начала теста показано на фиг.6, и состояние кристалла из сравнительного примера 1 по завершении теста показано на фиг.7.
[0108] Обнаружено, что кристалл β-типа, полученный из примера 3, имеет очевидно меньшее окрашивание и лучшую стабильность при хранении по сравнению с соединением из сравнительного примера 1.
[0109] Здесь далее описаны примеры, касающиеся получения 5-гидрокси-1H-имидазол-4-карбоксамид · 3/4 гидрата по настоящему изобретению.
[0110] Условия измерения порошковой рентгеновской дифракции:
Антикатод: Cu
Напряжение на трубке: 40 кВ
Ток в трубке: 40 мА
Ось сканирования: 2θ
Также характеристические пики на картине порошковой рентгеновской дифракции могут меняться в зависимости от условий определения. Вообще, имеет место ошибка 2θ в диапазоне ±0,2°. Таким образом, выражение "угол дифракции X°, представленный как 2θ" обозначает "угол дифракции от (X-0,2)° до (X+0,2)°, представленный как 2θ".
[0111] Использовали следующие реагенты.
2-Аминомалонамид: Tateyama Kasei (партия № 091026)
Триэтилортоформиат: NIPPOH CHEMICALS CO., LTD. (партия № OJ1401, чистота: 99,5%, при содержании в качестве примеси основного соединения, такого как триазин) (справочные примеры 1, 2 и 4; примеры 9 и 10 и сравнительные примеры 2, 3, 6 и 7).
Триэтилортоформиат: Wako Pure Chemical Industries, Ltd. (партия № CDM1714) (справочный пример 3 и пример 11).
[0112] Справочный пример 1
В атмосфере азота добавляли 10 г 2-аминомалонамида и 19,7 мг муравьиной кислоты к 200 мл 2-пропанола. После нагревания до 80°C к смеси добавляли по каплям 35,4 мл триэтилортоформиата в течение 5 мин. Затем реакционную смесь перемешивали в течение 8 ч при 80°C. По завершении взаимодействия реакционная жидкость имеет бледно-синий цвет. Далее после охлаждения до 56°C к реакционной смеси добавляли 10 мл воды, а затем 8 мл концентрированной соляной кислоты. После охлаждения водой кристаллы собирали фильтрованием и затем промывали 40 мл ацетона, получая 16 г дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида в виде бледных желтовато-зеленых кристаллов.
[0113] Справочный пример 2
В атмосфере азота добавляли 10 г 2-аминомалонамида и 38,4 мг щавелевой кислоты к 200 мл 2-пропанола. После нагревания до 80°C к смеси добавляли по каплям 35,4 мл триэтилортоформиата в течение 5 мин. Затем реакционную смесь перемешивали в течение 8 ч при 80°C. По завершении взаимодействия реакционная жидкость имеет бледно-желтый цвет. Далее после охлаждения до 53°C добавляли к реакционной смеси 10 мл воды, а затем 8 мл концентрированной соляной кислоты. После охлаждения водой кристаллы собирали фильтрованием и затем промывали 40 мл ацетона, получая 16 г дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида в виде бледно-желтых кристаллов.
[0114] Справочный пример 3
В атмосфере азота добавляли 10 г 2-аминомалонамида и 35,4 мл триэтилортоформиата к 200 мл 2-пропанола. После нагревания до 80°C реакционную смесь перемешивали в течение 8 ч при той же температуре. По завершении взаимодействия реакционная жидкость имеет бледно-желтый цвет. Далее после охлаждения до 57°C добавляли к реакционной смеси 10 мл воды, а затем 8 мл концентрированной соляной кислоты. После охлаждения водой кристаллы собирали фильтрованием и затем промывали 40 мл ацетона, получая 16 г дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида в виде бледно-желтых кристаллов.
[0115] Справочный пример 4
В атмосфере азота добавляли 10 г 2-аминомалонамида и 35,4 мл триэтилортоформиата к 200 мл 2-пропанола. После нагревания до 80°C реакционную смесь перемешивали в течение 13 ч при той же температуре. По завершении взаимодействия реакционная жидкость имеет бледно-синий цвет. Далее реакционную смесь охлаждали до 58°C, добавляли 10 мл воды, а затем 8 мл концентрированной соляной кислоты. После охлаждения реакционной смеси до 5°C кристаллы собирали фильтрованием и затем промывали 40 мл ацетона, получая 16 г дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида в виде зеленых кристаллов.
[0116] Пример 4
В атмосфере азота 20,0 г дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида, полученного способом по справочному примеру 2, добавляли к 240 мл соляной кислоты (0,45 моль/л) и растворяли в ней при нагревании до 50°C. При 50°C добавляли туда по каплям 40 мл водного раствора, содержащего 14,3 г формиата натрия, в течение 35 мин. Реакционную смесь охлаждали и перемешивали в течение 90 мин при внутренней температуре 5°C. Кристаллы собирали фильтрованием и затем промывали жидкой смесью, содержащей 20 мл ацетона и 40 мл воды, а затем 60 мл ацетона, получая 12,6 г 5-гидрокси-1H-имидазол-4-карбоксамид·3/4 гидрата в виде бледно-желтых кристаллов.
Содержание воды: 8,6% (метод Карла Фишера)
ИК (ATR) 1655, 1619, 1584, 1551 см-1
Картина порошковой рентгеновской дифракции показана на фиг.8 и в таблице 3, тогда как инфракрасный абсорбционный спектр (ATR-метод) показан на фиг.9.
[0117]
[0118] Пример 5
В атмосфере азота 20,0 г дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида, полученного способом по справочному примеру 2, добавляли к 240 мл 0,45 моль/л соляной кислоты и растворяли в ней при нагревании до 50°C. При 50°C добавляли туда по каплям 40 мл водного раствора, содержащего 14,3 г формиата натрия, в течение 33 мин. Реакционную смесь охлаждали. Кристаллы собирали фильтрованием и далее промывали жидкой смесью, содержащей 20 мл ацетона и 40 мл воды, а затем 60 мл ацетона, получая 12,8 г 5-гидрокси-1H-имидазол-4-карбоксамид·3/4 гидрата в виде бледно-желтых кристаллов. Инфракрасный абсорбционный спектр (ATR-метод) соответствует спектру из примера 4.
[0119] Пример 6
В атмосфере азота 1,5 г дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида, полученного способом по справочному примеру 2, добавляли к 18 мл 0,45 моль/л соляной кислоты и растворяли в ней при нагревании до 50°C. При 50°C добавляли туда по каплям 6 мл водного раствора, содержащего 1,9 г формиата натрия. pH реакционной смеси равен 3,1. Реакционную смесь охлаждали до комнатной температуры. Кристаллы собирали фильтрованием и далее промывали жидкой смесью, содержащей 1,5 мл ацетона и 3,0 мл воды, а затем 4,5 мл ацетона, получая 0,90 г 5-гидрокси-1H-имидазол-4-карбоксамид·3/4 гидрата в виде бледно-желтых кристаллов. Инфракрасный абсорбционный спектр (ATR-метод) соответствует спектру из примера 4.
[0120] Пример 7
В атмосфере азота 10,0 г дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида, полученного способом по справочному примеру 2, добавляли к 120 мл 0,45 моль/л соляной кислоты и растворяли в ней при нагревании до 40-50°C. При 50°C добавляли туда по каплям 25 мл водного раствора, содержащего 10,3 г ацетата натрия. pH реакционной смеси равен 4,0. По завершении капельного добавления реакционный раствор охлаждали до комнатной температуры и перемешивали в течение 1 ч. Кристаллы собирали фильтрованием и далее промывали жидкой смесью, содержащей 10 мл ацетона и 20 мл воды, а затем 30 мл ацетона с получением 6,1 г 5-гидрокси-1H-имидазол-4-карбоксамид·3/4 гидрата в виде серых кристаллов. Инфракрасный абсорбционный спектр (ATR-метод) соответствует спектру из примера 4.
[0121] Пример 8
В атмосфере азота 1,5 г дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида, полученного способом по справочному примеру 2, добавляли к 18 мл 0,45 моль/л соляной кислоты и растворяли в ней при нагревании до 50°C. При 50°C добавляли туда по каплям 6 мл водного раствора, содержащего 3,5 г малата натрия. pH реакционной смеси равен 2,6. Реакционную смесь охлаждали до комнатной температуры. Кристаллы собирали фильтрованием и далее промывали жидкой смесью, содержащей 1,5 мл ацетона и 3,0 мл воды, а затем 4,5 мл ацетона, получая 0,89 г 5-гидрокси-1H-имидазол-4-карбоксамид·3/4 гидрата в виде бледно-желтых кристаллов. Инфракрасный абсорбционный спектр (ATR-метод) соответствует спектру из примера 4.
[0122] Пример 9
(1) В атмосфере азота добавляли 30 г 2-аминомалонамида и 115 мг щавелевой кислоты к 600 мл 2-пропанола. После нагревания до 82°C добавляли к смеси по каплям 106 мл триэтилортоформиата в течение 10 мин. Затем реакционную смесь перемешивали в течение 7 ч и 30 мин при 84°C. Далее после охлаждения до 57°C добавляли к реакционной смеси 30 мл воды, а затем 24 мл концентрированной соляной кислоты. После охлаждения реакционной смеси до 5°C кристаллы собирали фильтрованием и затем промывали 120 мл ацетона, получая 49 г дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида в виде бледно-желтых кристаллов.
(2) В атмосфере азота добавляли 20,0 г полученного таким образом дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида к 240 мл 0,45 моль/л соляной кислоты и растворяли в ней при нагревании до 50°C. После этого добавляли туда по каплям в течение 33 мин 40 мл водного раствора, содержащего 14,3 г формиата натрия. Реакционную смесь охлаждали до 5°C. Кристаллы собирали фильтрованием и затем промывали жидкой смесью, содержащей 20 мл ацетона и 40 мл воды, а затем 60 мл ацетона, получая 12,8 г 5-гидрокси-1H-имидазол-4-карбоксамид·3/4 гидрата в виде бледно-желтых кристаллов.
Инфракрасный абсорбционный спектр (ATR-метод) соответствует спектру из примера 4.
[0123] Пример 10
(1) В атмосфере азота добавляли 5,00 г 2-аминомалонамида и 20 мг муравьиной кислоты к 80 мл 2-пропанола. После нагревания до 81°C добавляли к смеси в течение 14 мин 17,7 мл триэтилортоформиата. Затем реакционную смесь перемешивали в течение 6 ч и 33 мин при 83°C. Далее после охлаждения до 58°C к реакционной смеси добавляли по каплям 5 мл воды, а затем 4 мл концентрированной соляной кислоты. После охлаждения смеси до 20°C кристаллы собирали фильтрованием и затем промывали 20 мл ацетона, получая 8,05 г дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида в виде желтых кристаллов.
(2) В атмосфере азота добавляли 2,00 г полученного таким образом дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида к 22 мл 0,45 моль/л соляной кислоты и растворяли в ней при нагревании до 46-48°C. После нагревания до 65°C добавляли 2 мл 0,45 моль/л соляной кислоты. Раствор охлаждали до 50°C и добавляли туда по каплям водный раствор, приготовленный из 1,43 г формиата натрия и 4 мл воды. Реакционную смесь охлаждали до 5°C. Кристаллы собирали фильтрованием и затем промывали жидкой смесью, содержащей 2 мл ацетона и 4 мл воды, а затем 6 мл ацетона. После сушки при пониженном давлении получали 1,23 г 5-гидрокси-1H-имидазол-4-карбоксамид·3/4 гидрата в виде коричневых кристаллов. Инфракрасный абсорбционный спектр (ATR-метод) соответствует спектру из примера 4.
[0124] Пример 11
(1) В атмосфере азота добавляли 5,00 г 2-аминомалонамида к 100 мл 2-пропанола. После нагревания до 80°C добавляли к смеси по каплям 17,7 мл триэтилортоформиата в течение 30 мин. Затем реакционную смесь перемешивали в течение 7 ч и 50 мин при 83-84°C. Затем после охлаждения до 50-60°C добавляли к реакционной смеси водный раствор, приготовленный из 4 мл концентрированной соляной кислоты и 5 мл воды. После охлаждения смеси до 20-30°C кристалл собирали фильтрованием и затем промывали 25 мл ацетона, получая 8,04 г дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида в виде бледно-желтых кристаллов.
(2) В атмосфере азота добавляли 5,00 г полученного таким образом дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида к 50 мл 0,5 моль/л соляной кислоты и растворяли в ней при нагревании до 48-49°C. К полученному раствору добавляли по каплям в течение 18 мин водный раствор, приготовленный из 4,11 г ацетата натрия и 10 мл воды. Реакционную смесь охлаждали, кристаллы собирали фильтрованием и затем промывали жидкой смесью, содержащей 5 мл ацетона и 10 мл воды, а затем 15 мл ацетона. После сушки при пониженном давлении получали 3,21 г 5-гидрокси-1H-имидазол-4-карбоксамид·3/4 гидрата в виде бледно-желтых кристаллов. Инфракрасный абсорбционный спектр (ATR-метод) соответствует спектру из примера 4.
[0125] Пример 12
(1) В атмосфере азота добавляли 400 г дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида, полученного способом по примеру 9(1), к 4,40 л 0,45 моль/л соляной кислоты. После растворения при нагревании до 57°C добавляли туда 20,0 г активированного угля (TOKUSEI SHIRASAGI, производства Japan EnviroChemicals, Ltd.) и перемешивали в течение 45 мин. Активированный уголь удаляли фильтрованием и промывали 400 мл 0,45 моль/л соляной кислоты. Промывочную жидкость объединяли с фильтратом. Фильтрат охлаждали до 15°C и перемешивали в течение 1 ч после добавления 400 мл концентрированной соляной кислоты. Выпавшие в осадок кристаллы собирали фильтрованием и далее промывали 1,6 л 2,4 моль/л соляной кислоты, а затем 1,60 л ацетона. Выполняя сушку при пониженном давлении, получали 364 г дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида в виде бледно-желтых кристаллов.
(2) В атмосфере азота добавляли 320 г полученного таким образом дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида к 3,52 л 0,45 моль/л соляной кислоты и растворяли в ней при нагревании до 57°C. Реакционный раствор фильтровали, и остаток промывали 320 мл соляной кислоты (0,45 моль/л). Промывную жидкость объединяли с реакционным раствором и доводили до 50°C. После этого добавляли туда по каплям в течение 66 мин водный раствор, приготовленный из 229 г формиата натрия и 1,14 л воды, при 50°C. Реакционную смесь охлаждали до температуры 10°C или ниже и оставляли на ночь. Выпавшие в осадок кристаллы собирали фильтрованием и далее промывали жидкой смесью, содержащей 320 мл ацетона и 640 мл воды, а затем 960 мл ацетона. После сушки при пониженном давлении получали 198 г 5-гидрокси-1H-имидазол-4-карбоксамид·3/4 гидрата в виде бледно-желтых кристаллов. Инфракрасный абсорбционный спектр (ATR-метод) соответствует спектру из примера 4.
[0126] Пример 13
(1) В атмосфере азота добавляли 7,00 г дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида, полученного способом по примеру 10(1), к 77 мл 0,45 моль/л соляной кислоты. После растворения при нагревании до 60°C добавляли туда 350 мг активированного угля (TOKUSEI SHIRASAGI, производства Japan EnviroChemicals, Ltd.) и перемешивали в течение 30 мин при 60°C. Активированный уголь удаляли фильтрованием и промывали 7 мл 0,45 моль/л соляной кислоты. Промывную жидкость объединяли с фильтратом. Фильтрат охлаждали до 5-10°C и перемешивали при температуре от 5 до 10°C в течение 1 ч после добавления 7 мл концентрированной соляной кислоты. Выпавшие в осадок кристаллы собирали фильтрованием и далее промывали 28 мл 1,5 моль/л соляной кислоты, а затем 28 мл ацетона. Выполняя сушку при пониженном давлении, получали 6,44 г дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида в виде бледно-желтых кристаллов.
(2) В атмосфере азота добавляли 5,00 г полученного таким образом дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида к 60 мл 0,45 моль/л соляной кислоты и растворяли в ней при нагревании до 50°C. После этого добавляли туда по каплям водный раствор, приготовленный из 3,58 г формиата натрия и 20 мл воды. Реакционную смесь охлаждали до 5°C на бане со льдом и перемешивали в течение 30 мин. Выпавшие в осадок кристаллы собирали фильтрованием и далее промывали жидкой смесью, содержащей 5 мл ацетона и 10 мл воды, а затем 15 мл ацетона. После сушки при пониженном давлении получали 3,17 г 5-гидрокси-1H-имидазол-4-карбоксамид·3/4 гидрата в виде бледно-желтых кристаллов. Инфракрасный абсорбционный спектр (ATR-метод) соответствует спектру из примера 4.
[0127] Пример 14
(1) В атмосфере азота добавляли 7,00 г дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида, полученного способом по справочному примеру 3, к 77 мл 0,45 моль/л соляной кислоты. После растворения при нагревании до 60°C добавляли туда 350 мг активированного угля (TOKUSEI SHIRASAGI, производства Japan EnviroChemicals, Ltd.) и перемешивали в течение 30 мин при 60°C. Активированный уголь удаляли фильтрованием и промывали 7 мл 0,45 моль/л соляной кислоты. Промывную жидкость объединяли с фильтратом. Фильтрат охлаждали до 5-10°C и перемешивали при температуре от 5 до 10°C в течение 1 ч после добавления 7 мл концентрированной соляной кислоты. Выпавшие в осадок кристаллы собирали фильтрованием и далее промывали 28 мл 1,5 моль/л соляной кислоты, а затем 28 мл ацетона. Выполняя сушку при пониженном давлении, получали 6,44 г дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида в виде бледно-желтых кристаллов.
(2) В атмосфере азота добавляли 5,00 г полученного таким образом дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида к 60 мл 0,45 моль/л соляной кислоты и растворяли в ней при нагревании до 50°C. После этого добавляли туда по каплям водный раствор, приготовленный из 3,58 г формиата натрия и 20 мл воды. Реакционную смесь охлаждали до 5°C и перемешивали в течение 30 мин. Выпавшие в осадок кристаллы собирали фильтрованием и далее промывали жидкой смесью, содержащей 5 мл ацетона и 10 мл воды, а затем 15 мл ацетона. После сушки при пониженном давлении получали 3,22 г 5-гидрокси-1H-имидазол-4-карбоксамид·3/4 гидрата в виде бледно-желтых кристаллов. Инфракрасный абсорбционный спектр (ATR-метод) соответствует спектру из примера 4.
[0128] Сравнительный пример 2
В атмосфере азота добавляли 10,0 г 2-аминомалонамида и 81,2 мг моногидрата пара-толуолсульфоновой кислоты к 200 мл 2-пропанола. После нагревания до 82°C добавляли к смеси по каплям 35,4 мл триэтилортоформиата в течение 5 мин. Затем реакционную смесь перемешивали в течение 3 ч при 80°C. По завершении взаимодействия реакционная жидкость имеет глубокий синий цвет. Далее после охлаждения до 57°C добавляли к реакционной смеси 10 мл воды, а затем 8 мл концентрированной соляной кислоты. После охлаждения реакционной смеси до 5°C кристаллы собирали фильтрованием и затем промывали 40 мл ацетона, получая 15,6 г дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида в виде зеленых кристаллов.
[0129] Сравнительный пример 3
В атмосфере азота добавляли 10,0 г 2-аминомалонамида и 44 мг серной кислоты к 200 мл 2-пропанола. После нагревания до 80°C добавляли к смеси по каплям 35,4 мл триэтилортоформиата в течение 10 мин. Затем реакционную смесь перемешивали в течение 7 ч при 80°C. По завершении взаимодействия реакционная жидкость имеет глубокий синий цвет. Далее после охлаждения до 58°C добавляли к реакционной смеси 10 мл воды, а затем 8 мл концентрированной соляной кислоты. После охлаждения реакционной смеси до 5°C кристаллы собирали фильтрованием и затем промывали 40 мл ацетона, получая 15,6 г дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида в виде зеленых кристаллов.
[0130] Сравнительный пример 4
(1) Добавляли 100 г 5-гидрокси-1H-имидазол-4-карбоксамида, полученного со ссылкой на способ, описанный в примере 9 патентного документа 1 (международная публикация № 2009/035168), к 1456 г 7% соляной кислоты. После растворения при нагревании до 75°C добавляли туда 4,6 г активированного угля (TOKUSEI SHIRASAGI, производства Japan EnviroChemicals, Ltd.) и 14 г 7% соляной кислоты и перемешивали в течение 10 мин при 75°C. Активированный уголь удаляли фильтрованием и промывали 211 г 7% соляной кислоты. Промывную жидкость объединяли с фильтратом. Фильтрат охлаждали до 20-25°C и перемешивали в течение 1 ч. Выпавшие в осадок кристаллы собирали фильтрованием и затем дважды промывали 314 мл 2-пропанола. Выполняя сушку при пониженном давлении, получали 133 г дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида в виде бесцветных кристаллов.
(2) В атмосфере азота добавляли 1,5 г полученного таким образом дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида к 18 мл 0,45 моль/л соляной кислоты и растворяли в ней при нагревании до 40-50°C. К данному раствору добавляли по каплям в течение 10 мин 6 мл водного раствора, содержащего 0,63 г гидроксида натрия. pH реакционной смеси равен 6,6. По завершении капельного добавления реакционный раствор охлаждали до комнатной температуры и перемешивали в течение 1 ч. Кристаллы собирали фильтрованием и далее промывали жидкой смесью, содержащей 1,5 мл ацетона и 3,0 мл воды, а затем 4,5 мл ацетона, получая 0,52 г 5-гидрокси-1H-имидазол-4-карбоксамид·3/4 гидрата в виде синих кристаллов.
[0131] Сравнительный пример 5
В атмосфере азота добавляли 5,0 г дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида, полученного способом по примеру сравнения 4(1), к 60 мл 0,45 моль/л соляной кислоты и растворяли в ней при нагревании до 40-50°C. К данному раствору добавляли по каплям в течение 10 мин 3,9 мл 25% аммиачной воды. pH реакционной смеси равен 6,6. По завершении капельного добавления реакционный раствор охлаждали до комнатной температуры и перемешивали в течение 1 ч. Кристаллы собирали фильтрованием и далее промывали жидкой смесью, содержащей 5 мл ацетона и 10 мл воды, а затем 15 мл ацетона, получая 0,54 г 5-гидрокси-1H-имидазол-4-карбоксамид·3/4 гидрата в виде синих кристаллов.
[0132] Сравнительный пример 6
Со ссылкой на способ, описанный в примере 9 патентного документа 1 (международная публикация № 2009/035168), получали 5-гидрокси-1H-имидазол-4-карбоксамид в виде бледно-желтого твердого вещества.
По результатам 1H-ЯМР анализа обнаружено, что полученный 5-гидрокси-1H-имидазол-4-карбоксамид содержит примерно 0,15% бензойной кислоты.
[0133] Сравнительный пример 7
(1) В атмосфере азота добавляли 20 г 2-аминомалонамида и 325 мг моногидрата пара-толуолсульфоновой кислоты к 400 мл 2-пропанола. После нагревания до 82°C добавляли к смеси 56,7 мл триэтилортоформиата, поделив на порции, в течение 4 ч и 8 мин. Затем реакционную смесь перемешивали в течение 3 ч и 21 мин при 79°C. Далее после охлаждения до 51°C к реакционной смеси добавляли по каплям 20 мл воды, а затем 15,7 мл концентрированной соляной кислоты. После охлаждения реакционной смеси до комнатной температуры кристаллы собирали фильтрованием и затем промывали 100 мл ацетона, получая 30,56 г дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида в виде серых кристаллов.
(2) В атмосфере азота добавляли 1 г полученного таким образом дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида к 11 мл 0,45 моль/л соляной кислоты и растворяли в ней при нагревании до 46-48°C. После нагревания до 65°C добавляли туда 1 мл 0,45 моль/л соляной кислоты. После охлаждения полученного раствора до 50°C добавляли туда по каплям водный раствор, содержащий 0,72 г формиата натрия и 2 мл воды. Реакционную смесь охлаждали, кристаллы собирали фильтрованием и далее промывали жидкой смесью, содержащей 1 мл ацетона и 2 мл воды, а затем 3 мл ацетона. После сушки при пониженном давлении получали 0,63 г 5-гидрокси-1H-имидазол-4-карбоксамид·3/4 гидрата в виде коричневых кристаллов. Инфракрасный абсорбционный спектр (ATR-метод) соответствует спектру из примера 4.
[0134] Сравнительный пример 8
(1) В атмосфере азота добавляли 7,00 г дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида, полученного способом по примеру сравнения 7(1), к 77 мл 0,45 моль/л соляной кислоты. После растворения в ней при нагревании до 60°C добавляли туда 350 мг активированного угля (TOKUSEI SHIRASAGI, производства Japan EnviroChemicals. Ltd.) и перемешивали в течение 30 мин при 60°C. Активированный уголь удаляли фильтрованием и промывали 7 мл 0,45 моль/л соляной кислоты. Промывную жидкость объединяли с фильтратом. Фильтрат охлаждали до 5-10°C и перемешивали при 5-10°C в течение 1 ч после добавления 7 мл концентрированной соляной кислоты. Выпавшие в осадок кристаллы собирали фильтрованием и далее промывали 28 мл 1,5 моль/л соляной кислоты, а затем 28 мл ацетона. Выполняя сушку при пониженном давлении, получали 6,44 г дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида в виде бледно-желтых кристаллов.
(2) В атмосфере азота добавляли 5,00 г полученного таким образом дигидрата солянокислой соли 5-гидрокси-1H-имидазол-4-карбоксамида к 60 мл 0,45 моль/л соляной кислоты и растворяли в ней при нагревании до 50°C. После этого добавляли туда по каплям водный раствор, приготовленный из 3,58 г формиата натрия и 20 мл воды. Реакционную смесь охлаждали до 5°C и перемешивали в течение 30 мин. Выпавшие в осадок кристаллы собирали фильтрованием и далее промывали жидкой смесью, содержащей 5 мл ацетона и 10 мл воды, а затем 15 мл ацетона. После сушки при пониженном давлении получали 3,21 г 5-гидрокси-1H-имидазол-4-карбоксамид·3/4 гидрата в виде бледно-желтых кристаллов. Инфракрасный абсорбционный спектр (ATR-метод) соответствует спектру из примера 4.
[0135] Применимость данного изобретения объясняют, принимая во внимание следующие примеры исследования.
Между тем, в каждом примере исследования используют конечные соединения, получаемые из справочных примеров, примеров и сравнительных примеров.
[0136] Пример исследования 2
Осуществляли наблюдение цвета реакционной жидкости из справочных примеров 2-4 и сравнительных примеров 2 и 3. Реакционные жидкости в момент окончания взаимодействия показаны на фигурах 12-16, и результаты приведены в таблице 4.
[0137]
[0138] Реакционные жидкости из справочных примеров 2 и 3 имеют бледно-желтый цвет. Тогда как реакционная жидкость из справочного примера 4 имеет бледно-синий цвет, и реакционная жидкость из примеров сравнения 2 и 3 имеет глубокий синий цвет.
Таким образом, показано, что проводя синтез соединения A в отсутствие минеральной кислоты и в отсутствие сульфоновой кислоты, ингибируют окрашивание реакционной жидкости в синий цвет.
[0139] Пример исследования 3
Осуществляли наблюдение цвета кристаллов, получаемых в примерах 5 и 8 и сравнительных примерах 4 и 5. Результаты приведены в таблице 5.
[0140]
[0141] Реакционные жидкости из примеров 5 и 8 имеют бледно-желтый цвет. Тогда как реакционные жидкости из примеров сравнения 4 и 5 имеют синий цвет.
Таким образом, показано, что в кристаллах, получаемых в примерах 5 и 8, ингибируют окрашивание в синий цвет.
[0142] Пример исследования 4
В качестве испытываемого материала использовали соединения из примеров 9, 11 и 13, а также соединения из примеров 6 и 7.
Около 1 г испытываемого материала помещали в пакет для хранения, пакет для хранения готовили, вставляя один полиэтиленовый пакет в другой полиэтиленовый пакет, причем каждый полиэтиленовый пакет имеет толщину 0,04 мм, каждый полиэтиленовый пакет запечатывали и хранили испытываемый материал в течение 2 недель в условиях воздушной атмосферы при температуре 40°C и относительной влажности 75%. Оценивали изменение цвета кристалла до и после тестирования на основании цветового различия. Результаты приведены в таблице 6.
[0143] Колориметр: спектрофотометрический колориметр SE 2000, производства NIPPON DENSHOKU INDUSTRIES CO., LTD.
Метод измерения: отражательный метод.
[0144]
[0145] Соединение из сравнительного примера 7 демонстрирует меньшее цветовое различие по сравнению с соединением из сравнительного примера 6.
Соединения из примеров 9, 11 и 13 демонстрируют меньшее цветовое различие по сравнению с соединениями из сравнительных примеров 6 и 7.
Кроме того, при наблюдении невооруженным глазом соединение из примера 9 демонстрирует отсутствие окрашивания в синий цвет даже после хранения в течение 2 недель в условиях температуры 60°C и относительной влажности 75%.
Соединение из примера 9 до начала теста показано на фиг.10.
Соединение из примера 9 после хранения в течение 2 недель в условиях температуры 60°C и относительной влажности 75% показано на фиг.11.
Гидрат соединения A, который получали согласно способу получения по данному изобретению, имеет небольшое цветовое различие между кристаллом до хранения и кристаллом после хранения и обладает превосходной стабильностью при хранении.
[0146] Пример исследования 5
В качестве испытываемого материала использовали соединение из примера 12, а также соединение из сравнительного примера 8.
Около 1 г испытываемого материала помещали в пакет для хранения, при этом пакет для хранения готовили, вставляя один полиэтиленовый пакет в другой полиэтиленовый пакет, причем каждый полиэтиленовый пакет имеет толщину 0,04 мм, каждый полиэтиленовый пакет запечатывали и хранили испытываемый материал в течение 2 недель в условиях воздушной атмосферы при температуре 40°C и относительной влажности 75%. Оценивали изменение цвета кристалла до и после тестирования на основании цветового различия. Результаты приведены в таблице 7.
[0147] Колориметр: спектрофотометрический колориметр SE 2000 производства NIPPON DENSHOKU INDUSTRIES CO., LTD.
Метод измерения: отражательный метод.
[0148]
[0149] Соединение из сравнительного примера 8 получали, используя активированный уголь. Однако оно демонстрирует различие цветового тона. Кроме того, соединение из сравнительного примера 8 демонстрирует плохую стабильность при хранении по сравнению с соединением из примера 12. Тогда как соединение из примера 12 демонстрирует небольшое цветовое различие.
Гидрат соединения A, который получали способом получения по данному изобретению, имеет небольшое цветовое различие между кристаллом до хранения и кристаллом после хранения, и обладает превосходной стабильностью при хранении.
[0150] Пример исследования 6
Наблюдали тон (H) кристаллов из примеров 9, 11-14 в цветовой системе Манселла. Результаты приведены в таблице 8.
[0151] Колориметр: спектрофотометрический колориметр SE 2000, производства NIPPON DENSHOKU INDUSTRIES CO., LTD.
Метод измерения: отражательный метод
[0152]
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
[0153] Кристалл по настоящему изобретению имеет по меньшей мере одну из следующих характеристик: (1) он не содержит добавок, (2) его можно хранить в течение длительного периода времени, так как не происходит ухудшения качества, например, окрашивания, даже в условиях высокой температуры и высокой влажности, (3) он имеет мало примесей, (4) он легко поддается обработке, (5) его получают, используя растворитель, безопасный для человеческого организма, (6) его получают в условиях с низкими экологическими проблемами, и (7) его можно получать в крупном масштабе, и он пригоден в качестве исходного лекарственного вещества для фармацевтического средства.
[0154] Способ получения по изобретению очень подходит в качестве способа промышленного производства 5-гидрокси-1H-имидазол-4-карбоксамид·3/4 гидрата, при котором (1) он не содержит добавок, (2) цветовое различие между кристаллом до хранения и кристаллом после хранения мало, (3) он имеет превосходную стабильность при хранении, и (4) он имеет мало примесей.
[0155] Патентная заявка Японии № 2011-213501 от 28 сентября 2011 включена в настоящее описание в виде ссылки во всей своей полноте.







Изобретение относится к области органической химии, а именно к кристаллу гидрата 5-гидрокси-1H-имидазол-4-карбоксамида, где кристалл имеет дифракционные пики при углах дифракции 8,1, 12,6, 17,1, 19,3, 20,3 и 21,6°, представленных как 2θ на картине порошковой рентгеновской дифракции. Также изобретение относится к фармацевтической композиции на его основе. Технический результат: получен новый кристалл гидрата 5-гидрокси-1H-имидазол-4-карбоксамида, имеющий превосходную стабильность при хранении. 2 н.п. ф-лы, 16 ил., 8 табл., 27 пр.
1. Кристалл гидрата 5-гидрокси-1H-имидазол-4-карбоксамида, где кристалл имеет дифракционные пики при углах дифракции 8,1, 12,6, 17,1, 19,3, 20,3 и 21,6°, представленных как 2θ на картине порошковой рентгеновской дифракции.
2. Фармацевтическая композиция для ингибирования рака, содержащая эффективное количество кристалла гидрата 5-гидрокси-1H-имидазол-4-карбоксамида, где кристалл имеет дифракционные пики при углах дифракции 8,1, 12,6, 17,1, 19,3, 20,3 и 21,6°, представленных как 2θ на картине порошковой рентгеновской дифракции, и по меньшей мере один, выбранный из группы, состоящей из наполнителя, носителя или разбавителя.
| СПОСОБ ПРОИЗВОДСТВА ПИТАТЕЛЬНОЙ СМЕСИ ДЛЯ ДЕТСКОГО, ДИЕТИЧЕСКОГО ИЛИ ВНУТРИКИШЕЧНОГО ЗОНДОВОГО ПИТАНИЯ | 2001 |
|
RU2196460C1 |
| ПРОИЗВОДНЫЕ 1Н-ИМИДАЗОЛА КАК МОДУЛЯТОРЫ РЕЦЕПТОРА КАННАБИНОИДОВ СВ2 | 2006 |
|
RU2410377C2 |

